User login
Autoinflammatory diseases ‘not so rare after all,’ expert says
Not long ago, after all.

“Patients with autoinflammatory diseases are all around us, but many go several years without a diagnosis,” Dr. Dissanayake, a rheumatologist at the Autoinflammatory Disease Clinic at the Hospital for Sick Children, Toronto, said during the annual meeting of the Society for Pediatric Dermatology. “The median time to diagnosis has been estimated to be between 2.5 and 5 years. You can imagine that this type of delay can lead to significant issues, not only with quality of life but also morbidity due to unchecked inflammation that can cause organ damage, and in the most severe cases, can result in an early death.”
Effective treatment options such as biologic medications, however, can prevent these negative sequelae if the disease is recognized early. “Dermatologists are in a unique position because they will often be the first specialist to see these patients and therefore make the diagnosis early on and really alter the lives of these patients,” he said.
While it’s common to classify autoinflammatory diseases by presenting features, such as age of onset, associated symptoms, family history/ethnicity, and triggers/alleviating factors for episodes, Dr. Dissanayake prefers to classify them into one of three groups based on pathophysiology, the first being inflammasomopathies. “When activated, an inflammasome is responsible for processing cytokines from the [interleukin]-1 family from the pro form to the active form,” he explained. As a result, if there is dysregulation and overactivity of the inflammasome, there is excessive production of cytokines like IL-1 beta and IL-18 driving the disease.
Clinical characteristics include fevers and organ involvement, notably abdominal pain, nonvasculitic rashes, uveitis, arthritis, elevated white blood cell count/neutrophils, and highly elevated inflammatory markers. Potential treatments include IL-1 blockers.
The second category of autoinflammatory diseases are the interferonopathies, which are caused by overactivity of the antiviral side of the innate immune system. “For example, if you have overactivity of a sensor for a nucleic acid in your cytosol, the cell misinterprets this as a viral infection and will turn on type 1 interferon production,” said Dr. Dissanayake, who is also an assistant professor of pediatrics at the University of Toronto. “As a result, if you have dysregulation of these pathways, you will get excessive type 1 interferon that contributes to your disease manifestations.” Clinical characteristics include fevers and organ involvement, notably vasculitic rashes, interstitial lung disease, and intracranial calcifications. Inflammatory markers may not be as elevated, and autoantibodies may be present. Janus kinase inhibitors are a potential treatment, he said.
The third category of autoinflammatory diseases are the NF-kappaBopathies, which are caused by overactivity of the NF-kappaB signaling pathway. Clinical characteristics can include fevers with organ involvement that can be highly variable but may include mucocutaneous lesions or granulomatous disease as potential clues. Treatment options depend on the pathway that is involved but tumor necrosis factor blockers often play a role because of the importance of NF-KB in this signaling pathway.
From a skin perspective, most of the rashes Dr. Dissanayake and colleagues see in the rheumatology clinic consist of nonspecific dermohypodermatitis: macules, papules, patches, or plaques. The most common monogenic autoinflammatory disease is Familial Mediterranean Fever syndrome, which “commonly presents as an erysipelas-like rash of the lower extremities, typically below the knee, often over the malleolus,” he said.
Other monogenic autoinflammatory diseases with similar rashes include TNF receptor–associated periodic syndrome, Hyper-IgD syndrome, and systemic juvenile idiopathic arthritis.
Other patients present with urticarial rashes, most commonly cryopyrin-associated periodic syndrome (CAPS). “This is a neutrophilic urticaria, so it tends not to be pruritic and can actually sometimes be tender,” he said. “It also tends not to be as transient as your typical urticaria.” Urticarial rashes can also appear with NLRP12-associated autoinflammatory syndrome (familial cold autoinflammatory syndrome–2), PLCgamma2-associated antibody deficiency and immune dysregulation, and Schnitzler syndrome (monoclonal IgM gammopathy).
Patients can also present with pyogenic or pustular lesions, which can appear with pyoderma gangrenosum–related diseases, such as pyogenic arthritis, pyoderma gangrenosum, arthritis (PAPA) syndrome; pyrin-associated inflammation with neutrophilic dermatosis; deficiency of the IL-1 receptor antagonist; deficiency of IL-36 receptor antagonist; and Majeed syndrome, a mutation in the LPIN2 gene.
The mucocutaneous system can also be affected in autoinflammatory diseases, often presenting with symptoms such as periodic fever, aphthous stomatitis, and pharyngitis. Cervical adenitis syndrome is the most common autoinflammatory disease in childhood and can present with aphthous stomatitis, he said, while Behcet’s disease typically presents with oral and genital ulcers. “More recently, monogenic forms of Behcet’s disease have been described, with haploinsufficiency of A20 and RelA, which are both part of the NF-KB pathway,” he said.
Finally, the presence of vasculitic lesions often suggest interferonopathies such as STING-associated vasculopathy in infancy, proteasome-associated autoinflammatory syndrome and deficiency of adenosine deaminase 2.
Dr. Dissanayake noted that dermatologists should suspect an autoimmune disease if a patient has recurrent fevers, evidence of systemic inflammation on blood work, and if multiple organ systems are involved, especially the lungs, gut, joints, CNS system, and eyes. “Many of these patients have episodic and stereotypical attacks,” he said.
“One of the tools we use in the autoinflammatory clinic is to have patients and families keep a symptom diary where they track the dates of the various symptoms. We can review this during their appointment and try to come up with a diagnosis based on the pattern,” he said.
Since many of these diseases are due to a single gene defect, if there’s any evidence to suggest a monogenic cause, consider an autoinflammatory disease, he added. “If there’s a family history, if there’s consanguinity, or if there’s early age of onset – these may all lead you to think about monogenic autoinflammatory disease.”
During a question-and-answer session, a meeting attendee asked what type of workup he recommends when an autoinflammatory syndrome is suspected. “It partially depends on what organ systems you suspect to be involved,” Dr. Dissanayake said. “As a routine baseline, typically what we would check is CBC and differential, [erythrocyte sedimentation rate] and [C-reactive protein], and we screen for liver transaminases and creatinine to check for liver and kidney issues. A serum albumin will also tell you if the patient is hypoalbuminemic, that there’s been some chronic inflammation and they’re starting to leak the protein out. It’s good to check blood work during the flare and off the flare, to get a sense of the persistence of that inflammation.”
Dr. Dissanayake disclosed that he has received research finding from Gilead Sciences and speaker fees from Novartis.
*This story was updated on 9/20/2021.
Not long ago, after all.
“Patients with autoinflammatory diseases are all around us, but many go several years without a diagnosis,” Dr. Dissanayake, a rheumatologist at the Autoinflammatory Disease Clinic at the Hospital for Sick Children, Toronto, said during the annual meeting of the Society for Pediatric Dermatology. “The median time to diagnosis has been estimated to be between 2.5 and 5 years. You can imagine that this type of delay can lead to significant issues, not only with quality of life but also morbidity due to unchecked inflammation that can cause organ damage, and in the most severe cases, can result in an early death.”
Effective treatment options such as biologic medications, however, can prevent these negative sequelae if the disease is recognized early. “Dermatologists are in a unique position because they will often be the first specialist to see these patients and therefore make the diagnosis early on and really alter the lives of these patients,” he said.
While it’s common to classify autoinflammatory diseases by presenting features, such as age of onset, associated symptoms, family history/ethnicity, and triggers/alleviating factors for episodes, Dr. Dissanayake prefers to classify them into one of three groups based on pathophysiology, the first being inflammasomopathies. “When activated, an inflammasome is responsible for processing cytokines from the [interleukin]-1 family from the pro form to the active form,” he explained. As a result, if there is dysregulation and overactivity of the inflammasome, there is excessive production of cytokines like IL-1 beta and IL-18 driving the disease.
Clinical characteristics include fevers and organ involvement, notably abdominal pain, nonvasculitic rashes, uveitis, arthritis, elevated white blood cell count/neutrophils, and highly elevated inflammatory markers. Potential treatments include IL-1 blockers.
The second category of autoinflammatory diseases are the interferonopathies, which are caused by overactivity of the antiviral side of the innate immune system. “For example, if you have overactivity of a sensor for a nucleic acid in your cytosol, the cell misinterprets this as a viral infection and will turn on type 1 interferon production,” said Dr. Dissanayake, who is also an assistant professor of pediatrics at the University of Toronto. “As a result, if you have dysregulation of these pathways, you will get excessive type 1 interferon that contributes to your disease manifestations.” Clinical characteristics include fevers and organ involvement, notably vasculitic rashes, interstitial lung disease, and intracranial calcifications. Inflammatory markers may not be as elevated, and autoantibodies may be present. Janus kinase inhibitors are a potential treatment, he said.
The third category of autoinflammatory diseases are the NF-kappaBopathies, which are caused by overactivity of the NF-kappaB signaling pathway. Clinical characteristics can include fevers with organ involvement that can be highly variable but may include mucocutaneous lesions or granulomatous disease as potential clues. Treatment options depend on the pathway that is involved but tumor necrosis factor blockers often play a role because of the importance of NF-KB in this signaling pathway.
From a skin perspective, most of the rashes Dr. Dissanayake and colleagues see in the rheumatology clinic consist of nonspecific dermohypodermatitis: macules, papules, patches, or plaques. The most common monogenic autoinflammatory disease is Familial Mediterranean Fever syndrome, which “commonly presents as an erysipelas-like rash of the lower extremities, typically below the knee, often over the malleolus,” he said.
Other monogenic autoinflammatory diseases with similar rashes include TNF receptor–associated periodic syndrome, Hyper-IgD syndrome, and systemic juvenile idiopathic arthritis.
Other patients present with urticarial rashes, most commonly cryopyrin-associated periodic syndrome (CAPS). “This is a neutrophilic urticaria, so it tends not to be pruritic and can actually sometimes be tender,” he said. “It also tends not to be as transient as your typical urticaria.” Urticarial rashes can also appear with NLRP12-associated autoinflammatory syndrome (familial cold autoinflammatory syndrome–2), PLCgamma2-associated antibody deficiency and immune dysregulation, and Schnitzler syndrome (monoclonal IgM gammopathy).
Patients can also present with pyogenic or pustular lesions, which can appear with pyoderma gangrenosum–related diseases, such as pyogenic arthritis, pyoderma gangrenosum, arthritis (PAPA) syndrome; pyrin-associated inflammation with neutrophilic dermatosis; deficiency of the IL-1 receptor antagonist; deficiency of IL-36 receptor antagonist; and Majeed syndrome, a mutation in the LPIN2 gene.
The mucocutaneous system can also be affected in autoinflammatory diseases, often presenting with symptoms such as periodic fever, aphthous stomatitis, and pharyngitis. Cervical adenitis syndrome is the most common autoinflammatory disease in childhood and can present with aphthous stomatitis, he said, while Behcet’s disease typically presents with oral and genital ulcers. “More recently, monogenic forms of Behcet’s disease have been described, with haploinsufficiency of A20 and RelA, which are both part of the NF-KB pathway,” he said.
Finally, the presence of vasculitic lesions often suggest interferonopathies such as STING-associated vasculopathy in infancy, proteasome-associated autoinflammatory syndrome and deficiency of adenosine deaminase 2.
Dr. Dissanayake noted that dermatologists should suspect an autoimmune disease if a patient has recurrent fevers, evidence of systemic inflammation on blood work, and if multiple organ systems are involved, especially the lungs, gut, joints, CNS system, and eyes. “Many of these patients have episodic and stereotypical attacks,” he said.
“One of the tools we use in the autoinflammatory clinic is to have patients and families keep a symptom diary where they track the dates of the various symptoms. We can review this during their appointment and try to come up with a diagnosis based on the pattern,” he said.
Since many of these diseases are due to a single gene defect, if there’s any evidence to suggest a monogenic cause, consider an autoinflammatory disease, he added. “If there’s a family history, if there’s consanguinity, or if there’s early age of onset – these may all lead you to think about monogenic autoinflammatory disease.”
During a question-and-answer session, a meeting attendee asked what type of workup he recommends when an autoinflammatory syndrome is suspected. “It partially depends on what organ systems you suspect to be involved,” Dr. Dissanayake said. “As a routine baseline, typically what we would check is CBC and differential, [erythrocyte sedimentation rate] and [C-reactive protein], and we screen for liver transaminases and creatinine to check for liver and kidney issues. A serum albumin will also tell you if the patient is hypoalbuminemic, that there’s been some chronic inflammation and they’re starting to leak the protein out. It’s good to check blood work during the flare and off the flare, to get a sense of the persistence of that inflammation.”
Dr. Dissanayake disclosed that he has received research finding from Gilead Sciences and speaker fees from Novartis.
*This story was updated on 9/20/2021.
Not long ago, after all.
“Patients with autoinflammatory diseases are all around us, but many go several years without a diagnosis,” Dr. Dissanayake, a rheumatologist at the Autoinflammatory Disease Clinic at the Hospital for Sick Children, Toronto, said during the annual meeting of the Society for Pediatric Dermatology. “The median time to diagnosis has been estimated to be between 2.5 and 5 years. You can imagine that this type of delay can lead to significant issues, not only with quality of life but also morbidity due to unchecked inflammation that can cause organ damage, and in the most severe cases, can result in an early death.”
Effective treatment options such as biologic medications, however, can prevent these negative sequelae if the disease is recognized early. “Dermatologists are in a unique position because they will often be the first specialist to see these patients and therefore make the diagnosis early on and really alter the lives of these patients,” he said.
While it’s common to classify autoinflammatory diseases by presenting features, such as age of onset, associated symptoms, family history/ethnicity, and triggers/alleviating factors for episodes, Dr. Dissanayake prefers to classify them into one of three groups based on pathophysiology, the first being inflammasomopathies. “When activated, an inflammasome is responsible for processing cytokines from the [interleukin]-1 family from the pro form to the active form,” he explained. As a result, if there is dysregulation and overactivity of the inflammasome, there is excessive production of cytokines like IL-1 beta and IL-18 driving the disease.
Clinical characteristics include fevers and organ involvement, notably abdominal pain, nonvasculitic rashes, uveitis, arthritis, elevated white blood cell count/neutrophils, and highly elevated inflammatory markers. Potential treatments include IL-1 blockers.
The second category of autoinflammatory diseases are the interferonopathies, which are caused by overactivity of the antiviral side of the innate immune system. “For example, if you have overactivity of a sensor for a nucleic acid in your cytosol, the cell misinterprets this as a viral infection and will turn on type 1 interferon production,” said Dr. Dissanayake, who is also an assistant professor of pediatrics at the University of Toronto. “As a result, if you have dysregulation of these pathways, you will get excessive type 1 interferon that contributes to your disease manifestations.” Clinical characteristics include fevers and organ involvement, notably vasculitic rashes, interstitial lung disease, and intracranial calcifications. Inflammatory markers may not be as elevated, and autoantibodies may be present. Janus kinase inhibitors are a potential treatment, he said.
The third category of autoinflammatory diseases are the NF-kappaBopathies, which are caused by overactivity of the NF-kappaB signaling pathway. Clinical characteristics can include fevers with organ involvement that can be highly variable but may include mucocutaneous lesions or granulomatous disease as potential clues. Treatment options depend on the pathway that is involved but tumor necrosis factor blockers often play a role because of the importance of NF-KB in this signaling pathway.
From a skin perspective, most of the rashes Dr. Dissanayake and colleagues see in the rheumatology clinic consist of nonspecific dermohypodermatitis: macules, papules, patches, or plaques. The most common monogenic autoinflammatory disease is Familial Mediterranean Fever syndrome, which “commonly presents as an erysipelas-like rash of the lower extremities, typically below the knee, often over the malleolus,” he said.
Other monogenic autoinflammatory diseases with similar rashes include TNF receptor–associated periodic syndrome, Hyper-IgD syndrome, and systemic juvenile idiopathic arthritis.
Other patients present with urticarial rashes, most commonly cryopyrin-associated periodic syndrome (CAPS). “This is a neutrophilic urticaria, so it tends not to be pruritic and can actually sometimes be tender,” he said. “It also tends not to be as transient as your typical urticaria.” Urticarial rashes can also appear with NLRP12-associated autoinflammatory syndrome (familial cold autoinflammatory syndrome–2), PLCgamma2-associated antibody deficiency and immune dysregulation, and Schnitzler syndrome (monoclonal IgM gammopathy).
Patients can also present with pyogenic or pustular lesions, which can appear with pyoderma gangrenosum–related diseases, such as pyogenic arthritis, pyoderma gangrenosum, arthritis (PAPA) syndrome; pyrin-associated inflammation with neutrophilic dermatosis; deficiency of the IL-1 receptor antagonist; deficiency of IL-36 receptor antagonist; and Majeed syndrome, a mutation in the LPIN2 gene.
The mucocutaneous system can also be affected in autoinflammatory diseases, often presenting with symptoms such as periodic fever, aphthous stomatitis, and pharyngitis. Cervical adenitis syndrome is the most common autoinflammatory disease in childhood and can present with aphthous stomatitis, he said, while Behcet’s disease typically presents with oral and genital ulcers. “More recently, monogenic forms of Behcet’s disease have been described, with haploinsufficiency of A20 and RelA, which are both part of the NF-KB pathway,” he said.
Finally, the presence of vasculitic lesions often suggest interferonopathies such as STING-associated vasculopathy in infancy, proteasome-associated autoinflammatory syndrome and deficiency of adenosine deaminase 2.
Dr. Dissanayake noted that dermatologists should suspect an autoimmune disease if a patient has recurrent fevers, evidence of systemic inflammation on blood work, and if multiple organ systems are involved, especially the lungs, gut, joints, CNS system, and eyes. “Many of these patients have episodic and stereotypical attacks,” he said.
“One of the tools we use in the autoinflammatory clinic is to have patients and families keep a symptom diary where they track the dates of the various symptoms. We can review this during their appointment and try to come up with a diagnosis based on the pattern,” he said.
Since many of these diseases are due to a single gene defect, if there’s any evidence to suggest a monogenic cause, consider an autoinflammatory disease, he added. “If there’s a family history, if there’s consanguinity, or if there’s early age of onset – these may all lead you to think about monogenic autoinflammatory disease.”
During a question-and-answer session, a meeting attendee asked what type of workup he recommends when an autoinflammatory syndrome is suspected. “It partially depends on what organ systems you suspect to be involved,” Dr. Dissanayake said. “As a routine baseline, typically what we would check is CBC and differential, [erythrocyte sedimentation rate] and [C-reactive protein], and we screen for liver transaminases and creatinine to check for liver and kidney issues. A serum albumin will also tell you if the patient is hypoalbuminemic, that there’s been some chronic inflammation and they’re starting to leak the protein out. It’s good to check blood work during the flare and off the flare, to get a sense of the persistence of that inflammation.”
Dr. Dissanayake disclosed that he has received research finding from Gilead Sciences and speaker fees from Novartis.
*This story was updated on 9/20/2021.
FROM SPD 2021
Several uncommon skin disorders related to internal diseases reviewed
and may spawn misdiagnoses, a dermatologist told colleagues.
“Proper diagnosis can lead to an effective management in our patients,” said Jeffrey Callen, MD, professor of medicine and chief of dermatology at the University of Louisville (Ky.), who spoke at the Inaugural Symposium for Inflammatory Skin Disease.
Sarcoidosis
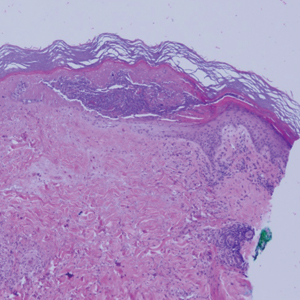
The cause of sarcoidosis, an inflammatory disease that tends to affect the lungs, “is unknown, but it’s probably an immunologic disorder,” Dr. Callen said, “and there probably is a genetic predisposition.” About 20%-25% of patients with sarcoidosis have skin lesions that are either “specific” (a biopsy that reveals a noncaseating – “naked” – granuloma) or “nonspecific” (most commonly, erythema nodosum, or EN).
The specific lesions in sarcoidosis may occur in parts of the body, such as the knees, which were injured earlier in life and may have taken in foreign bodies, Dr. Callen said. As for nonspecific lesions, about 20% of patients with EN have an acute, self-limiting form of sarcoidosis. “These patients will have bilateral hilar lymphadenopathy, anterior uveitis, and polyarthritis. It’s generally treated symptomatically because it goes away on its own.”
He cautioned colleagues to beware of indurated, infiltrative facial lesions known as lupus pernio that are commonly found on the nose. They’re more prevalent in Black patients and possibly women, who are at higher risk of manifestations outside the skin, he said. “If you have it along the nasal rim, you should look into the upper respiratory tract for involvement.”
Dr. Callen recommends an extensive workup in patients with suspected sarcoidosis, including biopsy (with the exception of EN lesions), cultures and special stains, and screening when appropriate, for disease in organs such as the eyes, lungs, heart, and kidneys.
As for treatment, “the disease is in the dermis, and some topical therapies are not highly effective,” he said. There are injections that can be given, including corticosteroids, and there are a variety of oral treatments that are all off label.” These include corticosteroids, antimalarials, allopurinol, and tetracyclines, among several others. Subcutaneous and intravenous treatments are also options, along with surgery and laser therapy to treat specific lesions.
Rosai-Dorfman disease
This rare disorder is caused by overproduction of certain white blood cells in the lymph nodes, which can cause nodular lesions. The disease most often appears in children and young adults, often Black individuals and males. It is fatal in as many as 11% of patients, justifying aggressive treatment in patients with aggressive disease, Dr. Callen said. When it’s limited to the skin, however, “nothing may need to be done.”
Dr. Callen highlighted consensus recommendations about diagnosis and treatment of Rosai-Dorfman disease published in 2018.
He also noted the existence of cutaneous Rosai-Dorfman disease, a “solitary process” that appears more commonly in females, and in people of Asian heritage, compared with White individuals. It is characterized by single, clustered or widespread lesions: They can be xanthomatous, erythematous, or red-brown papules, nodules, and plaques. They’re acneiform, pustular, giant granuloma annulare–like, subcutaneous, and vasculitis-like, he said.
While Rosai-Dorfman disease can be linked to lymphoma, hypothyroidism, and lupus erythematosus, “nothing necessarily needs to be done when it’s skin-limited since it can be self-resolving,” he noted. Other treatments include radiotherapy, cryotherapy, excision, topical and oral corticosteroids, thalidomide, and methotrexate.
The disease can be serious, and is fatal in 5% of cases. When a vital organ is threatened, Dr. Callen suggested surgery, chemotherapy, or radiation.
Erdheim-Chester disease
This disease – which is extremely rare, with just 500 cases noted before 2014 – occurs when the body overproduces macrophages. It’s most common in middle-aged people and in men, who make up 75% of cases. About a quarter of patients develop skin lesions: Red-brown to yellow nodules and xanthelasma-like indurated plaques on the eyelids, scalp, neck, trunk, and axillae, and “other cutaneous manifestations have been reported in patients,” Dr. Callen said.
The disease also frequently affects the bones, large vessels, heart, lungs, and central nervous system. Interferon-alpha is the first-line treatment, and there are several other alternative therapies, although 5-year survival (68%) is poor, and it is especially likely to be fatal in those with central nervous system involvement.
Eosinophilic fasciitis
Eosinophilic fasciitis (EF) “is a disorder of unknown etiology that causes sclerosis of the skin” without Raynaud’s phenomenon, Dr. Callen said. Look for erythema, swelling, and induration of the extremities that is accompanied by peripheral eosinophilia, and if necessary, confirm the diagnosis with full skin-to-muscle biopsy or MRI.
There are many possible triggers, including strenuous exercise, initiation with hemodialysis, radiation therapy and burns, and graft-versus-host disease. Other potential causes include exposure to medications such as statins, phenytoin, ramipril, subcutaneous heparin, and immune checkpoint inhibitor therapy. The disorder is also linked to autoimmune and hematologic disorders.
Dr. Callen, who highlighted EF guidelines published in 2018, said treatments include physical therapy, prednisone, methotrexate, mycophenolate, and hydroxychloroquine.
Metastatic Crohn’s disease
This is a rare granulomatous inflammation of skin that often affects the genitals, especially in children. It is noncontiguous with the GI tract, and severity of skin involvement does not always parallel the severity of the disease in the GI tract, Dr. Callen said. However, the condition can occur before or simultaneously with the development of GI disease, or after GI surgery.
He highlighted a review of metastatic Crohn’s disease, published in 2014, and noted that there are multiple treatments, including systemic corticosteroids, tumor necrosis factor–alpha inhibitors, and topical therapies.
Dr. Callen reported no relevant disclosures.
and may spawn misdiagnoses, a dermatologist told colleagues.
“Proper diagnosis can lead to an effective management in our patients,” said Jeffrey Callen, MD, professor of medicine and chief of dermatology at the University of Louisville (Ky.), who spoke at the Inaugural Symposium for Inflammatory Skin Disease.
Sarcoidosis
The cause of sarcoidosis, an inflammatory disease that tends to affect the lungs, “is unknown, but it’s probably an immunologic disorder,” Dr. Callen said, “and there probably is a genetic predisposition.” About 20%-25% of patients with sarcoidosis have skin lesions that are either “specific” (a biopsy that reveals a noncaseating – “naked” – granuloma) or “nonspecific” (most commonly, erythema nodosum, or EN).
The specific lesions in sarcoidosis may occur in parts of the body, such as the knees, which were injured earlier in life and may have taken in foreign bodies, Dr. Callen said. As for nonspecific lesions, about 20% of patients with EN have an acute, self-limiting form of sarcoidosis. “These patients will have bilateral hilar lymphadenopathy, anterior uveitis, and polyarthritis. It’s generally treated symptomatically because it goes away on its own.”
He cautioned colleagues to beware of indurated, infiltrative facial lesions known as lupus pernio that are commonly found on the nose. They’re more prevalent in Black patients and possibly women, who are at higher risk of manifestations outside the skin, he said. “If you have it along the nasal rim, you should look into the upper respiratory tract for involvement.”
Dr. Callen recommends an extensive workup in patients with suspected sarcoidosis, including biopsy (with the exception of EN lesions), cultures and special stains, and screening when appropriate, for disease in organs such as the eyes, lungs, heart, and kidneys.
As for treatment, “the disease is in the dermis, and some topical therapies are not highly effective,” he said. There are injections that can be given, including corticosteroids, and there are a variety of oral treatments that are all off label.” These include corticosteroids, antimalarials, allopurinol, and tetracyclines, among several others. Subcutaneous and intravenous treatments are also options, along with surgery and laser therapy to treat specific lesions.
Rosai-Dorfman disease
This rare disorder is caused by overproduction of certain white blood cells in the lymph nodes, which can cause nodular lesions. The disease most often appears in children and young adults, often Black individuals and males. It is fatal in as many as 11% of patients, justifying aggressive treatment in patients with aggressive disease, Dr. Callen said. When it’s limited to the skin, however, “nothing may need to be done.”
Dr. Callen highlighted consensus recommendations about diagnosis and treatment of Rosai-Dorfman disease published in 2018.
He also noted the existence of cutaneous Rosai-Dorfman disease, a “solitary process” that appears more commonly in females, and in people of Asian heritage, compared with White individuals. It is characterized by single, clustered or widespread lesions: They can be xanthomatous, erythematous, or red-brown papules, nodules, and plaques. They’re acneiform, pustular, giant granuloma annulare–like, subcutaneous, and vasculitis-like, he said.
While Rosai-Dorfman disease can be linked to lymphoma, hypothyroidism, and lupus erythematosus, “nothing necessarily needs to be done when it’s skin-limited since it can be self-resolving,” he noted. Other treatments include radiotherapy, cryotherapy, excision, topical and oral corticosteroids, thalidomide, and methotrexate.
The disease can be serious, and is fatal in 5% of cases. When a vital organ is threatened, Dr. Callen suggested surgery, chemotherapy, or radiation.
Erdheim-Chester disease
This disease – which is extremely rare, with just 500 cases noted before 2014 – occurs when the body overproduces macrophages. It’s most common in middle-aged people and in men, who make up 75% of cases. About a quarter of patients develop skin lesions: Red-brown to yellow nodules and xanthelasma-like indurated plaques on the eyelids, scalp, neck, trunk, and axillae, and “other cutaneous manifestations have been reported in patients,” Dr. Callen said.
The disease also frequently affects the bones, large vessels, heart, lungs, and central nervous system. Interferon-alpha is the first-line treatment, and there are several other alternative therapies, although 5-year survival (68%) is poor, and it is especially likely to be fatal in those with central nervous system involvement.
Eosinophilic fasciitis
Eosinophilic fasciitis (EF) “is a disorder of unknown etiology that causes sclerosis of the skin” without Raynaud’s phenomenon, Dr. Callen said. Look for erythema, swelling, and induration of the extremities that is accompanied by peripheral eosinophilia, and if necessary, confirm the diagnosis with full skin-to-muscle biopsy or MRI.
There are many possible triggers, including strenuous exercise, initiation with hemodialysis, radiation therapy and burns, and graft-versus-host disease. Other potential causes include exposure to medications such as statins, phenytoin, ramipril, subcutaneous heparin, and immune checkpoint inhibitor therapy. The disorder is also linked to autoimmune and hematologic disorders.
Dr. Callen, who highlighted EF guidelines published in 2018, said treatments include physical therapy, prednisone, methotrexate, mycophenolate, and hydroxychloroquine.
Metastatic Crohn’s disease
This is a rare granulomatous inflammation of skin that often affects the genitals, especially in children. It is noncontiguous with the GI tract, and severity of skin involvement does not always parallel the severity of the disease in the GI tract, Dr. Callen said. However, the condition can occur before or simultaneously with the development of GI disease, or after GI surgery.
He highlighted a review of metastatic Crohn’s disease, published in 2014, and noted that there are multiple treatments, including systemic corticosteroids, tumor necrosis factor–alpha inhibitors, and topical therapies.
Dr. Callen reported no relevant disclosures.
and may spawn misdiagnoses, a dermatologist told colleagues.
“Proper diagnosis can lead to an effective management in our patients,” said Jeffrey Callen, MD, professor of medicine and chief of dermatology at the University of Louisville (Ky.), who spoke at the Inaugural Symposium for Inflammatory Skin Disease.
Sarcoidosis
The cause of sarcoidosis, an inflammatory disease that tends to affect the lungs, “is unknown, but it’s probably an immunologic disorder,” Dr. Callen said, “and there probably is a genetic predisposition.” About 20%-25% of patients with sarcoidosis have skin lesions that are either “specific” (a biopsy that reveals a noncaseating – “naked” – granuloma) or “nonspecific” (most commonly, erythema nodosum, or EN).
The specific lesions in sarcoidosis may occur in parts of the body, such as the knees, which were injured earlier in life and may have taken in foreign bodies, Dr. Callen said. As for nonspecific lesions, about 20% of patients with EN have an acute, self-limiting form of sarcoidosis. “These patients will have bilateral hilar lymphadenopathy, anterior uveitis, and polyarthritis. It’s generally treated symptomatically because it goes away on its own.”
He cautioned colleagues to beware of indurated, infiltrative facial lesions known as lupus pernio that are commonly found on the nose. They’re more prevalent in Black patients and possibly women, who are at higher risk of manifestations outside the skin, he said. “If you have it along the nasal rim, you should look into the upper respiratory tract for involvement.”
Dr. Callen recommends an extensive workup in patients with suspected sarcoidosis, including biopsy (with the exception of EN lesions), cultures and special stains, and screening when appropriate, for disease in organs such as the eyes, lungs, heart, and kidneys.
As for treatment, “the disease is in the dermis, and some topical therapies are not highly effective,” he said. There are injections that can be given, including corticosteroids, and there are a variety of oral treatments that are all off label.” These include corticosteroids, antimalarials, allopurinol, and tetracyclines, among several others. Subcutaneous and intravenous treatments are also options, along with surgery and laser therapy to treat specific lesions.
Rosai-Dorfman disease
This rare disorder is caused by overproduction of certain white blood cells in the lymph nodes, which can cause nodular lesions. The disease most often appears in children and young adults, often Black individuals and males. It is fatal in as many as 11% of patients, justifying aggressive treatment in patients with aggressive disease, Dr. Callen said. When it’s limited to the skin, however, “nothing may need to be done.”
Dr. Callen highlighted consensus recommendations about diagnosis and treatment of Rosai-Dorfman disease published in 2018.
He also noted the existence of cutaneous Rosai-Dorfman disease, a “solitary process” that appears more commonly in females, and in people of Asian heritage, compared with White individuals. It is characterized by single, clustered or widespread lesions: They can be xanthomatous, erythematous, or red-brown papules, nodules, and plaques. They’re acneiform, pustular, giant granuloma annulare–like, subcutaneous, and vasculitis-like, he said.
While Rosai-Dorfman disease can be linked to lymphoma, hypothyroidism, and lupus erythematosus, “nothing necessarily needs to be done when it’s skin-limited since it can be self-resolving,” he noted. Other treatments include radiotherapy, cryotherapy, excision, topical and oral corticosteroids, thalidomide, and methotrexate.
The disease can be serious, and is fatal in 5% of cases. When a vital organ is threatened, Dr. Callen suggested surgery, chemotherapy, or radiation.
Erdheim-Chester disease
This disease – which is extremely rare, with just 500 cases noted before 2014 – occurs when the body overproduces macrophages. It’s most common in middle-aged people and in men, who make up 75% of cases. About a quarter of patients develop skin lesions: Red-brown to yellow nodules and xanthelasma-like indurated plaques on the eyelids, scalp, neck, trunk, and axillae, and “other cutaneous manifestations have been reported in patients,” Dr. Callen said.
The disease also frequently affects the bones, large vessels, heart, lungs, and central nervous system. Interferon-alpha is the first-line treatment, and there are several other alternative therapies, although 5-year survival (68%) is poor, and it is especially likely to be fatal in those with central nervous system involvement.
Eosinophilic fasciitis
Eosinophilic fasciitis (EF) “is a disorder of unknown etiology that causes sclerosis of the skin” without Raynaud’s phenomenon, Dr. Callen said. Look for erythema, swelling, and induration of the extremities that is accompanied by peripheral eosinophilia, and if necessary, confirm the diagnosis with full skin-to-muscle biopsy or MRI.
There are many possible triggers, including strenuous exercise, initiation with hemodialysis, radiation therapy and burns, and graft-versus-host disease. Other potential causes include exposure to medications such as statins, phenytoin, ramipril, subcutaneous heparin, and immune checkpoint inhibitor therapy. The disorder is also linked to autoimmune and hematologic disorders.
Dr. Callen, who highlighted EF guidelines published in 2018, said treatments include physical therapy, prednisone, methotrexate, mycophenolate, and hydroxychloroquine.
Metastatic Crohn’s disease
This is a rare granulomatous inflammation of skin that often affects the genitals, especially in children. It is noncontiguous with the GI tract, and severity of skin involvement does not always parallel the severity of the disease in the GI tract, Dr. Callen said. However, the condition can occur before or simultaneously with the development of GI disease, or after GI surgery.
He highlighted a review of metastatic Crohn’s disease, published in 2014, and noted that there are multiple treatments, including systemic corticosteroids, tumor necrosis factor–alpha inhibitors, and topical therapies.
Dr. Callen reported no relevant disclosures.
FROM SISD 2021
Novel gene therapy ‘reprograms’ cells to reverse neurologic deficits in children with rare disease
An experimental gene therapy produced marked clinical improvement in children with aromatic L-amino acid decarboxylase (AADC) deficiency, a rare genetic disorder that affects the synthesis of key neurotransmitters to cause severe developmental and motor disability.

In an article published July 12, 2021, in Nature Communications, a group of researchers based at the University of California, San Francisco, and Ohio State University, Columbus, described results from seven children ages 4-9 with AADC deficiency who underwent a novel form of surgery to deliver a viral vector expressing the human AADC gene to the midbrain.
Previous trials of this gene therapy in children with AADC deficiency targeted a different region of the brain, the putamen, with only slight clinical improvement. Here, investigators chose two midbrain regions – the substantia nigra pars compacta and the ventral tegmental area – in the hope of restoring healthy AADC enzyme activity in those neurons.
The study’s corresponding author, Krystof Bankiewicz, MD, PhD, professor and vice chair of research at Ohio State University, director of the Brain Health and Performance Center at Ohio State University, and professor emeritus and vice chair for research at UCSF, said in an interview that the brain regions chosen for this trial resulted from years of efforts to identify an ideal target in this disease.
“This particular vector undergoes axonal transport,” he said. “If you inject it into specific regions of the brain it will be transported into the terminals [of the nerve fibers]. And by looking at the imaging of these patients, we found that they still have the wiring in the brain that’s so critical. So we decided to aim at a much more difficult target, going directly to the source of the problem, which is the substantia nigra and the ventral tegmental area. This targets two critical pathways in the brain: one that drives motor responses and another that controls emotions.”
‘Surprising’ improvement seen
The children in the study – four girls and three boys – underwent surgery from 2016 to the end of 2018, and were divided into two dose cohorts, with one receiving three times the amount of vector as the other. Both groups, however, saw similar levels of improvement.
All but one child saw complete resolution of a hallmark symptom of the disease – oculogyric crises, or prolonged spasms of muscles controlling eye movement – within 3 months of surgery. Of the children followed at least 18 months, six attained head control within a year, two became able to eat and drink by mouth, and four gained the ability to sit up unaided in that time. At 18 months one child had learned to speak 50 words using an augmentative communication device.
One child died unexpectedly 7 months after the procedure, Dr. Bankiewicz said in an interview. This death appeared to be caused by cardiac complications of his disease, Dr. Bankiewicz said, which are common in AADC deficiency.
While the investigators are now looking at delivering the AADC gene therapy in younger children – who were excluded from this trial because of safety concerns surrounding the complex procedure – investigators were surprised by the level of improvement seen in older children.
“We initially didn’t believe – at least not all of us – that we could actually make an impact in the older patients, and that is not the case,” said Dr. Bankiewicz, who has since used the same gene therapy on a compassionate-use basis in Europe and seen durable clinical improvement in patients as old as 26. “The fact that we saw a response in that patient tells us something about how incredibly plastic the brain is.”
While the new study does not detail improvements in the children’s social and emotional well-being, Dr. Bankiewicz said these, too, were pronounced. “Kids fall into oculogyric crises in stress-inducing situation. They might be in a stroller being taken for a walk, and something in the environment would stress them. Sometimes they had to be kept in a dark room isolated from stress.” Following the gene therapy, “they’re laughing, they’re social, they can interact with their environment. It’s really touching to see them able to develop a bond now with their caregivers.”
Implication for other disorders
Dr. Bankiewicz and colleagues have previously used the same gene to boost AADC activity in patients with Parkinson’s disease. The group is also in trials to deliver a neuroprotective gene to the brains of people with early-stage Alzheimer’s disease, and a gene-silencing therapy in patients with Huntington’s disease. They will also continue recruiting pediatric patients for trials of the AADC gene therapy.
“We have been developing a method for safely treating younger children, so now we will go to 3 years old and maybe even below,” Dr. Bankiewicz said. “Earlier is probably better, but for technical and safety considerations we needed to be conservative first. It is hugely stressful to go into very sick patients with that type of therapy in that part of the brain. We had to get it right the first time, and it looks like we did.”
The study was funded by the National Institutes of Health, the AADC Research Trust, the Pediatric Neurotransmitter Disease Association, and Ohio State University, with materials and technical support donated by ClearPoint Neuro. Several coauthors disclosed financial relationships with producers of diagnostic tests or biotechnology firms. Dr. Bankiewicz is a founder and shareholder of Brain Neurotherapy Bio, a company that develops gene therapies for Parkinson’s and other diseases.
An experimental gene therapy produced marked clinical improvement in children with aromatic L-amino acid decarboxylase (AADC) deficiency, a rare genetic disorder that affects the synthesis of key neurotransmitters to cause severe developmental and motor disability.
In an article published July 12, 2021, in Nature Communications, a group of researchers based at the University of California, San Francisco, and Ohio State University, Columbus, described results from seven children ages 4-9 with AADC deficiency who underwent a novel form of surgery to deliver a viral vector expressing the human AADC gene to the midbrain.
Previous trials of this gene therapy in children with AADC deficiency targeted a different region of the brain, the putamen, with only slight clinical improvement. Here, investigators chose two midbrain regions – the substantia nigra pars compacta and the ventral tegmental area – in the hope of restoring healthy AADC enzyme activity in those neurons.
The study’s corresponding author, Krystof Bankiewicz, MD, PhD, professor and vice chair of research at Ohio State University, director of the Brain Health and Performance Center at Ohio State University, and professor emeritus and vice chair for research at UCSF, said in an interview that the brain regions chosen for this trial resulted from years of efforts to identify an ideal target in this disease.
“This particular vector undergoes axonal transport,” he said. “If you inject it into specific regions of the brain it will be transported into the terminals [of the nerve fibers]. And by looking at the imaging of these patients, we found that they still have the wiring in the brain that’s so critical. So we decided to aim at a much more difficult target, going directly to the source of the problem, which is the substantia nigra and the ventral tegmental area. This targets two critical pathways in the brain: one that drives motor responses and another that controls emotions.”
‘Surprising’ improvement seen
The children in the study – four girls and three boys – underwent surgery from 2016 to the end of 2018, and were divided into two dose cohorts, with one receiving three times the amount of vector as the other. Both groups, however, saw similar levels of improvement.
All but one child saw complete resolution of a hallmark symptom of the disease – oculogyric crises, or prolonged spasms of muscles controlling eye movement – within 3 months of surgery. Of the children followed at least 18 months, six attained head control within a year, two became able to eat and drink by mouth, and four gained the ability to sit up unaided in that time. At 18 months one child had learned to speak 50 words using an augmentative communication device.
One child died unexpectedly 7 months after the procedure, Dr. Bankiewicz said in an interview. This death appeared to be caused by cardiac complications of his disease, Dr. Bankiewicz said, which are common in AADC deficiency.
While the investigators are now looking at delivering the AADC gene therapy in younger children – who were excluded from this trial because of safety concerns surrounding the complex procedure – investigators were surprised by the level of improvement seen in older children.
“We initially didn’t believe – at least not all of us – that we could actually make an impact in the older patients, and that is not the case,” said Dr. Bankiewicz, who has since used the same gene therapy on a compassionate-use basis in Europe and seen durable clinical improvement in patients as old as 26. “The fact that we saw a response in that patient tells us something about how incredibly plastic the brain is.”
While the new study does not detail improvements in the children’s social and emotional well-being, Dr. Bankiewicz said these, too, were pronounced. “Kids fall into oculogyric crises in stress-inducing situation. They might be in a stroller being taken for a walk, and something in the environment would stress them. Sometimes they had to be kept in a dark room isolated from stress.” Following the gene therapy, “they’re laughing, they’re social, they can interact with their environment. It’s really touching to see them able to develop a bond now with their caregivers.”
Implication for other disorders
Dr. Bankiewicz and colleagues have previously used the same gene to boost AADC activity in patients with Parkinson’s disease. The group is also in trials to deliver a neuroprotective gene to the brains of people with early-stage Alzheimer’s disease, and a gene-silencing therapy in patients with Huntington’s disease. They will also continue recruiting pediatric patients for trials of the AADC gene therapy.
“We have been developing a method for safely treating younger children, so now we will go to 3 years old and maybe even below,” Dr. Bankiewicz said. “Earlier is probably better, but for technical and safety considerations we needed to be conservative first. It is hugely stressful to go into very sick patients with that type of therapy in that part of the brain. We had to get it right the first time, and it looks like we did.”
The study was funded by the National Institutes of Health, the AADC Research Trust, the Pediatric Neurotransmitter Disease Association, and Ohio State University, with materials and technical support donated by ClearPoint Neuro. Several coauthors disclosed financial relationships with producers of diagnostic tests or biotechnology firms. Dr. Bankiewicz is a founder and shareholder of Brain Neurotherapy Bio, a company that develops gene therapies for Parkinson’s and other diseases.
An experimental gene therapy produced marked clinical improvement in children with aromatic L-amino acid decarboxylase (AADC) deficiency, a rare genetic disorder that affects the synthesis of key neurotransmitters to cause severe developmental and motor disability.
In an article published July 12, 2021, in Nature Communications, a group of researchers based at the University of California, San Francisco, and Ohio State University, Columbus, described results from seven children ages 4-9 with AADC deficiency who underwent a novel form of surgery to deliver a viral vector expressing the human AADC gene to the midbrain.
Previous trials of this gene therapy in children with AADC deficiency targeted a different region of the brain, the putamen, with only slight clinical improvement. Here, investigators chose two midbrain regions – the substantia nigra pars compacta and the ventral tegmental area – in the hope of restoring healthy AADC enzyme activity in those neurons.
The study’s corresponding author, Krystof Bankiewicz, MD, PhD, professor and vice chair of research at Ohio State University, director of the Brain Health and Performance Center at Ohio State University, and professor emeritus and vice chair for research at UCSF, said in an interview that the brain regions chosen for this trial resulted from years of efforts to identify an ideal target in this disease.
“This particular vector undergoes axonal transport,” he said. “If you inject it into specific regions of the brain it will be transported into the terminals [of the nerve fibers]. And by looking at the imaging of these patients, we found that they still have the wiring in the brain that’s so critical. So we decided to aim at a much more difficult target, going directly to the source of the problem, which is the substantia nigra and the ventral tegmental area. This targets two critical pathways in the brain: one that drives motor responses and another that controls emotions.”
‘Surprising’ improvement seen
The children in the study – four girls and three boys – underwent surgery from 2016 to the end of 2018, and were divided into two dose cohorts, with one receiving three times the amount of vector as the other. Both groups, however, saw similar levels of improvement.
All but one child saw complete resolution of a hallmark symptom of the disease – oculogyric crises, or prolonged spasms of muscles controlling eye movement – within 3 months of surgery. Of the children followed at least 18 months, six attained head control within a year, two became able to eat and drink by mouth, and four gained the ability to sit up unaided in that time. At 18 months one child had learned to speak 50 words using an augmentative communication device.
One child died unexpectedly 7 months after the procedure, Dr. Bankiewicz said in an interview. This death appeared to be caused by cardiac complications of his disease, Dr. Bankiewicz said, which are common in AADC deficiency.
While the investigators are now looking at delivering the AADC gene therapy in younger children – who were excluded from this trial because of safety concerns surrounding the complex procedure – investigators were surprised by the level of improvement seen in older children.
“We initially didn’t believe – at least not all of us – that we could actually make an impact in the older patients, and that is not the case,” said Dr. Bankiewicz, who has since used the same gene therapy on a compassionate-use basis in Europe and seen durable clinical improvement in patients as old as 26. “The fact that we saw a response in that patient tells us something about how incredibly plastic the brain is.”
While the new study does not detail improvements in the children’s social and emotional well-being, Dr. Bankiewicz said these, too, were pronounced. “Kids fall into oculogyric crises in stress-inducing situation. They might be in a stroller being taken for a walk, and something in the environment would stress them. Sometimes they had to be kept in a dark room isolated from stress.” Following the gene therapy, “they’re laughing, they’re social, they can interact with their environment. It’s really touching to see them able to develop a bond now with their caregivers.”
Implication for other disorders
Dr. Bankiewicz and colleagues have previously used the same gene to boost AADC activity in patients with Parkinson’s disease. The group is also in trials to deliver a neuroprotective gene to the brains of people with early-stage Alzheimer’s disease, and a gene-silencing therapy in patients with Huntington’s disease. They will also continue recruiting pediatric patients for trials of the AADC gene therapy.
“We have been developing a method for safely treating younger children, so now we will go to 3 years old and maybe even below,” Dr. Bankiewicz said. “Earlier is probably better, but for technical and safety considerations we needed to be conservative first. It is hugely stressful to go into very sick patients with that type of therapy in that part of the brain. We had to get it right the first time, and it looks like we did.”
The study was funded by the National Institutes of Health, the AADC Research Trust, the Pediatric Neurotransmitter Disease Association, and Ohio State University, with materials and technical support donated by ClearPoint Neuro. Several coauthors disclosed financial relationships with producers of diagnostic tests or biotechnology firms. Dr. Bankiewicz is a founder and shareholder of Brain Neurotherapy Bio, a company that develops gene therapies for Parkinson’s and other diseases.
FROM NATURE COMMUNICATIONS
FDA approves intravenous immunoglobulin for dermatomyositis
, according to a statement from manufacturer Octapharma USA.

Dermatomyositis is a rare, idiopathic autoimmune disorder that affects approximately 10 out of every million people in the United States, mainly adults in their late 40s to early 60s, according to the company, but children aged 5-15 years can be affected. The disease is characterized by skin rashes, chronic muscle inflammation, progressive muscle weakness, and risk for mortality that is three times higher than for the general population.
There are no previously approved treatments for dermatomyositis prior to Octagam 10%, which also is indicated for chronic immune thrombocytopenic purpura in adults.
The approval for dermatomyositis was based on the results of a phase 3 randomized, double-blind, placebo-controlled clinical trial (the ProDERM trial) that included 95 adult patients at 36 sites worldwide, with 17 sites in the United States. In the trial, 78.7% of patients with dermatomyositis who were randomized to receive 2 g/kg of Octagam 10% every 4 weeks showed response at 16 weeks, compared with 43.8% of patients who received placebo. Response was based on the 2016 American College of Rheumatology/European Alliance of Associations for Rheumatology myositis response criteria. Placebo patients who switched to intravenous immunoglobulin (IVIG) during a trial extension had response rates at week 40 similar to the original patients at week 16.
“The study gives clinicians much more confidence in the efficacy and safety of intravenous immunoglobulin and provides valuable information about what type of patient is best suited for the treatment,” Rohit Aggarwal, MD, medical director of the Arthritis and Autoimmunity Center at the University of Pittsburgh and a member of the ProDERM study Steering Committee, said in the Octapharma statement.
Safety and tolerability were similar to profiles seen with other IVIG medications, according to the statement. The medication does carry a boxed warning from its chronic ITP approval, cautioning about the potential for thrombosis, renal dysfunction, and acute renal failure.
The most common adverse reactions reported by dermatomyositis patients in the ProDERM trial were headache, fever, nausea, vomiting, increased blood pressure, chills, musculoskeletal pain, increased heart rate, dyspnea, and reactions at the infusion sites.
Read the full prescribing information here.
, according to a statement from manufacturer Octapharma USA.
Dermatomyositis is a rare, idiopathic autoimmune disorder that affects approximately 10 out of every million people in the United States, mainly adults in their late 40s to early 60s, according to the company, but children aged 5-15 years can be affected. The disease is characterized by skin rashes, chronic muscle inflammation, progressive muscle weakness, and risk for mortality that is three times higher than for the general population.
There are no previously approved treatments for dermatomyositis prior to Octagam 10%, which also is indicated for chronic immune thrombocytopenic purpura in adults.
The approval for dermatomyositis was based on the results of a phase 3 randomized, double-blind, placebo-controlled clinical trial (the ProDERM trial) that included 95 adult patients at 36 sites worldwide, with 17 sites in the United States. In the trial, 78.7% of patients with dermatomyositis who were randomized to receive 2 g/kg of Octagam 10% every 4 weeks showed response at 16 weeks, compared with 43.8% of patients who received placebo. Response was based on the 2016 American College of Rheumatology/European Alliance of Associations for Rheumatology myositis response criteria. Placebo patients who switched to intravenous immunoglobulin (IVIG) during a trial extension had response rates at week 40 similar to the original patients at week 16.
“The study gives clinicians much more confidence in the efficacy and safety of intravenous immunoglobulin and provides valuable information about what type of patient is best suited for the treatment,” Rohit Aggarwal, MD, medical director of the Arthritis and Autoimmunity Center at the University of Pittsburgh and a member of the ProDERM study Steering Committee, said in the Octapharma statement.
Safety and tolerability were similar to profiles seen with other IVIG medications, according to the statement. The medication does carry a boxed warning from its chronic ITP approval, cautioning about the potential for thrombosis, renal dysfunction, and acute renal failure.
The most common adverse reactions reported by dermatomyositis patients in the ProDERM trial were headache, fever, nausea, vomiting, increased blood pressure, chills, musculoskeletal pain, increased heart rate, dyspnea, and reactions at the infusion sites.
Read the full prescribing information here.
, according to a statement from manufacturer Octapharma USA.
Dermatomyositis is a rare, idiopathic autoimmune disorder that affects approximately 10 out of every million people in the United States, mainly adults in their late 40s to early 60s, according to the company, but children aged 5-15 years can be affected. The disease is characterized by skin rashes, chronic muscle inflammation, progressive muscle weakness, and risk for mortality that is three times higher than for the general population.
There are no previously approved treatments for dermatomyositis prior to Octagam 10%, which also is indicated for chronic immune thrombocytopenic purpura in adults.
The approval for dermatomyositis was based on the results of a phase 3 randomized, double-blind, placebo-controlled clinical trial (the ProDERM trial) that included 95 adult patients at 36 sites worldwide, with 17 sites in the United States. In the trial, 78.7% of patients with dermatomyositis who were randomized to receive 2 g/kg of Octagam 10% every 4 weeks showed response at 16 weeks, compared with 43.8% of patients who received placebo. Response was based on the 2016 American College of Rheumatology/European Alliance of Associations for Rheumatology myositis response criteria. Placebo patients who switched to intravenous immunoglobulin (IVIG) during a trial extension had response rates at week 40 similar to the original patients at week 16.
“The study gives clinicians much more confidence in the efficacy and safety of intravenous immunoglobulin and provides valuable information about what type of patient is best suited for the treatment,” Rohit Aggarwal, MD, medical director of the Arthritis and Autoimmunity Center at the University of Pittsburgh and a member of the ProDERM study Steering Committee, said in the Octapharma statement.
Safety and tolerability were similar to profiles seen with other IVIG medications, according to the statement. The medication does carry a boxed warning from its chronic ITP approval, cautioning about the potential for thrombosis, renal dysfunction, and acute renal failure.
The most common adverse reactions reported by dermatomyositis patients in the ProDERM trial were headache, fever, nausea, vomiting, increased blood pressure, chills, musculoskeletal pain, increased heart rate, dyspnea, and reactions at the infusion sites.
Read the full prescribing information here.
27-year-old woman • postpartum seizures • PTSD • history of depression • Dx?
THE CASE
A 27-year-old woman presented to the family medicine clinic to establish care for a recent onset of seizures, for which she had previously been admitted, 4 months after delivering her first child. Her pregnancy was complicated by type 1 diabetes and poor glycemic control. Labor was induced at 37 weeks; however, vaginal delivery was impeded by arrest of dilation. An emergency cesarean section was performed under general anesthesia, resulting in a healthy newborn male.
Six weeks after giving birth, the patient was started on sertraline 50 mg/d for postpartum depression. Her history was significant for depression 8 years prior that was controlled with psychotherapy, and treated prior to coming to our clinic. She had not experienced any depressive symptoms during pregnancy.
Three months postpartum, she was hospitalized for recurrent syncopal episodes. They lasted about 2 minutes, with prodromal generalized weakness followed by loss of consciousness. There was no post-event confusion, tongue-biting, or incontinence. Physical exam, electroencephalogram (EEG), echocardiogram, and magnetic resonance imaging of the head and neck demonstrated no acute findings.
These episodes escalated in frequency weeks after they began, involving as many as 40 daily attacks, some of which lasted up to 45 minutes. During these events, the patient was nonresponsive but reported reliving the delivery of her child. Upon initial consultation with Neurology, no cause was found, and she was advised to wear a helmet, stop driving, and refrain from carrying her son. No antiepileptic medications were initiated because there were no EEG findings that supported seizure, and her mood had not improved, despite an increase in sertraline dosage, a switch to citalopram, and the addition of bupropion. She described anxiety, nightmares, and intrusive thoughts during psychotherapy sessions. Her psychiatrist gave her an additional diagnosis of posttraumatic stress disorder (PTSD) secondary to her delivery. The family medicine clinic assisted the patient and her family throughout her care by functioning as a home base for her.
Eight months following initial symptoms, repeat evaluation with a video-EEG revealed no evidence of EEG changes during seizure-like activity.
THE DIAGNOSIS
The patient was given a diagnosis of
DISCUSSION
With a prevalence of 5% to 10% and 20% to 40% in outpatient and inpatient epilepsy clinics respectively, PNES events have become of increasing interest to physicians.2 There are few cases of PNES in women during pregnancy reported in the literature.3,4 This is the first case report of PNES with postpartum onset.
Continue to: Epilepsy vs psychogenic nonepileptic seizures
Epilepsy vs psychogenic nonepileptic seizures
PNES episodes appear similar to epileptic seizures, but without a definitive neurobiologic source.2,3 However, recent literature suggests the root cause may be found in abnormalities in neurologic networks, such as dysfunction of frontal and parietal lobe connectivity and increased communication from emotional centers of the brain.2,5 There are no typical pathognomonic symptoms of PNES, leading to diagnostic difficulty.2 A definitive diagnosis may be made when a patient experiences seizures without EEG abnormalities.2 Further diagnostic brain imaging is unnecessary.
Trauma may be the underlying cause
A predominance of PNES in both women and young adults, with no definitive associated factors, has been reported in the literature.2 Studies suggest childhood sexual abuse, physical abuse, traumatic brain injury, and health-related trauma, such as distressing medical experiences and surgeries, may be risk factors, while depression, misdiagnosis, and mistreatment can heighten seizure activity.2,3
Treatment requires a multidisciplinary team
Effective management of PNES requires collaboration between the primary care physician, neurologist, psychiatrist, and psychotherapist, with an emphasis on evaluation and control of the underlying trigger(s).3 Randomized controlled trials have demonstrated the efficacy of cognitive behavioral therapy (CBT), supportive care, and patient education in reducing seizure frequency at the 6-month follow-up.3,6 Additional studies have reported the best prognostic factor in PNES management is patient employment of an internal locus of control—the patient’s belief that they control life events.7,8 Case series suggest electroconvulsive therapy (ECT) is an effective alternative mood stabilization and seizure reduction therapy when tolerated.9
Our patient tried several combinations of treatment to manage PNES and comorbid psychiatric conditions, including CBT, antidepressants, and anxiolytics. After about 5 treatment failures, she pursued ECT for treatment-resistant depression and PNES frequency reduction but failed to tolerate therapy. Currently, her PNES has been reduced to 1 to 2 weekly episodes with a 200 mg/d dose of lamotrigine as a mood stabilizer combined with CBT.
THE TAKEAWAY
Providers should investigate a patient’s history and psychologic disposition when the patient presents with seizure-like behavior without a neurobiologic source or with a negative video-EEG study. A history of depression, traumatic experience, PTSD, or other psychosocial triggers must be noted early to prevent a delay in treatment when PNES is part of the differential. Due to a delayed diagnosis of PNES in our patient, she went without full treatment for almost 12 months and experienced worsening episodes. The primary care physician plays an integral role in early identification and intervention through anticipatory guidance, initial work-up, and support for patients with suspected PNES (TABLE).

CORRESPONDENCE
Karim Hanna, MD, 13330 USF Laurel Drive, Tampa, FL; [email protected]
1. LaFrance WC Jr, Baker GA, Duncan R, et al. Minimum requirements for the diagnosis of psychogenic nonepileptic seizures: a staged approach: a report from the International League Against Epilepsy Nonepileptic Seizures Task Force. Epilepsia. 2013;54:2005-2018. doi: 10.1111/epi.12356
2. Asadi-Pooya AA, Sperling MR. Epidemiology of psychogenic nonepileptic seizures. Epilepsy Behav. 2015;46:60-65. doi: 10.1016/j.yebeh.2015.03.015
3. Devireddy VK, Sharma A. A case of psychogenic non-epileptic seizures, unresponsive type, in pregnancy. Prim Care Companion CNS Disord. 2014;16:PCC.13l01574. doi: 10.4088/PCC.13l01574
4. DeToledo JC, Lowe MR, Puig A. Nonepileptic seizures in pregnancy. Neurology. 2000;55:120-121. doi: 10.1212/wnl.55.1.120
5. Ding J-R, An D, Liao W, et al. Altered functional and structural connectivity networks in psychogenic non-epileptic seizures. PLoS One. 2013;8:e63850. doi: 10.1371/journal.pone.0063850
6. Goldstein LH, Chalder T, Chigwedere C, et al. Cognitive-behavioral therapy for psychogenic nonepileptic seizures: a pilot RCT. Neurology. 2010;74:1986-1994. doi: 0.1212/WNL.0b013e3181e39658
7. McLaughlin DP, Pachana NA, McFarland K. The impact of depression, seizure variables and locus of control on health related quality of life in a community dwelling sample of older adults. Seizure. 2010;19:232-236. doi: 10.1016/j.seizure.2010.02.008
8. Duncan R, Anderson J, Cullen B, et al. Predictors of 6-month and 3-year outcomes after psychological intervention for psychogenic non epileptic seizures. Seizure. 2016;36:22-26. doi: 10.1016/j.seizure.2015.12.016
9. Blumer D, Rice S, Adamolekun B. Electroconvulsive treatment for nonepileptic seizure disorders. Epilepsy Behav. 2009;15:382-387. doi: 10.1016/j.yebeh.2009.05.004
THE CASE
A 27-year-old woman presented to the family medicine clinic to establish care for a recent onset of seizures, for which she had previously been admitted, 4 months after delivering her first child. Her pregnancy was complicated by type 1 diabetes and poor glycemic control. Labor was induced at 37 weeks; however, vaginal delivery was impeded by arrest of dilation. An emergency cesarean section was performed under general anesthesia, resulting in a healthy newborn male.
Six weeks after giving birth, the patient was started on sertraline 50 mg/d for postpartum depression. Her history was significant for depression 8 years prior that was controlled with psychotherapy, and treated prior to coming to our clinic. She had not experienced any depressive symptoms during pregnancy.
Three months postpartum, she was hospitalized for recurrent syncopal episodes. They lasted about 2 minutes, with prodromal generalized weakness followed by loss of consciousness. There was no post-event confusion, tongue-biting, or incontinence. Physical exam, electroencephalogram (EEG), echocardiogram, and magnetic resonance imaging of the head and neck demonstrated no acute findings.
These episodes escalated in frequency weeks after they began, involving as many as 40 daily attacks, some of which lasted up to 45 minutes. During these events, the patient was nonresponsive but reported reliving the delivery of her child. Upon initial consultation with Neurology, no cause was found, and she was advised to wear a helmet, stop driving, and refrain from carrying her son. No antiepileptic medications were initiated because there were no EEG findings that supported seizure, and her mood had not improved, despite an increase in sertraline dosage, a switch to citalopram, and the addition of bupropion. She described anxiety, nightmares, and intrusive thoughts during psychotherapy sessions. Her psychiatrist gave her an additional diagnosis of posttraumatic stress disorder (PTSD) secondary to her delivery. The family medicine clinic assisted the patient and her family throughout her care by functioning as a home base for her.
Eight months following initial symptoms, repeat evaluation with a video-EEG revealed no evidence of EEG changes during seizure-like activity.
THE DIAGNOSIS
The patient was given a diagnosis of
DISCUSSION
With a prevalence of 5% to 10% and 20% to 40% in outpatient and inpatient epilepsy clinics respectively, PNES events have become of increasing interest to physicians.2 There are few cases of PNES in women during pregnancy reported in the literature.3,4 This is the first case report of PNES with postpartum onset.
Continue to: Epilepsy vs psychogenic nonepileptic seizures
Epilepsy vs psychogenic nonepileptic seizures
PNES episodes appear similar to epileptic seizures, but without a definitive neurobiologic source.2,3 However, recent literature suggests the root cause may be found in abnormalities in neurologic networks, such as dysfunction of frontal and parietal lobe connectivity and increased communication from emotional centers of the brain.2,5 There are no typical pathognomonic symptoms of PNES, leading to diagnostic difficulty.2 A definitive diagnosis may be made when a patient experiences seizures without EEG abnormalities.2 Further diagnostic brain imaging is unnecessary.
Trauma may be the underlying cause
A predominance of PNES in both women and young adults, with no definitive associated factors, has been reported in the literature.2 Studies suggest childhood sexual abuse, physical abuse, traumatic brain injury, and health-related trauma, such as distressing medical experiences and surgeries, may be risk factors, while depression, misdiagnosis, and mistreatment can heighten seizure activity.2,3
Treatment requires a multidisciplinary team
Effective management of PNES requires collaboration between the primary care physician, neurologist, psychiatrist, and psychotherapist, with an emphasis on evaluation and control of the underlying trigger(s).3 Randomized controlled trials have demonstrated the efficacy of cognitive behavioral therapy (CBT), supportive care, and patient education in reducing seizure frequency at the 6-month follow-up.3,6 Additional studies have reported the best prognostic factor in PNES management is patient employment of an internal locus of control—the patient’s belief that they control life events.7,8 Case series suggest electroconvulsive therapy (ECT) is an effective alternative mood stabilization and seizure reduction therapy when tolerated.9
Our patient tried several combinations of treatment to manage PNES and comorbid psychiatric conditions, including CBT, antidepressants, and anxiolytics. After about 5 treatment failures, she pursued ECT for treatment-resistant depression and PNES frequency reduction but failed to tolerate therapy. Currently, her PNES has been reduced to 1 to 2 weekly episodes with a 200 mg/d dose of lamotrigine as a mood stabilizer combined with CBT.
THE TAKEAWAY
Providers should investigate a patient’s history and psychologic disposition when the patient presents with seizure-like behavior without a neurobiologic source or with a negative video-EEG study. A history of depression, traumatic experience, PTSD, or other psychosocial triggers must be noted early to prevent a delay in treatment when PNES is part of the differential. Due to a delayed diagnosis of PNES in our patient, she went without full treatment for almost 12 months and experienced worsening episodes. The primary care physician plays an integral role in early identification and intervention through anticipatory guidance, initial work-up, and support for patients with suspected PNES (TABLE).
CORRESPONDENCE
Karim Hanna, MD, 13330 USF Laurel Drive, Tampa, FL; [email protected]
THE CASE
A 27-year-old woman presented to the family medicine clinic to establish care for a recent onset of seizures, for which she had previously been admitted, 4 months after delivering her first child. Her pregnancy was complicated by type 1 diabetes and poor glycemic control. Labor was induced at 37 weeks; however, vaginal delivery was impeded by arrest of dilation. An emergency cesarean section was performed under general anesthesia, resulting in a healthy newborn male.
Six weeks after giving birth, the patient was started on sertraline 50 mg/d for postpartum depression. Her history was significant for depression 8 years prior that was controlled with psychotherapy, and treated prior to coming to our clinic. She had not experienced any depressive symptoms during pregnancy.
Three months postpartum, she was hospitalized for recurrent syncopal episodes. They lasted about 2 minutes, with prodromal generalized weakness followed by loss of consciousness. There was no post-event confusion, tongue-biting, or incontinence. Physical exam, electroencephalogram (EEG), echocardiogram, and magnetic resonance imaging of the head and neck demonstrated no acute findings.
These episodes escalated in frequency weeks after they began, involving as many as 40 daily attacks, some of which lasted up to 45 minutes. During these events, the patient was nonresponsive but reported reliving the delivery of her child. Upon initial consultation with Neurology, no cause was found, and she was advised to wear a helmet, stop driving, and refrain from carrying her son. No antiepileptic medications were initiated because there were no EEG findings that supported seizure, and her mood had not improved, despite an increase in sertraline dosage, a switch to citalopram, and the addition of bupropion. She described anxiety, nightmares, and intrusive thoughts during psychotherapy sessions. Her psychiatrist gave her an additional diagnosis of posttraumatic stress disorder (PTSD) secondary to her delivery. The family medicine clinic assisted the patient and her family throughout her care by functioning as a home base for her.
Eight months following initial symptoms, repeat evaluation with a video-EEG revealed no evidence of EEG changes during seizure-like activity.
THE DIAGNOSIS
The patient was given a diagnosis of
DISCUSSION
With a prevalence of 5% to 10% and 20% to 40% in outpatient and inpatient epilepsy clinics respectively, PNES events have become of increasing interest to physicians.2 There are few cases of PNES in women during pregnancy reported in the literature.3,4 This is the first case report of PNES with postpartum onset.
Continue to: Epilepsy vs psychogenic nonepileptic seizures
Epilepsy vs psychogenic nonepileptic seizures
PNES episodes appear similar to epileptic seizures, but without a definitive neurobiologic source.2,3 However, recent literature suggests the root cause may be found in abnormalities in neurologic networks, such as dysfunction of frontal and parietal lobe connectivity and increased communication from emotional centers of the brain.2,5 There are no typical pathognomonic symptoms of PNES, leading to diagnostic difficulty.2 A definitive diagnosis may be made when a patient experiences seizures without EEG abnormalities.2 Further diagnostic brain imaging is unnecessary.
Trauma may be the underlying cause
A predominance of PNES in both women and young adults, with no definitive associated factors, has been reported in the literature.2 Studies suggest childhood sexual abuse, physical abuse, traumatic brain injury, and health-related trauma, such as distressing medical experiences and surgeries, may be risk factors, while depression, misdiagnosis, and mistreatment can heighten seizure activity.2,3
Treatment requires a multidisciplinary team
Effective management of PNES requires collaboration between the primary care physician, neurologist, psychiatrist, and psychotherapist, with an emphasis on evaluation and control of the underlying trigger(s).3 Randomized controlled trials have demonstrated the efficacy of cognitive behavioral therapy (CBT), supportive care, and patient education in reducing seizure frequency at the 6-month follow-up.3,6 Additional studies have reported the best prognostic factor in PNES management is patient employment of an internal locus of control—the patient’s belief that they control life events.7,8 Case series suggest electroconvulsive therapy (ECT) is an effective alternative mood stabilization and seizure reduction therapy when tolerated.9
Our patient tried several combinations of treatment to manage PNES and comorbid psychiatric conditions, including CBT, antidepressants, and anxiolytics. After about 5 treatment failures, she pursued ECT for treatment-resistant depression and PNES frequency reduction but failed to tolerate therapy. Currently, her PNES has been reduced to 1 to 2 weekly episodes with a 200 mg/d dose of lamotrigine as a mood stabilizer combined with CBT.
THE TAKEAWAY
Providers should investigate a patient’s history and psychologic disposition when the patient presents with seizure-like behavior without a neurobiologic source or with a negative video-EEG study. A history of depression, traumatic experience, PTSD, or other psychosocial triggers must be noted early to prevent a delay in treatment when PNES is part of the differential. Due to a delayed diagnosis of PNES in our patient, she went without full treatment for almost 12 months and experienced worsening episodes. The primary care physician plays an integral role in early identification and intervention through anticipatory guidance, initial work-up, and support for patients with suspected PNES (TABLE).
CORRESPONDENCE
Karim Hanna, MD, 13330 USF Laurel Drive, Tampa, FL; [email protected]
1. LaFrance WC Jr, Baker GA, Duncan R, et al. Minimum requirements for the diagnosis of psychogenic nonepileptic seizures: a staged approach: a report from the International League Against Epilepsy Nonepileptic Seizures Task Force. Epilepsia. 2013;54:2005-2018. doi: 10.1111/epi.12356
2. Asadi-Pooya AA, Sperling MR. Epidemiology of psychogenic nonepileptic seizures. Epilepsy Behav. 2015;46:60-65. doi: 10.1016/j.yebeh.2015.03.015
3. Devireddy VK, Sharma A. A case of psychogenic non-epileptic seizures, unresponsive type, in pregnancy. Prim Care Companion CNS Disord. 2014;16:PCC.13l01574. doi: 10.4088/PCC.13l01574
4. DeToledo JC, Lowe MR, Puig A. Nonepileptic seizures in pregnancy. Neurology. 2000;55:120-121. doi: 10.1212/wnl.55.1.120
5. Ding J-R, An D, Liao W, et al. Altered functional and structural connectivity networks in psychogenic non-epileptic seizures. PLoS One. 2013;8:e63850. doi: 10.1371/journal.pone.0063850
6. Goldstein LH, Chalder T, Chigwedere C, et al. Cognitive-behavioral therapy for psychogenic nonepileptic seizures: a pilot RCT. Neurology. 2010;74:1986-1994. doi: 0.1212/WNL.0b013e3181e39658
7. McLaughlin DP, Pachana NA, McFarland K. The impact of depression, seizure variables and locus of control on health related quality of life in a community dwelling sample of older adults. Seizure. 2010;19:232-236. doi: 10.1016/j.seizure.2010.02.008
8. Duncan R, Anderson J, Cullen B, et al. Predictors of 6-month and 3-year outcomes after psychological intervention for psychogenic non epileptic seizures. Seizure. 2016;36:22-26. doi: 10.1016/j.seizure.2015.12.016
9. Blumer D, Rice S, Adamolekun B. Electroconvulsive treatment for nonepileptic seizure disorders. Epilepsy Behav. 2009;15:382-387. doi: 10.1016/j.yebeh.2009.05.004
1. LaFrance WC Jr, Baker GA, Duncan R, et al. Minimum requirements for the diagnosis of psychogenic nonepileptic seizures: a staged approach: a report from the International League Against Epilepsy Nonepileptic Seizures Task Force. Epilepsia. 2013;54:2005-2018. doi: 10.1111/epi.12356
2. Asadi-Pooya AA, Sperling MR. Epidemiology of psychogenic nonepileptic seizures. Epilepsy Behav. 2015;46:60-65. doi: 10.1016/j.yebeh.2015.03.015
3. Devireddy VK, Sharma A. A case of psychogenic non-epileptic seizures, unresponsive type, in pregnancy. Prim Care Companion CNS Disord. 2014;16:PCC.13l01574. doi: 10.4088/PCC.13l01574
4. DeToledo JC, Lowe MR, Puig A. Nonepileptic seizures in pregnancy. Neurology. 2000;55:120-121. doi: 10.1212/wnl.55.1.120
5. Ding J-R, An D, Liao W, et al. Altered functional and structural connectivity networks in psychogenic non-epileptic seizures. PLoS One. 2013;8:e63850. doi: 10.1371/journal.pone.0063850
6. Goldstein LH, Chalder T, Chigwedere C, et al. Cognitive-behavioral therapy for psychogenic nonepileptic seizures: a pilot RCT. Neurology. 2010;74:1986-1994. doi: 0.1212/WNL.0b013e3181e39658
7. McLaughlin DP, Pachana NA, McFarland K. The impact of depression, seizure variables and locus of control on health related quality of life in a community dwelling sample of older adults. Seizure. 2010;19:232-236. doi: 10.1016/j.seizure.2010.02.008
8. Duncan R, Anderson J, Cullen B, et al. Predictors of 6-month and 3-year outcomes after psychological intervention for psychogenic non epileptic seizures. Seizure. 2016;36:22-26. doi: 10.1016/j.seizure.2015.12.016
9. Blumer D, Rice S, Adamolekun B. Electroconvulsive treatment for nonepileptic seizure disorders. Epilepsy Behav. 2009;15:382-387. doi: 10.1016/j.yebeh.2009.05.004

Two case reports identify Guillain-Barré variants after SARS-CoV-2 vaccination
Guillain-Barré syndrome, a rare peripheral nerve disorder that can occur after certain types of viral and bacterial infections, has not to date been definitively linked to infection by SARS-CoV-2 or with vaccination against the virus, despite surveillance searching for such associations.
Spikes in Guillain-Barré syndrome incidence have previously, but rarely, been associated with outbreaks of other viral diseases, including Zika, but not with vaccination, except for a 1976-1977 swine influenza vaccine campaign in the United States that was seen associated with a slight elevation in risk, and was halted when that risk became known. Since then, all sorts of vaccines in the European Union and United States have come with warnings about Guillain-Barré syndrome in their package inserts – a fact that some Guillain-Barré syndrome experts lament as perpetuating the notion that vaccines cause Guillain-Barré syndrome.
Epidemiologic studies in the United Kingdom and Singapore did not detect increases in Guillain-Barré syndrome incidence during the COVID-19 pandemic. And as mass vaccination against COVID-19 got underway early this year, experts cautioned against the temptation to attribute incident Guillain-Barré syndrome cases following vaccination to SARS-CoV-2 without careful statistical and epidemiological analysis. Until now reports of Guillain-Barré syndrome have been scant: clinical trials of a viral vector vaccine developed by Johnson & Johnson saw one in the placebo arm and another in the intervention arm, while another case was reported following administration of a Pfizer mRNA SARS-Cov-2 vaccine.
Recent case reports
None of the patients had evidence of current SARS-CoV-2 infection.
From India, Boby V. Maramattom, MD, of Aster Medcity in Kochi, India, and colleagues reported on seven severe cases of Guillain-Barré syndrome occurring between 10 and 14 days after a first dose of the AstraZeneca vaccine. All but one of the patients were women, all had bilateral facial paresis, all progressed to areflexic quadriplegia, and six required respiratory support. Patients’ ages ranged from 43 to 70. Four developed other cranial neuropathies, including abducens palsy and trigeminal sensory nerve involvement, which are rare in reports of Guillain-Barré syndrome from India, Dr. Maramattom and colleagues noted.
The authors argued that their findings “should prompt all physicians to be vigilant in recognizing Guillain-Barré syndrome in patients who have received the AstraZeneca vaccine. While the risk per patient (5.8 per million) may be relatively low, our observations suggest that this clinically distinct [Guillain-Barré syndrome] variant is more severe than usual and may require mechanical ventilation.”
The U.K. cases, reported by Christopher Martin Allen, MD, and colleagues at Nottingham (England) University Hospitals NHS Trust, describe bifacial weakness and normal facial sensation in four men between 11 and 22 days after their first doses of the Astra-Zeneca vaccine. This type of facial palsy, the authors wrote, was unusual Guillain-Barré syndrome variant that one rapid review found in 3 of 42 European patients diagnosed with Guillain-Barré syndrome following SARS-CoV-2 infection.
Dr. Allen and colleagues acknowledged that causality could not be assumed from the temporal relationship of immunization to onset of bifacial weakness in their report, but argued that their findings argued for “robust postvaccination surveillance” and that “the report of a similar syndrome in the setting of SARS-CoV-2 infection suggests an immunologic response to the spike protein.” If the link is casual, they wrote, “it could be due to a cross-reactive immune response to the SARS-CoV-2 spike protein and components of the peripheral immune system.”
‘The jury is still out’
Asked for comment, neurologist Anthony Amato, MD, of Brigham and Women’s Hospital, Boston, said that he did not see what the two new studies add to what is already known. “Guillain-Barré syndrome has already been reported temporally following COVID-19 along with accompanying editorials that such temporal occurrences do not imply causation and there is a need for surveillance and epidemiological studies.”
Robert Lisak, MD, of Wayne State University, Detroit, and a longtime adviser to the GBS-CIDP Foundation International, commented that “the relationship between vaccines and association with Guillain-Barré syndrome continues to be controversial in part because Guillain-Barré syndrome, a rare disorder, has many reported associated illnesses including infections. Many vaccines have been implicated but with the probable exception of the ‘swine flu’ vaccine in the 1970s, most have not stood up to scrutiny.”
With SARS-Cov-2 infection and vaccines, “the jury is still out,” Dr. Lisak said. “The report from the U.K. is intriguing since they report several cases of an uncommon variant, but the cases from India seem to be more of the usual forms of Guillain-Barré syndrome.”
Dr. Lisak noted that, even if an association turns out to be valid, “we are talking about a very low incidence of Guillain-Barré syndrome associated with COVID-19 vaccines,” one that would not justify avoiding them because of a possible association with Guillain-Barré syndrome.
The GBS-CIDP Foundation, which supports research into Guillain-Barré syndrome and related diseases, has likewise stressed the low risk presented by SARS-CoV-2 vaccines, noting on its website that “the risk of death or long-term complications from COVID in adults still far exceeds the risk of any possible risk of Guillain-Barré syndrome by several orders of magnitude.”
None of the study authors reported financial conflicts of interest related to their research. Dr. Amato is an adviser to the pharmaceutical firms Alexion and Argenx, while Dr. Lisak has received research support or honoraria from Alexion, Novartis, Hoffmann–La Roche, and others.
Guillain-Barré syndrome, a rare peripheral nerve disorder that can occur after certain types of viral and bacterial infections, has not to date been definitively linked to infection by SARS-CoV-2 or with vaccination against the virus, despite surveillance searching for such associations.
Spikes in Guillain-Barré syndrome incidence have previously, but rarely, been associated with outbreaks of other viral diseases, including Zika, but not with vaccination, except for a 1976-1977 swine influenza vaccine campaign in the United States that was seen associated with a slight elevation in risk, and was halted when that risk became known. Since then, all sorts of vaccines in the European Union and United States have come with warnings about Guillain-Barré syndrome in their package inserts – a fact that some Guillain-Barré syndrome experts lament as perpetuating the notion that vaccines cause Guillain-Barré syndrome.
Epidemiologic studies in the United Kingdom and Singapore did not detect increases in Guillain-Barré syndrome incidence during the COVID-19 pandemic. And as mass vaccination against COVID-19 got underway early this year, experts cautioned against the temptation to attribute incident Guillain-Barré syndrome cases following vaccination to SARS-CoV-2 without careful statistical and epidemiological analysis. Until now reports of Guillain-Barré syndrome have been scant: clinical trials of a viral vector vaccine developed by Johnson & Johnson saw one in the placebo arm and another in the intervention arm, while another case was reported following administration of a Pfizer mRNA SARS-Cov-2 vaccine.
Recent case reports
None of the patients had evidence of current SARS-CoV-2 infection.
From India, Boby V. Maramattom, MD, of Aster Medcity in Kochi, India, and colleagues reported on seven severe cases of Guillain-Barré syndrome occurring between 10 and 14 days after a first dose of the AstraZeneca vaccine. All but one of the patients were women, all had bilateral facial paresis, all progressed to areflexic quadriplegia, and six required respiratory support. Patients’ ages ranged from 43 to 70. Four developed other cranial neuropathies, including abducens palsy and trigeminal sensory nerve involvement, which are rare in reports of Guillain-Barré syndrome from India, Dr. Maramattom and colleagues noted.
The authors argued that their findings “should prompt all physicians to be vigilant in recognizing Guillain-Barré syndrome in patients who have received the AstraZeneca vaccine. While the risk per patient (5.8 per million) may be relatively low, our observations suggest that this clinically distinct [Guillain-Barré syndrome] variant is more severe than usual and may require mechanical ventilation.”
The U.K. cases, reported by Christopher Martin Allen, MD, and colleagues at Nottingham (England) University Hospitals NHS Trust, describe bifacial weakness and normal facial sensation in four men between 11 and 22 days after their first doses of the Astra-Zeneca vaccine. This type of facial palsy, the authors wrote, was unusual Guillain-Barré syndrome variant that one rapid review found in 3 of 42 European patients diagnosed with Guillain-Barré syndrome following SARS-CoV-2 infection.
Dr. Allen and colleagues acknowledged that causality could not be assumed from the temporal relationship of immunization to onset of bifacial weakness in their report, but argued that their findings argued for “robust postvaccination surveillance” and that “the report of a similar syndrome in the setting of SARS-CoV-2 infection suggests an immunologic response to the spike protein.” If the link is casual, they wrote, “it could be due to a cross-reactive immune response to the SARS-CoV-2 spike protein and components of the peripheral immune system.”
‘The jury is still out’
Asked for comment, neurologist Anthony Amato, MD, of Brigham and Women’s Hospital, Boston, said that he did not see what the two new studies add to what is already known. “Guillain-Barré syndrome has already been reported temporally following COVID-19 along with accompanying editorials that such temporal occurrences do not imply causation and there is a need for surveillance and epidemiological studies.”
Robert Lisak, MD, of Wayne State University, Detroit, and a longtime adviser to the GBS-CIDP Foundation International, commented that “the relationship between vaccines and association with Guillain-Barré syndrome continues to be controversial in part because Guillain-Barré syndrome, a rare disorder, has many reported associated illnesses including infections. Many vaccines have been implicated but with the probable exception of the ‘swine flu’ vaccine in the 1970s, most have not stood up to scrutiny.”
With SARS-Cov-2 infection and vaccines, “the jury is still out,” Dr. Lisak said. “The report from the U.K. is intriguing since they report several cases of an uncommon variant, but the cases from India seem to be more of the usual forms of Guillain-Barré syndrome.”
Dr. Lisak noted that, even if an association turns out to be valid, “we are talking about a very low incidence of Guillain-Barré syndrome associated with COVID-19 vaccines,” one that would not justify avoiding them because of a possible association with Guillain-Barré syndrome.
The GBS-CIDP Foundation, which supports research into Guillain-Barré syndrome and related diseases, has likewise stressed the low risk presented by SARS-CoV-2 vaccines, noting on its website that “the risk of death or long-term complications from COVID in adults still far exceeds the risk of any possible risk of Guillain-Barré syndrome by several orders of magnitude.”
None of the study authors reported financial conflicts of interest related to their research. Dr. Amato is an adviser to the pharmaceutical firms Alexion and Argenx, while Dr. Lisak has received research support or honoraria from Alexion, Novartis, Hoffmann–La Roche, and others.
Guillain-Barré syndrome, a rare peripheral nerve disorder that can occur after certain types of viral and bacterial infections, has not to date been definitively linked to infection by SARS-CoV-2 or with vaccination against the virus, despite surveillance searching for such associations.
Spikes in Guillain-Barré syndrome incidence have previously, but rarely, been associated with outbreaks of other viral diseases, including Zika, but not with vaccination, except for a 1976-1977 swine influenza vaccine campaign in the United States that was seen associated with a slight elevation in risk, and was halted when that risk became known. Since then, all sorts of vaccines in the European Union and United States have come with warnings about Guillain-Barré syndrome in their package inserts – a fact that some Guillain-Barré syndrome experts lament as perpetuating the notion that vaccines cause Guillain-Barré syndrome.
Epidemiologic studies in the United Kingdom and Singapore did not detect increases in Guillain-Barré syndrome incidence during the COVID-19 pandemic. And as mass vaccination against COVID-19 got underway early this year, experts cautioned against the temptation to attribute incident Guillain-Barré syndrome cases following vaccination to SARS-CoV-2 without careful statistical and epidemiological analysis. Until now reports of Guillain-Barré syndrome have been scant: clinical trials of a viral vector vaccine developed by Johnson & Johnson saw one in the placebo arm and another in the intervention arm, while another case was reported following administration of a Pfizer mRNA SARS-Cov-2 vaccine.
Recent case reports
None of the patients had evidence of current SARS-CoV-2 infection.
From India, Boby V. Maramattom, MD, of Aster Medcity in Kochi, India, and colleagues reported on seven severe cases of Guillain-Barré syndrome occurring between 10 and 14 days after a first dose of the AstraZeneca vaccine. All but one of the patients were women, all had bilateral facial paresis, all progressed to areflexic quadriplegia, and six required respiratory support. Patients’ ages ranged from 43 to 70. Four developed other cranial neuropathies, including abducens palsy and trigeminal sensory nerve involvement, which are rare in reports of Guillain-Barré syndrome from India, Dr. Maramattom and colleagues noted.
The authors argued that their findings “should prompt all physicians to be vigilant in recognizing Guillain-Barré syndrome in patients who have received the AstraZeneca vaccine. While the risk per patient (5.8 per million) may be relatively low, our observations suggest that this clinically distinct [Guillain-Barré syndrome] variant is more severe than usual and may require mechanical ventilation.”
The U.K. cases, reported by Christopher Martin Allen, MD, and colleagues at Nottingham (England) University Hospitals NHS Trust, describe bifacial weakness and normal facial sensation in four men between 11 and 22 days after their first doses of the Astra-Zeneca vaccine. This type of facial palsy, the authors wrote, was unusual Guillain-Barré syndrome variant that one rapid review found in 3 of 42 European patients diagnosed with Guillain-Barré syndrome following SARS-CoV-2 infection.
Dr. Allen and colleagues acknowledged that causality could not be assumed from the temporal relationship of immunization to onset of bifacial weakness in their report, but argued that their findings argued for “robust postvaccination surveillance” and that “the report of a similar syndrome in the setting of SARS-CoV-2 infection suggests an immunologic response to the spike protein.” If the link is casual, they wrote, “it could be due to a cross-reactive immune response to the SARS-CoV-2 spike protein and components of the peripheral immune system.”
‘The jury is still out’
Asked for comment, neurologist Anthony Amato, MD, of Brigham and Women’s Hospital, Boston, said that he did not see what the two new studies add to what is already known. “Guillain-Barré syndrome has already been reported temporally following COVID-19 along with accompanying editorials that such temporal occurrences do not imply causation and there is a need for surveillance and epidemiological studies.”
Robert Lisak, MD, of Wayne State University, Detroit, and a longtime adviser to the GBS-CIDP Foundation International, commented that “the relationship between vaccines and association with Guillain-Barré syndrome continues to be controversial in part because Guillain-Barré syndrome, a rare disorder, has many reported associated illnesses including infections. Many vaccines have been implicated but with the probable exception of the ‘swine flu’ vaccine in the 1970s, most have not stood up to scrutiny.”
With SARS-Cov-2 infection and vaccines, “the jury is still out,” Dr. Lisak said. “The report from the U.K. is intriguing since they report several cases of an uncommon variant, but the cases from India seem to be more of the usual forms of Guillain-Barré syndrome.”
Dr. Lisak noted that, even if an association turns out to be valid, “we are talking about a very low incidence of Guillain-Barré syndrome associated with COVID-19 vaccines,” one that would not justify avoiding them because of a possible association with Guillain-Barré syndrome.
The GBS-CIDP Foundation, which supports research into Guillain-Barré syndrome and related diseases, has likewise stressed the low risk presented by SARS-CoV-2 vaccines, noting on its website that “the risk of death or long-term complications from COVID in adults still far exceeds the risk of any possible risk of Guillain-Barré syndrome by several orders of magnitude.”
None of the study authors reported financial conflicts of interest related to their research. Dr. Amato is an adviser to the pharmaceutical firms Alexion and Argenx, while Dr. Lisak has received research support or honoraria from Alexion, Novartis, Hoffmann–La Roche, and others.
FROM ANNALS OF NEUROLOGY
U.S., international MIS-C studies yield disparate results
That requires rapid pragmatic evaluation of therapies. Two real-world observational studies published online June 16 in The New England Journal of Medicine do that, with differing results.
In the Overcoming COVID-19 study, investigators assessed initial therapy and outcomes for patients with MIS-C using surveillance data from 58 pediatric hospitals nationwide.
The results suggest that patients with MIS-C who were younger than 21 years of age and who were initially treated with intravenous immunoglobulin (IVIG) plus glucocorticoids fared better in terms of cardiovascular function.
The study included 518 children (median age, 8.7 years) who were admitted to the hospital between March and October 2020 and who received at least one immunomodulatory therapy. In a propensity score–matched analysis, those given IVIG plus glucocorticoids (n = 103) had a lower risk for the primary outcome of cardiovascular dysfunction on or after day 2 than those given IVIG alone (n = 103), at 17% versus 31% (risk ratio, 0.56; 95% confidence interval, 0.34-0.94).
Risks for individual aspects of the study’s composite outcome were also lower with IVIG plus glucocorticoids. Left ventricular dysfunction occurred in 8% and 17%, respectively (RR, 0.46; 95% CI, 0.19-1.15). Shock requiring vasopressor use emerged in 13% and 24%, respectively (RR, 0.54; 95% CI, 0.29-1.00).
In addition, there were fewer cases in which adjunctive therapy was given on day one among those who received combination therapy than among those who received IVIG alone, at 34% versus 70% (RR, 0.49; 95% CI, 0.36-0.65), but the risk for fever was not lower on or after day two (31% and 40%, respectively; RR, 0.78; 95% CI, 0.53-1.13).
Lead author Mary Beth F. Son, MD, director of the rheumatology program at Boston Children’s Hospital, who is also associate professor of pediatrics at Harvard Medical School, stressed that the study did not assess which MIS-C patients should receive treatment. “Rather, we studied children who had been treated with one of two initial regimens and then assessed short-term outcomes,” she told this news organization.
Going forward, it will be important to study which children should receive immunomodulatory treatment, Dr. Son said. “Specifically, can the less ill children receive IVIG alone or no treatment? This is an unanswered question at the moment, which could be addressed with a randomized controlled trial.”
Future directions, she added, will include assessing long-term cardiac outcomes for patients with MIS-C as well as studying outpatient regimens, especially those that involve steroids.
Earlier this year, French investigators found better outcomes with combined corticosteroids and IVIG than with IVIG alone. They suggested that combination therapy should be the standard of care, given the present state of therapeutic knowledge.
Maybe not so standard
Different results emerged, however, from an international study of MIS-C that compared three, rather than two, treatment approaches. Collaborators from the Best Available Treatment Study for MIS-C (BATS) evaluated data for 614 children with suspected MIS-C between June 2020 and February 2021 in 32 countries and found no substantial differences in recovery among children whose primary treatment was IVIG alone, IVIG plus glucocorticoids, or glucocorticoids alone.
The study by Andrew J. McArdle, MB BChir, MSC, a clinical research fellow at Imperial College London, and colleagues was published June 16 in The New England Journal of Medicine.
In the BATS cohort, 246 received IVIG alone, 208 received IVIG plus glucocorticoids, and 99 received glucocorticoids alone. Twenty-two patients received other combinations, including biologics, and 39 received no immunomodulatory therapy.
Among patients who were included in the primary analysis, death occurred or inotropic or ventilatory support was employed in 56 of 180 of the patients who received IVIG plus glucocorticoids, compared with 44 of 211 patients treated with IVIG alone, for an adjusted odds ratio (aOR) of 0.77 (95% CI, 0.33-1.82). Among those who received glucocorticoids alone, 17 of 83 met the primary endpoint of death or inotropic or ventilatory support, for an aOR relative to IVIG alone of 0.54 (95% CI, 0.22-1.33).
After adjustments, the likelihood for reduced disease severity was similar in the two groups relative to IVIG alone, at 0.90 for IVIG plus glucocorticoids and 0.93 for glucocorticoids alone. Time to reduction in disease severity was also comparable across all groups.
Some of the differences between the U.S. study and the global studies could be the result of the larger size of the international cohort and possibly a difference in the strains of virus in the United States and abroad, according to S. Sexson Tejtel, MD, PhD, MPH, a pediatric cardiologist at Texas Children’s Hospital and an assistant professor at Baylor College of Medicine, Houston, Texas. “Some strains make children sicker than others, and they’re going to need more treatment,” said Dr. Sexson Tejtel, who was not involved in either study.
Dr. Sexson Tejtel also noted that the U.S. researchers did not assess outcomes among children treated with steroids alone. “It would be interesting to know what steroids alone look like in the U.S. MIS-C population,” she said in an interview.
BATS corresponding author Michael Levin, MBE, PhD, FRCPCH, an Imperial College professor of pediatrics and international child health, told this news organization that the differing results may have arisen because of the international study’s three-treatment focus, its wider spectrum of patients, and its different endpoints: Death and inotropic support on or after day 2, versus echocardiographic left ventricular dysfunction or inotropic usage.
Regardless of the differences between the two studies, neither establishes the most effective single or combination treatment, writes Roberta L. DeBiasi, MD, of the Division of Pediatric Infectious Diseases at Children’s National Hospital and Research Institute and George Washington University, Washington, in an accompanying editorial. “Specifically, neither study was powered to include an evaluation of approaches that steer away from broad immunosuppression with glucocorticoids and that focus on more targeted and titratable treatments with biologic agents, such as anakinra and infliximab,” she writes.
Dr. DeBiasi adds that long-term follow-up studies of cardiac and noncardiac outcomes in these patients will launch soon. “Meanwhile, continued collaboration across centers is essential to decreasing the short-term incidence of death and complications,” she writes.
“It will be interesting as we apply results from these studies as they come out to see how they change our practice,” Dr. Sexson Tejtel said. “And it would be good to have some randomized clinical trials.”
For Dr. Levin, the bottom line is that all three treatments are associated with recovery for a majority of children. “This is good news for clinicians who have been guessing which treatment to use,” he said. “Both studies are attempts to provide doctors with some evidence on which to base treatment decisions and are not the final answer. Our study is ongoing, and with larger numbers of patients it may give clearer answers.”
The Overcoming COVID-19 study was funded by the U.S. Centers for Disease Control and Prevention. Several coauthors have reported support from industry outside of the submitted work. BATS was funded by the European Union’s Horizons 2020 Program. The study authors have disclosed no relevant financial relationships. One coauthor’s spouse is employed by GlaxoSmithKline. Dr. DeBiasi and Dr. Sexson Tejtel have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
That requires rapid pragmatic evaluation of therapies. Two real-world observational studies published online June 16 in The New England Journal of Medicine do that, with differing results.
In the Overcoming COVID-19 study, investigators assessed initial therapy and outcomes for patients with MIS-C using surveillance data from 58 pediatric hospitals nationwide.
The results suggest that patients with MIS-C who were younger than 21 years of age and who were initially treated with intravenous immunoglobulin (IVIG) plus glucocorticoids fared better in terms of cardiovascular function.
The study included 518 children (median age, 8.7 years) who were admitted to the hospital between March and October 2020 and who received at least one immunomodulatory therapy. In a propensity score–matched analysis, those given IVIG plus glucocorticoids (n = 103) had a lower risk for the primary outcome of cardiovascular dysfunction on or after day 2 than those given IVIG alone (n = 103), at 17% versus 31% (risk ratio, 0.56; 95% confidence interval, 0.34-0.94).
Risks for individual aspects of the study’s composite outcome were also lower with IVIG plus glucocorticoids. Left ventricular dysfunction occurred in 8% and 17%, respectively (RR, 0.46; 95% CI, 0.19-1.15). Shock requiring vasopressor use emerged in 13% and 24%, respectively (RR, 0.54; 95% CI, 0.29-1.00).
In addition, there were fewer cases in which adjunctive therapy was given on day one among those who received combination therapy than among those who received IVIG alone, at 34% versus 70% (RR, 0.49; 95% CI, 0.36-0.65), but the risk for fever was not lower on or after day two (31% and 40%, respectively; RR, 0.78; 95% CI, 0.53-1.13).
Lead author Mary Beth F. Son, MD, director of the rheumatology program at Boston Children’s Hospital, who is also associate professor of pediatrics at Harvard Medical School, stressed that the study did not assess which MIS-C patients should receive treatment. “Rather, we studied children who had been treated with one of two initial regimens and then assessed short-term outcomes,” she told this news organization.
Going forward, it will be important to study which children should receive immunomodulatory treatment, Dr. Son said. “Specifically, can the less ill children receive IVIG alone or no treatment? This is an unanswered question at the moment, which could be addressed with a randomized controlled trial.”
Future directions, she added, will include assessing long-term cardiac outcomes for patients with MIS-C as well as studying outpatient regimens, especially those that involve steroids.
Earlier this year, French investigators found better outcomes with combined corticosteroids and IVIG than with IVIG alone. They suggested that combination therapy should be the standard of care, given the present state of therapeutic knowledge.
Maybe not so standard
Different results emerged, however, from an international study of MIS-C that compared three, rather than two, treatment approaches. Collaborators from the Best Available Treatment Study for MIS-C (BATS) evaluated data for 614 children with suspected MIS-C between June 2020 and February 2021 in 32 countries and found no substantial differences in recovery among children whose primary treatment was IVIG alone, IVIG plus glucocorticoids, or glucocorticoids alone.
The study by Andrew J. McArdle, MB BChir, MSC, a clinical research fellow at Imperial College London, and colleagues was published June 16 in The New England Journal of Medicine.
In the BATS cohort, 246 received IVIG alone, 208 received IVIG plus glucocorticoids, and 99 received glucocorticoids alone. Twenty-two patients received other combinations, including biologics, and 39 received no immunomodulatory therapy.
Among patients who were included in the primary analysis, death occurred or inotropic or ventilatory support was employed in 56 of 180 of the patients who received IVIG plus glucocorticoids, compared with 44 of 211 patients treated with IVIG alone, for an adjusted odds ratio (aOR) of 0.77 (95% CI, 0.33-1.82). Among those who received glucocorticoids alone, 17 of 83 met the primary endpoint of death or inotropic or ventilatory support, for an aOR relative to IVIG alone of 0.54 (95% CI, 0.22-1.33).
After adjustments, the likelihood for reduced disease severity was similar in the two groups relative to IVIG alone, at 0.90 for IVIG plus glucocorticoids and 0.93 for glucocorticoids alone. Time to reduction in disease severity was also comparable across all groups.
Some of the differences between the U.S. study and the global studies could be the result of the larger size of the international cohort and possibly a difference in the strains of virus in the United States and abroad, according to S. Sexson Tejtel, MD, PhD, MPH, a pediatric cardiologist at Texas Children’s Hospital and an assistant professor at Baylor College of Medicine, Houston, Texas. “Some strains make children sicker than others, and they’re going to need more treatment,” said Dr. Sexson Tejtel, who was not involved in either study.
Dr. Sexson Tejtel also noted that the U.S. researchers did not assess outcomes among children treated with steroids alone. “It would be interesting to know what steroids alone look like in the U.S. MIS-C population,” she said in an interview.
BATS corresponding author Michael Levin, MBE, PhD, FRCPCH, an Imperial College professor of pediatrics and international child health, told this news organization that the differing results may have arisen because of the international study’s three-treatment focus, its wider spectrum of patients, and its different endpoints: Death and inotropic support on or after day 2, versus echocardiographic left ventricular dysfunction or inotropic usage.
Regardless of the differences between the two studies, neither establishes the most effective single or combination treatment, writes Roberta L. DeBiasi, MD, of the Division of Pediatric Infectious Diseases at Children’s National Hospital and Research Institute and George Washington University, Washington, in an accompanying editorial. “Specifically, neither study was powered to include an evaluation of approaches that steer away from broad immunosuppression with glucocorticoids and that focus on more targeted and titratable treatments with biologic agents, such as anakinra and infliximab,” she writes.
Dr. DeBiasi adds that long-term follow-up studies of cardiac and noncardiac outcomes in these patients will launch soon. “Meanwhile, continued collaboration across centers is essential to decreasing the short-term incidence of death and complications,” she writes.
“It will be interesting as we apply results from these studies as they come out to see how they change our practice,” Dr. Sexson Tejtel said. “And it would be good to have some randomized clinical trials.”
For Dr. Levin, the bottom line is that all three treatments are associated with recovery for a majority of children. “This is good news for clinicians who have been guessing which treatment to use,” he said. “Both studies are attempts to provide doctors with some evidence on which to base treatment decisions and are not the final answer. Our study is ongoing, and with larger numbers of patients it may give clearer answers.”
The Overcoming COVID-19 study was funded by the U.S. Centers for Disease Control and Prevention. Several coauthors have reported support from industry outside of the submitted work. BATS was funded by the European Union’s Horizons 2020 Program. The study authors have disclosed no relevant financial relationships. One coauthor’s spouse is employed by GlaxoSmithKline. Dr. DeBiasi and Dr. Sexson Tejtel have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
That requires rapid pragmatic evaluation of therapies. Two real-world observational studies published online June 16 in The New England Journal of Medicine do that, with differing results.
In the Overcoming COVID-19 study, investigators assessed initial therapy and outcomes for patients with MIS-C using surveillance data from 58 pediatric hospitals nationwide.
The results suggest that patients with MIS-C who were younger than 21 years of age and who were initially treated with intravenous immunoglobulin (IVIG) plus glucocorticoids fared better in terms of cardiovascular function.
The study included 518 children (median age, 8.7 years) who were admitted to the hospital between March and October 2020 and who received at least one immunomodulatory therapy. In a propensity score–matched analysis, those given IVIG plus glucocorticoids (n = 103) had a lower risk for the primary outcome of cardiovascular dysfunction on or after day 2 than those given IVIG alone (n = 103), at 17% versus 31% (risk ratio, 0.56; 95% confidence interval, 0.34-0.94).
Risks for individual aspects of the study’s composite outcome were also lower with IVIG plus glucocorticoids. Left ventricular dysfunction occurred in 8% and 17%, respectively (RR, 0.46; 95% CI, 0.19-1.15). Shock requiring vasopressor use emerged in 13% and 24%, respectively (RR, 0.54; 95% CI, 0.29-1.00).
In addition, there were fewer cases in which adjunctive therapy was given on day one among those who received combination therapy than among those who received IVIG alone, at 34% versus 70% (RR, 0.49; 95% CI, 0.36-0.65), but the risk for fever was not lower on or after day two (31% and 40%, respectively; RR, 0.78; 95% CI, 0.53-1.13).
Lead author Mary Beth F. Son, MD, director of the rheumatology program at Boston Children’s Hospital, who is also associate professor of pediatrics at Harvard Medical School, stressed that the study did not assess which MIS-C patients should receive treatment. “Rather, we studied children who had been treated with one of two initial regimens and then assessed short-term outcomes,” she told this news organization.
Going forward, it will be important to study which children should receive immunomodulatory treatment, Dr. Son said. “Specifically, can the less ill children receive IVIG alone or no treatment? This is an unanswered question at the moment, which could be addressed with a randomized controlled trial.”
Future directions, she added, will include assessing long-term cardiac outcomes for patients with MIS-C as well as studying outpatient regimens, especially those that involve steroids.
Earlier this year, French investigators found better outcomes with combined corticosteroids and IVIG than with IVIG alone. They suggested that combination therapy should be the standard of care, given the present state of therapeutic knowledge.
Maybe not so standard
Different results emerged, however, from an international study of MIS-C that compared three, rather than two, treatment approaches. Collaborators from the Best Available Treatment Study for MIS-C (BATS) evaluated data for 614 children with suspected MIS-C between June 2020 and February 2021 in 32 countries and found no substantial differences in recovery among children whose primary treatment was IVIG alone, IVIG plus glucocorticoids, or glucocorticoids alone.
The study by Andrew J. McArdle, MB BChir, MSC, a clinical research fellow at Imperial College London, and colleagues was published June 16 in The New England Journal of Medicine.
In the BATS cohort, 246 received IVIG alone, 208 received IVIG plus glucocorticoids, and 99 received glucocorticoids alone. Twenty-two patients received other combinations, including biologics, and 39 received no immunomodulatory therapy.
Among patients who were included in the primary analysis, death occurred or inotropic or ventilatory support was employed in 56 of 180 of the patients who received IVIG plus glucocorticoids, compared with 44 of 211 patients treated with IVIG alone, for an adjusted odds ratio (aOR) of 0.77 (95% CI, 0.33-1.82). Among those who received glucocorticoids alone, 17 of 83 met the primary endpoint of death or inotropic or ventilatory support, for an aOR relative to IVIG alone of 0.54 (95% CI, 0.22-1.33).
After adjustments, the likelihood for reduced disease severity was similar in the two groups relative to IVIG alone, at 0.90 for IVIG plus glucocorticoids and 0.93 for glucocorticoids alone. Time to reduction in disease severity was also comparable across all groups.
Some of the differences between the U.S. study and the global studies could be the result of the larger size of the international cohort and possibly a difference in the strains of virus in the United States and abroad, according to S. Sexson Tejtel, MD, PhD, MPH, a pediatric cardiologist at Texas Children’s Hospital and an assistant professor at Baylor College of Medicine, Houston, Texas. “Some strains make children sicker than others, and they’re going to need more treatment,” said Dr. Sexson Tejtel, who was not involved in either study.
Dr. Sexson Tejtel also noted that the U.S. researchers did not assess outcomes among children treated with steroids alone. “It would be interesting to know what steroids alone look like in the U.S. MIS-C population,” she said in an interview.
BATS corresponding author Michael Levin, MBE, PhD, FRCPCH, an Imperial College professor of pediatrics and international child health, told this news organization that the differing results may have arisen because of the international study’s three-treatment focus, its wider spectrum of patients, and its different endpoints: Death and inotropic support on or after day 2, versus echocardiographic left ventricular dysfunction or inotropic usage.
Regardless of the differences between the two studies, neither establishes the most effective single or combination treatment, writes Roberta L. DeBiasi, MD, of the Division of Pediatric Infectious Diseases at Children’s National Hospital and Research Institute and George Washington University, Washington, in an accompanying editorial. “Specifically, neither study was powered to include an evaluation of approaches that steer away from broad immunosuppression with glucocorticoids and that focus on more targeted and titratable treatments with biologic agents, such as anakinra and infliximab,” she writes.
Dr. DeBiasi adds that long-term follow-up studies of cardiac and noncardiac outcomes in these patients will launch soon. “Meanwhile, continued collaboration across centers is essential to decreasing the short-term incidence of death and complications,” she writes.
“It will be interesting as we apply results from these studies as they come out to see how they change our practice,” Dr. Sexson Tejtel said. “And it would be good to have some randomized clinical trials.”
For Dr. Levin, the bottom line is that all three treatments are associated with recovery for a majority of children. “This is good news for clinicians who have been guessing which treatment to use,” he said. “Both studies are attempts to provide doctors with some evidence on which to base treatment decisions and are not the final answer. Our study is ongoing, and with larger numbers of patients it may give clearer answers.”
The Overcoming COVID-19 study was funded by the U.S. Centers for Disease Control and Prevention. Several coauthors have reported support from industry outside of the submitted work. BATS was funded by the European Union’s Horizons 2020 Program. The study authors have disclosed no relevant financial relationships. One coauthor’s spouse is employed by GlaxoSmithKline. Dr. DeBiasi and Dr. Sexson Tejtel have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
New biomarkers may predict interstitial lung disease progression in patients with systemic sclerosis
Quantitative assessment of the extent of interstitial lung disease in patients with systemic sclerosis and levels of certain proteins in bronchoalveolar lavage samples have potential for predicting mortality and disease progression, according to two analyses of data from the Scleroderma Lung Study I and II.

The analyses, presented at the annual European Congress of Rheumatology, aim to improve current prognostic abilities in patients with systemic sclerosis–interstitial lung disease (SSc-ILD). Although forced vital capacity is commonly used as a biomarker for survival in many SSc-ILD trials, other factors can affect FVC, such as respiratory muscle weakness and skin fibrosis. Further, FVC correlates poorly with patient-reported outcomes, explained first author Elizabeth Volkmann, MD, director of the scleroderma program at the University of California, Los Angeles, and the founder and codirector of the UCLA connective tissue disease–related interstitial lung disease program.
Dr. Volkmann presented two studies that investigated the potential of radiographic and protein biomarkers for predicting mortality and identifying patients at risk for ILD progression. The biomarkers may also help to identify patients who would benefit most from immunosuppressive therapy.
The first study found that tracking the quantitative extent of ILD (QILD) over time with high-resolution CT (HRCT) predicted poorer outcomes and could therefore act as a surrogate endpoint for mortality among patients with SSc-ILD. The other study identified associations between specific proteins from bronchoalveolar lavage (BAL) and the likelihood of ILD progression, although some associations were treatment dependent.

Jacob M. van Laar, MD, PhD, professor of rheumatology at the University Medical Center Utrecht (the Netherlands), who was not involved in the study, found the results intriguing and noted the importance of further validation in research before these biomarkers are considered for clinical use.
“It would be wonderful if we can tailor therapy based on BAL biomarkers in the future, as clinicians often struggle to decide on selection, timing, and duration of immunosuppressive treatment,” Dr. van Laar told this news organization. “This has become even more relevant with the introduction of new drugs such as nintedanib.”
Extent of ILD progression as a surrogate for mortality
Scleroderma Lung Study I involved 158 patients with SSc-ILD who were randomly assigned to receive either cyclophosphamide or placebo for 12 months. Scleroderma Lung Study II included 142 patients with SSc-ILD who were randomly assigned to receive either mycophenolate for 24 months or cyclophosphamide for 12 months followed by placebo for 12 months.
The researchers calculated QILD in the whole lung at baseline, at 12 months in the first trial, and at 24 months in the second trial. However, only 82 participants from the first trial and 90 participants from the second trial underwent HRCT. Demographic and disease characteristics were similar between the two groups on follow-up scans.
Follow-up continued for 12 years for patients in the first trial and 8 years in the second. The researchers compared survival rates between the 41% of participants from the first study and 31% of participants from the second study who had poorer QILD scores (at least a 2% increase) with the participants who had stable or improved scores (less than 2% increase).
Participants from both trials had significantly poorer long-term survival if their QILD scores had increased by at least 2% at follow-up (P = .01 for I; P = .019 for II). The association was no longer significant after adjustment for baseline FVC, age, and modified Rodnan skin score in the first trial (hazard ratio, 1.98; P = .089), but it remained significant for participants of the second trial (HR, 3.86; P = .014).
“Data from two independent trial cohorts demonstrated that radiographic progression of SSc-ILD at 1 and 2 years is associated with worse long-term survival,” Dr. Volkmann told attendees.
However, FVC did not significantly predict risk of mortality in either trial.
“To me, the most striking finding from the first study was that change in QILD performed better as a predictor of survival than change in FVC,” Dr. van Laar said in an interview. “This indicates QILD is fit for purpose and worth including in future clinical trials.”
Limitations of the study included lack of HRCT for all participants in the trials and the difference in timing (1 year and 2 years) of HRCT assessment between the two trials. The greater hazard ratio for worsened QILD in the second trial may suggest that assessment at 2 years provides more reliable data as a biomarker, Dr. Volkmann said.
“QILD may represent a better proxy for how a patient feels, functions, and survives than FVC,” she said.
Treatment-dependent biomarkers for worsening lung fibrosis
In the second study, the researchers looked for any associations between changes in the radiographic extent of SSc-ILD and 68 proteins from BAL.
“Being able to risk-stratify patients with interstitial lung disease at the time of diagnosis and predict which patients are likely to have a stable versus progressive disease course is critical for making important treatment decisions for these patients,” Dr. Volkmann told attendees.
The second study she presented involved Scleroderma Lung Study I. Of the 158 participants, 144 underwent a bronchoscopy, yielding BAL protein samples from 103 participants. The researchers determined the extent of radiographic fibrosis in the whole lung with quantitative imaging analysis of HRCT of the chest at baseline and 12 months.
Although the researchers identified several statistically significant associations between certain proteins and changes in radiographic fibrosis, “baseline protein levels were differentially associated with the course of ILD based on treatment status,” she told attendees.
For example, increased levels of the following proteins were linked to poor radiographic fibrosis scores for patients who received placebo:
- Granulocyte-macrophage colony-stimulating factor
- Interleukin-1
- Monocyte chemoattractant protein–3
- Chemokine ligand–5
- Transforming growth factor–beta
- Hepatocyte growth factor
- Stem cell factor
- IL-4
- TGF-alpha
Yet increases in these proteins predicted improvement in radiographic fibrosis in patients who had taken cyclophosphamide.
Independently of treatment, the researchers also identified an association between higher levels of fractalkine and poorer radiographic fibrosis scores and between higher IL-7 levels and improved radiographic fibrosis scores.
After adjusting for treatment arm and baseline severity of ILD, significant associations remained between change in radiographic fibrosis score and IL-1, MCP-3, surfactant protein C, IL-7 and CCL-5 levels.
“Biomarker discovery is really central to our ability to risk stratify patients with SSc-ILD,” Dr. Volkmann told attendees. “Understanding how biomarkers predict outcomes in treated and untreated patients may improve personalized medicine to patients with SSc-ILD and could also reveal novel treatment targets.”
Dr. van Laar said in an interview that this study’s biggest strength lay in its large sample size and in the comprehensiveness of the biomarkers studied.
“The findings are interesting from a research perspective and potentially relevant for clinical practice, but the utility of measuring biomarkers in BAL should be further studied for predictive value on clinical endpoints,” Dr. van Laar said. “BAL is an invasive procedure [that] is not routinely done.”
The research was funded by the National Institutes of Health. Dr. Volkmann has consulted for Boehringer Ingelheim and received grant funding from Corbus, Forbius, and Kadmon. Dr. van Laar has received grant funding or personal fees from Arthrogen, Arxx Therapeutics, AstraZeneca, Bristol-Myers Squibb, Eli Lilly, Gesynta, Leadiant, Merck Sharp & Dohme, Roche, Sanofi, and Thermofisher.
A version of this article first appeared on Medscape.com.
Quantitative assessment of the extent of interstitial lung disease in patients with systemic sclerosis and levels of certain proteins in bronchoalveolar lavage samples have potential for predicting mortality and disease progression, according to two analyses of data from the Scleroderma Lung Study I and II.
The analyses, presented at the annual European Congress of Rheumatology, aim to improve current prognostic abilities in patients with systemic sclerosis–interstitial lung disease (SSc-ILD). Although forced vital capacity is commonly used as a biomarker for survival in many SSc-ILD trials, other factors can affect FVC, such as respiratory muscle weakness and skin fibrosis. Further, FVC correlates poorly with patient-reported outcomes, explained first author Elizabeth Volkmann, MD, director of the scleroderma program at the University of California, Los Angeles, and the founder and codirector of the UCLA connective tissue disease–related interstitial lung disease program.
Dr. Volkmann presented two studies that investigated the potential of radiographic and protein biomarkers for predicting mortality and identifying patients at risk for ILD progression. The biomarkers may also help to identify patients who would benefit most from immunosuppressive therapy.
The first study found that tracking the quantitative extent of ILD (QILD) over time with high-resolution CT (HRCT) predicted poorer outcomes and could therefore act as a surrogate endpoint for mortality among patients with SSc-ILD. The other study identified associations between specific proteins from bronchoalveolar lavage (BAL) and the likelihood of ILD progression, although some associations were treatment dependent.
Jacob M. van Laar, MD, PhD, professor of rheumatology at the University Medical Center Utrecht (the Netherlands), who was not involved in the study, found the results intriguing and noted the importance of further validation in research before these biomarkers are considered for clinical use.
“It would be wonderful if we can tailor therapy based on BAL biomarkers in the future, as clinicians often struggle to decide on selection, timing, and duration of immunosuppressive treatment,” Dr. van Laar told this news organization. “This has become even more relevant with the introduction of new drugs such as nintedanib.”
Extent of ILD progression as a surrogate for mortality
Scleroderma Lung Study I involved 158 patients with SSc-ILD who were randomly assigned to receive either cyclophosphamide or placebo for 12 months. Scleroderma Lung Study II included 142 patients with SSc-ILD who were randomly assigned to receive either mycophenolate for 24 months or cyclophosphamide for 12 months followed by placebo for 12 months.
The researchers calculated QILD in the whole lung at baseline, at 12 months in the first trial, and at 24 months in the second trial. However, only 82 participants from the first trial and 90 participants from the second trial underwent HRCT. Demographic and disease characteristics were similar between the two groups on follow-up scans.
Follow-up continued for 12 years for patients in the first trial and 8 years in the second. The researchers compared survival rates between the 41% of participants from the first study and 31% of participants from the second study who had poorer QILD scores (at least a 2% increase) with the participants who had stable or improved scores (less than 2% increase).
Participants from both trials had significantly poorer long-term survival if their QILD scores had increased by at least 2% at follow-up (P = .01 for I; P = .019 for II). The association was no longer significant after adjustment for baseline FVC, age, and modified Rodnan skin score in the first trial (hazard ratio, 1.98; P = .089), but it remained significant for participants of the second trial (HR, 3.86; P = .014).
“Data from two independent trial cohorts demonstrated that radiographic progression of SSc-ILD at 1 and 2 years is associated with worse long-term survival,” Dr. Volkmann told attendees.
However, FVC did not significantly predict risk of mortality in either trial.
“To me, the most striking finding from the first study was that change in QILD performed better as a predictor of survival than change in FVC,” Dr. van Laar said in an interview. “This indicates QILD is fit for purpose and worth including in future clinical trials.”
Limitations of the study included lack of HRCT for all participants in the trials and the difference in timing (1 year and 2 years) of HRCT assessment between the two trials. The greater hazard ratio for worsened QILD in the second trial may suggest that assessment at 2 years provides more reliable data as a biomarker, Dr. Volkmann said.
“QILD may represent a better proxy for how a patient feels, functions, and survives than FVC,” she said.
Treatment-dependent biomarkers for worsening lung fibrosis
In the second study, the researchers looked for any associations between changes in the radiographic extent of SSc-ILD and 68 proteins from BAL.
“Being able to risk-stratify patients with interstitial lung disease at the time of diagnosis and predict which patients are likely to have a stable versus progressive disease course is critical for making important treatment decisions for these patients,” Dr. Volkmann told attendees.
The second study she presented involved Scleroderma Lung Study I. Of the 158 participants, 144 underwent a bronchoscopy, yielding BAL protein samples from 103 participants. The researchers determined the extent of radiographic fibrosis in the whole lung with quantitative imaging analysis of HRCT of the chest at baseline and 12 months.
Although the researchers identified several statistically significant associations between certain proteins and changes in radiographic fibrosis, “baseline protein levels were differentially associated with the course of ILD based on treatment status,” she told attendees.
For example, increased levels of the following proteins were linked to poor radiographic fibrosis scores for patients who received placebo:
- Granulocyte-macrophage colony-stimulating factor
- Interleukin-1
- Monocyte chemoattractant protein–3
- Chemokine ligand–5
- Transforming growth factor–beta
- Hepatocyte growth factor
- Stem cell factor
- IL-4
- TGF-alpha
Yet increases in these proteins predicted improvement in radiographic fibrosis in patients who had taken cyclophosphamide.
Independently of treatment, the researchers also identified an association between higher levels of fractalkine and poorer radiographic fibrosis scores and between higher IL-7 levels and improved radiographic fibrosis scores.
After adjusting for treatment arm and baseline severity of ILD, significant associations remained between change in radiographic fibrosis score and IL-1, MCP-3, surfactant protein C, IL-7 and CCL-5 levels.
“Biomarker discovery is really central to our ability to risk stratify patients with SSc-ILD,” Dr. Volkmann told attendees. “Understanding how biomarkers predict outcomes in treated and untreated patients may improve personalized medicine to patients with SSc-ILD and could also reveal novel treatment targets.”
Dr. van Laar said in an interview that this study’s biggest strength lay in its large sample size and in the comprehensiveness of the biomarkers studied.
“The findings are interesting from a research perspective and potentially relevant for clinical practice, but the utility of measuring biomarkers in BAL should be further studied for predictive value on clinical endpoints,” Dr. van Laar said. “BAL is an invasive procedure [that] is not routinely done.”
The research was funded by the National Institutes of Health. Dr. Volkmann has consulted for Boehringer Ingelheim and received grant funding from Corbus, Forbius, and Kadmon. Dr. van Laar has received grant funding or personal fees from Arthrogen, Arxx Therapeutics, AstraZeneca, Bristol-Myers Squibb, Eli Lilly, Gesynta, Leadiant, Merck Sharp & Dohme, Roche, Sanofi, and Thermofisher.
A version of this article first appeared on Medscape.com.
Quantitative assessment of the extent of interstitial lung disease in patients with systemic sclerosis and levels of certain proteins in bronchoalveolar lavage samples have potential for predicting mortality and disease progression, according to two analyses of data from the Scleroderma Lung Study I and II.
The analyses, presented at the annual European Congress of Rheumatology, aim to improve current prognostic abilities in patients with systemic sclerosis–interstitial lung disease (SSc-ILD). Although forced vital capacity is commonly used as a biomarker for survival in many SSc-ILD trials, other factors can affect FVC, such as respiratory muscle weakness and skin fibrosis. Further, FVC correlates poorly with patient-reported outcomes, explained first author Elizabeth Volkmann, MD, director of the scleroderma program at the University of California, Los Angeles, and the founder and codirector of the UCLA connective tissue disease–related interstitial lung disease program.
Dr. Volkmann presented two studies that investigated the potential of radiographic and protein biomarkers for predicting mortality and identifying patients at risk for ILD progression. The biomarkers may also help to identify patients who would benefit most from immunosuppressive therapy.
The first study found that tracking the quantitative extent of ILD (QILD) over time with high-resolution CT (HRCT) predicted poorer outcomes and could therefore act as a surrogate endpoint for mortality among patients with SSc-ILD. The other study identified associations between specific proteins from bronchoalveolar lavage (BAL) and the likelihood of ILD progression, although some associations were treatment dependent.
Jacob M. van Laar, MD, PhD, professor of rheumatology at the University Medical Center Utrecht (the Netherlands), who was not involved in the study, found the results intriguing and noted the importance of further validation in research before these biomarkers are considered for clinical use.
“It would be wonderful if we can tailor therapy based on BAL biomarkers in the future, as clinicians often struggle to decide on selection, timing, and duration of immunosuppressive treatment,” Dr. van Laar told this news organization. “This has become even more relevant with the introduction of new drugs such as nintedanib.”
Extent of ILD progression as a surrogate for mortality
Scleroderma Lung Study I involved 158 patients with SSc-ILD who were randomly assigned to receive either cyclophosphamide or placebo for 12 months. Scleroderma Lung Study II included 142 patients with SSc-ILD who were randomly assigned to receive either mycophenolate for 24 months or cyclophosphamide for 12 months followed by placebo for 12 months.
The researchers calculated QILD in the whole lung at baseline, at 12 months in the first trial, and at 24 months in the second trial. However, only 82 participants from the first trial and 90 participants from the second trial underwent HRCT. Demographic and disease characteristics were similar between the two groups on follow-up scans.
Follow-up continued for 12 years for patients in the first trial and 8 years in the second. The researchers compared survival rates between the 41% of participants from the first study and 31% of participants from the second study who had poorer QILD scores (at least a 2% increase) with the participants who had stable or improved scores (less than 2% increase).
Participants from both trials had significantly poorer long-term survival if their QILD scores had increased by at least 2% at follow-up (P = .01 for I; P = .019 for II). The association was no longer significant after adjustment for baseline FVC, age, and modified Rodnan skin score in the first trial (hazard ratio, 1.98; P = .089), but it remained significant for participants of the second trial (HR, 3.86; P = .014).
“Data from two independent trial cohorts demonstrated that radiographic progression of SSc-ILD at 1 and 2 years is associated with worse long-term survival,” Dr. Volkmann told attendees.
However, FVC did not significantly predict risk of mortality in either trial.
“To me, the most striking finding from the first study was that change in QILD performed better as a predictor of survival than change in FVC,” Dr. van Laar said in an interview. “This indicates QILD is fit for purpose and worth including in future clinical trials.”
Limitations of the study included lack of HRCT for all participants in the trials and the difference in timing (1 year and 2 years) of HRCT assessment between the two trials. The greater hazard ratio for worsened QILD in the second trial may suggest that assessment at 2 years provides more reliable data as a biomarker, Dr. Volkmann said.
“QILD may represent a better proxy for how a patient feels, functions, and survives than FVC,” she said.
Treatment-dependent biomarkers for worsening lung fibrosis
In the second study, the researchers looked for any associations between changes in the radiographic extent of SSc-ILD and 68 proteins from BAL.
“Being able to risk-stratify patients with interstitial lung disease at the time of diagnosis and predict which patients are likely to have a stable versus progressive disease course is critical for making important treatment decisions for these patients,” Dr. Volkmann told attendees.
The second study she presented involved Scleroderma Lung Study I. Of the 158 participants, 144 underwent a bronchoscopy, yielding BAL protein samples from 103 participants. The researchers determined the extent of radiographic fibrosis in the whole lung with quantitative imaging analysis of HRCT of the chest at baseline and 12 months.
Although the researchers identified several statistically significant associations between certain proteins and changes in radiographic fibrosis, “baseline protein levels were differentially associated with the course of ILD based on treatment status,” she told attendees.
For example, increased levels of the following proteins were linked to poor radiographic fibrosis scores for patients who received placebo:
- Granulocyte-macrophage colony-stimulating factor
- Interleukin-1
- Monocyte chemoattractant protein–3
- Chemokine ligand–5
- Transforming growth factor–beta
- Hepatocyte growth factor
- Stem cell factor
- IL-4
- TGF-alpha
Yet increases in these proteins predicted improvement in radiographic fibrosis in patients who had taken cyclophosphamide.
Independently of treatment, the researchers also identified an association between higher levels of fractalkine and poorer radiographic fibrosis scores and between higher IL-7 levels and improved radiographic fibrosis scores.
After adjusting for treatment arm and baseline severity of ILD, significant associations remained between change in radiographic fibrosis score and IL-1, MCP-3, surfactant protein C, IL-7 and CCL-5 levels.
“Biomarker discovery is really central to our ability to risk stratify patients with SSc-ILD,” Dr. Volkmann told attendees. “Understanding how biomarkers predict outcomes in treated and untreated patients may improve personalized medicine to patients with SSc-ILD and could also reveal novel treatment targets.”
Dr. van Laar said in an interview that this study’s biggest strength lay in its large sample size and in the comprehensiveness of the biomarkers studied.
“The findings are interesting from a research perspective and potentially relevant for clinical practice, but the utility of measuring biomarkers in BAL should be further studied for predictive value on clinical endpoints,” Dr. van Laar said. “BAL is an invasive procedure [that] is not routinely done.”
The research was funded by the National Institutes of Health. Dr. Volkmann has consulted for Boehringer Ingelheim and received grant funding from Corbus, Forbius, and Kadmon. Dr. van Laar has received grant funding or personal fees from Arthrogen, Arxx Therapeutics, AstraZeneca, Bristol-Myers Squibb, Eli Lilly, Gesynta, Leadiant, Merck Sharp & Dohme, Roche, Sanofi, and Thermofisher.
A version of this article first appeared on Medscape.com.
Secukinumab provides clinical benefit in phase 3 juvenile arthritis trial
Favorable safety sustained at 104 weeks
Secukinumab (Cosentyx), an interleukin-17A inhibitor, is effective and reasonably well tolerated for treatment of enthesitis-related arthritis (ERA) and juvenile psoriatic arthritis (JPsA) in children and adolescents, according to a phase 3 trial presented at a late breaking abstracts session of the annual European Congress of Rheumatology.
On the primary outcome of time to flare, the curves for secukinumab and placebo separated almost immediately, with fewer than half the number of flares occurring in the experimental arm over the course of the study, according to Nicolino Ruperto, MD, senior research scientist at IRCCS Istituto Giannina Gaslini in Genoa, Italy.
The trial, called JUNIPERA, was conducted over 2 years and included an open-label treatment period (TP1) and then a randomized, placebo-controlled comparison (TP2). In TP1, 86 children were initiated on open-label secukinumab administered subcutaneously on weeks 1, 2, 3, 4, 8, and 12. The dose was 75 mg for children less than 50 kg and 150 kg for those heavier.
Average patient age was 13.1 years
Of these 86 children, 52 had ERA and 34 had JPsA. Disease duration of at least 6 months was required for entry. Patients up to the age of 18 years were permitted to enroll. The average age was 13.1 years. Most patients, two-thirds of whom were male, had received an immunomodulator prior to study entry.
At the end of TP1, 69.9% of patients had achieved 70% improvement in the Juvenile Idiopathic Arthritis American College of Rheumatology joint score (JIA ACR70). The 90.4% of patients who achieved JIA ACR30 were invited to enroll in TP2. A total of 75 patients did so.
At the end of TP2, response rates strongly favored secukinumab over placebo for JIA ACR30 (89.2% vs. 64.9%; P = .014) and JIA ACR70 (67.7% vs. 43.2%; P = .042). Higher but not statistically significant differences in response rates were seen for secukinumab over placebo for JIA ACR50 (78.4% vs. 62.2%; P = .152), JIA ACR90 (51.4% vs. 40.5%; P = .431) and JIA ACR100 (43.2% vs. 37.8%; P = .755).
During TP2, there were 10 flares in the group randomized to secukinumab versus 21 flares in the placebo group, translating by hazard ratio (HR) into a 72% risk reduction (HR, 0.28; P < .001).
Side effects similar to those in adults
The types and rates of serious adverse events were similar to those reported previously in adult patients, according to Dr. Ruperto. Although the rate of serious adverse events (14.6% vs. 10.6%) was only moderately higher in the experimental arm, more patients randomized to secukinumab than placebo discontinued therapy (13.2% vs. 6.3%) before the end of follow-up.
The side effects that occurred more commonly on secukinumab included gastrointestinal complaints, such as diarrhea (22.9% vs. 15.8%). Other adverse events occurring in more than 10% of patients included headache and nasopharyngitis, but most side effects were mild and resolved.
Although the proportion of patients with flare increased over time in both groups, Dr. Ruperto reported that protection against flares and relative improvement in clinical markers of disease activity relative to placebo “were sustained out to 2 years of follow-up.”
The submission of these data to regulatory agencies is anticipated. If secukinumab is given an indication for these forms of arthritis, it will join an indication for plaque psoriasis in children that was granted just a few days before these data were presented. The psoriasis indication is the only current use approved for children in the United States.
More biologics needed for JPsA
Additional biologics will be helpful for children with arthritis who are poorly controlled on available treatments, according to Natasha M. Ruth, MD, director of the division of pediatric rheumatology at the Medical University of South Carolina, Charleston. Dr. Ruth was senior author of a case study published 2 years ago in which secukinumab was used to control psoriatic arthritis and nail manifestations of psoriasis.
“It was a girl who had already failed to improve adequately to TNF inhibitors,” reported Dr. Ruth, who had said the child and her parent were very concerned about the nail appearance.
“The nail involvement completely resolved, so it was a very good result in a difficult situation,” Dr. Ruth explained. She said that the decision to try secukinumab was made collaboratively in a clinic in which dermatologists and rheumatologists at her institution work together on difficult cases.
“There is a need for more biologics with different mechanisms of action,” Dr. Ruth said. Based on her experience, secukinumab could be an important addition to treatment options.
Dr. Ruperto reported having financial relationships with more than 20 pharmaceutical companies, including Novartis, which provided financial support for this trial. Many coauthors had financial relationships with multiple companies, including Novartis, and some were employees of the company. Dr. Ruth reported having no potential conflicts of interest.
Favorable safety sustained at 104 weeks
Favorable safety sustained at 104 weeks
Secukinumab (Cosentyx), an interleukin-17A inhibitor, is effective and reasonably well tolerated for treatment of enthesitis-related arthritis (ERA) and juvenile psoriatic arthritis (JPsA) in children and adolescents, according to a phase 3 trial presented at a late breaking abstracts session of the annual European Congress of Rheumatology.
On the primary outcome of time to flare, the curves for secukinumab and placebo separated almost immediately, with fewer than half the number of flares occurring in the experimental arm over the course of the study, according to Nicolino Ruperto, MD, senior research scientist at IRCCS Istituto Giannina Gaslini in Genoa, Italy.
The trial, called JUNIPERA, was conducted over 2 years and included an open-label treatment period (TP1) and then a randomized, placebo-controlled comparison (TP2). In TP1, 86 children were initiated on open-label secukinumab administered subcutaneously on weeks 1, 2, 3, 4, 8, and 12. The dose was 75 mg for children less than 50 kg and 150 kg for those heavier.
Average patient age was 13.1 years
Of these 86 children, 52 had ERA and 34 had JPsA. Disease duration of at least 6 months was required for entry. Patients up to the age of 18 years were permitted to enroll. The average age was 13.1 years. Most patients, two-thirds of whom were male, had received an immunomodulator prior to study entry.
At the end of TP1, 69.9% of patients had achieved 70% improvement in the Juvenile Idiopathic Arthritis American College of Rheumatology joint score (JIA ACR70). The 90.4% of patients who achieved JIA ACR30 were invited to enroll in TP2. A total of 75 patients did so.
At the end of TP2, response rates strongly favored secukinumab over placebo for JIA ACR30 (89.2% vs. 64.9%; P = .014) and JIA ACR70 (67.7% vs. 43.2%; P = .042). Higher but not statistically significant differences in response rates were seen for secukinumab over placebo for JIA ACR50 (78.4% vs. 62.2%; P = .152), JIA ACR90 (51.4% vs. 40.5%; P = .431) and JIA ACR100 (43.2% vs. 37.8%; P = .755).
During TP2, there were 10 flares in the group randomized to secukinumab versus 21 flares in the placebo group, translating by hazard ratio (HR) into a 72% risk reduction (HR, 0.28; P < .001).
Side effects similar to those in adults
The types and rates of serious adverse events were similar to those reported previously in adult patients, according to Dr. Ruperto. Although the rate of serious adverse events (14.6% vs. 10.6%) was only moderately higher in the experimental arm, more patients randomized to secukinumab than placebo discontinued therapy (13.2% vs. 6.3%) before the end of follow-up.
The side effects that occurred more commonly on secukinumab included gastrointestinal complaints, such as diarrhea (22.9% vs. 15.8%). Other adverse events occurring in more than 10% of patients included headache and nasopharyngitis, but most side effects were mild and resolved.
Although the proportion of patients with flare increased over time in both groups, Dr. Ruperto reported that protection against flares and relative improvement in clinical markers of disease activity relative to placebo “were sustained out to 2 years of follow-up.”
The submission of these data to regulatory agencies is anticipated. If secukinumab is given an indication for these forms of arthritis, it will join an indication for plaque psoriasis in children that was granted just a few days before these data were presented. The psoriasis indication is the only current use approved for children in the United States.
More biologics needed for JPsA
Additional biologics will be helpful for children with arthritis who are poorly controlled on available treatments, according to Natasha M. Ruth, MD, director of the division of pediatric rheumatology at the Medical University of South Carolina, Charleston. Dr. Ruth was senior author of a case study published 2 years ago in which secukinumab was used to control psoriatic arthritis and nail manifestations of psoriasis.
“It was a girl who had already failed to improve adequately to TNF inhibitors,” reported Dr. Ruth, who had said the child and her parent were very concerned about the nail appearance.
“The nail involvement completely resolved, so it was a very good result in a difficult situation,” Dr. Ruth explained. She said that the decision to try secukinumab was made collaboratively in a clinic in which dermatologists and rheumatologists at her institution work together on difficult cases.
“There is a need for more biologics with different mechanisms of action,” Dr. Ruth said. Based on her experience, secukinumab could be an important addition to treatment options.
Dr. Ruperto reported having financial relationships with more than 20 pharmaceutical companies, including Novartis, which provided financial support for this trial. Many coauthors had financial relationships with multiple companies, including Novartis, and some were employees of the company. Dr. Ruth reported having no potential conflicts of interest.
Secukinumab (Cosentyx), an interleukin-17A inhibitor, is effective and reasonably well tolerated for treatment of enthesitis-related arthritis (ERA) and juvenile psoriatic arthritis (JPsA) in children and adolescents, according to a phase 3 trial presented at a late breaking abstracts session of the annual European Congress of Rheumatology.
On the primary outcome of time to flare, the curves for secukinumab and placebo separated almost immediately, with fewer than half the number of flares occurring in the experimental arm over the course of the study, according to Nicolino Ruperto, MD, senior research scientist at IRCCS Istituto Giannina Gaslini in Genoa, Italy.
The trial, called JUNIPERA, was conducted over 2 years and included an open-label treatment period (TP1) and then a randomized, placebo-controlled comparison (TP2). In TP1, 86 children were initiated on open-label secukinumab administered subcutaneously on weeks 1, 2, 3, 4, 8, and 12. The dose was 75 mg for children less than 50 kg and 150 kg for those heavier.
Average patient age was 13.1 years
Of these 86 children, 52 had ERA and 34 had JPsA. Disease duration of at least 6 months was required for entry. Patients up to the age of 18 years were permitted to enroll. The average age was 13.1 years. Most patients, two-thirds of whom were male, had received an immunomodulator prior to study entry.
At the end of TP1, 69.9% of patients had achieved 70% improvement in the Juvenile Idiopathic Arthritis American College of Rheumatology joint score (JIA ACR70). The 90.4% of patients who achieved JIA ACR30 were invited to enroll in TP2. A total of 75 patients did so.
At the end of TP2, response rates strongly favored secukinumab over placebo for JIA ACR30 (89.2% vs. 64.9%; P = .014) and JIA ACR70 (67.7% vs. 43.2%; P = .042). Higher but not statistically significant differences in response rates were seen for secukinumab over placebo for JIA ACR50 (78.4% vs. 62.2%; P = .152), JIA ACR90 (51.4% vs. 40.5%; P = .431) and JIA ACR100 (43.2% vs. 37.8%; P = .755).
During TP2, there were 10 flares in the group randomized to secukinumab versus 21 flares in the placebo group, translating by hazard ratio (HR) into a 72% risk reduction (HR, 0.28; P < .001).
Side effects similar to those in adults
The types and rates of serious adverse events were similar to those reported previously in adult patients, according to Dr. Ruperto. Although the rate of serious adverse events (14.6% vs. 10.6%) was only moderately higher in the experimental arm, more patients randomized to secukinumab than placebo discontinued therapy (13.2% vs. 6.3%) before the end of follow-up.
The side effects that occurred more commonly on secukinumab included gastrointestinal complaints, such as diarrhea (22.9% vs. 15.8%). Other adverse events occurring in more than 10% of patients included headache and nasopharyngitis, but most side effects were mild and resolved.
Although the proportion of patients with flare increased over time in both groups, Dr. Ruperto reported that protection against flares and relative improvement in clinical markers of disease activity relative to placebo “were sustained out to 2 years of follow-up.”
The submission of these data to regulatory agencies is anticipated. If secukinumab is given an indication for these forms of arthritis, it will join an indication for plaque psoriasis in children that was granted just a few days before these data were presented. The psoriasis indication is the only current use approved for children in the United States.
More biologics needed for JPsA
Additional biologics will be helpful for children with arthritis who are poorly controlled on available treatments, according to Natasha M. Ruth, MD, director of the division of pediatric rheumatology at the Medical University of South Carolina, Charleston. Dr. Ruth was senior author of a case study published 2 years ago in which secukinumab was used to control psoriatic arthritis and nail manifestations of psoriasis.
“It was a girl who had already failed to improve adequately to TNF inhibitors,” reported Dr. Ruth, who had said the child and her parent were very concerned about the nail appearance.
“The nail involvement completely resolved, so it was a very good result in a difficult situation,” Dr. Ruth explained. She said that the decision to try secukinumab was made collaboratively in a clinic in which dermatologists and rheumatologists at her institution work together on difficult cases.
“There is a need for more biologics with different mechanisms of action,” Dr. Ruth said. Based on her experience, secukinumab could be an important addition to treatment options.
Dr. Ruperto reported having financial relationships with more than 20 pharmaceutical companies, including Novartis, which provided financial support for this trial. Many coauthors had financial relationships with multiple companies, including Novartis, and some were employees of the company. Dr. Ruth reported having no potential conflicts of interest.
FROM THE EULAR 2021 CONGRESS
Atrophic Lesions in a Pregnant Woman
The Diagnosis: Degos Disease
The pathophysiology of Degos disease (malignant atrophic papulosis) is unknown.1 Histopathology demonstrates a wedge-shaped area of dermal necrosis with edema and mucin deposition extending from the papillary dermis to the deep reticular dermis. Occluded vessels, thrombosis, and perivascular lymphocytic infiltrates also may be seen, particularly at the dermal subcutaneous junction and at the periphery of the wedge-shaped infarction. The vascular damage that occurs may be the result of vasculitis, coagulopathy, or endothelial cell dysfunction.1
Patients typically present with small, round, erythematous papules that eventually develop atrophic porcelain white centers and telangiectatic rims. These lesions most commonly occur on the trunk and arms. In the benign form of atrophic papulosis, only the skin is involved; however, systemic involvement of the gastrointestinal tract and central nervous system can occur, resulting in bowel perforation and stroke, respectively.1 Although there is no definitive treatment of Degos disease, successful therapy with aspirin or dipyridamole has been reported.1 Eculizumab, a monoclonal antibody that binds C5, and treprostinil, a prostacyclin analog, are emerging treatment options.2,3 The differential diagnosis of Degos disease may include granuloma annulare, guttate extragenital lichen sclerosus, livedoid vasculopathy, and lymphomatoid papulosis.
Granuloma annulare may clinically mimic the erythematous papules seen in early Degos disease, and histopathology can be used to distinguish between these two disease processes. Localized granuloma annulare is the most common variant and clinically presents as pink papules and plaques in an annular configuration.4 Histopathology demonstrates an unremarkable epidermis; however, the dermis contains degenerated collagen surrounded by palisading histiocytes as well as lymphocytes. Similar to Degos disease, increased mucin is seen within these areas of degeneration, but occluded vessels and thrombosis typically are not seen (Figure 1).4,5
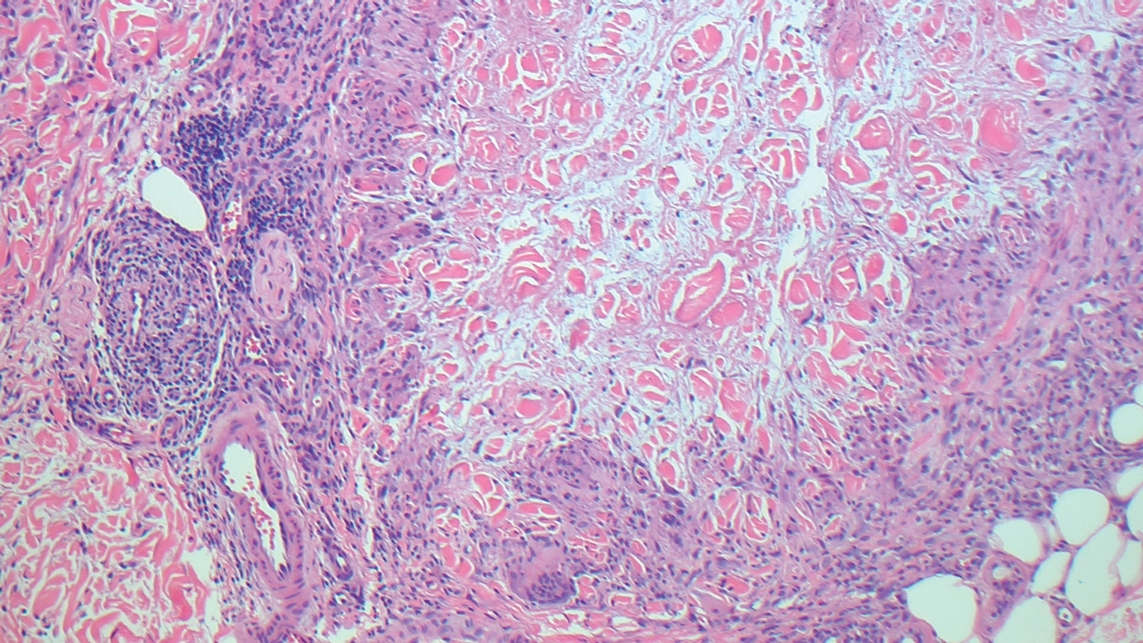
Guttate extragenital lichen sclerosus initially presents as polygonal, bluish white papules that coalesce into plaques.6 Over time, these lesions become more atrophic and may mimic Degos disease but appear differently on histopathology. Histopathology of lichen sclerosus classically demonstrates atrophy of the epidermis with loss of the rete ridges and vacuolar surface changes. Homogenization of the superficial/papillary dermis with an underlying bandlike lymphocytic infiltrate also is seen (Figure 2).6
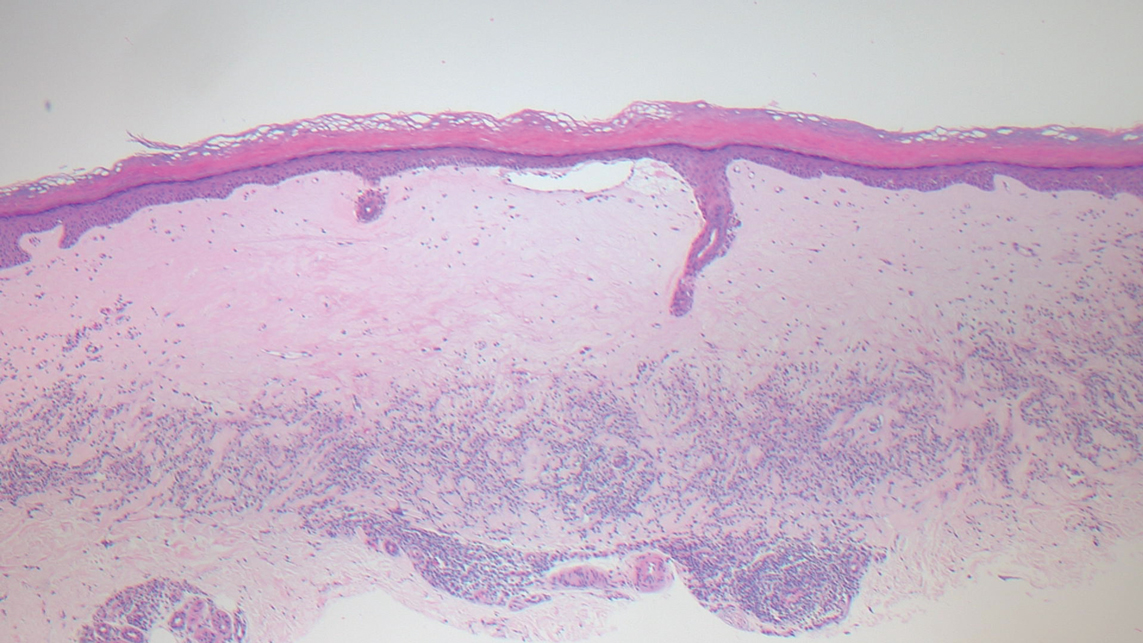
Livedoid vasculopathy is characterized by chronic recurrent ulceration of the legs secondary to thrombosis and subsequent ischemia. In the initial phase of this disease, livedo reticularis is seen followed by the development of ulcerations. As these ulcerations heal, they leave behind porcelain white scars referred to as atrophie blanche.7 The areas of scarring in livedoid vasculopathy are broad and angulated, differentiating them from the small, round, porcelain white macules in end-stage Degos disease. Histopathology demonstrates thrombosis and fibrin occlusion of the upper and mid dermal vessels. Very minimal perivascular infiltrate typically is seen, but when it is present, the infiltrate mostly is lymphocytic. Hyalinization of the vessel walls also is seen, particularly in the atrophie blanche stage (Figure 3).7
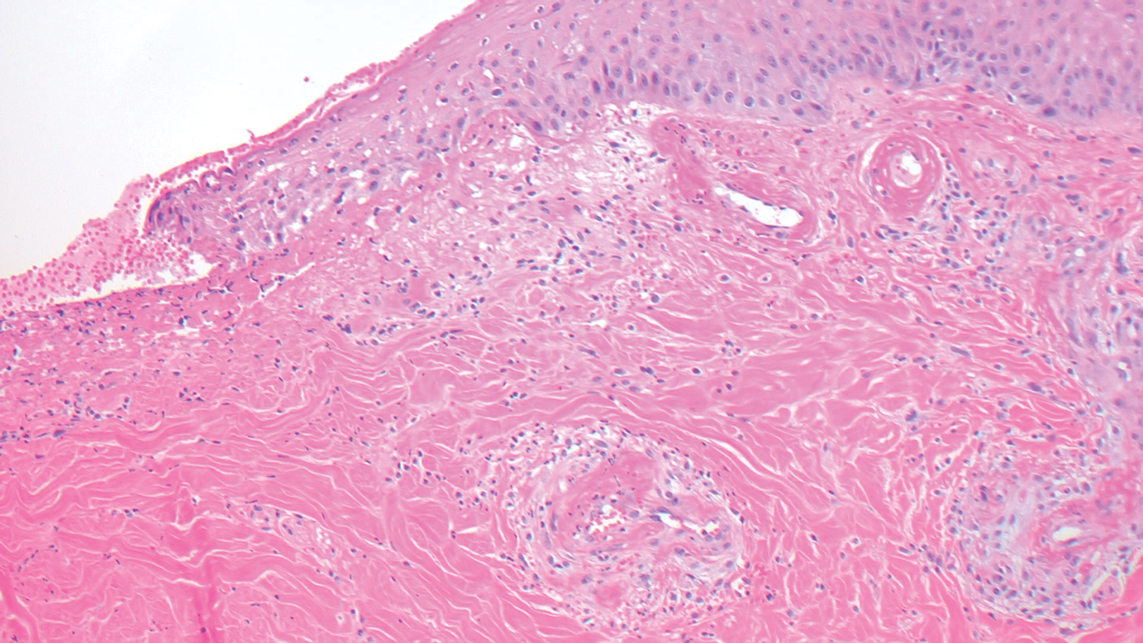
Lymphomatoid papulosis classically presents with pruritic red papules that often spontaneously involute. After resolution of the primary lesions, atrophic varioliform scars may be left behind that can resemble Degos disease.8 Classically, there are 5 histopathologic subtypes: A, B, C, D, and E. Type A is the most common type of lymphomatoid papulosis, and histopathology demonstrates a dermal lymphocytic infiltrate that consists of cells arranged in small clusters. Numerous medium- to large-sized atypical lymphocytes with prominent nucleoli and abundant cytoplasm are seen, and mitotic figures are common (Figure 4).8
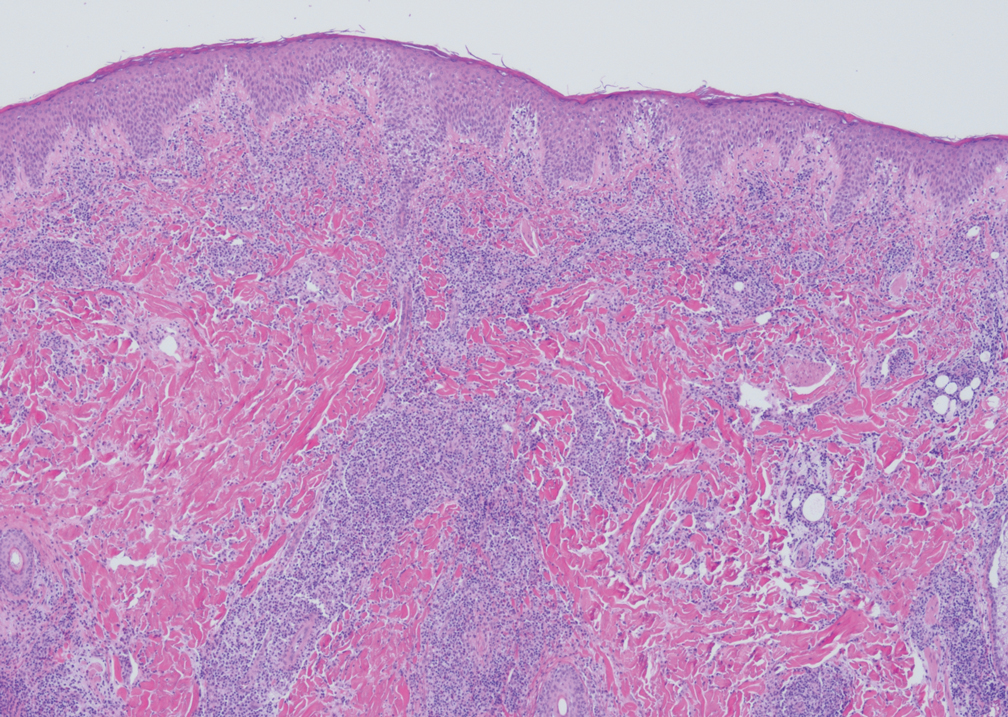
Our case was particularly interesting because the patient was 2 to 3 weeks pregnant. Degos disease in pregnancy appears to be quite exceptional. A PubMed search of articles indexed for MEDLINE using the terms Degos disease and pregnancy revealed only 4 other cases reported in the literature.9-12 With the exception of a single case that was complicated by severe abdominal pain requiring labor induction, the other reported cases resulted in uncomplicated pregnancies.9-12 Conversely, our patient's pregnancy was complicated by gestational hypertension and fetal hydrops requiring a preterm cesarean delivery. Furthermore, the infant had multiple complications, which were attributed to both placental insufficiency and a coagulopathic state.
Our patient also was found to have a heterozygous factor V Leiden mutation on workup. A PubMed search using the terms factor V Leiden mutation and Degos disease revealed 2 other cases of factor V Leiden mutation-associated Degos disease.13,14 The importance of factor V Leiden mutations in patients with Degos disease currently is unclear.
- Theodoridis A, Makrantonaki E, Zouboulis CC. Malignant atrophic papulosis (Köhlmeier-Degos disease)--a review. Orphanet J Rare Dis. 2013;8:10.
- Oliver B, Boehm M, Rosing DR, et al. Diffuse atrophic papules and plaques, intermittent abdominal pain, paresthesias, and cardiac abnormalities in a 55-year-old woman. J Am Acad Dermatol. 2016;75:1274-1277.
- Magro CM, Wang X, Garrett-Bakelman F, et al. The effects of eculizumab on the pathology of malignant atrophic papulosis. Orphanet J Rare Dis. 2013;8:185.
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016;75:457-465.
- Tronnier M, Mitteldorf C. Histologic features of granulomatous skin diseases. part 1: non-infectious granulomatous disorders. J Dtsch Dermatol Ges. 2015;13:211-216.
- Fistarol SK, Itin PH. Diagnosis and treatment of lichen sclerosus: an update. Am J Clin Dermatol. 2013;14:27-47.
- Vasudevan B, Neema S, Verma R. Livedoid vasculopathy: a review of pathogenesis and principles of management. Indian J Dermatol Venereol Leprol. 2016;82:478‐488.
- Martinez-Cabriales SA, Walsh S, Sade S, et al. Lymphomatoid papulosis: an update and review. J Eur Acad Dermatol Venereol. 2020;34:59-73.
- Moulin G, Barrut D, Franc MP, et al. Familial Degos' atrophic papulosis (mother-daughter). Ann Dermatol Venereol. 1984;111:149-155.
- Bogenrieder T, Kuske M, Landthaler M, et al. Benign Degos' disease developing during pregnancy and followed for 10 years. Acta Derm Venereol. 2002;82:284-287.
- Sharma S, Brennan B, Naden R, et al. A case of Degos disease in pregnancy. Obstet Med. 2016;9:167-168.
- Zhao Q, Zhang S, Dong A. An unusual case of abdominal pain. Gastroenterology. 2018;154:E1-E2.
- Darwich E, Guilabert A, Mascaró JM Jr, et al. Dermoscopic description of a patient with thrombocythemia and factor V Leiden mutation-associated Degos' disease. Int J Dermatol. 2011;50:604-606.
- Hohwy T, Jensen MG, Tøttrup A, et al. A fatal case of malignant atrophic papulosis (Degos' disease) in a man with factor V Leiden mutation and lupus anticoagulant. Acta Derm Venereol. 2006;86:245-247.
The Diagnosis: Degos Disease
The pathophysiology of Degos disease (malignant atrophic papulosis) is unknown.1 Histopathology demonstrates a wedge-shaped area of dermal necrosis with edema and mucin deposition extending from the papillary dermis to the deep reticular dermis. Occluded vessels, thrombosis, and perivascular lymphocytic infiltrates also may be seen, particularly at the dermal subcutaneous junction and at the periphery of the wedge-shaped infarction. The vascular damage that occurs may be the result of vasculitis, coagulopathy, or endothelial cell dysfunction.1
Patients typically present with small, round, erythematous papules that eventually develop atrophic porcelain white centers and telangiectatic rims. These lesions most commonly occur on the trunk and arms. In the benign form of atrophic papulosis, only the skin is involved; however, systemic involvement of the gastrointestinal tract and central nervous system can occur, resulting in bowel perforation and stroke, respectively.1 Although there is no definitive treatment of Degos disease, successful therapy with aspirin or dipyridamole has been reported.1 Eculizumab, a monoclonal antibody that binds C5, and treprostinil, a prostacyclin analog, are emerging treatment options.2,3 The differential diagnosis of Degos disease may include granuloma annulare, guttate extragenital lichen sclerosus, livedoid vasculopathy, and lymphomatoid papulosis.
Granuloma annulare may clinically mimic the erythematous papules seen in early Degos disease, and histopathology can be used to distinguish between these two disease processes. Localized granuloma annulare is the most common variant and clinically presents as pink papules and plaques in an annular configuration.4 Histopathology demonstrates an unremarkable epidermis; however, the dermis contains degenerated collagen surrounded by palisading histiocytes as well as lymphocytes. Similar to Degos disease, increased mucin is seen within these areas of degeneration, but occluded vessels and thrombosis typically are not seen (Figure 1).4,5
Guttate extragenital lichen sclerosus initially presents as polygonal, bluish white papules that coalesce into plaques.6 Over time, these lesions become more atrophic and may mimic Degos disease but appear differently on histopathology. Histopathology of lichen sclerosus classically demonstrates atrophy of the epidermis with loss of the rete ridges and vacuolar surface changes. Homogenization of the superficial/papillary dermis with an underlying bandlike lymphocytic infiltrate also is seen (Figure 2).6
Livedoid vasculopathy is characterized by chronic recurrent ulceration of the legs secondary to thrombosis and subsequent ischemia. In the initial phase of this disease, livedo reticularis is seen followed by the development of ulcerations. As these ulcerations heal, they leave behind porcelain white scars referred to as atrophie blanche.7 The areas of scarring in livedoid vasculopathy are broad and angulated, differentiating them from the small, round, porcelain white macules in end-stage Degos disease. Histopathology demonstrates thrombosis and fibrin occlusion of the upper and mid dermal vessels. Very minimal perivascular infiltrate typically is seen, but when it is present, the infiltrate mostly is lymphocytic. Hyalinization of the vessel walls also is seen, particularly in the atrophie blanche stage (Figure 3).7
Lymphomatoid papulosis classically presents with pruritic red papules that often spontaneously involute. After resolution of the primary lesions, atrophic varioliform scars may be left behind that can resemble Degos disease.8 Classically, there are 5 histopathologic subtypes: A, B, C, D, and E. Type A is the most common type of lymphomatoid papulosis, and histopathology demonstrates a dermal lymphocytic infiltrate that consists of cells arranged in small clusters. Numerous medium- to large-sized atypical lymphocytes with prominent nucleoli and abundant cytoplasm are seen, and mitotic figures are common (Figure 4).8
Our case was particularly interesting because the patient was 2 to 3 weeks pregnant. Degos disease in pregnancy appears to be quite exceptional. A PubMed search of articles indexed for MEDLINE using the terms Degos disease and pregnancy revealed only 4 other cases reported in the literature.9-12 With the exception of a single case that was complicated by severe abdominal pain requiring labor induction, the other reported cases resulted in uncomplicated pregnancies.9-12 Conversely, our patient's pregnancy was complicated by gestational hypertension and fetal hydrops requiring a preterm cesarean delivery. Furthermore, the infant had multiple complications, which were attributed to both placental insufficiency and a coagulopathic state.
Our patient also was found to have a heterozygous factor V Leiden mutation on workup. A PubMed search using the terms factor V Leiden mutation and Degos disease revealed 2 other cases of factor V Leiden mutation-associated Degos disease.13,14 The importance of factor V Leiden mutations in patients with Degos disease currently is unclear.
The Diagnosis: Degos Disease
The pathophysiology of Degos disease (malignant atrophic papulosis) is unknown.1 Histopathology demonstrates a wedge-shaped area of dermal necrosis with edema and mucin deposition extending from the papillary dermis to the deep reticular dermis. Occluded vessels, thrombosis, and perivascular lymphocytic infiltrates also may be seen, particularly at the dermal subcutaneous junction and at the periphery of the wedge-shaped infarction. The vascular damage that occurs may be the result of vasculitis, coagulopathy, or endothelial cell dysfunction.1
Patients typically present with small, round, erythematous papules that eventually develop atrophic porcelain white centers and telangiectatic rims. These lesions most commonly occur on the trunk and arms. In the benign form of atrophic papulosis, only the skin is involved; however, systemic involvement of the gastrointestinal tract and central nervous system can occur, resulting in bowel perforation and stroke, respectively.1 Although there is no definitive treatment of Degos disease, successful therapy with aspirin or dipyridamole has been reported.1 Eculizumab, a monoclonal antibody that binds C5, and treprostinil, a prostacyclin analog, are emerging treatment options.2,3 The differential diagnosis of Degos disease may include granuloma annulare, guttate extragenital lichen sclerosus, livedoid vasculopathy, and lymphomatoid papulosis.
Granuloma annulare may clinically mimic the erythematous papules seen in early Degos disease, and histopathology can be used to distinguish between these two disease processes. Localized granuloma annulare is the most common variant and clinically presents as pink papules and plaques in an annular configuration.4 Histopathology demonstrates an unremarkable epidermis; however, the dermis contains degenerated collagen surrounded by palisading histiocytes as well as lymphocytes. Similar to Degos disease, increased mucin is seen within these areas of degeneration, but occluded vessels and thrombosis typically are not seen (Figure 1).4,5
Guttate extragenital lichen sclerosus initially presents as polygonal, bluish white papules that coalesce into plaques.6 Over time, these lesions become more atrophic and may mimic Degos disease but appear differently on histopathology. Histopathology of lichen sclerosus classically demonstrates atrophy of the epidermis with loss of the rete ridges and vacuolar surface changes. Homogenization of the superficial/papillary dermis with an underlying bandlike lymphocytic infiltrate also is seen (Figure 2).6
Livedoid vasculopathy is characterized by chronic recurrent ulceration of the legs secondary to thrombosis and subsequent ischemia. In the initial phase of this disease, livedo reticularis is seen followed by the development of ulcerations. As these ulcerations heal, they leave behind porcelain white scars referred to as atrophie blanche.7 The areas of scarring in livedoid vasculopathy are broad and angulated, differentiating them from the small, round, porcelain white macules in end-stage Degos disease. Histopathology demonstrates thrombosis and fibrin occlusion of the upper and mid dermal vessels. Very minimal perivascular infiltrate typically is seen, but when it is present, the infiltrate mostly is lymphocytic. Hyalinization of the vessel walls also is seen, particularly in the atrophie blanche stage (Figure 3).7
Lymphomatoid papulosis classically presents with pruritic red papules that often spontaneously involute. After resolution of the primary lesions, atrophic varioliform scars may be left behind that can resemble Degos disease.8 Classically, there are 5 histopathologic subtypes: A, B, C, D, and E. Type A is the most common type of lymphomatoid papulosis, and histopathology demonstrates a dermal lymphocytic infiltrate that consists of cells arranged in small clusters. Numerous medium- to large-sized atypical lymphocytes with prominent nucleoli and abundant cytoplasm are seen, and mitotic figures are common (Figure 4).8
Our case was particularly interesting because the patient was 2 to 3 weeks pregnant. Degos disease in pregnancy appears to be quite exceptional. A PubMed search of articles indexed for MEDLINE using the terms Degos disease and pregnancy revealed only 4 other cases reported in the literature.9-12 With the exception of a single case that was complicated by severe abdominal pain requiring labor induction, the other reported cases resulted in uncomplicated pregnancies.9-12 Conversely, our patient's pregnancy was complicated by gestational hypertension and fetal hydrops requiring a preterm cesarean delivery. Furthermore, the infant had multiple complications, which were attributed to both placental insufficiency and a coagulopathic state.
Our patient also was found to have a heterozygous factor V Leiden mutation on workup. A PubMed search using the terms factor V Leiden mutation and Degos disease revealed 2 other cases of factor V Leiden mutation-associated Degos disease.13,14 The importance of factor V Leiden mutations in patients with Degos disease currently is unclear.
- Theodoridis A, Makrantonaki E, Zouboulis CC. Malignant atrophic papulosis (Köhlmeier-Degos disease)--a review. Orphanet J Rare Dis. 2013;8:10.
- Oliver B, Boehm M, Rosing DR, et al. Diffuse atrophic papules and plaques, intermittent abdominal pain, paresthesias, and cardiac abnormalities in a 55-year-old woman. J Am Acad Dermatol. 2016;75:1274-1277.
- Magro CM, Wang X, Garrett-Bakelman F, et al. The effects of eculizumab on the pathology of malignant atrophic papulosis. Orphanet J Rare Dis. 2013;8:185.
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016;75:457-465.
- Tronnier M, Mitteldorf C. Histologic features of granulomatous skin diseases. part 1: non-infectious granulomatous disorders. J Dtsch Dermatol Ges. 2015;13:211-216.
- Fistarol SK, Itin PH. Diagnosis and treatment of lichen sclerosus: an update. Am J Clin Dermatol. 2013;14:27-47.
- Vasudevan B, Neema S, Verma R. Livedoid vasculopathy: a review of pathogenesis and principles of management. Indian J Dermatol Venereol Leprol. 2016;82:478‐488.
- Martinez-Cabriales SA, Walsh S, Sade S, et al. Lymphomatoid papulosis: an update and review. J Eur Acad Dermatol Venereol. 2020;34:59-73.
- Moulin G, Barrut D, Franc MP, et al. Familial Degos' atrophic papulosis (mother-daughter). Ann Dermatol Venereol. 1984;111:149-155.
- Bogenrieder T, Kuske M, Landthaler M, et al. Benign Degos' disease developing during pregnancy and followed for 10 years. Acta Derm Venereol. 2002;82:284-287.
- Sharma S, Brennan B, Naden R, et al. A case of Degos disease in pregnancy. Obstet Med. 2016;9:167-168.
- Zhao Q, Zhang S, Dong A. An unusual case of abdominal pain. Gastroenterology. 2018;154:E1-E2.
- Darwich E, Guilabert A, Mascaró JM Jr, et al. Dermoscopic description of a patient with thrombocythemia and factor V Leiden mutation-associated Degos' disease. Int J Dermatol. 2011;50:604-606.
- Hohwy T, Jensen MG, Tøttrup A, et al. A fatal case of malignant atrophic papulosis (Degos' disease) in a man with factor V Leiden mutation and lupus anticoagulant. Acta Derm Venereol. 2006;86:245-247.
- Theodoridis A, Makrantonaki E, Zouboulis CC. Malignant atrophic papulosis (Köhlmeier-Degos disease)--a review. Orphanet J Rare Dis. 2013;8:10.
- Oliver B, Boehm M, Rosing DR, et al. Diffuse atrophic papules and plaques, intermittent abdominal pain, paresthesias, and cardiac abnormalities in a 55-year-old woman. J Am Acad Dermatol. 2016;75:1274-1277.
- Magro CM, Wang X, Garrett-Bakelman F, et al. The effects of eculizumab on the pathology of malignant atrophic papulosis. Orphanet J Rare Dis. 2013;8:185.
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016;75:457-465.
- Tronnier M, Mitteldorf C. Histologic features of granulomatous skin diseases. part 1: non-infectious granulomatous disorders. J Dtsch Dermatol Ges. 2015;13:211-216.
- Fistarol SK, Itin PH. Diagnosis and treatment of lichen sclerosus: an update. Am J Clin Dermatol. 2013;14:27-47.
- Vasudevan B, Neema S, Verma R. Livedoid vasculopathy: a review of pathogenesis and principles of management. Indian J Dermatol Venereol Leprol. 2016;82:478‐488.
- Martinez-Cabriales SA, Walsh S, Sade S, et al. Lymphomatoid papulosis: an update and review. J Eur Acad Dermatol Venereol. 2020;34:59-73.
- Moulin G, Barrut D, Franc MP, et al. Familial Degos' atrophic papulosis (mother-daughter). Ann Dermatol Venereol. 1984;111:149-155.
- Bogenrieder T, Kuske M, Landthaler M, et al. Benign Degos' disease developing during pregnancy and followed for 10 years. Acta Derm Venereol. 2002;82:284-287.
- Sharma S, Brennan B, Naden R, et al. A case of Degos disease in pregnancy. Obstet Med. 2016;9:167-168.
- Zhao Q, Zhang S, Dong A. An unusual case of abdominal pain. Gastroenterology. 2018;154:E1-E2.
- Darwich E, Guilabert A, Mascaró JM Jr, et al. Dermoscopic description of a patient with thrombocythemia and factor V Leiden mutation-associated Degos' disease. Int J Dermatol. 2011;50:604-606.
- Hohwy T, Jensen MG, Tøttrup A, et al. A fatal case of malignant atrophic papulosis (Degos' disease) in a man with factor V Leiden mutation and lupus anticoagulant. Acta Derm Venereol. 2006;86:245-247.
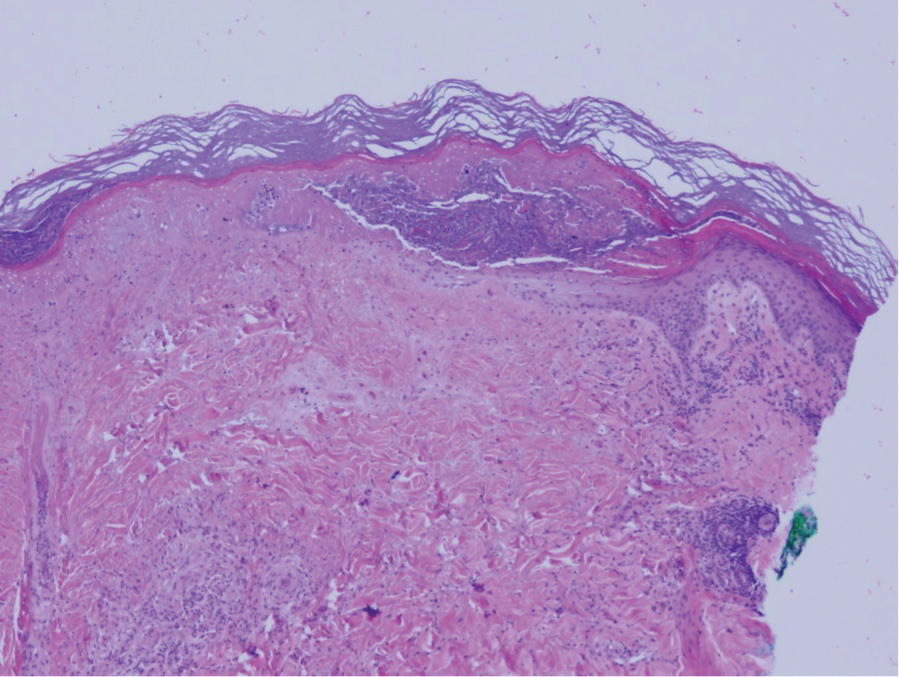
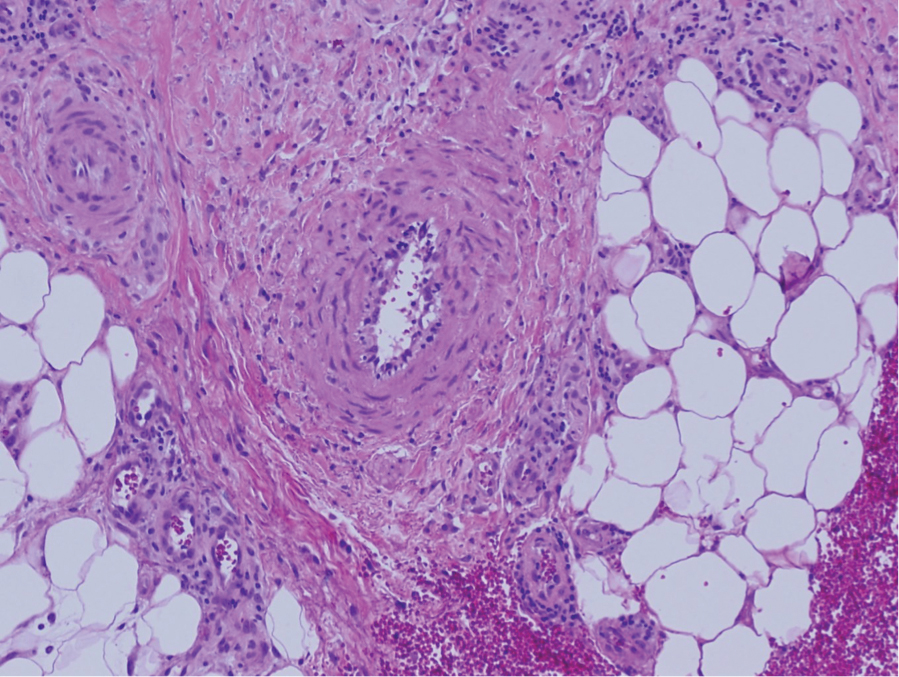
A 36-year-old pregnant woman presented with painful erythematous papules on the palms and fingers of 2 months’ duration. Similar lesions developed on the thighs and feet several weeks later. Two tender macules with central areas of porcelain white scarring rimmed by telangiectases on the right foot also were present. A punch biopsy of these lesions demonstrated a wedge-shaped area of ischemic necrosis associated with dermal mucin without associated necrobiosis. Fibrin thrombi were seen within several small dermal vessels and were associated with a perivascular lymphocytic infiltrate. Endotheliitis was observed within a deep dermal vessel. Laboratory workup including syphilis IgG, antinuclear antibodies, extractable nuclear antigen antibodies, anti–double-stranded DNA, antistreptolysin O antibodies, Russell viper venom time, cryoglobulin, hepatitis screening, perinuclear antineutrophil cytoplasmic antibodies (ANCA), and cytoplasmic ANCA was unremarkable. Hypercoagulable studies including prothrombin gene mutation, factor V Leiden, plasminogen, proteins C and S, antithrombin III, homocysteine, and antiphospholipid IgM and IgG antibodies were notable only for heterozygosity for factor V Leiden.