User login
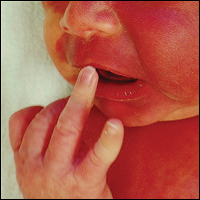
Cases link vitiligoid lichen sclerosus and darker skin
, said Margaret H. Dennin, of the University of Chicago, and her associates.
Vitiligoid lichen sclerosus is a superficial variant of lichen sclerosus (LS), in which the lesion clinically appears to be vitiligo, but histologically is consistent with LS.
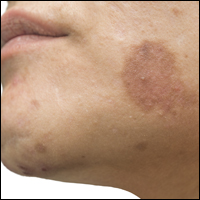
Seven dark-skinned girls aged 3-9 years had symptomatic (pruritus, pain, bleeding, constipation) depigmented patches of the vulvar or perianal region; three had purpuric lesions. None of the patients had atrophy or scarring, and they had no depigmentation anywhere else on their bodies. Follow-up was an average 2 years (range 3 months to 4 years).
Treatment with high-potency topical steroids, calcineurin inhibitors, or both resulted in improvement or resolution of their symptoms in all cases, but there was mild or no improvement in the depigmentation. Biopsies were not performed because of the patients’ young age and the location of the lesions, the investigators said.
The term vitiligoid lichen sclerosus was first coined in 1961 by Borda et al. when depigmented patches, as seen in both conditions, constituted the clinical appearance, but lacked the inflammation, atrophy, and sclerosis of typical LS. Histologically, these lesions were like LS, “based on the presence of a thin band of papillary dermal sclerosis,” Ms. Dennin and her associates said. Borda et al. suggested that vitiligoid lichen sclerosus might be limited to dark-skinned people, and recent reports support this. Alternatively, it may be that the depigmentation simply is more obvious on dark-skinned people, and asymptomatic cases go unnoticed on lighter-skinned people, the investigators surmised.
Both vitiligo and LS are autoimmune cutaneous disorders, and they both often affect the anogenital region. The conditions “may be linked through a common autoimmune response from exposed intracellular or altered cell surface antigens on damaged melanocytes,” the investigators said. “Histologic evidence demonstrates that development of vitiligo involves a preceding lichenoid inflammatory reaction that may trigger an autoimmune reaction to melanocytes, decreasing their number. Evolving vitiligo with a lichenoid reaction may result in epitope spreading and the development of LS.”
The study is limited by its retrospective nature, small sample size, and lack of biopsies, the researchers noted. Larger studies are needed to look at the overlap of the conditions, and “understand the true prevalence of vitiligoid lichen sclerosus,” Ms. Dennin and her associates said.
Read more in Pediatric Dermatology (2018. doi: 10.1111/pde.13399).
, said Margaret H. Dennin, of the University of Chicago, and her associates.
Vitiligoid lichen sclerosus is a superficial variant of lichen sclerosus (LS), in which the lesion clinically appears to be vitiligo, but histologically is consistent with LS.
Seven dark-skinned girls aged 3-9 years had symptomatic (pruritus, pain, bleeding, constipation) depigmented patches of the vulvar or perianal region; three had purpuric lesions. None of the patients had atrophy or scarring, and they had no depigmentation anywhere else on their bodies. Follow-up was an average 2 years (range 3 months to 4 years).
Treatment with high-potency topical steroids, calcineurin inhibitors, or both resulted in improvement or resolution of their symptoms in all cases, but there was mild or no improvement in the depigmentation. Biopsies were not performed because of the patients’ young age and the location of the lesions, the investigators said.
The term vitiligoid lichen sclerosus was first coined in 1961 by Borda et al. when depigmented patches, as seen in both conditions, constituted the clinical appearance, but lacked the inflammation, atrophy, and sclerosis of typical LS. Histologically, these lesions were like LS, “based on the presence of a thin band of papillary dermal sclerosis,” Ms. Dennin and her associates said. Borda et al. suggested that vitiligoid lichen sclerosus might be limited to dark-skinned people, and recent reports support this. Alternatively, it may be that the depigmentation simply is more obvious on dark-skinned people, and asymptomatic cases go unnoticed on lighter-skinned people, the investigators surmised.
Both vitiligo and LS are autoimmune cutaneous disorders, and they both often affect the anogenital region. The conditions “may be linked through a common autoimmune response from exposed intracellular or altered cell surface antigens on damaged melanocytes,” the investigators said. “Histologic evidence demonstrates that development of vitiligo involves a preceding lichenoid inflammatory reaction that may trigger an autoimmune reaction to melanocytes, decreasing their number. Evolving vitiligo with a lichenoid reaction may result in epitope spreading and the development of LS.”
The study is limited by its retrospective nature, small sample size, and lack of biopsies, the researchers noted. Larger studies are needed to look at the overlap of the conditions, and “understand the true prevalence of vitiligoid lichen sclerosus,” Ms. Dennin and her associates said.
Read more in Pediatric Dermatology (2018. doi: 10.1111/pde.13399).
, said Margaret H. Dennin, of the University of Chicago, and her associates.
Vitiligoid lichen sclerosus is a superficial variant of lichen sclerosus (LS), in which the lesion clinically appears to be vitiligo, but histologically is consistent with LS.
Seven dark-skinned girls aged 3-9 years had symptomatic (pruritus, pain, bleeding, constipation) depigmented patches of the vulvar or perianal region; three had purpuric lesions. None of the patients had atrophy or scarring, and they had no depigmentation anywhere else on their bodies. Follow-up was an average 2 years (range 3 months to 4 years).
Treatment with high-potency topical steroids, calcineurin inhibitors, or both resulted in improvement or resolution of their symptoms in all cases, but there was mild or no improvement in the depigmentation. Biopsies were not performed because of the patients’ young age and the location of the lesions, the investigators said.
The term vitiligoid lichen sclerosus was first coined in 1961 by Borda et al. when depigmented patches, as seen in both conditions, constituted the clinical appearance, but lacked the inflammation, atrophy, and sclerosis of typical LS. Histologically, these lesions were like LS, “based on the presence of a thin band of papillary dermal sclerosis,” Ms. Dennin and her associates said. Borda et al. suggested that vitiligoid lichen sclerosus might be limited to dark-skinned people, and recent reports support this. Alternatively, it may be that the depigmentation simply is more obvious on dark-skinned people, and asymptomatic cases go unnoticed on lighter-skinned people, the investigators surmised.
Both vitiligo and LS are autoimmune cutaneous disorders, and they both often affect the anogenital region. The conditions “may be linked through a common autoimmune response from exposed intracellular or altered cell surface antigens on damaged melanocytes,” the investigators said. “Histologic evidence demonstrates that development of vitiligo involves a preceding lichenoid inflammatory reaction that may trigger an autoimmune reaction to melanocytes, decreasing their number. Evolving vitiligo with a lichenoid reaction may result in epitope spreading and the development of LS.”
The study is limited by its retrospective nature, small sample size, and lack of biopsies, the researchers noted. Larger studies are needed to look at the overlap of the conditions, and “understand the true prevalence of vitiligoid lichen sclerosus,” Ms. Dennin and her associates said.
Read more in Pediatric Dermatology (2018. doi: 10.1111/pde.13399).
FROM PEDIATRIC DERMATOLOGY
Local Depigmentation of a Tattoo
The Diagnosis: Dermatofibroma
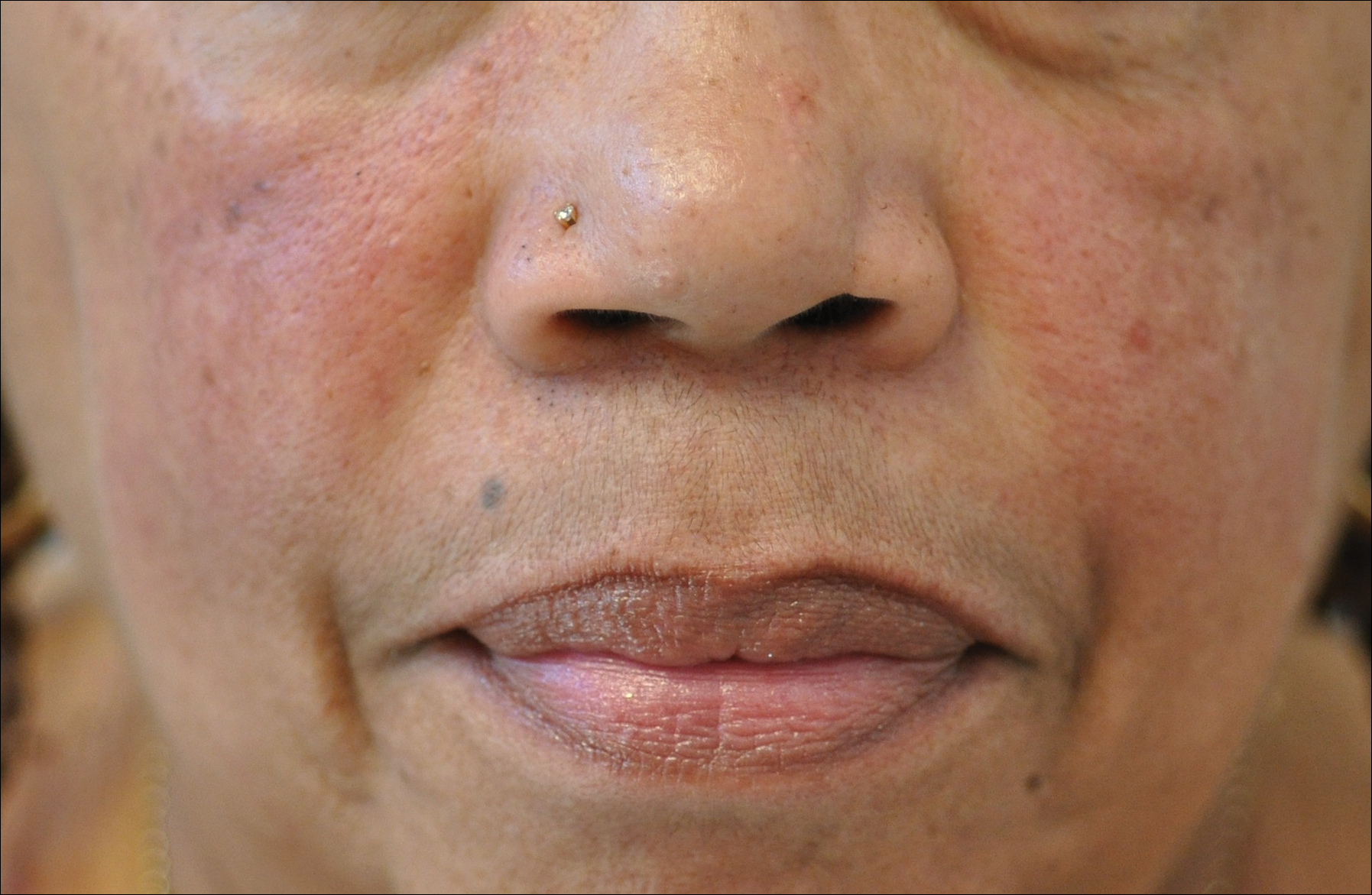
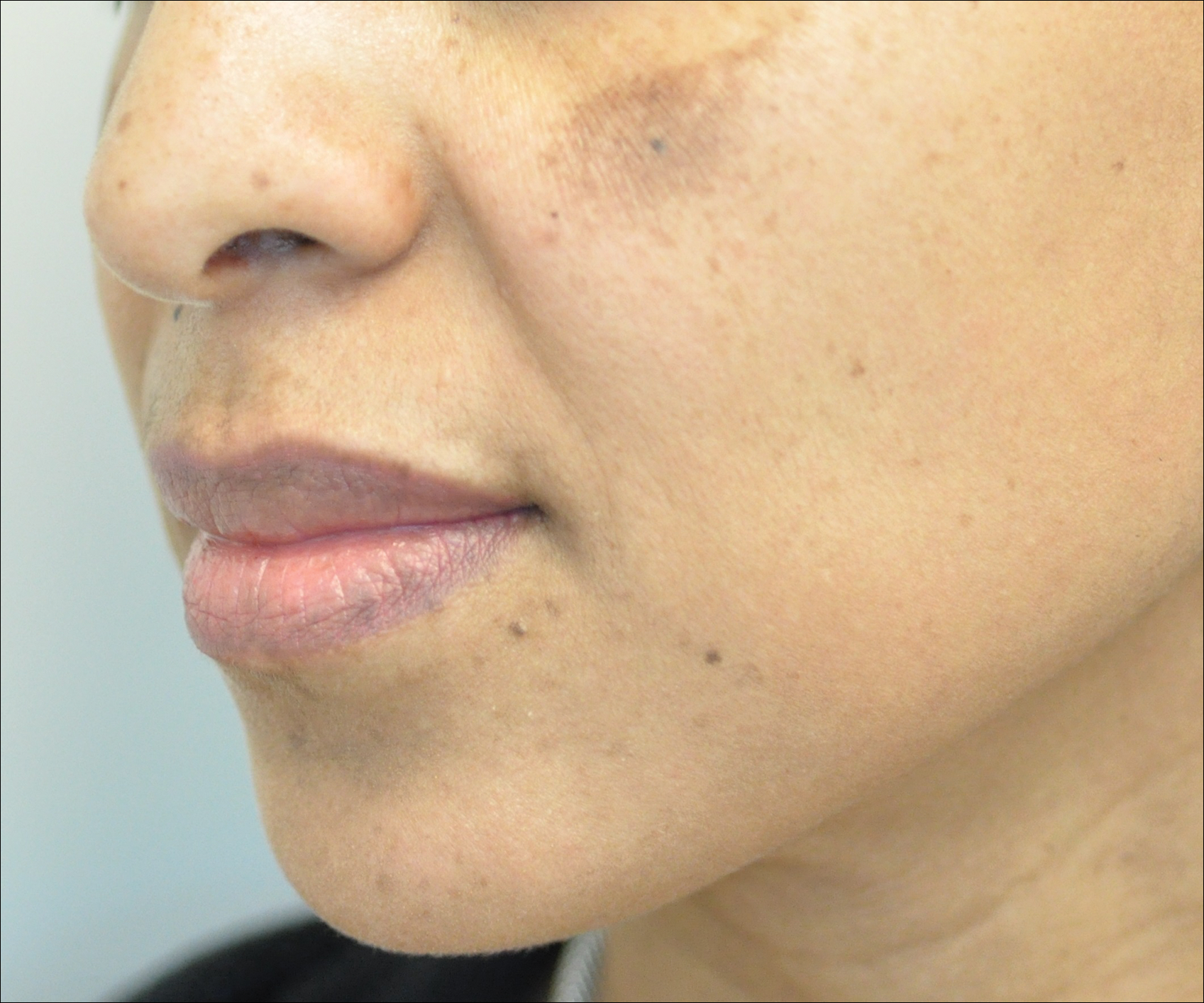
On dermoscopy, a central stellate, white, scarlike patch was seen (Figure). On both legs the patient had several additional brown 5- to 7-mm papules with similar dermoscopic features.
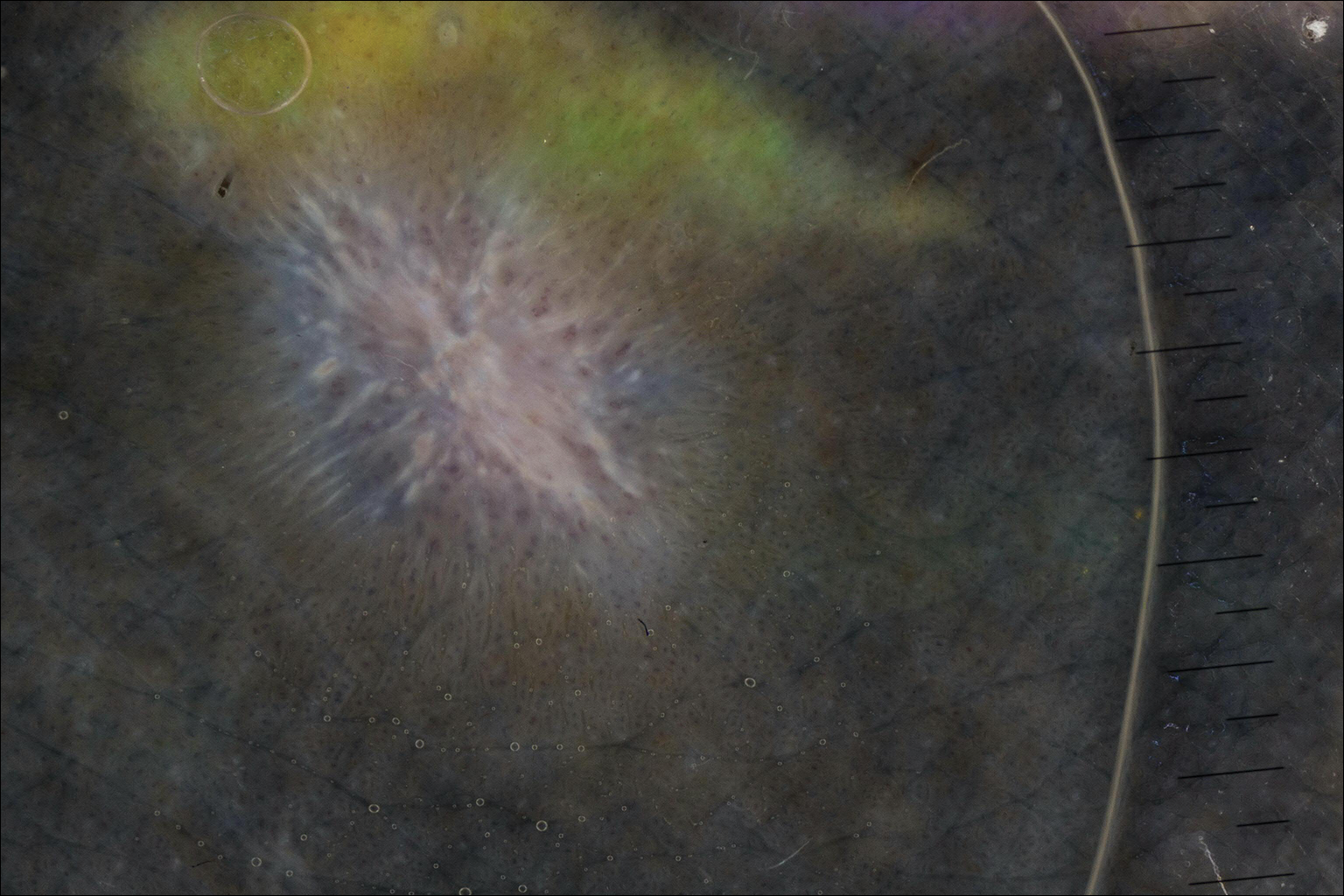
Dermatofibromas are common benign fibrosing tumors that appear as firm papules or plaques with variable color, commonly on the legs. Typically, lateral compression of a dermatofibroma causes downward displacement, called a positive dimple sign. On histology, fibroblasts and myofibroblasts can be seen as short intersecting fascicles with variable inflammatory cells and induction of adjacent structure hyperplasia. The etiology of dermatofibromas is unclear, though some are thought to be secondary to trauma or arthropod bites.1 Because these tumors are benign, the correct diagnosis can avoid unnecessary biopsies or other procedures.
The dermoscopic features of dermatofibromas have been well established.2 As perhaps the most easily identified structure, scarlike patches were seen in as many as 92% (22/24) of dermatofibromas in one study by Ferarri et al,3 while pigment networks also are commonly seen.2 In our case, given the surrounding dense tattoo deposition, it was difficult to ascertain any pigment network. However, the scarlike central patch was clearly apparent by dermoscopy.
Because dermatofibromas are hypothesized to be secondary to trauma, presumably applying tattoos also may cause dermatofibromas. Limited cases have described dermatofibromas arising in tattoos applied several months to years prior.4-6 No prior cases utilized dermoscopy. In our case, clinical examination and dermoscopy clearly demonstrated features consistent with a dermatofibroma, and the patient had more characteristic dermatofibromas scattered elsewhere on both legs. The patient was reassured that the lesions were benign and that the depigmentation was likely secondary to the process of dermatofibroma growth. She declined any treatment.
- Bolognia J, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Saunders; 2012.
- Zaballos P, Puig S, Llambrich A, et al. Dermoscopy of dermatofibromas: a prospective morphological study of 412 cases. Arch Dermatol. 2008;144:75-83.
- Ferrari A, Soyer HP, Peris K, et al. Central white scarlike patch: a dermatoscopic clue for the diagnosis of dermatofibroma. J Am Acad Dermatol. 2000;43:1123-1125.
- Kluger N, Cotten H, Magana C, et al. Dermatofibroma occurring within a tattoo: report of two cases. J Cutan Pathol. 2008;35:696-698.
- Lobato-Berezo A, Churruca-Grijelmo M, Martínez-Pérez M, et al. Dermatofibroma arising within a black tattoo [published online September 23, 2014]. Case Rep Dermatol Med. 2014;2014:745304.
- Bittencourt Mde J, Miranda MF, Parijós AM, et al. Dermatofibroma in a black tattoo: report of a case. An Bras Dermatol. 2013;88:614-616.
The Diagnosis: Dermatofibroma
On dermoscopy, a central stellate, white, scarlike patch was seen (Figure). On both legs the patient had several additional brown 5- to 7-mm papules with similar dermoscopic features.
Dermatofibromas are common benign fibrosing tumors that appear as firm papules or plaques with variable color, commonly on the legs. Typically, lateral compression of a dermatofibroma causes downward displacement, called a positive dimple sign. On histology, fibroblasts and myofibroblasts can be seen as short intersecting fascicles with variable inflammatory cells and induction of adjacent structure hyperplasia. The etiology of dermatofibromas is unclear, though some are thought to be secondary to trauma or arthropod bites.1 Because these tumors are benign, the correct diagnosis can avoid unnecessary biopsies or other procedures.
The dermoscopic features of dermatofibromas have been well established.2 As perhaps the most easily identified structure, scarlike patches were seen in as many as 92% (22/24) of dermatofibromas in one study by Ferarri et al,3 while pigment networks also are commonly seen.2 In our case, given the surrounding dense tattoo deposition, it was difficult to ascertain any pigment network. However, the scarlike central patch was clearly apparent by dermoscopy.
Because dermatofibromas are hypothesized to be secondary to trauma, presumably applying tattoos also may cause dermatofibromas. Limited cases have described dermatofibromas arising in tattoos applied several months to years prior.4-6 No prior cases utilized dermoscopy. In our case, clinical examination and dermoscopy clearly demonstrated features consistent with a dermatofibroma, and the patient had more characteristic dermatofibromas scattered elsewhere on both legs. The patient was reassured that the lesions were benign and that the depigmentation was likely secondary to the process of dermatofibroma growth. She declined any treatment.
The Diagnosis: Dermatofibroma
On dermoscopy, a central stellate, white, scarlike patch was seen (Figure). On both legs the patient had several additional brown 5- to 7-mm papules with similar dermoscopic features.
Dermatofibromas are common benign fibrosing tumors that appear as firm papules or plaques with variable color, commonly on the legs. Typically, lateral compression of a dermatofibroma causes downward displacement, called a positive dimple sign. On histology, fibroblasts and myofibroblasts can be seen as short intersecting fascicles with variable inflammatory cells and induction of adjacent structure hyperplasia. The etiology of dermatofibromas is unclear, though some are thought to be secondary to trauma or arthropod bites.1 Because these tumors are benign, the correct diagnosis can avoid unnecessary biopsies or other procedures.
The dermoscopic features of dermatofibromas have been well established.2 As perhaps the most easily identified structure, scarlike patches were seen in as many as 92% (22/24) of dermatofibromas in one study by Ferarri et al,3 while pigment networks also are commonly seen.2 In our case, given the surrounding dense tattoo deposition, it was difficult to ascertain any pigment network. However, the scarlike central patch was clearly apparent by dermoscopy.
Because dermatofibromas are hypothesized to be secondary to trauma, presumably applying tattoos also may cause dermatofibromas. Limited cases have described dermatofibromas arising in tattoos applied several months to years prior.4-6 No prior cases utilized dermoscopy. In our case, clinical examination and dermoscopy clearly demonstrated features consistent with a dermatofibroma, and the patient had more characteristic dermatofibromas scattered elsewhere on both legs. The patient was reassured that the lesions were benign and that the depigmentation was likely secondary to the process of dermatofibroma growth. She declined any treatment.
- Bolognia J, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Saunders; 2012.
- Zaballos P, Puig S, Llambrich A, et al. Dermoscopy of dermatofibromas: a prospective morphological study of 412 cases. Arch Dermatol. 2008;144:75-83.
- Ferrari A, Soyer HP, Peris K, et al. Central white scarlike patch: a dermatoscopic clue for the diagnosis of dermatofibroma. J Am Acad Dermatol. 2000;43:1123-1125.
- Kluger N, Cotten H, Magana C, et al. Dermatofibroma occurring within a tattoo: report of two cases. J Cutan Pathol. 2008;35:696-698.
- Lobato-Berezo A, Churruca-Grijelmo M, Martínez-Pérez M, et al. Dermatofibroma arising within a black tattoo [published online September 23, 2014]. Case Rep Dermatol Med. 2014;2014:745304.
- Bittencourt Mde J, Miranda MF, Parijós AM, et al. Dermatofibroma in a black tattoo: report of a case. An Bras Dermatol. 2013;88:614-616.
- Bolognia J, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Saunders; 2012.
- Zaballos P, Puig S, Llambrich A, et al. Dermoscopy of dermatofibromas: a prospective morphological study of 412 cases. Arch Dermatol. 2008;144:75-83.
- Ferrari A, Soyer HP, Peris K, et al. Central white scarlike patch: a dermatoscopic clue for the diagnosis of dermatofibroma. J Am Acad Dermatol. 2000;43:1123-1125.
- Kluger N, Cotten H, Magana C, et al. Dermatofibroma occurring within a tattoo: report of two cases. J Cutan Pathol. 2008;35:696-698.
- Lobato-Berezo A, Churruca-Grijelmo M, Martínez-Pérez M, et al. Dermatofibroma arising within a black tattoo [published online September 23, 2014]. Case Rep Dermatol Med. 2014;2014:745304.
- Bittencourt Mde J, Miranda MF, Parijós AM, et al. Dermatofibroma in a black tattoo: report of a case. An Bras Dermatol. 2013;88:614-616.
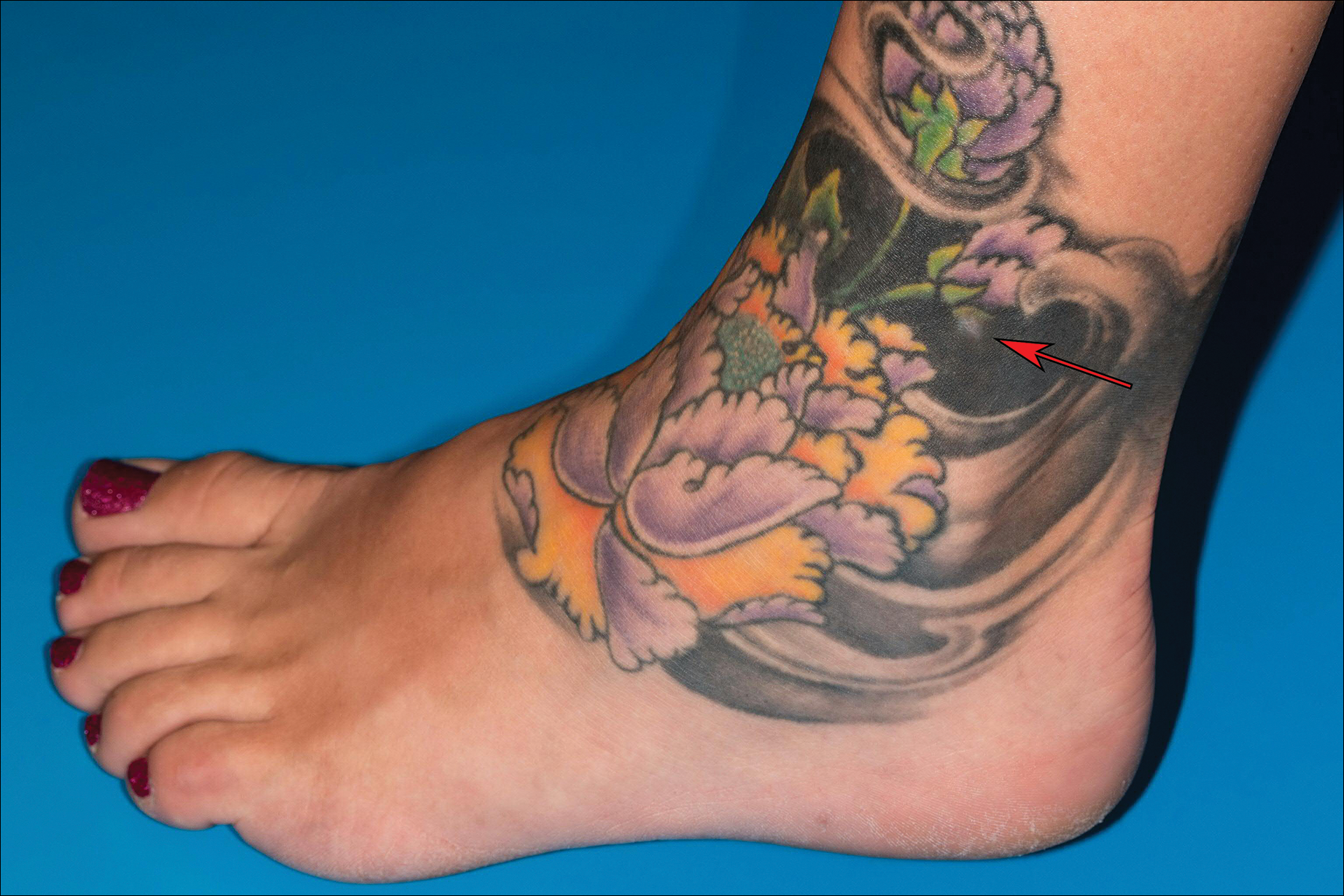
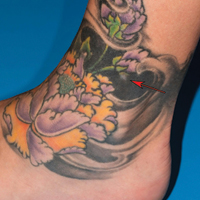
A 41-year-old woman presented with loss of pigment in a tattoo on the left ankle. The tattoo was initially placed several years prior to presentation. For an uncertain amount of time, she had noticed a small palpable whitish area with loss of tattoo pigment. There was no corresponding pain, pruritis, or other symptoms. Her dermatologic history was notable only for keratosis pilaris. Physical examination showed an approximately 7-mm whitish firm papule on the lateral aspect of the left ankle, clearly visible in an otherwise green-black area of the tattoo (arrow). The lesion displaced downward with lateral compression.
Cosmetic Corner: Dermatologists Weigh in on Pigment Correctors
To improve patient care and outcomes, leading dermatologists offered their recommendations on pigment correctors. Consideration must be given to:
- dEp Patch Full Face Mask
Activaderm, Inc
“This product uses microcurrent to push vitamin C into the skin. Vitamin C, a known antioxidant that usually has a difficult time passing through the stratum corneum, corrects pigmentary abnormalities. The product also comes with a botanical pigment corrector.”—Gary Goldenberg, MD, New York, New York
- De-Spot Skin Brightening Corrector
Peter Thomas Roth Labs LLC
“This product is a useful over-the-counter adjunct to prescription-strength hydroquinone, with niacinamide as one of the active ingredients.”—Shari Lipner, MD, PhD, New York, New York
- Glytone Dark Spot Corrector
Pierre Fabre Laboratories
“With 2% hydroquinone, glycolic acid, and kojic acid, you have a highly effective combination of ingredients that work synergistically to lighten areas of skin discoloration.”—Jeannette Graf, MD, Great Neck, New York
Cutis invites readers to send us their recommendations. Bar soap, lip plumper, and night cream will be featured in upcoming editions of Cosmetic Corner. Please e-mail your recommendation(s) to the Editorial Office.
Disclaimer: Opinions expressed herein do not necessarily reflect those of Cutis or Frontline Medical Communications Inc. and shall not be used for product endorsement purposes. Any reference made to a specific commercial product does not indicate or imply that Cutis or Frontline Medical Communications Inc. endorses, recommends, or favors the product mentioned. No guarantee is given to the effects of recommended products.
To improve patient care and outcomes, leading dermatologists offered their recommendations on pigment correctors. Consideration must be given to:
- dEp Patch Full Face Mask
Activaderm, Inc
“This product uses microcurrent to push vitamin C into the skin. Vitamin C, a known antioxidant that usually has a difficult time passing through the stratum corneum, corrects pigmentary abnormalities. The product also comes with a botanical pigment corrector.”—Gary Goldenberg, MD, New York, New York
- De-Spot Skin Brightening Corrector
Peter Thomas Roth Labs LLC
“This product is a useful over-the-counter adjunct to prescription-strength hydroquinone, with niacinamide as one of the active ingredients.”—Shari Lipner, MD, PhD, New York, New York
- Glytone Dark Spot Corrector
Pierre Fabre Laboratories
“With 2% hydroquinone, glycolic acid, and kojic acid, you have a highly effective combination of ingredients that work synergistically to lighten areas of skin discoloration.”—Jeannette Graf, MD, Great Neck, New York
Cutis invites readers to send us their recommendations. Bar soap, lip plumper, and night cream will be featured in upcoming editions of Cosmetic Corner. Please e-mail your recommendation(s) to the Editorial Office.
Disclaimer: Opinions expressed herein do not necessarily reflect those of Cutis or Frontline Medical Communications Inc. and shall not be used for product endorsement purposes. Any reference made to a specific commercial product does not indicate or imply that Cutis or Frontline Medical Communications Inc. endorses, recommends, or favors the product mentioned. No guarantee is given to the effects of recommended products.
To improve patient care and outcomes, leading dermatologists offered their recommendations on pigment correctors. Consideration must be given to:
- dEp Patch Full Face Mask
Activaderm, Inc
“This product uses microcurrent to push vitamin C into the skin. Vitamin C, a known antioxidant that usually has a difficult time passing through the stratum corneum, corrects pigmentary abnormalities. The product also comes with a botanical pigment corrector.”—Gary Goldenberg, MD, New York, New York
- De-Spot Skin Brightening Corrector
Peter Thomas Roth Labs LLC
“This product is a useful over-the-counter adjunct to prescription-strength hydroquinone, with niacinamide as one of the active ingredients.”—Shari Lipner, MD, PhD, New York, New York
- Glytone Dark Spot Corrector
Pierre Fabre Laboratories
“With 2% hydroquinone, glycolic acid, and kojic acid, you have a highly effective combination of ingredients that work synergistically to lighten areas of skin discoloration.”—Jeannette Graf, MD, Great Neck, New York
Cutis invites readers to send us their recommendations. Bar soap, lip plumper, and night cream will be featured in upcoming editions of Cosmetic Corner. Please e-mail your recommendation(s) to the Editorial Office.
Disclaimer: Opinions expressed herein do not necessarily reflect those of Cutis or Frontline Medical Communications Inc. and shall not be used for product endorsement purposes. Any reference made to a specific commercial product does not indicate or imply that Cutis or Frontline Medical Communications Inc. endorses, recommends, or favors the product mentioned. No guarantee is given to the effects of recommended products.
Approach to Treatment of Medical and Cosmetic Facial Concerns in Skin of Color Patients
The approach to the treatment of common skin disorders and cosmetic concerns in patients with skin of color (SOC) requires the clinician to understand the biological differences, nuances, and special considerations that are unique to patients with darker skin types.1-3 This article addresses 4 common facial concerns in SOC patients—acne, rosacea, facial hyperpigmentation, and cosmetic enhancement—and provides treatment recommendations and management pearls to assist the clinician with optimal outcomes for SOC patients.
Acne in SOC Patients
Acne vulgaris is one of the most common conditions that dermatologists treat and is estimated to affect 40 to 50 million individuals in the United States.1 Many of these acne patients are individuals with SOC.2-4 A study of 2835 females (aged 10–70 years) conducted in 4 different cities—Los Angeles, California; London, United Kingdom; Akita, Japan; and Rome, Italy—demonstrated acne prevalence of 37% in blacks, 32% in Hispanics, 30% in Asians, 24% in whites, and 23% in Continental Indians.5 Blacks, Hispanics, and Continental Indians demonstrated equal prevalence with comedonal and inflammatory acne. Asians displayed more inflammatory acne lesions than comedones. In contrast, whites demonstrated more comedones than inflammatory acne. Dyspigmentation, postinflammatory hyperpigmentation (PIH), and atrophic scars were more common in black and Hispanic females than other ethnicities.5 This study illustrated that acne-induced PIH is a common sequela in SOC patients and is the main reason they seek treatment.6,7
The pathogenesis of acne is the same in all racial and ethnic groups: (1) follicular hyperkeratinization and the formation of a microcomedone caused by abnormal desquamation of the keratinocytes within the sebaceous follicle, (2) production of sebum by circulating androgens, (3) proliferation of Propionibacterium acnes, and (4) inflammation. Subclinical inflammation is present throughout all stages of acne, including normal-appearing skin, inflammatory lesions, comedones, and scarring, and may contribute to PIH in acne patients with SOC (Figure 1).8 A thorough history should be obtained from acne patients, including answers to the following questions7:
- What skin and hair care products do you use?
- Do you use sunscreen daily?
- What cosmetic products or makeup do you use?
- Do you use any ethnic skin care products, including skin lightening creams?
- Do you have a history of keloids?

It is important to ask these questions to assess if the SOC patient has developed pomade acne,9 acne cosmetica,10 or a potential risk of skin irritation from the use of skin care practices. It is best to take total control of the patient’s skin care regimen and discontinue use of toners, astringents, witch hazel, exfoliants, and rubbing alcohol, which may lead to skin dryness and irritation, particularly when combined with topical acne medications.
Treatment
Treatment of acne in SOC patients is similar to generally recommended treatments, with special considerations. Consider the following key points when treating acne in SOC patients:
- Treat acne early and aggressively to prevent or minimize subsequent PIH and acne scarring.
- Balance aggressive treatment with nonirritating topical skin care.
- Most importantly, target PIH in addition to acne and choose a regimen that limits skin irritation that might exacerbate existing PIH.7
Develop a maintenance program to control future breakouts. Topical agents can be used as monotherapy or in fixed combinations and may include benzoyl peroxide, antibiotics, dapsone, azelaic acid (AZA), and retinoids. Similar to white patients, topical retinoids remain a first-line treatment for acne in patients with SOC.11,12
Tolerability must be managed in SOC acne patients. Therapeutic maneuvers that can be instituted should include a discussion on using gentle skin care, initiating therapy with a retinoid applied every other night starting with a low concentration and gradually titrating up, and applying a moisturizer before or after applying acne medication. Oral therapies consist of antibiotics (doxycycline, minocycline), retinoids (isotretinoin), and hormonal modulators (oral contraceptives, spironolactone). Isotretinoin, recommended for patients with nodulocystic acne, may play a possible role in treating acne-induced PIH.13
Two common procedural therapies for acne include comedone extraction and intralesional corticosteroid injection. A 6- to 8-week course of a topical retinoid prior to comedonal extraction may facilitate the procedure and is recommended in SOC patients to help reduce cutaneous trauma and PIH.11 Inflammatory acne lesions can be treated with intralesional injection of triamcinolone acetonide 2.5 or 5.0 mg/mL, which usually reduces inflammation within 2 to 5 days.11
Treatment of acne-induced PIH includes sun protection, topical and oral medications, chemical peels, lasers, and energy devices. Treatment of hypertrophic scarring and keloids involves intralesional injection of triamcinolone acetonide 20, 30, or 40 mg/mL every 4 weeks until the lesion is flat.11
Superficial chemical peels can be used to treat acne and PIH in SOC patients,14 such as salicylic acid (20%–30%), glycolic acid (20%–70%), trichloroacetic acid (15%–30%), and Jessner peels.
Acne Scarring
Surgical approaches to acne scarring in patients with SOC include elliptical excision, punch excision, punch elevation, punch autografting, dermal grafting, dermal planning, subcutaneous incision (subcision), dermabrasion, microneedling, fillers, and laser skin resurfacing. The treatment of choice depends on the size, type, and depth of the scar and the clinician’s preference.
Lasers
Fractional photothermolysis has emerged as a treatment option for acne scars in SOC patients. This procedure produces microscopic columns of thermal injury in the epidermis and dermis, sparing the surrounding tissue and minimizing downtime and adverse events. Because fractional photothermolysis does not target melanin and produces limited epidermal injury, darker Fitzpatrick skin types (IV–VI) can be safely and effectively treated with this procedure.15
Rosacea in SOC Patients
Rosacea is a chronic inflammatory disorder that affects the vasculature and pilosebaceous units of the face. It commonly is seen in Fitzpatrick skin types I and II; however, rosacea can occur in all skin types (Figure 2). Triggers include emotional stress, extreme environmental temperatures, hot and spicy foods, red wine or alcohol, and topical irritants or allergens found in common cosmetic products.16
Data suggest that 4% of rosacea patients in the United States are of African, Latino, or Asian descent.11 National Ambulatory Medical Care Survey data revealed that of 31.5 million rosacea visits, 2% of patients were black, 2.3% were Asian or Pacific Islander, and 3.9% were Hispanic or Latino. In a 5-year longitudinal study of 2587 rosacea patients enrolled in Medicaid in North Carolina who were prescribed at least 1 topical treatment for rosacea, 16.27% were black and 10% were of a race other than white.17
Although the pathogenesis of rosacea is unclear, hypotheses include immune system abnormalities, neurogenic dysregulation, presence of microorganisms (eg, Demodex folliculorum), UV damage, and skin barrier dysfunction.18
The 4 major subtypes of rosacea are erythematotelangiectatic, papulopustular, phymatous, and ocular rosacea.16 Interestingly, rosacea in SOC patients may present with hypopigmentation surrounding the borders of the facial erythema. For phymatous rosacea, isotretinoin may reduce incipient rhinophyma but must be carefully monitored and pregnancy must be excluded. Surgical or laser therapy may be indicated to recontour the nose if severe.
There are several skin conditions that can present with facial erythema in patients with SOC, including seborrheic dermatitis, systemic lupus erythematosus, and contact dermatitis. It is important to note that the detection of facial erythema in darker skin types may be difficult; therefore, laboratory evaluation (antinuclear antibodies), patch testing, and skin biopsy should be considered if the clinical diagnosis is unclear.
Treatment
Treatment of rosacea in SOC patients does not differ from other racial groups. Common strategies include gentle skin care, sun protection (sun protection factor 30+), and barrier repair creams. Topical agents include metronidazole, AZA, sodium sulfacetamide/sulfur, ivermectin, and retinoids.16 Oral treatments include antibiotics in the tetracycline family (eg, subantimicrobial dose doxycycline) and isotretinoin.16 Persistent erythema associated with rosacea can be treated with brimonidine19 and oxymetazoline.20 Vascular lasers and intense pulsed light may be used to address the vascular components of rosacea21; however, the latter is not recommended in Fitzpatrick skin types IV through VI.
Facial Hyperpigmentation in SOC Patients
Hyperpigmentation disorders can be divided into conditions that affect Fitzpatrick skin types I through III and IV though VI. Mottled hyperpigmentation (photodamage) and solar lentigines occur in patients with lighter skin types as compared to melasma, PIH, and age-related (UV-induced) hyperpigmentation, which occur more commonly in patients with darker skin types. Facial hyperpigmentation is a common concern in SOC patients. In a survey of cosmetic concerns of 100 women with SOC, hyperpigmentation or dark spots (86%) and blotchy uneven skin (80%) were the top concerns.22 In addition, facial hyperpigmentation has been shown to negatively impact quality of life.23
Postinflammatory hyperpigmentation occurs from a pathophysiological response to inflammation, cutaneous irritation or injury, and subsequent melanocyte lability. Postinflammatory hyperpigmentation is a common presenting concern in patients with SOC and is seen as a result of many inflammatory skin disorders (eg, acne, eczema) and dermatologic procedures (eg, adverse reaction to electrodesiccation, microdermabrasion, chemical peels, laser surgery).24
Melasma is an acquired idiopathic disorder of hyperpigmentation and often referred to as the mask of pregnancy (Figure 3). It occurs on sun-exposed areas of skin, mainly in women with Fitzpatrick skin types III through V. Associated factors or triggers include pregnancy, hormonal treatments, exposure to UV radiation, and medications.25 Hereditary factors play a role in more than 40% of cases.26
Other not-so-common facial dyschromias include contact dermatitis, acanthosis nigricans, exogenous ochronosis, lichen planus pigmentosus (associated with frontal fibrosing alopecia),27 drug-induced hyperpigmentation (associated with minocycline or diltiazem),28,29 and UV-induced (age-related) hyperpigmentation.
Treatment
The treatment of hyperpigmentation should provide the following: (1) protection from sun exposure; (2) inhibition of tyrosinase, the enzyme responsible for the conversion of tyrosine to melanin; (3) inhibition of melanosome transfer from the melanocyte to the keratinocyte; (4) removal of melanin from the epidermis through exfoliation; and (5) destruction or disruption of melanin in the dermis.30 Therapies for facial hyperpigmentation are listed in Table 1.

Topical therapies include prescription medications and nonprescription cosmeceuticals. Prescription medications include hydroquinone (HQ), topical retinoids, and AZA. Hydroquinone, a tyrosinase inhibitor, is the gold standard for skin lightening and often is used as a first-line therapy. It is used as a monotherapy (HQ 4%) or as a fixed combination with tretinoin 0.05% and fluocinolone 0.01%.31 Use caution with HQ in high concentrations (6% and higher) and low concentrations (2% [over-the-counter strength]) used long-term due to the potential risk of exogenous ochronosis.
Topical retinoids have been shown to be effective therapeutic agents for melasma and PIH. Tretinoin,32 tazarotene,33 and adapalene34 all have demonstrated efficacy for acne and acne-induced PIH in SOC patients. Patients must be monitored for the development of retinoid dermatitis and worsening of hyperpigmentation.
Azelaic acid is a naturally occurring dicarboxylic acid obtained from cultures of Malassezia furfur. Azelaic acid inhibits tyrosinase activity, DNA synthesis, and mitochondrial enzymes, thus blocking direct cytotoxic effects toward melanocytes. Azelaic acid is approved by the US Food and Drug Administration for acne in a 20% cream formulation and rosacea in 15% gel and foam formulations, and it is used off label for melasma and PIH.35
Oral tranexamic acid is currently used as a hemostatic agent due to its ability to inhibit the plasminogen-plasmin pathway. In melasma, it blocks the interaction between melanocytes and keratinocytes in the epidermis and modulates the vascular component of melasma in the dermis. In an open-label study, 561 Asian melasma patients were treated with oral tranexamic acid 250 mg twice daily for 4 months. Results demonstrated improvement in 90% of patients, and 7.1% reported adverse effects (eg, abdominal bloating and pain, nausea, vomiting, headache, tinnitus, numbness, menstrual irregularities).36 Coagulation screening should be monitored monthly, and any patient with a history of clotting abnormalities should be excluded from off-label treatment with oral tranexamic acid.
Nonprescription cosmeceuticals are available over-the-counter or are office dispensed.37 For optimal results, cosmeceutical agents for skin lightening are used in combination. Most of these combinations are HQ free and have additive benefits such as a multimodal skin lightening agent containing key ingredients that correct and prevent skin pigmentation via several pathways affecting melanogenesis.38 It is an excellent alternative to HQ for mottled and diffuse UV-induced hyperpigmentation and can be used for maintenance therapy in patients with melasma.
Photoprotection is an essential component of therapy for melasma and PIH, but there is a paucity of data on the benefits for SOC patients. Halder et al39 performed a randomized prospective study of 89 black and Hispanic patients who applied sunscreen with a sun protection factor of 30 or 60 daily for 8 weeks. Clinical grading, triplicate L*A*B chromameter, and clinical photography were taken at baseline and weeks 4 and 8. The results demonstrated skin lightening in both black and Hispanic patients and support the use of sunscreen in the prevention and management of dyschromia in SOC patients.39 Visible light also may play a role in melasma development, and thus use of sunscreens or makeup containing iron oxides are recommended.40
Procedural treatments for facial hyperpigmentation include microdermabrasion, chemical peels, lasers, energy-based devices, and microneedling. There are many types and formulations of chemical peeling agents available; however, superficial and medium-depth chemical peels are recommended for SOC patients (Table 2). Deep chemical peels are not recommended for SOC patients due to the potential increased risk for PIH and scarring.

Cosmetic Enhancement in SOC Patients
Cosmetic procedures are gaining popularity in the SOC population and account for more than 20% of cosmetic procedures in the United States.41 Facial cosmetic concerns in SOC include dyschromia, benign growths (dermatosis papulosa nigra), hyperkinetic facial lines, volume loss, and skin laxity.42 Key principles to consider when treating SOC patients are the impact of ethnicity on aging and facial structure, the patient’s desired cosmetic outcome, tissue reaction to anticipated treatments, and the patient’s expectations for recommended therapies.
Aging in SOC Patients
Skin aging can be classified as intrinsic aging or extrinsic aging. Intrinsic aging is genetic and involves subsurface changes such as volume loss, muscle atrophy, and resorption of bony structure. Extrinsic aging (or photoaging) involves surface changes of the epidermis/dermis and manifests as mottled pigmentation, textural changes, and fine wrinkling. Due to the photoprotection of melanin (black skin=SPF 13.4), skin aging in SOC patients is delayed by 10 to 20 years.43 In addition, SOC patients have more reactive collagen and can benefit from noninvasive cosmetic procedures such as fillers and skin-tightening procedures.42
Cosmetic Treatments and Procedures
Dermatosis papulosa nigra (benign growths of skin that have a genetic predisposition)44 occur mainly on the face but can involve the entire body. Treatment modalities include electrodesiccation, cryotherapy, scissor excision, and laser surgery.45
Treatment of hyperkinetic facial lines with botulinum toxin type A is a safe and effective procedure in patients with SOC. Grimes and Shabazz46 performed a 4-month, randomized, double-blind study that evaluated the treatment of glabellar lines in women with Fitzpatrick skin types V and VI. The results demonstrated that the duration of effects was the same in the patients who received either 20 or 30 U of botulinum toxin type A.46 Dynamic rhytides (furrows and frown/scowl lines arising from laughing, frowning, or smiling) can be treated safely in patients with SOC using botulinum toxin type A off label for relaxation of the upper and lower hyperkinetic muscles that result in these unwanted signs of aging. Botulinum toxin type A often is used for etched-in crow’s-feet, which rarely are evident in SOC patients.47 Facial shaping also can be accomplished by injecting botulinum toxin type A in combination with soft-tissue dermal fillers.47
Although black individuals do not experience perioral rhytides at the frequency of white individuals, they experience a variety of other cosmetic issues related to skin sagging and sinking. Currently available hyaluronic acid (HA) fillers have been shown to be safe in patients with Fitzpatrick skin types IV through VI.48 Two studies evaluated fillers in patients with SOC, specifically HA49 and calcium hydroxylapatite,50 focused on treatment of the nasolabial folds and the potential risk for dyspigmentation and keloidal scarring. Taylor et al49 noted that the risk of hyperpigmentation was 6% to 9% for large- and small-particle HA, respectively, and was associated with the serial or multiple puncture injection technique. No hypertrophic or keloidal scarring occurred in both studies.49,50
Facial contouring applications with fillers include glabellar lines, temples, nasal bridge, tear troughs, malar and submalar areas, nasolabial folds, radial lines, lips, marionette lines, mental crease, and chin. Hyaluronic acid fillers also can be used for lip enhancement.47 Although white women are looking to increase the size of their lips, black women are seeking augmentation to restore their lip size to that of their youth. Black individuals do not experience the same frequency of perioral rhytides as white patients, but they experience a variety of other issues related to skin sagging and sinking. Unlike white women, enhancement of the vermilion border rarely is performed in black women due to development of rhytides, predominantly in the body of the lip below the vermilion border in response to volume loss in the upper lip while the lower lip usually maintains its same appearance.47
Facial enhancement utilizing poly-L-lactic acid can be used safely in SOC patients.51 Poly-L-lactic acid microparticles induce collagen formation, leading to dermal thickening over 3 to 6 months; however, multiple sessions are required to achieve optimal aesthetic results.
Patients with more reactive collagen can benefit from noninvasive cosmetic procedures such as skin-tightening procedures.52 Radiofrequency and microfocused ultrasound are cosmetic procedures used to provide skin tightening and facial lifting. They are safe and effective treatments for patients with Fitzpatrick skin types IV to VI.53 Histologically, there is less thinning of collagen bundles and elastic tissue in ethnic skin. Due to stimulation of collagen by these procedures, most SOC patients will experience a more enhanced response, requiring fewer treatment sessions than white individuals.
Conclusion
Medical and aesthetic facial concerns in SOC patients vary and can be a source of emotional and psychological distress that can negatively impact quality of life. The approach to the treatment of SOC patients should be a balance between tolerability and efficacy, considering the potential risk for PIH.
- White GM. Recent findings in the epidemiologic evidence, classification, and subtypes of acne vulgaris. J Am Acad Dermatol. 1998;39(2 pt 3):S34-S37.
- Halder RM, Grimes PE, McLaurin CL, et al. Incidence of common dermatoses in a predominantly black dermatologic practice. Cutis. 1983;32:388, 390.
- Alexis AF, Sergay AB, Taylor SC. Common dermatologic disorders in skin of color: a comparative practice survey. Cutis. 2007;80:387-394.
- Davis SA, Narahari S, Feldman SR, et al. Top dermatologic conditions in patients of color: an analysis of nationally representative data. J Drugs Dermatol. 2012;11:466-473.
- Perkins AC, Cheng CE, Hillebrand GG, et al. Comparison of the epidemiology of acne vulgaris among Caucasians, Asian, Continental Indian and African American women. J Eur Acad Dermatol Venereol. 2011;25:1054-1060.
- Taylor SC, Cook-Bolden F, Rahman Z, et al. Acne vulgaris in skin of color. J Am Acad Dermatol. 2002;46(2 suppl):S98-S106.
- Davis EC, Callender VD. A review of acne in ethnic skin: pathogenesis, clinical manifestations, and management strategies. J Clin Aesthet Dermatol. 2010;3:24-38.
- Halder RM, Holmes YC, Bridgeman-Shah S, et al. A clinicohistologic study of acne vulgaris in black females (abstract). J Invest Dermatol. 1996;106:888.
- Plewig G, Fulton JE, Kligman AM. Pomade acne. Arch Dermatol. 1970;101:580-584.
- Kligman AM, Mills OH. Acne cosmetica. Arch Dermatol. 1972;106:893-897.
- Halder RM, Brooks HL, Callender VD. Acne in ethnic skin. Dermatol Clin. 2003;21:609-615.
- Callender VD. Acne in ethnic skin: special considerations for therapy. Dermatol Ther. 2004;17:184-195.
- Winhoven SM. Postinflammatory hyperpigmentation in an Asian patient. a dramatic response to oral isotretinoin (13-cis-retinoic acid). Br J Med. 2005;152:368-403.
- Sarkar R, Bansal S, Garg VK. Chemical peels for melasma in dark-skinned patients. J Cutan Aesthet Surg. 2012;5:247-253.
- Alexis AF, Coley MK, Nijhawan RI, et al. Nonablative fractional laser resurfacing for acne scarring in patients with Fitzpatrick skin phototypes IV-VI. Dermatol Surg. 2016;42:392-402.
- Culp B, Scheinfeld N. Rosacea: a review. P T. 2009;34:38-45.
- Al-Dabagh A, Davis SA, McMichael AJ, et al. Rosacea in skin of color: not a rare diagnosis. Dermatol Online J. 2014:20. pii:13030/qt1mv9r0ss.
- Del Rosso JQ. Advances in understanding and managing rosacea: part 1: connecting the dots between pathophysiological mechanisms and common clinical features of rosacea with emphasis on vascular changes and facial erythema. J Clin Aesthet Dermatol. 2012;5:16-25.
- Jackson JM, Knuckles M, Minni JP, et al. The role of brimonidine tartrate gel in the treatment of rosacea. Clin Cosmet Investig Dermatol. 2015;23:529-538.
- Patel NU, Shukla S, Zaki J, et al. Oxymetazoline hydrochloride cream for facial erythema associated with rosacea. Expert Rev Clin Pharmacol. 2017;10:104954.
- Weinkle AP, Doktor V, Emer J. Update on the management of rosacea. Clin Cosmet Investig Dermatol. 2015;8:159-177.
- Grimes PE. Skin and hair cosmetic issues in women of color. Dermatol Clin. 2000;19:659-665.
- Taylor A, Pawaskar M, Taylor SL, et al. Prevalence of pigmentary disorders and their impact on quality of life: a prospective cohort study. J Cosmet Dermatol. 2008;7:164-168.
- Davis EC, Callender VD. Postinflammatory hyperpigmentation: a review of the epidemiology, clinical features, and treatment options in skin of color. J Clin Aesthet Dermatol. 2010;3:20-31.
- Grimes PE. Melasma: etiologic and therapeutic considerations. Arch Dermatol. 1995;131:1453-1457.
- Handel AC, Miot LD, Miot HA. Melasma: a clinical and epidemiological review. An Bras Dermatol. 2014;89:771-782.
- Callender VD, Reid SD, Obayan O, et al. Diagnostic clues to frontal fibrosing alopecia in patients of African descent. J Clin Aesthet Dermatol. 2016;9:45-51.
- Narang T, Sawatkar GU, Kumaran MS, et al. Minocycline for recurrent and/or chronic erythema nodosum leprosum. JAMA Dermatol. 2015;151:1026-1028.
- Boyer M, Katta R, Markus R. Diltiazem-induced photodistributed hyperpigmentation. Dermatol Online J. 2003;9:10.
- Pandya AG, Guevara IL. Disorders of hyperpigmentation. Dermatol Clin. 2000;18:91-98.
- Taylor SC, Torok H, Jones T, et al. Efficacy and safety of a new triple-combination agent for the treatment of facial melasma. Cutis. 2003;72:67-72.
- Bulengo-Ransby SM. Topical tretinoin (retinoic acid) therapy for hyperpigmented lesions caused by inflammation of the skin in black patients. N Engl J Med. 1993;328:1438-1443.
- Grimes P, Callender V. Tazarotene cream for postinflammatory hyperpigmentation and acne vulgaris in darker skin: a double-blind, randomized, vehicle-controlled study. Cutis. 2006;77:45-50.
- Jacyk WK. Adapalene in the treatment of African patients. J Eur Acad Dermatol Venereol. 2001;15(suppl 3):37-42.
- Kircik LH. Efficacy and safety of azelaic acid (AzA) gel 15% in the treatment of postinflammatory hyperpigmentation and acne: a 16-week, baseline-controlled study. J Drugs Dermatol. 2011;10:586-590.
- Lee HC, Thng TG, Goh CL. Oral tranexamic acid (TA) in the treatment of melasma. J Am Acad Dermatol. 2016;75:385-392.
- Kindred C, Okereke U, Callender VD. Skin-lightening agents: an overview of prescription, office-dispensed, and over-the-counter products. Cosmet Dermatol. 2013;26:18-26.
- Makino ET, Kadoya K, Sigler ML, et al. Development and clinical assessment of a comprehensive product for pigmentation control in multiple ethnic populations. J Drugs Dermatol. 2016;15:1562-1570.
- Halder R, Rodney I, Munhutu M, et al. Evaluation and effectiveness of a photoprotection composition (sunscreen) on subjects of skin of color. J Am Acad Dermatol. 2015;72(suppl 1):AB215.
- Castanedo-Cazares JP, Hernandez-Blanco D, Carlos-Ortega B, et al. Near-visible light and UV photoprotection in the treatment of melasma: a double-blind randomized trial. Photodermatol Photoimmunol Photomed. 2014;30:35-42.
- American Society for Aesthetic Plastic Surgery. 2016 Cosmetic Surgery National Data Bank Statistics. https://www.surgery.org/sites/default/files/ASAPS-Stats2016.pdf. Accessed November 15, 2017.
- Burgess CM. Soft tissue augmentation in skin of color: market growth, available fillers and successful techniques. J Drugs Dermatol. 2007;6:51-55.
- Davis EC, Callender VD. Aesthetic dermatology for aging ethnic skin. Dermatol Surg. 2011;37:901-917.
- Grimes PE, Arora S, Minus HR, et al. Dermatosis papulosa nigra. Cutis. 1983;32:385-386.
- Lupo M. Dermatosis papulosa nigra: treatment options. J Drugs Dermatol. 2007;6:29-30.
- Grimes PE, Shabazz D. A four-month randomized, double-blind evaluation of the efficacy of botulinum toxin type A for the treatment of glabellar lines in women with skin types V and VI. Dermatol Surg. 2009;35:429-435.
- Burgess CM, Awosika O. Ethnic and gender considerations in the use of facial injectables: African-American patients. Plast Reconstr Surg. 2015;136(5 suppl):28S-31S.
- Taylor SC, Kelly AP, Lim HW, et al, eds. Taylor and Kelly’s Dermatology for Skin of Color. 2nd ed. New York, NY: McGraw-Hill Education; 2016.
- Taylor SC, Burgess CM, Callender VD. Safety of nonanimal stabilized hyaluronic acid dermal fillers in patients with skin of color: a randomized, evaluator-blinded comparative trial. Dermatol Surg. 2009;35(suppl 2):1653-1660.
- Marmur ES, Taylor SC, Grimes PE, et al. Six-month safety results of calcium hydroxylapatite for treatment of nasolabial folds in Fitzpatrick skin types IV to VI. Dermatol Surg. 2009;35(suppl 2):1641-1645.
- Hamilton TK, Burgess CM. Consideration for the use of injectable poly-L-lactic acid in people of color. J Drugs Dermatol. 2010;9:451-456.
- Fabi SG, Goldman MP. Retrospective evaluation of micro-focused ultrasound for lifting and tightening of the face and neck. Dermatol Surg. 2014;40:569-575.
- Harris MO, Sundaram HA. Safety of microfocused ultrasound with visualization in patients with Fitzpatrick skin phototypes III to VI. JAMA Facial Plast Surg. 2015;17:355-357.
The approach to the treatment of common skin disorders and cosmetic concerns in patients with skin of color (SOC) requires the clinician to understand the biological differences, nuances, and special considerations that are unique to patients with darker skin types.1-3 This article addresses 4 common facial concerns in SOC patients—acne, rosacea, facial hyperpigmentation, and cosmetic enhancement—and provides treatment recommendations and management pearls to assist the clinician with optimal outcomes for SOC patients.
Acne in SOC Patients
Acne vulgaris is one of the most common conditions that dermatologists treat and is estimated to affect 40 to 50 million individuals in the United States.1 Many of these acne patients are individuals with SOC.2-4 A study of 2835 females (aged 10–70 years) conducted in 4 different cities—Los Angeles, California; London, United Kingdom; Akita, Japan; and Rome, Italy—demonstrated acne prevalence of 37% in blacks, 32% in Hispanics, 30% in Asians, 24% in whites, and 23% in Continental Indians.5 Blacks, Hispanics, and Continental Indians demonstrated equal prevalence with comedonal and inflammatory acne. Asians displayed more inflammatory acne lesions than comedones. In contrast, whites demonstrated more comedones than inflammatory acne. Dyspigmentation, postinflammatory hyperpigmentation (PIH), and atrophic scars were more common in black and Hispanic females than other ethnicities.5 This study illustrated that acne-induced PIH is a common sequela in SOC patients and is the main reason they seek treatment.6,7
The pathogenesis of acne is the same in all racial and ethnic groups: (1) follicular hyperkeratinization and the formation of a microcomedone caused by abnormal desquamation of the keratinocytes within the sebaceous follicle, (2) production of sebum by circulating androgens, (3) proliferation of Propionibacterium acnes, and (4) inflammation. Subclinical inflammation is present throughout all stages of acne, including normal-appearing skin, inflammatory lesions, comedones, and scarring, and may contribute to PIH in acne patients with SOC (Figure 1).8 A thorough history should be obtained from acne patients, including answers to the following questions7:
- What skin and hair care products do you use?
- Do you use sunscreen daily?
- What cosmetic products or makeup do you use?
- Do you use any ethnic skin care products, including skin lightening creams?
- Do you have a history of keloids?
It is important to ask these questions to assess if the SOC patient has developed pomade acne,9 acne cosmetica,10 or a potential risk of skin irritation from the use of skin care practices. It is best to take total control of the patient’s skin care regimen and discontinue use of toners, astringents, witch hazel, exfoliants, and rubbing alcohol, which may lead to skin dryness and irritation, particularly when combined with topical acne medications.
Treatment
Treatment of acne in SOC patients is similar to generally recommended treatments, with special considerations. Consider the following key points when treating acne in SOC patients:
- Treat acne early and aggressively to prevent or minimize subsequent PIH and acne scarring.
- Balance aggressive treatment with nonirritating topical skin care.
- Most importantly, target PIH in addition to acne and choose a regimen that limits skin irritation that might exacerbate existing PIH.7
Develop a maintenance program to control future breakouts. Topical agents can be used as monotherapy or in fixed combinations and may include benzoyl peroxide, antibiotics, dapsone, azelaic acid (AZA), and retinoids. Similar to white patients, topical retinoids remain a first-line treatment for acne in patients with SOC.11,12
Tolerability must be managed in SOC acne patients. Therapeutic maneuvers that can be instituted should include a discussion on using gentle skin care, initiating therapy with a retinoid applied every other night starting with a low concentration and gradually titrating up, and applying a moisturizer before or after applying acne medication. Oral therapies consist of antibiotics (doxycycline, minocycline), retinoids (isotretinoin), and hormonal modulators (oral contraceptives, spironolactone). Isotretinoin, recommended for patients with nodulocystic acne, may play a possible role in treating acne-induced PIH.13
Two common procedural therapies for acne include comedone extraction and intralesional corticosteroid injection. A 6- to 8-week course of a topical retinoid prior to comedonal extraction may facilitate the procedure and is recommended in SOC patients to help reduce cutaneous trauma and PIH.11 Inflammatory acne lesions can be treated with intralesional injection of triamcinolone acetonide 2.5 or 5.0 mg/mL, which usually reduces inflammation within 2 to 5 days.11
Treatment of acne-induced PIH includes sun protection, topical and oral medications, chemical peels, lasers, and energy devices. Treatment of hypertrophic scarring and keloids involves intralesional injection of triamcinolone acetonide 20, 30, or 40 mg/mL every 4 weeks until the lesion is flat.11
Superficial chemical peels can be used to treat acne and PIH in SOC patients,14 such as salicylic acid (20%–30%), glycolic acid (20%–70%), trichloroacetic acid (15%–30%), and Jessner peels.
Acne Scarring
Surgical approaches to acne scarring in patients with SOC include elliptical excision, punch excision, punch elevation, punch autografting, dermal grafting, dermal planning, subcutaneous incision (subcision), dermabrasion, microneedling, fillers, and laser skin resurfacing. The treatment of choice depends on the size, type, and depth of the scar and the clinician’s preference.
Lasers
Fractional photothermolysis has emerged as a treatment option for acne scars in SOC patients. This procedure produces microscopic columns of thermal injury in the epidermis and dermis, sparing the surrounding tissue and minimizing downtime and adverse events. Because fractional photothermolysis does not target melanin and produces limited epidermal injury, darker Fitzpatrick skin types (IV–VI) can be safely and effectively treated with this procedure.15
Rosacea in SOC Patients
Rosacea is a chronic inflammatory disorder that affects the vasculature and pilosebaceous units of the face. It commonly is seen in Fitzpatrick skin types I and II; however, rosacea can occur in all skin types (Figure 2). Triggers include emotional stress, extreme environmental temperatures, hot and spicy foods, red wine or alcohol, and topical irritants or allergens found in common cosmetic products.16
Data suggest that 4% of rosacea patients in the United States are of African, Latino, or Asian descent.11 National Ambulatory Medical Care Survey data revealed that of 31.5 million rosacea visits, 2% of patients were black, 2.3% were Asian or Pacific Islander, and 3.9% were Hispanic or Latino. In a 5-year longitudinal study of 2587 rosacea patients enrolled in Medicaid in North Carolina who were prescribed at least 1 topical treatment for rosacea, 16.27% were black and 10% were of a race other than white.17
Although the pathogenesis of rosacea is unclear, hypotheses include immune system abnormalities, neurogenic dysregulation, presence of microorganisms (eg, Demodex folliculorum), UV damage, and skin barrier dysfunction.18
The 4 major subtypes of rosacea are erythematotelangiectatic, papulopustular, phymatous, and ocular rosacea.16 Interestingly, rosacea in SOC patients may present with hypopigmentation surrounding the borders of the facial erythema. For phymatous rosacea, isotretinoin may reduce incipient rhinophyma but must be carefully monitored and pregnancy must be excluded. Surgical or laser therapy may be indicated to recontour the nose if severe.
There are several skin conditions that can present with facial erythema in patients with SOC, including seborrheic dermatitis, systemic lupus erythematosus, and contact dermatitis. It is important to note that the detection of facial erythema in darker skin types may be difficult; therefore, laboratory evaluation (antinuclear antibodies), patch testing, and skin biopsy should be considered if the clinical diagnosis is unclear.
Treatment
Treatment of rosacea in SOC patients does not differ from other racial groups. Common strategies include gentle skin care, sun protection (sun protection factor 30+), and barrier repair creams. Topical agents include metronidazole, AZA, sodium sulfacetamide/sulfur, ivermectin, and retinoids.16 Oral treatments include antibiotics in the tetracycline family (eg, subantimicrobial dose doxycycline) and isotretinoin.16 Persistent erythema associated with rosacea can be treated with brimonidine19 and oxymetazoline.20 Vascular lasers and intense pulsed light may be used to address the vascular components of rosacea21; however, the latter is not recommended in Fitzpatrick skin types IV through VI.
Facial Hyperpigmentation in SOC Patients
Hyperpigmentation disorders can be divided into conditions that affect Fitzpatrick skin types I through III and IV though VI. Mottled hyperpigmentation (photodamage) and solar lentigines occur in patients with lighter skin types as compared to melasma, PIH, and age-related (UV-induced) hyperpigmentation, which occur more commonly in patients with darker skin types. Facial hyperpigmentation is a common concern in SOC patients. In a survey of cosmetic concerns of 100 women with SOC, hyperpigmentation or dark spots (86%) and blotchy uneven skin (80%) were the top concerns.22 In addition, facial hyperpigmentation has been shown to negatively impact quality of life.23
Postinflammatory hyperpigmentation occurs from a pathophysiological response to inflammation, cutaneous irritation or injury, and subsequent melanocyte lability. Postinflammatory hyperpigmentation is a common presenting concern in patients with SOC and is seen as a result of many inflammatory skin disorders (eg, acne, eczema) and dermatologic procedures (eg, adverse reaction to electrodesiccation, microdermabrasion, chemical peels, laser surgery).24
Melasma is an acquired idiopathic disorder of hyperpigmentation and often referred to as the mask of pregnancy (Figure 3). It occurs on sun-exposed areas of skin, mainly in women with Fitzpatrick skin types III through V. Associated factors or triggers include pregnancy, hormonal treatments, exposure to UV radiation, and medications.25 Hereditary factors play a role in more than 40% of cases.26
Other not-so-common facial dyschromias include contact dermatitis, acanthosis nigricans, exogenous ochronosis, lichen planus pigmentosus (associated with frontal fibrosing alopecia),27 drug-induced hyperpigmentation (associated with minocycline or diltiazem),28,29 and UV-induced (age-related) hyperpigmentation.
Treatment
The treatment of hyperpigmentation should provide the following: (1) protection from sun exposure; (2) inhibition of tyrosinase, the enzyme responsible for the conversion of tyrosine to melanin; (3) inhibition of melanosome transfer from the melanocyte to the keratinocyte; (4) removal of melanin from the epidermis through exfoliation; and (5) destruction or disruption of melanin in the dermis.30 Therapies for facial hyperpigmentation are listed in Table 1.
Topical therapies include prescription medications and nonprescription cosmeceuticals. Prescription medications include hydroquinone (HQ), topical retinoids, and AZA. Hydroquinone, a tyrosinase inhibitor, is the gold standard for skin lightening and often is used as a first-line therapy. It is used as a monotherapy (HQ 4%) or as a fixed combination with tretinoin 0.05% and fluocinolone 0.01%.31 Use caution with HQ in high concentrations (6% and higher) and low concentrations (2% [over-the-counter strength]) used long-term due to the potential risk of exogenous ochronosis.
Topical retinoids have been shown to be effective therapeutic agents for melasma and PIH. Tretinoin,32 tazarotene,33 and adapalene34 all have demonstrated efficacy for acne and acne-induced PIH in SOC patients. Patients must be monitored for the development of retinoid dermatitis and worsening of hyperpigmentation.
Azelaic acid is a naturally occurring dicarboxylic acid obtained from cultures of Malassezia furfur. Azelaic acid inhibits tyrosinase activity, DNA synthesis, and mitochondrial enzymes, thus blocking direct cytotoxic effects toward melanocytes. Azelaic acid is approved by the US Food and Drug Administration for acne in a 20% cream formulation and rosacea in 15% gel and foam formulations, and it is used off label for melasma and PIH.35
Oral tranexamic acid is currently used as a hemostatic agent due to its ability to inhibit the plasminogen-plasmin pathway. In melasma, it blocks the interaction between melanocytes and keratinocytes in the epidermis and modulates the vascular component of melasma in the dermis. In an open-label study, 561 Asian melasma patients were treated with oral tranexamic acid 250 mg twice daily for 4 months. Results demonstrated improvement in 90% of patients, and 7.1% reported adverse effects (eg, abdominal bloating and pain, nausea, vomiting, headache, tinnitus, numbness, menstrual irregularities).36 Coagulation screening should be monitored monthly, and any patient with a history of clotting abnormalities should be excluded from off-label treatment with oral tranexamic acid.
Nonprescription cosmeceuticals are available over-the-counter or are office dispensed.37 For optimal results, cosmeceutical agents for skin lightening are used in combination. Most of these combinations are HQ free and have additive benefits such as a multimodal skin lightening agent containing key ingredients that correct and prevent skin pigmentation via several pathways affecting melanogenesis.38 It is an excellent alternative to HQ for mottled and diffuse UV-induced hyperpigmentation and can be used for maintenance therapy in patients with melasma.
Photoprotection is an essential component of therapy for melasma and PIH, but there is a paucity of data on the benefits for SOC patients. Halder et al39 performed a randomized prospective study of 89 black and Hispanic patients who applied sunscreen with a sun protection factor of 30 or 60 daily for 8 weeks. Clinical grading, triplicate L*A*B chromameter, and clinical photography were taken at baseline and weeks 4 and 8. The results demonstrated skin lightening in both black and Hispanic patients and support the use of sunscreen in the prevention and management of dyschromia in SOC patients.39 Visible light also may play a role in melasma development, and thus use of sunscreens or makeup containing iron oxides are recommended.40
Procedural treatments for facial hyperpigmentation include microdermabrasion, chemical peels, lasers, energy-based devices, and microneedling. There are many types and formulations of chemical peeling agents available; however, superficial and medium-depth chemical peels are recommended for SOC patients (Table 2). Deep chemical peels are not recommended for SOC patients due to the potential increased risk for PIH and scarring.
Cosmetic Enhancement in SOC Patients
Cosmetic procedures are gaining popularity in the SOC population and account for more than 20% of cosmetic procedures in the United States.41 Facial cosmetic concerns in SOC include dyschromia, benign growths (dermatosis papulosa nigra), hyperkinetic facial lines, volume loss, and skin laxity.42 Key principles to consider when treating SOC patients are the impact of ethnicity on aging and facial structure, the patient’s desired cosmetic outcome, tissue reaction to anticipated treatments, and the patient’s expectations for recommended therapies.
Aging in SOC Patients
Skin aging can be classified as intrinsic aging or extrinsic aging. Intrinsic aging is genetic and involves subsurface changes such as volume loss, muscle atrophy, and resorption of bony structure. Extrinsic aging (or photoaging) involves surface changes of the epidermis/dermis and manifests as mottled pigmentation, textural changes, and fine wrinkling. Due to the photoprotection of melanin (black skin=SPF 13.4), skin aging in SOC patients is delayed by 10 to 20 years.43 In addition, SOC patients have more reactive collagen and can benefit from noninvasive cosmetic procedures such as fillers and skin-tightening procedures.42
Cosmetic Treatments and Procedures
Dermatosis papulosa nigra (benign growths of skin that have a genetic predisposition)44 occur mainly on the face but can involve the entire body. Treatment modalities include electrodesiccation, cryotherapy, scissor excision, and laser surgery.45
Treatment of hyperkinetic facial lines with botulinum toxin type A is a safe and effective procedure in patients with SOC. Grimes and Shabazz46 performed a 4-month, randomized, double-blind study that evaluated the treatment of glabellar lines in women with Fitzpatrick skin types V and VI. The results demonstrated that the duration of effects was the same in the patients who received either 20 or 30 U of botulinum toxin type A.46 Dynamic rhytides (furrows and frown/scowl lines arising from laughing, frowning, or smiling) can be treated safely in patients with SOC using botulinum toxin type A off label for relaxation of the upper and lower hyperkinetic muscles that result in these unwanted signs of aging. Botulinum toxin type A often is used for etched-in crow’s-feet, which rarely are evident in SOC patients.47 Facial shaping also can be accomplished by injecting botulinum toxin type A in combination with soft-tissue dermal fillers.47
Although black individuals do not experience perioral rhytides at the frequency of white individuals, they experience a variety of other cosmetic issues related to skin sagging and sinking. Currently available hyaluronic acid (HA) fillers have been shown to be safe in patients with Fitzpatrick skin types IV through VI.48 Two studies evaluated fillers in patients with SOC, specifically HA49 and calcium hydroxylapatite,50 focused on treatment of the nasolabial folds and the potential risk for dyspigmentation and keloidal scarring. Taylor et al49 noted that the risk of hyperpigmentation was 6% to 9% for large- and small-particle HA, respectively, and was associated with the serial or multiple puncture injection technique. No hypertrophic or keloidal scarring occurred in both studies.49,50
Facial contouring applications with fillers include glabellar lines, temples, nasal bridge, tear troughs, malar and submalar areas, nasolabial folds, radial lines, lips, marionette lines, mental crease, and chin. Hyaluronic acid fillers also can be used for lip enhancement.47 Although white women are looking to increase the size of their lips, black women are seeking augmentation to restore their lip size to that of their youth. Black individuals do not experience the same frequency of perioral rhytides as white patients, but they experience a variety of other issues related to skin sagging and sinking. Unlike white women, enhancement of the vermilion border rarely is performed in black women due to development of rhytides, predominantly in the body of the lip below the vermilion border in response to volume loss in the upper lip while the lower lip usually maintains its same appearance.47
Facial enhancement utilizing poly-L-lactic acid can be used safely in SOC patients.51 Poly-L-lactic acid microparticles induce collagen formation, leading to dermal thickening over 3 to 6 months; however, multiple sessions are required to achieve optimal aesthetic results.
Patients with more reactive collagen can benefit from noninvasive cosmetic procedures such as skin-tightening procedures.52 Radiofrequency and microfocused ultrasound are cosmetic procedures used to provide skin tightening and facial lifting. They are safe and effective treatments for patients with Fitzpatrick skin types IV to VI.53 Histologically, there is less thinning of collagen bundles and elastic tissue in ethnic skin. Due to stimulation of collagen by these procedures, most SOC patients will experience a more enhanced response, requiring fewer treatment sessions than white individuals.
Conclusion
Medical and aesthetic facial concerns in SOC patients vary and can be a source of emotional and psychological distress that can negatively impact quality of life. The approach to the treatment of SOC patients should be a balance between tolerability and efficacy, considering the potential risk for PIH.
The approach to the treatment of common skin disorders and cosmetic concerns in patients with skin of color (SOC) requires the clinician to understand the biological differences, nuances, and special considerations that are unique to patients with darker skin types.1-3 This article addresses 4 common facial concerns in SOC patients—acne, rosacea, facial hyperpigmentation, and cosmetic enhancement—and provides treatment recommendations and management pearls to assist the clinician with optimal outcomes for SOC patients.
Acne in SOC Patients
Acne vulgaris is one of the most common conditions that dermatologists treat and is estimated to affect 40 to 50 million individuals in the United States.1 Many of these acne patients are individuals with SOC.2-4 A study of 2835 females (aged 10–70 years) conducted in 4 different cities—Los Angeles, California; London, United Kingdom; Akita, Japan; and Rome, Italy—demonstrated acne prevalence of 37% in blacks, 32% in Hispanics, 30% in Asians, 24% in whites, and 23% in Continental Indians.5 Blacks, Hispanics, and Continental Indians demonstrated equal prevalence with comedonal and inflammatory acne. Asians displayed more inflammatory acne lesions than comedones. In contrast, whites demonstrated more comedones than inflammatory acne. Dyspigmentation, postinflammatory hyperpigmentation (PIH), and atrophic scars were more common in black and Hispanic females than other ethnicities.5 This study illustrated that acne-induced PIH is a common sequela in SOC patients and is the main reason they seek treatment.6,7
The pathogenesis of acne is the same in all racial and ethnic groups: (1) follicular hyperkeratinization and the formation of a microcomedone caused by abnormal desquamation of the keratinocytes within the sebaceous follicle, (2) production of sebum by circulating androgens, (3) proliferation of Propionibacterium acnes, and (4) inflammation. Subclinical inflammation is present throughout all stages of acne, including normal-appearing skin, inflammatory lesions, comedones, and scarring, and may contribute to PIH in acne patients with SOC (Figure 1).8 A thorough history should be obtained from acne patients, including answers to the following questions7:
- What skin and hair care products do you use?
- Do you use sunscreen daily?
- What cosmetic products or makeup do you use?
- Do you use any ethnic skin care products, including skin lightening creams?
- Do you have a history of keloids?
It is important to ask these questions to assess if the SOC patient has developed pomade acne,9 acne cosmetica,10 or a potential risk of skin irritation from the use of skin care practices. It is best to take total control of the patient’s skin care regimen and discontinue use of toners, astringents, witch hazel, exfoliants, and rubbing alcohol, which may lead to skin dryness and irritation, particularly when combined with topical acne medications.
Treatment
Treatment of acne in SOC patients is similar to generally recommended treatments, with special considerations. Consider the following key points when treating acne in SOC patients:
- Treat acne early and aggressively to prevent or minimize subsequent PIH and acne scarring.
- Balance aggressive treatment with nonirritating topical skin care.
- Most importantly, target PIH in addition to acne and choose a regimen that limits skin irritation that might exacerbate existing PIH.7
Develop a maintenance program to control future breakouts. Topical agents can be used as monotherapy or in fixed combinations and may include benzoyl peroxide, antibiotics, dapsone, azelaic acid (AZA), and retinoids. Similar to white patients, topical retinoids remain a first-line treatment for acne in patients with SOC.11,12
Tolerability must be managed in SOC acne patients. Therapeutic maneuvers that can be instituted should include a discussion on using gentle skin care, initiating therapy with a retinoid applied every other night starting with a low concentration and gradually titrating up, and applying a moisturizer before or after applying acne medication. Oral therapies consist of antibiotics (doxycycline, minocycline), retinoids (isotretinoin), and hormonal modulators (oral contraceptives, spironolactone). Isotretinoin, recommended for patients with nodulocystic acne, may play a possible role in treating acne-induced PIH.13
Two common procedural therapies for acne include comedone extraction and intralesional corticosteroid injection. A 6- to 8-week course of a topical retinoid prior to comedonal extraction may facilitate the procedure and is recommended in SOC patients to help reduce cutaneous trauma and PIH.11 Inflammatory acne lesions can be treated with intralesional injection of triamcinolone acetonide 2.5 or 5.0 mg/mL, which usually reduces inflammation within 2 to 5 days.11
Treatment of acne-induced PIH includes sun protection, topical and oral medications, chemical peels, lasers, and energy devices. Treatment of hypertrophic scarring and keloids involves intralesional injection of triamcinolone acetonide 20, 30, or 40 mg/mL every 4 weeks until the lesion is flat.11
Superficial chemical peels can be used to treat acne and PIH in SOC patients,14 such as salicylic acid (20%–30%), glycolic acid (20%–70%), trichloroacetic acid (15%–30%), and Jessner peels.
Acne Scarring
Surgical approaches to acne scarring in patients with SOC include elliptical excision, punch excision, punch elevation, punch autografting, dermal grafting, dermal planning, subcutaneous incision (subcision), dermabrasion, microneedling, fillers, and laser skin resurfacing. The treatment of choice depends on the size, type, and depth of the scar and the clinician’s preference.
Lasers
Fractional photothermolysis has emerged as a treatment option for acne scars in SOC patients. This procedure produces microscopic columns of thermal injury in the epidermis and dermis, sparing the surrounding tissue and minimizing downtime and adverse events. Because fractional photothermolysis does not target melanin and produces limited epidermal injury, darker Fitzpatrick skin types (IV–VI) can be safely and effectively treated with this procedure.15
Rosacea in SOC Patients
Rosacea is a chronic inflammatory disorder that affects the vasculature and pilosebaceous units of the face. It commonly is seen in Fitzpatrick skin types I and II; however, rosacea can occur in all skin types (Figure 2). Triggers include emotional stress, extreme environmental temperatures, hot and spicy foods, red wine or alcohol, and topical irritants or allergens found in common cosmetic products.16
Data suggest that 4% of rosacea patients in the United States are of African, Latino, or Asian descent.11 National Ambulatory Medical Care Survey data revealed that of 31.5 million rosacea visits, 2% of patients were black, 2.3% were Asian or Pacific Islander, and 3.9% were Hispanic or Latino. In a 5-year longitudinal study of 2587 rosacea patients enrolled in Medicaid in North Carolina who were prescribed at least 1 topical treatment for rosacea, 16.27% were black and 10% were of a race other than white.17
Although the pathogenesis of rosacea is unclear, hypotheses include immune system abnormalities, neurogenic dysregulation, presence of microorganisms (eg, Demodex folliculorum), UV damage, and skin barrier dysfunction.18
The 4 major subtypes of rosacea are erythematotelangiectatic, papulopustular, phymatous, and ocular rosacea.16 Interestingly, rosacea in SOC patients may present with hypopigmentation surrounding the borders of the facial erythema. For phymatous rosacea, isotretinoin may reduce incipient rhinophyma but must be carefully monitored and pregnancy must be excluded. Surgical or laser therapy may be indicated to recontour the nose if severe.
There are several skin conditions that can present with facial erythema in patients with SOC, including seborrheic dermatitis, systemic lupus erythematosus, and contact dermatitis. It is important to note that the detection of facial erythema in darker skin types may be difficult; therefore, laboratory evaluation (antinuclear antibodies), patch testing, and skin biopsy should be considered if the clinical diagnosis is unclear.
Treatment
Treatment of rosacea in SOC patients does not differ from other racial groups. Common strategies include gentle skin care, sun protection (sun protection factor 30+), and barrier repair creams. Topical agents include metronidazole, AZA, sodium sulfacetamide/sulfur, ivermectin, and retinoids.16 Oral treatments include antibiotics in the tetracycline family (eg, subantimicrobial dose doxycycline) and isotretinoin.16 Persistent erythema associated with rosacea can be treated with brimonidine19 and oxymetazoline.20 Vascular lasers and intense pulsed light may be used to address the vascular components of rosacea21; however, the latter is not recommended in Fitzpatrick skin types IV through VI.
Facial Hyperpigmentation in SOC Patients
Hyperpigmentation disorders can be divided into conditions that affect Fitzpatrick skin types I through III and IV though VI. Mottled hyperpigmentation (photodamage) and solar lentigines occur in patients with lighter skin types as compared to melasma, PIH, and age-related (UV-induced) hyperpigmentation, which occur more commonly in patients with darker skin types. Facial hyperpigmentation is a common concern in SOC patients. In a survey of cosmetic concerns of 100 women with SOC, hyperpigmentation or dark spots (86%) and blotchy uneven skin (80%) were the top concerns.22 In addition, facial hyperpigmentation has been shown to negatively impact quality of life.23
Postinflammatory hyperpigmentation occurs from a pathophysiological response to inflammation, cutaneous irritation or injury, and subsequent melanocyte lability. Postinflammatory hyperpigmentation is a common presenting concern in patients with SOC and is seen as a result of many inflammatory skin disorders (eg, acne, eczema) and dermatologic procedures (eg, adverse reaction to electrodesiccation, microdermabrasion, chemical peels, laser surgery).24
Melasma is an acquired idiopathic disorder of hyperpigmentation and often referred to as the mask of pregnancy (Figure 3). It occurs on sun-exposed areas of skin, mainly in women with Fitzpatrick skin types III through V. Associated factors or triggers include pregnancy, hormonal treatments, exposure to UV radiation, and medications.25 Hereditary factors play a role in more than 40% of cases.26
Other not-so-common facial dyschromias include contact dermatitis, acanthosis nigricans, exogenous ochronosis, lichen planus pigmentosus (associated with frontal fibrosing alopecia),27 drug-induced hyperpigmentation (associated with minocycline or diltiazem),28,29 and UV-induced (age-related) hyperpigmentation.
Treatment
The treatment of hyperpigmentation should provide the following: (1) protection from sun exposure; (2) inhibition of tyrosinase, the enzyme responsible for the conversion of tyrosine to melanin; (3) inhibition of melanosome transfer from the melanocyte to the keratinocyte; (4) removal of melanin from the epidermis through exfoliation; and (5) destruction or disruption of melanin in the dermis.30 Therapies for facial hyperpigmentation are listed in Table 1.
Topical therapies include prescription medications and nonprescription cosmeceuticals. Prescription medications include hydroquinone (HQ), topical retinoids, and AZA. Hydroquinone, a tyrosinase inhibitor, is the gold standard for skin lightening and often is used as a first-line therapy. It is used as a monotherapy (HQ 4%) or as a fixed combination with tretinoin 0.05% and fluocinolone 0.01%.31 Use caution with HQ in high concentrations (6% and higher) and low concentrations (2% [over-the-counter strength]) used long-term due to the potential risk of exogenous ochronosis.
Topical retinoids have been shown to be effective therapeutic agents for melasma and PIH. Tretinoin,32 tazarotene,33 and adapalene34 all have demonstrated efficacy for acne and acne-induced PIH in SOC patients. Patients must be monitored for the development of retinoid dermatitis and worsening of hyperpigmentation.
Azelaic acid is a naturally occurring dicarboxylic acid obtained from cultures of Malassezia furfur. Azelaic acid inhibits tyrosinase activity, DNA synthesis, and mitochondrial enzymes, thus blocking direct cytotoxic effects toward melanocytes. Azelaic acid is approved by the US Food and Drug Administration for acne in a 20% cream formulation and rosacea in 15% gel and foam formulations, and it is used off label for melasma and PIH.35
Oral tranexamic acid is currently used as a hemostatic agent due to its ability to inhibit the plasminogen-plasmin pathway. In melasma, it blocks the interaction between melanocytes and keratinocytes in the epidermis and modulates the vascular component of melasma in the dermis. In an open-label study, 561 Asian melasma patients were treated with oral tranexamic acid 250 mg twice daily for 4 months. Results demonstrated improvement in 90% of patients, and 7.1% reported adverse effects (eg, abdominal bloating and pain, nausea, vomiting, headache, tinnitus, numbness, menstrual irregularities).36 Coagulation screening should be monitored monthly, and any patient with a history of clotting abnormalities should be excluded from off-label treatment with oral tranexamic acid.
Nonprescription cosmeceuticals are available over-the-counter or are office dispensed.37 For optimal results, cosmeceutical agents for skin lightening are used in combination. Most of these combinations are HQ free and have additive benefits such as a multimodal skin lightening agent containing key ingredients that correct and prevent skin pigmentation via several pathways affecting melanogenesis.38 It is an excellent alternative to HQ for mottled and diffuse UV-induced hyperpigmentation and can be used for maintenance therapy in patients with melasma.
Photoprotection is an essential component of therapy for melasma and PIH, but there is a paucity of data on the benefits for SOC patients. Halder et al39 performed a randomized prospective study of 89 black and Hispanic patients who applied sunscreen with a sun protection factor of 30 or 60 daily for 8 weeks. Clinical grading, triplicate L*A*B chromameter, and clinical photography were taken at baseline and weeks 4 and 8. The results demonstrated skin lightening in both black and Hispanic patients and support the use of sunscreen in the prevention and management of dyschromia in SOC patients.39 Visible light also may play a role in melasma development, and thus use of sunscreens or makeup containing iron oxides are recommended.40
Procedural treatments for facial hyperpigmentation include microdermabrasion, chemical peels, lasers, energy-based devices, and microneedling. There are many types and formulations of chemical peeling agents available; however, superficial and medium-depth chemical peels are recommended for SOC patients (Table 2). Deep chemical peels are not recommended for SOC patients due to the potential increased risk for PIH and scarring.
Cosmetic Enhancement in SOC Patients
Cosmetic procedures are gaining popularity in the SOC population and account for more than 20% of cosmetic procedures in the United States.41 Facial cosmetic concerns in SOC include dyschromia, benign growths (dermatosis papulosa nigra), hyperkinetic facial lines, volume loss, and skin laxity.42 Key principles to consider when treating SOC patients are the impact of ethnicity on aging and facial structure, the patient’s desired cosmetic outcome, tissue reaction to anticipated treatments, and the patient’s expectations for recommended therapies.
Aging in SOC Patients
Skin aging can be classified as intrinsic aging or extrinsic aging. Intrinsic aging is genetic and involves subsurface changes such as volume loss, muscle atrophy, and resorption of bony structure. Extrinsic aging (or photoaging) involves surface changes of the epidermis/dermis and manifests as mottled pigmentation, textural changes, and fine wrinkling. Due to the photoprotection of melanin (black skin=SPF 13.4), skin aging in SOC patients is delayed by 10 to 20 years.43 In addition, SOC patients have more reactive collagen and can benefit from noninvasive cosmetic procedures such as fillers and skin-tightening procedures.42
Cosmetic Treatments and Procedures
Dermatosis papulosa nigra (benign growths of skin that have a genetic predisposition)44 occur mainly on the face but can involve the entire body. Treatment modalities include electrodesiccation, cryotherapy, scissor excision, and laser surgery.45
Treatment of hyperkinetic facial lines with botulinum toxin type A is a safe and effective procedure in patients with SOC. Grimes and Shabazz46 performed a 4-month, randomized, double-blind study that evaluated the treatment of glabellar lines in women with Fitzpatrick skin types V and VI. The results demonstrated that the duration of effects was the same in the patients who received either 20 or 30 U of botulinum toxin type A.46 Dynamic rhytides (furrows and frown/scowl lines arising from laughing, frowning, or smiling) can be treated safely in patients with SOC using botulinum toxin type A off label for relaxation of the upper and lower hyperkinetic muscles that result in these unwanted signs of aging. Botulinum toxin type A often is used for etched-in crow’s-feet, which rarely are evident in SOC patients.47 Facial shaping also can be accomplished by injecting botulinum toxin type A in combination with soft-tissue dermal fillers.47
Although black individuals do not experience perioral rhytides at the frequency of white individuals, they experience a variety of other cosmetic issues related to skin sagging and sinking. Currently available hyaluronic acid (HA) fillers have been shown to be safe in patients with Fitzpatrick skin types IV through VI.48 Two studies evaluated fillers in patients with SOC, specifically HA49 and calcium hydroxylapatite,50 focused on treatment of the nasolabial folds and the potential risk for dyspigmentation and keloidal scarring. Taylor et al49 noted that the risk of hyperpigmentation was 6% to 9% for large- and small-particle HA, respectively, and was associated with the serial or multiple puncture injection technique. No hypertrophic or keloidal scarring occurred in both studies.49,50
Facial contouring applications with fillers include glabellar lines, temples, nasal bridge, tear troughs, malar and submalar areas, nasolabial folds, radial lines, lips, marionette lines, mental crease, and chin. Hyaluronic acid fillers also can be used for lip enhancement.47 Although white women are looking to increase the size of their lips, black women are seeking augmentation to restore their lip size to that of their youth. Black individuals do not experience the same frequency of perioral rhytides as white patients, but they experience a variety of other issues related to skin sagging and sinking. Unlike white women, enhancement of the vermilion border rarely is performed in black women due to development of rhytides, predominantly in the body of the lip below the vermilion border in response to volume loss in the upper lip while the lower lip usually maintains its same appearance.47
Facial enhancement utilizing poly-L-lactic acid can be used safely in SOC patients.51 Poly-L-lactic acid microparticles induce collagen formation, leading to dermal thickening over 3 to 6 months; however, multiple sessions are required to achieve optimal aesthetic results.
Patients with more reactive collagen can benefit from noninvasive cosmetic procedures such as skin-tightening procedures.52 Radiofrequency and microfocused ultrasound are cosmetic procedures used to provide skin tightening and facial lifting. They are safe and effective treatments for patients with Fitzpatrick skin types IV to VI.53 Histologically, there is less thinning of collagen bundles and elastic tissue in ethnic skin. Due to stimulation of collagen by these procedures, most SOC patients will experience a more enhanced response, requiring fewer treatment sessions than white individuals.
Conclusion
Medical and aesthetic facial concerns in SOC patients vary and can be a source of emotional and psychological distress that can negatively impact quality of life. The approach to the treatment of SOC patients should be a balance between tolerability and efficacy, considering the potential risk for PIH.
- White GM. Recent findings in the epidemiologic evidence, classification, and subtypes of acne vulgaris. J Am Acad Dermatol. 1998;39(2 pt 3):S34-S37.
- Halder RM, Grimes PE, McLaurin CL, et al. Incidence of common dermatoses in a predominantly black dermatologic practice. Cutis. 1983;32:388, 390.
- Alexis AF, Sergay AB, Taylor SC. Common dermatologic disorders in skin of color: a comparative practice survey. Cutis. 2007;80:387-394.
- Davis SA, Narahari S, Feldman SR, et al. Top dermatologic conditions in patients of color: an analysis of nationally representative data. J Drugs Dermatol. 2012;11:466-473.
- Perkins AC, Cheng CE, Hillebrand GG, et al. Comparison of the epidemiology of acne vulgaris among Caucasians, Asian, Continental Indian and African American women. J Eur Acad Dermatol Venereol. 2011;25:1054-1060.
- Taylor SC, Cook-Bolden F, Rahman Z, et al. Acne vulgaris in skin of color. J Am Acad Dermatol. 2002;46(2 suppl):S98-S106.
- Davis EC, Callender VD. A review of acne in ethnic skin: pathogenesis, clinical manifestations, and management strategies. J Clin Aesthet Dermatol. 2010;3:24-38.
- Halder RM, Holmes YC, Bridgeman-Shah S, et al. A clinicohistologic study of acne vulgaris in black females (abstract). J Invest Dermatol. 1996;106:888.
- Plewig G, Fulton JE, Kligman AM. Pomade acne. Arch Dermatol. 1970;101:580-584.
- Kligman AM, Mills OH. Acne cosmetica. Arch Dermatol. 1972;106:893-897.
- Halder RM, Brooks HL, Callender VD. Acne in ethnic skin. Dermatol Clin. 2003;21:609-615.
- Callender VD. Acne in ethnic skin: special considerations for therapy. Dermatol Ther. 2004;17:184-195.
- Winhoven SM. Postinflammatory hyperpigmentation in an Asian patient. a dramatic response to oral isotretinoin (13-cis-retinoic acid). Br J Med. 2005;152:368-403.
- Sarkar R, Bansal S, Garg VK. Chemical peels for melasma in dark-skinned patients. J Cutan Aesthet Surg. 2012;5:247-253.
- Alexis AF, Coley MK, Nijhawan RI, et al. Nonablative fractional laser resurfacing for acne scarring in patients with Fitzpatrick skin phototypes IV-VI. Dermatol Surg. 2016;42:392-402.
- Culp B, Scheinfeld N. Rosacea: a review. P T. 2009;34:38-45.
- Al-Dabagh A, Davis SA, McMichael AJ, et al. Rosacea in skin of color: not a rare diagnosis. Dermatol Online J. 2014:20. pii:13030/qt1mv9r0ss.
- Del Rosso JQ. Advances in understanding and managing rosacea: part 1: connecting the dots between pathophysiological mechanisms and common clinical features of rosacea with emphasis on vascular changes and facial erythema. J Clin Aesthet Dermatol. 2012;5:16-25.
- Jackson JM, Knuckles M, Minni JP, et al. The role of brimonidine tartrate gel in the treatment of rosacea. Clin Cosmet Investig Dermatol. 2015;23:529-538.
- Patel NU, Shukla S, Zaki J, et al. Oxymetazoline hydrochloride cream for facial erythema associated with rosacea. Expert Rev Clin Pharmacol. 2017;10:104954.
- Weinkle AP, Doktor V, Emer J. Update on the management of rosacea. Clin Cosmet Investig Dermatol. 2015;8:159-177.
- Grimes PE. Skin and hair cosmetic issues in women of color. Dermatol Clin. 2000;19:659-665.
- Taylor A, Pawaskar M, Taylor SL, et al. Prevalence of pigmentary disorders and their impact on quality of life: a prospective cohort study. J Cosmet Dermatol. 2008;7:164-168.
- Davis EC, Callender VD. Postinflammatory hyperpigmentation: a review of the epidemiology, clinical features, and treatment options in skin of color. J Clin Aesthet Dermatol. 2010;3:20-31.
- Grimes PE. Melasma: etiologic and therapeutic considerations. Arch Dermatol. 1995;131:1453-1457.
- Handel AC, Miot LD, Miot HA. Melasma: a clinical and epidemiological review. An Bras Dermatol. 2014;89:771-782.
- Callender VD, Reid SD, Obayan O, et al. Diagnostic clues to frontal fibrosing alopecia in patients of African descent. J Clin Aesthet Dermatol. 2016;9:45-51.
- Narang T, Sawatkar GU, Kumaran MS, et al. Minocycline for recurrent and/or chronic erythema nodosum leprosum. JAMA Dermatol. 2015;151:1026-1028.
- Boyer M, Katta R, Markus R. Diltiazem-induced photodistributed hyperpigmentation. Dermatol Online J. 2003;9:10.
- Pandya AG, Guevara IL. Disorders of hyperpigmentation. Dermatol Clin. 2000;18:91-98.
- Taylor SC, Torok H, Jones T, et al. Efficacy and safety of a new triple-combination agent for the treatment of facial melasma. Cutis. 2003;72:67-72.
- Bulengo-Ransby SM. Topical tretinoin (retinoic acid) therapy for hyperpigmented lesions caused by inflammation of the skin in black patients. N Engl J Med. 1993;328:1438-1443.
- Grimes P, Callender V. Tazarotene cream for postinflammatory hyperpigmentation and acne vulgaris in darker skin: a double-blind, randomized, vehicle-controlled study. Cutis. 2006;77:45-50.
- Jacyk WK. Adapalene in the treatment of African patients. J Eur Acad Dermatol Venereol. 2001;15(suppl 3):37-42.
- Kircik LH. Efficacy and safety of azelaic acid (AzA) gel 15% in the treatment of postinflammatory hyperpigmentation and acne: a 16-week, baseline-controlled study. J Drugs Dermatol. 2011;10:586-590.
- Lee HC, Thng TG, Goh CL. Oral tranexamic acid (TA) in the treatment of melasma. J Am Acad Dermatol. 2016;75:385-392.
- Kindred C, Okereke U, Callender VD. Skin-lightening agents: an overview of prescription, office-dispensed, and over-the-counter products. Cosmet Dermatol. 2013;26:18-26.
- Makino ET, Kadoya K, Sigler ML, et al. Development and clinical assessment of a comprehensive product for pigmentation control in multiple ethnic populations. J Drugs Dermatol. 2016;15:1562-1570.
- Halder R, Rodney I, Munhutu M, et al. Evaluation and effectiveness of a photoprotection composition (sunscreen) on subjects of skin of color. J Am Acad Dermatol. 2015;72(suppl 1):AB215.
- Castanedo-Cazares JP, Hernandez-Blanco D, Carlos-Ortega B, et al. Near-visible light and UV photoprotection in the treatment of melasma: a double-blind randomized trial. Photodermatol Photoimmunol Photomed. 2014;30:35-42.
- American Society for Aesthetic Plastic Surgery. 2016 Cosmetic Surgery National Data Bank Statistics. https://www.surgery.org/sites/default/files/ASAPS-Stats2016.pdf. Accessed November 15, 2017.
- Burgess CM. Soft tissue augmentation in skin of color: market growth, available fillers and successful techniques. J Drugs Dermatol. 2007;6:51-55.
- Davis EC, Callender VD. Aesthetic dermatology for aging ethnic skin. Dermatol Surg. 2011;37:901-917.
- Grimes PE, Arora S, Minus HR, et al. Dermatosis papulosa nigra. Cutis. 1983;32:385-386.
- Lupo M. Dermatosis papulosa nigra: treatment options. J Drugs Dermatol. 2007;6:29-30.
- Grimes PE, Shabazz D. A four-month randomized, double-blind evaluation of the efficacy of botulinum toxin type A for the treatment of glabellar lines in women with skin types V and VI. Dermatol Surg. 2009;35:429-435.
- Burgess CM, Awosika O. Ethnic and gender considerations in the use of facial injectables: African-American patients. Plast Reconstr Surg. 2015;136(5 suppl):28S-31S.
- Taylor SC, Kelly AP, Lim HW, et al, eds. Taylor and Kelly’s Dermatology for Skin of Color. 2nd ed. New York, NY: McGraw-Hill Education; 2016.
- Taylor SC, Burgess CM, Callender VD. Safety of nonanimal stabilized hyaluronic acid dermal fillers in patients with skin of color: a randomized, evaluator-blinded comparative trial. Dermatol Surg. 2009;35(suppl 2):1653-1660.
- Marmur ES, Taylor SC, Grimes PE, et al. Six-month safety results of calcium hydroxylapatite for treatment of nasolabial folds in Fitzpatrick skin types IV to VI. Dermatol Surg. 2009;35(suppl 2):1641-1645.
- Hamilton TK, Burgess CM. Consideration for the use of injectable poly-L-lactic acid in people of color. J Drugs Dermatol. 2010;9:451-456.
- Fabi SG, Goldman MP. Retrospective evaluation of micro-focused ultrasound for lifting and tightening of the face and neck. Dermatol Surg. 2014;40:569-575.
- Harris MO, Sundaram HA. Safety of microfocused ultrasound with visualization in patients with Fitzpatrick skin phototypes III to VI. JAMA Facial Plast Surg. 2015;17:355-357.
- White GM. Recent findings in the epidemiologic evidence, classification, and subtypes of acne vulgaris. J Am Acad Dermatol. 1998;39(2 pt 3):S34-S37.
- Halder RM, Grimes PE, McLaurin CL, et al. Incidence of common dermatoses in a predominantly black dermatologic practice. Cutis. 1983;32:388, 390.
- Alexis AF, Sergay AB, Taylor SC. Common dermatologic disorders in skin of color: a comparative practice survey. Cutis. 2007;80:387-394.
- Davis SA, Narahari S, Feldman SR, et al. Top dermatologic conditions in patients of color: an analysis of nationally representative data. J Drugs Dermatol. 2012;11:466-473.
- Perkins AC, Cheng CE, Hillebrand GG, et al. Comparison of the epidemiology of acne vulgaris among Caucasians, Asian, Continental Indian and African American women. J Eur Acad Dermatol Venereol. 2011;25:1054-1060.
- Taylor SC, Cook-Bolden F, Rahman Z, et al. Acne vulgaris in skin of color. J Am Acad Dermatol. 2002;46(2 suppl):S98-S106.
- Davis EC, Callender VD. A review of acne in ethnic skin: pathogenesis, clinical manifestations, and management strategies. J Clin Aesthet Dermatol. 2010;3:24-38.
- Halder RM, Holmes YC, Bridgeman-Shah S, et al. A clinicohistologic study of acne vulgaris in black females (abstract). J Invest Dermatol. 1996;106:888.
- Plewig G, Fulton JE, Kligman AM. Pomade acne. Arch Dermatol. 1970;101:580-584.
- Kligman AM, Mills OH. Acne cosmetica. Arch Dermatol. 1972;106:893-897.
- Halder RM, Brooks HL, Callender VD. Acne in ethnic skin. Dermatol Clin. 2003;21:609-615.
- Callender VD. Acne in ethnic skin: special considerations for therapy. Dermatol Ther. 2004;17:184-195.
- Winhoven SM. Postinflammatory hyperpigmentation in an Asian patient. a dramatic response to oral isotretinoin (13-cis-retinoic acid). Br J Med. 2005;152:368-403.
- Sarkar R, Bansal S, Garg VK. Chemical peels for melasma in dark-skinned patients. J Cutan Aesthet Surg. 2012;5:247-253.
- Alexis AF, Coley MK, Nijhawan RI, et al. Nonablative fractional laser resurfacing for acne scarring in patients with Fitzpatrick skin phototypes IV-VI. Dermatol Surg. 2016;42:392-402.
- Culp B, Scheinfeld N. Rosacea: a review. P T. 2009;34:38-45.
- Al-Dabagh A, Davis SA, McMichael AJ, et al. Rosacea in skin of color: not a rare diagnosis. Dermatol Online J. 2014:20. pii:13030/qt1mv9r0ss.
- Del Rosso JQ. Advances in understanding and managing rosacea: part 1: connecting the dots between pathophysiological mechanisms and common clinical features of rosacea with emphasis on vascular changes and facial erythema. J Clin Aesthet Dermatol. 2012;5:16-25.
- Jackson JM, Knuckles M, Minni JP, et al. The role of brimonidine tartrate gel in the treatment of rosacea. Clin Cosmet Investig Dermatol. 2015;23:529-538.
- Patel NU, Shukla S, Zaki J, et al. Oxymetazoline hydrochloride cream for facial erythema associated with rosacea. Expert Rev Clin Pharmacol. 2017;10:104954.
- Weinkle AP, Doktor V, Emer J. Update on the management of rosacea. Clin Cosmet Investig Dermatol. 2015;8:159-177.
- Grimes PE. Skin and hair cosmetic issues in women of color. Dermatol Clin. 2000;19:659-665.
- Taylor A, Pawaskar M, Taylor SL, et al. Prevalence of pigmentary disorders and their impact on quality of life: a prospective cohort study. J Cosmet Dermatol. 2008;7:164-168.
- Davis EC, Callender VD. Postinflammatory hyperpigmentation: a review of the epidemiology, clinical features, and treatment options in skin of color. J Clin Aesthet Dermatol. 2010;3:20-31.
- Grimes PE. Melasma: etiologic and therapeutic considerations. Arch Dermatol. 1995;131:1453-1457.
- Handel AC, Miot LD, Miot HA. Melasma: a clinical and epidemiological review. An Bras Dermatol. 2014;89:771-782.
- Callender VD, Reid SD, Obayan O, et al. Diagnostic clues to frontal fibrosing alopecia in patients of African descent. J Clin Aesthet Dermatol. 2016;9:45-51.
- Narang T, Sawatkar GU, Kumaran MS, et al. Minocycline for recurrent and/or chronic erythema nodosum leprosum. JAMA Dermatol. 2015;151:1026-1028.
- Boyer M, Katta R, Markus R. Diltiazem-induced photodistributed hyperpigmentation. Dermatol Online J. 2003;9:10.
- Pandya AG, Guevara IL. Disorders of hyperpigmentation. Dermatol Clin. 2000;18:91-98.
- Taylor SC, Torok H, Jones T, et al. Efficacy and safety of a new triple-combination agent for the treatment of facial melasma. Cutis. 2003;72:67-72.
- Bulengo-Ransby SM. Topical tretinoin (retinoic acid) therapy for hyperpigmented lesions caused by inflammation of the skin in black patients. N Engl J Med. 1993;328:1438-1443.
- Grimes P, Callender V. Tazarotene cream for postinflammatory hyperpigmentation and acne vulgaris in darker skin: a double-blind, randomized, vehicle-controlled study. Cutis. 2006;77:45-50.
- Jacyk WK. Adapalene in the treatment of African patients. J Eur Acad Dermatol Venereol. 2001;15(suppl 3):37-42.
- Kircik LH. Efficacy and safety of azelaic acid (AzA) gel 15% in the treatment of postinflammatory hyperpigmentation and acne: a 16-week, baseline-controlled study. J Drugs Dermatol. 2011;10:586-590.
- Lee HC, Thng TG, Goh CL. Oral tranexamic acid (TA) in the treatment of melasma. J Am Acad Dermatol. 2016;75:385-392.
- Kindred C, Okereke U, Callender VD. Skin-lightening agents: an overview of prescription, office-dispensed, and over-the-counter products. Cosmet Dermatol. 2013;26:18-26.
- Makino ET, Kadoya K, Sigler ML, et al. Development and clinical assessment of a comprehensive product for pigmentation control in multiple ethnic populations. J Drugs Dermatol. 2016;15:1562-1570.
- Halder R, Rodney I, Munhutu M, et al. Evaluation and effectiveness of a photoprotection composition (sunscreen) on subjects of skin of color. J Am Acad Dermatol. 2015;72(suppl 1):AB215.
- Castanedo-Cazares JP, Hernandez-Blanco D, Carlos-Ortega B, et al. Near-visible light and UV photoprotection in the treatment of melasma: a double-blind randomized trial. Photodermatol Photoimmunol Photomed. 2014;30:35-42.
- American Society for Aesthetic Plastic Surgery. 2016 Cosmetic Surgery National Data Bank Statistics. https://www.surgery.org/sites/default/files/ASAPS-Stats2016.pdf. Accessed November 15, 2017.
- Burgess CM. Soft tissue augmentation in skin of color: market growth, available fillers and successful techniques. J Drugs Dermatol. 2007;6:51-55.
- Davis EC, Callender VD. Aesthetic dermatology for aging ethnic skin. Dermatol Surg. 2011;37:901-917.
- Grimes PE, Arora S, Minus HR, et al. Dermatosis papulosa nigra. Cutis. 1983;32:385-386.
- Lupo M. Dermatosis papulosa nigra: treatment options. J Drugs Dermatol. 2007;6:29-30.
- Grimes PE, Shabazz D. A four-month randomized, double-blind evaluation of the efficacy of botulinum toxin type A for the treatment of glabellar lines in women with skin types V and VI. Dermatol Surg. 2009;35:429-435.
- Burgess CM, Awosika O. Ethnic and gender considerations in the use of facial injectables: African-American patients. Plast Reconstr Surg. 2015;136(5 suppl):28S-31S.
- Taylor SC, Kelly AP, Lim HW, et al, eds. Taylor and Kelly’s Dermatology for Skin of Color. 2nd ed. New York, NY: McGraw-Hill Education; 2016.
- Taylor SC, Burgess CM, Callender VD. Safety of nonanimal stabilized hyaluronic acid dermal fillers in patients with skin of color: a randomized, evaluator-blinded comparative trial. Dermatol Surg. 2009;35(suppl 2):1653-1660.
- Marmur ES, Taylor SC, Grimes PE, et al. Six-month safety results of calcium hydroxylapatite for treatment of nasolabial folds in Fitzpatrick skin types IV to VI. Dermatol Surg. 2009;35(suppl 2):1641-1645.
- Hamilton TK, Burgess CM. Consideration for the use of injectable poly-L-lactic acid in people of color. J Drugs Dermatol. 2010;9:451-456.
- Fabi SG, Goldman MP. Retrospective evaluation of micro-focused ultrasound for lifting and tightening of the face and neck. Dermatol Surg. 2014;40:569-575.
- Harris MO, Sundaram HA. Safety of microfocused ultrasound with visualization in patients with Fitzpatrick skin phototypes III to VI. JAMA Facial Plast Surg. 2015;17:355-357.
Practice Points
- Treat acne in skin of color (SOC) patients early and aggressively to prevent or minimize subsequent postinflammatory hyperpigmentation (PIH) and acne scarring.
- Vascular lasers and intense pulsed light may be used to address the vascular components of rosacea; however, the latter is not recommended in Fitzpatrick skin types IV to VI.
- Hydroquinone is the gold standard for skin lightening and is often used as a first-line therapy for melasma and PIH.
- Photoprotection is an essential component of therapy for hyperpigmented skin disorders.
- Cosmetic procedures are gaining popularity in the SOC population. When treating SOC patients, consider the impact of ethnicity on aging and facial structure, the patient's desired cosmetic outcome, tissue reaction to anticipated treatments, and the patient's expectations for recommended therapies.
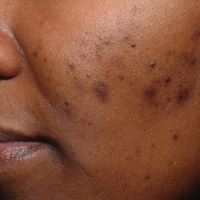
How to decide which ‘birthmarks’ spell trouble
When evaluating lumps and bumps in infants, categorizing them can help determine whether they need immediate attention, said James R. Treat, MD, a pediatric dermatologist at Children’s Hospital of Philadelphia, Pennsylvania.
“Divide ‘birthmarks’ based on appearance, “then decide when to worry,” he said in a presentation at Skin Disease Education Foundation’s Women’s & Pediatric Dermatology Seminar.
For example, Spitz nevi occur in patients younger than 20 years, most often on the face and lower extremities, but most are benign, Dr. Treat said. However, he recommends a biopsy if the patient is pubertal or older, or if the lesions are larger than 8 mm, amelanotic, or show asymmetry, ulceration, or excessive growth.
By contrast, neurocutaneous melanosis is a rare but serious skin condition that occurs in children and can be fatal if it progresses to melanoma, he pointed out. The condition involves the migration of melanocytes into the spinal canal and cerebrospinal fluid during development. Symptoms may include headache, seizures, and paralysis, and clinicians should keep them in mind when seeing children with melanocytic nevi, he noted. The highest risk for melanoma transformation is increased for individuals with more than 20 congenital moles, and “the second-highest risk is having a giant nevus lying overtop of the midline spine or scalp,” he said.
In some cases, yellow or tan lesions in children are benign and will resolve on their own, Dr. Treat said.
Juvenile xanthogranuloma (JXG), characterized by yellow-brown asymptomatic papules and nodules, develops most often within the first year of life, but the lesions usually resolve spontaneously by school age, he added.
Mastocytosis, localized collections of mast cells, presents as yellow/tan lesions that develop within the first 2 years of life. The condition can be systemic; patients may experience flushing and diarrhea because of localized release of histamines, and those with a history of weight loss, easy bruising or bleeding, hepatosplenomegaly, or lymphadenopathy may have systemic disease, Dr. Treat explained.
Subcutaneous fat necrosis can present within the first 2 weeks of life as firm, red-purple tender nodules that may be disturbing to parents. These lesions are most likely to appear on the cheeks, arms, back, and thighs, and are related to hypoxia or trauma, he added. The lesions usually resolve spontaneously within a period of weeks to months, although they may heal with some atrophy and scarring, he said. Subcutaneous fat necrosis is associated with hypercalcemia, so “it is important to check frequently, as hypercalcemia can occur weeks after the nodules resolve,” he commented.
Dr. Treat disclosed serving as a consultant to Procter & Gamble. SDEF and this news organization are owned by Frontline Medical Communications.
When evaluating lumps and bumps in infants, categorizing them can help determine whether they need immediate attention, said James R. Treat, MD, a pediatric dermatologist at Children’s Hospital of Philadelphia, Pennsylvania.
“Divide ‘birthmarks’ based on appearance, “then decide when to worry,” he said in a presentation at Skin Disease Education Foundation’s Women’s & Pediatric Dermatology Seminar.
For example, Spitz nevi occur in patients younger than 20 years, most often on the face and lower extremities, but most are benign, Dr. Treat said. However, he recommends a biopsy if the patient is pubertal or older, or if the lesions are larger than 8 mm, amelanotic, or show asymmetry, ulceration, or excessive growth.
By contrast, neurocutaneous melanosis is a rare but serious skin condition that occurs in children and can be fatal if it progresses to melanoma, he pointed out. The condition involves the migration of melanocytes into the spinal canal and cerebrospinal fluid during development. Symptoms may include headache, seizures, and paralysis, and clinicians should keep them in mind when seeing children with melanocytic nevi, he noted. The highest risk for melanoma transformation is increased for individuals with more than 20 congenital moles, and “the second-highest risk is having a giant nevus lying overtop of the midline spine or scalp,” he said.
In some cases, yellow or tan lesions in children are benign and will resolve on their own, Dr. Treat said.
Juvenile xanthogranuloma (JXG), characterized by yellow-brown asymptomatic papules and nodules, develops most often within the first year of life, but the lesions usually resolve spontaneously by school age, he added.
Mastocytosis, localized collections of mast cells, presents as yellow/tan lesions that develop within the first 2 years of life. The condition can be systemic; patients may experience flushing and diarrhea because of localized release of histamines, and those with a history of weight loss, easy bruising or bleeding, hepatosplenomegaly, or lymphadenopathy may have systemic disease, Dr. Treat explained.
Subcutaneous fat necrosis can present within the first 2 weeks of life as firm, red-purple tender nodules that may be disturbing to parents. These lesions are most likely to appear on the cheeks, arms, back, and thighs, and are related to hypoxia or trauma, he added. The lesions usually resolve spontaneously within a period of weeks to months, although they may heal with some atrophy and scarring, he said. Subcutaneous fat necrosis is associated with hypercalcemia, so “it is important to check frequently, as hypercalcemia can occur weeks after the nodules resolve,” he commented.
Dr. Treat disclosed serving as a consultant to Procter & Gamble. SDEF and this news organization are owned by Frontline Medical Communications.
When evaluating lumps and bumps in infants, categorizing them can help determine whether they need immediate attention, said James R. Treat, MD, a pediatric dermatologist at Children’s Hospital of Philadelphia, Pennsylvania.
“Divide ‘birthmarks’ based on appearance, “then decide when to worry,” he said in a presentation at Skin Disease Education Foundation’s Women’s & Pediatric Dermatology Seminar.
For example, Spitz nevi occur in patients younger than 20 years, most often on the face and lower extremities, but most are benign, Dr. Treat said. However, he recommends a biopsy if the patient is pubertal or older, or if the lesions are larger than 8 mm, amelanotic, or show asymmetry, ulceration, or excessive growth.
By contrast, neurocutaneous melanosis is a rare but serious skin condition that occurs in children and can be fatal if it progresses to melanoma, he pointed out. The condition involves the migration of melanocytes into the spinal canal and cerebrospinal fluid during development. Symptoms may include headache, seizures, and paralysis, and clinicians should keep them in mind when seeing children with melanocytic nevi, he noted. The highest risk for melanoma transformation is increased for individuals with more than 20 congenital moles, and “the second-highest risk is having a giant nevus lying overtop of the midline spine or scalp,” he said.
In some cases, yellow or tan lesions in children are benign and will resolve on their own, Dr. Treat said.
Juvenile xanthogranuloma (JXG), characterized by yellow-brown asymptomatic papules and nodules, develops most often within the first year of life, but the lesions usually resolve spontaneously by school age, he added.
Mastocytosis, localized collections of mast cells, presents as yellow/tan lesions that develop within the first 2 years of life. The condition can be systemic; patients may experience flushing and diarrhea because of localized release of histamines, and those with a history of weight loss, easy bruising or bleeding, hepatosplenomegaly, or lymphadenopathy may have systemic disease, Dr. Treat explained.
Subcutaneous fat necrosis can present within the first 2 weeks of life as firm, red-purple tender nodules that may be disturbing to parents. These lesions are most likely to appear on the cheeks, arms, back, and thighs, and are related to hypoxia or trauma, he added. The lesions usually resolve spontaneously within a period of weeks to months, although they may heal with some atrophy and scarring, he said. Subcutaneous fat necrosis is associated with hypercalcemia, so “it is important to check frequently, as hypercalcemia can occur weeks after the nodules resolve,” he commented.
Dr. Treat disclosed serving as a consultant to Procter & Gamble. SDEF and this news organization are owned by Frontline Medical Communications.
FROM SDEF WOMEN’S & PEDIATRIC DERMATOLOGY SEMINAR
Asymptomatic Pink Plaque on the Scapula
The Diagnosis: Primary Cutaneous Follicle Center Lymphoma
Immunohistochemistry revealed a nodular infiltrate consisting of small to large atypical lymphocytes forming an irregular germinal center with notably thinned mantle zones and lack of polarization (Figure, A). Atypical cells stained positively with Bcl-6, and CD20 was diffusely positive (Figure, B-D). Bcl-2 and CD3 colocalized to the reactive T-cell infiltrate, and CD10 was largely negative. Further workup with bone marrow biopsy and full-body positron emission tomography-computed tomography was unremarkable. Given these findings, a diagnosis of primary cutaneous follicle center lymphoma (FCL) was made. At 1 month following radiation therapy, complete clinical clearance of the lymphoma was achieved.
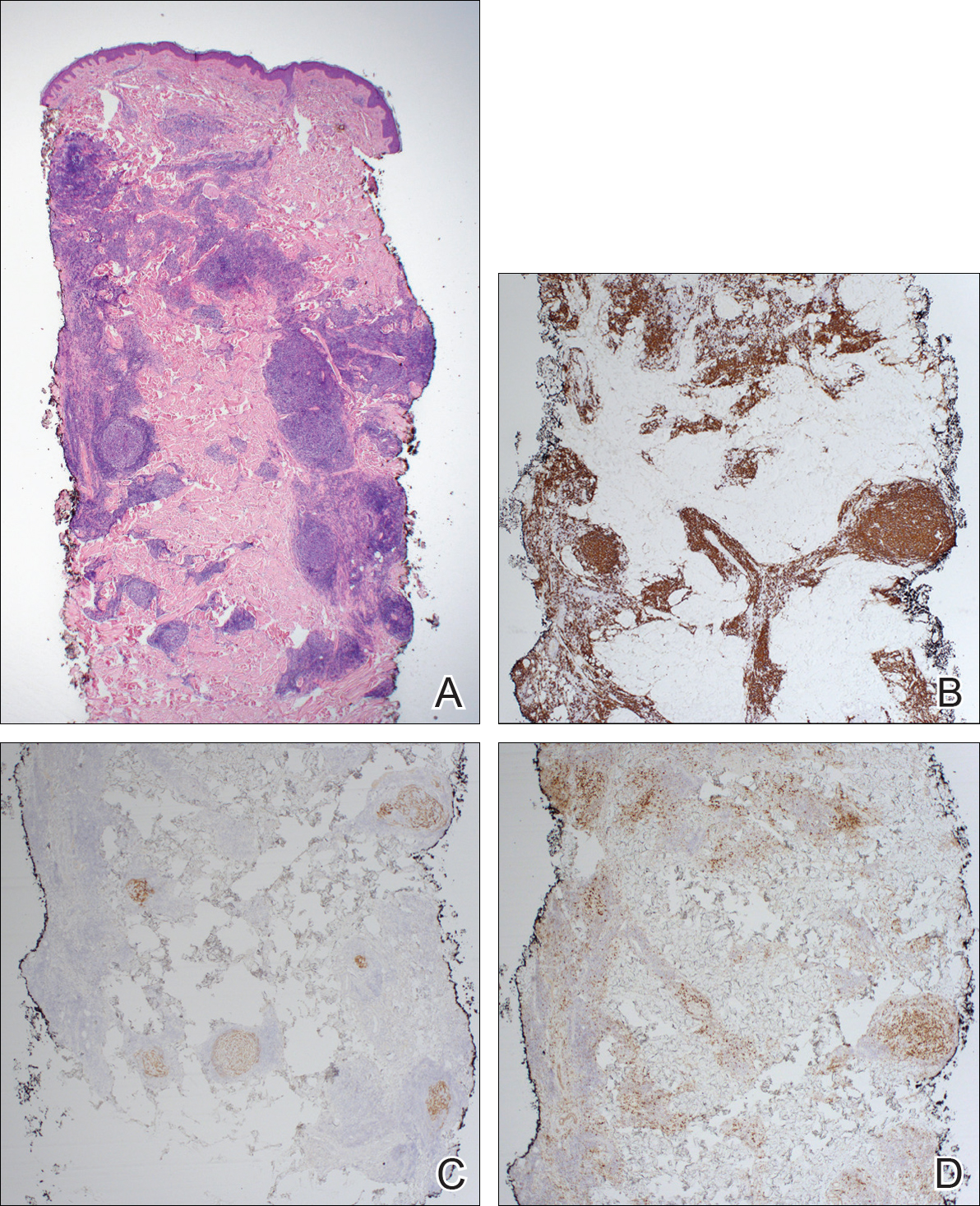
Follicle center lymphoma, also known as cutaneous follicular lymphoma, is the most common subtype of primary cutaneous B-cell lymphomas, representing approximately 57% of cases.1 Follicle center lymphoma typically affects older, non-Hispanic white adults with a median age of onset of 60 years. It has a predilection for the head, neck, and trunk.2 Lesions present as solitary erythematous to violaceous papules, plaques, or nodules, but they can more rarely be multifocal.3 Clinical diagnosis of FCL can be difficult, with papular lesions resembling acne, rosacea, folliculitis, or arthropod assault.4,5 As such, diagnosis of FCL typically relies on histopathologic analysis.
Histologically, FCL can present in several different patterns including follicular, nodular, diffuse, or a pleomorphic mix of these.2,6 The cells are comprised of germinal center B cells, staining positively for Bcl-6, CD20, and CD79a.7 Tumor cells do not exhibit the t(14;18) translocation seen in nodal follicular lymphomas.2,8 Unlike marginal zone lymphoma, FCL stains negatively for Bcl-2 and multiple myeloma 1/interferon regulatory factor 4 (MUM1/IRF-4).2,9 Forkhead box P1 (FOXP1) also is usually negative, but its presence can indicate a poorer prognosis.2 It is important to distinguish primary cutaneous B-cell lymphomas from systemic B-cell lymphoma with secondary cutaneous involvement, as they have a different clinical prognosis and management course. Further workup includes bone marrow biopsy, serum analysis for clonal involvement, and positron emission tomography-computed tomography imaging. Follicle center lymphoma generally has an indolent disease course with a favorable 5-year survival rate of approximately 95%.6,8
Untreated lesions may enlarge slowly or even spontaneously involute.10 The histologic growth pattern and number of lesions do not affect prognosis, but presence on the legs has a 5-year survival rate of 41%.2 Extracutaneous dissemination can occur in 5% to 10% of cases.2 Given the slow progression of FCL, conservative management with observation is an option. However, curative treatment can be reasonably attempted for solitary lesions by excision or radiation. Treatment of FCL often can be complicated by its predilection for the head and neck. Other treatment modalities include topical steroids, imiquimod, nitrogen mustard, and bexarotene.10 More generalized involvement may require systemic therapy with rituximab or chemotherapy. Recurrence after therapy is common, reported in 46.5% of patients, but does not affect prognosis.2
- Zinzani PL, Quaglino P, Pimpinelli N, et al. Prognostic factors in primary cutaneous B-cell lymphoma: The Italian Study Group for Cutaneous Lymphomas. J Clin Oncol. 2006;24:1376-1382.
- Suárez AL, Pulitzer M, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part I. clinical features, diagnosis, and classification. J Am Acad Dermatol. 2013;69:1-13.
- Grange F, Bekkenk MW, Wechsler J, et al. Prognostic factors in primary cutaneous large B-cell lymphomas: a European multicenter study. J Clin Oncol. 2001;19:3602-3610.
- Soon CW, Pincus LB, Ai WZ, et al. Acneiform presentation of primary cutaneous follicle center lymphoma. J Am Acad Dermatol. 2011;65:887-889.
- Massone C, Fink-Puches R, Laimer M, et al. Miliary and agminated-type primary cutaneous follicle center lymphoma: a report of 18 cases. J Am Acad Dermatol. 2011;65:749-755.
- Wilcox RA. CME information: cutaneous B-cell lymphomas: 2015 update on diagnosis, risk-stratification, and management. Am J Hematol. 2015;90:73-76.
- Franco R, Fernandez-Vazquez A, Rodriguez-Peralto JL, et al. Cutaneous follicular B-cell lymphoma: description of a series of 18 cases. Am J Surg Pathol. 2001;25:875-883.
- Kempf W, Denisjuk N, Kerl K, et al. Primary cutaneous B-cell lymphomas. J Dtsch Dermatol Ges. 2012;10:12-22; quiz 23.
- de Leval L HN, Longtine J, Ferry JA, et al. Cutaneous B-cell lymphomas of follicular and marginal zone types: use of Bcl-6, CD10, Bcl-2, and CD21 in differential diagnosis and classification. Am J Surg Pathol. 2001;25:732-741.
- Suárez AL, Querfeld C, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part II. therapy and future directions. J Am Acad Dermatol. 2013;69:1-11.
The Diagnosis: Primary Cutaneous Follicle Center Lymphoma
Immunohistochemistry revealed a nodular infiltrate consisting of small to large atypical lymphocytes forming an irregular germinal center with notably thinned mantle zones and lack of polarization (Figure, A). Atypical cells stained positively with Bcl-6, and CD20 was diffusely positive (Figure, B-D). Bcl-2 and CD3 colocalized to the reactive T-cell infiltrate, and CD10 was largely negative. Further workup with bone marrow biopsy and full-body positron emission tomography-computed tomography was unremarkable. Given these findings, a diagnosis of primary cutaneous follicle center lymphoma (FCL) was made. At 1 month following radiation therapy, complete clinical clearance of the lymphoma was achieved.
Follicle center lymphoma, also known as cutaneous follicular lymphoma, is the most common subtype of primary cutaneous B-cell lymphomas, representing approximately 57% of cases.1 Follicle center lymphoma typically affects older, non-Hispanic white adults with a median age of onset of 60 years. It has a predilection for the head, neck, and trunk.2 Lesions present as solitary erythematous to violaceous papules, plaques, or nodules, but they can more rarely be multifocal.3 Clinical diagnosis of FCL can be difficult, with papular lesions resembling acne, rosacea, folliculitis, or arthropod assault.4,5 As such, diagnosis of FCL typically relies on histopathologic analysis.
Histologically, FCL can present in several different patterns including follicular, nodular, diffuse, or a pleomorphic mix of these.2,6 The cells are comprised of germinal center B cells, staining positively for Bcl-6, CD20, and CD79a.7 Tumor cells do not exhibit the t(14;18) translocation seen in nodal follicular lymphomas.2,8 Unlike marginal zone lymphoma, FCL stains negatively for Bcl-2 and multiple myeloma 1/interferon regulatory factor 4 (MUM1/IRF-4).2,9 Forkhead box P1 (FOXP1) also is usually negative, but its presence can indicate a poorer prognosis.2 It is important to distinguish primary cutaneous B-cell lymphomas from systemic B-cell lymphoma with secondary cutaneous involvement, as they have a different clinical prognosis and management course. Further workup includes bone marrow biopsy, serum analysis for clonal involvement, and positron emission tomography-computed tomography imaging. Follicle center lymphoma generally has an indolent disease course with a favorable 5-year survival rate of approximately 95%.6,8
Untreated lesions may enlarge slowly or even spontaneously involute.10 The histologic growth pattern and number of lesions do not affect prognosis, but presence on the legs has a 5-year survival rate of 41%.2 Extracutaneous dissemination can occur in 5% to 10% of cases.2 Given the slow progression of FCL, conservative management with observation is an option. However, curative treatment can be reasonably attempted for solitary lesions by excision or radiation. Treatment of FCL often can be complicated by its predilection for the head and neck. Other treatment modalities include topical steroids, imiquimod, nitrogen mustard, and bexarotene.10 More generalized involvement may require systemic therapy with rituximab or chemotherapy. Recurrence after therapy is common, reported in 46.5% of patients, but does not affect prognosis.2
The Diagnosis: Primary Cutaneous Follicle Center Lymphoma
Immunohistochemistry revealed a nodular infiltrate consisting of small to large atypical lymphocytes forming an irregular germinal center with notably thinned mantle zones and lack of polarization (Figure, A). Atypical cells stained positively with Bcl-6, and CD20 was diffusely positive (Figure, B-D). Bcl-2 and CD3 colocalized to the reactive T-cell infiltrate, and CD10 was largely negative. Further workup with bone marrow biopsy and full-body positron emission tomography-computed tomography was unremarkable. Given these findings, a diagnosis of primary cutaneous follicle center lymphoma (FCL) was made. At 1 month following radiation therapy, complete clinical clearance of the lymphoma was achieved.
Follicle center lymphoma, also known as cutaneous follicular lymphoma, is the most common subtype of primary cutaneous B-cell lymphomas, representing approximately 57% of cases.1 Follicle center lymphoma typically affects older, non-Hispanic white adults with a median age of onset of 60 years. It has a predilection for the head, neck, and trunk.2 Lesions present as solitary erythematous to violaceous papules, plaques, or nodules, but they can more rarely be multifocal.3 Clinical diagnosis of FCL can be difficult, with papular lesions resembling acne, rosacea, folliculitis, or arthropod assault.4,5 As such, diagnosis of FCL typically relies on histopathologic analysis.
Histologically, FCL can present in several different patterns including follicular, nodular, diffuse, or a pleomorphic mix of these.2,6 The cells are comprised of germinal center B cells, staining positively for Bcl-6, CD20, and CD79a.7 Tumor cells do not exhibit the t(14;18) translocation seen in nodal follicular lymphomas.2,8 Unlike marginal zone lymphoma, FCL stains negatively for Bcl-2 and multiple myeloma 1/interferon regulatory factor 4 (MUM1/IRF-4).2,9 Forkhead box P1 (FOXP1) also is usually negative, but its presence can indicate a poorer prognosis.2 It is important to distinguish primary cutaneous B-cell lymphomas from systemic B-cell lymphoma with secondary cutaneous involvement, as they have a different clinical prognosis and management course. Further workup includes bone marrow biopsy, serum analysis for clonal involvement, and positron emission tomography-computed tomography imaging. Follicle center lymphoma generally has an indolent disease course with a favorable 5-year survival rate of approximately 95%.6,8
Untreated lesions may enlarge slowly or even spontaneously involute.10 The histologic growth pattern and number of lesions do not affect prognosis, but presence on the legs has a 5-year survival rate of 41%.2 Extracutaneous dissemination can occur in 5% to 10% of cases.2 Given the slow progression of FCL, conservative management with observation is an option. However, curative treatment can be reasonably attempted for solitary lesions by excision or radiation. Treatment of FCL often can be complicated by its predilection for the head and neck. Other treatment modalities include topical steroids, imiquimod, nitrogen mustard, and bexarotene.10 More generalized involvement may require systemic therapy with rituximab or chemotherapy. Recurrence after therapy is common, reported in 46.5% of patients, but does not affect prognosis.2
- Zinzani PL, Quaglino P, Pimpinelli N, et al. Prognostic factors in primary cutaneous B-cell lymphoma: The Italian Study Group for Cutaneous Lymphomas. J Clin Oncol. 2006;24:1376-1382.
- Suárez AL, Pulitzer M, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part I. clinical features, diagnosis, and classification. J Am Acad Dermatol. 2013;69:1-13.
- Grange F, Bekkenk MW, Wechsler J, et al. Prognostic factors in primary cutaneous large B-cell lymphomas: a European multicenter study. J Clin Oncol. 2001;19:3602-3610.
- Soon CW, Pincus LB, Ai WZ, et al. Acneiform presentation of primary cutaneous follicle center lymphoma. J Am Acad Dermatol. 2011;65:887-889.
- Massone C, Fink-Puches R, Laimer M, et al. Miliary and agminated-type primary cutaneous follicle center lymphoma: a report of 18 cases. J Am Acad Dermatol. 2011;65:749-755.
- Wilcox RA. CME information: cutaneous B-cell lymphomas: 2015 update on diagnosis, risk-stratification, and management. Am J Hematol. 2015;90:73-76.
- Franco R, Fernandez-Vazquez A, Rodriguez-Peralto JL, et al. Cutaneous follicular B-cell lymphoma: description of a series of 18 cases. Am J Surg Pathol. 2001;25:875-883.
- Kempf W, Denisjuk N, Kerl K, et al. Primary cutaneous B-cell lymphomas. J Dtsch Dermatol Ges. 2012;10:12-22; quiz 23.
- de Leval L HN, Longtine J, Ferry JA, et al. Cutaneous B-cell lymphomas of follicular and marginal zone types: use of Bcl-6, CD10, Bcl-2, and CD21 in differential diagnosis and classification. Am J Surg Pathol. 2001;25:732-741.
- Suárez AL, Querfeld C, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part II. therapy and future directions. J Am Acad Dermatol. 2013;69:1-11.
- Zinzani PL, Quaglino P, Pimpinelli N, et al. Prognostic factors in primary cutaneous B-cell lymphoma: The Italian Study Group for Cutaneous Lymphomas. J Clin Oncol. 2006;24:1376-1382.
- Suárez AL, Pulitzer M, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part I. clinical features, diagnosis, and classification. J Am Acad Dermatol. 2013;69:1-13.
- Grange F, Bekkenk MW, Wechsler J, et al. Prognostic factors in primary cutaneous large B-cell lymphomas: a European multicenter study. J Clin Oncol. 2001;19:3602-3610.
- Soon CW, Pincus LB, Ai WZ, et al. Acneiform presentation of primary cutaneous follicle center lymphoma. J Am Acad Dermatol. 2011;65:887-889.
- Massone C, Fink-Puches R, Laimer M, et al. Miliary and agminated-type primary cutaneous follicle center lymphoma: a report of 18 cases. J Am Acad Dermatol. 2011;65:749-755.
- Wilcox RA. CME information: cutaneous B-cell lymphomas: 2015 update on diagnosis, risk-stratification, and management. Am J Hematol. 2015;90:73-76.
- Franco R, Fernandez-Vazquez A, Rodriguez-Peralto JL, et al. Cutaneous follicular B-cell lymphoma: description of a series of 18 cases. Am J Surg Pathol. 2001;25:875-883.
- Kempf W, Denisjuk N, Kerl K, et al. Primary cutaneous B-cell lymphomas. J Dtsch Dermatol Ges. 2012;10:12-22; quiz 23.
- de Leval L HN, Longtine J, Ferry JA, et al. Cutaneous B-cell lymphomas of follicular and marginal zone types: use of Bcl-6, CD10, Bcl-2, and CD21 in differential diagnosis and classification. Am J Surg Pathol. 2001;25:732-741.
- Suárez AL, Querfeld C, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part II. therapy and future directions. J Am Acad Dermatol. 2013;69:1-11.
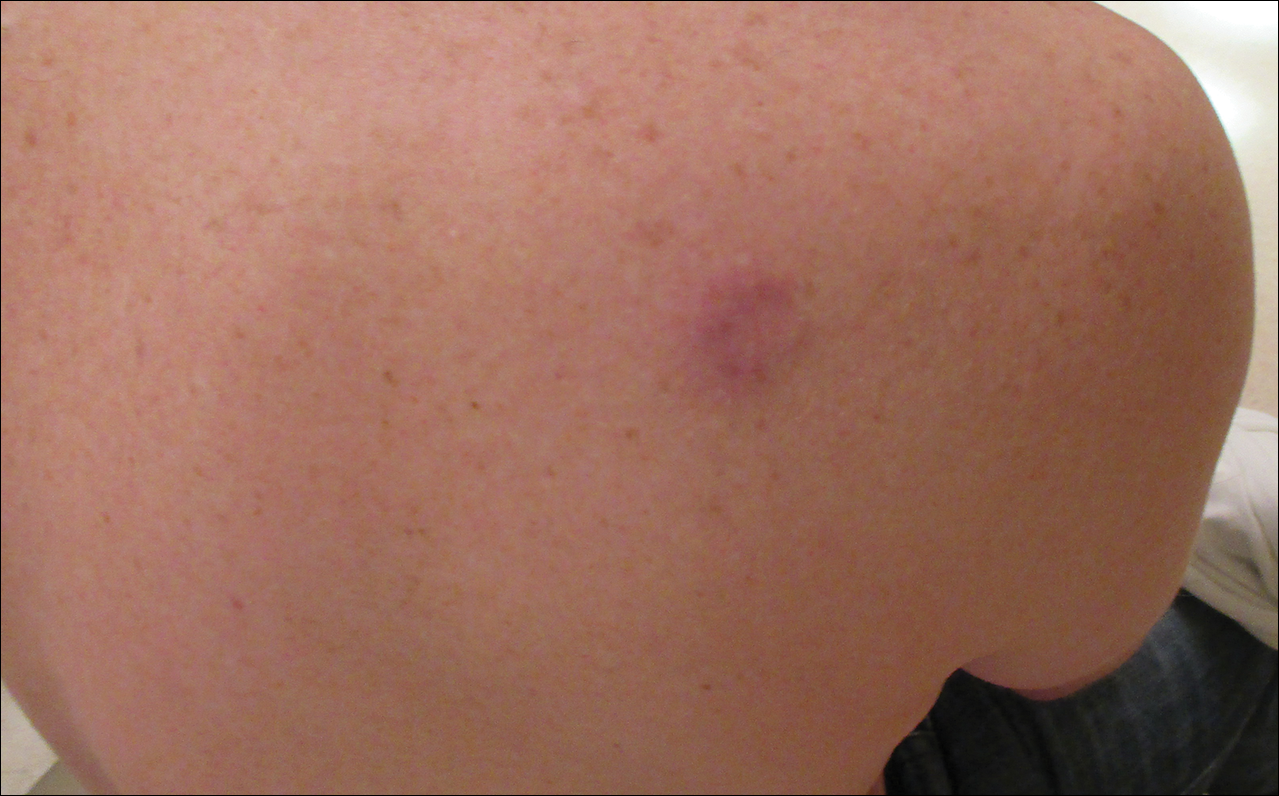
A 36-year-old man presented with a pink plaque on the right side of the scapula of 1 year's duration. The plaque had not grown and was completely asymptomatic. Physical examination revealed a violaceous, pink, 2-cm nodule with overlying telangiectasia. No other concerning lesions were identified on total-body skin examination. A punch biopsy was obtained.
Early referral recommended for high-risk port-wine stain cases
In high-risk port-wine stain phenotypes – forehead, hemifacial, and median – early referral to a pediatric neurologist is the best way to enable early symptom recognition of Sturge-Weber syndrome (SWS), according to results of a literature review.
If imaging is desired, electroencephalography (EEG) is cheaper and more minimally invasive than MRI (Pediatr Dermatol. 2017 Oct 16. doi: 10.1111/pde.13304).
Of the 34 studies analyzed, none examined the correlation between early detection of presymptomatic SWS and earlier seizure detection. There were also no data to verify improved outcomes with early detection, Michaela Zallmann, MD, of the department of dermatology at Eastern Health, Monash University, Box Hill, Australia, and her coauthors reported. While this indicates a need for further studies, it also shows a need for improved education and clinical monitoring as standard practice.
Additionally, negative imaging results do not obviate the possibility of SWS diagnosis, the investigators reported. In the studies that recorded false negatives, MRI was negative in 3%-6% of infants who later became symptomatic. Of the seven infants with false-negative results, four were less than 6 months old when the initial imaging was conducted. The investigators noted that it is not known at what age a negative MRI can reliably exclude SWS.
When imaging was used for early detection, EEG was shown to be safer and less expensive than MRI, according to the review. Of children who were not anesthetized for MRI, 30%-50% showed considerable distress, while EEG was shown to be minimally invasive. Findings from a retrospective chart review show that 14 MRIs were performed to detect one asymptomatic case of SWS, costing $11,768. In comparison, EEG costs $87 for a routine outpatient study.
“Demonstrating brain involvement on MRI in infants with high-risk PWS may facilitate more stringent counseling and monitoring, but … a negative MRI does not obviate the need for neurologic counseling and monitoring,” the investigators wrote. “Allaying anxiety about diagnostic uncertainty is not achieved using a scan but through detailed education, appropriate clinical monitoring, and nuanced reassurance.”
The investigators did not report any financial disclosures.
In high-risk port-wine stain phenotypes – forehead, hemifacial, and median – early referral to a pediatric neurologist is the best way to enable early symptom recognition of Sturge-Weber syndrome (SWS), according to results of a literature review.
If imaging is desired, electroencephalography (EEG) is cheaper and more minimally invasive than MRI (Pediatr Dermatol. 2017 Oct 16. doi: 10.1111/pde.13304).
Of the 34 studies analyzed, none examined the correlation between early detection of presymptomatic SWS and earlier seizure detection. There were also no data to verify improved outcomes with early detection, Michaela Zallmann, MD, of the department of dermatology at Eastern Health, Monash University, Box Hill, Australia, and her coauthors reported. While this indicates a need for further studies, it also shows a need for improved education and clinical monitoring as standard practice.
Additionally, negative imaging results do not obviate the possibility of SWS diagnosis, the investigators reported. In the studies that recorded false negatives, MRI was negative in 3%-6% of infants who later became symptomatic. Of the seven infants with false-negative results, four were less than 6 months old when the initial imaging was conducted. The investigators noted that it is not known at what age a negative MRI can reliably exclude SWS.
When imaging was used for early detection, EEG was shown to be safer and less expensive than MRI, according to the review. Of children who were not anesthetized for MRI, 30%-50% showed considerable distress, while EEG was shown to be minimally invasive. Findings from a retrospective chart review show that 14 MRIs were performed to detect one asymptomatic case of SWS, costing $11,768. In comparison, EEG costs $87 for a routine outpatient study.
“Demonstrating brain involvement on MRI in infants with high-risk PWS may facilitate more stringent counseling and monitoring, but … a negative MRI does not obviate the need for neurologic counseling and monitoring,” the investigators wrote. “Allaying anxiety about diagnostic uncertainty is not achieved using a scan but through detailed education, appropriate clinical monitoring, and nuanced reassurance.”
The investigators did not report any financial disclosures.
In high-risk port-wine stain phenotypes – forehead, hemifacial, and median – early referral to a pediatric neurologist is the best way to enable early symptom recognition of Sturge-Weber syndrome (SWS), according to results of a literature review.
If imaging is desired, electroencephalography (EEG) is cheaper and more minimally invasive than MRI (Pediatr Dermatol. 2017 Oct 16. doi: 10.1111/pde.13304).
Of the 34 studies analyzed, none examined the correlation between early detection of presymptomatic SWS and earlier seizure detection. There were also no data to verify improved outcomes with early detection, Michaela Zallmann, MD, of the department of dermatology at Eastern Health, Monash University, Box Hill, Australia, and her coauthors reported. While this indicates a need for further studies, it also shows a need for improved education and clinical monitoring as standard practice.
Additionally, negative imaging results do not obviate the possibility of SWS diagnosis, the investigators reported. In the studies that recorded false negatives, MRI was negative in 3%-6% of infants who later became symptomatic. Of the seven infants with false-negative results, four were less than 6 months old when the initial imaging was conducted. The investigators noted that it is not known at what age a negative MRI can reliably exclude SWS.
When imaging was used for early detection, EEG was shown to be safer and less expensive than MRI, according to the review. Of children who were not anesthetized for MRI, 30%-50% showed considerable distress, while EEG was shown to be minimally invasive. Findings from a retrospective chart review show that 14 MRIs were performed to detect one asymptomatic case of SWS, costing $11,768. In comparison, EEG costs $87 for a routine outpatient study.
“Demonstrating brain involvement on MRI in infants with high-risk PWS may facilitate more stringent counseling and monitoring, but … a negative MRI does not obviate the need for neurologic counseling and monitoring,” the investigators wrote. “Allaying anxiety about diagnostic uncertainty is not achieved using a scan but through detailed education, appropriate clinical monitoring, and nuanced reassurance.”
The investigators did not report any financial disclosures.
Key clinical point:
Major finding: To identify one case of Sturge-Weber syndrome, MRI cost $11,768, compared with $87 for a routine outpatient EEG.
Data source: A literature review of 34 articles.
Disclosures: The investigators did not report any financial disclosures.
Sunburn Purpura
To the Editor:
Chronic UV exposure has been linked to increased skin fragility and the development of purpuric lesions, a benign condition known as actinic purpura and commonly seen in elderly patients. Petechial skin changes acutely following intense sun exposure is a rare phenomenon referred to as sunburn purpura, photolocalized purpura, or solar purpura.
A 19-year-old woman presented with red and purple spots on the pretibial region of both legs extending to the thigh. One week prior to presentation she had a severe sunburn affecting most of the body, which resolved without blistering. Two days later, the spots appeared within the most severely sunburned areas of both legs. The patient reported that the lesions were mildly painful to palpation, but she was more concerned about the appearance. She denied any history of similar skin changes associated with sun exposure. The patient was otherwise healthy and denied any recent illnesses. She noted a history of mild bruising and bleeding with a resulting unremarkable workup by her primary care physician. The only medication taken was etonogestrel-ethinyl estradiol vaginal ring.
The scalp, face, arms, trunk, and legs were examined, and nonpalpable petechial changes were noted on the anterior aspect of the legs (Figure 1), with changes more prominent on the distal aspect of the legs. Mild superficial epidermal exfoliation was noted on both anterior thighs. The area of the lesions was not warm. The lesions were mildly tender to palpation. The remainder of the physical examination was unremarkable.
Given the timing of onset, preceding sun exposure, and the morphologic characteristics of the lesions, sunburn purpura was suspected. A punch biopsy of the anterior aspect of the left thigh was performed to rule out vasculitis. Microscopic examination revealed reactive epidermal changes with mild vascular ectasia and erythrocyte extravasation not associated with appreciable inflammation or evidence of vascular injury (Figure 2). Biopsy exposure to fluorescein-labeled antibodies directed against IgG, IgM, IgA, C3, and polyvalent immunoglobulins (IgG, IgM, and IgA) yielded no immunofluorescence. These biopsy results were consistent with sunburn purpura. Given the patient's normal platelet count, a diagnosis of idiopathic sunburn purpura was made. The patient was informed of the biopsy results and advised that the petechiae should resolve without treatment in 1 to 2 weeks, which occurred.
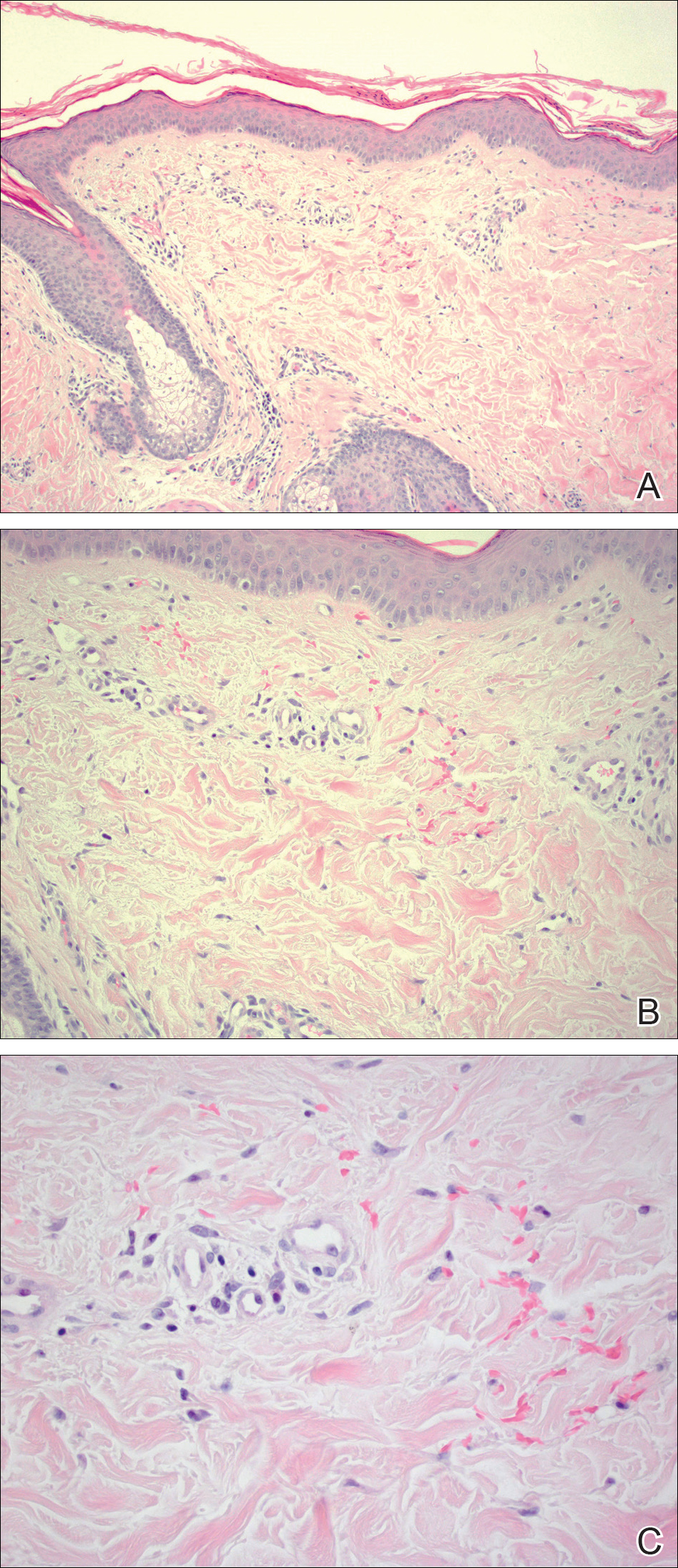
Sunburn purpura remains a rare phenomenon in which a petechial or purpuric rash develops acutely after intense sun exposure. We prefer the term sunburn purpura because it reflects the acuity of the phenomenon, as opposed to the previous labels solar purpura or photolocalized purpura, which also could suggest causality from chronic sun exposure. It has been proposed that sunburn purpura is a finding associated with a number of conditions rather than a unique entity.1 The following characteristics can be helpful in describing the development of sunburn purpura: delay following UV exposure, gross morphology, histologic findings, and possible associated medical conditions.1 Our case represents an important addition to the literature, as it differs from previously reported cases. Most importantly, the nonspecific biopsy findings and unremarkable laboratory findings associated with our case may represent primary or idiopathic sunburn purpura.
Previously reported cases of sunburn purpura have occurred in patients aged 10 to 66 years. It has been seen following UV exposure, vigorous exercise and high-dose aspirin, or concurrent fluoroquinolone therapy, or in the setting of erythropoietic protoporphyria, idiopathic thrombocytopenic purpura, or polymorphous light eruption.2-8 When performed, histology has revealed capillaritis, solar elastosis, perivascular infiltrate, lymphocytic perivascular infiltrate with dermal edema, or leukocytoclastic vasculitis.1,2,7-9 Our patient did not have a history of erythropoietic protoporphyria, polymorphous light eruption, or idiopathic thrombocytopenic purpura. She had not recently exercised, was not thrombocytopenic, and was not taking antiplatelet medications. She had no recent history of fluoroquinolone use. On histologic examination, our patient's biopsy demonstrated nonspecific petechial changes without signs of chronic UV exposure, dermal edema, vasculitis, lymphocytic infiltrate, or capillaritis.
Idiopathic sunburn purpura should only be diagnosed after other conditions are excluded. When evaluating a patient who presents with new-onset petechial rash following sun exposure, it is important to rule out vasculitis or thrombocytopenia as the cause, which is best achieved through skin biopsy and a platelet count, respectively. If there are no associated symptoms or thrombocytopenia and biopsy shows nonspecific vascular ectasia and erythrocyte extravasation, the physician should consider the diagnosis of idiopathic sunburn (solar or photolocalized) purpura. Along with regular UV protection, the physician should advise that the rash typically resolves without treatment in 1 to 2 weeks.
- Waters AJ, Sandhu DR, Green CM, et al. Solar capillaritis as a cause of solar purpura. Clin Exp Dermatol. 2009;34:E821-E824.
- Latenser BA, Hempstead RW. Exercise-associated solar purpura in an atypical location. Cutis. 1985;35:365-366.
- Rubegni P, Feci L, Pellegrino M, et al. Photolocalized purpura during levofloxacin therapy. Photodermatol Photoimmunol Photomed. 2012;28:105-107.
- Urbina F, Barrios M, Sudy E. Photolocalized purpura during ciprofloxacin therapy. Photodermatol Photoimmunol Photomed. 2006;22:111-112.
- Torinuki W, Miura T. Erythropoietic protoporphyria showing solar purpura. Dermatologica. 1983;167:220-222.
- Leung AK. Purpura associated with exposure to sunlight. J R Soc Med. 1986;79:423-424.
- Kalivas J, Kalivas L. Solar purpura appearing in a patient with polymorphous light eruption. Photodermatol Photoimmunol Photomed. 1995;11:31-32.
- Ros AM. Solar purpura--an unusual manifestation of polymorphous light eruption. Photodermatol. 1988;5:47-48.
- Guarrera M, Parodi A, Rebora A. Solar purpura is not related to polymorphous light eruption. Photodermatol. 1989;6:293-294.
To the Editor:
Chronic UV exposure has been linked to increased skin fragility and the development of purpuric lesions, a benign condition known as actinic purpura and commonly seen in elderly patients. Petechial skin changes acutely following intense sun exposure is a rare phenomenon referred to as sunburn purpura, photolocalized purpura, or solar purpura.
A 19-year-old woman presented with red and purple spots on the pretibial region of both legs extending to the thigh. One week prior to presentation she had a severe sunburn affecting most of the body, which resolved without blistering. Two days later, the spots appeared within the most severely sunburned areas of both legs. The patient reported that the lesions were mildly painful to palpation, but she was more concerned about the appearance. She denied any history of similar skin changes associated with sun exposure. The patient was otherwise healthy and denied any recent illnesses. She noted a history of mild bruising and bleeding with a resulting unremarkable workup by her primary care physician. The only medication taken was etonogestrel-ethinyl estradiol vaginal ring.
The scalp, face, arms, trunk, and legs were examined, and nonpalpable petechial changes were noted on the anterior aspect of the legs (Figure 1), with changes more prominent on the distal aspect of the legs. Mild superficial epidermal exfoliation was noted on both anterior thighs. The area of the lesions was not warm. The lesions were mildly tender to palpation. The remainder of the physical examination was unremarkable.
Given the timing of onset, preceding sun exposure, and the morphologic characteristics of the lesions, sunburn purpura was suspected. A punch biopsy of the anterior aspect of the left thigh was performed to rule out vasculitis. Microscopic examination revealed reactive epidermal changes with mild vascular ectasia and erythrocyte extravasation not associated with appreciable inflammation or evidence of vascular injury (Figure 2). Biopsy exposure to fluorescein-labeled antibodies directed against IgG, IgM, IgA, C3, and polyvalent immunoglobulins (IgG, IgM, and IgA) yielded no immunofluorescence. These biopsy results were consistent with sunburn purpura. Given the patient's normal platelet count, a diagnosis of idiopathic sunburn purpura was made. The patient was informed of the biopsy results and advised that the petechiae should resolve without treatment in 1 to 2 weeks, which occurred.
Sunburn purpura remains a rare phenomenon in which a petechial or purpuric rash develops acutely after intense sun exposure. We prefer the term sunburn purpura because it reflects the acuity of the phenomenon, as opposed to the previous labels solar purpura or photolocalized purpura, which also could suggest causality from chronic sun exposure. It has been proposed that sunburn purpura is a finding associated with a number of conditions rather than a unique entity.1 The following characteristics can be helpful in describing the development of sunburn purpura: delay following UV exposure, gross morphology, histologic findings, and possible associated medical conditions.1 Our case represents an important addition to the literature, as it differs from previously reported cases. Most importantly, the nonspecific biopsy findings and unremarkable laboratory findings associated with our case may represent primary or idiopathic sunburn purpura.
Previously reported cases of sunburn purpura have occurred in patients aged 10 to 66 years. It has been seen following UV exposure, vigorous exercise and high-dose aspirin, or concurrent fluoroquinolone therapy, or in the setting of erythropoietic protoporphyria, idiopathic thrombocytopenic purpura, or polymorphous light eruption.2-8 When performed, histology has revealed capillaritis, solar elastosis, perivascular infiltrate, lymphocytic perivascular infiltrate with dermal edema, or leukocytoclastic vasculitis.1,2,7-9 Our patient did not have a history of erythropoietic protoporphyria, polymorphous light eruption, or idiopathic thrombocytopenic purpura. She had not recently exercised, was not thrombocytopenic, and was not taking antiplatelet medications. She had no recent history of fluoroquinolone use. On histologic examination, our patient's biopsy demonstrated nonspecific petechial changes without signs of chronic UV exposure, dermal edema, vasculitis, lymphocytic infiltrate, or capillaritis.
Idiopathic sunburn purpura should only be diagnosed after other conditions are excluded. When evaluating a patient who presents with new-onset petechial rash following sun exposure, it is important to rule out vasculitis or thrombocytopenia as the cause, which is best achieved through skin biopsy and a platelet count, respectively. If there are no associated symptoms or thrombocytopenia and biopsy shows nonspecific vascular ectasia and erythrocyte extravasation, the physician should consider the diagnosis of idiopathic sunburn (solar or photolocalized) purpura. Along with regular UV protection, the physician should advise that the rash typically resolves without treatment in 1 to 2 weeks.
To the Editor:
Chronic UV exposure has been linked to increased skin fragility and the development of purpuric lesions, a benign condition known as actinic purpura and commonly seen in elderly patients. Petechial skin changes acutely following intense sun exposure is a rare phenomenon referred to as sunburn purpura, photolocalized purpura, or solar purpura.
A 19-year-old woman presented with red and purple spots on the pretibial region of both legs extending to the thigh. One week prior to presentation she had a severe sunburn affecting most of the body, which resolved without blistering. Two days later, the spots appeared within the most severely sunburned areas of both legs. The patient reported that the lesions were mildly painful to palpation, but she was more concerned about the appearance. She denied any history of similar skin changes associated with sun exposure. The patient was otherwise healthy and denied any recent illnesses. She noted a history of mild bruising and bleeding with a resulting unremarkable workup by her primary care physician. The only medication taken was etonogestrel-ethinyl estradiol vaginal ring.
The scalp, face, arms, trunk, and legs were examined, and nonpalpable petechial changes were noted on the anterior aspect of the legs (Figure 1), with changes more prominent on the distal aspect of the legs. Mild superficial epidermal exfoliation was noted on both anterior thighs. The area of the lesions was not warm. The lesions were mildly tender to palpation. The remainder of the physical examination was unremarkable.
Given the timing of onset, preceding sun exposure, and the morphologic characteristics of the lesions, sunburn purpura was suspected. A punch biopsy of the anterior aspect of the left thigh was performed to rule out vasculitis. Microscopic examination revealed reactive epidermal changes with mild vascular ectasia and erythrocyte extravasation not associated with appreciable inflammation or evidence of vascular injury (Figure 2). Biopsy exposure to fluorescein-labeled antibodies directed against IgG, IgM, IgA, C3, and polyvalent immunoglobulins (IgG, IgM, and IgA) yielded no immunofluorescence. These biopsy results were consistent with sunburn purpura. Given the patient's normal platelet count, a diagnosis of idiopathic sunburn purpura was made. The patient was informed of the biopsy results and advised that the petechiae should resolve without treatment in 1 to 2 weeks, which occurred.
Sunburn purpura remains a rare phenomenon in which a petechial or purpuric rash develops acutely after intense sun exposure. We prefer the term sunburn purpura because it reflects the acuity of the phenomenon, as opposed to the previous labels solar purpura or photolocalized purpura, which also could suggest causality from chronic sun exposure. It has been proposed that sunburn purpura is a finding associated with a number of conditions rather than a unique entity.1 The following characteristics can be helpful in describing the development of sunburn purpura: delay following UV exposure, gross morphology, histologic findings, and possible associated medical conditions.1 Our case represents an important addition to the literature, as it differs from previously reported cases. Most importantly, the nonspecific biopsy findings and unremarkable laboratory findings associated with our case may represent primary or idiopathic sunburn purpura.
Previously reported cases of sunburn purpura have occurred in patients aged 10 to 66 years. It has been seen following UV exposure, vigorous exercise and high-dose aspirin, or concurrent fluoroquinolone therapy, or in the setting of erythropoietic protoporphyria, idiopathic thrombocytopenic purpura, or polymorphous light eruption.2-8 When performed, histology has revealed capillaritis, solar elastosis, perivascular infiltrate, lymphocytic perivascular infiltrate with dermal edema, or leukocytoclastic vasculitis.1,2,7-9 Our patient did not have a history of erythropoietic protoporphyria, polymorphous light eruption, or idiopathic thrombocytopenic purpura. She had not recently exercised, was not thrombocytopenic, and was not taking antiplatelet medications. She had no recent history of fluoroquinolone use. On histologic examination, our patient's biopsy demonstrated nonspecific petechial changes without signs of chronic UV exposure, dermal edema, vasculitis, lymphocytic infiltrate, or capillaritis.
Idiopathic sunburn purpura should only be diagnosed after other conditions are excluded. When evaluating a patient who presents with new-onset petechial rash following sun exposure, it is important to rule out vasculitis or thrombocytopenia as the cause, which is best achieved through skin biopsy and a platelet count, respectively. If there are no associated symptoms or thrombocytopenia and biopsy shows nonspecific vascular ectasia and erythrocyte extravasation, the physician should consider the diagnosis of idiopathic sunburn (solar or photolocalized) purpura. Along with regular UV protection, the physician should advise that the rash typically resolves without treatment in 1 to 2 weeks.
- Waters AJ, Sandhu DR, Green CM, et al. Solar capillaritis as a cause of solar purpura. Clin Exp Dermatol. 2009;34:E821-E824.
- Latenser BA, Hempstead RW. Exercise-associated solar purpura in an atypical location. Cutis. 1985;35:365-366.
- Rubegni P, Feci L, Pellegrino M, et al. Photolocalized purpura during levofloxacin therapy. Photodermatol Photoimmunol Photomed. 2012;28:105-107.
- Urbina F, Barrios M, Sudy E. Photolocalized purpura during ciprofloxacin therapy. Photodermatol Photoimmunol Photomed. 2006;22:111-112.
- Torinuki W, Miura T. Erythropoietic protoporphyria showing solar purpura. Dermatologica. 1983;167:220-222.
- Leung AK. Purpura associated with exposure to sunlight. J R Soc Med. 1986;79:423-424.
- Kalivas J, Kalivas L. Solar purpura appearing in a patient with polymorphous light eruption. Photodermatol Photoimmunol Photomed. 1995;11:31-32.
- Ros AM. Solar purpura--an unusual manifestation of polymorphous light eruption. Photodermatol. 1988;5:47-48.
- Guarrera M, Parodi A, Rebora A. Solar purpura is not related to polymorphous light eruption. Photodermatol. 1989;6:293-294.
- Waters AJ, Sandhu DR, Green CM, et al. Solar capillaritis as a cause of solar purpura. Clin Exp Dermatol. 2009;34:E821-E824.
- Latenser BA, Hempstead RW. Exercise-associated solar purpura in an atypical location. Cutis. 1985;35:365-366.
- Rubegni P, Feci L, Pellegrino M, et al. Photolocalized purpura during levofloxacin therapy. Photodermatol Photoimmunol Photomed. 2012;28:105-107.
- Urbina F, Barrios M, Sudy E. Photolocalized purpura during ciprofloxacin therapy. Photodermatol Photoimmunol Photomed. 2006;22:111-112.
- Torinuki W, Miura T. Erythropoietic protoporphyria showing solar purpura. Dermatologica. 1983;167:220-222.
- Leung AK. Purpura associated with exposure to sunlight. J R Soc Med. 1986;79:423-424.
- Kalivas J, Kalivas L. Solar purpura appearing in a patient with polymorphous light eruption. Photodermatol Photoimmunol Photomed. 1995;11:31-32.
- Ros AM. Solar purpura--an unusual manifestation of polymorphous light eruption. Photodermatol. 1988;5:47-48.
- Guarrera M, Parodi A, Rebora A. Solar purpura is not related to polymorphous light eruption. Photodermatol. 1989;6:293-294.
Practice Points
- Petechial skin changes acutely following intense sun exposure is a rare phenomenon referred to as sunburn purpura, photolocalized purpura, or solar purpura.
- Idiopathic sunburn purpura should only be diagnosed after vasculitis and/or thrombocytopenia is ruled out, which is best achieved through skin biopsy and a platelet count, respectively.
- The rash typically resolves without treatment in 1 to 2 weeks; however, a variety of UV protection modalities and education should be offered to the patient.
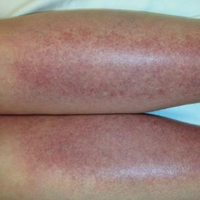
Levofloxacin-Induced Purpura Annularis Telangiectodes of Majocchi
To the Editor:
Purpura annularis telangiectodes of Majocchi (PATM) is a type of pigmented purpuric dermatosis (PPD). Patients present with nonblanchable, annular, symmetric, purpuric, and telangiectatic patches, often on the legs, with histology revealing a perivascular lymphocytic infiltrate and extravasated erythrocytes.1,2 A variety of medications have been linked to the development of PPD. We describe a case of levofloxacin-induced PATM.
RELATED ARTICLE: Granulomatous Changes Associated With Pigmented Purpuric Dermatosis
A 42-year-old man presented with a rash on the arms, trunk, abdomen, and legs of 1 month’s duration. He reported no associated itching, bleeding, or pain, and no history of a similar rash. He had a history of hypothyroidism and had been taking levothyroxine for years. He had no known allergies and no history of childhood eczema, asthma, or allergic rhinitis. Notably, the rash started shortly after the patient finished a 2-week course of levofloxacin, an antibiotic he had not taken in the past. The patient resided with his wife, 3 children, and a pet dog, and no family members had the rash. Prior to presentation, the patient had tried econazole cream and then triamcinolone acetonide cream 0.5% without any clinical improvement.
A complete review of systems was unremarkable. Physical examination revealed scattered, reddish brown, annular, nonscaly patches on the back, abdomen (Figure 1), arms, and legs with nonblanching petechiae within the patches.
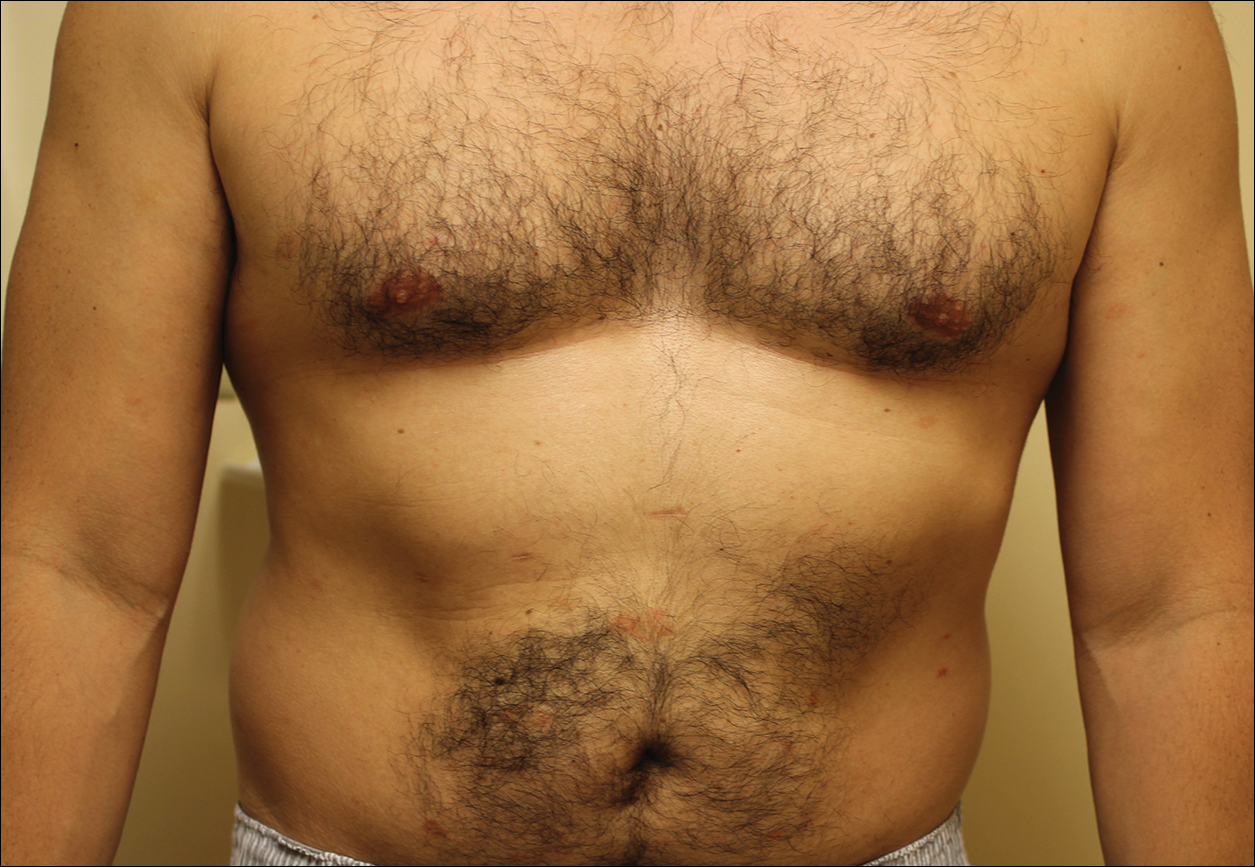
A punch biopsy of the left inner thigh demonstrated patchy interface dermatitis, superficial perivascular inflammation, and numerous extravasated red blood cells in the papillary dermis (Figure 2). The histologic features were compatible with the clinical impression of PATM. The patient presented for a follow-up visit 2 weeks later with no new lesions and the old lesions were rapidly fading (Figure 3).
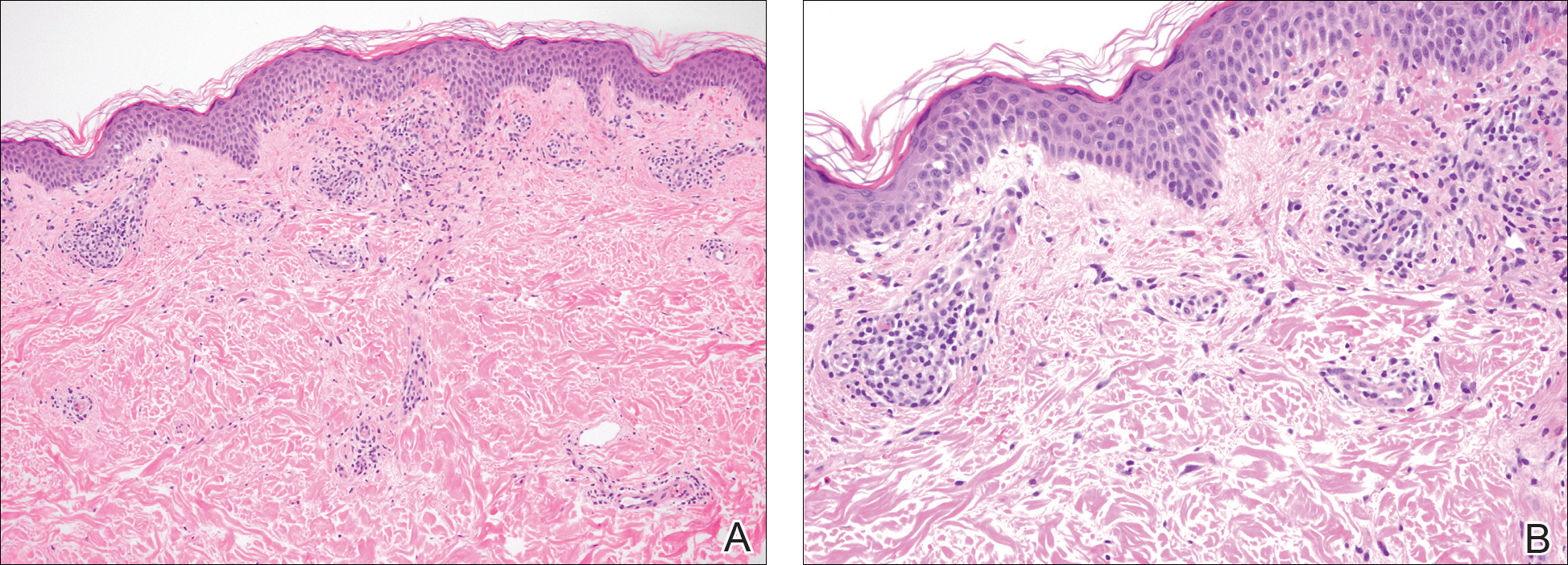
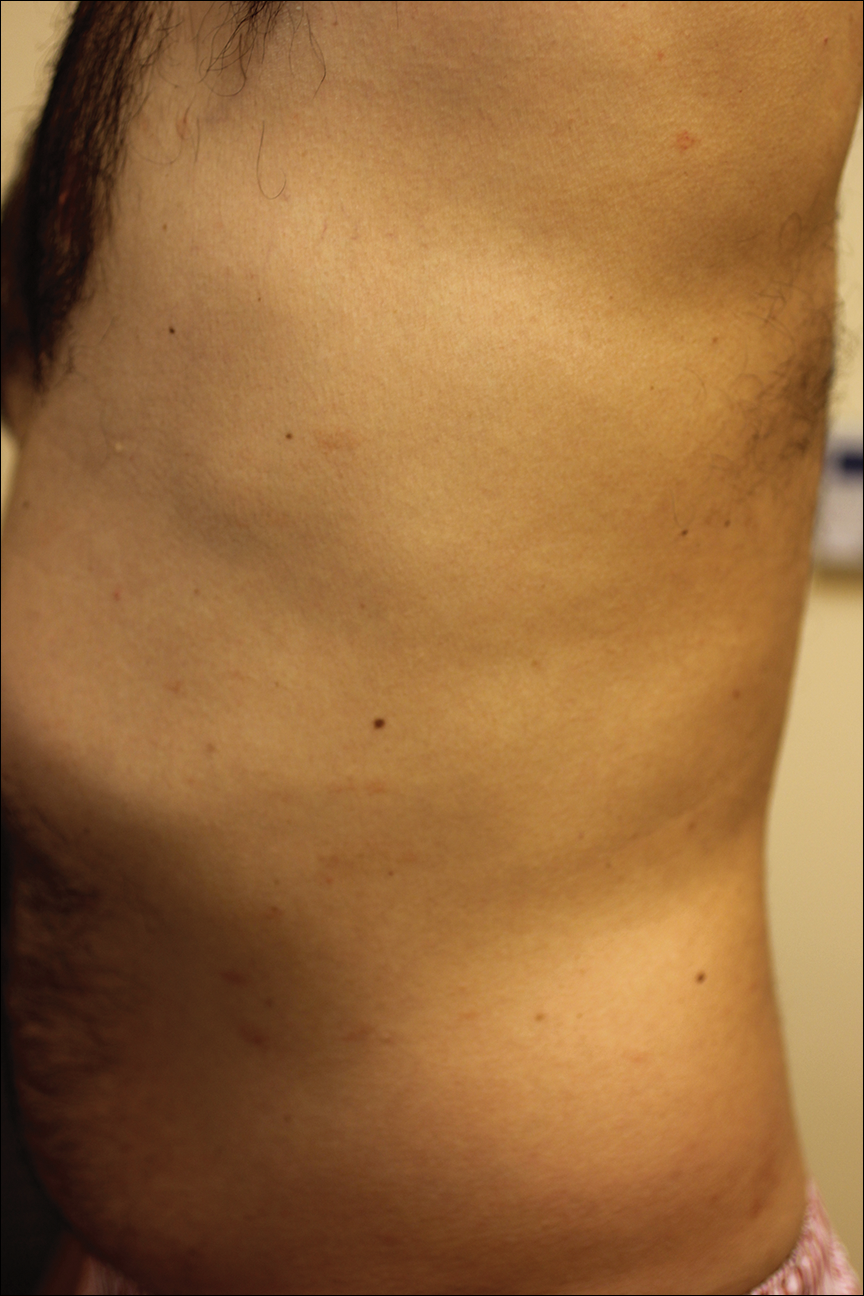
Pigmented purpuric dermatoses are a group of conditions that have different clinical morphologies but similar histopathologic examinations.2 All PPDs are characterized by nonblanching, nonpalpable, purpuric lesions that often are bilaterally symmetrical and present on the legs.2,3 Although the precise etiology of these conditions is not known, most cases include a perivascular lymphocytic infiltrate along with the presence of extravasated erythrocytes and hemosiderin deposition in the dermis.2 Of note, PATM often is idiopathic and patients usually present with no associated comorbidities.3 The currently established PPDs include progressive pigmentary dermatosis (Schamberg disease), PATM, pigmented purpuric lichenoid dermatosis of Gougerot and Blum, lichen aureus, and eczematidlike purpura of Doucas and Kapetanakis.2,4
RELATED ARTICLE: Granulomatous Pigmented Purpuric Dermatosis
The lesions of PATM are symmetrically distributed on the bilateral legs and may be symptomatic in most cases, with severe pruritus being reported in several drug-induced PATM cases.3,5 Although the exact etiology of PPDs currently is unknown, some contributing factors that are thought to play a role include exercise, venous stasis, gravitational dependence, capillary fragility, hypertension, drugs, chemical exposure or ingestions, and contact allergy to dyes.3 Some of the drugs known to cause drug-induced PPDs fall into the class of sedatives, stimulants, antibiotics, cardiovascular drugs, vitamins, and nutritional supplements.3,6 Some medications that have been reported to cause PPDs include acetaminophen, aspirin, carbamazepine, diltiazem, furosemide, glipizide, hydralazine, infliximab, isotretinoin, lorazepam, minocycline, nitroglycerine, and sildenafil.3,7-15
Although the mechanism of drug-induced PPD is not completely understood, it is thought that the ingested substance leads to an immunologic response in the capillary endothelium, which results in a cell-mediated immune response causing vascular damage.3 The ingested substance may act as a hapten, stimulating antibody formation and immune-mediated injury, leading to the clinical presentation of nonblanching, symmetric, purpuric, telangiectatic, and atrophic patches at the site of injury.1,3
Levofloxacin is a broad-spectrum antibiotic that has activity against both gram-positive and gram-negative bacteria. It inhibits the enzymes DNA gyrase and topoisomerase IV, preventing bacteria from undergoing proper DNA synthesis.16 Our patient’s rash began shortly after a 2-week course of levofloxacin and faded within a few weeks of discontinuing the drug; the clinical presentation, time course, and histologic appearance of the lesions were consistent with the diagnosis of drug-induced PPD. Of note, solar capillaritis has been reported following a phototoxic reaction induced by levofloxacin.17 Our case differs in that our patient had annular lesions on both photoprotected and photoexposed skin.
The first-line interventions for the treatment of PPDs are nonpharmacologic, such as discontinuation of an offending drug or allergen or wearing supportive stockings if there are signs of venous stasis. Other interventions include the use of a medium- or high-potency topical corticosteroid once to twice daily to affected areas for 4 to 6 weeks.18 Some case series also have shown improvement with narrowband UVB treatment after 24 to 28 treatment sessions or with psoralen plus UVA phototherapy within 7 to 20 treatments.19,20 If the above measures are unsuccessful in resolving symptoms, other treatment alternatives may include pentoxifylline, griseofulvin, colchicine, cyclosporine, and methotrexate. The potential benefit of treatment must be weighed against the side-effect profile of these medications.2,21-24 Of note, oral rutoside (50 mg twice daily) and ascorbic acid (500 mg twice daily) were administered to 3 patients with chronic progressive pigmented purpura. At the end of the 4-week treatment period, complete clearance of skin lesions was seen in all patients with no adverse reactions noted.25
Despite these treatment options, PATM does not necessitate treatment given its benign course and often self-resolving nature.26 In cases of drug-induced PPD such as in our patient, discontinuation of the offending drug often may lead to resolution.
In summary, PATM is a PPD that has been associated with different etiologic factors. If PATM is suspected to be caused by a drug, discontinuation of the offending agent usually results in resolution of symptoms, as it did in our case with fading of lesions within a few weeks after the patient was no longer taking levofloxacin.
- Hale EK. Purpura annularis telangiectodes of Majocchi. Dermatol Online J. 2003;9:17.
- Hoesly FJ, Huerter CJ, Shehan JM. Purpura annularis telangiectodes of Majocchi: case report and review of the literature. Int J Dermatol. 2009;48:1129-1133.
- Kaplan R, Meehan SA, Leger M. A case of isotretinoin-induced purpura annularis telangiectodes of Majocchi and review of substance-induced pigmented purpuric dermatosis. JAMA Dermatol. 2014;150:182-184.
- Newton RC, Raimer SS. Pigmented purpuric eruptions. Dermatol Clin. 1985;3:165-169.
- Ratnam KV, Su WP, Peters MS. Purpura simplex (inflammatory purpura without vasculitis): a clinicopathologic study of 174 cases. J Am Acad Dermatol. 1991;25:642-647.
- Pang BK, Su D, Ratnam KV. Drug-induced purpura simplex: clinical and histological characteristics. Ann Acad Med Singapore. 1993;22:870-872.
- Abeck D, Gross GE, Kuwert C, et al. Acetaminophen-induced progressive pigmentary purpura (Schamberg’s disease). J Am Acad Dermatol. 1992;27:123-124.
- Lipsker D, Cribier B, Heid E, et al. Cutaneous lymphoma manifesting as pigmented, purpuric capillaries [in French]. Ann Dermatol Venereol. 1999;126:321-326.
- Peterson WC Jr, Manick KP. Purpuric eruptions associated with use of carbromal and meprobamate. Arch Dermatol. 1967;95:40-42.
- Nishioka K, Katayama I, Masuzawa M, et al. Drug-induced chronic pigmented purpura. J Dermatol. 1989;16:220-222.
- Voelter WW. Pigmented purpuric dermatosis-like reaction to topical fluorouracil. Arch Dermatol. 1983;119:875-876.
- Adams BB, Gadenne AS. Glipizide-induced pigmented purpuric dermatosis. J Am Acad Dermatol. 1999;41(5, pt 2):827-829.
- Tsao H, Lerner LH. Pigmented purpuric eruption associated with injection medroxyprogesterone acetate. J Am Acad Dermatol. 2000;43(2, pt 1):308-310.
- Koçak AY, Akay BN, Heper AO. Sildenafil-induced pigmented purpuric dermatosis. Cutan Ocul Toxicol. 2013;32:91-92.
- Nishioka K, Sarashi C, Katayama I. Chronic pigmented purpura induced by chemical substances. Clin Exp Dermatol. 1980;5:213-218.
- Drlica K, Zhao X. DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiol Mol Biol Rev. 1997;61:377-392.
- Rubegni P, Feci L, Pellegrino M, et al. Photolocalized purpura during levofloxacin therapy. Photodermatol Photoimmunol Photomed. 2012;28:105-107.
- Sardana K, Sarkar R, Sehgal VN. Pigmented purpuric dermatoses: an overview. Int J Dermatol. 2004;43:482-488.
- Fathy H, Abdelgaber S. Treatment of pigmented purpuric dermatoses with narrow-band UVB: a report of six cases. J Eur Acad Dermatol Venereol. 2011;25:603-606.
- Krizsa J, Hunyadi J, Dobozy A. PUVA treatment of pigmented purpuric lichenoid dermatitis (Gougerot-Blum). J Am Acad Dermatol. 1992;27(5, pt 1):778-780.
- Panda S, Malakar S, Lahiri K. Oral pentoxifylline vs topical betamethasone in Schamberg disease: a comparative randomized investigator-blinded parallel-group trial. Arch Dermatol. 2004;140:491-493.
- Tamaki K, Yasaka N, Osada A, et al. Successful treatment of pigmented purpuric dermatosis with griseofulvin. Br J Dermatol. 1995;132:159-160.
- Geller M. Benefit of colchicine in the treatment of Schamberg’s disease. Ann Allergy Asthma Immunol. 2000;85:246.
- Okada K, Ishikawa O, Miyachi Y. Purpura pigmentosa chronica successfully treated with oral cyclosporin A. Br J Dermatol. 1996;134:180-181.
- Reinhold U, Seiter S, Ugurel S, et al. Treatment of progressive pigmented purpura with oral bioflavonoids and ascorbic acid: an open pilot study in 3 patients. J Am Acad Dermatol. 1999;41(2, pt 1):207-208.
- Wang A, Shuja F, Chan A, et al. Unilateral purpura annularis telangiectodes of Majocchi in an elderly male: an atypical presentation. Dermatol Online J. 2013;19:19263.
To the Editor:
Purpura annularis telangiectodes of Majocchi (PATM) is a type of pigmented purpuric dermatosis (PPD). Patients present with nonblanchable, annular, symmetric, purpuric, and telangiectatic patches, often on the legs, with histology revealing a perivascular lymphocytic infiltrate and extravasated erythrocytes.1,2 A variety of medications have been linked to the development of PPD. We describe a case of levofloxacin-induced PATM.
RELATED ARTICLE: Granulomatous Changes Associated With Pigmented Purpuric Dermatosis
A 42-year-old man presented with a rash on the arms, trunk, abdomen, and legs of 1 month’s duration. He reported no associated itching, bleeding, or pain, and no history of a similar rash. He had a history of hypothyroidism and had been taking levothyroxine for years. He had no known allergies and no history of childhood eczema, asthma, or allergic rhinitis. Notably, the rash started shortly after the patient finished a 2-week course of levofloxacin, an antibiotic he had not taken in the past. The patient resided with his wife, 3 children, and a pet dog, and no family members had the rash. Prior to presentation, the patient had tried econazole cream and then triamcinolone acetonide cream 0.5% without any clinical improvement.
A complete review of systems was unremarkable. Physical examination revealed scattered, reddish brown, annular, nonscaly patches on the back, abdomen (Figure 1), arms, and legs with nonblanching petechiae within the patches.
A punch biopsy of the left inner thigh demonstrated patchy interface dermatitis, superficial perivascular inflammation, and numerous extravasated red blood cells in the papillary dermis (Figure 2). The histologic features were compatible with the clinical impression of PATM. The patient presented for a follow-up visit 2 weeks later with no new lesions and the old lesions were rapidly fading (Figure 3).
Pigmented purpuric dermatoses are a group of conditions that have different clinical morphologies but similar histopathologic examinations.2 All PPDs are characterized by nonblanching, nonpalpable, purpuric lesions that often are bilaterally symmetrical and present on the legs.2,3 Although the precise etiology of these conditions is not known, most cases include a perivascular lymphocytic infiltrate along with the presence of extravasated erythrocytes and hemosiderin deposition in the dermis.2 Of note, PATM often is idiopathic and patients usually present with no associated comorbidities.3 The currently established PPDs include progressive pigmentary dermatosis (Schamberg disease), PATM, pigmented purpuric lichenoid dermatosis of Gougerot and Blum, lichen aureus, and eczematidlike purpura of Doucas and Kapetanakis.2,4
RELATED ARTICLE: Granulomatous Pigmented Purpuric Dermatosis
The lesions of PATM are symmetrically distributed on the bilateral legs and may be symptomatic in most cases, with severe pruritus being reported in several drug-induced PATM cases.3,5 Although the exact etiology of PPDs currently is unknown, some contributing factors that are thought to play a role include exercise, venous stasis, gravitational dependence, capillary fragility, hypertension, drugs, chemical exposure or ingestions, and contact allergy to dyes.3 Some of the drugs known to cause drug-induced PPDs fall into the class of sedatives, stimulants, antibiotics, cardiovascular drugs, vitamins, and nutritional supplements.3,6 Some medications that have been reported to cause PPDs include acetaminophen, aspirin, carbamazepine, diltiazem, furosemide, glipizide, hydralazine, infliximab, isotretinoin, lorazepam, minocycline, nitroglycerine, and sildenafil.3,7-15
Although the mechanism of drug-induced PPD is not completely understood, it is thought that the ingested substance leads to an immunologic response in the capillary endothelium, which results in a cell-mediated immune response causing vascular damage.3 The ingested substance may act as a hapten, stimulating antibody formation and immune-mediated injury, leading to the clinical presentation of nonblanching, symmetric, purpuric, telangiectatic, and atrophic patches at the site of injury.1,3
Levofloxacin is a broad-spectrum antibiotic that has activity against both gram-positive and gram-negative bacteria. It inhibits the enzymes DNA gyrase and topoisomerase IV, preventing bacteria from undergoing proper DNA synthesis.16 Our patient’s rash began shortly after a 2-week course of levofloxacin and faded within a few weeks of discontinuing the drug; the clinical presentation, time course, and histologic appearance of the lesions were consistent with the diagnosis of drug-induced PPD. Of note, solar capillaritis has been reported following a phototoxic reaction induced by levofloxacin.17 Our case differs in that our patient had annular lesions on both photoprotected and photoexposed skin.
The first-line interventions for the treatment of PPDs are nonpharmacologic, such as discontinuation of an offending drug or allergen or wearing supportive stockings if there are signs of venous stasis. Other interventions include the use of a medium- or high-potency topical corticosteroid once to twice daily to affected areas for 4 to 6 weeks.18 Some case series also have shown improvement with narrowband UVB treatment after 24 to 28 treatment sessions or with psoralen plus UVA phototherapy within 7 to 20 treatments.19,20 If the above measures are unsuccessful in resolving symptoms, other treatment alternatives may include pentoxifylline, griseofulvin, colchicine, cyclosporine, and methotrexate. The potential benefit of treatment must be weighed against the side-effect profile of these medications.2,21-24 Of note, oral rutoside (50 mg twice daily) and ascorbic acid (500 mg twice daily) were administered to 3 patients with chronic progressive pigmented purpura. At the end of the 4-week treatment period, complete clearance of skin lesions was seen in all patients with no adverse reactions noted.25
Despite these treatment options, PATM does not necessitate treatment given its benign course and often self-resolving nature.26 In cases of drug-induced PPD such as in our patient, discontinuation of the offending drug often may lead to resolution.
In summary, PATM is a PPD that has been associated with different etiologic factors. If PATM is suspected to be caused by a drug, discontinuation of the offending agent usually results in resolution of symptoms, as it did in our case with fading of lesions within a few weeks after the patient was no longer taking levofloxacin.
To the Editor:
Purpura annularis telangiectodes of Majocchi (PATM) is a type of pigmented purpuric dermatosis (PPD). Patients present with nonblanchable, annular, symmetric, purpuric, and telangiectatic patches, often on the legs, with histology revealing a perivascular lymphocytic infiltrate and extravasated erythrocytes.1,2 A variety of medications have been linked to the development of PPD. We describe a case of levofloxacin-induced PATM.
RELATED ARTICLE: Granulomatous Changes Associated With Pigmented Purpuric Dermatosis
A 42-year-old man presented with a rash on the arms, trunk, abdomen, and legs of 1 month’s duration. He reported no associated itching, bleeding, or pain, and no history of a similar rash. He had a history of hypothyroidism and had been taking levothyroxine for years. He had no known allergies and no history of childhood eczema, asthma, or allergic rhinitis. Notably, the rash started shortly after the patient finished a 2-week course of levofloxacin, an antibiotic he had not taken in the past. The patient resided with his wife, 3 children, and a pet dog, and no family members had the rash. Prior to presentation, the patient had tried econazole cream and then triamcinolone acetonide cream 0.5% without any clinical improvement.
A complete review of systems was unremarkable. Physical examination revealed scattered, reddish brown, annular, nonscaly patches on the back, abdomen (Figure 1), arms, and legs with nonblanching petechiae within the patches.
A punch biopsy of the left inner thigh demonstrated patchy interface dermatitis, superficial perivascular inflammation, and numerous extravasated red blood cells in the papillary dermis (Figure 2). The histologic features were compatible with the clinical impression of PATM. The patient presented for a follow-up visit 2 weeks later with no new lesions and the old lesions were rapidly fading (Figure 3).
Pigmented purpuric dermatoses are a group of conditions that have different clinical morphologies but similar histopathologic examinations.2 All PPDs are characterized by nonblanching, nonpalpable, purpuric lesions that often are bilaterally symmetrical and present on the legs.2,3 Although the precise etiology of these conditions is not known, most cases include a perivascular lymphocytic infiltrate along with the presence of extravasated erythrocytes and hemosiderin deposition in the dermis.2 Of note, PATM often is idiopathic and patients usually present with no associated comorbidities.3 The currently established PPDs include progressive pigmentary dermatosis (Schamberg disease), PATM, pigmented purpuric lichenoid dermatosis of Gougerot and Blum, lichen aureus, and eczematidlike purpura of Doucas and Kapetanakis.2,4
RELATED ARTICLE: Granulomatous Pigmented Purpuric Dermatosis
The lesions of PATM are symmetrically distributed on the bilateral legs and may be symptomatic in most cases, with severe pruritus being reported in several drug-induced PATM cases.3,5 Although the exact etiology of PPDs currently is unknown, some contributing factors that are thought to play a role include exercise, venous stasis, gravitational dependence, capillary fragility, hypertension, drugs, chemical exposure or ingestions, and contact allergy to dyes.3 Some of the drugs known to cause drug-induced PPDs fall into the class of sedatives, stimulants, antibiotics, cardiovascular drugs, vitamins, and nutritional supplements.3,6 Some medications that have been reported to cause PPDs include acetaminophen, aspirin, carbamazepine, diltiazem, furosemide, glipizide, hydralazine, infliximab, isotretinoin, lorazepam, minocycline, nitroglycerine, and sildenafil.3,7-15
Although the mechanism of drug-induced PPD is not completely understood, it is thought that the ingested substance leads to an immunologic response in the capillary endothelium, which results in a cell-mediated immune response causing vascular damage.3 The ingested substance may act as a hapten, stimulating antibody formation and immune-mediated injury, leading to the clinical presentation of nonblanching, symmetric, purpuric, telangiectatic, and atrophic patches at the site of injury.1,3
Levofloxacin is a broad-spectrum antibiotic that has activity against both gram-positive and gram-negative bacteria. It inhibits the enzymes DNA gyrase and topoisomerase IV, preventing bacteria from undergoing proper DNA synthesis.16 Our patient’s rash began shortly after a 2-week course of levofloxacin and faded within a few weeks of discontinuing the drug; the clinical presentation, time course, and histologic appearance of the lesions were consistent with the diagnosis of drug-induced PPD. Of note, solar capillaritis has been reported following a phototoxic reaction induced by levofloxacin.17 Our case differs in that our patient had annular lesions on both photoprotected and photoexposed skin.
The first-line interventions for the treatment of PPDs are nonpharmacologic, such as discontinuation of an offending drug or allergen or wearing supportive stockings if there are signs of venous stasis. Other interventions include the use of a medium- or high-potency topical corticosteroid once to twice daily to affected areas for 4 to 6 weeks.18 Some case series also have shown improvement with narrowband UVB treatment after 24 to 28 treatment sessions or with psoralen plus UVA phototherapy within 7 to 20 treatments.19,20 If the above measures are unsuccessful in resolving symptoms, other treatment alternatives may include pentoxifylline, griseofulvin, colchicine, cyclosporine, and methotrexate. The potential benefit of treatment must be weighed against the side-effect profile of these medications.2,21-24 Of note, oral rutoside (50 mg twice daily) and ascorbic acid (500 mg twice daily) were administered to 3 patients with chronic progressive pigmented purpura. At the end of the 4-week treatment period, complete clearance of skin lesions was seen in all patients with no adverse reactions noted.25
Despite these treatment options, PATM does not necessitate treatment given its benign course and often self-resolving nature.26 In cases of drug-induced PPD such as in our patient, discontinuation of the offending drug often may lead to resolution.
In summary, PATM is a PPD that has been associated with different etiologic factors. If PATM is suspected to be caused by a drug, discontinuation of the offending agent usually results in resolution of symptoms, as it did in our case with fading of lesions within a few weeks after the patient was no longer taking levofloxacin.
- Hale EK. Purpura annularis telangiectodes of Majocchi. Dermatol Online J. 2003;9:17.
- Hoesly FJ, Huerter CJ, Shehan JM. Purpura annularis telangiectodes of Majocchi: case report and review of the literature. Int J Dermatol. 2009;48:1129-1133.
- Kaplan R, Meehan SA, Leger M. A case of isotretinoin-induced purpura annularis telangiectodes of Majocchi and review of substance-induced pigmented purpuric dermatosis. JAMA Dermatol. 2014;150:182-184.
- Newton RC, Raimer SS. Pigmented purpuric eruptions. Dermatol Clin. 1985;3:165-169.
- Ratnam KV, Su WP, Peters MS. Purpura simplex (inflammatory purpura without vasculitis): a clinicopathologic study of 174 cases. J Am Acad Dermatol. 1991;25:642-647.
- Pang BK, Su D, Ratnam KV. Drug-induced purpura simplex: clinical and histological characteristics. Ann Acad Med Singapore. 1993;22:870-872.
- Abeck D, Gross GE, Kuwert C, et al. Acetaminophen-induced progressive pigmentary purpura (Schamberg’s disease). J Am Acad Dermatol. 1992;27:123-124.
- Lipsker D, Cribier B, Heid E, et al. Cutaneous lymphoma manifesting as pigmented, purpuric capillaries [in French]. Ann Dermatol Venereol. 1999;126:321-326.
- Peterson WC Jr, Manick KP. Purpuric eruptions associated with use of carbromal and meprobamate. Arch Dermatol. 1967;95:40-42.
- Nishioka K, Katayama I, Masuzawa M, et al. Drug-induced chronic pigmented purpura. J Dermatol. 1989;16:220-222.
- Voelter WW. Pigmented purpuric dermatosis-like reaction to topical fluorouracil. Arch Dermatol. 1983;119:875-876.
- Adams BB, Gadenne AS. Glipizide-induced pigmented purpuric dermatosis. J Am Acad Dermatol. 1999;41(5, pt 2):827-829.
- Tsao H, Lerner LH. Pigmented purpuric eruption associated with injection medroxyprogesterone acetate. J Am Acad Dermatol. 2000;43(2, pt 1):308-310.
- Koçak AY, Akay BN, Heper AO. Sildenafil-induced pigmented purpuric dermatosis. Cutan Ocul Toxicol. 2013;32:91-92.
- Nishioka K, Sarashi C, Katayama I. Chronic pigmented purpura induced by chemical substances. Clin Exp Dermatol. 1980;5:213-218.
- Drlica K, Zhao X. DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiol Mol Biol Rev. 1997;61:377-392.
- Rubegni P, Feci L, Pellegrino M, et al. Photolocalized purpura during levofloxacin therapy. Photodermatol Photoimmunol Photomed. 2012;28:105-107.
- Sardana K, Sarkar R, Sehgal VN. Pigmented purpuric dermatoses: an overview. Int J Dermatol. 2004;43:482-488.
- Fathy H, Abdelgaber S. Treatment of pigmented purpuric dermatoses with narrow-band UVB: a report of six cases. J Eur Acad Dermatol Venereol. 2011;25:603-606.
- Krizsa J, Hunyadi J, Dobozy A. PUVA treatment of pigmented purpuric lichenoid dermatitis (Gougerot-Blum). J Am Acad Dermatol. 1992;27(5, pt 1):778-780.
- Panda S, Malakar S, Lahiri K. Oral pentoxifylline vs topical betamethasone in Schamberg disease: a comparative randomized investigator-blinded parallel-group trial. Arch Dermatol. 2004;140:491-493.
- Tamaki K, Yasaka N, Osada A, et al. Successful treatment of pigmented purpuric dermatosis with griseofulvin. Br J Dermatol. 1995;132:159-160.
- Geller M. Benefit of colchicine in the treatment of Schamberg’s disease. Ann Allergy Asthma Immunol. 2000;85:246.
- Okada K, Ishikawa O, Miyachi Y. Purpura pigmentosa chronica successfully treated with oral cyclosporin A. Br J Dermatol. 1996;134:180-181.
- Reinhold U, Seiter S, Ugurel S, et al. Treatment of progressive pigmented purpura with oral bioflavonoids and ascorbic acid: an open pilot study in 3 patients. J Am Acad Dermatol. 1999;41(2, pt 1):207-208.
- Wang A, Shuja F, Chan A, et al. Unilateral purpura annularis telangiectodes of Majocchi in an elderly male: an atypical presentation. Dermatol Online J. 2013;19:19263.
- Hale EK. Purpura annularis telangiectodes of Majocchi. Dermatol Online J. 2003;9:17.
- Hoesly FJ, Huerter CJ, Shehan JM. Purpura annularis telangiectodes of Majocchi: case report and review of the literature. Int J Dermatol. 2009;48:1129-1133.
- Kaplan R, Meehan SA, Leger M. A case of isotretinoin-induced purpura annularis telangiectodes of Majocchi and review of substance-induced pigmented purpuric dermatosis. JAMA Dermatol. 2014;150:182-184.
- Newton RC, Raimer SS. Pigmented purpuric eruptions. Dermatol Clin. 1985;3:165-169.
- Ratnam KV, Su WP, Peters MS. Purpura simplex (inflammatory purpura without vasculitis): a clinicopathologic study of 174 cases. J Am Acad Dermatol. 1991;25:642-647.
- Pang BK, Su D, Ratnam KV. Drug-induced purpura simplex: clinical and histological characteristics. Ann Acad Med Singapore. 1993;22:870-872.
- Abeck D, Gross GE, Kuwert C, et al. Acetaminophen-induced progressive pigmentary purpura (Schamberg’s disease). J Am Acad Dermatol. 1992;27:123-124.
- Lipsker D, Cribier B, Heid E, et al. Cutaneous lymphoma manifesting as pigmented, purpuric capillaries [in French]. Ann Dermatol Venereol. 1999;126:321-326.
- Peterson WC Jr, Manick KP. Purpuric eruptions associated with use of carbromal and meprobamate. Arch Dermatol. 1967;95:40-42.
- Nishioka K, Katayama I, Masuzawa M, et al. Drug-induced chronic pigmented purpura. J Dermatol. 1989;16:220-222.
- Voelter WW. Pigmented purpuric dermatosis-like reaction to topical fluorouracil. Arch Dermatol. 1983;119:875-876.
- Adams BB, Gadenne AS. Glipizide-induced pigmented purpuric dermatosis. J Am Acad Dermatol. 1999;41(5, pt 2):827-829.
- Tsao H, Lerner LH. Pigmented purpuric eruption associated with injection medroxyprogesterone acetate. J Am Acad Dermatol. 2000;43(2, pt 1):308-310.
- Koçak AY, Akay BN, Heper AO. Sildenafil-induced pigmented purpuric dermatosis. Cutan Ocul Toxicol. 2013;32:91-92.
- Nishioka K, Sarashi C, Katayama I. Chronic pigmented purpura induced by chemical substances. Clin Exp Dermatol. 1980;5:213-218.
- Drlica K, Zhao X. DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiol Mol Biol Rev. 1997;61:377-392.
- Rubegni P, Feci L, Pellegrino M, et al. Photolocalized purpura during levofloxacin therapy. Photodermatol Photoimmunol Photomed. 2012;28:105-107.
- Sardana K, Sarkar R, Sehgal VN. Pigmented purpuric dermatoses: an overview. Int J Dermatol. 2004;43:482-488.
- Fathy H, Abdelgaber S. Treatment of pigmented purpuric dermatoses with narrow-band UVB: a report of six cases. J Eur Acad Dermatol Venereol. 2011;25:603-606.
- Krizsa J, Hunyadi J, Dobozy A. PUVA treatment of pigmented purpuric lichenoid dermatitis (Gougerot-Blum). J Am Acad Dermatol. 1992;27(5, pt 1):778-780.
- Panda S, Malakar S, Lahiri K. Oral pentoxifylline vs topical betamethasone in Schamberg disease: a comparative randomized investigator-blinded parallel-group trial. Arch Dermatol. 2004;140:491-493.
- Tamaki K, Yasaka N, Osada A, et al. Successful treatment of pigmented purpuric dermatosis with griseofulvin. Br J Dermatol. 1995;132:159-160.
- Geller M. Benefit of colchicine in the treatment of Schamberg’s disease. Ann Allergy Asthma Immunol. 2000;85:246.
- Okada K, Ishikawa O, Miyachi Y. Purpura pigmentosa chronica successfully treated with oral cyclosporin A. Br J Dermatol. 1996;134:180-181.
- Reinhold U, Seiter S, Ugurel S, et al. Treatment of progressive pigmented purpura with oral bioflavonoids and ascorbic acid: an open pilot study in 3 patients. J Am Acad Dermatol. 1999;41(2, pt 1):207-208.
- Wang A, Shuja F, Chan A, et al. Unilateral purpura annularis telangiectodes of Majocchi in an elderly male: an atypical presentation. Dermatol Online J. 2013;19:19263.
Practice Point
- Purpura annularis telangiectodes of Majocchi, a type of pigmented purpuric dermatosis, may on occasion be triggered by a medication; therefore, a careful medication history may prove to be an important part of the workup for this eruption.
Irregular Erythematous Patch on the Face of an Infant
The Diagnosis: Phakomatosis Pigmentovascularis With Sturge-Weber Syndrome
The erythematous patches were identified as capillary malformations (port-wine stains) and the slate gray pigmentary changes as dermal melanocytosis (Mongolian spots)(Figure). In fact, the diagnosis of phakomatosis pigmentovascularis (PPV) type II requires dermal melanocytosis and capillary malformation with and without nevus anemicus.1 In one case series, 46% (7/15) of patients with PPV had nevus anemicus2 but our patient did not.

Phakomatosis pigmentovascularis was divided into 4 types in 1985,3 then later 5 types.4 Subcategories of the 5 types include type A, which denotes a lack of extracutaneous involvement, and type B, which is used when internal manifestations have been exhibited. Since 1947, approximately 222 cases of PPV have been described in the literature.2
A case of PPV associated with Sturge-Weber syndrome (SWS) was reported in 1997.5 Since then, PPV occasionally has been linked with SWS,5-9 though there have been other syndromic associations including Klippel-Trenaunay-Weber syndrome and melanosis oculi.2 The incidence and prevalence of overlap of PPV and SWS is unknown but is likely to be rare. In our case, magnetic resonance imaging of the patient's brain did not reveal the characteristic tram-track appearance of SWS; however, the diagnosis of SWS type II only requires facial angioma with or without glaucoma.9,10 Most cases of PPV originate from Japan, Argentina, and Mexico.2 Interestingly, our patient's parents were both of Mexican ancestry. Phakomatosis pigmentovascularis type IIb is the most common, followed by type IIa.2 Most cases have been described as sporadic, though our patient's mother also exhibited a port-wine stain on the right neck, suggesting a possible genetic association.
The etiology of PPV has been postulated as twin spotting or didymosis (Greek for twin), most commonly seen in plants and animals. A previous review defined twin spotting as 2 mutant tissues situated adjacent to one another and unique from the normal tissue surrounding both of them.2 When the cell loses its heterozygosity, this phenomenon appears. An alternative etiology supplants that a drug or virus toxic to the nervous system causes aberrant angioblasts and melanoblasts.11,12 The etiology of SWS also is unknown, though vasomotor instability has been postulated as a cause.6,13
It is important to exclude associated internal organ involvement with both of these syndromes because approximately 50% of PPV cases have extracutaneous organ involvement.2,14 In fact, PPV is known to involve the brain, skeletal system, and eye, potentially manifesting as deafness, hydrocephalus, extremity overgrowth, scoliosis, cataracts, and more.2 Patients with SWS often exhibit brain and eye symptoms including seizures.1 To screen for extracutaneous involvement, multiple imaging studies should be performed. In our patient, an echocardiogram revealed a patent foramen ovale and normal cardiac anatomy for his age. Brain imaging revealed a hypoplastic left sigmoid and transverse sinus without venous thrombosis and unremarkable appearance of the brain. An ultrasound of the liver, spleen, kidneys, and pancreas revealed no evidence of solid, cystic, or vascular lesions, though the gallbladder exhibited hyperechoic areas.
To manage the skin lesions, some authors recommend Q-switched lasers for pigmented lesions and pulsed dye lasers for capillary malformations.15 Paller and Mancini1 cited evidence that pulsed dye laser treatment before the age of 1 year may offer a psychological advantage, while other views have been offered.16 Some physicians believe that no urgent treatment of capillary malformations is needed unless internal organs are involved.2,15
- Paller AS, Mancini AJ. Hurwitz Clinical Pediatric Dermatology: A Textbook of Skin Disorders of Childhood and Adolescence. 4th ed. New York, NY: Elsevier/Saunders; 2011.
- Fernández-Guarino M, Boixeda P, de Las Heras E, et al. Phakomatosis pigmentovascularis: clinical findings in 15 patients and review of the literature. J Am Acad Dermatol. 2008;58:88-93.
- Hasegawa Y, Yasuhara M. Phakomatosis pigmentovascularis type VIa. Arch Dermatol. 1985;121:651-655.
- Torrelo A, Zambrano A, Happle R. Cutis marmorata telangiectatica congenita and extensive Mongolian spots: type V phacomatosis pigmentovascularis. Br J Dermatol. 2003;148:342-345.
- Teekhasaenee C, Ritch R. Glaucoma in phakomatosis pigmentovascularis. Ophthalmology. 1997;104:150-157.
- Patil B, Sinha G, Nayak B, et al. Bilateral Sturge-Weber and phakomatosis pigmentovascularis with glaucoma, an overlap syndrome [published online May 6, 2015]. Case Rep Ophthalmol Med. 2015;2015:106932.
- Hagiwara K, Uezato H, Nonaka S. Phacomatosis pigmentovascularis type IIb associated with Sturge-Weber syndrome and pyogenic granuloma. J Dermatol. 1998;25:721-729.
- Al Robaee A, Banka N, Alfadley A. Phakomatosis pigmentovascularis type IIb associated with Sturge-Weber syndrome. Pediatr Dermatol. 2004;21:642-645.
- Yang Y, Guo X, Xu J, et al. Phakomatosis pigmentovascularis associated with Sturge-Weber syndrome, ota nevus, and congenital glaucoma. Medicine (Baltimore). 2015;94:E1025.
- Roach ES. Neurocutaneous syndromes. Pediatr Clin North Am. 1992;39:591-620.
- Happle R. Mosaicism in human skin, understanding the patterns and mechanisms. Arch Dermatol. 1993;129:1460-1470.
- Happle R. Loss of heterozygosity in human skin. J Am Acad Dermatol. 1999;85:355-358.
- Comi AM. Pathophysiology of Sturge-Weber syndrome. J Child Neurol. 2003;18:509-516.
- Kim YC, Park HJ, Cinn YW. Phakomatosis pigmentovascularis type IIa with generalized vitiligo. Br J Dermatol. 2002;147:1028-1029.
- Brittain P, Walsh EJ, Smidt AC. Blotchy baby: a case of phakomatosis pigmentovascularis [published online February 1, 2013]. J Pediatr. 2013;162:1293.
- Van der Horst CM, Koster PH, de Borgie CA, et al. Effect of the timing of treatment of port-wine stains with the flash-lamp-pumped pulsed-dye laser. N Engl J Med. 1998;338:1028-1033.
The Diagnosis: Phakomatosis Pigmentovascularis With Sturge-Weber Syndrome
The erythematous patches were identified as capillary malformations (port-wine stains) and the slate gray pigmentary changes as dermal melanocytosis (Mongolian spots)(Figure). In fact, the diagnosis of phakomatosis pigmentovascularis (PPV) type II requires dermal melanocytosis and capillary malformation with and without nevus anemicus.1 In one case series, 46% (7/15) of patients with PPV had nevus anemicus2 but our patient did not.
Phakomatosis pigmentovascularis was divided into 4 types in 1985,3 then later 5 types.4 Subcategories of the 5 types include type A, which denotes a lack of extracutaneous involvement, and type B, which is used when internal manifestations have been exhibited. Since 1947, approximately 222 cases of PPV have been described in the literature.2
A case of PPV associated with Sturge-Weber syndrome (SWS) was reported in 1997.5 Since then, PPV occasionally has been linked with SWS,5-9 though there have been other syndromic associations including Klippel-Trenaunay-Weber syndrome and melanosis oculi.2 The incidence and prevalence of overlap of PPV and SWS is unknown but is likely to be rare. In our case, magnetic resonance imaging of the patient's brain did not reveal the characteristic tram-track appearance of SWS; however, the diagnosis of SWS type II only requires facial angioma with or without glaucoma.9,10 Most cases of PPV originate from Japan, Argentina, and Mexico.2 Interestingly, our patient's parents were both of Mexican ancestry. Phakomatosis pigmentovascularis type IIb is the most common, followed by type IIa.2 Most cases have been described as sporadic, though our patient's mother also exhibited a port-wine stain on the right neck, suggesting a possible genetic association.
The etiology of PPV has been postulated as twin spotting or didymosis (Greek for twin), most commonly seen in plants and animals. A previous review defined twin spotting as 2 mutant tissues situated adjacent to one another and unique from the normal tissue surrounding both of them.2 When the cell loses its heterozygosity, this phenomenon appears. An alternative etiology supplants that a drug or virus toxic to the nervous system causes aberrant angioblasts and melanoblasts.11,12 The etiology of SWS also is unknown, though vasomotor instability has been postulated as a cause.6,13
It is important to exclude associated internal organ involvement with both of these syndromes because approximately 50% of PPV cases have extracutaneous organ involvement.2,14 In fact, PPV is known to involve the brain, skeletal system, and eye, potentially manifesting as deafness, hydrocephalus, extremity overgrowth, scoliosis, cataracts, and more.2 Patients with SWS often exhibit brain and eye symptoms including seizures.1 To screen for extracutaneous involvement, multiple imaging studies should be performed. In our patient, an echocardiogram revealed a patent foramen ovale and normal cardiac anatomy for his age. Brain imaging revealed a hypoplastic left sigmoid and transverse sinus without venous thrombosis and unremarkable appearance of the brain. An ultrasound of the liver, spleen, kidneys, and pancreas revealed no evidence of solid, cystic, or vascular lesions, though the gallbladder exhibited hyperechoic areas.
To manage the skin lesions, some authors recommend Q-switched lasers for pigmented lesions and pulsed dye lasers for capillary malformations.15 Paller and Mancini1 cited evidence that pulsed dye laser treatment before the age of 1 year may offer a psychological advantage, while other views have been offered.16 Some physicians believe that no urgent treatment of capillary malformations is needed unless internal organs are involved.2,15
The Diagnosis: Phakomatosis Pigmentovascularis With Sturge-Weber Syndrome
The erythematous patches were identified as capillary malformations (port-wine stains) and the slate gray pigmentary changes as dermal melanocytosis (Mongolian spots)(Figure). In fact, the diagnosis of phakomatosis pigmentovascularis (PPV) type II requires dermal melanocytosis and capillary malformation with and without nevus anemicus.1 In one case series, 46% (7/15) of patients with PPV had nevus anemicus2 but our patient did not.
Phakomatosis pigmentovascularis was divided into 4 types in 1985,3 then later 5 types.4 Subcategories of the 5 types include type A, which denotes a lack of extracutaneous involvement, and type B, which is used when internal manifestations have been exhibited. Since 1947, approximately 222 cases of PPV have been described in the literature.2
A case of PPV associated with Sturge-Weber syndrome (SWS) was reported in 1997.5 Since then, PPV occasionally has been linked with SWS,5-9 though there have been other syndromic associations including Klippel-Trenaunay-Weber syndrome and melanosis oculi.2 The incidence and prevalence of overlap of PPV and SWS is unknown but is likely to be rare. In our case, magnetic resonance imaging of the patient's brain did not reveal the characteristic tram-track appearance of SWS; however, the diagnosis of SWS type II only requires facial angioma with or without glaucoma.9,10 Most cases of PPV originate from Japan, Argentina, and Mexico.2 Interestingly, our patient's parents were both of Mexican ancestry. Phakomatosis pigmentovascularis type IIb is the most common, followed by type IIa.2 Most cases have been described as sporadic, though our patient's mother also exhibited a port-wine stain on the right neck, suggesting a possible genetic association.
The etiology of PPV has been postulated as twin spotting or didymosis (Greek for twin), most commonly seen in plants and animals. A previous review defined twin spotting as 2 mutant tissues situated adjacent to one another and unique from the normal tissue surrounding both of them.2 When the cell loses its heterozygosity, this phenomenon appears. An alternative etiology supplants that a drug or virus toxic to the nervous system causes aberrant angioblasts and melanoblasts.11,12 The etiology of SWS also is unknown, though vasomotor instability has been postulated as a cause.6,13
It is important to exclude associated internal organ involvement with both of these syndromes because approximately 50% of PPV cases have extracutaneous organ involvement.2,14 In fact, PPV is known to involve the brain, skeletal system, and eye, potentially manifesting as deafness, hydrocephalus, extremity overgrowth, scoliosis, cataracts, and more.2 Patients with SWS often exhibit brain and eye symptoms including seizures.1 To screen for extracutaneous involvement, multiple imaging studies should be performed. In our patient, an echocardiogram revealed a patent foramen ovale and normal cardiac anatomy for his age. Brain imaging revealed a hypoplastic left sigmoid and transverse sinus without venous thrombosis and unremarkable appearance of the brain. An ultrasound of the liver, spleen, kidneys, and pancreas revealed no evidence of solid, cystic, or vascular lesions, though the gallbladder exhibited hyperechoic areas.
To manage the skin lesions, some authors recommend Q-switched lasers for pigmented lesions and pulsed dye lasers for capillary malformations.15 Paller and Mancini1 cited evidence that pulsed dye laser treatment before the age of 1 year may offer a psychological advantage, while other views have been offered.16 Some physicians believe that no urgent treatment of capillary malformations is needed unless internal organs are involved.2,15
- Paller AS, Mancini AJ. Hurwitz Clinical Pediatric Dermatology: A Textbook of Skin Disorders of Childhood and Adolescence. 4th ed. New York, NY: Elsevier/Saunders; 2011.
- Fernández-Guarino M, Boixeda P, de Las Heras E, et al. Phakomatosis pigmentovascularis: clinical findings in 15 patients and review of the literature. J Am Acad Dermatol. 2008;58:88-93.
- Hasegawa Y, Yasuhara M. Phakomatosis pigmentovascularis type VIa. Arch Dermatol. 1985;121:651-655.
- Torrelo A, Zambrano A, Happle R. Cutis marmorata telangiectatica congenita and extensive Mongolian spots: type V phacomatosis pigmentovascularis. Br J Dermatol. 2003;148:342-345.
- Teekhasaenee C, Ritch R. Glaucoma in phakomatosis pigmentovascularis. Ophthalmology. 1997;104:150-157.
- Patil B, Sinha G, Nayak B, et al. Bilateral Sturge-Weber and phakomatosis pigmentovascularis with glaucoma, an overlap syndrome [published online May 6, 2015]. Case Rep Ophthalmol Med. 2015;2015:106932.
- Hagiwara K, Uezato H, Nonaka S. Phacomatosis pigmentovascularis type IIb associated with Sturge-Weber syndrome and pyogenic granuloma. J Dermatol. 1998;25:721-729.
- Al Robaee A, Banka N, Alfadley A. Phakomatosis pigmentovascularis type IIb associated with Sturge-Weber syndrome. Pediatr Dermatol. 2004;21:642-645.
- Yang Y, Guo X, Xu J, et al. Phakomatosis pigmentovascularis associated with Sturge-Weber syndrome, ota nevus, and congenital glaucoma. Medicine (Baltimore). 2015;94:E1025.
- Roach ES. Neurocutaneous syndromes. Pediatr Clin North Am. 1992;39:591-620.
- Happle R. Mosaicism in human skin, understanding the patterns and mechanisms. Arch Dermatol. 1993;129:1460-1470.
- Happle R. Loss of heterozygosity in human skin. J Am Acad Dermatol. 1999;85:355-358.
- Comi AM. Pathophysiology of Sturge-Weber syndrome. J Child Neurol. 2003;18:509-516.
- Kim YC, Park HJ, Cinn YW. Phakomatosis pigmentovascularis type IIa with generalized vitiligo. Br J Dermatol. 2002;147:1028-1029.
- Brittain P, Walsh EJ, Smidt AC. Blotchy baby: a case of phakomatosis pigmentovascularis [published online February 1, 2013]. J Pediatr. 2013;162:1293.
- Van der Horst CM, Koster PH, de Borgie CA, et al. Effect of the timing of treatment of port-wine stains with the flash-lamp-pumped pulsed-dye laser. N Engl J Med. 1998;338:1028-1033.
- Paller AS, Mancini AJ. Hurwitz Clinical Pediatric Dermatology: A Textbook of Skin Disorders of Childhood and Adolescence. 4th ed. New York, NY: Elsevier/Saunders; 2011.
- Fernández-Guarino M, Boixeda P, de Las Heras E, et al. Phakomatosis pigmentovascularis: clinical findings in 15 patients and review of the literature. J Am Acad Dermatol. 2008;58:88-93.
- Hasegawa Y, Yasuhara M. Phakomatosis pigmentovascularis type VIa. Arch Dermatol. 1985;121:651-655.
- Torrelo A, Zambrano A, Happle R. Cutis marmorata telangiectatica congenita and extensive Mongolian spots: type V phacomatosis pigmentovascularis. Br J Dermatol. 2003;148:342-345.
- Teekhasaenee C, Ritch R. Glaucoma in phakomatosis pigmentovascularis. Ophthalmology. 1997;104:150-157.
- Patil B, Sinha G, Nayak B, et al. Bilateral Sturge-Weber and phakomatosis pigmentovascularis with glaucoma, an overlap syndrome [published online May 6, 2015]. Case Rep Ophthalmol Med. 2015;2015:106932.
- Hagiwara K, Uezato H, Nonaka S. Phacomatosis pigmentovascularis type IIb associated with Sturge-Weber syndrome and pyogenic granuloma. J Dermatol. 1998;25:721-729.
- Al Robaee A, Banka N, Alfadley A. Phakomatosis pigmentovascularis type IIb associated with Sturge-Weber syndrome. Pediatr Dermatol. 2004;21:642-645.
- Yang Y, Guo X, Xu J, et al. Phakomatosis pigmentovascularis associated with Sturge-Weber syndrome, ota nevus, and congenital glaucoma. Medicine (Baltimore). 2015;94:E1025.
- Roach ES. Neurocutaneous syndromes. Pediatr Clin North Am. 1992;39:591-620.
- Happle R. Mosaicism in human skin, understanding the patterns and mechanisms. Arch Dermatol. 1993;129:1460-1470.
- Happle R. Loss of heterozygosity in human skin. J Am Acad Dermatol. 1999;85:355-358.
- Comi AM. Pathophysiology of Sturge-Weber syndrome. J Child Neurol. 2003;18:509-516.
- Kim YC, Park HJ, Cinn YW. Phakomatosis pigmentovascularis type IIa with generalized vitiligo. Br J Dermatol. 2002;147:1028-1029.
- Brittain P, Walsh EJ, Smidt AC. Blotchy baby: a case of phakomatosis pigmentovascularis [published online February 1, 2013]. J Pediatr. 2013;162:1293.
- Van der Horst CM, Koster PH, de Borgie CA, et al. Effect of the timing of treatment of port-wine stains with the flash-lamp-pumped pulsed-dye laser. N Engl J Med. 1998;338:1028-1033.
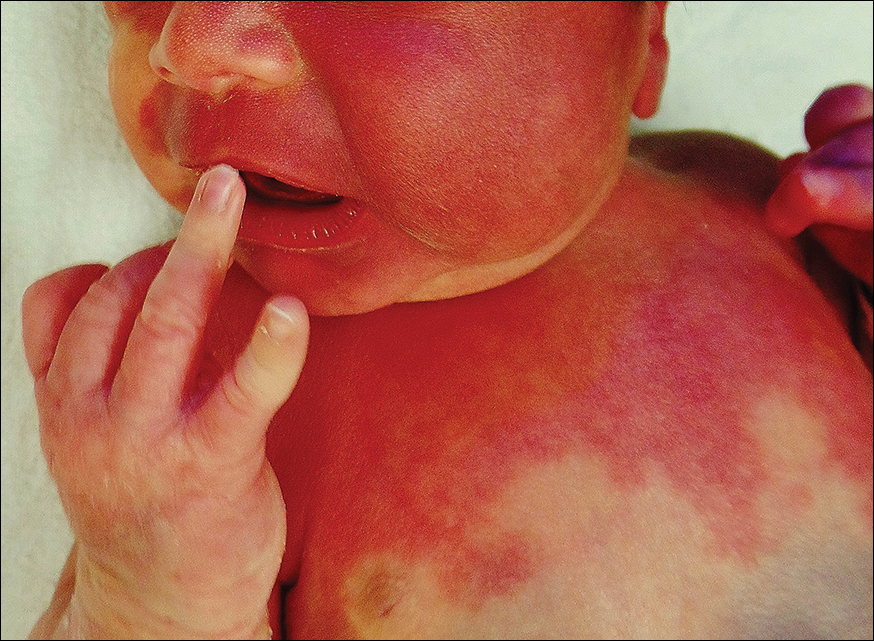
A newborn presented with an irregular and well-demarcated erythematous patch on the face, trunk, buttocks, and toes on the left foot. Another red patch was present on the right side of the face, while a slate gray patch covered the flanks and back. The limbs appeared symmetric and he exhibited no gross deformities. On close physical examination, he was noted to have a cloudy left eye. An ophthalmology consultation revealed a choroidal hemangioma and congenital glaucoma in the left eye.