User login
VIDEO: Is the FDA’s black box on estrogen too ‘alarming’?
NATIONAL HARBOR, MD. – Does the Food and Drug Administration’s black-box warning on estrogen contribute to noncompliance – or worse, to clinicians being unwilling to prescribe estrogen treatments – leaving many women to suffer untreated menopause symptoms?
The leadership of the North American Menopause Society thinks so, and the organization has started a campaign to get the FDA to reconsider how it labels estrogen.
“We would like the label for low-dose, vaginal estrogen to better reflect the true safety and risk profile,” said Dr. JoAnn Manson, chair of this year’s NAMS scientific committee.
In a video interview, Dr. Manson discusses how the current black-box labeling may impede effective treatment, and why revised, more-nuanced estrogen labeling could improve outcomes for many women.
On Twitter @whitneymcknight
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
NATIONAL HARBOR, MD. – Does the Food and Drug Administration’s black-box warning on estrogen contribute to noncompliance – or worse, to clinicians being unwilling to prescribe estrogen treatments – leaving many women to suffer untreated menopause symptoms?
The leadership of the North American Menopause Society thinks so, and the organization has started a campaign to get the FDA to reconsider how it labels estrogen.
“We would like the label for low-dose, vaginal estrogen to better reflect the true safety and risk profile,” said Dr. JoAnn Manson, chair of this year’s NAMS scientific committee.
In a video interview, Dr. Manson discusses how the current black-box labeling may impede effective treatment, and why revised, more-nuanced estrogen labeling could improve outcomes for many women.
On Twitter @whitneymcknight
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
NATIONAL HARBOR, MD. – Does the Food and Drug Administration’s black-box warning on estrogen contribute to noncompliance – or worse, to clinicians being unwilling to prescribe estrogen treatments – leaving many women to suffer untreated menopause symptoms?
The leadership of the North American Menopause Society thinks so, and the organization has started a campaign to get the FDA to reconsider how it labels estrogen.
“We would like the label for low-dose, vaginal estrogen to better reflect the true safety and risk profile,” said Dr. JoAnn Manson, chair of this year’s NAMS scientific committee.
In a video interview, Dr. Manson discusses how the current black-box labeling may impede effective treatment, and why revised, more-nuanced estrogen labeling could improve outcomes for many women.
On Twitter @whitneymcknight
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel

PODCAST: Alternatives to hormone therapy exist for women with moderate to severe menopause symptoms
NATIONAL HARBOR, MD. – “The good news is that, if someone presents with hot flashes but isn’t a candidate for hormone therapy, you can offer them a range of options,” Dr. JoAnne Pinkerton, professor of ob.gyn. at the University of Virginia, Charlottesville, said at the annual meeting of the North American Menopause Society.
In addition to hypnosis or products such as phytoestrogens that have a demonstrated strong placebo effect, Dr. Pinkerton recommends in this audio interview that physicians familiarize themselves with a Food and Drug Administration–approved paroxetine salt, certain doses of a variety of antidepressants, and other off-label treatments that can help alleviate menopause symptoms, including hot flashes, insomnia, and night sweats.
On Twitter @whitneymcknight
NATIONAL HARBOR, MD. – “The good news is that, if someone presents with hot flashes but isn’t a candidate for hormone therapy, you can offer them a range of options,” Dr. JoAnne Pinkerton, professor of ob.gyn. at the University of Virginia, Charlottesville, said at the annual meeting of the North American Menopause Society.
In addition to hypnosis or products such as phytoestrogens that have a demonstrated strong placebo effect, Dr. Pinkerton recommends in this audio interview that physicians familiarize themselves with a Food and Drug Administration–approved paroxetine salt, certain doses of a variety of antidepressants, and other off-label treatments that can help alleviate menopause symptoms, including hot flashes, insomnia, and night sweats.
On Twitter @whitneymcknight
NATIONAL HARBOR, MD. – “The good news is that, if someone presents with hot flashes but isn’t a candidate for hormone therapy, you can offer them a range of options,” Dr. JoAnne Pinkerton, professor of ob.gyn. at the University of Virginia, Charlottesville, said at the annual meeting of the North American Menopause Society.
In addition to hypnosis or products such as phytoestrogens that have a demonstrated strong placebo effect, Dr. Pinkerton recommends in this audio interview that physicians familiarize themselves with a Food and Drug Administration–approved paroxetine salt, certain doses of a variety of antidepressants, and other off-label treatments that can help alleviate menopause symptoms, including hot flashes, insomnia, and night sweats.
On Twitter @whitneymcknight
Ideal patient selection for this new approach to HT
Dr. Moore and Dr. Pinkerton focus on a new estrogen therapy: the combination conjugated estrogen and bazedoxifene (CE/BZA) for the treatment of moderate to severe hot flashes due to menopause and the prevention of menopausal osteoporosis.
Which patients can benefit most? What is CE/BZA’s safety profile? How do traditional estrogen-progestin therapies compare with CE/BZA therapy? Tune in for answers to these questions and more.
Read Cases in Menopause: Conjugated estrogen plus bazedoxifene—a new approach to estrogen therapy, from Dr. Anne Moore and Dr. JoAnn Pinkerton (October 2014).
Dr. Moore and Dr. Pinkerton focus on a new estrogen therapy: the combination conjugated estrogen and bazedoxifene (CE/BZA) for the treatment of moderate to severe hot flashes due to menopause and the prevention of menopausal osteoporosis.
Which patients can benefit most? What is CE/BZA’s safety profile? How do traditional estrogen-progestin therapies compare with CE/BZA therapy? Tune in for answers to these questions and more.
Read Cases in Menopause: Conjugated estrogen plus bazedoxifene—a new approach to estrogen therapy, from Dr. Anne Moore and Dr. JoAnn Pinkerton (October 2014).
Dr. Moore and Dr. Pinkerton focus on a new estrogen therapy: the combination conjugated estrogen and bazedoxifene (CE/BZA) for the treatment of moderate to severe hot flashes due to menopause and the prevention of menopausal osteoporosis.
Which patients can benefit most? What is CE/BZA’s safety profile? How do traditional estrogen-progestin therapies compare with CE/BZA therapy? Tune in for answers to these questions and more.
Read Cases in Menopause: Conjugated estrogen plus bazedoxifene—a new approach to estrogen therapy, from Dr. Anne Moore and Dr. JoAnn Pinkerton (October 2014).

New mobile app assists clinicians in assessing menopausal patients
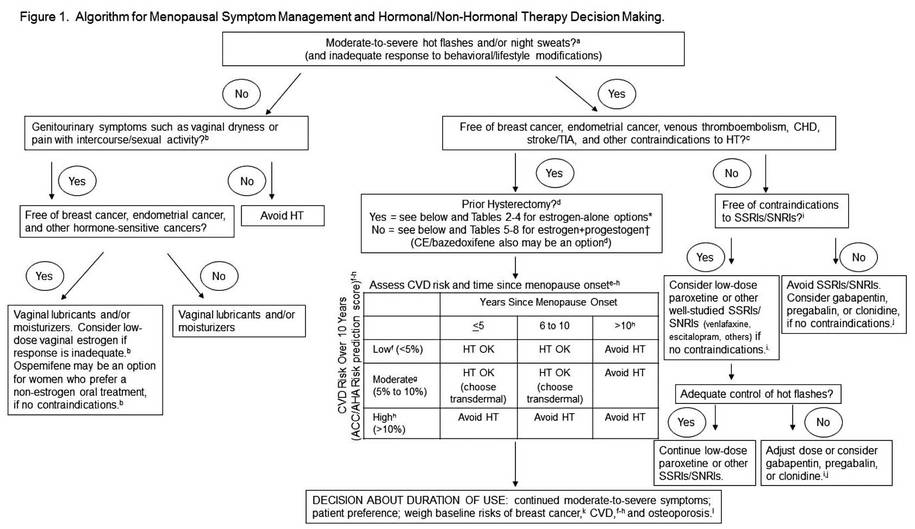
A new mobile app for iPhone and iPad enables both clinicians and patients to make decisions about menopausal therapies for moderate to severe hot flashes, night sweats, and/or genitourinary symptoms. The app also aids in assessing the patient’s risk of cardiovascular disease, breast cancer, and fracture.

|

|
|
The MenoPro app, developed in association with the North American Menopause Society (NAMS), is available free of charge from Apple. The app is designed to aid in the assessment and management of bothersome menopausal symptoms in women aged 45 and older.
Designed for both clinician and patient
A novel feature of the app is its two modes—one for the clinician and another for the patient. The clinician mode enables risk assessment and decision-making to determine whether hormonal therapy might be indicated and to determine the formulation and dosage of the therapy selected. It also features assessment of the patient’s 10-year risk of cardiovascular disease, her risk of breast cancer using the Gail model, and her fracture risk using the FRAX tool. When hormonal therapies are not appropriate, the app steers the clinician to nonhormonal options.
The patient can make use of the app to learn about her different treatment options, including lifestyle modifications. The app guides her through a self-assessment to gauge how far along she is in the menopausal transition, the severity of her symptoms, and her interest in hormonal or nonhormonal therapy. The app begins by recommending lifestyle changes and behavioral factors that can reduce menopausal symptoms. After a 3-month trial of these modifications, the patient is prompted to visit her health-care provider if further relief is needed.
Only FDA-approved drugs are recommended
“The app is completely up to date in terms of information about the newest medications that have been approved by the US Food and Drug Administration,” says JoAnn E. Manson, MD, DrPH, current chair of the NAMS Scientific Program and a past president of NAMS. Dr. Manson is Chief of the Division of Preventive Medicine at Brigham and Women’s Hospital in Boston. She also is Professor of Medicine and the Michael and Lee Bell Professor of Women’s Health at Harvard Medical School.
“The app focuses on FDA-approved medications, including off-label use of medications that may be commonly prescribed in practice to treat hot flashes, such as selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs),” she says.
“I think another big advantage is that very often clinicians who are managing patients during the menopausal transition or in early menopause may not be thinking that much about cardiovascular risk or even know how to evaluate it or make use of a 10-year risk score. So the app really helps them to become very familiar with the evaluation of cardiovascular risk, breast cancer risk, and fracture risk, and provides them with the resources to make use of the information.”
An algorithm is available within the app
The app is based on an algorithm that can be accessed within the app by choosing the “About” button. Another feature: the clinician can email a summary of the patient’s assessment directly to her, along with links to resources on a variety of relevant topics.
“In the future, there is a plan to have the app available for other mobile phones and tablet devices in addition to the iPhone and iPad,” says Dr. Manson. “We also hope to have it incorporated into electronic health records, where it could be used for clinical decision-making within the record.”
The app is not intended to replace clinical judgment, she adds. “I think clinicians are really familiar with the concept that, when you’re using an app, clinical judgment remains paramount. The app is not going to replace the clinician’s own discernment of what is going on with the patient.”
For detailed information, see an article on the app in the journal Menopause, available at http://www.menopause.org/docs/default-source/professional/our-new-paper.pdf
Share your thoughts on this article! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
A new mobile app for iPhone and iPad enables both clinicians and patients to make decisions about menopausal therapies for moderate to severe hot flashes, night sweats, and/or genitourinary symptoms. The app also aids in assessing the patient’s risk of cardiovascular disease, breast cancer, and fracture.
|
|
|
|
|
|
The MenoPro app, developed in association with the North American Menopause Society (NAMS), is available free of charge from Apple. The app is designed to aid in the assessment and management of bothersome menopausal symptoms in women aged 45 and older.
Designed for both clinician and patient
A novel feature of the app is its two modes—one for the clinician and another for the patient. The clinician mode enables risk assessment and decision-making to determine whether hormonal therapy might be indicated and to determine the formulation and dosage of the therapy selected. It also features assessment of the patient’s 10-year risk of cardiovascular disease, her risk of breast cancer using the Gail model, and her fracture risk using the FRAX tool. When hormonal therapies are not appropriate, the app steers the clinician to nonhormonal options.
The patient can make use of the app to learn about her different treatment options, including lifestyle modifications. The app guides her through a self-assessment to gauge how far along she is in the menopausal transition, the severity of her symptoms, and her interest in hormonal or nonhormonal therapy. The app begins by recommending lifestyle changes and behavioral factors that can reduce menopausal symptoms. After a 3-month trial of these modifications, the patient is prompted to visit her health-care provider if further relief is needed.
Only FDA-approved drugs are recommended
“The app is completely up to date in terms of information about the newest medications that have been approved by the US Food and Drug Administration,” says JoAnn E. Manson, MD, DrPH, current chair of the NAMS Scientific Program and a past president of NAMS. Dr. Manson is Chief of the Division of Preventive Medicine at Brigham and Women’s Hospital in Boston. She also is Professor of Medicine and the Michael and Lee Bell Professor of Women’s Health at Harvard Medical School.
“The app focuses on FDA-approved medications, including off-label use of medications that may be commonly prescribed in practice to treat hot flashes, such as selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs),” she says.
“I think another big advantage is that very often clinicians who are managing patients during the menopausal transition or in early menopause may not be thinking that much about cardiovascular risk or even know how to evaluate it or make use of a 10-year risk score. So the app really helps them to become very familiar with the evaluation of cardiovascular risk, breast cancer risk, and fracture risk, and provides them with the resources to make use of the information.”
An algorithm is available within the app
The app is based on an algorithm that can be accessed within the app by choosing the “About” button. Another feature: the clinician can email a summary of the patient’s assessment directly to her, along with links to resources on a variety of relevant topics.
“In the future, there is a plan to have the app available for other mobile phones and tablet devices in addition to the iPhone and iPad,” says Dr. Manson. “We also hope to have it incorporated into electronic health records, where it could be used for clinical decision-making within the record.”
The app is not intended to replace clinical judgment, she adds. “I think clinicians are really familiar with the concept that, when you’re using an app, clinical judgment remains paramount. The app is not going to replace the clinician’s own discernment of what is going on with the patient.”
For detailed information, see an article on the app in the journal Menopause, available at http://www.menopause.org/docs/default-source/professional/our-new-paper.pdf
Share your thoughts on this article! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
A new mobile app for iPhone and iPad enables both clinicians and patients to make decisions about menopausal therapies for moderate to severe hot flashes, night sweats, and/or genitourinary symptoms. The app also aids in assessing the patient’s risk of cardiovascular disease, breast cancer, and fracture.
|
|
|
|
|
|
The MenoPro app, developed in association with the North American Menopause Society (NAMS), is available free of charge from Apple. The app is designed to aid in the assessment and management of bothersome menopausal symptoms in women aged 45 and older.
Designed for both clinician and patient
A novel feature of the app is its two modes—one for the clinician and another for the patient. The clinician mode enables risk assessment and decision-making to determine whether hormonal therapy might be indicated and to determine the formulation and dosage of the therapy selected. It also features assessment of the patient’s 10-year risk of cardiovascular disease, her risk of breast cancer using the Gail model, and her fracture risk using the FRAX tool. When hormonal therapies are not appropriate, the app steers the clinician to nonhormonal options.
The patient can make use of the app to learn about her different treatment options, including lifestyle modifications. The app guides her through a self-assessment to gauge how far along she is in the menopausal transition, the severity of her symptoms, and her interest in hormonal or nonhormonal therapy. The app begins by recommending lifestyle changes and behavioral factors that can reduce menopausal symptoms. After a 3-month trial of these modifications, the patient is prompted to visit her health-care provider if further relief is needed.
Only FDA-approved drugs are recommended
“The app is completely up to date in terms of information about the newest medications that have been approved by the US Food and Drug Administration,” says JoAnn E. Manson, MD, DrPH, current chair of the NAMS Scientific Program and a past president of NAMS. Dr. Manson is Chief of the Division of Preventive Medicine at Brigham and Women’s Hospital in Boston. She also is Professor of Medicine and the Michael and Lee Bell Professor of Women’s Health at Harvard Medical School.
“The app focuses on FDA-approved medications, including off-label use of medications that may be commonly prescribed in practice to treat hot flashes, such as selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs),” she says.
“I think another big advantage is that very often clinicians who are managing patients during the menopausal transition or in early menopause may not be thinking that much about cardiovascular risk or even know how to evaluate it or make use of a 10-year risk score. So the app really helps them to become very familiar with the evaluation of cardiovascular risk, breast cancer risk, and fracture risk, and provides them with the resources to make use of the information.”
An algorithm is available within the app
The app is based on an algorithm that can be accessed within the app by choosing the “About” button. Another feature: the clinician can email a summary of the patient’s assessment directly to her, along with links to resources on a variety of relevant topics.
“In the future, there is a plan to have the app available for other mobile phones and tablet devices in addition to the iPhone and iPad,” says Dr. Manson. “We also hope to have it incorporated into electronic health records, where it could be used for clinical decision-making within the record.”
The app is not intended to replace clinical judgment, she adds. “I think clinicians are really familiar with the concept that, when you’re using an app, clinical judgment remains paramount. The app is not going to replace the clinician’s own discernment of what is going on with the patient.”
For detailed information, see an article on the app in the journal Menopause, available at http://www.menopause.org/docs/default-source/professional/our-new-paper.pdf
Share your thoughts on this article! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Conjugated estrogen plus bazedoxifene—a new approach to estrogen therapy
In this special installment of Cases in Menopause, I interview series contributor and menopause expert JoAnn V. Pinkerton, MD. We discuss a fairly new therapy: the combination conjugated estrogen and bazedoxifene (CE/BZA; Duavee) for the treatment of moderate to severe hot flashes due to menopause and the prevention of menopausal osteoporosis.
Much of my practice has focused on the treatment of menopausal women, but which of my patients can benefit from this particular combination of CE 0.45 mg plus BZA 20 mg? I asked Dr. Pinkerton this question, and more.
Which patients can benefit most?
Dr. Pinkerton CE/BZA was tested in healthy postmenopausal women with a uterus at risk for bone loss who were reporting 50 or more moderate to severe hot flashes per week. The combination of CE and BZA is a good choice for women who have bothersome menopausal symptoms: hot flashes, night sweats, and sleep disruption or symptomatic vulvovaginal atrophy (VVA)—although it’s not approved for VVA.
Efficacy and safety data show that compared with placebo:
- CE/BZA decreases the frequency and severity of hot flashes at 12 weeks, and those decreases are maintained at 12 months.1,2
- Women taking CE/BZA have greater improvements in sleep, with both decreased sleep disturbance and time to fall asleep.3
- CE/BZA maintained or prevented lumbar spine and hip bone loss in postmenopausal women at risk for osteoporosis. 1,4,5
Although fracture data were not captured and the drug was not tested in osteoporotic women, study results showed bone loss prevention at 12 months, which was sustained at 24 months. The improvement in bone mineral density from baseline was about 1% to 1.5%. This was compared with a bone loss of 1.8% in women taking placebo (P<.01).
In clinical studies, women taking CE/BZA versus placebo also have reported a lower incidence of painful intercourse,6 and some improvement in health-related quality of life and treatment satisfaction.7,8
In short, CE/BZA is a good option for symptomatic menopausal women with a uterus who have bothersome hot flashes, night sweats, and sleep disruptions and want to prevent bone loss.
What about adverse effects?
Dr. Pinkerton In general, CE/BZA has a favorable safety and tolerability profile, with an overall incidence of adverse events similar to placebo. The rates of cardiovascular and cerebrovascular events, cancers (breast, endometrial, and ovarian), and mortality are comparable to placebo in 2-year trials. These data are limited; studies have been conducted in healthy postmenopausal women. Future studies need to define the full risk profile, particularly among overweight or obese women and different ethnic groups and for longer-term use.
Is there a role among women with breast cancer?
Dr. Pinkerton CE/BZA has not been tested in women at risk for or with prior breast cancer. In preclinical trials of up to 2 years, involving healthy postmenopausal women, the rates for breast cancer with CE/BZA were similar to placebo. There are no long-term data, however, and there are no data in women at risk for breast cancer. I recommend that women who have or are at high risk for breast cancer consider nonhormonal treatment options.9–11
Has there been an associated increase in breast density with CE/BZA?
Dr. Pinkerton No. Data from two randomized clinical trials showed that the breast density changes with 12-month CE/BZA treatment was similar to placebo—which is markedly different from comparisons of placebo and combination estrogen-progestin therapy (EPT), where EPT increased breast density. If indeed this lack of an association translates into fewer breast cancers, it would be wonderful, but we do not have long-term data. We can tell our patients that using CE/BZA has not been shown to increase the risk of breast cancer, at least up to 2 years.
What makes CE/BZA different from traditional EPT?
Dr. Pinkerton There are two exciting differences:
- The incidences of breast pain and tenderness were found to be similar to placebo, and were significantly less than those with the comparator EPT (conjugated estrogens 0.45 mg plus medroxyprogesterone acetate [CE/MPA] 1.5 mg).9,10,12
- Bleeding and spotting rates were significantly less than those found with CE/MPA.13
In addition, high rates of amenorrhea have been found—comparable to placebo.13
CE/BZA is similar to traditional EPT in several ways. For instance, compared with placebo, at 2 years, CE/BZA was not found to increase the incidence of endometrial hyperplasia, endometrial thickness (increase from baseline was <1 mm and comparable to placebo), or endometrial cancers.14 Lastly, similar to EPT, there is probably a twofold risk of venous thromboembolism (VTE) with BZA 20 mg alone.15 Importantly, there has been no additive effect on VTE risk when combining CE with BZA; however, we will need longer studies, in older women, to fully evaluate this risk.1
Overall, in symptomatic postmenopausal women with a uterus, randomized controlled data show the same improvement with CE/BZA as that seen with traditional oral EPTs, with improvements in hot flashes; night sweats, with fewer sleep disruptions; and prevention of bone loss. In addition, the changes in cholesterol (an increase in triglyceride levels) and effect on the vagina are the same. Yet, CE/BZA appears to have a neutral effect on the breast and protects against endometrial hyperplasia and endometrial cancer without causing bleeding.9,10 CE/BZA’s VTE and stroke risks are expected to be similar to traditional oral EPT.
Therefore, the major benefit of CE/BZA for women who have a uterus is the lack of significant breast tenderness, lack of changes in breast density, and lack of vaginal bleeding that is often seen with traditional EPT.12
Then, is progestogen the harmful agent in traditional HT options?
Dr. Pinkerton There is evidence that estrogen plus progestogen therapy has more risk for breast cancer than estrogen alone. But in women who have a uterus, you need to protect against uterine cancer so, up until now, the only option was to add progestogen. Some studies suggest the risk of breast cancer may differ depending on the type of progestogen. So it’s a laudable goal to try to protect the endometrium without using a progestogen.
Given its safety profile, do you see CE/BZA being indicated for women without a uterus?
Dr. Pinkerton CE/BZA has been tested only in women with a uterus; there is no indication for using it in hysterectomized women. In the future, unless trial data show a benefit to hysterectomized women—by a reduction in breast cancer compared with estrogen alone—there would be no reason to add BZA to the CE for these women. You would just use CE or another type of estrogen alone.
Do you anticipate BZA being used alone?
Dr. Pinkerton For treating osteoporosis in postmenopausal women at increased fracture risk, BZA alone has greater benefits than risks. It is approved in other countries to prevent or treat osteoporosis. In 2008, Wyeth received an approval letter from the US Food and Drug Administration for BZA alone but, for whatever reason, the drug was not brought to market. BZA reduces the number of new lumbar spine fractures by 4% (vs 2% for placebo), with efficacy better in those with a higher risk of fractures. Like raloxifene, it has not been shown effective at reducing nonvertebral fractures, although it maintains spinal bone density.16
BZA available as monotherapy could tempt clinicians to pair it with other estrogens. We must recognize that the combination of the specific estrogen and BZA dose and type need to be balanced to provide endometrial hyperplasia protection. It would not be safe or effective to take BZA as a selective estrogen-receptor modulator and pair it with any other untested systemic estrogen. I do not anticipate, in this country, that BZA will become available as monotherapy.
New options are welcome
Dr. Moore Novel strategies for clinicians to optimally treat menopausal symptoms are always welcome. I look forward to more data from the SMART trials on CE/BZA and to moving forward as we gain experience with using this new treatment option.
Share your thoughts on this article! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
1. Lobo RA, Pinkerton JV, Gass ML, et al. Evaluation of bazedoxifene/conjugated estrogens for the treatment of menopausal symptoms and effects on metabolic bone parameters and overall safety profile. Fertil Steril. 2009;92(3):1025–1038.
2. Pinkerton JV, Utian WH, Constantine GD, Olivier S, Pickar JH. Relief of vasomotor symptoms with the tissue-selective estrogen complex containing bazedoxifene/conjugated estrogens. Menopause. 2009;16:(6)1116–1124.
3. Pinkerton JV, Pan K, Abraham L, et al. Sleep parameters and health-related quality of life with bazedoxifene/conjugated estrogens. Menopause. 2014;21(3):252–259.
4. Lindsay R, Gallagher JC, Kagan R, Pickar JH, Constantine G. Efficacy of tissue-selective estrogen complex (TSEC) of bazedoxifene/conjugated estrogens (BZA/CE) for osteoporosis prevention in at-risk postmenopausal women. Fertil Steril. 2009;92(3):1045–1052.
5. Pinkerton JV, Harvey JA, Lindsay R, et al; SMART-5 Investigators. Effects of bazedoxifene/conjugated estrogens on the endometrium and bone. J Clin Endocrinol Metab. 2014;99(2):E189–E198.
6. Kagan R, Williams RS, Pan K, Mirkin S, Pickar JH. A randomized, placebo- and active-controlled trial of bazedoxifene/conjugated estrogens (BZA/CE) for treatment of moderate to severe vulvar/vaginal atrophy in postmenopausal women. Menopause. 2010;17(2):281–289.
7. Utian W, Yu H, Bobula J, Mirkin S, Olivier S, Pickar JH. Bazedoxifene/conjugated estrogens and quality of life in postmenopausal women. Maturitas. 2009;63:(4)329–335.
8. Abraham L, Pinkerton JV, Messig M, Ryan KA, Komm BS, Mirkin S. Menopause-specific quality of life across varying menopausal populations with conjugated estrogens/bazedoxifene. Maturitas. 2014;78(3):212–218.
9. Harvey JA, Pinkerton JV, Baracat EC, Shi H, Chines AA, Mirkin S. Breast density changes in a randomized controlled trial evaluating bazedoxifene/conjugated estrogens. Menopause. 2013;20:(2)138–145.
10. Pinkerton JV, Harvey JA, Pan K, et al. Breast effects of bazedoxifene-conjugated estrogens. Obstet Gynecol. 2013;121(5):959–968.
11. Kaunitz AM. When should a menopausal woman discontinue hormone therapy? OBG Manag. 2014;26(2):59–65.
12. Pinkerton JV, Pickar JH, Racketa J, Mirkin S. Bazedoxifene/conjugated estrogens for menopausal symptom treatment and osteoporosis prevention. Climacteric. 2012;15:(5)411–418.
13. Archer DF, Lewis V, Carr BR, Olivier S, Pickar JH. Bazedoxifene/conjugated estrogens (BZA/CE): incidence of uterine bleeding in postmenopausal women. Fertil Steril. 2009;92:1039–1044.
14. Pickar JH, Yeh I-T, Bachmann G, Speroff L. Endometrial effects of a tissue selective estrogen complex (TSEC) containing bazedoxifene/conjugated estrogens as a menopausal therapy. Fertil Steril. 2009; 92(3):1018–1024.
15. Mirkin S, Komm BS. Tissue-selective estrogen complexes for postmenopausal women. Maturitas. 2013;76(3):213–220.
16. Ellis AG, Reginster JY, Luo X, et al. Bazedoxifene versus oral bisphosphonates for the prevention of nonvertebral fractures in postmenopausal women with osteoporosis at higher risk of fracture. Value Health. 2014;17(4):424–432.
Anne A. Moore, DNP, APN
Brought to you by the menopause experts


Disclosures
Dr. Moore reports no financial relationships relevant to this article. Dr. Pinkerton reports that her institution receives consulting fees from DepoMed, Noven, NovoNordisk, Pfizer, and Shionogi; current grant or research support from Therapeutics MD, prior support from DepoMed, Bionova, and Endoceutics, and, several years ago, support from Pfizer; and travel funds from DepoMed, Noven, NovoNordisk, Pfizer, Therapeutics MD, and Shionogi.
Anne A. Moore, DNP, APN
Brought to you by the menopause experts
Disclosures
Dr. Moore reports no financial relationships relevant to this article. Dr. Pinkerton reports that her institution receives consulting fees from DepoMed, Noven, NovoNordisk, Pfizer, and Shionogi; current grant or research support from Therapeutics MD, prior support from DepoMed, Bionova, and Endoceutics, and, several years ago, support from Pfizer; and travel funds from DepoMed, Noven, NovoNordisk, Pfizer, Therapeutics MD, and Shionogi.
Anne A. Moore, DNP, APN
Brought to you by the menopause experts
Disclosures
Dr. Moore reports no financial relationships relevant to this article. Dr. Pinkerton reports that her institution receives consulting fees from DepoMed, Noven, NovoNordisk, Pfizer, and Shionogi; current grant or research support from Therapeutics MD, prior support from DepoMed, Bionova, and Endoceutics, and, several years ago, support from Pfizer; and travel funds from DepoMed, Noven, NovoNordisk, Pfizer, Therapeutics MD, and Shionogi.
In this special installment of Cases in Menopause, I interview series contributor and menopause expert JoAnn V. Pinkerton, MD. We discuss a fairly new therapy: the combination conjugated estrogen and bazedoxifene (CE/BZA; Duavee) for the treatment of moderate to severe hot flashes due to menopause and the prevention of menopausal osteoporosis.
Much of my practice has focused on the treatment of menopausal women, but which of my patients can benefit from this particular combination of CE 0.45 mg plus BZA 20 mg? I asked Dr. Pinkerton this question, and more.
Which patients can benefit most?
Dr. Pinkerton CE/BZA was tested in healthy postmenopausal women with a uterus at risk for bone loss who were reporting 50 or more moderate to severe hot flashes per week. The combination of CE and BZA is a good choice for women who have bothersome menopausal symptoms: hot flashes, night sweats, and sleep disruption or symptomatic vulvovaginal atrophy (VVA)—although it’s not approved for VVA.
Efficacy and safety data show that compared with placebo:
- CE/BZA decreases the frequency and severity of hot flashes at 12 weeks, and those decreases are maintained at 12 months.1,2
- Women taking CE/BZA have greater improvements in sleep, with both decreased sleep disturbance and time to fall asleep.3
- CE/BZA maintained or prevented lumbar spine and hip bone loss in postmenopausal women at risk for osteoporosis. 1,4,5
Although fracture data were not captured and the drug was not tested in osteoporotic women, study results showed bone loss prevention at 12 months, which was sustained at 24 months. The improvement in bone mineral density from baseline was about 1% to 1.5%. This was compared with a bone loss of 1.8% in women taking placebo (P<.01).
In clinical studies, women taking CE/BZA versus placebo also have reported a lower incidence of painful intercourse,6 and some improvement in health-related quality of life and treatment satisfaction.7,8
In short, CE/BZA is a good option for symptomatic menopausal women with a uterus who have bothersome hot flashes, night sweats, and sleep disruptions and want to prevent bone loss.
What about adverse effects?
Dr. Pinkerton In general, CE/BZA has a favorable safety and tolerability profile, with an overall incidence of adverse events similar to placebo. The rates of cardiovascular and cerebrovascular events, cancers (breast, endometrial, and ovarian), and mortality are comparable to placebo in 2-year trials. These data are limited; studies have been conducted in healthy postmenopausal women. Future studies need to define the full risk profile, particularly among overweight or obese women and different ethnic groups and for longer-term use.
Is there a role among women with breast cancer?
Dr. Pinkerton CE/BZA has not been tested in women at risk for or with prior breast cancer. In preclinical trials of up to 2 years, involving healthy postmenopausal women, the rates for breast cancer with CE/BZA were similar to placebo. There are no long-term data, however, and there are no data in women at risk for breast cancer. I recommend that women who have or are at high risk for breast cancer consider nonhormonal treatment options.9–11
Has there been an associated increase in breast density with CE/BZA?
Dr. Pinkerton No. Data from two randomized clinical trials showed that the breast density changes with 12-month CE/BZA treatment was similar to placebo—which is markedly different from comparisons of placebo and combination estrogen-progestin therapy (EPT), where EPT increased breast density. If indeed this lack of an association translates into fewer breast cancers, it would be wonderful, but we do not have long-term data. We can tell our patients that using CE/BZA has not been shown to increase the risk of breast cancer, at least up to 2 years.
What makes CE/BZA different from traditional EPT?
Dr. Pinkerton There are two exciting differences:
- The incidences of breast pain and tenderness were found to be similar to placebo, and were significantly less than those with the comparator EPT (conjugated estrogens 0.45 mg plus medroxyprogesterone acetate [CE/MPA] 1.5 mg).9,10,12
- Bleeding and spotting rates were significantly less than those found with CE/MPA.13
In addition, high rates of amenorrhea have been found—comparable to placebo.13
CE/BZA is similar to traditional EPT in several ways. For instance, compared with placebo, at 2 years, CE/BZA was not found to increase the incidence of endometrial hyperplasia, endometrial thickness (increase from baseline was <1 mm and comparable to placebo), or endometrial cancers.14 Lastly, similar to EPT, there is probably a twofold risk of venous thromboembolism (VTE) with BZA 20 mg alone.15 Importantly, there has been no additive effect on VTE risk when combining CE with BZA; however, we will need longer studies, in older women, to fully evaluate this risk.1
Overall, in symptomatic postmenopausal women with a uterus, randomized controlled data show the same improvement with CE/BZA as that seen with traditional oral EPTs, with improvements in hot flashes; night sweats, with fewer sleep disruptions; and prevention of bone loss. In addition, the changes in cholesterol (an increase in triglyceride levels) and effect on the vagina are the same. Yet, CE/BZA appears to have a neutral effect on the breast and protects against endometrial hyperplasia and endometrial cancer without causing bleeding.9,10 CE/BZA’s VTE and stroke risks are expected to be similar to traditional oral EPT.
Therefore, the major benefit of CE/BZA for women who have a uterus is the lack of significant breast tenderness, lack of changes in breast density, and lack of vaginal bleeding that is often seen with traditional EPT.12
Then, is progestogen the harmful agent in traditional HT options?
Dr. Pinkerton There is evidence that estrogen plus progestogen therapy has more risk for breast cancer than estrogen alone. But in women who have a uterus, you need to protect against uterine cancer so, up until now, the only option was to add progestogen. Some studies suggest the risk of breast cancer may differ depending on the type of progestogen. So it’s a laudable goal to try to protect the endometrium without using a progestogen.
Given its safety profile, do you see CE/BZA being indicated for women without a uterus?
Dr. Pinkerton CE/BZA has been tested only in women with a uterus; there is no indication for using it in hysterectomized women. In the future, unless trial data show a benefit to hysterectomized women—by a reduction in breast cancer compared with estrogen alone—there would be no reason to add BZA to the CE for these women. You would just use CE or another type of estrogen alone.
Do you anticipate BZA being used alone?
Dr. Pinkerton For treating osteoporosis in postmenopausal women at increased fracture risk, BZA alone has greater benefits than risks. It is approved in other countries to prevent or treat osteoporosis. In 2008, Wyeth received an approval letter from the US Food and Drug Administration for BZA alone but, for whatever reason, the drug was not brought to market. BZA reduces the number of new lumbar spine fractures by 4% (vs 2% for placebo), with efficacy better in those with a higher risk of fractures. Like raloxifene, it has not been shown effective at reducing nonvertebral fractures, although it maintains spinal bone density.16
BZA available as monotherapy could tempt clinicians to pair it with other estrogens. We must recognize that the combination of the specific estrogen and BZA dose and type need to be balanced to provide endometrial hyperplasia protection. It would not be safe or effective to take BZA as a selective estrogen-receptor modulator and pair it with any other untested systemic estrogen. I do not anticipate, in this country, that BZA will become available as monotherapy.
New options are welcome
Dr. Moore Novel strategies for clinicians to optimally treat menopausal symptoms are always welcome. I look forward to more data from the SMART trials on CE/BZA and to moving forward as we gain experience with using this new treatment option.
Share your thoughts on this article! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
In this special installment of Cases in Menopause, I interview series contributor and menopause expert JoAnn V. Pinkerton, MD. We discuss a fairly new therapy: the combination conjugated estrogen and bazedoxifene (CE/BZA; Duavee) for the treatment of moderate to severe hot flashes due to menopause and the prevention of menopausal osteoporosis.
Much of my practice has focused on the treatment of menopausal women, but which of my patients can benefit from this particular combination of CE 0.45 mg plus BZA 20 mg? I asked Dr. Pinkerton this question, and more.
Which patients can benefit most?
Dr. Pinkerton CE/BZA was tested in healthy postmenopausal women with a uterus at risk for bone loss who were reporting 50 or more moderate to severe hot flashes per week. The combination of CE and BZA is a good choice for women who have bothersome menopausal symptoms: hot flashes, night sweats, and sleep disruption or symptomatic vulvovaginal atrophy (VVA)—although it’s not approved for VVA.
Efficacy and safety data show that compared with placebo:
- CE/BZA decreases the frequency and severity of hot flashes at 12 weeks, and those decreases are maintained at 12 months.1,2
- Women taking CE/BZA have greater improvements in sleep, with both decreased sleep disturbance and time to fall asleep.3
- CE/BZA maintained or prevented lumbar spine and hip bone loss in postmenopausal women at risk for osteoporosis. 1,4,5
Although fracture data were not captured and the drug was not tested in osteoporotic women, study results showed bone loss prevention at 12 months, which was sustained at 24 months. The improvement in bone mineral density from baseline was about 1% to 1.5%. This was compared with a bone loss of 1.8% in women taking placebo (P<.01).
In clinical studies, women taking CE/BZA versus placebo also have reported a lower incidence of painful intercourse,6 and some improvement in health-related quality of life and treatment satisfaction.7,8
In short, CE/BZA is a good option for symptomatic menopausal women with a uterus who have bothersome hot flashes, night sweats, and sleep disruptions and want to prevent bone loss.
What about adverse effects?
Dr. Pinkerton In general, CE/BZA has a favorable safety and tolerability profile, with an overall incidence of adverse events similar to placebo. The rates of cardiovascular and cerebrovascular events, cancers (breast, endometrial, and ovarian), and mortality are comparable to placebo in 2-year trials. These data are limited; studies have been conducted in healthy postmenopausal women. Future studies need to define the full risk profile, particularly among overweight or obese women and different ethnic groups and for longer-term use.
Is there a role among women with breast cancer?
Dr. Pinkerton CE/BZA has not been tested in women at risk for or with prior breast cancer. In preclinical trials of up to 2 years, involving healthy postmenopausal women, the rates for breast cancer with CE/BZA were similar to placebo. There are no long-term data, however, and there are no data in women at risk for breast cancer. I recommend that women who have or are at high risk for breast cancer consider nonhormonal treatment options.9–11
Has there been an associated increase in breast density with CE/BZA?
Dr. Pinkerton No. Data from two randomized clinical trials showed that the breast density changes with 12-month CE/BZA treatment was similar to placebo—which is markedly different from comparisons of placebo and combination estrogen-progestin therapy (EPT), where EPT increased breast density. If indeed this lack of an association translates into fewer breast cancers, it would be wonderful, but we do not have long-term data. We can tell our patients that using CE/BZA has not been shown to increase the risk of breast cancer, at least up to 2 years.
What makes CE/BZA different from traditional EPT?
Dr. Pinkerton There are two exciting differences:
- The incidences of breast pain and tenderness were found to be similar to placebo, and were significantly less than those with the comparator EPT (conjugated estrogens 0.45 mg plus medroxyprogesterone acetate [CE/MPA] 1.5 mg).9,10,12
- Bleeding and spotting rates were significantly less than those found with CE/MPA.13
In addition, high rates of amenorrhea have been found—comparable to placebo.13
CE/BZA is similar to traditional EPT in several ways. For instance, compared with placebo, at 2 years, CE/BZA was not found to increase the incidence of endometrial hyperplasia, endometrial thickness (increase from baseline was <1 mm and comparable to placebo), or endometrial cancers.14 Lastly, similar to EPT, there is probably a twofold risk of venous thromboembolism (VTE) with BZA 20 mg alone.15 Importantly, there has been no additive effect on VTE risk when combining CE with BZA; however, we will need longer studies, in older women, to fully evaluate this risk.1
Overall, in symptomatic postmenopausal women with a uterus, randomized controlled data show the same improvement with CE/BZA as that seen with traditional oral EPTs, with improvements in hot flashes; night sweats, with fewer sleep disruptions; and prevention of bone loss. In addition, the changes in cholesterol (an increase in triglyceride levels) and effect on the vagina are the same. Yet, CE/BZA appears to have a neutral effect on the breast and protects against endometrial hyperplasia and endometrial cancer without causing bleeding.9,10 CE/BZA’s VTE and stroke risks are expected to be similar to traditional oral EPT.
Therefore, the major benefit of CE/BZA for women who have a uterus is the lack of significant breast tenderness, lack of changes in breast density, and lack of vaginal bleeding that is often seen with traditional EPT.12
Then, is progestogen the harmful agent in traditional HT options?
Dr. Pinkerton There is evidence that estrogen plus progestogen therapy has more risk for breast cancer than estrogen alone. But in women who have a uterus, you need to protect against uterine cancer so, up until now, the only option was to add progestogen. Some studies suggest the risk of breast cancer may differ depending on the type of progestogen. So it’s a laudable goal to try to protect the endometrium without using a progestogen.
Given its safety profile, do you see CE/BZA being indicated for women without a uterus?
Dr. Pinkerton CE/BZA has been tested only in women with a uterus; there is no indication for using it in hysterectomized women. In the future, unless trial data show a benefit to hysterectomized women—by a reduction in breast cancer compared with estrogen alone—there would be no reason to add BZA to the CE for these women. You would just use CE or another type of estrogen alone.
Do you anticipate BZA being used alone?
Dr. Pinkerton For treating osteoporosis in postmenopausal women at increased fracture risk, BZA alone has greater benefits than risks. It is approved in other countries to prevent or treat osteoporosis. In 2008, Wyeth received an approval letter from the US Food and Drug Administration for BZA alone but, for whatever reason, the drug was not brought to market. BZA reduces the number of new lumbar spine fractures by 4% (vs 2% for placebo), with efficacy better in those with a higher risk of fractures. Like raloxifene, it has not been shown effective at reducing nonvertebral fractures, although it maintains spinal bone density.16
BZA available as monotherapy could tempt clinicians to pair it with other estrogens. We must recognize that the combination of the specific estrogen and BZA dose and type need to be balanced to provide endometrial hyperplasia protection. It would not be safe or effective to take BZA as a selective estrogen-receptor modulator and pair it with any other untested systemic estrogen. I do not anticipate, in this country, that BZA will become available as monotherapy.
New options are welcome
Dr. Moore Novel strategies for clinicians to optimally treat menopausal symptoms are always welcome. I look forward to more data from the SMART trials on CE/BZA and to moving forward as we gain experience with using this new treatment option.
Share your thoughts on this article! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
1. Lobo RA, Pinkerton JV, Gass ML, et al. Evaluation of bazedoxifene/conjugated estrogens for the treatment of menopausal symptoms and effects on metabolic bone parameters and overall safety profile. Fertil Steril. 2009;92(3):1025–1038.
2. Pinkerton JV, Utian WH, Constantine GD, Olivier S, Pickar JH. Relief of vasomotor symptoms with the tissue-selective estrogen complex containing bazedoxifene/conjugated estrogens. Menopause. 2009;16:(6)1116–1124.
3. Pinkerton JV, Pan K, Abraham L, et al. Sleep parameters and health-related quality of life with bazedoxifene/conjugated estrogens. Menopause. 2014;21(3):252–259.
4. Lindsay R, Gallagher JC, Kagan R, Pickar JH, Constantine G. Efficacy of tissue-selective estrogen complex (TSEC) of bazedoxifene/conjugated estrogens (BZA/CE) for osteoporosis prevention in at-risk postmenopausal women. Fertil Steril. 2009;92(3):1045–1052.
5. Pinkerton JV, Harvey JA, Lindsay R, et al; SMART-5 Investigators. Effects of bazedoxifene/conjugated estrogens on the endometrium and bone. J Clin Endocrinol Metab. 2014;99(2):E189–E198.
6. Kagan R, Williams RS, Pan K, Mirkin S, Pickar JH. A randomized, placebo- and active-controlled trial of bazedoxifene/conjugated estrogens (BZA/CE) for treatment of moderate to severe vulvar/vaginal atrophy in postmenopausal women. Menopause. 2010;17(2):281–289.
7. Utian W, Yu H, Bobula J, Mirkin S, Olivier S, Pickar JH. Bazedoxifene/conjugated estrogens and quality of life in postmenopausal women. Maturitas. 2009;63:(4)329–335.
8. Abraham L, Pinkerton JV, Messig M, Ryan KA, Komm BS, Mirkin S. Menopause-specific quality of life across varying menopausal populations with conjugated estrogens/bazedoxifene. Maturitas. 2014;78(3):212–218.
9. Harvey JA, Pinkerton JV, Baracat EC, Shi H, Chines AA, Mirkin S. Breast density changes in a randomized controlled trial evaluating bazedoxifene/conjugated estrogens. Menopause. 2013;20:(2)138–145.
10. Pinkerton JV, Harvey JA, Pan K, et al. Breast effects of bazedoxifene-conjugated estrogens. Obstet Gynecol. 2013;121(5):959–968.
11. Kaunitz AM. When should a menopausal woman discontinue hormone therapy? OBG Manag. 2014;26(2):59–65.
12. Pinkerton JV, Pickar JH, Racketa J, Mirkin S. Bazedoxifene/conjugated estrogens for menopausal symptom treatment and osteoporosis prevention. Climacteric. 2012;15:(5)411–418.
13. Archer DF, Lewis V, Carr BR, Olivier S, Pickar JH. Bazedoxifene/conjugated estrogens (BZA/CE): incidence of uterine bleeding in postmenopausal women. Fertil Steril. 2009;92:1039–1044.
14. Pickar JH, Yeh I-T, Bachmann G, Speroff L. Endometrial effects of a tissue selective estrogen complex (TSEC) containing bazedoxifene/conjugated estrogens as a menopausal therapy. Fertil Steril. 2009; 92(3):1018–1024.
15. Mirkin S, Komm BS. Tissue-selective estrogen complexes for postmenopausal women. Maturitas. 2013;76(3):213–220.
16. Ellis AG, Reginster JY, Luo X, et al. Bazedoxifene versus oral bisphosphonates for the prevention of nonvertebral fractures in postmenopausal women with osteoporosis at higher risk of fracture. Value Health. 2014;17(4):424–432.
1. Lobo RA, Pinkerton JV, Gass ML, et al. Evaluation of bazedoxifene/conjugated estrogens for the treatment of menopausal symptoms and effects on metabolic bone parameters and overall safety profile. Fertil Steril. 2009;92(3):1025–1038.
2. Pinkerton JV, Utian WH, Constantine GD, Olivier S, Pickar JH. Relief of vasomotor symptoms with the tissue-selective estrogen complex containing bazedoxifene/conjugated estrogens. Menopause. 2009;16:(6)1116–1124.
3. Pinkerton JV, Pan K, Abraham L, et al. Sleep parameters and health-related quality of life with bazedoxifene/conjugated estrogens. Menopause. 2014;21(3):252–259.
4. Lindsay R, Gallagher JC, Kagan R, Pickar JH, Constantine G. Efficacy of tissue-selective estrogen complex (TSEC) of bazedoxifene/conjugated estrogens (BZA/CE) for osteoporosis prevention in at-risk postmenopausal women. Fertil Steril. 2009;92(3):1045–1052.
5. Pinkerton JV, Harvey JA, Lindsay R, et al; SMART-5 Investigators. Effects of bazedoxifene/conjugated estrogens on the endometrium and bone. J Clin Endocrinol Metab. 2014;99(2):E189–E198.
6. Kagan R, Williams RS, Pan K, Mirkin S, Pickar JH. A randomized, placebo- and active-controlled trial of bazedoxifene/conjugated estrogens (BZA/CE) for treatment of moderate to severe vulvar/vaginal atrophy in postmenopausal women. Menopause. 2010;17(2):281–289.
7. Utian W, Yu H, Bobula J, Mirkin S, Olivier S, Pickar JH. Bazedoxifene/conjugated estrogens and quality of life in postmenopausal women. Maturitas. 2009;63:(4)329–335.
8. Abraham L, Pinkerton JV, Messig M, Ryan KA, Komm BS, Mirkin S. Menopause-specific quality of life across varying menopausal populations with conjugated estrogens/bazedoxifene. Maturitas. 2014;78(3):212–218.
9. Harvey JA, Pinkerton JV, Baracat EC, Shi H, Chines AA, Mirkin S. Breast density changes in a randomized controlled trial evaluating bazedoxifene/conjugated estrogens. Menopause. 2013;20:(2)138–145.
10. Pinkerton JV, Harvey JA, Pan K, et al. Breast effects of bazedoxifene-conjugated estrogens. Obstet Gynecol. 2013;121(5):959–968.
11. Kaunitz AM. When should a menopausal woman discontinue hormone therapy? OBG Manag. 2014;26(2):59–65.
12. Pinkerton JV, Pickar JH, Racketa J, Mirkin S. Bazedoxifene/conjugated estrogens for menopausal symptom treatment and osteoporosis prevention. Climacteric. 2012;15:(5)411–418.
13. Archer DF, Lewis V, Carr BR, Olivier S, Pickar JH. Bazedoxifene/conjugated estrogens (BZA/CE): incidence of uterine bleeding in postmenopausal women. Fertil Steril. 2009;92:1039–1044.
14. Pickar JH, Yeh I-T, Bachmann G, Speroff L. Endometrial effects of a tissue selective estrogen complex (TSEC) containing bazedoxifene/conjugated estrogens as a menopausal therapy. Fertil Steril. 2009; 92(3):1018–1024.
15. Mirkin S, Komm BS. Tissue-selective estrogen complexes for postmenopausal women. Maturitas. 2013;76(3):213–220.
16. Ellis AG, Reginster JY, Luo X, et al. Bazedoxifene versus oral bisphosphonates for the prevention of nonvertebral fractures in postmenopausal women with osteoporosis at higher risk of fracture. Value Health. 2014;17(4):424–432.
After 3-year stumble, new weight-loss drug wins FDA approval
After a delay of more than 3 years, the Food and Drug Administration has approved the nation’s third weight-loss drug, a combination of naltrexone and bupropion.
The extended release tablets (Contrave; Orexigen and Takeda) are approved for use in adults who have a body mass index of at least 30 kg/m2, or those with a BMI of at least 27 kg/m2 and at least one additional weight-related condition such as hypertension, type 2 diabetes, or dyslipidemia. The agency recommended that Contrave be used in addition to caloric restriction and increased physical activity.

Dr. Timothy Garvey, chair of the American Association of Clinical Endocrinologists’ scientific committee, lauded the approval.
"We have a new tool now to treat obesity – and that is very good news," he said in an interview.
He said Contrave will be a valuable addition to the existing weight-loss medications: the phentermine/topiramate combo (Qsymia; Vivus) and lorcaserin (Belviq; Arena).
"There are no head-to-head trials with the other drugs, so we really can’t say much about relative efficacy," said Dr. Garvey. "But when you look at the placebo-subtracted weight loss in all the phase III data, it looks like Contrave is in the middle, with about a 6% loss over lifestyle interventions alone. So it’s not as effective as the topiramate combination, but more effective than lorcaserin."
In the pivotal, 56-week phase III trials, those taking Contrave lost 5%-8% of their baseline body weight, compared with a loss of 1%-2% in those on placebo. The proportion of those who lost at least 5% of their baseline body weight ranged from 45%-56% of those on the proposed dose, compared with 16%-43% of those on placebo.
The FDA guidance on weight-loss drugs suggests a 12-week efficacy evaluation – if the patient has not lost at least 5% of total body weight by then, the drug should be discontinued and another started.
It’s not possible to predict who will respond best to which drug, although there are some things to consider when choosing, said Dr. Garvey, who is chair of the department of nutrition at the University of Alabama at Birmingham.
"For example, women of childbearing age need to take precautions against becoming pregnant if they take the topiramate combination, and should stop it right away if they do become pregnant. And since lorcaserin is a serotonergic drug, it has to be used very cautiously in patients who are on other serotonergic medications. We definitely need more safety data there."
Additionally, none of the weight-loss drugs should be used in children or teens until more studies confirm their safety for those patients, "All of the companies are planning these trials, and we hope they will complete them expeditiously," Dr. Garvey said. "Childhood and adolescent obesity is a huge problem, and we really need some good treatment options there."
Because it contains bupropion, an antidepressant that has been associated with an increased risk of suicidal thoughts and actions, the drug carries a black box warning. Bupropion is also known to lower seizure threshold, so the drug should not be used in patients with seizure disorders. If a seizure occurs while taking on the medication, it should be permanently discontinued. Nor should it be used in patients with uncontrolled hypertension.
Orexigen and Takeda originally brought the drug forward in December 2011. It was not approved at that time because of concerns about its effect on blood pressure – an unexpected move, and one that Orexigen management called "a big setback."
About a quarter of those in the 56-week pivotal phase III trial experienced significant blood pressure increases of at least 10% above their baseline, compared with about 20% of those in the control arm. Increases of diastolic blood pressure of at least 5 mm Hg over baseline occurred in 37% of those on the combination, compared with 29% of those on placebo. About a quarter in the active arm also had heart rate increases of at least 10 beats per minute, compared with 19% of those taking placebo.
Because of these concerns, the FDA required the drug companies to conduct a large, double-blinded, randomized, placebo-controlled trial to investigate the risk of major cardiovascular events. Takeda and Orexigen then launched the 4-year, 8,900 patient Light study, which is still ongoing. Endpoints are major adverse cardiovascular events (cardiovascular death, nonfatal myocardial infarction, and nonfatal stroke) in overweight and obese subjects who have concomitant diabetes and/or other cardiovascular risk factors.
In June, citing encouraging preliminary results, Takeda and Orexigen brought Contrave to the FDA once more – only to be shot down again, at least temporarily. The agency required a review extension in order to come to agreement on the final form of postmarketing surveillance, said Denise Powell, a spokeswoman for Orexigen.
"At that time, FDA said the data looked good," she said in an interview. "We just needed more time to work out the postmarketing requirements."
Those will include:
• A cardiovascular outcomes trial to assess the cardiovascular risk associated with Contrave use.
• Two efficacy, safety, and clinical pharmacology studies in pediatric patients (one in patients 12-17 years old, and one in patients 7-11 years old).
• A juvenile animal toxicity study with a particular focus on growth and development as well as behavior, learning, and memory.
• A cardiac conduction study.
• Clinical trials to evaluate dosing in patients with hepatic or renal impairment.
• A clinical trial to evaluate the potential for interactions between the medication and other drugs.
Contrave contains an extended-release formulation of 8 mg naltrexone and 90 mg bupropion. It is to be administered in an in a 4-week upward titration schedule, with a single morning tablet during week 1; a single tablet at morning and evening during week 2; two tablets in the morning and one in the evening during week 3; and two tablets both morning and evening from week 4 and onward.
Dr. Garvey is a consultant for Daiichi Sankyo, LipoScience, Takeda, Vivus, Boehringer Ingelheim, Janssen, Eisai, and Novo Nordisk. He has received research funding from Merck, AstraZeneca, Weight Watchers, Eisai, and Sanofi.
On Twitter @alz_gal
After a delay of more than 3 years, the Food and Drug Administration has approved the nation’s third weight-loss drug, a combination of naltrexone and bupropion.
The extended release tablets (Contrave; Orexigen and Takeda) are approved for use in adults who have a body mass index of at least 30 kg/m2, or those with a BMI of at least 27 kg/m2 and at least one additional weight-related condition such as hypertension, type 2 diabetes, or dyslipidemia. The agency recommended that Contrave be used in addition to caloric restriction and increased physical activity.
Dr. Timothy Garvey, chair of the American Association of Clinical Endocrinologists’ scientific committee, lauded the approval.
"We have a new tool now to treat obesity – and that is very good news," he said in an interview.
He said Contrave will be a valuable addition to the existing weight-loss medications: the phentermine/topiramate combo (Qsymia; Vivus) and lorcaserin (Belviq; Arena).
"There are no head-to-head trials with the other drugs, so we really can’t say much about relative efficacy," said Dr. Garvey. "But when you look at the placebo-subtracted weight loss in all the phase III data, it looks like Contrave is in the middle, with about a 6% loss over lifestyle interventions alone. So it’s not as effective as the topiramate combination, but more effective than lorcaserin."
In the pivotal, 56-week phase III trials, those taking Contrave lost 5%-8% of their baseline body weight, compared with a loss of 1%-2% in those on placebo. The proportion of those who lost at least 5% of their baseline body weight ranged from 45%-56% of those on the proposed dose, compared with 16%-43% of those on placebo.
The FDA guidance on weight-loss drugs suggests a 12-week efficacy evaluation – if the patient has not lost at least 5% of total body weight by then, the drug should be discontinued and another started.
It’s not possible to predict who will respond best to which drug, although there are some things to consider when choosing, said Dr. Garvey, who is chair of the department of nutrition at the University of Alabama at Birmingham.
"For example, women of childbearing age need to take precautions against becoming pregnant if they take the topiramate combination, and should stop it right away if they do become pregnant. And since lorcaserin is a serotonergic drug, it has to be used very cautiously in patients who are on other serotonergic medications. We definitely need more safety data there."
Additionally, none of the weight-loss drugs should be used in children or teens until more studies confirm their safety for those patients, "All of the companies are planning these trials, and we hope they will complete them expeditiously," Dr. Garvey said. "Childhood and adolescent obesity is a huge problem, and we really need some good treatment options there."
Because it contains bupropion, an antidepressant that has been associated with an increased risk of suicidal thoughts and actions, the drug carries a black box warning. Bupropion is also known to lower seizure threshold, so the drug should not be used in patients with seizure disorders. If a seizure occurs while taking on the medication, it should be permanently discontinued. Nor should it be used in patients with uncontrolled hypertension.
Orexigen and Takeda originally brought the drug forward in December 2011. It was not approved at that time because of concerns about its effect on blood pressure – an unexpected move, and one that Orexigen management called "a big setback."
About a quarter of those in the 56-week pivotal phase III trial experienced significant blood pressure increases of at least 10% above their baseline, compared with about 20% of those in the control arm. Increases of diastolic blood pressure of at least 5 mm Hg over baseline occurred in 37% of those on the combination, compared with 29% of those on placebo. About a quarter in the active arm also had heart rate increases of at least 10 beats per minute, compared with 19% of those taking placebo.
Because of these concerns, the FDA required the drug companies to conduct a large, double-blinded, randomized, placebo-controlled trial to investigate the risk of major cardiovascular events. Takeda and Orexigen then launched the 4-year, 8,900 patient Light study, which is still ongoing. Endpoints are major adverse cardiovascular events (cardiovascular death, nonfatal myocardial infarction, and nonfatal stroke) in overweight and obese subjects who have concomitant diabetes and/or other cardiovascular risk factors.
In June, citing encouraging preliminary results, Takeda and Orexigen brought Contrave to the FDA once more – only to be shot down again, at least temporarily. The agency required a review extension in order to come to agreement on the final form of postmarketing surveillance, said Denise Powell, a spokeswoman for Orexigen.
"At that time, FDA said the data looked good," she said in an interview. "We just needed more time to work out the postmarketing requirements."
Those will include:
• A cardiovascular outcomes trial to assess the cardiovascular risk associated with Contrave use.
• Two efficacy, safety, and clinical pharmacology studies in pediatric patients (one in patients 12-17 years old, and one in patients 7-11 years old).
• A juvenile animal toxicity study with a particular focus on growth and development as well as behavior, learning, and memory.
• A cardiac conduction study.
• Clinical trials to evaluate dosing in patients with hepatic or renal impairment.
• A clinical trial to evaluate the potential for interactions between the medication and other drugs.
Contrave contains an extended-release formulation of 8 mg naltrexone and 90 mg bupropion. It is to be administered in an in a 4-week upward titration schedule, with a single morning tablet during week 1; a single tablet at morning and evening during week 2; two tablets in the morning and one in the evening during week 3; and two tablets both morning and evening from week 4 and onward.
Dr. Garvey is a consultant for Daiichi Sankyo, LipoScience, Takeda, Vivus, Boehringer Ingelheim, Janssen, Eisai, and Novo Nordisk. He has received research funding from Merck, AstraZeneca, Weight Watchers, Eisai, and Sanofi.
On Twitter @alz_gal
After a delay of more than 3 years, the Food and Drug Administration has approved the nation’s third weight-loss drug, a combination of naltrexone and bupropion.
The extended release tablets (Contrave; Orexigen and Takeda) are approved for use in adults who have a body mass index of at least 30 kg/m2, or those with a BMI of at least 27 kg/m2 and at least one additional weight-related condition such as hypertension, type 2 diabetes, or dyslipidemia. The agency recommended that Contrave be used in addition to caloric restriction and increased physical activity.
Dr. Timothy Garvey, chair of the American Association of Clinical Endocrinologists’ scientific committee, lauded the approval.
"We have a new tool now to treat obesity – and that is very good news," he said in an interview.
He said Contrave will be a valuable addition to the existing weight-loss medications: the phentermine/topiramate combo (Qsymia; Vivus) and lorcaserin (Belviq; Arena).
"There are no head-to-head trials with the other drugs, so we really can’t say much about relative efficacy," said Dr. Garvey. "But when you look at the placebo-subtracted weight loss in all the phase III data, it looks like Contrave is in the middle, with about a 6% loss over lifestyle interventions alone. So it’s not as effective as the topiramate combination, but more effective than lorcaserin."
In the pivotal, 56-week phase III trials, those taking Contrave lost 5%-8% of their baseline body weight, compared with a loss of 1%-2% in those on placebo. The proportion of those who lost at least 5% of their baseline body weight ranged from 45%-56% of those on the proposed dose, compared with 16%-43% of those on placebo.
The FDA guidance on weight-loss drugs suggests a 12-week efficacy evaluation – if the patient has not lost at least 5% of total body weight by then, the drug should be discontinued and another started.
It’s not possible to predict who will respond best to which drug, although there are some things to consider when choosing, said Dr. Garvey, who is chair of the department of nutrition at the University of Alabama at Birmingham.
"For example, women of childbearing age need to take precautions against becoming pregnant if they take the topiramate combination, and should stop it right away if they do become pregnant. And since lorcaserin is a serotonergic drug, it has to be used very cautiously in patients who are on other serotonergic medications. We definitely need more safety data there."
Additionally, none of the weight-loss drugs should be used in children or teens until more studies confirm their safety for those patients, "All of the companies are planning these trials, and we hope they will complete them expeditiously," Dr. Garvey said. "Childhood and adolescent obesity is a huge problem, and we really need some good treatment options there."
Because it contains bupropion, an antidepressant that has been associated with an increased risk of suicidal thoughts and actions, the drug carries a black box warning. Bupropion is also known to lower seizure threshold, so the drug should not be used in patients with seizure disorders. If a seizure occurs while taking on the medication, it should be permanently discontinued. Nor should it be used in patients with uncontrolled hypertension.
Orexigen and Takeda originally brought the drug forward in December 2011. It was not approved at that time because of concerns about its effect on blood pressure – an unexpected move, and one that Orexigen management called "a big setback."
About a quarter of those in the 56-week pivotal phase III trial experienced significant blood pressure increases of at least 10% above their baseline, compared with about 20% of those in the control arm. Increases of diastolic blood pressure of at least 5 mm Hg over baseline occurred in 37% of those on the combination, compared with 29% of those on placebo. About a quarter in the active arm also had heart rate increases of at least 10 beats per minute, compared with 19% of those taking placebo.
Because of these concerns, the FDA required the drug companies to conduct a large, double-blinded, randomized, placebo-controlled trial to investigate the risk of major cardiovascular events. Takeda and Orexigen then launched the 4-year, 8,900 patient Light study, which is still ongoing. Endpoints are major adverse cardiovascular events (cardiovascular death, nonfatal myocardial infarction, and nonfatal stroke) in overweight and obese subjects who have concomitant diabetes and/or other cardiovascular risk factors.
In June, citing encouraging preliminary results, Takeda and Orexigen brought Contrave to the FDA once more – only to be shot down again, at least temporarily. The agency required a review extension in order to come to agreement on the final form of postmarketing surveillance, said Denise Powell, a spokeswoman for Orexigen.
"At that time, FDA said the data looked good," she said in an interview. "We just needed more time to work out the postmarketing requirements."
Those will include:
• A cardiovascular outcomes trial to assess the cardiovascular risk associated with Contrave use.
• Two efficacy, safety, and clinical pharmacology studies in pediatric patients (one in patients 12-17 years old, and one in patients 7-11 years old).
• A juvenile animal toxicity study with a particular focus on growth and development as well as behavior, learning, and memory.
• A cardiac conduction study.
• Clinical trials to evaluate dosing in patients with hepatic or renal impairment.
• A clinical trial to evaluate the potential for interactions between the medication and other drugs.
Contrave contains an extended-release formulation of 8 mg naltrexone and 90 mg bupropion. It is to be administered in an in a 4-week upward titration schedule, with a single morning tablet during week 1; a single tablet at morning and evening during week 2; two tablets in the morning and one in the evening during week 3; and two tablets both morning and evening from week 4 and onward.
Dr. Garvey is a consultant for Daiichi Sankyo, LipoScience, Takeda, Vivus, Boehringer Ingelheim, Janssen, Eisai, and Novo Nordisk. He has received research funding from Merck, AstraZeneca, Weight Watchers, Eisai, and Sanofi.
On Twitter @alz_gal

2014 Update on sexual dysfunction
Since the last installment of this Update on Sexual Dysfunction, three new drugs have been added to the armamentarium for menopausal symptoms and dyspareunia:
- paroxetine 7.5 mg (Brisdelle)
- conjugated estrogens and bazedoxifene (Duavee)
- ospemifene (Osphena).
In this article, I present a case-based approach to incorporating these drugs into practice and restoring sexual function in the setting of vulvovaginal atrophy and dyspareunia. As is often the case, decision-making requires sifting through multiple layers of information.
How to “tease out” the problem and help the patient regain sexual function
Simon JA, Portman DJ, Kazempour K, Mekonnen H, Bhaskar S, Lippman J. Safety profile of paroxetine 7.5 mg in women with moderate-to-severe vasomotor symptoms. Obstet Gynecol. 2014;123(suppl 1):132S–133S.
Conjugated estrogens/bazedoxifene (Duavee) for menopausal symptoms and prevention of osteoporosis. Med Lett Drugs Ther. 2014;56(1441):33–34.
DeGregorio MW, Zerbe RL, Wurz GT. Ospemifene: a first-in-class, non-hormonal selective estrogen receptor modulator approved for the treatment of dyspareunia associated with vulvar and vaginal atrophy [published online ahead of print August 1, 2014]. Steroids. doi:10.1016/j.steroids.2014.07.012.
Goldstein SR, Archer DF, Simon JA, Constantine G. Endometrial safety of ospemifene and the ability of transvaginal ultrasonography to detect small changes in endometrial thickness. Obstet Gynecol. 2014;123(suppl 1):96S–97S.
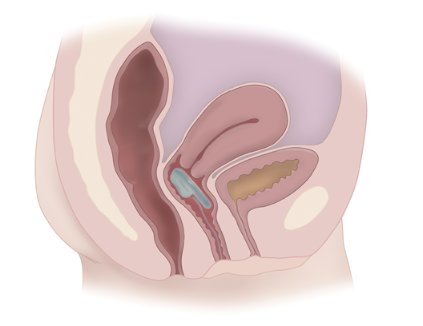
CASE: LOW DESIRE AND DISCOMFORT DURING INTERCOURSE
Your 58-year-old patient, G2P2, mentions during her annual visit that she’s not that interested in sex anymore. Her children are grown, she’s been happily married for 28 years, and she enjoys her job and denies any symptoms of depression. She says her relationship with her husband is good and, aside from her low desire, she has no worries about the marriage. Her only medication is paroxetine 7.5 mg/day (Brisdelle) for management of her moderate hot flashes, which she initiated at her last annual visit. She reports improvement in her sleep and menopausal symptoms as a result. She has an intact uterus.
You perform a pelvic exam and find atrophic vulva and vagina with mild erythema, and thinned epithelium. When you ask if she has experienced any discomfort, she reports that she needs to use lubrication for intercourse and that, even with lubrication, she has pain upon penetration and a burning sensation that continues throughout intercourse. She also reports that it seems to take her much longer to achieve arousal than in the past, and she often fails to reach orgasm.
How would you manage this patient?
As always, begin with the history
The transition to menopause creates multiple layers of potential symptoms and problems for our patients, and sometimes medical therapy can generate additional ones.
In a patient reporting the onset of low desire and dyspareunia, you would want to first consider her medication history, despite the clear evidence of vaginal atrophy. Begin by asking whether she is taking any new medications prescribed by another provider. In some cases, antihypertensive drugs, psychotropic agents, and other medications can affect sexual function.
This patient has been taking Brisdelle for 1 year and is happy with its effect on her sleep and hot flashes. Simon and colleagues found this nonhormonal agent for moderate to severe vasomotor symptoms to produce no notable effects in weight, libido, or sleep, compared with placebo.
Nevertheless, in this case, because selective serotonin reuptake inhibitors (SSRIs) such as paroxetine can affect arousal and orgasm, it is unclear whether the ultra-low dose of paroxetine she is taking is contributing to her problems. If you were to discontinue the drug to find out, her vasomotor symptoms and sleep disruption would likely recur.
Your decision-making is important here and should involve the patient in an extensive discussion. If there is not enough time for this discussion at the current visit, schedule a follow-up to address her issues fully.
Vulvovaginal atrophy has its own timeline
In many cases, vasomotor symptoms such as hot flashes occur years before the skin begins to atrophy in the vulva and vagina, particularly in women who enter menopause naturally. Among menopausal women who continue to have intercourse on a regular basis, however, these skin changes often are much less troublesome than they are for women who have sex more rarely.
In this patient, one possible scenario is that paroxetine caused a slight reduction in sexual interest, and the frequency of intercourse went down as a result. In women who have little or no intercourse, the vagina begins to shrink and the tissues lose elasticity. This patient may have been undergoing the natural process of menopause, and that process may have been compounded by a decrease in the frequency of sex.
If you were to discontinue the paroxetine, it would still be necessary to treat the vulvovaginal skin and work on manual techniques to gently dilate the introitus.
Option 1: Systemic hormone therapy
Systemic estrogen is the most effective treatment for menopausal vasomotor symptoms, reducing hot flashes by 50% to 100% within 4 weeks of initiation. However, because our patient has an intact uterus, any systemic estrogen she opts to use must be opposed by a progestin for safety reasons.
In terms of estrogen, her options are oral or nonoral formulations. Not only would estrogen manage our patient’s hot flashes but, over time, it would improve her sexual problems and atrophy, which might or might not improve her current complaint of low desire. You likely would need to add a short regimen of topical estrogen and perhaps even a dilator to restore her sexual function completely, however.
Since our patient chose the nonhormonal agent Brisdelle to manage her menopausal symptoms, she may be worried about the increased risk of breast cancer associated with use of a progestin in combination with estrogen. One hormonal option now available that eliminates the need for a progestin is conjugated estrogens and bazedoxefine (Duavee). Bazedoxefine is a third-
generation selective estrogen receptor modulator (SERM). This drug has estrogen-like effects on bone and antiestrogen effects on the uterus.
Duavee is indicated for use in women with a uterus for treatment of:
- moderate to severe vasomotor symptoms of menopause
- prevention of postmenopausal osteoporosis.
Among the risks are an increased risk of venous thromboembolism (VTE) and stroke. It is not approved specifically for the treatment of dyspareunia.
Another hormonal option is ospemifene (Osphena), an estrogen agonist/antagonist indicated for the treatment of moderate to severe dyspareunia in menopausal women. Among the drugs in its class, such as tamoxifen and raloxifene, ospemifene is the only agent that maintains a full estrogenic effect on vaginal tissues. Its risks include VTE and stroke.
Although the labeling includes a warning about the risk of endometrial hyperplasia associated with its use, Goldstein and colleagues found no significant difference in the rate of endometrial thickening greater than 5 mm between women taking ospemifene and those taking placebo after 1 year of daily oral treatment. No carcinomas were found in either group.
Option 2: Local estrogen
If our patient declines all systemic hormone therapy, the topical approach should resolve her vulvovaginal symptoms, and she could continue taking Brisdelle for her menopausal symptoms. Vaginal estrogen would address the skin problems, provided the patient applies it correctly. Many women are afraid to use estrogen creams and compensate by applying them only to the vulva, thinking that, by limiting their use to external tissues, they are avoiding any associated risks.
If she opts for the local approach, this patient should be encouraged to use transvaginal estrogen in small doses to increase the elasticity of the vulvovaginal tissue, even though it may require daily use for a week or two to improve her symptoms, after which once- or twice-weekly administration should suffice.
The use of low-dose vaginal cream for a short duration is unlikely to increase her risks in any way.
Local estrogen is available as a tablet, cream, or ring.
Option 3: A nonhormonal approach
If the patient refuses any hormonal agent—even topical estrogen—I would recommend the use of silicone-based lubricants and a dilator and prescribe more frequent penetration to increase elasticity and reduce pain.
Brisdelle could be continued to address her menopausal symptoms.
Don’t overlook behavioral techniques
Before this patient leaves your office with the option of her choice, a bit of counseling is necessary to instruct her about methods of restoring full sexual function.
Pain is a powerful aversive stimulus. This patient clearly states that she has had less frequent intercourse as a result of dyspareunia. It is not unusual for patients to develop a “habit” of avoidance in response to the behavior that causes their pain.
One recommendation is to talk to this patient about putting sex back into her life by encouraging her to increase sexual activity without penetration until she begins to arouse easily again. Arousal produces physiologic effects, increasing the caliber and length of the vagina as well as lubrication. The use of fingers or dilators may help restore caliber.
The patient can be encouraged to engage in snuggling and cuddling to regain those activities without the fear of pain associated with penetration. Follow-up after 2 weeks of this therapy can confirm the restoration of tissue elasticity, and the green light can be given for penetration to begin again. Couples can be encouraged to plan a “honeymoon weekend” and put some fun back into their sex lives so that this phase of healing doesn’t become an onerous task.
CASE RESOLVED After a discussion of her options, the patient chooses to stick with Brisdelle and use behavioral therapy alone to resolve her dyspareunia. At her follow-up visit 2 weeks later, she reports that she has enjoyed the period of pain-free “sex” and feels ready to add penetration into her activities.
You encourage her to continue sexual intercourse on a regular, relatively frequent basis to prevent a recurrence of dyspareunia. She continues to use silicone-based lubricants.
Share your thoughts on this article! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.

Barbara S. Levy, MD
Dr. Levy is Vice President of Health Policy for the American College of Obstetricians and Gynecologists.
The author reports no financial relationships relevant to this article.
Barbara S. Levy, MD
Dr. Levy is Vice President of Health Policy for the American College of Obstetricians and Gynecologists.
The author reports no financial relationships relevant to this article.
Barbara S. Levy, MD
Dr. Levy is Vice President of Health Policy for the American College of Obstetricians and Gynecologists.
The author reports no financial relationships relevant to this article.
Since the last installment of this Update on Sexual Dysfunction, three new drugs have been added to the armamentarium for menopausal symptoms and dyspareunia:
- paroxetine 7.5 mg (Brisdelle)
- conjugated estrogens and bazedoxifene (Duavee)
- ospemifene (Osphena).
In this article, I present a case-based approach to incorporating these drugs into practice and restoring sexual function in the setting of vulvovaginal atrophy and dyspareunia. As is often the case, decision-making requires sifting through multiple layers of information.
How to “tease out” the problem and help the patient regain sexual function
Simon JA, Portman DJ, Kazempour K, Mekonnen H, Bhaskar S, Lippman J. Safety profile of paroxetine 7.5 mg in women with moderate-to-severe vasomotor symptoms. Obstet Gynecol. 2014;123(suppl 1):132S–133S.
Conjugated estrogens/bazedoxifene (Duavee) for menopausal symptoms and prevention of osteoporosis. Med Lett Drugs Ther. 2014;56(1441):33–34.
DeGregorio MW, Zerbe RL, Wurz GT. Ospemifene: a first-in-class, non-hormonal selective estrogen receptor modulator approved for the treatment of dyspareunia associated with vulvar and vaginal atrophy [published online ahead of print August 1, 2014]. Steroids. doi:10.1016/j.steroids.2014.07.012.
Goldstein SR, Archer DF, Simon JA, Constantine G. Endometrial safety of ospemifene and the ability of transvaginal ultrasonography to detect small changes in endometrial thickness. Obstet Gynecol. 2014;123(suppl 1):96S–97S.
CASE: LOW DESIRE AND DISCOMFORT DURING INTERCOURSE
Your 58-year-old patient, G2P2, mentions during her annual visit that she’s not that interested in sex anymore. Her children are grown, she’s been happily married for 28 years, and she enjoys her job and denies any symptoms of depression. She says her relationship with her husband is good and, aside from her low desire, she has no worries about the marriage. Her only medication is paroxetine 7.5 mg/day (Brisdelle) for management of her moderate hot flashes, which she initiated at her last annual visit. She reports improvement in her sleep and menopausal symptoms as a result. She has an intact uterus.
You perform a pelvic exam and find atrophic vulva and vagina with mild erythema, and thinned epithelium. When you ask if she has experienced any discomfort, she reports that she needs to use lubrication for intercourse and that, even with lubrication, she has pain upon penetration and a burning sensation that continues throughout intercourse. She also reports that it seems to take her much longer to achieve arousal than in the past, and she often fails to reach orgasm.
How would you manage this patient?
As always, begin with the history
The transition to menopause creates multiple layers of potential symptoms and problems for our patients, and sometimes medical therapy can generate additional ones.
In a patient reporting the onset of low desire and dyspareunia, you would want to first consider her medication history, despite the clear evidence of vaginal atrophy. Begin by asking whether she is taking any new medications prescribed by another provider. In some cases, antihypertensive drugs, psychotropic agents, and other medications can affect sexual function.
This patient has been taking Brisdelle for 1 year and is happy with its effect on her sleep and hot flashes. Simon and colleagues found this nonhormonal agent for moderate to severe vasomotor symptoms to produce no notable effects in weight, libido, or sleep, compared with placebo.
Nevertheless, in this case, because selective serotonin reuptake inhibitors (SSRIs) such as paroxetine can affect arousal and orgasm, it is unclear whether the ultra-low dose of paroxetine she is taking is contributing to her problems. If you were to discontinue the drug to find out, her vasomotor symptoms and sleep disruption would likely recur.
Your decision-making is important here and should involve the patient in an extensive discussion. If there is not enough time for this discussion at the current visit, schedule a follow-up to address her issues fully.
Vulvovaginal atrophy has its own timeline
In many cases, vasomotor symptoms such as hot flashes occur years before the skin begins to atrophy in the vulva and vagina, particularly in women who enter menopause naturally. Among menopausal women who continue to have intercourse on a regular basis, however, these skin changes often are much less troublesome than they are for women who have sex more rarely.
In this patient, one possible scenario is that paroxetine caused a slight reduction in sexual interest, and the frequency of intercourse went down as a result. In women who have little or no intercourse, the vagina begins to shrink and the tissues lose elasticity. This patient may have been undergoing the natural process of menopause, and that process may have been compounded by a decrease in the frequency of sex.
If you were to discontinue the paroxetine, it would still be necessary to treat the vulvovaginal skin and work on manual techniques to gently dilate the introitus.
Option 1: Systemic hormone therapy
Systemic estrogen is the most effective treatment for menopausal vasomotor symptoms, reducing hot flashes by 50% to 100% within 4 weeks of initiation. However, because our patient has an intact uterus, any systemic estrogen she opts to use must be opposed by a progestin for safety reasons.
In terms of estrogen, her options are oral or nonoral formulations. Not only would estrogen manage our patient’s hot flashes but, over time, it would improve her sexual problems and atrophy, which might or might not improve her current complaint of low desire. You likely would need to add a short regimen of topical estrogen and perhaps even a dilator to restore her sexual function completely, however.
Since our patient chose the nonhormonal agent Brisdelle to manage her menopausal symptoms, she may be worried about the increased risk of breast cancer associated with use of a progestin in combination with estrogen. One hormonal option now available that eliminates the need for a progestin is conjugated estrogens and bazedoxefine (Duavee). Bazedoxefine is a third-
generation selective estrogen receptor modulator (SERM). This drug has estrogen-like effects on bone and antiestrogen effects on the uterus.
Duavee is indicated for use in women with a uterus for treatment of:
- moderate to severe vasomotor symptoms of menopause
- prevention of postmenopausal osteoporosis.
Among the risks are an increased risk of venous thromboembolism (VTE) and stroke. It is not approved specifically for the treatment of dyspareunia.
Another hormonal option is ospemifene (Osphena), an estrogen agonist/antagonist indicated for the treatment of moderate to severe dyspareunia in menopausal women. Among the drugs in its class, such as tamoxifen and raloxifene, ospemifene is the only agent that maintains a full estrogenic effect on vaginal tissues. Its risks include VTE and stroke.
Although the labeling includes a warning about the risk of endometrial hyperplasia associated with its use, Goldstein and colleagues found no significant difference in the rate of endometrial thickening greater than 5 mm between women taking ospemifene and those taking placebo after 1 year of daily oral treatment. No carcinomas were found in either group.
Option 2: Local estrogen
If our patient declines all systemic hormone therapy, the topical approach should resolve her vulvovaginal symptoms, and she could continue taking Brisdelle for her menopausal symptoms. Vaginal estrogen would address the skin problems, provided the patient applies it correctly. Many women are afraid to use estrogen creams and compensate by applying them only to the vulva, thinking that, by limiting their use to external tissues, they are avoiding any associated risks.
If she opts for the local approach, this patient should be encouraged to use transvaginal estrogen in small doses to increase the elasticity of the vulvovaginal tissue, even though it may require daily use for a week or two to improve her symptoms, after which once- or twice-weekly administration should suffice.
The use of low-dose vaginal cream for a short duration is unlikely to increase her risks in any way.
Local estrogen is available as a tablet, cream, or ring.
Option 3: A nonhormonal approach
If the patient refuses any hormonal agent—even topical estrogen—I would recommend the use of silicone-based lubricants and a dilator and prescribe more frequent penetration to increase elasticity and reduce pain.
Brisdelle could be continued to address her menopausal symptoms.
Don’t overlook behavioral techniques
Before this patient leaves your office with the option of her choice, a bit of counseling is necessary to instruct her about methods of restoring full sexual function.
Pain is a powerful aversive stimulus. This patient clearly states that she has had less frequent intercourse as a result of dyspareunia. It is not unusual for patients to develop a “habit” of avoidance in response to the behavior that causes their pain.
One recommendation is to talk to this patient about putting sex back into her life by encouraging her to increase sexual activity without penetration until she begins to arouse easily again. Arousal produces physiologic effects, increasing the caliber and length of the vagina as well as lubrication. The use of fingers or dilators may help restore caliber.
The patient can be encouraged to engage in snuggling and cuddling to regain those activities without the fear of pain associated with penetration. Follow-up after 2 weeks of this therapy can confirm the restoration of tissue elasticity, and the green light can be given for penetration to begin again. Couples can be encouraged to plan a “honeymoon weekend” and put some fun back into their sex lives so that this phase of healing doesn’t become an onerous task.
CASE RESOLVED After a discussion of her options, the patient chooses to stick with Brisdelle and use behavioral therapy alone to resolve her dyspareunia. At her follow-up visit 2 weeks later, she reports that she has enjoyed the period of pain-free “sex” and feels ready to add penetration into her activities.
You encourage her to continue sexual intercourse on a regular, relatively frequent basis to prevent a recurrence of dyspareunia. She continues to use silicone-based lubricants.
Share your thoughts on this article! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Since the last installment of this Update on Sexual Dysfunction, three new drugs have been added to the armamentarium for menopausal symptoms and dyspareunia:
- paroxetine 7.5 mg (Brisdelle)
- conjugated estrogens and bazedoxifene (Duavee)
- ospemifene (Osphena).
In this article, I present a case-based approach to incorporating these drugs into practice and restoring sexual function in the setting of vulvovaginal atrophy and dyspareunia. As is often the case, decision-making requires sifting through multiple layers of information.
How to “tease out” the problem and help the patient regain sexual function
Simon JA, Portman DJ, Kazempour K, Mekonnen H, Bhaskar S, Lippman J. Safety profile of paroxetine 7.5 mg in women with moderate-to-severe vasomotor symptoms. Obstet Gynecol. 2014;123(suppl 1):132S–133S.
Conjugated estrogens/bazedoxifene (Duavee) for menopausal symptoms and prevention of osteoporosis. Med Lett Drugs Ther. 2014;56(1441):33–34.
DeGregorio MW, Zerbe RL, Wurz GT. Ospemifene: a first-in-class, non-hormonal selective estrogen receptor modulator approved for the treatment of dyspareunia associated with vulvar and vaginal atrophy [published online ahead of print August 1, 2014]. Steroids. doi:10.1016/j.steroids.2014.07.012.
Goldstein SR, Archer DF, Simon JA, Constantine G. Endometrial safety of ospemifene and the ability of transvaginal ultrasonography to detect small changes in endometrial thickness. Obstet Gynecol. 2014;123(suppl 1):96S–97S.
CASE: LOW DESIRE AND DISCOMFORT DURING INTERCOURSE
Your 58-year-old patient, G2P2, mentions during her annual visit that she’s not that interested in sex anymore. Her children are grown, she’s been happily married for 28 years, and she enjoys her job and denies any symptoms of depression. She says her relationship with her husband is good and, aside from her low desire, she has no worries about the marriage. Her only medication is paroxetine 7.5 mg/day (Brisdelle) for management of her moderate hot flashes, which she initiated at her last annual visit. She reports improvement in her sleep and menopausal symptoms as a result. She has an intact uterus.
You perform a pelvic exam and find atrophic vulva and vagina with mild erythema, and thinned epithelium. When you ask if she has experienced any discomfort, she reports that she needs to use lubrication for intercourse and that, even with lubrication, she has pain upon penetration and a burning sensation that continues throughout intercourse. She also reports that it seems to take her much longer to achieve arousal than in the past, and she often fails to reach orgasm.
How would you manage this patient?
As always, begin with the history
The transition to menopause creates multiple layers of potential symptoms and problems for our patients, and sometimes medical therapy can generate additional ones.
In a patient reporting the onset of low desire and dyspareunia, you would want to first consider her medication history, despite the clear evidence of vaginal atrophy. Begin by asking whether she is taking any new medications prescribed by another provider. In some cases, antihypertensive drugs, psychotropic agents, and other medications can affect sexual function.
This patient has been taking Brisdelle for 1 year and is happy with its effect on her sleep and hot flashes. Simon and colleagues found this nonhormonal agent for moderate to severe vasomotor symptoms to produce no notable effects in weight, libido, or sleep, compared with placebo.
Nevertheless, in this case, because selective serotonin reuptake inhibitors (SSRIs) such as paroxetine can affect arousal and orgasm, it is unclear whether the ultra-low dose of paroxetine she is taking is contributing to her problems. If you were to discontinue the drug to find out, her vasomotor symptoms and sleep disruption would likely recur.
Your decision-making is important here and should involve the patient in an extensive discussion. If there is not enough time for this discussion at the current visit, schedule a follow-up to address her issues fully.
Vulvovaginal atrophy has its own timeline
In many cases, vasomotor symptoms such as hot flashes occur years before the skin begins to atrophy in the vulva and vagina, particularly in women who enter menopause naturally. Among menopausal women who continue to have intercourse on a regular basis, however, these skin changes often are much less troublesome than they are for women who have sex more rarely.
In this patient, one possible scenario is that paroxetine caused a slight reduction in sexual interest, and the frequency of intercourse went down as a result. In women who have little or no intercourse, the vagina begins to shrink and the tissues lose elasticity. This patient may have been undergoing the natural process of menopause, and that process may have been compounded by a decrease in the frequency of sex.
If you were to discontinue the paroxetine, it would still be necessary to treat the vulvovaginal skin and work on manual techniques to gently dilate the introitus.
Option 1: Systemic hormone therapy
Systemic estrogen is the most effective treatment for menopausal vasomotor symptoms, reducing hot flashes by 50% to 100% within 4 weeks of initiation. However, because our patient has an intact uterus, any systemic estrogen she opts to use must be opposed by a progestin for safety reasons.
In terms of estrogen, her options are oral or nonoral formulations. Not only would estrogen manage our patient’s hot flashes but, over time, it would improve her sexual problems and atrophy, which might or might not improve her current complaint of low desire. You likely would need to add a short regimen of topical estrogen and perhaps even a dilator to restore her sexual function completely, however.
Since our patient chose the nonhormonal agent Brisdelle to manage her menopausal symptoms, she may be worried about the increased risk of breast cancer associated with use of a progestin in combination with estrogen. One hormonal option now available that eliminates the need for a progestin is conjugated estrogens and bazedoxefine (Duavee). Bazedoxefine is a third-
generation selective estrogen receptor modulator (SERM). This drug has estrogen-like effects on bone and antiestrogen effects on the uterus.
Duavee is indicated for use in women with a uterus for treatment of:
- moderate to severe vasomotor symptoms of menopause
- prevention of postmenopausal osteoporosis.
Among the risks are an increased risk of venous thromboembolism (VTE) and stroke. It is not approved specifically for the treatment of dyspareunia.
Another hormonal option is ospemifene (Osphena), an estrogen agonist/antagonist indicated for the treatment of moderate to severe dyspareunia in menopausal women. Among the drugs in its class, such as tamoxifen and raloxifene, ospemifene is the only agent that maintains a full estrogenic effect on vaginal tissues. Its risks include VTE and stroke.
Although the labeling includes a warning about the risk of endometrial hyperplasia associated with its use, Goldstein and colleagues found no significant difference in the rate of endometrial thickening greater than 5 mm between women taking ospemifene and those taking placebo after 1 year of daily oral treatment. No carcinomas were found in either group.
Option 2: Local estrogen
If our patient declines all systemic hormone therapy, the topical approach should resolve her vulvovaginal symptoms, and she could continue taking Brisdelle for her menopausal symptoms. Vaginal estrogen would address the skin problems, provided the patient applies it correctly. Many women are afraid to use estrogen creams and compensate by applying them only to the vulva, thinking that, by limiting their use to external tissues, they are avoiding any associated risks.
If she opts for the local approach, this patient should be encouraged to use transvaginal estrogen in small doses to increase the elasticity of the vulvovaginal tissue, even though it may require daily use for a week or two to improve her symptoms, after which once- or twice-weekly administration should suffice.
The use of low-dose vaginal cream for a short duration is unlikely to increase her risks in any way.
Local estrogen is available as a tablet, cream, or ring.
Option 3: A nonhormonal approach
If the patient refuses any hormonal agent—even topical estrogen—I would recommend the use of silicone-based lubricants and a dilator and prescribe more frequent penetration to increase elasticity and reduce pain.
Brisdelle could be continued to address her menopausal symptoms.
Don’t overlook behavioral techniques
Before this patient leaves your office with the option of her choice, a bit of counseling is necessary to instruct her about methods of restoring full sexual function.
Pain is a powerful aversive stimulus. This patient clearly states that she has had less frequent intercourse as a result of dyspareunia. It is not unusual for patients to develop a “habit” of avoidance in response to the behavior that causes their pain.
One recommendation is to talk to this patient about putting sex back into her life by encouraging her to increase sexual activity without penetration until she begins to arouse easily again. Arousal produces physiologic effects, increasing the caliber and length of the vagina as well as lubrication. The use of fingers or dilators may help restore caliber.
The patient can be encouraged to engage in snuggling and cuddling to regain those activities without the fear of pain associated with penetration. Follow-up after 2 weeks of this therapy can confirm the restoration of tissue elasticity, and the green light can be given for penetration to begin again. Couples can be encouraged to plan a “honeymoon weekend” and put some fun back into their sex lives so that this phase of healing doesn’t become an onerous task.
CASE RESOLVED After a discussion of her options, the patient chooses to stick with Brisdelle and use behavioral therapy alone to resolve her dyspareunia. At her follow-up visit 2 weeks later, she reports that she has enjoyed the period of pain-free “sex” and feels ready to add penetration into her activities.
You encourage her to continue sexual intercourse on a regular, relatively frequent basis to prevent a recurrence of dyspareunia. She continues to use silicone-based lubricants.
Share your thoughts on this article! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Laparoscopic dual-port contained power morcellation: An offered solution
Minimally invasive surgery utilizing laparoscopy for hysterectomy and myomectomy has become more common in women with gynecologic pathology. The benefits of this approach compared with laparotomy include decreased hospital stay, shorter recovery and, in experienced hands, significantly decreased morbidity.1–3
Approximately 600,000 hysterectomies are performed annually in the United States—30% of which are performed laparoscopically.4 The primary indication for surgical intervention is uterine leiomyoma. This pathology accounts for 40% of procedures.5 During these surgeries, electromechanical morcellation (EMM), or open “power” morcellation, is commonly used to cut large tissue specimens into small pieces for removal and thereby avoid a larger incision. Concerns have been raised regarding the use of open power morcellation because of the risk of spreading an unrecognized malignancy.
Based on case reports and retrospective studies, the FDA issued a statement in April of this year discouraging the use of EMM for hysterectomy and myomectomy in women with uterine fibroids.6 The concern for inadvertent spread of an occult malignancy was the reasoning for the communication. Since that time, the FDA’s Obstetrics and Gynecology Devices Panel of the Medical Devices Advisory Committee held a public meeting in which the panel heard comments from patients, societies, and industry regarding their positions on the safety of laparoscopic power morcellation. The panel made several recommendations to the FDA but, at the time of this writing, the FDA has yet to issue a final decision.
Reaction to FDA’s action/inaction
The FDA’s “safety” communication was in response to the concern of a few who experienced a bad outcome believed to be secondary to open power morcellation of enlarged uteri or fibroid tumors. In its statement, the FDA estimated the risk of an occult sarcoma to be about 1 in 350 and stated that the risk of disseminating a sarcoma with morcellation is substantial. The FDA discouraged the use of the power morcellator during hysterectomy or myomectomy for uterine fibroids.
Many organizations, including the Society of Gynecologic Oncology, The American Association of Gynecologic Laparoscopists (AAGL), and the American College of Obstetricians and Gynecologists, issued less stringent statements regarding this technology.7–9 These organizations stated generally that there were too few data to make a statement at that time, advocated the collection of more data, and encouraged detailed informed consent to be given to patients undergoing these procedures.
However, the FDA’s statement, and lack of a timely follow-up to clarify the role of the laparoscopic power morcellator in gynecologic surgery, has effectively stopped the use of this technology in its current form. In fact, in response to the statement, Ethicon Endosurgery has discontinued the distribution and sales of its power morcellator and many institutions have severely or completely restricted the use of this technology. The reason for these restrictions is that the medicolegal consequences of an adverse outcome would be very difficult to defend given the current, albeit premature, recommendations of the FDA. This statement makes it difficult to defend any adverse outcome that may occur in association with the use of the laparoscopic power morcellator. Furthermore, this statement by the FDA has largely prevented the medical community at large from collecting additional useful information to allow for a data-driven determination.
Power morcellation is not without risks. In fact, we outline them in this article. However, we believe that minimally invasive surgery should be allowed to continue to advance. In that vein, here we describe a technique of dual-port contained EMM. This surgical approach is performed under direct visualization—which solves the problem of poor visualization that hinders other contained EMM techniques.
Risks of power morcellation
The potential for inadvertent spread of occult malignancy is not the only risk of open EMM. Reports of disseminated leiomyomatosis, adenomyosis, and endometriosis also have been described from inadvertent tissue dispersion during open EMM with resulting ectopic reperitonealization.10–12
The procedure itself is not without risks. A recent systematic review documented 55 major and minor complications from EMM.13 Multiple organ systems were injured including bowel, urinary, vascular, and others, resulting in six deaths from these complications. The investigators concluded that “laparoscopic morcellator–related injuries continue to increase and short- and long-term complications are emerging in both the medical literature and device-related databases. Surgeon inexperience is descriptively identified as one of the most common contributing factors.”
All of the above risks must be weighed against the known benefits of laparoscopic surgery and presented to each patient to assist in deciding which route of surgery should be performed.
Tissue extraction options for large specimens
Large specimen extraction options during gynecologic surgery include:
Vaginal coring. Delivery through the vagina or colpotomy during vaginal or laparoscopic hysterectomy uses the technique of coring, which has long been established in our field.
Manual morcellation through a single incision. Mini-laparotomy or laparoendoscopic single-site surgery (LESS) incisions provide another option of removal with manual morcellation after laparoscopic hysterectomy or myomectomy. One study revealed that specimens up to 22 weeks in size can be placed in a large EndoCatch bag and morcellated extracorporeally by circumferentially coring with a scalpel.14
Contained power morcellation through a single port. Finally, the technique of contained EMM was recently described.15 This technique uses a large containment bag placed through a LESS incision with EMM being performed in an artificially created pneumoperitoneum. This technique isolates the specimen so that it can be morcellated without risk of exposing the patient to any malignant cells that might be unrecognized within the specimen.
Each of these techniques allows many patients to consider a minimally invasive option for their surgery. However, the ability to safely morcellate a very large uterus or myoma may be limited by visualization, and the experience of the surgeon is often critical in the successful performance of these procedures.16
Therefore, at Washington Universitywe have developed a technique using dual ports, with isolation of the uterus or myomas to improve visualization and prevent spillage of malignant tumor or dispersion of other benign tissue.
Dual-port EMM: Technique, tips, and tricks
Our technique of dual-port contained EMM allows the removal of large fibroids or uteri much larger than 20 weeks in size safely under direct visualization through a 15-mm incision. The technique uses:
- Karl Storz Rotocut tissue morcellator with spacers (FIGURE 1)
- 15-mm trocar
- 5-mm balloon trocar
- 20320-inch containment bag (FIGURE 2).


Containment bag placement
Once the specimen is free, we place it to the right or left side of the abdomen. The 15-mm trocar is placed through the umbilicus while visualizing from a lateral trocar site. We then fan-fold the containment bag and introduce it through the 15-mm trocar, keeping the bag oriented with the opening anterior (FIGURE 3). The bag is then grasped at the opening along the drawstring with an atraumatic grasper.
Tip: Care must be taken when introducing the bag in order to avoid tearing or making a small hole in it.
The leading edge is then introduced into the deepest part of the pelvis, and the remainder of the bag (left outside of the abdomen) is then fed cephalad into the abdomen.
Once the bag is completely in the abdomen, we orient the bag with the opening as wide as possible. This allows placement of a very large specimen. Once the specimen is within the containment bag, the drawstring is pulled tight and the mouth of the bag is removed through the 15-mm trocar site at the umbilicus.
The abdominal lateral gas port is opened to allow the intra-abdominal pneumoperitoneum to escape. A 5-mm trocar is placed into the bag through the opening at the umbilicus and the containment bag is insufflated with carbon dioxide and the insufflation pressure is set to 30 mm. The laparoscope placed through this trocar allows the artificial pneumoperitoneum being created to be observed (VIDEO).

Tip: The containment bag covers the entire abdominal cavity and should be fully distended. If it does not distend fully, a hole in the bag may be present and the bag must be replaced.
At this point, we place a balloon trocar at the lateral trocar site and into the bag under direct visualization. The balloon tip is inflated and pulled up tightly against the bag and abdominal wall (FIGURE 4). This allows a tight seal so there is no gas leak or spillage of the morcellated specimen. The laparoscope is placed through this trocar and the insufflation tubing is moved to this port.
Morcellator insertion
The morcellator is introduced through the umbilicus under direct visualization using the short morcellator blade in most instances. Spacers are used to set the length of the morcellator within the containment bag. The tip of the morcellator should be approximately 3 cm to 4 cm within the bag but well away from the retroperitoneum. Remember, any bag will be cut easily by the morcellator and should be thought of as peritoneum only and not a tough barrier. Serious injuries could otherwise develop.
At this point, place the patient flat or out of Trendelenburg position. Morcellation may now proceed.
Tip: Morcellation is best performed with the morcellator perpendicular to the abdomen under direct visualization using a 30° laparoscope to optimize the view. Morcellation in this position uses gravity to facilitate “peeling” of the specimen during morcellation and allows for faster removal.
Before removing the morcellator, inspect the containment bag for any large pieces that may have been dispersed during the morcellation process and remove them. Once there are only small fragments remaining, remove the morcellator, allowing the carbon dioxide to escape. Deflate the balloon tip on the trocar.
Now the containment bag with the remaining specimen may be removed through the umbilicus, while simultaneously removing the balloon-tip trocar from the bag.
A safe minimally invasive approach
This technique has allowed us to safely remove specimens larger than 1,500 g while keeping them in a contained environment with no spill of tissue within the abdomen.
Tracking and adaptation needed
The FDA safety communication has severely limited the practice of morcellation in the minimally invasive gynecologic surgical setting. Many hospitals around the country have reacted by placing significant restrictions on the use of EMM or banned it outright. This action may reverse the national trend of increasing rates of laparoscopic hysterectomy and force many practitioners to return to open surgery.
Currently, it is unclear what the true risk of tissue extraction is whether it is performed via EMM or manually. Large national databases including the BOLD database from the Surgical Review Corporation, as well as AAGL, must be utilized to track these cases and their outcomes to guide therapy. In the meantime, in order to continue to offer a minimally invasive approach to gynecologic surgery, new techniques and instrumentation in the operating room will need to be modified to adapt to these new guidelines. This is vital to maintain or even reduce the rates of open hysterectomy and associated morbidity while diminishing the potential risks of inadvertent benign as well as malignant tissue dispersion with tissue extraction.
Share your thoughts on this article! Send your Letter to the Editor to: [email protected]
1. Nieboer TE, Johnson N, Lethaby A, et al. Surgical approach to hysterectomy for benign gynaecological disease. Cochrane Database Syst Rev. 2009;(3):CD003677. doi:10.1002/14651858.CD003677.pub4.
2. Wright KN, Jonsdottir GM, Jorgensen S, Shah N, Einarsson JI. Costs and outcomes of abdominal, vaginal, laparoscopic and robotic hysterectomies. JSLS. 2012;16(4):519–524.
3. Wiser A, Holcroft CA, Tolandi T, Abenhaim HA. Abdominal versus laparoscopic hysterectomies for benign diseases: evaluation of morbidity and mortality among 465,798 cases. Gynecol Surg. 2013;10(2):117–122.
4. Wright JD, Ananth CV, Lewin SN, et al. Robotically assisted vs laparoscopic hysterectomy among women with benign gynecologic disease. JAMA. 2013;309(7):689–698.
5. Whiteman MK, Hillis SD, Jamieson DJ, et al. Inpatient hysterectomy surveillance in the United States, 2000-2004. Am J Obstet Gynecol. 2008;198(1):34.e1–e7.
6. U S Food and Drug Administration. Quantitative assessment of the prevalence of unsuspected uterine sarcoma in women undergoing treatment of uterine fibroids: Summary and key findings. Silver Spring, Maryland: FDA. http://www.fda.gov/downloads/MedicalDevices/Safety/AlertsandNotices/UCM393589.pdf. Published April 17, 2014. Accessed August 19, 2014.
7. Society of Gynecologic Oncology (SGO). SGO Position Statement: Morcellation. https://www.sgo.org/newsroom/position-statements-2/morcellation. Published December 2013. Accessed March 1, 2014.
8. AAGL. Member Update: Disseminated leiomyosarcoma with power morcellation (Update #2). https://www.aagl.org/aaglnews/member-update-disseminated-leiomyosarcoma-with-power-morcellation-update-2/. Published July 11, 2014. Accessed August 19, 2014.
9. American College of Obstetricians and Gynecologists. Power morcellation and occult malignancy in gynecologic surgery. http://www.acog.org/Resources_And_Publications/Task_Force_and_Work_Group_Reports/Power_Morcellation_and_Occult_Malignancy_in_Gynecologic_Surgery. Published May 2014. Accessed August 19, 2014.
10. Sepilian V, Della Badia C. Iatrogenic endometriosis caused by uterine morcellation during a supracervical hysterectomy. Obstet Gynecol. 2003;102(5 Pt 2):1125–1127.
11. Takeda A, Mori M, Sakai K, Mitsui T, Nakamura H. Parasitic peritoneal leiomyomatosis diagnosed 6 years after laparoscopic myomectomy with electric tissue morcellation: report of a case and review of the literature. J Minim Invasive Gynecol. 2007;14(6):770–775.
12. Donnez O, Squifflet J, Leconte I, Jadoul P, Donnez J. Posthysterectomy pelvic adenomyotic masses observed in 8 cases out of a series of 1405 laparoscopic subtotal hysterectomies. J Minim Invasive Gynecol. 2007;14(2):156–160.
13. Milad MP, Milad EA. Laparoscopic morcellator-related complications. J Minim Invasive Gynecol. 2014;21(3):486–491.
14. Serur E, Lakhi N. Laparoscopic hysterectomy with manual morcellation of the uterus: an original technique that permits the safe and quick removal of a large uterus. Am J. Obstet Gynecol. 2011;204(6):566.e1–e2.
15. Shibley KA. Feasibilty of intra-abdominal tissue isolation and extraction with an artificially created pneumoperitoneum, at laparoscopy for gynecologic procedures. J Min Invasive Gynecol. 2012;19(6):S75.
Scott W. Biest, MD, and David G. Mutch, MD

Dr. Biest is Assistant Professor, Department of Obstetrics and Gynecology, and Director, Division of Minimally Invasive Gynecology, Washington University School of Medicine in St Louis, Missouri.

Dr. Mutch is Ira C. and Judith Gall Professor of Obstetrics and Gynecology and Chief of Gynecologic Oncology at Washington University School of Medicine in St. Louis. He serves on the OBG Management Board of Editors.
The authors report no financial relationships relevant to this article.
Scott W. Biest, MD, and David G. Mutch, MD
Dr. Biest is Assistant Professor, Department of Obstetrics and Gynecology, and Director, Division of Minimally Invasive Gynecology, Washington University School of Medicine in St Louis, Missouri.
Dr. Mutch is Ira C. and Judith Gall Professor of Obstetrics and Gynecology and Chief of Gynecologic Oncology at Washington University School of Medicine in St. Louis. He serves on the OBG Management Board of Editors.
The authors report no financial relationships relevant to this article.
Scott W. Biest, MD, and David G. Mutch, MD
Dr. Biest is Assistant Professor, Department of Obstetrics and Gynecology, and Director, Division of Minimally Invasive Gynecology, Washington University School of Medicine in St Louis, Missouri.
Dr. Mutch is Ira C. and Judith Gall Professor of Obstetrics and Gynecology and Chief of Gynecologic Oncology at Washington University School of Medicine in St. Louis. He serves on the OBG Management Board of Editors.
The authors report no financial relationships relevant to this article.
Minimally invasive surgery utilizing laparoscopy for hysterectomy and myomectomy has become more common in women with gynecologic pathology. The benefits of this approach compared with laparotomy include decreased hospital stay, shorter recovery and, in experienced hands, significantly decreased morbidity.1–3
Approximately 600,000 hysterectomies are performed annually in the United States—30% of which are performed laparoscopically.4 The primary indication for surgical intervention is uterine leiomyoma. This pathology accounts for 40% of procedures.5 During these surgeries, electromechanical morcellation (EMM), or open “power” morcellation, is commonly used to cut large tissue specimens into small pieces for removal and thereby avoid a larger incision. Concerns have been raised regarding the use of open power morcellation because of the risk of spreading an unrecognized malignancy.
Based on case reports and retrospective studies, the FDA issued a statement in April of this year discouraging the use of EMM for hysterectomy and myomectomy in women with uterine fibroids.6 The concern for inadvertent spread of an occult malignancy was the reasoning for the communication. Since that time, the FDA’s Obstetrics and Gynecology Devices Panel of the Medical Devices Advisory Committee held a public meeting in which the panel heard comments from patients, societies, and industry regarding their positions on the safety of laparoscopic power morcellation. The panel made several recommendations to the FDA but, at the time of this writing, the FDA has yet to issue a final decision.
Reaction to FDA’s action/inaction
The FDA’s “safety” communication was in response to the concern of a few who experienced a bad outcome believed to be secondary to open power morcellation of enlarged uteri or fibroid tumors. In its statement, the FDA estimated the risk of an occult sarcoma to be about 1 in 350 and stated that the risk of disseminating a sarcoma with morcellation is substantial. The FDA discouraged the use of the power morcellator during hysterectomy or myomectomy for uterine fibroids.
Many organizations, including the Society of Gynecologic Oncology, The American Association of Gynecologic Laparoscopists (AAGL), and the American College of Obstetricians and Gynecologists, issued less stringent statements regarding this technology.7–9 These organizations stated generally that there were too few data to make a statement at that time, advocated the collection of more data, and encouraged detailed informed consent to be given to patients undergoing these procedures.
However, the FDA’s statement, and lack of a timely follow-up to clarify the role of the laparoscopic power morcellator in gynecologic surgery, has effectively stopped the use of this technology in its current form. In fact, in response to the statement, Ethicon Endosurgery has discontinued the distribution and sales of its power morcellator and many institutions have severely or completely restricted the use of this technology. The reason for these restrictions is that the medicolegal consequences of an adverse outcome would be very difficult to defend given the current, albeit premature, recommendations of the FDA. This statement makes it difficult to defend any adverse outcome that may occur in association with the use of the laparoscopic power morcellator. Furthermore, this statement by the FDA has largely prevented the medical community at large from collecting additional useful information to allow for a data-driven determination.
Power morcellation is not without risks. In fact, we outline them in this article. However, we believe that minimally invasive surgery should be allowed to continue to advance. In that vein, here we describe a technique of dual-port contained EMM. This surgical approach is performed under direct visualization—which solves the problem of poor visualization that hinders other contained EMM techniques.
Risks of power morcellation
The potential for inadvertent spread of occult malignancy is not the only risk of open EMM. Reports of disseminated leiomyomatosis, adenomyosis, and endometriosis also have been described from inadvertent tissue dispersion during open EMM with resulting ectopic reperitonealization.10–12
The procedure itself is not without risks. A recent systematic review documented 55 major and minor complications from EMM.13 Multiple organ systems were injured including bowel, urinary, vascular, and others, resulting in six deaths from these complications. The investigators concluded that “laparoscopic morcellator–related injuries continue to increase and short- and long-term complications are emerging in both the medical literature and device-related databases. Surgeon inexperience is descriptively identified as one of the most common contributing factors.”
All of the above risks must be weighed against the known benefits of laparoscopic surgery and presented to each patient to assist in deciding which route of surgery should be performed.
Tissue extraction options for large specimens
Large specimen extraction options during gynecologic surgery include:
Vaginal coring. Delivery through the vagina or colpotomy during vaginal or laparoscopic hysterectomy uses the technique of coring, which has long been established in our field.
Manual morcellation through a single incision. Mini-laparotomy or laparoendoscopic single-site surgery (LESS) incisions provide another option of removal with manual morcellation after laparoscopic hysterectomy or myomectomy. One study revealed that specimens up to 22 weeks in size can be placed in a large EndoCatch bag and morcellated extracorporeally by circumferentially coring with a scalpel.14
Contained power morcellation through a single port. Finally, the technique of contained EMM was recently described.15 This technique uses a large containment bag placed through a LESS incision with EMM being performed in an artificially created pneumoperitoneum. This technique isolates the specimen so that it can be morcellated without risk of exposing the patient to any malignant cells that might be unrecognized within the specimen.
Each of these techniques allows many patients to consider a minimally invasive option for their surgery. However, the ability to safely morcellate a very large uterus or myoma may be limited by visualization, and the experience of the surgeon is often critical in the successful performance of these procedures.16
Therefore, at Washington Universitywe have developed a technique using dual ports, with isolation of the uterus or myomas to improve visualization and prevent spillage of malignant tumor or dispersion of other benign tissue.
Dual-port EMM: Technique, tips, and tricks
Our technique of dual-port contained EMM allows the removal of large fibroids or uteri much larger than 20 weeks in size safely under direct visualization through a 15-mm incision. The technique uses:
- Karl Storz Rotocut tissue morcellator with spacers (FIGURE 1)
- 15-mm trocar
- 5-mm balloon trocar
- 20320-inch containment bag (FIGURE 2).
Containment bag placement
Once the specimen is free, we place it to the right or left side of the abdomen. The 15-mm trocar is placed through the umbilicus while visualizing from a lateral trocar site. We then fan-fold the containment bag and introduce it through the 15-mm trocar, keeping the bag oriented with the opening anterior (FIGURE 3). The bag is then grasped at the opening along the drawstring with an atraumatic grasper.
Tip: Care must be taken when introducing the bag in order to avoid tearing or making a small hole in it.
The leading edge is then introduced into the deepest part of the pelvis, and the remainder of the bag (left outside of the abdomen) is then fed cephalad into the abdomen.
Once the bag is completely in the abdomen, we orient the bag with the opening as wide as possible. This allows placement of a very large specimen. Once the specimen is within the containment bag, the drawstring is pulled tight and the mouth of the bag is removed through the 15-mm trocar site at the umbilicus.
The abdominal lateral gas port is opened to allow the intra-abdominal pneumoperitoneum to escape. A 5-mm trocar is placed into the bag through the opening at the umbilicus and the containment bag is insufflated with carbon dioxide and the insufflation pressure is set to 30 mm. The laparoscope placed through this trocar allows the artificial pneumoperitoneum being created to be observed (VIDEO).
Tip: The containment bag covers the entire abdominal cavity and should be fully distended. If it does not distend fully, a hole in the bag may be present and the bag must be replaced.
At this point, we place a balloon trocar at the lateral trocar site and into the bag under direct visualization. The balloon tip is inflated and pulled up tightly against the bag and abdominal wall (FIGURE 4). This allows a tight seal so there is no gas leak or spillage of the morcellated specimen. The laparoscope is placed through this trocar and the insufflation tubing is moved to this port.
Morcellator insertion
The morcellator is introduced through the umbilicus under direct visualization using the short morcellator blade in most instances. Spacers are used to set the length of the morcellator within the containment bag. The tip of the morcellator should be approximately 3 cm to 4 cm within the bag but well away from the retroperitoneum. Remember, any bag will be cut easily by the morcellator and should be thought of as peritoneum only and not a tough barrier. Serious injuries could otherwise develop.
At this point, place the patient flat or out of Trendelenburg position. Morcellation may now proceed.
Tip: Morcellation is best performed with the morcellator perpendicular to the abdomen under direct visualization using a 30° laparoscope to optimize the view. Morcellation in this position uses gravity to facilitate “peeling” of the specimen during morcellation and allows for faster removal.
Before removing the morcellator, inspect the containment bag for any large pieces that may have been dispersed during the morcellation process and remove them. Once there are only small fragments remaining, remove the morcellator, allowing the carbon dioxide to escape. Deflate the balloon tip on the trocar.
Now the containment bag with the remaining specimen may be removed through the umbilicus, while simultaneously removing the balloon-tip trocar from the bag.
A safe minimally invasive approach
This technique has allowed us to safely remove specimens larger than 1,500 g while keeping them in a contained environment with no spill of tissue within the abdomen.
Tracking and adaptation needed
The FDA safety communication has severely limited the practice of morcellation in the minimally invasive gynecologic surgical setting. Many hospitals around the country have reacted by placing significant restrictions on the use of EMM or banned it outright. This action may reverse the national trend of increasing rates of laparoscopic hysterectomy and force many practitioners to return to open surgery.
Currently, it is unclear what the true risk of tissue extraction is whether it is performed via EMM or manually. Large national databases including the BOLD database from the Surgical Review Corporation, as well as AAGL, must be utilized to track these cases and their outcomes to guide therapy. In the meantime, in order to continue to offer a minimally invasive approach to gynecologic surgery, new techniques and instrumentation in the operating room will need to be modified to adapt to these new guidelines. This is vital to maintain or even reduce the rates of open hysterectomy and associated morbidity while diminishing the potential risks of inadvertent benign as well as malignant tissue dispersion with tissue extraction.
Share your thoughts on this article! Send your Letter to the Editor to: [email protected]
Minimally invasive surgery utilizing laparoscopy for hysterectomy and myomectomy has become more common in women with gynecologic pathology. The benefits of this approach compared with laparotomy include decreased hospital stay, shorter recovery and, in experienced hands, significantly decreased morbidity.1–3
Approximately 600,000 hysterectomies are performed annually in the United States—30% of which are performed laparoscopically.4 The primary indication for surgical intervention is uterine leiomyoma. This pathology accounts for 40% of procedures.5 During these surgeries, electromechanical morcellation (EMM), or open “power” morcellation, is commonly used to cut large tissue specimens into small pieces for removal and thereby avoid a larger incision. Concerns have been raised regarding the use of open power morcellation because of the risk of spreading an unrecognized malignancy.
Based on case reports and retrospective studies, the FDA issued a statement in April of this year discouraging the use of EMM for hysterectomy and myomectomy in women with uterine fibroids.6 The concern for inadvertent spread of an occult malignancy was the reasoning for the communication. Since that time, the FDA’s Obstetrics and Gynecology Devices Panel of the Medical Devices Advisory Committee held a public meeting in which the panel heard comments from patients, societies, and industry regarding their positions on the safety of laparoscopic power morcellation. The panel made several recommendations to the FDA but, at the time of this writing, the FDA has yet to issue a final decision.
Reaction to FDA’s action/inaction
The FDA’s “safety” communication was in response to the concern of a few who experienced a bad outcome believed to be secondary to open power morcellation of enlarged uteri or fibroid tumors. In its statement, the FDA estimated the risk of an occult sarcoma to be about 1 in 350 and stated that the risk of disseminating a sarcoma with morcellation is substantial. The FDA discouraged the use of the power morcellator during hysterectomy or myomectomy for uterine fibroids.
Many organizations, including the Society of Gynecologic Oncology, The American Association of Gynecologic Laparoscopists (AAGL), and the American College of Obstetricians and Gynecologists, issued less stringent statements regarding this technology.7–9 These organizations stated generally that there were too few data to make a statement at that time, advocated the collection of more data, and encouraged detailed informed consent to be given to patients undergoing these procedures.
However, the FDA’s statement, and lack of a timely follow-up to clarify the role of the laparoscopic power morcellator in gynecologic surgery, has effectively stopped the use of this technology in its current form. In fact, in response to the statement, Ethicon Endosurgery has discontinued the distribution and sales of its power morcellator and many institutions have severely or completely restricted the use of this technology. The reason for these restrictions is that the medicolegal consequences of an adverse outcome would be very difficult to defend given the current, albeit premature, recommendations of the FDA. This statement makes it difficult to defend any adverse outcome that may occur in association with the use of the laparoscopic power morcellator. Furthermore, this statement by the FDA has largely prevented the medical community at large from collecting additional useful information to allow for a data-driven determination.
Power morcellation is not without risks. In fact, we outline them in this article. However, we believe that minimally invasive surgery should be allowed to continue to advance. In that vein, here we describe a technique of dual-port contained EMM. This surgical approach is performed under direct visualization—which solves the problem of poor visualization that hinders other contained EMM techniques.
Risks of power morcellation
The potential for inadvertent spread of occult malignancy is not the only risk of open EMM. Reports of disseminated leiomyomatosis, adenomyosis, and endometriosis also have been described from inadvertent tissue dispersion during open EMM with resulting ectopic reperitonealization.10–12
The procedure itself is not without risks. A recent systematic review documented 55 major and minor complications from EMM.13 Multiple organ systems were injured including bowel, urinary, vascular, and others, resulting in six deaths from these complications. The investigators concluded that “laparoscopic morcellator–related injuries continue to increase and short- and long-term complications are emerging in both the medical literature and device-related databases. Surgeon inexperience is descriptively identified as one of the most common contributing factors.”
All of the above risks must be weighed against the known benefits of laparoscopic surgery and presented to each patient to assist in deciding which route of surgery should be performed.
Tissue extraction options for large specimens
Large specimen extraction options during gynecologic surgery include:
Vaginal coring. Delivery through the vagina or colpotomy during vaginal or laparoscopic hysterectomy uses the technique of coring, which has long been established in our field.
Manual morcellation through a single incision. Mini-laparotomy or laparoendoscopic single-site surgery (LESS) incisions provide another option of removal with manual morcellation after laparoscopic hysterectomy or myomectomy. One study revealed that specimens up to 22 weeks in size can be placed in a large EndoCatch bag and morcellated extracorporeally by circumferentially coring with a scalpel.14
Contained power morcellation through a single port. Finally, the technique of contained EMM was recently described.15 This technique uses a large containment bag placed through a LESS incision with EMM being performed in an artificially created pneumoperitoneum. This technique isolates the specimen so that it can be morcellated without risk of exposing the patient to any malignant cells that might be unrecognized within the specimen.
Each of these techniques allows many patients to consider a minimally invasive option for their surgery. However, the ability to safely morcellate a very large uterus or myoma may be limited by visualization, and the experience of the surgeon is often critical in the successful performance of these procedures.16
Therefore, at Washington Universitywe have developed a technique using dual ports, with isolation of the uterus or myomas to improve visualization and prevent spillage of malignant tumor or dispersion of other benign tissue.
Dual-port EMM: Technique, tips, and tricks
Our technique of dual-port contained EMM allows the removal of large fibroids or uteri much larger than 20 weeks in size safely under direct visualization through a 15-mm incision. The technique uses:
- Karl Storz Rotocut tissue morcellator with spacers (FIGURE 1)
- 15-mm trocar
- 5-mm balloon trocar
- 20320-inch containment bag (FIGURE 2).
Containment bag placement
Once the specimen is free, we place it to the right or left side of the abdomen. The 15-mm trocar is placed through the umbilicus while visualizing from a lateral trocar site. We then fan-fold the containment bag and introduce it through the 15-mm trocar, keeping the bag oriented with the opening anterior (FIGURE 3). The bag is then grasped at the opening along the drawstring with an atraumatic grasper.
Tip: Care must be taken when introducing the bag in order to avoid tearing or making a small hole in it.
The leading edge is then introduced into the deepest part of the pelvis, and the remainder of the bag (left outside of the abdomen) is then fed cephalad into the abdomen.
Once the bag is completely in the abdomen, we orient the bag with the opening as wide as possible. This allows placement of a very large specimen. Once the specimen is within the containment bag, the drawstring is pulled tight and the mouth of the bag is removed through the 15-mm trocar site at the umbilicus.
The abdominal lateral gas port is opened to allow the intra-abdominal pneumoperitoneum to escape. A 5-mm trocar is placed into the bag through the opening at the umbilicus and the containment bag is insufflated with carbon dioxide and the insufflation pressure is set to 30 mm. The laparoscope placed through this trocar allows the artificial pneumoperitoneum being created to be observed (VIDEO).
Tip: The containment bag covers the entire abdominal cavity and should be fully distended. If it does not distend fully, a hole in the bag may be present and the bag must be replaced.
At this point, we place a balloon trocar at the lateral trocar site and into the bag under direct visualization. The balloon tip is inflated and pulled up tightly against the bag and abdominal wall (FIGURE 4). This allows a tight seal so there is no gas leak or spillage of the morcellated specimen. The laparoscope is placed through this trocar and the insufflation tubing is moved to this port.
Morcellator insertion
The morcellator is introduced through the umbilicus under direct visualization using the short morcellator blade in most instances. Spacers are used to set the length of the morcellator within the containment bag. The tip of the morcellator should be approximately 3 cm to 4 cm within the bag but well away from the retroperitoneum. Remember, any bag will be cut easily by the morcellator and should be thought of as peritoneum only and not a tough barrier. Serious injuries could otherwise develop.
At this point, place the patient flat or out of Trendelenburg position. Morcellation may now proceed.
Tip: Morcellation is best performed with the morcellator perpendicular to the abdomen under direct visualization using a 30° laparoscope to optimize the view. Morcellation in this position uses gravity to facilitate “peeling” of the specimen during morcellation and allows for faster removal.
Before removing the morcellator, inspect the containment bag for any large pieces that may have been dispersed during the morcellation process and remove them. Once there are only small fragments remaining, remove the morcellator, allowing the carbon dioxide to escape. Deflate the balloon tip on the trocar.
Now the containment bag with the remaining specimen may be removed through the umbilicus, while simultaneously removing the balloon-tip trocar from the bag.
A safe minimally invasive approach
This technique has allowed us to safely remove specimens larger than 1,500 g while keeping them in a contained environment with no spill of tissue within the abdomen.
Tracking and adaptation needed
The FDA safety communication has severely limited the practice of morcellation in the minimally invasive gynecologic surgical setting. Many hospitals around the country have reacted by placing significant restrictions on the use of EMM or banned it outright. This action may reverse the national trend of increasing rates of laparoscopic hysterectomy and force many practitioners to return to open surgery.
Currently, it is unclear what the true risk of tissue extraction is whether it is performed via EMM or manually. Large national databases including the BOLD database from the Surgical Review Corporation, as well as AAGL, must be utilized to track these cases and their outcomes to guide therapy. In the meantime, in order to continue to offer a minimally invasive approach to gynecologic surgery, new techniques and instrumentation in the operating room will need to be modified to adapt to these new guidelines. This is vital to maintain or even reduce the rates of open hysterectomy and associated morbidity while diminishing the potential risks of inadvertent benign as well as malignant tissue dispersion with tissue extraction.
Share your thoughts on this article! Send your Letter to the Editor to: [email protected]
1. Nieboer TE, Johnson N, Lethaby A, et al. Surgical approach to hysterectomy for benign gynaecological disease. Cochrane Database Syst Rev. 2009;(3):CD003677. doi:10.1002/14651858.CD003677.pub4.
2. Wright KN, Jonsdottir GM, Jorgensen S, Shah N, Einarsson JI. Costs and outcomes of abdominal, vaginal, laparoscopic and robotic hysterectomies. JSLS. 2012;16(4):519–524.
3. Wiser A, Holcroft CA, Tolandi T, Abenhaim HA. Abdominal versus laparoscopic hysterectomies for benign diseases: evaluation of morbidity and mortality among 465,798 cases. Gynecol Surg. 2013;10(2):117–122.
4. Wright JD, Ananth CV, Lewin SN, et al. Robotically assisted vs laparoscopic hysterectomy among women with benign gynecologic disease. JAMA. 2013;309(7):689–698.
5. Whiteman MK, Hillis SD, Jamieson DJ, et al. Inpatient hysterectomy surveillance in the United States, 2000-2004. Am J Obstet Gynecol. 2008;198(1):34.e1–e7.
6. U S Food and Drug Administration. Quantitative assessment of the prevalence of unsuspected uterine sarcoma in women undergoing treatment of uterine fibroids: Summary and key findings. Silver Spring, Maryland: FDA. http://www.fda.gov/downloads/MedicalDevices/Safety/AlertsandNotices/UCM393589.pdf. Published April 17, 2014. Accessed August 19, 2014.
7. Society of Gynecologic Oncology (SGO). SGO Position Statement: Morcellation. https://www.sgo.org/newsroom/position-statements-2/morcellation. Published December 2013. Accessed March 1, 2014.
8. AAGL. Member Update: Disseminated leiomyosarcoma with power morcellation (Update #2). https://www.aagl.org/aaglnews/member-update-disseminated-leiomyosarcoma-with-power-morcellation-update-2/. Published July 11, 2014. Accessed August 19, 2014.
9. American College of Obstetricians and Gynecologists. Power morcellation and occult malignancy in gynecologic surgery. http://www.acog.org/Resources_And_Publications/Task_Force_and_Work_Group_Reports/Power_Morcellation_and_Occult_Malignancy_in_Gynecologic_Surgery. Published May 2014. Accessed August 19, 2014.
10. Sepilian V, Della Badia C. Iatrogenic endometriosis caused by uterine morcellation during a supracervical hysterectomy. Obstet Gynecol. 2003;102(5 Pt 2):1125–1127.
11. Takeda A, Mori M, Sakai K, Mitsui T, Nakamura H. Parasitic peritoneal leiomyomatosis diagnosed 6 years after laparoscopic myomectomy with electric tissue morcellation: report of a case and review of the literature. J Minim Invasive Gynecol. 2007;14(6):770–775.
12. Donnez O, Squifflet J, Leconte I, Jadoul P, Donnez J. Posthysterectomy pelvic adenomyotic masses observed in 8 cases out of a series of 1405 laparoscopic subtotal hysterectomies. J Minim Invasive Gynecol. 2007;14(2):156–160.
13. Milad MP, Milad EA. Laparoscopic morcellator-related complications. J Minim Invasive Gynecol. 2014;21(3):486–491.
14. Serur E, Lakhi N. Laparoscopic hysterectomy with manual morcellation of the uterus: an original technique that permits the safe and quick removal of a large uterus. Am J. Obstet Gynecol. 2011;204(6):566.e1–e2.
15. Shibley KA. Feasibilty of intra-abdominal tissue isolation and extraction with an artificially created pneumoperitoneum, at laparoscopy for gynecologic procedures. J Min Invasive Gynecol. 2012;19(6):S75.
1. Nieboer TE, Johnson N, Lethaby A, et al. Surgical approach to hysterectomy for benign gynaecological disease. Cochrane Database Syst Rev. 2009;(3):CD003677. doi:10.1002/14651858.CD003677.pub4.
2. Wright KN, Jonsdottir GM, Jorgensen S, Shah N, Einarsson JI. Costs and outcomes of abdominal, vaginal, laparoscopic and robotic hysterectomies. JSLS. 2012;16(4):519–524.
3. Wiser A, Holcroft CA, Tolandi T, Abenhaim HA. Abdominal versus laparoscopic hysterectomies for benign diseases: evaluation of morbidity and mortality among 465,798 cases. Gynecol Surg. 2013;10(2):117–122.
4. Wright JD, Ananth CV, Lewin SN, et al. Robotically assisted vs laparoscopic hysterectomy among women with benign gynecologic disease. JAMA. 2013;309(7):689–698.
5. Whiteman MK, Hillis SD, Jamieson DJ, et al. Inpatient hysterectomy surveillance in the United States, 2000-2004. Am J Obstet Gynecol. 2008;198(1):34.e1–e7.
6. U S Food and Drug Administration. Quantitative assessment of the prevalence of unsuspected uterine sarcoma in women undergoing treatment of uterine fibroids: Summary and key findings. Silver Spring, Maryland: FDA. http://www.fda.gov/downloads/MedicalDevices/Safety/AlertsandNotices/UCM393589.pdf. Published April 17, 2014. Accessed August 19, 2014.
7. Society of Gynecologic Oncology (SGO). SGO Position Statement: Morcellation. https://www.sgo.org/newsroom/position-statements-2/morcellation. Published December 2013. Accessed March 1, 2014.
8. AAGL. Member Update: Disseminated leiomyosarcoma with power morcellation (Update #2). https://www.aagl.org/aaglnews/member-update-disseminated-leiomyosarcoma-with-power-morcellation-update-2/. Published July 11, 2014. Accessed August 19, 2014.
9. American College of Obstetricians and Gynecologists. Power morcellation and occult malignancy in gynecologic surgery. http://www.acog.org/Resources_And_Publications/Task_Force_and_Work_Group_Reports/Power_Morcellation_and_Occult_Malignancy_in_Gynecologic_Surgery. Published May 2014. Accessed August 19, 2014.
10. Sepilian V, Della Badia C. Iatrogenic endometriosis caused by uterine morcellation during a supracervical hysterectomy. Obstet Gynecol. 2003;102(5 Pt 2):1125–1127.
11. Takeda A, Mori M, Sakai K, Mitsui T, Nakamura H. Parasitic peritoneal leiomyomatosis diagnosed 6 years after laparoscopic myomectomy with electric tissue morcellation: report of a case and review of the literature. J Minim Invasive Gynecol. 2007;14(6):770–775.
12. Donnez O, Squifflet J, Leconte I, Jadoul P, Donnez J. Posthysterectomy pelvic adenomyotic masses observed in 8 cases out of a series of 1405 laparoscopic subtotal hysterectomies. J Minim Invasive Gynecol. 2007;14(2):156–160.
13. Milad MP, Milad EA. Laparoscopic morcellator-related complications. J Minim Invasive Gynecol. 2014;21(3):486–491.
14. Serur E, Lakhi N. Laparoscopic hysterectomy with manual morcellation of the uterus: an original technique that permits the safe and quick removal of a large uterus. Am J. Obstet Gynecol. 2011;204(6):566.e1–e2.
15. Shibley KA. Feasibilty of intra-abdominal tissue isolation and extraction with an artificially created pneumoperitoneum, at laparoscopy for gynecologic procedures. J Min Invasive Gynecol. 2012;19(6):S75.
Estrogen does not cause breast cancer
“OOPHORECTOMY OR SALPINGECTOMY—WHICH MAKES MORE SENSE?”
William H. Parker, MD (March 2014)
Estrogen does not cause breast cancer
Excellent article by Dr. Parker, with one exception—he continues to promulgate the erroneous misconception that estrogen is what causes breast cancer. He repeats the inaccurate early conclusion of the Women’s Health Initiative (WHI) in 2002 that reported an increased risk of breast cancer. Yet review of that early data suggested that progestin was responsible for the increase, because patients taking estrogen alone had a lower breast cancer risk. Indeed, final and comprehensive review of the WHI studies in 2011 indicates that the estrogen-only arm of the study demonstrated lower breast cancer risks than the placebo group; that distinction continued in the post-intervention period as well.
We need to stop perpetuating the misconception that estrogen causes breast cancer, as the idea continues to be a major deterrent for patients to take estrogen during menopause, to the detriment of their health.
Rafael Haciski, MD
Naples, Florida
Dr. Parker responds:
Dr. Haciski is absolutely right about the fact that the estrogen-only arm of the WHI found a lower risk of breast cancer. The point I was trying to make, perhaps unsuccessfully, was that, in response to the 2002 WHI publication findings of an increased risk of breast cancer after estrogen and progesterone administration, the rate of oophorectomy declined. One interpretation of this trend toward ovarian conservation is that it reflected women deciding to keep their own ovarian hormones, rather than take exogenous hormones associated with health-related risks. While the 2004 WHI estrogen-only publication found a trend toward lower breast cancer risk, this association was not fully confirmed until 2012.1 Even now, many women find the WHI estrogen-progestin and the estrogen-only publications confusing. It was not my intention to add to that confusion.
Reference
1. Anderson GL, Chlebowski RT, Aragaki AK, et al. Conjugated equine oestrogen and breast cancer incidence and mortality in postmenopausal women with hysterectomy: extended follow-up of the Women’s Health Initiative randomised placebo-controlled trial. Lancet Oncol. 2012;13(5):476–486.
“UPDATE ON MINIMALLY INVASIVE GYNECOLOGY”
Amy Garcia, MD (April 2014)
Terminology is important: A cesarean scar pregnancy is not an ectopic pregnancy
I read with great interest the excellent “Update on minimally invasive gynecology,” by Amy Garcia, MD, co-member of OBG Management’s Board of Editors. In it she gives an excellent definition and discussion of an increase in the epidemic of cesarean scar defects (CSD). Her update focuses on intermenstrual bleeding when the menstrual blood presumably collects in the defect and comes out externally, and unpredictably, as old dark blood. I strongly agree with how she managed her clinical case, as I too have had success in such cases using the lowest dosed birth control pills.
My concern, however, is for a potentially fatal outcome because of our use of the term “cesarean scar ectopic pregnancy,” which she mentions as being an additional clinical outcome as described in the review article by Tower and colleagues.1 The strictest definition of ectopic pregnancy is “a pregnancy that occurs outside the uterus.” Many clinicians, however, refer to any pregnancy outside the normal endometrial cavity as being “ectopic.” Regardless of the definition used, I am aware of cases when this nomenclature has been responsible (at least in part) for maternal mortality.
Here is a scenario: A clinician gets an imaging report that there is a cesarean scar ectopic pregnancy. The patient is then treated with a methotrexate protocol2 for the ectopic pregnancy, even though the sac size is small and there is no embryo or cardiac activity.
This patient did not actually have a cesarean scar ectopic pregnancy. Ninety-eight percent of ectopic pregnancies are tubal and these are the ones in which methotrexate has been studied adequately and used. Cesarean scar pregnancies (and, in my opinion, cervical pregnancies as well as cornual pregnancies) are very different from “garden variety” tubal ectopic pregnancies, and should not automatically be plugged into existing methotrexate protocols. In my experience, the existing methotrexate protocols do not work for cesarean scar pregnancies, and place these women at risk for potential harm, such as severe hemorrhage.
We should be meticulous in referring to these as cesarean scar pregnancies—not cesarean scar ectopic pregnancies—to help keep well-meaning clinicians from being misled.
Steven R. Goldstein, MD
Professor, Department of Obstetrics and Gynecology
New York University School of Medicine, New York, New York
References
1. Tower AM, Frishman GN. Cesarean scar defects: an underrecognized cause of abnormal uterine bleeding and other gynecologic complications.
J Minim Invasive Gynecol. 2013;20(5):562–572.
2. American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 94: Medical management of ectopic pregnancy. Obstet Gynecol. 2008;111(6):1479–1485.
Dr. Garcia responds:
While I can appreciate the comments from my co-editor, Dr. Goldstein, I believe he is defocusing the real issue regarding pregnancies occurring in scar defects caused by previous cesarean section. Rather than creating an issue with the semantics of “ectopic,” I would have preferred to read that Dr. Goldstein was emphasizing the potentially significant, if not fatal, complications and management of cesarean scar pregnancies as he did with co-authors Dr. Timor-Trisch and Dr. Monteagudo in a recent OBG Management article.1
To clarify the terminology, I used “cesarean scar ectopic pregnancy” as it was quoted from the work of Tower and colleagues.2 This terminology adequately describes the location of the pregnancy, as many cesarean scar defects are not just located in the isthmus of the uterus but in the cervix itself.3 And by all accounts a pregnancy at this location would be considered to be a cervical pregnancy, which ACOG defines as ectopic. The following definition of ectopic pregnancy is taken from Dr. Goldstein’s reference to ACOG Practice Bulletin #94: “Nearly all ectopic pregnancies (97%) are implanted within the fallopian tube, although implantation can occur within the abdomen, cervix, ovary, or uterine cornua.”4 Indeed if ACOG uses “ectopic” to describe a pregnancy within the cervix, then “cesarean scar ectopic pregnancy” should be adequate to describe the location of a potentially dangerous pregnancy.
Dr. Goldstein is concerned that our colleagues may become confused by the terminology “cesarean scar ectopic pregnancy” because the word “ectopic” might denote a less clinically significant scenario. Yet I say that anyone confused by the location of the pregnancy has not understood the part of the descriptive term that is “cesarean scar” regardless of the use of “ectopic.” Any clinician treating a patient with a cesarean scar pregnancy would benefit from Dr. Goldstein and his colleagues’ article for diagnosis and management of this critical and potentially life-threatening scenario.
References
1. Timor-Trisch IE, Monteagudo A, Goldstein SR. How to identify and manage cesarean scar pregnancy. OBG Manag. 2014;26(6):19–27.
2. Tower AM, Frishman GN. Cesarean scar defects: an underrecognized cause of abnormal uterine bleeding and other gynecologic complications. J Minim Invasive Gynecol. 2013;20(5):562–572.
3. Garcia A. Update on minimally invasive gynecology. OBG Manag. 2014;26(4):18–32.
4. American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 94: Medical management of ectopic pregnancy. Obstet Gynecol. 2008;111(6):1479–1485.
Totally painless birth: A new concept in obstetrical anesthesia and labor management
A recent review on epidural anesthesia for pain management during labor, which was published in the New England Journal of Medicine,1 starts with the following case presentation: “A 30-year-old nulliparous woman at 39 weeks gestation is undergoing induction of labor for premature rupture of membranes. She is currently receiving an oxytocin infusion, and her cervical dilatation is 1 cm. Her obstetrician has ordered intermittent intravenous administration of Fentanyl for pain relief, but she feels nauseated, has been unable to rest, and describes her pain as 9 on scale of 10.”
The case we present in this letter sounds totally different: A 26-year-old multiparous woman at 39 weeks’ gestation is scheduled for induction of labor because of a decreased amount of amniotic fluid detected on prenatal ultrasonography. On admission, her cervix is 1-cm dilated and 60% effaced. She has no uterine contractions, and she describes her pain as 0 on the scale of 10. Anticipating a long induction and unwilling to experience any pain, the patient requests and is offered epidural anesthesia prior to oxytocin administration. Labor takes longer than expected but ultimately results in the birth of a healthy child. At no time during the course of labor and delivery does the mother experience any pain or discomfort, and she reports an extremely satisfying experience.
Traditionally, epidural anesthesia is administered upon maternal request when labor pain becomes difficult or intolerable. Many obstetricians discourage the use of epidural anesthesia prior to the onset of the active stage of labor, where the cervix is 4- to 6-cm dilated and the patient is in a great deal of pain.
This concept of administering an analgesic after the onset of pain is very different from the one where anesthesia is provided to patients prior to surgical procedures. No physician would begin a scheduled surgical procedure before confirming administration of adequate anesthesia. Why should labor be any different?
The pain of labor caused by uterine contractions and cervical dilatation is transmitted through visceral afferent nerves entering the spinal cord from T10 through L1. Perineal stretching transmits pain through the pudendal and sacral nerves. Epidural analgesia for labor and delivery involves injection of a local anesthetic agent and an opioid analgesic into the lumbar epidural space. The injected agents gradually diffuse across the dura into the subarachnoid cavity. In spinal analgesia, which is often combined with epidural analgesia, the medication is injected directly into the subarachnoid cavity, resulting in a more rapid effect.2
The American Society of Anesthesiologists and the American College of Obstetrics and Gynecology jointly state, “Labor causes severe pain for many women. There is no other circumstance where it is considered acceptable for an individual to experience severe pain, untreated. In the absence of medical contraindications, maternal request is a sufficient medical indication for pain relief during labor.”3
We decided to stretch this concept by allowing epidural anesthesia to be administered upon maternal consent in anticipation of pain, prior to onset. Our extensive literature search failed to find any comprehensive studies on the use of epidural anesthesia in anticipation of labor pain.
In theory, neuraxial local anesthetic in clinically relevant doses affect only skeletal muscles, not smooth muscles; therefore, this agent should not significantly decrease the amplitude or frequency of contractions in the myometrium.4 Randomized controlled trials of the effects of analgesia during labor do not exist since it is impossible to randomly assign women to a placebo group (no pain relief). Most trials have compared the use of epidural analgesia with that of systemic narcotics. In one large trial, 992 multiparous women were randomly assigned to either epidural analgesia or midwifery support supplemented by intramuscular narcotic injections. When pain was rated on a scale of 0–100 (100 is high), the median score before the study interventions was 80 in the group of patients who did not receive an epidural analgesia. With the administration of epidural analgesia, the median score was 27.5
Clinicians and patients also have been concerned about whether the use of epidural analgesia increases the risk of cesarean delivery. The results of three randomized controlled trials demonstrated that early administration of epidural analgesia does not increase the rate of cesarean delivery among women with spontaneous or induced labor as compared with early initiation of opioids.6–8 The fact that early epidural placement does not appear to increase the incidence of cesarean delivery is encouraging for our ongoing study but doesn’t necessarily mean that epidural analgesia prior to the onset of pain (preventive epidural) will have the same outcome. More research is needed.
Boris M. Petrikovsky, MD, PhD; Elya Kozlov, MD; Matthew Ackert, MD; Ralph Ruggiero, MD; and Crystal Behofsits, DO
Wyckoff Heights Medical Center, Brooklyn NY
References
1. Haukins GL. Epidural analgesia for labor and delivery. N Engl J Med. 2010;362(16):1502–1510.
2. Catterall WA, Mackie K. Local anesthetics. In: Brunton LL, Lazo JS, Parker KL, eds. Goodman & Gilman’s The Pharmacological Basis of Therapeutics. 11th ed. New York, New York: McGraw-Hill; 2005;369–386.
3. American College of Obstetricians and Gynecologists. ACOG Committee Opinion #295: pain relief during labor. Obstet Gynecol. 2004;104(1):213.
4. Fanning RA, Campion DP, Collins CB, et al. A comparison of the inhibitory effects of bupivacaine and levobupivacaine on isolated human pregnant myometrium contractility. Anesth Analg. 2008;107(4):1303–1307.
5. Dickinson JE, Paech MJ, McDonald SJ, Evans SF. Maternal satisfaction with childbirth and intrapartum analgesia in nulliparous labor. Aust N Z J Obstet Gynecol. 2003;43(6):463–468.
6. Wong CA, Scavone BM, Peaceman AM, et al. The risk of cesarean delivery with neuraxial analgesia in labor. N Engl J Med. 2005;352(7):655–665.
7. Ohel G, Gonen R, Vaida S, Barak S, Gaitini L. Early versus late initiation of epidural analgesia in labor: does it increase the risk of cesarean section? A randomized trial. Am J Obstet Gynecol. 2006:194(3):600–605.
8. Wong CA, McCarthy RJ, Sullivan JT, Scavone BM, Gerber SE, Yaghmour EA. Early compared with late neuraxial analgesia in nulliparous labor induction: a randomized controlled trial. Obstet Gynecol. 2009;113(5):1066–1074.
Share your thoughts on this article! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Boris M. Petrikovsky,Elya Kozlov,Matthew Ackert,Ralph Ruggiero,Crystal Behofsits,totally painless birth,obstetrical anesthesia,labor management,labor and delivery,New England Journal of Medicine,epidural anesthesia,pain management during labor,nulliparous,premature rupture of membranes,multiparous,prenatal ultrasonography,uterine contractions,cervical dilation,visceral afferent nerves,American Society of Anesthesiologists,American College of Obstetricians and Gynecologists,
“OOPHORECTOMY OR SALPINGECTOMY—WHICH MAKES MORE SENSE?”
William H. Parker, MD (March 2014)
Estrogen does not cause breast cancer
Excellent article by Dr. Parker, with one exception—he continues to promulgate the erroneous misconception that estrogen is what causes breast cancer. He repeats the inaccurate early conclusion of the Women’s Health Initiative (WHI) in 2002 that reported an increased risk of breast cancer. Yet review of that early data suggested that progestin was responsible for the increase, because patients taking estrogen alone had a lower breast cancer risk. Indeed, final and comprehensive review of the WHI studies in 2011 indicates that the estrogen-only arm of the study demonstrated lower breast cancer risks than the placebo group; that distinction continued in the post-intervention period as well.
We need to stop perpetuating the misconception that estrogen causes breast cancer, as the idea continues to be a major deterrent for patients to take estrogen during menopause, to the detriment of their health.
Rafael Haciski, MD
Naples, Florida
Dr. Parker responds:
Dr. Haciski is absolutely right about the fact that the estrogen-only arm of the WHI found a lower risk of breast cancer. The point I was trying to make, perhaps unsuccessfully, was that, in response to the 2002 WHI publication findings of an increased risk of breast cancer after estrogen and progesterone administration, the rate of oophorectomy declined. One interpretation of this trend toward ovarian conservation is that it reflected women deciding to keep their own ovarian hormones, rather than take exogenous hormones associated with health-related risks. While the 2004 WHI estrogen-only publication found a trend toward lower breast cancer risk, this association was not fully confirmed until 2012.1 Even now, many women find the WHI estrogen-progestin and the estrogen-only publications confusing. It was not my intention to add to that confusion.
Reference
1. Anderson GL, Chlebowski RT, Aragaki AK, et al. Conjugated equine oestrogen and breast cancer incidence and mortality in postmenopausal women with hysterectomy: extended follow-up of the Women’s Health Initiative randomised placebo-controlled trial. Lancet Oncol. 2012;13(5):476–486.
“UPDATE ON MINIMALLY INVASIVE GYNECOLOGY”
Amy Garcia, MD (April 2014)
Terminology is important: A cesarean scar pregnancy is not an ectopic pregnancy
I read with great interest the excellent “Update on minimally invasive gynecology,” by Amy Garcia, MD, co-member of OBG Management’s Board of Editors. In it she gives an excellent definition and discussion of an increase in the epidemic of cesarean scar defects (CSD). Her update focuses on intermenstrual bleeding when the menstrual blood presumably collects in the defect and comes out externally, and unpredictably, as old dark blood. I strongly agree with how she managed her clinical case, as I too have had success in such cases using the lowest dosed birth control pills.
My concern, however, is for a potentially fatal outcome because of our use of the term “cesarean scar ectopic pregnancy,” which she mentions as being an additional clinical outcome as described in the review article by Tower and colleagues.1 The strictest definition of ectopic pregnancy is “a pregnancy that occurs outside the uterus.” Many clinicians, however, refer to any pregnancy outside the normal endometrial cavity as being “ectopic.” Regardless of the definition used, I am aware of cases when this nomenclature has been responsible (at least in part) for maternal mortality.
Here is a scenario: A clinician gets an imaging report that there is a cesarean scar ectopic pregnancy. The patient is then treated with a methotrexate protocol2 for the ectopic pregnancy, even though the sac size is small and there is no embryo or cardiac activity.
This patient did not actually have a cesarean scar ectopic pregnancy. Ninety-eight percent of ectopic pregnancies are tubal and these are the ones in which methotrexate has been studied adequately and used. Cesarean scar pregnancies (and, in my opinion, cervical pregnancies as well as cornual pregnancies) are very different from “garden variety” tubal ectopic pregnancies, and should not automatically be plugged into existing methotrexate protocols. In my experience, the existing methotrexate protocols do not work for cesarean scar pregnancies, and place these women at risk for potential harm, such as severe hemorrhage.
We should be meticulous in referring to these as cesarean scar pregnancies—not cesarean scar ectopic pregnancies—to help keep well-meaning clinicians from being misled.
Steven R. Goldstein, MD
Professor, Department of Obstetrics and Gynecology
New York University School of Medicine, New York, New York
References
1. Tower AM, Frishman GN. Cesarean scar defects: an underrecognized cause of abnormal uterine bleeding and other gynecologic complications.
J Minim Invasive Gynecol. 2013;20(5):562–572.
2. American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 94: Medical management of ectopic pregnancy. Obstet Gynecol. 2008;111(6):1479–1485.
Dr. Garcia responds:
While I can appreciate the comments from my co-editor, Dr. Goldstein, I believe he is defocusing the real issue regarding pregnancies occurring in scar defects caused by previous cesarean section. Rather than creating an issue with the semantics of “ectopic,” I would have preferred to read that Dr. Goldstein was emphasizing the potentially significant, if not fatal, complications and management of cesarean scar pregnancies as he did with co-authors Dr. Timor-Trisch and Dr. Monteagudo in a recent OBG Management article.1
To clarify the terminology, I used “cesarean scar ectopic pregnancy” as it was quoted from the work of Tower and colleagues.2 This terminology adequately describes the location of the pregnancy, as many cesarean scar defects are not just located in the isthmus of the uterus but in the cervix itself.3 And by all accounts a pregnancy at this location would be considered to be a cervical pregnancy, which ACOG defines as ectopic. The following definition of ectopic pregnancy is taken from Dr. Goldstein’s reference to ACOG Practice Bulletin #94: “Nearly all ectopic pregnancies (97%) are implanted within the fallopian tube, although implantation can occur within the abdomen, cervix, ovary, or uterine cornua.”4 Indeed if ACOG uses “ectopic” to describe a pregnancy within the cervix, then “cesarean scar ectopic pregnancy” should be adequate to describe the location of a potentially dangerous pregnancy.
Dr. Goldstein is concerned that our colleagues may become confused by the terminology “cesarean scar ectopic pregnancy” because the word “ectopic” might denote a less clinically significant scenario. Yet I say that anyone confused by the location of the pregnancy has not understood the part of the descriptive term that is “cesarean scar” regardless of the use of “ectopic.” Any clinician treating a patient with a cesarean scar pregnancy would benefit from Dr. Goldstein and his colleagues’ article for diagnosis and management of this critical and potentially life-threatening scenario.
References
1. Timor-Trisch IE, Monteagudo A, Goldstein SR. How to identify and manage cesarean scar pregnancy. OBG Manag. 2014;26(6):19–27.
2. Tower AM, Frishman GN. Cesarean scar defects: an underrecognized cause of abnormal uterine bleeding and other gynecologic complications. J Minim Invasive Gynecol. 2013;20(5):562–572.
3. Garcia A. Update on minimally invasive gynecology. OBG Manag. 2014;26(4):18–32.
4. American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 94: Medical management of ectopic pregnancy. Obstet Gynecol. 2008;111(6):1479–1485.
Totally painless birth: A new concept in obstetrical anesthesia and labor management
A recent review on epidural anesthesia for pain management during labor, which was published in the New England Journal of Medicine,1 starts with the following case presentation: “A 30-year-old nulliparous woman at 39 weeks gestation is undergoing induction of labor for premature rupture of membranes. She is currently receiving an oxytocin infusion, and her cervical dilatation is 1 cm. Her obstetrician has ordered intermittent intravenous administration of Fentanyl for pain relief, but she feels nauseated, has been unable to rest, and describes her pain as 9 on scale of 10.”
The case we present in this letter sounds totally different: A 26-year-old multiparous woman at 39 weeks’ gestation is scheduled for induction of labor because of a decreased amount of amniotic fluid detected on prenatal ultrasonography. On admission, her cervix is 1-cm dilated and 60% effaced. She has no uterine contractions, and she describes her pain as 0 on the scale of 10. Anticipating a long induction and unwilling to experience any pain, the patient requests and is offered epidural anesthesia prior to oxytocin administration. Labor takes longer than expected but ultimately results in the birth of a healthy child. At no time during the course of labor and delivery does the mother experience any pain or discomfort, and she reports an extremely satisfying experience.
Traditionally, epidural anesthesia is administered upon maternal request when labor pain becomes difficult or intolerable. Many obstetricians discourage the use of epidural anesthesia prior to the onset of the active stage of labor, where the cervix is 4- to 6-cm dilated and the patient is in a great deal of pain.
This concept of administering an analgesic after the onset of pain is very different from the one where anesthesia is provided to patients prior to surgical procedures. No physician would begin a scheduled surgical procedure before confirming administration of adequate anesthesia. Why should labor be any different?
The pain of labor caused by uterine contractions and cervical dilatation is transmitted through visceral afferent nerves entering the spinal cord from T10 through L1. Perineal stretching transmits pain through the pudendal and sacral nerves. Epidural analgesia for labor and delivery involves injection of a local anesthetic agent and an opioid analgesic into the lumbar epidural space. The injected agents gradually diffuse across the dura into the subarachnoid cavity. In spinal analgesia, which is often combined with epidural analgesia, the medication is injected directly into the subarachnoid cavity, resulting in a more rapid effect.2
The American Society of Anesthesiologists and the American College of Obstetrics and Gynecology jointly state, “Labor causes severe pain for many women. There is no other circumstance where it is considered acceptable for an individual to experience severe pain, untreated. In the absence of medical contraindications, maternal request is a sufficient medical indication for pain relief during labor.”3
We decided to stretch this concept by allowing epidural anesthesia to be administered upon maternal consent in anticipation of pain, prior to onset. Our extensive literature search failed to find any comprehensive studies on the use of epidural anesthesia in anticipation of labor pain.
In theory, neuraxial local anesthetic in clinically relevant doses affect only skeletal muscles, not smooth muscles; therefore, this agent should not significantly decrease the amplitude or frequency of contractions in the myometrium.4 Randomized controlled trials of the effects of analgesia during labor do not exist since it is impossible to randomly assign women to a placebo group (no pain relief). Most trials have compared the use of epidural analgesia with that of systemic narcotics. In one large trial, 992 multiparous women were randomly assigned to either epidural analgesia or midwifery support supplemented by intramuscular narcotic injections. When pain was rated on a scale of 0–100 (100 is high), the median score before the study interventions was 80 in the group of patients who did not receive an epidural analgesia. With the administration of epidural analgesia, the median score was 27.5
Clinicians and patients also have been concerned about whether the use of epidural analgesia increases the risk of cesarean delivery. The results of three randomized controlled trials demonstrated that early administration of epidural analgesia does not increase the rate of cesarean delivery among women with spontaneous or induced labor as compared with early initiation of opioids.6–8 The fact that early epidural placement does not appear to increase the incidence of cesarean delivery is encouraging for our ongoing study but doesn’t necessarily mean that epidural analgesia prior to the onset of pain (preventive epidural) will have the same outcome. More research is needed.
Boris M. Petrikovsky, MD, PhD; Elya Kozlov, MD; Matthew Ackert, MD; Ralph Ruggiero, MD; and Crystal Behofsits, DO
Wyckoff Heights Medical Center, Brooklyn NY
References
1. Haukins GL. Epidural analgesia for labor and delivery. N Engl J Med. 2010;362(16):1502–1510.
2. Catterall WA, Mackie K. Local anesthetics. In: Brunton LL, Lazo JS, Parker KL, eds. Goodman & Gilman’s The Pharmacological Basis of Therapeutics. 11th ed. New York, New York: McGraw-Hill; 2005;369–386.
3. American College of Obstetricians and Gynecologists. ACOG Committee Opinion #295: pain relief during labor. Obstet Gynecol. 2004;104(1):213.
4. Fanning RA, Campion DP, Collins CB, et al. A comparison of the inhibitory effects of bupivacaine and levobupivacaine on isolated human pregnant myometrium contractility. Anesth Analg. 2008;107(4):1303–1307.
5. Dickinson JE, Paech MJ, McDonald SJ, Evans SF. Maternal satisfaction with childbirth and intrapartum analgesia in nulliparous labor. Aust N Z J Obstet Gynecol. 2003;43(6):463–468.
6. Wong CA, Scavone BM, Peaceman AM, et al. The risk of cesarean delivery with neuraxial analgesia in labor. N Engl J Med. 2005;352(7):655–665.
7. Ohel G, Gonen R, Vaida S, Barak S, Gaitini L. Early versus late initiation of epidural analgesia in labor: does it increase the risk of cesarean section? A randomized trial. Am J Obstet Gynecol. 2006:194(3):600–605.
8. Wong CA, McCarthy RJ, Sullivan JT, Scavone BM, Gerber SE, Yaghmour EA. Early compared with late neuraxial analgesia in nulliparous labor induction: a randomized controlled trial. Obstet Gynecol. 2009;113(5):1066–1074.
Share your thoughts on this article! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
“OOPHORECTOMY OR SALPINGECTOMY—WHICH MAKES MORE SENSE?”
William H. Parker, MD (March 2014)
Estrogen does not cause breast cancer
Excellent article by Dr. Parker, with one exception—he continues to promulgate the erroneous misconception that estrogen is what causes breast cancer. He repeats the inaccurate early conclusion of the Women’s Health Initiative (WHI) in 2002 that reported an increased risk of breast cancer. Yet review of that early data suggested that progestin was responsible for the increase, because patients taking estrogen alone had a lower breast cancer risk. Indeed, final and comprehensive review of the WHI studies in 2011 indicates that the estrogen-only arm of the study demonstrated lower breast cancer risks than the placebo group; that distinction continued in the post-intervention period as well.
We need to stop perpetuating the misconception that estrogen causes breast cancer, as the idea continues to be a major deterrent for patients to take estrogen during menopause, to the detriment of their health.
Rafael Haciski, MD
Naples, Florida
Dr. Parker responds:
Dr. Haciski is absolutely right about the fact that the estrogen-only arm of the WHI found a lower risk of breast cancer. The point I was trying to make, perhaps unsuccessfully, was that, in response to the 2002 WHI publication findings of an increased risk of breast cancer after estrogen and progesterone administration, the rate of oophorectomy declined. One interpretation of this trend toward ovarian conservation is that it reflected women deciding to keep their own ovarian hormones, rather than take exogenous hormones associated with health-related risks. While the 2004 WHI estrogen-only publication found a trend toward lower breast cancer risk, this association was not fully confirmed until 2012.1 Even now, many women find the WHI estrogen-progestin and the estrogen-only publications confusing. It was not my intention to add to that confusion.
Reference
1. Anderson GL, Chlebowski RT, Aragaki AK, et al. Conjugated equine oestrogen and breast cancer incidence and mortality in postmenopausal women with hysterectomy: extended follow-up of the Women’s Health Initiative randomised placebo-controlled trial. Lancet Oncol. 2012;13(5):476–486.
“UPDATE ON MINIMALLY INVASIVE GYNECOLOGY”
Amy Garcia, MD (April 2014)
Terminology is important: A cesarean scar pregnancy is not an ectopic pregnancy
I read with great interest the excellent “Update on minimally invasive gynecology,” by Amy Garcia, MD, co-member of OBG Management’s Board of Editors. In it she gives an excellent definition and discussion of an increase in the epidemic of cesarean scar defects (CSD). Her update focuses on intermenstrual bleeding when the menstrual blood presumably collects in the defect and comes out externally, and unpredictably, as old dark blood. I strongly agree with how she managed her clinical case, as I too have had success in such cases using the lowest dosed birth control pills.
My concern, however, is for a potentially fatal outcome because of our use of the term “cesarean scar ectopic pregnancy,” which she mentions as being an additional clinical outcome as described in the review article by Tower and colleagues.1 The strictest definition of ectopic pregnancy is “a pregnancy that occurs outside the uterus.” Many clinicians, however, refer to any pregnancy outside the normal endometrial cavity as being “ectopic.” Regardless of the definition used, I am aware of cases when this nomenclature has been responsible (at least in part) for maternal mortality.
Here is a scenario: A clinician gets an imaging report that there is a cesarean scar ectopic pregnancy. The patient is then treated with a methotrexate protocol2 for the ectopic pregnancy, even though the sac size is small and there is no embryo or cardiac activity.
This patient did not actually have a cesarean scar ectopic pregnancy. Ninety-eight percent of ectopic pregnancies are tubal and these are the ones in which methotrexate has been studied adequately and used. Cesarean scar pregnancies (and, in my opinion, cervical pregnancies as well as cornual pregnancies) are very different from “garden variety” tubal ectopic pregnancies, and should not automatically be plugged into existing methotrexate protocols. In my experience, the existing methotrexate protocols do not work for cesarean scar pregnancies, and place these women at risk for potential harm, such as severe hemorrhage.
We should be meticulous in referring to these as cesarean scar pregnancies—not cesarean scar ectopic pregnancies—to help keep well-meaning clinicians from being misled.
Steven R. Goldstein, MD
Professor, Department of Obstetrics and Gynecology
New York University School of Medicine, New York, New York
References
1. Tower AM, Frishman GN. Cesarean scar defects: an underrecognized cause of abnormal uterine bleeding and other gynecologic complications.
J Minim Invasive Gynecol. 2013;20(5):562–572.
2. American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 94: Medical management of ectopic pregnancy. Obstet Gynecol. 2008;111(6):1479–1485.
Dr. Garcia responds:
While I can appreciate the comments from my co-editor, Dr. Goldstein, I believe he is defocusing the real issue regarding pregnancies occurring in scar defects caused by previous cesarean section. Rather than creating an issue with the semantics of “ectopic,” I would have preferred to read that Dr. Goldstein was emphasizing the potentially significant, if not fatal, complications and management of cesarean scar pregnancies as he did with co-authors Dr. Timor-Trisch and Dr. Monteagudo in a recent OBG Management article.1
To clarify the terminology, I used “cesarean scar ectopic pregnancy” as it was quoted from the work of Tower and colleagues.2 This terminology adequately describes the location of the pregnancy, as many cesarean scar defects are not just located in the isthmus of the uterus but in the cervix itself.3 And by all accounts a pregnancy at this location would be considered to be a cervical pregnancy, which ACOG defines as ectopic. The following definition of ectopic pregnancy is taken from Dr. Goldstein’s reference to ACOG Practice Bulletin #94: “Nearly all ectopic pregnancies (97%) are implanted within the fallopian tube, although implantation can occur within the abdomen, cervix, ovary, or uterine cornua.”4 Indeed if ACOG uses “ectopic” to describe a pregnancy within the cervix, then “cesarean scar ectopic pregnancy” should be adequate to describe the location of a potentially dangerous pregnancy.
Dr. Goldstein is concerned that our colleagues may become confused by the terminology “cesarean scar ectopic pregnancy” because the word “ectopic” might denote a less clinically significant scenario. Yet I say that anyone confused by the location of the pregnancy has not understood the part of the descriptive term that is “cesarean scar” regardless of the use of “ectopic.” Any clinician treating a patient with a cesarean scar pregnancy would benefit from Dr. Goldstein and his colleagues’ article for diagnosis and management of this critical and potentially life-threatening scenario.
References
1. Timor-Trisch IE, Monteagudo A, Goldstein SR. How to identify and manage cesarean scar pregnancy. OBG Manag. 2014;26(6):19–27.
2. Tower AM, Frishman GN. Cesarean scar defects: an underrecognized cause of abnormal uterine bleeding and other gynecologic complications. J Minim Invasive Gynecol. 2013;20(5):562–572.
3. Garcia A. Update on minimally invasive gynecology. OBG Manag. 2014;26(4):18–32.
4. American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 94: Medical management of ectopic pregnancy. Obstet Gynecol. 2008;111(6):1479–1485.
Totally painless birth: A new concept in obstetrical anesthesia and labor management
A recent review on epidural anesthesia for pain management during labor, which was published in the New England Journal of Medicine,1 starts with the following case presentation: “A 30-year-old nulliparous woman at 39 weeks gestation is undergoing induction of labor for premature rupture of membranes. She is currently receiving an oxytocin infusion, and her cervical dilatation is 1 cm. Her obstetrician has ordered intermittent intravenous administration of Fentanyl for pain relief, but she feels nauseated, has been unable to rest, and describes her pain as 9 on scale of 10.”
The case we present in this letter sounds totally different: A 26-year-old multiparous woman at 39 weeks’ gestation is scheduled for induction of labor because of a decreased amount of amniotic fluid detected on prenatal ultrasonography. On admission, her cervix is 1-cm dilated and 60% effaced. She has no uterine contractions, and she describes her pain as 0 on the scale of 10. Anticipating a long induction and unwilling to experience any pain, the patient requests and is offered epidural anesthesia prior to oxytocin administration. Labor takes longer than expected but ultimately results in the birth of a healthy child. At no time during the course of labor and delivery does the mother experience any pain or discomfort, and she reports an extremely satisfying experience.
Traditionally, epidural anesthesia is administered upon maternal request when labor pain becomes difficult or intolerable. Many obstetricians discourage the use of epidural anesthesia prior to the onset of the active stage of labor, where the cervix is 4- to 6-cm dilated and the patient is in a great deal of pain.
This concept of administering an analgesic after the onset of pain is very different from the one where anesthesia is provided to patients prior to surgical procedures. No physician would begin a scheduled surgical procedure before confirming administration of adequate anesthesia. Why should labor be any different?
The pain of labor caused by uterine contractions and cervical dilatation is transmitted through visceral afferent nerves entering the spinal cord from T10 through L1. Perineal stretching transmits pain through the pudendal and sacral nerves. Epidural analgesia for labor and delivery involves injection of a local anesthetic agent and an opioid analgesic into the lumbar epidural space. The injected agents gradually diffuse across the dura into the subarachnoid cavity. In spinal analgesia, which is often combined with epidural analgesia, the medication is injected directly into the subarachnoid cavity, resulting in a more rapid effect.2
The American Society of Anesthesiologists and the American College of Obstetrics and Gynecology jointly state, “Labor causes severe pain for many women. There is no other circumstance where it is considered acceptable for an individual to experience severe pain, untreated. In the absence of medical contraindications, maternal request is a sufficient medical indication for pain relief during labor.”3
We decided to stretch this concept by allowing epidural anesthesia to be administered upon maternal consent in anticipation of pain, prior to onset. Our extensive literature search failed to find any comprehensive studies on the use of epidural anesthesia in anticipation of labor pain.
In theory, neuraxial local anesthetic in clinically relevant doses affect only skeletal muscles, not smooth muscles; therefore, this agent should not significantly decrease the amplitude or frequency of contractions in the myometrium.4 Randomized controlled trials of the effects of analgesia during labor do not exist since it is impossible to randomly assign women to a placebo group (no pain relief). Most trials have compared the use of epidural analgesia with that of systemic narcotics. In one large trial, 992 multiparous women were randomly assigned to either epidural analgesia or midwifery support supplemented by intramuscular narcotic injections. When pain was rated on a scale of 0–100 (100 is high), the median score before the study interventions was 80 in the group of patients who did not receive an epidural analgesia. With the administration of epidural analgesia, the median score was 27.5
Clinicians and patients also have been concerned about whether the use of epidural analgesia increases the risk of cesarean delivery. The results of three randomized controlled trials demonstrated that early administration of epidural analgesia does not increase the rate of cesarean delivery among women with spontaneous or induced labor as compared with early initiation of opioids.6–8 The fact that early epidural placement does not appear to increase the incidence of cesarean delivery is encouraging for our ongoing study but doesn’t necessarily mean that epidural analgesia prior to the onset of pain (preventive epidural) will have the same outcome. More research is needed.
Boris M. Petrikovsky, MD, PhD; Elya Kozlov, MD; Matthew Ackert, MD; Ralph Ruggiero, MD; and Crystal Behofsits, DO
Wyckoff Heights Medical Center, Brooklyn NY
References
1. Haukins GL. Epidural analgesia for labor and delivery. N Engl J Med. 2010;362(16):1502–1510.
2. Catterall WA, Mackie K. Local anesthetics. In: Brunton LL, Lazo JS, Parker KL, eds. Goodman & Gilman’s The Pharmacological Basis of Therapeutics. 11th ed. New York, New York: McGraw-Hill; 2005;369–386.
3. American College of Obstetricians and Gynecologists. ACOG Committee Opinion #295: pain relief during labor. Obstet Gynecol. 2004;104(1):213.
4. Fanning RA, Campion DP, Collins CB, et al. A comparison of the inhibitory effects of bupivacaine and levobupivacaine on isolated human pregnant myometrium contractility. Anesth Analg. 2008;107(4):1303–1307.
5. Dickinson JE, Paech MJ, McDonald SJ, Evans SF. Maternal satisfaction with childbirth and intrapartum analgesia in nulliparous labor. Aust N Z J Obstet Gynecol. 2003;43(6):463–468.
6. Wong CA, Scavone BM, Peaceman AM, et al. The risk of cesarean delivery with neuraxial analgesia in labor. N Engl J Med. 2005;352(7):655–665.
7. Ohel G, Gonen R, Vaida S, Barak S, Gaitini L. Early versus late initiation of epidural analgesia in labor: does it increase the risk of cesarean section? A randomized trial. Am J Obstet Gynecol. 2006:194(3):600–605.
8. Wong CA, McCarthy RJ, Sullivan JT, Scavone BM, Gerber SE, Yaghmour EA. Early compared with late neuraxial analgesia in nulliparous labor induction: a randomized controlled trial. Obstet Gynecol. 2009;113(5):1066–1074.
Share your thoughts on this article! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Boris M. Petrikovsky,Elya Kozlov,Matthew Ackert,Ralph Ruggiero,Crystal Behofsits,totally painless birth,obstetrical anesthesia,labor management,labor and delivery,New England Journal of Medicine,epidural anesthesia,pain management during labor,nulliparous,premature rupture of membranes,multiparous,prenatal ultrasonography,uterine contractions,cervical dilation,visceral afferent nerves,American Society of Anesthesiologists,American College of Obstetricians and Gynecologists,
Boris M. Petrikovsky,Elya Kozlov,Matthew Ackert,Ralph Ruggiero,Crystal Behofsits,totally painless birth,obstetrical anesthesia,labor management,labor and delivery,New England Journal of Medicine,epidural anesthesia,pain management during labor,nulliparous,premature rupture of membranes,multiparous,prenatal ultrasonography,uterine contractions,cervical dilation,visceral afferent nerves,American Society of Anesthesiologists,American College of Obstetricians and Gynecologists,
Vaginal bowel control device shown safe, effective in pivotal trial
WASHINGTON – Use of a vaginal bowel control device currently under review by the Food and Drug Administration resulted in a significant reduction of fecal incontinence episodes and significant improvement in quality of life, with only mild and transient adverse events, a pivotal study has shown.
The device – the Vaginal Bowel Control (VBC) System made by Pelvalon (Sunnyvale, Calif.) – consists of a silicone base and a posteriorly directed balloon. The patient controls the inflation pump, inflating the balloon to deflect the rectovaginal septum and interrupt stool passage, and deflating it to allow bowel movements.
"It provides [the patient with] dynamic control of the rectum," said Holly E. Richter, Ph.D., M.D., who served as the national principal investigator of the study. "It’s a new paradigm for treating patients with fecal incontinence."
At the 1-month primary endpoint, 79% of the 61 patients in the study’s intent-to-treat cohort – and 86% of patients in the per-protocol cohort – had experienced treatment success, which was defined as a 50% or greater reduction in fecal incontinence episodes, Dr. Richter reported at the scientific meetings of the American Urogynecologic Society and the International Urogynecological Association.
Mean weekly fecal incontinence episodes were reduced from 5.9 per week at baseline to 1.1 at 1 month.
After the 1-month treatment period, women were given the option of continuing with an additional 2-month study period. The rate of treatment success held steady among the 44 women who continued, with 86% seeing a 50% or greater reduction in fecal incontinence at 3 months, said Dr. Richter, director of the division of urogynecology and pelvic reconstructive surgery at the University of Alabama at Birmingham.
Total continence was achieved in approximately 40% of women in the 1-month and 3-month per-protocol cohorts, she noted.
Scores on the Fecal Incontinence Quality of Life survey and the Modified Manchester Health Questionnaire improved significantly across all subscales. At 1 month, 86% reported their fecal incontinence to be "very much better" (57%) or "much better" (29%). Almost all – 96% – reported their insert to be comfortable, and half said they could not feel it.
To qualify for the six-center study, women had to have a history of fecal incontinence of at least 6 months, with 4 or more incontinence episodes recorded during a 2-week baseline bowel diary.
Study participants had a mean age of 61 years and a mean of 5.9 weekly fecal incontinence episodes (or 12 in their diaries).Thirty percent were obese. Nearly 50% had prior hysterectomy, 8% had prior pelvic prolapse surgery, and 15% had prior urinary incontinence surgery.
There were no device-related serious adverse events. The most common complaint was cramping or discomfort, which most often occurred during the fitting period. There were approximately a half-dozen reported changes in urinary incontinence or overactive bladder symptoms.
"We believe that the Vaginal Bowel Control System can be tried early in the algorithm of treatment for fecal incontinence," Dr. Richter said.
It won’t be a good option for everyone, however. After being fitted with the device (it is made with three base sizes and two balloon sizes), women in the study were instructed to try it for a week before entering the 1-month treatment period. Approximately 40 women who were screened and fitted did not continue. This rate of discomfort or nonacceptance is similar to that associated with pessaries, Dr. Richter noted.
The initial fitting procedure for the new bowel control device "requires a little more provider and patient training, beyond [what is needed] for use of a traditional vaginal pessary," she said.
"Patients in the study could put the device in and take it out as they wished. Most patients used it all day long, and some took it out at night," she said. Patients were instructed to "take it out once or twice a week to wash it."
Dr. Richter is a paid consultant for Pelvalon. Her coinvestigators reported consulting for Pelvalon and other companies.
Fecal incontinence is one of the most emotionally devastating of all nonfatal medical conditions, and there is a need for new and effective strategies when medical therapy fails. The vaginal bowel control device appears to hold promise for some women with fecal incontinence by mechanically impeding stool passage to the anorectum by means of an inflatable balloon inserted into the vagina. The premise is that it can be used during times when the patient is not near to toileting facilities, can be inserted and removed at will, and is not a safety concern.
It is important to emphasize that this was not a randomized, placebo-controlled trial but rather a proof-of-concept study. The 40% complete continence response is 1) similar to that observed with the sacral nerve stimulation trial that resulted in Food and Drug Administration approval in the absence of a double-blind, controlled trial (Ann. Surg. 2010;1251:441), and 2) superior to the injectable bulking agent (6% complete continence), which was approved on the basis of a randomized, double-blind, sham-controlled trial (Lancet 2011;377:997).
However, complete continence is not necessary to improve quality of life in patients with incontinence. All three of these techniques require future studies to determine which incontinent patients are likely to benefit from each of these modalities. It must be recognized that there are many causes of incontinence, which often require different therapeutic approaches.
I look forward to reading a peer-reviewed paper on this new technique and to future randomized, controlled studies on this innovative approach to a condition with a large unmet need.
Dr. Arnold Wald is professor of medicine at the University of Wisconsin School of Medicine and Public Health, Madison.
Fecal incontinence is one of the most emotionally devastating of all nonfatal medical conditions, and there is a need for new and effective strategies when medical therapy fails. The vaginal bowel control device appears to hold promise for some women with fecal incontinence by mechanically impeding stool passage to the anorectum by means of an inflatable balloon inserted into the vagina. The premise is that it can be used during times when the patient is not near to toileting facilities, can be inserted and removed at will, and is not a safety concern.
It is important to emphasize that this was not a randomized, placebo-controlled trial but rather a proof-of-concept study. The 40% complete continence response is 1) similar to that observed with the sacral nerve stimulation trial that resulted in Food and Drug Administration approval in the absence of a double-blind, controlled trial (Ann. Surg. 2010;1251:441), and 2) superior to the injectable bulking agent (6% complete continence), which was approved on the basis of a randomized, double-blind, sham-controlled trial (Lancet 2011;377:997).
However, complete continence is not necessary to improve quality of life in patients with incontinence. All three of these techniques require future studies to determine which incontinent patients are likely to benefit from each of these modalities. It must be recognized that there are many causes of incontinence, which often require different therapeutic approaches.
I look forward to reading a peer-reviewed paper on this new technique and to future randomized, controlled studies on this innovative approach to a condition with a large unmet need.
Dr. Arnold Wald is professor of medicine at the University of Wisconsin School of Medicine and Public Health, Madison.
Fecal incontinence is one of the most emotionally devastating of all nonfatal medical conditions, and there is a need for new and effective strategies when medical therapy fails. The vaginal bowel control device appears to hold promise for some women with fecal incontinence by mechanically impeding stool passage to the anorectum by means of an inflatable balloon inserted into the vagina. The premise is that it can be used during times when the patient is not near to toileting facilities, can be inserted and removed at will, and is not a safety concern.
It is important to emphasize that this was not a randomized, placebo-controlled trial but rather a proof-of-concept study. The 40% complete continence response is 1) similar to that observed with the sacral nerve stimulation trial that resulted in Food and Drug Administration approval in the absence of a double-blind, controlled trial (Ann. Surg. 2010;1251:441), and 2) superior to the injectable bulking agent (6% complete continence), which was approved on the basis of a randomized, double-blind, sham-controlled trial (Lancet 2011;377:997).
However, complete continence is not necessary to improve quality of life in patients with incontinence. All three of these techniques require future studies to determine which incontinent patients are likely to benefit from each of these modalities. It must be recognized that there are many causes of incontinence, which often require different therapeutic approaches.
I look forward to reading a peer-reviewed paper on this new technique and to future randomized, controlled studies on this innovative approach to a condition with a large unmet need.
Dr. Arnold Wald is professor of medicine at the University of Wisconsin School of Medicine and Public Health, Madison.
WASHINGTON – Use of a vaginal bowel control device currently under review by the Food and Drug Administration resulted in a significant reduction of fecal incontinence episodes and significant improvement in quality of life, with only mild and transient adverse events, a pivotal study has shown.
The device – the Vaginal Bowel Control (VBC) System made by Pelvalon (Sunnyvale, Calif.) – consists of a silicone base and a posteriorly directed balloon. The patient controls the inflation pump, inflating the balloon to deflect the rectovaginal septum and interrupt stool passage, and deflating it to allow bowel movements.
"It provides [the patient with] dynamic control of the rectum," said Holly E. Richter, Ph.D., M.D., who served as the national principal investigator of the study. "It’s a new paradigm for treating patients with fecal incontinence."
At the 1-month primary endpoint, 79% of the 61 patients in the study’s intent-to-treat cohort – and 86% of patients in the per-protocol cohort – had experienced treatment success, which was defined as a 50% or greater reduction in fecal incontinence episodes, Dr. Richter reported at the scientific meetings of the American Urogynecologic Society and the International Urogynecological Association.
Mean weekly fecal incontinence episodes were reduced from 5.9 per week at baseline to 1.1 at 1 month.
After the 1-month treatment period, women were given the option of continuing with an additional 2-month study period. The rate of treatment success held steady among the 44 women who continued, with 86% seeing a 50% or greater reduction in fecal incontinence at 3 months, said Dr. Richter, director of the division of urogynecology and pelvic reconstructive surgery at the University of Alabama at Birmingham.
Total continence was achieved in approximately 40% of women in the 1-month and 3-month per-protocol cohorts, she noted.
Scores on the Fecal Incontinence Quality of Life survey and the Modified Manchester Health Questionnaire improved significantly across all subscales. At 1 month, 86% reported their fecal incontinence to be "very much better" (57%) or "much better" (29%). Almost all – 96% – reported their insert to be comfortable, and half said they could not feel it.
To qualify for the six-center study, women had to have a history of fecal incontinence of at least 6 months, with 4 or more incontinence episodes recorded during a 2-week baseline bowel diary.
Study participants had a mean age of 61 years and a mean of 5.9 weekly fecal incontinence episodes (or 12 in their diaries).Thirty percent were obese. Nearly 50% had prior hysterectomy, 8% had prior pelvic prolapse surgery, and 15% had prior urinary incontinence surgery.
There were no device-related serious adverse events. The most common complaint was cramping or discomfort, which most often occurred during the fitting period. There were approximately a half-dozen reported changes in urinary incontinence or overactive bladder symptoms.
"We believe that the Vaginal Bowel Control System can be tried early in the algorithm of treatment for fecal incontinence," Dr. Richter said.
It won’t be a good option for everyone, however. After being fitted with the device (it is made with three base sizes and two balloon sizes), women in the study were instructed to try it for a week before entering the 1-month treatment period. Approximately 40 women who were screened and fitted did not continue. This rate of discomfort or nonacceptance is similar to that associated with pessaries, Dr. Richter noted.
The initial fitting procedure for the new bowel control device "requires a little more provider and patient training, beyond [what is needed] for use of a traditional vaginal pessary," she said.
"Patients in the study could put the device in and take it out as they wished. Most patients used it all day long, and some took it out at night," she said. Patients were instructed to "take it out once or twice a week to wash it."
Dr. Richter is a paid consultant for Pelvalon. Her coinvestigators reported consulting for Pelvalon and other companies.
WASHINGTON – Use of a vaginal bowel control device currently under review by the Food and Drug Administration resulted in a significant reduction of fecal incontinence episodes and significant improvement in quality of life, with only mild and transient adverse events, a pivotal study has shown.
The device – the Vaginal Bowel Control (VBC) System made by Pelvalon (Sunnyvale, Calif.) – consists of a silicone base and a posteriorly directed balloon. The patient controls the inflation pump, inflating the balloon to deflect the rectovaginal septum and interrupt stool passage, and deflating it to allow bowel movements.
"It provides [the patient with] dynamic control of the rectum," said Holly E. Richter, Ph.D., M.D., who served as the national principal investigator of the study. "It’s a new paradigm for treating patients with fecal incontinence."
At the 1-month primary endpoint, 79% of the 61 patients in the study’s intent-to-treat cohort – and 86% of patients in the per-protocol cohort – had experienced treatment success, which was defined as a 50% or greater reduction in fecal incontinence episodes, Dr. Richter reported at the scientific meetings of the American Urogynecologic Society and the International Urogynecological Association.
Mean weekly fecal incontinence episodes were reduced from 5.9 per week at baseline to 1.1 at 1 month.
After the 1-month treatment period, women were given the option of continuing with an additional 2-month study period. The rate of treatment success held steady among the 44 women who continued, with 86% seeing a 50% or greater reduction in fecal incontinence at 3 months, said Dr. Richter, director of the division of urogynecology and pelvic reconstructive surgery at the University of Alabama at Birmingham.
Total continence was achieved in approximately 40% of women in the 1-month and 3-month per-protocol cohorts, she noted.
Scores on the Fecal Incontinence Quality of Life survey and the Modified Manchester Health Questionnaire improved significantly across all subscales. At 1 month, 86% reported their fecal incontinence to be "very much better" (57%) or "much better" (29%). Almost all – 96% – reported their insert to be comfortable, and half said they could not feel it.
To qualify for the six-center study, women had to have a history of fecal incontinence of at least 6 months, with 4 or more incontinence episodes recorded during a 2-week baseline bowel diary.
Study participants had a mean age of 61 years and a mean of 5.9 weekly fecal incontinence episodes (or 12 in their diaries).Thirty percent were obese. Nearly 50% had prior hysterectomy, 8% had prior pelvic prolapse surgery, and 15% had prior urinary incontinence surgery.
There were no device-related serious adverse events. The most common complaint was cramping or discomfort, which most often occurred during the fitting period. There were approximately a half-dozen reported changes in urinary incontinence or overactive bladder symptoms.
"We believe that the Vaginal Bowel Control System can be tried early in the algorithm of treatment for fecal incontinence," Dr. Richter said.
It won’t be a good option for everyone, however. After being fitted with the device (it is made with three base sizes and two balloon sizes), women in the study were instructed to try it for a week before entering the 1-month treatment period. Approximately 40 women who were screened and fitted did not continue. This rate of discomfort or nonacceptance is similar to that associated with pessaries, Dr. Richter noted.
The initial fitting procedure for the new bowel control device "requires a little more provider and patient training, beyond [what is needed] for use of a traditional vaginal pessary," she said.
"Patients in the study could put the device in and take it out as they wished. Most patients used it all day long, and some took it out at night," she said. Patients were instructed to "take it out once or twice a week to wash it."
Dr. Richter is a paid consultant for Pelvalon. Her coinvestigators reported consulting for Pelvalon and other companies.
AT AUGS/IUGA 2014
Key clinical finding: A controllable vaginal approach to bowel continence may be useful in the future.
Major finding: Almost 80% of 61 patients in an intent-to-treat cohort saw a 50% or greater reduction in fecal incontinence episodes with use of the Vaginal Bowel Control System, a device placed in the vagina to restore bowel control.
Data source: A multicenter, prospective, open-label study of women with fecal incontinence.
Disclosures: Dr. Richter is a paid consultant for Pelvalon. Her coinvestigators reported consulting for Pelvalon and other companies.