User login
Skin ulcers can pose tricky diagnostic challenges
In the clinical opinion of Alex G. Ortega-Loayza, MD, MCR, few absolutes drive the initial assessment of patients who present with skin ulcers.

The causes can be neoplastic, infectious, inflammatory, vasculopathic, external, and genetic. “Sometimes they can be of mixed etiology, which make them even more complicated to heal,” Dr. Ortega-Loayza, of the department of dermatology at Oregon Health & Science University, Portland, said during the annual meeting of the Pacific Dermatologic Association.
In a study published in 2019, he and his colleagues at four academic hospitals evaluated characteristics and diagnoses of ulcers in 274 patients with skin ulcers in inpatient dermatology consultation services between July 2015 and July 2018. Most primary teams requesting the consultation (93%) were from nonsurgical specialties. The median age of these patients was 54 years, 45% were male, and 50% had lower-extremity ulcers. Nearly two-thirds of the ulcers (62%) were chronic in nature, while the remaining 38% were acute. The skin ulcer was the chief reason for admission in 49% of cases and 66% were admitted through the ED. In addition, 11% had a superinfected skin ulcer.
The top three etiologies rendered by dermatologists after assessing these patients were pyoderma gangrenosum (17%), infection (13%), and exogenous causes (12%); another 12% remained diagnostically inconclusive after consultation. Diagnostic agreements between the primary team requesting the consultation and the dermatologist were poor to modest.
These data highlights the role of the dermatologists in the workup of skin ulcers of unknown etiology.
“The diagnosis of skin ulcers can be challenging,” Dr. Ortega-Loayza said. “Subjective factors playing a role in the diagnosis of skin ulcers include the type of level of training/experience you’ve had and general awareness and education about skin ulcers.” In addition, there is also a lack of gold-standard diagnostic criteria for atypical/inflammatory ulcers and a lack of specificity of ancillary testing, such as for pyoderma gangrenosum.
Dr. Ortega-Loayza’s basic workup is based on the review of systems and the patient’s comorbidities. Blood work may include CBC, comprehensive metabolic panel, erythrocyte sedimentation rate/C-reactive protein, glucose-6-phosphate dehydrogenase, albumin/prealbumin, autoimmune panels, and hypercoagulable panels. He may order a skin biopsy with H&E staining and microbiological studies, superficial bacterial wound cultures, and vascular studies, such as ankle brachial index (ABI) and chronic venous reflux tests, and Doppler ultrasound, and he might consider an angiogram for certain type of ulcers. Additional imaging studies may include x-ray, CT scan, and/or MRI.
The four key factors to control in patients with skin ulcers, he continued, include effective management of edema (such as compression garments depending on the results of the vascular studies); infection (with topical/oral antibiotics and debridement); the wound microenvironment (with wound dressings), and pain (mainly with nonopioids). “In my practice, we tend to do multilayered compression,” he said. “This can be two- or four-layer. I do light compression if the patient has peripheral arterial disease. I always bring in the patient 2 days later to check on them, or do a telehealth visit, to make sure they are not developing any worsening of the ulcers.”
Infections can be managed with topical antimicrobials such as metronidazole 1% gel and cadexomer iodine. “Iodine can also help dry the wound when you need to do so,” said Dr. Ortega-Loayza, who directs a pyoderma gangrenosum clinic at OHSU. “Debridement can be done with a curette or with commercially available enzymatic products such as Collagenase, PluroGel, and MediHoney.”
When the ulcer is in an active phase (characterized by significant amount of drainage and erythema), he uses one or more of the following products to control the wound microenvironment: zinc oxide, an antimicrobial dressing, a hyperabsorbent dressing, an abdominal pad, and compression.
During the healing phase, with evidence of re-epithelization, he tends to use more foam dressings and continues with compression. His preferred options for managing pain associated with ulcers are medications to control neuropathic pain including initially gabapentin (100 mg-300 mg at bedtime), pregabalin (75 mg twice a day), or duloxetine (extended release, 30 mg once a day). All of these medications can be titrated up based on patients’ needs. Foam dressings with ibuprofen can also provide comfort, he said.
Dr. Ortega-Loayza also provided a few clinical pearls highlighting the role and utility of interleukin-23 inhibitors in the management of patients with pyoderma gangrenosum, oral vitamin K in patients with calciphylaxis, and stanozolol for lipodermatosclerosis. He is also leading the first open-label trial testing a Janus kinase inhibitor – baricitinib – as a treatment for patients with pyoderma gangrenosum.
Dr. Ortega-Loayza disclosed that he is a consultant to Genentech and Guidepoint and is a member of the advisory board for Bristol-Myers Squibb, Boehringer Ingelheim, and Janssen. He also has received research support from Lilly.
In the clinical opinion of Alex G. Ortega-Loayza, MD, MCR, few absolutes drive the initial assessment of patients who present with skin ulcers.
The causes can be neoplastic, infectious, inflammatory, vasculopathic, external, and genetic. “Sometimes they can be of mixed etiology, which make them even more complicated to heal,” Dr. Ortega-Loayza, of the department of dermatology at Oregon Health & Science University, Portland, said during the annual meeting of the Pacific Dermatologic Association.
In a study published in 2019, he and his colleagues at four academic hospitals evaluated characteristics and diagnoses of ulcers in 274 patients with skin ulcers in inpatient dermatology consultation services between July 2015 and July 2018. Most primary teams requesting the consultation (93%) were from nonsurgical specialties. The median age of these patients was 54 years, 45% were male, and 50% had lower-extremity ulcers. Nearly two-thirds of the ulcers (62%) were chronic in nature, while the remaining 38% were acute. The skin ulcer was the chief reason for admission in 49% of cases and 66% were admitted through the ED. In addition, 11% had a superinfected skin ulcer.
The top three etiologies rendered by dermatologists after assessing these patients were pyoderma gangrenosum (17%), infection (13%), and exogenous causes (12%); another 12% remained diagnostically inconclusive after consultation. Diagnostic agreements between the primary team requesting the consultation and the dermatologist were poor to modest.
These data highlights the role of the dermatologists in the workup of skin ulcers of unknown etiology.
“The diagnosis of skin ulcers can be challenging,” Dr. Ortega-Loayza said. “Subjective factors playing a role in the diagnosis of skin ulcers include the type of level of training/experience you’ve had and general awareness and education about skin ulcers.” In addition, there is also a lack of gold-standard diagnostic criteria for atypical/inflammatory ulcers and a lack of specificity of ancillary testing, such as for pyoderma gangrenosum.
Dr. Ortega-Loayza’s basic workup is based on the review of systems and the patient’s comorbidities. Blood work may include CBC, comprehensive metabolic panel, erythrocyte sedimentation rate/C-reactive protein, glucose-6-phosphate dehydrogenase, albumin/prealbumin, autoimmune panels, and hypercoagulable panels. He may order a skin biopsy with H&E staining and microbiological studies, superficial bacterial wound cultures, and vascular studies, such as ankle brachial index (ABI) and chronic venous reflux tests, and Doppler ultrasound, and he might consider an angiogram for certain type of ulcers. Additional imaging studies may include x-ray, CT scan, and/or MRI.
The four key factors to control in patients with skin ulcers, he continued, include effective management of edema (such as compression garments depending on the results of the vascular studies); infection (with topical/oral antibiotics and debridement); the wound microenvironment (with wound dressings), and pain (mainly with nonopioids). “In my practice, we tend to do multilayered compression,” he said. “This can be two- or four-layer. I do light compression if the patient has peripheral arterial disease. I always bring in the patient 2 days later to check on them, or do a telehealth visit, to make sure they are not developing any worsening of the ulcers.”
Infections can be managed with topical antimicrobials such as metronidazole 1% gel and cadexomer iodine. “Iodine can also help dry the wound when you need to do so,” said Dr. Ortega-Loayza, who directs a pyoderma gangrenosum clinic at OHSU. “Debridement can be done with a curette or with commercially available enzymatic products such as Collagenase, PluroGel, and MediHoney.”
When the ulcer is in an active phase (characterized by significant amount of drainage and erythema), he uses one or more of the following products to control the wound microenvironment: zinc oxide, an antimicrobial dressing, a hyperabsorbent dressing, an abdominal pad, and compression.
During the healing phase, with evidence of re-epithelization, he tends to use more foam dressings and continues with compression. His preferred options for managing pain associated with ulcers are medications to control neuropathic pain including initially gabapentin (100 mg-300 mg at bedtime), pregabalin (75 mg twice a day), or duloxetine (extended release, 30 mg once a day). All of these medications can be titrated up based on patients’ needs. Foam dressings with ibuprofen can also provide comfort, he said.
Dr. Ortega-Loayza also provided a few clinical pearls highlighting the role and utility of interleukin-23 inhibitors in the management of patients with pyoderma gangrenosum, oral vitamin K in patients with calciphylaxis, and stanozolol for lipodermatosclerosis. He is also leading the first open-label trial testing a Janus kinase inhibitor – baricitinib – as a treatment for patients with pyoderma gangrenosum.
Dr. Ortega-Loayza disclosed that he is a consultant to Genentech and Guidepoint and is a member of the advisory board for Bristol-Myers Squibb, Boehringer Ingelheim, and Janssen. He also has received research support from Lilly.
In the clinical opinion of Alex G. Ortega-Loayza, MD, MCR, few absolutes drive the initial assessment of patients who present with skin ulcers.
The causes can be neoplastic, infectious, inflammatory, vasculopathic, external, and genetic. “Sometimes they can be of mixed etiology, which make them even more complicated to heal,” Dr. Ortega-Loayza, of the department of dermatology at Oregon Health & Science University, Portland, said during the annual meeting of the Pacific Dermatologic Association.
In a study published in 2019, he and his colleagues at four academic hospitals evaluated characteristics and diagnoses of ulcers in 274 patients with skin ulcers in inpatient dermatology consultation services between July 2015 and July 2018. Most primary teams requesting the consultation (93%) were from nonsurgical specialties. The median age of these patients was 54 years, 45% were male, and 50% had lower-extremity ulcers. Nearly two-thirds of the ulcers (62%) were chronic in nature, while the remaining 38% were acute. The skin ulcer was the chief reason for admission in 49% of cases and 66% were admitted through the ED. In addition, 11% had a superinfected skin ulcer.
The top three etiologies rendered by dermatologists after assessing these patients were pyoderma gangrenosum (17%), infection (13%), and exogenous causes (12%); another 12% remained diagnostically inconclusive after consultation. Diagnostic agreements between the primary team requesting the consultation and the dermatologist were poor to modest.
These data highlights the role of the dermatologists in the workup of skin ulcers of unknown etiology.
“The diagnosis of skin ulcers can be challenging,” Dr. Ortega-Loayza said. “Subjective factors playing a role in the diagnosis of skin ulcers include the type of level of training/experience you’ve had and general awareness and education about skin ulcers.” In addition, there is also a lack of gold-standard diagnostic criteria for atypical/inflammatory ulcers and a lack of specificity of ancillary testing, such as for pyoderma gangrenosum.
Dr. Ortega-Loayza’s basic workup is based on the review of systems and the patient’s comorbidities. Blood work may include CBC, comprehensive metabolic panel, erythrocyte sedimentation rate/C-reactive protein, glucose-6-phosphate dehydrogenase, albumin/prealbumin, autoimmune panels, and hypercoagulable panels. He may order a skin biopsy with H&E staining and microbiological studies, superficial bacterial wound cultures, and vascular studies, such as ankle brachial index (ABI) and chronic venous reflux tests, and Doppler ultrasound, and he might consider an angiogram for certain type of ulcers. Additional imaging studies may include x-ray, CT scan, and/or MRI.
The four key factors to control in patients with skin ulcers, he continued, include effective management of edema (such as compression garments depending on the results of the vascular studies); infection (with topical/oral antibiotics and debridement); the wound microenvironment (with wound dressings), and pain (mainly with nonopioids). “In my practice, we tend to do multilayered compression,” he said. “This can be two- or four-layer. I do light compression if the patient has peripheral arterial disease. I always bring in the patient 2 days later to check on them, or do a telehealth visit, to make sure they are not developing any worsening of the ulcers.”
Infections can be managed with topical antimicrobials such as metronidazole 1% gel and cadexomer iodine. “Iodine can also help dry the wound when you need to do so,” said Dr. Ortega-Loayza, who directs a pyoderma gangrenosum clinic at OHSU. “Debridement can be done with a curette or with commercially available enzymatic products such as Collagenase, PluroGel, and MediHoney.”
When the ulcer is in an active phase (characterized by significant amount of drainage and erythema), he uses one or more of the following products to control the wound microenvironment: zinc oxide, an antimicrobial dressing, a hyperabsorbent dressing, an abdominal pad, and compression.
During the healing phase, with evidence of re-epithelization, he tends to use more foam dressings and continues with compression. His preferred options for managing pain associated with ulcers are medications to control neuropathic pain including initially gabapentin (100 mg-300 mg at bedtime), pregabalin (75 mg twice a day), or duloxetine (extended release, 30 mg once a day). All of these medications can be titrated up based on patients’ needs. Foam dressings with ibuprofen can also provide comfort, he said.
Dr. Ortega-Loayza also provided a few clinical pearls highlighting the role and utility of interleukin-23 inhibitors in the management of patients with pyoderma gangrenosum, oral vitamin K in patients with calciphylaxis, and stanozolol for lipodermatosclerosis. He is also leading the first open-label trial testing a Janus kinase inhibitor – baricitinib – as a treatment for patients with pyoderma gangrenosum.
Dr. Ortega-Loayza disclosed that he is a consultant to Genentech and Guidepoint and is a member of the advisory board for Bristol-Myers Squibb, Boehringer Ingelheim, and Janssen. He also has received research support from Lilly.
FROM PDA 2021
Severe skin reactions with enfortumab vedotin
The cases came to light during routine surveillance, say staff from the division of pharmacovigilance of the Food and Drug Administration in a research letter published online Sept. 8, 2021, in JAMA Dermatology.
Eight cases of serious skin reactions characterized as SJS/TEN were identified from the FDA’s Adverse Event Reporting System (FAERS). In five of these cases, the diagnosis of SJS/TEN was confirmed by a dermatologist and/or biopsy findings.
The median time to onset of SJS/TEN was 11 days (range, 9-21 days) from the start of treatment.
In the eight cases, serious outcomes were reported. In four cases, deaths that were attributed to SJS/TEN occurred. “Other serious outcomes included admission to the burn unit in four cases,” the researchers wrote.
First-in-class agent
Enfortumab vedotin is a first-in-class agent directed against cell adhesion molecule nectin-4, which is located on the surface of cells and is highly expressed in bladder cancer. The product is an antibody conjugate, in which the antibody directs the product to these cells and then releases the cytoxic monomethyl auristantin E. It is administered intravenously.
The product was granted accelerated approval by the FDA in December 2019. This approval was based on response data from the EV-201 study, a phase 2 clinical trial that involved 125 patients with locally advanced or metastatic urothelial cancer who received prior treatment with a PD-1 or PD-L1 inhibitor and platinum-based chemotherapy.
The results were presented in June 2019 at the annual meeting of the American Society of Clinical Oncology. The overall response rate was 44%; 12% of patients achieved a complete response, and 32% had a partial response. The median duration of response was 7.6 months.
At the meeting, Daniel P. Petrylak, MD, professor of medicine (medical oncology) and urology at Yale Cancer Center, New Haven, Conn., noted that there is a “high unmet need” among patients with advanced and metastatic urothelial cancer. There has been a flurry of new drug approvals for this disease. Five immune checkpoint inhibitor drugs have been approved in recent years. Most patients (75%-80%) experience disease progression after receiving immunotherapy.
Enfortumab vedotin is the “first novel therapeutic to demonstrate substantial clinical activity” in patients whose disease has progressed after platinum chemotherapy and immunotherapies, commented Dr. Petrylak.
At the time, maculopapular rash of grade 3 or higher was reported in 4% of the cohort. That was the only serious dermatologic adverse event noted.
Clinically significant findings
The cases of severe skin reactions now being reported come from postmarketing surveillance, noted the authors, led by Michelle Nadeau Nguyen, PharmD, BCOP, BCPS. They reviewed data from FAERS, PubMed, and Embase from Dec. 18, 2019, the date the product was approved, to Oct. 7, 2020.
Other than the eight cases reported to FAERS, no additional cases were identified from PubMed or Embase.
The authors noted that, because cases of SJS/TEN are rare but serious, these well-documented postmarketing reports are clinically significant. “Moreover, we find the rapid accumulation of cases over an approximate 12-month marketing period a concerning observation,” they wrote.
The rate at which these reactions were reported is higher than would be expected, they commented.
The annual incidence of locally advanced urothelial cancer, the disease most likely to be treated with this drug, is around 12,494-40,000 cases per year in the United States. The expected incidence rate of SJS/TEN is about 1-7 cases per 1,000,000 patients. The team calculated from the reports that, among patients who received enfortumab vedotin, the rate was 20 cases per 1,000,000 patients.
This reporting rate is likely to be underestimated, inasmuch as underreporting is known to be a limitation of spontaneous reporting systems such as FAERS, the authors noted.
The mechanism for toxic skin effects with enfortumab vedotin is as yet unknown, but it may be related to the inhibitory effects of the drug on nectin-4 expression, they suggest. Nectin-4 is expressed by epithelial tissues, including skin.
Dr. Nguyen and colleagues noted that, on approval, the U.S. prescribing information for the drug noted that skin reactions were seen in 55% of patients in clinical trials.
The prescribing information was recently revised to include SJS/TEN and to recommend permanent discontinuance of the drug if cases of SJS/TEN are suspected.
“This revision is intended to increase clinicians’ awareness of the risk for SJS/TEN and mitigate serious outcomes by improving the likelihood of early identification and intervention,” they added.
The authors also encouraged continued reporting of adverse events with enfortumab vedotin to the FDA via the MedWatch portal.
The authors disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The cases came to light during routine surveillance, say staff from the division of pharmacovigilance of the Food and Drug Administration in a research letter published online Sept. 8, 2021, in JAMA Dermatology.
Eight cases of serious skin reactions characterized as SJS/TEN were identified from the FDA’s Adverse Event Reporting System (FAERS). In five of these cases, the diagnosis of SJS/TEN was confirmed by a dermatologist and/or biopsy findings.
The median time to onset of SJS/TEN was 11 days (range, 9-21 days) from the start of treatment.
In the eight cases, serious outcomes were reported. In four cases, deaths that were attributed to SJS/TEN occurred. “Other serious outcomes included admission to the burn unit in four cases,” the researchers wrote.
First-in-class agent
Enfortumab vedotin is a first-in-class agent directed against cell adhesion molecule nectin-4, which is located on the surface of cells and is highly expressed in bladder cancer. The product is an antibody conjugate, in which the antibody directs the product to these cells and then releases the cytoxic monomethyl auristantin E. It is administered intravenously.
The product was granted accelerated approval by the FDA in December 2019. This approval was based on response data from the EV-201 study, a phase 2 clinical trial that involved 125 patients with locally advanced or metastatic urothelial cancer who received prior treatment with a PD-1 or PD-L1 inhibitor and platinum-based chemotherapy.
The results were presented in June 2019 at the annual meeting of the American Society of Clinical Oncology. The overall response rate was 44%; 12% of patients achieved a complete response, and 32% had a partial response. The median duration of response was 7.6 months.
At the meeting, Daniel P. Petrylak, MD, professor of medicine (medical oncology) and urology at Yale Cancer Center, New Haven, Conn., noted that there is a “high unmet need” among patients with advanced and metastatic urothelial cancer. There has been a flurry of new drug approvals for this disease. Five immune checkpoint inhibitor drugs have been approved in recent years. Most patients (75%-80%) experience disease progression after receiving immunotherapy.
Enfortumab vedotin is the “first novel therapeutic to demonstrate substantial clinical activity” in patients whose disease has progressed after platinum chemotherapy and immunotherapies, commented Dr. Petrylak.
At the time, maculopapular rash of grade 3 or higher was reported in 4% of the cohort. That was the only serious dermatologic adverse event noted.
Clinically significant findings
The cases of severe skin reactions now being reported come from postmarketing surveillance, noted the authors, led by Michelle Nadeau Nguyen, PharmD, BCOP, BCPS. They reviewed data from FAERS, PubMed, and Embase from Dec. 18, 2019, the date the product was approved, to Oct. 7, 2020.
Other than the eight cases reported to FAERS, no additional cases were identified from PubMed or Embase.
The authors noted that, because cases of SJS/TEN are rare but serious, these well-documented postmarketing reports are clinically significant. “Moreover, we find the rapid accumulation of cases over an approximate 12-month marketing period a concerning observation,” they wrote.
The rate at which these reactions were reported is higher than would be expected, they commented.
The annual incidence of locally advanced urothelial cancer, the disease most likely to be treated with this drug, is around 12,494-40,000 cases per year in the United States. The expected incidence rate of SJS/TEN is about 1-7 cases per 1,000,000 patients. The team calculated from the reports that, among patients who received enfortumab vedotin, the rate was 20 cases per 1,000,000 patients.
This reporting rate is likely to be underestimated, inasmuch as underreporting is known to be a limitation of spontaneous reporting systems such as FAERS, the authors noted.
The mechanism for toxic skin effects with enfortumab vedotin is as yet unknown, but it may be related to the inhibitory effects of the drug on nectin-4 expression, they suggest. Nectin-4 is expressed by epithelial tissues, including skin.
Dr. Nguyen and colleagues noted that, on approval, the U.S. prescribing information for the drug noted that skin reactions were seen in 55% of patients in clinical trials.
The prescribing information was recently revised to include SJS/TEN and to recommend permanent discontinuance of the drug if cases of SJS/TEN are suspected.
“This revision is intended to increase clinicians’ awareness of the risk for SJS/TEN and mitigate serious outcomes by improving the likelihood of early identification and intervention,” they added.
The authors also encouraged continued reporting of adverse events with enfortumab vedotin to the FDA via the MedWatch portal.
The authors disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The cases came to light during routine surveillance, say staff from the division of pharmacovigilance of the Food and Drug Administration in a research letter published online Sept. 8, 2021, in JAMA Dermatology.
Eight cases of serious skin reactions characterized as SJS/TEN were identified from the FDA’s Adverse Event Reporting System (FAERS). In five of these cases, the diagnosis of SJS/TEN was confirmed by a dermatologist and/or biopsy findings.
The median time to onset of SJS/TEN was 11 days (range, 9-21 days) from the start of treatment.
In the eight cases, serious outcomes were reported. In four cases, deaths that were attributed to SJS/TEN occurred. “Other serious outcomes included admission to the burn unit in four cases,” the researchers wrote.
First-in-class agent
Enfortumab vedotin is a first-in-class agent directed against cell adhesion molecule nectin-4, which is located on the surface of cells and is highly expressed in bladder cancer. The product is an antibody conjugate, in which the antibody directs the product to these cells and then releases the cytoxic monomethyl auristantin E. It is administered intravenously.
The product was granted accelerated approval by the FDA in December 2019. This approval was based on response data from the EV-201 study, a phase 2 clinical trial that involved 125 patients with locally advanced or metastatic urothelial cancer who received prior treatment with a PD-1 or PD-L1 inhibitor and platinum-based chemotherapy.
The results were presented in June 2019 at the annual meeting of the American Society of Clinical Oncology. The overall response rate was 44%; 12% of patients achieved a complete response, and 32% had a partial response. The median duration of response was 7.6 months.
At the meeting, Daniel P. Petrylak, MD, professor of medicine (medical oncology) and urology at Yale Cancer Center, New Haven, Conn., noted that there is a “high unmet need” among patients with advanced and metastatic urothelial cancer. There has been a flurry of new drug approvals for this disease. Five immune checkpoint inhibitor drugs have been approved in recent years. Most patients (75%-80%) experience disease progression after receiving immunotherapy.
Enfortumab vedotin is the “first novel therapeutic to demonstrate substantial clinical activity” in patients whose disease has progressed after platinum chemotherapy and immunotherapies, commented Dr. Petrylak.
At the time, maculopapular rash of grade 3 or higher was reported in 4% of the cohort. That was the only serious dermatologic adverse event noted.
Clinically significant findings
The cases of severe skin reactions now being reported come from postmarketing surveillance, noted the authors, led by Michelle Nadeau Nguyen, PharmD, BCOP, BCPS. They reviewed data from FAERS, PubMed, and Embase from Dec. 18, 2019, the date the product was approved, to Oct. 7, 2020.
Other than the eight cases reported to FAERS, no additional cases were identified from PubMed or Embase.
The authors noted that, because cases of SJS/TEN are rare but serious, these well-documented postmarketing reports are clinically significant. “Moreover, we find the rapid accumulation of cases over an approximate 12-month marketing period a concerning observation,” they wrote.
The rate at which these reactions were reported is higher than would be expected, they commented.
The annual incidence of locally advanced urothelial cancer, the disease most likely to be treated with this drug, is around 12,494-40,000 cases per year in the United States. The expected incidence rate of SJS/TEN is about 1-7 cases per 1,000,000 patients. The team calculated from the reports that, among patients who received enfortumab vedotin, the rate was 20 cases per 1,000,000 patients.
This reporting rate is likely to be underestimated, inasmuch as underreporting is known to be a limitation of spontaneous reporting systems such as FAERS, the authors noted.
The mechanism for toxic skin effects with enfortumab vedotin is as yet unknown, but it may be related to the inhibitory effects of the drug on nectin-4 expression, they suggest. Nectin-4 is expressed by epithelial tissues, including skin.
Dr. Nguyen and colleagues noted that, on approval, the U.S. prescribing information for the drug noted that skin reactions were seen in 55% of patients in clinical trials.
The prescribing information was recently revised to include SJS/TEN and to recommend permanent discontinuance of the drug if cases of SJS/TEN are suspected.
“This revision is intended to increase clinicians’ awareness of the risk for SJS/TEN and mitigate serious outcomes by improving the likelihood of early identification and intervention,” they added.
The authors also encouraged continued reporting of adverse events with enfortumab vedotin to the FDA via the MedWatch portal.
The authors disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
What’s under my toenail?
After the teledermatology consultation, an x-ray was recommended. The x-ray showed an elongated irregular radiopaque mass projecting from the anterior medial aspect of the midshaft of the distal phalanx of the great toe (Picture 3). With these findings, subungual exostosis was suspected, and she was referred to orthopedic surgery for excision of the lesion. Histopathology showed a stack of trabecular bone with a fibrocartilaginous cap, confirming the diagnosis of subungual exostosis.

Subungual exostosis is a benign osteocartilaginous tumor, first described by Dupuytren in 1874. These lesions are rare and are seen mainly in children and young adults. Females appear to be affected more often than males.1 In a systematic review by DaCambra and colleagues, 55% of the cases occur in patients aged younger than 18 years, and the hallux was the most commonly affected digit, though any finger or toe can be affected.2 There are reported case of congenital multiple exostosis delineated to translocation t(X;6)(q22;q13-14).3
The exact cause of these lesions is unknown, but there are multiple theories, which include a reactive process secondary to trauma, infection, or genetic causes. Pathologic examination of the lesions shows an osseous center covered by a fibrocartilaginous cap. There is proliferation of spindle cells that generate cartilage, which later forms trabecular bone.4
On physical examination, subungual exostosis appear like a firm, fixed nodule with a hyperkeratotic smooth surface at the distal end of the nail bed, that slowly grows and can distort and lift up the nail. Dermoscopy features of these lesions include vascular ectasia, hyperkeratosis, onycholysis, and ulceration.

The differential diagnosis of subungual growths includes osteochondromas, which can present in a similar way but are rarer. Pathologic examination is usually required to differentiate between both lesions.5 In exostoses, bone is formed directly from fibrous tissue, whereas in osteochondromas they derive from enchondral ossification.6 The cartilaginous cap of this lesion is what helps to differentiate it in histopathology. In subungual exostosis, the cap is composed of fibrocartilage, while in osteochondromas it is made of hyaline cartilage similar to what is seen in normal growing epiphysis.5 Subungual exostosis can be confused with pyogenic granulomas and verruca, and often are treated as such, which delays appropriate surgical management.
Firm, slow-growing tumors in the fingers or toes of children should raise suspicion for underlying bony lesions like subungual exostosis and osteochondromas. X-rays of the lesion should be performed in order to clarify the diagnosis. Referral to orthopedic surgery is needed for definitive surgical management.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
1. Zhang W et al. JAAD Case Rep. 2020 Jun 1;6(8):725-6.
2. DaCambra MP et al. Clin Orthop Relat Res. 2014 Apr;472(4):1251-9.
3. Torlazzi C et al. Int J Cancer. 2006;118:1972-6.
4. Calonje E et al. McKee’s pathology of the skin: With clinical correlations. (4th ed.) Philadelphia: Elsevier/Saunders, 2012.
5. Lee SK et al. Foot Ankle Int. 2007 May;28(5):595-601.
6. Mavrogenis A et al. Orthopedics. 2008 Oct;31(10).
After the teledermatology consultation, an x-ray was recommended. The x-ray showed an elongated irregular radiopaque mass projecting from the anterior medial aspect of the midshaft of the distal phalanx of the great toe (Picture 3). With these findings, subungual exostosis was suspected, and she was referred to orthopedic surgery for excision of the lesion. Histopathology showed a stack of trabecular bone with a fibrocartilaginous cap, confirming the diagnosis of subungual exostosis.
Subungual exostosis is a benign osteocartilaginous tumor, first described by Dupuytren in 1874. These lesions are rare and are seen mainly in children and young adults. Females appear to be affected more often than males.1 In a systematic review by DaCambra and colleagues, 55% of the cases occur in patients aged younger than 18 years, and the hallux was the most commonly affected digit, though any finger or toe can be affected.2 There are reported case of congenital multiple exostosis delineated to translocation t(X;6)(q22;q13-14).3
The exact cause of these lesions is unknown, but there are multiple theories, which include a reactive process secondary to trauma, infection, or genetic causes. Pathologic examination of the lesions shows an osseous center covered by a fibrocartilaginous cap. There is proliferation of spindle cells that generate cartilage, which later forms trabecular bone.4
On physical examination, subungual exostosis appear like a firm, fixed nodule with a hyperkeratotic smooth surface at the distal end of the nail bed, that slowly grows and can distort and lift up the nail. Dermoscopy features of these lesions include vascular ectasia, hyperkeratosis, onycholysis, and ulceration.
The differential diagnosis of subungual growths includes osteochondromas, which can present in a similar way but are rarer. Pathologic examination is usually required to differentiate between both lesions.5 In exostoses, bone is formed directly from fibrous tissue, whereas in osteochondromas they derive from enchondral ossification.6 The cartilaginous cap of this lesion is what helps to differentiate it in histopathology. In subungual exostosis, the cap is composed of fibrocartilage, while in osteochondromas it is made of hyaline cartilage similar to what is seen in normal growing epiphysis.5 Subungual exostosis can be confused with pyogenic granulomas and verruca, and often are treated as such, which delays appropriate surgical management.
Firm, slow-growing tumors in the fingers or toes of children should raise suspicion for underlying bony lesions like subungual exostosis and osteochondromas. X-rays of the lesion should be performed in order to clarify the diagnosis. Referral to orthopedic surgery is needed for definitive surgical management.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
1. Zhang W et al. JAAD Case Rep. 2020 Jun 1;6(8):725-6.
2. DaCambra MP et al. Clin Orthop Relat Res. 2014 Apr;472(4):1251-9.
3. Torlazzi C et al. Int J Cancer. 2006;118:1972-6.
4. Calonje E et al. McKee’s pathology of the skin: With clinical correlations. (4th ed.) Philadelphia: Elsevier/Saunders, 2012.
5. Lee SK et al. Foot Ankle Int. 2007 May;28(5):595-601.
6. Mavrogenis A et al. Orthopedics. 2008 Oct;31(10).
After the teledermatology consultation, an x-ray was recommended. The x-ray showed an elongated irregular radiopaque mass projecting from the anterior medial aspect of the midshaft of the distal phalanx of the great toe (Picture 3). With these findings, subungual exostosis was suspected, and she was referred to orthopedic surgery for excision of the lesion. Histopathology showed a stack of trabecular bone with a fibrocartilaginous cap, confirming the diagnosis of subungual exostosis.
Subungual exostosis is a benign osteocartilaginous tumor, first described by Dupuytren in 1874. These lesions are rare and are seen mainly in children and young adults. Females appear to be affected more often than males.1 In a systematic review by DaCambra and colleagues, 55% of the cases occur in patients aged younger than 18 years, and the hallux was the most commonly affected digit, though any finger or toe can be affected.2 There are reported case of congenital multiple exostosis delineated to translocation t(X;6)(q22;q13-14).3
The exact cause of these lesions is unknown, but there are multiple theories, which include a reactive process secondary to trauma, infection, or genetic causes. Pathologic examination of the lesions shows an osseous center covered by a fibrocartilaginous cap. There is proliferation of spindle cells that generate cartilage, which later forms trabecular bone.4
On physical examination, subungual exostosis appear like a firm, fixed nodule with a hyperkeratotic smooth surface at the distal end of the nail bed, that slowly grows and can distort and lift up the nail. Dermoscopy features of these lesions include vascular ectasia, hyperkeratosis, onycholysis, and ulceration.
The differential diagnosis of subungual growths includes osteochondromas, which can present in a similar way but are rarer. Pathologic examination is usually required to differentiate between both lesions.5 In exostoses, bone is formed directly from fibrous tissue, whereas in osteochondromas they derive from enchondral ossification.6 The cartilaginous cap of this lesion is what helps to differentiate it in histopathology. In subungual exostosis, the cap is composed of fibrocartilage, while in osteochondromas it is made of hyaline cartilage similar to what is seen in normal growing epiphysis.5 Subungual exostosis can be confused with pyogenic granulomas and verruca, and often are treated as such, which delays appropriate surgical management.
Firm, slow-growing tumors in the fingers or toes of children should raise suspicion for underlying bony lesions like subungual exostosis and osteochondromas. X-rays of the lesion should be performed in order to clarify the diagnosis. Referral to orthopedic surgery is needed for definitive surgical management.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
1. Zhang W et al. JAAD Case Rep. 2020 Jun 1;6(8):725-6.
2. DaCambra MP et al. Clin Orthop Relat Res. 2014 Apr;472(4):1251-9.
3. Torlazzi C et al. Int J Cancer. 2006;118:1972-6.
4. Calonje E et al. McKee’s pathology of the skin: With clinical correlations. (4th ed.) Philadelphia: Elsevier/Saunders, 2012.
5. Lee SK et al. Foot Ankle Int. 2007 May;28(5):595-601.
6. Mavrogenis A et al. Orthopedics. 2008 Oct;31(10).
A 13-year-old female was seen by her pediatrician for a lesion that had been on her right toe for about 6 months. She is unaware of any trauma to the area. The lesion has been growing slowly and recently it started lifting up the nail, became tender, and was bleeding, which is the reason why she sought care.
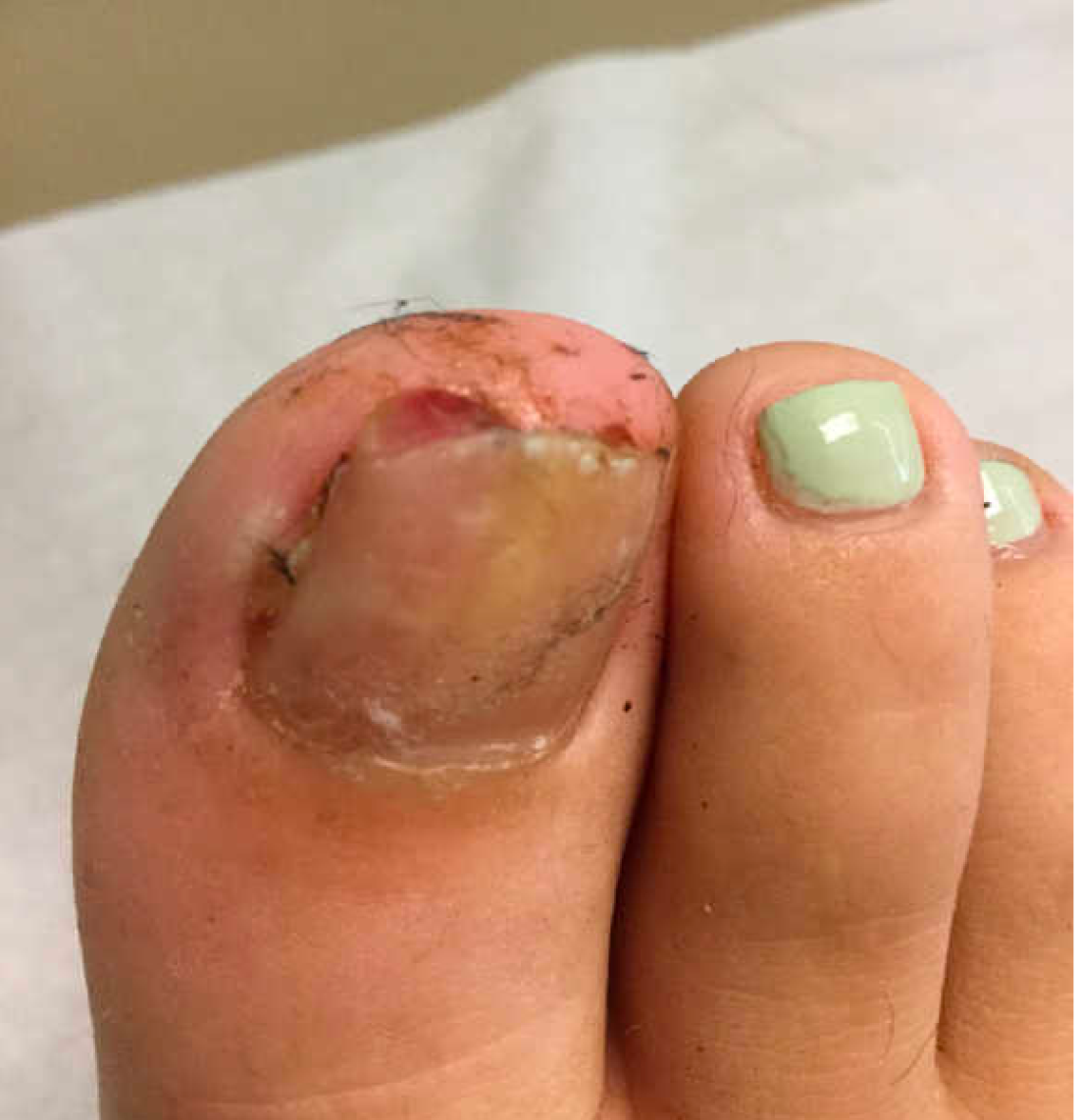
At the pediatrician's office, he noted a pink crusted papule under the nail. The nail was lifting up and was tender to the touch. She is a healthy girl who is not taking any medications and has no allergies. There is no family history of similar lesions.
The pediatrician took a picture of the lesion and he send it to our pediatric teledermatology service for consultation.
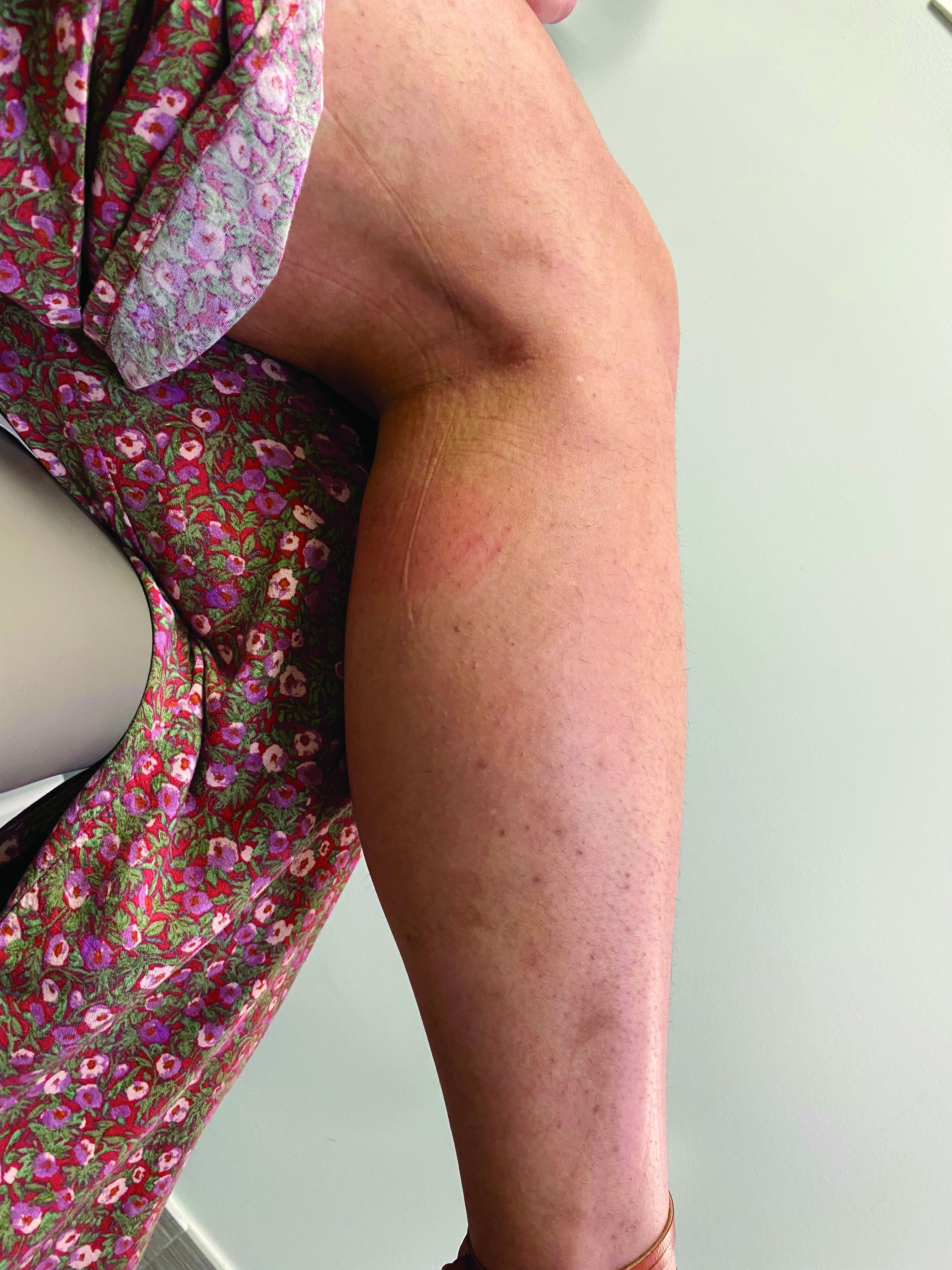
A 35-year-old with erythematous, dusky patches on both lower extremities
Zinc deficiency may be inherited or acquired. Acrodermatitis enteropathica is an autosomal recessive genetic disorder caused by a mutation in the gene that encodes a zinc transporter. It presents in infancy with the classic triad of diarrhea, dermatitis, and alopecia. Acquired zinc deficiency is due to causes such as alcoholism, malabsorption disorders like cystic fibrosis, inflammatory disease, gastrointestinal surgery, metabolic stress following general surgery, eating disorders, infections, malignancy, or occasionally in pregnancy. Classically, the face, groin, and extremities are affected (often acral), with erythematous, scaly patches. Pustules and bullae may be present. Angular cheilitis is often seen.
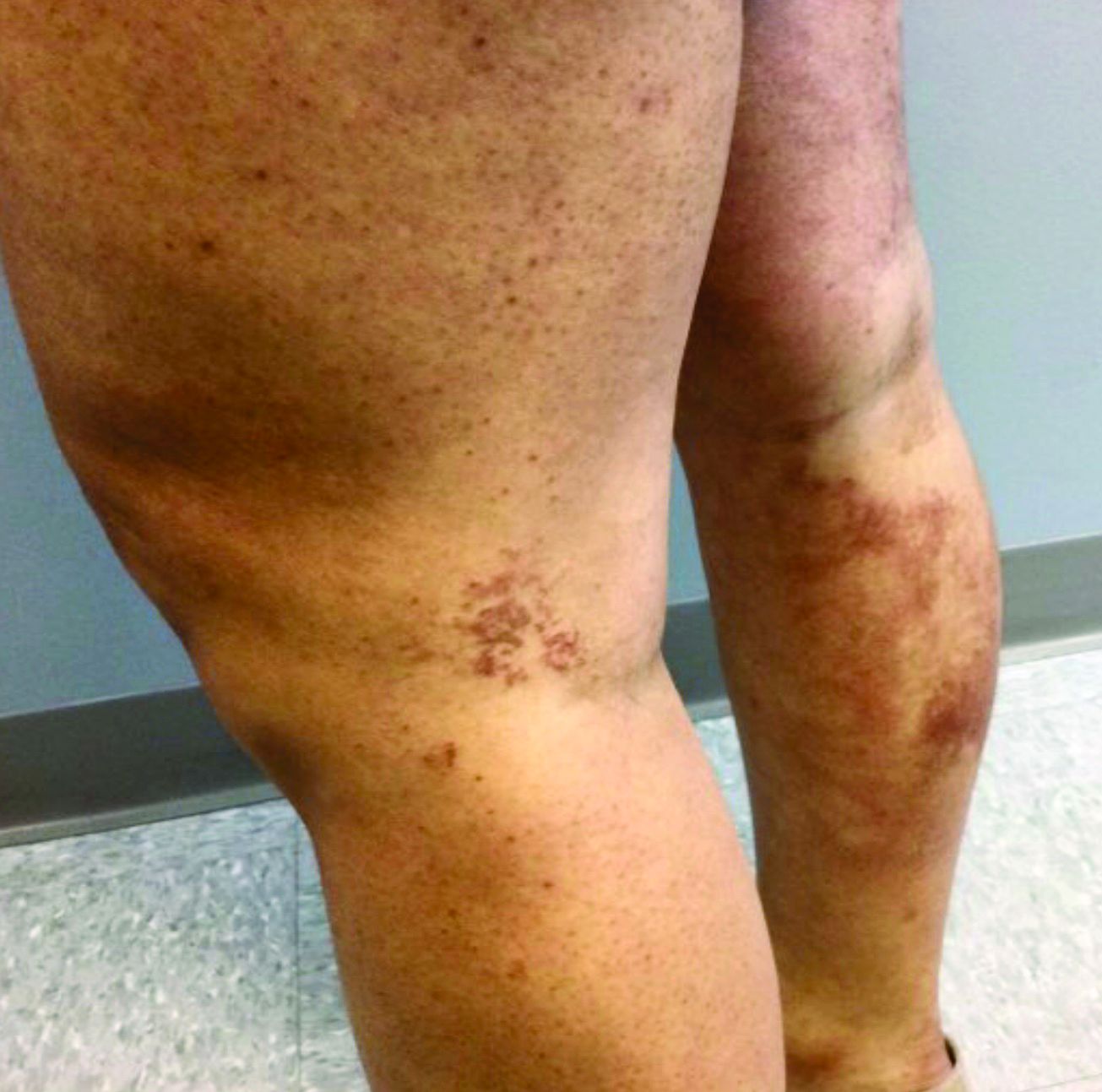
Necrolytic migratory erythema, or glucagonoma syndrome, is a very rare syndrome that presents as annular, erythematous patches with blisters that erode on the lower extremities and groin. The condition results from a cancerous tumor in the alpha cells of the pancreas called a glucagonoma, which secretes the hormone glucagon. It is often associated with diabetes and hyperglycemia.
Necrolytic acral erythema resembles acrodermatitis enteropathica and necrolytic migratory erythema clinically, however, it is associated with hepatitis C infection. Lesions are plaques with well defined borders distributed acrally. Treatment of the hepatitis C often improves the dermatitis.

Our patient’s blood work was consistent with nutritional deficiency and revealed low levels of zinc, vitamin A, ceruloplasmin, albumin and prealbumin, total protein, calcium, selenium, vitamin E, vitamin K, and vitamin C. Her hemoglobin A1C was under 4. Her hepatitis serologies were negative. The patient received total parenteral nutrition with subsequent complete resolution of her rash. Follow up for gastric bypass patients should be performed long term as they are at risk for nutritional deficiencies.
Dr. Bilu Martin, and Andrew Harris, DO, Mount Sinai Medical Center, Aventura, Fla., provided the case and photos.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
Dermatol Online J. 2016 Nov 15; 22(11):13030.
Andrews’ Disease of the Skin: Clinical Dermatology. Philadelphia: Saunders Elsevier, 2006.
Bolognia et al. Dermatology. St. Louis: Mosby/Elsevier, 2008.
Zinc deficiency may be inherited or acquired. Acrodermatitis enteropathica is an autosomal recessive genetic disorder caused by a mutation in the gene that encodes a zinc transporter. It presents in infancy with the classic triad of diarrhea, dermatitis, and alopecia. Acquired zinc deficiency is due to causes such as alcoholism, malabsorption disorders like cystic fibrosis, inflammatory disease, gastrointestinal surgery, metabolic stress following general surgery, eating disorders, infections, malignancy, or occasionally in pregnancy. Classically, the face, groin, and extremities are affected (often acral), with erythematous, scaly patches. Pustules and bullae may be present. Angular cheilitis is often seen.
Necrolytic migratory erythema, or glucagonoma syndrome, is a very rare syndrome that presents as annular, erythematous patches with blisters that erode on the lower extremities and groin. The condition results from a cancerous tumor in the alpha cells of the pancreas called a glucagonoma, which secretes the hormone glucagon. It is often associated with diabetes and hyperglycemia.
Necrolytic acral erythema resembles acrodermatitis enteropathica and necrolytic migratory erythema clinically, however, it is associated with hepatitis C infection. Lesions are plaques with well defined borders distributed acrally. Treatment of the hepatitis C often improves the dermatitis.
Our patient’s blood work was consistent with nutritional deficiency and revealed low levels of zinc, vitamin A, ceruloplasmin, albumin and prealbumin, total protein, calcium, selenium, vitamin E, vitamin K, and vitamin C. Her hemoglobin A1C was under 4. Her hepatitis serologies were negative. The patient received total parenteral nutrition with subsequent complete resolution of her rash. Follow up for gastric bypass patients should be performed long term as they are at risk for nutritional deficiencies.
Dr. Bilu Martin, and Andrew Harris, DO, Mount Sinai Medical Center, Aventura, Fla., provided the case and photos.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
Dermatol Online J. 2016 Nov 15; 22(11):13030.
Andrews’ Disease of the Skin: Clinical Dermatology. Philadelphia: Saunders Elsevier, 2006.
Bolognia et al. Dermatology. St. Louis: Mosby/Elsevier, 2008.
Zinc deficiency may be inherited or acquired. Acrodermatitis enteropathica is an autosomal recessive genetic disorder caused by a mutation in the gene that encodes a zinc transporter. It presents in infancy with the classic triad of diarrhea, dermatitis, and alopecia. Acquired zinc deficiency is due to causes such as alcoholism, malabsorption disorders like cystic fibrosis, inflammatory disease, gastrointestinal surgery, metabolic stress following general surgery, eating disorders, infections, malignancy, or occasionally in pregnancy. Classically, the face, groin, and extremities are affected (often acral), with erythematous, scaly patches. Pustules and bullae may be present. Angular cheilitis is often seen.
Necrolytic migratory erythema, or glucagonoma syndrome, is a very rare syndrome that presents as annular, erythematous patches with blisters that erode on the lower extremities and groin. The condition results from a cancerous tumor in the alpha cells of the pancreas called a glucagonoma, which secretes the hormone glucagon. It is often associated with diabetes and hyperglycemia.
Necrolytic acral erythema resembles acrodermatitis enteropathica and necrolytic migratory erythema clinically, however, it is associated with hepatitis C infection. Lesions are plaques with well defined borders distributed acrally. Treatment of the hepatitis C often improves the dermatitis.
Our patient’s blood work was consistent with nutritional deficiency and revealed low levels of zinc, vitamin A, ceruloplasmin, albumin and prealbumin, total protein, calcium, selenium, vitamin E, vitamin K, and vitamin C. Her hemoglobin A1C was under 4. Her hepatitis serologies were negative. The patient received total parenteral nutrition with subsequent complete resolution of her rash. Follow up for gastric bypass patients should be performed long term as they are at risk for nutritional deficiencies.
Dr. Bilu Martin, and Andrew Harris, DO, Mount Sinai Medical Center, Aventura, Fla., provided the case and photos.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
Dermatol Online J. 2016 Nov 15; 22(11):13030.
Andrews’ Disease of the Skin: Clinical Dermatology. Philadelphia: Saunders Elsevier, 2006.
Bolognia et al. Dermatology. St. Louis: Mosby/Elsevier, 2008.
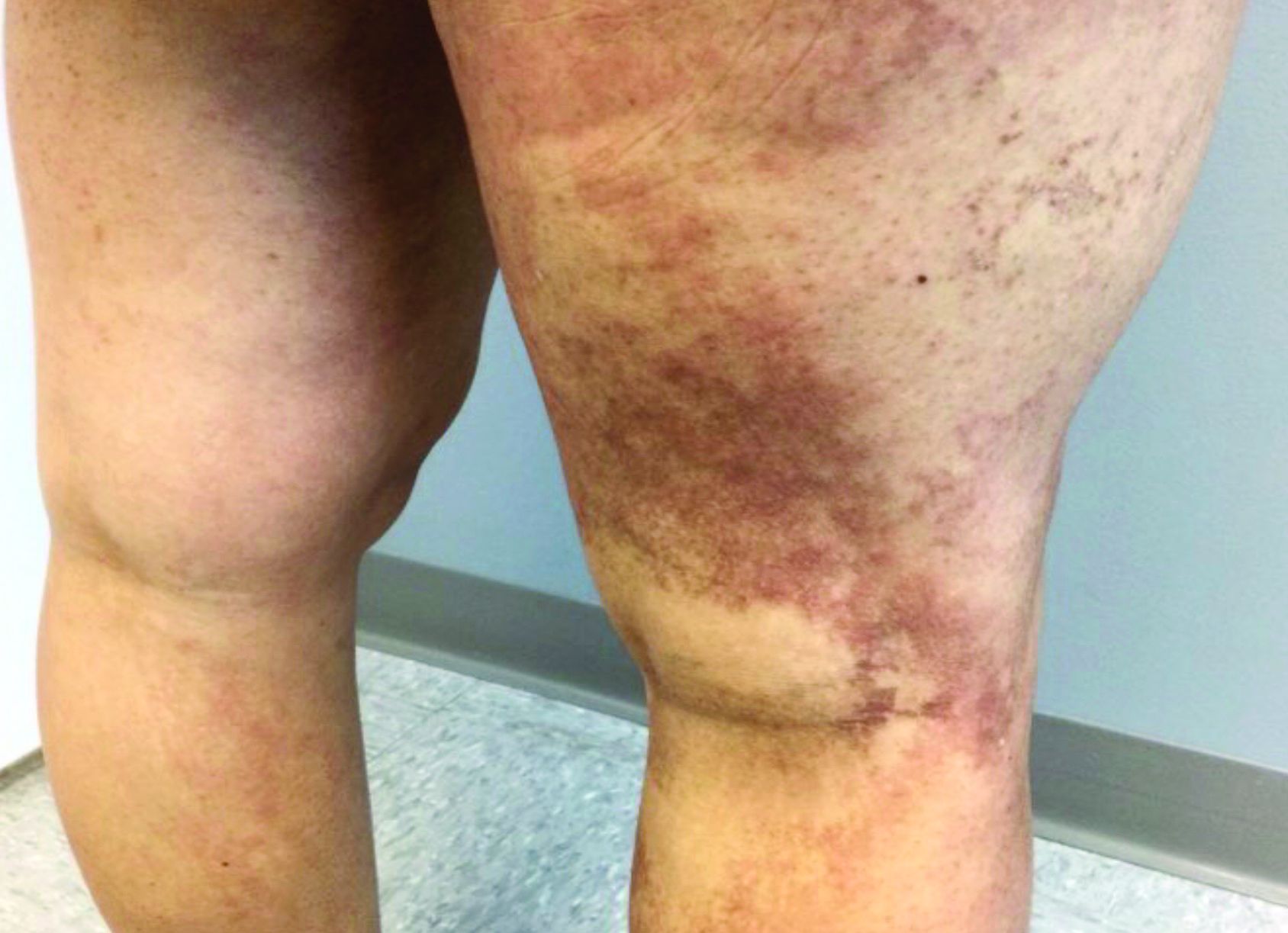
Dyspigmentation common in SOC patients with bullous pemphigoid
Patients of skin of color (SOC) with bullous pemphigoid presented significantly more often with dyspigmentation than did White patients in a retrospective observational study of patients diagnosed with BP at New York University Langone Health and Bellevue Hospital, also in New York.
“Dyspigmentation in the skin-of-color patient population is important to recognize not only for an objective evaluation of the disease process, but also from a quality of life perspective ... to ensure there is timely diagnosis and initiation of treatment in the skin-of-color population,” said medical student Payal Shah, BS, of New York University, in presenting the findings at the annual Skin of Color Society symposium.
Ms. Shah and coresearchers identified 94 cases of BP through retrospective view of electronic health records – 59 in White patients and 35 in SOC patients. The physical examination features most commonly found at initial presentation were bullae or vesicles in both White patients (64.4% ) and SOC patients (80%). Erosions or ulcers were also commonly found in both groups (42.4% of White patients and 60% of SOC patients).
Erythema was more commonly found in White patients at initial presentation: 35.6% vs. 14.3% of SOC patients (P = .032). Dyspigmentation, defined as areas of hyper- or hypopigmentation, was more commonly found in SOC patients: 54.3% versus 10.2% in White patients (P < .001). The difference in erythema of inflammatory bullae in BP may stem from the fact that erythema is more difficult to discern in patients with darker skin types, Ms. Shah said.
SOC patients also were significantly younger at the time of initial presentation; their mean age was 63 years, compared with 77 years in the White population (P < .001).
The time to diagnosis, defined as the time from initial symptoms to dermatologic diagnosis, was greater for the SOC population –7.6 months vs. 6.2 months for white patients –though the difference was not statistically significant, they said in the abstract .
Dyspigmentation has been shown to be among the top dermatologic concerns of Black patients and has important quality of life implications. “Early diagnosis to prevent difficult-to-treat dyspigmentation is therefore of utmost importance,” they said in the abstract.
Prior research has demonstrated that non-White populations are at greater risk for hospitalization secondary to BP and have a greater risk of disease mortality, Ms. Shah noted in her presentation.
Patients of skin of color (SOC) with bullous pemphigoid presented significantly more often with dyspigmentation than did White patients in a retrospective observational study of patients diagnosed with BP at New York University Langone Health and Bellevue Hospital, also in New York.
“Dyspigmentation in the skin-of-color patient population is important to recognize not only for an objective evaluation of the disease process, but also from a quality of life perspective ... to ensure there is timely diagnosis and initiation of treatment in the skin-of-color population,” said medical student Payal Shah, BS, of New York University, in presenting the findings at the annual Skin of Color Society symposium.
Ms. Shah and coresearchers identified 94 cases of BP through retrospective view of electronic health records – 59 in White patients and 35 in SOC patients. The physical examination features most commonly found at initial presentation were bullae or vesicles in both White patients (64.4% ) and SOC patients (80%). Erosions or ulcers were also commonly found in both groups (42.4% of White patients and 60% of SOC patients).
Erythema was more commonly found in White patients at initial presentation: 35.6% vs. 14.3% of SOC patients (P = .032). Dyspigmentation, defined as areas of hyper- or hypopigmentation, was more commonly found in SOC patients: 54.3% versus 10.2% in White patients (P < .001). The difference in erythema of inflammatory bullae in BP may stem from the fact that erythema is more difficult to discern in patients with darker skin types, Ms. Shah said.
SOC patients also were significantly younger at the time of initial presentation; their mean age was 63 years, compared with 77 years in the White population (P < .001).
The time to diagnosis, defined as the time from initial symptoms to dermatologic diagnosis, was greater for the SOC population –7.6 months vs. 6.2 months for white patients –though the difference was not statistically significant, they said in the abstract .
Dyspigmentation has been shown to be among the top dermatologic concerns of Black patients and has important quality of life implications. “Early diagnosis to prevent difficult-to-treat dyspigmentation is therefore of utmost importance,” they said in the abstract.
Prior research has demonstrated that non-White populations are at greater risk for hospitalization secondary to BP and have a greater risk of disease mortality, Ms. Shah noted in her presentation.
Patients of skin of color (SOC) with bullous pemphigoid presented significantly more often with dyspigmentation than did White patients in a retrospective observational study of patients diagnosed with BP at New York University Langone Health and Bellevue Hospital, also in New York.
“Dyspigmentation in the skin-of-color patient population is important to recognize not only for an objective evaluation of the disease process, but also from a quality of life perspective ... to ensure there is timely diagnosis and initiation of treatment in the skin-of-color population,” said medical student Payal Shah, BS, of New York University, in presenting the findings at the annual Skin of Color Society symposium.
Ms. Shah and coresearchers identified 94 cases of BP through retrospective view of electronic health records – 59 in White patients and 35 in SOC patients. The physical examination features most commonly found at initial presentation were bullae or vesicles in both White patients (64.4% ) and SOC patients (80%). Erosions or ulcers were also commonly found in both groups (42.4% of White patients and 60% of SOC patients).
Erythema was more commonly found in White patients at initial presentation: 35.6% vs. 14.3% of SOC patients (P = .032). Dyspigmentation, defined as areas of hyper- or hypopigmentation, was more commonly found in SOC patients: 54.3% versus 10.2% in White patients (P < .001). The difference in erythema of inflammatory bullae in BP may stem from the fact that erythema is more difficult to discern in patients with darker skin types, Ms. Shah said.
SOC patients also were significantly younger at the time of initial presentation; their mean age was 63 years, compared with 77 years in the White population (P < .001).
The time to diagnosis, defined as the time from initial symptoms to dermatologic diagnosis, was greater for the SOC population –7.6 months vs. 6.2 months for white patients –though the difference was not statistically significant, they said in the abstract .
Dyspigmentation has been shown to be among the top dermatologic concerns of Black patients and has important quality of life implications. “Early diagnosis to prevent difficult-to-treat dyspigmentation is therefore of utmost importance,” they said in the abstract.
Prior research has demonstrated that non-White populations are at greater risk for hospitalization secondary to BP and have a greater risk of disease mortality, Ms. Shah noted in her presentation.
FROM SOC 2021
Expert proposes rethinking the classification of SJS/TEN
In the opinion of Neil H. Shear, MD, a stepwise approach is the best way to diagnose possible drug-induced skin disease and determine the root cause.

“Often, we need to think of more than one cause,” he said during the annual meeting of the Society for Pediatric Dermatology. “It could be drug X. It could be drug Y. It could be contrast media. We must think broadly and pay special attention to skin of color, overlapping syndromes, and the changing diagnostic assessment over time.”
His suggested diagnostic triangle includes appearance of the rash or lesion(s), systemic impact, and histology. “The first is the appearance,” said Dr. Shear, professor emeritus of dermatology, clinical pharmacology and toxicology, and medicine at the University of Toronto. “Is it exanthem? Is it blistering? Don’t just say drug ‘rash.’ That doesn’t work. You need to know if there are systemic features, and sometimes histologic information can change your approach or diagnosis, but not as often as one might think,” he said, noting that, in his view, the two main factors are appearance and systemic impact.
The presence of fever is a hallmark of systemic problems, he continued, “so if you see fever, you know you’re probably going to be dealing with a complex reaction, so we need to know the morphology.” Consider whether it is simple exanthem (a mild, uncomplicated rash) or complex exanthem (drug rash with eosinophilia and systemic symptoms or fever, malaise, and adenopathy).
As for other morphologies, urticarial lesions could be urticaria or a serum sickness-like reaction, pustular lesions could be acneiform or acute generalized exanthematous pustulosis, while
Dr. Shear considers SJS/TEN as a spectrum of blistering disease, “because there’s not a single diagnosis,” he said. “There’s a spectrum, if you will, depending on how advanced people are in their disease.” He coauthored a 1991 report describing eight cases of mycoplasma and Stevens-Johnson syndrome. “I was surprised at how long that stood up as about the only paper in that area,” he said. “But there’s much more happening now with a proliferation of terms,” he added, referring to MIRM (Mycoplasma pneumonia–induced rash and mucositis), RIME (reactive infectious mucocutaneous eruption); and Fuchs syndrome, or SJS without skin lesions.
What was not appreciated in the early classification of SJS, he continued, was a “side basket” of bullous erythema multiforme. “We didn’t know what to call it,” he said. “At one point we called it bullous erythema multiforme. At another point we called it erythema multiforme major. We just didn’t know what it was.”
The appearance and systemic effects of SJS comprise what he termed SJS type 2 – or the early stages of TEN. Taken together, he refers to these two conditions as TEN Spectrum, or TENS. “One of the traps is that TENS can look like varicella, and vice versa, especially in very dark brown or black skin,” Dr. Shear said. “You have to be careful. A biopsy might be worthwhile. Acute lupus has the pathology of TENS but the patients are not as systemically ill as true TENS.”
In 2011, Japanese researchers reported on 38 cases of SJS associated with M. pneumoniae, and 78 cases of drug-induced SJS. They found that 66% of adult patients with M. pneumoniae–associated SJS developed mucocutaneous lesions and fever/respiratory symptoms on the same day, mostly shortness of breath and cough. In contrast, most of the patients aged under 20 years developed fever/respiratory symptoms before mucocutaneous involvement.
“The big clinical differentiator between drug-induced SJS and mycoplasma-induced SJS was respiratory disorder,” said Dr. Shear, who was not affiliated with the study. “That means you’re probably looking at something that’s mycoplasma related [when respiratory problems are present]. Even if you can’t prove it’s mycoplasma related, that probably needs to be the target of your therapy. The idea ... is to make sure it’s clear at the end. One, so they get better, and two, so that we’re not giving drugs needlessly when it was really mycoplasma.”
Noting that HLA-B*15:02 is a marker for carbamazepine-induced SJS and TEN, he said, “a positive HLA test can support the diagnosis, confirm the suspected offending drug, and is valuable for familial genetic counseling.”
As for treatment of SJS, TEN, and other cytotoxic T-lymphocyte–mediated severe cutaneous adverse reactions, a randomized Japanese clinical trial evaluating prednisolone 1-1.5 mg/kg/day IV versus etanercept 25-50 mg subcutaneously twice per week in 96 patients with SJS-TEN found that etanercept decreased the mortality rate by 8.3%. In addition, etanercept reduced skin healing time, when compared with prednisolone (a median of 14 vs. 19 days, respectively; P = .010), and was associated with a lower incidence of GI hemorrhage (2.6% vs. 18.2%, respectively; P = .03).
Dr. Shear said that he would like to see better therapeutics for severe, complex patients. “After leaving the hospital, people with SJS or people with TEN need to have ongoing care, consultation, and explanation so they and their families know what drugs are safe in the future.”
Dr. Shear disclosed that he has been a consultant to AbbVie, Amgen, Bausch Medicine, Novartis, Sanofi-Genzyme, UCB, LEO Pharma, Otsuka, Janssen, Alpha Laboratories, Lilly, ChemoCentryx, Vivoryon, Galderma, Innovaderm, Chromocell, and Kyowa Kirin.
In the opinion of Neil H. Shear, MD, a stepwise approach is the best way to diagnose possible drug-induced skin disease and determine the root cause.
“Often, we need to think of more than one cause,” he said during the annual meeting of the Society for Pediatric Dermatology. “It could be drug X. It could be drug Y. It could be contrast media. We must think broadly and pay special attention to skin of color, overlapping syndromes, and the changing diagnostic assessment over time.”
His suggested diagnostic triangle includes appearance of the rash or lesion(s), systemic impact, and histology. “The first is the appearance,” said Dr. Shear, professor emeritus of dermatology, clinical pharmacology and toxicology, and medicine at the University of Toronto. “Is it exanthem? Is it blistering? Don’t just say drug ‘rash.’ That doesn’t work. You need to know if there are systemic features, and sometimes histologic information can change your approach or diagnosis, but not as often as one might think,” he said, noting that, in his view, the two main factors are appearance and systemic impact.
The presence of fever is a hallmark of systemic problems, he continued, “so if you see fever, you know you’re probably going to be dealing with a complex reaction, so we need to know the morphology.” Consider whether it is simple exanthem (a mild, uncomplicated rash) or complex exanthem (drug rash with eosinophilia and systemic symptoms or fever, malaise, and adenopathy).
As for other morphologies, urticarial lesions could be urticaria or a serum sickness-like reaction, pustular lesions could be acneiform or acute generalized exanthematous pustulosis, while
Dr. Shear considers SJS/TEN as a spectrum of blistering disease, “because there’s not a single diagnosis,” he said. “There’s a spectrum, if you will, depending on how advanced people are in their disease.” He coauthored a 1991 report describing eight cases of mycoplasma and Stevens-Johnson syndrome. “I was surprised at how long that stood up as about the only paper in that area,” he said. “But there’s much more happening now with a proliferation of terms,” he added, referring to MIRM (Mycoplasma pneumonia–induced rash and mucositis), RIME (reactive infectious mucocutaneous eruption); and Fuchs syndrome, or SJS without skin lesions.
What was not appreciated in the early classification of SJS, he continued, was a “side basket” of bullous erythema multiforme. “We didn’t know what to call it,” he said. “At one point we called it bullous erythema multiforme. At another point we called it erythema multiforme major. We just didn’t know what it was.”
The appearance and systemic effects of SJS comprise what he termed SJS type 2 – or the early stages of TEN. Taken together, he refers to these two conditions as TEN Spectrum, or TENS. “One of the traps is that TENS can look like varicella, and vice versa, especially in very dark brown or black skin,” Dr. Shear said. “You have to be careful. A biopsy might be worthwhile. Acute lupus has the pathology of TENS but the patients are not as systemically ill as true TENS.”
In 2011, Japanese researchers reported on 38 cases of SJS associated with M. pneumoniae, and 78 cases of drug-induced SJS. They found that 66% of adult patients with M. pneumoniae–associated SJS developed mucocutaneous lesions and fever/respiratory symptoms on the same day, mostly shortness of breath and cough. In contrast, most of the patients aged under 20 years developed fever/respiratory symptoms before mucocutaneous involvement.
“The big clinical differentiator between drug-induced SJS and mycoplasma-induced SJS was respiratory disorder,” said Dr. Shear, who was not affiliated with the study. “That means you’re probably looking at something that’s mycoplasma related [when respiratory problems are present]. Even if you can’t prove it’s mycoplasma related, that probably needs to be the target of your therapy. The idea ... is to make sure it’s clear at the end. One, so they get better, and two, so that we’re not giving drugs needlessly when it was really mycoplasma.”
Noting that HLA-B*15:02 is a marker for carbamazepine-induced SJS and TEN, he said, “a positive HLA test can support the diagnosis, confirm the suspected offending drug, and is valuable for familial genetic counseling.”
As for treatment of SJS, TEN, and other cytotoxic T-lymphocyte–mediated severe cutaneous adverse reactions, a randomized Japanese clinical trial evaluating prednisolone 1-1.5 mg/kg/day IV versus etanercept 25-50 mg subcutaneously twice per week in 96 patients with SJS-TEN found that etanercept decreased the mortality rate by 8.3%. In addition, etanercept reduced skin healing time, when compared with prednisolone (a median of 14 vs. 19 days, respectively; P = .010), and was associated with a lower incidence of GI hemorrhage (2.6% vs. 18.2%, respectively; P = .03).
Dr. Shear said that he would like to see better therapeutics for severe, complex patients. “After leaving the hospital, people with SJS or people with TEN need to have ongoing care, consultation, and explanation so they and their families know what drugs are safe in the future.”
Dr. Shear disclosed that he has been a consultant to AbbVie, Amgen, Bausch Medicine, Novartis, Sanofi-Genzyme, UCB, LEO Pharma, Otsuka, Janssen, Alpha Laboratories, Lilly, ChemoCentryx, Vivoryon, Galderma, Innovaderm, Chromocell, and Kyowa Kirin.
In the opinion of Neil H. Shear, MD, a stepwise approach is the best way to diagnose possible drug-induced skin disease and determine the root cause.
“Often, we need to think of more than one cause,” he said during the annual meeting of the Society for Pediatric Dermatology. “It could be drug X. It could be drug Y. It could be contrast media. We must think broadly and pay special attention to skin of color, overlapping syndromes, and the changing diagnostic assessment over time.”
His suggested diagnostic triangle includes appearance of the rash or lesion(s), systemic impact, and histology. “The first is the appearance,” said Dr. Shear, professor emeritus of dermatology, clinical pharmacology and toxicology, and medicine at the University of Toronto. “Is it exanthem? Is it blistering? Don’t just say drug ‘rash.’ That doesn’t work. You need to know if there are systemic features, and sometimes histologic information can change your approach or diagnosis, but not as often as one might think,” he said, noting that, in his view, the two main factors are appearance and systemic impact.
The presence of fever is a hallmark of systemic problems, he continued, “so if you see fever, you know you’re probably going to be dealing with a complex reaction, so we need to know the morphology.” Consider whether it is simple exanthem (a mild, uncomplicated rash) or complex exanthem (drug rash with eosinophilia and systemic symptoms or fever, malaise, and adenopathy).
As for other morphologies, urticarial lesions could be urticaria or a serum sickness-like reaction, pustular lesions could be acneiform or acute generalized exanthematous pustulosis, while
Dr. Shear considers SJS/TEN as a spectrum of blistering disease, “because there’s not a single diagnosis,” he said. “There’s a spectrum, if you will, depending on how advanced people are in their disease.” He coauthored a 1991 report describing eight cases of mycoplasma and Stevens-Johnson syndrome. “I was surprised at how long that stood up as about the only paper in that area,” he said. “But there’s much more happening now with a proliferation of terms,” he added, referring to MIRM (Mycoplasma pneumonia–induced rash and mucositis), RIME (reactive infectious mucocutaneous eruption); and Fuchs syndrome, or SJS without skin lesions.
What was not appreciated in the early classification of SJS, he continued, was a “side basket” of bullous erythema multiforme. “We didn’t know what to call it,” he said. “At one point we called it bullous erythema multiforme. At another point we called it erythema multiforme major. We just didn’t know what it was.”
The appearance and systemic effects of SJS comprise what he termed SJS type 2 – or the early stages of TEN. Taken together, he refers to these two conditions as TEN Spectrum, or TENS. “One of the traps is that TENS can look like varicella, and vice versa, especially in very dark brown or black skin,” Dr. Shear said. “You have to be careful. A biopsy might be worthwhile. Acute lupus has the pathology of TENS but the patients are not as systemically ill as true TENS.”
In 2011, Japanese researchers reported on 38 cases of SJS associated with M. pneumoniae, and 78 cases of drug-induced SJS. They found that 66% of adult patients with M. pneumoniae–associated SJS developed mucocutaneous lesions and fever/respiratory symptoms on the same day, mostly shortness of breath and cough. In contrast, most of the patients aged under 20 years developed fever/respiratory symptoms before mucocutaneous involvement.
“The big clinical differentiator between drug-induced SJS and mycoplasma-induced SJS was respiratory disorder,” said Dr. Shear, who was not affiliated with the study. “That means you’re probably looking at something that’s mycoplasma related [when respiratory problems are present]. Even if you can’t prove it’s mycoplasma related, that probably needs to be the target of your therapy. The idea ... is to make sure it’s clear at the end. One, so they get better, and two, so that we’re not giving drugs needlessly when it was really mycoplasma.”
Noting that HLA-B*15:02 is a marker for carbamazepine-induced SJS and TEN, he said, “a positive HLA test can support the diagnosis, confirm the suspected offending drug, and is valuable for familial genetic counseling.”
As for treatment of SJS, TEN, and other cytotoxic T-lymphocyte–mediated severe cutaneous adverse reactions, a randomized Japanese clinical trial evaluating prednisolone 1-1.5 mg/kg/day IV versus etanercept 25-50 mg subcutaneously twice per week in 96 patients with SJS-TEN found that etanercept decreased the mortality rate by 8.3%. In addition, etanercept reduced skin healing time, when compared with prednisolone (a median of 14 vs. 19 days, respectively; P = .010), and was associated with a lower incidence of GI hemorrhage (2.6% vs. 18.2%, respectively; P = .03).
Dr. Shear said that he would like to see better therapeutics for severe, complex patients. “After leaving the hospital, people with SJS or people with TEN need to have ongoing care, consultation, and explanation so they and their families know what drugs are safe in the future.”
Dr. Shear disclosed that he has been a consultant to AbbVie, Amgen, Bausch Medicine, Novartis, Sanofi-Genzyme, UCB, LEO Pharma, Otsuka, Janssen, Alpha Laboratories, Lilly, ChemoCentryx, Vivoryon, Galderma, Innovaderm, Chromocell, and Kyowa Kirin.
FROM SPD 2021
Several uncommon skin disorders related to internal diseases reviewed
and may spawn misdiagnoses, a dermatologist told colleagues.
“Proper diagnosis can lead to an effective management in our patients,” said Jeffrey Callen, MD, professor of medicine and chief of dermatology at the University of Louisville (Ky.), who spoke at the Inaugural Symposium for Inflammatory Skin Disease.
Sarcoidosis
The cause of sarcoidosis, an inflammatory disease that tends to affect the lungs, “is unknown, but it’s probably an immunologic disorder,” Dr. Callen said, “and there probably is a genetic predisposition.” About 20%-25% of patients with sarcoidosis have skin lesions that are either “specific” (a biopsy that reveals a noncaseating – “naked” – granuloma) or “nonspecific” (most commonly, erythema nodosum, or EN).
The specific lesions in sarcoidosis may occur in parts of the body, such as the knees, which were injured earlier in life and may have taken in foreign bodies, Dr. Callen said. As for nonspecific lesions, about 20% of patients with EN have an acute, self-limiting form of sarcoidosis. “These patients will have bilateral hilar lymphadenopathy, anterior uveitis, and polyarthritis. It’s generally treated symptomatically because it goes away on its own.”
He cautioned colleagues to beware of indurated, infiltrative facial lesions known as lupus pernio that are commonly found on the nose. They’re more prevalent in Black patients and possibly women, who are at higher risk of manifestations outside the skin, he said. “If you have it along the nasal rim, you should look into the upper respiratory tract for involvement.”
Dr. Callen recommends an extensive workup in patients with suspected sarcoidosis, including biopsy (with the exception of EN lesions), cultures and special stains, and screening when appropriate, for disease in organs such as the eyes, lungs, heart, and kidneys.
As for treatment, “the disease is in the dermis, and some topical therapies are not highly effective,” he said. There are injections that can be given, including corticosteroids, and there are a variety of oral treatments that are all off label.” These include corticosteroids, antimalarials, allopurinol, and tetracyclines, among several others. Subcutaneous and intravenous treatments are also options, along with surgery and laser therapy to treat specific lesions.
Rosai-Dorfman disease
This rare disorder is caused by overproduction of certain white blood cells in the lymph nodes, which can cause nodular lesions. The disease most often appears in children and young adults, often Black individuals and males. It is fatal in as many as 11% of patients, justifying aggressive treatment in patients with aggressive disease, Dr. Callen said. When it’s limited to the skin, however, “nothing may need to be done.”
Dr. Callen highlighted consensus recommendations about diagnosis and treatment of Rosai-Dorfman disease published in 2018.
He also noted the existence of cutaneous Rosai-Dorfman disease, a “solitary process” that appears more commonly in females, and in people of Asian heritage, compared with White individuals. It is characterized by single, clustered or widespread lesions: They can be xanthomatous, erythematous, or red-brown papules, nodules, and plaques. They’re acneiform, pustular, giant granuloma annulare–like, subcutaneous, and vasculitis-like, he said.
While Rosai-Dorfman disease can be linked to lymphoma, hypothyroidism, and lupus erythematosus, “nothing necessarily needs to be done when it’s skin-limited since it can be self-resolving,” he noted. Other treatments include radiotherapy, cryotherapy, excision, topical and oral corticosteroids, thalidomide, and methotrexate.
The disease can be serious, and is fatal in 5% of cases. When a vital organ is threatened, Dr. Callen suggested surgery, chemotherapy, or radiation.
Erdheim-Chester disease
This disease – which is extremely rare, with just 500 cases noted before 2014 – occurs when the body overproduces macrophages. It’s most common in middle-aged people and in men, who make up 75% of cases. About a quarter of patients develop skin lesions: Red-brown to yellow nodules and xanthelasma-like indurated plaques on the eyelids, scalp, neck, trunk, and axillae, and “other cutaneous manifestations have been reported in patients,” Dr. Callen said.
The disease also frequently affects the bones, large vessels, heart, lungs, and central nervous system. Interferon-alpha is the first-line treatment, and there are several other alternative therapies, although 5-year survival (68%) is poor, and it is especially likely to be fatal in those with central nervous system involvement.
Eosinophilic fasciitis
Eosinophilic fasciitis (EF) “is a disorder of unknown etiology that causes sclerosis of the skin” without Raynaud’s phenomenon, Dr. Callen said. Look for erythema, swelling, and induration of the extremities that is accompanied by peripheral eosinophilia, and if necessary, confirm the diagnosis with full skin-to-muscle biopsy or MRI.
There are many possible triggers, including strenuous exercise, initiation with hemodialysis, radiation therapy and burns, and graft-versus-host disease. Other potential causes include exposure to medications such as statins, phenytoin, ramipril, subcutaneous heparin, and immune checkpoint inhibitor therapy. The disorder is also linked to autoimmune and hematologic disorders.
Dr. Callen, who highlighted EF guidelines published in 2018, said treatments include physical therapy, prednisone, methotrexate, mycophenolate, and hydroxychloroquine.
Metastatic Crohn’s disease
This is a rare granulomatous inflammation of skin that often affects the genitals, especially in children. It is noncontiguous with the GI tract, and severity of skin involvement does not always parallel the severity of the disease in the GI tract, Dr. Callen said. However, the condition can occur before or simultaneously with the development of GI disease, or after GI surgery.
He highlighted a review of metastatic Crohn’s disease, published in 2014, and noted that there are multiple treatments, including systemic corticosteroids, tumor necrosis factor–alpha inhibitors, and topical therapies.
Dr. Callen reported no relevant disclosures.
and may spawn misdiagnoses, a dermatologist told colleagues.
“Proper diagnosis can lead to an effective management in our patients,” said Jeffrey Callen, MD, professor of medicine and chief of dermatology at the University of Louisville (Ky.), who spoke at the Inaugural Symposium for Inflammatory Skin Disease.
Sarcoidosis
The cause of sarcoidosis, an inflammatory disease that tends to affect the lungs, “is unknown, but it’s probably an immunologic disorder,” Dr. Callen said, “and there probably is a genetic predisposition.” About 20%-25% of patients with sarcoidosis have skin lesions that are either “specific” (a biopsy that reveals a noncaseating – “naked” – granuloma) or “nonspecific” (most commonly, erythema nodosum, or EN).
The specific lesions in sarcoidosis may occur in parts of the body, such as the knees, which were injured earlier in life and may have taken in foreign bodies, Dr. Callen said. As for nonspecific lesions, about 20% of patients with EN have an acute, self-limiting form of sarcoidosis. “These patients will have bilateral hilar lymphadenopathy, anterior uveitis, and polyarthritis. It’s generally treated symptomatically because it goes away on its own.”
He cautioned colleagues to beware of indurated, infiltrative facial lesions known as lupus pernio that are commonly found on the nose. They’re more prevalent in Black patients and possibly women, who are at higher risk of manifestations outside the skin, he said. “If you have it along the nasal rim, you should look into the upper respiratory tract for involvement.”
Dr. Callen recommends an extensive workup in patients with suspected sarcoidosis, including biopsy (with the exception of EN lesions), cultures and special stains, and screening when appropriate, for disease in organs such as the eyes, lungs, heart, and kidneys.
As for treatment, “the disease is in the dermis, and some topical therapies are not highly effective,” he said. There are injections that can be given, including corticosteroids, and there are a variety of oral treatments that are all off label.” These include corticosteroids, antimalarials, allopurinol, and tetracyclines, among several others. Subcutaneous and intravenous treatments are also options, along with surgery and laser therapy to treat specific lesions.
Rosai-Dorfman disease
This rare disorder is caused by overproduction of certain white blood cells in the lymph nodes, which can cause nodular lesions. The disease most often appears in children and young adults, often Black individuals and males. It is fatal in as many as 11% of patients, justifying aggressive treatment in patients with aggressive disease, Dr. Callen said. When it’s limited to the skin, however, “nothing may need to be done.”
Dr. Callen highlighted consensus recommendations about diagnosis and treatment of Rosai-Dorfman disease published in 2018.
He also noted the existence of cutaneous Rosai-Dorfman disease, a “solitary process” that appears more commonly in females, and in people of Asian heritage, compared with White individuals. It is characterized by single, clustered or widespread lesions: They can be xanthomatous, erythematous, or red-brown papules, nodules, and plaques. They’re acneiform, pustular, giant granuloma annulare–like, subcutaneous, and vasculitis-like, he said.
While Rosai-Dorfman disease can be linked to lymphoma, hypothyroidism, and lupus erythematosus, “nothing necessarily needs to be done when it’s skin-limited since it can be self-resolving,” he noted. Other treatments include radiotherapy, cryotherapy, excision, topical and oral corticosteroids, thalidomide, and methotrexate.
The disease can be serious, and is fatal in 5% of cases. When a vital organ is threatened, Dr. Callen suggested surgery, chemotherapy, or radiation.
Erdheim-Chester disease
This disease – which is extremely rare, with just 500 cases noted before 2014 – occurs when the body overproduces macrophages. It’s most common in middle-aged people and in men, who make up 75% of cases. About a quarter of patients develop skin lesions: Red-brown to yellow nodules and xanthelasma-like indurated plaques on the eyelids, scalp, neck, trunk, and axillae, and “other cutaneous manifestations have been reported in patients,” Dr. Callen said.
The disease also frequently affects the bones, large vessels, heart, lungs, and central nervous system. Interferon-alpha is the first-line treatment, and there are several other alternative therapies, although 5-year survival (68%) is poor, and it is especially likely to be fatal in those with central nervous system involvement.
Eosinophilic fasciitis
Eosinophilic fasciitis (EF) “is a disorder of unknown etiology that causes sclerosis of the skin” without Raynaud’s phenomenon, Dr. Callen said. Look for erythema, swelling, and induration of the extremities that is accompanied by peripheral eosinophilia, and if necessary, confirm the diagnosis with full skin-to-muscle biopsy or MRI.
There are many possible triggers, including strenuous exercise, initiation with hemodialysis, radiation therapy and burns, and graft-versus-host disease. Other potential causes include exposure to medications such as statins, phenytoin, ramipril, subcutaneous heparin, and immune checkpoint inhibitor therapy. The disorder is also linked to autoimmune and hematologic disorders.
Dr. Callen, who highlighted EF guidelines published in 2018, said treatments include physical therapy, prednisone, methotrexate, mycophenolate, and hydroxychloroquine.
Metastatic Crohn’s disease
This is a rare granulomatous inflammation of skin that often affects the genitals, especially in children. It is noncontiguous with the GI tract, and severity of skin involvement does not always parallel the severity of the disease in the GI tract, Dr. Callen said. However, the condition can occur before or simultaneously with the development of GI disease, or after GI surgery.
He highlighted a review of metastatic Crohn’s disease, published in 2014, and noted that there are multiple treatments, including systemic corticosteroids, tumor necrosis factor–alpha inhibitors, and topical therapies.
Dr. Callen reported no relevant disclosures.
and may spawn misdiagnoses, a dermatologist told colleagues.
“Proper diagnosis can lead to an effective management in our patients,” said Jeffrey Callen, MD, professor of medicine and chief of dermatology at the University of Louisville (Ky.), who spoke at the Inaugural Symposium for Inflammatory Skin Disease.
Sarcoidosis
The cause of sarcoidosis, an inflammatory disease that tends to affect the lungs, “is unknown, but it’s probably an immunologic disorder,” Dr. Callen said, “and there probably is a genetic predisposition.” About 20%-25% of patients with sarcoidosis have skin lesions that are either “specific” (a biopsy that reveals a noncaseating – “naked” – granuloma) or “nonspecific” (most commonly, erythema nodosum, or EN).
The specific lesions in sarcoidosis may occur in parts of the body, such as the knees, which were injured earlier in life and may have taken in foreign bodies, Dr. Callen said. As for nonspecific lesions, about 20% of patients with EN have an acute, self-limiting form of sarcoidosis. “These patients will have bilateral hilar lymphadenopathy, anterior uveitis, and polyarthritis. It’s generally treated symptomatically because it goes away on its own.”
He cautioned colleagues to beware of indurated, infiltrative facial lesions known as lupus pernio that are commonly found on the nose. They’re more prevalent in Black patients and possibly women, who are at higher risk of manifestations outside the skin, he said. “If you have it along the nasal rim, you should look into the upper respiratory tract for involvement.”
Dr. Callen recommends an extensive workup in patients with suspected sarcoidosis, including biopsy (with the exception of EN lesions), cultures and special stains, and screening when appropriate, for disease in organs such as the eyes, lungs, heart, and kidneys.
As for treatment, “the disease is in the dermis, and some topical therapies are not highly effective,” he said. There are injections that can be given, including corticosteroids, and there are a variety of oral treatments that are all off label.” These include corticosteroids, antimalarials, allopurinol, and tetracyclines, among several others. Subcutaneous and intravenous treatments are also options, along with surgery and laser therapy to treat specific lesions.
Rosai-Dorfman disease
This rare disorder is caused by overproduction of certain white blood cells in the lymph nodes, which can cause nodular lesions. The disease most often appears in children and young adults, often Black individuals and males. It is fatal in as many as 11% of patients, justifying aggressive treatment in patients with aggressive disease, Dr. Callen said. When it’s limited to the skin, however, “nothing may need to be done.”
Dr. Callen highlighted consensus recommendations about diagnosis and treatment of Rosai-Dorfman disease published in 2018.
He also noted the existence of cutaneous Rosai-Dorfman disease, a “solitary process” that appears more commonly in females, and in people of Asian heritage, compared with White individuals. It is characterized by single, clustered or widespread lesions: They can be xanthomatous, erythematous, or red-brown papules, nodules, and plaques. They’re acneiform, pustular, giant granuloma annulare–like, subcutaneous, and vasculitis-like, he said.
While Rosai-Dorfman disease can be linked to lymphoma, hypothyroidism, and lupus erythematosus, “nothing necessarily needs to be done when it’s skin-limited since it can be self-resolving,” he noted. Other treatments include radiotherapy, cryotherapy, excision, topical and oral corticosteroids, thalidomide, and methotrexate.
The disease can be serious, and is fatal in 5% of cases. When a vital organ is threatened, Dr. Callen suggested surgery, chemotherapy, or radiation.
Erdheim-Chester disease
This disease – which is extremely rare, with just 500 cases noted before 2014 – occurs when the body overproduces macrophages. It’s most common in middle-aged people and in men, who make up 75% of cases. About a quarter of patients develop skin lesions: Red-brown to yellow nodules and xanthelasma-like indurated plaques on the eyelids, scalp, neck, trunk, and axillae, and “other cutaneous manifestations have been reported in patients,” Dr. Callen said.
The disease also frequently affects the bones, large vessels, heart, lungs, and central nervous system. Interferon-alpha is the first-line treatment, and there are several other alternative therapies, although 5-year survival (68%) is poor, and it is especially likely to be fatal in those with central nervous system involvement.
Eosinophilic fasciitis
Eosinophilic fasciitis (EF) “is a disorder of unknown etiology that causes sclerosis of the skin” without Raynaud’s phenomenon, Dr. Callen said. Look for erythema, swelling, and induration of the extremities that is accompanied by peripheral eosinophilia, and if necessary, confirm the diagnosis with full skin-to-muscle biopsy or MRI.
There are many possible triggers, including strenuous exercise, initiation with hemodialysis, radiation therapy and burns, and graft-versus-host disease. Other potential causes include exposure to medications such as statins, phenytoin, ramipril, subcutaneous heparin, and immune checkpoint inhibitor therapy. The disorder is also linked to autoimmune and hematologic disorders.
Dr. Callen, who highlighted EF guidelines published in 2018, said treatments include physical therapy, prednisone, methotrexate, mycophenolate, and hydroxychloroquine.
Metastatic Crohn’s disease
This is a rare granulomatous inflammation of skin that often affects the genitals, especially in children. It is noncontiguous with the GI tract, and severity of skin involvement does not always parallel the severity of the disease in the GI tract, Dr. Callen said. However, the condition can occur before or simultaneously with the development of GI disease, or after GI surgery.
He highlighted a review of metastatic Crohn’s disease, published in 2014, and noted that there are multiple treatments, including systemic corticosteroids, tumor necrosis factor–alpha inhibitors, and topical therapies.
Dr. Callen reported no relevant disclosures.
FROM SISD 2021
HIFEM procedure helped to improve UI and female sexual function
Using high-intensity focused electromagnetic (HIFEM) technology to strengthen pelvic floor muscles for the improvement of urinary incontinence (UI) and female sexual function was safe and effective at 9 months follow-up, results from a multicenter study showed.
“The pelvic floor consists of three pairs of muscles: the pubococcygeus, the iliococcygeus, and the puborectalis,” lead study author Joseph Berenholz, MD, and a diplomate of the American Board of Obstetics & Gynecology, said during the annual conference of the American Society for Laser Medicine and Surgery. “They control continence through support of pelvic organs. The urethra, the vagina, and the rectum pass through that diaphragm. It also contributes to sexual sensation and arousal. A deconditioning of the pelvic floor is usually the result of child-bearing years or aging, which usually results in urinary incontinence and impairment of sexual function. The noninvasive strengthening of the pelvic floor muscles helps to regain muscle tone and strength.”
In a prospective, open-label, single-arm study conducted at four sites, Dr. Berenholz, medical director of the Michigan Center for Women’s Health in Farmington Hills, and colleagues investigated the long-term effectiveness of HIFEM-induced pelvic floor muscle (PFM) strengthening for improvement of UI and sexual function. HIFEM selectively targets neuromuscular tissue and induces supramaximal PFM contractions that cannot be achieved voluntarily, he said, causing muscle strengthening due to muscle fiber hypertrophy, which helps patients to better isolate and command their muscles.
The study population consisted of 33 females with a mean age of 49 years who had UI and UI-related problems in sexual life. They received six 28-minute HIFEM treatments of the pelvic floor with the BTL Emsella, which is FDA cleared for both stress and urge incontinence. The frequency of visits was two treatments per week and the intensity of HIFEM was adjusted between 0% and 100% based on the patient’s tolerance threshold. Evaluations were conducted at baseline, after the last treatment, at 1, 3, 6, and 9 months. The primary outcomes were change in urine leakage based on the International Consultation on Incontinence Questionnaire–Short Form (ICIQ-UI-SF) and change in sexual function based on the Female Sexual Function Index (FSFI) and the Pelvic Organ Prolapse/Urinary Incontinence Sexual Questionnaire (PISQ-12). Secondary endpoints were adverse events and the comfort of therapy based on a 7-point Likert scale.
Dr. Berenholz reported that from baseline the severity of UI based on the ICIQ-SF significantly decreased 60% by a mean of 8.1 points between baseline and 9 months (P < .001). At 1 month, the FSFI score improved 32% by a mean of 7.1 points (P < .001) and was sustained throughout the study. The most prominent changes were seen in the subdomains of desire, arousal, lubrication, and orgasm response.
The PISQ-12 score incrementally increased 25% to a mean improvement of 8.2 points at 9 months (P < .001). Subjects improved most in the emotive subdomain, reporting more frequent orgasms, increased desire, and sexual excitement. The minimal important difference was 6 points.
“This is a true paradigm shift in the treatment of incontinence and sexual dysfunction,” Dr. Berenholz said. “The therapy was safe, comfortable, no adverse events emerged, and 31 subjects (94%) described the therapy as comfortable. Interim data suggest that treatment effect was maintained for 9 months, and there were no significant declines in scores in the long term. The upcoming 12-month follow-up data will let us know if more maintenance therapy is needed.”
During a question-and-answer session, one of the abstract section chairs, Albert Wolkerstorfer, MD, PhD, wondered about the potential for combination treatments in this patient population. “I can imagine that something that is working on the muscle tone has a totally different mechanism than something that is working on the mucosa and the underlying tissue without really affecting the muscle,” said Dr. Wolkerstorfer, a dermatologist at the Netherlands Institute for Pigment Disorders, department of dermatology, University of Amsterdam. “Would a combination be the way to go?”
Dr. Berenholz said that he sometimes combines HIFEM with the ULTRA Femme 360, a radiofrequency thermal energy device. “We thought this addresses two issues,” he said. “One is fascial muscle, which is the underlying structural issue for incontinence. The other is thermal energy to aid in incontinence prevention by inducing production of elastin and collagen in the midurethra, but also to promote lubrication and heightened sensitivity in the patient who’s either menopausal or has undergone chemotherapy for breast cancer.”
Dr. Berenholz reported having no financial disclosures. Dr. Wolkerstorfer disclosed that he has received consulting fees from Lumenis and InCyte and equipment from Humeca and PerfAction Technologies. He has also received grant funding from Novartis and InCyte and he is a member of InCyte’s advisory board.
Using high-intensity focused electromagnetic (HIFEM) technology to strengthen pelvic floor muscles for the improvement of urinary incontinence (UI) and female sexual function was safe and effective at 9 months follow-up, results from a multicenter study showed.
“The pelvic floor consists of three pairs of muscles: the pubococcygeus, the iliococcygeus, and the puborectalis,” lead study author Joseph Berenholz, MD, and a diplomate of the American Board of Obstetics & Gynecology, said during the annual conference of the American Society for Laser Medicine and Surgery. “They control continence through support of pelvic organs. The urethra, the vagina, and the rectum pass through that diaphragm. It also contributes to sexual sensation and arousal. A deconditioning of the pelvic floor is usually the result of child-bearing years or aging, which usually results in urinary incontinence and impairment of sexual function. The noninvasive strengthening of the pelvic floor muscles helps to regain muscle tone and strength.”
In a prospective, open-label, single-arm study conducted at four sites, Dr. Berenholz, medical director of the Michigan Center for Women’s Health in Farmington Hills, and colleagues investigated the long-term effectiveness of HIFEM-induced pelvic floor muscle (PFM) strengthening for improvement of UI and sexual function. HIFEM selectively targets neuromuscular tissue and induces supramaximal PFM contractions that cannot be achieved voluntarily, he said, causing muscle strengthening due to muscle fiber hypertrophy, which helps patients to better isolate and command their muscles.
The study population consisted of 33 females with a mean age of 49 years who had UI and UI-related problems in sexual life. They received six 28-minute HIFEM treatments of the pelvic floor with the BTL Emsella, which is FDA cleared for both stress and urge incontinence. The frequency of visits was two treatments per week and the intensity of HIFEM was adjusted between 0% and 100% based on the patient’s tolerance threshold. Evaluations were conducted at baseline, after the last treatment, at 1, 3, 6, and 9 months. The primary outcomes were change in urine leakage based on the International Consultation on Incontinence Questionnaire–Short Form (ICIQ-UI-SF) and change in sexual function based on the Female Sexual Function Index (FSFI) and the Pelvic Organ Prolapse/Urinary Incontinence Sexual Questionnaire (PISQ-12). Secondary endpoints were adverse events and the comfort of therapy based on a 7-point Likert scale.
Dr. Berenholz reported that from baseline the severity of UI based on the ICIQ-SF significantly decreased 60% by a mean of 8.1 points between baseline and 9 months (P < .001). At 1 month, the FSFI score improved 32% by a mean of 7.1 points (P < .001) and was sustained throughout the study. The most prominent changes were seen in the subdomains of desire, arousal, lubrication, and orgasm response.
The PISQ-12 score incrementally increased 25% to a mean improvement of 8.2 points at 9 months (P < .001). Subjects improved most in the emotive subdomain, reporting more frequent orgasms, increased desire, and sexual excitement. The minimal important difference was 6 points.
“This is a true paradigm shift in the treatment of incontinence and sexual dysfunction,” Dr. Berenholz said. “The therapy was safe, comfortable, no adverse events emerged, and 31 subjects (94%) described the therapy as comfortable. Interim data suggest that treatment effect was maintained for 9 months, and there were no significant declines in scores in the long term. The upcoming 12-month follow-up data will let us know if more maintenance therapy is needed.”
During a question-and-answer session, one of the abstract section chairs, Albert Wolkerstorfer, MD, PhD, wondered about the potential for combination treatments in this patient population. “I can imagine that something that is working on the muscle tone has a totally different mechanism than something that is working on the mucosa and the underlying tissue without really affecting the muscle,” said Dr. Wolkerstorfer, a dermatologist at the Netherlands Institute for Pigment Disorders, department of dermatology, University of Amsterdam. “Would a combination be the way to go?”
Dr. Berenholz said that he sometimes combines HIFEM with the ULTRA Femme 360, a radiofrequency thermal energy device. “We thought this addresses two issues,” he said. “One is fascial muscle, which is the underlying structural issue for incontinence. The other is thermal energy to aid in incontinence prevention by inducing production of elastin and collagen in the midurethra, but also to promote lubrication and heightened sensitivity in the patient who’s either menopausal or has undergone chemotherapy for breast cancer.”
Dr. Berenholz reported having no financial disclosures. Dr. Wolkerstorfer disclosed that he has received consulting fees from Lumenis and InCyte and equipment from Humeca and PerfAction Technologies. He has also received grant funding from Novartis and InCyte and he is a member of InCyte’s advisory board.
Using high-intensity focused electromagnetic (HIFEM) technology to strengthen pelvic floor muscles for the improvement of urinary incontinence (UI) and female sexual function was safe and effective at 9 months follow-up, results from a multicenter study showed.
“The pelvic floor consists of three pairs of muscles: the pubococcygeus, the iliococcygeus, and the puborectalis,” lead study author Joseph Berenholz, MD, and a diplomate of the American Board of Obstetics & Gynecology, said during the annual conference of the American Society for Laser Medicine and Surgery. “They control continence through support of pelvic organs. The urethra, the vagina, and the rectum pass through that diaphragm. It also contributes to sexual sensation and arousal. A deconditioning of the pelvic floor is usually the result of child-bearing years or aging, which usually results in urinary incontinence and impairment of sexual function. The noninvasive strengthening of the pelvic floor muscles helps to regain muscle tone and strength.”
In a prospective, open-label, single-arm study conducted at four sites, Dr. Berenholz, medical director of the Michigan Center for Women’s Health in Farmington Hills, and colleagues investigated the long-term effectiveness of HIFEM-induced pelvic floor muscle (PFM) strengthening for improvement of UI and sexual function. HIFEM selectively targets neuromuscular tissue and induces supramaximal PFM contractions that cannot be achieved voluntarily, he said, causing muscle strengthening due to muscle fiber hypertrophy, which helps patients to better isolate and command their muscles.
The study population consisted of 33 females with a mean age of 49 years who had UI and UI-related problems in sexual life. They received six 28-minute HIFEM treatments of the pelvic floor with the BTL Emsella, which is FDA cleared for both stress and urge incontinence. The frequency of visits was two treatments per week and the intensity of HIFEM was adjusted between 0% and 100% based on the patient’s tolerance threshold. Evaluations were conducted at baseline, after the last treatment, at 1, 3, 6, and 9 months. The primary outcomes were change in urine leakage based on the International Consultation on Incontinence Questionnaire–Short Form (ICIQ-UI-SF) and change in sexual function based on the Female Sexual Function Index (FSFI) and the Pelvic Organ Prolapse/Urinary Incontinence Sexual Questionnaire (PISQ-12). Secondary endpoints were adverse events and the comfort of therapy based on a 7-point Likert scale.
Dr. Berenholz reported that from baseline the severity of UI based on the ICIQ-SF significantly decreased 60% by a mean of 8.1 points between baseline and 9 months (P < .001). At 1 month, the FSFI score improved 32% by a mean of 7.1 points (P < .001) and was sustained throughout the study. The most prominent changes were seen in the subdomains of desire, arousal, lubrication, and orgasm response.
The PISQ-12 score incrementally increased 25% to a mean improvement of 8.2 points at 9 months (P < .001). Subjects improved most in the emotive subdomain, reporting more frequent orgasms, increased desire, and sexual excitement. The minimal important difference was 6 points.
“This is a true paradigm shift in the treatment of incontinence and sexual dysfunction,” Dr. Berenholz said. “The therapy was safe, comfortable, no adverse events emerged, and 31 subjects (94%) described the therapy as comfortable. Interim data suggest that treatment effect was maintained for 9 months, and there were no significant declines in scores in the long term. The upcoming 12-month follow-up data will let us know if more maintenance therapy is needed.”
During a question-and-answer session, one of the abstract section chairs, Albert Wolkerstorfer, MD, PhD, wondered about the potential for combination treatments in this patient population. “I can imagine that something that is working on the muscle tone has a totally different mechanism than something that is working on the mucosa and the underlying tissue without really affecting the muscle,” said Dr. Wolkerstorfer, a dermatologist at the Netherlands Institute for Pigment Disorders, department of dermatology, University of Amsterdam. “Would a combination be the way to go?”
Dr. Berenholz said that he sometimes combines HIFEM with the ULTRA Femme 360, a radiofrequency thermal energy device. “We thought this addresses two issues,” he said. “One is fascial muscle, which is the underlying structural issue for incontinence. The other is thermal energy to aid in incontinence prevention by inducing production of elastin and collagen in the midurethra, but also to promote lubrication and heightened sensitivity in the patient who’s either menopausal or has undergone chemotherapy for breast cancer.”
Dr. Berenholz reported having no financial disclosures. Dr. Wolkerstorfer disclosed that he has received consulting fees from Lumenis and InCyte and equipment from Humeca and PerfAction Technologies. He has also received grant funding from Novartis and InCyte and he is a member of InCyte’s advisory board.
FROM ASLMS 2021
Inpatient care for HS higher for Black and Hispanic patients
National Inpatient Sample.
The differences occurred despite Black and Hispanic patients being younger at the time of admission than White patients, and may reflect increased disease severity and management challenges in these patients with skin of color, Nishadh Sutaria, BS, a medical student at Tufts University, Boston, said at the annual Skin of Color Society symposium. “They may also reflect social inequities in access to dermatologists, with racial and ethnic minorities using inpatient services in lieu of outpatient care.”
Mr. Sutaria and coinvestigators, led by Shawn Kwatra, MD, of Johns Hopkins University, Baltimore, identified 8,040 HS admissions for White patients, 16,490 Black patients, and 2,405 for Hispanic patients during the 5-year period.
Black and Hispanic patients were significantly younger than White patients, with a mean age of 38.1 years and 35 years, respectively, compared with 42 years for White patients (P < .001 in each case). Compared with White patients, Black patients had more procedures (2.03 vs. 1.84, P = .006), a longer length of stay (5.82 days vs. 4.97 days, P = .001), and higher cost of care ($46,119 vs. $39,862, P = .010). Compared with White patients, Hispanic patients had higher cost of care ($52,334 vs. $39,862, P = .004).
“In these models, Black patients stayed almost a full day longer and accrued a charge of $8,000 more than White patients, and Hispanic patients stayed about a half-day longer and accrued a charge of almost $15,000 more than White patients,” Mr. Sutaria said.
In a multilinear regression analysis adjusting for age, sex, and insurance type, Black race correlated with more procedures, higher length of stay, and higher cost of care, and Hispanic ethnicity with more procedures and higher cost of care.
Prior research has shown that Black patients may be disproportionately affected by HS. A 2017 analysis of electronic health record data for tens of millions of patients nationally, for instance, showed an incidence of HS that was over 2.5 times greater in Blacks than Whites. And a recent analysis of electronic data in Wisconsin for patients with an HS diagnosis and 3 or more encounters for the disease showed that Blacks are more likely to have HS that is Hurley Stage 3, the most severe type.
Increased severity “has not been explicitly shown in Hispanic patients,” Dr. Kwatra said in an interview, “[but] there is a strong relationship between obesity/metabolic syndrome with HS. Because Hispanic patients have higher rates of obesity and metabolic syndrome, it’s [thought] that they may have more severe HS.”
HS patients with skin of color are underrepresented in clinical trials, he said. “Severe HS can be difficult to treat because there are few effective treatments,” he said, noting that adalimumab is the only Food and Drug Administration–approved therapy.
The National Inpatient Sample is a publicly available, all-payer inpatient care database developed for the Agency for Healthcare Research and Quality’s Healthcare Cost and Utilization Project.
Mr. Sutaria is a dermatology research fellow working under the guidance of Dr. Kwatra.
National Inpatient Sample.
The differences occurred despite Black and Hispanic patients being younger at the time of admission than White patients, and may reflect increased disease severity and management challenges in these patients with skin of color, Nishadh Sutaria, BS, a medical student at Tufts University, Boston, said at the annual Skin of Color Society symposium. “They may also reflect social inequities in access to dermatologists, with racial and ethnic minorities using inpatient services in lieu of outpatient care.”
Mr. Sutaria and coinvestigators, led by Shawn Kwatra, MD, of Johns Hopkins University, Baltimore, identified 8,040 HS admissions for White patients, 16,490 Black patients, and 2,405 for Hispanic patients during the 5-year period.
Black and Hispanic patients were significantly younger than White patients, with a mean age of 38.1 years and 35 years, respectively, compared with 42 years for White patients (P < .001 in each case). Compared with White patients, Black patients had more procedures (2.03 vs. 1.84, P = .006), a longer length of stay (5.82 days vs. 4.97 days, P = .001), and higher cost of care ($46,119 vs. $39,862, P = .010). Compared with White patients, Hispanic patients had higher cost of care ($52,334 vs. $39,862, P = .004).
“In these models, Black patients stayed almost a full day longer and accrued a charge of $8,000 more than White patients, and Hispanic patients stayed about a half-day longer and accrued a charge of almost $15,000 more than White patients,” Mr. Sutaria said.
In a multilinear regression analysis adjusting for age, sex, and insurance type, Black race correlated with more procedures, higher length of stay, and higher cost of care, and Hispanic ethnicity with more procedures and higher cost of care.
Prior research has shown that Black patients may be disproportionately affected by HS. A 2017 analysis of electronic health record data for tens of millions of patients nationally, for instance, showed an incidence of HS that was over 2.5 times greater in Blacks than Whites. And a recent analysis of electronic data in Wisconsin for patients with an HS diagnosis and 3 or more encounters for the disease showed that Blacks are more likely to have HS that is Hurley Stage 3, the most severe type.
Increased severity “has not been explicitly shown in Hispanic patients,” Dr. Kwatra said in an interview, “[but] there is a strong relationship between obesity/metabolic syndrome with HS. Because Hispanic patients have higher rates of obesity and metabolic syndrome, it’s [thought] that they may have more severe HS.”
HS patients with skin of color are underrepresented in clinical trials, he said. “Severe HS can be difficult to treat because there are few effective treatments,” he said, noting that adalimumab is the only Food and Drug Administration–approved therapy.
The National Inpatient Sample is a publicly available, all-payer inpatient care database developed for the Agency for Healthcare Research and Quality’s Healthcare Cost and Utilization Project.
Mr. Sutaria is a dermatology research fellow working under the guidance of Dr. Kwatra.
National Inpatient Sample.
The differences occurred despite Black and Hispanic patients being younger at the time of admission than White patients, and may reflect increased disease severity and management challenges in these patients with skin of color, Nishadh Sutaria, BS, a medical student at Tufts University, Boston, said at the annual Skin of Color Society symposium. “They may also reflect social inequities in access to dermatologists, with racial and ethnic minorities using inpatient services in lieu of outpatient care.”
Mr. Sutaria and coinvestigators, led by Shawn Kwatra, MD, of Johns Hopkins University, Baltimore, identified 8,040 HS admissions for White patients, 16,490 Black patients, and 2,405 for Hispanic patients during the 5-year period.
Black and Hispanic patients were significantly younger than White patients, with a mean age of 38.1 years and 35 years, respectively, compared with 42 years for White patients (P < .001 in each case). Compared with White patients, Black patients had more procedures (2.03 vs. 1.84, P = .006), a longer length of stay (5.82 days vs. 4.97 days, P = .001), and higher cost of care ($46,119 vs. $39,862, P = .010). Compared with White patients, Hispanic patients had higher cost of care ($52,334 vs. $39,862, P = .004).
“In these models, Black patients stayed almost a full day longer and accrued a charge of $8,000 more than White patients, and Hispanic patients stayed about a half-day longer and accrued a charge of almost $15,000 more than White patients,” Mr. Sutaria said.
In a multilinear regression analysis adjusting for age, sex, and insurance type, Black race correlated with more procedures, higher length of stay, and higher cost of care, and Hispanic ethnicity with more procedures and higher cost of care.
Prior research has shown that Black patients may be disproportionately affected by HS. A 2017 analysis of electronic health record data for tens of millions of patients nationally, for instance, showed an incidence of HS that was over 2.5 times greater in Blacks than Whites. And a recent analysis of electronic data in Wisconsin for patients with an HS diagnosis and 3 or more encounters for the disease showed that Blacks are more likely to have HS that is Hurley Stage 3, the most severe type.
Increased severity “has not been explicitly shown in Hispanic patients,” Dr. Kwatra said in an interview, “[but] there is a strong relationship between obesity/metabolic syndrome with HS. Because Hispanic patients have higher rates of obesity and metabolic syndrome, it’s [thought] that they may have more severe HS.”
HS patients with skin of color are underrepresented in clinical trials, he said. “Severe HS can be difficult to treat because there are few effective treatments,” he said, noting that adalimumab is the only Food and Drug Administration–approved therapy.
The National Inpatient Sample is a publicly available, all-payer inpatient care database developed for the Agency for Healthcare Research and Quality’s Healthcare Cost and Utilization Project.
Mr. Sutaria is a dermatology research fellow working under the guidance of Dr. Kwatra.
FROM SOC SOCIETY 2021
Pyoderma gangrenosum: Understanding the difficult diagnosis
Pyoderma gangrenosum (PG), a rare and painful ulcerative skin disorder, requires special care because “it’s a challenging diagnosis to make” and is frequently linked to serious comorbid conditions, a dermatologist told colleagues.
PG is also challenging to manage, said Jeffrey Callen, MD, professor and chief of the division of dermatology at the University of Louisville (Ky.), who spoke at the Inaugural Symposium for Inflammatory Skin Disease. “There are multiple treatments, but few have a high level of evidence to document their efficacy.”
PG is a neutrophilic dermatosis that usually occurs with a small lesion, often pustular, that spreads, and is a diagnosis of exclusion. “There’s no way you can possibly exclude everything, but the major things that have to be excluded are infection and malignancies,” he said. “Doing a good history and physical examination is critical, and a biopsy should be done in the vast majority of patients.”
Cultures and routine labs should be obtained, said Dr. Callen, highlighting tests that measure immunofixation (IFE), antineutrophil cytoplasmic antibodies (ANCE), anticardiolipin antibody (aCL), and lupus anticoagulant (LA).
Bowel and bone marrow tests may be appropriate in some patients, he said, noting that about half of PG cases are linked to comorbid conditions such as inflammatory bowel disease (IBD), arthritis, and hematologic diseases.
Dr. Callen also made the following points about making the diagnosis:
- Several clinical variants exist: classic, peristomal, and atypical.
- Pathergy – hyperreactivity of skin to injury – occurs in about a third of patients.
- Neutrophilic infiltrates may occur in other organs.
- Numerous drugs, including isotretinoin, can cause PG.
- PG may be misdiagnosed as necrotizing fasciitis.
- Several diagnostic frameworks exist: the Su Criteria, the PARACELCUS Score, and the Delphi Consensus Criteria. The Delphi criteria identified the highest percentage of cases (89%) in a study comparing the three, published in 2020. The frameworks “are helpful in the clinic, but they are not to be used as criteria for diagnosis. They’re really for classification,” Dr. Callen said.
Once the diagnosis has been made, he said, focus on healing the wound, which he said “can be done as any other wound would be healed,” and calming the inflammation.
“Patients who have mild disease might be treated with lower doses of prednisone, topical medications, or intralesional injections,” he said. “Corticosteroids are never wrong in the beginning.” Some patients may have genetic abnormalities related to PG, he added, and medications that target them may be appropriate.
Antibiotics and biologic agents, particularly TNF-alpha inhibitors, are possible treatments, Dr. Callen said. He highlighted a 2018 systematic review that evaluated treatments and found the most evidence supported systemic corticosteroids, cyclosporine, and TNF-alpha inhibitors. However, the quality of studies was limited, and the authors noted that the lesions frequently failed to respond or recurred.
When appropriate, surgery can be performed, he said.
Dr. Callen reported no relevant disclosures.
Pyoderma gangrenosum (PG), a rare and painful ulcerative skin disorder, requires special care because “it’s a challenging diagnosis to make” and is frequently linked to serious comorbid conditions, a dermatologist told colleagues.
PG is also challenging to manage, said Jeffrey Callen, MD, professor and chief of the division of dermatology at the University of Louisville (Ky.), who spoke at the Inaugural Symposium for Inflammatory Skin Disease. “There are multiple treatments, but few have a high level of evidence to document their efficacy.”
PG is a neutrophilic dermatosis that usually occurs with a small lesion, often pustular, that spreads, and is a diagnosis of exclusion. “There’s no way you can possibly exclude everything, but the major things that have to be excluded are infection and malignancies,” he said. “Doing a good history and physical examination is critical, and a biopsy should be done in the vast majority of patients.”
Cultures and routine labs should be obtained, said Dr. Callen, highlighting tests that measure immunofixation (IFE), antineutrophil cytoplasmic antibodies (ANCE), anticardiolipin antibody (aCL), and lupus anticoagulant (LA).
Bowel and bone marrow tests may be appropriate in some patients, he said, noting that about half of PG cases are linked to comorbid conditions such as inflammatory bowel disease (IBD), arthritis, and hematologic diseases.
Dr. Callen also made the following points about making the diagnosis:
- Several clinical variants exist: classic, peristomal, and atypical.
- Pathergy – hyperreactivity of skin to injury – occurs in about a third of patients.
- Neutrophilic infiltrates may occur in other organs.
- Numerous drugs, including isotretinoin, can cause PG.
- PG may be misdiagnosed as necrotizing fasciitis.
- Several diagnostic frameworks exist: the Su Criteria, the PARACELCUS Score, and the Delphi Consensus Criteria. The Delphi criteria identified the highest percentage of cases (89%) in a study comparing the three, published in 2020. The frameworks “are helpful in the clinic, but they are not to be used as criteria for diagnosis. They’re really for classification,” Dr. Callen said.
Once the diagnosis has been made, he said, focus on healing the wound, which he said “can be done as any other wound would be healed,” and calming the inflammation.
“Patients who have mild disease might be treated with lower doses of prednisone, topical medications, or intralesional injections,” he said. “Corticosteroids are never wrong in the beginning.” Some patients may have genetic abnormalities related to PG, he added, and medications that target them may be appropriate.
Antibiotics and biologic agents, particularly TNF-alpha inhibitors, are possible treatments, Dr. Callen said. He highlighted a 2018 systematic review that evaluated treatments and found the most evidence supported systemic corticosteroids, cyclosporine, and TNF-alpha inhibitors. However, the quality of studies was limited, and the authors noted that the lesions frequently failed to respond or recurred.
When appropriate, surgery can be performed, he said.
Dr. Callen reported no relevant disclosures.
Pyoderma gangrenosum (PG), a rare and painful ulcerative skin disorder, requires special care because “it’s a challenging diagnosis to make” and is frequently linked to serious comorbid conditions, a dermatologist told colleagues.
PG is also challenging to manage, said Jeffrey Callen, MD, professor and chief of the division of dermatology at the University of Louisville (Ky.), who spoke at the Inaugural Symposium for Inflammatory Skin Disease. “There are multiple treatments, but few have a high level of evidence to document their efficacy.”
PG is a neutrophilic dermatosis that usually occurs with a small lesion, often pustular, that spreads, and is a diagnosis of exclusion. “There’s no way you can possibly exclude everything, but the major things that have to be excluded are infection and malignancies,” he said. “Doing a good history and physical examination is critical, and a biopsy should be done in the vast majority of patients.”
Cultures and routine labs should be obtained, said Dr. Callen, highlighting tests that measure immunofixation (IFE), antineutrophil cytoplasmic antibodies (ANCE), anticardiolipin antibody (aCL), and lupus anticoagulant (LA).
Bowel and bone marrow tests may be appropriate in some patients, he said, noting that about half of PG cases are linked to comorbid conditions such as inflammatory bowel disease (IBD), arthritis, and hematologic diseases.
Dr. Callen also made the following points about making the diagnosis:
- Several clinical variants exist: classic, peristomal, and atypical.
- Pathergy – hyperreactivity of skin to injury – occurs in about a third of patients.
- Neutrophilic infiltrates may occur in other organs.
- Numerous drugs, including isotretinoin, can cause PG.
- PG may be misdiagnosed as necrotizing fasciitis.
- Several diagnostic frameworks exist: the Su Criteria, the PARACELCUS Score, and the Delphi Consensus Criteria. The Delphi criteria identified the highest percentage of cases (89%) in a study comparing the three, published in 2020. The frameworks “are helpful in the clinic, but they are not to be used as criteria for diagnosis. They’re really for classification,” Dr. Callen said.
Once the diagnosis has been made, he said, focus on healing the wound, which he said “can be done as any other wound would be healed,” and calming the inflammation.
“Patients who have mild disease might be treated with lower doses of prednisone, topical medications, or intralesional injections,” he said. “Corticosteroids are never wrong in the beginning.” Some patients may have genetic abnormalities related to PG, he added, and medications that target them may be appropriate.
Antibiotics and biologic agents, particularly TNF-alpha inhibitors, are possible treatments, Dr. Callen said. He highlighted a 2018 systematic review that evaluated treatments and found the most evidence supported systemic corticosteroids, cyclosporine, and TNF-alpha inhibitors. However, the quality of studies was limited, and the authors noted that the lesions frequently failed to respond or recurred.
When appropriate, surgery can be performed, he said.
Dr. Callen reported no relevant disclosures.
FROM SISD 2021