User login
Bell Palsy Mimics
Facial paralysis is a common medical complaint—one that has fascinated ancient and contemporary physicians alike.1 An idiopathic facial nerve paresis involving the lower motor neuron was described in 1821 by Sir Charles Bell. This entity became known as a Bell’s palsy, the hallmark of which was weakness or complete paralysis of the muscles of one side of the face, with no sparing of the muscles of the forehead. However, not all facial paralysis is due to Bell’s palsy.
We present a case of a patient with a Bell’s palsy mimic to facilitate and guide the differential diagnosis and distinguish conditions from the classical presentation that Bell first described to the more concerning symptoms that may not be immediately obvious. Our case further underscores the importance of performing a thorough assessment to determine the presence of other neurological findings.
Case
A 61-year-old woman presented to the ED for evaluation of right facial droop and sensation of “room spinning.” The patient stated both symptoms began approximately 36 hours prior to presentation, upon awakening.
The patient denied any headache, neck or chest pain, extremity numbness, or weakness, but stated that she felt like she was going to fall toward her right side whenever she attempted to walk. The patient’s medical history was significant for hypertension, for which she was taking losartan. Her surgical history was notable for a left oophorectomy secondary to an ovarian cyst. Regarding the social history, the patient admitted to smoking 90 packs of cigarettes per year, but denied alcohol or illicit drug use.
Upon arrival at the ED, the patient’s vital signs were: blood pressure, 164/86 mm Hg: pulse, 89 beats/min; respiratory rate, 18 breaths/min; and temperature, 98.6°F. Oxygen saturation was 98% on room air.
Physical examination revealed the patient had a right facial droop consistent with right facial palsy. She was unable to wrinkle her right forehead or fully close her right eye. There were no field cuts on confrontation. The patient’s speech was noticeable for a mild dysarthria. The motor examination revealed mild weakness of the left upper extremity and impaired right facial sensation. There were no rashes noted on the face, head, or ears. The patient had slightly impaired hearing in the right ear, which was new in onset. The remainder of the physical examination was unremarkable.
Although the patient exhibited the classic signs of Bell’s palsy, including complete paralysis of the muscles of one side of the face, inability to wrinkle the muscle of the right forehead, and inability to fully close the right eye, she also had concerning symptoms of vertigo, dysarthria, and contralateral upper extremity weakness.
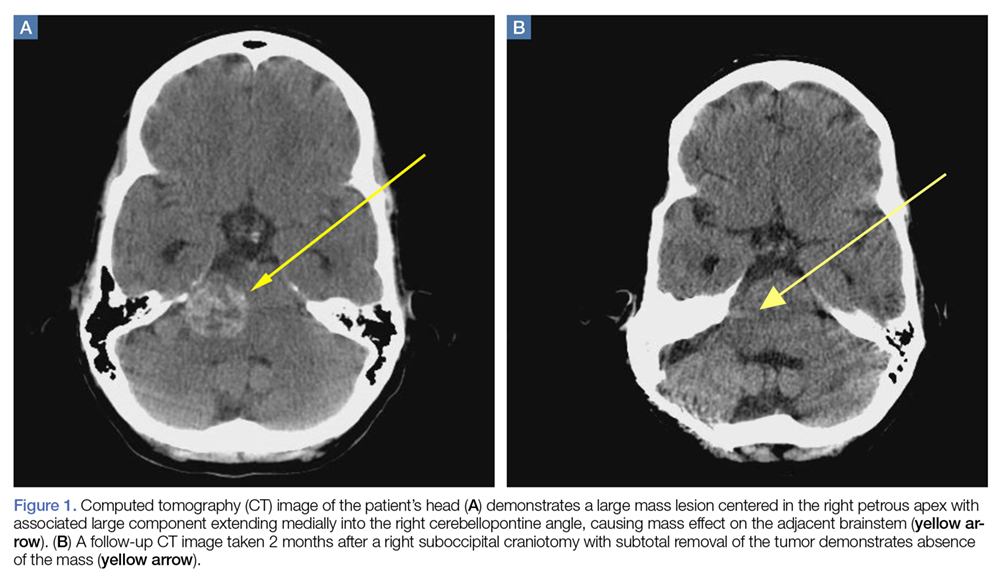
A computed tomography (CT) scan of the head was ordered, which revealed a large mass lesion centered in the right petrous apex, with an associated large component extending medially into the right cerebellopontine angle (CPA) that caused a mass effect on the adjacent brainstem (Figures 1a and 1b).
Upon these findings, the patient was transferred to another facility for neurosurgical evaluation. Magnetic resonance imaging (MRI) studies performed at the receiving hospital demonstrated a large expansile heterogeneous mass lesion centered in the right petrous apex with an associated large, probable hemorrhagic soft-tissue component extending medially into the right CPA, causing a mass effect on the adjacent brainstem and mild obstructive hydrocephalus (Figures 2a and 2b).

The patient was given dexamethasone 10 mg intravenously and taken to the operating room for a right suboccipital craniotomy with subtotal tumor removal. Intraoperative high-voltage stimulation of the fifth to eighth cranial nerves showed no response, indicating significant impairment.
While there were no intraoperative complications, the patient had significant postoperative dysphagia and resultant aspiration. A tracheostomy and percutaneous endoscopic gastrostomy tube were subsequently placed. Results of a biopsy taken during surgery identified an atypical meningioma. The patient remained in the hospital for 4 weeks, after which she was discharged to a long-term care (LTC) and rehabilitation facility.
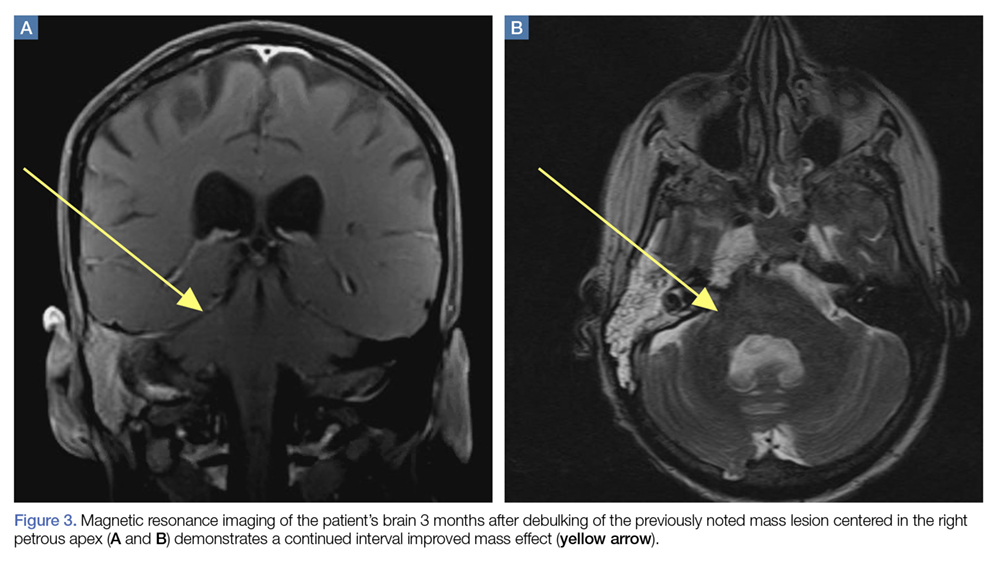
A repeat CT scan taken 2 months after surgery demonstrated absence of the previously identified large mass (Figure 1b). Three months after discharge from the LTC-rehabilitation facility, MRI of the brain showed continued interval improvement of the previously noted mass centered in the right petrous apex (Figures 3a and 3b).
Discussion
Accounts of facial paralysis and facial nerve disorders have been noted throughout history and include accounts of the condition by Hippocrates.1 Bell’s palsy was named after surgeon Sir Charles Bell, who described a peripheral-nerve paralysis of the facial nerve in 1821. Bell’s work helped to elucidate the anatomy and functional role of the facial nerve.1,2
Signs and Symptoms
The classic presentation of Bell’s palsy is weakness or complete paralysis of the muscles of one side of the face, with no sparing of the muscles of the forehead. The eyelid on the affected side generally does not close, which can result in ocular irritation due to ineffective lubrication.
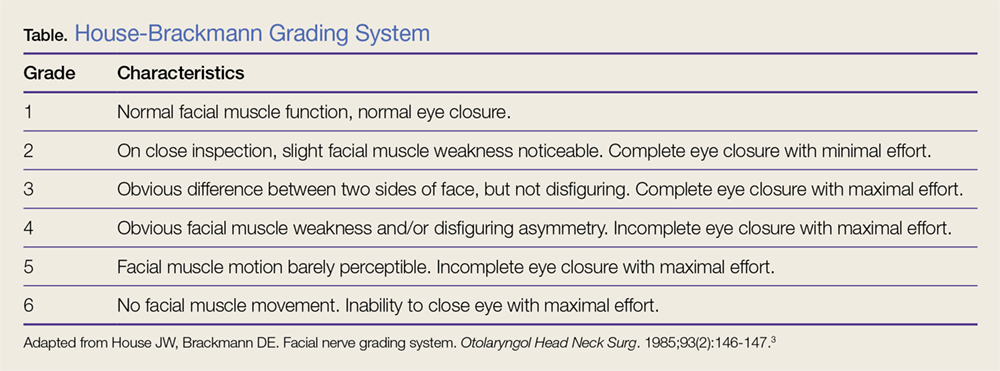
A scoring system has been developed by House and Brackmann which grades the degree impairment based on such characteristics as facial muscle function and eye closure.3,4 Approximately 96% of patients with a Bell’s palsy will improve to a House-Brackmann score of 2 or better within 1 year from diagnosis,5 and 85% of patients with Bell’s palsy will show at least some improvement within 3 weeks of onset (Table).2 Although the classic description of Bell’s palsy notes the condition as idiopathic, there is an increasing body of evidence in the literature showing a link to herpes simplex virus 1.5-7
Ramsey-Hunt Syndrome
The relationship between Bell’s palsy and Ramsey-Hunt syndrome is complex and controversial. Ramsey-Hunt syndrome is a constellation of possible complications from varicella-virus infection. Symptoms of Ramsey-Hunt syndrome include facial paralysis, tinnitus, hearing loss, vertigo, hyperacusis (increased sensitivity to certain frequencies and volume ranges of sound), and decreased ocular tearing.8 Due to the nature of symptoms associated with Ramsey-Hunt syndrome, it is apparent that the condition involves more than the seventh cranial nerve. In fact, studies have shown that Ramsey-Hunt syndrome can affect the fifth, sixth, eighth, and ninth cranial nerves.8
Ramsey-Hunt syndrome, which can present in the absence of cutaneous rash (referred to as zoster sine herpete), is estimated to occur in 8% to 20% of unilateral facial nerve palsies in adult patients.8,9 Regardless of the etiology of Bell’s palsy, a review of the literature makes it clear that facial nerve paralysis is not synonymous with Bell’s palsy.10 In one example, Yetter et al10 describe the case of a patient who, though initially diagnosed with Bell’s palsy, ultimately was found to have a facial palsy due to a parotid gland malignancy.
Likewise, Stomeo11 describes a case of a patient with facial paralysis and profound ipsilateral hearing loss who ultimately was found to have a mucoepithelial carcinoma of the parotid gland. In their report, the authors note that approximately 80% of facial nerve paralysis is due to Bell’s palsy, while 5% is due to malignancy.
In another report, Clemis12 describes a case in which a patient who initially was diagnosed with Bell’s palsy eventually was found to have an adenoid cystic carcinoma of the parotid. Thus, the authors appropriately emphasize in their report that “all that palsies is not Bell’s.”
Differential Diagnosis
Historical factors, including timing and duration of symptom onset, help to distinguish a Bell’s palsy from other disorders that can mimic this condition. In their study, Brach VanSwewaringen13 highlight the fact that “not all facial paralysis is Bell’s palsy.” In their review, the authors describe clues to help distinguish conditions that mimic Bell’s palsy. For example, maximal weakness from Bell’s Palsy typically occurs within 3 to 7 days from symptom onset, and that a more gradual onset of symptoms, with slow or negligible improvement over 6 to 12 months, is more indicative of a space-occupying lesion than Bell’s palsy.13It is, however, important to note that although the patient in our case had a central lesion, she experienced an acute onset of symptoms.
The presence of additional symptoms may also suggest an alternative diagnosis. Brach and VanSwearingen13 further noted that symptoms associated with the eighth nerve, such as vertigo, tinnitus, and hearing loss may be found in patients with a CPA tumor. In patients with larger tumors, ninth and 10th nerve symptoms, including the impaired hearing noted in our patient, may be present. Some patients with ninth and 10th nerve symptoms may perceive a sense of facial numbness, but actual sensory changes in the facial nerve distribution are unlikely in Bell’s palsy. Gustatory changes, however, are consistent with Bell’s palsy.
Ear pain is consistent with Bell’s palsy and is a signal to be vigilant for the possible emergence of an ear rash, which would suggest the diagnosis of herpes zoster oticus along the trajectory of Ramsey-Hunt syndrome. Facial pain in the area of the facial nerve is inconsistent with Bell’s palsy, while hyperacusis is consistent with Bell’s palsy. Hearing loss is an eighth nerve symptom that is inconsistent with Bell’s palsy.
Similarly, there are physical examination findings that can help distinguish a true Bell’s palsy from a mimic. Changes in tear production are consistent with Bell’s palsy, but imbalance and disequilibrium are not.14
As previously noted, the patient in this case had difficulty walking and felt as if she was falling toward her right side.
One way to organize the causes of facial paralysis has been proposed by Adour et al.15 In this system, etiologies are listed as either acute paralysis or chronic, progressive paralysis. Acute paralysis (ie, the sudden onset of symptoms with maximal severity within 2 weeks), of which Bell’s palsy is the most common, can be seen in cases of polyneuritis.
A new case of Bell’s palsy has been estimated to occur in the United States every 10 minutes.8 Guillain-Barré syndrome and Lyme disease are also in this category, as is Ramsey-Hunt syndrome. Patients with Lyme disease may have a history of a tick bite or rash.14
Trauma can also cause acute facial nerve paralysis (eg, blunt trauma-associated facial fracture, penetrating trauma, birth trauma). Unilateral central facial weakness can have a neurological cause, such as a lesion to the contralateral cortex, subcortical white matter, or internal capsule.2,15 Otitis media can sometimes cause facial paralysis.16 A cholesteatoma can cause acute facial paralysis.2 Malignancies cause 5% of all cases of facial paralysis. Primary parotid tumors of various types are in this category. Metastatic disease from breast, lung, skin, colon, and kidney may cause facial paralysis. As our case illustrates, CPA tumors can cause facial paralysis.15 It is important to also note that a patient can have both a Bell’s palsy and a concurrent disease. There are a number of case reports in the literature that describe acute onset of facial paralysis as a presenting symptom of malignancy.17 In addition, there are cases wherein a neurological finding on imaging, such as an acoustic neuroma, was presumed to be the cause of facial paralysis, yet the patient’s symptoms resolved in a manner consistent with Bell’s palsy.18
For example, Lagman et al19 described a patient in which a CPA lipoma was presumed to be the cause of the facial paralysis, but the eventual outcome showed the lipoma to have been an incidentaloma.
Conclusion
This case demonstrates a presenting symptom of facial palsy and the presence of a CPA tumor. The presence of vertigo along with other historical and physical examination findings inconsistent with Bell’s palsy prompted the CT scan of the head. A review of the literature suggests a number of important findings in patients with facial palsy to assist the clinician in distinguishing true Bell’s palsy from other diseases that can mimic this condition. This case serves as a reminder of the need to perform a thorough and diligent workup to determine the presence or absence of other neurologic findings prior to closing on the diagnosis of Bell’s palsy.
1. Glicenstein J. Ann Chir Plast Esthet. 2015;60(5):347-362. doi:10.1016/j.anplas.2015.05.007.
2. Tiemstra JD, Khatkhate N. Bell’s palsy: diagnosis and management. Am Fam Physician. 2007;76(7):997-1002.
3. House JW, Brackmann DE. Facial nerve grading system. Otolaryngol Head Neck Surg. 1985;93(2):146-147. doi:10.1177/019459988509300202.
4. Reitzen SD, Babb JS, Lalwani AK. Significance and reliability of the House-Brackmann grading system for regional facial nerve function. Otolaryngol Head Neck Surg. 2009;140(2):154-158. doi:10.1016/j.otohns.2008.11.021.
5. Yeo SW, Lee DH, Jun BC, Chang KH, Park YS. Analysis of prognostic factors in Bell’s palsy and Ramsay Hunt syndrome. Auris Nasus Larynx. 2007;34(2):159-164. doi:10.1016/j.anl.2006.09.005.
6. Ahmed A. When is facial paralysis Bell palsy? Current diagnosis and treatment. Cleve Clin J Med. 2005;72(5):398-401, 405.
7. Gilden DH. Clinical practice. Bell’s palsy. N Engl J Med. 2004;351(13):1323-1331. doi:10.1056/NEJMcp041120.
8. Adour KK. Otological complications of herpes zoster. Ann Neurol. 1994;35:Suppl:S62-S64.
9. Furuta Y, Ohtani F, Mesuda Y, Fukuda S, Inuyama Y. Early diagnosis of zoster sine herpete and antiviral therapy for the treatment of facial palsy. Neurology. 2000;55(5):708-710.
10. Yetter MF, Ogren FP, Moore GF, Yonkers AJ. Bell’s palsy: a facial nerve paralysis diagnosis of exclusion. Nebr Med J. 1990;75(5):109-116.
11. Stomeo F. Possibilities of diagnostic errors in paralysis of the 7th cranial nerve. Acta Otorhinolaryngol Ital. 1989;9(6):629-633.
12. Clemis JD. All that palsies is not Bell’s: Bell’s palsy due to adenoid cystic carcinoma of the parotid. Am J Otol. 1991;12(5):397.
13. Brach JS, VanSwearingen JM. Not all facial paralysis is Bell’s palsy: a case report. Arch Phys Med Rehabil. 1999;80(7):857-859.
14. Albers JR, Tamang S. Common questions about Bell palsy. Am Fam Physician. 2014;89(3):209-212.
15. Adour KK, Hilsinger RL Jr, Callan EJ. Facial paralysis and Bell’s palsy: a protocol for differential diagnosis. Am J Otol. 1985;Suppl:68-73.
16. Morrow MJ. Bell’s palsy and herpes zoster. Curr Treat Options Neurol. 2000;2(5):407-416.
17. Quesnel AM, Lindsay RW, Hadlock TA. When the bell tolls on Bell’s palsy: finding occult malignancy in acute-onset facial paralysis. Am J Otolaryngol. 2010;31(5):339-342. doi:10.1016/j.amjoto.2009.04.003.
18. Kaushal A, Curran WJ Jr. For whom the Bell’s palsy tolls? Am J Clin Oncol. 2009;32(4):450-451. doi:10.1097/01.coc.0000239141.22916.22.
19. Lagman C, Choy W, Lee SJ, et al. A Case of Bell’s palsy with an incidental finding of a cerebellopontine angle lipoma. Cureus. 2016;8(8):e747. doi:10.7759/cureus.747.
Facial paralysis is a common medical complaint—one that has fascinated ancient and contemporary physicians alike.1 An idiopathic facial nerve paresis involving the lower motor neuron was described in 1821 by Sir Charles Bell. This entity became known as a Bell’s palsy, the hallmark of which was weakness or complete paralysis of the muscles of one side of the face, with no sparing of the muscles of the forehead. However, not all facial paralysis is due to Bell’s palsy.
We present a case of a patient with a Bell’s palsy mimic to facilitate and guide the differential diagnosis and distinguish conditions from the classical presentation that Bell first described to the more concerning symptoms that may not be immediately obvious. Our case further underscores the importance of performing a thorough assessment to determine the presence of other neurological findings.
Case
A 61-year-old woman presented to the ED for evaluation of right facial droop and sensation of “room spinning.” The patient stated both symptoms began approximately 36 hours prior to presentation, upon awakening.
The patient denied any headache, neck or chest pain, extremity numbness, or weakness, but stated that she felt like she was going to fall toward her right side whenever she attempted to walk. The patient’s medical history was significant for hypertension, for which she was taking losartan. Her surgical history was notable for a left oophorectomy secondary to an ovarian cyst. Regarding the social history, the patient admitted to smoking 90 packs of cigarettes per year, but denied alcohol or illicit drug use.
Upon arrival at the ED, the patient’s vital signs were: blood pressure, 164/86 mm Hg: pulse, 89 beats/min; respiratory rate, 18 breaths/min; and temperature, 98.6°F. Oxygen saturation was 98% on room air.
Physical examination revealed the patient had a right facial droop consistent with right facial palsy. She was unable to wrinkle her right forehead or fully close her right eye. There were no field cuts on confrontation. The patient’s speech was noticeable for a mild dysarthria. The motor examination revealed mild weakness of the left upper extremity and impaired right facial sensation. There were no rashes noted on the face, head, or ears. The patient had slightly impaired hearing in the right ear, which was new in onset. The remainder of the physical examination was unremarkable.
Although the patient exhibited the classic signs of Bell’s palsy, including complete paralysis of the muscles of one side of the face, inability to wrinkle the muscle of the right forehead, and inability to fully close the right eye, she also had concerning symptoms of vertigo, dysarthria, and contralateral upper extremity weakness.
A computed tomography (CT) scan of the head was ordered, which revealed a large mass lesion centered in the right petrous apex, with an associated large component extending medially into the right cerebellopontine angle (CPA) that caused a mass effect on the adjacent brainstem (Figures 1a and 1b).
Upon these findings, the patient was transferred to another facility for neurosurgical evaluation. Magnetic resonance imaging (MRI) studies performed at the receiving hospital demonstrated a large expansile heterogeneous mass lesion centered in the right petrous apex with an associated large, probable hemorrhagic soft-tissue component extending medially into the right CPA, causing a mass effect on the adjacent brainstem and mild obstructive hydrocephalus (Figures 2a and 2b).
The patient was given dexamethasone 10 mg intravenously and taken to the operating room for a right suboccipital craniotomy with subtotal tumor removal. Intraoperative high-voltage stimulation of the fifth to eighth cranial nerves showed no response, indicating significant impairment.
While there were no intraoperative complications, the patient had significant postoperative dysphagia and resultant aspiration. A tracheostomy and percutaneous endoscopic gastrostomy tube were subsequently placed. Results of a biopsy taken during surgery identified an atypical meningioma. The patient remained in the hospital for 4 weeks, after which she was discharged to a long-term care (LTC) and rehabilitation facility.
A repeat CT scan taken 2 months after surgery demonstrated absence of the previously identified large mass (Figure 1b). Three months after discharge from the LTC-rehabilitation facility, MRI of the brain showed continued interval improvement of the previously noted mass centered in the right petrous apex (Figures 3a and 3b).
Discussion
Accounts of facial paralysis and facial nerve disorders have been noted throughout history and include accounts of the condition by Hippocrates.1 Bell’s palsy was named after surgeon Sir Charles Bell, who described a peripheral-nerve paralysis of the facial nerve in 1821. Bell’s work helped to elucidate the anatomy and functional role of the facial nerve.1,2
Signs and Symptoms
The classic presentation of Bell’s palsy is weakness or complete paralysis of the muscles of one side of the face, with no sparing of the muscles of the forehead. The eyelid on the affected side generally does not close, which can result in ocular irritation due to ineffective lubrication.
A scoring system has been developed by House and Brackmann which grades the degree impairment based on such characteristics as facial muscle function and eye closure.3,4 Approximately 96% of patients with a Bell’s palsy will improve to a House-Brackmann score of 2 or better within 1 year from diagnosis,5 and 85% of patients with Bell’s palsy will show at least some improvement within 3 weeks of onset (Table).2 Although the classic description of Bell’s palsy notes the condition as idiopathic, there is an increasing body of evidence in the literature showing a link to herpes simplex virus 1.5-7
Ramsey-Hunt Syndrome
The relationship between Bell’s palsy and Ramsey-Hunt syndrome is complex and controversial. Ramsey-Hunt syndrome is a constellation of possible complications from varicella-virus infection. Symptoms of Ramsey-Hunt syndrome include facial paralysis, tinnitus, hearing loss, vertigo, hyperacusis (increased sensitivity to certain frequencies and volume ranges of sound), and decreased ocular tearing.8 Due to the nature of symptoms associated with Ramsey-Hunt syndrome, it is apparent that the condition involves more than the seventh cranial nerve. In fact, studies have shown that Ramsey-Hunt syndrome can affect the fifth, sixth, eighth, and ninth cranial nerves.8
Ramsey-Hunt syndrome, which can present in the absence of cutaneous rash (referred to as zoster sine herpete), is estimated to occur in 8% to 20% of unilateral facial nerve palsies in adult patients.8,9 Regardless of the etiology of Bell’s palsy, a review of the literature makes it clear that facial nerve paralysis is not synonymous with Bell’s palsy.10 In one example, Yetter et al10 describe the case of a patient who, though initially diagnosed with Bell’s palsy, ultimately was found to have a facial palsy due to a parotid gland malignancy.
Likewise, Stomeo11 describes a case of a patient with facial paralysis and profound ipsilateral hearing loss who ultimately was found to have a mucoepithelial carcinoma of the parotid gland. In their report, the authors note that approximately 80% of facial nerve paralysis is due to Bell’s palsy, while 5% is due to malignancy.
In another report, Clemis12 describes a case in which a patient who initially was diagnosed with Bell’s palsy eventually was found to have an adenoid cystic carcinoma of the parotid. Thus, the authors appropriately emphasize in their report that “all that palsies is not Bell’s.”
Differential Diagnosis
Historical factors, including timing and duration of symptom onset, help to distinguish a Bell’s palsy from other disorders that can mimic this condition. In their study, Brach VanSwewaringen13 highlight the fact that “not all facial paralysis is Bell’s palsy.” In their review, the authors describe clues to help distinguish conditions that mimic Bell’s palsy. For example, maximal weakness from Bell’s Palsy typically occurs within 3 to 7 days from symptom onset, and that a more gradual onset of symptoms, with slow or negligible improvement over 6 to 12 months, is more indicative of a space-occupying lesion than Bell’s palsy.13It is, however, important to note that although the patient in our case had a central lesion, she experienced an acute onset of symptoms.
The presence of additional symptoms may also suggest an alternative diagnosis. Brach and VanSwearingen13 further noted that symptoms associated with the eighth nerve, such as vertigo, tinnitus, and hearing loss may be found in patients with a CPA tumor. In patients with larger tumors, ninth and 10th nerve symptoms, including the impaired hearing noted in our patient, may be present. Some patients with ninth and 10th nerve symptoms may perceive a sense of facial numbness, but actual sensory changes in the facial nerve distribution are unlikely in Bell’s palsy. Gustatory changes, however, are consistent with Bell’s palsy.
Ear pain is consistent with Bell’s palsy and is a signal to be vigilant for the possible emergence of an ear rash, which would suggest the diagnosis of herpes zoster oticus along the trajectory of Ramsey-Hunt syndrome. Facial pain in the area of the facial nerve is inconsistent with Bell’s palsy, while hyperacusis is consistent with Bell’s palsy. Hearing loss is an eighth nerve symptom that is inconsistent with Bell’s palsy.
Similarly, there are physical examination findings that can help distinguish a true Bell’s palsy from a mimic. Changes in tear production are consistent with Bell’s palsy, but imbalance and disequilibrium are not.14
As previously noted, the patient in this case had difficulty walking and felt as if she was falling toward her right side.
One way to organize the causes of facial paralysis has been proposed by Adour et al.15 In this system, etiologies are listed as either acute paralysis or chronic, progressive paralysis. Acute paralysis (ie, the sudden onset of symptoms with maximal severity within 2 weeks), of which Bell’s palsy is the most common, can be seen in cases of polyneuritis.
A new case of Bell’s palsy has been estimated to occur in the United States every 10 minutes.8 Guillain-Barré syndrome and Lyme disease are also in this category, as is Ramsey-Hunt syndrome. Patients with Lyme disease may have a history of a tick bite or rash.14
Trauma can also cause acute facial nerve paralysis (eg, blunt trauma-associated facial fracture, penetrating trauma, birth trauma). Unilateral central facial weakness can have a neurological cause, such as a lesion to the contralateral cortex, subcortical white matter, or internal capsule.2,15 Otitis media can sometimes cause facial paralysis.16 A cholesteatoma can cause acute facial paralysis.2 Malignancies cause 5% of all cases of facial paralysis. Primary parotid tumors of various types are in this category. Metastatic disease from breast, lung, skin, colon, and kidney may cause facial paralysis. As our case illustrates, CPA tumors can cause facial paralysis.15 It is important to also note that a patient can have both a Bell’s palsy and a concurrent disease. There are a number of case reports in the literature that describe acute onset of facial paralysis as a presenting symptom of malignancy.17 In addition, there are cases wherein a neurological finding on imaging, such as an acoustic neuroma, was presumed to be the cause of facial paralysis, yet the patient’s symptoms resolved in a manner consistent with Bell’s palsy.18
For example, Lagman et al19 described a patient in which a CPA lipoma was presumed to be the cause of the facial paralysis, but the eventual outcome showed the lipoma to have been an incidentaloma.
Conclusion
This case demonstrates a presenting symptom of facial palsy and the presence of a CPA tumor. The presence of vertigo along with other historical and physical examination findings inconsistent with Bell’s palsy prompted the CT scan of the head. A review of the literature suggests a number of important findings in patients with facial palsy to assist the clinician in distinguishing true Bell’s palsy from other diseases that can mimic this condition. This case serves as a reminder of the need to perform a thorough and diligent workup to determine the presence or absence of other neurologic findings prior to closing on the diagnosis of Bell’s palsy.
Facial paralysis is a common medical complaint—one that has fascinated ancient and contemporary physicians alike.1 An idiopathic facial nerve paresis involving the lower motor neuron was described in 1821 by Sir Charles Bell. This entity became known as a Bell’s palsy, the hallmark of which was weakness or complete paralysis of the muscles of one side of the face, with no sparing of the muscles of the forehead. However, not all facial paralysis is due to Bell’s palsy.
We present a case of a patient with a Bell’s palsy mimic to facilitate and guide the differential diagnosis and distinguish conditions from the classical presentation that Bell first described to the more concerning symptoms that may not be immediately obvious. Our case further underscores the importance of performing a thorough assessment to determine the presence of other neurological findings.
Case
A 61-year-old woman presented to the ED for evaluation of right facial droop and sensation of “room spinning.” The patient stated both symptoms began approximately 36 hours prior to presentation, upon awakening.
The patient denied any headache, neck or chest pain, extremity numbness, or weakness, but stated that she felt like she was going to fall toward her right side whenever she attempted to walk. The patient’s medical history was significant for hypertension, for which she was taking losartan. Her surgical history was notable for a left oophorectomy secondary to an ovarian cyst. Regarding the social history, the patient admitted to smoking 90 packs of cigarettes per year, but denied alcohol or illicit drug use.
Upon arrival at the ED, the patient’s vital signs were: blood pressure, 164/86 mm Hg: pulse, 89 beats/min; respiratory rate, 18 breaths/min; and temperature, 98.6°F. Oxygen saturation was 98% on room air.
Physical examination revealed the patient had a right facial droop consistent with right facial palsy. She was unable to wrinkle her right forehead or fully close her right eye. There were no field cuts on confrontation. The patient’s speech was noticeable for a mild dysarthria. The motor examination revealed mild weakness of the left upper extremity and impaired right facial sensation. There were no rashes noted on the face, head, or ears. The patient had slightly impaired hearing in the right ear, which was new in onset. The remainder of the physical examination was unremarkable.
Although the patient exhibited the classic signs of Bell’s palsy, including complete paralysis of the muscles of one side of the face, inability to wrinkle the muscle of the right forehead, and inability to fully close the right eye, she also had concerning symptoms of vertigo, dysarthria, and contralateral upper extremity weakness.
A computed tomography (CT) scan of the head was ordered, which revealed a large mass lesion centered in the right petrous apex, with an associated large component extending medially into the right cerebellopontine angle (CPA) that caused a mass effect on the adjacent brainstem (Figures 1a and 1b).
Upon these findings, the patient was transferred to another facility for neurosurgical evaluation. Magnetic resonance imaging (MRI) studies performed at the receiving hospital demonstrated a large expansile heterogeneous mass lesion centered in the right petrous apex with an associated large, probable hemorrhagic soft-tissue component extending medially into the right CPA, causing a mass effect on the adjacent brainstem and mild obstructive hydrocephalus (Figures 2a and 2b).
The patient was given dexamethasone 10 mg intravenously and taken to the operating room for a right suboccipital craniotomy with subtotal tumor removal. Intraoperative high-voltage stimulation of the fifth to eighth cranial nerves showed no response, indicating significant impairment.
While there were no intraoperative complications, the patient had significant postoperative dysphagia and resultant aspiration. A tracheostomy and percutaneous endoscopic gastrostomy tube were subsequently placed. Results of a biopsy taken during surgery identified an atypical meningioma. The patient remained in the hospital for 4 weeks, after which she was discharged to a long-term care (LTC) and rehabilitation facility.
A repeat CT scan taken 2 months after surgery demonstrated absence of the previously identified large mass (Figure 1b). Three months after discharge from the LTC-rehabilitation facility, MRI of the brain showed continued interval improvement of the previously noted mass centered in the right petrous apex (Figures 3a and 3b).
Discussion
Accounts of facial paralysis and facial nerve disorders have been noted throughout history and include accounts of the condition by Hippocrates.1 Bell’s palsy was named after surgeon Sir Charles Bell, who described a peripheral-nerve paralysis of the facial nerve in 1821. Bell’s work helped to elucidate the anatomy and functional role of the facial nerve.1,2
Signs and Symptoms
The classic presentation of Bell’s palsy is weakness or complete paralysis of the muscles of one side of the face, with no sparing of the muscles of the forehead. The eyelid on the affected side generally does not close, which can result in ocular irritation due to ineffective lubrication.
A scoring system has been developed by House and Brackmann which grades the degree impairment based on such characteristics as facial muscle function and eye closure.3,4 Approximately 96% of patients with a Bell’s palsy will improve to a House-Brackmann score of 2 or better within 1 year from diagnosis,5 and 85% of patients with Bell’s palsy will show at least some improvement within 3 weeks of onset (Table).2 Although the classic description of Bell’s palsy notes the condition as idiopathic, there is an increasing body of evidence in the literature showing a link to herpes simplex virus 1.5-7
Ramsey-Hunt Syndrome
The relationship between Bell’s palsy and Ramsey-Hunt syndrome is complex and controversial. Ramsey-Hunt syndrome is a constellation of possible complications from varicella-virus infection. Symptoms of Ramsey-Hunt syndrome include facial paralysis, tinnitus, hearing loss, vertigo, hyperacusis (increased sensitivity to certain frequencies and volume ranges of sound), and decreased ocular tearing.8 Due to the nature of symptoms associated with Ramsey-Hunt syndrome, it is apparent that the condition involves more than the seventh cranial nerve. In fact, studies have shown that Ramsey-Hunt syndrome can affect the fifth, sixth, eighth, and ninth cranial nerves.8
Ramsey-Hunt syndrome, which can present in the absence of cutaneous rash (referred to as zoster sine herpete), is estimated to occur in 8% to 20% of unilateral facial nerve palsies in adult patients.8,9 Regardless of the etiology of Bell’s palsy, a review of the literature makes it clear that facial nerve paralysis is not synonymous with Bell’s palsy.10 In one example, Yetter et al10 describe the case of a patient who, though initially diagnosed with Bell’s palsy, ultimately was found to have a facial palsy due to a parotid gland malignancy.
Likewise, Stomeo11 describes a case of a patient with facial paralysis and profound ipsilateral hearing loss who ultimately was found to have a mucoepithelial carcinoma of the parotid gland. In their report, the authors note that approximately 80% of facial nerve paralysis is due to Bell’s palsy, while 5% is due to malignancy.
In another report, Clemis12 describes a case in which a patient who initially was diagnosed with Bell’s palsy eventually was found to have an adenoid cystic carcinoma of the parotid. Thus, the authors appropriately emphasize in their report that “all that palsies is not Bell’s.”
Differential Diagnosis
Historical factors, including timing and duration of symptom onset, help to distinguish a Bell’s palsy from other disorders that can mimic this condition. In their study, Brach VanSwewaringen13 highlight the fact that “not all facial paralysis is Bell’s palsy.” In their review, the authors describe clues to help distinguish conditions that mimic Bell’s palsy. For example, maximal weakness from Bell’s Palsy typically occurs within 3 to 7 days from symptom onset, and that a more gradual onset of symptoms, with slow or negligible improvement over 6 to 12 months, is more indicative of a space-occupying lesion than Bell’s palsy.13It is, however, important to note that although the patient in our case had a central lesion, she experienced an acute onset of symptoms.
The presence of additional symptoms may also suggest an alternative diagnosis. Brach and VanSwearingen13 further noted that symptoms associated with the eighth nerve, such as vertigo, tinnitus, and hearing loss may be found in patients with a CPA tumor. In patients with larger tumors, ninth and 10th nerve symptoms, including the impaired hearing noted in our patient, may be present. Some patients with ninth and 10th nerve symptoms may perceive a sense of facial numbness, but actual sensory changes in the facial nerve distribution are unlikely in Bell’s palsy. Gustatory changes, however, are consistent with Bell’s palsy.
Ear pain is consistent with Bell’s palsy and is a signal to be vigilant for the possible emergence of an ear rash, which would suggest the diagnosis of herpes zoster oticus along the trajectory of Ramsey-Hunt syndrome. Facial pain in the area of the facial nerve is inconsistent with Bell’s palsy, while hyperacusis is consistent with Bell’s palsy. Hearing loss is an eighth nerve symptom that is inconsistent with Bell’s palsy.
Similarly, there are physical examination findings that can help distinguish a true Bell’s palsy from a mimic. Changes in tear production are consistent with Bell’s palsy, but imbalance and disequilibrium are not.14
As previously noted, the patient in this case had difficulty walking and felt as if she was falling toward her right side.
One way to organize the causes of facial paralysis has been proposed by Adour et al.15 In this system, etiologies are listed as either acute paralysis or chronic, progressive paralysis. Acute paralysis (ie, the sudden onset of symptoms with maximal severity within 2 weeks), of which Bell’s palsy is the most common, can be seen in cases of polyneuritis.
A new case of Bell’s palsy has been estimated to occur in the United States every 10 minutes.8 Guillain-Barré syndrome and Lyme disease are also in this category, as is Ramsey-Hunt syndrome. Patients with Lyme disease may have a history of a tick bite or rash.14
Trauma can also cause acute facial nerve paralysis (eg, blunt trauma-associated facial fracture, penetrating trauma, birth trauma). Unilateral central facial weakness can have a neurological cause, such as a lesion to the contralateral cortex, subcortical white matter, or internal capsule.2,15 Otitis media can sometimes cause facial paralysis.16 A cholesteatoma can cause acute facial paralysis.2 Malignancies cause 5% of all cases of facial paralysis. Primary parotid tumors of various types are in this category. Metastatic disease from breast, lung, skin, colon, and kidney may cause facial paralysis. As our case illustrates, CPA tumors can cause facial paralysis.15 It is important to also note that a patient can have both a Bell’s palsy and a concurrent disease. There are a number of case reports in the literature that describe acute onset of facial paralysis as a presenting symptom of malignancy.17 In addition, there are cases wherein a neurological finding on imaging, such as an acoustic neuroma, was presumed to be the cause of facial paralysis, yet the patient’s symptoms resolved in a manner consistent with Bell’s palsy.18
For example, Lagman et al19 described a patient in which a CPA lipoma was presumed to be the cause of the facial paralysis, but the eventual outcome showed the lipoma to have been an incidentaloma.
Conclusion
This case demonstrates a presenting symptom of facial palsy and the presence of a CPA tumor. The presence of vertigo along with other historical and physical examination findings inconsistent with Bell’s palsy prompted the CT scan of the head. A review of the literature suggests a number of important findings in patients with facial palsy to assist the clinician in distinguishing true Bell’s palsy from other diseases that can mimic this condition. This case serves as a reminder of the need to perform a thorough and diligent workup to determine the presence or absence of other neurologic findings prior to closing on the diagnosis of Bell’s palsy.
1. Glicenstein J. Ann Chir Plast Esthet. 2015;60(5):347-362. doi:10.1016/j.anplas.2015.05.007.
2. Tiemstra JD, Khatkhate N. Bell’s palsy: diagnosis and management. Am Fam Physician. 2007;76(7):997-1002.
3. House JW, Brackmann DE. Facial nerve grading system. Otolaryngol Head Neck Surg. 1985;93(2):146-147. doi:10.1177/019459988509300202.
4. Reitzen SD, Babb JS, Lalwani AK. Significance and reliability of the House-Brackmann grading system for regional facial nerve function. Otolaryngol Head Neck Surg. 2009;140(2):154-158. doi:10.1016/j.otohns.2008.11.021.
5. Yeo SW, Lee DH, Jun BC, Chang KH, Park YS. Analysis of prognostic factors in Bell’s palsy and Ramsay Hunt syndrome. Auris Nasus Larynx. 2007;34(2):159-164. doi:10.1016/j.anl.2006.09.005.
6. Ahmed A. When is facial paralysis Bell palsy? Current diagnosis and treatment. Cleve Clin J Med. 2005;72(5):398-401, 405.
7. Gilden DH. Clinical practice. Bell’s palsy. N Engl J Med. 2004;351(13):1323-1331. doi:10.1056/NEJMcp041120.
8. Adour KK. Otological complications of herpes zoster. Ann Neurol. 1994;35:Suppl:S62-S64.
9. Furuta Y, Ohtani F, Mesuda Y, Fukuda S, Inuyama Y. Early diagnosis of zoster sine herpete and antiviral therapy for the treatment of facial palsy. Neurology. 2000;55(5):708-710.
10. Yetter MF, Ogren FP, Moore GF, Yonkers AJ. Bell’s palsy: a facial nerve paralysis diagnosis of exclusion. Nebr Med J. 1990;75(5):109-116.
11. Stomeo F. Possibilities of diagnostic errors in paralysis of the 7th cranial nerve. Acta Otorhinolaryngol Ital. 1989;9(6):629-633.
12. Clemis JD. All that palsies is not Bell’s: Bell’s palsy due to adenoid cystic carcinoma of the parotid. Am J Otol. 1991;12(5):397.
13. Brach JS, VanSwearingen JM. Not all facial paralysis is Bell’s palsy: a case report. Arch Phys Med Rehabil. 1999;80(7):857-859.
14. Albers JR, Tamang S. Common questions about Bell palsy. Am Fam Physician. 2014;89(3):209-212.
15. Adour KK, Hilsinger RL Jr, Callan EJ. Facial paralysis and Bell’s palsy: a protocol for differential diagnosis. Am J Otol. 1985;Suppl:68-73.
16. Morrow MJ. Bell’s palsy and herpes zoster. Curr Treat Options Neurol. 2000;2(5):407-416.
17. Quesnel AM, Lindsay RW, Hadlock TA. When the bell tolls on Bell’s palsy: finding occult malignancy in acute-onset facial paralysis. Am J Otolaryngol. 2010;31(5):339-342. doi:10.1016/j.amjoto.2009.04.003.
18. Kaushal A, Curran WJ Jr. For whom the Bell’s palsy tolls? Am J Clin Oncol. 2009;32(4):450-451. doi:10.1097/01.coc.0000239141.22916.22.
19. Lagman C, Choy W, Lee SJ, et al. A Case of Bell’s palsy with an incidental finding of a cerebellopontine angle lipoma. Cureus. 2016;8(8):e747. doi:10.7759/cureus.747.
1. Glicenstein J. Ann Chir Plast Esthet. 2015;60(5):347-362. doi:10.1016/j.anplas.2015.05.007.
2. Tiemstra JD, Khatkhate N. Bell’s palsy: diagnosis and management. Am Fam Physician. 2007;76(7):997-1002.
3. House JW, Brackmann DE. Facial nerve grading system. Otolaryngol Head Neck Surg. 1985;93(2):146-147. doi:10.1177/019459988509300202.
4. Reitzen SD, Babb JS, Lalwani AK. Significance and reliability of the House-Brackmann grading system for regional facial nerve function. Otolaryngol Head Neck Surg. 2009;140(2):154-158. doi:10.1016/j.otohns.2008.11.021.
5. Yeo SW, Lee DH, Jun BC, Chang KH, Park YS. Analysis of prognostic factors in Bell’s palsy and Ramsay Hunt syndrome. Auris Nasus Larynx. 2007;34(2):159-164. doi:10.1016/j.anl.2006.09.005.
6. Ahmed A. When is facial paralysis Bell palsy? Current diagnosis and treatment. Cleve Clin J Med. 2005;72(5):398-401, 405.
7. Gilden DH. Clinical practice. Bell’s palsy. N Engl J Med. 2004;351(13):1323-1331. doi:10.1056/NEJMcp041120.
8. Adour KK. Otological complications of herpes zoster. Ann Neurol. 1994;35:Suppl:S62-S64.
9. Furuta Y, Ohtani F, Mesuda Y, Fukuda S, Inuyama Y. Early diagnosis of zoster sine herpete and antiviral therapy for the treatment of facial palsy. Neurology. 2000;55(5):708-710.
10. Yetter MF, Ogren FP, Moore GF, Yonkers AJ. Bell’s palsy: a facial nerve paralysis diagnosis of exclusion. Nebr Med J. 1990;75(5):109-116.
11. Stomeo F. Possibilities of diagnostic errors in paralysis of the 7th cranial nerve. Acta Otorhinolaryngol Ital. 1989;9(6):629-633.
12. Clemis JD. All that palsies is not Bell’s: Bell’s palsy due to adenoid cystic carcinoma of the parotid. Am J Otol. 1991;12(5):397.
13. Brach JS, VanSwearingen JM. Not all facial paralysis is Bell’s palsy: a case report. Arch Phys Med Rehabil. 1999;80(7):857-859.
14. Albers JR, Tamang S. Common questions about Bell palsy. Am Fam Physician. 2014;89(3):209-212.
15. Adour KK, Hilsinger RL Jr, Callan EJ. Facial paralysis and Bell’s palsy: a protocol for differential diagnosis. Am J Otol. 1985;Suppl:68-73.
16. Morrow MJ. Bell’s palsy and herpes zoster. Curr Treat Options Neurol. 2000;2(5):407-416.
17. Quesnel AM, Lindsay RW, Hadlock TA. When the bell tolls on Bell’s palsy: finding occult malignancy in acute-onset facial paralysis. Am J Otolaryngol. 2010;31(5):339-342. doi:10.1016/j.amjoto.2009.04.003.
18. Kaushal A, Curran WJ Jr. For whom the Bell’s palsy tolls? Am J Clin Oncol. 2009;32(4):450-451. doi:10.1097/01.coc.0000239141.22916.22.
19. Lagman C, Choy W, Lee SJ, et al. A Case of Bell’s palsy with an incidental finding of a cerebellopontine angle lipoma. Cureus. 2016;8(8):e747. doi:10.7759/cureus.747.
Use of a Core Reamer for the Resection of a Central Distal Femoral Physeal Bone Bridge: A Novel Technique with 3-Year Follow-up
ABSTRACT
A central distal femoral physeal bone bridge in a boy aged 5 years and 7 months was resected with a fluoroscopically guided core reamer placed through a lateral parapatellar approach. At 3-year follow-up, the boy’s leg-length discrepancy was 3.0 cm (3.9 cm preoperatively), and the physeal bone bridge did not recur. The patient had full function and no pain or other patellofemoral complaints. This technique provided direct access to the physeal bone bridge, and complete resection was performed without injury to the adjacent physeal cartilage in the medial and lateral columns of the distal femur, which is expected to grow normally in the absence of the bridge.
A physeal bone bridge is an osseous connection that forms across a physis. It may cause partial premature physeal arrest. Angular deformity and limb-length discrepancy are the main complications caused by physeal bone bridges.1-4 The indications for the treatment of physeal bridges are well documented.1-5 Trauma and infection are common causes of distal femoral physeal bone bridges. Arkader and colleagues6 showed that among different types of physeal bridges, the Salter-Harris type is significantly associated with complications, among which growth arrest is the most common and occurs in 27.4% of all patients.
The treatment of distal femoral physeal bone bridges is technically difficult and provides variable results. Poor results are reported in 13% to 40% of patients.7-10 Procedure failure has been attributed to incomplete resection with the persistent tethering and dislodgement of the graft.11 Methods with improved efficacy for the removal of central physeal bridges will help prevent reformation after treatment. We have used a novel technique that allows the direct resection of a central physeal bone bridge in the distal femur through the use of a fluoroscopically guided core reamer. This technique enables the complete removal of the bone bridge and the direct visual assessment of the remaining physis. The patient’s parents provided written informed consent for print and electronic publication of this case report.
CASE
A 3-year-old boy with a history of hemifacial microsomia presented for the evaluation of genu valgum and leg-length discrepancy. His intermalleolar distance at that time was 8 cm. A standing radiograph of his lower extremities demonstrated changes consistent with physiologic genu valgum. He had no history of knee trauma, infection, or pain.
At the age of 5 years and 7 months, the patient returned for a repeat evaluation and was noted to exhibit the progressive valgus deformity of the right leg and a leg-length discrepancy of 3.9 cm (Figure 1). 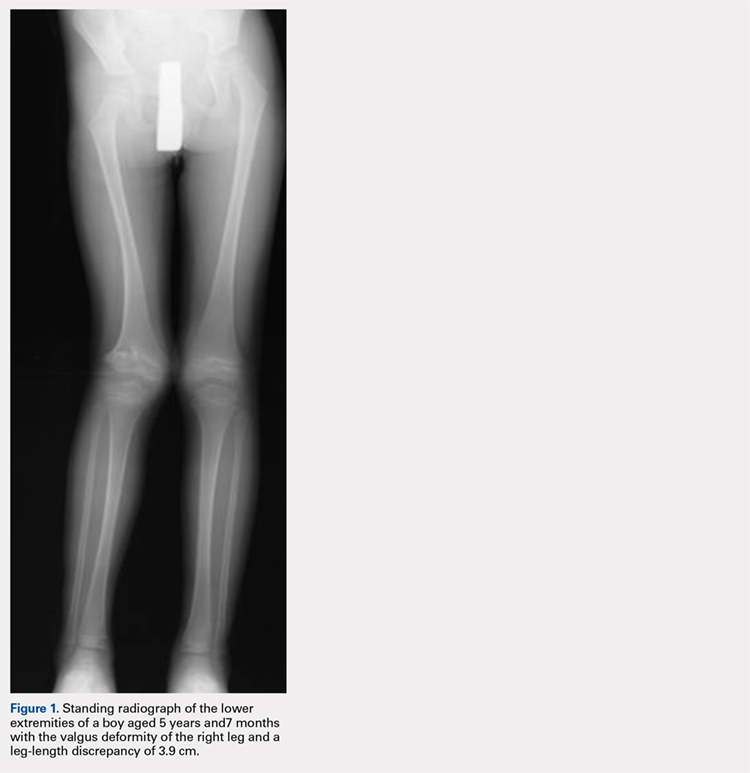
Continue to: With the patient supine on the operating...
OPERATIVE TECHNIQUE
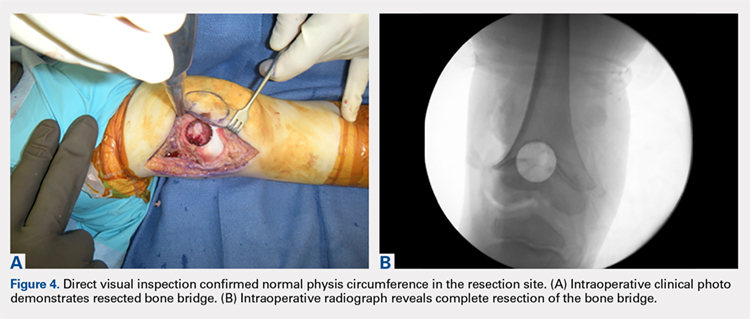
With the patient supine on the operating table and after the administration of general anesthesia, 3-dimensional (3-D) fluoroscopy was used to localize the bone bridge, which confirmed the fluoroscopic location that was previously visualized through preoperative 3-D imaging. The leg was elevated, and a tourniquet was applied and inflated. A lateral parapatellar approach was used to isolate the distal femoral physis anteriorly because the bone bridge was centered just lateral to the central portion of the distal femoral physis. A Kirschner wire was placed in the center of the bridge under anteroposterior and lateral fluoroscopic imaging (Figures 3A-3E). 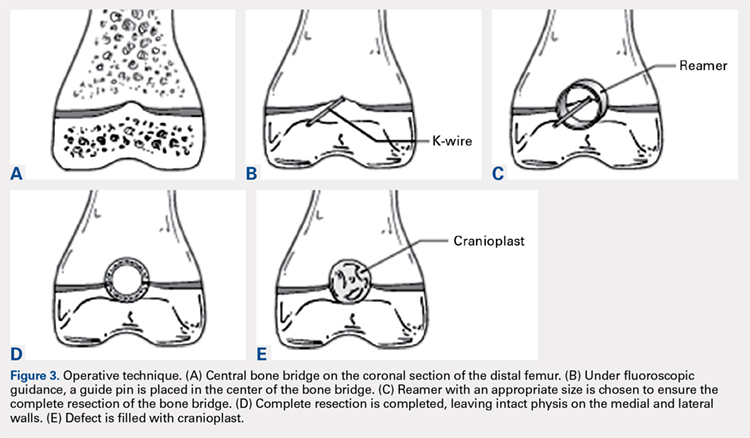
OUTCOME
The patient healed uneventfully, and early range-of-motion exercises were started 6 weeks postoperatively. At 6-month follow-up, his leg-length discrepancy was 2.7 cm, and the bone bridge did not recur. At 3-year follow-up, his leg-length discrepancy was 3.0 cm, and the bone bridge did not recur. Over the 3 years postoperatively, the patient exhibited 9.8 cm of growth on his operative side and 9.5 cm on his nonoperative side (Figure 5). 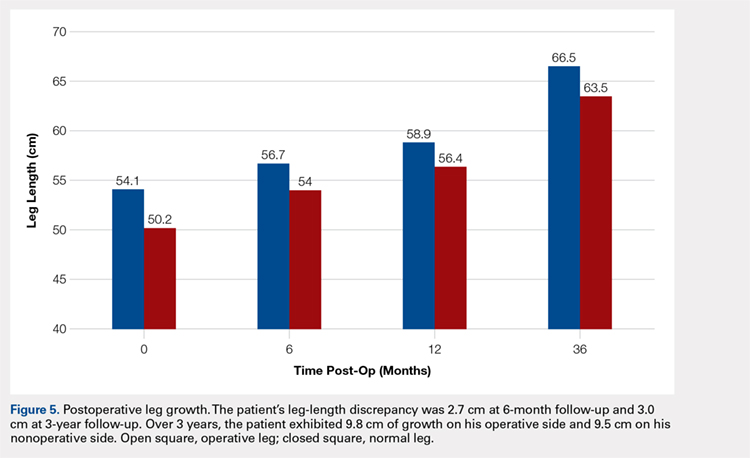
DISCUSSION
Given the considerable growth potential of the distal femoral physis,1,14-16 an injury to the distal femoral physis and the formation of a physeal bone bridge can have a profound effect on a young patient in terms of leg-length discrepancy and angular deformity. Fracture from trauma or infection is a common cause of physeal bone bridges.6,17-19 The etiology of our patient’s distal femoral physeal bone bridge is idiopathic, which is considerably less common than other etiologies, and the incidence of idiopathic physeal bone bridge formation is not well established in the literature. Hresko and Kasser21 identified atraumatic physeal bone bridge formations in 7 patients. Among the 13 patients with physeal bone bridges described by Broughton and colleagues,20 the cause of bridge formation is unknown in 1.
Physeal bone bridges that form centrally are particularly challenging because they are difficult to visualize through a peripheral approach. A number of methods for resecting central physeal bone bridges have been described. These methods have varying degrees of success. In 1981, Langenskiöld7 first described the creation of a metaphyseal mirror and the use of a dental mirror for visualization. This technique, however, yielded unfavorable results in 16% of patients. Williamson and Staheli9 reported poor results in 23% of patients. Loraas and Schmale4 described the use of an endoscope, termed an osteoscope, for visualization, citing advantages of superior illumination and potential for image magnification and capture. Marsh and Polzhofer8 also showed this technique to have low morbidity but poor results in 13% of patients, whereas Moreta and colleagues10 reported poor results in 2 out of 5 patients. The rate of poor results of these methods may be related to the technical difficulty of using dental mirrors and arthroscopes and can be improved by highly efficient direct methods with improved visualization, such as the method described in this article.
Continue to: Proper imaging is necessary for...
Proper imaging is necessary for the accurate quantification of bone bridges to determine resectability and to identify the best surgical approach to resection. MRI with software for the generation of 3-D physeal maps is a reproducible method with good interobserver reliability.22,23 Intraoperative computer-assisted imaging also is beneficial for determining the extent and location of the resection to ensure complete bone bridge removal.24
To our knowledge, a direct approach through parapatellar arthrotomy for the resection of a centrally located distal femoral physeal bone bridge has not been previously described. This novel technique provided direct access to the physeal bone bridge and was performed without injuring the adjacent physeal cartilage in the medial and lateral columns of the distal femur, which may grow normally in the absence of the bridge. Instead of using a lateral or medial approach with a metaphyseal window,4 we directly approached this central bar through a parapatellar approach and were able to completely resect it under direct visualization. This obviated the need for an arthroscope or dental mirror. To remove the entire physeal bone bridge, we needed to resect completely from the anterior cortex to the posterior cortex. Although this technique potentially increased the risk of iatrogenic fracture, we believed that this risk would not differ greatly from that of disrupting the medial or lateral metaphysis and would be more stable with either axial and torsion load. At 3-year follow-up, the patient exhibited restored normal growth in his operative limb relative to that in his nonoperative limb, had not developed angular deformity, and had maintained his previously developed limb-length discrepancy that could be corrected with the epiphysiodesis of his opposite limb at a later date.
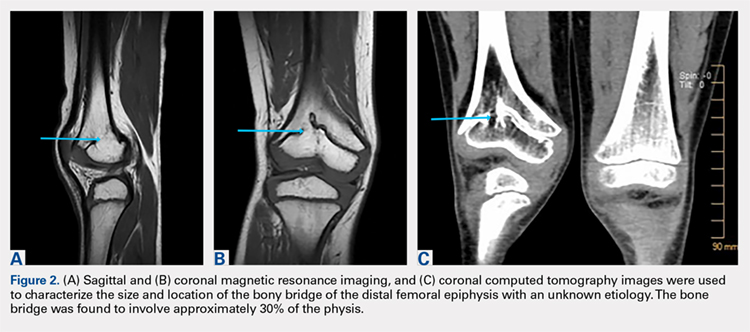
The limitations to this technique include the fact that it may be most effective with small-to moderate-sized central physeal bone bridges, although resection has shown good results with up to 70% physeal involvement.8 In this patient, the bone bridge was moderately sized (30% of the physis), centrally located, and clearly visible on fluoroscopy. These characteristics increased the technical safety and ease of the procedure. The resection of large, peripheral bridges may destabilize the distal femur. The destabilization of the distal femur, in turn, can lead to fracture. Patellofemoral mechanics may also be affected during the treatment of distal femoral physeal bone bridges. This patient has not experienced any patellofemoral dysfunction or symptoms. Given the patient’s age and significant amount of remaining growth, he will need close monitoring until he reaches skeletal maturity.
This paper will be judged for the Resident Writer’s Award.
1. Murphy GA. Disorders of tendons and fascia and adolescent and adult pes planus. In: Canale ST, Beaty JH, eds. Campbell’s Operative Orthopaedics. 12th edition. Philadelphia, PA: Mosby-Elsevier; 2013:3966-3972.
2. Khoshhal KI, Kiefer GN. Physeal bridge resection. J Am Acad Orthop Surg. 2005;13(1):47-58. doi:10.5435/00124635-200501000-00007.
3. Stans AA. Excision of physeal bar. In: Wiesel SW, ed. Operative Techniques in Orthopaedic Surgery. Philadelphia, PA: Lippincott Williams & Wilkins; 2011:1244-1249.
4. Loraas EK, Schmale GA. Endoscopically aided physeal bar takedown and guided growth for the treatment of angular limb deformity. J Pediatr Orthop B. 2012;21(4):348-351. doi:10.1097/BPB.0b013e328346d308.
5. Inoue T, Naito M, Fuhii T, Akiyoshi Y, Yoshimura I, Takamura K. Partial physeal growth arrest treated by bridge resection and artificial dura substitute interposition. J Pediatr Orthop B. 2006;15(1):65-69. doi:10.1097/01202412-200601000-00014.
6. Arkader A, Warner WC Jr, Horn BD, Shaw RN, Wells L. Predicting the outcome of physeal fractures of the distal femur. J Pediatr Orthop. 2007;27(6):703-708. doi:10.1097/BPO.0b013e3180dca0e5.
7. Langenskiöld A. Surgical treatment of partial closure of the growth plate. J Pediatr Orthop. 1981;1(1):3-11. doi:10.1097/01241398-198101010-00002.
8. Marsh JS, Polzhofer GK. Arthroscopically assisted central physeal bar resection. J Pediatr Orthop. 2006;26(2):255-259. doi:10.1097/01.bpo.0000218533.43986.e1.
9. Williamson RV, Staheli LT. Partial physeal growth arrest: treatment by bridge resection and fat interposition. J Pediatr Orthop. 1990;10(6):769-776. doi:10.1097/01241398-199011000-00012.
10. Moreta J, Abril JC, Miranda C. Arthroscopy-assisted resection-interposition of post-traumatic central physeal bridges. Rev Esp Cir Orthop Traumatol. 2013;57(5):333-339. doi:10.1016/j.recot.2013.07.004.
11. Hasler CC, Foster BK. Secondary tethers after physeal bar resection: a common source of failure? Clin Orthop Relat Res. 2002;405:242-249.
12. Paley D, Bhave A, Herzenberg JE, Bowen JR. Multiplier method for predicting limb-length discrepancy. J Bone Joint Surg Am. 2000;82(10):1432-1446. doi:10.2106/00004623-200010000-00010.
13. Khoshhal KI, Kiefer GN. Physeal bridge resection. J Am Acad Orthop Surg. 2005;13(1):47-58. doi:10.5435/00124635-200501000-00007.
14. Rathjen KE, Kim HKW. Physeal injuries and growth disturbances. In: Flynn JM, Skaggs DL, Waters PM, eds. Rockwood and Wilkins’ Fractures in Children. 8th edition. Philadelphia, PA: Wolters-Kluwer; 2015:135-137.
15. Peterson CA, Peterson HA. Analysis of the incidence of injuries to the epiphyseal growth plate. J Trauma. 1972;12(4):275-281. doi:10.1097/00005373-197204000-00002.
16. Pritchett JW. Longitudinal growth and growth-plate activity in the lower extremity. Clin Orthop Relat Res. 1992;275:274-279.
17. Cassebaum WH, Patterson AH. Fracture of the distal femoral epiphysis. Clin Orthop Relat Res. 1965;41:79-91. doi:10.1097/00003086-196500410-00009.
18. Dahl WJ, Silva S, Vanderhave KL. Distal femoral physeal fixation: are smooth pins really safe? J Pedatir Orthop. 2014;34(2):134-138. doi:10.1097/BPO.0000000000000083.
19. Roberts J. Fracture separation of the distal femoral epiphyseal growth line. J Bone Joint Surg Am. 1973;55:1324.
20. Broughton NS, Dickens DR, Cole WG, Menelaus MB. Epiphyseolysis for partial growth plate arrest. Results after four years or at maturity. J Bone Joint Surg Br. 1989;71(1):13-16. doi:10.1302/0301-620X.71B1.2914983.
21. Hresko MT, Kasser JR. Physeal arrest about the knee associated with non-physeal fractures in the lower extremity. J Bone Joint Surg Am. 1989;71(5):698-703. doi:10.2106/00004623-198971050-00009.
22. Lurie B, Koff MF, Shah P, et al. Three-dimensional magnetic resonance imaging of physeal injury: reliability and clinical utility. J Pediatr Orthop. 2014;34(3):239-245. doi:10.1097/BPO.0000000000000104.
23. Sailhan F, Chotel F, Guibal AL, et al. Three-dimensional MR imaging in the assessment of physeal growth arrest. Eur Radiol. 2004;14(9):1600-1608. doi:10.1007/s00330-004-2319-z.
24. Kang HG, Yoon SJ, Kim JR. Resection of a physeal bar under computer-assisted guidance. J Bone Joint Surg Br. 2010;92(10):1452-1455. doi:10.1302/0301-620X.92B10.24587.
ABSTRACT
A central distal femoral physeal bone bridge in a boy aged 5 years and 7 months was resected with a fluoroscopically guided core reamer placed through a lateral parapatellar approach. At 3-year follow-up, the boy’s leg-length discrepancy was 3.0 cm (3.9 cm preoperatively), and the physeal bone bridge did not recur. The patient had full function and no pain or other patellofemoral complaints. This technique provided direct access to the physeal bone bridge, and complete resection was performed without injury to the adjacent physeal cartilage in the medial and lateral columns of the distal femur, which is expected to grow normally in the absence of the bridge.
A physeal bone bridge is an osseous connection that forms across a physis. It may cause partial premature physeal arrest. Angular deformity and limb-length discrepancy are the main complications caused by physeal bone bridges.1-4 The indications for the treatment of physeal bridges are well documented.1-5 Trauma and infection are common causes of distal femoral physeal bone bridges. Arkader and colleagues6 showed that among different types of physeal bridges, the Salter-Harris type is significantly associated with complications, among which growth arrest is the most common and occurs in 27.4% of all patients.
The treatment of distal femoral physeal bone bridges is technically difficult and provides variable results. Poor results are reported in 13% to 40% of patients.7-10 Procedure failure has been attributed to incomplete resection with the persistent tethering and dislodgement of the graft.11 Methods with improved efficacy for the removal of central physeal bridges will help prevent reformation after treatment. We have used a novel technique that allows the direct resection of a central physeal bone bridge in the distal femur through the use of a fluoroscopically guided core reamer. This technique enables the complete removal of the bone bridge and the direct visual assessment of the remaining physis. The patient’s parents provided written informed consent for print and electronic publication of this case report.
CASE
A 3-year-old boy with a history of hemifacial microsomia presented for the evaluation of genu valgum and leg-length discrepancy. His intermalleolar distance at that time was 8 cm. A standing radiograph of his lower extremities demonstrated changes consistent with physiologic genu valgum. He had no history of knee trauma, infection, or pain.
At the age of 5 years and 7 months, the patient returned for a repeat evaluation and was noted to exhibit the progressive valgus deformity of the right leg and a leg-length discrepancy of 3.9 cm (Figure 1).
Continue to: With the patient supine on the operating...
OPERATIVE TECHNIQUE
With the patient supine on the operating table and after the administration of general anesthesia, 3-dimensional (3-D) fluoroscopy was used to localize the bone bridge, which confirmed the fluoroscopic location that was previously visualized through preoperative 3-D imaging. The leg was elevated, and a tourniquet was applied and inflated. A lateral parapatellar approach was used to isolate the distal femoral physis anteriorly because the bone bridge was centered just lateral to the central portion of the distal femoral physis. A Kirschner wire was placed in the center of the bridge under anteroposterior and lateral fluoroscopic imaging (Figures 3A-3E).
OUTCOME
The patient healed uneventfully, and early range-of-motion exercises were started 6 weeks postoperatively. At 6-month follow-up, his leg-length discrepancy was 2.7 cm, and the bone bridge did not recur. At 3-year follow-up, his leg-length discrepancy was 3.0 cm, and the bone bridge did not recur. Over the 3 years postoperatively, the patient exhibited 9.8 cm of growth on his operative side and 9.5 cm on his nonoperative side (Figure 5).
DISCUSSION
Given the considerable growth potential of the distal femoral physis,1,14-16 an injury to the distal femoral physis and the formation of a physeal bone bridge can have a profound effect on a young patient in terms of leg-length discrepancy and angular deformity. Fracture from trauma or infection is a common cause of physeal bone bridges.6,17-19 The etiology of our patient’s distal femoral physeal bone bridge is idiopathic, which is considerably less common than other etiologies, and the incidence of idiopathic physeal bone bridge formation is not well established in the literature. Hresko and Kasser21 identified atraumatic physeal bone bridge formations in 7 patients. Among the 13 patients with physeal bone bridges described by Broughton and colleagues,20 the cause of bridge formation is unknown in 1.
Physeal bone bridges that form centrally are particularly challenging because they are difficult to visualize through a peripheral approach. A number of methods for resecting central physeal bone bridges have been described. These methods have varying degrees of success. In 1981, Langenskiöld7 first described the creation of a metaphyseal mirror and the use of a dental mirror for visualization. This technique, however, yielded unfavorable results in 16% of patients. Williamson and Staheli9 reported poor results in 23% of patients. Loraas and Schmale4 described the use of an endoscope, termed an osteoscope, for visualization, citing advantages of superior illumination and potential for image magnification and capture. Marsh and Polzhofer8 also showed this technique to have low morbidity but poor results in 13% of patients, whereas Moreta and colleagues10 reported poor results in 2 out of 5 patients. The rate of poor results of these methods may be related to the technical difficulty of using dental mirrors and arthroscopes and can be improved by highly efficient direct methods with improved visualization, such as the method described in this article.
Continue to: Proper imaging is necessary for...
Proper imaging is necessary for the accurate quantification of bone bridges to determine resectability and to identify the best surgical approach to resection. MRI with software for the generation of 3-D physeal maps is a reproducible method with good interobserver reliability.22,23 Intraoperative computer-assisted imaging also is beneficial for determining the extent and location of the resection to ensure complete bone bridge removal.24
To our knowledge, a direct approach through parapatellar arthrotomy for the resection of a centrally located distal femoral physeal bone bridge has not been previously described. This novel technique provided direct access to the physeal bone bridge and was performed without injuring the adjacent physeal cartilage in the medial and lateral columns of the distal femur, which may grow normally in the absence of the bridge. Instead of using a lateral or medial approach with a metaphyseal window,4 we directly approached this central bar through a parapatellar approach and were able to completely resect it under direct visualization. This obviated the need for an arthroscope or dental mirror. To remove the entire physeal bone bridge, we needed to resect completely from the anterior cortex to the posterior cortex. Although this technique potentially increased the risk of iatrogenic fracture, we believed that this risk would not differ greatly from that of disrupting the medial or lateral metaphysis and would be more stable with either axial and torsion load. At 3-year follow-up, the patient exhibited restored normal growth in his operative limb relative to that in his nonoperative limb, had not developed angular deformity, and had maintained his previously developed limb-length discrepancy that could be corrected with the epiphysiodesis of his opposite limb at a later date.
The limitations to this technique include the fact that it may be most effective with small-to moderate-sized central physeal bone bridges, although resection has shown good results with up to 70% physeal involvement.8 In this patient, the bone bridge was moderately sized (30% of the physis), centrally located, and clearly visible on fluoroscopy. These characteristics increased the technical safety and ease of the procedure. The resection of large, peripheral bridges may destabilize the distal femur. The destabilization of the distal femur, in turn, can lead to fracture. Patellofemoral mechanics may also be affected during the treatment of distal femoral physeal bone bridges. This patient has not experienced any patellofemoral dysfunction or symptoms. Given the patient’s age and significant amount of remaining growth, he will need close monitoring until he reaches skeletal maturity.
This paper will be judged for the Resident Writer’s Award.
ABSTRACT
A central distal femoral physeal bone bridge in a boy aged 5 years and 7 months was resected with a fluoroscopically guided core reamer placed through a lateral parapatellar approach. At 3-year follow-up, the boy’s leg-length discrepancy was 3.0 cm (3.9 cm preoperatively), and the physeal bone bridge did not recur. The patient had full function and no pain or other patellofemoral complaints. This technique provided direct access to the physeal bone bridge, and complete resection was performed without injury to the adjacent physeal cartilage in the medial and lateral columns of the distal femur, which is expected to grow normally in the absence of the bridge.
A physeal bone bridge is an osseous connection that forms across a physis. It may cause partial premature physeal arrest. Angular deformity and limb-length discrepancy are the main complications caused by physeal bone bridges.1-4 The indications for the treatment of physeal bridges are well documented.1-5 Trauma and infection are common causes of distal femoral physeal bone bridges. Arkader and colleagues6 showed that among different types of physeal bridges, the Salter-Harris type is significantly associated with complications, among which growth arrest is the most common and occurs in 27.4% of all patients.
The treatment of distal femoral physeal bone bridges is technically difficult and provides variable results. Poor results are reported in 13% to 40% of patients.7-10 Procedure failure has been attributed to incomplete resection with the persistent tethering and dislodgement of the graft.11 Methods with improved efficacy for the removal of central physeal bridges will help prevent reformation after treatment. We have used a novel technique that allows the direct resection of a central physeal bone bridge in the distal femur through the use of a fluoroscopically guided core reamer. This technique enables the complete removal of the bone bridge and the direct visual assessment of the remaining physis. The patient’s parents provided written informed consent for print and electronic publication of this case report.
CASE
A 3-year-old boy with a history of hemifacial microsomia presented for the evaluation of genu valgum and leg-length discrepancy. His intermalleolar distance at that time was 8 cm. A standing radiograph of his lower extremities demonstrated changes consistent with physiologic genu valgum. He had no history of knee trauma, infection, or pain.
At the age of 5 years and 7 months, the patient returned for a repeat evaluation and was noted to exhibit the progressive valgus deformity of the right leg and a leg-length discrepancy of 3.9 cm (Figure 1).
Continue to: With the patient supine on the operating...
OPERATIVE TECHNIQUE
With the patient supine on the operating table and after the administration of general anesthesia, 3-dimensional (3-D) fluoroscopy was used to localize the bone bridge, which confirmed the fluoroscopic location that was previously visualized through preoperative 3-D imaging. The leg was elevated, and a tourniquet was applied and inflated. A lateral parapatellar approach was used to isolate the distal femoral physis anteriorly because the bone bridge was centered just lateral to the central portion of the distal femoral physis. A Kirschner wire was placed in the center of the bridge under anteroposterior and lateral fluoroscopic imaging (Figures 3A-3E).
OUTCOME
The patient healed uneventfully, and early range-of-motion exercises were started 6 weeks postoperatively. At 6-month follow-up, his leg-length discrepancy was 2.7 cm, and the bone bridge did not recur. At 3-year follow-up, his leg-length discrepancy was 3.0 cm, and the bone bridge did not recur. Over the 3 years postoperatively, the patient exhibited 9.8 cm of growth on his operative side and 9.5 cm on his nonoperative side (Figure 5).
DISCUSSION
Given the considerable growth potential of the distal femoral physis,1,14-16 an injury to the distal femoral physis and the formation of a physeal bone bridge can have a profound effect on a young patient in terms of leg-length discrepancy and angular deformity. Fracture from trauma or infection is a common cause of physeal bone bridges.6,17-19 The etiology of our patient’s distal femoral physeal bone bridge is idiopathic, which is considerably less common than other etiologies, and the incidence of idiopathic physeal bone bridge formation is not well established in the literature. Hresko and Kasser21 identified atraumatic physeal bone bridge formations in 7 patients. Among the 13 patients with physeal bone bridges described by Broughton and colleagues,20 the cause of bridge formation is unknown in 1.
Physeal bone bridges that form centrally are particularly challenging because they are difficult to visualize through a peripheral approach. A number of methods for resecting central physeal bone bridges have been described. These methods have varying degrees of success. In 1981, Langenskiöld7 first described the creation of a metaphyseal mirror and the use of a dental mirror for visualization. This technique, however, yielded unfavorable results in 16% of patients. Williamson and Staheli9 reported poor results in 23% of patients. Loraas and Schmale4 described the use of an endoscope, termed an osteoscope, for visualization, citing advantages of superior illumination and potential for image magnification and capture. Marsh and Polzhofer8 also showed this technique to have low morbidity but poor results in 13% of patients, whereas Moreta and colleagues10 reported poor results in 2 out of 5 patients. The rate of poor results of these methods may be related to the technical difficulty of using dental mirrors and arthroscopes and can be improved by highly efficient direct methods with improved visualization, such as the method described in this article.
Continue to: Proper imaging is necessary for...
Proper imaging is necessary for the accurate quantification of bone bridges to determine resectability and to identify the best surgical approach to resection. MRI with software for the generation of 3-D physeal maps is a reproducible method with good interobserver reliability.22,23 Intraoperative computer-assisted imaging also is beneficial for determining the extent and location of the resection to ensure complete bone bridge removal.24
To our knowledge, a direct approach through parapatellar arthrotomy for the resection of a centrally located distal femoral physeal bone bridge has not been previously described. This novel technique provided direct access to the physeal bone bridge and was performed without injuring the adjacent physeal cartilage in the medial and lateral columns of the distal femur, which may grow normally in the absence of the bridge. Instead of using a lateral or medial approach with a metaphyseal window,4 we directly approached this central bar through a parapatellar approach and were able to completely resect it under direct visualization. This obviated the need for an arthroscope or dental mirror. To remove the entire physeal bone bridge, we needed to resect completely from the anterior cortex to the posterior cortex. Although this technique potentially increased the risk of iatrogenic fracture, we believed that this risk would not differ greatly from that of disrupting the medial or lateral metaphysis and would be more stable with either axial and torsion load. At 3-year follow-up, the patient exhibited restored normal growth in his operative limb relative to that in his nonoperative limb, had not developed angular deformity, and had maintained his previously developed limb-length discrepancy that could be corrected with the epiphysiodesis of his opposite limb at a later date.
The limitations to this technique include the fact that it may be most effective with small-to moderate-sized central physeal bone bridges, although resection has shown good results with up to 70% physeal involvement.8 In this patient, the bone bridge was moderately sized (30% of the physis), centrally located, and clearly visible on fluoroscopy. These characteristics increased the technical safety and ease of the procedure. The resection of large, peripheral bridges may destabilize the distal femur. The destabilization of the distal femur, in turn, can lead to fracture. Patellofemoral mechanics may also be affected during the treatment of distal femoral physeal bone bridges. This patient has not experienced any patellofemoral dysfunction or symptoms. Given the patient’s age and significant amount of remaining growth, he will need close monitoring until he reaches skeletal maturity.
This paper will be judged for the Resident Writer’s Award.
1. Murphy GA. Disorders of tendons and fascia and adolescent and adult pes planus. In: Canale ST, Beaty JH, eds. Campbell’s Operative Orthopaedics. 12th edition. Philadelphia, PA: Mosby-Elsevier; 2013:3966-3972.
2. Khoshhal KI, Kiefer GN. Physeal bridge resection. J Am Acad Orthop Surg. 2005;13(1):47-58. doi:10.5435/00124635-200501000-00007.
3. Stans AA. Excision of physeal bar. In: Wiesel SW, ed. Operative Techniques in Orthopaedic Surgery. Philadelphia, PA: Lippincott Williams & Wilkins; 2011:1244-1249.
4. Loraas EK, Schmale GA. Endoscopically aided physeal bar takedown and guided growth for the treatment of angular limb deformity. J Pediatr Orthop B. 2012;21(4):348-351. doi:10.1097/BPB.0b013e328346d308.
5. Inoue T, Naito M, Fuhii T, Akiyoshi Y, Yoshimura I, Takamura K. Partial physeal growth arrest treated by bridge resection and artificial dura substitute interposition. J Pediatr Orthop B. 2006;15(1):65-69. doi:10.1097/01202412-200601000-00014.
6. Arkader A, Warner WC Jr, Horn BD, Shaw RN, Wells L. Predicting the outcome of physeal fractures of the distal femur. J Pediatr Orthop. 2007;27(6):703-708. doi:10.1097/BPO.0b013e3180dca0e5.
7. Langenskiöld A. Surgical treatment of partial closure of the growth plate. J Pediatr Orthop. 1981;1(1):3-11. doi:10.1097/01241398-198101010-00002.
8. Marsh JS, Polzhofer GK. Arthroscopically assisted central physeal bar resection. J Pediatr Orthop. 2006;26(2):255-259. doi:10.1097/01.bpo.0000218533.43986.e1.
9. Williamson RV, Staheli LT. Partial physeal growth arrest: treatment by bridge resection and fat interposition. J Pediatr Orthop. 1990;10(6):769-776. doi:10.1097/01241398-199011000-00012.
10. Moreta J, Abril JC, Miranda C. Arthroscopy-assisted resection-interposition of post-traumatic central physeal bridges. Rev Esp Cir Orthop Traumatol. 2013;57(5):333-339. doi:10.1016/j.recot.2013.07.004.
11. Hasler CC, Foster BK. Secondary tethers after physeal bar resection: a common source of failure? Clin Orthop Relat Res. 2002;405:242-249.
12. Paley D, Bhave A, Herzenberg JE, Bowen JR. Multiplier method for predicting limb-length discrepancy. J Bone Joint Surg Am. 2000;82(10):1432-1446. doi:10.2106/00004623-200010000-00010.
13. Khoshhal KI, Kiefer GN. Physeal bridge resection. J Am Acad Orthop Surg. 2005;13(1):47-58. doi:10.5435/00124635-200501000-00007.
14. Rathjen KE, Kim HKW. Physeal injuries and growth disturbances. In: Flynn JM, Skaggs DL, Waters PM, eds. Rockwood and Wilkins’ Fractures in Children. 8th edition. Philadelphia, PA: Wolters-Kluwer; 2015:135-137.
15. Peterson CA, Peterson HA. Analysis of the incidence of injuries to the epiphyseal growth plate. J Trauma. 1972;12(4):275-281. doi:10.1097/00005373-197204000-00002.
16. Pritchett JW. Longitudinal growth and growth-plate activity in the lower extremity. Clin Orthop Relat Res. 1992;275:274-279.
17. Cassebaum WH, Patterson AH. Fracture of the distal femoral epiphysis. Clin Orthop Relat Res. 1965;41:79-91. doi:10.1097/00003086-196500410-00009.
18. Dahl WJ, Silva S, Vanderhave KL. Distal femoral physeal fixation: are smooth pins really safe? J Pedatir Orthop. 2014;34(2):134-138. doi:10.1097/BPO.0000000000000083.
19. Roberts J. Fracture separation of the distal femoral epiphyseal growth line. J Bone Joint Surg Am. 1973;55:1324.
20. Broughton NS, Dickens DR, Cole WG, Menelaus MB. Epiphyseolysis for partial growth plate arrest. Results after four years or at maturity. J Bone Joint Surg Br. 1989;71(1):13-16. doi:10.1302/0301-620X.71B1.2914983.
21. Hresko MT, Kasser JR. Physeal arrest about the knee associated with non-physeal fractures in the lower extremity. J Bone Joint Surg Am. 1989;71(5):698-703. doi:10.2106/00004623-198971050-00009.
22. Lurie B, Koff MF, Shah P, et al. Three-dimensional magnetic resonance imaging of physeal injury: reliability and clinical utility. J Pediatr Orthop. 2014;34(3):239-245. doi:10.1097/BPO.0000000000000104.
23. Sailhan F, Chotel F, Guibal AL, et al. Three-dimensional MR imaging in the assessment of physeal growth arrest. Eur Radiol. 2004;14(9):1600-1608. doi:10.1007/s00330-004-2319-z.
24. Kang HG, Yoon SJ, Kim JR. Resection of a physeal bar under computer-assisted guidance. J Bone Joint Surg Br. 2010;92(10):1452-1455. doi:10.1302/0301-620X.92B10.24587.
1. Murphy GA. Disorders of tendons and fascia and adolescent and adult pes planus. In: Canale ST, Beaty JH, eds. Campbell’s Operative Orthopaedics. 12th edition. Philadelphia, PA: Mosby-Elsevier; 2013:3966-3972.
2. Khoshhal KI, Kiefer GN. Physeal bridge resection. J Am Acad Orthop Surg. 2005;13(1):47-58. doi:10.5435/00124635-200501000-00007.
3. Stans AA. Excision of physeal bar. In: Wiesel SW, ed. Operative Techniques in Orthopaedic Surgery. Philadelphia, PA: Lippincott Williams & Wilkins; 2011:1244-1249.
4. Loraas EK, Schmale GA. Endoscopically aided physeal bar takedown and guided growth for the treatment of angular limb deformity. J Pediatr Orthop B. 2012;21(4):348-351. doi:10.1097/BPB.0b013e328346d308.
5. Inoue T, Naito M, Fuhii T, Akiyoshi Y, Yoshimura I, Takamura K. Partial physeal growth arrest treated by bridge resection and artificial dura substitute interposition. J Pediatr Orthop B. 2006;15(1):65-69. doi:10.1097/01202412-200601000-00014.
6. Arkader A, Warner WC Jr, Horn BD, Shaw RN, Wells L. Predicting the outcome of physeal fractures of the distal femur. J Pediatr Orthop. 2007;27(6):703-708. doi:10.1097/BPO.0b013e3180dca0e5.
7. Langenskiöld A. Surgical treatment of partial closure of the growth plate. J Pediatr Orthop. 1981;1(1):3-11. doi:10.1097/01241398-198101010-00002.
8. Marsh JS, Polzhofer GK. Arthroscopically assisted central physeal bar resection. J Pediatr Orthop. 2006;26(2):255-259. doi:10.1097/01.bpo.0000218533.43986.e1.
9. Williamson RV, Staheli LT. Partial physeal growth arrest: treatment by bridge resection and fat interposition. J Pediatr Orthop. 1990;10(6):769-776. doi:10.1097/01241398-199011000-00012.
10. Moreta J, Abril JC, Miranda C. Arthroscopy-assisted resection-interposition of post-traumatic central physeal bridges. Rev Esp Cir Orthop Traumatol. 2013;57(5):333-339. doi:10.1016/j.recot.2013.07.004.
11. Hasler CC, Foster BK. Secondary tethers after physeal bar resection: a common source of failure? Clin Orthop Relat Res. 2002;405:242-249.
12. Paley D, Bhave A, Herzenberg JE, Bowen JR. Multiplier method for predicting limb-length discrepancy. J Bone Joint Surg Am. 2000;82(10):1432-1446. doi:10.2106/00004623-200010000-00010.
13. Khoshhal KI, Kiefer GN. Physeal bridge resection. J Am Acad Orthop Surg. 2005;13(1):47-58. doi:10.5435/00124635-200501000-00007.
14. Rathjen KE, Kim HKW. Physeal injuries and growth disturbances. In: Flynn JM, Skaggs DL, Waters PM, eds. Rockwood and Wilkins’ Fractures in Children. 8th edition. Philadelphia, PA: Wolters-Kluwer; 2015:135-137.
15. Peterson CA, Peterson HA. Analysis of the incidence of injuries to the epiphyseal growth plate. J Trauma. 1972;12(4):275-281. doi:10.1097/00005373-197204000-00002.
16. Pritchett JW. Longitudinal growth and growth-plate activity in the lower extremity. Clin Orthop Relat Res. 1992;275:274-279.
17. Cassebaum WH, Patterson AH. Fracture of the distal femoral epiphysis. Clin Orthop Relat Res. 1965;41:79-91. doi:10.1097/00003086-196500410-00009.
18. Dahl WJ, Silva S, Vanderhave KL. Distal femoral physeal fixation: are smooth pins really safe? J Pedatir Orthop. 2014;34(2):134-138. doi:10.1097/BPO.0000000000000083.
19. Roberts J. Fracture separation of the distal femoral epiphyseal growth line. J Bone Joint Surg Am. 1973;55:1324.
20. Broughton NS, Dickens DR, Cole WG, Menelaus MB. Epiphyseolysis for partial growth plate arrest. Results after four years or at maturity. J Bone Joint Surg Br. 1989;71(1):13-16. doi:10.1302/0301-620X.71B1.2914983.
21. Hresko MT, Kasser JR. Physeal arrest about the knee associated with non-physeal fractures in the lower extremity. J Bone Joint Surg Am. 1989;71(5):698-703. doi:10.2106/00004623-198971050-00009.
22. Lurie B, Koff MF, Shah P, et al. Three-dimensional magnetic resonance imaging of physeal injury: reliability and clinical utility. J Pediatr Orthop. 2014;34(3):239-245. doi:10.1097/BPO.0000000000000104.
23. Sailhan F, Chotel F, Guibal AL, et al. Three-dimensional MR imaging in the assessment of physeal growth arrest. Eur Radiol. 2004;14(9):1600-1608. doi:10.1007/s00330-004-2319-z.
24. Kang HG, Yoon SJ, Kim JR. Resection of a physeal bar under computer-assisted guidance. J Bone Joint Surg Br. 2010;92(10):1452-1455. doi:10.1302/0301-620X.92B10.24587.
TAKE-HOME POINTS
- Central physeal arrest of the distal femur is challenging, but this surgical technique provides an option for treatment.
- Partial bone bridges can be resected, but advanced imaging with MRI or CT, or both, is helpful in preoperative planning.
- Regardless of the type of physeal bar resection that is chosen, it is unlikely that complete, normal bone growth will be restored and closed follow up will be needed.
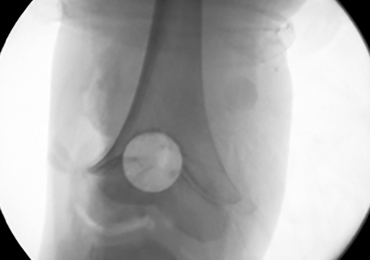
Recurrence of Extranodal Natural Killer/T-cell Lymphoma Presenting as Tarsal Tunnel Syndrome
ABSTRACT
This case report is a rare form of lymphoma recurrence which presented as tarsal tunnel syndrome. The patient had been previously treated for the malignancy and was presumed to be in remission; however, standard radiology imaging protocols failed to include the distal extremities on these scans. The patient presented to the orthopedic clinic with tarsal tunnel symptoms and a mass in the tarsal tunnel. A complete evaluation resulted in a diagnosis of recurrence of the malignancy. This case illustrates the importance of a thorough medical history and personal review of imaging studies, and how a systematic approach can produce the correct diagnosis for any unknown lesion. Furthermore, this case may prompt oncologists to consider obtaining whole-body fluorodeoxyglucose positron emission tomography computed tomography when evaluating for recurrence in patients.
Nasal-type, extranodal natural killer/T-cell lymphoma (ENKTL) is a rare form of non-Hodgkin lymphoma (NHL). Malignancies account for only 10% of NHL in Asian and South American populations. However, in Caucasians, it represents <1% of all cases. In addition, at 3:1 male to female ratio, the disease most commonly affects male patients who are 50 to 59 years old.1-3 The etiology of this malignancy is strongly related to prior infection with Epstein-Barr virus (EBV) as EBV-encoded early small ribonucleic acid on in situ hybridization of lymphoma cells is positive in 95% of cases.4-6
Typical sites of involvement include the nasal cavity, nasopharynx, and sinuses, causing patients to present with nasal obstruction, chronic sinusitis, or epistaxis. Additionally, ENKTL can occur primarily in the skin, gastrointestinal tract, spleen, and testis, whereas the bone marrow may be involved in 10% of cases. Although rare, unusual sites, including muscle, adrenals, and ovaries, have been published.7,8
Staging is best performed using the T-staging system, which accounts for the extent of local tumor involvement. Higher stages, such as T3 /T4, equate to locally advanced disease and imply a worse prognosis.9,10 Computed tomography (CT) and magnetic resonance imaging (MRI) help define local soft tissues and bony involvement. Furthermore, CT of the chest, abdomen, and pelvis as well as bone marrow biopsy are performed as part of the staging process. Lastly, fluorine-18 fluorodeoxyglucose positron emission tomography CT (18-FDG PET-CT) is often used to detect extranodal spread, define the extent of involvement, differentiate between lymphoma and inflammatory masses, and monitor for recurrence.11
Treatment for local ENKTL involves concurrent chemoradiotherapy followed by 3 cycles of etoposide, ifosfamide, cisplatin, and dexamethasone, which results in a complete response rate of 80%, and is the most favorable when comparing treatment modalities.12 Unfortunately, recurrence rates reach as high as 50%, whereas the 5-year survival rate is 59%.13,14 For recurrent or disseminated disease, high-dose chemotherapy and hematopoietic stem cell transplantation remain as alternative treatments for patients who have undergone 2 complete remissions and can be curative in some instances.13,15
Continue to: In summary, ENKTL is a rare form...
In summary, ENKTL is a rare form of NHL which classically presents in the nasal cavity; however, this type of lymphoma may present in a variety of extranodal sites.7,8 Despite the numerous published reports on ENKTL, no study has reported either primary or recurrent ENKTL in the feet or hands. To our knowledge, this is one of the first published cases of a patient who developed a rare and recurring ENKTL in the foot and ankle. The patient provided written informed consent for print and electronic publication of this case report.
CASE
A 59-year-old Caucasian woman was referred to the orthopedic foot and ankle clinic by her primary care physician for right medial ankle pain, skin ulceration, and numbness over the plantar aspect of her right foot. Upon questioning, the patient noted that the pain and numbness were present for almost 6 months. She denied trauma to the concerned area. Previously, the patient was observed and treated elsewhere for plantar fasciitis and was prescribed a brace before being immobilized in a controlled ankle motion (CAM) boot for 6 weeks. At follow-up with her outside provider, the patient had developed skin breakdown over the medial aspect of the right ankle, and this condition was presumed to be caused by the boot. After local wound care failed to improve her skin ulceration, she returned to her primary care physician, who ordered an MRI of the area and referred her to our specialty clinic.
Upon review, the patient’s past medical history included a diagnosis of nasal-type ENKTL. Her malignancy was treated with chemoradiotherapy 2 years prior to her consultation with the foot and ankle clinic.
The patient was noted by her medical oncologist and interventional radiologist to be in complete stage 4 remission since being treated. She underwent routine MRI and CT scans of the head and neck at 6-month intervals and FDG PET-CT scans at 3-month intervals, as per institutional protocol. The examinations showed no evidence of malignancy or metabolically active disease. The last imaging study occurred 2 months prior to admission to the foot and ankle clinic.
The patient consulted her medical oncologist 1 month prior to presenting to our clinic and was noted to exhibit an “excellent response to chemoradiotherapy” and “continues to remain disease free at 2 years.” She was instructed to continue routine follow-up. However, the office notes mentioned no ankle pain and non-healing wounds.
During physical examination, the patient presented an antalgic gait on the right side. Inspection demonstrated an increased circumference of the right ankle compared with the left, with a soft, palpable mass over the medial aspect of her right ankle. A 3 cm × 2 cm, grade 2 abrasion of the skin was observed over the medial mass just posterior to her medial malleolus. Range of motion was within normal limits. The patient exhibited a palpable posterior tibial artery pulse and full strength upon muscle testing of the lower extremities. She featured a positive Tinel’s sign and discomfort over the mass itself, with the pain radiating down to the plantar aspect of her foot and diffuse numbness over the plantar aspect of the foot.
Continue to: Review of her plain radiographs...
Review of her plain radiographs demonstrated no bony abnormalities, fractures, nor visible deformity (Figures 1A, 1B).
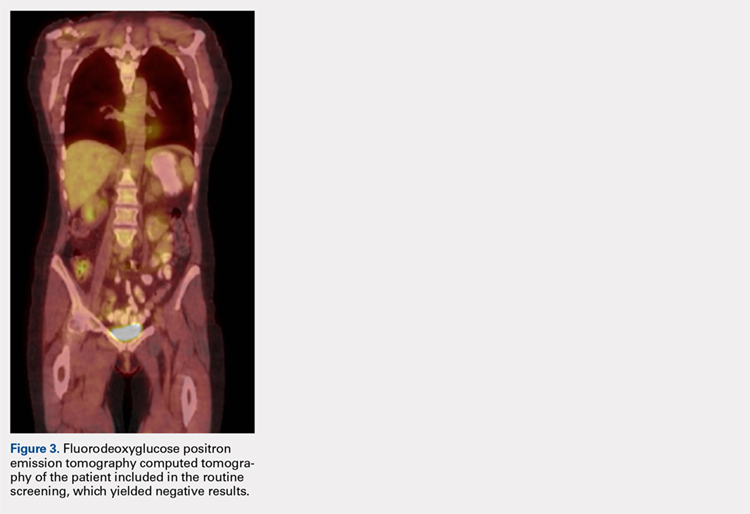
At presentation, our differential diagnosis included recurrence of the malignancy, secondary malignancy, infection, and inflammatory disease. After a lengthy discussion with the patient and consultation with our institution’s musculoskeletal oncologist, the decision was made to perform a right-ankle mass biopsy and marginal excision with wound irrigation and débridement and tarsal tunnel release.
The patient was placed in the supine position with standard prepping and draping. The medial eschar was excised in an elliptical fashion, and a curvilinear, longitudinal approach was performed within the compartment to access the mass along the posteromedial aspect of the ankle. Although no evidence of infection was observed, the tissue was thickened with areas of necrosis down to the flexor retinaculum. Once the flexor retinaculum was opened, a fibrous, plaque-like mass was observed, and it was encased with flexor tendons and neurovascular structures of the tarsal tunnel. After mass excision, a complete tarsal tunnel release was performed until the neurovascular bundle was free. Irrigation and débridement of the ulcer were performed along with complicated wound closure, and the patient was placed in a well-padded postoperative splint.
Pathology was finalized as a recurrent, EBV-positive, and nasal-type ENKTL. The patient underwent bone marrow biopsy, which yielded negative results. CT of the chest, abdomen, and pelvis were negative for the disease. FDG PET-CT, which included the extremities, was performed and demonstrated increased uptake in the right ankle, consistent with the malignancy (Figure 4). 
DISCUSSION
ENKTL is an uncommon form of lymphoma and is exceedingly rare in Caucasian females.1-3 Although the patient’s primary occurrence was in the nasal cavity, recurrence in the foot and ankle must still be described.7,8 To our knowledge, this article is one of the first published cases of a patient who developed a rare-recurrence ENKTL about the foot and ankle. Occurrence in extremities is extremely rare that the staging protocol does not include FDG PET-CT of these areas. The patient’s “negative” scans led many providers to neglect the symptoms in her right ankle until the lesion had ulcerated through the skin. If one would have relied on imaging reports and outside records alone, the diagnosis would have been delayed longer or missed all together. This case illustrates the importance of a thorough medical history and personal review of imaging studies, and how a systematic approach can produce the correct diagnosis for any unknown lesion. Furthermore, this case may prompt oncologists to consider obtaining whole-body FDG PET-CT when evaluating for recurrence in patients.
1. Quintanilla-Martinez L, Kremer M, Keller G, et al. p53 mutations in nasal natural killer/T-cell lymphoma from Mexico: association with large cell morphology and advanced disease. Am J Pathol. 2001;159(6):2095-2105. doi:10.1016/S0002-9440(10)63061-1.
2. Au WY, Ma SY, Chim CS, et al. Clinicopathologic features and treatment outcome of mature T-cell and natural killer-cell lymphomas diagnosed according to the World Health Organization classification scheme: a single center experience of 10 years. Ann Oncol. 2005;16(2):206-214. doi:10.1093/annonc/mdi037.
3. Armitage JO. A clinical evaluation of the International Lymphoma Study Group classification of non-Hodgkin’s lymphoma. Blood. 1997;89(11):3909-3918.
4. Medeiros LJ, Peiper SC, Elwood L, Yano T, Raffeld M, Jaffe ES. Angiocentric immunoproliferative lesions: a molecular analysis of eight cases. Hum Pathol. 1991;22(11):1150-1157. doi:10.1016/0046-8177(91)90269-U.
5. Ho FC, Srivastava G, Loke SL, et al. Presence of Epstein-Barr virus DNA in nasal lymphomas of B and ‘T’ cell type. Hematol Oncol. 1990;8(5):271-281. doi:10.1002/hon.2900080505.
6. Gelb AB, van de Rijn M, Regula DP Jr, et al. Epstein-Barr virus-associated natural killer-large granular lymphocyte leukemia. Hum Pathol. 1994;25(9):953-960. doi:10.1016/0046-8177(94)90018-3.
7. Petrella T, Delfau-Larue MH, Caillot D, et al. Nasopharyngeal lymphomas: further evidence for a natural killer cell origin. Hum Pathol. 1996;27(8):827-833. doi:10.1016/S0046-8177(96)90457-8.
8. Hasserjian RP, Harris NL. NK-cell lymphomas and leukemias: a spectrum of tumors with variable manifestations and immunophenotype. Am J Clin Pathol. 2007;127(6):860-868. doi:10.1309/2F39NX1AL3L54WU8.
9. Robbins KT, Fuller LM, Vlasak M. Primary lymphomas of the nasal cavity and paranasal sinuses. Cancer. 1985;56(4):814-819. doi:10.1002/1097-0142(19850815)56.
10. Ooi GC, Chim CS, Liang R, Tsang KW, Kwong YL. Nasal T-cell/natural killer cell lymphoma: CT and MR imaging features of a new clinicopathologic entity. Am J Roentgenol. 2000;174(4):1141-1145. doi:10.2214/ajr.174.4.1741141.
11. Khong PL, Pang CB, Liang R, Kwong YL, Au WY. Fluorine-18 fluorodeoxyglucose positron emission tomography in mature T-cell and natural killer cell malignancies. Ann Hematol. 2008;87(8):613-621. doi:10.1007/s00277-008-0494-8.
12. Kim SJ, Kim K, Kim BS, et al. Phase II trial of concurrent radiation and weekly cisplatin followed by VIPD chemotherapy in newly diagnosed, stage IE to IIE, nasal, extranodal NK/T-cell lymphoma: consortium for improving survival of lymphoma study. J Clin Oncol. 2009;27(35):6027-6032. doi:10.1200/JCO.2009.23.8592.
13. Kwong YL. Natural killer-cell malignancies: diagnosis and treatment. Leukemia. 2005;19(12):2186-2194. doi:10.1038/sj.leu.2403955.
14. Liang R. Advances in the management and monitoring of extranodal NK/T-cell lymphoma, nasal type. Br J Haematol. 2009;147(1):13-21. doi:10.1111/j.1365-2141.2009.07802.x.
15. Yokoyama H, Yamamoto J, Tohmiya Y, et al. Allogeneic hematopoietic stem cell transplant following chemotherapy containing l-asparaginase as a promising treatment for patients with relapsed or refractory extranodal natural killer/T cell lymphoma, nasal type. Leuk Lymphoma. 2010;51(8):1509-1512. doi:10.3109/10428194.2010.487958.
ABSTRACT
This case report is a rare form of lymphoma recurrence which presented as tarsal tunnel syndrome. The patient had been previously treated for the malignancy and was presumed to be in remission; however, standard radiology imaging protocols failed to include the distal extremities on these scans. The patient presented to the orthopedic clinic with tarsal tunnel symptoms and a mass in the tarsal tunnel. A complete evaluation resulted in a diagnosis of recurrence of the malignancy. This case illustrates the importance of a thorough medical history and personal review of imaging studies, and how a systematic approach can produce the correct diagnosis for any unknown lesion. Furthermore, this case may prompt oncologists to consider obtaining whole-body fluorodeoxyglucose positron emission tomography computed tomography when evaluating for recurrence in patients.
Nasal-type, extranodal natural killer/T-cell lymphoma (ENKTL) is a rare form of non-Hodgkin lymphoma (NHL). Malignancies account for only 10% of NHL in Asian and South American populations. However, in Caucasians, it represents <1% of all cases. In addition, at 3:1 male to female ratio, the disease most commonly affects male patients who are 50 to 59 years old.1-3 The etiology of this malignancy is strongly related to prior infection with Epstein-Barr virus (EBV) as EBV-encoded early small ribonucleic acid on in situ hybridization of lymphoma cells is positive in 95% of cases.4-6
Typical sites of involvement include the nasal cavity, nasopharynx, and sinuses, causing patients to present with nasal obstruction, chronic sinusitis, or epistaxis. Additionally, ENKTL can occur primarily in the skin, gastrointestinal tract, spleen, and testis, whereas the bone marrow may be involved in 10% of cases. Although rare, unusual sites, including muscle, adrenals, and ovaries, have been published.7,8
Staging is best performed using the T-staging system, which accounts for the extent of local tumor involvement. Higher stages, such as T3 /T4, equate to locally advanced disease and imply a worse prognosis.9,10 Computed tomography (CT) and magnetic resonance imaging (MRI) help define local soft tissues and bony involvement. Furthermore, CT of the chest, abdomen, and pelvis as well as bone marrow biopsy are performed as part of the staging process. Lastly, fluorine-18 fluorodeoxyglucose positron emission tomography CT (18-FDG PET-CT) is often used to detect extranodal spread, define the extent of involvement, differentiate between lymphoma and inflammatory masses, and monitor for recurrence.11
Treatment for local ENKTL involves concurrent chemoradiotherapy followed by 3 cycles of etoposide, ifosfamide, cisplatin, and dexamethasone, which results in a complete response rate of 80%, and is the most favorable when comparing treatment modalities.12 Unfortunately, recurrence rates reach as high as 50%, whereas the 5-year survival rate is 59%.13,14 For recurrent or disseminated disease, high-dose chemotherapy and hematopoietic stem cell transplantation remain as alternative treatments for patients who have undergone 2 complete remissions and can be curative in some instances.13,15
Continue to: In summary, ENKTL is a rare form...
In summary, ENKTL is a rare form of NHL which classically presents in the nasal cavity; however, this type of lymphoma may present in a variety of extranodal sites.7,8 Despite the numerous published reports on ENKTL, no study has reported either primary or recurrent ENKTL in the feet or hands. To our knowledge, this is one of the first published cases of a patient who developed a rare and recurring ENKTL in the foot and ankle. The patient provided written informed consent for print and electronic publication of this case report.
CASE
A 59-year-old Caucasian woman was referred to the orthopedic foot and ankle clinic by her primary care physician for right medial ankle pain, skin ulceration, and numbness over the plantar aspect of her right foot. Upon questioning, the patient noted that the pain and numbness were present for almost 6 months. She denied trauma to the concerned area. Previously, the patient was observed and treated elsewhere for plantar fasciitis and was prescribed a brace before being immobilized in a controlled ankle motion (CAM) boot for 6 weeks. At follow-up with her outside provider, the patient had developed skin breakdown over the medial aspect of the right ankle, and this condition was presumed to be caused by the boot. After local wound care failed to improve her skin ulceration, she returned to her primary care physician, who ordered an MRI of the area and referred her to our specialty clinic.
Upon review, the patient’s past medical history included a diagnosis of nasal-type ENKTL. Her malignancy was treated with chemoradiotherapy 2 years prior to her consultation with the foot and ankle clinic.
The patient was noted by her medical oncologist and interventional radiologist to be in complete stage 4 remission since being treated. She underwent routine MRI and CT scans of the head and neck at 6-month intervals and FDG PET-CT scans at 3-month intervals, as per institutional protocol. The examinations showed no evidence of malignancy or metabolically active disease. The last imaging study occurred 2 months prior to admission to the foot and ankle clinic.
The patient consulted her medical oncologist 1 month prior to presenting to our clinic and was noted to exhibit an “excellent response to chemoradiotherapy” and “continues to remain disease free at 2 years.” She was instructed to continue routine follow-up. However, the office notes mentioned no ankle pain and non-healing wounds.
During physical examination, the patient presented an antalgic gait on the right side. Inspection demonstrated an increased circumference of the right ankle compared with the left, with a soft, palpable mass over the medial aspect of her right ankle. A 3 cm × 2 cm, grade 2 abrasion of the skin was observed over the medial mass just posterior to her medial malleolus. Range of motion was within normal limits. The patient exhibited a palpable posterior tibial artery pulse and full strength upon muscle testing of the lower extremities. She featured a positive Tinel’s sign and discomfort over the mass itself, with the pain radiating down to the plantar aspect of her foot and diffuse numbness over the plantar aspect of the foot.
Continue to: Review of her plain radiographs...
Review of her plain radiographs demonstrated no bony abnormalities, fractures, nor visible deformity (Figures 1A, 1B).
At presentation, our differential diagnosis included recurrence of the malignancy, secondary malignancy, infection, and inflammatory disease. After a lengthy discussion with the patient and consultation with our institution’s musculoskeletal oncologist, the decision was made to perform a right-ankle mass biopsy and marginal excision with wound irrigation and débridement and tarsal tunnel release.
The patient was placed in the supine position with standard prepping and draping. The medial eschar was excised in an elliptical fashion, and a curvilinear, longitudinal approach was performed within the compartment to access the mass along the posteromedial aspect of the ankle. Although no evidence of infection was observed, the tissue was thickened with areas of necrosis down to the flexor retinaculum. Once the flexor retinaculum was opened, a fibrous, plaque-like mass was observed, and it was encased with flexor tendons and neurovascular structures of the tarsal tunnel. After mass excision, a complete tarsal tunnel release was performed until the neurovascular bundle was free. Irrigation and débridement of the ulcer were performed along with complicated wound closure, and the patient was placed in a well-padded postoperative splint.
Pathology was finalized as a recurrent, EBV-positive, and nasal-type ENKTL. The patient underwent bone marrow biopsy, which yielded negative results. CT of the chest, abdomen, and pelvis were negative for the disease. FDG PET-CT, which included the extremities, was performed and demonstrated increased uptake in the right ankle, consistent with the malignancy (Figure 4).
DISCUSSION
ENKTL is an uncommon form of lymphoma and is exceedingly rare in Caucasian females.1-3 Although the patient’s primary occurrence was in the nasal cavity, recurrence in the foot and ankle must still be described.7,8 To our knowledge, this article is one of the first published cases of a patient who developed a rare-recurrence ENKTL about the foot and ankle. Occurrence in extremities is extremely rare that the staging protocol does not include FDG PET-CT of these areas. The patient’s “negative” scans led many providers to neglect the symptoms in her right ankle until the lesion had ulcerated through the skin. If one would have relied on imaging reports and outside records alone, the diagnosis would have been delayed longer or missed all together. This case illustrates the importance of a thorough medical history and personal review of imaging studies, and how a systematic approach can produce the correct diagnosis for any unknown lesion. Furthermore, this case may prompt oncologists to consider obtaining whole-body FDG PET-CT when evaluating for recurrence in patients.
ABSTRACT
This case report is a rare form of lymphoma recurrence which presented as tarsal tunnel syndrome. The patient had been previously treated for the malignancy and was presumed to be in remission; however, standard radiology imaging protocols failed to include the distal extremities on these scans. The patient presented to the orthopedic clinic with tarsal tunnel symptoms and a mass in the tarsal tunnel. A complete evaluation resulted in a diagnosis of recurrence of the malignancy. This case illustrates the importance of a thorough medical history and personal review of imaging studies, and how a systematic approach can produce the correct diagnosis for any unknown lesion. Furthermore, this case may prompt oncologists to consider obtaining whole-body fluorodeoxyglucose positron emission tomography computed tomography when evaluating for recurrence in patients.
Nasal-type, extranodal natural killer/T-cell lymphoma (ENKTL) is a rare form of non-Hodgkin lymphoma (NHL). Malignancies account for only 10% of NHL in Asian and South American populations. However, in Caucasians, it represents <1% of all cases. In addition, at 3:1 male to female ratio, the disease most commonly affects male patients who are 50 to 59 years old.1-3 The etiology of this malignancy is strongly related to prior infection with Epstein-Barr virus (EBV) as EBV-encoded early small ribonucleic acid on in situ hybridization of lymphoma cells is positive in 95% of cases.4-6
Typical sites of involvement include the nasal cavity, nasopharynx, and sinuses, causing patients to present with nasal obstruction, chronic sinusitis, or epistaxis. Additionally, ENKTL can occur primarily in the skin, gastrointestinal tract, spleen, and testis, whereas the bone marrow may be involved in 10% of cases. Although rare, unusual sites, including muscle, adrenals, and ovaries, have been published.7,8
Staging is best performed using the T-staging system, which accounts for the extent of local tumor involvement. Higher stages, such as T3 /T4, equate to locally advanced disease and imply a worse prognosis.9,10 Computed tomography (CT) and magnetic resonance imaging (MRI) help define local soft tissues and bony involvement. Furthermore, CT of the chest, abdomen, and pelvis as well as bone marrow biopsy are performed as part of the staging process. Lastly, fluorine-18 fluorodeoxyglucose positron emission tomography CT (18-FDG PET-CT) is often used to detect extranodal spread, define the extent of involvement, differentiate between lymphoma and inflammatory masses, and monitor for recurrence.11
Treatment for local ENKTL involves concurrent chemoradiotherapy followed by 3 cycles of etoposide, ifosfamide, cisplatin, and dexamethasone, which results in a complete response rate of 80%, and is the most favorable when comparing treatment modalities.12 Unfortunately, recurrence rates reach as high as 50%, whereas the 5-year survival rate is 59%.13,14 For recurrent or disseminated disease, high-dose chemotherapy and hematopoietic stem cell transplantation remain as alternative treatments for patients who have undergone 2 complete remissions and can be curative in some instances.13,15
Continue to: In summary, ENKTL is a rare form...
In summary, ENKTL is a rare form of NHL which classically presents in the nasal cavity; however, this type of lymphoma may present in a variety of extranodal sites.7,8 Despite the numerous published reports on ENKTL, no study has reported either primary or recurrent ENKTL in the feet or hands. To our knowledge, this is one of the first published cases of a patient who developed a rare and recurring ENKTL in the foot and ankle. The patient provided written informed consent for print and electronic publication of this case report.
CASE
A 59-year-old Caucasian woman was referred to the orthopedic foot and ankle clinic by her primary care physician for right medial ankle pain, skin ulceration, and numbness over the plantar aspect of her right foot. Upon questioning, the patient noted that the pain and numbness were present for almost 6 months. She denied trauma to the concerned area. Previously, the patient was observed and treated elsewhere for plantar fasciitis and was prescribed a brace before being immobilized in a controlled ankle motion (CAM) boot for 6 weeks. At follow-up with her outside provider, the patient had developed skin breakdown over the medial aspect of the right ankle, and this condition was presumed to be caused by the boot. After local wound care failed to improve her skin ulceration, she returned to her primary care physician, who ordered an MRI of the area and referred her to our specialty clinic.
Upon review, the patient’s past medical history included a diagnosis of nasal-type ENKTL. Her malignancy was treated with chemoradiotherapy 2 years prior to her consultation with the foot and ankle clinic.
The patient was noted by her medical oncologist and interventional radiologist to be in complete stage 4 remission since being treated. She underwent routine MRI and CT scans of the head and neck at 6-month intervals and FDG PET-CT scans at 3-month intervals, as per institutional protocol. The examinations showed no evidence of malignancy or metabolically active disease. The last imaging study occurred 2 months prior to admission to the foot and ankle clinic.
The patient consulted her medical oncologist 1 month prior to presenting to our clinic and was noted to exhibit an “excellent response to chemoradiotherapy” and “continues to remain disease free at 2 years.” She was instructed to continue routine follow-up. However, the office notes mentioned no ankle pain and non-healing wounds.
During physical examination, the patient presented an antalgic gait on the right side. Inspection demonstrated an increased circumference of the right ankle compared with the left, with a soft, palpable mass over the medial aspect of her right ankle. A 3 cm × 2 cm, grade 2 abrasion of the skin was observed over the medial mass just posterior to her medial malleolus. Range of motion was within normal limits. The patient exhibited a palpable posterior tibial artery pulse and full strength upon muscle testing of the lower extremities. She featured a positive Tinel’s sign and discomfort over the mass itself, with the pain radiating down to the plantar aspect of her foot and diffuse numbness over the plantar aspect of the foot.
Continue to: Review of her plain radiographs...
Review of her plain radiographs demonstrated no bony abnormalities, fractures, nor visible deformity (Figures 1A, 1B).
At presentation, our differential diagnosis included recurrence of the malignancy, secondary malignancy, infection, and inflammatory disease. After a lengthy discussion with the patient and consultation with our institution’s musculoskeletal oncologist, the decision was made to perform a right-ankle mass biopsy and marginal excision with wound irrigation and débridement and tarsal tunnel release.
The patient was placed in the supine position with standard prepping and draping. The medial eschar was excised in an elliptical fashion, and a curvilinear, longitudinal approach was performed within the compartment to access the mass along the posteromedial aspect of the ankle. Although no evidence of infection was observed, the tissue was thickened with areas of necrosis down to the flexor retinaculum. Once the flexor retinaculum was opened, a fibrous, plaque-like mass was observed, and it was encased with flexor tendons and neurovascular structures of the tarsal tunnel. After mass excision, a complete tarsal tunnel release was performed until the neurovascular bundle was free. Irrigation and débridement of the ulcer were performed along with complicated wound closure, and the patient was placed in a well-padded postoperative splint.
Pathology was finalized as a recurrent, EBV-positive, and nasal-type ENKTL. The patient underwent bone marrow biopsy, which yielded negative results. CT of the chest, abdomen, and pelvis were negative for the disease. FDG PET-CT, which included the extremities, was performed and demonstrated increased uptake in the right ankle, consistent with the malignancy (Figure 4).
DISCUSSION
ENKTL is an uncommon form of lymphoma and is exceedingly rare in Caucasian females.1-3 Although the patient’s primary occurrence was in the nasal cavity, recurrence in the foot and ankle must still be described.7,8 To our knowledge, this article is one of the first published cases of a patient who developed a rare-recurrence ENKTL about the foot and ankle. Occurrence in extremities is extremely rare that the staging protocol does not include FDG PET-CT of these areas. The patient’s “negative” scans led many providers to neglect the symptoms in her right ankle until the lesion had ulcerated through the skin. If one would have relied on imaging reports and outside records alone, the diagnosis would have been delayed longer or missed all together. This case illustrates the importance of a thorough medical history and personal review of imaging studies, and how a systematic approach can produce the correct diagnosis for any unknown lesion. Furthermore, this case may prompt oncologists to consider obtaining whole-body FDG PET-CT when evaluating for recurrence in patients.
1. Quintanilla-Martinez L, Kremer M, Keller G, et al. p53 mutations in nasal natural killer/T-cell lymphoma from Mexico: association with large cell morphology and advanced disease. Am J Pathol. 2001;159(6):2095-2105. doi:10.1016/S0002-9440(10)63061-1.
2. Au WY, Ma SY, Chim CS, et al. Clinicopathologic features and treatment outcome of mature T-cell and natural killer-cell lymphomas diagnosed according to the World Health Organization classification scheme: a single center experience of 10 years. Ann Oncol. 2005;16(2):206-214. doi:10.1093/annonc/mdi037.
3. Armitage JO. A clinical evaluation of the International Lymphoma Study Group classification of non-Hodgkin’s lymphoma. Blood. 1997;89(11):3909-3918.
4. Medeiros LJ, Peiper SC, Elwood L, Yano T, Raffeld M, Jaffe ES. Angiocentric immunoproliferative lesions: a molecular analysis of eight cases. Hum Pathol. 1991;22(11):1150-1157. doi:10.1016/0046-8177(91)90269-U.
5. Ho FC, Srivastava G, Loke SL, et al. Presence of Epstein-Barr virus DNA in nasal lymphomas of B and ‘T’ cell type. Hematol Oncol. 1990;8(5):271-281. doi:10.1002/hon.2900080505.
6. Gelb AB, van de Rijn M, Regula DP Jr, et al. Epstein-Barr virus-associated natural killer-large granular lymphocyte leukemia. Hum Pathol. 1994;25(9):953-960. doi:10.1016/0046-8177(94)90018-3.
7. Petrella T, Delfau-Larue MH, Caillot D, et al. Nasopharyngeal lymphomas: further evidence for a natural killer cell origin. Hum Pathol. 1996;27(8):827-833. doi:10.1016/S0046-8177(96)90457-8.
8. Hasserjian RP, Harris NL. NK-cell lymphomas and leukemias: a spectrum of tumors with variable manifestations and immunophenotype. Am J Clin Pathol. 2007;127(6):860-868. doi:10.1309/2F39NX1AL3L54WU8.
9. Robbins KT, Fuller LM, Vlasak M. Primary lymphomas of the nasal cavity and paranasal sinuses. Cancer. 1985;56(4):814-819. doi:10.1002/1097-0142(19850815)56.
10. Ooi GC, Chim CS, Liang R, Tsang KW, Kwong YL. Nasal T-cell/natural killer cell lymphoma: CT and MR imaging features of a new clinicopathologic entity. Am J Roentgenol. 2000;174(4):1141-1145. doi:10.2214/ajr.174.4.1741141.
11. Khong PL, Pang CB, Liang R, Kwong YL, Au WY. Fluorine-18 fluorodeoxyglucose positron emission tomography in mature T-cell and natural killer cell malignancies. Ann Hematol. 2008;87(8):613-621. doi:10.1007/s00277-008-0494-8.
12. Kim SJ, Kim K, Kim BS, et al. Phase II trial of concurrent radiation and weekly cisplatin followed by VIPD chemotherapy in newly diagnosed, stage IE to IIE, nasal, extranodal NK/T-cell lymphoma: consortium for improving survival of lymphoma study. J Clin Oncol. 2009;27(35):6027-6032. doi:10.1200/JCO.2009.23.8592.
13. Kwong YL. Natural killer-cell malignancies: diagnosis and treatment. Leukemia. 2005;19(12):2186-2194. doi:10.1038/sj.leu.2403955.
14. Liang R. Advances in the management and monitoring of extranodal NK/T-cell lymphoma, nasal type. Br J Haematol. 2009;147(1):13-21. doi:10.1111/j.1365-2141.2009.07802.x.
15. Yokoyama H, Yamamoto J, Tohmiya Y, et al. Allogeneic hematopoietic stem cell transplant following chemotherapy containing l-asparaginase as a promising treatment for patients with relapsed or refractory extranodal natural killer/T cell lymphoma, nasal type. Leuk Lymphoma. 2010;51(8):1509-1512. doi:10.3109/10428194.2010.487958.
1. Quintanilla-Martinez L, Kremer M, Keller G, et al. p53 mutations in nasal natural killer/T-cell lymphoma from Mexico: association with large cell morphology and advanced disease. Am J Pathol. 2001;159(6):2095-2105. doi:10.1016/S0002-9440(10)63061-1.
2. Au WY, Ma SY, Chim CS, et al. Clinicopathologic features and treatment outcome of mature T-cell and natural killer-cell lymphomas diagnosed according to the World Health Organization classification scheme: a single center experience of 10 years. Ann Oncol. 2005;16(2):206-214. doi:10.1093/annonc/mdi037.
3. Armitage JO. A clinical evaluation of the International Lymphoma Study Group classification of non-Hodgkin’s lymphoma. Blood. 1997;89(11):3909-3918.
4. Medeiros LJ, Peiper SC, Elwood L, Yano T, Raffeld M, Jaffe ES. Angiocentric immunoproliferative lesions: a molecular analysis of eight cases. Hum Pathol. 1991;22(11):1150-1157. doi:10.1016/0046-8177(91)90269-U.
5. Ho FC, Srivastava G, Loke SL, et al. Presence of Epstein-Barr virus DNA in nasal lymphomas of B and ‘T’ cell type. Hematol Oncol. 1990;8(5):271-281. doi:10.1002/hon.2900080505.
6. Gelb AB, van de Rijn M, Regula DP Jr, et al. Epstein-Barr virus-associated natural killer-large granular lymphocyte leukemia. Hum Pathol. 1994;25(9):953-960. doi:10.1016/0046-8177(94)90018-3.
7. Petrella T, Delfau-Larue MH, Caillot D, et al. Nasopharyngeal lymphomas: further evidence for a natural killer cell origin. Hum Pathol. 1996;27(8):827-833. doi:10.1016/S0046-8177(96)90457-8.
8. Hasserjian RP, Harris NL. NK-cell lymphomas and leukemias: a spectrum of tumors with variable manifestations and immunophenotype. Am J Clin Pathol. 2007;127(6):860-868. doi:10.1309/2F39NX1AL3L54WU8.
9. Robbins KT, Fuller LM, Vlasak M. Primary lymphomas of the nasal cavity and paranasal sinuses. Cancer. 1985;56(4):814-819. doi:10.1002/1097-0142(19850815)56.
10. Ooi GC, Chim CS, Liang R, Tsang KW, Kwong YL. Nasal T-cell/natural killer cell lymphoma: CT and MR imaging features of a new clinicopathologic entity. Am J Roentgenol. 2000;174(4):1141-1145. doi:10.2214/ajr.174.4.1741141.
11. Khong PL, Pang CB, Liang R, Kwong YL, Au WY. Fluorine-18 fluorodeoxyglucose positron emission tomography in mature T-cell and natural killer cell malignancies. Ann Hematol. 2008;87(8):613-621. doi:10.1007/s00277-008-0494-8.
12. Kim SJ, Kim K, Kim BS, et al. Phase II trial of concurrent radiation and weekly cisplatin followed by VIPD chemotherapy in newly diagnosed, stage IE to IIE, nasal, extranodal NK/T-cell lymphoma: consortium for improving survival of lymphoma study. J Clin Oncol. 2009;27(35):6027-6032. doi:10.1200/JCO.2009.23.8592.
13. Kwong YL. Natural killer-cell malignancies: diagnosis and treatment. Leukemia. 2005;19(12):2186-2194. doi:10.1038/sj.leu.2403955.
14. Liang R. Advances in the management and monitoring of extranodal NK/T-cell lymphoma, nasal type. Br J Haematol. 2009;147(1):13-21. doi:10.1111/j.1365-2141.2009.07802.x.
15. Yokoyama H, Yamamoto J, Tohmiya Y, et al. Allogeneic hematopoietic stem cell transplant following chemotherapy containing l-asparaginase as a promising treatment for patients with relapsed or refractory extranodal natural killer/T cell lymphoma, nasal type. Leuk Lymphoma. 2010;51(8):1509-1512. doi:10.3109/10428194.2010.487958.
TAKE-HOME POINTS
- A thorough review of systems, physical examination, and personal review of a patient’s advanced imaging is critical to avoid missed diagnosis or delays in diagnosis.
- Any mass lesion encountered in clinical practice, no matter how benign appearing, should be presumed malignant until proven otherwise.
- Fluorine-18 fluorodeoxyglucose positron emission tomography CT (18-FDG PET-CT) should include whole-body scans when evaluating patients for recurrence of malignancy.
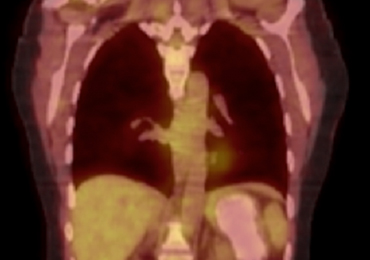
Gone but Not Forgotten: Acute Appendicitis Postappendectomy
Acute appendicitis is a common condition emergency physicians (EPs) encounter in the ED, and it is also one of the most common general surgeries.1Although stump appendicitis is a rare, long-term complication of appendectomy, it should always be included in the differential diagnosis of patients presenting with right-sided abdominal pain and a history of appendectomy. Delays in diagnosing stump appendicitis can lead to perforation, gangrene, and sepsis.2
Case
A 33-year-old previously healthy man, whose medical history was significant for an appendectomy 6 months earlier, presented to the ED with progressive and worsening right lower quadrant abdominal pain that radiated to his right testicle. The patient stated that the pain started 3 days prior while he was lifting a bale of hay. He further noted having a fever of 102oF, nausea, and vomiting hours prior to his arrival at the ED.
Upon presentation, the patient’s vital signs were: heart rate, 89 beats/min; respiratory rate, 17 breaths/min; blood pressure, 132/84 mm Hg; and temperature, 98.9°F. Oxygen saturation was 98% on room air. Physical examination revealed exquisite tenderness in the right lower quadrant and suprapubic region. The testicular examination and the remainder of the physical examination were normal. Laboratory evaluation included a complete blood count and urinalysis, the results of which were significant for an elevated white blood cell count of 17 x 109/Lmicroscopic hematuria, trace leukocyte esterase, and ketones.

A computed tomography (CT) scan of the abdomen and pelvis with intravenous (IV) and oral contrast demonstrated a phlegmonous process surrounding the surgical site, which was concerning for stump appendicitis. The terminal ilium and colon were noted to be normal (Figures 1 and 2).

The patient was started on IV fluids and IV antibiotics, and received Zosyn in the ED. Surgical service was consulted, and the patient was admitted to the hospital where he continued nonoperative treatment with IV ciprofloxacin and metronidazole. The patient was discharged home on hospital day 3 without further complication. A repeat CT scan was taken of the abdomen and pelvis 3 weeks after discharge, and demonstrated complete resolution of the inflammatory process at the appendiceal stump with chronic scarring.
Discussion
Approximately 7% of patients who present to the ED with abdominal pain are diagnosed with appendicitis.3 Although appendectomy is one of the most common surgical procedures, stump appendicitis is a rare postsurgical complication, with a reported incidence of 1 in 50,000 cases.4,5
Stump appendicitis is an acute inflammation of the residual appendicular stump; the incidence of stump perforation is approximately 60% to 70%.4,6 Thus, stump appendicitis has a high morbidity and complication rate. Unfortunately, though stump appendicitis is a condition in which timely diagnosis and intervention are essential to prevent morbidity, due to its rarity and low occurrence, there is often a delay in diagnosis. It is therefore important that EPs include stump appendicitis in the differential diagnosis of patients presenting with right-sided abdominal pain and a history of appendectomy.
Stump appendicitis was initially described by Rose et al in 1945.2 This condition is underreported, and the exact causes are still unclear.Of the reported cases of stump appendicitis, approximately 66% developed following an open surgical appendectomy;5 therefore, complicated surgery or difficult dissection of the appendix is considered a risk factor for stump appendicitis. Conversely, adequate visualization of the appendiceal base during appendectomy and a stump measuring less than 3 to 5 mm1,4 are associated with a lower risk for stump appendicitis.
Stump appendicitis can develop as early as a few days postappendectomy or as late as 50 years postappendectomy. Patients with stump appendicitis present with signs and symptoms similar to that of acute appendicitis.2,4,7 Diagnosis can be made through ultrasound or CT studies, though CT is the preferred modality due to its higher specificity and ability to exclude other causes of right-sided abdominal pain.4
Management
Surgical intervention to remove the appendiceal stump is typically the preferred treatment. However, as with our patient, cases of successful and uncomplicated medical management have been reported.1,2,4
Conclusion
While stump appendicitis is rare, there has been a rise in the number of reported cases over the past few years due to the increasing use and availability of CT.4 The diagnosis of stump appendicitis is time-critical to prevent associated complications of stump perforation, gangrene, and sepsis. It is therefore imperative that EPs consider this
1. Shah T, Gupta RK, Karkee RJ, Agarwal CS. Recurrent pain abdomen following appendectomy: stump appendicitis, a surgeon’s dilemma. Clin Case Rep. 2017;5(3):215-217. doi:10.1002/ccr3.781.
2. Giwa A, Reyes M. Three times a charm…a case of repeat appendicitis status post two prior appendectomies. Am J Emerg Med. 2018;36(3):528.e1-528.e2. doi:10.1016/j.ajem.2017.12.024.
3. Addiss DG, Shaffer N, Fowler BS, Tauxe RV. The epidemiology of appendicitis and appendectomy in the United States. Am J Epidemiol. 1990;132(5):910-925.
4. Hendahewa R, Shekhar A, Ratnayake S. The dilemma of stump appendicitis—a case report and literature review. Int J Surg. Case Rep. 2015;14:101-103. doi:10.1016/j.ijscr.2015.07.017.
5. Liang MK, Lo HG, Marks JL. Stump appendicitis: a comprehensive review of literature. Am Surg. 2006;72(2):162-166.
6. Parthsarathi R, Jankar SV, Chittawadgi B, et al. Laraposcopic management of symptomatic residual appendicular tip: a rare case report. J Minim Access Surg. 2017;13(2):154-156. doi:10.4103/0972-9941.199610.
7. Kanona H, Al Samaraee A, Nice C, Bhattacharya V. Stump appendicitis: a review. Int J Surg. 2012;10(9):425-428. doi:10.1016/j.ijsu.2012.07.007.
Acute appendicitis is a common condition emergency physicians (EPs) encounter in the ED, and it is also one of the most common general surgeries.1Although stump appendicitis is a rare, long-term complication of appendectomy, it should always be included in the differential diagnosis of patients presenting with right-sided abdominal pain and a history of appendectomy. Delays in diagnosing stump appendicitis can lead to perforation, gangrene, and sepsis.2
Case
A 33-year-old previously healthy man, whose medical history was significant for an appendectomy 6 months earlier, presented to the ED with progressive and worsening right lower quadrant abdominal pain that radiated to his right testicle. The patient stated that the pain started 3 days prior while he was lifting a bale of hay. He further noted having a fever of 102oF, nausea, and vomiting hours prior to his arrival at the ED.
Upon presentation, the patient’s vital signs were: heart rate, 89 beats/min; respiratory rate, 17 breaths/min; blood pressure, 132/84 mm Hg; and temperature, 98.9°F. Oxygen saturation was 98% on room air. Physical examination revealed exquisite tenderness in the right lower quadrant and suprapubic region. The testicular examination and the remainder of the physical examination were normal. Laboratory evaluation included a complete blood count and urinalysis, the results of which were significant for an elevated white blood cell count of 17 x 109/Lmicroscopic hematuria, trace leukocyte esterase, and ketones.
A computed tomography (CT) scan of the abdomen and pelvis with intravenous (IV) and oral contrast demonstrated a phlegmonous process surrounding the surgical site, which was concerning for stump appendicitis. The terminal ilium and colon were noted to be normal (Figures 1 and 2).
The patient was started on IV fluids and IV antibiotics, and received Zosyn in the ED. Surgical service was consulted, and the patient was admitted to the hospital where he continued nonoperative treatment with IV ciprofloxacin and metronidazole. The patient was discharged home on hospital day 3 without further complication. A repeat CT scan was taken of the abdomen and pelvis 3 weeks after discharge, and demonstrated complete resolution of the inflammatory process at the appendiceal stump with chronic scarring.
Discussion
Approximately 7% of patients who present to the ED with abdominal pain are diagnosed with appendicitis.3 Although appendectomy is one of the most common surgical procedures, stump appendicitis is a rare postsurgical complication, with a reported incidence of 1 in 50,000 cases.4,5
Stump appendicitis is an acute inflammation of the residual appendicular stump; the incidence of stump perforation is approximately 60% to 70%.4,6 Thus, stump appendicitis has a high morbidity and complication rate. Unfortunately, though stump appendicitis is a condition in which timely diagnosis and intervention are essential to prevent morbidity, due to its rarity and low occurrence, there is often a delay in diagnosis. It is therefore important that EPs include stump appendicitis in the differential diagnosis of patients presenting with right-sided abdominal pain and a history of appendectomy.
Stump appendicitis was initially described by Rose et al in 1945.2 This condition is underreported, and the exact causes are still unclear.Of the reported cases of stump appendicitis, approximately 66% developed following an open surgical appendectomy;5 therefore, complicated surgery or difficult dissection of the appendix is considered a risk factor for stump appendicitis. Conversely, adequate visualization of the appendiceal base during appendectomy and a stump measuring less than 3 to 5 mm1,4 are associated with a lower risk for stump appendicitis.
Stump appendicitis can develop as early as a few days postappendectomy or as late as 50 years postappendectomy. Patients with stump appendicitis present with signs and symptoms similar to that of acute appendicitis.2,4,7 Diagnosis can be made through ultrasound or CT studies, though CT is the preferred modality due to its higher specificity and ability to exclude other causes of right-sided abdominal pain.4
Management
Surgical intervention to remove the appendiceal stump is typically the preferred treatment. However, as with our patient, cases of successful and uncomplicated medical management have been reported.1,2,4
Conclusion
While stump appendicitis is rare, there has been a rise in the number of reported cases over the past few years due to the increasing use and availability of CT.4 The diagnosis of stump appendicitis is time-critical to prevent associated complications of stump perforation, gangrene, and sepsis. It is therefore imperative that EPs consider this
Acute appendicitis is a common condition emergency physicians (EPs) encounter in the ED, and it is also one of the most common general surgeries.1Although stump appendicitis is a rare, long-term complication of appendectomy, it should always be included in the differential diagnosis of patients presenting with right-sided abdominal pain and a history of appendectomy. Delays in diagnosing stump appendicitis can lead to perforation, gangrene, and sepsis.2
Case
A 33-year-old previously healthy man, whose medical history was significant for an appendectomy 6 months earlier, presented to the ED with progressive and worsening right lower quadrant abdominal pain that radiated to his right testicle. The patient stated that the pain started 3 days prior while he was lifting a bale of hay. He further noted having a fever of 102oF, nausea, and vomiting hours prior to his arrival at the ED.
Upon presentation, the patient’s vital signs were: heart rate, 89 beats/min; respiratory rate, 17 breaths/min; blood pressure, 132/84 mm Hg; and temperature, 98.9°F. Oxygen saturation was 98% on room air. Physical examination revealed exquisite tenderness in the right lower quadrant and suprapubic region. The testicular examination and the remainder of the physical examination were normal. Laboratory evaluation included a complete blood count and urinalysis, the results of which were significant for an elevated white blood cell count of 17 x 109/Lmicroscopic hematuria, trace leukocyte esterase, and ketones.
A computed tomography (CT) scan of the abdomen and pelvis with intravenous (IV) and oral contrast demonstrated a phlegmonous process surrounding the surgical site, which was concerning for stump appendicitis. The terminal ilium and colon were noted to be normal (Figures 1 and 2).
The patient was started on IV fluids and IV antibiotics, and received Zosyn in the ED. Surgical service was consulted, and the patient was admitted to the hospital where he continued nonoperative treatment with IV ciprofloxacin and metronidazole. The patient was discharged home on hospital day 3 without further complication. A repeat CT scan was taken of the abdomen and pelvis 3 weeks after discharge, and demonstrated complete resolution of the inflammatory process at the appendiceal stump with chronic scarring.
Discussion
Approximately 7% of patients who present to the ED with abdominal pain are diagnosed with appendicitis.3 Although appendectomy is one of the most common surgical procedures, stump appendicitis is a rare postsurgical complication, with a reported incidence of 1 in 50,000 cases.4,5
Stump appendicitis is an acute inflammation of the residual appendicular stump; the incidence of stump perforation is approximately 60% to 70%.4,6 Thus, stump appendicitis has a high morbidity and complication rate. Unfortunately, though stump appendicitis is a condition in which timely diagnosis and intervention are essential to prevent morbidity, due to its rarity and low occurrence, there is often a delay in diagnosis. It is therefore important that EPs include stump appendicitis in the differential diagnosis of patients presenting with right-sided abdominal pain and a history of appendectomy.
Stump appendicitis was initially described by Rose et al in 1945.2 This condition is underreported, and the exact causes are still unclear.Of the reported cases of stump appendicitis, approximately 66% developed following an open surgical appendectomy;5 therefore, complicated surgery or difficult dissection of the appendix is considered a risk factor for stump appendicitis. Conversely, adequate visualization of the appendiceal base during appendectomy and a stump measuring less than 3 to 5 mm1,4 are associated with a lower risk for stump appendicitis.
Stump appendicitis can develop as early as a few days postappendectomy or as late as 50 years postappendectomy. Patients with stump appendicitis present with signs and symptoms similar to that of acute appendicitis.2,4,7 Diagnosis can be made through ultrasound or CT studies, though CT is the preferred modality due to its higher specificity and ability to exclude other causes of right-sided abdominal pain.4
Management
Surgical intervention to remove the appendiceal stump is typically the preferred treatment. However, as with our patient, cases of successful and uncomplicated medical management have been reported.1,2,4
Conclusion
While stump appendicitis is rare, there has been a rise in the number of reported cases over the past few years due to the increasing use and availability of CT.4 The diagnosis of stump appendicitis is time-critical to prevent associated complications of stump perforation, gangrene, and sepsis. It is therefore imperative that EPs consider this
1. Shah T, Gupta RK, Karkee RJ, Agarwal CS. Recurrent pain abdomen following appendectomy: stump appendicitis, a surgeon’s dilemma. Clin Case Rep. 2017;5(3):215-217. doi:10.1002/ccr3.781.
2. Giwa A, Reyes M. Three times a charm…a case of repeat appendicitis status post two prior appendectomies. Am J Emerg Med. 2018;36(3):528.e1-528.e2. doi:10.1016/j.ajem.2017.12.024.
3. Addiss DG, Shaffer N, Fowler BS, Tauxe RV. The epidemiology of appendicitis and appendectomy in the United States. Am J Epidemiol. 1990;132(5):910-925.
4. Hendahewa R, Shekhar A, Ratnayake S. The dilemma of stump appendicitis—a case report and literature review. Int J Surg. Case Rep. 2015;14:101-103. doi:10.1016/j.ijscr.2015.07.017.
5. Liang MK, Lo HG, Marks JL. Stump appendicitis: a comprehensive review of literature. Am Surg. 2006;72(2):162-166.
6. Parthsarathi R, Jankar SV, Chittawadgi B, et al. Laraposcopic management of symptomatic residual appendicular tip: a rare case report. J Minim Access Surg. 2017;13(2):154-156. doi:10.4103/0972-9941.199610.
7. Kanona H, Al Samaraee A, Nice C, Bhattacharya V. Stump appendicitis: a review. Int J Surg. 2012;10(9):425-428. doi:10.1016/j.ijsu.2012.07.007.
1. Shah T, Gupta RK, Karkee RJ, Agarwal CS. Recurrent pain abdomen following appendectomy: stump appendicitis, a surgeon’s dilemma. Clin Case Rep. 2017;5(3):215-217. doi:10.1002/ccr3.781.
2. Giwa A, Reyes M. Three times a charm…a case of repeat appendicitis status post two prior appendectomies. Am J Emerg Med. 2018;36(3):528.e1-528.e2. doi:10.1016/j.ajem.2017.12.024.
3. Addiss DG, Shaffer N, Fowler BS, Tauxe RV. The epidemiology of appendicitis and appendectomy in the United States. Am J Epidemiol. 1990;132(5):910-925.
4. Hendahewa R, Shekhar A, Ratnayake S. The dilemma of stump appendicitis—a case report and literature review. Int J Surg. Case Rep. 2015;14:101-103. doi:10.1016/j.ijscr.2015.07.017.
5. Liang MK, Lo HG, Marks JL. Stump appendicitis: a comprehensive review of literature. Am Surg. 2006;72(2):162-166.
6. Parthsarathi R, Jankar SV, Chittawadgi B, et al. Laraposcopic management of symptomatic residual appendicular tip: a rare case report. J Minim Access Surg. 2017;13(2):154-156. doi:10.4103/0972-9941.199610.
7. Kanona H, Al Samaraee A, Nice C, Bhattacharya V. Stump appendicitis: a review. Int J Surg. 2012;10(9):425-428. doi:10.1016/j.ijsu.2012.07.007.
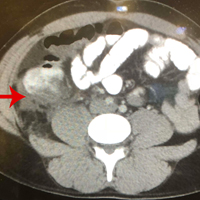
Hypertrophic cardiomyopathy: A complex disease
Hypertrophic cardiomyopathy (HCM) is a complex disease. Most people who carry the mutations that cause it are never affected at any point in their life, but some are affected at a young age. And in rare but tragic cases, some die suddenly while competing in sports. With such a wide range of phenotypic expressions, a single therapy does not fit all.
HCM is more common than once thought. Since the discovery of its genetic predisposition in 1960, it has come to be recognized as the most common heritable cardiovascular disease.1 Although earlier epidemiologic studies had estimated a prevalence of 1 in 500 (0.2%) of the general population, genetic testing and cardiac magnetic resonance imaging (MRI) now show that up to 1 in 200 (0.5%) of all people may be affected.1,2 Its prevalence is significant in all ethnic groups.
This review outlines our expanding knowledge of the pathophysiology, diagnosis, and clinical management of HCM.
A PLETHORA OF MUTATIONS IN CARDIAC SARCOMERIC GENES
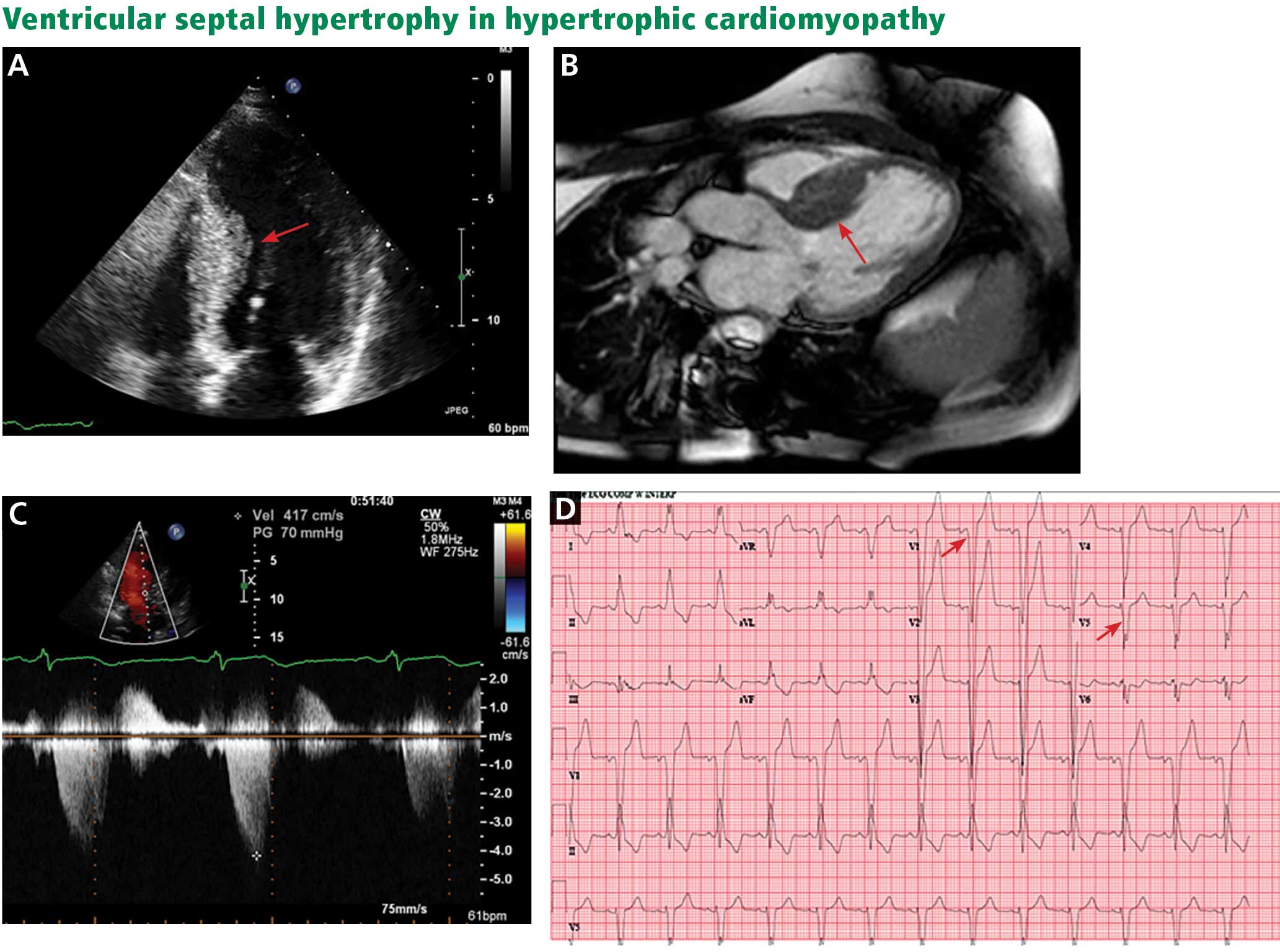
The genetic differences within HCM result in varying degrees and locations of left ventricular hypertrophy. Any segment of the ventricle can be involved, although HCM is classically asymmetric and mainly involves the septum (Figure 1). A variant form of HCM involves the apex of the heart (Figure 2).
LEFT VENTRICULAR OUTFLOW TRACT OBSTRUCTION
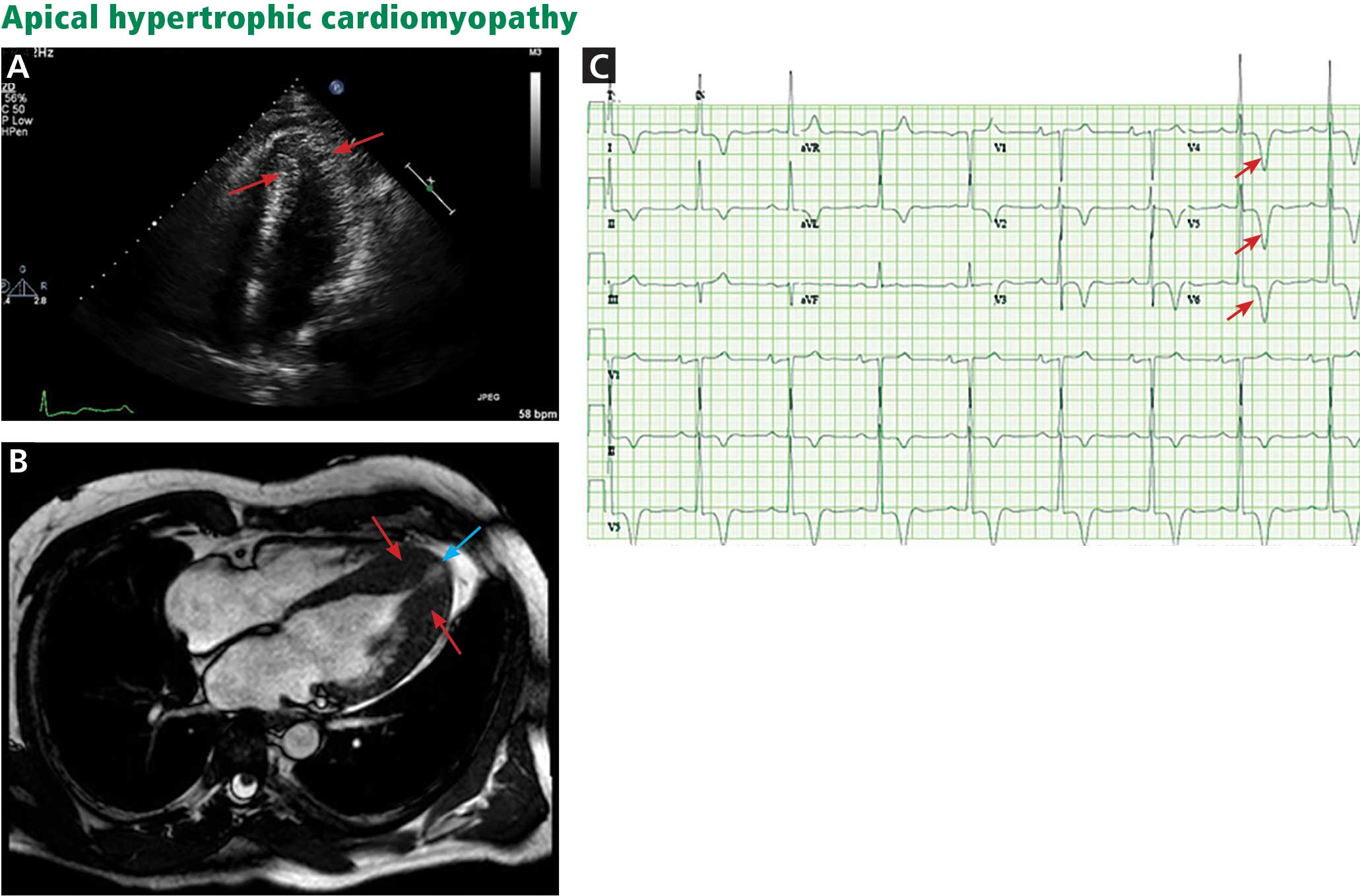
Only in the last decade has the significance of left ventricular outflow tract obstruction in HCM been truly appreciated. The degree of obstruction in HCM is dynamic, as opposed to the fixed obstruction in patients with aortic stenosis or congenital subvalvular membranes. Therefore, in HCM, exercise or drugs (eg, dobutamine) that increase cardiac contractility increase the obstruction, as do maneuvers or drugs (the Valsalva maneuver, nitrates) that reduce filling of the left ventricle.
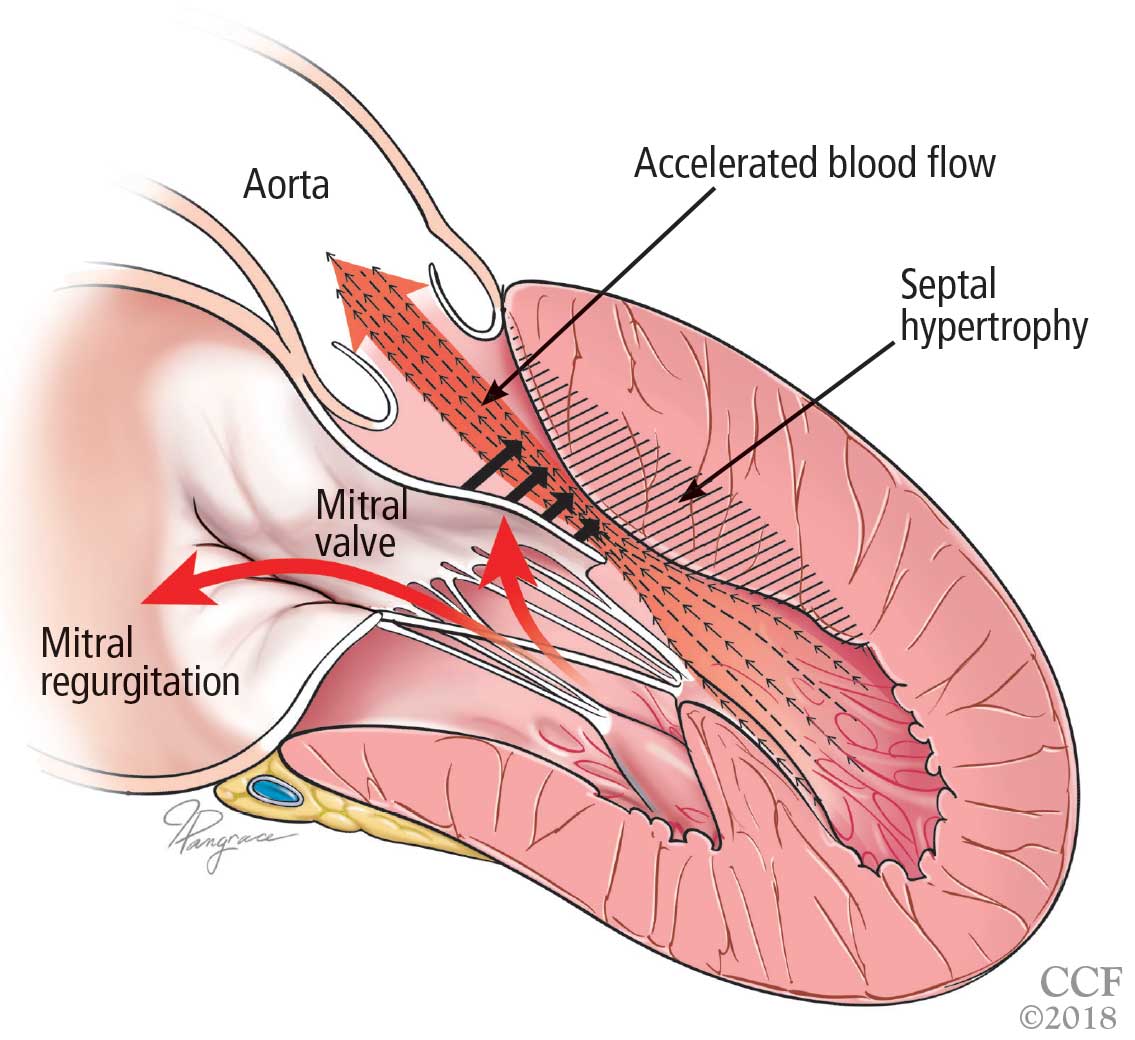
A less common source of dynamic obstruction is the papillary muscles (Figure 4). Hypertrophy of the papillary muscles can result in obstruction by these muscles themselves, which is visible on echocardiography. Anatomic variations include anteroapical displacement or bifid papillary muscles, and these variants can be associated with dynamic left ventricular outflow tract obstruction, even with no evidence of septal thickening (Figure 5).7,8 Recognizing this patient subset has important implications for management, as discussed below.
DIAGNOSTIC EVALUATION
The clinical presentation varies
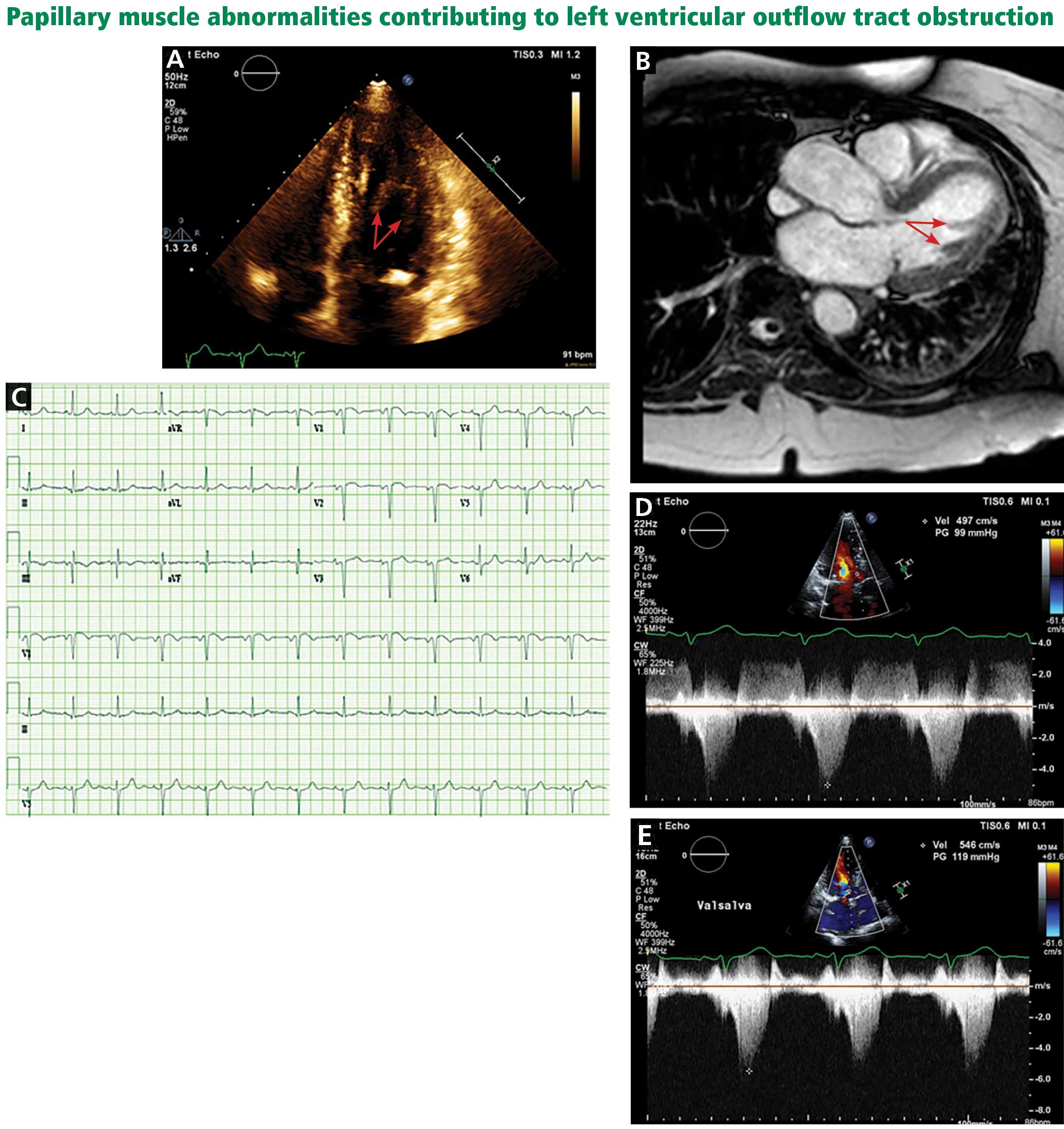
Even if patients harbor the same genetic variant, the clinical presentation can differ widely. Although the most feared presentation is sudden cardiac death, particularly in young athletes, most patients have no symptoms and can anticipate a normal life expectancy. The annual incidence of sudden cardiac death in all HCM patients is estimated at less than 1%.10 Sudden cardiac death in HCM patients is most often due to ventricular tachyarrhythmias and most often occurs in asymptomatic patients under age 35.
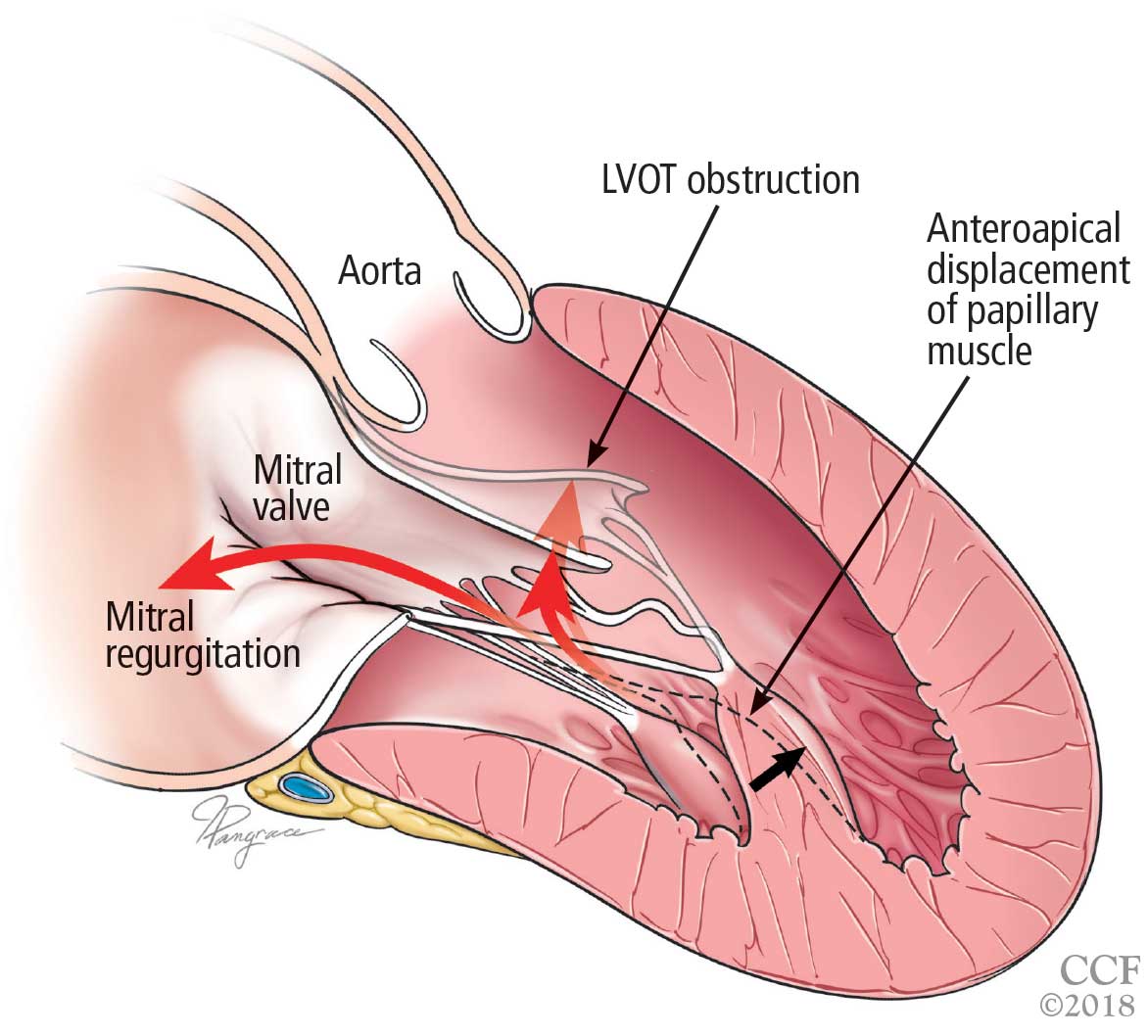
Physical findings are nonspecific

It can be difficult to distinguish the murmur of left ventricular outflow tract obstruction in HCM from a murmur related to aortic stenosis by auscultation alone. The simplest clinical method for telling them apart involves the Valsalva maneuver: bearing down creates a positive intrathoracic pressure and limits venous return, thus decreasing intracardiac filling pressure. This in turn results in less separation between the mitral valve and the ventricular septum in HCM, which increases obstruction and therefore makes the murmur louder. In contrast, in patients with fixed obstruction due to aortic stenosis, the murmur will decrease in intensity owing to the reduced flow associated with reduced preload.
Laboratory testing for phenocopies of HCM

A metabolic panel will show derangements in liver function and glucose levels in patients with glycogen storage disorders such as Pompe disease.
Serum creatinine. Renal dysfunction will be seen in patients with Fabry disease or amyloidosis.
Creatine kinase may be elevated in patients with Danon disease.
Electrocardiographic findings are common
More than 90% of HCM patients have electrocardiographic abnormalities. Although the findings can vary widely, common manifestations include:
- Left ventricular hypertrophy
- A pseudoinfarct pattern with Q waves in the anterolateral leads
- Repolarization changes such as T-wave inversions and horizontal or down-sloping ST segments.
Apical HCM, seen mainly in Asian populations, often presents with giant T-wave inversion (> 10 mm) in the anterolateral leads, most prominent in V4, V5, and V6.
Notably, the degree of electrocardiographic abnormalities does not correlate with the severity or pattern of hypertrophy.9 Electrocardiography lacks specificity for definitive diagnosis, and further diagnostic testing should therefore be pursued.
Echocardiography: Initial imaging test
Transthoracic echocardiography is the initial imaging test in patients with suspected HCM, allowing for cost-effective quantitative and qualitative assessment of left ventricular morphology and function. Left ventricular hypertrophy is considered pathologic if wall thickness is 15 mm or greater without a known cause. Transthoracic echocardiography also allows for evaluation of left atrial volume and mitral valve anatomy and function.
Speckle tracking imaging is an advanced echocardiographic technique that measures strain. Its major advantage is in identifying early abnormalities in genotype-positive, phenotype-negative HCM patients, ie, people who harbor mutations but who have no clinical symptoms or signs of HCM, potentially allowing for modification of the natural history of HCM.12 Strain imaging can also differentiate between physiologic hypertrophy (“athlete’s heart”) and hypertension and HCM.13,14
The utility of echocardiography in HCM is heavily influenced by the sonographer’s experience in obtaining adequate acoustic windows. This may be more difficult in obese patients, patients with advanced obstructive lung disease or pleural effusions, and women with breast implants.
Magnetic resonance imaging
MRI has an emerging role in both diagnosing and predicting risk in HCM, and is routinely done as an adjunct to transthoracic echocardiography on initial diagnosis in our tertiary referral center. It is particularly useful in patients suspected of having apical hypertrophy (Figure 2), in whom the diagnosis may be missed in up to 10% on transthoracic echocardiography alone.15 MRI can also enhance the assessment of left ventricular hypertrophy and has been shown to improve the diagnostic classification of HCM.16 It is the best way to assess myocardial tissue abnormalities, and late gadolinium enhancement to detect interstitial fibrosis can be used for further prognostication. While historically the primary role of MRI in HCM has been in phenotype classification, there is currently much interest in its role in risk stratification of HCM patients for ICD implantation.
MRI with late gadolinium enhancement provides insight into the location, pattern, and extent of myocardial fibrosis; the extent of fibrosis has been shown to be a strong independent predictor of poor outcomes, including sudden cardiac death.17–20 However, late gadolinium enhancement can be technically challenging, as variations in the timing of postcontrast imaging, sequences for measuring late gadolinium enhancement, or detection thresholds can result in widely variable image quality. Cardiac MRI should therefore be performed at an experienced center with standardized imaging protocols in place.
Current guidelines recommend considering cardiac MRI if a patient’s risk of sudden cardiac death remains inconclusive after conventional risk stratification, as discussed below.9,21
Stress testing for risk stratification
Exercise stress electrocardiography. Treadmill exercise stress testing with electrocardiography and hemodynamic monitoring was one of the first tools used for risk stratification in HCM.
Although systolic blood pressure normally increases by at least 20 mm Hg with exercise, one-quarter of HCM patients have either a blunted response (failure of systolic blood pressure to increase by at least 20 mm Hg) or a hypotensive response (a drop in systolic blood pressure of 20 mm Hg or more, either continuously or after an initial increase). Studies have shown that HCM patients who have abnormal blood pressure responses during exercise have a higher risk of sudden cardiac death.22–24
Exercise stress echocardiography can be useful to evaluate for provoked increases in the left ventricular outflow tract gradient, which may contribute to a patient’s symptoms even if the resting left ventricular outflow tract gradient is normal. Exercise testing is preferred over pharmacologic stimulation because it can provide functional assessment of whether a patient’s clinical symptoms are truly related to hemodynamic changes due to the hypertrophied ventricle, or whether alternative mechanisms should be explored.
Cardiopulmonary stress testing can readily add prognostic value with additional measurements of functional capacity. HCM patients who cannot achieve their predicted maximal exercise value such as peak rate of oxygen consumption, ventilation efficiency, or anaerobic threshold have higher rates of morbidity and mortality.25,26 Stress testing can also be useful for risk stratification in asymptomatic patients, with one study showing that those who achieve more than 100% of their age- and sex-predicted metabolic equivalents have a low event rate.27
Ambulatory electrocardiographic monitoring in all patients at diagnosis
Ambulatory electrocardiographic monitoring for 24 to 48 hours is recommended for all HCM patients at the time of diagnosis, even if they have no symptoms. Any evidence of nonsustained ventricular tachycardia suggests a substantially higher risk of sudden cardiac death.28,29
In patients with no symptoms or history of arrhythmia, current guidelines suggest ambulatory electrocardiographic monitoring every 1 to 2 years.9,21
Two risk-stratification models

The North American model was the first risk-stratification tool and considers 5 risk factors.9 However, if this algorithm were strictly followed, up to 60% of HCM patients would be candidates for cardioverter-defibrillator implantation.
The European model. This concern led to the development of the HCM Risk-SCD (sudden cardiac death), a risk-stratification tool introduced in the 2014 European Society of Cardiology HCM guidelines.30 This web-based calculator estimates a patient’s 5-year risk of sudden cardiac death using a complex calculation based on 7 clinical risk factors. If a patient’s calculated 5-year risk of sudden cardiac death is 6% or higher, cardioverter-defibrillator implantation is recommended for primary prevention.
The HCM Risk-SCD calculator was validated and compared with classic risk factors alone in a retrospective cohort study in 48 HCM patients.30 Compared with the North American model, the European model results in a lower rate of cardioverter-defibrillator implantation (20% to 26%).31,32
Despite the better specificity of the European model, a large retrospective cohort analysis showed that a significant number of patients stratified as being at low risk for sudden cardiac death were ultimately found to be at high risk in clinical practice.31 Further research is needed to find the optimal risk-stratification approach in HCM patients at low to intermediate risk.
GENETIC TESTING, COUNSELING, AND FAMILY SCREENING
Genetic testing is becoming more widely available and has rapidly expanded in clinical practice. Genetic counseling must be performed alongside genetic testing and requires professionals trained to handle the clinical and social implications of genetic testing. With this in mind, genetic testing can provide a definitive means of identifying family members at risk of HCM.
Given the autosomal dominant nature of HCM, screening for HCM is recommended in all first-degree relatives of an affected patient. Genetic testing may be a means to achieve this if a pathogenic mutation has been identified in the affected patient. However, serial electrocardiographic and transthoracic echocardiographic monitoring is an acceptable alternative in those without a clear genetic mutation association or in those who do not want to undergo genetic testing. If these first-degree relatives who do not undergo genetic testing are adult athletes or adolescents, they should undergo surveillance monitoring, with echocardiography and electrocardiography, whereas adults not participating in athletics should be monitored every 5 years.9,21
As genetic counseling and testing become more widely available, more patients are being found who harbor a mutation but have no phenotypic manifestations of HCM on initial presentation. Clinical expression varies, so continued monitoring of these patients is important. Expert guidelines again recommend serial electrocardiography, transthoracic echocardiography, and clinical assessment every 5 years for adults.9
Recent data suggest that up to 40% of HCM cases are nonfamilial, ie, their inheritance is sporadic with no known family history and no sarcomeric gene mutation evident on screening.33,34 The clinical course in this subgroup seems to be more benign, with later clinical presentations (age > 40) and lower risk of major adverse cardiovascular events.
MANAGEMENT
Conservative management
Asymptomatic HCM can usually be managed with lifestyle modifications.
Avoiding high-risk physical activities is the most important modification. All HCM patients should be counseled on the risk of sudden cardiac death and advised against participating in competitive sports or intense physical activity.35 Aerobic exercise is preferable to isometric exercises such as weightlifting, which may prompt the Valsalva maneuver with worsening of left ventricular outflow tract obstruction leading to syncope. A recent study showed that moderate-intensity aerobic exercise can safely improve exercise capacity, which may ultimately improve functional status and quality of life.36
Avoiding dehydration and excessive alcohol intake are also important in maintaining adequate preload to prevent an increasing left ventricular outflow tract gradient, given the dynamic nature of the left ventricular outflow tract obstruction in HCM.
Medical management: Beta-blockers, then calcium channel blockers
Beta-blockers are the first-line therapy for symptomatic HCM related to left ventricular outflow tract obstruction. Their negative inotropic effect reduces the contractile force of the ventricle, effectively reducing the pressure gradient across the outflow tract. Reduced contractility also means that the overall myocardial workload is less, which ultimately translates to a reduced oxygen demand. With their negative chronotropic effect, beta-blockers lower the heart rate and thereby lengthen the diastolic filling phase, allowing for optimization of preload conditions to help prevent increasing the left ventricular outflow tract gradient.37,38
Beta-blockers can be titrated according to the patient’s symptoms and tolerance. Fatigue and loss of libido are among the most common side effects.
Nondihydropyridine calcium channel blockers can be a second-line therapy in patients who cannot tolerate beta-blockers. Several studies have shown improvement in surrogate outcomes such as estimated left ventricular mass and QRS amplitude on electrocardiography, but currently no available data show that these drugs improve symptoms.28,39,40 They should be avoided in those with severe left ventricular outflow tract obstruction (gradient ≥ 100 mm Hg), as they can lead to critical outflow tract obstruction owing to their peripheral vasodilatory effect.
Dihydropyridine calcium channel blockers should be avoided altogether, as they produce even more peripheral vasodilation and afterload reduction than nondihydropyridine calcium channel blockers.
Disopyramide, a class IA antiarrhythmic, has been shown to effectively reduce outflow gradients and relieve symptoms. However, in view of its adverse effects, it is a third-line therapy, given to those for whom beta-blockers and calcium channel blockers have failed. Its most worrisome adverse effect is QT prolongation, and the QT interval should therefore be closely monitored at the start of treatment. Anticholinergic effects are common and include dry eyes and mouth, urinary retention, and drowsiness.
Disopyramide is usually used in combination with beta-blockers for symptom control as a bridge to a planned invasive intervention.41
Use with caution
Any medication that causes afterload reduction, peripheral vasodilation, intravascular volume depletion, or positive inotropy can worsen the dynamic left ventricular outflow tract obstruction in a patient with HCM and should be avoided.
Angiotensin-converting enzyme (ACE) inhibitors, angiotensin II receptor blockers (ARBs), and nitrates must be used with extreme caution in these patients.
Diuretics. Even restrained use of diuretics can cause significant hemodynamic compromise in patients with obstructive physiology. Therefore, diuretics should be used sparingly in these patients.
Digoxin should not be used for managing atrial fibrillation in these patients, as its positive inotropic effect increases contractility and increases the left ventricular outflow tract gradient.
Norepinephrine and inotropic agents such as dobutamine and dopamine should be avoided for the same reason as digoxin. In patients with circulatory shock requiring vasopressor support, pure alpha-agonists such as phenylephrine are preferred, as they increase peripheral resistance without an inotropic effect.
Anticoagulation for atrial tachyarrhythmias
The risk of systemic thromboembolic events is significantly increased in HCM patients with atrial fibrillation or flutter, regardless of their estimated risk using conventional risk-stratification tools such as the CHADS2 score.42–44 In accordance with current American Heart Association and American College of Cardiology guidelines, we recommend anticoagulation therapy for all HCM patients with a history of atrial fibrillation or flutter. Warfarin is the preferred anticoagulant; direct oral anticoagulants can be considered, but there are currently no data on their use in HCM.9
Standard heart failure treatments
End-stage systolic heart failure is a consequence of HCM but affects only 3% to 4% of patients.45 While most randomized controlled trials of heart failure treatment have excluded HCM patients, current guidelines recommend the same evidence-based medical therapies used in other patients who have heart failure with reduced ejection fraction. This includes ACE inhibitors, ARBs, beta-blockers, and aldosterone antagonists if indicated.9,21
Heart transplant should be considered in patients with class III or IV New York Heart Association functional status despite optimization of their HCM treatment regimen. Heart transplant outcomes for HCM patients are comparable to outcomes for patients who receive a transplant for non-HCM cardiovascular disease.45,46
Septal reduction therapy
If medical therapy fails or is not tolerated in patients with severe symptoms, surgery can be considered for obstructive HCM.
Ventricular septal myectomy has been the long-standing gold standard of invasive therapy. Multiple studies have demonstrated long-term survival after myectomy to be equivalent to that in the general population and better than that of HCM patients who do not undergo this surgery.47–50 Factors that may be associated with better surgical outcomes include age younger than 50, left atrial size less than 46 mm, and resolution of atrial fibrillation during follow-up.51
Septal reduction therapy may also be considered in patients at high risk of sudden cardiac death based on a history of recurrent ventricular tachycardia or risk-stratification models as described above. Retrospective analyses have shown that surgical myectomy can markedly reduce the incidence of appropriate implantable cardioverter-defibrillator discharges and the risk of sudden cardiac death.52
Alcohol septal ablation is an alternative. This percutaneous procedure, first described in the mid-1990s, consists of injecting a small amount of alcohol into the artery supplying the septum to induce myocardial necrosis, ultimately leading to scarring and widening of the left ventricular outflow tract.53
Up to 50% of patients develop right bundle branch block after alcohol septal ablation, and the risk of complete heart block is highest in those with preexisting left bundle branch block. Nevertheless, studies have shown significant symptomatic improvement after alcohol septal ablation, with long-term survival comparable to that in the general population.53–56
Several meta-analyses compared alcohol septal ablation and septal myectomy and found that the rates of functional improvement and long-term mortality were similar.57–59 However, the less-invasive approach with alcohol septal ablation comes at the cost of a higher incidence of conduction abnormalities and higher left ventricular outflow tract gradients afterward. One meta-analysis found that alcohol septal ablation patients may have 5 times the risk of needing additional septal reduction therapy compared with their myectomy counterparts.
Current US guidelines recommend septal myectomy, performed at an experienced center, as the first-line interventional treatment, leaving alcohol septal ablation to be considered in those who have contraindications to myectomy.9 The treatment strategy should ultimately be individualized based on a patient’s comorbidities and personal preferences following informed consent.
A nationwide database study recently suggested that postmyectomy mortality rates may be as high as 5.9%,60 although earlier studies at high-volume centers showed much lower mortality rates (< 1%).50–52,61 This discrepancy highlights the critical role of expert centers in optimizing surgical management of these patients. Regardless of the approach, interventional therapies for HCM should be performed by a multidisciplinary team at a medical center able to handle the complexity of these cases.
Additional surgical procedures
A handful of other procedures may benefit specific patient subgroups.

Apical myectomy has been shown to improve functional status in patients with isolated apical hypertrophy by reducing left ventricular end-diastolic pressure and thereby allowing for improved diastolic filling.63
Mitral valve surgery may need to be considered at the time of myectomy in patients with degenerative valve disease. As in the general population, mitral valve repair is preferred to replacement if possible.
- Maron BJ. Hypertrophic cardiomyopathy: an important global disease. Am J Med 2004; 116(1):63–65. pmid:14706671
- Semsarian C, Ingles J, Maron MS, Maron BJ. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol 2015; 65(12):1249–1254. doi:10.1016/j.jacc.2015.01.019
- Maron BJ, Maron MS, Semsarian C. Genetics of hypertrophic cardiomyopathy after 20 years: clinical perspectives. J Am Coll Cardiol 2012; 60(8):705–715. doi:10.1016/j.jacc.2012.02.068
- Shirani J, Pick R, Roberts WC, Maron BJ. Morphology and significance of the left ventricular collagen network in young patients with hypertrophic cardiomyopathy and sudden cardiac death. J Am Coll Cardiol 2000; 35(1):36–44. pmid:10636256
- Sherrid MV, Chu CK, Delia E, Mogtader A, Dwyer EM Jr. An echocardiographic study of the fluid mechanics of obstruction in hypertrophic cardiomyopathy. J Am Coll Cardiol 1993; 22(3):816–825. pmid:8354817
- Ro R, Halpern D, Sahn DJ, et al. Vector flow mapping in obstructive hypertrophic cardiomyopathy to assess the relationship of early systolic left ventricular flow and the mitral valve. J Am Coll Cardiol 2014; 64(19):1984–1995. doi:10.1016/j.jacc.2014.04.090
- Kwon DH, Setser RM, Thamilarasan M, et al. Abnormal papillary muscle morphology is independently associated with increased left ventricular outflow tract obstruction in hypertrophic cardiomyopathy. Heart 2008; 94(10):1295–1301. doi:10.1136/hrt.2007.118018
- Patel P, Dhillon A, Popovic ZB, et al. Left ventricular outflow tract obstruction in hypertrophic cardiomyopathy patients without severe septal hypertrophy: implications of mitral valve and papillary muscle abnormalities assessed using cardiac magnetic resonance and echocardiography. Circ Cardiovasc Imaging 2015; 8(7):e003132. doi:10.1161/CIRCIMAGING.115.003132
- Gersh BJ, Maron BJ, Bonow RO, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Thorac Cardiovasc Surg 2011; 142(6):e153-e203. doi:10.1016/j.jtcvs.2011.10.020
- Elliott PM, Gimeno JR, Thaman R, et al. Historical trends in reported survival rates in patients with hypertrophic cardiomyopathy. Heart 2006; 92(6):785–791. doi:10.1136/hrt.2005.068577
- Maron MS, Olivotto I, Zenovich AG, et al. Hypertrophic cardiomyopathy is predominantly a disease of left ventricular outflow tract obstruction. Circulation 2006; 114(21):2232–2239. doi:10.1161/CIRCULATIONAHA.106.644682
- Ho CY, Carlsen C, Thune JJ, et al. Echocardiographic strain imaging to assess early and late consequences of sarcomere mutations in hypertrophic cardiomyopathy. Circ Cardiovasc Genet 2009; 2(4):314–321. doi:10.1161/CIRCGENETICS.109.862128
- Wasfy MM, Weiner RB. Differentiating the athlete’s heart from hypertrophic cardiomyopathy. Curr Opin Cardiol 2015; 30(5):500–505. doi:10.1097/HCO.0000000000000203
- Palka P, Lange A, Fleming AD, et al. Differences in myocardial velocity gradient measured throughout the cardiac cycle in patients with hypertrophic cardiomyopathy, athletes and patients with left ventricular hypertrophy due to hypertension. J Am Coll Cardiol 1997; 30(3):760–768. pmid:9283537
- Eriksson MJ, Sonnenberg B, Woo A, et al. Long-term outcome in patients with apical hypertrophic cardiomyopathy. J Am Coll Cardiol 2002; 39(4):638–645. pmid:11849863
- Rickers C, Wilke NM, Jerosch-Herold M, et al. Utility of cardiac magnetic resonance imaging in the diagnosis of hypertrophic cardiomyopathy. Circulation 2005; 112(6):855–861. doi:10.1161/CIRCULATIONAHA.104.507723
- Kwon DH, Setser RM, Popovic ZB, et al. Association of myocardial fibrosis, electrocardiography and ventricular tachyarrhythmia in hypertrophic cardiomyopathy: a delayed contrast enhanced MRI study. Int J Cardiovasc Imaging 2008; 24(6):617–625. doi:10.1007/s10554-008-9292-6
- Rubinshtein R, Glockner JF, Ommen SR, et al. Characteristics and clinical significance of late gadolinium enhancement by contrast-enhanced magnetic resonance imaging in patients with hypertrophic cardiomyopathy. Circ Heart Fail 2010; 3(1):51–58. doi:10.1161/CIRCHEARTFAILURE.109.854026
- O’Hanlon R, Grasso A, Roughton M, et al. Prognostic significance of myocardial fibrosis in hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 56(11):867–874. doi:10.1016/j.jacc.2010.05.010
- Bruder O, Wagner A, Jensen CJ, et al. Myocardial scar visualized by cardiovascular magnetic resonance imaging predicts major adverse events in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 56(11):875–887. doi:10.1016/j.jacc.2010.05.007
- Authors/Task Force members, Elliott PM, Anastasakis A, Borger MA, et al. 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J 2014; 35(39):2733–2779. doi:10.1093/eurheartj/ehu284
- Olivotto I, Maron BJ, Montereggi A, Mazzuoli F, Dolara A, Cecchi F. Prognostic value of systemic blood pressure response during exercise in a community-based patient population with hypertrophic cardiomyopathy. J Am Coll Cardiol 1999; 33(7):2044–2051. pmid:10362212
- Sadoul N, Prasad K, Elliott PM, Bannerjee S, Frenneaux MP, McKenna WJ. Prospective prognostic assessment of blood pressure response during exercise in patients with hypertrophic cardiomyopathy. Circulation 1997; 96(9):2987–2991. pmid:9386166
- Elliott PM, Poloniecki J, Dickie S, et al. Sudden death in hypertrophic cardiomyopathy: identification of high risk patients. J Am Coll Cardiol 2000; 36(7):2212–2218. pmid:11127463
- Masri A, Pierson LM, Smedira NG, et al. Predictors of long-term outcomes in patients with hypertrophic cardiomyopathy undergoing cardiopulmonary stress testing and echocardiography. Am Heart J 2015; 169(5):684–692.e1. doi:10.1016/j.ahj.2015.02.006
- Coats CJ, Rantell K, Bartnik A, et al. Cardiopulmonary exercise testing and prognosis in hypertrophic cardiomyopathy. Circ Heart Fail 2015; 8(6):1022–1031. doi:10.1161/CIRCHEARTFAILURE.114.002248
- Desai MY, Bhonsale A, Patel P, et al. Exercise echocardiography in asymptomatic HCM: exercise capacity, and not LV outflow tract gradient predicts long-term outcomes. JACC Cardiovasc Imaging 2014; 7(1):26–36. doi:10.1016/j.jcmg.2013.08.010
- Spirito P, Seidman CE, McKenna WJ, Maron BJ. The management of hypertrophic cardiomyopathy. N Engl J Med 1997; 336(11):775–785. doi:10.1056/NEJM199703133361107
- Wang W, Lian Z, Rowin EJ, Maron BJ, Maron MS, Link MS. Prognostic implications of nonsustained ventricular tachycardia in high-risk patients with hypertrophic cardiomyopathy. Circ Arrhythm Electrophysiol 2017; 10(3)e004604. doi:10.1161/CIRCEP.116.004604
- O’Mahony C, Jichi F, Pavlou M, et al. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM risk-SCD). Eur Heart J 2014; 35(30):2010–2020. doi:10.1093/eurheartj/eht439
- Maron BJ, Casey SA, Chan RH, Garberich RF, Rowin EJ, Maron MS. Independent assessment of the European Society of Cardiology sudden death risk model for hypertrophic cardiomyopathy. Am J Cardiol 2015; 116(5):757–764. doi:10.1016/j.amjcard.2015.05.047
- Jahnlová D, Tomašov P, Zemánek D, Veselka J. Transatlantic differences in assessment of risk of sudden cardiac death in patients with hypertrophic cardiomyopathy. Int J Cardiol 2015; 186:3–4. doi:10.1016/j.ijcard.2015.03.207
- Ingles J, Burns C, Bagnall RD, et al. Nonfamilial hypertrophic cardiomyopathy: prevalence, natural history, and clinical implications. Circ Cardiovasc Genet 2017; 10(2)e001620. doi:10.1161/CIRCGENETICS.116.001620
- Ko C, Arscott P, Concannon M, et al. Genetic testing impacts the utility of prospective familial screening in hypertrophic cardiomyopathy through identification of a nonfamilial subgroup. Genet Med 2017; 20(1):69–75. doi:10.1038/gim.2017.79
- Maron BJ, Chaitman BR, Ackerman MJ, et al. Recommendations for physical activity and recreational sports participation for young patients with genetic cardiovascular diseases. Circulation 2004; 109(22):2807–2816. doi:10.1161/01.CIR.0000128363.85581.E1
- Saberi S, Wheeler M, Bragg-Gresham J, et al. Effect of moderate-intensity exercise training on peak oxygen consumption in patients with hypertrophic cardiomyopathy: a randomized clinical trial. JAMA 2017; 317(13):1349–1357. doi:10.1001/jama.2017.2503
- Bourmayan C, Razavi A, Fournier C, et al. Effect of propranolol on left ventricular relaxation in hypertrophic cardiomyopathy: an echographic study. Am Heart J 1985; 109(6):1311–1316. pmid:4039882
- Spoladore R, Maron MS, D’Amato R, Camici PG, Olivotto I. Pharmacological treatment options for hypertrophic cardiomyopathy: high time for evidence. Eur Heart J 2012; 33(14):1724–1733. doi:10.1093/eurheartj/ehs150
- Choudhury L, Elliott P, Rimoldi O, et al. Transmural myocardial blood flow distribution in hypertrophic cardiomyopathy and effect of treatment. Basic Res Cardiol 1999; 94(1):49–59. pmid:10097830
- Kaltenbach M, Hopf R, Kober G, Bussmann WD, Keller M, Petersen Y. Treatment of hypertrophic obstructive cardiomyopathy with verapamil. Br Heart J 1979; 42(1):35–42. doi:10.1136/hrt.42.1.35
- Sherrid MV, Shetty A, Winson G, et al. Treatment of obstructive hypertrophic cardiomyopathy symptoms and gradient resistant to first-line therapy with beta-blockade or verapamil. Circ Heart Fail 2013; 6(4):694–702. doi:10.1161/CIRCHEARTFAILURE.112.000122
- Guttmann OP, Rahman MS, O’Mahony C, Anastasakis A, Elliott PM. Atrial fibrillation and thromboembolism in patients with hypertrophic cardiomyopathy: systematic review. Heart 2014; 100(6):465–472. doi:10.1136/heartjnl-2013-304276
- Olivotto I, Cecchi F, Casey SA, Dolara A, Traverse JH, Maron BJ. Impact of atrial fibrillation on the clinical course of hypertrophic cardiomyopathy. Circulation 2001; 104(21):2517–2524. pmid:11714644
- Maron BJ, Olivotto I, Spirito P, et al. Epidemiology of hypertrophic cardiomyopathy-related death: revisited in a large non-referral-based patient population. Circulation 2000; 102(8):858–864. pmid:10952953
- Harris KM, Spirito P, Maron MS, et al. Prevalence, clinical profile, and significance of left ventricular remodeling in the end-stage phase of hypertrophic cardiomyopathy. Circulation 2006; 114(3):216-225. doi:10.1161/CIRCULATIONAHA.105.583500
- Maron MS, Kalsmith BM, Udelson JE, Li W, DeNofrio D. Survival after cardiac transplantation in patients with hypertrophic cardiomyopathy. Circ Heart Fail 2010; 3(5):574–579. doi:10.1161/CIRCHEARTFAILURE.109.922872
- Smedira NG, Lytle BW, Lever HM, et al. Current effectiveness and risks of isolated septal myectomy for hypertrophic obstructive cardiomyopathy. Ann Thorac Surg 2008; 85(1):127–133. doi:10.1016/j.athoracsur.2007.07.063
- Robbins RC, Stinson EB. Long-term results of left ventricular myotomy and myectomy for obstructive hypertrophic cardiomyopathy. J Thorac Cardiovasc Surg 1996; 111(3):586–594. pmid:8601973
- Heric B, Lytle BW, Miller DP, Rosenkranz ER, Lever HM, Cosgrove DM. Surgical management of hypertrophic obstructive cardiomyopathy. Early and late results. J Thorac Cardiovasc Surg 1995; 110(1):195–208. pmid:7609544
- Ommen SR, Maron BJ, Olivotto I, et al. Long-term effects of surgical septal myectomy on survival in patients with obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol 2005; 46(3):470–476. doi:10.1016/j.jacc.2005.02.090
- Desai MY, Bhonsale A, Smedira NG, et al. Predictors of long-term outcomes in symptomatic hypertrophic obstructive cardiomyopathy patients undergoing surgical relief of left ventricular outflow tract obstruction. Circulation 2013; 128(3):209–216. doi:10.1161/CIRCULATIONAHA.112.000849
- McLeod CJ, Ommen SR, Ackerman MJ, et al. Surgical septal myectomy decreases the risk for appropriate implantable cardioverter defibrillator discharge in obstructive hypertrophic cardiomyopathy. Eur Heart J 2007; 28(21):2583–2588. doi:10.1093/eurheartj/ehm117
- Veselka J, Tomasov P, Zemanek D. Long-term effects of varying alcohol dosing in percutaneous septal ablation for obstructive hypertrophic cardiomyopathy: a randomized study with a follow-up up to 11 years. Can J Cardiol 2011; 27(6):763–767. doi:10.1016/j.cjca.2011.09.001
- Veselka J, Jensen MK, Liebregts M, et al. Low procedure-related mortality achieved with alcohol septal ablation in European patients. Int J Cardiol 2016; 209:194–195. doi:10.1016/j.ijcard.2016.02.077
- Veselka J, Krejci J, Tomašov P, Zemánek D. Long-term survival after alcohol septal ablation for hypertrophic obstructive cardiomyopathy: a comparison with general population. Eur Heart J 2014; 35(30):2040–2045. doi:10.1093/eurheartj/eht495
- Sorajja P, Ommen SR, Holmes DR Jr, et al. Survival after alcohol septal ablation for obstructive hypertrophic cardiomyopathy. Circulation 2012; 126(20):2374–2380. doi:10.1161/CIRCULATIONAHA.111.076257
- Agarwal S, Tuzcu EM, Desai MY, et al. Updated meta-analysis of septal alcohol ablation versus myectomy for hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 55(8):823–834. doi:10.1016/j.jacc.2009.09.047
- Leonardi RA, Kransdorf EP, Simel DL, Wang A. Meta-analyses of septal reduction therapies for obstructive hypertrophic cardiomyopathy: comparative rates of overall mortality and sudden cardiac death after treatment. Circ Cardiovasc Interv 2010; 3(2):97–104. doi:10.1161/CIRCINTERVENTIONS.109.916676
- Liebregts M, Vriesendorp PA, Mahmoodi BK, Schinkel AF, Michels M, ten Berg JM. A systematic review and meta-analysis of long-term outcomes after septal reduction therapy in patients with hypertrophic cardiomyopathy. JACC Heart Fail 2015; 3(11):896–905. doi:10.1016/j.jchf.2015.06.011
- Panaich SS, Badheka AO, Chothani A, et al. Results of ventricular septal myectomy and hypertrophic cardiomyopathy (from Nationwide Inpatient Sample [1998-2010]). Am J Cardiol 2014; 114(9):1390–1395. doi:10.1016/j.amjcard.2014.07.075
- Maron BJ, Dearani JA, Ommen SR, et al. Low operative mortality achieved with surgical septal myectomy: role of dedicated hypertrophic cardiomyopathy centers in the management of dynamic subaortic obstruction. J Am Coll Cardiol 2015; 66(11):1307–1308. doi:10.1016/j.jacc.2015.06.1333
- Kwon DH, Smedira NG, Thamilarasan M, Lytle BW, Lever H, Desai MY. Characteristics and surgical outcomes of symptomatic patients with hypertrophic cardiomyopathy with abnormal papillary muscle morphology undergoing papillary muscle reorientation. J Thorac Cardiovasc Surg 2010; 140(2):317–324. doi:10.1016/j.jtcvs.2009.10.045
- Schaff HV, Brown ML, Dearani JA, et al. Apical myectomy: a new surgical technique for management of severely symptomatic patients with apical hypertrophic cardiomyopathy. J Thorac Cardiovasc Surg 2010; 139(3):634–640. doi:10.1016/j.jtcvs.2009.07.079
Hypertrophic cardiomyopathy (HCM) is a complex disease. Most people who carry the mutations that cause it are never affected at any point in their life, but some are affected at a young age. And in rare but tragic cases, some die suddenly while competing in sports. With such a wide range of phenotypic expressions, a single therapy does not fit all.
HCM is more common than once thought. Since the discovery of its genetic predisposition in 1960, it has come to be recognized as the most common heritable cardiovascular disease.1 Although earlier epidemiologic studies had estimated a prevalence of 1 in 500 (0.2%) of the general population, genetic testing and cardiac magnetic resonance imaging (MRI) now show that up to 1 in 200 (0.5%) of all people may be affected.1,2 Its prevalence is significant in all ethnic groups.
This review outlines our expanding knowledge of the pathophysiology, diagnosis, and clinical management of HCM.
A PLETHORA OF MUTATIONS IN CARDIAC SARCOMERIC GENES
The genetic differences within HCM result in varying degrees and locations of left ventricular hypertrophy. Any segment of the ventricle can be involved, although HCM is classically asymmetric and mainly involves the septum (Figure 1). A variant form of HCM involves the apex of the heart (Figure 2).
LEFT VENTRICULAR OUTFLOW TRACT OBSTRUCTION
Only in the last decade has the significance of left ventricular outflow tract obstruction in HCM been truly appreciated. The degree of obstruction in HCM is dynamic, as opposed to the fixed obstruction in patients with aortic stenosis or congenital subvalvular membranes. Therefore, in HCM, exercise or drugs (eg, dobutamine) that increase cardiac contractility increase the obstruction, as do maneuvers or drugs (the Valsalva maneuver, nitrates) that reduce filling of the left ventricle.
A less common source of dynamic obstruction is the papillary muscles (Figure 4). Hypertrophy of the papillary muscles can result in obstruction by these muscles themselves, which is visible on echocardiography. Anatomic variations include anteroapical displacement or bifid papillary muscles, and these variants can be associated with dynamic left ventricular outflow tract obstruction, even with no evidence of septal thickening (Figure 5).7,8 Recognizing this patient subset has important implications for management, as discussed below.
DIAGNOSTIC EVALUATION
The clinical presentation varies
Even if patients harbor the same genetic variant, the clinical presentation can differ widely. Although the most feared presentation is sudden cardiac death, particularly in young athletes, most patients have no symptoms and can anticipate a normal life expectancy. The annual incidence of sudden cardiac death in all HCM patients is estimated at less than 1%.10 Sudden cardiac death in HCM patients is most often due to ventricular tachyarrhythmias and most often occurs in asymptomatic patients under age 35.
Physical findings are nonspecific
It can be difficult to distinguish the murmur of left ventricular outflow tract obstruction in HCM from a murmur related to aortic stenosis by auscultation alone. The simplest clinical method for telling them apart involves the Valsalva maneuver: bearing down creates a positive intrathoracic pressure and limits venous return, thus decreasing intracardiac filling pressure. This in turn results in less separation between the mitral valve and the ventricular septum in HCM, which increases obstruction and therefore makes the murmur louder. In contrast, in patients with fixed obstruction due to aortic stenosis, the murmur will decrease in intensity owing to the reduced flow associated with reduced preload.
Laboratory testing for phenocopies of HCM
A metabolic panel will show derangements in liver function and glucose levels in patients with glycogen storage disorders such as Pompe disease.
Serum creatinine. Renal dysfunction will be seen in patients with Fabry disease or amyloidosis.
Creatine kinase may be elevated in patients with Danon disease.
Electrocardiographic findings are common
More than 90% of HCM patients have electrocardiographic abnormalities. Although the findings can vary widely, common manifestations include:
- Left ventricular hypertrophy
- A pseudoinfarct pattern with Q waves in the anterolateral leads
- Repolarization changes such as T-wave inversions and horizontal or down-sloping ST segments.
Apical HCM, seen mainly in Asian populations, often presents with giant T-wave inversion (> 10 mm) in the anterolateral leads, most prominent in V4, V5, and V6.
Notably, the degree of electrocardiographic abnormalities does not correlate with the severity or pattern of hypertrophy.9 Electrocardiography lacks specificity for definitive diagnosis, and further diagnostic testing should therefore be pursued.
Echocardiography: Initial imaging test
Transthoracic echocardiography is the initial imaging test in patients with suspected HCM, allowing for cost-effective quantitative and qualitative assessment of left ventricular morphology and function. Left ventricular hypertrophy is considered pathologic if wall thickness is 15 mm or greater without a known cause. Transthoracic echocardiography also allows for evaluation of left atrial volume and mitral valve anatomy and function.
Speckle tracking imaging is an advanced echocardiographic technique that measures strain. Its major advantage is in identifying early abnormalities in genotype-positive, phenotype-negative HCM patients, ie, people who harbor mutations but who have no clinical symptoms or signs of HCM, potentially allowing for modification of the natural history of HCM.12 Strain imaging can also differentiate between physiologic hypertrophy (“athlete’s heart”) and hypertension and HCM.13,14
The utility of echocardiography in HCM is heavily influenced by the sonographer’s experience in obtaining adequate acoustic windows. This may be more difficult in obese patients, patients with advanced obstructive lung disease or pleural effusions, and women with breast implants.
Magnetic resonance imaging
MRI has an emerging role in both diagnosing and predicting risk in HCM, and is routinely done as an adjunct to transthoracic echocardiography on initial diagnosis in our tertiary referral center. It is particularly useful in patients suspected of having apical hypertrophy (Figure 2), in whom the diagnosis may be missed in up to 10% on transthoracic echocardiography alone.15 MRI can also enhance the assessment of left ventricular hypertrophy and has been shown to improve the diagnostic classification of HCM.16 It is the best way to assess myocardial tissue abnormalities, and late gadolinium enhancement to detect interstitial fibrosis can be used for further prognostication. While historically the primary role of MRI in HCM has been in phenotype classification, there is currently much interest in its role in risk stratification of HCM patients for ICD implantation.
MRI with late gadolinium enhancement provides insight into the location, pattern, and extent of myocardial fibrosis; the extent of fibrosis has been shown to be a strong independent predictor of poor outcomes, including sudden cardiac death.17–20 However, late gadolinium enhancement can be technically challenging, as variations in the timing of postcontrast imaging, sequences for measuring late gadolinium enhancement, or detection thresholds can result in widely variable image quality. Cardiac MRI should therefore be performed at an experienced center with standardized imaging protocols in place.
Current guidelines recommend considering cardiac MRI if a patient’s risk of sudden cardiac death remains inconclusive after conventional risk stratification, as discussed below.9,21
Stress testing for risk stratification
Exercise stress electrocardiography. Treadmill exercise stress testing with electrocardiography and hemodynamic monitoring was one of the first tools used for risk stratification in HCM.
Although systolic blood pressure normally increases by at least 20 mm Hg with exercise, one-quarter of HCM patients have either a blunted response (failure of systolic blood pressure to increase by at least 20 mm Hg) or a hypotensive response (a drop in systolic blood pressure of 20 mm Hg or more, either continuously or after an initial increase). Studies have shown that HCM patients who have abnormal blood pressure responses during exercise have a higher risk of sudden cardiac death.22–24
Exercise stress echocardiography can be useful to evaluate for provoked increases in the left ventricular outflow tract gradient, which may contribute to a patient’s symptoms even if the resting left ventricular outflow tract gradient is normal. Exercise testing is preferred over pharmacologic stimulation because it can provide functional assessment of whether a patient’s clinical symptoms are truly related to hemodynamic changes due to the hypertrophied ventricle, or whether alternative mechanisms should be explored.
Cardiopulmonary stress testing can readily add prognostic value with additional measurements of functional capacity. HCM patients who cannot achieve their predicted maximal exercise value such as peak rate of oxygen consumption, ventilation efficiency, or anaerobic threshold have higher rates of morbidity and mortality.25,26 Stress testing can also be useful for risk stratification in asymptomatic patients, with one study showing that those who achieve more than 100% of their age- and sex-predicted metabolic equivalents have a low event rate.27
Ambulatory electrocardiographic monitoring in all patients at diagnosis
Ambulatory electrocardiographic monitoring for 24 to 48 hours is recommended for all HCM patients at the time of diagnosis, even if they have no symptoms. Any evidence of nonsustained ventricular tachycardia suggests a substantially higher risk of sudden cardiac death.28,29
In patients with no symptoms or history of arrhythmia, current guidelines suggest ambulatory electrocardiographic monitoring every 1 to 2 years.9,21
Two risk-stratification models
The North American model was the first risk-stratification tool and considers 5 risk factors.9 However, if this algorithm were strictly followed, up to 60% of HCM patients would be candidates for cardioverter-defibrillator implantation.
The European model. This concern led to the development of the HCM Risk-SCD (sudden cardiac death), a risk-stratification tool introduced in the 2014 European Society of Cardiology HCM guidelines.30 This web-based calculator estimates a patient’s 5-year risk of sudden cardiac death using a complex calculation based on 7 clinical risk factors. If a patient’s calculated 5-year risk of sudden cardiac death is 6% or higher, cardioverter-defibrillator implantation is recommended for primary prevention.
The HCM Risk-SCD calculator was validated and compared with classic risk factors alone in a retrospective cohort study in 48 HCM patients.30 Compared with the North American model, the European model results in a lower rate of cardioverter-defibrillator implantation (20% to 26%).31,32
Despite the better specificity of the European model, a large retrospective cohort analysis showed that a significant number of patients stratified as being at low risk for sudden cardiac death were ultimately found to be at high risk in clinical practice.31 Further research is needed to find the optimal risk-stratification approach in HCM patients at low to intermediate risk.
GENETIC TESTING, COUNSELING, AND FAMILY SCREENING
Genetic testing is becoming more widely available and has rapidly expanded in clinical practice. Genetic counseling must be performed alongside genetic testing and requires professionals trained to handle the clinical and social implications of genetic testing. With this in mind, genetic testing can provide a definitive means of identifying family members at risk of HCM.
Given the autosomal dominant nature of HCM, screening for HCM is recommended in all first-degree relatives of an affected patient. Genetic testing may be a means to achieve this if a pathogenic mutation has been identified in the affected patient. However, serial electrocardiographic and transthoracic echocardiographic monitoring is an acceptable alternative in those without a clear genetic mutation association or in those who do not want to undergo genetic testing. If these first-degree relatives who do not undergo genetic testing are adult athletes or adolescents, they should undergo surveillance monitoring, with echocardiography and electrocardiography, whereas adults not participating in athletics should be monitored every 5 years.9,21
As genetic counseling and testing become more widely available, more patients are being found who harbor a mutation but have no phenotypic manifestations of HCM on initial presentation. Clinical expression varies, so continued monitoring of these patients is important. Expert guidelines again recommend serial electrocardiography, transthoracic echocardiography, and clinical assessment every 5 years for adults.9
Recent data suggest that up to 40% of HCM cases are nonfamilial, ie, their inheritance is sporadic with no known family history and no sarcomeric gene mutation evident on screening.33,34 The clinical course in this subgroup seems to be more benign, with later clinical presentations (age > 40) and lower risk of major adverse cardiovascular events.
MANAGEMENT
Conservative management
Asymptomatic HCM can usually be managed with lifestyle modifications.
Avoiding high-risk physical activities is the most important modification. All HCM patients should be counseled on the risk of sudden cardiac death and advised against participating in competitive sports or intense physical activity.35 Aerobic exercise is preferable to isometric exercises such as weightlifting, which may prompt the Valsalva maneuver with worsening of left ventricular outflow tract obstruction leading to syncope. A recent study showed that moderate-intensity aerobic exercise can safely improve exercise capacity, which may ultimately improve functional status and quality of life.36
Avoiding dehydration and excessive alcohol intake are also important in maintaining adequate preload to prevent an increasing left ventricular outflow tract gradient, given the dynamic nature of the left ventricular outflow tract obstruction in HCM.
Medical management: Beta-blockers, then calcium channel blockers
Beta-blockers are the first-line therapy for symptomatic HCM related to left ventricular outflow tract obstruction. Their negative inotropic effect reduces the contractile force of the ventricle, effectively reducing the pressure gradient across the outflow tract. Reduced contractility also means that the overall myocardial workload is less, which ultimately translates to a reduced oxygen demand. With their negative chronotropic effect, beta-blockers lower the heart rate and thereby lengthen the diastolic filling phase, allowing for optimization of preload conditions to help prevent increasing the left ventricular outflow tract gradient.37,38
Beta-blockers can be titrated according to the patient’s symptoms and tolerance. Fatigue and loss of libido are among the most common side effects.
Nondihydropyridine calcium channel blockers can be a second-line therapy in patients who cannot tolerate beta-blockers. Several studies have shown improvement in surrogate outcomes such as estimated left ventricular mass and QRS amplitude on electrocardiography, but currently no available data show that these drugs improve symptoms.28,39,40 They should be avoided in those with severe left ventricular outflow tract obstruction (gradient ≥ 100 mm Hg), as they can lead to critical outflow tract obstruction owing to their peripheral vasodilatory effect.
Dihydropyridine calcium channel blockers should be avoided altogether, as they produce even more peripheral vasodilation and afterload reduction than nondihydropyridine calcium channel blockers.
Disopyramide, a class IA antiarrhythmic, has been shown to effectively reduce outflow gradients and relieve symptoms. However, in view of its adverse effects, it is a third-line therapy, given to those for whom beta-blockers and calcium channel blockers have failed. Its most worrisome adverse effect is QT prolongation, and the QT interval should therefore be closely monitored at the start of treatment. Anticholinergic effects are common and include dry eyes and mouth, urinary retention, and drowsiness.
Disopyramide is usually used in combination with beta-blockers for symptom control as a bridge to a planned invasive intervention.41
Use with caution
Any medication that causes afterload reduction, peripheral vasodilation, intravascular volume depletion, or positive inotropy can worsen the dynamic left ventricular outflow tract obstruction in a patient with HCM and should be avoided.
Angiotensin-converting enzyme (ACE) inhibitors, angiotensin II receptor blockers (ARBs), and nitrates must be used with extreme caution in these patients.
Diuretics. Even restrained use of diuretics can cause significant hemodynamic compromise in patients with obstructive physiology. Therefore, diuretics should be used sparingly in these patients.
Digoxin should not be used for managing atrial fibrillation in these patients, as its positive inotropic effect increases contractility and increases the left ventricular outflow tract gradient.
Norepinephrine and inotropic agents such as dobutamine and dopamine should be avoided for the same reason as digoxin. In patients with circulatory shock requiring vasopressor support, pure alpha-agonists such as phenylephrine are preferred, as they increase peripheral resistance without an inotropic effect.
Anticoagulation for atrial tachyarrhythmias
The risk of systemic thromboembolic events is significantly increased in HCM patients with atrial fibrillation or flutter, regardless of their estimated risk using conventional risk-stratification tools such as the CHADS2 score.42–44 In accordance with current American Heart Association and American College of Cardiology guidelines, we recommend anticoagulation therapy for all HCM patients with a history of atrial fibrillation or flutter. Warfarin is the preferred anticoagulant; direct oral anticoagulants can be considered, but there are currently no data on their use in HCM.9
Standard heart failure treatments
End-stage systolic heart failure is a consequence of HCM but affects only 3% to 4% of patients.45 While most randomized controlled trials of heart failure treatment have excluded HCM patients, current guidelines recommend the same evidence-based medical therapies used in other patients who have heart failure with reduced ejection fraction. This includes ACE inhibitors, ARBs, beta-blockers, and aldosterone antagonists if indicated.9,21
Heart transplant should be considered in patients with class III or IV New York Heart Association functional status despite optimization of their HCM treatment regimen. Heart transplant outcomes for HCM patients are comparable to outcomes for patients who receive a transplant for non-HCM cardiovascular disease.45,46
Septal reduction therapy
If medical therapy fails or is not tolerated in patients with severe symptoms, surgery can be considered for obstructive HCM.
Ventricular septal myectomy has been the long-standing gold standard of invasive therapy. Multiple studies have demonstrated long-term survival after myectomy to be equivalent to that in the general population and better than that of HCM patients who do not undergo this surgery.47–50 Factors that may be associated with better surgical outcomes include age younger than 50, left atrial size less than 46 mm, and resolution of atrial fibrillation during follow-up.51
Septal reduction therapy may also be considered in patients at high risk of sudden cardiac death based on a history of recurrent ventricular tachycardia or risk-stratification models as described above. Retrospective analyses have shown that surgical myectomy can markedly reduce the incidence of appropriate implantable cardioverter-defibrillator discharges and the risk of sudden cardiac death.52
Alcohol septal ablation is an alternative. This percutaneous procedure, first described in the mid-1990s, consists of injecting a small amount of alcohol into the artery supplying the septum to induce myocardial necrosis, ultimately leading to scarring and widening of the left ventricular outflow tract.53
Up to 50% of patients develop right bundle branch block after alcohol septal ablation, and the risk of complete heart block is highest in those with preexisting left bundle branch block. Nevertheless, studies have shown significant symptomatic improvement after alcohol septal ablation, with long-term survival comparable to that in the general population.53–56
Several meta-analyses compared alcohol septal ablation and septal myectomy and found that the rates of functional improvement and long-term mortality were similar.57–59 However, the less-invasive approach with alcohol septal ablation comes at the cost of a higher incidence of conduction abnormalities and higher left ventricular outflow tract gradients afterward. One meta-analysis found that alcohol septal ablation patients may have 5 times the risk of needing additional septal reduction therapy compared with their myectomy counterparts.
Current US guidelines recommend septal myectomy, performed at an experienced center, as the first-line interventional treatment, leaving alcohol septal ablation to be considered in those who have contraindications to myectomy.9 The treatment strategy should ultimately be individualized based on a patient’s comorbidities and personal preferences following informed consent.
A nationwide database study recently suggested that postmyectomy mortality rates may be as high as 5.9%,60 although earlier studies at high-volume centers showed much lower mortality rates (< 1%).50–52,61 This discrepancy highlights the critical role of expert centers in optimizing surgical management of these patients. Regardless of the approach, interventional therapies for HCM should be performed by a multidisciplinary team at a medical center able to handle the complexity of these cases.
Additional surgical procedures
A handful of other procedures may benefit specific patient subgroups.
Apical myectomy has been shown to improve functional status in patients with isolated apical hypertrophy by reducing left ventricular end-diastolic pressure and thereby allowing for improved diastolic filling.63
Mitral valve surgery may need to be considered at the time of myectomy in patients with degenerative valve disease. As in the general population, mitral valve repair is preferred to replacement if possible.
Hypertrophic cardiomyopathy (HCM) is a complex disease. Most people who carry the mutations that cause it are never affected at any point in their life, but some are affected at a young age. And in rare but tragic cases, some die suddenly while competing in sports. With such a wide range of phenotypic expressions, a single therapy does not fit all.
HCM is more common than once thought. Since the discovery of its genetic predisposition in 1960, it has come to be recognized as the most common heritable cardiovascular disease.1 Although earlier epidemiologic studies had estimated a prevalence of 1 in 500 (0.2%) of the general population, genetic testing and cardiac magnetic resonance imaging (MRI) now show that up to 1 in 200 (0.5%) of all people may be affected.1,2 Its prevalence is significant in all ethnic groups.
This review outlines our expanding knowledge of the pathophysiology, diagnosis, and clinical management of HCM.
A PLETHORA OF MUTATIONS IN CARDIAC SARCOMERIC GENES
The genetic differences within HCM result in varying degrees and locations of left ventricular hypertrophy. Any segment of the ventricle can be involved, although HCM is classically asymmetric and mainly involves the septum (Figure 1). A variant form of HCM involves the apex of the heart (Figure 2).
LEFT VENTRICULAR OUTFLOW TRACT OBSTRUCTION
Only in the last decade has the significance of left ventricular outflow tract obstruction in HCM been truly appreciated. The degree of obstruction in HCM is dynamic, as opposed to the fixed obstruction in patients with aortic stenosis or congenital subvalvular membranes. Therefore, in HCM, exercise or drugs (eg, dobutamine) that increase cardiac contractility increase the obstruction, as do maneuvers or drugs (the Valsalva maneuver, nitrates) that reduce filling of the left ventricle.
A less common source of dynamic obstruction is the papillary muscles (Figure 4). Hypertrophy of the papillary muscles can result in obstruction by these muscles themselves, which is visible on echocardiography. Anatomic variations include anteroapical displacement or bifid papillary muscles, and these variants can be associated with dynamic left ventricular outflow tract obstruction, even with no evidence of septal thickening (Figure 5).7,8 Recognizing this patient subset has important implications for management, as discussed below.
DIAGNOSTIC EVALUATION
The clinical presentation varies
Even if patients harbor the same genetic variant, the clinical presentation can differ widely. Although the most feared presentation is sudden cardiac death, particularly in young athletes, most patients have no symptoms and can anticipate a normal life expectancy. The annual incidence of sudden cardiac death in all HCM patients is estimated at less than 1%.10 Sudden cardiac death in HCM patients is most often due to ventricular tachyarrhythmias and most often occurs in asymptomatic patients under age 35.
Physical findings are nonspecific
It can be difficult to distinguish the murmur of left ventricular outflow tract obstruction in HCM from a murmur related to aortic stenosis by auscultation alone. The simplest clinical method for telling them apart involves the Valsalva maneuver: bearing down creates a positive intrathoracic pressure and limits venous return, thus decreasing intracardiac filling pressure. This in turn results in less separation between the mitral valve and the ventricular septum in HCM, which increases obstruction and therefore makes the murmur louder. In contrast, in patients with fixed obstruction due to aortic stenosis, the murmur will decrease in intensity owing to the reduced flow associated with reduced preload.
Laboratory testing for phenocopies of HCM
A metabolic panel will show derangements in liver function and glucose levels in patients with glycogen storage disorders such as Pompe disease.
Serum creatinine. Renal dysfunction will be seen in patients with Fabry disease or amyloidosis.
Creatine kinase may be elevated in patients with Danon disease.
Electrocardiographic findings are common
More than 90% of HCM patients have electrocardiographic abnormalities. Although the findings can vary widely, common manifestations include:
- Left ventricular hypertrophy
- A pseudoinfarct pattern with Q waves in the anterolateral leads
- Repolarization changes such as T-wave inversions and horizontal or down-sloping ST segments.
Apical HCM, seen mainly in Asian populations, often presents with giant T-wave inversion (> 10 mm) in the anterolateral leads, most prominent in V4, V5, and V6.
Notably, the degree of electrocardiographic abnormalities does not correlate with the severity or pattern of hypertrophy.9 Electrocardiography lacks specificity for definitive diagnosis, and further diagnostic testing should therefore be pursued.
Echocardiography: Initial imaging test
Transthoracic echocardiography is the initial imaging test in patients with suspected HCM, allowing for cost-effective quantitative and qualitative assessment of left ventricular morphology and function. Left ventricular hypertrophy is considered pathologic if wall thickness is 15 mm or greater without a known cause. Transthoracic echocardiography also allows for evaluation of left atrial volume and mitral valve anatomy and function.
Speckle tracking imaging is an advanced echocardiographic technique that measures strain. Its major advantage is in identifying early abnormalities in genotype-positive, phenotype-negative HCM patients, ie, people who harbor mutations but who have no clinical symptoms or signs of HCM, potentially allowing for modification of the natural history of HCM.12 Strain imaging can also differentiate between physiologic hypertrophy (“athlete’s heart”) and hypertension and HCM.13,14
The utility of echocardiography in HCM is heavily influenced by the sonographer’s experience in obtaining adequate acoustic windows. This may be more difficult in obese patients, patients with advanced obstructive lung disease or pleural effusions, and women with breast implants.
Magnetic resonance imaging
MRI has an emerging role in both diagnosing and predicting risk in HCM, and is routinely done as an adjunct to transthoracic echocardiography on initial diagnosis in our tertiary referral center. It is particularly useful in patients suspected of having apical hypertrophy (Figure 2), in whom the diagnosis may be missed in up to 10% on transthoracic echocardiography alone.15 MRI can also enhance the assessment of left ventricular hypertrophy and has been shown to improve the diagnostic classification of HCM.16 It is the best way to assess myocardial tissue abnormalities, and late gadolinium enhancement to detect interstitial fibrosis can be used for further prognostication. While historically the primary role of MRI in HCM has been in phenotype classification, there is currently much interest in its role in risk stratification of HCM patients for ICD implantation.
MRI with late gadolinium enhancement provides insight into the location, pattern, and extent of myocardial fibrosis; the extent of fibrosis has been shown to be a strong independent predictor of poor outcomes, including sudden cardiac death.17–20 However, late gadolinium enhancement can be technically challenging, as variations in the timing of postcontrast imaging, sequences for measuring late gadolinium enhancement, or detection thresholds can result in widely variable image quality. Cardiac MRI should therefore be performed at an experienced center with standardized imaging protocols in place.
Current guidelines recommend considering cardiac MRI if a patient’s risk of sudden cardiac death remains inconclusive after conventional risk stratification, as discussed below.9,21
Stress testing for risk stratification
Exercise stress electrocardiography. Treadmill exercise stress testing with electrocardiography and hemodynamic monitoring was one of the first tools used for risk stratification in HCM.
Although systolic blood pressure normally increases by at least 20 mm Hg with exercise, one-quarter of HCM patients have either a blunted response (failure of systolic blood pressure to increase by at least 20 mm Hg) or a hypotensive response (a drop in systolic blood pressure of 20 mm Hg or more, either continuously or after an initial increase). Studies have shown that HCM patients who have abnormal blood pressure responses during exercise have a higher risk of sudden cardiac death.22–24
Exercise stress echocardiography can be useful to evaluate for provoked increases in the left ventricular outflow tract gradient, which may contribute to a patient’s symptoms even if the resting left ventricular outflow tract gradient is normal. Exercise testing is preferred over pharmacologic stimulation because it can provide functional assessment of whether a patient’s clinical symptoms are truly related to hemodynamic changes due to the hypertrophied ventricle, or whether alternative mechanisms should be explored.
Cardiopulmonary stress testing can readily add prognostic value with additional measurements of functional capacity. HCM patients who cannot achieve their predicted maximal exercise value such as peak rate of oxygen consumption, ventilation efficiency, or anaerobic threshold have higher rates of morbidity and mortality.25,26 Stress testing can also be useful for risk stratification in asymptomatic patients, with one study showing that those who achieve more than 100% of their age- and sex-predicted metabolic equivalents have a low event rate.27
Ambulatory electrocardiographic monitoring in all patients at diagnosis
Ambulatory electrocardiographic monitoring for 24 to 48 hours is recommended for all HCM patients at the time of diagnosis, even if they have no symptoms. Any evidence of nonsustained ventricular tachycardia suggests a substantially higher risk of sudden cardiac death.28,29
In patients with no symptoms or history of arrhythmia, current guidelines suggest ambulatory electrocardiographic monitoring every 1 to 2 years.9,21
Two risk-stratification models
The North American model was the first risk-stratification tool and considers 5 risk factors.9 However, if this algorithm were strictly followed, up to 60% of HCM patients would be candidates for cardioverter-defibrillator implantation.
The European model. This concern led to the development of the HCM Risk-SCD (sudden cardiac death), a risk-stratification tool introduced in the 2014 European Society of Cardiology HCM guidelines.30 This web-based calculator estimates a patient’s 5-year risk of sudden cardiac death using a complex calculation based on 7 clinical risk factors. If a patient’s calculated 5-year risk of sudden cardiac death is 6% or higher, cardioverter-defibrillator implantation is recommended for primary prevention.
The HCM Risk-SCD calculator was validated and compared with classic risk factors alone in a retrospective cohort study in 48 HCM patients.30 Compared with the North American model, the European model results in a lower rate of cardioverter-defibrillator implantation (20% to 26%).31,32
Despite the better specificity of the European model, a large retrospective cohort analysis showed that a significant number of patients stratified as being at low risk for sudden cardiac death were ultimately found to be at high risk in clinical practice.31 Further research is needed to find the optimal risk-stratification approach in HCM patients at low to intermediate risk.
GENETIC TESTING, COUNSELING, AND FAMILY SCREENING
Genetic testing is becoming more widely available and has rapidly expanded in clinical practice. Genetic counseling must be performed alongside genetic testing and requires professionals trained to handle the clinical and social implications of genetic testing. With this in mind, genetic testing can provide a definitive means of identifying family members at risk of HCM.
Given the autosomal dominant nature of HCM, screening for HCM is recommended in all first-degree relatives of an affected patient. Genetic testing may be a means to achieve this if a pathogenic mutation has been identified in the affected patient. However, serial electrocardiographic and transthoracic echocardiographic monitoring is an acceptable alternative in those without a clear genetic mutation association or in those who do not want to undergo genetic testing. If these first-degree relatives who do not undergo genetic testing are adult athletes or adolescents, they should undergo surveillance monitoring, with echocardiography and electrocardiography, whereas adults not participating in athletics should be monitored every 5 years.9,21
As genetic counseling and testing become more widely available, more patients are being found who harbor a mutation but have no phenotypic manifestations of HCM on initial presentation. Clinical expression varies, so continued monitoring of these patients is important. Expert guidelines again recommend serial electrocardiography, transthoracic echocardiography, and clinical assessment every 5 years for adults.9
Recent data suggest that up to 40% of HCM cases are nonfamilial, ie, their inheritance is sporadic with no known family history and no sarcomeric gene mutation evident on screening.33,34 The clinical course in this subgroup seems to be more benign, with later clinical presentations (age > 40) and lower risk of major adverse cardiovascular events.
MANAGEMENT
Conservative management
Asymptomatic HCM can usually be managed with lifestyle modifications.
Avoiding high-risk physical activities is the most important modification. All HCM patients should be counseled on the risk of sudden cardiac death and advised against participating in competitive sports or intense physical activity.35 Aerobic exercise is preferable to isometric exercises such as weightlifting, which may prompt the Valsalva maneuver with worsening of left ventricular outflow tract obstruction leading to syncope. A recent study showed that moderate-intensity aerobic exercise can safely improve exercise capacity, which may ultimately improve functional status and quality of life.36
Avoiding dehydration and excessive alcohol intake are also important in maintaining adequate preload to prevent an increasing left ventricular outflow tract gradient, given the dynamic nature of the left ventricular outflow tract obstruction in HCM.
Medical management: Beta-blockers, then calcium channel blockers
Beta-blockers are the first-line therapy for symptomatic HCM related to left ventricular outflow tract obstruction. Their negative inotropic effect reduces the contractile force of the ventricle, effectively reducing the pressure gradient across the outflow tract. Reduced contractility also means that the overall myocardial workload is less, which ultimately translates to a reduced oxygen demand. With their negative chronotropic effect, beta-blockers lower the heart rate and thereby lengthen the diastolic filling phase, allowing for optimization of preload conditions to help prevent increasing the left ventricular outflow tract gradient.37,38
Beta-blockers can be titrated according to the patient’s symptoms and tolerance. Fatigue and loss of libido are among the most common side effects.
Nondihydropyridine calcium channel blockers can be a second-line therapy in patients who cannot tolerate beta-blockers. Several studies have shown improvement in surrogate outcomes such as estimated left ventricular mass and QRS amplitude on electrocardiography, but currently no available data show that these drugs improve symptoms.28,39,40 They should be avoided in those with severe left ventricular outflow tract obstruction (gradient ≥ 100 mm Hg), as they can lead to critical outflow tract obstruction owing to their peripheral vasodilatory effect.
Dihydropyridine calcium channel blockers should be avoided altogether, as they produce even more peripheral vasodilation and afterload reduction than nondihydropyridine calcium channel blockers.
Disopyramide, a class IA antiarrhythmic, has been shown to effectively reduce outflow gradients and relieve symptoms. However, in view of its adverse effects, it is a third-line therapy, given to those for whom beta-blockers and calcium channel blockers have failed. Its most worrisome adverse effect is QT prolongation, and the QT interval should therefore be closely monitored at the start of treatment. Anticholinergic effects are common and include dry eyes and mouth, urinary retention, and drowsiness.
Disopyramide is usually used in combination with beta-blockers for symptom control as a bridge to a planned invasive intervention.41
Use with caution
Any medication that causes afterload reduction, peripheral vasodilation, intravascular volume depletion, or positive inotropy can worsen the dynamic left ventricular outflow tract obstruction in a patient with HCM and should be avoided.
Angiotensin-converting enzyme (ACE) inhibitors, angiotensin II receptor blockers (ARBs), and nitrates must be used with extreme caution in these patients.
Diuretics. Even restrained use of diuretics can cause significant hemodynamic compromise in patients with obstructive physiology. Therefore, diuretics should be used sparingly in these patients.
Digoxin should not be used for managing atrial fibrillation in these patients, as its positive inotropic effect increases contractility and increases the left ventricular outflow tract gradient.
Norepinephrine and inotropic agents such as dobutamine and dopamine should be avoided for the same reason as digoxin. In patients with circulatory shock requiring vasopressor support, pure alpha-agonists such as phenylephrine are preferred, as they increase peripheral resistance without an inotropic effect.
Anticoagulation for atrial tachyarrhythmias
The risk of systemic thromboembolic events is significantly increased in HCM patients with atrial fibrillation or flutter, regardless of their estimated risk using conventional risk-stratification tools such as the CHADS2 score.42–44 In accordance with current American Heart Association and American College of Cardiology guidelines, we recommend anticoagulation therapy for all HCM patients with a history of atrial fibrillation or flutter. Warfarin is the preferred anticoagulant; direct oral anticoagulants can be considered, but there are currently no data on their use in HCM.9
Standard heart failure treatments
End-stage systolic heart failure is a consequence of HCM but affects only 3% to 4% of patients.45 While most randomized controlled trials of heart failure treatment have excluded HCM patients, current guidelines recommend the same evidence-based medical therapies used in other patients who have heart failure with reduced ejection fraction. This includes ACE inhibitors, ARBs, beta-blockers, and aldosterone antagonists if indicated.9,21
Heart transplant should be considered in patients with class III or IV New York Heart Association functional status despite optimization of their HCM treatment regimen. Heart transplant outcomes for HCM patients are comparable to outcomes for patients who receive a transplant for non-HCM cardiovascular disease.45,46
Septal reduction therapy
If medical therapy fails or is not tolerated in patients with severe symptoms, surgery can be considered for obstructive HCM.
Ventricular septal myectomy has been the long-standing gold standard of invasive therapy. Multiple studies have demonstrated long-term survival after myectomy to be equivalent to that in the general population and better than that of HCM patients who do not undergo this surgery.47–50 Factors that may be associated with better surgical outcomes include age younger than 50, left atrial size less than 46 mm, and resolution of atrial fibrillation during follow-up.51
Septal reduction therapy may also be considered in patients at high risk of sudden cardiac death based on a history of recurrent ventricular tachycardia or risk-stratification models as described above. Retrospective analyses have shown that surgical myectomy can markedly reduce the incidence of appropriate implantable cardioverter-defibrillator discharges and the risk of sudden cardiac death.52
Alcohol septal ablation is an alternative. This percutaneous procedure, first described in the mid-1990s, consists of injecting a small amount of alcohol into the artery supplying the septum to induce myocardial necrosis, ultimately leading to scarring and widening of the left ventricular outflow tract.53
Up to 50% of patients develop right bundle branch block after alcohol septal ablation, and the risk of complete heart block is highest in those with preexisting left bundle branch block. Nevertheless, studies have shown significant symptomatic improvement after alcohol septal ablation, with long-term survival comparable to that in the general population.53–56
Several meta-analyses compared alcohol septal ablation and septal myectomy and found that the rates of functional improvement and long-term mortality were similar.57–59 However, the less-invasive approach with alcohol septal ablation comes at the cost of a higher incidence of conduction abnormalities and higher left ventricular outflow tract gradients afterward. One meta-analysis found that alcohol septal ablation patients may have 5 times the risk of needing additional septal reduction therapy compared with their myectomy counterparts.
Current US guidelines recommend septal myectomy, performed at an experienced center, as the first-line interventional treatment, leaving alcohol septal ablation to be considered in those who have contraindications to myectomy.9 The treatment strategy should ultimately be individualized based on a patient’s comorbidities and personal preferences following informed consent.
A nationwide database study recently suggested that postmyectomy mortality rates may be as high as 5.9%,60 although earlier studies at high-volume centers showed much lower mortality rates (< 1%).50–52,61 This discrepancy highlights the critical role of expert centers in optimizing surgical management of these patients. Regardless of the approach, interventional therapies for HCM should be performed by a multidisciplinary team at a medical center able to handle the complexity of these cases.
Additional surgical procedures
A handful of other procedures may benefit specific patient subgroups.
Apical myectomy has been shown to improve functional status in patients with isolated apical hypertrophy by reducing left ventricular end-diastolic pressure and thereby allowing for improved diastolic filling.63
Mitral valve surgery may need to be considered at the time of myectomy in patients with degenerative valve disease. As in the general population, mitral valve repair is preferred to replacement if possible.
- Maron BJ. Hypertrophic cardiomyopathy: an important global disease. Am J Med 2004; 116(1):63–65. pmid:14706671
- Semsarian C, Ingles J, Maron MS, Maron BJ. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol 2015; 65(12):1249–1254. doi:10.1016/j.jacc.2015.01.019
- Maron BJ, Maron MS, Semsarian C. Genetics of hypertrophic cardiomyopathy after 20 years: clinical perspectives. J Am Coll Cardiol 2012; 60(8):705–715. doi:10.1016/j.jacc.2012.02.068
- Shirani J, Pick R, Roberts WC, Maron BJ. Morphology and significance of the left ventricular collagen network in young patients with hypertrophic cardiomyopathy and sudden cardiac death. J Am Coll Cardiol 2000; 35(1):36–44. pmid:10636256
- Sherrid MV, Chu CK, Delia E, Mogtader A, Dwyer EM Jr. An echocardiographic study of the fluid mechanics of obstruction in hypertrophic cardiomyopathy. J Am Coll Cardiol 1993; 22(3):816–825. pmid:8354817
- Ro R, Halpern D, Sahn DJ, et al. Vector flow mapping in obstructive hypertrophic cardiomyopathy to assess the relationship of early systolic left ventricular flow and the mitral valve. J Am Coll Cardiol 2014; 64(19):1984–1995. doi:10.1016/j.jacc.2014.04.090
- Kwon DH, Setser RM, Thamilarasan M, et al. Abnormal papillary muscle morphology is independently associated with increased left ventricular outflow tract obstruction in hypertrophic cardiomyopathy. Heart 2008; 94(10):1295–1301. doi:10.1136/hrt.2007.118018
- Patel P, Dhillon A, Popovic ZB, et al. Left ventricular outflow tract obstruction in hypertrophic cardiomyopathy patients without severe septal hypertrophy: implications of mitral valve and papillary muscle abnormalities assessed using cardiac magnetic resonance and echocardiography. Circ Cardiovasc Imaging 2015; 8(7):e003132. doi:10.1161/CIRCIMAGING.115.003132
- Gersh BJ, Maron BJ, Bonow RO, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Thorac Cardiovasc Surg 2011; 142(6):e153-e203. doi:10.1016/j.jtcvs.2011.10.020
- Elliott PM, Gimeno JR, Thaman R, et al. Historical trends in reported survival rates in patients with hypertrophic cardiomyopathy. Heart 2006; 92(6):785–791. doi:10.1136/hrt.2005.068577
- Maron MS, Olivotto I, Zenovich AG, et al. Hypertrophic cardiomyopathy is predominantly a disease of left ventricular outflow tract obstruction. Circulation 2006; 114(21):2232–2239. doi:10.1161/CIRCULATIONAHA.106.644682
- Ho CY, Carlsen C, Thune JJ, et al. Echocardiographic strain imaging to assess early and late consequences of sarcomere mutations in hypertrophic cardiomyopathy. Circ Cardiovasc Genet 2009; 2(4):314–321. doi:10.1161/CIRCGENETICS.109.862128
- Wasfy MM, Weiner RB. Differentiating the athlete’s heart from hypertrophic cardiomyopathy. Curr Opin Cardiol 2015; 30(5):500–505. doi:10.1097/HCO.0000000000000203
- Palka P, Lange A, Fleming AD, et al. Differences in myocardial velocity gradient measured throughout the cardiac cycle in patients with hypertrophic cardiomyopathy, athletes and patients with left ventricular hypertrophy due to hypertension. J Am Coll Cardiol 1997; 30(3):760–768. pmid:9283537
- Eriksson MJ, Sonnenberg B, Woo A, et al. Long-term outcome in patients with apical hypertrophic cardiomyopathy. J Am Coll Cardiol 2002; 39(4):638–645. pmid:11849863
- Rickers C, Wilke NM, Jerosch-Herold M, et al. Utility of cardiac magnetic resonance imaging in the diagnosis of hypertrophic cardiomyopathy. Circulation 2005; 112(6):855–861. doi:10.1161/CIRCULATIONAHA.104.507723
- Kwon DH, Setser RM, Popovic ZB, et al. Association of myocardial fibrosis, electrocardiography and ventricular tachyarrhythmia in hypertrophic cardiomyopathy: a delayed contrast enhanced MRI study. Int J Cardiovasc Imaging 2008; 24(6):617–625. doi:10.1007/s10554-008-9292-6
- Rubinshtein R, Glockner JF, Ommen SR, et al. Characteristics and clinical significance of late gadolinium enhancement by contrast-enhanced magnetic resonance imaging in patients with hypertrophic cardiomyopathy. Circ Heart Fail 2010; 3(1):51–58. doi:10.1161/CIRCHEARTFAILURE.109.854026
- O’Hanlon R, Grasso A, Roughton M, et al. Prognostic significance of myocardial fibrosis in hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 56(11):867–874. doi:10.1016/j.jacc.2010.05.010
- Bruder O, Wagner A, Jensen CJ, et al. Myocardial scar visualized by cardiovascular magnetic resonance imaging predicts major adverse events in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 56(11):875–887. doi:10.1016/j.jacc.2010.05.007
- Authors/Task Force members, Elliott PM, Anastasakis A, Borger MA, et al. 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J 2014; 35(39):2733–2779. doi:10.1093/eurheartj/ehu284
- Olivotto I, Maron BJ, Montereggi A, Mazzuoli F, Dolara A, Cecchi F. Prognostic value of systemic blood pressure response during exercise in a community-based patient population with hypertrophic cardiomyopathy. J Am Coll Cardiol 1999; 33(7):2044–2051. pmid:10362212
- Sadoul N, Prasad K, Elliott PM, Bannerjee S, Frenneaux MP, McKenna WJ. Prospective prognostic assessment of blood pressure response during exercise in patients with hypertrophic cardiomyopathy. Circulation 1997; 96(9):2987–2991. pmid:9386166
- Elliott PM, Poloniecki J, Dickie S, et al. Sudden death in hypertrophic cardiomyopathy: identification of high risk patients. J Am Coll Cardiol 2000; 36(7):2212–2218. pmid:11127463
- Masri A, Pierson LM, Smedira NG, et al. Predictors of long-term outcomes in patients with hypertrophic cardiomyopathy undergoing cardiopulmonary stress testing and echocardiography. Am Heart J 2015; 169(5):684–692.e1. doi:10.1016/j.ahj.2015.02.006
- Coats CJ, Rantell K, Bartnik A, et al. Cardiopulmonary exercise testing and prognosis in hypertrophic cardiomyopathy. Circ Heart Fail 2015; 8(6):1022–1031. doi:10.1161/CIRCHEARTFAILURE.114.002248
- Desai MY, Bhonsale A, Patel P, et al. Exercise echocardiography in asymptomatic HCM: exercise capacity, and not LV outflow tract gradient predicts long-term outcomes. JACC Cardiovasc Imaging 2014; 7(1):26–36. doi:10.1016/j.jcmg.2013.08.010
- Spirito P, Seidman CE, McKenna WJ, Maron BJ. The management of hypertrophic cardiomyopathy. N Engl J Med 1997; 336(11):775–785. doi:10.1056/NEJM199703133361107
- Wang W, Lian Z, Rowin EJ, Maron BJ, Maron MS, Link MS. Prognostic implications of nonsustained ventricular tachycardia in high-risk patients with hypertrophic cardiomyopathy. Circ Arrhythm Electrophysiol 2017; 10(3)e004604. doi:10.1161/CIRCEP.116.004604
- O’Mahony C, Jichi F, Pavlou M, et al. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM risk-SCD). Eur Heart J 2014; 35(30):2010–2020. doi:10.1093/eurheartj/eht439
- Maron BJ, Casey SA, Chan RH, Garberich RF, Rowin EJ, Maron MS. Independent assessment of the European Society of Cardiology sudden death risk model for hypertrophic cardiomyopathy. Am J Cardiol 2015; 116(5):757–764. doi:10.1016/j.amjcard.2015.05.047
- Jahnlová D, Tomašov P, Zemánek D, Veselka J. Transatlantic differences in assessment of risk of sudden cardiac death in patients with hypertrophic cardiomyopathy. Int J Cardiol 2015; 186:3–4. doi:10.1016/j.ijcard.2015.03.207
- Ingles J, Burns C, Bagnall RD, et al. Nonfamilial hypertrophic cardiomyopathy: prevalence, natural history, and clinical implications. Circ Cardiovasc Genet 2017; 10(2)e001620. doi:10.1161/CIRCGENETICS.116.001620
- Ko C, Arscott P, Concannon M, et al. Genetic testing impacts the utility of prospective familial screening in hypertrophic cardiomyopathy through identification of a nonfamilial subgroup. Genet Med 2017; 20(1):69–75. doi:10.1038/gim.2017.79
- Maron BJ, Chaitman BR, Ackerman MJ, et al. Recommendations for physical activity and recreational sports participation for young patients with genetic cardiovascular diseases. Circulation 2004; 109(22):2807–2816. doi:10.1161/01.CIR.0000128363.85581.E1
- Saberi S, Wheeler M, Bragg-Gresham J, et al. Effect of moderate-intensity exercise training on peak oxygen consumption in patients with hypertrophic cardiomyopathy: a randomized clinical trial. JAMA 2017; 317(13):1349–1357. doi:10.1001/jama.2017.2503
- Bourmayan C, Razavi A, Fournier C, et al. Effect of propranolol on left ventricular relaxation in hypertrophic cardiomyopathy: an echographic study. Am Heart J 1985; 109(6):1311–1316. pmid:4039882
- Spoladore R, Maron MS, D’Amato R, Camici PG, Olivotto I. Pharmacological treatment options for hypertrophic cardiomyopathy: high time for evidence. Eur Heart J 2012; 33(14):1724–1733. doi:10.1093/eurheartj/ehs150
- Choudhury L, Elliott P, Rimoldi O, et al. Transmural myocardial blood flow distribution in hypertrophic cardiomyopathy and effect of treatment. Basic Res Cardiol 1999; 94(1):49–59. pmid:10097830
- Kaltenbach M, Hopf R, Kober G, Bussmann WD, Keller M, Petersen Y. Treatment of hypertrophic obstructive cardiomyopathy with verapamil. Br Heart J 1979; 42(1):35–42. doi:10.1136/hrt.42.1.35
- Sherrid MV, Shetty A, Winson G, et al. Treatment of obstructive hypertrophic cardiomyopathy symptoms and gradient resistant to first-line therapy with beta-blockade or verapamil. Circ Heart Fail 2013; 6(4):694–702. doi:10.1161/CIRCHEARTFAILURE.112.000122
- Guttmann OP, Rahman MS, O’Mahony C, Anastasakis A, Elliott PM. Atrial fibrillation and thromboembolism in patients with hypertrophic cardiomyopathy: systematic review. Heart 2014; 100(6):465–472. doi:10.1136/heartjnl-2013-304276
- Olivotto I, Cecchi F, Casey SA, Dolara A, Traverse JH, Maron BJ. Impact of atrial fibrillation on the clinical course of hypertrophic cardiomyopathy. Circulation 2001; 104(21):2517–2524. pmid:11714644
- Maron BJ, Olivotto I, Spirito P, et al. Epidemiology of hypertrophic cardiomyopathy-related death: revisited in a large non-referral-based patient population. Circulation 2000; 102(8):858–864. pmid:10952953
- Harris KM, Spirito P, Maron MS, et al. Prevalence, clinical profile, and significance of left ventricular remodeling in the end-stage phase of hypertrophic cardiomyopathy. Circulation 2006; 114(3):216-225. doi:10.1161/CIRCULATIONAHA.105.583500
- Maron MS, Kalsmith BM, Udelson JE, Li W, DeNofrio D. Survival after cardiac transplantation in patients with hypertrophic cardiomyopathy. Circ Heart Fail 2010; 3(5):574–579. doi:10.1161/CIRCHEARTFAILURE.109.922872
- Smedira NG, Lytle BW, Lever HM, et al. Current effectiveness and risks of isolated septal myectomy for hypertrophic obstructive cardiomyopathy. Ann Thorac Surg 2008; 85(1):127–133. doi:10.1016/j.athoracsur.2007.07.063
- Robbins RC, Stinson EB. Long-term results of left ventricular myotomy and myectomy for obstructive hypertrophic cardiomyopathy. J Thorac Cardiovasc Surg 1996; 111(3):586–594. pmid:8601973
- Heric B, Lytle BW, Miller DP, Rosenkranz ER, Lever HM, Cosgrove DM. Surgical management of hypertrophic obstructive cardiomyopathy. Early and late results. J Thorac Cardiovasc Surg 1995; 110(1):195–208. pmid:7609544
- Ommen SR, Maron BJ, Olivotto I, et al. Long-term effects of surgical septal myectomy on survival in patients with obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol 2005; 46(3):470–476. doi:10.1016/j.jacc.2005.02.090
- Desai MY, Bhonsale A, Smedira NG, et al. Predictors of long-term outcomes in symptomatic hypertrophic obstructive cardiomyopathy patients undergoing surgical relief of left ventricular outflow tract obstruction. Circulation 2013; 128(3):209–216. doi:10.1161/CIRCULATIONAHA.112.000849
- McLeod CJ, Ommen SR, Ackerman MJ, et al. Surgical septal myectomy decreases the risk for appropriate implantable cardioverter defibrillator discharge in obstructive hypertrophic cardiomyopathy. Eur Heart J 2007; 28(21):2583–2588. doi:10.1093/eurheartj/ehm117
- Veselka J, Tomasov P, Zemanek D. Long-term effects of varying alcohol dosing in percutaneous septal ablation for obstructive hypertrophic cardiomyopathy: a randomized study with a follow-up up to 11 years. Can J Cardiol 2011; 27(6):763–767. doi:10.1016/j.cjca.2011.09.001
- Veselka J, Jensen MK, Liebregts M, et al. Low procedure-related mortality achieved with alcohol septal ablation in European patients. Int J Cardiol 2016; 209:194–195. doi:10.1016/j.ijcard.2016.02.077
- Veselka J, Krejci J, Tomašov P, Zemánek D. Long-term survival after alcohol septal ablation for hypertrophic obstructive cardiomyopathy: a comparison with general population. Eur Heart J 2014; 35(30):2040–2045. doi:10.1093/eurheartj/eht495
- Sorajja P, Ommen SR, Holmes DR Jr, et al. Survival after alcohol septal ablation for obstructive hypertrophic cardiomyopathy. Circulation 2012; 126(20):2374–2380. doi:10.1161/CIRCULATIONAHA.111.076257
- Agarwal S, Tuzcu EM, Desai MY, et al. Updated meta-analysis of septal alcohol ablation versus myectomy for hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 55(8):823–834. doi:10.1016/j.jacc.2009.09.047
- Leonardi RA, Kransdorf EP, Simel DL, Wang A. Meta-analyses of septal reduction therapies for obstructive hypertrophic cardiomyopathy: comparative rates of overall mortality and sudden cardiac death after treatment. Circ Cardiovasc Interv 2010; 3(2):97–104. doi:10.1161/CIRCINTERVENTIONS.109.916676
- Liebregts M, Vriesendorp PA, Mahmoodi BK, Schinkel AF, Michels M, ten Berg JM. A systematic review and meta-analysis of long-term outcomes after septal reduction therapy in patients with hypertrophic cardiomyopathy. JACC Heart Fail 2015; 3(11):896–905. doi:10.1016/j.jchf.2015.06.011
- Panaich SS, Badheka AO, Chothani A, et al. Results of ventricular septal myectomy and hypertrophic cardiomyopathy (from Nationwide Inpatient Sample [1998-2010]). Am J Cardiol 2014; 114(9):1390–1395. doi:10.1016/j.amjcard.2014.07.075
- Maron BJ, Dearani JA, Ommen SR, et al. Low operative mortality achieved with surgical septal myectomy: role of dedicated hypertrophic cardiomyopathy centers in the management of dynamic subaortic obstruction. J Am Coll Cardiol 2015; 66(11):1307–1308. doi:10.1016/j.jacc.2015.06.1333
- Kwon DH, Smedira NG, Thamilarasan M, Lytle BW, Lever H, Desai MY. Characteristics and surgical outcomes of symptomatic patients with hypertrophic cardiomyopathy with abnormal papillary muscle morphology undergoing papillary muscle reorientation. J Thorac Cardiovasc Surg 2010; 140(2):317–324. doi:10.1016/j.jtcvs.2009.10.045
- Schaff HV, Brown ML, Dearani JA, et al. Apical myectomy: a new surgical technique for management of severely symptomatic patients with apical hypertrophic cardiomyopathy. J Thorac Cardiovasc Surg 2010; 139(3):634–640. doi:10.1016/j.jtcvs.2009.07.079
- Maron BJ. Hypertrophic cardiomyopathy: an important global disease. Am J Med 2004; 116(1):63–65. pmid:14706671
- Semsarian C, Ingles J, Maron MS, Maron BJ. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol 2015; 65(12):1249–1254. doi:10.1016/j.jacc.2015.01.019
- Maron BJ, Maron MS, Semsarian C. Genetics of hypertrophic cardiomyopathy after 20 years: clinical perspectives. J Am Coll Cardiol 2012; 60(8):705–715. doi:10.1016/j.jacc.2012.02.068
- Shirani J, Pick R, Roberts WC, Maron BJ. Morphology and significance of the left ventricular collagen network in young patients with hypertrophic cardiomyopathy and sudden cardiac death. J Am Coll Cardiol 2000; 35(1):36–44. pmid:10636256
- Sherrid MV, Chu CK, Delia E, Mogtader A, Dwyer EM Jr. An echocardiographic study of the fluid mechanics of obstruction in hypertrophic cardiomyopathy. J Am Coll Cardiol 1993; 22(3):816–825. pmid:8354817
- Ro R, Halpern D, Sahn DJ, et al. Vector flow mapping in obstructive hypertrophic cardiomyopathy to assess the relationship of early systolic left ventricular flow and the mitral valve. J Am Coll Cardiol 2014; 64(19):1984–1995. doi:10.1016/j.jacc.2014.04.090
- Kwon DH, Setser RM, Thamilarasan M, et al. Abnormal papillary muscle morphology is independently associated with increased left ventricular outflow tract obstruction in hypertrophic cardiomyopathy. Heart 2008; 94(10):1295–1301. doi:10.1136/hrt.2007.118018
- Patel P, Dhillon A, Popovic ZB, et al. Left ventricular outflow tract obstruction in hypertrophic cardiomyopathy patients without severe septal hypertrophy: implications of mitral valve and papillary muscle abnormalities assessed using cardiac magnetic resonance and echocardiography. Circ Cardiovasc Imaging 2015; 8(7):e003132. doi:10.1161/CIRCIMAGING.115.003132
- Gersh BJ, Maron BJ, Bonow RO, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Thorac Cardiovasc Surg 2011; 142(6):e153-e203. doi:10.1016/j.jtcvs.2011.10.020
- Elliott PM, Gimeno JR, Thaman R, et al. Historical trends in reported survival rates in patients with hypertrophic cardiomyopathy. Heart 2006; 92(6):785–791. doi:10.1136/hrt.2005.068577
- Maron MS, Olivotto I, Zenovich AG, et al. Hypertrophic cardiomyopathy is predominantly a disease of left ventricular outflow tract obstruction. Circulation 2006; 114(21):2232–2239. doi:10.1161/CIRCULATIONAHA.106.644682
- Ho CY, Carlsen C, Thune JJ, et al. Echocardiographic strain imaging to assess early and late consequences of sarcomere mutations in hypertrophic cardiomyopathy. Circ Cardiovasc Genet 2009; 2(4):314–321. doi:10.1161/CIRCGENETICS.109.862128
- Wasfy MM, Weiner RB. Differentiating the athlete’s heart from hypertrophic cardiomyopathy. Curr Opin Cardiol 2015; 30(5):500–505. doi:10.1097/HCO.0000000000000203
- Palka P, Lange A, Fleming AD, et al. Differences in myocardial velocity gradient measured throughout the cardiac cycle in patients with hypertrophic cardiomyopathy, athletes and patients with left ventricular hypertrophy due to hypertension. J Am Coll Cardiol 1997; 30(3):760–768. pmid:9283537
- Eriksson MJ, Sonnenberg B, Woo A, et al. Long-term outcome in patients with apical hypertrophic cardiomyopathy. J Am Coll Cardiol 2002; 39(4):638–645. pmid:11849863
- Rickers C, Wilke NM, Jerosch-Herold M, et al. Utility of cardiac magnetic resonance imaging in the diagnosis of hypertrophic cardiomyopathy. Circulation 2005; 112(6):855–861. doi:10.1161/CIRCULATIONAHA.104.507723
- Kwon DH, Setser RM, Popovic ZB, et al. Association of myocardial fibrosis, electrocardiography and ventricular tachyarrhythmia in hypertrophic cardiomyopathy: a delayed contrast enhanced MRI study. Int J Cardiovasc Imaging 2008; 24(6):617–625. doi:10.1007/s10554-008-9292-6
- Rubinshtein R, Glockner JF, Ommen SR, et al. Characteristics and clinical significance of late gadolinium enhancement by contrast-enhanced magnetic resonance imaging in patients with hypertrophic cardiomyopathy. Circ Heart Fail 2010; 3(1):51–58. doi:10.1161/CIRCHEARTFAILURE.109.854026
- O’Hanlon R, Grasso A, Roughton M, et al. Prognostic significance of myocardial fibrosis in hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 56(11):867–874. doi:10.1016/j.jacc.2010.05.010
- Bruder O, Wagner A, Jensen CJ, et al. Myocardial scar visualized by cardiovascular magnetic resonance imaging predicts major adverse events in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 56(11):875–887. doi:10.1016/j.jacc.2010.05.007
- Authors/Task Force members, Elliott PM, Anastasakis A, Borger MA, et al. 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J 2014; 35(39):2733–2779. doi:10.1093/eurheartj/ehu284
- Olivotto I, Maron BJ, Montereggi A, Mazzuoli F, Dolara A, Cecchi F. Prognostic value of systemic blood pressure response during exercise in a community-based patient population with hypertrophic cardiomyopathy. J Am Coll Cardiol 1999; 33(7):2044–2051. pmid:10362212
- Sadoul N, Prasad K, Elliott PM, Bannerjee S, Frenneaux MP, McKenna WJ. Prospective prognostic assessment of blood pressure response during exercise in patients with hypertrophic cardiomyopathy. Circulation 1997; 96(9):2987–2991. pmid:9386166
- Elliott PM, Poloniecki J, Dickie S, et al. Sudden death in hypertrophic cardiomyopathy: identification of high risk patients. J Am Coll Cardiol 2000; 36(7):2212–2218. pmid:11127463
- Masri A, Pierson LM, Smedira NG, et al. Predictors of long-term outcomes in patients with hypertrophic cardiomyopathy undergoing cardiopulmonary stress testing and echocardiography. Am Heart J 2015; 169(5):684–692.e1. doi:10.1016/j.ahj.2015.02.006
- Coats CJ, Rantell K, Bartnik A, et al. Cardiopulmonary exercise testing and prognosis in hypertrophic cardiomyopathy. Circ Heart Fail 2015; 8(6):1022–1031. doi:10.1161/CIRCHEARTFAILURE.114.002248
- Desai MY, Bhonsale A, Patel P, et al. Exercise echocardiography in asymptomatic HCM: exercise capacity, and not LV outflow tract gradient predicts long-term outcomes. JACC Cardiovasc Imaging 2014; 7(1):26–36. doi:10.1016/j.jcmg.2013.08.010
- Spirito P, Seidman CE, McKenna WJ, Maron BJ. The management of hypertrophic cardiomyopathy. N Engl J Med 1997; 336(11):775–785. doi:10.1056/NEJM199703133361107
- Wang W, Lian Z, Rowin EJ, Maron BJ, Maron MS, Link MS. Prognostic implications of nonsustained ventricular tachycardia in high-risk patients with hypertrophic cardiomyopathy. Circ Arrhythm Electrophysiol 2017; 10(3)e004604. doi:10.1161/CIRCEP.116.004604
- O’Mahony C, Jichi F, Pavlou M, et al. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM risk-SCD). Eur Heart J 2014; 35(30):2010–2020. doi:10.1093/eurheartj/eht439
- Maron BJ, Casey SA, Chan RH, Garberich RF, Rowin EJ, Maron MS. Independent assessment of the European Society of Cardiology sudden death risk model for hypertrophic cardiomyopathy. Am J Cardiol 2015; 116(5):757–764. doi:10.1016/j.amjcard.2015.05.047
- Jahnlová D, Tomašov P, Zemánek D, Veselka J. Transatlantic differences in assessment of risk of sudden cardiac death in patients with hypertrophic cardiomyopathy. Int J Cardiol 2015; 186:3–4. doi:10.1016/j.ijcard.2015.03.207
- Ingles J, Burns C, Bagnall RD, et al. Nonfamilial hypertrophic cardiomyopathy: prevalence, natural history, and clinical implications. Circ Cardiovasc Genet 2017; 10(2)e001620. doi:10.1161/CIRCGENETICS.116.001620
- Ko C, Arscott P, Concannon M, et al. Genetic testing impacts the utility of prospective familial screening in hypertrophic cardiomyopathy through identification of a nonfamilial subgroup. Genet Med 2017; 20(1):69–75. doi:10.1038/gim.2017.79
- Maron BJ, Chaitman BR, Ackerman MJ, et al. Recommendations for physical activity and recreational sports participation for young patients with genetic cardiovascular diseases. Circulation 2004; 109(22):2807–2816. doi:10.1161/01.CIR.0000128363.85581.E1
- Saberi S, Wheeler M, Bragg-Gresham J, et al. Effect of moderate-intensity exercise training on peak oxygen consumption in patients with hypertrophic cardiomyopathy: a randomized clinical trial. JAMA 2017; 317(13):1349–1357. doi:10.1001/jama.2017.2503
- Bourmayan C, Razavi A, Fournier C, et al. Effect of propranolol on left ventricular relaxation in hypertrophic cardiomyopathy: an echographic study. Am Heart J 1985; 109(6):1311–1316. pmid:4039882
- Spoladore R, Maron MS, D’Amato R, Camici PG, Olivotto I. Pharmacological treatment options for hypertrophic cardiomyopathy: high time for evidence. Eur Heart J 2012; 33(14):1724–1733. doi:10.1093/eurheartj/ehs150
- Choudhury L, Elliott P, Rimoldi O, et al. Transmural myocardial blood flow distribution in hypertrophic cardiomyopathy and effect of treatment. Basic Res Cardiol 1999; 94(1):49–59. pmid:10097830
- Kaltenbach M, Hopf R, Kober G, Bussmann WD, Keller M, Petersen Y. Treatment of hypertrophic obstructive cardiomyopathy with verapamil. Br Heart J 1979; 42(1):35–42. doi:10.1136/hrt.42.1.35
- Sherrid MV, Shetty A, Winson G, et al. Treatment of obstructive hypertrophic cardiomyopathy symptoms and gradient resistant to first-line therapy with beta-blockade or verapamil. Circ Heart Fail 2013; 6(4):694–702. doi:10.1161/CIRCHEARTFAILURE.112.000122
- Guttmann OP, Rahman MS, O’Mahony C, Anastasakis A, Elliott PM. Atrial fibrillation and thromboembolism in patients with hypertrophic cardiomyopathy: systematic review. Heart 2014; 100(6):465–472. doi:10.1136/heartjnl-2013-304276
- Olivotto I, Cecchi F, Casey SA, Dolara A, Traverse JH, Maron BJ. Impact of atrial fibrillation on the clinical course of hypertrophic cardiomyopathy. Circulation 2001; 104(21):2517–2524. pmid:11714644
- Maron BJ, Olivotto I, Spirito P, et al. Epidemiology of hypertrophic cardiomyopathy-related death: revisited in a large non-referral-based patient population. Circulation 2000; 102(8):858–864. pmid:10952953
- Harris KM, Spirito P, Maron MS, et al. Prevalence, clinical profile, and significance of left ventricular remodeling in the end-stage phase of hypertrophic cardiomyopathy. Circulation 2006; 114(3):216-225. doi:10.1161/CIRCULATIONAHA.105.583500
- Maron MS, Kalsmith BM, Udelson JE, Li W, DeNofrio D. Survival after cardiac transplantation in patients with hypertrophic cardiomyopathy. Circ Heart Fail 2010; 3(5):574–579. doi:10.1161/CIRCHEARTFAILURE.109.922872
- Smedira NG, Lytle BW, Lever HM, et al. Current effectiveness and risks of isolated septal myectomy for hypertrophic obstructive cardiomyopathy. Ann Thorac Surg 2008; 85(1):127–133. doi:10.1016/j.athoracsur.2007.07.063
- Robbins RC, Stinson EB. Long-term results of left ventricular myotomy and myectomy for obstructive hypertrophic cardiomyopathy. J Thorac Cardiovasc Surg 1996; 111(3):586–594. pmid:8601973
- Heric B, Lytle BW, Miller DP, Rosenkranz ER, Lever HM, Cosgrove DM. Surgical management of hypertrophic obstructive cardiomyopathy. Early and late results. J Thorac Cardiovasc Surg 1995; 110(1):195–208. pmid:7609544
- Ommen SR, Maron BJ, Olivotto I, et al. Long-term effects of surgical septal myectomy on survival in patients with obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol 2005; 46(3):470–476. doi:10.1016/j.jacc.2005.02.090
- Desai MY, Bhonsale A, Smedira NG, et al. Predictors of long-term outcomes in symptomatic hypertrophic obstructive cardiomyopathy patients undergoing surgical relief of left ventricular outflow tract obstruction. Circulation 2013; 128(3):209–216. doi:10.1161/CIRCULATIONAHA.112.000849
- McLeod CJ, Ommen SR, Ackerman MJ, et al. Surgical septal myectomy decreases the risk for appropriate implantable cardioverter defibrillator discharge in obstructive hypertrophic cardiomyopathy. Eur Heart J 2007; 28(21):2583–2588. doi:10.1093/eurheartj/ehm117
- Veselka J, Tomasov P, Zemanek D. Long-term effects of varying alcohol dosing in percutaneous septal ablation for obstructive hypertrophic cardiomyopathy: a randomized study with a follow-up up to 11 years. Can J Cardiol 2011; 27(6):763–767. doi:10.1016/j.cjca.2011.09.001
- Veselka J, Jensen MK, Liebregts M, et al. Low procedure-related mortality achieved with alcohol septal ablation in European patients. Int J Cardiol 2016; 209:194–195. doi:10.1016/j.ijcard.2016.02.077
- Veselka J, Krejci J, Tomašov P, Zemánek D. Long-term survival after alcohol septal ablation for hypertrophic obstructive cardiomyopathy: a comparison with general population. Eur Heart J 2014; 35(30):2040–2045. doi:10.1093/eurheartj/eht495
- Sorajja P, Ommen SR, Holmes DR Jr, et al. Survival after alcohol septal ablation for obstructive hypertrophic cardiomyopathy. Circulation 2012; 126(20):2374–2380. doi:10.1161/CIRCULATIONAHA.111.076257
- Agarwal S, Tuzcu EM, Desai MY, et al. Updated meta-analysis of septal alcohol ablation versus myectomy for hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 55(8):823–834. doi:10.1016/j.jacc.2009.09.047
- Leonardi RA, Kransdorf EP, Simel DL, Wang A. Meta-analyses of septal reduction therapies for obstructive hypertrophic cardiomyopathy: comparative rates of overall mortality and sudden cardiac death after treatment. Circ Cardiovasc Interv 2010; 3(2):97–104. doi:10.1161/CIRCINTERVENTIONS.109.916676
- Liebregts M, Vriesendorp PA, Mahmoodi BK, Schinkel AF, Michels M, ten Berg JM. A systematic review and meta-analysis of long-term outcomes after septal reduction therapy in patients with hypertrophic cardiomyopathy. JACC Heart Fail 2015; 3(11):896–905. doi:10.1016/j.jchf.2015.06.011
- Panaich SS, Badheka AO, Chothani A, et al. Results of ventricular septal myectomy and hypertrophic cardiomyopathy (from Nationwide Inpatient Sample [1998-2010]). Am J Cardiol 2014; 114(9):1390–1395. doi:10.1016/j.amjcard.2014.07.075
- Maron BJ, Dearani JA, Ommen SR, et al. Low operative mortality achieved with surgical septal myectomy: role of dedicated hypertrophic cardiomyopathy centers in the management of dynamic subaortic obstruction. J Am Coll Cardiol 2015; 66(11):1307–1308. doi:10.1016/j.jacc.2015.06.1333
- Kwon DH, Smedira NG, Thamilarasan M, Lytle BW, Lever H, Desai MY. Characteristics and surgical outcomes of symptomatic patients with hypertrophic cardiomyopathy with abnormal papillary muscle morphology undergoing papillary muscle reorientation. J Thorac Cardiovasc Surg 2010; 140(2):317–324. doi:10.1016/j.jtcvs.2009.10.045
- Schaff HV, Brown ML, Dearani JA, et al. Apical myectomy: a new surgical technique for management of severely symptomatic patients with apical hypertrophic cardiomyopathy. J Thorac Cardiovasc Surg 2010; 139(3):634–640. doi:10.1016/j.jtcvs.2009.07.079
KEY POINTS
- Obstruction of the left ventricular outflow tract is a key pathophysiologic mechanism in HCM.
- Because most of the genetic variants that contribute to HCM are autosomal dominant, genetic counseling and testing are suggested for patients and their first-degree relatives.
- Transthoracic echocardiography is the first-line imaging test, followed by magnetic resonance imaging.
- Beta-blockers are the first-line drugs for treating symptoms of HCM.
- An implantable cardioverter-defibrillator can be considered for patients at risk of sudden cardiac death.
- When medical therapy fails or is not tolerated in patients with severe symptoms of obstructive HCM, surgery to reduce the size of the ventricular septum can be considered. Alcohol septal ablation is an alternative.

Idiopathic pulmonary fibrosis: What primary care physicians need to know
Idiopathic pulmonary fibrosis (IPF) is a devastating and fatal lung disease that generally affects older adults. It is characterized by a radiographic and histopathologic pattern of usual interstitial pneumonia (UIP) that has no other known etiology.
Accurate diagnosis of IPF is crucial. We recommend early referral to a center specializing in interstitial lung disease to confirm the diagnosis, start appropriate therapy, advise the patient on prognosis and enrollment in disease registries and clinical trials, and determine candidacy for lung transplant.
Primary care physicians are uniquely positioned to encounter patients with IPF, whether because of a patient complaint or as an incidental finding on computed tomography. The goal of this article is to delineate the features of IPF so that it may be recognized early and thus expedite referral to a center with expertise in interstitial lung disease for a thorough evaluation and appropriate management.
WHAT IS IDIOPATHIC PULMONARY FIBROSIS?

MORE COMMON THAN ONCE THOUGHT
The true incidence and prevalence of IPF are difficult to assess. IPF is generally considered a rare disease, but it is more common than once thought. In 2011, Raghu et al3 estimated the prevalence in Medicare beneficiaries to be 495 cases per 100,000. Based on this estimate and the current US population, up to 160,000 Americans could have IPF.4 Raghu et al also showed that IPF more often affects adults over age 65, which suggests that as the US population ages, the incidence of IPF may rise. Studies have also reported an increased incidence of IPF worldwide.5
Further, with the rising use of low-dose computed tomography to screen for lung cancer, more incidental cases of IPF will likely be found.6–8
Older data showed a lag from symptom onset to accurate diagnosis of 1 to 2 years.9 A more recent study found a lag in referral of patients with IPF to tertiary care centers, and this delay was associated with a higher rate of death independent of disease severity.10
TYPICALLY PROGRESSIVE, OFTEN FATAL
IPF is typically progressive and limited to the lungs, and it portends a poor prognosis. The median survival is commonly cited as 2 to 5 years from diagnosis, although this is based on older observations that may not reflect current best practice and newer therapies. More recent studies suggest higher survival rates if patients have preserved lung function.11
As the name indicates, the etiology of IPF is unknown, but studies have indicated genetic underpinnings in a notable proportion of cases.12 Regardless of the cause, the pathogenesis and progression of IPF are thought to be the result of an abnormal and persistent wound-repair response. The progressive deposition of scar tissue disrupts normal lung architecture and function, eventually causing clinical disease.13
SYMPTOMS AND KEY FEATURES
Patients with IPF typically present with the insidious onset of dyspnea on exertion, with or without chronic cough. Risk factors include male sex, increasing age, and a history of smoking. Patients with undiagnosed IPF who present with dyspnea and a history of smoking are often treated empirically for chronic obstructive pulmonary disease (COPD).
Rales are a common finding on auscultation in IPF, and this can lead to an exhaustive cardiac evaluation and empiric treatment for heart failure. Digital clubbing is also relatively common.14 Hypoxemia with exertion is another common feature that also often correlates with disease severity and prognosis. Resting hypoxemia is more common in advanced disease.
On spirometry, patients with IPF typically demonstrate restrictive physiology, suggested by a normal or elevated ratio of the forced expiratory volume in 1 second to the forced vital capacity (FEV1/FVC) (> 70% predicted or above the lower limit of normal) combined with a lower than normal FVC. Restrictive physiology is definitively demonstrated by a decreased total lung capacity (< 80% predicted or below the lower limit of normal) on plethysmography. Impaired gas exchange, manifested by a decreased diffusing capacity of the lungs for carbon monoxide (DLCO) on pulmonary function testing, is also common. Because pulmonary perfusion is higher in the lung bases, where IPF is also predominant, the DLCO is often reduced to a greater extent than the FVC.
PROGNOSTIC INDICATORS

Clinicians typically view IPF as a relentless and progressive process, but its course is variable and can be uncertain in an individual patient (Figure 1).15,16 Nevertheless, over time, most patients have a decline in lung function leading to respiratory failure. Respiratory failure, often preceded by a subacute deterioration (over weeks to months) or an acute deterioration (< 4 weeks), is the most common cause of death, but comorbid diseases such as lung cancer, infection, and heart failure are also common causes of death in these patients.17,18
Predictors of mortality include worsening FVC, DLCO, symptoms, and physiologic impairment, manifested by a decline in the 6-minute walking test or worsening exertional hypoxemia.19–22 Other common comorbidities linked with impaired quality of life and poor prognosis include obstructive sleep apnea, gastroesophageal reflux disease, and depression.16,23 Retrospective studies suggest that most IPF patients die 2 to 5 years after symptom onset. With the lag from symptom onset to final diagnosis, the average life expectancy is as little as 2 years from the time of diagnosis.9,18,24,25
Two staging systems have been developed to predict short-term and long-term mortality risk based on sex, age, and physiologic parameters.23,24 The GAP (gender, age, physiology) index provides an estimate of the risk of death for a cohort of patients: a score of 0 to 8 is calculated, and the score is then categorized as stage I, II, or III. Each stage is associated with 1-, 2-, and 3-year mortality rates, with stage III having the highest rates. The GAP calculator (www.acponline.org/journals/annals/extras/gap) provides an estimate of the risk of death for an individual patient. The application of these tools for the management of IPF is evolving; however, they may be helpful for counseling patients about disease prognosis.
CLUES TO DIAGNOSIS
Histologic patterns
")
UIP on histologic study is also seen in fibrotic lung diseases other than IPF, including connective tissue disease-associated interstitial lung disease, inhalational or occupational interstitial lung disease, and chronic hypersensitivity pneumonitis.26–29 Consequently, the diagnosis of IPF requires exclusion of other known causes of UIP.
According to the 2011 guidelines,16 the histology of interstitial lung disease can be categorized as definite UIP, probable UIP, or possible UIP, or as an atypical pattern suggesting another diagnosis. If no definite cause of the interstitial lung abnormality is found, the level of certainty of the histopathologic pattern of UIP helps formulate the clinical diagnosis and management plan.
Clues on computed tomography
The UIP nomenclature also describes patterns on high-resolution computed tomography (HRCT). HRCT is done without contrast and produces thin-sliced images (usually < 1.5 mm) in inspiratory, expiratory, and prone views; this allows detection of air trapping, which may indicate an airway-centric alternative diagnosis.
On HRCT, UIP appears as reticular opacities, often with traction bronchiectasis or bronchiolectasis, usually with a basilar and peripheral predominance. Honeycombing is a key feature and appears as clustered cystic spaces with well-defined walls in the periphery of the lung parenchyma. Ground-glass opacities are not a prominent feature of UIP, and although they do not exclude a UIP pattern, they should spur consideration of other diagnoses.16 Reactive mediastinal and hilar lymphadenopathy is another common feature of UIP.

When evaluating results of HRCT for UIP, the radiologist categorizes the pattern as definite UIP, possible UIP, or inconsistent. The definite pattern meets all the above features and has none of the features suggesting an alternative diagnosis (Figure 3). The possible pattern includes all the above features with the exception of honeycombing. If the predominant features on HRCT include any atypical finding listed above, then the study is considered inconsistent with UIP. If the pattern on HRCT is considered definite, evaluation of pathology is not necessary. If the pattern is categorized as possible or is inconsistent, then surgical lung biopsy-confirmed UIP is necessary for the definitive diagnosis of IPF.
However, evidence is emerging that in the correct clinical scenario, possible UIP behaves similarly to definite UIP and may be sufficient to make the clinical diagnosis of IPF even without surgical biopsy confirmation.30
A DIAGNOSTIC ALGORITHM FOR IPF
Given the multitude of interstitial lung diseases, their complexities, and the lack of a gold standard definitive diagnostic test, the diagnosis of IPF can be difficult, requiring the integration of clinical, radiologic, and, if necessary, pathologic findings.
")
Demographic features
The patient’s demographic features and risk factors dictate the initial clinical suspicion of IPF compared with other interstitial lung diseases. The incidence of IPF increases with age, and IPF is more common in men. A history of smoking is another risk factor.31 A 45-year-old never-smoker is much less likely to have IPF than a 70-year-old former smoker, and a 70-year-old man is more likely to have IPF than a woman of the same age. Thus, the finding of interstitial lung disease in a patient with a demographic profile that is not typical (ie, a younger woman who never smoked) should prompt an exhaustive investigation for another diagnosis such as hypersensitivity pneumonitis or connective tissue disease.
Key elements of the history
After considering the demographic profile and risk factors, the next step in the evaluation is a thorough and accurate medical history. This should include assessment of the severity of dyspnea and cough, signs and symptoms of connective tissue disease (eg, arthralgias, sicca symptoms, Raynaud phenomenon, difficulty swallowing), and gastroesophageal reflux disease, which can be associated with connective tissue disease and, independently, with IPF.
It is also important to identify any environmental exposures that suggest pneumoconiosis or chronic hypersensitivity pneumonitis. The most common risk factors for hypersensitivity pneumonitis are birds and bird feathers, molds, fungi, hot tub use, and some industrial chemicals.32
A medication history is important. Many medications are associated with interstitial lung disease, but amiodarone, bleomycin, methotrexate, and nitrofurantoin are among the common offenders.33
A thorough family history is necessary, as there are familial forms of IPF.
Focus of the physical examination
The physical examination must include careful auscultation for rales. While rales are not specific for IPF, they are the most common pulmonary abnormality. Detailed skin, musculoskeletal, and cardiovascular examinations are also important to evaluate for rheumatologic signs, clubbing, or evidence of heart failure or pulmonary hypertension.
Laboratory tests
Laboratory testing should include a serologic autoantibody panel to evaluate for connective tissue diseases that can manifest as interstitial lung disease, including rheumatoid arthritis, dermatopolymyositis, scleroderma, Sjögren syndrome, and undifferentiated or mixed connective tissue disease. Typical preliminary laboratory tests are antinuclear antibody, rheumatoid factor, erythrocyte sedimentation rate, and C-reactive protein. Others may include anticyclic citrullinated peptide (anti-CCP), anti-Scl-70, anti-RNP, anti-SS-A, anti-SS-B, and anti-Jo-1.16 The breadth of the panel should depend on patient demographics and findings in the history or physical examination that increase or decrease the likelihood of a connective tissue disease.
Lung function testing
Assessing the patient’s pulmonary physiology should include spirometry, DLCO, and body plethysmography (lung volumes). In most cases, IPF manifests with restrictive physiology. Once restrictive physiology is confirmed with a low total lung capacity, FVC testing can be used as a longitudinal surrogate, as it is less expensive and easier for the patient to perform. In general, a lower total lung capacity or FVC indicates more severe impairment.
The DLCO serves as another marker of severity but is less reliable due to baseline variability and difficulties performing the maneuver.
A 6-minute walk test is another crucial physiologic assessment tool that can quantify exertional hypoxemia and functional status (ie, distance walked), and can assist in prognosis.
Imaging
Most patients undergo chest radiography in the workup for undiagnosed dyspnea. However, chest radiography is not adequate to formulate an accurate diagnosis in suspected interstitial lung disease, and a normal radiograph cannot exclude changes that might reflect early phases of the disease. As the disease progresses, the plain radiograph can show reticulonodular opacities and honeycombing in the peripheral and lower lung zones (Figure 3).34
The decision whether to order HRCT in the workup for a patient who has dyspnea and a normal chest radiograph is challenging. We recommend cross-sectional imaging when physiologic testing shows restriction or low DLCO, or when there is a high index of suspicion for underlying lung disease as the cause of symptoms.
Expert consultation can aid with this decision, especially when the underlying cause of dyspnea remains unclear after initial studies have been completed. Otherwise, HRCT is an essential test in the evaluation of interstitial lung disease.
Bronchoscopy’s role controversial
If the pattern on HRCT is nondiagnostic, then surgical biopsy is necessary, and the diagnosis of IPF requires a histologic pattern of UIP as described above.16,35
Although bronchoscopy can be valuable if an alternative diagnosis such as sarcoidosis or chronic hypersensitivity pneumonitis is suspected, the role of bronchoscopic biopsy in the workup of IPF is controversial. The patchy nature of UIP does not lend itself to the relatively small biopsy samples obtained through bronchoscopy.36,37
Surgical biopsy options
The favored biopsy approach is surgical, using either an open or a video-assisted thoracoscopic technique. The latter is preferred as it is less invasive, requires a shorter length of hospital stay, and allows a faster recovery.38 Bronchoscopic cryobiopsy, currently under investigation, is a potentially valuable tool whose role in diagnosing IPF is evolving.
Frequently, neither HRCT nor surgical lung biopsy demonstrates UIP, making the definitive diagnosis of IPF difficult. Moreover, some patients with nondiagnostic HRCT results are unable to tolerate surgical lung biopsy because of severely impaired lung function or other comorbidities.
The role of multidisciplinary discussions
When surgical lung biopsy is not possible, current practice at leading centers uses a multidisciplinary approach to allow for a confident diagnosis.30,39 Discussions take place between pulmonologists, pathologists, radiologists, and other specialists to collectively consider all aspects of a case before rendering a consensus opinion on the diagnosis and the management plan. If the discussion leads to a consensus diagnosis of IPF, then the patient’s clinician can move forward with treatment options. If not, the group can recommend further workup or alternative diagnoses and treatment regimens. The multidisciplinary group is also well positioned to consider the relative risks and benefits of moving forward with surgical lung biopsy for individual patients.
This approach illustrates the importance of referring these patients to centers of excellence in diagnosing and managing complex cases of interstitial lung disease, including IPF.40
TREATMENT OF IPF
Antifibrotic therapy
Antifibrotic therapy is a choice between pirfenidone and nintedanib.
Pirfenidone, which has an undefined molecular target, was approved based on the results of 3 trials.41,42 Pooled analyses from these trials showed a reduction in the decline from baseline in FVC percent predicted and improved progression-free survival.43 Pooled and meta-analyses of pirfenidone clinical trials have shown a mortality benefit, although no individual study has shown such an effect on mortality rates.44
The major adverse effects of pirfenidone are gastrointestinal distress and photosensitivity rash.
Nintedanib is a triple tyrosine kinase inhibitor that broadly targets fibroblast growth factor, vascular endothelial growth factor, and platelet-derived growth factor receptors. Combined analysis of 2 concurrent trials45 showed that nintedanib reduced the decline in FVC, similarly to pirfenidone. The major adverse event associated with nintedanib was diarrhea. Since it inhibits vascular endothelial growth factor, there is a risk of hematologic complications such as bleeding or clotting events.
Because pirfenidone and nintedanib can increase aminotransferase levels, regular monitoring is recommended.
To date, no trial has compared pirfenidone and nintedanib in terms of their efficacy and tolerability. Therefore, the choice of agent is based on the patient’s preference after a discussion of potential risks and expected benefits, a review of each drug’s side effects, and consideration of comorbid conditions and physician experience.
Patients need to understand that these drugs slow the rate of decline in FVC but have not been shown to improve symptoms or functional status.
Corticosteroids are not routine
Corticosteroids should not be used routinely in the treatment of IPF. Although steroids, alone or in combination with other immunosuppressive medications, were commonly used for IPF in the past, such use was not based on results of randomized controlled trials.46 Retrospective controlled studies have failed to show that corticosteroids improve mortality rates in IPF; indeed, they have shown that corticosteroids confer substantial morbidity.47,48 In addition, a randomized controlled trial combining corticosteroids with N-acetylcysteine and azathioprine was stopped early due to an increased risk of death and hospitalization.49 Collectively, these data suggest that corticosteroids confer no benefit and are potentially harmful. Their use in IPF is discouraged, and the joint international guidelines recommend against immunosuppression to treat IPF.16
Other treatments
The guidelines offer additional suggestions for the management of IPF.
Preliminary evidence suggests that microaspiration associated with abnormal gastroesophageal acid reflux is a risk factor for IPF. As such, there is a weak recommendation for aggressive treatment of reflux disease.50 However, because evidence suggests that proton-pump inhibitor therapy may be associated with adverse renal or central nervous system effects, this recommendation bears caution. It is hoped that ongoing studies will provide further insight into the role of acid-suppression in the management of IPF.51,52
Further treatment recommendations include best supportive management such as supplemental oxygen, pulmonary rehabilitation, and vaccinations.

Prompt referral for lung transplant is imperative. IPF is now the most common indication for lung transplant, and given the poor overall prognosis of advanced IPF, transplant confers a survival benefit in appropriately selected patients.53,54 Table 2 provides an evidence-based checklist for the workup and management of IPF.
ACUTE EXACERBATIONS OF IPF
The unpredictable nature of IPF can manifest in the form of acute exacerbations without an identifiable cause. The loosely defined diagnostic criteria for the diagnosis of acute exacerbations are a previous or new diagnosis of IPF, worsening or development of dyspnea in the last 30 days, and new bilateral ground-glass or consolidative changes with a background of UIP on HRCT.16
A new definition has been proposed55 to facilitate research in the characterization and treatment of acute exacerbations of IPF. The new definition includes all causes of respiratory deterioration except for heart failure and volume overload. It is less strict about the 30-day time frame. This newer definition is based on the lack of evidence differentiating outcomes when an acute deterioration is associated with known or unknown etiologies.55
The incidence of acute exacerbations is variable, with a 1- and 3-year incidence ranging between 8.6% and 23.9% depending on the criteria used.56 In general, acute exacerbations carry a grim prognosis, with a median life expectancy of 2.2 months.57
There is no approved therapy for exacerbations of IPF. Rather, treatment is mainly supportive with supplemental oxygen and mechanical ventilation. Current guidelines have a weak recommendation for the use of corticosteroids, but there are no recommendations regarding dose, route, or duration of therapy. Other treatments, primarily immunomodulatory agents, have been suggested but lack evidence of benefit.
Acknowledgments: Pathology images were provided by Carol Farver, MD, Pathology Institute, Cleveland Clinic. Radiology images were provided by Ruchi Yadav, MD, Imaging Institute, Cleveland Clinic.
- Brown KK, Raghu G. Medical treatment for pulmonary fibrosis: current trends, concepts, and prospects. Clin Chest Med 2004; 25(4):759–772, vii. doi:10.1016/j.ccm.2004.08.003
- Ryerson CJ, Collard HR. Update on the diagnosis and classification of ILD. Curr Opin Pulm Med 2013; 19(5):453–459. doi:10.1097/MCP.0b013e328363f48d
- Raghu G, Chen SY, Yeh WS, et al. Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001-11. Lancet Respir Med 2014; 2(7):566–572. doi:10.1016/S2213-2600(14)70101-8
- Nalysnyk L, Cid-Ruzafa J, Rotella P, Esser D. Incidence and prevalence of idiopathic pulmonary fibrosis: review of the literature. Eur Respir Rev 2012; 21(126):355–361. doi:10.1183/09059180.00002512
- Hutchinson J, Fogarty A, Hubbard R, McKeever T. Global incidence and mortality of idiopathic pulmonary fibrosis: a systematic review. Eur Respir J 2015; 46(3):795–806. doi:10.1183/09031936.00185114
- National Lung Screening Trial Research Team; Aberle DR, Adams AM, Berg CD, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med 2011; 365(5):395–409. doi:10.1056/NEJMoa1102873
- Jin GY, Lynch D, Chawla A, et al. Interstitial lung abnormalities in a CT lung cancer screening population: prevalence and progression rate. Radiology 2013; 268(2):563–571. doi:10.1148/radiol.13120816
- Southern BD, Scheraga RG, Yadav R. Managing interstitial lung disease detected on CT during lung cancer screening. Cleve Clin J Med 2016; 83(1):55–65. doi:10.3949/ccjm.83a.14157
- King TE Jr, Schwarz MI, Brown K, et al. Idiopathic pulmonary fibrosis: relationship between histopathologic features and mortality. Am J Respir Crit Care Med 2001; 164(5):1025–1032. doi:10.1164/ajrccm.164.6.2001056
- Lamas DJ, Kawut SM, Bagiella E, et al. Delayed access and survival in idiopathic pulmonary fibrosis: a cohort study. Am J Respir Crit Care Med 2011; 184(7):842–847. doi:10.1164/rccm.201104-0668OC
- Jo HE, Glaspole I, Moodley Y, et al. Disease progression in idiopathic pulmonary fibrosis with mild physiological impairment: analysis from the Australian IPF registry. BMC Pulm Med 2018; 18(1):19. doi:10.1186/s12890-018-0575-y
- Yang IV, Schwartz DA. Epigenetics of idiopathic pulmonary fibrosis. Transl Res 2015; 165(1):48–60. doi:10.1016/j.trsl.2014.03.011
- King TE Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet 2011; 378(9807):1949–1961. doi:10.1016/S0140-6736(11)60052-4
- Meltzer EB, Noble PW. Idiopathic pulmonary fibrosis. Orphanet J Rare Dis 2008; 3:8. doi:10.1186/1750-1172-3-8
- Raghu G. Idiopathic pulmonary fibrosis. A rational clinical approach. Chest 1987; 92(1):148–154. doi:10.1378/chest.92.1.148
- Raghu G, Collard HR, Egan JJ, et al; ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183(6):788–824. doi:10.1164/rccm.2009-040GL
- Panos RJ, Mortenson RL, Niccoli SA, King TE Jr. Clinical deterioration in patients with idiopathic pulmonary fibrosis: causes and assessment. Am J Med 1990; 88(4):396–404. doi:10.1016/0002-9343(90)90495-Y
- Ley B, Collard HR, King TE Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011; 183(4):431–440. doi:10.1164/rccm.201006-0894CI
- Collard HR, King TE Jr, Bartelson BB, Vourlekis JS, Schwarz MI, Brown KK. Changes in clinical and physiologic variables predict survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2003; 168(5):538–542. doi:10.1164/rccm.200211-1311OC
- Flaherty KR, Andrei AC, Murray S, et al. Idiopathic pulmonary fibrosis: prognostic value of changes in physiology and six-minute-walk test. Am J Respir Crit Care Med 2006; 174(7):803–809. doi:10.1164/rccm.200604-488OC
- Jegal Y, Kim DS, Shim TS, et al. Physiology is a stronger predictor of survival than pathology in fibrotic interstitial pneumonia. Am J Respir Crit Care Med 2005; 171(6):639–644. doi:10.1164/rccm.200403-331OC
- Latsi PI, du Bois RM, Nicholson AG, et al. Fibrotic idiopathic interstitial pneumonia: the prognostic value of longitudinal functional trends. Am J Respir Crit Care Med 2003; 168(5):531–537. doi:10.1164/rccm.200210-1245OC
- King CS, Nathan SD. Idiopathic pulmonary fibrosis: effects and optimal management of comorbidities. Lancet Respir Med 2017; 5(1):72–84. doi:10.1016/S2213-2600(16)30222-3
- Ley B, Ryerson CJ, Vittinghoff E, et al. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med 2012; 156(1):684–691. doi:10.7326/0003-4819-156-10-201205150-00004
- Rudd RM, Prescott RJ, Chalmers JC, Johnston ID; Fibrosing Alveolitis Subcommittee of the Research Committee of the British Thoracic Society. British Thoracic Society study on cryptogenic fibrosing alveolitis: response to treatment and survival. Thorax 2007; 62(1):62–66. doi:10.1136/thx.2005.045591
- Gutsche M, Rosen GD, Swigris JJ. Connective tissue disease-associated interstitial lung disease: a review. Curr Respir Care Rep 2012; 1:224–232. doi:10.1007/s13665-012-0028-7
- Park JH, Kim DS, Park IN, et al. Prognosis of fibrotic interstitial pneumonia: idiopathic versus collagen vascular disease-related subtypes. Am J Respir Crit Care Med 2007; 175(7):705–711. doi:10.1164/rccm.200607-912OC
- Taskar VS, Coultas DB. Is idiopathic pulmonary fibrosis an environmental disease? Proc Am Thorac Soc 2006; 3(4):293–298. doi:10.1513/pats.200512-131TK
- Vourlekis JS, Schwarz MI, Cherniack RM, et al. The effect of pulmonary fibrosis on survival in patients with hypersensitivity pneumonitis. Am J Med 2004; 116(10):662–668. doi:10.1016/j.amjmed.2003.12.030
- Brownell R, Moua T, Henry TS, et al. The use of pretest probability increases the value of high-resolution CT in diagnosing usual interstitial pneumonia. Thorax 2017; 72(5):424–429. doi:10.1136/thoraxjnl-2016-209671
- Baumgartner KB, Samet JM, Stidley CA, Colby TV, Waldron JA. Cigarette smoking: a risk factor for idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 1997; 155(1):242–248. doi:10.1164/ajrccm.155.1.9001319
- Selman M, Pardo A, King TE Jr. Hypersensitivity pneumonitis: insights in diagnosis and pathobiology. Am J Respir Crit Care Med 2012; 186(4):314–324. doi:10.1164/rccm.201203-0513CI
- Schwaiblmair M, Behr W, Haeckel T, Markl B, Foerg W, Berghaus T. Drug induced interstitial lung disease. Open Respir Med J 2012; 6:63–74. doi:10.2174/1874306401206010063
- Grenier P, Valeyre D, Cluzel P, Brauner MW, Lenoir S, Chastang C. Chronic diffuse interstitial lung disease: diagnostic value of chest radiography and high-resolution CT. Radiology 1991; 179(1):123–132. doi:10.1148/radiology.179.1.2006262
- Lynch JP 3rd, Huynh RH, Fishbein MC, Saggar R, Belperio JA, Weigt SS. Idiopathic pulmonary fibrosis: epidemiology, clinical features, prognosis, and management. Semin Respir Crit Care Med 2016; 37(3):331–357. doi:10.1055/s-0036-1582011
- Berbescu EA, Katzenstein AL, Snow JL, Zisman DA. Transbronchial biopsy in usual interstitial pneumonia. Chest 2006; 129(5):1126–1131. doi:10.1378/chest.129.5.1126
- Ohshimo S, Bonella F, Cui A, et al. Significance of bronchoalveolar lavage for the diagnosis of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2009; 179(11):1043–1047. doi:10.1164/rccm.200808-1313OC
- Oparka J, Yan TD, Ryan E, Dunning J. Does video-assisted thoracic surgery provide a safe alternative to conventional techniques in patients with limited pulmonary function who are otherwise suitable for lung resection? Interact Cardiovasc Thorac Surg 2013; 17(1):159–162. doi:10.1093/icvts/ivt097
- Flaherty KR, King TE Jr, Raghu G, et al. Idiopathic interstitial pneumonia: what is the effect of a multidisciplinary approach to diagnosis? Am J Respir Crit Care Med 2004; 170(8):904–910. doi:10.1164/rccm.200402-147OC
- Walsh SL, Wells AU, Desai SR, et al. Multicentre evaluation of multidisciplinary team meeting agreement on diagnosis in diffuse parenchymal lung disease: a case-cohort study. Lancet Respir Med 2016; 4(7):557–565. doi:10.1016/S2213-2600(16)30033-9
- King TE Jr, Bradford WZ, Castro-Bernardini S, et al; ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22):2083–2092. doi:10.1056/NEJMoa1402582
- Noble PW, Albera C, Bradford WZ, et al; CAPACITY Study Group. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet 2011; 377(9779):1760–1769. doi:10.1016/S0140-6736(11)60405-4
- Noble PW, Albera C, Bradford WZ, et al. Pirfenidone for idiopathic pulmonary fibrosis: analysis of pooled data from three multinational phase 3 trials. Eur Respir J 2016; 47(1):243–253. doi:10.1183/13993003.00026-2015
- Nathan SD, Albera C, Bradford WZ, et al. Effect of pirfenidone on mortality: pooled analyses and meta-analyses of clinical trials in idiopathic pulmonary fibrosis. Lancet Respir Med 2017; 5(1):33–41. doi:10.1016/S2213-2600(16)30326-5
- Richeldi L, du Bois RM, Raghu G, et al; INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22):2071–2082. doi:10.1056/NEJMoa1402584
- Richeldi L, Davies HR, Ferrara G, Franco F. Corticosteroids for idiopathic pulmonary fibrosis. Cochrane Database Syst Rev 2003: 3:CD002880. doi:10.1002/14651858.CD002880
- Douglas WW, Ryu JH, Schroeder DR. Idiopathic pulmonary fibrosis: impact of oxygen and colchicine, prednisone, or no therapy on survival. Am J Respir Crit Care Med 2000; 161(4 pt 1):1172–1178. doi:10.1164/ajrccm.161.4.9907002
- Gay SE, Kazerooni EA, Toews GB, et al. Idiopathic pulmonary fibrosis: predicting response to therapy and survival. Am J Respir Crit Care Med 1998; 157(4 pt 1):1063–1072. doi:10.1164/ajrccm.157.4.9703022
- Idiopathic Pulmonary Fibrosis Clinical Research Network; Raghu G, Anstrom KJ, King TE Jr, Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med 2012; 366(21):1968–1977. doi:10.1056/NEJMoa1113354
- Raghu G, Freudenberger TD, Yang S, et al. High prevalence of abnormal acid gastro-oesophageal reflux in idiopathic pulmonary fibrosis. Eur Respir J 2006; 27(1):136–142. doi:10.1183/09031936.06.00037005
- Gomm W, von Holt K, Thome F, et al. Association of proton pump inhibitors with risk of dementia: a pharmacoepidemiological claims data analysis. JAMA Neurol 2016; 73(4):410–416. doi:10.1001/jamaneurol.2015.4791
- Xie Y, Bowe B, Li T, Xian H, Balasubramanian S, Al-Aly Z. Proton pump inhibitors and risk of incident CKD and progression to ESRD. J Am Soc Nephrol 2016; 27(10):3153–3163. doi:10.1681/ASN.2015121377
- Thabut G, Mal H, Castier Y, et al. Survival benefit of lung transplantation for patients with idiopathic pulmonary fibrosis. J Thorac Cardiovasc Surg 2003; 126(2):469–475. doi:10.1016/S0022-5223(03)00600-7
- Valapour M, Skeans MA, Smith JM, et al. Lung. Am J Transplant 2016; 16(suppl 2):141–168. doi:10.1111/ajt.13671
- Collard HR, Ryerson CJ, Corte TJ, et al. Acute exacerbation of idiopathic pulmonary fibrosis. An International Working Group Report. Am J Respir Crit Care Med 2016; 194(3):265–275. doi:10.1164/rccm.201604-0801CI
- Kondoh Y, Taniguchi H, Katsuta T, et al. Risk factors of acute exacerbation of idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis 2010; 27(2):103–110. doi:10.1016/j.resinv.2015.04.005
- Song JW, Hong SB, Lim CM, Koh Y, Kim DS. Acute exacerbation of idiopathic pulmonary fibrosis: incidence, risk factors and outcome. Eur Respir J 2011; 37(2):356–363. doi:10.1183/09031936.00159709
Idiopathic pulmonary fibrosis (IPF) is a devastating and fatal lung disease that generally affects older adults. It is characterized by a radiographic and histopathologic pattern of usual interstitial pneumonia (UIP) that has no other known etiology.
Accurate diagnosis of IPF is crucial. We recommend early referral to a center specializing in interstitial lung disease to confirm the diagnosis, start appropriate therapy, advise the patient on prognosis and enrollment in disease registries and clinical trials, and determine candidacy for lung transplant.
Primary care physicians are uniquely positioned to encounter patients with IPF, whether because of a patient complaint or as an incidental finding on computed tomography. The goal of this article is to delineate the features of IPF so that it may be recognized early and thus expedite referral to a center with expertise in interstitial lung disease for a thorough evaluation and appropriate management.
WHAT IS IDIOPATHIC PULMONARY FIBROSIS?
MORE COMMON THAN ONCE THOUGHT
The true incidence and prevalence of IPF are difficult to assess. IPF is generally considered a rare disease, but it is more common than once thought. In 2011, Raghu et al3 estimated the prevalence in Medicare beneficiaries to be 495 cases per 100,000. Based on this estimate and the current US population, up to 160,000 Americans could have IPF.4 Raghu et al also showed that IPF more often affects adults over age 65, which suggests that as the US population ages, the incidence of IPF may rise. Studies have also reported an increased incidence of IPF worldwide.5
Further, with the rising use of low-dose computed tomography to screen for lung cancer, more incidental cases of IPF will likely be found.6–8
Older data showed a lag from symptom onset to accurate diagnosis of 1 to 2 years.9 A more recent study found a lag in referral of patients with IPF to tertiary care centers, and this delay was associated with a higher rate of death independent of disease severity.10
TYPICALLY PROGRESSIVE, OFTEN FATAL
IPF is typically progressive and limited to the lungs, and it portends a poor prognosis. The median survival is commonly cited as 2 to 5 years from diagnosis, although this is based on older observations that may not reflect current best practice and newer therapies. More recent studies suggest higher survival rates if patients have preserved lung function.11
As the name indicates, the etiology of IPF is unknown, but studies have indicated genetic underpinnings in a notable proportion of cases.12 Regardless of the cause, the pathogenesis and progression of IPF are thought to be the result of an abnormal and persistent wound-repair response. The progressive deposition of scar tissue disrupts normal lung architecture and function, eventually causing clinical disease.13
SYMPTOMS AND KEY FEATURES
Patients with IPF typically present with the insidious onset of dyspnea on exertion, with or without chronic cough. Risk factors include male sex, increasing age, and a history of smoking. Patients with undiagnosed IPF who present with dyspnea and a history of smoking are often treated empirically for chronic obstructive pulmonary disease (COPD).
Rales are a common finding on auscultation in IPF, and this can lead to an exhaustive cardiac evaluation and empiric treatment for heart failure. Digital clubbing is also relatively common.14 Hypoxemia with exertion is another common feature that also often correlates with disease severity and prognosis. Resting hypoxemia is more common in advanced disease.
On spirometry, patients with IPF typically demonstrate restrictive physiology, suggested by a normal or elevated ratio of the forced expiratory volume in 1 second to the forced vital capacity (FEV1/FVC) (> 70% predicted or above the lower limit of normal) combined with a lower than normal FVC. Restrictive physiology is definitively demonstrated by a decreased total lung capacity (< 80% predicted or below the lower limit of normal) on plethysmography. Impaired gas exchange, manifested by a decreased diffusing capacity of the lungs for carbon monoxide (DLCO) on pulmonary function testing, is also common. Because pulmonary perfusion is higher in the lung bases, where IPF is also predominant, the DLCO is often reduced to a greater extent than the FVC.
PROGNOSTIC INDICATORS
Clinicians typically view IPF as a relentless and progressive process, but its course is variable and can be uncertain in an individual patient (Figure 1).15,16 Nevertheless, over time, most patients have a decline in lung function leading to respiratory failure. Respiratory failure, often preceded by a subacute deterioration (over weeks to months) or an acute deterioration (< 4 weeks), is the most common cause of death, but comorbid diseases such as lung cancer, infection, and heart failure are also common causes of death in these patients.17,18
Predictors of mortality include worsening FVC, DLCO, symptoms, and physiologic impairment, manifested by a decline in the 6-minute walking test or worsening exertional hypoxemia.19–22 Other common comorbidities linked with impaired quality of life and poor prognosis include obstructive sleep apnea, gastroesophageal reflux disease, and depression.16,23 Retrospective studies suggest that most IPF patients die 2 to 5 years after symptom onset. With the lag from symptom onset to final diagnosis, the average life expectancy is as little as 2 years from the time of diagnosis.9,18,24,25
Two staging systems have been developed to predict short-term and long-term mortality risk based on sex, age, and physiologic parameters.23,24 The GAP (gender, age, physiology) index provides an estimate of the risk of death for a cohort of patients: a score of 0 to 8 is calculated, and the score is then categorized as stage I, II, or III. Each stage is associated with 1-, 2-, and 3-year mortality rates, with stage III having the highest rates. The GAP calculator (www.acponline.org/journals/annals/extras/gap) provides an estimate of the risk of death for an individual patient. The application of these tools for the management of IPF is evolving; however, they may be helpful for counseling patients about disease prognosis.
CLUES TO DIAGNOSIS
Histologic patterns
UIP on histologic study is also seen in fibrotic lung diseases other than IPF, including connective tissue disease-associated interstitial lung disease, inhalational or occupational interstitial lung disease, and chronic hypersensitivity pneumonitis.26–29 Consequently, the diagnosis of IPF requires exclusion of other known causes of UIP.
According to the 2011 guidelines,16 the histology of interstitial lung disease can be categorized as definite UIP, probable UIP, or possible UIP, or as an atypical pattern suggesting another diagnosis. If no definite cause of the interstitial lung abnormality is found, the level of certainty of the histopathologic pattern of UIP helps formulate the clinical diagnosis and management plan.
Clues on computed tomography
The UIP nomenclature also describes patterns on high-resolution computed tomography (HRCT). HRCT is done without contrast and produces thin-sliced images (usually < 1.5 mm) in inspiratory, expiratory, and prone views; this allows detection of air trapping, which may indicate an airway-centric alternative diagnosis.
On HRCT, UIP appears as reticular opacities, often with traction bronchiectasis or bronchiolectasis, usually with a basilar and peripheral predominance. Honeycombing is a key feature and appears as clustered cystic spaces with well-defined walls in the periphery of the lung parenchyma. Ground-glass opacities are not a prominent feature of UIP, and although they do not exclude a UIP pattern, they should spur consideration of other diagnoses.16 Reactive mediastinal and hilar lymphadenopathy is another common feature of UIP.
When evaluating results of HRCT for UIP, the radiologist categorizes the pattern as definite UIP, possible UIP, or inconsistent. The definite pattern meets all the above features and has none of the features suggesting an alternative diagnosis (Figure 3). The possible pattern includes all the above features with the exception of honeycombing. If the predominant features on HRCT include any atypical finding listed above, then the study is considered inconsistent with UIP. If the pattern on HRCT is considered definite, evaluation of pathology is not necessary. If the pattern is categorized as possible or is inconsistent, then surgical lung biopsy-confirmed UIP is necessary for the definitive diagnosis of IPF.
However, evidence is emerging that in the correct clinical scenario, possible UIP behaves similarly to definite UIP and may be sufficient to make the clinical diagnosis of IPF even without surgical biopsy confirmation.30
A DIAGNOSTIC ALGORITHM FOR IPF
Given the multitude of interstitial lung diseases, their complexities, and the lack of a gold standard definitive diagnostic test, the diagnosis of IPF can be difficult, requiring the integration of clinical, radiologic, and, if necessary, pathologic findings.
Demographic features
The patient’s demographic features and risk factors dictate the initial clinical suspicion of IPF compared with other interstitial lung diseases. The incidence of IPF increases with age, and IPF is more common in men. A history of smoking is another risk factor.31 A 45-year-old never-smoker is much less likely to have IPF than a 70-year-old former smoker, and a 70-year-old man is more likely to have IPF than a woman of the same age. Thus, the finding of interstitial lung disease in a patient with a demographic profile that is not typical (ie, a younger woman who never smoked) should prompt an exhaustive investigation for another diagnosis such as hypersensitivity pneumonitis or connective tissue disease.
Key elements of the history
After considering the demographic profile and risk factors, the next step in the evaluation is a thorough and accurate medical history. This should include assessment of the severity of dyspnea and cough, signs and symptoms of connective tissue disease (eg, arthralgias, sicca symptoms, Raynaud phenomenon, difficulty swallowing), and gastroesophageal reflux disease, which can be associated with connective tissue disease and, independently, with IPF.
It is also important to identify any environmental exposures that suggest pneumoconiosis or chronic hypersensitivity pneumonitis. The most common risk factors for hypersensitivity pneumonitis are birds and bird feathers, molds, fungi, hot tub use, and some industrial chemicals.32
A medication history is important. Many medications are associated with interstitial lung disease, but amiodarone, bleomycin, methotrexate, and nitrofurantoin are among the common offenders.33
A thorough family history is necessary, as there are familial forms of IPF.
Focus of the physical examination
The physical examination must include careful auscultation for rales. While rales are not specific for IPF, they are the most common pulmonary abnormality. Detailed skin, musculoskeletal, and cardiovascular examinations are also important to evaluate for rheumatologic signs, clubbing, or evidence of heart failure or pulmonary hypertension.
Laboratory tests
Laboratory testing should include a serologic autoantibody panel to evaluate for connective tissue diseases that can manifest as interstitial lung disease, including rheumatoid arthritis, dermatopolymyositis, scleroderma, Sjögren syndrome, and undifferentiated or mixed connective tissue disease. Typical preliminary laboratory tests are antinuclear antibody, rheumatoid factor, erythrocyte sedimentation rate, and C-reactive protein. Others may include anticyclic citrullinated peptide (anti-CCP), anti-Scl-70, anti-RNP, anti-SS-A, anti-SS-B, and anti-Jo-1.16 The breadth of the panel should depend on patient demographics and findings in the history or physical examination that increase or decrease the likelihood of a connective tissue disease.
Lung function testing
Assessing the patient’s pulmonary physiology should include spirometry, DLCO, and body plethysmography (lung volumes). In most cases, IPF manifests with restrictive physiology. Once restrictive physiology is confirmed with a low total lung capacity, FVC testing can be used as a longitudinal surrogate, as it is less expensive and easier for the patient to perform. In general, a lower total lung capacity or FVC indicates more severe impairment.
The DLCO serves as another marker of severity but is less reliable due to baseline variability and difficulties performing the maneuver.
A 6-minute walk test is another crucial physiologic assessment tool that can quantify exertional hypoxemia and functional status (ie, distance walked), and can assist in prognosis.
Imaging
Most patients undergo chest radiography in the workup for undiagnosed dyspnea. However, chest radiography is not adequate to formulate an accurate diagnosis in suspected interstitial lung disease, and a normal radiograph cannot exclude changes that might reflect early phases of the disease. As the disease progresses, the plain radiograph can show reticulonodular opacities and honeycombing in the peripheral and lower lung zones (Figure 3).34
The decision whether to order HRCT in the workup for a patient who has dyspnea and a normal chest radiograph is challenging. We recommend cross-sectional imaging when physiologic testing shows restriction or low DLCO, or when there is a high index of suspicion for underlying lung disease as the cause of symptoms.
Expert consultation can aid with this decision, especially when the underlying cause of dyspnea remains unclear after initial studies have been completed. Otherwise, HRCT is an essential test in the evaluation of interstitial lung disease.
Bronchoscopy’s role controversial
If the pattern on HRCT is nondiagnostic, then surgical biopsy is necessary, and the diagnosis of IPF requires a histologic pattern of UIP as described above.16,35
Although bronchoscopy can be valuable if an alternative diagnosis such as sarcoidosis or chronic hypersensitivity pneumonitis is suspected, the role of bronchoscopic biopsy in the workup of IPF is controversial. The patchy nature of UIP does not lend itself to the relatively small biopsy samples obtained through bronchoscopy.36,37
Surgical biopsy options
The favored biopsy approach is surgical, using either an open or a video-assisted thoracoscopic technique. The latter is preferred as it is less invasive, requires a shorter length of hospital stay, and allows a faster recovery.38 Bronchoscopic cryobiopsy, currently under investigation, is a potentially valuable tool whose role in diagnosing IPF is evolving.
Frequently, neither HRCT nor surgical lung biopsy demonstrates UIP, making the definitive diagnosis of IPF difficult. Moreover, some patients with nondiagnostic HRCT results are unable to tolerate surgical lung biopsy because of severely impaired lung function or other comorbidities.
The role of multidisciplinary discussions
When surgical lung biopsy is not possible, current practice at leading centers uses a multidisciplinary approach to allow for a confident diagnosis.30,39 Discussions take place between pulmonologists, pathologists, radiologists, and other specialists to collectively consider all aspects of a case before rendering a consensus opinion on the diagnosis and the management plan. If the discussion leads to a consensus diagnosis of IPF, then the patient’s clinician can move forward with treatment options. If not, the group can recommend further workup or alternative diagnoses and treatment regimens. The multidisciplinary group is also well positioned to consider the relative risks and benefits of moving forward with surgical lung biopsy for individual patients.
This approach illustrates the importance of referring these patients to centers of excellence in diagnosing and managing complex cases of interstitial lung disease, including IPF.40
TREATMENT OF IPF
Antifibrotic therapy
Antifibrotic therapy is a choice between pirfenidone and nintedanib.
Pirfenidone, which has an undefined molecular target, was approved based on the results of 3 trials.41,42 Pooled analyses from these trials showed a reduction in the decline from baseline in FVC percent predicted and improved progression-free survival.43 Pooled and meta-analyses of pirfenidone clinical trials have shown a mortality benefit, although no individual study has shown such an effect on mortality rates.44
The major adverse effects of pirfenidone are gastrointestinal distress and photosensitivity rash.
Nintedanib is a triple tyrosine kinase inhibitor that broadly targets fibroblast growth factor, vascular endothelial growth factor, and platelet-derived growth factor receptors. Combined analysis of 2 concurrent trials45 showed that nintedanib reduced the decline in FVC, similarly to pirfenidone. The major adverse event associated with nintedanib was diarrhea. Since it inhibits vascular endothelial growth factor, there is a risk of hematologic complications such as bleeding or clotting events.
Because pirfenidone and nintedanib can increase aminotransferase levels, regular monitoring is recommended.
To date, no trial has compared pirfenidone and nintedanib in terms of their efficacy and tolerability. Therefore, the choice of agent is based on the patient’s preference after a discussion of potential risks and expected benefits, a review of each drug’s side effects, and consideration of comorbid conditions and physician experience.
Patients need to understand that these drugs slow the rate of decline in FVC but have not been shown to improve symptoms or functional status.
Corticosteroids are not routine
Corticosteroids should not be used routinely in the treatment of IPF. Although steroids, alone or in combination with other immunosuppressive medications, were commonly used for IPF in the past, such use was not based on results of randomized controlled trials.46 Retrospective controlled studies have failed to show that corticosteroids improve mortality rates in IPF; indeed, they have shown that corticosteroids confer substantial morbidity.47,48 In addition, a randomized controlled trial combining corticosteroids with N-acetylcysteine and azathioprine was stopped early due to an increased risk of death and hospitalization.49 Collectively, these data suggest that corticosteroids confer no benefit and are potentially harmful. Their use in IPF is discouraged, and the joint international guidelines recommend against immunosuppression to treat IPF.16
Other treatments
The guidelines offer additional suggestions for the management of IPF.
Preliminary evidence suggests that microaspiration associated with abnormal gastroesophageal acid reflux is a risk factor for IPF. As such, there is a weak recommendation for aggressive treatment of reflux disease.50 However, because evidence suggests that proton-pump inhibitor therapy may be associated with adverse renal or central nervous system effects, this recommendation bears caution. It is hoped that ongoing studies will provide further insight into the role of acid-suppression in the management of IPF.51,52
Further treatment recommendations include best supportive management such as supplemental oxygen, pulmonary rehabilitation, and vaccinations.
Prompt referral for lung transplant is imperative. IPF is now the most common indication for lung transplant, and given the poor overall prognosis of advanced IPF, transplant confers a survival benefit in appropriately selected patients.53,54 Table 2 provides an evidence-based checklist for the workup and management of IPF.
ACUTE EXACERBATIONS OF IPF
The unpredictable nature of IPF can manifest in the form of acute exacerbations without an identifiable cause. The loosely defined diagnostic criteria for the diagnosis of acute exacerbations are a previous or new diagnosis of IPF, worsening or development of dyspnea in the last 30 days, and new bilateral ground-glass or consolidative changes with a background of UIP on HRCT.16
A new definition has been proposed55 to facilitate research in the characterization and treatment of acute exacerbations of IPF. The new definition includes all causes of respiratory deterioration except for heart failure and volume overload. It is less strict about the 30-day time frame. This newer definition is based on the lack of evidence differentiating outcomes when an acute deterioration is associated with known or unknown etiologies.55
The incidence of acute exacerbations is variable, with a 1- and 3-year incidence ranging between 8.6% and 23.9% depending on the criteria used.56 In general, acute exacerbations carry a grim prognosis, with a median life expectancy of 2.2 months.57
There is no approved therapy for exacerbations of IPF. Rather, treatment is mainly supportive with supplemental oxygen and mechanical ventilation. Current guidelines have a weak recommendation for the use of corticosteroids, but there are no recommendations regarding dose, route, or duration of therapy. Other treatments, primarily immunomodulatory agents, have been suggested but lack evidence of benefit.
Acknowledgments: Pathology images were provided by Carol Farver, MD, Pathology Institute, Cleveland Clinic. Radiology images were provided by Ruchi Yadav, MD, Imaging Institute, Cleveland Clinic.
Idiopathic pulmonary fibrosis (IPF) is a devastating and fatal lung disease that generally affects older adults. It is characterized by a radiographic and histopathologic pattern of usual interstitial pneumonia (UIP) that has no other known etiology.
Accurate diagnosis of IPF is crucial. We recommend early referral to a center specializing in interstitial lung disease to confirm the diagnosis, start appropriate therapy, advise the patient on prognosis and enrollment in disease registries and clinical trials, and determine candidacy for lung transplant.
Primary care physicians are uniquely positioned to encounter patients with IPF, whether because of a patient complaint or as an incidental finding on computed tomography. The goal of this article is to delineate the features of IPF so that it may be recognized early and thus expedite referral to a center with expertise in interstitial lung disease for a thorough evaluation and appropriate management.
WHAT IS IDIOPATHIC PULMONARY FIBROSIS?
MORE COMMON THAN ONCE THOUGHT
The true incidence and prevalence of IPF are difficult to assess. IPF is generally considered a rare disease, but it is more common than once thought. In 2011, Raghu et al3 estimated the prevalence in Medicare beneficiaries to be 495 cases per 100,000. Based on this estimate and the current US population, up to 160,000 Americans could have IPF.4 Raghu et al also showed that IPF more often affects adults over age 65, which suggests that as the US population ages, the incidence of IPF may rise. Studies have also reported an increased incidence of IPF worldwide.5
Further, with the rising use of low-dose computed tomography to screen for lung cancer, more incidental cases of IPF will likely be found.6–8
Older data showed a lag from symptom onset to accurate diagnosis of 1 to 2 years.9 A more recent study found a lag in referral of patients with IPF to tertiary care centers, and this delay was associated with a higher rate of death independent of disease severity.10
TYPICALLY PROGRESSIVE, OFTEN FATAL
IPF is typically progressive and limited to the lungs, and it portends a poor prognosis. The median survival is commonly cited as 2 to 5 years from diagnosis, although this is based on older observations that may not reflect current best practice and newer therapies. More recent studies suggest higher survival rates if patients have preserved lung function.11
As the name indicates, the etiology of IPF is unknown, but studies have indicated genetic underpinnings in a notable proportion of cases.12 Regardless of the cause, the pathogenesis and progression of IPF are thought to be the result of an abnormal and persistent wound-repair response. The progressive deposition of scar tissue disrupts normal lung architecture and function, eventually causing clinical disease.13
SYMPTOMS AND KEY FEATURES
Patients with IPF typically present with the insidious onset of dyspnea on exertion, with or without chronic cough. Risk factors include male sex, increasing age, and a history of smoking. Patients with undiagnosed IPF who present with dyspnea and a history of smoking are often treated empirically for chronic obstructive pulmonary disease (COPD).
Rales are a common finding on auscultation in IPF, and this can lead to an exhaustive cardiac evaluation and empiric treatment for heart failure. Digital clubbing is also relatively common.14 Hypoxemia with exertion is another common feature that also often correlates with disease severity and prognosis. Resting hypoxemia is more common in advanced disease.
On spirometry, patients with IPF typically demonstrate restrictive physiology, suggested by a normal or elevated ratio of the forced expiratory volume in 1 second to the forced vital capacity (FEV1/FVC) (> 70% predicted or above the lower limit of normal) combined with a lower than normal FVC. Restrictive physiology is definitively demonstrated by a decreased total lung capacity (< 80% predicted or below the lower limit of normal) on plethysmography. Impaired gas exchange, manifested by a decreased diffusing capacity of the lungs for carbon monoxide (DLCO) on pulmonary function testing, is also common. Because pulmonary perfusion is higher in the lung bases, where IPF is also predominant, the DLCO is often reduced to a greater extent than the FVC.
PROGNOSTIC INDICATORS
Clinicians typically view IPF as a relentless and progressive process, but its course is variable and can be uncertain in an individual patient (Figure 1).15,16 Nevertheless, over time, most patients have a decline in lung function leading to respiratory failure. Respiratory failure, often preceded by a subacute deterioration (over weeks to months) or an acute deterioration (< 4 weeks), is the most common cause of death, but comorbid diseases such as lung cancer, infection, and heart failure are also common causes of death in these patients.17,18
Predictors of mortality include worsening FVC, DLCO, symptoms, and physiologic impairment, manifested by a decline in the 6-minute walking test or worsening exertional hypoxemia.19–22 Other common comorbidities linked with impaired quality of life and poor prognosis include obstructive sleep apnea, gastroesophageal reflux disease, and depression.16,23 Retrospective studies suggest that most IPF patients die 2 to 5 years after symptom onset. With the lag from symptom onset to final diagnosis, the average life expectancy is as little as 2 years from the time of diagnosis.9,18,24,25
Two staging systems have been developed to predict short-term and long-term mortality risk based on sex, age, and physiologic parameters.23,24 The GAP (gender, age, physiology) index provides an estimate of the risk of death for a cohort of patients: a score of 0 to 8 is calculated, and the score is then categorized as stage I, II, or III. Each stage is associated with 1-, 2-, and 3-year mortality rates, with stage III having the highest rates. The GAP calculator (www.acponline.org/journals/annals/extras/gap) provides an estimate of the risk of death for an individual patient. The application of these tools for the management of IPF is evolving; however, they may be helpful for counseling patients about disease prognosis.
CLUES TO DIAGNOSIS
Histologic patterns
UIP on histologic study is also seen in fibrotic lung diseases other than IPF, including connective tissue disease-associated interstitial lung disease, inhalational or occupational interstitial lung disease, and chronic hypersensitivity pneumonitis.26–29 Consequently, the diagnosis of IPF requires exclusion of other known causes of UIP.
According to the 2011 guidelines,16 the histology of interstitial lung disease can be categorized as definite UIP, probable UIP, or possible UIP, or as an atypical pattern suggesting another diagnosis. If no definite cause of the interstitial lung abnormality is found, the level of certainty of the histopathologic pattern of UIP helps formulate the clinical diagnosis and management plan.
Clues on computed tomography
The UIP nomenclature also describes patterns on high-resolution computed tomography (HRCT). HRCT is done without contrast and produces thin-sliced images (usually < 1.5 mm) in inspiratory, expiratory, and prone views; this allows detection of air trapping, which may indicate an airway-centric alternative diagnosis.
On HRCT, UIP appears as reticular opacities, often with traction bronchiectasis or bronchiolectasis, usually with a basilar and peripheral predominance. Honeycombing is a key feature and appears as clustered cystic spaces with well-defined walls in the periphery of the lung parenchyma. Ground-glass opacities are not a prominent feature of UIP, and although they do not exclude a UIP pattern, they should spur consideration of other diagnoses.16 Reactive mediastinal and hilar lymphadenopathy is another common feature of UIP.
When evaluating results of HRCT for UIP, the radiologist categorizes the pattern as definite UIP, possible UIP, or inconsistent. The definite pattern meets all the above features and has none of the features suggesting an alternative diagnosis (Figure 3). The possible pattern includes all the above features with the exception of honeycombing. If the predominant features on HRCT include any atypical finding listed above, then the study is considered inconsistent with UIP. If the pattern on HRCT is considered definite, evaluation of pathology is not necessary. If the pattern is categorized as possible or is inconsistent, then surgical lung biopsy-confirmed UIP is necessary for the definitive diagnosis of IPF.
However, evidence is emerging that in the correct clinical scenario, possible UIP behaves similarly to definite UIP and may be sufficient to make the clinical diagnosis of IPF even without surgical biopsy confirmation.30
A DIAGNOSTIC ALGORITHM FOR IPF
Given the multitude of interstitial lung diseases, their complexities, and the lack of a gold standard definitive diagnostic test, the diagnosis of IPF can be difficult, requiring the integration of clinical, radiologic, and, if necessary, pathologic findings.
Demographic features
The patient’s demographic features and risk factors dictate the initial clinical suspicion of IPF compared with other interstitial lung diseases. The incidence of IPF increases with age, and IPF is more common in men. A history of smoking is another risk factor.31 A 45-year-old never-smoker is much less likely to have IPF than a 70-year-old former smoker, and a 70-year-old man is more likely to have IPF than a woman of the same age. Thus, the finding of interstitial lung disease in a patient with a demographic profile that is not typical (ie, a younger woman who never smoked) should prompt an exhaustive investigation for another diagnosis such as hypersensitivity pneumonitis or connective tissue disease.
Key elements of the history
After considering the demographic profile and risk factors, the next step in the evaluation is a thorough and accurate medical history. This should include assessment of the severity of dyspnea and cough, signs and symptoms of connective tissue disease (eg, arthralgias, sicca symptoms, Raynaud phenomenon, difficulty swallowing), and gastroesophageal reflux disease, which can be associated with connective tissue disease and, independently, with IPF.
It is also important to identify any environmental exposures that suggest pneumoconiosis or chronic hypersensitivity pneumonitis. The most common risk factors for hypersensitivity pneumonitis are birds and bird feathers, molds, fungi, hot tub use, and some industrial chemicals.32
A medication history is important. Many medications are associated with interstitial lung disease, but amiodarone, bleomycin, methotrexate, and nitrofurantoin are among the common offenders.33
A thorough family history is necessary, as there are familial forms of IPF.
Focus of the physical examination
The physical examination must include careful auscultation for rales. While rales are not specific for IPF, they are the most common pulmonary abnormality. Detailed skin, musculoskeletal, and cardiovascular examinations are also important to evaluate for rheumatologic signs, clubbing, or evidence of heart failure or pulmonary hypertension.
Laboratory tests
Laboratory testing should include a serologic autoantibody panel to evaluate for connective tissue diseases that can manifest as interstitial lung disease, including rheumatoid arthritis, dermatopolymyositis, scleroderma, Sjögren syndrome, and undifferentiated or mixed connective tissue disease. Typical preliminary laboratory tests are antinuclear antibody, rheumatoid factor, erythrocyte sedimentation rate, and C-reactive protein. Others may include anticyclic citrullinated peptide (anti-CCP), anti-Scl-70, anti-RNP, anti-SS-A, anti-SS-B, and anti-Jo-1.16 The breadth of the panel should depend on patient demographics and findings in the history or physical examination that increase or decrease the likelihood of a connective tissue disease.
Lung function testing
Assessing the patient’s pulmonary physiology should include spirometry, DLCO, and body plethysmography (lung volumes). In most cases, IPF manifests with restrictive physiology. Once restrictive physiology is confirmed with a low total lung capacity, FVC testing can be used as a longitudinal surrogate, as it is less expensive and easier for the patient to perform. In general, a lower total lung capacity or FVC indicates more severe impairment.
The DLCO serves as another marker of severity but is less reliable due to baseline variability and difficulties performing the maneuver.
A 6-minute walk test is another crucial physiologic assessment tool that can quantify exertional hypoxemia and functional status (ie, distance walked), and can assist in prognosis.
Imaging
Most patients undergo chest radiography in the workup for undiagnosed dyspnea. However, chest radiography is not adequate to formulate an accurate diagnosis in suspected interstitial lung disease, and a normal radiograph cannot exclude changes that might reflect early phases of the disease. As the disease progresses, the plain radiograph can show reticulonodular opacities and honeycombing in the peripheral and lower lung zones (Figure 3).34
The decision whether to order HRCT in the workup for a patient who has dyspnea and a normal chest radiograph is challenging. We recommend cross-sectional imaging when physiologic testing shows restriction or low DLCO, or when there is a high index of suspicion for underlying lung disease as the cause of symptoms.
Expert consultation can aid with this decision, especially when the underlying cause of dyspnea remains unclear after initial studies have been completed. Otherwise, HRCT is an essential test in the evaluation of interstitial lung disease.
Bronchoscopy’s role controversial
If the pattern on HRCT is nondiagnostic, then surgical biopsy is necessary, and the diagnosis of IPF requires a histologic pattern of UIP as described above.16,35
Although bronchoscopy can be valuable if an alternative diagnosis such as sarcoidosis or chronic hypersensitivity pneumonitis is suspected, the role of bronchoscopic biopsy in the workup of IPF is controversial. The patchy nature of UIP does not lend itself to the relatively small biopsy samples obtained through bronchoscopy.36,37
Surgical biopsy options
The favored biopsy approach is surgical, using either an open or a video-assisted thoracoscopic technique. The latter is preferred as it is less invasive, requires a shorter length of hospital stay, and allows a faster recovery.38 Bronchoscopic cryobiopsy, currently under investigation, is a potentially valuable tool whose role in diagnosing IPF is evolving.
Frequently, neither HRCT nor surgical lung biopsy demonstrates UIP, making the definitive diagnosis of IPF difficult. Moreover, some patients with nondiagnostic HRCT results are unable to tolerate surgical lung biopsy because of severely impaired lung function or other comorbidities.
The role of multidisciplinary discussions
When surgical lung biopsy is not possible, current practice at leading centers uses a multidisciplinary approach to allow for a confident diagnosis.30,39 Discussions take place between pulmonologists, pathologists, radiologists, and other specialists to collectively consider all aspects of a case before rendering a consensus opinion on the diagnosis and the management plan. If the discussion leads to a consensus diagnosis of IPF, then the patient’s clinician can move forward with treatment options. If not, the group can recommend further workup or alternative diagnoses and treatment regimens. The multidisciplinary group is also well positioned to consider the relative risks and benefits of moving forward with surgical lung biopsy for individual patients.
This approach illustrates the importance of referring these patients to centers of excellence in diagnosing and managing complex cases of interstitial lung disease, including IPF.40
TREATMENT OF IPF
Antifibrotic therapy
Antifibrotic therapy is a choice between pirfenidone and nintedanib.
Pirfenidone, which has an undefined molecular target, was approved based on the results of 3 trials.41,42 Pooled analyses from these trials showed a reduction in the decline from baseline in FVC percent predicted and improved progression-free survival.43 Pooled and meta-analyses of pirfenidone clinical trials have shown a mortality benefit, although no individual study has shown such an effect on mortality rates.44
The major adverse effects of pirfenidone are gastrointestinal distress and photosensitivity rash.
Nintedanib is a triple tyrosine kinase inhibitor that broadly targets fibroblast growth factor, vascular endothelial growth factor, and platelet-derived growth factor receptors. Combined analysis of 2 concurrent trials45 showed that nintedanib reduced the decline in FVC, similarly to pirfenidone. The major adverse event associated with nintedanib was diarrhea. Since it inhibits vascular endothelial growth factor, there is a risk of hematologic complications such as bleeding or clotting events.
Because pirfenidone and nintedanib can increase aminotransferase levels, regular monitoring is recommended.
To date, no trial has compared pirfenidone and nintedanib in terms of their efficacy and tolerability. Therefore, the choice of agent is based on the patient’s preference after a discussion of potential risks and expected benefits, a review of each drug’s side effects, and consideration of comorbid conditions and physician experience.
Patients need to understand that these drugs slow the rate of decline in FVC but have not been shown to improve symptoms or functional status.
Corticosteroids are not routine
Corticosteroids should not be used routinely in the treatment of IPF. Although steroids, alone or in combination with other immunosuppressive medications, were commonly used for IPF in the past, such use was not based on results of randomized controlled trials.46 Retrospective controlled studies have failed to show that corticosteroids improve mortality rates in IPF; indeed, they have shown that corticosteroids confer substantial morbidity.47,48 In addition, a randomized controlled trial combining corticosteroids with N-acetylcysteine and azathioprine was stopped early due to an increased risk of death and hospitalization.49 Collectively, these data suggest that corticosteroids confer no benefit and are potentially harmful. Their use in IPF is discouraged, and the joint international guidelines recommend against immunosuppression to treat IPF.16
Other treatments
The guidelines offer additional suggestions for the management of IPF.
Preliminary evidence suggests that microaspiration associated with abnormal gastroesophageal acid reflux is a risk factor for IPF. As such, there is a weak recommendation for aggressive treatment of reflux disease.50 However, because evidence suggests that proton-pump inhibitor therapy may be associated with adverse renal or central nervous system effects, this recommendation bears caution. It is hoped that ongoing studies will provide further insight into the role of acid-suppression in the management of IPF.51,52
Further treatment recommendations include best supportive management such as supplemental oxygen, pulmonary rehabilitation, and vaccinations.
Prompt referral for lung transplant is imperative. IPF is now the most common indication for lung transplant, and given the poor overall prognosis of advanced IPF, transplant confers a survival benefit in appropriately selected patients.53,54 Table 2 provides an evidence-based checklist for the workup and management of IPF.
ACUTE EXACERBATIONS OF IPF
The unpredictable nature of IPF can manifest in the form of acute exacerbations without an identifiable cause. The loosely defined diagnostic criteria for the diagnosis of acute exacerbations are a previous or new diagnosis of IPF, worsening or development of dyspnea in the last 30 days, and new bilateral ground-glass or consolidative changes with a background of UIP on HRCT.16
A new definition has been proposed55 to facilitate research in the characterization and treatment of acute exacerbations of IPF. The new definition includes all causes of respiratory deterioration except for heart failure and volume overload. It is less strict about the 30-day time frame. This newer definition is based on the lack of evidence differentiating outcomes when an acute deterioration is associated with known or unknown etiologies.55
The incidence of acute exacerbations is variable, with a 1- and 3-year incidence ranging between 8.6% and 23.9% depending on the criteria used.56 In general, acute exacerbations carry a grim prognosis, with a median life expectancy of 2.2 months.57
There is no approved therapy for exacerbations of IPF. Rather, treatment is mainly supportive with supplemental oxygen and mechanical ventilation. Current guidelines have a weak recommendation for the use of corticosteroids, but there are no recommendations regarding dose, route, or duration of therapy. Other treatments, primarily immunomodulatory agents, have been suggested but lack evidence of benefit.
Acknowledgments: Pathology images were provided by Carol Farver, MD, Pathology Institute, Cleveland Clinic. Radiology images were provided by Ruchi Yadav, MD, Imaging Institute, Cleveland Clinic.
- Brown KK, Raghu G. Medical treatment for pulmonary fibrosis: current trends, concepts, and prospects. Clin Chest Med 2004; 25(4):759–772, vii. doi:10.1016/j.ccm.2004.08.003
- Ryerson CJ, Collard HR. Update on the diagnosis and classification of ILD. Curr Opin Pulm Med 2013; 19(5):453–459. doi:10.1097/MCP.0b013e328363f48d
- Raghu G, Chen SY, Yeh WS, et al. Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001-11. Lancet Respir Med 2014; 2(7):566–572. doi:10.1016/S2213-2600(14)70101-8
- Nalysnyk L, Cid-Ruzafa J, Rotella P, Esser D. Incidence and prevalence of idiopathic pulmonary fibrosis: review of the literature. Eur Respir Rev 2012; 21(126):355–361. doi:10.1183/09059180.00002512
- Hutchinson J, Fogarty A, Hubbard R, McKeever T. Global incidence and mortality of idiopathic pulmonary fibrosis: a systematic review. Eur Respir J 2015; 46(3):795–806. doi:10.1183/09031936.00185114
- National Lung Screening Trial Research Team; Aberle DR, Adams AM, Berg CD, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med 2011; 365(5):395–409. doi:10.1056/NEJMoa1102873
- Jin GY, Lynch D, Chawla A, et al. Interstitial lung abnormalities in a CT lung cancer screening population: prevalence and progression rate. Radiology 2013; 268(2):563–571. doi:10.1148/radiol.13120816
- Southern BD, Scheraga RG, Yadav R. Managing interstitial lung disease detected on CT during lung cancer screening. Cleve Clin J Med 2016; 83(1):55–65. doi:10.3949/ccjm.83a.14157
- King TE Jr, Schwarz MI, Brown K, et al. Idiopathic pulmonary fibrosis: relationship between histopathologic features and mortality. Am J Respir Crit Care Med 2001; 164(5):1025–1032. doi:10.1164/ajrccm.164.6.2001056
- Lamas DJ, Kawut SM, Bagiella E, et al. Delayed access and survival in idiopathic pulmonary fibrosis: a cohort study. Am J Respir Crit Care Med 2011; 184(7):842–847. doi:10.1164/rccm.201104-0668OC
- Jo HE, Glaspole I, Moodley Y, et al. Disease progression in idiopathic pulmonary fibrosis with mild physiological impairment: analysis from the Australian IPF registry. BMC Pulm Med 2018; 18(1):19. doi:10.1186/s12890-018-0575-y
- Yang IV, Schwartz DA. Epigenetics of idiopathic pulmonary fibrosis. Transl Res 2015; 165(1):48–60. doi:10.1016/j.trsl.2014.03.011
- King TE Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet 2011; 378(9807):1949–1961. doi:10.1016/S0140-6736(11)60052-4
- Meltzer EB, Noble PW. Idiopathic pulmonary fibrosis. Orphanet J Rare Dis 2008; 3:8. doi:10.1186/1750-1172-3-8
- Raghu G. Idiopathic pulmonary fibrosis. A rational clinical approach. Chest 1987; 92(1):148–154. doi:10.1378/chest.92.1.148
- Raghu G, Collard HR, Egan JJ, et al; ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183(6):788–824. doi:10.1164/rccm.2009-040GL
- Panos RJ, Mortenson RL, Niccoli SA, King TE Jr. Clinical deterioration in patients with idiopathic pulmonary fibrosis: causes and assessment. Am J Med 1990; 88(4):396–404. doi:10.1016/0002-9343(90)90495-Y
- Ley B, Collard HR, King TE Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011; 183(4):431–440. doi:10.1164/rccm.201006-0894CI
- Collard HR, King TE Jr, Bartelson BB, Vourlekis JS, Schwarz MI, Brown KK. Changes in clinical and physiologic variables predict survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2003; 168(5):538–542. doi:10.1164/rccm.200211-1311OC
- Flaherty KR, Andrei AC, Murray S, et al. Idiopathic pulmonary fibrosis: prognostic value of changes in physiology and six-minute-walk test. Am J Respir Crit Care Med 2006; 174(7):803–809. doi:10.1164/rccm.200604-488OC
- Jegal Y, Kim DS, Shim TS, et al. Physiology is a stronger predictor of survival than pathology in fibrotic interstitial pneumonia. Am J Respir Crit Care Med 2005; 171(6):639–644. doi:10.1164/rccm.200403-331OC
- Latsi PI, du Bois RM, Nicholson AG, et al. Fibrotic idiopathic interstitial pneumonia: the prognostic value of longitudinal functional trends. Am J Respir Crit Care Med 2003; 168(5):531–537. doi:10.1164/rccm.200210-1245OC
- King CS, Nathan SD. Idiopathic pulmonary fibrosis: effects and optimal management of comorbidities. Lancet Respir Med 2017; 5(1):72–84. doi:10.1016/S2213-2600(16)30222-3
- Ley B, Ryerson CJ, Vittinghoff E, et al. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med 2012; 156(1):684–691. doi:10.7326/0003-4819-156-10-201205150-00004
- Rudd RM, Prescott RJ, Chalmers JC, Johnston ID; Fibrosing Alveolitis Subcommittee of the Research Committee of the British Thoracic Society. British Thoracic Society study on cryptogenic fibrosing alveolitis: response to treatment and survival. Thorax 2007; 62(1):62–66. doi:10.1136/thx.2005.045591
- Gutsche M, Rosen GD, Swigris JJ. Connective tissue disease-associated interstitial lung disease: a review. Curr Respir Care Rep 2012; 1:224–232. doi:10.1007/s13665-012-0028-7
- Park JH, Kim DS, Park IN, et al. Prognosis of fibrotic interstitial pneumonia: idiopathic versus collagen vascular disease-related subtypes. Am J Respir Crit Care Med 2007; 175(7):705–711. doi:10.1164/rccm.200607-912OC
- Taskar VS, Coultas DB. Is idiopathic pulmonary fibrosis an environmental disease? Proc Am Thorac Soc 2006; 3(4):293–298. doi:10.1513/pats.200512-131TK
- Vourlekis JS, Schwarz MI, Cherniack RM, et al. The effect of pulmonary fibrosis on survival in patients with hypersensitivity pneumonitis. Am J Med 2004; 116(10):662–668. doi:10.1016/j.amjmed.2003.12.030
- Brownell R, Moua T, Henry TS, et al. The use of pretest probability increases the value of high-resolution CT in diagnosing usual interstitial pneumonia. Thorax 2017; 72(5):424–429. doi:10.1136/thoraxjnl-2016-209671
- Baumgartner KB, Samet JM, Stidley CA, Colby TV, Waldron JA. Cigarette smoking: a risk factor for idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 1997; 155(1):242–248. doi:10.1164/ajrccm.155.1.9001319
- Selman M, Pardo A, King TE Jr. Hypersensitivity pneumonitis: insights in diagnosis and pathobiology. Am J Respir Crit Care Med 2012; 186(4):314–324. doi:10.1164/rccm.201203-0513CI
- Schwaiblmair M, Behr W, Haeckel T, Markl B, Foerg W, Berghaus T. Drug induced interstitial lung disease. Open Respir Med J 2012; 6:63–74. doi:10.2174/1874306401206010063
- Grenier P, Valeyre D, Cluzel P, Brauner MW, Lenoir S, Chastang C. Chronic diffuse interstitial lung disease: diagnostic value of chest radiography and high-resolution CT. Radiology 1991; 179(1):123–132. doi:10.1148/radiology.179.1.2006262
- Lynch JP 3rd, Huynh RH, Fishbein MC, Saggar R, Belperio JA, Weigt SS. Idiopathic pulmonary fibrosis: epidemiology, clinical features, prognosis, and management. Semin Respir Crit Care Med 2016; 37(3):331–357. doi:10.1055/s-0036-1582011
- Berbescu EA, Katzenstein AL, Snow JL, Zisman DA. Transbronchial biopsy in usual interstitial pneumonia. Chest 2006; 129(5):1126–1131. doi:10.1378/chest.129.5.1126
- Ohshimo S, Bonella F, Cui A, et al. Significance of bronchoalveolar lavage for the diagnosis of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2009; 179(11):1043–1047. doi:10.1164/rccm.200808-1313OC
- Oparka J, Yan TD, Ryan E, Dunning J. Does video-assisted thoracic surgery provide a safe alternative to conventional techniques in patients with limited pulmonary function who are otherwise suitable for lung resection? Interact Cardiovasc Thorac Surg 2013; 17(1):159–162. doi:10.1093/icvts/ivt097
- Flaherty KR, King TE Jr, Raghu G, et al. Idiopathic interstitial pneumonia: what is the effect of a multidisciplinary approach to diagnosis? Am J Respir Crit Care Med 2004; 170(8):904–910. doi:10.1164/rccm.200402-147OC
- Walsh SL, Wells AU, Desai SR, et al. Multicentre evaluation of multidisciplinary team meeting agreement on diagnosis in diffuse parenchymal lung disease: a case-cohort study. Lancet Respir Med 2016; 4(7):557–565. doi:10.1016/S2213-2600(16)30033-9
- King TE Jr, Bradford WZ, Castro-Bernardini S, et al; ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22):2083–2092. doi:10.1056/NEJMoa1402582
- Noble PW, Albera C, Bradford WZ, et al; CAPACITY Study Group. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet 2011; 377(9779):1760–1769. doi:10.1016/S0140-6736(11)60405-4
- Noble PW, Albera C, Bradford WZ, et al. Pirfenidone for idiopathic pulmonary fibrosis: analysis of pooled data from three multinational phase 3 trials. Eur Respir J 2016; 47(1):243–253. doi:10.1183/13993003.00026-2015
- Nathan SD, Albera C, Bradford WZ, et al. Effect of pirfenidone on mortality: pooled analyses and meta-analyses of clinical trials in idiopathic pulmonary fibrosis. Lancet Respir Med 2017; 5(1):33–41. doi:10.1016/S2213-2600(16)30326-5
- Richeldi L, du Bois RM, Raghu G, et al; INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22):2071–2082. doi:10.1056/NEJMoa1402584
- Richeldi L, Davies HR, Ferrara G, Franco F. Corticosteroids for idiopathic pulmonary fibrosis. Cochrane Database Syst Rev 2003: 3:CD002880. doi:10.1002/14651858.CD002880
- Douglas WW, Ryu JH, Schroeder DR. Idiopathic pulmonary fibrosis: impact of oxygen and colchicine, prednisone, or no therapy on survival. Am J Respir Crit Care Med 2000; 161(4 pt 1):1172–1178. doi:10.1164/ajrccm.161.4.9907002
- Gay SE, Kazerooni EA, Toews GB, et al. Idiopathic pulmonary fibrosis: predicting response to therapy and survival. Am J Respir Crit Care Med 1998; 157(4 pt 1):1063–1072. doi:10.1164/ajrccm.157.4.9703022
- Idiopathic Pulmonary Fibrosis Clinical Research Network; Raghu G, Anstrom KJ, King TE Jr, Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med 2012; 366(21):1968–1977. doi:10.1056/NEJMoa1113354
- Raghu G, Freudenberger TD, Yang S, et al. High prevalence of abnormal acid gastro-oesophageal reflux in idiopathic pulmonary fibrosis. Eur Respir J 2006; 27(1):136–142. doi:10.1183/09031936.06.00037005
- Gomm W, von Holt K, Thome F, et al. Association of proton pump inhibitors with risk of dementia: a pharmacoepidemiological claims data analysis. JAMA Neurol 2016; 73(4):410–416. doi:10.1001/jamaneurol.2015.4791
- Xie Y, Bowe B, Li T, Xian H, Balasubramanian S, Al-Aly Z. Proton pump inhibitors and risk of incident CKD and progression to ESRD. J Am Soc Nephrol 2016; 27(10):3153–3163. doi:10.1681/ASN.2015121377
- Thabut G, Mal H, Castier Y, et al. Survival benefit of lung transplantation for patients with idiopathic pulmonary fibrosis. J Thorac Cardiovasc Surg 2003; 126(2):469–475. doi:10.1016/S0022-5223(03)00600-7
- Valapour M, Skeans MA, Smith JM, et al. Lung. Am J Transplant 2016; 16(suppl 2):141–168. doi:10.1111/ajt.13671
- Collard HR, Ryerson CJ, Corte TJ, et al. Acute exacerbation of idiopathic pulmonary fibrosis. An International Working Group Report. Am J Respir Crit Care Med 2016; 194(3):265–275. doi:10.1164/rccm.201604-0801CI
- Kondoh Y, Taniguchi H, Katsuta T, et al. Risk factors of acute exacerbation of idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis 2010; 27(2):103–110. doi:10.1016/j.resinv.2015.04.005
- Song JW, Hong SB, Lim CM, Koh Y, Kim DS. Acute exacerbation of idiopathic pulmonary fibrosis: incidence, risk factors and outcome. Eur Respir J 2011; 37(2):356–363. doi:10.1183/09031936.00159709
- Brown KK, Raghu G. Medical treatment for pulmonary fibrosis: current trends, concepts, and prospects. Clin Chest Med 2004; 25(4):759–772, vii. doi:10.1016/j.ccm.2004.08.003
- Ryerson CJ, Collard HR. Update on the diagnosis and classification of ILD. Curr Opin Pulm Med 2013; 19(5):453–459. doi:10.1097/MCP.0b013e328363f48d
- Raghu G, Chen SY, Yeh WS, et al. Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001-11. Lancet Respir Med 2014; 2(7):566–572. doi:10.1016/S2213-2600(14)70101-8
- Nalysnyk L, Cid-Ruzafa J, Rotella P, Esser D. Incidence and prevalence of idiopathic pulmonary fibrosis: review of the literature. Eur Respir Rev 2012; 21(126):355–361. doi:10.1183/09059180.00002512
- Hutchinson J, Fogarty A, Hubbard R, McKeever T. Global incidence and mortality of idiopathic pulmonary fibrosis: a systematic review. Eur Respir J 2015; 46(3):795–806. doi:10.1183/09031936.00185114
- National Lung Screening Trial Research Team; Aberle DR, Adams AM, Berg CD, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med 2011; 365(5):395–409. doi:10.1056/NEJMoa1102873
- Jin GY, Lynch D, Chawla A, et al. Interstitial lung abnormalities in a CT lung cancer screening population: prevalence and progression rate. Radiology 2013; 268(2):563–571. doi:10.1148/radiol.13120816
- Southern BD, Scheraga RG, Yadav R. Managing interstitial lung disease detected on CT during lung cancer screening. Cleve Clin J Med 2016; 83(1):55–65. doi:10.3949/ccjm.83a.14157
- King TE Jr, Schwarz MI, Brown K, et al. Idiopathic pulmonary fibrosis: relationship between histopathologic features and mortality. Am J Respir Crit Care Med 2001; 164(5):1025–1032. doi:10.1164/ajrccm.164.6.2001056
- Lamas DJ, Kawut SM, Bagiella E, et al. Delayed access and survival in idiopathic pulmonary fibrosis: a cohort study. Am J Respir Crit Care Med 2011; 184(7):842–847. doi:10.1164/rccm.201104-0668OC
- Jo HE, Glaspole I, Moodley Y, et al. Disease progression in idiopathic pulmonary fibrosis with mild physiological impairment: analysis from the Australian IPF registry. BMC Pulm Med 2018; 18(1):19. doi:10.1186/s12890-018-0575-y
- Yang IV, Schwartz DA. Epigenetics of idiopathic pulmonary fibrosis. Transl Res 2015; 165(1):48–60. doi:10.1016/j.trsl.2014.03.011
- King TE Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet 2011; 378(9807):1949–1961. doi:10.1016/S0140-6736(11)60052-4
- Meltzer EB, Noble PW. Idiopathic pulmonary fibrosis. Orphanet J Rare Dis 2008; 3:8. doi:10.1186/1750-1172-3-8
- Raghu G. Idiopathic pulmonary fibrosis. A rational clinical approach. Chest 1987; 92(1):148–154. doi:10.1378/chest.92.1.148
- Raghu G, Collard HR, Egan JJ, et al; ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183(6):788–824. doi:10.1164/rccm.2009-040GL
- Panos RJ, Mortenson RL, Niccoli SA, King TE Jr. Clinical deterioration in patients with idiopathic pulmonary fibrosis: causes and assessment. Am J Med 1990; 88(4):396–404. doi:10.1016/0002-9343(90)90495-Y
- Ley B, Collard HR, King TE Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011; 183(4):431–440. doi:10.1164/rccm.201006-0894CI
- Collard HR, King TE Jr, Bartelson BB, Vourlekis JS, Schwarz MI, Brown KK. Changes in clinical and physiologic variables predict survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2003; 168(5):538–542. doi:10.1164/rccm.200211-1311OC
- Flaherty KR, Andrei AC, Murray S, et al. Idiopathic pulmonary fibrosis: prognostic value of changes in physiology and six-minute-walk test. Am J Respir Crit Care Med 2006; 174(7):803–809. doi:10.1164/rccm.200604-488OC
- Jegal Y, Kim DS, Shim TS, et al. Physiology is a stronger predictor of survival than pathology in fibrotic interstitial pneumonia. Am J Respir Crit Care Med 2005; 171(6):639–644. doi:10.1164/rccm.200403-331OC
- Latsi PI, du Bois RM, Nicholson AG, et al. Fibrotic idiopathic interstitial pneumonia: the prognostic value of longitudinal functional trends. Am J Respir Crit Care Med 2003; 168(5):531–537. doi:10.1164/rccm.200210-1245OC
- King CS, Nathan SD. Idiopathic pulmonary fibrosis: effects and optimal management of comorbidities. Lancet Respir Med 2017; 5(1):72–84. doi:10.1016/S2213-2600(16)30222-3
- Ley B, Ryerson CJ, Vittinghoff E, et al. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med 2012; 156(1):684–691. doi:10.7326/0003-4819-156-10-201205150-00004
- Rudd RM, Prescott RJ, Chalmers JC, Johnston ID; Fibrosing Alveolitis Subcommittee of the Research Committee of the British Thoracic Society. British Thoracic Society study on cryptogenic fibrosing alveolitis: response to treatment and survival. Thorax 2007; 62(1):62–66. doi:10.1136/thx.2005.045591
- Gutsche M, Rosen GD, Swigris JJ. Connective tissue disease-associated interstitial lung disease: a review. Curr Respir Care Rep 2012; 1:224–232. doi:10.1007/s13665-012-0028-7
- Park JH, Kim DS, Park IN, et al. Prognosis of fibrotic interstitial pneumonia: idiopathic versus collagen vascular disease-related subtypes. Am J Respir Crit Care Med 2007; 175(7):705–711. doi:10.1164/rccm.200607-912OC
- Taskar VS, Coultas DB. Is idiopathic pulmonary fibrosis an environmental disease? Proc Am Thorac Soc 2006; 3(4):293–298. doi:10.1513/pats.200512-131TK
- Vourlekis JS, Schwarz MI, Cherniack RM, et al. The effect of pulmonary fibrosis on survival in patients with hypersensitivity pneumonitis. Am J Med 2004; 116(10):662–668. doi:10.1016/j.amjmed.2003.12.030
- Brownell R, Moua T, Henry TS, et al. The use of pretest probability increases the value of high-resolution CT in diagnosing usual interstitial pneumonia. Thorax 2017; 72(5):424–429. doi:10.1136/thoraxjnl-2016-209671
- Baumgartner KB, Samet JM, Stidley CA, Colby TV, Waldron JA. Cigarette smoking: a risk factor for idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 1997; 155(1):242–248. doi:10.1164/ajrccm.155.1.9001319
- Selman M, Pardo A, King TE Jr. Hypersensitivity pneumonitis: insights in diagnosis and pathobiology. Am J Respir Crit Care Med 2012; 186(4):314–324. doi:10.1164/rccm.201203-0513CI
- Schwaiblmair M, Behr W, Haeckel T, Markl B, Foerg W, Berghaus T. Drug induced interstitial lung disease. Open Respir Med J 2012; 6:63–74. doi:10.2174/1874306401206010063
- Grenier P, Valeyre D, Cluzel P, Brauner MW, Lenoir S, Chastang C. Chronic diffuse interstitial lung disease: diagnostic value of chest radiography and high-resolution CT. Radiology 1991; 179(1):123–132. doi:10.1148/radiology.179.1.2006262
- Lynch JP 3rd, Huynh RH, Fishbein MC, Saggar R, Belperio JA, Weigt SS. Idiopathic pulmonary fibrosis: epidemiology, clinical features, prognosis, and management. Semin Respir Crit Care Med 2016; 37(3):331–357. doi:10.1055/s-0036-1582011
- Berbescu EA, Katzenstein AL, Snow JL, Zisman DA. Transbronchial biopsy in usual interstitial pneumonia. Chest 2006; 129(5):1126–1131. doi:10.1378/chest.129.5.1126
- Ohshimo S, Bonella F, Cui A, et al. Significance of bronchoalveolar lavage for the diagnosis of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2009; 179(11):1043–1047. doi:10.1164/rccm.200808-1313OC
- Oparka J, Yan TD, Ryan E, Dunning J. Does video-assisted thoracic surgery provide a safe alternative to conventional techniques in patients with limited pulmonary function who are otherwise suitable for lung resection? Interact Cardiovasc Thorac Surg 2013; 17(1):159–162. doi:10.1093/icvts/ivt097
- Flaherty KR, King TE Jr, Raghu G, et al. Idiopathic interstitial pneumonia: what is the effect of a multidisciplinary approach to diagnosis? Am J Respir Crit Care Med 2004; 170(8):904–910. doi:10.1164/rccm.200402-147OC
- Walsh SL, Wells AU, Desai SR, et al. Multicentre evaluation of multidisciplinary team meeting agreement on diagnosis in diffuse parenchymal lung disease: a case-cohort study. Lancet Respir Med 2016; 4(7):557–565. doi:10.1016/S2213-2600(16)30033-9
- King TE Jr, Bradford WZ, Castro-Bernardini S, et al; ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22):2083–2092. doi:10.1056/NEJMoa1402582
- Noble PW, Albera C, Bradford WZ, et al; CAPACITY Study Group. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet 2011; 377(9779):1760–1769. doi:10.1016/S0140-6736(11)60405-4
- Noble PW, Albera C, Bradford WZ, et al. Pirfenidone for idiopathic pulmonary fibrosis: analysis of pooled data from three multinational phase 3 trials. Eur Respir J 2016; 47(1):243–253. doi:10.1183/13993003.00026-2015
- Nathan SD, Albera C, Bradford WZ, et al. Effect of pirfenidone on mortality: pooled analyses and meta-analyses of clinical trials in idiopathic pulmonary fibrosis. Lancet Respir Med 2017; 5(1):33–41. doi:10.1016/S2213-2600(16)30326-5
- Richeldi L, du Bois RM, Raghu G, et al; INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22):2071–2082. doi:10.1056/NEJMoa1402584
- Richeldi L, Davies HR, Ferrara G, Franco F. Corticosteroids for idiopathic pulmonary fibrosis. Cochrane Database Syst Rev 2003: 3:CD002880. doi:10.1002/14651858.CD002880
- Douglas WW, Ryu JH, Schroeder DR. Idiopathic pulmonary fibrosis: impact of oxygen and colchicine, prednisone, or no therapy on survival. Am J Respir Crit Care Med 2000; 161(4 pt 1):1172–1178. doi:10.1164/ajrccm.161.4.9907002
- Gay SE, Kazerooni EA, Toews GB, et al. Idiopathic pulmonary fibrosis: predicting response to therapy and survival. Am J Respir Crit Care Med 1998; 157(4 pt 1):1063–1072. doi:10.1164/ajrccm.157.4.9703022
- Idiopathic Pulmonary Fibrosis Clinical Research Network; Raghu G, Anstrom KJ, King TE Jr, Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med 2012; 366(21):1968–1977. doi:10.1056/NEJMoa1113354
- Raghu G, Freudenberger TD, Yang S, et al. High prevalence of abnormal acid gastro-oesophageal reflux in idiopathic pulmonary fibrosis. Eur Respir J 2006; 27(1):136–142. doi:10.1183/09031936.06.00037005
- Gomm W, von Holt K, Thome F, et al. Association of proton pump inhibitors with risk of dementia: a pharmacoepidemiological claims data analysis. JAMA Neurol 2016; 73(4):410–416. doi:10.1001/jamaneurol.2015.4791
- Xie Y, Bowe B, Li T, Xian H, Balasubramanian S, Al-Aly Z. Proton pump inhibitors and risk of incident CKD and progression to ESRD. J Am Soc Nephrol 2016; 27(10):3153–3163. doi:10.1681/ASN.2015121377
- Thabut G, Mal H, Castier Y, et al. Survival benefit of lung transplantation for patients with idiopathic pulmonary fibrosis. J Thorac Cardiovasc Surg 2003; 126(2):469–475. doi:10.1016/S0022-5223(03)00600-7
- Valapour M, Skeans MA, Smith JM, et al. Lung. Am J Transplant 2016; 16(suppl 2):141–168. doi:10.1111/ajt.13671
- Collard HR, Ryerson CJ, Corte TJ, et al. Acute exacerbation of idiopathic pulmonary fibrosis. An International Working Group Report. Am J Respir Crit Care Med 2016; 194(3):265–275. doi:10.1164/rccm.201604-0801CI
- Kondoh Y, Taniguchi H, Katsuta T, et al. Risk factors of acute exacerbation of idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis 2010; 27(2):103–110. doi:10.1016/j.resinv.2015.04.005
- Song JW, Hong SB, Lim CM, Koh Y, Kim DS. Acute exacerbation of idiopathic pulmonary fibrosis: incidence, risk factors and outcome. Eur Respir J 2011; 37(2):356–363. doi:10.1183/09031936.00159709
KEY POINTS
- IPF is characterized by a pattern of usual interstitial pneumonia on imaging and histopathology without another known etiology.
- We recommend early referral to a center specializing in interstitial lung disease to confirm the diagnosis and to initiate appropriate therapy.
- Specialized centers offer advice on prognosis, enrollment in disease registries and clinical trials, and candidacy for lung transplant.
It takes a village to care for the patient with idiopathic pulmonary fibrosis
Idiopathic pulmonary fibrosis (IPF) is a devastating progressive fibrosing interstitial lung disease associated with a high burden of morbidity and death.1 A clinical diagnosis of IPF is made only after careful interpretation of integrated clinical, radiologic, and often histopathologic data.
Interstitial lung disease encompasses a broad spectrum of parenchymal lung diseases, and a classification of IPF is restricted to a lung injury pattern of usual interstitial pneumonia (UIP) based on high-resolution computed tomography or surgical lung biopsy, after all known causes of UIP have been excluded.1
However, a lung injury pattern of UIP is not synonymous with IPF, as UIP can be seen with connective tissue disease, chronic hypersensitivity pneumonitis, drug toxicity, and sarcoidosis.1 As such, rendering a diagnosis of IPF requires a thorough evaluation to exclude such diverse potential etiologies.
In this issue of the Cleveland Clinic Journal of Medicine, Tolle and colleagues2 provide an up-to-date, broad overview of IPF focused on what the primary care provider needs to know about the disease. Their review is timely and serves as a useful primer for the practicing clinician.
The field of IPF is actively evolving, as this era has been witness to a recent paradigm shift in pharmacologic management. Immunosuppression is no longer recommended3 and may even be harmful.4 And the US Food and Drug Administration has approved 2 antifibrotic drugs—pirfenidone and nintedanib—that have been shown to delay progression of IPF.5,6
Primary care providers have a unique opportunity to play an integral role in the evaluation and care of patients with IPF, in particular with earlier disease recognition, initial disease assessment, and timely specialty consultative referral—as well as implementing a comprehensive longitudinal care plan.
EARLIER DISEASE RECOGNITION
IPF is a rare disease primarily affecting men over the age of 65.1 It is reasonable to presume that many or most of these individuals ultimately diagnosed with IPF are already seeking routine care for existing common medical conditions such as hypertension or dyslipidemia—or at least having periodic routine health maintenance assessments. Such evaluations may offer an opportunity for earlier recognition of an underlying fibrotic lung disease that may be subclinical in nature.
IPF has a lower-lung zone predominance. The importance of chest auscultation, particularly listening carefully to the lung bases, is poignantly highlighted in a recent editorial: “It is time that the stethoscope draped around the neck of physicians, which tends to be used for identification purposes rather than for medical diagnosis, be also the (presently only) genuine tool for an earlier diagnosis of IPF.”7
Advances in imaging also provide an opportunity for earlier diagnosis. Many patients undergo screening computed tomography for coronary calcium scoring or lung cancer surveillance, and these studies may incidentally identify subtle interstitial lung abnormalities. These incidental findings should lead to further investigation, as they have been shown to be functionally important and carry risk of progression to clinical interstitial lung disease.8
INITIAL ASSESSMENT, TIMELY REFERRAL
But whether evidence of interstitial lung disease is detected incidentally or during testing for respiratory symptoms, further evaluation is necessary. Primary care providers are uniquely positioned to initiate the assessment and to expedite and guide further evaluation and specialty referral consultation to ensure an accurate diagnosis. They can also help grade the severity of the disease with pulmonary function testing, oxygen assessments at rest and with ambulation, and ordering thoracic high-resolution computed tomography to provide valuable information about disease extent and interstitial lung disease pattern.
General practitioners may assess for features suggesting connective tissue disease that would warrant specific serologic testing and dedicated rheumatologic consultation.
Finally, given the rarity, complexity, and challenges of interstitial lung disease, an effective multidisciplinary team consisting of clinicians, radiologists, and pathologists enhances diagnostic accuracy.9 This may also help general practitioners deviate from normal patterns of referral to general pulmonary providers, and instead refer patients to specialized centers with dedicated clinical and research expertise in interstitial lung disease.
IMPLEMENTING A COMPREHENSIVE, LONGITUDINAL CARE PLAN
The primary care practitioner often has developed long-term relationships with patients ultimately diagnosed with IPF, and because of this is particularly well positioned to help implement a collaborative and comprehensive care plan. Logistical realities such as distance to a specialty center, limited insurance coverage for specialty visits, and limited specialty availability all reinforce the central role that primary care practitioners play in ensuring that patients adhere to a comprehensive treatment program.
Primary providers may be very experienced and more inclined to manage a number of the common and often important comorbid conditions seen in patients with IPF, such as gastroesophageal reflux disease, obstructive sleep apnea, and depression. Reinforcing to the patient the need to adhere to adjunctive therapies such as supplemental oxygen and pulmonary rehabilitation is another key opportunity to actively engage in the management of patients with IPF.
Primary providers may also play a central role in IPF care through prevention strategies such as smoking cessation and ensuring appropriate immunization against seasonal influenza, pneumococcal pneumonia, and pertussis, among other age-appropriate vaccinations.
With the introduction and expansion of use of nintedanib and pirfenidone for IPF over the past few years, general practitioners may be called on to help manage common gastrointestinal side effects associated with pirfenidone (primarily nausea) and nintedanib (primarily diarrhea), and to be aware of potential drug-drug interactions and other medication-related toxicities.
Finally, as IPF remains a progressive disease, primary care practitioners are often well positioned to help implement palliative care, hospice care, and end-of-life care.
Despite recent advances in treatment, IPF remains a devastating lung disease with a high degree of morbidity and mortality. It takes a village to help care for the IPF patient. And as key members of the healthcare team, primary care providers have unique and important opportunities to help in the early recognition, thorough assessment, and comprehensive management of patients with IPF.
- Raghu G, Collard HR, Egan JJ, et al; ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183(6):788–824. doi:10.1164/rccm.2009-040GL
- Tolle L, Southern BD, Culver D, Horowitz JC. Idiopathic pulmonary fibrosis: what primary care physicians need to know. Cleve Clin J Med 2018; 85(5):377–386. doi:10.3949/ccjm.85a.17018
- Raghu G, Richeldi L. Current approaches to the management of idiopathic pulmonary fibrosis. Respir Med 2017; 129:24–30. doi:10.1016/j.rmed.2017.05.017
- Idiopathic Pulmonary Fibrosis Clinical Research Network; Raghu G, Anstrom KJ, King TE Jr, Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med 2012; 366(21):1968–1977. doi:10.1056/NEJMoa1113354
- King TE Jr, Bradford WZ, Castro-Bernardini S, et al; ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22):2083–2092. doi:10.1056/NEJMoa1402582
- Richeldi L, du Bois RM, Raghu G, et al; INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22):2071–2082. doi:10.1056/NEJMoa1402584
- Cottin V, Cordier JF. Velcro crackles: the key for early diagnosis of idiopathic pulmonary fibrosis? Eur Respir J 2012; 40(3):519–521. doi:10.1183/09031936.00001612
- Doyle TJ, Hunninghake GM, Rosas IO. Subclinical interstitial lung disease: why you should care. Am J Respir Crit Care Med 2012; 185(11):1147–1153. doi:10.1164/rccm.201108-1420PP
- Walsh SLF, Maher TM, Kolb M, et al; IPF Project Consortium. Diagnostic accuracy of a clinical diagnosis of idiopathic pulmonary fibrosis: an international case-cohort study. Eur Respir J 2017; 50(2):1700936. doi:10.1183/13993003.00936-2017
Idiopathic pulmonary fibrosis (IPF) is a devastating progressive fibrosing interstitial lung disease associated with a high burden of morbidity and death.1 A clinical diagnosis of IPF is made only after careful interpretation of integrated clinical, radiologic, and often histopathologic data.
Interstitial lung disease encompasses a broad spectrum of parenchymal lung diseases, and a classification of IPF is restricted to a lung injury pattern of usual interstitial pneumonia (UIP) based on high-resolution computed tomography or surgical lung biopsy, after all known causes of UIP have been excluded.1
However, a lung injury pattern of UIP is not synonymous with IPF, as UIP can be seen with connective tissue disease, chronic hypersensitivity pneumonitis, drug toxicity, and sarcoidosis.1 As such, rendering a diagnosis of IPF requires a thorough evaluation to exclude such diverse potential etiologies.
In this issue of the Cleveland Clinic Journal of Medicine, Tolle and colleagues2 provide an up-to-date, broad overview of IPF focused on what the primary care provider needs to know about the disease. Their review is timely and serves as a useful primer for the practicing clinician.
The field of IPF is actively evolving, as this era has been witness to a recent paradigm shift in pharmacologic management. Immunosuppression is no longer recommended3 and may even be harmful.4 And the US Food and Drug Administration has approved 2 antifibrotic drugs—pirfenidone and nintedanib—that have been shown to delay progression of IPF.5,6
Primary care providers have a unique opportunity to play an integral role in the evaluation and care of patients with IPF, in particular with earlier disease recognition, initial disease assessment, and timely specialty consultative referral—as well as implementing a comprehensive longitudinal care plan.
EARLIER DISEASE RECOGNITION
IPF is a rare disease primarily affecting men over the age of 65.1 It is reasonable to presume that many or most of these individuals ultimately diagnosed with IPF are already seeking routine care for existing common medical conditions such as hypertension or dyslipidemia—or at least having periodic routine health maintenance assessments. Such evaluations may offer an opportunity for earlier recognition of an underlying fibrotic lung disease that may be subclinical in nature.
IPF has a lower-lung zone predominance. The importance of chest auscultation, particularly listening carefully to the lung bases, is poignantly highlighted in a recent editorial: “It is time that the stethoscope draped around the neck of physicians, which tends to be used for identification purposes rather than for medical diagnosis, be also the (presently only) genuine tool for an earlier diagnosis of IPF.”7
Advances in imaging also provide an opportunity for earlier diagnosis. Many patients undergo screening computed tomography for coronary calcium scoring or lung cancer surveillance, and these studies may incidentally identify subtle interstitial lung abnormalities. These incidental findings should lead to further investigation, as they have been shown to be functionally important and carry risk of progression to clinical interstitial lung disease.8
INITIAL ASSESSMENT, TIMELY REFERRAL
But whether evidence of interstitial lung disease is detected incidentally or during testing for respiratory symptoms, further evaluation is necessary. Primary care providers are uniquely positioned to initiate the assessment and to expedite and guide further evaluation and specialty referral consultation to ensure an accurate diagnosis. They can also help grade the severity of the disease with pulmonary function testing, oxygen assessments at rest and with ambulation, and ordering thoracic high-resolution computed tomography to provide valuable information about disease extent and interstitial lung disease pattern.
General practitioners may assess for features suggesting connective tissue disease that would warrant specific serologic testing and dedicated rheumatologic consultation.
Finally, given the rarity, complexity, and challenges of interstitial lung disease, an effective multidisciplinary team consisting of clinicians, radiologists, and pathologists enhances diagnostic accuracy.9 This may also help general practitioners deviate from normal patterns of referral to general pulmonary providers, and instead refer patients to specialized centers with dedicated clinical and research expertise in interstitial lung disease.
IMPLEMENTING A COMPREHENSIVE, LONGITUDINAL CARE PLAN
The primary care practitioner often has developed long-term relationships with patients ultimately diagnosed with IPF, and because of this is particularly well positioned to help implement a collaborative and comprehensive care plan. Logistical realities such as distance to a specialty center, limited insurance coverage for specialty visits, and limited specialty availability all reinforce the central role that primary care practitioners play in ensuring that patients adhere to a comprehensive treatment program.
Primary providers may be very experienced and more inclined to manage a number of the common and often important comorbid conditions seen in patients with IPF, such as gastroesophageal reflux disease, obstructive sleep apnea, and depression. Reinforcing to the patient the need to adhere to adjunctive therapies such as supplemental oxygen and pulmonary rehabilitation is another key opportunity to actively engage in the management of patients with IPF.
Primary providers may also play a central role in IPF care through prevention strategies such as smoking cessation and ensuring appropriate immunization against seasonal influenza, pneumococcal pneumonia, and pertussis, among other age-appropriate vaccinations.
With the introduction and expansion of use of nintedanib and pirfenidone for IPF over the past few years, general practitioners may be called on to help manage common gastrointestinal side effects associated with pirfenidone (primarily nausea) and nintedanib (primarily diarrhea), and to be aware of potential drug-drug interactions and other medication-related toxicities.
Finally, as IPF remains a progressive disease, primary care practitioners are often well positioned to help implement palliative care, hospice care, and end-of-life care.
Despite recent advances in treatment, IPF remains a devastating lung disease with a high degree of morbidity and mortality. It takes a village to help care for the IPF patient. And as key members of the healthcare team, primary care providers have unique and important opportunities to help in the early recognition, thorough assessment, and comprehensive management of patients with IPF.
Idiopathic pulmonary fibrosis (IPF) is a devastating progressive fibrosing interstitial lung disease associated with a high burden of morbidity and death.1 A clinical diagnosis of IPF is made only after careful interpretation of integrated clinical, radiologic, and often histopathologic data.
Interstitial lung disease encompasses a broad spectrum of parenchymal lung diseases, and a classification of IPF is restricted to a lung injury pattern of usual interstitial pneumonia (UIP) based on high-resolution computed tomography or surgical lung biopsy, after all known causes of UIP have been excluded.1
However, a lung injury pattern of UIP is not synonymous with IPF, as UIP can be seen with connective tissue disease, chronic hypersensitivity pneumonitis, drug toxicity, and sarcoidosis.1 As such, rendering a diagnosis of IPF requires a thorough evaluation to exclude such diverse potential etiologies.
In this issue of the Cleveland Clinic Journal of Medicine, Tolle and colleagues2 provide an up-to-date, broad overview of IPF focused on what the primary care provider needs to know about the disease. Their review is timely and serves as a useful primer for the practicing clinician.
The field of IPF is actively evolving, as this era has been witness to a recent paradigm shift in pharmacologic management. Immunosuppression is no longer recommended3 and may even be harmful.4 And the US Food and Drug Administration has approved 2 antifibrotic drugs—pirfenidone and nintedanib—that have been shown to delay progression of IPF.5,6
Primary care providers have a unique opportunity to play an integral role in the evaluation and care of patients with IPF, in particular with earlier disease recognition, initial disease assessment, and timely specialty consultative referral—as well as implementing a comprehensive longitudinal care plan.
EARLIER DISEASE RECOGNITION
IPF is a rare disease primarily affecting men over the age of 65.1 It is reasonable to presume that many or most of these individuals ultimately diagnosed with IPF are already seeking routine care for existing common medical conditions such as hypertension or dyslipidemia—or at least having periodic routine health maintenance assessments. Such evaluations may offer an opportunity for earlier recognition of an underlying fibrotic lung disease that may be subclinical in nature.
IPF has a lower-lung zone predominance. The importance of chest auscultation, particularly listening carefully to the lung bases, is poignantly highlighted in a recent editorial: “It is time that the stethoscope draped around the neck of physicians, which tends to be used for identification purposes rather than for medical diagnosis, be also the (presently only) genuine tool for an earlier diagnosis of IPF.”7
Advances in imaging also provide an opportunity for earlier diagnosis. Many patients undergo screening computed tomography for coronary calcium scoring or lung cancer surveillance, and these studies may incidentally identify subtle interstitial lung abnormalities. These incidental findings should lead to further investigation, as they have been shown to be functionally important and carry risk of progression to clinical interstitial lung disease.8
INITIAL ASSESSMENT, TIMELY REFERRAL
But whether evidence of interstitial lung disease is detected incidentally or during testing for respiratory symptoms, further evaluation is necessary. Primary care providers are uniquely positioned to initiate the assessment and to expedite and guide further evaluation and specialty referral consultation to ensure an accurate diagnosis. They can also help grade the severity of the disease with pulmonary function testing, oxygen assessments at rest and with ambulation, and ordering thoracic high-resolution computed tomography to provide valuable information about disease extent and interstitial lung disease pattern.
General practitioners may assess for features suggesting connective tissue disease that would warrant specific serologic testing and dedicated rheumatologic consultation.
Finally, given the rarity, complexity, and challenges of interstitial lung disease, an effective multidisciplinary team consisting of clinicians, radiologists, and pathologists enhances diagnostic accuracy.9 This may also help general practitioners deviate from normal patterns of referral to general pulmonary providers, and instead refer patients to specialized centers with dedicated clinical and research expertise in interstitial lung disease.
IMPLEMENTING A COMPREHENSIVE, LONGITUDINAL CARE PLAN
The primary care practitioner often has developed long-term relationships with patients ultimately diagnosed with IPF, and because of this is particularly well positioned to help implement a collaborative and comprehensive care plan. Logistical realities such as distance to a specialty center, limited insurance coverage for specialty visits, and limited specialty availability all reinforce the central role that primary care practitioners play in ensuring that patients adhere to a comprehensive treatment program.
Primary providers may be very experienced and more inclined to manage a number of the common and often important comorbid conditions seen in patients with IPF, such as gastroesophageal reflux disease, obstructive sleep apnea, and depression. Reinforcing to the patient the need to adhere to adjunctive therapies such as supplemental oxygen and pulmonary rehabilitation is another key opportunity to actively engage in the management of patients with IPF.
Primary providers may also play a central role in IPF care through prevention strategies such as smoking cessation and ensuring appropriate immunization against seasonal influenza, pneumococcal pneumonia, and pertussis, among other age-appropriate vaccinations.
With the introduction and expansion of use of nintedanib and pirfenidone for IPF over the past few years, general practitioners may be called on to help manage common gastrointestinal side effects associated with pirfenidone (primarily nausea) and nintedanib (primarily diarrhea), and to be aware of potential drug-drug interactions and other medication-related toxicities.
Finally, as IPF remains a progressive disease, primary care practitioners are often well positioned to help implement palliative care, hospice care, and end-of-life care.
Despite recent advances in treatment, IPF remains a devastating lung disease with a high degree of morbidity and mortality. It takes a village to help care for the IPF patient. And as key members of the healthcare team, primary care providers have unique and important opportunities to help in the early recognition, thorough assessment, and comprehensive management of patients with IPF.
- Raghu G, Collard HR, Egan JJ, et al; ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183(6):788–824. doi:10.1164/rccm.2009-040GL
- Tolle L, Southern BD, Culver D, Horowitz JC. Idiopathic pulmonary fibrosis: what primary care physicians need to know. Cleve Clin J Med 2018; 85(5):377–386. doi:10.3949/ccjm.85a.17018
- Raghu G, Richeldi L. Current approaches to the management of idiopathic pulmonary fibrosis. Respir Med 2017; 129:24–30. doi:10.1016/j.rmed.2017.05.017
- Idiopathic Pulmonary Fibrosis Clinical Research Network; Raghu G, Anstrom KJ, King TE Jr, Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med 2012; 366(21):1968–1977. doi:10.1056/NEJMoa1113354
- King TE Jr, Bradford WZ, Castro-Bernardini S, et al; ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22):2083–2092. doi:10.1056/NEJMoa1402582
- Richeldi L, du Bois RM, Raghu G, et al; INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22):2071–2082. doi:10.1056/NEJMoa1402584
- Cottin V, Cordier JF. Velcro crackles: the key for early diagnosis of idiopathic pulmonary fibrosis? Eur Respir J 2012; 40(3):519–521. doi:10.1183/09031936.00001612
- Doyle TJ, Hunninghake GM, Rosas IO. Subclinical interstitial lung disease: why you should care. Am J Respir Crit Care Med 2012; 185(11):1147–1153. doi:10.1164/rccm.201108-1420PP
- Walsh SLF, Maher TM, Kolb M, et al; IPF Project Consortium. Diagnostic accuracy of a clinical diagnosis of idiopathic pulmonary fibrosis: an international case-cohort study. Eur Respir J 2017; 50(2):1700936. doi:10.1183/13993003.00936-2017
- Raghu G, Collard HR, Egan JJ, et al; ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183(6):788–824. doi:10.1164/rccm.2009-040GL
- Tolle L, Southern BD, Culver D, Horowitz JC. Idiopathic pulmonary fibrosis: what primary care physicians need to know. Cleve Clin J Med 2018; 85(5):377–386. doi:10.3949/ccjm.85a.17018
- Raghu G, Richeldi L. Current approaches to the management of idiopathic pulmonary fibrosis. Respir Med 2017; 129:24–30. doi:10.1016/j.rmed.2017.05.017
- Idiopathic Pulmonary Fibrosis Clinical Research Network; Raghu G, Anstrom KJ, King TE Jr, Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med 2012; 366(21):1968–1977. doi:10.1056/NEJMoa1113354
- King TE Jr, Bradford WZ, Castro-Bernardini S, et al; ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22):2083–2092. doi:10.1056/NEJMoa1402582
- Richeldi L, du Bois RM, Raghu G, et al; INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22):2071–2082. doi:10.1056/NEJMoa1402584
- Cottin V, Cordier JF. Velcro crackles: the key for early diagnosis of idiopathic pulmonary fibrosis? Eur Respir J 2012; 40(3):519–521. doi:10.1183/09031936.00001612
- Doyle TJ, Hunninghake GM, Rosas IO. Subclinical interstitial lung disease: why you should care. Am J Respir Crit Care Med 2012; 185(11):1147–1153. doi:10.1164/rccm.201108-1420PP
- Walsh SLF, Maher TM, Kolb M, et al; IPF Project Consortium. Diagnostic accuracy of a clinical diagnosis of idiopathic pulmonary fibrosis: an international case-cohort study. Eur Respir J 2017; 50(2):1700936. doi:10.1183/13993003.00936-2017

An unusual complication of peritoneal dialysis
A 45-year-old man with end-stage renal disease secondary to hypertension presented with abdominal pain, nausea, vomiting, and fever. He had been on peritoneal dialysis for 15 years.
Results of initial laboratory testing were as follows:
- Sodium 137 mmol/L (reference range 136–144)
- Potassium 3.7 mmol/L (3.5–5.0)
- Bicarbonate 31 mmol/L (22–30)
- Creatinine 17.5 mg/dL (0.58–0.96)
- Blood urea nitrogen 57 mg/dL (7–21)
- Lactic acid 1.7 mmol/L (0.5–2.2)
- White blood cell count 14.34 × 109/L (3.70–11.0).
Blood cultures were negative. Peritoneal fluid analysis showed a white blood cell count of 1.2 × 109/L (reference range < 0.5 × 109/L) with 89% neutrophils, and an amylase level less than 3 U/L (reference range < 100). Fluid cultures were positive for coagulase-negative staphylococci and Staphylococcus epidermidis.
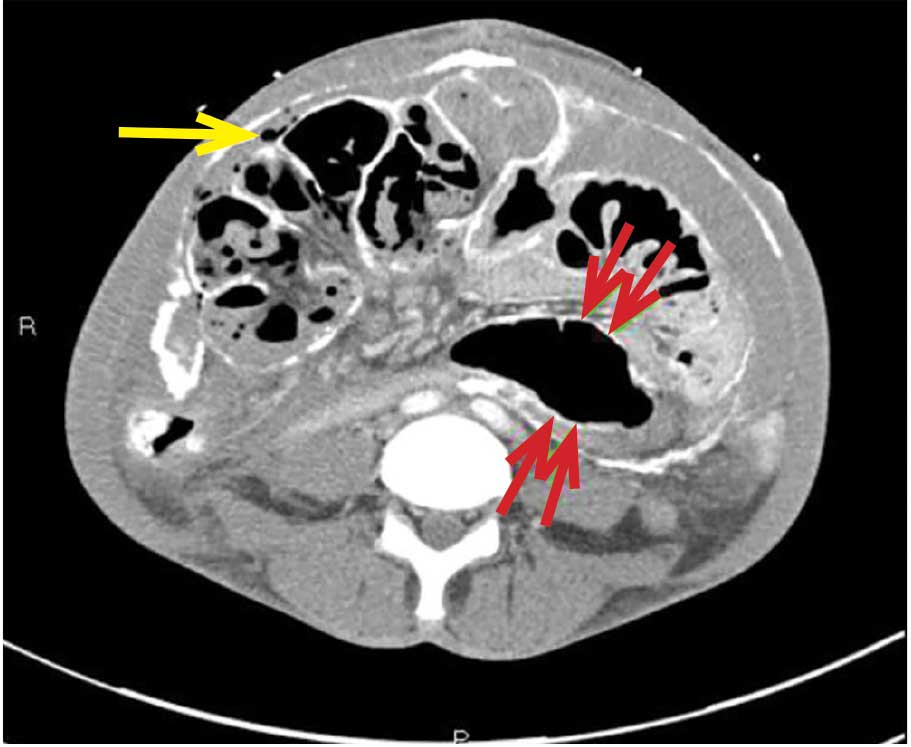
CAUSES AND CLINICAL FEATURES
Encapsulating peritoneal sclerosis is a devastating complication of peritoneal dialysis, occurring in 3% of patients on peritoneal dialysis. The mortality rate is above 40%.1,2 It is characterized by an initial inflammatory phase followed by extensive intraperitoneal fibrosis and encasement of bowel. Causes include prolonged exposure to peritoneal dialysis or glucose degradation products, a history of severe peritonitis, use of acetate as a dialysate buffer, and reaction to medications such as beta-blockers.3
Clinical features result from inflammation, ileus, and peritoneal adhesions and include abdominal pain, nausea, and vomiting. A high peritoneal transport rate, which often heralds development of encapsulating peritoneal sclerosis, leads to failure of ultrafiltration and to fluid retention.
CT is recommended for diagnosis and demonstrates peritoneal calcification with bowel thickening and dilation.
TREATMENT
Treatment entails stopping peritoneal dialysis, changing to hemodialysis, bowel rest, and corticosteroids. Successful treatment has been reported with a combination of corticosteroids and azathioprine.4,5 A retrospective study showed that adding the antifibrotic agent tamoxifen was associated with a decrease in the mortality rate.6 Bowel obstruction is a common complication, and surgery may be indicated. Enterolysis is a new surgical technique that has shown improved outcomes.7
- Kawaguchi Y, Saito A, Kawanishi H, et al. Recommendations on the management of encapsulating peritoneal sclerosis in Japan, 2005: diagnosis, predictive markers, treatment, and preventive measures. Perit Dial Int 2005; 25(suppl 4):S83–S95. pmid:16300277
- Lee HY, Kim BS, Choi HY, et al. Sclerosing encapsulating peritonitis as a complication of long-term continuous ambulatory peritoneal dialysis in Korea. Nephrology (Carlton) 2003; 8(suppl 2):S33–S39. doi:10.1046/J.1440-1797.8.S.11.X
- Kawaguchi Y, Tranaeus A. A historical review of encapsulating peritoneal sclerosis. Perit Dial Int 2005; 25(suppl 4):S7–S13. pmid:16300267
- Martins LS, Rodrigues AS, Cabrita AN, Guimaraes S. Sclerosing encapsulating peritonitis: a case successfully treated with immunosuppression. Perit Dial Int 1999; 19(5):478–481. pmid:11379862
- Wong CF, Beshir S, Khalil A, Pai P, Ahmad R. Successful treatment of encapsulating peritoneal sclerosis with azathioprine and prednisolone. Perit Dial Int 2005; 25(3):285–287. pmid:15981777
- Korte MR, Fieren MW, Sampimon DE, Lingsma HF, Weimar W, Betjes MG; investigators of the Dutch Multicentre EPS Study. Tamoxifen is associated with lower mortality of encapsulating peritoneal sclerosis: results of the Dutch Multicentre EPS Study. Nephrol Dial Transplant 2011; 26(2):691–697. doi:10.1093/ndt/gfq362
- Kawanishi H, Watanabe H, Moriishi M, Tsuchiya S. Successful surgical management of encapsulating peritoneal sclerosis. Perit Dial Int 2005; 25(suppl 4):S39–S47. pmid:16300271
A 45-year-old man with end-stage renal disease secondary to hypertension presented with abdominal pain, nausea, vomiting, and fever. He had been on peritoneal dialysis for 15 years.
Results of initial laboratory testing were as follows:
- Sodium 137 mmol/L (reference range 136–144)
- Potassium 3.7 mmol/L (3.5–5.0)
- Bicarbonate 31 mmol/L (22–30)
- Creatinine 17.5 mg/dL (0.58–0.96)
- Blood urea nitrogen 57 mg/dL (7–21)
- Lactic acid 1.7 mmol/L (0.5–2.2)
- White blood cell count 14.34 × 109/L (3.70–11.0).
Blood cultures were negative. Peritoneal fluid analysis showed a white blood cell count of 1.2 × 109/L (reference range < 0.5 × 109/L) with 89% neutrophils, and an amylase level less than 3 U/L (reference range < 100). Fluid cultures were positive for coagulase-negative staphylococci and Staphylococcus epidermidis.
CAUSES AND CLINICAL FEATURES
Encapsulating peritoneal sclerosis is a devastating complication of peritoneal dialysis, occurring in 3% of patients on peritoneal dialysis. The mortality rate is above 40%.1,2 It is characterized by an initial inflammatory phase followed by extensive intraperitoneal fibrosis and encasement of bowel. Causes include prolonged exposure to peritoneal dialysis or glucose degradation products, a history of severe peritonitis, use of acetate as a dialysate buffer, and reaction to medications such as beta-blockers.3
Clinical features result from inflammation, ileus, and peritoneal adhesions and include abdominal pain, nausea, and vomiting. A high peritoneal transport rate, which often heralds development of encapsulating peritoneal sclerosis, leads to failure of ultrafiltration and to fluid retention.
CT is recommended for diagnosis and demonstrates peritoneal calcification with bowel thickening and dilation.
TREATMENT
Treatment entails stopping peritoneal dialysis, changing to hemodialysis, bowel rest, and corticosteroids. Successful treatment has been reported with a combination of corticosteroids and azathioprine.4,5 A retrospective study showed that adding the antifibrotic agent tamoxifen was associated with a decrease in the mortality rate.6 Bowel obstruction is a common complication, and surgery may be indicated. Enterolysis is a new surgical technique that has shown improved outcomes.7
A 45-year-old man with end-stage renal disease secondary to hypertension presented with abdominal pain, nausea, vomiting, and fever. He had been on peritoneal dialysis for 15 years.
Results of initial laboratory testing were as follows:
- Sodium 137 mmol/L (reference range 136–144)
- Potassium 3.7 mmol/L (3.5–5.0)
- Bicarbonate 31 mmol/L (22–30)
- Creatinine 17.5 mg/dL (0.58–0.96)
- Blood urea nitrogen 57 mg/dL (7–21)
- Lactic acid 1.7 mmol/L (0.5–2.2)
- White blood cell count 14.34 × 109/L (3.70–11.0).
Blood cultures were negative. Peritoneal fluid analysis showed a white blood cell count of 1.2 × 109/L (reference range < 0.5 × 109/L) with 89% neutrophils, and an amylase level less than 3 U/L (reference range < 100). Fluid cultures were positive for coagulase-negative staphylococci and Staphylococcus epidermidis.
CAUSES AND CLINICAL FEATURES
Encapsulating peritoneal sclerosis is a devastating complication of peritoneal dialysis, occurring in 3% of patients on peritoneal dialysis. The mortality rate is above 40%.1,2 It is characterized by an initial inflammatory phase followed by extensive intraperitoneal fibrosis and encasement of bowel. Causes include prolonged exposure to peritoneal dialysis or glucose degradation products, a history of severe peritonitis, use of acetate as a dialysate buffer, and reaction to medications such as beta-blockers.3
Clinical features result from inflammation, ileus, and peritoneal adhesions and include abdominal pain, nausea, and vomiting. A high peritoneal transport rate, which often heralds development of encapsulating peritoneal sclerosis, leads to failure of ultrafiltration and to fluid retention.
CT is recommended for diagnosis and demonstrates peritoneal calcification with bowel thickening and dilation.
TREATMENT
Treatment entails stopping peritoneal dialysis, changing to hemodialysis, bowel rest, and corticosteroids. Successful treatment has been reported with a combination of corticosteroids and azathioprine.4,5 A retrospective study showed that adding the antifibrotic agent tamoxifen was associated with a decrease in the mortality rate.6 Bowel obstruction is a common complication, and surgery may be indicated. Enterolysis is a new surgical technique that has shown improved outcomes.7
- Kawaguchi Y, Saito A, Kawanishi H, et al. Recommendations on the management of encapsulating peritoneal sclerosis in Japan, 2005: diagnosis, predictive markers, treatment, and preventive measures. Perit Dial Int 2005; 25(suppl 4):S83–S95. pmid:16300277
- Lee HY, Kim BS, Choi HY, et al. Sclerosing encapsulating peritonitis as a complication of long-term continuous ambulatory peritoneal dialysis in Korea. Nephrology (Carlton) 2003; 8(suppl 2):S33–S39. doi:10.1046/J.1440-1797.8.S.11.X
- Kawaguchi Y, Tranaeus A. A historical review of encapsulating peritoneal sclerosis. Perit Dial Int 2005; 25(suppl 4):S7–S13. pmid:16300267
- Martins LS, Rodrigues AS, Cabrita AN, Guimaraes S. Sclerosing encapsulating peritonitis: a case successfully treated with immunosuppression. Perit Dial Int 1999; 19(5):478–481. pmid:11379862
- Wong CF, Beshir S, Khalil A, Pai P, Ahmad R. Successful treatment of encapsulating peritoneal sclerosis with azathioprine and prednisolone. Perit Dial Int 2005; 25(3):285–287. pmid:15981777
- Korte MR, Fieren MW, Sampimon DE, Lingsma HF, Weimar W, Betjes MG; investigators of the Dutch Multicentre EPS Study. Tamoxifen is associated with lower mortality of encapsulating peritoneal sclerosis: results of the Dutch Multicentre EPS Study. Nephrol Dial Transplant 2011; 26(2):691–697. doi:10.1093/ndt/gfq362
- Kawanishi H, Watanabe H, Moriishi M, Tsuchiya S. Successful surgical management of encapsulating peritoneal sclerosis. Perit Dial Int 2005; 25(suppl 4):S39–S47. pmid:16300271
- Kawaguchi Y, Saito A, Kawanishi H, et al. Recommendations on the management of encapsulating peritoneal sclerosis in Japan, 2005: diagnosis, predictive markers, treatment, and preventive measures. Perit Dial Int 2005; 25(suppl 4):S83–S95. pmid:16300277
- Lee HY, Kim BS, Choi HY, et al. Sclerosing encapsulating peritonitis as a complication of long-term continuous ambulatory peritoneal dialysis in Korea. Nephrology (Carlton) 2003; 8(suppl 2):S33–S39. doi:10.1046/J.1440-1797.8.S.11.X
- Kawaguchi Y, Tranaeus A. A historical review of encapsulating peritoneal sclerosis. Perit Dial Int 2005; 25(suppl 4):S7–S13. pmid:16300267
- Martins LS, Rodrigues AS, Cabrita AN, Guimaraes S. Sclerosing encapsulating peritonitis: a case successfully treated with immunosuppression. Perit Dial Int 1999; 19(5):478–481. pmid:11379862
- Wong CF, Beshir S, Khalil A, Pai P, Ahmad R. Successful treatment of encapsulating peritoneal sclerosis with azathioprine and prednisolone. Perit Dial Int 2005; 25(3):285–287. pmid:15981777
- Korte MR, Fieren MW, Sampimon DE, Lingsma HF, Weimar W, Betjes MG; investigators of the Dutch Multicentre EPS Study. Tamoxifen is associated with lower mortality of encapsulating peritoneal sclerosis: results of the Dutch Multicentre EPS Study. Nephrol Dial Transplant 2011; 26(2):691–697. doi:10.1093/ndt/gfq362
- Kawanishi H, Watanabe H, Moriishi M, Tsuchiya S. Successful surgical management of encapsulating peritoneal sclerosis. Perit Dial Int 2005; 25(suppl 4):S39–S47. pmid:16300271
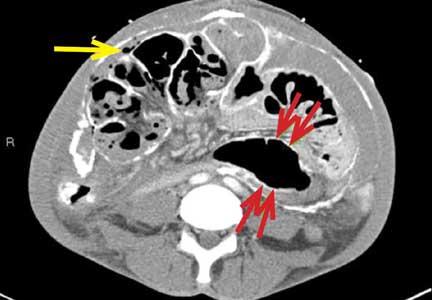
Proximal Humerus Fracture 3-D Modeling
ABSTRACT
The objective of this study is to determine the reproducibility and feasibility of using 3-dimensional (3-D) computer simulation of proximal humerus fracture computed tomography (CT) scans for fracture reduction. We hypothesized that anatomic reconstruction with 3-D models would be anatomically accurate and reproducible.
Preoperative CT scans of 28 patients with 3- and 4-part (AO classification 11-B1, 11-B2, 11-C1, 11-C2) proximal humerus fractures who were treated by hemiarthroplasty were converted into 3-D computer models. The displaced fractured fragments were anatomically reduced with computer simulation by 2 fellowship-trained shoulder surgeons, and measurements were made of the reconstructed proximal humerus.
The measurements of the reconstructed models had very good to excellent interobserver and intraobserver reliability. The reconstructions of these humerus fractures showed interclass correlation coefficients ranging from 0.71 to 0.93 between 1 observer and from 0.82 to 0.98 between 2 different observers. The fracture reduction was judged against normal proximal humerus geometry to determine reduction accuracy.
The 3-D modeling techniques used to reconstruct 3- and 4-part proximal humerus fractures were reliable and accurate. This technique of modeling and reconstructing proximal humerus fractures could be used to enhance the preoperative planning of open reduction and internal fixation or hemiarthroplasty for 3- and 4-part proximal humerus fractures.
The treatment of proximal humerus fractures is influenced by multiple factors, including patient age, associated injuries, bone quality, and fracture pattern. Three- and 4-part fractures are among the more severe of these fractures, which may result in vascular compromise to the humeral head, leading to avascular necrosis. Surgical goals for the management of these fractures are to optimize functional outcomes by re-creating a stable construct with a functional rotator cuff by open reduction and internal fixation (ORIF), hemiarthroplasty with tuberosity ORIF, or reverse shoulder replacement. Achieving a good outcome following hemiarthroplasty is dependent on many factors, including anatomic tuberosity healing and component positioning.1,2,3 Repairing the greater tuberosity in a near-anatomic position has been shown to greatly affect the results of hemiarthroplasty for fracture.3,4
Continue to: Three-dimensional (3-D) modeling...
Three-dimensional (3-D) modeling is increasingly being used in preoperative planning of shoulder arthroplasty and determining proper proximal humeral fracture treatment. 5 However, no studies have examined the reconstruction of a fractured proximal humerus into native anatomy using computer simulation. The purpose of this study is to determine the accuracy and reliability of anatomically reconstructing the preinjury proximal humerus using 3-D computer models created from postinjury computed tomography (CT) scans. The results of this study could lead to useful techniques employing CT–based models for patient-specific preoperative planning of proximal humeral fracture ORIF and during tuberosity reduction and fixation during hemiarthroplasty for fracture. We hypothesize that it is feasible to reconstruct the original anatomy of the proximal humerus by using 3-D computer modeling of proximal humerus fractures with high reliability based on interobserver and intraobserver review.
METHODS
After Institutional Review Board approval was obtained, we reviewed the medical records of consecutive patients with a diagnosis of proximal humeral fracture and the treatment codes for hemiarthroplasty from 2000 to 2013. Inclusion criteria included 3- and 4-part fractures (AO classifications 11-B1, 11-B2, 11-C1, 11-C2). CT scans with insufficient quality to differentiate bone from soft tissue (inadequate signal-to-noise ratio) were excluded from the study. A total of 28 patients with adequate CT scans met the criteria for inclusion in this study.
The CT scan protocol included 0.5-mm axial cuts with inclusion of the proximal humerus in the Digital Imaging and Communications in Medicine format. These CT scans were converted into patient-specific 3-D computer models of the shoulder using Mimics software (Materialise Inc.). The use of this software to produce anatomically accurate models has previously been verified in a shoulder model.6,7 The tuberosity fragments were then individually separated from each other using the voxel-selecting capabilities of 3-D software and manipulated with translation and rotation for anatomic reduction (Figures 1A-1D, Figure 2). 
The de-identified anatomically reconstructed shoulder models were then uploaded into Materialise’s Magics rapid prototyping software, and a user-defined humeral Cartesian coordinate system was defined with anatomic landmarks as reference points to standardize the position of each model (Figure 3).8,9 
A series of measurements were made on these models to assess the validity and reliability of the reassembly. The bicipital groove at the anatomic neck was used to measure humeral head version as described by Kummer and colleagues.10 The head-shaft angle, humeral head-greater tuberosity distance, humeral head-bicipital groove angle, and posterior and medial humeral head offset were measured directly on the reconstructed humerus.
Continue to: Two fellowship-trained shoulder...
Two fellowship-trained shoulder surgeons independently reassembled these fracture fragments via computer simulation. Interobserver reliability testing was conducted on these reconstructions by measuring the geometry between the 2 different surgeons’ reconstructions. Intraobserver reliability testing was conducted by 1 surgeon repeating the reconstructions with 4-week intervals between trials and measuring the geometry between the 2 different trials. The average dimensions of the reconstructed proximal humerus fractures were compared with the geometry of normal humeri reported in previously conducted anatomic studies.11,12,13
STATISTICS
The measured dimensions of the 28 reassembled proximal humeri models were averaged across all trials between the 2 fellowship-trained surgeons and compared with the range of normal dimensions of a healthy proximal humerus using the 2 one-sided tests (TOST) method for equivalence between 2 means given a range. The interobserver and intraobserver reliabilities were quantified using the interclass correlation coefficient. An excellent correlation was defined as a correlation coefficient >0.81; very good was defined as 0.61 to 0.80; and good was defined as 0.41 to 0.60.
RESULTS
Of the patients studied, 9 (32.1%) were male, and the average age at the time of CT scanning was 72 years. Of the 28 patients with fracture, 18 (64.2%) had 3-part fractures (AO classifications 11-B1, 11-B2), and 10 (35.8%) had 4-part fractures (AO classifications 11-C1, 11-C2). When examining the location of the intertubercular fracture line, we found that 13 (46.4%) fractures went through the bicipital groove. Of the remaining fracture lines, 9 (32.1%) extended into the greater tuberosity and 6 (21.4%) extended into the lesser tuberosity.
All users were able to reconstruct all 28 fractures using this technique. The average measured dimensions fell within the range of dimensions of a normal healthy proximal humerus specified in the literature to within a 95% confidence interval using the TOST for equivalence, in which we compared measured values with ranges reported in the literature (Table).11,12,13
Table. Dimensions of Proximal Humerus Geometry
| Normal Parameters | Average Dimensions From Trials | Dimensions From Literature |
| Head shaft angle | 43.5° ± 1° | 42.5° ± 12.5° |
| Head to greater tuberosity distance | 4.9 mm ± 0.4 mm | 8 mm ± 3.2 mm |
Head to bicipital groove angle (anatomic neck) | 26.4° ± 2° | 27.3° ± 14° |
| Posterior humeral head offset | 1.6 mm ± 0.3 mm | 4 mm ± 6 mm |
| Medial humeral head offset | 4.5 mm ± 0.3 mm | 9 mm ± 5 mm |
The reconstructions of these humerus fractures showed intraclass correlation coefficients ranging from 0.71 to 0.93 in 1 observer and interclass correlation coefficients from 0.82 to 0.98 between 2 different observers (Table).
DISCUSSION
This study demonstrates that it is feasible to reliably and accurately reconstruct the original anatomy of the proximal humerus by using 3-D computer modeling of proximal humerus fractures. Poor outcomes after hemiarthroplasty for proximal humerus fractures are mostly related to tuberosity malpositioning, resorption, or failure of fixation and resultant dysfunction of the rotator cuff.14,15,16 These studies highlight the importance of accurate tuberosity reduction during surgical care of these fractures.
Continue to: The 3-D computer model...
The 3-D computer model reconstruction of 3- and 4-part proximal humerus fractures were reliable and valid. The interclass correlation coefficients showed very good to excellent interobserver and intraobserver reliability for all measurements conducted. The averaged dimensions from all trials fell within the appropriate range of dimensions for a normal healthy humerus reported in the literature, as verified by the TOST method.11,12,13 The 3-D modeling capabilities demonstrated in this study allowed a greater understanding of the fracture patterns present in 3- and 4-part (AO classifications 11-B1, 11-B2, 11-C1, 11-C2) humerus fractures.
Overreduction of greater tuberosity to create cortical overlap with the lateral shaft may be used to promote bony union. As a result of this distalization, there may be extra strains placed on the rotator cuff, making the patient more prone to rotator cuff tear, as well as improperly balancing the dynamic stabilizers of the shoulder. Poor clinical outcomes in hemiarthroplasty for proximal humerus fractures have been correlated with a greater tuberosity placed distal relative to the humeral head by 1 cm in a study2 and by 2 cm in another.3
This study has several limitations. The first is the assumption that our injured patients had preinjury proximal humerus geometry within the range of normal dimensions of a healthy humerus. Unfortunately, because we were unable to obtain CT scans of the contralateral shoulder, we had to use standard proximal humerus geometry as the control. Another limitation, inherent in the technique, is that only cortical and dense trabecular bone was modeled, so that comminuted or osteoporotic bone was not well modeled. This study did not correlate the findings from these models with clinical outcomes. A prospective study is needed to evaluate the impact of this 3-D modeling on fracture reductions and clinical outcomes.
This study demonstrates that patient-specific modeling of proximal humerus fracture 3-D CT scans may help surgeons reliably and accurately reconstruct fractures. This technique may have utility in the preoperative planning of tuberosity fracture reduction and hemiarthroplasty. It gives surgeons the ability to visualize fracture fragments, and the process of reconstructing the fragments may help surgeons understand the required maneuvers for reduction at the time of surgery. This technique also provides dimensions of the patient’s native humerus, thus potentially improving the anatomic accuracy of the reduction or hemiarthroplasty reconstruction. With the new trend toward patient-specific instrumentation, this study also provides a means of planning the size of the humeral prostheses as well as the version relative to the biceps groove and intertubercular fracture line.
CONCLUSION
This study demonstrates the feasibility of using 3-D computer modeling of complex proximal humerus fractures in anatomic reconstruction. These techniques of computer-simulated 3-D models are valid and reliable. We believe that this technique of modeling and reconstructing proximal humerus fractures could be used to enhance the preoperative planning of hemiarthroplasty for 3- and 4-part proximal humerus fractures by providing improved understanding of the patient’s native humeral geometry and tuberosity reduction.
1. Boileau P, Krishnan SG, Tinsi L, Walch G, Coste JS, Mole D. Tuberosity malposition and migration: reasons for poor outcomes after hemiarthroplasty for displaced fractures of the proximal humerus. J Shoulder Elbow Surg. 2002;11(5):401-412. doi:10.1067/mse.2002.124527.
2. Mighell MA, Kolm GP, Collinge CA, Frankle MA. Outcomes of hemiarthroplasty for fractures of the proximal humerus. J Shoulder Elbow Surg. 2003;12(6):569-577. doi:10.1016/S1058274603002131.
3. Greiner SH, Kaab MJ, Kroning I, Scheibel M, Perka C. Reconstruction of humeral length and centering of the prosthetic head in hemiarthroplasty for proximal humeral fractures. J Shoulder Elbow Surg. 2008;17(5):709-714. doi:10.1016/j.jse.2008.03.004.
4. Smith AM, Mardones RM, Sperling JW, Cofield RH. Early complications of operatively treated proximal humeral fractures. J Shoulder Elbow Surg. 2007;16(1):14-24. doi:10.1016/j.jse.2006.05.008.
5. Scalise JJ, Codsi MJ, Bryan J, Iannotti JP. The three-dimensional glenoid vault model can estimate normal glenoid version in osteoarthritis. J Shoulder Elbow Surg. 2008;17(3):487-491. doi:10.1016/j.jse.2007.09.006.
6. Bryce CD, Pennypacker JL, Kulkarni N, et al. Validation of three-dimensional models of in situ scapulae. J Shoulder Elbow Surg. 2008;17(5):825-832. doi:10.1016/j.jse.2008.01.141.
7. Yongpravat C, Kim HM, Gardner TR, Bigliani LU, Levine WN, Ahmad CS. Glenoid implant orientation and cement failure in total shoulder arthroplasty: a finite element analysis. J Shoulder Elbow Surg. 2013;22(7):940-947. doi:10.1016/j.jse.2012.09.007.
8. Boileau P, Walch G. The three-dimensional geometry of the proximal humerus. Implications for surgical technique and prosthetic design. J Bone Joint Surg Br. 1997;79(5):857-865. doi:10.1302/0301-620X.79B5.0790857.
9. Wu G, van der Helm FC, Veeger HE, et al. ISB recommendation on definitions of joint coordinate systems of various joints for the reporting of human joint motion--Part II: shoulder, elbow, wrist and hand. J Biomech. 2005;38(5):981-992.
10. Kummer FJ, Perkins R, Zuckerman JD. The use of the bicipital groove for alignment of the humeral stem in shoulder arthroplasty. J Shoulder Elbow Surg. 1998;7(2):144-146. doi:10.1016/S1058-2746(98)90225-7.
11. Iannotti JP, Gabriel JP, Schneck SL, Evans BG, Misra S. The normal glenohumeral relationships. An anatomical study of one hundred and forty shoulders. J Bone Joint Surg Am. 1992;74(4):491-500.
12. Pearl ML, Volk AG. Coronal plane geometry of the proximal humerus relevant to prosthetic arthroplasty. J Shoulder Elbow Surg. 1996;5(4):320-326. doi:10.1016/S1058-2746(96)80060-7.
13. Pearl ML. Proximal humeral anatomy in shoulder arthroplasty: Implications for prosthetic design and surgical technique. J Shoulder Elbow Surg. 2005;14(1 Suppl S):99S-104S. doi:10.1016/j.jse.2004.09.025.
14. Prakash U, McGurty DW, Dent JA. Hemiarthroplasty for severe fractures of the proximal humerus. J Shoulder Elbow Surg. 2002;11(5):428-430. doi:10.1067/mse.2002.126615.
15. Robinson CM, Page RS, Hill RM, Sanders DL, Court-Brown CM, Wakefield AE. Primary hemiarthroplasty for treatment of proximal humeral fractures. J Bone Joint Surg Am. 2003;85-A(7):1215-1223.
16. Zyto K, Wallace WA, Frostick SP, Preston BJ. Outcome after hemiarthroplasty for three- and four-part fractures of the proximal humerus. J Shoulder Elbow Surg. 1998;7(2):85-89. doi:10.1016/S1058-2746(98)90215-4.
ABSTRACT
The objective of this study is to determine the reproducibility and feasibility of using 3-dimensional (3-D) computer simulation of proximal humerus fracture computed tomography (CT) scans for fracture reduction. We hypothesized that anatomic reconstruction with 3-D models would be anatomically accurate and reproducible.
Preoperative CT scans of 28 patients with 3- and 4-part (AO classification 11-B1, 11-B2, 11-C1, 11-C2) proximal humerus fractures who were treated by hemiarthroplasty were converted into 3-D computer models. The displaced fractured fragments were anatomically reduced with computer simulation by 2 fellowship-trained shoulder surgeons, and measurements were made of the reconstructed proximal humerus.
The measurements of the reconstructed models had very good to excellent interobserver and intraobserver reliability. The reconstructions of these humerus fractures showed interclass correlation coefficients ranging from 0.71 to 0.93 between 1 observer and from 0.82 to 0.98 between 2 different observers. The fracture reduction was judged against normal proximal humerus geometry to determine reduction accuracy.
The 3-D modeling techniques used to reconstruct 3- and 4-part proximal humerus fractures were reliable and accurate. This technique of modeling and reconstructing proximal humerus fractures could be used to enhance the preoperative planning of open reduction and internal fixation or hemiarthroplasty for 3- and 4-part proximal humerus fractures.
The treatment of proximal humerus fractures is influenced by multiple factors, including patient age, associated injuries, bone quality, and fracture pattern. Three- and 4-part fractures are among the more severe of these fractures, which may result in vascular compromise to the humeral head, leading to avascular necrosis. Surgical goals for the management of these fractures are to optimize functional outcomes by re-creating a stable construct with a functional rotator cuff by open reduction and internal fixation (ORIF), hemiarthroplasty with tuberosity ORIF, or reverse shoulder replacement. Achieving a good outcome following hemiarthroplasty is dependent on many factors, including anatomic tuberosity healing and component positioning.1,2,3 Repairing the greater tuberosity in a near-anatomic position has been shown to greatly affect the results of hemiarthroplasty for fracture.3,4
Continue to: Three-dimensional (3-D) modeling...
Three-dimensional (3-D) modeling is increasingly being used in preoperative planning of shoulder arthroplasty and determining proper proximal humeral fracture treatment. 5 However, no studies have examined the reconstruction of a fractured proximal humerus into native anatomy using computer simulation. The purpose of this study is to determine the accuracy and reliability of anatomically reconstructing the preinjury proximal humerus using 3-D computer models created from postinjury computed tomography (CT) scans. The results of this study could lead to useful techniques employing CT–based models for patient-specific preoperative planning of proximal humeral fracture ORIF and during tuberosity reduction and fixation during hemiarthroplasty for fracture. We hypothesize that it is feasible to reconstruct the original anatomy of the proximal humerus by using 3-D computer modeling of proximal humerus fractures with high reliability based on interobserver and intraobserver review.
METHODS
After Institutional Review Board approval was obtained, we reviewed the medical records of consecutive patients with a diagnosis of proximal humeral fracture and the treatment codes for hemiarthroplasty from 2000 to 2013. Inclusion criteria included 3- and 4-part fractures (AO classifications 11-B1, 11-B2, 11-C1, 11-C2). CT scans with insufficient quality to differentiate bone from soft tissue (inadequate signal-to-noise ratio) were excluded from the study. A total of 28 patients with adequate CT scans met the criteria for inclusion in this study.
The CT scan protocol included 0.5-mm axial cuts with inclusion of the proximal humerus in the Digital Imaging and Communications in Medicine format. These CT scans were converted into patient-specific 3-D computer models of the shoulder using Mimics software (Materialise Inc.). The use of this software to produce anatomically accurate models has previously been verified in a shoulder model.6,7 The tuberosity fragments were then individually separated from each other using the voxel-selecting capabilities of 3-D software and manipulated with translation and rotation for anatomic reduction (Figures 1A-1D, Figure 2).
The de-identified anatomically reconstructed shoulder models were then uploaded into Materialise’s Magics rapid prototyping software, and a user-defined humeral Cartesian coordinate system was defined with anatomic landmarks as reference points to standardize the position of each model (Figure 3).8,9
A series of measurements were made on these models to assess the validity and reliability of the reassembly. The bicipital groove at the anatomic neck was used to measure humeral head version as described by Kummer and colleagues.10 The head-shaft angle, humeral head-greater tuberosity distance, humeral head-bicipital groove angle, and posterior and medial humeral head offset were measured directly on the reconstructed humerus.
Continue to: Two fellowship-trained shoulder...
Two fellowship-trained shoulder surgeons independently reassembled these fracture fragments via computer simulation. Interobserver reliability testing was conducted on these reconstructions by measuring the geometry between the 2 different surgeons’ reconstructions. Intraobserver reliability testing was conducted by 1 surgeon repeating the reconstructions with 4-week intervals between trials and measuring the geometry between the 2 different trials. The average dimensions of the reconstructed proximal humerus fractures were compared with the geometry of normal humeri reported in previously conducted anatomic studies.11,12,13
STATISTICS
The measured dimensions of the 28 reassembled proximal humeri models were averaged across all trials between the 2 fellowship-trained surgeons and compared with the range of normal dimensions of a healthy proximal humerus using the 2 one-sided tests (TOST) method for equivalence between 2 means given a range. The interobserver and intraobserver reliabilities were quantified using the interclass correlation coefficient. An excellent correlation was defined as a correlation coefficient >0.81; very good was defined as 0.61 to 0.80; and good was defined as 0.41 to 0.60.
RESULTS
Of the patients studied, 9 (32.1%) were male, and the average age at the time of CT scanning was 72 years. Of the 28 patients with fracture, 18 (64.2%) had 3-part fractures (AO classifications 11-B1, 11-B2), and 10 (35.8%) had 4-part fractures (AO classifications 11-C1, 11-C2). When examining the location of the intertubercular fracture line, we found that 13 (46.4%) fractures went through the bicipital groove. Of the remaining fracture lines, 9 (32.1%) extended into the greater tuberosity and 6 (21.4%) extended into the lesser tuberosity.
All users were able to reconstruct all 28 fractures using this technique. The average measured dimensions fell within the range of dimensions of a normal healthy proximal humerus specified in the literature to within a 95% confidence interval using the TOST for equivalence, in which we compared measured values with ranges reported in the literature (Table).11,12,13
Table. Dimensions of Proximal Humerus Geometry
| Normal Parameters | Average Dimensions From Trials | Dimensions From Literature |
| Head shaft angle | 43.5° ± 1° | 42.5° ± 12.5° |
| Head to greater tuberosity distance | 4.9 mm ± 0.4 mm | 8 mm ± 3.2 mm |
Head to bicipital groove angle (anatomic neck) | 26.4° ± 2° | 27.3° ± 14° |
| Posterior humeral head offset | 1.6 mm ± 0.3 mm | 4 mm ± 6 mm |
| Medial humeral head offset | 4.5 mm ± 0.3 mm | 9 mm ± 5 mm |
The reconstructions of these humerus fractures showed intraclass correlation coefficients ranging from 0.71 to 0.93 in 1 observer and interclass correlation coefficients from 0.82 to 0.98 between 2 different observers (Table).
DISCUSSION
This study demonstrates that it is feasible to reliably and accurately reconstruct the original anatomy of the proximal humerus by using 3-D computer modeling of proximal humerus fractures. Poor outcomes after hemiarthroplasty for proximal humerus fractures are mostly related to tuberosity malpositioning, resorption, or failure of fixation and resultant dysfunction of the rotator cuff.14,15,16 These studies highlight the importance of accurate tuberosity reduction during surgical care of these fractures.
Continue to: The 3-D computer model...
The 3-D computer model reconstruction of 3- and 4-part proximal humerus fractures were reliable and valid. The interclass correlation coefficients showed very good to excellent interobserver and intraobserver reliability for all measurements conducted. The averaged dimensions from all trials fell within the appropriate range of dimensions for a normal healthy humerus reported in the literature, as verified by the TOST method.11,12,13 The 3-D modeling capabilities demonstrated in this study allowed a greater understanding of the fracture patterns present in 3- and 4-part (AO classifications 11-B1, 11-B2, 11-C1, 11-C2) humerus fractures.
Overreduction of greater tuberosity to create cortical overlap with the lateral shaft may be used to promote bony union. As a result of this distalization, there may be extra strains placed on the rotator cuff, making the patient more prone to rotator cuff tear, as well as improperly balancing the dynamic stabilizers of the shoulder. Poor clinical outcomes in hemiarthroplasty for proximal humerus fractures have been correlated with a greater tuberosity placed distal relative to the humeral head by 1 cm in a study2 and by 2 cm in another.3
This study has several limitations. The first is the assumption that our injured patients had preinjury proximal humerus geometry within the range of normal dimensions of a healthy humerus. Unfortunately, because we were unable to obtain CT scans of the contralateral shoulder, we had to use standard proximal humerus geometry as the control. Another limitation, inherent in the technique, is that only cortical and dense trabecular bone was modeled, so that comminuted or osteoporotic bone was not well modeled. This study did not correlate the findings from these models with clinical outcomes. A prospective study is needed to evaluate the impact of this 3-D modeling on fracture reductions and clinical outcomes.
This study demonstrates that patient-specific modeling of proximal humerus fracture 3-D CT scans may help surgeons reliably and accurately reconstruct fractures. This technique may have utility in the preoperative planning of tuberosity fracture reduction and hemiarthroplasty. It gives surgeons the ability to visualize fracture fragments, and the process of reconstructing the fragments may help surgeons understand the required maneuvers for reduction at the time of surgery. This technique also provides dimensions of the patient’s native humerus, thus potentially improving the anatomic accuracy of the reduction or hemiarthroplasty reconstruction. With the new trend toward patient-specific instrumentation, this study also provides a means of planning the size of the humeral prostheses as well as the version relative to the biceps groove and intertubercular fracture line.
CONCLUSION
This study demonstrates the feasibility of using 3-D computer modeling of complex proximal humerus fractures in anatomic reconstruction. These techniques of computer-simulated 3-D models are valid and reliable. We believe that this technique of modeling and reconstructing proximal humerus fractures could be used to enhance the preoperative planning of hemiarthroplasty for 3- and 4-part proximal humerus fractures by providing improved understanding of the patient’s native humeral geometry and tuberosity reduction.
ABSTRACT
The objective of this study is to determine the reproducibility and feasibility of using 3-dimensional (3-D) computer simulation of proximal humerus fracture computed tomography (CT) scans for fracture reduction. We hypothesized that anatomic reconstruction with 3-D models would be anatomically accurate and reproducible.
Preoperative CT scans of 28 patients with 3- and 4-part (AO classification 11-B1, 11-B2, 11-C1, 11-C2) proximal humerus fractures who were treated by hemiarthroplasty were converted into 3-D computer models. The displaced fractured fragments were anatomically reduced with computer simulation by 2 fellowship-trained shoulder surgeons, and measurements were made of the reconstructed proximal humerus.
The measurements of the reconstructed models had very good to excellent interobserver and intraobserver reliability. The reconstructions of these humerus fractures showed interclass correlation coefficients ranging from 0.71 to 0.93 between 1 observer and from 0.82 to 0.98 between 2 different observers. The fracture reduction was judged against normal proximal humerus geometry to determine reduction accuracy.
The 3-D modeling techniques used to reconstruct 3- and 4-part proximal humerus fractures were reliable and accurate. This technique of modeling and reconstructing proximal humerus fractures could be used to enhance the preoperative planning of open reduction and internal fixation or hemiarthroplasty for 3- and 4-part proximal humerus fractures.
The treatment of proximal humerus fractures is influenced by multiple factors, including patient age, associated injuries, bone quality, and fracture pattern. Three- and 4-part fractures are among the more severe of these fractures, which may result in vascular compromise to the humeral head, leading to avascular necrosis. Surgical goals for the management of these fractures are to optimize functional outcomes by re-creating a stable construct with a functional rotator cuff by open reduction and internal fixation (ORIF), hemiarthroplasty with tuberosity ORIF, or reverse shoulder replacement. Achieving a good outcome following hemiarthroplasty is dependent on many factors, including anatomic tuberosity healing and component positioning.1,2,3 Repairing the greater tuberosity in a near-anatomic position has been shown to greatly affect the results of hemiarthroplasty for fracture.3,4
Continue to: Three-dimensional (3-D) modeling...
Three-dimensional (3-D) modeling is increasingly being used in preoperative planning of shoulder arthroplasty and determining proper proximal humeral fracture treatment. 5 However, no studies have examined the reconstruction of a fractured proximal humerus into native anatomy using computer simulation. The purpose of this study is to determine the accuracy and reliability of anatomically reconstructing the preinjury proximal humerus using 3-D computer models created from postinjury computed tomography (CT) scans. The results of this study could lead to useful techniques employing CT–based models for patient-specific preoperative planning of proximal humeral fracture ORIF and during tuberosity reduction and fixation during hemiarthroplasty for fracture. We hypothesize that it is feasible to reconstruct the original anatomy of the proximal humerus by using 3-D computer modeling of proximal humerus fractures with high reliability based on interobserver and intraobserver review.
METHODS
After Institutional Review Board approval was obtained, we reviewed the medical records of consecutive patients with a diagnosis of proximal humeral fracture and the treatment codes for hemiarthroplasty from 2000 to 2013. Inclusion criteria included 3- and 4-part fractures (AO classifications 11-B1, 11-B2, 11-C1, 11-C2). CT scans with insufficient quality to differentiate bone from soft tissue (inadequate signal-to-noise ratio) were excluded from the study. A total of 28 patients with adequate CT scans met the criteria for inclusion in this study.
The CT scan protocol included 0.5-mm axial cuts with inclusion of the proximal humerus in the Digital Imaging and Communications in Medicine format. These CT scans were converted into patient-specific 3-D computer models of the shoulder using Mimics software (Materialise Inc.). The use of this software to produce anatomically accurate models has previously been verified in a shoulder model.6,7 The tuberosity fragments were then individually separated from each other using the voxel-selecting capabilities of 3-D software and manipulated with translation and rotation for anatomic reduction (Figures 1A-1D, Figure 2).
The de-identified anatomically reconstructed shoulder models were then uploaded into Materialise’s Magics rapid prototyping software, and a user-defined humeral Cartesian coordinate system was defined with anatomic landmarks as reference points to standardize the position of each model (Figure 3).8,9
A series of measurements were made on these models to assess the validity and reliability of the reassembly. The bicipital groove at the anatomic neck was used to measure humeral head version as described by Kummer and colleagues.10 The head-shaft angle, humeral head-greater tuberosity distance, humeral head-bicipital groove angle, and posterior and medial humeral head offset were measured directly on the reconstructed humerus.
Continue to: Two fellowship-trained shoulder...
Two fellowship-trained shoulder surgeons independently reassembled these fracture fragments via computer simulation. Interobserver reliability testing was conducted on these reconstructions by measuring the geometry between the 2 different surgeons’ reconstructions. Intraobserver reliability testing was conducted by 1 surgeon repeating the reconstructions with 4-week intervals between trials and measuring the geometry between the 2 different trials. The average dimensions of the reconstructed proximal humerus fractures were compared with the geometry of normal humeri reported in previously conducted anatomic studies.11,12,13
STATISTICS
The measured dimensions of the 28 reassembled proximal humeri models were averaged across all trials between the 2 fellowship-trained surgeons and compared with the range of normal dimensions of a healthy proximal humerus using the 2 one-sided tests (TOST) method for equivalence between 2 means given a range. The interobserver and intraobserver reliabilities were quantified using the interclass correlation coefficient. An excellent correlation was defined as a correlation coefficient >0.81; very good was defined as 0.61 to 0.80; and good was defined as 0.41 to 0.60.
RESULTS
Of the patients studied, 9 (32.1%) were male, and the average age at the time of CT scanning was 72 years. Of the 28 patients with fracture, 18 (64.2%) had 3-part fractures (AO classifications 11-B1, 11-B2), and 10 (35.8%) had 4-part fractures (AO classifications 11-C1, 11-C2). When examining the location of the intertubercular fracture line, we found that 13 (46.4%) fractures went through the bicipital groove. Of the remaining fracture lines, 9 (32.1%) extended into the greater tuberosity and 6 (21.4%) extended into the lesser tuberosity.
All users were able to reconstruct all 28 fractures using this technique. The average measured dimensions fell within the range of dimensions of a normal healthy proximal humerus specified in the literature to within a 95% confidence interval using the TOST for equivalence, in which we compared measured values with ranges reported in the literature (Table).11,12,13
Table. Dimensions of Proximal Humerus Geometry
| Normal Parameters | Average Dimensions From Trials | Dimensions From Literature |
| Head shaft angle | 43.5° ± 1° | 42.5° ± 12.5° |
| Head to greater tuberosity distance | 4.9 mm ± 0.4 mm | 8 mm ± 3.2 mm |
Head to bicipital groove angle (anatomic neck) | 26.4° ± 2° | 27.3° ± 14° |
| Posterior humeral head offset | 1.6 mm ± 0.3 mm | 4 mm ± 6 mm |
| Medial humeral head offset | 4.5 mm ± 0.3 mm | 9 mm ± 5 mm |
The reconstructions of these humerus fractures showed intraclass correlation coefficients ranging from 0.71 to 0.93 in 1 observer and interclass correlation coefficients from 0.82 to 0.98 between 2 different observers (Table).
DISCUSSION
This study demonstrates that it is feasible to reliably and accurately reconstruct the original anatomy of the proximal humerus by using 3-D computer modeling of proximal humerus fractures. Poor outcomes after hemiarthroplasty for proximal humerus fractures are mostly related to tuberosity malpositioning, resorption, or failure of fixation and resultant dysfunction of the rotator cuff.14,15,16 These studies highlight the importance of accurate tuberosity reduction during surgical care of these fractures.
Continue to: The 3-D computer model...
The 3-D computer model reconstruction of 3- and 4-part proximal humerus fractures were reliable and valid. The interclass correlation coefficients showed very good to excellent interobserver and intraobserver reliability for all measurements conducted. The averaged dimensions from all trials fell within the appropriate range of dimensions for a normal healthy humerus reported in the literature, as verified by the TOST method.11,12,13 The 3-D modeling capabilities demonstrated in this study allowed a greater understanding of the fracture patterns present in 3- and 4-part (AO classifications 11-B1, 11-B2, 11-C1, 11-C2) humerus fractures.
Overreduction of greater tuberosity to create cortical overlap with the lateral shaft may be used to promote bony union. As a result of this distalization, there may be extra strains placed on the rotator cuff, making the patient more prone to rotator cuff tear, as well as improperly balancing the dynamic stabilizers of the shoulder. Poor clinical outcomes in hemiarthroplasty for proximal humerus fractures have been correlated with a greater tuberosity placed distal relative to the humeral head by 1 cm in a study2 and by 2 cm in another.3
This study has several limitations. The first is the assumption that our injured patients had preinjury proximal humerus geometry within the range of normal dimensions of a healthy humerus. Unfortunately, because we were unable to obtain CT scans of the contralateral shoulder, we had to use standard proximal humerus geometry as the control. Another limitation, inherent in the technique, is that only cortical and dense trabecular bone was modeled, so that comminuted or osteoporotic bone was not well modeled. This study did not correlate the findings from these models with clinical outcomes. A prospective study is needed to evaluate the impact of this 3-D modeling on fracture reductions and clinical outcomes.
This study demonstrates that patient-specific modeling of proximal humerus fracture 3-D CT scans may help surgeons reliably and accurately reconstruct fractures. This technique may have utility in the preoperative planning of tuberosity fracture reduction and hemiarthroplasty. It gives surgeons the ability to visualize fracture fragments, and the process of reconstructing the fragments may help surgeons understand the required maneuvers for reduction at the time of surgery. This technique also provides dimensions of the patient’s native humerus, thus potentially improving the anatomic accuracy of the reduction or hemiarthroplasty reconstruction. With the new trend toward patient-specific instrumentation, this study also provides a means of planning the size of the humeral prostheses as well as the version relative to the biceps groove and intertubercular fracture line.
CONCLUSION
This study demonstrates the feasibility of using 3-D computer modeling of complex proximal humerus fractures in anatomic reconstruction. These techniques of computer-simulated 3-D models are valid and reliable. We believe that this technique of modeling and reconstructing proximal humerus fractures could be used to enhance the preoperative planning of hemiarthroplasty for 3- and 4-part proximal humerus fractures by providing improved understanding of the patient’s native humeral geometry and tuberosity reduction.
1. Boileau P, Krishnan SG, Tinsi L, Walch G, Coste JS, Mole D. Tuberosity malposition and migration: reasons for poor outcomes after hemiarthroplasty for displaced fractures of the proximal humerus. J Shoulder Elbow Surg. 2002;11(5):401-412. doi:10.1067/mse.2002.124527.
2. Mighell MA, Kolm GP, Collinge CA, Frankle MA. Outcomes of hemiarthroplasty for fractures of the proximal humerus. J Shoulder Elbow Surg. 2003;12(6):569-577. doi:10.1016/S1058274603002131.
3. Greiner SH, Kaab MJ, Kroning I, Scheibel M, Perka C. Reconstruction of humeral length and centering of the prosthetic head in hemiarthroplasty for proximal humeral fractures. J Shoulder Elbow Surg. 2008;17(5):709-714. doi:10.1016/j.jse.2008.03.004.
4. Smith AM, Mardones RM, Sperling JW, Cofield RH. Early complications of operatively treated proximal humeral fractures. J Shoulder Elbow Surg. 2007;16(1):14-24. doi:10.1016/j.jse.2006.05.008.
5. Scalise JJ, Codsi MJ, Bryan J, Iannotti JP. The three-dimensional glenoid vault model can estimate normal glenoid version in osteoarthritis. J Shoulder Elbow Surg. 2008;17(3):487-491. doi:10.1016/j.jse.2007.09.006.
6. Bryce CD, Pennypacker JL, Kulkarni N, et al. Validation of three-dimensional models of in situ scapulae. J Shoulder Elbow Surg. 2008;17(5):825-832. doi:10.1016/j.jse.2008.01.141.
7. Yongpravat C, Kim HM, Gardner TR, Bigliani LU, Levine WN, Ahmad CS. Glenoid implant orientation and cement failure in total shoulder arthroplasty: a finite element analysis. J Shoulder Elbow Surg. 2013;22(7):940-947. doi:10.1016/j.jse.2012.09.007.
8. Boileau P, Walch G. The three-dimensional geometry of the proximal humerus. Implications for surgical technique and prosthetic design. J Bone Joint Surg Br. 1997;79(5):857-865. doi:10.1302/0301-620X.79B5.0790857.
9. Wu G, van der Helm FC, Veeger HE, et al. ISB recommendation on definitions of joint coordinate systems of various joints for the reporting of human joint motion--Part II: shoulder, elbow, wrist and hand. J Biomech. 2005;38(5):981-992.
10. Kummer FJ, Perkins R, Zuckerman JD. The use of the bicipital groove for alignment of the humeral stem in shoulder arthroplasty. J Shoulder Elbow Surg. 1998;7(2):144-146. doi:10.1016/S1058-2746(98)90225-7.
11. Iannotti JP, Gabriel JP, Schneck SL, Evans BG, Misra S. The normal glenohumeral relationships. An anatomical study of one hundred and forty shoulders. J Bone Joint Surg Am. 1992;74(4):491-500.
12. Pearl ML, Volk AG. Coronal plane geometry of the proximal humerus relevant to prosthetic arthroplasty. J Shoulder Elbow Surg. 1996;5(4):320-326. doi:10.1016/S1058-2746(96)80060-7.
13. Pearl ML. Proximal humeral anatomy in shoulder arthroplasty: Implications for prosthetic design and surgical technique. J Shoulder Elbow Surg. 2005;14(1 Suppl S):99S-104S. doi:10.1016/j.jse.2004.09.025.
14. Prakash U, McGurty DW, Dent JA. Hemiarthroplasty for severe fractures of the proximal humerus. J Shoulder Elbow Surg. 2002;11(5):428-430. doi:10.1067/mse.2002.126615.
15. Robinson CM, Page RS, Hill RM, Sanders DL, Court-Brown CM, Wakefield AE. Primary hemiarthroplasty for treatment of proximal humeral fractures. J Bone Joint Surg Am. 2003;85-A(7):1215-1223.
16. Zyto K, Wallace WA, Frostick SP, Preston BJ. Outcome after hemiarthroplasty for three- and four-part fractures of the proximal humerus. J Shoulder Elbow Surg. 1998;7(2):85-89. doi:10.1016/S1058-2746(98)90215-4.
1. Boileau P, Krishnan SG, Tinsi L, Walch G, Coste JS, Mole D. Tuberosity malposition and migration: reasons for poor outcomes after hemiarthroplasty for displaced fractures of the proximal humerus. J Shoulder Elbow Surg. 2002;11(5):401-412. doi:10.1067/mse.2002.124527.
2. Mighell MA, Kolm GP, Collinge CA, Frankle MA. Outcomes of hemiarthroplasty for fractures of the proximal humerus. J Shoulder Elbow Surg. 2003;12(6):569-577. doi:10.1016/S1058274603002131.
3. Greiner SH, Kaab MJ, Kroning I, Scheibel M, Perka C. Reconstruction of humeral length and centering of the prosthetic head in hemiarthroplasty for proximal humeral fractures. J Shoulder Elbow Surg. 2008;17(5):709-714. doi:10.1016/j.jse.2008.03.004.
4. Smith AM, Mardones RM, Sperling JW, Cofield RH. Early complications of operatively treated proximal humeral fractures. J Shoulder Elbow Surg. 2007;16(1):14-24. doi:10.1016/j.jse.2006.05.008.
5. Scalise JJ, Codsi MJ, Bryan J, Iannotti JP. The three-dimensional glenoid vault model can estimate normal glenoid version in osteoarthritis. J Shoulder Elbow Surg. 2008;17(3):487-491. doi:10.1016/j.jse.2007.09.006.
6. Bryce CD, Pennypacker JL, Kulkarni N, et al. Validation of three-dimensional models of in situ scapulae. J Shoulder Elbow Surg. 2008;17(5):825-832. doi:10.1016/j.jse.2008.01.141.
7. Yongpravat C, Kim HM, Gardner TR, Bigliani LU, Levine WN, Ahmad CS. Glenoid implant orientation and cement failure in total shoulder arthroplasty: a finite element analysis. J Shoulder Elbow Surg. 2013;22(7):940-947. doi:10.1016/j.jse.2012.09.007.
8. Boileau P, Walch G. The three-dimensional geometry of the proximal humerus. Implications for surgical technique and prosthetic design. J Bone Joint Surg Br. 1997;79(5):857-865. doi:10.1302/0301-620X.79B5.0790857.
9. Wu G, van der Helm FC, Veeger HE, et al. ISB recommendation on definitions of joint coordinate systems of various joints for the reporting of human joint motion--Part II: shoulder, elbow, wrist and hand. J Biomech. 2005;38(5):981-992.
10. Kummer FJ, Perkins R, Zuckerman JD. The use of the bicipital groove for alignment of the humeral stem in shoulder arthroplasty. J Shoulder Elbow Surg. 1998;7(2):144-146. doi:10.1016/S1058-2746(98)90225-7.
11. Iannotti JP, Gabriel JP, Schneck SL, Evans BG, Misra S. The normal glenohumeral relationships. An anatomical study of one hundred and forty shoulders. J Bone Joint Surg Am. 1992;74(4):491-500.
12. Pearl ML, Volk AG. Coronal plane geometry of the proximal humerus relevant to prosthetic arthroplasty. J Shoulder Elbow Surg. 1996;5(4):320-326. doi:10.1016/S1058-2746(96)80060-7.
13. Pearl ML. Proximal humeral anatomy in shoulder arthroplasty: Implications for prosthetic design and surgical technique. J Shoulder Elbow Surg. 2005;14(1 Suppl S):99S-104S. doi:10.1016/j.jse.2004.09.025.
14. Prakash U, McGurty DW, Dent JA. Hemiarthroplasty for severe fractures of the proximal humerus. J Shoulder Elbow Surg. 2002;11(5):428-430. doi:10.1067/mse.2002.126615.
15. Robinson CM, Page RS, Hill RM, Sanders DL, Court-Brown CM, Wakefield AE. Primary hemiarthroplasty for treatment of proximal humeral fractures. J Bone Joint Surg Am. 2003;85-A(7):1215-1223.
16. Zyto K, Wallace WA, Frostick SP, Preston BJ. Outcome after hemiarthroplasty for three- and four-part fractures of the proximal humerus. J Shoulder Elbow Surg. 1998;7(2):85-89. doi:10.1016/S1058-2746(98)90215-4.
TAKE-HOME POINTS
- Proximal humerus fractures may be better understood with 3-D CT imaging.
- 3-D computer modeling of complex proximal humerus fractures allows an understanding of tuebroisty reduction durring ORIF or hemiarthroplasty.
- 3-D modeling enhances preoperative planning for hemiarthroplasty implant size and position relative to the repaired tuberosity fragments.
- 3-D modeling of fracture reduction can help surgeons understand the patient’s native humeral geometry and anatomy.
- Preoperative evaluation of fracture characteristics and fragment reduction help surgeons better understand surgical solutions.
Providers’ prior imaging patterns and ownership of equipment are strong predictors of low-value imaging
Background: For many common conditions, expert guidelines such as Choosing Wisely recommend against ordering specific low-value tests, yet overuse of these tests remains widespread, and unnecessary care may account for up to a third of all medical expenditures. Studies have demonstrated that there is considerable geographic variation in health care usage and higher overall imaging among clinicians who own imaging equipment. No prior study has assessed whether prior ordering patterns of low-value care predict future ordering patterns or whether providers who order low-value imaging in one clinical scenario are more likely to do so in another scenario.
Study design: Retrospective analysis of insurance claims data.
Setting: Medical claims data from a large U.S. commercial health insurer, inclusive of 29 million commercially insured members across all 50 states from January 2010 to December 2014.
Synopsis: Using the claims database, researchers created three unique study samples to examine clinician predictors of low-value imaging. The study involved outpatient visits by patients aged 18-64 years without red-flag symptoms. The first included 1,007,392 visits across 878,720 patients with acute, uncomplicated low-back pain. Physicians who ordered imaging for the prior patient with back pain were 1.8 times more likely to do so again. Similarly, in 492,804 visits by 417,010 patients with headache, clinicians who ordered imaging on the prior patient demonstrated a twofold higher odds of imaging. Physicians who practiced low-value ordering for one condition were 1.8 times more likely to do so for the other. Across all studies, imaging ownership was an independent predictor (odds ratio, 1.8).
As this study analyzed only claims data, some patients may have warranted imaging based on red-flag symptoms that were not documented. Hospitalists designing initiatives to reduce low-value testing should consider targeting specific providers with a history of low-value test use.
Bottom line: Among commercially insured patients, a clinician’s prior imaging pattern and ownership of imaging equipment were strong independent predictors of low-value back pain and headache imaging.
Citation: Hong AS et al. Clinician-level predictors for ordering low-value imaging. JAMA Intern Med. 2017 Nov 1;177(11):1577-85.

Background: For many common conditions, expert guidelines such as Choosing Wisely recommend against ordering specific low-value tests, yet overuse of these tests remains widespread, and unnecessary care may account for up to a third of all medical expenditures. Studies have demonstrated that there is considerable geographic variation in health care usage and higher overall imaging among clinicians who own imaging equipment. No prior study has assessed whether prior ordering patterns of low-value care predict future ordering patterns or whether providers who order low-value imaging in one clinical scenario are more likely to do so in another scenario.
Study design: Retrospective analysis of insurance claims data.
Setting: Medical claims data from a large U.S. commercial health insurer, inclusive of 29 million commercially insured members across all 50 states from January 2010 to December 2014.
Synopsis: Using the claims database, researchers created three unique study samples to examine clinician predictors of low-value imaging. The study involved outpatient visits by patients aged 18-64 years without red-flag symptoms. The first included 1,007,392 visits across 878,720 patients with acute, uncomplicated low-back pain. Physicians who ordered imaging for the prior patient with back pain were 1.8 times more likely to do so again. Similarly, in 492,804 visits by 417,010 patients with headache, clinicians who ordered imaging on the prior patient demonstrated a twofold higher odds of imaging. Physicians who practiced low-value ordering for one condition were 1.8 times more likely to do so for the other. Across all studies, imaging ownership was an independent predictor (odds ratio, 1.8).
As this study analyzed only claims data, some patients may have warranted imaging based on red-flag symptoms that were not documented. Hospitalists designing initiatives to reduce low-value testing should consider targeting specific providers with a history of low-value test use.
Bottom line: Among commercially insured patients, a clinician’s prior imaging pattern and ownership of imaging equipment were strong independent predictors of low-value back pain and headache imaging.
Citation: Hong AS et al. Clinician-level predictors for ordering low-value imaging. JAMA Intern Med. 2017 Nov 1;177(11):1577-85.
Background: For many common conditions, expert guidelines such as Choosing Wisely recommend against ordering specific low-value tests, yet overuse of these tests remains widespread, and unnecessary care may account for up to a third of all medical expenditures. Studies have demonstrated that there is considerable geographic variation in health care usage and higher overall imaging among clinicians who own imaging equipment. No prior study has assessed whether prior ordering patterns of low-value care predict future ordering patterns or whether providers who order low-value imaging in one clinical scenario are more likely to do so in another scenario.
Study design: Retrospective analysis of insurance claims data.
Setting: Medical claims data from a large U.S. commercial health insurer, inclusive of 29 million commercially insured members across all 50 states from January 2010 to December 2014.
Synopsis: Using the claims database, researchers created three unique study samples to examine clinician predictors of low-value imaging. The study involved outpatient visits by patients aged 18-64 years without red-flag symptoms. The first included 1,007,392 visits across 878,720 patients with acute, uncomplicated low-back pain. Physicians who ordered imaging for the prior patient with back pain were 1.8 times more likely to do so again. Similarly, in 492,804 visits by 417,010 patients with headache, clinicians who ordered imaging on the prior patient demonstrated a twofold higher odds of imaging. Physicians who practiced low-value ordering for one condition were 1.8 times more likely to do so for the other. Across all studies, imaging ownership was an independent predictor (odds ratio, 1.8).
As this study analyzed only claims data, some patients may have warranted imaging based on red-flag symptoms that were not documented. Hospitalists designing initiatives to reduce low-value testing should consider targeting specific providers with a history of low-value test use.
Bottom line: Among commercially insured patients, a clinician’s prior imaging pattern and ownership of imaging equipment were strong independent predictors of low-value back pain and headache imaging.
Citation: Hong AS et al. Clinician-level predictors for ordering low-value imaging. JAMA Intern Med. 2017 Nov 1;177(11):1577-85.