User login
Imaging recommendations issued for COVID-19 patients
A consensus statement on the role of imaging during the acute work-up of COVID-19 patients called for liberal use in patients with moderate to severe clinical features indicative of infection, regardless of their COVID-19 test results, but limited use in patients who present with mild symptoms or are asymptomatic.

The consensus statement on The Role of Imaging in Patient Management during the COVID-19 Pandemic released by the Fleischner Society on April 7 was designed to highlight the “key decision points around imaging” in COVID-19 patients.
“We developed the statement to be applicable across settings” so that each clinic or hospital managing COVID-19 patients could decide the situations where chest radiography (CXR) or CT would work best, said Geoffrey D. Rubin, MD, professor of cardiovascular research, radiology, and bioengineering at Duke University in Durham, N.C., and lead author of the statement.
Written by 15 thoracic radiologists and 10 pulmonologists/intensivists including an anesthesiologist, a pathologist, and additional experts in emergency medicine, infection control, and laboratory medicine, and with members from any of 10 countries on three continents, the panel arrived at agreement by more than 70% for each of the 14 questions.
“I was impressed and a little surprised that consensus was achieved for every question” posed to the panel by the Fleischner Society for Thoracic Imaging and Diagnosis, Dr. Rubin said in an interview. The panel also placed their 14 decisions about imaging within the context of three distinct clinical scenarios chosen to mirror common real-world situations: mild COVID-19 features, moderate to severe features with no critical-resource constraints, and moderate to severe features with constrained resources. The statement also summarized its conclusions as five main recommendations and three additional recommendations.
Main recommendations
- Imaging is not routinely indicated for COVID-19 screening in asymptomatic people.
- Imaging is not indicated for patients with mild features of COVID-19 unless they are at risk for disease progression.
- Imaging is indicated for patients with features of moderate to severe COVID-19 regardless of COVID-19 test results.
- Imaging is indicated for patients with COVID-19 and evidence of worsening respiratory status.
- When access to CT is limited, chest radiography may be preferred for COVID-19 patients unless features of respiratory worsening warrant using CT.
Additional recommendations
- Daily chest radiographs are not indicated in stable, intubated patients with COVID-19.
- CT is indicated in patients with functional impairment, hypoxemia, or both, after COVID-19 recovery.
- COVID-19 testing is warranted in patients incidentally found to have findings suggestive of COVID-19 on a CT scan.
The statement particularly called out one of its recommendations – that a COVID-19 diagnosis “may be presumed when imaging findings are strongly suggestive of COVID-19 despite negative COVID-19 testing” in a patient who has moderate to severe clinical features of COVID-19 and whose pretest probability is high. The panel voted unanimously in favor of this concept, that imaging is “indicated” in hospitalized patients with moderate to severe symptoms consistent with COVID-19 despite a negative COVID-19 test result. “This guidance represents variance from other published recommendations which advise against the use of imaging for the initial diagnosis of COVID-19,” the statement acknowledged and specifically cited the recommendations issued in March 2020 by the American College of Radiology. Despite that, the ACR and Fleischner recommendations “are not at odds with one another,” maintained Dr. Rubin. The panel based its take on this question on the “direct experience” of its members caring for COVID-19 patients, according to the statement.

“I wholeheartedly agree with the suggested uses of imaging outlined by the panel,” commented Sachin Gupta, MD, FCCP, a pulmonologist and critical care physician in San Francisco. “The consensus statement brings a practical way to consider obtaining imaging. It leaves the door open to local standards and best judgment for using CXR or CT. Many physicians are unclear whether to image low-risk and mildly symptomatic patients. This statement gives support to a watchful waiting approach.” Another recommendation advises against daily CXR in stable, intubated COVID-19 patients. This “now gives backing from an important society and thought leaders while giving an explanation” for why daily imaging is problematic, he noted in an interview. The daily CXR in these patients adds no value, and skipping unneeded imaging minimizes SARS-CoV-2 exposure to radiology personnel, and conserves personal protection equipment, said the statement.
“The Fleischner Society is known worldwide for its recommendations. Having the society lend its weight on triage with imaging for COVID-19 patients is important. I suspect it will help standardize practice.”
Dr. Gupta also highlighted that lung imaging with a portable ultrasound unit has quickly become recognized as a very useful imaging tool with increasing use as the pandemic has unfolded, an option not covered by the Fleischner statement. Study results have “confirmed excellent sensitivity, specificity, and reproducibility” with lung ultrasound, and it’s also “easy to use,” Dr. Gupta said.
Ultrasound chest imaging of COVID-19 patients did not get included in the statement despite the reliance some U.S. sites have already placed on it largely because few on the panel had direct experience using it. “We didn’t feel we could contribute” to a discussion of ultrasound, Dr. Rubin said.
The statement’s recommendations appear to have already begun influencing practice. “The feedback I’ve gotten is that people are relying on them,” said Dr. Rubin, and some programs have sent him screen shots of the recommendations embedded in their local electronic health record.
The Radiological Society of North America is hosting a webinar on the statement on April 17.
A consensus statement on the role of imaging during the acute work-up of COVID-19 patients called for liberal use in patients with moderate to severe clinical features indicative of infection, regardless of their COVID-19 test results, but limited use in patients who present with mild symptoms or are asymptomatic.
The consensus statement on The Role of Imaging in Patient Management during the COVID-19 Pandemic released by the Fleischner Society on April 7 was designed to highlight the “key decision points around imaging” in COVID-19 patients.
“We developed the statement to be applicable across settings” so that each clinic or hospital managing COVID-19 patients could decide the situations where chest radiography (CXR) or CT would work best, said Geoffrey D. Rubin, MD, professor of cardiovascular research, radiology, and bioengineering at Duke University in Durham, N.C., and lead author of the statement.
Written by 15 thoracic radiologists and 10 pulmonologists/intensivists including an anesthesiologist, a pathologist, and additional experts in emergency medicine, infection control, and laboratory medicine, and with members from any of 10 countries on three continents, the panel arrived at agreement by more than 70% for each of the 14 questions.
“I was impressed and a little surprised that consensus was achieved for every question” posed to the panel by the Fleischner Society for Thoracic Imaging and Diagnosis, Dr. Rubin said in an interview. The panel also placed their 14 decisions about imaging within the context of three distinct clinical scenarios chosen to mirror common real-world situations: mild COVID-19 features, moderate to severe features with no critical-resource constraints, and moderate to severe features with constrained resources. The statement also summarized its conclusions as five main recommendations and three additional recommendations.
Main recommendations
- Imaging is not routinely indicated for COVID-19 screening in asymptomatic people.
- Imaging is not indicated for patients with mild features of COVID-19 unless they are at risk for disease progression.
- Imaging is indicated for patients with features of moderate to severe COVID-19 regardless of COVID-19 test results.
- Imaging is indicated for patients with COVID-19 and evidence of worsening respiratory status.
- When access to CT is limited, chest radiography may be preferred for COVID-19 patients unless features of respiratory worsening warrant using CT.
Additional recommendations
- Daily chest radiographs are not indicated in stable, intubated patients with COVID-19.
- CT is indicated in patients with functional impairment, hypoxemia, or both, after COVID-19 recovery.
- COVID-19 testing is warranted in patients incidentally found to have findings suggestive of COVID-19 on a CT scan.
The statement particularly called out one of its recommendations – that a COVID-19 diagnosis “may be presumed when imaging findings are strongly suggestive of COVID-19 despite negative COVID-19 testing” in a patient who has moderate to severe clinical features of COVID-19 and whose pretest probability is high. The panel voted unanimously in favor of this concept, that imaging is “indicated” in hospitalized patients with moderate to severe symptoms consistent with COVID-19 despite a negative COVID-19 test result. “This guidance represents variance from other published recommendations which advise against the use of imaging for the initial diagnosis of COVID-19,” the statement acknowledged and specifically cited the recommendations issued in March 2020 by the American College of Radiology. Despite that, the ACR and Fleischner recommendations “are not at odds with one another,” maintained Dr. Rubin. The panel based its take on this question on the “direct experience” of its members caring for COVID-19 patients, according to the statement.
“I wholeheartedly agree with the suggested uses of imaging outlined by the panel,” commented Sachin Gupta, MD, FCCP, a pulmonologist and critical care physician in San Francisco. “The consensus statement brings a practical way to consider obtaining imaging. It leaves the door open to local standards and best judgment for using CXR or CT. Many physicians are unclear whether to image low-risk and mildly symptomatic patients. This statement gives support to a watchful waiting approach.” Another recommendation advises against daily CXR in stable, intubated COVID-19 patients. This “now gives backing from an important society and thought leaders while giving an explanation” for why daily imaging is problematic, he noted in an interview. The daily CXR in these patients adds no value, and skipping unneeded imaging minimizes SARS-CoV-2 exposure to radiology personnel, and conserves personal protection equipment, said the statement.
“The Fleischner Society is known worldwide for its recommendations. Having the society lend its weight on triage with imaging for COVID-19 patients is important. I suspect it will help standardize practice.”
Dr. Gupta also highlighted that lung imaging with a portable ultrasound unit has quickly become recognized as a very useful imaging tool with increasing use as the pandemic has unfolded, an option not covered by the Fleischner statement. Study results have “confirmed excellent sensitivity, specificity, and reproducibility” with lung ultrasound, and it’s also “easy to use,” Dr. Gupta said.
Ultrasound chest imaging of COVID-19 patients did not get included in the statement despite the reliance some U.S. sites have already placed on it largely because few on the panel had direct experience using it. “We didn’t feel we could contribute” to a discussion of ultrasound, Dr. Rubin said.
The statement’s recommendations appear to have already begun influencing practice. “The feedback I’ve gotten is that people are relying on them,” said Dr. Rubin, and some programs have sent him screen shots of the recommendations embedded in their local electronic health record.
The Radiological Society of North America is hosting a webinar on the statement on April 17.
A consensus statement on the role of imaging during the acute work-up of COVID-19 patients called for liberal use in patients with moderate to severe clinical features indicative of infection, regardless of their COVID-19 test results, but limited use in patients who present with mild symptoms or are asymptomatic.
The consensus statement on The Role of Imaging in Patient Management during the COVID-19 Pandemic released by the Fleischner Society on April 7 was designed to highlight the “key decision points around imaging” in COVID-19 patients.
“We developed the statement to be applicable across settings” so that each clinic or hospital managing COVID-19 patients could decide the situations where chest radiography (CXR) or CT would work best, said Geoffrey D. Rubin, MD, professor of cardiovascular research, radiology, and bioengineering at Duke University in Durham, N.C., and lead author of the statement.
Written by 15 thoracic radiologists and 10 pulmonologists/intensivists including an anesthesiologist, a pathologist, and additional experts in emergency medicine, infection control, and laboratory medicine, and with members from any of 10 countries on three continents, the panel arrived at agreement by more than 70% for each of the 14 questions.
“I was impressed and a little surprised that consensus was achieved for every question” posed to the panel by the Fleischner Society for Thoracic Imaging and Diagnosis, Dr. Rubin said in an interview. The panel also placed their 14 decisions about imaging within the context of three distinct clinical scenarios chosen to mirror common real-world situations: mild COVID-19 features, moderate to severe features with no critical-resource constraints, and moderate to severe features with constrained resources. The statement also summarized its conclusions as five main recommendations and three additional recommendations.
Main recommendations
- Imaging is not routinely indicated for COVID-19 screening in asymptomatic people.
- Imaging is not indicated for patients with mild features of COVID-19 unless they are at risk for disease progression.
- Imaging is indicated for patients with features of moderate to severe COVID-19 regardless of COVID-19 test results.
- Imaging is indicated for patients with COVID-19 and evidence of worsening respiratory status.
- When access to CT is limited, chest radiography may be preferred for COVID-19 patients unless features of respiratory worsening warrant using CT.
Additional recommendations
- Daily chest radiographs are not indicated in stable, intubated patients with COVID-19.
- CT is indicated in patients with functional impairment, hypoxemia, or both, after COVID-19 recovery.
- COVID-19 testing is warranted in patients incidentally found to have findings suggestive of COVID-19 on a CT scan.
The statement particularly called out one of its recommendations – that a COVID-19 diagnosis “may be presumed when imaging findings are strongly suggestive of COVID-19 despite negative COVID-19 testing” in a patient who has moderate to severe clinical features of COVID-19 and whose pretest probability is high. The panel voted unanimously in favor of this concept, that imaging is “indicated” in hospitalized patients with moderate to severe symptoms consistent with COVID-19 despite a negative COVID-19 test result. “This guidance represents variance from other published recommendations which advise against the use of imaging for the initial diagnosis of COVID-19,” the statement acknowledged and specifically cited the recommendations issued in March 2020 by the American College of Radiology. Despite that, the ACR and Fleischner recommendations “are not at odds with one another,” maintained Dr. Rubin. The panel based its take on this question on the “direct experience” of its members caring for COVID-19 patients, according to the statement.
“I wholeheartedly agree with the suggested uses of imaging outlined by the panel,” commented Sachin Gupta, MD, FCCP, a pulmonologist and critical care physician in San Francisco. “The consensus statement brings a practical way to consider obtaining imaging. It leaves the door open to local standards and best judgment for using CXR or CT. Many physicians are unclear whether to image low-risk and mildly symptomatic patients. This statement gives support to a watchful waiting approach.” Another recommendation advises against daily CXR in stable, intubated COVID-19 patients. This “now gives backing from an important society and thought leaders while giving an explanation” for why daily imaging is problematic, he noted in an interview. The daily CXR in these patients adds no value, and skipping unneeded imaging minimizes SARS-CoV-2 exposure to radiology personnel, and conserves personal protection equipment, said the statement.
“The Fleischner Society is known worldwide for its recommendations. Having the society lend its weight on triage with imaging for COVID-19 patients is important. I suspect it will help standardize practice.”
Dr. Gupta also highlighted that lung imaging with a portable ultrasound unit has quickly become recognized as a very useful imaging tool with increasing use as the pandemic has unfolded, an option not covered by the Fleischner statement. Study results have “confirmed excellent sensitivity, specificity, and reproducibility” with lung ultrasound, and it’s also “easy to use,” Dr. Gupta said.
Ultrasound chest imaging of COVID-19 patients did not get included in the statement despite the reliance some U.S. sites have already placed on it largely because few on the panel had direct experience using it. “We didn’t feel we could contribute” to a discussion of ultrasound, Dr. Rubin said.
The statement’s recommendations appear to have already begun influencing practice. “The feedback I’ve gotten is that people are relying on them,” said Dr. Rubin, and some programs have sent him screen shots of the recommendations embedded in their local electronic health record.
The Radiological Society of North America is hosting a webinar on the statement on April 17.
FROM CHEST
Can this patient get IV contrast?
A 59-year-old man is admitted with abdominal pain. He has a history of pancreatitis. A contrast CT scan is ordered. He reports a history of severe shellfish allergy when the radiology tech checks him in for the procedure. You are paged regarding what to do:
A) Continue with scan as ordered.
B) Switch to MRI scan.
C) Switch to MRI scan with gadolinium.
D) Continue with CT with contrast, give dose of Solu-Medrol.
E) Continue with CT with contrast give IV diphenhydramine.
The correct answer here is A, This patient can receive his scan and receive contrast as ordered.
The mistaken thought was that shellfish contains iodine, so allergy to shellfish was likely to portend allergy to iodine.

Allergy to shellfish is caused by individual proteins that are definitely not in iodine-containing contrast.1 Beaty et al. studied the prevalence of the belief that allergy to shellfish is tied to iodine allergy in a survey given to 231 faculty radiologists and interventional cardiologists.2 Almost 70% responded that they inquire about seafood allergy before procedures that require iodine contrast, and 37% reported they would withhold the contrast or premedicate patients if they had a seafood allergy.
In a more recent study, Westermann-Clark and colleagues surveyed 252 health professionals before and after an educational intervention to dispel the myth of shellfish allergy and iodinated contrast reactions.3 Before the intervention, 66% of participants felt it was important to ask about shellfish allergies and 93% felt it was important to ask about iodine allergies; 26% responded that they would withhold iodinated contrast material in patients with a shellfish allergy, and 56% would withhold in patients with an iodine allergy. A total of 62% reported they would premedicate patients with a shellfish allergy and 75% would premedicate patients with an iodine allergy. The numbers declined dramatically after the educational intervention.
Patients who have seafood allergy have a higher rate of reactions to iodinated contrast, but not at a higher rate than do patients with other food allergies or asthma.4 Most radiology departments do not screen for other food allergies despite the fact these allergies have the same increased risk as for patients with a seafood/shellfish allergy. These patients are more allergic, and in general, are more likely to have reactions. The American Academy of Allergy, Asthma, and Immunology recommends not routinely ordering low- or iso-osmolar radiocontrast media or pretreating with either antihistamines or steroids in patients with a history of seafood allergy.5

There is no evidence that iodine causes allergic reactions. It makes sense that iodine does not cause allergic reactions, as it is an essential component in the human body, in thyroid hormone and in amino acids.6 Patients with dermatitis following topical application of iodine preparations such as povidone-iodide are not reacting to the iodine.
Van Ketel and van den Berg patch-tested patients with a history of dermatitis after exposure to povidone-iodine.7 All patients reacted to patch testing with povidone-iodine, but none reacted to direct testing to iodine (0/5 with patch testing of potassium iodide and 0/3 with testing with iodine tincture).
Take home points:
- It is unnecessary and unhelpful to ask patients about seafood allergies before ordering radiologic studies involving contrast.
- Iodine allergy does not exist.
Dr. Paauw is professor of medicine in the division of general internal medicine at the University of Washington, Seattle, and he serves as third-year medical student clerkship director at the University of Washington. Contact Dr. Paauw at [email protected].
References
1. Narayan AK et al. Avoiding contrast-enhanced computed tomography scans in patients with shellfish allergies. J Hosp Med. 2016 Jun;11(6):435-7.
2. Beaty AD et al. Seafood allergy and radiocontrast media: Are physicians propagating a myth? Am J Med. 2008 Feb;121(2):158.e1-4.
3. Westermann-Clark E et al. Debunking myths about “allergy” to radiocontrast media in an academic institution. Postgrad Med. 2015 Apr;127(3):295-300.
4. Coakley FV and DM Panicek. Iodine allergy: An oyster without a pearl? AJR Am J Roentgenol. 1997 Oct;169(4):951-2.
5. American Academy of Allergy, Asthma & Immunology recommendations on low- or iso-osmolar radiocontrast media.
6. Schabelman E and M Witting. The relationship of radiocontrast, iodine, and seafood allergies: A medical myth exposed. J Emerg Med. 2010 Nov;39(5):701-7.
7. van Ketel WG and WH van den Berg. Sensitization to povidone-iodine. Dermatol Clin. 1990 Jan;8(1):107-9.
A 59-year-old man is admitted with abdominal pain. He has a history of pancreatitis. A contrast CT scan is ordered. He reports a history of severe shellfish allergy when the radiology tech checks him in for the procedure. You are paged regarding what to do:
A) Continue with scan as ordered.
B) Switch to MRI scan.
C) Switch to MRI scan with gadolinium.
D) Continue with CT with contrast, give dose of Solu-Medrol.
E) Continue with CT with contrast give IV diphenhydramine.
The correct answer here is A, This patient can receive his scan and receive contrast as ordered.
The mistaken thought was that shellfish contains iodine, so allergy to shellfish was likely to portend allergy to iodine.
Allergy to shellfish is caused by individual proteins that are definitely not in iodine-containing contrast.1 Beaty et al. studied the prevalence of the belief that allergy to shellfish is tied to iodine allergy in a survey given to 231 faculty radiologists and interventional cardiologists.2 Almost 70% responded that they inquire about seafood allergy before procedures that require iodine contrast, and 37% reported they would withhold the contrast or premedicate patients if they had a seafood allergy.
In a more recent study, Westermann-Clark and colleagues surveyed 252 health professionals before and after an educational intervention to dispel the myth of shellfish allergy and iodinated contrast reactions.3 Before the intervention, 66% of participants felt it was important to ask about shellfish allergies and 93% felt it was important to ask about iodine allergies; 26% responded that they would withhold iodinated contrast material in patients with a shellfish allergy, and 56% would withhold in patients with an iodine allergy. A total of 62% reported they would premedicate patients with a shellfish allergy and 75% would premedicate patients with an iodine allergy. The numbers declined dramatically after the educational intervention.
Patients who have seafood allergy have a higher rate of reactions to iodinated contrast, but not at a higher rate than do patients with other food allergies or asthma.4 Most radiology departments do not screen for other food allergies despite the fact these allergies have the same increased risk as for patients with a seafood/shellfish allergy. These patients are more allergic, and in general, are more likely to have reactions. The American Academy of Allergy, Asthma, and Immunology recommends not routinely ordering low- or iso-osmolar radiocontrast media or pretreating with either antihistamines or steroids in patients with a history of seafood allergy.5
There is no evidence that iodine causes allergic reactions. It makes sense that iodine does not cause allergic reactions, as it is an essential component in the human body, in thyroid hormone and in amino acids.6 Patients with dermatitis following topical application of iodine preparations such as povidone-iodide are not reacting to the iodine.
Van Ketel and van den Berg patch-tested patients with a history of dermatitis after exposure to povidone-iodine.7 All patients reacted to patch testing with povidone-iodine, but none reacted to direct testing to iodine (0/5 with patch testing of potassium iodide and 0/3 with testing with iodine tincture).
Take home points:
- It is unnecessary and unhelpful to ask patients about seafood allergies before ordering radiologic studies involving contrast.
- Iodine allergy does not exist.
Dr. Paauw is professor of medicine in the division of general internal medicine at the University of Washington, Seattle, and he serves as third-year medical student clerkship director at the University of Washington. Contact Dr. Paauw at [email protected].
References
1. Narayan AK et al. Avoiding contrast-enhanced computed tomography scans in patients with shellfish allergies. J Hosp Med. 2016 Jun;11(6):435-7.
2. Beaty AD et al. Seafood allergy and radiocontrast media: Are physicians propagating a myth? Am J Med. 2008 Feb;121(2):158.e1-4.
3. Westermann-Clark E et al. Debunking myths about “allergy” to radiocontrast media in an academic institution. Postgrad Med. 2015 Apr;127(3):295-300.
4. Coakley FV and DM Panicek. Iodine allergy: An oyster without a pearl? AJR Am J Roentgenol. 1997 Oct;169(4):951-2.
5. American Academy of Allergy, Asthma & Immunology recommendations on low- or iso-osmolar radiocontrast media.
6. Schabelman E and M Witting. The relationship of radiocontrast, iodine, and seafood allergies: A medical myth exposed. J Emerg Med. 2010 Nov;39(5):701-7.
7. van Ketel WG and WH van den Berg. Sensitization to povidone-iodine. Dermatol Clin. 1990 Jan;8(1):107-9.
A 59-year-old man is admitted with abdominal pain. He has a history of pancreatitis. A contrast CT scan is ordered. He reports a history of severe shellfish allergy when the radiology tech checks him in for the procedure. You are paged regarding what to do:
A) Continue with scan as ordered.
B) Switch to MRI scan.
C) Switch to MRI scan with gadolinium.
D) Continue with CT with contrast, give dose of Solu-Medrol.
E) Continue with CT with contrast give IV diphenhydramine.
The correct answer here is A, This patient can receive his scan and receive contrast as ordered.
The mistaken thought was that shellfish contains iodine, so allergy to shellfish was likely to portend allergy to iodine.
Allergy to shellfish is caused by individual proteins that are definitely not in iodine-containing contrast.1 Beaty et al. studied the prevalence of the belief that allergy to shellfish is tied to iodine allergy in a survey given to 231 faculty radiologists and interventional cardiologists.2 Almost 70% responded that they inquire about seafood allergy before procedures that require iodine contrast, and 37% reported they would withhold the contrast or premedicate patients if they had a seafood allergy.
In a more recent study, Westermann-Clark and colleagues surveyed 252 health professionals before and after an educational intervention to dispel the myth of shellfish allergy and iodinated contrast reactions.3 Before the intervention, 66% of participants felt it was important to ask about shellfish allergies and 93% felt it was important to ask about iodine allergies; 26% responded that they would withhold iodinated contrast material in patients with a shellfish allergy, and 56% would withhold in patients with an iodine allergy. A total of 62% reported they would premedicate patients with a shellfish allergy and 75% would premedicate patients with an iodine allergy. The numbers declined dramatically after the educational intervention.
Patients who have seafood allergy have a higher rate of reactions to iodinated contrast, but not at a higher rate than do patients with other food allergies or asthma.4 Most radiology departments do not screen for other food allergies despite the fact these allergies have the same increased risk as for patients with a seafood/shellfish allergy. These patients are more allergic, and in general, are more likely to have reactions. The American Academy of Allergy, Asthma, and Immunology recommends not routinely ordering low- or iso-osmolar radiocontrast media or pretreating with either antihistamines or steroids in patients with a history of seafood allergy.5
There is no evidence that iodine causes allergic reactions. It makes sense that iodine does not cause allergic reactions, as it is an essential component in the human body, in thyroid hormone and in amino acids.6 Patients with dermatitis following topical application of iodine preparations such as povidone-iodide are not reacting to the iodine.
Van Ketel and van den Berg patch-tested patients with a history of dermatitis after exposure to povidone-iodine.7 All patients reacted to patch testing with povidone-iodine, but none reacted to direct testing to iodine (0/5 with patch testing of potassium iodide and 0/3 with testing with iodine tincture).
Take home points:
- It is unnecessary and unhelpful to ask patients about seafood allergies before ordering radiologic studies involving contrast.
- Iodine allergy does not exist.
Dr. Paauw is professor of medicine in the division of general internal medicine at the University of Washington, Seattle, and he serves as third-year medical student clerkship director at the University of Washington. Contact Dr. Paauw at [email protected].
References
1. Narayan AK et al. Avoiding contrast-enhanced computed tomography scans in patients with shellfish allergies. J Hosp Med. 2016 Jun;11(6):435-7.
2. Beaty AD et al. Seafood allergy and radiocontrast media: Are physicians propagating a myth? Am J Med. 2008 Feb;121(2):158.e1-4.
3. Westermann-Clark E et al. Debunking myths about “allergy” to radiocontrast media in an academic institution. Postgrad Med. 2015 Apr;127(3):295-300.
4. Coakley FV and DM Panicek. Iodine allergy: An oyster without a pearl? AJR Am J Roentgenol. 1997 Oct;169(4):951-2.
5. American Academy of Allergy, Asthma & Immunology recommendations on low- or iso-osmolar radiocontrast media.
6. Schabelman E and M Witting. The relationship of radiocontrast, iodine, and seafood allergies: A medical myth exposed. J Emerg Med. 2010 Nov;39(5):701-7.
7. van Ketel WG and WH van den Berg. Sensitization to povidone-iodine. Dermatol Clin. 1990 Jan;8(1):107-9.
Consider PET/CT when infectious source is a puzzler
CHICAGO – Dual positron emission tomography-computed tomography (PET/CT) scans changed the treatment course of nearly half of patients whose scans were positive for infection. In a single-center systematic review of 18fluorodeoxyglucose (FDG)–PET/CT scans, 55 of the 138 scans (40%) changed clinical management.

Presenting the findings at the annual meeting of the Radiological Society of North America, Benjamin Viglianti, MD, PhD, said that PET/CT had particular utility in cases of bacteremia and endocarditis, in which the scans changed treatment in 46% of those cases.
Dr. Viglianti, a radiologist at the University of Michigan, Ann Arbor, explained that medical student and first author Anitha Menon, himself, and their collaborators deliberately used a broad definition of clinical management change. The management course was considered to change not only if an unknown infection site was discovered or if a new intervention was initiated after the scan, but also if antibiotic choice or duration was changed or an additional specialty was consulted.
Scans were included in the study if an infectious etiology was found in the scan and if the patient received an infectious disease consult. Bacteremia and endocarditis were the most frequent indications for scans and also the indications for which management was most frequently changed. When a vascular cause was the indication for the scan, management changed 41% of the time. For fevers of unknown origin, the scan changed management in 30% of the cases, while for osteomyelitis, management was changed for 28% of patients.
The investigators identified several broad themes from their review that pointed toward when clinicians might consider FDG-PET/CT imaging in infectious disease management.
The first, said Dr. Viglianti, was that “for patients with suspected vascular graft infection, PET/CT using FDG may be a good first-choice imaging modality.” He pointed to an illustrative case of a patient who was 1 month out from open repair of a thoracoabdominal aortic aneurysm. The patient had abdominal pain, epigastric tenderness and nausea, as well as an erythematous incision site. A CT scan just revealed an abdominal fluid collection, but the PET/CT scan showed radiotracer uptake at the prior repair site, indicating infection.
For patients with bacteremia, the investigators judged that FDG-PET/CT might be particularly useful in patients who have a graft, prosthetic valve, or cardiac device. Here, Dr. Viglianti and his collaborators highlighted the scan of a woman with DiGeorge syndrome who had received aortic root replacement for truncus arteriosis. She had been found to have persistent enterococcal bacteremia at high levels, but had been symptom free. To take a close look at the suspected infectious nidus, a transesophageal echocardiogram had been obtained, but this study didn’t turn up any clear masses or vegetations. The PET/CT scan, though, revealed avid FDG uptake in the area of the prosthesis.
Management course was not likely to be changed for patients with fever of unknown origin, but the investigators did note that whole-body PET/CT was useful to distinguish infectious etiologies from hematologic and oncologic processes. Their review included a patient who had Crohn’s disease and fever, myalgias, and upper abdominal pain, as well as liver enzyme elevation. The PET/CT showed radiotracer uptake within the spleen, which was enlarged. The scan also showed bone marrow uptake; these findings pointed toward hemophagocytic lymphohistiocytosis rather than an infectious etiology.
For osteomyelitis, said Dr. Viglianti, FDG-PET may have limited utility; it might be most useful when MRI is contraindicated. Within the study population, the investigators identified a patient who had chills and fever along with focal tenderness over the lumbar spine in the context of recent pyelonephritis of a graft kidney. Here, MRI findings were suspicious for osteomyelitis and diskitis, and the FDG uptake at the L4-L5 vertebral levels confirmed the MRI results.
When a patient with a prosthetic valve is suspected of having endocarditis, “cardiac PET/CT may be of high diagnostic value,” said Dr. Viglianti. For patients with endocarditis of native valves, though, a full-body FDG-PET/CT scan may spot septic emboli. A patient identified in the investigators’ review had been admitted for methicillin-resistant Staphylococcus aureus endocarditis. The patient, who had a history of intravenous drug use, received a transesophageal echocardiogram that found severe tricuspid valve regurgitation and vegetations. The whole-body PET/CT scan, though, revealed avid uptake in both buttocks, as well as thigh, ankle and calf muscles – a pattern “suspicious for infectious myositis,” said the researchers.
In discussion during the poster session, Dr. Viglianti said that, although reimbursement for PET/CT scans for infectious etiologies might not be feasible, it can still be a reasonable and even cost-effective choice. At his institution, he said, the requisite radioisotope is made in-house, twice daily, so it’s relatively easy to arrange scans. Since PET/CT scans can be acquired relatively quickly and there’s no delay while waiting for radiotracer uptake, clinical decisions can be made more quickly than when waiting for bone uptake for a technetium-99 scan, he said. This can have the effect of saving a night of hospitalization in many cases.
Dr. Viglianti and Ms. Menon reported that they had no relevant conflicts of interest. No outside sources of funding were reported.
SOURCE: Menon A et al. RSNA 2019, Abstract NM203-SDSUB1.
CHICAGO – Dual positron emission tomography-computed tomography (PET/CT) scans changed the treatment course of nearly half of patients whose scans were positive for infection. In a single-center systematic review of 18fluorodeoxyglucose (FDG)–PET/CT scans, 55 of the 138 scans (40%) changed clinical management.
Presenting the findings at the annual meeting of the Radiological Society of North America, Benjamin Viglianti, MD, PhD, said that PET/CT had particular utility in cases of bacteremia and endocarditis, in which the scans changed treatment in 46% of those cases.
Dr. Viglianti, a radiologist at the University of Michigan, Ann Arbor, explained that medical student and first author Anitha Menon, himself, and their collaborators deliberately used a broad definition of clinical management change. The management course was considered to change not only if an unknown infection site was discovered or if a new intervention was initiated after the scan, but also if antibiotic choice or duration was changed or an additional specialty was consulted.
Scans were included in the study if an infectious etiology was found in the scan and if the patient received an infectious disease consult. Bacteremia and endocarditis were the most frequent indications for scans and also the indications for which management was most frequently changed. When a vascular cause was the indication for the scan, management changed 41% of the time. For fevers of unknown origin, the scan changed management in 30% of the cases, while for osteomyelitis, management was changed for 28% of patients.
The investigators identified several broad themes from their review that pointed toward when clinicians might consider FDG-PET/CT imaging in infectious disease management.
The first, said Dr. Viglianti, was that “for patients with suspected vascular graft infection, PET/CT using FDG may be a good first-choice imaging modality.” He pointed to an illustrative case of a patient who was 1 month out from open repair of a thoracoabdominal aortic aneurysm. The patient had abdominal pain, epigastric tenderness and nausea, as well as an erythematous incision site. A CT scan just revealed an abdominal fluid collection, but the PET/CT scan showed radiotracer uptake at the prior repair site, indicating infection.
For patients with bacteremia, the investigators judged that FDG-PET/CT might be particularly useful in patients who have a graft, prosthetic valve, or cardiac device. Here, Dr. Viglianti and his collaborators highlighted the scan of a woman with DiGeorge syndrome who had received aortic root replacement for truncus arteriosis. She had been found to have persistent enterococcal bacteremia at high levels, but had been symptom free. To take a close look at the suspected infectious nidus, a transesophageal echocardiogram had been obtained, but this study didn’t turn up any clear masses or vegetations. The PET/CT scan, though, revealed avid FDG uptake in the area of the prosthesis.
Management course was not likely to be changed for patients with fever of unknown origin, but the investigators did note that whole-body PET/CT was useful to distinguish infectious etiologies from hematologic and oncologic processes. Their review included a patient who had Crohn’s disease and fever, myalgias, and upper abdominal pain, as well as liver enzyme elevation. The PET/CT showed radiotracer uptake within the spleen, which was enlarged. The scan also showed bone marrow uptake; these findings pointed toward hemophagocytic lymphohistiocytosis rather than an infectious etiology.
For osteomyelitis, said Dr. Viglianti, FDG-PET may have limited utility; it might be most useful when MRI is contraindicated. Within the study population, the investigators identified a patient who had chills and fever along with focal tenderness over the lumbar spine in the context of recent pyelonephritis of a graft kidney. Here, MRI findings were suspicious for osteomyelitis and diskitis, and the FDG uptake at the L4-L5 vertebral levels confirmed the MRI results.
When a patient with a prosthetic valve is suspected of having endocarditis, “cardiac PET/CT may be of high diagnostic value,” said Dr. Viglianti. For patients with endocarditis of native valves, though, a full-body FDG-PET/CT scan may spot septic emboli. A patient identified in the investigators’ review had been admitted for methicillin-resistant Staphylococcus aureus endocarditis. The patient, who had a history of intravenous drug use, received a transesophageal echocardiogram that found severe tricuspid valve regurgitation and vegetations. The whole-body PET/CT scan, though, revealed avid uptake in both buttocks, as well as thigh, ankle and calf muscles – a pattern “suspicious for infectious myositis,” said the researchers.
In discussion during the poster session, Dr. Viglianti said that, although reimbursement for PET/CT scans for infectious etiologies might not be feasible, it can still be a reasonable and even cost-effective choice. At his institution, he said, the requisite radioisotope is made in-house, twice daily, so it’s relatively easy to arrange scans. Since PET/CT scans can be acquired relatively quickly and there’s no delay while waiting for radiotracer uptake, clinical decisions can be made more quickly than when waiting for bone uptake for a technetium-99 scan, he said. This can have the effect of saving a night of hospitalization in many cases.
Dr. Viglianti and Ms. Menon reported that they had no relevant conflicts of interest. No outside sources of funding were reported.
SOURCE: Menon A et al. RSNA 2019, Abstract NM203-SDSUB1.
CHICAGO – Dual positron emission tomography-computed tomography (PET/CT) scans changed the treatment course of nearly half of patients whose scans were positive for infection. In a single-center systematic review of 18fluorodeoxyglucose (FDG)–PET/CT scans, 55 of the 138 scans (40%) changed clinical management.
Presenting the findings at the annual meeting of the Radiological Society of North America, Benjamin Viglianti, MD, PhD, said that PET/CT had particular utility in cases of bacteremia and endocarditis, in which the scans changed treatment in 46% of those cases.
Dr. Viglianti, a radiologist at the University of Michigan, Ann Arbor, explained that medical student and first author Anitha Menon, himself, and their collaborators deliberately used a broad definition of clinical management change. The management course was considered to change not only if an unknown infection site was discovered or if a new intervention was initiated after the scan, but also if antibiotic choice or duration was changed or an additional specialty was consulted.
Scans were included in the study if an infectious etiology was found in the scan and if the patient received an infectious disease consult. Bacteremia and endocarditis were the most frequent indications for scans and also the indications for which management was most frequently changed. When a vascular cause was the indication for the scan, management changed 41% of the time. For fevers of unknown origin, the scan changed management in 30% of the cases, while for osteomyelitis, management was changed for 28% of patients.
The investigators identified several broad themes from their review that pointed toward when clinicians might consider FDG-PET/CT imaging in infectious disease management.
The first, said Dr. Viglianti, was that “for patients with suspected vascular graft infection, PET/CT using FDG may be a good first-choice imaging modality.” He pointed to an illustrative case of a patient who was 1 month out from open repair of a thoracoabdominal aortic aneurysm. The patient had abdominal pain, epigastric tenderness and nausea, as well as an erythematous incision site. A CT scan just revealed an abdominal fluid collection, but the PET/CT scan showed radiotracer uptake at the prior repair site, indicating infection.
For patients with bacteremia, the investigators judged that FDG-PET/CT might be particularly useful in patients who have a graft, prosthetic valve, or cardiac device. Here, Dr. Viglianti and his collaborators highlighted the scan of a woman with DiGeorge syndrome who had received aortic root replacement for truncus arteriosis. She had been found to have persistent enterococcal bacteremia at high levels, but had been symptom free. To take a close look at the suspected infectious nidus, a transesophageal echocardiogram had been obtained, but this study didn’t turn up any clear masses or vegetations. The PET/CT scan, though, revealed avid FDG uptake in the area of the prosthesis.
Management course was not likely to be changed for patients with fever of unknown origin, but the investigators did note that whole-body PET/CT was useful to distinguish infectious etiologies from hematologic and oncologic processes. Their review included a patient who had Crohn’s disease and fever, myalgias, and upper abdominal pain, as well as liver enzyme elevation. The PET/CT showed radiotracer uptake within the spleen, which was enlarged. The scan also showed bone marrow uptake; these findings pointed toward hemophagocytic lymphohistiocytosis rather than an infectious etiology.
For osteomyelitis, said Dr. Viglianti, FDG-PET may have limited utility; it might be most useful when MRI is contraindicated. Within the study population, the investigators identified a patient who had chills and fever along with focal tenderness over the lumbar spine in the context of recent pyelonephritis of a graft kidney. Here, MRI findings were suspicious for osteomyelitis and diskitis, and the FDG uptake at the L4-L5 vertebral levels confirmed the MRI results.
When a patient with a prosthetic valve is suspected of having endocarditis, “cardiac PET/CT may be of high diagnostic value,” said Dr. Viglianti. For patients with endocarditis of native valves, though, a full-body FDG-PET/CT scan may spot septic emboli. A patient identified in the investigators’ review had been admitted for methicillin-resistant Staphylococcus aureus endocarditis. The patient, who had a history of intravenous drug use, received a transesophageal echocardiogram that found severe tricuspid valve regurgitation and vegetations. The whole-body PET/CT scan, though, revealed avid uptake in both buttocks, as well as thigh, ankle and calf muscles – a pattern “suspicious for infectious myositis,” said the researchers.
In discussion during the poster session, Dr. Viglianti said that, although reimbursement for PET/CT scans for infectious etiologies might not be feasible, it can still be a reasonable and even cost-effective choice. At his institution, he said, the requisite radioisotope is made in-house, twice daily, so it’s relatively easy to arrange scans. Since PET/CT scans can be acquired relatively quickly and there’s no delay while waiting for radiotracer uptake, clinical decisions can be made more quickly than when waiting for bone uptake for a technetium-99 scan, he said. This can have the effect of saving a night of hospitalization in many cases.
Dr. Viglianti and Ms. Menon reported that they had no relevant conflicts of interest. No outside sources of funding were reported.
SOURCE: Menon A et al. RSNA 2019, Abstract NM203-SDSUB1.
REPORTING FROM RSNA 2019
Decreasing Overutilization of Echocardiograms and Abdominal Imaging in the Evaluation of Children with Fungemia
From the University of Miami, Department of Pediatrics and Department of Medicine, Miami, FL.
Abstract
- Objective: Pediatric fungemia is associated with a low risk of fungal endocarditis and renal infections. The majority of current guidelines do not recommend routine abdominal imaging/echocardiograms in the evaluation of fungemia, but such imaging has been routinely ordered for patients on the pediatric gastroenterology service at our institution. Our goals were to assess the financial impact of this deviation from current clinical guidelines and redefine the standard work to reduce overutilization of abdominal ultrasounds and echocardiograms. Specifically, our goal was to reduce imaging by 50% by 18 months.
- Methods: Root cause analysis showed a lack of familiarity with current evidence. Using this data, countermeasures were implemented, including practitioner education of guidelines and creation of a readily accessible clinical pathway and an electronic order set for pediatric fungemia management. Balancing measures were missed episodes of fungal endocarditis and renal infection.
- Results: During the period January 1, 2016 to November 19, 2017, 18 of 21 episodes of fungemia in our pediatric institution occurred in patients admitted to the pediatric gastroenterology service. Abdominal imaging and echocardiograms were done 100% of the time, with no positive findings and an estimated cost of approximately $58,000. Post-intervention from November 20, 2017 to April 3, 2019, 7 of 13 episodes of fungemia occurred on this service. Frequency of abdominal imaging and echocardiograms decreased to 43% and 57%, respectively. No episodes of fungal endocarditis or renal infection were identified.
- Conclusion: Overutilization of abdominal imaging and echocardiograms in pediatric fungemia evaluation can be safely decreased.
Keywords: guidelines; cost; candidemia; endocarditis.
Practitioners may remain under the impression that routine abdominal ultrasounds (US) and echocardiograms (echo) are indicated in fungemia to evaluate for fungal endocarditis and renal infection, although these conditions are rare and limited to a subset of the population.1-10 Risk factors include prematurity, immunosuppression, prior bacterial endocarditis, abnormal cardiac valves, and previous urogenital surgeries.11
The 2016 Infectious Diseases Society of America (IDSA) guidelines do not recommend routine US or echo but rather provide scenarios in which Candida endocarditis should be suspected, and these include: persistently positive blood cultures, persistent fevers despite appropriate therapy, and clinical signs that may suggest endocarditis, such as a new heart murmur, heart failure, or embolic phenomena.11 IDSA recommends abdominal imaging in neonates with persistently positive blood cultures to evaluate the urogenital system, in addition to the liver and spleen. They also recommend abdominal imaging in symptomatic ascending Candida pyelonephritis beyond the neonatal period and in chronic disseminated candidiasis; the latter is uncommon and seen almost exclusively in patients recovering from neutropenia with a hematologic malignancy.11
We also reviewed guidelines on fungemia originating outside the United States. The 2010 Canadian clinical guidelines on invasive candidiasis do not explicitly recommend routine imaging, but rather state that various imaging studies, including US and echo among others, may be helpful.12 The German Speaking Mycological Society and the Paul-Ehrlich-Society for Chemotherapy published a joint recommendation against routine US and echo in uncomplicated candidemia in 2011.13
The European Society for Clinical Microbiology and Infectious Diseases is the only society that recommends routine echo. Their 2012 guidelines on candidiasis recommend transesophageal echo in adults14 and echocardiography in children,15 as well as abdominal imaging in the diagnosis of chronic disseminated candidiasis in adults with hematological malignancies/hematopoietic stem cell transplantation.16
The 2013 Brazilian guidelines explicitly recommend against routine abdominal imaging and echo because of the low frequency of visceral lesions in adults with candidemia and recommend reserving imaging for those with persistently positive blood cultures or with clinical signs/symptoms suggestive of endocarditis/abdominal infection or clinical deterioration.17 The 2014 Japanese guidelines recommend ruling out chronic disseminated candidiasis in these patients with symptoms during the neutrophil recovery phase, but do not mention routinely imaging other patients. They do not address the role of echocardiography.18
Although physicians in the United Sates typically follow IDSA guidelines, abdominal US and echo were ordered routinely for patients with fungemia on the pediatric gastroenterology service at our institution, leading to higher medical costs and waste of medical resources. Our goals were to assess the current standard work for fungemia evaluation on this service, assess the impact of its deviation from current clinical guidelines, and redefine the standard work by (1) presenting current evidence to practitioners taking care of patients on this service, (2) providing a clinical pathway that allowed for variations where appropriate, and (3) providing a plan for pediatric fungemia management. Our SMART (Specific, Measurable, Attainable, Relevant and Timely) goal was to reduce overutilization of abdominal US and echo in pediatric patients with fungemia on the pediatric gastroenterology service by 50%.
Methods
Study, Setting, and Participants
We executed this quality improvement project at a quaternary care pediatric hospital affiliated with a school of medicine. The project scope consisted of inpatient pediatric patients with fungemia on the pediatric gastroenterology service admitted to the wards or pediatric critical care unit at this institution, along with the practitioners caring for these patients. The project was part of an institutional quality improvement initiative program. The quality improvement team included quality improvement experts from the departments of medicine and pediatrics, a pediatric resident and student, and physicians from the divisions of pediatric infectious disease, pediatric critical care, and pediatric gastroenterology. This study qualified for Institutional Review Board (IRB) exemption based on the University’s IRB stipulations.
Current Condition
Root cause analysis was performed by creating a process map of the current standard work and a fishbone diagram (Figure 1). We incorporated feedback from voice of the customer in the root cause analysis. In this analysis, the voice of the customer came from the bedside floor nurses, ultrasound clerk and sonographer, echo technician, cardiology fellow, and microbiology medical technician. We got their feedback on our process map, its accuracy and ways to expand, their thoughts on the problem and why we have this problem, and any solutions they could offer to help improve the problem. Some of the key points obtained were: echos were not routinely done on the floors and were not considered urgent as they often did not change management; the sonographer and those from the cardiology department felt imaging was often overutilized because of misconceptions and lack of available hospital guidelines. Suggested solutions included provider education with reference to Duke’s criteria and establishing a clinical pathway approved by all concerned departments.

Prior to education, we surveyed current practices of practitioners on teams caring for these patients, which included physicians of all levels (attendings, fellows, residents) as well as nurse practitioners and medical students from the department of pediatrics and divisions of pediatric gastroenterology, pediatric infectious disease, and pediatric critical care medicine.
Countermeasures
Practitioner Education. In October 2017 practitioners were given a 20-minute presentation on the latest international guidelines on fungemia. Fifty-nine practitioners completed pre- and post-test surveys. Eight respondents were excluded due to incomplete surveys. We compared self-reported frequencies of ordering abdominal imaging and echo before the presentation with intention to order post education. Intention to change clinical practice after the presentation was also surveyed.
Clinical Pathway. Education alone may not result in sustainability, and thus we provided a readily accessible clinical pathway and an electronic order set for pediatric fungemia management. Inter-department buy-in was also necessary for success. It was important to get the input from the various teams (infectious disease, cardiology, gastroenterology, and critical care), which was done by incorporating members from those divisions in the project or getting their feedback through voice of the customer analysis.
We redefined standard work based on current evidence and created a clinical pathway during March 2018 that included variations when appropriate (Figure 2). We presented the clinical pathway to practitioners and distributed it via email. We also made it available to pediatric residents and fellows on their mobile institutional work resource application.

Electronic Order Set. We created an electronic order set for pediatric fungemia management and made it available in the electronic health record May 2018.
Measurement
Cases of fungemia were identified through the electronic health record pre-intervention (January 1, 2016 through November 19, 2017) and post-intervention (November 20, 2019 through April 3, 2019). An episode of fungemia was defined as an encounter with 1 or more positive blood culture(s) for Candida species or Cryptococcus species. We manually identified patients belonging to the pediatric gastroenterology service and reviewed these charts to determine the presenting complaint, organism isolated, transplant status, central lines status, risk factors, if abdominal imaging or echocardiography were done for the episode of fungemia, and their corresponding results. We calculated overall and per patient medical charges by using the average charges at our institution of US and echocardiography with a cardiology consult. These average charges were provided by patient financial services and the pediatric cardiology department, respectively. To address non-technical expenditures, we calculated the average time taken for transport to and from radiology and the echo suite for each identified patient. We identified missed fungal endocarditis and fungal balls as balancing measures.
Results
Survey
Among the 51 practitioners surveyed, 36% were performing routine echo and 22% self-reported performing routine abdominal imaging. After education, no respondents planned to routinely do echo or abdominal imaging. All but 1 respondent planned to change their practice for evaluation of fungemia patients based on the presentation (eFigure 1).

Baseline Data
Over the 23-month period from January 1, 2016 to November 19, 2017, there were 21 episodes of fungemia, 18 of which occurred in patients on the pediatric gastroenterology service (2 of the 18 were transplant recipients). For the 18 episodes on this service, abdominal imaging and echo were done 100% of the time, with 0 positive findings (eFigure 2).

Of those 18 episodes, the average age was 4.6 years, with two-thirds of the population being male. There were 3 patients with multiple episodes that accounted for 8 of the episodes (3, 3, and 2 episodes each). Fever was the most common presenting complaint. The most common organism was Candida parapsilosis (6 of the 18 episodes). All episodes but one involved a central line, and all central lines were removed when present except for one case. Of the risk factors, 3 episodes occurred in neutropenic patients, and for 1 episode the patient had a questionable history of fungal endocarditis (and was on fungal prophylaxis). There were no patients with recent cardiac/urogenital surgery or prior fungal balls. No episodes had clinical symptoms suggestive of fungal endocarditis or fungal balls.
Post-Intervention Data
Over the subsequent 17-month period (November 11, 2017 to April 3, 2019), there were 13 episodes of candidemia. There were no episodes of Cryptococcus fungemia. Seven episodes occurred in patients on the pediatric gastroenterology service (2 of the 7 occurred in transplant recipients). Abdominal imaging was done in 3 of these episodes (43%), and in 2 of these 3 episodes, imaging was done at an outside institution prior to arrival, with no positive results (eFigure 2).
Echocardiography was done 57% of the time (n = 4), with echo being done at an outside institution prior to arrival half of the time (n = 2), with no endocarditis identified. The cases of abdominal imaging and echo done at outside institutions prior to arrival were not impacted by the countermeasures. Excluding those 2 patients who had both abdominal imaging and echocardiography done prior to arrival, the overall rate of imaging (both abdominal imaging and echo) done after countermeasures were instituted was 30% (Figure 3).
 was done.")
Of those 7 episodes, the average age was 6.8 years (57% female). There were no patients with multiple episodes. The most common presenting complaint was fever. The most common organism was Candida albicans (3 of the 7 episodes). All episodes involved a central line, which was removed in all cases except for one. Of the risk factors, 2 episodes were in neutropenic patients, and 1 episode had a history of bacterial endocarditis (not related to fungemia). No episodes occurred in patients with prior fungal renal infection, urogenital malformations, or recent cardiac/urogenital surgery. No episodes had clinical symptoms suggestive of fungal endocarditis or renal infection. No episodes of fungal endocarditis or renal infection were identified.
On average, a patient at our institution undergoing abdominal US and echo with a cardiology consult results in medical waste of approximately $3200 per patient. This cost does not take into account other miscellaneous charges possibly incurred, such as the radiologist interpreting the findings and transportation. Baseline data calculations show that patients waste on average 55 minutes in physical transport, and this does not take into account wait times.
Discussion
Candidemia contributes to 10% of central-line associated blood stream infections (CLABSI).19 Increased usage of indwelling central catheters for administration of parenteral nutrition will inevitably result in practitioners encountering cases of candidemia when caring for this population. As seen from our results, the majority of episodes of candidemia at our institution occurred on the pediatric gastroenterology service, and thus redefining standard work on this service will be impactful.
Candida parapsilosis and Candida albicans were the most common causative agents before and after intervention, respectively, but overall the most common organism was Candida albicans, which is in keeping with that of CLABSI in the literature.19 Growth of Candida parapsilosis has been particularly linked to CLABSI.19 The third most common organism in our study was Candida glabrata, which is the second most common cause of candidemia in CLABSI.19
The cases of positive abdominal imaging in fungemia in the literature are limited to the neonatal population1-4 and chronic disseminated candidiasis in patients with hematologic malignancies/neutropenia/immunosuppression.5,6 In fungal endocarditis, the reported cases were generally in neonates,1,3,7 critically ill patients,8 patients with hematologic malignancies/neutropenia/immunosuppression,6,9 or those with a cardiac history.9,10 This population differs from the patient population on the pediatric gastroenterology service. Patients on this service may not need US or echo. Performing abdominal US and echo in fungemia patients in whom such imaging is not indicated may result in medical waste of approximately $3200 per patient. There is also a waste of medical resources and time.
We found almost all practitioners are willing to change clinical practice once provided with current guidelines. Face-to-face oral presentations allowed for questions and interaction, making this form of information dissemination better than e-mails or handouts.
Though the numbers were small over the short study period, we were able to decrease overutilization of abdominal imaging and echo after implementing countermeasures. Frequency decreased from 100% to 43% and 57% for abdominal imaging and echo, respectively. Imaging that was done after the countermeasures were implemented was mainly attributed to imaging patients underwent prior to presenting to our institution. This reinforces the need for education at other institutions as well. Of the balancing measures assessed, there were no missed cases of fungal balls or fungal endocarditis. Additionally,
The findings from this quality improvement project underscore current recommendations that, despite common misconceptions, routine abdominal US and echo are not indicated in all cases of fungemia. Case-by-case assessment based on the clinical scenario remains key to management of fungemia to avoid unnecessary medical interventions.
Corresponding author: Donna Cheung, MBBS, 200 Hawkins Drive, BT 1120-G, Iowa City, IA 52242; [email protected].
Financial support: None.
1. Benjamin DK Jr, Poole C, Steinbach WJ, et al. Neonatal candidemia and end-organ damage: a critical appraisal of the literature using meta-analytic techniques. Pediatrics. 2003;112:634-640.
2. Wynn JL, Tan S, Gantz MG, et al. Outcomes following candiduria in extremely low birth weight infants. Clin Infect Dis. 2012;54:331-339.
3. Noyola DE, Fernandez M, Moylett EH, et al. Ophthalmologic, visceral, and cardiac involvement in neonates with candidemia. Clin Infect Dis. 2001;32:1018-1023.
4. Phillips JR, Karlowicz MG Prevalence of Candida species in hospital-acquired urinary tract infections in a neonatal intensive care unit. Pediatr Infect Dis J. 1997;16:190-194.
5. Pagano L, Mele L, Fianchi L, et al. Chronic disseminated candidiasis in patients with hematologic malignancies. Clinical features and outcome of 29 episodes. Haematologica. 2002;87:535-541.
6. Zaoutis TE, Greves HM, Lautenbach E, et al. Risk factors for disseminated candidiasis in children with candidemia. Pediatr Infect Dis J. 2004;23:635-641.
7. Levy I, Shalit I, Birk E, et al. Candida endocarditis in neonates: report of five cases and review of the literature. Mycoses. 2006;49:43-48.
8. Aspesberro F, Beghetti M, Oberhansli I, et al. Fungal endocarditis in critically ill children. Eur J Pediatr. 1999;158:275-280.
9. Fernandez-Cruz A, Cruz Menarguez M, Munoz P, et al. The search for endocarditis in patients with candidemia: a systematic recommendation for echocardiography? A prospective cohort. Eur J Clin Microbiol Infect Dis. 2015;34:1543-1549.
10. Hernandez-Torres A, Garcia-Vazquez E, Laso-Ortiz A, et al. [Candida sp endocarditis. Experience in a third-level hospital and review of the literature]. Rev Esp Quimioter. 2013;26:51-55.
11. Pappas PG, Kauffman CA, Andes DR, et al. Clinical practice guideline for the management of candidiasis: 2016 Update by the Infectious Diseases Society of America. Clin Infect Dis. 2016;62:e1-50.
12. Bow EJ, Evans G, Fuller J, et al. Canadian clinical practice guidelines for invasive candidiasis in adults. Can J Infect Dis Med Microbiol. 2010;21:e122-50.
13. Ruhnke M, Rickerts V, Cornely OA, et al. Diagnosis and therapy of Candida infections: joint recommendations of the German Speaking Mycological Society and the Paul-Ehrlich-Society for Chemotherapy. Mycoses. 2011;54:279-310.
14. Cornely OA, Bassetti M, Calandra T, et al. ESCMID* guideline for the diagnosis and management of Candida diseases 2012: non-neutropenic adult patients. Clin Microbiol Infect. 2012;18 Suppl 7:19-37.
15. Hope WW, Castagnola E, Groll AH, et al. ESCMID* guideline for the diagnosis and management of Candida diseases 2012: prevention and management of invasive infections in neonates and children caused by Candida spp. Clin Microbiol Infect. 2012;18 Suppl 7:38-52.
16. Ullmann AJ, Akova M, Herbrecht R, et al. ESCMID* guideline for the diagnosis and management of Candida diseases 2012: adults with haematological malignancies and after haematopoietic stem cell transplantation (HCT). Clin Microbiol Infect. 2012;18 Suppl 7:53-67.
17. Colombo AL, Guimaraes T, Camargo LF, et al. Brazilian guidelines for the management of candidiasis - a joint meeting report of three medical societies: Sociedade Brasileira de Infectologia, Sociedade Paulista de Infectologia and Sociedade Brasileira de Medicina Tropical. Braz J Infect Dis. 2013;17:283-312.
18. Kohno S, Tamura K, Niki Y, et al. Executive Summary of Japanese Domestic guidelines for management of deep-seated mycosis 2014. Med Mycol J. 2016;57:E117-E163.
19. Kojic EM, Darouiche RO. Candida infections of medical devices. Clin Microbiology Rev. 2004;17:255-267.
From the University of Miami, Department of Pediatrics and Department of Medicine, Miami, FL.
Abstract
- Objective: Pediatric fungemia is associated with a low risk of fungal endocarditis and renal infections. The majority of current guidelines do not recommend routine abdominal imaging/echocardiograms in the evaluation of fungemia, but such imaging has been routinely ordered for patients on the pediatric gastroenterology service at our institution. Our goals were to assess the financial impact of this deviation from current clinical guidelines and redefine the standard work to reduce overutilization of abdominal ultrasounds and echocardiograms. Specifically, our goal was to reduce imaging by 50% by 18 months.
- Methods: Root cause analysis showed a lack of familiarity with current evidence. Using this data, countermeasures were implemented, including practitioner education of guidelines and creation of a readily accessible clinical pathway and an electronic order set for pediatric fungemia management. Balancing measures were missed episodes of fungal endocarditis and renal infection.
- Results: During the period January 1, 2016 to November 19, 2017, 18 of 21 episodes of fungemia in our pediatric institution occurred in patients admitted to the pediatric gastroenterology service. Abdominal imaging and echocardiograms were done 100% of the time, with no positive findings and an estimated cost of approximately $58,000. Post-intervention from November 20, 2017 to April 3, 2019, 7 of 13 episodes of fungemia occurred on this service. Frequency of abdominal imaging and echocardiograms decreased to 43% and 57%, respectively. No episodes of fungal endocarditis or renal infection were identified.
- Conclusion: Overutilization of abdominal imaging and echocardiograms in pediatric fungemia evaluation can be safely decreased.
Keywords: guidelines; cost; candidemia; endocarditis.
Practitioners may remain under the impression that routine abdominal ultrasounds (US) and echocardiograms (echo) are indicated in fungemia to evaluate for fungal endocarditis and renal infection, although these conditions are rare and limited to a subset of the population.1-10 Risk factors include prematurity, immunosuppression, prior bacterial endocarditis, abnormal cardiac valves, and previous urogenital surgeries.11
The 2016 Infectious Diseases Society of America (IDSA) guidelines do not recommend routine US or echo but rather provide scenarios in which Candida endocarditis should be suspected, and these include: persistently positive blood cultures, persistent fevers despite appropriate therapy, and clinical signs that may suggest endocarditis, such as a new heart murmur, heart failure, or embolic phenomena.11 IDSA recommends abdominal imaging in neonates with persistently positive blood cultures to evaluate the urogenital system, in addition to the liver and spleen. They also recommend abdominal imaging in symptomatic ascending Candida pyelonephritis beyond the neonatal period and in chronic disseminated candidiasis; the latter is uncommon and seen almost exclusively in patients recovering from neutropenia with a hematologic malignancy.11
We also reviewed guidelines on fungemia originating outside the United States. The 2010 Canadian clinical guidelines on invasive candidiasis do not explicitly recommend routine imaging, but rather state that various imaging studies, including US and echo among others, may be helpful.12 The German Speaking Mycological Society and the Paul-Ehrlich-Society for Chemotherapy published a joint recommendation against routine US and echo in uncomplicated candidemia in 2011.13
The European Society for Clinical Microbiology and Infectious Diseases is the only society that recommends routine echo. Their 2012 guidelines on candidiasis recommend transesophageal echo in adults14 and echocardiography in children,15 as well as abdominal imaging in the diagnosis of chronic disseminated candidiasis in adults with hematological malignancies/hematopoietic stem cell transplantation.16
The 2013 Brazilian guidelines explicitly recommend against routine abdominal imaging and echo because of the low frequency of visceral lesions in adults with candidemia and recommend reserving imaging for those with persistently positive blood cultures or with clinical signs/symptoms suggestive of endocarditis/abdominal infection or clinical deterioration.17 The 2014 Japanese guidelines recommend ruling out chronic disseminated candidiasis in these patients with symptoms during the neutrophil recovery phase, but do not mention routinely imaging other patients. They do not address the role of echocardiography.18
Although physicians in the United Sates typically follow IDSA guidelines, abdominal US and echo were ordered routinely for patients with fungemia on the pediatric gastroenterology service at our institution, leading to higher medical costs and waste of medical resources. Our goals were to assess the current standard work for fungemia evaluation on this service, assess the impact of its deviation from current clinical guidelines, and redefine the standard work by (1) presenting current evidence to practitioners taking care of patients on this service, (2) providing a clinical pathway that allowed for variations where appropriate, and (3) providing a plan for pediatric fungemia management. Our SMART (Specific, Measurable, Attainable, Relevant and Timely) goal was to reduce overutilization of abdominal US and echo in pediatric patients with fungemia on the pediatric gastroenterology service by 50%.
Methods
Study, Setting, and Participants
We executed this quality improvement project at a quaternary care pediatric hospital affiliated with a school of medicine. The project scope consisted of inpatient pediatric patients with fungemia on the pediatric gastroenterology service admitted to the wards or pediatric critical care unit at this institution, along with the practitioners caring for these patients. The project was part of an institutional quality improvement initiative program. The quality improvement team included quality improvement experts from the departments of medicine and pediatrics, a pediatric resident and student, and physicians from the divisions of pediatric infectious disease, pediatric critical care, and pediatric gastroenterology. This study qualified for Institutional Review Board (IRB) exemption based on the University’s IRB stipulations.
Current Condition
Root cause analysis was performed by creating a process map of the current standard work and a fishbone diagram (Figure 1). We incorporated feedback from voice of the customer in the root cause analysis. In this analysis, the voice of the customer came from the bedside floor nurses, ultrasound clerk and sonographer, echo technician, cardiology fellow, and microbiology medical technician. We got their feedback on our process map, its accuracy and ways to expand, their thoughts on the problem and why we have this problem, and any solutions they could offer to help improve the problem. Some of the key points obtained were: echos were not routinely done on the floors and were not considered urgent as they often did not change management; the sonographer and those from the cardiology department felt imaging was often overutilized because of misconceptions and lack of available hospital guidelines. Suggested solutions included provider education with reference to Duke’s criteria and establishing a clinical pathway approved by all concerned departments.
Prior to education, we surveyed current practices of practitioners on teams caring for these patients, which included physicians of all levels (attendings, fellows, residents) as well as nurse practitioners and medical students from the department of pediatrics and divisions of pediatric gastroenterology, pediatric infectious disease, and pediatric critical care medicine.
Countermeasures
Practitioner Education. In October 2017 practitioners were given a 20-minute presentation on the latest international guidelines on fungemia. Fifty-nine practitioners completed pre- and post-test surveys. Eight respondents were excluded due to incomplete surveys. We compared self-reported frequencies of ordering abdominal imaging and echo before the presentation with intention to order post education. Intention to change clinical practice after the presentation was also surveyed.
Clinical Pathway. Education alone may not result in sustainability, and thus we provided a readily accessible clinical pathway and an electronic order set for pediatric fungemia management. Inter-department buy-in was also necessary for success. It was important to get the input from the various teams (infectious disease, cardiology, gastroenterology, and critical care), which was done by incorporating members from those divisions in the project or getting their feedback through voice of the customer analysis.
We redefined standard work based on current evidence and created a clinical pathway during March 2018 that included variations when appropriate (Figure 2). We presented the clinical pathway to practitioners and distributed it via email. We also made it available to pediatric residents and fellows on their mobile institutional work resource application.
Electronic Order Set. We created an electronic order set for pediatric fungemia management and made it available in the electronic health record May 2018.
Measurement
Cases of fungemia were identified through the electronic health record pre-intervention (January 1, 2016 through November 19, 2017) and post-intervention (November 20, 2019 through April 3, 2019). An episode of fungemia was defined as an encounter with 1 or more positive blood culture(s) for Candida species or Cryptococcus species. We manually identified patients belonging to the pediatric gastroenterology service and reviewed these charts to determine the presenting complaint, organism isolated, transplant status, central lines status, risk factors, if abdominal imaging or echocardiography were done for the episode of fungemia, and their corresponding results. We calculated overall and per patient medical charges by using the average charges at our institution of US and echocardiography with a cardiology consult. These average charges were provided by patient financial services and the pediatric cardiology department, respectively. To address non-technical expenditures, we calculated the average time taken for transport to and from radiology and the echo suite for each identified patient. We identified missed fungal endocarditis and fungal balls as balancing measures.
Results
Survey
Among the 51 practitioners surveyed, 36% were performing routine echo and 22% self-reported performing routine abdominal imaging. After education, no respondents planned to routinely do echo or abdominal imaging. All but 1 respondent planned to change their practice for evaluation of fungemia patients based on the presentation (eFigure 1).
Baseline Data
Over the 23-month period from January 1, 2016 to November 19, 2017, there were 21 episodes of fungemia, 18 of which occurred in patients on the pediatric gastroenterology service (2 of the 18 were transplant recipients). For the 18 episodes on this service, abdominal imaging and echo were done 100% of the time, with 0 positive findings (eFigure 2).
Of those 18 episodes, the average age was 4.6 years, with two-thirds of the population being male. There were 3 patients with multiple episodes that accounted for 8 of the episodes (3, 3, and 2 episodes each). Fever was the most common presenting complaint. The most common organism was Candida parapsilosis (6 of the 18 episodes). All episodes but one involved a central line, and all central lines were removed when present except for one case. Of the risk factors, 3 episodes occurred in neutropenic patients, and for 1 episode the patient had a questionable history of fungal endocarditis (and was on fungal prophylaxis). There were no patients with recent cardiac/urogenital surgery or prior fungal balls. No episodes had clinical symptoms suggestive of fungal endocarditis or fungal balls.
Post-Intervention Data
Over the subsequent 17-month period (November 11, 2017 to April 3, 2019), there were 13 episodes of candidemia. There were no episodes of Cryptococcus fungemia. Seven episodes occurred in patients on the pediatric gastroenterology service (2 of the 7 occurred in transplant recipients). Abdominal imaging was done in 3 of these episodes (43%), and in 2 of these 3 episodes, imaging was done at an outside institution prior to arrival, with no positive results (eFigure 2).
Echocardiography was done 57% of the time (n = 4), with echo being done at an outside institution prior to arrival half of the time (n = 2), with no endocarditis identified. The cases of abdominal imaging and echo done at outside institutions prior to arrival were not impacted by the countermeasures. Excluding those 2 patients who had both abdominal imaging and echocardiography done prior to arrival, the overall rate of imaging (both abdominal imaging and echo) done after countermeasures were instituted was 30% (Figure 3).
Of those 7 episodes, the average age was 6.8 years (57% female). There were no patients with multiple episodes. The most common presenting complaint was fever. The most common organism was Candida albicans (3 of the 7 episodes). All episodes involved a central line, which was removed in all cases except for one. Of the risk factors, 2 episodes were in neutropenic patients, and 1 episode had a history of bacterial endocarditis (not related to fungemia). No episodes occurred in patients with prior fungal renal infection, urogenital malformations, or recent cardiac/urogenital surgery. No episodes had clinical symptoms suggestive of fungal endocarditis or renal infection. No episodes of fungal endocarditis or renal infection were identified.
On average, a patient at our institution undergoing abdominal US and echo with a cardiology consult results in medical waste of approximately $3200 per patient. This cost does not take into account other miscellaneous charges possibly incurred, such as the radiologist interpreting the findings and transportation. Baseline data calculations show that patients waste on average 55 minutes in physical transport, and this does not take into account wait times.
Discussion
Candidemia contributes to 10% of central-line associated blood stream infections (CLABSI).19 Increased usage of indwelling central catheters for administration of parenteral nutrition will inevitably result in practitioners encountering cases of candidemia when caring for this population. As seen from our results, the majority of episodes of candidemia at our institution occurred on the pediatric gastroenterology service, and thus redefining standard work on this service will be impactful.
Candida parapsilosis and Candida albicans were the most common causative agents before and after intervention, respectively, but overall the most common organism was Candida albicans, which is in keeping with that of CLABSI in the literature.19 Growth of Candida parapsilosis has been particularly linked to CLABSI.19 The third most common organism in our study was Candida glabrata, which is the second most common cause of candidemia in CLABSI.19
The cases of positive abdominal imaging in fungemia in the literature are limited to the neonatal population1-4 and chronic disseminated candidiasis in patients with hematologic malignancies/neutropenia/immunosuppression.5,6 In fungal endocarditis, the reported cases were generally in neonates,1,3,7 critically ill patients,8 patients with hematologic malignancies/neutropenia/immunosuppression,6,9 or those with a cardiac history.9,10 This population differs from the patient population on the pediatric gastroenterology service. Patients on this service may not need US or echo. Performing abdominal US and echo in fungemia patients in whom such imaging is not indicated may result in medical waste of approximately $3200 per patient. There is also a waste of medical resources and time.
We found almost all practitioners are willing to change clinical practice once provided with current guidelines. Face-to-face oral presentations allowed for questions and interaction, making this form of information dissemination better than e-mails or handouts.
Though the numbers were small over the short study period, we were able to decrease overutilization of abdominal imaging and echo after implementing countermeasures. Frequency decreased from 100% to 43% and 57% for abdominal imaging and echo, respectively. Imaging that was done after the countermeasures were implemented was mainly attributed to imaging patients underwent prior to presenting to our institution. This reinforces the need for education at other institutions as well. Of the balancing measures assessed, there were no missed cases of fungal balls or fungal endocarditis. Additionally,
The findings from this quality improvement project underscore current recommendations that, despite common misconceptions, routine abdominal US and echo are not indicated in all cases of fungemia. Case-by-case assessment based on the clinical scenario remains key to management of fungemia to avoid unnecessary medical interventions.
Corresponding author: Donna Cheung, MBBS, 200 Hawkins Drive, BT 1120-G, Iowa City, IA 52242; [email protected].
Financial support: None.
From the University of Miami, Department of Pediatrics and Department of Medicine, Miami, FL.
Abstract
- Objective: Pediatric fungemia is associated with a low risk of fungal endocarditis and renal infections. The majority of current guidelines do not recommend routine abdominal imaging/echocardiograms in the evaluation of fungemia, but such imaging has been routinely ordered for patients on the pediatric gastroenterology service at our institution. Our goals were to assess the financial impact of this deviation from current clinical guidelines and redefine the standard work to reduce overutilization of abdominal ultrasounds and echocardiograms. Specifically, our goal was to reduce imaging by 50% by 18 months.
- Methods: Root cause analysis showed a lack of familiarity with current evidence. Using this data, countermeasures were implemented, including practitioner education of guidelines and creation of a readily accessible clinical pathway and an electronic order set for pediatric fungemia management. Balancing measures were missed episodes of fungal endocarditis and renal infection.
- Results: During the period January 1, 2016 to November 19, 2017, 18 of 21 episodes of fungemia in our pediatric institution occurred in patients admitted to the pediatric gastroenterology service. Abdominal imaging and echocardiograms were done 100% of the time, with no positive findings and an estimated cost of approximately $58,000. Post-intervention from November 20, 2017 to April 3, 2019, 7 of 13 episodes of fungemia occurred on this service. Frequency of abdominal imaging and echocardiograms decreased to 43% and 57%, respectively. No episodes of fungal endocarditis or renal infection were identified.
- Conclusion: Overutilization of abdominal imaging and echocardiograms in pediatric fungemia evaluation can be safely decreased.
Keywords: guidelines; cost; candidemia; endocarditis.
Practitioners may remain under the impression that routine abdominal ultrasounds (US) and echocardiograms (echo) are indicated in fungemia to evaluate for fungal endocarditis and renal infection, although these conditions are rare and limited to a subset of the population.1-10 Risk factors include prematurity, immunosuppression, prior bacterial endocarditis, abnormal cardiac valves, and previous urogenital surgeries.11
The 2016 Infectious Diseases Society of America (IDSA) guidelines do not recommend routine US or echo but rather provide scenarios in which Candida endocarditis should be suspected, and these include: persistently positive blood cultures, persistent fevers despite appropriate therapy, and clinical signs that may suggest endocarditis, such as a new heart murmur, heart failure, or embolic phenomena.11 IDSA recommends abdominal imaging in neonates with persistently positive blood cultures to evaluate the urogenital system, in addition to the liver and spleen. They also recommend abdominal imaging in symptomatic ascending Candida pyelonephritis beyond the neonatal period and in chronic disseminated candidiasis; the latter is uncommon and seen almost exclusively in patients recovering from neutropenia with a hematologic malignancy.11
We also reviewed guidelines on fungemia originating outside the United States. The 2010 Canadian clinical guidelines on invasive candidiasis do not explicitly recommend routine imaging, but rather state that various imaging studies, including US and echo among others, may be helpful.12 The German Speaking Mycological Society and the Paul-Ehrlich-Society for Chemotherapy published a joint recommendation against routine US and echo in uncomplicated candidemia in 2011.13
The European Society for Clinical Microbiology and Infectious Diseases is the only society that recommends routine echo. Their 2012 guidelines on candidiasis recommend transesophageal echo in adults14 and echocardiography in children,15 as well as abdominal imaging in the diagnosis of chronic disseminated candidiasis in adults with hematological malignancies/hematopoietic stem cell transplantation.16
The 2013 Brazilian guidelines explicitly recommend against routine abdominal imaging and echo because of the low frequency of visceral lesions in adults with candidemia and recommend reserving imaging for those with persistently positive blood cultures or with clinical signs/symptoms suggestive of endocarditis/abdominal infection or clinical deterioration.17 The 2014 Japanese guidelines recommend ruling out chronic disseminated candidiasis in these patients with symptoms during the neutrophil recovery phase, but do not mention routinely imaging other patients. They do not address the role of echocardiography.18
Although physicians in the United Sates typically follow IDSA guidelines, abdominal US and echo were ordered routinely for patients with fungemia on the pediatric gastroenterology service at our institution, leading to higher medical costs and waste of medical resources. Our goals were to assess the current standard work for fungemia evaluation on this service, assess the impact of its deviation from current clinical guidelines, and redefine the standard work by (1) presenting current evidence to practitioners taking care of patients on this service, (2) providing a clinical pathway that allowed for variations where appropriate, and (3) providing a plan for pediatric fungemia management. Our SMART (Specific, Measurable, Attainable, Relevant and Timely) goal was to reduce overutilization of abdominal US and echo in pediatric patients with fungemia on the pediatric gastroenterology service by 50%.
Methods
Study, Setting, and Participants
We executed this quality improvement project at a quaternary care pediatric hospital affiliated with a school of medicine. The project scope consisted of inpatient pediatric patients with fungemia on the pediatric gastroenterology service admitted to the wards or pediatric critical care unit at this institution, along with the practitioners caring for these patients. The project was part of an institutional quality improvement initiative program. The quality improvement team included quality improvement experts from the departments of medicine and pediatrics, a pediatric resident and student, and physicians from the divisions of pediatric infectious disease, pediatric critical care, and pediatric gastroenterology. This study qualified for Institutional Review Board (IRB) exemption based on the University’s IRB stipulations.
Current Condition
Root cause analysis was performed by creating a process map of the current standard work and a fishbone diagram (Figure 1). We incorporated feedback from voice of the customer in the root cause analysis. In this analysis, the voice of the customer came from the bedside floor nurses, ultrasound clerk and sonographer, echo technician, cardiology fellow, and microbiology medical technician. We got their feedback on our process map, its accuracy and ways to expand, their thoughts on the problem and why we have this problem, and any solutions they could offer to help improve the problem. Some of the key points obtained were: echos were not routinely done on the floors and were not considered urgent as they often did not change management; the sonographer and those from the cardiology department felt imaging was often overutilized because of misconceptions and lack of available hospital guidelines. Suggested solutions included provider education with reference to Duke’s criteria and establishing a clinical pathway approved by all concerned departments.
Prior to education, we surveyed current practices of practitioners on teams caring for these patients, which included physicians of all levels (attendings, fellows, residents) as well as nurse practitioners and medical students from the department of pediatrics and divisions of pediatric gastroenterology, pediatric infectious disease, and pediatric critical care medicine.
Countermeasures
Practitioner Education. In October 2017 practitioners were given a 20-minute presentation on the latest international guidelines on fungemia. Fifty-nine practitioners completed pre- and post-test surveys. Eight respondents were excluded due to incomplete surveys. We compared self-reported frequencies of ordering abdominal imaging and echo before the presentation with intention to order post education. Intention to change clinical practice after the presentation was also surveyed.
Clinical Pathway. Education alone may not result in sustainability, and thus we provided a readily accessible clinical pathway and an electronic order set for pediatric fungemia management. Inter-department buy-in was also necessary for success. It was important to get the input from the various teams (infectious disease, cardiology, gastroenterology, and critical care), which was done by incorporating members from those divisions in the project or getting their feedback through voice of the customer analysis.
We redefined standard work based on current evidence and created a clinical pathway during March 2018 that included variations when appropriate (Figure 2). We presented the clinical pathway to practitioners and distributed it via email. We also made it available to pediatric residents and fellows on their mobile institutional work resource application.
Electronic Order Set. We created an electronic order set for pediatric fungemia management and made it available in the electronic health record May 2018.
Measurement
Cases of fungemia were identified through the electronic health record pre-intervention (January 1, 2016 through November 19, 2017) and post-intervention (November 20, 2019 through April 3, 2019). An episode of fungemia was defined as an encounter with 1 or more positive blood culture(s) for Candida species or Cryptococcus species. We manually identified patients belonging to the pediatric gastroenterology service and reviewed these charts to determine the presenting complaint, organism isolated, transplant status, central lines status, risk factors, if abdominal imaging or echocardiography were done for the episode of fungemia, and their corresponding results. We calculated overall and per patient medical charges by using the average charges at our institution of US and echocardiography with a cardiology consult. These average charges were provided by patient financial services and the pediatric cardiology department, respectively. To address non-technical expenditures, we calculated the average time taken for transport to and from radiology and the echo suite for each identified patient. We identified missed fungal endocarditis and fungal balls as balancing measures.
Results
Survey
Among the 51 practitioners surveyed, 36% were performing routine echo and 22% self-reported performing routine abdominal imaging. After education, no respondents planned to routinely do echo or abdominal imaging. All but 1 respondent planned to change their practice for evaluation of fungemia patients based on the presentation (eFigure 1).
Baseline Data
Over the 23-month period from January 1, 2016 to November 19, 2017, there were 21 episodes of fungemia, 18 of which occurred in patients on the pediatric gastroenterology service (2 of the 18 were transplant recipients). For the 18 episodes on this service, abdominal imaging and echo were done 100% of the time, with 0 positive findings (eFigure 2).
Of those 18 episodes, the average age was 4.6 years, with two-thirds of the population being male. There were 3 patients with multiple episodes that accounted for 8 of the episodes (3, 3, and 2 episodes each). Fever was the most common presenting complaint. The most common organism was Candida parapsilosis (6 of the 18 episodes). All episodes but one involved a central line, and all central lines were removed when present except for one case. Of the risk factors, 3 episodes occurred in neutropenic patients, and for 1 episode the patient had a questionable history of fungal endocarditis (and was on fungal prophylaxis). There were no patients with recent cardiac/urogenital surgery or prior fungal balls. No episodes had clinical symptoms suggestive of fungal endocarditis or fungal balls.
Post-Intervention Data
Over the subsequent 17-month period (November 11, 2017 to April 3, 2019), there were 13 episodes of candidemia. There were no episodes of Cryptococcus fungemia. Seven episodes occurred in patients on the pediatric gastroenterology service (2 of the 7 occurred in transplant recipients). Abdominal imaging was done in 3 of these episodes (43%), and in 2 of these 3 episodes, imaging was done at an outside institution prior to arrival, with no positive results (eFigure 2).
Echocardiography was done 57% of the time (n = 4), with echo being done at an outside institution prior to arrival half of the time (n = 2), with no endocarditis identified. The cases of abdominal imaging and echo done at outside institutions prior to arrival were not impacted by the countermeasures. Excluding those 2 patients who had both abdominal imaging and echocardiography done prior to arrival, the overall rate of imaging (both abdominal imaging and echo) done after countermeasures were instituted was 30% (Figure 3).
Of those 7 episodes, the average age was 6.8 years (57% female). There were no patients with multiple episodes. The most common presenting complaint was fever. The most common organism was Candida albicans (3 of the 7 episodes). All episodes involved a central line, which was removed in all cases except for one. Of the risk factors, 2 episodes were in neutropenic patients, and 1 episode had a history of bacterial endocarditis (not related to fungemia). No episodes occurred in patients with prior fungal renal infection, urogenital malformations, or recent cardiac/urogenital surgery. No episodes had clinical symptoms suggestive of fungal endocarditis or renal infection. No episodes of fungal endocarditis or renal infection were identified.
On average, a patient at our institution undergoing abdominal US and echo with a cardiology consult results in medical waste of approximately $3200 per patient. This cost does not take into account other miscellaneous charges possibly incurred, such as the radiologist interpreting the findings and transportation. Baseline data calculations show that patients waste on average 55 minutes in physical transport, and this does not take into account wait times.
Discussion
Candidemia contributes to 10% of central-line associated blood stream infections (CLABSI).19 Increased usage of indwelling central catheters for administration of parenteral nutrition will inevitably result in practitioners encountering cases of candidemia when caring for this population. As seen from our results, the majority of episodes of candidemia at our institution occurred on the pediatric gastroenterology service, and thus redefining standard work on this service will be impactful.
Candida parapsilosis and Candida albicans were the most common causative agents before and after intervention, respectively, but overall the most common organism was Candida albicans, which is in keeping with that of CLABSI in the literature.19 Growth of Candida parapsilosis has been particularly linked to CLABSI.19 The third most common organism in our study was Candida glabrata, which is the second most common cause of candidemia in CLABSI.19
The cases of positive abdominal imaging in fungemia in the literature are limited to the neonatal population1-4 and chronic disseminated candidiasis in patients with hematologic malignancies/neutropenia/immunosuppression.5,6 In fungal endocarditis, the reported cases were generally in neonates,1,3,7 critically ill patients,8 patients with hematologic malignancies/neutropenia/immunosuppression,6,9 or those with a cardiac history.9,10 This population differs from the patient population on the pediatric gastroenterology service. Patients on this service may not need US or echo. Performing abdominal US and echo in fungemia patients in whom such imaging is not indicated may result in medical waste of approximately $3200 per patient. There is also a waste of medical resources and time.
We found almost all practitioners are willing to change clinical practice once provided with current guidelines. Face-to-face oral presentations allowed for questions and interaction, making this form of information dissemination better than e-mails or handouts.
Though the numbers were small over the short study period, we were able to decrease overutilization of abdominal imaging and echo after implementing countermeasures. Frequency decreased from 100% to 43% and 57% for abdominal imaging and echo, respectively. Imaging that was done after the countermeasures were implemented was mainly attributed to imaging patients underwent prior to presenting to our institution. This reinforces the need for education at other institutions as well. Of the balancing measures assessed, there were no missed cases of fungal balls or fungal endocarditis. Additionally,
The findings from this quality improvement project underscore current recommendations that, despite common misconceptions, routine abdominal US and echo are not indicated in all cases of fungemia. Case-by-case assessment based on the clinical scenario remains key to management of fungemia to avoid unnecessary medical interventions.
Corresponding author: Donna Cheung, MBBS, 200 Hawkins Drive, BT 1120-G, Iowa City, IA 52242; [email protected].
Financial support: None.
1. Benjamin DK Jr, Poole C, Steinbach WJ, et al. Neonatal candidemia and end-organ damage: a critical appraisal of the literature using meta-analytic techniques. Pediatrics. 2003;112:634-640.
2. Wynn JL, Tan S, Gantz MG, et al. Outcomes following candiduria in extremely low birth weight infants. Clin Infect Dis. 2012;54:331-339.
3. Noyola DE, Fernandez M, Moylett EH, et al. Ophthalmologic, visceral, and cardiac involvement in neonates with candidemia. Clin Infect Dis. 2001;32:1018-1023.
4. Phillips JR, Karlowicz MG Prevalence of Candida species in hospital-acquired urinary tract infections in a neonatal intensive care unit. Pediatr Infect Dis J. 1997;16:190-194.
5. Pagano L, Mele L, Fianchi L, et al. Chronic disseminated candidiasis in patients with hematologic malignancies. Clinical features and outcome of 29 episodes. Haematologica. 2002;87:535-541.
6. Zaoutis TE, Greves HM, Lautenbach E, et al. Risk factors for disseminated candidiasis in children with candidemia. Pediatr Infect Dis J. 2004;23:635-641.
7. Levy I, Shalit I, Birk E, et al. Candida endocarditis in neonates: report of five cases and review of the literature. Mycoses. 2006;49:43-48.
8. Aspesberro F, Beghetti M, Oberhansli I, et al. Fungal endocarditis in critically ill children. Eur J Pediatr. 1999;158:275-280.
9. Fernandez-Cruz A, Cruz Menarguez M, Munoz P, et al. The search for endocarditis in patients with candidemia: a systematic recommendation for echocardiography? A prospective cohort. Eur J Clin Microbiol Infect Dis. 2015;34:1543-1549.
10. Hernandez-Torres A, Garcia-Vazquez E, Laso-Ortiz A, et al. [Candida sp endocarditis. Experience in a third-level hospital and review of the literature]. Rev Esp Quimioter. 2013;26:51-55.
11. Pappas PG, Kauffman CA, Andes DR, et al. Clinical practice guideline for the management of candidiasis: 2016 Update by the Infectious Diseases Society of America. Clin Infect Dis. 2016;62:e1-50.
12. Bow EJ, Evans G, Fuller J, et al. Canadian clinical practice guidelines for invasive candidiasis in adults. Can J Infect Dis Med Microbiol. 2010;21:e122-50.
13. Ruhnke M, Rickerts V, Cornely OA, et al. Diagnosis and therapy of Candida infections: joint recommendations of the German Speaking Mycological Society and the Paul-Ehrlich-Society for Chemotherapy. Mycoses. 2011;54:279-310.
14. Cornely OA, Bassetti M, Calandra T, et al. ESCMID* guideline for the diagnosis and management of Candida diseases 2012: non-neutropenic adult patients. Clin Microbiol Infect. 2012;18 Suppl 7:19-37.
15. Hope WW, Castagnola E, Groll AH, et al. ESCMID* guideline for the diagnosis and management of Candida diseases 2012: prevention and management of invasive infections in neonates and children caused by Candida spp. Clin Microbiol Infect. 2012;18 Suppl 7:38-52.
16. Ullmann AJ, Akova M, Herbrecht R, et al. ESCMID* guideline for the diagnosis and management of Candida diseases 2012: adults with haematological malignancies and after haematopoietic stem cell transplantation (HCT). Clin Microbiol Infect. 2012;18 Suppl 7:53-67.
17. Colombo AL, Guimaraes T, Camargo LF, et al. Brazilian guidelines for the management of candidiasis - a joint meeting report of three medical societies: Sociedade Brasileira de Infectologia, Sociedade Paulista de Infectologia and Sociedade Brasileira de Medicina Tropical. Braz J Infect Dis. 2013;17:283-312.
18. Kohno S, Tamura K, Niki Y, et al. Executive Summary of Japanese Domestic guidelines for management of deep-seated mycosis 2014. Med Mycol J. 2016;57:E117-E163.
19. Kojic EM, Darouiche RO. Candida infections of medical devices. Clin Microbiology Rev. 2004;17:255-267.
1. Benjamin DK Jr, Poole C, Steinbach WJ, et al. Neonatal candidemia and end-organ damage: a critical appraisal of the literature using meta-analytic techniques. Pediatrics. 2003;112:634-640.
2. Wynn JL, Tan S, Gantz MG, et al. Outcomes following candiduria in extremely low birth weight infants. Clin Infect Dis. 2012;54:331-339.
3. Noyola DE, Fernandez M, Moylett EH, et al. Ophthalmologic, visceral, and cardiac involvement in neonates with candidemia. Clin Infect Dis. 2001;32:1018-1023.
4. Phillips JR, Karlowicz MG Prevalence of Candida species in hospital-acquired urinary tract infections in a neonatal intensive care unit. Pediatr Infect Dis J. 1997;16:190-194.
5. Pagano L, Mele L, Fianchi L, et al. Chronic disseminated candidiasis in patients with hematologic malignancies. Clinical features and outcome of 29 episodes. Haematologica. 2002;87:535-541.
6. Zaoutis TE, Greves HM, Lautenbach E, et al. Risk factors for disseminated candidiasis in children with candidemia. Pediatr Infect Dis J. 2004;23:635-641.
7. Levy I, Shalit I, Birk E, et al. Candida endocarditis in neonates: report of five cases and review of the literature. Mycoses. 2006;49:43-48.
8. Aspesberro F, Beghetti M, Oberhansli I, et al. Fungal endocarditis in critically ill children. Eur J Pediatr. 1999;158:275-280.
9. Fernandez-Cruz A, Cruz Menarguez M, Munoz P, et al. The search for endocarditis in patients with candidemia: a systematic recommendation for echocardiography? A prospective cohort. Eur J Clin Microbiol Infect Dis. 2015;34:1543-1549.
10. Hernandez-Torres A, Garcia-Vazquez E, Laso-Ortiz A, et al. [Candida sp endocarditis. Experience in a third-level hospital and review of the literature]. Rev Esp Quimioter. 2013;26:51-55.
11. Pappas PG, Kauffman CA, Andes DR, et al. Clinical practice guideline for the management of candidiasis: 2016 Update by the Infectious Diseases Society of America. Clin Infect Dis. 2016;62:e1-50.
12. Bow EJ, Evans G, Fuller J, et al. Canadian clinical practice guidelines for invasive candidiasis in adults. Can J Infect Dis Med Microbiol. 2010;21:e122-50.
13. Ruhnke M, Rickerts V, Cornely OA, et al. Diagnosis and therapy of Candida infections: joint recommendations of the German Speaking Mycological Society and the Paul-Ehrlich-Society for Chemotherapy. Mycoses. 2011;54:279-310.
14. Cornely OA, Bassetti M, Calandra T, et al. ESCMID* guideline for the diagnosis and management of Candida diseases 2012: non-neutropenic adult patients. Clin Microbiol Infect. 2012;18 Suppl 7:19-37.
15. Hope WW, Castagnola E, Groll AH, et al. ESCMID* guideline for the diagnosis and management of Candida diseases 2012: prevention and management of invasive infections in neonates and children caused by Candida spp. Clin Microbiol Infect. 2012;18 Suppl 7:38-52.
16. Ullmann AJ, Akova M, Herbrecht R, et al. ESCMID* guideline for the diagnosis and management of Candida diseases 2012: adults with haematological malignancies and after haematopoietic stem cell transplantation (HCT). Clin Microbiol Infect. 2012;18 Suppl 7:53-67.
17. Colombo AL, Guimaraes T, Camargo LF, et al. Brazilian guidelines for the management of candidiasis - a joint meeting report of three medical societies: Sociedade Brasileira de Infectologia, Sociedade Paulista de Infectologia and Sociedade Brasileira de Medicina Tropical. Braz J Infect Dis. 2013;17:283-312.
18. Kohno S, Tamura K, Niki Y, et al. Executive Summary of Japanese Domestic guidelines for management of deep-seated mycosis 2014. Med Mycol J. 2016;57:E117-E163.
19. Kojic EM, Darouiche RO. Candida infections of medical devices. Clin Microbiology Rev. 2004;17:255-267.
Atraumatic splenic rupture in acute myeloid leukemia
A 50-year-old man with acute myeloid leukemia (AML) with a complex karyotype was admitted to the hospital with several days of dull, left-sided abdominal pain. His most recent bone marrow biopsy showed 30% blasts, and immunophenotyping was suggestive of persistent AML (CD13+, CD34+, CD117+, CD33+, CD7+, MPO–). He was on treatment with venetoclax and cytarabine after induction therapy had failed.
On admission, his heart rate was 101 beats per minute and his blood pressure was 122/85 mm Hg. Abdominal examination revealed mild distention, hepatomegaly, and previously known massive splenomegaly, with the splenic tip extending to the umbilicus, and mild tenderness.
Results of laboratory testing revealed persistent pancytopenia:
- Hemoglobin level 6.8 g/dL (reference range 13.0–17.0)
- Total white blood cell count 0.8 × 109/L (4.5–11.0)
- Platelet count 8 × 109/L (150–400).
 and transverse (right) views on initial computed tomography of the abdomen without contrast showed massive splenomegaly (white arrow).")
The next day, he developed severe, acute-onset left-sided abdominal pain. A check of vital signs showed worsening sinus tachycardia at 132 beats per minute and a drop in blood pressure to 90/56 mm Hg. He had worsening diffuse abdominal tenderness with sluggish bowel sounds. His hemoglobin concentration was 6.4 g/dL and platelet count 12 × 109/L.
 and transverse (right) views showed irregular splenic margins (red arrows), intraparenchymal hemorrhages (black arrows), and hemoperitoneum (white arrows).")
He received supportive transfusions of blood products. Surgical exploration was deemed risky, given his overall condition and severe thrombocytopenia. Splenic angiography showed no evidence of pseudoaneurysm or focal contrast extravasation. He underwent empiric embolization of the midsplenic artery, after which his hemodynamic status stabilized. He died 4 weeks later of acute respiratory failure from pneumonia.
SPLENIC RUPTURE IN AML
Atraumatic splenic rupture is rare but potentially life-threatening, especially if the diagnosis is delayed. Conditions that can cause splenomegaly and predispose to rupture include infection (infectious mononucleosis, malaria), malignant hematologic disorders (leukemia, lymphoma), other neoplasms, and amyloidosis.1
The literature includes a few reports of splenic rupture in patients with AML.2–4 The proposed mechanisms include bleeding from infarction sites or tumor foci, dysregulated hemostasis, and leukostasis.
The classic presentation of splenic rupture is acute-onset left-sided abdominal pain associated with hypotension and decreasing hemoglobin levels. CT of the abdomen is confirmatory, and resuscitation with crystalloids and blood products is a vital initial step in management. Choice of treatment depends on the patient’s surgical risk and hemodynamic status; options include conservative medical management, splenic artery embolization, and exploratory laparotomy.
In patients with AML and splenomegaly presenting with acute abdominal pain, clinicians need to be aware of this potential hematologic emergency.
- Renzulli P, Hostettler A, Schoepfer AM, Gloor B, Candinas D. Systematic review of atraumatic splenic rupture. Br J Surg 2009; 96(10):1114–1121. doi:10.1002/bjs.6737
- Gardner JA, Bao L, Ornstein DL. Spontaneous splenic rupture in acute myeloid leukemia with mixed-lineage leukemia gene rearrangement. Med Rep Case Stud 2016; 1:119. doi:10.4172/2572-5130.1000119
- Zeidan AM, Mitchell M, Khatri R, et al. Spontaneous splenic rupture during induction chemotherapy for acute myeloid leukemia. Leuk Lymphoma 2014; 55(1):209–212. doi:10.3109/10428194.2013.796060
- Fahmi Y, Elabbasi T, Khaiz D, et al. Splenic spontaneous rupture associated with acute myeloïd leukemia: report of a case and literature review. Surgery Curr Res 2014; 4:170. doi:10.4172/2161-1076.1000170
A 50-year-old man with acute myeloid leukemia (AML) with a complex karyotype was admitted to the hospital with several days of dull, left-sided abdominal pain. His most recent bone marrow biopsy showed 30% blasts, and immunophenotyping was suggestive of persistent AML (CD13+, CD34+, CD117+, CD33+, CD7+, MPO–). He was on treatment with venetoclax and cytarabine after induction therapy had failed.
On admission, his heart rate was 101 beats per minute and his blood pressure was 122/85 mm Hg. Abdominal examination revealed mild distention, hepatomegaly, and previously known massive splenomegaly, with the splenic tip extending to the umbilicus, and mild tenderness.
Results of laboratory testing revealed persistent pancytopenia:
- Hemoglobin level 6.8 g/dL (reference range 13.0–17.0)
- Total white blood cell count 0.8 × 109/L (4.5–11.0)
- Platelet count 8 × 109/L (150–400).
The next day, he developed severe, acute-onset left-sided abdominal pain. A check of vital signs showed worsening sinus tachycardia at 132 beats per minute and a drop in blood pressure to 90/56 mm Hg. He had worsening diffuse abdominal tenderness with sluggish bowel sounds. His hemoglobin concentration was 6.4 g/dL and platelet count 12 × 109/L.
He received supportive transfusions of blood products. Surgical exploration was deemed risky, given his overall condition and severe thrombocytopenia. Splenic angiography showed no evidence of pseudoaneurysm or focal contrast extravasation. He underwent empiric embolization of the midsplenic artery, after which his hemodynamic status stabilized. He died 4 weeks later of acute respiratory failure from pneumonia.
SPLENIC RUPTURE IN AML
Atraumatic splenic rupture is rare but potentially life-threatening, especially if the diagnosis is delayed. Conditions that can cause splenomegaly and predispose to rupture include infection (infectious mononucleosis, malaria), malignant hematologic disorders (leukemia, lymphoma), other neoplasms, and amyloidosis.1
The literature includes a few reports of splenic rupture in patients with AML.2–4 The proposed mechanisms include bleeding from infarction sites or tumor foci, dysregulated hemostasis, and leukostasis.
The classic presentation of splenic rupture is acute-onset left-sided abdominal pain associated with hypotension and decreasing hemoglobin levels. CT of the abdomen is confirmatory, and resuscitation with crystalloids and blood products is a vital initial step in management. Choice of treatment depends on the patient’s surgical risk and hemodynamic status; options include conservative medical management, splenic artery embolization, and exploratory laparotomy.
In patients with AML and splenomegaly presenting with acute abdominal pain, clinicians need to be aware of this potential hematologic emergency.
A 50-year-old man with acute myeloid leukemia (AML) with a complex karyotype was admitted to the hospital with several days of dull, left-sided abdominal pain. His most recent bone marrow biopsy showed 30% blasts, and immunophenotyping was suggestive of persistent AML (CD13+, CD34+, CD117+, CD33+, CD7+, MPO–). He was on treatment with venetoclax and cytarabine after induction therapy had failed.
On admission, his heart rate was 101 beats per minute and his blood pressure was 122/85 mm Hg. Abdominal examination revealed mild distention, hepatomegaly, and previously known massive splenomegaly, with the splenic tip extending to the umbilicus, and mild tenderness.
Results of laboratory testing revealed persistent pancytopenia:
- Hemoglobin level 6.8 g/dL (reference range 13.0–17.0)
- Total white blood cell count 0.8 × 109/L (4.5–11.0)
- Platelet count 8 × 109/L (150–400).
The next day, he developed severe, acute-onset left-sided abdominal pain. A check of vital signs showed worsening sinus tachycardia at 132 beats per minute and a drop in blood pressure to 90/56 mm Hg. He had worsening diffuse abdominal tenderness with sluggish bowel sounds. His hemoglobin concentration was 6.4 g/dL and platelet count 12 × 109/L.
He received supportive transfusions of blood products. Surgical exploration was deemed risky, given his overall condition and severe thrombocytopenia. Splenic angiography showed no evidence of pseudoaneurysm or focal contrast extravasation. He underwent empiric embolization of the midsplenic artery, after which his hemodynamic status stabilized. He died 4 weeks later of acute respiratory failure from pneumonia.
SPLENIC RUPTURE IN AML
Atraumatic splenic rupture is rare but potentially life-threatening, especially if the diagnosis is delayed. Conditions that can cause splenomegaly and predispose to rupture include infection (infectious mononucleosis, malaria), malignant hematologic disorders (leukemia, lymphoma), other neoplasms, and amyloidosis.1
The literature includes a few reports of splenic rupture in patients with AML.2–4 The proposed mechanisms include bleeding from infarction sites or tumor foci, dysregulated hemostasis, and leukostasis.
The classic presentation of splenic rupture is acute-onset left-sided abdominal pain associated with hypotension and decreasing hemoglobin levels. CT of the abdomen is confirmatory, and resuscitation with crystalloids and blood products is a vital initial step in management. Choice of treatment depends on the patient’s surgical risk and hemodynamic status; options include conservative medical management, splenic artery embolization, and exploratory laparotomy.
In patients with AML and splenomegaly presenting with acute abdominal pain, clinicians need to be aware of this potential hematologic emergency.
- Renzulli P, Hostettler A, Schoepfer AM, Gloor B, Candinas D. Systematic review of atraumatic splenic rupture. Br J Surg 2009; 96(10):1114–1121. doi:10.1002/bjs.6737
- Gardner JA, Bao L, Ornstein DL. Spontaneous splenic rupture in acute myeloid leukemia with mixed-lineage leukemia gene rearrangement. Med Rep Case Stud 2016; 1:119. doi:10.4172/2572-5130.1000119
- Zeidan AM, Mitchell M, Khatri R, et al. Spontaneous splenic rupture during induction chemotherapy for acute myeloid leukemia. Leuk Lymphoma 2014; 55(1):209–212. doi:10.3109/10428194.2013.796060
- Fahmi Y, Elabbasi T, Khaiz D, et al. Splenic spontaneous rupture associated with acute myeloïd leukemia: report of a case and literature review. Surgery Curr Res 2014; 4:170. doi:10.4172/2161-1076.1000170
- Renzulli P, Hostettler A, Schoepfer AM, Gloor B, Candinas D. Systematic review of atraumatic splenic rupture. Br J Surg 2009; 96(10):1114–1121. doi:10.1002/bjs.6737
- Gardner JA, Bao L, Ornstein DL. Spontaneous splenic rupture in acute myeloid leukemia with mixed-lineage leukemia gene rearrangement. Med Rep Case Stud 2016; 1:119. doi:10.4172/2572-5130.1000119
- Zeidan AM, Mitchell M, Khatri R, et al. Spontaneous splenic rupture during induction chemotherapy for acute myeloid leukemia. Leuk Lymphoma 2014; 55(1):209–212. doi:10.3109/10428194.2013.796060
- Fahmi Y, Elabbasi T, Khaiz D, et al. Splenic spontaneous rupture associated with acute myeloïd leukemia: report of a case and literature review. Surgery Curr Res 2014; 4:170. doi:10.4172/2161-1076.1000170
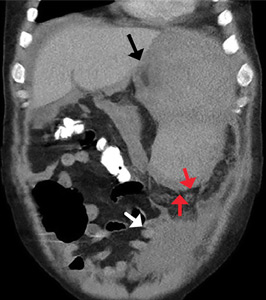
Severe hypercalcemia in a 54-year-old woman
A morbidly obese 54-year-old woman presented to the emergency department after experiencing generalized abdominal pain for 3 days. She rated the pain as 5 on a scale of 10 and described it as dull, cramping, waxing and waning, not radiating, and not relieved with changes of position—in fact, not alleviated by anything she had tried. Her pain was associated with nausea and 1 episode of vomiting. She also experienced constipation before the onset of pain.
She denied recent trauma, recent travel, diarrhea, fevers, weakness, shortness of breath, chest pain, other muscle pains, or recent changes in diet. She also denied having this pain in the past. She said she had unintentionally lost some weight but was not certain how much. She denied tobacco, alcohol, or illicit drug use. She had no history of surgery.
Her medical history included hypertension, anemia, and uterine fibroids. Her current medications included losartan, hydrochlorothiazide, and albuterol. She had no family history of significant disease.
INITIAL EVALUATION AND MANAGEMENT
On admission, her temperature was 97.8°F (36.6°C), heart rate 100 beats per minute, blood pressure 136/64 mm Hg, respiratory rate 18 breaths per minute, oxygen saturation 97% on room air, weight 130.6 kg, and body mass index 35 kg/m2.
She was alert and oriented to person, place, and time. She was in mild discomfort but no distress. Her lungs were clear to auscultation, with no wheezing or crackles. Heart rate and rhythm were regular, with no extra heart sounds or murmurs. Bowel sounds were normal in all 4 quadrants, with tenderness to palpation of the epigastric area, but with no guarding or rebound tenderness.
Laboratory test results
Notable results of blood testing at presentation were as follows:
- Hemoglobin 8.2 g/dL (reference range 12.3–15.3)
- Hematocrit 26% (41–50)
- Mean corpuscular volume 107 fL (80–100)
- Blood urea nitrogen 33 mg/dL (8–21); 6 months earlier it was 16
- Serum creatinine 3.6 mg/dL (0.58–0.96); 6 months earlier, it was 0.75
- Albumin 3.3 g/dL (3.5–5)
- Calcium 18.4 mg/dL (8.4–10.2); 6 months earlier, it was 9.6
- Corrected calcium 19 mg/dL.
Findings on imaging, electrocardiography
Chest radiography showed no acute cardiopulmonary abnormalities. Abdominal computed tomography without contrast showed no abnormalities within the pancreas and no evidence of inflammation or obstruction. Electrocardiography showed sinus tachycardia.
DIFFERENTIAL DIAGNOSIS
1. Which is the most likely cause of this patient’s symptoms?
- Primary hyperparathyroidism
- Malignancy
- Her drug therapy
- Familial hypercalcemic hypocalciuria

In total, her laboratory results were consistent with macrocytic anemia, severe hypercalcemia, and acute kidney injury, and she had generalized symptoms.
Primary hyperparathyroidism
A main cause of hypercalcemia is primary hyperparathyroidism, and this needs to be ruled out. Benign adenomas are the most common cause of primary hyperparathyroidism, and a risk factor for benign adenoma is exposure to therapeutic levels of radiation.3
In hyperparathyroidism, there is an increased secretion of parathyroid hormone (PTH), which has multiple effects including increased reabsorption of calcium from the urine, increased excretion of phosphate, and increased expression of 1,25-hydroxyvitamin D hydroxylase to activate vitamin D. PTH also stimulates osteoclasts to increase their expression of receptor activator of nuclear factor kappa B ligand (RANKL), which has a downstream effect on osteoclast precursors to cause bone reabsorption.3
Inherited primary hyperparathyroidism tends to present at a younger age, with multiple overactive parathyroid glands.3 Given our patient’s age, inherited primary hyparathyroidism is thus less likely.
Malignancy
The probability that malignancy is causing the hypercalcemia increases with calcium levels greater than 13 mg/dL. Epidemiologically, in hospitalized patients with hypercalcemia, the source tends to be malignancy.4 Typically, patients who develop hypercalcemia from malignancy have a worse prognosis.5
Solid tumors and leukemias can cause hypercalcemia. The mechanisms include humoral factors secreted by the malignancy, local osteolysis due to tumor invasion of bone, and excessive absorption of calcium due to excess vitamin D produced by malignancies.5 The cancers that most frequently cause an increase in calcium resorption are lung cancer, renal cancer, breast cancer, and multiple myeloma.1
Solid tumors with no bone metastasis and non-Hodgkin lymphoma that release PTH-related protein (PTHrP) cause humoral hypercalcemia in malignancy. The patient is typically in an advanced stage of disease. PTHrP increases serum calcium levels by decreasing the kidney’s ability to excrete calcium and by increasing bone turnover. It has no effect on intestinal absorption because of its inability to stimulate activated vitamin D3. Thus, the increase in systemic calcium comes directly from breakdown of bone and inability to excrete the excess.
PTHrP has a unique role in breast cancer: it is released locally in areas where cancer cells have metastasized to bone, but it does not cause a systemic effect. Bone resorption occurs in areas of metastasis and results from an increase in expression of RANKL and RANK in osteoclasts in response to the effects of PTHrP, leading to an increase in the production of osteoclastic cells.1
Tamoxifen, an endocrine therapy often used in breast cancer, also causes a release of bone-reabsorbing factors from tumor cells, which can partially contribute to hypercalcemia.5
Myeloma cells secrete RANKL, which stimulates osteoclastic activity, and they also release interleukin 6 (IL-6) and activating macrophage inflammatory protein alpha. Serum testing usually shows low or normal intact PTH, PTHrP, and 1,25-dihydroxyvitamin D.1
Patients with multiple myeloma have a worse prognosis if they have a high red blood cell distribution width, a condition shown to correlate with malnutrition, leading to deficiencies in vitamin B12 and to poor response to treatment.6 Up to 14% of patients with multiple myeloma have vitamin B12 deficiency.7
Our patient’s recent weight loss and severe hypercalcemia raise suspicion of malignancy. Further, her obesity makes proper routine breast examination difficult and thus increases the chance of undiagnosed breast cancer.8 Her decrease in renal function and her anemia complicated by hypercalcemia also raise suspicion of multiple myeloma.
Hypercalcemia due to drug therapy
Thiazide diuretics, lithium, teriparatide, and vitamin A in excessive amounts can raise the serum calcium concentration.5 Our patient was taking a thiazide for hypertension, but her extremely high calcium level places drug-induced hypercalcemia as the sole cause lower on the differential list.
Familial hypercalcemic hypocalciuria
Familial hypercalcemic hypocalciuria is a rare autosomal-dominant cause of hypercalcemia in which the ability of the body (and especially the kidneys) to sense levels of calcium is impaired, leading to a decrease in excretion of calcium in the urine.3 Very high calcium levels are rare in hypercalcemic hypocalciuria.3 In our patient with a corrected calcium concentration of nearly 19 mg/dL, familial hypercalcemic hypocalciuria is very unlikely to be the cause of the hypercalcemia.
WHAT ARE THE NEXT STEPS IN THE WORKUP?
As hypercalcemia has been confirmed, the intact PTH level should be checked to determine whether the patient’s condition is PTH-mediated. If the PTH level is in the upper range of normal or is minimally elevated, primary hyperparathyroidism is likely. Elevated PTH confirms primary hyperparathyroidism. A low-normal or low intact PTH confirms a non-PTH-mediated process, and once this is confirmed, PTHrP levels should be checked. An elevated PTHrP suggests humoral hypercalcemia of malignancy. Serum protein electrophoresis, urine protein electrophoresis, and a serum light chain assay should be performed to rule out multiple myeloma.
Vitamin D toxicity is associated with high concentrations of 1,25-dihydroxyvitamin D and 25-hydroxyvitamin D metabolites. These levels should be checked in this patient.
Other disorders that cause hypercalcemia are vitamin A toxicity and hyperthyroidism, so vitamin A and thyroid-stimulating hormone levels should also be checked.5
CASE CONTINUED
After further questioning, the patient said that she had had lower back pain about 1 to 2 weeks before coming to the emergency room; her primary care doctor had said the pain was likely from muscle strain. The pain had almost resolved but was still present.
The results of further laboratory testing were as follows:
- Serum PTH 11 pg/mL (15–65)
- PTHrP 3.4 pmol/L (< 2.0)
- Protein electrophoresis showed a monoclonal (M) spike of 0.2 g/dL (0)
- Activated vitamin D < 5 ng/mL (19.9–79.3)
- Vitamin A 7.2 mg/dL (33.1–100)
- Vitamin B12 194 pg/mL (239–931)
- Thyroid-stimulating hormone 1.21 mIU/ L (0.47–4.68
- Free thyroxine 1.27 ng/dL (0.78–2.19)
- Iron 103 µg/dL (37–170)
- Total iron-binding capacity 335 µg/dL (265–497)
- Transferrin 248 mg/dL (206–381)
- Ferritin 66 ng/mL (11.1–264)
- Urine protein (random) 100 mg/dL (0–20)
- Urine microalbumin (random) 5.9 mg/dL (0–1.6)
- Urine creatinine clearance 88.5 mL/min (88–128)
- Urine albumin-creatinine ratio 66.66 mg/g (< 30).
Imaging reports
A nuclear bone scan showed increased bone uptake in the hip and both shoulders, consistent with arthritis, and increased activity in 2 of the lower left ribs, associated with rib fractures secondary to lytic lesions. A skeletal survey at a later date showed multiple well-circumscribed “punched-out” lytic lesions in both forearms and both femurs.
2. What should be the next step in this patient’s management?
- Intravenous (IV) fluids
- Calcitonin
- Bisphosphonate treatment
- Denosumab
- Hemodialysis
Initial treatment of severe hypercalcemia includes the following:
Start IV isotonic fluids at a rate of 150 mL/h (if the patient is making urine) to maintain urine output at more than 100 mL/h. Closely monitor urine output.
Give calcitonin 4 IU/kg in combination with IV fluids to reduce calcium levels within the first 12 to 48 hours of treatment.
Give a bisphosphonate, eg, zoledronic acid 4 mg over 15 minutes, or pamidronate 60 to 90 mg over 2 hours. Zoledronic acid is preferred in malignancy-induced hypercalcemia because it is more potent. Doses should be adjusted in patients with renal failure.
Give denosumab if hypercalcemia is refractory to bisphosphonates, or when bisphosphonates cannot be used in renal failure.9
Hemodialysis is performed in patients who have significant neurologic symptoms irrespective of acute renal insufficiency.
Our patient was started on 0.9% sodium chloride at a rate of 150 mL/h for severe hypercalcemia. Zoledronic acid 4 mg IV was given once. These measures lowered her calcium level and lessened her acute kidney injury.
ADDITIONAL FINDINGS
Urine testing was positive for Bence Jones protein. Immune electrophoresis, performed because of suspicion of multiple myeloma, showed an elevated level of kappa light chains at 806.7 mg/dL (0.33–1.94) and normal lambda light chains at 0.62 mg/dL (0.57–2.63). The immunoglobulin G level was low at 496 mg/dL (610–1,660). In patients with severe hypercalcemia, these results point to a diagnosis of malignancy. Bone marrow aspiration study showed greater than 10% plasma cells, confirming multiple myeloma.
MULTIPLE MYELOMA
The diagnosis of multiple myeloma is based in part on the presence of 10% or more of clonal bone marrow plasma cells10 and of specific end-organ damage (anemia, hypercalcemia, renal insufficiency, or bone lesions).9
Bone marrow clonality can be shown by the ratio of kappa to lambda light chains as detected with immunohistochemistry, immunofluorescence, or flow cytometry.11 The normal ratio is 0.26 to 1.65 for a patient with normal kidney function. In this patient, however, the ratio was 1,301.08 (806.67 kappa to 0.62 lambda), which was extremely out of range. The patient’s bone marrow biopsy results revealed the presence of 15% clonal bone marrow plasma cells.
Multiple myeloma causes osteolytic lesions through increased activation of osteoclast activating factor that stimulates the growth of osteoclast precursors. At the same time, it inhibits osteoblast formation via multiple pathways, including the action of sclerostin.11 Our patient had lytic lesions in 2 left lower ribs and in both forearms and femurs.
Hypercalcemia in multiple myeloma is attributed to 2 main factors: bone breakdown and macrophage overactivation. Multiple myeloma cells increase the release of macrophage inflammatory protein 1-alpha and tumor necrosis factor, which are inflammatory proteins that cause an increase in macrophages, which cause an increase in calcitriol.11 As noted, our patient’s calcium level at presentation was 18.4 mg/dL uncorrected and 18.96 mg/dL corrected.
Cast nephropathy can occur in the distal tubules from the increased free light chains circulating and combining with Tamm-Horsfall protein, which in turn causes obstruction and local inflammation,12 leading to a rise in creatinine levels and resulting in acute kidney injury,12 as in our patient.
TREATMENT CONSIDERATIONS IN MULTIPLE MYELOMA
Our patient was referred to an oncologist for management.
In the management of multiple myeloma, the patient’s quality of life needs to be considered. With the development of new agents to combat the damages of the osteolytic effects, there is hope for improving quality of life.13,14 New agents under study include anabolic agents such as antisclerostin and anti-Dickkopf-1, which promote osteoblastogenesis, leading to bone formation, with the possibility of repairing existing damage.15
TAKE-HOME POINTS
- If hypercalcemia is mild to moderate, consider primary hyperparathyroidism.
- Identify patients with severe symptoms of hypercalcemia such as volume depletion, acute kidney injury, arrhythmia, or seizures.
- Confirm severe cases of hypercalcemia and treat severe cases effectively.
- Severe hypercalcemia may need further investigation into a potential underlying malignancy.
- Sternlicht H, Glezerman IG. Hypercalcemia of malignancy and new treatment options. Ther Clin Risk Manag 2015; 11:1779–1788. doi:10.2147/TCRM.S83681
- Ahmed R, Hashiba K. Reliability of QT intervals as indicators of clinical hypercalcemia. Clin Cardiol 1988; 11(6):395–400. doi:10.1002/clc.4960110607
- Bilezikian JP, Cusano NE, Khan AA, Liu JM, Marcocci C, Bandeira F. Primary hyperparathyroidism. Nat Rev Dis Primers 2016; 2:16033. doi:10.1038/nrdp.2016.33
- Kuchay MS, Kaur P, Mishra SK, Mithal A. The changing profile of hypercalcemia in a tertiary care setting in North India: an 18-month retrospective study. Clin Cases Miner Bone Metab 2017; 14(2):131–135. doi:10.11138/ccmbm/2017.14.1.131
- Rosner MH, Dalkin AC. Onco-nephrology: the pathophysiology and treatment of malignancy-associated hypercalcemia. Clin J Am Soc Nephrol 2012; 7(10):1722–1729. doi:10.2215/CJN.02470312
- Ai L, Mu S, Hu Y. Prognostic role of RDW in hematological malignancies: a systematic review and meta-analysis. Cancer Cell Int 2018; 18:61. doi:10.1186/s12935-018-0558-3
- Baz R, Alemany C, Green R, Hussein MA. Prevalence of vitamin B12 deficiency in patients with plasma cell dyscrasias: a retrospective review. Cancer 2004; 101(4):790–795. doi:10.1002/cncr.20441
- Elmore JG, Carney PA, Abraham LA, et al. The association between obesity and screening mammography accuracy. Arch Intern Med 2004; 164(10):1140–1147. doi:10.1001/archinte.164.10.1140
- Gerecke C, Fuhrmann S, Strifler S, Schmidt-Hieber M, Einsele H, Knop S. The diagnosis and treatment of multiple myeloma. Dtsch Arztebl Int 2016; 113(27–28):470–476. doi:10.3238/arztebl.2016.0470
- Rajkumar SV. Multiple myeloma: 2016 update on diagnosis, risk-stratification, and management. Am J Hematol 2016; 91(7):719–734. doi:10.1002/ajh.24402
- Silbermann R, Roodman GD. Myeloma bone disease: pathophysiology and management. J Bone Oncol 2013; 2(2):59–69. doi:10.1016/j.jbo.2013.04.001
- Doshi M, Lahoti A, Danesh FR, Batuman V, Sanders PW; American Society of Nephrology Onco-Nephrology Forum. Paraprotein-related kidney disease: kidney injury from paraproteins—what determines the site of injury? Clin J Am Soc Nephrol 2016; 11(12):2288–2294. doi:10.2215/CJN.02560316
- Reece D. Update on the initial therapy of multiple myeloma. Am Soc Clin Oncol Educ Book 2013. doi:10.1200/EdBook_AM.2013.33.e307
- Nishida H. Bone-targeted agents in multiple myeloma. Hematol Rep 2018; 10(1):7401. doi:10.4081/hr.2018.7401
- Ring ES, Lawson MA, Snowden JA, Jolley I, Chantry AD. New agents in the treatment of myeloma bone disease. Calcif Tissue Int 2018; 102(2):196–209. doi:10.1007/s00223-017-0351-7
A morbidly obese 54-year-old woman presented to the emergency department after experiencing generalized abdominal pain for 3 days. She rated the pain as 5 on a scale of 10 and described it as dull, cramping, waxing and waning, not radiating, and not relieved with changes of position—in fact, not alleviated by anything she had tried. Her pain was associated with nausea and 1 episode of vomiting. She also experienced constipation before the onset of pain.
She denied recent trauma, recent travel, diarrhea, fevers, weakness, shortness of breath, chest pain, other muscle pains, or recent changes in diet. She also denied having this pain in the past. She said she had unintentionally lost some weight but was not certain how much. She denied tobacco, alcohol, or illicit drug use. She had no history of surgery.
Her medical history included hypertension, anemia, and uterine fibroids. Her current medications included losartan, hydrochlorothiazide, and albuterol. She had no family history of significant disease.
INITIAL EVALUATION AND MANAGEMENT
On admission, her temperature was 97.8°F (36.6°C), heart rate 100 beats per minute, blood pressure 136/64 mm Hg, respiratory rate 18 breaths per minute, oxygen saturation 97% on room air, weight 130.6 kg, and body mass index 35 kg/m2.
She was alert and oriented to person, place, and time. She was in mild discomfort but no distress. Her lungs were clear to auscultation, with no wheezing or crackles. Heart rate and rhythm were regular, with no extra heart sounds or murmurs. Bowel sounds were normal in all 4 quadrants, with tenderness to palpation of the epigastric area, but with no guarding or rebound tenderness.
Laboratory test results
Notable results of blood testing at presentation were as follows:
- Hemoglobin 8.2 g/dL (reference range 12.3–15.3)
- Hematocrit 26% (41–50)
- Mean corpuscular volume 107 fL (80–100)
- Blood urea nitrogen 33 mg/dL (8–21); 6 months earlier it was 16
- Serum creatinine 3.6 mg/dL (0.58–0.96); 6 months earlier, it was 0.75
- Albumin 3.3 g/dL (3.5–5)
- Calcium 18.4 mg/dL (8.4–10.2); 6 months earlier, it was 9.6
- Corrected calcium 19 mg/dL.
Findings on imaging, electrocardiography
Chest radiography showed no acute cardiopulmonary abnormalities. Abdominal computed tomography without contrast showed no abnormalities within the pancreas and no evidence of inflammation or obstruction. Electrocardiography showed sinus tachycardia.
DIFFERENTIAL DIAGNOSIS
1. Which is the most likely cause of this patient’s symptoms?
- Primary hyperparathyroidism
- Malignancy
- Her drug therapy
- Familial hypercalcemic hypocalciuria
In total, her laboratory results were consistent with macrocytic anemia, severe hypercalcemia, and acute kidney injury, and she had generalized symptoms.
Primary hyperparathyroidism
A main cause of hypercalcemia is primary hyperparathyroidism, and this needs to be ruled out. Benign adenomas are the most common cause of primary hyperparathyroidism, and a risk factor for benign adenoma is exposure to therapeutic levels of radiation.3
In hyperparathyroidism, there is an increased secretion of parathyroid hormone (PTH), which has multiple effects including increased reabsorption of calcium from the urine, increased excretion of phosphate, and increased expression of 1,25-hydroxyvitamin D hydroxylase to activate vitamin D. PTH also stimulates osteoclasts to increase their expression of receptor activator of nuclear factor kappa B ligand (RANKL), which has a downstream effect on osteoclast precursors to cause bone reabsorption.3
Inherited primary hyperparathyroidism tends to present at a younger age, with multiple overactive parathyroid glands.3 Given our patient’s age, inherited primary hyparathyroidism is thus less likely.
Malignancy
The probability that malignancy is causing the hypercalcemia increases with calcium levels greater than 13 mg/dL. Epidemiologically, in hospitalized patients with hypercalcemia, the source tends to be malignancy.4 Typically, patients who develop hypercalcemia from malignancy have a worse prognosis.5
Solid tumors and leukemias can cause hypercalcemia. The mechanisms include humoral factors secreted by the malignancy, local osteolysis due to tumor invasion of bone, and excessive absorption of calcium due to excess vitamin D produced by malignancies.5 The cancers that most frequently cause an increase in calcium resorption are lung cancer, renal cancer, breast cancer, and multiple myeloma.1
Solid tumors with no bone metastasis and non-Hodgkin lymphoma that release PTH-related protein (PTHrP) cause humoral hypercalcemia in malignancy. The patient is typically in an advanced stage of disease. PTHrP increases serum calcium levels by decreasing the kidney’s ability to excrete calcium and by increasing bone turnover. It has no effect on intestinal absorption because of its inability to stimulate activated vitamin D3. Thus, the increase in systemic calcium comes directly from breakdown of bone and inability to excrete the excess.
PTHrP has a unique role in breast cancer: it is released locally in areas where cancer cells have metastasized to bone, but it does not cause a systemic effect. Bone resorption occurs in areas of metastasis and results from an increase in expression of RANKL and RANK in osteoclasts in response to the effects of PTHrP, leading to an increase in the production of osteoclastic cells.1
Tamoxifen, an endocrine therapy often used in breast cancer, also causes a release of bone-reabsorbing factors from tumor cells, which can partially contribute to hypercalcemia.5
Myeloma cells secrete RANKL, which stimulates osteoclastic activity, and they also release interleukin 6 (IL-6) and activating macrophage inflammatory protein alpha. Serum testing usually shows low or normal intact PTH, PTHrP, and 1,25-dihydroxyvitamin D.1
Patients with multiple myeloma have a worse prognosis if they have a high red blood cell distribution width, a condition shown to correlate with malnutrition, leading to deficiencies in vitamin B12 and to poor response to treatment.6 Up to 14% of patients with multiple myeloma have vitamin B12 deficiency.7
Our patient’s recent weight loss and severe hypercalcemia raise suspicion of malignancy. Further, her obesity makes proper routine breast examination difficult and thus increases the chance of undiagnosed breast cancer.8 Her decrease in renal function and her anemia complicated by hypercalcemia also raise suspicion of multiple myeloma.
Hypercalcemia due to drug therapy
Thiazide diuretics, lithium, teriparatide, and vitamin A in excessive amounts can raise the serum calcium concentration.5 Our patient was taking a thiazide for hypertension, but her extremely high calcium level places drug-induced hypercalcemia as the sole cause lower on the differential list.
Familial hypercalcemic hypocalciuria
Familial hypercalcemic hypocalciuria is a rare autosomal-dominant cause of hypercalcemia in which the ability of the body (and especially the kidneys) to sense levels of calcium is impaired, leading to a decrease in excretion of calcium in the urine.3 Very high calcium levels are rare in hypercalcemic hypocalciuria.3 In our patient with a corrected calcium concentration of nearly 19 mg/dL, familial hypercalcemic hypocalciuria is very unlikely to be the cause of the hypercalcemia.
WHAT ARE THE NEXT STEPS IN THE WORKUP?
As hypercalcemia has been confirmed, the intact PTH level should be checked to determine whether the patient’s condition is PTH-mediated. If the PTH level is in the upper range of normal or is minimally elevated, primary hyperparathyroidism is likely. Elevated PTH confirms primary hyperparathyroidism. A low-normal or low intact PTH confirms a non-PTH-mediated process, and once this is confirmed, PTHrP levels should be checked. An elevated PTHrP suggests humoral hypercalcemia of malignancy. Serum protein electrophoresis, urine protein electrophoresis, and a serum light chain assay should be performed to rule out multiple myeloma.
Vitamin D toxicity is associated with high concentrations of 1,25-dihydroxyvitamin D and 25-hydroxyvitamin D metabolites. These levels should be checked in this patient.
Other disorders that cause hypercalcemia are vitamin A toxicity and hyperthyroidism, so vitamin A and thyroid-stimulating hormone levels should also be checked.5
CASE CONTINUED
After further questioning, the patient said that she had had lower back pain about 1 to 2 weeks before coming to the emergency room; her primary care doctor had said the pain was likely from muscle strain. The pain had almost resolved but was still present.
The results of further laboratory testing were as follows:
- Serum PTH 11 pg/mL (15–65)
- PTHrP 3.4 pmol/L (< 2.0)
- Protein electrophoresis showed a monoclonal (M) spike of 0.2 g/dL (0)
- Activated vitamin D < 5 ng/mL (19.9–79.3)
- Vitamin A 7.2 mg/dL (33.1–100)
- Vitamin B12 194 pg/mL (239–931)
- Thyroid-stimulating hormone 1.21 mIU/ L (0.47–4.68
- Free thyroxine 1.27 ng/dL (0.78–2.19)
- Iron 103 µg/dL (37–170)
- Total iron-binding capacity 335 µg/dL (265–497)
- Transferrin 248 mg/dL (206–381)
- Ferritin 66 ng/mL (11.1–264)
- Urine protein (random) 100 mg/dL (0–20)
- Urine microalbumin (random) 5.9 mg/dL (0–1.6)
- Urine creatinine clearance 88.5 mL/min (88–128)
- Urine albumin-creatinine ratio 66.66 mg/g (< 30).
Imaging reports
A nuclear bone scan showed increased bone uptake in the hip and both shoulders, consistent with arthritis, and increased activity in 2 of the lower left ribs, associated with rib fractures secondary to lytic lesions. A skeletal survey at a later date showed multiple well-circumscribed “punched-out” lytic lesions in both forearms and both femurs.
2. What should be the next step in this patient’s management?
- Intravenous (IV) fluids
- Calcitonin
- Bisphosphonate treatment
- Denosumab
- Hemodialysis
Initial treatment of severe hypercalcemia includes the following:
Start IV isotonic fluids at a rate of 150 mL/h (if the patient is making urine) to maintain urine output at more than 100 mL/h. Closely monitor urine output.
Give calcitonin 4 IU/kg in combination with IV fluids to reduce calcium levels within the first 12 to 48 hours of treatment.
Give a bisphosphonate, eg, zoledronic acid 4 mg over 15 minutes, or pamidronate 60 to 90 mg over 2 hours. Zoledronic acid is preferred in malignancy-induced hypercalcemia because it is more potent. Doses should be adjusted in patients with renal failure.
Give denosumab if hypercalcemia is refractory to bisphosphonates, or when bisphosphonates cannot be used in renal failure.9
Hemodialysis is performed in patients who have significant neurologic symptoms irrespective of acute renal insufficiency.
Our patient was started on 0.9% sodium chloride at a rate of 150 mL/h for severe hypercalcemia. Zoledronic acid 4 mg IV was given once. These measures lowered her calcium level and lessened her acute kidney injury.
ADDITIONAL FINDINGS
Urine testing was positive for Bence Jones protein. Immune electrophoresis, performed because of suspicion of multiple myeloma, showed an elevated level of kappa light chains at 806.7 mg/dL (0.33–1.94) and normal lambda light chains at 0.62 mg/dL (0.57–2.63). The immunoglobulin G level was low at 496 mg/dL (610–1,660). In patients with severe hypercalcemia, these results point to a diagnosis of malignancy. Bone marrow aspiration study showed greater than 10% plasma cells, confirming multiple myeloma.
MULTIPLE MYELOMA
The diagnosis of multiple myeloma is based in part on the presence of 10% or more of clonal bone marrow plasma cells10 and of specific end-organ damage (anemia, hypercalcemia, renal insufficiency, or bone lesions).9
Bone marrow clonality can be shown by the ratio of kappa to lambda light chains as detected with immunohistochemistry, immunofluorescence, or flow cytometry.11 The normal ratio is 0.26 to 1.65 for a patient with normal kidney function. In this patient, however, the ratio was 1,301.08 (806.67 kappa to 0.62 lambda), which was extremely out of range. The patient’s bone marrow biopsy results revealed the presence of 15% clonal bone marrow plasma cells.
Multiple myeloma causes osteolytic lesions through increased activation of osteoclast activating factor that stimulates the growth of osteoclast precursors. At the same time, it inhibits osteoblast formation via multiple pathways, including the action of sclerostin.11 Our patient had lytic lesions in 2 left lower ribs and in both forearms and femurs.
Hypercalcemia in multiple myeloma is attributed to 2 main factors: bone breakdown and macrophage overactivation. Multiple myeloma cells increase the release of macrophage inflammatory protein 1-alpha and tumor necrosis factor, which are inflammatory proteins that cause an increase in macrophages, which cause an increase in calcitriol.11 As noted, our patient’s calcium level at presentation was 18.4 mg/dL uncorrected and 18.96 mg/dL corrected.
Cast nephropathy can occur in the distal tubules from the increased free light chains circulating and combining with Tamm-Horsfall protein, which in turn causes obstruction and local inflammation,12 leading to a rise in creatinine levels and resulting in acute kidney injury,12 as in our patient.
TREATMENT CONSIDERATIONS IN MULTIPLE MYELOMA
Our patient was referred to an oncologist for management.
In the management of multiple myeloma, the patient’s quality of life needs to be considered. With the development of new agents to combat the damages of the osteolytic effects, there is hope for improving quality of life.13,14 New agents under study include anabolic agents such as antisclerostin and anti-Dickkopf-1, which promote osteoblastogenesis, leading to bone formation, with the possibility of repairing existing damage.15
TAKE-HOME POINTS
- If hypercalcemia is mild to moderate, consider primary hyperparathyroidism.
- Identify patients with severe symptoms of hypercalcemia such as volume depletion, acute kidney injury, arrhythmia, or seizures.
- Confirm severe cases of hypercalcemia and treat severe cases effectively.
- Severe hypercalcemia may need further investigation into a potential underlying malignancy.
A morbidly obese 54-year-old woman presented to the emergency department after experiencing generalized abdominal pain for 3 days. She rated the pain as 5 on a scale of 10 and described it as dull, cramping, waxing and waning, not radiating, and not relieved with changes of position—in fact, not alleviated by anything she had tried. Her pain was associated with nausea and 1 episode of vomiting. She also experienced constipation before the onset of pain.
She denied recent trauma, recent travel, diarrhea, fevers, weakness, shortness of breath, chest pain, other muscle pains, or recent changes in diet. She also denied having this pain in the past. She said she had unintentionally lost some weight but was not certain how much. She denied tobacco, alcohol, or illicit drug use. She had no history of surgery.
Her medical history included hypertension, anemia, and uterine fibroids. Her current medications included losartan, hydrochlorothiazide, and albuterol. She had no family history of significant disease.
INITIAL EVALUATION AND MANAGEMENT
On admission, her temperature was 97.8°F (36.6°C), heart rate 100 beats per minute, blood pressure 136/64 mm Hg, respiratory rate 18 breaths per minute, oxygen saturation 97% on room air, weight 130.6 kg, and body mass index 35 kg/m2.
She was alert and oriented to person, place, and time. She was in mild discomfort but no distress. Her lungs were clear to auscultation, with no wheezing or crackles. Heart rate and rhythm were regular, with no extra heart sounds or murmurs. Bowel sounds were normal in all 4 quadrants, with tenderness to palpation of the epigastric area, but with no guarding or rebound tenderness.
Laboratory test results
Notable results of blood testing at presentation were as follows:
- Hemoglobin 8.2 g/dL (reference range 12.3–15.3)
- Hematocrit 26% (41–50)
- Mean corpuscular volume 107 fL (80–100)
- Blood urea nitrogen 33 mg/dL (8–21); 6 months earlier it was 16
- Serum creatinine 3.6 mg/dL (0.58–0.96); 6 months earlier, it was 0.75
- Albumin 3.3 g/dL (3.5–5)
- Calcium 18.4 mg/dL (8.4–10.2); 6 months earlier, it was 9.6
- Corrected calcium 19 mg/dL.
Findings on imaging, electrocardiography
Chest radiography showed no acute cardiopulmonary abnormalities. Abdominal computed tomography without contrast showed no abnormalities within the pancreas and no evidence of inflammation or obstruction. Electrocardiography showed sinus tachycardia.
DIFFERENTIAL DIAGNOSIS
1. Which is the most likely cause of this patient’s symptoms?
- Primary hyperparathyroidism
- Malignancy
- Her drug therapy
- Familial hypercalcemic hypocalciuria
In total, her laboratory results were consistent with macrocytic anemia, severe hypercalcemia, and acute kidney injury, and she had generalized symptoms.
Primary hyperparathyroidism
A main cause of hypercalcemia is primary hyperparathyroidism, and this needs to be ruled out. Benign adenomas are the most common cause of primary hyperparathyroidism, and a risk factor for benign adenoma is exposure to therapeutic levels of radiation.3
In hyperparathyroidism, there is an increased secretion of parathyroid hormone (PTH), which has multiple effects including increased reabsorption of calcium from the urine, increased excretion of phosphate, and increased expression of 1,25-hydroxyvitamin D hydroxylase to activate vitamin D. PTH also stimulates osteoclasts to increase their expression of receptor activator of nuclear factor kappa B ligand (RANKL), which has a downstream effect on osteoclast precursors to cause bone reabsorption.3
Inherited primary hyperparathyroidism tends to present at a younger age, with multiple overactive parathyroid glands.3 Given our patient’s age, inherited primary hyparathyroidism is thus less likely.
Malignancy
The probability that malignancy is causing the hypercalcemia increases with calcium levels greater than 13 mg/dL. Epidemiologically, in hospitalized patients with hypercalcemia, the source tends to be malignancy.4 Typically, patients who develop hypercalcemia from malignancy have a worse prognosis.5
Solid tumors and leukemias can cause hypercalcemia. The mechanisms include humoral factors secreted by the malignancy, local osteolysis due to tumor invasion of bone, and excessive absorption of calcium due to excess vitamin D produced by malignancies.5 The cancers that most frequently cause an increase in calcium resorption are lung cancer, renal cancer, breast cancer, and multiple myeloma.1
Solid tumors with no bone metastasis and non-Hodgkin lymphoma that release PTH-related protein (PTHrP) cause humoral hypercalcemia in malignancy. The patient is typically in an advanced stage of disease. PTHrP increases serum calcium levels by decreasing the kidney’s ability to excrete calcium and by increasing bone turnover. It has no effect on intestinal absorption because of its inability to stimulate activated vitamin D3. Thus, the increase in systemic calcium comes directly from breakdown of bone and inability to excrete the excess.
PTHrP has a unique role in breast cancer: it is released locally in areas where cancer cells have metastasized to bone, but it does not cause a systemic effect. Bone resorption occurs in areas of metastasis and results from an increase in expression of RANKL and RANK in osteoclasts in response to the effects of PTHrP, leading to an increase in the production of osteoclastic cells.1
Tamoxifen, an endocrine therapy often used in breast cancer, also causes a release of bone-reabsorbing factors from tumor cells, which can partially contribute to hypercalcemia.5
Myeloma cells secrete RANKL, which stimulates osteoclastic activity, and they also release interleukin 6 (IL-6) and activating macrophage inflammatory protein alpha. Serum testing usually shows low or normal intact PTH, PTHrP, and 1,25-dihydroxyvitamin D.1
Patients with multiple myeloma have a worse prognosis if they have a high red blood cell distribution width, a condition shown to correlate with malnutrition, leading to deficiencies in vitamin B12 and to poor response to treatment.6 Up to 14% of patients with multiple myeloma have vitamin B12 deficiency.7
Our patient’s recent weight loss and severe hypercalcemia raise suspicion of malignancy. Further, her obesity makes proper routine breast examination difficult and thus increases the chance of undiagnosed breast cancer.8 Her decrease in renal function and her anemia complicated by hypercalcemia also raise suspicion of multiple myeloma.
Hypercalcemia due to drug therapy
Thiazide diuretics, lithium, teriparatide, and vitamin A in excessive amounts can raise the serum calcium concentration.5 Our patient was taking a thiazide for hypertension, but her extremely high calcium level places drug-induced hypercalcemia as the sole cause lower on the differential list.
Familial hypercalcemic hypocalciuria
Familial hypercalcemic hypocalciuria is a rare autosomal-dominant cause of hypercalcemia in which the ability of the body (and especially the kidneys) to sense levels of calcium is impaired, leading to a decrease in excretion of calcium in the urine.3 Very high calcium levels are rare in hypercalcemic hypocalciuria.3 In our patient with a corrected calcium concentration of nearly 19 mg/dL, familial hypercalcemic hypocalciuria is very unlikely to be the cause of the hypercalcemia.
WHAT ARE THE NEXT STEPS IN THE WORKUP?
As hypercalcemia has been confirmed, the intact PTH level should be checked to determine whether the patient’s condition is PTH-mediated. If the PTH level is in the upper range of normal or is minimally elevated, primary hyperparathyroidism is likely. Elevated PTH confirms primary hyperparathyroidism. A low-normal or low intact PTH confirms a non-PTH-mediated process, and once this is confirmed, PTHrP levels should be checked. An elevated PTHrP suggests humoral hypercalcemia of malignancy. Serum protein electrophoresis, urine protein electrophoresis, and a serum light chain assay should be performed to rule out multiple myeloma.
Vitamin D toxicity is associated with high concentrations of 1,25-dihydroxyvitamin D and 25-hydroxyvitamin D metabolites. These levels should be checked in this patient.
Other disorders that cause hypercalcemia are vitamin A toxicity and hyperthyroidism, so vitamin A and thyroid-stimulating hormone levels should also be checked.5
CASE CONTINUED
After further questioning, the patient said that she had had lower back pain about 1 to 2 weeks before coming to the emergency room; her primary care doctor had said the pain was likely from muscle strain. The pain had almost resolved but was still present.
The results of further laboratory testing were as follows:
- Serum PTH 11 pg/mL (15–65)
- PTHrP 3.4 pmol/L (< 2.0)
- Protein electrophoresis showed a monoclonal (M) spike of 0.2 g/dL (0)
- Activated vitamin D < 5 ng/mL (19.9–79.3)
- Vitamin A 7.2 mg/dL (33.1–100)
- Vitamin B12 194 pg/mL (239–931)
- Thyroid-stimulating hormone 1.21 mIU/ L (0.47–4.68
- Free thyroxine 1.27 ng/dL (0.78–2.19)
- Iron 103 µg/dL (37–170)
- Total iron-binding capacity 335 µg/dL (265–497)
- Transferrin 248 mg/dL (206–381)
- Ferritin 66 ng/mL (11.1–264)
- Urine protein (random) 100 mg/dL (0–20)
- Urine microalbumin (random) 5.9 mg/dL (0–1.6)
- Urine creatinine clearance 88.5 mL/min (88–128)
- Urine albumin-creatinine ratio 66.66 mg/g (< 30).
Imaging reports
A nuclear bone scan showed increased bone uptake in the hip and both shoulders, consistent with arthritis, and increased activity in 2 of the lower left ribs, associated with rib fractures secondary to lytic lesions. A skeletal survey at a later date showed multiple well-circumscribed “punched-out” lytic lesions in both forearms and both femurs.
2. What should be the next step in this patient’s management?
- Intravenous (IV) fluids
- Calcitonin
- Bisphosphonate treatment
- Denosumab
- Hemodialysis
Initial treatment of severe hypercalcemia includes the following:
Start IV isotonic fluids at a rate of 150 mL/h (if the patient is making urine) to maintain urine output at more than 100 mL/h. Closely monitor urine output.
Give calcitonin 4 IU/kg in combination with IV fluids to reduce calcium levels within the first 12 to 48 hours of treatment.
Give a bisphosphonate, eg, zoledronic acid 4 mg over 15 minutes, or pamidronate 60 to 90 mg over 2 hours. Zoledronic acid is preferred in malignancy-induced hypercalcemia because it is more potent. Doses should be adjusted in patients with renal failure.
Give denosumab if hypercalcemia is refractory to bisphosphonates, or when bisphosphonates cannot be used in renal failure.9
Hemodialysis is performed in patients who have significant neurologic symptoms irrespective of acute renal insufficiency.
Our patient was started on 0.9% sodium chloride at a rate of 150 mL/h for severe hypercalcemia. Zoledronic acid 4 mg IV was given once. These measures lowered her calcium level and lessened her acute kidney injury.
ADDITIONAL FINDINGS
Urine testing was positive for Bence Jones protein. Immune electrophoresis, performed because of suspicion of multiple myeloma, showed an elevated level of kappa light chains at 806.7 mg/dL (0.33–1.94) and normal lambda light chains at 0.62 mg/dL (0.57–2.63). The immunoglobulin G level was low at 496 mg/dL (610–1,660). In patients with severe hypercalcemia, these results point to a diagnosis of malignancy. Bone marrow aspiration study showed greater than 10% plasma cells, confirming multiple myeloma.
MULTIPLE MYELOMA
The diagnosis of multiple myeloma is based in part on the presence of 10% or more of clonal bone marrow plasma cells10 and of specific end-organ damage (anemia, hypercalcemia, renal insufficiency, or bone lesions).9
Bone marrow clonality can be shown by the ratio of kappa to lambda light chains as detected with immunohistochemistry, immunofluorescence, or flow cytometry.11 The normal ratio is 0.26 to 1.65 for a patient with normal kidney function. In this patient, however, the ratio was 1,301.08 (806.67 kappa to 0.62 lambda), which was extremely out of range. The patient’s bone marrow biopsy results revealed the presence of 15% clonal bone marrow plasma cells.
Multiple myeloma causes osteolytic lesions through increased activation of osteoclast activating factor that stimulates the growth of osteoclast precursors. At the same time, it inhibits osteoblast formation via multiple pathways, including the action of sclerostin.11 Our patient had lytic lesions in 2 left lower ribs and in both forearms and femurs.
Hypercalcemia in multiple myeloma is attributed to 2 main factors: bone breakdown and macrophage overactivation. Multiple myeloma cells increase the release of macrophage inflammatory protein 1-alpha and tumor necrosis factor, which are inflammatory proteins that cause an increase in macrophages, which cause an increase in calcitriol.11 As noted, our patient’s calcium level at presentation was 18.4 mg/dL uncorrected and 18.96 mg/dL corrected.
Cast nephropathy can occur in the distal tubules from the increased free light chains circulating and combining with Tamm-Horsfall protein, which in turn causes obstruction and local inflammation,12 leading to a rise in creatinine levels and resulting in acute kidney injury,12 as in our patient.
TREATMENT CONSIDERATIONS IN MULTIPLE MYELOMA
Our patient was referred to an oncologist for management.
In the management of multiple myeloma, the patient’s quality of life needs to be considered. With the development of new agents to combat the damages of the osteolytic effects, there is hope for improving quality of life.13,14 New agents under study include anabolic agents such as antisclerostin and anti-Dickkopf-1, which promote osteoblastogenesis, leading to bone formation, with the possibility of repairing existing damage.15
TAKE-HOME POINTS
- If hypercalcemia is mild to moderate, consider primary hyperparathyroidism.
- Identify patients with severe symptoms of hypercalcemia such as volume depletion, acute kidney injury, arrhythmia, or seizures.
- Confirm severe cases of hypercalcemia and treat severe cases effectively.
- Severe hypercalcemia may need further investigation into a potential underlying malignancy.
- Sternlicht H, Glezerman IG. Hypercalcemia of malignancy and new treatment options. Ther Clin Risk Manag 2015; 11:1779–1788. doi:10.2147/TCRM.S83681
- Ahmed R, Hashiba K. Reliability of QT intervals as indicators of clinical hypercalcemia. Clin Cardiol 1988; 11(6):395–400. doi:10.1002/clc.4960110607
- Bilezikian JP, Cusano NE, Khan AA, Liu JM, Marcocci C, Bandeira F. Primary hyperparathyroidism. Nat Rev Dis Primers 2016; 2:16033. doi:10.1038/nrdp.2016.33
- Kuchay MS, Kaur P, Mishra SK, Mithal A. The changing profile of hypercalcemia in a tertiary care setting in North India: an 18-month retrospective study. Clin Cases Miner Bone Metab 2017; 14(2):131–135. doi:10.11138/ccmbm/2017.14.1.131
- Rosner MH, Dalkin AC. Onco-nephrology: the pathophysiology and treatment of malignancy-associated hypercalcemia. Clin J Am Soc Nephrol 2012; 7(10):1722–1729. doi:10.2215/CJN.02470312
- Ai L, Mu S, Hu Y. Prognostic role of RDW in hematological malignancies: a systematic review and meta-analysis. Cancer Cell Int 2018; 18:61. doi:10.1186/s12935-018-0558-3
- Baz R, Alemany C, Green R, Hussein MA. Prevalence of vitamin B12 deficiency in patients with plasma cell dyscrasias: a retrospective review. Cancer 2004; 101(4):790–795. doi:10.1002/cncr.20441
- Elmore JG, Carney PA, Abraham LA, et al. The association between obesity and screening mammography accuracy. Arch Intern Med 2004; 164(10):1140–1147. doi:10.1001/archinte.164.10.1140
- Gerecke C, Fuhrmann S, Strifler S, Schmidt-Hieber M, Einsele H, Knop S. The diagnosis and treatment of multiple myeloma. Dtsch Arztebl Int 2016; 113(27–28):470–476. doi:10.3238/arztebl.2016.0470
- Rajkumar SV. Multiple myeloma: 2016 update on diagnosis, risk-stratification, and management. Am J Hematol 2016; 91(7):719–734. doi:10.1002/ajh.24402
- Silbermann R, Roodman GD. Myeloma bone disease: pathophysiology and management. J Bone Oncol 2013; 2(2):59–69. doi:10.1016/j.jbo.2013.04.001
- Doshi M, Lahoti A, Danesh FR, Batuman V, Sanders PW; American Society of Nephrology Onco-Nephrology Forum. Paraprotein-related kidney disease: kidney injury from paraproteins—what determines the site of injury? Clin J Am Soc Nephrol 2016; 11(12):2288–2294. doi:10.2215/CJN.02560316
- Reece D. Update on the initial therapy of multiple myeloma. Am Soc Clin Oncol Educ Book 2013. doi:10.1200/EdBook_AM.2013.33.e307
- Nishida H. Bone-targeted agents in multiple myeloma. Hematol Rep 2018; 10(1):7401. doi:10.4081/hr.2018.7401
- Ring ES, Lawson MA, Snowden JA, Jolley I, Chantry AD. New agents in the treatment of myeloma bone disease. Calcif Tissue Int 2018; 102(2):196–209. doi:10.1007/s00223-017-0351-7
- Sternlicht H, Glezerman IG. Hypercalcemia of malignancy and new treatment options. Ther Clin Risk Manag 2015; 11:1779–1788. doi:10.2147/TCRM.S83681
- Ahmed R, Hashiba K. Reliability of QT intervals as indicators of clinical hypercalcemia. Clin Cardiol 1988; 11(6):395–400. doi:10.1002/clc.4960110607
- Bilezikian JP, Cusano NE, Khan AA, Liu JM, Marcocci C, Bandeira F. Primary hyperparathyroidism. Nat Rev Dis Primers 2016; 2:16033. doi:10.1038/nrdp.2016.33
- Kuchay MS, Kaur P, Mishra SK, Mithal A. The changing profile of hypercalcemia in a tertiary care setting in North India: an 18-month retrospective study. Clin Cases Miner Bone Metab 2017; 14(2):131–135. doi:10.11138/ccmbm/2017.14.1.131
- Rosner MH, Dalkin AC. Onco-nephrology: the pathophysiology and treatment of malignancy-associated hypercalcemia. Clin J Am Soc Nephrol 2012; 7(10):1722–1729. doi:10.2215/CJN.02470312
- Ai L, Mu S, Hu Y. Prognostic role of RDW in hematological malignancies: a systematic review and meta-analysis. Cancer Cell Int 2018; 18:61. doi:10.1186/s12935-018-0558-3
- Baz R, Alemany C, Green R, Hussein MA. Prevalence of vitamin B12 deficiency in patients with plasma cell dyscrasias: a retrospective review. Cancer 2004; 101(4):790–795. doi:10.1002/cncr.20441
- Elmore JG, Carney PA, Abraham LA, et al. The association between obesity and screening mammography accuracy. Arch Intern Med 2004; 164(10):1140–1147. doi:10.1001/archinte.164.10.1140
- Gerecke C, Fuhrmann S, Strifler S, Schmidt-Hieber M, Einsele H, Knop S. The diagnosis and treatment of multiple myeloma. Dtsch Arztebl Int 2016; 113(27–28):470–476. doi:10.3238/arztebl.2016.0470
- Rajkumar SV. Multiple myeloma: 2016 update on diagnosis, risk-stratification, and management. Am J Hematol 2016; 91(7):719–734. doi:10.1002/ajh.24402
- Silbermann R, Roodman GD. Myeloma bone disease: pathophysiology and management. J Bone Oncol 2013; 2(2):59–69. doi:10.1016/j.jbo.2013.04.001
- Doshi M, Lahoti A, Danesh FR, Batuman V, Sanders PW; American Society of Nephrology Onco-Nephrology Forum. Paraprotein-related kidney disease: kidney injury from paraproteins—what determines the site of injury? Clin J Am Soc Nephrol 2016; 11(12):2288–2294. doi:10.2215/CJN.02560316
- Reece D. Update on the initial therapy of multiple myeloma. Am Soc Clin Oncol Educ Book 2013. doi:10.1200/EdBook_AM.2013.33.e307
- Nishida H. Bone-targeted agents in multiple myeloma. Hematol Rep 2018; 10(1):7401. doi:10.4081/hr.2018.7401
- Ring ES, Lawson MA, Snowden JA, Jolley I, Chantry AD. New agents in the treatment of myeloma bone disease. Calcif Tissue Int 2018; 102(2):196–209. doi:10.1007/s00223-017-0351-7

MRI saves money, better than CT in acute stroke
ST. LOUIS – , according to a review from Johns Hopkins University, Baltimore.

MRI as the first scan leads to “a definitive diagnoses sooner and helps you manage the person more rapidly and appropriately, without negatively affecting outcomes even in stroke patients who receive endovascular therapy,” said neurologist and senior investigator Argye Hillis, MD, director of the Center of Excellence in Stroke Detection and Diagnosis at Hopkins. “Consider skipping the CT and getting an MRI, and get the MRI while they are still in the emergency room.”
Almost all emergency departments in the United States are set up to get a CT first, but MRI is known to be the better study, according to the researchers. MRI is much more sensitive to stroke, especially in the first 24 hours, and pinpoints the location and extent of the damage. It can detect causes of stroke invisible to CT, with no radiation, and rule out stroke entirely, whereas CT can rule out only intracranial bleeding. Increasingly in Europe, MRI is the first study in suspected stroke, and new EDs in the United States are being designed with an in-house MRI, or one nearby.
The ED at Hopkins’ main campus in downtown Baltimore already has an MRI, and uses it first whenever possible. The problem has been that MRI techs are available only during weekdays, so physicians have to default back to CT at night and on weekends. The impetus for the review, presented at the annual meeting of the American Neurological Association, was to see if savings from unnecessary admissions prevented by MRI would be enough to offset the cost of around-the-clock staffing for the MRI scanner.
Dr. Hillis and her team reviewed 320 patients with suspected ischemic stroke who were seen at the main campus in 2018 and had CT in the ED, and then definitive diagnosis by MRI, which is the usual approach in most U.S. hospitals.
A total of 134 patients had a final diagnosis on MRI that did not justify admission; techs were available to give 75 of them MRIs in the ED after the CT, and those patients were sent home. Techs were not available, however, for 59 patients and since the CT was not able to rule out stroke, those patients were admitted. The cost of those 59 admissions was $814,016.
The cost of the noncontrast CTs for the 75 patients who were sent home after definitive MRI imaging was $28,050, plus an additional $46,072 for those who had CT neck/head angiograms. Altogether, skipping the CT and going straight to the MRI would have saved Hopkins $888,138 in 2018, enough to cover round-the-clock MRI staffing in the ED, which is now the plan at the main campus.
Once the facility moves to 24-and-7 MRI coverage, the next step in the project is to compare stroke outcomes with Johns Hopkins Bayview Medical Center, also in Baltimore, which will continue to do CT first. “We know MRI first is cheaper. We want to see if we have better outcomes. If we find they’re much better, I think many hospitals will say it’s worth the 5 minutes longer it takes to get to the MRI scanner,” Dr. Hillis said.
Stroke mimics among the 134 patients included peripheral nerve palsy and migraine, but also people simply faking it for a hot meal and a warm bed. “Its pretty common, unfortunately,” she said.
The average age for stroke admissions at Hopkins is 55 years, with as many men as women.
There was no industry funding, and Dr. Hillis didn’t have any relevant disclosures.
ST. LOUIS – , according to a review from Johns Hopkins University, Baltimore.
MRI as the first scan leads to “a definitive diagnoses sooner and helps you manage the person more rapidly and appropriately, without negatively affecting outcomes even in stroke patients who receive endovascular therapy,” said neurologist and senior investigator Argye Hillis, MD, director of the Center of Excellence in Stroke Detection and Diagnosis at Hopkins. “Consider skipping the CT and getting an MRI, and get the MRI while they are still in the emergency room.”
Almost all emergency departments in the United States are set up to get a CT first, but MRI is known to be the better study, according to the researchers. MRI is much more sensitive to stroke, especially in the first 24 hours, and pinpoints the location and extent of the damage. It can detect causes of stroke invisible to CT, with no radiation, and rule out stroke entirely, whereas CT can rule out only intracranial bleeding. Increasingly in Europe, MRI is the first study in suspected stroke, and new EDs in the United States are being designed with an in-house MRI, or one nearby.
The ED at Hopkins’ main campus in downtown Baltimore already has an MRI, and uses it first whenever possible. The problem has been that MRI techs are available only during weekdays, so physicians have to default back to CT at night and on weekends. The impetus for the review, presented at the annual meeting of the American Neurological Association, was to see if savings from unnecessary admissions prevented by MRI would be enough to offset the cost of around-the-clock staffing for the MRI scanner.
Dr. Hillis and her team reviewed 320 patients with suspected ischemic stroke who were seen at the main campus in 2018 and had CT in the ED, and then definitive diagnosis by MRI, which is the usual approach in most U.S. hospitals.
A total of 134 patients had a final diagnosis on MRI that did not justify admission; techs were available to give 75 of them MRIs in the ED after the CT, and those patients were sent home. Techs were not available, however, for 59 patients and since the CT was not able to rule out stroke, those patients were admitted. The cost of those 59 admissions was $814,016.
The cost of the noncontrast CTs for the 75 patients who were sent home after definitive MRI imaging was $28,050, plus an additional $46,072 for those who had CT neck/head angiograms. Altogether, skipping the CT and going straight to the MRI would have saved Hopkins $888,138 in 2018, enough to cover round-the-clock MRI staffing in the ED, which is now the plan at the main campus.
Once the facility moves to 24-and-7 MRI coverage, the next step in the project is to compare stroke outcomes with Johns Hopkins Bayview Medical Center, also in Baltimore, which will continue to do CT first. “We know MRI first is cheaper. We want to see if we have better outcomes. If we find they’re much better, I think many hospitals will say it’s worth the 5 minutes longer it takes to get to the MRI scanner,” Dr. Hillis said.
Stroke mimics among the 134 patients included peripheral nerve palsy and migraine, but also people simply faking it for a hot meal and a warm bed. “Its pretty common, unfortunately,” she said.
The average age for stroke admissions at Hopkins is 55 years, with as many men as women.
There was no industry funding, and Dr. Hillis didn’t have any relevant disclosures.
ST. LOUIS – , according to a review from Johns Hopkins University, Baltimore.
MRI as the first scan leads to “a definitive diagnoses sooner and helps you manage the person more rapidly and appropriately, without negatively affecting outcomes even in stroke patients who receive endovascular therapy,” said neurologist and senior investigator Argye Hillis, MD, director of the Center of Excellence in Stroke Detection and Diagnosis at Hopkins. “Consider skipping the CT and getting an MRI, and get the MRI while they are still in the emergency room.”
Almost all emergency departments in the United States are set up to get a CT first, but MRI is known to be the better study, according to the researchers. MRI is much more sensitive to stroke, especially in the first 24 hours, and pinpoints the location and extent of the damage. It can detect causes of stroke invisible to CT, with no radiation, and rule out stroke entirely, whereas CT can rule out only intracranial bleeding. Increasingly in Europe, MRI is the first study in suspected stroke, and new EDs in the United States are being designed with an in-house MRI, or one nearby.
The ED at Hopkins’ main campus in downtown Baltimore already has an MRI, and uses it first whenever possible. The problem has been that MRI techs are available only during weekdays, so physicians have to default back to CT at night and on weekends. The impetus for the review, presented at the annual meeting of the American Neurological Association, was to see if savings from unnecessary admissions prevented by MRI would be enough to offset the cost of around-the-clock staffing for the MRI scanner.
Dr. Hillis and her team reviewed 320 patients with suspected ischemic stroke who were seen at the main campus in 2018 and had CT in the ED, and then definitive diagnosis by MRI, which is the usual approach in most U.S. hospitals.
A total of 134 patients had a final diagnosis on MRI that did not justify admission; techs were available to give 75 of them MRIs in the ED after the CT, and those patients were sent home. Techs were not available, however, for 59 patients and since the CT was not able to rule out stroke, those patients were admitted. The cost of those 59 admissions was $814,016.
The cost of the noncontrast CTs for the 75 patients who were sent home after definitive MRI imaging was $28,050, plus an additional $46,072 for those who had CT neck/head angiograms. Altogether, skipping the CT and going straight to the MRI would have saved Hopkins $888,138 in 2018, enough to cover round-the-clock MRI staffing in the ED, which is now the plan at the main campus.
Once the facility moves to 24-and-7 MRI coverage, the next step in the project is to compare stroke outcomes with Johns Hopkins Bayview Medical Center, also in Baltimore, which will continue to do CT first. “We know MRI first is cheaper. We want to see if we have better outcomes. If we find they’re much better, I think many hospitals will say it’s worth the 5 minutes longer it takes to get to the MRI scanner,” Dr. Hillis said.
Stroke mimics among the 134 patients included peripheral nerve palsy and migraine, but also people simply faking it for a hot meal and a warm bed. “Its pretty common, unfortunately,” she said.
The average age for stroke admissions at Hopkins is 55 years, with as many men as women.
There was no industry funding, and Dr. Hillis didn’t have any relevant disclosures.
REPORTING FROM ANA 2019
Key clinical point: Getting an MRI first for suspected stroke, instead of a CT, saves money by avoiding unnecessary admissions and might lead to better outcomes.
Major finding: An MRI-first approach at a busy ED in downtown Baltimore would have saved $888,138 in 1 year.
Study details: Review of 320 patients with suspected ischemic strokes.
Disclosures: There was no industry funding, and the senior investigator did not have any relevant disclosures.
Source: Sherry E et al. ANA 2019. Abstract M123.
Cardiovascular complications of systemic sclerosis: What to look for
Autoimmune rheumatic diseases increase the risk of cardiovascular disease. In rheumatoid arthritis and systemic lupus erythematosus, the risk is driven primarily by the inflammatory milieu, leading to accelerated coronary and cerebrovascular atherosclerosis independent of traditional atherosclerotic risk factors.1–3 The extent of cardiovascular involvement in other rheumatologic diseases has been less well characterized but is an area of growing interest.
In this review, we focus on the cardiovascular complications of systemic sclerosis and review recommendations for monitoring these patients in clinical practice.
SYSTEMIC SCLEROSIS, AN AUTOIMMUNE RHEUMATIC DISEASE
Systemic sclerosis is an autoimmune rheumatic disease characterized by excessive extracellular matrix deposition leading to diffuse fibrosis, endothelial dysfunction, and microvascular injury. It is most common in North America, Southern Europe, and Australia,4,5 and it affects women more than men in ratios ranging from 3:1 to 14:1.6 The mean age at diagnosis is around 50.
The disease can affect the lungs (interstitial lung disease and pulmonary hypertension), the heart, the kidneys, and the gastrointestinal tract.
Systemic sclerosis has 2 main subtypes: limited cutaneous systemic sclerosis, formerly called CREST syndrome) and diffuse cutaneous systemic sclerosis. The limited cutaneous subtype is characterized by tightening of the skin of the distal extremities (below the elbows and knees) and face, while diffuse cutaneous systemic sclerosis can manifest as more extensive skin tightening also involving proximal extremities and the trunk. Both subtypes can have an effect on the cardiovascular system.
Some cardiovascular risk factors such as dyslipidemia, diabetes mellitus, and high body mass index are less common in patients with systemic sclerosis than in patients with rheumatoid arthritis, while the rates of arterial hypertension, smoking, chronic obstructive pulmonary disease, osteoporosis, and neoplasms are similar between the 2 groups.7
HEART INVOLVEMENT HAS SERIOUS CONSEQUENCES
Overt cardiac involvement in systemic sclerosis is associated with a mortality rate of up to 70% over 5 years,8,9 and about one-fourth of deaths in patients with systemic sclerosis are from cardiac causes.10,11 Studies in Europe10,12 showed that many patients with systemic sclerosis have cardiac involvement detectable by magnetic resonance imaging even if they do not have clinical disease. Pulmonary arterial hypertension (PAH) is a complication of both subtypes of systemic sclerosis and portends a higher risk of death.8
Thus, it is critical for clinicians to understand the potential comorbid conditions associated with systemic sclerosis, particularly the cardiovascular ones, and to work closely with cardiologists to help optimize the evaluation and management.
MECHANISMS OF CARDIAC DISEASE IN SYSTEMIC SCLEROSIS

Abnormal vasoreactivity, a consequence of an imbalance between endothelium-derived vasoconstrictors and vasodilators, defective angiogenesis, and endothelial injury, leads to tissue ischemia and vascular endothelial growth factor expression, which initiates injury and fibrosis in the myocardium and in other organs.14–17 Fibrosis involves the myocardium, pericardium, and conduction system.13,18
Myocardial involvement in systemic sclerosis is thought to be due mainly to abnormal vasoreactivity and microvascular abnormalities such as transient coronary artery spasm leading to repeated focal ischemia.19,20 Abnormal vasoreactivity has been demonstrated during cardiac catheterization21: while mean coronary sinus blood flow in systemic sclerosis patients was normal at rest, vasodilator reserve was significantly reduced in patients with diffuse cutaneous systemic sclerosis after maximal vasodilation with dipyridamole. Additionally, endomyocardial biopsy showed fibrosis and concentric intimal hypertrophy with normal epicardial coronary arteries.21
More research into other mechanisms of cardiovascular disease in systemic sclerosis is needed to allow for better preventive care for these patients.
PULMONARY ARTERIAL HYPERTENSION
Systemic sclerosis can be associated with World Health Organization (WHO) groups 1, 2, 3, and 4 pulmonary hypertension. WHO group 1, called pulmonary arterial hypertension or PAH, is one of the most common cardiac complications of systemic sclerosis, with a reported prevalence as high as 12%.22 Systemic sclerosis-associated PAH carries a high mortality rate, with a mean survival of only 3 years.23
With advances in treatments for other complications of systemic sclerosis, the percentage of systemic sclerosis patients who die of PAH has increased from 6% to 33%.24
Compared with patients with idiopathic PAH, those with systemic sclerosis get less of a response from therapy and have poorer outcomes despite lower mean pulmonary artery pressures and similar reductions in cardiac index. However, recent studies have suggested that with aggressive treatment, patients with systemic sclerosis-related PAH can achieve outcomes similar to those with idiopathic PAH.25 Thus, recognizing this condition early is imperative.
Pulmonary arterial hypertension defined
PAH is defined as the combination of all of the following26:
- Mean pulmonary artery pressure > 20 mm Hg at rest
- Normal pulmonary capillary wedge pressure (≤ 15 mm Hg)
- Pulmonary vascular resistance ≥ 3 Wood units on right heart catheterization.
Other causes of pulmonary hypertension such as interstitial lung disease, chronic pulmonary thromboembolic disease, and left heart disease must be excluded.24,27
Remodeling in the pulmonary arteries
The events that lead to PAH in systemic sclerosis remain unclear but are believed to involve initial inflammation or endothelial injury that leads to a dysequilibrium between proliferative mediators and antiproliferative vasodilators. This dysequilibrium, along with endothelial dysfunction, causes an obliterative vasculopathy in the pulmonary artery branches and arterioles. Sympathetic overactivity, hypoxemia, and ischemia-reperfusion injury additionally promote vascular proliferation, fibrosis, and remodeling, leading to increased pulmonary vascular resistance, PAH, and increased right ventricular pressures.23,27
The subtype of systemic sclerosis is an important factor in the development and progression of PAH. PAH appears to be the major cause of death in limited cutaneous systemic sclerosis, while interstitial lung disease is the major cause of death in diffuse cutaneous systemic sclerosis.28
Pulmonary arterial hypertension is a late complication of systemic sclerosis
Data from the South Australian Scleroderma Registry29 revealed that PAH tends to be a late complication of systemic sclerosis, occurring around 20 years after disease onset. In this study of 608 patients, no patient with diffuse cutaneous systemic sclerosis developed PAH.
Systemic sclerosis-related PAH initially follows an indolent course with few symptoms until right ventricular function deteriorates. Early in the disease, patients may experience nonspecific symptoms of fatigue, lightheadedness, and dyspnea on exertion.23 As it progresses, they tend to have worsening dyspnea and may experience exertional syncope, palpitations, and chest pain.
Physical findings may suggest elevated right ventricular pressure and right ventricular failure; these include a loud P2, a prominent jugular a wave, a tricuspid regurgitant murmur, jugular venous distention, and lower-extremity edema.27
Screening for pulmonary arterial hypertension in systemic sclerosis
Significant signs and symptoms usually occur late in the disease; thus, it is important to appropriately screen patients who are at risk so that they can begin aggressive treatment.
Doppler echocardiography is recommended by European and American guidelines to screen for PAH in patients who have systemic sclerosis, and most agree that screening is appropriate even if the patient has no symptoms.30 European consensus documents recommend that transthoracic echocardiography be done annually for the first 5 years of disease and be continued every year in patients at high risk, ie, those with anticentromere antibodies, anti-Th/To antibodies, or interstitial lung disease. Patients not at high risk of developing pulmonary hypertension should also have regular transthoracic echocardiography, though the exact timing is not defined.31 While American societies have not issued corresponding recommendations, many experts follow the European recommendations.
Worrisome features on echocardiography in asymptomatic patients should be followed up with right heart catheterization to assess mean right ventricular pressure. These include:
- Estimated right ventricular systolic pressure ≥ 40 mm Hg
- Tricuspid regurgitant jet velocity > 2.8 m/s
- Right atrial enlargement > 53 mm
- Right ventricular enlargement (mid-cavity dimension > 35 mm).32
Although echocardiography is the most common form of screening, it gives only an estimate of right ventricular systolic pressure, which is imprecise. Other noninvasive markers are helpful and necessary to appropriately screen this population.
Diffusion capacity. The Itinerair study33 found that a diffusing capacity for carbon monoxide (DLCO) of 60% or higher has a high specificity in excluding PAH.
Uric acid has been found to be elevated in patients with systemic sclerosis-related PAH, and levels inversely correlate with 6-minute walking distance.34
Other predictors. N-terminal pro-B-type natriuretic peptide (NT-proBNP), left atrial volume, and the right ventricular myocardial performance index have also been shown to be independent predictors of PAH in patients with systemic sclerosis.35
An algorithm. The DETECT study36 enrolled patients at increased risk who had had systemic sclerosis longer than 3 years and a DLCO less than 60%. The investigators developed a 2-step algorithm to determine which patients should be referred for right heart catheterization to try to detect PAH earlier while minimizing the number of missed diagnoses and optimizing the use of invasive diagnostic right heart catheterization.
The first step was to assess serum values of anticentromere antibodies, NT-proBNP, and urate, and clinical features (telangiectasias), forced vital capacity, and electrocardiographic changes of right axis deviation to derive a prediction score. The second step was to assess surface echocardiographic features of the right atrial area and tricuspid regurgitation velocity.
This approach led to right heart catheterization in 62% of patients and was associated with a false-negative rate of 4%. Importantly, of the patients with PAH, 1 in 5 had no symptoms, and 33% had tricuspid regurgitation velocity less than 2.8 m/s. No single measurement performed well in isolation in this study.37
Thus, we recommend that, in addition to routine surface echocardiography, a multimodal approach be used that includes laboratory testing, clinical features, and electrocardiographic findings when screening this high-risk patient population.
ATHEROSCLEROTIC DISEASES
Although macrovascular disease has not typically been regarded as a significant systemic feature in systemic sclerosis, myocardial infarction and stroke are more common in patients with systemic sclerosis than in controls.38,39
Coronary artery disease in systemic sclerosis
Man et al38 reported that the incidence of myocardial infarction in patients with systemic sclerosis was 4.4 per 1,000 persons per year, and the incidence of stroke was 4.8 per 1,000 persons per year, compared with 2.5 per 1,000 persons per year for both myocardial infarction and stroke in healthy controls matched for age, sex, and time of entry.
The Australian Scleroderma Cohort Study39 found a 3-fold higher prevalence of coronary artery disease in systemic sclerosis patients than in controls after factoring in traditional risk factors.
Aviña-Zubieta et al,40 in a cohort of 1,239 systemic sclerosis patients, estimated a hazard ratio (HR) of 3.49 for myocardial infarction and 2.35 for stroke compared with age- and sex-matched controls. Not all of these events were related to macrovascular atherosclerosis—vasospasm and microvascular ischemia may have played significant roles in the etiology of clinical manifestations.
Studies of coronary atherosclerosis in systemic sclerosis are limited. An autopsy study41 of 58 patients with systemic sclerosis and 58 controls matched for age, sex, and ethnicity found that the prevalence of atherosclerosis of small coronary arteries and arterioles was significantly higher in systemic sclerosis patients than in controls (17% vs 2%, P < .01). However, the prevalence of medium-vessel coronary atherosclerosis was similar (48% vs 43%).
Why patients with systemic sclerosis develop atherosclerosis has not yet been determined. Traditional risk factors such as hypertension, dyslipidemia, diabetes mellitus, and obesity are typically no more prevalent in systemic sclerosis patients than in controls,38,42 and thus do not explain the increased risk of atherosclerotic cardiovascular disease. There is some evidence that novel markers of atherosclerotic risk such as homocysteine,43 lipoprotein[a],44 and oxidized low-density lipoprotein45 are more prevalent in systemic sclerosis, but these results have not been substantiated in more extensive studies.
Peripheral artery disease
It remains unclear whether peripheral artery disease is more prevalent in systemic sclerosis patients than in controls.
Individual studies have shown mixed results in comparing carotid artery stenosis between systemic sclerosis patients and controls using carotid duplex ultrasonography,46 the ankle-brachial index,46–48 carotid intima-media thickness,49–54 and brachial flow-mediated dilation.51,53,55–58 A meta-analysis found that the carotid intima and media are significantly thicker in systemic sclerosis patients than in controls,59 and the magnitude of difference is similar to that in other groups at increased cardiovascular risk, such as those with rheumatoid arthritis, diabetes, and familial hypercholesterolemia.60–63
A meta-analysis of brachial artery findings showed significantly lower flow-mediated dilation in systemic sclerosis patients than in controls.64
Overall, given the inconsistency of study results, systemic sclerosis patients should be screened and managed as in other patients with peripheral artery disease, but the clinician should be aware that there may be a higher risk of peripheral artery disease in these patients.
RIGHT AND LEFT VENTRICULAR DYSFUNCTION
Many patients with systemic sclerosis have right ventricular dysfunction as a consequence of PAH.65 It is important to detect diastolic dysfunction in this population, as it may be an even stronger predictor of death than pulmonary hypertension on right heart catheterization (HR 3.7 vs 2.0).66
Fewer patients have left ventricular dysfunction. In a multicenter study of 570 systemic sclerosis patients, only 1.4% had left ventricular systolic dysfunction on echocardiography, though 22.6% had left ventricular hypertrophy and 17.7% had left ventricular diastolic dysfunction.67 In the European League Against Rheumatism (EULAR) database, the prevalence of reduced left ventricular ejection fraction was 5.4%.68
Though traditional echocardiographic screening suggests the prevalence of left ventricular dysfunction in systemic sclerosis patients is low, cardiac magnetic resonance imaging (MRI) may be more sensitive than echocardiography for detecting subclinical myocardial involvement. Cardiac MRI has been shown to detect evidence of myocardial pathology (increased T2 signal, left ventricular thinning, pericardial effusion, reduced left ventricular and right ventricular ejection fraction, left ventricular diastolic dysfunction, and delayed myocardial contrast enhancement) in up to 75% of systemic sclerosis cases studied.69
Patients with systemic sclerosis should already be undergoing echocardiography every year to screen for PAH, and screening should also include tissue Doppler imaging to detect various forms of left and right ventricular systolic and diastolic dysfunction that may not be clinically apparent.
Though cardiac MRI can provide useful additional information, it is not currently recommended for routine screening in patients with systemic sclerosis.
ARRHYTHMIAS AND CONDUCTION DEFECTS
Patients with systemic sclerosis are prone to arrhythmias due to both conduction system fibrosis and myocardial damage.
Arrhythmias accounted for 6% of the deaths in the EULAR Scleroderma Trials and Research (EUSTAR) database.11
In the Genetics Versus Environment in Scleroderma Outcome Study (GENISOS),70 250 patients who had had systemic sclerosis for at least 3 years were studied during a period of approximately 6 years, during which there were 52 deaths, 29 of which were directly attributable to systemic sclerosis. Multivariable Cox modeling showed that 7 variables predicted mortality:
- Body mass index < 18.5 kg/m2
- Age ≥ 65
- Forced vital capacity < 50% predicted
- Systolic blood pressure ≥ 140 or diastolic blood pressure ≥ 90 mm Hg
- Pulmonary fibrosis
- Positive anticentromere antibodies
- Cardiac arrhythmias.
The hazard ratio for death in patients with arrhythmias in this model was 2.18 (95% CI 1.05–4.50, P = .035). Thus, finding arrhythmias in systemic sclerosis patients can provide important prognostic information.
While resting electrocardiography in patients with systemic sclerosis most commonly shows sinus rhythm, 24-hour electrocardiographic monitoring has revealed nonsustained supraventricular and ventricular arrhythmias in a significant percentage.71,72 Although difficult to quantify in routine practice, parameters controlled by the autonomic nervous system including heart rate variability and heart rate turbulence have been shown to be impaired in systemic sclerosis, and these measures are associated with an increased risk of malignant arrhythmias and sudden cardiac death.73,74
Conduction abnormalities
Conduction abnormalities occur in one-fifth to one-third of patients with systemic sclerosis.75,76 The most common abnormal conduction finding is left bundle branch block, followed by first-degree atrioventricular block. High-degree atrioventricular block is uncommon,76 though a few case reports of complete heart block thought to be related to systemic sclerosis have been published.77–79 An autopsy study showed that the conduction system is relatively spared from myocardial changes seen in systemic sclerosis patients, and thus it is speculated that the conduction disturbances are a consequence of damaged myocardium rather than damage to conduction tissue.80
Given the array of electrophysiologic abnormalities that systemic sclerosis patients can have, it is critical to monitor all patients with routine (annual or biannual) electrocardiography; to take possible arrhythmia-related symptoms seriously; and to evaluate them with further workup such as Holter monitoring for 24 hours or even longer, event monitoring, exercise testing, or tilt-table testing.
PERICARDIAL DISEASE
Pericardial disease is clinically apparent in 5% to 16% of patients with systemic sclerosis81; patients with limited cutaneous systemic sclerosis have more pericardial disease than those with diffuse cutaneous systemic sclerosis (30% vs 16%).82 Forty-one percent of systemic sclerosis patients have been shown to have pericardial effusion by echocardiography,81 but the effusions are typically small and rarely cause tamponade, though tamponade is associated with a poor prognosis.
Large pericardial effusions can develop before skin thickening and diagnosis of systemic sclerosis.81,83,84 Thus, systemic sclerosis should be considered in patients with pericardial effusions of unknown etiology.
In a small study,85 the pericardial fluid in systemic sclerosis was typically exudative, with lactate dehydrogenase greater than 200 U/L, a fluid-serum lactate dehydrogenase ratio greater than 0.6, and a fluid-serum total protein ratio greater than 0.5.
Pericardial effusion can be a sign of impending scleroderma renal crisis,86 and thus renal function should be carefully monitored in systemic sclerosis patients with pericardial effusion. Constrictive pericarditis and restrictive cardiomyopathy can rarely occur in systemic sclerosis and may more commonly present with symptoms.
Pericardial disease in systemic sclerosis should be treated in a standard fashion with nonsteroidal anti-inflammatory drugs. Corticosteroids are generally of limited benefit and should be avoided, especially in the setting of scleroderma renal crisis.81
VALVULAR HEART DISEASE
Based on limited studies, the prevalence of significant valvular heart disease in systemic sclerosis patients does not seem to be higher than that in the general population. While patients with systemic sclerosis and CREST syndrome (calcinosis, Raynaud phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasia) have been shown to have a higher frequency of mitral valve prolapse and mild mitral regurgitation,87,88 these abnormalities do not often progress in severity, and thus their clinical significance is limited.
RECOMMENDATIONS FOR CARE OF SYSTEMIC SCLEROSIS PATIENTS
It is important for physicians caring for patients with systemic sclerosis to be aware of its most common cardiac manifestations, including left and right ventricular systolic and diastolic dysfunction, pulmonary hypertension, conduction abnormalities, arrhythmias, and cardiomyopathy.
Look for volume overload
On clinical examination, assess for clinical markers of volume overload such as distended neck veins, peripheral edema, or an abnormal blood pressure response to the Valsalva maneuver. These findings should prompt measurement of NT-proBNP,89 and may warrant prescription of a diuretic.
Electrocardiography to investigate arrhythmias
Electrocardiography should be done if patients describe symptoms of palpitations, and should also include continuous rhythm monitoring with Holter or event monitoring, depending on the frequency of symptoms. Otherwise, patients should routinely undergo electrocardiography once or twice a year.
Q waves are common in systemic sclerosis patients (especially those with diffuse cutaneous systemic sclerosis), notably in the precordial leads, and can occur without coronary artery disease.90 Symptoms such as presyncope should be further investigated with Holter monitoring and tilt-table testing.
Assess, modify traditional risk factors
Subclinical atherosclerosis as detected by carotid intima-media thickness is as common in systemic sclerosis as in rheumatoid arthritis.61 However, traditional risk indices such as SCORE (Systematic Coronary Risk Evaluation), QRISK2, and the American College of Cardiology/American Heart Association indices may underestimate risk in patients who have systemic sclerosis.
Strict hypertension control should be the goal for all systemic sclerosis patients. Though there are no specific guidelines on which antihypertensive medications are preferred, calcium channel blockers or angiotensin II receptor blockers, which are typically used to treat systemic sclerosis-related Raynaud phenomenon, may be appropriate.
Statins reduce vascular complications and are generally well tolerated in patients with systemic sclerosis.91,92
Aspirin is not recommended for routine primary prevention in view of data suggesting that its benefits in diabetic patients are counterbalanced by increased bleeding risk.93
Echocardiography to detect pulmonary arterial hypertension
At this time, guidelines for monitoring for cardiovascular manifestations in systemic sclerosis patients are limited. The only well-defined ones are European consensus guidelines, which suggest annual transthoracic echocardiography for the first 5 years after systemic sclerosis is diagnosed and continued annual screening in patients at risk of developing PAH.31
We support this strategy, with annual screening for the first 5 years followed by surveillance echocardiography every 2 to 3 years unless there is a high risk of PAH. Specific attention should be paid to right ventricular diastolic function, right atrial volume, and right ventricular myocardial performance index.
Emerging data suggest that the addition of global longitudinal strain of ventricles to routine echocardiography can help detect subclinical cardiac risk.94 Although further study is needed into the predictive value of global longitudinal strain, it is a low-cost and noninvasive addition to standard echocardiography that can help guide risk stratification, and thus we recommend that it be part of the echocardiographic examination for all systemic sclerosis patients.
Pulmonary function testing. In addition to screening for PAH with echocardiography, we recommend obtaining baseline pulmonary function tests, including DLCO, at the time systemic sclerosis is diagnosed, with repeat testing annually.
Magnetic resonance imaging
While echocardiography is the gold standard for monitoring systemic sclerosis patients, cardiovascular MRI may have a role in identifying those at higher risk of dangerous arrhythmias such as ventricular tachycardia and ventricular fibrillation. In addition to assessing ventricular function, MRI can detect myocardial inflammation, ischemia, and fibrosis that may predispose a patient to develop ventricular tachycardia or fibrillation.95 Variables such as T1/T2 mapping, extracellular volume fraction, T2 signal ratio, and early vs late gadolinium enhancement can help identify patients who had past ventricular tachycardia or fibrillation.96
Finding an increased risk of arrhythmias may prompt a conversation between the patient and the physician about the need for an implantable cardiac defibrillator.
If cardiac MRI is available and is reimbursed by the patient’s insurance carrier, physicians should strongly consider obtaining at least one baseline scan in systemic sclerosis patients to identify those at risk of highly fatal arrhythmias.
Teamwork is needed
Systemic sclerosis has not traditionally been associated with cardiovascular disease to the extent of other rheumatic conditions, but the cardiovascular system can be affected in various ways that can ultimately lead to an early death. These manifestations may be asymptomatic for long periods, and overt clinical disease portends a poorer prognosis.
Primary care physicians managing these patients should be aware of the cardiovascular complications of systemic sclerosis and should implement appropriate screening tests in conjunction with rheumatologists and cardiologists. It is also essential for general and subspecialty cardiologists to understand the broad spectrum of organ system involvement that can affect systemic sclerosis patients and to tailor their investigation and management recommendations accordingly. By designing a multidisciplinary approach to the treatment of systemic sclerosis patients, physicians can help to optimize cardiovascular risk modification in this vulnerable population.
- Maradit-Kremers H, Crowson CS, Nicola PJ, et al. Increased unrecognized coronary heart disease and sudden deaths in rheumatoid arthritis: a population-based cohort study. Arthritis Rheum 2005; 52(2):402–411. doi:10.1002/art.20853
- Naranjo A, Sokka T, Descalzo MA, et al; QUEST-RA Group. Cardiovascular disease in patients with rheumatoid arthritis: results from the QUEST-RA study. Arthritis Res Ther 2008; 10(2):R30. doi:10.1186/ar2383
- Innala L, Möller B, Ljung L, et al. Cardiovascular events in early RA are a result of inflammatory burden and traditional risk factors: a five year prospective study. Arthritis Res Ther 2011; 13(4):R131. doi:10.1186/ar3442
- Barnes J, Mayes MD. Epidemiology of systemic sclerosis: incidence, prevalence, survival, risk factors, malignancy, and environmental triggers. Curr Opin Rheumatol 2012; 24(2):165–170. doi:10.1097/BOR.0b013e32834ff2e8
- Chifflot H, Fautrel B, Sordet C, Chatelus E, Sibilia J. Incidence and prevalence of systemic sclerosis: a systematic literature review. Semin Arthritis Rheum 2008; 37(4):223–235 doi:10.1016/j.semarthrit.2007.05.003
- Gabrielli A, Avvedimento EV, Krieg T. Scleroderma. N Engl J Med 2009; 360(19):1989–2003. doi:10.1056/NEJMra0806188
- Panopoulos S, Tektonidou M, Drosos AA, et al. Prevalence of comorbidities in systemic sclerosis versus rheumatoid arthritis: a comparative, multicenter, matched-cohort study. Arthritis Res Ther 2018; 20(1):267. doi:10.1186/s13075-018-1771-0
- Ferri C, Valentini G, Cozzi F, et al. Systemic sclerosis: demographic, clinical, and serologic features and survival in 1,012 Italian patients. Medicine (Baltimore) 2002; 81(8):139–153. doi:10.1097/00005792-200203000-00004
- Steen VD, Medsger TA Jr. Severe organ involvement in systemic sclerosis with diffuse scleroderma. Arthritis Rheum 2000; 43(11):2437–2444. doi:10.1002/1529-0131(200011)43:11<2437::AID-ANR10>3.0.CO;2-U
- Hachulla AL, Launay D, Gaxotte V, et al. Cardiac magnetic resonance imaging in systemic sclerosis: a cross-sectional observational study of 52 patients. Ann Rheum Dis 2009; 68(12):1878–1884. doi:10.1136/ard.2008.095836
- Tyndall AJ, Bannert B, Vonk M, et al. Causes and risk factors for death in systemic sclerosis: a study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann Rheum Dis 2010; 69(10):1809–1815. doi:10.1136/ard.2009.114264
- Nassenstein K, Breuckmann F, Huger M, et al. Detection of myocardial fibrosis in systemic sclerosis by contrast-enhanced magnetic resonance imaging. Rofo 2008; 180(12):1054–1060. doi:10.1055/s-2008-1027864
- Psarras A, Soulaidopoulos S, Garyfallos A, Kitas G, Dimitroulas T. A critical view on cardiovascular risk in systemic sclerosis. Rheumatol Int 2017; 37(1):85–95. doi:10.1007/s00296-016-3530-3
- Lekakis J, Mavrikakis M, Emmanuel M, et al. Cold-induced coronary Raynaud’s phenomenon in patients with systemic sclerosis. Clin Exp Rheumatol 1998; 16(2):135–140. pmid:9536388
- Altorok N, Wang Y, Kahaleh B. Endothelial dysfunction in systemic sclerosis. Curr Opin Rheumatol 2014; 26(6):615–620. doi:10.1097/BOR.0000000000000112
- Fleming JN, Nash RA, Mahoney WM Jr, Schwartz SM. Is scleroderma a vasculopathy? Curr Rheumatol Rep 2009; 11(2):103–110. pmid:19296882
- Maurer B, Distler A, Suliman YA, et al. Vascular endothelial growth factor aggravates fibrosis and vasculopathy in experimental models of systemic sclerosis. Ann Rheum Dis 2014; 73(10):1880–1887. doi:10.1136/annrheumdis-2013-203535
- Meune C, Vignaux O, Kahan A, Allanore Y. Heart involvement in systemic sclerosis: evolving concept and diagnostic methodologies. Arch Cardiovasc Dis 2010; 103(1):46–52. doi:10.1016/j.acvd.2009.06.009
- Dimitroulas T, Giannakoulas G, Karvounis H, Garyfallos A, Settas L, Kitas GD. Micro- and macrovascular treatment targets in scleroderma heart disease. Curr Pharm Des 2014; 20(4):536–544. pmid:23565639
- Allanore Y, Meune C. Primary myocardial involvement in systemic sclerosis: evidence for a microvascular origin. Clin Exp Rheumatol 2010; 28(5 suppl 62):S48–S53. pmid:21050545
- Kahan A, Nitenberg A, Foult JM, et al. Decreased coronary reserve in primary scleroderma myocardial disease. Arthritis Rheum 1985; 28(6):637–646. pmid:4004974
- Morrisroe K, Stevens W, Sahhar J, et al. Epidemiology and disease characteristics of systemic sclerosis-related pulmonary arterial hypertension: results from a real-life screening program. Arthritis Res Ther 2017; 19(1):42. doi:10.1186/s13075-017-1250-z
- Chaisson NF, Hassoun PM. Systemic sclerosis-associated pulmonary arterial hypertension. Chest 2013; 144(4):1346–1356. doi:10.1378/chest.12-2396
- Steen VD, Medsger TA. Changes in causes of death in systemic sclerosis, 1972–2002. Ann Rheum Dis 2007; 66(7):940–944. doi:10.1136/ard.2006.066068
- Coghlan JG, Galiè N, Barberà JA, et al; AMBITION investigators. Initial combination therapy with ambrisentan and tadalafil in connective tissue disease-associated pulmonary arterial hypertension (CTD-PAH): subgroup analysis from the AMBITION trial. Ann Rheum Dis 2017; 76(7):1219–1227. doi:10.1136/annrheumdis-2016-210236
- Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J 2019; 53(1):1801913. doi:10.1183/13993003.01913-2018
- Chatterjee S. Pulmonary hypertension in systemic sclerosis. Semin Arthritis Rheum 2011; 41(1):19–37. doi:10.1016/j.semarthrit.2010.08.004
- Sweiss NJ, Hushaw L, Thenappan T, et al. Diagnosis and management of pulmonary hypertension in systemic sclerosis. Curr Rheumatol Rep 2010; 12(1):8–18. doi:10.1007/s11926-009-0078-1
- Cox SR, Walker JG, Coleman M, et al. Isolated pulmonary hypertension in scleroderma. Intern Med J 2005; 35(1):28–33. doi:10.1111/j.1445-5994.2004.00646.x
- Sánchez-Román J, Opitz CF, Kowal-Bielecka O, García-Hernández FJ, Castillo-Palma MJ, Pittrow D; EPOSS-OMERACT Group. Screening for PAH in patients with systemic sclerosis: focus on Doppler echocardiography. Rheumatology (Oxford) 2008; 47(suppl 5):v33–v35. doi:10.1093/rheumatology/ken306
- Walker KM, Pope J; Scleroderma Clinical Trials Consortium; Canadian Scleroderma Research Group. Expert agreement on EULAR/EUSTAR recommendations for the management of systemic sclerosis. J Rheumatol 2011; 38(7):1326–1328. doi:10.3899/jrheum.101262
- Khanna D, Gladue H, Channick R, et al; Scleroderma Foundation and Pulmonary Hypertension Association. Recommendations for screening and detection of connective tissue disease-associated pulmonary arterial hypertension. Arthritis Rheum 2013; 65(12):3194–3201. doi:10.1002/art.38172
- Hachulla E, Gressin V, Guillevin L, et al. Early detection of pulmonary arterial hypertension in systemic sclerosis: a French nationwide prospective multicenter study. Arthritis Rheum 2005; 52(12):3792–3800. doi:10.1002/art.21433
- Dimitroulas T, Giannakoulas G, Dimitroula H, et al. Significance of serum uric acid in pulmonary hypertension due to systemic sclerosis: a pilot study. Rheumatol Int 2011; 31(2):263–267. doi:10.1007/s00296-010-1557-4
- Dimitroulas T, Giannakoulas G, Papadopoulou K, et al. Left atrial volume and N-terminal pro-B type natriuretic peptide are associated with elevated pulmonary artery pressure in patients with systemic sclerosis. Clin Rheumatol 2010; 29(9):957–964. doi:10.1007/s10067-010-1494-3
- Coghlan JG, Denton CP, Grünig E, et al; DETECT study group. Evidence-based detection of pulmonary arterial hypertension in systemic sclerosis: the DETECT study. Ann Rheum Dis 2014; 73(7):1340–1349. doi:10.1136/annrheumdis-2013-203301
- Schwaiger JP, Khanna D, Gerry Coghlan J. Screening patients with scleroderma for pulmonary arterial hypertension and implications for other at-risk populations. Eur Respir Rev 2013; 22(130):515–525. doi:10.1183/09059180.00006013
- Man A, Zhu Y, Zhang Y, et al. The risk of cardiovascular disease in systemic sclerosis: a population-based cohort study. Ann Rheum Dis 2013; 72(7):1188–1193. doi:10.1136/annrheumdis-2012-202007
- Ngian G-S, Sahhar J, Proudman SM, Stevens W, Wicks IP, Van Doornum S. Prevalence of coronary heart disease and cardiovascular risk factors in a national cross-sectional cohort study of systemic sclerosis. Ann Rheum Dis 2012; 71(12):1980–1983. doi:10.1136/annrheumdis-2011-201176
- Aviña-Zubieta JA, Man A, Yurkovich M, Huang K, Sayre EC, Choi HK. Early cardiovascular disease after the diagnosis of systemic sclerosis. Am J Med 2016; 29(3):324–331. doi:10.1016/j.amjmed.2015.10.037
- D’Angelo WA, Fries JF, Masi AT, Shulman LE. Pathologic observations in systemic sclerosis (scleroderma). A study of fifty-eight autopsy cases and fifty-eight matched controls. Am J Med 1969; 46(3):428–440. doi:10.1016/0002-9343(69)90044-8
- Ngian GS, Sahhar J, Proudman SM, Stevens W, Wicks IP, Van Doornum S. Prevalence of coronary heart disease and cardiovascular risk factors in a national cross-sectional cohort study of systemic sclerosis. Ann Rheum Dis 2012; 71(12):1980–1983. doi:10.1136/annrheumdis-2011-201176
- Khurma V, Meyer C, Park GS, et al. A pilot study of subclinical coronary atherosclerosis in systemic sclerosis: coronary artery calcification in cases and controls. Arthritis Rheum 2008; 59(4):591–597. doi:10.1002/art.23540
- Lippi G, Caramaschi P, Montagnana M, Salvagno GL, Volpe A, Guidi G. Lipoprotein[a] and the lipid profile in patients with systemic sclerosis. Clin Chim Acta 2006; 364(1–2):345–348. doi:10.1016/j.cca.2005.07.015
- Palinski W, Hörkkö S, Miller E, et al. Cloning of monoclonal autoantibodies to epitopes of oxidized lipoproteins from apolipoprotein E-deficient mice. Demonstration of epitopes of oxidized low density lipoprotein in human plasma. J Clin Invest 1996; 98(3):800–814. doi:10.1172/JCI118853
- Ho M, Veale D, Eastmond C, Nuki G, Belch J. Macrovascular disease and systemic sclerosis. Ann Rheum Dis 2000; 59(1):39–43. doi:10.1136/ard.59.1.39
- Kaloudi O, Basta G, Perfetto F, et al. Circulating levels of Ne-(carboxymethyl)lysine are increased in systemic sclerosis. Rheumatology (Oxford) 2007; 46(3):412–416. doi:10.1093/rheumatology/kel076
- Muro Y, Sugiura K, Morita Y, Tomita Y. An evaluation of the efficacy of the toe brachial index measuring vascular involvement in systemic sclerosis and other connective tissue diseases. Clin Exp Rheumatol 2009; 27(3 suppl 54):26–31. pmid:19796558
- Cheng K-S, Tiwari A, Boutin A, et al. Differentiation of primary and secondary Raynaud’s disease by carotid arterial stiffness. Eur J Vasc Endovasc Surg 2003; 25(4):336–341. doi:10.1053/ejvs.2002.1845
- Kawasaki M, Ito Y, Yokoyama H, et al. Assessment of arterial medial characteristics in human carotid arteries using integrated backscatter ultrasound and its histological implications. Atherosclerosis 2005; 180(1):145–154. doi:10.1016/j.atherosclerosis.2004.11.018
- Szucs G, Tímár O, Szekanecz Z, et al. Endothelial dysfunction precedes atherosclerosis in systemic sclerosis—relevance for prevention of vascular complications. Rheumatology (Oxford) 2007; 46(5):759–762. doi:10.1093/rheumatology/kel426
- Hettema ME, Zhang D, de Leeuw K, et al. Early atherosclerosis in systemic sclerosis and its relation to disease or traditional risk factors. Arthritis Res Ther 2008;10(2):R49. doi:10.1186/ar2408
- Roustit M, Simmons GH, Baguet JP, Carpentier P, Cracowski JL. Discrepancy between simultaneous digital skin microvascular and brachial artery macrovascular post-occlusive hyperemia in systemic sclerosis. J Rheumatol 2008; 35(8):1576–1583. pmid:18597404
- Vettori S, Maresca L, Cuomo G, Abbadessa S, Leonardo G, Valentini G. Clinical and subclinical atherosclerosis in systemic sclerosis: consequences of previous corticosteroid treatment. Scand J Rheumatol 2010; 39(6):485–489. doi:10.3109/03009741003781985
- Lekakis J, Mavrikakis M, Papamichael C, et al. Short-term estrogen administration improves abnormal endothelial function in women with systemic sclerosis and Raynaud’s phenomenon. Am Heart J 1998; 136(5):905–912. doi:10.1016/s0002-8703(98)70137-1
- Bartoli F, Blagojevic J, Bacci M, et al. Flow-mediated vasodilation and carotid intima-media thickness in systemic sclerosis. Ann N Y Acad Sci 2007; 1108:283–290. doi:10.1196/annals.1422.030
- Rollando D, Bezante GP, Sulli A, et al. Brachial artery endothelial-dependent flow-mediated dilation identifies early-stage endothelial dysfunction in systemic sclerosis and correlates with nailfold microvascular impairment. J Rheumatol 2010; 37(6):1168–1173. doi:10.3899/jrheum.091116
- Andersen GN, Mincheva-Nilsson L, Kazzam E, et al. Assessment of vascular function in systemic sclerosis: indications of the development of nitrate tolerance as a result of enhanced endothelial nitric oxide production. Arthritis Rheum 2002; 46(5):1324–1332. doi:10.1002/art.10191
- Au K, Singh MK, Bodukam V, et al. Atherosclerosis in systemic sclerosis: a systematic review and meta-analysis. Arthritis Rheum 2011; 63(7):2078–2090. doi:10.1002/art.30380
- van Sijl AM, Peters MJ, Knol DK, et al. Carotid intima media thickness in rheumatoid arthritis as compared to control subjects: a meta-analysis. Semin Arthritis Rheum 2011; 40(5):389–397. doi:10.1016/j.semarthrit.2010.06.006
- Brohall G, Odén A, Fagerberg B. Carotid artery intima-media thickness in patients with type 2 diabetes mellitus and impaired glucose tolerance: a systematic review. Diabet Med 2006; 23(6):609–616. doi:10.1111/j.1464-5491.2005.01725.x
- Masoura C, Pitsavos C, Aznaouridis K, Skoumas I, Vlachopoulos C, Stefanadis C. Arterial endothelial function and wall thickness in familial hypercholesterolemia and familial combined hyperlipidemia and the effect of statins. A systematic review and meta-analysis. Atherosclerosis 2011; 214(1):129–138. doi:10.1016/j.atherosclerosis.2010.10.008
- Ozen G, Inanc N, Unal AU, et al. Subclinical atherosclerosis in systemic sclerosis: not less frequent than rheumatoid arthritis and not detected with cardiovascular risk indices. Arthritis Care Res (Hoboken) 2016; 68(10):1538–1546. doi:10.1002/acr.22852
- Inaba Y, Chen JA, Bergmann SR. Prediction of future cardiovascular outcomes by flow-mediated vasodilatation of brachial artery: a meta-analysis. Int J Cardiovasc Imaging 2010; 26(6):631–640. doi:10.1007/s10554-010-9616-1
- Meune C, Avouac J, Wahbi K, et al. Cardiac involvement in systemic sclerosis assessed by tissue-doppler echocardiography during routine care: a controlled study of 100 consecutive patients. Arthritis Rheum 2008; 58(6):1803–1809. doi:10.1002/art.23463
- Tennøe AH, Murbræch K, Andreassen JC, et al. Left ventricular diastolic dysfunction predicts mortality in patients with systemic sclerosis. J Am Coll Cardiol 2018; 72(15):1804–1813. doi:10.1016/j.jacc.2018.07.068
- de Groote P, Gressin V, Hachulla E, et al; ItinerAIR-Scleroderma Investigators. Evaluation of cardiac abnormalities by Doppler echocardiography in a large nationwide multicentric cohort of patients with systemic sclerosis. Ann Rheum Dis 2008; 67(1):31–36. doi:10.1136/ard.2006.057760
- Allanore Y, Meune C, Vonk MC, et al; EUSTAR co-authors. Prevalence and factors associated with left ventricular dysfunction in the EULAR Scleroderma Trial and Research group (EUSTAR) database of patients with systemic sclerosis. Ann Rheum Dis 2010; 69(1):218–221. doi:10.1136/ard.2008.103382
- Hachulla AL, Launay D, Gaxotte V, et al. Cardiac magnetic resonance imaging in systemic sclerosis: a cross-sectional observational study of 52 patients. Ann Rheum Dis 2009; 68(12):1878–1884. doi:10.1136/ard.2008.095836
- Assassi S, Del Junco D, Sutter K, et al. Clinical and genetic factors predictive of mortality in early systemic sclerosis. Arthritis Rheum 2009; 61(10):1403–1411. doi:10.1002/art.24734
- Rokas S, Mavrikakis M, Agrios N, Mylonas D, Antoniadou L, Moulopoulos S. Electrophysiologic abnormalities of cardiac function in progressive systemic sclerosis. J Electrocardiol 1996; 29(1):17–25. pmid:8808521
- Kostis JB, Seibold JR, Turkevich D, et al. Prognostic importance of cardiac arrhythmias in systemic sclerosis. Am J Med 1988; 84(6):1007–1015. doi:10.1016/0002-9343(88)90305-1
- Biełous-Wilk A, Poreba M, Staniszewska-Marszałek E, et al. Electrocardiographic evaluation in patients with systemic scleroderma and without clinically evident heart disease. Ann Noninvasive Electrocardiol 2009; 14(3):251–257. doi:10.1111/j.1542-474X.2009.00306.x
- Bienias P, Ciurzynski M, Glinska-Wielochowska M, et al. Heart rate turbulence assessment in systemic sclerosis: the role for the detection of cardiac autonomic nervous system dysfunction. Rheumatology (Oxford) 2010; 49(2):355–360. doi:10.1093/rheumatology/kep394
- Ferri C, Bernini L, Bongiorni MG, et al. Noninvasive evaluation of cardiac dysrhythmias, and their relationship with multisystemic symptoms, in progressive systemic sclerosis patients. Arthritis Rheum 1985; 28(11):1259–1266. pmid:4063000
- Roberts NK, Cabeen WR, Moss J, Clements PJ, Furst DE. The prevalence of conduction defects and cardiac arrhythmias in progressive systemic sclerosis. Ann Intern Med 1981; 94(1):38–40. doi:10.7326/0003-4819-94-1-38
- Wang Q, Shang Y, Li S, Wu Y, Wang C, Yan X. Complete heart block in systemic sclerosis: a case report and literature review. Medicine (Baltimore) 2018; 97(46):e13226. doi:10.1097/MD.0000000000013226
- Summerfield BJ. Progressive systemic sclerosis with complete heart block. Br Heart J 1975; 37(12):1308–1310. doi:10.1136/hrt.37.12.1308
- Moyssakis I, Papadopoulos DP, Tzioufas AG, Votteas V. Complete heart block in a patient with systemic sclerosis. Clin Rheumatol 2006; 25(4):551–552. doi:10.1007/s10067-005-0068-2
- Ridolfi RL, Bulkley BH, Hutchins GM. The cardiac conduction system in progressive systemic sclerosis. Clinical and pathologic features of 35 patients. Am J Med 1976; 61(3):361–366. doi:10.1016/0002-9343(76)90373-9
- Champion HC. The heart in scleroderma. Rheum Dis Clin North Am 2008; 34(1):181–190. doi:10.1016/j.rdc.2007.12.002
- Gowda RM, Khan IA, Sacchi TJ, Vasavada BC. Scleroderma pericardial disease presented with a large pericardial effusion—a case report. Angiology 2001; 52(1):59–62. doi:10.1177/000331970105200108
- Meier FMP, Frommer KW, Dinser R, et al; EUSTAR Co-authors. Update on the profile of the EUSTAR cohort: an analysis of the EULAR scleroderma trials and research group database. Ann Rheum Dis 2012; 71(8):1355–1360. doi:10.1136/annrheumdis-2011-200742
- Subramanian SR, Akram R, Velayati A, Chadow H. New development of cardiac tamponade on underlying effusive-constrictive pericarditis: an uncommon initial presentation of scleroderma. BMJ Case Rep 2013; 2013. doi:10.1136/bcr-2013-010254
- Kitchongcharoenying P, Foocharoen C, Mahakkanukrauh A, Suwannaroj S, Nanagara R. Pericardial fluid profiles of pericardial effusion in systemic sclerosis patients. Asian Pac J Allergy Immunol 2013; 31(4):314–319. doi:10.12932/AP0305.31.4.2013
- McWhorter JE, LeRoy EC. Pericardial disease in scleroderma (systemic sclerosis). Am J Med 1974; 57(4):566–575. doi:10.1016/0002-9343(74)90008-4
- Comens SM, Alpert MA, Sharp GC, et al. Frequency of mitral valve prolapse in systemic lupus erythematosus, progressive systemic sclerosis and mixed connective tissue disease. Am J Cardiol 1989; 63(5):369–370. doi:10.1016/0002-9149(89)90351-2
- Candell-Riera J, Armadans-Gil L, Simeón CP, et al. Comprehensive noninvasive assessment of cardiac involvement in limited systemic sclerosis. Arthritis Rheum 1996; 39(7):1138–1145. pmid:8670322
- Caforio ALP, Adler Y, Agostini C, et al. Diagnosis and management of myocardial involvement in systemic immune-mediated diseases: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Disease. Eur Heart J 2017; 38(35):2649–2662. doi:10.1093/eurheartj/ehx321
- Mavrogeni S, Karabela G, Koutsogeorgopoulou L, et al. Pseudo-infarction pattern in diffuse systemic sclerosis. Evaluation using cardiovascular magnetic resonance. Int J Cardiol 2016; 214:465–468. doi:10.1016/j.ijcard.2016.03.235
- Ladak K, Pope JE. A review of the effects of statins in systemic sclerosis. Semin Arthritis Rheum 2016; 45(6):698–705. doi:10.1016/j.semarthrit.2015.10.013
- Abou-Raya A, Abou-Raya S, Helmii M. Statins: potentially useful in therapy of systemic sclerosis-related Raynaud’s phenomenon and digital ulcers. J Rheumatol 2008; 35(9):1801–1808. pmid:18709692
- ASCEND Study Collaborative Group; Bowman L, Mafham M, Wallendszus K, et al. Effects of aspirin for primary prevention in persons with diabetes mellitus. N Engl J Med 2018; 379(16):1529–1539. doi:10.1056/NEJMoa1804988
- Guerra F, Stronati G, Fischietti C, et al. Global longitudinal strain measured by speckle tracking identifies subclinical heart involvement in patients with systemic sclerosis. Eur J Prev Cardiol 2018; 25(15):1598–1606. doi:10.1177/2047487318786315
- Mavrogeni SI, Sfikakis PP, Dimitroulas T, et al. Prospects of using cardiovascular magnetic resonance in the identification of arrhythmogenic substrate in autoimmune rheumatic diseases. Rheumatol Int 2018; 38(9):1615–1621. doi:10.1007/s00296-018-4110-5
- Mavrogeni SI, Sfikakis PP, Markousis-Mavrogenis G, et al. Cardiovascular magnetic resonance imaging pattern in patients with autoimmune rheumatic diseases and ventricular tachycardia with preserved ejection fraction. Int J Cardiol 2019; 284:105–109. doi:10.1016/j.ijcard.2018.10.067
Autoimmune rheumatic diseases increase the risk of cardiovascular disease. In rheumatoid arthritis and systemic lupus erythematosus, the risk is driven primarily by the inflammatory milieu, leading to accelerated coronary and cerebrovascular atherosclerosis independent of traditional atherosclerotic risk factors.1–3 The extent of cardiovascular involvement in other rheumatologic diseases has been less well characterized but is an area of growing interest.
In this review, we focus on the cardiovascular complications of systemic sclerosis and review recommendations for monitoring these patients in clinical practice.
SYSTEMIC SCLEROSIS, AN AUTOIMMUNE RHEUMATIC DISEASE
Systemic sclerosis is an autoimmune rheumatic disease characterized by excessive extracellular matrix deposition leading to diffuse fibrosis, endothelial dysfunction, and microvascular injury. It is most common in North America, Southern Europe, and Australia,4,5 and it affects women more than men in ratios ranging from 3:1 to 14:1.6 The mean age at diagnosis is around 50.
The disease can affect the lungs (interstitial lung disease and pulmonary hypertension), the heart, the kidneys, and the gastrointestinal tract.
Systemic sclerosis has 2 main subtypes: limited cutaneous systemic sclerosis, formerly called CREST syndrome) and diffuse cutaneous systemic sclerosis. The limited cutaneous subtype is characterized by tightening of the skin of the distal extremities (below the elbows and knees) and face, while diffuse cutaneous systemic sclerosis can manifest as more extensive skin tightening also involving proximal extremities and the trunk. Both subtypes can have an effect on the cardiovascular system.
Some cardiovascular risk factors such as dyslipidemia, diabetes mellitus, and high body mass index are less common in patients with systemic sclerosis than in patients with rheumatoid arthritis, while the rates of arterial hypertension, smoking, chronic obstructive pulmonary disease, osteoporosis, and neoplasms are similar between the 2 groups.7
HEART INVOLVEMENT HAS SERIOUS CONSEQUENCES
Overt cardiac involvement in systemic sclerosis is associated with a mortality rate of up to 70% over 5 years,8,9 and about one-fourth of deaths in patients with systemic sclerosis are from cardiac causes.10,11 Studies in Europe10,12 showed that many patients with systemic sclerosis have cardiac involvement detectable by magnetic resonance imaging even if they do not have clinical disease. Pulmonary arterial hypertension (PAH) is a complication of both subtypes of systemic sclerosis and portends a higher risk of death.8
Thus, it is critical for clinicians to understand the potential comorbid conditions associated with systemic sclerosis, particularly the cardiovascular ones, and to work closely with cardiologists to help optimize the evaluation and management.
MECHANISMS OF CARDIAC DISEASE IN SYSTEMIC SCLEROSIS
Abnormal vasoreactivity, a consequence of an imbalance between endothelium-derived vasoconstrictors and vasodilators, defective angiogenesis, and endothelial injury, leads to tissue ischemia and vascular endothelial growth factor expression, which initiates injury and fibrosis in the myocardium and in other organs.14–17 Fibrosis involves the myocardium, pericardium, and conduction system.13,18
Myocardial involvement in systemic sclerosis is thought to be due mainly to abnormal vasoreactivity and microvascular abnormalities such as transient coronary artery spasm leading to repeated focal ischemia.19,20 Abnormal vasoreactivity has been demonstrated during cardiac catheterization21: while mean coronary sinus blood flow in systemic sclerosis patients was normal at rest, vasodilator reserve was significantly reduced in patients with diffuse cutaneous systemic sclerosis after maximal vasodilation with dipyridamole. Additionally, endomyocardial biopsy showed fibrosis and concentric intimal hypertrophy with normal epicardial coronary arteries.21
More research into other mechanisms of cardiovascular disease in systemic sclerosis is needed to allow for better preventive care for these patients.
PULMONARY ARTERIAL HYPERTENSION
Systemic sclerosis can be associated with World Health Organization (WHO) groups 1, 2, 3, and 4 pulmonary hypertension. WHO group 1, called pulmonary arterial hypertension or PAH, is one of the most common cardiac complications of systemic sclerosis, with a reported prevalence as high as 12%.22 Systemic sclerosis-associated PAH carries a high mortality rate, with a mean survival of only 3 years.23
With advances in treatments for other complications of systemic sclerosis, the percentage of systemic sclerosis patients who die of PAH has increased from 6% to 33%.24
Compared with patients with idiopathic PAH, those with systemic sclerosis get less of a response from therapy and have poorer outcomes despite lower mean pulmonary artery pressures and similar reductions in cardiac index. However, recent studies have suggested that with aggressive treatment, patients with systemic sclerosis-related PAH can achieve outcomes similar to those with idiopathic PAH.25 Thus, recognizing this condition early is imperative.
Pulmonary arterial hypertension defined
PAH is defined as the combination of all of the following26:
- Mean pulmonary artery pressure > 20 mm Hg at rest
- Normal pulmonary capillary wedge pressure (≤ 15 mm Hg)
- Pulmonary vascular resistance ≥ 3 Wood units on right heart catheterization.
Other causes of pulmonary hypertension such as interstitial lung disease, chronic pulmonary thromboembolic disease, and left heart disease must be excluded.24,27
Remodeling in the pulmonary arteries
The events that lead to PAH in systemic sclerosis remain unclear but are believed to involve initial inflammation or endothelial injury that leads to a dysequilibrium between proliferative mediators and antiproliferative vasodilators. This dysequilibrium, along with endothelial dysfunction, causes an obliterative vasculopathy in the pulmonary artery branches and arterioles. Sympathetic overactivity, hypoxemia, and ischemia-reperfusion injury additionally promote vascular proliferation, fibrosis, and remodeling, leading to increased pulmonary vascular resistance, PAH, and increased right ventricular pressures.23,27
The subtype of systemic sclerosis is an important factor in the development and progression of PAH. PAH appears to be the major cause of death in limited cutaneous systemic sclerosis, while interstitial lung disease is the major cause of death in diffuse cutaneous systemic sclerosis.28
Pulmonary arterial hypertension is a late complication of systemic sclerosis
Data from the South Australian Scleroderma Registry29 revealed that PAH tends to be a late complication of systemic sclerosis, occurring around 20 years after disease onset. In this study of 608 patients, no patient with diffuse cutaneous systemic sclerosis developed PAH.
Systemic sclerosis-related PAH initially follows an indolent course with few symptoms until right ventricular function deteriorates. Early in the disease, patients may experience nonspecific symptoms of fatigue, lightheadedness, and dyspnea on exertion.23 As it progresses, they tend to have worsening dyspnea and may experience exertional syncope, palpitations, and chest pain.
Physical findings may suggest elevated right ventricular pressure and right ventricular failure; these include a loud P2, a prominent jugular a wave, a tricuspid regurgitant murmur, jugular venous distention, and lower-extremity edema.27
Screening for pulmonary arterial hypertension in systemic sclerosis
Significant signs and symptoms usually occur late in the disease; thus, it is important to appropriately screen patients who are at risk so that they can begin aggressive treatment.
Doppler echocardiography is recommended by European and American guidelines to screen for PAH in patients who have systemic sclerosis, and most agree that screening is appropriate even if the patient has no symptoms.30 European consensus documents recommend that transthoracic echocardiography be done annually for the first 5 years of disease and be continued every year in patients at high risk, ie, those with anticentromere antibodies, anti-Th/To antibodies, or interstitial lung disease. Patients not at high risk of developing pulmonary hypertension should also have regular transthoracic echocardiography, though the exact timing is not defined.31 While American societies have not issued corresponding recommendations, many experts follow the European recommendations.
Worrisome features on echocardiography in asymptomatic patients should be followed up with right heart catheterization to assess mean right ventricular pressure. These include:
- Estimated right ventricular systolic pressure ≥ 40 mm Hg
- Tricuspid regurgitant jet velocity > 2.8 m/s
- Right atrial enlargement > 53 mm
- Right ventricular enlargement (mid-cavity dimension > 35 mm).32
Although echocardiography is the most common form of screening, it gives only an estimate of right ventricular systolic pressure, which is imprecise. Other noninvasive markers are helpful and necessary to appropriately screen this population.
Diffusion capacity. The Itinerair study33 found that a diffusing capacity for carbon monoxide (DLCO) of 60% or higher has a high specificity in excluding PAH.
Uric acid has been found to be elevated in patients with systemic sclerosis-related PAH, and levels inversely correlate with 6-minute walking distance.34
Other predictors. N-terminal pro-B-type natriuretic peptide (NT-proBNP), left atrial volume, and the right ventricular myocardial performance index have also been shown to be independent predictors of PAH in patients with systemic sclerosis.35
An algorithm. The DETECT study36 enrolled patients at increased risk who had had systemic sclerosis longer than 3 years and a DLCO less than 60%. The investigators developed a 2-step algorithm to determine which patients should be referred for right heart catheterization to try to detect PAH earlier while minimizing the number of missed diagnoses and optimizing the use of invasive diagnostic right heart catheterization.
The first step was to assess serum values of anticentromere antibodies, NT-proBNP, and urate, and clinical features (telangiectasias), forced vital capacity, and electrocardiographic changes of right axis deviation to derive a prediction score. The second step was to assess surface echocardiographic features of the right atrial area and tricuspid regurgitation velocity.
This approach led to right heart catheterization in 62% of patients and was associated with a false-negative rate of 4%. Importantly, of the patients with PAH, 1 in 5 had no symptoms, and 33% had tricuspid regurgitation velocity less than 2.8 m/s. No single measurement performed well in isolation in this study.37
Thus, we recommend that, in addition to routine surface echocardiography, a multimodal approach be used that includes laboratory testing, clinical features, and electrocardiographic findings when screening this high-risk patient population.
ATHEROSCLEROTIC DISEASES
Although macrovascular disease has not typically been regarded as a significant systemic feature in systemic sclerosis, myocardial infarction and stroke are more common in patients with systemic sclerosis than in controls.38,39
Coronary artery disease in systemic sclerosis
Man et al38 reported that the incidence of myocardial infarction in patients with systemic sclerosis was 4.4 per 1,000 persons per year, and the incidence of stroke was 4.8 per 1,000 persons per year, compared with 2.5 per 1,000 persons per year for both myocardial infarction and stroke in healthy controls matched for age, sex, and time of entry.
The Australian Scleroderma Cohort Study39 found a 3-fold higher prevalence of coronary artery disease in systemic sclerosis patients than in controls after factoring in traditional risk factors.
Aviña-Zubieta et al,40 in a cohort of 1,239 systemic sclerosis patients, estimated a hazard ratio (HR) of 3.49 for myocardial infarction and 2.35 for stroke compared with age- and sex-matched controls. Not all of these events were related to macrovascular atherosclerosis—vasospasm and microvascular ischemia may have played significant roles in the etiology of clinical manifestations.
Studies of coronary atherosclerosis in systemic sclerosis are limited. An autopsy study41 of 58 patients with systemic sclerosis and 58 controls matched for age, sex, and ethnicity found that the prevalence of atherosclerosis of small coronary arteries and arterioles was significantly higher in systemic sclerosis patients than in controls (17% vs 2%, P < .01). However, the prevalence of medium-vessel coronary atherosclerosis was similar (48% vs 43%).
Why patients with systemic sclerosis develop atherosclerosis has not yet been determined. Traditional risk factors such as hypertension, dyslipidemia, diabetes mellitus, and obesity are typically no more prevalent in systemic sclerosis patients than in controls,38,42 and thus do not explain the increased risk of atherosclerotic cardiovascular disease. There is some evidence that novel markers of atherosclerotic risk such as homocysteine,43 lipoprotein[a],44 and oxidized low-density lipoprotein45 are more prevalent in systemic sclerosis, but these results have not been substantiated in more extensive studies.
Peripheral artery disease
It remains unclear whether peripheral artery disease is more prevalent in systemic sclerosis patients than in controls.
Individual studies have shown mixed results in comparing carotid artery stenosis between systemic sclerosis patients and controls using carotid duplex ultrasonography,46 the ankle-brachial index,46–48 carotid intima-media thickness,49–54 and brachial flow-mediated dilation.51,53,55–58 A meta-analysis found that the carotid intima and media are significantly thicker in systemic sclerosis patients than in controls,59 and the magnitude of difference is similar to that in other groups at increased cardiovascular risk, such as those with rheumatoid arthritis, diabetes, and familial hypercholesterolemia.60–63
A meta-analysis of brachial artery findings showed significantly lower flow-mediated dilation in systemic sclerosis patients than in controls.64
Overall, given the inconsistency of study results, systemic sclerosis patients should be screened and managed as in other patients with peripheral artery disease, but the clinician should be aware that there may be a higher risk of peripheral artery disease in these patients.
RIGHT AND LEFT VENTRICULAR DYSFUNCTION
Many patients with systemic sclerosis have right ventricular dysfunction as a consequence of PAH.65 It is important to detect diastolic dysfunction in this population, as it may be an even stronger predictor of death than pulmonary hypertension on right heart catheterization (HR 3.7 vs 2.0).66
Fewer patients have left ventricular dysfunction. In a multicenter study of 570 systemic sclerosis patients, only 1.4% had left ventricular systolic dysfunction on echocardiography, though 22.6% had left ventricular hypertrophy and 17.7% had left ventricular diastolic dysfunction.67 In the European League Against Rheumatism (EULAR) database, the prevalence of reduced left ventricular ejection fraction was 5.4%.68
Though traditional echocardiographic screening suggests the prevalence of left ventricular dysfunction in systemic sclerosis patients is low, cardiac magnetic resonance imaging (MRI) may be more sensitive than echocardiography for detecting subclinical myocardial involvement. Cardiac MRI has been shown to detect evidence of myocardial pathology (increased T2 signal, left ventricular thinning, pericardial effusion, reduced left ventricular and right ventricular ejection fraction, left ventricular diastolic dysfunction, and delayed myocardial contrast enhancement) in up to 75% of systemic sclerosis cases studied.69
Patients with systemic sclerosis should already be undergoing echocardiography every year to screen for PAH, and screening should also include tissue Doppler imaging to detect various forms of left and right ventricular systolic and diastolic dysfunction that may not be clinically apparent.
Though cardiac MRI can provide useful additional information, it is not currently recommended for routine screening in patients with systemic sclerosis.
ARRHYTHMIAS AND CONDUCTION DEFECTS
Patients with systemic sclerosis are prone to arrhythmias due to both conduction system fibrosis and myocardial damage.
Arrhythmias accounted for 6% of the deaths in the EULAR Scleroderma Trials and Research (EUSTAR) database.11
In the Genetics Versus Environment in Scleroderma Outcome Study (GENISOS),70 250 patients who had had systemic sclerosis for at least 3 years were studied during a period of approximately 6 years, during which there were 52 deaths, 29 of which were directly attributable to systemic sclerosis. Multivariable Cox modeling showed that 7 variables predicted mortality:
- Body mass index < 18.5 kg/m2
- Age ≥ 65
- Forced vital capacity < 50% predicted
- Systolic blood pressure ≥ 140 or diastolic blood pressure ≥ 90 mm Hg
- Pulmonary fibrosis
- Positive anticentromere antibodies
- Cardiac arrhythmias.
The hazard ratio for death in patients with arrhythmias in this model was 2.18 (95% CI 1.05–4.50, P = .035). Thus, finding arrhythmias in systemic sclerosis patients can provide important prognostic information.
While resting electrocardiography in patients with systemic sclerosis most commonly shows sinus rhythm, 24-hour electrocardiographic monitoring has revealed nonsustained supraventricular and ventricular arrhythmias in a significant percentage.71,72 Although difficult to quantify in routine practice, parameters controlled by the autonomic nervous system including heart rate variability and heart rate turbulence have been shown to be impaired in systemic sclerosis, and these measures are associated with an increased risk of malignant arrhythmias and sudden cardiac death.73,74
Conduction abnormalities
Conduction abnormalities occur in one-fifth to one-third of patients with systemic sclerosis.75,76 The most common abnormal conduction finding is left bundle branch block, followed by first-degree atrioventricular block. High-degree atrioventricular block is uncommon,76 though a few case reports of complete heart block thought to be related to systemic sclerosis have been published.77–79 An autopsy study showed that the conduction system is relatively spared from myocardial changes seen in systemic sclerosis patients, and thus it is speculated that the conduction disturbances are a consequence of damaged myocardium rather than damage to conduction tissue.80
Given the array of electrophysiologic abnormalities that systemic sclerosis patients can have, it is critical to monitor all patients with routine (annual or biannual) electrocardiography; to take possible arrhythmia-related symptoms seriously; and to evaluate them with further workup such as Holter monitoring for 24 hours or even longer, event monitoring, exercise testing, or tilt-table testing.
PERICARDIAL DISEASE
Pericardial disease is clinically apparent in 5% to 16% of patients with systemic sclerosis81; patients with limited cutaneous systemic sclerosis have more pericardial disease than those with diffuse cutaneous systemic sclerosis (30% vs 16%).82 Forty-one percent of systemic sclerosis patients have been shown to have pericardial effusion by echocardiography,81 but the effusions are typically small and rarely cause tamponade, though tamponade is associated with a poor prognosis.
Large pericardial effusions can develop before skin thickening and diagnosis of systemic sclerosis.81,83,84 Thus, systemic sclerosis should be considered in patients with pericardial effusions of unknown etiology.
In a small study,85 the pericardial fluid in systemic sclerosis was typically exudative, with lactate dehydrogenase greater than 200 U/L, a fluid-serum lactate dehydrogenase ratio greater than 0.6, and a fluid-serum total protein ratio greater than 0.5.
Pericardial effusion can be a sign of impending scleroderma renal crisis,86 and thus renal function should be carefully monitored in systemic sclerosis patients with pericardial effusion. Constrictive pericarditis and restrictive cardiomyopathy can rarely occur in systemic sclerosis and may more commonly present with symptoms.
Pericardial disease in systemic sclerosis should be treated in a standard fashion with nonsteroidal anti-inflammatory drugs. Corticosteroids are generally of limited benefit and should be avoided, especially in the setting of scleroderma renal crisis.81
VALVULAR HEART DISEASE
Based on limited studies, the prevalence of significant valvular heart disease in systemic sclerosis patients does not seem to be higher than that in the general population. While patients with systemic sclerosis and CREST syndrome (calcinosis, Raynaud phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasia) have been shown to have a higher frequency of mitral valve prolapse and mild mitral regurgitation,87,88 these abnormalities do not often progress in severity, and thus their clinical significance is limited.
RECOMMENDATIONS FOR CARE OF SYSTEMIC SCLEROSIS PATIENTS
It is important for physicians caring for patients with systemic sclerosis to be aware of its most common cardiac manifestations, including left and right ventricular systolic and diastolic dysfunction, pulmonary hypertension, conduction abnormalities, arrhythmias, and cardiomyopathy.
Look for volume overload
On clinical examination, assess for clinical markers of volume overload such as distended neck veins, peripheral edema, or an abnormal blood pressure response to the Valsalva maneuver. These findings should prompt measurement of NT-proBNP,89 and may warrant prescription of a diuretic.
Electrocardiography to investigate arrhythmias
Electrocardiography should be done if patients describe symptoms of palpitations, and should also include continuous rhythm monitoring with Holter or event monitoring, depending on the frequency of symptoms. Otherwise, patients should routinely undergo electrocardiography once or twice a year.
Q waves are common in systemic sclerosis patients (especially those with diffuse cutaneous systemic sclerosis), notably in the precordial leads, and can occur without coronary artery disease.90 Symptoms such as presyncope should be further investigated with Holter monitoring and tilt-table testing.
Assess, modify traditional risk factors
Subclinical atherosclerosis as detected by carotid intima-media thickness is as common in systemic sclerosis as in rheumatoid arthritis.61 However, traditional risk indices such as SCORE (Systematic Coronary Risk Evaluation), QRISK2, and the American College of Cardiology/American Heart Association indices may underestimate risk in patients who have systemic sclerosis.
Strict hypertension control should be the goal for all systemic sclerosis patients. Though there are no specific guidelines on which antihypertensive medications are preferred, calcium channel blockers or angiotensin II receptor blockers, which are typically used to treat systemic sclerosis-related Raynaud phenomenon, may be appropriate.
Statins reduce vascular complications and are generally well tolerated in patients with systemic sclerosis.91,92
Aspirin is not recommended for routine primary prevention in view of data suggesting that its benefits in diabetic patients are counterbalanced by increased bleeding risk.93
Echocardiography to detect pulmonary arterial hypertension
At this time, guidelines for monitoring for cardiovascular manifestations in systemic sclerosis patients are limited. The only well-defined ones are European consensus guidelines, which suggest annual transthoracic echocardiography for the first 5 years after systemic sclerosis is diagnosed and continued annual screening in patients at risk of developing PAH.31
We support this strategy, with annual screening for the first 5 years followed by surveillance echocardiography every 2 to 3 years unless there is a high risk of PAH. Specific attention should be paid to right ventricular diastolic function, right atrial volume, and right ventricular myocardial performance index.
Emerging data suggest that the addition of global longitudinal strain of ventricles to routine echocardiography can help detect subclinical cardiac risk.94 Although further study is needed into the predictive value of global longitudinal strain, it is a low-cost and noninvasive addition to standard echocardiography that can help guide risk stratification, and thus we recommend that it be part of the echocardiographic examination for all systemic sclerosis patients.
Pulmonary function testing. In addition to screening for PAH with echocardiography, we recommend obtaining baseline pulmonary function tests, including DLCO, at the time systemic sclerosis is diagnosed, with repeat testing annually.
Magnetic resonance imaging
While echocardiography is the gold standard for monitoring systemic sclerosis patients, cardiovascular MRI may have a role in identifying those at higher risk of dangerous arrhythmias such as ventricular tachycardia and ventricular fibrillation. In addition to assessing ventricular function, MRI can detect myocardial inflammation, ischemia, and fibrosis that may predispose a patient to develop ventricular tachycardia or fibrillation.95 Variables such as T1/T2 mapping, extracellular volume fraction, T2 signal ratio, and early vs late gadolinium enhancement can help identify patients who had past ventricular tachycardia or fibrillation.96
Finding an increased risk of arrhythmias may prompt a conversation between the patient and the physician about the need for an implantable cardiac defibrillator.
If cardiac MRI is available and is reimbursed by the patient’s insurance carrier, physicians should strongly consider obtaining at least one baseline scan in systemic sclerosis patients to identify those at risk of highly fatal arrhythmias.
Teamwork is needed
Systemic sclerosis has not traditionally been associated with cardiovascular disease to the extent of other rheumatic conditions, but the cardiovascular system can be affected in various ways that can ultimately lead to an early death. These manifestations may be asymptomatic for long periods, and overt clinical disease portends a poorer prognosis.
Primary care physicians managing these patients should be aware of the cardiovascular complications of systemic sclerosis and should implement appropriate screening tests in conjunction with rheumatologists and cardiologists. It is also essential for general and subspecialty cardiologists to understand the broad spectrum of organ system involvement that can affect systemic sclerosis patients and to tailor their investigation and management recommendations accordingly. By designing a multidisciplinary approach to the treatment of systemic sclerosis patients, physicians can help to optimize cardiovascular risk modification in this vulnerable population.
Autoimmune rheumatic diseases increase the risk of cardiovascular disease. In rheumatoid arthritis and systemic lupus erythematosus, the risk is driven primarily by the inflammatory milieu, leading to accelerated coronary and cerebrovascular atherosclerosis independent of traditional atherosclerotic risk factors.1–3 The extent of cardiovascular involvement in other rheumatologic diseases has been less well characterized but is an area of growing interest.
In this review, we focus on the cardiovascular complications of systemic sclerosis and review recommendations for monitoring these patients in clinical practice.
SYSTEMIC SCLEROSIS, AN AUTOIMMUNE RHEUMATIC DISEASE
Systemic sclerosis is an autoimmune rheumatic disease characterized by excessive extracellular matrix deposition leading to diffuse fibrosis, endothelial dysfunction, and microvascular injury. It is most common in North America, Southern Europe, and Australia,4,5 and it affects women more than men in ratios ranging from 3:1 to 14:1.6 The mean age at diagnosis is around 50.
The disease can affect the lungs (interstitial lung disease and pulmonary hypertension), the heart, the kidneys, and the gastrointestinal tract.
Systemic sclerosis has 2 main subtypes: limited cutaneous systemic sclerosis, formerly called CREST syndrome) and diffuse cutaneous systemic sclerosis. The limited cutaneous subtype is characterized by tightening of the skin of the distal extremities (below the elbows and knees) and face, while diffuse cutaneous systemic sclerosis can manifest as more extensive skin tightening also involving proximal extremities and the trunk. Both subtypes can have an effect on the cardiovascular system.
Some cardiovascular risk factors such as dyslipidemia, diabetes mellitus, and high body mass index are less common in patients with systemic sclerosis than in patients with rheumatoid arthritis, while the rates of arterial hypertension, smoking, chronic obstructive pulmonary disease, osteoporosis, and neoplasms are similar between the 2 groups.7
HEART INVOLVEMENT HAS SERIOUS CONSEQUENCES
Overt cardiac involvement in systemic sclerosis is associated with a mortality rate of up to 70% over 5 years,8,9 and about one-fourth of deaths in patients with systemic sclerosis are from cardiac causes.10,11 Studies in Europe10,12 showed that many patients with systemic sclerosis have cardiac involvement detectable by magnetic resonance imaging even if they do not have clinical disease. Pulmonary arterial hypertension (PAH) is a complication of both subtypes of systemic sclerosis and portends a higher risk of death.8
Thus, it is critical for clinicians to understand the potential comorbid conditions associated with systemic sclerosis, particularly the cardiovascular ones, and to work closely with cardiologists to help optimize the evaluation and management.
MECHANISMS OF CARDIAC DISEASE IN SYSTEMIC SCLEROSIS
Abnormal vasoreactivity, a consequence of an imbalance between endothelium-derived vasoconstrictors and vasodilators, defective angiogenesis, and endothelial injury, leads to tissue ischemia and vascular endothelial growth factor expression, which initiates injury and fibrosis in the myocardium and in other organs.14–17 Fibrosis involves the myocardium, pericardium, and conduction system.13,18
Myocardial involvement in systemic sclerosis is thought to be due mainly to abnormal vasoreactivity and microvascular abnormalities such as transient coronary artery spasm leading to repeated focal ischemia.19,20 Abnormal vasoreactivity has been demonstrated during cardiac catheterization21: while mean coronary sinus blood flow in systemic sclerosis patients was normal at rest, vasodilator reserve was significantly reduced in patients with diffuse cutaneous systemic sclerosis after maximal vasodilation with dipyridamole. Additionally, endomyocardial biopsy showed fibrosis and concentric intimal hypertrophy with normal epicardial coronary arteries.21
More research into other mechanisms of cardiovascular disease in systemic sclerosis is needed to allow for better preventive care for these patients.
PULMONARY ARTERIAL HYPERTENSION
Systemic sclerosis can be associated with World Health Organization (WHO) groups 1, 2, 3, and 4 pulmonary hypertension. WHO group 1, called pulmonary arterial hypertension or PAH, is one of the most common cardiac complications of systemic sclerosis, with a reported prevalence as high as 12%.22 Systemic sclerosis-associated PAH carries a high mortality rate, with a mean survival of only 3 years.23
With advances in treatments for other complications of systemic sclerosis, the percentage of systemic sclerosis patients who die of PAH has increased from 6% to 33%.24
Compared with patients with idiopathic PAH, those with systemic sclerosis get less of a response from therapy and have poorer outcomes despite lower mean pulmonary artery pressures and similar reductions in cardiac index. However, recent studies have suggested that with aggressive treatment, patients with systemic sclerosis-related PAH can achieve outcomes similar to those with idiopathic PAH.25 Thus, recognizing this condition early is imperative.
Pulmonary arterial hypertension defined
PAH is defined as the combination of all of the following26:
- Mean pulmonary artery pressure > 20 mm Hg at rest
- Normal pulmonary capillary wedge pressure (≤ 15 mm Hg)
- Pulmonary vascular resistance ≥ 3 Wood units on right heart catheterization.
Other causes of pulmonary hypertension such as interstitial lung disease, chronic pulmonary thromboembolic disease, and left heart disease must be excluded.24,27
Remodeling in the pulmonary arteries
The events that lead to PAH in systemic sclerosis remain unclear but are believed to involve initial inflammation or endothelial injury that leads to a dysequilibrium between proliferative mediators and antiproliferative vasodilators. This dysequilibrium, along with endothelial dysfunction, causes an obliterative vasculopathy in the pulmonary artery branches and arterioles. Sympathetic overactivity, hypoxemia, and ischemia-reperfusion injury additionally promote vascular proliferation, fibrosis, and remodeling, leading to increased pulmonary vascular resistance, PAH, and increased right ventricular pressures.23,27
The subtype of systemic sclerosis is an important factor in the development and progression of PAH. PAH appears to be the major cause of death in limited cutaneous systemic sclerosis, while interstitial lung disease is the major cause of death in diffuse cutaneous systemic sclerosis.28
Pulmonary arterial hypertension is a late complication of systemic sclerosis
Data from the South Australian Scleroderma Registry29 revealed that PAH tends to be a late complication of systemic sclerosis, occurring around 20 years after disease onset. In this study of 608 patients, no patient with diffuse cutaneous systemic sclerosis developed PAH.
Systemic sclerosis-related PAH initially follows an indolent course with few symptoms until right ventricular function deteriorates. Early in the disease, patients may experience nonspecific symptoms of fatigue, lightheadedness, and dyspnea on exertion.23 As it progresses, they tend to have worsening dyspnea and may experience exertional syncope, palpitations, and chest pain.
Physical findings may suggest elevated right ventricular pressure and right ventricular failure; these include a loud P2, a prominent jugular a wave, a tricuspid regurgitant murmur, jugular venous distention, and lower-extremity edema.27
Screening for pulmonary arterial hypertension in systemic sclerosis
Significant signs and symptoms usually occur late in the disease; thus, it is important to appropriately screen patients who are at risk so that they can begin aggressive treatment.
Doppler echocardiography is recommended by European and American guidelines to screen for PAH in patients who have systemic sclerosis, and most agree that screening is appropriate even if the patient has no symptoms.30 European consensus documents recommend that transthoracic echocardiography be done annually for the first 5 years of disease and be continued every year in patients at high risk, ie, those with anticentromere antibodies, anti-Th/To antibodies, or interstitial lung disease. Patients not at high risk of developing pulmonary hypertension should also have regular transthoracic echocardiography, though the exact timing is not defined.31 While American societies have not issued corresponding recommendations, many experts follow the European recommendations.
Worrisome features on echocardiography in asymptomatic patients should be followed up with right heart catheterization to assess mean right ventricular pressure. These include:
- Estimated right ventricular systolic pressure ≥ 40 mm Hg
- Tricuspid regurgitant jet velocity > 2.8 m/s
- Right atrial enlargement > 53 mm
- Right ventricular enlargement (mid-cavity dimension > 35 mm).32
Although echocardiography is the most common form of screening, it gives only an estimate of right ventricular systolic pressure, which is imprecise. Other noninvasive markers are helpful and necessary to appropriately screen this population.
Diffusion capacity. The Itinerair study33 found that a diffusing capacity for carbon monoxide (DLCO) of 60% or higher has a high specificity in excluding PAH.
Uric acid has been found to be elevated in patients with systemic sclerosis-related PAH, and levels inversely correlate with 6-minute walking distance.34
Other predictors. N-terminal pro-B-type natriuretic peptide (NT-proBNP), left atrial volume, and the right ventricular myocardial performance index have also been shown to be independent predictors of PAH in patients with systemic sclerosis.35
An algorithm. The DETECT study36 enrolled patients at increased risk who had had systemic sclerosis longer than 3 years and a DLCO less than 60%. The investigators developed a 2-step algorithm to determine which patients should be referred for right heart catheterization to try to detect PAH earlier while minimizing the number of missed diagnoses and optimizing the use of invasive diagnostic right heart catheterization.
The first step was to assess serum values of anticentromere antibodies, NT-proBNP, and urate, and clinical features (telangiectasias), forced vital capacity, and electrocardiographic changes of right axis deviation to derive a prediction score. The second step was to assess surface echocardiographic features of the right atrial area and tricuspid regurgitation velocity.
This approach led to right heart catheterization in 62% of patients and was associated with a false-negative rate of 4%. Importantly, of the patients with PAH, 1 in 5 had no symptoms, and 33% had tricuspid regurgitation velocity less than 2.8 m/s. No single measurement performed well in isolation in this study.37
Thus, we recommend that, in addition to routine surface echocardiography, a multimodal approach be used that includes laboratory testing, clinical features, and electrocardiographic findings when screening this high-risk patient population.
ATHEROSCLEROTIC DISEASES
Although macrovascular disease has not typically been regarded as a significant systemic feature in systemic sclerosis, myocardial infarction and stroke are more common in patients with systemic sclerosis than in controls.38,39
Coronary artery disease in systemic sclerosis
Man et al38 reported that the incidence of myocardial infarction in patients with systemic sclerosis was 4.4 per 1,000 persons per year, and the incidence of stroke was 4.8 per 1,000 persons per year, compared with 2.5 per 1,000 persons per year for both myocardial infarction and stroke in healthy controls matched for age, sex, and time of entry.
The Australian Scleroderma Cohort Study39 found a 3-fold higher prevalence of coronary artery disease in systemic sclerosis patients than in controls after factoring in traditional risk factors.
Aviña-Zubieta et al,40 in a cohort of 1,239 systemic sclerosis patients, estimated a hazard ratio (HR) of 3.49 for myocardial infarction and 2.35 for stroke compared with age- and sex-matched controls. Not all of these events were related to macrovascular atherosclerosis—vasospasm and microvascular ischemia may have played significant roles in the etiology of clinical manifestations.
Studies of coronary atherosclerosis in systemic sclerosis are limited. An autopsy study41 of 58 patients with systemic sclerosis and 58 controls matched for age, sex, and ethnicity found that the prevalence of atherosclerosis of small coronary arteries and arterioles was significantly higher in systemic sclerosis patients than in controls (17% vs 2%, P < .01). However, the prevalence of medium-vessel coronary atherosclerosis was similar (48% vs 43%).
Why patients with systemic sclerosis develop atherosclerosis has not yet been determined. Traditional risk factors such as hypertension, dyslipidemia, diabetes mellitus, and obesity are typically no more prevalent in systemic sclerosis patients than in controls,38,42 and thus do not explain the increased risk of atherosclerotic cardiovascular disease. There is some evidence that novel markers of atherosclerotic risk such as homocysteine,43 lipoprotein[a],44 and oxidized low-density lipoprotein45 are more prevalent in systemic sclerosis, but these results have not been substantiated in more extensive studies.
Peripheral artery disease
It remains unclear whether peripheral artery disease is more prevalent in systemic sclerosis patients than in controls.
Individual studies have shown mixed results in comparing carotid artery stenosis between systemic sclerosis patients and controls using carotid duplex ultrasonography,46 the ankle-brachial index,46–48 carotid intima-media thickness,49–54 and brachial flow-mediated dilation.51,53,55–58 A meta-analysis found that the carotid intima and media are significantly thicker in systemic sclerosis patients than in controls,59 and the magnitude of difference is similar to that in other groups at increased cardiovascular risk, such as those with rheumatoid arthritis, diabetes, and familial hypercholesterolemia.60–63
A meta-analysis of brachial artery findings showed significantly lower flow-mediated dilation in systemic sclerosis patients than in controls.64
Overall, given the inconsistency of study results, systemic sclerosis patients should be screened and managed as in other patients with peripheral artery disease, but the clinician should be aware that there may be a higher risk of peripheral artery disease in these patients.
RIGHT AND LEFT VENTRICULAR DYSFUNCTION
Many patients with systemic sclerosis have right ventricular dysfunction as a consequence of PAH.65 It is important to detect diastolic dysfunction in this population, as it may be an even stronger predictor of death than pulmonary hypertension on right heart catheterization (HR 3.7 vs 2.0).66
Fewer patients have left ventricular dysfunction. In a multicenter study of 570 systemic sclerosis patients, only 1.4% had left ventricular systolic dysfunction on echocardiography, though 22.6% had left ventricular hypertrophy and 17.7% had left ventricular diastolic dysfunction.67 In the European League Against Rheumatism (EULAR) database, the prevalence of reduced left ventricular ejection fraction was 5.4%.68
Though traditional echocardiographic screening suggests the prevalence of left ventricular dysfunction in systemic sclerosis patients is low, cardiac magnetic resonance imaging (MRI) may be more sensitive than echocardiography for detecting subclinical myocardial involvement. Cardiac MRI has been shown to detect evidence of myocardial pathology (increased T2 signal, left ventricular thinning, pericardial effusion, reduced left ventricular and right ventricular ejection fraction, left ventricular diastolic dysfunction, and delayed myocardial contrast enhancement) in up to 75% of systemic sclerosis cases studied.69
Patients with systemic sclerosis should already be undergoing echocardiography every year to screen for PAH, and screening should also include tissue Doppler imaging to detect various forms of left and right ventricular systolic and diastolic dysfunction that may not be clinically apparent.
Though cardiac MRI can provide useful additional information, it is not currently recommended for routine screening in patients with systemic sclerosis.
ARRHYTHMIAS AND CONDUCTION DEFECTS
Patients with systemic sclerosis are prone to arrhythmias due to both conduction system fibrosis and myocardial damage.
Arrhythmias accounted for 6% of the deaths in the EULAR Scleroderma Trials and Research (EUSTAR) database.11
In the Genetics Versus Environment in Scleroderma Outcome Study (GENISOS),70 250 patients who had had systemic sclerosis for at least 3 years were studied during a period of approximately 6 years, during which there were 52 deaths, 29 of which were directly attributable to systemic sclerosis. Multivariable Cox modeling showed that 7 variables predicted mortality:
- Body mass index < 18.5 kg/m2
- Age ≥ 65
- Forced vital capacity < 50% predicted
- Systolic blood pressure ≥ 140 or diastolic blood pressure ≥ 90 mm Hg
- Pulmonary fibrosis
- Positive anticentromere antibodies
- Cardiac arrhythmias.
The hazard ratio for death in patients with arrhythmias in this model was 2.18 (95% CI 1.05–4.50, P = .035). Thus, finding arrhythmias in systemic sclerosis patients can provide important prognostic information.
While resting electrocardiography in patients with systemic sclerosis most commonly shows sinus rhythm, 24-hour electrocardiographic monitoring has revealed nonsustained supraventricular and ventricular arrhythmias in a significant percentage.71,72 Although difficult to quantify in routine practice, parameters controlled by the autonomic nervous system including heart rate variability and heart rate turbulence have been shown to be impaired in systemic sclerosis, and these measures are associated with an increased risk of malignant arrhythmias and sudden cardiac death.73,74
Conduction abnormalities
Conduction abnormalities occur in one-fifth to one-third of patients with systemic sclerosis.75,76 The most common abnormal conduction finding is left bundle branch block, followed by first-degree atrioventricular block. High-degree atrioventricular block is uncommon,76 though a few case reports of complete heart block thought to be related to systemic sclerosis have been published.77–79 An autopsy study showed that the conduction system is relatively spared from myocardial changes seen in systemic sclerosis patients, and thus it is speculated that the conduction disturbances are a consequence of damaged myocardium rather than damage to conduction tissue.80
Given the array of electrophysiologic abnormalities that systemic sclerosis patients can have, it is critical to monitor all patients with routine (annual or biannual) electrocardiography; to take possible arrhythmia-related symptoms seriously; and to evaluate them with further workup such as Holter monitoring for 24 hours or even longer, event monitoring, exercise testing, or tilt-table testing.
PERICARDIAL DISEASE
Pericardial disease is clinically apparent in 5% to 16% of patients with systemic sclerosis81; patients with limited cutaneous systemic sclerosis have more pericardial disease than those with diffuse cutaneous systemic sclerosis (30% vs 16%).82 Forty-one percent of systemic sclerosis patients have been shown to have pericardial effusion by echocardiography,81 but the effusions are typically small and rarely cause tamponade, though tamponade is associated with a poor prognosis.
Large pericardial effusions can develop before skin thickening and diagnosis of systemic sclerosis.81,83,84 Thus, systemic sclerosis should be considered in patients with pericardial effusions of unknown etiology.
In a small study,85 the pericardial fluid in systemic sclerosis was typically exudative, with lactate dehydrogenase greater than 200 U/L, a fluid-serum lactate dehydrogenase ratio greater than 0.6, and a fluid-serum total protein ratio greater than 0.5.
Pericardial effusion can be a sign of impending scleroderma renal crisis,86 and thus renal function should be carefully monitored in systemic sclerosis patients with pericardial effusion. Constrictive pericarditis and restrictive cardiomyopathy can rarely occur in systemic sclerosis and may more commonly present with symptoms.
Pericardial disease in systemic sclerosis should be treated in a standard fashion with nonsteroidal anti-inflammatory drugs. Corticosteroids are generally of limited benefit and should be avoided, especially in the setting of scleroderma renal crisis.81
VALVULAR HEART DISEASE
Based on limited studies, the prevalence of significant valvular heart disease in systemic sclerosis patients does not seem to be higher than that in the general population. While patients with systemic sclerosis and CREST syndrome (calcinosis, Raynaud phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasia) have been shown to have a higher frequency of mitral valve prolapse and mild mitral regurgitation,87,88 these abnormalities do not often progress in severity, and thus their clinical significance is limited.
RECOMMENDATIONS FOR CARE OF SYSTEMIC SCLEROSIS PATIENTS
It is important for physicians caring for patients with systemic sclerosis to be aware of its most common cardiac manifestations, including left and right ventricular systolic and diastolic dysfunction, pulmonary hypertension, conduction abnormalities, arrhythmias, and cardiomyopathy.
Look for volume overload
On clinical examination, assess for clinical markers of volume overload such as distended neck veins, peripheral edema, or an abnormal blood pressure response to the Valsalva maneuver. These findings should prompt measurement of NT-proBNP,89 and may warrant prescription of a diuretic.
Electrocardiography to investigate arrhythmias
Electrocardiography should be done if patients describe symptoms of palpitations, and should also include continuous rhythm monitoring with Holter or event monitoring, depending on the frequency of symptoms. Otherwise, patients should routinely undergo electrocardiography once or twice a year.
Q waves are common in systemic sclerosis patients (especially those with diffuse cutaneous systemic sclerosis), notably in the precordial leads, and can occur without coronary artery disease.90 Symptoms such as presyncope should be further investigated with Holter monitoring and tilt-table testing.
Assess, modify traditional risk factors
Subclinical atherosclerosis as detected by carotid intima-media thickness is as common in systemic sclerosis as in rheumatoid arthritis.61 However, traditional risk indices such as SCORE (Systematic Coronary Risk Evaluation), QRISK2, and the American College of Cardiology/American Heart Association indices may underestimate risk in patients who have systemic sclerosis.
Strict hypertension control should be the goal for all systemic sclerosis patients. Though there are no specific guidelines on which antihypertensive medications are preferred, calcium channel blockers or angiotensin II receptor blockers, which are typically used to treat systemic sclerosis-related Raynaud phenomenon, may be appropriate.
Statins reduce vascular complications and are generally well tolerated in patients with systemic sclerosis.91,92
Aspirin is not recommended for routine primary prevention in view of data suggesting that its benefits in diabetic patients are counterbalanced by increased bleeding risk.93
Echocardiography to detect pulmonary arterial hypertension
At this time, guidelines for monitoring for cardiovascular manifestations in systemic sclerosis patients are limited. The only well-defined ones are European consensus guidelines, which suggest annual transthoracic echocardiography for the first 5 years after systemic sclerosis is diagnosed and continued annual screening in patients at risk of developing PAH.31
We support this strategy, with annual screening for the first 5 years followed by surveillance echocardiography every 2 to 3 years unless there is a high risk of PAH. Specific attention should be paid to right ventricular diastolic function, right atrial volume, and right ventricular myocardial performance index.
Emerging data suggest that the addition of global longitudinal strain of ventricles to routine echocardiography can help detect subclinical cardiac risk.94 Although further study is needed into the predictive value of global longitudinal strain, it is a low-cost and noninvasive addition to standard echocardiography that can help guide risk stratification, and thus we recommend that it be part of the echocardiographic examination for all systemic sclerosis patients.
Pulmonary function testing. In addition to screening for PAH with echocardiography, we recommend obtaining baseline pulmonary function tests, including DLCO, at the time systemic sclerosis is diagnosed, with repeat testing annually.
Magnetic resonance imaging
While echocardiography is the gold standard for monitoring systemic sclerosis patients, cardiovascular MRI may have a role in identifying those at higher risk of dangerous arrhythmias such as ventricular tachycardia and ventricular fibrillation. In addition to assessing ventricular function, MRI can detect myocardial inflammation, ischemia, and fibrosis that may predispose a patient to develop ventricular tachycardia or fibrillation.95 Variables such as T1/T2 mapping, extracellular volume fraction, T2 signal ratio, and early vs late gadolinium enhancement can help identify patients who had past ventricular tachycardia or fibrillation.96
Finding an increased risk of arrhythmias may prompt a conversation between the patient and the physician about the need for an implantable cardiac defibrillator.
If cardiac MRI is available and is reimbursed by the patient’s insurance carrier, physicians should strongly consider obtaining at least one baseline scan in systemic sclerosis patients to identify those at risk of highly fatal arrhythmias.
Teamwork is needed
Systemic sclerosis has not traditionally been associated with cardiovascular disease to the extent of other rheumatic conditions, but the cardiovascular system can be affected in various ways that can ultimately lead to an early death. These manifestations may be asymptomatic for long periods, and overt clinical disease portends a poorer prognosis.
Primary care physicians managing these patients should be aware of the cardiovascular complications of systemic sclerosis and should implement appropriate screening tests in conjunction with rheumatologists and cardiologists. It is also essential for general and subspecialty cardiologists to understand the broad spectrum of organ system involvement that can affect systemic sclerosis patients and to tailor their investigation and management recommendations accordingly. By designing a multidisciplinary approach to the treatment of systemic sclerosis patients, physicians can help to optimize cardiovascular risk modification in this vulnerable population.
- Maradit-Kremers H, Crowson CS, Nicola PJ, et al. Increased unrecognized coronary heart disease and sudden deaths in rheumatoid arthritis: a population-based cohort study. Arthritis Rheum 2005; 52(2):402–411. doi:10.1002/art.20853
- Naranjo A, Sokka T, Descalzo MA, et al; QUEST-RA Group. Cardiovascular disease in patients with rheumatoid arthritis: results from the QUEST-RA study. Arthritis Res Ther 2008; 10(2):R30. doi:10.1186/ar2383
- Innala L, Möller B, Ljung L, et al. Cardiovascular events in early RA are a result of inflammatory burden and traditional risk factors: a five year prospective study. Arthritis Res Ther 2011; 13(4):R131. doi:10.1186/ar3442
- Barnes J, Mayes MD. Epidemiology of systemic sclerosis: incidence, prevalence, survival, risk factors, malignancy, and environmental triggers. Curr Opin Rheumatol 2012; 24(2):165–170. doi:10.1097/BOR.0b013e32834ff2e8
- Chifflot H, Fautrel B, Sordet C, Chatelus E, Sibilia J. Incidence and prevalence of systemic sclerosis: a systematic literature review. Semin Arthritis Rheum 2008; 37(4):223–235 doi:10.1016/j.semarthrit.2007.05.003
- Gabrielli A, Avvedimento EV, Krieg T. Scleroderma. N Engl J Med 2009; 360(19):1989–2003. doi:10.1056/NEJMra0806188
- Panopoulos S, Tektonidou M, Drosos AA, et al. Prevalence of comorbidities in systemic sclerosis versus rheumatoid arthritis: a comparative, multicenter, matched-cohort study. Arthritis Res Ther 2018; 20(1):267. doi:10.1186/s13075-018-1771-0
- Ferri C, Valentini G, Cozzi F, et al. Systemic sclerosis: demographic, clinical, and serologic features and survival in 1,012 Italian patients. Medicine (Baltimore) 2002; 81(8):139–153. doi:10.1097/00005792-200203000-00004
- Steen VD, Medsger TA Jr. Severe organ involvement in systemic sclerosis with diffuse scleroderma. Arthritis Rheum 2000; 43(11):2437–2444. doi:10.1002/1529-0131(200011)43:11<2437::AID-ANR10>3.0.CO;2-U
- Hachulla AL, Launay D, Gaxotte V, et al. Cardiac magnetic resonance imaging in systemic sclerosis: a cross-sectional observational study of 52 patients. Ann Rheum Dis 2009; 68(12):1878–1884. doi:10.1136/ard.2008.095836
- Tyndall AJ, Bannert B, Vonk M, et al. Causes and risk factors for death in systemic sclerosis: a study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann Rheum Dis 2010; 69(10):1809–1815. doi:10.1136/ard.2009.114264
- Nassenstein K, Breuckmann F, Huger M, et al. Detection of myocardial fibrosis in systemic sclerosis by contrast-enhanced magnetic resonance imaging. Rofo 2008; 180(12):1054–1060. doi:10.1055/s-2008-1027864
- Psarras A, Soulaidopoulos S, Garyfallos A, Kitas G, Dimitroulas T. A critical view on cardiovascular risk in systemic sclerosis. Rheumatol Int 2017; 37(1):85–95. doi:10.1007/s00296-016-3530-3
- Lekakis J, Mavrikakis M, Emmanuel M, et al. Cold-induced coronary Raynaud’s phenomenon in patients with systemic sclerosis. Clin Exp Rheumatol 1998; 16(2):135–140. pmid:9536388
- Altorok N, Wang Y, Kahaleh B. Endothelial dysfunction in systemic sclerosis. Curr Opin Rheumatol 2014; 26(6):615–620. doi:10.1097/BOR.0000000000000112
- Fleming JN, Nash RA, Mahoney WM Jr, Schwartz SM. Is scleroderma a vasculopathy? Curr Rheumatol Rep 2009; 11(2):103–110. pmid:19296882
- Maurer B, Distler A, Suliman YA, et al. Vascular endothelial growth factor aggravates fibrosis and vasculopathy in experimental models of systemic sclerosis. Ann Rheum Dis 2014; 73(10):1880–1887. doi:10.1136/annrheumdis-2013-203535
- Meune C, Vignaux O, Kahan A, Allanore Y. Heart involvement in systemic sclerosis: evolving concept and diagnostic methodologies. Arch Cardiovasc Dis 2010; 103(1):46–52. doi:10.1016/j.acvd.2009.06.009
- Dimitroulas T, Giannakoulas G, Karvounis H, Garyfallos A, Settas L, Kitas GD. Micro- and macrovascular treatment targets in scleroderma heart disease. Curr Pharm Des 2014; 20(4):536–544. pmid:23565639
- Allanore Y, Meune C. Primary myocardial involvement in systemic sclerosis: evidence for a microvascular origin. Clin Exp Rheumatol 2010; 28(5 suppl 62):S48–S53. pmid:21050545
- Kahan A, Nitenberg A, Foult JM, et al. Decreased coronary reserve in primary scleroderma myocardial disease. Arthritis Rheum 1985; 28(6):637–646. pmid:4004974
- Morrisroe K, Stevens W, Sahhar J, et al. Epidemiology and disease characteristics of systemic sclerosis-related pulmonary arterial hypertension: results from a real-life screening program. Arthritis Res Ther 2017; 19(1):42. doi:10.1186/s13075-017-1250-z
- Chaisson NF, Hassoun PM. Systemic sclerosis-associated pulmonary arterial hypertension. Chest 2013; 144(4):1346–1356. doi:10.1378/chest.12-2396
- Steen VD, Medsger TA. Changes in causes of death in systemic sclerosis, 1972–2002. Ann Rheum Dis 2007; 66(7):940–944. doi:10.1136/ard.2006.066068
- Coghlan JG, Galiè N, Barberà JA, et al; AMBITION investigators. Initial combination therapy with ambrisentan and tadalafil in connective tissue disease-associated pulmonary arterial hypertension (CTD-PAH): subgroup analysis from the AMBITION trial. Ann Rheum Dis 2017; 76(7):1219–1227. doi:10.1136/annrheumdis-2016-210236
- Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J 2019; 53(1):1801913. doi:10.1183/13993003.01913-2018
- Chatterjee S. Pulmonary hypertension in systemic sclerosis. Semin Arthritis Rheum 2011; 41(1):19–37. doi:10.1016/j.semarthrit.2010.08.004
- Sweiss NJ, Hushaw L, Thenappan T, et al. Diagnosis and management of pulmonary hypertension in systemic sclerosis. Curr Rheumatol Rep 2010; 12(1):8–18. doi:10.1007/s11926-009-0078-1
- Cox SR, Walker JG, Coleman M, et al. Isolated pulmonary hypertension in scleroderma. Intern Med J 2005; 35(1):28–33. doi:10.1111/j.1445-5994.2004.00646.x
- Sánchez-Román J, Opitz CF, Kowal-Bielecka O, García-Hernández FJ, Castillo-Palma MJ, Pittrow D; EPOSS-OMERACT Group. Screening for PAH in patients with systemic sclerosis: focus on Doppler echocardiography. Rheumatology (Oxford) 2008; 47(suppl 5):v33–v35. doi:10.1093/rheumatology/ken306
- Walker KM, Pope J; Scleroderma Clinical Trials Consortium; Canadian Scleroderma Research Group. Expert agreement on EULAR/EUSTAR recommendations for the management of systemic sclerosis. J Rheumatol 2011; 38(7):1326–1328. doi:10.3899/jrheum.101262
- Khanna D, Gladue H, Channick R, et al; Scleroderma Foundation and Pulmonary Hypertension Association. Recommendations for screening and detection of connective tissue disease-associated pulmonary arterial hypertension. Arthritis Rheum 2013; 65(12):3194–3201. doi:10.1002/art.38172
- Hachulla E, Gressin V, Guillevin L, et al. Early detection of pulmonary arterial hypertension in systemic sclerosis: a French nationwide prospective multicenter study. Arthritis Rheum 2005; 52(12):3792–3800. doi:10.1002/art.21433
- Dimitroulas T, Giannakoulas G, Dimitroula H, et al. Significance of serum uric acid in pulmonary hypertension due to systemic sclerosis: a pilot study. Rheumatol Int 2011; 31(2):263–267. doi:10.1007/s00296-010-1557-4
- Dimitroulas T, Giannakoulas G, Papadopoulou K, et al. Left atrial volume and N-terminal pro-B type natriuretic peptide are associated with elevated pulmonary artery pressure in patients with systemic sclerosis. Clin Rheumatol 2010; 29(9):957–964. doi:10.1007/s10067-010-1494-3
- Coghlan JG, Denton CP, Grünig E, et al; DETECT study group. Evidence-based detection of pulmonary arterial hypertension in systemic sclerosis: the DETECT study. Ann Rheum Dis 2014; 73(7):1340–1349. doi:10.1136/annrheumdis-2013-203301
- Schwaiger JP, Khanna D, Gerry Coghlan J. Screening patients with scleroderma for pulmonary arterial hypertension and implications for other at-risk populations. Eur Respir Rev 2013; 22(130):515–525. doi:10.1183/09059180.00006013
- Man A, Zhu Y, Zhang Y, et al. The risk of cardiovascular disease in systemic sclerosis: a population-based cohort study. Ann Rheum Dis 2013; 72(7):1188–1193. doi:10.1136/annrheumdis-2012-202007
- Ngian G-S, Sahhar J, Proudman SM, Stevens W, Wicks IP, Van Doornum S. Prevalence of coronary heart disease and cardiovascular risk factors in a national cross-sectional cohort study of systemic sclerosis. Ann Rheum Dis 2012; 71(12):1980–1983. doi:10.1136/annrheumdis-2011-201176
- Aviña-Zubieta JA, Man A, Yurkovich M, Huang K, Sayre EC, Choi HK. Early cardiovascular disease after the diagnosis of systemic sclerosis. Am J Med 2016; 29(3):324–331. doi:10.1016/j.amjmed.2015.10.037
- D’Angelo WA, Fries JF, Masi AT, Shulman LE. Pathologic observations in systemic sclerosis (scleroderma). A study of fifty-eight autopsy cases and fifty-eight matched controls. Am J Med 1969; 46(3):428–440. doi:10.1016/0002-9343(69)90044-8
- Ngian GS, Sahhar J, Proudman SM, Stevens W, Wicks IP, Van Doornum S. Prevalence of coronary heart disease and cardiovascular risk factors in a national cross-sectional cohort study of systemic sclerosis. Ann Rheum Dis 2012; 71(12):1980–1983. doi:10.1136/annrheumdis-2011-201176
- Khurma V, Meyer C, Park GS, et al. A pilot study of subclinical coronary atherosclerosis in systemic sclerosis: coronary artery calcification in cases and controls. Arthritis Rheum 2008; 59(4):591–597. doi:10.1002/art.23540
- Lippi G, Caramaschi P, Montagnana M, Salvagno GL, Volpe A, Guidi G. Lipoprotein[a] and the lipid profile in patients with systemic sclerosis. Clin Chim Acta 2006; 364(1–2):345–348. doi:10.1016/j.cca.2005.07.015
- Palinski W, Hörkkö S, Miller E, et al. Cloning of monoclonal autoantibodies to epitopes of oxidized lipoproteins from apolipoprotein E-deficient mice. Demonstration of epitopes of oxidized low density lipoprotein in human plasma. J Clin Invest 1996; 98(3):800–814. doi:10.1172/JCI118853
- Ho M, Veale D, Eastmond C, Nuki G, Belch J. Macrovascular disease and systemic sclerosis. Ann Rheum Dis 2000; 59(1):39–43. doi:10.1136/ard.59.1.39
- Kaloudi O, Basta G, Perfetto F, et al. Circulating levels of Ne-(carboxymethyl)lysine are increased in systemic sclerosis. Rheumatology (Oxford) 2007; 46(3):412–416. doi:10.1093/rheumatology/kel076
- Muro Y, Sugiura K, Morita Y, Tomita Y. An evaluation of the efficacy of the toe brachial index measuring vascular involvement in systemic sclerosis and other connective tissue diseases. Clin Exp Rheumatol 2009; 27(3 suppl 54):26–31. pmid:19796558
- Cheng K-S, Tiwari A, Boutin A, et al. Differentiation of primary and secondary Raynaud’s disease by carotid arterial stiffness. Eur J Vasc Endovasc Surg 2003; 25(4):336–341. doi:10.1053/ejvs.2002.1845
- Kawasaki M, Ito Y, Yokoyama H, et al. Assessment of arterial medial characteristics in human carotid arteries using integrated backscatter ultrasound and its histological implications. Atherosclerosis 2005; 180(1):145–154. doi:10.1016/j.atherosclerosis.2004.11.018
- Szucs G, Tímár O, Szekanecz Z, et al. Endothelial dysfunction precedes atherosclerosis in systemic sclerosis—relevance for prevention of vascular complications. Rheumatology (Oxford) 2007; 46(5):759–762. doi:10.1093/rheumatology/kel426
- Hettema ME, Zhang D, de Leeuw K, et al. Early atherosclerosis in systemic sclerosis and its relation to disease or traditional risk factors. Arthritis Res Ther 2008;10(2):R49. doi:10.1186/ar2408
- Roustit M, Simmons GH, Baguet JP, Carpentier P, Cracowski JL. Discrepancy between simultaneous digital skin microvascular and brachial artery macrovascular post-occlusive hyperemia in systemic sclerosis. J Rheumatol 2008; 35(8):1576–1583. pmid:18597404
- Vettori S, Maresca L, Cuomo G, Abbadessa S, Leonardo G, Valentini G. Clinical and subclinical atherosclerosis in systemic sclerosis: consequences of previous corticosteroid treatment. Scand J Rheumatol 2010; 39(6):485–489. doi:10.3109/03009741003781985
- Lekakis J, Mavrikakis M, Papamichael C, et al. Short-term estrogen administration improves abnormal endothelial function in women with systemic sclerosis and Raynaud’s phenomenon. Am Heart J 1998; 136(5):905–912. doi:10.1016/s0002-8703(98)70137-1
- Bartoli F, Blagojevic J, Bacci M, et al. Flow-mediated vasodilation and carotid intima-media thickness in systemic sclerosis. Ann N Y Acad Sci 2007; 1108:283–290. doi:10.1196/annals.1422.030
- Rollando D, Bezante GP, Sulli A, et al. Brachial artery endothelial-dependent flow-mediated dilation identifies early-stage endothelial dysfunction in systemic sclerosis and correlates with nailfold microvascular impairment. J Rheumatol 2010; 37(6):1168–1173. doi:10.3899/jrheum.091116
- Andersen GN, Mincheva-Nilsson L, Kazzam E, et al. Assessment of vascular function in systemic sclerosis: indications of the development of nitrate tolerance as a result of enhanced endothelial nitric oxide production. Arthritis Rheum 2002; 46(5):1324–1332. doi:10.1002/art.10191
- Au K, Singh MK, Bodukam V, et al. Atherosclerosis in systemic sclerosis: a systematic review and meta-analysis. Arthritis Rheum 2011; 63(7):2078–2090. doi:10.1002/art.30380
- van Sijl AM, Peters MJ, Knol DK, et al. Carotid intima media thickness in rheumatoid arthritis as compared to control subjects: a meta-analysis. Semin Arthritis Rheum 2011; 40(5):389–397. doi:10.1016/j.semarthrit.2010.06.006
- Brohall G, Odén A, Fagerberg B. Carotid artery intima-media thickness in patients with type 2 diabetes mellitus and impaired glucose tolerance: a systematic review. Diabet Med 2006; 23(6):609–616. doi:10.1111/j.1464-5491.2005.01725.x
- Masoura C, Pitsavos C, Aznaouridis K, Skoumas I, Vlachopoulos C, Stefanadis C. Arterial endothelial function and wall thickness in familial hypercholesterolemia and familial combined hyperlipidemia and the effect of statins. A systematic review and meta-analysis. Atherosclerosis 2011; 214(1):129–138. doi:10.1016/j.atherosclerosis.2010.10.008
- Ozen G, Inanc N, Unal AU, et al. Subclinical atherosclerosis in systemic sclerosis: not less frequent than rheumatoid arthritis and not detected with cardiovascular risk indices. Arthritis Care Res (Hoboken) 2016; 68(10):1538–1546. doi:10.1002/acr.22852
- Inaba Y, Chen JA, Bergmann SR. Prediction of future cardiovascular outcomes by flow-mediated vasodilatation of brachial artery: a meta-analysis. Int J Cardiovasc Imaging 2010; 26(6):631–640. doi:10.1007/s10554-010-9616-1
- Meune C, Avouac J, Wahbi K, et al. Cardiac involvement in systemic sclerosis assessed by tissue-doppler echocardiography during routine care: a controlled study of 100 consecutive patients. Arthritis Rheum 2008; 58(6):1803–1809. doi:10.1002/art.23463
- Tennøe AH, Murbræch K, Andreassen JC, et al. Left ventricular diastolic dysfunction predicts mortality in patients with systemic sclerosis. J Am Coll Cardiol 2018; 72(15):1804–1813. doi:10.1016/j.jacc.2018.07.068
- de Groote P, Gressin V, Hachulla E, et al; ItinerAIR-Scleroderma Investigators. Evaluation of cardiac abnormalities by Doppler echocardiography in a large nationwide multicentric cohort of patients with systemic sclerosis. Ann Rheum Dis 2008; 67(1):31–36. doi:10.1136/ard.2006.057760
- Allanore Y, Meune C, Vonk MC, et al; EUSTAR co-authors. Prevalence and factors associated with left ventricular dysfunction in the EULAR Scleroderma Trial and Research group (EUSTAR) database of patients with systemic sclerosis. Ann Rheum Dis 2010; 69(1):218–221. doi:10.1136/ard.2008.103382
- Hachulla AL, Launay D, Gaxotte V, et al. Cardiac magnetic resonance imaging in systemic sclerosis: a cross-sectional observational study of 52 patients. Ann Rheum Dis 2009; 68(12):1878–1884. doi:10.1136/ard.2008.095836
- Assassi S, Del Junco D, Sutter K, et al. Clinical and genetic factors predictive of mortality in early systemic sclerosis. Arthritis Rheum 2009; 61(10):1403–1411. doi:10.1002/art.24734
- Rokas S, Mavrikakis M, Agrios N, Mylonas D, Antoniadou L, Moulopoulos S. Electrophysiologic abnormalities of cardiac function in progressive systemic sclerosis. J Electrocardiol 1996; 29(1):17–25. pmid:8808521
- Kostis JB, Seibold JR, Turkevich D, et al. Prognostic importance of cardiac arrhythmias in systemic sclerosis. Am J Med 1988; 84(6):1007–1015. doi:10.1016/0002-9343(88)90305-1
- Biełous-Wilk A, Poreba M, Staniszewska-Marszałek E, et al. Electrocardiographic evaluation in patients with systemic scleroderma and without clinically evident heart disease. Ann Noninvasive Electrocardiol 2009; 14(3):251–257. doi:10.1111/j.1542-474X.2009.00306.x
- Bienias P, Ciurzynski M, Glinska-Wielochowska M, et al. Heart rate turbulence assessment in systemic sclerosis: the role for the detection of cardiac autonomic nervous system dysfunction. Rheumatology (Oxford) 2010; 49(2):355–360. doi:10.1093/rheumatology/kep394
- Ferri C, Bernini L, Bongiorni MG, et al. Noninvasive evaluation of cardiac dysrhythmias, and their relationship with multisystemic symptoms, in progressive systemic sclerosis patients. Arthritis Rheum 1985; 28(11):1259–1266. pmid:4063000
- Roberts NK, Cabeen WR, Moss J, Clements PJ, Furst DE. The prevalence of conduction defects and cardiac arrhythmias in progressive systemic sclerosis. Ann Intern Med 1981; 94(1):38–40. doi:10.7326/0003-4819-94-1-38
- Wang Q, Shang Y, Li S, Wu Y, Wang C, Yan X. Complete heart block in systemic sclerosis: a case report and literature review. Medicine (Baltimore) 2018; 97(46):e13226. doi:10.1097/MD.0000000000013226
- Summerfield BJ. Progressive systemic sclerosis with complete heart block. Br Heart J 1975; 37(12):1308–1310. doi:10.1136/hrt.37.12.1308
- Moyssakis I, Papadopoulos DP, Tzioufas AG, Votteas V. Complete heart block in a patient with systemic sclerosis. Clin Rheumatol 2006; 25(4):551–552. doi:10.1007/s10067-005-0068-2
- Ridolfi RL, Bulkley BH, Hutchins GM. The cardiac conduction system in progressive systemic sclerosis. Clinical and pathologic features of 35 patients. Am J Med 1976; 61(3):361–366. doi:10.1016/0002-9343(76)90373-9
- Champion HC. The heart in scleroderma. Rheum Dis Clin North Am 2008; 34(1):181–190. doi:10.1016/j.rdc.2007.12.002
- Gowda RM, Khan IA, Sacchi TJ, Vasavada BC. Scleroderma pericardial disease presented with a large pericardial effusion—a case report. Angiology 2001; 52(1):59–62. doi:10.1177/000331970105200108
- Meier FMP, Frommer KW, Dinser R, et al; EUSTAR Co-authors. Update on the profile of the EUSTAR cohort: an analysis of the EULAR scleroderma trials and research group database. Ann Rheum Dis 2012; 71(8):1355–1360. doi:10.1136/annrheumdis-2011-200742
- Subramanian SR, Akram R, Velayati A, Chadow H. New development of cardiac tamponade on underlying effusive-constrictive pericarditis: an uncommon initial presentation of scleroderma. BMJ Case Rep 2013; 2013. doi:10.1136/bcr-2013-010254
- Kitchongcharoenying P, Foocharoen C, Mahakkanukrauh A, Suwannaroj S, Nanagara R. Pericardial fluid profiles of pericardial effusion in systemic sclerosis patients. Asian Pac J Allergy Immunol 2013; 31(4):314–319. doi:10.12932/AP0305.31.4.2013
- McWhorter JE, LeRoy EC. Pericardial disease in scleroderma (systemic sclerosis). Am J Med 1974; 57(4):566–575. doi:10.1016/0002-9343(74)90008-4
- Comens SM, Alpert MA, Sharp GC, et al. Frequency of mitral valve prolapse in systemic lupus erythematosus, progressive systemic sclerosis and mixed connective tissue disease. Am J Cardiol 1989; 63(5):369–370. doi:10.1016/0002-9149(89)90351-2
- Candell-Riera J, Armadans-Gil L, Simeón CP, et al. Comprehensive noninvasive assessment of cardiac involvement in limited systemic sclerosis. Arthritis Rheum 1996; 39(7):1138–1145. pmid:8670322
- Caforio ALP, Adler Y, Agostini C, et al. Diagnosis and management of myocardial involvement in systemic immune-mediated diseases: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Disease. Eur Heart J 2017; 38(35):2649–2662. doi:10.1093/eurheartj/ehx321
- Mavrogeni S, Karabela G, Koutsogeorgopoulou L, et al. Pseudo-infarction pattern in diffuse systemic sclerosis. Evaluation using cardiovascular magnetic resonance. Int J Cardiol 2016; 214:465–468. doi:10.1016/j.ijcard.2016.03.235
- Ladak K, Pope JE. A review of the effects of statins in systemic sclerosis. Semin Arthritis Rheum 2016; 45(6):698–705. doi:10.1016/j.semarthrit.2015.10.013
- Abou-Raya A, Abou-Raya S, Helmii M. Statins: potentially useful in therapy of systemic sclerosis-related Raynaud’s phenomenon and digital ulcers. J Rheumatol 2008; 35(9):1801–1808. pmid:18709692
- ASCEND Study Collaborative Group; Bowman L, Mafham M, Wallendszus K, et al. Effects of aspirin for primary prevention in persons with diabetes mellitus. N Engl J Med 2018; 379(16):1529–1539. doi:10.1056/NEJMoa1804988
- Guerra F, Stronati G, Fischietti C, et al. Global longitudinal strain measured by speckle tracking identifies subclinical heart involvement in patients with systemic sclerosis. Eur J Prev Cardiol 2018; 25(15):1598–1606. doi:10.1177/2047487318786315
- Mavrogeni SI, Sfikakis PP, Dimitroulas T, et al. Prospects of using cardiovascular magnetic resonance in the identification of arrhythmogenic substrate in autoimmune rheumatic diseases. Rheumatol Int 2018; 38(9):1615–1621. doi:10.1007/s00296-018-4110-5
- Mavrogeni SI, Sfikakis PP, Markousis-Mavrogenis G, et al. Cardiovascular magnetic resonance imaging pattern in patients with autoimmune rheumatic diseases and ventricular tachycardia with preserved ejection fraction. Int J Cardiol 2019; 284:105–109. doi:10.1016/j.ijcard.2018.10.067
- Maradit-Kremers H, Crowson CS, Nicola PJ, et al. Increased unrecognized coronary heart disease and sudden deaths in rheumatoid arthritis: a population-based cohort study. Arthritis Rheum 2005; 52(2):402–411. doi:10.1002/art.20853
- Naranjo A, Sokka T, Descalzo MA, et al; QUEST-RA Group. Cardiovascular disease in patients with rheumatoid arthritis: results from the QUEST-RA study. Arthritis Res Ther 2008; 10(2):R30. doi:10.1186/ar2383
- Innala L, Möller B, Ljung L, et al. Cardiovascular events in early RA are a result of inflammatory burden and traditional risk factors: a five year prospective study. Arthritis Res Ther 2011; 13(4):R131. doi:10.1186/ar3442
- Barnes J, Mayes MD. Epidemiology of systemic sclerosis: incidence, prevalence, survival, risk factors, malignancy, and environmental triggers. Curr Opin Rheumatol 2012; 24(2):165–170. doi:10.1097/BOR.0b013e32834ff2e8
- Chifflot H, Fautrel B, Sordet C, Chatelus E, Sibilia J. Incidence and prevalence of systemic sclerosis: a systematic literature review. Semin Arthritis Rheum 2008; 37(4):223–235 doi:10.1016/j.semarthrit.2007.05.003
- Gabrielli A, Avvedimento EV, Krieg T. Scleroderma. N Engl J Med 2009; 360(19):1989–2003. doi:10.1056/NEJMra0806188
- Panopoulos S, Tektonidou M, Drosos AA, et al. Prevalence of comorbidities in systemic sclerosis versus rheumatoid arthritis: a comparative, multicenter, matched-cohort study. Arthritis Res Ther 2018; 20(1):267. doi:10.1186/s13075-018-1771-0
- Ferri C, Valentini G, Cozzi F, et al. Systemic sclerosis: demographic, clinical, and serologic features and survival in 1,012 Italian patients. Medicine (Baltimore) 2002; 81(8):139–153. doi:10.1097/00005792-200203000-00004
- Steen VD, Medsger TA Jr. Severe organ involvement in systemic sclerosis with diffuse scleroderma. Arthritis Rheum 2000; 43(11):2437–2444. doi:10.1002/1529-0131(200011)43:11<2437::AID-ANR10>3.0.CO;2-U
- Hachulla AL, Launay D, Gaxotte V, et al. Cardiac magnetic resonance imaging in systemic sclerosis: a cross-sectional observational study of 52 patients. Ann Rheum Dis 2009; 68(12):1878–1884. doi:10.1136/ard.2008.095836
- Tyndall AJ, Bannert B, Vonk M, et al. Causes and risk factors for death in systemic sclerosis: a study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann Rheum Dis 2010; 69(10):1809–1815. doi:10.1136/ard.2009.114264
- Nassenstein K, Breuckmann F, Huger M, et al. Detection of myocardial fibrosis in systemic sclerosis by contrast-enhanced magnetic resonance imaging. Rofo 2008; 180(12):1054–1060. doi:10.1055/s-2008-1027864
- Psarras A, Soulaidopoulos S, Garyfallos A, Kitas G, Dimitroulas T. A critical view on cardiovascular risk in systemic sclerosis. Rheumatol Int 2017; 37(1):85–95. doi:10.1007/s00296-016-3530-3
- Lekakis J, Mavrikakis M, Emmanuel M, et al. Cold-induced coronary Raynaud’s phenomenon in patients with systemic sclerosis. Clin Exp Rheumatol 1998; 16(2):135–140. pmid:9536388
- Altorok N, Wang Y, Kahaleh B. Endothelial dysfunction in systemic sclerosis. Curr Opin Rheumatol 2014; 26(6):615–620. doi:10.1097/BOR.0000000000000112
- Fleming JN, Nash RA, Mahoney WM Jr, Schwartz SM. Is scleroderma a vasculopathy? Curr Rheumatol Rep 2009; 11(2):103–110. pmid:19296882
- Maurer B, Distler A, Suliman YA, et al. Vascular endothelial growth factor aggravates fibrosis and vasculopathy in experimental models of systemic sclerosis. Ann Rheum Dis 2014; 73(10):1880–1887. doi:10.1136/annrheumdis-2013-203535
- Meune C, Vignaux O, Kahan A, Allanore Y. Heart involvement in systemic sclerosis: evolving concept and diagnostic methodologies. Arch Cardiovasc Dis 2010; 103(1):46–52. doi:10.1016/j.acvd.2009.06.009
- Dimitroulas T, Giannakoulas G, Karvounis H, Garyfallos A, Settas L, Kitas GD. Micro- and macrovascular treatment targets in scleroderma heart disease. Curr Pharm Des 2014; 20(4):536–544. pmid:23565639
- Allanore Y, Meune C. Primary myocardial involvement in systemic sclerosis: evidence for a microvascular origin. Clin Exp Rheumatol 2010; 28(5 suppl 62):S48–S53. pmid:21050545
- Kahan A, Nitenberg A, Foult JM, et al. Decreased coronary reserve in primary scleroderma myocardial disease. Arthritis Rheum 1985; 28(6):637–646. pmid:4004974
- Morrisroe K, Stevens W, Sahhar J, et al. Epidemiology and disease characteristics of systemic sclerosis-related pulmonary arterial hypertension: results from a real-life screening program. Arthritis Res Ther 2017; 19(1):42. doi:10.1186/s13075-017-1250-z
- Chaisson NF, Hassoun PM. Systemic sclerosis-associated pulmonary arterial hypertension. Chest 2013; 144(4):1346–1356. doi:10.1378/chest.12-2396
- Steen VD, Medsger TA. Changes in causes of death in systemic sclerosis, 1972–2002. Ann Rheum Dis 2007; 66(7):940–944. doi:10.1136/ard.2006.066068
- Coghlan JG, Galiè N, Barberà JA, et al; AMBITION investigators. Initial combination therapy with ambrisentan and tadalafil in connective tissue disease-associated pulmonary arterial hypertension (CTD-PAH): subgroup analysis from the AMBITION trial. Ann Rheum Dis 2017; 76(7):1219–1227. doi:10.1136/annrheumdis-2016-210236
- Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J 2019; 53(1):1801913. doi:10.1183/13993003.01913-2018
- Chatterjee S. Pulmonary hypertension in systemic sclerosis. Semin Arthritis Rheum 2011; 41(1):19–37. doi:10.1016/j.semarthrit.2010.08.004
- Sweiss NJ, Hushaw L, Thenappan T, et al. Diagnosis and management of pulmonary hypertension in systemic sclerosis. Curr Rheumatol Rep 2010; 12(1):8–18. doi:10.1007/s11926-009-0078-1
- Cox SR, Walker JG, Coleman M, et al. Isolated pulmonary hypertension in scleroderma. Intern Med J 2005; 35(1):28–33. doi:10.1111/j.1445-5994.2004.00646.x
- Sánchez-Román J, Opitz CF, Kowal-Bielecka O, García-Hernández FJ, Castillo-Palma MJ, Pittrow D; EPOSS-OMERACT Group. Screening for PAH in patients with systemic sclerosis: focus on Doppler echocardiography. Rheumatology (Oxford) 2008; 47(suppl 5):v33–v35. doi:10.1093/rheumatology/ken306
- Walker KM, Pope J; Scleroderma Clinical Trials Consortium; Canadian Scleroderma Research Group. Expert agreement on EULAR/EUSTAR recommendations for the management of systemic sclerosis. J Rheumatol 2011; 38(7):1326–1328. doi:10.3899/jrheum.101262
- Khanna D, Gladue H, Channick R, et al; Scleroderma Foundation and Pulmonary Hypertension Association. Recommendations for screening and detection of connective tissue disease-associated pulmonary arterial hypertension. Arthritis Rheum 2013; 65(12):3194–3201. doi:10.1002/art.38172
- Hachulla E, Gressin V, Guillevin L, et al. Early detection of pulmonary arterial hypertension in systemic sclerosis: a French nationwide prospective multicenter study. Arthritis Rheum 2005; 52(12):3792–3800. doi:10.1002/art.21433
- Dimitroulas T, Giannakoulas G, Dimitroula H, et al. Significance of serum uric acid in pulmonary hypertension due to systemic sclerosis: a pilot study. Rheumatol Int 2011; 31(2):263–267. doi:10.1007/s00296-010-1557-4
- Dimitroulas T, Giannakoulas G, Papadopoulou K, et al. Left atrial volume and N-terminal pro-B type natriuretic peptide are associated with elevated pulmonary artery pressure in patients with systemic sclerosis. Clin Rheumatol 2010; 29(9):957–964. doi:10.1007/s10067-010-1494-3
- Coghlan JG, Denton CP, Grünig E, et al; DETECT study group. Evidence-based detection of pulmonary arterial hypertension in systemic sclerosis: the DETECT study. Ann Rheum Dis 2014; 73(7):1340–1349. doi:10.1136/annrheumdis-2013-203301
- Schwaiger JP, Khanna D, Gerry Coghlan J. Screening patients with scleroderma for pulmonary arterial hypertension and implications for other at-risk populations. Eur Respir Rev 2013; 22(130):515–525. doi:10.1183/09059180.00006013
- Man A, Zhu Y, Zhang Y, et al. The risk of cardiovascular disease in systemic sclerosis: a population-based cohort study. Ann Rheum Dis 2013; 72(7):1188–1193. doi:10.1136/annrheumdis-2012-202007
- Ngian G-S, Sahhar J, Proudman SM, Stevens W, Wicks IP, Van Doornum S. Prevalence of coronary heart disease and cardiovascular risk factors in a national cross-sectional cohort study of systemic sclerosis. Ann Rheum Dis 2012; 71(12):1980–1983. doi:10.1136/annrheumdis-2011-201176
- Aviña-Zubieta JA, Man A, Yurkovich M, Huang K, Sayre EC, Choi HK. Early cardiovascular disease after the diagnosis of systemic sclerosis. Am J Med 2016; 29(3):324–331. doi:10.1016/j.amjmed.2015.10.037
- D’Angelo WA, Fries JF, Masi AT, Shulman LE. Pathologic observations in systemic sclerosis (scleroderma). A study of fifty-eight autopsy cases and fifty-eight matched controls. Am J Med 1969; 46(3):428–440. doi:10.1016/0002-9343(69)90044-8
- Ngian GS, Sahhar J, Proudman SM, Stevens W, Wicks IP, Van Doornum S. Prevalence of coronary heart disease and cardiovascular risk factors in a national cross-sectional cohort study of systemic sclerosis. Ann Rheum Dis 2012; 71(12):1980–1983. doi:10.1136/annrheumdis-2011-201176
- Khurma V, Meyer C, Park GS, et al. A pilot study of subclinical coronary atherosclerosis in systemic sclerosis: coronary artery calcification in cases and controls. Arthritis Rheum 2008; 59(4):591–597. doi:10.1002/art.23540
- Lippi G, Caramaschi P, Montagnana M, Salvagno GL, Volpe A, Guidi G. Lipoprotein[a] and the lipid profile in patients with systemic sclerosis. Clin Chim Acta 2006; 364(1–2):345–348. doi:10.1016/j.cca.2005.07.015
- Palinski W, Hörkkö S, Miller E, et al. Cloning of monoclonal autoantibodies to epitopes of oxidized lipoproteins from apolipoprotein E-deficient mice. Demonstration of epitopes of oxidized low density lipoprotein in human plasma. J Clin Invest 1996; 98(3):800–814. doi:10.1172/JCI118853
- Ho M, Veale D, Eastmond C, Nuki G, Belch J. Macrovascular disease and systemic sclerosis. Ann Rheum Dis 2000; 59(1):39–43. doi:10.1136/ard.59.1.39
- Kaloudi O, Basta G, Perfetto F, et al. Circulating levels of Ne-(carboxymethyl)lysine are increased in systemic sclerosis. Rheumatology (Oxford) 2007; 46(3):412–416. doi:10.1093/rheumatology/kel076
- Muro Y, Sugiura K, Morita Y, Tomita Y. An evaluation of the efficacy of the toe brachial index measuring vascular involvement in systemic sclerosis and other connective tissue diseases. Clin Exp Rheumatol 2009; 27(3 suppl 54):26–31. pmid:19796558
- Cheng K-S, Tiwari A, Boutin A, et al. Differentiation of primary and secondary Raynaud’s disease by carotid arterial stiffness. Eur J Vasc Endovasc Surg 2003; 25(4):336–341. doi:10.1053/ejvs.2002.1845
- Kawasaki M, Ito Y, Yokoyama H, et al. Assessment of arterial medial characteristics in human carotid arteries using integrated backscatter ultrasound and its histological implications. Atherosclerosis 2005; 180(1):145–154. doi:10.1016/j.atherosclerosis.2004.11.018
- Szucs G, Tímár O, Szekanecz Z, et al. Endothelial dysfunction precedes atherosclerosis in systemic sclerosis—relevance for prevention of vascular complications. Rheumatology (Oxford) 2007; 46(5):759–762. doi:10.1093/rheumatology/kel426
- Hettema ME, Zhang D, de Leeuw K, et al. Early atherosclerosis in systemic sclerosis and its relation to disease or traditional risk factors. Arthritis Res Ther 2008;10(2):R49. doi:10.1186/ar2408
- Roustit M, Simmons GH, Baguet JP, Carpentier P, Cracowski JL. Discrepancy between simultaneous digital skin microvascular and brachial artery macrovascular post-occlusive hyperemia in systemic sclerosis. J Rheumatol 2008; 35(8):1576–1583. pmid:18597404
- Vettori S, Maresca L, Cuomo G, Abbadessa S, Leonardo G, Valentini G. Clinical and subclinical atherosclerosis in systemic sclerosis: consequences of previous corticosteroid treatment. Scand J Rheumatol 2010; 39(6):485–489. doi:10.3109/03009741003781985
- Lekakis J, Mavrikakis M, Papamichael C, et al. Short-term estrogen administration improves abnormal endothelial function in women with systemic sclerosis and Raynaud’s phenomenon. Am Heart J 1998; 136(5):905–912. doi:10.1016/s0002-8703(98)70137-1
- Bartoli F, Blagojevic J, Bacci M, et al. Flow-mediated vasodilation and carotid intima-media thickness in systemic sclerosis. Ann N Y Acad Sci 2007; 1108:283–290. doi:10.1196/annals.1422.030
- Rollando D, Bezante GP, Sulli A, et al. Brachial artery endothelial-dependent flow-mediated dilation identifies early-stage endothelial dysfunction in systemic sclerosis and correlates with nailfold microvascular impairment. J Rheumatol 2010; 37(6):1168–1173. doi:10.3899/jrheum.091116
- Andersen GN, Mincheva-Nilsson L, Kazzam E, et al. Assessment of vascular function in systemic sclerosis: indications of the development of nitrate tolerance as a result of enhanced endothelial nitric oxide production. Arthritis Rheum 2002; 46(5):1324–1332. doi:10.1002/art.10191
- Au K, Singh MK, Bodukam V, et al. Atherosclerosis in systemic sclerosis: a systematic review and meta-analysis. Arthritis Rheum 2011; 63(7):2078–2090. doi:10.1002/art.30380
- van Sijl AM, Peters MJ, Knol DK, et al. Carotid intima media thickness in rheumatoid arthritis as compared to control subjects: a meta-analysis. Semin Arthritis Rheum 2011; 40(5):389–397. doi:10.1016/j.semarthrit.2010.06.006
- Brohall G, Odén A, Fagerberg B. Carotid artery intima-media thickness in patients with type 2 diabetes mellitus and impaired glucose tolerance: a systematic review. Diabet Med 2006; 23(6):609–616. doi:10.1111/j.1464-5491.2005.01725.x
- Masoura C, Pitsavos C, Aznaouridis K, Skoumas I, Vlachopoulos C, Stefanadis C. Arterial endothelial function and wall thickness in familial hypercholesterolemia and familial combined hyperlipidemia and the effect of statins. A systematic review and meta-analysis. Atherosclerosis 2011; 214(1):129–138. doi:10.1016/j.atherosclerosis.2010.10.008
- Ozen G, Inanc N, Unal AU, et al. Subclinical atherosclerosis in systemic sclerosis: not less frequent than rheumatoid arthritis and not detected with cardiovascular risk indices. Arthritis Care Res (Hoboken) 2016; 68(10):1538–1546. doi:10.1002/acr.22852
- Inaba Y, Chen JA, Bergmann SR. Prediction of future cardiovascular outcomes by flow-mediated vasodilatation of brachial artery: a meta-analysis. Int J Cardiovasc Imaging 2010; 26(6):631–640. doi:10.1007/s10554-010-9616-1
- Meune C, Avouac J, Wahbi K, et al. Cardiac involvement in systemic sclerosis assessed by tissue-doppler echocardiography during routine care: a controlled study of 100 consecutive patients. Arthritis Rheum 2008; 58(6):1803–1809. doi:10.1002/art.23463
- Tennøe AH, Murbræch K, Andreassen JC, et al. Left ventricular diastolic dysfunction predicts mortality in patients with systemic sclerosis. J Am Coll Cardiol 2018; 72(15):1804–1813. doi:10.1016/j.jacc.2018.07.068
- de Groote P, Gressin V, Hachulla E, et al; ItinerAIR-Scleroderma Investigators. Evaluation of cardiac abnormalities by Doppler echocardiography in a large nationwide multicentric cohort of patients with systemic sclerosis. Ann Rheum Dis 2008; 67(1):31–36. doi:10.1136/ard.2006.057760
- Allanore Y, Meune C, Vonk MC, et al; EUSTAR co-authors. Prevalence and factors associated with left ventricular dysfunction in the EULAR Scleroderma Trial and Research group (EUSTAR) database of patients with systemic sclerosis. Ann Rheum Dis 2010; 69(1):218–221. doi:10.1136/ard.2008.103382
- Hachulla AL, Launay D, Gaxotte V, et al. Cardiac magnetic resonance imaging in systemic sclerosis: a cross-sectional observational study of 52 patients. Ann Rheum Dis 2009; 68(12):1878–1884. doi:10.1136/ard.2008.095836
- Assassi S, Del Junco D, Sutter K, et al. Clinical and genetic factors predictive of mortality in early systemic sclerosis. Arthritis Rheum 2009; 61(10):1403–1411. doi:10.1002/art.24734
- Rokas S, Mavrikakis M, Agrios N, Mylonas D, Antoniadou L, Moulopoulos S. Electrophysiologic abnormalities of cardiac function in progressive systemic sclerosis. J Electrocardiol 1996; 29(1):17–25. pmid:8808521
- Kostis JB, Seibold JR, Turkevich D, et al. Prognostic importance of cardiac arrhythmias in systemic sclerosis. Am J Med 1988; 84(6):1007–1015. doi:10.1016/0002-9343(88)90305-1
- Biełous-Wilk A, Poreba M, Staniszewska-Marszałek E, et al. Electrocardiographic evaluation in patients with systemic scleroderma and without clinically evident heart disease. Ann Noninvasive Electrocardiol 2009; 14(3):251–257. doi:10.1111/j.1542-474X.2009.00306.x
- Bienias P, Ciurzynski M, Glinska-Wielochowska M, et al. Heart rate turbulence assessment in systemic sclerosis: the role for the detection of cardiac autonomic nervous system dysfunction. Rheumatology (Oxford) 2010; 49(2):355–360. doi:10.1093/rheumatology/kep394
- Ferri C, Bernini L, Bongiorni MG, et al. Noninvasive evaluation of cardiac dysrhythmias, and their relationship with multisystemic symptoms, in progressive systemic sclerosis patients. Arthritis Rheum 1985; 28(11):1259–1266. pmid:4063000
- Roberts NK, Cabeen WR, Moss J, Clements PJ, Furst DE. The prevalence of conduction defects and cardiac arrhythmias in progressive systemic sclerosis. Ann Intern Med 1981; 94(1):38–40. doi:10.7326/0003-4819-94-1-38
- Wang Q, Shang Y, Li S, Wu Y, Wang C, Yan X. Complete heart block in systemic sclerosis: a case report and literature review. Medicine (Baltimore) 2018; 97(46):e13226. doi:10.1097/MD.0000000000013226
- Summerfield BJ. Progressive systemic sclerosis with complete heart block. Br Heart J 1975; 37(12):1308–1310. doi:10.1136/hrt.37.12.1308
- Moyssakis I, Papadopoulos DP, Tzioufas AG, Votteas V. Complete heart block in a patient with systemic sclerosis. Clin Rheumatol 2006; 25(4):551–552. doi:10.1007/s10067-005-0068-2
- Ridolfi RL, Bulkley BH, Hutchins GM. The cardiac conduction system in progressive systemic sclerosis. Clinical and pathologic features of 35 patients. Am J Med 1976; 61(3):361–366. doi:10.1016/0002-9343(76)90373-9
- Champion HC. The heart in scleroderma. Rheum Dis Clin North Am 2008; 34(1):181–190. doi:10.1016/j.rdc.2007.12.002
- Gowda RM, Khan IA, Sacchi TJ, Vasavada BC. Scleroderma pericardial disease presented with a large pericardial effusion—a case report. Angiology 2001; 52(1):59–62. doi:10.1177/000331970105200108
- Meier FMP, Frommer KW, Dinser R, et al; EUSTAR Co-authors. Update on the profile of the EUSTAR cohort: an analysis of the EULAR scleroderma trials and research group database. Ann Rheum Dis 2012; 71(8):1355–1360. doi:10.1136/annrheumdis-2011-200742
- Subramanian SR, Akram R, Velayati A, Chadow H. New development of cardiac tamponade on underlying effusive-constrictive pericarditis: an uncommon initial presentation of scleroderma. BMJ Case Rep 2013; 2013. doi:10.1136/bcr-2013-010254
- Kitchongcharoenying P, Foocharoen C, Mahakkanukrauh A, Suwannaroj S, Nanagara R. Pericardial fluid profiles of pericardial effusion in systemic sclerosis patients. Asian Pac J Allergy Immunol 2013; 31(4):314–319. doi:10.12932/AP0305.31.4.2013
- McWhorter JE, LeRoy EC. Pericardial disease in scleroderma (systemic sclerosis). Am J Med 1974; 57(4):566–575. doi:10.1016/0002-9343(74)90008-4
- Comens SM, Alpert MA, Sharp GC, et al. Frequency of mitral valve prolapse in systemic lupus erythematosus, progressive systemic sclerosis and mixed connective tissue disease. Am J Cardiol 1989; 63(5):369–370. doi:10.1016/0002-9149(89)90351-2
- Candell-Riera J, Armadans-Gil L, Simeón CP, et al. Comprehensive noninvasive assessment of cardiac involvement in limited systemic sclerosis. Arthritis Rheum 1996; 39(7):1138–1145. pmid:8670322
- Caforio ALP, Adler Y, Agostini C, et al. Diagnosis and management of myocardial involvement in systemic immune-mediated diseases: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Disease. Eur Heart J 2017; 38(35):2649–2662. doi:10.1093/eurheartj/ehx321
- Mavrogeni S, Karabela G, Koutsogeorgopoulou L, et al. Pseudo-infarction pattern in diffuse systemic sclerosis. Evaluation using cardiovascular magnetic resonance. Int J Cardiol 2016; 214:465–468. doi:10.1016/j.ijcard.2016.03.235
- Ladak K, Pope JE. A review of the effects of statins in systemic sclerosis. Semin Arthritis Rheum 2016; 45(6):698–705. doi:10.1016/j.semarthrit.2015.10.013
- Abou-Raya A, Abou-Raya S, Helmii M. Statins: potentially useful in therapy of systemic sclerosis-related Raynaud’s phenomenon and digital ulcers. J Rheumatol 2008; 35(9):1801–1808. pmid:18709692
- ASCEND Study Collaborative Group; Bowman L, Mafham M, Wallendszus K, et al. Effects of aspirin for primary prevention in persons with diabetes mellitus. N Engl J Med 2018; 379(16):1529–1539. doi:10.1056/NEJMoa1804988
- Guerra F, Stronati G, Fischietti C, et al. Global longitudinal strain measured by speckle tracking identifies subclinical heart involvement in patients with systemic sclerosis. Eur J Prev Cardiol 2018; 25(15):1598–1606. doi:10.1177/2047487318786315
- Mavrogeni SI, Sfikakis PP, Dimitroulas T, et al. Prospects of using cardiovascular magnetic resonance in the identification of arrhythmogenic substrate in autoimmune rheumatic diseases. Rheumatol Int 2018; 38(9):1615–1621. doi:10.1007/s00296-018-4110-5
- Mavrogeni SI, Sfikakis PP, Markousis-Mavrogenis G, et al. Cardiovascular magnetic resonance imaging pattern in patients with autoimmune rheumatic diseases and ventricular tachycardia with preserved ejection fraction. Int J Cardiol 2019; 284:105–109. doi:10.1016/j.ijcard.2018.10.067
KEY POINTS
- Pulmonary hypertension is common in systemic sclerosis and carries a poor prognosis. Patients with systemic sclerosis should be screened regularly with echocardiography, followed, when necessary, by right heart catheterization to detect it early.
- Myocardial infarction and stroke are more common in patients with systemic sclerosis, and preventive measures are the same as for the general population.
- Right ventricular dysfunction secondary to pulmonary hypertension is common in systemic sclerosis; left ventricular dysfunction is less so. Routine echocardiography should include assessment of right and left ventricular function.
- Electrocardiography should be performed periodically, and urgently when indicated, to look for potentially dangerous arrhythmias.
A complication of enoxaparin injection
A 78-year-old woman presented to the emergency department with shortness of breath and palpitations and was found to have atrial fibrillation with rapid ventricular response. Medical therapy with drug therapy and cardioversion proved ineffective. She then underwent atrioventricular node ablation and placement of a pacemaker.
At the time of admission, anticoagulation was started with full-dose enoxaparin, injected subcutaneously on the left side of the abdominal wall, as her CHA2DS2-VASc score (http://chadvasc.org) was 5, due to age, female sex, and history of heart failure and hypertension.
Four days after admission, she reported lower abdominal pain, and her urine output was minimal. A bladder scan showed more than 500 mL of residual urine. She was hemodynamically stable, but physical examination revealed mild abdominal distention and tenderness in the suprapubic region. Laboratory testing showed a sharp rise in serum creatinine and a drop in hematocrit.

The patient was initially managed conservatively with serial physical examinations, monitoring of the hematocrit, serial imaging studies, and discontinuation of anticoagulation, but the pain and anuria persisted. Repeat computed tomography 15 days after admission showed that the hematoma had expanded, and she now had hydronephrosis on the right side as well, requiring urologic intervention with bilateral nephrostomy tube placement.
The size of the hematoma was evaluated with serial abdominal and pelvic examinations. After several days, her urine output had improved, the nephrostomy tubes were removed, and she was discharged.
RECTUS SHEATH HEMATOMA
Our patient had a giant pelvic hematoma, probably arising from the rectus sheath. This uncommon problem can arise from trauma, anticoagulation, or increased intra-abdominal pressure, but it can also occur spontaneously.1
In rectus sheath hematoma, a branch of the inferior epigastric artery is injured at its insertion into the rectus abdominis muscle. Symptoms arise if bleeding does not stop spontaneously from a tamponade effect.2
We speculate that in our patient, deep injection of enoxaparin into the abdominal wall injured the inferior epigastric artery, which started the hematoma, and the bleeding was exacerbated by the anticoagulation effect of the enoxaparin.
Another form of pelvic hematoma is retroperitoneal. It is most commonly caused by trauma but can occur due to rupture of the aorta, compression from tumors, or, infrequently, anticoagulation therapy.3
The role of anticoagulation
Spontaneous pelvic hematoma is usually missed as a cause of abdominal pain in patients on anticoagulation therapy and is mistaken for common acute conditions such as ulcer, diverticulitis, appendicitis, ovarian cyst torsion, and tumor.4 It usually develops within 5 days of starting anticoagulation therapy. Symptoms vary depending on the location of the hematoma and are best diagnosed with abdominal computed tomography, with sensitivity as high as 100%.
MANAGEMENT
Conservative management, reserved for patients in stable condition, includes temporarily stopping and reevaluating the risks and benefits of anticoagulation and antiplatelet agents, giving blood transfusions, and controlling pain. If conservative measures fail, options are arterial embolization, stent grafting, and blood vessel ligation.5 If these measures fail, patients should undergo surgical evacuation of the hematoma and ligation of bleeding vessels.6
TAKE-HOME MESSAGE
Subcutaneous injections, especially of anticoagulants, into the abdominal wall can increase the risk of hematoma. Other risk factors are older age, female sex, and thin body habitus with less abdominal fat.7 Healthcare professionals should avoid deep injections into the abdomen and should counsel patients and their caregivers about this, as well. The deltoid region could be a safer alternative.
- Cherry WB, Mueller PS. Rectus sheath hematoma: review of 126 cases at a single institution. Medicine (Baltimore) 2006; 85(2):105–110. doi:10.1097/01.md.0000216818.13067.5a
- Hatjipetrou A, Anyfantakis D, Kastanakis M. Rectus sheath hematoma: a review of the literature. Int J Surg 2015; 13:267–271. doi:10.1016/j.ijsu.2014.12.015
- Haq MM, Taimur SDM, Khan SR, Rahman MA. Retroperitoneal hematoma following enoxaparin treatment in an elderly woman—a case report. Cardiovasc J 2010; 3(1):94–97. doi:10.3329/cardio.v3i1.6434
- Luhmann A, Williams EV. Rectus sheath hematoma: a series of unfortunate events. World J Surg 2006; 30(11):2050–2055. doi:10.1007/s00268-005-0702-9
- Pace F, Colombo GM, Del Vecchio LR, et al. Low molecular weight heparin and fatal spontaneous extraperitoneal hematoma in the elderly. Geriatr Gerontol Int 2012; 12(1):172–174. doi:10.1111/j.1447-0594.2011.00742.x
- Velicki L, Cemerlic-Adic N, Bogdanovic D, Mrdanin T. Rectus sheath haematoma: enoxaparin-related complication. Acta Clin Belg 2013; 68(2):147–149. doi:10.2143/ACB.68.2.3213
- Sheth HS, Kumar R, DiNella J, Janov C, Kaldas H, Smith RE. Evaluation of risk factors for rectus sheath hematoma. Clin Appl Thromb Hemost 2016; 22(3):292–296. doi:10.1177/1076029614553024
A 78-year-old woman presented to the emergency department with shortness of breath and palpitations and was found to have atrial fibrillation with rapid ventricular response. Medical therapy with drug therapy and cardioversion proved ineffective. She then underwent atrioventricular node ablation and placement of a pacemaker.
At the time of admission, anticoagulation was started with full-dose enoxaparin, injected subcutaneously on the left side of the abdominal wall, as her CHA2DS2-VASc score (http://chadvasc.org) was 5, due to age, female sex, and history of heart failure and hypertension.
Four days after admission, she reported lower abdominal pain, and her urine output was minimal. A bladder scan showed more than 500 mL of residual urine. She was hemodynamically stable, but physical examination revealed mild abdominal distention and tenderness in the suprapubic region. Laboratory testing showed a sharp rise in serum creatinine and a drop in hematocrit.
The patient was initially managed conservatively with serial physical examinations, monitoring of the hematocrit, serial imaging studies, and discontinuation of anticoagulation, but the pain and anuria persisted. Repeat computed tomography 15 days after admission showed that the hematoma had expanded, and she now had hydronephrosis on the right side as well, requiring urologic intervention with bilateral nephrostomy tube placement.
The size of the hematoma was evaluated with serial abdominal and pelvic examinations. After several days, her urine output had improved, the nephrostomy tubes were removed, and she was discharged.
RECTUS SHEATH HEMATOMA
Our patient had a giant pelvic hematoma, probably arising from the rectus sheath. This uncommon problem can arise from trauma, anticoagulation, or increased intra-abdominal pressure, but it can also occur spontaneously.1
In rectus sheath hematoma, a branch of the inferior epigastric artery is injured at its insertion into the rectus abdominis muscle. Symptoms arise if bleeding does not stop spontaneously from a tamponade effect.2
We speculate that in our patient, deep injection of enoxaparin into the abdominal wall injured the inferior epigastric artery, which started the hematoma, and the bleeding was exacerbated by the anticoagulation effect of the enoxaparin.
Another form of pelvic hematoma is retroperitoneal. It is most commonly caused by trauma but can occur due to rupture of the aorta, compression from tumors, or, infrequently, anticoagulation therapy.3
The role of anticoagulation
Spontaneous pelvic hematoma is usually missed as a cause of abdominal pain in patients on anticoagulation therapy and is mistaken for common acute conditions such as ulcer, diverticulitis, appendicitis, ovarian cyst torsion, and tumor.4 It usually develops within 5 days of starting anticoagulation therapy. Symptoms vary depending on the location of the hematoma and are best diagnosed with abdominal computed tomography, with sensitivity as high as 100%.
MANAGEMENT
Conservative management, reserved for patients in stable condition, includes temporarily stopping and reevaluating the risks and benefits of anticoagulation and antiplatelet agents, giving blood transfusions, and controlling pain. If conservative measures fail, options are arterial embolization, stent grafting, and blood vessel ligation.5 If these measures fail, patients should undergo surgical evacuation of the hematoma and ligation of bleeding vessels.6
TAKE-HOME MESSAGE
Subcutaneous injections, especially of anticoagulants, into the abdominal wall can increase the risk of hematoma. Other risk factors are older age, female sex, and thin body habitus with less abdominal fat.7 Healthcare professionals should avoid deep injections into the abdomen and should counsel patients and their caregivers about this, as well. The deltoid region could be a safer alternative.
A 78-year-old woman presented to the emergency department with shortness of breath and palpitations and was found to have atrial fibrillation with rapid ventricular response. Medical therapy with drug therapy and cardioversion proved ineffective. She then underwent atrioventricular node ablation and placement of a pacemaker.
At the time of admission, anticoagulation was started with full-dose enoxaparin, injected subcutaneously on the left side of the abdominal wall, as her CHA2DS2-VASc score (http://chadvasc.org) was 5, due to age, female sex, and history of heart failure and hypertension.
Four days after admission, she reported lower abdominal pain, and her urine output was minimal. A bladder scan showed more than 500 mL of residual urine. She was hemodynamically stable, but physical examination revealed mild abdominal distention and tenderness in the suprapubic region. Laboratory testing showed a sharp rise in serum creatinine and a drop in hematocrit.
The patient was initially managed conservatively with serial physical examinations, monitoring of the hematocrit, serial imaging studies, and discontinuation of anticoagulation, but the pain and anuria persisted. Repeat computed tomography 15 days after admission showed that the hematoma had expanded, and she now had hydronephrosis on the right side as well, requiring urologic intervention with bilateral nephrostomy tube placement.
The size of the hematoma was evaluated with serial abdominal and pelvic examinations. After several days, her urine output had improved, the nephrostomy tubes were removed, and she was discharged.
RECTUS SHEATH HEMATOMA
Our patient had a giant pelvic hematoma, probably arising from the rectus sheath. This uncommon problem can arise from trauma, anticoagulation, or increased intra-abdominal pressure, but it can also occur spontaneously.1
In rectus sheath hematoma, a branch of the inferior epigastric artery is injured at its insertion into the rectus abdominis muscle. Symptoms arise if bleeding does not stop spontaneously from a tamponade effect.2
We speculate that in our patient, deep injection of enoxaparin into the abdominal wall injured the inferior epigastric artery, which started the hematoma, and the bleeding was exacerbated by the anticoagulation effect of the enoxaparin.
Another form of pelvic hematoma is retroperitoneal. It is most commonly caused by trauma but can occur due to rupture of the aorta, compression from tumors, or, infrequently, anticoagulation therapy.3
The role of anticoagulation
Spontaneous pelvic hematoma is usually missed as a cause of abdominal pain in patients on anticoagulation therapy and is mistaken for common acute conditions such as ulcer, diverticulitis, appendicitis, ovarian cyst torsion, and tumor.4 It usually develops within 5 days of starting anticoagulation therapy. Symptoms vary depending on the location of the hematoma and are best diagnosed with abdominal computed tomography, with sensitivity as high as 100%.
MANAGEMENT
Conservative management, reserved for patients in stable condition, includes temporarily stopping and reevaluating the risks and benefits of anticoagulation and antiplatelet agents, giving blood transfusions, and controlling pain. If conservative measures fail, options are arterial embolization, stent grafting, and blood vessel ligation.5 If these measures fail, patients should undergo surgical evacuation of the hematoma and ligation of bleeding vessels.6
TAKE-HOME MESSAGE
Subcutaneous injections, especially of anticoagulants, into the abdominal wall can increase the risk of hematoma. Other risk factors are older age, female sex, and thin body habitus with less abdominal fat.7 Healthcare professionals should avoid deep injections into the abdomen and should counsel patients and their caregivers about this, as well. The deltoid region could be a safer alternative.
- Cherry WB, Mueller PS. Rectus sheath hematoma: review of 126 cases at a single institution. Medicine (Baltimore) 2006; 85(2):105–110. doi:10.1097/01.md.0000216818.13067.5a
- Hatjipetrou A, Anyfantakis D, Kastanakis M. Rectus sheath hematoma: a review of the literature. Int J Surg 2015; 13:267–271. doi:10.1016/j.ijsu.2014.12.015
- Haq MM, Taimur SDM, Khan SR, Rahman MA. Retroperitoneal hematoma following enoxaparin treatment in an elderly woman—a case report. Cardiovasc J 2010; 3(1):94–97. doi:10.3329/cardio.v3i1.6434
- Luhmann A, Williams EV. Rectus sheath hematoma: a series of unfortunate events. World J Surg 2006; 30(11):2050–2055. doi:10.1007/s00268-005-0702-9
- Pace F, Colombo GM, Del Vecchio LR, et al. Low molecular weight heparin and fatal spontaneous extraperitoneal hematoma in the elderly. Geriatr Gerontol Int 2012; 12(1):172–174. doi:10.1111/j.1447-0594.2011.00742.x
- Velicki L, Cemerlic-Adic N, Bogdanovic D, Mrdanin T. Rectus sheath haematoma: enoxaparin-related complication. Acta Clin Belg 2013; 68(2):147–149. doi:10.2143/ACB.68.2.3213
- Sheth HS, Kumar R, DiNella J, Janov C, Kaldas H, Smith RE. Evaluation of risk factors for rectus sheath hematoma. Clin Appl Thromb Hemost 2016; 22(3):292–296. doi:10.1177/1076029614553024
- Cherry WB, Mueller PS. Rectus sheath hematoma: review of 126 cases at a single institution. Medicine (Baltimore) 2006; 85(2):105–110. doi:10.1097/01.md.0000216818.13067.5a
- Hatjipetrou A, Anyfantakis D, Kastanakis M. Rectus sheath hematoma: a review of the literature. Int J Surg 2015; 13:267–271. doi:10.1016/j.ijsu.2014.12.015
- Haq MM, Taimur SDM, Khan SR, Rahman MA. Retroperitoneal hematoma following enoxaparin treatment in an elderly woman—a case report. Cardiovasc J 2010; 3(1):94–97. doi:10.3329/cardio.v3i1.6434
- Luhmann A, Williams EV. Rectus sheath hematoma: a series of unfortunate events. World J Surg 2006; 30(11):2050–2055. doi:10.1007/s00268-005-0702-9
- Pace F, Colombo GM, Del Vecchio LR, et al. Low molecular weight heparin and fatal spontaneous extraperitoneal hematoma in the elderly. Geriatr Gerontol Int 2012; 12(1):172–174. doi:10.1111/j.1447-0594.2011.00742.x
- Velicki L, Cemerlic-Adic N, Bogdanovic D, Mrdanin T. Rectus sheath haematoma: enoxaparin-related complication. Acta Clin Belg 2013; 68(2):147–149. doi:10.2143/ACB.68.2.3213
- Sheth HS, Kumar R, DiNella J, Janov C, Kaldas H, Smith RE. Evaluation of risk factors for rectus sheath hematoma. Clin Appl Thromb Hemost 2016; 22(3):292–296. doi:10.1177/1076029614553024
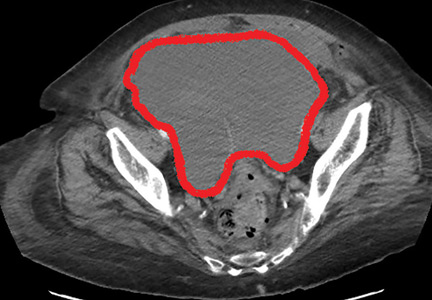
POCUS for hospitalists: The SHM position statement
Background: POCUS is becoming more prevalent in the daily practice of hospitalists; however, there are currently no established standards or guidelines for the use of POCUS for hospitalists.

Study design: Position statement.
Setting: SHM Executive Committee and Multi-Institutional POCUS faculty meeting through the Society of Hospital Medicine 2018 Annual Conference reviewed and approved this statement.
Synopsis: In contrast to the comprehensive ultrasound exam, POCUS is used by hospitalists to answer focused questions, by the same clinician who is generating the clinical question, to evaluate multiple body systems, or to serially investigate changes clinical status or evaluate responses to therapy.
This position statement provides guidance on the use of POCUS by hospitalists and the administrators who oversee it by outlining POCUS in terms of common diagnostic and procedural applications; training; assessments by the categories of basic knowledge, image acquisition, interpretation, clinical integration, and certification and maintenance of skills; and program management.
Bottom line: This position statement by the SHM provides guidance for hospitalists and administrators on the use and oversight of POCUS.
Citation: Soni NJ et al. Point-of-care ultrasound for hospitalists: A position statement of the Society of Hospital Medicine. J Hosp Med. 2019 Jan 2;14:E1-E6.
Dr. Wang is an associate professor of medicine in the division of general and hospital medicine at UT Health San Antonio and a hospitalist at South Texas Veterans Health Care System.
Background: POCUS is becoming more prevalent in the daily practice of hospitalists; however, there are currently no established standards or guidelines for the use of POCUS for hospitalists.
Study design: Position statement.
Setting: SHM Executive Committee and Multi-Institutional POCUS faculty meeting through the Society of Hospital Medicine 2018 Annual Conference reviewed and approved this statement.
Synopsis: In contrast to the comprehensive ultrasound exam, POCUS is used by hospitalists to answer focused questions, by the same clinician who is generating the clinical question, to evaluate multiple body systems, or to serially investigate changes clinical status or evaluate responses to therapy.
This position statement provides guidance on the use of POCUS by hospitalists and the administrators who oversee it by outlining POCUS in terms of common diagnostic and procedural applications; training; assessments by the categories of basic knowledge, image acquisition, interpretation, clinical integration, and certification and maintenance of skills; and program management.
Bottom line: This position statement by the SHM provides guidance for hospitalists and administrators on the use and oversight of POCUS.
Citation: Soni NJ et al. Point-of-care ultrasound for hospitalists: A position statement of the Society of Hospital Medicine. J Hosp Med. 2019 Jan 2;14:E1-E6.
Dr. Wang is an associate professor of medicine in the division of general and hospital medicine at UT Health San Antonio and a hospitalist at South Texas Veterans Health Care System.
Background: POCUS is becoming more prevalent in the daily practice of hospitalists; however, there are currently no established standards or guidelines for the use of POCUS for hospitalists.
Study design: Position statement.
Setting: SHM Executive Committee and Multi-Institutional POCUS faculty meeting through the Society of Hospital Medicine 2018 Annual Conference reviewed and approved this statement.
Synopsis: In contrast to the comprehensive ultrasound exam, POCUS is used by hospitalists to answer focused questions, by the same clinician who is generating the clinical question, to evaluate multiple body systems, or to serially investigate changes clinical status or evaluate responses to therapy.
This position statement provides guidance on the use of POCUS by hospitalists and the administrators who oversee it by outlining POCUS in terms of common diagnostic and procedural applications; training; assessments by the categories of basic knowledge, image acquisition, interpretation, clinical integration, and certification and maintenance of skills; and program management.
Bottom line: This position statement by the SHM provides guidance for hospitalists and administrators on the use and oversight of POCUS.
Citation: Soni NJ et al. Point-of-care ultrasound for hospitalists: A position statement of the Society of Hospital Medicine. J Hosp Med. 2019 Jan 2;14:E1-E6.
Dr. Wang is an associate professor of medicine in the division of general and hospital medicine at UT Health San Antonio and a hospitalist at South Texas Veterans Health Care System.