User login
Septic shock: The initial moments and beyond
Considerably fewer patients who develop sepsis are dying of it now, thanks to a number of studies of how to reverse sepsis-induced tissue hypoxia.1 The greatest strides in improving outcomes have been attributed to better early management, which includes prompt recognition of sepsis, rapid initiation of antimicrobial therapy, elimination of the source of infection, and early goal-directed therapy. Thus, even though the incidence of severe sepsis and septic shock is increasing,2,3 the Surviving Sepsis Campaign has documented a significant decrease in unadjusted mortality rates (37% to 30.8%) associated with the bundled approach in the management of sepsis.4 (We will talk about this later in the article.)
This review will summarize the evidence for the early management of septic shock and will evaluate the various treatment decisions beyond the initial phases of resuscitation.
INFLAMMATION AND VASODILATION
Sepsis syndrome starts with an infection that leads to a proinflammatory state with a complex interaction between anti-inflammatory and proinflammatory mediators, enhanced coagulation, and impaired fibrinolysis.5,6
Sepsis induces vasodilation by way of inappropriate activation of vasodilatory mechanisms (increased synthesis of nitric oxide and vasopressin deficiency) and failure of vasoconstrictor mechanisms (activation of ATP-sensitive potassium channels in vascular smooth muscle).7 Thus, the hemodynamic abnormalities are multifactorial, and the resultant tissue hypoperfusion further contributes to the proinflammatory and procoagulant state, precipitating multiorgan dysfunction and, often, death.
DEFINITIONS
- Sepsis—infection together with systemic manifestation of inflammatory response
- Severe sepsis—sepsis plus induced organ dysfunction or evidence of tissue hypoperfusion
- Septic shock—sepsis-induced hypotension persisting despite adequate fluid resuscitation.
EARLY MANAGEMENT OF SEPTIC SHOCK
Early in the course of septic shock, the physician’s job is to:
- Recognize it promptly
- Begin empiric antibiotic therapy quickly
- Eliminate the source of infection, if applicable, eg, by removing an infected central venous catheter
- Give fluid resuscitation, titrated to specific goals
- Give vasopressor therapy to maintain blood pressure, organ perfusion, and oxygen delivery (Table 1).

The line between “early” and “late” is not clear. Traditionally, it has been drawn at 6 hours from presentation, and this cutoff was used in some of the studies we will discuss here.
Recognizing severe sepsis early in its course
The diagnosis of severe sepsis may be challenging, since up to 40% of patients may present with cryptic shock. These patients may not be hemodynamically compromised but may show evidence of tissue hypoxia, eg, an elevated serum lactate concentration or a low central venous oxygen saturation (Scvo2), or both.8 In view of this, much effort has gone into finding a biomarker that, in addition to clinical features, can help identify patients in an early stage of sepsis.
Procalcitonin levels rise in response to severe bacterial infection,9 and they correlate with sepsis-related organ failure scores and outcomes.10,11 Thus, the serum procalcitonin level may help in assessing the severity of sepsis, especially when combined with standard clinical and laboratory variables. However, controversy exists about the threshold to use in making decisions about antibiotic therapy and the value of this test in differentiating severe noninfectious inflammatory reactions from infectious causes of shock.12 Therefore, it is not widely used in clinical practice.
Serum lactate has been used for decades as a marker of tissue hypoperfusion. It is typically elevated in patients with severe sepsis and septic shock, and although the hyperlactatemia could be a result of global hypoperfusion, it can also be secondary to sepsis-induced mitochondrial dysfunction,13 impaired pyruvate dehydrogenase activity,14 increased aerobic glycolysis by catecholamine-stimulated sodium-potassium pump hyperactivity,15 and even impaired clearance.16
But whatever the mechanism, elevated lactate in severe sepsis and septic shock predicts a poor outcome and may help guide aggressive resuscitation. In fact, early lactate clearance (ie, normalization of an elevated value on repeat testing within the first 6 hours) is associated with better outcomes in patients with severe sepsis and septic shock.17,18
Panels of biomarkers. A literature search revealed over 3,000 papers on 178 different biomarkers in sepsis.19 Many of these biomarkers lack sufficient specificity and sensitivity for clinical use, and thus some investigators have suggested using a panel of them to enhance their predictive ability. Shapiro et al20 evaluated 971 patients admitted to the emergency department with suspected infection and discovered that a panel of three biomarkers (neutrophil gelatinase-associated lipocalin, protein C, and interleukin-1 receptor antagonist) was highly predictive of severe sepsis, septic shock, and death.
Starting empiric antibiotic therapy early
As soon as severe sepsis and septic shock are recognized, it is imperative that adequate empiric antibiotic treatment be started, along with infectious source control if applicable.21 The Surviving Sepsis Campaign guidelines recommend starting intravenous antibiotics as early as possible—within the first hour of recognition of severe sepsis with or without septic shock.22
Kumar et al,23 in a multicenter retrospective study of patients with septic shock, found that each hour of delay in giving appropriate antimicrobial agents in the first 6 hours from the onset of hypotension was associated with a 7.6% decrease in the in-hospital survival rate.
In a similar study,24 the same investigators analyzed data from 5,715 septic shock patients regarding the impact of starting the right antimicrobial therapy. Appropriate antimicrobial agents (ie, those having in vitro activity against the isolated pathogens) were given in 80.1% of cases, and the survival rate in those who received appropriate antibiotics was drastically higher than in those who received inappropriate ones (52.0% vs 10.3%, P < .0001).
In addition, two recent studies evaluated the importance of early empiric antibiotic therapy in conjunction with resuscitative protocols.25,26 In a preplanned analysis of early antimicrobial use in a study comparing lactate clearance and Scvo2 as goals of therapy, Puskarich et al26 found that fewer patients who received antibiotics before shock was recognized (according to formal criteria) died. Similarly, in a retrospective study in patients presenting to the emergency department and treated with early goal-directed therapy (defined below), Gaieski et al25 found that the mortality rate was drastically lower when antibiotics were started within 1 hour of either triage or initiation of early goal-directed therapy.
In short, it is imperative to promptly start the most appropriate broad-spectrum antibiotics to target the most likely pathogens based on site of infection, patient risk of multidrug-resistant pathogens, and local susceptibility patterns.
Goal-directed resuscitative therapy
As with antimicrobial therapy, resuscitative therapy should be started early and directed at defined goals.
Rivers et al27 conducted a randomized, controlled study in patients with severe sepsis or septic shock presenting to an emergency department of an urban teaching hospital. The patients were at high risk and had either persistent hypotension after a fluid challenge or serum lactate levels of 4 mmol/L or higher.
Two hundred sixty patients were randomized to receive either early goal-directed therapy in a protocol aimed at maximizing the intravascular volume and correcting global tissue hypoxia or standard therapy in the first 6 hours after presentation. The goals in the goal-directed therapy group were:
- Central venous pressure 8 to 12 mm Hg (achieved with aggressive fluid resuscitation with crystalloids)
- Mean arterial blood pressure greater than 65 mm Hg (maintained with vasoactive drugs, if necessary)
- Scvo2 above 70%. To achieve this third goal, packed red blood cells were infused to reach a target hematocrit of greater than 30%. For patients with a hematocrit higher than 30% but still with an Scvo2 less than 70%, inotropic agents were added and titrated to the Scvo2 goal of 70%.
Goal-directed therapy reduced the in-hospital mortality rate by 16% (the mortality rates were 30.5% in the goal-directed group and 46.5% in the standard therapy group, P = .009) and also reduced the 28- and 60-day mortality rates by similar proportions.27
Subsequent studies of a protocol for early recognition and treatment of sepsis have concluded that early aggressive fluid resuscitation decreases the ensuing need for vasopressor support.28 A resuscitation strategy based on early goal-directed therapy is a major component of the initial resuscitation bundle recommended by the Surviving Sepsis Campaign.22 (A “bundle” refers to the implementation of a core set of recommendations involving the simultaneous adaptation of a number of interventions.)
Areas of debate. However, concerns have been raised about the design of the study by Rivers et al and the mortality rate in the control group, which was higher than one would expect from the patients’ Acute Physiology and Chronic Health Evaluation II (APACHE II) scores.29 In particular, the bundled approach they used precludes the ability to differentiate which interventions were responsible for the outcome benefits. Indeed, there were two major interventions in the early goal-directed therapy group: a protocol for achieving the goals described and the use of Scvo2 as a goal.
Aggressive fluid resuscitation is considered the most critical aspect of all the major interventions, and there is little argument on its value. The debate centers on central venous pressure as a preload marker, since after the publication of the early goal-directed therapy trial,27 several studies showed that central venous pressure may not be a valid measure to predict fluid responsiveness (discussed later in this paper).30,31
The choice of colloids or crystalloids for fluid resuscitation is another area of debate. Clinical evidence suggests that albumin is equivalent to normal saline in a heterogeneous intensive care unit population,32 but subgroup analyses suggest albumin may be superior in patients with septic shock.33 Studies are ongoing (NCT00707122, NCT01337934, and NCT00318942). The use of hydroxyethyl starch in severe sepsis is associated with higher rates of acute renal failure and need for renal replacement therapy than Ringer’s lactate,34 and is generally not recommended. This is further substantiated by two recent randomized controlled studies, which found that the use of hydroxyethyl starch for fluid resuscitation in severe sepsis, compared with crystalloids, did not reduce the mortality rate (and even increased it in one study), and was associated with more need for renal replacement therapy.35,36
The use of Scvo2 is yet another topic of debate, and other monitoring variables have been evaluated. A recent study assessed the noninferiority of incorporating venous lactate clearance into the early goal-directed therapy protocol vs Scvo2.37 Both groups had identical goals for central venous pressure and mean arterial pressure but differed in the use of lactate clearance (defined as at least a 10% decline) or Scvo2 (> 70%) as the goal for improving tissue hypoxia. There were no significant differences between groups in their in-hospital mortality rates (17% in the lactate clearance group vs 23% in the Scvo2 group; criteria for noninferiority met). This suggests that lactate may be an alternative to Scvo2 as a goal in early goal-directed therapy. However, a secondary analysis of the data revealed a lack of concordance in achieving lactate clearance and Scvo2 goals, which suggests that these parameters may be measuring distinct physiologic processes.38 Since the hemodynamic profiles of septic shock patients are complex, it may be prudent to use both of these markers of resuscitation until further studies are completed.
Given the debate, a number of prospective randomized trials are under way to evaluate resuscitative interventions. These include the Protocolized Care for Early Septic Shock trial (NCT00510835), the Australasian Resuscitation in Sepsis Evaluation trial (NCT00975793), and the Protocolised Management of Sepsis (ProMISe) trial in the United Kingdom (ISRCTN 36307479). These three trials will evaluate, collectively, close to 4,000 patients and will provide considerable insights into resuscitative interventions in septic shock.
Vasopressors: Which one to use?
If fluid therapy does not restore perfusion, vasopressors should be promptly initiated, as the longer that hypotension goes on, the lower the survival rate.39
But which vasopressor should be used? The early goal-directed therapy protocol used in the study by Rivers et al27 did not specify which vasopressor should be used to keep the mean arterial pressure above 65 mm Hg.
The Surviving Sepsis Campaign22 recommends norepinephrine as the first-choice vasopressor, with dopamine as an alternative only in selected patients, such as those with absolute or relative bradycardia.
The guidelines also recommend epinephrine to be added to or substituted for norepinephrine when an additional catecholamine is needed to maintain adequate blood pressure.22 Furthermore, vasopressin at a dose of 0.03 units/min can be added to norepinephrine with the intent of raising the blood pressure or decreasing the norepinephrine requirement. Higher doses of vasopressin should be reserved for salvage therapy.
Regarding phenylephrine, the guidelines recommend against its use except when norepinephrine use is associated with significant tachyarrhythmias, cardiac output is known to be higher, or as a salvage therapy.22
This is a topic of debate, with recent clinical studies offering further insight.
De Backer et al40 compared the effects of dopamine vs norepinephrine for the treatment of shock in 1,679 patients, 62% of whom had septic shock. Overall, there was a trend towards better outcomes with norepinephrine, but no significant difference in mortality rates at 28 days (52.5% with dopamine vs 48.5% with norepinephrine, P = .10). Importantly, fewer patients who were randomized to norepinephrine developed arrhythmias (12.4% vs 24.1%, P < .001), and the norepinephrine group required fewer days of study drug (11.0 vs 12.5, P = .01) and open-label vasopressors (12.6 vs 14.2, P = .007). Of note, patients with cardiogenic shock randomized to norepinephrine had a significantly lower mortality rate than those randomized to dopamine. Although no significant difference in outcome was found between the two vasopressors in the subgroup of patients with septic shock, the overall improvements in secondary surrogate markers suggest that norepinephrine should be the first-line agent.
Norepinephrine has also been compared with “secondary” vasopressors. Annane et al,41 in a prospective multicenter randomized controlled study, evaluated the effect of norepinephrine plus dobutamine vs epinephrine alone in managing septic shock. There was no significant difference in the primary outcome measure of 28-day mortality (34% with norepinephrine plus dobutamine vs 40% with epinephrine alone, P = .31). However, the study was powered to evaluate for an absolute risk reduction of 20% in the mortality rate, which would be a big reduction. A smaller reduction in the mortality rate, which would not have been statistically significant in this study, might still be considered clinically significant. Furthermore, the group randomized to norepinephrine plus dobutamine had more vasopressor-free days (20 days vs 22 days, P = .05) and less acidosis on days 1 to 4 than the group randomized to epinephrine.
Norepinephrine was also compared with phenylephrine as a first-line vasopressor in a randomized controlled trial in 32 patients with septic shock. No difference was found in cardiopulmonary performance, global oxygen transport, or regional hemodynamics between phenylephrine and norepinephrine.42
While encouraging, these preliminary data need to be verified in a larger randomized controlled trial with concrete outcome measures before being clinically adapted. Taken together, the above studies suggest that norepinephrine should be the initial vasopressor of choice for patients with septic shock.
CONTINUED MANAGEMENT OF SEPTIC SHOCK
How to manage septic shock after the initial stages is much less defined.

Uncertainty persists about the importance of achieving the early goals of resuscitation in patients who did not reach them in the initial 6 hours of treatment. Although there are data suggesting that extending the goals beyond the initial 6 hours may be beneficial, clinicians should use caution when interpreting these results in light of the observational design of the studies.43,44 For the purpose of this discussion, “continued management” of septic shock will mean after the first 6 hours and after all the early goals are met.
The clinical decisions necessary after the initial stages of resuscitation include:
- Whether further fluid resuscitation is needed
- Assessment for further and additional hemodynamic therapies
- Consideration of adjunctive therapies
- Reevaluation of antibiotic choices (Table 2).
Is more fluid needed? How can we tell?
There is considerable debate about the ideal method for assessing fluid responsiveness. In fact, one of the criticisms of the early goal-directed therapy study27 was that it used central venous pressure as a marker of fluid responsiveness.
Several studies have shown that central venous pressure or pulmonary artery occlusion pressure may not be valid measures of fluid responsiveness.45 In fact, in a retrospective study of 150 volume challenges, the area under the receiver-operating-characteristics curve of central venous pressure as a marker of fluid responsiveness was only 0.58. (Recall that the closer the area under the curve is to 1.0, the better the test; a value of 0.50 is the same as chance.) The area under the curve for pulmonary artery occlusion pressure was 0.63.46
In contrast, several dynamic indices have been proposed to better guide fluid resuscitation in mechanically ventilated patients.31 These are based on changes in stroke volume, aortic blood flow, or arterial pulse pressure in response to the ventilator cycle or passive leg-raising. A detailed review of these markers can be found elsewhere,31 but taken together, they have a sensitivity and specificity of over 90% for predicting fluid responsiveness. Clinicians may consider using dynamic markers of fluid responsiveness to determine when to give additional fluids, particularly after the first 6 hours of shock, in which data supporting the use of central venous pressure are lacking.
Optimal use of fluids is particularly important, since some studies suggest that “overresuscitation” has negative consequences. In a multicenter observational study of 1,177 patients with sepsis, after adjusting for a number of comorbidities and baseline severity of illness, the cumulative fluid balance in the first 72 hours after the onset of sepsis was independently associated with a worse mortality rate.47
Furthermore, in a retrospective analysis of a randomized controlled trial of vasopressin in conjunction with norepinephrine for septic shock, patients in the highest quartile of fluid balance (more fluid in than out) at 12 hours and 4 days after presentation had significantly higher mortality rates than those in the lowest two quartiles.48 The worse outcome with a positive fluid balance might be explained by worsening oxygenation and prolonged mechanical ventilation, as demonstrated by the Fluid and Catheter Treatment Trial in patients with acute lung injury or acute respiratory distress syndrome (ALI/ARDS).49 Indeed, when fluid balance in patients with septic shockinduced ALI/ARDS was evaluated, patients with both adequate initial fluid resuscitation and conservative late fluid management had a lower mortality rate than those with either one alone.50
In view of these findings, especially beyond the initial hours of resuscitation, clinicians should remember that further unnecessary fluid administration may have detrimental effects. Therefore, given the superior predictive abilities of dynamic markers of fluid responsiveness, these should be used to determine the need for further fluid boluses.
In cases in which patients are no longer fluid-responsive and need increasing levels of hemodynamic support, clinicians still have a number of options. These include increasing the current vasopressor dose or starting an additional therapy such as an alternative catecholamine vasopressor, vasopressin, inotropic therapy, or an adjunctive therapy such as a corticosteroid. The intervention could also be a combination of the above choices.
Adding catecholamines
The optimal time point or vasopressor dose at which to consider initiating additional therapies is unknown. However, the Vasopressin and Septic Shock Trial (VASST) provides some insight.51
This study compared two strategies: escalating doses of norepinephrine vs adding vasopressin to norepinephrine. Overall, adding vasopressin showed no benefit in terms of a lower mortality rate. However, in the subgroup of patients with norepinephrine requirements of 5 to 14 μg/min at study enrollment (ie, a low dose, reflecting less-severe sepsis) vasopressin was associated with a lower 28-day mortality rate (26.5% vs 35.7%, P = .05) and 90-day mortality rate (35.8% vs 46.1%, P = .04). Benefit was also noted in patients with other markers of lower disease severity such as low lactate levels or having received a single vasopressor at baseline.51
Although subgroup analyses should not generally be used to guide treatment decisions, a prospective trial may never be done to evaluate adding vasopressin to catecholamines earlier vs later. Thus, clinicians who choose to use vasopressin may consider starting this therapy when catecholamine doses are relatively low or before profound hyperlactatemia from prolonged tissue hypoxia has developed.
There is less evidence to guide clinicians who are considering adding a different catecholamine. The theoretical concerns of splanchnic ischemia and cardiac arrhythmia associated with higher doses of catecholamines are usually the impetus to limit a single catecholamine to a “maximum” dose. However, studies that have evaluated combination catecholamine therapies have generally studied combinations of vasopressors with inotropes and lacked standardization in their protocols, thus making them difficult to interpret.52–54 One could also argue that additional catecholamine therapies, which all function similarly, may have additive effects and cause even more adverse effects. As such, adding another vasopressor should be reserved for patients experiencing noticeable adverse effects (such as tachycardia) on first-line therapy.
Inotropic support
Left ventricular function should be assessed in all patients who continue to be hypotensive despite adequate fluid resuscitation and vasopressor therapy. In a study of patients with septic shock in whom echocardiography was performed daily for the first 3 days of hemodynamic support, new-onset left ventricular hypokinesia was found in 26 (39%) of 67 patients on presentation and in an additional 14 patients (21%) after at least 24 hours of norepinephrine.55 Adding inotropic support with dobutamine or epinephrine led to decreases in vasopressor dose and enhanced left ventricular ejection fraction.
In short, left ventricular hypokinesia is common in septic shock, may occur at presentation or after a period of vasopressor support, and is usually correctable with the addition of inotropic support.
Corticosteroids
Beyond hemodynamic support with fluids and catecholamines or vasopressin (or both), clinicians should also consider adjunctive corticosteroid therapy. However, for many years the issue has been controversial for patients with severe sepsis and septic shock.
Annane et al56 conducted a large, multicenter, randomized, double-blind, placebocontrolled trial to assess the effect of low doses of corticosteroids in patients with refractory septic shock. Overall, the 28-day mortality rate was 61% in the treatment group and 55% in the placebo group, which was not statistically significant (adjusted odds ratio 0.65, 95% confidence interval 0.39–1.07, P value .09). However, when separated by response to cosyntropin stimulation, those with a change in cortisol of 9 ug/dL or less (nonresponders) randomized to receive corticosteroids had significantly higher survival rates in the short term (28 days) and the long term (1 year). The positive results of this study led to the adoption of low-dose hydrocortisone as standard practice in most patients with septic shock.57
But then, to evaluate the effects of corticosteroids in a broader intensive-care population with septic shock, another trial was designed: the Corticosteroid Therapy of Septic Shock (CORTICUS) trial.58 Surprisingly, this multicenter, randomized, double-blind, placebo-controlled trial found no significant difference in survival between the group that received hydrocortisone and the placebo group, regardless of response to a cosyntropin stimulation test.
Taking into account the above studies and other randomized controlled trials, the 2012 Surviving Sepsis Campaign guidelines and the International Task Force for the Diagnosis and Management of Corticosteroid Insufficiency in Critically Ill Adult Patients recommend intravenous hydrocortisone therapy in adults with septic shock whose blood pressure responds poorly to fluid resuscitation and vasopressor therapy. These consensus statements do not recommend the cosyntropin stimulation test to identify patients with septic shock who should receive corticosteroids.22,59 The guidelines, however, do not explicitly define poor response to initial therapy.
Of note, in the Annane study, which found a lower mortality rate with corticosteroids, the patients were severely ill, with a mean baseline norepinephrine dose of 1.1 μg/kg/min. In contrast, in the CORTICUS study (which found no benefit of hydrocortisone), patients had lower baseline vasopressor doses, with a mean norepinephrine dose of 0.5 μg/kg/min.
While corticosteroids are associated with a higher rate of shock reversal 7 days after initiation, 59 this has not translated into a consistent reduction in the death rate. If a clinician is considering adding corticosteroids to decrease the risk of death, it would seem prudent to add this therapy in patients receiving norepinephrine in doses above 0.5 μg/kg/min.
The ideal sequence and combination of the above therapies including fluids, catecholamine vasopressors, vasopressin, inotropes, and vasopressors have not been elucidated. However, some preliminary evidence suggests an advantage with the combination of vasopressin and corticosteroids. In a subgroup analysis of the VASST study, in patients who received corticosteroids, the combination of vasopressin plus norepinephrine was associated with a lower 28-day mortality rate than with norepinephrine alone (35.9% vs 44.7%, P = .03).60 These findings have been replicated in other studies,61,62 prompting suggestions for a study of vasopressin with and without corticosteroids in patients on norepinephrine to elucidate the role of each therapy individually and in combination.
Tight glycemic control
As with corticosteroids, the pendulum for tight glycemic control in critically ill patients has swung widely in recent years. Enthusiasm was high at first after the publication of a study by van den Berghe et al, which described a 3.4% absolute reduction in mortality with intensive insulin therapy to maintain blood glucose at or below 110 mg/dL.63 However, the significant benefits found in this study were never replicated.
In fact, recent evidence suggests that tight glycemic control is associated with no benefit and a higher risk of hypoglycemia.34,64 In the largest randomized controlled trial of this topic, with more than 6,000 patients, intensive insulin therapy with a target blood glucose level of 81 to 108 mg/dL was associated with a significantly higher mortality rate (odds ratio 1.14, 95% confidence interval 1.02–1.28, P = .02) than with a target glucose level of less than 180 mg/dL.65 Furthermore, in a recent follow-up analysis,66 moderate hypoglycemia (serum glucose 41–70 mg/dL) and severe hypoglycemia (serum glucose < 41 mg/dL) were associated with a higher rate of death in a dose-response relationship.66
Taking this information together, clinicians should be aware that there is no additional benefit in lowering blood glucose below the range of 140 to 180 mg/dL, and that doing so may be harmful.
Drotecogin alfa
Drotecogin alfa (Xigris) was another adjunctive therapy that has fallen from favor. It was approved for the treatment of severe sepsis in light of promising findings in initial studies.67
However, on October 25, 2011, drotecogin alfa was voluntarily withdrawn from the market by the manufacturer after another study found no beneficial effect on the mortality rates at 28 days or at 90 days.68 Furthermore, no difference could be found regarding any predetermined primary or secondary outcome measures.
Continued antibiotic therapy
The decision whether to continue initial empiric antimicrobial coverage, broaden it, or de-escalate must be faced for all patients with septic shock, and is ultimately clinical.
The serum procalcitonin level has been proposed to guide antibiotic discontinuation in several clinical settings, although there are still questions about the safety of such an approach. The largest randomized trial published to date reported that a procalcitoninguided strategy to treat suspected bacterial infections in nonsurgical patients could reduce antibiotic exposure with no apparent adverse outcomes.69 On the other hand, other data discourage the use of procalcitonin-guided antimicrobial escalation, as this approach did not improve survival and worsened organ function and length of stay in the intensive care unit.70
The Surviving Sepsis Campaign guidelines recommend combination antibiotic therapy for no longer than 3 to 5 days and limiting the duration of antibiotics in most cases to 7 to 10 days.22
TRIALS ARE ONGOING
The understanding of the pathophysiology and treatment of sepsis has greatly advanced over the last decade. Adoption of evidence-based protocols for managing patients with septic shock has improved outcomes. Nevertheless, many multicenter trials are being conducted worldwide to look into some of the most controversial therapies, and their results will guide therapy in the future.
- Kumar G, Kumar N, Taneja A, et al. Nationwide trends of severe sepsis in the 21st century (2000–2007). Chest 2011; 140:1223–1231.
- Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med 2001; 29:1303–1310.
- Annane D, Aegerter P, Jars-Guincestre MC, Guidet B. Current epidemiology of septic shock: the CUB-Rea Network. Am J Respir Crit Care Med 2003; 168:165–172.
- Levy MM, Dellinger RP, Townsend SR, et al. The Surviving Sepsis Campaign: results of an international guideline-based performance improvement program targeting severe sepsis. Intensive Care Med 2010; 36:222–231.
- Amaral A, Opal SM, Vincent JL. Coagulation in sepsis. Intensive Care Med 2004; 30:1032–1040.
- Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med 2003; 348:138–150.
- Landry DW, Oliver JA. The pathogenesis of vasodilatory shock. N Engl J Med 2001; 345:588–595.
- Rady MY, Rivers EP, Nowak RM. Resuscitation of the critically ill in the ED: responses of blood pressure, heart rate, shock index, central venous oxygen saturation, and lactate. Am J Emerg Med 1996; 14:218–225.
- Assicot M, Gendrel D, Carsin H, Raymond J, Guilbaud J, Bohuon C. High serum procalcitonin concentrations in patients with sepsis and infection. Lancet 1993; 34:515–518.
- Muller B, Becker KL, Schachinger H, et al. Calcitonin precursors are reliable markers of sepsis in a medical intensive care unit. Crit Care Med 2000; 28:977–983.
- Meisner M, Tschaikowsky K, Palmaers T, Schmidt J. Comparison of procalcitonin (PCT) and C-reactive protein (CRP) plasma concentrations at different SOFA scores during the course of sepsis and MODS. Crit Care (London, England) 1999; 3:45–50.
- Tang BM, Eslick GD, Craig JC, McLean AS. Accuracy of procalcitonin for sepsis diagnosis in critically ill patients: systematic review and meta-analysis. Lancet Infect Dis 2007; 7:210–217.
- Brealey D, Brand M, Hargreaves I, et al. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 2002; 360:219–223.
- Vary TC. Sepsis-induced alterations in pyruvate dehydrogenase complex activity in rat skeletal muscle: effects on plasma lactate. Shock 1996; 6:89–94.
- Levy B, Gibot S, Franck P, Cravoisy A, Bollaert PE. Relation between muscle Na+K+ ATPase activity and raised lactate concentrations in septic shock: a prospective study. Lancet 2005; 365:871–875.
- Levraut J, Ciebiera JP, Chave S, et al. Mild hyperlactatemia in stable septic patients is due to impaired lactate clearance rather than over-production. Am J Respir Crit Care Med 1998; 157:1021–1026.
- Arnold RC, Shapiro NI, Jones AE, et al. Multicenter study of early lactate clearance as a determinant of survival in patients with presumed sepsis. Shock 2009; 32:35–39.
- Nguyen HB, Rivers EP, Knoblich BP, et al. Early lactate clearance is associated with improved outcome in severe sepsis and septic shock. Crit Care Med 2004; 32:1637–1642.
- Pierrakos C, Vincent JL. Sepsis biomarkers: a review. Crit Care 2010; 14:R15.
- Shapiro NI, Trzeciak S, Hollander JE, et al. A prospective, multicenter derivation of a biomarker panel to assess risk of organ dysfunction, shock, and death in emergency department patients with suspected sepsis. Crit Care Med 2009; 37:96–104.
- Marshall JC, al Naqbi A. Principles of source control in the management of sepsis. Crit Care Clin 2009; 25:753–768,viii–ix.
- Dellinger RP, Levy MM, Rhodes A, et al. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock: 2012. Crit Care Med 2013; 41:580–637.
- Kumar A, Roberts D, Wood KE, et al. Duration of hypotension before initiation of effective antimicrobial therapy is the critical determinant of survival in human septic shock. Crit Care Med 2006; 34:1589–1596.
- Kumar A, Ellis P, Arabi Y, et al. Initiation of inappropriate antimicrobial therapy results in a fivefold reduction of survival in human septic shock. Chest 2009; 136:1237–1248.
- Gaieski DF, Mikkelsen ME, Band RA, et al. Impact of time to antibiotics on survival in patients with severe sepsis or septic shock in whom early goal-directed therapy was initiated in the emergency department. Crit Care Med 2010; 38:1045–1053.
- Puskarich MA, Trzeciak S, Shapiro NI, et al. Association between timing of antibiotic administration and mortality from septic shock in patients treated with a quantitative resuscitation protocol. Crit Care Med 2011; 39:2066–2071.
- Rivers E, Nguyen B, Havstad S, et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med 2001; 345:1368–1377.
- Micek ST, Roubinian N, Heuring T, et al. Before-after study of a standardized hospital order set for the management of septic shock. Crit Care Med 2006; 34:2707–2713.
- Schmidt GA. Counterpoint: adherence to early goal-directed therapy: does it really matter? No. Both risks and benefits require further study. Chest 2010; 138:480–483; discussion 483–484.
- Jain RK, Antonio BL, Bowton DL, Houle TT, MacGregor DA. Variability in central venous pressure measurements and the potential impact on fluid management. Shock 2009; 33:253–257.
- Durairaj L, Schmidt GA. Fluid therapy in resuscitated sepsis: less is more. Chest 2008; 133:252–263.
- Finfer S, Bellomo R, Boyce N, French J, Myburgh J, Norton R. A comparison of albumin and saline for fluid resuscitation in the intensive care unit. N Engl J Med 2004; 350:2247–2256.
- Finfer S, McEvoy S, Bellomo R, McArthur C, Myburgh J, Norton R. Impact of albumin compared to saline on organ function and mortality of patients with severe sepsis. Intensive Care Med 2011; 37:86–96.
- Brunkhorst FM, Engel C, Bloos F, et al. Intensive insulin therapy and pentastarch resuscitation in severe sepsis. N Engl J Med 2008; 358:125–139.
- Perner A, Haase N, Guttormsen AB, et al. Hydroxyethyl starch 130/0.42 versus Ringer’s acetate in severe sepsis. N Engl J Med 2012; 367:124–134.
- Myburgh JA, Finfer S, Bellomo R, et al. Hydroxyethyl starch or saline for fluid resuscitation in intensive care. N Engl J Med 2012; 367:1901–1911.
- Jones AE, Shapiro NI, Trzeciak S, Arnold RC, Claremont HA, Kline JA. Lactate clearance vs central venous oxygen saturation as goals of early sepsis therapy: a randomized clinical trial. JAMA 2010; 303:739–746.
- Puskarich MA, Trzciak S, Shapiro NI, Kline JA, Jones AE. Concordance and prognostic value of central venous oxygen saturation and lactate clearance in emergency department patients with septic shock. Acad Emerg Med 2011; 19:S159–S160.
- Dunser MW, Takala J, Ulmer H, et al. Arterial blood pressure during early sepsis and outcome. Intensive Care Med 2009; 35:1225–1233.
- De Backer D, Biston P, Devriendt J, et al. Comparison of dopamine and norepinephrine in the treatment of shock. N Engl J Med 2010; 362:779–789.
- Annane D, Vignon P, Renault A, et al. Norepinephrine plus dobutamine versus epinephrine alone for management of septic shock: a randomised trial. Lancet 2007; 370:676–684.
- Morelli A, Ertmer C, Rehberg S, et al. Phenylephrine versus norepinephrine for initial hemodynamic support of patients with septic shock: a randomized, controlled trial. Crit Care (London, England) 2008; 12:R143.
- Coba V, Whitmill M, Mooney R, et al. Resuscitation bundle compliance in severe sepsis and septic shock: improves survival, is better late than never. J Intensive Care Med 2011 Jan 10[Epub ahead of print].
- Castellanos-Ortega A, Suberviola B, Garcia-Astudillo LA, Ortiz F, Llorca J, Delgado-Rodriguez M. Late compliance with the sepsis resuscitation bundle: impact on mortality. Shock 2011; 36:542–547.
- Marik PE, Baram M, Vahid B. Does central venous pressure predict fluid responsiveness? A systematic review of the literature and the tale of seven mares. Chest 2008; 134:172–178.
- Osman D, Ridel C, Ray P, et al. Cardiac filling pressures are not appropriate to predict hemodynamic response to volume challenge. Crit Care Med 2007; 35:64–68.
- Vincent JL, Sakr Y, Sprung CL, et al. Sepsis in European intensive care units: results of the SOAP study. Crit Care Med 2006; 34:344–353.
- Boyd JH, Forbes J, Nakada TA, Walley KR, Russell JA. Fluid resuscitation in septic shock: a positive fluid balance and elevated central venous pressure are associated with increased mortality. Crit Care Med 2011; 39:259–265.
- Wiedemann HP, Wheeler AP, Bernard GR, et al. Comparison of two fluid-management strategies in acute lung injury. N Engl J Med 2006; 354:2564–2575.
- Murphy CV, Schramm GE, Doherty JA, et al. The importance of fluid management in acute lung injury secondary to septic shock. Chest 2009; 136:102–109.
- Russell JA, Walley KR, Singer J, et al. Vasopressin versus norepinephrine infusion in patients with septic shock. N Engl J Med 2008; 358:877–887.
- Vincent JL, Roman A, Kahn RJ. Dobutamine administration in septic shock: addition to a standard protocol. Crit Care Med 1990; 18:689–693.
- Levy B, Bollaert PE, Charpentier C, et al. Comparison of norepinephrine and dobutamine to epinephrine for hemodynamics, lactate metabolism, and gastric tonometric variables in septic shock: a prospective, randomized study. Intensive Care Med 1997; 23:282–287.
- Redl-Wenzl EM, Armbruster C, Edelmann G, et al. The effects of norepinephrine on hemodynamics and renal function in severe septic shock states. Intensive Care Med 1993; 19:151–154.
- Vieillard-Baron A, Caille V, Charron C, Belliard G, Page B, Jardin F. Actual incidence of global left ventricular hypokinesia in adult septic shock. Crit Care Med 2008; 36:1701–1706.
- Annane D, Sebille V, Charpentier C, et al. Effect of treatment with low doses of hydrocortisone and fludrocortisone on mortality in patients with septic shock. JAMA 2002; 288:862–871.
- Dellinger RP, Carlet JM, Masur H, et al. Surviving Sepsis Campaign guidelines for management of severe sepsis and septic shock. Crit Care Med 2004; 32:858–873.
- Sprung CL, Annane D, Keh D, et al. Hydrocortisone therapy for patients with septic shock. N Engl J Med 2008; 358:111–124.
- Marik PE, Pastores SM, Annane D, et al. Recommendations for the diagnosis and management of corticosteroid insufficiency in critically ill adult patients: consensus statements from an international task force by the American College of Critical Care Medicine. Crit Care Med 2008; 36:1937–1949.
- Russell JA, Walley KR, Gordon AC, et al. Interaction of vasopressin infusion, corticosteroid treatment, and mortality of septic shock. Crit Care Med 2009; 37:811–818.
- Bauer SR, Lam SW, Cha SS, Oyen LJ. Effect of corticosteroids on arginine vasopressin-containing vasopressor therapy for septic shock: a case control study. J Crit Care 2008; 23:500–506.
- Torgersen C, Luckner G, Schroder DC, et al. Concomitant arginine-vasopressin and hydrocortisone therapy in severe septic shock: association with mortality. Intensive Care Med 2011; 37:1432–1437.
- van den Berghe G, Wouters P, Weekers F, et al. Intensive insulin therapy in critically ill patients. N Engl J Med 2001; 345:1359–1367.
- Preiser JC, Devos P, Ruiz-Santana S, et al. A prospective randomised multi-centre controlled trial on tight glucose control by intensive insulin therapy in adult intensive care units: the Glucontrol study. Intensive Care Med 2009; 35:1738–1748.
- Finfer S, Chittock DR, Su SY, et al. Intensive versus conventional glucose control in critically ill patients. N Engl J Med 2009; 360:1283–1297.
- Finfer S, Liu B, Chittock DR, et al. Hypoglycemia and risk of death in critically ill patients. N Engl J Med 2012; 367:1108–1118.
- Bernard GR, Vincent JL, Laterre PF, et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med 2001; 344:699–709.
- Ranieri VM, Thompson BT, Barie PS, et al. Drotrecogin alfa (activated) in adults with septic shock. N Engl J Med 2012; 366:2055–2064.
- Bouadma L, Luyt CE, Tubach F, et al. Use of procalcitonin to reduce patients’ exposure to antibiotics in intensive care units (PRORATA trial): a multicentre randomised controlled trial. Lancet 2009; 375:463–474.
- Jensen JU, Hein L, Lundgren B, et al. Procalcitonin-guided interventions against infections to increase early appropriate antibiotics and improve survival in the intensive care unit: a randomized trial. Crit Care Med 2011; 39:2048–2058.
Considerably fewer patients who develop sepsis are dying of it now, thanks to a number of studies of how to reverse sepsis-induced tissue hypoxia.1 The greatest strides in improving outcomes have been attributed to better early management, which includes prompt recognition of sepsis, rapid initiation of antimicrobial therapy, elimination of the source of infection, and early goal-directed therapy. Thus, even though the incidence of severe sepsis and septic shock is increasing,2,3 the Surviving Sepsis Campaign has documented a significant decrease in unadjusted mortality rates (37% to 30.8%) associated with the bundled approach in the management of sepsis.4 (We will talk about this later in the article.)
This review will summarize the evidence for the early management of septic shock and will evaluate the various treatment decisions beyond the initial phases of resuscitation.
INFLAMMATION AND VASODILATION
Sepsis syndrome starts with an infection that leads to a proinflammatory state with a complex interaction between anti-inflammatory and proinflammatory mediators, enhanced coagulation, and impaired fibrinolysis.5,6
Sepsis induces vasodilation by way of inappropriate activation of vasodilatory mechanisms (increased synthesis of nitric oxide and vasopressin deficiency) and failure of vasoconstrictor mechanisms (activation of ATP-sensitive potassium channels in vascular smooth muscle).7 Thus, the hemodynamic abnormalities are multifactorial, and the resultant tissue hypoperfusion further contributes to the proinflammatory and procoagulant state, precipitating multiorgan dysfunction and, often, death.
DEFINITIONS
- Sepsis—infection together with systemic manifestation of inflammatory response
- Severe sepsis—sepsis plus induced organ dysfunction or evidence of tissue hypoperfusion
- Septic shock—sepsis-induced hypotension persisting despite adequate fluid resuscitation.
EARLY MANAGEMENT OF SEPTIC SHOCK
Early in the course of septic shock, the physician’s job is to:
- Recognize it promptly
- Begin empiric antibiotic therapy quickly
- Eliminate the source of infection, if applicable, eg, by removing an infected central venous catheter
- Give fluid resuscitation, titrated to specific goals
- Give vasopressor therapy to maintain blood pressure, organ perfusion, and oxygen delivery (Table 1).
The line between “early” and “late” is not clear. Traditionally, it has been drawn at 6 hours from presentation, and this cutoff was used in some of the studies we will discuss here.
Recognizing severe sepsis early in its course
The diagnosis of severe sepsis may be challenging, since up to 40% of patients may present with cryptic shock. These patients may not be hemodynamically compromised but may show evidence of tissue hypoxia, eg, an elevated serum lactate concentration or a low central venous oxygen saturation (Scvo2), or both.8 In view of this, much effort has gone into finding a biomarker that, in addition to clinical features, can help identify patients in an early stage of sepsis.
Procalcitonin levels rise in response to severe bacterial infection,9 and they correlate with sepsis-related organ failure scores and outcomes.10,11 Thus, the serum procalcitonin level may help in assessing the severity of sepsis, especially when combined with standard clinical and laboratory variables. However, controversy exists about the threshold to use in making decisions about antibiotic therapy and the value of this test in differentiating severe noninfectious inflammatory reactions from infectious causes of shock.12 Therefore, it is not widely used in clinical practice.
Serum lactate has been used for decades as a marker of tissue hypoperfusion. It is typically elevated in patients with severe sepsis and septic shock, and although the hyperlactatemia could be a result of global hypoperfusion, it can also be secondary to sepsis-induced mitochondrial dysfunction,13 impaired pyruvate dehydrogenase activity,14 increased aerobic glycolysis by catecholamine-stimulated sodium-potassium pump hyperactivity,15 and even impaired clearance.16
But whatever the mechanism, elevated lactate in severe sepsis and septic shock predicts a poor outcome and may help guide aggressive resuscitation. In fact, early lactate clearance (ie, normalization of an elevated value on repeat testing within the first 6 hours) is associated with better outcomes in patients with severe sepsis and septic shock.17,18
Panels of biomarkers. A literature search revealed over 3,000 papers on 178 different biomarkers in sepsis.19 Many of these biomarkers lack sufficient specificity and sensitivity for clinical use, and thus some investigators have suggested using a panel of them to enhance their predictive ability. Shapiro et al20 evaluated 971 patients admitted to the emergency department with suspected infection and discovered that a panel of three biomarkers (neutrophil gelatinase-associated lipocalin, protein C, and interleukin-1 receptor antagonist) was highly predictive of severe sepsis, septic shock, and death.
Starting empiric antibiotic therapy early
As soon as severe sepsis and septic shock are recognized, it is imperative that adequate empiric antibiotic treatment be started, along with infectious source control if applicable.21 The Surviving Sepsis Campaign guidelines recommend starting intravenous antibiotics as early as possible—within the first hour of recognition of severe sepsis with or without septic shock.22
Kumar et al,23 in a multicenter retrospective study of patients with septic shock, found that each hour of delay in giving appropriate antimicrobial agents in the first 6 hours from the onset of hypotension was associated with a 7.6% decrease in the in-hospital survival rate.
In a similar study,24 the same investigators analyzed data from 5,715 septic shock patients regarding the impact of starting the right antimicrobial therapy. Appropriate antimicrobial agents (ie, those having in vitro activity against the isolated pathogens) were given in 80.1% of cases, and the survival rate in those who received appropriate antibiotics was drastically higher than in those who received inappropriate ones (52.0% vs 10.3%, P < .0001).
In addition, two recent studies evaluated the importance of early empiric antibiotic therapy in conjunction with resuscitative protocols.25,26 In a preplanned analysis of early antimicrobial use in a study comparing lactate clearance and Scvo2 as goals of therapy, Puskarich et al26 found that fewer patients who received antibiotics before shock was recognized (according to formal criteria) died. Similarly, in a retrospective study in patients presenting to the emergency department and treated with early goal-directed therapy (defined below), Gaieski et al25 found that the mortality rate was drastically lower when antibiotics were started within 1 hour of either triage or initiation of early goal-directed therapy.
In short, it is imperative to promptly start the most appropriate broad-spectrum antibiotics to target the most likely pathogens based on site of infection, patient risk of multidrug-resistant pathogens, and local susceptibility patterns.
Goal-directed resuscitative therapy
As with antimicrobial therapy, resuscitative therapy should be started early and directed at defined goals.
Rivers et al27 conducted a randomized, controlled study in patients with severe sepsis or septic shock presenting to an emergency department of an urban teaching hospital. The patients were at high risk and had either persistent hypotension after a fluid challenge or serum lactate levels of 4 mmol/L or higher.
Two hundred sixty patients were randomized to receive either early goal-directed therapy in a protocol aimed at maximizing the intravascular volume and correcting global tissue hypoxia or standard therapy in the first 6 hours after presentation. The goals in the goal-directed therapy group were:
- Central venous pressure 8 to 12 mm Hg (achieved with aggressive fluid resuscitation with crystalloids)
- Mean arterial blood pressure greater than 65 mm Hg (maintained with vasoactive drugs, if necessary)
- Scvo2 above 70%. To achieve this third goal, packed red blood cells were infused to reach a target hematocrit of greater than 30%. For patients with a hematocrit higher than 30% but still with an Scvo2 less than 70%, inotropic agents were added and titrated to the Scvo2 goal of 70%.
Goal-directed therapy reduced the in-hospital mortality rate by 16% (the mortality rates were 30.5% in the goal-directed group and 46.5% in the standard therapy group, P = .009) and also reduced the 28- and 60-day mortality rates by similar proportions.27
Subsequent studies of a protocol for early recognition and treatment of sepsis have concluded that early aggressive fluid resuscitation decreases the ensuing need for vasopressor support.28 A resuscitation strategy based on early goal-directed therapy is a major component of the initial resuscitation bundle recommended by the Surviving Sepsis Campaign.22 (A “bundle” refers to the implementation of a core set of recommendations involving the simultaneous adaptation of a number of interventions.)
Areas of debate. However, concerns have been raised about the design of the study by Rivers et al and the mortality rate in the control group, which was higher than one would expect from the patients’ Acute Physiology and Chronic Health Evaluation II (APACHE II) scores.29 In particular, the bundled approach they used precludes the ability to differentiate which interventions were responsible for the outcome benefits. Indeed, there were two major interventions in the early goal-directed therapy group: a protocol for achieving the goals described and the use of Scvo2 as a goal.
Aggressive fluid resuscitation is considered the most critical aspect of all the major interventions, and there is little argument on its value. The debate centers on central venous pressure as a preload marker, since after the publication of the early goal-directed therapy trial,27 several studies showed that central venous pressure may not be a valid measure to predict fluid responsiveness (discussed later in this paper).30,31
The choice of colloids or crystalloids for fluid resuscitation is another area of debate. Clinical evidence suggests that albumin is equivalent to normal saline in a heterogeneous intensive care unit population,32 but subgroup analyses suggest albumin may be superior in patients with septic shock.33 Studies are ongoing (NCT00707122, NCT01337934, and NCT00318942). The use of hydroxyethyl starch in severe sepsis is associated with higher rates of acute renal failure and need for renal replacement therapy than Ringer’s lactate,34 and is generally not recommended. This is further substantiated by two recent randomized controlled studies, which found that the use of hydroxyethyl starch for fluid resuscitation in severe sepsis, compared with crystalloids, did not reduce the mortality rate (and even increased it in one study), and was associated with more need for renal replacement therapy.35,36
The use of Scvo2 is yet another topic of debate, and other monitoring variables have been evaluated. A recent study assessed the noninferiority of incorporating venous lactate clearance into the early goal-directed therapy protocol vs Scvo2.37 Both groups had identical goals for central venous pressure and mean arterial pressure but differed in the use of lactate clearance (defined as at least a 10% decline) or Scvo2 (> 70%) as the goal for improving tissue hypoxia. There were no significant differences between groups in their in-hospital mortality rates (17% in the lactate clearance group vs 23% in the Scvo2 group; criteria for noninferiority met). This suggests that lactate may be an alternative to Scvo2 as a goal in early goal-directed therapy. However, a secondary analysis of the data revealed a lack of concordance in achieving lactate clearance and Scvo2 goals, which suggests that these parameters may be measuring distinct physiologic processes.38 Since the hemodynamic profiles of septic shock patients are complex, it may be prudent to use both of these markers of resuscitation until further studies are completed.
Given the debate, a number of prospective randomized trials are under way to evaluate resuscitative interventions. These include the Protocolized Care for Early Septic Shock trial (NCT00510835), the Australasian Resuscitation in Sepsis Evaluation trial (NCT00975793), and the Protocolised Management of Sepsis (ProMISe) trial in the United Kingdom (ISRCTN 36307479). These three trials will evaluate, collectively, close to 4,000 patients and will provide considerable insights into resuscitative interventions in septic shock.
Vasopressors: Which one to use?
If fluid therapy does not restore perfusion, vasopressors should be promptly initiated, as the longer that hypotension goes on, the lower the survival rate.39
But which vasopressor should be used? The early goal-directed therapy protocol used in the study by Rivers et al27 did not specify which vasopressor should be used to keep the mean arterial pressure above 65 mm Hg.
The Surviving Sepsis Campaign22 recommends norepinephrine as the first-choice vasopressor, with dopamine as an alternative only in selected patients, such as those with absolute or relative bradycardia.
The guidelines also recommend epinephrine to be added to or substituted for norepinephrine when an additional catecholamine is needed to maintain adequate blood pressure.22 Furthermore, vasopressin at a dose of 0.03 units/min can be added to norepinephrine with the intent of raising the blood pressure or decreasing the norepinephrine requirement. Higher doses of vasopressin should be reserved for salvage therapy.
Regarding phenylephrine, the guidelines recommend against its use except when norepinephrine use is associated with significant tachyarrhythmias, cardiac output is known to be higher, or as a salvage therapy.22
This is a topic of debate, with recent clinical studies offering further insight.
De Backer et al40 compared the effects of dopamine vs norepinephrine for the treatment of shock in 1,679 patients, 62% of whom had septic shock. Overall, there was a trend towards better outcomes with norepinephrine, but no significant difference in mortality rates at 28 days (52.5% with dopamine vs 48.5% with norepinephrine, P = .10). Importantly, fewer patients who were randomized to norepinephrine developed arrhythmias (12.4% vs 24.1%, P < .001), and the norepinephrine group required fewer days of study drug (11.0 vs 12.5, P = .01) and open-label vasopressors (12.6 vs 14.2, P = .007). Of note, patients with cardiogenic shock randomized to norepinephrine had a significantly lower mortality rate than those randomized to dopamine. Although no significant difference in outcome was found between the two vasopressors in the subgroup of patients with septic shock, the overall improvements in secondary surrogate markers suggest that norepinephrine should be the first-line agent.
Norepinephrine has also been compared with “secondary” vasopressors. Annane et al,41 in a prospective multicenter randomized controlled study, evaluated the effect of norepinephrine plus dobutamine vs epinephrine alone in managing septic shock. There was no significant difference in the primary outcome measure of 28-day mortality (34% with norepinephrine plus dobutamine vs 40% with epinephrine alone, P = .31). However, the study was powered to evaluate for an absolute risk reduction of 20% in the mortality rate, which would be a big reduction. A smaller reduction in the mortality rate, which would not have been statistically significant in this study, might still be considered clinically significant. Furthermore, the group randomized to norepinephrine plus dobutamine had more vasopressor-free days (20 days vs 22 days, P = .05) and less acidosis on days 1 to 4 than the group randomized to epinephrine.
Norepinephrine was also compared with phenylephrine as a first-line vasopressor in a randomized controlled trial in 32 patients with septic shock. No difference was found in cardiopulmonary performance, global oxygen transport, or regional hemodynamics between phenylephrine and norepinephrine.42
While encouraging, these preliminary data need to be verified in a larger randomized controlled trial with concrete outcome measures before being clinically adapted. Taken together, the above studies suggest that norepinephrine should be the initial vasopressor of choice for patients with septic shock.
CONTINUED MANAGEMENT OF SEPTIC SHOCK
How to manage septic shock after the initial stages is much less defined.
Uncertainty persists about the importance of achieving the early goals of resuscitation in patients who did not reach them in the initial 6 hours of treatment. Although there are data suggesting that extending the goals beyond the initial 6 hours may be beneficial, clinicians should use caution when interpreting these results in light of the observational design of the studies.43,44 For the purpose of this discussion, “continued management” of septic shock will mean after the first 6 hours and after all the early goals are met.
The clinical decisions necessary after the initial stages of resuscitation include:
- Whether further fluid resuscitation is needed
- Assessment for further and additional hemodynamic therapies
- Consideration of adjunctive therapies
- Reevaluation of antibiotic choices (Table 2).
Is more fluid needed? How can we tell?
There is considerable debate about the ideal method for assessing fluid responsiveness. In fact, one of the criticisms of the early goal-directed therapy study27 was that it used central venous pressure as a marker of fluid responsiveness.
Several studies have shown that central venous pressure or pulmonary artery occlusion pressure may not be valid measures of fluid responsiveness.45 In fact, in a retrospective study of 150 volume challenges, the area under the receiver-operating-characteristics curve of central venous pressure as a marker of fluid responsiveness was only 0.58. (Recall that the closer the area under the curve is to 1.0, the better the test; a value of 0.50 is the same as chance.) The area under the curve for pulmonary artery occlusion pressure was 0.63.46
In contrast, several dynamic indices have been proposed to better guide fluid resuscitation in mechanically ventilated patients.31 These are based on changes in stroke volume, aortic blood flow, or arterial pulse pressure in response to the ventilator cycle or passive leg-raising. A detailed review of these markers can be found elsewhere,31 but taken together, they have a sensitivity and specificity of over 90% for predicting fluid responsiveness. Clinicians may consider using dynamic markers of fluid responsiveness to determine when to give additional fluids, particularly after the first 6 hours of shock, in which data supporting the use of central venous pressure are lacking.
Optimal use of fluids is particularly important, since some studies suggest that “overresuscitation” has negative consequences. In a multicenter observational study of 1,177 patients with sepsis, after adjusting for a number of comorbidities and baseline severity of illness, the cumulative fluid balance in the first 72 hours after the onset of sepsis was independently associated with a worse mortality rate.47
Furthermore, in a retrospective analysis of a randomized controlled trial of vasopressin in conjunction with norepinephrine for septic shock, patients in the highest quartile of fluid balance (more fluid in than out) at 12 hours and 4 days after presentation had significantly higher mortality rates than those in the lowest two quartiles.48 The worse outcome with a positive fluid balance might be explained by worsening oxygenation and prolonged mechanical ventilation, as demonstrated by the Fluid and Catheter Treatment Trial in patients with acute lung injury or acute respiratory distress syndrome (ALI/ARDS).49 Indeed, when fluid balance in patients with septic shockinduced ALI/ARDS was evaluated, patients with both adequate initial fluid resuscitation and conservative late fluid management had a lower mortality rate than those with either one alone.50
In view of these findings, especially beyond the initial hours of resuscitation, clinicians should remember that further unnecessary fluid administration may have detrimental effects. Therefore, given the superior predictive abilities of dynamic markers of fluid responsiveness, these should be used to determine the need for further fluid boluses.
In cases in which patients are no longer fluid-responsive and need increasing levels of hemodynamic support, clinicians still have a number of options. These include increasing the current vasopressor dose or starting an additional therapy such as an alternative catecholamine vasopressor, vasopressin, inotropic therapy, or an adjunctive therapy such as a corticosteroid. The intervention could also be a combination of the above choices.
Adding catecholamines
The optimal time point or vasopressor dose at which to consider initiating additional therapies is unknown. However, the Vasopressin and Septic Shock Trial (VASST) provides some insight.51
This study compared two strategies: escalating doses of norepinephrine vs adding vasopressin to norepinephrine. Overall, adding vasopressin showed no benefit in terms of a lower mortality rate. However, in the subgroup of patients with norepinephrine requirements of 5 to 14 μg/min at study enrollment (ie, a low dose, reflecting less-severe sepsis) vasopressin was associated with a lower 28-day mortality rate (26.5% vs 35.7%, P = .05) and 90-day mortality rate (35.8% vs 46.1%, P = .04). Benefit was also noted in patients with other markers of lower disease severity such as low lactate levels or having received a single vasopressor at baseline.51
Although subgroup analyses should not generally be used to guide treatment decisions, a prospective trial may never be done to evaluate adding vasopressin to catecholamines earlier vs later. Thus, clinicians who choose to use vasopressin may consider starting this therapy when catecholamine doses are relatively low or before profound hyperlactatemia from prolonged tissue hypoxia has developed.
There is less evidence to guide clinicians who are considering adding a different catecholamine. The theoretical concerns of splanchnic ischemia and cardiac arrhythmia associated with higher doses of catecholamines are usually the impetus to limit a single catecholamine to a “maximum” dose. However, studies that have evaluated combination catecholamine therapies have generally studied combinations of vasopressors with inotropes and lacked standardization in their protocols, thus making them difficult to interpret.52–54 One could also argue that additional catecholamine therapies, which all function similarly, may have additive effects and cause even more adverse effects. As such, adding another vasopressor should be reserved for patients experiencing noticeable adverse effects (such as tachycardia) on first-line therapy.
Inotropic support
Left ventricular function should be assessed in all patients who continue to be hypotensive despite adequate fluid resuscitation and vasopressor therapy. In a study of patients with septic shock in whom echocardiography was performed daily for the first 3 days of hemodynamic support, new-onset left ventricular hypokinesia was found in 26 (39%) of 67 patients on presentation and in an additional 14 patients (21%) after at least 24 hours of norepinephrine.55 Adding inotropic support with dobutamine or epinephrine led to decreases in vasopressor dose and enhanced left ventricular ejection fraction.
In short, left ventricular hypokinesia is common in septic shock, may occur at presentation or after a period of vasopressor support, and is usually correctable with the addition of inotropic support.
Corticosteroids
Beyond hemodynamic support with fluids and catecholamines or vasopressin (or both), clinicians should also consider adjunctive corticosteroid therapy. However, for many years the issue has been controversial for patients with severe sepsis and septic shock.
Annane et al56 conducted a large, multicenter, randomized, double-blind, placebocontrolled trial to assess the effect of low doses of corticosteroids in patients with refractory septic shock. Overall, the 28-day mortality rate was 61% in the treatment group and 55% in the placebo group, which was not statistically significant (adjusted odds ratio 0.65, 95% confidence interval 0.39–1.07, P value .09). However, when separated by response to cosyntropin stimulation, those with a change in cortisol of 9 ug/dL or less (nonresponders) randomized to receive corticosteroids had significantly higher survival rates in the short term (28 days) and the long term (1 year). The positive results of this study led to the adoption of low-dose hydrocortisone as standard practice in most patients with septic shock.57
But then, to evaluate the effects of corticosteroids in a broader intensive-care population with septic shock, another trial was designed: the Corticosteroid Therapy of Septic Shock (CORTICUS) trial.58 Surprisingly, this multicenter, randomized, double-blind, placebo-controlled trial found no significant difference in survival between the group that received hydrocortisone and the placebo group, regardless of response to a cosyntropin stimulation test.
Taking into account the above studies and other randomized controlled trials, the 2012 Surviving Sepsis Campaign guidelines and the International Task Force for the Diagnosis and Management of Corticosteroid Insufficiency in Critically Ill Adult Patients recommend intravenous hydrocortisone therapy in adults with septic shock whose blood pressure responds poorly to fluid resuscitation and vasopressor therapy. These consensus statements do not recommend the cosyntropin stimulation test to identify patients with septic shock who should receive corticosteroids.22,59 The guidelines, however, do not explicitly define poor response to initial therapy.
Of note, in the Annane study, which found a lower mortality rate with corticosteroids, the patients were severely ill, with a mean baseline norepinephrine dose of 1.1 μg/kg/min. In contrast, in the CORTICUS study (which found no benefit of hydrocortisone), patients had lower baseline vasopressor doses, with a mean norepinephrine dose of 0.5 μg/kg/min.
While corticosteroids are associated with a higher rate of shock reversal 7 days after initiation, 59 this has not translated into a consistent reduction in the death rate. If a clinician is considering adding corticosteroids to decrease the risk of death, it would seem prudent to add this therapy in patients receiving norepinephrine in doses above 0.5 μg/kg/min.
The ideal sequence and combination of the above therapies including fluids, catecholamine vasopressors, vasopressin, inotropes, and vasopressors have not been elucidated. However, some preliminary evidence suggests an advantage with the combination of vasopressin and corticosteroids. In a subgroup analysis of the VASST study, in patients who received corticosteroids, the combination of vasopressin plus norepinephrine was associated with a lower 28-day mortality rate than with norepinephrine alone (35.9% vs 44.7%, P = .03).60 These findings have been replicated in other studies,61,62 prompting suggestions for a study of vasopressin with and without corticosteroids in patients on norepinephrine to elucidate the role of each therapy individually and in combination.
Tight glycemic control
As with corticosteroids, the pendulum for tight glycemic control in critically ill patients has swung widely in recent years. Enthusiasm was high at first after the publication of a study by van den Berghe et al, which described a 3.4% absolute reduction in mortality with intensive insulin therapy to maintain blood glucose at or below 110 mg/dL.63 However, the significant benefits found in this study were never replicated.
In fact, recent evidence suggests that tight glycemic control is associated with no benefit and a higher risk of hypoglycemia.34,64 In the largest randomized controlled trial of this topic, with more than 6,000 patients, intensive insulin therapy with a target blood glucose level of 81 to 108 mg/dL was associated with a significantly higher mortality rate (odds ratio 1.14, 95% confidence interval 1.02–1.28, P = .02) than with a target glucose level of less than 180 mg/dL.65 Furthermore, in a recent follow-up analysis,66 moderate hypoglycemia (serum glucose 41–70 mg/dL) and severe hypoglycemia (serum glucose < 41 mg/dL) were associated with a higher rate of death in a dose-response relationship.66
Taking this information together, clinicians should be aware that there is no additional benefit in lowering blood glucose below the range of 140 to 180 mg/dL, and that doing so may be harmful.
Drotecogin alfa
Drotecogin alfa (Xigris) was another adjunctive therapy that has fallen from favor. It was approved for the treatment of severe sepsis in light of promising findings in initial studies.67
However, on October 25, 2011, drotecogin alfa was voluntarily withdrawn from the market by the manufacturer after another study found no beneficial effect on the mortality rates at 28 days or at 90 days.68 Furthermore, no difference could be found regarding any predetermined primary or secondary outcome measures.
Continued antibiotic therapy
The decision whether to continue initial empiric antimicrobial coverage, broaden it, or de-escalate must be faced for all patients with septic shock, and is ultimately clinical.
The serum procalcitonin level has been proposed to guide antibiotic discontinuation in several clinical settings, although there are still questions about the safety of such an approach. The largest randomized trial published to date reported that a procalcitoninguided strategy to treat suspected bacterial infections in nonsurgical patients could reduce antibiotic exposure with no apparent adverse outcomes.69 On the other hand, other data discourage the use of procalcitonin-guided antimicrobial escalation, as this approach did not improve survival and worsened organ function and length of stay in the intensive care unit.70
The Surviving Sepsis Campaign guidelines recommend combination antibiotic therapy for no longer than 3 to 5 days and limiting the duration of antibiotics in most cases to 7 to 10 days.22
TRIALS ARE ONGOING
The understanding of the pathophysiology and treatment of sepsis has greatly advanced over the last decade. Adoption of evidence-based protocols for managing patients with septic shock has improved outcomes. Nevertheless, many multicenter trials are being conducted worldwide to look into some of the most controversial therapies, and their results will guide therapy in the future.
Considerably fewer patients who develop sepsis are dying of it now, thanks to a number of studies of how to reverse sepsis-induced tissue hypoxia.1 The greatest strides in improving outcomes have been attributed to better early management, which includes prompt recognition of sepsis, rapid initiation of antimicrobial therapy, elimination of the source of infection, and early goal-directed therapy. Thus, even though the incidence of severe sepsis and septic shock is increasing,2,3 the Surviving Sepsis Campaign has documented a significant decrease in unadjusted mortality rates (37% to 30.8%) associated with the bundled approach in the management of sepsis.4 (We will talk about this later in the article.)
This review will summarize the evidence for the early management of septic shock and will evaluate the various treatment decisions beyond the initial phases of resuscitation.
INFLAMMATION AND VASODILATION
Sepsis syndrome starts with an infection that leads to a proinflammatory state with a complex interaction between anti-inflammatory and proinflammatory mediators, enhanced coagulation, and impaired fibrinolysis.5,6
Sepsis induces vasodilation by way of inappropriate activation of vasodilatory mechanisms (increased synthesis of nitric oxide and vasopressin deficiency) and failure of vasoconstrictor mechanisms (activation of ATP-sensitive potassium channels in vascular smooth muscle).7 Thus, the hemodynamic abnormalities are multifactorial, and the resultant tissue hypoperfusion further contributes to the proinflammatory and procoagulant state, precipitating multiorgan dysfunction and, often, death.
DEFINITIONS
- Sepsis—infection together with systemic manifestation of inflammatory response
- Severe sepsis—sepsis plus induced organ dysfunction or evidence of tissue hypoperfusion
- Septic shock—sepsis-induced hypotension persisting despite adequate fluid resuscitation.
EARLY MANAGEMENT OF SEPTIC SHOCK
Early in the course of septic shock, the physician’s job is to:
- Recognize it promptly
- Begin empiric antibiotic therapy quickly
- Eliminate the source of infection, if applicable, eg, by removing an infected central venous catheter
- Give fluid resuscitation, titrated to specific goals
- Give vasopressor therapy to maintain blood pressure, organ perfusion, and oxygen delivery (Table 1).
The line between “early” and “late” is not clear. Traditionally, it has been drawn at 6 hours from presentation, and this cutoff was used in some of the studies we will discuss here.
Recognizing severe sepsis early in its course
The diagnosis of severe sepsis may be challenging, since up to 40% of patients may present with cryptic shock. These patients may not be hemodynamically compromised but may show evidence of tissue hypoxia, eg, an elevated serum lactate concentration or a low central venous oxygen saturation (Scvo2), or both.8 In view of this, much effort has gone into finding a biomarker that, in addition to clinical features, can help identify patients in an early stage of sepsis.
Procalcitonin levels rise in response to severe bacterial infection,9 and they correlate with sepsis-related organ failure scores and outcomes.10,11 Thus, the serum procalcitonin level may help in assessing the severity of sepsis, especially when combined with standard clinical and laboratory variables. However, controversy exists about the threshold to use in making decisions about antibiotic therapy and the value of this test in differentiating severe noninfectious inflammatory reactions from infectious causes of shock.12 Therefore, it is not widely used in clinical practice.
Serum lactate has been used for decades as a marker of tissue hypoperfusion. It is typically elevated in patients with severe sepsis and septic shock, and although the hyperlactatemia could be a result of global hypoperfusion, it can also be secondary to sepsis-induced mitochondrial dysfunction,13 impaired pyruvate dehydrogenase activity,14 increased aerobic glycolysis by catecholamine-stimulated sodium-potassium pump hyperactivity,15 and even impaired clearance.16
But whatever the mechanism, elevated lactate in severe sepsis and septic shock predicts a poor outcome and may help guide aggressive resuscitation. In fact, early lactate clearance (ie, normalization of an elevated value on repeat testing within the first 6 hours) is associated with better outcomes in patients with severe sepsis and septic shock.17,18
Panels of biomarkers. A literature search revealed over 3,000 papers on 178 different biomarkers in sepsis.19 Many of these biomarkers lack sufficient specificity and sensitivity for clinical use, and thus some investigators have suggested using a panel of them to enhance their predictive ability. Shapiro et al20 evaluated 971 patients admitted to the emergency department with suspected infection and discovered that a panel of three biomarkers (neutrophil gelatinase-associated lipocalin, protein C, and interleukin-1 receptor antagonist) was highly predictive of severe sepsis, septic shock, and death.
Starting empiric antibiotic therapy early
As soon as severe sepsis and septic shock are recognized, it is imperative that adequate empiric antibiotic treatment be started, along with infectious source control if applicable.21 The Surviving Sepsis Campaign guidelines recommend starting intravenous antibiotics as early as possible—within the first hour of recognition of severe sepsis with or without septic shock.22
Kumar et al,23 in a multicenter retrospective study of patients with septic shock, found that each hour of delay in giving appropriate antimicrobial agents in the first 6 hours from the onset of hypotension was associated with a 7.6% decrease in the in-hospital survival rate.
In a similar study,24 the same investigators analyzed data from 5,715 septic shock patients regarding the impact of starting the right antimicrobial therapy. Appropriate antimicrobial agents (ie, those having in vitro activity against the isolated pathogens) were given in 80.1% of cases, and the survival rate in those who received appropriate antibiotics was drastically higher than in those who received inappropriate ones (52.0% vs 10.3%, P < .0001).
In addition, two recent studies evaluated the importance of early empiric antibiotic therapy in conjunction with resuscitative protocols.25,26 In a preplanned analysis of early antimicrobial use in a study comparing lactate clearance and Scvo2 as goals of therapy, Puskarich et al26 found that fewer patients who received antibiotics before shock was recognized (according to formal criteria) died. Similarly, in a retrospective study in patients presenting to the emergency department and treated with early goal-directed therapy (defined below), Gaieski et al25 found that the mortality rate was drastically lower when antibiotics were started within 1 hour of either triage or initiation of early goal-directed therapy.
In short, it is imperative to promptly start the most appropriate broad-spectrum antibiotics to target the most likely pathogens based on site of infection, patient risk of multidrug-resistant pathogens, and local susceptibility patterns.
Goal-directed resuscitative therapy
As with antimicrobial therapy, resuscitative therapy should be started early and directed at defined goals.
Rivers et al27 conducted a randomized, controlled study in patients with severe sepsis or septic shock presenting to an emergency department of an urban teaching hospital. The patients were at high risk and had either persistent hypotension after a fluid challenge or serum lactate levels of 4 mmol/L or higher.
Two hundred sixty patients were randomized to receive either early goal-directed therapy in a protocol aimed at maximizing the intravascular volume and correcting global tissue hypoxia or standard therapy in the first 6 hours after presentation. The goals in the goal-directed therapy group were:
- Central venous pressure 8 to 12 mm Hg (achieved with aggressive fluid resuscitation with crystalloids)
- Mean arterial blood pressure greater than 65 mm Hg (maintained with vasoactive drugs, if necessary)
- Scvo2 above 70%. To achieve this third goal, packed red blood cells were infused to reach a target hematocrit of greater than 30%. For patients with a hematocrit higher than 30% but still with an Scvo2 less than 70%, inotropic agents were added and titrated to the Scvo2 goal of 70%.
Goal-directed therapy reduced the in-hospital mortality rate by 16% (the mortality rates were 30.5% in the goal-directed group and 46.5% in the standard therapy group, P = .009) and also reduced the 28- and 60-day mortality rates by similar proportions.27
Subsequent studies of a protocol for early recognition and treatment of sepsis have concluded that early aggressive fluid resuscitation decreases the ensuing need for vasopressor support.28 A resuscitation strategy based on early goal-directed therapy is a major component of the initial resuscitation bundle recommended by the Surviving Sepsis Campaign.22 (A “bundle” refers to the implementation of a core set of recommendations involving the simultaneous adaptation of a number of interventions.)
Areas of debate. However, concerns have been raised about the design of the study by Rivers et al and the mortality rate in the control group, which was higher than one would expect from the patients’ Acute Physiology and Chronic Health Evaluation II (APACHE II) scores.29 In particular, the bundled approach they used precludes the ability to differentiate which interventions were responsible for the outcome benefits. Indeed, there were two major interventions in the early goal-directed therapy group: a protocol for achieving the goals described and the use of Scvo2 as a goal.
Aggressive fluid resuscitation is considered the most critical aspect of all the major interventions, and there is little argument on its value. The debate centers on central venous pressure as a preload marker, since after the publication of the early goal-directed therapy trial,27 several studies showed that central venous pressure may not be a valid measure to predict fluid responsiveness (discussed later in this paper).30,31
The choice of colloids or crystalloids for fluid resuscitation is another area of debate. Clinical evidence suggests that albumin is equivalent to normal saline in a heterogeneous intensive care unit population,32 but subgroup analyses suggest albumin may be superior in patients with septic shock.33 Studies are ongoing (NCT00707122, NCT01337934, and NCT00318942). The use of hydroxyethyl starch in severe sepsis is associated with higher rates of acute renal failure and need for renal replacement therapy than Ringer’s lactate,34 and is generally not recommended. This is further substantiated by two recent randomized controlled studies, which found that the use of hydroxyethyl starch for fluid resuscitation in severe sepsis, compared with crystalloids, did not reduce the mortality rate (and even increased it in one study), and was associated with more need for renal replacement therapy.35,36
The use of Scvo2 is yet another topic of debate, and other monitoring variables have been evaluated. A recent study assessed the noninferiority of incorporating venous lactate clearance into the early goal-directed therapy protocol vs Scvo2.37 Both groups had identical goals for central venous pressure and mean arterial pressure but differed in the use of lactate clearance (defined as at least a 10% decline) or Scvo2 (> 70%) as the goal for improving tissue hypoxia. There were no significant differences between groups in their in-hospital mortality rates (17% in the lactate clearance group vs 23% in the Scvo2 group; criteria for noninferiority met). This suggests that lactate may be an alternative to Scvo2 as a goal in early goal-directed therapy. However, a secondary analysis of the data revealed a lack of concordance in achieving lactate clearance and Scvo2 goals, which suggests that these parameters may be measuring distinct physiologic processes.38 Since the hemodynamic profiles of septic shock patients are complex, it may be prudent to use both of these markers of resuscitation until further studies are completed.
Given the debate, a number of prospective randomized trials are under way to evaluate resuscitative interventions. These include the Protocolized Care for Early Septic Shock trial (NCT00510835), the Australasian Resuscitation in Sepsis Evaluation trial (NCT00975793), and the Protocolised Management of Sepsis (ProMISe) trial in the United Kingdom (ISRCTN 36307479). These three trials will evaluate, collectively, close to 4,000 patients and will provide considerable insights into resuscitative interventions in septic shock.
Vasopressors: Which one to use?
If fluid therapy does not restore perfusion, vasopressors should be promptly initiated, as the longer that hypotension goes on, the lower the survival rate.39
But which vasopressor should be used? The early goal-directed therapy protocol used in the study by Rivers et al27 did not specify which vasopressor should be used to keep the mean arterial pressure above 65 mm Hg.
The Surviving Sepsis Campaign22 recommends norepinephrine as the first-choice vasopressor, with dopamine as an alternative only in selected patients, such as those with absolute or relative bradycardia.
The guidelines also recommend epinephrine to be added to or substituted for norepinephrine when an additional catecholamine is needed to maintain adequate blood pressure.22 Furthermore, vasopressin at a dose of 0.03 units/min can be added to norepinephrine with the intent of raising the blood pressure or decreasing the norepinephrine requirement. Higher doses of vasopressin should be reserved for salvage therapy.
Regarding phenylephrine, the guidelines recommend against its use except when norepinephrine use is associated with significant tachyarrhythmias, cardiac output is known to be higher, or as a salvage therapy.22
This is a topic of debate, with recent clinical studies offering further insight.
De Backer et al40 compared the effects of dopamine vs norepinephrine for the treatment of shock in 1,679 patients, 62% of whom had septic shock. Overall, there was a trend towards better outcomes with norepinephrine, but no significant difference in mortality rates at 28 days (52.5% with dopamine vs 48.5% with norepinephrine, P = .10). Importantly, fewer patients who were randomized to norepinephrine developed arrhythmias (12.4% vs 24.1%, P < .001), and the norepinephrine group required fewer days of study drug (11.0 vs 12.5, P = .01) and open-label vasopressors (12.6 vs 14.2, P = .007). Of note, patients with cardiogenic shock randomized to norepinephrine had a significantly lower mortality rate than those randomized to dopamine. Although no significant difference in outcome was found between the two vasopressors in the subgroup of patients with septic shock, the overall improvements in secondary surrogate markers suggest that norepinephrine should be the first-line agent.
Norepinephrine has also been compared with “secondary” vasopressors. Annane et al,41 in a prospective multicenter randomized controlled study, evaluated the effect of norepinephrine plus dobutamine vs epinephrine alone in managing septic shock. There was no significant difference in the primary outcome measure of 28-day mortality (34% with norepinephrine plus dobutamine vs 40% with epinephrine alone, P = .31). However, the study was powered to evaluate for an absolute risk reduction of 20% in the mortality rate, which would be a big reduction. A smaller reduction in the mortality rate, which would not have been statistically significant in this study, might still be considered clinically significant. Furthermore, the group randomized to norepinephrine plus dobutamine had more vasopressor-free days (20 days vs 22 days, P = .05) and less acidosis on days 1 to 4 than the group randomized to epinephrine.
Norepinephrine was also compared with phenylephrine as a first-line vasopressor in a randomized controlled trial in 32 patients with septic shock. No difference was found in cardiopulmonary performance, global oxygen transport, or regional hemodynamics between phenylephrine and norepinephrine.42
While encouraging, these preliminary data need to be verified in a larger randomized controlled trial with concrete outcome measures before being clinically adapted. Taken together, the above studies suggest that norepinephrine should be the initial vasopressor of choice for patients with septic shock.
CONTINUED MANAGEMENT OF SEPTIC SHOCK
How to manage septic shock after the initial stages is much less defined.
Uncertainty persists about the importance of achieving the early goals of resuscitation in patients who did not reach them in the initial 6 hours of treatment. Although there are data suggesting that extending the goals beyond the initial 6 hours may be beneficial, clinicians should use caution when interpreting these results in light of the observational design of the studies.43,44 For the purpose of this discussion, “continued management” of septic shock will mean after the first 6 hours and after all the early goals are met.
The clinical decisions necessary after the initial stages of resuscitation include:
- Whether further fluid resuscitation is needed
- Assessment for further and additional hemodynamic therapies
- Consideration of adjunctive therapies
- Reevaluation of antibiotic choices (Table 2).
Is more fluid needed? How can we tell?
There is considerable debate about the ideal method for assessing fluid responsiveness. In fact, one of the criticisms of the early goal-directed therapy study27 was that it used central venous pressure as a marker of fluid responsiveness.
Several studies have shown that central venous pressure or pulmonary artery occlusion pressure may not be valid measures of fluid responsiveness.45 In fact, in a retrospective study of 150 volume challenges, the area under the receiver-operating-characteristics curve of central venous pressure as a marker of fluid responsiveness was only 0.58. (Recall that the closer the area under the curve is to 1.0, the better the test; a value of 0.50 is the same as chance.) The area under the curve for pulmonary artery occlusion pressure was 0.63.46
In contrast, several dynamic indices have been proposed to better guide fluid resuscitation in mechanically ventilated patients.31 These are based on changes in stroke volume, aortic blood flow, or arterial pulse pressure in response to the ventilator cycle or passive leg-raising. A detailed review of these markers can be found elsewhere,31 but taken together, they have a sensitivity and specificity of over 90% for predicting fluid responsiveness. Clinicians may consider using dynamic markers of fluid responsiveness to determine when to give additional fluids, particularly after the first 6 hours of shock, in which data supporting the use of central venous pressure are lacking.
Optimal use of fluids is particularly important, since some studies suggest that “overresuscitation” has negative consequences. In a multicenter observational study of 1,177 patients with sepsis, after adjusting for a number of comorbidities and baseline severity of illness, the cumulative fluid balance in the first 72 hours after the onset of sepsis was independently associated with a worse mortality rate.47
Furthermore, in a retrospective analysis of a randomized controlled trial of vasopressin in conjunction with norepinephrine for septic shock, patients in the highest quartile of fluid balance (more fluid in than out) at 12 hours and 4 days after presentation had significantly higher mortality rates than those in the lowest two quartiles.48 The worse outcome with a positive fluid balance might be explained by worsening oxygenation and prolonged mechanical ventilation, as demonstrated by the Fluid and Catheter Treatment Trial in patients with acute lung injury or acute respiratory distress syndrome (ALI/ARDS).49 Indeed, when fluid balance in patients with septic shockinduced ALI/ARDS was evaluated, patients with both adequate initial fluid resuscitation and conservative late fluid management had a lower mortality rate than those with either one alone.50
In view of these findings, especially beyond the initial hours of resuscitation, clinicians should remember that further unnecessary fluid administration may have detrimental effects. Therefore, given the superior predictive abilities of dynamic markers of fluid responsiveness, these should be used to determine the need for further fluid boluses.
In cases in which patients are no longer fluid-responsive and need increasing levels of hemodynamic support, clinicians still have a number of options. These include increasing the current vasopressor dose or starting an additional therapy such as an alternative catecholamine vasopressor, vasopressin, inotropic therapy, or an adjunctive therapy such as a corticosteroid. The intervention could also be a combination of the above choices.
Adding catecholamines
The optimal time point or vasopressor dose at which to consider initiating additional therapies is unknown. However, the Vasopressin and Septic Shock Trial (VASST) provides some insight.51
This study compared two strategies: escalating doses of norepinephrine vs adding vasopressin to norepinephrine. Overall, adding vasopressin showed no benefit in terms of a lower mortality rate. However, in the subgroup of patients with norepinephrine requirements of 5 to 14 μg/min at study enrollment (ie, a low dose, reflecting less-severe sepsis) vasopressin was associated with a lower 28-day mortality rate (26.5% vs 35.7%, P = .05) and 90-day mortality rate (35.8% vs 46.1%, P = .04). Benefit was also noted in patients with other markers of lower disease severity such as low lactate levels or having received a single vasopressor at baseline.51
Although subgroup analyses should not generally be used to guide treatment decisions, a prospective trial may never be done to evaluate adding vasopressin to catecholamines earlier vs later. Thus, clinicians who choose to use vasopressin may consider starting this therapy when catecholamine doses are relatively low or before profound hyperlactatemia from prolonged tissue hypoxia has developed.
There is less evidence to guide clinicians who are considering adding a different catecholamine. The theoretical concerns of splanchnic ischemia and cardiac arrhythmia associated with higher doses of catecholamines are usually the impetus to limit a single catecholamine to a “maximum” dose. However, studies that have evaluated combination catecholamine therapies have generally studied combinations of vasopressors with inotropes and lacked standardization in their protocols, thus making them difficult to interpret.52–54 One could also argue that additional catecholamine therapies, which all function similarly, may have additive effects and cause even more adverse effects. As such, adding another vasopressor should be reserved for patients experiencing noticeable adverse effects (such as tachycardia) on first-line therapy.
Inotropic support
Left ventricular function should be assessed in all patients who continue to be hypotensive despite adequate fluid resuscitation and vasopressor therapy. In a study of patients with septic shock in whom echocardiography was performed daily for the first 3 days of hemodynamic support, new-onset left ventricular hypokinesia was found in 26 (39%) of 67 patients on presentation and in an additional 14 patients (21%) after at least 24 hours of norepinephrine.55 Adding inotropic support with dobutamine or epinephrine led to decreases in vasopressor dose and enhanced left ventricular ejection fraction.
In short, left ventricular hypokinesia is common in septic shock, may occur at presentation or after a period of vasopressor support, and is usually correctable with the addition of inotropic support.
Corticosteroids
Beyond hemodynamic support with fluids and catecholamines or vasopressin (or both), clinicians should also consider adjunctive corticosteroid therapy. However, for many years the issue has been controversial for patients with severe sepsis and septic shock.
Annane et al56 conducted a large, multicenter, randomized, double-blind, placebocontrolled trial to assess the effect of low doses of corticosteroids in patients with refractory septic shock. Overall, the 28-day mortality rate was 61% in the treatment group and 55% in the placebo group, which was not statistically significant (adjusted odds ratio 0.65, 95% confidence interval 0.39–1.07, P value .09). However, when separated by response to cosyntropin stimulation, those with a change in cortisol of 9 ug/dL or less (nonresponders) randomized to receive corticosteroids had significantly higher survival rates in the short term (28 days) and the long term (1 year). The positive results of this study led to the adoption of low-dose hydrocortisone as standard practice in most patients with septic shock.57
But then, to evaluate the effects of corticosteroids in a broader intensive-care population with septic shock, another trial was designed: the Corticosteroid Therapy of Septic Shock (CORTICUS) trial.58 Surprisingly, this multicenter, randomized, double-blind, placebo-controlled trial found no significant difference in survival between the group that received hydrocortisone and the placebo group, regardless of response to a cosyntropin stimulation test.
Taking into account the above studies and other randomized controlled trials, the 2012 Surviving Sepsis Campaign guidelines and the International Task Force for the Diagnosis and Management of Corticosteroid Insufficiency in Critically Ill Adult Patients recommend intravenous hydrocortisone therapy in adults with septic shock whose blood pressure responds poorly to fluid resuscitation and vasopressor therapy. These consensus statements do not recommend the cosyntropin stimulation test to identify patients with septic shock who should receive corticosteroids.22,59 The guidelines, however, do not explicitly define poor response to initial therapy.
Of note, in the Annane study, which found a lower mortality rate with corticosteroids, the patients were severely ill, with a mean baseline norepinephrine dose of 1.1 μg/kg/min. In contrast, in the CORTICUS study (which found no benefit of hydrocortisone), patients had lower baseline vasopressor doses, with a mean norepinephrine dose of 0.5 μg/kg/min.
While corticosteroids are associated with a higher rate of shock reversal 7 days after initiation, 59 this has not translated into a consistent reduction in the death rate. If a clinician is considering adding corticosteroids to decrease the risk of death, it would seem prudent to add this therapy in patients receiving norepinephrine in doses above 0.5 μg/kg/min.
The ideal sequence and combination of the above therapies including fluids, catecholamine vasopressors, vasopressin, inotropes, and vasopressors have not been elucidated. However, some preliminary evidence suggests an advantage with the combination of vasopressin and corticosteroids. In a subgroup analysis of the VASST study, in patients who received corticosteroids, the combination of vasopressin plus norepinephrine was associated with a lower 28-day mortality rate than with norepinephrine alone (35.9% vs 44.7%, P = .03).60 These findings have been replicated in other studies,61,62 prompting suggestions for a study of vasopressin with and without corticosteroids in patients on norepinephrine to elucidate the role of each therapy individually and in combination.
Tight glycemic control
As with corticosteroids, the pendulum for tight glycemic control in critically ill patients has swung widely in recent years. Enthusiasm was high at first after the publication of a study by van den Berghe et al, which described a 3.4% absolute reduction in mortality with intensive insulin therapy to maintain blood glucose at or below 110 mg/dL.63 However, the significant benefits found in this study were never replicated.
In fact, recent evidence suggests that tight glycemic control is associated with no benefit and a higher risk of hypoglycemia.34,64 In the largest randomized controlled trial of this topic, with more than 6,000 patients, intensive insulin therapy with a target blood glucose level of 81 to 108 mg/dL was associated with a significantly higher mortality rate (odds ratio 1.14, 95% confidence interval 1.02–1.28, P = .02) than with a target glucose level of less than 180 mg/dL.65 Furthermore, in a recent follow-up analysis,66 moderate hypoglycemia (serum glucose 41–70 mg/dL) and severe hypoglycemia (serum glucose < 41 mg/dL) were associated with a higher rate of death in a dose-response relationship.66
Taking this information together, clinicians should be aware that there is no additional benefit in lowering blood glucose below the range of 140 to 180 mg/dL, and that doing so may be harmful.
Drotecogin alfa
Drotecogin alfa (Xigris) was another adjunctive therapy that has fallen from favor. It was approved for the treatment of severe sepsis in light of promising findings in initial studies.67
However, on October 25, 2011, drotecogin alfa was voluntarily withdrawn from the market by the manufacturer after another study found no beneficial effect on the mortality rates at 28 days or at 90 days.68 Furthermore, no difference could be found regarding any predetermined primary or secondary outcome measures.
Continued antibiotic therapy
The decision whether to continue initial empiric antimicrobial coverage, broaden it, or de-escalate must be faced for all patients with septic shock, and is ultimately clinical.
The serum procalcitonin level has been proposed to guide antibiotic discontinuation in several clinical settings, although there are still questions about the safety of such an approach. The largest randomized trial published to date reported that a procalcitoninguided strategy to treat suspected bacterial infections in nonsurgical patients could reduce antibiotic exposure with no apparent adverse outcomes.69 On the other hand, other data discourage the use of procalcitonin-guided antimicrobial escalation, as this approach did not improve survival and worsened organ function and length of stay in the intensive care unit.70
The Surviving Sepsis Campaign guidelines recommend combination antibiotic therapy for no longer than 3 to 5 days and limiting the duration of antibiotics in most cases to 7 to 10 days.22
TRIALS ARE ONGOING
The understanding of the pathophysiology and treatment of sepsis has greatly advanced over the last decade. Adoption of evidence-based protocols for managing patients with septic shock has improved outcomes. Nevertheless, many multicenter trials are being conducted worldwide to look into some of the most controversial therapies, and their results will guide therapy in the future.
- Kumar G, Kumar N, Taneja A, et al. Nationwide trends of severe sepsis in the 21st century (2000–2007). Chest 2011; 140:1223–1231.
- Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med 2001; 29:1303–1310.
- Annane D, Aegerter P, Jars-Guincestre MC, Guidet B. Current epidemiology of septic shock: the CUB-Rea Network. Am J Respir Crit Care Med 2003; 168:165–172.
- Levy MM, Dellinger RP, Townsend SR, et al. The Surviving Sepsis Campaign: results of an international guideline-based performance improvement program targeting severe sepsis. Intensive Care Med 2010; 36:222–231.
- Amaral A, Opal SM, Vincent JL. Coagulation in sepsis. Intensive Care Med 2004; 30:1032–1040.
- Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med 2003; 348:138–150.
- Landry DW, Oliver JA. The pathogenesis of vasodilatory shock. N Engl J Med 2001; 345:588–595.
- Rady MY, Rivers EP, Nowak RM. Resuscitation of the critically ill in the ED: responses of blood pressure, heart rate, shock index, central venous oxygen saturation, and lactate. Am J Emerg Med 1996; 14:218–225.
- Assicot M, Gendrel D, Carsin H, Raymond J, Guilbaud J, Bohuon C. High serum procalcitonin concentrations in patients with sepsis and infection. Lancet 1993; 34:515–518.
- Muller B, Becker KL, Schachinger H, et al. Calcitonin precursors are reliable markers of sepsis in a medical intensive care unit. Crit Care Med 2000; 28:977–983.
- Meisner M, Tschaikowsky K, Palmaers T, Schmidt J. Comparison of procalcitonin (PCT) and C-reactive protein (CRP) plasma concentrations at different SOFA scores during the course of sepsis and MODS. Crit Care (London, England) 1999; 3:45–50.
- Tang BM, Eslick GD, Craig JC, McLean AS. Accuracy of procalcitonin for sepsis diagnosis in critically ill patients: systematic review and meta-analysis. Lancet Infect Dis 2007; 7:210–217.
- Brealey D, Brand M, Hargreaves I, et al. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 2002; 360:219–223.
- Vary TC. Sepsis-induced alterations in pyruvate dehydrogenase complex activity in rat skeletal muscle: effects on plasma lactate. Shock 1996; 6:89–94.
- Levy B, Gibot S, Franck P, Cravoisy A, Bollaert PE. Relation between muscle Na+K+ ATPase activity and raised lactate concentrations in septic shock: a prospective study. Lancet 2005; 365:871–875.
- Levraut J, Ciebiera JP, Chave S, et al. Mild hyperlactatemia in stable septic patients is due to impaired lactate clearance rather than over-production. Am J Respir Crit Care Med 1998; 157:1021–1026.
- Arnold RC, Shapiro NI, Jones AE, et al. Multicenter study of early lactate clearance as a determinant of survival in patients with presumed sepsis. Shock 2009; 32:35–39.
- Nguyen HB, Rivers EP, Knoblich BP, et al. Early lactate clearance is associated with improved outcome in severe sepsis and septic shock. Crit Care Med 2004; 32:1637–1642.
- Pierrakos C, Vincent JL. Sepsis biomarkers: a review. Crit Care 2010; 14:R15.
- Shapiro NI, Trzeciak S, Hollander JE, et al. A prospective, multicenter derivation of a biomarker panel to assess risk of organ dysfunction, shock, and death in emergency department patients with suspected sepsis. Crit Care Med 2009; 37:96–104.
- Marshall JC, al Naqbi A. Principles of source control in the management of sepsis. Crit Care Clin 2009; 25:753–768,viii–ix.
- Dellinger RP, Levy MM, Rhodes A, et al. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock: 2012. Crit Care Med 2013; 41:580–637.
- Kumar A, Roberts D, Wood KE, et al. Duration of hypotension before initiation of effective antimicrobial therapy is the critical determinant of survival in human septic shock. Crit Care Med 2006; 34:1589–1596.
- Kumar A, Ellis P, Arabi Y, et al. Initiation of inappropriate antimicrobial therapy results in a fivefold reduction of survival in human septic shock. Chest 2009; 136:1237–1248.
- Gaieski DF, Mikkelsen ME, Band RA, et al. Impact of time to antibiotics on survival in patients with severe sepsis or septic shock in whom early goal-directed therapy was initiated in the emergency department. Crit Care Med 2010; 38:1045–1053.
- Puskarich MA, Trzeciak S, Shapiro NI, et al. Association between timing of antibiotic administration and mortality from septic shock in patients treated with a quantitative resuscitation protocol. Crit Care Med 2011; 39:2066–2071.
- Rivers E, Nguyen B, Havstad S, et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med 2001; 345:1368–1377.
- Micek ST, Roubinian N, Heuring T, et al. Before-after study of a standardized hospital order set for the management of septic shock. Crit Care Med 2006; 34:2707–2713.
- Schmidt GA. Counterpoint: adherence to early goal-directed therapy: does it really matter? No. Both risks and benefits require further study. Chest 2010; 138:480–483; discussion 483–484.
- Jain RK, Antonio BL, Bowton DL, Houle TT, MacGregor DA. Variability in central venous pressure measurements and the potential impact on fluid management. Shock 2009; 33:253–257.
- Durairaj L, Schmidt GA. Fluid therapy in resuscitated sepsis: less is more. Chest 2008; 133:252–263.
- Finfer S, Bellomo R, Boyce N, French J, Myburgh J, Norton R. A comparison of albumin and saline for fluid resuscitation in the intensive care unit. N Engl J Med 2004; 350:2247–2256.
- Finfer S, McEvoy S, Bellomo R, McArthur C, Myburgh J, Norton R. Impact of albumin compared to saline on organ function and mortality of patients with severe sepsis. Intensive Care Med 2011; 37:86–96.
- Brunkhorst FM, Engel C, Bloos F, et al. Intensive insulin therapy and pentastarch resuscitation in severe sepsis. N Engl J Med 2008; 358:125–139.
- Perner A, Haase N, Guttormsen AB, et al. Hydroxyethyl starch 130/0.42 versus Ringer’s acetate in severe sepsis. N Engl J Med 2012; 367:124–134.
- Myburgh JA, Finfer S, Bellomo R, et al. Hydroxyethyl starch or saline for fluid resuscitation in intensive care. N Engl J Med 2012; 367:1901–1911.
- Jones AE, Shapiro NI, Trzeciak S, Arnold RC, Claremont HA, Kline JA. Lactate clearance vs central venous oxygen saturation as goals of early sepsis therapy: a randomized clinical trial. JAMA 2010; 303:739–746.
- Puskarich MA, Trzciak S, Shapiro NI, Kline JA, Jones AE. Concordance and prognostic value of central venous oxygen saturation and lactate clearance in emergency department patients with septic shock. Acad Emerg Med 2011; 19:S159–S160.
- Dunser MW, Takala J, Ulmer H, et al. Arterial blood pressure during early sepsis and outcome. Intensive Care Med 2009; 35:1225–1233.
- De Backer D, Biston P, Devriendt J, et al. Comparison of dopamine and norepinephrine in the treatment of shock. N Engl J Med 2010; 362:779–789.
- Annane D, Vignon P, Renault A, et al. Norepinephrine plus dobutamine versus epinephrine alone for management of septic shock: a randomised trial. Lancet 2007; 370:676–684.
- Morelli A, Ertmer C, Rehberg S, et al. Phenylephrine versus norepinephrine for initial hemodynamic support of patients with septic shock: a randomized, controlled trial. Crit Care (London, England) 2008; 12:R143.
- Coba V, Whitmill M, Mooney R, et al. Resuscitation bundle compliance in severe sepsis and septic shock: improves survival, is better late than never. J Intensive Care Med 2011 Jan 10[Epub ahead of print].
- Castellanos-Ortega A, Suberviola B, Garcia-Astudillo LA, Ortiz F, Llorca J, Delgado-Rodriguez M. Late compliance with the sepsis resuscitation bundle: impact on mortality. Shock 2011; 36:542–547.
- Marik PE, Baram M, Vahid B. Does central venous pressure predict fluid responsiveness? A systematic review of the literature and the tale of seven mares. Chest 2008; 134:172–178.
- Osman D, Ridel C, Ray P, et al. Cardiac filling pressures are not appropriate to predict hemodynamic response to volume challenge. Crit Care Med 2007; 35:64–68.
- Vincent JL, Sakr Y, Sprung CL, et al. Sepsis in European intensive care units: results of the SOAP study. Crit Care Med 2006; 34:344–353.
- Boyd JH, Forbes J, Nakada TA, Walley KR, Russell JA. Fluid resuscitation in septic shock: a positive fluid balance and elevated central venous pressure are associated with increased mortality. Crit Care Med 2011; 39:259–265.
- Wiedemann HP, Wheeler AP, Bernard GR, et al. Comparison of two fluid-management strategies in acute lung injury. N Engl J Med 2006; 354:2564–2575.
- Murphy CV, Schramm GE, Doherty JA, et al. The importance of fluid management in acute lung injury secondary to septic shock. Chest 2009; 136:102–109.
- Russell JA, Walley KR, Singer J, et al. Vasopressin versus norepinephrine infusion in patients with septic shock. N Engl J Med 2008; 358:877–887.
- Vincent JL, Roman A, Kahn RJ. Dobutamine administration in septic shock: addition to a standard protocol. Crit Care Med 1990; 18:689–693.
- Levy B, Bollaert PE, Charpentier C, et al. Comparison of norepinephrine and dobutamine to epinephrine for hemodynamics, lactate metabolism, and gastric tonometric variables in septic shock: a prospective, randomized study. Intensive Care Med 1997; 23:282–287.
- Redl-Wenzl EM, Armbruster C, Edelmann G, et al. The effects of norepinephrine on hemodynamics and renal function in severe septic shock states. Intensive Care Med 1993; 19:151–154.
- Vieillard-Baron A, Caille V, Charron C, Belliard G, Page B, Jardin F. Actual incidence of global left ventricular hypokinesia in adult septic shock. Crit Care Med 2008; 36:1701–1706.
- Annane D, Sebille V, Charpentier C, et al. Effect of treatment with low doses of hydrocortisone and fludrocortisone on mortality in patients with septic shock. JAMA 2002; 288:862–871.
- Dellinger RP, Carlet JM, Masur H, et al. Surviving Sepsis Campaign guidelines for management of severe sepsis and septic shock. Crit Care Med 2004; 32:858–873.
- Sprung CL, Annane D, Keh D, et al. Hydrocortisone therapy for patients with septic shock. N Engl J Med 2008; 358:111–124.
- Marik PE, Pastores SM, Annane D, et al. Recommendations for the diagnosis and management of corticosteroid insufficiency in critically ill adult patients: consensus statements from an international task force by the American College of Critical Care Medicine. Crit Care Med 2008; 36:1937–1949.
- Russell JA, Walley KR, Gordon AC, et al. Interaction of vasopressin infusion, corticosteroid treatment, and mortality of septic shock. Crit Care Med 2009; 37:811–818.
- Bauer SR, Lam SW, Cha SS, Oyen LJ. Effect of corticosteroids on arginine vasopressin-containing vasopressor therapy for septic shock: a case control study. J Crit Care 2008; 23:500–506.
- Torgersen C, Luckner G, Schroder DC, et al. Concomitant arginine-vasopressin and hydrocortisone therapy in severe septic shock: association with mortality. Intensive Care Med 2011; 37:1432–1437.
- van den Berghe G, Wouters P, Weekers F, et al. Intensive insulin therapy in critically ill patients. N Engl J Med 2001; 345:1359–1367.
- Preiser JC, Devos P, Ruiz-Santana S, et al. A prospective randomised multi-centre controlled trial on tight glucose control by intensive insulin therapy in adult intensive care units: the Glucontrol study. Intensive Care Med 2009; 35:1738–1748.
- Finfer S, Chittock DR, Su SY, et al. Intensive versus conventional glucose control in critically ill patients. N Engl J Med 2009; 360:1283–1297.
- Finfer S, Liu B, Chittock DR, et al. Hypoglycemia and risk of death in critically ill patients. N Engl J Med 2012; 367:1108–1118.
- Bernard GR, Vincent JL, Laterre PF, et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med 2001; 344:699–709.
- Ranieri VM, Thompson BT, Barie PS, et al. Drotrecogin alfa (activated) in adults with septic shock. N Engl J Med 2012; 366:2055–2064.
- Bouadma L, Luyt CE, Tubach F, et al. Use of procalcitonin to reduce patients’ exposure to antibiotics in intensive care units (PRORATA trial): a multicentre randomised controlled trial. Lancet 2009; 375:463–474.
- Jensen JU, Hein L, Lundgren B, et al. Procalcitonin-guided interventions against infections to increase early appropriate antibiotics and improve survival in the intensive care unit: a randomized trial. Crit Care Med 2011; 39:2048–2058.
- Kumar G, Kumar N, Taneja A, et al. Nationwide trends of severe sepsis in the 21st century (2000–2007). Chest 2011; 140:1223–1231.
- Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med 2001; 29:1303–1310.
- Annane D, Aegerter P, Jars-Guincestre MC, Guidet B. Current epidemiology of septic shock: the CUB-Rea Network. Am J Respir Crit Care Med 2003; 168:165–172.
- Levy MM, Dellinger RP, Townsend SR, et al. The Surviving Sepsis Campaign: results of an international guideline-based performance improvement program targeting severe sepsis. Intensive Care Med 2010; 36:222–231.
- Amaral A, Opal SM, Vincent JL. Coagulation in sepsis. Intensive Care Med 2004; 30:1032–1040.
- Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med 2003; 348:138–150.
- Landry DW, Oliver JA. The pathogenesis of vasodilatory shock. N Engl J Med 2001; 345:588–595.
- Rady MY, Rivers EP, Nowak RM. Resuscitation of the critically ill in the ED: responses of blood pressure, heart rate, shock index, central venous oxygen saturation, and lactate. Am J Emerg Med 1996; 14:218–225.
- Assicot M, Gendrel D, Carsin H, Raymond J, Guilbaud J, Bohuon C. High serum procalcitonin concentrations in patients with sepsis and infection. Lancet 1993; 34:515–518.
- Muller B, Becker KL, Schachinger H, et al. Calcitonin precursors are reliable markers of sepsis in a medical intensive care unit. Crit Care Med 2000; 28:977–983.
- Meisner M, Tschaikowsky K, Palmaers T, Schmidt J. Comparison of procalcitonin (PCT) and C-reactive protein (CRP) plasma concentrations at different SOFA scores during the course of sepsis and MODS. Crit Care (London, England) 1999; 3:45–50.
- Tang BM, Eslick GD, Craig JC, McLean AS. Accuracy of procalcitonin for sepsis diagnosis in critically ill patients: systematic review and meta-analysis. Lancet Infect Dis 2007; 7:210–217.
- Brealey D, Brand M, Hargreaves I, et al. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 2002; 360:219–223.
- Vary TC. Sepsis-induced alterations in pyruvate dehydrogenase complex activity in rat skeletal muscle: effects on plasma lactate. Shock 1996; 6:89–94.
- Levy B, Gibot S, Franck P, Cravoisy A, Bollaert PE. Relation between muscle Na+K+ ATPase activity and raised lactate concentrations in septic shock: a prospective study. Lancet 2005; 365:871–875.
- Levraut J, Ciebiera JP, Chave S, et al. Mild hyperlactatemia in stable septic patients is due to impaired lactate clearance rather than over-production. Am J Respir Crit Care Med 1998; 157:1021–1026.
- Arnold RC, Shapiro NI, Jones AE, et al. Multicenter study of early lactate clearance as a determinant of survival in patients with presumed sepsis. Shock 2009; 32:35–39.
- Nguyen HB, Rivers EP, Knoblich BP, et al. Early lactate clearance is associated with improved outcome in severe sepsis and septic shock. Crit Care Med 2004; 32:1637–1642.
- Pierrakos C, Vincent JL. Sepsis biomarkers: a review. Crit Care 2010; 14:R15.
- Shapiro NI, Trzeciak S, Hollander JE, et al. A prospective, multicenter derivation of a biomarker panel to assess risk of organ dysfunction, shock, and death in emergency department patients with suspected sepsis. Crit Care Med 2009; 37:96–104.
- Marshall JC, al Naqbi A. Principles of source control in the management of sepsis. Crit Care Clin 2009; 25:753–768,viii–ix.
- Dellinger RP, Levy MM, Rhodes A, et al. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock: 2012. Crit Care Med 2013; 41:580–637.
- Kumar A, Roberts D, Wood KE, et al. Duration of hypotension before initiation of effective antimicrobial therapy is the critical determinant of survival in human septic shock. Crit Care Med 2006; 34:1589–1596.
- Kumar A, Ellis P, Arabi Y, et al. Initiation of inappropriate antimicrobial therapy results in a fivefold reduction of survival in human septic shock. Chest 2009; 136:1237–1248.
- Gaieski DF, Mikkelsen ME, Band RA, et al. Impact of time to antibiotics on survival in patients with severe sepsis or septic shock in whom early goal-directed therapy was initiated in the emergency department. Crit Care Med 2010; 38:1045–1053.
- Puskarich MA, Trzeciak S, Shapiro NI, et al. Association between timing of antibiotic administration and mortality from septic shock in patients treated with a quantitative resuscitation protocol. Crit Care Med 2011; 39:2066–2071.
- Rivers E, Nguyen B, Havstad S, et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med 2001; 345:1368–1377.
- Micek ST, Roubinian N, Heuring T, et al. Before-after study of a standardized hospital order set for the management of septic shock. Crit Care Med 2006; 34:2707–2713.
- Schmidt GA. Counterpoint: adherence to early goal-directed therapy: does it really matter? No. Both risks and benefits require further study. Chest 2010; 138:480–483; discussion 483–484.
- Jain RK, Antonio BL, Bowton DL, Houle TT, MacGregor DA. Variability in central venous pressure measurements and the potential impact on fluid management. Shock 2009; 33:253–257.
- Durairaj L, Schmidt GA. Fluid therapy in resuscitated sepsis: less is more. Chest 2008; 133:252–263.
- Finfer S, Bellomo R, Boyce N, French J, Myburgh J, Norton R. A comparison of albumin and saline for fluid resuscitation in the intensive care unit. N Engl J Med 2004; 350:2247–2256.
- Finfer S, McEvoy S, Bellomo R, McArthur C, Myburgh J, Norton R. Impact of albumin compared to saline on organ function and mortality of patients with severe sepsis. Intensive Care Med 2011; 37:86–96.
- Brunkhorst FM, Engel C, Bloos F, et al. Intensive insulin therapy and pentastarch resuscitation in severe sepsis. N Engl J Med 2008; 358:125–139.
- Perner A, Haase N, Guttormsen AB, et al. Hydroxyethyl starch 130/0.42 versus Ringer’s acetate in severe sepsis. N Engl J Med 2012; 367:124–134.
- Myburgh JA, Finfer S, Bellomo R, et al. Hydroxyethyl starch or saline for fluid resuscitation in intensive care. N Engl J Med 2012; 367:1901–1911.
- Jones AE, Shapiro NI, Trzeciak S, Arnold RC, Claremont HA, Kline JA. Lactate clearance vs central venous oxygen saturation as goals of early sepsis therapy: a randomized clinical trial. JAMA 2010; 303:739–746.
- Puskarich MA, Trzciak S, Shapiro NI, Kline JA, Jones AE. Concordance and prognostic value of central venous oxygen saturation and lactate clearance in emergency department patients with septic shock. Acad Emerg Med 2011; 19:S159–S160.
- Dunser MW, Takala J, Ulmer H, et al. Arterial blood pressure during early sepsis and outcome. Intensive Care Med 2009; 35:1225–1233.
- De Backer D, Biston P, Devriendt J, et al. Comparison of dopamine and norepinephrine in the treatment of shock. N Engl J Med 2010; 362:779–789.
- Annane D, Vignon P, Renault A, et al. Norepinephrine plus dobutamine versus epinephrine alone for management of septic shock: a randomised trial. Lancet 2007; 370:676–684.
- Morelli A, Ertmer C, Rehberg S, et al. Phenylephrine versus norepinephrine for initial hemodynamic support of patients with septic shock: a randomized, controlled trial. Crit Care (London, England) 2008; 12:R143.
- Coba V, Whitmill M, Mooney R, et al. Resuscitation bundle compliance in severe sepsis and septic shock: improves survival, is better late than never. J Intensive Care Med 2011 Jan 10[Epub ahead of print].
- Castellanos-Ortega A, Suberviola B, Garcia-Astudillo LA, Ortiz F, Llorca J, Delgado-Rodriguez M. Late compliance with the sepsis resuscitation bundle: impact on mortality. Shock 2011; 36:542–547.
- Marik PE, Baram M, Vahid B. Does central venous pressure predict fluid responsiveness? A systematic review of the literature and the tale of seven mares. Chest 2008; 134:172–178.
- Osman D, Ridel C, Ray P, et al. Cardiac filling pressures are not appropriate to predict hemodynamic response to volume challenge. Crit Care Med 2007; 35:64–68.
- Vincent JL, Sakr Y, Sprung CL, et al. Sepsis in European intensive care units: results of the SOAP study. Crit Care Med 2006; 34:344–353.
- Boyd JH, Forbes J, Nakada TA, Walley KR, Russell JA. Fluid resuscitation in septic shock: a positive fluid balance and elevated central venous pressure are associated with increased mortality. Crit Care Med 2011; 39:259–265.
- Wiedemann HP, Wheeler AP, Bernard GR, et al. Comparison of two fluid-management strategies in acute lung injury. N Engl J Med 2006; 354:2564–2575.
- Murphy CV, Schramm GE, Doherty JA, et al. The importance of fluid management in acute lung injury secondary to septic shock. Chest 2009; 136:102–109.
- Russell JA, Walley KR, Singer J, et al. Vasopressin versus norepinephrine infusion in patients with septic shock. N Engl J Med 2008; 358:877–887.
- Vincent JL, Roman A, Kahn RJ. Dobutamine administration in septic shock: addition to a standard protocol. Crit Care Med 1990; 18:689–693.
- Levy B, Bollaert PE, Charpentier C, et al. Comparison of norepinephrine and dobutamine to epinephrine for hemodynamics, lactate metabolism, and gastric tonometric variables in septic shock: a prospective, randomized study. Intensive Care Med 1997; 23:282–287.
- Redl-Wenzl EM, Armbruster C, Edelmann G, et al. The effects of norepinephrine on hemodynamics and renal function in severe septic shock states. Intensive Care Med 1993; 19:151–154.
- Vieillard-Baron A, Caille V, Charron C, Belliard G, Page B, Jardin F. Actual incidence of global left ventricular hypokinesia in adult septic shock. Crit Care Med 2008; 36:1701–1706.
- Annane D, Sebille V, Charpentier C, et al. Effect of treatment with low doses of hydrocortisone and fludrocortisone on mortality in patients with septic shock. JAMA 2002; 288:862–871.
- Dellinger RP, Carlet JM, Masur H, et al. Surviving Sepsis Campaign guidelines for management of severe sepsis and septic shock. Crit Care Med 2004; 32:858–873.
- Sprung CL, Annane D, Keh D, et al. Hydrocortisone therapy for patients with septic shock. N Engl J Med 2008; 358:111–124.
- Marik PE, Pastores SM, Annane D, et al. Recommendations for the diagnosis and management of corticosteroid insufficiency in critically ill adult patients: consensus statements from an international task force by the American College of Critical Care Medicine. Crit Care Med 2008; 36:1937–1949.
- Russell JA, Walley KR, Gordon AC, et al. Interaction of vasopressin infusion, corticosteroid treatment, and mortality of septic shock. Crit Care Med 2009; 37:811–818.
- Bauer SR, Lam SW, Cha SS, Oyen LJ. Effect of corticosteroids on arginine vasopressin-containing vasopressor therapy for septic shock: a case control study. J Crit Care 2008; 23:500–506.
- Torgersen C, Luckner G, Schroder DC, et al. Concomitant arginine-vasopressin and hydrocortisone therapy in severe septic shock: association with mortality. Intensive Care Med 2011; 37:1432–1437.
- van den Berghe G, Wouters P, Weekers F, et al. Intensive insulin therapy in critically ill patients. N Engl J Med 2001; 345:1359–1367.
- Preiser JC, Devos P, Ruiz-Santana S, et al. A prospective randomised multi-centre controlled trial on tight glucose control by intensive insulin therapy in adult intensive care units: the Glucontrol study. Intensive Care Med 2009; 35:1738–1748.
- Finfer S, Chittock DR, Su SY, et al. Intensive versus conventional glucose control in critically ill patients. N Engl J Med 2009; 360:1283–1297.
- Finfer S, Liu B, Chittock DR, et al. Hypoglycemia and risk of death in critically ill patients. N Engl J Med 2012; 367:1108–1118.
- Bernard GR, Vincent JL, Laterre PF, et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med 2001; 344:699–709.
- Ranieri VM, Thompson BT, Barie PS, et al. Drotrecogin alfa (activated) in adults with septic shock. N Engl J Med 2012; 366:2055–2064.
- Bouadma L, Luyt CE, Tubach F, et al. Use of procalcitonin to reduce patients’ exposure to antibiotics in intensive care units (PRORATA trial): a multicentre randomised controlled trial. Lancet 2009; 375:463–474.
- Jensen JU, Hein L, Lundgren B, et al. Procalcitonin-guided interventions against infections to increase early appropriate antibiotics and improve survival in the intensive care unit: a randomized trial. Crit Care Med 2011; 39:2048–2058.
KEY POINTS
- Managing septic shock in the first 6 hours involves prompt recognition, empiric antibiotic therapy, elimination of the source of infection (if applicable), fluid resuscitation titrated to specific goals, and vasopressor therapy.
- A number of biomarkers have been proposed to help recognize septic shock early in its course.
- A delay in starting appropriate antibiotic treatment is associated with higher risk of death.
- The ideal measure of the adequacy of fluid resuscitation remains a topic of study and debate.
- Preliminary studies suggest that norepinephrine should be the initial vasopressor.
- Management after the first 6 hours is less well defined. Decisions in this period include whether to give further fluid resuscitation, further and additional hemodynamic therapies, adjunctive therapies, and antibiotics.
Cardiac tamponade: 12 pearls in diagnosis and management
Cardiac tamponade is a life-threatening condition that can be palliated or cured, depending on its cause and on the timeliness of treatment. Making a timely diagnosis and providing the appropriate treatment can be gratifying for both patient and physician.
Cardiac tamponade occurs when fluid in the pericardial space reaches a pressure exceeding central venous pressure. This leads to jugular venous distention, visceral organ engorgement, edema, and elevated pulmonary venous pressure that causes dyspnea. Despite compensatory tachycardia, the decrease in cardiac filling leads to a fall in cardiac output and to arterial hypoperfusion of vital organs.
PEARL 1: SLOW ACCUMULATION LEADS TO EDEMA
The rate at which pericardial fluid accumulates influences the clinical presentation of cardiac tamponade, in particular whether or not there is edema. Whereas rapid accumulation is characterized more by hypotension than by edema, the slow accumulation of pericardial fluid affords the patient time to drink enough liquid to keep the central venous pressure higher than the rising pericardial pressure. Thus, edema and dyspnea are more prominent features of cardiac tamponade when there is a slow rise in pericardial pressure.
PEARL 2: EDEMA IS NOT ALWAYS TREATED WITH A DIURETIC
Edema is not always treated with a diuretic. In a patient who has a pericardial effusion that has developed slowly and who has been drinking enough fluid to keep the central venous pressure higher than the pericardial pressure, a diuretic can remove enough volume from the circulation to lower the central venous pressure below the intrapericardial pressure and thus convert a benign pericardial effusion to potentially lethal cardiac tamponade.
One must understand the cause of edema or low urine output before treating it. This underscores the importance of the history and the physical examination. All of the following must be assessed:
- Symptoms and time course of the illness
- Concurrent medical illnesses
- Neck veins
- Blood pressure and its response to inspiration
- Heart sounds
- Heart rate and rhythm
- Abdominal organ engorgement
- Edema (or its absence).
PEARL 3: UNDERSTANDING THE CAUSE IS ESSENTIAL
Understanding the cause of cardiac tamponade is essential.
A trauma patient first encountered in the emergency department may have an underlying disease, but the focus is squarely on the effects of trauma or violent injury. In a patient with multiple trauma, hypotension and tachycardia that do not respond to intravenous volume replacement when there is an obvious rise in central venous pressure should be clues to cardiac tamponade.1
If the patient has recently undergone a cardiac procedure (for example, cardiac surgery, myocardial biopsy, coronary intervention, electrophysiologic study with intracardiac electrodes, transvenous pacemaker placement, pacemaker lead extraction, or radiofrequency ablation), knowing about the procedure narrows the differential diagnosis when hypotension, tachycardia, and jugular venous distention develop.
PEARL 4: CARDIAC OR AORTIC RUPTURE REQUIRES SURGERY
When the etiology of cardiac tamponade is cardiac or aortic rupture, the treatment is surgical.
Painful sudden causes of cardiac tamponade include hemopericardium due to rupture of the free wall after myocardial infarction, and spontaneous or posttraumatic dissection and rupture of the ascending aorta. Prompt diagnosis is necessary, but since these lesions will not close and heal spontaneously, the definitive treatment should be surgery. Moreover, needle removal of intrapericardial blood that has been opposing further bleeding is sure to permit bleeding to recur, often with lethal consequences.2
Causes of cardiac tamponade that have a less-acute onset are likely to be complications of medical problems. Medical illnesses known to be associated with cardiac tamponade include:
- Infectious disease (idiopathic or viral, associated with smallpox vaccination, mycobacterial, purulent bacterial, fungal)
- Metastatic cancer (lung, breast, esophagus, lymphoma, pancreas, liver, leukemia, stomach, melanoma)3
- Connective tissue disease (rheumatoid arthritis, systemic lupus erythematosus, ankylosing spondylitis, scleroderma, Wegener granulomatosis, acute rheumatic fever)
- Endocrine disease (hypothyroidism)
- Drug side effects (procainamide, isoniazid, hydralazine, minoxidil, phenytoin, anticoagulants, methysergide)
- Inflammatory bowel disease (Crohn disease, ulcerative colitis)
- Congestive heart failure
- Uremia
- Radiation therapy
- Postmyocardial infarction syndrome (Dressler syndrome)
- Postpericardiotomy syndrome.
PEARL 5: REVIEW IMAGING BEFORE DIAGNOSING
What often brings a patient with cardiac tamponade to the attention of the physician is a finding on echocardiography, computed tomography, or magnetic resonance imaging of the chest.
Always review the imaging studies before making the diagnosis of cardiac tamponade. These tests must be reviewed to assess the anatomy and the size and location of the effusion. Particularly, one must look for atrial and right ventricular collapse and inferior vena caval plethora, which are echocardiographic signs of cardiac tamponade.4 Figures 1, 2, and 3 show imaging studies in a patient who presented with worsening cough 2 weeks after undergoing a cardiac procedure and who was found to have cardiac tamponade.
When the history and these imaging studies place cardiac tamponade high in the differential diagnosis as the cause of edema or dyspnea, it is time to reexamine the patient. The best first step is to measure pulsus paradoxus.
HOW PULSUS PARADOXUS OCCURS
To fully appreciate the subtleties of the next pearls, it is necessary to understand the pathophysiology of cardiac tamponade.
When pericardial fluid accumulation raises the pericardial pressure above the central venous pressure and pulmonary venous pressure (intravascular pressure), blood will not passively return to the right side of the heart from the venae cavae nor to the left side of the heart from the pulmonary veins unless it is influenced by the effects of respiration on intrathoracic pressure. During respiration, the right and left sides of the heart are alternately filled and deprived of their respective venous return.
During inspiration, as the intrathoracic pressure decreases, blood in the venae cavae empties into the right side of the heart, while blood in the pulmonary veins preferentially remains in the pulmonary veins, underfilling the left side of the heart. Since the right ventricle is more filled than the left ventricle during inspiration, the ventricular septum shifts from right to left, further opposing pulmonary venous return. As a result, during cardiac tamponade, the systemic blood pressure falls with inspiration.
During expiration the opposite occurs. Expiration decreases the intrathoracic volume, so the intrathoracic pressure rises. This tends to oppose vena caval return to the right side of the heart and to favor pulmonary venous return to the left side of the heart. The ventricular septum shifts from left to right, further accommodating left ventricular filling, raising stroke volume, and increasing blood pressure. This exaggerated alternate filling of the right and left sides of the heart during cardiac tamponade is what accounts for pulsus paradoxus, an inspiratory fall in systolic blood pressure of greater than 10 mm Hg.
If intravascular pressure is low (due to hemorrhage, dehydration, or diuretic therapy), the pressure in the pericardial space needed to oppose venous return is much less. In this low-pressure scenario, the results are low cardiac output and hypotension, which are treated by giving intravenous fluids to maintain intravascular volume.
PEARL 6: MEASURE PULSUS PARADOXUS
When cardiac tamponade is considered, one must always measure the pulsus paradoxus.
The term pulsus paradoxus was coined by Adolph Kussmaul in 1873, before physicians could even measure blood pressure. All they could do at that time was palpate the pulse and listen to the heart. Kussmaul described his observation as a conspicuous discrepancy between the cardiac action and the arterial pulse.
Although not described by Kussmaul, another explanation for this finding might be more suited to the use of the word “paradoxical.” When the pulse is palpated in a normal patient, with inspiration the pulse rate will increase via the Bainbridge reflex, and with expiration it will decrease. But in a patient with cardiac tamponade, there is a paradoxical inspiratory slowing of the pulse (because the decreased magnitude of the pulse at times makes it imperceptible) and an expiratory increase in pulse rate as the magnitude of the pulse again makes it palpable.
The magnitude of the fall in systolic blood pressure during inspiration has been used to estimate the level of hemodynamic impairment resulting from pericardial effusion.5 A rapidly accumulating pericardial effusion can have more hemodynamic impact than a much larger one that accumulates slowly. Thus, the intrapericardial pressure must be considered more than the volume of pericardial fluid.
When there is severe cardiac tamponade and overt pulsus paradoxus, simple palpation of a proximal arterial pulse can detect a marked inspiratory decrease or loss of the pulse, which returns with expiration. Tachycardia is almost always present, unless the cause is hypothyroidism.6
How to measure pulsus paradoxus with a manual sphygmomanometer
A stethoscope and manual sphygmomanometer are all that is needed to measure pulsus paradoxus. A noninvasive blood pressure monitor that averages multiple measurements cannot detect or quantify pulsus paradoxus.
The patient should be supine with the head elevated 30° to 45°, and the examiner should be seated comfortably at the patient’s side. The manometer should be on the opposite side of the patient in plain view of the examiner. Position the cuff on the arm above the elbow and place your stethoscope on the antecubital fossa. Then:
- Inflate the cuff 20 mm Hg above the highest systolic pressure previously auscultated.
- Slowly decrease the manometer pressure by 5 mm Hg and hold it there through two or three respiratory cycles while listening for the first Korotkoff (systolic) sound. Repeat this until you can hear the systolic sound (but only during expiration) and mentally note the pressure.
- Continue to decrease the manometer pressure by 5-mm Hg increments while listening. When the Korotkoff sounds no longer disappear with inspiration, mentally note this second value as well. The pulsus paradoxus is the difference between these values.
- When the Korotkoff sounds disappear as the manometer pressure is decreased, note this final value. This is the diastolic blood pressure.
PEARL 7: THE PLETHYSMOGRAM WAVE-FORM PARALLELS PULSUS PARADOXUS
Manual measurement of blood pressure and pulsus paradoxus can be difficult, especially in an obese patient or one with a fat-distorted arm on which the cuff does not maintain its position. In such patients, increased girth of the neck and abdomen also make it difficult to assess the jugular venous distention and visceral organ engorgement that characterize cardiac tamponade.
When the use of a sphygmomanometer is not possible, an arterial catheter can be inserted to demonstrate pulsus paradoxus. Simpler, however, is the novel use of another noninvasive instrument to detect and coarsely quantify pulsus paradoxus.7 The waveform on finger pulse oximetry can demonstrate pulsus paradoxus. The plethysmogram of the finger pulse oximeter can demonstrate the decrease in magnitude of the waveform with each inspiration (Figure 4).
Caution must be taken when interpreting this waveform, as with any measurement of pulsus paradoxus, to exclude a concomitant arrhythmia.
PEARL 8: PULSUS PARADOXUS WITHOUT CARDIAC TAMPONADE
Pulsus paradoxus can be present in the absence of cardiac tamponade. Once pulsus paradoxus of more than 10 mm Hg is measured, one must be sure the patient does not have a condition that can cause pulsus paradoxus without cardiac tamponade. Most of these are pulmonary conditions that necessitate an exaggerated inspiratory effort that can lower intrathoracic pressure sufficiently to oppose pulmonary venous return and cause a fall in systemic blood pressure:
- Chronic bronchitis
- Emphysema
- Mucus plug
- Pneumothorax
- Pulmonary embolism
- Stridor.
In these, there may be pulsus paradoxus, but not due to cardiac tamponade.
PEARL 9: CARDIAC TAMPONADE CAN BE PRESENT WITHOUT PULSUS PARADOXUS
Cardiac tamponade can be present without pulsus paradoxus. This occurs when certain conditions prevent inspiratory underfilling of the left ventricle relative to the filling of the right ventricle.8
How does this work? In cardiac tamponade, factors that drive the exaggerated fall in arterial pressure with inspiration (pulsus paradoxus) are the augmented right ventricular filling and the decreased left ventricular filling, both due to the lowering of the intrathoracic pressure. As the vena caval emptying is augmented, the right ventricular filling is increased, the ventricular septum shifts to the left, and pulmonary venous return to the heart is decreased.
Factors that can oppose pulsus paradoxus:
- Positive pressure ventilation prevents pulsus paradoxus by preventing the fall in intrathoracic pressure.
- Severe aortic regurgitation does not permit underfilling of the left ventricle during inspiration.
- An atrial septal defect will always equalize the right and left atrial pressures, preventing differential right ventricular and left ventricular filling with inspiration.
- Severe left ventricular hypertrophy does not permit the inspiratory shift of the ventricular septum from right to left that would otherwise lead to decreased left ventricular filling.
- Severe left ventricular dysfunction, with its low stroke volume and severe elevation of left ventricular end-diastolic pressure, never permits underfilling of the left ventricle, despite cardiac tamponade and an inspiratory decrease in intrathoracic pressure.
- Intravascular volume depletion due to hemorrhage, hemodialysis, or mistaken use of diuretics to treat edema can cause marked hypotension, making pulsus paradoxus impossible to detect.
Knowledge of underlying medical conditions, the likelihood of their causing cardiac tamponade, and the appearance of the echocardiogram prompt the physician to look further when the presence or absence of pulsus paradoxus does not fit with the working diagnosis.
The echocardiogram can give hints to the etiology of a pericardial effusion, such as clotted blood after trauma or a cardiac-perforating procedure, tumor studding of the epicardium,9 or fibrin strands indicating chronicity or an inflammatory process.10 Diastolic collapse of the right ventricle, more than collapse of the right atrium or left atrium, speaks for the severity of cardiac tamponade. With hemodynamically significant pericardial effusion and cardiac tamponade, the inferior vena cava is distended and does not decrease in size with inspiration unless there is severe intravascular volume depletion, at which time the inferior vena cava is underfilled throughout the respiratory cycle.
PEARL 10: PLAN HOW TO DRAIN
The size and location of the pericardial effusion and the patient’s hemodynamics must be integrated when deciding how to relieve cardiac tamponade. When cardiac tamponade is indeed severe and the patient and physician agree that it must be drained, the options are percutaneous needle aspiration (pericardiocentesis) and surgical pericardiostomy (creation of a pericardial window). Here again, as assessed by echocardiography, the access to the pericardial fluid should influence the choice.
Pericardiocentesis can be safely done if certain criteria are met. The patient must be able to lie still in the supine position, perhaps with the head of the bed elevated 30 degrees. Anticoagulation must be reversed or allowed time to resolve if drainage is not an emergency.
Pericardiocentesis can be risky or unsuccessful if there is not enough pericardial fluid to permit respiratory cardiac motion without perforating the heart with the needle; if the effusion is loculated (confined to a pocket) posteriorly; or if it is too far from the skin to permit precise control and placement of a spinal needle into the pericardial space. In cases of cardiac tamponade in which the anatomy indicates surgical pericardiostomy but severe hypotension prevents the induction of anesthesia and positive-pressure ventilation—which can result in profound, irreversible hypotension—percutaneous needle drainage (pericardiocentesis) should be performed in the operating room to relieve the tamponade before the induction of anesthesia and the surgical drainage.11
To reiterate, a suspected cardiac or aortic rupture that causes cardiac tamponade is usually large and not apt to self-seal. In such cases, the halt in the accumulation of pericardial blood is due to hypotension and not due to spontaneous resolution. Open surgical drainage is required from the outset because an initial success of pericardiocentesis yields to the recurrence of cardiac tamponade.
PEARL 11: ANTICIPATE WHAT THE FLUID SHOULD LOOK LIKE
Before performing pericardiocentesis, anticipate the appearance of the pericardial fluid on the basis of the presumed etiology, ie:
- Sanguinous—trauma, heart surgery, cardiac perforation from a procedure, anticoagulation, uremia, or malignancy
- Serous—congestive heart failure, acute radiation therapy
- Purulent—infections (natural or postoperative)
- Turbid (like gold paint)—mycobacterial infection, rheumatoid arthritis, myxedema
- Chylous—pericardium fistulized to the thoracic duct by a natural or postsurgical cause.
Sanguinous pericardial effusion encountered during a pericardiocentesis, if not anticipated, can be daunting and can cause the operator to question if it is the result of inadvertent needle placement in a cardiac chamber. If the needle is indeed in the heart, blood often surges out under pressure in pulses, which strongly suggests that the needle is not in the pericardial space and should be removed; but if confirmation of the location is needed before removing the needle, it can be done by injecting 2 mL of agitated sterile saline through the pericardiocentesis needle during echocardiographic imaging.12
Before inserting the needle, the ideal access location and needle angle must be determined by the operator with echocardiographic transducer in hand. The distance from skin to a point just through the parietal pericardium can also be measured at this time.
Once the needle is in the pericardial fluid (and you are confident of its placement), removal of 50 to 100 mL of the fluid with a large syringe can be enough to afford the patient easier breathing, higher blood pressure, and lower pulsus paradoxus—and even the physician will breathe easier. The same syringe can be filled and emptied multiple times. Less traumatic and more complete removal of pericardial fluid requires insertion of a multihole pigtail catheter over a J-tipped guidewire that is introduced through the needle.
PEARL 12: DRAIN SLOWLY TO AVOID PULMONARY EDEMA
Pulmonary edema is an uncommon complication of pericardiocentesis that might be avoidable. Heralded by sudden coughing and pink, frothy sputum, it can rapidly deteriorate into respiratory failure. The mechanism has been attributed to a sudden increase in right ventricular stroke volume and resultant left ventricular filling after the excess pericardial fluid has been removed, before the systemic arteries, which constrict to keep the systemic blood pressure up during cardiac tamponade, have had time to relax.13
To avoid this complication, if the volume of pericardial fluid responsible for cardiac tamponade is large, it should be removed slowly,14 stopping for a several-minute rest after each 250 mL. Catheter removal of pericardial fluid by gravity drainage over 24 hours has been suggested.15 A drawback to this approach is catheter clotting or sludging before all the fluid has been removed. It is helpful to keep the drainage catheter close to the patient’s body temperature to make the fluid less viscous. Output should be monitored hourly.
When the pericardial fluid has been completely drained, one must decide how long to leave the catheter in. One reason to remove the catheter at this time is that it causes pleuritic pain; another is to avoid introducing infection. A reason to leave the catheter in is to observe the effect of medical treatment on the hourly pericardial fluid output. Nonsteroidal anti-inflammatory drugs are the drugs of first choice when treating pericardial inflammation and suppressing production of pericardial fluid.16 In most cases the catheter should not be left in place for more than 3 days.
Laboratory analysis of the pericardial fluid should shed light on its suspected cause. Analysis usually involves chemistry testing, microscopic inspection of blood cell smears, cytology, microbiologic stains and cultures, and immunologic tests. Results often take days. Meyers and colleagues17 expound on this subject.
- Schiavone WA, Ghumrawi BK, Catalano DR, et al. The use of echocardiography in the emergency management of nonpenetrating traumatic cardiac rupture. Ann Emerg Med 1991; 20:1248–1250.
- Manuchehry A, Fontana GP, Gurudevan S, Marchevsky AM, Siegel RJ. Missed diagnosis of limited ascending aortic dissection by multiple imaging modalities leading to fatal cardiac tamponade and aortic rupture. Echocardiography 2011; 28:E187–E190.
- Lam KY, Dickens P, Chan AC. Tumors of the heart. A 20-year experience with a review of 12,485 consecutive autopsies. Arch Pathol Lab Med 1993; 117:1027–1031.
- Tsang TS, Oh JK, Seward JB, Tajik AJ. Diagnostic value of echocardiography in cardiac tamponade. Herz 2000; 25:734–740.
- Curtiss EI, Reddy PS, Uretsky BF, Cecchetti AA. Pulsus paradoxus: definition and relation to the severity of cardiac tamponade. Am Heart J 1988; 115:391–398.
- Wang JL, Hsieh MJ, Lee CH, et al. Hypothyroid cardiac tamponade: clinical features, electrocardiography, pericardial fluid and management. Am J Med Sci 2010; 340:276–281.
- Tamburro RF, Ring JC, Womback K. Detection of pulsus paradoxus associated with large pericardial effusions in pediatric patients by analysis of the pulse-oximetry waveform. Pediatrics 2002; 109:673–677.
- Spodick DH. Pulsus paradoxus. In:Spodick DH, editor. The Pericardium: A Comprehensive Textbook. New York, NY: Marcel Dekker; 1997:191–199.
- Burke A, Jeudy J, Virmani R. Cardiac tumors. In:Topol EJ, editor. Textbook of Cardiovascular Medicine. 3rd ed. Philadelphia, PA: Lippincott, Williams & Wilkins; 2007:710–720.
- Roberts WC. Pericardial heart disease: Its morphologic features and its causes. Proc (Bayl Univ Med Cent) 2005; 18:38–55.
- Stoelting RK, Miller RD, editors. Basics of Anesthesia. 4th ed. New York, NY: Churchill Livingstone; 2000:264–265.
- Ainsworth CD, Salehian O. Echo-guided pericardiocentesis: let the bubbles show the way. Circulation 2011; 123:e210–e211.
- Maisch B, Seferovic PM, Ristic AD, et al; Task Force on the Diagnosis and Management of Pericardial Diseases of the European Society of Cardiology. Guidelines on the diagnosis and management of pericardial diseases executive summary; The Task Force on the Diagnosis and Management of Pericardial Diseases of the European Society of Cardiology. Eur Heart J 2004; 25:587–610.
- Vandyke WH, Cure J, Chakko CS, Gheorghiade M. Pulmonary edema after pericardiocentesis for cardiac tamponade. N Engl J Med 1983; 309:595–596.
- Bernal JM, Pradhan J, Li T, Tchokonte R, Afonso L. Acute pulmonary edema following pericardiocentesis for cardiac tamponade. Can J Cardiol 2007; 23:1155–1156.
- Sagristà-Sauleda J, Mercé AS, Soler-Soler J. Diagnosis and management of pericardial effusion. World J Cardiol 2011; 3:135–143.
- Meyers DG, Meyers RE, Prendergast TW. The usefulness of diagnostic tests on pericardial fluid. Chest 1997; 111:1213–1221.
Cardiac tamponade is a life-threatening condition that can be palliated or cured, depending on its cause and on the timeliness of treatment. Making a timely diagnosis and providing the appropriate treatment can be gratifying for both patient and physician.
Cardiac tamponade occurs when fluid in the pericardial space reaches a pressure exceeding central venous pressure. This leads to jugular venous distention, visceral organ engorgement, edema, and elevated pulmonary venous pressure that causes dyspnea. Despite compensatory tachycardia, the decrease in cardiac filling leads to a fall in cardiac output and to arterial hypoperfusion of vital organs.
PEARL 1: SLOW ACCUMULATION LEADS TO EDEMA
The rate at which pericardial fluid accumulates influences the clinical presentation of cardiac tamponade, in particular whether or not there is edema. Whereas rapid accumulation is characterized more by hypotension than by edema, the slow accumulation of pericardial fluid affords the patient time to drink enough liquid to keep the central venous pressure higher than the rising pericardial pressure. Thus, edema and dyspnea are more prominent features of cardiac tamponade when there is a slow rise in pericardial pressure.
PEARL 2: EDEMA IS NOT ALWAYS TREATED WITH A DIURETIC
Edema is not always treated with a diuretic. In a patient who has a pericardial effusion that has developed slowly and who has been drinking enough fluid to keep the central venous pressure higher than the pericardial pressure, a diuretic can remove enough volume from the circulation to lower the central venous pressure below the intrapericardial pressure and thus convert a benign pericardial effusion to potentially lethal cardiac tamponade.
One must understand the cause of edema or low urine output before treating it. This underscores the importance of the history and the physical examination. All of the following must be assessed:
- Symptoms and time course of the illness
- Concurrent medical illnesses
- Neck veins
- Blood pressure and its response to inspiration
- Heart sounds
- Heart rate and rhythm
- Abdominal organ engorgement
- Edema (or its absence).
PEARL 3: UNDERSTANDING THE CAUSE IS ESSENTIAL
Understanding the cause of cardiac tamponade is essential.
A trauma patient first encountered in the emergency department may have an underlying disease, but the focus is squarely on the effects of trauma or violent injury. In a patient with multiple trauma, hypotension and tachycardia that do not respond to intravenous volume replacement when there is an obvious rise in central venous pressure should be clues to cardiac tamponade.1
If the patient has recently undergone a cardiac procedure (for example, cardiac surgery, myocardial biopsy, coronary intervention, electrophysiologic study with intracardiac electrodes, transvenous pacemaker placement, pacemaker lead extraction, or radiofrequency ablation), knowing about the procedure narrows the differential diagnosis when hypotension, tachycardia, and jugular venous distention develop.
PEARL 4: CARDIAC OR AORTIC RUPTURE REQUIRES SURGERY
When the etiology of cardiac tamponade is cardiac or aortic rupture, the treatment is surgical.
Painful sudden causes of cardiac tamponade include hemopericardium due to rupture of the free wall after myocardial infarction, and spontaneous or posttraumatic dissection and rupture of the ascending aorta. Prompt diagnosis is necessary, but since these lesions will not close and heal spontaneously, the definitive treatment should be surgery. Moreover, needle removal of intrapericardial blood that has been opposing further bleeding is sure to permit bleeding to recur, often with lethal consequences.2
Causes of cardiac tamponade that have a less-acute onset are likely to be complications of medical problems. Medical illnesses known to be associated with cardiac tamponade include:
- Infectious disease (idiopathic or viral, associated with smallpox vaccination, mycobacterial, purulent bacterial, fungal)
- Metastatic cancer (lung, breast, esophagus, lymphoma, pancreas, liver, leukemia, stomach, melanoma)3
- Connective tissue disease (rheumatoid arthritis, systemic lupus erythematosus, ankylosing spondylitis, scleroderma, Wegener granulomatosis, acute rheumatic fever)
- Endocrine disease (hypothyroidism)
- Drug side effects (procainamide, isoniazid, hydralazine, minoxidil, phenytoin, anticoagulants, methysergide)
- Inflammatory bowel disease (Crohn disease, ulcerative colitis)
- Congestive heart failure
- Uremia
- Radiation therapy
- Postmyocardial infarction syndrome (Dressler syndrome)
- Postpericardiotomy syndrome.
PEARL 5: REVIEW IMAGING BEFORE DIAGNOSING
What often brings a patient with cardiac tamponade to the attention of the physician is a finding on echocardiography, computed tomography, or magnetic resonance imaging of the chest.
Always review the imaging studies before making the diagnosis of cardiac tamponade. These tests must be reviewed to assess the anatomy and the size and location of the effusion. Particularly, one must look for atrial and right ventricular collapse and inferior vena caval plethora, which are echocardiographic signs of cardiac tamponade.4 Figures 1, 2, and 3 show imaging studies in a patient who presented with worsening cough 2 weeks after undergoing a cardiac procedure and who was found to have cardiac tamponade.
When the history and these imaging studies place cardiac tamponade high in the differential diagnosis as the cause of edema or dyspnea, it is time to reexamine the patient. The best first step is to measure pulsus paradoxus.
HOW PULSUS PARADOXUS OCCURS
To fully appreciate the subtleties of the next pearls, it is necessary to understand the pathophysiology of cardiac tamponade.
When pericardial fluid accumulation raises the pericardial pressure above the central venous pressure and pulmonary venous pressure (intravascular pressure), blood will not passively return to the right side of the heart from the venae cavae nor to the left side of the heart from the pulmonary veins unless it is influenced by the effects of respiration on intrathoracic pressure. During respiration, the right and left sides of the heart are alternately filled and deprived of their respective venous return.
During inspiration, as the intrathoracic pressure decreases, blood in the venae cavae empties into the right side of the heart, while blood in the pulmonary veins preferentially remains in the pulmonary veins, underfilling the left side of the heart. Since the right ventricle is more filled than the left ventricle during inspiration, the ventricular septum shifts from right to left, further opposing pulmonary venous return. As a result, during cardiac tamponade, the systemic blood pressure falls with inspiration.
During expiration the opposite occurs. Expiration decreases the intrathoracic volume, so the intrathoracic pressure rises. This tends to oppose vena caval return to the right side of the heart and to favor pulmonary venous return to the left side of the heart. The ventricular septum shifts from left to right, further accommodating left ventricular filling, raising stroke volume, and increasing blood pressure. This exaggerated alternate filling of the right and left sides of the heart during cardiac tamponade is what accounts for pulsus paradoxus, an inspiratory fall in systolic blood pressure of greater than 10 mm Hg.
If intravascular pressure is low (due to hemorrhage, dehydration, or diuretic therapy), the pressure in the pericardial space needed to oppose venous return is much less. In this low-pressure scenario, the results are low cardiac output and hypotension, which are treated by giving intravenous fluids to maintain intravascular volume.
PEARL 6: MEASURE PULSUS PARADOXUS
When cardiac tamponade is considered, one must always measure the pulsus paradoxus.
The term pulsus paradoxus was coined by Adolph Kussmaul in 1873, before physicians could even measure blood pressure. All they could do at that time was palpate the pulse and listen to the heart. Kussmaul described his observation as a conspicuous discrepancy between the cardiac action and the arterial pulse.
Although not described by Kussmaul, another explanation for this finding might be more suited to the use of the word “paradoxical.” When the pulse is palpated in a normal patient, with inspiration the pulse rate will increase via the Bainbridge reflex, and with expiration it will decrease. But in a patient with cardiac tamponade, there is a paradoxical inspiratory slowing of the pulse (because the decreased magnitude of the pulse at times makes it imperceptible) and an expiratory increase in pulse rate as the magnitude of the pulse again makes it palpable.
The magnitude of the fall in systolic blood pressure during inspiration has been used to estimate the level of hemodynamic impairment resulting from pericardial effusion.5 A rapidly accumulating pericardial effusion can have more hemodynamic impact than a much larger one that accumulates slowly. Thus, the intrapericardial pressure must be considered more than the volume of pericardial fluid.
When there is severe cardiac tamponade and overt pulsus paradoxus, simple palpation of a proximal arterial pulse can detect a marked inspiratory decrease or loss of the pulse, which returns with expiration. Tachycardia is almost always present, unless the cause is hypothyroidism.6
How to measure pulsus paradoxus with a manual sphygmomanometer
A stethoscope and manual sphygmomanometer are all that is needed to measure pulsus paradoxus. A noninvasive blood pressure monitor that averages multiple measurements cannot detect or quantify pulsus paradoxus.
The patient should be supine with the head elevated 30° to 45°, and the examiner should be seated comfortably at the patient’s side. The manometer should be on the opposite side of the patient in plain view of the examiner. Position the cuff on the arm above the elbow and place your stethoscope on the antecubital fossa. Then:
- Inflate the cuff 20 mm Hg above the highest systolic pressure previously auscultated.
- Slowly decrease the manometer pressure by 5 mm Hg and hold it there through two or three respiratory cycles while listening for the first Korotkoff (systolic) sound. Repeat this until you can hear the systolic sound (but only during expiration) and mentally note the pressure.
- Continue to decrease the manometer pressure by 5-mm Hg increments while listening. When the Korotkoff sounds no longer disappear with inspiration, mentally note this second value as well. The pulsus paradoxus is the difference between these values.
- When the Korotkoff sounds disappear as the manometer pressure is decreased, note this final value. This is the diastolic blood pressure.
PEARL 7: THE PLETHYSMOGRAM WAVE-FORM PARALLELS PULSUS PARADOXUS
Manual measurement of blood pressure and pulsus paradoxus can be difficult, especially in an obese patient or one with a fat-distorted arm on which the cuff does not maintain its position. In such patients, increased girth of the neck and abdomen also make it difficult to assess the jugular venous distention and visceral organ engorgement that characterize cardiac tamponade.
When the use of a sphygmomanometer is not possible, an arterial catheter can be inserted to demonstrate pulsus paradoxus. Simpler, however, is the novel use of another noninvasive instrument to detect and coarsely quantify pulsus paradoxus.7 The waveform on finger pulse oximetry can demonstrate pulsus paradoxus. The plethysmogram of the finger pulse oximeter can demonstrate the decrease in magnitude of the waveform with each inspiration (Figure 4).
Caution must be taken when interpreting this waveform, as with any measurement of pulsus paradoxus, to exclude a concomitant arrhythmia.
PEARL 8: PULSUS PARADOXUS WITHOUT CARDIAC TAMPONADE
Pulsus paradoxus can be present in the absence of cardiac tamponade. Once pulsus paradoxus of more than 10 mm Hg is measured, one must be sure the patient does not have a condition that can cause pulsus paradoxus without cardiac tamponade. Most of these are pulmonary conditions that necessitate an exaggerated inspiratory effort that can lower intrathoracic pressure sufficiently to oppose pulmonary venous return and cause a fall in systemic blood pressure:
- Chronic bronchitis
- Emphysema
- Mucus plug
- Pneumothorax
- Pulmonary embolism
- Stridor.
In these, there may be pulsus paradoxus, but not due to cardiac tamponade.
PEARL 9: CARDIAC TAMPONADE CAN BE PRESENT WITHOUT PULSUS PARADOXUS
Cardiac tamponade can be present without pulsus paradoxus. This occurs when certain conditions prevent inspiratory underfilling of the left ventricle relative to the filling of the right ventricle.8
How does this work? In cardiac tamponade, factors that drive the exaggerated fall in arterial pressure with inspiration (pulsus paradoxus) are the augmented right ventricular filling and the decreased left ventricular filling, both due to the lowering of the intrathoracic pressure. As the vena caval emptying is augmented, the right ventricular filling is increased, the ventricular septum shifts to the left, and pulmonary venous return to the heart is decreased.
Factors that can oppose pulsus paradoxus:
- Positive pressure ventilation prevents pulsus paradoxus by preventing the fall in intrathoracic pressure.
- Severe aortic regurgitation does not permit underfilling of the left ventricle during inspiration.
- An atrial septal defect will always equalize the right and left atrial pressures, preventing differential right ventricular and left ventricular filling with inspiration.
- Severe left ventricular hypertrophy does not permit the inspiratory shift of the ventricular septum from right to left that would otherwise lead to decreased left ventricular filling.
- Severe left ventricular dysfunction, with its low stroke volume and severe elevation of left ventricular end-diastolic pressure, never permits underfilling of the left ventricle, despite cardiac tamponade and an inspiratory decrease in intrathoracic pressure.
- Intravascular volume depletion due to hemorrhage, hemodialysis, or mistaken use of diuretics to treat edema can cause marked hypotension, making pulsus paradoxus impossible to detect.
Knowledge of underlying medical conditions, the likelihood of their causing cardiac tamponade, and the appearance of the echocardiogram prompt the physician to look further when the presence or absence of pulsus paradoxus does not fit with the working diagnosis.
The echocardiogram can give hints to the etiology of a pericardial effusion, such as clotted blood after trauma or a cardiac-perforating procedure, tumor studding of the epicardium,9 or fibrin strands indicating chronicity or an inflammatory process.10 Diastolic collapse of the right ventricle, more than collapse of the right atrium or left atrium, speaks for the severity of cardiac tamponade. With hemodynamically significant pericardial effusion and cardiac tamponade, the inferior vena cava is distended and does not decrease in size with inspiration unless there is severe intravascular volume depletion, at which time the inferior vena cava is underfilled throughout the respiratory cycle.
PEARL 10: PLAN HOW TO DRAIN
The size and location of the pericardial effusion and the patient’s hemodynamics must be integrated when deciding how to relieve cardiac tamponade. When cardiac tamponade is indeed severe and the patient and physician agree that it must be drained, the options are percutaneous needle aspiration (pericardiocentesis) and surgical pericardiostomy (creation of a pericardial window). Here again, as assessed by echocardiography, the access to the pericardial fluid should influence the choice.
Pericardiocentesis can be safely done if certain criteria are met. The patient must be able to lie still in the supine position, perhaps with the head of the bed elevated 30 degrees. Anticoagulation must be reversed or allowed time to resolve if drainage is not an emergency.
Pericardiocentesis can be risky or unsuccessful if there is not enough pericardial fluid to permit respiratory cardiac motion without perforating the heart with the needle; if the effusion is loculated (confined to a pocket) posteriorly; or if it is too far from the skin to permit precise control and placement of a spinal needle into the pericardial space. In cases of cardiac tamponade in which the anatomy indicates surgical pericardiostomy but severe hypotension prevents the induction of anesthesia and positive-pressure ventilation—which can result in profound, irreversible hypotension—percutaneous needle drainage (pericardiocentesis) should be performed in the operating room to relieve the tamponade before the induction of anesthesia and the surgical drainage.11
To reiterate, a suspected cardiac or aortic rupture that causes cardiac tamponade is usually large and not apt to self-seal. In such cases, the halt in the accumulation of pericardial blood is due to hypotension and not due to spontaneous resolution. Open surgical drainage is required from the outset because an initial success of pericardiocentesis yields to the recurrence of cardiac tamponade.
PEARL 11: ANTICIPATE WHAT THE FLUID SHOULD LOOK LIKE
Before performing pericardiocentesis, anticipate the appearance of the pericardial fluid on the basis of the presumed etiology, ie:
- Sanguinous—trauma, heart surgery, cardiac perforation from a procedure, anticoagulation, uremia, or malignancy
- Serous—congestive heart failure, acute radiation therapy
- Purulent—infections (natural or postoperative)
- Turbid (like gold paint)—mycobacterial infection, rheumatoid arthritis, myxedema
- Chylous—pericardium fistulized to the thoracic duct by a natural or postsurgical cause.
Sanguinous pericardial effusion encountered during a pericardiocentesis, if not anticipated, can be daunting and can cause the operator to question if it is the result of inadvertent needle placement in a cardiac chamber. If the needle is indeed in the heart, blood often surges out under pressure in pulses, which strongly suggests that the needle is not in the pericardial space and should be removed; but if confirmation of the location is needed before removing the needle, it can be done by injecting 2 mL of agitated sterile saline through the pericardiocentesis needle during echocardiographic imaging.12
Before inserting the needle, the ideal access location and needle angle must be determined by the operator with echocardiographic transducer in hand. The distance from skin to a point just through the parietal pericardium can also be measured at this time.
Once the needle is in the pericardial fluid (and you are confident of its placement), removal of 50 to 100 mL of the fluid with a large syringe can be enough to afford the patient easier breathing, higher blood pressure, and lower pulsus paradoxus—and even the physician will breathe easier. The same syringe can be filled and emptied multiple times. Less traumatic and more complete removal of pericardial fluid requires insertion of a multihole pigtail catheter over a J-tipped guidewire that is introduced through the needle.
PEARL 12: DRAIN SLOWLY TO AVOID PULMONARY EDEMA
Pulmonary edema is an uncommon complication of pericardiocentesis that might be avoidable. Heralded by sudden coughing and pink, frothy sputum, it can rapidly deteriorate into respiratory failure. The mechanism has been attributed to a sudden increase in right ventricular stroke volume and resultant left ventricular filling after the excess pericardial fluid has been removed, before the systemic arteries, which constrict to keep the systemic blood pressure up during cardiac tamponade, have had time to relax.13
To avoid this complication, if the volume of pericardial fluid responsible for cardiac tamponade is large, it should be removed slowly,14 stopping for a several-minute rest after each 250 mL. Catheter removal of pericardial fluid by gravity drainage over 24 hours has been suggested.15 A drawback to this approach is catheter clotting or sludging before all the fluid has been removed. It is helpful to keep the drainage catheter close to the patient’s body temperature to make the fluid less viscous. Output should be monitored hourly.
When the pericardial fluid has been completely drained, one must decide how long to leave the catheter in. One reason to remove the catheter at this time is that it causes pleuritic pain; another is to avoid introducing infection. A reason to leave the catheter in is to observe the effect of medical treatment on the hourly pericardial fluid output. Nonsteroidal anti-inflammatory drugs are the drugs of first choice when treating pericardial inflammation and suppressing production of pericardial fluid.16 In most cases the catheter should not be left in place for more than 3 days.
Laboratory analysis of the pericardial fluid should shed light on its suspected cause. Analysis usually involves chemistry testing, microscopic inspection of blood cell smears, cytology, microbiologic stains and cultures, and immunologic tests. Results often take days. Meyers and colleagues17 expound on this subject.
Cardiac tamponade is a life-threatening condition that can be palliated or cured, depending on its cause and on the timeliness of treatment. Making a timely diagnosis and providing the appropriate treatment can be gratifying for both patient and physician.
Cardiac tamponade occurs when fluid in the pericardial space reaches a pressure exceeding central venous pressure. This leads to jugular venous distention, visceral organ engorgement, edema, and elevated pulmonary venous pressure that causes dyspnea. Despite compensatory tachycardia, the decrease in cardiac filling leads to a fall in cardiac output and to arterial hypoperfusion of vital organs.
PEARL 1: SLOW ACCUMULATION LEADS TO EDEMA
The rate at which pericardial fluid accumulates influences the clinical presentation of cardiac tamponade, in particular whether or not there is edema. Whereas rapid accumulation is characterized more by hypotension than by edema, the slow accumulation of pericardial fluid affords the patient time to drink enough liquid to keep the central venous pressure higher than the rising pericardial pressure. Thus, edema and dyspnea are more prominent features of cardiac tamponade when there is a slow rise in pericardial pressure.
PEARL 2: EDEMA IS NOT ALWAYS TREATED WITH A DIURETIC
Edema is not always treated with a diuretic. In a patient who has a pericardial effusion that has developed slowly and who has been drinking enough fluid to keep the central venous pressure higher than the pericardial pressure, a diuretic can remove enough volume from the circulation to lower the central venous pressure below the intrapericardial pressure and thus convert a benign pericardial effusion to potentially lethal cardiac tamponade.
One must understand the cause of edema or low urine output before treating it. This underscores the importance of the history and the physical examination. All of the following must be assessed:
- Symptoms and time course of the illness
- Concurrent medical illnesses
- Neck veins
- Blood pressure and its response to inspiration
- Heart sounds
- Heart rate and rhythm
- Abdominal organ engorgement
- Edema (or its absence).
PEARL 3: UNDERSTANDING THE CAUSE IS ESSENTIAL
Understanding the cause of cardiac tamponade is essential.
A trauma patient first encountered in the emergency department may have an underlying disease, but the focus is squarely on the effects of trauma or violent injury. In a patient with multiple trauma, hypotension and tachycardia that do not respond to intravenous volume replacement when there is an obvious rise in central venous pressure should be clues to cardiac tamponade.1
If the patient has recently undergone a cardiac procedure (for example, cardiac surgery, myocardial biopsy, coronary intervention, electrophysiologic study with intracardiac electrodes, transvenous pacemaker placement, pacemaker lead extraction, or radiofrequency ablation), knowing about the procedure narrows the differential diagnosis when hypotension, tachycardia, and jugular venous distention develop.
PEARL 4: CARDIAC OR AORTIC RUPTURE REQUIRES SURGERY
When the etiology of cardiac tamponade is cardiac or aortic rupture, the treatment is surgical.
Painful sudden causes of cardiac tamponade include hemopericardium due to rupture of the free wall after myocardial infarction, and spontaneous or posttraumatic dissection and rupture of the ascending aorta. Prompt diagnosis is necessary, but since these lesions will not close and heal spontaneously, the definitive treatment should be surgery. Moreover, needle removal of intrapericardial blood that has been opposing further bleeding is sure to permit bleeding to recur, often with lethal consequences.2
Causes of cardiac tamponade that have a less-acute onset are likely to be complications of medical problems. Medical illnesses known to be associated with cardiac tamponade include:
- Infectious disease (idiopathic or viral, associated with smallpox vaccination, mycobacterial, purulent bacterial, fungal)
- Metastatic cancer (lung, breast, esophagus, lymphoma, pancreas, liver, leukemia, stomach, melanoma)3
- Connective tissue disease (rheumatoid arthritis, systemic lupus erythematosus, ankylosing spondylitis, scleroderma, Wegener granulomatosis, acute rheumatic fever)
- Endocrine disease (hypothyroidism)
- Drug side effects (procainamide, isoniazid, hydralazine, minoxidil, phenytoin, anticoagulants, methysergide)
- Inflammatory bowel disease (Crohn disease, ulcerative colitis)
- Congestive heart failure
- Uremia
- Radiation therapy
- Postmyocardial infarction syndrome (Dressler syndrome)
- Postpericardiotomy syndrome.
PEARL 5: REVIEW IMAGING BEFORE DIAGNOSING
What often brings a patient with cardiac tamponade to the attention of the physician is a finding on echocardiography, computed tomography, or magnetic resonance imaging of the chest.
Always review the imaging studies before making the diagnosis of cardiac tamponade. These tests must be reviewed to assess the anatomy and the size and location of the effusion. Particularly, one must look for atrial and right ventricular collapse and inferior vena caval plethora, which are echocardiographic signs of cardiac tamponade.4 Figures 1, 2, and 3 show imaging studies in a patient who presented with worsening cough 2 weeks after undergoing a cardiac procedure and who was found to have cardiac tamponade.
When the history and these imaging studies place cardiac tamponade high in the differential diagnosis as the cause of edema or dyspnea, it is time to reexamine the patient. The best first step is to measure pulsus paradoxus.
HOW PULSUS PARADOXUS OCCURS
To fully appreciate the subtleties of the next pearls, it is necessary to understand the pathophysiology of cardiac tamponade.
When pericardial fluid accumulation raises the pericardial pressure above the central venous pressure and pulmonary venous pressure (intravascular pressure), blood will not passively return to the right side of the heart from the venae cavae nor to the left side of the heart from the pulmonary veins unless it is influenced by the effects of respiration on intrathoracic pressure. During respiration, the right and left sides of the heart are alternately filled and deprived of their respective venous return.
During inspiration, as the intrathoracic pressure decreases, blood in the venae cavae empties into the right side of the heart, while blood in the pulmonary veins preferentially remains in the pulmonary veins, underfilling the left side of the heart. Since the right ventricle is more filled than the left ventricle during inspiration, the ventricular septum shifts from right to left, further opposing pulmonary venous return. As a result, during cardiac tamponade, the systemic blood pressure falls with inspiration.
During expiration the opposite occurs. Expiration decreases the intrathoracic volume, so the intrathoracic pressure rises. This tends to oppose vena caval return to the right side of the heart and to favor pulmonary venous return to the left side of the heart. The ventricular septum shifts from left to right, further accommodating left ventricular filling, raising stroke volume, and increasing blood pressure. This exaggerated alternate filling of the right and left sides of the heart during cardiac tamponade is what accounts for pulsus paradoxus, an inspiratory fall in systolic blood pressure of greater than 10 mm Hg.
If intravascular pressure is low (due to hemorrhage, dehydration, or diuretic therapy), the pressure in the pericardial space needed to oppose venous return is much less. In this low-pressure scenario, the results are low cardiac output and hypotension, which are treated by giving intravenous fluids to maintain intravascular volume.
PEARL 6: MEASURE PULSUS PARADOXUS
When cardiac tamponade is considered, one must always measure the pulsus paradoxus.
The term pulsus paradoxus was coined by Adolph Kussmaul in 1873, before physicians could even measure blood pressure. All they could do at that time was palpate the pulse and listen to the heart. Kussmaul described his observation as a conspicuous discrepancy between the cardiac action and the arterial pulse.
Although not described by Kussmaul, another explanation for this finding might be more suited to the use of the word “paradoxical.” When the pulse is palpated in a normal patient, with inspiration the pulse rate will increase via the Bainbridge reflex, and with expiration it will decrease. But in a patient with cardiac tamponade, there is a paradoxical inspiratory slowing of the pulse (because the decreased magnitude of the pulse at times makes it imperceptible) and an expiratory increase in pulse rate as the magnitude of the pulse again makes it palpable.
The magnitude of the fall in systolic blood pressure during inspiration has been used to estimate the level of hemodynamic impairment resulting from pericardial effusion.5 A rapidly accumulating pericardial effusion can have more hemodynamic impact than a much larger one that accumulates slowly. Thus, the intrapericardial pressure must be considered more than the volume of pericardial fluid.
When there is severe cardiac tamponade and overt pulsus paradoxus, simple palpation of a proximal arterial pulse can detect a marked inspiratory decrease or loss of the pulse, which returns with expiration. Tachycardia is almost always present, unless the cause is hypothyroidism.6
How to measure pulsus paradoxus with a manual sphygmomanometer
A stethoscope and manual sphygmomanometer are all that is needed to measure pulsus paradoxus. A noninvasive blood pressure monitor that averages multiple measurements cannot detect or quantify pulsus paradoxus.
The patient should be supine with the head elevated 30° to 45°, and the examiner should be seated comfortably at the patient’s side. The manometer should be on the opposite side of the patient in plain view of the examiner. Position the cuff on the arm above the elbow and place your stethoscope on the antecubital fossa. Then:
- Inflate the cuff 20 mm Hg above the highest systolic pressure previously auscultated.
- Slowly decrease the manometer pressure by 5 mm Hg and hold it there through two or three respiratory cycles while listening for the first Korotkoff (systolic) sound. Repeat this until you can hear the systolic sound (but only during expiration) and mentally note the pressure.
- Continue to decrease the manometer pressure by 5-mm Hg increments while listening. When the Korotkoff sounds no longer disappear with inspiration, mentally note this second value as well. The pulsus paradoxus is the difference between these values.
- When the Korotkoff sounds disappear as the manometer pressure is decreased, note this final value. This is the diastolic blood pressure.
PEARL 7: THE PLETHYSMOGRAM WAVE-FORM PARALLELS PULSUS PARADOXUS
Manual measurement of blood pressure and pulsus paradoxus can be difficult, especially in an obese patient or one with a fat-distorted arm on which the cuff does not maintain its position. In such patients, increased girth of the neck and abdomen also make it difficult to assess the jugular venous distention and visceral organ engorgement that characterize cardiac tamponade.
When the use of a sphygmomanometer is not possible, an arterial catheter can be inserted to demonstrate pulsus paradoxus. Simpler, however, is the novel use of another noninvasive instrument to detect and coarsely quantify pulsus paradoxus.7 The waveform on finger pulse oximetry can demonstrate pulsus paradoxus. The plethysmogram of the finger pulse oximeter can demonstrate the decrease in magnitude of the waveform with each inspiration (Figure 4).
Caution must be taken when interpreting this waveform, as with any measurement of pulsus paradoxus, to exclude a concomitant arrhythmia.
PEARL 8: PULSUS PARADOXUS WITHOUT CARDIAC TAMPONADE
Pulsus paradoxus can be present in the absence of cardiac tamponade. Once pulsus paradoxus of more than 10 mm Hg is measured, one must be sure the patient does not have a condition that can cause pulsus paradoxus without cardiac tamponade. Most of these are pulmonary conditions that necessitate an exaggerated inspiratory effort that can lower intrathoracic pressure sufficiently to oppose pulmonary venous return and cause a fall in systemic blood pressure:
- Chronic bronchitis
- Emphysema
- Mucus plug
- Pneumothorax
- Pulmonary embolism
- Stridor.
In these, there may be pulsus paradoxus, but not due to cardiac tamponade.
PEARL 9: CARDIAC TAMPONADE CAN BE PRESENT WITHOUT PULSUS PARADOXUS
Cardiac tamponade can be present without pulsus paradoxus. This occurs when certain conditions prevent inspiratory underfilling of the left ventricle relative to the filling of the right ventricle.8
How does this work? In cardiac tamponade, factors that drive the exaggerated fall in arterial pressure with inspiration (pulsus paradoxus) are the augmented right ventricular filling and the decreased left ventricular filling, both due to the lowering of the intrathoracic pressure. As the vena caval emptying is augmented, the right ventricular filling is increased, the ventricular septum shifts to the left, and pulmonary venous return to the heart is decreased.
Factors that can oppose pulsus paradoxus:
- Positive pressure ventilation prevents pulsus paradoxus by preventing the fall in intrathoracic pressure.
- Severe aortic regurgitation does not permit underfilling of the left ventricle during inspiration.
- An atrial septal defect will always equalize the right and left atrial pressures, preventing differential right ventricular and left ventricular filling with inspiration.
- Severe left ventricular hypertrophy does not permit the inspiratory shift of the ventricular septum from right to left that would otherwise lead to decreased left ventricular filling.
- Severe left ventricular dysfunction, with its low stroke volume and severe elevation of left ventricular end-diastolic pressure, never permits underfilling of the left ventricle, despite cardiac tamponade and an inspiratory decrease in intrathoracic pressure.
- Intravascular volume depletion due to hemorrhage, hemodialysis, or mistaken use of diuretics to treat edema can cause marked hypotension, making pulsus paradoxus impossible to detect.
Knowledge of underlying medical conditions, the likelihood of their causing cardiac tamponade, and the appearance of the echocardiogram prompt the physician to look further when the presence or absence of pulsus paradoxus does not fit with the working diagnosis.
The echocardiogram can give hints to the etiology of a pericardial effusion, such as clotted blood after trauma or a cardiac-perforating procedure, tumor studding of the epicardium,9 or fibrin strands indicating chronicity or an inflammatory process.10 Diastolic collapse of the right ventricle, more than collapse of the right atrium or left atrium, speaks for the severity of cardiac tamponade. With hemodynamically significant pericardial effusion and cardiac tamponade, the inferior vena cava is distended and does not decrease in size with inspiration unless there is severe intravascular volume depletion, at which time the inferior vena cava is underfilled throughout the respiratory cycle.
PEARL 10: PLAN HOW TO DRAIN
The size and location of the pericardial effusion and the patient’s hemodynamics must be integrated when deciding how to relieve cardiac tamponade. When cardiac tamponade is indeed severe and the patient and physician agree that it must be drained, the options are percutaneous needle aspiration (pericardiocentesis) and surgical pericardiostomy (creation of a pericardial window). Here again, as assessed by echocardiography, the access to the pericardial fluid should influence the choice.
Pericardiocentesis can be safely done if certain criteria are met. The patient must be able to lie still in the supine position, perhaps with the head of the bed elevated 30 degrees. Anticoagulation must be reversed or allowed time to resolve if drainage is not an emergency.
Pericardiocentesis can be risky or unsuccessful if there is not enough pericardial fluid to permit respiratory cardiac motion without perforating the heart with the needle; if the effusion is loculated (confined to a pocket) posteriorly; or if it is too far from the skin to permit precise control and placement of a spinal needle into the pericardial space. In cases of cardiac tamponade in which the anatomy indicates surgical pericardiostomy but severe hypotension prevents the induction of anesthesia and positive-pressure ventilation—which can result in profound, irreversible hypotension—percutaneous needle drainage (pericardiocentesis) should be performed in the operating room to relieve the tamponade before the induction of anesthesia and the surgical drainage.11
To reiterate, a suspected cardiac or aortic rupture that causes cardiac tamponade is usually large and not apt to self-seal. In such cases, the halt in the accumulation of pericardial blood is due to hypotension and not due to spontaneous resolution. Open surgical drainage is required from the outset because an initial success of pericardiocentesis yields to the recurrence of cardiac tamponade.
PEARL 11: ANTICIPATE WHAT THE FLUID SHOULD LOOK LIKE
Before performing pericardiocentesis, anticipate the appearance of the pericardial fluid on the basis of the presumed etiology, ie:
- Sanguinous—trauma, heart surgery, cardiac perforation from a procedure, anticoagulation, uremia, or malignancy
- Serous—congestive heart failure, acute radiation therapy
- Purulent—infections (natural or postoperative)
- Turbid (like gold paint)—mycobacterial infection, rheumatoid arthritis, myxedema
- Chylous—pericardium fistulized to the thoracic duct by a natural or postsurgical cause.
Sanguinous pericardial effusion encountered during a pericardiocentesis, if not anticipated, can be daunting and can cause the operator to question if it is the result of inadvertent needle placement in a cardiac chamber. If the needle is indeed in the heart, blood often surges out under pressure in pulses, which strongly suggests that the needle is not in the pericardial space and should be removed; but if confirmation of the location is needed before removing the needle, it can be done by injecting 2 mL of agitated sterile saline through the pericardiocentesis needle during echocardiographic imaging.12
Before inserting the needle, the ideal access location and needle angle must be determined by the operator with echocardiographic transducer in hand. The distance from skin to a point just through the parietal pericardium can also be measured at this time.
Once the needle is in the pericardial fluid (and you are confident of its placement), removal of 50 to 100 mL of the fluid with a large syringe can be enough to afford the patient easier breathing, higher blood pressure, and lower pulsus paradoxus—and even the physician will breathe easier. The same syringe can be filled and emptied multiple times. Less traumatic and more complete removal of pericardial fluid requires insertion of a multihole pigtail catheter over a J-tipped guidewire that is introduced through the needle.
PEARL 12: DRAIN SLOWLY TO AVOID PULMONARY EDEMA
Pulmonary edema is an uncommon complication of pericardiocentesis that might be avoidable. Heralded by sudden coughing and pink, frothy sputum, it can rapidly deteriorate into respiratory failure. The mechanism has been attributed to a sudden increase in right ventricular stroke volume and resultant left ventricular filling after the excess pericardial fluid has been removed, before the systemic arteries, which constrict to keep the systemic blood pressure up during cardiac tamponade, have had time to relax.13
To avoid this complication, if the volume of pericardial fluid responsible for cardiac tamponade is large, it should be removed slowly,14 stopping for a several-minute rest after each 250 mL. Catheter removal of pericardial fluid by gravity drainage over 24 hours has been suggested.15 A drawback to this approach is catheter clotting or sludging before all the fluid has been removed. It is helpful to keep the drainage catheter close to the patient’s body temperature to make the fluid less viscous. Output should be monitored hourly.
When the pericardial fluid has been completely drained, one must decide how long to leave the catheter in. One reason to remove the catheter at this time is that it causes pleuritic pain; another is to avoid introducing infection. A reason to leave the catheter in is to observe the effect of medical treatment on the hourly pericardial fluid output. Nonsteroidal anti-inflammatory drugs are the drugs of first choice when treating pericardial inflammation and suppressing production of pericardial fluid.16 In most cases the catheter should not be left in place for more than 3 days.
Laboratory analysis of the pericardial fluid should shed light on its suspected cause. Analysis usually involves chemistry testing, microscopic inspection of blood cell smears, cytology, microbiologic stains and cultures, and immunologic tests. Results often take days. Meyers and colleagues17 expound on this subject.
- Schiavone WA, Ghumrawi BK, Catalano DR, et al. The use of echocardiography in the emergency management of nonpenetrating traumatic cardiac rupture. Ann Emerg Med 1991; 20:1248–1250.
- Manuchehry A, Fontana GP, Gurudevan S, Marchevsky AM, Siegel RJ. Missed diagnosis of limited ascending aortic dissection by multiple imaging modalities leading to fatal cardiac tamponade and aortic rupture. Echocardiography 2011; 28:E187–E190.
- Lam KY, Dickens P, Chan AC. Tumors of the heart. A 20-year experience with a review of 12,485 consecutive autopsies. Arch Pathol Lab Med 1993; 117:1027–1031.
- Tsang TS, Oh JK, Seward JB, Tajik AJ. Diagnostic value of echocardiography in cardiac tamponade. Herz 2000; 25:734–740.
- Curtiss EI, Reddy PS, Uretsky BF, Cecchetti AA. Pulsus paradoxus: definition and relation to the severity of cardiac tamponade. Am Heart J 1988; 115:391–398.
- Wang JL, Hsieh MJ, Lee CH, et al. Hypothyroid cardiac tamponade: clinical features, electrocardiography, pericardial fluid and management. Am J Med Sci 2010; 340:276–281.
- Tamburro RF, Ring JC, Womback K. Detection of pulsus paradoxus associated with large pericardial effusions in pediatric patients by analysis of the pulse-oximetry waveform. Pediatrics 2002; 109:673–677.
- Spodick DH. Pulsus paradoxus. In:Spodick DH, editor. The Pericardium: A Comprehensive Textbook. New York, NY: Marcel Dekker; 1997:191–199.
- Burke A, Jeudy J, Virmani R. Cardiac tumors. In:Topol EJ, editor. Textbook of Cardiovascular Medicine. 3rd ed. Philadelphia, PA: Lippincott, Williams & Wilkins; 2007:710–720.
- Roberts WC. Pericardial heart disease: Its morphologic features and its causes. Proc (Bayl Univ Med Cent) 2005; 18:38–55.
- Stoelting RK, Miller RD, editors. Basics of Anesthesia. 4th ed. New York, NY: Churchill Livingstone; 2000:264–265.
- Ainsworth CD, Salehian O. Echo-guided pericardiocentesis: let the bubbles show the way. Circulation 2011; 123:e210–e211.
- Maisch B, Seferovic PM, Ristic AD, et al; Task Force on the Diagnosis and Management of Pericardial Diseases of the European Society of Cardiology. Guidelines on the diagnosis and management of pericardial diseases executive summary; The Task Force on the Diagnosis and Management of Pericardial Diseases of the European Society of Cardiology. Eur Heart J 2004; 25:587–610.
- Vandyke WH, Cure J, Chakko CS, Gheorghiade M. Pulmonary edema after pericardiocentesis for cardiac tamponade. N Engl J Med 1983; 309:595–596.
- Bernal JM, Pradhan J, Li T, Tchokonte R, Afonso L. Acute pulmonary edema following pericardiocentesis for cardiac tamponade. Can J Cardiol 2007; 23:1155–1156.
- Sagristà-Sauleda J, Mercé AS, Soler-Soler J. Diagnosis and management of pericardial effusion. World J Cardiol 2011; 3:135–143.
- Meyers DG, Meyers RE, Prendergast TW. The usefulness of diagnostic tests on pericardial fluid. Chest 1997; 111:1213–1221.
- Schiavone WA, Ghumrawi BK, Catalano DR, et al. The use of echocardiography in the emergency management of nonpenetrating traumatic cardiac rupture. Ann Emerg Med 1991; 20:1248–1250.
- Manuchehry A, Fontana GP, Gurudevan S, Marchevsky AM, Siegel RJ. Missed diagnosis of limited ascending aortic dissection by multiple imaging modalities leading to fatal cardiac tamponade and aortic rupture. Echocardiography 2011; 28:E187–E190.
- Lam KY, Dickens P, Chan AC. Tumors of the heart. A 20-year experience with a review of 12,485 consecutive autopsies. Arch Pathol Lab Med 1993; 117:1027–1031.
- Tsang TS, Oh JK, Seward JB, Tajik AJ. Diagnostic value of echocardiography in cardiac tamponade. Herz 2000; 25:734–740.
- Curtiss EI, Reddy PS, Uretsky BF, Cecchetti AA. Pulsus paradoxus: definition and relation to the severity of cardiac tamponade. Am Heart J 1988; 115:391–398.
- Wang JL, Hsieh MJ, Lee CH, et al. Hypothyroid cardiac tamponade: clinical features, electrocardiography, pericardial fluid and management. Am J Med Sci 2010; 340:276–281.
- Tamburro RF, Ring JC, Womback K. Detection of pulsus paradoxus associated with large pericardial effusions in pediatric patients by analysis of the pulse-oximetry waveform. Pediatrics 2002; 109:673–677.
- Spodick DH. Pulsus paradoxus. In:Spodick DH, editor. The Pericardium: A Comprehensive Textbook. New York, NY: Marcel Dekker; 1997:191–199.
- Burke A, Jeudy J, Virmani R. Cardiac tumors. In:Topol EJ, editor. Textbook of Cardiovascular Medicine. 3rd ed. Philadelphia, PA: Lippincott, Williams & Wilkins; 2007:710–720.
- Roberts WC. Pericardial heart disease: Its morphologic features and its causes. Proc (Bayl Univ Med Cent) 2005; 18:38–55.
- Stoelting RK, Miller RD, editors. Basics of Anesthesia. 4th ed. New York, NY: Churchill Livingstone; 2000:264–265.
- Ainsworth CD, Salehian O. Echo-guided pericardiocentesis: let the bubbles show the way. Circulation 2011; 123:e210–e211.
- Maisch B, Seferovic PM, Ristic AD, et al; Task Force on the Diagnosis and Management of Pericardial Diseases of the European Society of Cardiology. Guidelines on the diagnosis and management of pericardial diseases executive summary; The Task Force on the Diagnosis and Management of Pericardial Diseases of the European Society of Cardiology. Eur Heart J 2004; 25:587–610.
- Vandyke WH, Cure J, Chakko CS, Gheorghiade M. Pulmonary edema after pericardiocentesis for cardiac tamponade. N Engl J Med 1983; 309:595–596.
- Bernal JM, Pradhan J, Li T, Tchokonte R, Afonso L. Acute pulmonary edema following pericardiocentesis for cardiac tamponade. Can J Cardiol 2007; 23:1155–1156.
- Sagristà-Sauleda J, Mercé AS, Soler-Soler J. Diagnosis and management of pericardial effusion. World J Cardiol 2011; 3:135–143.
- Meyers DG, Meyers RE, Prendergast TW. The usefulness of diagnostic tests on pericardial fluid. Chest 1997; 111:1213–1221.
KEY POINTS
- Slow accumulation of pericardial fluid can result in edema, whereas rapid accumulation leads to hypotension.
- Diuretics can worsen tamponade by removing enough volume from the circulation to lower the central venous pressure below the intrapericardial pressure.
- Try to determine why cardiac tamponade has occurred. Cardiac or aortic rupture requires surgery. If the gross appearance of the pericardial fluid does not match the presumed etiology, reconsider your diagnosis.
- Always review imaging studies before making the diagnosis of cardiac tamponade.
- When cardiac tamponade is considered, pulsus paradoxus must be measured, and if present, integrated with other physical findings and the echocardiogram. However, pulsus paradoxus can be present in the absence of cardiac tamponade, and vice versa.
- Consider the size and location of the pericardial effusion and the patient’s hemodynamic status when deciding between surgery and needle aspiration.
A short story of the short QT syndrome
Sudden cardiac death in a young person is a devastating event that has puzzled physicians for decades. In recent years, many of the underlying cardiac pathologies have been identified. These include structural abnormalities such as hypertrophic cardiomyopathy and nonstructural disorders associated with unstable rhythms that lead to sudden cardiac death.
The best known of these “channelopathies” are the long QT syndromes, which result from abnormal potassium and sodium channels in myocytes. Recently, interest has been growing in a disorder that may carry a similarly grim prognosis but that has an opposite finding on electrocardiography (ECG).
Short QT syndrome is a recently described heterogeneous genetic channelopathy that causes both atrial and ventricular arrhythmias and that has been documented to cause sudden cardiac death.
In 1996, a 37-year-old woman from Spain died suddenly; ECG several days earlier had shown a short QT interval of 266 ms.1 Two years later, an unrelated 17-year-old American woman undergoing laparoscopic cholecystectomy suddenly developed atrial fibrillation with a rapid ventricular response.1 Her QT interval was 225 ms. Her brother had a QT interval of 240 ms, and her mother’s was 230 ms. The patient’s maternal grandfather had a history of atrial fibrillation, and his QT interval was 245 ms. These cases led to the description of this new clinical syndrome (see below).2
CLINICAL FEATURES
Short QT syndrome has been associated with both atrial and ventricular arrhythmias. Atrial fibrillation, polymorphic ventricular tachycardia, and ventricular fibrillation have all been well described. Patients who have symptoms usually present with palpitations, presyncope, syncope, or sudden or aborted cardiac death.3,4
ELECTROCARDIOGRAPHIC FEATURES
The primary finding on ECG is a short QT interval. However, others have been noted (Figure 1):
Short or absent ST segment
This finding is not merely a consequence of the short QT interval. In 10 patients with short QT syndrome, the distance from the J point to the peak T wave ranged from 80 to 120 ms. In 12 healthy people whose QT interval was less than 320 ms, this distance ranged from 150 ms to 240 ms.5
Tall and peaked T wave
A tall and peaked T wave is a common feature in short QT syndrome. However, it was also evident in people with short QT intervals who had no other features of the syndrome.5
QT response to heart rate
Normally, the QT interval is inversely related to the heart rate, but this is not true in short QT syndrome: the QT interval remains relatively fixed with changes in heart rate.6,7 This feature is less helpful in the office setting but may be found with Holter monitoring by measuring the QT interval at different heart rates.
BUT WHAT IS CONSIDERED A SHORT QT INTERVAL?
In clinical practice, the QT interval is corrected for the heart rate by the Bazett formula:
Corrected QT (QTc) = [QT interval/square root of the RR interval]
Review of ECGs from large populations in Finland (n = 10,822), Japan (n = 12,149), the United States (n = 79,743), and Switzerland (n = 41,676) revealed that a QTc value of 350 ms in males and 365 ms in females was 2.0 standard deviations (SD) below the mean.8–11 However, a QTc less than the 2.0 SD cutoff did not necessarily equal arrhythmogenic potential. This was illustrated in a 29-year follow-up study of Finnish patients with QTc values as short as 320 ms, in whom no arrhythmias were documented.8 Conversely, some patients with purported short QT syndrome had QTc intervals as long as 381 ms.12
Similar problems with uncertainty of values have plagued the diagnosis of long QT syndrome.13 The lack of reference ranges and the overlap between healthy and affected people called for the development of a scoring system that involves criteria based on ECG and on the clinical evaluation.14,15
ESTABLISHING THE DIAGNOSIS OF SHORT QT SYNDROME
Clearly, the diagnosis of short QT syndrome can be challenging to establish. The first step is to rule out other causes of a short QT interval.
Differential diagnosis of short QT interval
In addition to genetic channelopathies, other causes of short QT interval must be ruled out before entertaining the diagnosis of short QT syndrome.
- Hypercalcemia is the most important of these: there is usually an accompanying prolonged PR interval and a wide QRS complex16
- Hyperkalemia17
- Acidosis17
- Increased vagal tone17
- After ventricular fibrillation (thought to be related to increased intracellular calcium)18
- Digitalis use19
- Androgen use.20
Interestingly, a shorter-than-expected QT interval was noted in patients with chronic fatigue syndrome.21
Which interval to use: QT or QTc?
Unfortunately, most population-based studies that searched for a short QT interval on ECG have used QTc as the main search parameter.8–11 As already mentioned, in patients with short QT syndrome, the QT interval is, uniquely, not shortened if the heart beats faster. In contrast, the QTc often overestimates the QT interval in patients with short QT syndrome, especially when the heart rate is in the 80s to 90s.16
In a review of cases of short QT syndrome worldwide, Bjerregaard et al22 found that the QT interval ranged from 210 ms to 340 ms with a mean ± 2 SD of 282 ± 62 ms. On the other hand, the QTc ranged from 248 ms to 345 ms with a mean ± 2 SD of 305 ± 42 ms.
Therefore, correction formulas (such as the Bazett formula) do not perform well in ruling in the diagnosis of short QT syndrome—and they do even worse in ruling it out.16,22
To establish a diagnosis of short QT syndrome in someone with prior evidence of atrial or ventricular fibrillation, a QT interval less than 340 ms or a QTc less than 345 ms is usually sufficient.22 In borderline cases in which the QT interval is slightly longer, some experts recommend other tests, although strong evidence validating their predictive value does not exist. These tests include genotyping, analysis of T wave morphology, and electrophysiologic studies.16
Recently, Gollob et al23 proposed a scoring system for short QT syndrome (Table 1). After reviewing the literature and comparing the diagnostic markers, the investigators determined diagnostic criteria that, when applied to the previously reported cases, were able to identify 58 (95.08%) of 61 patients with short QT syndrome (ie, a sensitivity of 95%).
For patients with intermediate probability, the authors recommended continued medical and ECG surveillance as well as ECGs for first-degree relatives, to further clarify the diagnosis.
Again, a principal caveat about this system is that it relies on the QTc interval rather than the QT interval to diagnose short QT syndrome.
THE SCOPE OF THE DISEASE
In a recent review of the literature, Gollob et al23 found a total of 61 cases of short QT syndrome reported in English. The cohort was predominantly male (75.4%), and most of the symptomatic patients presented during late adolescence and early adulthood. However, there have been reports of infants (4 and 8 months old), and of a man who presented for the first time at the age of 70. Of note, the authors only considered short QT syndrome types 1, 2, and 3 (see below) in their search for cases.
Whether the syndrome is truly this rare or, rather, whether many physicians are not aware of it is still to be determined. In addition, it is possible that incorrectly measuring the QT interval contributes to the lack of identification of this entity. Both of these factors were implicated in the rarity of reported long QT syndrome early after its discovery.14,15
MUTATIONS IN CARDIAC ION CHANNELS
Five distinct genetic defects have been associated with short QT syndrome. As in long QT syndrome, these give rise to subtypes of short QT syndrome, which are numbered 1 to 5 (see below).
The cardiac action potential
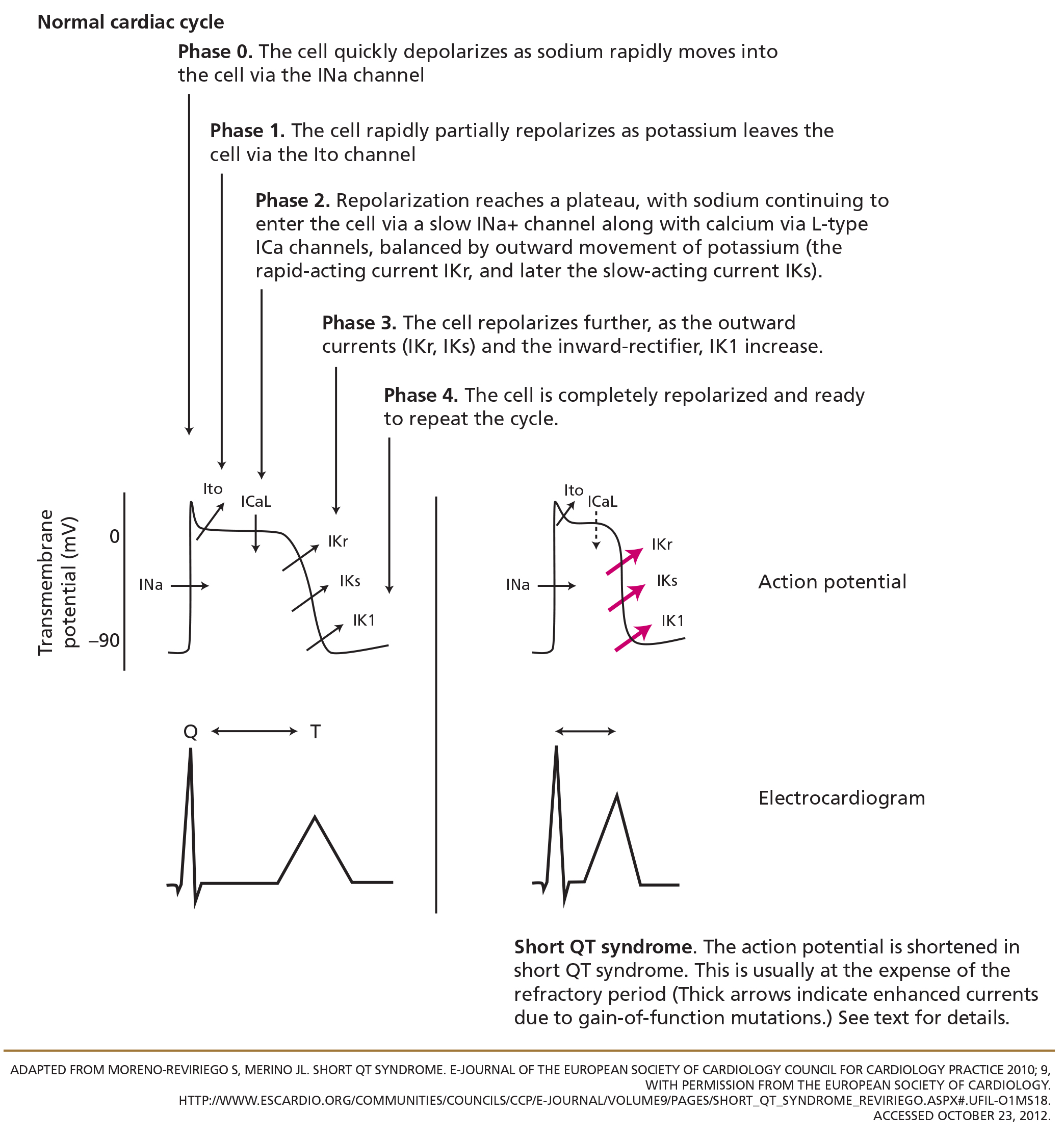
To understand how the mutations shorten the QT interval, we will briefly review of the cardiac myocyte action potential.24 In nonpacemaker cells of the heart, the activation of the cell membrane initiates a series of changes in ion channels that allow the movement of ions along an electrical gradient. This movement occurs in five phases and is repeated with every cardiac cycle (Figure 2).
In phase 0, the cardiac cell rapidly depolarizes.
Repolarization occurs in phases 1, 2, and 3 and is largely a function of potassium ions leaving the cell. During phase 2, calcium and sodium ions enter the cell and balance the outward potassium flow, creating the “flat” portion of the repolarization curve. Phase 3 is the main phase of repolarization in which the membrane potential rapidly falls back to its resting state (–90 mV). During phases 1 and 2, the cell membrane is completely refractory to stimulation, whereas phase 3 is divided into three parts:
- The effective refractory period: the cell is able to generate a potential that is too weak to be propagated
- The relative refractory period: the cell can respond to a stimulus that is stronger than normal
- The supernormal phase: the last small portion of phase 3, in which a less-than-normal stimulus can yield a response in the cell.
In phase 4, the cell is completely repolarized, and the cycle can start again.
Five types of short QT syndrome
Short QT syndrome 1. In 2004, Brugada et al25 identified the first mutation that causes abnormal shortening of the action potential duration. In contrast to the mutations that underlie long QT syndrome, this mutation actually causes a gain of function in the gene coding the rapidly acting delayed potassium current (IKr) channel proteins KCNH2 or HERG. Potassium leaving at a more rapid rate causes the cell to repolarize more quickly and shortens the QT interval. The clinical syndrome associated with KCNH2 gene gain-of-function mutation is called short QT syndrome 1.
Short QT syndromes 2 and 3. Other IK (potassium channel) proteins have been implicated as well. Gain-of-function mutations in the KCNQ1 and KCNJ2 genes are believed to account for short QT syndromes 2 and 3, respectively. KCNQ1 codes for the IKs protein, and KCNJ2 codes for the IK1 protein.26,27
Short QT syndromes 4 and 5 were identified by Antzelevitch et al,28 who described several patients who had a combination of channel abnormalities and ECG findings. Their ECGs showed “Brugada-syndrome-like” ST elevation in the right precordial leads, but with a short QT interval. These new syndromes were found to be associated with genetic abnormalities distinct from those of Brugada syndrome and other short QT syndromes. These abnormalities involved loss-of-function mutations in the CACNA1C gene (which codes for the alpha-1 subunit of the L-type cardiac calcium channel) and in the CACNB2 gene (which codes for the beta-2b subunit of the same channel). The two defects correspond to the clinical syndromes short QT syndrome 4 and short QT syndrome 5, respectively.28
MECHANISM OF ARRHYTHMOGENESIS IN SHORT QT SYNDROME
The myocardium is made of different layers: the epicardium, the endocardium, and the middle layer of myocytes composed mainly of M cells. Cells in the different layers differ in the concentration of their channels and can be affected differently in various syndromes. When cells in one or two of the layers repolarize at a rate different from cells in another layer, they create different degrees of refractoriness, which establishes the potential for reentry circuits to form.
It is believed that in short QT syndrome the endocardial cells and M cells repolarize faster than the epicardial cells, predisposing to reentry and arrhythmias. This accentuation of “transmural dispersion of repolarization” accounts for arrhythmogenesis in short QT syndrome as well as in long QT syndrome and the Brugada syndromes. The difference between these syndromes appears to be the layer or area of the myocardium that is affected more by the channelopathy (the M cells in long QT syndrome and the epicardium of the right ventricle in the Brugada syndrome).29
WHEN TO THINK OF SHORT QT SYNDROME
In any survivor of sudden cardiac death, the QT interval should be thoroughly scrutinized, and family members should undergo ECG. Patients in whom a short QT interval is incidentally discovered and for which other reasons are ruled out (see differential diagnosis) should be encouraged to have family members undergo ECG. Other potential patients are young people who develop atrial fibrillation and patients who have idiopathic ventricular fibrillation.4
TREATMENT AND PROGNOSIS
Evidence-based recommendations for the management of short QT syndrome do not yet exist, mainly because the number of patients identified to date is small.
Implantable cardioverter-defibrillators
Although placing an implantable cardioverter-defibrillator (ICD) seems to be warranted in patients who experience ventricular fibrillation, ventricular tachycardia, or aborted cardiac death, or in patients who have a family history of the same symptoms, the best management option is less clear for patients who have no symptoms and no family history.30 In addition, some patients may not want an ICD or may even not qualify for this therapy.
A unique problem with ICDs in short QT syndrome stems from one of the syndrome’s main features on ECG: the tall and peaked T wave that closely follows the R wave can sometimes be interpreted as a short R-R interval, provoking an inappropriate shock from the ICD.31
For the above reasons, we strongly encourage consulting a center with expertise in QT-interval-related disorders before placing an ICD in a patient suspected of having short QT syndrome.
Antiarrhythmic drugs
Prolongation of the QT interval (and the effective refractory period) with drugs has been an interesting area of research. Gaita et al32 studied the effect of four antiarrhythmics—flecainide (Tambocor), sotalol (Betapace), ibutilide (Corvert), and quinidine—in six patients with short QT syndrome. Only quinidine was associated with significant QT prolongation, from 263 ± 12 ms to 362 ms ± 25 ms. This resulted in a longer ventricular effective refractory period (> 200 ms), and ventricular fibrillation was no longer inducible during provocative testing.
In a recent study of long-term outcomes of 53 patients with short QT syndrome, Giustetto et al33 noticed that none of the patients taking quinidine, including those with a history of cardiac arrest, had any further arrhythmsic events. On the other hand, the incidence of arrhythmic events during the follow-up was 4.9% per year in patients not taking this drug. Quinidine had a stronger effect on the QT interval in patients with the HERG mutation than in those without.
RESEARCH MAY LEAD TO A BETTER UNDERSTANDING OF OTHER DISEASES
The short QT syndrome is one of the most recently recognized cardiac channelopathies associated with malignant arrhythmias. As with long QT syndrome, research in short QT syndrome may lead to a better understanding of the pathogenesis of more common but still poorly understood arrhythmias such as lone atrial fibrillation and idiopathic ventricular fibrillation.
- The Short QT Syndrome http://www.shortqtsyndrome.org/short_qt_history.htm. Accessed October 30, 2012.
- Gussak I, Brugada P, Brugada J, et al. Idiopathic short QT interval: a new clinical syndrome? Cardiology 2000; 94:99–102.
- Giustetto C, Di Monte F, Wolpert C, et al. Short QT syndrome: clinical findings and diagnostic-therapeutic implications. Eur Heart J 2006; 27:2440–2447.
- Viskin S, Zeltser D, Ish-Shalom M, et al. Is idiopathic ventricular fibrillation a short QT syndrome? Comparison of QT intervals of patients with idiopathic ventricular fibrillation and healthy controls. Heart Rhythm 2004; 1:587–591.
- Anttonen O, Junttila MJ, Maury P, et al. Differences in twelve-lead electrocardiogram between symptomatic and asymptomatic subjects with short QT interval. Heart Rhythm 2009; 6:267–271.
- Redpath CJ, Green MS, Birnie DH, Gollob MH. Rapid genetic testing facilitating the diagnosis of short QT syndrome. Can J Cardiol 2009; 25:e133–e135.
- Wolpert C, Schimpf R, Giustetto C, et al. Further insights into the effect of quinidine in short QT syndrome caused by a mutation in HERG. J Cardiovasc Electrophysiol 2005; 16:54–58.
- Anttonen O, Junttila MJ, Rissanen H, Reunanen A, Viitasalo M, Huikuri HV. Prevalence and prognostic significance of short QT interval in a middle-aged Finnish population. Circulation 2007; 116:714–720.
- Funada A, Hayashi K, Ino H, et al. Assessment of QT intervals and prevalence of short QT syndrome in Japan. Clin Cardiol 2008; 31:270–274.
- Mason JW, Ramseth DJ, Chanter DO, Moon TE, Goodman DB, Mendzelevski B. Electrocardiographic reference ranges derived from 79,743 ambulatory subjects. J Electrocardiol 2007; 40:228–234.
- Kobza R, Roos M, Niggli B, et al. Prevalence of long and short QT in a young population of 41,767 predominantly male Swiss conscripts. Heart Rhythm 2009; 6:652–657.
- Itoh H, Sakaguchi T, Ashihara T, et al. A novel KCNH2 mutation as a modifier for short QT interval. Int J Cardiol 2009; 137:83–85.
- Vincent GM, Timothy KW, Leppert M, Keating M. The spectrum of symptoms and QT intervals in carriers of the gene for the long-QT syndrome. N Engl J Med 1992; 327:846–852.
- Schwartz PJ. Idiopathic long QT syndrome: progress and questions. Am Heart J 1985; 109:399–411.
- Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Diagnostic criteria for the long QT syndrome. An update. Circulation 1993; 88:782–784.
- Bjerregaard P, Nallapaneni H, Gussak I. Short QT interval in clinical practice. J Electrocardiol 2010; 43:390–395.
- Maury P, Extramiana F, Sbragia P, et al. Short QT syndrome. Update on a recent entity. Arch Cardiovasc Dis 2008; 101:779–786.
- Kontny F, Dale J. Self-terminating idiopathic ventricular fibrillation presenting as syncope: a 40-year follow-up report. J Intern Med 1990; 227:211–213.
- Cheng TO. Digitalis administration: an underappreciated but common cause of short QT interval. Circulation 2004; 109:e152.
- Hancox JC, Choisy SC, James AF. Short QT interval linked to androgen misuse: wider significance and possible basis. Ann Noninvasive Electrocardiol 2009; 14:311–312.
- Naschitz J, Fields M, Isseroff H, Sharif D, Sabo E, Rosner I. Shortened QT interval: a distinctive feature of the dysautonomia of chronic fatigue syndrome. J Electrocardiol 2006; 39:389–394.
- Bjerregaard P, Collier JL, Gussak I. Upper limits of QT/QTc intervals in the short QT syndrome. Review of the world-wide short QT syndrome population and 3 new USA families. Heart Rhythm 2008; 5:AB43.
- Gollob MH, Redpath CJ, Roberts JD. The short QT syndrome: proposed diagnostic criteria. J Am Coll Cardiol 2011; 57:802–812.
- Shih HT. Anatomy of the action potential in the heart. Tex Heart Inst J 1994; 21:30–41.
- Brugada R, Hong K, Dumaine R, et al. Sudden death associated with short-QT syndrome linked to mutations in HERG. Circulation 2004; 109:30–35.
- Bellocq C, van Ginneken AC, Bezzina CR, et al. Mutation in the KCNQ1 gene leading to the short QT-interval syndrome. Circulation 2004; 109:2394–2397.
- Priori SG, Pandit SV, Rivolta I, et al. A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ Res 2005; 96:800–807.
- Antzelevitch C, Pollevick GD, Cordeiro JM, et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation 2007; 115:442–449.
- Antzelevitch C. Heterogeneity and cardiac arrhythmias: an overview. Heart Rhythm 2007; 4:964–972.
- Lunati M, Bongiorni MG, Boriani G, et al. Linee guida AIAC 2006 all’impianto di pacemaker, dispositivi per la resincronizzazione cardiaca (CRT) e defibrillatori automatici impiantabili (ICD). GIAC 2005; 8:1–58.
- Schimpf R, Wolpert C, Bianchi F, et al. Congenital short QT syndrome and implantable cardioverter defibrillator treatment: inherent risk for inappropriate shock delivery. J Cardiovasc Electrophysiol 2003; 14:1273–1277.
- Gaita F, Giustetto C, Bianchi F, et al. Short QT syndrome: pharmacological treatment. J Am Coll Cardiol 2004; 43:1494–1499.
- Giustetto C, Schimpf R, Mazzanti A, et al. Long-term follow-up of patients with short QT syndrome. J Am Coll Cardiol 2011; 58:587–595.
Sudden cardiac death in a young person is a devastating event that has puzzled physicians for decades. In recent years, many of the underlying cardiac pathologies have been identified. These include structural abnormalities such as hypertrophic cardiomyopathy and nonstructural disorders associated with unstable rhythms that lead to sudden cardiac death.
The best known of these “channelopathies” are the long QT syndromes, which result from abnormal potassium and sodium channels in myocytes. Recently, interest has been growing in a disorder that may carry a similarly grim prognosis but that has an opposite finding on electrocardiography (ECG).
Short QT syndrome is a recently described heterogeneous genetic channelopathy that causes both atrial and ventricular arrhythmias and that has been documented to cause sudden cardiac death.
In 1996, a 37-year-old woman from Spain died suddenly; ECG several days earlier had shown a short QT interval of 266 ms.1 Two years later, an unrelated 17-year-old American woman undergoing laparoscopic cholecystectomy suddenly developed atrial fibrillation with a rapid ventricular response.1 Her QT interval was 225 ms. Her brother had a QT interval of 240 ms, and her mother’s was 230 ms. The patient’s maternal grandfather had a history of atrial fibrillation, and his QT interval was 245 ms. These cases led to the description of this new clinical syndrome (see below).2
CLINICAL FEATURES
Short QT syndrome has been associated with both atrial and ventricular arrhythmias. Atrial fibrillation, polymorphic ventricular tachycardia, and ventricular fibrillation have all been well described. Patients who have symptoms usually present with palpitations, presyncope, syncope, or sudden or aborted cardiac death.3,4
ELECTROCARDIOGRAPHIC FEATURES
The primary finding on ECG is a short QT interval. However, others have been noted (Figure 1):
Short or absent ST segment
This finding is not merely a consequence of the short QT interval. In 10 patients with short QT syndrome, the distance from the J point to the peak T wave ranged from 80 to 120 ms. In 12 healthy people whose QT interval was less than 320 ms, this distance ranged from 150 ms to 240 ms.5
Tall and peaked T wave
A tall and peaked T wave is a common feature in short QT syndrome. However, it was also evident in people with short QT intervals who had no other features of the syndrome.5
QT response to heart rate
Normally, the QT interval is inversely related to the heart rate, but this is not true in short QT syndrome: the QT interval remains relatively fixed with changes in heart rate.6,7 This feature is less helpful in the office setting but may be found with Holter monitoring by measuring the QT interval at different heart rates.
BUT WHAT IS CONSIDERED A SHORT QT INTERVAL?
In clinical practice, the QT interval is corrected for the heart rate by the Bazett formula:
Corrected QT (QTc) = [QT interval/square root of the RR interval]
Review of ECGs from large populations in Finland (n = 10,822), Japan (n = 12,149), the United States (n = 79,743), and Switzerland (n = 41,676) revealed that a QTc value of 350 ms in males and 365 ms in females was 2.0 standard deviations (SD) below the mean.8–11 However, a QTc less than the 2.0 SD cutoff did not necessarily equal arrhythmogenic potential. This was illustrated in a 29-year follow-up study of Finnish patients with QTc values as short as 320 ms, in whom no arrhythmias were documented.8 Conversely, some patients with purported short QT syndrome had QTc intervals as long as 381 ms.12
Similar problems with uncertainty of values have plagued the diagnosis of long QT syndrome.13 The lack of reference ranges and the overlap between healthy and affected people called for the development of a scoring system that involves criteria based on ECG and on the clinical evaluation.14,15
ESTABLISHING THE DIAGNOSIS OF SHORT QT SYNDROME
Clearly, the diagnosis of short QT syndrome can be challenging to establish. The first step is to rule out other causes of a short QT interval.
Differential diagnosis of short QT interval
In addition to genetic channelopathies, other causes of short QT interval must be ruled out before entertaining the diagnosis of short QT syndrome.
- Hypercalcemia is the most important of these: there is usually an accompanying prolonged PR interval and a wide QRS complex16
- Hyperkalemia17
- Acidosis17
- Increased vagal tone17
- After ventricular fibrillation (thought to be related to increased intracellular calcium)18
- Digitalis use19
- Androgen use.20
Interestingly, a shorter-than-expected QT interval was noted in patients with chronic fatigue syndrome.21
Which interval to use: QT or QTc?
Unfortunately, most population-based studies that searched for a short QT interval on ECG have used QTc as the main search parameter.8–11 As already mentioned, in patients with short QT syndrome, the QT interval is, uniquely, not shortened if the heart beats faster. In contrast, the QTc often overestimates the QT interval in patients with short QT syndrome, especially when the heart rate is in the 80s to 90s.16
In a review of cases of short QT syndrome worldwide, Bjerregaard et al22 found that the QT interval ranged from 210 ms to 340 ms with a mean ± 2 SD of 282 ± 62 ms. On the other hand, the QTc ranged from 248 ms to 345 ms with a mean ± 2 SD of 305 ± 42 ms.
Therefore, correction formulas (such as the Bazett formula) do not perform well in ruling in the diagnosis of short QT syndrome—and they do even worse in ruling it out.16,22
To establish a diagnosis of short QT syndrome in someone with prior evidence of atrial or ventricular fibrillation, a QT interval less than 340 ms or a QTc less than 345 ms is usually sufficient.22 In borderline cases in which the QT interval is slightly longer, some experts recommend other tests, although strong evidence validating their predictive value does not exist. These tests include genotyping, analysis of T wave morphology, and electrophysiologic studies.16
Recently, Gollob et al23 proposed a scoring system for short QT syndrome (Table 1). After reviewing the literature and comparing the diagnostic markers, the investigators determined diagnostic criteria that, when applied to the previously reported cases, were able to identify 58 (95.08%) of 61 patients with short QT syndrome (ie, a sensitivity of 95%).
For patients with intermediate probability, the authors recommended continued medical and ECG surveillance as well as ECGs for first-degree relatives, to further clarify the diagnosis.
Again, a principal caveat about this system is that it relies on the QTc interval rather than the QT interval to diagnose short QT syndrome.
THE SCOPE OF THE DISEASE
In a recent review of the literature, Gollob et al23 found a total of 61 cases of short QT syndrome reported in English. The cohort was predominantly male (75.4%), and most of the symptomatic patients presented during late adolescence and early adulthood. However, there have been reports of infants (4 and 8 months old), and of a man who presented for the first time at the age of 70. Of note, the authors only considered short QT syndrome types 1, 2, and 3 (see below) in their search for cases.
Whether the syndrome is truly this rare or, rather, whether many physicians are not aware of it is still to be determined. In addition, it is possible that incorrectly measuring the QT interval contributes to the lack of identification of this entity. Both of these factors were implicated in the rarity of reported long QT syndrome early after its discovery.14,15
MUTATIONS IN CARDIAC ION CHANNELS
Five distinct genetic defects have been associated with short QT syndrome. As in long QT syndrome, these give rise to subtypes of short QT syndrome, which are numbered 1 to 5 (see below).
The cardiac action potential
To understand how the mutations shorten the QT interval, we will briefly review of the cardiac myocyte action potential.24 In nonpacemaker cells of the heart, the activation of the cell membrane initiates a series of changes in ion channels that allow the movement of ions along an electrical gradient. This movement occurs in five phases and is repeated with every cardiac cycle (Figure 2).
In phase 0, the cardiac cell rapidly depolarizes.
Repolarization occurs in phases 1, 2, and 3 and is largely a function of potassium ions leaving the cell. During phase 2, calcium and sodium ions enter the cell and balance the outward potassium flow, creating the “flat” portion of the repolarization curve. Phase 3 is the main phase of repolarization in which the membrane potential rapidly falls back to its resting state (–90 mV). During phases 1 and 2, the cell membrane is completely refractory to stimulation, whereas phase 3 is divided into three parts:
- The effective refractory period: the cell is able to generate a potential that is too weak to be propagated
- The relative refractory period: the cell can respond to a stimulus that is stronger than normal
- The supernormal phase: the last small portion of phase 3, in which a less-than-normal stimulus can yield a response in the cell.
In phase 4, the cell is completely repolarized, and the cycle can start again.
Five types of short QT syndrome
Short QT syndrome 1. In 2004, Brugada et al25 identified the first mutation that causes abnormal shortening of the action potential duration. In contrast to the mutations that underlie long QT syndrome, this mutation actually causes a gain of function in the gene coding the rapidly acting delayed potassium current (IKr) channel proteins KCNH2 or HERG. Potassium leaving at a more rapid rate causes the cell to repolarize more quickly and shortens the QT interval. The clinical syndrome associated with KCNH2 gene gain-of-function mutation is called short QT syndrome 1.
Short QT syndromes 2 and 3. Other IK (potassium channel) proteins have been implicated as well. Gain-of-function mutations in the KCNQ1 and KCNJ2 genes are believed to account for short QT syndromes 2 and 3, respectively. KCNQ1 codes for the IKs protein, and KCNJ2 codes for the IK1 protein.26,27
Short QT syndromes 4 and 5 were identified by Antzelevitch et al,28 who described several patients who had a combination of channel abnormalities and ECG findings. Their ECGs showed “Brugada-syndrome-like” ST elevation in the right precordial leads, but with a short QT interval. These new syndromes were found to be associated with genetic abnormalities distinct from those of Brugada syndrome and other short QT syndromes. These abnormalities involved loss-of-function mutations in the CACNA1C gene (which codes for the alpha-1 subunit of the L-type cardiac calcium channel) and in the CACNB2 gene (which codes for the beta-2b subunit of the same channel). The two defects correspond to the clinical syndromes short QT syndrome 4 and short QT syndrome 5, respectively.28
MECHANISM OF ARRHYTHMOGENESIS IN SHORT QT SYNDROME
The myocardium is made of different layers: the epicardium, the endocardium, and the middle layer of myocytes composed mainly of M cells. Cells in the different layers differ in the concentration of their channels and can be affected differently in various syndromes. When cells in one or two of the layers repolarize at a rate different from cells in another layer, they create different degrees of refractoriness, which establishes the potential for reentry circuits to form.
It is believed that in short QT syndrome the endocardial cells and M cells repolarize faster than the epicardial cells, predisposing to reentry and arrhythmias. This accentuation of “transmural dispersion of repolarization” accounts for arrhythmogenesis in short QT syndrome as well as in long QT syndrome and the Brugada syndromes. The difference between these syndromes appears to be the layer or area of the myocardium that is affected more by the channelopathy (the M cells in long QT syndrome and the epicardium of the right ventricle in the Brugada syndrome).29
WHEN TO THINK OF SHORT QT SYNDROME
In any survivor of sudden cardiac death, the QT interval should be thoroughly scrutinized, and family members should undergo ECG. Patients in whom a short QT interval is incidentally discovered and for which other reasons are ruled out (see differential diagnosis) should be encouraged to have family members undergo ECG. Other potential patients are young people who develop atrial fibrillation and patients who have idiopathic ventricular fibrillation.4
TREATMENT AND PROGNOSIS
Evidence-based recommendations for the management of short QT syndrome do not yet exist, mainly because the number of patients identified to date is small.
Implantable cardioverter-defibrillators
Although placing an implantable cardioverter-defibrillator (ICD) seems to be warranted in patients who experience ventricular fibrillation, ventricular tachycardia, or aborted cardiac death, or in patients who have a family history of the same symptoms, the best management option is less clear for patients who have no symptoms and no family history.30 In addition, some patients may not want an ICD or may even not qualify for this therapy.
A unique problem with ICDs in short QT syndrome stems from one of the syndrome’s main features on ECG: the tall and peaked T wave that closely follows the R wave can sometimes be interpreted as a short R-R interval, provoking an inappropriate shock from the ICD.31
For the above reasons, we strongly encourage consulting a center with expertise in QT-interval-related disorders before placing an ICD in a patient suspected of having short QT syndrome.
Antiarrhythmic drugs
Prolongation of the QT interval (and the effective refractory period) with drugs has been an interesting area of research. Gaita et al32 studied the effect of four antiarrhythmics—flecainide (Tambocor), sotalol (Betapace), ibutilide (Corvert), and quinidine—in six patients with short QT syndrome. Only quinidine was associated with significant QT prolongation, from 263 ± 12 ms to 362 ms ± 25 ms. This resulted in a longer ventricular effective refractory period (> 200 ms), and ventricular fibrillation was no longer inducible during provocative testing.
In a recent study of long-term outcomes of 53 patients with short QT syndrome, Giustetto et al33 noticed that none of the patients taking quinidine, including those with a history of cardiac arrest, had any further arrhythmsic events. On the other hand, the incidence of arrhythmic events during the follow-up was 4.9% per year in patients not taking this drug. Quinidine had a stronger effect on the QT interval in patients with the HERG mutation than in those without.
RESEARCH MAY LEAD TO A BETTER UNDERSTANDING OF OTHER DISEASES
The short QT syndrome is one of the most recently recognized cardiac channelopathies associated with malignant arrhythmias. As with long QT syndrome, research in short QT syndrome may lead to a better understanding of the pathogenesis of more common but still poorly understood arrhythmias such as lone atrial fibrillation and idiopathic ventricular fibrillation.
Sudden cardiac death in a young person is a devastating event that has puzzled physicians for decades. In recent years, many of the underlying cardiac pathologies have been identified. These include structural abnormalities such as hypertrophic cardiomyopathy and nonstructural disorders associated with unstable rhythms that lead to sudden cardiac death.
The best known of these “channelopathies” are the long QT syndromes, which result from abnormal potassium and sodium channels in myocytes. Recently, interest has been growing in a disorder that may carry a similarly grim prognosis but that has an opposite finding on electrocardiography (ECG).
Short QT syndrome is a recently described heterogeneous genetic channelopathy that causes both atrial and ventricular arrhythmias and that has been documented to cause sudden cardiac death.
In 1996, a 37-year-old woman from Spain died suddenly; ECG several days earlier had shown a short QT interval of 266 ms.1 Two years later, an unrelated 17-year-old American woman undergoing laparoscopic cholecystectomy suddenly developed atrial fibrillation with a rapid ventricular response.1 Her QT interval was 225 ms. Her brother had a QT interval of 240 ms, and her mother’s was 230 ms. The patient’s maternal grandfather had a history of atrial fibrillation, and his QT interval was 245 ms. These cases led to the description of this new clinical syndrome (see below).2
CLINICAL FEATURES
Short QT syndrome has been associated with both atrial and ventricular arrhythmias. Atrial fibrillation, polymorphic ventricular tachycardia, and ventricular fibrillation have all been well described. Patients who have symptoms usually present with palpitations, presyncope, syncope, or sudden or aborted cardiac death.3,4
ELECTROCARDIOGRAPHIC FEATURES
The primary finding on ECG is a short QT interval. However, others have been noted (Figure 1):
Short or absent ST segment
This finding is not merely a consequence of the short QT interval. In 10 patients with short QT syndrome, the distance from the J point to the peak T wave ranged from 80 to 120 ms. In 12 healthy people whose QT interval was less than 320 ms, this distance ranged from 150 ms to 240 ms.5
Tall and peaked T wave
A tall and peaked T wave is a common feature in short QT syndrome. However, it was also evident in people with short QT intervals who had no other features of the syndrome.5
QT response to heart rate
Normally, the QT interval is inversely related to the heart rate, but this is not true in short QT syndrome: the QT interval remains relatively fixed with changes in heart rate.6,7 This feature is less helpful in the office setting but may be found with Holter monitoring by measuring the QT interval at different heart rates.
BUT WHAT IS CONSIDERED A SHORT QT INTERVAL?
In clinical practice, the QT interval is corrected for the heart rate by the Bazett formula:
Corrected QT (QTc) = [QT interval/square root of the RR interval]
Review of ECGs from large populations in Finland (n = 10,822), Japan (n = 12,149), the United States (n = 79,743), and Switzerland (n = 41,676) revealed that a QTc value of 350 ms in males and 365 ms in females was 2.0 standard deviations (SD) below the mean.8–11 However, a QTc less than the 2.0 SD cutoff did not necessarily equal arrhythmogenic potential. This was illustrated in a 29-year follow-up study of Finnish patients with QTc values as short as 320 ms, in whom no arrhythmias were documented.8 Conversely, some patients with purported short QT syndrome had QTc intervals as long as 381 ms.12
Similar problems with uncertainty of values have plagued the diagnosis of long QT syndrome.13 The lack of reference ranges and the overlap between healthy and affected people called for the development of a scoring system that involves criteria based on ECG and on the clinical evaluation.14,15
ESTABLISHING THE DIAGNOSIS OF SHORT QT SYNDROME
Clearly, the diagnosis of short QT syndrome can be challenging to establish. The first step is to rule out other causes of a short QT interval.
Differential diagnosis of short QT interval
In addition to genetic channelopathies, other causes of short QT interval must be ruled out before entertaining the diagnosis of short QT syndrome.
- Hypercalcemia is the most important of these: there is usually an accompanying prolonged PR interval and a wide QRS complex16
- Hyperkalemia17
- Acidosis17
- Increased vagal tone17
- After ventricular fibrillation (thought to be related to increased intracellular calcium)18
- Digitalis use19
- Androgen use.20
Interestingly, a shorter-than-expected QT interval was noted in patients with chronic fatigue syndrome.21
Which interval to use: QT or QTc?
Unfortunately, most population-based studies that searched for a short QT interval on ECG have used QTc as the main search parameter.8–11 As already mentioned, in patients with short QT syndrome, the QT interval is, uniquely, not shortened if the heart beats faster. In contrast, the QTc often overestimates the QT interval in patients with short QT syndrome, especially when the heart rate is in the 80s to 90s.16
In a review of cases of short QT syndrome worldwide, Bjerregaard et al22 found that the QT interval ranged from 210 ms to 340 ms with a mean ± 2 SD of 282 ± 62 ms. On the other hand, the QTc ranged from 248 ms to 345 ms with a mean ± 2 SD of 305 ± 42 ms.
Therefore, correction formulas (such as the Bazett formula) do not perform well in ruling in the diagnosis of short QT syndrome—and they do even worse in ruling it out.16,22
To establish a diagnosis of short QT syndrome in someone with prior evidence of atrial or ventricular fibrillation, a QT interval less than 340 ms or a QTc less than 345 ms is usually sufficient.22 In borderline cases in which the QT interval is slightly longer, some experts recommend other tests, although strong evidence validating their predictive value does not exist. These tests include genotyping, analysis of T wave morphology, and electrophysiologic studies.16
Recently, Gollob et al23 proposed a scoring system for short QT syndrome (Table 1). After reviewing the literature and comparing the diagnostic markers, the investigators determined diagnostic criteria that, when applied to the previously reported cases, were able to identify 58 (95.08%) of 61 patients with short QT syndrome (ie, a sensitivity of 95%).
For patients with intermediate probability, the authors recommended continued medical and ECG surveillance as well as ECGs for first-degree relatives, to further clarify the diagnosis.
Again, a principal caveat about this system is that it relies on the QTc interval rather than the QT interval to diagnose short QT syndrome.
THE SCOPE OF THE DISEASE
In a recent review of the literature, Gollob et al23 found a total of 61 cases of short QT syndrome reported in English. The cohort was predominantly male (75.4%), and most of the symptomatic patients presented during late adolescence and early adulthood. However, there have been reports of infants (4 and 8 months old), and of a man who presented for the first time at the age of 70. Of note, the authors only considered short QT syndrome types 1, 2, and 3 (see below) in their search for cases.
Whether the syndrome is truly this rare or, rather, whether many physicians are not aware of it is still to be determined. In addition, it is possible that incorrectly measuring the QT interval contributes to the lack of identification of this entity. Both of these factors were implicated in the rarity of reported long QT syndrome early after its discovery.14,15
MUTATIONS IN CARDIAC ION CHANNELS
Five distinct genetic defects have been associated with short QT syndrome. As in long QT syndrome, these give rise to subtypes of short QT syndrome, which are numbered 1 to 5 (see below).
The cardiac action potential
To understand how the mutations shorten the QT interval, we will briefly review of the cardiac myocyte action potential.24 In nonpacemaker cells of the heart, the activation of the cell membrane initiates a series of changes in ion channels that allow the movement of ions along an electrical gradient. This movement occurs in five phases and is repeated with every cardiac cycle (Figure 2).
In phase 0, the cardiac cell rapidly depolarizes.
Repolarization occurs in phases 1, 2, and 3 and is largely a function of potassium ions leaving the cell. During phase 2, calcium and sodium ions enter the cell and balance the outward potassium flow, creating the “flat” portion of the repolarization curve. Phase 3 is the main phase of repolarization in which the membrane potential rapidly falls back to its resting state (–90 mV). During phases 1 and 2, the cell membrane is completely refractory to stimulation, whereas phase 3 is divided into three parts:
- The effective refractory period: the cell is able to generate a potential that is too weak to be propagated
- The relative refractory period: the cell can respond to a stimulus that is stronger than normal
- The supernormal phase: the last small portion of phase 3, in which a less-than-normal stimulus can yield a response in the cell.
In phase 4, the cell is completely repolarized, and the cycle can start again.
Five types of short QT syndrome
Short QT syndrome 1. In 2004, Brugada et al25 identified the first mutation that causes abnormal shortening of the action potential duration. In contrast to the mutations that underlie long QT syndrome, this mutation actually causes a gain of function in the gene coding the rapidly acting delayed potassium current (IKr) channel proteins KCNH2 or HERG. Potassium leaving at a more rapid rate causes the cell to repolarize more quickly and shortens the QT interval. The clinical syndrome associated with KCNH2 gene gain-of-function mutation is called short QT syndrome 1.
Short QT syndromes 2 and 3. Other IK (potassium channel) proteins have been implicated as well. Gain-of-function mutations in the KCNQ1 and KCNJ2 genes are believed to account for short QT syndromes 2 and 3, respectively. KCNQ1 codes for the IKs protein, and KCNJ2 codes for the IK1 protein.26,27
Short QT syndromes 4 and 5 were identified by Antzelevitch et al,28 who described several patients who had a combination of channel abnormalities and ECG findings. Their ECGs showed “Brugada-syndrome-like” ST elevation in the right precordial leads, but with a short QT interval. These new syndromes were found to be associated with genetic abnormalities distinct from those of Brugada syndrome and other short QT syndromes. These abnormalities involved loss-of-function mutations in the CACNA1C gene (which codes for the alpha-1 subunit of the L-type cardiac calcium channel) and in the CACNB2 gene (which codes for the beta-2b subunit of the same channel). The two defects correspond to the clinical syndromes short QT syndrome 4 and short QT syndrome 5, respectively.28
MECHANISM OF ARRHYTHMOGENESIS IN SHORT QT SYNDROME
The myocardium is made of different layers: the epicardium, the endocardium, and the middle layer of myocytes composed mainly of M cells. Cells in the different layers differ in the concentration of their channels and can be affected differently in various syndromes. When cells in one or two of the layers repolarize at a rate different from cells in another layer, they create different degrees of refractoriness, which establishes the potential for reentry circuits to form.
It is believed that in short QT syndrome the endocardial cells and M cells repolarize faster than the epicardial cells, predisposing to reentry and arrhythmias. This accentuation of “transmural dispersion of repolarization” accounts for arrhythmogenesis in short QT syndrome as well as in long QT syndrome and the Brugada syndromes. The difference between these syndromes appears to be the layer or area of the myocardium that is affected more by the channelopathy (the M cells in long QT syndrome and the epicardium of the right ventricle in the Brugada syndrome).29
WHEN TO THINK OF SHORT QT SYNDROME
In any survivor of sudden cardiac death, the QT interval should be thoroughly scrutinized, and family members should undergo ECG. Patients in whom a short QT interval is incidentally discovered and for which other reasons are ruled out (see differential diagnosis) should be encouraged to have family members undergo ECG. Other potential patients are young people who develop atrial fibrillation and patients who have idiopathic ventricular fibrillation.4
TREATMENT AND PROGNOSIS
Evidence-based recommendations for the management of short QT syndrome do not yet exist, mainly because the number of patients identified to date is small.
Implantable cardioverter-defibrillators
Although placing an implantable cardioverter-defibrillator (ICD) seems to be warranted in patients who experience ventricular fibrillation, ventricular tachycardia, or aborted cardiac death, or in patients who have a family history of the same symptoms, the best management option is less clear for patients who have no symptoms and no family history.30 In addition, some patients may not want an ICD or may even not qualify for this therapy.
A unique problem with ICDs in short QT syndrome stems from one of the syndrome’s main features on ECG: the tall and peaked T wave that closely follows the R wave can sometimes be interpreted as a short R-R interval, provoking an inappropriate shock from the ICD.31
For the above reasons, we strongly encourage consulting a center with expertise in QT-interval-related disorders before placing an ICD in a patient suspected of having short QT syndrome.
Antiarrhythmic drugs
Prolongation of the QT interval (and the effective refractory period) with drugs has been an interesting area of research. Gaita et al32 studied the effect of four antiarrhythmics—flecainide (Tambocor), sotalol (Betapace), ibutilide (Corvert), and quinidine—in six patients with short QT syndrome. Only quinidine was associated with significant QT prolongation, from 263 ± 12 ms to 362 ms ± 25 ms. This resulted in a longer ventricular effective refractory period (> 200 ms), and ventricular fibrillation was no longer inducible during provocative testing.
In a recent study of long-term outcomes of 53 patients with short QT syndrome, Giustetto et al33 noticed that none of the patients taking quinidine, including those with a history of cardiac arrest, had any further arrhythmsic events. On the other hand, the incidence of arrhythmic events during the follow-up was 4.9% per year in patients not taking this drug. Quinidine had a stronger effect on the QT interval in patients with the HERG mutation than in those without.
RESEARCH MAY LEAD TO A BETTER UNDERSTANDING OF OTHER DISEASES
The short QT syndrome is one of the most recently recognized cardiac channelopathies associated with malignant arrhythmias. As with long QT syndrome, research in short QT syndrome may lead to a better understanding of the pathogenesis of more common but still poorly understood arrhythmias such as lone atrial fibrillation and idiopathic ventricular fibrillation.
- The Short QT Syndrome http://www.shortqtsyndrome.org/short_qt_history.htm. Accessed October 30, 2012.
- Gussak I, Brugada P, Brugada J, et al. Idiopathic short QT interval: a new clinical syndrome? Cardiology 2000; 94:99–102.
- Giustetto C, Di Monte F, Wolpert C, et al. Short QT syndrome: clinical findings and diagnostic-therapeutic implications. Eur Heart J 2006; 27:2440–2447.
- Viskin S, Zeltser D, Ish-Shalom M, et al. Is idiopathic ventricular fibrillation a short QT syndrome? Comparison of QT intervals of patients with idiopathic ventricular fibrillation and healthy controls. Heart Rhythm 2004; 1:587–591.
- Anttonen O, Junttila MJ, Maury P, et al. Differences in twelve-lead electrocardiogram between symptomatic and asymptomatic subjects with short QT interval. Heart Rhythm 2009; 6:267–271.
- Redpath CJ, Green MS, Birnie DH, Gollob MH. Rapid genetic testing facilitating the diagnosis of short QT syndrome. Can J Cardiol 2009; 25:e133–e135.
- Wolpert C, Schimpf R, Giustetto C, et al. Further insights into the effect of quinidine in short QT syndrome caused by a mutation in HERG. J Cardiovasc Electrophysiol 2005; 16:54–58.
- Anttonen O, Junttila MJ, Rissanen H, Reunanen A, Viitasalo M, Huikuri HV. Prevalence and prognostic significance of short QT interval in a middle-aged Finnish population. Circulation 2007; 116:714–720.
- Funada A, Hayashi K, Ino H, et al. Assessment of QT intervals and prevalence of short QT syndrome in Japan. Clin Cardiol 2008; 31:270–274.
- Mason JW, Ramseth DJ, Chanter DO, Moon TE, Goodman DB, Mendzelevski B. Electrocardiographic reference ranges derived from 79,743 ambulatory subjects. J Electrocardiol 2007; 40:228–234.
- Kobza R, Roos M, Niggli B, et al. Prevalence of long and short QT in a young population of 41,767 predominantly male Swiss conscripts. Heart Rhythm 2009; 6:652–657.
- Itoh H, Sakaguchi T, Ashihara T, et al. A novel KCNH2 mutation as a modifier for short QT interval. Int J Cardiol 2009; 137:83–85.
- Vincent GM, Timothy KW, Leppert M, Keating M. The spectrum of symptoms and QT intervals in carriers of the gene for the long-QT syndrome. N Engl J Med 1992; 327:846–852.
- Schwartz PJ. Idiopathic long QT syndrome: progress and questions. Am Heart J 1985; 109:399–411.
- Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Diagnostic criteria for the long QT syndrome. An update. Circulation 1993; 88:782–784.
- Bjerregaard P, Nallapaneni H, Gussak I. Short QT interval in clinical practice. J Electrocardiol 2010; 43:390–395.
- Maury P, Extramiana F, Sbragia P, et al. Short QT syndrome. Update on a recent entity. Arch Cardiovasc Dis 2008; 101:779–786.
- Kontny F, Dale J. Self-terminating idiopathic ventricular fibrillation presenting as syncope: a 40-year follow-up report. J Intern Med 1990; 227:211–213.
- Cheng TO. Digitalis administration: an underappreciated but common cause of short QT interval. Circulation 2004; 109:e152.
- Hancox JC, Choisy SC, James AF. Short QT interval linked to androgen misuse: wider significance and possible basis. Ann Noninvasive Electrocardiol 2009; 14:311–312.
- Naschitz J, Fields M, Isseroff H, Sharif D, Sabo E, Rosner I. Shortened QT interval: a distinctive feature of the dysautonomia of chronic fatigue syndrome. J Electrocardiol 2006; 39:389–394.
- Bjerregaard P, Collier JL, Gussak I. Upper limits of QT/QTc intervals in the short QT syndrome. Review of the world-wide short QT syndrome population and 3 new USA families. Heart Rhythm 2008; 5:AB43.
- Gollob MH, Redpath CJ, Roberts JD. The short QT syndrome: proposed diagnostic criteria. J Am Coll Cardiol 2011; 57:802–812.
- Shih HT. Anatomy of the action potential in the heart. Tex Heart Inst J 1994; 21:30–41.
- Brugada R, Hong K, Dumaine R, et al. Sudden death associated with short-QT syndrome linked to mutations in HERG. Circulation 2004; 109:30–35.
- Bellocq C, van Ginneken AC, Bezzina CR, et al. Mutation in the KCNQ1 gene leading to the short QT-interval syndrome. Circulation 2004; 109:2394–2397.
- Priori SG, Pandit SV, Rivolta I, et al. A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ Res 2005; 96:800–807.
- Antzelevitch C, Pollevick GD, Cordeiro JM, et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation 2007; 115:442–449.
- Antzelevitch C. Heterogeneity and cardiac arrhythmias: an overview. Heart Rhythm 2007; 4:964–972.
- Lunati M, Bongiorni MG, Boriani G, et al. Linee guida AIAC 2006 all’impianto di pacemaker, dispositivi per la resincronizzazione cardiaca (CRT) e defibrillatori automatici impiantabili (ICD). GIAC 2005; 8:1–58.
- Schimpf R, Wolpert C, Bianchi F, et al. Congenital short QT syndrome and implantable cardioverter defibrillator treatment: inherent risk for inappropriate shock delivery. J Cardiovasc Electrophysiol 2003; 14:1273–1277.
- Gaita F, Giustetto C, Bianchi F, et al. Short QT syndrome: pharmacological treatment. J Am Coll Cardiol 2004; 43:1494–1499.
- Giustetto C, Schimpf R, Mazzanti A, et al. Long-term follow-up of patients with short QT syndrome. J Am Coll Cardiol 2011; 58:587–595.
- The Short QT Syndrome http://www.shortqtsyndrome.org/short_qt_history.htm. Accessed October 30, 2012.
- Gussak I, Brugada P, Brugada J, et al. Idiopathic short QT interval: a new clinical syndrome? Cardiology 2000; 94:99–102.
- Giustetto C, Di Monte F, Wolpert C, et al. Short QT syndrome: clinical findings and diagnostic-therapeutic implications. Eur Heart J 2006; 27:2440–2447.
- Viskin S, Zeltser D, Ish-Shalom M, et al. Is idiopathic ventricular fibrillation a short QT syndrome? Comparison of QT intervals of patients with idiopathic ventricular fibrillation and healthy controls. Heart Rhythm 2004; 1:587–591.
- Anttonen O, Junttila MJ, Maury P, et al. Differences in twelve-lead electrocardiogram between symptomatic and asymptomatic subjects with short QT interval. Heart Rhythm 2009; 6:267–271.
- Redpath CJ, Green MS, Birnie DH, Gollob MH. Rapid genetic testing facilitating the diagnosis of short QT syndrome. Can J Cardiol 2009; 25:e133–e135.
- Wolpert C, Schimpf R, Giustetto C, et al. Further insights into the effect of quinidine in short QT syndrome caused by a mutation in HERG. J Cardiovasc Electrophysiol 2005; 16:54–58.
- Anttonen O, Junttila MJ, Rissanen H, Reunanen A, Viitasalo M, Huikuri HV. Prevalence and prognostic significance of short QT interval in a middle-aged Finnish population. Circulation 2007; 116:714–720.
- Funada A, Hayashi K, Ino H, et al. Assessment of QT intervals and prevalence of short QT syndrome in Japan. Clin Cardiol 2008; 31:270–274.
- Mason JW, Ramseth DJ, Chanter DO, Moon TE, Goodman DB, Mendzelevski B. Electrocardiographic reference ranges derived from 79,743 ambulatory subjects. J Electrocardiol 2007; 40:228–234.
- Kobza R, Roos M, Niggli B, et al. Prevalence of long and short QT in a young population of 41,767 predominantly male Swiss conscripts. Heart Rhythm 2009; 6:652–657.
- Itoh H, Sakaguchi T, Ashihara T, et al. A novel KCNH2 mutation as a modifier for short QT interval. Int J Cardiol 2009; 137:83–85.
- Vincent GM, Timothy KW, Leppert M, Keating M. The spectrum of symptoms and QT intervals in carriers of the gene for the long-QT syndrome. N Engl J Med 1992; 327:846–852.
- Schwartz PJ. Idiopathic long QT syndrome: progress and questions. Am Heart J 1985; 109:399–411.
- Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Diagnostic criteria for the long QT syndrome. An update. Circulation 1993; 88:782–784.
- Bjerregaard P, Nallapaneni H, Gussak I. Short QT interval in clinical practice. J Electrocardiol 2010; 43:390–395.
- Maury P, Extramiana F, Sbragia P, et al. Short QT syndrome. Update on a recent entity. Arch Cardiovasc Dis 2008; 101:779–786.
- Kontny F, Dale J. Self-terminating idiopathic ventricular fibrillation presenting as syncope: a 40-year follow-up report. J Intern Med 1990; 227:211–213.
- Cheng TO. Digitalis administration: an underappreciated but common cause of short QT interval. Circulation 2004; 109:e152.
- Hancox JC, Choisy SC, James AF. Short QT interval linked to androgen misuse: wider significance and possible basis. Ann Noninvasive Electrocardiol 2009; 14:311–312.
- Naschitz J, Fields M, Isseroff H, Sharif D, Sabo E, Rosner I. Shortened QT interval: a distinctive feature of the dysautonomia of chronic fatigue syndrome. J Electrocardiol 2006; 39:389–394.
- Bjerregaard P, Collier JL, Gussak I. Upper limits of QT/QTc intervals in the short QT syndrome. Review of the world-wide short QT syndrome population and 3 new USA families. Heart Rhythm 2008; 5:AB43.
- Gollob MH, Redpath CJ, Roberts JD. The short QT syndrome: proposed diagnostic criteria. J Am Coll Cardiol 2011; 57:802–812.
- Shih HT. Anatomy of the action potential in the heart. Tex Heart Inst J 1994; 21:30–41.
- Brugada R, Hong K, Dumaine R, et al. Sudden death associated with short-QT syndrome linked to mutations in HERG. Circulation 2004; 109:30–35.
- Bellocq C, van Ginneken AC, Bezzina CR, et al. Mutation in the KCNQ1 gene leading to the short QT-interval syndrome. Circulation 2004; 109:2394–2397.
- Priori SG, Pandit SV, Rivolta I, et al. A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ Res 2005; 96:800–807.
- Antzelevitch C, Pollevick GD, Cordeiro JM, et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation 2007; 115:442–449.
- Antzelevitch C. Heterogeneity and cardiac arrhythmias: an overview. Heart Rhythm 2007; 4:964–972.
- Lunati M, Bongiorni MG, Boriani G, et al. Linee guida AIAC 2006 all’impianto di pacemaker, dispositivi per la resincronizzazione cardiaca (CRT) e defibrillatori automatici impiantabili (ICD). GIAC 2005; 8:1–58.
- Schimpf R, Wolpert C, Bianchi F, et al. Congenital short QT syndrome and implantable cardioverter defibrillator treatment: inherent risk for inappropriate shock delivery. J Cardiovasc Electrophysiol 2003; 14:1273–1277.
- Gaita F, Giustetto C, Bianchi F, et al. Short QT syndrome: pharmacological treatment. J Am Coll Cardiol 2004; 43:1494–1499.
- Giustetto C, Schimpf R, Mazzanti A, et al. Long-term follow-up of patients with short QT syndrome. J Am Coll Cardiol 2011; 58:587–595.
KEY POINTS
- Short QT syndrome is a genetic disease described initially in young patients who had atrial fibrillation or who died suddenly with no apparent structural heart disease.
- The diagnosis is established by the finding of a short QT interval. However, other factors including personal and family history are also important in establishing the diagnosis.
- The current recommendations for managing patients with short QT syndrome are not evidence-based. We encourage consultation with centers that have special interest in QT-interval-related disorders.
- Placement of an implantable cardioverter-defibrillator is considered the standard of care, especially in survivors of sudden cardiac death, ventricular fibrillation, or ventricular tachycardia. Unfortunately, a higher incidence of inappropriate shocks adds to the challenges of managing this potentially deadly disease.
Advanced heart failure: Transplantation, LVADs, and beyond
Patients with advanced heart failure far outnumber the hearts available for transplantation. Partly as a consequence of this shortage, left-ventricular assist devices (LVADs) are being used more widely.
This article is an update on options for managing severe, advanced heart failure, with special attention to new developments and continuing challenges in heart transplantation and LVADs.
THE PREVALENCE OF HEART FAILURE
About 2.6% of the US population age 20 and older have heart failure—some 5.8 million people. Of these, about half have systolic heart failure.1 Patients with systolic heart failure can be classified by degree of severity under two systems:
The New York Heart Association (NYHA) classifies patients by their functional status, from I (no limitation in activities) to IV (symptoms at rest). NYHA class III (symptoms with minimal exertion) is sometimes further broken down into IIIa and IIIb, with the latter defined as having a recent history of dyspnea at rest.
The joint American College of Cardiology and American Heart Association (ACC/AHA) classification uses four stages, from A (high risk of developing heart failure, ie, having risk factors such as family history of heart disease, hypertension, or diabetes) to D (advanced heart disease despite treatment). Patients in stage D tend to be recurrently hospitalized despite cardiac resynchronization therapy and drug therapy, and they cannot be safely discharged without specialized interventions. The options for these patients are limited: either end-of-life care or extraordinary measures such as heart transplantation, long-term treatment with inotropic drugs, permanent mechanical circulatory support, or experimental therapies.2
The estimated number of people in ACC/AHA stage D or NYHA class IV is 15,600 to 156,000. The approximate number of heart transplants performed in the United States each year is 2,100.3
WHICH AMBULATORY PATIENTS ARE MOST AT RISK?
The range for the estimated number of patients with advanced heart failure (NYHA class IIIb or IV) is wide (see above) because these patients may be hard to recognize. The most debilitated patients are obvious: they tend to be in the intensive care unit with end-organ failure. However, it is a challenge to recognize patients at extremely high risk who are still ambulatory.
The European Society of Cardiology4 developed a definition of advanced chronic heart failure that can help identify patients who are candidates for the transplant list and for an LVAD. All the following features must be present despite optimal therapy that includes diuretics, inhibitors of the renin-angiotensin-aldosterone system, and beta-blockers, unless these are poorly tolerated or contraindicated, and cardiac resynchronization therapy if indicated:
- Severe symptoms, with dyspnea or fatigue at rest or with minimal exertion (NYHA class III or IV)
- Episodes of fluid retention (pulmonary or systemic congestion, peripheral edema) or of reduced cardiac output at rest (peripheral hypoperfusion)
- Objective evidence of severe cardiac dysfunction (at least one of the following): left ventricular ejection fraction less than 30%, pseudonormal or restrictive mitral inflow pattern on Doppler echocardiography, high left or right ventricular filling pressure (or both left and right filling pressures), and elevated B-type natriuretic peptides
- Severely impaired functional capacity demonstrated by one of the following: inability to exercise, 6-minute walk test distance less than 300 m (or less in women or patients who are age 75 and older), or peak oxygen intake less than 12 to 14 mL/kg/min
- One or more hospitalizations for heart failure in the past 6 months.
Treadmill exercise time is an easily performed test. Hsich et al5 found that the longer patients can walk, the lower their risk of death, and that this variable is about as predictive of survival in patients with systolic left ventricular dysfunction as peak oxygen consumption, which is much more cumbersome to measure.
The Seattle Heart Failure Model gives an estimate of prognosis for ambulatory patients with advanced heart failure. Available at http://depts.washington.edu/shfm/, it is based on age, sex, NYHA class, weight, ejection fraction, blood pressure, medications, a few laboratory values, and other clinical information. The model has been validated in numerous cohorts,6 but it may underestimate risk and is currently being tested in clinical trials (REVIVE-IT and ROADMAP; see at www.clinicaltrials.gov).
Recurrent hospitalization is a simple predictor of risk. A study of about 7,000 patients worldwide found that after hospitalization with acute decompensated heart failure, the strongest predictor of death within 6 months was readmission for any reason within 30 days of the index hospitalization (Starling RC, unpublished observation, 2011). Any patient with heart failure who is repeatedly hospitalized should have a consultation with a heart failure specialist.
INOTROPIC THERAPY FOR BRIDGING
Inotropic drugs, which include intravenous dobutamine (Dobutrex) and milrinone (Primacor), are used to help maintain end-organ function until a patient can obtain a heart transplant or LVAD.
Inotropic therapy should not be viewed as an alternative to heart transplantation or device implantation. We inform patients that inotropic therapy is purely palliative and may actually increase the risk of death, which is about 50% at 6 months and nearly 100% at 1 year. A patient on inotropic therapy who is not a candidate for a transplant or for an assist device should be referred to a hospice program.7
CARDIAC TRANSPLANTATION: SUCCESSES, CHALLENGES
Survival rates after heart transplantation are now excellent. The 1-year survival rate is about 90%, the 5-year rate is about 70%, but only about 20% survive 20 years or longer.8,9 The prognosis is not as good as for combined heart-lung transplantation patients.
Age is an important factor and is a contentious issue: some medical centers will not offer transplantation to patients over age 65. Others regard age as just another risk factor, like renal dysfunction or diabetes.
Quality of life after heart transplantation is excellent: patients are usually able to return to work, regardless of their profession.
The leading cause of death after heart transplantation is malignancy, followed by coronary artery vasculopathy, then by graft failure. Some patients develop left ventricular dysfunction and heart failure of unknown cause. Others develop antibody-mediated rejection; in recent years this has been more promptly recognized, but treatment remains a challenge.
Acute rejection, which used to be one of the main causes of death, now has an extremely low incidence because of modern drug therapies. In a US observational study currently being conducted in about 200 patients receiving a heart transplant (details on CTOT-05 at www.clinicaltrials.gov), the incidence of moderate rejection during the first year is less than 10% (Starling RC, unpublished observation). But several concerns remain.
Adverse effects of immunosuppressive drugs continue to be problematic. These include infection, malignancy, osteoporosis, chronic kidney toxicity, hypertension, and neuropathy.
Renal dysfunction is one of the largest issues. About 10% of heart transplant recipients develop stage 4 kidney disease (with a glomerular filtration rate < 30 mL/min) and need kidney transplantation or renal replacement therapy because of the use of calcineurin inhibitors for immunosuppression.10
Coronary artery vasculopathy was the largest problem when heart transplantation began and continues to be a major concern and focus of research.11,12 Case 1 (below) is an example of the problem.
Case 1: Poor outcome despite an ideal scenario
A 57-year-old businessman had dilated cardiomyopathy and progressive heart failure for 10 years. He was listed for transplantation in 2008 and was given an LVAD (HeartMate II, Thoratec Corp, Pleasanton, CA) as a bridge until a donor heart became available.
In 2009, he received a heart transplant under ideal conditions: the donor was a large 30-year-old man who died of a gunshot wound to the head in the same city in which the patient and transplant hospital were located. Cardiopulmonary resuscitation was not performed, and the cold ischemic time was just a little more than 3 hours. Immune indicators were ideal with a negative prospective cross-match.
Laboratory results after transplantation included creatinine 1.7 mg/dL (normal 0.6–1.2 mg/dL), low-density lipoprotein cholesterol 75 mg/dL, high-density lipoprotein cholesterol 64 mg/dL, and triglycerides 90 mg/dL.
The patient was given immunosuppressive therapy with cyclosporine (Neoral), mycophenolate (CellCept), and prednisone. Because his creatinine level was high, he was also perioperatively given basiliximab (Simulect), a monoclonal antibody to the alpha chain (CD25) of the interleukin-2 receptor. (In a patient who has poor renal function, basilixumab may help by enabling us to delay the use of calcineurin inhibitors.) He also received simvastatin (Zocor) 10 mg.
Per Cleveland Clinic protocol, he underwent 13 biopsy procedures during his first year, and each was normal (grade 0 or 1R). Evaluation by cardiac catheterization at 1 year showed some irregularities in the left anterior descending artery, but a stent was not deemed necessary. Also, per protocol, he underwent intravascular ultrasonography, which revealed abnormal thickness in the intima and media, indicating that coronary artery disease was developing, although it was nonobstructive.
Two months after this checkup, the patient collapsed and suddenly died while shopping. At autopsy, his left anterior descending artery was found to be severely obstructed.
Coronary artery vasculopathy is still a major problem
This case shows that coronary artery vasculopathy may develop despite an ideal transplantation scenario. It remains a large concern and a focus of research.
Coronary vasculopathy develops in 30% to 40% of heart transplant recipients within 5 years, and the incidence has not been reduced by much over the years. However, probably fewer than 5% of these patients die or even need bypass surgery or stenting, and the problem is managed the same as native atherosclerosis. We perform routine annual cardiac catheterizations or stress tests, or both, and place stents in severely blocked arteries.
THE DONOR SHORTAGE: CHANGING HOW HEARTS ARE ALLOCATED
The number of patients receiving a heart transplant in the United States—about 2,000 per year—has not increased in the past decade. The European Union also has great difficulty obtaining hearts for people in need, and almost every transplant candidate there gets mechanical support for some time. The gap between those listed for transplant and the number transplanted each year continues to widen in both the United States and Europe.
All transplant candidates are assigned a status by the United Network of Organ Sharing (UNOS) based on their medical condition. The highest status, 1A, goes to patients who are seriously ill, in the hospital, on high doses of inotropic drugs (specific dosages are defined) and mechanical circulatory support such as an LVAD, and expected to live less than 1 month without a transplant. Status 1B patients are stable on lower-dose inotropic therapy or on mechanical support, and can be in the hospital or at home. Status 2 patients are stable and ambulatory and are not on inotropic drugs.
In July 2006, UNOS changed the rules on how patients are prioritized for obtaining an organ. The new rules are based both on severity of illness (see above) and geographic proximity to the donor heart—local, within 500 miles (“zone A”) or within 500 to 1,000 miles (“zone B”). The order of priority for donor hearts is:
- Local, status 1A
- Local, status 1B
- Zone A, status 1A
- Zone A, status 1B
- Local, status 2
- Zone B, status 1A
- Zone B, status 1B
- Zone A, status 2.
As a result of the change, donor hearts that become available in a particular hospital do not necessarily go to a patient in that state. Another result is that status 2 patients, who were previously the most common transplant recipients, now have much less access because all status 1 patients within 500 miles are given higher priority. Since the change, only 8% of hearts nationwide go to status 2 patients, which is 67% fewer than before. At the same time, organs allocated to status 1A patients have increased by 26%, and their death rates have fallen.3
The new allocation system is a positive change for the sickest patients, providing quicker access and a reduction in waiting-list mortality.13 The drawback is that status 2 patients who are less ill are less likely to ever receive an organ until their condition worsens.
Heart transplant outcomes are publicly reported
The Scientific Registry of Transplant Recipients publicly reports heart transplant outcomes (www.srtr.org). For any transplant center, the public can learn the number of patients waiting for a transplant, the death rate on the waiting list, the number of transplants performed in the previous 12 months, the waiting time in months, and observed and risk-adjusted expected survival rates. A center that deviates from the expected survival rates by 10% or more may be audited and could lose its certification.
Also listed on the Web site is the percentage of patients who receive a support device before receiving a transplant. This can vary widely between institutions and may reflect the organ availability at the transplant center (waiting times) or the preferences and expertise of the transplantation team. We believe that the mortality rate on the waiting list will be reduced with appropriate use of LVADs as a bridge to transplantation when indicated. We have now transitioned to the use of the improved continuous-flow LVADs and rarely maintain patients on continuous inotropic therapy at home to await a donor organ.
MECHANICAL CIRCULATORY SUPPORT: BRIDGE OR DESTINATION?
Mechanical circulatory support devices are increasingly being used to sustain patients with advanced heart failure. Currently at Cleveland Clinic, more LVADs are implanted than hearts are transplanted.
Mechanical circulatory support is indicated for patients who are listed for transplant to keep them functioning as well as possible while they are waiting (bridge to transplant). For others it is “destination therapy”: they are not candidates for a transplant, but a device may improve and prolong the rest of their life.
Case 2: A good outcome despite a poor prognosis
A 71-year-old man was rejected for transplantation by his local hospital because of his age and also because he had pulmonary artery hypertension (78/42 mm Hg; reference range 15–30/5–15 mm Hg) and creatinine elevation (3.0 mg/dL; reference range 0.6–1.5 mg/dL). Nevertheless, he did well on a mechanical device and was accepted for transplantation by Cleveland Clinic. He received a transplant and is still alive and active 14 years later.
Comment. Determining that a patient is not a good transplantation candidate is often impossible. Putting the patient on mechanical support for a period of time can often help clarify whether transplantation is advisable. Probably most patients who receive mechanical support do so as a bridge to decision: most are acutely ill and many have organ dysfunction, pulmonary hypertension, and renal insufficiency. After a period of support, they can be assessed for suitability for transplantation.
LVADs continue to improve
Many devices are available for mechanical circulatory support.14 In addition to LVADs, there are right-ventricular assist devices (RVADs), and devices that simultaneously support both ventricles (BiVADs). Total artificial hearts are also available, as are acute temporary percutaneous devices. These temporary devices—TandemHeart (CardiacAssist, Pittsburgh, PA) and Impella (Abiomed, Danvers, MD)—can be used before a long-term mechanical device can be surgically implanted.
LVADs are of three types:
- Pulsatile volume-displacement pumps, which mimic the pumping action of the natural heart. These early devices were large and placed in the abdomen.
- Continuous axial-flow pumps, which do not have a “heartbeat.” These are quieter and lighter than the early pumps, and use a turbine that spins at 8,000 to 10,000 rpm.
- Continuous centrifugal-flow pumps. These have a rotor spinning at 2,000 to 3,000 rpm, and most of them are magnetically powered and suspended.
The superiority of LVADs over medical therapy was clearly shown even in early studies that used pulsatile LVADs.15 The results of such studies and the increased durability of the devices have led to their rapidly expanded use.
The newer continuous-flow pumps offer significant improvements over the old pulsatile-flow pumps, being smaller, lighter, quieter, and more durable (Table 1). A 2007 study of 133 patients on a continuous axial-flow LVAD (HeartMate II) found that 76% were still alive after 6 months, and patients had significant improvement in functional status and quality of life.16 In a postapproval study based on registry data, HeartMate II was found superior to pulsatile pumps in terms of survival up to 12 months, percentage of patients reaching transplant, and cardiac recovery. Adverse event rates were similar or lower for HeartMate II.17
Another study compared a continuousflow with a pulsatile-flow LVAD for patients who were ineligible for transplantation. Survival at 2 years was 58% with the continuousflow device vs 24% with the pulsatile-flow device (P = .008).18 Since then, postmarket data of patients who received an LVAD showed that 85% are still alive at 1 year.19 This study can be viewed as supporting the use of LVADs as destination therapy.
Quality of life for patients receiving an LVAD has been excellent. When biventricular pacemakers for resynchronization therapy first became available, distances on the 6-minute walk test improved by 39 m, which was deemed a big improvement. LVAD devices have increased the 6-minute walk distance by 156 m.20
Adverse events with LVADs have improved, but continue to be of concern
Infections can arise in the blood stream, in the device pocket, or especially where the driveline exits the skin. As devices have become smaller, driveline diameters have become smaller as well, allowing for a better seal at the skin and making this less of a problem. Some centers report the incidence of driveline infections as less than 20%, but they continue to be a focus of concern.18
Stroke rates continue to improve, although patients still require intensive lifelong anticoagulation. The target international normalized ratio varies by device manufacturer, ranging from 1.7 to 2.5.
Bleeding. Acquired von Willebrand syndrome can develop in patients who have an LVAD, with the gastrointestinal system being the most frequent site of bleeding.21
Device thrombosis occurs very rarely (2%–3%) but is very serious and may require pump exchange.
Mechanical malfunction. As duration of therapy lengthens, problems are arising with aging devices, such as broken wires or short circuits. New-generation pumps have markedly improved durability and reliability.
Good data are kept on device outcomes
The Interagency for Mechanically Assisted Circulatory Support (INTERMACS) maintains a national registry of patients with a mechanical circulatory support device to treat advanced heart failure. It was jointly established in 2006 by the National Heart, Lung, and Blood Institute, Centers for Medicare and Medicaid Services (CMS), the US Food and Drug Administration, and others. Reporting to INTERMACS is required for CMS reimbursement.
The INTERMACS database now has about 4,500 patients at 126 medical centers and is yielding useful information that is published in annual reports.22 The 2011 report focused on the experience with mechanical circulatory support as destination therapy and showed that patients who receive continuousflow pumps have significantly better survival rates than those with pulsatile-flow pumps.23 An earlier report showed that the level of illness at the time of implantation predicts survival24; this information now drives cardiologists to try to improve patient status with a temporary support device or intra-aortic balloon pump before implanting a durable device. The sickest patients (INTERMACS level 1) have the poorest outcomes, and centers now do fewer implantations in patients in this category. We have learned this important lesson from the INTERMACS registry.
The new devices have received a lot of media attention, and patient accrual has increased steadily as the devices have been approved.
On November 20, 2012, the US Food and Drug Administration approved the HeartWare Ventricular Assist System (HeartWare, Framingham, MA) for heart failure patients awaiting a transplant.
FUTURE DIRECTIONS
PROCEED II is an ongoing global clinical trial comparing the outcomes with donor hearts transported in standard cold storage to those transported in an experimental transport device that pumps the heart under physiologic conditions. If proven effective, this device could allow long-distance transport of donor hearts and expand the donor population.
A prospective, randomized study is now enrolling patients to evaluate induction therapy with rituximab (Rituxan) plus conventional immunosuppression (tacrolimus [Prograf], mycophenolate, steroid taper) vs placebo induction plus conventional immunosuppression. The study will enroll 400 patients (200 to each treatment arm) at 25 sites and will have a 36-month accrual period with 12-month follow-up (see http://clinicaltrials.gov/show/NCT01278745). The study is based on data in primates that found that eliminating B cells with an anti-CD20 drug before transplantation markedly reduced the incidence of coronary artery vasculopathy.
- Lloyd-Jones D, Adams RJ, Brown TM, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2010 update: a report from the American Heart Association. Circulation 2010; 1221:e46–e215.
- Jessup M, Abraham WT, Casey DE, et al. 2009 focused update: ACCF/AHA Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation 2009; 119:1977–2016.
- 2009 Annual Report of the U.S. Organ Procurement and Transplantation Network and the Scientific Registry of Transplant Recipients: Transplant Data 1999–2008. U.S. Department of Health and Human Services, Health Resources and Services Administration, Healthcare Systems Bureau, Division of Transplantation, Rockville, MD.
- Metra M, Ponikowski P, Dickstein K, et al; Heart Failure Association of the European Society of Cardiology. Advanced chronic heart failure: a position statement from the Study Group on Advanced Heart Failure of the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail 2007; 9:684–694.
- Hsich E, Gorodeski EZ, Starling RC, Blackstone EH, Ishwaran H, Lauer MS. Importance of treadmill exercise time as an initial prognostic screening tool in patients with systolic left ventricular dysfunction. Circulation 2009; 119:3189–3197.
- Gorodeski EZ, Chu EC, Chow CH, Levy WC, Hsich E, Starling RC. Application of the Seattle Heart Failure Model in ambulatory patients presented to an advanced heart failure therapeutics committee. Circ Heart Fail 2010; 3:706–714.
- Gorodeski EZ, Chu EC, Reese JR, Shishehbor MH, Hsich E, Starling RC. Prognosis on chronic dobutamine or milrinone infusions for stage D heart failure. Circ Heart Fail 2009; 2:320–324.
- Taylor DO, Stehlik J, Edwards LB, et al. Registry of the International Society for Heart and Lung Transplantation: Twenty-sixth official adult heart transplant report—2009. J Heart Lung Transplant 2009; 28:1007–1022.
- Stehlik J, Edwards LB, Kucheryavaya AY, et al. The registry of the International Society for Heart and Lung Transplantation: twenty-seventh official adult heart transplant report—2010. J Heart Lung Transplant 2010; 29:1089–1103.
- Ojo AO, Held PJ, Port FK, et al. Chronic renal failure after transplantation of a nonrenal organ. N Engl J Med 2003; 349:931–940.
- Kobashigawa JA, Katznelson S, Laks H, et al. Effect of pravastatin on outcomes after cardiac transplantation. N Engl J Med 1999; 340:272–277. Erratum in: N Engl J Med 1999; 340:976.
- Eisen HJ, Tuzcu EM, Dorent R, et al. Everolimus for the prevention of allograft rejection and vasculopathy in cardiactransplant recipients. N Engl J Med 2003; 349:847–858.
- Singh TP, Almond CS, Taylor DO, Graham DA. Decline in heart transplant wait list mortality in the United States following broader regional sharing of donor hearts. Circ Heart Fail 2012; 5:249–258.
- Baughman KL, Jarcho JA. Bridge to life—cardiac mechanical support. N Engl J Med 2007; 357:846–849.
- Rose EA, Gelijns AC, Moskowitz AJ, et al; Randomized Evaluation of Mechanical Assistance for the Treatment of Congestive Heart Failure (REMATCH) Study Group. Long-term use of a left ventricular assist device for end-stage heart failure. N Engl J Med 2001; 345:1435–1443.
- Miller LW, Pagani FD, Russell SD, et al; HeartMate II Clinical Investigators. Use of a continuous-flow device in patients awaiting heart transplantation. N Engl J Med 2007; 357:885–896.
- Starling RC, Naka Y, Boyle AJ, et al. Results of the post-U.S. Food and Drug Administration-approval study with a continuous flow left ventricular assist device as a bridge to heart transplantation: a prospective study using the INTERMACS (Interagency Registry for Mechanically Assisted Circulatory Support). J Am Coll Cardiol 2011; 57:1890–1898.
- Slaughter MS, Rogers JG, Milano CA, et al; HeartMate II Investigators. Advanced heart failure treated with continuous-flow left ventricular assist device. N Engl J Med 2009; 361:2241–2251.
- John R, Naka Y, Smedira NG, et al. Continuous flow left ventricular assist device outcomes in commercial use compared with the prior clinical trial. Ann Thorac Surg 2011; 92:1406–1413.
- Starling RC. Improved quantity and quality of life: a winning combination to treat advanced heart failure. J Am Coll Cardiol 2010; 55:1835–1836.
- Uriel N, Pak SW, Jorde UP, et al. Acquired von Willebrand syndrome after continuous-flow mechanical device support contributes to a high prevalence of bleeding during long-term support and at the time of transplantation. J Am Coll Cardiol 2010; 56:1207–1213.
- Kirklin JK, Naftel DC, Kormos RL, et al. The fourth INTERMACS annual report: 4,000 implants and counting. J Heart Lung Transplant 2012; 31:117–126.
- Kirklin JK, Naftel DC, Kormos RL, et al. Third INTERMACS Annual Report: the evolution of destination therapy in the United States. J Heart Lung Transplant 2011; 30:115–123.
- Kirklin JK, Naftel DC, Kormos RL, et al. Second INTERMACS annual report: more than 1,000 primary left ventricular assist device implants. J Heart Lung Transplant 2010; 29:1–10.
SUGGESTED READING
Costanzo MR, Dipchand A, Starling R, et al; International Society of Heart and Lung Transplantation Guidelines. The International Society of Heart and Lung Transplantation guidelines for the care of heart transplant recipients. J Heart Lung Transplant 2010; 29:914–956.
Mehra MR, Kobashigawa J, Starling R, et al. Listing criteria for heart transplantation: International Society for Heart and Lung Transplantation guidelines for the care of cardiac transplant candidates–2006. J Heart Lung Transplant 2006; 25:1024–1042.
Slaughter MS, Pagani FD, Rogers JG, et al; HeartMate II Clinical Investigators. Clinical management of continuous flow left ventricular assist devices in advanced heart failure. J Heart Lung Transplant 2010; 29(4 suppl):S1–S39.
Patients with advanced heart failure far outnumber the hearts available for transplantation. Partly as a consequence of this shortage, left-ventricular assist devices (LVADs) are being used more widely.
This article is an update on options for managing severe, advanced heart failure, with special attention to new developments and continuing challenges in heart transplantation and LVADs.
THE PREVALENCE OF HEART FAILURE
About 2.6% of the US population age 20 and older have heart failure—some 5.8 million people. Of these, about half have systolic heart failure.1 Patients with systolic heart failure can be classified by degree of severity under two systems:
The New York Heart Association (NYHA) classifies patients by their functional status, from I (no limitation in activities) to IV (symptoms at rest). NYHA class III (symptoms with minimal exertion) is sometimes further broken down into IIIa and IIIb, with the latter defined as having a recent history of dyspnea at rest.
The joint American College of Cardiology and American Heart Association (ACC/AHA) classification uses four stages, from A (high risk of developing heart failure, ie, having risk factors such as family history of heart disease, hypertension, or diabetes) to D (advanced heart disease despite treatment). Patients in stage D tend to be recurrently hospitalized despite cardiac resynchronization therapy and drug therapy, and they cannot be safely discharged without specialized interventions. The options for these patients are limited: either end-of-life care or extraordinary measures such as heart transplantation, long-term treatment with inotropic drugs, permanent mechanical circulatory support, or experimental therapies.2
The estimated number of people in ACC/AHA stage D or NYHA class IV is 15,600 to 156,000. The approximate number of heart transplants performed in the United States each year is 2,100.3
WHICH AMBULATORY PATIENTS ARE MOST AT RISK?
The range for the estimated number of patients with advanced heart failure (NYHA class IIIb or IV) is wide (see above) because these patients may be hard to recognize. The most debilitated patients are obvious: they tend to be in the intensive care unit with end-organ failure. However, it is a challenge to recognize patients at extremely high risk who are still ambulatory.
The European Society of Cardiology4 developed a definition of advanced chronic heart failure that can help identify patients who are candidates for the transplant list and for an LVAD. All the following features must be present despite optimal therapy that includes diuretics, inhibitors of the renin-angiotensin-aldosterone system, and beta-blockers, unless these are poorly tolerated or contraindicated, and cardiac resynchronization therapy if indicated:
- Severe symptoms, with dyspnea or fatigue at rest or with minimal exertion (NYHA class III or IV)
- Episodes of fluid retention (pulmonary or systemic congestion, peripheral edema) or of reduced cardiac output at rest (peripheral hypoperfusion)
- Objective evidence of severe cardiac dysfunction (at least one of the following): left ventricular ejection fraction less than 30%, pseudonormal or restrictive mitral inflow pattern on Doppler echocardiography, high left or right ventricular filling pressure (or both left and right filling pressures), and elevated B-type natriuretic peptides
- Severely impaired functional capacity demonstrated by one of the following: inability to exercise, 6-minute walk test distance less than 300 m (or less in women or patients who are age 75 and older), or peak oxygen intake less than 12 to 14 mL/kg/min
- One or more hospitalizations for heart failure in the past 6 months.
Treadmill exercise time is an easily performed test. Hsich et al5 found that the longer patients can walk, the lower their risk of death, and that this variable is about as predictive of survival in patients with systolic left ventricular dysfunction as peak oxygen consumption, which is much more cumbersome to measure.
The Seattle Heart Failure Model gives an estimate of prognosis for ambulatory patients with advanced heart failure. Available at http://depts.washington.edu/shfm/, it is based on age, sex, NYHA class, weight, ejection fraction, blood pressure, medications, a few laboratory values, and other clinical information. The model has been validated in numerous cohorts,6 but it may underestimate risk and is currently being tested in clinical trials (REVIVE-IT and ROADMAP; see at www.clinicaltrials.gov).
Recurrent hospitalization is a simple predictor of risk. A study of about 7,000 patients worldwide found that after hospitalization with acute decompensated heart failure, the strongest predictor of death within 6 months was readmission for any reason within 30 days of the index hospitalization (Starling RC, unpublished observation, 2011). Any patient with heart failure who is repeatedly hospitalized should have a consultation with a heart failure specialist.
INOTROPIC THERAPY FOR BRIDGING
Inotropic drugs, which include intravenous dobutamine (Dobutrex) and milrinone (Primacor), are used to help maintain end-organ function until a patient can obtain a heart transplant or LVAD.
Inotropic therapy should not be viewed as an alternative to heart transplantation or device implantation. We inform patients that inotropic therapy is purely palliative and may actually increase the risk of death, which is about 50% at 6 months and nearly 100% at 1 year. A patient on inotropic therapy who is not a candidate for a transplant or for an assist device should be referred to a hospice program.7
CARDIAC TRANSPLANTATION: SUCCESSES, CHALLENGES
Survival rates after heart transplantation are now excellent. The 1-year survival rate is about 90%, the 5-year rate is about 70%, but only about 20% survive 20 years or longer.8,9 The prognosis is not as good as for combined heart-lung transplantation patients.
Age is an important factor and is a contentious issue: some medical centers will not offer transplantation to patients over age 65. Others regard age as just another risk factor, like renal dysfunction or diabetes.
Quality of life after heart transplantation is excellent: patients are usually able to return to work, regardless of their profession.
The leading cause of death after heart transplantation is malignancy, followed by coronary artery vasculopathy, then by graft failure. Some patients develop left ventricular dysfunction and heart failure of unknown cause. Others develop antibody-mediated rejection; in recent years this has been more promptly recognized, but treatment remains a challenge.
Acute rejection, which used to be one of the main causes of death, now has an extremely low incidence because of modern drug therapies. In a US observational study currently being conducted in about 200 patients receiving a heart transplant (details on CTOT-05 at www.clinicaltrials.gov), the incidence of moderate rejection during the first year is less than 10% (Starling RC, unpublished observation). But several concerns remain.
Adverse effects of immunosuppressive drugs continue to be problematic. These include infection, malignancy, osteoporosis, chronic kidney toxicity, hypertension, and neuropathy.
Renal dysfunction is one of the largest issues. About 10% of heart transplant recipients develop stage 4 kidney disease (with a glomerular filtration rate < 30 mL/min) and need kidney transplantation or renal replacement therapy because of the use of calcineurin inhibitors for immunosuppression.10
Coronary artery vasculopathy was the largest problem when heart transplantation began and continues to be a major concern and focus of research.11,12 Case 1 (below) is an example of the problem.
Case 1: Poor outcome despite an ideal scenario
A 57-year-old businessman had dilated cardiomyopathy and progressive heart failure for 10 years. He was listed for transplantation in 2008 and was given an LVAD (HeartMate II, Thoratec Corp, Pleasanton, CA) as a bridge until a donor heart became available.
In 2009, he received a heart transplant under ideal conditions: the donor was a large 30-year-old man who died of a gunshot wound to the head in the same city in which the patient and transplant hospital were located. Cardiopulmonary resuscitation was not performed, and the cold ischemic time was just a little more than 3 hours. Immune indicators were ideal with a negative prospective cross-match.
Laboratory results after transplantation included creatinine 1.7 mg/dL (normal 0.6–1.2 mg/dL), low-density lipoprotein cholesterol 75 mg/dL, high-density lipoprotein cholesterol 64 mg/dL, and triglycerides 90 mg/dL.
The patient was given immunosuppressive therapy with cyclosporine (Neoral), mycophenolate (CellCept), and prednisone. Because his creatinine level was high, he was also perioperatively given basiliximab (Simulect), a monoclonal antibody to the alpha chain (CD25) of the interleukin-2 receptor. (In a patient who has poor renal function, basilixumab may help by enabling us to delay the use of calcineurin inhibitors.) He also received simvastatin (Zocor) 10 mg.
Per Cleveland Clinic protocol, he underwent 13 biopsy procedures during his first year, and each was normal (grade 0 or 1R). Evaluation by cardiac catheterization at 1 year showed some irregularities in the left anterior descending artery, but a stent was not deemed necessary. Also, per protocol, he underwent intravascular ultrasonography, which revealed abnormal thickness in the intima and media, indicating that coronary artery disease was developing, although it was nonobstructive.
Two months after this checkup, the patient collapsed and suddenly died while shopping. At autopsy, his left anterior descending artery was found to be severely obstructed.
Coronary artery vasculopathy is still a major problem
This case shows that coronary artery vasculopathy may develop despite an ideal transplantation scenario. It remains a large concern and a focus of research.
Coronary vasculopathy develops in 30% to 40% of heart transplant recipients within 5 years, and the incidence has not been reduced by much over the years. However, probably fewer than 5% of these patients die or even need bypass surgery or stenting, and the problem is managed the same as native atherosclerosis. We perform routine annual cardiac catheterizations or stress tests, or both, and place stents in severely blocked arteries.
THE DONOR SHORTAGE: CHANGING HOW HEARTS ARE ALLOCATED
The number of patients receiving a heart transplant in the United States—about 2,000 per year—has not increased in the past decade. The European Union also has great difficulty obtaining hearts for people in need, and almost every transplant candidate there gets mechanical support for some time. The gap between those listed for transplant and the number transplanted each year continues to widen in both the United States and Europe.
All transplant candidates are assigned a status by the United Network of Organ Sharing (UNOS) based on their medical condition. The highest status, 1A, goes to patients who are seriously ill, in the hospital, on high doses of inotropic drugs (specific dosages are defined) and mechanical circulatory support such as an LVAD, and expected to live less than 1 month without a transplant. Status 1B patients are stable on lower-dose inotropic therapy or on mechanical support, and can be in the hospital or at home. Status 2 patients are stable and ambulatory and are not on inotropic drugs.
In July 2006, UNOS changed the rules on how patients are prioritized for obtaining an organ. The new rules are based both on severity of illness (see above) and geographic proximity to the donor heart—local, within 500 miles (“zone A”) or within 500 to 1,000 miles (“zone B”). The order of priority for donor hearts is:
- Local, status 1A
- Local, status 1B
- Zone A, status 1A
- Zone A, status 1B
- Local, status 2
- Zone B, status 1A
- Zone B, status 1B
- Zone A, status 2.
As a result of the change, donor hearts that become available in a particular hospital do not necessarily go to a patient in that state. Another result is that status 2 patients, who were previously the most common transplant recipients, now have much less access because all status 1 patients within 500 miles are given higher priority. Since the change, only 8% of hearts nationwide go to status 2 patients, which is 67% fewer than before. At the same time, organs allocated to status 1A patients have increased by 26%, and their death rates have fallen.3
The new allocation system is a positive change for the sickest patients, providing quicker access and a reduction in waiting-list mortality.13 The drawback is that status 2 patients who are less ill are less likely to ever receive an organ until their condition worsens.
Heart transplant outcomes are publicly reported
The Scientific Registry of Transplant Recipients publicly reports heart transplant outcomes (www.srtr.org). For any transplant center, the public can learn the number of patients waiting for a transplant, the death rate on the waiting list, the number of transplants performed in the previous 12 months, the waiting time in months, and observed and risk-adjusted expected survival rates. A center that deviates from the expected survival rates by 10% or more may be audited and could lose its certification.
Also listed on the Web site is the percentage of patients who receive a support device before receiving a transplant. This can vary widely between institutions and may reflect the organ availability at the transplant center (waiting times) or the preferences and expertise of the transplantation team. We believe that the mortality rate on the waiting list will be reduced with appropriate use of LVADs as a bridge to transplantation when indicated. We have now transitioned to the use of the improved continuous-flow LVADs and rarely maintain patients on continuous inotropic therapy at home to await a donor organ.
MECHANICAL CIRCULATORY SUPPORT: BRIDGE OR DESTINATION?
Mechanical circulatory support devices are increasingly being used to sustain patients with advanced heart failure. Currently at Cleveland Clinic, more LVADs are implanted than hearts are transplanted.
Mechanical circulatory support is indicated for patients who are listed for transplant to keep them functioning as well as possible while they are waiting (bridge to transplant). For others it is “destination therapy”: they are not candidates for a transplant, but a device may improve and prolong the rest of their life.
Case 2: A good outcome despite a poor prognosis
A 71-year-old man was rejected for transplantation by his local hospital because of his age and also because he had pulmonary artery hypertension (78/42 mm Hg; reference range 15–30/5–15 mm Hg) and creatinine elevation (3.0 mg/dL; reference range 0.6–1.5 mg/dL). Nevertheless, he did well on a mechanical device and was accepted for transplantation by Cleveland Clinic. He received a transplant and is still alive and active 14 years later.
Comment. Determining that a patient is not a good transplantation candidate is often impossible. Putting the patient on mechanical support for a period of time can often help clarify whether transplantation is advisable. Probably most patients who receive mechanical support do so as a bridge to decision: most are acutely ill and many have organ dysfunction, pulmonary hypertension, and renal insufficiency. After a period of support, they can be assessed for suitability for transplantation.
LVADs continue to improve
Many devices are available for mechanical circulatory support.14 In addition to LVADs, there are right-ventricular assist devices (RVADs), and devices that simultaneously support both ventricles (BiVADs). Total artificial hearts are also available, as are acute temporary percutaneous devices. These temporary devices—TandemHeart (CardiacAssist, Pittsburgh, PA) and Impella (Abiomed, Danvers, MD)—can be used before a long-term mechanical device can be surgically implanted.
LVADs are of three types:
- Pulsatile volume-displacement pumps, which mimic the pumping action of the natural heart. These early devices were large and placed in the abdomen.
- Continuous axial-flow pumps, which do not have a “heartbeat.” These are quieter and lighter than the early pumps, and use a turbine that spins at 8,000 to 10,000 rpm.
- Continuous centrifugal-flow pumps. These have a rotor spinning at 2,000 to 3,000 rpm, and most of them are magnetically powered and suspended.
The superiority of LVADs over medical therapy was clearly shown even in early studies that used pulsatile LVADs.15 The results of such studies and the increased durability of the devices have led to their rapidly expanded use.
The newer continuous-flow pumps offer significant improvements over the old pulsatile-flow pumps, being smaller, lighter, quieter, and more durable (Table 1). A 2007 study of 133 patients on a continuous axial-flow LVAD (HeartMate II) found that 76% were still alive after 6 months, and patients had significant improvement in functional status and quality of life.16 In a postapproval study based on registry data, HeartMate II was found superior to pulsatile pumps in terms of survival up to 12 months, percentage of patients reaching transplant, and cardiac recovery. Adverse event rates were similar or lower for HeartMate II.17
Another study compared a continuousflow with a pulsatile-flow LVAD for patients who were ineligible for transplantation. Survival at 2 years was 58% with the continuousflow device vs 24% with the pulsatile-flow device (P = .008).18 Since then, postmarket data of patients who received an LVAD showed that 85% are still alive at 1 year.19 This study can be viewed as supporting the use of LVADs as destination therapy.
Quality of life for patients receiving an LVAD has been excellent. When biventricular pacemakers for resynchronization therapy first became available, distances on the 6-minute walk test improved by 39 m, which was deemed a big improvement. LVAD devices have increased the 6-minute walk distance by 156 m.20
Adverse events with LVADs have improved, but continue to be of concern
Infections can arise in the blood stream, in the device pocket, or especially where the driveline exits the skin. As devices have become smaller, driveline diameters have become smaller as well, allowing for a better seal at the skin and making this less of a problem. Some centers report the incidence of driveline infections as less than 20%, but they continue to be a focus of concern.18
Stroke rates continue to improve, although patients still require intensive lifelong anticoagulation. The target international normalized ratio varies by device manufacturer, ranging from 1.7 to 2.5.
Bleeding. Acquired von Willebrand syndrome can develop in patients who have an LVAD, with the gastrointestinal system being the most frequent site of bleeding.21
Device thrombosis occurs very rarely (2%–3%) but is very serious and may require pump exchange.
Mechanical malfunction. As duration of therapy lengthens, problems are arising with aging devices, such as broken wires or short circuits. New-generation pumps have markedly improved durability and reliability.
Good data are kept on device outcomes
The Interagency for Mechanically Assisted Circulatory Support (INTERMACS) maintains a national registry of patients with a mechanical circulatory support device to treat advanced heart failure. It was jointly established in 2006 by the National Heart, Lung, and Blood Institute, Centers for Medicare and Medicaid Services (CMS), the US Food and Drug Administration, and others. Reporting to INTERMACS is required for CMS reimbursement.
The INTERMACS database now has about 4,500 patients at 126 medical centers and is yielding useful information that is published in annual reports.22 The 2011 report focused on the experience with mechanical circulatory support as destination therapy and showed that patients who receive continuousflow pumps have significantly better survival rates than those with pulsatile-flow pumps.23 An earlier report showed that the level of illness at the time of implantation predicts survival24; this information now drives cardiologists to try to improve patient status with a temporary support device or intra-aortic balloon pump before implanting a durable device. The sickest patients (INTERMACS level 1) have the poorest outcomes, and centers now do fewer implantations in patients in this category. We have learned this important lesson from the INTERMACS registry.
The new devices have received a lot of media attention, and patient accrual has increased steadily as the devices have been approved.
On November 20, 2012, the US Food and Drug Administration approved the HeartWare Ventricular Assist System (HeartWare, Framingham, MA) for heart failure patients awaiting a transplant.
FUTURE DIRECTIONS
PROCEED II is an ongoing global clinical trial comparing the outcomes with donor hearts transported in standard cold storage to those transported in an experimental transport device that pumps the heart under physiologic conditions. If proven effective, this device could allow long-distance transport of donor hearts and expand the donor population.
A prospective, randomized study is now enrolling patients to evaluate induction therapy with rituximab (Rituxan) plus conventional immunosuppression (tacrolimus [Prograf], mycophenolate, steroid taper) vs placebo induction plus conventional immunosuppression. The study will enroll 400 patients (200 to each treatment arm) at 25 sites and will have a 36-month accrual period with 12-month follow-up (see http://clinicaltrials.gov/show/NCT01278745). The study is based on data in primates that found that eliminating B cells with an anti-CD20 drug before transplantation markedly reduced the incidence of coronary artery vasculopathy.
Patients with advanced heart failure far outnumber the hearts available for transplantation. Partly as a consequence of this shortage, left-ventricular assist devices (LVADs) are being used more widely.
This article is an update on options for managing severe, advanced heart failure, with special attention to new developments and continuing challenges in heart transplantation and LVADs.
THE PREVALENCE OF HEART FAILURE
About 2.6% of the US population age 20 and older have heart failure—some 5.8 million people. Of these, about half have systolic heart failure.1 Patients with systolic heart failure can be classified by degree of severity under two systems:
The New York Heart Association (NYHA) classifies patients by their functional status, from I (no limitation in activities) to IV (symptoms at rest). NYHA class III (symptoms with minimal exertion) is sometimes further broken down into IIIa and IIIb, with the latter defined as having a recent history of dyspnea at rest.
The joint American College of Cardiology and American Heart Association (ACC/AHA) classification uses four stages, from A (high risk of developing heart failure, ie, having risk factors such as family history of heart disease, hypertension, or diabetes) to D (advanced heart disease despite treatment). Patients in stage D tend to be recurrently hospitalized despite cardiac resynchronization therapy and drug therapy, and they cannot be safely discharged without specialized interventions. The options for these patients are limited: either end-of-life care or extraordinary measures such as heart transplantation, long-term treatment with inotropic drugs, permanent mechanical circulatory support, or experimental therapies.2
The estimated number of people in ACC/AHA stage D or NYHA class IV is 15,600 to 156,000. The approximate number of heart transplants performed in the United States each year is 2,100.3
WHICH AMBULATORY PATIENTS ARE MOST AT RISK?
The range for the estimated number of patients with advanced heart failure (NYHA class IIIb or IV) is wide (see above) because these patients may be hard to recognize. The most debilitated patients are obvious: they tend to be in the intensive care unit with end-organ failure. However, it is a challenge to recognize patients at extremely high risk who are still ambulatory.
The European Society of Cardiology4 developed a definition of advanced chronic heart failure that can help identify patients who are candidates for the transplant list and for an LVAD. All the following features must be present despite optimal therapy that includes diuretics, inhibitors of the renin-angiotensin-aldosterone system, and beta-blockers, unless these are poorly tolerated or contraindicated, and cardiac resynchronization therapy if indicated:
- Severe symptoms, with dyspnea or fatigue at rest or with minimal exertion (NYHA class III or IV)
- Episodes of fluid retention (pulmonary or systemic congestion, peripheral edema) or of reduced cardiac output at rest (peripheral hypoperfusion)
- Objective evidence of severe cardiac dysfunction (at least one of the following): left ventricular ejection fraction less than 30%, pseudonormal or restrictive mitral inflow pattern on Doppler echocardiography, high left or right ventricular filling pressure (or both left and right filling pressures), and elevated B-type natriuretic peptides
- Severely impaired functional capacity demonstrated by one of the following: inability to exercise, 6-minute walk test distance less than 300 m (or less in women or patients who are age 75 and older), or peak oxygen intake less than 12 to 14 mL/kg/min
- One or more hospitalizations for heart failure in the past 6 months.
Treadmill exercise time is an easily performed test. Hsich et al5 found that the longer patients can walk, the lower their risk of death, and that this variable is about as predictive of survival in patients with systolic left ventricular dysfunction as peak oxygen consumption, which is much more cumbersome to measure.
The Seattle Heart Failure Model gives an estimate of prognosis for ambulatory patients with advanced heart failure. Available at http://depts.washington.edu/shfm/, it is based on age, sex, NYHA class, weight, ejection fraction, blood pressure, medications, a few laboratory values, and other clinical information. The model has been validated in numerous cohorts,6 but it may underestimate risk and is currently being tested in clinical trials (REVIVE-IT and ROADMAP; see at www.clinicaltrials.gov).
Recurrent hospitalization is a simple predictor of risk. A study of about 7,000 patients worldwide found that after hospitalization with acute decompensated heart failure, the strongest predictor of death within 6 months was readmission for any reason within 30 days of the index hospitalization (Starling RC, unpublished observation, 2011). Any patient with heart failure who is repeatedly hospitalized should have a consultation with a heart failure specialist.
INOTROPIC THERAPY FOR BRIDGING
Inotropic drugs, which include intravenous dobutamine (Dobutrex) and milrinone (Primacor), are used to help maintain end-organ function until a patient can obtain a heart transplant or LVAD.
Inotropic therapy should not be viewed as an alternative to heart transplantation or device implantation. We inform patients that inotropic therapy is purely palliative and may actually increase the risk of death, which is about 50% at 6 months and nearly 100% at 1 year. A patient on inotropic therapy who is not a candidate for a transplant or for an assist device should be referred to a hospice program.7
CARDIAC TRANSPLANTATION: SUCCESSES, CHALLENGES
Survival rates after heart transplantation are now excellent. The 1-year survival rate is about 90%, the 5-year rate is about 70%, but only about 20% survive 20 years or longer.8,9 The prognosis is not as good as for combined heart-lung transplantation patients.
Age is an important factor and is a contentious issue: some medical centers will not offer transplantation to patients over age 65. Others regard age as just another risk factor, like renal dysfunction or diabetes.
Quality of life after heart transplantation is excellent: patients are usually able to return to work, regardless of their profession.
The leading cause of death after heart transplantation is malignancy, followed by coronary artery vasculopathy, then by graft failure. Some patients develop left ventricular dysfunction and heart failure of unknown cause. Others develop antibody-mediated rejection; in recent years this has been more promptly recognized, but treatment remains a challenge.
Acute rejection, which used to be one of the main causes of death, now has an extremely low incidence because of modern drug therapies. In a US observational study currently being conducted in about 200 patients receiving a heart transplant (details on CTOT-05 at www.clinicaltrials.gov), the incidence of moderate rejection during the first year is less than 10% (Starling RC, unpublished observation). But several concerns remain.
Adverse effects of immunosuppressive drugs continue to be problematic. These include infection, malignancy, osteoporosis, chronic kidney toxicity, hypertension, and neuropathy.
Renal dysfunction is one of the largest issues. About 10% of heart transplant recipients develop stage 4 kidney disease (with a glomerular filtration rate < 30 mL/min) and need kidney transplantation or renal replacement therapy because of the use of calcineurin inhibitors for immunosuppression.10
Coronary artery vasculopathy was the largest problem when heart transplantation began and continues to be a major concern and focus of research.11,12 Case 1 (below) is an example of the problem.
Case 1: Poor outcome despite an ideal scenario
A 57-year-old businessman had dilated cardiomyopathy and progressive heart failure for 10 years. He was listed for transplantation in 2008 and was given an LVAD (HeartMate II, Thoratec Corp, Pleasanton, CA) as a bridge until a donor heart became available.
In 2009, he received a heart transplant under ideal conditions: the donor was a large 30-year-old man who died of a gunshot wound to the head in the same city in which the patient and transplant hospital were located. Cardiopulmonary resuscitation was not performed, and the cold ischemic time was just a little more than 3 hours. Immune indicators were ideal with a negative prospective cross-match.
Laboratory results after transplantation included creatinine 1.7 mg/dL (normal 0.6–1.2 mg/dL), low-density lipoprotein cholesterol 75 mg/dL, high-density lipoprotein cholesterol 64 mg/dL, and triglycerides 90 mg/dL.
The patient was given immunosuppressive therapy with cyclosporine (Neoral), mycophenolate (CellCept), and prednisone. Because his creatinine level was high, he was also perioperatively given basiliximab (Simulect), a monoclonal antibody to the alpha chain (CD25) of the interleukin-2 receptor. (In a patient who has poor renal function, basilixumab may help by enabling us to delay the use of calcineurin inhibitors.) He also received simvastatin (Zocor) 10 mg.
Per Cleveland Clinic protocol, he underwent 13 biopsy procedures during his first year, and each was normal (grade 0 or 1R). Evaluation by cardiac catheterization at 1 year showed some irregularities in the left anterior descending artery, but a stent was not deemed necessary. Also, per protocol, he underwent intravascular ultrasonography, which revealed abnormal thickness in the intima and media, indicating that coronary artery disease was developing, although it was nonobstructive.
Two months after this checkup, the patient collapsed and suddenly died while shopping. At autopsy, his left anterior descending artery was found to be severely obstructed.
Coronary artery vasculopathy is still a major problem
This case shows that coronary artery vasculopathy may develop despite an ideal transplantation scenario. It remains a large concern and a focus of research.
Coronary vasculopathy develops in 30% to 40% of heart transplant recipients within 5 years, and the incidence has not been reduced by much over the years. However, probably fewer than 5% of these patients die or even need bypass surgery or stenting, and the problem is managed the same as native atherosclerosis. We perform routine annual cardiac catheterizations or stress tests, or both, and place stents in severely blocked arteries.
THE DONOR SHORTAGE: CHANGING HOW HEARTS ARE ALLOCATED
The number of patients receiving a heart transplant in the United States—about 2,000 per year—has not increased in the past decade. The European Union also has great difficulty obtaining hearts for people in need, and almost every transplant candidate there gets mechanical support for some time. The gap between those listed for transplant and the number transplanted each year continues to widen in both the United States and Europe.
All transplant candidates are assigned a status by the United Network of Organ Sharing (UNOS) based on their medical condition. The highest status, 1A, goes to patients who are seriously ill, in the hospital, on high doses of inotropic drugs (specific dosages are defined) and mechanical circulatory support such as an LVAD, and expected to live less than 1 month without a transplant. Status 1B patients are stable on lower-dose inotropic therapy or on mechanical support, and can be in the hospital or at home. Status 2 patients are stable and ambulatory and are not on inotropic drugs.
In July 2006, UNOS changed the rules on how patients are prioritized for obtaining an organ. The new rules are based both on severity of illness (see above) and geographic proximity to the donor heart—local, within 500 miles (“zone A”) or within 500 to 1,000 miles (“zone B”). The order of priority for donor hearts is:
- Local, status 1A
- Local, status 1B
- Zone A, status 1A
- Zone A, status 1B
- Local, status 2
- Zone B, status 1A
- Zone B, status 1B
- Zone A, status 2.
As a result of the change, donor hearts that become available in a particular hospital do not necessarily go to a patient in that state. Another result is that status 2 patients, who were previously the most common transplant recipients, now have much less access because all status 1 patients within 500 miles are given higher priority. Since the change, only 8% of hearts nationwide go to status 2 patients, which is 67% fewer than before. At the same time, organs allocated to status 1A patients have increased by 26%, and their death rates have fallen.3
The new allocation system is a positive change for the sickest patients, providing quicker access and a reduction in waiting-list mortality.13 The drawback is that status 2 patients who are less ill are less likely to ever receive an organ until their condition worsens.
Heart transplant outcomes are publicly reported
The Scientific Registry of Transplant Recipients publicly reports heart transplant outcomes (www.srtr.org). For any transplant center, the public can learn the number of patients waiting for a transplant, the death rate on the waiting list, the number of transplants performed in the previous 12 months, the waiting time in months, and observed and risk-adjusted expected survival rates. A center that deviates from the expected survival rates by 10% or more may be audited and could lose its certification.
Also listed on the Web site is the percentage of patients who receive a support device before receiving a transplant. This can vary widely between institutions and may reflect the organ availability at the transplant center (waiting times) or the preferences and expertise of the transplantation team. We believe that the mortality rate on the waiting list will be reduced with appropriate use of LVADs as a bridge to transplantation when indicated. We have now transitioned to the use of the improved continuous-flow LVADs and rarely maintain patients on continuous inotropic therapy at home to await a donor organ.
MECHANICAL CIRCULATORY SUPPORT: BRIDGE OR DESTINATION?
Mechanical circulatory support devices are increasingly being used to sustain patients with advanced heart failure. Currently at Cleveland Clinic, more LVADs are implanted than hearts are transplanted.
Mechanical circulatory support is indicated for patients who are listed for transplant to keep them functioning as well as possible while they are waiting (bridge to transplant). For others it is “destination therapy”: they are not candidates for a transplant, but a device may improve and prolong the rest of their life.
Case 2: A good outcome despite a poor prognosis
A 71-year-old man was rejected for transplantation by his local hospital because of his age and also because he had pulmonary artery hypertension (78/42 mm Hg; reference range 15–30/5–15 mm Hg) and creatinine elevation (3.0 mg/dL; reference range 0.6–1.5 mg/dL). Nevertheless, he did well on a mechanical device and was accepted for transplantation by Cleveland Clinic. He received a transplant and is still alive and active 14 years later.
Comment. Determining that a patient is not a good transplantation candidate is often impossible. Putting the patient on mechanical support for a period of time can often help clarify whether transplantation is advisable. Probably most patients who receive mechanical support do so as a bridge to decision: most are acutely ill and many have organ dysfunction, pulmonary hypertension, and renal insufficiency. After a period of support, they can be assessed for suitability for transplantation.
LVADs continue to improve
Many devices are available for mechanical circulatory support.14 In addition to LVADs, there are right-ventricular assist devices (RVADs), and devices that simultaneously support both ventricles (BiVADs). Total artificial hearts are also available, as are acute temporary percutaneous devices. These temporary devices—TandemHeart (CardiacAssist, Pittsburgh, PA) and Impella (Abiomed, Danvers, MD)—can be used before a long-term mechanical device can be surgically implanted.
LVADs are of three types:
- Pulsatile volume-displacement pumps, which mimic the pumping action of the natural heart. These early devices were large and placed in the abdomen.
- Continuous axial-flow pumps, which do not have a “heartbeat.” These are quieter and lighter than the early pumps, and use a turbine that spins at 8,000 to 10,000 rpm.
- Continuous centrifugal-flow pumps. These have a rotor spinning at 2,000 to 3,000 rpm, and most of them are magnetically powered and suspended.
The superiority of LVADs over medical therapy was clearly shown even in early studies that used pulsatile LVADs.15 The results of such studies and the increased durability of the devices have led to their rapidly expanded use.
The newer continuous-flow pumps offer significant improvements over the old pulsatile-flow pumps, being smaller, lighter, quieter, and more durable (Table 1). A 2007 study of 133 patients on a continuous axial-flow LVAD (HeartMate II) found that 76% were still alive after 6 months, and patients had significant improvement in functional status and quality of life.16 In a postapproval study based on registry data, HeartMate II was found superior to pulsatile pumps in terms of survival up to 12 months, percentage of patients reaching transplant, and cardiac recovery. Adverse event rates were similar or lower for HeartMate II.17
Another study compared a continuousflow with a pulsatile-flow LVAD for patients who were ineligible for transplantation. Survival at 2 years was 58% with the continuousflow device vs 24% with the pulsatile-flow device (P = .008).18 Since then, postmarket data of patients who received an LVAD showed that 85% are still alive at 1 year.19 This study can be viewed as supporting the use of LVADs as destination therapy.
Quality of life for patients receiving an LVAD has been excellent. When biventricular pacemakers for resynchronization therapy first became available, distances on the 6-minute walk test improved by 39 m, which was deemed a big improvement. LVAD devices have increased the 6-minute walk distance by 156 m.20
Adverse events with LVADs have improved, but continue to be of concern
Infections can arise in the blood stream, in the device pocket, or especially where the driveline exits the skin. As devices have become smaller, driveline diameters have become smaller as well, allowing for a better seal at the skin and making this less of a problem. Some centers report the incidence of driveline infections as less than 20%, but they continue to be a focus of concern.18
Stroke rates continue to improve, although patients still require intensive lifelong anticoagulation. The target international normalized ratio varies by device manufacturer, ranging from 1.7 to 2.5.
Bleeding. Acquired von Willebrand syndrome can develop in patients who have an LVAD, with the gastrointestinal system being the most frequent site of bleeding.21
Device thrombosis occurs very rarely (2%–3%) but is very serious and may require pump exchange.
Mechanical malfunction. As duration of therapy lengthens, problems are arising with aging devices, such as broken wires or short circuits. New-generation pumps have markedly improved durability and reliability.
Good data are kept on device outcomes
The Interagency for Mechanically Assisted Circulatory Support (INTERMACS) maintains a national registry of patients with a mechanical circulatory support device to treat advanced heart failure. It was jointly established in 2006 by the National Heart, Lung, and Blood Institute, Centers for Medicare and Medicaid Services (CMS), the US Food and Drug Administration, and others. Reporting to INTERMACS is required for CMS reimbursement.
The INTERMACS database now has about 4,500 patients at 126 medical centers and is yielding useful information that is published in annual reports.22 The 2011 report focused on the experience with mechanical circulatory support as destination therapy and showed that patients who receive continuousflow pumps have significantly better survival rates than those with pulsatile-flow pumps.23 An earlier report showed that the level of illness at the time of implantation predicts survival24; this information now drives cardiologists to try to improve patient status with a temporary support device or intra-aortic balloon pump before implanting a durable device. The sickest patients (INTERMACS level 1) have the poorest outcomes, and centers now do fewer implantations in patients in this category. We have learned this important lesson from the INTERMACS registry.
The new devices have received a lot of media attention, and patient accrual has increased steadily as the devices have been approved.
On November 20, 2012, the US Food and Drug Administration approved the HeartWare Ventricular Assist System (HeartWare, Framingham, MA) for heart failure patients awaiting a transplant.
FUTURE DIRECTIONS
PROCEED II is an ongoing global clinical trial comparing the outcomes with donor hearts transported in standard cold storage to those transported in an experimental transport device that pumps the heart under physiologic conditions. If proven effective, this device could allow long-distance transport of donor hearts and expand the donor population.
A prospective, randomized study is now enrolling patients to evaluate induction therapy with rituximab (Rituxan) plus conventional immunosuppression (tacrolimus [Prograf], mycophenolate, steroid taper) vs placebo induction plus conventional immunosuppression. The study will enroll 400 patients (200 to each treatment arm) at 25 sites and will have a 36-month accrual period with 12-month follow-up (see http://clinicaltrials.gov/show/NCT01278745). The study is based on data in primates that found that eliminating B cells with an anti-CD20 drug before transplantation markedly reduced the incidence of coronary artery vasculopathy.
- Lloyd-Jones D, Adams RJ, Brown TM, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2010 update: a report from the American Heart Association. Circulation 2010; 1221:e46–e215.
- Jessup M, Abraham WT, Casey DE, et al. 2009 focused update: ACCF/AHA Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation 2009; 119:1977–2016.
- 2009 Annual Report of the U.S. Organ Procurement and Transplantation Network and the Scientific Registry of Transplant Recipients: Transplant Data 1999–2008. U.S. Department of Health and Human Services, Health Resources and Services Administration, Healthcare Systems Bureau, Division of Transplantation, Rockville, MD.
- Metra M, Ponikowski P, Dickstein K, et al; Heart Failure Association of the European Society of Cardiology. Advanced chronic heart failure: a position statement from the Study Group on Advanced Heart Failure of the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail 2007; 9:684–694.
- Hsich E, Gorodeski EZ, Starling RC, Blackstone EH, Ishwaran H, Lauer MS. Importance of treadmill exercise time as an initial prognostic screening tool in patients with systolic left ventricular dysfunction. Circulation 2009; 119:3189–3197.
- Gorodeski EZ, Chu EC, Chow CH, Levy WC, Hsich E, Starling RC. Application of the Seattle Heart Failure Model in ambulatory patients presented to an advanced heart failure therapeutics committee. Circ Heart Fail 2010; 3:706–714.
- Gorodeski EZ, Chu EC, Reese JR, Shishehbor MH, Hsich E, Starling RC. Prognosis on chronic dobutamine or milrinone infusions for stage D heart failure. Circ Heart Fail 2009; 2:320–324.
- Taylor DO, Stehlik J, Edwards LB, et al. Registry of the International Society for Heart and Lung Transplantation: Twenty-sixth official adult heart transplant report—2009. J Heart Lung Transplant 2009; 28:1007–1022.
- Stehlik J, Edwards LB, Kucheryavaya AY, et al. The registry of the International Society for Heart and Lung Transplantation: twenty-seventh official adult heart transplant report—2010. J Heart Lung Transplant 2010; 29:1089–1103.
- Ojo AO, Held PJ, Port FK, et al. Chronic renal failure after transplantation of a nonrenal organ. N Engl J Med 2003; 349:931–940.
- Kobashigawa JA, Katznelson S, Laks H, et al. Effect of pravastatin on outcomes after cardiac transplantation. N Engl J Med 1999; 340:272–277. Erratum in: N Engl J Med 1999; 340:976.
- Eisen HJ, Tuzcu EM, Dorent R, et al. Everolimus for the prevention of allograft rejection and vasculopathy in cardiactransplant recipients. N Engl J Med 2003; 349:847–858.
- Singh TP, Almond CS, Taylor DO, Graham DA. Decline in heart transplant wait list mortality in the United States following broader regional sharing of donor hearts. Circ Heart Fail 2012; 5:249–258.
- Baughman KL, Jarcho JA. Bridge to life—cardiac mechanical support. N Engl J Med 2007; 357:846–849.
- Rose EA, Gelijns AC, Moskowitz AJ, et al; Randomized Evaluation of Mechanical Assistance for the Treatment of Congestive Heart Failure (REMATCH) Study Group. Long-term use of a left ventricular assist device for end-stage heart failure. N Engl J Med 2001; 345:1435–1443.
- Miller LW, Pagani FD, Russell SD, et al; HeartMate II Clinical Investigators. Use of a continuous-flow device in patients awaiting heart transplantation. N Engl J Med 2007; 357:885–896.
- Starling RC, Naka Y, Boyle AJ, et al. Results of the post-U.S. Food and Drug Administration-approval study with a continuous flow left ventricular assist device as a bridge to heart transplantation: a prospective study using the INTERMACS (Interagency Registry for Mechanically Assisted Circulatory Support). J Am Coll Cardiol 2011; 57:1890–1898.
- Slaughter MS, Rogers JG, Milano CA, et al; HeartMate II Investigators. Advanced heart failure treated with continuous-flow left ventricular assist device. N Engl J Med 2009; 361:2241–2251.
- John R, Naka Y, Smedira NG, et al. Continuous flow left ventricular assist device outcomes in commercial use compared with the prior clinical trial. Ann Thorac Surg 2011; 92:1406–1413.
- Starling RC. Improved quantity and quality of life: a winning combination to treat advanced heart failure. J Am Coll Cardiol 2010; 55:1835–1836.
- Uriel N, Pak SW, Jorde UP, et al. Acquired von Willebrand syndrome after continuous-flow mechanical device support contributes to a high prevalence of bleeding during long-term support and at the time of transplantation. J Am Coll Cardiol 2010; 56:1207–1213.
- Kirklin JK, Naftel DC, Kormos RL, et al. The fourth INTERMACS annual report: 4,000 implants and counting. J Heart Lung Transplant 2012; 31:117–126.
- Kirklin JK, Naftel DC, Kormos RL, et al. Third INTERMACS Annual Report: the evolution of destination therapy in the United States. J Heart Lung Transplant 2011; 30:115–123.
- Kirklin JK, Naftel DC, Kormos RL, et al. Second INTERMACS annual report: more than 1,000 primary left ventricular assist device implants. J Heart Lung Transplant 2010; 29:1–10.
SUGGESTED READING
Costanzo MR, Dipchand A, Starling R, et al; International Society of Heart and Lung Transplantation Guidelines. The International Society of Heart and Lung Transplantation guidelines for the care of heart transplant recipients. J Heart Lung Transplant 2010; 29:914–956.
Mehra MR, Kobashigawa J, Starling R, et al. Listing criteria for heart transplantation: International Society for Heart and Lung Transplantation guidelines for the care of cardiac transplant candidates–2006. J Heart Lung Transplant 2006; 25:1024–1042.
Slaughter MS, Pagani FD, Rogers JG, et al; HeartMate II Clinical Investigators. Clinical management of continuous flow left ventricular assist devices in advanced heart failure. J Heart Lung Transplant 2010; 29(4 suppl):S1–S39.
- Lloyd-Jones D, Adams RJ, Brown TM, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2010 update: a report from the American Heart Association. Circulation 2010; 1221:e46–e215.
- Jessup M, Abraham WT, Casey DE, et al. 2009 focused update: ACCF/AHA Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation 2009; 119:1977–2016.
- 2009 Annual Report of the U.S. Organ Procurement and Transplantation Network and the Scientific Registry of Transplant Recipients: Transplant Data 1999–2008. U.S. Department of Health and Human Services, Health Resources and Services Administration, Healthcare Systems Bureau, Division of Transplantation, Rockville, MD.
- Metra M, Ponikowski P, Dickstein K, et al; Heart Failure Association of the European Society of Cardiology. Advanced chronic heart failure: a position statement from the Study Group on Advanced Heart Failure of the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail 2007; 9:684–694.
- Hsich E, Gorodeski EZ, Starling RC, Blackstone EH, Ishwaran H, Lauer MS. Importance of treadmill exercise time as an initial prognostic screening tool in patients with systolic left ventricular dysfunction. Circulation 2009; 119:3189–3197.
- Gorodeski EZ, Chu EC, Chow CH, Levy WC, Hsich E, Starling RC. Application of the Seattle Heart Failure Model in ambulatory patients presented to an advanced heart failure therapeutics committee. Circ Heart Fail 2010; 3:706–714.
- Gorodeski EZ, Chu EC, Reese JR, Shishehbor MH, Hsich E, Starling RC. Prognosis on chronic dobutamine or milrinone infusions for stage D heart failure. Circ Heart Fail 2009; 2:320–324.
- Taylor DO, Stehlik J, Edwards LB, et al. Registry of the International Society for Heart and Lung Transplantation: Twenty-sixth official adult heart transplant report—2009. J Heart Lung Transplant 2009; 28:1007–1022.
- Stehlik J, Edwards LB, Kucheryavaya AY, et al. The registry of the International Society for Heart and Lung Transplantation: twenty-seventh official adult heart transplant report—2010. J Heart Lung Transplant 2010; 29:1089–1103.
- Ojo AO, Held PJ, Port FK, et al. Chronic renal failure after transplantation of a nonrenal organ. N Engl J Med 2003; 349:931–940.
- Kobashigawa JA, Katznelson S, Laks H, et al. Effect of pravastatin on outcomes after cardiac transplantation. N Engl J Med 1999; 340:272–277. Erratum in: N Engl J Med 1999; 340:976.
- Eisen HJ, Tuzcu EM, Dorent R, et al. Everolimus for the prevention of allograft rejection and vasculopathy in cardiactransplant recipients. N Engl J Med 2003; 349:847–858.
- Singh TP, Almond CS, Taylor DO, Graham DA. Decline in heart transplant wait list mortality in the United States following broader regional sharing of donor hearts. Circ Heart Fail 2012; 5:249–258.
- Baughman KL, Jarcho JA. Bridge to life—cardiac mechanical support. N Engl J Med 2007; 357:846–849.
- Rose EA, Gelijns AC, Moskowitz AJ, et al; Randomized Evaluation of Mechanical Assistance for the Treatment of Congestive Heart Failure (REMATCH) Study Group. Long-term use of a left ventricular assist device for end-stage heart failure. N Engl J Med 2001; 345:1435–1443.
- Miller LW, Pagani FD, Russell SD, et al; HeartMate II Clinical Investigators. Use of a continuous-flow device in patients awaiting heart transplantation. N Engl J Med 2007; 357:885–896.
- Starling RC, Naka Y, Boyle AJ, et al. Results of the post-U.S. Food and Drug Administration-approval study with a continuous flow left ventricular assist device as a bridge to heart transplantation: a prospective study using the INTERMACS (Interagency Registry for Mechanically Assisted Circulatory Support). J Am Coll Cardiol 2011; 57:1890–1898.
- Slaughter MS, Rogers JG, Milano CA, et al; HeartMate II Investigators. Advanced heart failure treated with continuous-flow left ventricular assist device. N Engl J Med 2009; 361:2241–2251.
- John R, Naka Y, Smedira NG, et al. Continuous flow left ventricular assist device outcomes in commercial use compared with the prior clinical trial. Ann Thorac Surg 2011; 92:1406–1413.
- Starling RC. Improved quantity and quality of life: a winning combination to treat advanced heart failure. J Am Coll Cardiol 2010; 55:1835–1836.
- Uriel N, Pak SW, Jorde UP, et al. Acquired von Willebrand syndrome after continuous-flow mechanical device support contributes to a high prevalence of bleeding during long-term support and at the time of transplantation. J Am Coll Cardiol 2010; 56:1207–1213.
- Kirklin JK, Naftel DC, Kormos RL, et al. The fourth INTERMACS annual report: 4,000 implants and counting. J Heart Lung Transplant 2012; 31:117–126.
- Kirklin JK, Naftel DC, Kormos RL, et al. Third INTERMACS Annual Report: the evolution of destination therapy in the United States. J Heart Lung Transplant 2011; 30:115–123.
- Kirklin JK, Naftel DC, Kormos RL, et al. Second INTERMACS annual report: more than 1,000 primary left ventricular assist device implants. J Heart Lung Transplant 2010; 29:1–10.
SUGGESTED READING
Costanzo MR, Dipchand A, Starling R, et al; International Society of Heart and Lung Transplantation Guidelines. The International Society of Heart and Lung Transplantation guidelines for the care of heart transplant recipients. J Heart Lung Transplant 2010; 29:914–956.
Mehra MR, Kobashigawa J, Starling R, et al. Listing criteria for heart transplantation: International Society for Heart and Lung Transplantation guidelines for the care of cardiac transplant candidates–2006. J Heart Lung Transplant 2006; 25:1024–1042.
Slaughter MS, Pagani FD, Rogers JG, et al; HeartMate II Clinical Investigators. Clinical management of continuous flow left ventricular assist devices in advanced heart failure. J Heart Lung Transplant 2010; 29(4 suppl):S1–S39.
KEY POINTS
- After heart transplantation, survival rates are high and quality of life is excellent, although coronary artery disease, renal dysfunction, and the need for immunosuppressive drugs are ongoing challenges.
- Changes in donor heart allocation made in 2006 more strongly favor the sickest patients and have reduced the rate of mortality on the waiting list.
- Continuous-flow left-ventricular assist devices offer many advantages over the older pulsatile-flow devices, including improved outcomes, smaller size, less noise, and greater durability.
- Inotropic therapy is purely palliative and should not be viewed as an alternative to heart transplantation or device implantation.
A 47-year-old man with chest and neck pain
A 47-year-old man presented with acute shortness of breath and chest and neck pain, which began after he heard popping sounds while boarding a bus. The pain was right-sided, sharp, worse with deep breathing, and associated with a sensation of fullness over the right chest.
His medical conditions included controlled hypertension, gastroesophageal reflux disease, and chronic obstructive pulmonary disease (COPD). The COPD was managed with an albuterol inhaler only. He had a 50-pack-year history of smoking, and he drank alcohol occasionally.
On arrival, he was in mild respiratory distress, but his vital signs were stable. We could hear wheezing on both sides of his chest and feel subcutaneous crepitation on both sides of his chest and neck, the latter more on the right side. The rest of the examination was unremarkable.
Results of a complete blood cell count and metabolic panel were within normal limits. Because of the above findings, nasopharyngeal radiogragraphy was ordered (Figures 1 and 2).
Q: What is the most likely cause of this presentation?
- Esophageal rupture
- Gas gangrene
- Asthma exacerbation
- Ruptured emphysematous bullae
A: This patient had a history of COPD, which put him at risk of developing bullous emphysematous bullae that can rupture and cause subcutaneous emphysema. His nasopharyngeal radiograph (Figure1) showed bilateral extensive subcutaneous emphysema. His lateral nasopharyngeal radiograph (Figure 2) showed air-tracking within the mediastinum and into the retropharyngeal space (arrow). Computed tomography (Figure 3) showed extensive subcutaneous emphysema in the right lateral chest wall (arrow) and large bullae in the right upper lobe (arrow heads). As for the other possibilities:
Esophageal ruptures and tears are iatrogenic in most cases and usually occur after endoscopic procedures, but they can also occur in patients with intractable vomiting. Computed tomography often shows esophageal thickening, periesophageal fluid, mediastinal widening, and extraluminal air. However, in most cases, it is seen as pneumomediastinum and subcutaneous emphysema.1
Gas gangrene is a life-threatening soft-tissue and muscle infection caused by Clostridium perfringens in most cases.2 The pain is out of proportion to the findings on physical examination. Patients usually have toxic signs and symptoms such as fever and hypotension. Our patient was hemodynamically stable, with no changes in skin color.
Severe exacerbations of asthma can lead to alveolar rupture, pneumothorax, and subcutaneous emphysema, although this is a rare complication. Air can dissect along the bronchovascular sheaths into the neck and cause subcutaneous emphysema, or into the pleural space and cause pneumothorax. Our patient had no history of asthma and plainly had emphysematous bullae.3
SUBCUTANEOUS EMPHYSEMA
Subcutaneous emphysema is a collection of air within subcutaneous tissues. It usually presents as bloating of the skin around the neck and the chest wall. It is often seen in patients with pneumothorax.
The most common cause of subcutaneous emphysema is traumatic injury to the chest wall, such as from a motor vehicle accident or a stab wound,4 but it can also occur spontaneously in patients who have severe emphysema with large bullae. As the emphysema progresses, the bullae can easily rupture, and this can lead to pneumothorax, which can lead to subcutaneous emphysema. Primary spontaneous pneumothorax and subcutaneous emphysema can occur in people who have unrecognized lung disease and genetic disorders such as Marfan syndrome and Ehler-Danlos syndrome.5 Other causes include iatrogenic injury, Pneumocystis jirovecii pneumonia (common in patients with human immunodeficiency virus infection), and cystic fibrosis. Pneumothorax occurs in about 30% of cases of P jirovecii pneumonia,6 and in about 6% of patients with cystic fibrosis.7 Bronchocutaneous fistula is an extremely rare complication of lung cancer and can cause subcutaneous emphysema.8 Tuberculosis is another possible cause.9
Subcutaneous emphysema mainly presents with chest or neck pain and wheezing. In severe cases, air can track to the face, causing facial swelling and difficulty breathing due to compression of the larynx. Also, it can track down to the thighs, causing leg pain and swelling.10
On examination, subcutaneous emphysema can be detected by palpating the chest wall, which causes the air bubble to move and produce crackling sounds. Most cases of subcutaneous emphysema are diagnosed clinically. Chest radiography and computed tomography help identify the source of air leak. Ultrasonography is usually used in cases of blunt trauma to the chest as part of the Focal Assessment With Sonography for Trauma protocol.11
Subcutaneous emphysema can resolve spontaneously, requiring only pain management and supplemental oxygen.12 In severe cases, air collection can lead to what is called “massive subcutaneous emphysema,” which requires surgical drainage.
Our patient had large emphysematous bullae in the apical region of the right lung that ruptured and led to subcutaneous emphysema. After placement of a chest tube, he underwent right-sided thoracotomy with bullectomy. His postoperative course was uneventful, and he was discharged a few days later. Three weeks later, repeated chest radiography showed resolution of his subcutaneous emphysema (Figure 4).
- White CS, Templeton PA, Attar S. Esophageal perforation: CT findings. AJR Am J Roentgenol 1993; 160:767–770.
- Aggelidakis J, Lasithiotakis K, Topalidou A, Koutroumpas J, Kouvidis G, Katonis P. Limb salvage after gas gangrene: a case report and review of the literature. World J Emerg Surg 2011; 6:28.
- Romero KJ, Trujillo MH. Spontaneous pneumomediastinum and subcutaneous emphysema in asthma exacerbation: the Macklin effect. Heart Lung 2010; 39:444–447.
- Peart O. Subcutaneous emphysema. Radiol Technol 2006; 77:296.
- Chiu HT, Garcia CK. Familial spontaneous pneumothorax. Curr Opin Pulm Med 2006; 12:268–272.
- Sepkowitz KA, Telzak EE, Gold JW, et al. Pneumothorax in AIDS. Ann Intern Med 1991; 114:455–459.
- Flume PA, Strange C, Ye X, Ebeling M, Hulsey T, Clark LL. Pneumothorax in cystic fibrosis. Chest 2005; 128:720–728.
- Yalçinkaya S, Vural AH, Göncü MT, Özyazicioglu AF. Cavitary lung cancer presenting as subcutaneous emphysema on the contralateral side. Interact Cardiovasc Thorac Surg 2012; 14:338–339.
- Shamaei M, Tabarsi P, Pojhan S, et al. Tuberculosis-associated secondary pneumothorax: a retrospective study of 53 patients. Respir Care 2011; 56:298–302.
- Sherif HM, Ott DA. The use of subcutaneous drains to manage subcutaneous emphysema. Tex Heart Inst J 1999; 26:129–131.
- Wilkerson RG, Stone MB. Sensitivity of bedside ultrasound and supine anteroposterior chest radiographs for the identification of pneumothorax after blunt trauma. Acad Emerg Med 2010; 17:11–17.
- Mattox KL, Allen MK. Systematic approach to pneumothorax, haemothorax, pneumomediastinum and subcutaneous emphysema. Injury 1986; 17:309–312.
A 47-year-old man presented with acute shortness of breath and chest and neck pain, which began after he heard popping sounds while boarding a bus. The pain was right-sided, sharp, worse with deep breathing, and associated with a sensation of fullness over the right chest.
His medical conditions included controlled hypertension, gastroesophageal reflux disease, and chronic obstructive pulmonary disease (COPD). The COPD was managed with an albuterol inhaler only. He had a 50-pack-year history of smoking, and he drank alcohol occasionally.
On arrival, he was in mild respiratory distress, but his vital signs were stable. We could hear wheezing on both sides of his chest and feel subcutaneous crepitation on both sides of his chest and neck, the latter more on the right side. The rest of the examination was unremarkable.
Results of a complete blood cell count and metabolic panel were within normal limits. Because of the above findings, nasopharyngeal radiogragraphy was ordered (Figures 1 and 2).
Q: What is the most likely cause of this presentation?
- Esophageal rupture
- Gas gangrene
- Asthma exacerbation
- Ruptured emphysematous bullae
A: This patient had a history of COPD, which put him at risk of developing bullous emphysematous bullae that can rupture and cause subcutaneous emphysema. His nasopharyngeal radiograph (Figure1) showed bilateral extensive subcutaneous emphysema. His lateral nasopharyngeal radiograph (Figure 2) showed air-tracking within the mediastinum and into the retropharyngeal space (arrow). Computed tomography (Figure 3) showed extensive subcutaneous emphysema in the right lateral chest wall (arrow) and large bullae in the right upper lobe (arrow heads). As for the other possibilities:
Esophageal ruptures and tears are iatrogenic in most cases and usually occur after endoscopic procedures, but they can also occur in patients with intractable vomiting. Computed tomography often shows esophageal thickening, periesophageal fluid, mediastinal widening, and extraluminal air. However, in most cases, it is seen as pneumomediastinum and subcutaneous emphysema.1
Gas gangrene is a life-threatening soft-tissue and muscle infection caused by Clostridium perfringens in most cases.2 The pain is out of proportion to the findings on physical examination. Patients usually have toxic signs and symptoms such as fever and hypotension. Our patient was hemodynamically stable, with no changes in skin color.
Severe exacerbations of asthma can lead to alveolar rupture, pneumothorax, and subcutaneous emphysema, although this is a rare complication. Air can dissect along the bronchovascular sheaths into the neck and cause subcutaneous emphysema, or into the pleural space and cause pneumothorax. Our patient had no history of asthma and plainly had emphysematous bullae.3
SUBCUTANEOUS EMPHYSEMA
Subcutaneous emphysema is a collection of air within subcutaneous tissues. It usually presents as bloating of the skin around the neck and the chest wall. It is often seen in patients with pneumothorax.
The most common cause of subcutaneous emphysema is traumatic injury to the chest wall, such as from a motor vehicle accident or a stab wound,4 but it can also occur spontaneously in patients who have severe emphysema with large bullae. As the emphysema progresses, the bullae can easily rupture, and this can lead to pneumothorax, which can lead to subcutaneous emphysema. Primary spontaneous pneumothorax and subcutaneous emphysema can occur in people who have unrecognized lung disease and genetic disorders such as Marfan syndrome and Ehler-Danlos syndrome.5 Other causes include iatrogenic injury, Pneumocystis jirovecii pneumonia (common in patients with human immunodeficiency virus infection), and cystic fibrosis. Pneumothorax occurs in about 30% of cases of P jirovecii pneumonia,6 and in about 6% of patients with cystic fibrosis.7 Bronchocutaneous fistula is an extremely rare complication of lung cancer and can cause subcutaneous emphysema.8 Tuberculosis is another possible cause.9
Subcutaneous emphysema mainly presents with chest or neck pain and wheezing. In severe cases, air can track to the face, causing facial swelling and difficulty breathing due to compression of the larynx. Also, it can track down to the thighs, causing leg pain and swelling.10
On examination, subcutaneous emphysema can be detected by palpating the chest wall, which causes the air bubble to move and produce crackling sounds. Most cases of subcutaneous emphysema are diagnosed clinically. Chest radiography and computed tomography help identify the source of air leak. Ultrasonography is usually used in cases of blunt trauma to the chest as part of the Focal Assessment With Sonography for Trauma protocol.11
Subcutaneous emphysema can resolve spontaneously, requiring only pain management and supplemental oxygen.12 In severe cases, air collection can lead to what is called “massive subcutaneous emphysema,” which requires surgical drainage.
Our patient had large emphysematous bullae in the apical region of the right lung that ruptured and led to subcutaneous emphysema. After placement of a chest tube, he underwent right-sided thoracotomy with bullectomy. His postoperative course was uneventful, and he was discharged a few days later. Three weeks later, repeated chest radiography showed resolution of his subcutaneous emphysema (Figure 4).
A 47-year-old man presented with acute shortness of breath and chest and neck pain, which began after he heard popping sounds while boarding a bus. The pain was right-sided, sharp, worse with deep breathing, and associated with a sensation of fullness over the right chest.
His medical conditions included controlled hypertension, gastroesophageal reflux disease, and chronic obstructive pulmonary disease (COPD). The COPD was managed with an albuterol inhaler only. He had a 50-pack-year history of smoking, and he drank alcohol occasionally.
On arrival, he was in mild respiratory distress, but his vital signs were stable. We could hear wheezing on both sides of his chest and feel subcutaneous crepitation on both sides of his chest and neck, the latter more on the right side. The rest of the examination was unremarkable.
Results of a complete blood cell count and metabolic panel were within normal limits. Because of the above findings, nasopharyngeal radiogragraphy was ordered (Figures 1 and 2).
Q: What is the most likely cause of this presentation?
- Esophageal rupture
- Gas gangrene
- Asthma exacerbation
- Ruptured emphysematous bullae
A: This patient had a history of COPD, which put him at risk of developing bullous emphysematous bullae that can rupture and cause subcutaneous emphysema. His nasopharyngeal radiograph (Figure1) showed bilateral extensive subcutaneous emphysema. His lateral nasopharyngeal radiograph (Figure 2) showed air-tracking within the mediastinum and into the retropharyngeal space (arrow). Computed tomography (Figure 3) showed extensive subcutaneous emphysema in the right lateral chest wall (arrow) and large bullae in the right upper lobe (arrow heads). As for the other possibilities:
Esophageal ruptures and tears are iatrogenic in most cases and usually occur after endoscopic procedures, but they can also occur in patients with intractable vomiting. Computed tomography often shows esophageal thickening, periesophageal fluid, mediastinal widening, and extraluminal air. However, in most cases, it is seen as pneumomediastinum and subcutaneous emphysema.1
Gas gangrene is a life-threatening soft-tissue and muscle infection caused by Clostridium perfringens in most cases.2 The pain is out of proportion to the findings on physical examination. Patients usually have toxic signs and symptoms such as fever and hypotension. Our patient was hemodynamically stable, with no changes in skin color.
Severe exacerbations of asthma can lead to alveolar rupture, pneumothorax, and subcutaneous emphysema, although this is a rare complication. Air can dissect along the bronchovascular sheaths into the neck and cause subcutaneous emphysema, or into the pleural space and cause pneumothorax. Our patient had no history of asthma and plainly had emphysematous bullae.3
SUBCUTANEOUS EMPHYSEMA
Subcutaneous emphysema is a collection of air within subcutaneous tissues. It usually presents as bloating of the skin around the neck and the chest wall. It is often seen in patients with pneumothorax.
The most common cause of subcutaneous emphysema is traumatic injury to the chest wall, such as from a motor vehicle accident or a stab wound,4 but it can also occur spontaneously in patients who have severe emphysema with large bullae. As the emphysema progresses, the bullae can easily rupture, and this can lead to pneumothorax, which can lead to subcutaneous emphysema. Primary spontaneous pneumothorax and subcutaneous emphysema can occur in people who have unrecognized lung disease and genetic disorders such as Marfan syndrome and Ehler-Danlos syndrome.5 Other causes include iatrogenic injury, Pneumocystis jirovecii pneumonia (common in patients with human immunodeficiency virus infection), and cystic fibrosis. Pneumothorax occurs in about 30% of cases of P jirovecii pneumonia,6 and in about 6% of patients with cystic fibrosis.7 Bronchocutaneous fistula is an extremely rare complication of lung cancer and can cause subcutaneous emphysema.8 Tuberculosis is another possible cause.9
Subcutaneous emphysema mainly presents with chest or neck pain and wheezing. In severe cases, air can track to the face, causing facial swelling and difficulty breathing due to compression of the larynx. Also, it can track down to the thighs, causing leg pain and swelling.10
On examination, subcutaneous emphysema can be detected by palpating the chest wall, which causes the air bubble to move and produce crackling sounds. Most cases of subcutaneous emphysema are diagnosed clinically. Chest radiography and computed tomography help identify the source of air leak. Ultrasonography is usually used in cases of blunt trauma to the chest as part of the Focal Assessment With Sonography for Trauma protocol.11
Subcutaneous emphysema can resolve spontaneously, requiring only pain management and supplemental oxygen.12 In severe cases, air collection can lead to what is called “massive subcutaneous emphysema,” which requires surgical drainage.
Our patient had large emphysematous bullae in the apical region of the right lung that ruptured and led to subcutaneous emphysema. After placement of a chest tube, he underwent right-sided thoracotomy with bullectomy. His postoperative course was uneventful, and he was discharged a few days later. Three weeks later, repeated chest radiography showed resolution of his subcutaneous emphysema (Figure 4).
- White CS, Templeton PA, Attar S. Esophageal perforation: CT findings. AJR Am J Roentgenol 1993; 160:767–770.
- Aggelidakis J, Lasithiotakis K, Topalidou A, Koutroumpas J, Kouvidis G, Katonis P. Limb salvage after gas gangrene: a case report and review of the literature. World J Emerg Surg 2011; 6:28.
- Romero KJ, Trujillo MH. Spontaneous pneumomediastinum and subcutaneous emphysema in asthma exacerbation: the Macklin effect. Heart Lung 2010; 39:444–447.
- Peart O. Subcutaneous emphysema. Radiol Technol 2006; 77:296.
- Chiu HT, Garcia CK. Familial spontaneous pneumothorax. Curr Opin Pulm Med 2006; 12:268–272.
- Sepkowitz KA, Telzak EE, Gold JW, et al. Pneumothorax in AIDS. Ann Intern Med 1991; 114:455–459.
- Flume PA, Strange C, Ye X, Ebeling M, Hulsey T, Clark LL. Pneumothorax in cystic fibrosis. Chest 2005; 128:720–728.
- Yalçinkaya S, Vural AH, Göncü MT, Özyazicioglu AF. Cavitary lung cancer presenting as subcutaneous emphysema on the contralateral side. Interact Cardiovasc Thorac Surg 2012; 14:338–339.
- Shamaei M, Tabarsi P, Pojhan S, et al. Tuberculosis-associated secondary pneumothorax: a retrospective study of 53 patients. Respir Care 2011; 56:298–302.
- Sherif HM, Ott DA. The use of subcutaneous drains to manage subcutaneous emphysema. Tex Heart Inst J 1999; 26:129–131.
- Wilkerson RG, Stone MB. Sensitivity of bedside ultrasound and supine anteroposterior chest radiographs for the identification of pneumothorax after blunt trauma. Acad Emerg Med 2010; 17:11–17.
- Mattox KL, Allen MK. Systematic approach to pneumothorax, haemothorax, pneumomediastinum and subcutaneous emphysema. Injury 1986; 17:309–312.
- White CS, Templeton PA, Attar S. Esophageal perforation: CT findings. AJR Am J Roentgenol 1993; 160:767–770.
- Aggelidakis J, Lasithiotakis K, Topalidou A, Koutroumpas J, Kouvidis G, Katonis P. Limb salvage after gas gangrene: a case report and review of the literature. World J Emerg Surg 2011; 6:28.
- Romero KJ, Trujillo MH. Spontaneous pneumomediastinum and subcutaneous emphysema in asthma exacerbation: the Macklin effect. Heart Lung 2010; 39:444–447.
- Peart O. Subcutaneous emphysema. Radiol Technol 2006; 77:296.
- Chiu HT, Garcia CK. Familial spontaneous pneumothorax. Curr Opin Pulm Med 2006; 12:268–272.
- Sepkowitz KA, Telzak EE, Gold JW, et al. Pneumothorax in AIDS. Ann Intern Med 1991; 114:455–459.
- Flume PA, Strange C, Ye X, Ebeling M, Hulsey T, Clark LL. Pneumothorax in cystic fibrosis. Chest 2005; 128:720–728.
- Yalçinkaya S, Vural AH, Göncü MT, Özyazicioglu AF. Cavitary lung cancer presenting as subcutaneous emphysema on the contralateral side. Interact Cardiovasc Thorac Surg 2012; 14:338–339.
- Shamaei M, Tabarsi P, Pojhan S, et al. Tuberculosis-associated secondary pneumothorax: a retrospective study of 53 patients. Respir Care 2011; 56:298–302.
- Sherif HM, Ott DA. The use of subcutaneous drains to manage subcutaneous emphysema. Tex Heart Inst J 1999; 26:129–131.
- Wilkerson RG, Stone MB. Sensitivity of bedside ultrasound and supine anteroposterior chest radiographs for the identification of pneumothorax after blunt trauma. Acad Emerg Med 2010; 17:11–17.
- Mattox KL, Allen MK. Systematic approach to pneumothorax, haemothorax, pneumomediastinum and subcutaneous emphysema. Injury 1986; 17:309–312.
Should N-acetylcysteine be used routinely to prevent contrast-induced acute kidney injury?
No. Using N-acetylcysteine (NAC) routinely to prevent contrast-induced acute kidney injury is not supported by the evidence at this time.1,2 However, there is evidence to suggest using it for patients at high risk, ie, those with significant baseline renal dysfunction.3,4
INCIDENCE AND IMPACT OF ACUTE KIDNEY INJURY
Intraarterial use of contrast is associated with a higher risk of acute kidney injury than intravenous use. Most studies of NAC for the prevention of contrast-induced acute kidney injury have focused on patients receiving contrast intraarterially. The reported rates of contrast-induced acute kidney injury also vary depending on how acute kidney injury was defined.
Although the incidence is low (1% to 2%) in patients with normal renal function, it can be as high as 25% in patients with renal impairment or a chronic condition such as diabetes or congestive heart failure, or in elderly patients.5
The development of acute kidney injury after percutaneous coronary intervention is associated with a longer hospital stay, a higher cost of care, and higher rates of morbidity and death.6
RATIONALE FOR USING N-ACETYLCYSTEINE
Contrast-induced acute kidney injury is thought to involve vasoconstriction and medullary ischemia mediated by reactive oxygen species.5 As an antioxidant and a scavenger of free radicals, NAC showed early promise in reducing the risk of this complication, but subsequent trials raised doubts about its efficacy. 1,2 In clinical practice, the drug is often used to prevent acute kidney injury because it is easy to give, cheap, and has few side effects. Recently, however, there have been suggestions that giving it intravenously may be associated with adverse effects that include anaphylactoid reactions.7
THE POSITIVE TRIALS
Tepel et al3 performed one of the earliest trials that found that NAC prevented contrast-induced acute kidney injury. The trial included 83 patients with stable chronic kidney disease (mean serum creatinine 2.4 mg/dL) who underwent computed tomography with about 75 mL of a nonionic, low-osmolality contrast agent. Participants were randomized to receive either NAC (600 mg orally twice daily) and 0.45% saline intravenously or placebo and saline. Acute kidney injury was defined as an increase of at least 0.5 mg/dL in the serum creatinine level 48 hours after the contrast dye was given.
The rate of acute kidney injury was significantly lower in the treatment group (2% vs 21%, P = .01). None of the patients who developed acute kidney injury needed hemodialysis.
Shyu et al4 studied 121 patients with chronic kidney disease (mean serum creatinine 2.8 mg/dL) who underwent a coronary procedure. Patients were randomized to receive NAC 400 mg orally twice daily or placebo in addition to 0.45% saline in both groups. Two (3.3%) of the 60 patients in the treated group and 15 (24.6%) of the 61 patients in the control group had an increase in creatinine concentration greater than 0.5 mg/dL at 48 hours (P < .001).
Both of these single-center studies were limited by small sample sizes and very short follow-up. Further, the impact of the drug on important clinical outcomes such as death and progression of chronic kidney disease was not reported.
Marenzi et al8 randomized 354 patients undergoing coronary angioplasty as the primary treatment for acute myocardial infarction to one of three treatment groups:
- NAC in a standard dosage (a 600-mg intravenous bolus before the procedure and then 600 mg orally twice daily for 48 hours afterward)
- NAC in a high dosage (a 1,200-mg intravenous bolus and then 1,200 mg orally twice daily for 48 hours)
- Placebo.
The two treatment groups had significantly lower rates of acute kidney injury than the placebo group. In addition, the hospital mortality rate and the rate of a composite end point of death, need for renal replacement therapy, or need for mechanical ventilation were significantly lower in the treated groups. However, the number of events was small, and a beneficial effect on the death rate has not been confirmed by other studies.5
THE NEGATIVE TRIALS
Several studies found that NAC did not prevent contrast-induced acute kidney injury.1,2,9
The Acetylcysteine for Contrast-induced Nephropathy Trial (ACT), published in 2011,1 was the largest of these trials. It included 2,308 patients undergoing an angiographic procedure who had at least one risk factor for contrast-induced acute kidney injury (age > 70, renal failure, diabetes mellitus, heart failure, or hypotension). Patients were randomly assigned to receive the drug (1,200 mg by mouth) or placebo.
The incidence of contrast-induced acute kidney injury was 12.7% in the treated group and 12.7% in the control group (relative risk 1.00; 95% confidence interval 0.81–1.25; P = .97). The rate of a combined end point of death or need for dialysis at 30 days was also similar in both groups (2.2% with treatment vs 2.3% with placebo).
Importantly, only about 15% of patients had a baseline serum creatinine greater than 1.5 mg/dL. Of these, most had an estimated glomerular filtration rate between 45 and 60 mL/min. Indeed, most patients in the ACT were at low risk of contrast-induced acute kidney injury. As a result, there were low event rates and, not surprisingly, no differences between the control and treatment groups.
Subgroup analysis did not suggest a benefit of treatment in those with a baseline serum creatinine greater than 1.5 mg/dL. However, as the authors pointed out, this subgroup was small, so definitive statistically powered conclusions cannot be drawn. There was no significant difference in the primary end point among several other predefined subgroups (age > 70, female sex, diabetes).1
The ACT differed from the “positive” study by Marenzi et al8 in several ways. The ACT patients were at lower risk, the coronary catheterizations were being done mainly for diagnosis rather than intervention, a lower volume of contrast dye was used (100 mL in the ACT vs 250 mL in the Marenzi study), and patients with ST-elevation myocardial infarction were excluded. Other weaknesses of the ACT include use of a baseline serum creatinine within 3 months of study entry, variations in the hydration protocol, and the use of a high-osmolar contrast agent in some patients.
Webb et al2 found, in a large, randomized trial, that intravenous NAC did not prevent contrast-induced acute kidney injury. Patients with renal dysfunction (mean serum creatinine around 1.6 mg/dL) undergoing cardiac catheterization were randomly assigned to receive either NAC 500 mg or placebo immediately before the procedure. All patients first received isotonic saline 200 mL, then 1.5 mL/kg per hour for 6 hours, unless contraindicated. The study was terminated early because of a determination of futility.
Gurm et al9 found that a database of 90,578 consecutive patients undergoing nonemergency coronary angiography from 2006 to 2009 did not show differences in the rate of contrast-induced acute kidney injury between patients who received NAC and those who did not (5.5% vs 5.5%, P = .99). There was also no difference in the rate of death or the need for dialysis. These negative findings were consistent across many prespecified subgroups.
MIXED RESULTS IN META-ANALYSES
Results from meta-analyses have been mixed,10,11 mainly because of study heterogeneity (eg, baseline risk, end points, dose of the drug) and publication bias. None of the previous meta-analyses included the recent negative results from the ACT.
CURRENT GUIDELINES
After the publication of the ACT, the joint guidelines of the American College of Cardiology and the American Heart Association were updated, designating NAC as class III (no benefit) and level of evidence A.12
However, recently published guidelines from the Kidney Disease: Improving Global Outcomes Acute Kidney Injury Working Group recommend using the drug together with intravenous isotonic crystalloids in patients at high risk of contrast-induced acute kidney injury, although the level of evidence is 2D (2 = suggestion, D = quality of evidence very low).5
WHAT WE RECOMMEND
The routine use of NAC to prevent contrast-induced acute kidney injury is not supported by the current evidence. However, clarification of its efficacy in high-risk patients is needed, especially those with baseline renal dysfunction and diabetes mellitus.
The Prevention of Serious Adverse Events Following Angiography (PRESERVE) study (ClinTrials.gov identifier NCT01467466) may clarify the role of this drug in a high-risk cohort using the important clinical outcomes of death, need for acute dialysis, or persistent decline in kidney function after angiography. This important study was set to begin in July 2012, with an anticipated enrollment of more than 8,000 patients who have glomerular filtration rates of 15 to 59 mL/min/1.73 m2.
In the meantime, we recommend the following in patients at high risk of contrast-induced acute kidney injury:
- Clarify whether contrast is truly needed
- When possible, limit the volume of contrast, avoid repeated doses over a short time frame, and use an iso-osmolar or low-osmolar contrast agent
- Discontinue nephrotoxic agents
- Provide an evidence-based intravenous crystalloid regimen with isotonic sodium bicarbonate or saline
- Although it is not strictly evidence-based, use NAC in patients with significant baseline renal dysfunction (glomerular filtration rate < 45 mL/min/1.73 m2), multiple concurrent risk factors such as hypotension, diabetes, preexisting kidney injury, or congestive heart failure that limits the use of intravenous fluids, or who need a high volume of contrast dye
- Avoid using intravenous NAC, given its lack of benefit and risk of anaphylactoid reactions.7,13
We do not yet have clear evidence on the optimal dosing regimen. But based on the limited data, we recommend 600 to 1,200 mg twice a day for 1 day before and 1 day after the dye is given.
- ACT Investigators. Acetylcysteine for prevention of renal outcomes in patients undergoing coronary and peripheral vascular angiography: main results from the randomized Acetylcysteine for Contrast-induced nephropathy Trial (ACT). Circulation 2011; 124:1250–1259.
- Webb JG, Pate GE, Humphries KH, et al. A randomized controlled trial of intravenous N-acetylcysteine for the prevention of contrast-induced nephropathy after cardiac catheterization: lack of effect. Am Heart J 2004; 148:422–429.
- Tepel M, van der Giet M, Schwarzfeld C, Laufer U, Liermann D, Zidek W. Prevention of radiographic-contrast-agent-induced reductions in renal function by acetylcysteine. N Engl J Med 2000; 343:180–184.
- Shyu KG, Cheng JJ, Kuan P. Acetylcysteine protects against acute renal damage in patients with abnormal renal function undergoing a coronary procedure. J Am Coll Cardiol 2002; 40:1383–1388.
- Kidney Disease: Improving Global Outcomes (KDIGO) Acute Kidney Injury Work Group. KDIGO clinical practice guideline for acute kidney injury. Kidney Int 2012; 2(suppl 1):1–138.
- Rihal CS, Textor SC, Grill DE, et al. Incidence and prognostic importance of acute renal failure after percutaneous coronary intervention. Circulation 2002; 105:2259–2264.
- Baker CS, Wragg A, Kumar S, De Palma R, Baker LR, Knight CJ. A rapid protocol for the prevention of contrast-induced renal dysfunction: the RAPPID study. J Am Coll Cardiol 2003; 41:2114–2118.
- Marenzi G, Assanelli E, Marana I, et al. N-acetylcysteine and contrast-induced nephropathy in primary angioplasty. N Engl J Med 2006; 354:2773–2782.
- Gurm HS, Smith DE, Berwanger O, et al; BMC2 (Blue Cross Blue Shield of Michigan Cardiovascular Consortium). Contemporary use and effectiveness of N-acetylcysteine in preventing contrast-induced nephropathy among patients undergoing percutaneous coronary intervention. JACC Cardiovasc Interv 2012; 5:98–104.
- Duong MH, MacKenzie TA, Malenka DJ. N-acetylcysteine prophylaxis significantly reduces the risk of radiocontrast-induced nephropathy: comprehensive meta-analysis. Catheter Cardiovasc Interv 2005; 64:471–479.
- Gonzales DA, Norsworthy KJ, Kern SJ, et al. A meta-analysis of N-acetylcysteine in contrast-induced nephrotoxicity: unsupervised clustering to resolve heterogeneity. BMC Med 2007; 5:32.
- Levine GN, Bates ER, Blankenship JC, et al. 2011 ACCF/AHA/SCAI Guideline for Percutaneous Coronary Intervention: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines and the Society for Cardiovascular Angiography and Interventions. Circulation 2011; 124:e574–e651.
- Kanter MZ. Comparison of oral and i.v. acetylcysteine in the treatment of acetaminophen poisoning. Am J Health Syst Pharm 2006; 63:1821–1827.
No. Using N-acetylcysteine (NAC) routinely to prevent contrast-induced acute kidney injury is not supported by the evidence at this time.1,2 However, there is evidence to suggest using it for patients at high risk, ie, those with significant baseline renal dysfunction.3,4
INCIDENCE AND IMPACT OF ACUTE KIDNEY INJURY
Intraarterial use of contrast is associated with a higher risk of acute kidney injury than intravenous use. Most studies of NAC for the prevention of contrast-induced acute kidney injury have focused on patients receiving contrast intraarterially. The reported rates of contrast-induced acute kidney injury also vary depending on how acute kidney injury was defined.
Although the incidence is low (1% to 2%) in patients with normal renal function, it can be as high as 25% in patients with renal impairment or a chronic condition such as diabetes or congestive heart failure, or in elderly patients.5
The development of acute kidney injury after percutaneous coronary intervention is associated with a longer hospital stay, a higher cost of care, and higher rates of morbidity and death.6
RATIONALE FOR USING N-ACETYLCYSTEINE
Contrast-induced acute kidney injury is thought to involve vasoconstriction and medullary ischemia mediated by reactive oxygen species.5 As an antioxidant and a scavenger of free radicals, NAC showed early promise in reducing the risk of this complication, but subsequent trials raised doubts about its efficacy. 1,2 In clinical practice, the drug is often used to prevent acute kidney injury because it is easy to give, cheap, and has few side effects. Recently, however, there have been suggestions that giving it intravenously may be associated with adverse effects that include anaphylactoid reactions.7
THE POSITIVE TRIALS
Tepel et al3 performed one of the earliest trials that found that NAC prevented contrast-induced acute kidney injury. The trial included 83 patients with stable chronic kidney disease (mean serum creatinine 2.4 mg/dL) who underwent computed tomography with about 75 mL of a nonionic, low-osmolality contrast agent. Participants were randomized to receive either NAC (600 mg orally twice daily) and 0.45% saline intravenously or placebo and saline. Acute kidney injury was defined as an increase of at least 0.5 mg/dL in the serum creatinine level 48 hours after the contrast dye was given.
The rate of acute kidney injury was significantly lower in the treatment group (2% vs 21%, P = .01). None of the patients who developed acute kidney injury needed hemodialysis.
Shyu et al4 studied 121 patients with chronic kidney disease (mean serum creatinine 2.8 mg/dL) who underwent a coronary procedure. Patients were randomized to receive NAC 400 mg orally twice daily or placebo in addition to 0.45% saline in both groups. Two (3.3%) of the 60 patients in the treated group and 15 (24.6%) of the 61 patients in the control group had an increase in creatinine concentration greater than 0.5 mg/dL at 48 hours (P < .001).
Both of these single-center studies were limited by small sample sizes and very short follow-up. Further, the impact of the drug on important clinical outcomes such as death and progression of chronic kidney disease was not reported.
Marenzi et al8 randomized 354 patients undergoing coronary angioplasty as the primary treatment for acute myocardial infarction to one of three treatment groups:
- NAC in a standard dosage (a 600-mg intravenous bolus before the procedure and then 600 mg orally twice daily for 48 hours afterward)
- NAC in a high dosage (a 1,200-mg intravenous bolus and then 1,200 mg orally twice daily for 48 hours)
- Placebo.
The two treatment groups had significantly lower rates of acute kidney injury than the placebo group. In addition, the hospital mortality rate and the rate of a composite end point of death, need for renal replacement therapy, or need for mechanical ventilation were significantly lower in the treated groups. However, the number of events was small, and a beneficial effect on the death rate has not been confirmed by other studies.5
THE NEGATIVE TRIALS
Several studies found that NAC did not prevent contrast-induced acute kidney injury.1,2,9
The Acetylcysteine for Contrast-induced Nephropathy Trial (ACT), published in 2011,1 was the largest of these trials. It included 2,308 patients undergoing an angiographic procedure who had at least one risk factor for contrast-induced acute kidney injury (age > 70, renal failure, diabetes mellitus, heart failure, or hypotension). Patients were randomly assigned to receive the drug (1,200 mg by mouth) or placebo.
The incidence of contrast-induced acute kidney injury was 12.7% in the treated group and 12.7% in the control group (relative risk 1.00; 95% confidence interval 0.81–1.25; P = .97). The rate of a combined end point of death or need for dialysis at 30 days was also similar in both groups (2.2% with treatment vs 2.3% with placebo).
Importantly, only about 15% of patients had a baseline serum creatinine greater than 1.5 mg/dL. Of these, most had an estimated glomerular filtration rate between 45 and 60 mL/min. Indeed, most patients in the ACT were at low risk of contrast-induced acute kidney injury. As a result, there were low event rates and, not surprisingly, no differences between the control and treatment groups.
Subgroup analysis did not suggest a benefit of treatment in those with a baseline serum creatinine greater than 1.5 mg/dL. However, as the authors pointed out, this subgroup was small, so definitive statistically powered conclusions cannot be drawn. There was no significant difference in the primary end point among several other predefined subgroups (age > 70, female sex, diabetes).1
The ACT differed from the “positive” study by Marenzi et al8 in several ways. The ACT patients were at lower risk, the coronary catheterizations were being done mainly for diagnosis rather than intervention, a lower volume of contrast dye was used (100 mL in the ACT vs 250 mL in the Marenzi study), and patients with ST-elevation myocardial infarction were excluded. Other weaknesses of the ACT include use of a baseline serum creatinine within 3 months of study entry, variations in the hydration protocol, and the use of a high-osmolar contrast agent in some patients.
Webb et al2 found, in a large, randomized trial, that intravenous NAC did not prevent contrast-induced acute kidney injury. Patients with renal dysfunction (mean serum creatinine around 1.6 mg/dL) undergoing cardiac catheterization were randomly assigned to receive either NAC 500 mg or placebo immediately before the procedure. All patients first received isotonic saline 200 mL, then 1.5 mL/kg per hour for 6 hours, unless contraindicated. The study was terminated early because of a determination of futility.
Gurm et al9 found that a database of 90,578 consecutive patients undergoing nonemergency coronary angiography from 2006 to 2009 did not show differences in the rate of contrast-induced acute kidney injury between patients who received NAC and those who did not (5.5% vs 5.5%, P = .99). There was also no difference in the rate of death or the need for dialysis. These negative findings were consistent across many prespecified subgroups.
MIXED RESULTS IN META-ANALYSES
Results from meta-analyses have been mixed,10,11 mainly because of study heterogeneity (eg, baseline risk, end points, dose of the drug) and publication bias. None of the previous meta-analyses included the recent negative results from the ACT.
CURRENT GUIDELINES
After the publication of the ACT, the joint guidelines of the American College of Cardiology and the American Heart Association were updated, designating NAC as class III (no benefit) and level of evidence A.12
However, recently published guidelines from the Kidney Disease: Improving Global Outcomes Acute Kidney Injury Working Group recommend using the drug together with intravenous isotonic crystalloids in patients at high risk of contrast-induced acute kidney injury, although the level of evidence is 2D (2 = suggestion, D = quality of evidence very low).5
WHAT WE RECOMMEND
The routine use of NAC to prevent contrast-induced acute kidney injury is not supported by the current evidence. However, clarification of its efficacy in high-risk patients is needed, especially those with baseline renal dysfunction and diabetes mellitus.
The Prevention of Serious Adverse Events Following Angiography (PRESERVE) study (ClinTrials.gov identifier NCT01467466) may clarify the role of this drug in a high-risk cohort using the important clinical outcomes of death, need for acute dialysis, or persistent decline in kidney function after angiography. This important study was set to begin in July 2012, with an anticipated enrollment of more than 8,000 patients who have glomerular filtration rates of 15 to 59 mL/min/1.73 m2.
In the meantime, we recommend the following in patients at high risk of contrast-induced acute kidney injury:
- Clarify whether contrast is truly needed
- When possible, limit the volume of contrast, avoid repeated doses over a short time frame, and use an iso-osmolar or low-osmolar contrast agent
- Discontinue nephrotoxic agents
- Provide an evidence-based intravenous crystalloid regimen with isotonic sodium bicarbonate or saline
- Although it is not strictly evidence-based, use NAC in patients with significant baseline renal dysfunction (glomerular filtration rate < 45 mL/min/1.73 m2), multiple concurrent risk factors such as hypotension, diabetes, preexisting kidney injury, or congestive heart failure that limits the use of intravenous fluids, or who need a high volume of contrast dye
- Avoid using intravenous NAC, given its lack of benefit and risk of anaphylactoid reactions.7,13
We do not yet have clear evidence on the optimal dosing regimen. But based on the limited data, we recommend 600 to 1,200 mg twice a day for 1 day before and 1 day after the dye is given.
No. Using N-acetylcysteine (NAC) routinely to prevent contrast-induced acute kidney injury is not supported by the evidence at this time.1,2 However, there is evidence to suggest using it for patients at high risk, ie, those with significant baseline renal dysfunction.3,4
INCIDENCE AND IMPACT OF ACUTE KIDNEY INJURY
Intraarterial use of contrast is associated with a higher risk of acute kidney injury than intravenous use. Most studies of NAC for the prevention of contrast-induced acute kidney injury have focused on patients receiving contrast intraarterially. The reported rates of contrast-induced acute kidney injury also vary depending on how acute kidney injury was defined.
Although the incidence is low (1% to 2%) in patients with normal renal function, it can be as high as 25% in patients with renal impairment or a chronic condition such as diabetes or congestive heart failure, or in elderly patients.5
The development of acute kidney injury after percutaneous coronary intervention is associated with a longer hospital stay, a higher cost of care, and higher rates of morbidity and death.6
RATIONALE FOR USING N-ACETYLCYSTEINE
Contrast-induced acute kidney injury is thought to involve vasoconstriction and medullary ischemia mediated by reactive oxygen species.5 As an antioxidant and a scavenger of free radicals, NAC showed early promise in reducing the risk of this complication, but subsequent trials raised doubts about its efficacy. 1,2 In clinical practice, the drug is often used to prevent acute kidney injury because it is easy to give, cheap, and has few side effects. Recently, however, there have been suggestions that giving it intravenously may be associated with adverse effects that include anaphylactoid reactions.7
THE POSITIVE TRIALS
Tepel et al3 performed one of the earliest trials that found that NAC prevented contrast-induced acute kidney injury. The trial included 83 patients with stable chronic kidney disease (mean serum creatinine 2.4 mg/dL) who underwent computed tomography with about 75 mL of a nonionic, low-osmolality contrast agent. Participants were randomized to receive either NAC (600 mg orally twice daily) and 0.45% saline intravenously or placebo and saline. Acute kidney injury was defined as an increase of at least 0.5 mg/dL in the serum creatinine level 48 hours after the contrast dye was given.
The rate of acute kidney injury was significantly lower in the treatment group (2% vs 21%, P = .01). None of the patients who developed acute kidney injury needed hemodialysis.
Shyu et al4 studied 121 patients with chronic kidney disease (mean serum creatinine 2.8 mg/dL) who underwent a coronary procedure. Patients were randomized to receive NAC 400 mg orally twice daily or placebo in addition to 0.45% saline in both groups. Two (3.3%) of the 60 patients in the treated group and 15 (24.6%) of the 61 patients in the control group had an increase in creatinine concentration greater than 0.5 mg/dL at 48 hours (P < .001).
Both of these single-center studies were limited by small sample sizes and very short follow-up. Further, the impact of the drug on important clinical outcomes such as death and progression of chronic kidney disease was not reported.
Marenzi et al8 randomized 354 patients undergoing coronary angioplasty as the primary treatment for acute myocardial infarction to one of three treatment groups:
- NAC in a standard dosage (a 600-mg intravenous bolus before the procedure and then 600 mg orally twice daily for 48 hours afterward)
- NAC in a high dosage (a 1,200-mg intravenous bolus and then 1,200 mg orally twice daily for 48 hours)
- Placebo.
The two treatment groups had significantly lower rates of acute kidney injury than the placebo group. In addition, the hospital mortality rate and the rate of a composite end point of death, need for renal replacement therapy, or need for mechanical ventilation were significantly lower in the treated groups. However, the number of events was small, and a beneficial effect on the death rate has not been confirmed by other studies.5
THE NEGATIVE TRIALS
Several studies found that NAC did not prevent contrast-induced acute kidney injury.1,2,9
The Acetylcysteine for Contrast-induced Nephropathy Trial (ACT), published in 2011,1 was the largest of these trials. It included 2,308 patients undergoing an angiographic procedure who had at least one risk factor for contrast-induced acute kidney injury (age > 70, renal failure, diabetes mellitus, heart failure, or hypotension). Patients were randomly assigned to receive the drug (1,200 mg by mouth) or placebo.
The incidence of contrast-induced acute kidney injury was 12.7% in the treated group and 12.7% in the control group (relative risk 1.00; 95% confidence interval 0.81–1.25; P = .97). The rate of a combined end point of death or need for dialysis at 30 days was also similar in both groups (2.2% with treatment vs 2.3% with placebo).
Importantly, only about 15% of patients had a baseline serum creatinine greater than 1.5 mg/dL. Of these, most had an estimated glomerular filtration rate between 45 and 60 mL/min. Indeed, most patients in the ACT were at low risk of contrast-induced acute kidney injury. As a result, there were low event rates and, not surprisingly, no differences between the control and treatment groups.
Subgroup analysis did not suggest a benefit of treatment in those with a baseline serum creatinine greater than 1.5 mg/dL. However, as the authors pointed out, this subgroup was small, so definitive statistically powered conclusions cannot be drawn. There was no significant difference in the primary end point among several other predefined subgroups (age > 70, female sex, diabetes).1
The ACT differed from the “positive” study by Marenzi et al8 in several ways. The ACT patients were at lower risk, the coronary catheterizations were being done mainly for diagnosis rather than intervention, a lower volume of contrast dye was used (100 mL in the ACT vs 250 mL in the Marenzi study), and patients with ST-elevation myocardial infarction were excluded. Other weaknesses of the ACT include use of a baseline serum creatinine within 3 months of study entry, variations in the hydration protocol, and the use of a high-osmolar contrast agent in some patients.
Webb et al2 found, in a large, randomized trial, that intravenous NAC did not prevent contrast-induced acute kidney injury. Patients with renal dysfunction (mean serum creatinine around 1.6 mg/dL) undergoing cardiac catheterization were randomly assigned to receive either NAC 500 mg or placebo immediately before the procedure. All patients first received isotonic saline 200 mL, then 1.5 mL/kg per hour for 6 hours, unless contraindicated. The study was terminated early because of a determination of futility.
Gurm et al9 found that a database of 90,578 consecutive patients undergoing nonemergency coronary angiography from 2006 to 2009 did not show differences in the rate of contrast-induced acute kidney injury between patients who received NAC and those who did not (5.5% vs 5.5%, P = .99). There was also no difference in the rate of death or the need for dialysis. These negative findings were consistent across many prespecified subgroups.
MIXED RESULTS IN META-ANALYSES
Results from meta-analyses have been mixed,10,11 mainly because of study heterogeneity (eg, baseline risk, end points, dose of the drug) and publication bias. None of the previous meta-analyses included the recent negative results from the ACT.
CURRENT GUIDELINES
After the publication of the ACT, the joint guidelines of the American College of Cardiology and the American Heart Association were updated, designating NAC as class III (no benefit) and level of evidence A.12
However, recently published guidelines from the Kidney Disease: Improving Global Outcomes Acute Kidney Injury Working Group recommend using the drug together with intravenous isotonic crystalloids in patients at high risk of contrast-induced acute kidney injury, although the level of evidence is 2D (2 = suggestion, D = quality of evidence very low).5
WHAT WE RECOMMEND
The routine use of NAC to prevent contrast-induced acute kidney injury is not supported by the current evidence. However, clarification of its efficacy in high-risk patients is needed, especially those with baseline renal dysfunction and diabetes mellitus.
The Prevention of Serious Adverse Events Following Angiography (PRESERVE) study (ClinTrials.gov identifier NCT01467466) may clarify the role of this drug in a high-risk cohort using the important clinical outcomes of death, need for acute dialysis, or persistent decline in kidney function after angiography. This important study was set to begin in July 2012, with an anticipated enrollment of more than 8,000 patients who have glomerular filtration rates of 15 to 59 mL/min/1.73 m2.
In the meantime, we recommend the following in patients at high risk of contrast-induced acute kidney injury:
- Clarify whether contrast is truly needed
- When possible, limit the volume of contrast, avoid repeated doses over a short time frame, and use an iso-osmolar or low-osmolar contrast agent
- Discontinue nephrotoxic agents
- Provide an evidence-based intravenous crystalloid regimen with isotonic sodium bicarbonate or saline
- Although it is not strictly evidence-based, use NAC in patients with significant baseline renal dysfunction (glomerular filtration rate < 45 mL/min/1.73 m2), multiple concurrent risk factors such as hypotension, diabetes, preexisting kidney injury, or congestive heart failure that limits the use of intravenous fluids, or who need a high volume of contrast dye
- Avoid using intravenous NAC, given its lack of benefit and risk of anaphylactoid reactions.7,13
We do not yet have clear evidence on the optimal dosing regimen. But based on the limited data, we recommend 600 to 1,200 mg twice a day for 1 day before and 1 day after the dye is given.
- ACT Investigators. Acetylcysteine for prevention of renal outcomes in patients undergoing coronary and peripheral vascular angiography: main results from the randomized Acetylcysteine for Contrast-induced nephropathy Trial (ACT). Circulation 2011; 124:1250–1259.
- Webb JG, Pate GE, Humphries KH, et al. A randomized controlled trial of intravenous N-acetylcysteine for the prevention of contrast-induced nephropathy after cardiac catheterization: lack of effect. Am Heart J 2004; 148:422–429.
- Tepel M, van der Giet M, Schwarzfeld C, Laufer U, Liermann D, Zidek W. Prevention of radiographic-contrast-agent-induced reductions in renal function by acetylcysteine. N Engl J Med 2000; 343:180–184.
- Shyu KG, Cheng JJ, Kuan P. Acetylcysteine protects against acute renal damage in patients with abnormal renal function undergoing a coronary procedure. J Am Coll Cardiol 2002; 40:1383–1388.
- Kidney Disease: Improving Global Outcomes (KDIGO) Acute Kidney Injury Work Group. KDIGO clinical practice guideline for acute kidney injury. Kidney Int 2012; 2(suppl 1):1–138.
- Rihal CS, Textor SC, Grill DE, et al. Incidence and prognostic importance of acute renal failure after percutaneous coronary intervention. Circulation 2002; 105:2259–2264.
- Baker CS, Wragg A, Kumar S, De Palma R, Baker LR, Knight CJ. A rapid protocol for the prevention of contrast-induced renal dysfunction: the RAPPID study. J Am Coll Cardiol 2003; 41:2114–2118.
- Marenzi G, Assanelli E, Marana I, et al. N-acetylcysteine and contrast-induced nephropathy in primary angioplasty. N Engl J Med 2006; 354:2773–2782.
- Gurm HS, Smith DE, Berwanger O, et al; BMC2 (Blue Cross Blue Shield of Michigan Cardiovascular Consortium). Contemporary use and effectiveness of N-acetylcysteine in preventing contrast-induced nephropathy among patients undergoing percutaneous coronary intervention. JACC Cardiovasc Interv 2012; 5:98–104.
- Duong MH, MacKenzie TA, Malenka DJ. N-acetylcysteine prophylaxis significantly reduces the risk of radiocontrast-induced nephropathy: comprehensive meta-analysis. Catheter Cardiovasc Interv 2005; 64:471–479.
- Gonzales DA, Norsworthy KJ, Kern SJ, et al. A meta-analysis of N-acetylcysteine in contrast-induced nephrotoxicity: unsupervised clustering to resolve heterogeneity. BMC Med 2007; 5:32.
- Levine GN, Bates ER, Blankenship JC, et al. 2011 ACCF/AHA/SCAI Guideline for Percutaneous Coronary Intervention: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines and the Society for Cardiovascular Angiography and Interventions. Circulation 2011; 124:e574–e651.
- Kanter MZ. Comparison of oral and i.v. acetylcysteine in the treatment of acetaminophen poisoning. Am J Health Syst Pharm 2006; 63:1821–1827.
- ACT Investigators. Acetylcysteine for prevention of renal outcomes in patients undergoing coronary and peripheral vascular angiography: main results from the randomized Acetylcysteine for Contrast-induced nephropathy Trial (ACT). Circulation 2011; 124:1250–1259.
- Webb JG, Pate GE, Humphries KH, et al. A randomized controlled trial of intravenous N-acetylcysteine for the prevention of contrast-induced nephropathy after cardiac catheterization: lack of effect. Am Heart J 2004; 148:422–429.
- Tepel M, van der Giet M, Schwarzfeld C, Laufer U, Liermann D, Zidek W. Prevention of radiographic-contrast-agent-induced reductions in renal function by acetylcysteine. N Engl J Med 2000; 343:180–184.
- Shyu KG, Cheng JJ, Kuan P. Acetylcysteine protects against acute renal damage in patients with abnormal renal function undergoing a coronary procedure. J Am Coll Cardiol 2002; 40:1383–1388.
- Kidney Disease: Improving Global Outcomes (KDIGO) Acute Kidney Injury Work Group. KDIGO clinical practice guideline for acute kidney injury. Kidney Int 2012; 2(suppl 1):1–138.
- Rihal CS, Textor SC, Grill DE, et al. Incidence and prognostic importance of acute renal failure after percutaneous coronary intervention. Circulation 2002; 105:2259–2264.
- Baker CS, Wragg A, Kumar S, De Palma R, Baker LR, Knight CJ. A rapid protocol for the prevention of contrast-induced renal dysfunction: the RAPPID study. J Am Coll Cardiol 2003; 41:2114–2118.
- Marenzi G, Assanelli E, Marana I, et al. N-acetylcysteine and contrast-induced nephropathy in primary angioplasty. N Engl J Med 2006; 354:2773–2782.
- Gurm HS, Smith DE, Berwanger O, et al; BMC2 (Blue Cross Blue Shield of Michigan Cardiovascular Consortium). Contemporary use and effectiveness of N-acetylcysteine in preventing contrast-induced nephropathy among patients undergoing percutaneous coronary intervention. JACC Cardiovasc Interv 2012; 5:98–104.
- Duong MH, MacKenzie TA, Malenka DJ. N-acetylcysteine prophylaxis significantly reduces the risk of radiocontrast-induced nephropathy: comprehensive meta-analysis. Catheter Cardiovasc Interv 2005; 64:471–479.
- Gonzales DA, Norsworthy KJ, Kern SJ, et al. A meta-analysis of N-acetylcysteine in contrast-induced nephrotoxicity: unsupervised clustering to resolve heterogeneity. BMC Med 2007; 5:32.
- Levine GN, Bates ER, Blankenship JC, et al. 2011 ACCF/AHA/SCAI Guideline for Percutaneous Coronary Intervention: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines and the Society for Cardiovascular Angiography and Interventions. Circulation 2011; 124:e574–e651.
- Kanter MZ. Comparison of oral and i.v. acetylcysteine in the treatment of acetaminophen poisoning. Am J Health Syst Pharm 2006; 63:1821–1827.
Cognitive impairment in ICU survivors: Assessment and therapy
Intensive care medicine has dramatically evolved over the last 15 years, after reports from many landmark trials.1 Updated strategies for mechanical ventilation2 and “bundles” of strategies to optimize hemodynamic therapy3 have reduced the rates of morbidity and death from deadly critical conditions such as the adult respiratory distress syndrome (ARDS) and sepsis.
Despite these important improvements in short-term outcomes, it is increasingly recognized that intensive care unit (ICU) survivors suffer considerable long-term complications that affect their usual functioning.4 Recently, the Society of Critical Care Medicine convened a conference in which these long-term complications were named the “post-intensive care syndrome.”5
Quality of life, particularly its physical component, is considerably lower after a stay in the medical or surgical ICU.6–8 Posttraumatic stress disorder, depression, and sexual dysfunction are consistently reported years after ICU discharge.9–13
Perhaps the most frequently unrecognized complication in ICU survivors is cognitive impairment. Current data suggest that neurocognitive impairment after an ICU stay is common and that it persists 6 years or more after hospital discharge.
Hopkins et al14,15 analyzed 10 cohort studies of long-term cognitive impairment after an ICU stay; 5 of them focused on patients with ARDS. The prevalence of cognitive impairment was as high as 78% at hospital discharge, 46% at 1 year, and 25% 6 years after discharge.15,16 Of the cognitive domains compromised, memory was the most often affected, followed by executive function and attention.14,17
Interestingly, data suggest that cognition may improve somewhat in the first 6 to 12 months after ICU discharge.15 Therefore, if we can detect it early on and promptly refer patients for cognitive therapy, we may eventually improve the prognosis of this disabling complication.
This review will focus on how to evaluate, prevent, and treat cognitive impairment in patients who survive an ICU stay.
COGNITIVE IMPAIRMENT AFTER A STAY IN THE ICU
The association between ICU stay and neurocognitive dysfunction is poorly understood. Potential causes include hypoxemia,18 hypotension, 19 hyperglycemia,14 and—an area of growing interest and evolving research—sedation and delirium.20
Patients on mechanical ventilation are commonly given sedatives and analgesics to prevent anxiety and pain.21 However, these medications are strongly associated with delirium.22 In fact, recent studies found that benzodiazepines have an independent, dose-related, temporal association with delirium, with some reports describing a 20% increase in delirium per milligram of benzodiazepine.23 In another study, which included medical and surgical ICU patients, use of morphine was the strongest predictor of delirium, with a sixfold increase in odds over a period of 5 months.24
Delirium is important to prevent, diagnose, and treat, since it has a direct association with the development of long-term cognitive impairment.22,25 A review of studies that included 1,885 medical and surgical patients found that those who developed delirium during an ICU stay were three times more likely to have cognitive dysfunction when assessed 3 years later.20
Whether delirium is a primary disorder associated with cognitive impairment or if it only represents an underlying process leading to poor cognitive outcomes is unknown. As delirious patients are more likely to be older, to be mechanically ventilated, to require more sedation, and, in particular, to be sicker, the association between delirium and cognitive impairment may reflect the relationship between these risk factors and poor cognitive outcomes.26
Glucose and its relationship with cognitive function is another topic of investigation. A secondary analysis of a study that included ARDS survivors revealed that blood glucose values higher than 153 mg/dL, higher glucose variability, and duration of mechanical ventilation were associated with cognitive sequelae.27,28
Other studies focused on mechanical ventilation. In one study,29 one-third of patients who had been mechanically ventilated showed signs of neurocognitive impairment when they were evaluated 6 months after hospital discharge.
Mild cognitive impairment differs from cognitive impairment after an ICU stay
Cognitive impairment after ICU discharge does not follow the same pattern as mild cognitive impairment, and some authors consider these two types of cognitive impairment to be unrelated.
While mild cognitive impairment is progressive and associated with aging, cognitive impairment in ICU survivors develops rapidly after acute illness and is usually related to numerous pathologic and neurochemical pathways.
For example, the neurotransmitter acetylcholine is thought to be involved in cognitive function as well as neuroplasticity of the motor cortex. In a model of cognitive impairment after stroke, activity of the cholinergic system was reduced.30,31 Further, in a study in rats, Baskerville et al32 showed that experience-dependent plasticity could be completely blocked by damaging the cholinergic neurons in the nucleus basalis of Meynert, thereby affecting memory and other functions supported by this pathway.
Another implicated pathway involves dopamine. Of interest, dopamine augmentation has been shown to enhance simple motor memories and to improve procedural learning. Understanding of these neurochemical alterations opens opportunities for investigation of drug therapies.
ASSESSMENT TOOLS
Cognitive impairment is important to detect in ICU survivors because it predicts poor outcomes from rehabilitation. A study of stroke patients found that those with cognitive alterations immediately after the stroke were less likely to be discharged home or to be living at home 6 months after discharge.33
A possible explanation may be that affected patients cannot fully participate in rehabilitation activities, owing to impairment in executive function, inability to remember therapy instructions, or disruption of implicit and explicit learning. Indeed, some authors consider cognitive impairment after acquired brain injury to be the most relevant surrogate marker of rehabilitation potential. Consequently, manipulation or enhancement of cognition may directly affect rehabilitation outcomes.34
Disagreement about terminology and diagnostic criteria creates a problem for health care providers working with patients with potential cognitive impairment. Numerous systems have been proposed to define this condition; in fact, Stephan et al35 reviewed the literature and found no fewer than 17. None of them is specific for cognitive impairment after an ICU stay.
Petersen et al36 in 1999 proposed initial criteria for mild cognitive impairment that included the following:
- A memory complaint
- Normal general cognitive functioning
- Normal activities of daily living
- Memory impairment in relation to age and education
- No dementia.
Later, other areas of impairment besides memory were recognized, such as language, attention, perception, reasoning, and motor planning.37 Therefore, mild cognitive impairment is currently classified into subtypes, which include amnestic (affecting single or multiple domains) and nonamnestic (also affecting single or multiple domains).38
In clinical practice, impairment of specific cognitive domains may be challenging to detect, and neuropsychological testing is often needed. Cognitive screening tests can detect impairment across a restricted range of cognitive abilities, while more comprehensive assessments address each of the primary domains of cognition.39 Formal testing provides normative and validated data on cognition performance and severity.
The Montreal Cognitive Assessment40 is popular, comprehensive, used in a variety of professions in diverse types of facilities (acute care, rehabilitation, and skilled care facilities), and brief (taking 11 minutes to administer). It evaluates orientation, memory, language, attention, reasoning, and visual-constructional abilities. The maximum score is 30; cognitive impairment is defined as a score of less than 26. It has a sensitivity of 90% and a specificity of 87%.
The Folstein Mini-Mental State Examination (MMSE) is the most commonly used of the noncomprehensive tests in clinical practice.41 It assesses orientation, memory, language, attention, and praxis. It has a maximum score of 30 points; the cutoff score for cognitive impairment is 24 points or less.
A limitation of the MMSE is that its sensitivity is very low, ranging from 1% to 49%.42,43 The MMSE scores of patients with cognitive impairment overlap considerably with those of age-matched healthy controls.39 Conversely, the MMSE’s specificity is usually high, ranging from 85% to 100%.42
Moreover, the MMSE poses copyright issues, an important consideration when selecting a test. In 2001, the authors of the MMSE transferred all intellectual property rights to Psychological Assessment Resources, which has exclusive rights to publish, license, and manage all intellectual property rights in all media and languages. Photocopying and using the MMSE without applying for permission from and paying this company ($1.23 per use) constitutes copyright infringement. Therefore, health care providers and researchers have been using other tests to evaluate cognition.
Other tests of cognition assess individual domains. Interestingly, studies of long-term cognitive impairment after ICU admission used these tests to define outcomes.25 Specific tests include:
- The Digit Span and the Trailmaking Test A (used to assess attention and orientation)25
- The Rey Auditory Verbal Learning Test (used to evaluate verbal memory)
- The Complex Figure Test (helpful in defining visual-spatial construction and delayed visual memory)
- The Trailmaking Test B (also included in the Montreal Cognitive Assessment; assesses executive functioning).
Besides formal testing, an informal battery is often recommended to provide additional information. An informal evaluation includes word definition, reading and verbal fluency, reading comprehension, and performance of instrumental activities of daily living. Observing as patients perform tasks of daily living provides therapists with a vast amount of information, as these tasks require using multiple cognitive processes. Therefore, if a functional breakdown occurs during this assessment, the clinician needs to identify the domain or specific level of cognitive dysfunction involved in that deficit.44
PREVENTIVE STRATEGIES
Strategies for minimizing the long-term effects of cognitive impairment have mostly focused on preventing it.
During the ICU stay, optimizing hemodynamic, glucose, and oxygenation levels may prevent future long-term complications.18
Also, the association between sedation, delirium, and consequent cognitive impairment (see above) has led many investigators to apply the “ABCDE” bundle of strategies.25,45,46 Specifically, ABCDE stands for awakening and breathing, choice of sedatives with fewer adverse effects, daily delirium monitoring, and early mobility exercise. These strategies have been shown in randomized controlled trials to prevent delirium; however, they have not been proved to prevent cognitive impairment.
Awakening and breathing
In the Awakening and Breathing Controlled Trial,47 patients in the intervention group (ie, those who had their sedatives interrupted every morning to see if they would awaken, and if so, if they could breathe on their own) were extubated 3 days sooner than those in the control group (who underwent daily trials of spontaneous breathing, if deemed safe). Also, ICU and hospital length of stay were shorter by 4 days. Best of all, over 1 year, the mortality rate was lower by 14 absolute percentage points.
Choice of sedatives
Often, mechanically ventilated patients are given benzodiazepines, opiates, and propofol (Diprivan).21 Dexmedetomidine (Precedex), a newer agent, is an alpha-2 agonist and may offer advantages over the others.
To date, three randomized controlled trials have assessed the effect of dexmedetomidine in terms of outcomes associated with delirium, and one trial evaluated its association with intellectual capacity in ICU patients.
The Maximizing Efficacy of Targeted Sedation and Reducing Neurological Dysfunction (MENDS) trial randomized patients on mechanical ventilation to receive either dexmedetomidine or lorazepam (Ativan).48 Dexmedetomidine-treated patients had 4 more days alive without delirium or coma (7 vs 3 days, P = .01).
Subsequently, the Safety and Efficacy of Dexmedetomidine Compared With Midazolam (SEDCOM) trial compared dexmedetomidine and midazolam (Versed) in mechanically ventilated patients. Those who received dexmedetomidine had a lower incidence of delirium (54% vs 76%, P < .001), and 2 fewer days on mechanical ventilation.49
Reade et al50 evaluated time to extubation in already delirious patients randomized to receive either dexmedetomidine or haloperidol (Haldol). Those receiving dexmedetomidine had a shorter time to extubation as well as a shorter ICU length of stay.
The Acute Neuroscience Intensive Care Sedation Trial51 evaluated intellectual capacity in neurological ICU patients sedated with either dexmedetomidine or propofol. This randomized, double-blind trial included 18 brain-injured and 12 non-brain-injured intubated patients. In a crossover protocol, each received the combination of fentanyl (Sublimaze) and propofol and the combination of fentanyl and dexmedetomidine.
Cognition was evaluated using the Adapted Cognitive Exam (ACE), which assesses intellectual capacity through orientation, language, registration, attention, calculation, and recall. This 10-minute examination does not require verbal communication, as it relies on the ability to respond to yes-or-no questions and perform simple motor tasks. The maximum possible score is 100 points.
Interestingly, while on propofol, the patients’ adjusted ACE scores went down by a mean of 12.4 points, whereas they went up by 6.8 points while on dexmedetomidine. Even though brain-injured patients required less sedation than non-brain-injured patients, the effect of dexmedetomidine and propofol did not change.51
In summary, these studies suggest that all sedatives are not the same in their short-term and intermediate-term outcomes.
In our practice, we use dexmedetomidine as our first-line sedation therapy. In patients with hemodynamic instability, we use benzodiazepines. We reserve propofol for very short periods of intubation or for hemodynamically stable patients who cannot be sedated with dexmedetomidine.
Daily delirium monitoring
As mentioned above, delirium affects many patients on mechanical ventilation, and it is highly underrecognized if valid tests are not used.52 Therefore, it is critically important to be familiar with the tests for assessing delirium. Of these, the Confusion Assessment Method for the ICU is probably the one with the best performance, with a sensitivity of 93% to 100% and a specificity of 98% to 100%.53,54
Early mobilization
A landmark study paired the awakening and breathing strategy with early mobilization through physical and occupational therapy in the ICU.55 Patients in the intervention group had a higher rate of return to independent functional status upon hospital discharge and a shorter duration of mechanical ventilation and delirium.
In conclusion, even though direct prevention of cognitive dysfunction is a challenging task, the ABCDE approach targets individual risk factors for delirium, which is an important contributor to cognitive impairment. Whether the ABCDE bundle directly affects the development of cognitive impairment requires further investigation.
COGNITIVE THERAPIES
The cognition-focused intervention most often described is cognitive training. Cognitive training is delivered in individual or group sessions in which the patient practices tasks targeting different domains, such as memory, language, and attention. Outcomes are often assessed in terms of improvement in test scores or effects on everyday functioning. Unfortunately, because of heterogeneity among cognitive training interventions and studied populations, we cannot yet make strong evidence-based recommendations for clinical practice.
Martin et al56 in 2011 reviewed cognition-based interventions for healthy older people and people with mild cognitive impairment and found 36 relevant studies. Of these, only 3 were in patients with mild cognitive impairment, while the rest were in healthy older people.56–58 Overall, the only available data were related to the memory domain, and outcomes were mostly associated with immediate recall of words, paragraphs, and stories. Based on this, cognitive therapy is currently considered justified, as most patients with cognitive impairment after an ICU stay have memory problems.
Zelinski et al59 conducted a randomized, controlled, double-blind study comparing outcomes in an intervention group that underwent a computerized cognitive training program with those in a control group that viewed videos on a variety of topics such as literature, art, and history. The intervention, based on brain plasticity, aimed to improve the speed and accuracy of auditory information processing and to engage neuromodulatory systems. Some of the secondary outcomes favored the intervention group. These outcomes were related mostly to measures of overall memory, such as immediate and delayed recall, but also to a composite outcome that included letter-number sequencing and the digit span backwards test.
Despite these encouraging results, it is worth mentioning that these studies were not performed in patients with cognitive impairment associated with ICU admission. Therefore, the applicability and effectiveness of such therapies in post-ICU patients remains unknown.
Patients with posttraumatic brain injury and stroke have also been extensively studied in regard to the development of cognitive impairment.34 These patients probably represent a better standard for comparison, as their cognitive impairment does not necessarily progress.
The effect of cognitive rehabilitation on the recovery in these patients depends on adaptation and remediation. Adaptation describes a patient’s ability to compensate for functional impairment.34 This can be divided into internal and external adaptation. Internal adaptation requires the patient to recognize his or her cognitive limitation in order to adapt the to the environment accordingly. External adaptation entails getting help from devices or relatives (eg, phone calls) to achieve desired goals (eg, taking medication at scheduled times). Again, to adapt, the patient needs to be able to recognize his or her affected cognitive domain. Unfortunately, this is not always the case.
Remediation refers to the actual regaining of a lost ability. To stimulate neural plasticity, the patient is required to experience and repeat targeted skill-building activities.38 There is evidence that patients are more likely to regain lost ability by repeating the practice frequently during a short period of time.60
From the physician’s perspective, evaluating and identifying deficits in particular cognitive domains may help in designing a remediation plan in partnership with a cognitive therapist.
Cognitive rehabilitation in ICU survivors
The Returning to Everyday Tasks Utilizing Rehabilitation Networks (RETURN) study focused on cognitive and physical rehabilitation in post-ICU patients.61 This pilot study included 21 ICU survivors with cognitive or functional impairment at hospital discharge. Eight patients received usual care and 13 received a combination of in-home cognitive, physical, and functional rehabilitation over a 3-month period with a social worker or a master’s-level psychology technician.
Interventions included six in-person visits for cognitive rehabilitation and six televisits for physical and functional rehabilitation. Cognitive training was based on the goal-management training (GMT) protocol.62 This strategy attempts to improve executive function by increasing goal-directed behavior and by helping patients learn to be reflective before making decisions and executing tasks. The GMT model consists of sessions that build on one another to increase the rehabilitation intensity. During each session, goals are explained and participants perform increasingly challenging cognitive tasks.
Cognitive outcomes were evaluated using the Delis-Kaplan Tower Test to evaluate executive function by assessing the ability to plan and strategize efficiently. The patient is required to move disks across three pegs until a tower is built. The object is to use the fewest moves possible while adhering to two rules: larger disks cannot be placed on top of smaller ones, and disks must be moved one at a time, using only one hand.
At 3 months there was a significant difference between groups, with the intervention group earning higher tower test scores than controls did (median of 13 vs 7.5).
The Activity and Cognitive Therapy in the Intensive Care Unit (ACT-ICU) trial is another pilot study that will attempt to assess the feasibility of early cognitive rehabilitation in ICU survivors. This study will combine early mobilization with a cognitive intervention, and its primary outcome is executive function (with the tower test) at 3 months after discharge.63
DRUG THERAPY
Some medications have been tested to assess whether they reduce the risk of progression from adult traumatic brain injury to cognitive impairment. These drugs augment dopamine and acetylcholine activity.
Methylphenidate (Ritalin), a dopaminergic drug, was studied in two trials. The first was a double-blind trial in 18 patients with posttraumatic brain injury. Memory was found to improve, based on the Working Memory Task Test. However, due to the small number of participants, no further conclusions were obtained.64
The second trial, in 19 patients with posttraumatic brain injury, had a double-blind crossover design. Attention, evaluated by the Distraction Task Test, improved with the use of methylphenidate.65 Again, the small number of patients precludes generalization of these results.
Donepezil (Aricept), a cholinergic drug, was evaluated in four clinical trials in posttraumatic brain injury patients66–69; each trial included 21 to 180 patients. The trials evaluated the drug’s effect on memory and attention through a variety of tools (Paced Auditory Serial Addition Test; Wechsler Memory Scale; Boston Naming Test; Rey Auditory Verbal Learning Test; Complex Figure Test; and Reaction Time–Dual Task). Interestingly, donepezil was associated with large improvements in objective assessments of attention and memory. Despite methodologic flaws, such as a lack of blinding in one of these studies69 and an open-label design in two of them,66,68 of the drugs available, donepezil presents the strongest evidence for use in cognitive impairment after traumatic brain injury.70
- Diaz-Guzman E, Sanchez J, Arroliga AC. Update in intensive care medicine: studies that challenged our practice in the last 5 years. Cleve Clin J Med 2011; 78:665–674.
- Acute Respiratory Distress Syndrome Network. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med 2000; 342:1301–1308.
- Rivers E, Nguyen B, Havstad S, et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med 2001; 345:1368–1377.
- Oeyen SG, Vandijck DM, Benoit DD, Annemans L, Decruyenaere JM. Quality of life after intensive care: a systematic review of the literature. Crit Care Med 2010; 38:2386–2400.
- Needham DM, Davidson J, Cohen H, et al. Improving long-term outcomes after discharge from intensive care unit: report from a stakeholders’ conference. Crit Care Med 2012; 40:502–509.
- Herridge MS, Cheung AM, Tansey CM, et al. One-year outcomes in survivors of the acute respiratory distress syndrome. N Engl J Med 2003; 348:683–693.
- Herridge MS, Tansey CM, Matte A, et al. Functional disability 5 years after acute respiratory distress syndrome. N Engl J Med 2011; 364:1293–1304.
- Timmers TK, Verhofstad MH, Moons KG, van Beeck EF, Leenen LP. Long-term quality of life after surgical intensive care admission. Arch Surg 2011; 146:412–418.
- Michaels AJ, Michaels CE, Moon CH, et al. Posttraumatic stress disorder after injury: impact on general health outcome and early risk assessment. J Trauma 1999; 47:460–466; discussion466–467.
- Stoll C, Schelling G, Goetz AE, et al. Health-related quality of life and post-traumatic stress disorder in patients after cardiac surgery and intensive care treatment. J Thorac Cardiovasc Surg 2000; 120:505–512.
- Jones C, Skirrow P, Griffiths RD, et al Post-traumatic stress disorder-related symptoms in relatives of patients following intensive care. Intensive Care Med 2004; 30:456–460.
- Griffiths J, Gager M, Alder N, Fawcett D, Waldmann C, Quinlan J. A self-report-based study of the incidence and associations of sexual dysfunction in survivors of intensive care treatment. Intensive Care Med 2006; 32:445–451.
- Griffiths J, Waldmann C, Quinlan J. Sexual dysfunction in intensive care survivors. Br J Hosp Med (Lond) 2007; 68:470–473.
- Hopkins RO, Jackson JC. Long-term neurocognitive function after critical illness. Chest 2006; 130:869–878.
- Hopkins RO, Weaver LK, Collingridge D, Parkinson RB, Chan KJ, Orme JF. Two-year cognitive, emotional, and quality-of-life outcomes in acute respiratory distress syndrome. Am J Respir Crit Care Med 2005; 171:340–347.
- Rothenhausler HB, Ehrentraut S, Stoll C, Schelling G, Kapfhammer HP. The relationship between cognitive performance and employment and health status in long-term survivors of the acute respiratory distress syndrome: results of an exploratory study. Gen Hosp Psychiatry 2001; 23:90–96.
- Sukantarat KT, Burgess PW, Williamson RC, Brett SJ. Prolonged cognitive dysfunction in survivors of critical illness. Anaesthesia 2005; 60:847–853.
- Hopkins RO, Weaver LK, Pope D, Orme JF, Bigler ED, Larson LV. Neuropsychological sequelae and impaired health status in survivors of severe acute respiratory distress syndrome. Am J Respir Crit Care Med 1999; 160:50–56.
- Hopkins RO, Weaver LK, Chan KJ, Orme JF. Quality of life, emotional, and cognitive function following acute respiratory distress syndrome. J Int Neuropsychol Soc 2004; 10:1005–1017.
- Jackson JC, Gordon SM, Hart RP, Hopkins RO, Ely EW. The association between delirium and cognitive decline: a review of the empirical literature. Neuropsychol Rev 2004; 14:87–98.
- Arroliga AC, Thompson BT, Ancukiewicz M, et al. Use of sedatives, opioids, and neuromuscular blocking agents in patients with acute lung injury and acute respiratory distress syndrome. Crit Care Med 2008; 36:1083–1088.
- Miller RR, Ely EW. Delirium and cognitive dysfunction in the intensive care unit. Semin Respir Crit Care Med 2006; 27:210–220.
- Pandharipande P, Shintani A, Peterson J, et al. Lorazepam is an independent risk factor for transitioning to delirium in intensive care unit patients. Anesthesiology 2006; 104:21–26.
- Dubois MJ, Bergeron N, Dumont M, Dial S, Skrobik Y. Delirium in an intensive care unit: a study of risk factors. Intensive Care Med 2001; 27:1297–1304.
- Girard TD, Jackson JC, Pandharipande PP, et al. Delirium as a predictor of long-term cognitive impairment in survivors of critical illness. Crit Care Med 2010; 38:1513–1520.
- Miller RR, Ely EW. Delirium and cognitive dysfunction in the intensive care unit. Curr Psychiatry Rep 2007; 9:26–34.
- Hopkins RO, Suchyta MR, Snow GL, Jephson A, Weaver LK, Orme JF. Blood glucose dysregulation and cognitive outcome in ARDS survivors. Brain Inj 2010; 24:1478–1484.
- Hough CL, Herridge MS. Long-term outcome after acute lung injury. Curr Opin Crit Care 2012; 18:8–15.
- Jackson JC, Hart RP, Gordon SM, et al. Six-month neuropsychological outcome of medical intensive care unit patients. Crit Care Med 2003; 31:1226–1234.
- Court JA, Perry EK. Neurotransmitter abnormalities in vascular dementia. Int Psychogeriatr 2003; 15(suppl 1):81–87.
- Gottfries CG, Blennow K, Karlsson I, Wallin A. The neurochemistry of vascular dementia. Dementia 1994; 5:163–167.
- Baskerville KA, Schweitzer JB, Herron P. Effects of cholinergic depletion on experience-dependent plasticity in the cortex of the rat. Neuroscience 1997; 80:1159–1169.
- Henon H, Lebert F, Durieu I, et al. Confusional state in stroke: relation to preexisting dementia, patient characteristics, and outcome. Stroke 1999; 30:773–779.
- Whyte E, Skidmore E, Aizenstein H, Ricker J, Butters M. Cognitive impairment in acquired brain injury: a predictor of rehabilitation outcomes and an opportunity for novel interventions. PMR 2011; 3(suppl 1):S45–S51.
- Stephan BC, Matthews FE, McKeith IG, Bond J, Brayne C. Early cognitive change in the general population: how do different definitions work? J Am Geriatr Soc 2007; 55:1534–1540.
- Petersen RC, Smith GE, Waring SC, Ivnik RJ, Tangalos EG, Kokmen E. Mild cognitive impairment: clinical characterization and outcome. Arch Neurol 1999; 56:303–308.
- Palmer K, Fratiglioni L, Winblad B. What is mild cognitive impairment? Variations in definitions and evolution of nondemented persons with cognitive impairment. Acta Neurol Scand Suppl 2003; 179:14–20.
- Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med 2004; 256:183–194.
- Lonie JA, Tierney KM, Ebmeier KP. Screening for mild cognitive impairment: a systematic review. Int J Geriatr Psychiatry 2009; 24:902–915.
- Nasreddine ZS, Phillips NA, Bedirian V, et al. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc 2005; 53:695–699.
- Folstein MF, Folstein SE, McHugh PR. “Mini-mental state.” A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975; 12:189–198.
- Sager MA, Hermann BP, La Rue A, Woodard JL. Screening for dementia in community-based memory clinics. WMJ 2006; 105:25–29.
- Ravaglia G, Forti P, Maioli F, et al. Screening for mild cognitive impairment in elderly ambulatory patients with cognitive complaints. Aging Clin Exp Res 2005; 17:374–379.
- Vogenthaler DR. An overview of head injury: its consequences and rehabilitation. Brain Inj 1987; 1:113–127.
- van den Boogaard M, Schoonhoven L, Evers AW, van der Hoeven JG, van Achterberg T, Pickkers P. Delirium in critically ill patients: impact on long-term health-related quality of life and cognitive functioning. Crit Care Med 2012; 40:112–118.
- Morandi A, Brummel NE, Ely EW. Sedation, delirium and mechanical ventilation: the ‘ABCDE’ approach. Curr Opin Crit Care 2011; 17:43–49.
- Girard TD, Kress JP, Fuchs BD, et al. Efficacy and safety of a paired sedation and ventilator weaning protocol for mechanically ventilated patients in intensive care (Awakening and Breathing Controlled trial): a randomised controlled trial. Lancet 2008; 371:126–134.
- Pandharipande PP, Pun BT, Herr DL, et al. Effect of sedation with dexmedetomidine vs lorazepam on acute brain dysfunction in mechanically ventilated patients: the MENDS randomized controlled trial. JAMA 2007; 298:2644–2653.
- Riker RR, Shehabi Y, Bokesch PM, et al. Dexmedetomidine vs midazolam for sedation of critically ill patients: a randomized trial. JAMA 2009; 301:489–499.
- Reade MC, O’Sullivan K, Bates S, Goldsmith D, Ainslie WR, Bellomo R. Dexmedetomidine vs. haloperidol in delirious, agitated, intubated patients: a randomised open-label trial. Crit Care 2009; 13:R75.
- Mirski MA, Lewin JJ, Ledroux S, et al. Cognitive improvement during continuous sedation in critically ill, awake and responsive patients: the Acute Neurological ICU Sedation Trial (ANIST). Intensive Care Med 2010; 36:1505–1513.
- Spronk PE, Riekerk B, Hofhuis J, Rommes JH. Occurrence of delirium is severely underestimated in the ICU during daily care. Intensive Care Med 2009; 35:1276–1280.
- Ely EW, Inouye SK, Bernard GR, et al. Delirium in mechanically ventilated patients: validity and reliability of the confusion assessment method for the intensive care unit (CAM-ICU). JAMA 2001; 286:2703–2710.
- Luetz A, Heymann A, Radtke FM, et al. Different assessment tools for intensive care unit delirium: which score to use? Crit Care Med 2010; 38:409–418.
- Schweickert WD, Pohlman MC, Pohlman AS, et al. Early physical and occupational therapy in mechanically ventilated, critically ill patients: a randomised controlled trial. Lancet 2009; 373:1874–1882.
- Martin M, Clare L, Altgassen AM, Cameron MH, Zehnder F. Cognition-based interventions for healthy older people and people with mild cognitive impairment. Cochrane Database Syst Rev 2011(1):CD006220.
- Rozzini L, Costardi D, Chilovi BV, Franzoni S, Trabucchi M, Padovani A. Efficacy of cognitive rehabilitation in patients with mild cognitive impairment treated with cholinesterase inhibitors. Int J Geriatr Psychiatry 2007; 22:356–360.
- Jean L, Bergeron ME, Thivierge S, Simard M. Cognitive intervention programs for individuals with mild cognitive impairment: systematic review of the literature. Am J Geriatr Psychiatry 2010; 18:281–296.
- Zelinski EM, Spina LM, Yaffe K, et al. Improvement in memory with plasticity-based adaptive cognitive training: results of the 3-month follow-up. J Am Geriatr Soc 2011; 59:258–265.
- Cicerone KD, Dahlberg C, Malec JF, et al. Evidence-based cognitive rehabilitation: updated review of the literature from 1998 through 2002. Arch Phys Med Rehabil 2005; 86:1681–1692.
- Jackson JC, Ely EW, Morey MC, et al. Cognitive and physical rehabilitation of intensive care unit survivors: results of the RETURN randomized controlled pilot investigation. Crit Care Med 2012; 40:1088–1097.
- Levine B, Stuss DT, Winocur G, et al. Cognitive rehabilitation in the elderly: effects on strategic behavior in relation to goal management. J Int Neuropsychol Soc 2007; 13:143–152.
- ACT-ICU Study: Activity and Cognitive Therapy in the Intensive Care Unit. http://clinicaltrials.gov/ct2/show/NCT01270269. Accessed August 9, 2012.
- Kim YH, Ko MH, Na SY, Park SH, Kim KW. Effects of single-dose methylphenidate on cognitive performance in patients with traumatic brain injury: a double-blind placebo-controlled study. Clin Rehabil 2006; 20:24–30.
- Whyte J, Hart T, Schuster K, Fleming M, Polansky M, Coslett HB. Effects of methylphenidate on attentional function after traumatic brain injury. A randomized, placebo-controlled trial. Am J Phys Med Rehabil 1997; 76:440–450.
- Masanic CA, Bayley MT, VanReekum R, Simard M. Open-label study of donepezil in traumatic brain injury. Arch Phys Med Rehabil 2001; 82:896–901.
- Zhang L, Plotkin RC, Wang G, Sandel ME, Lee S. Cholinergic augmentation with donepezil enhances recovery in short-term memory and sustained attention after traumatic brain injury. Arch Phys Med Rehabil 2004; 85:1050–1055.
- Khateb A, Ammann J, Annoni JM, Diserens K. Cognition-enhancing effects of donepezil in traumatic brain injury. Eur Neurol 2005; 54:39–45.
- Kim YW, Kim DY, Shin JC, Park CI, Lee JD. The changes of cortical metabolism associated with the clinical response to donepezil therapy in traumatic brain injury. Clin Neuropharmacol 2009; 32:63–68.
- Wheaton P, Mathias JL, Vink R. Impact of pharmacological treatments on cognitive and behavioral outcome in the postacute stages of adult traumatic brain injury: a meta-analysis. J Clin Psychopharmacol 2011; 31:745–757.
Intensive care medicine has dramatically evolved over the last 15 years, after reports from many landmark trials.1 Updated strategies for mechanical ventilation2 and “bundles” of strategies to optimize hemodynamic therapy3 have reduced the rates of morbidity and death from deadly critical conditions such as the adult respiratory distress syndrome (ARDS) and sepsis.
Despite these important improvements in short-term outcomes, it is increasingly recognized that intensive care unit (ICU) survivors suffer considerable long-term complications that affect their usual functioning.4 Recently, the Society of Critical Care Medicine convened a conference in which these long-term complications were named the “post-intensive care syndrome.”5
Quality of life, particularly its physical component, is considerably lower after a stay in the medical or surgical ICU.6–8 Posttraumatic stress disorder, depression, and sexual dysfunction are consistently reported years after ICU discharge.9–13
Perhaps the most frequently unrecognized complication in ICU survivors is cognitive impairment. Current data suggest that neurocognitive impairment after an ICU stay is common and that it persists 6 years or more after hospital discharge.
Hopkins et al14,15 analyzed 10 cohort studies of long-term cognitive impairment after an ICU stay; 5 of them focused on patients with ARDS. The prevalence of cognitive impairment was as high as 78% at hospital discharge, 46% at 1 year, and 25% 6 years after discharge.15,16 Of the cognitive domains compromised, memory was the most often affected, followed by executive function and attention.14,17
Interestingly, data suggest that cognition may improve somewhat in the first 6 to 12 months after ICU discharge.15 Therefore, if we can detect it early on and promptly refer patients for cognitive therapy, we may eventually improve the prognosis of this disabling complication.
This review will focus on how to evaluate, prevent, and treat cognitive impairment in patients who survive an ICU stay.
COGNITIVE IMPAIRMENT AFTER A STAY IN THE ICU
The association between ICU stay and neurocognitive dysfunction is poorly understood. Potential causes include hypoxemia,18 hypotension, 19 hyperglycemia,14 and—an area of growing interest and evolving research—sedation and delirium.20
Patients on mechanical ventilation are commonly given sedatives and analgesics to prevent anxiety and pain.21 However, these medications are strongly associated with delirium.22 In fact, recent studies found that benzodiazepines have an independent, dose-related, temporal association with delirium, with some reports describing a 20% increase in delirium per milligram of benzodiazepine.23 In another study, which included medical and surgical ICU patients, use of morphine was the strongest predictor of delirium, with a sixfold increase in odds over a period of 5 months.24
Delirium is important to prevent, diagnose, and treat, since it has a direct association with the development of long-term cognitive impairment.22,25 A review of studies that included 1,885 medical and surgical patients found that those who developed delirium during an ICU stay were three times more likely to have cognitive dysfunction when assessed 3 years later.20
Whether delirium is a primary disorder associated with cognitive impairment or if it only represents an underlying process leading to poor cognitive outcomes is unknown. As delirious patients are more likely to be older, to be mechanically ventilated, to require more sedation, and, in particular, to be sicker, the association between delirium and cognitive impairment may reflect the relationship between these risk factors and poor cognitive outcomes.26
Glucose and its relationship with cognitive function is another topic of investigation. A secondary analysis of a study that included ARDS survivors revealed that blood glucose values higher than 153 mg/dL, higher glucose variability, and duration of mechanical ventilation were associated with cognitive sequelae.27,28
Other studies focused on mechanical ventilation. In one study,29 one-third of patients who had been mechanically ventilated showed signs of neurocognitive impairment when they were evaluated 6 months after hospital discharge.
Mild cognitive impairment differs from cognitive impairment after an ICU stay
Cognitive impairment after ICU discharge does not follow the same pattern as mild cognitive impairment, and some authors consider these two types of cognitive impairment to be unrelated.
While mild cognitive impairment is progressive and associated with aging, cognitive impairment in ICU survivors develops rapidly after acute illness and is usually related to numerous pathologic and neurochemical pathways.
For example, the neurotransmitter acetylcholine is thought to be involved in cognitive function as well as neuroplasticity of the motor cortex. In a model of cognitive impairment after stroke, activity of the cholinergic system was reduced.30,31 Further, in a study in rats, Baskerville et al32 showed that experience-dependent plasticity could be completely blocked by damaging the cholinergic neurons in the nucleus basalis of Meynert, thereby affecting memory and other functions supported by this pathway.
Another implicated pathway involves dopamine. Of interest, dopamine augmentation has been shown to enhance simple motor memories and to improve procedural learning. Understanding of these neurochemical alterations opens opportunities for investigation of drug therapies.
ASSESSMENT TOOLS
Cognitive impairment is important to detect in ICU survivors because it predicts poor outcomes from rehabilitation. A study of stroke patients found that those with cognitive alterations immediately after the stroke were less likely to be discharged home or to be living at home 6 months after discharge.33
A possible explanation may be that affected patients cannot fully participate in rehabilitation activities, owing to impairment in executive function, inability to remember therapy instructions, or disruption of implicit and explicit learning. Indeed, some authors consider cognitive impairment after acquired brain injury to be the most relevant surrogate marker of rehabilitation potential. Consequently, manipulation or enhancement of cognition may directly affect rehabilitation outcomes.34
Disagreement about terminology and diagnostic criteria creates a problem for health care providers working with patients with potential cognitive impairment. Numerous systems have been proposed to define this condition; in fact, Stephan et al35 reviewed the literature and found no fewer than 17. None of them is specific for cognitive impairment after an ICU stay.
Petersen et al36 in 1999 proposed initial criteria for mild cognitive impairment that included the following:
- A memory complaint
- Normal general cognitive functioning
- Normal activities of daily living
- Memory impairment in relation to age and education
- No dementia.
Later, other areas of impairment besides memory were recognized, such as language, attention, perception, reasoning, and motor planning.37 Therefore, mild cognitive impairment is currently classified into subtypes, which include amnestic (affecting single or multiple domains) and nonamnestic (also affecting single or multiple domains).38
In clinical practice, impairment of specific cognitive domains may be challenging to detect, and neuropsychological testing is often needed. Cognitive screening tests can detect impairment across a restricted range of cognitive abilities, while more comprehensive assessments address each of the primary domains of cognition.39 Formal testing provides normative and validated data on cognition performance and severity.
The Montreal Cognitive Assessment40 is popular, comprehensive, used in a variety of professions in diverse types of facilities (acute care, rehabilitation, and skilled care facilities), and brief (taking 11 minutes to administer). It evaluates orientation, memory, language, attention, reasoning, and visual-constructional abilities. The maximum score is 30; cognitive impairment is defined as a score of less than 26. It has a sensitivity of 90% and a specificity of 87%.
The Folstein Mini-Mental State Examination (MMSE) is the most commonly used of the noncomprehensive tests in clinical practice.41 It assesses orientation, memory, language, attention, and praxis. It has a maximum score of 30 points; the cutoff score for cognitive impairment is 24 points or less.
A limitation of the MMSE is that its sensitivity is very low, ranging from 1% to 49%.42,43 The MMSE scores of patients with cognitive impairment overlap considerably with those of age-matched healthy controls.39 Conversely, the MMSE’s specificity is usually high, ranging from 85% to 100%.42
Moreover, the MMSE poses copyright issues, an important consideration when selecting a test. In 2001, the authors of the MMSE transferred all intellectual property rights to Psychological Assessment Resources, which has exclusive rights to publish, license, and manage all intellectual property rights in all media and languages. Photocopying and using the MMSE without applying for permission from and paying this company ($1.23 per use) constitutes copyright infringement. Therefore, health care providers and researchers have been using other tests to evaluate cognition.
Other tests of cognition assess individual domains. Interestingly, studies of long-term cognitive impairment after ICU admission used these tests to define outcomes.25 Specific tests include:
- The Digit Span and the Trailmaking Test A (used to assess attention and orientation)25
- The Rey Auditory Verbal Learning Test (used to evaluate verbal memory)
- The Complex Figure Test (helpful in defining visual-spatial construction and delayed visual memory)
- The Trailmaking Test B (also included in the Montreal Cognitive Assessment; assesses executive functioning).
Besides formal testing, an informal battery is often recommended to provide additional information. An informal evaluation includes word definition, reading and verbal fluency, reading comprehension, and performance of instrumental activities of daily living. Observing as patients perform tasks of daily living provides therapists with a vast amount of information, as these tasks require using multiple cognitive processes. Therefore, if a functional breakdown occurs during this assessment, the clinician needs to identify the domain or specific level of cognitive dysfunction involved in that deficit.44
PREVENTIVE STRATEGIES
Strategies for minimizing the long-term effects of cognitive impairment have mostly focused on preventing it.
During the ICU stay, optimizing hemodynamic, glucose, and oxygenation levels may prevent future long-term complications.18
Also, the association between sedation, delirium, and consequent cognitive impairment (see above) has led many investigators to apply the “ABCDE” bundle of strategies.25,45,46 Specifically, ABCDE stands for awakening and breathing, choice of sedatives with fewer adverse effects, daily delirium monitoring, and early mobility exercise. These strategies have been shown in randomized controlled trials to prevent delirium; however, they have not been proved to prevent cognitive impairment.
Awakening and breathing
In the Awakening and Breathing Controlled Trial,47 patients in the intervention group (ie, those who had their sedatives interrupted every morning to see if they would awaken, and if so, if they could breathe on their own) were extubated 3 days sooner than those in the control group (who underwent daily trials of spontaneous breathing, if deemed safe). Also, ICU and hospital length of stay were shorter by 4 days. Best of all, over 1 year, the mortality rate was lower by 14 absolute percentage points.
Choice of sedatives
Often, mechanically ventilated patients are given benzodiazepines, opiates, and propofol (Diprivan).21 Dexmedetomidine (Precedex), a newer agent, is an alpha-2 agonist and may offer advantages over the others.
To date, three randomized controlled trials have assessed the effect of dexmedetomidine in terms of outcomes associated with delirium, and one trial evaluated its association with intellectual capacity in ICU patients.
The Maximizing Efficacy of Targeted Sedation and Reducing Neurological Dysfunction (MENDS) trial randomized patients on mechanical ventilation to receive either dexmedetomidine or lorazepam (Ativan).48 Dexmedetomidine-treated patients had 4 more days alive without delirium or coma (7 vs 3 days, P = .01).
Subsequently, the Safety and Efficacy of Dexmedetomidine Compared With Midazolam (SEDCOM) trial compared dexmedetomidine and midazolam (Versed) in mechanically ventilated patients. Those who received dexmedetomidine had a lower incidence of delirium (54% vs 76%, P < .001), and 2 fewer days on mechanical ventilation.49
Reade et al50 evaluated time to extubation in already delirious patients randomized to receive either dexmedetomidine or haloperidol (Haldol). Those receiving dexmedetomidine had a shorter time to extubation as well as a shorter ICU length of stay.
The Acute Neuroscience Intensive Care Sedation Trial51 evaluated intellectual capacity in neurological ICU patients sedated with either dexmedetomidine or propofol. This randomized, double-blind trial included 18 brain-injured and 12 non-brain-injured intubated patients. In a crossover protocol, each received the combination of fentanyl (Sublimaze) and propofol and the combination of fentanyl and dexmedetomidine.
Cognition was evaluated using the Adapted Cognitive Exam (ACE), which assesses intellectual capacity through orientation, language, registration, attention, calculation, and recall. This 10-minute examination does not require verbal communication, as it relies on the ability to respond to yes-or-no questions and perform simple motor tasks. The maximum possible score is 100 points.
Interestingly, while on propofol, the patients’ adjusted ACE scores went down by a mean of 12.4 points, whereas they went up by 6.8 points while on dexmedetomidine. Even though brain-injured patients required less sedation than non-brain-injured patients, the effect of dexmedetomidine and propofol did not change.51
In summary, these studies suggest that all sedatives are not the same in their short-term and intermediate-term outcomes.
In our practice, we use dexmedetomidine as our first-line sedation therapy. In patients with hemodynamic instability, we use benzodiazepines. We reserve propofol for very short periods of intubation or for hemodynamically stable patients who cannot be sedated with dexmedetomidine.
Daily delirium monitoring
As mentioned above, delirium affects many patients on mechanical ventilation, and it is highly underrecognized if valid tests are not used.52 Therefore, it is critically important to be familiar with the tests for assessing delirium. Of these, the Confusion Assessment Method for the ICU is probably the one with the best performance, with a sensitivity of 93% to 100% and a specificity of 98% to 100%.53,54
Early mobilization
A landmark study paired the awakening and breathing strategy with early mobilization through physical and occupational therapy in the ICU.55 Patients in the intervention group had a higher rate of return to independent functional status upon hospital discharge and a shorter duration of mechanical ventilation and delirium.
In conclusion, even though direct prevention of cognitive dysfunction is a challenging task, the ABCDE approach targets individual risk factors for delirium, which is an important contributor to cognitive impairment. Whether the ABCDE bundle directly affects the development of cognitive impairment requires further investigation.
COGNITIVE THERAPIES
The cognition-focused intervention most often described is cognitive training. Cognitive training is delivered in individual or group sessions in which the patient practices tasks targeting different domains, such as memory, language, and attention. Outcomes are often assessed in terms of improvement in test scores or effects on everyday functioning. Unfortunately, because of heterogeneity among cognitive training interventions and studied populations, we cannot yet make strong evidence-based recommendations for clinical practice.
Martin et al56 in 2011 reviewed cognition-based interventions for healthy older people and people with mild cognitive impairment and found 36 relevant studies. Of these, only 3 were in patients with mild cognitive impairment, while the rest were in healthy older people.56–58 Overall, the only available data were related to the memory domain, and outcomes were mostly associated with immediate recall of words, paragraphs, and stories. Based on this, cognitive therapy is currently considered justified, as most patients with cognitive impairment after an ICU stay have memory problems.
Zelinski et al59 conducted a randomized, controlled, double-blind study comparing outcomes in an intervention group that underwent a computerized cognitive training program with those in a control group that viewed videos on a variety of topics such as literature, art, and history. The intervention, based on brain plasticity, aimed to improve the speed and accuracy of auditory information processing and to engage neuromodulatory systems. Some of the secondary outcomes favored the intervention group. These outcomes were related mostly to measures of overall memory, such as immediate and delayed recall, but also to a composite outcome that included letter-number sequencing and the digit span backwards test.
Despite these encouraging results, it is worth mentioning that these studies were not performed in patients with cognitive impairment associated with ICU admission. Therefore, the applicability and effectiveness of such therapies in post-ICU patients remains unknown.
Patients with posttraumatic brain injury and stroke have also been extensively studied in regard to the development of cognitive impairment.34 These patients probably represent a better standard for comparison, as their cognitive impairment does not necessarily progress.
The effect of cognitive rehabilitation on the recovery in these patients depends on adaptation and remediation. Adaptation describes a patient’s ability to compensate for functional impairment.34 This can be divided into internal and external adaptation. Internal adaptation requires the patient to recognize his or her cognitive limitation in order to adapt the to the environment accordingly. External adaptation entails getting help from devices or relatives (eg, phone calls) to achieve desired goals (eg, taking medication at scheduled times). Again, to adapt, the patient needs to be able to recognize his or her affected cognitive domain. Unfortunately, this is not always the case.
Remediation refers to the actual regaining of a lost ability. To stimulate neural plasticity, the patient is required to experience and repeat targeted skill-building activities.38 There is evidence that patients are more likely to regain lost ability by repeating the practice frequently during a short period of time.60
From the physician’s perspective, evaluating and identifying deficits in particular cognitive domains may help in designing a remediation plan in partnership with a cognitive therapist.
Cognitive rehabilitation in ICU survivors
The Returning to Everyday Tasks Utilizing Rehabilitation Networks (RETURN) study focused on cognitive and physical rehabilitation in post-ICU patients.61 This pilot study included 21 ICU survivors with cognitive or functional impairment at hospital discharge. Eight patients received usual care and 13 received a combination of in-home cognitive, physical, and functional rehabilitation over a 3-month period with a social worker or a master’s-level psychology technician.
Interventions included six in-person visits for cognitive rehabilitation and six televisits for physical and functional rehabilitation. Cognitive training was based on the goal-management training (GMT) protocol.62 This strategy attempts to improve executive function by increasing goal-directed behavior and by helping patients learn to be reflective before making decisions and executing tasks. The GMT model consists of sessions that build on one another to increase the rehabilitation intensity. During each session, goals are explained and participants perform increasingly challenging cognitive tasks.
Cognitive outcomes were evaluated using the Delis-Kaplan Tower Test to evaluate executive function by assessing the ability to plan and strategize efficiently. The patient is required to move disks across three pegs until a tower is built. The object is to use the fewest moves possible while adhering to two rules: larger disks cannot be placed on top of smaller ones, and disks must be moved one at a time, using only one hand.
At 3 months there was a significant difference between groups, with the intervention group earning higher tower test scores than controls did (median of 13 vs 7.5).
The Activity and Cognitive Therapy in the Intensive Care Unit (ACT-ICU) trial is another pilot study that will attempt to assess the feasibility of early cognitive rehabilitation in ICU survivors. This study will combine early mobilization with a cognitive intervention, and its primary outcome is executive function (with the tower test) at 3 months after discharge.63
DRUG THERAPY
Some medications have been tested to assess whether they reduce the risk of progression from adult traumatic brain injury to cognitive impairment. These drugs augment dopamine and acetylcholine activity.
Methylphenidate (Ritalin), a dopaminergic drug, was studied in two trials. The first was a double-blind trial in 18 patients with posttraumatic brain injury. Memory was found to improve, based on the Working Memory Task Test. However, due to the small number of participants, no further conclusions were obtained.64
The second trial, in 19 patients with posttraumatic brain injury, had a double-blind crossover design. Attention, evaluated by the Distraction Task Test, improved with the use of methylphenidate.65 Again, the small number of patients precludes generalization of these results.
Donepezil (Aricept), a cholinergic drug, was evaluated in four clinical trials in posttraumatic brain injury patients66–69; each trial included 21 to 180 patients. The trials evaluated the drug’s effect on memory and attention through a variety of tools (Paced Auditory Serial Addition Test; Wechsler Memory Scale; Boston Naming Test; Rey Auditory Verbal Learning Test; Complex Figure Test; and Reaction Time–Dual Task). Interestingly, donepezil was associated with large improvements in objective assessments of attention and memory. Despite methodologic flaws, such as a lack of blinding in one of these studies69 and an open-label design in two of them,66,68 of the drugs available, donepezil presents the strongest evidence for use in cognitive impairment after traumatic brain injury.70
Intensive care medicine has dramatically evolved over the last 15 years, after reports from many landmark trials.1 Updated strategies for mechanical ventilation2 and “bundles” of strategies to optimize hemodynamic therapy3 have reduced the rates of morbidity and death from deadly critical conditions such as the adult respiratory distress syndrome (ARDS) and sepsis.
Despite these important improvements in short-term outcomes, it is increasingly recognized that intensive care unit (ICU) survivors suffer considerable long-term complications that affect their usual functioning.4 Recently, the Society of Critical Care Medicine convened a conference in which these long-term complications were named the “post-intensive care syndrome.”5
Quality of life, particularly its physical component, is considerably lower after a stay in the medical or surgical ICU.6–8 Posttraumatic stress disorder, depression, and sexual dysfunction are consistently reported years after ICU discharge.9–13
Perhaps the most frequently unrecognized complication in ICU survivors is cognitive impairment. Current data suggest that neurocognitive impairment after an ICU stay is common and that it persists 6 years or more after hospital discharge.
Hopkins et al14,15 analyzed 10 cohort studies of long-term cognitive impairment after an ICU stay; 5 of them focused on patients with ARDS. The prevalence of cognitive impairment was as high as 78% at hospital discharge, 46% at 1 year, and 25% 6 years after discharge.15,16 Of the cognitive domains compromised, memory was the most often affected, followed by executive function and attention.14,17
Interestingly, data suggest that cognition may improve somewhat in the first 6 to 12 months after ICU discharge.15 Therefore, if we can detect it early on and promptly refer patients for cognitive therapy, we may eventually improve the prognosis of this disabling complication.
This review will focus on how to evaluate, prevent, and treat cognitive impairment in patients who survive an ICU stay.
COGNITIVE IMPAIRMENT AFTER A STAY IN THE ICU
The association between ICU stay and neurocognitive dysfunction is poorly understood. Potential causes include hypoxemia,18 hypotension, 19 hyperglycemia,14 and—an area of growing interest and evolving research—sedation and delirium.20
Patients on mechanical ventilation are commonly given sedatives and analgesics to prevent anxiety and pain.21 However, these medications are strongly associated with delirium.22 In fact, recent studies found that benzodiazepines have an independent, dose-related, temporal association with delirium, with some reports describing a 20% increase in delirium per milligram of benzodiazepine.23 In another study, which included medical and surgical ICU patients, use of morphine was the strongest predictor of delirium, with a sixfold increase in odds over a period of 5 months.24
Delirium is important to prevent, diagnose, and treat, since it has a direct association with the development of long-term cognitive impairment.22,25 A review of studies that included 1,885 medical and surgical patients found that those who developed delirium during an ICU stay were three times more likely to have cognitive dysfunction when assessed 3 years later.20
Whether delirium is a primary disorder associated with cognitive impairment or if it only represents an underlying process leading to poor cognitive outcomes is unknown. As delirious patients are more likely to be older, to be mechanically ventilated, to require more sedation, and, in particular, to be sicker, the association between delirium and cognitive impairment may reflect the relationship between these risk factors and poor cognitive outcomes.26
Glucose and its relationship with cognitive function is another topic of investigation. A secondary analysis of a study that included ARDS survivors revealed that blood glucose values higher than 153 mg/dL, higher glucose variability, and duration of mechanical ventilation were associated with cognitive sequelae.27,28
Other studies focused on mechanical ventilation. In one study,29 one-third of patients who had been mechanically ventilated showed signs of neurocognitive impairment when they were evaluated 6 months after hospital discharge.
Mild cognitive impairment differs from cognitive impairment after an ICU stay
Cognitive impairment after ICU discharge does not follow the same pattern as mild cognitive impairment, and some authors consider these two types of cognitive impairment to be unrelated.
While mild cognitive impairment is progressive and associated with aging, cognitive impairment in ICU survivors develops rapidly after acute illness and is usually related to numerous pathologic and neurochemical pathways.
For example, the neurotransmitter acetylcholine is thought to be involved in cognitive function as well as neuroplasticity of the motor cortex. In a model of cognitive impairment after stroke, activity of the cholinergic system was reduced.30,31 Further, in a study in rats, Baskerville et al32 showed that experience-dependent plasticity could be completely blocked by damaging the cholinergic neurons in the nucleus basalis of Meynert, thereby affecting memory and other functions supported by this pathway.
Another implicated pathway involves dopamine. Of interest, dopamine augmentation has been shown to enhance simple motor memories and to improve procedural learning. Understanding of these neurochemical alterations opens opportunities for investigation of drug therapies.
ASSESSMENT TOOLS
Cognitive impairment is important to detect in ICU survivors because it predicts poor outcomes from rehabilitation. A study of stroke patients found that those with cognitive alterations immediately after the stroke were less likely to be discharged home or to be living at home 6 months after discharge.33
A possible explanation may be that affected patients cannot fully participate in rehabilitation activities, owing to impairment in executive function, inability to remember therapy instructions, or disruption of implicit and explicit learning. Indeed, some authors consider cognitive impairment after acquired brain injury to be the most relevant surrogate marker of rehabilitation potential. Consequently, manipulation or enhancement of cognition may directly affect rehabilitation outcomes.34
Disagreement about terminology and diagnostic criteria creates a problem for health care providers working with patients with potential cognitive impairment. Numerous systems have been proposed to define this condition; in fact, Stephan et al35 reviewed the literature and found no fewer than 17. None of them is specific for cognitive impairment after an ICU stay.
Petersen et al36 in 1999 proposed initial criteria for mild cognitive impairment that included the following:
- A memory complaint
- Normal general cognitive functioning
- Normal activities of daily living
- Memory impairment in relation to age and education
- No dementia.
Later, other areas of impairment besides memory were recognized, such as language, attention, perception, reasoning, and motor planning.37 Therefore, mild cognitive impairment is currently classified into subtypes, which include amnestic (affecting single or multiple domains) and nonamnestic (also affecting single or multiple domains).38
In clinical practice, impairment of specific cognitive domains may be challenging to detect, and neuropsychological testing is often needed. Cognitive screening tests can detect impairment across a restricted range of cognitive abilities, while more comprehensive assessments address each of the primary domains of cognition.39 Formal testing provides normative and validated data on cognition performance and severity.
The Montreal Cognitive Assessment40 is popular, comprehensive, used in a variety of professions in diverse types of facilities (acute care, rehabilitation, and skilled care facilities), and brief (taking 11 minutes to administer). It evaluates orientation, memory, language, attention, reasoning, and visual-constructional abilities. The maximum score is 30; cognitive impairment is defined as a score of less than 26. It has a sensitivity of 90% and a specificity of 87%.
The Folstein Mini-Mental State Examination (MMSE) is the most commonly used of the noncomprehensive tests in clinical practice.41 It assesses orientation, memory, language, attention, and praxis. It has a maximum score of 30 points; the cutoff score for cognitive impairment is 24 points or less.
A limitation of the MMSE is that its sensitivity is very low, ranging from 1% to 49%.42,43 The MMSE scores of patients with cognitive impairment overlap considerably with those of age-matched healthy controls.39 Conversely, the MMSE’s specificity is usually high, ranging from 85% to 100%.42
Moreover, the MMSE poses copyright issues, an important consideration when selecting a test. In 2001, the authors of the MMSE transferred all intellectual property rights to Psychological Assessment Resources, which has exclusive rights to publish, license, and manage all intellectual property rights in all media and languages. Photocopying and using the MMSE without applying for permission from and paying this company ($1.23 per use) constitutes copyright infringement. Therefore, health care providers and researchers have been using other tests to evaluate cognition.
Other tests of cognition assess individual domains. Interestingly, studies of long-term cognitive impairment after ICU admission used these tests to define outcomes.25 Specific tests include:
- The Digit Span and the Trailmaking Test A (used to assess attention and orientation)25
- The Rey Auditory Verbal Learning Test (used to evaluate verbal memory)
- The Complex Figure Test (helpful in defining visual-spatial construction and delayed visual memory)
- The Trailmaking Test B (also included in the Montreal Cognitive Assessment; assesses executive functioning).
Besides formal testing, an informal battery is often recommended to provide additional information. An informal evaluation includes word definition, reading and verbal fluency, reading comprehension, and performance of instrumental activities of daily living. Observing as patients perform tasks of daily living provides therapists with a vast amount of information, as these tasks require using multiple cognitive processes. Therefore, if a functional breakdown occurs during this assessment, the clinician needs to identify the domain or specific level of cognitive dysfunction involved in that deficit.44
PREVENTIVE STRATEGIES
Strategies for minimizing the long-term effects of cognitive impairment have mostly focused on preventing it.
During the ICU stay, optimizing hemodynamic, glucose, and oxygenation levels may prevent future long-term complications.18
Also, the association between sedation, delirium, and consequent cognitive impairment (see above) has led many investigators to apply the “ABCDE” bundle of strategies.25,45,46 Specifically, ABCDE stands for awakening and breathing, choice of sedatives with fewer adverse effects, daily delirium monitoring, and early mobility exercise. These strategies have been shown in randomized controlled trials to prevent delirium; however, they have not been proved to prevent cognitive impairment.
Awakening and breathing
In the Awakening and Breathing Controlled Trial,47 patients in the intervention group (ie, those who had their sedatives interrupted every morning to see if they would awaken, and if so, if they could breathe on their own) were extubated 3 days sooner than those in the control group (who underwent daily trials of spontaneous breathing, if deemed safe). Also, ICU and hospital length of stay were shorter by 4 days. Best of all, over 1 year, the mortality rate was lower by 14 absolute percentage points.
Choice of sedatives
Often, mechanically ventilated patients are given benzodiazepines, opiates, and propofol (Diprivan).21 Dexmedetomidine (Precedex), a newer agent, is an alpha-2 agonist and may offer advantages over the others.
To date, three randomized controlled trials have assessed the effect of dexmedetomidine in terms of outcomes associated with delirium, and one trial evaluated its association with intellectual capacity in ICU patients.
The Maximizing Efficacy of Targeted Sedation and Reducing Neurological Dysfunction (MENDS) trial randomized patients on mechanical ventilation to receive either dexmedetomidine or lorazepam (Ativan).48 Dexmedetomidine-treated patients had 4 more days alive without delirium or coma (7 vs 3 days, P = .01).
Subsequently, the Safety and Efficacy of Dexmedetomidine Compared With Midazolam (SEDCOM) trial compared dexmedetomidine and midazolam (Versed) in mechanically ventilated patients. Those who received dexmedetomidine had a lower incidence of delirium (54% vs 76%, P < .001), and 2 fewer days on mechanical ventilation.49
Reade et al50 evaluated time to extubation in already delirious patients randomized to receive either dexmedetomidine or haloperidol (Haldol). Those receiving dexmedetomidine had a shorter time to extubation as well as a shorter ICU length of stay.
The Acute Neuroscience Intensive Care Sedation Trial51 evaluated intellectual capacity in neurological ICU patients sedated with either dexmedetomidine or propofol. This randomized, double-blind trial included 18 brain-injured and 12 non-brain-injured intubated patients. In a crossover protocol, each received the combination of fentanyl (Sublimaze) and propofol and the combination of fentanyl and dexmedetomidine.
Cognition was evaluated using the Adapted Cognitive Exam (ACE), which assesses intellectual capacity through orientation, language, registration, attention, calculation, and recall. This 10-minute examination does not require verbal communication, as it relies on the ability to respond to yes-or-no questions and perform simple motor tasks. The maximum possible score is 100 points.
Interestingly, while on propofol, the patients’ adjusted ACE scores went down by a mean of 12.4 points, whereas they went up by 6.8 points while on dexmedetomidine. Even though brain-injured patients required less sedation than non-brain-injured patients, the effect of dexmedetomidine and propofol did not change.51
In summary, these studies suggest that all sedatives are not the same in their short-term and intermediate-term outcomes.
In our practice, we use dexmedetomidine as our first-line sedation therapy. In patients with hemodynamic instability, we use benzodiazepines. We reserve propofol for very short periods of intubation or for hemodynamically stable patients who cannot be sedated with dexmedetomidine.
Daily delirium monitoring
As mentioned above, delirium affects many patients on mechanical ventilation, and it is highly underrecognized if valid tests are not used.52 Therefore, it is critically important to be familiar with the tests for assessing delirium. Of these, the Confusion Assessment Method for the ICU is probably the one with the best performance, with a sensitivity of 93% to 100% and a specificity of 98% to 100%.53,54
Early mobilization
A landmark study paired the awakening and breathing strategy with early mobilization through physical and occupational therapy in the ICU.55 Patients in the intervention group had a higher rate of return to independent functional status upon hospital discharge and a shorter duration of mechanical ventilation and delirium.
In conclusion, even though direct prevention of cognitive dysfunction is a challenging task, the ABCDE approach targets individual risk factors for delirium, which is an important contributor to cognitive impairment. Whether the ABCDE bundle directly affects the development of cognitive impairment requires further investigation.
COGNITIVE THERAPIES
The cognition-focused intervention most often described is cognitive training. Cognitive training is delivered in individual or group sessions in which the patient practices tasks targeting different domains, such as memory, language, and attention. Outcomes are often assessed in terms of improvement in test scores or effects on everyday functioning. Unfortunately, because of heterogeneity among cognitive training interventions and studied populations, we cannot yet make strong evidence-based recommendations for clinical practice.
Martin et al56 in 2011 reviewed cognition-based interventions for healthy older people and people with mild cognitive impairment and found 36 relevant studies. Of these, only 3 were in patients with mild cognitive impairment, while the rest were in healthy older people.56–58 Overall, the only available data were related to the memory domain, and outcomes were mostly associated with immediate recall of words, paragraphs, and stories. Based on this, cognitive therapy is currently considered justified, as most patients with cognitive impairment after an ICU stay have memory problems.
Zelinski et al59 conducted a randomized, controlled, double-blind study comparing outcomes in an intervention group that underwent a computerized cognitive training program with those in a control group that viewed videos on a variety of topics such as literature, art, and history. The intervention, based on brain plasticity, aimed to improve the speed and accuracy of auditory information processing and to engage neuromodulatory systems. Some of the secondary outcomes favored the intervention group. These outcomes were related mostly to measures of overall memory, such as immediate and delayed recall, but also to a composite outcome that included letter-number sequencing and the digit span backwards test.
Despite these encouraging results, it is worth mentioning that these studies were not performed in patients with cognitive impairment associated with ICU admission. Therefore, the applicability and effectiveness of such therapies in post-ICU patients remains unknown.
Patients with posttraumatic brain injury and stroke have also been extensively studied in regard to the development of cognitive impairment.34 These patients probably represent a better standard for comparison, as their cognitive impairment does not necessarily progress.
The effect of cognitive rehabilitation on the recovery in these patients depends on adaptation and remediation. Adaptation describes a patient’s ability to compensate for functional impairment.34 This can be divided into internal and external adaptation. Internal adaptation requires the patient to recognize his or her cognitive limitation in order to adapt the to the environment accordingly. External adaptation entails getting help from devices or relatives (eg, phone calls) to achieve desired goals (eg, taking medication at scheduled times). Again, to adapt, the patient needs to be able to recognize his or her affected cognitive domain. Unfortunately, this is not always the case.
Remediation refers to the actual regaining of a lost ability. To stimulate neural plasticity, the patient is required to experience and repeat targeted skill-building activities.38 There is evidence that patients are more likely to regain lost ability by repeating the practice frequently during a short period of time.60
From the physician’s perspective, evaluating and identifying deficits in particular cognitive domains may help in designing a remediation plan in partnership with a cognitive therapist.
Cognitive rehabilitation in ICU survivors
The Returning to Everyday Tasks Utilizing Rehabilitation Networks (RETURN) study focused on cognitive and physical rehabilitation in post-ICU patients.61 This pilot study included 21 ICU survivors with cognitive or functional impairment at hospital discharge. Eight patients received usual care and 13 received a combination of in-home cognitive, physical, and functional rehabilitation over a 3-month period with a social worker or a master’s-level psychology technician.
Interventions included six in-person visits for cognitive rehabilitation and six televisits for physical and functional rehabilitation. Cognitive training was based on the goal-management training (GMT) protocol.62 This strategy attempts to improve executive function by increasing goal-directed behavior and by helping patients learn to be reflective before making decisions and executing tasks. The GMT model consists of sessions that build on one another to increase the rehabilitation intensity. During each session, goals are explained and participants perform increasingly challenging cognitive tasks.
Cognitive outcomes were evaluated using the Delis-Kaplan Tower Test to evaluate executive function by assessing the ability to plan and strategize efficiently. The patient is required to move disks across three pegs until a tower is built. The object is to use the fewest moves possible while adhering to two rules: larger disks cannot be placed on top of smaller ones, and disks must be moved one at a time, using only one hand.
At 3 months there was a significant difference between groups, with the intervention group earning higher tower test scores than controls did (median of 13 vs 7.5).
The Activity and Cognitive Therapy in the Intensive Care Unit (ACT-ICU) trial is another pilot study that will attempt to assess the feasibility of early cognitive rehabilitation in ICU survivors. This study will combine early mobilization with a cognitive intervention, and its primary outcome is executive function (with the tower test) at 3 months after discharge.63
DRUG THERAPY
Some medications have been tested to assess whether they reduce the risk of progression from adult traumatic brain injury to cognitive impairment. These drugs augment dopamine and acetylcholine activity.
Methylphenidate (Ritalin), a dopaminergic drug, was studied in two trials. The first was a double-blind trial in 18 patients with posttraumatic brain injury. Memory was found to improve, based on the Working Memory Task Test. However, due to the small number of participants, no further conclusions were obtained.64
The second trial, in 19 patients with posttraumatic brain injury, had a double-blind crossover design. Attention, evaluated by the Distraction Task Test, improved with the use of methylphenidate.65 Again, the small number of patients precludes generalization of these results.
Donepezil (Aricept), a cholinergic drug, was evaluated in four clinical trials in posttraumatic brain injury patients66–69; each trial included 21 to 180 patients. The trials evaluated the drug’s effect on memory and attention through a variety of tools (Paced Auditory Serial Addition Test; Wechsler Memory Scale; Boston Naming Test; Rey Auditory Verbal Learning Test; Complex Figure Test; and Reaction Time–Dual Task). Interestingly, donepezil was associated with large improvements in objective assessments of attention and memory. Despite methodologic flaws, such as a lack of blinding in one of these studies69 and an open-label design in two of them,66,68 of the drugs available, donepezil presents the strongest evidence for use in cognitive impairment after traumatic brain injury.70
- Diaz-Guzman E, Sanchez J, Arroliga AC. Update in intensive care medicine: studies that challenged our practice in the last 5 years. Cleve Clin J Med 2011; 78:665–674.
- Acute Respiratory Distress Syndrome Network. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med 2000; 342:1301–1308.
- Rivers E, Nguyen B, Havstad S, et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med 2001; 345:1368–1377.
- Oeyen SG, Vandijck DM, Benoit DD, Annemans L, Decruyenaere JM. Quality of life after intensive care: a systematic review of the literature. Crit Care Med 2010; 38:2386–2400.
- Needham DM, Davidson J, Cohen H, et al. Improving long-term outcomes after discharge from intensive care unit: report from a stakeholders’ conference. Crit Care Med 2012; 40:502–509.
- Herridge MS, Cheung AM, Tansey CM, et al. One-year outcomes in survivors of the acute respiratory distress syndrome. N Engl J Med 2003; 348:683–693.
- Herridge MS, Tansey CM, Matte A, et al. Functional disability 5 years after acute respiratory distress syndrome. N Engl J Med 2011; 364:1293–1304.
- Timmers TK, Verhofstad MH, Moons KG, van Beeck EF, Leenen LP. Long-term quality of life after surgical intensive care admission. Arch Surg 2011; 146:412–418.
- Michaels AJ, Michaels CE, Moon CH, et al. Posttraumatic stress disorder after injury: impact on general health outcome and early risk assessment. J Trauma 1999; 47:460–466; discussion466–467.
- Stoll C, Schelling G, Goetz AE, et al. Health-related quality of life and post-traumatic stress disorder in patients after cardiac surgery and intensive care treatment. J Thorac Cardiovasc Surg 2000; 120:505–512.
- Jones C, Skirrow P, Griffiths RD, et al Post-traumatic stress disorder-related symptoms in relatives of patients following intensive care. Intensive Care Med 2004; 30:456–460.
- Griffiths J, Gager M, Alder N, Fawcett D, Waldmann C, Quinlan J. A self-report-based study of the incidence and associations of sexual dysfunction in survivors of intensive care treatment. Intensive Care Med 2006; 32:445–451.
- Griffiths J, Waldmann C, Quinlan J. Sexual dysfunction in intensive care survivors. Br J Hosp Med (Lond) 2007; 68:470–473.
- Hopkins RO, Jackson JC. Long-term neurocognitive function after critical illness. Chest 2006; 130:869–878.
- Hopkins RO, Weaver LK, Collingridge D, Parkinson RB, Chan KJ, Orme JF. Two-year cognitive, emotional, and quality-of-life outcomes in acute respiratory distress syndrome. Am J Respir Crit Care Med 2005; 171:340–347.
- Rothenhausler HB, Ehrentraut S, Stoll C, Schelling G, Kapfhammer HP. The relationship between cognitive performance and employment and health status in long-term survivors of the acute respiratory distress syndrome: results of an exploratory study. Gen Hosp Psychiatry 2001; 23:90–96.
- Sukantarat KT, Burgess PW, Williamson RC, Brett SJ. Prolonged cognitive dysfunction in survivors of critical illness. Anaesthesia 2005; 60:847–853.
- Hopkins RO, Weaver LK, Pope D, Orme JF, Bigler ED, Larson LV. Neuropsychological sequelae and impaired health status in survivors of severe acute respiratory distress syndrome. Am J Respir Crit Care Med 1999; 160:50–56.
- Hopkins RO, Weaver LK, Chan KJ, Orme JF. Quality of life, emotional, and cognitive function following acute respiratory distress syndrome. J Int Neuropsychol Soc 2004; 10:1005–1017.
- Jackson JC, Gordon SM, Hart RP, Hopkins RO, Ely EW. The association between delirium and cognitive decline: a review of the empirical literature. Neuropsychol Rev 2004; 14:87–98.
- Arroliga AC, Thompson BT, Ancukiewicz M, et al. Use of sedatives, opioids, and neuromuscular blocking agents in patients with acute lung injury and acute respiratory distress syndrome. Crit Care Med 2008; 36:1083–1088.
- Miller RR, Ely EW. Delirium and cognitive dysfunction in the intensive care unit. Semin Respir Crit Care Med 2006; 27:210–220.
- Pandharipande P, Shintani A, Peterson J, et al. Lorazepam is an independent risk factor for transitioning to delirium in intensive care unit patients. Anesthesiology 2006; 104:21–26.
- Dubois MJ, Bergeron N, Dumont M, Dial S, Skrobik Y. Delirium in an intensive care unit: a study of risk factors. Intensive Care Med 2001; 27:1297–1304.
- Girard TD, Jackson JC, Pandharipande PP, et al. Delirium as a predictor of long-term cognitive impairment in survivors of critical illness. Crit Care Med 2010; 38:1513–1520.
- Miller RR, Ely EW. Delirium and cognitive dysfunction in the intensive care unit. Curr Psychiatry Rep 2007; 9:26–34.
- Hopkins RO, Suchyta MR, Snow GL, Jephson A, Weaver LK, Orme JF. Blood glucose dysregulation and cognitive outcome in ARDS survivors. Brain Inj 2010; 24:1478–1484.
- Hough CL, Herridge MS. Long-term outcome after acute lung injury. Curr Opin Crit Care 2012; 18:8–15.
- Jackson JC, Hart RP, Gordon SM, et al. Six-month neuropsychological outcome of medical intensive care unit patients. Crit Care Med 2003; 31:1226–1234.
- Court JA, Perry EK. Neurotransmitter abnormalities in vascular dementia. Int Psychogeriatr 2003; 15(suppl 1):81–87.
- Gottfries CG, Blennow K, Karlsson I, Wallin A. The neurochemistry of vascular dementia. Dementia 1994; 5:163–167.
- Baskerville KA, Schweitzer JB, Herron P. Effects of cholinergic depletion on experience-dependent plasticity in the cortex of the rat. Neuroscience 1997; 80:1159–1169.
- Henon H, Lebert F, Durieu I, et al. Confusional state in stroke: relation to preexisting dementia, patient characteristics, and outcome. Stroke 1999; 30:773–779.
- Whyte E, Skidmore E, Aizenstein H, Ricker J, Butters M. Cognitive impairment in acquired brain injury: a predictor of rehabilitation outcomes and an opportunity for novel interventions. PMR 2011; 3(suppl 1):S45–S51.
- Stephan BC, Matthews FE, McKeith IG, Bond J, Brayne C. Early cognitive change in the general population: how do different definitions work? J Am Geriatr Soc 2007; 55:1534–1540.
- Petersen RC, Smith GE, Waring SC, Ivnik RJ, Tangalos EG, Kokmen E. Mild cognitive impairment: clinical characterization and outcome. Arch Neurol 1999; 56:303–308.
- Palmer K, Fratiglioni L, Winblad B. What is mild cognitive impairment? Variations in definitions and evolution of nondemented persons with cognitive impairment. Acta Neurol Scand Suppl 2003; 179:14–20.
- Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med 2004; 256:183–194.
- Lonie JA, Tierney KM, Ebmeier KP. Screening for mild cognitive impairment: a systematic review. Int J Geriatr Psychiatry 2009; 24:902–915.
- Nasreddine ZS, Phillips NA, Bedirian V, et al. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc 2005; 53:695–699.
- Folstein MF, Folstein SE, McHugh PR. “Mini-mental state.” A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975; 12:189–198.
- Sager MA, Hermann BP, La Rue A, Woodard JL. Screening for dementia in community-based memory clinics. WMJ 2006; 105:25–29.
- Ravaglia G, Forti P, Maioli F, et al. Screening for mild cognitive impairment in elderly ambulatory patients with cognitive complaints. Aging Clin Exp Res 2005; 17:374–379.
- Vogenthaler DR. An overview of head injury: its consequences and rehabilitation. Brain Inj 1987; 1:113–127.
- van den Boogaard M, Schoonhoven L, Evers AW, van der Hoeven JG, van Achterberg T, Pickkers P. Delirium in critically ill patients: impact on long-term health-related quality of life and cognitive functioning. Crit Care Med 2012; 40:112–118.
- Morandi A, Brummel NE, Ely EW. Sedation, delirium and mechanical ventilation: the ‘ABCDE’ approach. Curr Opin Crit Care 2011; 17:43–49.
- Girard TD, Kress JP, Fuchs BD, et al. Efficacy and safety of a paired sedation and ventilator weaning protocol for mechanically ventilated patients in intensive care (Awakening and Breathing Controlled trial): a randomised controlled trial. Lancet 2008; 371:126–134.
- Pandharipande PP, Pun BT, Herr DL, et al. Effect of sedation with dexmedetomidine vs lorazepam on acute brain dysfunction in mechanically ventilated patients: the MENDS randomized controlled trial. JAMA 2007; 298:2644–2653.
- Riker RR, Shehabi Y, Bokesch PM, et al. Dexmedetomidine vs midazolam for sedation of critically ill patients: a randomized trial. JAMA 2009; 301:489–499.
- Reade MC, O’Sullivan K, Bates S, Goldsmith D, Ainslie WR, Bellomo R. Dexmedetomidine vs. haloperidol in delirious, agitated, intubated patients: a randomised open-label trial. Crit Care 2009; 13:R75.
- Mirski MA, Lewin JJ, Ledroux S, et al. Cognitive improvement during continuous sedation in critically ill, awake and responsive patients: the Acute Neurological ICU Sedation Trial (ANIST). Intensive Care Med 2010; 36:1505–1513.
- Spronk PE, Riekerk B, Hofhuis J, Rommes JH. Occurrence of delirium is severely underestimated in the ICU during daily care. Intensive Care Med 2009; 35:1276–1280.
- Ely EW, Inouye SK, Bernard GR, et al. Delirium in mechanically ventilated patients: validity and reliability of the confusion assessment method for the intensive care unit (CAM-ICU). JAMA 2001; 286:2703–2710.
- Luetz A, Heymann A, Radtke FM, et al. Different assessment tools for intensive care unit delirium: which score to use? Crit Care Med 2010; 38:409–418.
- Schweickert WD, Pohlman MC, Pohlman AS, et al. Early physical and occupational therapy in mechanically ventilated, critically ill patients: a randomised controlled trial. Lancet 2009; 373:1874–1882.
- Martin M, Clare L, Altgassen AM, Cameron MH, Zehnder F. Cognition-based interventions for healthy older people and people with mild cognitive impairment. Cochrane Database Syst Rev 2011(1):CD006220.
- Rozzini L, Costardi D, Chilovi BV, Franzoni S, Trabucchi M, Padovani A. Efficacy of cognitive rehabilitation in patients with mild cognitive impairment treated with cholinesterase inhibitors. Int J Geriatr Psychiatry 2007; 22:356–360.
- Jean L, Bergeron ME, Thivierge S, Simard M. Cognitive intervention programs for individuals with mild cognitive impairment: systematic review of the literature. Am J Geriatr Psychiatry 2010; 18:281–296.
- Zelinski EM, Spina LM, Yaffe K, et al. Improvement in memory with plasticity-based adaptive cognitive training: results of the 3-month follow-up. J Am Geriatr Soc 2011; 59:258–265.
- Cicerone KD, Dahlberg C, Malec JF, et al. Evidence-based cognitive rehabilitation: updated review of the literature from 1998 through 2002. Arch Phys Med Rehabil 2005; 86:1681–1692.
- Jackson JC, Ely EW, Morey MC, et al. Cognitive and physical rehabilitation of intensive care unit survivors: results of the RETURN randomized controlled pilot investigation. Crit Care Med 2012; 40:1088–1097.
- Levine B, Stuss DT, Winocur G, et al. Cognitive rehabilitation in the elderly: effects on strategic behavior in relation to goal management. J Int Neuropsychol Soc 2007; 13:143–152.
- ACT-ICU Study: Activity and Cognitive Therapy in the Intensive Care Unit. http://clinicaltrials.gov/ct2/show/NCT01270269. Accessed August 9, 2012.
- Kim YH, Ko MH, Na SY, Park SH, Kim KW. Effects of single-dose methylphenidate on cognitive performance in patients with traumatic brain injury: a double-blind placebo-controlled study. Clin Rehabil 2006; 20:24–30.
- Whyte J, Hart T, Schuster K, Fleming M, Polansky M, Coslett HB. Effects of methylphenidate on attentional function after traumatic brain injury. A randomized, placebo-controlled trial. Am J Phys Med Rehabil 1997; 76:440–450.
- Masanic CA, Bayley MT, VanReekum R, Simard M. Open-label study of donepezil in traumatic brain injury. Arch Phys Med Rehabil 2001; 82:896–901.
- Zhang L, Plotkin RC, Wang G, Sandel ME, Lee S. Cholinergic augmentation with donepezil enhances recovery in short-term memory and sustained attention after traumatic brain injury. Arch Phys Med Rehabil 2004; 85:1050–1055.
- Khateb A, Ammann J, Annoni JM, Diserens K. Cognition-enhancing effects of donepezil in traumatic brain injury. Eur Neurol 2005; 54:39–45.
- Kim YW, Kim DY, Shin JC, Park CI, Lee JD. The changes of cortical metabolism associated with the clinical response to donepezil therapy in traumatic brain injury. Clin Neuropharmacol 2009; 32:63–68.
- Wheaton P, Mathias JL, Vink R. Impact of pharmacological treatments on cognitive and behavioral outcome in the postacute stages of adult traumatic brain injury: a meta-analysis. J Clin Psychopharmacol 2011; 31:745–757.
- Diaz-Guzman E, Sanchez J, Arroliga AC. Update in intensive care medicine: studies that challenged our practice in the last 5 years. Cleve Clin J Med 2011; 78:665–674.
- Acute Respiratory Distress Syndrome Network. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med 2000; 342:1301–1308.
- Rivers E, Nguyen B, Havstad S, et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med 2001; 345:1368–1377.
- Oeyen SG, Vandijck DM, Benoit DD, Annemans L, Decruyenaere JM. Quality of life after intensive care: a systematic review of the literature. Crit Care Med 2010; 38:2386–2400.
- Needham DM, Davidson J, Cohen H, et al. Improving long-term outcomes after discharge from intensive care unit: report from a stakeholders’ conference. Crit Care Med 2012; 40:502–509.
- Herridge MS, Cheung AM, Tansey CM, et al. One-year outcomes in survivors of the acute respiratory distress syndrome. N Engl J Med 2003; 348:683–693.
- Herridge MS, Tansey CM, Matte A, et al. Functional disability 5 years after acute respiratory distress syndrome. N Engl J Med 2011; 364:1293–1304.
- Timmers TK, Verhofstad MH, Moons KG, van Beeck EF, Leenen LP. Long-term quality of life after surgical intensive care admission. Arch Surg 2011; 146:412–418.
- Michaels AJ, Michaels CE, Moon CH, et al. Posttraumatic stress disorder after injury: impact on general health outcome and early risk assessment. J Trauma 1999; 47:460–466; discussion466–467.
- Stoll C, Schelling G, Goetz AE, et al. Health-related quality of life and post-traumatic stress disorder in patients after cardiac surgery and intensive care treatment. J Thorac Cardiovasc Surg 2000; 120:505–512.
- Jones C, Skirrow P, Griffiths RD, et al Post-traumatic stress disorder-related symptoms in relatives of patients following intensive care. Intensive Care Med 2004; 30:456–460.
- Griffiths J, Gager M, Alder N, Fawcett D, Waldmann C, Quinlan J. A self-report-based study of the incidence and associations of sexual dysfunction in survivors of intensive care treatment. Intensive Care Med 2006; 32:445–451.
- Griffiths J, Waldmann C, Quinlan J. Sexual dysfunction in intensive care survivors. Br J Hosp Med (Lond) 2007; 68:470–473.
- Hopkins RO, Jackson JC. Long-term neurocognitive function after critical illness. Chest 2006; 130:869–878.
- Hopkins RO, Weaver LK, Collingridge D, Parkinson RB, Chan KJ, Orme JF. Two-year cognitive, emotional, and quality-of-life outcomes in acute respiratory distress syndrome. Am J Respir Crit Care Med 2005; 171:340–347.
- Rothenhausler HB, Ehrentraut S, Stoll C, Schelling G, Kapfhammer HP. The relationship between cognitive performance and employment and health status in long-term survivors of the acute respiratory distress syndrome: results of an exploratory study. Gen Hosp Psychiatry 2001; 23:90–96.
- Sukantarat KT, Burgess PW, Williamson RC, Brett SJ. Prolonged cognitive dysfunction in survivors of critical illness. Anaesthesia 2005; 60:847–853.
- Hopkins RO, Weaver LK, Pope D, Orme JF, Bigler ED, Larson LV. Neuropsychological sequelae and impaired health status in survivors of severe acute respiratory distress syndrome. Am J Respir Crit Care Med 1999; 160:50–56.
- Hopkins RO, Weaver LK, Chan KJ, Orme JF. Quality of life, emotional, and cognitive function following acute respiratory distress syndrome. J Int Neuropsychol Soc 2004; 10:1005–1017.
- Jackson JC, Gordon SM, Hart RP, Hopkins RO, Ely EW. The association between delirium and cognitive decline: a review of the empirical literature. Neuropsychol Rev 2004; 14:87–98.
- Arroliga AC, Thompson BT, Ancukiewicz M, et al. Use of sedatives, opioids, and neuromuscular blocking agents in patients with acute lung injury and acute respiratory distress syndrome. Crit Care Med 2008; 36:1083–1088.
- Miller RR, Ely EW. Delirium and cognitive dysfunction in the intensive care unit. Semin Respir Crit Care Med 2006; 27:210–220.
- Pandharipande P, Shintani A, Peterson J, et al. Lorazepam is an independent risk factor for transitioning to delirium in intensive care unit patients. Anesthesiology 2006; 104:21–26.
- Dubois MJ, Bergeron N, Dumont M, Dial S, Skrobik Y. Delirium in an intensive care unit: a study of risk factors. Intensive Care Med 2001; 27:1297–1304.
- Girard TD, Jackson JC, Pandharipande PP, et al. Delirium as a predictor of long-term cognitive impairment in survivors of critical illness. Crit Care Med 2010; 38:1513–1520.
- Miller RR, Ely EW. Delirium and cognitive dysfunction in the intensive care unit. Curr Psychiatry Rep 2007; 9:26–34.
- Hopkins RO, Suchyta MR, Snow GL, Jephson A, Weaver LK, Orme JF. Blood glucose dysregulation and cognitive outcome in ARDS survivors. Brain Inj 2010; 24:1478–1484.
- Hough CL, Herridge MS. Long-term outcome after acute lung injury. Curr Opin Crit Care 2012; 18:8–15.
- Jackson JC, Hart RP, Gordon SM, et al. Six-month neuropsychological outcome of medical intensive care unit patients. Crit Care Med 2003; 31:1226–1234.
- Court JA, Perry EK. Neurotransmitter abnormalities in vascular dementia. Int Psychogeriatr 2003; 15(suppl 1):81–87.
- Gottfries CG, Blennow K, Karlsson I, Wallin A. The neurochemistry of vascular dementia. Dementia 1994; 5:163–167.
- Baskerville KA, Schweitzer JB, Herron P. Effects of cholinergic depletion on experience-dependent plasticity in the cortex of the rat. Neuroscience 1997; 80:1159–1169.
- Henon H, Lebert F, Durieu I, et al. Confusional state in stroke: relation to preexisting dementia, patient characteristics, and outcome. Stroke 1999; 30:773–779.
- Whyte E, Skidmore E, Aizenstein H, Ricker J, Butters M. Cognitive impairment in acquired brain injury: a predictor of rehabilitation outcomes and an opportunity for novel interventions. PMR 2011; 3(suppl 1):S45–S51.
- Stephan BC, Matthews FE, McKeith IG, Bond J, Brayne C. Early cognitive change in the general population: how do different definitions work? J Am Geriatr Soc 2007; 55:1534–1540.
- Petersen RC, Smith GE, Waring SC, Ivnik RJ, Tangalos EG, Kokmen E. Mild cognitive impairment: clinical characterization and outcome. Arch Neurol 1999; 56:303–308.
- Palmer K, Fratiglioni L, Winblad B. What is mild cognitive impairment? Variations in definitions and evolution of nondemented persons with cognitive impairment. Acta Neurol Scand Suppl 2003; 179:14–20.
- Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med 2004; 256:183–194.
- Lonie JA, Tierney KM, Ebmeier KP. Screening for mild cognitive impairment: a systematic review. Int J Geriatr Psychiatry 2009; 24:902–915.
- Nasreddine ZS, Phillips NA, Bedirian V, et al. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc 2005; 53:695–699.
- Folstein MF, Folstein SE, McHugh PR. “Mini-mental state.” A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975; 12:189–198.
- Sager MA, Hermann BP, La Rue A, Woodard JL. Screening for dementia in community-based memory clinics. WMJ 2006; 105:25–29.
- Ravaglia G, Forti P, Maioli F, et al. Screening for mild cognitive impairment in elderly ambulatory patients with cognitive complaints. Aging Clin Exp Res 2005; 17:374–379.
- Vogenthaler DR. An overview of head injury: its consequences and rehabilitation. Brain Inj 1987; 1:113–127.
- van den Boogaard M, Schoonhoven L, Evers AW, van der Hoeven JG, van Achterberg T, Pickkers P. Delirium in critically ill patients: impact on long-term health-related quality of life and cognitive functioning. Crit Care Med 2012; 40:112–118.
- Morandi A, Brummel NE, Ely EW. Sedation, delirium and mechanical ventilation: the ‘ABCDE’ approach. Curr Opin Crit Care 2011; 17:43–49.
- Girard TD, Kress JP, Fuchs BD, et al. Efficacy and safety of a paired sedation and ventilator weaning protocol for mechanically ventilated patients in intensive care (Awakening and Breathing Controlled trial): a randomised controlled trial. Lancet 2008; 371:126–134.
- Pandharipande PP, Pun BT, Herr DL, et al. Effect of sedation with dexmedetomidine vs lorazepam on acute brain dysfunction in mechanically ventilated patients: the MENDS randomized controlled trial. JAMA 2007; 298:2644–2653.
- Riker RR, Shehabi Y, Bokesch PM, et al. Dexmedetomidine vs midazolam for sedation of critically ill patients: a randomized trial. JAMA 2009; 301:489–499.
- Reade MC, O’Sullivan K, Bates S, Goldsmith D, Ainslie WR, Bellomo R. Dexmedetomidine vs. haloperidol in delirious, agitated, intubated patients: a randomised open-label trial. Crit Care 2009; 13:R75.
- Mirski MA, Lewin JJ, Ledroux S, et al. Cognitive improvement during continuous sedation in critically ill, awake and responsive patients: the Acute Neurological ICU Sedation Trial (ANIST). Intensive Care Med 2010; 36:1505–1513.
- Spronk PE, Riekerk B, Hofhuis J, Rommes JH. Occurrence of delirium is severely underestimated in the ICU during daily care. Intensive Care Med 2009; 35:1276–1280.
- Ely EW, Inouye SK, Bernard GR, et al. Delirium in mechanically ventilated patients: validity and reliability of the confusion assessment method for the intensive care unit (CAM-ICU). JAMA 2001; 286:2703–2710.
- Luetz A, Heymann A, Radtke FM, et al. Different assessment tools for intensive care unit delirium: which score to use? Crit Care Med 2010; 38:409–418.
- Schweickert WD, Pohlman MC, Pohlman AS, et al. Early physical and occupational therapy in mechanically ventilated, critically ill patients: a randomised controlled trial. Lancet 2009; 373:1874–1882.
- Martin M, Clare L, Altgassen AM, Cameron MH, Zehnder F. Cognition-based interventions for healthy older people and people with mild cognitive impairment. Cochrane Database Syst Rev 2011(1):CD006220.
- Rozzini L, Costardi D, Chilovi BV, Franzoni S, Trabucchi M, Padovani A. Efficacy of cognitive rehabilitation in patients with mild cognitive impairment treated with cholinesterase inhibitors. Int J Geriatr Psychiatry 2007; 22:356–360.
- Jean L, Bergeron ME, Thivierge S, Simard M. Cognitive intervention programs for individuals with mild cognitive impairment: systematic review of the literature. Am J Geriatr Psychiatry 2010; 18:281–296.
- Zelinski EM, Spina LM, Yaffe K, et al. Improvement in memory with plasticity-based adaptive cognitive training: results of the 3-month follow-up. J Am Geriatr Soc 2011; 59:258–265.
- Cicerone KD, Dahlberg C, Malec JF, et al. Evidence-based cognitive rehabilitation: updated review of the literature from 1998 through 2002. Arch Phys Med Rehabil 2005; 86:1681–1692.
- Jackson JC, Ely EW, Morey MC, et al. Cognitive and physical rehabilitation of intensive care unit survivors: results of the RETURN randomized controlled pilot investigation. Crit Care Med 2012; 40:1088–1097.
- Levine B, Stuss DT, Winocur G, et al. Cognitive rehabilitation in the elderly: effects on strategic behavior in relation to goal management. J Int Neuropsychol Soc 2007; 13:143–152.
- ACT-ICU Study: Activity and Cognitive Therapy in the Intensive Care Unit. http://clinicaltrials.gov/ct2/show/NCT01270269. Accessed August 9, 2012.
- Kim YH, Ko MH, Na SY, Park SH, Kim KW. Effects of single-dose methylphenidate on cognitive performance in patients with traumatic brain injury: a double-blind placebo-controlled study. Clin Rehabil 2006; 20:24–30.
- Whyte J, Hart T, Schuster K, Fleming M, Polansky M, Coslett HB. Effects of methylphenidate on attentional function after traumatic brain injury. A randomized, placebo-controlled trial. Am J Phys Med Rehabil 1997; 76:440–450.
- Masanic CA, Bayley MT, VanReekum R, Simard M. Open-label study of donepezil in traumatic brain injury. Arch Phys Med Rehabil 2001; 82:896–901.
- Zhang L, Plotkin RC, Wang G, Sandel ME, Lee S. Cholinergic augmentation with donepezil enhances recovery in short-term memory and sustained attention after traumatic brain injury. Arch Phys Med Rehabil 2004; 85:1050–1055.
- Khateb A, Ammann J, Annoni JM, Diserens K. Cognition-enhancing effects of donepezil in traumatic brain injury. Eur Neurol 2005; 54:39–45.
- Kim YW, Kim DY, Shin JC, Park CI, Lee JD. The changes of cortical metabolism associated with the clinical response to donepezil therapy in traumatic brain injury. Clin Neuropharmacol 2009; 32:63–68.
- Wheaton P, Mathias JL, Vink R. Impact of pharmacological treatments on cognitive and behavioral outcome in the postacute stages of adult traumatic brain injury: a meta-analysis. J Clin Psychopharmacol 2011; 31:745–757.
KEY POINTS
- The development of cognitive impairment during hospitalization has been associated with complications such as hypotension, hyperglycemia, hypoxemia, and delirium.
- The “ABCDE” strategy is used to prevent delirium, although its effect on cognitive impairment has not been proven. ABCD stands for awakening and early spontaneous breathing, choice of sedatives with fewer adverse effects (ie, avoidance of benzodiazepines and opioids), daily delirium monitoring, and early mobility exercise.
- Cognitive impairment is usually diagnosed using restrictive or comprehensive evaluation tools. The Montreal Cognitive Assessment is probably the one most often used since it is readily available, simple, and reliable.
- Most of the evidence on treating cognitive impairment after an ICU stay is extrapolated from studies in patients with mild cognitive impairment or traumatic brain injury. Cognitive training has shown positive results, mostly in improvement of memory, particularly immediate recall.
A 60-year-old man with abdominal bruising
A 60-year-old man with hepatocellular carcinoma was admitted to the hospital with pulmonary emboli secondary to inferior vena caval thrombosis that extended to the right atrium.
He became hypotensive on the second day, with a heart rate of 124 per minute, respiratory rate 44 per minute, pulse oxygen saturation 79% on room air, and systolic blood pressure 70 mm Hg. Physical examination revealed abdominal ecchymoses resembling the Cullen sign and flank ecchymoses resembling the Grey Turner sign (Figures 1 and 2).
He was given a bolus of normal saline followed by infusion of fresh frozen plasma and packed red blood cells. His lactate level was 17.52 mg/dL (reference range 0.1–2.2). His hemoglobin and hematocrit decreased precipitously—the hemoglobin from 10.5 g/dL to 5.9 (reference range 14.0–17.5), and the hematocrit from 30.3% to 17.8% (reference range 41–50). He was transferred to the intensive care unit. A do-not-resuscitate order was instituted, and he died 12 hours later.
CONDITIONS RESULTING IN THE CULLEN AND GREY TURNER SIGNS
The Cullen sign, a bluish discoloration of the periumbilical skin, was originally described in 1918 by the gynecologist Thomas Cullen, MD, in a patient with a ruptured ectopic pregnancy.1 The Grey Turner sign, an ecchymotic discoloration of the lateral abdominal wall or flank, was first reported in 1920 by a surgeon, Dr. George Grey Turner, in a patient with acute pancreatitis.2
The signs occur in about 1% of patients with acute pancreatitis and predict a poor prognosis, with a reported death rate of 37%.3
The appearance of ecchymoses in the periumbilical area or flank has been taught as a hallmark of acute pancreatitis.4 However, the original patient described with the Cullen sign did not have pancreatitis,4,5 and it has been reported with many other conditions, including ruptured aortic aneurysm, splenic rupture, and rectus sheath hematoma, as a complication of anticoagulation or perforated duodenal ulcer, and, as in our patient, as a manifestation of liver disease.
How they occur
The common pathway leading to the occurrence of these subcutaneous ecchymoses is retroperitoneal bleeding followed by tracking of blood from the retroperitoneum through a defect in the transversalis fascia to the abdominal wall musculature and then to the periumbilical subcutaneous tissue. In the Cullen sign, blood diffuses from the retroperitoneum along the gastrohepatic and falciform ligaments to the umbilicus. In the Grey Turner sign, blood diffuses from the posterior pararenal space to the lateral edge of the quadratus lumborum muscle.6–8 Blood from a retroperitoneal hemorrhage may also diffuse and pool at the inguinal ligament (Fox sign) or at the scrotum (Bryant sign).5
The time to the appearance of the Cullen or the Grey Turner sign is thought to be at least 24 hours after the onset of retroperitoneal bleeding and averages about 3 days after the onset of pancreatitis.3
- Cullen TS. A new sign in ruptured extrauterine pregnancy. Am J Obstet Gynecol 1918; 78:457.
- Grey Turner G. Local discoloration of the abdominal wall as a sign of acute pancreatitis. Br J Surg 1910; 7:394–395.
- Dickson AP, Imrie CW. The incidence and prognosis of body wall ecchymosis in acute pancreatitis. Surg Gynecol Obstet 1984; 159:343–347.
- Harris S, Naina HV. Cullen’s sign revisited. Am J Med 2008; 121:682–683.
- Bosmann M, Schreiner O, Galle PR. Coexistence of Cullen’s and Grey Turner’s signs in acute pancreatitis. Am J Med 2009; 122:333–334.
- Bem J, Bradley EL. Subcutaneous manifestations of severe acute pancreatitis. Pancreas 1998; 16:551–555.
- Meyers MA, Feldberg MA, Oliphant M. Grey Turner’s sign and Cullen’s sign in acute pancreatitis. Gastrointest Radiol 1989; 14:31–37.
- Mabin TA, Gelfand M. Cullen’s sign, a feature in liver disease. Br Med J 1974; 1:493–494.
A 60-year-old man with hepatocellular carcinoma was admitted to the hospital with pulmonary emboli secondary to inferior vena caval thrombosis that extended to the right atrium.
He became hypotensive on the second day, with a heart rate of 124 per minute, respiratory rate 44 per minute, pulse oxygen saturation 79% on room air, and systolic blood pressure 70 mm Hg. Physical examination revealed abdominal ecchymoses resembling the Cullen sign and flank ecchymoses resembling the Grey Turner sign (Figures 1 and 2).
He was given a bolus of normal saline followed by infusion of fresh frozen plasma and packed red blood cells. His lactate level was 17.52 mg/dL (reference range 0.1–2.2). His hemoglobin and hematocrit decreased precipitously—the hemoglobin from 10.5 g/dL to 5.9 (reference range 14.0–17.5), and the hematocrit from 30.3% to 17.8% (reference range 41–50). He was transferred to the intensive care unit. A do-not-resuscitate order was instituted, and he died 12 hours later.
CONDITIONS RESULTING IN THE CULLEN AND GREY TURNER SIGNS
The Cullen sign, a bluish discoloration of the periumbilical skin, was originally described in 1918 by the gynecologist Thomas Cullen, MD, in a patient with a ruptured ectopic pregnancy.1 The Grey Turner sign, an ecchymotic discoloration of the lateral abdominal wall or flank, was first reported in 1920 by a surgeon, Dr. George Grey Turner, in a patient with acute pancreatitis.2
The signs occur in about 1% of patients with acute pancreatitis and predict a poor prognosis, with a reported death rate of 37%.3
The appearance of ecchymoses in the periumbilical area or flank has been taught as a hallmark of acute pancreatitis.4 However, the original patient described with the Cullen sign did not have pancreatitis,4,5 and it has been reported with many other conditions, including ruptured aortic aneurysm, splenic rupture, and rectus sheath hematoma, as a complication of anticoagulation or perforated duodenal ulcer, and, as in our patient, as a manifestation of liver disease.
How they occur
The common pathway leading to the occurrence of these subcutaneous ecchymoses is retroperitoneal bleeding followed by tracking of blood from the retroperitoneum through a defect in the transversalis fascia to the abdominal wall musculature and then to the periumbilical subcutaneous tissue. In the Cullen sign, blood diffuses from the retroperitoneum along the gastrohepatic and falciform ligaments to the umbilicus. In the Grey Turner sign, blood diffuses from the posterior pararenal space to the lateral edge of the quadratus lumborum muscle.6–8 Blood from a retroperitoneal hemorrhage may also diffuse and pool at the inguinal ligament (Fox sign) or at the scrotum (Bryant sign).5
The time to the appearance of the Cullen or the Grey Turner sign is thought to be at least 24 hours after the onset of retroperitoneal bleeding and averages about 3 days after the onset of pancreatitis.3
A 60-year-old man with hepatocellular carcinoma was admitted to the hospital with pulmonary emboli secondary to inferior vena caval thrombosis that extended to the right atrium.
He became hypotensive on the second day, with a heart rate of 124 per minute, respiratory rate 44 per minute, pulse oxygen saturation 79% on room air, and systolic blood pressure 70 mm Hg. Physical examination revealed abdominal ecchymoses resembling the Cullen sign and flank ecchymoses resembling the Grey Turner sign (Figures 1 and 2).
He was given a bolus of normal saline followed by infusion of fresh frozen plasma and packed red blood cells. His lactate level was 17.52 mg/dL (reference range 0.1–2.2). His hemoglobin and hematocrit decreased precipitously—the hemoglobin from 10.5 g/dL to 5.9 (reference range 14.0–17.5), and the hematocrit from 30.3% to 17.8% (reference range 41–50). He was transferred to the intensive care unit. A do-not-resuscitate order was instituted, and he died 12 hours later.
CONDITIONS RESULTING IN THE CULLEN AND GREY TURNER SIGNS
The Cullen sign, a bluish discoloration of the periumbilical skin, was originally described in 1918 by the gynecologist Thomas Cullen, MD, in a patient with a ruptured ectopic pregnancy.1 The Grey Turner sign, an ecchymotic discoloration of the lateral abdominal wall or flank, was first reported in 1920 by a surgeon, Dr. George Grey Turner, in a patient with acute pancreatitis.2
The signs occur in about 1% of patients with acute pancreatitis and predict a poor prognosis, with a reported death rate of 37%.3
The appearance of ecchymoses in the periumbilical area or flank has been taught as a hallmark of acute pancreatitis.4 However, the original patient described with the Cullen sign did not have pancreatitis,4,5 and it has been reported with many other conditions, including ruptured aortic aneurysm, splenic rupture, and rectus sheath hematoma, as a complication of anticoagulation or perforated duodenal ulcer, and, as in our patient, as a manifestation of liver disease.
How they occur
The common pathway leading to the occurrence of these subcutaneous ecchymoses is retroperitoneal bleeding followed by tracking of blood from the retroperitoneum through a defect in the transversalis fascia to the abdominal wall musculature and then to the periumbilical subcutaneous tissue. In the Cullen sign, blood diffuses from the retroperitoneum along the gastrohepatic and falciform ligaments to the umbilicus. In the Grey Turner sign, blood diffuses from the posterior pararenal space to the lateral edge of the quadratus lumborum muscle.6–8 Blood from a retroperitoneal hemorrhage may also diffuse and pool at the inguinal ligament (Fox sign) or at the scrotum (Bryant sign).5
The time to the appearance of the Cullen or the Grey Turner sign is thought to be at least 24 hours after the onset of retroperitoneal bleeding and averages about 3 days after the onset of pancreatitis.3
- Cullen TS. A new sign in ruptured extrauterine pregnancy. Am J Obstet Gynecol 1918; 78:457.
- Grey Turner G. Local discoloration of the abdominal wall as a sign of acute pancreatitis. Br J Surg 1910; 7:394–395.
- Dickson AP, Imrie CW. The incidence and prognosis of body wall ecchymosis in acute pancreatitis. Surg Gynecol Obstet 1984; 159:343–347.
- Harris S, Naina HV. Cullen’s sign revisited. Am J Med 2008; 121:682–683.
- Bosmann M, Schreiner O, Galle PR. Coexistence of Cullen’s and Grey Turner’s signs in acute pancreatitis. Am J Med 2009; 122:333–334.
- Bem J, Bradley EL. Subcutaneous manifestations of severe acute pancreatitis. Pancreas 1998; 16:551–555.
- Meyers MA, Feldberg MA, Oliphant M. Grey Turner’s sign and Cullen’s sign in acute pancreatitis. Gastrointest Radiol 1989; 14:31–37.
- Mabin TA, Gelfand M. Cullen’s sign, a feature in liver disease. Br Med J 1974; 1:493–494.
- Cullen TS. A new sign in ruptured extrauterine pregnancy. Am J Obstet Gynecol 1918; 78:457.
- Grey Turner G. Local discoloration of the abdominal wall as a sign of acute pancreatitis. Br J Surg 1910; 7:394–395.
- Dickson AP, Imrie CW. The incidence and prognosis of body wall ecchymosis in acute pancreatitis. Surg Gynecol Obstet 1984; 159:343–347.
- Harris S, Naina HV. Cullen’s sign revisited. Am J Med 2008; 121:682–683.
- Bosmann M, Schreiner O, Galle PR. Coexistence of Cullen’s and Grey Turner’s signs in acute pancreatitis. Am J Med 2009; 122:333–334.
- Bem J, Bradley EL. Subcutaneous manifestations of severe acute pancreatitis. Pancreas 1998; 16:551–555.
- Meyers MA, Feldberg MA, Oliphant M. Grey Turner’s sign and Cullen’s sign in acute pancreatitis. Gastrointest Radiol 1989; 14:31–37.
- Mabin TA, Gelfand M. Cullen’s sign, a feature in liver disease. Br Med J 1974; 1:493–494.
Flashing lights, floaters, and reduced vision
A 62-year-old woman has had flashing lights and floaters in her left eye with progressive loss of vision over the past month. She has not had recent trauma. She does not smoke.
She was referred for an ophthalmologic evaluation. Her visual acuity was 20/20 in the right eye, but she could only count fingers with the left. The anterior segment appeared normal in both eyes. Funduscopic examination of the left eye revealed numerous lobulated, yellowish, choroidal lesions in the posterior pole with overlying subretinal fluid. The lesions involved the fovea, accounting for the poor visual acuity. There were two similar but smaller lesions in the right eye (Figure 1). Ultrasonography confirmed the choroidal location of the lesions (Figure 2).
Q: Which is the most likely diagnosis?
- Retinal detachment
- Choroidal melanoma
- Uveitis
- Uveal metastatic tumor
A: Uveal metastatic tumor is the correct diagnosis. Funduscopic findings of bilateral yellow choroidal lesions are consistent with metastatic cancer.
The patient was admitted to the hospital for a thorough evaluation. Computed tomography of the chest showed a 2.1-by-4.5-cm mass in the lower lobe of the left lung, highly suspicious for malignancy and associated with left hilar lymphadenopathy and right acute pulmonary embolism. Bronchoscopy showed an endobronchial tumor completely occluding the left lower lobe and the lingular orifices.
Pathologic specimens from the endobronchial tumor confirmed adenocarcinoma, consistent with a primary lung cancer.
THE OTHER DIAGNOSTIC CHOICES
Detachment or separation of the retina from the underlying pigment epithelium is one of the most commonly encountered eye emergencies.1 It requires urgent attention, since delay in treatment can cause permanent vision loss.
Retinal detachment differs from uveal metastatic tumor in that it presents and progresses rapidly. The common signs and symptoms are floaters in the center of the visual axis, a sensation of flashing lights (related to retinal traction), and, eventually, loss of vision. The detachment most often represents a break or tear (rhegmatogenous retinal detachment), but it is also a common sequela of neglected diabetic retinopathy. Exudative retinal detachment is usually secondary to uveal inflammation or a uveal tumor.
Choroidal melanoma, the most common primary intraocular malignancy, arises from melanocytes within the choroid. In most cases, it develops from preexisting melanocytic nevi.2 It may present as blurred vision, a paracentral scotoma, painless and progressive visual field loss, and floaters. Choroidal melanoma is usually pigmented (dark brown) and is invariably unilateral.
Uveitis is an inflammation of the uveal tract, which includes the iris, ciliary body, and choroid. It is classified as anterior, intermediate, or posterior uveitis or as panuveitis.3
Although flashing lights, floaters, and reduced vision can occur in uveitis, its other important presenting symptoms (ie, pain, redness, and photophobia) were absent in this patient. The absence of anterior chamber cells and corneal inflammatory deposits (keratic precipitates) also made uveitis less likely.4 However, granulomatous uveitis such as sarcoidosis can present as nodular thickening of the uvea, mimicking an intraocular tumor.5
THE MOST COMMON INTRAOCULAR MALIGNANCY
Uveal metastasis is the most common intraocular malignancy6 and is found on autopsy in up to 12% of people who die of cancer; it involves both eyes in 4.4% of cases. Multiple metastases are seen in one eye in up to 20% of cases.7
The tumors are most often in the choroid, probably because of its extensive blood supply. Breast cancer (in women) and lung cancer (in men) are the most common cancers with uveal metastasis.8 Uveal metastasis from cancers of the prostate, kidney, thyroid, and gastrointestinal tract and from lymphoma and leukemia is less common.8
Patients with choroidal metastasis can see flashing lights, floating spots, and distortion of their vision. In such patients, a careful history and physical examination can uncover signs and symptoms of the hidden cancer, especially of lung cancer.9
Once uveal metastasis is suspected, both eyes and orbits and the central nervous system should be examined, as this disease tends to present bilaterally and to involve the central nervous system.10 Uveal metastases respond to chemotherapy and radiotherapy, depending on the nature of the primary tumor. In general, treatment is based on the extent of the metastasis, prior treatments, and the patient’s overall functional status.
- Hatten B, Browne V. Retinal detachment. Emerg Med J 2011; 28:83.
- Factors predictive of growth and treatment of small choroidal melanoma: COMS Report No. 5. The Collaborative Ocular Melanoma Study Group. Arch Ophthalmol 1997; 115:1537–1544.
- Jabs DA, Nussenblatt RB, Rosenbaum JT; Standardization of Uveitis Nomenclature (SUN) Working Group. Standardization of uveitis nomenclature for reporting clinical data. Results of the First International Workshop. Am J Ophthalmol 2005; 140:509–516.
- Wertheim MS, Mathers WD, Planck SJ, et al. In vivo confocal microscopy of keratic precipitates. Arch Ophthalmol 2004; 122:1773–1781.
- Desai UR, Tawansy KA, Joondeph BC, Schiffman RM. Choroidal granulomas in systemic sarcoidosis. Retina 2001; 21:40–47.
- Singh AD, Damato BE, Pe’er J, Murphree AL, Perry JD, eds. Uveal metastatic tumors. In: Clinical Ophthalmic Oncology. Philadelphia, PA: Saunders-Elsevier; 2007:322–327.
- Eliassi-Rad B, Albert DM, Green WR. Frequency of ocular metastases in patients dying of cancer in eye bank populations. Br J Ophthalmol 1996; 80:125–128.
- Shields CL, Shields JA, Gross NE, Schwartz GP, Lally SE. Survey of 520 eyes with uveal metastases. Ophthalmology 1997; 104:1265–1276.
- Herrag M, Lahmiti S, Yazidi AA, Le Lez ML, Diot P. Choroidal metastasis revealing a lung adenocarcinoma. Ann Thorac Surg 2010; 89:1013–1014.
- Kanthan GL, Jayamohan J, Yip D, Conway RM. Management of metastatic carcinoma of the uveal tract: an evidence-based analysis. Clin Exp Ophthalmol 2007; 35:553–565.
A 62-year-old woman has had flashing lights and floaters in her left eye with progressive loss of vision over the past month. She has not had recent trauma. She does not smoke.
She was referred for an ophthalmologic evaluation. Her visual acuity was 20/20 in the right eye, but she could only count fingers with the left. The anterior segment appeared normal in both eyes. Funduscopic examination of the left eye revealed numerous lobulated, yellowish, choroidal lesions in the posterior pole with overlying subretinal fluid. The lesions involved the fovea, accounting for the poor visual acuity. There were two similar but smaller lesions in the right eye (Figure 1). Ultrasonography confirmed the choroidal location of the lesions (Figure 2).
Q: Which is the most likely diagnosis?
- Retinal detachment
- Choroidal melanoma
- Uveitis
- Uveal metastatic tumor
A: Uveal metastatic tumor is the correct diagnosis. Funduscopic findings of bilateral yellow choroidal lesions are consistent with metastatic cancer.
The patient was admitted to the hospital for a thorough evaluation. Computed tomography of the chest showed a 2.1-by-4.5-cm mass in the lower lobe of the left lung, highly suspicious for malignancy and associated with left hilar lymphadenopathy and right acute pulmonary embolism. Bronchoscopy showed an endobronchial tumor completely occluding the left lower lobe and the lingular orifices.
Pathologic specimens from the endobronchial tumor confirmed adenocarcinoma, consistent with a primary lung cancer.
THE OTHER DIAGNOSTIC CHOICES
Detachment or separation of the retina from the underlying pigment epithelium is one of the most commonly encountered eye emergencies.1 It requires urgent attention, since delay in treatment can cause permanent vision loss.
Retinal detachment differs from uveal metastatic tumor in that it presents and progresses rapidly. The common signs and symptoms are floaters in the center of the visual axis, a sensation of flashing lights (related to retinal traction), and, eventually, loss of vision. The detachment most often represents a break or tear (rhegmatogenous retinal detachment), but it is also a common sequela of neglected diabetic retinopathy. Exudative retinal detachment is usually secondary to uveal inflammation or a uveal tumor.
Choroidal melanoma, the most common primary intraocular malignancy, arises from melanocytes within the choroid. In most cases, it develops from preexisting melanocytic nevi.2 It may present as blurred vision, a paracentral scotoma, painless and progressive visual field loss, and floaters. Choroidal melanoma is usually pigmented (dark brown) and is invariably unilateral.
Uveitis is an inflammation of the uveal tract, which includes the iris, ciliary body, and choroid. It is classified as anterior, intermediate, or posterior uveitis or as panuveitis.3
Although flashing lights, floaters, and reduced vision can occur in uveitis, its other important presenting symptoms (ie, pain, redness, and photophobia) were absent in this patient. The absence of anterior chamber cells and corneal inflammatory deposits (keratic precipitates) also made uveitis less likely.4 However, granulomatous uveitis such as sarcoidosis can present as nodular thickening of the uvea, mimicking an intraocular tumor.5
THE MOST COMMON INTRAOCULAR MALIGNANCY
Uveal metastasis is the most common intraocular malignancy6 and is found on autopsy in up to 12% of people who die of cancer; it involves both eyes in 4.4% of cases. Multiple metastases are seen in one eye in up to 20% of cases.7
The tumors are most often in the choroid, probably because of its extensive blood supply. Breast cancer (in women) and lung cancer (in men) are the most common cancers with uveal metastasis.8 Uveal metastasis from cancers of the prostate, kidney, thyroid, and gastrointestinal tract and from lymphoma and leukemia is less common.8
Patients with choroidal metastasis can see flashing lights, floating spots, and distortion of their vision. In such patients, a careful history and physical examination can uncover signs and symptoms of the hidden cancer, especially of lung cancer.9
Once uveal metastasis is suspected, both eyes and orbits and the central nervous system should be examined, as this disease tends to present bilaterally and to involve the central nervous system.10 Uveal metastases respond to chemotherapy and radiotherapy, depending on the nature of the primary tumor. In general, treatment is based on the extent of the metastasis, prior treatments, and the patient’s overall functional status.
A 62-year-old woman has had flashing lights and floaters in her left eye with progressive loss of vision over the past month. She has not had recent trauma. She does not smoke.
She was referred for an ophthalmologic evaluation. Her visual acuity was 20/20 in the right eye, but she could only count fingers with the left. The anterior segment appeared normal in both eyes. Funduscopic examination of the left eye revealed numerous lobulated, yellowish, choroidal lesions in the posterior pole with overlying subretinal fluid. The lesions involved the fovea, accounting for the poor visual acuity. There were two similar but smaller lesions in the right eye (Figure 1). Ultrasonography confirmed the choroidal location of the lesions (Figure 2).
Q: Which is the most likely diagnosis?
- Retinal detachment
- Choroidal melanoma
- Uveitis
- Uveal metastatic tumor
A: Uveal metastatic tumor is the correct diagnosis. Funduscopic findings of bilateral yellow choroidal lesions are consistent with metastatic cancer.
The patient was admitted to the hospital for a thorough evaluation. Computed tomography of the chest showed a 2.1-by-4.5-cm mass in the lower lobe of the left lung, highly suspicious for malignancy and associated with left hilar lymphadenopathy and right acute pulmonary embolism. Bronchoscopy showed an endobronchial tumor completely occluding the left lower lobe and the lingular orifices.
Pathologic specimens from the endobronchial tumor confirmed adenocarcinoma, consistent with a primary lung cancer.
THE OTHER DIAGNOSTIC CHOICES
Detachment or separation of the retina from the underlying pigment epithelium is one of the most commonly encountered eye emergencies.1 It requires urgent attention, since delay in treatment can cause permanent vision loss.
Retinal detachment differs from uveal metastatic tumor in that it presents and progresses rapidly. The common signs and symptoms are floaters in the center of the visual axis, a sensation of flashing lights (related to retinal traction), and, eventually, loss of vision. The detachment most often represents a break or tear (rhegmatogenous retinal detachment), but it is also a common sequela of neglected diabetic retinopathy. Exudative retinal detachment is usually secondary to uveal inflammation or a uveal tumor.
Choroidal melanoma, the most common primary intraocular malignancy, arises from melanocytes within the choroid. In most cases, it develops from preexisting melanocytic nevi.2 It may present as blurred vision, a paracentral scotoma, painless and progressive visual field loss, and floaters. Choroidal melanoma is usually pigmented (dark brown) and is invariably unilateral.
Uveitis is an inflammation of the uveal tract, which includes the iris, ciliary body, and choroid. It is classified as anterior, intermediate, or posterior uveitis or as panuveitis.3
Although flashing lights, floaters, and reduced vision can occur in uveitis, its other important presenting symptoms (ie, pain, redness, and photophobia) were absent in this patient. The absence of anterior chamber cells and corneal inflammatory deposits (keratic precipitates) also made uveitis less likely.4 However, granulomatous uveitis such as sarcoidosis can present as nodular thickening of the uvea, mimicking an intraocular tumor.5
THE MOST COMMON INTRAOCULAR MALIGNANCY
Uveal metastasis is the most common intraocular malignancy6 and is found on autopsy in up to 12% of people who die of cancer; it involves both eyes in 4.4% of cases. Multiple metastases are seen in one eye in up to 20% of cases.7
The tumors are most often in the choroid, probably because of its extensive blood supply. Breast cancer (in women) and lung cancer (in men) are the most common cancers with uveal metastasis.8 Uveal metastasis from cancers of the prostate, kidney, thyroid, and gastrointestinal tract and from lymphoma and leukemia is less common.8
Patients with choroidal metastasis can see flashing lights, floating spots, and distortion of their vision. In such patients, a careful history and physical examination can uncover signs and symptoms of the hidden cancer, especially of lung cancer.9
Once uveal metastasis is suspected, both eyes and orbits and the central nervous system should be examined, as this disease tends to present bilaterally and to involve the central nervous system.10 Uveal metastases respond to chemotherapy and radiotherapy, depending on the nature of the primary tumor. In general, treatment is based on the extent of the metastasis, prior treatments, and the patient’s overall functional status.
- Hatten B, Browne V. Retinal detachment. Emerg Med J 2011; 28:83.
- Factors predictive of growth and treatment of small choroidal melanoma: COMS Report No. 5. The Collaborative Ocular Melanoma Study Group. Arch Ophthalmol 1997; 115:1537–1544.
- Jabs DA, Nussenblatt RB, Rosenbaum JT; Standardization of Uveitis Nomenclature (SUN) Working Group. Standardization of uveitis nomenclature for reporting clinical data. Results of the First International Workshop. Am J Ophthalmol 2005; 140:509–516.
- Wertheim MS, Mathers WD, Planck SJ, et al. In vivo confocal microscopy of keratic precipitates. Arch Ophthalmol 2004; 122:1773–1781.
- Desai UR, Tawansy KA, Joondeph BC, Schiffman RM. Choroidal granulomas in systemic sarcoidosis. Retina 2001; 21:40–47.
- Singh AD, Damato BE, Pe’er J, Murphree AL, Perry JD, eds. Uveal metastatic tumors. In: Clinical Ophthalmic Oncology. Philadelphia, PA: Saunders-Elsevier; 2007:322–327.
- Eliassi-Rad B, Albert DM, Green WR. Frequency of ocular metastases in patients dying of cancer in eye bank populations. Br J Ophthalmol 1996; 80:125–128.
- Shields CL, Shields JA, Gross NE, Schwartz GP, Lally SE. Survey of 520 eyes with uveal metastases. Ophthalmology 1997; 104:1265–1276.
- Herrag M, Lahmiti S, Yazidi AA, Le Lez ML, Diot P. Choroidal metastasis revealing a lung adenocarcinoma. Ann Thorac Surg 2010; 89:1013–1014.
- Kanthan GL, Jayamohan J, Yip D, Conway RM. Management of metastatic carcinoma of the uveal tract: an evidence-based analysis. Clin Exp Ophthalmol 2007; 35:553–565.
- Hatten B, Browne V. Retinal detachment. Emerg Med J 2011; 28:83.
- Factors predictive of growth and treatment of small choroidal melanoma: COMS Report No. 5. The Collaborative Ocular Melanoma Study Group. Arch Ophthalmol 1997; 115:1537–1544.
- Jabs DA, Nussenblatt RB, Rosenbaum JT; Standardization of Uveitis Nomenclature (SUN) Working Group. Standardization of uveitis nomenclature for reporting clinical data. Results of the First International Workshop. Am J Ophthalmol 2005; 140:509–516.
- Wertheim MS, Mathers WD, Planck SJ, et al. In vivo confocal microscopy of keratic precipitates. Arch Ophthalmol 2004; 122:1773–1781.
- Desai UR, Tawansy KA, Joondeph BC, Schiffman RM. Choroidal granulomas in systemic sarcoidosis. Retina 2001; 21:40–47.
- Singh AD, Damato BE, Pe’er J, Murphree AL, Perry JD, eds. Uveal metastatic tumors. In: Clinical Ophthalmic Oncology. Philadelphia, PA: Saunders-Elsevier; 2007:322–327.
- Eliassi-Rad B, Albert DM, Green WR. Frequency of ocular metastases in patients dying of cancer in eye bank populations. Br J Ophthalmol 1996; 80:125–128.
- Shields CL, Shields JA, Gross NE, Schwartz GP, Lally SE. Survey of 520 eyes with uveal metastases. Ophthalmology 1997; 104:1265–1276.
- Herrag M, Lahmiti S, Yazidi AA, Le Lez ML, Diot P. Choroidal metastasis revealing a lung adenocarcinoma. Ann Thorac Surg 2010; 89:1013–1014.
- Kanthan GL, Jayamohan J, Yip D, Conway RM. Management of metastatic carcinoma of the uveal tract: an evidence-based analysis. Clin Exp Ophthalmol 2007; 35:553–565.
POLST: An improvement over traditional advance directives
An 89-year-old woman with advanced dementia is living in a nursing home and is fully dependent in all aspects of personal care, including feeding. She has a health care proxy and a living will.
Her husband is her health care agent and has established that the primary goal of her care should be to keep her comfortable. He has repeatedly discussed this goal with her attending physician and the nursing-home staff and has reiterated that when his wife had capacity, she wanted “no heroics,” “no feeding tube,” and no life-sustaining treatment that would prolong her dying. He has requested that she not be transferred to the hospital and that she receive all further care at the nursing home. These preferences are consistent with her living will.
One evening, she becomes somnolent and febrile, with rapid breathing. The physician covering for the attending physician does not know the patient, cannot reach her husband, and sends her to the hospital, where she is admitted with aspiration pneumonia.
Her level of alertness improves with hydration. However, the hospital nurses have a difficult time feeding her. She does not seem to want to eat, “pockets” food in her cheeks, is slow to swallow, and sometimes coughs during feeding. This is nothing new—at the nursing home, her feeding pattern had been the same for nearly 6 months. During this time she always had a cough; fevers came and went. She has slowly lost weight; she now weighs 100 lb (45 kg), down 30 lb (14 kg) in 3 years.
With treatment, her respiratory distress and fever resolve. The physician orders a swallowing evaluation by a speech therapist, who determines that she needs a feeding tube. After that, a meeting is scheduled with her husband and physician to discuss the speech therapist’s assessment. The patient’s husband emphatically refuses the feeding tube and is upset that she was transferred to the hospital against his expressed wishes.
Why did this happen?
TRADITIONAL ADVANCE DIRECTIVES ARE OFTEN NOT ENOUGH
Even when patients fill out advance directives in accordance with state law, their preferences for care at the end of life are not consistently followed.
Problems with living wills
Living wills state patients’ wishes about medical care in the event that they develop an irreversible condition that prevents them from making their own medical decisions. The living will becomes effective if they become terminally ill, permanently unconscious, or minimally conscious due to brain damage and will never regain the ability to make decisions. People who want to indicate under what set of circumstances they favor or object to receiving any specific treatments use a living will.
The Patient Self-Determination Act of 1990 states that on admission to a hospital or nursing home, patients have to be informed of their rights, including the right to accept or refuse treatment.1 However, the current system of communicating wishes about end-of-life care using solely traditional advance directives such as the living will has proven insufficient. This is because traditional advance directives, being general statements of patients’ preferences, need to be carried out through specifications in medical orders when the need arises.2
Further, traditional advance directives require patients to recognize the importance of advance care planning, understand medical interventions, evaluate their own personal values and beliefs, and communicate their wishes to their agents, loved ones, physicians, and health care providers. Moreover, these documents apply to future circumstances, require further interpretation by the agent and health care professionals, and do not result in actionable medical orders. Decisions about care depend on interpreting earlier conversations, the physician’s estimates of prognosis, and, possibly, the personal convictions of the physician, agent, and loved ones, even though ethically, all involved need to focus on the patient’s stated wishes or best interest. A living will does not help clarify the patient’s wishes in the absence of antecedent conversation with the family, close friends, and the patient’s personal physician. And living wills cannot be read and interpreted in an emergency.
The situation is further complicated by difficulty in defining “terminal” or “irreversible” conditions and accounting for the different perspective that physicians, agents, and loved ones bring to the situation. For example, imagine a patient with dementia nearing the end of life who eats less, has difficulty managing secretions, aspirates, and develops pneumonia. While end-stage dementia is terminal, pneumonia may be reversible.
Increasingly, therefore, people are being counseled to appoint a health care agent (see below).3
The importance of a health care proxy (durable power of attorney for health care)
In a health care proxy document (also known as durable power of attorney for health care), the patient names a health care agent. This person has authority to make decisions about the patient’s medical care, including life-sustaining treatment. In other words, you the patient appoint someone to speak for you in the event you are unable to make your own medical decisions (not only at the end of life).
Since anyone may face a sudden and unexpected acute illness or injury with the risk of becoming incapacitated and unable to make medical decisions, everyone age 18 and older should be encouraged to complete a health care proxy document and to engage in advance care planning discussions with family and loved ones. Physicians can initiate this process as a wellness initiative and can help patients and families understand advance care planning. In all health care settings, trained and qualified health care professionals can provide education on advance care planning to patients, families, and loved ones.
A key issue when naming a health care agent is choosing the right one, someone who will make decisions in accordance with the person’s current values and beliefs and who can separate his or her personal values from the patient’s values. Another key issue: people need to have proactive discussions about their personal values, beliefs, and goals of care, which many are reluctant to do, and the health care agent must be willing to talk about sensitive issues ahead of time. Even when a health care agent is available in an emergency, emergency medical services personnel cannot follow directions from a health care agent. Most importantly, a health care agent must be able to handle potential conflicts between family and providers.
POLST ENSURES PATIENT PREFERENCES ARE HONORED AT THE END OF LIFE
Approximately 20 years ago, a team of health care professionals at the University of Oregon recognized these problems and realized that physicians needed to be more involved in discussions with patients about end-of-life care and in translating the patient’s preferences and values into concrete medical orders. The result was the Physician Orders for Life-Sustaining Treatment (POLST) Paradigm Program.4
What is POLST?
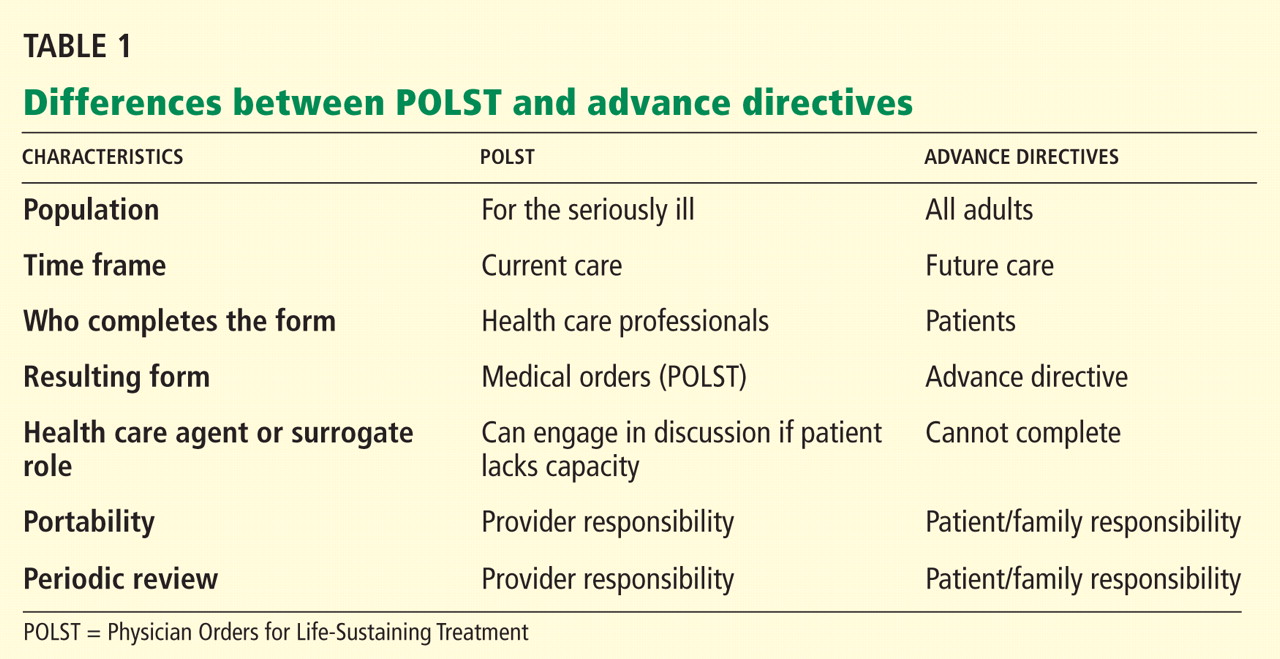
POLST is an end-of-life-care transitions program that focuses on patient-centered goals for care and shared informed medical decision-making.5,6 It offers a mechanism to communicate the wishes of seriously ill patients to have or to limit medical treatment as they move from one care setting to another. Table 1 lists the differences between traditional advance directives and POLST.

The aim is to improve the quality of care that seriously ill patients receive at the end of life. POLST is based on effective communication of the patient’s wishes, with actionable medical orders documented on a brightly colored form (www.ohsu.edu/polst/programs/sample-forms.htm; Figure 1) and a promise by health care professionals to honor these wishes.7 Key features of the program include education, training, and a quality-improvement process.
Who is POLST for?
POLST is for patients with serious life-limiting illness who have a life expectancy of less than 1 year, or anyone of advanced age interested in defining their end-of-life care wishes. Qualified and trained health care professionals (physicians, physician’s assistants, nurse practitioners, and social workers) participate in discussions leading to the completion of a POLST form in all settings, particularly along the long-term care continuum and for home hospice.
The key element of the POLST process: Shared, informed medical decision-making

Health care professionals working as an interdisciplinary team play a key role in educating patients and their families about advance care planning and shared, informed medical decision-making, as well as in resolving conflict. To be effective, shared medical decision-making must be well-informed. The decision-maker (patient, health care agent, or surrogate) must weigh the following questions (Table 2):
- Will treatment make a difference?
- Do the burdens of treatment outweigh its benefits?
- Is there hope of recovery? If so, what will life be like afterward?
- What does the patient value? What is the patient’s goal for his or her care?

In-depth discussions with patients, family members, and surrogates are needed, and these people are often reluctant to ask these questions and afraid to discuss the dying process. Even if they are informed of their diagnosis and prognosis, they may not know what they mean in terms of their everyday experience and future.
Health care professionals engaging in these conversations can use the eight-step POLST protocol (Table 3) to elicit their preferences at the end of life. Table 4 lists tools and resources to enhance the understanding of advance care planning and POLST.
What does the POLST form cover?

The POLST form (Figure 1) provides instructions about resuscitation if the patient has no pulse and is not breathing. Additionally, the medical orders indicate decisions about the level of medical intervention that the patient wants or does not want, eg, intubation, mechanical ventilation, transport to the hospital, intensive care, artificial nutrition and hydration, and antibiotics.
Thus, POLST is outcome-neutral and can be used either to limit medical interventions or to clarify a request for any or all medically indicated treatments.
Both the practitioner and the patient or patient’s surrogate sign the form. The original goes into the patient’s chart, and a copy should accompany the patient if he or she is transferred or discharged. Additionally, if the state has a POLST registry, the POLST information should be entered into the registry.
POLST is expanding across the country
The use of POLST has been expanding across the United States, with POLST programs now implemented in all or part of at least 30 states. There are endorsed programs in 14 states, and programs are being developed in 26 more. Requirements for endorsement are found at www.polst.org. Figure 2 shows the status of POLST in the 50 states.
Oregon’s POLST form is the original model for other forms designed to meet specific legislative or regulatory requirements in other states. POLST-like programs are known by different names in different states: eg, New York’s Medical Orders for Life-Sustaining Treatment (MOLST) and West Virginia’s Physicians Orders for Scope of Treatment (POST), but all endorsed programs share common core elements.
POLST research
A number of studies in the past 10 years have shown that POLST improves the documentation and honoring of patient preferences, whatever they may be.4,8–16
Emergency medical technicians in Oregon reported that the POLST form provides clear instructions about patient preferences and is useful when deciding which treatments to provide. In contrast to the single-intervention focus of out-of-hospital do-not-resuscitate orders, the POLST form provides patients the opportunity to document treatment goals and preferences for interventions across a range of treatment options, thus permitting greater individualization.13
Comfort care is not sacrificed if a POLST document is in place. Most hospice patients choose at least one life-sustaining treatment on their POLST form.14
In a multistate study published in 2010, the medical records of residents in 90 randomly chosen Medicaid-eligible nursing homes were reviewed.15 POLST was compared with traditional advance care planning in terms of the effect on the presence of medical orders reflecting treatment preferences, symptom management, and use of life-sustaining treatments. The study found that residents with POLST forms had significantly more medical orders about life-sustaining treatments than residents with traditional advance directives. There were no differences between residents with or without POLST forms on symptom assessment or management measures. POLST was more effective than traditional advance planning at limiting unwanted life-sustaining treatments. The study suggests that POLST offers significant advantages over traditional advance directives in nursing facilities.15,16
In summary, more than a decade of research has shown that the POLST Paradigm Program serves as an emerging national model for implementing shared, informed medical decision-making. Furthermore, POLST more accurately conveys end-of-life care preferences for patients with advanced chronic illness and for dying patients than traditional advance directives and yields higher adherence by medical professionals.
CLINICAL CASE REVISITED
Let’s consider if the physician for our 89-year-old woman with dementia had completed a POLST form with orders indicating “do not attempt resuscitation (DNR/no CPR)” and “comfort measures only, do not transfer to hospital for life-sustaining treatment and transfer if comfort needs cannot be met in current location.”
The patient’s respiratory distress and fever would have been treated at her nursing home with medication and oxygen. She would have been transferred to the hospital only if her comfort needs would not have been met at the nursing home. Unwanted life-sustaining treatment would have been avoided. The wishes of the patient, based on her values and careful consideration of options, would have been respected.
- Dunn PM, Tolle SW, Moss AH, Black JS. The POLST paradigm: respecting the wishes of patients and families. Ann Long-Term Care 2007; 15:33–40.
- Patient Self-Determination Act of 1990. Pub. L. No. 101-508, ss 4206, 104 Stat. 1388.
- Bomba PA, Sabatino CP. POLST: an emerging model for end-of-life care planning. The ElderLaw Report 2009; 20:1–5.
- Karp Sabatino C. AARP Public Policy Institute, Improving advance illness care: the evolution of state POLST programs 2011. http://assets.aarp.org/rgcenter/ppi/cons-prot/POLST-Report-04-11.pdf. Accessed May 30, 2012.
- Bomba PA. Discussing patient p and end of life care, Journal of the Monroe County Medical Society, 7th District Branch, MSSNY 2011;12–15. www.compassionandsupport.org/index.php/research_/. Accessed May 30, 2012.
- Citko J, Moss AH, Carley M, Tolle SW. The National POLST Paradigm Initiative, 2ND ed. Fast Facts and Concepts 2010;178. www.eperc.mcw.edu/fastfact/ff_178.htm. Accessed May 30, 2012.
- Center for Ethics in Health Care, Oregon Health & Science University. www.ohsu.edu/polst/. Accessed May 30, 2012.
- Lee MA, Brummel-Smith K, Meyer J, Drew N, London MR. Physician orders for life-sustaining treatment (POLST): outcomes in a PACE program. Program of All-Inclusive Care for the Elderly. J Am Geriatr Soc 2000; 48:1219–1225.
- Meyers JL, Moore C, McGrory A, Sparr J, Ahern M. Physician orders for life-sustaining treatment form: honoring end-of-life directives for nursing home residents. J Gerontol Nurs 2004; 30:37–46.
- Dunn PM, Schmidt TA, Carley MM, Donius M, Weinstein MA, Dull VT. A method to communicate patient p about medically indicated life-sustaining treatment in the out-of-hospital setting. J Am Geriatr Soc 1996; 44:785–791.
- Cantor MD. Improving advance care planning: lessons from POLST. Physician Orders for Life-Sustaining Treatment (comment). J Am Geriatr Soc 2000; 48:1343–1344.
- Tolle SW, Tilden VP, Nelson CA, Dunn PM. A prospective study of the efficacy of the physician order form for life-sustaining treatment. J Am Geriatr Soc 1998; 46:1097–1102.
- Schmidt TA, Hickman SE, Tolle SW, Brooks HS. The Physician Orders for Life-Sustaining Treatment program: Oregon emergency medical technicians’ practical experiences and attitudes. J Am Geriatr Soc 2004; 52:1430–1434.
- Hickman SE, Nelson CA, Moss AH, et al. Use of the Physician Orders for Life-Sustaining Treatment (POLST) paradigm program in the hospice setting. J Palliat Med 2009; 12:133–141.
- Hickman SE, Nelson CA, Perrin NA, Moss AH, Hammes BJ, Tolle SW. A comparison of methods to communicate treatment p in nursing facilities: traditional practices versus the physician orders for life-sustaining treatment program. J Am Geriatr Soc 2010; 58:1241–1248.
- Hickman SE, Nelson CA, Moss AH, Tolle SW, Perrin NA, Hammes BJ. The consistency between treatments provided to nursing facility residents and orders on the physician orders for life-sustaining treatment form. J Am Geriatr Soc 2011; 59:2091–2099.
An 89-year-old woman with advanced dementia is living in a nursing home and is fully dependent in all aspects of personal care, including feeding. She has a health care proxy and a living will.
Her husband is her health care agent and has established that the primary goal of her care should be to keep her comfortable. He has repeatedly discussed this goal with her attending physician and the nursing-home staff and has reiterated that when his wife had capacity, she wanted “no heroics,” “no feeding tube,” and no life-sustaining treatment that would prolong her dying. He has requested that she not be transferred to the hospital and that she receive all further care at the nursing home. These preferences are consistent with her living will.
One evening, she becomes somnolent and febrile, with rapid breathing. The physician covering for the attending physician does not know the patient, cannot reach her husband, and sends her to the hospital, where she is admitted with aspiration pneumonia.
Her level of alertness improves with hydration. However, the hospital nurses have a difficult time feeding her. She does not seem to want to eat, “pockets” food in her cheeks, is slow to swallow, and sometimes coughs during feeding. This is nothing new—at the nursing home, her feeding pattern had been the same for nearly 6 months. During this time she always had a cough; fevers came and went. She has slowly lost weight; she now weighs 100 lb (45 kg), down 30 lb (14 kg) in 3 years.
With treatment, her respiratory distress and fever resolve. The physician orders a swallowing evaluation by a speech therapist, who determines that she needs a feeding tube. After that, a meeting is scheduled with her husband and physician to discuss the speech therapist’s assessment. The patient’s husband emphatically refuses the feeding tube and is upset that she was transferred to the hospital against his expressed wishes.
Why did this happen?
TRADITIONAL ADVANCE DIRECTIVES ARE OFTEN NOT ENOUGH
Even when patients fill out advance directives in accordance with state law, their preferences for care at the end of life are not consistently followed.
Problems with living wills
Living wills state patients’ wishes about medical care in the event that they develop an irreversible condition that prevents them from making their own medical decisions. The living will becomes effective if they become terminally ill, permanently unconscious, or minimally conscious due to brain damage and will never regain the ability to make decisions. People who want to indicate under what set of circumstances they favor or object to receiving any specific treatments use a living will.
The Patient Self-Determination Act of 1990 states that on admission to a hospital or nursing home, patients have to be informed of their rights, including the right to accept or refuse treatment.1 However, the current system of communicating wishes about end-of-life care using solely traditional advance directives such as the living will has proven insufficient. This is because traditional advance directives, being general statements of patients’ preferences, need to be carried out through specifications in medical orders when the need arises.2
Further, traditional advance directives require patients to recognize the importance of advance care planning, understand medical interventions, evaluate their own personal values and beliefs, and communicate their wishes to their agents, loved ones, physicians, and health care providers. Moreover, these documents apply to future circumstances, require further interpretation by the agent and health care professionals, and do not result in actionable medical orders. Decisions about care depend on interpreting earlier conversations, the physician’s estimates of prognosis, and, possibly, the personal convictions of the physician, agent, and loved ones, even though ethically, all involved need to focus on the patient’s stated wishes or best interest. A living will does not help clarify the patient’s wishes in the absence of antecedent conversation with the family, close friends, and the patient’s personal physician. And living wills cannot be read and interpreted in an emergency.
The situation is further complicated by difficulty in defining “terminal” or “irreversible” conditions and accounting for the different perspective that physicians, agents, and loved ones bring to the situation. For example, imagine a patient with dementia nearing the end of life who eats less, has difficulty managing secretions, aspirates, and develops pneumonia. While end-stage dementia is terminal, pneumonia may be reversible.
Increasingly, therefore, people are being counseled to appoint a health care agent (see below).3
The importance of a health care proxy (durable power of attorney for health care)
In a health care proxy document (also known as durable power of attorney for health care), the patient names a health care agent. This person has authority to make decisions about the patient’s medical care, including life-sustaining treatment. In other words, you the patient appoint someone to speak for you in the event you are unable to make your own medical decisions (not only at the end of life).
Since anyone may face a sudden and unexpected acute illness or injury with the risk of becoming incapacitated and unable to make medical decisions, everyone age 18 and older should be encouraged to complete a health care proxy document and to engage in advance care planning discussions with family and loved ones. Physicians can initiate this process as a wellness initiative and can help patients and families understand advance care planning. In all health care settings, trained and qualified health care professionals can provide education on advance care planning to patients, families, and loved ones.
A key issue when naming a health care agent is choosing the right one, someone who will make decisions in accordance with the person’s current values and beliefs and who can separate his or her personal values from the patient’s values. Another key issue: people need to have proactive discussions about their personal values, beliefs, and goals of care, which many are reluctant to do, and the health care agent must be willing to talk about sensitive issues ahead of time. Even when a health care agent is available in an emergency, emergency medical services personnel cannot follow directions from a health care agent. Most importantly, a health care agent must be able to handle potential conflicts between family and providers.
POLST ENSURES PATIENT PREFERENCES ARE HONORED AT THE END OF LIFE
Approximately 20 years ago, a team of health care professionals at the University of Oregon recognized these problems and realized that physicians needed to be more involved in discussions with patients about end-of-life care and in translating the patient’s preferences and values into concrete medical orders. The result was the Physician Orders for Life-Sustaining Treatment (POLST) Paradigm Program.4
What is POLST?
POLST is an end-of-life-care transitions program that focuses on patient-centered goals for care and shared informed medical decision-making.5,6 It offers a mechanism to communicate the wishes of seriously ill patients to have or to limit medical treatment as they move from one care setting to another. Table 1 lists the differences between traditional advance directives and POLST.
The aim is to improve the quality of care that seriously ill patients receive at the end of life. POLST is based on effective communication of the patient’s wishes, with actionable medical orders documented on a brightly colored form (www.ohsu.edu/polst/programs/sample-forms.htm; Figure 1) and a promise by health care professionals to honor these wishes.7 Key features of the program include education, training, and a quality-improvement process.
Who is POLST for?
POLST is for patients with serious life-limiting illness who have a life expectancy of less than 1 year, or anyone of advanced age interested in defining their end-of-life care wishes. Qualified and trained health care professionals (physicians, physician’s assistants, nurse practitioners, and social workers) participate in discussions leading to the completion of a POLST form in all settings, particularly along the long-term care continuum and for home hospice.
The key element of the POLST process: Shared, informed medical decision-making
Health care professionals working as an interdisciplinary team play a key role in educating patients and their families about advance care planning and shared, informed medical decision-making, as well as in resolving conflict. To be effective, shared medical decision-making must be well-informed. The decision-maker (patient, health care agent, or surrogate) must weigh the following questions (Table 2):
- Will treatment make a difference?
- Do the burdens of treatment outweigh its benefits?
- Is there hope of recovery? If so, what will life be like afterward?
- What does the patient value? What is the patient’s goal for his or her care?
In-depth discussions with patients, family members, and surrogates are needed, and these people are often reluctant to ask these questions and afraid to discuss the dying process. Even if they are informed of their diagnosis and prognosis, they may not know what they mean in terms of their everyday experience and future.
Health care professionals engaging in these conversations can use the eight-step POLST protocol (Table 3) to elicit their preferences at the end of life. Table 4 lists tools and resources to enhance the understanding of advance care planning and POLST.
What does the POLST form cover?
The POLST form (Figure 1) provides instructions about resuscitation if the patient has no pulse and is not breathing. Additionally, the medical orders indicate decisions about the level of medical intervention that the patient wants or does not want, eg, intubation, mechanical ventilation, transport to the hospital, intensive care, artificial nutrition and hydration, and antibiotics.
Thus, POLST is outcome-neutral and can be used either to limit medical interventions or to clarify a request for any or all medically indicated treatments.
Both the practitioner and the patient or patient’s surrogate sign the form. The original goes into the patient’s chart, and a copy should accompany the patient if he or she is transferred or discharged. Additionally, if the state has a POLST registry, the POLST information should be entered into the registry.
POLST is expanding across the country
The use of POLST has been expanding across the United States, with POLST programs now implemented in all or part of at least 30 states. There are endorsed programs in 14 states, and programs are being developed in 26 more. Requirements for endorsement are found at www.polst.org. Figure 2 shows the status of POLST in the 50 states.
Oregon’s POLST form is the original model for other forms designed to meet specific legislative or regulatory requirements in other states. POLST-like programs are known by different names in different states: eg, New York’s Medical Orders for Life-Sustaining Treatment (MOLST) and West Virginia’s Physicians Orders for Scope of Treatment (POST), but all endorsed programs share common core elements.
POLST research
A number of studies in the past 10 years have shown that POLST improves the documentation and honoring of patient preferences, whatever they may be.4,8–16
Emergency medical technicians in Oregon reported that the POLST form provides clear instructions about patient preferences and is useful when deciding which treatments to provide. In contrast to the single-intervention focus of out-of-hospital do-not-resuscitate orders, the POLST form provides patients the opportunity to document treatment goals and preferences for interventions across a range of treatment options, thus permitting greater individualization.13
Comfort care is not sacrificed if a POLST document is in place. Most hospice patients choose at least one life-sustaining treatment on their POLST form.14
In a multistate study published in 2010, the medical records of residents in 90 randomly chosen Medicaid-eligible nursing homes were reviewed.15 POLST was compared with traditional advance care planning in terms of the effect on the presence of medical orders reflecting treatment preferences, symptom management, and use of life-sustaining treatments. The study found that residents with POLST forms had significantly more medical orders about life-sustaining treatments than residents with traditional advance directives. There were no differences between residents with or without POLST forms on symptom assessment or management measures. POLST was more effective than traditional advance planning at limiting unwanted life-sustaining treatments. The study suggests that POLST offers significant advantages over traditional advance directives in nursing facilities.15,16
In summary, more than a decade of research has shown that the POLST Paradigm Program serves as an emerging national model for implementing shared, informed medical decision-making. Furthermore, POLST more accurately conveys end-of-life care preferences for patients with advanced chronic illness and for dying patients than traditional advance directives and yields higher adherence by medical professionals.
CLINICAL CASE REVISITED
Let’s consider if the physician for our 89-year-old woman with dementia had completed a POLST form with orders indicating “do not attempt resuscitation (DNR/no CPR)” and “comfort measures only, do not transfer to hospital for life-sustaining treatment and transfer if comfort needs cannot be met in current location.”
The patient’s respiratory distress and fever would have been treated at her nursing home with medication and oxygen. She would have been transferred to the hospital only if her comfort needs would not have been met at the nursing home. Unwanted life-sustaining treatment would have been avoided. The wishes of the patient, based on her values and careful consideration of options, would have been respected.
An 89-year-old woman with advanced dementia is living in a nursing home and is fully dependent in all aspects of personal care, including feeding. She has a health care proxy and a living will.
Her husband is her health care agent and has established that the primary goal of her care should be to keep her comfortable. He has repeatedly discussed this goal with her attending physician and the nursing-home staff and has reiterated that when his wife had capacity, she wanted “no heroics,” “no feeding tube,” and no life-sustaining treatment that would prolong her dying. He has requested that she not be transferred to the hospital and that she receive all further care at the nursing home. These preferences are consistent with her living will.
One evening, she becomes somnolent and febrile, with rapid breathing. The physician covering for the attending physician does not know the patient, cannot reach her husband, and sends her to the hospital, where she is admitted with aspiration pneumonia.
Her level of alertness improves with hydration. However, the hospital nurses have a difficult time feeding her. She does not seem to want to eat, “pockets” food in her cheeks, is slow to swallow, and sometimes coughs during feeding. This is nothing new—at the nursing home, her feeding pattern had been the same for nearly 6 months. During this time she always had a cough; fevers came and went. She has slowly lost weight; she now weighs 100 lb (45 kg), down 30 lb (14 kg) in 3 years.
With treatment, her respiratory distress and fever resolve. The physician orders a swallowing evaluation by a speech therapist, who determines that she needs a feeding tube. After that, a meeting is scheduled with her husband and physician to discuss the speech therapist’s assessment. The patient’s husband emphatically refuses the feeding tube and is upset that she was transferred to the hospital against his expressed wishes.
Why did this happen?
TRADITIONAL ADVANCE DIRECTIVES ARE OFTEN NOT ENOUGH
Even when patients fill out advance directives in accordance with state law, their preferences for care at the end of life are not consistently followed.
Problems with living wills
Living wills state patients’ wishes about medical care in the event that they develop an irreversible condition that prevents them from making their own medical decisions. The living will becomes effective if they become terminally ill, permanently unconscious, or minimally conscious due to brain damage and will never regain the ability to make decisions. People who want to indicate under what set of circumstances they favor or object to receiving any specific treatments use a living will.
The Patient Self-Determination Act of 1990 states that on admission to a hospital or nursing home, patients have to be informed of their rights, including the right to accept or refuse treatment.1 However, the current system of communicating wishes about end-of-life care using solely traditional advance directives such as the living will has proven insufficient. This is because traditional advance directives, being general statements of patients’ preferences, need to be carried out through specifications in medical orders when the need arises.2
Further, traditional advance directives require patients to recognize the importance of advance care planning, understand medical interventions, evaluate their own personal values and beliefs, and communicate their wishes to their agents, loved ones, physicians, and health care providers. Moreover, these documents apply to future circumstances, require further interpretation by the agent and health care professionals, and do not result in actionable medical orders. Decisions about care depend on interpreting earlier conversations, the physician’s estimates of prognosis, and, possibly, the personal convictions of the physician, agent, and loved ones, even though ethically, all involved need to focus on the patient’s stated wishes or best interest. A living will does not help clarify the patient’s wishes in the absence of antecedent conversation with the family, close friends, and the patient’s personal physician. And living wills cannot be read and interpreted in an emergency.
The situation is further complicated by difficulty in defining “terminal” or “irreversible” conditions and accounting for the different perspective that physicians, agents, and loved ones bring to the situation. For example, imagine a patient with dementia nearing the end of life who eats less, has difficulty managing secretions, aspirates, and develops pneumonia. While end-stage dementia is terminal, pneumonia may be reversible.
Increasingly, therefore, people are being counseled to appoint a health care agent (see below).3
The importance of a health care proxy (durable power of attorney for health care)
In a health care proxy document (also known as durable power of attorney for health care), the patient names a health care agent. This person has authority to make decisions about the patient’s medical care, including life-sustaining treatment. In other words, you the patient appoint someone to speak for you in the event you are unable to make your own medical decisions (not only at the end of life).
Since anyone may face a sudden and unexpected acute illness or injury with the risk of becoming incapacitated and unable to make medical decisions, everyone age 18 and older should be encouraged to complete a health care proxy document and to engage in advance care planning discussions with family and loved ones. Physicians can initiate this process as a wellness initiative and can help patients and families understand advance care planning. In all health care settings, trained and qualified health care professionals can provide education on advance care planning to patients, families, and loved ones.
A key issue when naming a health care agent is choosing the right one, someone who will make decisions in accordance with the person’s current values and beliefs and who can separate his or her personal values from the patient’s values. Another key issue: people need to have proactive discussions about their personal values, beliefs, and goals of care, which many are reluctant to do, and the health care agent must be willing to talk about sensitive issues ahead of time. Even when a health care agent is available in an emergency, emergency medical services personnel cannot follow directions from a health care agent. Most importantly, a health care agent must be able to handle potential conflicts between family and providers.
POLST ENSURES PATIENT PREFERENCES ARE HONORED AT THE END OF LIFE
Approximately 20 years ago, a team of health care professionals at the University of Oregon recognized these problems and realized that physicians needed to be more involved in discussions with patients about end-of-life care and in translating the patient’s preferences and values into concrete medical orders. The result was the Physician Orders for Life-Sustaining Treatment (POLST) Paradigm Program.4
What is POLST?
POLST is an end-of-life-care transitions program that focuses on patient-centered goals for care and shared informed medical decision-making.5,6 It offers a mechanism to communicate the wishes of seriously ill patients to have or to limit medical treatment as they move from one care setting to another. Table 1 lists the differences between traditional advance directives and POLST.
The aim is to improve the quality of care that seriously ill patients receive at the end of life. POLST is based on effective communication of the patient’s wishes, with actionable medical orders documented on a brightly colored form (www.ohsu.edu/polst/programs/sample-forms.htm; Figure 1) and a promise by health care professionals to honor these wishes.7 Key features of the program include education, training, and a quality-improvement process.
Who is POLST for?
POLST is for patients with serious life-limiting illness who have a life expectancy of less than 1 year, or anyone of advanced age interested in defining their end-of-life care wishes. Qualified and trained health care professionals (physicians, physician’s assistants, nurse practitioners, and social workers) participate in discussions leading to the completion of a POLST form in all settings, particularly along the long-term care continuum and for home hospice.
The key element of the POLST process: Shared, informed medical decision-making
Health care professionals working as an interdisciplinary team play a key role in educating patients and their families about advance care planning and shared, informed medical decision-making, as well as in resolving conflict. To be effective, shared medical decision-making must be well-informed. The decision-maker (patient, health care agent, or surrogate) must weigh the following questions (Table 2):
- Will treatment make a difference?
- Do the burdens of treatment outweigh its benefits?
- Is there hope of recovery? If so, what will life be like afterward?
- What does the patient value? What is the patient’s goal for his or her care?
In-depth discussions with patients, family members, and surrogates are needed, and these people are often reluctant to ask these questions and afraid to discuss the dying process. Even if they are informed of their diagnosis and prognosis, they may not know what they mean in terms of their everyday experience and future.
Health care professionals engaging in these conversations can use the eight-step POLST protocol (Table 3) to elicit their preferences at the end of life. Table 4 lists tools and resources to enhance the understanding of advance care planning and POLST.
What does the POLST form cover?
The POLST form (Figure 1) provides instructions about resuscitation if the patient has no pulse and is not breathing. Additionally, the medical orders indicate decisions about the level of medical intervention that the patient wants or does not want, eg, intubation, mechanical ventilation, transport to the hospital, intensive care, artificial nutrition and hydration, and antibiotics.
Thus, POLST is outcome-neutral and can be used either to limit medical interventions or to clarify a request for any or all medically indicated treatments.
Both the practitioner and the patient or patient’s surrogate sign the form. The original goes into the patient’s chart, and a copy should accompany the patient if he or she is transferred or discharged. Additionally, if the state has a POLST registry, the POLST information should be entered into the registry.
POLST is expanding across the country
The use of POLST has been expanding across the United States, with POLST programs now implemented in all or part of at least 30 states. There are endorsed programs in 14 states, and programs are being developed in 26 more. Requirements for endorsement are found at www.polst.org. Figure 2 shows the status of POLST in the 50 states.
Oregon’s POLST form is the original model for other forms designed to meet specific legislative or regulatory requirements in other states. POLST-like programs are known by different names in different states: eg, New York’s Medical Orders for Life-Sustaining Treatment (MOLST) and West Virginia’s Physicians Orders for Scope of Treatment (POST), but all endorsed programs share common core elements.
POLST research
A number of studies in the past 10 years have shown that POLST improves the documentation and honoring of patient preferences, whatever they may be.4,8–16
Emergency medical technicians in Oregon reported that the POLST form provides clear instructions about patient preferences and is useful when deciding which treatments to provide. In contrast to the single-intervention focus of out-of-hospital do-not-resuscitate orders, the POLST form provides patients the opportunity to document treatment goals and preferences for interventions across a range of treatment options, thus permitting greater individualization.13
Comfort care is not sacrificed if a POLST document is in place. Most hospice patients choose at least one life-sustaining treatment on their POLST form.14
In a multistate study published in 2010, the medical records of residents in 90 randomly chosen Medicaid-eligible nursing homes were reviewed.15 POLST was compared with traditional advance care planning in terms of the effect on the presence of medical orders reflecting treatment preferences, symptom management, and use of life-sustaining treatments. The study found that residents with POLST forms had significantly more medical orders about life-sustaining treatments than residents with traditional advance directives. There were no differences between residents with or without POLST forms on symptom assessment or management measures. POLST was more effective than traditional advance planning at limiting unwanted life-sustaining treatments. The study suggests that POLST offers significant advantages over traditional advance directives in nursing facilities.15,16
In summary, more than a decade of research has shown that the POLST Paradigm Program serves as an emerging national model for implementing shared, informed medical decision-making. Furthermore, POLST more accurately conveys end-of-life care preferences for patients with advanced chronic illness and for dying patients than traditional advance directives and yields higher adherence by medical professionals.
CLINICAL CASE REVISITED
Let’s consider if the physician for our 89-year-old woman with dementia had completed a POLST form with orders indicating “do not attempt resuscitation (DNR/no CPR)” and “comfort measures only, do not transfer to hospital for life-sustaining treatment and transfer if comfort needs cannot be met in current location.”
The patient’s respiratory distress and fever would have been treated at her nursing home with medication and oxygen. She would have been transferred to the hospital only if her comfort needs would not have been met at the nursing home. Unwanted life-sustaining treatment would have been avoided. The wishes of the patient, based on her values and careful consideration of options, would have been respected.
- Dunn PM, Tolle SW, Moss AH, Black JS. The POLST paradigm: respecting the wishes of patients and families. Ann Long-Term Care 2007; 15:33–40.
- Patient Self-Determination Act of 1990. Pub. L. No. 101-508, ss 4206, 104 Stat. 1388.
- Bomba PA, Sabatino CP. POLST: an emerging model for end-of-life care planning. The ElderLaw Report 2009; 20:1–5.
- Karp Sabatino C. AARP Public Policy Institute, Improving advance illness care: the evolution of state POLST programs 2011. http://assets.aarp.org/rgcenter/ppi/cons-prot/POLST-Report-04-11.pdf. Accessed May 30, 2012.
- Bomba PA. Discussing patient p and end of life care, Journal of the Monroe County Medical Society, 7th District Branch, MSSNY 2011;12–15. www.compassionandsupport.org/index.php/research_/. Accessed May 30, 2012.
- Citko J, Moss AH, Carley M, Tolle SW. The National POLST Paradigm Initiative, 2ND ed. Fast Facts and Concepts 2010;178. www.eperc.mcw.edu/fastfact/ff_178.htm. Accessed May 30, 2012.
- Center for Ethics in Health Care, Oregon Health & Science University. www.ohsu.edu/polst/. Accessed May 30, 2012.
- Lee MA, Brummel-Smith K, Meyer J, Drew N, London MR. Physician orders for life-sustaining treatment (POLST): outcomes in a PACE program. Program of All-Inclusive Care for the Elderly. J Am Geriatr Soc 2000; 48:1219–1225.
- Meyers JL, Moore C, McGrory A, Sparr J, Ahern M. Physician orders for life-sustaining treatment form: honoring end-of-life directives for nursing home residents. J Gerontol Nurs 2004; 30:37–46.
- Dunn PM, Schmidt TA, Carley MM, Donius M, Weinstein MA, Dull VT. A method to communicate patient p about medically indicated life-sustaining treatment in the out-of-hospital setting. J Am Geriatr Soc 1996; 44:785–791.
- Cantor MD. Improving advance care planning: lessons from POLST. Physician Orders for Life-Sustaining Treatment (comment). J Am Geriatr Soc 2000; 48:1343–1344.
- Tolle SW, Tilden VP, Nelson CA, Dunn PM. A prospective study of the efficacy of the physician order form for life-sustaining treatment. J Am Geriatr Soc 1998; 46:1097–1102.
- Schmidt TA, Hickman SE, Tolle SW, Brooks HS. The Physician Orders for Life-Sustaining Treatment program: Oregon emergency medical technicians’ practical experiences and attitudes. J Am Geriatr Soc 2004; 52:1430–1434.
- Hickman SE, Nelson CA, Moss AH, et al. Use of the Physician Orders for Life-Sustaining Treatment (POLST) paradigm program in the hospice setting. J Palliat Med 2009; 12:133–141.
- Hickman SE, Nelson CA, Perrin NA, Moss AH, Hammes BJ, Tolle SW. A comparison of methods to communicate treatment p in nursing facilities: traditional practices versus the physician orders for life-sustaining treatment program. J Am Geriatr Soc 2010; 58:1241–1248.
- Hickman SE, Nelson CA, Moss AH, Tolle SW, Perrin NA, Hammes BJ. The consistency between treatments provided to nursing facility residents and orders on the physician orders for life-sustaining treatment form. J Am Geriatr Soc 2011; 59:2091–2099.
- Dunn PM, Tolle SW, Moss AH, Black JS. The POLST paradigm: respecting the wishes of patients and families. Ann Long-Term Care 2007; 15:33–40.
- Patient Self-Determination Act of 1990. Pub. L. No. 101-508, ss 4206, 104 Stat. 1388.
- Bomba PA, Sabatino CP. POLST: an emerging model for end-of-life care planning. The ElderLaw Report 2009; 20:1–5.
- Karp Sabatino C. AARP Public Policy Institute, Improving advance illness care: the evolution of state POLST programs 2011. http://assets.aarp.org/rgcenter/ppi/cons-prot/POLST-Report-04-11.pdf. Accessed May 30, 2012.
- Bomba PA. Discussing patient p and end of life care, Journal of the Monroe County Medical Society, 7th District Branch, MSSNY 2011;12–15. www.compassionandsupport.org/index.php/research_/. Accessed May 30, 2012.
- Citko J, Moss AH, Carley M, Tolle SW. The National POLST Paradigm Initiative, 2ND ed. Fast Facts and Concepts 2010;178. www.eperc.mcw.edu/fastfact/ff_178.htm. Accessed May 30, 2012.
- Center for Ethics in Health Care, Oregon Health & Science University. www.ohsu.edu/polst/. Accessed May 30, 2012.
- Lee MA, Brummel-Smith K, Meyer J, Drew N, London MR. Physician orders for life-sustaining treatment (POLST): outcomes in a PACE program. Program of All-Inclusive Care for the Elderly. J Am Geriatr Soc 2000; 48:1219–1225.
- Meyers JL, Moore C, McGrory A, Sparr J, Ahern M. Physician orders for life-sustaining treatment form: honoring end-of-life directives for nursing home residents. J Gerontol Nurs 2004; 30:37–46.
- Dunn PM, Schmidt TA, Carley MM, Donius M, Weinstein MA, Dull VT. A method to communicate patient p about medically indicated life-sustaining treatment in the out-of-hospital setting. J Am Geriatr Soc 1996; 44:785–791.
- Cantor MD. Improving advance care planning: lessons from POLST. Physician Orders for Life-Sustaining Treatment (comment). J Am Geriatr Soc 2000; 48:1343–1344.
- Tolle SW, Tilden VP, Nelson CA, Dunn PM. A prospective study of the efficacy of the physician order form for life-sustaining treatment. J Am Geriatr Soc 1998; 46:1097–1102.
- Schmidt TA, Hickman SE, Tolle SW, Brooks HS. The Physician Orders for Life-Sustaining Treatment program: Oregon emergency medical technicians’ practical experiences and attitudes. J Am Geriatr Soc 2004; 52:1430–1434.
- Hickman SE, Nelson CA, Moss AH, et al. Use of the Physician Orders for Life-Sustaining Treatment (POLST) paradigm program in the hospice setting. J Palliat Med 2009; 12:133–141.
- Hickman SE, Nelson CA, Perrin NA, Moss AH, Hammes BJ, Tolle SW. A comparison of methods to communicate treatment p in nursing facilities: traditional practices versus the physician orders for life-sustaining treatment program. J Am Geriatr Soc 2010; 58:1241–1248.
- Hickman SE, Nelson CA, Moss AH, Tolle SW, Perrin NA, Hammes BJ. The consistency between treatments provided to nursing facility residents and orders on the physician orders for life-sustaining treatment form. J Am Geriatr Soc 2011; 59:2091–2099.
KEY POINTS
- Failures and opportunities for improvement in current advance care planning processes highlight the need for change.
- Differences exist between traditional advance directives and actionable medical orders.
- Advance care planning discussions can be initiated by physicians as a wellness initiative for everyone 18 years of age and older and can help patients and families understand advance care planning.
- POLST is outcome-neutral and may be used either to limit medical interventions or to clarify a request for any or all medically indicated treatments.
- Shared, informed medical decision-making is an essential element of the POLST process.