User login
Case Studies in Toxicology: A Common Procedure, an Uncommon Complication
Case
A 35-year-old woman underwent an elective hysteroscopic myomectomy to remove a symptomatic 2.7-cm uterine leiomyoma. The procedure was uncomplicated, and the patient awoke in the postanesthesia care unit (PACU) in good condition. Two hours later, however, she developed severe shortness of breath and required bilevel positive airway pressure ventilation. Her vital signs in the PACU were: blood pressure (BP), 110/70 mm Hg; heart rate, 90 beats/minute; respiratory rate, 12 breaths/minute; temperature, 98.4°F. Oxygen saturation was 94% on room air. She was diaphoretic and tachycardic on physical examination, but her pulmonary, abdominal, and gynecologic examinations were normal. During the examination, she complained of nausea, vomited, and then became increasingly lethargic and confused.
How can this patient’s clinical presentation be explained?
Uterine fibroids are the most common pelvic tumor in women.1 Hysteroscopic myomectomy is a minimally invasive surgical procedure commonly performed to resect submucosal fibroids. The procedure takes about 60 minutes, and is often performed on an outpatient basis under general anesthesia. During the procedure, an electrosurgery device called a resectoscope is inserted through the cervix. The uterine cavity is then distended with a large volume of irrigating solution. Maneuvering the resectoscope, the surgeon then shaves the protruding fibroid layer-by-layer until it aligns with the surrounding myometrium.
Surgical complications of hysteroscopic myomectomy may produce life-threatening effects. Excessive resection of the myometrium may increase blood loss, which can cause chest pain, shortness of breath, diaphoresis, lethargy, and confusion. Uterine perforation may produce hypotension, abdominal pain and distention, infection, and vaginal bleeding.
Venous Thromboembolism
Venous thromboembolism (VTE) is a common postoperative complication, with pulmonary embolism accounting for the most common preventable cause of hospital death in the United States.2 Gynecologic surgery, especially brief procedures, are associated with among the lowest rates of VTE, however, making this an unlikely explanation in this case.3 Additionally, VTE is not expected to produce the neurological findings observed in this patient.
Negative Pressure Pulmonary Edema
An uncommon but life-threatening complication for patients undergoing general anesthesia is negative pressure pulmonary edema, or “postextubation pulmonary edema,” which is estimated to occur in up to 1 in 1,000 procedures involving mechanical ventilation. During extubation, forced inspiration against a closed glottis causes intravascular fluid to be drawn into the interstitial space leading to pulmonary edema.4
Hyponatremia
An unusual but well described complication of endoscopic surgery is hyponatremia from systemic absorption of the irrigating fluid. Fluid overload may result in pulmonary edema, and dilutional hyponatremia may cause altered mental status or seizures.
Case Continuation
A chest X-ray performed after the patient became symptomatic revealed mild bilateral pulmonary edema. Her postoperative laboratory values were: sodium, 112 mEq/L; potassium, 3.3 mEq/L; chloride, 81 mEq/L; bicarbonate, 25 mEq/L; blood urea nitrogen, 18 mg/dL; creatinine, 0.6 mg/dL. Her ammonia level was 24 mmol/L (normal range, 11-35 mmol/L). An endotracheal tube was placed after her level of consciousness declined further. Her neurological examination revealed bilateral fixed and dilated pupils. An emergent computed tomography (CT) scan of the brain revealed severe generalized swelling of the brain.
What is the cause of this patient’s hyponatremia?
Monopolar electrosurgical devices such as the resectoscope cannot be used with electrolyte-containing irrigation fluids (eg, isotonic saline or lactated Ringer’s solution). Nonconductive, nonelectrolyte solutions such as glycine 1.5%, sorbitol 3%, or mannitol 5%, are the most common irrigating fluids employed to dilate the operating field and to wash away debris and blood.5
A dilutional hyponatremia can occur when the irrigating fluid is absorbed systemically. As it was first described following transurethral resection of the prostate procedures in the 1950s, the syndrome is referred to as “TURP” syndrome. Since then, several hundred life-threatening and even fatal cases of TURP syndrome have been reported.6 The syndrome occurs with other operations including transcervical resection of the endometrium (TCRE).5 The irrigating fluid is most frequently absorbed directly into the vascular system when a vein has been severed during the electrosurgery, particularly when the infusion pressure exceeds the venous pressure.6 Additionally, extravasation of the irrigating fluid into the intraperitoneal space can occur after instrument perforation of the uterine wall in TCRE, or via the fallopian tubes.6
What are the signs and symptoms of TURP syndrome?
Mild-to-moderate TURP syndrome occurs in 1% to 8% of TURP procedures performed. Fluid absorption is slightly more common during TCRE, and occurs more often during the resection of fibroids.6 The dilutional hyponatremia can result in brain edema, as well as pharmacological effects specific to the irrigant solutes.
Symptoms of TURP syndrome are primarily neurological, with nausea being the earliest sign of a mild syndrome. A “mini” mental-status test may show transient confusion with smaller absorption volumes.7 As the fluid absorption increases, the hyponatremia worsens, resulting in cerebral edema. This manifests as encephalopathy, which includes disorientation, twitching, and seizures. Hypotension is uncommon, since the fluid is being absorbed intravascularly.6 Shortness of breath, uneasiness, chest pain, and pulmonary edema may develop from systemic fluid overload. The intra-abdominal extravasation of fluid can result in abdominal pain. Other symptoms are specific to the irrigant.
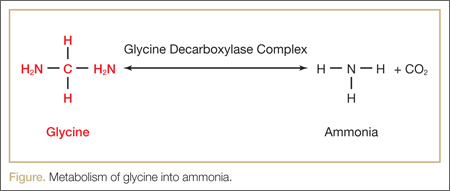
Glycine
Glycine 1.5% is the most common irrigant solution used; as such, it produces the highest incidence of TURP syndrome.8 This solution is hypoosmotic (osmolality of 200 mosm/kg) compared with the normal serum (osmolality of 280 to 296 mosm/kg).5 In addition, glycine may cause visual disturbances.8 The metabolism of glycine produces ammonia, serine, and oxalate (Figure), and 10% of patients who absorb glycine show a marked hyperammonemia, further exacerbating the neurological effects.9,10
Sorbitol and mannitol
Sorbitol and mannitol irrigation fluids are used less frequently than glycine. Sorbitol 3% is metabolized to fructose and glucose, and has an osmolality of 165 mosm/kg.6 When absorbed systemically, sorbitol’s effects are similar to those of glycine, except that it does not induce visual symptoms. Mannitol 5% solution has the advantage of being isosmotic (275 mosm/kg). It is not metabolized and is excreted entirely in the urine. The excretion of mannitol creates an osmotic diuresis, thereby preventing hyponatremia from occurring.9Sorbitol and Mannitol
What are the treatment options for TURP Syndrome? Can it be prevented?
Patients with TURP syndrome in its mildest form can be asymptomatic, but severe cases can be life threatening or fatal. Unlike the treatment for hyponatremia caused by psychogenic polydipsia or the syndrome of inappropriate antidiuretic hormone, which calls for fluid restriction, plasma volume expansion is indicated in TURP syndrome, as hypovolemia and low-cardiac output develop as soon as irrigation is discontinued.
Hypertonic saline is indicated for neurological symptoms, or if the serum sodium concentration is <120mEq/L.6 Although furosemide has been used to treat acute pulmonary edema, no studies support its routine use in the treatment of fluid absorption,6 and its use may aggravate hyponatremia and hypovolemia.
Bipolar electrosurgical systems, unlike monopolar systems, permit the use of electrolyte solutions such as isotonic saline, thereby significantly reducing the risk of hyponatremia. For hysteroscopic procedures, the American College of Obstetricians and Gynecologists recommends the use of an automated fluid pump and monitoring system, thus minimizing the fluid pressure and halting or terminating the procedure before absorption thresholds are exceeded.11
Case Conclusion
The patient was immediately given a 1 mL/kg bolus of hypertonic saline. Two hours later, her serum sodium improved to 114 mEq/L and serum sodium concentration normalized over the next 24 hours. Her cardiovascular and neurological examinations worsened, however, and she required vasopressors. Her pupils remained fixed and dilated, and she lost her corneal and gag reflexes. A repeat CT of the brain showed persistent cerebral edema with signs of herniation, and she did not recover.
Dr Nguyen is a medical toxicology fellow in the department of emergency medicine at New York University Langone Medical Center. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Buttram VC Jr, Reiter RC. Uterine leiomyomata: etiology, symptomatology, and management. Fertil Steril. 1981;36(4):433-445.
- Horlander KT, Mannino DM, Leeper KV. Pulmonary embolism mortality in the United States, 1979-1998: an analysis using multiple-cause mortality data. Arch Intern Med. 2003;163(14):1711-1717.
- White RH, Zhou H, Romano PS. Incidence of symptomatic venous thromboembolism after different elective or urgent surgical procedures. Thromb Haemost. 2003;90(3):446-455.
- McConkey PP. Postobstructive Pulmonary Oedema—a case series and review. Anaest Intensive Care. 2000;28(1):72-76.
- Charney AN, Hoffman RS. Fluid, Electrolyte, and Acid-Base Principles. In: Nelson LS, Lewin NA, Howland MA, Hoffman RS, Goldfrank LR, Flomenbaum NE, eds. Goldfrank’s Toxicological Emergencies. 9th ed. New York, NY: McGraw Hill; 2010:249-264.
- Hahn RG. Fluid absorption in endoscopic surgery. Br J Anaesth. 2006;96(1):8-20.
- Nilsson A, Hahn RG. Mental status after transurethral resection of the prostate. Eur Urol. 1994;26(1):1-5.
- Hahn RG. Glycine 1.5% for irrigation should be abandoned. Urol Int. 2013;91(3):249-255.
- Phillips DR, Milim SJ, Nathanson HG, Phillips RE, Haselkorn JS. Preventing hyponatremic encephalopathy: comparison of serum sodium and osmolality during operative hysteroscopy with 5.0% mannitol and 1.5% glycine distention media. J Am Assoc Gynecol Laparosc. 1997;4(5):567-576.
- Ghanem AN, Ward JP. Osmotic and metabolic sequelae of volumetric overload in relation to the TUR syndrome. Br J Urol. 1990;66(1):71-78.
- American College of Obstetricians and Gynecologists. ACOG technology assessment in obstetrics and gynecology, number 4, August 2005: hysteroscopy. Obstet Gynecol. 2005;106(2):439-442.
Case
A 35-year-old woman underwent an elective hysteroscopic myomectomy to remove a symptomatic 2.7-cm uterine leiomyoma. The procedure was uncomplicated, and the patient awoke in the postanesthesia care unit (PACU) in good condition. Two hours later, however, she developed severe shortness of breath and required bilevel positive airway pressure ventilation. Her vital signs in the PACU were: blood pressure (BP), 110/70 mm Hg; heart rate, 90 beats/minute; respiratory rate, 12 breaths/minute; temperature, 98.4°F. Oxygen saturation was 94% on room air. She was diaphoretic and tachycardic on physical examination, but her pulmonary, abdominal, and gynecologic examinations were normal. During the examination, she complained of nausea, vomited, and then became increasingly lethargic and confused.
How can this patient’s clinical presentation be explained?
Uterine fibroids are the most common pelvic tumor in women.1 Hysteroscopic myomectomy is a minimally invasive surgical procedure commonly performed to resect submucosal fibroids. The procedure takes about 60 minutes, and is often performed on an outpatient basis under general anesthesia. During the procedure, an electrosurgery device called a resectoscope is inserted through the cervix. The uterine cavity is then distended with a large volume of irrigating solution. Maneuvering the resectoscope, the surgeon then shaves the protruding fibroid layer-by-layer until it aligns with the surrounding myometrium.
Surgical complications of hysteroscopic myomectomy may produce life-threatening effects. Excessive resection of the myometrium may increase blood loss, which can cause chest pain, shortness of breath, diaphoresis, lethargy, and confusion. Uterine perforation may produce hypotension, abdominal pain and distention, infection, and vaginal bleeding.
Venous Thromboembolism
Venous thromboembolism (VTE) is a common postoperative complication, with pulmonary embolism accounting for the most common preventable cause of hospital death in the United States.2 Gynecologic surgery, especially brief procedures, are associated with among the lowest rates of VTE, however, making this an unlikely explanation in this case.3 Additionally, VTE is not expected to produce the neurological findings observed in this patient.
Negative Pressure Pulmonary Edema
An uncommon but life-threatening complication for patients undergoing general anesthesia is negative pressure pulmonary edema, or “postextubation pulmonary edema,” which is estimated to occur in up to 1 in 1,000 procedures involving mechanical ventilation. During extubation, forced inspiration against a closed glottis causes intravascular fluid to be drawn into the interstitial space leading to pulmonary edema.4
Hyponatremia
An unusual but well described complication of endoscopic surgery is hyponatremia from systemic absorption of the irrigating fluid. Fluid overload may result in pulmonary edema, and dilutional hyponatremia may cause altered mental status or seizures.
Case Continuation
A chest X-ray performed after the patient became symptomatic revealed mild bilateral pulmonary edema. Her postoperative laboratory values were: sodium, 112 mEq/L; potassium, 3.3 mEq/L; chloride, 81 mEq/L; bicarbonate, 25 mEq/L; blood urea nitrogen, 18 mg/dL; creatinine, 0.6 mg/dL. Her ammonia level was 24 mmol/L (normal range, 11-35 mmol/L). An endotracheal tube was placed after her level of consciousness declined further. Her neurological examination revealed bilateral fixed and dilated pupils. An emergent computed tomography (CT) scan of the brain revealed severe generalized swelling of the brain.
What is the cause of this patient’s hyponatremia?
Monopolar electrosurgical devices such as the resectoscope cannot be used with electrolyte-containing irrigation fluids (eg, isotonic saline or lactated Ringer’s solution). Nonconductive, nonelectrolyte solutions such as glycine 1.5%, sorbitol 3%, or mannitol 5%, are the most common irrigating fluids employed to dilate the operating field and to wash away debris and blood.5
A dilutional hyponatremia can occur when the irrigating fluid is absorbed systemically. As it was first described following transurethral resection of the prostate procedures in the 1950s, the syndrome is referred to as “TURP” syndrome. Since then, several hundred life-threatening and even fatal cases of TURP syndrome have been reported.6 The syndrome occurs with other operations including transcervical resection of the endometrium (TCRE).5 The irrigating fluid is most frequently absorbed directly into the vascular system when a vein has been severed during the electrosurgery, particularly when the infusion pressure exceeds the venous pressure.6 Additionally, extravasation of the irrigating fluid into the intraperitoneal space can occur after instrument perforation of the uterine wall in TCRE, or via the fallopian tubes.6
What are the signs and symptoms of TURP syndrome?
Mild-to-moderate TURP syndrome occurs in 1% to 8% of TURP procedures performed. Fluid absorption is slightly more common during TCRE, and occurs more often during the resection of fibroids.6 The dilutional hyponatremia can result in brain edema, as well as pharmacological effects specific to the irrigant solutes.
Symptoms of TURP syndrome are primarily neurological, with nausea being the earliest sign of a mild syndrome. A “mini” mental-status test may show transient confusion with smaller absorption volumes.7 As the fluid absorption increases, the hyponatremia worsens, resulting in cerebral edema. This manifests as encephalopathy, which includes disorientation, twitching, and seizures. Hypotension is uncommon, since the fluid is being absorbed intravascularly.6 Shortness of breath, uneasiness, chest pain, and pulmonary edema may develop from systemic fluid overload. The intra-abdominal extravasation of fluid can result in abdominal pain. Other symptoms are specific to the irrigant.
Glycine
Glycine 1.5% is the most common irrigant solution used; as such, it produces the highest incidence of TURP syndrome.8 This solution is hypoosmotic (osmolality of 200 mosm/kg) compared with the normal serum (osmolality of 280 to 296 mosm/kg).5 In addition, glycine may cause visual disturbances.8 The metabolism of glycine produces ammonia, serine, and oxalate (Figure), and 10% of patients who absorb glycine show a marked hyperammonemia, further exacerbating the neurological effects.9,10
Sorbitol and mannitol
Sorbitol and mannitol irrigation fluids are used less frequently than glycine. Sorbitol 3% is metabolized to fructose and glucose, and has an osmolality of 165 mosm/kg.6 When absorbed systemically, sorbitol’s effects are similar to those of glycine, except that it does not induce visual symptoms. Mannitol 5% solution has the advantage of being isosmotic (275 mosm/kg). It is not metabolized and is excreted entirely in the urine. The excretion of mannitol creates an osmotic diuresis, thereby preventing hyponatremia from occurring.9Sorbitol and Mannitol
What are the treatment options for TURP Syndrome? Can it be prevented?
Patients with TURP syndrome in its mildest form can be asymptomatic, but severe cases can be life threatening or fatal. Unlike the treatment for hyponatremia caused by psychogenic polydipsia or the syndrome of inappropriate antidiuretic hormone, which calls for fluid restriction, plasma volume expansion is indicated in TURP syndrome, as hypovolemia and low-cardiac output develop as soon as irrigation is discontinued.
Hypertonic saline is indicated for neurological symptoms, or if the serum sodium concentration is <120mEq/L.6 Although furosemide has been used to treat acute pulmonary edema, no studies support its routine use in the treatment of fluid absorption,6 and its use may aggravate hyponatremia and hypovolemia.
Bipolar electrosurgical systems, unlike monopolar systems, permit the use of electrolyte solutions such as isotonic saline, thereby significantly reducing the risk of hyponatremia. For hysteroscopic procedures, the American College of Obstetricians and Gynecologists recommends the use of an automated fluid pump and monitoring system, thus minimizing the fluid pressure and halting or terminating the procedure before absorption thresholds are exceeded.11
Case Conclusion
The patient was immediately given a 1 mL/kg bolus of hypertonic saline. Two hours later, her serum sodium improved to 114 mEq/L and serum sodium concentration normalized over the next 24 hours. Her cardiovascular and neurological examinations worsened, however, and she required vasopressors. Her pupils remained fixed and dilated, and she lost her corneal and gag reflexes. A repeat CT of the brain showed persistent cerebral edema with signs of herniation, and she did not recover.
Dr Nguyen is a medical toxicology fellow in the department of emergency medicine at New York University Langone Medical Center. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
Case
A 35-year-old woman underwent an elective hysteroscopic myomectomy to remove a symptomatic 2.7-cm uterine leiomyoma. The procedure was uncomplicated, and the patient awoke in the postanesthesia care unit (PACU) in good condition. Two hours later, however, she developed severe shortness of breath and required bilevel positive airway pressure ventilation. Her vital signs in the PACU were: blood pressure (BP), 110/70 mm Hg; heart rate, 90 beats/minute; respiratory rate, 12 breaths/minute; temperature, 98.4°F. Oxygen saturation was 94% on room air. She was diaphoretic and tachycardic on physical examination, but her pulmonary, abdominal, and gynecologic examinations were normal. During the examination, she complained of nausea, vomited, and then became increasingly lethargic and confused.
How can this patient’s clinical presentation be explained?
Uterine fibroids are the most common pelvic tumor in women.1 Hysteroscopic myomectomy is a minimally invasive surgical procedure commonly performed to resect submucosal fibroids. The procedure takes about 60 minutes, and is often performed on an outpatient basis under general anesthesia. During the procedure, an electrosurgery device called a resectoscope is inserted through the cervix. The uterine cavity is then distended with a large volume of irrigating solution. Maneuvering the resectoscope, the surgeon then shaves the protruding fibroid layer-by-layer until it aligns with the surrounding myometrium.
Surgical complications of hysteroscopic myomectomy may produce life-threatening effects. Excessive resection of the myometrium may increase blood loss, which can cause chest pain, shortness of breath, diaphoresis, lethargy, and confusion. Uterine perforation may produce hypotension, abdominal pain and distention, infection, and vaginal bleeding.
Venous Thromboembolism
Venous thromboembolism (VTE) is a common postoperative complication, with pulmonary embolism accounting for the most common preventable cause of hospital death in the United States.2 Gynecologic surgery, especially brief procedures, are associated with among the lowest rates of VTE, however, making this an unlikely explanation in this case.3 Additionally, VTE is not expected to produce the neurological findings observed in this patient.
Negative Pressure Pulmonary Edema
An uncommon but life-threatening complication for patients undergoing general anesthesia is negative pressure pulmonary edema, or “postextubation pulmonary edema,” which is estimated to occur in up to 1 in 1,000 procedures involving mechanical ventilation. During extubation, forced inspiration against a closed glottis causes intravascular fluid to be drawn into the interstitial space leading to pulmonary edema.4
Hyponatremia
An unusual but well described complication of endoscopic surgery is hyponatremia from systemic absorption of the irrigating fluid. Fluid overload may result in pulmonary edema, and dilutional hyponatremia may cause altered mental status or seizures.
Case Continuation
A chest X-ray performed after the patient became symptomatic revealed mild bilateral pulmonary edema. Her postoperative laboratory values were: sodium, 112 mEq/L; potassium, 3.3 mEq/L; chloride, 81 mEq/L; bicarbonate, 25 mEq/L; blood urea nitrogen, 18 mg/dL; creatinine, 0.6 mg/dL. Her ammonia level was 24 mmol/L (normal range, 11-35 mmol/L). An endotracheal tube was placed after her level of consciousness declined further. Her neurological examination revealed bilateral fixed and dilated pupils. An emergent computed tomography (CT) scan of the brain revealed severe generalized swelling of the brain.
What is the cause of this patient’s hyponatremia?
Monopolar electrosurgical devices such as the resectoscope cannot be used with electrolyte-containing irrigation fluids (eg, isotonic saline or lactated Ringer’s solution). Nonconductive, nonelectrolyte solutions such as glycine 1.5%, sorbitol 3%, or mannitol 5%, are the most common irrigating fluids employed to dilate the operating field and to wash away debris and blood.5
A dilutional hyponatremia can occur when the irrigating fluid is absorbed systemically. As it was first described following transurethral resection of the prostate procedures in the 1950s, the syndrome is referred to as “TURP” syndrome. Since then, several hundred life-threatening and even fatal cases of TURP syndrome have been reported.6 The syndrome occurs with other operations including transcervical resection of the endometrium (TCRE).5 The irrigating fluid is most frequently absorbed directly into the vascular system when a vein has been severed during the electrosurgery, particularly when the infusion pressure exceeds the venous pressure.6 Additionally, extravasation of the irrigating fluid into the intraperitoneal space can occur after instrument perforation of the uterine wall in TCRE, or via the fallopian tubes.6
What are the signs and symptoms of TURP syndrome?
Mild-to-moderate TURP syndrome occurs in 1% to 8% of TURP procedures performed. Fluid absorption is slightly more common during TCRE, and occurs more often during the resection of fibroids.6 The dilutional hyponatremia can result in brain edema, as well as pharmacological effects specific to the irrigant solutes.
Symptoms of TURP syndrome are primarily neurological, with nausea being the earliest sign of a mild syndrome. A “mini” mental-status test may show transient confusion with smaller absorption volumes.7 As the fluid absorption increases, the hyponatremia worsens, resulting in cerebral edema. This manifests as encephalopathy, which includes disorientation, twitching, and seizures. Hypotension is uncommon, since the fluid is being absorbed intravascularly.6 Shortness of breath, uneasiness, chest pain, and pulmonary edema may develop from systemic fluid overload. The intra-abdominal extravasation of fluid can result in abdominal pain. Other symptoms are specific to the irrigant.
Glycine
Glycine 1.5% is the most common irrigant solution used; as such, it produces the highest incidence of TURP syndrome.8 This solution is hypoosmotic (osmolality of 200 mosm/kg) compared with the normal serum (osmolality of 280 to 296 mosm/kg).5 In addition, glycine may cause visual disturbances.8 The metabolism of glycine produces ammonia, serine, and oxalate (Figure), and 10% of patients who absorb glycine show a marked hyperammonemia, further exacerbating the neurological effects.9,10
Sorbitol and mannitol
Sorbitol and mannitol irrigation fluids are used less frequently than glycine. Sorbitol 3% is metabolized to fructose and glucose, and has an osmolality of 165 mosm/kg.6 When absorbed systemically, sorbitol’s effects are similar to those of glycine, except that it does not induce visual symptoms. Mannitol 5% solution has the advantage of being isosmotic (275 mosm/kg). It is not metabolized and is excreted entirely in the urine. The excretion of mannitol creates an osmotic diuresis, thereby preventing hyponatremia from occurring.9Sorbitol and Mannitol
What are the treatment options for TURP Syndrome? Can it be prevented?
Patients with TURP syndrome in its mildest form can be asymptomatic, but severe cases can be life threatening or fatal. Unlike the treatment for hyponatremia caused by psychogenic polydipsia or the syndrome of inappropriate antidiuretic hormone, which calls for fluid restriction, plasma volume expansion is indicated in TURP syndrome, as hypovolemia and low-cardiac output develop as soon as irrigation is discontinued.
Hypertonic saline is indicated for neurological symptoms, or if the serum sodium concentration is <120mEq/L.6 Although furosemide has been used to treat acute pulmonary edema, no studies support its routine use in the treatment of fluid absorption,6 and its use may aggravate hyponatremia and hypovolemia.
Bipolar electrosurgical systems, unlike monopolar systems, permit the use of electrolyte solutions such as isotonic saline, thereby significantly reducing the risk of hyponatremia. For hysteroscopic procedures, the American College of Obstetricians and Gynecologists recommends the use of an automated fluid pump and monitoring system, thus minimizing the fluid pressure and halting or terminating the procedure before absorption thresholds are exceeded.11
Case Conclusion
The patient was immediately given a 1 mL/kg bolus of hypertonic saline. Two hours later, her serum sodium improved to 114 mEq/L and serum sodium concentration normalized over the next 24 hours. Her cardiovascular and neurological examinations worsened, however, and she required vasopressors. Her pupils remained fixed and dilated, and she lost her corneal and gag reflexes. A repeat CT of the brain showed persistent cerebral edema with signs of herniation, and she did not recover.
Dr Nguyen is a medical toxicology fellow in the department of emergency medicine at New York University Langone Medical Center. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Buttram VC Jr, Reiter RC. Uterine leiomyomata: etiology, symptomatology, and management. Fertil Steril. 1981;36(4):433-445.
- Horlander KT, Mannino DM, Leeper KV. Pulmonary embolism mortality in the United States, 1979-1998: an analysis using multiple-cause mortality data. Arch Intern Med. 2003;163(14):1711-1717.
- White RH, Zhou H, Romano PS. Incidence of symptomatic venous thromboembolism after different elective or urgent surgical procedures. Thromb Haemost. 2003;90(3):446-455.
- McConkey PP. Postobstructive Pulmonary Oedema—a case series and review. Anaest Intensive Care. 2000;28(1):72-76.
- Charney AN, Hoffman RS. Fluid, Electrolyte, and Acid-Base Principles. In: Nelson LS, Lewin NA, Howland MA, Hoffman RS, Goldfrank LR, Flomenbaum NE, eds. Goldfrank’s Toxicological Emergencies. 9th ed. New York, NY: McGraw Hill; 2010:249-264.
- Hahn RG. Fluid absorption in endoscopic surgery. Br J Anaesth. 2006;96(1):8-20.
- Nilsson A, Hahn RG. Mental status after transurethral resection of the prostate. Eur Urol. 1994;26(1):1-5.
- Hahn RG. Glycine 1.5% for irrigation should be abandoned. Urol Int. 2013;91(3):249-255.
- Phillips DR, Milim SJ, Nathanson HG, Phillips RE, Haselkorn JS. Preventing hyponatremic encephalopathy: comparison of serum sodium and osmolality during operative hysteroscopy with 5.0% mannitol and 1.5% glycine distention media. J Am Assoc Gynecol Laparosc. 1997;4(5):567-576.
- Ghanem AN, Ward JP. Osmotic and metabolic sequelae of volumetric overload in relation to the TUR syndrome. Br J Urol. 1990;66(1):71-78.
- American College of Obstetricians and Gynecologists. ACOG technology assessment in obstetrics and gynecology, number 4, August 2005: hysteroscopy. Obstet Gynecol. 2005;106(2):439-442.
- Buttram VC Jr, Reiter RC. Uterine leiomyomata: etiology, symptomatology, and management. Fertil Steril. 1981;36(4):433-445.
- Horlander KT, Mannino DM, Leeper KV. Pulmonary embolism mortality in the United States, 1979-1998: an analysis using multiple-cause mortality data. Arch Intern Med. 2003;163(14):1711-1717.
- White RH, Zhou H, Romano PS. Incidence of symptomatic venous thromboembolism after different elective or urgent surgical procedures. Thromb Haemost. 2003;90(3):446-455.
- McConkey PP. Postobstructive Pulmonary Oedema—a case series and review. Anaest Intensive Care. 2000;28(1):72-76.
- Charney AN, Hoffman RS. Fluid, Electrolyte, and Acid-Base Principles. In: Nelson LS, Lewin NA, Howland MA, Hoffman RS, Goldfrank LR, Flomenbaum NE, eds. Goldfrank’s Toxicological Emergencies. 9th ed. New York, NY: McGraw Hill; 2010:249-264.
- Hahn RG. Fluid absorption in endoscopic surgery. Br J Anaesth. 2006;96(1):8-20.
- Nilsson A, Hahn RG. Mental status after transurethral resection of the prostate. Eur Urol. 1994;26(1):1-5.
- Hahn RG. Glycine 1.5% for irrigation should be abandoned. Urol Int. 2013;91(3):249-255.
- Phillips DR, Milim SJ, Nathanson HG, Phillips RE, Haselkorn JS. Preventing hyponatremic encephalopathy: comparison of serum sodium and osmolality during operative hysteroscopy with 5.0% mannitol and 1.5% glycine distention media. J Am Assoc Gynecol Laparosc. 1997;4(5):567-576.
- Ghanem AN, Ward JP. Osmotic and metabolic sequelae of volumetric overload in relation to the TUR syndrome. Br J Urol. 1990;66(1):71-78.
- American College of Obstetricians and Gynecologists. ACOG technology assessment in obstetrics and gynecology, number 4, August 2005: hysteroscopy. Obstet Gynecol. 2005;106(2):439-442.

Nausea, Vomiting, and Worsening Pain
ANSWER
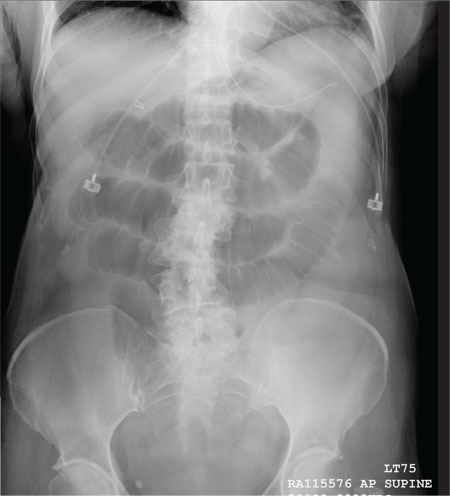
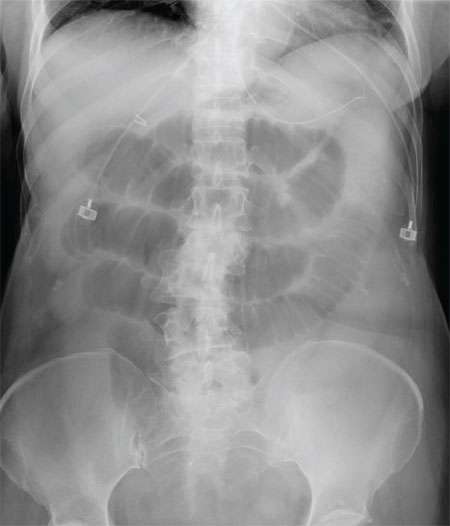
The radiograph shows multiple stacked dilated loops of small bowel. The colon does not appear distended. (A nasogastric tube is also present, and there are degenerative changes in the spine.) Such a finding is typically associated with at least a partial small bowel obstruction, since no definite air fluid levels are noted.
The patient was admitted and made npo. Nasogastric decompression was started, and general surgery consultation was obtained.
ANSWER
The radiograph shows multiple stacked dilated loops of small bowel. The colon does not appear distended. (A nasogastric tube is also present, and there are degenerative changes in the spine.) Such a finding is typically associated with at least a partial small bowel obstruction, since no definite air fluid levels are noted.
The patient was admitted and made npo. Nasogastric decompression was started, and general surgery consultation was obtained.
ANSWER
The radiograph shows multiple stacked dilated loops of small bowel. The colon does not appear distended. (A nasogastric tube is also present, and there are degenerative changes in the spine.) Such a finding is typically associated with at least a partial small bowel obstruction, since no definite air fluid levels are noted.
The patient was admitted and made npo. Nasogastric decompression was started, and general surgery consultation was obtained.
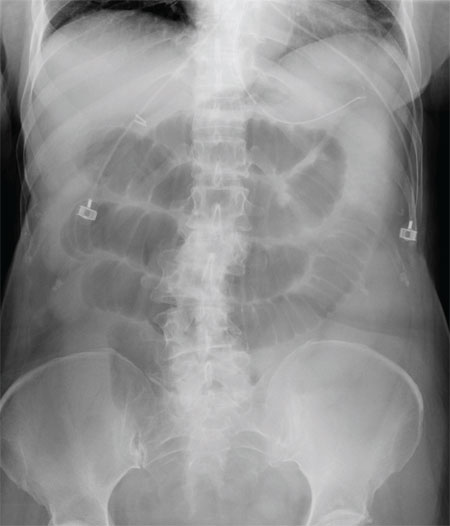
A 75-year-old woman presents to the emergency department with a three-day history of abdominal pain. She does not recall eating anything unusual. She reports having nausea and vomiting and states that her pain is progressively worsening. Her medical history is significant for hypertension. Surgical history is significant for previous cholecystectomy and total abdominal hysterectomy. She is afebrile, and her vital signs are within normal limits. Her abdomen is soft and diffusely tender, with slightly decreased bowel sounds. No rebound or guarding is present. The rest of her physical examination overall is within normal limits. During the exam, she experiences a couple episodes of bilious vomiting. You order some laboratory studies as well as an abdominal radiograph (shown). What is your impression?
'Allergic to the sun'
A 54-year-old white man presents to the emergency department with burning pain in his left upper arm for the past 2 to 3 days. His medical history includes seizure disorder, for which he takes levetiracetam (Keppra); hypertension, for which he takes metoprolol succinate (Toprol); and in the remote past, a gunshot wound to the head that left his right arm with residual contracture and weakness.
He says he is homeless, has been “allergic to the sun for a while,” and has had dark-colored urine and intermittent abdominal pain. He states that he does not use illicit substances but that he drinks 6 to 12 beers per night and smokes 1 pack of cigarettes per day.
Initial vital signs:
- Temperature 37.7°C (99.9°F)
- Blood pressure 217/114 mm Hg
- Heart rate 82 bpm
- Respiratory rate 18 per minute
- Capillary oxygen saturation 98% while breathing room air.
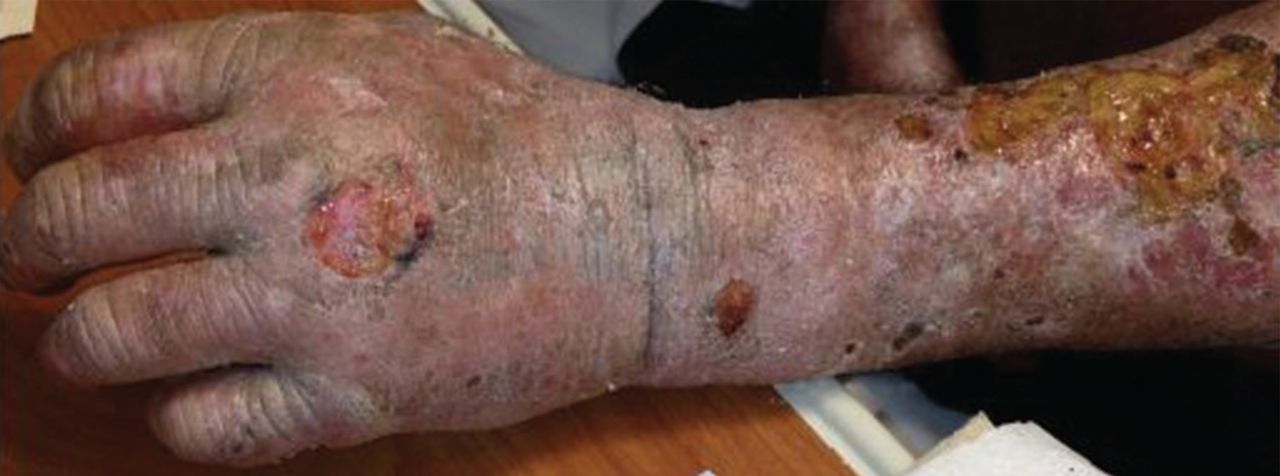
On examination, his right arm is significantly weak and contracted. His left arm has decreased sensation to pinprick and light touch from elbow to fingers. His face and both arms show hyperpigmentation alternating with atrophic scarring, which also affects his lips. There is no overt mucosal involvement. His hands and forearms have a sclerotic texture and patchy hair loss. Several small bullae are present on the dorsum of the left forearm and hand. There is a 6-inch, irregular, open lesion on the left forearm and a 1-inch lesion on the left hand (Figure 1).
Initial laboratory studies show:
- Chemistries and complete blood cell count within normal limits
- Platelet count 305 × 109/L (reference range 150–350)
- Orange-colored urine
- Hepatitis C virus (HCV) antibody positive (new finding)
- Human immunodeficiency virus antibody, hepatitis B surface antigen, and antinuclear antibody negative
- Phenytoin and urine drug screen negative
- Aspartate aminotransferase 70 U/L (reference range 5–34)
- Alanine aminotransferase 73 U/L (reference range 0–55)
- Prothrombin time 10.8 seconds (reference range 8.3–13.0), international normalized ratio 0.98 (reference range 0.8–1.2)
- Iron studies within normal limits.
The patient is admitted to the hospital and is started on cefazolin and clindamycin. Urine is collected for a porphyrin screen, and punch-biopsy samples from the forearms are sent for study. Ultrasonography shows splenomegaly, as well as increased echogenicity of the liver without structural abnormalities. Blood and urine cultures, drawn upon admission, are negative by discharge.
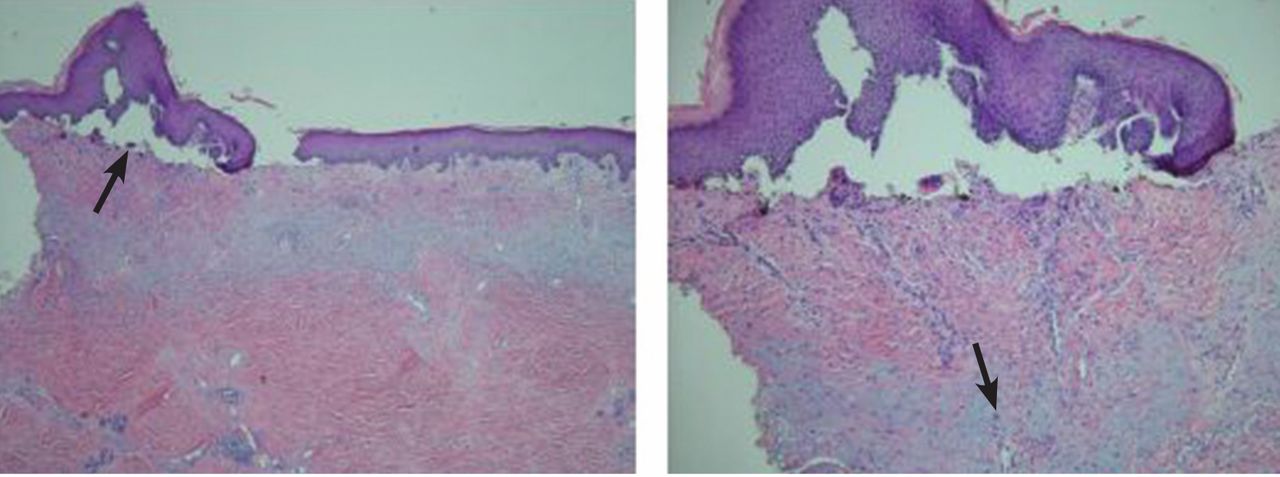
Pathologic study of the punch-biopsy specimens (Figure 2) shows the formation of subepidermal vesicles with extensive reticular and dermal fibrosis.
DIAGNOSIS: PORPHYRIA CUTANEA TARDA
Because of the patient’s history, examination, and pathology results, he was preliminarily diagnosed with porphyria cutanea tarda (PCT).1,2 The diagnosis was confirmed after he was discharged when his urine uroporphyrin level was found to be 157.5 μmol/mol of creatinine (reference range < 4) and his urine heptacarboxylporphyrin level was 118.0 μmol/mol of creatinine (reference range < 2).
This patient’s clinical presentation is classic for sporadic (ie, type 1) PCT. Sporadic PCT is an acquired deficiency of uroporphyrinogen decarboxylase, an enzyme that catalyzes the fifth step in heme metabolism.3 The deficiency of this enzyme is exclusively hepatic and is strongly associated with chronic hepatitis C infection. Mutations of the hemochromatosis gene (HFE), human immunodeficiency virus infection, alcohol use, and smoking are also risk factors.4 The prevalence in the United States is about 1:25,000; nearly 80% of cases are sporadic (type 1), and 20% are familial (type 2).5
Manifestations of PCT include photosensitive dermatitis, facial hypertrichosis, and orange urine.3 The photosensitivity dermatitis heals slowly and leads to sclerosis and hyperpigmentation.
Repeated phlebotomy is the first-line treatment, and hydroxychloroquine (Plaquenil) is the second-line treatment.6 Patients with PCT and hepatitis C should be considered for antiviral therapy according to standard guidelines. Treatment of hepatitis C may reduce the symptoms of PCT, even without a sustained viral response. However, not enough evidence exists to make treatment recommendations for this group.7
Because we were uncertain that the patient would return for follow-up, we did not start phlebotomy or treatment for hepatitis C. However, we did prescribe hydroxychloroquine 100 mg three times a week and instructed him to cover his skin when outside and to use effective sunblock. An outpatient visit was scheduled prior to discharge. Unfortunately, the patient was lost to follow-up.
Acknowledgment: The authors would like to personally thank Dr. Karen DeSouza from the University of Tennessee, Graduate School of Medicine, Department of Pathology, for her clinical expertise and kind advice.
- The University of Iowa, Department of Pathology, Laboratory Services Handbook. Porphyrins & Porphobilinogen, Urine (24 hr or random). www.healthcare.uiowa.edu/path_handbook/handbook/test2893.html. Accessed August 8, 2014.
- Maynard B, Peters MS. Histologic and immunofluorescence study of cutaneous porphyrias. J Cutan Pathol 1992; 19:40–47.
- Thunell S, Harper P. Porphyrins, porphyrin metabolism, porphyrias. III. Diagnosis, care and monitoring in porphyria cutanea tarda—suggestions for a handling programme. Scand J Clin Lab Invest 2000; 60:561–579.
- Lambrecht RW, Thapar M, Bonkovsky HL. Genetic aspects of porphyria cutanea tarda. Semin Liver Dis 2007; 27:99–108.
- Kushner JP, Barbuto AJ, Lee GR. An inherited enzymatic defect in porphyria cutanea tarda: decreased uroporphyrinogen decarboxylase activity. J Clin Invest 1976; 58:1089–1097.
- Singal AK, Kormos-Hallberg C, Lee C, et al. Low-dose hydroxychloroquine is as effective as phlebotomy in treatment of patients with porphyria cutanea tarda. Clin Gastroenterol Hepatol 2012; 10:1402–1409.
- Ryan Caballes F, Sendi H, Bonkovsky HL. Hepatitis C, porphyria cutanea tarda and liver iron: an update. Liver Int 2012; 32:880–893.
A 54-year-old white man presents to the emergency department with burning pain in his left upper arm for the past 2 to 3 days. His medical history includes seizure disorder, for which he takes levetiracetam (Keppra); hypertension, for which he takes metoprolol succinate (Toprol); and in the remote past, a gunshot wound to the head that left his right arm with residual contracture and weakness.
He says he is homeless, has been “allergic to the sun for a while,” and has had dark-colored urine and intermittent abdominal pain. He states that he does not use illicit substances but that he drinks 6 to 12 beers per night and smokes 1 pack of cigarettes per day.
Initial vital signs:
- Temperature 37.7°C (99.9°F)
- Blood pressure 217/114 mm Hg
- Heart rate 82 bpm
- Respiratory rate 18 per minute
- Capillary oxygen saturation 98% while breathing room air.
On examination, his right arm is significantly weak and contracted. His left arm has decreased sensation to pinprick and light touch from elbow to fingers. His face and both arms show hyperpigmentation alternating with atrophic scarring, which also affects his lips. There is no overt mucosal involvement. His hands and forearms have a sclerotic texture and patchy hair loss. Several small bullae are present on the dorsum of the left forearm and hand. There is a 6-inch, irregular, open lesion on the left forearm and a 1-inch lesion on the left hand (Figure 1).
Initial laboratory studies show:
- Chemistries and complete blood cell count within normal limits
- Platelet count 305 × 109/L (reference range 150–350)
- Orange-colored urine
- Hepatitis C virus (HCV) antibody positive (new finding)
- Human immunodeficiency virus antibody, hepatitis B surface antigen, and antinuclear antibody negative
- Phenytoin and urine drug screen negative
- Aspartate aminotransferase 70 U/L (reference range 5–34)
- Alanine aminotransferase 73 U/L (reference range 0–55)
- Prothrombin time 10.8 seconds (reference range 8.3–13.0), international normalized ratio 0.98 (reference range 0.8–1.2)
- Iron studies within normal limits.
The patient is admitted to the hospital and is started on cefazolin and clindamycin. Urine is collected for a porphyrin screen, and punch-biopsy samples from the forearms are sent for study. Ultrasonography shows splenomegaly, as well as increased echogenicity of the liver without structural abnormalities. Blood and urine cultures, drawn upon admission, are negative by discharge.
Pathologic study of the punch-biopsy specimens (Figure 2) shows the formation of subepidermal vesicles with extensive reticular and dermal fibrosis.
DIAGNOSIS: PORPHYRIA CUTANEA TARDA
Because of the patient’s history, examination, and pathology results, he was preliminarily diagnosed with porphyria cutanea tarda (PCT).1,2 The diagnosis was confirmed after he was discharged when his urine uroporphyrin level was found to be 157.5 μmol/mol of creatinine (reference range < 4) and his urine heptacarboxylporphyrin level was 118.0 μmol/mol of creatinine (reference range < 2).
This patient’s clinical presentation is classic for sporadic (ie, type 1) PCT. Sporadic PCT is an acquired deficiency of uroporphyrinogen decarboxylase, an enzyme that catalyzes the fifth step in heme metabolism.3 The deficiency of this enzyme is exclusively hepatic and is strongly associated with chronic hepatitis C infection. Mutations of the hemochromatosis gene (HFE), human immunodeficiency virus infection, alcohol use, and smoking are also risk factors.4 The prevalence in the United States is about 1:25,000; nearly 80% of cases are sporadic (type 1), and 20% are familial (type 2).5
Manifestations of PCT include photosensitive dermatitis, facial hypertrichosis, and orange urine.3 The photosensitivity dermatitis heals slowly and leads to sclerosis and hyperpigmentation.
Repeated phlebotomy is the first-line treatment, and hydroxychloroquine (Plaquenil) is the second-line treatment.6 Patients with PCT and hepatitis C should be considered for antiviral therapy according to standard guidelines. Treatment of hepatitis C may reduce the symptoms of PCT, even without a sustained viral response. However, not enough evidence exists to make treatment recommendations for this group.7
Because we were uncertain that the patient would return for follow-up, we did not start phlebotomy or treatment for hepatitis C. However, we did prescribe hydroxychloroquine 100 mg three times a week and instructed him to cover his skin when outside and to use effective sunblock. An outpatient visit was scheduled prior to discharge. Unfortunately, the patient was lost to follow-up.
Acknowledgment: The authors would like to personally thank Dr. Karen DeSouza from the University of Tennessee, Graduate School of Medicine, Department of Pathology, for her clinical expertise and kind advice.
A 54-year-old white man presents to the emergency department with burning pain in his left upper arm for the past 2 to 3 days. His medical history includes seizure disorder, for which he takes levetiracetam (Keppra); hypertension, for which he takes metoprolol succinate (Toprol); and in the remote past, a gunshot wound to the head that left his right arm with residual contracture and weakness.
He says he is homeless, has been “allergic to the sun for a while,” and has had dark-colored urine and intermittent abdominal pain. He states that he does not use illicit substances but that he drinks 6 to 12 beers per night and smokes 1 pack of cigarettes per day.
Initial vital signs:
- Temperature 37.7°C (99.9°F)
- Blood pressure 217/114 mm Hg
- Heart rate 82 bpm
- Respiratory rate 18 per minute
- Capillary oxygen saturation 98% while breathing room air.
On examination, his right arm is significantly weak and contracted. His left arm has decreased sensation to pinprick and light touch from elbow to fingers. His face and both arms show hyperpigmentation alternating with atrophic scarring, which also affects his lips. There is no overt mucosal involvement. His hands and forearms have a sclerotic texture and patchy hair loss. Several small bullae are present on the dorsum of the left forearm and hand. There is a 6-inch, irregular, open lesion on the left forearm and a 1-inch lesion on the left hand (Figure 1).
Initial laboratory studies show:
- Chemistries and complete blood cell count within normal limits
- Platelet count 305 × 109/L (reference range 150–350)
- Orange-colored urine
- Hepatitis C virus (HCV) antibody positive (new finding)
- Human immunodeficiency virus antibody, hepatitis B surface antigen, and antinuclear antibody negative
- Phenytoin and urine drug screen negative
- Aspartate aminotransferase 70 U/L (reference range 5–34)
- Alanine aminotransferase 73 U/L (reference range 0–55)
- Prothrombin time 10.8 seconds (reference range 8.3–13.0), international normalized ratio 0.98 (reference range 0.8–1.2)
- Iron studies within normal limits.
The patient is admitted to the hospital and is started on cefazolin and clindamycin. Urine is collected for a porphyrin screen, and punch-biopsy samples from the forearms are sent for study. Ultrasonography shows splenomegaly, as well as increased echogenicity of the liver without structural abnormalities. Blood and urine cultures, drawn upon admission, are negative by discharge.
Pathologic study of the punch-biopsy specimens (Figure 2) shows the formation of subepidermal vesicles with extensive reticular and dermal fibrosis.
DIAGNOSIS: PORPHYRIA CUTANEA TARDA
Because of the patient’s history, examination, and pathology results, he was preliminarily diagnosed with porphyria cutanea tarda (PCT).1,2 The diagnosis was confirmed after he was discharged when his urine uroporphyrin level was found to be 157.5 μmol/mol of creatinine (reference range < 4) and his urine heptacarboxylporphyrin level was 118.0 μmol/mol of creatinine (reference range < 2).
This patient’s clinical presentation is classic for sporadic (ie, type 1) PCT. Sporadic PCT is an acquired deficiency of uroporphyrinogen decarboxylase, an enzyme that catalyzes the fifth step in heme metabolism.3 The deficiency of this enzyme is exclusively hepatic and is strongly associated with chronic hepatitis C infection. Mutations of the hemochromatosis gene (HFE), human immunodeficiency virus infection, alcohol use, and smoking are also risk factors.4 The prevalence in the United States is about 1:25,000; nearly 80% of cases are sporadic (type 1), and 20% are familial (type 2).5
Manifestations of PCT include photosensitive dermatitis, facial hypertrichosis, and orange urine.3 The photosensitivity dermatitis heals slowly and leads to sclerosis and hyperpigmentation.
Repeated phlebotomy is the first-line treatment, and hydroxychloroquine (Plaquenil) is the second-line treatment.6 Patients with PCT and hepatitis C should be considered for antiviral therapy according to standard guidelines. Treatment of hepatitis C may reduce the symptoms of PCT, even without a sustained viral response. However, not enough evidence exists to make treatment recommendations for this group.7
Because we were uncertain that the patient would return for follow-up, we did not start phlebotomy or treatment for hepatitis C. However, we did prescribe hydroxychloroquine 100 mg three times a week and instructed him to cover his skin when outside and to use effective sunblock. An outpatient visit was scheduled prior to discharge. Unfortunately, the patient was lost to follow-up.
Acknowledgment: The authors would like to personally thank Dr. Karen DeSouza from the University of Tennessee, Graduate School of Medicine, Department of Pathology, for her clinical expertise and kind advice.
- The University of Iowa, Department of Pathology, Laboratory Services Handbook. Porphyrins & Porphobilinogen, Urine (24 hr or random). www.healthcare.uiowa.edu/path_handbook/handbook/test2893.html. Accessed August 8, 2014.
- Maynard B, Peters MS. Histologic and immunofluorescence study of cutaneous porphyrias. J Cutan Pathol 1992; 19:40–47.
- Thunell S, Harper P. Porphyrins, porphyrin metabolism, porphyrias. III. Diagnosis, care and monitoring in porphyria cutanea tarda—suggestions for a handling programme. Scand J Clin Lab Invest 2000; 60:561–579.
- Lambrecht RW, Thapar M, Bonkovsky HL. Genetic aspects of porphyria cutanea tarda. Semin Liver Dis 2007; 27:99–108.
- Kushner JP, Barbuto AJ, Lee GR. An inherited enzymatic defect in porphyria cutanea tarda: decreased uroporphyrinogen decarboxylase activity. J Clin Invest 1976; 58:1089–1097.
- Singal AK, Kormos-Hallberg C, Lee C, et al. Low-dose hydroxychloroquine is as effective as phlebotomy in treatment of patients with porphyria cutanea tarda. Clin Gastroenterol Hepatol 2012; 10:1402–1409.
- Ryan Caballes F, Sendi H, Bonkovsky HL. Hepatitis C, porphyria cutanea tarda and liver iron: an update. Liver Int 2012; 32:880–893.
- The University of Iowa, Department of Pathology, Laboratory Services Handbook. Porphyrins & Porphobilinogen, Urine (24 hr or random). www.healthcare.uiowa.edu/path_handbook/handbook/test2893.html. Accessed August 8, 2014.
- Maynard B, Peters MS. Histologic and immunofluorescence study of cutaneous porphyrias. J Cutan Pathol 1992; 19:40–47.
- Thunell S, Harper P. Porphyrins, porphyrin metabolism, porphyrias. III. Diagnosis, care and monitoring in porphyria cutanea tarda—suggestions for a handling programme. Scand J Clin Lab Invest 2000; 60:561–579.
- Lambrecht RW, Thapar M, Bonkovsky HL. Genetic aspects of porphyria cutanea tarda. Semin Liver Dis 2007; 27:99–108.
- Kushner JP, Barbuto AJ, Lee GR. An inherited enzymatic defect in porphyria cutanea tarda: decreased uroporphyrinogen decarboxylase activity. J Clin Invest 1976; 58:1089–1097.
- Singal AK, Kormos-Hallberg C, Lee C, et al. Low-dose hydroxychloroquine is as effective as phlebotomy in treatment of patients with porphyria cutanea tarda. Clin Gastroenterol Hepatol 2012; 10:1402–1409.
- Ryan Caballes F, Sendi H, Bonkovsky HL. Hepatitis C, porphyria cutanea tarda and liver iron: an update. Liver Int 2012; 32:880–893.
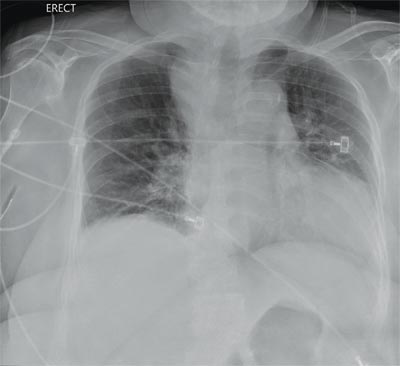
“Something Abnormal” on a Chest X-ray
ANSWER
The radiograph demonstrates a fairly large (4 x 6 cm) right paratracheal mass of unclear etiology. This type of finding warrants further evaluation with contrasted CT.
Fortunately for this patient, a subsequent study demonstrated a slightly enlarged thyroid gland. This correlated with the radiographic
finding.

ANSWER
The radiograph demonstrates a fairly large (4 x 6 cm) right paratracheal mass of unclear etiology. This type of finding warrants further evaluation with contrasted CT.
Fortunately for this patient, a subsequent study demonstrated a slightly enlarged thyroid gland. This correlated with the radiographic
finding.
ANSWER
The radiograph demonstrates a fairly large (4 x 6 cm) right paratracheal mass of unclear etiology. This type of finding warrants further evaluation with contrasted CT.
Fortunately for this patient, a subsequent study demonstrated a slightly enlarged thyroid gland. This correlated with the radiographic
finding.
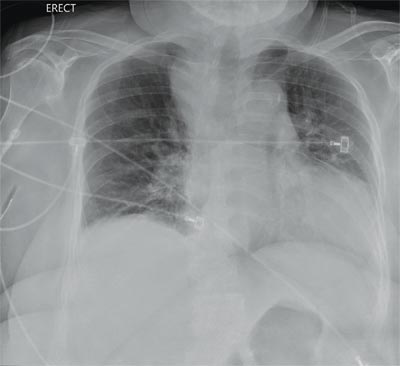
You are doing preoperative orders on a patient scheduled for surgery tomorrow morning. The patient is a 75-year-old woman who was admitted with an acute left subdural hematoma after sustaining a ground-level fall. Her medical history is significant for hypertension and diabetes. Social history is unremarkable. She is neurologically intact except for occasional confusion and aphasia. She moves all her extremities well. As you review her lab results, one of the nurses mentions that the radiology department called about “something abnormal” on the patient’s chest radiograph. You pull up the patient’s portable chest radiograph on the computer to review. What is your impression?
Case Studies in Toxicology: Hot as a Hare and Red as a Beet
A previously healthy 11-month-old boy was brought to the ED after his parents discovered him with an open bottle of nonprescription diphenhydramine. On initial presentation, the child was irritable with diffuse skin redness and dry mucous membranes. He was tremulous and making nonpurposeful reaching movements with his arms. He had roving eye movements and markedly dilated pupils that were minimally reactive. Initial vital signs were: blood pressure, 140/95 mm Hg; heart rate, 220 beats/minute; respiratory rate, 30 breaths/minute; temperature, 100.6ºF. Capillary glucose was 120 mg/dL, and oxygen saturation was 100% on room air. An electrocardiogram (ECG) revealed sinus tachycardia with normal QRS and QTc intervals.
What is the toxicological differential diagnosis?
Toxicity from several different classes of drugs may cause an altered level of consciousness, tachycardia, and hyperthermia. Serotonin agonists, such as selective serotonin reuptake inhibitors, may result in serotonin toxicity—a syndrome that includes altered cognition, autonomic changes (eg, tachycardia, hyperthermia), and neuromuscular effects (eg, rigidity, clonus), along with mydriasis and diaphoresis. Neuroleptic malignant syndrome (NMS) occurs following exposure to dopamine antagonists, such as antipsychotic medications.
Neuroleptic malignant syndrome presents in a similar manner to serotonin toxicity but tends to have a more indolent course compared with the abrupt onset and resolution of serotonin toxicity. Sympathomimetic medications (eg, methylphenidate) or drugs of abuse (eg, cocaine, methamphetamines) result in catecholamine effects including tachycardia, hypertension, diaphoresis, and mydriasis. Acetylsalicylic-acid (aspirin) toxicity (salicylism) often causes tinnitus, hyperpnea, and gastrointestinal (GI) effects following exposure. Severe toxicity may cause altered level of consciousness and hyperthermia; however, these are ominous and late findings. Mydriasis is not common.
What is the anticholinergic toxidrome?
Acetylcholine is a neurotransmitter present both in the central and peripheral nervous systems. In the periphery, acetylcholine acts at both the sympathetic and parasympathetic components of the autonomic nervous system and at somatic motor fibers. Acetylcholine acts at two classes of receptors, namely, nicotinic and muscarinic types. Muscarinic receptors are found in the central nervous system (CNS) (specifically the brain) and peripherally on effector cells of the parasympathetic nervous system and on sympathetically innervated sweat glands.1 Anticholinergic toxicity results from antagonism of muscarinic receptors and is more appropriately referred to as antimuscarinic poisoning, though the terms are used interchangeably. Nicotinic receptor antagonists are used primarily for neuromuscular blockade and do not cause this syndrome.
- “Hot as a hare” (anhidrosis with temperature elevation);
- “Red as a beet” (vasodilation with skin hyperemia);
- “Blind as a bat” (pupillary dilation with loss of accommodation);
- “Dry as a bone” (drying of mucosal surfaces and skin);
- “Full as a flask” (urinary retention); “Stuffed as a pepper” (constipation); and
- “Mad as a hatter” (describing the central anticholinergic effects that are often present—eg, altered mental status manifested as agitation, delirium, hallucinations, abnormal picking movements, rarely seizures).
Elderly patients and those with underlying medical illness or psychiatric disorders may be more prone to the CNS manifestations of anticholinergic medications. Anticholinergic effects can occur through ingestion, smoking, inhalation, and topical absorption (including transdermal or ophthalmic routes). Delayed or prolonged effects may occur due to slow gastric emptying and prolonged GI absorption. The duration of effects is variable and central anticholinergic manifestations of confusion or agitation may be present for several days, even after peripheral manifestations have resolved (termed the central anticholinergic syndrome).
What are common causes of anticholinergic toxicity?
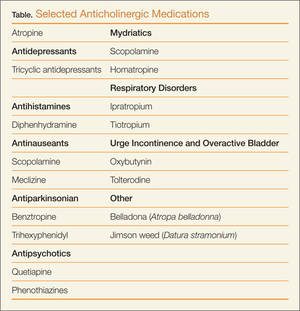
Although anticholinergic effects are often described in terms of “toxicity,” these effects are often used for therapeutic benefit. Such roles of anticholinergic agents include the following:
- Atropine to treat bradycardia;
- Ipratropium bromide to manage asthma;
- Antinauseants (eg, scopolamine, meclizine) for symptom relief;
- Tolterodine to treat urge incontinence and overactive bladder; and
- Ophthalmologic medications (eg, scopolamine, homatropine) to inhibit ciliary spasm in patients with iritis.
Although the above medications are being used for a specific anticholinergic property, other unintended and troublesome anticholinergic effects are often seen. Similarly, many other medications often have unintended anticholinergic effects (see Table). Anticholinergic “toxicity” is simply an extension of the effects that occur with therapeutic use.
What is the treatment for patients with anticholinergic toxicity?
Most patients with anticholinergic toxicity do well with supportive management. Benzodiazepines are the treatment of choice for agitation. Haloperidol and other antipsychotics are relatively contraindicated for treatment of agitation as they may impair temperature regulation and lead to hyperthermia. Although likely of limited overall benefit, oral activated charcoal may reduce the amount of drug absorbed.
Antidotal therapy with physostigmine should be considered for select patients presenting with altered mental status due to an anticholinergic. Physostigmine is an acetylcholinesterase inhibitor that prevents the breakdown of acetylcholine in the synaptic cleft, thus antagonizing the effects of anticholinergic drugs. A retrospective study noted a lower incidence of complications and shorter time to recovery with the use of physostigmine compared with benzodiazepines in patients with anticholinergic toxicity.2 The use of physostigmine in select patients may obviate the need for a further delirium workup, which often includes computed tomography or lumbar puncture.
When administering physostigmine, atropine should be present at the bedside with airway equipment readily available as cholinergic effects may develop (specifically bronchospasm, bronchorrhea, or bradycardia). Dosing of physostigmine in adult patients is 1 to 2 mg via slow intravenous (IV) push, in aliquots of 0.2 to 0.3 mg each, over 5 minutes; pediatric dosing is 20 mcg/kg to maximum 0.5 mg. Onset of effects can be expected within minutes of administration.3 Since the duration of physostigmine is less than that of many anticholinergic drugs, recurrence of anticholinergic effects should be anticipated.
Historically, physostigmine was included in the “coma cocktail,” along with thiamine, dextrose, and naloxone for treating undifferentiated patients with altered level of consciousness. Concern for its ubiquitous use arose following reports of asystole in two patients who presented with tricyclic antidepressant (TCA) overdose, although these patients actually had more complicated multidrug overdoses.4 Nevertheless, an ECG should be performed in all patients for whom physostigmine is being considered, and it should not be administered (or perhaps only extremely cautiously) if the ECG demonstrates a QRS complex duration >100 ms.3 Relative contraindications include reactive airways disease, peripheral vascular disease, or intestinal or bladder-outlet obstruction.
Prolongation of the QRS interval is not always indicative of TCA ingestion as certain other antimuscarinic drugs, such as diphenhydramine, may cause sodium-channel blockade. Based on extrapolation from TCA literature,5 if the QRS >100 ms, a bolus of 1 to 2 mEq/kg sodium bicarbonate should be given with monitoring of the QRS interval for narrowing.
Case conclusion
The clinicians at the bedside felt that the infant’s presentation was consistent with anticholinergic toxicity. Physostigmine was administered by slow IV push for a total dose of 1.5 mg. The patient had immediate improvement of symptoms, including decreased skin redness, decreased agitation, and improved vital signs (BP, 118/80 mm Hg and HR, 160 beats/minute). He was admitted to the pediatric intensive care unit for monitoring and was subsequently discharged home with complete symptom resolution 2 days later.
- Gerretsen P, Pollock BG. Drugs with anticholinergic properties: a current perspective on use and safety. Expert Opin Drug Saf. 2011;10(5):751-765.
- Burns MJ, Linden CH, Graudins A, Brown RM, Fletcher KE. A comparison of physostigmine and benzodiazepines for the treatment of anticholinergic poisoning. Ann Emerg Med. 2000;35(4):374-381.
- Howland MA. Physostigmine salicylate. In: Nelson LS, Lewin NA, Howland MA, Hoffman RS, Goldfrank LR, Flomenbaum NE, eds. Goldfrank’s Toxicologic Emergencies. 9th ed. New York, NY: McGraw Hill; 2011:759-762.
- Pentel P, Peterson CD. Asystole complicating physostigmine treatment of tricyclic antidepressant overdose. Ann Emerg Med. 1980;9(11):588-590.
- Boehnert MT, Lovejoy FH, Jr. Value of the QRS duration versus the serum drug level in predicting seizures and ventricular arrhythmias after an acute overdose of tricyclic antidepressants. N Engl J Med. 1985;313(8):474-479.
A previously healthy 11-month-old boy was brought to the ED after his parents discovered him with an open bottle of nonprescription diphenhydramine. On initial presentation, the child was irritable with diffuse skin redness and dry mucous membranes. He was tremulous and making nonpurposeful reaching movements with his arms. He had roving eye movements and markedly dilated pupils that were minimally reactive. Initial vital signs were: blood pressure, 140/95 mm Hg; heart rate, 220 beats/minute; respiratory rate, 30 breaths/minute; temperature, 100.6ºF. Capillary glucose was 120 mg/dL, and oxygen saturation was 100% on room air. An electrocardiogram (ECG) revealed sinus tachycardia with normal QRS and QTc intervals.
What is the toxicological differential diagnosis?
Toxicity from several different classes of drugs may cause an altered level of consciousness, tachycardia, and hyperthermia. Serotonin agonists, such as selective serotonin reuptake inhibitors, may result in serotonin toxicity—a syndrome that includes altered cognition, autonomic changes (eg, tachycardia, hyperthermia), and neuromuscular effects (eg, rigidity, clonus), along with mydriasis and diaphoresis. Neuroleptic malignant syndrome (NMS) occurs following exposure to dopamine antagonists, such as antipsychotic medications.
Neuroleptic malignant syndrome presents in a similar manner to serotonin toxicity but tends to have a more indolent course compared with the abrupt onset and resolution of serotonin toxicity. Sympathomimetic medications (eg, methylphenidate) or drugs of abuse (eg, cocaine, methamphetamines) result in catecholamine effects including tachycardia, hypertension, diaphoresis, and mydriasis. Acetylsalicylic-acid (aspirin) toxicity (salicylism) often causes tinnitus, hyperpnea, and gastrointestinal (GI) effects following exposure. Severe toxicity may cause altered level of consciousness and hyperthermia; however, these are ominous and late findings. Mydriasis is not common.
What is the anticholinergic toxidrome?
Acetylcholine is a neurotransmitter present both in the central and peripheral nervous systems. In the periphery, acetylcholine acts at both the sympathetic and parasympathetic components of the autonomic nervous system and at somatic motor fibers. Acetylcholine acts at two classes of receptors, namely, nicotinic and muscarinic types. Muscarinic receptors are found in the central nervous system (CNS) (specifically the brain) and peripherally on effector cells of the parasympathetic nervous system and on sympathetically innervated sweat glands.1 Anticholinergic toxicity results from antagonism of muscarinic receptors and is more appropriately referred to as antimuscarinic poisoning, though the terms are used interchangeably. Nicotinic receptor antagonists are used primarily for neuromuscular blockade and do not cause this syndrome.
- “Hot as a hare” (anhidrosis with temperature elevation);
- “Red as a beet” (vasodilation with skin hyperemia);
- “Blind as a bat” (pupillary dilation with loss of accommodation);
- “Dry as a bone” (drying of mucosal surfaces and skin);
- “Full as a flask” (urinary retention); “Stuffed as a pepper” (constipation); and
- “Mad as a hatter” (describing the central anticholinergic effects that are often present—eg, altered mental status manifested as agitation, delirium, hallucinations, abnormal picking movements, rarely seizures).
Elderly patients and those with underlying medical illness or psychiatric disorders may be more prone to the CNS manifestations of anticholinergic medications. Anticholinergic effects can occur through ingestion, smoking, inhalation, and topical absorption (including transdermal or ophthalmic routes). Delayed or prolonged effects may occur due to slow gastric emptying and prolonged GI absorption. The duration of effects is variable and central anticholinergic manifestations of confusion or agitation may be present for several days, even after peripheral manifestations have resolved (termed the central anticholinergic syndrome).
What are common causes of anticholinergic toxicity?
Although anticholinergic effects are often described in terms of “toxicity,” these effects are often used for therapeutic benefit. Such roles of anticholinergic agents include the following:
- Atropine to treat bradycardia;
- Ipratropium bromide to manage asthma;
- Antinauseants (eg, scopolamine, meclizine) for symptom relief;
- Tolterodine to treat urge incontinence and overactive bladder; and
- Ophthalmologic medications (eg, scopolamine, homatropine) to inhibit ciliary spasm in patients with iritis.
Although the above medications are being used for a specific anticholinergic property, other unintended and troublesome anticholinergic effects are often seen. Similarly, many other medications often have unintended anticholinergic effects (see Table). Anticholinergic “toxicity” is simply an extension of the effects that occur with therapeutic use.
What is the treatment for patients with anticholinergic toxicity?
Most patients with anticholinergic toxicity do well with supportive management. Benzodiazepines are the treatment of choice for agitation. Haloperidol and other antipsychotics are relatively contraindicated for treatment of agitation as they may impair temperature regulation and lead to hyperthermia. Although likely of limited overall benefit, oral activated charcoal may reduce the amount of drug absorbed.
Antidotal therapy with physostigmine should be considered for select patients presenting with altered mental status due to an anticholinergic. Physostigmine is an acetylcholinesterase inhibitor that prevents the breakdown of acetylcholine in the synaptic cleft, thus antagonizing the effects of anticholinergic drugs. A retrospective study noted a lower incidence of complications and shorter time to recovery with the use of physostigmine compared with benzodiazepines in patients with anticholinergic toxicity.2 The use of physostigmine in select patients may obviate the need for a further delirium workup, which often includes computed tomography or lumbar puncture.
When administering physostigmine, atropine should be present at the bedside with airway equipment readily available as cholinergic effects may develop (specifically bronchospasm, bronchorrhea, or bradycardia). Dosing of physostigmine in adult patients is 1 to 2 mg via slow intravenous (IV) push, in aliquots of 0.2 to 0.3 mg each, over 5 minutes; pediatric dosing is 20 mcg/kg to maximum 0.5 mg. Onset of effects can be expected within minutes of administration.3 Since the duration of physostigmine is less than that of many anticholinergic drugs, recurrence of anticholinergic effects should be anticipated.
Historically, physostigmine was included in the “coma cocktail,” along with thiamine, dextrose, and naloxone for treating undifferentiated patients with altered level of consciousness. Concern for its ubiquitous use arose following reports of asystole in two patients who presented with tricyclic antidepressant (TCA) overdose, although these patients actually had more complicated multidrug overdoses.4 Nevertheless, an ECG should be performed in all patients for whom physostigmine is being considered, and it should not be administered (or perhaps only extremely cautiously) if the ECG demonstrates a QRS complex duration >100 ms.3 Relative contraindications include reactive airways disease, peripheral vascular disease, or intestinal or bladder-outlet obstruction.
Prolongation of the QRS interval is not always indicative of TCA ingestion as certain other antimuscarinic drugs, such as diphenhydramine, may cause sodium-channel blockade. Based on extrapolation from TCA literature,5 if the QRS >100 ms, a bolus of 1 to 2 mEq/kg sodium bicarbonate should be given with monitoring of the QRS interval for narrowing.
Case conclusion
The clinicians at the bedside felt that the infant’s presentation was consistent with anticholinergic toxicity. Physostigmine was administered by slow IV push for a total dose of 1.5 mg. The patient had immediate improvement of symptoms, including decreased skin redness, decreased agitation, and improved vital signs (BP, 118/80 mm Hg and HR, 160 beats/minute). He was admitted to the pediatric intensive care unit for monitoring and was subsequently discharged home with complete symptom resolution 2 days later.
A previously healthy 11-month-old boy was brought to the ED after his parents discovered him with an open bottle of nonprescription diphenhydramine. On initial presentation, the child was irritable with diffuse skin redness and dry mucous membranes. He was tremulous and making nonpurposeful reaching movements with his arms. He had roving eye movements and markedly dilated pupils that were minimally reactive. Initial vital signs were: blood pressure, 140/95 mm Hg; heart rate, 220 beats/minute; respiratory rate, 30 breaths/minute; temperature, 100.6ºF. Capillary glucose was 120 mg/dL, and oxygen saturation was 100% on room air. An electrocardiogram (ECG) revealed sinus tachycardia with normal QRS and QTc intervals.
What is the toxicological differential diagnosis?
Toxicity from several different classes of drugs may cause an altered level of consciousness, tachycardia, and hyperthermia. Serotonin agonists, such as selective serotonin reuptake inhibitors, may result in serotonin toxicity—a syndrome that includes altered cognition, autonomic changes (eg, tachycardia, hyperthermia), and neuromuscular effects (eg, rigidity, clonus), along with mydriasis and diaphoresis. Neuroleptic malignant syndrome (NMS) occurs following exposure to dopamine antagonists, such as antipsychotic medications.
Neuroleptic malignant syndrome presents in a similar manner to serotonin toxicity but tends to have a more indolent course compared with the abrupt onset and resolution of serotonin toxicity. Sympathomimetic medications (eg, methylphenidate) or drugs of abuse (eg, cocaine, methamphetamines) result in catecholamine effects including tachycardia, hypertension, diaphoresis, and mydriasis. Acetylsalicylic-acid (aspirin) toxicity (salicylism) often causes tinnitus, hyperpnea, and gastrointestinal (GI) effects following exposure. Severe toxicity may cause altered level of consciousness and hyperthermia; however, these are ominous and late findings. Mydriasis is not common.
What is the anticholinergic toxidrome?
Acetylcholine is a neurotransmitter present both in the central and peripheral nervous systems. In the periphery, acetylcholine acts at both the sympathetic and parasympathetic components of the autonomic nervous system and at somatic motor fibers. Acetylcholine acts at two classes of receptors, namely, nicotinic and muscarinic types. Muscarinic receptors are found in the central nervous system (CNS) (specifically the brain) and peripherally on effector cells of the parasympathetic nervous system and on sympathetically innervated sweat glands.1 Anticholinergic toxicity results from antagonism of muscarinic receptors and is more appropriately referred to as antimuscarinic poisoning, though the terms are used interchangeably. Nicotinic receptor antagonists are used primarily for neuromuscular blockade and do not cause this syndrome.
- “Hot as a hare” (anhidrosis with temperature elevation);
- “Red as a beet” (vasodilation with skin hyperemia);
- “Blind as a bat” (pupillary dilation with loss of accommodation);
- “Dry as a bone” (drying of mucosal surfaces and skin);
- “Full as a flask” (urinary retention); “Stuffed as a pepper” (constipation); and
- “Mad as a hatter” (describing the central anticholinergic effects that are often present—eg, altered mental status manifested as agitation, delirium, hallucinations, abnormal picking movements, rarely seizures).
Elderly patients and those with underlying medical illness or psychiatric disorders may be more prone to the CNS manifestations of anticholinergic medications. Anticholinergic effects can occur through ingestion, smoking, inhalation, and topical absorption (including transdermal or ophthalmic routes). Delayed or prolonged effects may occur due to slow gastric emptying and prolonged GI absorption. The duration of effects is variable and central anticholinergic manifestations of confusion or agitation may be present for several days, even after peripheral manifestations have resolved (termed the central anticholinergic syndrome).
What are common causes of anticholinergic toxicity?
Although anticholinergic effects are often described in terms of “toxicity,” these effects are often used for therapeutic benefit. Such roles of anticholinergic agents include the following:
- Atropine to treat bradycardia;
- Ipratropium bromide to manage asthma;
- Antinauseants (eg, scopolamine, meclizine) for symptom relief;
- Tolterodine to treat urge incontinence and overactive bladder; and
- Ophthalmologic medications (eg, scopolamine, homatropine) to inhibit ciliary spasm in patients with iritis.
Although the above medications are being used for a specific anticholinergic property, other unintended and troublesome anticholinergic effects are often seen. Similarly, many other medications often have unintended anticholinergic effects (see Table). Anticholinergic “toxicity” is simply an extension of the effects that occur with therapeutic use.
What is the treatment for patients with anticholinergic toxicity?
Most patients with anticholinergic toxicity do well with supportive management. Benzodiazepines are the treatment of choice for agitation. Haloperidol and other antipsychotics are relatively contraindicated for treatment of agitation as they may impair temperature regulation and lead to hyperthermia. Although likely of limited overall benefit, oral activated charcoal may reduce the amount of drug absorbed.
Antidotal therapy with physostigmine should be considered for select patients presenting with altered mental status due to an anticholinergic. Physostigmine is an acetylcholinesterase inhibitor that prevents the breakdown of acetylcholine in the synaptic cleft, thus antagonizing the effects of anticholinergic drugs. A retrospective study noted a lower incidence of complications and shorter time to recovery with the use of physostigmine compared with benzodiazepines in patients with anticholinergic toxicity.2 The use of physostigmine in select patients may obviate the need for a further delirium workup, which often includes computed tomography or lumbar puncture.
When administering physostigmine, atropine should be present at the bedside with airway equipment readily available as cholinergic effects may develop (specifically bronchospasm, bronchorrhea, or bradycardia). Dosing of physostigmine in adult patients is 1 to 2 mg via slow intravenous (IV) push, in aliquots of 0.2 to 0.3 mg each, over 5 minutes; pediatric dosing is 20 mcg/kg to maximum 0.5 mg. Onset of effects can be expected within minutes of administration.3 Since the duration of physostigmine is less than that of many anticholinergic drugs, recurrence of anticholinergic effects should be anticipated.
Historically, physostigmine was included in the “coma cocktail,” along with thiamine, dextrose, and naloxone for treating undifferentiated patients with altered level of consciousness. Concern for its ubiquitous use arose following reports of asystole in two patients who presented with tricyclic antidepressant (TCA) overdose, although these patients actually had more complicated multidrug overdoses.4 Nevertheless, an ECG should be performed in all patients for whom physostigmine is being considered, and it should not be administered (or perhaps only extremely cautiously) if the ECG demonstrates a QRS complex duration >100 ms.3 Relative contraindications include reactive airways disease, peripheral vascular disease, or intestinal or bladder-outlet obstruction.
Prolongation of the QRS interval is not always indicative of TCA ingestion as certain other antimuscarinic drugs, such as diphenhydramine, may cause sodium-channel blockade. Based on extrapolation from TCA literature,5 if the QRS >100 ms, a bolus of 1 to 2 mEq/kg sodium bicarbonate should be given with monitoring of the QRS interval for narrowing.
Case conclusion
The clinicians at the bedside felt that the infant’s presentation was consistent with anticholinergic toxicity. Physostigmine was administered by slow IV push for a total dose of 1.5 mg. The patient had immediate improvement of symptoms, including decreased skin redness, decreased agitation, and improved vital signs (BP, 118/80 mm Hg and HR, 160 beats/minute). He was admitted to the pediatric intensive care unit for monitoring and was subsequently discharged home with complete symptom resolution 2 days later.
- Gerretsen P, Pollock BG. Drugs with anticholinergic properties: a current perspective on use and safety. Expert Opin Drug Saf. 2011;10(5):751-765.
- Burns MJ, Linden CH, Graudins A, Brown RM, Fletcher KE. A comparison of physostigmine and benzodiazepines for the treatment of anticholinergic poisoning. Ann Emerg Med. 2000;35(4):374-381.
- Howland MA. Physostigmine salicylate. In: Nelson LS, Lewin NA, Howland MA, Hoffman RS, Goldfrank LR, Flomenbaum NE, eds. Goldfrank’s Toxicologic Emergencies. 9th ed. New York, NY: McGraw Hill; 2011:759-762.
- Pentel P, Peterson CD. Asystole complicating physostigmine treatment of tricyclic antidepressant overdose. Ann Emerg Med. 1980;9(11):588-590.
- Boehnert MT, Lovejoy FH, Jr. Value of the QRS duration versus the serum drug level in predicting seizures and ventricular arrhythmias after an acute overdose of tricyclic antidepressants. N Engl J Med. 1985;313(8):474-479.
- Gerretsen P, Pollock BG. Drugs with anticholinergic properties: a current perspective on use and safety. Expert Opin Drug Saf. 2011;10(5):751-765.
- Burns MJ, Linden CH, Graudins A, Brown RM, Fletcher KE. A comparison of physostigmine and benzodiazepines for the treatment of anticholinergic poisoning. Ann Emerg Med. 2000;35(4):374-381.
- Howland MA. Physostigmine salicylate. In: Nelson LS, Lewin NA, Howland MA, Hoffman RS, Goldfrank LR, Flomenbaum NE, eds. Goldfrank’s Toxicologic Emergencies. 9th ed. New York, NY: McGraw Hill; 2011:759-762.
- Pentel P, Peterson CD. Asystole complicating physostigmine treatment of tricyclic antidepressant overdose. Ann Emerg Med. 1980;9(11):588-590.
- Boehnert MT, Lovejoy FH, Jr. Value of the QRS duration versus the serum drug level in predicting seizures and ventricular arrhythmias after an acute overdose of tricyclic antidepressants. N Engl J Med. 1985;313(8):474-479.

Woman With Blue-Gray Palate and Nail Beds
A 62-year-old African-American woman presented for evaluation of a bluish discoloration of the hard palate and nail beds, noticeable for several months. In addition, she had complaints of fatigue and arthralgia. She reported that she had been taking hydroxychloroquine 400 mg/d and quinacrine 100 mg/d for several years for the treatment of systemic lupus erythematosus (SLE). Her medical history was also significant for dry mouth syndrome treated with pilocarpine.
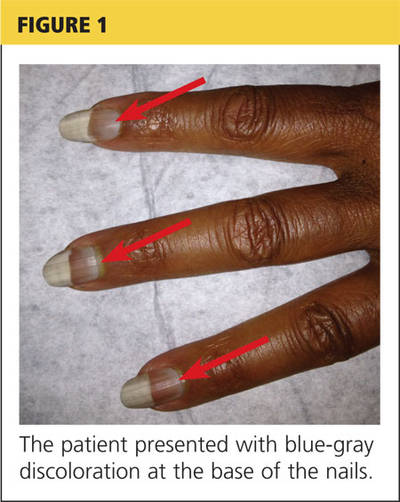
The patient’s vital signs included a temperature of 97°F;
respiratory rate, 15 breaths/min; pulse, 72 beats/min; and blood pressure, 130/80 mm Hg. Height was 62 in, weight was 189 lb, and BMI was 34.56. A bluish gray color was noted in the subungual areas of her nails (see Figure 1). There were several circumferential areas of skin hyperpigmentation resulting from healed lupus skin lesions on her arms. Nailfold capillaroscopy revealed several dilated blood vessels. The sclerae appeared dry, but no erythema or inflammation was noted.
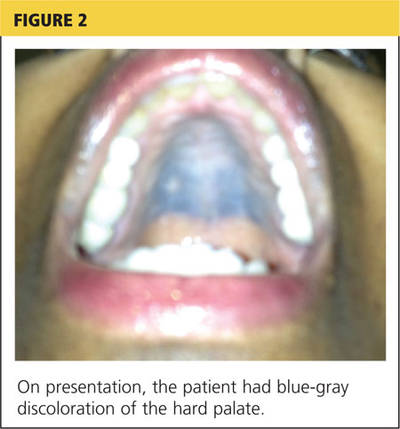
Examination of the mouth revealed a bluish discoloration of the hard palate (see Figure 2) and decreased salivary pool. Respiratory, cardiovascular, and abdominal examination findings were normal. Musculoskeletal examination was unremarkable for acute joint tenderness or synovitis. Crepitation and bony changes were noted in the left knee, without effusion or decreased range of motion.
Laboratory studies were ordered, and the results are listed in the table.
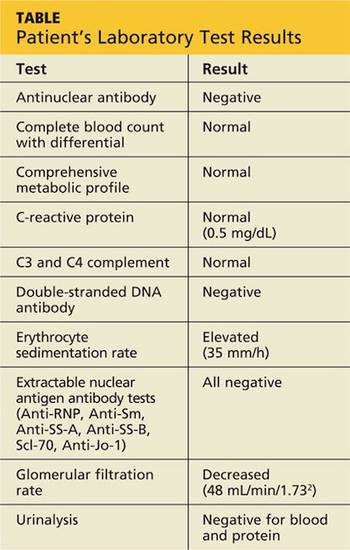
DISCUSSION
Hyperpigmentation of the oral mucosa can be associated with a number of conditions, including adrenal insufficiency, Peutz-Jeghers syndrome, hemochromatosis, polyostotic fibrous dysplasia, hyperparathyroidism, neurofibromatosis, and bronchogenic malignancy.1,2 Other causes of oral hyperpigmentation include physiologic pigmentary or postinflammatory changes, oral melanoacanthosis, blue nevus, and melanoma.2,3 While these diagnoses should be considered when encountering a mucosal lesion, they were unlikely in this patient because of the color changes in her nail beds.
Systemic skin and mucous membrane discoloration can also occur with the use of certain drugs and other substances, including chemotherapeutic agents, benzodiazepines, hormones, carotenoids, phenolphthalein, heavy metal salts, and several antimicrobial agents.1 In dark-skinned individuals, hyperpigmentation of the oral mucosa can be caused by a physiologic deposition of melanin.4
Pigmentary Changes
The use of antimalarial drugs, such as quinacrine, chloroquine, and hydroxychloroquine, has long been associated with pigmentary changes to the palatal mucosa and subungual areas.1,3 These drugs can stimulate melanin production and cause hemosiderin deposition, resulting in pigmentary changes.5 Skin discoloration is believed to be the result of the formation of a melanin-drug complex in areas with an elevated affinity for melanin.1 Besides malaria, these drugs are commonly used to treat SLE and discoid lupus erythematosus, rheumatoid arthritis, and other rheumatologic conditions.5
The diagnosis of drug-induced hyperpigmentation is generally clinical, supported by the patient’s history—which often includes the use of antimalarial drugs—and presentation.1 If a clear cause cannot be determined by clinical evaluation, then a biopsy to confirm a drug-induced cause may be necessary.2 A classic study by Tuffanelli et al reported that the onset of hyperpigmentation related to antimalarial drug therapy may not occur until 4 to 70 months after initiation of treatment.6 Once the offending drug is discontinued, pigmentation changes slowly fade but often do not completely resolve,7 and patients should be advised of this.
Ocular Retinopathy
While pigmentary changes associated with antimalarial drugs are benign,3 a rare but serious adverse effect of antimalarials is retinal toxicity. Ocular retinopathy related to chloroquine and hydroxychloroquine therapy has been well documented and may result in irreversible vision loss.8,9 The most recent recommendations from the American Academy of Ophthalmology suggest a baseline eye examination at initiation of antimalarial treatment and annual examinations starting after five years of therapy because the risk for toxicity relates to the cumulative dose.8 More frequent ophthalmologic evaluations are recommended for individuals at higher risk, such as those with preexisting retinal or macular disease.9
Outcome for the case patient >>
OUTCOME FOR THE CASE PATIENT
A biopsy of the roof of the patient’s mouth confirmed that the palatal hyperpigmentation was caused by her antimalarial medications. Since the patient displayed no evidence of active lupus skin lesions and laboratory results indicated that her SLE was inactive, one of the drugs, quinacrine, was discontinued.
The patient was referred for an ophthalmologic evaluation. No evidence of retinal toxicity was found.
Follow-up evaluations at two months and six months revealed no significant improvement in the discoloration of the patient’s oral mucosa or nail beds. At the six-month visit, her dosage of hydroxychloroquine was reevaluated.
The patient’s hydroxychloroquine dosage was determined based on 7.3 mg/kg/d. In the case of an overweight patient, especially one of shorter-than-average stature, hydroxychloroquine dosing should be based on ideal body weight to minimize the risk for overdosage; in general, a maximum dosage of 6.5 mg/kg/d is recommended.8,9 As a result, the patient’s dosage was decreased to 300 mg/d.
At her nine-month follow-up evaluation, the discoloration to the patient’s oral mucosa had faded but had not resolved completely (see Figure 3). No significant change was noted in the subungual discoloration. The patient had experienced no exacerbations of lupus-related symptoms since her medication adjustments.
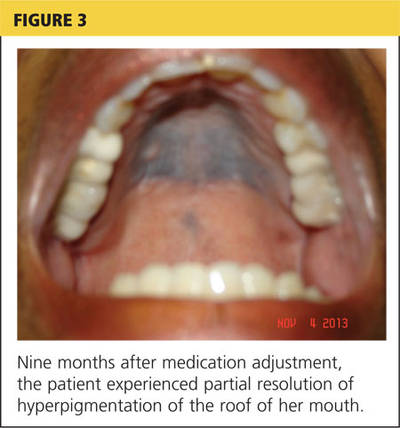
CONCLUSION
Although this patient’s hyperpigmentation was benign, staying alert to this potential adverse effect of antimalarial drugs is important in making a diagnosis. As with many skin lesions, if the clinical evaluation does not provide a clear cause, a biopsy may be needed. For anyone taking antimalarial drugs, regular ophthalmologic evaluations are recommended to facilitate early detection of the rare adverse effect of retinal toxicity. Nevertheless, with careful monitoring, antimalarial drugs are safe and effective for the treatment of inflammatory conditions such as SLE and rheumatoid arthritis.
REFERENCES
1. Kleinegger CL, Hammond HL, Finkelstein MW. Oral mucosal hyperpigmentation secondary to antimalarial drug therapy. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;90(2):189-194.
2. Gondak R-O, da Silva-Jorge R, Jorge J, et al. Oral pigmented lesions: clinicopathologic features and review of the literature. Med Oral Pathol Oral Cir Bucal. 2012;17(6):e919-e924.
3. Lerman MA, Karimbux N, Guze KA, Woo SB. Pigmentation of the hard palate. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2009;
107:8-12.
4. Kalampalikis A, Goetze S, Elsner P. Isolated hyperpigmentation of the oral mucosa due to hydroxychloroquine. J Dtsch Dermatol Ges. 2012; 10(12):921-922.
5. de Andrade BA, Fonseca FP, Pires FR, et al. Hard palate hyperpigmentation secondary to chronic chloroquine therapy: report of five cases.
J Cutan Pathol. 2013;40(9):833-838.
6. Tuffanelli D, Abraham RK, Dubois EI. Pigmentation from antimalarial therapy: its possible relationship to the ocular lesions. Arch Derm. 1963; 88:419-426.
7. Melikoglu MA, Melikoglu M, Gurbuz U, et al. Hydroxychloroquine-induced hyperpigmentation: a case report. J Clin Pharm Ther. 2008; 33(6):699-701.
8. Marmor MF, Kellner U, Lai YY, et al; American Academy of Ophthalmology. Revised recommendations on screening for chloroquine and hydroxychloroquine retinopathy. Ophthalmology. 2011;118(2):
415-422.
9. Screening for hydroxychloroquine retinopathy. Position statement, American College of Rheumatology. www.rheumatology.org/Practice/Clinical/Position/Position_Statements/. Accessed July 17, 2014.
A 62-year-old African-American woman presented for evaluation of a bluish discoloration of the hard palate and nail beds, noticeable for several months. In addition, she had complaints of fatigue and arthralgia. She reported that she had been taking hydroxychloroquine 400 mg/d and quinacrine 100 mg/d for several years for the treatment of systemic lupus erythematosus (SLE). Her medical history was also significant for dry mouth syndrome treated with pilocarpine.
The patient’s vital signs included a temperature of 97°F;
respiratory rate, 15 breaths/min; pulse, 72 beats/min; and blood pressure, 130/80 mm Hg. Height was 62 in, weight was 189 lb, and BMI was 34.56. A bluish gray color was noted in the subungual areas of her nails (see Figure 1). There were several circumferential areas of skin hyperpigmentation resulting from healed lupus skin lesions on her arms. Nailfold capillaroscopy revealed several dilated blood vessels. The sclerae appeared dry, but no erythema or inflammation was noted.
Examination of the mouth revealed a bluish discoloration of the hard palate (see Figure 2) and decreased salivary pool. Respiratory, cardiovascular, and abdominal examination findings were normal. Musculoskeletal examination was unremarkable for acute joint tenderness or synovitis. Crepitation and bony changes were noted in the left knee, without effusion or decreased range of motion.
Laboratory studies were ordered, and the results are listed in the table.
DISCUSSION
Hyperpigmentation of the oral mucosa can be associated with a number of conditions, including adrenal insufficiency, Peutz-Jeghers syndrome, hemochromatosis, polyostotic fibrous dysplasia, hyperparathyroidism, neurofibromatosis, and bronchogenic malignancy.1,2 Other causes of oral hyperpigmentation include physiologic pigmentary or postinflammatory changes, oral melanoacanthosis, blue nevus, and melanoma.2,3 While these diagnoses should be considered when encountering a mucosal lesion, they were unlikely in this patient because of the color changes in her nail beds.
Systemic skin and mucous membrane discoloration can also occur with the use of certain drugs and other substances, including chemotherapeutic agents, benzodiazepines, hormones, carotenoids, phenolphthalein, heavy metal salts, and several antimicrobial agents.1 In dark-skinned individuals, hyperpigmentation of the oral mucosa can be caused by a physiologic deposition of melanin.4
Pigmentary Changes
The use of antimalarial drugs, such as quinacrine, chloroquine, and hydroxychloroquine, has long been associated with pigmentary changes to the palatal mucosa and subungual areas.1,3 These drugs can stimulate melanin production and cause hemosiderin deposition, resulting in pigmentary changes.5 Skin discoloration is believed to be the result of the formation of a melanin-drug complex in areas with an elevated affinity for melanin.1 Besides malaria, these drugs are commonly used to treat SLE and discoid lupus erythematosus, rheumatoid arthritis, and other rheumatologic conditions.5
The diagnosis of drug-induced hyperpigmentation is generally clinical, supported by the patient’s history—which often includes the use of antimalarial drugs—and presentation.1 If a clear cause cannot be determined by clinical evaluation, then a biopsy to confirm a drug-induced cause may be necessary.2 A classic study by Tuffanelli et al reported that the onset of hyperpigmentation related to antimalarial drug therapy may not occur until 4 to 70 months after initiation of treatment.6 Once the offending drug is discontinued, pigmentation changes slowly fade but often do not completely resolve,7 and patients should be advised of this.
Ocular Retinopathy
While pigmentary changes associated with antimalarial drugs are benign,3 a rare but serious adverse effect of antimalarials is retinal toxicity. Ocular retinopathy related to chloroquine and hydroxychloroquine therapy has been well documented and may result in irreversible vision loss.8,9 The most recent recommendations from the American Academy of Ophthalmology suggest a baseline eye examination at initiation of antimalarial treatment and annual examinations starting after five years of therapy because the risk for toxicity relates to the cumulative dose.8 More frequent ophthalmologic evaluations are recommended for individuals at higher risk, such as those with preexisting retinal or macular disease.9
Outcome for the case patient >>
OUTCOME FOR THE CASE PATIENT
A biopsy of the roof of the patient’s mouth confirmed that the palatal hyperpigmentation was caused by her antimalarial medications. Since the patient displayed no evidence of active lupus skin lesions and laboratory results indicated that her SLE was inactive, one of the drugs, quinacrine, was discontinued.
The patient was referred for an ophthalmologic evaluation. No evidence of retinal toxicity was found.
Follow-up evaluations at two months and six months revealed no significant improvement in the discoloration of the patient’s oral mucosa or nail beds. At the six-month visit, her dosage of hydroxychloroquine was reevaluated.
The patient’s hydroxychloroquine dosage was determined based on 7.3 mg/kg/d. In the case of an overweight patient, especially one of shorter-than-average stature, hydroxychloroquine dosing should be based on ideal body weight to minimize the risk for overdosage; in general, a maximum dosage of 6.5 mg/kg/d is recommended.8,9 As a result, the patient’s dosage was decreased to 300 mg/d.
At her nine-month follow-up evaluation, the discoloration to the patient’s oral mucosa had faded but had not resolved completely (see Figure 3). No significant change was noted in the subungual discoloration. The patient had experienced no exacerbations of lupus-related symptoms since her medication adjustments.
CONCLUSION
Although this patient’s hyperpigmentation was benign, staying alert to this potential adverse effect of antimalarial drugs is important in making a diagnosis. As with many skin lesions, if the clinical evaluation does not provide a clear cause, a biopsy may be needed. For anyone taking antimalarial drugs, regular ophthalmologic evaluations are recommended to facilitate early detection of the rare adverse effect of retinal toxicity. Nevertheless, with careful monitoring, antimalarial drugs are safe and effective for the treatment of inflammatory conditions such as SLE and rheumatoid arthritis.
REFERENCES
1. Kleinegger CL, Hammond HL, Finkelstein MW. Oral mucosal hyperpigmentation secondary to antimalarial drug therapy. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;90(2):189-194.
2. Gondak R-O, da Silva-Jorge R, Jorge J, et al. Oral pigmented lesions: clinicopathologic features and review of the literature. Med Oral Pathol Oral Cir Bucal. 2012;17(6):e919-e924.
3. Lerman MA, Karimbux N, Guze KA, Woo SB. Pigmentation of the hard palate. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2009;
107:8-12.
4. Kalampalikis A, Goetze S, Elsner P. Isolated hyperpigmentation of the oral mucosa due to hydroxychloroquine. J Dtsch Dermatol Ges. 2012; 10(12):921-922.
5. de Andrade BA, Fonseca FP, Pires FR, et al. Hard palate hyperpigmentation secondary to chronic chloroquine therapy: report of five cases.
J Cutan Pathol. 2013;40(9):833-838.
6. Tuffanelli D, Abraham RK, Dubois EI. Pigmentation from antimalarial therapy: its possible relationship to the ocular lesions. Arch Derm. 1963; 88:419-426.
7. Melikoglu MA, Melikoglu M, Gurbuz U, et al. Hydroxychloroquine-induced hyperpigmentation: a case report. J Clin Pharm Ther. 2008; 33(6):699-701.
8. Marmor MF, Kellner U, Lai YY, et al; American Academy of Ophthalmology. Revised recommendations on screening for chloroquine and hydroxychloroquine retinopathy. Ophthalmology. 2011;118(2):
415-422.
9. Screening for hydroxychloroquine retinopathy. Position statement, American College of Rheumatology. www.rheumatology.org/Practice/Clinical/Position/Position_Statements/. Accessed July 17, 2014.
A 62-year-old African-American woman presented for evaluation of a bluish discoloration of the hard palate and nail beds, noticeable for several months. In addition, she had complaints of fatigue and arthralgia. She reported that she had been taking hydroxychloroquine 400 mg/d and quinacrine 100 mg/d for several years for the treatment of systemic lupus erythematosus (SLE). Her medical history was also significant for dry mouth syndrome treated with pilocarpine.
The patient’s vital signs included a temperature of 97°F;
respiratory rate, 15 breaths/min; pulse, 72 beats/min; and blood pressure, 130/80 mm Hg. Height was 62 in, weight was 189 lb, and BMI was 34.56. A bluish gray color was noted in the subungual areas of her nails (see Figure 1). There were several circumferential areas of skin hyperpigmentation resulting from healed lupus skin lesions on her arms. Nailfold capillaroscopy revealed several dilated blood vessels. The sclerae appeared dry, but no erythema or inflammation was noted.
Examination of the mouth revealed a bluish discoloration of the hard palate (see Figure 2) and decreased salivary pool. Respiratory, cardiovascular, and abdominal examination findings were normal. Musculoskeletal examination was unremarkable for acute joint tenderness or synovitis. Crepitation and bony changes were noted in the left knee, without effusion or decreased range of motion.
Laboratory studies were ordered, and the results are listed in the table.
DISCUSSION
Hyperpigmentation of the oral mucosa can be associated with a number of conditions, including adrenal insufficiency, Peutz-Jeghers syndrome, hemochromatosis, polyostotic fibrous dysplasia, hyperparathyroidism, neurofibromatosis, and bronchogenic malignancy.1,2 Other causes of oral hyperpigmentation include physiologic pigmentary or postinflammatory changes, oral melanoacanthosis, blue nevus, and melanoma.2,3 While these diagnoses should be considered when encountering a mucosal lesion, they were unlikely in this patient because of the color changes in her nail beds.
Systemic skin and mucous membrane discoloration can also occur with the use of certain drugs and other substances, including chemotherapeutic agents, benzodiazepines, hormones, carotenoids, phenolphthalein, heavy metal salts, and several antimicrobial agents.1 In dark-skinned individuals, hyperpigmentation of the oral mucosa can be caused by a physiologic deposition of melanin.4
Pigmentary Changes
The use of antimalarial drugs, such as quinacrine, chloroquine, and hydroxychloroquine, has long been associated with pigmentary changes to the palatal mucosa and subungual areas.1,3 These drugs can stimulate melanin production and cause hemosiderin deposition, resulting in pigmentary changes.5 Skin discoloration is believed to be the result of the formation of a melanin-drug complex in areas with an elevated affinity for melanin.1 Besides malaria, these drugs are commonly used to treat SLE and discoid lupus erythematosus, rheumatoid arthritis, and other rheumatologic conditions.5
The diagnosis of drug-induced hyperpigmentation is generally clinical, supported by the patient’s history—which often includes the use of antimalarial drugs—and presentation.1 If a clear cause cannot be determined by clinical evaluation, then a biopsy to confirm a drug-induced cause may be necessary.2 A classic study by Tuffanelli et al reported that the onset of hyperpigmentation related to antimalarial drug therapy may not occur until 4 to 70 months after initiation of treatment.6 Once the offending drug is discontinued, pigmentation changes slowly fade but often do not completely resolve,7 and patients should be advised of this.
Ocular Retinopathy
While pigmentary changes associated with antimalarial drugs are benign,3 a rare but serious adverse effect of antimalarials is retinal toxicity. Ocular retinopathy related to chloroquine and hydroxychloroquine therapy has been well documented and may result in irreversible vision loss.8,9 The most recent recommendations from the American Academy of Ophthalmology suggest a baseline eye examination at initiation of antimalarial treatment and annual examinations starting after five years of therapy because the risk for toxicity relates to the cumulative dose.8 More frequent ophthalmologic evaluations are recommended for individuals at higher risk, such as those with preexisting retinal or macular disease.9
Outcome for the case patient >>
OUTCOME FOR THE CASE PATIENT
A biopsy of the roof of the patient’s mouth confirmed that the palatal hyperpigmentation was caused by her antimalarial medications. Since the patient displayed no evidence of active lupus skin lesions and laboratory results indicated that her SLE was inactive, one of the drugs, quinacrine, was discontinued.
The patient was referred for an ophthalmologic evaluation. No evidence of retinal toxicity was found.
Follow-up evaluations at two months and six months revealed no significant improvement in the discoloration of the patient’s oral mucosa or nail beds. At the six-month visit, her dosage of hydroxychloroquine was reevaluated.
The patient’s hydroxychloroquine dosage was determined based on 7.3 mg/kg/d. In the case of an overweight patient, especially one of shorter-than-average stature, hydroxychloroquine dosing should be based on ideal body weight to minimize the risk for overdosage; in general, a maximum dosage of 6.5 mg/kg/d is recommended.8,9 As a result, the patient’s dosage was decreased to 300 mg/d.
At her nine-month follow-up evaluation, the discoloration to the patient’s oral mucosa had faded but had not resolved completely (see Figure 3). No significant change was noted in the subungual discoloration. The patient had experienced no exacerbations of lupus-related symptoms since her medication adjustments.
CONCLUSION
Although this patient’s hyperpigmentation was benign, staying alert to this potential adverse effect of antimalarial drugs is important in making a diagnosis. As with many skin lesions, if the clinical evaluation does not provide a clear cause, a biopsy may be needed. For anyone taking antimalarial drugs, regular ophthalmologic evaluations are recommended to facilitate early detection of the rare adverse effect of retinal toxicity. Nevertheless, with careful monitoring, antimalarial drugs are safe and effective for the treatment of inflammatory conditions such as SLE and rheumatoid arthritis.
REFERENCES
1. Kleinegger CL, Hammond HL, Finkelstein MW. Oral mucosal hyperpigmentation secondary to antimalarial drug therapy. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;90(2):189-194.
2. Gondak R-O, da Silva-Jorge R, Jorge J, et al. Oral pigmented lesions: clinicopathologic features and review of the literature. Med Oral Pathol Oral Cir Bucal. 2012;17(6):e919-e924.
3. Lerman MA, Karimbux N, Guze KA, Woo SB. Pigmentation of the hard palate. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2009;
107:8-12.
4. Kalampalikis A, Goetze S, Elsner P. Isolated hyperpigmentation of the oral mucosa due to hydroxychloroquine. J Dtsch Dermatol Ges. 2012; 10(12):921-922.
5. de Andrade BA, Fonseca FP, Pires FR, et al. Hard palate hyperpigmentation secondary to chronic chloroquine therapy: report of five cases.
J Cutan Pathol. 2013;40(9):833-838.
6. Tuffanelli D, Abraham RK, Dubois EI. Pigmentation from antimalarial therapy: its possible relationship to the ocular lesions. Arch Derm. 1963; 88:419-426.
7. Melikoglu MA, Melikoglu M, Gurbuz U, et al. Hydroxychloroquine-induced hyperpigmentation: a case report. J Clin Pharm Ther. 2008; 33(6):699-701.
8. Marmor MF, Kellner U, Lai YY, et al; American Academy of Ophthalmology. Revised recommendations on screening for chloroquine and hydroxychloroquine retinopathy. Ophthalmology. 2011;118(2):
415-422.
9. Screening for hydroxychloroquine retinopathy. Position statement, American College of Rheumatology. www.rheumatology.org/Practice/Clinical/Position/Position_Statements/. Accessed July 17, 2014.
Man Falls on Buttocks
ANSWER
There are degenerative changes present. Bilateral hip prostheses are noted. Within the coccyx, there is bone remodeling and angulation that are likely chronic and related to remote trauma or injury (arrow). Below this, some cortical lucency (circled) is noted, most likely consistent with an acute fracture. The patient was prescribed a nonsteroidal medication and a mild narcotic pain medication.
ANSWER
There are degenerative changes present. Bilateral hip prostheses are noted. Within the coccyx, there is bone remodeling and angulation that are likely chronic and related to remote trauma or injury (arrow). Below this, some cortical lucency (circled) is noted, most likely consistent with an acute fracture. The patient was prescribed a nonsteroidal medication and a mild narcotic pain medication.
ANSWER
There are degenerative changes present. Bilateral hip prostheses are noted. Within the coccyx, there is bone remodeling and angulation that are likely chronic and related to remote trauma or injury (arrow). Below this, some cortical lucency (circled) is noted, most likely consistent with an acute fracture. The patient was prescribed a nonsteroidal medication and a mild narcotic pain medication.
A 75-year-old man presents to the urgent care center for evaluation of pain in his buttocks after a fall. He states he was walking when his “legs gave out” and he hit the ground. He landed squarely on his buttocks, causing immediate pain. He was eventually able to get up with some assistance. He denies any current weakness or any bowel or bladder complaints. His medical/surgical history is significant for coronary artery disease, hypertension, and bilateral hip replacements. Physical exam reveals an elderly male who is uncomfortable but in no obvious distress. His vital signs are stable. He has moderate point tenderness over his sacrum but is able to move all his extremities well, with normal strength. Radiograph of his sacrum/coccyx is shown. What is your impression?
A Quality Improvement Initiative to Improve Emergency Department Care for Pediatric Patients with Sickle Cell Disease
From the Children’s Hospital & Research Center Oakland, Oakland, CA.
Abstract
- Objective: To determine whether a quality improvement (QI) initiative would result in more timely assessment and treatment of acute sickle cell–related pain for pediatric patients with sickle cell disease (SCD) treated in the emergency department (ED).
- Methods: We created and implemented a protocol for SCD pain management in the ED with the goals of improving (1) mean time from triage to first analgesic dose; (2) percentage of patients that received their first analgesic dose within 30 minutes of triage, and (3) percentage of patients who had pain assessment performed within 30 minutes of triage and who were re-assessed within 30 minutes after the first analgesic dose.
- Results: Significant improvements were achieved between baseline (55 patient visits) and post order set implementation (165 visits) in time from triage to administration of first analgesic (decreased from 89.9 ± 50.5 to 35.2 ± 22.8 minutes, P < 0.001); percentage of patient visits receiving pain medications within 30 minutes of triage (from 7% to 53%, P < 0.001); percentage of patient visits assessed within 30 minutes of triage (from 64% to 99.4%, P < 0.001); and percentage of patient visits re-assessed within 30 minutes of initial analgesic (from 54% to 86%, P < 0.001).
- Conclusions: Implementation of a QI initiative in the ED led to expeditious care for pediatric patients with SCD presenting with pain. A QI framework provided us with unique challenges but also invaluable lessons as we address our objective of decreasing the quality gap in SCD medical care.
Pain is the leading cause of emergency department (ED) visits for patients with sickle cell disease (SCD) [1]. In the United States, 78% of the nearly 200,000 annual ED visits for SCD are for a complaint of pain [1]. Guidelines for the management of sickle cell vaso-occlusive pain episodes (VOE) suggest prompt initiation of parenteral opioids, use of effective opioid doses, and repeat opioid doses at frequent intervals [2–4]. Adherence to guidelines is poor. Both pediatric and adult patients with SCD experience delays in the initiation of analgesics and are routinely undertreated with respect to opioid dosing [5–8]. Even after controlling for race, the delays in time to analgesic administration experienced by patients with SCD exceed the delays encountered by patients who present to the ED with other types of pain [5,9]. These disparities warrant efforts designed to improve the delivery of quality care to patients with SCD.
Barriers to rapid and appropriate care of VOE in the ED are multifactorial and include systems-based limitations, such as acuity of the ED census, staffing limitations (eg, nurse-to-patient ratios), and facility limitations (eg, room availability) [6]. Provider-based limitations may include lack of awareness of available guidelines [10]. Biases and misunderstandings amongst providers about sickle cell pain and adequate medication dosing may also play a role [11–13]. These provider biases often lead to undertreatment of the pain, which in turn can lead to pseudoaddiction (drug-seeking behavior due to inadequate treatment) and a cycle of increased ED and inpatient utilization [14,15].
Patient-specific barriers to effective ED management of pain are equally complex. Previous negative experiences in the ED can lead patients and families to delay seeking care or avoid the ED altogether despite severe VOE pain [16]. Patients report frustration with the lack of consideration that they receive for their reports of pain, perceived insensitivity of hospital staff, inadequate analgesic administration, staff preoccupation with concerns of drug addiction, and an overall lack of respect and trust [17–19]. Patients also perceive a lack of knowledge of SCD and its treatments on the part of ED staff [7]. Other barriers to effective management are technical in nature, such as difficulty in establishing timely intravenous (IV) access.
Gaps and variations in quality of care contribute to poor outcomes for patients with SCD [20,21]. To help address these inequities, the Working to Improve Sickle Cell Healthcare (WISCH) project began in 2010 to improve care and outcomes for patients with SCD. WISCH is a collaborative quality improvement (QI) project funded by the Health Resources and Services Administration (HRSA) that has the goal to use improvement science to improve outcomes for patients with SCD across the life course (Ed note: see Editorial by Oyeku et al in this issue). As one of the HRSA-WISCH grantee networks, we undertook a QI project designed to decrease the quality gap in SCD medical care by creating and implementing a protocol for ED pain management for pediatric patients. Goals of the project were to improve the timely and appropriate assessment and treatment of acute VOE in the ED.
Methods
Setting
This ED QI initiative was implemented at Children’s Hospital & Research Center Oakland, an urban free-standing pediatric hospital that serves a demographically diverse population. The hospital ED sees over 45,000 visits per year, with 250 visits per year for VOE. Residents in pediatrics, family medicine, and emergency medicine staff the ED. All attending physicians are subspecialists in pediatric emergency medicine. Study procedures were approved by the hospital’s institutional review board.
Intervention
A multidisciplinary team consisting of ED staff and sickle cell center staff drafted a nursing-driven protocol for the assessment and management of acute pain associated with VOE, incorporating elements from a protocol in use by another WISCH collaborative member. The protocol called for the immediate triage and assessment of all patients with SCD who presented with moderate to severe pain suggestive of VOE. Moderate to severe pain was defined as a pain score of ≥ 5 on a numeric scale of 0 to 10, where 0 = no pain and 10 = the worst pain imaginable. Exclusion criteria included a chief complaint of pain not considered secondary to VOE (eg, trauma, fracture). Patients were also excluded if they had been transferred from another facility. The protocol called for IV pain medication to be administered within 10 minutes of the patient being roomed, with re-evaluation at 20-minute intervals and re-dosing of pain medication based on the patient’s subsequent pain rating.
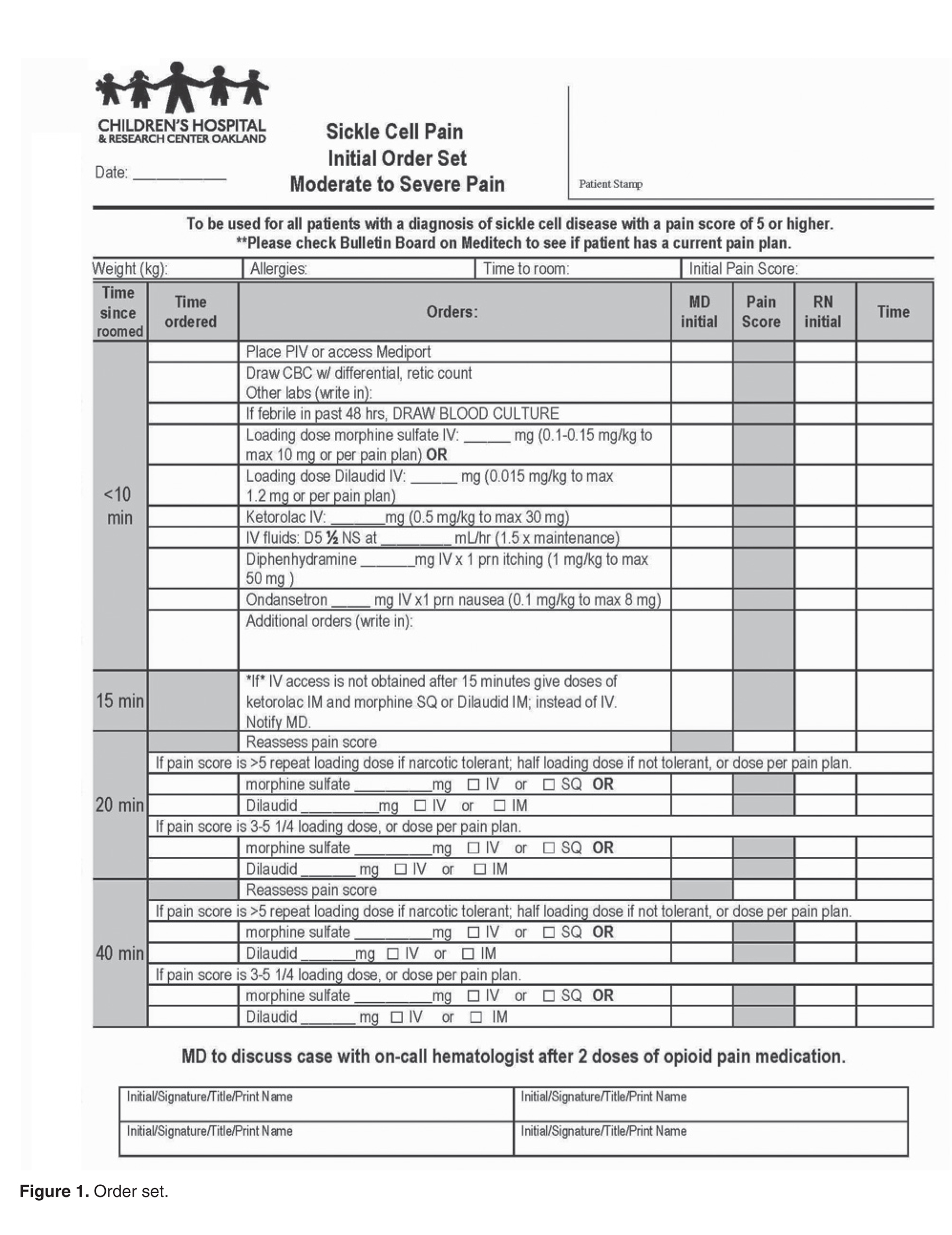
Measures
We selected performance measures from the bank developed by the WISCH team to track improvement and evaluate progress. These performance measures included (1) mean time from triage to first analgesic dose, (2) percentage of patients that received their first dose of analgesic within 30 minutes of triage, (3) percentage of patients who had a pain assessment performed within 30 minutes of triage, and (4) percentage of patients re-assessed within 30 minutes after the first dose of analgesic had been administered. Our aims were to have 80% of patients assessed and given pain medications within 30 minutes of triage, and to have 80% of patients re-assessed within 30 minutes after having received their first dose of an analgesic, within 12 months of implementing our intervention.
Data Collection and Analysis
The WISCH project coordinator reviewed records of visits to the ED for a baseline period of 6 months and post-order set implementaton. Demographic data (age, gender), clinical data (hemoglobin type), pain scores, utilization data (number of ED visits during the study period), and data pertaining to the metrics chosen from the WISCH measurement bank were extracted from each eligible patient’s ED chart after the visit was completed. If patients were admitted, their length of hospitalization was extracted from their inpatient medical record.
All biostatistical analyses were conducted using Stata 9.2 (StataCorp, College Station, TX). Descriptive statistics computed at 2 time-points (pre and post order set implementation) were utilized to examine means, standard deviations and percentages. The 2 time-points were initially compared at the visit level of measurement, using Student’s t tests corrected for unequal variances where necessary for continuous variables and chi-square analyses for categorical variables, to evaluate if there was an improvement in timely triage, assessment, and treatment of acute VOE pain for all ED visits pre and post order set implementation. To account for trends and possible correlations across the months post order set implementation, we ran a mixed linear model with repeated measures over time to compare visits during all months post order set implementation with the baseline months, for metric 1, time from triage to first pain medication. If significant differences were found, we used Dunnett’s method of multiple comparisons to determine which months differed from baseline. For metrics 2 through 4, we ran linear models with a binary outcome, a logit link function and using general estimating equations to determine trends and to account for correlations over time.
Secondary analyses were conducted to evaluate whether mean pain scores were significantly different over the course of the ED visit for the 78 unique patients seen post order set implementation. A multivariable mixed linear model, for the outcome of the third pain score, was used to assess the associations with prior scores and to control for potential covariates (age, gender, number of ED visits, hemoglobin type) that were determined in advance. A statistical significance level of 0.05 was used for all tests.
Results
Baseline data were collected from December 2011 to May 2012. The protocol was implemented in July 2012 and was utilized during 165 ED visits (91% of eligible visits) through April 2013. There were no statistically significant differences in demographic or clinical characteristics between the 55 patients whose charts were reviewed prior to implementing the order set and the 78 unique patients treated thereafter. Pre order set implementation, the mean age was 14.6 ± 6.4 years; 60% were female and the primary diagnosis was HgbSS disease (61.8% of diagnoses). Post order set implementation, the mean age was 16.0 ± 8.0 years; 51.3% were female and the primary diagnosis was HgbSS disease (61.5% of diagnoses). The mean number of visits was 1.5 visits per patient with a range of 1–8 visits, both pre and post order set implementation. Thirty-one patients had ED visits at both time periods.
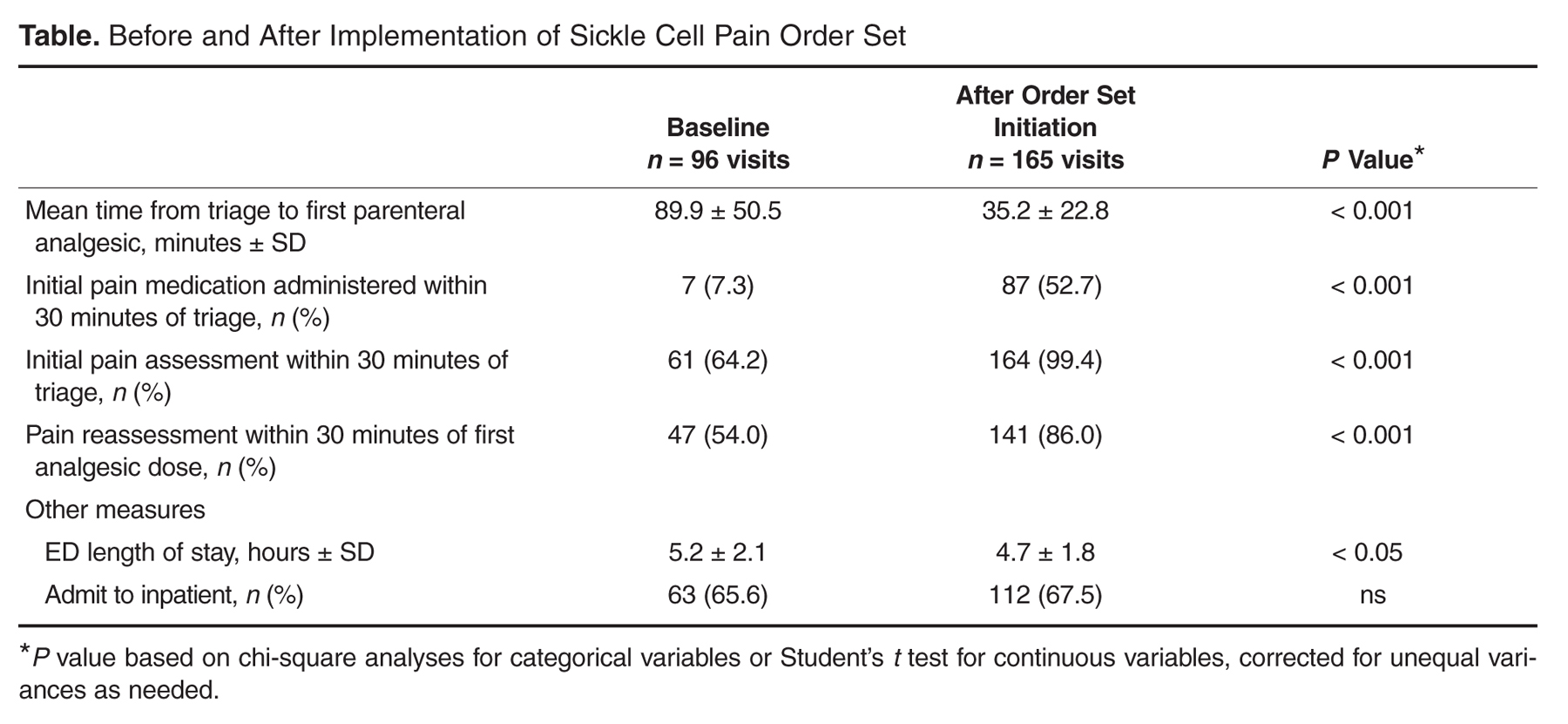
It can be seen in Figure 2 that staff performance on 3 of the 4 metrics (with the exception of initial analgesic within 30 minutes of triage) began to improve prior to implementing the order set. The mean length of ED stays decreased by 30 minutes, from a mean of 5.2 hours down to 4.7 hours (P < 0.05, Table). There was no significant change in the percentage of patients admitted to the inpatient unit.
We performed secondary analyses to determine if performance on our first metric, mean time from triage to first analgesic dose, was associated with any improvement on the third pain assessment for the patients enrolled post order set implementation. Looking at the first ED visit during the study period for the 78 unique patients, we found significant decreases in mean pain scores from the first to the second, from the second to the third, and from the first to the third assessment (P < 0.01). The mean pain scores were 8.3 ± 1.8, 5.9 ± 2.8, and 5.1 ± 3.0 on initial, second and third assessments, respectively. A multivariable model controlling for gender, hemoglobin type, number of ED visits and time to first pain medication showed that only the score at the second pain assessment (β = 0.88 ± 0.08, P < 0.001) was a significant predictor of the score at the third pain assessment.
Discussion
We demonstrated that a QI initiative to improve acute pain management resulted in more timely assessment and treatment of pain in pediatric patients with SCD. Significant improvements from baseline were achieved and sustained over a 10-month period in all 4 targeted metrics. We consistently exceeded our goal of having 80% of patients assessed within 30 minutes of triage, and our mean time to first pain medication (35.2 ± 22.8 minutes) came close to our goal of 30 minutes from triage. While we also achieved our goal to have 80% of patients re-assessed within 30 minutes after having received their first dose of an analgesic, we fell short in the percent who received their initial pain medication within 30 minutes of triage (52.7% versus goal of 80%). Although the length of stay in the ED decreased, no change was observed in the percentage of patients who required admission to the inpatient unit. A secondary analysis showed that mean pain scores significantly decreased over the course of the ED visit, from severe to moderate intensity.
The improvements that we observed began prior to implementation of the order set. We recognize that simply raising awareness and educating staff about the importance of timely and appropriate assessment and treatment of acute sickle cell related pain in the ED might be a potential confounder of our results. However, changes were sustained for 10 months post order set implementation and beyond, with no evidence that the performance on the target metrics is drifting back to baseline levels. Education and awareness-raising alone rarely result in sustained application of clinical practice guidelines [22]. We collaborated with NICHQ and other HRSA-WISCH grantees to systematically implement improvement science to ensure that the changes that we observed were indeed improvements and would be sustained [23] by first changing the system of care in the ED by introducing a standard order set [24,25]. We put a system into place to track use of the order set and to work with providers almost immediately if deviations were observed, to understand and overcome any barriers to the order set implementation. Systems in the ED and in the sickle cell center were aligned with the hospital’s QI initiatives [23].
Another strategy that we used to insure that the changes we observed would be sustained was to create a multidisciplinary team to build knowledge, skills, and new practices, including learning from other WISCH grantees and the NICHQ coordinating center [23]. We modified and adapted the intervention to our specific context [25]; although the outline of the order set was influenced by our WISCH colleagues, the final order set was structured to be consistent with other protocols within our institution. Finally, we included consumer input in the design of the project from the outset.
A previous study of a multi-institutional QI initiative aimed at improving acute SCD pain management for adult patients in the ED was unable to demonstrate an improvement in time to administration of initial analgesic [26]. Our study with pediatric patients was able to demonstrate a clinically meaningful decrease in the time to administration of first parenteral analgesic. The factors that account for the discrepant findings between these studies are likely multifactorial. Age (ie, pediatric vs. adult patients) may have played a role given that IV access may become increasingly difficult as patients with SCD age [26]. Education for providers should include the importance of alternative methods of administration of opioids, including subcutaneous and intranasal routes, to avoid delays when IV access is difficult. It is possible that negative provider attitudes converge with the documented increase in patient visits during the young adult years [27]. This may set up a challenging feedback loop wherein these vulnerable young adults are faced with greater stigma and consequently receive lower quality care, even when there is an attempt to carry out a standardized protocol.
We did not find that the QI intervention resulted in decreased admissions to the inpatient unit, with 68% of visits resulting in admission. In a recent pediatric SCD study, hospital admissions for pain control accounted for 78% of all admissions and 70% of readmissions within 30 days [28]. The investigators found that use of a SCD analgesic protocol including patient-controlled analgesia (PCA) improved quality of care as well as hospital readmission rates within 30 days (from 28% to 11%). Our ED QI protocol focused on only the first 90 minutes of the visit for pain. Our team has discussed the potential for starting the PCA in the ED and we should build on our success to focus on specific care that patients receive beyond their initial presentation. Further, we introduced pain action planning into outpatient care and need to continue to improve positive patient self-management strategies to ensure more seamless transition of pain management between home, ED, and inpatient settings.
Several valuable lessons were learned over the course of the ED QI initiative. Previous researchers [28] have emphasized the importance of coupling provider education with standardized order sets in efforts to improve the care of patients with SCD. Although we did not offer monthly formal education to our providers, the immediate follow-up when there were protocol deviations most likely served as teaching moments. These teaching moments also surfaced when some ED and hematology providers expressed concerns about the risk for oversedation with the rapid reassessment of pain and re-dosing of pain medications. Although rare, some parents also expressed that their child was being treated too vigorously with opioids. Our project highlighted the element of stigma that still accompanies the use of opioids for SCD pain management.
The project could not have been undertaken were it not for a small but determined multidisciplinary team of individuals who were personally invested in seeing the project come to fruition. The identification of physician and nurse champions who were enthusiastic about the project, invested in its conduct, and committed to its success was a cornerstone of the project’s success. These champions played an essential role in engaging staff interest in the project and oversaw the practicalities of implementing a new protocol in the ED. A spirit of collaboration, teamwork, and good communication between all involved parties was also critical. At the same time, we incorporated input from the treating ED and hematology clinicians using PDSA cycles as we were refining our protocol. We believe that our process enhanced buy-in from participating providers and clarified any issues that needed to be addressed in our setting, resulting in accelerated and sustained quality improvement.
Limitations
Although protocol-driven interventions are designed to provide a certain degree of uniformity of care, the protocol was not designed nor utilized in such a way that it superseded the best medical judgment of the treating clinicians. Deviations from the protocol were permissible when they were felt to be in the patient’s best interest. The study did not control for confounding variables such as disease severity, how long the patient had been in pain prior to coming to the ED, nor did we assess therapeutic interventions the patient had utilized at home prior to seeking out care in the ED. All of these factors could affect how well a patient might respond to treatment. We believe that sharing baseline data and monthly progress via run charts (graphs of data over time) with ED and sickle cell center staff and with consumer representatives enhanced the pace and focus of the project [23]. We had a dedicated person managing our data in real time through our HRSA funding, thus the project might not be generalizable to other institutions that do not have such staffing or access to the technology to allow project progress to be closely monitored by stakeholders.
Future Directions
With the goal of further reducing the time to administration of first analgesic dose in the ED setting, intranasal fentanyl will be utilized in our ED as the initial drug of choice for patients who do not object to or have a contraindication to its use. Collection of data from patients and family members is being undertaken to assess consumer satisfaction with the ED QI initiative. Recognizing that the ED management of acute pain addresses only one aspect of sickle cell pain, we are looking at ways to more comprehensively address pain. Individualized outpatient pain management plans are being created and patients and families are being encouraged and empowered to become active partners with their sickle cell providers in their own care. Although our initial efforts have focused on our pediatric patients, an additional aim of our project is to broaden the scope of our ED QI initiative to include community hospitals in the region that serve adult patients with SCD.
Conclusion
Implementation of a QI initiative in the ED has led to expeditious care for pediatric patients with SCD presenting with VOE. A multidisciplinary approach, ongoing staff education, and commitment to the initiative have been necessary to sustain the improvements. Our success can provide a template for other QI initiatives in the ED that translate to improved patient care for other diseases. A QI framework provided us with unique challenges but also invaluable lessons as we addressed our objective to improve outcomes for patients with SCD across the life course.
Acknowledgments: The authors wish to thank Theresa Freitas, RN, Lisa Hale, PNP, Carolyn Hoppe, MD, Ileana Mendez, RN, Helen Mitchell, Mary Rutherford, MD, Augusta Saulys, MD and the Children’s Hospital & Research Center Oakland Emergency Medicine Department and Sickle Cell Center for their support.
Corresponding author: Marsha Treadwell, PhD, Children’s Hospital & Research Center Oakland, 747 52nd St, Oakland, CA 94609, [email protected].
Funding/support: This research was conducted as part of the National Initiative for Children’s Healthcare Quality (NICHQ) Working to Improve Sickle Cell Healthcare (WISCH) project. Further support came from a grant from the Health Resources and Services Administration Sickle Cell Disease Treatment Demonstration Project Grant No. U1EMC16492 and from NIH CTSA grant UL1 RR024131. The views expressed in this publication do not necessarily reflect the views of WISCH, NICHQ, or HRSA.
1. Yusuf HR, Atrash HK, Grosse SD, et al. Emergency department visits made by patients with sickle cell disease: a descriptive study, 1999-2007. Am J Preventive Med 2010;38 (4 Suppl):S536–41.
2. Benjamin L, Dampier C, Jacox A, et al. Guideline for the management of acute and chronic pain in sickle cell disease. American Pain Society; 1999.
3. Rees DC, Olujohungbe AD, Parker NE, et al. Guidelines for the management of the acute painful crisis in sickle cell disease. Br J Haematology 2003;120:744–52.
4. Solomon LR. Pain management in adults with sickle cell disease in a medical center emergency department. J Nat Med Assoc 2010;102:1025–32.
5. Lazio MP, Costello HH, Courtney DM, et al. A comparison of analgesic management for emergency department patients with sickle cell disease and renal colic. Clin J Pain 2010;26:199–205.
6. Shenoi R, Ma L, Syblik D, Yusuf S. Emergency department crowding and analgesic delay in pediatric sickle cell pain crises. Ped Emerg Care 2011;27:911–7.
7. Tanabe P, Artz N, Mark Courtney D, et al. Adult emergency department patients with sickle cell pain crisis: a learning collaborative model to improve analgesic management. Acad Emerg Med 2010;17:399–407.
8. Zempsky WT. Evaluation and treatment of sickle cell pain in the emergency department: paths to a better future. Clin Ped Emerg Med 2010;11:265–73.
9. Haywood C Jr, Tanabe P, Naik R, et al. The impact of race and disease on sickle cell patient wait times in the emergency department. Am J Emerg Med 2013;31:651–6.
10. Solomon LR. Treatment and prevention of pain due to vaso-occlusive crises in adults with sickle cell disease: an educational void. Blood 2008;111:997–1003.
11. Ballas SK. New era dawns on sickle cell pain. Blood 2010;116:311–2.
12. Haywood C Jr, Lanzkron S, Ratanawongsa N, et al. The association of provider communication with trust among adults with sickle cell disease. J Gen Intern Med 2010;25:543–8.
13. Zempsky WT. Treatment of sickle cell pain: fostering trust and justice. JAMA 2009;302:2479–80.
14. Elander J, Lusher J, Bevan D, Telfer P. Pain management and symptoms of substance dependence among patients with sickle cell disease. Soc Sci Med 2003;57:1683–96.
15. Elander J, Lusher J, Bevan D, et al. Understanding the causes of problematic pain management in sickle cell disease: evidence that pseudoaddiction plays a more important role than genuine analgesic dependence. J Pain Sympt Manag 2004;27:156–69.
16. Smith WR, Penberthy LT, Bovbjerg VE, et al. Daily assessment of pain in adults with sickle cell disease. Ann Intern Med 2008;148:94–101.
17. Harris A, Parker N, Baker C. Adults with sickle cell. Psychol Health Med 1998;3:171–9.
18. Jenerette CM, Brewer C. Health-related stigma in young adults with sickle cell disease. J Nat Med Assoc 2010;102:1050–5.
19. Maxwell K, Streetly A, Bevan D. Experiences of hospital care and treatment seeking for pain from sickle cell disease: qualitative study. BMJ 1999;318:1585–90.
20. Oyeku SO, Wang CJ, Scoville R, et al. Hemoglobinopathy Learning Collaborative: using quality improvement (QI) to achieve equity in health care quality, coordination, and outcomes for sickle cell disease. J Health Care Poor Underserved 2012;23(3 Suppl):34–48.
21. Wang CJ, Kavanagh PL, Little AA, et al. Quality-of-care indicators for children with sickle cell disease. Pediatrics 2011;128:484–93.
22. Mansouri M, Lockyer J. A meta-analysis of continuing medical education effectiveness. J Contin Ed Health Prof 2007;27:6–15.
23. The breakthrough series: IHI’s collaborative model for achieving breakthrough improvement. Boston: Institute for Healthcare Improvement; 2003.
24. Berwick DM. Improvement, trust, and the healthcare workforce. Qual Safety Health Care 2003;12:448–52.
25. Hovlid E, Bukve O, Haug K, et al. Sustainability of healthcare improvement: what can we learn from learning theory? BMC Health Serv Res 2012;12:235.
26. Tanabe P, Hafner JW, Martinovich Z, Artz N. Adult emergency department patients with sickle cell pain crisis: results from a quality improvement learning collaborative model to improve analgesic management. Acad Emerg Med 2012;19:430–8.
27. Brousseau DC, Owens PL, Mosso AL, et al. Acute care utilization and rehospitalizations for sickle cell disease. JAMA 2010;303:1288–94.
28. Frei-Jones MJ, Field JJ, DeBaun MR. Multi-modal intervention and prospective implementation of standardized sickle cell pain admission orders reduces 30-day readmission rate. Pediatr Blood Cancer 2009;53:401–5.
From the Children’s Hospital & Research Center Oakland, Oakland, CA.
Abstract
- Objective: To determine whether a quality improvement (QI) initiative would result in more timely assessment and treatment of acute sickle cell–related pain for pediatric patients with sickle cell disease (SCD) treated in the emergency department (ED).
- Methods: We created and implemented a protocol for SCD pain management in the ED with the goals of improving (1) mean time from triage to first analgesic dose; (2) percentage of patients that received their first analgesic dose within 30 minutes of triage, and (3) percentage of patients who had pain assessment performed within 30 minutes of triage and who were re-assessed within 30 minutes after the first analgesic dose.
- Results: Significant improvements were achieved between baseline (55 patient visits) and post order set implementation (165 visits) in time from triage to administration of first analgesic (decreased from 89.9 ± 50.5 to 35.2 ± 22.8 minutes, P < 0.001); percentage of patient visits receiving pain medications within 30 minutes of triage (from 7% to 53%, P < 0.001); percentage of patient visits assessed within 30 minutes of triage (from 64% to 99.4%, P < 0.001); and percentage of patient visits re-assessed within 30 minutes of initial analgesic (from 54% to 86%, P < 0.001).
- Conclusions: Implementation of a QI initiative in the ED led to expeditious care for pediatric patients with SCD presenting with pain. A QI framework provided us with unique challenges but also invaluable lessons as we address our objective of decreasing the quality gap in SCD medical care.
Pain is the leading cause of emergency department (ED) visits for patients with sickle cell disease (SCD) [1]. In the United States, 78% of the nearly 200,000 annual ED visits for SCD are for a complaint of pain [1]. Guidelines for the management of sickle cell vaso-occlusive pain episodes (VOE) suggest prompt initiation of parenteral opioids, use of effective opioid doses, and repeat opioid doses at frequent intervals [2–4]. Adherence to guidelines is poor. Both pediatric and adult patients with SCD experience delays in the initiation of analgesics and are routinely undertreated with respect to opioid dosing [5–8]. Even after controlling for race, the delays in time to analgesic administration experienced by patients with SCD exceed the delays encountered by patients who present to the ED with other types of pain [5,9]. These disparities warrant efforts designed to improve the delivery of quality care to patients with SCD.
Barriers to rapid and appropriate care of VOE in the ED are multifactorial and include systems-based limitations, such as acuity of the ED census, staffing limitations (eg, nurse-to-patient ratios), and facility limitations (eg, room availability) [6]. Provider-based limitations may include lack of awareness of available guidelines [10]. Biases and misunderstandings amongst providers about sickle cell pain and adequate medication dosing may also play a role [11–13]. These provider biases often lead to undertreatment of the pain, which in turn can lead to pseudoaddiction (drug-seeking behavior due to inadequate treatment) and a cycle of increased ED and inpatient utilization [14,15].
Patient-specific barriers to effective ED management of pain are equally complex. Previous negative experiences in the ED can lead patients and families to delay seeking care or avoid the ED altogether despite severe VOE pain [16]. Patients report frustration with the lack of consideration that they receive for their reports of pain, perceived insensitivity of hospital staff, inadequate analgesic administration, staff preoccupation with concerns of drug addiction, and an overall lack of respect and trust [17–19]. Patients also perceive a lack of knowledge of SCD and its treatments on the part of ED staff [7]. Other barriers to effective management are technical in nature, such as difficulty in establishing timely intravenous (IV) access.
Gaps and variations in quality of care contribute to poor outcomes for patients with SCD [20,21]. To help address these inequities, the Working to Improve Sickle Cell Healthcare (WISCH) project began in 2010 to improve care and outcomes for patients with SCD. WISCH is a collaborative quality improvement (QI) project funded by the Health Resources and Services Administration (HRSA) that has the goal to use improvement science to improve outcomes for patients with SCD across the life course (Ed note: see Editorial by Oyeku et al in this issue). As one of the HRSA-WISCH grantee networks, we undertook a QI project designed to decrease the quality gap in SCD medical care by creating and implementing a protocol for ED pain management for pediatric patients. Goals of the project were to improve the timely and appropriate assessment and treatment of acute VOE in the ED.
Methods
Setting
This ED QI initiative was implemented at Children’s Hospital & Research Center Oakland, an urban free-standing pediatric hospital that serves a demographically diverse population. The hospital ED sees over 45,000 visits per year, with 250 visits per year for VOE. Residents in pediatrics, family medicine, and emergency medicine staff the ED. All attending physicians are subspecialists in pediatric emergency medicine. Study procedures were approved by the hospital’s institutional review board.
Intervention
A multidisciplinary team consisting of ED staff and sickle cell center staff drafted a nursing-driven protocol for the assessment and management of acute pain associated with VOE, incorporating elements from a protocol in use by another WISCH collaborative member. The protocol called for the immediate triage and assessment of all patients with SCD who presented with moderate to severe pain suggestive of VOE. Moderate to severe pain was defined as a pain score of ≥ 5 on a numeric scale of 0 to 10, where 0 = no pain and 10 = the worst pain imaginable. Exclusion criteria included a chief complaint of pain not considered secondary to VOE (eg, trauma, fracture). Patients were also excluded if they had been transferred from another facility. The protocol called for IV pain medication to be administered within 10 minutes of the patient being roomed, with re-evaluation at 20-minute intervals and re-dosing of pain medication based on the patient’s subsequent pain rating.
Measures
We selected performance measures from the bank developed by the WISCH team to track improvement and evaluate progress. These performance measures included (1) mean time from triage to first analgesic dose, (2) percentage of patients that received their first dose of analgesic within 30 minutes of triage, (3) percentage of patients who had a pain assessment performed within 30 minutes of triage, and (4) percentage of patients re-assessed within 30 minutes after the first dose of analgesic had been administered. Our aims were to have 80% of patients assessed and given pain medications within 30 minutes of triage, and to have 80% of patients re-assessed within 30 minutes after having received their first dose of an analgesic, within 12 months of implementing our intervention.
Data Collection and Analysis
The WISCH project coordinator reviewed records of visits to the ED for a baseline period of 6 months and post-order set implementaton. Demographic data (age, gender), clinical data (hemoglobin type), pain scores, utilization data (number of ED visits during the study period), and data pertaining to the metrics chosen from the WISCH measurement bank were extracted from each eligible patient’s ED chart after the visit was completed. If patients were admitted, their length of hospitalization was extracted from their inpatient medical record.
All biostatistical analyses were conducted using Stata 9.2 (StataCorp, College Station, TX). Descriptive statistics computed at 2 time-points (pre and post order set implementation) were utilized to examine means, standard deviations and percentages. The 2 time-points were initially compared at the visit level of measurement, using Student’s t tests corrected for unequal variances where necessary for continuous variables and chi-square analyses for categorical variables, to evaluate if there was an improvement in timely triage, assessment, and treatment of acute VOE pain for all ED visits pre and post order set implementation. To account for trends and possible correlations across the months post order set implementation, we ran a mixed linear model with repeated measures over time to compare visits during all months post order set implementation with the baseline months, for metric 1, time from triage to first pain medication. If significant differences were found, we used Dunnett’s method of multiple comparisons to determine which months differed from baseline. For metrics 2 through 4, we ran linear models with a binary outcome, a logit link function and using general estimating equations to determine trends and to account for correlations over time.
Secondary analyses were conducted to evaluate whether mean pain scores were significantly different over the course of the ED visit for the 78 unique patients seen post order set implementation. A multivariable mixed linear model, for the outcome of the third pain score, was used to assess the associations with prior scores and to control for potential covariates (age, gender, number of ED visits, hemoglobin type) that were determined in advance. A statistical significance level of 0.05 was used for all tests.
Results
Baseline data were collected from December 2011 to May 2012. The protocol was implemented in July 2012 and was utilized during 165 ED visits (91% of eligible visits) through April 2013. There were no statistically significant differences in demographic or clinical characteristics between the 55 patients whose charts were reviewed prior to implementing the order set and the 78 unique patients treated thereafter. Pre order set implementation, the mean age was 14.6 ± 6.4 years; 60% were female and the primary diagnosis was HgbSS disease (61.8% of diagnoses). Post order set implementation, the mean age was 16.0 ± 8.0 years; 51.3% were female and the primary diagnosis was HgbSS disease (61.5% of diagnoses). The mean number of visits was 1.5 visits per patient with a range of 1–8 visits, both pre and post order set implementation. Thirty-one patients had ED visits at both time periods.
It can be seen in Figure 2 that staff performance on 3 of the 4 metrics (with the exception of initial analgesic within 30 minutes of triage) began to improve prior to implementing the order set. The mean length of ED stays decreased by 30 minutes, from a mean of 5.2 hours down to 4.7 hours (P < 0.05, Table). There was no significant change in the percentage of patients admitted to the inpatient unit.
We performed secondary analyses to determine if performance on our first metric, mean time from triage to first analgesic dose, was associated with any improvement on the third pain assessment for the patients enrolled post order set implementation. Looking at the first ED visit during the study period for the 78 unique patients, we found significant decreases in mean pain scores from the first to the second, from the second to the third, and from the first to the third assessment (P < 0.01). The mean pain scores were 8.3 ± 1.8, 5.9 ± 2.8, and 5.1 ± 3.0 on initial, second and third assessments, respectively. A multivariable model controlling for gender, hemoglobin type, number of ED visits and time to first pain medication showed that only the score at the second pain assessment (β = 0.88 ± 0.08, P < 0.001) was a significant predictor of the score at the third pain assessment.
Discussion
We demonstrated that a QI initiative to improve acute pain management resulted in more timely assessment and treatment of pain in pediatric patients with SCD. Significant improvements from baseline were achieved and sustained over a 10-month period in all 4 targeted metrics. We consistently exceeded our goal of having 80% of patients assessed within 30 minutes of triage, and our mean time to first pain medication (35.2 ± 22.8 minutes) came close to our goal of 30 minutes from triage. While we also achieved our goal to have 80% of patients re-assessed within 30 minutes after having received their first dose of an analgesic, we fell short in the percent who received their initial pain medication within 30 minutes of triage (52.7% versus goal of 80%). Although the length of stay in the ED decreased, no change was observed in the percentage of patients who required admission to the inpatient unit. A secondary analysis showed that mean pain scores significantly decreased over the course of the ED visit, from severe to moderate intensity.
The improvements that we observed began prior to implementation of the order set. We recognize that simply raising awareness and educating staff about the importance of timely and appropriate assessment and treatment of acute sickle cell related pain in the ED might be a potential confounder of our results. However, changes were sustained for 10 months post order set implementation and beyond, with no evidence that the performance on the target metrics is drifting back to baseline levels. Education and awareness-raising alone rarely result in sustained application of clinical practice guidelines [22]. We collaborated with NICHQ and other HRSA-WISCH grantees to systematically implement improvement science to ensure that the changes that we observed were indeed improvements and would be sustained [23] by first changing the system of care in the ED by introducing a standard order set [24,25]. We put a system into place to track use of the order set and to work with providers almost immediately if deviations were observed, to understand and overcome any barriers to the order set implementation. Systems in the ED and in the sickle cell center were aligned with the hospital’s QI initiatives [23].
Another strategy that we used to insure that the changes we observed would be sustained was to create a multidisciplinary team to build knowledge, skills, and new practices, including learning from other WISCH grantees and the NICHQ coordinating center [23]. We modified and adapted the intervention to our specific context [25]; although the outline of the order set was influenced by our WISCH colleagues, the final order set was structured to be consistent with other protocols within our institution. Finally, we included consumer input in the design of the project from the outset.
A previous study of a multi-institutional QI initiative aimed at improving acute SCD pain management for adult patients in the ED was unable to demonstrate an improvement in time to administration of initial analgesic [26]. Our study with pediatric patients was able to demonstrate a clinically meaningful decrease in the time to administration of first parenteral analgesic. The factors that account for the discrepant findings between these studies are likely multifactorial. Age (ie, pediatric vs. adult patients) may have played a role given that IV access may become increasingly difficult as patients with SCD age [26]. Education for providers should include the importance of alternative methods of administration of opioids, including subcutaneous and intranasal routes, to avoid delays when IV access is difficult. It is possible that negative provider attitudes converge with the documented increase in patient visits during the young adult years [27]. This may set up a challenging feedback loop wherein these vulnerable young adults are faced with greater stigma and consequently receive lower quality care, even when there is an attempt to carry out a standardized protocol.
We did not find that the QI intervention resulted in decreased admissions to the inpatient unit, with 68% of visits resulting in admission. In a recent pediatric SCD study, hospital admissions for pain control accounted for 78% of all admissions and 70% of readmissions within 30 days [28]. The investigators found that use of a SCD analgesic protocol including patient-controlled analgesia (PCA) improved quality of care as well as hospital readmission rates within 30 days (from 28% to 11%). Our ED QI protocol focused on only the first 90 minutes of the visit for pain. Our team has discussed the potential for starting the PCA in the ED and we should build on our success to focus on specific care that patients receive beyond their initial presentation. Further, we introduced pain action planning into outpatient care and need to continue to improve positive patient self-management strategies to ensure more seamless transition of pain management between home, ED, and inpatient settings.
Several valuable lessons were learned over the course of the ED QI initiative. Previous researchers [28] have emphasized the importance of coupling provider education with standardized order sets in efforts to improve the care of patients with SCD. Although we did not offer monthly formal education to our providers, the immediate follow-up when there were protocol deviations most likely served as teaching moments. These teaching moments also surfaced when some ED and hematology providers expressed concerns about the risk for oversedation with the rapid reassessment of pain and re-dosing of pain medications. Although rare, some parents also expressed that their child was being treated too vigorously with opioids. Our project highlighted the element of stigma that still accompanies the use of opioids for SCD pain management.
The project could not have been undertaken were it not for a small but determined multidisciplinary team of individuals who were personally invested in seeing the project come to fruition. The identification of physician and nurse champions who were enthusiastic about the project, invested in its conduct, and committed to its success was a cornerstone of the project’s success. These champions played an essential role in engaging staff interest in the project and oversaw the practicalities of implementing a new protocol in the ED. A spirit of collaboration, teamwork, and good communication between all involved parties was also critical. At the same time, we incorporated input from the treating ED and hematology clinicians using PDSA cycles as we were refining our protocol. We believe that our process enhanced buy-in from participating providers and clarified any issues that needed to be addressed in our setting, resulting in accelerated and sustained quality improvement.
Limitations
Although protocol-driven interventions are designed to provide a certain degree of uniformity of care, the protocol was not designed nor utilized in such a way that it superseded the best medical judgment of the treating clinicians. Deviations from the protocol were permissible when they were felt to be in the patient’s best interest. The study did not control for confounding variables such as disease severity, how long the patient had been in pain prior to coming to the ED, nor did we assess therapeutic interventions the patient had utilized at home prior to seeking out care in the ED. All of these factors could affect how well a patient might respond to treatment. We believe that sharing baseline data and monthly progress via run charts (graphs of data over time) with ED and sickle cell center staff and with consumer representatives enhanced the pace and focus of the project [23]. We had a dedicated person managing our data in real time through our HRSA funding, thus the project might not be generalizable to other institutions that do not have such staffing or access to the technology to allow project progress to be closely monitored by stakeholders.
Future Directions
With the goal of further reducing the time to administration of first analgesic dose in the ED setting, intranasal fentanyl will be utilized in our ED as the initial drug of choice for patients who do not object to or have a contraindication to its use. Collection of data from patients and family members is being undertaken to assess consumer satisfaction with the ED QI initiative. Recognizing that the ED management of acute pain addresses only one aspect of sickle cell pain, we are looking at ways to more comprehensively address pain. Individualized outpatient pain management plans are being created and patients and families are being encouraged and empowered to become active partners with their sickle cell providers in their own care. Although our initial efforts have focused on our pediatric patients, an additional aim of our project is to broaden the scope of our ED QI initiative to include community hospitals in the region that serve adult patients with SCD.
Conclusion
Implementation of a QI initiative in the ED has led to expeditious care for pediatric patients with SCD presenting with VOE. A multidisciplinary approach, ongoing staff education, and commitment to the initiative have been necessary to sustain the improvements. Our success can provide a template for other QI initiatives in the ED that translate to improved patient care for other diseases. A QI framework provided us with unique challenges but also invaluable lessons as we addressed our objective to improve outcomes for patients with SCD across the life course.
Acknowledgments: The authors wish to thank Theresa Freitas, RN, Lisa Hale, PNP, Carolyn Hoppe, MD, Ileana Mendez, RN, Helen Mitchell, Mary Rutherford, MD, Augusta Saulys, MD and the Children’s Hospital & Research Center Oakland Emergency Medicine Department and Sickle Cell Center for their support.
Corresponding author: Marsha Treadwell, PhD, Children’s Hospital & Research Center Oakland, 747 52nd St, Oakland, CA 94609, [email protected].
Funding/support: This research was conducted as part of the National Initiative for Children’s Healthcare Quality (NICHQ) Working to Improve Sickle Cell Healthcare (WISCH) project. Further support came from a grant from the Health Resources and Services Administration Sickle Cell Disease Treatment Demonstration Project Grant No. U1EMC16492 and from NIH CTSA grant UL1 RR024131. The views expressed in this publication do not necessarily reflect the views of WISCH, NICHQ, or HRSA.
From the Children’s Hospital & Research Center Oakland, Oakland, CA.
Abstract
- Objective: To determine whether a quality improvement (QI) initiative would result in more timely assessment and treatment of acute sickle cell–related pain for pediatric patients with sickle cell disease (SCD) treated in the emergency department (ED).
- Methods: We created and implemented a protocol for SCD pain management in the ED with the goals of improving (1) mean time from triage to first analgesic dose; (2) percentage of patients that received their first analgesic dose within 30 minutes of triage, and (3) percentage of patients who had pain assessment performed within 30 minutes of triage and who were re-assessed within 30 minutes after the first analgesic dose.
- Results: Significant improvements were achieved between baseline (55 patient visits) and post order set implementation (165 visits) in time from triage to administration of first analgesic (decreased from 89.9 ± 50.5 to 35.2 ± 22.8 minutes, P < 0.001); percentage of patient visits receiving pain medications within 30 minutes of triage (from 7% to 53%, P < 0.001); percentage of patient visits assessed within 30 minutes of triage (from 64% to 99.4%, P < 0.001); and percentage of patient visits re-assessed within 30 minutes of initial analgesic (from 54% to 86%, P < 0.001).
- Conclusions: Implementation of a QI initiative in the ED led to expeditious care for pediatric patients with SCD presenting with pain. A QI framework provided us with unique challenges but also invaluable lessons as we address our objective of decreasing the quality gap in SCD medical care.
Pain is the leading cause of emergency department (ED) visits for patients with sickle cell disease (SCD) [1]. In the United States, 78% of the nearly 200,000 annual ED visits for SCD are for a complaint of pain [1]. Guidelines for the management of sickle cell vaso-occlusive pain episodes (VOE) suggest prompt initiation of parenteral opioids, use of effective opioid doses, and repeat opioid doses at frequent intervals [2–4]. Adherence to guidelines is poor. Both pediatric and adult patients with SCD experience delays in the initiation of analgesics and are routinely undertreated with respect to opioid dosing [5–8]. Even after controlling for race, the delays in time to analgesic administration experienced by patients with SCD exceed the delays encountered by patients who present to the ED with other types of pain [5,9]. These disparities warrant efforts designed to improve the delivery of quality care to patients with SCD.
Barriers to rapid and appropriate care of VOE in the ED are multifactorial and include systems-based limitations, such as acuity of the ED census, staffing limitations (eg, nurse-to-patient ratios), and facility limitations (eg, room availability) [6]. Provider-based limitations may include lack of awareness of available guidelines [10]. Biases and misunderstandings amongst providers about sickle cell pain and adequate medication dosing may also play a role [11–13]. These provider biases often lead to undertreatment of the pain, which in turn can lead to pseudoaddiction (drug-seeking behavior due to inadequate treatment) and a cycle of increased ED and inpatient utilization [14,15].
Patient-specific barriers to effective ED management of pain are equally complex. Previous negative experiences in the ED can lead patients and families to delay seeking care or avoid the ED altogether despite severe VOE pain [16]. Patients report frustration with the lack of consideration that they receive for their reports of pain, perceived insensitivity of hospital staff, inadequate analgesic administration, staff preoccupation with concerns of drug addiction, and an overall lack of respect and trust [17–19]. Patients also perceive a lack of knowledge of SCD and its treatments on the part of ED staff [7]. Other barriers to effective management are technical in nature, such as difficulty in establishing timely intravenous (IV) access.
Gaps and variations in quality of care contribute to poor outcomes for patients with SCD [20,21]. To help address these inequities, the Working to Improve Sickle Cell Healthcare (WISCH) project began in 2010 to improve care and outcomes for patients with SCD. WISCH is a collaborative quality improvement (QI) project funded by the Health Resources and Services Administration (HRSA) that has the goal to use improvement science to improve outcomes for patients with SCD across the life course (Ed note: see Editorial by Oyeku et al in this issue). As one of the HRSA-WISCH grantee networks, we undertook a QI project designed to decrease the quality gap in SCD medical care by creating and implementing a protocol for ED pain management for pediatric patients. Goals of the project were to improve the timely and appropriate assessment and treatment of acute VOE in the ED.
Methods
Setting
This ED QI initiative was implemented at Children’s Hospital & Research Center Oakland, an urban free-standing pediatric hospital that serves a demographically diverse population. The hospital ED sees over 45,000 visits per year, with 250 visits per year for VOE. Residents in pediatrics, family medicine, and emergency medicine staff the ED. All attending physicians are subspecialists in pediatric emergency medicine. Study procedures were approved by the hospital’s institutional review board.
Intervention
A multidisciplinary team consisting of ED staff and sickle cell center staff drafted a nursing-driven protocol for the assessment and management of acute pain associated with VOE, incorporating elements from a protocol in use by another WISCH collaborative member. The protocol called for the immediate triage and assessment of all patients with SCD who presented with moderate to severe pain suggestive of VOE. Moderate to severe pain was defined as a pain score of ≥ 5 on a numeric scale of 0 to 10, where 0 = no pain and 10 = the worst pain imaginable. Exclusion criteria included a chief complaint of pain not considered secondary to VOE (eg, trauma, fracture). Patients were also excluded if they had been transferred from another facility. The protocol called for IV pain medication to be administered within 10 minutes of the patient being roomed, with re-evaluation at 20-minute intervals and re-dosing of pain medication based on the patient’s subsequent pain rating.
Measures
We selected performance measures from the bank developed by the WISCH team to track improvement and evaluate progress. These performance measures included (1) mean time from triage to first analgesic dose, (2) percentage of patients that received their first dose of analgesic within 30 minutes of triage, (3) percentage of patients who had a pain assessment performed within 30 minutes of triage, and (4) percentage of patients re-assessed within 30 minutes after the first dose of analgesic had been administered. Our aims were to have 80% of patients assessed and given pain medications within 30 minutes of triage, and to have 80% of patients re-assessed within 30 minutes after having received their first dose of an analgesic, within 12 months of implementing our intervention.
Data Collection and Analysis
The WISCH project coordinator reviewed records of visits to the ED for a baseline period of 6 months and post-order set implementaton. Demographic data (age, gender), clinical data (hemoglobin type), pain scores, utilization data (number of ED visits during the study period), and data pertaining to the metrics chosen from the WISCH measurement bank were extracted from each eligible patient’s ED chart after the visit was completed. If patients were admitted, their length of hospitalization was extracted from their inpatient medical record.
All biostatistical analyses were conducted using Stata 9.2 (StataCorp, College Station, TX). Descriptive statistics computed at 2 time-points (pre and post order set implementation) were utilized to examine means, standard deviations and percentages. The 2 time-points were initially compared at the visit level of measurement, using Student’s t tests corrected for unequal variances where necessary for continuous variables and chi-square analyses for categorical variables, to evaluate if there was an improvement in timely triage, assessment, and treatment of acute VOE pain for all ED visits pre and post order set implementation. To account for trends and possible correlations across the months post order set implementation, we ran a mixed linear model with repeated measures over time to compare visits during all months post order set implementation with the baseline months, for metric 1, time from triage to first pain medication. If significant differences were found, we used Dunnett’s method of multiple comparisons to determine which months differed from baseline. For metrics 2 through 4, we ran linear models with a binary outcome, a logit link function and using general estimating equations to determine trends and to account for correlations over time.
Secondary analyses were conducted to evaluate whether mean pain scores were significantly different over the course of the ED visit for the 78 unique patients seen post order set implementation. A multivariable mixed linear model, for the outcome of the third pain score, was used to assess the associations with prior scores and to control for potential covariates (age, gender, number of ED visits, hemoglobin type) that were determined in advance. A statistical significance level of 0.05 was used for all tests.
Results
Baseline data were collected from December 2011 to May 2012. The protocol was implemented in July 2012 and was utilized during 165 ED visits (91% of eligible visits) through April 2013. There were no statistically significant differences in demographic or clinical characteristics between the 55 patients whose charts were reviewed prior to implementing the order set and the 78 unique patients treated thereafter. Pre order set implementation, the mean age was 14.6 ± 6.4 years; 60% were female and the primary diagnosis was HgbSS disease (61.8% of diagnoses). Post order set implementation, the mean age was 16.0 ± 8.0 years; 51.3% were female and the primary diagnosis was HgbSS disease (61.5% of diagnoses). The mean number of visits was 1.5 visits per patient with a range of 1–8 visits, both pre and post order set implementation. Thirty-one patients had ED visits at both time periods.
It can be seen in Figure 2 that staff performance on 3 of the 4 metrics (with the exception of initial analgesic within 30 minutes of triage) began to improve prior to implementing the order set. The mean length of ED stays decreased by 30 minutes, from a mean of 5.2 hours down to 4.7 hours (P < 0.05, Table). There was no significant change in the percentage of patients admitted to the inpatient unit.
We performed secondary analyses to determine if performance on our first metric, mean time from triage to first analgesic dose, was associated with any improvement on the third pain assessment for the patients enrolled post order set implementation. Looking at the first ED visit during the study period for the 78 unique patients, we found significant decreases in mean pain scores from the first to the second, from the second to the third, and from the first to the third assessment (P < 0.01). The mean pain scores were 8.3 ± 1.8, 5.9 ± 2.8, and 5.1 ± 3.0 on initial, second and third assessments, respectively. A multivariable model controlling for gender, hemoglobin type, number of ED visits and time to first pain medication showed that only the score at the second pain assessment (β = 0.88 ± 0.08, P < 0.001) was a significant predictor of the score at the third pain assessment.
Discussion
We demonstrated that a QI initiative to improve acute pain management resulted in more timely assessment and treatment of pain in pediatric patients with SCD. Significant improvements from baseline were achieved and sustained over a 10-month period in all 4 targeted metrics. We consistently exceeded our goal of having 80% of patients assessed within 30 minutes of triage, and our mean time to first pain medication (35.2 ± 22.8 minutes) came close to our goal of 30 minutes from triage. While we also achieved our goal to have 80% of patients re-assessed within 30 minutes after having received their first dose of an analgesic, we fell short in the percent who received their initial pain medication within 30 minutes of triage (52.7% versus goal of 80%). Although the length of stay in the ED decreased, no change was observed in the percentage of patients who required admission to the inpatient unit. A secondary analysis showed that mean pain scores significantly decreased over the course of the ED visit, from severe to moderate intensity.
The improvements that we observed began prior to implementation of the order set. We recognize that simply raising awareness and educating staff about the importance of timely and appropriate assessment and treatment of acute sickle cell related pain in the ED might be a potential confounder of our results. However, changes were sustained for 10 months post order set implementation and beyond, with no evidence that the performance on the target metrics is drifting back to baseline levels. Education and awareness-raising alone rarely result in sustained application of clinical practice guidelines [22]. We collaborated with NICHQ and other HRSA-WISCH grantees to systematically implement improvement science to ensure that the changes that we observed were indeed improvements and would be sustained [23] by first changing the system of care in the ED by introducing a standard order set [24,25]. We put a system into place to track use of the order set and to work with providers almost immediately if deviations were observed, to understand and overcome any barriers to the order set implementation. Systems in the ED and in the sickle cell center were aligned with the hospital’s QI initiatives [23].
Another strategy that we used to insure that the changes we observed would be sustained was to create a multidisciplinary team to build knowledge, skills, and new practices, including learning from other WISCH grantees and the NICHQ coordinating center [23]. We modified and adapted the intervention to our specific context [25]; although the outline of the order set was influenced by our WISCH colleagues, the final order set was structured to be consistent with other protocols within our institution. Finally, we included consumer input in the design of the project from the outset.
A previous study of a multi-institutional QI initiative aimed at improving acute SCD pain management for adult patients in the ED was unable to demonstrate an improvement in time to administration of initial analgesic [26]. Our study with pediatric patients was able to demonstrate a clinically meaningful decrease in the time to administration of first parenteral analgesic. The factors that account for the discrepant findings between these studies are likely multifactorial. Age (ie, pediatric vs. adult patients) may have played a role given that IV access may become increasingly difficult as patients with SCD age [26]. Education for providers should include the importance of alternative methods of administration of opioids, including subcutaneous and intranasal routes, to avoid delays when IV access is difficult. It is possible that negative provider attitudes converge with the documented increase in patient visits during the young adult years [27]. This may set up a challenging feedback loop wherein these vulnerable young adults are faced with greater stigma and consequently receive lower quality care, even when there is an attempt to carry out a standardized protocol.
We did not find that the QI intervention resulted in decreased admissions to the inpatient unit, with 68% of visits resulting in admission. In a recent pediatric SCD study, hospital admissions for pain control accounted for 78% of all admissions and 70% of readmissions within 30 days [28]. The investigators found that use of a SCD analgesic protocol including patient-controlled analgesia (PCA) improved quality of care as well as hospital readmission rates within 30 days (from 28% to 11%). Our ED QI protocol focused on only the first 90 minutes of the visit for pain. Our team has discussed the potential for starting the PCA in the ED and we should build on our success to focus on specific care that patients receive beyond their initial presentation. Further, we introduced pain action planning into outpatient care and need to continue to improve positive patient self-management strategies to ensure more seamless transition of pain management between home, ED, and inpatient settings.
Several valuable lessons were learned over the course of the ED QI initiative. Previous researchers [28] have emphasized the importance of coupling provider education with standardized order sets in efforts to improve the care of patients with SCD. Although we did not offer monthly formal education to our providers, the immediate follow-up when there were protocol deviations most likely served as teaching moments. These teaching moments also surfaced when some ED and hematology providers expressed concerns about the risk for oversedation with the rapid reassessment of pain and re-dosing of pain medications. Although rare, some parents also expressed that their child was being treated too vigorously with opioids. Our project highlighted the element of stigma that still accompanies the use of opioids for SCD pain management.
The project could not have been undertaken were it not for a small but determined multidisciplinary team of individuals who were personally invested in seeing the project come to fruition. The identification of physician and nurse champions who were enthusiastic about the project, invested in its conduct, and committed to its success was a cornerstone of the project’s success. These champions played an essential role in engaging staff interest in the project and oversaw the practicalities of implementing a new protocol in the ED. A spirit of collaboration, teamwork, and good communication between all involved parties was also critical. At the same time, we incorporated input from the treating ED and hematology clinicians using PDSA cycles as we were refining our protocol. We believe that our process enhanced buy-in from participating providers and clarified any issues that needed to be addressed in our setting, resulting in accelerated and sustained quality improvement.
Limitations
Although protocol-driven interventions are designed to provide a certain degree of uniformity of care, the protocol was not designed nor utilized in such a way that it superseded the best medical judgment of the treating clinicians. Deviations from the protocol were permissible when they were felt to be in the patient’s best interest. The study did not control for confounding variables such as disease severity, how long the patient had been in pain prior to coming to the ED, nor did we assess therapeutic interventions the patient had utilized at home prior to seeking out care in the ED. All of these factors could affect how well a patient might respond to treatment. We believe that sharing baseline data and monthly progress via run charts (graphs of data over time) with ED and sickle cell center staff and with consumer representatives enhanced the pace and focus of the project [23]. We had a dedicated person managing our data in real time through our HRSA funding, thus the project might not be generalizable to other institutions that do not have such staffing or access to the technology to allow project progress to be closely monitored by stakeholders.
Future Directions
With the goal of further reducing the time to administration of first analgesic dose in the ED setting, intranasal fentanyl will be utilized in our ED as the initial drug of choice for patients who do not object to or have a contraindication to its use. Collection of data from patients and family members is being undertaken to assess consumer satisfaction with the ED QI initiative. Recognizing that the ED management of acute pain addresses only one aspect of sickle cell pain, we are looking at ways to more comprehensively address pain. Individualized outpatient pain management plans are being created and patients and families are being encouraged and empowered to become active partners with their sickle cell providers in their own care. Although our initial efforts have focused on our pediatric patients, an additional aim of our project is to broaden the scope of our ED QI initiative to include community hospitals in the region that serve adult patients with SCD.
Conclusion
Implementation of a QI initiative in the ED has led to expeditious care for pediatric patients with SCD presenting with VOE. A multidisciplinary approach, ongoing staff education, and commitment to the initiative have been necessary to sustain the improvements. Our success can provide a template for other QI initiatives in the ED that translate to improved patient care for other diseases. A QI framework provided us with unique challenges but also invaluable lessons as we addressed our objective to improve outcomes for patients with SCD across the life course.
Acknowledgments: The authors wish to thank Theresa Freitas, RN, Lisa Hale, PNP, Carolyn Hoppe, MD, Ileana Mendez, RN, Helen Mitchell, Mary Rutherford, MD, Augusta Saulys, MD and the Children’s Hospital & Research Center Oakland Emergency Medicine Department and Sickle Cell Center for their support.
Corresponding author: Marsha Treadwell, PhD, Children’s Hospital & Research Center Oakland, 747 52nd St, Oakland, CA 94609, [email protected].
Funding/support: This research was conducted as part of the National Initiative for Children’s Healthcare Quality (NICHQ) Working to Improve Sickle Cell Healthcare (WISCH) project. Further support came from a grant from the Health Resources and Services Administration Sickle Cell Disease Treatment Demonstration Project Grant No. U1EMC16492 and from NIH CTSA grant UL1 RR024131. The views expressed in this publication do not necessarily reflect the views of WISCH, NICHQ, or HRSA.
1. Yusuf HR, Atrash HK, Grosse SD, et al. Emergency department visits made by patients with sickle cell disease: a descriptive study, 1999-2007. Am J Preventive Med 2010;38 (4 Suppl):S536–41.
2. Benjamin L, Dampier C, Jacox A, et al. Guideline for the management of acute and chronic pain in sickle cell disease. American Pain Society; 1999.
3. Rees DC, Olujohungbe AD, Parker NE, et al. Guidelines for the management of the acute painful crisis in sickle cell disease. Br J Haematology 2003;120:744–52.
4. Solomon LR. Pain management in adults with sickle cell disease in a medical center emergency department. J Nat Med Assoc 2010;102:1025–32.
5. Lazio MP, Costello HH, Courtney DM, et al. A comparison of analgesic management for emergency department patients with sickle cell disease and renal colic. Clin J Pain 2010;26:199–205.
6. Shenoi R, Ma L, Syblik D, Yusuf S. Emergency department crowding and analgesic delay in pediatric sickle cell pain crises. Ped Emerg Care 2011;27:911–7.
7. Tanabe P, Artz N, Mark Courtney D, et al. Adult emergency department patients with sickle cell pain crisis: a learning collaborative model to improve analgesic management. Acad Emerg Med 2010;17:399–407.
8. Zempsky WT. Evaluation and treatment of sickle cell pain in the emergency department: paths to a better future. Clin Ped Emerg Med 2010;11:265–73.
9. Haywood C Jr, Tanabe P, Naik R, et al. The impact of race and disease on sickle cell patient wait times in the emergency department. Am J Emerg Med 2013;31:651–6.
10. Solomon LR. Treatment and prevention of pain due to vaso-occlusive crises in adults with sickle cell disease: an educational void. Blood 2008;111:997–1003.
11. Ballas SK. New era dawns on sickle cell pain. Blood 2010;116:311–2.
12. Haywood C Jr, Lanzkron S, Ratanawongsa N, et al. The association of provider communication with trust among adults with sickle cell disease. J Gen Intern Med 2010;25:543–8.
13. Zempsky WT. Treatment of sickle cell pain: fostering trust and justice. JAMA 2009;302:2479–80.
14. Elander J, Lusher J, Bevan D, Telfer P. Pain management and symptoms of substance dependence among patients with sickle cell disease. Soc Sci Med 2003;57:1683–96.
15. Elander J, Lusher J, Bevan D, et al. Understanding the causes of problematic pain management in sickle cell disease: evidence that pseudoaddiction plays a more important role than genuine analgesic dependence. J Pain Sympt Manag 2004;27:156–69.
16. Smith WR, Penberthy LT, Bovbjerg VE, et al. Daily assessment of pain in adults with sickle cell disease. Ann Intern Med 2008;148:94–101.
17. Harris A, Parker N, Baker C. Adults with sickle cell. Psychol Health Med 1998;3:171–9.
18. Jenerette CM, Brewer C. Health-related stigma in young adults with sickle cell disease. J Nat Med Assoc 2010;102:1050–5.
19. Maxwell K, Streetly A, Bevan D. Experiences of hospital care and treatment seeking for pain from sickle cell disease: qualitative study. BMJ 1999;318:1585–90.
20. Oyeku SO, Wang CJ, Scoville R, et al. Hemoglobinopathy Learning Collaborative: using quality improvement (QI) to achieve equity in health care quality, coordination, and outcomes for sickle cell disease. J Health Care Poor Underserved 2012;23(3 Suppl):34–48.
21. Wang CJ, Kavanagh PL, Little AA, et al. Quality-of-care indicators for children with sickle cell disease. Pediatrics 2011;128:484–93.
22. Mansouri M, Lockyer J. A meta-analysis of continuing medical education effectiveness. J Contin Ed Health Prof 2007;27:6–15.
23. The breakthrough series: IHI’s collaborative model for achieving breakthrough improvement. Boston: Institute for Healthcare Improvement; 2003.
24. Berwick DM. Improvement, trust, and the healthcare workforce. Qual Safety Health Care 2003;12:448–52.
25. Hovlid E, Bukve O, Haug K, et al. Sustainability of healthcare improvement: what can we learn from learning theory? BMC Health Serv Res 2012;12:235.
26. Tanabe P, Hafner JW, Martinovich Z, Artz N. Adult emergency department patients with sickle cell pain crisis: results from a quality improvement learning collaborative model to improve analgesic management. Acad Emerg Med 2012;19:430–8.
27. Brousseau DC, Owens PL, Mosso AL, et al. Acute care utilization and rehospitalizations for sickle cell disease. JAMA 2010;303:1288–94.
28. Frei-Jones MJ, Field JJ, DeBaun MR. Multi-modal intervention and prospective implementation of standardized sickle cell pain admission orders reduces 30-day readmission rate. Pediatr Blood Cancer 2009;53:401–5.
1. Yusuf HR, Atrash HK, Grosse SD, et al. Emergency department visits made by patients with sickle cell disease: a descriptive study, 1999-2007. Am J Preventive Med 2010;38 (4 Suppl):S536–41.
2. Benjamin L, Dampier C, Jacox A, et al. Guideline for the management of acute and chronic pain in sickle cell disease. American Pain Society; 1999.
3. Rees DC, Olujohungbe AD, Parker NE, et al. Guidelines for the management of the acute painful crisis in sickle cell disease. Br J Haematology 2003;120:744–52.
4. Solomon LR. Pain management in adults with sickle cell disease in a medical center emergency department. J Nat Med Assoc 2010;102:1025–32.
5. Lazio MP, Costello HH, Courtney DM, et al. A comparison of analgesic management for emergency department patients with sickle cell disease and renal colic. Clin J Pain 2010;26:199–205.
6. Shenoi R, Ma L, Syblik D, Yusuf S. Emergency department crowding and analgesic delay in pediatric sickle cell pain crises. Ped Emerg Care 2011;27:911–7.
7. Tanabe P, Artz N, Mark Courtney D, et al. Adult emergency department patients with sickle cell pain crisis: a learning collaborative model to improve analgesic management. Acad Emerg Med 2010;17:399–407.
8. Zempsky WT. Evaluation and treatment of sickle cell pain in the emergency department: paths to a better future. Clin Ped Emerg Med 2010;11:265–73.
9. Haywood C Jr, Tanabe P, Naik R, et al. The impact of race and disease on sickle cell patient wait times in the emergency department. Am J Emerg Med 2013;31:651–6.
10. Solomon LR. Treatment and prevention of pain due to vaso-occlusive crises in adults with sickle cell disease: an educational void. Blood 2008;111:997–1003.
11. Ballas SK. New era dawns on sickle cell pain. Blood 2010;116:311–2.
12. Haywood C Jr, Lanzkron S, Ratanawongsa N, et al. The association of provider communication with trust among adults with sickle cell disease. J Gen Intern Med 2010;25:543–8.
13. Zempsky WT. Treatment of sickle cell pain: fostering trust and justice. JAMA 2009;302:2479–80.
14. Elander J, Lusher J, Bevan D, Telfer P. Pain management and symptoms of substance dependence among patients with sickle cell disease. Soc Sci Med 2003;57:1683–96.
15. Elander J, Lusher J, Bevan D, et al. Understanding the causes of problematic pain management in sickle cell disease: evidence that pseudoaddiction plays a more important role than genuine analgesic dependence. J Pain Sympt Manag 2004;27:156–69.
16. Smith WR, Penberthy LT, Bovbjerg VE, et al. Daily assessment of pain in adults with sickle cell disease. Ann Intern Med 2008;148:94–101.
17. Harris A, Parker N, Baker C. Adults with sickle cell. Psychol Health Med 1998;3:171–9.
18. Jenerette CM, Brewer C. Health-related stigma in young adults with sickle cell disease. J Nat Med Assoc 2010;102:1050–5.
19. Maxwell K, Streetly A, Bevan D. Experiences of hospital care and treatment seeking for pain from sickle cell disease: qualitative study. BMJ 1999;318:1585–90.
20. Oyeku SO, Wang CJ, Scoville R, et al. Hemoglobinopathy Learning Collaborative: using quality improvement (QI) to achieve equity in health care quality, coordination, and outcomes for sickle cell disease. J Health Care Poor Underserved 2012;23(3 Suppl):34–48.
21. Wang CJ, Kavanagh PL, Little AA, et al. Quality-of-care indicators for children with sickle cell disease. Pediatrics 2011;128:484–93.
22. Mansouri M, Lockyer J. A meta-analysis of continuing medical education effectiveness. J Contin Ed Health Prof 2007;27:6–15.
23. The breakthrough series: IHI’s collaborative model for achieving breakthrough improvement. Boston: Institute for Healthcare Improvement; 2003.
24. Berwick DM. Improvement, trust, and the healthcare workforce. Qual Safety Health Care 2003;12:448–52.
25. Hovlid E, Bukve O, Haug K, et al. Sustainability of healthcare improvement: what can we learn from learning theory? BMC Health Serv Res 2012;12:235.
26. Tanabe P, Hafner JW, Martinovich Z, Artz N. Adult emergency department patients with sickle cell pain crisis: results from a quality improvement learning collaborative model to improve analgesic management. Acad Emerg Med 2012;19:430–8.
27. Brousseau DC, Owens PL, Mosso AL, et al. Acute care utilization and rehospitalizations for sickle cell disease. JAMA 2010;303:1288–94.
28. Frei-Jones MJ, Field JJ, DeBaun MR. Multi-modal intervention and prospective implementation of standardized sickle cell pain admission orders reduces 30-day readmission rate. Pediatr Blood Cancer 2009;53:401–5.
Swelling and pain 2 weeks after a dog bite
A 48-year-old man with gout, multiple sclerosis, and previously treated methicillin-resistant Staphylococcus aureus (MRSA) infection presented to the emergency room with pain and significant swelling at the site of a dog bite on his left forearm. He had been bitten 2 weeks earlier by a friend’s dog, and the bite had punctured the skin. He also had red streaking on the skin of the left arm from the wrist to the elbow, and he reported feeling “feverish” and having night sweats.
At first, the bite had seemed to improve, then swelling and pain had developed and increased. He reported this to his primary care physician, along with the information that he had previously had an anaphylactic reaction to penicillin and a cephalosporin. His physician, considering a penicillin allergy, started him on ciprofloxacin (Cipro) plus clindamycin (Cleocin). The patient took this for 5 days, but without improvement. The appearance of the red streaking on his left forearm prompted his presentation to our emergency room.
ORGANISMS IN DOG BITES
1. Which is the most common cause of infected dog bite?
- Pasteurella canis
- Streptococci and S aureus
- Erysipelothrix rhusiopathiae
- Capnocytophaga canimorsus
- Eikenella corrodens
Streptococci (50%) and S aureus (20% to 40%) are the organisms most commonly responsible for infected dog bites, as they are for other skin and soft-tissue infections.1P canis is unique to dog bite infections but accounts for only 18%.2E rhusiopathiae is an unusual isolate from cat and dog bites and is more commonly isolated from the mouths of fish and aquatic mammals. C canimorsus is a normal inhabitant of the oral cavity of dogs and cats but an unusual cause of wound infection from a dog bite. It is notable for sepsis and central nervous system infections uniquely associated with veterinarians, dog owners, kennel workers, and mail carriers.3E corrodens infection is more common with human bites.4
THE EVALUATION BEGINS
On examination, the patient had marked edema of the left forearm and pain in the joints of the left hand. His temperature was 100.2°F (37.9°C). Because of the duration and severity of symptoms, the examining physician was concerned about septic arthritis of the wrist, and the patient was admitted to the hospital.
In the hospital, our patient was thermodynamically stable without documented fever or chills. There was no open wound to culture, and blood cultures were negative. Marked edema and joint involvement raised suspicion of erysipeloid. This “cousin” of erysipelas often involves the underlying joint, is associated with edema, and produces systemic manifestations of fever and arthralgia.
Radiography of the left forearm and hand demonstrated multiple foci of demineralization within the carpal bones and proximal radius, attributed to disuse. Magnetic resonance imaging (MRI) the next day showed multiple bone infarcts in the carpal bones and the distal radius, with synovitis and fluid in the carpal joints and without adjacent osteomyelitis. Fluid was also seen in the soft tissues in the ulnar aspect of the left wrist, and tenosynovitis involving the flexor carpi radialis tendon was noted.
Arthrocentesis of his left radiocarpal joint produced synovial fluid negative for crystals and negative on Gram stain; the fluid was also sent for culture. The patient’s tetanus immunization was current, and the dog was known to have been immunized against rabies.
ANTIBIOTICS FOR INFECTED DOG BITES
2. Which antibiotic regimen would you choose for this patient?
- Oral amoxicillin and clavulanate
- Meropenem
- Vancomycin, clindamycin, aztreonam
- Clindamycin and levofloxacin
- Clindamycin and trimethoprim-sulfamethoxazole
Oral amoxicillin and clavulanate (Augmentin) is a judicious choice for prophylactic treatment of deep bites in the early stages of infection. However, our patient’s wound was no longer in the early stages of infection, and he had a history of an adverse reaction to penicillin.
Meropenem (Merrem IV) cross-reacts minimally with penicillin allergy and is reported to be safe in patients with a history of anaphylactic reactions to penicillin,5 but overuse of carbapenems has led to the development of carbapenem-resistant strains of Klebsiella, Stenotrophomonas, and Acinetobacter organisms.
Given the rise of MRSA infections and the common involvement of staphylococci, streptococci, and anaerobic bacteria in complicated dog bites, the combination of vancomycin and clindamycin is a good choice, and aztreonam (Azactam) would add empiric coverage of gram-negative enteric organisms.
Levofloxacin (Levaquin) also covers gramnegative enteric organisms, but Fusobacterium canifelinum, a common anaerobe in the oral flora of dogs and cats, is intrinsically resistant to fluoroquinolones.
Clindamycin and levofloxacin would be a good step-down oral regimen. Pasteurella multocida has variable sensitivity to the commonly used agents dicloxacillin (Dynapen), cephalexin (Keflex), macrolides, and clindamycin, but it is a less likely pathogen at this late stage and could be covered with levofloxacin alone.
C canimorsus is resistant to trimethoprim-sulfamethoxazole (Bactrim) and cephalexin, but is well covered by clindamycin.6
CASE CONTINUED
Our patient was started on intravenous vancomycin, clindamycin, and aztreonam for coverage of dog-mouth flora. Blood cultures and cultures of synovial fluid of the left wrist were negative. Vancomycin was discontinued after 48 hours when blood cultures did not grow staphylococcal organisms, but clindamycin and aztreonam were continued for a total of 8 days to treat possible infection with anaerobic and gram-negative enteric pathogens.
To test for autonomic dysfunction, a plastic pen case drawn lightly across each forearm revealed a loss of tactile adherence (ie, areas where moist, sweaty skin impeded the movement of the pen case) on the affected forearm, a sign of underlying nerve injury. The affected forearm was sensitive to light touch, with pain out of proportion to the stimulus.
ARRIVING AT THE DIAGNOSIS
Based on the wide distribution of inflammation, autonomic dysfunction (shown by differences in temperature and sweating), radiographic evidence of demineralization, hyperesthesia, and lack of improvement in pain and swelling after two courses of antibiotics, the patient’s clinical course was determined to be consistent with complex regional pain syndrome type 1, previously referred to as reflex sympathetic dystrophy.
Symptoms of complex regional pain syndrome traditionally include pain, regional edema, joint stiffness, muscular atrophy, vasomotor disturbances (causing temperature variability and erythema), regional diaphoresis, and localized skeletal demineralization on radiography.
Complex regional pain syndrome type 1 occurs as regional pain and inflammation as an excessive sympathetic reaction to an often minor insult, without nerve injury. When the syndrome occurs in a patient with obvious partial nerve injury, it is categorized as type 2 (formerly known as causalgia). The two types are clinically indistinguishable and are not uncommon. About 10% of all patients with complex regional pain syndrome have obvious nerve injury (complex regional pain syndrome type 2). In a study of 109 patients with Colles fracture, 25% developed symptoms of complex regional pain syndrome.7
Complex regional pain syndrome is difficult to diagnose, as it resembles many other ailments, such as gout, infection, bone tumor, stress fracture, and arthritis. Its pathophysiology is poorly understood, but it is believed to result from a “short circuit” in the reflex arc between somatic afferent sensory fibers and autonomic sympathetic efferent fibers, and this is thought to explain the increased sympathetic stimulation.
Although the pathophysiology is likely the same in type 1 and type 2, electromyography with a nerve conduction study is a reliable way to detect nerve damage and thus distinguish between the two types of complex regional pain syndrome.8
Our understanding of this syndrome is evolving. A recent study using sensory testing showed that 33% of patients with type 1 had combinations of increased and decreased thresholds for the detection of thermal, vibratory, and mechanical stimuli in the distribution of discrete peripheral nerves, suggesting that the patients actually had type 2.9
CONFIRMING COMPLEX REGIONAL PAIN SYNDROME TYPE 1
3. Which of the following is the best way to confirm complex regional pain syndrome type 1?
- Erythrocyte sedimentation rate, C-reactive protein, and complete blood cell count
- Plain radiography of the hand and forearm
- Three-phase technetium bone scan
- The Budapest diagnostic criteria
- MRI
- Autonomic testing

Complex regional pain syndrome type 1 is a clinical diagnosis. Diagnostic studies lack sensitivity and specificity but may confirm complex regional pain syndrome type 1 or rule out other diagnoses. The Budapest diagnostic criteria10 (Table 1) may be the best way to confirm this diagnosis. The criteria are as follows: continuing pain disproportionate to an inciting event, coupled with three of four symptoms plus at least one sign from sensory, vasomotor, sudomotor, and motor-trophic categories.
Laboratory tests are not helpful because acute-phase reactants and blood counts remain normal in these patients.
Plain radiography is not sensitive in early diagnosis, but at 2 weeks it may show patchy areas of osteopenia in adjacent bones throughout the region, as well as subsequent diffuse demineralization.
Three-phase bone scanning is more sensitive than plain radiography, with 75% of patients showing regional disparities in blood flow in early sequences and increased bone uptake in the later sequences.
MRI is a sensitive early test, as it better defines focal areas of bone loss and increased T2 bone signal in adjacent bone, as well as early soft-tissue changes. Computed tomography does not show early specific changes in muscle, tendon, or bone and so is not recommended.
THE EVALUATION CONTINUES
The admitting diagnosis was septic arthritis, and our patient underwent computed tomography, which showed focal demineralization that could have represented bone infarcts or infection, confounding the diagnosis of complex regional pain syndrome.
Autonomic nerve testing can help distinguish complex regional pain syndrome from other disorders. Complex regional pain syndrome is characterized by increased sympathetic activity and results in increased sweat output. Autonomic testing includes resting sweat output, resting skin temperature, and quantitative sudomotor axon reflex testing. In one study, an increase in resting sweat output used in conjunction with quantitative sudomotor axon reflex testing predicted the diagnosis of complex regional pain syndrome with a specificity of 98%.11 However, autonomic testing is limited to academic centers and is not readily available.
TREATING COMPLEX REGIONAL PAIN SYNDROME TYPE 1
4. Which is the best first-line therapy for complex regional pain syndrome type 1?
- Stellate ganglion nerve block
- Occupational therapy to splint the wrist and forearm
- Oral corticosteroids
- Physical therapy to prevent loss of joint motion
- Tricyclic antidepressant drugs (eg, amitriptyline), pregabalin, and bisphosphonates
Physical therapy should be started early in all patients, with range-of-motion exercises to prevent contracture and enhance mobility.
Stellate ganglion nerve block has been used to counter severe sympathetic hyperactivity, but it also may aggravate symptoms of complex regional pain syndrome and so remains a controversial treatment.
Immobilization and splinting should be avoided, as this will augment edema, pain, and contracture of joints.
Corticosteroids do not shorten the course or assuage symptoms and may increase edema.
Amitriptyline (Elavil) and pregabalin (Lyrica) have been used successfully to counter extended courses of allodynia and hyperalgesia. Bisphosphonates may decrease bone loss and pain and may be needed should the course be complicated by myositis ossificans, a form of dystrophic bone formation in juxtaposed tendon and muscle related to neuroactivation of fibroblasts and osteoblasts.
THE COURSE OF COMPLEX REGIONAL PAIN SYNDROME
Traditionally, type 1 was divided into three stages—an early inflammatory stage, a dystrophic stage, and a late atrophic stage.12 Although there is no evidence to support a consistent three-stage evolution, the severity of symptoms may help determine the best approach to management.13
Patients initially exhibit burning or throbbing pain, diffuse aching, sensitivity to touch or cold (allodynia), localized edema, and vasomotor disturbances of variable intensity that may produce altered color and temperature. Topical capsaicin cream; a tricyclic antidepressant; an anticonvulsant such as gabapentin (Neurontin), pregabalin, or lamotrigine (Lamictal); or a nonsteroidal anti-inflammatory drug should be tried first. Some of these treatments are poorly tolerated in elderly patients. If pain persists, nasal calcitonin may be added. Trigger-point injections with an anesthetic or glucocorticoid may be tried.
The management of early complex regional pain syndrome is sometimes supplemented with systemic corticosteroids, but reviews of randomized controlled trials have failed to show efficacy.14
Later in the course, patients may suffer persistent soft-tissue edema, accompanied by thickening of the skin and periarticular soft tissues, muscle wasting, and the skin changes of brawny edema. Regional blockade of sympathetic ganglions, epidural administration of clonidine, implantable peripheral nerve stimulators, and spinal cord stimulators have all been applied by experts in pain management and may provide benefit. Progression of the syndrome may include cyanosis, mottling, increased sweating, abnormal hair growth, and diffuse swelling in nonarticular tissue.
It is always acceptable to refer to an experienced pain management specialist, and a multidisciplinary approach is recommended at the outset.12
OUR PATIENT’S CARE CONTINUED
Our patient’s forearm and wrist were placed in a sling to keep his left arm elevated when active. This helped control sympathetic vascular edema and throbbing pain. Physical therapy with range-of-motion exercises prevented contracture.
He was discharged home on limited oxycodone as needed, with close follow-up by his primary care physician to monitor his pain symptoms. The pain and swelling slowly improved over the next 2 months, but he suffered a fall, twisting his left wrist. This minor injury was followed by more intense pain and swelling of the forearm, hand, and wrist.
COMORBIDITIES
5. Which of the following statements about conditions associated with complex regional pain syndrome most likely applies to our patient?
- Gout is likely following minor trauma
- Minor trauma or surgical bone biopsy may reactivate complex regional pain syndrome
- Septic hip arthritis due to MRSA may have reemerged and seeded the wrist
- Patients with multiple sclerosis have a propensity for complex regional pain syndrome
- Complex regional pain syndrome type 1 begets type 2
Gout does follow minor injury, but our patient’s uric acid was well controlled on allopurinol (Zyloprim), and gout is unlikely to be causing polyarticular swelling of the hand, wrist, and forearm.
Minor trauma, sometimes inconsequential enough to have been completely forgotten, may either initiate complex regional pain syndrome or, as seen here, reactivate it. Bone changes seen on MRI sometimes trigger surgical bone biopsy, only to reactivate the dysesthesia and sympathetic vascular reaction. Surgery should be avoided. Trauma and surgery are causative rather than associative comorbidities.
Sepsis due to MRSA after total hip arthroplasty may be reactivated, especially in the setting of immunosuppressive treatment. But the diffuse bone changes seen in multiple carpal, radial, and ulnar bones suggest generalized vascular and sympathetic disarray, most consistent with complex regional pain syndrome type 1.
AN ASSOCIATION WITH MULTIPLE SCLEROSIS?
Multiple sclerosis and other central neuropathic conditions such as stroke are associated with complex regional pain syndrome type 1.15,16
A hypothetical cause for the higher prevalence of complex regional pain syndrome in patients with multiple sclerosis may be demyelination resulting in aberrant signaling and overreaction to distal pain receptors. Demyelination of neurons within the autonomic or spinothalamic tracts potentially increases susceptibility to development of the pain syndrome.
Our patient had an apparent stimulus for the development of the syndrome, ie, the initial dog bite, and the wrist injury later may have caused peripheral nerve injury. Such injury may lead to release of vasodilatory neuropeptides including substance P from stimulated cutaneous nerves with cell bodies in the dorsal root ganglia. Excessive vasodilation and increased vascular permeability result in the affected limb becoming edematous and causing cutaneous nerves to be further activated. Stimulated cutaneous neurons normally have an inhibitory influence on sympathetic activity at the level of entry of the dorsal root ganglia in the cord. In complex regional pain syndrome, this inhibition is lost, resulting in a hyperactive somatosympathetic reflex.17 Underlying multiple sclerosis may have contributed to the loss of inhibition by the cutaneous nerves on the sympathetic system.
CASE CONCLUDED
We continued to closely follow this patient, who was on a self-directed program of physical therapy. One year after the original dog bite, the complex regional pain syndrome had completely resolved.
- Talan DA, Citron DM, Abrahamian FM, Moran GJ, Goldstein EJ. Bacteriologic analysis of infected dog and cat bites. Emergency Medicine Animal Bite Infection Study Group. N Engl J Med 1999; 340:85–92.
- Holst E, Rollof J, Larsson L, Nielsen JP. Characterization and distribution of Pasteurella species recovered from infected humans. J Clin Microbiol 1992; 30:2984–2987.
- Jolivet-Gougeon A, Sixou JL, Tamanai-Shacoori Z, Bonnaure-Mallet M. Antimicrobial treatment of Capnocytophaga infections. Int J Antimicrob Agents 2007; 29:367–373.
- Paul K, Patel SS. Eikenella corrodens infections in children and adolescents: case reports and review of the literature. Clin Infect Dis 2001; 33:54–61.
- Cunha BA, Hamid NS, Krol V, Eisenstein L. Safety of meropenem in patients reporting penicillin allergy: lack of allergic cross reactions. J Chemother 2008; 20:233–237.
- Verghese A, Hamati F, Berk S, Franzus B, Berk S, Smith JK. Susceptibility of dysgonic fermenter 2 to antimicrobial agents in vitro. Antimicrob Agents Chemother 1988; 32:78–80.
- Atkins RM, Duckworth T, Kanis JA. Algodystrophy following Colles’ fracture. J Hand Surg Br 1989; 14:161–164.
- Rommel O, Malin JP, Zenz M, Jänig W. Quantitative sensory testing, neurophysiological and psychological examination in patients with complex regional pain syndrome and hemisensory deficits. Pain 2001; 93:279–293.
- Sethna NF, Meier PM, Zurakowski D, Berde CB. Cutaneous sensory abnormalities in children and adolescents with complex regional pain syndromes. Pain 2007; 131:153–161.
- Harden RN, Bruehl S, Stanton-Hicks M, Wilson PR. Proposed new diagnostic criteria for complex regional pain syndrome. Pain Med 2007; 8:326–331.
- Chelimsky TC, Low PA, Naessens JM, Wilson PR, Amadio PC, O’Brien PC. Value of autonomic testing in reflex sympathetic dystrophy. Mayo Clin Proc 1995; 70:1029–1040.
- Stanton-Hicks MD, Burton AW, Bruehl SP, et al. An updated interdisciplinary clinical pathway for CRPS: report of an expert panel. Pain Pract 2002; 2:1–16.
- Brummett CM, Cohen SP, eds. Managing pain: essentials of diagnosis and treatment. New York; Oxford University Press; 2013.
- Dirckx M, Stronks DL, Groeneweg G, Huygen FJ. Effect of immunomodulating medications in complex regional pain syndrome: a systematic review. Clin J Pain 2012; 28:355–363.
- Schwartzman RJ, Gurusinghe C, Gracely E. Prevalence of complex regional pain syndrome in a cohort of multiple sclerosis patients. Pain Physician 2008; 11:133–136.
- Sandroni P, Benrud-Larson LM, McClelland RL, Low PA. Complex regional pain syndrome type I: incidence and prevalence in Olmsted county, a population-based study. Pain 2003; 103:199–207.
- Kurvers HA, Jacobs MJ, Beuk RJ, et al. Reflex sympathetic dystrophy: evolution of microcirculatory disturbances in time. Pain 1995; 60:333–340.
A 48-year-old man with gout, multiple sclerosis, and previously treated methicillin-resistant Staphylococcus aureus (MRSA) infection presented to the emergency room with pain and significant swelling at the site of a dog bite on his left forearm. He had been bitten 2 weeks earlier by a friend’s dog, and the bite had punctured the skin. He also had red streaking on the skin of the left arm from the wrist to the elbow, and he reported feeling “feverish” and having night sweats.
At first, the bite had seemed to improve, then swelling and pain had developed and increased. He reported this to his primary care physician, along with the information that he had previously had an anaphylactic reaction to penicillin and a cephalosporin. His physician, considering a penicillin allergy, started him on ciprofloxacin (Cipro) plus clindamycin (Cleocin). The patient took this for 5 days, but without improvement. The appearance of the red streaking on his left forearm prompted his presentation to our emergency room.
ORGANISMS IN DOG BITES
1. Which is the most common cause of infected dog bite?
- Pasteurella canis
- Streptococci and S aureus
- Erysipelothrix rhusiopathiae
- Capnocytophaga canimorsus
- Eikenella corrodens
Streptococci (50%) and S aureus (20% to 40%) are the organisms most commonly responsible for infected dog bites, as they are for other skin and soft-tissue infections.1P canis is unique to dog bite infections but accounts for only 18%.2E rhusiopathiae is an unusual isolate from cat and dog bites and is more commonly isolated from the mouths of fish and aquatic mammals. C canimorsus is a normal inhabitant of the oral cavity of dogs and cats but an unusual cause of wound infection from a dog bite. It is notable for sepsis and central nervous system infections uniquely associated with veterinarians, dog owners, kennel workers, and mail carriers.3E corrodens infection is more common with human bites.4
THE EVALUATION BEGINS
On examination, the patient had marked edema of the left forearm and pain in the joints of the left hand. His temperature was 100.2°F (37.9°C). Because of the duration and severity of symptoms, the examining physician was concerned about septic arthritis of the wrist, and the patient was admitted to the hospital.
In the hospital, our patient was thermodynamically stable without documented fever or chills. There was no open wound to culture, and blood cultures were negative. Marked edema and joint involvement raised suspicion of erysipeloid. This “cousin” of erysipelas often involves the underlying joint, is associated with edema, and produces systemic manifestations of fever and arthralgia.
Radiography of the left forearm and hand demonstrated multiple foci of demineralization within the carpal bones and proximal radius, attributed to disuse. Magnetic resonance imaging (MRI) the next day showed multiple bone infarcts in the carpal bones and the distal radius, with synovitis and fluid in the carpal joints and without adjacent osteomyelitis. Fluid was also seen in the soft tissues in the ulnar aspect of the left wrist, and tenosynovitis involving the flexor carpi radialis tendon was noted.
Arthrocentesis of his left radiocarpal joint produced synovial fluid negative for crystals and negative on Gram stain; the fluid was also sent for culture. The patient’s tetanus immunization was current, and the dog was known to have been immunized against rabies.
ANTIBIOTICS FOR INFECTED DOG BITES
2. Which antibiotic regimen would you choose for this patient?
- Oral amoxicillin and clavulanate
- Meropenem
- Vancomycin, clindamycin, aztreonam
- Clindamycin and levofloxacin
- Clindamycin and trimethoprim-sulfamethoxazole
Oral amoxicillin and clavulanate (Augmentin) is a judicious choice for prophylactic treatment of deep bites in the early stages of infection. However, our patient’s wound was no longer in the early stages of infection, and he had a history of an adverse reaction to penicillin.
Meropenem (Merrem IV) cross-reacts minimally with penicillin allergy and is reported to be safe in patients with a history of anaphylactic reactions to penicillin,5 but overuse of carbapenems has led to the development of carbapenem-resistant strains of Klebsiella, Stenotrophomonas, and Acinetobacter organisms.
Given the rise of MRSA infections and the common involvement of staphylococci, streptococci, and anaerobic bacteria in complicated dog bites, the combination of vancomycin and clindamycin is a good choice, and aztreonam (Azactam) would add empiric coverage of gram-negative enteric organisms.
Levofloxacin (Levaquin) also covers gramnegative enteric organisms, but Fusobacterium canifelinum, a common anaerobe in the oral flora of dogs and cats, is intrinsically resistant to fluoroquinolones.
Clindamycin and levofloxacin would be a good step-down oral regimen. Pasteurella multocida has variable sensitivity to the commonly used agents dicloxacillin (Dynapen), cephalexin (Keflex), macrolides, and clindamycin, but it is a less likely pathogen at this late stage and could be covered with levofloxacin alone.
C canimorsus is resistant to trimethoprim-sulfamethoxazole (Bactrim) and cephalexin, but is well covered by clindamycin.6
CASE CONTINUED
Our patient was started on intravenous vancomycin, clindamycin, and aztreonam for coverage of dog-mouth flora. Blood cultures and cultures of synovial fluid of the left wrist were negative. Vancomycin was discontinued after 48 hours when blood cultures did not grow staphylococcal organisms, but clindamycin and aztreonam were continued for a total of 8 days to treat possible infection with anaerobic and gram-negative enteric pathogens.
To test for autonomic dysfunction, a plastic pen case drawn lightly across each forearm revealed a loss of tactile adherence (ie, areas where moist, sweaty skin impeded the movement of the pen case) on the affected forearm, a sign of underlying nerve injury. The affected forearm was sensitive to light touch, with pain out of proportion to the stimulus.
ARRIVING AT THE DIAGNOSIS
Based on the wide distribution of inflammation, autonomic dysfunction (shown by differences in temperature and sweating), radiographic evidence of demineralization, hyperesthesia, and lack of improvement in pain and swelling after two courses of antibiotics, the patient’s clinical course was determined to be consistent with complex regional pain syndrome type 1, previously referred to as reflex sympathetic dystrophy.
Symptoms of complex regional pain syndrome traditionally include pain, regional edema, joint stiffness, muscular atrophy, vasomotor disturbances (causing temperature variability and erythema), regional diaphoresis, and localized skeletal demineralization on radiography.
Complex regional pain syndrome type 1 occurs as regional pain and inflammation as an excessive sympathetic reaction to an often minor insult, without nerve injury. When the syndrome occurs in a patient with obvious partial nerve injury, it is categorized as type 2 (formerly known as causalgia). The two types are clinically indistinguishable and are not uncommon. About 10% of all patients with complex regional pain syndrome have obvious nerve injury (complex regional pain syndrome type 2). In a study of 109 patients with Colles fracture, 25% developed symptoms of complex regional pain syndrome.7
Complex regional pain syndrome is difficult to diagnose, as it resembles many other ailments, such as gout, infection, bone tumor, stress fracture, and arthritis. Its pathophysiology is poorly understood, but it is believed to result from a “short circuit” in the reflex arc between somatic afferent sensory fibers and autonomic sympathetic efferent fibers, and this is thought to explain the increased sympathetic stimulation.
Although the pathophysiology is likely the same in type 1 and type 2, electromyography with a nerve conduction study is a reliable way to detect nerve damage and thus distinguish between the two types of complex regional pain syndrome.8
Our understanding of this syndrome is evolving. A recent study using sensory testing showed that 33% of patients with type 1 had combinations of increased and decreased thresholds for the detection of thermal, vibratory, and mechanical stimuli in the distribution of discrete peripheral nerves, suggesting that the patients actually had type 2.9
CONFIRMING COMPLEX REGIONAL PAIN SYNDROME TYPE 1
3. Which of the following is the best way to confirm complex regional pain syndrome type 1?
- Erythrocyte sedimentation rate, C-reactive protein, and complete blood cell count
- Plain radiography of the hand and forearm
- Three-phase technetium bone scan
- The Budapest diagnostic criteria
- MRI
- Autonomic testing
Complex regional pain syndrome type 1 is a clinical diagnosis. Diagnostic studies lack sensitivity and specificity but may confirm complex regional pain syndrome type 1 or rule out other diagnoses. The Budapest diagnostic criteria10 (Table 1) may be the best way to confirm this diagnosis. The criteria are as follows: continuing pain disproportionate to an inciting event, coupled with three of four symptoms plus at least one sign from sensory, vasomotor, sudomotor, and motor-trophic categories.
Laboratory tests are not helpful because acute-phase reactants and blood counts remain normal in these patients.
Plain radiography is not sensitive in early diagnosis, but at 2 weeks it may show patchy areas of osteopenia in adjacent bones throughout the region, as well as subsequent diffuse demineralization.
Three-phase bone scanning is more sensitive than plain radiography, with 75% of patients showing regional disparities in blood flow in early sequences and increased bone uptake in the later sequences.
MRI is a sensitive early test, as it better defines focal areas of bone loss and increased T2 bone signal in adjacent bone, as well as early soft-tissue changes. Computed tomography does not show early specific changes in muscle, tendon, or bone and so is not recommended.
THE EVALUATION CONTINUES
The admitting diagnosis was septic arthritis, and our patient underwent computed tomography, which showed focal demineralization that could have represented bone infarcts or infection, confounding the diagnosis of complex regional pain syndrome.
Autonomic nerve testing can help distinguish complex regional pain syndrome from other disorders. Complex regional pain syndrome is characterized by increased sympathetic activity and results in increased sweat output. Autonomic testing includes resting sweat output, resting skin temperature, and quantitative sudomotor axon reflex testing. In one study, an increase in resting sweat output used in conjunction with quantitative sudomotor axon reflex testing predicted the diagnosis of complex regional pain syndrome with a specificity of 98%.11 However, autonomic testing is limited to academic centers and is not readily available.
TREATING COMPLEX REGIONAL PAIN SYNDROME TYPE 1
4. Which is the best first-line therapy for complex regional pain syndrome type 1?
- Stellate ganglion nerve block
- Occupational therapy to splint the wrist and forearm
- Oral corticosteroids
- Physical therapy to prevent loss of joint motion
- Tricyclic antidepressant drugs (eg, amitriptyline), pregabalin, and bisphosphonates
Physical therapy should be started early in all patients, with range-of-motion exercises to prevent contracture and enhance mobility.
Stellate ganglion nerve block has been used to counter severe sympathetic hyperactivity, but it also may aggravate symptoms of complex regional pain syndrome and so remains a controversial treatment.
Immobilization and splinting should be avoided, as this will augment edema, pain, and contracture of joints.
Corticosteroids do not shorten the course or assuage symptoms and may increase edema.
Amitriptyline (Elavil) and pregabalin (Lyrica) have been used successfully to counter extended courses of allodynia and hyperalgesia. Bisphosphonates may decrease bone loss and pain and may be needed should the course be complicated by myositis ossificans, a form of dystrophic bone formation in juxtaposed tendon and muscle related to neuroactivation of fibroblasts and osteoblasts.
THE COURSE OF COMPLEX REGIONAL PAIN SYNDROME
Traditionally, type 1 was divided into three stages—an early inflammatory stage, a dystrophic stage, and a late atrophic stage.12 Although there is no evidence to support a consistent three-stage evolution, the severity of symptoms may help determine the best approach to management.13
Patients initially exhibit burning or throbbing pain, diffuse aching, sensitivity to touch or cold (allodynia), localized edema, and vasomotor disturbances of variable intensity that may produce altered color and temperature. Topical capsaicin cream; a tricyclic antidepressant; an anticonvulsant such as gabapentin (Neurontin), pregabalin, or lamotrigine (Lamictal); or a nonsteroidal anti-inflammatory drug should be tried first. Some of these treatments are poorly tolerated in elderly patients. If pain persists, nasal calcitonin may be added. Trigger-point injections with an anesthetic or glucocorticoid may be tried.
The management of early complex regional pain syndrome is sometimes supplemented with systemic corticosteroids, but reviews of randomized controlled trials have failed to show efficacy.14
Later in the course, patients may suffer persistent soft-tissue edema, accompanied by thickening of the skin and periarticular soft tissues, muscle wasting, and the skin changes of brawny edema. Regional blockade of sympathetic ganglions, epidural administration of clonidine, implantable peripheral nerve stimulators, and spinal cord stimulators have all been applied by experts in pain management and may provide benefit. Progression of the syndrome may include cyanosis, mottling, increased sweating, abnormal hair growth, and diffuse swelling in nonarticular tissue.
It is always acceptable to refer to an experienced pain management specialist, and a multidisciplinary approach is recommended at the outset.12
OUR PATIENT’S CARE CONTINUED
Our patient’s forearm and wrist were placed in a sling to keep his left arm elevated when active. This helped control sympathetic vascular edema and throbbing pain. Physical therapy with range-of-motion exercises prevented contracture.
He was discharged home on limited oxycodone as needed, with close follow-up by his primary care physician to monitor his pain symptoms. The pain and swelling slowly improved over the next 2 months, but he suffered a fall, twisting his left wrist. This minor injury was followed by more intense pain and swelling of the forearm, hand, and wrist.
COMORBIDITIES
5. Which of the following statements about conditions associated with complex regional pain syndrome most likely applies to our patient?
- Gout is likely following minor trauma
- Minor trauma or surgical bone biopsy may reactivate complex regional pain syndrome
- Septic hip arthritis due to MRSA may have reemerged and seeded the wrist
- Patients with multiple sclerosis have a propensity for complex regional pain syndrome
- Complex regional pain syndrome type 1 begets type 2
Gout does follow minor injury, but our patient’s uric acid was well controlled on allopurinol (Zyloprim), and gout is unlikely to be causing polyarticular swelling of the hand, wrist, and forearm.
Minor trauma, sometimes inconsequential enough to have been completely forgotten, may either initiate complex regional pain syndrome or, as seen here, reactivate it. Bone changes seen on MRI sometimes trigger surgical bone biopsy, only to reactivate the dysesthesia and sympathetic vascular reaction. Surgery should be avoided. Trauma and surgery are causative rather than associative comorbidities.
Sepsis due to MRSA after total hip arthroplasty may be reactivated, especially in the setting of immunosuppressive treatment. But the diffuse bone changes seen in multiple carpal, radial, and ulnar bones suggest generalized vascular and sympathetic disarray, most consistent with complex regional pain syndrome type 1.
AN ASSOCIATION WITH MULTIPLE SCLEROSIS?
Multiple sclerosis and other central neuropathic conditions such as stroke are associated with complex regional pain syndrome type 1.15,16
A hypothetical cause for the higher prevalence of complex regional pain syndrome in patients with multiple sclerosis may be demyelination resulting in aberrant signaling and overreaction to distal pain receptors. Demyelination of neurons within the autonomic or spinothalamic tracts potentially increases susceptibility to development of the pain syndrome.
Our patient had an apparent stimulus for the development of the syndrome, ie, the initial dog bite, and the wrist injury later may have caused peripheral nerve injury. Such injury may lead to release of vasodilatory neuropeptides including substance P from stimulated cutaneous nerves with cell bodies in the dorsal root ganglia. Excessive vasodilation and increased vascular permeability result in the affected limb becoming edematous and causing cutaneous nerves to be further activated. Stimulated cutaneous neurons normally have an inhibitory influence on sympathetic activity at the level of entry of the dorsal root ganglia in the cord. In complex regional pain syndrome, this inhibition is lost, resulting in a hyperactive somatosympathetic reflex.17 Underlying multiple sclerosis may have contributed to the loss of inhibition by the cutaneous nerves on the sympathetic system.
CASE CONCLUDED
We continued to closely follow this patient, who was on a self-directed program of physical therapy. One year after the original dog bite, the complex regional pain syndrome had completely resolved.
A 48-year-old man with gout, multiple sclerosis, and previously treated methicillin-resistant Staphylococcus aureus (MRSA) infection presented to the emergency room with pain and significant swelling at the site of a dog bite on his left forearm. He had been bitten 2 weeks earlier by a friend’s dog, and the bite had punctured the skin. He also had red streaking on the skin of the left arm from the wrist to the elbow, and he reported feeling “feverish” and having night sweats.
At first, the bite had seemed to improve, then swelling and pain had developed and increased. He reported this to his primary care physician, along with the information that he had previously had an anaphylactic reaction to penicillin and a cephalosporin. His physician, considering a penicillin allergy, started him on ciprofloxacin (Cipro) plus clindamycin (Cleocin). The patient took this for 5 days, but without improvement. The appearance of the red streaking on his left forearm prompted his presentation to our emergency room.
ORGANISMS IN DOG BITES
1. Which is the most common cause of infected dog bite?
- Pasteurella canis
- Streptococci and S aureus
- Erysipelothrix rhusiopathiae
- Capnocytophaga canimorsus
- Eikenella corrodens
Streptococci (50%) and S aureus (20% to 40%) are the organisms most commonly responsible for infected dog bites, as they are for other skin and soft-tissue infections.1P canis is unique to dog bite infections but accounts for only 18%.2E rhusiopathiae is an unusual isolate from cat and dog bites and is more commonly isolated from the mouths of fish and aquatic mammals. C canimorsus is a normal inhabitant of the oral cavity of dogs and cats but an unusual cause of wound infection from a dog bite. It is notable for sepsis and central nervous system infections uniquely associated with veterinarians, dog owners, kennel workers, and mail carriers.3E corrodens infection is more common with human bites.4
THE EVALUATION BEGINS
On examination, the patient had marked edema of the left forearm and pain in the joints of the left hand. His temperature was 100.2°F (37.9°C). Because of the duration and severity of symptoms, the examining physician was concerned about septic arthritis of the wrist, and the patient was admitted to the hospital.
In the hospital, our patient was thermodynamically stable without documented fever or chills. There was no open wound to culture, and blood cultures were negative. Marked edema and joint involvement raised suspicion of erysipeloid. This “cousin” of erysipelas often involves the underlying joint, is associated with edema, and produces systemic manifestations of fever and arthralgia.
Radiography of the left forearm and hand demonstrated multiple foci of demineralization within the carpal bones and proximal radius, attributed to disuse. Magnetic resonance imaging (MRI) the next day showed multiple bone infarcts in the carpal bones and the distal radius, with synovitis and fluid in the carpal joints and without adjacent osteomyelitis. Fluid was also seen in the soft tissues in the ulnar aspect of the left wrist, and tenosynovitis involving the flexor carpi radialis tendon was noted.
Arthrocentesis of his left radiocarpal joint produced synovial fluid negative for crystals and negative on Gram stain; the fluid was also sent for culture. The patient’s tetanus immunization was current, and the dog was known to have been immunized against rabies.
ANTIBIOTICS FOR INFECTED DOG BITES
2. Which antibiotic regimen would you choose for this patient?
- Oral amoxicillin and clavulanate
- Meropenem
- Vancomycin, clindamycin, aztreonam
- Clindamycin and levofloxacin
- Clindamycin and trimethoprim-sulfamethoxazole
Oral amoxicillin and clavulanate (Augmentin) is a judicious choice for prophylactic treatment of deep bites in the early stages of infection. However, our patient’s wound was no longer in the early stages of infection, and he had a history of an adverse reaction to penicillin.
Meropenem (Merrem IV) cross-reacts minimally with penicillin allergy and is reported to be safe in patients with a history of anaphylactic reactions to penicillin,5 but overuse of carbapenems has led to the development of carbapenem-resistant strains of Klebsiella, Stenotrophomonas, and Acinetobacter organisms.
Given the rise of MRSA infections and the common involvement of staphylococci, streptococci, and anaerobic bacteria in complicated dog bites, the combination of vancomycin and clindamycin is a good choice, and aztreonam (Azactam) would add empiric coverage of gram-negative enteric organisms.
Levofloxacin (Levaquin) also covers gramnegative enteric organisms, but Fusobacterium canifelinum, a common anaerobe in the oral flora of dogs and cats, is intrinsically resistant to fluoroquinolones.
Clindamycin and levofloxacin would be a good step-down oral regimen. Pasteurella multocida has variable sensitivity to the commonly used agents dicloxacillin (Dynapen), cephalexin (Keflex), macrolides, and clindamycin, but it is a less likely pathogen at this late stage and could be covered with levofloxacin alone.
C canimorsus is resistant to trimethoprim-sulfamethoxazole (Bactrim) and cephalexin, but is well covered by clindamycin.6
CASE CONTINUED
Our patient was started on intravenous vancomycin, clindamycin, and aztreonam for coverage of dog-mouth flora. Blood cultures and cultures of synovial fluid of the left wrist were negative. Vancomycin was discontinued after 48 hours when blood cultures did not grow staphylococcal organisms, but clindamycin and aztreonam were continued for a total of 8 days to treat possible infection with anaerobic and gram-negative enteric pathogens.
To test for autonomic dysfunction, a plastic pen case drawn lightly across each forearm revealed a loss of tactile adherence (ie, areas where moist, sweaty skin impeded the movement of the pen case) on the affected forearm, a sign of underlying nerve injury. The affected forearm was sensitive to light touch, with pain out of proportion to the stimulus.
ARRIVING AT THE DIAGNOSIS
Based on the wide distribution of inflammation, autonomic dysfunction (shown by differences in temperature and sweating), radiographic evidence of demineralization, hyperesthesia, and lack of improvement in pain and swelling after two courses of antibiotics, the patient’s clinical course was determined to be consistent with complex regional pain syndrome type 1, previously referred to as reflex sympathetic dystrophy.
Symptoms of complex regional pain syndrome traditionally include pain, regional edema, joint stiffness, muscular atrophy, vasomotor disturbances (causing temperature variability and erythema), regional diaphoresis, and localized skeletal demineralization on radiography.
Complex regional pain syndrome type 1 occurs as regional pain and inflammation as an excessive sympathetic reaction to an often minor insult, without nerve injury. When the syndrome occurs in a patient with obvious partial nerve injury, it is categorized as type 2 (formerly known as causalgia). The two types are clinically indistinguishable and are not uncommon. About 10% of all patients with complex regional pain syndrome have obvious nerve injury (complex regional pain syndrome type 2). In a study of 109 patients with Colles fracture, 25% developed symptoms of complex regional pain syndrome.7
Complex regional pain syndrome is difficult to diagnose, as it resembles many other ailments, such as gout, infection, bone tumor, stress fracture, and arthritis. Its pathophysiology is poorly understood, but it is believed to result from a “short circuit” in the reflex arc between somatic afferent sensory fibers and autonomic sympathetic efferent fibers, and this is thought to explain the increased sympathetic stimulation.
Although the pathophysiology is likely the same in type 1 and type 2, electromyography with a nerve conduction study is a reliable way to detect nerve damage and thus distinguish between the two types of complex regional pain syndrome.8
Our understanding of this syndrome is evolving. A recent study using sensory testing showed that 33% of patients with type 1 had combinations of increased and decreased thresholds for the detection of thermal, vibratory, and mechanical stimuli in the distribution of discrete peripheral nerves, suggesting that the patients actually had type 2.9
CONFIRMING COMPLEX REGIONAL PAIN SYNDROME TYPE 1
3. Which of the following is the best way to confirm complex regional pain syndrome type 1?
- Erythrocyte sedimentation rate, C-reactive protein, and complete blood cell count
- Plain radiography of the hand and forearm
- Three-phase technetium bone scan
- The Budapest diagnostic criteria
- MRI
- Autonomic testing
Complex regional pain syndrome type 1 is a clinical diagnosis. Diagnostic studies lack sensitivity and specificity but may confirm complex regional pain syndrome type 1 or rule out other diagnoses. The Budapest diagnostic criteria10 (Table 1) may be the best way to confirm this diagnosis. The criteria are as follows: continuing pain disproportionate to an inciting event, coupled with three of four symptoms plus at least one sign from sensory, vasomotor, sudomotor, and motor-trophic categories.
Laboratory tests are not helpful because acute-phase reactants and blood counts remain normal in these patients.
Plain radiography is not sensitive in early diagnosis, but at 2 weeks it may show patchy areas of osteopenia in adjacent bones throughout the region, as well as subsequent diffuse demineralization.
Three-phase bone scanning is more sensitive than plain radiography, with 75% of patients showing regional disparities in blood flow in early sequences and increased bone uptake in the later sequences.
MRI is a sensitive early test, as it better defines focal areas of bone loss and increased T2 bone signal in adjacent bone, as well as early soft-tissue changes. Computed tomography does not show early specific changes in muscle, tendon, or bone and so is not recommended.
THE EVALUATION CONTINUES
The admitting diagnosis was septic arthritis, and our patient underwent computed tomography, which showed focal demineralization that could have represented bone infarcts or infection, confounding the diagnosis of complex regional pain syndrome.
Autonomic nerve testing can help distinguish complex regional pain syndrome from other disorders. Complex regional pain syndrome is characterized by increased sympathetic activity and results in increased sweat output. Autonomic testing includes resting sweat output, resting skin temperature, and quantitative sudomotor axon reflex testing. In one study, an increase in resting sweat output used in conjunction with quantitative sudomotor axon reflex testing predicted the diagnosis of complex regional pain syndrome with a specificity of 98%.11 However, autonomic testing is limited to academic centers and is not readily available.
TREATING COMPLEX REGIONAL PAIN SYNDROME TYPE 1
4. Which is the best first-line therapy for complex regional pain syndrome type 1?
- Stellate ganglion nerve block
- Occupational therapy to splint the wrist and forearm
- Oral corticosteroids
- Physical therapy to prevent loss of joint motion
- Tricyclic antidepressant drugs (eg, amitriptyline), pregabalin, and bisphosphonates
Physical therapy should be started early in all patients, with range-of-motion exercises to prevent contracture and enhance mobility.
Stellate ganglion nerve block has been used to counter severe sympathetic hyperactivity, but it also may aggravate symptoms of complex regional pain syndrome and so remains a controversial treatment.
Immobilization and splinting should be avoided, as this will augment edema, pain, and contracture of joints.
Corticosteroids do not shorten the course or assuage symptoms and may increase edema.
Amitriptyline (Elavil) and pregabalin (Lyrica) have been used successfully to counter extended courses of allodynia and hyperalgesia. Bisphosphonates may decrease bone loss and pain and may be needed should the course be complicated by myositis ossificans, a form of dystrophic bone formation in juxtaposed tendon and muscle related to neuroactivation of fibroblasts and osteoblasts.
THE COURSE OF COMPLEX REGIONAL PAIN SYNDROME
Traditionally, type 1 was divided into three stages—an early inflammatory stage, a dystrophic stage, and a late atrophic stage.12 Although there is no evidence to support a consistent three-stage evolution, the severity of symptoms may help determine the best approach to management.13
Patients initially exhibit burning or throbbing pain, diffuse aching, sensitivity to touch or cold (allodynia), localized edema, and vasomotor disturbances of variable intensity that may produce altered color and temperature. Topical capsaicin cream; a tricyclic antidepressant; an anticonvulsant such as gabapentin (Neurontin), pregabalin, or lamotrigine (Lamictal); or a nonsteroidal anti-inflammatory drug should be tried first. Some of these treatments are poorly tolerated in elderly patients. If pain persists, nasal calcitonin may be added. Trigger-point injections with an anesthetic or glucocorticoid may be tried.
The management of early complex regional pain syndrome is sometimes supplemented with systemic corticosteroids, but reviews of randomized controlled trials have failed to show efficacy.14
Later in the course, patients may suffer persistent soft-tissue edema, accompanied by thickening of the skin and periarticular soft tissues, muscle wasting, and the skin changes of brawny edema. Regional blockade of sympathetic ganglions, epidural administration of clonidine, implantable peripheral nerve stimulators, and spinal cord stimulators have all been applied by experts in pain management and may provide benefit. Progression of the syndrome may include cyanosis, mottling, increased sweating, abnormal hair growth, and diffuse swelling in nonarticular tissue.
It is always acceptable to refer to an experienced pain management specialist, and a multidisciplinary approach is recommended at the outset.12
OUR PATIENT’S CARE CONTINUED
Our patient’s forearm and wrist were placed in a sling to keep his left arm elevated when active. This helped control sympathetic vascular edema and throbbing pain. Physical therapy with range-of-motion exercises prevented contracture.
He was discharged home on limited oxycodone as needed, with close follow-up by his primary care physician to monitor his pain symptoms. The pain and swelling slowly improved over the next 2 months, but he suffered a fall, twisting his left wrist. This minor injury was followed by more intense pain and swelling of the forearm, hand, and wrist.
COMORBIDITIES
5. Which of the following statements about conditions associated with complex regional pain syndrome most likely applies to our patient?
- Gout is likely following minor trauma
- Minor trauma or surgical bone biopsy may reactivate complex regional pain syndrome
- Septic hip arthritis due to MRSA may have reemerged and seeded the wrist
- Patients with multiple sclerosis have a propensity for complex regional pain syndrome
- Complex regional pain syndrome type 1 begets type 2
Gout does follow minor injury, but our patient’s uric acid was well controlled on allopurinol (Zyloprim), and gout is unlikely to be causing polyarticular swelling of the hand, wrist, and forearm.
Minor trauma, sometimes inconsequential enough to have been completely forgotten, may either initiate complex regional pain syndrome or, as seen here, reactivate it. Bone changes seen on MRI sometimes trigger surgical bone biopsy, only to reactivate the dysesthesia and sympathetic vascular reaction. Surgery should be avoided. Trauma and surgery are causative rather than associative comorbidities.
Sepsis due to MRSA after total hip arthroplasty may be reactivated, especially in the setting of immunosuppressive treatment. But the diffuse bone changes seen in multiple carpal, radial, and ulnar bones suggest generalized vascular and sympathetic disarray, most consistent with complex regional pain syndrome type 1.
AN ASSOCIATION WITH MULTIPLE SCLEROSIS?
Multiple sclerosis and other central neuropathic conditions such as stroke are associated with complex regional pain syndrome type 1.15,16
A hypothetical cause for the higher prevalence of complex regional pain syndrome in patients with multiple sclerosis may be demyelination resulting in aberrant signaling and overreaction to distal pain receptors. Demyelination of neurons within the autonomic or spinothalamic tracts potentially increases susceptibility to development of the pain syndrome.
Our patient had an apparent stimulus for the development of the syndrome, ie, the initial dog bite, and the wrist injury later may have caused peripheral nerve injury. Such injury may lead to release of vasodilatory neuropeptides including substance P from stimulated cutaneous nerves with cell bodies in the dorsal root ganglia. Excessive vasodilation and increased vascular permeability result in the affected limb becoming edematous and causing cutaneous nerves to be further activated. Stimulated cutaneous neurons normally have an inhibitory influence on sympathetic activity at the level of entry of the dorsal root ganglia in the cord. In complex regional pain syndrome, this inhibition is lost, resulting in a hyperactive somatosympathetic reflex.17 Underlying multiple sclerosis may have contributed to the loss of inhibition by the cutaneous nerves on the sympathetic system.
CASE CONCLUDED
We continued to closely follow this patient, who was on a self-directed program of physical therapy. One year after the original dog bite, the complex regional pain syndrome had completely resolved.
- Talan DA, Citron DM, Abrahamian FM, Moran GJ, Goldstein EJ. Bacteriologic analysis of infected dog and cat bites. Emergency Medicine Animal Bite Infection Study Group. N Engl J Med 1999; 340:85–92.
- Holst E, Rollof J, Larsson L, Nielsen JP. Characterization and distribution of Pasteurella species recovered from infected humans. J Clin Microbiol 1992; 30:2984–2987.
- Jolivet-Gougeon A, Sixou JL, Tamanai-Shacoori Z, Bonnaure-Mallet M. Antimicrobial treatment of Capnocytophaga infections. Int J Antimicrob Agents 2007; 29:367–373.
- Paul K, Patel SS. Eikenella corrodens infections in children and adolescents: case reports and review of the literature. Clin Infect Dis 2001; 33:54–61.
- Cunha BA, Hamid NS, Krol V, Eisenstein L. Safety of meropenem in patients reporting penicillin allergy: lack of allergic cross reactions. J Chemother 2008; 20:233–237.
- Verghese A, Hamati F, Berk S, Franzus B, Berk S, Smith JK. Susceptibility of dysgonic fermenter 2 to antimicrobial agents in vitro. Antimicrob Agents Chemother 1988; 32:78–80.
- Atkins RM, Duckworth T, Kanis JA. Algodystrophy following Colles’ fracture. J Hand Surg Br 1989; 14:161–164.
- Rommel O, Malin JP, Zenz M, Jänig W. Quantitative sensory testing, neurophysiological and psychological examination in patients with complex regional pain syndrome and hemisensory deficits. Pain 2001; 93:279–293.
- Sethna NF, Meier PM, Zurakowski D, Berde CB. Cutaneous sensory abnormalities in children and adolescents with complex regional pain syndromes. Pain 2007; 131:153–161.
- Harden RN, Bruehl S, Stanton-Hicks M, Wilson PR. Proposed new diagnostic criteria for complex regional pain syndrome. Pain Med 2007; 8:326–331.
- Chelimsky TC, Low PA, Naessens JM, Wilson PR, Amadio PC, O’Brien PC. Value of autonomic testing in reflex sympathetic dystrophy. Mayo Clin Proc 1995; 70:1029–1040.
- Stanton-Hicks MD, Burton AW, Bruehl SP, et al. An updated interdisciplinary clinical pathway for CRPS: report of an expert panel. Pain Pract 2002; 2:1–16.
- Brummett CM, Cohen SP, eds. Managing pain: essentials of diagnosis and treatment. New York; Oxford University Press; 2013.
- Dirckx M, Stronks DL, Groeneweg G, Huygen FJ. Effect of immunomodulating medications in complex regional pain syndrome: a systematic review. Clin J Pain 2012; 28:355–363.
- Schwartzman RJ, Gurusinghe C, Gracely E. Prevalence of complex regional pain syndrome in a cohort of multiple sclerosis patients. Pain Physician 2008; 11:133–136.
- Sandroni P, Benrud-Larson LM, McClelland RL, Low PA. Complex regional pain syndrome type I: incidence and prevalence in Olmsted county, a population-based study. Pain 2003; 103:199–207.
- Kurvers HA, Jacobs MJ, Beuk RJ, et al. Reflex sympathetic dystrophy: evolution of microcirculatory disturbances in time. Pain 1995; 60:333–340.
- Talan DA, Citron DM, Abrahamian FM, Moran GJ, Goldstein EJ. Bacteriologic analysis of infected dog and cat bites. Emergency Medicine Animal Bite Infection Study Group. N Engl J Med 1999; 340:85–92.
- Holst E, Rollof J, Larsson L, Nielsen JP. Characterization and distribution of Pasteurella species recovered from infected humans. J Clin Microbiol 1992; 30:2984–2987.
- Jolivet-Gougeon A, Sixou JL, Tamanai-Shacoori Z, Bonnaure-Mallet M. Antimicrobial treatment of Capnocytophaga infections. Int J Antimicrob Agents 2007; 29:367–373.
- Paul K, Patel SS. Eikenella corrodens infections in children and adolescents: case reports and review of the literature. Clin Infect Dis 2001; 33:54–61.
- Cunha BA, Hamid NS, Krol V, Eisenstein L. Safety of meropenem in patients reporting penicillin allergy: lack of allergic cross reactions. J Chemother 2008; 20:233–237.
- Verghese A, Hamati F, Berk S, Franzus B, Berk S, Smith JK. Susceptibility of dysgonic fermenter 2 to antimicrobial agents in vitro. Antimicrob Agents Chemother 1988; 32:78–80.
- Atkins RM, Duckworth T, Kanis JA. Algodystrophy following Colles’ fracture. J Hand Surg Br 1989; 14:161–164.
- Rommel O, Malin JP, Zenz M, Jänig W. Quantitative sensory testing, neurophysiological and psychological examination in patients with complex regional pain syndrome and hemisensory deficits. Pain 2001; 93:279–293.
- Sethna NF, Meier PM, Zurakowski D, Berde CB. Cutaneous sensory abnormalities in children and adolescents with complex regional pain syndromes. Pain 2007; 131:153–161.
- Harden RN, Bruehl S, Stanton-Hicks M, Wilson PR. Proposed new diagnostic criteria for complex regional pain syndrome. Pain Med 2007; 8:326–331.
- Chelimsky TC, Low PA, Naessens JM, Wilson PR, Amadio PC, O’Brien PC. Value of autonomic testing in reflex sympathetic dystrophy. Mayo Clin Proc 1995; 70:1029–1040.
- Stanton-Hicks MD, Burton AW, Bruehl SP, et al. An updated interdisciplinary clinical pathway for CRPS: report of an expert panel. Pain Pract 2002; 2:1–16.
- Brummett CM, Cohen SP, eds. Managing pain: essentials of diagnosis and treatment. New York; Oxford University Press; 2013.
- Dirckx M, Stronks DL, Groeneweg G, Huygen FJ. Effect of immunomodulating medications in complex regional pain syndrome: a systematic review. Clin J Pain 2012; 28:355–363.
- Schwartzman RJ, Gurusinghe C, Gracely E. Prevalence of complex regional pain syndrome in a cohort of multiple sclerosis patients. Pain Physician 2008; 11:133–136.
- Sandroni P, Benrud-Larson LM, McClelland RL, Low PA. Complex regional pain syndrome type I: incidence and prevalence in Olmsted county, a population-based study. Pain 2003; 103:199–207.
- Kurvers HA, Jacobs MJ, Beuk RJ, et al. Reflex sympathetic dystrophy: evolution of microcirculatory disturbances in time. Pain 1995; 60:333–340.
Changes to practice may help avoid ‘double trouble’
Large-volume thoracentesis is defined as the drainage of more than 1 L of fluid. Inherent in this procedure is the removal of a large amount of fluid from a cavity with a rigid wall, which leads to changes in pleural pressure and to expansion of the lung. Two specific complications occur, pneumothorax and reexpansion pulmonary edema. The images submitted for the Clinical Picture article by Drs. Apter and Aronowitz in this issue of the Journal highlight these complications.
Retrospective studies have found an association between the amount of fluid drained and the incidence of pneumothorax.1,2 Although technical issues may account for it (eg, needle injury to the lung that leads to postprocedural pneumothorax), the available evidence suggests that it has more to do with the drainage of larger volumes than the lung can expand to fill.3,4 That is, the patient’s lung cannot expand,5 so drainage creates a vacuum, and air enters the pleural space3 through the lung parenchyma, or perhaps from around the drainage catheter.
In a series of patients who underwent therapeutic thoracentesis,3 23 (8.7%) of 265 patients had pneumothorax. Interestingly, some patients had only symptoms, some had only excessively negative pressures (< 25 cm H2O), some had both, and some had neither. Thus, there does not seem to be a reliable sign or symptom of an unexpanding lung, but pleural manometry may help increase its detection.6 This technique, however, is rarely used in clinical practice.
Another consequence of therapeutic thoracentesis is reexpansion pulmonary edema. This rare condition occurs only after large-volume thoracentesis or evacuation of a moderate to large pneumothorax.7 The pathophysiology behind this is controversial.8 As with pneumothorax, a large case series did not find a correlation between volume removed or pleural pressures and reexpansion pulmonary edema.7 Experimental data and analysis of case series8–10 suggest that the duration of lung collapse and the speed of drainage and negative pressure applied contribute to the development of edema. Vacuum bottles are often used to speed drainage and to contain the large amount of fluid drained. These bottles have an initial negative pressure of about −723 mm Hg (personal communication with Baxter Healthcare Product information line), which may lead to rapid changes in lung volume and perhaps to higher negative pleural pressures.
Given the risks discussed above, we believe it is appropriate to avoid vacuum bottles and instead to use the syringe and one-way valve supplied in most thoracentesis kits. Further, pleural manometry to detect changes in pressure that suggest an unexpandable lung may lead to the appropriate early termination of a planned large-volume thoracentesis.3 The complications reported by Drs. Apter and Aronowitz are relatively rare and, at this point, unpredictable; therefore, generating high-quality evidence for prediction or management will be difficult. In the meantime, understanding the physiologic changes in the lung and the pleural space when draining large effusions from the chest may help avoid double trouble.
- Josephson T, Nordenskjold CA, Larsson J, Rosenberg LU, Kaijser M. Amount drained at ultrasound-guided thoracentesis and risk of pneumothorax. Acta Radiol 2009; 50:42–47.
- Gordon CE, Feller-Kopman D, Balk EM, Smetana GW. Pneumothorax following thoracentesis: a systematic review and meta-analysis. Arch Intern Med 2010; 170:332–339.
- Heidecker J, Huggins JT, Sahn SA, Doelken P. Pathophysiology of pneumothorax following ultrasound-guided thoracentesis. Chest 2006; 130:1173–1184.
- Huggins JT, Sahn SA, Heidecker J, Ravenel JG, Doelken P. Characteristics of trapped lung: pleural fluid analysis, manometry, and air-contrast chest CT. Chest 2007; 131:206–213.
- Woodring JH, Baker MD, Stark P. Pneumothorax ex vacuo. Chest 1996; 110:1102–1105.
- Feller-Kopman D. Therapeutic thoracentesis: the role of ultrasound and pleural manometry. Curr Opin Pulmon Med 2007; 13:312–318.
- Feller-Kopman D, Berkowitz D, Boiselle P, Ernst A. Large-volume thoracentesis and the risk of reexpansion pulmonary edema. Ann Thorac Surg 2007; 84:1656–1661.
- Tarver RD, Broderick LS, Conces DJ, Jr. Reexpansion pulmonary edema. J Thorac Imag 1996; 11:198–209.
- Murphy K, Tomlanovich MC. Unilateral pulmonary edema after drainage of a spontaneous pneumothorax: case report and review of the world literature. J Emerg Med 1983; 1:29–36.
- Pavlin J, Cheney FW Unilateral pulmonary edema in rabbits after reexpansion of collapsed lung. J Appl Physiol Respir Environ Exerc Physiol 1979; 46:31–35.
Large-volume thoracentesis is defined as the drainage of more than 1 L of fluid. Inherent in this procedure is the removal of a large amount of fluid from a cavity with a rigid wall, which leads to changes in pleural pressure and to expansion of the lung. Two specific complications occur, pneumothorax and reexpansion pulmonary edema. The images submitted for the Clinical Picture article by Drs. Apter and Aronowitz in this issue of the Journal highlight these complications.
Retrospective studies have found an association between the amount of fluid drained and the incidence of pneumothorax.1,2 Although technical issues may account for it (eg, needle injury to the lung that leads to postprocedural pneumothorax), the available evidence suggests that it has more to do with the drainage of larger volumes than the lung can expand to fill.3,4 That is, the patient’s lung cannot expand,5 so drainage creates a vacuum, and air enters the pleural space3 through the lung parenchyma, or perhaps from around the drainage catheter.
In a series of patients who underwent therapeutic thoracentesis,3 23 (8.7%) of 265 patients had pneumothorax. Interestingly, some patients had only symptoms, some had only excessively negative pressures (< 25 cm H2O), some had both, and some had neither. Thus, there does not seem to be a reliable sign or symptom of an unexpanding lung, but pleural manometry may help increase its detection.6 This technique, however, is rarely used in clinical practice.
Another consequence of therapeutic thoracentesis is reexpansion pulmonary edema. This rare condition occurs only after large-volume thoracentesis or evacuation of a moderate to large pneumothorax.7 The pathophysiology behind this is controversial.8 As with pneumothorax, a large case series did not find a correlation between volume removed or pleural pressures and reexpansion pulmonary edema.7 Experimental data and analysis of case series8–10 suggest that the duration of lung collapse and the speed of drainage and negative pressure applied contribute to the development of edema. Vacuum bottles are often used to speed drainage and to contain the large amount of fluid drained. These bottles have an initial negative pressure of about −723 mm Hg (personal communication with Baxter Healthcare Product information line), which may lead to rapid changes in lung volume and perhaps to higher negative pleural pressures.
Given the risks discussed above, we believe it is appropriate to avoid vacuum bottles and instead to use the syringe and one-way valve supplied in most thoracentesis kits. Further, pleural manometry to detect changes in pressure that suggest an unexpandable lung may lead to the appropriate early termination of a planned large-volume thoracentesis.3 The complications reported by Drs. Apter and Aronowitz are relatively rare and, at this point, unpredictable; therefore, generating high-quality evidence for prediction or management will be difficult. In the meantime, understanding the physiologic changes in the lung and the pleural space when draining large effusions from the chest may help avoid double trouble.
Large-volume thoracentesis is defined as the drainage of more than 1 L of fluid. Inherent in this procedure is the removal of a large amount of fluid from a cavity with a rigid wall, which leads to changes in pleural pressure and to expansion of the lung. Two specific complications occur, pneumothorax and reexpansion pulmonary edema. The images submitted for the Clinical Picture article by Drs. Apter and Aronowitz in this issue of the Journal highlight these complications.
Retrospective studies have found an association between the amount of fluid drained and the incidence of pneumothorax.1,2 Although technical issues may account for it (eg, needle injury to the lung that leads to postprocedural pneumothorax), the available evidence suggests that it has more to do with the drainage of larger volumes than the lung can expand to fill.3,4 That is, the patient’s lung cannot expand,5 so drainage creates a vacuum, and air enters the pleural space3 through the lung parenchyma, or perhaps from around the drainage catheter.
In a series of patients who underwent therapeutic thoracentesis,3 23 (8.7%) of 265 patients had pneumothorax. Interestingly, some patients had only symptoms, some had only excessively negative pressures (< 25 cm H2O), some had both, and some had neither. Thus, there does not seem to be a reliable sign or symptom of an unexpanding lung, but pleural manometry may help increase its detection.6 This technique, however, is rarely used in clinical practice.
Another consequence of therapeutic thoracentesis is reexpansion pulmonary edema. This rare condition occurs only after large-volume thoracentesis or evacuation of a moderate to large pneumothorax.7 The pathophysiology behind this is controversial.8 As with pneumothorax, a large case series did not find a correlation between volume removed or pleural pressures and reexpansion pulmonary edema.7 Experimental data and analysis of case series8–10 suggest that the duration of lung collapse and the speed of drainage and negative pressure applied contribute to the development of edema. Vacuum bottles are often used to speed drainage and to contain the large amount of fluid drained. These bottles have an initial negative pressure of about −723 mm Hg (personal communication with Baxter Healthcare Product information line), which may lead to rapid changes in lung volume and perhaps to higher negative pleural pressures.
Given the risks discussed above, we believe it is appropriate to avoid vacuum bottles and instead to use the syringe and one-way valve supplied in most thoracentesis kits. Further, pleural manometry to detect changes in pressure that suggest an unexpandable lung may lead to the appropriate early termination of a planned large-volume thoracentesis.3 The complications reported by Drs. Apter and Aronowitz are relatively rare and, at this point, unpredictable; therefore, generating high-quality evidence for prediction or management will be difficult. In the meantime, understanding the physiologic changes in the lung and the pleural space when draining large effusions from the chest may help avoid double trouble.
- Josephson T, Nordenskjold CA, Larsson J, Rosenberg LU, Kaijser M. Amount drained at ultrasound-guided thoracentesis and risk of pneumothorax. Acta Radiol 2009; 50:42–47.
- Gordon CE, Feller-Kopman D, Balk EM, Smetana GW. Pneumothorax following thoracentesis: a systematic review and meta-analysis. Arch Intern Med 2010; 170:332–339.
- Heidecker J, Huggins JT, Sahn SA, Doelken P. Pathophysiology of pneumothorax following ultrasound-guided thoracentesis. Chest 2006; 130:1173–1184.
- Huggins JT, Sahn SA, Heidecker J, Ravenel JG, Doelken P. Characteristics of trapped lung: pleural fluid analysis, manometry, and air-contrast chest CT. Chest 2007; 131:206–213.
- Woodring JH, Baker MD, Stark P. Pneumothorax ex vacuo. Chest 1996; 110:1102–1105.
- Feller-Kopman D. Therapeutic thoracentesis: the role of ultrasound and pleural manometry. Curr Opin Pulmon Med 2007; 13:312–318.
- Feller-Kopman D, Berkowitz D, Boiselle P, Ernst A. Large-volume thoracentesis and the risk of reexpansion pulmonary edema. Ann Thorac Surg 2007; 84:1656–1661.
- Tarver RD, Broderick LS, Conces DJ, Jr. Reexpansion pulmonary edema. J Thorac Imag 1996; 11:198–209.
- Murphy K, Tomlanovich MC. Unilateral pulmonary edema after drainage of a spontaneous pneumothorax: case report and review of the world literature. J Emerg Med 1983; 1:29–36.
- Pavlin J, Cheney FW Unilateral pulmonary edema in rabbits after reexpansion of collapsed lung. J Appl Physiol Respir Environ Exerc Physiol 1979; 46:31–35.
- Josephson T, Nordenskjold CA, Larsson J, Rosenberg LU, Kaijser M. Amount drained at ultrasound-guided thoracentesis and risk of pneumothorax. Acta Radiol 2009; 50:42–47.
- Gordon CE, Feller-Kopman D, Balk EM, Smetana GW. Pneumothorax following thoracentesis: a systematic review and meta-analysis. Arch Intern Med 2010; 170:332–339.
- Heidecker J, Huggins JT, Sahn SA, Doelken P. Pathophysiology of pneumothorax following ultrasound-guided thoracentesis. Chest 2006; 130:1173–1184.
- Huggins JT, Sahn SA, Heidecker J, Ravenel JG, Doelken P. Characteristics of trapped lung: pleural fluid analysis, manometry, and air-contrast chest CT. Chest 2007; 131:206–213.
- Woodring JH, Baker MD, Stark P. Pneumothorax ex vacuo. Chest 1996; 110:1102–1105.
- Feller-Kopman D. Therapeutic thoracentesis: the role of ultrasound and pleural manometry. Curr Opin Pulmon Med 2007; 13:312–318.
- Feller-Kopman D, Berkowitz D, Boiselle P, Ernst A. Large-volume thoracentesis and the risk of reexpansion pulmonary edema. Ann Thorac Surg 2007; 84:1656–1661.
- Tarver RD, Broderick LS, Conces DJ, Jr. Reexpansion pulmonary edema. J Thorac Imag 1996; 11:198–209.
- Murphy K, Tomlanovich MC. Unilateral pulmonary edema after drainage of a spontaneous pneumothorax: case report and review of the world literature. J Emerg Med 1983; 1:29–36.
- Pavlin J, Cheney FW Unilateral pulmonary edema in rabbits after reexpansion of collapsed lung. J Appl Physiol Respir Environ Exerc Physiol 1979; 46:31–35.