User login
Case Studies in Toxicology: An Amazonian Herb Goes Mainstream
Case
A 23-year-old Hispanic woman with no past medical history is brought to the ED for the second time in one day. On her first presentation, which was for a fever and a headache, meningitis was excluded with normal laboratory tests that included a lumbar puncture. She was administered acetaminophen for fever and pain control, and was discharged with a diagnosis of viral illness. On this second visit, 10 hours after being discharged, she presented because her family noted convulsions that began 3 hours after taking an herbal headache remedy given to her by a naturopath.
The patient arrived to the ED with a persistent seizure that terminated following administration of 2 mg of lorazepam. Her initial vital signs were: blood pressure, 115/51 mm Hg; heart rate, 121 beats/minute; respiratory rate, 24 breaths/minute; temperature, 97.6oF. Oxygen (O2) saturation was 100% with 2 L of O2 administered via nasal cannula. Her neurological examination was significant for a depressed mental status, pupils that were 6 mm and minimally reactive, clonus, and hyperreflexia. Repeat laboratory evaluation found a leukocytosis of 22.0 x 103/µL, serum bicarbonate of 9 mEq/L, and an anion gap of 22 with a normal serum lactate.
What is the differential diagnosis of this patient?
The history of medicinal plant ingestion raises the possibility of a toxicologic etiology. However, because the patient took the “medication” to treat another disorder, a search for an alternate cause should be performed. The differential diagnosis of a toxin-induced seizure is broad and includes pharmaceuticals (eg, tramadol, antihistamines), which may be surreptitiously added to herbal medication to assure efficacy. Plants associated with seizures include those containing antimuscarinic tropane alkaloids such as Jimsonweed (though a rare side effect from this plant product) or the water hemlock (Cicuta maculata). Contaminants of the plant itself may include pesticides such as organophosphates.
Although unlikely in a 21 year old, withdrawal from benzodiazepines, ethanol, baclofen, or gamma hydroxybutyrate are other possible etiologies. In addition to pharmaceutical and plant-derived causes, carbon monoxide poisoning should be a consideration in any patient with headache and flu-like illness.
This patient also presented with a constellation of other findings that included hyperreflexia, clonus, tachycardia, and altered mental status. Together these signs are expected in patients with serotonin toxicity (also referred to as serotonin syndrome), neuroleptic malignant syndrome, exogenous thyrotoxicosis, and lithium poisoning.
Case Continuation
The naturopathic practitioner arrived at the ED concerned about the patient, informing the ED team that she had given the patient 2 ounces of ayahuasca tea.
What is ayahuasca? What is the mechanism by which it exerts toxic effects?
Ayahuasca is a plant-derived psychotropic beverage that is used for religious purposes by members of two Brazilian churches—Centro Espírita Beneficente União do Vegetal (UDV) and Santo Daime. The ayahuasca beverage consists of two pharmacologically active compounds that together, but not individually, are psychoactive. The desired active effects for church participants include hallucinations, and vomiting to bring about a “religious purge.”1
Ayahuasca is prepared by combining two plants indigenous to the Amazon Basin area: Banisteriopsis caapi and either Psychotria viridis or Diplopterys cabrerana. B caapi contains the β-carboline alkaloids harmine, harmaline, and tetrahydroharmine. These alkaloids act as reversible inhibitors of the monoamine oxidase A (MAO-A) enzyme. The bark and stems of B caapi are boiled along with either P viridis or D cabrerana, both of which contain the potent hallucinogen N-N dimethyltryptamine (DMT).2 Normally, DMT is not active orally because it is enzymatically metabolized by MAO-A. However, when taken in the presence of the B caapi-derived MAO-A–inhibiting harmine alkaloids, DMT reaches the systemic circulation and produces its clinical effects.3
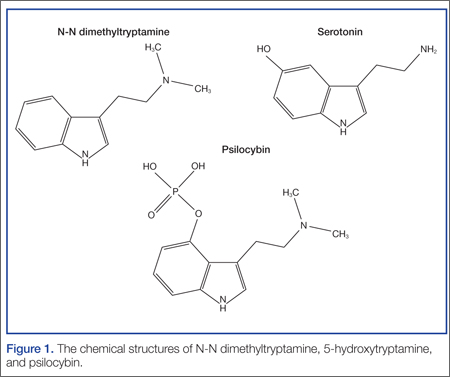
What are the clinical findings of serotonin toxicity?
Serotonin toxicity is a collection of clinical findings that fall under three main categories: autonomic hyperactivity, altered mental status, and muscle rigidity.5 The autonomic findings may include tachycardia, hypertension, hyperthermia, shivering, diaphoresis, or mydriasis. Altered mental status ranges from mild agitation and hypervigilance to agitated delirium to obtundation. Other neurological findings may include tremor, myoclonus, hyperreflexia, or seizures. The onset of these signs is rapid, usually occurring within minutes after exposure to one or more serotonergic compounds. Although rare, severe serotonin toxicity may be associated with hypotension and shock, leading to death.4
The diagnosis of serotonin toxicity is based on the history and physical examination of the patient. Diagnostic criteria that have been suggested include the following: (1) a recent addition or increase in a known serotonergic agent; (2) absence of other possible etiologies; (3) no recent increase or addition of a neuroleptic agent (suggesting neuroleptic malignant syndrome); and/or (4) at least 3 of the following symptoms—mental status changes, myoclonus, agitation, hyperreflexia, diaphoresis, shivering, tremor, diarrhea, incoordination, fever5 (Figure 2).
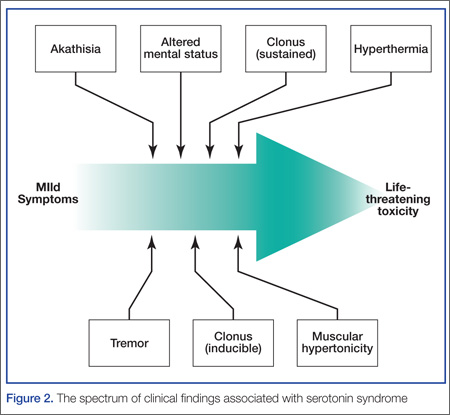
How should this patient be managed?
The management of serotonin toxicity is primarily supportive with aggressive control of hyperthermia and autonomic instability. The precipitating xenobiotic agent should be immediately discontinued. In general, treatment with intravenous fluids, cooling measures, benzodiazepines, and a nonspecific 5-HT antagonist such as cyproheptadine should greatly improve the patient’s clinical status. Patients with severe toxicity may require induced paralysis and intubation.4 It is not clear in this case if the serotonin hyperactivation was due to the DMT (5-HT2A is associated with serotonin toxicity) or another serotonergic agent (eg, dextromethorphan from a cough and cold preparation) in combination with the MAO-inhibiting harmine alkaloids.
What is the availability of ayahuasca in the United States? How is it used in its nonherbal form?
...[Ayahuasca] is currently available in the United States and is legal for use by members of the UDV and Santo Daime churches. Many clinicians are becoming increasingly familiar with this herbal preparation since the recreational use of ayahuasca is gaining popularity in the United States. Internet fora with information on how to safely use ayahuasca, such as avoiding aged cheeses, are becoming more prevalent.7 A recent article in the New York Times described an ayahuasca gathering in Brooklyn, New York, where participants use the herb in a communal fashion.8 This herbal product is also associated with the Hollywood social scene and has received celebrity endorsements.8
The National Survey on Drug Use and Health found that the number of people in the United States who have used DMT has gone up almost every year since 2006, from an estimated 688,000 in 2006 to 1,475,000 in 2012.9 When used alone (not as ayahuasca), DMT is almost exclusively insufflated as a nasal snuff, bypassing hepatic elimination. It has an onset of around 45 seconds and a duration of 5 to 10 minutes. Insufflating DMT was historically referred to as a “businessman’s trip” because users were able to have a brief hallucinogenic experience on a lunch break and recover rapidly to perform their normal work.10
International law declares that DMT is an illegal substance and its importation is banned. However, its use for religious purposes, as is allowed for mescaline found in peyote, remains controversial.7 The UDV brought suit in United States federal court to prevent interference with the church’s use of ayahuasca during religious ceremonies based on the Religious Freedom Restoration Act. This act states that the government should not cause substantial imposition on religious practices in the absence of a compelling government interest. The court sided with the UDV, finding that the government had not sufficiently proved the alleged health risks posed by ayahuasca and could not show a substantial risk that the drug would be abused recreationally.11 Thus it is currently available in the United States and is legal for use by members of the UDV and Santo Daime churches.
Ayahuasca is not regulated by the US Food and Drug Administration. Many different types of preparations with different ingredients as well as different concentrations may exist, and clinical variability should be expected. Understanding that ayahuasca is capable of inhibiting MAO is important in order to avoid foods and medications, such as dextromethorphan, that may trigger adverse effects.
Case Conclusion
The patient’s hospital course was complicated by an additional seizure 12 hours after her initial presentation. By 36 hours she was back to her baseline mental status with a normal neurological examination.
Dr Fil is a senior fellow in medical toxicology at North Shore University Hospital, Manhasset, New York. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Gable RS. Risk assessment of ritual use of oral dimethyltryptamine (DMT) and harmala alkaloids. Addiction. 2007;102(1):24-34.
- Riba J, McIlhenny EH, Valle M, Bouso JC, Barker SA. Metabolism and Disposition of N,N-dimethyltryptamine and harmala alkaloids after oral administration of ayahuasca. Drug Test Anal. 2012;4(7-8):610-616.
- Riba J, Valle M, Urbano G, Yritia M, Morte A, Barbanoj MJ. Human Pharmacology of Ayahuasca: Subjective and Cardiovascular Effects, Monoamine Metabolite Excretion and Pharmacokinetics. J Pharmacol Exp Ther. 2003;306(1):73-83
- Boyer EW, Shannon M. The serotonin syndrome. N Engl J Med. 2005;352(11);1112-1120.
- Sternbach H. The serotonin syndrome. Am J Psychiatry. 1991;148(6):6;705-713.
- Dunkley EJ, Isbister GK, Sibbritt D, Dawson AH, Whyte IM. The Hunter Serotonin Toxicity Criteria: simple and accurate diagnostic decision rules for serotonin toxicity. QJM. 2003;96(9):635-642.
- Erowid. Ayahuasca Vault. https://www.erowid.org/chemicals/ayahuasca/ayahuasca.shtml. Accessed November 25, 2014.
- Morris B. Ayahuasca: a strong cup of tea. New York Times. June 13, 2014. http://www.nytimes.com/2014/06/15/fashion/ayahuasca-a-strong-cup-of-tea.html. Accessed November 25, 2014.
- Quintanilla D. DMT: Hallucinogenic Drug Used in Shamanic Rituals Goes Mainstream. 10 Dec 2013. Available: http://www.opposingviews.com/i/health/dmt-hallucinogenic-drug-used-shamanic-rituals-goes-mainstream. Last accessed 11/14/14.
- Haroz R, Greenberg MI. Emerging drugs of abuse. Med Clin North Am. 2005;89(6):1259-1276.
- Gonzales v. O Centro Espirita Beneficente Uniao do Vegetal, 546 US 418 (2006). Available at http://scholar.google.com/scholar_case?case=7036734975431570669&hl=en&as_sdt=6&as_vis=1&oi=scholarr. Accessed November 25, 2014.
Case
A 23-year-old Hispanic woman with no past medical history is brought to the ED for the second time in one day. On her first presentation, which was for a fever and a headache, meningitis was excluded with normal laboratory tests that included a lumbar puncture. She was administered acetaminophen for fever and pain control, and was discharged with a diagnosis of viral illness. On this second visit, 10 hours after being discharged, she presented because her family noted convulsions that began 3 hours after taking an herbal headache remedy given to her by a naturopath.
The patient arrived to the ED with a persistent seizure that terminated following administration of 2 mg of lorazepam. Her initial vital signs were: blood pressure, 115/51 mm Hg; heart rate, 121 beats/minute; respiratory rate, 24 breaths/minute; temperature, 97.6oF. Oxygen (O2) saturation was 100% with 2 L of O2 administered via nasal cannula. Her neurological examination was significant for a depressed mental status, pupils that were 6 mm and minimally reactive, clonus, and hyperreflexia. Repeat laboratory evaluation found a leukocytosis of 22.0 x 103/µL, serum bicarbonate of 9 mEq/L, and an anion gap of 22 with a normal serum lactate.
What is the differential diagnosis of this patient?
The history of medicinal plant ingestion raises the possibility of a toxicologic etiology. However, because the patient took the “medication” to treat another disorder, a search for an alternate cause should be performed. The differential diagnosis of a toxin-induced seizure is broad and includes pharmaceuticals (eg, tramadol, antihistamines), which may be surreptitiously added to herbal medication to assure efficacy. Plants associated with seizures include those containing antimuscarinic tropane alkaloids such as Jimsonweed (though a rare side effect from this plant product) or the water hemlock (Cicuta maculata). Contaminants of the plant itself may include pesticides such as organophosphates.
Although unlikely in a 21 year old, withdrawal from benzodiazepines, ethanol, baclofen, or gamma hydroxybutyrate are other possible etiologies. In addition to pharmaceutical and plant-derived causes, carbon monoxide poisoning should be a consideration in any patient with headache and flu-like illness.
This patient also presented with a constellation of other findings that included hyperreflexia, clonus, tachycardia, and altered mental status. Together these signs are expected in patients with serotonin toxicity (also referred to as serotonin syndrome), neuroleptic malignant syndrome, exogenous thyrotoxicosis, and lithium poisoning.
Case Continuation
The naturopathic practitioner arrived at the ED concerned about the patient, informing the ED team that she had given the patient 2 ounces of ayahuasca tea.
What is ayahuasca? What is the mechanism by which it exerts toxic effects?
Ayahuasca is a plant-derived psychotropic beverage that is used for religious purposes by members of two Brazilian churches—Centro Espírita Beneficente União do Vegetal (UDV) and Santo Daime. The ayahuasca beverage consists of two pharmacologically active compounds that together, but not individually, are psychoactive. The desired active effects for church participants include hallucinations, and vomiting to bring about a “religious purge.”1
Ayahuasca is prepared by combining two plants indigenous to the Amazon Basin area: Banisteriopsis caapi and either Psychotria viridis or Diplopterys cabrerana. B caapi contains the β-carboline alkaloids harmine, harmaline, and tetrahydroharmine. These alkaloids act as reversible inhibitors of the monoamine oxidase A (MAO-A) enzyme. The bark and stems of B caapi are boiled along with either P viridis or D cabrerana, both of which contain the potent hallucinogen N-N dimethyltryptamine (DMT).2 Normally, DMT is not active orally because it is enzymatically metabolized by MAO-A. However, when taken in the presence of the B caapi-derived MAO-A–inhibiting harmine alkaloids, DMT reaches the systemic circulation and produces its clinical effects.3
What are the clinical findings of serotonin toxicity?
Serotonin toxicity is a collection of clinical findings that fall under three main categories: autonomic hyperactivity, altered mental status, and muscle rigidity.5 The autonomic findings may include tachycardia, hypertension, hyperthermia, shivering, diaphoresis, or mydriasis. Altered mental status ranges from mild agitation and hypervigilance to agitated delirium to obtundation. Other neurological findings may include tremor, myoclonus, hyperreflexia, or seizures. The onset of these signs is rapid, usually occurring within minutes after exposure to one or more serotonergic compounds. Although rare, severe serotonin toxicity may be associated with hypotension and shock, leading to death.4
The diagnosis of serotonin toxicity is based on the history and physical examination of the patient. Diagnostic criteria that have been suggested include the following: (1) a recent addition or increase in a known serotonergic agent; (2) absence of other possible etiologies; (3) no recent increase or addition of a neuroleptic agent (suggesting neuroleptic malignant syndrome); and/or (4) at least 3 of the following symptoms—mental status changes, myoclonus, agitation, hyperreflexia, diaphoresis, shivering, tremor, diarrhea, incoordination, fever5 (Figure 2).
How should this patient be managed?
The management of serotonin toxicity is primarily supportive with aggressive control of hyperthermia and autonomic instability. The precipitating xenobiotic agent should be immediately discontinued. In general, treatment with intravenous fluids, cooling measures, benzodiazepines, and a nonspecific 5-HT antagonist such as cyproheptadine should greatly improve the patient’s clinical status. Patients with severe toxicity may require induced paralysis and intubation.4 It is not clear in this case if the serotonin hyperactivation was due to the DMT (5-HT2A is associated with serotonin toxicity) or another serotonergic agent (eg, dextromethorphan from a cough and cold preparation) in combination with the MAO-inhibiting harmine alkaloids.
What is the availability of ayahuasca in the United States? How is it used in its nonherbal form?
...[Ayahuasca] is currently available in the United States and is legal for use by members of the UDV and Santo Daime churches. Many clinicians are becoming increasingly familiar with this herbal preparation since the recreational use of ayahuasca is gaining popularity in the United States. Internet fora with information on how to safely use ayahuasca, such as avoiding aged cheeses, are becoming more prevalent.7 A recent article in the New York Times described an ayahuasca gathering in Brooklyn, New York, where participants use the herb in a communal fashion.8 This herbal product is also associated with the Hollywood social scene and has received celebrity endorsements.8
The National Survey on Drug Use and Health found that the number of people in the United States who have used DMT has gone up almost every year since 2006, from an estimated 688,000 in 2006 to 1,475,000 in 2012.9 When used alone (not as ayahuasca), DMT is almost exclusively insufflated as a nasal snuff, bypassing hepatic elimination. It has an onset of around 45 seconds and a duration of 5 to 10 minutes. Insufflating DMT was historically referred to as a “businessman’s trip” because users were able to have a brief hallucinogenic experience on a lunch break and recover rapidly to perform their normal work.10
International law declares that DMT is an illegal substance and its importation is banned. However, its use for religious purposes, as is allowed for mescaline found in peyote, remains controversial.7 The UDV brought suit in United States federal court to prevent interference with the church’s use of ayahuasca during religious ceremonies based on the Religious Freedom Restoration Act. This act states that the government should not cause substantial imposition on religious practices in the absence of a compelling government interest. The court sided with the UDV, finding that the government had not sufficiently proved the alleged health risks posed by ayahuasca and could not show a substantial risk that the drug would be abused recreationally.11 Thus it is currently available in the United States and is legal for use by members of the UDV and Santo Daime churches.
Ayahuasca is not regulated by the US Food and Drug Administration. Many different types of preparations with different ingredients as well as different concentrations may exist, and clinical variability should be expected. Understanding that ayahuasca is capable of inhibiting MAO is important in order to avoid foods and medications, such as dextromethorphan, that may trigger adverse effects.
Case Conclusion
The patient’s hospital course was complicated by an additional seizure 12 hours after her initial presentation. By 36 hours she was back to her baseline mental status with a normal neurological examination.
Dr Fil is a senior fellow in medical toxicology at North Shore University Hospital, Manhasset, New York. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
Case
A 23-year-old Hispanic woman with no past medical history is brought to the ED for the second time in one day. On her first presentation, which was for a fever and a headache, meningitis was excluded with normal laboratory tests that included a lumbar puncture. She was administered acetaminophen for fever and pain control, and was discharged with a diagnosis of viral illness. On this second visit, 10 hours after being discharged, she presented because her family noted convulsions that began 3 hours after taking an herbal headache remedy given to her by a naturopath.
The patient arrived to the ED with a persistent seizure that terminated following administration of 2 mg of lorazepam. Her initial vital signs were: blood pressure, 115/51 mm Hg; heart rate, 121 beats/minute; respiratory rate, 24 breaths/minute; temperature, 97.6oF. Oxygen (O2) saturation was 100% with 2 L of O2 administered via nasal cannula. Her neurological examination was significant for a depressed mental status, pupils that were 6 mm and minimally reactive, clonus, and hyperreflexia. Repeat laboratory evaluation found a leukocytosis of 22.0 x 103/µL, serum bicarbonate of 9 mEq/L, and an anion gap of 22 with a normal serum lactate.
What is the differential diagnosis of this patient?
The history of medicinal plant ingestion raises the possibility of a toxicologic etiology. However, because the patient took the “medication” to treat another disorder, a search for an alternate cause should be performed. The differential diagnosis of a toxin-induced seizure is broad and includes pharmaceuticals (eg, tramadol, antihistamines), which may be surreptitiously added to herbal medication to assure efficacy. Plants associated with seizures include those containing antimuscarinic tropane alkaloids such as Jimsonweed (though a rare side effect from this plant product) or the water hemlock (Cicuta maculata). Contaminants of the plant itself may include pesticides such as organophosphates.
Although unlikely in a 21 year old, withdrawal from benzodiazepines, ethanol, baclofen, or gamma hydroxybutyrate are other possible etiologies. In addition to pharmaceutical and plant-derived causes, carbon monoxide poisoning should be a consideration in any patient with headache and flu-like illness.
This patient also presented with a constellation of other findings that included hyperreflexia, clonus, tachycardia, and altered mental status. Together these signs are expected in patients with serotonin toxicity (also referred to as serotonin syndrome), neuroleptic malignant syndrome, exogenous thyrotoxicosis, and lithium poisoning.
Case Continuation
The naturopathic practitioner arrived at the ED concerned about the patient, informing the ED team that she had given the patient 2 ounces of ayahuasca tea.
What is ayahuasca? What is the mechanism by which it exerts toxic effects?
Ayahuasca is a plant-derived psychotropic beverage that is used for religious purposes by members of two Brazilian churches—Centro Espírita Beneficente União do Vegetal (UDV) and Santo Daime. The ayahuasca beverage consists of two pharmacologically active compounds that together, but not individually, are psychoactive. The desired active effects for church participants include hallucinations, and vomiting to bring about a “religious purge.”1
Ayahuasca is prepared by combining two plants indigenous to the Amazon Basin area: Banisteriopsis caapi and either Psychotria viridis or Diplopterys cabrerana. B caapi contains the β-carboline alkaloids harmine, harmaline, and tetrahydroharmine. These alkaloids act as reversible inhibitors of the monoamine oxidase A (MAO-A) enzyme. The bark and stems of B caapi are boiled along with either P viridis or D cabrerana, both of which contain the potent hallucinogen N-N dimethyltryptamine (DMT).2 Normally, DMT is not active orally because it is enzymatically metabolized by MAO-A. However, when taken in the presence of the B caapi-derived MAO-A–inhibiting harmine alkaloids, DMT reaches the systemic circulation and produces its clinical effects.3
What are the clinical findings of serotonin toxicity?
Serotonin toxicity is a collection of clinical findings that fall under three main categories: autonomic hyperactivity, altered mental status, and muscle rigidity.5 The autonomic findings may include tachycardia, hypertension, hyperthermia, shivering, diaphoresis, or mydriasis. Altered mental status ranges from mild agitation and hypervigilance to agitated delirium to obtundation. Other neurological findings may include tremor, myoclonus, hyperreflexia, or seizures. The onset of these signs is rapid, usually occurring within minutes after exposure to one or more serotonergic compounds. Although rare, severe serotonin toxicity may be associated with hypotension and shock, leading to death.4
The diagnosis of serotonin toxicity is based on the history and physical examination of the patient. Diagnostic criteria that have been suggested include the following: (1) a recent addition or increase in a known serotonergic agent; (2) absence of other possible etiologies; (3) no recent increase or addition of a neuroleptic agent (suggesting neuroleptic malignant syndrome); and/or (4) at least 3 of the following symptoms—mental status changes, myoclonus, agitation, hyperreflexia, diaphoresis, shivering, tremor, diarrhea, incoordination, fever5 (Figure 2).
How should this patient be managed?
The management of serotonin toxicity is primarily supportive with aggressive control of hyperthermia and autonomic instability. The precipitating xenobiotic agent should be immediately discontinued. In general, treatment with intravenous fluids, cooling measures, benzodiazepines, and a nonspecific 5-HT antagonist such as cyproheptadine should greatly improve the patient’s clinical status. Patients with severe toxicity may require induced paralysis and intubation.4 It is not clear in this case if the serotonin hyperactivation was due to the DMT (5-HT2A is associated with serotonin toxicity) or another serotonergic agent (eg, dextromethorphan from a cough and cold preparation) in combination with the MAO-inhibiting harmine alkaloids.
What is the availability of ayahuasca in the United States? How is it used in its nonherbal form?
...[Ayahuasca] is currently available in the United States and is legal for use by members of the UDV and Santo Daime churches. Many clinicians are becoming increasingly familiar with this herbal preparation since the recreational use of ayahuasca is gaining popularity in the United States. Internet fora with information on how to safely use ayahuasca, such as avoiding aged cheeses, are becoming more prevalent.7 A recent article in the New York Times described an ayahuasca gathering in Brooklyn, New York, where participants use the herb in a communal fashion.8 This herbal product is also associated with the Hollywood social scene and has received celebrity endorsements.8
The National Survey on Drug Use and Health found that the number of people in the United States who have used DMT has gone up almost every year since 2006, from an estimated 688,000 in 2006 to 1,475,000 in 2012.9 When used alone (not as ayahuasca), DMT is almost exclusively insufflated as a nasal snuff, bypassing hepatic elimination. It has an onset of around 45 seconds and a duration of 5 to 10 minutes. Insufflating DMT was historically referred to as a “businessman’s trip” because users were able to have a brief hallucinogenic experience on a lunch break and recover rapidly to perform their normal work.10
International law declares that DMT is an illegal substance and its importation is banned. However, its use for religious purposes, as is allowed for mescaline found in peyote, remains controversial.7 The UDV brought suit in United States federal court to prevent interference with the church’s use of ayahuasca during religious ceremonies based on the Religious Freedom Restoration Act. This act states that the government should not cause substantial imposition on religious practices in the absence of a compelling government interest. The court sided with the UDV, finding that the government had not sufficiently proved the alleged health risks posed by ayahuasca and could not show a substantial risk that the drug would be abused recreationally.11 Thus it is currently available in the United States and is legal for use by members of the UDV and Santo Daime churches.
Ayahuasca is not regulated by the US Food and Drug Administration. Many different types of preparations with different ingredients as well as different concentrations may exist, and clinical variability should be expected. Understanding that ayahuasca is capable of inhibiting MAO is important in order to avoid foods and medications, such as dextromethorphan, that may trigger adverse effects.
Case Conclusion
The patient’s hospital course was complicated by an additional seizure 12 hours after her initial presentation. By 36 hours she was back to her baseline mental status with a normal neurological examination.
Dr Fil is a senior fellow in medical toxicology at North Shore University Hospital, Manhasset, New York. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Gable RS. Risk assessment of ritual use of oral dimethyltryptamine (DMT) and harmala alkaloids. Addiction. 2007;102(1):24-34.
- Riba J, McIlhenny EH, Valle M, Bouso JC, Barker SA. Metabolism and Disposition of N,N-dimethyltryptamine and harmala alkaloids after oral administration of ayahuasca. Drug Test Anal. 2012;4(7-8):610-616.
- Riba J, Valle M, Urbano G, Yritia M, Morte A, Barbanoj MJ. Human Pharmacology of Ayahuasca: Subjective and Cardiovascular Effects, Monoamine Metabolite Excretion and Pharmacokinetics. J Pharmacol Exp Ther. 2003;306(1):73-83
- Boyer EW, Shannon M. The serotonin syndrome. N Engl J Med. 2005;352(11);1112-1120.
- Sternbach H. The serotonin syndrome. Am J Psychiatry. 1991;148(6):6;705-713.
- Dunkley EJ, Isbister GK, Sibbritt D, Dawson AH, Whyte IM. The Hunter Serotonin Toxicity Criteria: simple and accurate diagnostic decision rules for serotonin toxicity. QJM. 2003;96(9):635-642.
- Erowid. Ayahuasca Vault. https://www.erowid.org/chemicals/ayahuasca/ayahuasca.shtml. Accessed November 25, 2014.
- Morris B. Ayahuasca: a strong cup of tea. New York Times. June 13, 2014. http://www.nytimes.com/2014/06/15/fashion/ayahuasca-a-strong-cup-of-tea.html. Accessed November 25, 2014.
- Quintanilla D. DMT: Hallucinogenic Drug Used in Shamanic Rituals Goes Mainstream. 10 Dec 2013. Available: http://www.opposingviews.com/i/health/dmt-hallucinogenic-drug-used-shamanic-rituals-goes-mainstream. Last accessed 11/14/14.
- Haroz R, Greenberg MI. Emerging drugs of abuse. Med Clin North Am. 2005;89(6):1259-1276.
- Gonzales v. O Centro Espirita Beneficente Uniao do Vegetal, 546 US 418 (2006). Available at http://scholar.google.com/scholar_case?case=7036734975431570669&hl=en&as_sdt=6&as_vis=1&oi=scholarr. Accessed November 25, 2014.
- Gable RS. Risk assessment of ritual use of oral dimethyltryptamine (DMT) and harmala alkaloids. Addiction. 2007;102(1):24-34.
- Riba J, McIlhenny EH, Valle M, Bouso JC, Barker SA. Metabolism and Disposition of N,N-dimethyltryptamine and harmala alkaloids after oral administration of ayahuasca. Drug Test Anal. 2012;4(7-8):610-616.
- Riba J, Valle M, Urbano G, Yritia M, Morte A, Barbanoj MJ. Human Pharmacology of Ayahuasca: Subjective and Cardiovascular Effects, Monoamine Metabolite Excretion and Pharmacokinetics. J Pharmacol Exp Ther. 2003;306(1):73-83
- Boyer EW, Shannon M. The serotonin syndrome. N Engl J Med. 2005;352(11);1112-1120.
- Sternbach H. The serotonin syndrome. Am J Psychiatry. 1991;148(6):6;705-713.
- Dunkley EJ, Isbister GK, Sibbritt D, Dawson AH, Whyte IM. The Hunter Serotonin Toxicity Criteria: simple and accurate diagnostic decision rules for serotonin toxicity. QJM. 2003;96(9):635-642.
- Erowid. Ayahuasca Vault. https://www.erowid.org/chemicals/ayahuasca/ayahuasca.shtml. Accessed November 25, 2014.
- Morris B. Ayahuasca: a strong cup of tea. New York Times. June 13, 2014. http://www.nytimes.com/2014/06/15/fashion/ayahuasca-a-strong-cup-of-tea.html. Accessed November 25, 2014.
- Quintanilla D. DMT: Hallucinogenic Drug Used in Shamanic Rituals Goes Mainstream. 10 Dec 2013. Available: http://www.opposingviews.com/i/health/dmt-hallucinogenic-drug-used-shamanic-rituals-goes-mainstream. Last accessed 11/14/14.
- Haroz R, Greenberg MI. Emerging drugs of abuse. Med Clin North Am. 2005;89(6):1259-1276.
- Gonzales v. O Centro Espirita Beneficente Uniao do Vegetal, 546 US 418 (2006). Available at http://scholar.google.com/scholar_case?case=7036734975431570669&hl=en&as_sdt=6&as_vis=1&oi=scholarr. Accessed November 25, 2014.

A 41-year-old man with abdominal pain
A 41-year-old man presented with pain in the left upper quadrant for 4 days. The pain was constant, was worse on inspiration, and did not radiate. He denied fevers, night sweats, nausea, vomiting, diarrhea, and urinary symptoms. He had been diagnosed with multiple sclerosis a few years earlier, and he had undergone aortofemoral bypass surgery on the left side 2 years ago. He denied smoking or using illicit drugs and described himself as a social drinker.
In the emergency room, he appeared comfortable. He was afebrile, blood pressure 136/69 mm Hg, pulse rate 98 per minute, and respiratory rate 16. All pulses were palpable and equal, the jugular venous pressure was not elevated, and no cardiac murmurs were heard. The abdomen was tender in the left upper quadrant, with no guarding or rigidity. Examination of the nervous, musculoskeletal, and respiratory systems was unremarkable. Skin examination revealed only scars from previous surgery.
LABORATORY AND IMAGING RESULTS
- White blood cell count 7.2 × 109/L (reference range 4.0–10.0) with a normal differential
- Hemoglobin 134 g/dL (140–180)
- Platelet count 167 × 109/L (150–400)
- Renal and liver panels were normal
- Erythrocyte sedimentation rate 30 mm/hour
- C-reactive protein level 14.3 mg/L
- D-dimer level 2,670 ng/mL (< 500)
- International normalized ratio (INR) 1.0 (0.9–1.3)
- Activated partial thromboplastin time (aPTT) 44 seconds (25–38)
- Fibrinogen level 3.0 g/L (1.8–3.5)
- Urinalysis negative for leukocytes and casts.
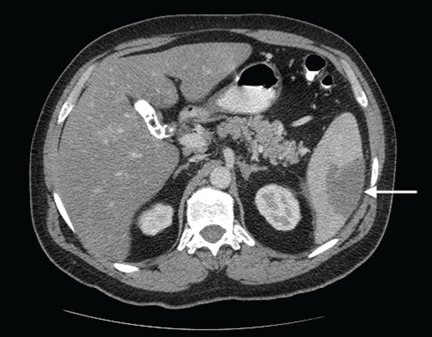
Computed tomography of the abdomen showed a wedge-shaped area of hypodensity along the inferolateral aspect of the spleen measuring 6 × 3.8 cm, consistent with a recent infarct (Figure 1). There was also evidence of a previous infarct in the posterolateral aspect of the spleen. Splenic, celiac, superior mesenteric, and inferior mesenteric arteries were patent.
1. Given these findings, which of the following diagnoses should be considered?
- Subacute infective endocarditis
- Inherited thrombophilia
- Antiphospholipid syndrome
All three diagnoses should be considered in this case.
Endocarditis
Embolism from a source in the heart caused by subacute bacterial endocarditis is more common than the other two conditions listed here and must be excluded.
Our patient lacks key features of this condition: he has no predisposing factors (artificial valve, cyanotic congenital heart disease, previous endocarditis, intravenous drug abuse); no constitutional symptoms of fever, night sweats, and weight loss; no findings on examination of skin and cardiovascular systems; and a normal white blood cell count. Nevertheless, even though the absence of these features makes bacterial endocarditis unlikely, it does not exclude it. Blood cultures and transesophageal echocardiography are indicated to rule out bacterial endocarditis.
We obtained serial blood cultures, which were negative, and transesophageal echocardiography showed normal valves and no evidence of thrombus or vegetation, thus excluding a cardiac source of emboli.
Thrombophilia
Our patient has a history of recurrent thromboembolic episodes, and this warrants testing to rule out an inherited thrombophilia. A family history of thromboembolic disease should also be sought.1
In our patient, tests for prothrombotic activity including protein C chromogen, activated protein C ratio, free protein S, functional protein S, antithrombin factor V Leiden, and the prothrombin 20210G>A mutation were either negative or within the reference range. A negative family history of thromboembolic disease and the negative laboratory tests make inherited thrombophilia unlikely in our patient.
Sickle cell disease, polycythemia vera, and essential thrombocythemia may also cause splenic infarction but can be ruled out in this patient on the basis of history and initial blood tests.
Antiphospholipid syndrome
A history of vascular disease (aortofemoral bypass surgery), a recent splenic infarct, and an elevated aPTT makes antiphospholipid syndrome the likeliest diagnosis in this patient.
Appropriate tests are for lupus anticoagulant, immunoglobulin G (IgG) or IgM cardiolipin antibody, and beta-2 glycoprotein 1 (beta-2 GP1) antibody, as well as the dilute Russell viper venom time (dRVVT) and the dRVVT ratio. The IgG and IgM cardiolipin antibody and beta-2 GP1 antibody tests have the same diagnostic value, and only medium to high titers should be considered positive.
Our patient’s IgG cardiolipin antibody level was in the normal range at 15 IgG phospholipid units (reference range 0–22); his IgM cardiolipin antibody level was high at 41 IgM phospholipid units (0–10). The dRVVT was 57 seconds (24–42), and the dRVVT ratio was 2.0 (0.0–1.3).
2. What further investigations are indicated before starting treatment?
- No further investigations required
- Repeat testing for phospholipid antibodies in 12 weeks
- Test for antinuclear antibodies
Antiphospholipid antibodies may appear transiently in certain infections, such as syphilis, Lyme disease, Epstein-Barr virus, cytomegalovirus, hepatitis C, and human immunodeficiency virus. Therefore, the presence of antiphospholipid antibodies must be confirmed over time, with two positive results at least 12 weeks apart.2
When repeated 12 weeks later, our patient’s IgG anticardiolipin antibody level was 14 GPL units, and the IgM anticardiolipin antibody level was 30 MPL units; the dRVVT was 55 seconds, and the dRVVT ratio was 1.8. These results, along with a history of recurrent arterial thrombosis, confirmed antiphospholipid syndrome.
The 2009 update of the International Society of Thrombosis and Haemostasis guidelines recommend two tests, the dRVVT and the aPTT, since no single test is 100% sensitive for lupus anticoagulant.3 The dRVVT has a high specificity for lupus anticoagulant in patients at high risk of thrombosis.
A SYNDROME WITH A WIDE RANGE OF EFFECTS AND COMPLICATIONS
Antiphospholipid syndrome is a systemic autoimmune disease that manifests as arterial and venous thrombosis and as obstetric complications. Thrombosis tends to be recurrent and may involve any site. For example, it can cause blurred vision in one or both eyes; amaurosis fugax; visual field defects; central or branch retinal artery or vein occlusion; deep vein thrombosis; pulmonary embolism; myocardial infarction; transient ischemic attack and stroke; cerebral vein thrombosis; and portal, renal, and mesenteric infarction involving veins or arteries.4 Pulmonary capillaritis may cause diffuse alveolar hemorrhage. Livedo reticularis, digital gangrene, cutaneous necrosis, splinter hemorrhages, chorea, and transverse myelopathy may also occur.
Obstetric complications of antiphospholipid syndrome include recurrent miscarriage and pregnancy loss at or after 10 weeks of gestation, eclampsia, preeclampsia, and placental insufficiency.5 The syndrome also has a potentially lethal variant characterized by multiorgan thrombosis affecting mainly small vessels.
The diagnosis of antiphospholipid syndrome requires relevant clinical features and symptoms and the presence of at least one of the antiphospholipid antibodies. Because the rate of false-positive tests for antiphospholipid antibodies ranges from 3% to 20% in the general population, asymptomatic patients should not be tested.6
Antiphospholipid syndrome may occur in the setting of other autoimmune diseases, most commonly systemic lupus erythematosus, when it is termed “secondary” antiphospholipid syndrome. Although only 40% of patients with lupus have antiphospholipid antibodies and less than 40% will have a thrombotic event, thrombotic antiphospholipid syndrome is a major adverse prognostic factor in these patients.7,8 Therefore, it is prudent to consider systemic lupus erythematosus and to do appropriate tests if the patient has other features suggestive of lupus, such as renal, skin, or musculoskeletal lesions.
In our patient, antinuclear antibody testing was positive, with a titer of 1:320, and showed a finely speckled staining pattern. Tests for antibodies to Sjögren syndrome A and B antigens were negative. The complement C3 level was 1.28 g/L (reference range 0.74–1.85) and the C4 level was 0.24 g/L (0.16–0.44). Although the speckled staining pattern can be seen in lupus, it is more common in Sjögren syndrome, mixed connective tissue disease, scleroderma, and CREST syndrome (calcinosis, Raynaud phenomenon, esophageal dysmotility, sclerodactyly, telangiectasia).9 Moreover, normal levels of complement C3 and C4, in the absence of clinical features, make lupus unlikely. Similarly, our patient had no clinical features of other connective tissue disorders. Therefore, he had primary antiphospholipid syndrome.
3. How should this patient be managed?
- Antiplatelet therapy
- Warfarin to maintain an INR between 2.0 and 3.0
- Warfarin to maintain an INR above 3.0
The risk of recurrent thrombosis is high in patients who test positive for lupus anticoagulant, and the risk is highest in patients who are also positive for anticardiolipin and anti-beta-2 GP1 antibodies: the incidence of thrombosis is 12.2% at 1 year, 26.1% at 5 years, 44.2% at 10 years.10
Since our patient is positive for lupus anticoagulant (prolonged aPTT and elevated dRVVT, both indicating lupus anticoagulant positivity) and for anticardiolipin antibodies (anti-beta-2 GP1 not tested), his risk of recurrent thrombosis is high, and he requires lifelong anticoagulation therapy.
The intensity of anticoagulation in different subgroups of patients is controversial. Based on retrospective trials, indefinite anticoagulation at an INR of 2.0 to 3.0 has been suggested for patients with antiphospholipid syndrome presenting with venous thrombosis, and more intense anticoagulation with an INR above 3.0 in patients with recurrent or arterial thrombosis.11 The combination of warfarin with an INR between 2.0 and 3.0 and aspirin 100 mg daily has also been proposed for patients with arterial thrombosis.12
Modifiable risk factors such as smoking, obesity, and use of estrogens should be addressed in all patients with antiphospholipid syndrome.
In pregnant women with complications such as preeclampsia, low-dose aspirin can be used, and in women with a history of miscarriage, the combination of low-dose aspirin and heparin is recommended throughout the prenatal period.4
In patients who have recurrent thrombosis despite adequate anticoagulation, an expert committee12 has proposed that alternative regimens could include long-term low-molecular-weight heparin instead of warfarin, the combination of warfarin and aspirin, or warfarin and hydroxychloroquine. Adding a statin can also be considered.
Treatment of catastrophic antiphospholipid syndrome is based on expert opinion. A combination of anticoagulation, corticosteroids, plasma exchange, intravenous immunoglobulins, and rituximab has been tried, but the mortality rate remains high.13
OUR PATIENT'S COURSE
Our patient was started on warfarin, with a target INR above 3.0, and was doing well at 6 months of follow-up.
- De Stefano V, Rossi E. Testing for inherited thrombophilia and consequences for antithrombotic prophylaxis in patients with venous thromboembolism and their relatives. A review of the Guidelines from Scientific Societies and Working Groups. Thromb Haemost 2013; 110:697–705.
- Galli M. Interpretation and recommended testing for antiphospholipid antibodies. Semin Thromb Hemost 2012; 38:348–352.
- Pengo V, Tripodi A, Reber G, et al; Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis. Update of the guidelines for lupus anticoagulant detection. Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis. J Thromb Haemost 2009; 7:1737–1740.
- Keeling D, Mackie I, Moore GW, Greer IA, Greaves M; British Committee for Standards in Haematology. Guidelines on the investigation and management of antiphospholipid syndrome. Br J Haematol 2012; 157:47–58.
- Misita CP, Moll S. Antiphospholipid antibodies. Circulation 2005; 112:e39–e44.
- Rand JH, Wolgast LR. Do’s and don’t’s in diagnosing antiphospholipid syndrome. Hematology Am Soc Hematol Educ Program 2012; 2012:455–459.
- Mok CC, Tang SS, To CH, Petri M. Incidence and risk factors of thromboembolism in systemic lupus erythematosus: a comparison of three ethnic groups. Arthritis Rheum 2005; 52:2774–2782.
- Ruiz-Irastorza G, Egurbide MV, Ugalde J, Aguirre C. High impact of antiphospholipid syndrome on irreversible organ damage and survival of patients with systemic lupus erythematosus. Arch Intern Med 2004; 164:77–82.
- Locht H, Pelck R, Manthorpe R. Clinical manifestations correlated to the prevalence of autoantibodies in a large (n = 321) cohort of patients with primary Sjögren’s syndrome: a comparison of patients initially diagnosed according to the Copenhagen classification criteria with the American-European consensus criteria. Autoimmun Rev 2005; 4:276–281.
- Pengo V, Ruffatti A, Legnani C, et al. Clinical course of high-risk patients diagnosed with antiphospholipid syndrome. J Thromb Haemost 2010; 8:237–242.
- Ruiz-Irastorza G, Crowther M, Branch W, Khamashta MA. Antiphospholipid syndrome. Lancet 2010; 376:1498–1509.
- Ruiz-Irastorza G, Cuadrado MJ, Ruiz-Arruza I, et al. Evidence-based recommendations for the prevention and long-term management of thrombosis in antiphospholipid antibody-positive patients: report of a task force at the 13th International Congress on antiphospholipid antibodies. Lupus 2011; 20:206–218.
- Cervera R. Update on the diagnosis, treatment, and prognosis of the catastrophic antiphospholipid syndrome. Curr Rheumatol Rep 2010; 12:70–76.
A 41-year-old man presented with pain in the left upper quadrant for 4 days. The pain was constant, was worse on inspiration, and did not radiate. He denied fevers, night sweats, nausea, vomiting, diarrhea, and urinary symptoms. He had been diagnosed with multiple sclerosis a few years earlier, and he had undergone aortofemoral bypass surgery on the left side 2 years ago. He denied smoking or using illicit drugs and described himself as a social drinker.
In the emergency room, he appeared comfortable. He was afebrile, blood pressure 136/69 mm Hg, pulse rate 98 per minute, and respiratory rate 16. All pulses were palpable and equal, the jugular venous pressure was not elevated, and no cardiac murmurs were heard. The abdomen was tender in the left upper quadrant, with no guarding or rigidity. Examination of the nervous, musculoskeletal, and respiratory systems was unremarkable. Skin examination revealed only scars from previous surgery.
LABORATORY AND IMAGING RESULTS
- White blood cell count 7.2 × 109/L (reference range 4.0–10.0) with a normal differential
- Hemoglobin 134 g/dL (140–180)
- Platelet count 167 × 109/L (150–400)
- Renal and liver panels were normal
- Erythrocyte sedimentation rate 30 mm/hour
- C-reactive protein level 14.3 mg/L
- D-dimer level 2,670 ng/mL (< 500)
- International normalized ratio (INR) 1.0 (0.9–1.3)
- Activated partial thromboplastin time (aPTT) 44 seconds (25–38)
- Fibrinogen level 3.0 g/L (1.8–3.5)
- Urinalysis negative for leukocytes and casts.
Computed tomography of the abdomen showed a wedge-shaped area of hypodensity along the inferolateral aspect of the spleen measuring 6 × 3.8 cm, consistent with a recent infarct (Figure 1). There was also evidence of a previous infarct in the posterolateral aspect of the spleen. Splenic, celiac, superior mesenteric, and inferior mesenteric arteries were patent.
1. Given these findings, which of the following diagnoses should be considered?
- Subacute infective endocarditis
- Inherited thrombophilia
- Antiphospholipid syndrome
All three diagnoses should be considered in this case.
Endocarditis
Embolism from a source in the heart caused by subacute bacterial endocarditis is more common than the other two conditions listed here and must be excluded.
Our patient lacks key features of this condition: he has no predisposing factors (artificial valve, cyanotic congenital heart disease, previous endocarditis, intravenous drug abuse); no constitutional symptoms of fever, night sweats, and weight loss; no findings on examination of skin and cardiovascular systems; and a normal white blood cell count. Nevertheless, even though the absence of these features makes bacterial endocarditis unlikely, it does not exclude it. Blood cultures and transesophageal echocardiography are indicated to rule out bacterial endocarditis.
We obtained serial blood cultures, which were negative, and transesophageal echocardiography showed normal valves and no evidence of thrombus or vegetation, thus excluding a cardiac source of emboli.
Thrombophilia
Our patient has a history of recurrent thromboembolic episodes, and this warrants testing to rule out an inherited thrombophilia. A family history of thromboembolic disease should also be sought.1
In our patient, tests for prothrombotic activity including protein C chromogen, activated protein C ratio, free protein S, functional protein S, antithrombin factor V Leiden, and the prothrombin 20210G>A mutation were either negative or within the reference range. A negative family history of thromboembolic disease and the negative laboratory tests make inherited thrombophilia unlikely in our patient.
Sickle cell disease, polycythemia vera, and essential thrombocythemia may also cause splenic infarction but can be ruled out in this patient on the basis of history and initial blood tests.
Antiphospholipid syndrome
A history of vascular disease (aortofemoral bypass surgery), a recent splenic infarct, and an elevated aPTT makes antiphospholipid syndrome the likeliest diagnosis in this patient.
Appropriate tests are for lupus anticoagulant, immunoglobulin G (IgG) or IgM cardiolipin antibody, and beta-2 glycoprotein 1 (beta-2 GP1) antibody, as well as the dilute Russell viper venom time (dRVVT) and the dRVVT ratio. The IgG and IgM cardiolipin antibody and beta-2 GP1 antibody tests have the same diagnostic value, and only medium to high titers should be considered positive.
Our patient’s IgG cardiolipin antibody level was in the normal range at 15 IgG phospholipid units (reference range 0–22); his IgM cardiolipin antibody level was high at 41 IgM phospholipid units (0–10). The dRVVT was 57 seconds (24–42), and the dRVVT ratio was 2.0 (0.0–1.3).
2. What further investigations are indicated before starting treatment?
- No further investigations required
- Repeat testing for phospholipid antibodies in 12 weeks
- Test for antinuclear antibodies
Antiphospholipid antibodies may appear transiently in certain infections, such as syphilis, Lyme disease, Epstein-Barr virus, cytomegalovirus, hepatitis C, and human immunodeficiency virus. Therefore, the presence of antiphospholipid antibodies must be confirmed over time, with two positive results at least 12 weeks apart.2
When repeated 12 weeks later, our patient’s IgG anticardiolipin antibody level was 14 GPL units, and the IgM anticardiolipin antibody level was 30 MPL units; the dRVVT was 55 seconds, and the dRVVT ratio was 1.8. These results, along with a history of recurrent arterial thrombosis, confirmed antiphospholipid syndrome.
The 2009 update of the International Society of Thrombosis and Haemostasis guidelines recommend two tests, the dRVVT and the aPTT, since no single test is 100% sensitive for lupus anticoagulant.3 The dRVVT has a high specificity for lupus anticoagulant in patients at high risk of thrombosis.
A SYNDROME WITH A WIDE RANGE OF EFFECTS AND COMPLICATIONS
Antiphospholipid syndrome is a systemic autoimmune disease that manifests as arterial and venous thrombosis and as obstetric complications. Thrombosis tends to be recurrent and may involve any site. For example, it can cause blurred vision in one or both eyes; amaurosis fugax; visual field defects; central or branch retinal artery or vein occlusion; deep vein thrombosis; pulmonary embolism; myocardial infarction; transient ischemic attack and stroke; cerebral vein thrombosis; and portal, renal, and mesenteric infarction involving veins or arteries.4 Pulmonary capillaritis may cause diffuse alveolar hemorrhage. Livedo reticularis, digital gangrene, cutaneous necrosis, splinter hemorrhages, chorea, and transverse myelopathy may also occur.
Obstetric complications of antiphospholipid syndrome include recurrent miscarriage and pregnancy loss at or after 10 weeks of gestation, eclampsia, preeclampsia, and placental insufficiency.5 The syndrome also has a potentially lethal variant characterized by multiorgan thrombosis affecting mainly small vessels.
The diagnosis of antiphospholipid syndrome requires relevant clinical features and symptoms and the presence of at least one of the antiphospholipid antibodies. Because the rate of false-positive tests for antiphospholipid antibodies ranges from 3% to 20% in the general population, asymptomatic patients should not be tested.6
Antiphospholipid syndrome may occur in the setting of other autoimmune diseases, most commonly systemic lupus erythematosus, when it is termed “secondary” antiphospholipid syndrome. Although only 40% of patients with lupus have antiphospholipid antibodies and less than 40% will have a thrombotic event, thrombotic antiphospholipid syndrome is a major adverse prognostic factor in these patients.7,8 Therefore, it is prudent to consider systemic lupus erythematosus and to do appropriate tests if the patient has other features suggestive of lupus, such as renal, skin, or musculoskeletal lesions.
In our patient, antinuclear antibody testing was positive, with a titer of 1:320, and showed a finely speckled staining pattern. Tests for antibodies to Sjögren syndrome A and B antigens were negative. The complement C3 level was 1.28 g/L (reference range 0.74–1.85) and the C4 level was 0.24 g/L (0.16–0.44). Although the speckled staining pattern can be seen in lupus, it is more common in Sjögren syndrome, mixed connective tissue disease, scleroderma, and CREST syndrome (calcinosis, Raynaud phenomenon, esophageal dysmotility, sclerodactyly, telangiectasia).9 Moreover, normal levels of complement C3 and C4, in the absence of clinical features, make lupus unlikely. Similarly, our patient had no clinical features of other connective tissue disorders. Therefore, he had primary antiphospholipid syndrome.
3. How should this patient be managed?
- Antiplatelet therapy
- Warfarin to maintain an INR between 2.0 and 3.0
- Warfarin to maintain an INR above 3.0
The risk of recurrent thrombosis is high in patients who test positive for lupus anticoagulant, and the risk is highest in patients who are also positive for anticardiolipin and anti-beta-2 GP1 antibodies: the incidence of thrombosis is 12.2% at 1 year, 26.1% at 5 years, 44.2% at 10 years.10
Since our patient is positive for lupus anticoagulant (prolonged aPTT and elevated dRVVT, both indicating lupus anticoagulant positivity) and for anticardiolipin antibodies (anti-beta-2 GP1 not tested), his risk of recurrent thrombosis is high, and he requires lifelong anticoagulation therapy.
The intensity of anticoagulation in different subgroups of patients is controversial. Based on retrospective trials, indefinite anticoagulation at an INR of 2.0 to 3.0 has been suggested for patients with antiphospholipid syndrome presenting with venous thrombosis, and more intense anticoagulation with an INR above 3.0 in patients with recurrent or arterial thrombosis.11 The combination of warfarin with an INR between 2.0 and 3.0 and aspirin 100 mg daily has also been proposed for patients with arterial thrombosis.12
Modifiable risk factors such as smoking, obesity, and use of estrogens should be addressed in all patients with antiphospholipid syndrome.
In pregnant women with complications such as preeclampsia, low-dose aspirin can be used, and in women with a history of miscarriage, the combination of low-dose aspirin and heparin is recommended throughout the prenatal period.4
In patients who have recurrent thrombosis despite adequate anticoagulation, an expert committee12 has proposed that alternative regimens could include long-term low-molecular-weight heparin instead of warfarin, the combination of warfarin and aspirin, or warfarin and hydroxychloroquine. Adding a statin can also be considered.
Treatment of catastrophic antiphospholipid syndrome is based on expert opinion. A combination of anticoagulation, corticosteroids, plasma exchange, intravenous immunoglobulins, and rituximab has been tried, but the mortality rate remains high.13
OUR PATIENT'S COURSE
Our patient was started on warfarin, with a target INR above 3.0, and was doing well at 6 months of follow-up.
A 41-year-old man presented with pain in the left upper quadrant for 4 days. The pain was constant, was worse on inspiration, and did not radiate. He denied fevers, night sweats, nausea, vomiting, diarrhea, and urinary symptoms. He had been diagnosed with multiple sclerosis a few years earlier, and he had undergone aortofemoral bypass surgery on the left side 2 years ago. He denied smoking or using illicit drugs and described himself as a social drinker.
In the emergency room, he appeared comfortable. He was afebrile, blood pressure 136/69 mm Hg, pulse rate 98 per minute, and respiratory rate 16. All pulses were palpable and equal, the jugular venous pressure was not elevated, and no cardiac murmurs were heard. The abdomen was tender in the left upper quadrant, with no guarding or rigidity. Examination of the nervous, musculoskeletal, and respiratory systems was unremarkable. Skin examination revealed only scars from previous surgery.
LABORATORY AND IMAGING RESULTS
- White blood cell count 7.2 × 109/L (reference range 4.0–10.0) with a normal differential
- Hemoglobin 134 g/dL (140–180)
- Platelet count 167 × 109/L (150–400)
- Renal and liver panels were normal
- Erythrocyte sedimentation rate 30 mm/hour
- C-reactive protein level 14.3 mg/L
- D-dimer level 2,670 ng/mL (< 500)
- International normalized ratio (INR) 1.0 (0.9–1.3)
- Activated partial thromboplastin time (aPTT) 44 seconds (25–38)
- Fibrinogen level 3.0 g/L (1.8–3.5)
- Urinalysis negative for leukocytes and casts.
Computed tomography of the abdomen showed a wedge-shaped area of hypodensity along the inferolateral aspect of the spleen measuring 6 × 3.8 cm, consistent with a recent infarct (Figure 1). There was also evidence of a previous infarct in the posterolateral aspect of the spleen. Splenic, celiac, superior mesenteric, and inferior mesenteric arteries were patent.
1. Given these findings, which of the following diagnoses should be considered?
- Subacute infective endocarditis
- Inherited thrombophilia
- Antiphospholipid syndrome
All three diagnoses should be considered in this case.
Endocarditis
Embolism from a source in the heart caused by subacute bacterial endocarditis is more common than the other two conditions listed here and must be excluded.
Our patient lacks key features of this condition: he has no predisposing factors (artificial valve, cyanotic congenital heart disease, previous endocarditis, intravenous drug abuse); no constitutional symptoms of fever, night sweats, and weight loss; no findings on examination of skin and cardiovascular systems; and a normal white blood cell count. Nevertheless, even though the absence of these features makes bacterial endocarditis unlikely, it does not exclude it. Blood cultures and transesophageal echocardiography are indicated to rule out bacterial endocarditis.
We obtained serial blood cultures, which were negative, and transesophageal echocardiography showed normal valves and no evidence of thrombus or vegetation, thus excluding a cardiac source of emboli.
Thrombophilia
Our patient has a history of recurrent thromboembolic episodes, and this warrants testing to rule out an inherited thrombophilia. A family history of thromboembolic disease should also be sought.1
In our patient, tests for prothrombotic activity including protein C chromogen, activated protein C ratio, free protein S, functional protein S, antithrombin factor V Leiden, and the prothrombin 20210G>A mutation were either negative or within the reference range. A negative family history of thromboembolic disease and the negative laboratory tests make inherited thrombophilia unlikely in our patient.
Sickle cell disease, polycythemia vera, and essential thrombocythemia may also cause splenic infarction but can be ruled out in this patient on the basis of history and initial blood tests.
Antiphospholipid syndrome
A history of vascular disease (aortofemoral bypass surgery), a recent splenic infarct, and an elevated aPTT makes antiphospholipid syndrome the likeliest diagnosis in this patient.
Appropriate tests are for lupus anticoagulant, immunoglobulin G (IgG) or IgM cardiolipin antibody, and beta-2 glycoprotein 1 (beta-2 GP1) antibody, as well as the dilute Russell viper venom time (dRVVT) and the dRVVT ratio. The IgG and IgM cardiolipin antibody and beta-2 GP1 antibody tests have the same diagnostic value, and only medium to high titers should be considered positive.
Our patient’s IgG cardiolipin antibody level was in the normal range at 15 IgG phospholipid units (reference range 0–22); his IgM cardiolipin antibody level was high at 41 IgM phospholipid units (0–10). The dRVVT was 57 seconds (24–42), and the dRVVT ratio was 2.0 (0.0–1.3).
2. What further investigations are indicated before starting treatment?
- No further investigations required
- Repeat testing for phospholipid antibodies in 12 weeks
- Test for antinuclear antibodies
Antiphospholipid antibodies may appear transiently in certain infections, such as syphilis, Lyme disease, Epstein-Barr virus, cytomegalovirus, hepatitis C, and human immunodeficiency virus. Therefore, the presence of antiphospholipid antibodies must be confirmed over time, with two positive results at least 12 weeks apart.2
When repeated 12 weeks later, our patient’s IgG anticardiolipin antibody level was 14 GPL units, and the IgM anticardiolipin antibody level was 30 MPL units; the dRVVT was 55 seconds, and the dRVVT ratio was 1.8. These results, along with a history of recurrent arterial thrombosis, confirmed antiphospholipid syndrome.
The 2009 update of the International Society of Thrombosis and Haemostasis guidelines recommend two tests, the dRVVT and the aPTT, since no single test is 100% sensitive for lupus anticoagulant.3 The dRVVT has a high specificity for lupus anticoagulant in patients at high risk of thrombosis.
A SYNDROME WITH A WIDE RANGE OF EFFECTS AND COMPLICATIONS
Antiphospholipid syndrome is a systemic autoimmune disease that manifests as arterial and venous thrombosis and as obstetric complications. Thrombosis tends to be recurrent and may involve any site. For example, it can cause blurred vision in one or both eyes; amaurosis fugax; visual field defects; central or branch retinal artery or vein occlusion; deep vein thrombosis; pulmonary embolism; myocardial infarction; transient ischemic attack and stroke; cerebral vein thrombosis; and portal, renal, and mesenteric infarction involving veins or arteries.4 Pulmonary capillaritis may cause diffuse alveolar hemorrhage. Livedo reticularis, digital gangrene, cutaneous necrosis, splinter hemorrhages, chorea, and transverse myelopathy may also occur.
Obstetric complications of antiphospholipid syndrome include recurrent miscarriage and pregnancy loss at or after 10 weeks of gestation, eclampsia, preeclampsia, and placental insufficiency.5 The syndrome also has a potentially lethal variant characterized by multiorgan thrombosis affecting mainly small vessels.
The diagnosis of antiphospholipid syndrome requires relevant clinical features and symptoms and the presence of at least one of the antiphospholipid antibodies. Because the rate of false-positive tests for antiphospholipid antibodies ranges from 3% to 20% in the general population, asymptomatic patients should not be tested.6
Antiphospholipid syndrome may occur in the setting of other autoimmune diseases, most commonly systemic lupus erythematosus, when it is termed “secondary” antiphospholipid syndrome. Although only 40% of patients with lupus have antiphospholipid antibodies and less than 40% will have a thrombotic event, thrombotic antiphospholipid syndrome is a major adverse prognostic factor in these patients.7,8 Therefore, it is prudent to consider systemic lupus erythematosus and to do appropriate tests if the patient has other features suggestive of lupus, such as renal, skin, or musculoskeletal lesions.
In our patient, antinuclear antibody testing was positive, with a titer of 1:320, and showed a finely speckled staining pattern. Tests for antibodies to Sjögren syndrome A and B antigens were negative. The complement C3 level was 1.28 g/L (reference range 0.74–1.85) and the C4 level was 0.24 g/L (0.16–0.44). Although the speckled staining pattern can be seen in lupus, it is more common in Sjögren syndrome, mixed connective tissue disease, scleroderma, and CREST syndrome (calcinosis, Raynaud phenomenon, esophageal dysmotility, sclerodactyly, telangiectasia).9 Moreover, normal levels of complement C3 and C4, in the absence of clinical features, make lupus unlikely. Similarly, our patient had no clinical features of other connective tissue disorders. Therefore, he had primary antiphospholipid syndrome.
3. How should this patient be managed?
- Antiplatelet therapy
- Warfarin to maintain an INR between 2.0 and 3.0
- Warfarin to maintain an INR above 3.0
The risk of recurrent thrombosis is high in patients who test positive for lupus anticoagulant, and the risk is highest in patients who are also positive for anticardiolipin and anti-beta-2 GP1 antibodies: the incidence of thrombosis is 12.2% at 1 year, 26.1% at 5 years, 44.2% at 10 years.10
Since our patient is positive for lupus anticoagulant (prolonged aPTT and elevated dRVVT, both indicating lupus anticoagulant positivity) and for anticardiolipin antibodies (anti-beta-2 GP1 not tested), his risk of recurrent thrombosis is high, and he requires lifelong anticoagulation therapy.
The intensity of anticoagulation in different subgroups of patients is controversial. Based on retrospective trials, indefinite anticoagulation at an INR of 2.0 to 3.0 has been suggested for patients with antiphospholipid syndrome presenting with venous thrombosis, and more intense anticoagulation with an INR above 3.0 in patients with recurrent or arterial thrombosis.11 The combination of warfarin with an INR between 2.0 and 3.0 and aspirin 100 mg daily has also been proposed for patients with arterial thrombosis.12
Modifiable risk factors such as smoking, obesity, and use of estrogens should be addressed in all patients with antiphospholipid syndrome.
In pregnant women with complications such as preeclampsia, low-dose aspirin can be used, and in women with a history of miscarriage, the combination of low-dose aspirin and heparin is recommended throughout the prenatal period.4
In patients who have recurrent thrombosis despite adequate anticoagulation, an expert committee12 has proposed that alternative regimens could include long-term low-molecular-weight heparin instead of warfarin, the combination of warfarin and aspirin, or warfarin and hydroxychloroquine. Adding a statin can also be considered.
Treatment of catastrophic antiphospholipid syndrome is based on expert opinion. A combination of anticoagulation, corticosteroids, plasma exchange, intravenous immunoglobulins, and rituximab has been tried, but the mortality rate remains high.13
OUR PATIENT'S COURSE
Our patient was started on warfarin, with a target INR above 3.0, and was doing well at 6 months of follow-up.
- De Stefano V, Rossi E. Testing for inherited thrombophilia and consequences for antithrombotic prophylaxis in patients with venous thromboembolism and their relatives. A review of the Guidelines from Scientific Societies and Working Groups. Thromb Haemost 2013; 110:697–705.
- Galli M. Interpretation and recommended testing for antiphospholipid antibodies. Semin Thromb Hemost 2012; 38:348–352.
- Pengo V, Tripodi A, Reber G, et al; Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis. Update of the guidelines for lupus anticoagulant detection. Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis. J Thromb Haemost 2009; 7:1737–1740.
- Keeling D, Mackie I, Moore GW, Greer IA, Greaves M; British Committee for Standards in Haematology. Guidelines on the investigation and management of antiphospholipid syndrome. Br J Haematol 2012; 157:47–58.
- Misita CP, Moll S. Antiphospholipid antibodies. Circulation 2005; 112:e39–e44.
- Rand JH, Wolgast LR. Do’s and don’t’s in diagnosing antiphospholipid syndrome. Hematology Am Soc Hematol Educ Program 2012; 2012:455–459.
- Mok CC, Tang SS, To CH, Petri M. Incidence and risk factors of thromboembolism in systemic lupus erythematosus: a comparison of three ethnic groups. Arthritis Rheum 2005; 52:2774–2782.
- Ruiz-Irastorza G, Egurbide MV, Ugalde J, Aguirre C. High impact of antiphospholipid syndrome on irreversible organ damage and survival of patients with systemic lupus erythematosus. Arch Intern Med 2004; 164:77–82.
- Locht H, Pelck R, Manthorpe R. Clinical manifestations correlated to the prevalence of autoantibodies in a large (n = 321) cohort of patients with primary Sjögren’s syndrome: a comparison of patients initially diagnosed according to the Copenhagen classification criteria with the American-European consensus criteria. Autoimmun Rev 2005; 4:276–281.
- Pengo V, Ruffatti A, Legnani C, et al. Clinical course of high-risk patients diagnosed with antiphospholipid syndrome. J Thromb Haemost 2010; 8:237–242.
- Ruiz-Irastorza G, Crowther M, Branch W, Khamashta MA. Antiphospholipid syndrome. Lancet 2010; 376:1498–1509.
- Ruiz-Irastorza G, Cuadrado MJ, Ruiz-Arruza I, et al. Evidence-based recommendations for the prevention and long-term management of thrombosis in antiphospholipid antibody-positive patients: report of a task force at the 13th International Congress on antiphospholipid antibodies. Lupus 2011; 20:206–218.
- Cervera R. Update on the diagnosis, treatment, and prognosis of the catastrophic antiphospholipid syndrome. Curr Rheumatol Rep 2010; 12:70–76.
- De Stefano V, Rossi E. Testing for inherited thrombophilia and consequences for antithrombotic prophylaxis in patients with venous thromboembolism and their relatives. A review of the Guidelines from Scientific Societies and Working Groups. Thromb Haemost 2013; 110:697–705.
- Galli M. Interpretation and recommended testing for antiphospholipid antibodies. Semin Thromb Hemost 2012; 38:348–352.
- Pengo V, Tripodi A, Reber G, et al; Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis. Update of the guidelines for lupus anticoagulant detection. Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis. J Thromb Haemost 2009; 7:1737–1740.
- Keeling D, Mackie I, Moore GW, Greer IA, Greaves M; British Committee for Standards in Haematology. Guidelines on the investigation and management of antiphospholipid syndrome. Br J Haematol 2012; 157:47–58.
- Misita CP, Moll S. Antiphospholipid antibodies. Circulation 2005; 112:e39–e44.
- Rand JH, Wolgast LR. Do’s and don’t’s in diagnosing antiphospholipid syndrome. Hematology Am Soc Hematol Educ Program 2012; 2012:455–459.
- Mok CC, Tang SS, To CH, Petri M. Incidence and risk factors of thromboembolism in systemic lupus erythematosus: a comparison of three ethnic groups. Arthritis Rheum 2005; 52:2774–2782.
- Ruiz-Irastorza G, Egurbide MV, Ugalde J, Aguirre C. High impact of antiphospholipid syndrome on irreversible organ damage and survival of patients with systemic lupus erythematosus. Arch Intern Med 2004; 164:77–82.
- Locht H, Pelck R, Manthorpe R. Clinical manifestations correlated to the prevalence of autoantibodies in a large (n = 321) cohort of patients with primary Sjögren’s syndrome: a comparison of patients initially diagnosed according to the Copenhagen classification criteria with the American-European consensus criteria. Autoimmun Rev 2005; 4:276–281.
- Pengo V, Ruffatti A, Legnani C, et al. Clinical course of high-risk patients diagnosed with antiphospholipid syndrome. J Thromb Haemost 2010; 8:237–242.
- Ruiz-Irastorza G, Crowther M, Branch W, Khamashta MA. Antiphospholipid syndrome. Lancet 2010; 376:1498–1509.
- Ruiz-Irastorza G, Cuadrado MJ, Ruiz-Arruza I, et al. Evidence-based recommendations for the prevention and long-term management of thrombosis in antiphospholipid antibody-positive patients: report of a task force at the 13th International Congress on antiphospholipid antibodies. Lupus 2011; 20:206–218.
- Cervera R. Update on the diagnosis, treatment, and prognosis of the catastrophic antiphospholipid syndrome. Curr Rheumatol Rep 2010; 12:70–76.
Ebola—lessons still to be learned

In this issue of the Journal, Dr. Kyle Brizendine reviews the basics of the Ebola virus and its natural history, diagnosis, and management.
Like many of you, I have followed the Ebola story with disquietude. So far, the disease has barely touched our country, with fewer than 10 confirmed cases on US soil, but it has had a big impact on our health care system and our national psyche.
The creation of specialized containment and management units may deplete some hospitals and their communities of intensive care beds. Specially trained caregivers will need to be diverted to staff these units, and the public’s fear may dissuade patients from undergoing elective procedures at hospitals caring for patients with Ebola. All of these pose a financial challenge to the hospitals most capable of dealing with these patients.
We have yet to hear about management guidelines dealing with renal replacement therapy and ventilator support, which may extend life but also pose extra risks to caregivers. Do we understand the disease well enough to know when advanced supportive therapies might be futile? Many lessons were learned from the Liberian patient who died of Ebola in Dallas, but many more clinical questions remain. I had hoped that in our sophisticated ICUs patients treated relatively early with aggressive supportive care would likely survive. We do not yet know if that is true. One death does not make it false, but it does give one pause.
About a half dozen other Ebola patients have survived with treatment here, but they were not African. Does genetic background play a role in disease severity and survival? Were the survivors treated sooner or differently in ways that matter? How much of the end-organ damage from the virus is from direct organ infection that cannot be reversed or prevented by even the best supportive treatment? Does the ability of the virus to suppress the immune system doom patients to opportunistic infections during prolonged supportive therapy? Is the viral-associated immunosuppression enough to prevent some patients from mounting an effective innate (interferon-based) or acquired (viral-specific T-cell or humoral) antiviral response? And is transfusing blood from survivors, presumably conferring passive immunity, actually efficacious?
I was relieved there were no new Ebola cases among the staff caring for Mr. Duncan at his second emergency room visit in Dallas, since at that time he was clearly quite ill, viremic, and contagious. Universal safety precautions must have helped. But how did the other nurses become infected, even though they presumably wore better protection? Hopefully, we will gain further understanding of transmissibility and resistance. We need this knowledge to inform safe and manageable protocols of care, particularly if successful vaccine development is delayed.
In this issue of the Journal, Dr. Kyle Brizendine reviews the basics of the Ebola virus and its natural history, diagnosis, and management.
Like many of you, I have followed the Ebola story with disquietude. So far, the disease has barely touched our country, with fewer than 10 confirmed cases on US soil, but it has had a big impact on our health care system and our national psyche.
The creation of specialized containment and management units may deplete some hospitals and their communities of intensive care beds. Specially trained caregivers will need to be diverted to staff these units, and the public’s fear may dissuade patients from undergoing elective procedures at hospitals caring for patients with Ebola. All of these pose a financial challenge to the hospitals most capable of dealing with these patients.
We have yet to hear about management guidelines dealing with renal replacement therapy and ventilator support, which may extend life but also pose extra risks to caregivers. Do we understand the disease well enough to know when advanced supportive therapies might be futile? Many lessons were learned from the Liberian patient who died of Ebola in Dallas, but many more clinical questions remain. I had hoped that in our sophisticated ICUs patients treated relatively early with aggressive supportive care would likely survive. We do not yet know if that is true. One death does not make it false, but it does give one pause.
About a half dozen other Ebola patients have survived with treatment here, but they were not African. Does genetic background play a role in disease severity and survival? Were the survivors treated sooner or differently in ways that matter? How much of the end-organ damage from the virus is from direct organ infection that cannot be reversed or prevented by even the best supportive treatment? Does the ability of the virus to suppress the immune system doom patients to opportunistic infections during prolonged supportive therapy? Is the viral-associated immunosuppression enough to prevent some patients from mounting an effective innate (interferon-based) or acquired (viral-specific T-cell or humoral) antiviral response? And is transfusing blood from survivors, presumably conferring passive immunity, actually efficacious?
I was relieved there were no new Ebola cases among the staff caring for Mr. Duncan at his second emergency room visit in Dallas, since at that time he was clearly quite ill, viremic, and contagious. Universal safety precautions must have helped. But how did the other nurses become infected, even though they presumably wore better protection? Hopefully, we will gain further understanding of transmissibility and resistance. We need this knowledge to inform safe and manageable protocols of care, particularly if successful vaccine development is delayed.
In this issue of the Journal, Dr. Kyle Brizendine reviews the basics of the Ebola virus and its natural history, diagnosis, and management.
Like many of you, I have followed the Ebola story with disquietude. So far, the disease has barely touched our country, with fewer than 10 confirmed cases on US soil, but it has had a big impact on our health care system and our national psyche.
The creation of specialized containment and management units may deplete some hospitals and their communities of intensive care beds. Specially trained caregivers will need to be diverted to staff these units, and the public’s fear may dissuade patients from undergoing elective procedures at hospitals caring for patients with Ebola. All of these pose a financial challenge to the hospitals most capable of dealing with these patients.
We have yet to hear about management guidelines dealing with renal replacement therapy and ventilator support, which may extend life but also pose extra risks to caregivers. Do we understand the disease well enough to know when advanced supportive therapies might be futile? Many lessons were learned from the Liberian patient who died of Ebola in Dallas, but many more clinical questions remain. I had hoped that in our sophisticated ICUs patients treated relatively early with aggressive supportive care would likely survive. We do not yet know if that is true. One death does not make it false, but it does give one pause.
About a half dozen other Ebola patients have survived with treatment here, but they were not African. Does genetic background play a role in disease severity and survival? Were the survivors treated sooner or differently in ways that matter? How much of the end-organ damage from the virus is from direct organ infection that cannot be reversed or prevented by even the best supportive treatment? Does the ability of the virus to suppress the immune system doom patients to opportunistic infections during prolonged supportive therapy? Is the viral-associated immunosuppression enough to prevent some patients from mounting an effective innate (interferon-based) or acquired (viral-specific T-cell or humoral) antiviral response? And is transfusing blood from survivors, presumably conferring passive immunity, actually efficacious?
I was relieved there were no new Ebola cases among the staff caring for Mr. Duncan at his second emergency room visit in Dallas, since at that time he was clearly quite ill, viremic, and contagious. Universal safety precautions must have helped. But how did the other nurses become infected, even though they presumably wore better protection? Hopefully, we will gain further understanding of transmissibility and resistance. We need this knowledge to inform safe and manageable protocols of care, particularly if successful vaccine development is delayed.
Ebola virus: Questions, answers, and more questions
A 50-year-old man who returned from a business trip to Nigeria 24 days ago presents with complaints of the sudden onset of fever, diarrhea, myalgia, and headache. He reports 10 bowel movements per day and has seen bloody stools.
During his trip he flew in to Murtala Muhammed International Airport in Lagos, ate meals only in his hotel, and attended meetings in Lagos central business district. He had no exposure to animals, mosquitoes, ticks, or sick people, and no sexual activity. After returning home, he felt well for the first 3 weeks.
The patient has a history of hypertension. He does not smoke, drink alcohol, or use injection drugs. He is married, works with commercial banks and financial institutions, and lives in Cleveland, OH.
On physical examination his temperature is 100.0˚F (37.8˚C), pulse 98, respirations 15, blood pressure 105/70 mm Hg, and weight 78 kg (172 lb). He appears comfortable but is a little diaphoretic. His abdomen is tender to palpation in the epigastrium and slightly to the right; he has no signs of peritonitis. His skin is without rash, bleeding, or bruising. The remainder of the examination is normal.
His white blood cell count is 17 × 109/L, hemoglobin 15 g/dL, hematocrit 41%, and platelet count 172 × 109/L. His sodium level is 126 mmol/L, potassium 3.8 mmol/L, chloride 95 mmol/L, carbon dioxide 20 mmol/L, blood urea nitrogen 11 mg/dL, creatinine 0.7 mg/dL, and glucose 130 mg/dL. His aminotransferase and alkaline phosphatase levels are normal.
Could this patient have Ebola virus disease?
With Ebola virus disease on the rise in West Africa, physicians who encounter patients like this one need to include it in the differential diagnosis. Because the disease is new, many questions are raised for which we as yet have no answers. Here, I will review what we know and do not know in an effort to remove some of the fear and uncertainty.
A NEW DISEASE

Ebola virus disease is a severe hemorrhagic fever caused by negative-sense single-stranded RNA viruses classified by the International Committee on Taxonomy of Viruses as belonging to the genus Ebolavirus in the family Filoviridae. Filoviruses get their name from the Latin filum, or thread-like structure.
The family Filoviridae was discovered in 1967 after inadvertent importation of infected monkeys from Uganda into Yugoslavia and Marburg, Germany. Outbreaks of severe illness occurred in workers at a vaccine plant who came into direct contact with the animals by killing them, removing their kidneys, or preparing primary cell cultures for polio vaccine production.
Ebola virus was discovered in 1976 by Peter Piot, who was working at the Institute of Tropical Medicine in Antwerp, Belgium. The blood of a Belgian woman who had been working in what is now the Democratic Republic of the Congo (formerly Zaire) had been sent to the institute; she and Mabalo Lokela, a school headmaster and the first recorded victim of Ebola virus, had been working near Yambuku, about 96 km from the Ebola River.
Before the 2014 outbreak, all known outbreaks had caused fewer than 2,400 cases across a dozen African countries over 3 decades.
Five species of Ebola virus
The genus Ebolavirus contains five species, each associated with a consistent case-fatality rate and a more or less well-identified endemic area.1
Zaire ebolavirus was recognized in 1976; it has caused multiple outbreaks, with high case-fatality rates.
Sudan ebolavirus was seen first in the 1970s; it has a 50% case-fatality rate.
Tai Forest ebolavirus has been found in only one person, an ethologist working with deceased chimpanzees.
Bundibugyo ebolavirus emerged in 2007 and has a 30% case-fatality rate.
Reston ebolavirus is maintained in an animal reservoir in the Philippines and is not found in Africa. It caused an outbreak of lethal infection in macaques imported into the United States in 1989. There is evidence that Reston ebolavirus can cause asymptomatic infection in humans. None of the caretakers of the macaques became ill, nor did farmers working with infected pigs, although both groups seroconverted.
A reservoir in bats?
A reservoir in nonhuman primates was initially suspected. However, studies subsequently showed that monkeys are susceptible to rapidly lethal filoviral disease, precluding any role as a host for persistent viral infection. It is likely that Ebola virus is maintained in small animals that serve as a source of infection for both humans and wild primates. A prominent suspect is fruit bats, which are consumed in soup in West Africa.
Transmission is person-to-person or nosocomial
Ebola virus is transmitted by direct contact with body fluids such as blood, urine, sweat, vomitus, semen, and breast milk. Filoviruses can initiate infection via ingestion, inhalation (although probably not Ebola), or passage through breaks in the skin. Droplet inoculation into the mouth or eyes has been shown to result from inadvertent transfer of virus from contaminated hands. Patients transmit the virus while febrile and through later stages of disease, as well as postmortem through contact with the body during funeral preparations. The virus has been isolated in semen for as many as 61 days after illness onset.
Ebola virus can also be spread nosocomially. In 1976, a 44-year-old teacher sought care for fever at the Yambuku Mission Hospital. He was given parenteral chloroquine as empiric treatment for presumed malaria, which was routine for all febrile patients. However, he had unrecognized Ebola virus infection. Moreover, syringes were rinsed in the same pan of water and reused, which spread the infection to nearly 100 people, all of whom developed fulminant Ebola virus disease and died. Infection then spread to family caregivers, the hospital staff, and those who prepared the bodies for burial.
Nosocomial transmission was also responsible for an outbreak of Lake Victoria Marburg virus in Uige Province in northern Angola in 2005, with 374 putative cases and 329 deaths. When teams from Médecins Sans Frontières started setting up the Marburg ward, there were five patients with hemorrhagic fever in a makeshift isolation room in the hospital, together with corpses that the hospital staff had been too afraid to remove. Healers found in many rural African communities were administering injections in homes or in makeshift clinics with reused needles or syringes.2
There is no evidence that filoviruses are carried by mosquitoes or other biting arthropods. Also, the risk of transmission via fomites appears to be low when currently recommended infection-control guidelines for the viral hemorrhagic fevers are followed.3 One primary human case generates only one to three secondary cases on average.
EBOLA IS AN IMMUNODEFICIENCY VIRUS
The main targets of infection are endothelial cells, mononuclear phagocytes, and hepatocytes. Ebola virus replicates at an unusually high rate. Macrophages infected with Zaire ebolavirus produce tumor necrosis factor alpha, interleukin (IL) 1 beta, IL-6, macrophage chemotactic protein 1, and nitric oxide. Virus-infected macrophages synthesize cell-surface tissue factor, triggering the extrinsic coagulation pathway.
Ebola is an immunodeficiency virus. Dendritic cells, which initiate adaptive immune responses, are a major site of filoviral replication. Infected cells cannot present antigens to naïve lymphocytes. Patients who die of Ebola virus disease do not develop antibodies to the virus. Lymphocytes remain uninfected, but undergo “bystander” apoptosis induced by inflammatory mediators.
CLINICAL MANIFESTATIONS
The incubation period is generally 5 to 7 days (range 2 to 28 days), during which the patient is not infectious. Symptoms begin abruptly, with fever, chills, general malaise, weakness, severe headache, and myalgia. By the time of case detection in West Africa, most patients also had nausea, vomiting, diarrhea, and abdominal pain. Once symptoms arise, patients have high levels of the virus in their blood and fluids and are infectious. Hemorrhagic symptoms have apparently been uncommon in West Africa, occurring in 1.0% to 5.7%, but “unexplained bleeding” has been documented in 18% of cases.4 Among those in whom the disease enters its hemorrhagic terminal phase, there is characteristic internal and subcutaneous bleeding, vomiting of blood, and subconjunctival hemorrhage.4
Laboratory findings include lymphocytopenia (often with counts as low as 1.0 × 109/L), thrombocytopenia (with counts in the range of 50 to 100 × 109/L), elevated aminotransferase levels (including aspartate aminotransferase levels 7 to 12 times higher than alanine aminotransferase in fatal cases), low total protein (due to capillary leak), and disseminated intravascular coagulation. Those who survive begin to improve in the second week, during which viremia resolves in association with the appearance of virus-specific antibodies.4
DIAGNOSIS
In symptomatic patients, Ebola virus infection is diagnosed by detection in blood or body fluids of viral antigens by enzyme-linked immunosorbent assay, or RNA sequences by reverse transcriptase polymerase chain reaction. The diagnosis is confirmed with cell culture (in a BSL-4 containment laboratory) showing characteristic viral particles by electron microscopy.
CARING FOR PATIENTS
The most detailed descriptions of the care of patients with Ebola virus disease have come from Dr. Bruce Ribner, of Emory University Hospital, in an October 2014 report of his experience caring for Ebola-infected patients at Emory University Hospital in Atlanta, GA.5 He described fluid losses of 5 to 10 L/day, profound hyponatremia, hypokalemia, and hypocalcemia, which were associated with cardiac arrhythmias and the need for intravenous and oral electrolyte repletion and hemodialysis. Intensive one-to-one nursing was critical, as was the coordination of many medical subspecialties. The Emory team arranged point-of-care testing near the unit and generally kept laboratory testing to a minimum. The team was surprised to learn that commercial carriers refused to transport specimens even when they were licensed for category A agents. Difficulties with the local water authority and waste disposal contractor required the hospital to dedicate an autoclave to process all materials used in clinical care.
TREATMENT: SUPPORTIVE AND EXPERIMENTAL
Treatment is supportive to maintain circulatory function and blood pressure and to correct coagulopathy. However, a variety of vaccines, antibodies, small-molecule agents, and antiviral agents are undergoing testing, mostly in animals at this point.
Vaccines. A therapeutic vaccine that worked only slightly was a live-attenuated recombinant vesicular stomatitis virus expressing Ebola virus transmembrane glycoproteins, which was tested in mice, guinea pigs, and rhesus macaques who had been exposed to Ebola virus.6
A preventive vaccine worked better. Stanley et al7 evaluated a replication-defective chimpanzee adenovirus 3-vectored vaccine that also contained Ebola virus glycoprotein. They gave macaques a single injection of this vaccine, and then 5 weeks later gave them a lethal dose of Ebola virus. All the vaccinated animals survived the infection, and half (2 of 4) survived when challenged 10 months later. With a prime-boost strategy (modified vaccinia virus Ankara, a poxvirus), all survived when challenged 10 months later.
KZ52, a neutralizing antibody, did not work. Oswald et al8 gave a human IgG monoclonal antibody against Zaire Ebola virus, designated KZ52, to four rhesus macaques, challenged them with the virus 24 hours later, and administered a second shot of KZ52 on day 4. All of them died.
ZMAb is a combination of three murine monoclonal antibodies, designated 1H3, 2G4, and 4G7. Ad-IFN is a human adenovirus, serotype 5, that expresses human interferon alpha. Qui et al9 gave ZMAb and Ad-IFN to macaques in several experiments. In experiment 1, eight macaques were infected and then were given ZMAb and Ad-IFN 3 days later, and ZMAb again on days 6 and 9. Seven of the eight survived. In a second experiment, Ad-IFN was given first, when the viral load was still less than the limit of detection of known assays, and then ZMAb was given upon detection of viremia and fever. Two of four macaques survived. Control animals had undetectable levels of IgG, whereas Ebola virus GP–specific IgG levels were detected in all survivors. IFN-gamma ELISpots showed high EBOV-GP–specific T-cell response in all survivors.
ZMapp is another cocktail of monoclonal antibodies, containing two from ZMab (2G4 and 4G7), plus a third, c13C6. In experiments in rhesus macaques, three groups of six animals each received three doses of ZMapp at varying times after being infected with Ebola virus: at 3, 6, and 9 days; at 4, 7, and 10 days, and at 5, 8, and 11 days. All 18 macaques treated with ZMapp survived. Thus, Zmapp extended the treatment window to 5 days postexposure.10 One of the American health care workers who contracted Ebola virus in Liberia received this medication.
HSPA5-PMO. Endoplasmic reticulum chaperone heat shock 70 kDa protein 5 (HSPA5) is instrumental in the maturation of envelope proteins in hepatitis C and influenza A virus. It plays a role in viral entry for coxsackievirus A9 and dengue virus serotype 2, and it may be involved in Ebola viral budding. Phosphorodiamidate morpholino oligomers (PMOs) are a class of antisense DNA nucleotide analogs.
Reid et al11 reported that mice treated with HSPA5–PMO were completely protected from lethal Ebola challenge. Therefore, HSPA5 appears to be a promising target for the development of antifilovirus countermeasures.
Favipiravir, an antiviral agent also known as T-705, is a pyrazinecarboxamide derivative. Invented in 2002 by Toyama Chemicals as an inhibitor of influenza virus replication, it acts as a nucleotide analog, selectively inhibiting the viral RNA-dependent RNA polymerase, or causes lethal mutagenesis upon incorporation into the virus RNA. Favipiravir suppresses Ebola virus replication by 4 log10 units in cell culture.12
Mice were challenged with intranasal inoculation of 1,000 focus-forming units of Ebola virus diluted in phosphate-buffered saline. Until the first day of treatment (postinfection day 6), all mice in the T-705 group lost weight similarly to control mice, developed viremia, and showed elevated serum levels of aspartate aminotransferase and alanine aminotransferase. Within 4 days of T-705 treatment (post-infection day 10), the animals had cleared the virus from blood. Surviving mice developed Ebola virus-specific antibodies and CD8+ T cells specific for the viral nucleoprotein.12
The authors hypothesized that suppression of virus replication by T-705 allowed the host to mount a virus-specific adaptive immune response, and concluded that T-705 was 100% effective in the treatment of Zaire Ebola virus infection up to postinfection day 6 but was hardly beneficial at the terminal stage of disease.12 Of note, favipiravir is undergoing phase 2 and phase 3 trials as an anti-influenza agent in Japan.
THE CURRENT OUTBREAK
The current outbreak is with Zaire ebolavirus. It seems to have started in a 2-year-old child who died in Meliandou in Guéckédou Prefecture, Guinea, on December 6, 2013. On March 21, 2014, the Guinea Ministry of Health reported the outbreak of an illness characterized by fever, severe diarrhea, vomiting, and a high case-fatality rate (59%) in 49 persons. On May 25, 2014, Kenema Government Hospital confirmed the first case of Ebola virus disease in Sierra Leone, probably brought there by a traditional healer who had treated Ebola patients from Guinea. Tracing led to 13 additional cases—all women who attended the burial.13
The Center for Systems Biology at Harvard University and the Broad Institute of Massachusetts Institute of Technology generated 99 Ebola virus genome sequences from 78 patients with confirmed disease, representing more than 70% of the patients diagnosed with the disease in Sierra Leone from May to mid-June 2014. They found genetic similarity across the sequenced 2014 samples, suggesting a single transmission from the natural reservoir, followed by human-to-human transmission during the outbreak. Continued human-reservoir exposure is unlikely to have contributed to the growth of this epidemic.14
As of October 14, 2014, there were 8,914 suspected and confirmed cases of Ebola virus infection, and 4,477 deaths.15
But how did Zaire Ebola virus make the 2,000-mile trek from Central Africa to Guinea in West Africa? There are two possibilities: it has always been present in the region but we just never noticed, or it was recently introduced. Bayesian phylogenetic analyses and sequence divergence studies suggest the virus has been present in bat populations in Guinea without previously infecting humans.
Why Guinea and why Guéckédou? Guinea is one of the poorest countries in the world, ranking 178th of 187 countries on the Human Development Index of the United Nations Development Programme, just behind Liberia (174th) and Sierra Leone (177th). In Guinea, the life expectancy is 56 years and the gross national income per capita is $440. The region has been systematically plundered and the forest decimated by clear-cut logging, leaving the Guinea Forest Region largely deforested, resulting in increased contact between humans and the small animals that serve as the source of infection.1
LIMITED CAPACITY, EVEN IN THE UNITED STATES
A few hospitals in the United States have dedicated units to handle serious infectious diseases such as Ebola: Emory University Hospital; Nebraska Medicine in Omaha; Providence St. Patrick Hospital in Missoula, MT; and the National Institutes of Health in Bethesda, MD. However, in total they have only 19 beds.
QUESTIONS, ANSWERS—AND MORE QUESTIONS
(The following is from a question-and-answer discussion that followed Dr. Brizendine’s Grand Rounds presentation.)
Q: Are there any differences between survivors and those who die of the disease? A: We do not know. Patient survival depends on early recognition and supportive care. There are disparities in the care of patients. Schieffelin et al16 analyzed the characteristics of patients who died or who survived in Sierra Leone and found that the mortality rate was higher in older patients and those with a higher viral load on presentation.
Q: Does the virus block production or release of interferon early in infection? A: Yes, it has been shown17 that Ebola virus protein VP24 inhibits signaling downstream of both interferon alpha/beta and interferon gamma by indirectly impairing the transport of a transcription factor termed STAT1. VP24 is also able to bind STAT1 directly. The resulting suppression of host interferon very early on in the incubation phase is key to the virulence of the virus.
Q: Does infection with one of the viral species confer immunity from other species? A: No, there is no cross-immunity.
Q: How soon do patients test positive? A: About 5 days after exposure, when they develop a fever. At this time patients are highly viremic, which PCR can detect.
Q: Before the virus is detectable in the blood, where is it? A: The liver, endothelial cells, antigen-presenting cells, and adrenal glands.
Q: Do we really need to quarantine ill patients and health care workers returning from Africa, per CDC recommendations? A: We don’t know everything, and some people do make bad decisions, such as traveling while symptomatic. I support a period of observation, although confinement is not reasonable, as it may pose a disincentive to cooperation.
Q: What is the role of giving plasma from survivors? A: Dr. Kent Brantly (see American citizens infected with Ebola) received the blood of a 14-year-old who survived. We don’t know. It is not proved. It did not result in improvement in animal models.
Q: Is the bleeding caused by a mechanism similar to that in enterohemorrhagic Escherichia coli infection? A: No. That is a bacterial toxin, whereas this is more like disseminated intravascular coagulation, with an intrinsic pathway anticoagulation cascade.
Q: How long does the virus remain viable outside the body? A: In one study,18 Ebola virus could not be recovered from experimentally contaminated surfaces (plastic, metal or glass) at room temperature. In another in which it was dried onto a surface,19 Ebola virus survived in the dark for several hours between 20 and 25°C. When dried in tissue culture media onto glass and stored at 4°C, it has survived for over 50 days.
Q: How long does the virus remain in breast milk? A: We know it has been detected 15 days after disease onset and think possibly as late as 28 days from symptom onset.3
Q: How are people actually infected? A: I believe people get the virus on their hands and then touch their face, eyes, or mouth. If you are wearing personal protective equipment, it must occur while doffing the equipment.
Q: Could we increase the sensitivity of the test so that we could detect the virus before the onset of symptoms? A: In theory it may be possible. The virus is somewhere in the body during the incubation period. Perhaps we could sample the right compartment in an enriched mononuclear cell line.
Q: When can patients who recover resume their normal activities? A: After their viral load returns to 0, I would still advise abstaining from unprotected sex and from breastfeeding for a few months. but as for other activities, no special precautions are needed.
Q: Does the virus appear to be mutating at a high rate? A: Looking back to 2004, mutations are occurring, but there is no sign that any of these mutations has contributed to the size of the outbreak by changing the characteristics of the Ebola virus. Can it become aerosolized? It has been suggested that the virus that caused the outbreak separated from those that caused past Ebola outbreaks but does not seem to be affecting the spread or efficacy of experimental drugs and vaccines. So, even though it is an RNA virus and mutations are occurring, no serious changes have emerged.14
BACK TO OUR PATIENT
The differential diagnosis for the patient described at the beginning of this paper includes travelers’ diarrhea, malaria, typhoid fever, yellow fever, meningococcal disease … and Ebola virus disease, although this is much less likely in view of the epidemiology and incubation period of this disease. When his stool was tested by enzyme immunoassay and culture, it was found to be positive for Campylobacter. He recovered with oral rehydration.
- Bausch DG, Schwarz L. Outbreak of ebola virus disease in Guinea: where ecology meets economy. PLoS Negl Trop Dis 2014; 8:e3056.
- Roddy P, Thomas SL, Jeffs B, et al. Factors associated with Marburg hemorrhagic fever: analysis of patient data from Uige, Angola. J Infect Dis 2010; 201:1909–1918.
- Bausch DG, Towner JS, Dowell SF, et al. Assessment of the risk of Ebola virus transmission from bodily fluids and fomites. J Infect Dis 2007; 196(suppl 2):S142–S147.
- WHO Ebola Response Team. Ebola virus disease in West Africa—the first 9 months of the epidemic and forward projections. N Engl J Med 2014; 371:1481–1495.
- Ribner BS. Treating patients with Ebola virus infections in the US: lessons learned. Presented at IDWeek, October 8, 2014. Philadelphia PA.
- Feldman H, Jones SM, Daddario-DiCaprio KM, et al. Effective post-exposure treatment of Ebola infection. PLoS Pathog 2007; 3:e2.
- Stanley DA, Honko AN, Asiedu C, et al. Chimpanzee adenovirus vaccine generates acute and durable protective immunity against ebolavirus challenge. Nat Med 2014; 20:1126–1129.
- Oswald WB, Geisbert TW, Davis KJ, et al. Neutralizing antibody fails to impact the course of Ebola virus infection in monkeys. PLos Pathog 2007; 3:e9.
- Qui X, Wong G, Fernando L, et al. mAbs and Ad-vectored IFN-a therapy rescue Ebola-infected nonhuman primates when administered after the detection of viremia and symptoms. Sci Transl Med 2013; 5:207ra143.
- Qui X, Wong G, Audet J, et al. Reversion of advanced Ebola virus disease in nonhuman primates with ZMapp. Nature 2014; 514:47–53.
- Reid SP, Shurtleff AC, Costantino JA, et al. HSPA5 is an essential host factor for Ebola virus infection. Antiviral Res 2014; 109:171–174.
- Oestereich L, Lüdtke A, Wurr S, Rieger T, Muñoz-Fontela C, Günther S. Successful treatment of advanced Ebola virus infection with T-705 (favipiravir) in a small animal model. Antiviral Res 2014; 105:17–21.
- Baize S, Pannetier D, Oestereich L, et al. Emergence of Zaire Ebola virus dsease in Guinea. N Engl J Med 2014; 371:1418–1425.
- Gire SK, Goba A, Andersen KG, et al. Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science 2014; 345:1369–1372.
- Chamary JV. 4000 deaths and counting: the Ebola epidemic in 4 charts. Forbes. http://www.forbes.com/sites/jvchamary/2014/10/13/ebola-trends. Accessed November 5, 2014.
- Schieffelin JS, Shaffer JG, Goba A, et al, for the KGH Lassa Fever Program, the Viral Hemorrhagic Fever Consortium, and the WHO Clinical Response Team. Clinical illness and outcomes in patients with Ebola in Sierra Leone. N Engl J Med 2014 Oct 29 [Epub ahead of print]. DOI: 10.1056/NEJMoa1411680.
- Zhang AP, Bornholdt ZA, Liu T, et al. The ebola virus interferon antagonist VP24 directly binds STAT1 and has a novel, pyramidal fold. PLoS Pathog 2012; 8:e1002550.
- Piercy TJ, Smither SJ, Steward JA, Eastaugh L, Lever MS. The survival of filoviruses in liquids, on solid substrates and in a dynamic aerosol. J Appl Microbiol 2010; 109:1531–1539.
- Sagripanti JL, Rom AM, Holland LE. Persistence in darkness of virulent alphaviruses, Ebola virus, and Lassa virus deposited on solid surfaces. Arch Virol 2010; 155:2035–2039.
A 50-year-old man who returned from a business trip to Nigeria 24 days ago presents with complaints of the sudden onset of fever, diarrhea, myalgia, and headache. He reports 10 bowel movements per day and has seen bloody stools.
During his trip he flew in to Murtala Muhammed International Airport in Lagos, ate meals only in his hotel, and attended meetings in Lagos central business district. He had no exposure to animals, mosquitoes, ticks, or sick people, and no sexual activity. After returning home, he felt well for the first 3 weeks.
The patient has a history of hypertension. He does not smoke, drink alcohol, or use injection drugs. He is married, works with commercial banks and financial institutions, and lives in Cleveland, OH.
On physical examination his temperature is 100.0˚F (37.8˚C), pulse 98, respirations 15, blood pressure 105/70 mm Hg, and weight 78 kg (172 lb). He appears comfortable but is a little diaphoretic. His abdomen is tender to palpation in the epigastrium and slightly to the right; he has no signs of peritonitis. His skin is without rash, bleeding, or bruising. The remainder of the examination is normal.
His white blood cell count is 17 × 109/L, hemoglobin 15 g/dL, hematocrit 41%, and platelet count 172 × 109/L. His sodium level is 126 mmol/L, potassium 3.8 mmol/L, chloride 95 mmol/L, carbon dioxide 20 mmol/L, blood urea nitrogen 11 mg/dL, creatinine 0.7 mg/dL, and glucose 130 mg/dL. His aminotransferase and alkaline phosphatase levels are normal.
Could this patient have Ebola virus disease?
With Ebola virus disease on the rise in West Africa, physicians who encounter patients like this one need to include it in the differential diagnosis. Because the disease is new, many questions are raised for which we as yet have no answers. Here, I will review what we know and do not know in an effort to remove some of the fear and uncertainty.
A NEW DISEASE
Ebola virus disease is a severe hemorrhagic fever caused by negative-sense single-stranded RNA viruses classified by the International Committee on Taxonomy of Viruses as belonging to the genus Ebolavirus in the family Filoviridae. Filoviruses get their name from the Latin filum, or thread-like structure.
The family Filoviridae was discovered in 1967 after inadvertent importation of infected monkeys from Uganda into Yugoslavia and Marburg, Germany. Outbreaks of severe illness occurred in workers at a vaccine plant who came into direct contact with the animals by killing them, removing their kidneys, or preparing primary cell cultures for polio vaccine production.
Ebola virus was discovered in 1976 by Peter Piot, who was working at the Institute of Tropical Medicine in Antwerp, Belgium. The blood of a Belgian woman who had been working in what is now the Democratic Republic of the Congo (formerly Zaire) had been sent to the institute; she and Mabalo Lokela, a school headmaster and the first recorded victim of Ebola virus, had been working near Yambuku, about 96 km from the Ebola River.
Before the 2014 outbreak, all known outbreaks had caused fewer than 2,400 cases across a dozen African countries over 3 decades.
Five species of Ebola virus
The genus Ebolavirus contains five species, each associated with a consistent case-fatality rate and a more or less well-identified endemic area.1
Zaire ebolavirus was recognized in 1976; it has caused multiple outbreaks, with high case-fatality rates.
Sudan ebolavirus was seen first in the 1970s; it has a 50% case-fatality rate.
Tai Forest ebolavirus has been found in only one person, an ethologist working with deceased chimpanzees.
Bundibugyo ebolavirus emerged in 2007 and has a 30% case-fatality rate.
Reston ebolavirus is maintained in an animal reservoir in the Philippines and is not found in Africa. It caused an outbreak of lethal infection in macaques imported into the United States in 1989. There is evidence that Reston ebolavirus can cause asymptomatic infection in humans. None of the caretakers of the macaques became ill, nor did farmers working with infected pigs, although both groups seroconverted.
A reservoir in bats?
A reservoir in nonhuman primates was initially suspected. However, studies subsequently showed that monkeys are susceptible to rapidly lethal filoviral disease, precluding any role as a host for persistent viral infection. It is likely that Ebola virus is maintained in small animals that serve as a source of infection for both humans and wild primates. A prominent suspect is fruit bats, which are consumed in soup in West Africa.
Transmission is person-to-person or nosocomial
Ebola virus is transmitted by direct contact with body fluids such as blood, urine, sweat, vomitus, semen, and breast milk. Filoviruses can initiate infection via ingestion, inhalation (although probably not Ebola), or passage through breaks in the skin. Droplet inoculation into the mouth or eyes has been shown to result from inadvertent transfer of virus from contaminated hands. Patients transmit the virus while febrile and through later stages of disease, as well as postmortem through contact with the body during funeral preparations. The virus has been isolated in semen for as many as 61 days after illness onset.
Ebola virus can also be spread nosocomially. In 1976, a 44-year-old teacher sought care for fever at the Yambuku Mission Hospital. He was given parenteral chloroquine as empiric treatment for presumed malaria, which was routine for all febrile patients. However, he had unrecognized Ebola virus infection. Moreover, syringes were rinsed in the same pan of water and reused, which spread the infection to nearly 100 people, all of whom developed fulminant Ebola virus disease and died. Infection then spread to family caregivers, the hospital staff, and those who prepared the bodies for burial.
Nosocomial transmission was also responsible for an outbreak of Lake Victoria Marburg virus in Uige Province in northern Angola in 2005, with 374 putative cases and 329 deaths. When teams from Médecins Sans Frontières started setting up the Marburg ward, there were five patients with hemorrhagic fever in a makeshift isolation room in the hospital, together with corpses that the hospital staff had been too afraid to remove. Healers found in many rural African communities were administering injections in homes or in makeshift clinics with reused needles or syringes.2
There is no evidence that filoviruses are carried by mosquitoes or other biting arthropods. Also, the risk of transmission via fomites appears to be low when currently recommended infection-control guidelines for the viral hemorrhagic fevers are followed.3 One primary human case generates only one to three secondary cases on average.
EBOLA IS AN IMMUNODEFICIENCY VIRUS
The main targets of infection are endothelial cells, mononuclear phagocytes, and hepatocytes. Ebola virus replicates at an unusually high rate. Macrophages infected with Zaire ebolavirus produce tumor necrosis factor alpha, interleukin (IL) 1 beta, IL-6, macrophage chemotactic protein 1, and nitric oxide. Virus-infected macrophages synthesize cell-surface tissue factor, triggering the extrinsic coagulation pathway.
Ebola is an immunodeficiency virus. Dendritic cells, which initiate adaptive immune responses, are a major site of filoviral replication. Infected cells cannot present antigens to naïve lymphocytes. Patients who die of Ebola virus disease do not develop antibodies to the virus. Lymphocytes remain uninfected, but undergo “bystander” apoptosis induced by inflammatory mediators.
CLINICAL MANIFESTATIONS
The incubation period is generally 5 to 7 days (range 2 to 28 days), during which the patient is not infectious. Symptoms begin abruptly, with fever, chills, general malaise, weakness, severe headache, and myalgia. By the time of case detection in West Africa, most patients also had nausea, vomiting, diarrhea, and abdominal pain. Once symptoms arise, patients have high levels of the virus in their blood and fluids and are infectious. Hemorrhagic symptoms have apparently been uncommon in West Africa, occurring in 1.0% to 5.7%, but “unexplained bleeding” has been documented in 18% of cases.4 Among those in whom the disease enters its hemorrhagic terminal phase, there is characteristic internal and subcutaneous bleeding, vomiting of blood, and subconjunctival hemorrhage.4
Laboratory findings include lymphocytopenia (often with counts as low as 1.0 × 109/L), thrombocytopenia (with counts in the range of 50 to 100 × 109/L), elevated aminotransferase levels (including aspartate aminotransferase levels 7 to 12 times higher than alanine aminotransferase in fatal cases), low total protein (due to capillary leak), and disseminated intravascular coagulation. Those who survive begin to improve in the second week, during which viremia resolves in association with the appearance of virus-specific antibodies.4
DIAGNOSIS
In symptomatic patients, Ebola virus infection is diagnosed by detection in blood or body fluids of viral antigens by enzyme-linked immunosorbent assay, or RNA sequences by reverse transcriptase polymerase chain reaction. The diagnosis is confirmed with cell culture (in a BSL-4 containment laboratory) showing characteristic viral particles by electron microscopy.
CARING FOR PATIENTS
The most detailed descriptions of the care of patients with Ebola virus disease have come from Dr. Bruce Ribner, of Emory University Hospital, in an October 2014 report of his experience caring for Ebola-infected patients at Emory University Hospital in Atlanta, GA.5 He described fluid losses of 5 to 10 L/day, profound hyponatremia, hypokalemia, and hypocalcemia, which were associated with cardiac arrhythmias and the need for intravenous and oral electrolyte repletion and hemodialysis. Intensive one-to-one nursing was critical, as was the coordination of many medical subspecialties. The Emory team arranged point-of-care testing near the unit and generally kept laboratory testing to a minimum. The team was surprised to learn that commercial carriers refused to transport specimens even when they were licensed for category A agents. Difficulties with the local water authority and waste disposal contractor required the hospital to dedicate an autoclave to process all materials used in clinical care.
TREATMENT: SUPPORTIVE AND EXPERIMENTAL
Treatment is supportive to maintain circulatory function and blood pressure and to correct coagulopathy. However, a variety of vaccines, antibodies, small-molecule agents, and antiviral agents are undergoing testing, mostly in animals at this point.
Vaccines. A therapeutic vaccine that worked only slightly was a live-attenuated recombinant vesicular stomatitis virus expressing Ebola virus transmembrane glycoproteins, which was tested in mice, guinea pigs, and rhesus macaques who had been exposed to Ebola virus.6
A preventive vaccine worked better. Stanley et al7 evaluated a replication-defective chimpanzee adenovirus 3-vectored vaccine that also contained Ebola virus glycoprotein. They gave macaques a single injection of this vaccine, and then 5 weeks later gave them a lethal dose of Ebola virus. All the vaccinated animals survived the infection, and half (2 of 4) survived when challenged 10 months later. With a prime-boost strategy (modified vaccinia virus Ankara, a poxvirus), all survived when challenged 10 months later.
KZ52, a neutralizing antibody, did not work. Oswald et al8 gave a human IgG monoclonal antibody against Zaire Ebola virus, designated KZ52, to four rhesus macaques, challenged them with the virus 24 hours later, and administered a second shot of KZ52 on day 4. All of them died.
ZMAb is a combination of three murine monoclonal antibodies, designated 1H3, 2G4, and 4G7. Ad-IFN is a human adenovirus, serotype 5, that expresses human interferon alpha. Qui et al9 gave ZMAb and Ad-IFN to macaques in several experiments. In experiment 1, eight macaques were infected and then were given ZMAb and Ad-IFN 3 days later, and ZMAb again on days 6 and 9. Seven of the eight survived. In a second experiment, Ad-IFN was given first, when the viral load was still less than the limit of detection of known assays, and then ZMAb was given upon detection of viremia and fever. Two of four macaques survived. Control animals had undetectable levels of IgG, whereas Ebola virus GP–specific IgG levels were detected in all survivors. IFN-gamma ELISpots showed high EBOV-GP–specific T-cell response in all survivors.
ZMapp is another cocktail of monoclonal antibodies, containing two from ZMab (2G4 and 4G7), plus a third, c13C6. In experiments in rhesus macaques, three groups of six animals each received three doses of ZMapp at varying times after being infected with Ebola virus: at 3, 6, and 9 days; at 4, 7, and 10 days, and at 5, 8, and 11 days. All 18 macaques treated with ZMapp survived. Thus, Zmapp extended the treatment window to 5 days postexposure.10 One of the American health care workers who contracted Ebola virus in Liberia received this medication.
HSPA5-PMO. Endoplasmic reticulum chaperone heat shock 70 kDa protein 5 (HSPA5) is instrumental in the maturation of envelope proteins in hepatitis C and influenza A virus. It plays a role in viral entry for coxsackievirus A9 and dengue virus serotype 2, and it may be involved in Ebola viral budding. Phosphorodiamidate morpholino oligomers (PMOs) are a class of antisense DNA nucleotide analogs.
Reid et al11 reported that mice treated with HSPA5–PMO were completely protected from lethal Ebola challenge. Therefore, HSPA5 appears to be a promising target for the development of antifilovirus countermeasures.
Favipiravir, an antiviral agent also known as T-705, is a pyrazinecarboxamide derivative. Invented in 2002 by Toyama Chemicals as an inhibitor of influenza virus replication, it acts as a nucleotide analog, selectively inhibiting the viral RNA-dependent RNA polymerase, or causes lethal mutagenesis upon incorporation into the virus RNA. Favipiravir suppresses Ebola virus replication by 4 log10 units in cell culture.12
Mice were challenged with intranasal inoculation of 1,000 focus-forming units of Ebola virus diluted in phosphate-buffered saline. Until the first day of treatment (postinfection day 6), all mice in the T-705 group lost weight similarly to control mice, developed viremia, and showed elevated serum levels of aspartate aminotransferase and alanine aminotransferase. Within 4 days of T-705 treatment (post-infection day 10), the animals had cleared the virus from blood. Surviving mice developed Ebola virus-specific antibodies and CD8+ T cells specific for the viral nucleoprotein.12
The authors hypothesized that suppression of virus replication by T-705 allowed the host to mount a virus-specific adaptive immune response, and concluded that T-705 was 100% effective in the treatment of Zaire Ebola virus infection up to postinfection day 6 but was hardly beneficial at the terminal stage of disease.12 Of note, favipiravir is undergoing phase 2 and phase 3 trials as an anti-influenza agent in Japan.
THE CURRENT OUTBREAK
The current outbreak is with Zaire ebolavirus. It seems to have started in a 2-year-old child who died in Meliandou in Guéckédou Prefecture, Guinea, on December 6, 2013. On March 21, 2014, the Guinea Ministry of Health reported the outbreak of an illness characterized by fever, severe diarrhea, vomiting, and a high case-fatality rate (59%) in 49 persons. On May 25, 2014, Kenema Government Hospital confirmed the first case of Ebola virus disease in Sierra Leone, probably brought there by a traditional healer who had treated Ebola patients from Guinea. Tracing led to 13 additional cases—all women who attended the burial.13
The Center for Systems Biology at Harvard University and the Broad Institute of Massachusetts Institute of Technology generated 99 Ebola virus genome sequences from 78 patients with confirmed disease, representing more than 70% of the patients diagnosed with the disease in Sierra Leone from May to mid-June 2014. They found genetic similarity across the sequenced 2014 samples, suggesting a single transmission from the natural reservoir, followed by human-to-human transmission during the outbreak. Continued human-reservoir exposure is unlikely to have contributed to the growth of this epidemic.14
As of October 14, 2014, there were 8,914 suspected and confirmed cases of Ebola virus infection, and 4,477 deaths.15
But how did Zaire Ebola virus make the 2,000-mile trek from Central Africa to Guinea in West Africa? There are two possibilities: it has always been present in the region but we just never noticed, or it was recently introduced. Bayesian phylogenetic analyses and sequence divergence studies suggest the virus has been present in bat populations in Guinea without previously infecting humans.
Why Guinea and why Guéckédou? Guinea is one of the poorest countries in the world, ranking 178th of 187 countries on the Human Development Index of the United Nations Development Programme, just behind Liberia (174th) and Sierra Leone (177th). In Guinea, the life expectancy is 56 years and the gross national income per capita is $440. The region has been systematically plundered and the forest decimated by clear-cut logging, leaving the Guinea Forest Region largely deforested, resulting in increased contact between humans and the small animals that serve as the source of infection.1
LIMITED CAPACITY, EVEN IN THE UNITED STATES
A few hospitals in the United States have dedicated units to handle serious infectious diseases such as Ebola: Emory University Hospital; Nebraska Medicine in Omaha; Providence St. Patrick Hospital in Missoula, MT; and the National Institutes of Health in Bethesda, MD. However, in total they have only 19 beds.
QUESTIONS, ANSWERS—AND MORE QUESTIONS
(The following is from a question-and-answer discussion that followed Dr. Brizendine’s Grand Rounds presentation.)
Q: Are there any differences between survivors and those who die of the disease? A: We do not know. Patient survival depends on early recognition and supportive care. There are disparities in the care of patients. Schieffelin et al16 analyzed the characteristics of patients who died or who survived in Sierra Leone and found that the mortality rate was higher in older patients and those with a higher viral load on presentation.
Q: Does the virus block production or release of interferon early in infection? A: Yes, it has been shown17 that Ebola virus protein VP24 inhibits signaling downstream of both interferon alpha/beta and interferon gamma by indirectly impairing the transport of a transcription factor termed STAT1. VP24 is also able to bind STAT1 directly. The resulting suppression of host interferon very early on in the incubation phase is key to the virulence of the virus.
Q: Does infection with one of the viral species confer immunity from other species? A: No, there is no cross-immunity.
Q: How soon do patients test positive? A: About 5 days after exposure, when they develop a fever. At this time patients are highly viremic, which PCR can detect.
Q: Before the virus is detectable in the blood, where is it? A: The liver, endothelial cells, antigen-presenting cells, and adrenal glands.
Q: Do we really need to quarantine ill patients and health care workers returning from Africa, per CDC recommendations? A: We don’t know everything, and some people do make bad decisions, such as traveling while symptomatic. I support a period of observation, although confinement is not reasonable, as it may pose a disincentive to cooperation.
Q: What is the role of giving plasma from survivors? A: Dr. Kent Brantly (see American citizens infected with Ebola) received the blood of a 14-year-old who survived. We don’t know. It is not proved. It did not result in improvement in animal models.
Q: Is the bleeding caused by a mechanism similar to that in enterohemorrhagic Escherichia coli infection? A: No. That is a bacterial toxin, whereas this is more like disseminated intravascular coagulation, with an intrinsic pathway anticoagulation cascade.
Q: How long does the virus remain viable outside the body? A: In one study,18 Ebola virus could not be recovered from experimentally contaminated surfaces (plastic, metal or glass) at room temperature. In another in which it was dried onto a surface,19 Ebola virus survived in the dark for several hours between 20 and 25°C. When dried in tissue culture media onto glass and stored at 4°C, it has survived for over 50 days.
Q: How long does the virus remain in breast milk? A: We know it has been detected 15 days after disease onset and think possibly as late as 28 days from symptom onset.3
Q: How are people actually infected? A: I believe people get the virus on their hands and then touch their face, eyes, or mouth. If you are wearing personal protective equipment, it must occur while doffing the equipment.
Q: Could we increase the sensitivity of the test so that we could detect the virus before the onset of symptoms? A: In theory it may be possible. The virus is somewhere in the body during the incubation period. Perhaps we could sample the right compartment in an enriched mononuclear cell line.
Q: When can patients who recover resume their normal activities? A: After their viral load returns to 0, I would still advise abstaining from unprotected sex and from breastfeeding for a few months. but as for other activities, no special precautions are needed.
Q: Does the virus appear to be mutating at a high rate? A: Looking back to 2004, mutations are occurring, but there is no sign that any of these mutations has contributed to the size of the outbreak by changing the characteristics of the Ebola virus. Can it become aerosolized? It has been suggested that the virus that caused the outbreak separated from those that caused past Ebola outbreaks but does not seem to be affecting the spread or efficacy of experimental drugs and vaccines. So, even though it is an RNA virus and mutations are occurring, no serious changes have emerged.14
BACK TO OUR PATIENT
The differential diagnosis for the patient described at the beginning of this paper includes travelers’ diarrhea, malaria, typhoid fever, yellow fever, meningococcal disease … and Ebola virus disease, although this is much less likely in view of the epidemiology and incubation period of this disease. When his stool was tested by enzyme immunoassay and culture, it was found to be positive for Campylobacter. He recovered with oral rehydration.
A 50-year-old man who returned from a business trip to Nigeria 24 days ago presents with complaints of the sudden onset of fever, diarrhea, myalgia, and headache. He reports 10 bowel movements per day and has seen bloody stools.
During his trip he flew in to Murtala Muhammed International Airport in Lagos, ate meals only in his hotel, and attended meetings in Lagos central business district. He had no exposure to animals, mosquitoes, ticks, or sick people, and no sexual activity. After returning home, he felt well for the first 3 weeks.
The patient has a history of hypertension. He does not smoke, drink alcohol, or use injection drugs. He is married, works with commercial banks and financial institutions, and lives in Cleveland, OH.
On physical examination his temperature is 100.0˚F (37.8˚C), pulse 98, respirations 15, blood pressure 105/70 mm Hg, and weight 78 kg (172 lb). He appears comfortable but is a little diaphoretic. His abdomen is tender to palpation in the epigastrium and slightly to the right; he has no signs of peritonitis. His skin is without rash, bleeding, or bruising. The remainder of the examination is normal.
His white blood cell count is 17 × 109/L, hemoglobin 15 g/dL, hematocrit 41%, and platelet count 172 × 109/L. His sodium level is 126 mmol/L, potassium 3.8 mmol/L, chloride 95 mmol/L, carbon dioxide 20 mmol/L, blood urea nitrogen 11 mg/dL, creatinine 0.7 mg/dL, and glucose 130 mg/dL. His aminotransferase and alkaline phosphatase levels are normal.
Could this patient have Ebola virus disease?
With Ebola virus disease on the rise in West Africa, physicians who encounter patients like this one need to include it in the differential diagnosis. Because the disease is new, many questions are raised for which we as yet have no answers. Here, I will review what we know and do not know in an effort to remove some of the fear and uncertainty.
A NEW DISEASE
Ebola virus disease is a severe hemorrhagic fever caused by negative-sense single-stranded RNA viruses classified by the International Committee on Taxonomy of Viruses as belonging to the genus Ebolavirus in the family Filoviridae. Filoviruses get their name from the Latin filum, or thread-like structure.
The family Filoviridae was discovered in 1967 after inadvertent importation of infected monkeys from Uganda into Yugoslavia and Marburg, Germany. Outbreaks of severe illness occurred in workers at a vaccine plant who came into direct contact with the animals by killing them, removing their kidneys, or preparing primary cell cultures for polio vaccine production.
Ebola virus was discovered in 1976 by Peter Piot, who was working at the Institute of Tropical Medicine in Antwerp, Belgium. The blood of a Belgian woman who had been working in what is now the Democratic Republic of the Congo (formerly Zaire) had been sent to the institute; she and Mabalo Lokela, a school headmaster and the first recorded victim of Ebola virus, had been working near Yambuku, about 96 km from the Ebola River.
Before the 2014 outbreak, all known outbreaks had caused fewer than 2,400 cases across a dozen African countries over 3 decades.
Five species of Ebola virus
The genus Ebolavirus contains five species, each associated with a consistent case-fatality rate and a more or less well-identified endemic area.1
Zaire ebolavirus was recognized in 1976; it has caused multiple outbreaks, with high case-fatality rates.
Sudan ebolavirus was seen first in the 1970s; it has a 50% case-fatality rate.
Tai Forest ebolavirus has been found in only one person, an ethologist working with deceased chimpanzees.
Bundibugyo ebolavirus emerged in 2007 and has a 30% case-fatality rate.
Reston ebolavirus is maintained in an animal reservoir in the Philippines and is not found in Africa. It caused an outbreak of lethal infection in macaques imported into the United States in 1989. There is evidence that Reston ebolavirus can cause asymptomatic infection in humans. None of the caretakers of the macaques became ill, nor did farmers working with infected pigs, although both groups seroconverted.
A reservoir in bats?
A reservoir in nonhuman primates was initially suspected. However, studies subsequently showed that monkeys are susceptible to rapidly lethal filoviral disease, precluding any role as a host for persistent viral infection. It is likely that Ebola virus is maintained in small animals that serve as a source of infection for both humans and wild primates. A prominent suspect is fruit bats, which are consumed in soup in West Africa.
Transmission is person-to-person or nosocomial
Ebola virus is transmitted by direct contact with body fluids such as blood, urine, sweat, vomitus, semen, and breast milk. Filoviruses can initiate infection via ingestion, inhalation (although probably not Ebola), or passage through breaks in the skin. Droplet inoculation into the mouth or eyes has been shown to result from inadvertent transfer of virus from contaminated hands. Patients transmit the virus while febrile and through later stages of disease, as well as postmortem through contact with the body during funeral preparations. The virus has been isolated in semen for as many as 61 days after illness onset.
Ebola virus can also be spread nosocomially. In 1976, a 44-year-old teacher sought care for fever at the Yambuku Mission Hospital. He was given parenteral chloroquine as empiric treatment for presumed malaria, which was routine for all febrile patients. However, he had unrecognized Ebola virus infection. Moreover, syringes were rinsed in the same pan of water and reused, which spread the infection to nearly 100 people, all of whom developed fulminant Ebola virus disease and died. Infection then spread to family caregivers, the hospital staff, and those who prepared the bodies for burial.
Nosocomial transmission was also responsible for an outbreak of Lake Victoria Marburg virus in Uige Province in northern Angola in 2005, with 374 putative cases and 329 deaths. When teams from Médecins Sans Frontières started setting up the Marburg ward, there were five patients with hemorrhagic fever in a makeshift isolation room in the hospital, together with corpses that the hospital staff had been too afraid to remove. Healers found in many rural African communities were administering injections in homes or in makeshift clinics with reused needles or syringes.2
There is no evidence that filoviruses are carried by mosquitoes or other biting arthropods. Also, the risk of transmission via fomites appears to be low when currently recommended infection-control guidelines for the viral hemorrhagic fevers are followed.3 One primary human case generates only one to three secondary cases on average.
EBOLA IS AN IMMUNODEFICIENCY VIRUS
The main targets of infection are endothelial cells, mononuclear phagocytes, and hepatocytes. Ebola virus replicates at an unusually high rate. Macrophages infected with Zaire ebolavirus produce tumor necrosis factor alpha, interleukin (IL) 1 beta, IL-6, macrophage chemotactic protein 1, and nitric oxide. Virus-infected macrophages synthesize cell-surface tissue factor, triggering the extrinsic coagulation pathway.
Ebola is an immunodeficiency virus. Dendritic cells, which initiate adaptive immune responses, are a major site of filoviral replication. Infected cells cannot present antigens to naïve lymphocytes. Patients who die of Ebola virus disease do not develop antibodies to the virus. Lymphocytes remain uninfected, but undergo “bystander” apoptosis induced by inflammatory mediators.
CLINICAL MANIFESTATIONS
The incubation period is generally 5 to 7 days (range 2 to 28 days), during which the patient is not infectious. Symptoms begin abruptly, with fever, chills, general malaise, weakness, severe headache, and myalgia. By the time of case detection in West Africa, most patients also had nausea, vomiting, diarrhea, and abdominal pain. Once symptoms arise, patients have high levels of the virus in their blood and fluids and are infectious. Hemorrhagic symptoms have apparently been uncommon in West Africa, occurring in 1.0% to 5.7%, but “unexplained bleeding” has been documented in 18% of cases.4 Among those in whom the disease enters its hemorrhagic terminal phase, there is characteristic internal and subcutaneous bleeding, vomiting of blood, and subconjunctival hemorrhage.4
Laboratory findings include lymphocytopenia (often with counts as low as 1.0 × 109/L), thrombocytopenia (with counts in the range of 50 to 100 × 109/L), elevated aminotransferase levels (including aspartate aminotransferase levels 7 to 12 times higher than alanine aminotransferase in fatal cases), low total protein (due to capillary leak), and disseminated intravascular coagulation. Those who survive begin to improve in the second week, during which viremia resolves in association with the appearance of virus-specific antibodies.4
DIAGNOSIS
In symptomatic patients, Ebola virus infection is diagnosed by detection in blood or body fluids of viral antigens by enzyme-linked immunosorbent assay, or RNA sequences by reverse transcriptase polymerase chain reaction. The diagnosis is confirmed with cell culture (in a BSL-4 containment laboratory) showing characteristic viral particles by electron microscopy.
CARING FOR PATIENTS
The most detailed descriptions of the care of patients with Ebola virus disease have come from Dr. Bruce Ribner, of Emory University Hospital, in an October 2014 report of his experience caring for Ebola-infected patients at Emory University Hospital in Atlanta, GA.5 He described fluid losses of 5 to 10 L/day, profound hyponatremia, hypokalemia, and hypocalcemia, which were associated with cardiac arrhythmias and the need for intravenous and oral electrolyte repletion and hemodialysis. Intensive one-to-one nursing was critical, as was the coordination of many medical subspecialties. The Emory team arranged point-of-care testing near the unit and generally kept laboratory testing to a minimum. The team was surprised to learn that commercial carriers refused to transport specimens even when they were licensed for category A agents. Difficulties with the local water authority and waste disposal contractor required the hospital to dedicate an autoclave to process all materials used in clinical care.
TREATMENT: SUPPORTIVE AND EXPERIMENTAL
Treatment is supportive to maintain circulatory function and blood pressure and to correct coagulopathy. However, a variety of vaccines, antibodies, small-molecule agents, and antiviral agents are undergoing testing, mostly in animals at this point.
Vaccines. A therapeutic vaccine that worked only slightly was a live-attenuated recombinant vesicular stomatitis virus expressing Ebola virus transmembrane glycoproteins, which was tested in mice, guinea pigs, and rhesus macaques who had been exposed to Ebola virus.6
A preventive vaccine worked better. Stanley et al7 evaluated a replication-defective chimpanzee adenovirus 3-vectored vaccine that also contained Ebola virus glycoprotein. They gave macaques a single injection of this vaccine, and then 5 weeks later gave them a lethal dose of Ebola virus. All the vaccinated animals survived the infection, and half (2 of 4) survived when challenged 10 months later. With a prime-boost strategy (modified vaccinia virus Ankara, a poxvirus), all survived when challenged 10 months later.
KZ52, a neutralizing antibody, did not work. Oswald et al8 gave a human IgG monoclonal antibody against Zaire Ebola virus, designated KZ52, to four rhesus macaques, challenged them with the virus 24 hours later, and administered a second shot of KZ52 on day 4. All of them died.
ZMAb is a combination of three murine monoclonal antibodies, designated 1H3, 2G4, and 4G7. Ad-IFN is a human adenovirus, serotype 5, that expresses human interferon alpha. Qui et al9 gave ZMAb and Ad-IFN to macaques in several experiments. In experiment 1, eight macaques were infected and then were given ZMAb and Ad-IFN 3 days later, and ZMAb again on days 6 and 9. Seven of the eight survived. In a second experiment, Ad-IFN was given first, when the viral load was still less than the limit of detection of known assays, and then ZMAb was given upon detection of viremia and fever. Two of four macaques survived. Control animals had undetectable levels of IgG, whereas Ebola virus GP–specific IgG levels were detected in all survivors. IFN-gamma ELISpots showed high EBOV-GP–specific T-cell response in all survivors.
ZMapp is another cocktail of monoclonal antibodies, containing two from ZMab (2G4 and 4G7), plus a third, c13C6. In experiments in rhesus macaques, three groups of six animals each received three doses of ZMapp at varying times after being infected with Ebola virus: at 3, 6, and 9 days; at 4, 7, and 10 days, and at 5, 8, and 11 days. All 18 macaques treated with ZMapp survived. Thus, Zmapp extended the treatment window to 5 days postexposure.10 One of the American health care workers who contracted Ebola virus in Liberia received this medication.
HSPA5-PMO. Endoplasmic reticulum chaperone heat shock 70 kDa protein 5 (HSPA5) is instrumental in the maturation of envelope proteins in hepatitis C and influenza A virus. It plays a role in viral entry for coxsackievirus A9 and dengue virus serotype 2, and it may be involved in Ebola viral budding. Phosphorodiamidate morpholino oligomers (PMOs) are a class of antisense DNA nucleotide analogs.
Reid et al11 reported that mice treated with HSPA5–PMO were completely protected from lethal Ebola challenge. Therefore, HSPA5 appears to be a promising target for the development of antifilovirus countermeasures.
Favipiravir, an antiviral agent also known as T-705, is a pyrazinecarboxamide derivative. Invented in 2002 by Toyama Chemicals as an inhibitor of influenza virus replication, it acts as a nucleotide analog, selectively inhibiting the viral RNA-dependent RNA polymerase, or causes lethal mutagenesis upon incorporation into the virus RNA. Favipiravir suppresses Ebola virus replication by 4 log10 units in cell culture.12
Mice were challenged with intranasal inoculation of 1,000 focus-forming units of Ebola virus diluted in phosphate-buffered saline. Until the first day of treatment (postinfection day 6), all mice in the T-705 group lost weight similarly to control mice, developed viremia, and showed elevated serum levels of aspartate aminotransferase and alanine aminotransferase. Within 4 days of T-705 treatment (post-infection day 10), the animals had cleared the virus from blood. Surviving mice developed Ebola virus-specific antibodies and CD8+ T cells specific for the viral nucleoprotein.12
The authors hypothesized that suppression of virus replication by T-705 allowed the host to mount a virus-specific adaptive immune response, and concluded that T-705 was 100% effective in the treatment of Zaire Ebola virus infection up to postinfection day 6 but was hardly beneficial at the terminal stage of disease.12 Of note, favipiravir is undergoing phase 2 and phase 3 trials as an anti-influenza agent in Japan.
THE CURRENT OUTBREAK
The current outbreak is with Zaire ebolavirus. It seems to have started in a 2-year-old child who died in Meliandou in Guéckédou Prefecture, Guinea, on December 6, 2013. On March 21, 2014, the Guinea Ministry of Health reported the outbreak of an illness characterized by fever, severe diarrhea, vomiting, and a high case-fatality rate (59%) in 49 persons. On May 25, 2014, Kenema Government Hospital confirmed the first case of Ebola virus disease in Sierra Leone, probably brought there by a traditional healer who had treated Ebola patients from Guinea. Tracing led to 13 additional cases—all women who attended the burial.13
The Center for Systems Biology at Harvard University and the Broad Institute of Massachusetts Institute of Technology generated 99 Ebola virus genome sequences from 78 patients with confirmed disease, representing more than 70% of the patients diagnosed with the disease in Sierra Leone from May to mid-June 2014. They found genetic similarity across the sequenced 2014 samples, suggesting a single transmission from the natural reservoir, followed by human-to-human transmission during the outbreak. Continued human-reservoir exposure is unlikely to have contributed to the growth of this epidemic.14
As of October 14, 2014, there were 8,914 suspected and confirmed cases of Ebola virus infection, and 4,477 deaths.15
But how did Zaire Ebola virus make the 2,000-mile trek from Central Africa to Guinea in West Africa? There are two possibilities: it has always been present in the region but we just never noticed, or it was recently introduced. Bayesian phylogenetic analyses and sequence divergence studies suggest the virus has been present in bat populations in Guinea without previously infecting humans.
Why Guinea and why Guéckédou? Guinea is one of the poorest countries in the world, ranking 178th of 187 countries on the Human Development Index of the United Nations Development Programme, just behind Liberia (174th) and Sierra Leone (177th). In Guinea, the life expectancy is 56 years and the gross national income per capita is $440. The region has been systematically plundered and the forest decimated by clear-cut logging, leaving the Guinea Forest Region largely deforested, resulting in increased contact between humans and the small animals that serve as the source of infection.1
LIMITED CAPACITY, EVEN IN THE UNITED STATES
A few hospitals in the United States have dedicated units to handle serious infectious diseases such as Ebola: Emory University Hospital; Nebraska Medicine in Omaha; Providence St. Patrick Hospital in Missoula, MT; and the National Institutes of Health in Bethesda, MD. However, in total they have only 19 beds.
QUESTIONS, ANSWERS—AND MORE QUESTIONS
(The following is from a question-and-answer discussion that followed Dr. Brizendine’s Grand Rounds presentation.)
Q: Are there any differences between survivors and those who die of the disease? A: We do not know. Patient survival depends on early recognition and supportive care. There are disparities in the care of patients. Schieffelin et al16 analyzed the characteristics of patients who died or who survived in Sierra Leone and found that the mortality rate was higher in older patients and those with a higher viral load on presentation.
Q: Does the virus block production or release of interferon early in infection? A: Yes, it has been shown17 that Ebola virus protein VP24 inhibits signaling downstream of both interferon alpha/beta and interferon gamma by indirectly impairing the transport of a transcription factor termed STAT1. VP24 is also able to bind STAT1 directly. The resulting suppression of host interferon very early on in the incubation phase is key to the virulence of the virus.
Q: Does infection with one of the viral species confer immunity from other species? A: No, there is no cross-immunity.
Q: How soon do patients test positive? A: About 5 days after exposure, when they develop a fever. At this time patients are highly viremic, which PCR can detect.
Q: Before the virus is detectable in the blood, where is it? A: The liver, endothelial cells, antigen-presenting cells, and adrenal glands.
Q: Do we really need to quarantine ill patients and health care workers returning from Africa, per CDC recommendations? A: We don’t know everything, and some people do make bad decisions, such as traveling while symptomatic. I support a period of observation, although confinement is not reasonable, as it may pose a disincentive to cooperation.
Q: What is the role of giving plasma from survivors? A: Dr. Kent Brantly (see American citizens infected with Ebola) received the blood of a 14-year-old who survived. We don’t know. It is not proved. It did not result in improvement in animal models.
Q: Is the bleeding caused by a mechanism similar to that in enterohemorrhagic Escherichia coli infection? A: No. That is a bacterial toxin, whereas this is more like disseminated intravascular coagulation, with an intrinsic pathway anticoagulation cascade.
Q: How long does the virus remain viable outside the body? A: In one study,18 Ebola virus could not be recovered from experimentally contaminated surfaces (plastic, metal or glass) at room temperature. In another in which it was dried onto a surface,19 Ebola virus survived in the dark for several hours between 20 and 25°C. When dried in tissue culture media onto glass and stored at 4°C, it has survived for over 50 days.
Q: How long does the virus remain in breast milk? A: We know it has been detected 15 days after disease onset and think possibly as late as 28 days from symptom onset.3
Q: How are people actually infected? A: I believe people get the virus on their hands and then touch their face, eyes, or mouth. If you are wearing personal protective equipment, it must occur while doffing the equipment.
Q: Could we increase the sensitivity of the test so that we could detect the virus before the onset of symptoms? A: In theory it may be possible. The virus is somewhere in the body during the incubation period. Perhaps we could sample the right compartment in an enriched mononuclear cell line.
Q: When can patients who recover resume their normal activities? A: After their viral load returns to 0, I would still advise abstaining from unprotected sex and from breastfeeding for a few months. but as for other activities, no special precautions are needed.
Q: Does the virus appear to be mutating at a high rate? A: Looking back to 2004, mutations are occurring, but there is no sign that any of these mutations has contributed to the size of the outbreak by changing the characteristics of the Ebola virus. Can it become aerosolized? It has been suggested that the virus that caused the outbreak separated from those that caused past Ebola outbreaks but does not seem to be affecting the spread or efficacy of experimental drugs and vaccines. So, even though it is an RNA virus and mutations are occurring, no serious changes have emerged.14
BACK TO OUR PATIENT
The differential diagnosis for the patient described at the beginning of this paper includes travelers’ diarrhea, malaria, typhoid fever, yellow fever, meningococcal disease … and Ebola virus disease, although this is much less likely in view of the epidemiology and incubation period of this disease. When his stool was tested by enzyme immunoassay and culture, it was found to be positive for Campylobacter. He recovered with oral rehydration.
- Bausch DG, Schwarz L. Outbreak of ebola virus disease in Guinea: where ecology meets economy. PLoS Negl Trop Dis 2014; 8:e3056.
- Roddy P, Thomas SL, Jeffs B, et al. Factors associated with Marburg hemorrhagic fever: analysis of patient data from Uige, Angola. J Infect Dis 2010; 201:1909–1918.
- Bausch DG, Towner JS, Dowell SF, et al. Assessment of the risk of Ebola virus transmission from bodily fluids and fomites. J Infect Dis 2007; 196(suppl 2):S142–S147.
- WHO Ebola Response Team. Ebola virus disease in West Africa—the first 9 months of the epidemic and forward projections. N Engl J Med 2014; 371:1481–1495.
- Ribner BS. Treating patients with Ebola virus infections in the US: lessons learned. Presented at IDWeek, October 8, 2014. Philadelphia PA.
- Feldman H, Jones SM, Daddario-DiCaprio KM, et al. Effective post-exposure treatment of Ebola infection. PLoS Pathog 2007; 3:e2.
- Stanley DA, Honko AN, Asiedu C, et al. Chimpanzee adenovirus vaccine generates acute and durable protective immunity against ebolavirus challenge. Nat Med 2014; 20:1126–1129.
- Oswald WB, Geisbert TW, Davis KJ, et al. Neutralizing antibody fails to impact the course of Ebola virus infection in monkeys. PLos Pathog 2007; 3:e9.
- Qui X, Wong G, Fernando L, et al. mAbs and Ad-vectored IFN-a therapy rescue Ebola-infected nonhuman primates when administered after the detection of viremia and symptoms. Sci Transl Med 2013; 5:207ra143.
- Qui X, Wong G, Audet J, et al. Reversion of advanced Ebola virus disease in nonhuman primates with ZMapp. Nature 2014; 514:47–53.
- Reid SP, Shurtleff AC, Costantino JA, et al. HSPA5 is an essential host factor for Ebola virus infection. Antiviral Res 2014; 109:171–174.
- Oestereich L, Lüdtke A, Wurr S, Rieger T, Muñoz-Fontela C, Günther S. Successful treatment of advanced Ebola virus infection with T-705 (favipiravir) in a small animal model. Antiviral Res 2014; 105:17–21.
- Baize S, Pannetier D, Oestereich L, et al. Emergence of Zaire Ebola virus dsease in Guinea. N Engl J Med 2014; 371:1418–1425.
- Gire SK, Goba A, Andersen KG, et al. Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science 2014; 345:1369–1372.
- Chamary JV. 4000 deaths and counting: the Ebola epidemic in 4 charts. Forbes. http://www.forbes.com/sites/jvchamary/2014/10/13/ebola-trends. Accessed November 5, 2014.
- Schieffelin JS, Shaffer JG, Goba A, et al, for the KGH Lassa Fever Program, the Viral Hemorrhagic Fever Consortium, and the WHO Clinical Response Team. Clinical illness and outcomes in patients with Ebola in Sierra Leone. N Engl J Med 2014 Oct 29 [Epub ahead of print]. DOI: 10.1056/NEJMoa1411680.
- Zhang AP, Bornholdt ZA, Liu T, et al. The ebola virus interferon antagonist VP24 directly binds STAT1 and has a novel, pyramidal fold. PLoS Pathog 2012; 8:e1002550.
- Piercy TJ, Smither SJ, Steward JA, Eastaugh L, Lever MS. The survival of filoviruses in liquids, on solid substrates and in a dynamic aerosol. J Appl Microbiol 2010; 109:1531–1539.
- Sagripanti JL, Rom AM, Holland LE. Persistence in darkness of virulent alphaviruses, Ebola virus, and Lassa virus deposited on solid surfaces. Arch Virol 2010; 155:2035–2039.
- Bausch DG, Schwarz L. Outbreak of ebola virus disease in Guinea: where ecology meets economy. PLoS Negl Trop Dis 2014; 8:e3056.
- Roddy P, Thomas SL, Jeffs B, et al. Factors associated with Marburg hemorrhagic fever: analysis of patient data from Uige, Angola. J Infect Dis 2010; 201:1909–1918.
- Bausch DG, Towner JS, Dowell SF, et al. Assessment of the risk of Ebola virus transmission from bodily fluids and fomites. J Infect Dis 2007; 196(suppl 2):S142–S147.
- WHO Ebola Response Team. Ebola virus disease in West Africa—the first 9 months of the epidemic and forward projections. N Engl J Med 2014; 371:1481–1495.
- Ribner BS. Treating patients with Ebola virus infections in the US: lessons learned. Presented at IDWeek, October 8, 2014. Philadelphia PA.
- Feldman H, Jones SM, Daddario-DiCaprio KM, et al. Effective post-exposure treatment of Ebola infection. PLoS Pathog 2007; 3:e2.
- Stanley DA, Honko AN, Asiedu C, et al. Chimpanzee adenovirus vaccine generates acute and durable protective immunity against ebolavirus challenge. Nat Med 2014; 20:1126–1129.
- Oswald WB, Geisbert TW, Davis KJ, et al. Neutralizing antibody fails to impact the course of Ebola virus infection in monkeys. PLos Pathog 2007; 3:e9.
- Qui X, Wong G, Fernando L, et al. mAbs and Ad-vectored IFN-a therapy rescue Ebola-infected nonhuman primates when administered after the detection of viremia and symptoms. Sci Transl Med 2013; 5:207ra143.
- Qui X, Wong G, Audet J, et al. Reversion of advanced Ebola virus disease in nonhuman primates with ZMapp. Nature 2014; 514:47–53.
- Reid SP, Shurtleff AC, Costantino JA, et al. HSPA5 is an essential host factor for Ebola virus infection. Antiviral Res 2014; 109:171–174.
- Oestereich L, Lüdtke A, Wurr S, Rieger T, Muñoz-Fontela C, Günther S. Successful treatment of advanced Ebola virus infection with T-705 (favipiravir) in a small animal model. Antiviral Res 2014; 105:17–21.
- Baize S, Pannetier D, Oestereich L, et al. Emergence of Zaire Ebola virus dsease in Guinea. N Engl J Med 2014; 371:1418–1425.
- Gire SK, Goba A, Andersen KG, et al. Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science 2014; 345:1369–1372.
- Chamary JV. 4000 deaths and counting: the Ebola epidemic in 4 charts. Forbes. http://www.forbes.com/sites/jvchamary/2014/10/13/ebola-trends. Accessed November 5, 2014.
- Schieffelin JS, Shaffer JG, Goba A, et al, for the KGH Lassa Fever Program, the Viral Hemorrhagic Fever Consortium, and the WHO Clinical Response Team. Clinical illness and outcomes in patients with Ebola in Sierra Leone. N Engl J Med 2014 Oct 29 [Epub ahead of print]. DOI: 10.1056/NEJMoa1411680.
- Zhang AP, Bornholdt ZA, Liu T, et al. The ebola virus interferon antagonist VP24 directly binds STAT1 and has a novel, pyramidal fold. PLoS Pathog 2012; 8:e1002550.
- Piercy TJ, Smither SJ, Steward JA, Eastaugh L, Lever MS. The survival of filoviruses in liquids, on solid substrates and in a dynamic aerosol. J Appl Microbiol 2010; 109:1531–1539.
- Sagripanti JL, Rom AM, Holland LE. Persistence in darkness of virulent alphaviruses, Ebola virus, and Lassa virus deposited on solid surfaces. Arch Virol 2010; 155:2035–2039.
KEY POINTS
- Ebola virus is spread by contact with body fluids, with no evidence to date that it is airborne.
- Ebola virus is likely maintained in a reservoir of small animals, possibly bats.
- The incubation period is about 5 to 7 days, during which the patient is not infectious.
- Symptoms begin abruptly, with fever, chills, and general malaise, which in some patients leads to weakness, severe headache, myalgia, nausea, vomiting, diarrhea, and abdominal pain.
- Once the disease is symptomatic, patients have high levels of virus in the blood and other body fluids and are therefore infectious.
- Survivors show improvement in the second week of illness, during which viremia resolves and virus-specific antibodies appear.
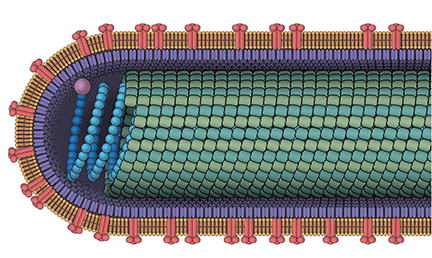
More to the Story Than a Skull Fracture
ANSWER
The radiograph shows two areas of concern: Within the apex of the right lung, there is a vague haziness that, in the setting of trauma, is suggestive of a contusion or even aspiration pneumonia. Another possibility is some sort of neoplasm. In addition, the patient has what appears to be a rounded density within the left lung, also suspicious for neoplasm. Additional work-up with contrast-enhanced CT is warranted.
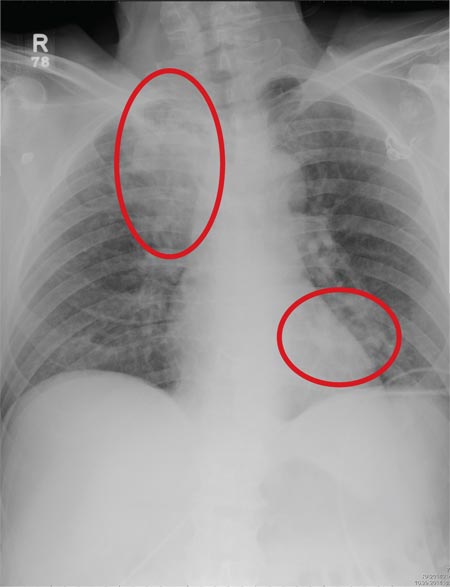
Through further questioning, the patient denies any current symptoms or previous/recent diagnosis of cancer. CT of the chest confirmed the presence of masses in the right upper and left lower lobes. Subsequent biopsy was consistent with a moderate to poorly differentiated squamous cell carcinoma.
ANSWER
The radiograph shows two areas of concern: Within the apex of the right lung, there is a vague haziness that, in the setting of trauma, is suggestive of a contusion or even aspiration pneumonia. Another possibility is some sort of neoplasm. In addition, the patient has what appears to be a rounded density within the left lung, also suspicious for neoplasm. Additional work-up with contrast-enhanced CT is warranted.
Through further questioning, the patient denies any current symptoms or previous/recent diagnosis of cancer. CT of the chest confirmed the presence of masses in the right upper and left lower lobes. Subsequent biopsy was consistent with a moderate to poorly differentiated squamous cell carcinoma.
ANSWER
The radiograph shows two areas of concern: Within the apex of the right lung, there is a vague haziness that, in the setting of trauma, is suggestive of a contusion or even aspiration pneumonia. Another possibility is some sort of neoplasm. In addition, the patient has what appears to be a rounded density within the left lung, also suspicious for neoplasm. Additional work-up with contrast-enhanced CT is warranted.
Through further questioning, the patient denies any current symptoms or previous/recent diagnosis of cancer. CT of the chest confirmed the presence of masses in the right upper and left lower lobes. Subsequent biopsy was consistent with a moderate to poorly differentiated squamous cell carcinoma.
A 63-year-old man is transferred to your facility with a skull fracture secondary to a fall. He thinks he tripped and fell, hitting his head. He does not recall experiencing dizziness or syncope. He states he was momentarily dazed but does not think he lost consciousness. He is complaining of a mild headache and has reported drainage from his left ear. He denies any noteworthy medical history and takes no medications regularly. He admits to smoking one to one-and-a-half packs of cigarettes per day. Initial assessment reveals an older-appearing male who is awake, alert, oriented, and in no obvious distress. His vital signs, including O2 saturation, are normal. His pupils are equal and react briskly. He does have obvious otorrhea from his left ear. He is moving all his extremities well and appears to have no deficits. You review his imaging studies, which include a chest radiograph (shown). What is your impression?
Case Studies in Toxicology: Sippin’ on Some “Sizzurp”
Case

A 19-year-old man was found unresponsive by his girlfriend. They both attended a party the previous night where a number of people were drinking alcohol and cough syrup to get “high.” When emergency medical technicians arrived at the patient’s house, they administered naloxone, which somewhat improved the patient’s level of consciousness; oxygen was also delivered via facemask.
Upon arrival to the ED, the patient complained of hearing loss and tinnitus. His initial vital signs were: blood pressure, 99/60 mm Hg; heart rate, 110 beats/minute; respiratory rate, 20 breaths/minute; temperature, 96.8°F. Oxygen saturation was 80% on room air. On examination, he was lethargic but responsive to voice and oriented to time, place, and person. His pupils were pinpoint; his hearing was decreased bilaterally; his breathing was shallow, with rales audible at both lung bases; his bowel sounds were hypoactive; and his skin was warm and moist. The rest of the examination was otherwise unremarkable.
What cough and cold products are commonly abused with the intent to get high?
Hundreds of nonprescription pharmaceutical products—each with the potential for misuse or abuse—are available to consumers in retail stores and online. These products can be classified by expected clinical effect, which helps clinicians with the diagnosis and management of these patients (Table).

Of the antitussive products currently available over the counter (OTC), those that contain dextromethorphan have the widest abuse potential. Referred to as “dex,” “DMX,” or “tuss,” this drug is widely abused among adolescents and young adults due to its easy availability. In therapeutic doses, dextromethorphan suppresses cough via the medullary cough center. Ingesting dextromethorphan at higher doses, a practice referred to as “Robo tripping,” can produce hallucinations and a dissociative state marked by alterations in consciousness and impaired motor control. Dextromethorphan is a structural analog of ketamine and phencyclidine, which accounts for their similar clinical effects.
Codeine
Codeine is another drug added to various cough medications for its antitussive properties. An opioid, it acts centrally to suppress cough and has mild analgesic properties. It is available only by prescription in the United States, but can be purchased as an OTC product in other countries. Recently, it has come into the media spotlight as the starting product to make “Krokodil” (see Emerg Med. 2014;46[2]:76-78).
Case Continuation
While undergoing his workup in the ED, the patient became increasingly lethargic with persistent hypoxia. Although initially
responsive to naloxone, his respirations became more labored, requiring intubation. Prior to intubation and while awake, the patient mentioned that he was drinking “sizzurp” the evening prior. He denied the use of other drugs or of having any suicidal intent. A postintubation chest X-ray revealed a left-sided retrocardiac infiltrate consistent with aspiration pneumonitis.
What is sizzurp?
Sizzurp is a slang term used to describe a beverage that is most frequently comprised of fruit-flavored soda, codeine/promethazine hydrochloride cough syrup (CPHCS), and hard candy (classically a Jolly Rancher).1 This combination is ingested by the user with the intent of achieving a unique high—attributable to the combined effects of codeine, an opioid, and promethazine, an antihistamine (with antipsychotic properties). According to user reports, CPHCS induces a deep sense of euphoria, relaxation, and a slowed sense of time.2 Additional slang terms used to describe this product include “lean,” “purple drank,” “purp,” “drank,” “syrup,” “barre,” and “Texas tea.”
According to one source, purple drank originated in Houston, Texas around the 1960s, when blues musicians would combine dextromethorphan with beer.3 Over time, the recipe was modified, and by the 1980s, when purple drank was adopted by hip-hop musicians from the same Houston neighborhoods, the name sizzurp took hold.
In the 1990s, one Houston-based hiphop artist, DJ Screw, developed a genre of music called “chopped and screwed,” inspired by the CPHCS high and notable for its slowed-down tempo that fit the sedation and decreased motor activity induced by the drug. As chopped and screwed music became popularized, so too did the recreational use of CPHCS. In 2000, “Sippin’ on Some Sizzurp,” a hit song by southern hip-hop group Three Six Mafia, introduced CPHCS to more mainstream hip-hop audiences.
Despite the CPHCS-related deaths of a number of hip-hop musicians, including DJ Screw, as well as the arrests of professional
football players linked to abusing the drug, CPHCS continues to be glorified by a number of hip-hop and pop musicians.
Unfortunately, media attention of these events often has the paradoxical effect of promoting use among adolescents and young adults, and CPHCS has become a drug of choice for black adolescents in many Texas communities.4 However, one study attempting to define a purple drank user profile among college students at a large public university in the southeastern United States revealed that use was most prevalent among urban male youth primarily from Hispanic, Native American, and white ethnic backgrounds—challenging the notion that it is confined to the black community.5
Although CPHCS is only available by prescription in the United States, its widespread abuse suggests easy access to this drug. In April 2014, Actavis, the pharmaceutical company that produces a promethazine/codeine product known as the “champagne of sizzurp,” made a bold decision to cease all production and sales of the product in direct response to the widespread media attention and glamorization of CPHCS. In its announcement, the company cited its “commitment to being a partner in the fight against prescription-drug abuse.”6 Despite Actavis’ cessation of manufacturing CPHC, at least four other companies continue to sell similar formulations.
What are the dangers of CPHCS use?
The effects produced by CPHCS are described as euphoric, which may be attributable to both codeine and promethazine. Codeine, or 3-methyl morphine, is an inactive opioid agonist and prodrug that requires metabolic activation via O-demethylation to morphine by CYP2D6. Onset of action occurs 30 to 45 minutes after ingestion, while peak effects are reached within 1 to 2 hours and last approximately 4 to 6 hours.7 Since approximately 5% to 7% of the white population lack CPY2D6 function, these individuals will experience no analgesic or euphoric effects from codeine.8 However, ultra-rapid CYP2D6 metabolizers can produce significant and potentially life-threatening concentrations of morphine.
Adverse effects of recreational codeine use are similar to that of any opioid and include central nervous system (CNS) depression, miosis, and hypoactive bowel sounds, with severe toxicity marked by coma, respiratory depression, hypotension, bradycardia, and/or death due to respiratory arrest. Aspiration pneumonitis and rhabdomyolysis are complications of impaired airway protection and prolonged immobility. Opioid-induced ototoxicity, resulting in either temporary or permanent hearing loss, is a rare complication, described largely in case reports.9 (See Emerg Med. 2012;44[11]:4-6).
Promethazine hydrochloride contributes to the unique effects experienced by the recreational user and likely acts synergistically with codeine to augment CNS depression. Both a histamine H1-receptor antagonist and the muscarinic dopamine (D2)-receptor antagonist promethazine is included in prescription cough syrups to produce its antihistamine, antiemetic, and sedative properties.7 It is well absorbed from the gastrointestinal (GI) tract with more limited oral bioavailability due to the first-pass effect. Onset of action occurs within 20 minutes of administration, and the duration of effect is approximately 4 to 6 hours. Adverse effects of promethazine include variable CNS effects, from obtundation to agitated delirium, and are often accompanied by anticholinergic effects such as hyperthermia, dry flushed skin, mydriasis, hypoactive bowel sounds, and urinary retention. Neurological manifestations, likely mediated by dopamine blockade, include muscle rigidity, athetosis, hyperreflexia, and other upper motor neuron signs. Severe toxicity can produce coma, respiratory depression, seizure, and/or death.
What are the treatment strategies?
Management of patients with CPHCS toxicity, as with all poisoned patients, begins with rapid evaluation and stabilization of the airway, breathing, and circulation. The benefits of GI decontamination are likely to be outweighed by the risks engendered by CNS depression. While supportive care is the mainstay, targeted therapies may include naloxone for the treatment of opioid-induced respiratory depression and physostigmine, when contraindications have been ruled out, for the reversal of the anticholinergic toxidrome.
Conclusion
The patient was admitted to the intensive care unit where he was treated for aspiration pneumonitis, acute respiratory distress syndrome, rhabdomyolysis, and acute renal failure. His hearing loss and tinnitus resolved. He was extubated on hospital day 9 and discharged from the hospital on day 14.
Dr Laskowski is a medical toxicology fellow in the department of emergency medicine at New York University Langone Medical Center. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Sizzurp. Urban Dictionary Web site. http://www.urbandictionary.com/define.php?term=sizzurp. Accessed October 15, 2014.
- Jodeine. Sippin’ purple drank: an experience with promethazine with codeine & cannabis. Erowid Web site. https://www.erowid.org/experiences/exp.php?ID=54165. Accessed October 15, 2014.
- Fergusen G. Sizzurp. KCRW Radio Web site. http://www.kcrw.com/news-culture/shows/good-food/butter-carving-the-last-supper-sizzurp-cheftestants. March 23, 2013. Accessed October 15, 2014.
- Elwood WN. Sticky business: patterns of procurement and misuse of prescription cough syrup in Houston. J Psychoactive Drugs. 2001;33(2):121-133.
- Agnich LE, Stogner JM, Miller BL, Marcum CD. Purple drank prevalence and characteristics of misusers of codeine cough syrup mixtures. Addict Behav. 2013;38(9):2445-2449.
- Hlavaty C. Drug company cites abuse, pop culture hype in ending cough syrup production. Houston Chronicle. April 24, 2014. http://blog.chron.com/thetexican/2014/04/drug-company-cites-abuse-pop-culture-hype-in-ending-cough-syrup-production/. Accessed October 15, 2014.
- Burns JM, Boyer EW. Antitussives and substance abuse. Subst Abuse Rehabil. 2013;4:75-82.
- Nelson LS, Olsen D. Opioids. In: Nelson LS, Lewin NA, Howland MA, Hoffman RS, Goldfrank LR, Flomenbaum NE, eds. Goldfrank’s Toxicologic Emergencies. 9th ed. New York, NY: McGraw Hill; 2011:559-578.
- Freeman SR, Bray ME, Amos CS, Gibson WP. The association of codeine, macrocytosis and bilateral sudden or rapidly progressive profound sensorineural deafness. Acta Otolaryngol. 2009;129(1):1061-1066.
Case
A 19-year-old man was found unresponsive by his girlfriend. They both attended a party the previous night where a number of people were drinking alcohol and cough syrup to get “high.” When emergency medical technicians arrived at the patient’s house, they administered naloxone, which somewhat improved the patient’s level of consciousness; oxygen was also delivered via facemask.
Upon arrival to the ED, the patient complained of hearing loss and tinnitus. His initial vital signs were: blood pressure, 99/60 mm Hg; heart rate, 110 beats/minute; respiratory rate, 20 breaths/minute; temperature, 96.8°F. Oxygen saturation was 80% on room air. On examination, he was lethargic but responsive to voice and oriented to time, place, and person. His pupils were pinpoint; his hearing was decreased bilaterally; his breathing was shallow, with rales audible at both lung bases; his bowel sounds were hypoactive; and his skin was warm and moist. The rest of the examination was otherwise unremarkable.
What cough and cold products are commonly abused with the intent to get high?
Hundreds of nonprescription pharmaceutical products—each with the potential for misuse or abuse—are available to consumers in retail stores and online. These products can be classified by expected clinical effect, which helps clinicians with the diagnosis and management of these patients (Table).
Of the antitussive products currently available over the counter (OTC), those that contain dextromethorphan have the widest abuse potential. Referred to as “dex,” “DMX,” or “tuss,” this drug is widely abused among adolescents and young adults due to its easy availability. In therapeutic doses, dextromethorphan suppresses cough via the medullary cough center. Ingesting dextromethorphan at higher doses, a practice referred to as “Robo tripping,” can produce hallucinations and a dissociative state marked by alterations in consciousness and impaired motor control. Dextromethorphan is a structural analog of ketamine and phencyclidine, which accounts for their similar clinical effects.
Codeine
Codeine is another drug added to various cough medications for its antitussive properties. An opioid, it acts centrally to suppress cough and has mild analgesic properties. It is available only by prescription in the United States, but can be purchased as an OTC product in other countries. Recently, it has come into the media spotlight as the starting product to make “Krokodil” (see Emerg Med. 2014;46[2]:76-78).
Case Continuation
While undergoing his workup in the ED, the patient became increasingly lethargic with persistent hypoxia. Although initially
responsive to naloxone, his respirations became more labored, requiring intubation. Prior to intubation and while awake, the patient mentioned that he was drinking “sizzurp” the evening prior. He denied the use of other drugs or of having any suicidal intent. A postintubation chest X-ray revealed a left-sided retrocardiac infiltrate consistent with aspiration pneumonitis.
What is sizzurp?
Sizzurp is a slang term used to describe a beverage that is most frequently comprised of fruit-flavored soda, codeine/promethazine hydrochloride cough syrup (CPHCS), and hard candy (classically a Jolly Rancher).1 This combination is ingested by the user with the intent of achieving a unique high—attributable to the combined effects of codeine, an opioid, and promethazine, an antihistamine (with antipsychotic properties). According to user reports, CPHCS induces a deep sense of euphoria, relaxation, and a slowed sense of time.2 Additional slang terms used to describe this product include “lean,” “purple drank,” “purp,” “drank,” “syrup,” “barre,” and “Texas tea.”
According to one source, purple drank originated in Houston, Texas around the 1960s, when blues musicians would combine dextromethorphan with beer.3 Over time, the recipe was modified, and by the 1980s, when purple drank was adopted by hip-hop musicians from the same Houston neighborhoods, the name sizzurp took hold.
In the 1990s, one Houston-based hiphop artist, DJ Screw, developed a genre of music called “chopped and screwed,” inspired by the CPHCS high and notable for its slowed-down tempo that fit the sedation and decreased motor activity induced by the drug. As chopped and screwed music became popularized, so too did the recreational use of CPHCS. In 2000, “Sippin’ on Some Sizzurp,” a hit song by southern hip-hop group Three Six Mafia, introduced CPHCS to more mainstream hip-hop audiences.
Despite the CPHCS-related deaths of a number of hip-hop musicians, including DJ Screw, as well as the arrests of professional
football players linked to abusing the drug, CPHCS continues to be glorified by a number of hip-hop and pop musicians.
Unfortunately, media attention of these events often has the paradoxical effect of promoting use among adolescents and young adults, and CPHCS has become a drug of choice for black adolescents in many Texas communities.4 However, one study attempting to define a purple drank user profile among college students at a large public university in the southeastern United States revealed that use was most prevalent among urban male youth primarily from Hispanic, Native American, and white ethnic backgrounds—challenging the notion that it is confined to the black community.5
Although CPHCS is only available by prescription in the United States, its widespread abuse suggests easy access to this drug. In April 2014, Actavis, the pharmaceutical company that produces a promethazine/codeine product known as the “champagne of sizzurp,” made a bold decision to cease all production and sales of the product in direct response to the widespread media attention and glamorization of CPHCS. In its announcement, the company cited its “commitment to being a partner in the fight against prescription-drug abuse.”6 Despite Actavis’ cessation of manufacturing CPHC, at least four other companies continue to sell similar formulations.
What are the dangers of CPHCS use?
The effects produced by CPHCS are described as euphoric, which may be attributable to both codeine and promethazine. Codeine, or 3-methyl morphine, is an inactive opioid agonist and prodrug that requires metabolic activation via O-demethylation to morphine by CYP2D6. Onset of action occurs 30 to 45 minutes after ingestion, while peak effects are reached within 1 to 2 hours and last approximately 4 to 6 hours.7 Since approximately 5% to 7% of the white population lack CPY2D6 function, these individuals will experience no analgesic or euphoric effects from codeine.8 However, ultra-rapid CYP2D6 metabolizers can produce significant and potentially life-threatening concentrations of morphine.
Adverse effects of recreational codeine use are similar to that of any opioid and include central nervous system (CNS) depression, miosis, and hypoactive bowel sounds, with severe toxicity marked by coma, respiratory depression, hypotension, bradycardia, and/or death due to respiratory arrest. Aspiration pneumonitis and rhabdomyolysis are complications of impaired airway protection and prolonged immobility. Opioid-induced ototoxicity, resulting in either temporary or permanent hearing loss, is a rare complication, described largely in case reports.9 (See Emerg Med. 2012;44[11]:4-6).
Promethazine hydrochloride contributes to the unique effects experienced by the recreational user and likely acts synergistically with codeine to augment CNS depression. Both a histamine H1-receptor antagonist and the muscarinic dopamine (D2)-receptor antagonist promethazine is included in prescription cough syrups to produce its antihistamine, antiemetic, and sedative properties.7 It is well absorbed from the gastrointestinal (GI) tract with more limited oral bioavailability due to the first-pass effect. Onset of action occurs within 20 minutes of administration, and the duration of effect is approximately 4 to 6 hours. Adverse effects of promethazine include variable CNS effects, from obtundation to agitated delirium, and are often accompanied by anticholinergic effects such as hyperthermia, dry flushed skin, mydriasis, hypoactive bowel sounds, and urinary retention. Neurological manifestations, likely mediated by dopamine blockade, include muscle rigidity, athetosis, hyperreflexia, and other upper motor neuron signs. Severe toxicity can produce coma, respiratory depression, seizure, and/or death.
What are the treatment strategies?
Management of patients with CPHCS toxicity, as with all poisoned patients, begins with rapid evaluation and stabilization of the airway, breathing, and circulation. The benefits of GI decontamination are likely to be outweighed by the risks engendered by CNS depression. While supportive care is the mainstay, targeted therapies may include naloxone for the treatment of opioid-induced respiratory depression and physostigmine, when contraindications have been ruled out, for the reversal of the anticholinergic toxidrome.
Conclusion
The patient was admitted to the intensive care unit where he was treated for aspiration pneumonitis, acute respiratory distress syndrome, rhabdomyolysis, and acute renal failure. His hearing loss and tinnitus resolved. He was extubated on hospital day 9 and discharged from the hospital on day 14.
Dr Laskowski is a medical toxicology fellow in the department of emergency medicine at New York University Langone Medical Center. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
Case
A 19-year-old man was found unresponsive by his girlfriend. They both attended a party the previous night where a number of people were drinking alcohol and cough syrup to get “high.” When emergency medical technicians arrived at the patient’s house, they administered naloxone, which somewhat improved the patient’s level of consciousness; oxygen was also delivered via facemask.
Upon arrival to the ED, the patient complained of hearing loss and tinnitus. His initial vital signs were: blood pressure, 99/60 mm Hg; heart rate, 110 beats/minute; respiratory rate, 20 breaths/minute; temperature, 96.8°F. Oxygen saturation was 80% on room air. On examination, he was lethargic but responsive to voice and oriented to time, place, and person. His pupils were pinpoint; his hearing was decreased bilaterally; his breathing was shallow, with rales audible at both lung bases; his bowel sounds were hypoactive; and his skin was warm and moist. The rest of the examination was otherwise unremarkable.
What cough and cold products are commonly abused with the intent to get high?
Hundreds of nonprescription pharmaceutical products—each with the potential for misuse or abuse—are available to consumers in retail stores and online. These products can be classified by expected clinical effect, which helps clinicians with the diagnosis and management of these patients (Table).
Of the antitussive products currently available over the counter (OTC), those that contain dextromethorphan have the widest abuse potential. Referred to as “dex,” “DMX,” or “tuss,” this drug is widely abused among adolescents and young adults due to its easy availability. In therapeutic doses, dextromethorphan suppresses cough via the medullary cough center. Ingesting dextromethorphan at higher doses, a practice referred to as “Robo tripping,” can produce hallucinations and a dissociative state marked by alterations in consciousness and impaired motor control. Dextromethorphan is a structural analog of ketamine and phencyclidine, which accounts for their similar clinical effects.
Codeine
Codeine is another drug added to various cough medications for its antitussive properties. An opioid, it acts centrally to suppress cough and has mild analgesic properties. It is available only by prescription in the United States, but can be purchased as an OTC product in other countries. Recently, it has come into the media spotlight as the starting product to make “Krokodil” (see Emerg Med. 2014;46[2]:76-78).
Case Continuation
While undergoing his workup in the ED, the patient became increasingly lethargic with persistent hypoxia. Although initially
responsive to naloxone, his respirations became more labored, requiring intubation. Prior to intubation and while awake, the patient mentioned that he was drinking “sizzurp” the evening prior. He denied the use of other drugs or of having any suicidal intent. A postintubation chest X-ray revealed a left-sided retrocardiac infiltrate consistent with aspiration pneumonitis.
What is sizzurp?
Sizzurp is a slang term used to describe a beverage that is most frequently comprised of fruit-flavored soda, codeine/promethazine hydrochloride cough syrup (CPHCS), and hard candy (classically a Jolly Rancher).1 This combination is ingested by the user with the intent of achieving a unique high—attributable to the combined effects of codeine, an opioid, and promethazine, an antihistamine (with antipsychotic properties). According to user reports, CPHCS induces a deep sense of euphoria, relaxation, and a slowed sense of time.2 Additional slang terms used to describe this product include “lean,” “purple drank,” “purp,” “drank,” “syrup,” “barre,” and “Texas tea.”
According to one source, purple drank originated in Houston, Texas around the 1960s, when blues musicians would combine dextromethorphan with beer.3 Over time, the recipe was modified, and by the 1980s, when purple drank was adopted by hip-hop musicians from the same Houston neighborhoods, the name sizzurp took hold.
In the 1990s, one Houston-based hiphop artist, DJ Screw, developed a genre of music called “chopped and screwed,” inspired by the CPHCS high and notable for its slowed-down tempo that fit the sedation and decreased motor activity induced by the drug. As chopped and screwed music became popularized, so too did the recreational use of CPHCS. In 2000, “Sippin’ on Some Sizzurp,” a hit song by southern hip-hop group Three Six Mafia, introduced CPHCS to more mainstream hip-hop audiences.
Despite the CPHCS-related deaths of a number of hip-hop musicians, including DJ Screw, as well as the arrests of professional
football players linked to abusing the drug, CPHCS continues to be glorified by a number of hip-hop and pop musicians.
Unfortunately, media attention of these events often has the paradoxical effect of promoting use among adolescents and young adults, and CPHCS has become a drug of choice for black adolescents in many Texas communities.4 However, one study attempting to define a purple drank user profile among college students at a large public university in the southeastern United States revealed that use was most prevalent among urban male youth primarily from Hispanic, Native American, and white ethnic backgrounds—challenging the notion that it is confined to the black community.5
Although CPHCS is only available by prescription in the United States, its widespread abuse suggests easy access to this drug. In April 2014, Actavis, the pharmaceutical company that produces a promethazine/codeine product known as the “champagne of sizzurp,” made a bold decision to cease all production and sales of the product in direct response to the widespread media attention and glamorization of CPHCS. In its announcement, the company cited its “commitment to being a partner in the fight against prescription-drug abuse.”6 Despite Actavis’ cessation of manufacturing CPHC, at least four other companies continue to sell similar formulations.
What are the dangers of CPHCS use?
The effects produced by CPHCS are described as euphoric, which may be attributable to both codeine and promethazine. Codeine, or 3-methyl morphine, is an inactive opioid agonist and prodrug that requires metabolic activation via O-demethylation to morphine by CYP2D6. Onset of action occurs 30 to 45 minutes after ingestion, while peak effects are reached within 1 to 2 hours and last approximately 4 to 6 hours.7 Since approximately 5% to 7% of the white population lack CPY2D6 function, these individuals will experience no analgesic or euphoric effects from codeine.8 However, ultra-rapid CYP2D6 metabolizers can produce significant and potentially life-threatening concentrations of morphine.
Adverse effects of recreational codeine use are similar to that of any opioid and include central nervous system (CNS) depression, miosis, and hypoactive bowel sounds, with severe toxicity marked by coma, respiratory depression, hypotension, bradycardia, and/or death due to respiratory arrest. Aspiration pneumonitis and rhabdomyolysis are complications of impaired airway protection and prolonged immobility. Opioid-induced ototoxicity, resulting in either temporary or permanent hearing loss, is a rare complication, described largely in case reports.9 (See Emerg Med. 2012;44[11]:4-6).
Promethazine hydrochloride contributes to the unique effects experienced by the recreational user and likely acts synergistically with codeine to augment CNS depression. Both a histamine H1-receptor antagonist and the muscarinic dopamine (D2)-receptor antagonist promethazine is included in prescription cough syrups to produce its antihistamine, antiemetic, and sedative properties.7 It is well absorbed from the gastrointestinal (GI) tract with more limited oral bioavailability due to the first-pass effect. Onset of action occurs within 20 minutes of administration, and the duration of effect is approximately 4 to 6 hours. Adverse effects of promethazine include variable CNS effects, from obtundation to agitated delirium, and are often accompanied by anticholinergic effects such as hyperthermia, dry flushed skin, mydriasis, hypoactive bowel sounds, and urinary retention. Neurological manifestations, likely mediated by dopamine blockade, include muscle rigidity, athetosis, hyperreflexia, and other upper motor neuron signs. Severe toxicity can produce coma, respiratory depression, seizure, and/or death.
What are the treatment strategies?
Management of patients with CPHCS toxicity, as with all poisoned patients, begins with rapid evaluation and stabilization of the airway, breathing, and circulation. The benefits of GI decontamination are likely to be outweighed by the risks engendered by CNS depression. While supportive care is the mainstay, targeted therapies may include naloxone for the treatment of opioid-induced respiratory depression and physostigmine, when contraindications have been ruled out, for the reversal of the anticholinergic toxidrome.
Conclusion
The patient was admitted to the intensive care unit where he was treated for aspiration pneumonitis, acute respiratory distress syndrome, rhabdomyolysis, and acute renal failure. His hearing loss and tinnitus resolved. He was extubated on hospital day 9 and discharged from the hospital on day 14.
Dr Laskowski is a medical toxicology fellow in the department of emergency medicine at New York University Langone Medical Center. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Sizzurp. Urban Dictionary Web site. http://www.urbandictionary.com/define.php?term=sizzurp. Accessed October 15, 2014.
- Jodeine. Sippin’ purple drank: an experience with promethazine with codeine & cannabis. Erowid Web site. https://www.erowid.org/experiences/exp.php?ID=54165. Accessed October 15, 2014.
- Fergusen G. Sizzurp. KCRW Radio Web site. http://www.kcrw.com/news-culture/shows/good-food/butter-carving-the-last-supper-sizzurp-cheftestants. March 23, 2013. Accessed October 15, 2014.
- Elwood WN. Sticky business: patterns of procurement and misuse of prescription cough syrup in Houston. J Psychoactive Drugs. 2001;33(2):121-133.
- Agnich LE, Stogner JM, Miller BL, Marcum CD. Purple drank prevalence and characteristics of misusers of codeine cough syrup mixtures. Addict Behav. 2013;38(9):2445-2449.
- Hlavaty C. Drug company cites abuse, pop culture hype in ending cough syrup production. Houston Chronicle. April 24, 2014. http://blog.chron.com/thetexican/2014/04/drug-company-cites-abuse-pop-culture-hype-in-ending-cough-syrup-production/. Accessed October 15, 2014.
- Burns JM, Boyer EW. Antitussives and substance abuse. Subst Abuse Rehabil. 2013;4:75-82.
- Nelson LS, Olsen D. Opioids. In: Nelson LS, Lewin NA, Howland MA, Hoffman RS, Goldfrank LR, Flomenbaum NE, eds. Goldfrank’s Toxicologic Emergencies. 9th ed. New York, NY: McGraw Hill; 2011:559-578.
- Freeman SR, Bray ME, Amos CS, Gibson WP. The association of codeine, macrocytosis and bilateral sudden or rapidly progressive profound sensorineural deafness. Acta Otolaryngol. 2009;129(1):1061-1066.
- Sizzurp. Urban Dictionary Web site. http://www.urbandictionary.com/define.php?term=sizzurp. Accessed October 15, 2014.
- Jodeine. Sippin’ purple drank: an experience with promethazine with codeine & cannabis. Erowid Web site. https://www.erowid.org/experiences/exp.php?ID=54165. Accessed October 15, 2014.
- Fergusen G. Sizzurp. KCRW Radio Web site. http://www.kcrw.com/news-culture/shows/good-food/butter-carving-the-last-supper-sizzurp-cheftestants. March 23, 2013. Accessed October 15, 2014.
- Elwood WN. Sticky business: patterns of procurement and misuse of prescription cough syrup in Houston. J Psychoactive Drugs. 2001;33(2):121-133.
- Agnich LE, Stogner JM, Miller BL, Marcum CD. Purple drank prevalence and characteristics of misusers of codeine cough syrup mixtures. Addict Behav. 2013;38(9):2445-2449.
- Hlavaty C. Drug company cites abuse, pop culture hype in ending cough syrup production. Houston Chronicle. April 24, 2014. http://blog.chron.com/thetexican/2014/04/drug-company-cites-abuse-pop-culture-hype-in-ending-cough-syrup-production/. Accessed October 15, 2014.
- Burns JM, Boyer EW. Antitussives and substance abuse. Subst Abuse Rehabil. 2013;4:75-82.
- Nelson LS, Olsen D. Opioids. In: Nelson LS, Lewin NA, Howland MA, Hoffman RS, Goldfrank LR, Flomenbaum NE, eds. Goldfrank’s Toxicologic Emergencies. 9th ed. New York, NY: McGraw Hill; 2011:559-578.
- Freeman SR, Bray ME, Amos CS, Gibson WP. The association of codeine, macrocytosis and bilateral sudden or rapidly progressive profound sensorineural deafness. Acta Otolaryngol. 2009;129(1):1061-1066.

A 61-year-old man with fluctuating hypertension
A 61-year-old man with type 2 diabetes mellitus on glimepiride therapy presented with somnolence and slurred speech. His capillary glucose level was 17 mg/dL and his serum glucose level was 28 mg/dL. He was treated with intravenous dextrose, and his glucose level promptly returned to normal.
He had been adherent to his medication regimen and denied overmedicating or accidental overdosing. Over the past 7 months, he had noted redness on his palms, a rash on his legs, intermittent moderate to severe headaches, weight loss, and decreased appetite. In addition, his blood pressure had been labile, which his physicians had attributed to autonomic instability. He had continued on the same dose of glimepiride despite losing weight.
His history included multivessel coronary artery disease treated with angioplasty and placement of multiple coronary stents; ischemic cardiomyopathy with a left ventricular ejection fraction of 28%; implantation of a cardioverter-defibrillator for secondary prevention of ventricular arrhythmia; an ischemic stroke; and multiple sclerosis complicated by bilateral blindness, with optic nerve involvement and autonomic instability, present for over a year and manifested by labile blood pressure. He was a long-time tobacco user. His daily medications included ticagrelor 90 mg, aspirin 81 mg, metoprolol 50 mg, ramipril 10 mg, simvastatin 20 mg, glimepiride 2 mg, and esomeprazole 40 mg. He needed help taking his medications.
At the time of hospital admission, his heart rate was 69 beats per minute with a regular rhythm, blood pressure 115/73 mm Hg, respiratory rate 11 breaths per minute with an oxygen saturation of 99% on room air, and oral temperature 34.7°C (94.5°F). He appeared to be in no distress.
Cardiovascular examination revealed no murmurs or gallops; there was mild nonpitting edema of the lower extremities. Pulmonary, abdominal, and neurologic examinations were unrevealing except for bilateral blindness. Vascular examination revealed no bruits. Results of a complete blood cell count and metabolic panel were normal except for a hemoglobin level of 9.9 g/dL (reference range 13.5–17.5) and a platelet count of 477 × 109/L (150–450).
Although he continued to receive the same medications he had been taking at home, his blood pressure fluctuated. On the second hospital day, it reached 186/135 mm Hg, at which time he also had palpitations, dyspnea, and crackles in the lower lobes of both lungs. Volume resuscitation on admission was suspected to have played a role, and he received furosemide, which improved his symptoms. But several hours later, his blood pressure rose again, and he became diaphoretic. Despite aggressive treatment with different antihypertensive agents, his blood pressure remained high and his symptoms persisted. Chest radiography showed no evidence of pulmonary edema. Because of his progressive dyspnea, the diagnosis of pulmonary embolism was entertained.
CAUSES OF RESISTANT HYPERTENSION
1. What could explain this patient’s high blood pressure?
- A drug effect
- Renovascular disease
- Excess circulating catecholamines
- Obstructive sleep apnea
- Primary aldosteronism
Sympathomimetic drugs such as epinephrine, norepinephrine, dopamine, and vasopressin, which are used when hemodynamic support is required, can raise both systolic and diastolic blood pressure. Nonsteroidal anti-inflammatory drugs and nasal decongestants are common culprits in the community. However, our patient was using none of these drugs.
Renovascular disease is one of many causes of resistant hypertension, accounting for 8% of all cases.1,2 Despite fluctuations, the blood pressure often remains chronically elevated, its changes are less paroxysmal than in our patient, and a precipitating factor such as a dietary indiscretion is sometimes identified.1
Excess circulating catecholamines can be a result of stress, exogenous administration, or endogenous oversecretion. Our patient’s clinical presentation is highly suspicious for a high-catecholamine state, and this should be further evaluated.
Obstructive sleep apnea is common in patients with resistant hypertension, with an estimated prevalence as high as 60% in this group.3,4
Primary aldosteronism has an estimated prevalence of about 20% in patients evaluated for resistant hypertension.5
AN ADRENAL MASS IS INCIDENTALLY DISCOVERED
Computed tomographic angiography of the chest revealed no evidence of pulmonary emboli. There was mild dilation of the central pulmonary arteries and an incidental, incompletely imaged 4.7-by-3.4-cm mass of mixed attenuation in the right adrenal gland, with macroscopic fat within the lesion.
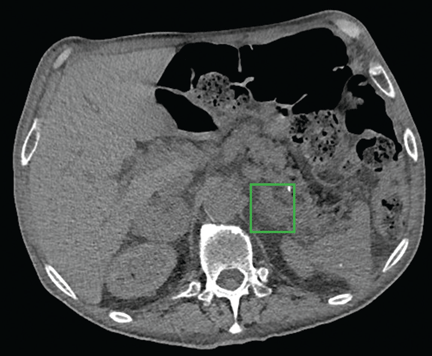
Computed tomography (CT) of the abdomen with dedicated cuts through the adrenal glands revealed a 4.7-cm heterogeneous right adrenal mass with a density of 34 Hounsfield units (HU). The left adrenal gland appeared diffusely enlarged without a discretely seen mass, consistent with hyperplasticity (Figure 1).
2. Based on the patient’s clinical presentation and findings on CT, what would be the most likely diagnosis for this incidentally found adrenal mass?
- Adrenocortical adenoma
- Adrenocortical carcinoma
- Metastatic mass
- Pheochromocytoma
Adrenocortical adenoma can present as a small homogeneous mass of variable size, with smooth margins, and rarely containing hemorrhagic tissue or calcifications. The typical density on nonenhanced CT is less than 10 HU. On enhanced CT, it is nonvascular. T2-weighted magnetic resonance imaging (MRI) shows a lesion of the same intensity as liver tissue.6
Adrenocortical adenoma is not classically associated with autologous activity and thus is less likely to explain our patient’s symptoms.
Adrenocortical carcinoma can present as a large heterogeneous mass, usually greater than 4 cm in diameter, with irregular margins and areas of necrosis, hemorrhage, or calcification. The typical density on nonenhanced CT is greater than 10 HU. On enhanced CT, the mass is usually vascular, and T2-weighted MRI will show a lesion more intense than liver tissue.6
Adrenocortical carcinoma is also not classically associated with autologous activity, and so is not likely to explain our patient’s symptoms.6
Metastatic disease can present with masses of variable size, often bilaterally, and occasionally with cysts or areas of hemorrhage. The typical density of metastatic lesions on nonenhanced CT is greater than 10 HU. On enhanced CT, they are usually vascular, and on T2-weighted MRI they are hyperintense.6 The characteristics of the mass and the absence of a primary malignancy on CT of the chest and abdomen do not support the diagnosis of metastatic disease.
Pheochromocytoma is a neuroendocrine tumor of the adrenal medulla that can present as a large heterogeneous mass, greater than 3 cm in diameter, with clear margins and cysts or areas of hemorrhage. Extra-adrenal neuroendocrine tumors are typically called paragangliomas and have features similar to those of pheochromocytoma. The typical density of pheochromocytoma on nonenhanced CT is greater than 10 HU. On enhanced CT, it is usually vascular, and T2-weighted MRI shows a hyperintense lesion. Pheochromocytoma can be biochemically active and thus can cause signs and symptoms that will lead to the diagnosis.6
Other imaging tests may play a role in the evaluation of adrenal masses but are not required for the diagnosis of pheochromocytoma. Functional positron emission tomography using metaiodobenzylguanidine labeled with iodine 123 or-iodine 131 or using the glucose analogue F-18 fluorodeoxyglucose has been used in the initial assessment of pheochromocytoma, with good sensitivity and specificity.7,8
Our patient’s pacemaker-defibrillator precluded him from undergoing MRI.
DIAGNOSIS: PHEOCHROMOCYTOMA
Pheochromocytoma was highly suspected on the basis of the patient’s clinical presentation, and metoprolol was immediately discontinued. He was started on the calcium channel blocker verapamil and the alpha-blocker phenoxybenzamine.
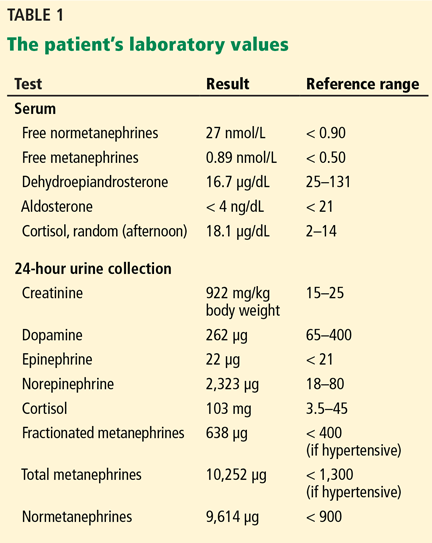
Serum samples were obtained to measure metanephrines, dehydroepiandrosterone, aldosterone, and cortisol, and a 24-hour urine collection was obtained to measure creatinine, dopamine, epinephrine, norepinephrine, cortisol, and metanephrines. Based on the results (Table 1) and on the findings on imaging, the patient was diagnosed with pheochromocytoma. A surgical consultation was obtained, and surgery was recommended.
WHEN DOES PHEOCHROMOCYTOMA CALL FOR SURGERY?
3. Which criterion is most important when determining the need for surgery for pheochromocytoma?
- Findings on fine-needle aspiration biopsy
- Biochemical activity
- Size of the mass
- Bilateral masses
Fine-needle aspiration biopsy can be done when a mass is found incidentally and no evidence of biochemical activity is detected, although it is not an essential part of the diagnostic workup.9 In most cases, the sampling from fine-needle aspiration is not sufficient to achieve a diagnosis.
Biochemical activity is the most important factor when determining the need for prompt surgical intervention. The excess circulating catecholamines have been associated with increased risk of cardiovascular morbidity and death independent of the morbidity associated with hypertension alone.10 Biochemical activity can be independent of the size of the mass, but larger masses typically present with symptoms.
Bilateral masses have been associated with metastatic disease.11 In retrospect, the patient’s history of hypertension and cerebrovascular accident could be associated with the development of a catecholamine-releasing tumor.
A GOOD OUTCOME FROM SURGERY
The patient was continued on phenoxybenzamine for 7 days and responded well to this therapy.
After this preoperative preparation, he underwent laparoscopic right adrenalectomy with excision of a retroperitoneal adrenal mass. His postoperative course was complicated by transient hypotension requiring low-dose vasopressin support for less than 24 hours. He was then restarted on his previous dosage of metoprolol and was discharged home on postoperative day 5 with stable blood pressure. Follow-up 24-hour urine collection 1 month after he was discharged showed normalization of metanephrine, normetanephrine, epinephrine, and norepinephrine levels.
Despite low suspicion for an underlying genetic syndrome, he was referred for genetic testing and was scheduled to have a repeat 24-hour urine collection and imaging in 6 months to follow his enlarged left adrenal gland, which did not appear to be metabolically hyperactive.
4. What is the most common perioperative complication of pheochromocytoma excision?
- Hypoglycemia
- Hypotension
- Hypocortisolism
- Hypertension
- Tachycardia
Hypoglycemia has been observed after removal of pheochromocytoma, as levels of catecholamines (which normally inhibit pancreatic beta cells) decrease and insulin secretion consequently increases.12 Our patient developed hypoglycemia before surgery, not after, and it was likely due to the combination of his antidiabetic therapy, weight loss, and decreased oral intake.
Hypotension is the most common complication in the perioperative period. It is associated with excessive loss of catecholamine secretion. It is usually short-lived but may require aggressive administration of intravenous fluids and use of sympathomimetic agents.
Hypocortisolism is unlikely in patients with pheochromocytoma, but it is likely after removal of adrenocortical adenoma.
Hypertension and tachycardia affect up to 40% of pheochromocytoma patients in some case series.12
PHEOCHROMOCYTOMA: A CATECHOLAMINE-SECRETING TUMOR
The pathophysiology of pheochromocytoma is complex. It is characterized by accelerated growth of cells producing catecholamines, which may produce symptoms when secreted into the bloodstream. The classic triad of symptoms is headache, hypertension, and hyperglycemia, although our patient had very low blood sugar levels. Other common symptoms are nausea, orthostasis, and tremor, although not all symptoms are invariably seen.
Genetic testing recommended
Genetic associations have been described and are thought to be responsible for 20% to 30% of cases of pheochromocytoma. All associated germline mutations are autosomal dominant, some with variable penetrance. These include:
- Succinate dehydrogenase subunit B, C, and D mutations
- von Hippel-Lindau syndrome
- Multiple endocrine neoplasia type 1 and type 2 syndromes
- Neurofibromatosis type 1.13,14
The succinate dehydrogenase subunit mutations have been associated with, but not limited to, extra-adrenal adenomas (paragangliomas) and carry a worse prognosis.
Some experts recommend genetic testing in all cases of pheochromocytoma, sporadic or familial, and this testing should be followed by counseling if a mutation is found.15 Others recommend genetic testing based on the patient’s age (under age 50), history, imaging, and biochemical features of the tumor (metanephrines predominate in multiple endocrine neoplasia syndromes, and normetanephrines in von Hippel-Lindau syndrome).13
Serious consequences
A thorough evaluation is recommended, since pheochromocytoma has been associated with increased cardiovascular morbidity. In a retrospective series, Stolk et al10 reported that patients with pheochromocytoma had a higher incidence of myocardial infarction, angina, and stroke in the years preceding the diagnosis than did patients with essential hypertension (13.8% vs 1.1%, P < .001).10
Catecholamine cardiomyopathy has been described and shares clinical features with Takotsubo or stress cardiomyopathy, with global left ventricular systolic and diastolic dysfunction that improve or resolve after the adrenergic insult is removed.16
Conditions that warrant further evaluation or that may suggest pheochromocytoma are malignant hypertension, hypertensive encephalopathy, ischemic stroke, subarachnoid hemorrhage, acute pulmonary edema, angina pectoris, myocardial infarction, aortic dissection, and kidney injury.
When to suspect pheochromocytoma
Pheochromocytoma should be suspected in a patient with resistant hypertension, family history, or imaging findings that suggest an adrenal mass with a heterogeneous appearance. The diagnostic algorithm follows the same pathway as for the evaluation of an incidentally found adrenal mass, with determination of its dimension and characteristics by CT or MRI, and with biochemical testing of urine catecholamines, plasma free metanephrines, renin, aldosterone, and cortisol.
The diagnosis of pheochromocytoma is established by obtaining fractionated metanephrines and catecholamines in a 24-hour urine collection (sensitivity 90%, specificity 98%). Analysis of plasma metanephrines has a higher sensitivity (97%) but lower specificity (85%).17 The combination of typical signs, symptoms, and laboratory findings makes the diagnosis likely, especially in combination with a unilateral adrenal mass.
Laparoscopic surgery after medical preparation for active tumors
If the mass appears benign and not biochemically hyperactive, then follow-up at 1 year is recommended, with repeat testing. Surgical evaluation and intervention is recommended for lesions that appear malignant or that are biochemically active and clinically symptomatic.9
Preoperative hemodynamic control is essential in the management of pheochromocytoma to prevent or minimize hemodynamic changes that can be driven by increased catecholamines. Control is typically achieved with initial alpha-blockade and then beta-blockade to avoid worsening hypertension and to prevent an acute hypertensive crisis during surgical intervention. Phenoxybenzamine, the mainstay of therapy, is a nonselective alpha-blocker with a long duration of action that requires titration over several days up to 3 weeks.
A selective alpha-1-blocker such as doxazosin can be used to control postoperative hypotension, as it has a shorter half-life than phenoxybenzamine. Alternative strategies include calcium channel blockers, centrally acting sympathetic blockers, and magnesium.18
Laparoscopic adrenalectomy by an experienced surgeon after excellent medical preparation is often considered the treatment of choice, but for larger or malignant masses, an open procedure is recommended. The risk of perioperative morbidity and death can be reduced by adequate medical management. With successful surgical resection, the long-term prognosis is favorable.
- Calhoun DA, Jones D, Textor S, et al. Resistant hypertension: diagnosis, evaluation, and treatment. A scientific statement from the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research. Hypertension 2008; 51:1403–1419.
- Kumar N, Calhoun DA, Dudenbostel T. Management of patients with resistant hypertension: current treatment options. Integr Blood Press Control 2013; 6:139–151.
- Pedrosa RP, Drager LF, Gonzaga CC, et al. Obstructive sleep apnea: the most common secondary cause of hypertension associated with resistant hypertension. Hypertension 2011; 58:811–817.
- Marcus JA, Pothineni A, Marcus CZ, Bisognano JD. The role of obesity and obstructive sleep apnea in the pathogenesis and treatment of resistant hypertension. Curr Hypertens Rep 2014; 16:411.
- Calhoun DA, Nishizaka MK, Zaman MA, Thakkar RB, Weissmann P. Hyperaldosteronism among black and white subjects with resistant hypertension. Hypertension 2002; 40:892–896.
- Young WF Clinical practice. The incidentally discovered adrenal mass. N Engl J Med 2007; 356:601–610.
- Lin M, Wong V, Yap J, Jin R, Leong P, Campbell P. FDG PET in the evaluation of phaeochromocytoma: a correlative study with MIBG scintigraphy and Ki-67 proliferative index. Clin Imaging 2013; 37:1084–1088.
- Raja A, Leung K, Stamm M, Girgis S, Low G. Multimodality imaging findings of pheochromocytoma with associated clinical and biochemical features in 53 patients with histologically confirmed tumors. AJR Am J Roentgenol 2013; 201:825–833.
- Nieman LK. Approach to the patient with an adrenal incidentaloma. J Clin Endocrinol Metab 2010; 95:4106–4113.
- Stolk RF, Bakx C, Mulder J, Timmers HJ, Lenders JW. Is the excess cardiovascular morbidity in pheochromocytoma related to blood pressure or to catecholamines? J Clin Endocrinol Metab 2013; 98:1100–1106.
- Grumbach MM, Biller BM, Braunstein GD, et al. Management of the clinically inapparent adrenal mass (‘incidentaloma’). Ann Intern Med 2003; 138:424–429.
- Lentschener C, Gaujoux S, Tesniere A, Dousset B. Point of controversy: perioperative care of patients undergoing pheochromocytoma removal—time for a reappraisal? Eur J Endocrinol 2011; 165:365–373.
- Bryant J, Farmer J, Kessler LJ, Townsend RR, Nathanson KL. Pheochromocytoma: the expanding genetic differential diagnosis. J Natl Cancer Inst 2003; 95:1196–1204.
- Lee P, Leonard J. Textbook on endocrinology. BMJ 1994; 308:1512.
- Fishbein L, Merrill S, Fraker DL, Cohen DL, Nathanson KL. Inherited mutations in pheochromocytoma and paraganglioma: why all patients should be offered genetic testing. Ann Surg Oncol 2013; 20:1444–1450.
- Agarwal G, Sadacharan D, Kapoor A, et al. Cardiovascular dysfunction and catecholamine cardiomyopathy in pheochromocytoma patients and their reversal following surgical cure: results of a prospective case-control study. Surgery 2011; 150:1202–1211.
- Sawka AM, Jaeschke R, Singh RJ, Young WF A comparison of biochemical tests for pheochromocytoma: measurement of fractionated plasma metanephrines compared with the combination of 24-hour urinary metanephrines and catecholamines. J Clin Endocrinol Metab 2003; 88:553–558.
- Domi R, Laho H. Management of pheochromocytoma: old ideas and new drugs. Niger J Clin Pract 2012; 15:253–257.
A 61-year-old man with type 2 diabetes mellitus on glimepiride therapy presented with somnolence and slurred speech. His capillary glucose level was 17 mg/dL and his serum glucose level was 28 mg/dL. He was treated with intravenous dextrose, and his glucose level promptly returned to normal.
He had been adherent to his medication regimen and denied overmedicating or accidental overdosing. Over the past 7 months, he had noted redness on his palms, a rash on his legs, intermittent moderate to severe headaches, weight loss, and decreased appetite. In addition, his blood pressure had been labile, which his physicians had attributed to autonomic instability. He had continued on the same dose of glimepiride despite losing weight.
His history included multivessel coronary artery disease treated with angioplasty and placement of multiple coronary stents; ischemic cardiomyopathy with a left ventricular ejection fraction of 28%; implantation of a cardioverter-defibrillator for secondary prevention of ventricular arrhythmia; an ischemic stroke; and multiple sclerosis complicated by bilateral blindness, with optic nerve involvement and autonomic instability, present for over a year and manifested by labile blood pressure. He was a long-time tobacco user. His daily medications included ticagrelor 90 mg, aspirin 81 mg, metoprolol 50 mg, ramipril 10 mg, simvastatin 20 mg, glimepiride 2 mg, and esomeprazole 40 mg. He needed help taking his medications.
At the time of hospital admission, his heart rate was 69 beats per minute with a regular rhythm, blood pressure 115/73 mm Hg, respiratory rate 11 breaths per minute with an oxygen saturation of 99% on room air, and oral temperature 34.7°C (94.5°F). He appeared to be in no distress.
Cardiovascular examination revealed no murmurs or gallops; there was mild nonpitting edema of the lower extremities. Pulmonary, abdominal, and neurologic examinations were unrevealing except for bilateral blindness. Vascular examination revealed no bruits. Results of a complete blood cell count and metabolic panel were normal except for a hemoglobin level of 9.9 g/dL (reference range 13.5–17.5) and a platelet count of 477 × 109/L (150–450).
Although he continued to receive the same medications he had been taking at home, his blood pressure fluctuated. On the second hospital day, it reached 186/135 mm Hg, at which time he also had palpitations, dyspnea, and crackles in the lower lobes of both lungs. Volume resuscitation on admission was suspected to have played a role, and he received furosemide, which improved his symptoms. But several hours later, his blood pressure rose again, and he became diaphoretic. Despite aggressive treatment with different antihypertensive agents, his blood pressure remained high and his symptoms persisted. Chest radiography showed no evidence of pulmonary edema. Because of his progressive dyspnea, the diagnosis of pulmonary embolism was entertained.
CAUSES OF RESISTANT HYPERTENSION
1. What could explain this patient’s high blood pressure?
- A drug effect
- Renovascular disease
- Excess circulating catecholamines
- Obstructive sleep apnea
- Primary aldosteronism
Sympathomimetic drugs such as epinephrine, norepinephrine, dopamine, and vasopressin, which are used when hemodynamic support is required, can raise both systolic and diastolic blood pressure. Nonsteroidal anti-inflammatory drugs and nasal decongestants are common culprits in the community. However, our patient was using none of these drugs.
Renovascular disease is one of many causes of resistant hypertension, accounting for 8% of all cases.1,2 Despite fluctuations, the blood pressure often remains chronically elevated, its changes are less paroxysmal than in our patient, and a precipitating factor such as a dietary indiscretion is sometimes identified.1
Excess circulating catecholamines can be a result of stress, exogenous administration, or endogenous oversecretion. Our patient’s clinical presentation is highly suspicious for a high-catecholamine state, and this should be further evaluated.
Obstructive sleep apnea is common in patients with resistant hypertension, with an estimated prevalence as high as 60% in this group.3,4
Primary aldosteronism has an estimated prevalence of about 20% in patients evaluated for resistant hypertension.5
AN ADRENAL MASS IS INCIDENTALLY DISCOVERED
Computed tomographic angiography of the chest revealed no evidence of pulmonary emboli. There was mild dilation of the central pulmonary arteries and an incidental, incompletely imaged 4.7-by-3.4-cm mass of mixed attenuation in the right adrenal gland, with macroscopic fat within the lesion.
Computed tomography (CT) of the abdomen with dedicated cuts through the adrenal glands revealed a 4.7-cm heterogeneous right adrenal mass with a density of 34 Hounsfield units (HU). The left adrenal gland appeared diffusely enlarged without a discretely seen mass, consistent with hyperplasticity (Figure 1).
2. Based on the patient’s clinical presentation and findings on CT, what would be the most likely diagnosis for this incidentally found adrenal mass?
- Adrenocortical adenoma
- Adrenocortical carcinoma
- Metastatic mass
- Pheochromocytoma
Adrenocortical adenoma can present as a small homogeneous mass of variable size, with smooth margins, and rarely containing hemorrhagic tissue or calcifications. The typical density on nonenhanced CT is less than 10 HU. On enhanced CT, it is nonvascular. T2-weighted magnetic resonance imaging (MRI) shows a lesion of the same intensity as liver tissue.6
Adrenocortical adenoma is not classically associated with autologous activity and thus is less likely to explain our patient’s symptoms.
Adrenocortical carcinoma can present as a large heterogeneous mass, usually greater than 4 cm in diameter, with irregular margins and areas of necrosis, hemorrhage, or calcification. The typical density on nonenhanced CT is greater than 10 HU. On enhanced CT, the mass is usually vascular, and T2-weighted MRI will show a lesion more intense than liver tissue.6
Adrenocortical carcinoma is also not classically associated with autologous activity, and so is not likely to explain our patient’s symptoms.6
Metastatic disease can present with masses of variable size, often bilaterally, and occasionally with cysts or areas of hemorrhage. The typical density of metastatic lesions on nonenhanced CT is greater than 10 HU. On enhanced CT, they are usually vascular, and on T2-weighted MRI they are hyperintense.6 The characteristics of the mass and the absence of a primary malignancy on CT of the chest and abdomen do not support the diagnosis of metastatic disease.
Pheochromocytoma is a neuroendocrine tumor of the adrenal medulla that can present as a large heterogeneous mass, greater than 3 cm in diameter, with clear margins and cysts or areas of hemorrhage. Extra-adrenal neuroendocrine tumors are typically called paragangliomas and have features similar to those of pheochromocytoma. The typical density of pheochromocytoma on nonenhanced CT is greater than 10 HU. On enhanced CT, it is usually vascular, and T2-weighted MRI shows a hyperintense lesion. Pheochromocytoma can be biochemically active and thus can cause signs and symptoms that will lead to the diagnosis.6
Other imaging tests may play a role in the evaluation of adrenal masses but are not required for the diagnosis of pheochromocytoma. Functional positron emission tomography using metaiodobenzylguanidine labeled with iodine 123 or-iodine 131 or using the glucose analogue F-18 fluorodeoxyglucose has been used in the initial assessment of pheochromocytoma, with good sensitivity and specificity.7,8
Our patient’s pacemaker-defibrillator precluded him from undergoing MRI.
DIAGNOSIS: PHEOCHROMOCYTOMA
Pheochromocytoma was highly suspected on the basis of the patient’s clinical presentation, and metoprolol was immediately discontinued. He was started on the calcium channel blocker verapamil and the alpha-blocker phenoxybenzamine.
Serum samples were obtained to measure metanephrines, dehydroepiandrosterone, aldosterone, and cortisol, and a 24-hour urine collection was obtained to measure creatinine, dopamine, epinephrine, norepinephrine, cortisol, and metanephrines. Based on the results (Table 1) and on the findings on imaging, the patient was diagnosed with pheochromocytoma. A surgical consultation was obtained, and surgery was recommended.
WHEN DOES PHEOCHROMOCYTOMA CALL FOR SURGERY?
3. Which criterion is most important when determining the need for surgery for pheochromocytoma?
- Findings on fine-needle aspiration biopsy
- Biochemical activity
- Size of the mass
- Bilateral masses
Fine-needle aspiration biopsy can be done when a mass is found incidentally and no evidence of biochemical activity is detected, although it is not an essential part of the diagnostic workup.9 In most cases, the sampling from fine-needle aspiration is not sufficient to achieve a diagnosis.
Biochemical activity is the most important factor when determining the need for prompt surgical intervention. The excess circulating catecholamines have been associated with increased risk of cardiovascular morbidity and death independent of the morbidity associated with hypertension alone.10 Biochemical activity can be independent of the size of the mass, but larger masses typically present with symptoms.
Bilateral masses have been associated with metastatic disease.11 In retrospect, the patient’s history of hypertension and cerebrovascular accident could be associated with the development of a catecholamine-releasing tumor.
A GOOD OUTCOME FROM SURGERY
The patient was continued on phenoxybenzamine for 7 days and responded well to this therapy.
After this preoperative preparation, he underwent laparoscopic right adrenalectomy with excision of a retroperitoneal adrenal mass. His postoperative course was complicated by transient hypotension requiring low-dose vasopressin support for less than 24 hours. He was then restarted on his previous dosage of metoprolol and was discharged home on postoperative day 5 with stable blood pressure. Follow-up 24-hour urine collection 1 month after he was discharged showed normalization of metanephrine, normetanephrine, epinephrine, and norepinephrine levels.
Despite low suspicion for an underlying genetic syndrome, he was referred for genetic testing and was scheduled to have a repeat 24-hour urine collection and imaging in 6 months to follow his enlarged left adrenal gland, which did not appear to be metabolically hyperactive.
4. What is the most common perioperative complication of pheochromocytoma excision?
- Hypoglycemia
- Hypotension
- Hypocortisolism
- Hypertension
- Tachycardia
Hypoglycemia has been observed after removal of pheochromocytoma, as levels of catecholamines (which normally inhibit pancreatic beta cells) decrease and insulin secretion consequently increases.12 Our patient developed hypoglycemia before surgery, not after, and it was likely due to the combination of his antidiabetic therapy, weight loss, and decreased oral intake.
Hypotension is the most common complication in the perioperative period. It is associated with excessive loss of catecholamine secretion. It is usually short-lived but may require aggressive administration of intravenous fluids and use of sympathomimetic agents.
Hypocortisolism is unlikely in patients with pheochromocytoma, but it is likely after removal of adrenocortical adenoma.
Hypertension and tachycardia affect up to 40% of pheochromocytoma patients in some case series.12
PHEOCHROMOCYTOMA: A CATECHOLAMINE-SECRETING TUMOR
The pathophysiology of pheochromocytoma is complex. It is characterized by accelerated growth of cells producing catecholamines, which may produce symptoms when secreted into the bloodstream. The classic triad of symptoms is headache, hypertension, and hyperglycemia, although our patient had very low blood sugar levels. Other common symptoms are nausea, orthostasis, and tremor, although not all symptoms are invariably seen.
Genetic testing recommended
Genetic associations have been described and are thought to be responsible for 20% to 30% of cases of pheochromocytoma. All associated germline mutations are autosomal dominant, some with variable penetrance. These include:
- Succinate dehydrogenase subunit B, C, and D mutations
- von Hippel-Lindau syndrome
- Multiple endocrine neoplasia type 1 and type 2 syndromes
- Neurofibromatosis type 1.13,14
The succinate dehydrogenase subunit mutations have been associated with, but not limited to, extra-adrenal adenomas (paragangliomas) and carry a worse prognosis.
Some experts recommend genetic testing in all cases of pheochromocytoma, sporadic or familial, and this testing should be followed by counseling if a mutation is found.15 Others recommend genetic testing based on the patient’s age (under age 50), history, imaging, and biochemical features of the tumor (metanephrines predominate in multiple endocrine neoplasia syndromes, and normetanephrines in von Hippel-Lindau syndrome).13
Serious consequences
A thorough evaluation is recommended, since pheochromocytoma has been associated with increased cardiovascular morbidity. In a retrospective series, Stolk et al10 reported that patients with pheochromocytoma had a higher incidence of myocardial infarction, angina, and stroke in the years preceding the diagnosis than did patients with essential hypertension (13.8% vs 1.1%, P < .001).10
Catecholamine cardiomyopathy has been described and shares clinical features with Takotsubo or stress cardiomyopathy, with global left ventricular systolic and diastolic dysfunction that improve or resolve after the adrenergic insult is removed.16
Conditions that warrant further evaluation or that may suggest pheochromocytoma are malignant hypertension, hypertensive encephalopathy, ischemic stroke, subarachnoid hemorrhage, acute pulmonary edema, angina pectoris, myocardial infarction, aortic dissection, and kidney injury.
When to suspect pheochromocytoma
Pheochromocytoma should be suspected in a patient with resistant hypertension, family history, or imaging findings that suggest an adrenal mass with a heterogeneous appearance. The diagnostic algorithm follows the same pathway as for the evaluation of an incidentally found adrenal mass, with determination of its dimension and characteristics by CT or MRI, and with biochemical testing of urine catecholamines, plasma free metanephrines, renin, aldosterone, and cortisol.
The diagnosis of pheochromocytoma is established by obtaining fractionated metanephrines and catecholamines in a 24-hour urine collection (sensitivity 90%, specificity 98%). Analysis of plasma metanephrines has a higher sensitivity (97%) but lower specificity (85%).17 The combination of typical signs, symptoms, and laboratory findings makes the diagnosis likely, especially in combination with a unilateral adrenal mass.
Laparoscopic surgery after medical preparation for active tumors
If the mass appears benign and not biochemically hyperactive, then follow-up at 1 year is recommended, with repeat testing. Surgical evaluation and intervention is recommended for lesions that appear malignant or that are biochemically active and clinically symptomatic.9
Preoperative hemodynamic control is essential in the management of pheochromocytoma to prevent or minimize hemodynamic changes that can be driven by increased catecholamines. Control is typically achieved with initial alpha-blockade and then beta-blockade to avoid worsening hypertension and to prevent an acute hypertensive crisis during surgical intervention. Phenoxybenzamine, the mainstay of therapy, is a nonselective alpha-blocker with a long duration of action that requires titration over several days up to 3 weeks.
A selective alpha-1-blocker such as doxazosin can be used to control postoperative hypotension, as it has a shorter half-life than phenoxybenzamine. Alternative strategies include calcium channel blockers, centrally acting sympathetic blockers, and magnesium.18
Laparoscopic adrenalectomy by an experienced surgeon after excellent medical preparation is often considered the treatment of choice, but for larger or malignant masses, an open procedure is recommended. The risk of perioperative morbidity and death can be reduced by adequate medical management. With successful surgical resection, the long-term prognosis is favorable.
A 61-year-old man with type 2 diabetes mellitus on glimepiride therapy presented with somnolence and slurred speech. His capillary glucose level was 17 mg/dL and his serum glucose level was 28 mg/dL. He was treated with intravenous dextrose, and his glucose level promptly returned to normal.
He had been adherent to his medication regimen and denied overmedicating or accidental overdosing. Over the past 7 months, he had noted redness on his palms, a rash on his legs, intermittent moderate to severe headaches, weight loss, and decreased appetite. In addition, his blood pressure had been labile, which his physicians had attributed to autonomic instability. He had continued on the same dose of glimepiride despite losing weight.
His history included multivessel coronary artery disease treated with angioplasty and placement of multiple coronary stents; ischemic cardiomyopathy with a left ventricular ejection fraction of 28%; implantation of a cardioverter-defibrillator for secondary prevention of ventricular arrhythmia; an ischemic stroke; and multiple sclerosis complicated by bilateral blindness, with optic nerve involvement and autonomic instability, present for over a year and manifested by labile blood pressure. He was a long-time tobacco user. His daily medications included ticagrelor 90 mg, aspirin 81 mg, metoprolol 50 mg, ramipril 10 mg, simvastatin 20 mg, glimepiride 2 mg, and esomeprazole 40 mg. He needed help taking his medications.
At the time of hospital admission, his heart rate was 69 beats per minute with a regular rhythm, blood pressure 115/73 mm Hg, respiratory rate 11 breaths per minute with an oxygen saturation of 99% on room air, and oral temperature 34.7°C (94.5°F). He appeared to be in no distress.
Cardiovascular examination revealed no murmurs or gallops; there was mild nonpitting edema of the lower extremities. Pulmonary, abdominal, and neurologic examinations were unrevealing except for bilateral blindness. Vascular examination revealed no bruits. Results of a complete blood cell count and metabolic panel were normal except for a hemoglobin level of 9.9 g/dL (reference range 13.5–17.5) and a platelet count of 477 × 109/L (150–450).
Although he continued to receive the same medications he had been taking at home, his blood pressure fluctuated. On the second hospital day, it reached 186/135 mm Hg, at which time he also had palpitations, dyspnea, and crackles in the lower lobes of both lungs. Volume resuscitation on admission was suspected to have played a role, and he received furosemide, which improved his symptoms. But several hours later, his blood pressure rose again, and he became diaphoretic. Despite aggressive treatment with different antihypertensive agents, his blood pressure remained high and his symptoms persisted. Chest radiography showed no evidence of pulmonary edema. Because of his progressive dyspnea, the diagnosis of pulmonary embolism was entertained.
CAUSES OF RESISTANT HYPERTENSION
1. What could explain this patient’s high blood pressure?
- A drug effect
- Renovascular disease
- Excess circulating catecholamines
- Obstructive sleep apnea
- Primary aldosteronism
Sympathomimetic drugs such as epinephrine, norepinephrine, dopamine, and vasopressin, which are used when hemodynamic support is required, can raise both systolic and diastolic blood pressure. Nonsteroidal anti-inflammatory drugs and nasal decongestants are common culprits in the community. However, our patient was using none of these drugs.
Renovascular disease is one of many causes of resistant hypertension, accounting for 8% of all cases.1,2 Despite fluctuations, the blood pressure often remains chronically elevated, its changes are less paroxysmal than in our patient, and a precipitating factor such as a dietary indiscretion is sometimes identified.1
Excess circulating catecholamines can be a result of stress, exogenous administration, or endogenous oversecretion. Our patient’s clinical presentation is highly suspicious for a high-catecholamine state, and this should be further evaluated.
Obstructive sleep apnea is common in patients with resistant hypertension, with an estimated prevalence as high as 60% in this group.3,4
Primary aldosteronism has an estimated prevalence of about 20% in patients evaluated for resistant hypertension.5
AN ADRENAL MASS IS INCIDENTALLY DISCOVERED
Computed tomographic angiography of the chest revealed no evidence of pulmonary emboli. There was mild dilation of the central pulmonary arteries and an incidental, incompletely imaged 4.7-by-3.4-cm mass of mixed attenuation in the right adrenal gland, with macroscopic fat within the lesion.
Computed tomography (CT) of the abdomen with dedicated cuts through the adrenal glands revealed a 4.7-cm heterogeneous right adrenal mass with a density of 34 Hounsfield units (HU). The left adrenal gland appeared diffusely enlarged without a discretely seen mass, consistent with hyperplasticity (Figure 1).
2. Based on the patient’s clinical presentation and findings on CT, what would be the most likely diagnosis for this incidentally found adrenal mass?
- Adrenocortical adenoma
- Adrenocortical carcinoma
- Metastatic mass
- Pheochromocytoma
Adrenocortical adenoma can present as a small homogeneous mass of variable size, with smooth margins, and rarely containing hemorrhagic tissue or calcifications. The typical density on nonenhanced CT is less than 10 HU. On enhanced CT, it is nonvascular. T2-weighted magnetic resonance imaging (MRI) shows a lesion of the same intensity as liver tissue.6
Adrenocortical adenoma is not classically associated with autologous activity and thus is less likely to explain our patient’s symptoms.
Adrenocortical carcinoma can present as a large heterogeneous mass, usually greater than 4 cm in diameter, with irregular margins and areas of necrosis, hemorrhage, or calcification. The typical density on nonenhanced CT is greater than 10 HU. On enhanced CT, the mass is usually vascular, and T2-weighted MRI will show a lesion more intense than liver tissue.6
Adrenocortical carcinoma is also not classically associated with autologous activity, and so is not likely to explain our patient’s symptoms.6
Metastatic disease can present with masses of variable size, often bilaterally, and occasionally with cysts or areas of hemorrhage. The typical density of metastatic lesions on nonenhanced CT is greater than 10 HU. On enhanced CT, they are usually vascular, and on T2-weighted MRI they are hyperintense.6 The characteristics of the mass and the absence of a primary malignancy on CT of the chest and abdomen do not support the diagnosis of metastatic disease.
Pheochromocytoma is a neuroendocrine tumor of the adrenal medulla that can present as a large heterogeneous mass, greater than 3 cm in diameter, with clear margins and cysts or areas of hemorrhage. Extra-adrenal neuroendocrine tumors are typically called paragangliomas and have features similar to those of pheochromocytoma. The typical density of pheochromocytoma on nonenhanced CT is greater than 10 HU. On enhanced CT, it is usually vascular, and T2-weighted MRI shows a hyperintense lesion. Pheochromocytoma can be biochemically active and thus can cause signs and symptoms that will lead to the diagnosis.6
Other imaging tests may play a role in the evaluation of adrenal masses but are not required for the diagnosis of pheochromocytoma. Functional positron emission tomography using metaiodobenzylguanidine labeled with iodine 123 or-iodine 131 or using the glucose analogue F-18 fluorodeoxyglucose has been used in the initial assessment of pheochromocytoma, with good sensitivity and specificity.7,8
Our patient’s pacemaker-defibrillator precluded him from undergoing MRI.
DIAGNOSIS: PHEOCHROMOCYTOMA
Pheochromocytoma was highly suspected on the basis of the patient’s clinical presentation, and metoprolol was immediately discontinued. He was started on the calcium channel blocker verapamil and the alpha-blocker phenoxybenzamine.
Serum samples were obtained to measure metanephrines, dehydroepiandrosterone, aldosterone, and cortisol, and a 24-hour urine collection was obtained to measure creatinine, dopamine, epinephrine, norepinephrine, cortisol, and metanephrines. Based on the results (Table 1) and on the findings on imaging, the patient was diagnosed with pheochromocytoma. A surgical consultation was obtained, and surgery was recommended.
WHEN DOES PHEOCHROMOCYTOMA CALL FOR SURGERY?
3. Which criterion is most important when determining the need for surgery for pheochromocytoma?
- Findings on fine-needle aspiration biopsy
- Biochemical activity
- Size of the mass
- Bilateral masses
Fine-needle aspiration biopsy can be done when a mass is found incidentally and no evidence of biochemical activity is detected, although it is not an essential part of the diagnostic workup.9 In most cases, the sampling from fine-needle aspiration is not sufficient to achieve a diagnosis.
Biochemical activity is the most important factor when determining the need for prompt surgical intervention. The excess circulating catecholamines have been associated with increased risk of cardiovascular morbidity and death independent of the morbidity associated with hypertension alone.10 Biochemical activity can be independent of the size of the mass, but larger masses typically present with symptoms.
Bilateral masses have been associated with metastatic disease.11 In retrospect, the patient’s history of hypertension and cerebrovascular accident could be associated with the development of a catecholamine-releasing tumor.
A GOOD OUTCOME FROM SURGERY
The patient was continued on phenoxybenzamine for 7 days and responded well to this therapy.
After this preoperative preparation, he underwent laparoscopic right adrenalectomy with excision of a retroperitoneal adrenal mass. His postoperative course was complicated by transient hypotension requiring low-dose vasopressin support for less than 24 hours. He was then restarted on his previous dosage of metoprolol and was discharged home on postoperative day 5 with stable blood pressure. Follow-up 24-hour urine collection 1 month after he was discharged showed normalization of metanephrine, normetanephrine, epinephrine, and norepinephrine levels.
Despite low suspicion for an underlying genetic syndrome, he was referred for genetic testing and was scheduled to have a repeat 24-hour urine collection and imaging in 6 months to follow his enlarged left adrenal gland, which did not appear to be metabolically hyperactive.
4. What is the most common perioperative complication of pheochromocytoma excision?
- Hypoglycemia
- Hypotension
- Hypocortisolism
- Hypertension
- Tachycardia
Hypoglycemia has been observed after removal of pheochromocytoma, as levels of catecholamines (which normally inhibit pancreatic beta cells) decrease and insulin secretion consequently increases.12 Our patient developed hypoglycemia before surgery, not after, and it was likely due to the combination of his antidiabetic therapy, weight loss, and decreased oral intake.
Hypotension is the most common complication in the perioperative period. It is associated with excessive loss of catecholamine secretion. It is usually short-lived but may require aggressive administration of intravenous fluids and use of sympathomimetic agents.
Hypocortisolism is unlikely in patients with pheochromocytoma, but it is likely after removal of adrenocortical adenoma.
Hypertension and tachycardia affect up to 40% of pheochromocytoma patients in some case series.12
PHEOCHROMOCYTOMA: A CATECHOLAMINE-SECRETING TUMOR
The pathophysiology of pheochromocytoma is complex. It is characterized by accelerated growth of cells producing catecholamines, which may produce symptoms when secreted into the bloodstream. The classic triad of symptoms is headache, hypertension, and hyperglycemia, although our patient had very low blood sugar levels. Other common symptoms are nausea, orthostasis, and tremor, although not all symptoms are invariably seen.
Genetic testing recommended
Genetic associations have been described and are thought to be responsible for 20% to 30% of cases of pheochromocytoma. All associated germline mutations are autosomal dominant, some with variable penetrance. These include:
- Succinate dehydrogenase subunit B, C, and D mutations
- von Hippel-Lindau syndrome
- Multiple endocrine neoplasia type 1 and type 2 syndromes
- Neurofibromatosis type 1.13,14
The succinate dehydrogenase subunit mutations have been associated with, but not limited to, extra-adrenal adenomas (paragangliomas) and carry a worse prognosis.
Some experts recommend genetic testing in all cases of pheochromocytoma, sporadic or familial, and this testing should be followed by counseling if a mutation is found.15 Others recommend genetic testing based on the patient’s age (under age 50), history, imaging, and biochemical features of the tumor (metanephrines predominate in multiple endocrine neoplasia syndromes, and normetanephrines in von Hippel-Lindau syndrome).13
Serious consequences
A thorough evaluation is recommended, since pheochromocytoma has been associated with increased cardiovascular morbidity. In a retrospective series, Stolk et al10 reported that patients with pheochromocytoma had a higher incidence of myocardial infarction, angina, and stroke in the years preceding the diagnosis than did patients with essential hypertension (13.8% vs 1.1%, P < .001).10
Catecholamine cardiomyopathy has been described and shares clinical features with Takotsubo or stress cardiomyopathy, with global left ventricular systolic and diastolic dysfunction that improve or resolve after the adrenergic insult is removed.16
Conditions that warrant further evaluation or that may suggest pheochromocytoma are malignant hypertension, hypertensive encephalopathy, ischemic stroke, subarachnoid hemorrhage, acute pulmonary edema, angina pectoris, myocardial infarction, aortic dissection, and kidney injury.
When to suspect pheochromocytoma
Pheochromocytoma should be suspected in a patient with resistant hypertension, family history, or imaging findings that suggest an adrenal mass with a heterogeneous appearance. The diagnostic algorithm follows the same pathway as for the evaluation of an incidentally found adrenal mass, with determination of its dimension and characteristics by CT or MRI, and with biochemical testing of urine catecholamines, plasma free metanephrines, renin, aldosterone, and cortisol.
The diagnosis of pheochromocytoma is established by obtaining fractionated metanephrines and catecholamines in a 24-hour urine collection (sensitivity 90%, specificity 98%). Analysis of plasma metanephrines has a higher sensitivity (97%) but lower specificity (85%).17 The combination of typical signs, symptoms, and laboratory findings makes the diagnosis likely, especially in combination with a unilateral adrenal mass.
Laparoscopic surgery after medical preparation for active tumors
If the mass appears benign and not biochemically hyperactive, then follow-up at 1 year is recommended, with repeat testing. Surgical evaluation and intervention is recommended for lesions that appear malignant or that are biochemically active and clinically symptomatic.9
Preoperative hemodynamic control is essential in the management of pheochromocytoma to prevent or minimize hemodynamic changes that can be driven by increased catecholamines. Control is typically achieved with initial alpha-blockade and then beta-blockade to avoid worsening hypertension and to prevent an acute hypertensive crisis during surgical intervention. Phenoxybenzamine, the mainstay of therapy, is a nonselective alpha-blocker with a long duration of action that requires titration over several days up to 3 weeks.
A selective alpha-1-blocker such as doxazosin can be used to control postoperative hypotension, as it has a shorter half-life than phenoxybenzamine. Alternative strategies include calcium channel blockers, centrally acting sympathetic blockers, and magnesium.18
Laparoscopic adrenalectomy by an experienced surgeon after excellent medical preparation is often considered the treatment of choice, but for larger or malignant masses, an open procedure is recommended. The risk of perioperative morbidity and death can be reduced by adequate medical management. With successful surgical resection, the long-term prognosis is favorable.
- Calhoun DA, Jones D, Textor S, et al. Resistant hypertension: diagnosis, evaluation, and treatment. A scientific statement from the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research. Hypertension 2008; 51:1403–1419.
- Kumar N, Calhoun DA, Dudenbostel T. Management of patients with resistant hypertension: current treatment options. Integr Blood Press Control 2013; 6:139–151.
- Pedrosa RP, Drager LF, Gonzaga CC, et al. Obstructive sleep apnea: the most common secondary cause of hypertension associated with resistant hypertension. Hypertension 2011; 58:811–817.
- Marcus JA, Pothineni A, Marcus CZ, Bisognano JD. The role of obesity and obstructive sleep apnea in the pathogenesis and treatment of resistant hypertension. Curr Hypertens Rep 2014; 16:411.
- Calhoun DA, Nishizaka MK, Zaman MA, Thakkar RB, Weissmann P. Hyperaldosteronism among black and white subjects with resistant hypertension. Hypertension 2002; 40:892–896.
- Young WF Clinical practice. The incidentally discovered adrenal mass. N Engl J Med 2007; 356:601–610.
- Lin M, Wong V, Yap J, Jin R, Leong P, Campbell P. FDG PET in the evaluation of phaeochromocytoma: a correlative study with MIBG scintigraphy and Ki-67 proliferative index. Clin Imaging 2013; 37:1084–1088.
- Raja A, Leung K, Stamm M, Girgis S, Low G. Multimodality imaging findings of pheochromocytoma with associated clinical and biochemical features in 53 patients with histologically confirmed tumors. AJR Am J Roentgenol 2013; 201:825–833.
- Nieman LK. Approach to the patient with an adrenal incidentaloma. J Clin Endocrinol Metab 2010; 95:4106–4113.
- Stolk RF, Bakx C, Mulder J, Timmers HJ, Lenders JW. Is the excess cardiovascular morbidity in pheochromocytoma related to blood pressure or to catecholamines? J Clin Endocrinol Metab 2013; 98:1100–1106.
- Grumbach MM, Biller BM, Braunstein GD, et al. Management of the clinically inapparent adrenal mass (‘incidentaloma’). Ann Intern Med 2003; 138:424–429.
- Lentschener C, Gaujoux S, Tesniere A, Dousset B. Point of controversy: perioperative care of patients undergoing pheochromocytoma removal—time for a reappraisal? Eur J Endocrinol 2011; 165:365–373.
- Bryant J, Farmer J, Kessler LJ, Townsend RR, Nathanson KL. Pheochromocytoma: the expanding genetic differential diagnosis. J Natl Cancer Inst 2003; 95:1196–1204.
- Lee P, Leonard J. Textbook on endocrinology. BMJ 1994; 308:1512.
- Fishbein L, Merrill S, Fraker DL, Cohen DL, Nathanson KL. Inherited mutations in pheochromocytoma and paraganglioma: why all patients should be offered genetic testing. Ann Surg Oncol 2013; 20:1444–1450.
- Agarwal G, Sadacharan D, Kapoor A, et al. Cardiovascular dysfunction and catecholamine cardiomyopathy in pheochromocytoma patients and their reversal following surgical cure: results of a prospective case-control study. Surgery 2011; 150:1202–1211.
- Sawka AM, Jaeschke R, Singh RJ, Young WF A comparison of biochemical tests for pheochromocytoma: measurement of fractionated plasma metanephrines compared with the combination of 24-hour urinary metanephrines and catecholamines. J Clin Endocrinol Metab 2003; 88:553–558.
- Domi R, Laho H. Management of pheochromocytoma: old ideas and new drugs. Niger J Clin Pract 2012; 15:253–257.
- Calhoun DA, Jones D, Textor S, et al. Resistant hypertension: diagnosis, evaluation, and treatment. A scientific statement from the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research. Hypertension 2008; 51:1403–1419.
- Kumar N, Calhoun DA, Dudenbostel T. Management of patients with resistant hypertension: current treatment options. Integr Blood Press Control 2013; 6:139–151.
- Pedrosa RP, Drager LF, Gonzaga CC, et al. Obstructive sleep apnea: the most common secondary cause of hypertension associated with resistant hypertension. Hypertension 2011; 58:811–817.
- Marcus JA, Pothineni A, Marcus CZ, Bisognano JD. The role of obesity and obstructive sleep apnea in the pathogenesis and treatment of resistant hypertension. Curr Hypertens Rep 2014; 16:411.
- Calhoun DA, Nishizaka MK, Zaman MA, Thakkar RB, Weissmann P. Hyperaldosteronism among black and white subjects with resistant hypertension. Hypertension 2002; 40:892–896.
- Young WF Clinical practice. The incidentally discovered adrenal mass. N Engl J Med 2007; 356:601–610.
- Lin M, Wong V, Yap J, Jin R, Leong P, Campbell P. FDG PET in the evaluation of phaeochromocytoma: a correlative study with MIBG scintigraphy and Ki-67 proliferative index. Clin Imaging 2013; 37:1084–1088.
- Raja A, Leung K, Stamm M, Girgis S, Low G. Multimodality imaging findings of pheochromocytoma with associated clinical and biochemical features in 53 patients with histologically confirmed tumors. AJR Am J Roentgenol 2013; 201:825–833.
- Nieman LK. Approach to the patient with an adrenal incidentaloma. J Clin Endocrinol Metab 2010; 95:4106–4113.
- Stolk RF, Bakx C, Mulder J, Timmers HJ, Lenders JW. Is the excess cardiovascular morbidity in pheochromocytoma related to blood pressure or to catecholamines? J Clin Endocrinol Metab 2013; 98:1100–1106.
- Grumbach MM, Biller BM, Braunstein GD, et al. Management of the clinically inapparent adrenal mass (‘incidentaloma’). Ann Intern Med 2003; 138:424–429.
- Lentschener C, Gaujoux S, Tesniere A, Dousset B. Point of controversy: perioperative care of patients undergoing pheochromocytoma removal—time for a reappraisal? Eur J Endocrinol 2011; 165:365–373.
- Bryant J, Farmer J, Kessler LJ, Townsend RR, Nathanson KL. Pheochromocytoma: the expanding genetic differential diagnosis. J Natl Cancer Inst 2003; 95:1196–1204.
- Lee P, Leonard J. Textbook on endocrinology. BMJ 1994; 308:1512.
- Fishbein L, Merrill S, Fraker DL, Cohen DL, Nathanson KL. Inherited mutations in pheochromocytoma and paraganglioma: why all patients should be offered genetic testing. Ann Surg Oncol 2013; 20:1444–1450.
- Agarwal G, Sadacharan D, Kapoor A, et al. Cardiovascular dysfunction and catecholamine cardiomyopathy in pheochromocytoma patients and their reversal following surgical cure: results of a prospective case-control study. Surgery 2011; 150:1202–1211.
- Sawka AM, Jaeschke R, Singh RJ, Young WF A comparison of biochemical tests for pheochromocytoma: measurement of fractionated plasma metanephrines compared with the combination of 24-hour urinary metanephrines and catecholamines. J Clin Endocrinol Metab 2003; 88:553–558.
- Domi R, Laho H. Management of pheochromocytoma: old ideas and new drugs. Niger J Clin Pract 2012; 15:253–257.
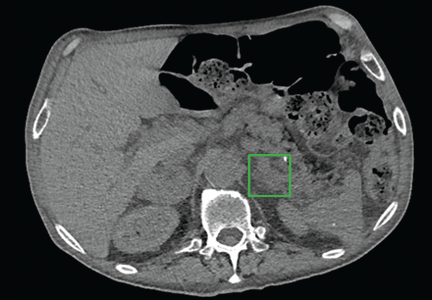
Does massive hemoptysis always merit diagnostic bronchoscopy?
Yes, all patients with massive hemoptysis should undergo diagnostic bronchoscopy. The procedure plays an important role in protecting the airway, maintaining ventilation, finding the site and underlying cause of the bleeding, and in some cases stopping the bleeding, either temporarily or definitively.

Frightening to patients, massive hemoptysis is a medical emergency and demands immediate attention by an experienced pulmonary team.1 Hemoptysis can be the initial presentation of an underlying infectious, autoimmune, or malignant disorder (Table 1).2 Fortunately, most cases of hemoptysis are not massive or life-threatening.1
WHAT IS ‘MASSIVE’ HEMOPTYSIS?
Numerous studies have defined massive hemoptysis on the basis of the volume of blood lost over time, eg, more than 1 L in 24 hours or more than 400 mL in 6 hours.
Ibrahim3 has proposed that we move away from using the word “massive,” which is not useful, and instead think in terms of “life-threatening” hemoptysis, defined as any of the following:
- More than 100 mL of blood lost in 24 hours (a low number, but blood loss is hard to estimate accurately)
- Causing abnormal gas exchange due to airway obstruction
- Causing hemodynamic instability.
In this article, we use the traditional “massive” terminology.
BRONCHOSCOPY IS SUPERIOR TO IMAGING FOR DIAGNOSIS
Radiography can help identify the side or the site of bleeding in 33% to 82% of patients, and computed tomography can in 70% to 88.5%.4 Magnetic resonance imaging may also have a role; one study found it useful in cases of thoracic endometriosis during the quiescent stage.5 However, transferring a patient who is actively bleeding out of the intensive care unit for imaging can be challenging.
Flexible bronchoscopy is superior to radiographic imaging in evaluating massive hemoptysis: it can be performed at the bed-side and can include therapeutic procedures to control the bleeding until the patient can undergo a definitive therapeutic procedure.6 It has been found helpful in identifying the side of bleeding in 73% to 93% of cases of massive hemoptysis.6
However, one should consider starting the procedure with a rigid bronchoscope, which protects the airway better and allows for better ventilation during the procedure than a flexible one. One can use it to isolate the nonbleeding lung and to apply pressure to the bleeding site if it is in the main bronchus.7 Measuring 12 mm in diameter, a rigid scope cannot go as far into the lung as a flexible bronchoscope (measuring 6.4 mm), but a flexible bronchoscope can be introduced through the rigid bronchoscope to go further in.
MANAGEMENT OPTIONS
The management team should include an anesthesiologist, an intensivist, a thoracic surgeon, an interventional radiologist, and an interventional pulmonologist.
In the intensive care unit, the patient should be placed in the lateral decubitus position on the bleeding side. To maintain ventilation, the nonbleeding lung should be intubated with a large-bore endotracheal tube (internal diameter 8.5–9.0 mm) or, ideally, with a rigid bronchoscope.6 Meanwhile, the patient’s circulatory status should be stabilized with adequate fluid resuscitation and transfusion of blood products, with close monitoring.
Once the bleeding site is found, a bronchoscopic treatment is selected based on the cause of the bleeding. Massive hemoptysis usually arises from high-pressure bronchial vessels (90%) or, less commonly, from non-bronchial vessels or capillaries (10%).8 A variety of agents (eg, cold saline lavage, epinephrine 1:20,000) can be instilled through the bronchoscope to slow the bleeding and offer better visualization of the airway.6
If a bleeding intrabronchial lesion is identified, such as a malignant tracheobronchial tumor, local coagulation therapy can be applied through the bronchoscope. Options include laser treatment, argon plasma coagulation, cryotherapy, and electrocautery (Figure 1).9,10
If the bleeding persists or cannot be localized to a particular subsegment, an endobronchial balloon plug can be placed proximally (Figure 2). This can be left in place to isolate the bleeding and apply tamponade until a definitive procedure can be performed, such as bronchial artery embolization, radiation therapy, or surgery.
- Jean-Baptiste E. Clinical assessment and management of massive hemoptysis. Crit Care Med 2000; 28:1642–1647.
- Abi Khalil S, Gourdier AL, Aoun N, et al. Cystic and cavitary lesions of the lung: imaging characteristics and differential diagnosis [in French]. J Radiol 2010; 91:465–473.
- Ibrahim WH. Massive haemoptysis: the definition should be revised. Eur Respir J 2008; 32:1131–1132.
- Khalil A, Soussan M, Mangiapan G, Fartoukh M, Parrot A, Carette MF. Utility of high-resolution chest CT scan in the emergency management of haemoptysis in the intensive care unit: severity, localization and aetiology. Br J Radiol 2007; 80:21–25.
- Cassina PC, Hauser M, Kacl G, Imthurn B, Schröder S, Weder W. Catamenial hemoptysis. Diagnosis with MRI. Chest 1997; 111:1447–1450.
- Sakr L, Dutau H. Massive hemoptysis: an update on the role of bronchoscopy in diagnosis and management. Respiration 2010; 80:38–58.
- Conlan AA, Hurwitz SS. Management of massive haemoptysis with the rigid bronchoscope and cold saline lavage. Thorax 1980; 35:901–904.
- Deffebach ME, Charan NB, Lakshminarayan S, Butler J. The bronchial circulation. Small, but a vital attribute of the lung. Am Rev Respir Dis 1987; 135:463–481.
- Morice RC, Ece T, Ece F, Keus L. Endobronchial argon plasma coagulation for treatment of hemoptysis and neoplastic airway obstruction. Chest 2001; 119:781–787.
- Sheski FD, Mathur PN. Cryotherapy, electrocautery, and brachytherapy. Clin Chest Med 1999; 20:123–138.
Yes, all patients with massive hemoptysis should undergo diagnostic bronchoscopy. The procedure plays an important role in protecting the airway, maintaining ventilation, finding the site and underlying cause of the bleeding, and in some cases stopping the bleeding, either temporarily or definitively.
Frightening to patients, massive hemoptysis is a medical emergency and demands immediate attention by an experienced pulmonary team.1 Hemoptysis can be the initial presentation of an underlying infectious, autoimmune, or malignant disorder (Table 1).2 Fortunately, most cases of hemoptysis are not massive or life-threatening.1
WHAT IS ‘MASSIVE’ HEMOPTYSIS?
Numerous studies have defined massive hemoptysis on the basis of the volume of blood lost over time, eg, more than 1 L in 24 hours or more than 400 mL in 6 hours.
Ibrahim3 has proposed that we move away from using the word “massive,” which is not useful, and instead think in terms of “life-threatening” hemoptysis, defined as any of the following:
- More than 100 mL of blood lost in 24 hours (a low number, but blood loss is hard to estimate accurately)
- Causing abnormal gas exchange due to airway obstruction
- Causing hemodynamic instability.
In this article, we use the traditional “massive” terminology.
BRONCHOSCOPY IS SUPERIOR TO IMAGING FOR DIAGNOSIS
Radiography can help identify the side or the site of bleeding in 33% to 82% of patients, and computed tomography can in 70% to 88.5%.4 Magnetic resonance imaging may also have a role; one study found it useful in cases of thoracic endometriosis during the quiescent stage.5 However, transferring a patient who is actively bleeding out of the intensive care unit for imaging can be challenging.
Flexible bronchoscopy is superior to radiographic imaging in evaluating massive hemoptysis: it can be performed at the bed-side and can include therapeutic procedures to control the bleeding until the patient can undergo a definitive therapeutic procedure.6 It has been found helpful in identifying the side of bleeding in 73% to 93% of cases of massive hemoptysis.6
However, one should consider starting the procedure with a rigid bronchoscope, which protects the airway better and allows for better ventilation during the procedure than a flexible one. One can use it to isolate the nonbleeding lung and to apply pressure to the bleeding site if it is in the main bronchus.7 Measuring 12 mm in diameter, a rigid scope cannot go as far into the lung as a flexible bronchoscope (measuring 6.4 mm), but a flexible bronchoscope can be introduced through the rigid bronchoscope to go further in.
MANAGEMENT OPTIONS
The management team should include an anesthesiologist, an intensivist, a thoracic surgeon, an interventional radiologist, and an interventional pulmonologist.
In the intensive care unit, the patient should be placed in the lateral decubitus position on the bleeding side. To maintain ventilation, the nonbleeding lung should be intubated with a large-bore endotracheal tube (internal diameter 8.5–9.0 mm) or, ideally, with a rigid bronchoscope.6 Meanwhile, the patient’s circulatory status should be stabilized with adequate fluid resuscitation and transfusion of blood products, with close monitoring.
Once the bleeding site is found, a bronchoscopic treatment is selected based on the cause of the bleeding. Massive hemoptysis usually arises from high-pressure bronchial vessels (90%) or, less commonly, from non-bronchial vessels or capillaries (10%).8 A variety of agents (eg, cold saline lavage, epinephrine 1:20,000) can be instilled through the bronchoscope to slow the bleeding and offer better visualization of the airway.6
If a bleeding intrabronchial lesion is identified, such as a malignant tracheobronchial tumor, local coagulation therapy can be applied through the bronchoscope. Options include laser treatment, argon plasma coagulation, cryotherapy, and electrocautery (Figure 1).9,10
If the bleeding persists or cannot be localized to a particular subsegment, an endobronchial balloon plug can be placed proximally (Figure 2). This can be left in place to isolate the bleeding and apply tamponade until a definitive procedure can be performed, such as bronchial artery embolization, radiation therapy, or surgery.
Yes, all patients with massive hemoptysis should undergo diagnostic bronchoscopy. The procedure plays an important role in protecting the airway, maintaining ventilation, finding the site and underlying cause of the bleeding, and in some cases stopping the bleeding, either temporarily or definitively.
Frightening to patients, massive hemoptysis is a medical emergency and demands immediate attention by an experienced pulmonary team.1 Hemoptysis can be the initial presentation of an underlying infectious, autoimmune, or malignant disorder (Table 1).2 Fortunately, most cases of hemoptysis are not massive or life-threatening.1
WHAT IS ‘MASSIVE’ HEMOPTYSIS?
Numerous studies have defined massive hemoptysis on the basis of the volume of blood lost over time, eg, more than 1 L in 24 hours or more than 400 mL in 6 hours.
Ibrahim3 has proposed that we move away from using the word “massive,” which is not useful, and instead think in terms of “life-threatening” hemoptysis, defined as any of the following:
- More than 100 mL of blood lost in 24 hours (a low number, but blood loss is hard to estimate accurately)
- Causing abnormal gas exchange due to airway obstruction
- Causing hemodynamic instability.
In this article, we use the traditional “massive” terminology.
BRONCHOSCOPY IS SUPERIOR TO IMAGING FOR DIAGNOSIS
Radiography can help identify the side or the site of bleeding in 33% to 82% of patients, and computed tomography can in 70% to 88.5%.4 Magnetic resonance imaging may also have a role; one study found it useful in cases of thoracic endometriosis during the quiescent stage.5 However, transferring a patient who is actively bleeding out of the intensive care unit for imaging can be challenging.
Flexible bronchoscopy is superior to radiographic imaging in evaluating massive hemoptysis: it can be performed at the bed-side and can include therapeutic procedures to control the bleeding until the patient can undergo a definitive therapeutic procedure.6 It has been found helpful in identifying the side of bleeding in 73% to 93% of cases of massive hemoptysis.6
However, one should consider starting the procedure with a rigid bronchoscope, which protects the airway better and allows for better ventilation during the procedure than a flexible one. One can use it to isolate the nonbleeding lung and to apply pressure to the bleeding site if it is in the main bronchus.7 Measuring 12 mm in diameter, a rigid scope cannot go as far into the lung as a flexible bronchoscope (measuring 6.4 mm), but a flexible bronchoscope can be introduced through the rigid bronchoscope to go further in.
MANAGEMENT OPTIONS
The management team should include an anesthesiologist, an intensivist, a thoracic surgeon, an interventional radiologist, and an interventional pulmonologist.
In the intensive care unit, the patient should be placed in the lateral decubitus position on the bleeding side. To maintain ventilation, the nonbleeding lung should be intubated with a large-bore endotracheal tube (internal diameter 8.5–9.0 mm) or, ideally, with a rigid bronchoscope.6 Meanwhile, the patient’s circulatory status should be stabilized with adequate fluid resuscitation and transfusion of blood products, with close monitoring.
Once the bleeding site is found, a bronchoscopic treatment is selected based on the cause of the bleeding. Massive hemoptysis usually arises from high-pressure bronchial vessels (90%) or, less commonly, from non-bronchial vessels or capillaries (10%).8 A variety of agents (eg, cold saline lavage, epinephrine 1:20,000) can be instilled through the bronchoscope to slow the bleeding and offer better visualization of the airway.6
If a bleeding intrabronchial lesion is identified, such as a malignant tracheobronchial tumor, local coagulation therapy can be applied through the bronchoscope. Options include laser treatment, argon plasma coagulation, cryotherapy, and electrocautery (Figure 1).9,10
If the bleeding persists or cannot be localized to a particular subsegment, an endobronchial balloon plug can be placed proximally (Figure 2). This can be left in place to isolate the bleeding and apply tamponade until a definitive procedure can be performed, such as bronchial artery embolization, radiation therapy, or surgery.
- Jean-Baptiste E. Clinical assessment and management of massive hemoptysis. Crit Care Med 2000; 28:1642–1647.
- Abi Khalil S, Gourdier AL, Aoun N, et al. Cystic and cavitary lesions of the lung: imaging characteristics and differential diagnosis [in French]. J Radiol 2010; 91:465–473.
- Ibrahim WH. Massive haemoptysis: the definition should be revised. Eur Respir J 2008; 32:1131–1132.
- Khalil A, Soussan M, Mangiapan G, Fartoukh M, Parrot A, Carette MF. Utility of high-resolution chest CT scan in the emergency management of haemoptysis in the intensive care unit: severity, localization and aetiology. Br J Radiol 2007; 80:21–25.
- Cassina PC, Hauser M, Kacl G, Imthurn B, Schröder S, Weder W. Catamenial hemoptysis. Diagnosis with MRI. Chest 1997; 111:1447–1450.
- Sakr L, Dutau H. Massive hemoptysis: an update on the role of bronchoscopy in diagnosis and management. Respiration 2010; 80:38–58.
- Conlan AA, Hurwitz SS. Management of massive haemoptysis with the rigid bronchoscope and cold saline lavage. Thorax 1980; 35:901–904.
- Deffebach ME, Charan NB, Lakshminarayan S, Butler J. The bronchial circulation. Small, but a vital attribute of the lung. Am Rev Respir Dis 1987; 135:463–481.
- Morice RC, Ece T, Ece F, Keus L. Endobronchial argon plasma coagulation for treatment of hemoptysis and neoplastic airway obstruction. Chest 2001; 119:781–787.
- Sheski FD, Mathur PN. Cryotherapy, electrocautery, and brachytherapy. Clin Chest Med 1999; 20:123–138.
- Jean-Baptiste E. Clinical assessment and management of massive hemoptysis. Crit Care Med 2000; 28:1642–1647.
- Abi Khalil S, Gourdier AL, Aoun N, et al. Cystic and cavitary lesions of the lung: imaging characteristics and differential diagnosis [in French]. J Radiol 2010; 91:465–473.
- Ibrahim WH. Massive haemoptysis: the definition should be revised. Eur Respir J 2008; 32:1131–1132.
- Khalil A, Soussan M, Mangiapan G, Fartoukh M, Parrot A, Carette MF. Utility of high-resolution chest CT scan in the emergency management of haemoptysis in the intensive care unit: severity, localization and aetiology. Br J Radiol 2007; 80:21–25.
- Cassina PC, Hauser M, Kacl G, Imthurn B, Schröder S, Weder W. Catamenial hemoptysis. Diagnosis with MRI. Chest 1997; 111:1447–1450.
- Sakr L, Dutau H. Massive hemoptysis: an update on the role of bronchoscopy in diagnosis and management. Respiration 2010; 80:38–58.
- Conlan AA, Hurwitz SS. Management of massive haemoptysis with the rigid bronchoscope and cold saline lavage. Thorax 1980; 35:901–904.
- Deffebach ME, Charan NB, Lakshminarayan S, Butler J. The bronchial circulation. Small, but a vital attribute of the lung. Am Rev Respir Dis 1987; 135:463–481.
- Morice RC, Ece T, Ece F, Keus L. Endobronchial argon plasma coagulation for treatment of hemoptysis and neoplastic airway obstruction. Chest 2001; 119:781–787.
- Sheski FD, Mathur PN. Cryotherapy, electrocautery, and brachytherapy. Clin Chest Med 1999; 20:123–138.
Confusion Follows Malaise and Pain
ANSWER
The radiograph demonstrates innumerable small lytic defects throughout the calvarium. The patient’s confusion is most likely secondary to profound metabolic abnormalities. However, in the setting of lytic bone lesions, metabolic abnormalities of renal insufficiency, severe hypercalcemia, and hypomagnesemia, one must be concerned about an occult myeloma, and appropriate work-up must be done.
ANSWER
The radiograph demonstrates innumerable small lytic defects throughout the calvarium. The patient’s confusion is most likely secondary to profound metabolic abnormalities. However, in the setting of lytic bone lesions, metabolic abnormalities of renal insufficiency, severe hypercalcemia, and hypomagnesemia, one must be concerned about an occult myeloma, and appropriate work-up must be done.
ANSWER
The radiograph demonstrates innumerable small lytic defects throughout the calvarium. The patient’s confusion is most likely secondary to profound metabolic abnormalities. However, in the setting of lytic bone lesions, metabolic abnormalities of renal insufficiency, severe hypercalcemia, and hypomagnesemia, one must be concerned about an occult myeloma, and appropriate work-up must be done.

A 70-year-old woman is brought to the emergency department by her family for evaluation of acute altered mental status. According to the family, the patient has been complaining of general malaise, back pain, and severe joint pain for the past few days. Her confusion has increased in the past 24 hours. Medical history is significant for hypertension. Physical exam reveals an elderly female who appears somewhat uncomfortable. Vital signs are normal. Overall, her exam is stable. She has tenderness throughout her back and several of her joints, but no abnormal effusion or swelling is noted. While the patient is in triage, baseline labwork is ordered. The results indicate a serum creatinine of 1.83 mg/dL; serum calcium, 16.7 mg/dL; and serum magnesium, 1.4 mEq/L. Radiograph of the skull is obtained. What is your impression?
Woman, 66, With Persistent Abdominal and Back Pain
A 66-year-old Latin American woman presented to the emergency department (ED) with persistent abdominal and back pain of about one month’s duration. She had visited another ED eight days earlier for similar symptoms and was discharged home with a mild opioid pain medication and a proton pump inhibitor. However, she said that she had received neither a diagnosis nor an explanation for her symptoms.
Medical history, obtained with the assistance of an interpreter because the patient was not fluent in English, included hypertension, coronary artery disease, and hyperlipidemia; these had gone untreated for at least two years. She denied any personal or family history of cancer or endocrine disorders. Surgical history included a cholecystectomy and a percutaneous coronary intervention for an unknown coronary artery lesion.
She had a 14-pack-year history of cigarette smoking. Her medications included only ibuprofen and hydrocodone, and she had no known drug allergies. The patient denied use of herbal preparations or vitamin supplements and unusual dietary practices.
Review of systems revealed occasional dizziness, constipation, decreased appetite, and some mild confusion noted by family members, but no fever, chills, palpitations, chest pain, shortness of breath, muscle spasm, or weakness. Vital signs were normal. Physical examination was remarkable for tenderness of the upper quadrants of the abdomen with deep palpation, without guarding or rebound. Bony tenderness at the right anterior costal margin of the rib cage was also noted.
Laboratory work-up revealed marked hypercalcemia (15.4 mg/dL), electrolyte abnormalities, anemia, impaired renal function, and elevated alkaline phosphatase and globulin levels (see Table 1). In addition, a plain abdominal x-ray series was negative for acute findings, but x-rays of the right ribs revealed a fracture of the sixth rib and osteopenia.

Continued >>
The patient was admitted to the hospital for treatment of hypercalcemia and hypokalemia and for work-up of elevated alkaline phosphatase and abdominal pain. Upon admission, serum ionized calcium measurement confirmed true hypercalcemia. Additional diagnostic tests were then ordered to help differentiate between parathyroid hormone (PTH)–mediated and non-PTH–mediated causes for the hypercalcemia (see Table 2).
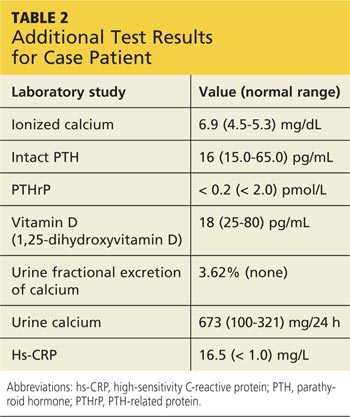
The patient’s PTH level was normal and the urine fractional excretion of calcium level was high, ruling out familial hypocalciuric hypercalcemia (FHH), in which urine calcium level is low. A measurement of PTH-related protein (PTHrP), secreted by some cancers, was normal, suggesting exclusion of solid tumor malignancy. Vitamin D toxicity was ruled out because the patient’s 1,25-dihydroxyvitamin D level was low.
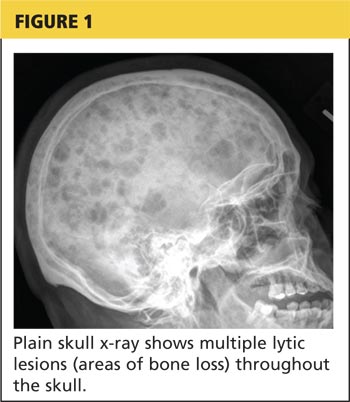
The patient continued to experience vague abdominal, back, and rib pain that seemed to migrate daily and worsened with movement. A skeletal x-ray was performed and revealed numerous lytic lesions of the skull (Figure 1), midright humerus (Figure 2), and distal left radius.
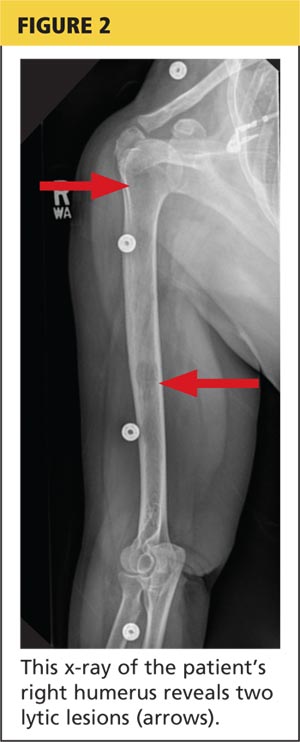
Continue for discussion >>
DISCUSSION
Hypercalcemia is a relatively common presentation in primary care. The most frequent causes are primary hyperparathyroidism and malignancy.1 One in 500 patients will be diagnosed incidentally with asymptomatic hypercalcemia caused by underlying hyperparathyroidism.1
Clinical manifestations of hypercalcemia can range from no symptoms to multisystem disease. Fatigue, nausea, vomiting, constipation, bone pain, osteoporosis, nephrolithiasis, mental status changes, hypertension, anemia, elevated creatinine, and cardiac arrhythmias are among the more common clinical conditions associated with hypercalcemia (hence the mnemonic “stones, bones, abdominal moans, and psychic groans” for its signs and symptoms).1
Diagnostic overview
Causes of hypercalcemia are numerous and can be broken down into two categories: PTH-mediated and non-PTH–mediated. PTH-mediated causes include primary and secondary hyperparathyroidism and FHH. Non-PTH–mediated causes include vitamin D toxicity, solid tumor malignancy with or without metastasis, multiple myeloma (MM) and other plasma cell dyscrasias, granulomatous disease such as sarcoid, and some medications.1
The differential diagnosis for hypercalcemia begins with measurement of the patient’s intact PTH level. An elevated or high-normal result indicates a PTH-mediated cause, so 24-hour measurement of excretion of urinary calcium is the next step. A low or low-normal PTH level (< 20 pg/mL), however, suggests the cause is non-PTH-mediated.2 The diagnostic approach in this situation is more challenging because testing to exclude or confirm various potential causes can be expensive and time-consuming. The degree of hypercalcemia, however, can aid in the diagnosis: Primary hyperparathyroidism is often associated with borderline or mild hypercalcemia, while calcium values > 13 mg/dL are more common in patients with malignancies.2
PTH elevates calcium levels in the blood when ionized (free) calcium levels are low by increasing gastrointestinal absorption, decreasing urinary excretion, and increasing bone resorption.3 With malignant tumors such as lung, breast, and renal cell, osteolytic metastases can destroy the bone, resulting in release of calcium. In other cases, solid-tumor cancers produce PTHrP, which increases serum calcium. This latter situation is referred to as humoral hypercalcemia of malignancy.3 Lymphoma and granulomatous disease, such as sarcoid, can be associated with excess production of 1,25-dihydroxyvitamin D.2 If vitamin D is elevated but PTH and PTHrP are normal, a chest x-ray should be obtained to evaluate the patient for sarcoid or lymphoma.
Hypercalcemia work-up
The work-up for suspected hypercalcemia begins with measurement of the patient’s calcium level. Because calcium is bound to albumin in the blood, the standard serum calcium test may not reflect the true calcium level. (If albumin is high, the calcium level will be high, and vice versa.)1 The true (serum ionized) calcium level (also known as corrected calcium level) should always be calculated to confirm true hypercalcemia. A formula commonly used to calculate the corrected calcium level is
Corrected calcium (mg/dL) = (measured calcium [mg/dL]) + 0.8 (4.0 – serum albumin [mg/dL])
Direct measurement of the serum ionized calcium level is not affected by the albumin level and can also confirm true hypercalcemia.4
Once hypercalcemia is confirmed, the next step is to measure the patient’s intact PTH level.
If intact PTH is elevated or high normal, consider primary hyperparathyroidism or FHH and confirm by obtaining the urine calcium level.
• If urine calcium level is high (> 200 mg/24 h), the diagnosis is primary hyperparathyroidism.
• If urine calcium level is low (< 100 mg/24 h), the diagnosis is FHH.
If intact PTH is low, consider non–PTH-mediated causes and confirm by obtaining PTHrP and vitamin D levels.
• If PTHrP level is elevated, scan for malignancy.
• If 1,25-dihydroxyvitamin D level is elevated, order a chest x-ray to rule out sarcoid or lymphoma.
• If both PTHrP and vitamin D levels are normal, order both serum protein electrophoresis (SPEP) and urine protein electrophoresis (UPEP) with immunofixation to rule out MM.
• If vitamin D level is elevated, check vitamin and herbal supplement use for excessive vitamin D intake.
Treatment of symptomatic hypercalcemia
The goals of treatment of symptomatic hypercalcemia are to reduce the serum calcium level to the normal range and to treat the underlying cause.1 Mild hypercalcemia (calcium level, 10-12 mg/dL) is typically asymptomatic and does not need to be treated. Moderate hypercalcemia (calcium level, 12-14 mg/dL) may not require treatment unless the patient is symptomatic and/or has had an acute rise in calcium level.1 In mild to moderate hypercalcemia, the serum calcium level should be monitored to establish a trend.
Treatment for symptomatic moderate and severe hypercalcemia (calcium level, > 14 mg/dL) typically involves a similar regimen:
• Volume expansion with isotonic saline at an initial rate of 2 to 4 L/d, which is then adjusted to achieve 200 mL/h of continuous urine output. IV furosemide can be used with caution (10-20 mg IV as needed) to promote diuresis if volume overload is a concern (furosemide promotes renal excretion of calcium).
• Administration of subcutaneous calcitonin (4-8 IU/kg, repeated every 6 h for 24 h). Calcitonin works rapidly to lower calcium levels in 4 to 6 h.
• Concurrent administration of IV bisphosphonate (zoledronic acid [4 mg over 15 min] or pamidronate [60-90 mg over 4 h]). Pamidronate is superior for reversal of malignancy-related hypercalcemia.1
Hypercalcemia and multiple myeloma
MM is a malignant neoplasm of plasma cells that accounts for approximately 1% of all cancers and about 10% of hematologic malignancies in the United States, with a median patient age of 70 at diagnosis.5-7 In MM, myeloma cells induce the secretion of cytokines and growth factors that alter plasma cells, activate osteoclasts, suppress osteoblasts, cause abnormal interactions between plasma cells and bone marrow, and stimulate aberrant angiogenesis.8 Osteoclastic bone resorption produces hypercalcemia as well as the lytic lesions seen on x-ray.7
Approximately 74% of patients present with typical MM symptoms of calcium elevation in the blood, renal insufficiency, anemia, and bone lesions, known as CRAB symptoms, but other myeloma-related manifestations may be present.9
Diagnostic criteria for MM include the following (all three must be present):10
• Monoclonal bone marrow plasma cells ≥ 10% and/or a biopsy-proven plasmacytoma
• Monoclonal protein in the serum and/or urine (if none is detected, disease is nonsecretory and diagnosis requires ≥ 30% bone marrow plasma cells and/or biopsy-proven plasmacytoma)
• Myeloma-related organ dysfunction, indicated by at least one of the CRAB symptoms.
In the absence of CRAB symptoms, an asymptomatic patient may have an MM precursor syndrome: monoclonal gammopathy of undetermined significance or smoldering (or indolent) MM.10
Treatment of multiple myeloma
In recent years, the use of induction therapy followed by autologous stem cell transplantation and the development of novel therapeutic agents have extended overall survival for patients with MM. These agents include proteasome inhibitors (bortezomib and the second-generation carfilzomib)11 and immunomodulators (thalidomide and the second-generation lenalidomide). Early diagnosis and treatment can improve progression-free survival as well as overall survival, including recovery of renal function for patients with renal failure.12 With survival ranging from one year or less—with aggressive disease—to 10 years or more for patients with responsive disease,7 there remains no cure for MM.
Outcome for the case patient >>
OUTCOME FOR THE CASE PATIENT
Further work-up included SPEP and UPEP with immunofixation, which revealed marked IgG free λ light chains with an M (monoclonal) component, making MM a very likely diagnosis. Confirmation by means of bone marrow biopsy was indicated, but the patient refused the procedure.
The patient’s hypercalcemia was treated by IV administration of calcitonin with isotonic saline. This reduced the serum calcium level from 15.4 mg/dL to 10.6 mg/dL within 48 hours. One dose of ergocalciferol (vitamin D2) was then administered to promote intestinal absorption of calcium and support bone mineralization, further lowering the patient’s serum calcium level to a normal 8.9 mg/dL. Hypokalemia was treated with oral potassium supplementation.
The patient, now stable, was referred to the hematology/oncology and bone mineral metabolism clinics and was discharged from the hospital. She did not keep those appointments and was lost to follow-up.
CONCLUSION
The most common causes of hypercalcemia are hyperparathyroidism and malignancy. Most cases do not require treatment unless the calcium level is >14 mg/dL and/or the patient is symptomatic. Red flag symptoms include weakness, abdominal pain, mental status changes, and coma.4 Primary care clinicians should suspect MM in older patients with laboratory findings of hypercalcemia, anemia, and renal dysfunction, with lytic lesions on x-ray.
REFERENCES
1. Carroll MF, Schade DS. A practical approach to hypercalcemia. Am Fam Physician. 2003;67(9):1959-1966.
2. Endres DB. Investigation of hypercalcemia. Clin Biochem. 2012;45(12):
954-963.
3. Moe SM. Disorders involving calcium, phosphorus, and magnesium. Prim Care. 2008;35(2):215-237.
4. Sharma B, Misicko NE. How should you evaluate elevated calcium in an asymptomatic patient? J Fam Pract. 2008;57(4):267-269.
5. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11-30.
6. Kyle RA, Gertz MA, Witzig TE, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc. 2003;78(1):21-33.
7. Shaheen SP, Talwalkar SS, Medeiros LJ. Multiple myeloma and immunosecretory disorders: an update. Adv Anat Pathol. 2008;15(4):196-210.
8. Palumbo A, Anderson K. Multiple myeloma. N Engl J Med. 2011;364(11): 1046-1060.
9. Talamo G, Farooq U, Zangari M, et al. Beyond the CRAB symptoms: a study of presenting clinical manifestations of multiple myeloma. Clin Lymphoma Myeloma Leuk. 2010;10(6):464-468.
10. Palumbo A, Sezer O, Kyle R, et al. International Myeloma Working Group guidelines for the management of multiple myeloma patients ineligible for standard high-dose chemotherapy with autologous stem cell transportation. Leukemia. 2009;23(10):1716-1730.
11. Kyprolis [package insert]. South San Francisco, CA: Onyx Pharmaceuticals, Inc; 2012.
12. Suyani E, Sucak GT, Erten Y, et al. Evaluation of multiple myeloma patients presenting with renal failure in a university hospital in the year 2010. Ren Fail. 2012;34(2):257-262.
A 66-year-old Latin American woman presented to the emergency department (ED) with persistent abdominal and back pain of about one month’s duration. She had visited another ED eight days earlier for similar symptoms and was discharged home with a mild opioid pain medication and a proton pump inhibitor. However, she said that she had received neither a diagnosis nor an explanation for her symptoms.
Medical history, obtained with the assistance of an interpreter because the patient was not fluent in English, included hypertension, coronary artery disease, and hyperlipidemia; these had gone untreated for at least two years. She denied any personal or family history of cancer or endocrine disorders. Surgical history included a cholecystectomy and a percutaneous coronary intervention for an unknown coronary artery lesion.
She had a 14-pack-year history of cigarette smoking. Her medications included only ibuprofen and hydrocodone, and she had no known drug allergies. The patient denied use of herbal preparations or vitamin supplements and unusual dietary practices.
Review of systems revealed occasional dizziness, constipation, decreased appetite, and some mild confusion noted by family members, but no fever, chills, palpitations, chest pain, shortness of breath, muscle spasm, or weakness. Vital signs were normal. Physical examination was remarkable for tenderness of the upper quadrants of the abdomen with deep palpation, without guarding or rebound. Bony tenderness at the right anterior costal margin of the rib cage was also noted.
Laboratory work-up revealed marked hypercalcemia (15.4 mg/dL), electrolyte abnormalities, anemia, impaired renal function, and elevated alkaline phosphatase and globulin levels (see Table 1). In addition, a plain abdominal x-ray series was negative for acute findings, but x-rays of the right ribs revealed a fracture of the sixth rib and osteopenia.
Continued >>
The patient was admitted to the hospital for treatment of hypercalcemia and hypokalemia and for work-up of elevated alkaline phosphatase and abdominal pain. Upon admission, serum ionized calcium measurement confirmed true hypercalcemia. Additional diagnostic tests were then ordered to help differentiate between parathyroid hormone (PTH)–mediated and non-PTH–mediated causes for the hypercalcemia (see Table 2).
The patient’s PTH level was normal and the urine fractional excretion of calcium level was high, ruling out familial hypocalciuric hypercalcemia (FHH), in which urine calcium level is low. A measurement of PTH-related protein (PTHrP), secreted by some cancers, was normal, suggesting exclusion of solid tumor malignancy. Vitamin D toxicity was ruled out because the patient’s 1,25-dihydroxyvitamin D level was low.
The patient continued to experience vague abdominal, back, and rib pain that seemed to migrate daily and worsened with movement. A skeletal x-ray was performed and revealed numerous lytic lesions of the skull (Figure 1), midright humerus (Figure 2), and distal left radius.
Continue for discussion >>
DISCUSSION
Hypercalcemia is a relatively common presentation in primary care. The most frequent causes are primary hyperparathyroidism and malignancy.1 One in 500 patients will be diagnosed incidentally with asymptomatic hypercalcemia caused by underlying hyperparathyroidism.1
Clinical manifestations of hypercalcemia can range from no symptoms to multisystem disease. Fatigue, nausea, vomiting, constipation, bone pain, osteoporosis, nephrolithiasis, mental status changes, hypertension, anemia, elevated creatinine, and cardiac arrhythmias are among the more common clinical conditions associated with hypercalcemia (hence the mnemonic “stones, bones, abdominal moans, and psychic groans” for its signs and symptoms).1
Diagnostic overview
Causes of hypercalcemia are numerous and can be broken down into two categories: PTH-mediated and non-PTH–mediated. PTH-mediated causes include primary and secondary hyperparathyroidism and FHH. Non-PTH–mediated causes include vitamin D toxicity, solid tumor malignancy with or without metastasis, multiple myeloma (MM) and other plasma cell dyscrasias, granulomatous disease such as sarcoid, and some medications.1
The differential diagnosis for hypercalcemia begins with measurement of the patient’s intact PTH level. An elevated or high-normal result indicates a PTH-mediated cause, so 24-hour measurement of excretion of urinary calcium is the next step. A low or low-normal PTH level (< 20 pg/mL), however, suggests the cause is non-PTH-mediated.2 The diagnostic approach in this situation is more challenging because testing to exclude or confirm various potential causes can be expensive and time-consuming. The degree of hypercalcemia, however, can aid in the diagnosis: Primary hyperparathyroidism is often associated with borderline or mild hypercalcemia, while calcium values > 13 mg/dL are more common in patients with malignancies.2
PTH elevates calcium levels in the blood when ionized (free) calcium levels are low by increasing gastrointestinal absorption, decreasing urinary excretion, and increasing bone resorption.3 With malignant tumors such as lung, breast, and renal cell, osteolytic metastases can destroy the bone, resulting in release of calcium. In other cases, solid-tumor cancers produce PTHrP, which increases serum calcium. This latter situation is referred to as humoral hypercalcemia of malignancy.3 Lymphoma and granulomatous disease, such as sarcoid, can be associated with excess production of 1,25-dihydroxyvitamin D.2 If vitamin D is elevated but PTH and PTHrP are normal, a chest x-ray should be obtained to evaluate the patient for sarcoid or lymphoma.
Hypercalcemia work-up
The work-up for suspected hypercalcemia begins with measurement of the patient’s calcium level. Because calcium is bound to albumin in the blood, the standard serum calcium test may not reflect the true calcium level. (If albumin is high, the calcium level will be high, and vice versa.)1 The true (serum ionized) calcium level (also known as corrected calcium level) should always be calculated to confirm true hypercalcemia. A formula commonly used to calculate the corrected calcium level is
Corrected calcium (mg/dL) = (measured calcium [mg/dL]) + 0.8 (4.0 – serum albumin [mg/dL])
Direct measurement of the serum ionized calcium level is not affected by the albumin level and can also confirm true hypercalcemia.4
Once hypercalcemia is confirmed, the next step is to measure the patient’s intact PTH level.
If intact PTH is elevated or high normal, consider primary hyperparathyroidism or FHH and confirm by obtaining the urine calcium level.
• If urine calcium level is high (> 200 mg/24 h), the diagnosis is primary hyperparathyroidism.
• If urine calcium level is low (< 100 mg/24 h), the diagnosis is FHH.
If intact PTH is low, consider non–PTH-mediated causes and confirm by obtaining PTHrP and vitamin D levels.
• If PTHrP level is elevated, scan for malignancy.
• If 1,25-dihydroxyvitamin D level is elevated, order a chest x-ray to rule out sarcoid or lymphoma.
• If both PTHrP and vitamin D levels are normal, order both serum protein electrophoresis (SPEP) and urine protein electrophoresis (UPEP) with immunofixation to rule out MM.
• If vitamin D level is elevated, check vitamin and herbal supplement use for excessive vitamin D intake.
Treatment of symptomatic hypercalcemia
The goals of treatment of symptomatic hypercalcemia are to reduce the serum calcium level to the normal range and to treat the underlying cause.1 Mild hypercalcemia (calcium level, 10-12 mg/dL) is typically asymptomatic and does not need to be treated. Moderate hypercalcemia (calcium level, 12-14 mg/dL) may not require treatment unless the patient is symptomatic and/or has had an acute rise in calcium level.1 In mild to moderate hypercalcemia, the serum calcium level should be monitored to establish a trend.
Treatment for symptomatic moderate and severe hypercalcemia (calcium level, > 14 mg/dL) typically involves a similar regimen:
• Volume expansion with isotonic saline at an initial rate of 2 to 4 L/d, which is then adjusted to achieve 200 mL/h of continuous urine output. IV furosemide can be used with caution (10-20 mg IV as needed) to promote diuresis if volume overload is a concern (furosemide promotes renal excretion of calcium).
• Administration of subcutaneous calcitonin (4-8 IU/kg, repeated every 6 h for 24 h). Calcitonin works rapidly to lower calcium levels in 4 to 6 h.
• Concurrent administration of IV bisphosphonate (zoledronic acid [4 mg over 15 min] or pamidronate [60-90 mg over 4 h]). Pamidronate is superior for reversal of malignancy-related hypercalcemia.1
Hypercalcemia and multiple myeloma
MM is a malignant neoplasm of plasma cells that accounts for approximately 1% of all cancers and about 10% of hematologic malignancies in the United States, with a median patient age of 70 at diagnosis.5-7 In MM, myeloma cells induce the secretion of cytokines and growth factors that alter plasma cells, activate osteoclasts, suppress osteoblasts, cause abnormal interactions between plasma cells and bone marrow, and stimulate aberrant angiogenesis.8 Osteoclastic bone resorption produces hypercalcemia as well as the lytic lesions seen on x-ray.7
Approximately 74% of patients present with typical MM symptoms of calcium elevation in the blood, renal insufficiency, anemia, and bone lesions, known as CRAB symptoms, but other myeloma-related manifestations may be present.9
Diagnostic criteria for MM include the following (all three must be present):10
• Monoclonal bone marrow plasma cells ≥ 10% and/or a biopsy-proven plasmacytoma
• Monoclonal protein in the serum and/or urine (if none is detected, disease is nonsecretory and diagnosis requires ≥ 30% bone marrow plasma cells and/or biopsy-proven plasmacytoma)
• Myeloma-related organ dysfunction, indicated by at least one of the CRAB symptoms.
In the absence of CRAB symptoms, an asymptomatic patient may have an MM precursor syndrome: monoclonal gammopathy of undetermined significance or smoldering (or indolent) MM.10
Treatment of multiple myeloma
In recent years, the use of induction therapy followed by autologous stem cell transplantation and the development of novel therapeutic agents have extended overall survival for patients with MM. These agents include proteasome inhibitors (bortezomib and the second-generation carfilzomib)11 and immunomodulators (thalidomide and the second-generation lenalidomide). Early diagnosis and treatment can improve progression-free survival as well as overall survival, including recovery of renal function for patients with renal failure.12 With survival ranging from one year or less—with aggressive disease—to 10 years or more for patients with responsive disease,7 there remains no cure for MM.
Outcome for the case patient >>
OUTCOME FOR THE CASE PATIENT
Further work-up included SPEP and UPEP with immunofixation, which revealed marked IgG free λ light chains with an M (monoclonal) component, making MM a very likely diagnosis. Confirmation by means of bone marrow biopsy was indicated, but the patient refused the procedure.
The patient’s hypercalcemia was treated by IV administration of calcitonin with isotonic saline. This reduced the serum calcium level from 15.4 mg/dL to 10.6 mg/dL within 48 hours. One dose of ergocalciferol (vitamin D2) was then administered to promote intestinal absorption of calcium and support bone mineralization, further lowering the patient’s serum calcium level to a normal 8.9 mg/dL. Hypokalemia was treated with oral potassium supplementation.
The patient, now stable, was referred to the hematology/oncology and bone mineral metabolism clinics and was discharged from the hospital. She did not keep those appointments and was lost to follow-up.
CONCLUSION
The most common causes of hypercalcemia are hyperparathyroidism and malignancy. Most cases do not require treatment unless the calcium level is >14 mg/dL and/or the patient is symptomatic. Red flag symptoms include weakness, abdominal pain, mental status changes, and coma.4 Primary care clinicians should suspect MM in older patients with laboratory findings of hypercalcemia, anemia, and renal dysfunction, with lytic lesions on x-ray.
REFERENCES
1. Carroll MF, Schade DS. A practical approach to hypercalcemia. Am Fam Physician. 2003;67(9):1959-1966.
2. Endres DB. Investigation of hypercalcemia. Clin Biochem. 2012;45(12):
954-963.
3. Moe SM. Disorders involving calcium, phosphorus, and magnesium. Prim Care. 2008;35(2):215-237.
4. Sharma B, Misicko NE. How should you evaluate elevated calcium in an asymptomatic patient? J Fam Pract. 2008;57(4):267-269.
5. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11-30.
6. Kyle RA, Gertz MA, Witzig TE, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc. 2003;78(1):21-33.
7. Shaheen SP, Talwalkar SS, Medeiros LJ. Multiple myeloma and immunosecretory disorders: an update. Adv Anat Pathol. 2008;15(4):196-210.
8. Palumbo A, Anderson K. Multiple myeloma. N Engl J Med. 2011;364(11): 1046-1060.
9. Talamo G, Farooq U, Zangari M, et al. Beyond the CRAB symptoms: a study of presenting clinical manifestations of multiple myeloma. Clin Lymphoma Myeloma Leuk. 2010;10(6):464-468.
10. Palumbo A, Sezer O, Kyle R, et al. International Myeloma Working Group guidelines for the management of multiple myeloma patients ineligible for standard high-dose chemotherapy with autologous stem cell transportation. Leukemia. 2009;23(10):1716-1730.
11. Kyprolis [package insert]. South San Francisco, CA: Onyx Pharmaceuticals, Inc; 2012.
12. Suyani E, Sucak GT, Erten Y, et al. Evaluation of multiple myeloma patients presenting with renal failure in a university hospital in the year 2010. Ren Fail. 2012;34(2):257-262.
A 66-year-old Latin American woman presented to the emergency department (ED) with persistent abdominal and back pain of about one month’s duration. She had visited another ED eight days earlier for similar symptoms and was discharged home with a mild opioid pain medication and a proton pump inhibitor. However, she said that she had received neither a diagnosis nor an explanation for her symptoms.
Medical history, obtained with the assistance of an interpreter because the patient was not fluent in English, included hypertension, coronary artery disease, and hyperlipidemia; these had gone untreated for at least two years. She denied any personal or family history of cancer or endocrine disorders. Surgical history included a cholecystectomy and a percutaneous coronary intervention for an unknown coronary artery lesion.
She had a 14-pack-year history of cigarette smoking. Her medications included only ibuprofen and hydrocodone, and she had no known drug allergies. The patient denied use of herbal preparations or vitamin supplements and unusual dietary practices.
Review of systems revealed occasional dizziness, constipation, decreased appetite, and some mild confusion noted by family members, but no fever, chills, palpitations, chest pain, shortness of breath, muscle spasm, or weakness. Vital signs were normal. Physical examination was remarkable for tenderness of the upper quadrants of the abdomen with deep palpation, without guarding or rebound. Bony tenderness at the right anterior costal margin of the rib cage was also noted.
Laboratory work-up revealed marked hypercalcemia (15.4 mg/dL), electrolyte abnormalities, anemia, impaired renal function, and elevated alkaline phosphatase and globulin levels (see Table 1). In addition, a plain abdominal x-ray series was negative for acute findings, but x-rays of the right ribs revealed a fracture of the sixth rib and osteopenia.
Continued >>
The patient was admitted to the hospital for treatment of hypercalcemia and hypokalemia and for work-up of elevated alkaline phosphatase and abdominal pain. Upon admission, serum ionized calcium measurement confirmed true hypercalcemia. Additional diagnostic tests were then ordered to help differentiate between parathyroid hormone (PTH)–mediated and non-PTH–mediated causes for the hypercalcemia (see Table 2).
The patient’s PTH level was normal and the urine fractional excretion of calcium level was high, ruling out familial hypocalciuric hypercalcemia (FHH), in which urine calcium level is low. A measurement of PTH-related protein (PTHrP), secreted by some cancers, was normal, suggesting exclusion of solid tumor malignancy. Vitamin D toxicity was ruled out because the patient’s 1,25-dihydroxyvitamin D level was low.
The patient continued to experience vague abdominal, back, and rib pain that seemed to migrate daily and worsened with movement. A skeletal x-ray was performed and revealed numerous lytic lesions of the skull (Figure 1), midright humerus (Figure 2), and distal left radius.
Continue for discussion >>
DISCUSSION
Hypercalcemia is a relatively common presentation in primary care. The most frequent causes are primary hyperparathyroidism and malignancy.1 One in 500 patients will be diagnosed incidentally with asymptomatic hypercalcemia caused by underlying hyperparathyroidism.1
Clinical manifestations of hypercalcemia can range from no symptoms to multisystem disease. Fatigue, nausea, vomiting, constipation, bone pain, osteoporosis, nephrolithiasis, mental status changes, hypertension, anemia, elevated creatinine, and cardiac arrhythmias are among the more common clinical conditions associated with hypercalcemia (hence the mnemonic “stones, bones, abdominal moans, and psychic groans” for its signs and symptoms).1
Diagnostic overview
Causes of hypercalcemia are numerous and can be broken down into two categories: PTH-mediated and non-PTH–mediated. PTH-mediated causes include primary and secondary hyperparathyroidism and FHH. Non-PTH–mediated causes include vitamin D toxicity, solid tumor malignancy with or without metastasis, multiple myeloma (MM) and other plasma cell dyscrasias, granulomatous disease such as sarcoid, and some medications.1
The differential diagnosis for hypercalcemia begins with measurement of the patient’s intact PTH level. An elevated or high-normal result indicates a PTH-mediated cause, so 24-hour measurement of excretion of urinary calcium is the next step. A low or low-normal PTH level (< 20 pg/mL), however, suggests the cause is non-PTH-mediated.2 The diagnostic approach in this situation is more challenging because testing to exclude or confirm various potential causes can be expensive and time-consuming. The degree of hypercalcemia, however, can aid in the diagnosis: Primary hyperparathyroidism is often associated with borderline or mild hypercalcemia, while calcium values > 13 mg/dL are more common in patients with malignancies.2
PTH elevates calcium levels in the blood when ionized (free) calcium levels are low by increasing gastrointestinal absorption, decreasing urinary excretion, and increasing bone resorption.3 With malignant tumors such as lung, breast, and renal cell, osteolytic metastases can destroy the bone, resulting in release of calcium. In other cases, solid-tumor cancers produce PTHrP, which increases serum calcium. This latter situation is referred to as humoral hypercalcemia of malignancy.3 Lymphoma and granulomatous disease, such as sarcoid, can be associated with excess production of 1,25-dihydroxyvitamin D.2 If vitamin D is elevated but PTH and PTHrP are normal, a chest x-ray should be obtained to evaluate the patient for sarcoid or lymphoma.
Hypercalcemia work-up
The work-up for suspected hypercalcemia begins with measurement of the patient’s calcium level. Because calcium is bound to albumin in the blood, the standard serum calcium test may not reflect the true calcium level. (If albumin is high, the calcium level will be high, and vice versa.)1 The true (serum ionized) calcium level (also known as corrected calcium level) should always be calculated to confirm true hypercalcemia. A formula commonly used to calculate the corrected calcium level is
Corrected calcium (mg/dL) = (measured calcium [mg/dL]) + 0.8 (4.0 – serum albumin [mg/dL])
Direct measurement of the serum ionized calcium level is not affected by the albumin level and can also confirm true hypercalcemia.4
Once hypercalcemia is confirmed, the next step is to measure the patient’s intact PTH level.
If intact PTH is elevated or high normal, consider primary hyperparathyroidism or FHH and confirm by obtaining the urine calcium level.
• If urine calcium level is high (> 200 mg/24 h), the diagnosis is primary hyperparathyroidism.
• If urine calcium level is low (< 100 mg/24 h), the diagnosis is FHH.
If intact PTH is low, consider non–PTH-mediated causes and confirm by obtaining PTHrP and vitamin D levels.
• If PTHrP level is elevated, scan for malignancy.
• If 1,25-dihydroxyvitamin D level is elevated, order a chest x-ray to rule out sarcoid or lymphoma.
• If both PTHrP and vitamin D levels are normal, order both serum protein electrophoresis (SPEP) and urine protein electrophoresis (UPEP) with immunofixation to rule out MM.
• If vitamin D level is elevated, check vitamin and herbal supplement use for excessive vitamin D intake.
Treatment of symptomatic hypercalcemia
The goals of treatment of symptomatic hypercalcemia are to reduce the serum calcium level to the normal range and to treat the underlying cause.1 Mild hypercalcemia (calcium level, 10-12 mg/dL) is typically asymptomatic and does not need to be treated. Moderate hypercalcemia (calcium level, 12-14 mg/dL) may not require treatment unless the patient is symptomatic and/or has had an acute rise in calcium level.1 In mild to moderate hypercalcemia, the serum calcium level should be monitored to establish a trend.
Treatment for symptomatic moderate and severe hypercalcemia (calcium level, > 14 mg/dL) typically involves a similar regimen:
• Volume expansion with isotonic saline at an initial rate of 2 to 4 L/d, which is then adjusted to achieve 200 mL/h of continuous urine output. IV furosemide can be used with caution (10-20 mg IV as needed) to promote diuresis if volume overload is a concern (furosemide promotes renal excretion of calcium).
• Administration of subcutaneous calcitonin (4-8 IU/kg, repeated every 6 h for 24 h). Calcitonin works rapidly to lower calcium levels in 4 to 6 h.
• Concurrent administration of IV bisphosphonate (zoledronic acid [4 mg over 15 min] or pamidronate [60-90 mg over 4 h]). Pamidronate is superior for reversal of malignancy-related hypercalcemia.1
Hypercalcemia and multiple myeloma
MM is a malignant neoplasm of plasma cells that accounts for approximately 1% of all cancers and about 10% of hematologic malignancies in the United States, with a median patient age of 70 at diagnosis.5-7 In MM, myeloma cells induce the secretion of cytokines and growth factors that alter plasma cells, activate osteoclasts, suppress osteoblasts, cause abnormal interactions between plasma cells and bone marrow, and stimulate aberrant angiogenesis.8 Osteoclastic bone resorption produces hypercalcemia as well as the lytic lesions seen on x-ray.7
Approximately 74% of patients present with typical MM symptoms of calcium elevation in the blood, renal insufficiency, anemia, and bone lesions, known as CRAB symptoms, but other myeloma-related manifestations may be present.9
Diagnostic criteria for MM include the following (all three must be present):10
• Monoclonal bone marrow plasma cells ≥ 10% and/or a biopsy-proven plasmacytoma
• Monoclonal protein in the serum and/or urine (if none is detected, disease is nonsecretory and diagnosis requires ≥ 30% bone marrow plasma cells and/or biopsy-proven plasmacytoma)
• Myeloma-related organ dysfunction, indicated by at least one of the CRAB symptoms.
In the absence of CRAB symptoms, an asymptomatic patient may have an MM precursor syndrome: monoclonal gammopathy of undetermined significance or smoldering (or indolent) MM.10
Treatment of multiple myeloma
In recent years, the use of induction therapy followed by autologous stem cell transplantation and the development of novel therapeutic agents have extended overall survival for patients with MM. These agents include proteasome inhibitors (bortezomib and the second-generation carfilzomib)11 and immunomodulators (thalidomide and the second-generation lenalidomide). Early diagnosis and treatment can improve progression-free survival as well as overall survival, including recovery of renal function for patients with renal failure.12 With survival ranging from one year or less—with aggressive disease—to 10 years or more for patients with responsive disease,7 there remains no cure for MM.
Outcome for the case patient >>
OUTCOME FOR THE CASE PATIENT
Further work-up included SPEP and UPEP with immunofixation, which revealed marked IgG free λ light chains with an M (monoclonal) component, making MM a very likely diagnosis. Confirmation by means of bone marrow biopsy was indicated, but the patient refused the procedure.
The patient’s hypercalcemia was treated by IV administration of calcitonin with isotonic saline. This reduced the serum calcium level from 15.4 mg/dL to 10.6 mg/dL within 48 hours. One dose of ergocalciferol (vitamin D2) was then administered to promote intestinal absorption of calcium and support bone mineralization, further lowering the patient’s serum calcium level to a normal 8.9 mg/dL. Hypokalemia was treated with oral potassium supplementation.
The patient, now stable, was referred to the hematology/oncology and bone mineral metabolism clinics and was discharged from the hospital. She did not keep those appointments and was lost to follow-up.
CONCLUSION
The most common causes of hypercalcemia are hyperparathyroidism and malignancy. Most cases do not require treatment unless the calcium level is >14 mg/dL and/or the patient is symptomatic. Red flag symptoms include weakness, abdominal pain, mental status changes, and coma.4 Primary care clinicians should suspect MM in older patients with laboratory findings of hypercalcemia, anemia, and renal dysfunction, with lytic lesions on x-ray.
REFERENCES
1. Carroll MF, Schade DS. A practical approach to hypercalcemia. Am Fam Physician. 2003;67(9):1959-1966.
2. Endres DB. Investigation of hypercalcemia. Clin Biochem. 2012;45(12):
954-963.
3. Moe SM. Disorders involving calcium, phosphorus, and magnesium. Prim Care. 2008;35(2):215-237.
4. Sharma B, Misicko NE. How should you evaluate elevated calcium in an asymptomatic patient? J Fam Pract. 2008;57(4):267-269.
5. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11-30.
6. Kyle RA, Gertz MA, Witzig TE, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc. 2003;78(1):21-33.
7. Shaheen SP, Talwalkar SS, Medeiros LJ. Multiple myeloma and immunosecretory disorders: an update. Adv Anat Pathol. 2008;15(4):196-210.
8. Palumbo A, Anderson K. Multiple myeloma. N Engl J Med. 2011;364(11): 1046-1060.
9. Talamo G, Farooq U, Zangari M, et al. Beyond the CRAB symptoms: a study of presenting clinical manifestations of multiple myeloma. Clin Lymphoma Myeloma Leuk. 2010;10(6):464-468.
10. Palumbo A, Sezer O, Kyle R, et al. International Myeloma Working Group guidelines for the management of multiple myeloma patients ineligible for standard high-dose chemotherapy with autologous stem cell transportation. Leukemia. 2009;23(10):1716-1730.
11. Kyprolis [package insert]. South San Francisco, CA: Onyx Pharmaceuticals, Inc; 2012.
12. Suyani E, Sucak GT, Erten Y, et al. Evaluation of multiple myeloma patients presenting with renal failure in a university hospital in the year 2010. Ren Fail. 2012;34(2):257-262.