User login
Does this patient need ultrasonography of the leg to evaluate for deep vein thrombosis?
A 38-year-old woman presents to the emergency department after experiencing several days of swelling and mild discomfort in her left calf. She denies chest pain or shortness of breath. She does not recall antecedent trauma, is a nonsmoker, is healthy, and takes no medications apart from a multivitamin. She has not undergone any surgical procedure, has not been hospitalized recently, and has no history of venous thromboembolic disease. She says she started an aerobics program 1 week ago.
On examination, her left lower leg is mildly swollen, but the difference in calf circumference between the right and left legs is less than 1 cm. There is no erythema, no pitting edema, and only mild and rather diffuse tenderness of the calf. A urine pregnancy test is negative and her D-dimer level is 350 ng/mL (reference range < 500 ng/mL). Does she require ultrasonography of the left leg to evaluate for deep vein thrombosis (DVT)?
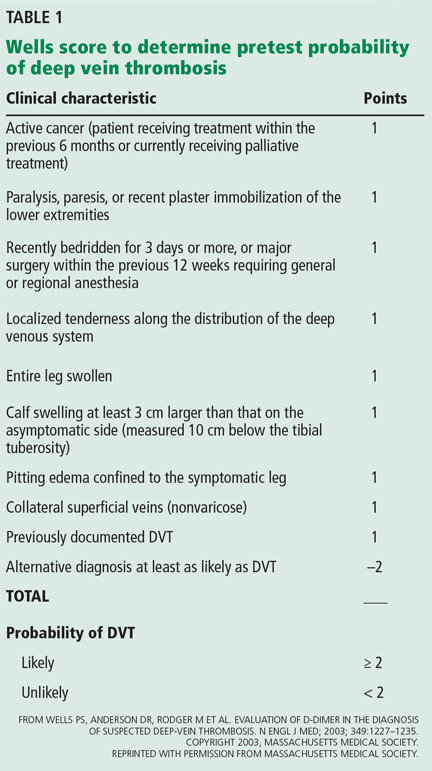
This patient does not need confirmatory ultrasonography, as her normal D-dimer level of 350 ng/mL is enough to rule out DVT. Her low probability of having DVT is further supported by her Wells score (Table 1), a tool that can help rule out DVT and reduce the need for further testing. DVT is unlikely if a patient’s Wells score is less than 2, and this patient’s score is –1. She receives 1 point for swelling of her left lower leg, but injury from her recent aerobic exercise is at least as likely as DVT to account for her symptoms (–2 points).
GUIDELINES AND CHOOSING WISELY
Compression ultrasonography is the study most commonly used to evaluate for DVT. The diagnosis is made if either the femoral or popliteal vein is noncompressible.1 In a patient with no history of DVT, the sensitivity of compression ultrasonography is 94%, and its specificity is 98%.
Several guidelines recommend using a clinical decision rule to establish the probability of venous thromboembolic disease before any additional diagnostic testing such as D-dimer measurement or ultrasonography.2–4 A number of clinical decision rules exist for DVT, but the Wells score is the most studied and validated.1 It incorporates the patient’s risk factors, symptoms, and signs to categorize the probability of DVT as low, moderate, or high and has been further modified to classify the risk as either likely or unlikely (Table 1).5
Guidelines from the American College of Chest Physicians (2012), Scottish Intercollegiate Guidelines Network (2010), and American Academy of Family Physicians and American College of Physicians (2007) recommend against performing imaging if a high-sensitivity D-dimer test is negative in a patient in whom the pretest probability of DVT is unlikely.2–4 Enzyme-linked immunofluorescence assays, microplate enzyme-linked immunosorbent assays, and latex quantitative assays are considered high-sensitivity D-dimer tests, having 96%, 94%, and 93% sensitivity, respectively, in ruling out DVT.1 Other D-dimer tests have lower sensitivity and cannot comfortably rule out DVT even if the results are negative.
Since D-dimer measurement is a sensitive but not specific test, it should be used only to rule out DVT—not to rule it in. Moreover, compression ultrasonography may be indicated to rule out other causes of the patient’s symptoms.

The guidelines caution against D-dimer testing if the patient has a comorbid condition that can by itself raise or lower the D-dimer level, leading one to falsely conclude the patient has or does not have DVT (Table 2).1–4 In these instances, the pretest probability of DVT may be higher than calculated by a clinical prediction rule, and compression ultrasonography may be an appropriate initial test.4 Compression ultrasonography is also recommended as a confirmatory test in low-risk patients who have a positive D-dimer test or as an initial test in patients at higher risk for DVT.2–4
If a patient has a low pretest probability of DVT as defined by the Wells score and a normal high-sensitivity D-dimer measurement, then ordering imaging studies is a questionable practice according to statements by the American College of Physicians, American College of Emergency Physicians, European Society of Cardiology, American Academy of Family Physicians, and Scottish Intercollegiate Guidelines Network.
HARMS OF ULTRASONOGRAPHY
Although ultrasonography is generally well tolerated, it may be unnecessary. Combining a prediction rule (to assess the probability) with D-dimer testing (to rule out DVT) can significantly reduce the use of ultrasonography and the associated cost.
Wells et al5 calculated that clinicians could cut back on ultrasonographic testing by 39% by not doing it in those who had a low pretest probability and a negative D-dimer test result.5 In that patient population, fewer than 1% of patients were later found to have DVT.
Ordering compression ultrasonography as additional testing may lead to a false-positive result and to additional unnecessary testing and treatments that would inconvenience the patient, increase the risk of serious complications such as bleeding, and incur increased costs. Cost considerations should include not only the cost of the test and its interpretation, but also the workup and treatment of false-positive results, patient time missed from work while being tested, and potential associated costs for patients who need to be evaluated in the emergency department to obtain same-day testing.
THE CLINICAL BOTTOM LINE
Our patient’s Wells score indicates that DVT is unlikely. A negative D-dimer test is sufficient to rule out DVT, and further testing is unnecessary.
- Huisman MV, Klok FA. Diagnostic management of acute deep vein thrombosis and pulmonary embolism. J Thromb Haemost 2013; 11:412–422.
- Bates SM, Jaeschke R, Stevens EM, et al. Antithrombotic therapy and prevention of thrombosis, 9th edition: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(2 suppl):e351S–e418S.
- Scottish Intercollegiate Guidelines Network (SIGN). Prevention and management of venous thromboembolism. A national clinical guideline. Edinburgh (Scotland): Scottish Intercollegiate Guidelines Network (SIGN); 2010: http://sign.ac.uk/guidelines/fulltext/122/index.html. Accessed February 6, 2015.
- Qaseem A, Snow V, Barry P, et al. Current diagnosis of venous thromboembolism in primary care: a clinical practice guideline from the American Academy of Family Physicians and the American College of Physicians. Ann Intern Med 2007; 146:454–458.
- Wells PS, Anderson DR, Rodger M, et al. Evaluation of D-dimer in the diagnosis of suspected deep-vein thrombosis. N Engl J Med 2003; 349:1227–1235.
A 38-year-old woman presents to the emergency department after experiencing several days of swelling and mild discomfort in her left calf. She denies chest pain or shortness of breath. She does not recall antecedent trauma, is a nonsmoker, is healthy, and takes no medications apart from a multivitamin. She has not undergone any surgical procedure, has not been hospitalized recently, and has no history of venous thromboembolic disease. She says she started an aerobics program 1 week ago.
On examination, her left lower leg is mildly swollen, but the difference in calf circumference between the right and left legs is less than 1 cm. There is no erythema, no pitting edema, and only mild and rather diffuse tenderness of the calf. A urine pregnancy test is negative and her D-dimer level is 350 ng/mL (reference range < 500 ng/mL). Does she require ultrasonography of the left leg to evaluate for deep vein thrombosis (DVT)?
This patient does not need confirmatory ultrasonography, as her normal D-dimer level of 350 ng/mL is enough to rule out DVT. Her low probability of having DVT is further supported by her Wells score (Table 1), a tool that can help rule out DVT and reduce the need for further testing. DVT is unlikely if a patient’s Wells score is less than 2, and this patient’s score is –1. She receives 1 point for swelling of her left lower leg, but injury from her recent aerobic exercise is at least as likely as DVT to account for her symptoms (–2 points).
GUIDELINES AND CHOOSING WISELY
Compression ultrasonography is the study most commonly used to evaluate for DVT. The diagnosis is made if either the femoral or popliteal vein is noncompressible.1 In a patient with no history of DVT, the sensitivity of compression ultrasonography is 94%, and its specificity is 98%.
Several guidelines recommend using a clinical decision rule to establish the probability of venous thromboembolic disease before any additional diagnostic testing such as D-dimer measurement or ultrasonography.2–4 A number of clinical decision rules exist for DVT, but the Wells score is the most studied and validated.1 It incorporates the patient’s risk factors, symptoms, and signs to categorize the probability of DVT as low, moderate, or high and has been further modified to classify the risk as either likely or unlikely (Table 1).5
Guidelines from the American College of Chest Physicians (2012), Scottish Intercollegiate Guidelines Network (2010), and American Academy of Family Physicians and American College of Physicians (2007) recommend against performing imaging if a high-sensitivity D-dimer test is negative in a patient in whom the pretest probability of DVT is unlikely.2–4 Enzyme-linked immunofluorescence assays, microplate enzyme-linked immunosorbent assays, and latex quantitative assays are considered high-sensitivity D-dimer tests, having 96%, 94%, and 93% sensitivity, respectively, in ruling out DVT.1 Other D-dimer tests have lower sensitivity and cannot comfortably rule out DVT even if the results are negative.
Since D-dimer measurement is a sensitive but not specific test, it should be used only to rule out DVT—not to rule it in. Moreover, compression ultrasonography may be indicated to rule out other causes of the patient’s symptoms.
The guidelines caution against D-dimer testing if the patient has a comorbid condition that can by itself raise or lower the D-dimer level, leading one to falsely conclude the patient has or does not have DVT (Table 2).1–4 In these instances, the pretest probability of DVT may be higher than calculated by a clinical prediction rule, and compression ultrasonography may be an appropriate initial test.4 Compression ultrasonography is also recommended as a confirmatory test in low-risk patients who have a positive D-dimer test or as an initial test in patients at higher risk for DVT.2–4
If a patient has a low pretest probability of DVT as defined by the Wells score and a normal high-sensitivity D-dimer measurement, then ordering imaging studies is a questionable practice according to statements by the American College of Physicians, American College of Emergency Physicians, European Society of Cardiology, American Academy of Family Physicians, and Scottish Intercollegiate Guidelines Network.
HARMS OF ULTRASONOGRAPHY
Although ultrasonography is generally well tolerated, it may be unnecessary. Combining a prediction rule (to assess the probability) with D-dimer testing (to rule out DVT) can significantly reduce the use of ultrasonography and the associated cost.
Wells et al5 calculated that clinicians could cut back on ultrasonographic testing by 39% by not doing it in those who had a low pretest probability and a negative D-dimer test result.5 In that patient population, fewer than 1% of patients were later found to have DVT.
Ordering compression ultrasonography as additional testing may lead to a false-positive result and to additional unnecessary testing and treatments that would inconvenience the patient, increase the risk of serious complications such as bleeding, and incur increased costs. Cost considerations should include not only the cost of the test and its interpretation, but also the workup and treatment of false-positive results, patient time missed from work while being tested, and potential associated costs for patients who need to be evaluated in the emergency department to obtain same-day testing.
THE CLINICAL BOTTOM LINE
Our patient’s Wells score indicates that DVT is unlikely. A negative D-dimer test is sufficient to rule out DVT, and further testing is unnecessary.
A 38-year-old woman presents to the emergency department after experiencing several days of swelling and mild discomfort in her left calf. She denies chest pain or shortness of breath. She does not recall antecedent trauma, is a nonsmoker, is healthy, and takes no medications apart from a multivitamin. She has not undergone any surgical procedure, has not been hospitalized recently, and has no history of venous thromboembolic disease. She says she started an aerobics program 1 week ago.
On examination, her left lower leg is mildly swollen, but the difference in calf circumference between the right and left legs is less than 1 cm. There is no erythema, no pitting edema, and only mild and rather diffuse tenderness of the calf. A urine pregnancy test is negative and her D-dimer level is 350 ng/mL (reference range < 500 ng/mL). Does she require ultrasonography of the left leg to evaluate for deep vein thrombosis (DVT)?
This patient does not need confirmatory ultrasonography, as her normal D-dimer level of 350 ng/mL is enough to rule out DVT. Her low probability of having DVT is further supported by her Wells score (Table 1), a tool that can help rule out DVT and reduce the need for further testing. DVT is unlikely if a patient’s Wells score is less than 2, and this patient’s score is –1. She receives 1 point for swelling of her left lower leg, but injury from her recent aerobic exercise is at least as likely as DVT to account for her symptoms (–2 points).
GUIDELINES AND CHOOSING WISELY
Compression ultrasonography is the study most commonly used to evaluate for DVT. The diagnosis is made if either the femoral or popliteal vein is noncompressible.1 In a patient with no history of DVT, the sensitivity of compression ultrasonography is 94%, and its specificity is 98%.
Several guidelines recommend using a clinical decision rule to establish the probability of venous thromboembolic disease before any additional diagnostic testing such as D-dimer measurement or ultrasonography.2–4 A number of clinical decision rules exist for DVT, but the Wells score is the most studied and validated.1 It incorporates the patient’s risk factors, symptoms, and signs to categorize the probability of DVT as low, moderate, or high and has been further modified to classify the risk as either likely or unlikely (Table 1).5
Guidelines from the American College of Chest Physicians (2012), Scottish Intercollegiate Guidelines Network (2010), and American Academy of Family Physicians and American College of Physicians (2007) recommend against performing imaging if a high-sensitivity D-dimer test is negative in a patient in whom the pretest probability of DVT is unlikely.2–4 Enzyme-linked immunofluorescence assays, microplate enzyme-linked immunosorbent assays, and latex quantitative assays are considered high-sensitivity D-dimer tests, having 96%, 94%, and 93% sensitivity, respectively, in ruling out DVT.1 Other D-dimer tests have lower sensitivity and cannot comfortably rule out DVT even if the results are negative.
Since D-dimer measurement is a sensitive but not specific test, it should be used only to rule out DVT—not to rule it in. Moreover, compression ultrasonography may be indicated to rule out other causes of the patient’s symptoms.
The guidelines caution against D-dimer testing if the patient has a comorbid condition that can by itself raise or lower the D-dimer level, leading one to falsely conclude the patient has or does not have DVT (Table 2).1–4 In these instances, the pretest probability of DVT may be higher than calculated by a clinical prediction rule, and compression ultrasonography may be an appropriate initial test.4 Compression ultrasonography is also recommended as a confirmatory test in low-risk patients who have a positive D-dimer test or as an initial test in patients at higher risk for DVT.2–4
If a patient has a low pretest probability of DVT as defined by the Wells score and a normal high-sensitivity D-dimer measurement, then ordering imaging studies is a questionable practice according to statements by the American College of Physicians, American College of Emergency Physicians, European Society of Cardiology, American Academy of Family Physicians, and Scottish Intercollegiate Guidelines Network.
HARMS OF ULTRASONOGRAPHY
Although ultrasonography is generally well tolerated, it may be unnecessary. Combining a prediction rule (to assess the probability) with D-dimer testing (to rule out DVT) can significantly reduce the use of ultrasonography and the associated cost.
Wells et al5 calculated that clinicians could cut back on ultrasonographic testing by 39% by not doing it in those who had a low pretest probability and a negative D-dimer test result.5 In that patient population, fewer than 1% of patients were later found to have DVT.
Ordering compression ultrasonography as additional testing may lead to a false-positive result and to additional unnecessary testing and treatments that would inconvenience the patient, increase the risk of serious complications such as bleeding, and incur increased costs. Cost considerations should include not only the cost of the test and its interpretation, but also the workup and treatment of false-positive results, patient time missed from work while being tested, and potential associated costs for patients who need to be evaluated in the emergency department to obtain same-day testing.
THE CLINICAL BOTTOM LINE
Our patient’s Wells score indicates that DVT is unlikely. A negative D-dimer test is sufficient to rule out DVT, and further testing is unnecessary.
- Huisman MV, Klok FA. Diagnostic management of acute deep vein thrombosis and pulmonary embolism. J Thromb Haemost 2013; 11:412–422.
- Bates SM, Jaeschke R, Stevens EM, et al. Antithrombotic therapy and prevention of thrombosis, 9th edition: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(2 suppl):e351S–e418S.
- Scottish Intercollegiate Guidelines Network (SIGN). Prevention and management of venous thromboembolism. A national clinical guideline. Edinburgh (Scotland): Scottish Intercollegiate Guidelines Network (SIGN); 2010: http://sign.ac.uk/guidelines/fulltext/122/index.html. Accessed February 6, 2015.
- Qaseem A, Snow V, Barry P, et al. Current diagnosis of venous thromboembolism in primary care: a clinical practice guideline from the American Academy of Family Physicians and the American College of Physicians. Ann Intern Med 2007; 146:454–458.
- Wells PS, Anderson DR, Rodger M, et al. Evaluation of D-dimer in the diagnosis of suspected deep-vein thrombosis. N Engl J Med 2003; 349:1227–1235.
- Huisman MV, Klok FA. Diagnostic management of acute deep vein thrombosis and pulmonary embolism. J Thromb Haemost 2013; 11:412–422.
- Bates SM, Jaeschke R, Stevens EM, et al. Antithrombotic therapy and prevention of thrombosis, 9th edition: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(2 suppl):e351S–e418S.
- Scottish Intercollegiate Guidelines Network (SIGN). Prevention and management of venous thromboembolism. A national clinical guideline. Edinburgh (Scotland): Scottish Intercollegiate Guidelines Network (SIGN); 2010: http://sign.ac.uk/guidelines/fulltext/122/index.html. Accessed February 6, 2015.
- Qaseem A, Snow V, Barry P, et al. Current diagnosis of venous thromboembolism in primary care: a clinical practice guideline from the American Academy of Family Physicians and the American College of Physicians. Ann Intern Med 2007; 146:454–458.
- Wells PS, Anderson DR, Rodger M, et al. Evaluation of D-dimer in the diagnosis of suspected deep-vein thrombosis. N Engl J Med 2003; 349:1227–1235.
An Incidental Finding During Neuro Evaluation
ANSWER
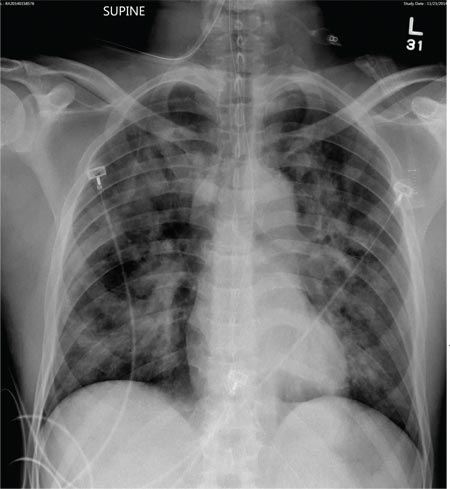
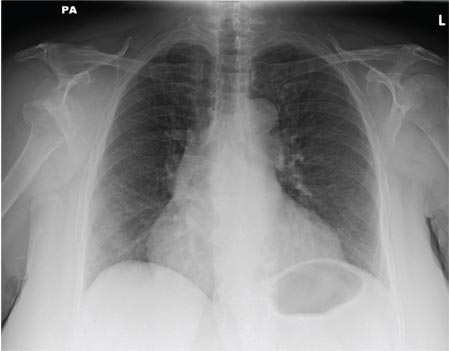
The radiograph shows a normal-appearing chest. Of note, though, is an anterior dislocation of the right shoulder. In addition, there is a fracture within the greater tuberosity of the right humerus.
Prompt orthopedic evaluation is obtained. In further discussion with the family, it was revealed that the patient had been experiencing falls recently; this injury was most likely the result of one.
ANSWER
The radiograph shows a normal-appearing chest. Of note, though, is an anterior dislocation of the right shoulder. In addition, there is a fracture within the greater tuberosity of the right humerus.
Prompt orthopedic evaluation is obtained. In further discussion with the family, it was revealed that the patient had been experiencing falls recently; this injury was most likely the result of one.
ANSWER
The radiograph shows a normal-appearing chest. Of note, though, is an anterior dislocation of the right shoulder. In addition, there is a fracture within the greater tuberosity of the right humerus.
Prompt orthopedic evaluation is obtained. In further discussion with the family, it was revealed that the patient had been experiencing falls recently; this injury was most likely the result of one.
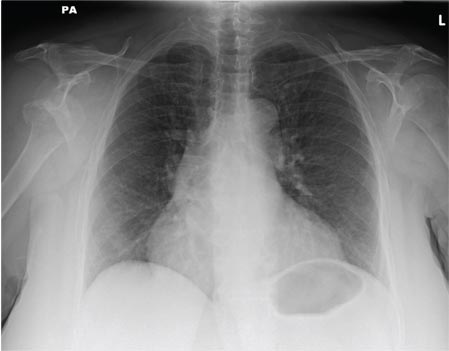
A 65-year-old woman is transferred to your facility from an outlying hospital for evaluation of a brain tumor. Family members found the patient sitting on the sofa, with a decreased level of consciousness. There was reported “seizure-type activity.” When she arrived at the outlying hospital, the patient was noted to have right-side weakness. Stat CT of the head demonstrated a fairly large parasagittal mass, and the patient was urgently transferred to your facility for neurosurgical evaluation. Primary survey on arrival shows an older female who is awake, alert, and in no obvious distress. Vital signs are normal. She has fairly pronounced right upper extremity weakness, more proximally than distally. Otherwise, the exam grossly appears normal. The patient’s initial imaging studies were sent with her on a CD. As you are trying to view the images of the brain, a chest radiograph pops up on your screen. What is your impression?
Woman, 64, With Eye Pain, Swelling, and Tearing
A 64-year-old woman presented to the clinic with a two-to-three-week history of significant pain, swelling, and excessive tearing of the left eye. The patient had a persistent cough but denied wheezing or shortness of breath.
Medical history was remarkable for uveitis, severe recurrent sinusitis, and allergic rhinitis. The patient reported that she had been exposed to benzene and burning paint fumes about 10 years ago but had no known symptoms or problems at the time.
Vital signs included a temperature of 97.0°F; respiratory rate, 18 breaths/min; pulse, 100 beats/min; and blood pressure, 144/80 mm Hg. Her height was 65 in; weight, 122 lb; and O2 saturation, 100% on room air.
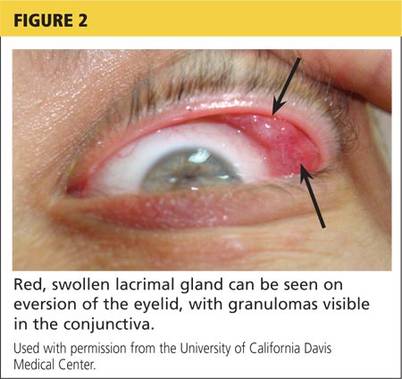
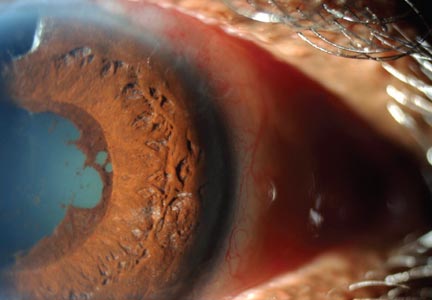
Physical examination revealed a left palpebral lacrimal mass with an enlarged lacrimal gland. The left lacrimal gland and conjunctiva were mildly erythematous, with a cobblestone appearance. The right eye was stable, with no significant inflammation. Pupils were equal, round, and reactive to light and accommodation. Extraocular movements were intact. Nasal turbinates were swollen and mildly erythematous. Oropharynx was stable and tonsils absent. Left parotid gland was slightly swollen and tender.
The neck was supple with no jugular venous distension. Palpable cervical and supraclavicular lymphadenopathy, measuring approximately 1.5 x 1.5 cm bilaterally, was present. The lungs were clear to auscultation and percussion. The heart rate and rhythm were regular, with normal S1 and S2 sounds. The abdomen was soft, nontender, and without hepatosplenomegaly. Extremities were stable, with no rashes, lesions, or cutaneous skin nodules.
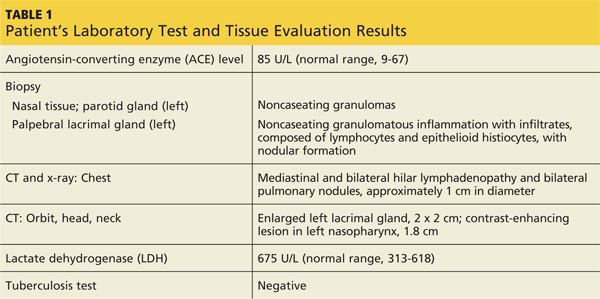
The patient was referred to a specialist for a complete ophthalmologic examination and further work-up. This included a complete blood count, comprehensive metabolic panel, tissue biopsies of the affected lacrimal gland and parotid gland, CT, and x-rays; results are shown in Table 1. In addition, the patient’s persistent nasosinus congestion was determined, by otolaryngologic consultation, to be the result of a deviated septum, for which she underwent endoscopic nasal septal repair with tissue biopsy.
The lacrimal gland biopsy led to a diagnosis of chronic noncaseating granulomatous dacryoadenitis, with an extensive area of necrosis. Significant findings included histiocytes and discrete nodules in the gland. Biopsies of the parotid gland and nasal tissue also identified noncaseating granulomas.
The patient’s test results suggested several possible diagnoses, including
• Granulomatosis with polyangiitis
• Tuberculosis (TB) or similar pulmonary infectious disease
• Sarcoidosis (ocular and/or pulmonary)
Continue for differential diagnosis >>
DISCUSSION
Differential diagnosis
Granulomatosis with polyangiitis. GPA, also known as Wegener granulomatosis, is characterized by necrotizing granulomatous inflammation with necrotizing vasculitis, usually of small and medium vessels; ocular involvement is frequent.1 Ocular granulomas of GPA can be mistaken for those caused by other diseases, such as mycobacterial or syphilitic infection or idiopathic uveitis.2
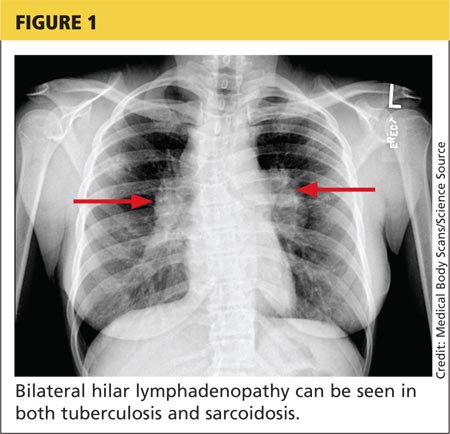
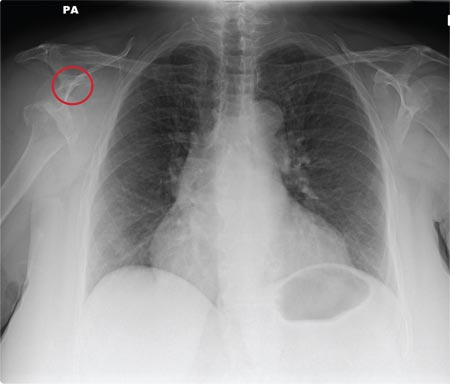
Tuberculosis. Common symptoms of TB include fever, cough, dyspnea, weight loss, malaise, and fatigue. Granulomas are typically necrotizing but are occasionally nonnecrotizing.3 TB can manifest with hilar and diffuse lymphadenopathy,4 which the patient’s chest imaging revealed (see Figure 1). Granulomas produced by Mycobacterium tuberculosis and atypical mycobacteria are similar histopathologically to sarcoidosis granulomas, complicating the diagnostic process.5
Next page: Sarcoidosis >>
Sarcoidosis. Sarcoidosis is a multisystem inflammatory disease characterized by noncaseating epithelioid granulomas in affected organs.6 More than 90% of patients with sarcoidosis present with pulmonary symptoms, including shortness of breath, cough, and pleuritic chest pain.6-8 Ocular manifestations, such as uveitis, iritis, or conjunctivitis, are less common, developing in 30% to 60% of patients.2,9,10 In addition, rashes, lesions, or cutaneous skin nodules, including erythema nodosum and lupus pernio, are seen in 25% to 35% of patients.2,6
In up to two-thirds of patients, sarcoidosis resolves spontaneously2; in others, it may become chronic and progressive.4 Patients may have few or no symptoms; some require no treatment, while others may be severely affected by the disease.
Ocular involvement in sarcoidosis generally manifests as uveitis, most commonly in the anterior chamber. Uveitis is a potentially vision-threatening inflammatory disease involving both the uveal tract and adjacent structures.11 In a review of records for 2,619 patients with uveitis, 59.9% had anterior disease, of whom 2.1% were diagnosed with sarcoidosis.11
While the etiology of sarcoidosis continues to be studied,7 the prevailing theory is that, in genetically predisposed individuals, sarcoidosis is a cell-mediated immune response to as-yet unknown antigen triggers that leads to granuloma formation.3,6,7
CD4+ activated T-cells stimulate the immune reaction against an antigen, producing cytokines that activate immune cells (eg, B cells, macrophages, monocytes, and neutrophils).2 Immune cells accumulate and aggregate at antigen sites in an exaggerated response, resulting in the formation of granulomas (see Figure 2).7,12,13
Infectious agents have long been investigated as possible causative agents in sarcoidosis, with Mycobacterium species most frequently identified.5 Additional possibilities include Propionibacterium acnes (found predominantly in skin lesions) and herpesviruses, although viruses are not known to cause epitheliod granulomas.14
Environmental triggers have also been explored. One large study found a possible association between exposure to insecticides, agricultural environments, and microbial bioaerosols and sarcoidosis.15
The difficulty of pinpointing a single etiology for sarcoidosis—with its varying clinical manifestations, severity, and disease course—suggests that sarcoidosis may be a spectrum of disorders caused by the interaction of genetic, immunologic, infectious, and environmental factors.14
Next page: Diagnosis of sarcoidosis >>
Diagnosis
The diagnosis of sarcoidosis is based on clinical and radiologic features, histologic evidence of noncaseating granulomas, and exclusion of other possible causes of granulomas.2,12 In addition, when ocular sarcoidosis is suspected, other possible causes of uveitis must be excluded.
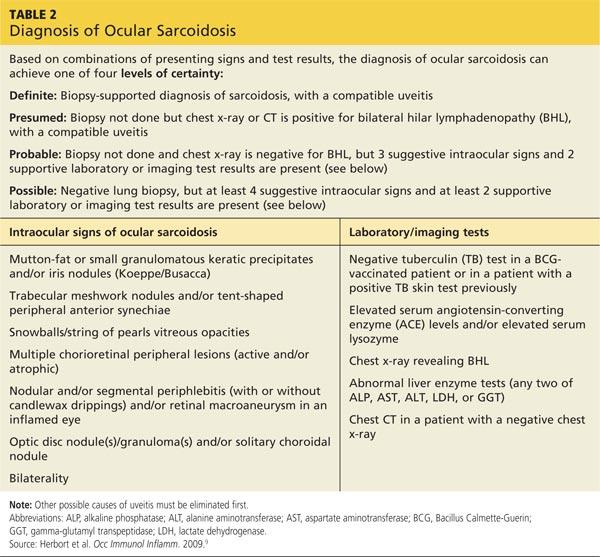
In an effort to address these challenges, the International Workshop on Ocular Sarcoidosis (IWOS) developed a standardized approach to diagnosis.9 The group first identified seven intraocular signs of ocular sarcoidosis and then five laboratory or imaging tests that are of value in making the diagnosis in patients with these signs. Last, they established four levels of certainty for the diagnosis of ocular sarcoidosis, based on these signs, tests, and biopsy results, if available (see Table 2).
Treatment
Anterior uveitis in sarcoidosis is usually treated initially with a topical corticosteroid (eg, prednisolone or difluprednate drops), particularly if the patient’s symptoms are mild. In more severe cases (eg, posterior or bilateral uveitis) or when topical corticosteroids are ineffective, systemic (oral) corticosteroids (eg, prednisone) may be initiated. Topical therapy can also be added to an oral regimen as a means of decreasing the oral dosage and thereby reducing the adverse effects of systemic corticosteroids. When the patient’s disease is refractory to corticosteroids or there are concerns about long-term adverse effects, chronic cases may be treated with immunosuppressive agents (eg, methotrexate, azathioprine, mycophenolate mofetil). Finally, refractory cases of ocular sarcoidosis may be treated with anti–tumor necrosis factor α (TNF-α) biologic agents such as infliximab and adalimumab.10,17
Continue for case patient outcome >>
OUTCOME FOR THE CASE PATIENT
Histologic evaluation of tissue from the lacrimal gland, parotid gland, and sinus cavity revealed inflammatory noncaseating granulomas, strongly suggestive of sarcoidosis. Diagnosis of ocular sarcoidosis was based on the noncaseating granulomas in the lacrimal gland.9,16 Pulmonary sarcoidosis was also diagnosed, based on the presence of hilar and mediastinal lymphadenopathy.7
The mass in the patient’s lacrimal gland was surgically removed. She was treated with a combination of topical and oral corticosteroids tapered over two weeks, which induced remission of her ocular disease. The patient will be seen annually by an ophthalmologic specialist and was advised to contact her clinician immediately if acute ocular symptoms recurred.10,17
The patient’s persistent cough was determined to be secondary to acute bronchitis, rather than to her pulmonary sarcoidosis, which required no treatment. She received a short course of antibiotics and antitussives for her bronchitis. Systemic corticosteroid treatment of her ocular sarcoidosis also had the benefit of decreasing the size of her pulmonary nodules. She will be followed with annual CT and chest x-rays to monitor the status of her hilar and mediastinal lymphadenopathy and the nodules.3 Periodic pulmonary function testing will also be performed.7
Continue for conclusion >>
CONCLUSION
The elusive nature of the diagnosis of sarcoidosis is well documented in the medical literature. In this case, histologic evaluation of biopsied tissue, correlated with clinical symptoms and radiographic findings, were essential in making the diagnosis.
Primary care providers may be the first to evaluate patients with ocular sarcoidosis and will oversee long-term management. Patients who present with symptoms of eye pain, visual disturbances, abnormal inflammatory ocular features, or swollen lacrimal glands should be referred to an ophthalmologic specialist for further evaluation.
REFERENCES
1. Jennette JC, Falk RJ, Bacon PA, et al. 2012 Revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013;65(1):1-11.
2. Culver DA. Sarcoidosis. Immunol Allergy Clin North Am. 2012;32(4):487-511.
3. Spagnolo P, Luppi F, Roversi P, et al. Sarcoidosis: challenging diagnostic aspects of an old disease. Am J Med. 2012;125(2):118-125.
4. Dempsey OJ, Peterson EW, Kerr KM, Denison AR. Sarcoidosis. BMJ. 2009;339:620-625.
5. Brownell I, Ramirez-Valle F, Sanchez M, Prystowsky S. Evidence for mycobacteria in sarcoidosis. Am J Respir Cell Mol Biol. 2011;45(5):899-905.
6. Iannuzzi MC, Fontana JR. Sarcoidosis: clinical presentation, immunopathogenesis, and therapeutics. JAMA. 2011;305(4):391-399.
7. Baughman MD, Culver DA, Judson MA. A concise review of pulmonary sarcoidosis. Am J Respir Crit Care Med. 2011;183(5):573-581.
8. Koyama T, Ueda H, Togashi K, et al. Radiologic manifestations of sarcoidosis in various organs. Radiographics. 2004;24(1):87-104.
9. Herbort CP, Rao NA, Mochizuki M; for the Scientific Committee of First International Workshop on Ocular Sarcoidosis. International criteria for the diagnosis of ocular sarcoidosis: results of the first International Workshop on Ocular Sarcoidosis (IWOS). Ocul Immunol Inflamm. 2009; 17(3):160-169.
10. Jamilloux Y, Kodjikian L, Broussolle C, Seve P. Sarcoidosis and uveitis. Autoimmun Rev. 2014;13(8):840-849.
11. Barisani-Asenbauer T, Maca SM, Mejdoubi L, et al. Uveitis—a rare disease often associated with systemic diseases and infections—a systematic review of 2619 patients. Orphanet J Rare Dis. 2012;7:57.
12. Iannuzzi MC, Rybicki BA, Teirstein AS. Sarcoidosis. New Engl J Med. 2007;357(21):2153-2165.
13. Fontenot A, King T. Pathogenesis of sarcoidosis. www.uptodate.com/contents/pathogenesis-of-sarcoidosis?source=search_result&search=Pathogenesis+of+sarcoidosis&selectedTitle=1%7E150. Accessed February 17, 2015.
14. Saidha S, Sotirchos ES, Eckstein C. Etiology of sarcoidosis: does infection play a role? Yale J Biol Med. 2012;85(1):133-141.
15. Newman LS, Rose CS, Bresnitz EA, et al; for the ACCESS Research Group. A case control etiologic study of sarcoidosis. Environmental and occupational risk factors. Am J Respir Crit Care Med. 2004;170:1324-1330.
16. Kawaguchi T, Hanada A, Horie S, et al. Evaluation of characteristic ocular signs and systemic investigations in ocular sarcoidosis patients. Jpn J Opthalmol. 2007;51(2):121-126.
17. Bodaghi B, Touitou V, Fardeau C, et al. Ocular sarcoidosis. Presse Med. 2012;41(6 Pt 2):e349-e354.
A 64-year-old woman presented to the clinic with a two-to-three-week history of significant pain, swelling, and excessive tearing of the left eye. The patient had a persistent cough but denied wheezing or shortness of breath.
Medical history was remarkable for uveitis, severe recurrent sinusitis, and allergic rhinitis. The patient reported that she had been exposed to benzene and burning paint fumes about 10 years ago but had no known symptoms or problems at the time.
Vital signs included a temperature of 97.0°F; respiratory rate, 18 breaths/min; pulse, 100 beats/min; and blood pressure, 144/80 mm Hg. Her height was 65 in; weight, 122 lb; and O2 saturation, 100% on room air.
Physical examination revealed a left palpebral lacrimal mass with an enlarged lacrimal gland. The left lacrimal gland and conjunctiva were mildly erythematous, with a cobblestone appearance. The right eye was stable, with no significant inflammation. Pupils were equal, round, and reactive to light and accommodation. Extraocular movements were intact. Nasal turbinates were swollen and mildly erythematous. Oropharynx was stable and tonsils absent. Left parotid gland was slightly swollen and tender.
The neck was supple with no jugular venous distension. Palpable cervical and supraclavicular lymphadenopathy, measuring approximately 1.5 x 1.5 cm bilaterally, was present. The lungs were clear to auscultation and percussion. The heart rate and rhythm were regular, with normal S1 and S2 sounds. The abdomen was soft, nontender, and without hepatosplenomegaly. Extremities were stable, with no rashes, lesions, or cutaneous skin nodules.
The patient was referred to a specialist for a complete ophthalmologic examination and further work-up. This included a complete blood count, comprehensive metabolic panel, tissue biopsies of the affected lacrimal gland and parotid gland, CT, and x-rays; results are shown in Table 1. In addition, the patient’s persistent nasosinus congestion was determined, by otolaryngologic consultation, to be the result of a deviated septum, for which she underwent endoscopic nasal septal repair with tissue biopsy.
The lacrimal gland biopsy led to a diagnosis of chronic noncaseating granulomatous dacryoadenitis, with an extensive area of necrosis. Significant findings included histiocytes and discrete nodules in the gland. Biopsies of the parotid gland and nasal tissue also identified noncaseating granulomas.
The patient’s test results suggested several possible diagnoses, including
• Granulomatosis with polyangiitis
• Tuberculosis (TB) or similar pulmonary infectious disease
• Sarcoidosis (ocular and/or pulmonary)
Continue for differential diagnosis >>
DISCUSSION
Differential diagnosis
Granulomatosis with polyangiitis. GPA, also known as Wegener granulomatosis, is characterized by necrotizing granulomatous inflammation with necrotizing vasculitis, usually of small and medium vessels; ocular involvement is frequent.1 Ocular granulomas of GPA can be mistaken for those caused by other diseases, such as mycobacterial or syphilitic infection or idiopathic uveitis.2
Tuberculosis. Common symptoms of TB include fever, cough, dyspnea, weight loss, malaise, and fatigue. Granulomas are typically necrotizing but are occasionally nonnecrotizing.3 TB can manifest with hilar and diffuse lymphadenopathy,4 which the patient’s chest imaging revealed (see Figure 1). Granulomas produced by Mycobacterium tuberculosis and atypical mycobacteria are similar histopathologically to sarcoidosis granulomas, complicating the diagnostic process.5
Next page: Sarcoidosis >>
Sarcoidosis. Sarcoidosis is a multisystem inflammatory disease characterized by noncaseating epithelioid granulomas in affected organs.6 More than 90% of patients with sarcoidosis present with pulmonary symptoms, including shortness of breath, cough, and pleuritic chest pain.6-8 Ocular manifestations, such as uveitis, iritis, or conjunctivitis, are less common, developing in 30% to 60% of patients.2,9,10 In addition, rashes, lesions, or cutaneous skin nodules, including erythema nodosum and lupus pernio, are seen in 25% to 35% of patients.2,6
In up to two-thirds of patients, sarcoidosis resolves spontaneously2; in others, it may become chronic and progressive.4 Patients may have few or no symptoms; some require no treatment, while others may be severely affected by the disease.
Ocular involvement in sarcoidosis generally manifests as uveitis, most commonly in the anterior chamber. Uveitis is a potentially vision-threatening inflammatory disease involving both the uveal tract and adjacent structures.11 In a review of records for 2,619 patients with uveitis, 59.9% had anterior disease, of whom 2.1% were diagnosed with sarcoidosis.11
While the etiology of sarcoidosis continues to be studied,7 the prevailing theory is that, in genetically predisposed individuals, sarcoidosis is a cell-mediated immune response to as-yet unknown antigen triggers that leads to granuloma formation.3,6,7
CD4+ activated T-cells stimulate the immune reaction against an antigen, producing cytokines that activate immune cells (eg, B cells, macrophages, monocytes, and neutrophils).2 Immune cells accumulate and aggregate at antigen sites in an exaggerated response, resulting in the formation of granulomas (see Figure 2).7,12,13
Infectious agents have long been investigated as possible causative agents in sarcoidosis, with Mycobacterium species most frequently identified.5 Additional possibilities include Propionibacterium acnes (found predominantly in skin lesions) and herpesviruses, although viruses are not known to cause epitheliod granulomas.14
Environmental triggers have also been explored. One large study found a possible association between exposure to insecticides, agricultural environments, and microbial bioaerosols and sarcoidosis.15
The difficulty of pinpointing a single etiology for sarcoidosis—with its varying clinical manifestations, severity, and disease course—suggests that sarcoidosis may be a spectrum of disorders caused by the interaction of genetic, immunologic, infectious, and environmental factors.14
Next page: Diagnosis of sarcoidosis >>
Diagnosis
The diagnosis of sarcoidosis is based on clinical and radiologic features, histologic evidence of noncaseating granulomas, and exclusion of other possible causes of granulomas.2,12 In addition, when ocular sarcoidosis is suspected, other possible causes of uveitis must be excluded.
In an effort to address these challenges, the International Workshop on Ocular Sarcoidosis (IWOS) developed a standardized approach to diagnosis.9 The group first identified seven intraocular signs of ocular sarcoidosis and then five laboratory or imaging tests that are of value in making the diagnosis in patients with these signs. Last, they established four levels of certainty for the diagnosis of ocular sarcoidosis, based on these signs, tests, and biopsy results, if available (see Table 2).
Treatment
Anterior uveitis in sarcoidosis is usually treated initially with a topical corticosteroid (eg, prednisolone or difluprednate drops), particularly if the patient’s symptoms are mild. In more severe cases (eg, posterior or bilateral uveitis) or when topical corticosteroids are ineffective, systemic (oral) corticosteroids (eg, prednisone) may be initiated. Topical therapy can also be added to an oral regimen as a means of decreasing the oral dosage and thereby reducing the adverse effects of systemic corticosteroids. When the patient’s disease is refractory to corticosteroids or there are concerns about long-term adverse effects, chronic cases may be treated with immunosuppressive agents (eg, methotrexate, azathioprine, mycophenolate mofetil). Finally, refractory cases of ocular sarcoidosis may be treated with anti–tumor necrosis factor α (TNF-α) biologic agents such as infliximab and adalimumab.10,17
Continue for case patient outcome >>
OUTCOME FOR THE CASE PATIENT
Histologic evaluation of tissue from the lacrimal gland, parotid gland, and sinus cavity revealed inflammatory noncaseating granulomas, strongly suggestive of sarcoidosis. Diagnosis of ocular sarcoidosis was based on the noncaseating granulomas in the lacrimal gland.9,16 Pulmonary sarcoidosis was also diagnosed, based on the presence of hilar and mediastinal lymphadenopathy.7
The mass in the patient’s lacrimal gland was surgically removed. She was treated with a combination of topical and oral corticosteroids tapered over two weeks, which induced remission of her ocular disease. The patient will be seen annually by an ophthalmologic specialist and was advised to contact her clinician immediately if acute ocular symptoms recurred.10,17
The patient’s persistent cough was determined to be secondary to acute bronchitis, rather than to her pulmonary sarcoidosis, which required no treatment. She received a short course of antibiotics and antitussives for her bronchitis. Systemic corticosteroid treatment of her ocular sarcoidosis also had the benefit of decreasing the size of her pulmonary nodules. She will be followed with annual CT and chest x-rays to monitor the status of her hilar and mediastinal lymphadenopathy and the nodules.3 Periodic pulmonary function testing will also be performed.7
Continue for conclusion >>
CONCLUSION
The elusive nature of the diagnosis of sarcoidosis is well documented in the medical literature. In this case, histologic evaluation of biopsied tissue, correlated with clinical symptoms and radiographic findings, were essential in making the diagnosis.
Primary care providers may be the first to evaluate patients with ocular sarcoidosis and will oversee long-term management. Patients who present with symptoms of eye pain, visual disturbances, abnormal inflammatory ocular features, or swollen lacrimal glands should be referred to an ophthalmologic specialist for further evaluation.
REFERENCES
1. Jennette JC, Falk RJ, Bacon PA, et al. 2012 Revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013;65(1):1-11.
2. Culver DA. Sarcoidosis. Immunol Allergy Clin North Am. 2012;32(4):487-511.
3. Spagnolo P, Luppi F, Roversi P, et al. Sarcoidosis: challenging diagnostic aspects of an old disease. Am J Med. 2012;125(2):118-125.
4. Dempsey OJ, Peterson EW, Kerr KM, Denison AR. Sarcoidosis. BMJ. 2009;339:620-625.
5. Brownell I, Ramirez-Valle F, Sanchez M, Prystowsky S. Evidence for mycobacteria in sarcoidosis. Am J Respir Cell Mol Biol. 2011;45(5):899-905.
6. Iannuzzi MC, Fontana JR. Sarcoidosis: clinical presentation, immunopathogenesis, and therapeutics. JAMA. 2011;305(4):391-399.
7. Baughman MD, Culver DA, Judson MA. A concise review of pulmonary sarcoidosis. Am J Respir Crit Care Med. 2011;183(5):573-581.
8. Koyama T, Ueda H, Togashi K, et al. Radiologic manifestations of sarcoidosis in various organs. Radiographics. 2004;24(1):87-104.
9. Herbort CP, Rao NA, Mochizuki M; for the Scientific Committee of First International Workshop on Ocular Sarcoidosis. International criteria for the diagnosis of ocular sarcoidosis: results of the first International Workshop on Ocular Sarcoidosis (IWOS). Ocul Immunol Inflamm. 2009; 17(3):160-169.
10. Jamilloux Y, Kodjikian L, Broussolle C, Seve P. Sarcoidosis and uveitis. Autoimmun Rev. 2014;13(8):840-849.
11. Barisani-Asenbauer T, Maca SM, Mejdoubi L, et al. Uveitis—a rare disease often associated with systemic diseases and infections—a systematic review of 2619 patients. Orphanet J Rare Dis. 2012;7:57.
12. Iannuzzi MC, Rybicki BA, Teirstein AS. Sarcoidosis. New Engl J Med. 2007;357(21):2153-2165.
13. Fontenot A, King T. Pathogenesis of sarcoidosis. www.uptodate.com/contents/pathogenesis-of-sarcoidosis?source=search_result&search=Pathogenesis+of+sarcoidosis&selectedTitle=1%7E150. Accessed February 17, 2015.
14. Saidha S, Sotirchos ES, Eckstein C. Etiology of sarcoidosis: does infection play a role? Yale J Biol Med. 2012;85(1):133-141.
15. Newman LS, Rose CS, Bresnitz EA, et al; for the ACCESS Research Group. A case control etiologic study of sarcoidosis. Environmental and occupational risk factors. Am J Respir Crit Care Med. 2004;170:1324-1330.
16. Kawaguchi T, Hanada A, Horie S, et al. Evaluation of characteristic ocular signs and systemic investigations in ocular sarcoidosis patients. Jpn J Opthalmol. 2007;51(2):121-126.
17. Bodaghi B, Touitou V, Fardeau C, et al. Ocular sarcoidosis. Presse Med. 2012;41(6 Pt 2):e349-e354.
A 64-year-old woman presented to the clinic with a two-to-three-week history of significant pain, swelling, and excessive tearing of the left eye. The patient had a persistent cough but denied wheezing or shortness of breath.
Medical history was remarkable for uveitis, severe recurrent sinusitis, and allergic rhinitis. The patient reported that she had been exposed to benzene and burning paint fumes about 10 years ago but had no known symptoms or problems at the time.
Vital signs included a temperature of 97.0°F; respiratory rate, 18 breaths/min; pulse, 100 beats/min; and blood pressure, 144/80 mm Hg. Her height was 65 in; weight, 122 lb; and O2 saturation, 100% on room air.
Physical examination revealed a left palpebral lacrimal mass with an enlarged lacrimal gland. The left lacrimal gland and conjunctiva were mildly erythematous, with a cobblestone appearance. The right eye was stable, with no significant inflammation. Pupils were equal, round, and reactive to light and accommodation. Extraocular movements were intact. Nasal turbinates were swollen and mildly erythematous. Oropharynx was stable and tonsils absent. Left parotid gland was slightly swollen and tender.
The neck was supple with no jugular venous distension. Palpable cervical and supraclavicular lymphadenopathy, measuring approximately 1.5 x 1.5 cm bilaterally, was present. The lungs were clear to auscultation and percussion. The heart rate and rhythm were regular, with normal S1 and S2 sounds. The abdomen was soft, nontender, and without hepatosplenomegaly. Extremities were stable, with no rashes, lesions, or cutaneous skin nodules.
The patient was referred to a specialist for a complete ophthalmologic examination and further work-up. This included a complete blood count, comprehensive metabolic panel, tissue biopsies of the affected lacrimal gland and parotid gland, CT, and x-rays; results are shown in Table 1. In addition, the patient’s persistent nasosinus congestion was determined, by otolaryngologic consultation, to be the result of a deviated septum, for which she underwent endoscopic nasal septal repair with tissue biopsy.
The lacrimal gland biopsy led to a diagnosis of chronic noncaseating granulomatous dacryoadenitis, with an extensive area of necrosis. Significant findings included histiocytes and discrete nodules in the gland. Biopsies of the parotid gland and nasal tissue also identified noncaseating granulomas.
The patient’s test results suggested several possible diagnoses, including
• Granulomatosis with polyangiitis
• Tuberculosis (TB) or similar pulmonary infectious disease
• Sarcoidosis (ocular and/or pulmonary)
Continue for differential diagnosis >>
DISCUSSION
Differential diagnosis
Granulomatosis with polyangiitis. GPA, also known as Wegener granulomatosis, is characterized by necrotizing granulomatous inflammation with necrotizing vasculitis, usually of small and medium vessels; ocular involvement is frequent.1 Ocular granulomas of GPA can be mistaken for those caused by other diseases, such as mycobacterial or syphilitic infection or idiopathic uveitis.2
Tuberculosis. Common symptoms of TB include fever, cough, dyspnea, weight loss, malaise, and fatigue. Granulomas are typically necrotizing but are occasionally nonnecrotizing.3 TB can manifest with hilar and diffuse lymphadenopathy,4 which the patient’s chest imaging revealed (see Figure 1). Granulomas produced by Mycobacterium tuberculosis and atypical mycobacteria are similar histopathologically to sarcoidosis granulomas, complicating the diagnostic process.5
Next page: Sarcoidosis >>
Sarcoidosis. Sarcoidosis is a multisystem inflammatory disease characterized by noncaseating epithelioid granulomas in affected organs.6 More than 90% of patients with sarcoidosis present with pulmonary symptoms, including shortness of breath, cough, and pleuritic chest pain.6-8 Ocular manifestations, such as uveitis, iritis, or conjunctivitis, are less common, developing in 30% to 60% of patients.2,9,10 In addition, rashes, lesions, or cutaneous skin nodules, including erythema nodosum and lupus pernio, are seen in 25% to 35% of patients.2,6
In up to two-thirds of patients, sarcoidosis resolves spontaneously2; in others, it may become chronic and progressive.4 Patients may have few or no symptoms; some require no treatment, while others may be severely affected by the disease.
Ocular involvement in sarcoidosis generally manifests as uveitis, most commonly in the anterior chamber. Uveitis is a potentially vision-threatening inflammatory disease involving both the uveal tract and adjacent structures.11 In a review of records for 2,619 patients with uveitis, 59.9% had anterior disease, of whom 2.1% were diagnosed with sarcoidosis.11
While the etiology of sarcoidosis continues to be studied,7 the prevailing theory is that, in genetically predisposed individuals, sarcoidosis is a cell-mediated immune response to as-yet unknown antigen triggers that leads to granuloma formation.3,6,7
CD4+ activated T-cells stimulate the immune reaction against an antigen, producing cytokines that activate immune cells (eg, B cells, macrophages, monocytes, and neutrophils).2 Immune cells accumulate and aggregate at antigen sites in an exaggerated response, resulting in the formation of granulomas (see Figure 2).7,12,13
Infectious agents have long been investigated as possible causative agents in sarcoidosis, with Mycobacterium species most frequently identified.5 Additional possibilities include Propionibacterium acnes (found predominantly in skin lesions) and herpesviruses, although viruses are not known to cause epitheliod granulomas.14
Environmental triggers have also been explored. One large study found a possible association between exposure to insecticides, agricultural environments, and microbial bioaerosols and sarcoidosis.15
The difficulty of pinpointing a single etiology for sarcoidosis—with its varying clinical manifestations, severity, and disease course—suggests that sarcoidosis may be a spectrum of disorders caused by the interaction of genetic, immunologic, infectious, and environmental factors.14
Next page: Diagnosis of sarcoidosis >>
Diagnosis
The diagnosis of sarcoidosis is based on clinical and radiologic features, histologic evidence of noncaseating granulomas, and exclusion of other possible causes of granulomas.2,12 In addition, when ocular sarcoidosis is suspected, other possible causes of uveitis must be excluded.
In an effort to address these challenges, the International Workshop on Ocular Sarcoidosis (IWOS) developed a standardized approach to diagnosis.9 The group first identified seven intraocular signs of ocular sarcoidosis and then five laboratory or imaging tests that are of value in making the diagnosis in patients with these signs. Last, they established four levels of certainty for the diagnosis of ocular sarcoidosis, based on these signs, tests, and biopsy results, if available (see Table 2).
Treatment
Anterior uveitis in sarcoidosis is usually treated initially with a topical corticosteroid (eg, prednisolone or difluprednate drops), particularly if the patient’s symptoms are mild. In more severe cases (eg, posterior or bilateral uveitis) or when topical corticosteroids are ineffective, systemic (oral) corticosteroids (eg, prednisone) may be initiated. Topical therapy can also be added to an oral regimen as a means of decreasing the oral dosage and thereby reducing the adverse effects of systemic corticosteroids. When the patient’s disease is refractory to corticosteroids or there are concerns about long-term adverse effects, chronic cases may be treated with immunosuppressive agents (eg, methotrexate, azathioprine, mycophenolate mofetil). Finally, refractory cases of ocular sarcoidosis may be treated with anti–tumor necrosis factor α (TNF-α) biologic agents such as infliximab and adalimumab.10,17
Continue for case patient outcome >>
OUTCOME FOR THE CASE PATIENT
Histologic evaluation of tissue from the lacrimal gland, parotid gland, and sinus cavity revealed inflammatory noncaseating granulomas, strongly suggestive of sarcoidosis. Diagnosis of ocular sarcoidosis was based on the noncaseating granulomas in the lacrimal gland.9,16 Pulmonary sarcoidosis was also diagnosed, based on the presence of hilar and mediastinal lymphadenopathy.7
The mass in the patient’s lacrimal gland was surgically removed. She was treated with a combination of topical and oral corticosteroids tapered over two weeks, which induced remission of her ocular disease. The patient will be seen annually by an ophthalmologic specialist and was advised to contact her clinician immediately if acute ocular symptoms recurred.10,17
The patient’s persistent cough was determined to be secondary to acute bronchitis, rather than to her pulmonary sarcoidosis, which required no treatment. She received a short course of antibiotics and antitussives for her bronchitis. Systemic corticosteroid treatment of her ocular sarcoidosis also had the benefit of decreasing the size of her pulmonary nodules. She will be followed with annual CT and chest x-rays to monitor the status of her hilar and mediastinal lymphadenopathy and the nodules.3 Periodic pulmonary function testing will also be performed.7
Continue for conclusion >>
CONCLUSION
The elusive nature of the diagnosis of sarcoidosis is well documented in the medical literature. In this case, histologic evaluation of biopsied tissue, correlated with clinical symptoms and radiographic findings, were essential in making the diagnosis.
Primary care providers may be the first to evaluate patients with ocular sarcoidosis and will oversee long-term management. Patients who present with symptoms of eye pain, visual disturbances, abnormal inflammatory ocular features, or swollen lacrimal glands should be referred to an ophthalmologic specialist for further evaluation.
REFERENCES
1. Jennette JC, Falk RJ, Bacon PA, et al. 2012 Revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013;65(1):1-11.
2. Culver DA. Sarcoidosis. Immunol Allergy Clin North Am. 2012;32(4):487-511.
3. Spagnolo P, Luppi F, Roversi P, et al. Sarcoidosis: challenging diagnostic aspects of an old disease. Am J Med. 2012;125(2):118-125.
4. Dempsey OJ, Peterson EW, Kerr KM, Denison AR. Sarcoidosis. BMJ. 2009;339:620-625.
5. Brownell I, Ramirez-Valle F, Sanchez M, Prystowsky S. Evidence for mycobacteria in sarcoidosis. Am J Respir Cell Mol Biol. 2011;45(5):899-905.
6. Iannuzzi MC, Fontana JR. Sarcoidosis: clinical presentation, immunopathogenesis, and therapeutics. JAMA. 2011;305(4):391-399.
7. Baughman MD, Culver DA, Judson MA. A concise review of pulmonary sarcoidosis. Am J Respir Crit Care Med. 2011;183(5):573-581.
8. Koyama T, Ueda H, Togashi K, et al. Radiologic manifestations of sarcoidosis in various organs. Radiographics. 2004;24(1):87-104.
9. Herbort CP, Rao NA, Mochizuki M; for the Scientific Committee of First International Workshop on Ocular Sarcoidosis. International criteria for the diagnosis of ocular sarcoidosis: results of the first International Workshop on Ocular Sarcoidosis (IWOS). Ocul Immunol Inflamm. 2009; 17(3):160-169.
10. Jamilloux Y, Kodjikian L, Broussolle C, Seve P. Sarcoidosis and uveitis. Autoimmun Rev. 2014;13(8):840-849.
11. Barisani-Asenbauer T, Maca SM, Mejdoubi L, et al. Uveitis—a rare disease often associated with systemic diseases and infections—a systematic review of 2619 patients. Orphanet J Rare Dis. 2012;7:57.
12. Iannuzzi MC, Rybicki BA, Teirstein AS. Sarcoidosis. New Engl J Med. 2007;357(21):2153-2165.
13. Fontenot A, King T. Pathogenesis of sarcoidosis. www.uptodate.com/contents/pathogenesis-of-sarcoidosis?source=search_result&search=Pathogenesis+of+sarcoidosis&selectedTitle=1%7E150. Accessed February 17, 2015.
14. Saidha S, Sotirchos ES, Eckstein C. Etiology of sarcoidosis: does infection play a role? Yale J Biol Med. 2012;85(1):133-141.
15. Newman LS, Rose CS, Bresnitz EA, et al; for the ACCESS Research Group. A case control etiologic study of sarcoidosis. Environmental and occupational risk factors. Am J Respir Crit Care Med. 2004;170:1324-1330.
16. Kawaguchi T, Hanada A, Horie S, et al. Evaluation of characteristic ocular signs and systemic investigations in ocular sarcoidosis patients. Jpn J Opthalmol. 2007;51(2):121-126.
17. Bodaghi B, Touitou V, Fardeau C, et al. Ocular sarcoidosis. Presse Med. 2012;41(6 Pt 2):e349-e354.
75-Year-Old Woman With Elevated Liver Enzymes
A 75-year-old woman, Gladys, was brought to the psychiatric clinic in a manic state by her concerned sister. The patient was disheveled, dehydrated, and having difficulty expressing her thoughts. Vital signs included a blood pressure of 200/94 mm Hg; pulse, 88 beats/min; temperature, 98.4°F; and respiratory rate, 20 breaths/min. Psychiatric history included a diagnosis of schizoaffective disorder with inconsistent adherence to treatment regimens, particularly mood stabilizers; and attention-deficit/hyperactivity disorder, for which she took methylphenidate regularly. Medical history was significant for asthma, osteoporosis, hypertension, type 2 diabetes, and hypothyroidism.
Gladys tended to become involved in personal relationships that adversely affected her mental health. This, in fact, had just happened: A “friend” had taken advantage of her kindness and then abruptly moved away, triggering the patient’s current decompensation. She was referred for admission for psychiatric evaluation and treatment.
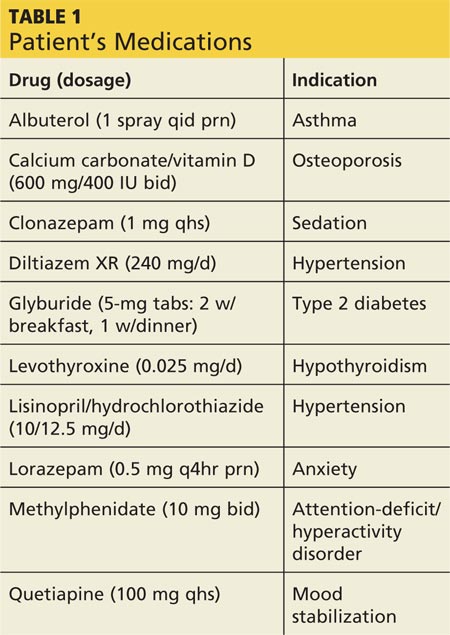
During the three-week hospitalization, Gladys was diagnosed with bipolar I disorder. She agreed to take mood-stabilizing medication primarily to alleviate her insomnia during manic episodes. She was discharged on a multidrug regimen for her coexisting conditions (see Table 1). Of note, her blood pressure at discharge was 148/66 mm Hg.
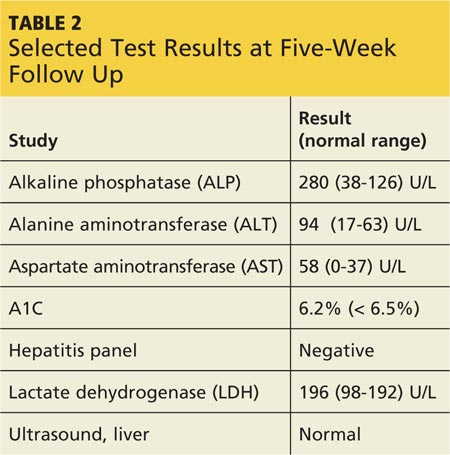
At outpatient follow-up five days later, the patient reported feeling better and stronger. However, five weeks after discharge, Gladys returned with complaints of tiredness during the day (though sleeping well at night), severe dry mouth, aching joints, and poor appetite. Her blood pressure was 100/50 mm Hg. She denied abdominal pain or change in the color of her urine or stool. She also denied use of alcohol, illicit drugs, or OTC medications. Laboratory results revealed elevated levels of several liver enzymes (see Table 2), all of which had been normal when she was admitted to the hospital two months earlier.
Continue for discussion >>
DISCUSSION
Elevations in alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels may result from a variety of factors. Mild elevations are commonly caused by alcohol consumption, hemochromatosis, medications, nonalcoholic fatty liver disease, and viral hepatitis (with which elevations may range from mild to marked).1 Moderate to marked elevations of ALT and AST are commonly seen with acute biliary obstruction, alcoholic hepatitis, toxic injury, and ischemic injury.2
Abnormal liver enzyme levels are common with use of psychotropic drugs, such as antipsychotics and mood stabilizers.3 In a systematic review that examined the effects of antipsychotics on liver function tests, a median 4% of patients experienced elevated ALT, AST, or gamma-glutamyl transferase (GGT) levels (defined as more than triple the normal level) or alkaline phosphatase (ALP) level (defined as more than twice the normal level).3 Of the studies reviewed, five noted an interval of one to six weeks between initiation of antipsychotic drugs and detection of liver function test abnormalities. None of the included studies reported severe or fatal hepatic injury.
For the atypical antipsychotic quetiapine, elevations in ALT and AST occurred in about 5% and 3% of patients, respectively, in clinical trials of the drug as monotherapy for schizophrenia or bipolar mania.4 These elevations were usually transient, occurring within the first three weeks of treatment initiation and subsiding with continued treatment.
There are rare published reports, however, of serious and even fatal hepatotoxicity induced by quetiapine. One 59-year-old woman developed fulminant hepatic failure (FHF) six weeks after she began taking quetiapine in addition to carbidopa/levodopa for Parkinson disease. She reported nausea, vomiting, poor appetite, and abdominal pain and required a six-week hospitalization, with multidrug treatment that continued after discharge. Liver biopsy identified acute hepatitis with confluent bridging necrosis, a sign that the liver injury was drug-induced. The authors concluded that, because drug-induced hepatotoxicity is the most common cause of FHF in many parts of the world, clinicians should evaluate a patient’s medications for a potential cause.5
In another case report, elevated liver enzymes were identified one month after a 58-year-old woman taking several other medications began treatment with quetiapine (100 mg/d). She developed liver failure and died after a three-week hospitalization. The authors concluded that liver failure was caused by an idiosyncratic reaction to a relatively low dose of quetiapine. This case supports the advisability of close monitoring of liver enzyme levels during quetiapine treatment.6
Naharci et al reported a case of a 77-year-old woman treated with quetiapine (12.5 mg bid for nine days). She developed acute hepatic failure leading to multi-organ system failure and died eight days later. Liver failure was attributed to an idiosyncratic reaction to low-dose quetiapine. The authors concluded that liver function monitoring is essential with quetiapine administration, especially in elderly or fragile patients.7
The initial recommended dosage of quetiapine for elderly patients (defined as age 65 or older) is 50 mg/d, with the dose increased in increments of 50 mg/d, based on clinical response and tolerability. In clinical trials, the mean plasma clearance of quetiapine was reduced by 30% to 50% in the elderly, so dosing adjustments may be necessary in this age-group.4 Gareri et al recommended that atypical antipsychotics be prescribed for elderly patients for the shortest necessary duration and at the lowest effective dose.8
For hepatically impaired patients, recommended initial dosing is 25 mg/d, with increases of 25 to 50 mg/d until an effective and tolerable dose is reached.4 Further, because quetiapine is primarily metabolized via the cytochrome P450 liver enzymes CYP3A4 and CYP2D6,9 when the clinician prescribes a potent CYP3A4 inhibitor (eg, ketoconazole) to a patient taking quetiapine, the quetiapine dosage needs to be reduced. Conversely, when prescribing a CYP3A4 inducer (eg, phenytoin), the quetiapine dosage should be adjusted upward.4
Even when an apparently well-tolerated, effective quetiapine dosage has been reached, clinicians and patients should remain alert to the warning signs of potentially serious events. Adverse effects of atypical antipsychotics, including quetiapine, were summarized by Gareri et al and rated on a scale ranging from no effect to severe effect.8 The most severe adverse effects for quetiapine were hypotension and prolonged QTc interval. Weight gain was identified as a moderate effect, and sedation, gastrointestinal problems (nausea, vomiting, and constipation), and anticholinergic effects as mild. Some effects—tardive dyskinesia, seizures, and hepatic—were deemed “uncertain”; this rating suggests the need for careful monitoring of patients (who should be informed of signs and symptoms that should be reported to the clinician).8
Atasoy et al reviewed the records of 110 patients to assess the effect of atypical antipsychotics on liver function tests. The patients’ records included both baseline liver function tests and repeat testing at six months. Forty-eight patients received quetiapine; 33 patients, olanzapine; and 29 patients, risperidone. Liver enzymes were elevated in 27.1% of the quetiapine group, 30.3% of the olanzapine group, and 27.6% of the risperidone group. In two patients taking olanzapine, liver enzyme levels reached three to four times normal but returned to normal when treatment was stopped. The authors concluded that baseline liver enzyme studies should be done prior to initiation of treatment with atypical antipsychotics, as well as periodically thereafter, especially for patients with preexisting hepatic disorders, those being treated with other potentially hepatotoxic drugs, or those who exhibit signs or symptoms of hepatic impairment.10
Continue for patient outcome >>
PATIENT OUTCOME
Gladys’s ALT and AST levels were mildly elevated. One of the more common causes for this pattern is medication. In addition, her ALP level of more than twice the upper limit of normal further pointed to a viral, alcohol-related, or drug toxicity cause. Since her hepatitis panel was negative and she did not use alcohol, it was determined that elevated liver enzymes were related to the recent addition of quetiapine, which was discontinued. In addition, in light of Gladys’s hypotension (which is also a potential adverse effect of quetiapine8), her dose of lisinopril/hydrochlorothiazide was decreased by half.
One week later, liver enzyme levels were returning to normal. Gladys, however, began having more difficulty sleeping and more trouble remaining focused and getting things done, despite taking methylphenidate. In place of quetiapine, risperidone (0.5 mg at bedtime) was initiated. At first, Gladys was concerned with her continuing dry mouth symptoms, but when she skipped doses of risperidone, she noticed that she functioned less well. Further, her liver enzyme levels were being closely monitored and were normal. With this reassurance, Gladys remained adherent to risperidone for mood stabilization.
CONCLUSION
Atypical antipsychotic drugs such as quetiapine can cause elevated liver enzyme levels, especially in the elderly, patients with hepatic impairment, or patients on polypharmacotherapy. Rarely, quetiapine has been reported to cause serious hepatotoxicity and even death. Patients taking these drugs should be informed of possible symptoms of liver toxicity, including fatigue, nausea, vomiting, abdominal pain, and change in color of urine or stools. Particularly in more vulnerable patients, liver enzyme levels should be monitored carefully to confirm the continued safety of antipsychotic treatment.
REFERENCES
1. Oh RC, Hustead TR. Causes and evaluation of mildly elevated liver transaminase levels. Am Fam Physician. 2011;84(9):1003-1008.
2. Giannini EG, Testa R, Savarino V. Liver enzyme elevation: a guide for clinicians. CMAJ. 2005;172(3):367-379.
3. Marwick KFM, Taylor M, Walker SW. Antipsychotics and abnormal liver function tests: Systematic review. Clin Neuropharmacol. 2012;35(5):244-253.
4. Seroquel [package insert]. Wilmington, DE: AstraZeneca Pharmaceuticals LP; 2013.
5. Al Mutairi F, Dwivedi G, Al Ameel T. Fulminant hepatic failure in association with quetiapine: A case report. J Med Case Rep. 2012;6:418.
6. El Hajj L, Sharara A, Rockey, DC. Subfulminant liver failure associated with quetiapine. Eur J Gastroenterol Hepatol. 2004;16(12):1415-1418.
7. Naharci MI, Karadurmus N, Demir O, et al. Fatal hepatotoxicity in an elderly patient receiving low-dose quetiapine. Am J Psychiatry. 2011;168(2):212-213.
8. Gareri P, Segura-Garcia C, Manfredi VG, et al. Use of atypical antipsychotics in the elderly: a clinical review. Clin Interv Aging. 2014;16(9):1363-1373.
9. Lin S, Chang Y, Moody DE, Foltz RL. A liquid chromatographic-electrospray-tandem mass spectrometric method for quanititation of quetiapine in human plasma and liver microsomes: application to a study of in vitro metabolism. J Anal Toxicol. 2004;28(6):443-446.
10. Atasoy N, Erdogan A, Yalug I, et al. A review of liver function tests during treatment with atypical antipsychotic drugs: a chart review study. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31(6):1255-1260.
A 75-year-old woman, Gladys, was brought to the psychiatric clinic in a manic state by her concerned sister. The patient was disheveled, dehydrated, and having difficulty expressing her thoughts. Vital signs included a blood pressure of 200/94 mm Hg; pulse, 88 beats/min; temperature, 98.4°F; and respiratory rate, 20 breaths/min. Psychiatric history included a diagnosis of schizoaffective disorder with inconsistent adherence to treatment regimens, particularly mood stabilizers; and attention-deficit/hyperactivity disorder, for which she took methylphenidate regularly. Medical history was significant for asthma, osteoporosis, hypertension, type 2 diabetes, and hypothyroidism.
Gladys tended to become involved in personal relationships that adversely affected her mental health. This, in fact, had just happened: A “friend” had taken advantage of her kindness and then abruptly moved away, triggering the patient’s current decompensation. She was referred for admission for psychiatric evaluation and treatment.
During the three-week hospitalization, Gladys was diagnosed with bipolar I disorder. She agreed to take mood-stabilizing medication primarily to alleviate her insomnia during manic episodes. She was discharged on a multidrug regimen for her coexisting conditions (see Table 1). Of note, her blood pressure at discharge was 148/66 mm Hg.
At outpatient follow-up five days later, the patient reported feeling better and stronger. However, five weeks after discharge, Gladys returned with complaints of tiredness during the day (though sleeping well at night), severe dry mouth, aching joints, and poor appetite. Her blood pressure was 100/50 mm Hg. She denied abdominal pain or change in the color of her urine or stool. She also denied use of alcohol, illicit drugs, or OTC medications. Laboratory results revealed elevated levels of several liver enzymes (see Table 2), all of which had been normal when she was admitted to the hospital two months earlier.
Continue for discussion >>
DISCUSSION
Elevations in alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels may result from a variety of factors. Mild elevations are commonly caused by alcohol consumption, hemochromatosis, medications, nonalcoholic fatty liver disease, and viral hepatitis (with which elevations may range from mild to marked).1 Moderate to marked elevations of ALT and AST are commonly seen with acute biliary obstruction, alcoholic hepatitis, toxic injury, and ischemic injury.2
Abnormal liver enzyme levels are common with use of psychotropic drugs, such as antipsychotics and mood stabilizers.3 In a systematic review that examined the effects of antipsychotics on liver function tests, a median 4% of patients experienced elevated ALT, AST, or gamma-glutamyl transferase (GGT) levels (defined as more than triple the normal level) or alkaline phosphatase (ALP) level (defined as more than twice the normal level).3 Of the studies reviewed, five noted an interval of one to six weeks between initiation of antipsychotic drugs and detection of liver function test abnormalities. None of the included studies reported severe or fatal hepatic injury.
For the atypical antipsychotic quetiapine, elevations in ALT and AST occurred in about 5% and 3% of patients, respectively, in clinical trials of the drug as monotherapy for schizophrenia or bipolar mania.4 These elevations were usually transient, occurring within the first three weeks of treatment initiation and subsiding with continued treatment.
There are rare published reports, however, of serious and even fatal hepatotoxicity induced by quetiapine. One 59-year-old woman developed fulminant hepatic failure (FHF) six weeks after she began taking quetiapine in addition to carbidopa/levodopa for Parkinson disease. She reported nausea, vomiting, poor appetite, and abdominal pain and required a six-week hospitalization, with multidrug treatment that continued after discharge. Liver biopsy identified acute hepatitis with confluent bridging necrosis, a sign that the liver injury was drug-induced. The authors concluded that, because drug-induced hepatotoxicity is the most common cause of FHF in many parts of the world, clinicians should evaluate a patient’s medications for a potential cause.5
In another case report, elevated liver enzymes were identified one month after a 58-year-old woman taking several other medications began treatment with quetiapine (100 mg/d). She developed liver failure and died after a three-week hospitalization. The authors concluded that liver failure was caused by an idiosyncratic reaction to a relatively low dose of quetiapine. This case supports the advisability of close monitoring of liver enzyme levels during quetiapine treatment.6
Naharci et al reported a case of a 77-year-old woman treated with quetiapine (12.5 mg bid for nine days). She developed acute hepatic failure leading to multi-organ system failure and died eight days later. Liver failure was attributed to an idiosyncratic reaction to low-dose quetiapine. The authors concluded that liver function monitoring is essential with quetiapine administration, especially in elderly or fragile patients.7
The initial recommended dosage of quetiapine for elderly patients (defined as age 65 or older) is 50 mg/d, with the dose increased in increments of 50 mg/d, based on clinical response and tolerability. In clinical trials, the mean plasma clearance of quetiapine was reduced by 30% to 50% in the elderly, so dosing adjustments may be necessary in this age-group.4 Gareri et al recommended that atypical antipsychotics be prescribed for elderly patients for the shortest necessary duration and at the lowest effective dose.8
For hepatically impaired patients, recommended initial dosing is 25 mg/d, with increases of 25 to 50 mg/d until an effective and tolerable dose is reached.4 Further, because quetiapine is primarily metabolized via the cytochrome P450 liver enzymes CYP3A4 and CYP2D6,9 when the clinician prescribes a potent CYP3A4 inhibitor (eg, ketoconazole) to a patient taking quetiapine, the quetiapine dosage needs to be reduced. Conversely, when prescribing a CYP3A4 inducer (eg, phenytoin), the quetiapine dosage should be adjusted upward.4
Even when an apparently well-tolerated, effective quetiapine dosage has been reached, clinicians and patients should remain alert to the warning signs of potentially serious events. Adverse effects of atypical antipsychotics, including quetiapine, were summarized by Gareri et al and rated on a scale ranging from no effect to severe effect.8 The most severe adverse effects for quetiapine were hypotension and prolonged QTc interval. Weight gain was identified as a moderate effect, and sedation, gastrointestinal problems (nausea, vomiting, and constipation), and anticholinergic effects as mild. Some effects—tardive dyskinesia, seizures, and hepatic—were deemed “uncertain”; this rating suggests the need for careful monitoring of patients (who should be informed of signs and symptoms that should be reported to the clinician).8
Atasoy et al reviewed the records of 110 patients to assess the effect of atypical antipsychotics on liver function tests. The patients’ records included both baseline liver function tests and repeat testing at six months. Forty-eight patients received quetiapine; 33 patients, olanzapine; and 29 patients, risperidone. Liver enzymes were elevated in 27.1% of the quetiapine group, 30.3% of the olanzapine group, and 27.6% of the risperidone group. In two patients taking olanzapine, liver enzyme levels reached three to four times normal but returned to normal when treatment was stopped. The authors concluded that baseline liver enzyme studies should be done prior to initiation of treatment with atypical antipsychotics, as well as periodically thereafter, especially for patients with preexisting hepatic disorders, those being treated with other potentially hepatotoxic drugs, or those who exhibit signs or symptoms of hepatic impairment.10
Continue for patient outcome >>
PATIENT OUTCOME
Gladys’s ALT and AST levels were mildly elevated. One of the more common causes for this pattern is medication. In addition, her ALP level of more than twice the upper limit of normal further pointed to a viral, alcohol-related, or drug toxicity cause. Since her hepatitis panel was negative and she did not use alcohol, it was determined that elevated liver enzymes were related to the recent addition of quetiapine, which was discontinued. In addition, in light of Gladys’s hypotension (which is also a potential adverse effect of quetiapine8), her dose of lisinopril/hydrochlorothiazide was decreased by half.
One week later, liver enzyme levels were returning to normal. Gladys, however, began having more difficulty sleeping and more trouble remaining focused and getting things done, despite taking methylphenidate. In place of quetiapine, risperidone (0.5 mg at bedtime) was initiated. At first, Gladys was concerned with her continuing dry mouth symptoms, but when she skipped doses of risperidone, she noticed that she functioned less well. Further, her liver enzyme levels were being closely monitored and were normal. With this reassurance, Gladys remained adherent to risperidone for mood stabilization.
CONCLUSION
Atypical antipsychotic drugs such as quetiapine can cause elevated liver enzyme levels, especially in the elderly, patients with hepatic impairment, or patients on polypharmacotherapy. Rarely, quetiapine has been reported to cause serious hepatotoxicity and even death. Patients taking these drugs should be informed of possible symptoms of liver toxicity, including fatigue, nausea, vomiting, abdominal pain, and change in color of urine or stools. Particularly in more vulnerable patients, liver enzyme levels should be monitored carefully to confirm the continued safety of antipsychotic treatment.
REFERENCES
1. Oh RC, Hustead TR. Causes and evaluation of mildly elevated liver transaminase levels. Am Fam Physician. 2011;84(9):1003-1008.
2. Giannini EG, Testa R, Savarino V. Liver enzyme elevation: a guide for clinicians. CMAJ. 2005;172(3):367-379.
3. Marwick KFM, Taylor M, Walker SW. Antipsychotics and abnormal liver function tests: Systematic review. Clin Neuropharmacol. 2012;35(5):244-253.
4. Seroquel [package insert]. Wilmington, DE: AstraZeneca Pharmaceuticals LP; 2013.
5. Al Mutairi F, Dwivedi G, Al Ameel T. Fulminant hepatic failure in association with quetiapine: A case report. J Med Case Rep. 2012;6:418.
6. El Hajj L, Sharara A, Rockey, DC. Subfulminant liver failure associated with quetiapine. Eur J Gastroenterol Hepatol. 2004;16(12):1415-1418.
7. Naharci MI, Karadurmus N, Demir O, et al. Fatal hepatotoxicity in an elderly patient receiving low-dose quetiapine. Am J Psychiatry. 2011;168(2):212-213.
8. Gareri P, Segura-Garcia C, Manfredi VG, et al. Use of atypical antipsychotics in the elderly: a clinical review. Clin Interv Aging. 2014;16(9):1363-1373.
9. Lin S, Chang Y, Moody DE, Foltz RL. A liquid chromatographic-electrospray-tandem mass spectrometric method for quanititation of quetiapine in human plasma and liver microsomes: application to a study of in vitro metabolism. J Anal Toxicol. 2004;28(6):443-446.
10. Atasoy N, Erdogan A, Yalug I, et al. A review of liver function tests during treatment with atypical antipsychotic drugs: a chart review study. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31(6):1255-1260.
A 75-year-old woman, Gladys, was brought to the psychiatric clinic in a manic state by her concerned sister. The patient was disheveled, dehydrated, and having difficulty expressing her thoughts. Vital signs included a blood pressure of 200/94 mm Hg; pulse, 88 beats/min; temperature, 98.4°F; and respiratory rate, 20 breaths/min. Psychiatric history included a diagnosis of schizoaffective disorder with inconsistent adherence to treatment regimens, particularly mood stabilizers; and attention-deficit/hyperactivity disorder, for which she took methylphenidate regularly. Medical history was significant for asthma, osteoporosis, hypertension, type 2 diabetes, and hypothyroidism.
Gladys tended to become involved in personal relationships that adversely affected her mental health. This, in fact, had just happened: A “friend” had taken advantage of her kindness and then abruptly moved away, triggering the patient’s current decompensation. She was referred for admission for psychiatric evaluation and treatment.
During the three-week hospitalization, Gladys was diagnosed with bipolar I disorder. She agreed to take mood-stabilizing medication primarily to alleviate her insomnia during manic episodes. She was discharged on a multidrug regimen for her coexisting conditions (see Table 1). Of note, her blood pressure at discharge was 148/66 mm Hg.
At outpatient follow-up five days later, the patient reported feeling better and stronger. However, five weeks after discharge, Gladys returned with complaints of tiredness during the day (though sleeping well at night), severe dry mouth, aching joints, and poor appetite. Her blood pressure was 100/50 mm Hg. She denied abdominal pain or change in the color of her urine or stool. She also denied use of alcohol, illicit drugs, or OTC medications. Laboratory results revealed elevated levels of several liver enzymes (see Table 2), all of which had been normal when she was admitted to the hospital two months earlier.
Continue for discussion >>
DISCUSSION
Elevations in alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels may result from a variety of factors. Mild elevations are commonly caused by alcohol consumption, hemochromatosis, medications, nonalcoholic fatty liver disease, and viral hepatitis (with which elevations may range from mild to marked).1 Moderate to marked elevations of ALT and AST are commonly seen with acute biliary obstruction, alcoholic hepatitis, toxic injury, and ischemic injury.2
Abnormal liver enzyme levels are common with use of psychotropic drugs, such as antipsychotics and mood stabilizers.3 In a systematic review that examined the effects of antipsychotics on liver function tests, a median 4% of patients experienced elevated ALT, AST, or gamma-glutamyl transferase (GGT) levels (defined as more than triple the normal level) or alkaline phosphatase (ALP) level (defined as more than twice the normal level).3 Of the studies reviewed, five noted an interval of one to six weeks between initiation of antipsychotic drugs and detection of liver function test abnormalities. None of the included studies reported severe or fatal hepatic injury.
For the atypical antipsychotic quetiapine, elevations in ALT and AST occurred in about 5% and 3% of patients, respectively, in clinical trials of the drug as monotherapy for schizophrenia or bipolar mania.4 These elevations were usually transient, occurring within the first three weeks of treatment initiation and subsiding with continued treatment.
There are rare published reports, however, of serious and even fatal hepatotoxicity induced by quetiapine. One 59-year-old woman developed fulminant hepatic failure (FHF) six weeks after she began taking quetiapine in addition to carbidopa/levodopa for Parkinson disease. She reported nausea, vomiting, poor appetite, and abdominal pain and required a six-week hospitalization, with multidrug treatment that continued after discharge. Liver biopsy identified acute hepatitis with confluent bridging necrosis, a sign that the liver injury was drug-induced. The authors concluded that, because drug-induced hepatotoxicity is the most common cause of FHF in many parts of the world, clinicians should evaluate a patient’s medications for a potential cause.5
In another case report, elevated liver enzymes were identified one month after a 58-year-old woman taking several other medications began treatment with quetiapine (100 mg/d). She developed liver failure and died after a three-week hospitalization. The authors concluded that liver failure was caused by an idiosyncratic reaction to a relatively low dose of quetiapine. This case supports the advisability of close monitoring of liver enzyme levels during quetiapine treatment.6
Naharci et al reported a case of a 77-year-old woman treated with quetiapine (12.5 mg bid for nine days). She developed acute hepatic failure leading to multi-organ system failure and died eight days later. Liver failure was attributed to an idiosyncratic reaction to low-dose quetiapine. The authors concluded that liver function monitoring is essential with quetiapine administration, especially in elderly or fragile patients.7
The initial recommended dosage of quetiapine for elderly patients (defined as age 65 or older) is 50 mg/d, with the dose increased in increments of 50 mg/d, based on clinical response and tolerability. In clinical trials, the mean plasma clearance of quetiapine was reduced by 30% to 50% in the elderly, so dosing adjustments may be necessary in this age-group.4 Gareri et al recommended that atypical antipsychotics be prescribed for elderly patients for the shortest necessary duration and at the lowest effective dose.8
For hepatically impaired patients, recommended initial dosing is 25 mg/d, with increases of 25 to 50 mg/d until an effective and tolerable dose is reached.4 Further, because quetiapine is primarily metabolized via the cytochrome P450 liver enzymes CYP3A4 and CYP2D6,9 when the clinician prescribes a potent CYP3A4 inhibitor (eg, ketoconazole) to a patient taking quetiapine, the quetiapine dosage needs to be reduced. Conversely, when prescribing a CYP3A4 inducer (eg, phenytoin), the quetiapine dosage should be adjusted upward.4
Even when an apparently well-tolerated, effective quetiapine dosage has been reached, clinicians and patients should remain alert to the warning signs of potentially serious events. Adverse effects of atypical antipsychotics, including quetiapine, were summarized by Gareri et al and rated on a scale ranging from no effect to severe effect.8 The most severe adverse effects for quetiapine were hypotension and prolonged QTc interval. Weight gain was identified as a moderate effect, and sedation, gastrointestinal problems (nausea, vomiting, and constipation), and anticholinergic effects as mild. Some effects—tardive dyskinesia, seizures, and hepatic—were deemed “uncertain”; this rating suggests the need for careful monitoring of patients (who should be informed of signs and symptoms that should be reported to the clinician).8
Atasoy et al reviewed the records of 110 patients to assess the effect of atypical antipsychotics on liver function tests. The patients’ records included both baseline liver function tests and repeat testing at six months. Forty-eight patients received quetiapine; 33 patients, olanzapine; and 29 patients, risperidone. Liver enzymes were elevated in 27.1% of the quetiapine group, 30.3% of the olanzapine group, and 27.6% of the risperidone group. In two patients taking olanzapine, liver enzyme levels reached three to four times normal but returned to normal when treatment was stopped. The authors concluded that baseline liver enzyme studies should be done prior to initiation of treatment with atypical antipsychotics, as well as periodically thereafter, especially for patients with preexisting hepatic disorders, those being treated with other potentially hepatotoxic drugs, or those who exhibit signs or symptoms of hepatic impairment.10
Continue for patient outcome >>
PATIENT OUTCOME
Gladys’s ALT and AST levels were mildly elevated. One of the more common causes for this pattern is medication. In addition, her ALP level of more than twice the upper limit of normal further pointed to a viral, alcohol-related, or drug toxicity cause. Since her hepatitis panel was negative and she did not use alcohol, it was determined that elevated liver enzymes were related to the recent addition of quetiapine, which was discontinued. In addition, in light of Gladys’s hypotension (which is also a potential adverse effect of quetiapine8), her dose of lisinopril/hydrochlorothiazide was decreased by half.
One week later, liver enzyme levels were returning to normal. Gladys, however, began having more difficulty sleeping and more trouble remaining focused and getting things done, despite taking methylphenidate. In place of quetiapine, risperidone (0.5 mg at bedtime) was initiated. At first, Gladys was concerned with her continuing dry mouth symptoms, but when she skipped doses of risperidone, she noticed that she functioned less well. Further, her liver enzyme levels were being closely monitored and were normal. With this reassurance, Gladys remained adherent to risperidone for mood stabilization.
CONCLUSION
Atypical antipsychotic drugs such as quetiapine can cause elevated liver enzyme levels, especially in the elderly, patients with hepatic impairment, or patients on polypharmacotherapy. Rarely, quetiapine has been reported to cause serious hepatotoxicity and even death. Patients taking these drugs should be informed of possible symptoms of liver toxicity, including fatigue, nausea, vomiting, abdominal pain, and change in color of urine or stools. Particularly in more vulnerable patients, liver enzyme levels should be monitored carefully to confirm the continued safety of antipsychotic treatment.
REFERENCES
1. Oh RC, Hustead TR. Causes and evaluation of mildly elevated liver transaminase levels. Am Fam Physician. 2011;84(9):1003-1008.
2. Giannini EG, Testa R, Savarino V. Liver enzyme elevation: a guide for clinicians. CMAJ. 2005;172(3):367-379.
3. Marwick KFM, Taylor M, Walker SW. Antipsychotics and abnormal liver function tests: Systematic review. Clin Neuropharmacol. 2012;35(5):244-253.
4. Seroquel [package insert]. Wilmington, DE: AstraZeneca Pharmaceuticals LP; 2013.
5. Al Mutairi F, Dwivedi G, Al Ameel T. Fulminant hepatic failure in association with quetiapine: A case report. J Med Case Rep. 2012;6:418.
6. El Hajj L, Sharara A, Rockey, DC. Subfulminant liver failure associated with quetiapine. Eur J Gastroenterol Hepatol. 2004;16(12):1415-1418.
7. Naharci MI, Karadurmus N, Demir O, et al. Fatal hepatotoxicity in an elderly patient receiving low-dose quetiapine. Am J Psychiatry. 2011;168(2):212-213.
8. Gareri P, Segura-Garcia C, Manfredi VG, et al. Use of atypical antipsychotics in the elderly: a clinical review. Clin Interv Aging. 2014;16(9):1363-1373.
9. Lin S, Chang Y, Moody DE, Foltz RL. A liquid chromatographic-electrospray-tandem mass spectrometric method for quanititation of quetiapine in human plasma and liver microsomes: application to a study of in vitro metabolism. J Anal Toxicol. 2004;28(6):443-446.
10. Atasoy N, Erdogan A, Yalug I, et al. A review of liver function tests during treatment with atypical antipsychotic drugs: a chart review study. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31(6):1255-1260.
Left Arm Pain, Numbness, and Weakness
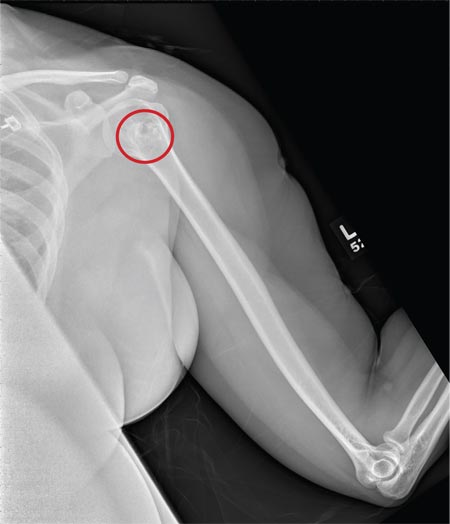
ANSWER
The radiograph shows no evidence of a fracture. However, there is a 2-cm focal sclerotic area noted within the juncture of the humeral neck and head. This finding could represent an enchondroma, a bone cyst, or a bone infarct. Additional imaging, including MRI and bone scan, is warranted, as is orthopedic evaluation. This finding is likely incidental, as the patient’s clinical exam is suggestive of a cervical radiculitis referable to the herniated disc in her neck.
ANSWER
The radiograph shows no evidence of a fracture. However, there is a 2-cm focal sclerotic area noted within the juncture of the humeral neck and head. This finding could represent an enchondroma, a bone cyst, or a bone infarct. Additional imaging, including MRI and bone scan, is warranted, as is orthopedic evaluation. This finding is likely incidental, as the patient’s clinical exam is suggestive of a cervical radiculitis referable to the herniated disc in her neck.
ANSWER
The radiograph shows no evidence of a fracture. However, there is a 2-cm focal sclerotic area noted within the juncture of the humeral neck and head. This finding could represent an enchondroma, a bone cyst, or a bone infarct. Additional imaging, including MRI and bone scan, is warranted, as is orthopedic evaluation. This finding is likely incidental, as the patient’s clinical exam is suggestive of a cervical radiculitis referable to the herniated disc in her neck.
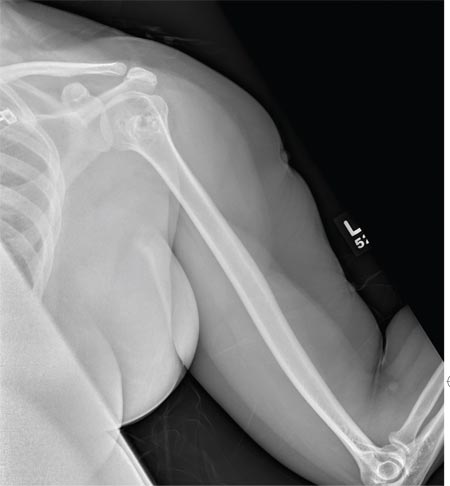
A 40-year-old woman presents to the urgent care clinic complaining of left arm pain with associated numbness and weakness. She denies any injury or trauma, adding that the pain manifested several months ago but has recently progressed. She has already undergone outpatient MRI of her neck; she was told she had some “herniated discs” and would need to see a specialist. Her medical history is significant for hypertension. On physical examination, the patient appears uncomfortable but in no obvious distress. Vital signs are normal. Tenderness is present at the left trapezius and the left shoulder. Mild weakness is present in the left arm; strength is 4/5 and grip strength, 3/5. Pulses are normal, and sensation is intact. Available medical records include a report from her recent MRI of the cervical spine. Findings include a moderate left-sided disc osteophyte at the C6-C7 level and resultant cervical stenosis. A radiograph of the left shoulder is obtained. What is your impression?
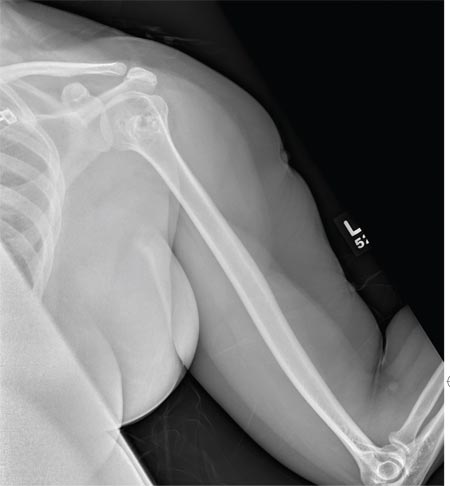
Case Studies in Toxicology: You Can’t See Dragonfly or Hear NBOMe, but They Can Still Hurt You
Case
A 24-year-old man was brought to the ED by emergency medical services (EMS) for altered mental status. The EMS crew reported they had picked up the patient at a nearby arts festival and concert series. A bystander at the event reported that the patient had taken something called “dragonfly.”
Initial assessment revealed the patient to be disoriented, with nonlinear thought patterns and an inability to follow commands. His vital signs were: blood pressure, 160/100 mm Hg; heart rate, 120 beats/minute; respiratory rate, 24 breaths/minute; and temperature, 102.2˚F. Oxygen saturation was 99% on room air. He was diaphoretic and agitated, and the nursing staff was concerned he would become aggressive and potentially violent. A quick Web search revealed that the agent the bystander mentioned was most likely Bromo-DragonFLY (BDF).
What is Bromo-DragonFLY?
In the 1960s, an American chemist named Alexander Shulgin ushered in a new era of psychedelic drug use by establishing a simple synthesis of 3,4-methylenedioxy-methamphetamine (MDMA). Following this discovery, he suggested a therapist friend use the drug therapeutically.1 Shulgin then began a process of homologation (ie, creating novel compounds by slightly altering existing ones in an organized fashion) and developed systems for rating the drug experiences and naming the drugs in shorthand, both of which are still in use. The chemical structure common to nearly all of the drugs he studied is phenylethylamine. The Figure shows the structures of several phenylethylamine derivatives that were created by adding functional groups to the phenylethylamine backbone. Although the popularity of psychedelic drugs surged during this time period, 2,5-dimethoxy-N-(2-methoxybenzyl)phenylethylamine) (NBOMe), one of a number of newly popular psychedelics, only became available in 2003.
What is known about the pharmacology of Bromo-DragonFLY and NBOMe?
The major target of psychedelic drugs is the serotonin (5-HT2) receptor, specifically the central 5-HT2A subtype. Bromo-DragonFLY is a classic example of designer pharmacology in that the it was intended to potently exert its effect at this specific receptor site.
As its name suggests, BDF adds the “wings of the fly” to the phenylethylamine backbone furanyl rings at positions 2 and 5, and a halogen (bromine) at position 4. The furanyl ring impairs enzymatic clearance of the drug,2 resulting in a duration of action of up to 3 days.3 The addition of halogens increases drug potency, but the mechanism is not clear. The psychedelic agent NBOMe results from chemical additions of methoxy groups at position 2 and 5, and the halogen moiety (iodine in this case) at position 4 of the phenyl ring of the phenylethylamine structure.4
Through the work of Shulgin, some of his colleagues, and many disparate street chemists, a vast family of substituted phenylethylamines have been synthesized and used. Shulgin’s semiautobiographical book PiHKAL: A Chemical Love Story includes his laboratory notes for the synthesis and initial test-dose experience of 179 compounds1; this does not include research done by others or any work since its publication in 1995.
Notable popular drugs chemically similar to NBOMe and BDF are mescaline (found in peyote), cathinones (“bath salts”), and MDMA (found in ecstasy) (Figure). Naturally occurring (and more complex) compounds with similar effects include ayahuasca, a plant-derived beverage consisting of Banisteriopsis caapi and either Psychotria viridis or Diplopterys cabrerana from the Brazilian rainforest (see Emerg Med. 2014;46[12]:553-556); psilocybin (“magic mushrooms”); and lysergic acid diethylamide.
How are these drugs used and what are their clinical effects?
Most phenylethylamine compounds are well absorbed across the buccal mucosa, which is why BDF and NBOMe are commonly used in liquid form or on blotter paper. Dosing guides also exist for insufflation and claim equipotent dosing for this route.5 Regardless of delivery route, given the high potency, inadvertent exposures to these drugs should be expected.
Users simply seeking to hallucinate may not be aware of the significant risks associated with these potent serotonergic agents, which include both life- and limb-threatening effects.6 The high 5-HT2A potency results both in vasoconstriction and promotion of clot formation due to the presence of 5HT2A receptors on small blood vessels and platelets, respectively. Ergotism, historically called Saint Anthony’s fire, is an example of serotonergic vasoconstriction and hallucination.7 Chronic users of substituted amphetamines can develop necrotic ulcers in distal vascular beds such as the hands and feet; these ulcers may progress to amputation despite treatment attempts with vasodilators.
In addition to the vasoconstrictive properties, there are multiple reports of serotonin toxicity (serotonin syndrome) associated with use of these designer serotonergic amphetamines. This syndrome includes severe psychomotor agitation that can lead to personal injury, along with muscle rigidity, tremor, hyperthermia, rhabdomyolysis, and seizures.8
How are patients with phenylethylamine exposures managed?
Management of a patient with a substituted phenylethylamine exposure is similar to management of those with cocaine overdose. Attention to the life-threatening clinical effects of psychomotor agitation, hyperthermia, and seizures is paramount. Appropriate supportive care includes intravenous (IV) benzodiazepines to control agitation and muscle rigidity, replacement of lost volume with crystalloids, and active cooling measures. Failure of benzodiazepines (preferably in conjunction with continuous electroencephalogram monitoring) to control rigidity may lead to the need for propofol and/or result in paralysis. Similar to patients with cocaine intoxication, some may experience ischemic chest pain, and the usual protocol of sedation, nitroglycerin, morphine, and an antiplatelet drug is appropriate.
Identification of phenylethylamines typically requires specialized laboratory testing since most will not trigger a positive result on a standard urine immunoassay. Many specialized laboratories have test catalogs on their Web sites listing under the “stimulants panel” which drugs can be identified. However, none of these assays is likely truly comprehensive, and minor alterations or substitutions to the compounds result in new analogs that may not be in the reference laboratory’s identification library.
The patient was initially restrained and given 5 mg IV diazepam, which was followed by escalating doses every 5 minutes to a total of 35 mg for effect. He had a rectal temperature of 102.5˚F and was externally cooled after sedation. After 20 minutes, he had a generalized convulsion; an additional 10 mg of IV diazepam terminated the seizure, but he remained hyperthermic at 104˚F. The patient was intubated, placed on a propofol infusion, and admitted to the intensive care unit where his temperature was carefully monitored. The following day his temperature had normalized and he was weaned from the ventilator and discharged to the floor for monitoring. On hospital day 3, he was discharged in stable condition.
Mr Waldrop is a fourth-year medical student at the State University of New York, Upstate Medical University, Syracuse. Dr Nacca is a fellow in medical toxicology, department of emergency medicine, State University of New York, Upstate Medical University, Syracuse. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine, and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Shulgin A, Shulgin A. PiHKAL: A Chemical Love Story. Berkeley, CA: Transform Press; 1995.
- Andreasen MF, Telving R, Birkler RI, Schumacher B, Johannsen M. A fatal poisoning involving bromo-dragonfly. Forensic Sci Int. 2009;183(1-3):91-96.
- Hill SL, Thomas SH. Clinical toxicology of newer recreational drugs. Clin Toxicol (Phila). 2011;49(8):705-719.
- Gentry CL, Egleton RD, Gillespie T, et al. The effect of halogenation on blood-brain barrier permeability of a novel peptide drug. Peptides. 1999;20(10):1229-1238.
- Erowid. Bromo-Dragonfly Dosage. http://www.erowid.org/chemicals/bromo_dragonfly/bromo_dragonfly_dose.shtml. Accessed January 14, 2015.
- Baumann MH, Ayestas MA Jr, Partilla JS, et al. The designer methcathinone analogs, mephedrone and methylone, are substrates for monoamine transporters in brain tissue. Neuropsychopharmacology. 2012;37(5):1192-1203.
- Walterscheid JP, Phillips GT, Lopez AE, Gonsoulin ML, Chen HH, Sanchez LA. Pathological findings in 2 cases of fatal 25I-NBOMe toxicity. Am J Forensic Med Pathol. 2014;35(1):20-25.
- Wood DM, Looker JJ, Shaikh L, et al. Delayed onset of seizures and toxicity associated with recreational use of Bromo-dragonFLY. J Med Toxicol. 2009;5(4):226-229.
Case
A 24-year-old man was brought to the ED by emergency medical services (EMS) for altered mental status. The EMS crew reported they had picked up the patient at a nearby arts festival and concert series. A bystander at the event reported that the patient had taken something called “dragonfly.”
Initial assessment revealed the patient to be disoriented, with nonlinear thought patterns and an inability to follow commands. His vital signs were: blood pressure, 160/100 mm Hg; heart rate, 120 beats/minute; respiratory rate, 24 breaths/minute; and temperature, 102.2˚F. Oxygen saturation was 99% on room air. He was diaphoretic and agitated, and the nursing staff was concerned he would become aggressive and potentially violent. A quick Web search revealed that the agent the bystander mentioned was most likely Bromo-DragonFLY (BDF).
What is Bromo-DragonFLY?
In the 1960s, an American chemist named Alexander Shulgin ushered in a new era of psychedelic drug use by establishing a simple synthesis of 3,4-methylenedioxy-methamphetamine (MDMA). Following this discovery, he suggested a therapist friend use the drug therapeutically.1 Shulgin then began a process of homologation (ie, creating novel compounds by slightly altering existing ones in an organized fashion) and developed systems for rating the drug experiences and naming the drugs in shorthand, both of which are still in use. The chemical structure common to nearly all of the drugs he studied is phenylethylamine. The Figure shows the structures of several phenylethylamine derivatives that were created by adding functional groups to the phenylethylamine backbone. Although the popularity of psychedelic drugs surged during this time period, 2,5-dimethoxy-N-(2-methoxybenzyl)phenylethylamine) (NBOMe), one of a number of newly popular psychedelics, only became available in 2003.
What is known about the pharmacology of Bromo-DragonFLY and NBOMe?
The major target of psychedelic drugs is the serotonin (5-HT2) receptor, specifically the central 5-HT2A subtype. Bromo-DragonFLY is a classic example of designer pharmacology in that the it was intended to potently exert its effect at this specific receptor site.
As its name suggests, BDF adds the “wings of the fly” to the phenylethylamine backbone furanyl rings at positions 2 and 5, and a halogen (bromine) at position 4. The furanyl ring impairs enzymatic clearance of the drug,2 resulting in a duration of action of up to 3 days.3 The addition of halogens increases drug potency, but the mechanism is not clear. The psychedelic agent NBOMe results from chemical additions of methoxy groups at position 2 and 5, and the halogen moiety (iodine in this case) at position 4 of the phenyl ring of the phenylethylamine structure.4
Through the work of Shulgin, some of his colleagues, and many disparate street chemists, a vast family of substituted phenylethylamines have been synthesized and used. Shulgin’s semiautobiographical book PiHKAL: A Chemical Love Story includes his laboratory notes for the synthesis and initial test-dose experience of 179 compounds1; this does not include research done by others or any work since its publication in 1995.
Notable popular drugs chemically similar to NBOMe and BDF are mescaline (found in peyote), cathinones (“bath salts”), and MDMA (found in ecstasy) (Figure). Naturally occurring (and more complex) compounds with similar effects include ayahuasca, a plant-derived beverage consisting of Banisteriopsis caapi and either Psychotria viridis or Diplopterys cabrerana from the Brazilian rainforest (see Emerg Med. 2014;46[12]:553-556); psilocybin (“magic mushrooms”); and lysergic acid diethylamide.
How are these drugs used and what are their clinical effects?
Most phenylethylamine compounds are well absorbed across the buccal mucosa, which is why BDF and NBOMe are commonly used in liquid form or on blotter paper. Dosing guides also exist for insufflation and claim equipotent dosing for this route.5 Regardless of delivery route, given the high potency, inadvertent exposures to these drugs should be expected.
Users simply seeking to hallucinate may not be aware of the significant risks associated with these potent serotonergic agents, which include both life- and limb-threatening effects.6 The high 5-HT2A potency results both in vasoconstriction and promotion of clot formation due to the presence of 5HT2A receptors on small blood vessels and platelets, respectively. Ergotism, historically called Saint Anthony’s fire, is an example of serotonergic vasoconstriction and hallucination.7 Chronic users of substituted amphetamines can develop necrotic ulcers in distal vascular beds such as the hands and feet; these ulcers may progress to amputation despite treatment attempts with vasodilators.
In addition to the vasoconstrictive properties, there are multiple reports of serotonin toxicity (serotonin syndrome) associated with use of these designer serotonergic amphetamines. This syndrome includes severe psychomotor agitation that can lead to personal injury, along with muscle rigidity, tremor, hyperthermia, rhabdomyolysis, and seizures.8
How are patients with phenylethylamine exposures managed?
Management of a patient with a substituted phenylethylamine exposure is similar to management of those with cocaine overdose. Attention to the life-threatening clinical effects of psychomotor agitation, hyperthermia, and seizures is paramount. Appropriate supportive care includes intravenous (IV) benzodiazepines to control agitation and muscle rigidity, replacement of lost volume with crystalloids, and active cooling measures. Failure of benzodiazepines (preferably in conjunction with continuous electroencephalogram monitoring) to control rigidity may lead to the need for propofol and/or result in paralysis. Similar to patients with cocaine intoxication, some may experience ischemic chest pain, and the usual protocol of sedation, nitroglycerin, morphine, and an antiplatelet drug is appropriate.
Identification of phenylethylamines typically requires specialized laboratory testing since most will not trigger a positive result on a standard urine immunoassay. Many specialized laboratories have test catalogs on their Web sites listing under the “stimulants panel” which drugs can be identified. However, none of these assays is likely truly comprehensive, and minor alterations or substitutions to the compounds result in new analogs that may not be in the reference laboratory’s identification library.
The patient was initially restrained and given 5 mg IV diazepam, which was followed by escalating doses every 5 minutes to a total of 35 mg for effect. He had a rectal temperature of 102.5˚F and was externally cooled after sedation. After 20 minutes, he had a generalized convulsion; an additional 10 mg of IV diazepam terminated the seizure, but he remained hyperthermic at 104˚F. The patient was intubated, placed on a propofol infusion, and admitted to the intensive care unit where his temperature was carefully monitored. The following day his temperature had normalized and he was weaned from the ventilator and discharged to the floor for monitoring. On hospital day 3, he was discharged in stable condition.
Mr Waldrop is a fourth-year medical student at the State University of New York, Upstate Medical University, Syracuse. Dr Nacca is a fellow in medical toxicology, department of emergency medicine, State University of New York, Upstate Medical University, Syracuse. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine, and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
Case
A 24-year-old man was brought to the ED by emergency medical services (EMS) for altered mental status. The EMS crew reported they had picked up the patient at a nearby arts festival and concert series. A bystander at the event reported that the patient had taken something called “dragonfly.”
Initial assessment revealed the patient to be disoriented, with nonlinear thought patterns and an inability to follow commands. His vital signs were: blood pressure, 160/100 mm Hg; heart rate, 120 beats/minute; respiratory rate, 24 breaths/minute; and temperature, 102.2˚F. Oxygen saturation was 99% on room air. He was diaphoretic and agitated, and the nursing staff was concerned he would become aggressive and potentially violent. A quick Web search revealed that the agent the bystander mentioned was most likely Bromo-DragonFLY (BDF).
What is Bromo-DragonFLY?
In the 1960s, an American chemist named Alexander Shulgin ushered in a new era of psychedelic drug use by establishing a simple synthesis of 3,4-methylenedioxy-methamphetamine (MDMA). Following this discovery, he suggested a therapist friend use the drug therapeutically.1 Shulgin then began a process of homologation (ie, creating novel compounds by slightly altering existing ones in an organized fashion) and developed systems for rating the drug experiences and naming the drugs in shorthand, both of which are still in use. The chemical structure common to nearly all of the drugs he studied is phenylethylamine. The Figure shows the structures of several phenylethylamine derivatives that were created by adding functional groups to the phenylethylamine backbone. Although the popularity of psychedelic drugs surged during this time period, 2,5-dimethoxy-N-(2-methoxybenzyl)phenylethylamine) (NBOMe), one of a number of newly popular psychedelics, only became available in 2003.
What is known about the pharmacology of Bromo-DragonFLY and NBOMe?
The major target of psychedelic drugs is the serotonin (5-HT2) receptor, specifically the central 5-HT2A subtype. Bromo-DragonFLY is a classic example of designer pharmacology in that the it was intended to potently exert its effect at this specific receptor site.
As its name suggests, BDF adds the “wings of the fly” to the phenylethylamine backbone furanyl rings at positions 2 and 5, and a halogen (bromine) at position 4. The furanyl ring impairs enzymatic clearance of the drug,2 resulting in a duration of action of up to 3 days.3 The addition of halogens increases drug potency, but the mechanism is not clear. The psychedelic agent NBOMe results from chemical additions of methoxy groups at position 2 and 5, and the halogen moiety (iodine in this case) at position 4 of the phenyl ring of the phenylethylamine structure.4
Through the work of Shulgin, some of his colleagues, and many disparate street chemists, a vast family of substituted phenylethylamines have been synthesized and used. Shulgin’s semiautobiographical book PiHKAL: A Chemical Love Story includes his laboratory notes for the synthesis and initial test-dose experience of 179 compounds1; this does not include research done by others or any work since its publication in 1995.
Notable popular drugs chemically similar to NBOMe and BDF are mescaline (found in peyote), cathinones (“bath salts”), and MDMA (found in ecstasy) (Figure). Naturally occurring (and more complex) compounds with similar effects include ayahuasca, a plant-derived beverage consisting of Banisteriopsis caapi and either Psychotria viridis or Diplopterys cabrerana from the Brazilian rainforest (see Emerg Med. 2014;46[12]:553-556); psilocybin (“magic mushrooms”); and lysergic acid diethylamide.
How are these drugs used and what are their clinical effects?
Most phenylethylamine compounds are well absorbed across the buccal mucosa, which is why BDF and NBOMe are commonly used in liquid form or on blotter paper. Dosing guides also exist for insufflation and claim equipotent dosing for this route.5 Regardless of delivery route, given the high potency, inadvertent exposures to these drugs should be expected.
Users simply seeking to hallucinate may not be aware of the significant risks associated with these potent serotonergic agents, which include both life- and limb-threatening effects.6 The high 5-HT2A potency results both in vasoconstriction and promotion of clot formation due to the presence of 5HT2A receptors on small blood vessels and platelets, respectively. Ergotism, historically called Saint Anthony’s fire, is an example of serotonergic vasoconstriction and hallucination.7 Chronic users of substituted amphetamines can develop necrotic ulcers in distal vascular beds such as the hands and feet; these ulcers may progress to amputation despite treatment attempts with vasodilators.
In addition to the vasoconstrictive properties, there are multiple reports of serotonin toxicity (serotonin syndrome) associated with use of these designer serotonergic amphetamines. This syndrome includes severe psychomotor agitation that can lead to personal injury, along with muscle rigidity, tremor, hyperthermia, rhabdomyolysis, and seizures.8
How are patients with phenylethylamine exposures managed?
Management of a patient with a substituted phenylethylamine exposure is similar to management of those with cocaine overdose. Attention to the life-threatening clinical effects of psychomotor agitation, hyperthermia, and seizures is paramount. Appropriate supportive care includes intravenous (IV) benzodiazepines to control agitation and muscle rigidity, replacement of lost volume with crystalloids, and active cooling measures. Failure of benzodiazepines (preferably in conjunction with continuous electroencephalogram monitoring) to control rigidity may lead to the need for propofol and/or result in paralysis. Similar to patients with cocaine intoxication, some may experience ischemic chest pain, and the usual protocol of sedation, nitroglycerin, morphine, and an antiplatelet drug is appropriate.
Identification of phenylethylamines typically requires specialized laboratory testing since most will not trigger a positive result on a standard urine immunoassay. Many specialized laboratories have test catalogs on their Web sites listing under the “stimulants panel” which drugs can be identified. However, none of these assays is likely truly comprehensive, and minor alterations or substitutions to the compounds result in new analogs that may not be in the reference laboratory’s identification library.
The patient was initially restrained and given 5 mg IV diazepam, which was followed by escalating doses every 5 minutes to a total of 35 mg for effect. He had a rectal temperature of 102.5˚F and was externally cooled after sedation. After 20 minutes, he had a generalized convulsion; an additional 10 mg of IV diazepam terminated the seizure, but he remained hyperthermic at 104˚F. The patient was intubated, placed on a propofol infusion, and admitted to the intensive care unit where his temperature was carefully monitored. The following day his temperature had normalized and he was weaned from the ventilator and discharged to the floor for monitoring. On hospital day 3, he was discharged in stable condition.
Mr Waldrop is a fourth-year medical student at the State University of New York, Upstate Medical University, Syracuse. Dr Nacca is a fellow in medical toxicology, department of emergency medicine, State University of New York, Upstate Medical University, Syracuse. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine, and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Shulgin A, Shulgin A. PiHKAL: A Chemical Love Story. Berkeley, CA: Transform Press; 1995.
- Andreasen MF, Telving R, Birkler RI, Schumacher B, Johannsen M. A fatal poisoning involving bromo-dragonfly. Forensic Sci Int. 2009;183(1-3):91-96.
- Hill SL, Thomas SH. Clinical toxicology of newer recreational drugs. Clin Toxicol (Phila). 2011;49(8):705-719.
- Gentry CL, Egleton RD, Gillespie T, et al. The effect of halogenation on blood-brain barrier permeability of a novel peptide drug. Peptides. 1999;20(10):1229-1238.
- Erowid. Bromo-Dragonfly Dosage. http://www.erowid.org/chemicals/bromo_dragonfly/bromo_dragonfly_dose.shtml. Accessed January 14, 2015.
- Baumann MH, Ayestas MA Jr, Partilla JS, et al. The designer methcathinone analogs, mephedrone and methylone, are substrates for monoamine transporters in brain tissue. Neuropsychopharmacology. 2012;37(5):1192-1203.
- Walterscheid JP, Phillips GT, Lopez AE, Gonsoulin ML, Chen HH, Sanchez LA. Pathological findings in 2 cases of fatal 25I-NBOMe toxicity. Am J Forensic Med Pathol. 2014;35(1):20-25.
- Wood DM, Looker JJ, Shaikh L, et al. Delayed onset of seizures and toxicity associated with recreational use of Bromo-dragonFLY. J Med Toxicol. 2009;5(4):226-229.
- Shulgin A, Shulgin A. PiHKAL: A Chemical Love Story. Berkeley, CA: Transform Press; 1995.
- Andreasen MF, Telving R, Birkler RI, Schumacher B, Johannsen M. A fatal poisoning involving bromo-dragonfly. Forensic Sci Int. 2009;183(1-3):91-96.
- Hill SL, Thomas SH. Clinical toxicology of newer recreational drugs. Clin Toxicol (Phila). 2011;49(8):705-719.
- Gentry CL, Egleton RD, Gillespie T, et al. The effect of halogenation on blood-brain barrier permeability of a novel peptide drug. Peptides. 1999;20(10):1229-1238.
- Erowid. Bromo-Dragonfly Dosage. http://www.erowid.org/chemicals/bromo_dragonfly/bromo_dragonfly_dose.shtml. Accessed January 14, 2015.
- Baumann MH, Ayestas MA Jr, Partilla JS, et al. The designer methcathinone analogs, mephedrone and methylone, are substrates for monoamine transporters in brain tissue. Neuropsychopharmacology. 2012;37(5):1192-1203.
- Walterscheid JP, Phillips GT, Lopez AE, Gonsoulin ML, Chen HH, Sanchez LA. Pathological findings in 2 cases of fatal 25I-NBOMe toxicity. Am J Forensic Med Pathol. 2014;35(1):20-25.
- Wood DM, Looker JJ, Shaikh L, et al. Delayed onset of seizures and toxicity associated with recreational use of Bromo-dragonFLY. J Med Toxicol. 2009;5(4):226-229.

Enterovirus D68: A clinically important respiratory enterovirus
In the fall of 2014, the United States experienced an outbreak of severe respiratory illness due to a virus of emerging importance, enterovirus D68 (EV-D68). Here, we review the features of this virus and related viruses, the clinical syndromes this virus causes, the epidemiology of the recent outbreak, and its diagnosis and treatment.
THE ENTEROVIRUSES: AN OVERVIEW
Originally identified in 1962 from the throat swab of a child with pneumonia, human EV-D68 has unique genetic and clinical features that blur the typical division between human enteroviruses and rhinoviruses.1–4 Enteroviruses and rhinoviruses are closely related species within the Picornaviridae family that are now classified together within the genus Enterovirus.5 Picornaviruses are small, nonenveloped, positive-stranded RNA viruses of medical significance.
Poliovirus: The first enterovirus discovered
The first human enterovirus to be discovered was poliovirus.6 Although sporadic cases of “infantile paralysis” occurred before the late 19th century, epidemic poliomyelitis abruptly appeared in Europe and the United States beginning around 1880. Before the introduction in 1955 of the inactivated poliovirus vaccine and then the oral poliovirus vaccine, polio was one of the most feared illnesses in the developed world. Outbreaks occurred primarily in cities during summer months. At its peak, epidemic polio killed or paralyzed more than half a million people a year.
One hypothesis to explain the sudden emergence of epidemic polio is that improved personal hygiene and public sanitation delayed the age at which children acquired this enteric infection.7 Infections acquired after infancy occurred in the absence of maternal antibodies that may have protected against the virus’s propensity to invade the nervous system.
Nonpolio human enteroviruses
In the decades since poliovirus was discovered, more than 100 nonpolio human enteroviruses have been recognized.8 This group includes the coxsackieviruses, echoviruses, and the newer numbered nonpolio human enteroviruses classified into four species, designated Human enterovirus A, B, C, and D. The last of these, Human enterovirus D, includes three serotypes known to cause disease in humans: EV-D68, EV-D70, and EV-D94.9
As with poliovirus infection, most people infected with a nonpolio human enterovirus have a mild illness without distinctive features.5 In temperate climates, enteroviral infections are most common during the summer and fall and are an important cause of the “summer cold.” In tropical climates, the seasonal pattern is absent, and infections may occur throughout the year.
The clinical syndromes associated with a nonpolio human enterovirus can include nonspecific febrile illness; upper respiratory tract infection; pharyngitis; herpangina; hand, foot, and mouth syndrome; various skin exanthems; bronchiolitis; asthma exacerbation; gastrointestinal manifestations such as diarrhea and vomiting (which are especially common); more serious clinical syndromes such as hepatitis, pancreatitis, and cardiomyopathy; and neurologic illness, including aseptic meningitis, encephalitis, and polio-like paralytic disease.
Outbreaks caused by nonpolio human enteroviruses occur on a regular basis, may vary by strain from year to year, and often occur within a geographic region; multiple strains may circulate simultaneously. Occasionally, as with EV-D68 in August 2014 in the United States, epidemics can emerge suddenly and spread rapidly across the world, causing disease in hundreds or thousands of people, demonstrating the breadth of illness associated with particular strains.10
ENTEROVIRUS D68: AN EMERGING PATHOGEN
EV-D68 was first isolated in the United States from four children in Berkeley, California, who had lower respiratory tract symptoms (bronchiolitis and pneumonia) in 1962. The finding was published in the medical literature in 1967.1 Since its initial identification, EV-D68 was infrequently reported as a cause of human disease, with the US Centers for Disease Control and Prevention (CDC) listing only 26 cases in the 36 years from 1970 through 2005.11
However, the past decade has seen EV-D68 emerge as a significant respiratory pathogen, with more reports of acute respiratory illness associated with it in North America, Europe, and Asia, especially in children.12–17 A seasonal pattern may exist; a longitudinal survey of samples collected from New York City detected a focal outbreak in the fall of 2009.18
The observation that recent EV-D68 outbreaks have primarily been in children suggests that most adults have immunity to it. In this regard, seroepidemiologic studies from Finland demonstrated that most adults have neutralizing antibodies from previous infection.9
The blurred line between enteroviruses and rhinoviruses
Enteroviruses and rhinoviruses are typically distinguished on the basis of the temperature at which they grow best (rhinoviruses grow better at lower temperatures, allowing them to replicate in the nose) and their sensitivity to acidity (enteroviruses are more resistant, enabling them to survive in the stomach).
The original (“Fermon”) strain of EV-D68 isolated in 1962 was first classified as an enterovirus because it was resistant to low pH.1 However, when molecular sequencing became available, EV-D68 was found to be identical to human rhinovirus 87 (HRV87), a phylogenetic outlier among the rhinoviruses that binds to cells at a receptor site distinct from that of other human rhinoviruses.19
Thereafter, further testing showed that both EV-D68 and HRV87 isolates were sensitive to acid treatment by two different methods.4 Moreover, unlike most enteroviruses, EV-D68 behaves like a rhinovirus and grows preferentially at 33°C, the temperature of the nose.2
How enterovirus D68 enters cells
Viral surface proteins, including hemagglutinin, from certain respiratory viruses have the ability to bind sugars on cells in the nose and lungs, which facilitates viral entry and replication. EV-D68 binds specifically to alpha 2-6 sialic acid, the predominant sialic acid found in the human upper respiratory tract.19,20 The absence of EV-D68 binding affinity for alpha 2-3 sialic acid, present in ciliated epithelial cells of the lower tract, suggests that alternative mechanisms may be responsible for the severe lower respiratory disease associated with this virus.
Entry of EV-D68 into cells requires additional mediators. EV-D70 belongs to the same genetic cluster as EV-D68 and enters HeLa cells using decay-accelerating factor (DAF).21 Evidence that EV-D68 also uses DAF for cell entry comes from experiments showing that monoclonal antibodies against DAF inhibit the cytopathic effects of this virus.4 Virus-receptor interactions have been more thoroughly characterized for other enteroviruses.22 In this regard, coxsackieviruses of group B use DAF as a coreceptor. Since DAF is expressed at high levels in both epithelial and endothelial cells, it may play an important role in the induction of the viremia that precedes the infection of specific tissues such as the heart or pancreas.
Different strains exist
EV-D68 strains can be divided into three genetic groups based on the sequence of the capsid-coding VP1 region, the most variable genome region of enteroviruses.23
Investigators have explored whether emergent EV-D68 strains differ in their anti-
genicity and receptor-binding properties in comparison to the Fermon strain isolated in 1962.20 Using antisera generated from various strains of EV-D68, significant differences were observed in terms of hemagglutination inhibition and neutralization titers both between emergent strains and the original Fermon strain and among the emergent strains.
Viremia in systemic disease
Like other enteroviruses, EV-D68 has the ability to infect lymphocytes.9 This may provide a mechanism by which the virus is transported during the viremic phase to secondary target organs. Indeed, EV-D68 was detected in the serum of 12 (43%) of 28 pediatric patients with pneumonia and positive nasopharyngeal swabs.24
Interestingly, whether EV-D68 was detected in the serum varied with age. Viremia was not detected in the serum of children younger than 1 year, an observation suggesting that maternal antibodies protect against viremia.
The role of viremia in systemic disease associated with EV-D68 is intriguing, especially since delayed acquisition of polio infection beyond infancy is hypothesized to have contributed to disease severity.7
ENTEROVIRUS D68 CAUSES SEVERE LOWER RESPIRATORY DISEASE
While identification of large numbers of patients with respiratory illnesses due to EV-D68 in a single season is unique to 2014, clusters of EV-D68-related respiratory illnesses have previously been recognized.25,26
As with EV-D68 outbreaks in other parts of the world, the outbreak in the US Midwest in August 2014 primarily involved children, many of whom needed to be admitted to the hospital because of severe lower respiratory symptoms.10 In the 30 children admitted to two children’s hospitals described in the initial report, difficulty breathing, hypoxemia, and wheezing were common. A minority of patients (23%) presented with fever. Of hospitalized children, 67% required admission to the intensive care unit. Two patients required intubation, including one who required extracorporeal membrane oxygenation. Six required bilevel positive airway pressure therapy.
Cleveland Clinic experience
At Cleveland Clinic during the same time, nearly 45% of patients identified with a respiratory enterovirus infection required intensive care.
For patients previously diagnosed with asthma, chronic lung disease, or wheezing, essential supportive care measures included continuing the inhaled steroids the patients were already taking, early use of short-acting beta agonists, and, in those with previously diagnosed asthma, consideration of a systemic steroid. Many of our patients with previously diagnosed asthma had an unusually long prodrome of an increase in mild symptoms, followed by a rapid and severe decline in respiratory status.
At the later phase, supportive care measures that were needed included maintenance of hydration and monitoring of oxyhemoglobin saturation with use of supplemental oxygen as necessary, as well as close observation of clinical indicators of respiratory distress, such as development of crackles, asymmetric air exchange, and progression in wheezing or in use of accessory muscles. In an attempt to avoid invasive ventilatory support in patients with asthma or other comorbid conditions, some patients were treated with aerosolized epinephrine, ipratropium, heliox, and noninvasive positive pressure ventilatory support.
NEUROLOGIC DISEASE: ACUTE FLACCID PARALYSIS
Although EV-D68 causes primarily respiratory illness, systemic disease occurs, especially neurologic involvement.
Before the recent outbreak of EV-D68, two cases of neurologic involvement from EV-D68 were reported. The first of these, mentioned in a 2006 enterovirus surveillance report issued by the CDC, was in a young adult with acute flaccid paralysis and EV-D68 isolated from the cerebral spinal fluid.11 In the second case, from 2010, a 5-year-old boy developed fatal meningomyeloencephalitis. The child had presented with pneumonia and acute flaccid paralysis. EV-D68 was identified in his cerebral spinal fluid by polymerase chain reaction (PCR), and histopathologic study of the meninges, cerebellum, midbrain, pons, medulla, and cervical cord demonstrated extensive T-cell lymphocytic meningomyelitis and encephalitis, characterized by prominent neuronophagia in motor nuclei.27
At the same time as the recent outbreak of EV-D68 respiratory disease, neurologists throughout the United States observed an increase in the number of children with polio-like acute flaccid paralysis. On September 26, 2014, the CDC issued an alert describing acute neurologic illness with focal limb weakness of unknown etiology in children, possibly associated with EV-D68.28 The report described nine cases of an acute neurologic illness in children ages 1 through 18 years (median age, 10) hospitalized in Colorado between August 9 and September 17, 2014. Common clinical features included acute focal limb weakness and paralysis and acute cranial nerve dysfunction, with no altered mental status or seizures. Pain before the onset of weakness was also identified as a common complaint.
Specific findings on magnetic resonance imaging of the spinal cord consisted of nonenhancing lesions largely restricted to the gray matter and in most cases spanning more than one level of the spinal cord. In patients with cranial nerve dysfunction, correlating nonenhancing brainstem lesions were observed.
Most children experienced a febrile respiratory illness in the 2 weeks preceding the onset of neurologic symptoms. In most cases, cerebrospinal fluid analyses demonstrated mild or moderate pleocytosis consistent with an inflammatory or infectious process, with normal to mildly elevated protein and normal glucose levels. In six of the eight patients tested, nasopharyngeal specimens were positive for rhinovirus-enterovirus. Of the six positive specimens, at least four were typed as EV-D68.
The CDC also reported a second cluster of cases of acute flaccid paralysis with anterior myelitis on magnetic resonance imaging, in 23 children (mean age 10 years) in California from June 2012 to June 2014.29 No common cause was identified, although clinical and laboratory findings supported a viral etiology. Two patients tested positive for EV-D68 from upper respiratory tract specimens. Common features among the clinical presentations included an upper respiratory or gastrointestinal prodrome less than 10 days before the onset of the paralysis (83%), cerebrospinal fluid pleocytosis (83%), and absence of sensory deficits (78%). Ten patients (43%) also had concomitant mental status changes, and eight (34%) had cranial nerve abnormalities.
Details regarding outcomes from these paralytic illnesses remain unclear, although it would appear that time to recovery has been prolonged in many cases, and the degree of recovery remains uncertain.
TREATMENT IS SUPPORTIVE
The treatment of EV-D68 infection is mainly supportive, as no specific antiviral therapy is currently available for any of the enteroviruses. Critically ill patients require organ-specific supportive care.
Potential targets for novel antienteroviral therapies exist; some of the experimental compounds were initially evaluated for their activity against polioviruses or rhinoviruses.30
TESTING MAY HAVE A ROLE
In general, testing does not play a role in the management of patients with mild disease, but it may be indicated for epidemiologic purposes or for specific diagnosis in critically ill patients. Molecular techniques are commonly used to detect respiratory viruses from clinical samples, either as discrete tests or as a multiplex viral panel.
Since patients with EV-D68 infection typically have respiratory symptoms, the virus is generally tested for in nasal wash samples. However, depending on the clinical presentation, it may be appropriate to attempt to detect the virus from other sites using either PCR or culture.
Many clinical laboratories use real-time PCR assays designed to detect both rhinoviruses and enteroviruses, but these tests do not distinguish between the species. While more specific real-time PCR assays are available that generally distinguish rhinoviruses from enteroviruses,31 during the recent outbreak our laboratory observed that confirmed EV-D68 samples cross-reacted with rhinovirus. Most clinical laboratories do not routinely perform viral sequence analysis to specifically identify EV-D68, but this test may be obtained through state health departments and the CDC on a case-by-case basis.
Recently, the CDC’s enterovirus laboratory announced the development of a real-time PCR assay specifically for EV-D68, which may make specific detection more readily available.
INFECTION PREVENTION
The routes by which EV-D68 is transmitted are not fully understood. In contrast to most enteroviruses, which are spread in a fecal-oral manner, it is possible that EV-D68 is also spread through close respiratory or mucous contact.
For this reason, interim infection prevention guidelines issued by the CDC recommend that hospitals use droplet precautions along with contact or standard precautions, depending on the scenario.32 In our children’s hospital, we use droplet and contact precautions for hospitalized patients.
- Schieble JH, Fox VL, Lennette EH. A probable new human picornavirus associated with respiratory diseases. Am J Epidemiol 1967; 85:297–310.
- Oberste MS, Maher K, Schnurr D, et al. Enterovirus 68 is associated with respiratory illness and shares biological features with both the enteroviruses and the rhinoviruses. J Gen Virol 2004; 85:2577–2584.
- Ishiko H, Miura R, Shimada Y, et al. Human rhinovirus 87 identified as human enterovirus 68 by VP4-based molecular diagnosis. Intervirology 2002; 45:136–141.
- Blomqvist S, Savolainen C, Raman L, Roivainen M, Hovi T. Human rhinovirus 87 and enterovirus 68 represent a unique serotype with rhinovirus and enterovirus features. J Clin Microbiol 2002; 40:4218–4223.
- Cherry JD, Krogstad P. Enterovirus, parechoviruses, and Saffold viruses. In: Cherry JD, Harrison GJ, Kaplan SL, Steinbach WJ, Hoetez PJ, editors. Feigin and Cherry’s Textbook of Pediatric Infectious Diseases. Vol 2. Seventh ed. Philadelphia: Elsevier Saunders; 2014:2051–2109.
- Rotbart HA. Enteroviral infections of the central nervous system. Clin Infect Dis 1995; 20:971–981.
- Nathanson N, Kew OM. From emergence to eradication: the epidemiology of poliomyelitis deconstructed. Am J Epidemiol 2010; 172:1213–1229.
- Santti J, Vainionpää R, Hyypiä T. Molecular detection and typing of human picornaviruses. Virus Res 1999; 62:177–183.
- Smura T, Ylipaasto P, Klemola P, et al. Cellular tropism of human enterovirus D species serotypes EV-94, EV-70, and EV-68 in vitro: implications for pathogenesis. J Med Virol 2010; 82:1940–1949.
- Midgley CM, Jackson MA, Selvarangan R, et al. Severe respiratory illness associated with enterovirus d68 - Missouri and Illinois, 2014. MMWR Morb Mortal Wkly Rep 2014; 63:798–799.
- Khetsuriani N, Lamonte-Fowlkes A, Oberst S, Pallansch MA. Enterovirus surveillance—United States, 1970–2005. MMWR Surveill Summ 2006; 55:1–20.
- Tokarz R, Firth C, Madhi SA, et al. Worldwide emergence of multiple clades of enterovirus 68. J Gen Virol 2012; 93:1952–1958.
- Clusters of acute respiratory illness associated with human enterovirus 68—Asia, Europe, and United States, 2008–2010. MMWR Morb Mortal Wkly Rep 2011; 60:1301–1304.
- Lauinger IL, Bible JM, Halligan EP, Aarons EJ, MacMahon E, Tong CY. Lineages, sub-lineages and variants of enterovirus 68 in recent outbreaks. PLoS One 2012; 7:e36005.
- Lu QB, Wo Y, Wang HY, et al. Detection of enterovirus 68 as one of the commonest types of enterovirus found in patients with acute respiratory tract infection in China. J Med Microbiol 2014; 63:408–414.
- Meijer A, van der Sanden S, Snijders BE, et al. Emergence and epidemic occurrence of enterovirus 68 respiratory infections in The Netherlands in 2010. Virology 2012; 423:49–57.
- Rahamat-Langendoen J, Riezebos-Brilman A, Borger R, et al. Upsurge of human enterovirus 68 infections in patients with severe respiratory tract infections. J Clin Virol 2011; 52:103–106.
- Tokarz R, Kapoor V, Wu W, et al. Longitudinal molecular microbial analysis of influenza-like illness in New York City, May 2009 through May 2010. Virol J 2011; 8:288.
- Uncapher CR, DeWitt CM, Colonno RJ. The major and minor group receptor families contain all but one human rhinovirus serotype. Virology 1991; 180:814–817.
- Imamura T, Okamoto M, Nakakita S, et al. Antigenic and receptor binding properties of enterovirus 68. J Virol 2014; 88:2374–2384.
- Karnauchow TM, Tolson DL, Harrison BA, Altman E, Lublin DM, Dimock K. The HeLa cell receptor for enterovirus 70 is decay-accelerating factor (CD55). J Virol 1996; 70:5143–5152.
- Selinka HC, Wolde A, Sauter M, Kandolf R, Klingel K. Virus-receptor interactions of coxsackie B viruses and their putative influence on cardiotropism. Med Microbiol Immunol 2004; 193:127–131.
- Piralla A, Girello A, Grignani M, et al. Phylogenetic characterization of enterovirus 68 strains in patients with respiratory syndromes in Italy. J Med Virol 2014; 86:1590–1593.
- Imamura T, Suzuki A, Lupisan S, et al. Detection of enterovirus 68 in serum from pediatric patients with pneumonia and their clinical outcomes. Influenza Other Respir Viruses 2014; 8:21–24.
- Imamura T, Fuji N, Suzuki A, et al. Enterovirus 68 among children with severe acute respiratory infection, the Philippines. Emerg Infect Dis 2011; 17:1430–1435.
- Kaida A, Kubo H, Sekiguchi J, et al. Enterovirus 68 in children with acute respiratory tract infections, Osaka, Japan. Emerg Infect Dis 2011; 17:1494–1497.
- Kreuter JD, Barnes A, McCarthy JE, et al. A fatal central nervous system enterovirus 68 infection. Arch Pathol Lab Med 2011; 135:793–796.
- Pastula DM, Aliabadi N, Haynes AK, et al. Acute neurologic illness of unknown etiology in children—Colorado, August-September 2014. MMWR Morb Mortal Wkly Rep 2014; 63:901–902.
- Ayscue P, Haren KV, Sheriff H, et al. Acute flaccid paralysis with anterior myelitis—California, June 2012–June 2014. MMWR Morb Mortal Wkly Rep 2014; 63:903–906.
- Abzug MJ. The enteroviruses: problems in need of treatments. J Infect 2014; 68(suppl 1):S108–S114.
- Pierce VM, Hodinka RL. Comparison of the GenMark Diagnostics eSensor respiratory viral panel to real-time PCR for detection of respiratory viruses in children. J Clin Microbiol 2012; 50:3458–3465.
- Non-polio enterovirus infection: enterovirus D68 (EV-D68)-CDC. 2014; www.cdc.gov/non-polio-enterovirus/about/ev-d68.html. Accessed November 21, 2014.
In the fall of 2014, the United States experienced an outbreak of severe respiratory illness due to a virus of emerging importance, enterovirus D68 (EV-D68). Here, we review the features of this virus and related viruses, the clinical syndromes this virus causes, the epidemiology of the recent outbreak, and its diagnosis and treatment.
THE ENTEROVIRUSES: AN OVERVIEW
Originally identified in 1962 from the throat swab of a child with pneumonia, human EV-D68 has unique genetic and clinical features that blur the typical division between human enteroviruses and rhinoviruses.1–4 Enteroviruses and rhinoviruses are closely related species within the Picornaviridae family that are now classified together within the genus Enterovirus.5 Picornaviruses are small, nonenveloped, positive-stranded RNA viruses of medical significance.
Poliovirus: The first enterovirus discovered
The first human enterovirus to be discovered was poliovirus.6 Although sporadic cases of “infantile paralysis” occurred before the late 19th century, epidemic poliomyelitis abruptly appeared in Europe and the United States beginning around 1880. Before the introduction in 1955 of the inactivated poliovirus vaccine and then the oral poliovirus vaccine, polio was one of the most feared illnesses in the developed world. Outbreaks occurred primarily in cities during summer months. At its peak, epidemic polio killed or paralyzed more than half a million people a year.
One hypothesis to explain the sudden emergence of epidemic polio is that improved personal hygiene and public sanitation delayed the age at which children acquired this enteric infection.7 Infections acquired after infancy occurred in the absence of maternal antibodies that may have protected against the virus’s propensity to invade the nervous system.
Nonpolio human enteroviruses
In the decades since poliovirus was discovered, more than 100 nonpolio human enteroviruses have been recognized.8 This group includes the coxsackieviruses, echoviruses, and the newer numbered nonpolio human enteroviruses classified into four species, designated Human enterovirus A, B, C, and D. The last of these, Human enterovirus D, includes three serotypes known to cause disease in humans: EV-D68, EV-D70, and EV-D94.9
As with poliovirus infection, most people infected with a nonpolio human enterovirus have a mild illness without distinctive features.5 In temperate climates, enteroviral infections are most common during the summer and fall and are an important cause of the “summer cold.” In tropical climates, the seasonal pattern is absent, and infections may occur throughout the year.
The clinical syndromes associated with a nonpolio human enterovirus can include nonspecific febrile illness; upper respiratory tract infection; pharyngitis; herpangina; hand, foot, and mouth syndrome; various skin exanthems; bronchiolitis; asthma exacerbation; gastrointestinal manifestations such as diarrhea and vomiting (which are especially common); more serious clinical syndromes such as hepatitis, pancreatitis, and cardiomyopathy; and neurologic illness, including aseptic meningitis, encephalitis, and polio-like paralytic disease.
Outbreaks caused by nonpolio human enteroviruses occur on a regular basis, may vary by strain from year to year, and often occur within a geographic region; multiple strains may circulate simultaneously. Occasionally, as with EV-D68 in August 2014 in the United States, epidemics can emerge suddenly and spread rapidly across the world, causing disease in hundreds or thousands of people, demonstrating the breadth of illness associated with particular strains.10
ENTEROVIRUS D68: AN EMERGING PATHOGEN
EV-D68 was first isolated in the United States from four children in Berkeley, California, who had lower respiratory tract symptoms (bronchiolitis and pneumonia) in 1962. The finding was published in the medical literature in 1967.1 Since its initial identification, EV-D68 was infrequently reported as a cause of human disease, with the US Centers for Disease Control and Prevention (CDC) listing only 26 cases in the 36 years from 1970 through 2005.11
However, the past decade has seen EV-D68 emerge as a significant respiratory pathogen, with more reports of acute respiratory illness associated with it in North America, Europe, and Asia, especially in children.12–17 A seasonal pattern may exist; a longitudinal survey of samples collected from New York City detected a focal outbreak in the fall of 2009.18
The observation that recent EV-D68 outbreaks have primarily been in children suggests that most adults have immunity to it. In this regard, seroepidemiologic studies from Finland demonstrated that most adults have neutralizing antibodies from previous infection.9
The blurred line between enteroviruses and rhinoviruses
Enteroviruses and rhinoviruses are typically distinguished on the basis of the temperature at which they grow best (rhinoviruses grow better at lower temperatures, allowing them to replicate in the nose) and their sensitivity to acidity (enteroviruses are more resistant, enabling them to survive in the stomach).
The original (“Fermon”) strain of EV-D68 isolated in 1962 was first classified as an enterovirus because it was resistant to low pH.1 However, when molecular sequencing became available, EV-D68 was found to be identical to human rhinovirus 87 (HRV87), a phylogenetic outlier among the rhinoviruses that binds to cells at a receptor site distinct from that of other human rhinoviruses.19
Thereafter, further testing showed that both EV-D68 and HRV87 isolates were sensitive to acid treatment by two different methods.4 Moreover, unlike most enteroviruses, EV-D68 behaves like a rhinovirus and grows preferentially at 33°C, the temperature of the nose.2
How enterovirus D68 enters cells
Viral surface proteins, including hemagglutinin, from certain respiratory viruses have the ability to bind sugars on cells in the nose and lungs, which facilitates viral entry and replication. EV-D68 binds specifically to alpha 2-6 sialic acid, the predominant sialic acid found in the human upper respiratory tract.19,20 The absence of EV-D68 binding affinity for alpha 2-3 sialic acid, present in ciliated epithelial cells of the lower tract, suggests that alternative mechanisms may be responsible for the severe lower respiratory disease associated with this virus.
Entry of EV-D68 into cells requires additional mediators. EV-D70 belongs to the same genetic cluster as EV-D68 and enters HeLa cells using decay-accelerating factor (DAF).21 Evidence that EV-D68 also uses DAF for cell entry comes from experiments showing that monoclonal antibodies against DAF inhibit the cytopathic effects of this virus.4 Virus-receptor interactions have been more thoroughly characterized for other enteroviruses.22 In this regard, coxsackieviruses of group B use DAF as a coreceptor. Since DAF is expressed at high levels in both epithelial and endothelial cells, it may play an important role in the induction of the viremia that precedes the infection of specific tissues such as the heart or pancreas.
Different strains exist
EV-D68 strains can be divided into three genetic groups based on the sequence of the capsid-coding VP1 region, the most variable genome region of enteroviruses.23
Investigators have explored whether emergent EV-D68 strains differ in their anti-
genicity and receptor-binding properties in comparison to the Fermon strain isolated in 1962.20 Using antisera generated from various strains of EV-D68, significant differences were observed in terms of hemagglutination inhibition and neutralization titers both between emergent strains and the original Fermon strain and among the emergent strains.
Viremia in systemic disease
Like other enteroviruses, EV-D68 has the ability to infect lymphocytes.9 This may provide a mechanism by which the virus is transported during the viremic phase to secondary target organs. Indeed, EV-D68 was detected in the serum of 12 (43%) of 28 pediatric patients with pneumonia and positive nasopharyngeal swabs.24
Interestingly, whether EV-D68 was detected in the serum varied with age. Viremia was not detected in the serum of children younger than 1 year, an observation suggesting that maternal antibodies protect against viremia.
The role of viremia in systemic disease associated with EV-D68 is intriguing, especially since delayed acquisition of polio infection beyond infancy is hypothesized to have contributed to disease severity.7
ENTEROVIRUS D68 CAUSES SEVERE LOWER RESPIRATORY DISEASE
While identification of large numbers of patients with respiratory illnesses due to EV-D68 in a single season is unique to 2014, clusters of EV-D68-related respiratory illnesses have previously been recognized.25,26
As with EV-D68 outbreaks in other parts of the world, the outbreak in the US Midwest in August 2014 primarily involved children, many of whom needed to be admitted to the hospital because of severe lower respiratory symptoms.10 In the 30 children admitted to two children’s hospitals described in the initial report, difficulty breathing, hypoxemia, and wheezing were common. A minority of patients (23%) presented with fever. Of hospitalized children, 67% required admission to the intensive care unit. Two patients required intubation, including one who required extracorporeal membrane oxygenation. Six required bilevel positive airway pressure therapy.
Cleveland Clinic experience
At Cleveland Clinic during the same time, nearly 45% of patients identified with a respiratory enterovirus infection required intensive care.
For patients previously diagnosed with asthma, chronic lung disease, or wheezing, essential supportive care measures included continuing the inhaled steroids the patients were already taking, early use of short-acting beta agonists, and, in those with previously diagnosed asthma, consideration of a systemic steroid. Many of our patients with previously diagnosed asthma had an unusually long prodrome of an increase in mild symptoms, followed by a rapid and severe decline in respiratory status.
At the later phase, supportive care measures that were needed included maintenance of hydration and monitoring of oxyhemoglobin saturation with use of supplemental oxygen as necessary, as well as close observation of clinical indicators of respiratory distress, such as development of crackles, asymmetric air exchange, and progression in wheezing or in use of accessory muscles. In an attempt to avoid invasive ventilatory support in patients with asthma or other comorbid conditions, some patients were treated with aerosolized epinephrine, ipratropium, heliox, and noninvasive positive pressure ventilatory support.
NEUROLOGIC DISEASE: ACUTE FLACCID PARALYSIS
Although EV-D68 causes primarily respiratory illness, systemic disease occurs, especially neurologic involvement.
Before the recent outbreak of EV-D68, two cases of neurologic involvement from EV-D68 were reported. The first of these, mentioned in a 2006 enterovirus surveillance report issued by the CDC, was in a young adult with acute flaccid paralysis and EV-D68 isolated from the cerebral spinal fluid.11 In the second case, from 2010, a 5-year-old boy developed fatal meningomyeloencephalitis. The child had presented with pneumonia and acute flaccid paralysis. EV-D68 was identified in his cerebral spinal fluid by polymerase chain reaction (PCR), and histopathologic study of the meninges, cerebellum, midbrain, pons, medulla, and cervical cord demonstrated extensive T-cell lymphocytic meningomyelitis and encephalitis, characterized by prominent neuronophagia in motor nuclei.27
At the same time as the recent outbreak of EV-D68 respiratory disease, neurologists throughout the United States observed an increase in the number of children with polio-like acute flaccid paralysis. On September 26, 2014, the CDC issued an alert describing acute neurologic illness with focal limb weakness of unknown etiology in children, possibly associated with EV-D68.28 The report described nine cases of an acute neurologic illness in children ages 1 through 18 years (median age, 10) hospitalized in Colorado between August 9 and September 17, 2014. Common clinical features included acute focal limb weakness and paralysis and acute cranial nerve dysfunction, with no altered mental status or seizures. Pain before the onset of weakness was also identified as a common complaint.
Specific findings on magnetic resonance imaging of the spinal cord consisted of nonenhancing lesions largely restricted to the gray matter and in most cases spanning more than one level of the spinal cord. In patients with cranial nerve dysfunction, correlating nonenhancing brainstem lesions were observed.
Most children experienced a febrile respiratory illness in the 2 weeks preceding the onset of neurologic symptoms. In most cases, cerebrospinal fluid analyses demonstrated mild or moderate pleocytosis consistent with an inflammatory or infectious process, with normal to mildly elevated protein and normal glucose levels. In six of the eight patients tested, nasopharyngeal specimens were positive for rhinovirus-enterovirus. Of the six positive specimens, at least four were typed as EV-D68.
The CDC also reported a second cluster of cases of acute flaccid paralysis with anterior myelitis on magnetic resonance imaging, in 23 children (mean age 10 years) in California from June 2012 to June 2014.29 No common cause was identified, although clinical and laboratory findings supported a viral etiology. Two patients tested positive for EV-D68 from upper respiratory tract specimens. Common features among the clinical presentations included an upper respiratory or gastrointestinal prodrome less than 10 days before the onset of the paralysis (83%), cerebrospinal fluid pleocytosis (83%), and absence of sensory deficits (78%). Ten patients (43%) also had concomitant mental status changes, and eight (34%) had cranial nerve abnormalities.
Details regarding outcomes from these paralytic illnesses remain unclear, although it would appear that time to recovery has been prolonged in many cases, and the degree of recovery remains uncertain.
TREATMENT IS SUPPORTIVE
The treatment of EV-D68 infection is mainly supportive, as no specific antiviral therapy is currently available for any of the enteroviruses. Critically ill patients require organ-specific supportive care.
Potential targets for novel antienteroviral therapies exist; some of the experimental compounds were initially evaluated for their activity against polioviruses or rhinoviruses.30
TESTING MAY HAVE A ROLE
In general, testing does not play a role in the management of patients with mild disease, but it may be indicated for epidemiologic purposes or for specific diagnosis in critically ill patients. Molecular techniques are commonly used to detect respiratory viruses from clinical samples, either as discrete tests or as a multiplex viral panel.
Since patients with EV-D68 infection typically have respiratory symptoms, the virus is generally tested for in nasal wash samples. However, depending on the clinical presentation, it may be appropriate to attempt to detect the virus from other sites using either PCR or culture.
Many clinical laboratories use real-time PCR assays designed to detect both rhinoviruses and enteroviruses, but these tests do not distinguish between the species. While more specific real-time PCR assays are available that generally distinguish rhinoviruses from enteroviruses,31 during the recent outbreak our laboratory observed that confirmed EV-D68 samples cross-reacted with rhinovirus. Most clinical laboratories do not routinely perform viral sequence analysis to specifically identify EV-D68, but this test may be obtained through state health departments and the CDC on a case-by-case basis.
Recently, the CDC’s enterovirus laboratory announced the development of a real-time PCR assay specifically for EV-D68, which may make specific detection more readily available.
INFECTION PREVENTION
The routes by which EV-D68 is transmitted are not fully understood. In contrast to most enteroviruses, which are spread in a fecal-oral manner, it is possible that EV-D68 is also spread through close respiratory or mucous contact.
For this reason, interim infection prevention guidelines issued by the CDC recommend that hospitals use droplet precautions along with contact or standard precautions, depending on the scenario.32 In our children’s hospital, we use droplet and contact precautions for hospitalized patients.
In the fall of 2014, the United States experienced an outbreak of severe respiratory illness due to a virus of emerging importance, enterovirus D68 (EV-D68). Here, we review the features of this virus and related viruses, the clinical syndromes this virus causes, the epidemiology of the recent outbreak, and its diagnosis and treatment.
THE ENTEROVIRUSES: AN OVERVIEW
Originally identified in 1962 from the throat swab of a child with pneumonia, human EV-D68 has unique genetic and clinical features that blur the typical division between human enteroviruses and rhinoviruses.1–4 Enteroviruses and rhinoviruses are closely related species within the Picornaviridae family that are now classified together within the genus Enterovirus.5 Picornaviruses are small, nonenveloped, positive-stranded RNA viruses of medical significance.
Poliovirus: The first enterovirus discovered
The first human enterovirus to be discovered was poliovirus.6 Although sporadic cases of “infantile paralysis” occurred before the late 19th century, epidemic poliomyelitis abruptly appeared in Europe and the United States beginning around 1880. Before the introduction in 1955 of the inactivated poliovirus vaccine and then the oral poliovirus vaccine, polio was one of the most feared illnesses in the developed world. Outbreaks occurred primarily in cities during summer months. At its peak, epidemic polio killed or paralyzed more than half a million people a year.
One hypothesis to explain the sudden emergence of epidemic polio is that improved personal hygiene and public sanitation delayed the age at which children acquired this enteric infection.7 Infections acquired after infancy occurred in the absence of maternal antibodies that may have protected against the virus’s propensity to invade the nervous system.
Nonpolio human enteroviruses
In the decades since poliovirus was discovered, more than 100 nonpolio human enteroviruses have been recognized.8 This group includes the coxsackieviruses, echoviruses, and the newer numbered nonpolio human enteroviruses classified into four species, designated Human enterovirus A, B, C, and D. The last of these, Human enterovirus D, includes three serotypes known to cause disease in humans: EV-D68, EV-D70, and EV-D94.9
As with poliovirus infection, most people infected with a nonpolio human enterovirus have a mild illness without distinctive features.5 In temperate climates, enteroviral infections are most common during the summer and fall and are an important cause of the “summer cold.” In tropical climates, the seasonal pattern is absent, and infections may occur throughout the year.
The clinical syndromes associated with a nonpolio human enterovirus can include nonspecific febrile illness; upper respiratory tract infection; pharyngitis; herpangina; hand, foot, and mouth syndrome; various skin exanthems; bronchiolitis; asthma exacerbation; gastrointestinal manifestations such as diarrhea and vomiting (which are especially common); more serious clinical syndromes such as hepatitis, pancreatitis, and cardiomyopathy; and neurologic illness, including aseptic meningitis, encephalitis, and polio-like paralytic disease.
Outbreaks caused by nonpolio human enteroviruses occur on a regular basis, may vary by strain from year to year, and often occur within a geographic region; multiple strains may circulate simultaneously. Occasionally, as with EV-D68 in August 2014 in the United States, epidemics can emerge suddenly and spread rapidly across the world, causing disease in hundreds or thousands of people, demonstrating the breadth of illness associated with particular strains.10
ENTEROVIRUS D68: AN EMERGING PATHOGEN
EV-D68 was first isolated in the United States from four children in Berkeley, California, who had lower respiratory tract symptoms (bronchiolitis and pneumonia) in 1962. The finding was published in the medical literature in 1967.1 Since its initial identification, EV-D68 was infrequently reported as a cause of human disease, with the US Centers for Disease Control and Prevention (CDC) listing only 26 cases in the 36 years from 1970 through 2005.11
However, the past decade has seen EV-D68 emerge as a significant respiratory pathogen, with more reports of acute respiratory illness associated with it in North America, Europe, and Asia, especially in children.12–17 A seasonal pattern may exist; a longitudinal survey of samples collected from New York City detected a focal outbreak in the fall of 2009.18
The observation that recent EV-D68 outbreaks have primarily been in children suggests that most adults have immunity to it. In this regard, seroepidemiologic studies from Finland demonstrated that most adults have neutralizing antibodies from previous infection.9
The blurred line between enteroviruses and rhinoviruses
Enteroviruses and rhinoviruses are typically distinguished on the basis of the temperature at which they grow best (rhinoviruses grow better at lower temperatures, allowing them to replicate in the nose) and their sensitivity to acidity (enteroviruses are more resistant, enabling them to survive in the stomach).
The original (“Fermon”) strain of EV-D68 isolated in 1962 was first classified as an enterovirus because it was resistant to low pH.1 However, when molecular sequencing became available, EV-D68 was found to be identical to human rhinovirus 87 (HRV87), a phylogenetic outlier among the rhinoviruses that binds to cells at a receptor site distinct from that of other human rhinoviruses.19
Thereafter, further testing showed that both EV-D68 and HRV87 isolates were sensitive to acid treatment by two different methods.4 Moreover, unlike most enteroviruses, EV-D68 behaves like a rhinovirus and grows preferentially at 33°C, the temperature of the nose.2
How enterovirus D68 enters cells
Viral surface proteins, including hemagglutinin, from certain respiratory viruses have the ability to bind sugars on cells in the nose and lungs, which facilitates viral entry and replication. EV-D68 binds specifically to alpha 2-6 sialic acid, the predominant sialic acid found in the human upper respiratory tract.19,20 The absence of EV-D68 binding affinity for alpha 2-3 sialic acid, present in ciliated epithelial cells of the lower tract, suggests that alternative mechanisms may be responsible for the severe lower respiratory disease associated with this virus.
Entry of EV-D68 into cells requires additional mediators. EV-D70 belongs to the same genetic cluster as EV-D68 and enters HeLa cells using decay-accelerating factor (DAF).21 Evidence that EV-D68 also uses DAF for cell entry comes from experiments showing that monoclonal antibodies against DAF inhibit the cytopathic effects of this virus.4 Virus-receptor interactions have been more thoroughly characterized for other enteroviruses.22 In this regard, coxsackieviruses of group B use DAF as a coreceptor. Since DAF is expressed at high levels in both epithelial and endothelial cells, it may play an important role in the induction of the viremia that precedes the infection of specific tissues such as the heart or pancreas.
Different strains exist
EV-D68 strains can be divided into three genetic groups based on the sequence of the capsid-coding VP1 region, the most variable genome region of enteroviruses.23
Investigators have explored whether emergent EV-D68 strains differ in their anti-
genicity and receptor-binding properties in comparison to the Fermon strain isolated in 1962.20 Using antisera generated from various strains of EV-D68, significant differences were observed in terms of hemagglutination inhibition and neutralization titers both between emergent strains and the original Fermon strain and among the emergent strains.
Viremia in systemic disease
Like other enteroviruses, EV-D68 has the ability to infect lymphocytes.9 This may provide a mechanism by which the virus is transported during the viremic phase to secondary target organs. Indeed, EV-D68 was detected in the serum of 12 (43%) of 28 pediatric patients with pneumonia and positive nasopharyngeal swabs.24
Interestingly, whether EV-D68 was detected in the serum varied with age. Viremia was not detected in the serum of children younger than 1 year, an observation suggesting that maternal antibodies protect against viremia.
The role of viremia in systemic disease associated with EV-D68 is intriguing, especially since delayed acquisition of polio infection beyond infancy is hypothesized to have contributed to disease severity.7
ENTEROVIRUS D68 CAUSES SEVERE LOWER RESPIRATORY DISEASE
While identification of large numbers of patients with respiratory illnesses due to EV-D68 in a single season is unique to 2014, clusters of EV-D68-related respiratory illnesses have previously been recognized.25,26
As with EV-D68 outbreaks in other parts of the world, the outbreak in the US Midwest in August 2014 primarily involved children, many of whom needed to be admitted to the hospital because of severe lower respiratory symptoms.10 In the 30 children admitted to two children’s hospitals described in the initial report, difficulty breathing, hypoxemia, and wheezing were common. A minority of patients (23%) presented with fever. Of hospitalized children, 67% required admission to the intensive care unit. Two patients required intubation, including one who required extracorporeal membrane oxygenation. Six required bilevel positive airway pressure therapy.
Cleveland Clinic experience
At Cleveland Clinic during the same time, nearly 45% of patients identified with a respiratory enterovirus infection required intensive care.
For patients previously diagnosed with asthma, chronic lung disease, or wheezing, essential supportive care measures included continuing the inhaled steroids the patients were already taking, early use of short-acting beta agonists, and, in those with previously diagnosed asthma, consideration of a systemic steroid. Many of our patients with previously diagnosed asthma had an unusually long prodrome of an increase in mild symptoms, followed by a rapid and severe decline in respiratory status.
At the later phase, supportive care measures that were needed included maintenance of hydration and monitoring of oxyhemoglobin saturation with use of supplemental oxygen as necessary, as well as close observation of clinical indicators of respiratory distress, such as development of crackles, asymmetric air exchange, and progression in wheezing or in use of accessory muscles. In an attempt to avoid invasive ventilatory support in patients with asthma or other comorbid conditions, some patients were treated with aerosolized epinephrine, ipratropium, heliox, and noninvasive positive pressure ventilatory support.
NEUROLOGIC DISEASE: ACUTE FLACCID PARALYSIS
Although EV-D68 causes primarily respiratory illness, systemic disease occurs, especially neurologic involvement.
Before the recent outbreak of EV-D68, two cases of neurologic involvement from EV-D68 were reported. The first of these, mentioned in a 2006 enterovirus surveillance report issued by the CDC, was in a young adult with acute flaccid paralysis and EV-D68 isolated from the cerebral spinal fluid.11 In the second case, from 2010, a 5-year-old boy developed fatal meningomyeloencephalitis. The child had presented with pneumonia and acute flaccid paralysis. EV-D68 was identified in his cerebral spinal fluid by polymerase chain reaction (PCR), and histopathologic study of the meninges, cerebellum, midbrain, pons, medulla, and cervical cord demonstrated extensive T-cell lymphocytic meningomyelitis and encephalitis, characterized by prominent neuronophagia in motor nuclei.27
At the same time as the recent outbreak of EV-D68 respiratory disease, neurologists throughout the United States observed an increase in the number of children with polio-like acute flaccid paralysis. On September 26, 2014, the CDC issued an alert describing acute neurologic illness with focal limb weakness of unknown etiology in children, possibly associated with EV-D68.28 The report described nine cases of an acute neurologic illness in children ages 1 through 18 years (median age, 10) hospitalized in Colorado between August 9 and September 17, 2014. Common clinical features included acute focal limb weakness and paralysis and acute cranial nerve dysfunction, with no altered mental status or seizures. Pain before the onset of weakness was also identified as a common complaint.
Specific findings on magnetic resonance imaging of the spinal cord consisted of nonenhancing lesions largely restricted to the gray matter and in most cases spanning more than one level of the spinal cord. In patients with cranial nerve dysfunction, correlating nonenhancing brainstem lesions were observed.
Most children experienced a febrile respiratory illness in the 2 weeks preceding the onset of neurologic symptoms. In most cases, cerebrospinal fluid analyses demonstrated mild or moderate pleocytosis consistent with an inflammatory or infectious process, with normal to mildly elevated protein and normal glucose levels. In six of the eight patients tested, nasopharyngeal specimens were positive for rhinovirus-enterovirus. Of the six positive specimens, at least four were typed as EV-D68.
The CDC also reported a second cluster of cases of acute flaccid paralysis with anterior myelitis on magnetic resonance imaging, in 23 children (mean age 10 years) in California from June 2012 to June 2014.29 No common cause was identified, although clinical and laboratory findings supported a viral etiology. Two patients tested positive for EV-D68 from upper respiratory tract specimens. Common features among the clinical presentations included an upper respiratory or gastrointestinal prodrome less than 10 days before the onset of the paralysis (83%), cerebrospinal fluid pleocytosis (83%), and absence of sensory deficits (78%). Ten patients (43%) also had concomitant mental status changes, and eight (34%) had cranial nerve abnormalities.
Details regarding outcomes from these paralytic illnesses remain unclear, although it would appear that time to recovery has been prolonged in many cases, and the degree of recovery remains uncertain.
TREATMENT IS SUPPORTIVE
The treatment of EV-D68 infection is mainly supportive, as no specific antiviral therapy is currently available for any of the enteroviruses. Critically ill patients require organ-specific supportive care.
Potential targets for novel antienteroviral therapies exist; some of the experimental compounds were initially evaluated for their activity against polioviruses or rhinoviruses.30
TESTING MAY HAVE A ROLE
In general, testing does not play a role in the management of patients with mild disease, but it may be indicated for epidemiologic purposes or for specific diagnosis in critically ill patients. Molecular techniques are commonly used to detect respiratory viruses from clinical samples, either as discrete tests or as a multiplex viral panel.
Since patients with EV-D68 infection typically have respiratory symptoms, the virus is generally tested for in nasal wash samples. However, depending on the clinical presentation, it may be appropriate to attempt to detect the virus from other sites using either PCR or culture.
Many clinical laboratories use real-time PCR assays designed to detect both rhinoviruses and enteroviruses, but these tests do not distinguish between the species. While more specific real-time PCR assays are available that generally distinguish rhinoviruses from enteroviruses,31 during the recent outbreak our laboratory observed that confirmed EV-D68 samples cross-reacted with rhinovirus. Most clinical laboratories do not routinely perform viral sequence analysis to specifically identify EV-D68, but this test may be obtained through state health departments and the CDC on a case-by-case basis.
Recently, the CDC’s enterovirus laboratory announced the development of a real-time PCR assay specifically for EV-D68, which may make specific detection more readily available.
INFECTION PREVENTION
The routes by which EV-D68 is transmitted are not fully understood. In contrast to most enteroviruses, which are spread in a fecal-oral manner, it is possible that EV-D68 is also spread through close respiratory or mucous contact.
For this reason, interim infection prevention guidelines issued by the CDC recommend that hospitals use droplet precautions along with contact or standard precautions, depending on the scenario.32 In our children’s hospital, we use droplet and contact precautions for hospitalized patients.
- Schieble JH, Fox VL, Lennette EH. A probable new human picornavirus associated with respiratory diseases. Am J Epidemiol 1967; 85:297–310.
- Oberste MS, Maher K, Schnurr D, et al. Enterovirus 68 is associated with respiratory illness and shares biological features with both the enteroviruses and the rhinoviruses. J Gen Virol 2004; 85:2577–2584.
- Ishiko H, Miura R, Shimada Y, et al. Human rhinovirus 87 identified as human enterovirus 68 by VP4-based molecular diagnosis. Intervirology 2002; 45:136–141.
- Blomqvist S, Savolainen C, Raman L, Roivainen M, Hovi T. Human rhinovirus 87 and enterovirus 68 represent a unique serotype with rhinovirus and enterovirus features. J Clin Microbiol 2002; 40:4218–4223.
- Cherry JD, Krogstad P. Enterovirus, parechoviruses, and Saffold viruses. In: Cherry JD, Harrison GJ, Kaplan SL, Steinbach WJ, Hoetez PJ, editors. Feigin and Cherry’s Textbook of Pediatric Infectious Diseases. Vol 2. Seventh ed. Philadelphia: Elsevier Saunders; 2014:2051–2109.
- Rotbart HA. Enteroviral infections of the central nervous system. Clin Infect Dis 1995; 20:971–981.
- Nathanson N, Kew OM. From emergence to eradication: the epidemiology of poliomyelitis deconstructed. Am J Epidemiol 2010; 172:1213–1229.
- Santti J, Vainionpää R, Hyypiä T. Molecular detection and typing of human picornaviruses. Virus Res 1999; 62:177–183.
- Smura T, Ylipaasto P, Klemola P, et al. Cellular tropism of human enterovirus D species serotypes EV-94, EV-70, and EV-68 in vitro: implications for pathogenesis. J Med Virol 2010; 82:1940–1949.
- Midgley CM, Jackson MA, Selvarangan R, et al. Severe respiratory illness associated with enterovirus d68 - Missouri and Illinois, 2014. MMWR Morb Mortal Wkly Rep 2014; 63:798–799.
- Khetsuriani N, Lamonte-Fowlkes A, Oberst S, Pallansch MA. Enterovirus surveillance—United States, 1970–2005. MMWR Surveill Summ 2006; 55:1–20.
- Tokarz R, Firth C, Madhi SA, et al. Worldwide emergence of multiple clades of enterovirus 68. J Gen Virol 2012; 93:1952–1958.
- Clusters of acute respiratory illness associated with human enterovirus 68—Asia, Europe, and United States, 2008–2010. MMWR Morb Mortal Wkly Rep 2011; 60:1301–1304.
- Lauinger IL, Bible JM, Halligan EP, Aarons EJ, MacMahon E, Tong CY. Lineages, sub-lineages and variants of enterovirus 68 in recent outbreaks. PLoS One 2012; 7:e36005.
- Lu QB, Wo Y, Wang HY, et al. Detection of enterovirus 68 as one of the commonest types of enterovirus found in patients with acute respiratory tract infection in China. J Med Microbiol 2014; 63:408–414.
- Meijer A, van der Sanden S, Snijders BE, et al. Emergence and epidemic occurrence of enterovirus 68 respiratory infections in The Netherlands in 2010. Virology 2012; 423:49–57.
- Rahamat-Langendoen J, Riezebos-Brilman A, Borger R, et al. Upsurge of human enterovirus 68 infections in patients with severe respiratory tract infections. J Clin Virol 2011; 52:103–106.
- Tokarz R, Kapoor V, Wu W, et al. Longitudinal molecular microbial analysis of influenza-like illness in New York City, May 2009 through May 2010. Virol J 2011; 8:288.
- Uncapher CR, DeWitt CM, Colonno RJ. The major and minor group receptor families contain all but one human rhinovirus serotype. Virology 1991; 180:814–817.
- Imamura T, Okamoto M, Nakakita S, et al. Antigenic and receptor binding properties of enterovirus 68. J Virol 2014; 88:2374–2384.
- Karnauchow TM, Tolson DL, Harrison BA, Altman E, Lublin DM, Dimock K. The HeLa cell receptor for enterovirus 70 is decay-accelerating factor (CD55). J Virol 1996; 70:5143–5152.
- Selinka HC, Wolde A, Sauter M, Kandolf R, Klingel K. Virus-receptor interactions of coxsackie B viruses and their putative influence on cardiotropism. Med Microbiol Immunol 2004; 193:127–131.
- Piralla A, Girello A, Grignani M, et al. Phylogenetic characterization of enterovirus 68 strains in patients with respiratory syndromes in Italy. J Med Virol 2014; 86:1590–1593.
- Imamura T, Suzuki A, Lupisan S, et al. Detection of enterovirus 68 in serum from pediatric patients with pneumonia and their clinical outcomes. Influenza Other Respir Viruses 2014; 8:21–24.
- Imamura T, Fuji N, Suzuki A, et al. Enterovirus 68 among children with severe acute respiratory infection, the Philippines. Emerg Infect Dis 2011; 17:1430–1435.
- Kaida A, Kubo H, Sekiguchi J, et al. Enterovirus 68 in children with acute respiratory tract infections, Osaka, Japan. Emerg Infect Dis 2011; 17:1494–1497.
- Kreuter JD, Barnes A, McCarthy JE, et al. A fatal central nervous system enterovirus 68 infection. Arch Pathol Lab Med 2011; 135:793–796.
- Pastula DM, Aliabadi N, Haynes AK, et al. Acute neurologic illness of unknown etiology in children—Colorado, August-September 2014. MMWR Morb Mortal Wkly Rep 2014; 63:901–902.
- Ayscue P, Haren KV, Sheriff H, et al. Acute flaccid paralysis with anterior myelitis—California, June 2012–June 2014. MMWR Morb Mortal Wkly Rep 2014; 63:903–906.
- Abzug MJ. The enteroviruses: problems in need of treatments. J Infect 2014; 68(suppl 1):S108–S114.
- Pierce VM, Hodinka RL. Comparison of the GenMark Diagnostics eSensor respiratory viral panel to real-time PCR for detection of respiratory viruses in children. J Clin Microbiol 2012; 50:3458–3465.
- Non-polio enterovirus infection: enterovirus D68 (EV-D68)-CDC. 2014; www.cdc.gov/non-polio-enterovirus/about/ev-d68.html. Accessed November 21, 2014.
- Schieble JH, Fox VL, Lennette EH. A probable new human picornavirus associated with respiratory diseases. Am J Epidemiol 1967; 85:297–310.
- Oberste MS, Maher K, Schnurr D, et al. Enterovirus 68 is associated with respiratory illness and shares biological features with both the enteroviruses and the rhinoviruses. J Gen Virol 2004; 85:2577–2584.
- Ishiko H, Miura R, Shimada Y, et al. Human rhinovirus 87 identified as human enterovirus 68 by VP4-based molecular diagnosis. Intervirology 2002; 45:136–141.
- Blomqvist S, Savolainen C, Raman L, Roivainen M, Hovi T. Human rhinovirus 87 and enterovirus 68 represent a unique serotype with rhinovirus and enterovirus features. J Clin Microbiol 2002; 40:4218–4223.
- Cherry JD, Krogstad P. Enterovirus, parechoviruses, and Saffold viruses. In: Cherry JD, Harrison GJ, Kaplan SL, Steinbach WJ, Hoetez PJ, editors. Feigin and Cherry’s Textbook of Pediatric Infectious Diseases. Vol 2. Seventh ed. Philadelphia: Elsevier Saunders; 2014:2051–2109.
- Rotbart HA. Enteroviral infections of the central nervous system. Clin Infect Dis 1995; 20:971–981.
- Nathanson N, Kew OM. From emergence to eradication: the epidemiology of poliomyelitis deconstructed. Am J Epidemiol 2010; 172:1213–1229.
- Santti J, Vainionpää R, Hyypiä T. Molecular detection and typing of human picornaviruses. Virus Res 1999; 62:177–183.
- Smura T, Ylipaasto P, Klemola P, et al. Cellular tropism of human enterovirus D species serotypes EV-94, EV-70, and EV-68 in vitro: implications for pathogenesis. J Med Virol 2010; 82:1940–1949.
- Midgley CM, Jackson MA, Selvarangan R, et al. Severe respiratory illness associated with enterovirus d68 - Missouri and Illinois, 2014. MMWR Morb Mortal Wkly Rep 2014; 63:798–799.
- Khetsuriani N, Lamonte-Fowlkes A, Oberst S, Pallansch MA. Enterovirus surveillance—United States, 1970–2005. MMWR Surveill Summ 2006; 55:1–20.
- Tokarz R, Firth C, Madhi SA, et al. Worldwide emergence of multiple clades of enterovirus 68. J Gen Virol 2012; 93:1952–1958.
- Clusters of acute respiratory illness associated with human enterovirus 68—Asia, Europe, and United States, 2008–2010. MMWR Morb Mortal Wkly Rep 2011; 60:1301–1304.
- Lauinger IL, Bible JM, Halligan EP, Aarons EJ, MacMahon E, Tong CY. Lineages, sub-lineages and variants of enterovirus 68 in recent outbreaks. PLoS One 2012; 7:e36005.
- Lu QB, Wo Y, Wang HY, et al. Detection of enterovirus 68 as one of the commonest types of enterovirus found in patients with acute respiratory tract infection in China. J Med Microbiol 2014; 63:408–414.
- Meijer A, van der Sanden S, Snijders BE, et al. Emergence and epidemic occurrence of enterovirus 68 respiratory infections in The Netherlands in 2010. Virology 2012; 423:49–57.
- Rahamat-Langendoen J, Riezebos-Brilman A, Borger R, et al. Upsurge of human enterovirus 68 infections in patients with severe respiratory tract infections. J Clin Virol 2011; 52:103–106.
- Tokarz R, Kapoor V, Wu W, et al. Longitudinal molecular microbial analysis of influenza-like illness in New York City, May 2009 through May 2010. Virol J 2011; 8:288.
- Uncapher CR, DeWitt CM, Colonno RJ. The major and minor group receptor families contain all but one human rhinovirus serotype. Virology 1991; 180:814–817.
- Imamura T, Okamoto M, Nakakita S, et al. Antigenic and receptor binding properties of enterovirus 68. J Virol 2014; 88:2374–2384.
- Karnauchow TM, Tolson DL, Harrison BA, Altman E, Lublin DM, Dimock K. The HeLa cell receptor for enterovirus 70 is decay-accelerating factor (CD55). J Virol 1996; 70:5143–5152.
- Selinka HC, Wolde A, Sauter M, Kandolf R, Klingel K. Virus-receptor interactions of coxsackie B viruses and their putative influence on cardiotropism. Med Microbiol Immunol 2004; 193:127–131.
- Piralla A, Girello A, Grignani M, et al. Phylogenetic characterization of enterovirus 68 strains in patients with respiratory syndromes in Italy. J Med Virol 2014; 86:1590–1593.
- Imamura T, Suzuki A, Lupisan S, et al. Detection of enterovirus 68 in serum from pediatric patients with pneumonia and their clinical outcomes. Influenza Other Respir Viruses 2014; 8:21–24.
- Imamura T, Fuji N, Suzuki A, et al. Enterovirus 68 among children with severe acute respiratory infection, the Philippines. Emerg Infect Dis 2011; 17:1430–1435.
- Kaida A, Kubo H, Sekiguchi J, et al. Enterovirus 68 in children with acute respiratory tract infections, Osaka, Japan. Emerg Infect Dis 2011; 17:1494–1497.
- Kreuter JD, Barnes A, McCarthy JE, et al. A fatal central nervous system enterovirus 68 infection. Arch Pathol Lab Med 2011; 135:793–796.
- Pastula DM, Aliabadi N, Haynes AK, et al. Acute neurologic illness of unknown etiology in children—Colorado, August-September 2014. MMWR Morb Mortal Wkly Rep 2014; 63:901–902.
- Ayscue P, Haren KV, Sheriff H, et al. Acute flaccid paralysis with anterior myelitis—California, June 2012–June 2014. MMWR Morb Mortal Wkly Rep 2014; 63:903–906.
- Abzug MJ. The enteroviruses: problems in need of treatments. J Infect 2014; 68(suppl 1):S108–S114.
- Pierce VM, Hodinka RL. Comparison of the GenMark Diagnostics eSensor respiratory viral panel to real-time PCR for detection of respiratory viruses in children. J Clin Microbiol 2012; 50:3458–3465.
- Non-polio enterovirus infection: enterovirus D68 (EV-D68)-CDC. 2014; www.cdc.gov/non-polio-enterovirus/about/ev-d68.html. Accessed November 21, 2014.
KEY POINTS
- EV-D68 is a respiratory virus that has genetic and biologic features that blur the distinction between the rhinoviruses and enteroviruses.
- Recognition of EV-D68 as an important cause of viral lower respiratory tract illness in children underscores the role of specific strain typing in advancing our understanding of the epidemiology of respiratory virus infections.
- Given the inability of commonly used clinical tests for rhinovirus to distinguish EV-D68 in the absence of strain-specific sequence data, caution needs to be used in attributing severe or acute lower respiratory illness to rhinovirus and in interpreting epidemiologic associations between asthma and rhinovirus.
- Emerging data suggest that, in addition to its important role in pediatric respiratory illness, EV-D68 may cause systemic disease, especially acute neurologic disease.
Retroperitoneal cyst hemorrhage in polycystic kidney disease
A 59-year-old man with autosomal dominant polycystic kidney disease (ADPKD), end-stage renal disease on hemodialysis, hypertension, and diverticulosis presented with acute pain in the left lower abdomen. The pain began 4 days previously, was dull and nonradiating, was relieved partially with hydrocodone-acetaminophen, and had no clear exacerbating factors. Two days before presentation, he developed a fever with chills. He reported no recent dysuria, diarrhea, hematuria, hematochezia, or melena. He had not been taking anticoagulants or nonsteroidal anti-inflammatory drugs, and he had no history of heavy lifting or trauma.
His temperature was 38.5˚C (101.3˚F), blood pressure 141/60 mm Hg (normal for this patient). On examination, his left lower quadrant was tender with voluntary guarding. Also present was a reducible ventral hernia, which was not new.
His hemoglobin level was 10.6 g/dL (reference range 13.0–17.0), which had dropped from a previous value of 13.7 g/dL.
Computed tomography of the abdomen and pelvis revealed a ruptured retroperitoneal hemorrhagic cyst (Figure 1) in the inferior aspect of the left kidney extending into the fascia of Gerota.
Since his vital signs were stable, he was managed supportively during his hospitalization with intravenous fluids, serial hemoglobin checks, and analgesia. He was eventually discharged home in good condition.
CYST HEMORRHAGE IN POLYCYSTIC KIDNEY DISEASE
ADPKD is a relatively common, inherited systemic disease that leads to cyst formation, primarily in the kidneys but also in the liver (94%), seminal vesicles (40%), pancreas (9%), arachnoid membrane (8%), and spinal meningeal area (2%).1
In addition to cyst formation in multiple organs, ADPKD can have extrarenal manifestations such as connective-tissue abnormalities (including mitral valve prolapse) (25%), abdominal hernia (10%), and intracranial aneurysm (8%).1 Management of extrarenal complications of ADPKD is discussed in detail elsewhere.2
The estimated prevalence of ADPKD is 1 of every 400 to 1,000 live births. However, given that ADPKD is often clinically silent, it is diagnosed during the lifetime of fewer than half of people who have it.3
Most ADPKD cases are caused by mutations in either the PKD1 or PKD2 gene.4,5 Although the mechanism of cyst formation in ADPKD is still unclear, it is known that PKD1 and PKD2 encode proteins called polycystin-1 and polycystin-2, respectively. Polycystin-1 is a membrane protein found in renal tubular epithelia, hepatic bile ductules, and pancreatic ducts. Polycystin-2 is involved in cell calcium signaling and has been identified in the renal distal tubules, collecting duct, and thick ascending limb. Mutations in PKD1 and PKD2 are thought to contribute to cyst formation, with PKD1 mutations associated with earlier onset and more severe development of renal and extrarenal cysts.
Cyst hemorrhage
Hemorrhage of renal cysts is a well-known complication, occurring in up to 70% of patients with ADPKD.6 Renal cyst hemorrhage often presents clinically as flank pain with point tenderness or hematuria, or both. Flank pain results from hemorrhage into a cyst with consequent distention of the renal capsule, whereas hematuria results from rupture of a cyst into the collecting system.
Spontaneous nonfatal retroperitoneal cyst hemorrhage, as in our patient, is rare. Indeed, in one series reviewing the abdominal computed tomographic findings of 66 patients with ADPKD, only 2 patients (3%) had perinephric hematomas in the absence of recent trauma.6
Management of cyst hemorrhage is primarily conservative. Pain associated with cyst hemorrhage is managed conservatively with bed rest, intravenous hydration, and analgesics (but not nonsteroidal anti-inflammatory drugs).
Hematuria is also managed conservatively with bedrest and intravenous hydration, and most episodes of hematuria are self-limiting and last 2 to 7 days. However, if excessive bleeding occurs, the patient may be at risk of urinary tract obstruction from clot formation. If obstruction occurs and persists beyond 2 weeks, then ureteral stenting may be necessary. In rare cases of prolonged, severe bleeding with extensive subcapsular or retroperitoneal hematomas, patients require hospitalization, transfusion, or percutaneous transcatheter embolization of the renal artery. If such efforts are not successful, surgery, including nephrectomy, may be required to control the hemorrhage.2
Other causes of abdominal pain
In addition to renal cyst hemorrhage, the differential diagnosis of abdominal pain in a patient with ADPKD includes cyst enlargement causing stretching of the renal capsule or traction on the renal pedicle, cyst infection, nephrolithiasis, pyelonephritis, and rarely, tumors including renal cell carcinoma.
Unlike cyst rupture and hemorrhage, which are associated with point tenderness, cyst infection often manifests as diffuse, usually unilateral flank pain with associated fever, nausea, malaise, and leukocytosis. Our patient had none of these except for fever, which can also occur in cyst hemorrhage.
Nephrolithiasis occurs in up to 35% of patients with ADPKD,7 but no kidney stones were seen on computed tomography in our patient.
Pyelonephritis was unlikely in our patient, given that he had no significant white blood cells in his urinalysis and no leukocytosis.
Abdominal and pelvic imaging did not reveal any tumors in our patient.
- Pirson Y. Extrarenal manifestations of autosomal dominant polycystic kidney disease. Adv Chronic Kidney Dis 2010; 17:173–180.
- Harris PC, Torres VE. Polycystic kidney disease, autosomal dominant. In: Pagon RA, Adam MP, Bird TD, et al, editors. GeneReviews. Seattle, WA: University of Washington, Seattle; 1993–2014.
- Grantham JJ. Clinical practice. Autosomal dominant polycystic kidney disease. N Engl J Med 2008; 359:1477–1485.
- Peters DJ, Spruit L, Saris JJ, et al. Chromosome 4 localization of a second gene for autosomal dominant polycystic kidney disease. Nat Genet 1993; 5:359–362.
- Rossetti S, Consugar MB, Chapman AB, et al; CRISP Consortium. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2007; 18:2143–2160.
- Levine E, Grantham JJ. Perinephric hemorrhage in autosomal dominant polycystic kidney disease: CT and MR findings. J Comput Assist Tomogr 1987; 11:108–111.
- Delaney VB, Adler S, Bruns FJ, Licinia M, Segel DP, Fraley DS. Autosomal dominant polycystic kidney disease: presentation, complications, and prognosis. Am J Kidney Dis 1985; 5:104–111.
A 59-year-old man with autosomal dominant polycystic kidney disease (ADPKD), end-stage renal disease on hemodialysis, hypertension, and diverticulosis presented with acute pain in the left lower abdomen. The pain began 4 days previously, was dull and nonradiating, was relieved partially with hydrocodone-acetaminophen, and had no clear exacerbating factors. Two days before presentation, he developed a fever with chills. He reported no recent dysuria, diarrhea, hematuria, hematochezia, or melena. He had not been taking anticoagulants or nonsteroidal anti-inflammatory drugs, and he had no history of heavy lifting or trauma.
His temperature was 38.5˚C (101.3˚F), blood pressure 141/60 mm Hg (normal for this patient). On examination, his left lower quadrant was tender with voluntary guarding. Also present was a reducible ventral hernia, which was not new.
His hemoglobin level was 10.6 g/dL (reference range 13.0–17.0), which had dropped from a previous value of 13.7 g/dL.
Computed tomography of the abdomen and pelvis revealed a ruptured retroperitoneal hemorrhagic cyst (Figure 1) in the inferior aspect of the left kidney extending into the fascia of Gerota.
Since his vital signs were stable, he was managed supportively during his hospitalization with intravenous fluids, serial hemoglobin checks, and analgesia. He was eventually discharged home in good condition.
CYST HEMORRHAGE IN POLYCYSTIC KIDNEY DISEASE
ADPKD is a relatively common, inherited systemic disease that leads to cyst formation, primarily in the kidneys but also in the liver (94%), seminal vesicles (40%), pancreas (9%), arachnoid membrane (8%), and spinal meningeal area (2%).1
In addition to cyst formation in multiple organs, ADPKD can have extrarenal manifestations such as connective-tissue abnormalities (including mitral valve prolapse) (25%), abdominal hernia (10%), and intracranial aneurysm (8%).1 Management of extrarenal complications of ADPKD is discussed in detail elsewhere.2
The estimated prevalence of ADPKD is 1 of every 400 to 1,000 live births. However, given that ADPKD is often clinically silent, it is diagnosed during the lifetime of fewer than half of people who have it.3
Most ADPKD cases are caused by mutations in either the PKD1 or PKD2 gene.4,5 Although the mechanism of cyst formation in ADPKD is still unclear, it is known that PKD1 and PKD2 encode proteins called polycystin-1 and polycystin-2, respectively. Polycystin-1 is a membrane protein found in renal tubular epithelia, hepatic bile ductules, and pancreatic ducts. Polycystin-2 is involved in cell calcium signaling and has been identified in the renal distal tubules, collecting duct, and thick ascending limb. Mutations in PKD1 and PKD2 are thought to contribute to cyst formation, with PKD1 mutations associated with earlier onset and more severe development of renal and extrarenal cysts.
Cyst hemorrhage
Hemorrhage of renal cysts is a well-known complication, occurring in up to 70% of patients with ADPKD.6 Renal cyst hemorrhage often presents clinically as flank pain with point tenderness or hematuria, or both. Flank pain results from hemorrhage into a cyst with consequent distention of the renal capsule, whereas hematuria results from rupture of a cyst into the collecting system.
Spontaneous nonfatal retroperitoneal cyst hemorrhage, as in our patient, is rare. Indeed, in one series reviewing the abdominal computed tomographic findings of 66 patients with ADPKD, only 2 patients (3%) had perinephric hematomas in the absence of recent trauma.6
Management of cyst hemorrhage is primarily conservative. Pain associated with cyst hemorrhage is managed conservatively with bed rest, intravenous hydration, and analgesics (but not nonsteroidal anti-inflammatory drugs).
Hematuria is also managed conservatively with bedrest and intravenous hydration, and most episodes of hematuria are self-limiting and last 2 to 7 days. However, if excessive bleeding occurs, the patient may be at risk of urinary tract obstruction from clot formation. If obstruction occurs and persists beyond 2 weeks, then ureteral stenting may be necessary. In rare cases of prolonged, severe bleeding with extensive subcapsular or retroperitoneal hematomas, patients require hospitalization, transfusion, or percutaneous transcatheter embolization of the renal artery. If such efforts are not successful, surgery, including nephrectomy, may be required to control the hemorrhage.2
Other causes of abdominal pain
In addition to renal cyst hemorrhage, the differential diagnosis of abdominal pain in a patient with ADPKD includes cyst enlargement causing stretching of the renal capsule or traction on the renal pedicle, cyst infection, nephrolithiasis, pyelonephritis, and rarely, tumors including renal cell carcinoma.
Unlike cyst rupture and hemorrhage, which are associated with point tenderness, cyst infection often manifests as diffuse, usually unilateral flank pain with associated fever, nausea, malaise, and leukocytosis. Our patient had none of these except for fever, which can also occur in cyst hemorrhage.
Nephrolithiasis occurs in up to 35% of patients with ADPKD,7 but no kidney stones were seen on computed tomography in our patient.
Pyelonephritis was unlikely in our patient, given that he had no significant white blood cells in his urinalysis and no leukocytosis.
Abdominal and pelvic imaging did not reveal any tumors in our patient.
A 59-year-old man with autosomal dominant polycystic kidney disease (ADPKD), end-stage renal disease on hemodialysis, hypertension, and diverticulosis presented with acute pain in the left lower abdomen. The pain began 4 days previously, was dull and nonradiating, was relieved partially with hydrocodone-acetaminophen, and had no clear exacerbating factors. Two days before presentation, he developed a fever with chills. He reported no recent dysuria, diarrhea, hematuria, hematochezia, or melena. He had not been taking anticoagulants or nonsteroidal anti-inflammatory drugs, and he had no history of heavy lifting or trauma.
His temperature was 38.5˚C (101.3˚F), blood pressure 141/60 mm Hg (normal for this patient). On examination, his left lower quadrant was tender with voluntary guarding. Also present was a reducible ventral hernia, which was not new.
His hemoglobin level was 10.6 g/dL (reference range 13.0–17.0), which had dropped from a previous value of 13.7 g/dL.
Computed tomography of the abdomen and pelvis revealed a ruptured retroperitoneal hemorrhagic cyst (Figure 1) in the inferior aspect of the left kidney extending into the fascia of Gerota.
Since his vital signs were stable, he was managed supportively during his hospitalization with intravenous fluids, serial hemoglobin checks, and analgesia. He was eventually discharged home in good condition.
CYST HEMORRHAGE IN POLYCYSTIC KIDNEY DISEASE
ADPKD is a relatively common, inherited systemic disease that leads to cyst formation, primarily in the kidneys but also in the liver (94%), seminal vesicles (40%), pancreas (9%), arachnoid membrane (8%), and spinal meningeal area (2%).1
In addition to cyst formation in multiple organs, ADPKD can have extrarenal manifestations such as connective-tissue abnormalities (including mitral valve prolapse) (25%), abdominal hernia (10%), and intracranial aneurysm (8%).1 Management of extrarenal complications of ADPKD is discussed in detail elsewhere.2
The estimated prevalence of ADPKD is 1 of every 400 to 1,000 live births. However, given that ADPKD is often clinically silent, it is diagnosed during the lifetime of fewer than half of people who have it.3
Most ADPKD cases are caused by mutations in either the PKD1 or PKD2 gene.4,5 Although the mechanism of cyst formation in ADPKD is still unclear, it is known that PKD1 and PKD2 encode proteins called polycystin-1 and polycystin-2, respectively. Polycystin-1 is a membrane protein found in renal tubular epithelia, hepatic bile ductules, and pancreatic ducts. Polycystin-2 is involved in cell calcium signaling and has been identified in the renal distal tubules, collecting duct, and thick ascending limb. Mutations in PKD1 and PKD2 are thought to contribute to cyst formation, with PKD1 mutations associated with earlier onset and more severe development of renal and extrarenal cysts.
Cyst hemorrhage
Hemorrhage of renal cysts is a well-known complication, occurring in up to 70% of patients with ADPKD.6 Renal cyst hemorrhage often presents clinically as flank pain with point tenderness or hematuria, or both. Flank pain results from hemorrhage into a cyst with consequent distention of the renal capsule, whereas hematuria results from rupture of a cyst into the collecting system.
Spontaneous nonfatal retroperitoneal cyst hemorrhage, as in our patient, is rare. Indeed, in one series reviewing the abdominal computed tomographic findings of 66 patients with ADPKD, only 2 patients (3%) had perinephric hematomas in the absence of recent trauma.6
Management of cyst hemorrhage is primarily conservative. Pain associated with cyst hemorrhage is managed conservatively with bed rest, intravenous hydration, and analgesics (but not nonsteroidal anti-inflammatory drugs).
Hematuria is also managed conservatively with bedrest and intravenous hydration, and most episodes of hematuria are self-limiting and last 2 to 7 days. However, if excessive bleeding occurs, the patient may be at risk of urinary tract obstruction from clot formation. If obstruction occurs and persists beyond 2 weeks, then ureteral stenting may be necessary. In rare cases of prolonged, severe bleeding with extensive subcapsular or retroperitoneal hematomas, patients require hospitalization, transfusion, or percutaneous transcatheter embolization of the renal artery. If such efforts are not successful, surgery, including nephrectomy, may be required to control the hemorrhage.2
Other causes of abdominal pain
In addition to renal cyst hemorrhage, the differential diagnosis of abdominal pain in a patient with ADPKD includes cyst enlargement causing stretching of the renal capsule or traction on the renal pedicle, cyst infection, nephrolithiasis, pyelonephritis, and rarely, tumors including renal cell carcinoma.
Unlike cyst rupture and hemorrhage, which are associated with point tenderness, cyst infection often manifests as diffuse, usually unilateral flank pain with associated fever, nausea, malaise, and leukocytosis. Our patient had none of these except for fever, which can also occur in cyst hemorrhage.
Nephrolithiasis occurs in up to 35% of patients with ADPKD,7 but no kidney stones were seen on computed tomography in our patient.
Pyelonephritis was unlikely in our patient, given that he had no significant white blood cells in his urinalysis and no leukocytosis.
Abdominal and pelvic imaging did not reveal any tumors in our patient.
- Pirson Y. Extrarenal manifestations of autosomal dominant polycystic kidney disease. Adv Chronic Kidney Dis 2010; 17:173–180.
- Harris PC, Torres VE. Polycystic kidney disease, autosomal dominant. In: Pagon RA, Adam MP, Bird TD, et al, editors. GeneReviews. Seattle, WA: University of Washington, Seattle; 1993–2014.
- Grantham JJ. Clinical practice. Autosomal dominant polycystic kidney disease. N Engl J Med 2008; 359:1477–1485.
- Peters DJ, Spruit L, Saris JJ, et al. Chromosome 4 localization of a second gene for autosomal dominant polycystic kidney disease. Nat Genet 1993; 5:359–362.
- Rossetti S, Consugar MB, Chapman AB, et al; CRISP Consortium. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2007; 18:2143–2160.
- Levine E, Grantham JJ. Perinephric hemorrhage in autosomal dominant polycystic kidney disease: CT and MR findings. J Comput Assist Tomogr 1987; 11:108–111.
- Delaney VB, Adler S, Bruns FJ, Licinia M, Segel DP, Fraley DS. Autosomal dominant polycystic kidney disease: presentation, complications, and prognosis. Am J Kidney Dis 1985; 5:104–111.
- Pirson Y. Extrarenal manifestations of autosomal dominant polycystic kidney disease. Adv Chronic Kidney Dis 2010; 17:173–180.
- Harris PC, Torres VE. Polycystic kidney disease, autosomal dominant. In: Pagon RA, Adam MP, Bird TD, et al, editors. GeneReviews. Seattle, WA: University of Washington, Seattle; 1993–2014.
- Grantham JJ. Clinical practice. Autosomal dominant polycystic kidney disease. N Engl J Med 2008; 359:1477–1485.
- Peters DJ, Spruit L, Saris JJ, et al. Chromosome 4 localization of a second gene for autosomal dominant polycystic kidney disease. Nat Genet 1993; 5:359–362.
- Rossetti S, Consugar MB, Chapman AB, et al; CRISP Consortium. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2007; 18:2143–2160.
- Levine E, Grantham JJ. Perinephric hemorrhage in autosomal dominant polycystic kidney disease: CT and MR findings. J Comput Assist Tomogr 1987; 11:108–111.
- Delaney VB, Adler S, Bruns FJ, Licinia M, Segel DP, Fraley DS. Autosomal dominant polycystic kidney disease: presentation, complications, and prognosis. Am J Kidney Dis 1985; 5:104–111.
Woman, 32, With Crusty Red Blisters
A 32-year-old Korean woman presented with a rash on her scalp, face, palms, soles, and genital region and with sores in the oral cavity. The blisters were red and flat with some crusting, particularly on the scalp and face. The patient described the blisters as very painful, adding that it hurt to walk, grasp objects, and drink fluids. Associated symptoms included painful urination, sore throat, malaise, and fever of up to 103°F. She was taking acetaminophen and ibuprofen to alleviate the fever and pain.
Medical history was unremarkable. Social history was negative for recent changes in sexual partner or travel to foreign countries.

Physical examination revealed numerous flat, erythematous lesions. Lesions on the face and scalp had developed a weeping, honey-colored crust (see Figure 1 and Figure 2). The lesions were tender to the touch, particularly on the palms and soles.
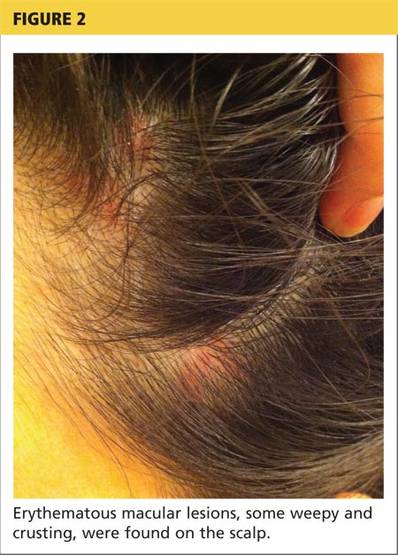
Further questioning revealed that the patient’s 18-month-old son had exhibited similar symptoms two to three days prior to her illness.
Continue for differential diagnosis >>
DIFFERENTIAL DIAGNOSIS
Because multiple bacterial and viral diseases manifest in this fashion, the differential diagnosis included the following disorders:
Erythema multiforme. This skin condition may result from an allergic or hypersensitivity reaction to certain drugs or from infections. Infections that can cause erythema multiforme include herpes simplex virus and mycoplasma. Patients present with lesions on the palms (see Figure 3), soles, extremities, face, or trunk. The lesions can appear as a nodule, papule, macule, or vesicle. Initially, in the mild form, the lesions may appear as hives or target-shaped rashes, occurring on the face and acral surfaces. A severe form of erythema multiforme, known as Stevens-Johnson syndrome, is characterized by rash, mucosal involvement, and systemic symptoms.1
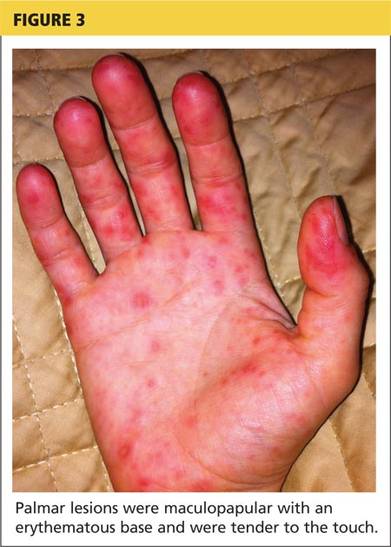
Herpes zoster. This viral infection is caused by the varicella-zoster virus, which also causes chicken pox. The virus lies dormant within a single sensory ganglion and may reappear as shingles along the dermatome of that nerve. Patients may experience burning or shooting pain with tingling or itching before the rash appears; vesicular lesions with erythematous bases appear days later. The rash occurs unilaterally on the body or face and does not cross the midline. A viral culture may be obtained for identification.2
Herpetic gingivostomatitis. This infection is most commonly caused by herpes simplex virus type 1, the same virus that causes cold sores. Patients may present with ulcerations along the buccal mucosa and gums. The infection manifests with systemic symptoms, including malaise, fever, irritability, and cervical adenopathy. A viral culture will identify the etiology.3
Impetigo. This skin infection is typically caused by bacteria, predominantly Staphylococcus aureus, Streptococcus pyogenes, or a combination. Infections generally occur after a break in the skin surface. The most common presentation is a rash that spreads to different parts of the body after scratching. Skin lesions can occur on the face, lips, or extremities. Initially vesicular, the lesions generally form a honey-colored crust after fluid discharge. The clinician should take a skin or fluid sample from the lesion to culture, which may identify the pathogen.4
Syphilis. This sexually transmitted infection is caused by the spirochete Treponema pallidum. In primary syphilis, patients can develop a painless sore, or chancre, on the genitals, rectal area, or mouth. If left untreated, the disease can progress to secondary syphilis, manifesting as a pale pink or reddish maculopapular rash on the palms and soles. The rash can be associated with fever, sore throat, myalgia, and fatigue. It is important to rule out syphilis because, left untreated, it can lead to cardiac and neurologic complications. Screening tests include VDRL and the rapid plasma reagin (RPR) test, both of which assess for antibodies to the organism, and dark field microscopy of ulcerations to identify the organism.5
Also included in the differential diagnosis for the patient’s symptoms was hand-foot-mouth disease (HFMD), discussed below.
Next page: Discussion >>
DISCUSSION
HFMD is an acute viral illness most often affecting children younger than 5 and occurring in summer to early fall months. HFMD manifests with fever and papulovesicular eruptions; lesions often appear in the oral cavity first, spreading to the palms, soles, and buttocks. Route of transmission is usually fecal-oral or through respiratory droplets, oral secretions, or direct contact with fluid-filled vesicles.6-8 The highly contagious nature of the virus causes it to spread to close contacts and family members and leads to outbreaks in schools and daycare centers.9,10
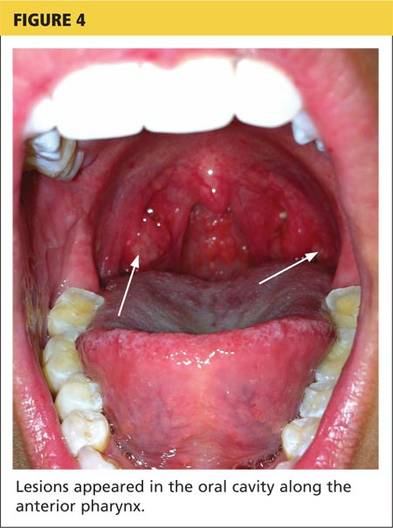
In the United States, the most common etiology of HFMD is coxsackievirus A16.7 Another causative agent, enterovirus 71 (EV71), has been found responsible for HFMD epidemics in southeast Asia and Australia.11,12 Recently, the coxsackievirus A6 (CV A6) strain has been linked to outbreaks of HFMD.7,9 This strain may produce an atypical manifestation of skin lesions on the face, trunk, and extremities. The lesions may also appear larger than usual and have a vesiculobullous rather than the more typical papulovesicular appearance. The course of the illness differs in severity depending on the strain of the virus causing HFMD.9
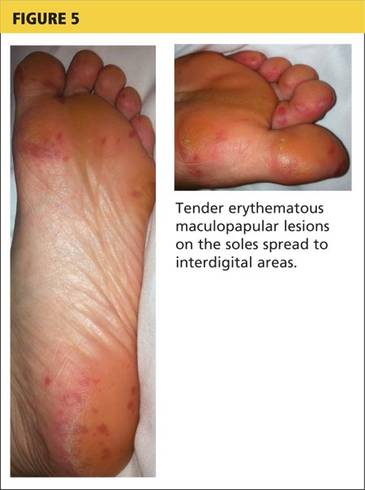
CLINICAL PRESENTATION
The acute phase of HFMD typically begins with prodromal symptoms such as fever, malaise, and sore throat. Erythematous ulcerations usually appear in the oral cavity first (see Figure 4) and often cause symptoms such as sore throat, dysphagia, or dryness. As the disease progresses, cutaneous lesions spread to the face, extremities, interdigital areas (see Figure 5), trunk, and perianal area (which may cause dysuria). The lesions may initially appear as erythematous macules or papules, transforming to vesicles as the disease progresses. The mucocutaneous lesions are usually asymptomatic but can be tender to touch or pressure and may leak fluid.7,10
In only a few cases—caused by CV A6—have lesions in the scalp been reported; the mechanism of action is unknown.10 Instances of lesions invading the nails have been reported, causing desquamation and shedding. This condition is known as onychomadesis.9,11
Continue for diagnosis >>
DIAGNOSIS
Serologic testing and viral cultures can identify the exact strain of virus causing HFMD and are particularly useful in unusual presentations. Polymerase chain reaction (PCR) testing yields a high sensitivity and specificity for the causative agent. Histologic examination of skin biopsies may show lymphocytic infiltrates and areas of degeneration along the epidermis. However, most cases are diagnosed based on clinical presentation alone.6,12
TREATMENT AND MANAGEMENT
Management of HFMD is primarily symptomatic, consisting of supportive care that includes use of antipyretics, NSAIDs, and adequate fluid intake to prevent dehydration. The disease is usually self-limited, resolving within seven to 10 days without sequelae.6,9 Aseptic meningitis and other severe complications (especially pulmonary and neurologic), most often associated with EV71 infection, can occur in vulnerable populations, including elderly, pregnant, and immunocompromised patients.9,11 Because the virus is excreted directly from palmar lesions onto the hands, proper hygiene and handwashing techniques offer an exceptionally strong protective effect, preventing transmission and reducing morbidity.8
PATIENT OUTCOME
Based on clinical findings and patient history, the patient was diagnosed with HFMD, which she contracted from her son. Laboratory testing and viral cultures were deemed unnecessary in this case. Treatment was symptomatic, and her skin lesions resolved in one to two weeks.
At follow-up, the patient stated her skin lesions resolved completely without leaving any scars. She also indicated that her nails peeled and shed approximately four weeks after diagnosis, but began to regrow normally four months after diagnosis.
CONCLUSION
Clinicians need to recognize that, although it is uncommon outside the pediatric population, HFMD may occur in adults with intact immune systems. The presentation of HFMD in adults may be atypical, including cutaneous lesions in the scalp and shedding of the nails several weeks after diagnosis. Depending on the viral strain involved, adult patients may have more severe illness and may take longer to recover. Therefore, early diagnosis is important to help prevent the spread of infection and reduce the severity of complications.
REFERENCES
1. Patel NN, Patel DN. Erythema multiforme syndrome. Am J Med. 2009;122(7):623-625.
2. Bader MS. Herpes zoster: diagnostic, therapeutic, and preventive approaches. Postgrad Med. 2013;125(5):78-91.
3. Avci O, Ertam I. Viral infections of the face. Clin Derm. 2014;32:715-733.
4. Hartman-Adams H, Banvard C, Juckett G. Impetigo: diagnosis and treatment. Am Fam Physician. 2014;90(4):229-235.
5. Markle W, Conti T, Kad M. Sexually transmitted diseases. Prim Care Clin Office Pract. 2013;40:557-587.
6. Shin JU, Oh SH, Lee JH. A case of hand-foot-mouth disease in an immunocompetent adult. Ann Dermatol. 2010;22(2):216-218.
7. CDC. Notes from the field: severe hand, foot, and mouth disease associated with coxsackievirus A6 - Alabama, Connecticut, California, and Nevada, November 2011-February 2012. MMWR Morb Mortal Wkly Rep. 2012;61(12):213-214.
8. Ruan F, Yang T, Ma H, et al. Risk factors for hand, foot, and mouth disease and herpangina and the preventive effect of hand-washing. Pediatrics. 2011;127(4):e898-e904.
9. Kaminska K, Martinetti G, Lucchini R, et al. Coxsackievirus A6 and hand, foot and mouth Disease: three case reports of familial child-to-immunocompetent adult transmission and a literature review. Case Rep Dermatol. 2013;5(2):203-209.
10. Lønnberg AS, Elberling J, Fischer TK, Skov L. Two cases of hand, foot, and mouth disease involving the scalp. Acta Derm Venereol. 2013;93(4):467-468.
11. Osterback R, Vuorinen T, Linna M, et al. Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerging Infect Dis. 2009;15(9):1485-1488.
12. Shea YF, Chan CY, Hung IFN, Chan KH. Hand, foot and mouth disease in an immunocompetent adult due to Coxsackievirus A6. Hong Kong Med J. 2013;19(3):262-264.
A 32-year-old Korean woman presented with a rash on her scalp, face, palms, soles, and genital region and with sores in the oral cavity. The blisters were red and flat with some crusting, particularly on the scalp and face. The patient described the blisters as very painful, adding that it hurt to walk, grasp objects, and drink fluids. Associated symptoms included painful urination, sore throat, malaise, and fever of up to 103°F. She was taking acetaminophen and ibuprofen to alleviate the fever and pain.
Medical history was unremarkable. Social history was negative for recent changes in sexual partner or travel to foreign countries.
Physical examination revealed numerous flat, erythematous lesions. Lesions on the face and scalp had developed a weeping, honey-colored crust (see Figure 1 and Figure 2). The lesions were tender to the touch, particularly on the palms and soles.
Further questioning revealed that the patient’s 18-month-old son had exhibited similar symptoms two to three days prior to her illness.
Continue for differential diagnosis >>
DIFFERENTIAL DIAGNOSIS
Because multiple bacterial and viral diseases manifest in this fashion, the differential diagnosis included the following disorders:
Erythema multiforme. This skin condition may result from an allergic or hypersensitivity reaction to certain drugs or from infections. Infections that can cause erythema multiforme include herpes simplex virus and mycoplasma. Patients present with lesions on the palms (see Figure 3), soles, extremities, face, or trunk. The lesions can appear as a nodule, papule, macule, or vesicle. Initially, in the mild form, the lesions may appear as hives or target-shaped rashes, occurring on the face and acral surfaces. A severe form of erythema multiforme, known as Stevens-Johnson syndrome, is characterized by rash, mucosal involvement, and systemic symptoms.1
Herpes zoster. This viral infection is caused by the varicella-zoster virus, which also causes chicken pox. The virus lies dormant within a single sensory ganglion and may reappear as shingles along the dermatome of that nerve. Patients may experience burning or shooting pain with tingling or itching before the rash appears; vesicular lesions with erythematous bases appear days later. The rash occurs unilaterally on the body or face and does not cross the midline. A viral culture may be obtained for identification.2
Herpetic gingivostomatitis. This infection is most commonly caused by herpes simplex virus type 1, the same virus that causes cold sores. Patients may present with ulcerations along the buccal mucosa and gums. The infection manifests with systemic symptoms, including malaise, fever, irritability, and cervical adenopathy. A viral culture will identify the etiology.3
Impetigo. This skin infection is typically caused by bacteria, predominantly Staphylococcus aureus, Streptococcus pyogenes, or a combination. Infections generally occur after a break in the skin surface. The most common presentation is a rash that spreads to different parts of the body after scratching. Skin lesions can occur on the face, lips, or extremities. Initially vesicular, the lesions generally form a honey-colored crust after fluid discharge. The clinician should take a skin or fluid sample from the lesion to culture, which may identify the pathogen.4
Syphilis. This sexually transmitted infection is caused by the spirochete Treponema pallidum. In primary syphilis, patients can develop a painless sore, or chancre, on the genitals, rectal area, or mouth. If left untreated, the disease can progress to secondary syphilis, manifesting as a pale pink or reddish maculopapular rash on the palms and soles. The rash can be associated with fever, sore throat, myalgia, and fatigue. It is important to rule out syphilis because, left untreated, it can lead to cardiac and neurologic complications. Screening tests include VDRL and the rapid plasma reagin (RPR) test, both of which assess for antibodies to the organism, and dark field microscopy of ulcerations to identify the organism.5
Also included in the differential diagnosis for the patient’s symptoms was hand-foot-mouth disease (HFMD), discussed below.
Next page: Discussion >>
DISCUSSION
HFMD is an acute viral illness most often affecting children younger than 5 and occurring in summer to early fall months. HFMD manifests with fever and papulovesicular eruptions; lesions often appear in the oral cavity first, spreading to the palms, soles, and buttocks. Route of transmission is usually fecal-oral or through respiratory droplets, oral secretions, or direct contact with fluid-filled vesicles.6-8 The highly contagious nature of the virus causes it to spread to close contacts and family members and leads to outbreaks in schools and daycare centers.9,10
In the United States, the most common etiology of HFMD is coxsackievirus A16.7 Another causative agent, enterovirus 71 (EV71), has been found responsible for HFMD epidemics in southeast Asia and Australia.11,12 Recently, the coxsackievirus A6 (CV A6) strain has been linked to outbreaks of HFMD.7,9 This strain may produce an atypical manifestation of skin lesions on the face, trunk, and extremities. The lesions may also appear larger than usual and have a vesiculobullous rather than the more typical papulovesicular appearance. The course of the illness differs in severity depending on the strain of the virus causing HFMD.9
CLINICAL PRESENTATION
The acute phase of HFMD typically begins with prodromal symptoms such as fever, malaise, and sore throat. Erythematous ulcerations usually appear in the oral cavity first (see Figure 4) and often cause symptoms such as sore throat, dysphagia, or dryness. As the disease progresses, cutaneous lesions spread to the face, extremities, interdigital areas (see Figure 5), trunk, and perianal area (which may cause dysuria). The lesions may initially appear as erythematous macules or papules, transforming to vesicles as the disease progresses. The mucocutaneous lesions are usually asymptomatic but can be tender to touch or pressure and may leak fluid.7,10
In only a few cases—caused by CV A6—have lesions in the scalp been reported; the mechanism of action is unknown.10 Instances of lesions invading the nails have been reported, causing desquamation and shedding. This condition is known as onychomadesis.9,11
Continue for diagnosis >>
DIAGNOSIS
Serologic testing and viral cultures can identify the exact strain of virus causing HFMD and are particularly useful in unusual presentations. Polymerase chain reaction (PCR) testing yields a high sensitivity and specificity for the causative agent. Histologic examination of skin biopsies may show lymphocytic infiltrates and areas of degeneration along the epidermis. However, most cases are diagnosed based on clinical presentation alone.6,12
TREATMENT AND MANAGEMENT
Management of HFMD is primarily symptomatic, consisting of supportive care that includes use of antipyretics, NSAIDs, and adequate fluid intake to prevent dehydration. The disease is usually self-limited, resolving within seven to 10 days without sequelae.6,9 Aseptic meningitis and other severe complications (especially pulmonary and neurologic), most often associated with EV71 infection, can occur in vulnerable populations, including elderly, pregnant, and immunocompromised patients.9,11 Because the virus is excreted directly from palmar lesions onto the hands, proper hygiene and handwashing techniques offer an exceptionally strong protective effect, preventing transmission and reducing morbidity.8
PATIENT OUTCOME
Based on clinical findings and patient history, the patient was diagnosed with HFMD, which she contracted from her son. Laboratory testing and viral cultures were deemed unnecessary in this case. Treatment was symptomatic, and her skin lesions resolved in one to two weeks.
At follow-up, the patient stated her skin lesions resolved completely without leaving any scars. She also indicated that her nails peeled and shed approximately four weeks after diagnosis, but began to regrow normally four months after diagnosis.
CONCLUSION
Clinicians need to recognize that, although it is uncommon outside the pediatric population, HFMD may occur in adults with intact immune systems. The presentation of HFMD in adults may be atypical, including cutaneous lesions in the scalp and shedding of the nails several weeks after diagnosis. Depending on the viral strain involved, adult patients may have more severe illness and may take longer to recover. Therefore, early diagnosis is important to help prevent the spread of infection and reduce the severity of complications.
REFERENCES
1. Patel NN, Patel DN. Erythema multiforme syndrome. Am J Med. 2009;122(7):623-625.
2. Bader MS. Herpes zoster: diagnostic, therapeutic, and preventive approaches. Postgrad Med. 2013;125(5):78-91.
3. Avci O, Ertam I. Viral infections of the face. Clin Derm. 2014;32:715-733.
4. Hartman-Adams H, Banvard C, Juckett G. Impetigo: diagnosis and treatment. Am Fam Physician. 2014;90(4):229-235.
5. Markle W, Conti T, Kad M. Sexually transmitted diseases. Prim Care Clin Office Pract. 2013;40:557-587.
6. Shin JU, Oh SH, Lee JH. A case of hand-foot-mouth disease in an immunocompetent adult. Ann Dermatol. 2010;22(2):216-218.
7. CDC. Notes from the field: severe hand, foot, and mouth disease associated with coxsackievirus A6 - Alabama, Connecticut, California, and Nevada, November 2011-February 2012. MMWR Morb Mortal Wkly Rep. 2012;61(12):213-214.
8. Ruan F, Yang T, Ma H, et al. Risk factors for hand, foot, and mouth disease and herpangina and the preventive effect of hand-washing. Pediatrics. 2011;127(4):e898-e904.
9. Kaminska K, Martinetti G, Lucchini R, et al. Coxsackievirus A6 and hand, foot and mouth Disease: three case reports of familial child-to-immunocompetent adult transmission and a literature review. Case Rep Dermatol. 2013;5(2):203-209.
10. Lønnberg AS, Elberling J, Fischer TK, Skov L. Two cases of hand, foot, and mouth disease involving the scalp. Acta Derm Venereol. 2013;93(4):467-468.
11. Osterback R, Vuorinen T, Linna M, et al. Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerging Infect Dis. 2009;15(9):1485-1488.
12. Shea YF, Chan CY, Hung IFN, Chan KH. Hand, foot and mouth disease in an immunocompetent adult due to Coxsackievirus A6. Hong Kong Med J. 2013;19(3):262-264.
A 32-year-old Korean woman presented with a rash on her scalp, face, palms, soles, and genital region and with sores in the oral cavity. The blisters were red and flat with some crusting, particularly on the scalp and face. The patient described the blisters as very painful, adding that it hurt to walk, grasp objects, and drink fluids. Associated symptoms included painful urination, sore throat, malaise, and fever of up to 103°F. She was taking acetaminophen and ibuprofen to alleviate the fever and pain.
Medical history was unremarkable. Social history was negative for recent changes in sexual partner or travel to foreign countries.
Physical examination revealed numerous flat, erythematous lesions. Lesions on the face and scalp had developed a weeping, honey-colored crust (see Figure 1 and Figure 2). The lesions were tender to the touch, particularly on the palms and soles.
Further questioning revealed that the patient’s 18-month-old son had exhibited similar symptoms two to three days prior to her illness.
Continue for differential diagnosis >>
DIFFERENTIAL DIAGNOSIS
Because multiple bacterial and viral diseases manifest in this fashion, the differential diagnosis included the following disorders:
Erythema multiforme. This skin condition may result from an allergic or hypersensitivity reaction to certain drugs or from infections. Infections that can cause erythema multiforme include herpes simplex virus and mycoplasma. Patients present with lesions on the palms (see Figure 3), soles, extremities, face, or trunk. The lesions can appear as a nodule, papule, macule, or vesicle. Initially, in the mild form, the lesions may appear as hives or target-shaped rashes, occurring on the face and acral surfaces. A severe form of erythema multiforme, known as Stevens-Johnson syndrome, is characterized by rash, mucosal involvement, and systemic symptoms.1
Herpes zoster. This viral infection is caused by the varicella-zoster virus, which also causes chicken pox. The virus lies dormant within a single sensory ganglion and may reappear as shingles along the dermatome of that nerve. Patients may experience burning or shooting pain with tingling or itching before the rash appears; vesicular lesions with erythematous bases appear days later. The rash occurs unilaterally on the body or face and does not cross the midline. A viral culture may be obtained for identification.2
Herpetic gingivostomatitis. This infection is most commonly caused by herpes simplex virus type 1, the same virus that causes cold sores. Patients may present with ulcerations along the buccal mucosa and gums. The infection manifests with systemic symptoms, including malaise, fever, irritability, and cervical adenopathy. A viral culture will identify the etiology.3
Impetigo. This skin infection is typically caused by bacteria, predominantly Staphylococcus aureus, Streptococcus pyogenes, or a combination. Infections generally occur after a break in the skin surface. The most common presentation is a rash that spreads to different parts of the body after scratching. Skin lesions can occur on the face, lips, or extremities. Initially vesicular, the lesions generally form a honey-colored crust after fluid discharge. The clinician should take a skin or fluid sample from the lesion to culture, which may identify the pathogen.4
Syphilis. This sexually transmitted infection is caused by the spirochete Treponema pallidum. In primary syphilis, patients can develop a painless sore, or chancre, on the genitals, rectal area, or mouth. If left untreated, the disease can progress to secondary syphilis, manifesting as a pale pink or reddish maculopapular rash on the palms and soles. The rash can be associated with fever, sore throat, myalgia, and fatigue. It is important to rule out syphilis because, left untreated, it can lead to cardiac and neurologic complications. Screening tests include VDRL and the rapid plasma reagin (RPR) test, both of which assess for antibodies to the organism, and dark field microscopy of ulcerations to identify the organism.5
Also included in the differential diagnosis for the patient’s symptoms was hand-foot-mouth disease (HFMD), discussed below.
Next page: Discussion >>
DISCUSSION
HFMD is an acute viral illness most often affecting children younger than 5 and occurring in summer to early fall months. HFMD manifests with fever and papulovesicular eruptions; lesions often appear in the oral cavity first, spreading to the palms, soles, and buttocks. Route of transmission is usually fecal-oral or through respiratory droplets, oral secretions, or direct contact with fluid-filled vesicles.6-8 The highly contagious nature of the virus causes it to spread to close contacts and family members and leads to outbreaks in schools and daycare centers.9,10
In the United States, the most common etiology of HFMD is coxsackievirus A16.7 Another causative agent, enterovirus 71 (EV71), has been found responsible for HFMD epidemics in southeast Asia and Australia.11,12 Recently, the coxsackievirus A6 (CV A6) strain has been linked to outbreaks of HFMD.7,9 This strain may produce an atypical manifestation of skin lesions on the face, trunk, and extremities. The lesions may also appear larger than usual and have a vesiculobullous rather than the more typical papulovesicular appearance. The course of the illness differs in severity depending on the strain of the virus causing HFMD.9
CLINICAL PRESENTATION
The acute phase of HFMD typically begins with prodromal symptoms such as fever, malaise, and sore throat. Erythematous ulcerations usually appear in the oral cavity first (see Figure 4) and often cause symptoms such as sore throat, dysphagia, or dryness. As the disease progresses, cutaneous lesions spread to the face, extremities, interdigital areas (see Figure 5), trunk, and perianal area (which may cause dysuria). The lesions may initially appear as erythematous macules or papules, transforming to vesicles as the disease progresses. The mucocutaneous lesions are usually asymptomatic but can be tender to touch or pressure and may leak fluid.7,10
In only a few cases—caused by CV A6—have lesions in the scalp been reported; the mechanism of action is unknown.10 Instances of lesions invading the nails have been reported, causing desquamation and shedding. This condition is known as onychomadesis.9,11
Continue for diagnosis >>
DIAGNOSIS
Serologic testing and viral cultures can identify the exact strain of virus causing HFMD and are particularly useful in unusual presentations. Polymerase chain reaction (PCR) testing yields a high sensitivity and specificity for the causative agent. Histologic examination of skin biopsies may show lymphocytic infiltrates and areas of degeneration along the epidermis. However, most cases are diagnosed based on clinical presentation alone.6,12
TREATMENT AND MANAGEMENT
Management of HFMD is primarily symptomatic, consisting of supportive care that includes use of antipyretics, NSAIDs, and adequate fluid intake to prevent dehydration. The disease is usually self-limited, resolving within seven to 10 days without sequelae.6,9 Aseptic meningitis and other severe complications (especially pulmonary and neurologic), most often associated with EV71 infection, can occur in vulnerable populations, including elderly, pregnant, and immunocompromised patients.9,11 Because the virus is excreted directly from palmar lesions onto the hands, proper hygiene and handwashing techniques offer an exceptionally strong protective effect, preventing transmission and reducing morbidity.8
PATIENT OUTCOME
Based on clinical findings and patient history, the patient was diagnosed with HFMD, which she contracted from her son. Laboratory testing and viral cultures were deemed unnecessary in this case. Treatment was symptomatic, and her skin lesions resolved in one to two weeks.
At follow-up, the patient stated her skin lesions resolved completely without leaving any scars. She also indicated that her nails peeled and shed approximately four weeks after diagnosis, but began to regrow normally four months after diagnosis.
CONCLUSION
Clinicians need to recognize that, although it is uncommon outside the pediatric population, HFMD may occur in adults with intact immune systems. The presentation of HFMD in adults may be atypical, including cutaneous lesions in the scalp and shedding of the nails several weeks after diagnosis. Depending on the viral strain involved, adult patients may have more severe illness and may take longer to recover. Therefore, early diagnosis is important to help prevent the spread of infection and reduce the severity of complications.
REFERENCES
1. Patel NN, Patel DN. Erythema multiforme syndrome. Am J Med. 2009;122(7):623-625.
2. Bader MS. Herpes zoster: diagnostic, therapeutic, and preventive approaches. Postgrad Med. 2013;125(5):78-91.
3. Avci O, Ertam I. Viral infections of the face. Clin Derm. 2014;32:715-733.
4. Hartman-Adams H, Banvard C, Juckett G. Impetigo: diagnosis and treatment. Am Fam Physician. 2014;90(4):229-235.
5. Markle W, Conti T, Kad M. Sexually transmitted diseases. Prim Care Clin Office Pract. 2013;40:557-587.
6. Shin JU, Oh SH, Lee JH. A case of hand-foot-mouth disease in an immunocompetent adult. Ann Dermatol. 2010;22(2):216-218.
7. CDC. Notes from the field: severe hand, foot, and mouth disease associated with coxsackievirus A6 - Alabama, Connecticut, California, and Nevada, November 2011-February 2012. MMWR Morb Mortal Wkly Rep. 2012;61(12):213-214.
8. Ruan F, Yang T, Ma H, et al. Risk factors for hand, foot, and mouth disease and herpangina and the preventive effect of hand-washing. Pediatrics. 2011;127(4):e898-e904.
9. Kaminska K, Martinetti G, Lucchini R, et al. Coxsackievirus A6 and hand, foot and mouth Disease: three case reports of familial child-to-immunocompetent adult transmission and a literature review. Case Rep Dermatol. 2013;5(2):203-209.
10. Lønnberg AS, Elberling J, Fischer TK, Skov L. Two cases of hand, foot, and mouth disease involving the scalp. Acta Derm Venereol. 2013;93(4):467-468.
11. Osterback R, Vuorinen T, Linna M, et al. Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerging Infect Dis. 2009;15(9):1485-1488.
12. Shea YF, Chan CY, Hung IFN, Chan KH. Hand, foot and mouth disease in an immunocompetent adult due to Coxsackievirus A6. Hong Kong Med J. 2013;19(3):262-264.
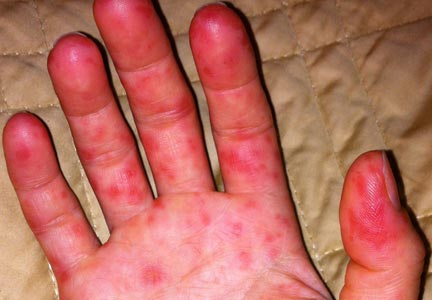
Man Unresponsive After Being Struck by Car
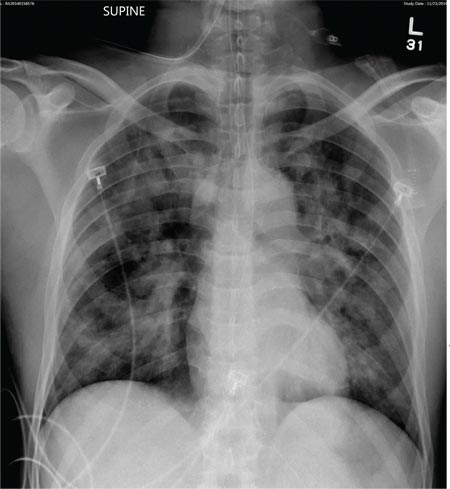
ANSWER
The radiograph demonstrates bilateral patchy, fluffy infiltrates as well as what is sometimes referred to as ground-glass opacities. In the setting of trauma and respiratory compromise, these areas are most suggestive of pulmonary contusions and early acute respiratory distress syndrome. Other possibilities in the differential diagnosis include pulmonary edema, atypical pneumonia, and pulmonary metastases.
ANSWER
The radiograph demonstrates bilateral patchy, fluffy infiltrates as well as what is sometimes referred to as ground-glass opacities. In the setting of trauma and respiratory compromise, these areas are most suggestive of pulmonary contusions and early acute respiratory distress syndrome. Other possibilities in the differential diagnosis include pulmonary edema, atypical pneumonia, and pulmonary metastases.
ANSWER
The radiograph demonstrates bilateral patchy, fluffy infiltrates as well as what is sometimes referred to as ground-glass opacities. In the setting of trauma and respiratory compromise, these areas are most suggestive of pulmonary contusions and early acute respiratory distress syndrome. Other possibilities in the differential diagnosis include pulmonary edema, atypical pneumonia, and pulmonary metastases.
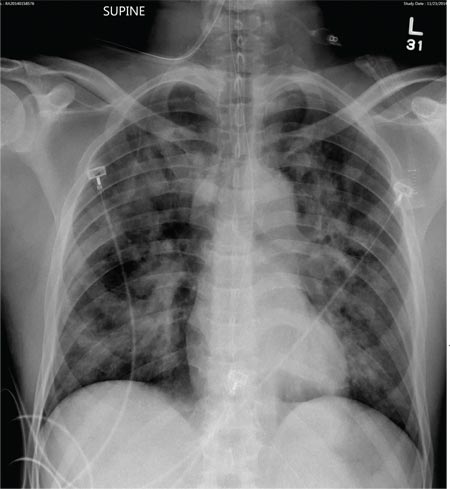
A 50-year-old man is transferred to your facility from an outlying community hospital. He is purportedly a pedestrian who was struck by a car. EMS personnel reported him to be unresponsive at the scene. He was intubated for airway protection and stabilized at the outside facility prior to transfer. Upon arrival at your facility, he is still intubated and unresponsive, and his Glasgow Coma Scale score is 3T. His heart rate is 150 beats/min and his blood pressure, 105/56 mm Hg. No additional history is available. Primary survey reveals a large scalp laceration with currently controlled bleeding. His pupils are nonreactive bilaterally. The patient is tachycardic with bilateral crackles. He also has a laceration and deformity of his right lower extremity. No imaging was provided in the transfer, so you obtain a portable chest radiograph. What is your impression?