User login
Light-headedness and bradycardia in a 72-year-old woman
A 72-year-old woman came to the emergency department because of persistent light-headedness. Her medical history included end-stage renal disease, hypertension, peripheral vascular disease, and diabetes mellitus. She said she had experienced similar symptoms before, but they had gone away.
She reported no visual changes, no loss of consciousness, and no history of seizures, syncope, chest pain, palpitations, or diaphoresis. She was not taking a beta-blocker, calcium channel blocker, or digoxin.
Her blood pressure was 75/44 mm Hg, heart rate 44 beats per minute, respiratory rate 16 breaths per minute, and oxygen saturation 97% while receiving oxygen at 3 L per minute. An electrolyte panel was normal except for an elevated creatinine level secondary to end-stage renal disease.
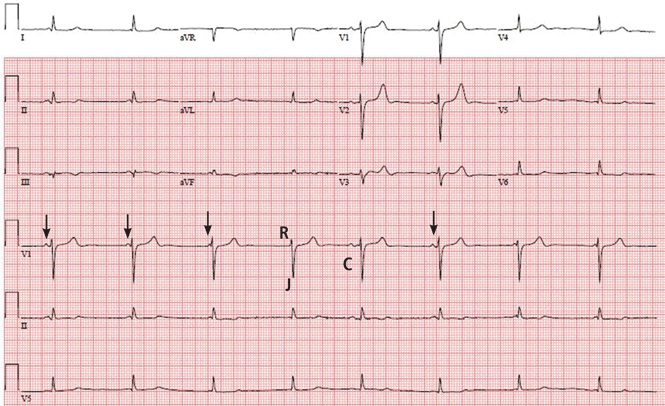
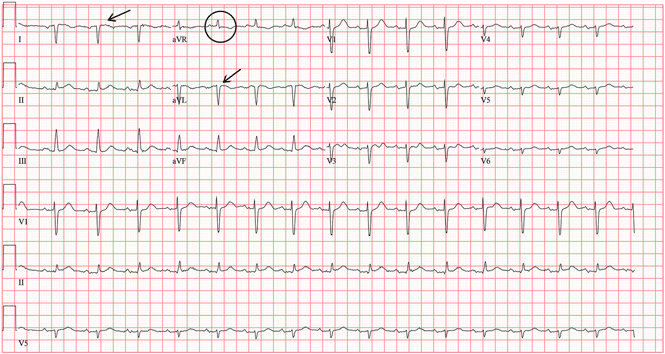
In view of her symptoms and bradycardia, she was admitted to the hospital. The initial electrocardiogram (Figure 1) showed an atrial rate of approximately 46 beats per minute, a ventricular rate of approximately 48 beats per minute, and a P wave in the refractory period caused by a junction impulse.
These findings pointed to atrioventricular (AV) dissociation, a term commonly applied to arrhythmias in which the atria and ventricles are rhythmically detached.
ATRIOVENTRICULAR DISSOCIATION
AV dissociation is often used interchangeably with complete heart block, but this is incorrect1; though complete heart block is a form of AV dissociation, not all AV dissociation is complete heart block. In complete heart block, there is no rhythmic relationship between the atria and ventricles, as they beat independently with no influence on each other. On the other hand, when a “blockade” is created by the physiologic refractory period of the atria (sinus node or atrial ectopic focus) and ventricles, interference dissociation can result.2 In this condition, when the ventricles are not in a refractory period, an atrial impulse may be conducted through the AV node, resulting in an atrial-driven beat. Simply put, a P wave has the potential to be conducted in AV dissociation if there is an opportunity, but in complete heart block it does not.1
AV dissociation is a secondary manifestation of a primary disorder or rhythm disturbance. In general, any rhythm that competes against an atrial impulse and inhibits its conduction through the AV node can cause AV dissociation. Common examples include junctional escape or accelerated rhythms, premature ventricular beats or ventricular tachycardia, and accelerated idioventricular rhythms. It also can be caused by drugs (eg, digoxin) or an increase in vagal tone.2
In normal myocardium, the sinus node has a higher impulse rate than the subordinate pacemaker (AV node or ventricular pacemaker). Generally, the atrial rate is higher than the ventricular rate in complete heart block, whereas in AV dissociation the ventricular rate is higher than the atrial rate.3
Thus, AV dissociation can result from one of the following mechanisms4:
- Slowing of the dominant pacemaker (sinus or atrial pacemaker)
- Acceleration or overtaking of the sinus node (or atrial focus) by a subordinate pacemaker (eg, a junctional or ventricular pacemaker)
- A block within the AV node that prevents an impulse generated by the dominant pacemaker (sinus or atrial focus) from crossing the AV node
- A combination of these mechanisms.
Another form of AV dissociation is isorhythmic dissociation. In this subtype, atrial and ventricular impulses occur at the same rate. This type of dissociation is most commonly confused with third-degree (or complete) heart block. It may be difficult to distinguish one from the other, but at higher sinus (or atrial) rates the difference becomes obvious—properly timed P waves may be conducted through the AV node in isorhythmic dissociation.1
The prevalence of AV dissociation is thought to be 0.48% to 0.68%,3 but it could be more common since it is underdiagnosed.5
Treatment should be directed at the primary disorder.4 The need for a pacemaker depends on the condition causing the AV dissociation. In conditions that slow the sinus node, such as increased vagal tone, patients may benefit from medications that decrease parasympathetic activity or increase adrenergic activity in the AV node (eg, isoproterenol, atropine).6
OUR PATIENT
Our patient’s electrocardiogram showed interference dissociation from competing junctional rhythms. Possibly, she had sinus node disease, explaining why the sinus node was not the dominant pacemaker. She had symptomatic hypotension, requiring dopamine for pressure support. She was started on intravenous isoproterenol, which eventually restored sinus rhythm.
During the same hospitalization, she was diagnosed with osteomyelitis of the left foot, without bacteremia. She was treated for her infection and later received a pacemaker. She was discharged to a rehabilitation facility.
TAKE-AWAY POINTS
- When an occasional impulse is conducted through the AV node, AV dissociation is most likely interference dissociation.
- AV dissociations are often confused with complete heart block.
- In AV dissociation, the ventricular rate is higher than the atrial rate.
- Complete heart block is a form of AV dissociation, but not all AV dissociation is complete heart block.
- AV dissociation can be caused by three main mechanisms or by a combination of them.
- AV dissociation is secondary to a primary rhythm disorder.
- Adrenergic drugs may help to correct the AV dissociation, but not always completely.
- Goldberger AL. Atrioventricular conduction abnormalities: delays, blocks, and dissociation syndromes. In: Goldberger AL, Goldberger ZD, Shvilkin A, eds. Clinical Electrocardiography: A Simplified Approach. 8th ed. Philadelphia, PA: Elsevier/Saunders; 2012:159–169.
- Wang K, Benditt DG. AV dissociation, an inevitable response. Ann Noninvasive Electrocardiol 2011; 16:227–231.
- Harrigan RA, Perron AD, Brady WJ. Atrioventricular dissociation. Am J Emerg Med 2001; 19:218–222.
- Jeffrey O, Zipes DP. Specific arrhythmias: diagnosis and treatment. In: Bonow RO, Mann DL, Zipes DP, Libby P, eds. Braunwald's Heart Disease: A Textbook of Cardiovascular Medicine. 9th ed. Philadelphia, PA: Elsevier/Saunders; 2011.
- Singh GD, Wong GB, Southard JA, Amsterdam EA. Food for thought: atrioventricular dissociation. Am J Med 2013; 126:1050–1053.
- Vavetsi S, Nikolaou N, Tsarouhas K, et al. Consecutive administration of atropine and isoproterenol for the evaluation of asymptomatic sinus bradycardia. Europace 2008; 10:1176–1181.
A 72-year-old woman came to the emergency department because of persistent light-headedness. Her medical history included end-stage renal disease, hypertension, peripheral vascular disease, and diabetes mellitus. She said she had experienced similar symptoms before, but they had gone away.
She reported no visual changes, no loss of consciousness, and no history of seizures, syncope, chest pain, palpitations, or diaphoresis. She was not taking a beta-blocker, calcium channel blocker, or digoxin.
Her blood pressure was 75/44 mm Hg, heart rate 44 beats per minute, respiratory rate 16 breaths per minute, and oxygen saturation 97% while receiving oxygen at 3 L per minute. An electrolyte panel was normal except for an elevated creatinine level secondary to end-stage renal disease.
In view of her symptoms and bradycardia, she was admitted to the hospital. The initial electrocardiogram (Figure 1) showed an atrial rate of approximately 46 beats per minute, a ventricular rate of approximately 48 beats per minute, and a P wave in the refractory period caused by a junction impulse.
These findings pointed to atrioventricular (AV) dissociation, a term commonly applied to arrhythmias in which the atria and ventricles are rhythmically detached.
ATRIOVENTRICULAR DISSOCIATION
AV dissociation is often used interchangeably with complete heart block, but this is incorrect1; though complete heart block is a form of AV dissociation, not all AV dissociation is complete heart block. In complete heart block, there is no rhythmic relationship between the atria and ventricles, as they beat independently with no influence on each other. On the other hand, when a “blockade” is created by the physiologic refractory period of the atria (sinus node or atrial ectopic focus) and ventricles, interference dissociation can result.2 In this condition, when the ventricles are not in a refractory period, an atrial impulse may be conducted through the AV node, resulting in an atrial-driven beat. Simply put, a P wave has the potential to be conducted in AV dissociation if there is an opportunity, but in complete heart block it does not.1
AV dissociation is a secondary manifestation of a primary disorder or rhythm disturbance. In general, any rhythm that competes against an atrial impulse and inhibits its conduction through the AV node can cause AV dissociation. Common examples include junctional escape or accelerated rhythms, premature ventricular beats or ventricular tachycardia, and accelerated idioventricular rhythms. It also can be caused by drugs (eg, digoxin) or an increase in vagal tone.2
In normal myocardium, the sinus node has a higher impulse rate than the subordinate pacemaker (AV node or ventricular pacemaker). Generally, the atrial rate is higher than the ventricular rate in complete heart block, whereas in AV dissociation the ventricular rate is higher than the atrial rate.3
Thus, AV dissociation can result from one of the following mechanisms4:
- Slowing of the dominant pacemaker (sinus or atrial pacemaker)
- Acceleration or overtaking of the sinus node (or atrial focus) by a subordinate pacemaker (eg, a junctional or ventricular pacemaker)
- A block within the AV node that prevents an impulse generated by the dominant pacemaker (sinus or atrial focus) from crossing the AV node
- A combination of these mechanisms.
Another form of AV dissociation is isorhythmic dissociation. In this subtype, atrial and ventricular impulses occur at the same rate. This type of dissociation is most commonly confused with third-degree (or complete) heart block. It may be difficult to distinguish one from the other, but at higher sinus (or atrial) rates the difference becomes obvious—properly timed P waves may be conducted through the AV node in isorhythmic dissociation.1
The prevalence of AV dissociation is thought to be 0.48% to 0.68%,3 but it could be more common since it is underdiagnosed.5
Treatment should be directed at the primary disorder.4 The need for a pacemaker depends on the condition causing the AV dissociation. In conditions that slow the sinus node, such as increased vagal tone, patients may benefit from medications that decrease parasympathetic activity or increase adrenergic activity in the AV node (eg, isoproterenol, atropine).6
OUR PATIENT
Our patient’s electrocardiogram showed interference dissociation from competing junctional rhythms. Possibly, she had sinus node disease, explaining why the sinus node was not the dominant pacemaker. She had symptomatic hypotension, requiring dopamine for pressure support. She was started on intravenous isoproterenol, which eventually restored sinus rhythm.
During the same hospitalization, she was diagnosed with osteomyelitis of the left foot, without bacteremia. She was treated for her infection and later received a pacemaker. She was discharged to a rehabilitation facility.
TAKE-AWAY POINTS
- When an occasional impulse is conducted through the AV node, AV dissociation is most likely interference dissociation.
- AV dissociations are often confused with complete heart block.
- In AV dissociation, the ventricular rate is higher than the atrial rate.
- Complete heart block is a form of AV dissociation, but not all AV dissociation is complete heart block.
- AV dissociation can be caused by three main mechanisms or by a combination of them.
- AV dissociation is secondary to a primary rhythm disorder.
- Adrenergic drugs may help to correct the AV dissociation, but not always completely.
A 72-year-old woman came to the emergency department because of persistent light-headedness. Her medical history included end-stage renal disease, hypertension, peripheral vascular disease, and diabetes mellitus. She said she had experienced similar symptoms before, but they had gone away.
She reported no visual changes, no loss of consciousness, and no history of seizures, syncope, chest pain, palpitations, or diaphoresis. She was not taking a beta-blocker, calcium channel blocker, or digoxin.
Her blood pressure was 75/44 mm Hg, heart rate 44 beats per minute, respiratory rate 16 breaths per minute, and oxygen saturation 97% while receiving oxygen at 3 L per minute. An electrolyte panel was normal except for an elevated creatinine level secondary to end-stage renal disease.
In view of her symptoms and bradycardia, she was admitted to the hospital. The initial electrocardiogram (Figure 1) showed an atrial rate of approximately 46 beats per minute, a ventricular rate of approximately 48 beats per minute, and a P wave in the refractory period caused by a junction impulse.
These findings pointed to atrioventricular (AV) dissociation, a term commonly applied to arrhythmias in which the atria and ventricles are rhythmically detached.
ATRIOVENTRICULAR DISSOCIATION
AV dissociation is often used interchangeably with complete heart block, but this is incorrect1; though complete heart block is a form of AV dissociation, not all AV dissociation is complete heart block. In complete heart block, there is no rhythmic relationship between the atria and ventricles, as they beat independently with no influence on each other. On the other hand, when a “blockade” is created by the physiologic refractory period of the atria (sinus node or atrial ectopic focus) and ventricles, interference dissociation can result.2 In this condition, when the ventricles are not in a refractory period, an atrial impulse may be conducted through the AV node, resulting in an atrial-driven beat. Simply put, a P wave has the potential to be conducted in AV dissociation if there is an opportunity, but in complete heart block it does not.1
AV dissociation is a secondary manifestation of a primary disorder or rhythm disturbance. In general, any rhythm that competes against an atrial impulse and inhibits its conduction through the AV node can cause AV dissociation. Common examples include junctional escape or accelerated rhythms, premature ventricular beats or ventricular tachycardia, and accelerated idioventricular rhythms. It also can be caused by drugs (eg, digoxin) or an increase in vagal tone.2
In normal myocardium, the sinus node has a higher impulse rate than the subordinate pacemaker (AV node or ventricular pacemaker). Generally, the atrial rate is higher than the ventricular rate in complete heart block, whereas in AV dissociation the ventricular rate is higher than the atrial rate.3
Thus, AV dissociation can result from one of the following mechanisms4:
- Slowing of the dominant pacemaker (sinus or atrial pacemaker)
- Acceleration or overtaking of the sinus node (or atrial focus) by a subordinate pacemaker (eg, a junctional or ventricular pacemaker)
- A block within the AV node that prevents an impulse generated by the dominant pacemaker (sinus or atrial focus) from crossing the AV node
- A combination of these mechanisms.
Another form of AV dissociation is isorhythmic dissociation. In this subtype, atrial and ventricular impulses occur at the same rate. This type of dissociation is most commonly confused with third-degree (or complete) heart block. It may be difficult to distinguish one from the other, but at higher sinus (or atrial) rates the difference becomes obvious—properly timed P waves may be conducted through the AV node in isorhythmic dissociation.1
The prevalence of AV dissociation is thought to be 0.48% to 0.68%,3 but it could be more common since it is underdiagnosed.5
Treatment should be directed at the primary disorder.4 The need for a pacemaker depends on the condition causing the AV dissociation. In conditions that slow the sinus node, such as increased vagal tone, patients may benefit from medications that decrease parasympathetic activity or increase adrenergic activity in the AV node (eg, isoproterenol, atropine).6
OUR PATIENT
Our patient’s electrocardiogram showed interference dissociation from competing junctional rhythms. Possibly, she had sinus node disease, explaining why the sinus node was not the dominant pacemaker. She had symptomatic hypotension, requiring dopamine for pressure support. She was started on intravenous isoproterenol, which eventually restored sinus rhythm.
During the same hospitalization, she was diagnosed with osteomyelitis of the left foot, without bacteremia. She was treated for her infection and later received a pacemaker. She was discharged to a rehabilitation facility.
TAKE-AWAY POINTS
- When an occasional impulse is conducted through the AV node, AV dissociation is most likely interference dissociation.
- AV dissociations are often confused with complete heart block.
- In AV dissociation, the ventricular rate is higher than the atrial rate.
- Complete heart block is a form of AV dissociation, but not all AV dissociation is complete heart block.
- AV dissociation can be caused by three main mechanisms or by a combination of them.
- AV dissociation is secondary to a primary rhythm disorder.
- Adrenergic drugs may help to correct the AV dissociation, but not always completely.
- Goldberger AL. Atrioventricular conduction abnormalities: delays, blocks, and dissociation syndromes. In: Goldberger AL, Goldberger ZD, Shvilkin A, eds. Clinical Electrocardiography: A Simplified Approach. 8th ed. Philadelphia, PA: Elsevier/Saunders; 2012:159–169.
- Wang K, Benditt DG. AV dissociation, an inevitable response. Ann Noninvasive Electrocardiol 2011; 16:227–231.
- Harrigan RA, Perron AD, Brady WJ. Atrioventricular dissociation. Am J Emerg Med 2001; 19:218–222.
- Jeffrey O, Zipes DP. Specific arrhythmias: diagnosis and treatment. In: Bonow RO, Mann DL, Zipes DP, Libby P, eds. Braunwald's Heart Disease: A Textbook of Cardiovascular Medicine. 9th ed. Philadelphia, PA: Elsevier/Saunders; 2011.
- Singh GD, Wong GB, Southard JA, Amsterdam EA. Food for thought: atrioventricular dissociation. Am J Med 2013; 126:1050–1053.
- Vavetsi S, Nikolaou N, Tsarouhas K, et al. Consecutive administration of atropine and isoproterenol for the evaluation of asymptomatic sinus bradycardia. Europace 2008; 10:1176–1181.
- Goldberger AL. Atrioventricular conduction abnormalities: delays, blocks, and dissociation syndromes. In: Goldberger AL, Goldberger ZD, Shvilkin A, eds. Clinical Electrocardiography: A Simplified Approach. 8th ed. Philadelphia, PA: Elsevier/Saunders; 2012:159–169.
- Wang K, Benditt DG. AV dissociation, an inevitable response. Ann Noninvasive Electrocardiol 2011; 16:227–231.
- Harrigan RA, Perron AD, Brady WJ. Atrioventricular dissociation. Am J Emerg Med 2001; 19:218–222.
- Jeffrey O, Zipes DP. Specific arrhythmias: diagnosis and treatment. In: Bonow RO, Mann DL, Zipes DP, Libby P, eds. Braunwald's Heart Disease: A Textbook of Cardiovascular Medicine. 9th ed. Philadelphia, PA: Elsevier/Saunders; 2011.
- Singh GD, Wong GB, Southard JA, Amsterdam EA. Food for thought: atrioventricular dissociation. Am J Med 2013; 126:1050–1053.
- Vavetsi S, Nikolaou N, Tsarouhas K, et al. Consecutive administration of atropine and isoproterenol for the evaluation of asymptomatic sinus bradycardia. Europace 2008; 10:1176–1181.
A lump in the umbilicus
A 60-year-old man presented to the emergency department with abdominal pain. The pain was dull and constant, with no radiation and no aggravating or relieving factors. He also reported decreased appetite, weight loss, and constipation over the past 3 months.
He had no history of significant medical problems and was not taking any medications. He had no fever and no evidence of gastrointestinal bleeding.
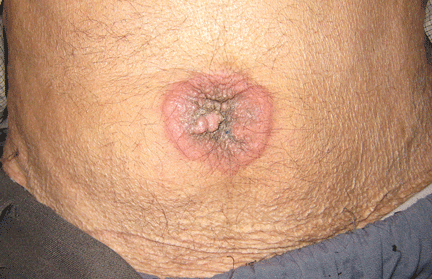
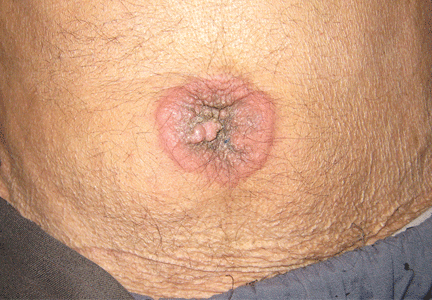
Physical examination showed mild tenderness around the umbilicus and a painless, small nodule (15 mm by 6 mm) protruding through the umbilicus with surrounding erythematous discoloration (Figure 1). A digital rectal examination was normal. Laboratory studies showed only mild normocytic anemia.
The patient underwent abdominal ultrasonography, which showed free fluid in the abdominopelvic cavity. This was followed by computed tomography of the abdominopelvic cavity, which revealed ascites and a small mass in the umbilicus. Punch biopsy of the umbilical lesion was performed, and histologic study indicated a diagnosis of adenocarcinoma.
Based on the biopsy results and the patient’s history of gastrointestinal symptoms, colonoscopy was performed, which showed an exophytic tumor of the transverse colon. The tumor was biopsied, and pathologic evaluation confirmed adenocarcinoma. A diagnosis of metastatic colon cancer was made. The patient received chemotherapy and underwent surgery to relieve the bowel obstruction.
SISTER MARY JOSEPH NODULE
A periumbilical nodule representing metastatic cancer, also known as Sister Mary Joseph nodule,1 is typically associated with intra-abdominal malignancy. An estimated 1% to 3% of patients with abdominopelvic malignancy present with this nodule,2 most often from gastrointestinal cancer but also from gynecologic malignancies. In about 15% to 30% of cases, no origin is identified.3
How these cancers spread to the umbilicus is not known. Proposed mechanisms include direct transperitoneal, lymphatic, or hematogenous spread, and even iatrogenic spread during laparotomy.4,5
The differential diagnosis includes umbilical hernia, cutaneous endometriosis, lymphangioma, melanoma, pilonidal sinus, and pyogenic granuloma. It is usually described as a painful nodule with irregular margins and a mean diameter of 2 to 3 cm.2 The condition is always a sign of metastatic cancer. Although it can be useful for diagnosing advanced disease, whether this would lead to earlier diagnosis is doubtful. Palliative treatment is generally most appropriate.
- Albano EA, Kanter J. Images in clinical medicine. Sister Mary Joseph’s nodule. N Engl J Med 2005; 352:1913.
- Iavazzo C, Madhuri K, Essapen S, Akrivos N, Tailor A, Butler-Manuel S. Sister Mary Joseph’s nodule as a first manifestation of primary peritoneal cancer. Case Rep Obstet Gynecol 2012; 2012:467240.
- Gabriele R, Borghese M, Conte M, Basso L. Sister Mary Joseph’s nodule as a first sign of cancer of the cecum: report of a case. Dis Colon Rectum 2004; 47:115–117.
- Dar IH, Kamili MA, Dar SH, Kuchaai FA. Sister Mary Joseph nodule—a case report with review of literature. J Res Med Sci 2009; 14:385–387.
- Martínez-Palones JM, Gil-Moreno A, Pérez-Benavente MA, Garcia-Giménez A, Xercavins J. Umbilical metastasis after laparoscopic retroperitoneal paraaortic lymphadenectomy for cervical cancer: a true port-site metastasis? Gynecol Oncol 2005; 97:292–295.
A 60-year-old man presented to the emergency department with abdominal pain. The pain was dull and constant, with no radiation and no aggravating or relieving factors. He also reported decreased appetite, weight loss, and constipation over the past 3 months.
He had no history of significant medical problems and was not taking any medications. He had no fever and no evidence of gastrointestinal bleeding.
Physical examination showed mild tenderness around the umbilicus and a painless, small nodule (15 mm by 6 mm) protruding through the umbilicus with surrounding erythematous discoloration (Figure 1). A digital rectal examination was normal. Laboratory studies showed only mild normocytic anemia.
The patient underwent abdominal ultrasonography, which showed free fluid in the abdominopelvic cavity. This was followed by computed tomography of the abdominopelvic cavity, which revealed ascites and a small mass in the umbilicus. Punch biopsy of the umbilical lesion was performed, and histologic study indicated a diagnosis of adenocarcinoma.
Based on the biopsy results and the patient’s history of gastrointestinal symptoms, colonoscopy was performed, which showed an exophytic tumor of the transverse colon. The tumor was biopsied, and pathologic evaluation confirmed adenocarcinoma. A diagnosis of metastatic colon cancer was made. The patient received chemotherapy and underwent surgery to relieve the bowel obstruction.
SISTER MARY JOSEPH NODULE
A periumbilical nodule representing metastatic cancer, also known as Sister Mary Joseph nodule,1 is typically associated with intra-abdominal malignancy. An estimated 1% to 3% of patients with abdominopelvic malignancy present with this nodule,2 most often from gastrointestinal cancer but also from gynecologic malignancies. In about 15% to 30% of cases, no origin is identified.3
How these cancers spread to the umbilicus is not known. Proposed mechanisms include direct transperitoneal, lymphatic, or hematogenous spread, and even iatrogenic spread during laparotomy.4,5
The differential diagnosis includes umbilical hernia, cutaneous endometriosis, lymphangioma, melanoma, pilonidal sinus, and pyogenic granuloma. It is usually described as a painful nodule with irregular margins and a mean diameter of 2 to 3 cm.2 The condition is always a sign of metastatic cancer. Although it can be useful for diagnosing advanced disease, whether this would lead to earlier diagnosis is doubtful. Palliative treatment is generally most appropriate.
A 60-year-old man presented to the emergency department with abdominal pain. The pain was dull and constant, with no radiation and no aggravating or relieving factors. He also reported decreased appetite, weight loss, and constipation over the past 3 months.
He had no history of significant medical problems and was not taking any medications. He had no fever and no evidence of gastrointestinal bleeding.
Physical examination showed mild tenderness around the umbilicus and a painless, small nodule (15 mm by 6 mm) protruding through the umbilicus with surrounding erythematous discoloration (Figure 1). A digital rectal examination was normal. Laboratory studies showed only mild normocytic anemia.
The patient underwent abdominal ultrasonography, which showed free fluid in the abdominopelvic cavity. This was followed by computed tomography of the abdominopelvic cavity, which revealed ascites and a small mass in the umbilicus. Punch biopsy of the umbilical lesion was performed, and histologic study indicated a diagnosis of adenocarcinoma.
Based on the biopsy results and the patient’s history of gastrointestinal symptoms, colonoscopy was performed, which showed an exophytic tumor of the transverse colon. The tumor was biopsied, and pathologic evaluation confirmed adenocarcinoma. A diagnosis of metastatic colon cancer was made. The patient received chemotherapy and underwent surgery to relieve the bowel obstruction.
SISTER MARY JOSEPH NODULE
A periumbilical nodule representing metastatic cancer, also known as Sister Mary Joseph nodule,1 is typically associated with intra-abdominal malignancy. An estimated 1% to 3% of patients with abdominopelvic malignancy present with this nodule,2 most often from gastrointestinal cancer but also from gynecologic malignancies. In about 15% to 30% of cases, no origin is identified.3
How these cancers spread to the umbilicus is not known. Proposed mechanisms include direct transperitoneal, lymphatic, or hematogenous spread, and even iatrogenic spread during laparotomy.4,5
The differential diagnosis includes umbilical hernia, cutaneous endometriosis, lymphangioma, melanoma, pilonidal sinus, and pyogenic granuloma. It is usually described as a painful nodule with irregular margins and a mean diameter of 2 to 3 cm.2 The condition is always a sign of metastatic cancer. Although it can be useful for diagnosing advanced disease, whether this would lead to earlier diagnosis is doubtful. Palliative treatment is generally most appropriate.
- Albano EA, Kanter J. Images in clinical medicine. Sister Mary Joseph’s nodule. N Engl J Med 2005; 352:1913.
- Iavazzo C, Madhuri K, Essapen S, Akrivos N, Tailor A, Butler-Manuel S. Sister Mary Joseph’s nodule as a first manifestation of primary peritoneal cancer. Case Rep Obstet Gynecol 2012; 2012:467240.
- Gabriele R, Borghese M, Conte M, Basso L. Sister Mary Joseph’s nodule as a first sign of cancer of the cecum: report of a case. Dis Colon Rectum 2004; 47:115–117.
- Dar IH, Kamili MA, Dar SH, Kuchaai FA. Sister Mary Joseph nodule—a case report with review of literature. J Res Med Sci 2009; 14:385–387.
- Martínez-Palones JM, Gil-Moreno A, Pérez-Benavente MA, Garcia-Giménez A, Xercavins J. Umbilical metastasis after laparoscopic retroperitoneal paraaortic lymphadenectomy for cervical cancer: a true port-site metastasis? Gynecol Oncol 2005; 97:292–295.
- Albano EA, Kanter J. Images in clinical medicine. Sister Mary Joseph’s nodule. N Engl J Med 2005; 352:1913.
- Iavazzo C, Madhuri K, Essapen S, Akrivos N, Tailor A, Butler-Manuel S. Sister Mary Joseph’s nodule as a first manifestation of primary peritoneal cancer. Case Rep Obstet Gynecol 2012; 2012:467240.
- Gabriele R, Borghese M, Conte M, Basso L. Sister Mary Joseph’s nodule as a first sign of cancer of the cecum: report of a case. Dis Colon Rectum 2004; 47:115–117.
- Dar IH, Kamili MA, Dar SH, Kuchaai FA. Sister Mary Joseph nodule—a case report with review of literature. J Res Med Sci 2009; 14:385–387.
- Martínez-Palones JM, Gil-Moreno A, Pérez-Benavente MA, Garcia-Giménez A, Xercavins J. Umbilical metastasis after laparoscopic retroperitoneal paraaortic lymphadenectomy for cervical cancer: a true port-site metastasis? Gynecol Oncol 2005; 97:292–295.
Pericarditis as a window into the mind of the internist

In this issue of the Journal, Alraies et al comment on how extensively we should look for the cause of an initial episode of pericarditis.
The pericardium, like the pleura, peritoneum, and synovium, can be affected in a number of inflammatory and infectious disorders. The mechanisms by which these tissues are affected are not fully understood, nor is the process by which different diseases seem to selectively target the joint or pericardium. Why are the joints only minimally inflamed in systemic lupus erythematosus (SLE), while lupus pericarditis, in the uncommon occurrence of significant effusion, is often quite inflammatory, with a neutrophil predominance in the fluid? Why is pericardial involvement so often demonstrable by imaging in patients with SLE and rheumatoid arthritis, yet an acute pericarditis presentation with audible pericardial rubs is so seldom recognized?
Although nuances like these are not well understood, in medical school we all learned the association between connective tissue disease and pericarditis. The importance of recalling these associations is repeatedly reinforced during residency and in disease-focused review articles. During my training, woe to the resident who presented a patient at rounds who was admitted with unexplained pericarditis and was not evaluated for SLE with at least an antinuclear antibody (ANA) test, even if there were no other features to suggest the disease. Ordering the test reflected that we knew that, occasionally, pericardial disease is the sole presenting manifestation of lupus.
Such is the plight of the internist. Pericarditis can be the initial manifestation of an autoimmune or inflammatory disease, but this is more often relevant on certification examinations and in medical education than in everyday practice. We are now charged with ordering tests in a more cost-effective manner than in the past. This means that we should not order tests simply because of an epidemiologic association, but only when the result is likely to influence decisions about testing or treatment. But that creates the intellectual dissonance of knowing of a potential relationship (which someone, someday, may challenge us about) but not looking for it. There is an inherent conflict between satisfying intellectual curiosity and the need to be thorough while at the same time containing costs and avoiding the potential harm inherent in overtesting.
A partial solution is to try to define the immediate risk of not recognizing a life- or organ-threatening disease process that can be suggested by a positive nonspecific test (eg, ANA), and to refine the pretest likelihood of specific diagnoses by obtaining an accurate and complete history and performing a focused physical examination. For example, if we suspect that SLE may be the cause of an initial episode of symptomatic pericarditis, our initial evaluation should focus on the patient’s clinical picture. Is there bitemporal hair-thinning? New-onset Raynaud symptoms? Mild generalized adenopathy or lymphopenia? A borderline-low platelet count, or any proteinuria or microhematuria (which should warrant a prompt examination of a fresh urine sediment sample by a physician at the point of care to look for cellular casts indicative of glomerulonephritis)?
As internists, we should try to fulfill our need to be thorough and compulsive by using our honed skills as careful observers and historians—taking a careful history from the patient and family, performing a focused physical examination, and appropriately using disease-defining or staging tests before ordering less specific serologic or other tests. Practicing medicine in a conscientious and compulsive manner does not mean that every diagnostic possibility must be tested for at initial presentation.
Reading how experienced clinicians approach the problem of pericarditis in a specialized clinic provides a useful prompt to self-assess how we approach analogous clinical scenarios.
In this issue of the Journal, Alraies et al comment on how extensively we should look for the cause of an initial episode of pericarditis.
The pericardium, like the pleura, peritoneum, and synovium, can be affected in a number of inflammatory and infectious disorders. The mechanisms by which these tissues are affected are not fully understood, nor is the process by which different diseases seem to selectively target the joint or pericardium. Why are the joints only minimally inflamed in systemic lupus erythematosus (SLE), while lupus pericarditis, in the uncommon occurrence of significant effusion, is often quite inflammatory, with a neutrophil predominance in the fluid? Why is pericardial involvement so often demonstrable by imaging in patients with SLE and rheumatoid arthritis, yet an acute pericarditis presentation with audible pericardial rubs is so seldom recognized?
Although nuances like these are not well understood, in medical school we all learned the association between connective tissue disease and pericarditis. The importance of recalling these associations is repeatedly reinforced during residency and in disease-focused review articles. During my training, woe to the resident who presented a patient at rounds who was admitted with unexplained pericarditis and was not evaluated for SLE with at least an antinuclear antibody (ANA) test, even if there were no other features to suggest the disease. Ordering the test reflected that we knew that, occasionally, pericardial disease is the sole presenting manifestation of lupus.
Such is the plight of the internist. Pericarditis can be the initial manifestation of an autoimmune or inflammatory disease, but this is more often relevant on certification examinations and in medical education than in everyday practice. We are now charged with ordering tests in a more cost-effective manner than in the past. This means that we should not order tests simply because of an epidemiologic association, but only when the result is likely to influence decisions about testing or treatment. But that creates the intellectual dissonance of knowing of a potential relationship (which someone, someday, may challenge us about) but not looking for it. There is an inherent conflict between satisfying intellectual curiosity and the need to be thorough while at the same time containing costs and avoiding the potential harm inherent in overtesting.
A partial solution is to try to define the immediate risk of not recognizing a life- or organ-threatening disease process that can be suggested by a positive nonspecific test (eg, ANA), and to refine the pretest likelihood of specific diagnoses by obtaining an accurate and complete history and performing a focused physical examination. For example, if we suspect that SLE may be the cause of an initial episode of symptomatic pericarditis, our initial evaluation should focus on the patient’s clinical picture. Is there bitemporal hair-thinning? New-onset Raynaud symptoms? Mild generalized adenopathy or lymphopenia? A borderline-low platelet count, or any proteinuria or microhematuria (which should warrant a prompt examination of a fresh urine sediment sample by a physician at the point of care to look for cellular casts indicative of glomerulonephritis)?
As internists, we should try to fulfill our need to be thorough and compulsive by using our honed skills as careful observers and historians—taking a careful history from the patient and family, performing a focused physical examination, and appropriately using disease-defining or staging tests before ordering less specific serologic or other tests. Practicing medicine in a conscientious and compulsive manner does not mean that every diagnostic possibility must be tested for at initial presentation.
Reading how experienced clinicians approach the problem of pericarditis in a specialized clinic provides a useful prompt to self-assess how we approach analogous clinical scenarios.
In this issue of the Journal, Alraies et al comment on how extensively we should look for the cause of an initial episode of pericarditis.
The pericardium, like the pleura, peritoneum, and synovium, can be affected in a number of inflammatory and infectious disorders. The mechanisms by which these tissues are affected are not fully understood, nor is the process by which different diseases seem to selectively target the joint or pericardium. Why are the joints only minimally inflamed in systemic lupus erythematosus (SLE), while lupus pericarditis, in the uncommon occurrence of significant effusion, is often quite inflammatory, with a neutrophil predominance in the fluid? Why is pericardial involvement so often demonstrable by imaging in patients with SLE and rheumatoid arthritis, yet an acute pericarditis presentation with audible pericardial rubs is so seldom recognized?
Although nuances like these are not well understood, in medical school we all learned the association between connective tissue disease and pericarditis. The importance of recalling these associations is repeatedly reinforced during residency and in disease-focused review articles. During my training, woe to the resident who presented a patient at rounds who was admitted with unexplained pericarditis and was not evaluated for SLE with at least an antinuclear antibody (ANA) test, even if there were no other features to suggest the disease. Ordering the test reflected that we knew that, occasionally, pericardial disease is the sole presenting manifestation of lupus.
Such is the plight of the internist. Pericarditis can be the initial manifestation of an autoimmune or inflammatory disease, but this is more often relevant on certification examinations and in medical education than in everyday practice. We are now charged with ordering tests in a more cost-effective manner than in the past. This means that we should not order tests simply because of an epidemiologic association, but only when the result is likely to influence decisions about testing or treatment. But that creates the intellectual dissonance of knowing of a potential relationship (which someone, someday, may challenge us about) but not looking for it. There is an inherent conflict between satisfying intellectual curiosity and the need to be thorough while at the same time containing costs and avoiding the potential harm inherent in overtesting.
A partial solution is to try to define the immediate risk of not recognizing a life- or organ-threatening disease process that can be suggested by a positive nonspecific test (eg, ANA), and to refine the pretest likelihood of specific diagnoses by obtaining an accurate and complete history and performing a focused physical examination. For example, if we suspect that SLE may be the cause of an initial episode of symptomatic pericarditis, our initial evaluation should focus on the patient’s clinical picture. Is there bitemporal hair-thinning? New-onset Raynaud symptoms? Mild generalized adenopathy or lymphopenia? A borderline-low platelet count, or any proteinuria or microhematuria (which should warrant a prompt examination of a fresh urine sediment sample by a physician at the point of care to look for cellular casts indicative of glomerulonephritis)?
As internists, we should try to fulfill our need to be thorough and compulsive by using our honed skills as careful observers and historians—taking a careful history from the patient and family, performing a focused physical examination, and appropriately using disease-defining or staging tests before ordering less specific serologic or other tests. Practicing medicine in a conscientious and compulsive manner does not mean that every diagnostic possibility must be tested for at initial presentation.
Reading how experienced clinicians approach the problem of pericarditis in a specialized clinic provides a useful prompt to self-assess how we approach analogous clinical scenarios.
Eruptive xanthoma
An obese 50-year-old man with hypertension, hyperlipidemia, recently diagnosed diabetes, and a history of grand mal seizures presented to the emergency room complaining of skin rash for 1 week. He denied having fever, chills, myalgia, abdominal pain, visual changes, recent changes in medications, or contact with anyone with similar symptoms.
He was a smoker, with a history of 20 pack-years; he denied abusing alcohol and taking illicit drugs.
He had no family history of diabetes, peripheral vascular disease, or coronary artery disease. His medications included lisinopril, simvastatin, niacin, metformin, and phenytoin.
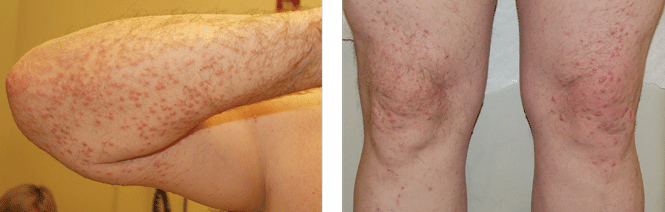
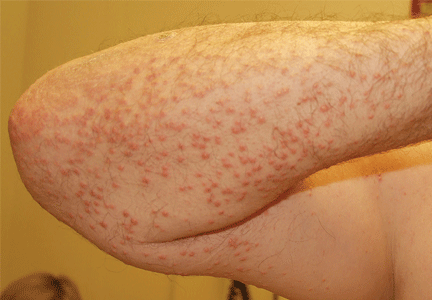
On physical examination, the lesions were small, reddish-yellow, nonpruritic tender papules covering the extensor surfaces of the knees, the forearms, the abdomen, and the back (Figure 1). Laboratory test results:
- Total cholesterol 1,045 mg/dL (reference range 100–199)
- Triglycerides 7,855 mg/dL (30–149)
- Thyroid-stimulating hormone 0.52 mIU/L (0.4–5.5)
- Fasting blood glucose 441 mg/dL (65–100)
- Hemoglobin A1c 12.6% (4.0–6.0)
- Total protein 7.2 g/dL (6.0–8.4)
- Albumin 4 g/dL (3.5–5.0)
- Creatinine 1 mg/dL (0.70–1.40)
- Glomerular filtration rate 79 mL/min/1.73 m2 (> 60)
- Urinalysis showed no proteinuria.
Histologic analysis of a lesion-biopsy specimen showed dermal foamy macrophages and loose lipids, which confirmed the suspicion of eruptive xanthoma.

The patient was started on strict glycemic and lipid control. Metformin and statin doses were increased and insulin was added. Three months later, laboratory results showed total cholesterol 128 mg/dL, triglycerides 164 mg/dL, fasting blood glucose 88 mg/dL, and hemoglobin A1c 5.5%. This was accompanied by marked improvement of the skin lesions (Figure 2).
CAUSES AND DIFFERENTIAL DIAGNOSIS
Eruptive xanthoma is a cutaneous disease most commonly arising over the extensor surfaces of the extremities and on the buttocks and shoulders, and it can be caused by high levels of serum triglycerides and uncontrolled diabetes mellitus.1 Hypothyroidism, end-stage renal disease, and nephrotic syndrome can cause secondary hypertriglyceridemia,2 which can cause eruptive xanthoma in severe cases. Patients with eruptive xanthoma may also have ophthalmologic and gastrointestinal involvement, such as lipemia retinalis (salmon-colored retina with creamy-white retinal vessels), abdominal pain, and hepatosplenomegaly.3
Other types of xanthoma associated with dyslipidemia include tuberous, tendinous, and plane xanthoma. Tuberous xanthoma is a firm, painless, deeper, red-yellow, larger nodular lesion, and the size may vary.4 Tendinous xanthoma is a slowly enlarging subcutaneous nodule typically located near tendons or ligaments in the hands, feet, and the Achilles tendon. Plane xanthoma is a flat papule or patch that can occur anywhere on the body.
The differential diagnosis includes disseminated granuloma annulare, non-Langerhans cell histiocytosis (xanthoma disseminatum, micronodular form of juvenile xanthogranuloma), and generalized eruptive histiocytoma. Eruptive xanthoma is differentiated from disseminated granuloma annulare by the abundance of perivascular histiocytes and xanthomized histiocytes, the presence of lipid deposits, and the deposition of hyaluronic acid on the edges.5 Xanthoma disseminatum consists of numerous, small, red-brown papules that are evenly spread on the face, skin-folds, trunk, and proximal extremities.6 Juvenile xanthogranuloma occurs mostly in children and is characterized by discrete orange-yellow nodules, which commonly appear on the scalp, face, and upper trunk. It is in most cases a solitary lesion, but multiple lesions may occur.7 Lesions of generalized eruptive histiocytoma are firm, erythematous or brownish papules that appear in successive crops over the face, trunk, and proximal surfaces of the limbs.
TREATMENT
Treatment of eruptive xanthoma involves dietary restriction, exercise, and drug therapy to control the hyperlipidemia and the diabetes.2 Early recognition and proper control of hypertriglyceridemia can prevent sequelae such as acute pancreatitis.3
- Durrington P. Dyslipidaemia. Lancet 2003; 362:717–731.
- Brunzell JD. Clinical practice. Hypertriglyceridemia. N Engl J Med 2007; 357:1009–1017.
- Leaf DA. Chylomicronemia and the chylomicronemia syndrome: a practical approach to management. Am J Med 2008; 121:10–12.
- Siddi GM, Pes GM, Errigo A, Corraduzza G, Ena P. Multiple tuberous xanthomas as the first manifestation of autosomal recessive hypercholesterolemia. J Eur Acad Dermatol Venereol 2006; 20:1376–1378.
- Cooper PH. Eruptive xanthoma: a microscopic simulant of granuloma annulare. J Cutan Pathol 1986; 13:207–215.
- Rupec RA, Schaller M. Xanthoma disseminatum. Int J Dermatol 2002; 41:911–913.
- Ferrari F, Masurel A, Olivier-Faivre L, Vabres P. Juvenile xanthogranuloma and nevus anemicus in the diagnosis of neurofibromatosis type 1. JAMA Dermatol 2014; 150:42–46.
An obese 50-year-old man with hypertension, hyperlipidemia, recently diagnosed diabetes, and a history of grand mal seizures presented to the emergency room complaining of skin rash for 1 week. He denied having fever, chills, myalgia, abdominal pain, visual changes, recent changes in medications, or contact with anyone with similar symptoms.
He was a smoker, with a history of 20 pack-years; he denied abusing alcohol and taking illicit drugs.
He had no family history of diabetes, peripheral vascular disease, or coronary artery disease. His medications included lisinopril, simvastatin, niacin, metformin, and phenytoin.
On physical examination, the lesions were small, reddish-yellow, nonpruritic tender papules covering the extensor surfaces of the knees, the forearms, the abdomen, and the back (Figure 1). Laboratory test results:
- Total cholesterol 1,045 mg/dL (reference range 100–199)
- Triglycerides 7,855 mg/dL (30–149)
- Thyroid-stimulating hormone 0.52 mIU/L (0.4–5.5)
- Fasting blood glucose 441 mg/dL (65–100)
- Hemoglobin A1c 12.6% (4.0–6.0)
- Total protein 7.2 g/dL (6.0–8.4)
- Albumin 4 g/dL (3.5–5.0)
- Creatinine 1 mg/dL (0.70–1.40)
- Glomerular filtration rate 79 mL/min/1.73 m2 (> 60)
- Urinalysis showed no proteinuria.
Histologic analysis of a lesion-biopsy specimen showed dermal foamy macrophages and loose lipids, which confirmed the suspicion of eruptive xanthoma.
The patient was started on strict glycemic and lipid control. Metformin and statin doses were increased and insulin was added. Three months later, laboratory results showed total cholesterol 128 mg/dL, triglycerides 164 mg/dL, fasting blood glucose 88 mg/dL, and hemoglobin A1c 5.5%. This was accompanied by marked improvement of the skin lesions (Figure 2).
CAUSES AND DIFFERENTIAL DIAGNOSIS
Eruptive xanthoma is a cutaneous disease most commonly arising over the extensor surfaces of the extremities and on the buttocks and shoulders, and it can be caused by high levels of serum triglycerides and uncontrolled diabetes mellitus.1 Hypothyroidism, end-stage renal disease, and nephrotic syndrome can cause secondary hypertriglyceridemia,2 which can cause eruptive xanthoma in severe cases. Patients with eruptive xanthoma may also have ophthalmologic and gastrointestinal involvement, such as lipemia retinalis (salmon-colored retina with creamy-white retinal vessels), abdominal pain, and hepatosplenomegaly.3
Other types of xanthoma associated with dyslipidemia include tuberous, tendinous, and plane xanthoma. Tuberous xanthoma is a firm, painless, deeper, red-yellow, larger nodular lesion, and the size may vary.4 Tendinous xanthoma is a slowly enlarging subcutaneous nodule typically located near tendons or ligaments in the hands, feet, and the Achilles tendon. Plane xanthoma is a flat papule or patch that can occur anywhere on the body.
The differential diagnosis includes disseminated granuloma annulare, non-Langerhans cell histiocytosis (xanthoma disseminatum, micronodular form of juvenile xanthogranuloma), and generalized eruptive histiocytoma. Eruptive xanthoma is differentiated from disseminated granuloma annulare by the abundance of perivascular histiocytes and xanthomized histiocytes, the presence of lipid deposits, and the deposition of hyaluronic acid on the edges.5 Xanthoma disseminatum consists of numerous, small, red-brown papules that are evenly spread on the face, skin-folds, trunk, and proximal extremities.6 Juvenile xanthogranuloma occurs mostly in children and is characterized by discrete orange-yellow nodules, which commonly appear on the scalp, face, and upper trunk. It is in most cases a solitary lesion, but multiple lesions may occur.7 Lesions of generalized eruptive histiocytoma are firm, erythematous or brownish papules that appear in successive crops over the face, trunk, and proximal surfaces of the limbs.
TREATMENT
Treatment of eruptive xanthoma involves dietary restriction, exercise, and drug therapy to control the hyperlipidemia and the diabetes.2 Early recognition and proper control of hypertriglyceridemia can prevent sequelae such as acute pancreatitis.3
An obese 50-year-old man with hypertension, hyperlipidemia, recently diagnosed diabetes, and a history of grand mal seizures presented to the emergency room complaining of skin rash for 1 week. He denied having fever, chills, myalgia, abdominal pain, visual changes, recent changes in medications, or contact with anyone with similar symptoms.
He was a smoker, with a history of 20 pack-years; he denied abusing alcohol and taking illicit drugs.
He had no family history of diabetes, peripheral vascular disease, or coronary artery disease. His medications included lisinopril, simvastatin, niacin, metformin, and phenytoin.
On physical examination, the lesions were small, reddish-yellow, nonpruritic tender papules covering the extensor surfaces of the knees, the forearms, the abdomen, and the back (Figure 1). Laboratory test results:
- Total cholesterol 1,045 mg/dL (reference range 100–199)
- Triglycerides 7,855 mg/dL (30–149)
- Thyroid-stimulating hormone 0.52 mIU/L (0.4–5.5)
- Fasting blood glucose 441 mg/dL (65–100)
- Hemoglobin A1c 12.6% (4.0–6.0)
- Total protein 7.2 g/dL (6.0–8.4)
- Albumin 4 g/dL (3.5–5.0)
- Creatinine 1 mg/dL (0.70–1.40)
- Glomerular filtration rate 79 mL/min/1.73 m2 (> 60)
- Urinalysis showed no proteinuria.
Histologic analysis of a lesion-biopsy specimen showed dermal foamy macrophages and loose lipids, which confirmed the suspicion of eruptive xanthoma.
The patient was started on strict glycemic and lipid control. Metformin and statin doses were increased and insulin was added. Three months later, laboratory results showed total cholesterol 128 mg/dL, triglycerides 164 mg/dL, fasting blood glucose 88 mg/dL, and hemoglobin A1c 5.5%. This was accompanied by marked improvement of the skin lesions (Figure 2).
CAUSES AND DIFFERENTIAL DIAGNOSIS
Eruptive xanthoma is a cutaneous disease most commonly arising over the extensor surfaces of the extremities and on the buttocks and shoulders, and it can be caused by high levels of serum triglycerides and uncontrolled diabetes mellitus.1 Hypothyroidism, end-stage renal disease, and nephrotic syndrome can cause secondary hypertriglyceridemia,2 which can cause eruptive xanthoma in severe cases. Patients with eruptive xanthoma may also have ophthalmologic and gastrointestinal involvement, such as lipemia retinalis (salmon-colored retina with creamy-white retinal vessels), abdominal pain, and hepatosplenomegaly.3
Other types of xanthoma associated with dyslipidemia include tuberous, tendinous, and plane xanthoma. Tuberous xanthoma is a firm, painless, deeper, red-yellow, larger nodular lesion, and the size may vary.4 Tendinous xanthoma is a slowly enlarging subcutaneous nodule typically located near tendons or ligaments in the hands, feet, and the Achilles tendon. Plane xanthoma is a flat papule or patch that can occur anywhere on the body.
The differential diagnosis includes disseminated granuloma annulare, non-Langerhans cell histiocytosis (xanthoma disseminatum, micronodular form of juvenile xanthogranuloma), and generalized eruptive histiocytoma. Eruptive xanthoma is differentiated from disseminated granuloma annulare by the abundance of perivascular histiocytes and xanthomized histiocytes, the presence of lipid deposits, and the deposition of hyaluronic acid on the edges.5 Xanthoma disseminatum consists of numerous, small, red-brown papules that are evenly spread on the face, skin-folds, trunk, and proximal extremities.6 Juvenile xanthogranuloma occurs mostly in children and is characterized by discrete orange-yellow nodules, which commonly appear on the scalp, face, and upper trunk. It is in most cases a solitary lesion, but multiple lesions may occur.7 Lesions of generalized eruptive histiocytoma are firm, erythematous or brownish papules that appear in successive crops over the face, trunk, and proximal surfaces of the limbs.
TREATMENT
Treatment of eruptive xanthoma involves dietary restriction, exercise, and drug therapy to control the hyperlipidemia and the diabetes.2 Early recognition and proper control of hypertriglyceridemia can prevent sequelae such as acute pancreatitis.3
- Durrington P. Dyslipidaemia. Lancet 2003; 362:717–731.
- Brunzell JD. Clinical practice. Hypertriglyceridemia. N Engl J Med 2007; 357:1009–1017.
- Leaf DA. Chylomicronemia and the chylomicronemia syndrome: a practical approach to management. Am J Med 2008; 121:10–12.
- Siddi GM, Pes GM, Errigo A, Corraduzza G, Ena P. Multiple tuberous xanthomas as the first manifestation of autosomal recessive hypercholesterolemia. J Eur Acad Dermatol Venereol 2006; 20:1376–1378.
- Cooper PH. Eruptive xanthoma: a microscopic simulant of granuloma annulare. J Cutan Pathol 1986; 13:207–215.
- Rupec RA, Schaller M. Xanthoma disseminatum. Int J Dermatol 2002; 41:911–913.
- Ferrari F, Masurel A, Olivier-Faivre L, Vabres P. Juvenile xanthogranuloma and nevus anemicus in the diagnosis of neurofibromatosis type 1. JAMA Dermatol 2014; 150:42–46.
- Durrington P. Dyslipidaemia. Lancet 2003; 362:717–731.
- Brunzell JD. Clinical practice. Hypertriglyceridemia. N Engl J Med 2007; 357:1009–1017.
- Leaf DA. Chylomicronemia and the chylomicronemia syndrome: a practical approach to management. Am J Med 2008; 121:10–12.
- Siddi GM, Pes GM, Errigo A, Corraduzza G, Ena P. Multiple tuberous xanthomas as the first manifestation of autosomal recessive hypercholesterolemia. J Eur Acad Dermatol Venereol 2006; 20:1376–1378.
- Cooper PH. Eruptive xanthoma: a microscopic simulant of granuloma annulare. J Cutan Pathol 1986; 13:207–215.
- Rupec RA, Schaller M. Xanthoma disseminatum. Int J Dermatol 2002; 41:911–913.
- Ferrari F, Masurel A, Olivier-Faivre L, Vabres P. Juvenile xanthogranuloma and nevus anemicus in the diagnosis of neurofibromatosis type 1. JAMA Dermatol 2014; 150:42–46.
Heart on the right may sometimes be ‘right’
A 76-year-old man presented to the emergency department with right-sided exertional chest pain radiating to the right shoulder and arm associated with shortness of breath. His vital signs were normal. On clinical examination, the cardiac apex was palpated on the right side, 9 cm from the midsternal line in the fifth intercostal space.
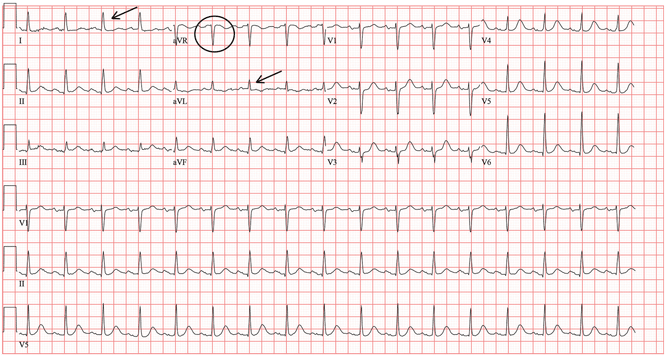
A standard left-sided 12-lead electrocardiogram (ECG) showed right-axis deviation and inverted P, QRS, and T waves in leads I and aVL (Figure 1). Although these changes are also seen when the right and left arm electrode wires are transposed, the precordial lead morphology in such a situation would usually be normal. In our patient, the precordial leads showed the absence or even slight reversal of R-wave progression, a feature indicative of dextrocardia.1,2
In patients with dextrocardia, right-sided hookup of the electrodes is usually necessary for proper interpretation of the ECG. When this was done in our patient, the ECG showed a normal cardiac axis, a negative QRS complex in lead aVR, a positive P wave and other complexes in lead I, and normal R-wave progression in the precordial leads—findings suggestive of dextrocardia (Figure 2).
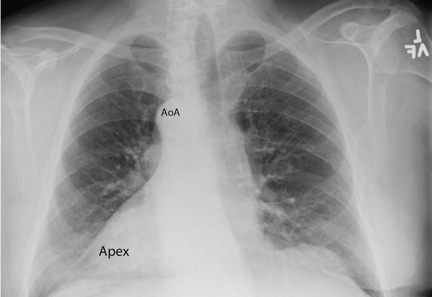
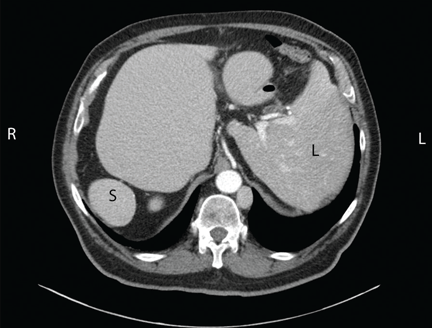
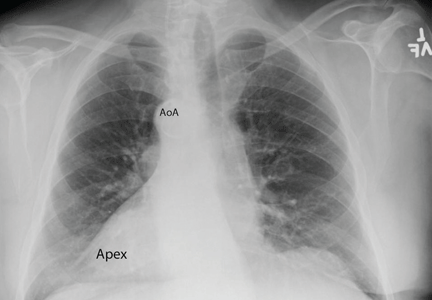
Chest radiography showed a right-sided cardiac silhouette (Figure 3), and computed tomography of the abdomen (Figure 4) revealed the liver positioned on the left side and the spleen on the right, confirming the diagnosis of situs inversus totalis. The ECG showed dextrocardia, but no other abnormalities. The patient eventually underwent coronary angiography, which showed nonobstructive coronary artery disease.
DEXTROCARDIA, OTHER CONGENITAL CARDIOVASCULAR MALFORMATIONS
Dextrocardia was first described in early 17th century.1 Situs solitus is the normal position of the heart and viscera, whereas situs inversus is a mirror-image anatomic arrangement of the organs. Situs inversus with dextrocardia, also called situs inversus totalis, is a rare condition (with a prevalence of 1 in 8,000) in which the heart and descending aorta are on the right and the thoracic and abdominal viscera are usually mirror images of the normal morphology.1,3,4 A mirror-image sinus node lies at the junction of the left superior vena cava and the left-sided (morphologic right) atrium.1 People with situs inversus with dextrocardia are usually asymptomatic and have a normal life expectancy.1,2 Situs inversus with levocardia is a rare condition in which the heart is in the normal position but the viscera are in the dextro-position. This anomaly has a prevalence of 1 in 22,000.5
Atrial situs almost always corresponds to visceral situs. However, when the alignment of the atria and viscera is inconsistent and situs cannot be determined clearly because of the malpositioning of organs, the condition is called “situs ambiguous.” This is very rare, with a prevalence of 1 in 40,000.6
Risk factors
The cause of congenital cardiovascular malformations such as these is not known, but risk factors include positive family history, maternal diabetes, and cocaine use in the first trimester.7
The prevalence of congenital heart disease in patients with situs inversus with dextrocardia is low and ranges from 2% to 5%. This is in contrast to situs solitus with dextrocardia (isolated dextrocardia), which is almost always associated with cardiovascular anomalies.2,4 Kartagener syndrome—the triad of situs inversus, sinusitis, and bronchiectasis—occurs in 25% of people with situs inversus with dextrocardia.4 Situs inversus with levocardia is also frequently associated with cardiac anomalies.5
The major features of dextrocardia on ECG are:
- Negative P wave, QRS complex, and T wave in lead I
- Positive QRS complex in aVR
- Right-axis deviation
- Reversal of R-wave progression in the precordial leads.
Ventricular activation and repolarization are reversed, resulting in a negative QRS complex and an inverted T wave in lead I. The absence of R-wave progression in the precordial leads helps differentiate mirror-image dextrocardia from erroneously reversed limb-electrode placement, which shows normal R-wave progression from V1 to V6 while showing similar features to those seen in dextrocardia in the limb leads.2 In right-sided hookup, the limb electrodes are reversed, and the chest electrodes are recorded from the right precordium.
CORONARY INTERVENTIONS REQUIRE SPECIAL CONSIDERATION
In patients with dextrocardia, coronary interventions can be challenging because of the mirror-image position of the coronary ostia and the aortic arch.8 These patients also need careful imaging, consideration of other associated congenital cardiac abnormalities, and detailed planning before cardiac surgery, including coronary artery bypass grafting.9
Patients with dextrocardia may present with cardiac symptoms localized to the right side of the body and have confusing clinical and diagnostic findings. Keeping dextrocardia and other such anomalies in mind can prevent delay in appropriately directed interventions. In a patient such as ours, the heart on the right side of the chest may indeed be “right.” Still, diagnostic tests to look for disorders encountered with dextrocardia may be necessary.
- Perloff JK. The cardiac malpositions. Am J Cardiol 2011; 108:1352–1361.
- Tanawuttiwat T, Vasaiwala S, Dia M. ECG image of the month. Mirror mirror. Am J Med 2010; 123:34–36.
- Douard R, Feldman A, Bargy F, Loric S, Delmas V. Anomalies of lateralization in man: a case of total situs in-versus. Surg Radiol Anat 2000; 22:293–297.
- Maldjian PD, Saric M. Approach to dextrocardia in adults: review. AJR Am J Roentgenol 2007; 188(suppl 6):S39–S49.
- Gindes L, Hegesh J, Barkai G, Jacobson JM, Achiron R. Isolated levocardia: prenatal diagnosis, clinical im-portance, and literature review. J Ultrasound Med 2007; 26:361–365.
- Abut E, Arman A, Güveli H, et al. Malposition of internal organs: a case of situs ambiguous anomaly in an adult. Turk J Gastroenterol 2003; 14:151–155.
- Kuehl KS, Loffredo C. Risk factors for heart disease associated with abnormal sidedness. Teratology 2002; 66:242–248.
- Aksoy S, Cam N, Gurkan U, Altay S, Bozbay M, Agirbasli M. Primary percutaneous intervention: for acute myo-cardial infarction in a patient with dextrocardia and situs inversus. Tex Heart Inst J 2012; 39:140–141.
- Murtuza B, Gupta P, Goli G, Lall KS. Coronary revascularization in adults with dextrocardia: surgical implications of the anatomic variants. Tex Heart Inst J 2010; 37:633–640.
A 76-year-old man presented to the emergency department with right-sided exertional chest pain radiating to the right shoulder and arm associated with shortness of breath. His vital signs were normal. On clinical examination, the cardiac apex was palpated on the right side, 9 cm from the midsternal line in the fifth intercostal space.
A standard left-sided 12-lead electrocardiogram (ECG) showed right-axis deviation and inverted P, QRS, and T waves in leads I and aVL (Figure 1). Although these changes are also seen when the right and left arm electrode wires are transposed, the precordial lead morphology in such a situation would usually be normal. In our patient, the precordial leads showed the absence or even slight reversal of R-wave progression, a feature indicative of dextrocardia.1,2
In patients with dextrocardia, right-sided hookup of the electrodes is usually necessary for proper interpretation of the ECG. When this was done in our patient, the ECG showed a normal cardiac axis, a negative QRS complex in lead aVR, a positive P wave and other complexes in lead I, and normal R-wave progression in the precordial leads—findings suggestive of dextrocardia (Figure 2).
Chest radiography showed a right-sided cardiac silhouette (Figure 3), and computed tomography of the abdomen (Figure 4) revealed the liver positioned on the left side and the spleen on the right, confirming the diagnosis of situs inversus totalis. The ECG showed dextrocardia, but no other abnormalities. The patient eventually underwent coronary angiography, which showed nonobstructive coronary artery disease.
DEXTROCARDIA, OTHER CONGENITAL CARDIOVASCULAR MALFORMATIONS
Dextrocardia was first described in early 17th century.1 Situs solitus is the normal position of the heart and viscera, whereas situs inversus is a mirror-image anatomic arrangement of the organs. Situs inversus with dextrocardia, also called situs inversus totalis, is a rare condition (with a prevalence of 1 in 8,000) in which the heart and descending aorta are on the right and the thoracic and abdominal viscera are usually mirror images of the normal morphology.1,3,4 A mirror-image sinus node lies at the junction of the left superior vena cava and the left-sided (morphologic right) atrium.1 People with situs inversus with dextrocardia are usually asymptomatic and have a normal life expectancy.1,2 Situs inversus with levocardia is a rare condition in which the heart is in the normal position but the viscera are in the dextro-position. This anomaly has a prevalence of 1 in 22,000.5
Atrial situs almost always corresponds to visceral situs. However, when the alignment of the atria and viscera is inconsistent and situs cannot be determined clearly because of the malpositioning of organs, the condition is called “situs ambiguous.” This is very rare, with a prevalence of 1 in 40,000.6
Risk factors
The cause of congenital cardiovascular malformations such as these is not known, but risk factors include positive family history, maternal diabetes, and cocaine use in the first trimester.7
The prevalence of congenital heart disease in patients with situs inversus with dextrocardia is low and ranges from 2% to 5%. This is in contrast to situs solitus with dextrocardia (isolated dextrocardia), which is almost always associated with cardiovascular anomalies.2,4 Kartagener syndrome—the triad of situs inversus, sinusitis, and bronchiectasis—occurs in 25% of people with situs inversus with dextrocardia.4 Situs inversus with levocardia is also frequently associated with cardiac anomalies.5
The major features of dextrocardia on ECG are:
- Negative P wave, QRS complex, and T wave in lead I
- Positive QRS complex in aVR
- Right-axis deviation
- Reversal of R-wave progression in the precordial leads.
Ventricular activation and repolarization are reversed, resulting in a negative QRS complex and an inverted T wave in lead I. The absence of R-wave progression in the precordial leads helps differentiate mirror-image dextrocardia from erroneously reversed limb-electrode placement, which shows normal R-wave progression from V1 to V6 while showing similar features to those seen in dextrocardia in the limb leads.2 In right-sided hookup, the limb electrodes are reversed, and the chest electrodes are recorded from the right precordium.
CORONARY INTERVENTIONS REQUIRE SPECIAL CONSIDERATION
In patients with dextrocardia, coronary interventions can be challenging because of the mirror-image position of the coronary ostia and the aortic arch.8 These patients also need careful imaging, consideration of other associated congenital cardiac abnormalities, and detailed planning before cardiac surgery, including coronary artery bypass grafting.9
Patients with dextrocardia may present with cardiac symptoms localized to the right side of the body and have confusing clinical and diagnostic findings. Keeping dextrocardia and other such anomalies in mind can prevent delay in appropriately directed interventions. In a patient such as ours, the heart on the right side of the chest may indeed be “right.” Still, diagnostic tests to look for disorders encountered with dextrocardia may be necessary.
A 76-year-old man presented to the emergency department with right-sided exertional chest pain radiating to the right shoulder and arm associated with shortness of breath. His vital signs were normal. On clinical examination, the cardiac apex was palpated on the right side, 9 cm from the midsternal line in the fifth intercostal space.
A standard left-sided 12-lead electrocardiogram (ECG) showed right-axis deviation and inverted P, QRS, and T waves in leads I and aVL (Figure 1). Although these changes are also seen when the right and left arm electrode wires are transposed, the precordial lead morphology in such a situation would usually be normal. In our patient, the precordial leads showed the absence or even slight reversal of R-wave progression, a feature indicative of dextrocardia.1,2
In patients with dextrocardia, right-sided hookup of the electrodes is usually necessary for proper interpretation of the ECG. When this was done in our patient, the ECG showed a normal cardiac axis, a negative QRS complex in lead aVR, a positive P wave and other complexes in lead I, and normal R-wave progression in the precordial leads—findings suggestive of dextrocardia (Figure 2).
Chest radiography showed a right-sided cardiac silhouette (Figure 3), and computed tomography of the abdomen (Figure 4) revealed the liver positioned on the left side and the spleen on the right, confirming the diagnosis of situs inversus totalis. The ECG showed dextrocardia, but no other abnormalities. The patient eventually underwent coronary angiography, which showed nonobstructive coronary artery disease.
DEXTROCARDIA, OTHER CONGENITAL CARDIOVASCULAR MALFORMATIONS
Dextrocardia was first described in early 17th century.1 Situs solitus is the normal position of the heart and viscera, whereas situs inversus is a mirror-image anatomic arrangement of the organs. Situs inversus with dextrocardia, also called situs inversus totalis, is a rare condition (with a prevalence of 1 in 8,000) in which the heart and descending aorta are on the right and the thoracic and abdominal viscera are usually mirror images of the normal morphology.1,3,4 A mirror-image sinus node lies at the junction of the left superior vena cava and the left-sided (morphologic right) atrium.1 People with situs inversus with dextrocardia are usually asymptomatic and have a normal life expectancy.1,2 Situs inversus with levocardia is a rare condition in which the heart is in the normal position but the viscera are in the dextro-position. This anomaly has a prevalence of 1 in 22,000.5
Atrial situs almost always corresponds to visceral situs. However, when the alignment of the atria and viscera is inconsistent and situs cannot be determined clearly because of the malpositioning of organs, the condition is called “situs ambiguous.” This is very rare, with a prevalence of 1 in 40,000.6
Risk factors
The cause of congenital cardiovascular malformations such as these is not known, but risk factors include positive family history, maternal diabetes, and cocaine use in the first trimester.7
The prevalence of congenital heart disease in patients with situs inversus with dextrocardia is low and ranges from 2% to 5%. This is in contrast to situs solitus with dextrocardia (isolated dextrocardia), which is almost always associated with cardiovascular anomalies.2,4 Kartagener syndrome—the triad of situs inversus, sinusitis, and bronchiectasis—occurs in 25% of people with situs inversus with dextrocardia.4 Situs inversus with levocardia is also frequently associated with cardiac anomalies.5
The major features of dextrocardia on ECG are:
- Negative P wave, QRS complex, and T wave in lead I
- Positive QRS complex in aVR
- Right-axis deviation
- Reversal of R-wave progression in the precordial leads.
Ventricular activation and repolarization are reversed, resulting in a negative QRS complex and an inverted T wave in lead I. The absence of R-wave progression in the precordial leads helps differentiate mirror-image dextrocardia from erroneously reversed limb-electrode placement, which shows normal R-wave progression from V1 to V6 while showing similar features to those seen in dextrocardia in the limb leads.2 In right-sided hookup, the limb electrodes are reversed, and the chest electrodes are recorded from the right precordium.
CORONARY INTERVENTIONS REQUIRE SPECIAL CONSIDERATION
In patients with dextrocardia, coronary interventions can be challenging because of the mirror-image position of the coronary ostia and the aortic arch.8 These patients also need careful imaging, consideration of other associated congenital cardiac abnormalities, and detailed planning before cardiac surgery, including coronary artery bypass grafting.9
Patients with dextrocardia may present with cardiac symptoms localized to the right side of the body and have confusing clinical and diagnostic findings. Keeping dextrocardia and other such anomalies in mind can prevent delay in appropriately directed interventions. In a patient such as ours, the heart on the right side of the chest may indeed be “right.” Still, diagnostic tests to look for disorders encountered with dextrocardia may be necessary.
- Perloff JK. The cardiac malpositions. Am J Cardiol 2011; 108:1352–1361.
- Tanawuttiwat T, Vasaiwala S, Dia M. ECG image of the month. Mirror mirror. Am J Med 2010; 123:34–36.
- Douard R, Feldman A, Bargy F, Loric S, Delmas V. Anomalies of lateralization in man: a case of total situs in-versus. Surg Radiol Anat 2000; 22:293–297.
- Maldjian PD, Saric M. Approach to dextrocardia in adults: review. AJR Am J Roentgenol 2007; 188(suppl 6):S39–S49.
- Gindes L, Hegesh J, Barkai G, Jacobson JM, Achiron R. Isolated levocardia: prenatal diagnosis, clinical im-portance, and literature review. J Ultrasound Med 2007; 26:361–365.
- Abut E, Arman A, Güveli H, et al. Malposition of internal organs: a case of situs ambiguous anomaly in an adult. Turk J Gastroenterol 2003; 14:151–155.
- Kuehl KS, Loffredo C. Risk factors for heart disease associated with abnormal sidedness. Teratology 2002; 66:242–248.
- Aksoy S, Cam N, Gurkan U, Altay S, Bozbay M, Agirbasli M. Primary percutaneous intervention: for acute myo-cardial infarction in a patient with dextrocardia and situs inversus. Tex Heart Inst J 2012; 39:140–141.
- Murtuza B, Gupta P, Goli G, Lall KS. Coronary revascularization in adults with dextrocardia: surgical implications of the anatomic variants. Tex Heart Inst J 2010; 37:633–640.
- Perloff JK. The cardiac malpositions. Am J Cardiol 2011; 108:1352–1361.
- Tanawuttiwat T, Vasaiwala S, Dia M. ECG image of the month. Mirror mirror. Am J Med 2010; 123:34–36.
- Douard R, Feldman A, Bargy F, Loric S, Delmas V. Anomalies of lateralization in man: a case of total situs in-versus. Surg Radiol Anat 2000; 22:293–297.
- Maldjian PD, Saric M. Approach to dextrocardia in adults: review. AJR Am J Roentgenol 2007; 188(suppl 6):S39–S49.
- Gindes L, Hegesh J, Barkai G, Jacobson JM, Achiron R. Isolated levocardia: prenatal diagnosis, clinical im-portance, and literature review. J Ultrasound Med 2007; 26:361–365.
- Abut E, Arman A, Güveli H, et al. Malposition of internal organs: a case of situs ambiguous anomaly in an adult. Turk J Gastroenterol 2003; 14:151–155.
- Kuehl KS, Loffredo C. Risk factors for heart disease associated with abnormal sidedness. Teratology 2002; 66:242–248.
- Aksoy S, Cam N, Gurkan U, Altay S, Bozbay M, Agirbasli M. Primary percutaneous intervention: for acute myo-cardial infarction in a patient with dextrocardia and situs inversus. Tex Heart Inst J 2012; 39:140–141.
- Murtuza B, Gupta P, Goli G, Lall KS. Coronary revascularization in adults with dextrocardia: surgical implications of the anatomic variants. Tex Heart Inst J 2010; 37:633–640.
A Not-So-Old Football Injury
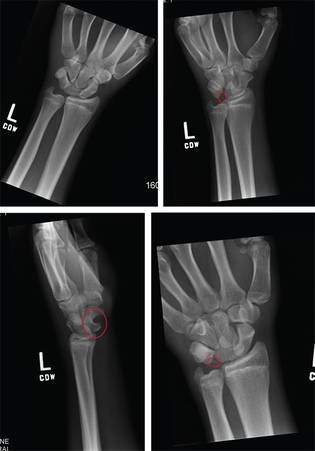
ANSWER
Imaging shows a ventral dislocation of the lunate. There is also a tiny avulsion fracture on the ulnar aspect of the adjacent triquetrum.
The patient was referred to orthopedics for a perilunate dislocation of the left wrist. He underwent successful closed reduction and was placed in a short arm cast for four weeks.
ANSWER
Imaging shows a ventral dislocation of the lunate. There is also a tiny avulsion fracture on the ulnar aspect of the adjacent triquetrum.
The patient was referred to orthopedics for a perilunate dislocation of the left wrist. He underwent successful closed reduction and was placed in a short arm cast for four weeks.
ANSWER
Imaging shows a ventral dislocation of the lunate. There is also a tiny avulsion fracture on the ulnar aspect of the adjacent triquetrum.
The patient was referred to orthopedics for a perilunate dislocation of the left wrist. He underwent successful closed reduction and was placed in a short arm cast for four weeks.
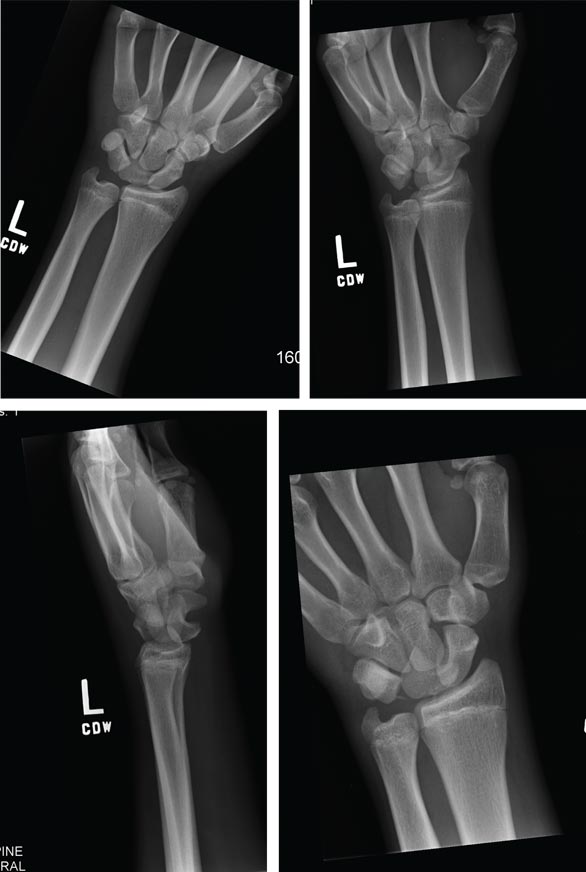
A 15-year-old boy presents for evaluation of left wrist pain. He says that two days ago, while playing football, he fell onto his outstretched left hand, which twisted upon impact with the ground. Immediately after the fall, he experienced severe pain. Since then, the pain has been constant, although it lessens to a moderate dull ache at rest and sharpens with activity. He denies any numbness or tingling in the affected hand and wrist. There are no other areas of injury from the fall, nor is there significant medical history. Physical exam identifies moderate left wrist swelling with focal tenderness over the volar aspect of the distal radius, extending to the wrist. The patient has limited active and passive flexion and extension of the left hand and wrist, along with reduced grip strength due to pain. There is mild navicular tenderness. Radial pulse is 2+, the hand is warm to the touch, and the skin is intact. Sensation is intact in all of the digits, which also demonstrate brisk capillary refill. Radiographs of the left wrist are obtained. What is your impression?
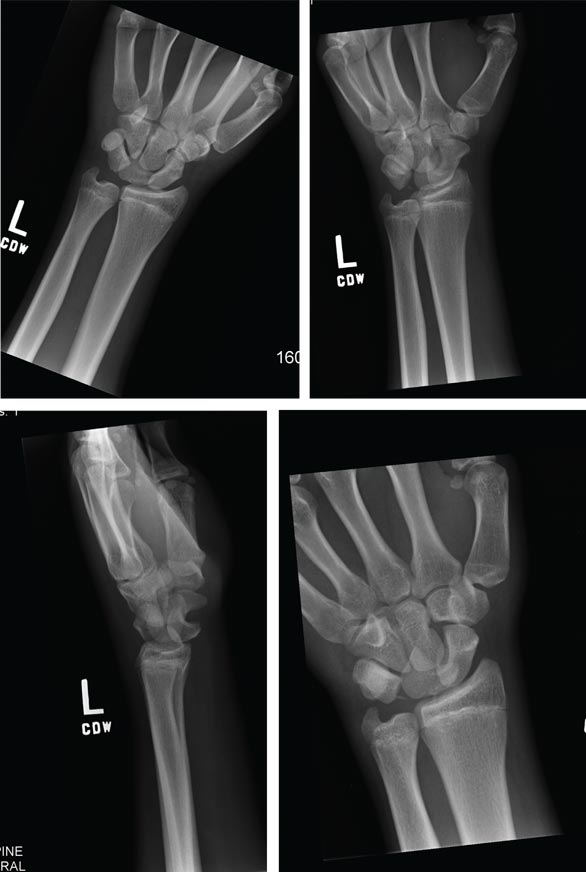
Postoperative Patient Suddenly Worsens
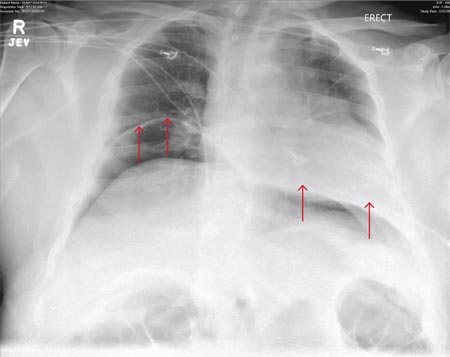
ANSWER
The radiograph demonstrates bilateral elevated diaphragm with a moderate amount of visible free air. With no history of recent abdominal procedures, the primary concern is a perforated viscus.
Urgent surgical consultation, as well as CT of the abdomen and pelvis, was obtained. The imaging confirmed the free air but provided no clear etiology. The patient underwent emergent laparotomy later that day and was found to have a perforated colon.
ANSWER
The radiograph demonstrates bilateral elevated diaphragm with a moderate amount of visible free air. With no history of recent abdominal procedures, the primary concern is a perforated viscus.
Urgent surgical consultation, as well as CT of the abdomen and pelvis, was obtained. The imaging confirmed the free air but provided no clear etiology. The patient underwent emergent laparotomy later that day and was found to have a perforated colon.
ANSWER
The radiograph demonstrates bilateral elevated diaphragm with a moderate amount of visible free air. With no history of recent abdominal procedures, the primary concern is a perforated viscus.
Urgent surgical consultation, as well as CT of the abdomen and pelvis, was obtained. The imaging confirmed the free air but provided no clear etiology. The patient underwent emergent laparotomy later that day and was found to have a perforated colon.
A 55-year-old man undergoes an elective craniotomy for tumor resection, with uneventful preoperative and intraoperative stages. Immediately postoperative, however, he experiences seizures. Noncontrast CT of the head is negative except for postoperative changes. The patient is placed in the ICU for close monitoring. He is slowly improving when, on the fifth postoperative day, tachypnea and dyspnea are observed. The patient is afebrile. His blood pressure is 116/70 mm Hg; pulse, 90 beats/min; respiratory rate, 30 breaths/min; and O2 saturation, 98%. A stat portable chest radiograph is obtained. What is your impression?
Case Studies in Toxicology: Double Take—Is Re-exposure Necessary to Explain Delayed Recurrent Opioid Toxicity?
Case
A previously healthy 10-month-old girl was brought to the ED by her mother, who noted that the child had been excessively drowsy throughout the day. She reported that her husband had dropped an unknown amount of his morphine sulfate extended-release 60-mg tablets and oxycodone 10-mg/acetaminophen 325-mg tablets on the floor 5 days earlier. Although unsure of how many tablets he had dropped, the father believed he had located all of them. The mother, however, found some of the tablets around the crib in their daughter’s room.

When the child arrived to the ED, her vital signs were: blood pressure, 95/60 mm Hg; heart rate, 102 beats/minute; respiratory rate (RR), 18 breaths/minute; and temperature, 98.4°F. Oxygen saturation was 98% on room air. On physical examination, the child was lethargic, her pupils were less than 1 mm in diameter, and her bowel sounds were absent. After the administration of intravenous (IV) naloxone 0.4 mg, the patient became less drowsy and her RR normalized. Approximately 1 hour later, though, the child again became lethargic; she was given a repeat dose of IV naloxone 0.4 mg, and a naloxone infusion was initiated at 0.3 mg/h. Over approximately 20 hours, the infusion was tapered and discontinued. Three hours after the infusion was stopped, the child’s vital signs and behavior were both normal. After a social worker and representative from the Administration for Children’s Services reviewed the patient’s case, she was discharged home with her parents.
Less than 1 hour later, however, the mother returned to the ED with the child, who was again unresponsive. Although the girl’s RR was normal, she had pinpoint pupils. After she was given IV naloxone 0.4 mg, the child awoke and remained responsive for 20 minutes before returning to a somnolent state. Another IV dose of naloxone 0.4 mg was administered, which showed partial improvement in responsiveness. A naloxone infusion was then initiated and titrated up to 1 mg/h to maintain wakefulness and ventilation. In the pediatric intensive care unit, the child required titration of the naloxone infusion to 2 mg/h to which she responded well. Over the next 12 hours, the infusion was tapered off and the child was discharged home with her parents.
Blood samples from both the initial visit and the return visit were sent for toxicologic analysis by gas chromatography-mass spectrometry (GC-MS). Serum from the first visit contained morphine at a concentration of 3,000 ng/mL; serum from the second visit contained morphine at 420 ng/mL. Both samples were negative for oxycodone or any of the other substances checked on the extended GC-MS screen.
What is the toxicologic differential?
Although this patient’s extreme somnolence was suspected to be opioid-induced, and was confirmed by an appropriate response to naloxone, children may present to the ED somnolent for a variety of unknown reasons. Even with a fairly clear history, the clinician should also consider metabolic, neurological, infectious, traumatic, and psychiatric causes of altered mental status.1 The toxicologic causes of altered mental status are expansive and include the effects of many medications used therapeutically or in overdose. Opioids, benzodiazepines, barbiturates, α-2 agonists (eg, clonidine), sleep aids (eg, zolpidem, diphenhydramine), and ethanol are common causes of induced an altered mental status. When taking a toxicologic history, it is important to inquire not only about the patient’s medications but also the medications of other members of the household to which the patient may have access. This includes not only prescription medications but also over-the-counter, complementary, and herbal preparations.
Why did this child have delayed recurrent opioid toxicity?
When used as directed, opioids cause analgesia and euphoria. Analgesia is mediated by agonism at the μ- , κ-, and δ-opioid receptors throughout the brain and spinal cord. The majority of morphine’s analgesic activity comes from activation of the μ-opioid receptors.2 In overdose, opioids classically cause a toxidrome characterized by miosis, coma, decreased bowel sounds, and respiratory depression. These signs can give clues to a patient’s exposure.
Supportive care is the cornerstone of treatment for patients with opioid toxicity, and maintaining the airway and monitoring the respiratory status are extremely important. When ventilation decreases due to the actions of opioids (typically denoted by a RR of <12 breaths/minute in adults, but may be marked by a reduction in depth of breathing as well), the use of an opioid antagonist is appropriate.4 The most commonly used antagonist is naloxone, an antidote with antagonism at all opioid receptor subtypes.5
In patients who are not dependent on opioids, IV naloxone 0.4 mg is an appropriate initial dose—regardless of patient size or specifics of the exposure. Patients with opioid dependency (eg, patients taking opioids for chronic pain or palliative care, or in those with suspected or confirmed opioid abuse), should receive smaller initial doses of naloxone (eg, 0.04 mg); the dose should be titrated up to effect to avoid precipitating acute opioid withdrawal. The goal of opioid antagonism is to allow the patient to breathe spontaneously and at an appropriate rate and depth without precipitating withdrawal. The duration of action of naloxone is 20 to 90 minutes in adults.
Patients presenting with heroin overdose should be monitored for at least 2 hours after naloxone administration (some suggest 3 hours) to determine whether or not additional dosing will be necessary. After oral opioid exposures, particularly with extended-release or long-acting formulations, longer periods of observation are required (this is unrelated to the naloxone pharmacokinetics, but rather to the slow rise in blood levels from some of these formulations). If repeated opioid toxicity occurs in adults, a naloxone infusion may be helpful to reduce the need for repetitive re-dosing. Initially, an hourly infusion equal to two-thirds of the dose of naloxone that reversed the patient’s respiratory depression is suggested6
Naloxone is eliminated by conjugation with glucuronic acid before is it excreted from the body. Due to decreased hepatic conjugation and prolonged metabolization of drugs in pediatric patients, naloxone may have a longer half-life in children—especially neonates and infants7; in children, the half-life of naloxone may extend up to three times that of adults.8 This extended half-life can lead to a false sense of assurance that a child is free of opioid effects 120 minutes after receiving naloxone—the time by which an adult patient would likely be without significant systemic effects of naloxone—when in fact the effect of naloxone has not yet sufficiently waned. This in turn may prompt discharge before sufficient time has passed to exclude recrudescence of opioid toxicity: The presence of persistent opioid agonist concentrations in the blood, even at consequential amounts, remains masked by the persistent presence of naloxone.
The goal of opioid antagonism is to allow the patient to breathe spontaneously and at an appropriate rate and depth without precipitating withdrawal. In this patient, it is not surprising that the the ingestion of an extended-relief form of morphine should produce a prolonged opioid effect. At therapeutic concentrations in children (~10 ng/mL), the half-life of morphine is slightly longer than in adults (~3 hours vs 2 hours) and is likely even longer with very high serum concentrations. It is metabolized to morphine 6-glucuronide, which is active and longer lasting than the parent compound. This may account for additional clinical effects beyond the time that the serum morphine concentration falls, and is particularly relevant following immediate-release morphine overdose.
In this case it is also important to consider whether or not the patient was re-exposed to an opioid between the first and second ED visit. The dramatically elevated initial serum morphine concentrations and the relatively appropriate fall in magnitude of the second sample suggest that the recurrence of respiratory depression was not the result of re-exposure. The patient’s recurrent effects, even a day out from exposure, can be explained by the immediate-release morphine exposure and the discharge prior to waning of the naloxone. In children with opioid toxicity, another potential option, though not directly studied, is to administer the long-acting opioid antagonist naltrexone to the patient prior to discharge.
Case Conclusion
When used appropriately and under the correct circumstances, naloxone is safe and effective for the reversal of opioid toxicity. As with any antidote, patients must be appropriately monitored for any adverse effects or recurrence of toxicity. Moreover, the clinician should be mindful of the pharmacokinetic differences between adults and young children and the possibility of a later-than-expected recurrence of opioid toxicity in pediatric patients.
This case is a reminder of the importance of safe medication storage. Infants and young children who are crawling and exploring their environment are especially vulnerable to toxicity from medications found on the floor. Regardless of age, quick recognition of opioid-induced respiratory depression and appropriate use of naloxone can help to decrease the morbidity associated with excessive opioid exposures in all patients.
Dr Berman is a senior medical toxicology fellow at North Shore-Long Island Jewish Medical Center, New York. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board. Dr Majlesi is the director of medical toxicology at Staten Island University Hospital, New York.
- Lehman RK, Mink J. Altered mental status. Clin Pediatr Emerg Med. 2008;9:68-75.
- Chang SH, Maney KM, Phillips JP, Langford RM, Mehta V. A comparison of the respiratory effects of oxycodone versus morphine: a randomised, double-blind, placebo-controlled investigation. Anaesthesia. 2010;65(10):1007-1012.
- Holstege CP, Borek HA. Toxidromes. Crit Care Clin. 2012;28(4):479-498.
- Hoffman JR, Schriger DL, Luo JS. The empiric use of naloxone in patients with altered mental status: a reappraisal. Ann Emerg Men. 1991;20(3):246-252.
- Howland MA, Nelson LS. Chapter A6. Opioid antagonists. In: Nelson LS, Lewin NA, Howland MA, Hoffman RS, Goldfrank LR, Flomenbaum NE, eds. Goldfrank’s Toxicologic Emergencies. 9th ed. New York, NY: McGraw Hill; 2011:579-585.
- Goldfrank L, Weisman RS, Errick JK, Lo MW. A dosing nomogram for continuous infusion intravenous naloxone. Ann Emerg Med. 1986;15(5):566-570.
- Moreland TA, Brice JE, Walker CH, Parija AC. Naloxone pharmacokinetics in the newborn. Br J Clin Pharmacol. 1980;9(6):609-612.
- Ngai SH, Berkowitz BA, Yang JC, et al. Pharmacokinetics of naloxone in rats and in man: basis for its potency and short duration of action. Anesthesiology. 1976;44(5):398-401.
Case
A previously healthy 10-month-old girl was brought to the ED by her mother, who noted that the child had been excessively drowsy throughout the day. She reported that her husband had dropped an unknown amount of his morphine sulfate extended-release 60-mg tablets and oxycodone 10-mg/acetaminophen 325-mg tablets on the floor 5 days earlier. Although unsure of how many tablets he had dropped, the father believed he had located all of them. The mother, however, found some of the tablets around the crib in their daughter’s room.
When the child arrived to the ED, her vital signs were: blood pressure, 95/60 mm Hg; heart rate, 102 beats/minute; respiratory rate (RR), 18 breaths/minute; and temperature, 98.4°F. Oxygen saturation was 98% on room air. On physical examination, the child was lethargic, her pupils were less than 1 mm in diameter, and her bowel sounds were absent. After the administration of intravenous (IV) naloxone 0.4 mg, the patient became less drowsy and her RR normalized. Approximately 1 hour later, though, the child again became lethargic; she was given a repeat dose of IV naloxone 0.4 mg, and a naloxone infusion was initiated at 0.3 mg/h. Over approximately 20 hours, the infusion was tapered and discontinued. Three hours after the infusion was stopped, the child’s vital signs and behavior were both normal. After a social worker and representative from the Administration for Children’s Services reviewed the patient’s case, she was discharged home with her parents.
Less than 1 hour later, however, the mother returned to the ED with the child, who was again unresponsive. Although the girl’s RR was normal, she had pinpoint pupils. After she was given IV naloxone 0.4 mg, the child awoke and remained responsive for 20 minutes before returning to a somnolent state. Another IV dose of naloxone 0.4 mg was administered, which showed partial improvement in responsiveness. A naloxone infusion was then initiated and titrated up to 1 mg/h to maintain wakefulness and ventilation. In the pediatric intensive care unit, the child required titration of the naloxone infusion to 2 mg/h to which she responded well. Over the next 12 hours, the infusion was tapered off and the child was discharged home with her parents.
Blood samples from both the initial visit and the return visit were sent for toxicologic analysis by gas chromatography-mass spectrometry (GC-MS). Serum from the first visit contained morphine at a concentration of 3,000 ng/mL; serum from the second visit contained morphine at 420 ng/mL. Both samples were negative for oxycodone or any of the other substances checked on the extended GC-MS screen.
What is the toxicologic differential?
Although this patient’s extreme somnolence was suspected to be opioid-induced, and was confirmed by an appropriate response to naloxone, children may present to the ED somnolent for a variety of unknown reasons. Even with a fairly clear history, the clinician should also consider metabolic, neurological, infectious, traumatic, and psychiatric causes of altered mental status.1 The toxicologic causes of altered mental status are expansive and include the effects of many medications used therapeutically or in overdose. Opioids, benzodiazepines, barbiturates, α-2 agonists (eg, clonidine), sleep aids (eg, zolpidem, diphenhydramine), and ethanol are common causes of induced an altered mental status. When taking a toxicologic history, it is important to inquire not only about the patient’s medications but also the medications of other members of the household to which the patient may have access. This includes not only prescription medications but also over-the-counter, complementary, and herbal preparations.
Why did this child have delayed recurrent opioid toxicity?
When used as directed, opioids cause analgesia and euphoria. Analgesia is mediated by agonism at the μ- , κ-, and δ-opioid receptors throughout the brain and spinal cord. The majority of morphine’s analgesic activity comes from activation of the μ-opioid receptors.2 In overdose, opioids classically cause a toxidrome characterized by miosis, coma, decreased bowel sounds, and respiratory depression. These signs can give clues to a patient’s exposure.
Supportive care is the cornerstone of treatment for patients with opioid toxicity, and maintaining the airway and monitoring the respiratory status are extremely important. When ventilation decreases due to the actions of opioids (typically denoted by a RR of <12 breaths/minute in adults, but may be marked by a reduction in depth of breathing as well), the use of an opioid antagonist is appropriate.4 The most commonly used antagonist is naloxone, an antidote with antagonism at all opioid receptor subtypes.5
In patients who are not dependent on opioids, IV naloxone 0.4 mg is an appropriate initial dose—regardless of patient size or specifics of the exposure. Patients with opioid dependency (eg, patients taking opioids for chronic pain or palliative care, or in those with suspected or confirmed opioid abuse), should receive smaller initial doses of naloxone (eg, 0.04 mg); the dose should be titrated up to effect to avoid precipitating acute opioid withdrawal. The goal of opioid antagonism is to allow the patient to breathe spontaneously and at an appropriate rate and depth without precipitating withdrawal. The duration of action of naloxone is 20 to 90 minutes in adults.
Patients presenting with heroin overdose should be monitored for at least 2 hours after naloxone administration (some suggest 3 hours) to determine whether or not additional dosing will be necessary. After oral opioid exposures, particularly with extended-release or long-acting formulations, longer periods of observation are required (this is unrelated to the naloxone pharmacokinetics, but rather to the slow rise in blood levels from some of these formulations). If repeated opioid toxicity occurs in adults, a naloxone infusion may be helpful to reduce the need for repetitive re-dosing. Initially, an hourly infusion equal to two-thirds of the dose of naloxone that reversed the patient’s respiratory depression is suggested6
Naloxone is eliminated by conjugation with glucuronic acid before is it excreted from the body. Due to decreased hepatic conjugation and prolonged metabolization of drugs in pediatric patients, naloxone may have a longer half-life in children—especially neonates and infants7; in children, the half-life of naloxone may extend up to three times that of adults.8 This extended half-life can lead to a false sense of assurance that a child is free of opioid effects 120 minutes after receiving naloxone—the time by which an adult patient would likely be without significant systemic effects of naloxone—when in fact the effect of naloxone has not yet sufficiently waned. This in turn may prompt discharge before sufficient time has passed to exclude recrudescence of opioid toxicity: The presence of persistent opioid agonist concentrations in the blood, even at consequential amounts, remains masked by the persistent presence of naloxone.
The goal of opioid antagonism is to allow the patient to breathe spontaneously and at an appropriate rate and depth without precipitating withdrawal. In this patient, it is not surprising that the the ingestion of an extended-relief form of morphine should produce a prolonged opioid effect. At therapeutic concentrations in children (~10 ng/mL), the half-life of morphine is slightly longer than in adults (~3 hours vs 2 hours) and is likely even longer with very high serum concentrations. It is metabolized to morphine 6-glucuronide, which is active and longer lasting than the parent compound. This may account for additional clinical effects beyond the time that the serum morphine concentration falls, and is particularly relevant following immediate-release morphine overdose.
In this case it is also important to consider whether or not the patient was re-exposed to an opioid between the first and second ED visit. The dramatically elevated initial serum morphine concentrations and the relatively appropriate fall in magnitude of the second sample suggest that the recurrence of respiratory depression was not the result of re-exposure. The patient’s recurrent effects, even a day out from exposure, can be explained by the immediate-release morphine exposure and the discharge prior to waning of the naloxone. In children with opioid toxicity, another potential option, though not directly studied, is to administer the long-acting opioid antagonist naltrexone to the patient prior to discharge.
Case Conclusion
When used appropriately and under the correct circumstances, naloxone is safe and effective for the reversal of opioid toxicity. As with any antidote, patients must be appropriately monitored for any adverse effects or recurrence of toxicity. Moreover, the clinician should be mindful of the pharmacokinetic differences between adults and young children and the possibility of a later-than-expected recurrence of opioid toxicity in pediatric patients.
This case is a reminder of the importance of safe medication storage. Infants and young children who are crawling and exploring their environment are especially vulnerable to toxicity from medications found on the floor. Regardless of age, quick recognition of opioid-induced respiratory depression and appropriate use of naloxone can help to decrease the morbidity associated with excessive opioid exposures in all patients.
Dr Berman is a senior medical toxicology fellow at North Shore-Long Island Jewish Medical Center, New York. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board. Dr Majlesi is the director of medical toxicology at Staten Island University Hospital, New York.
Case
A previously healthy 10-month-old girl was brought to the ED by her mother, who noted that the child had been excessively drowsy throughout the day. She reported that her husband had dropped an unknown amount of his morphine sulfate extended-release 60-mg tablets and oxycodone 10-mg/acetaminophen 325-mg tablets on the floor 5 days earlier. Although unsure of how many tablets he had dropped, the father believed he had located all of them. The mother, however, found some of the tablets around the crib in their daughter’s room.
When the child arrived to the ED, her vital signs were: blood pressure, 95/60 mm Hg; heart rate, 102 beats/minute; respiratory rate (RR), 18 breaths/minute; and temperature, 98.4°F. Oxygen saturation was 98% on room air. On physical examination, the child was lethargic, her pupils were less than 1 mm in diameter, and her bowel sounds were absent. After the administration of intravenous (IV) naloxone 0.4 mg, the patient became less drowsy and her RR normalized. Approximately 1 hour later, though, the child again became lethargic; she was given a repeat dose of IV naloxone 0.4 mg, and a naloxone infusion was initiated at 0.3 mg/h. Over approximately 20 hours, the infusion was tapered and discontinued. Three hours after the infusion was stopped, the child’s vital signs and behavior were both normal. After a social worker and representative from the Administration for Children’s Services reviewed the patient’s case, she was discharged home with her parents.
Less than 1 hour later, however, the mother returned to the ED with the child, who was again unresponsive. Although the girl’s RR was normal, she had pinpoint pupils. After she was given IV naloxone 0.4 mg, the child awoke and remained responsive for 20 minutes before returning to a somnolent state. Another IV dose of naloxone 0.4 mg was administered, which showed partial improvement in responsiveness. A naloxone infusion was then initiated and titrated up to 1 mg/h to maintain wakefulness and ventilation. In the pediatric intensive care unit, the child required titration of the naloxone infusion to 2 mg/h to which she responded well. Over the next 12 hours, the infusion was tapered off and the child was discharged home with her parents.
Blood samples from both the initial visit and the return visit were sent for toxicologic analysis by gas chromatography-mass spectrometry (GC-MS). Serum from the first visit contained morphine at a concentration of 3,000 ng/mL; serum from the second visit contained morphine at 420 ng/mL. Both samples were negative for oxycodone or any of the other substances checked on the extended GC-MS screen.
What is the toxicologic differential?
Although this patient’s extreme somnolence was suspected to be opioid-induced, and was confirmed by an appropriate response to naloxone, children may present to the ED somnolent for a variety of unknown reasons. Even with a fairly clear history, the clinician should also consider metabolic, neurological, infectious, traumatic, and psychiatric causes of altered mental status.1 The toxicologic causes of altered mental status are expansive and include the effects of many medications used therapeutically or in overdose. Opioids, benzodiazepines, barbiturates, α-2 agonists (eg, clonidine), sleep aids (eg, zolpidem, diphenhydramine), and ethanol are common causes of induced an altered mental status. When taking a toxicologic history, it is important to inquire not only about the patient’s medications but also the medications of other members of the household to which the patient may have access. This includes not only prescription medications but also over-the-counter, complementary, and herbal preparations.
Why did this child have delayed recurrent opioid toxicity?
When used as directed, opioids cause analgesia and euphoria. Analgesia is mediated by agonism at the μ- , κ-, and δ-opioid receptors throughout the brain and spinal cord. The majority of morphine’s analgesic activity comes from activation of the μ-opioid receptors.2 In overdose, opioids classically cause a toxidrome characterized by miosis, coma, decreased bowel sounds, and respiratory depression. These signs can give clues to a patient’s exposure.
Supportive care is the cornerstone of treatment for patients with opioid toxicity, and maintaining the airway and monitoring the respiratory status are extremely important. When ventilation decreases due to the actions of opioids (typically denoted by a RR of <12 breaths/minute in adults, but may be marked by a reduction in depth of breathing as well), the use of an opioid antagonist is appropriate.4 The most commonly used antagonist is naloxone, an antidote with antagonism at all opioid receptor subtypes.5
In patients who are not dependent on opioids, IV naloxone 0.4 mg is an appropriate initial dose—regardless of patient size or specifics of the exposure. Patients with opioid dependency (eg, patients taking opioids for chronic pain or palliative care, or in those with suspected or confirmed opioid abuse), should receive smaller initial doses of naloxone (eg, 0.04 mg); the dose should be titrated up to effect to avoid precipitating acute opioid withdrawal. The goal of opioid antagonism is to allow the patient to breathe spontaneously and at an appropriate rate and depth without precipitating withdrawal. The duration of action of naloxone is 20 to 90 minutes in adults.
Patients presenting with heroin overdose should be monitored for at least 2 hours after naloxone administration (some suggest 3 hours) to determine whether or not additional dosing will be necessary. After oral opioid exposures, particularly with extended-release or long-acting formulations, longer periods of observation are required (this is unrelated to the naloxone pharmacokinetics, but rather to the slow rise in blood levels from some of these formulations). If repeated opioid toxicity occurs in adults, a naloxone infusion may be helpful to reduce the need for repetitive re-dosing. Initially, an hourly infusion equal to two-thirds of the dose of naloxone that reversed the patient’s respiratory depression is suggested6
Naloxone is eliminated by conjugation with glucuronic acid before is it excreted from the body. Due to decreased hepatic conjugation and prolonged metabolization of drugs in pediatric patients, naloxone may have a longer half-life in children—especially neonates and infants7; in children, the half-life of naloxone may extend up to three times that of adults.8 This extended half-life can lead to a false sense of assurance that a child is free of opioid effects 120 minutes after receiving naloxone—the time by which an adult patient would likely be without significant systemic effects of naloxone—when in fact the effect of naloxone has not yet sufficiently waned. This in turn may prompt discharge before sufficient time has passed to exclude recrudescence of opioid toxicity: The presence of persistent opioid agonist concentrations in the blood, even at consequential amounts, remains masked by the persistent presence of naloxone.
The goal of opioid antagonism is to allow the patient to breathe spontaneously and at an appropriate rate and depth without precipitating withdrawal. In this patient, it is not surprising that the the ingestion of an extended-relief form of morphine should produce a prolonged opioid effect. At therapeutic concentrations in children (~10 ng/mL), the half-life of morphine is slightly longer than in adults (~3 hours vs 2 hours) and is likely even longer with very high serum concentrations. It is metabolized to morphine 6-glucuronide, which is active and longer lasting than the parent compound. This may account for additional clinical effects beyond the time that the serum morphine concentration falls, and is particularly relevant following immediate-release morphine overdose.
In this case it is also important to consider whether or not the patient was re-exposed to an opioid between the first and second ED visit. The dramatically elevated initial serum morphine concentrations and the relatively appropriate fall in magnitude of the second sample suggest that the recurrence of respiratory depression was not the result of re-exposure. The patient’s recurrent effects, even a day out from exposure, can be explained by the immediate-release morphine exposure and the discharge prior to waning of the naloxone. In children with opioid toxicity, another potential option, though not directly studied, is to administer the long-acting opioid antagonist naltrexone to the patient prior to discharge.
Case Conclusion
When used appropriately and under the correct circumstances, naloxone is safe and effective for the reversal of opioid toxicity. As with any antidote, patients must be appropriately monitored for any adverse effects or recurrence of toxicity. Moreover, the clinician should be mindful of the pharmacokinetic differences between adults and young children and the possibility of a later-than-expected recurrence of opioid toxicity in pediatric patients.
This case is a reminder of the importance of safe medication storage. Infants and young children who are crawling and exploring their environment are especially vulnerable to toxicity from medications found on the floor. Regardless of age, quick recognition of opioid-induced respiratory depression and appropriate use of naloxone can help to decrease the morbidity associated with excessive opioid exposures in all patients.
Dr Berman is a senior medical toxicology fellow at North Shore-Long Island Jewish Medical Center, New York. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board. Dr Majlesi is the director of medical toxicology at Staten Island University Hospital, New York.
- Lehman RK, Mink J. Altered mental status. Clin Pediatr Emerg Med. 2008;9:68-75.
- Chang SH, Maney KM, Phillips JP, Langford RM, Mehta V. A comparison of the respiratory effects of oxycodone versus morphine: a randomised, double-blind, placebo-controlled investigation. Anaesthesia. 2010;65(10):1007-1012.
- Holstege CP, Borek HA. Toxidromes. Crit Care Clin. 2012;28(4):479-498.
- Hoffman JR, Schriger DL, Luo JS. The empiric use of naloxone in patients with altered mental status: a reappraisal. Ann Emerg Men. 1991;20(3):246-252.
- Howland MA, Nelson LS. Chapter A6. Opioid antagonists. In: Nelson LS, Lewin NA, Howland MA, Hoffman RS, Goldfrank LR, Flomenbaum NE, eds. Goldfrank’s Toxicologic Emergencies. 9th ed. New York, NY: McGraw Hill; 2011:579-585.
- Goldfrank L, Weisman RS, Errick JK, Lo MW. A dosing nomogram for continuous infusion intravenous naloxone. Ann Emerg Med. 1986;15(5):566-570.
- Moreland TA, Brice JE, Walker CH, Parija AC. Naloxone pharmacokinetics in the newborn. Br J Clin Pharmacol. 1980;9(6):609-612.
- Ngai SH, Berkowitz BA, Yang JC, et al. Pharmacokinetics of naloxone in rats and in man: basis for its potency and short duration of action. Anesthesiology. 1976;44(5):398-401.
- Lehman RK, Mink J. Altered mental status. Clin Pediatr Emerg Med. 2008;9:68-75.
- Chang SH, Maney KM, Phillips JP, Langford RM, Mehta V. A comparison of the respiratory effects of oxycodone versus morphine: a randomised, double-blind, placebo-controlled investigation. Anaesthesia. 2010;65(10):1007-1012.
- Holstege CP, Borek HA. Toxidromes. Crit Care Clin. 2012;28(4):479-498.
- Hoffman JR, Schriger DL, Luo JS. The empiric use of naloxone in patients with altered mental status: a reappraisal. Ann Emerg Men. 1991;20(3):246-252.
- Howland MA, Nelson LS. Chapter A6. Opioid antagonists. In: Nelson LS, Lewin NA, Howland MA, Hoffman RS, Goldfrank LR, Flomenbaum NE, eds. Goldfrank’s Toxicologic Emergencies. 9th ed. New York, NY: McGraw Hill; 2011:579-585.
- Goldfrank L, Weisman RS, Errick JK, Lo MW. A dosing nomogram for continuous infusion intravenous naloxone. Ann Emerg Med. 1986;15(5):566-570.
- Moreland TA, Brice JE, Walker CH, Parija AC. Naloxone pharmacokinetics in the newborn. Br J Clin Pharmacol. 1980;9(6):609-612.
- Ngai SH, Berkowitz BA, Yang JC, et al. Pharmacokinetics of naloxone in rats and in man: basis for its potency and short duration of action. Anesthesiology. 1976;44(5):398-401.

Managing aneurysmal subarachnoid hemorrhage: It takes a team
Aneurysmal subarachnoid hemorrhage is a devastating condition, with an estimated death rate of 30% during the initial episode.1,2 Approximately the same number of patients survive but leave the hospital with disabling neurologic deficits.3
However, better outcomes can be achieved by systems that are able to work as a team on the collective goal of quick intervention to secure the ruptured aneurysm, followed by the implementation of measures to minimize secondary brain injury. Although the search for new diagnostic, prognostic, and therapeutic modalities continues, it is clear that there exists no “silver bullet” that will help all patients. Instead, it is the systematic integration and application of small advances that will ultimately maximize the patient’s chances of survival and neurologic recovery.
This review focuses on the management of aneurysmal subarachnoid hemorrhage and its systemic and neurologic complications.
ANEURYSM IS THE MOST COMMON CAUSE OF SUBARACHNOID BLEEDING
Aneurysmal subarachnoid hemorrhage, ie, rupture of an intracranial aneurysm, flooding the subarachnoid space with blood, affects about 24,000 Americans each year.1,2 A ruptured aneurysm is the most common cause of subarachnoid hemorrhage, accounting for about 85% of cases. Less common causes include idiopathic benign perimesencephalic hemorrhage, arteriovenous malformation, dural arteriovenous fistula, and hemorrhagic mycotic aneurysm. These have their own natural history, pathophysiology, and specific treatment, and will not be addressed in this article.
Risk factors for aneurysmal subarachnoid hemorrhage include having a first-degree relative who had the disease, hypertension, smoking, and consuming more than 150 g of alcohol per week.4
CLINICAL PRESENTATION AND DIAGNOSIS
The key symptom of aneurysmal subarachnoid hemorrhage is the abrupt onset of severe headache that peaks in intensity over 1 hour,5 often described as “the worst headache of my life.” Headache is accompanied by brief loss of consciousness in 53% of cases (conversely, nearly half of patients maintain normal mental status), by nausea or vomiting in 77%, and by meningismus (neck pain or stiffness) in 35%.6
These clinical manifestations and risk factors have been incorporated into a decision rule:
Obtain brain imaging if the patient has acute headache reaching maximal intensity within 1 hour, associated with any of the following factors:
- Age 40 or older
- Neck pain or stiffness
- Witnessed loss of consciousness
- Onset during exertion
- “Thunderclap” headache (ie, instantly peaking pain)
- Limited neck flexion on examination.5
This decision rule has nearly 100% sensitivity for aneurysmal subarachnoid hemorrhage in clinical practice.5 All patients require brain imaging if they have a severe headache plus either abnormal neurologic findings (eg, a focal neurologic deficit) or a history of cerebral aneurysm.
Emergency physicians should have a low threshold for ordering noncontrast computed tomography (CT) of the head in patients with even mild symptoms suggesting aneurysmal subarachnoid hemorrhage. Failure to order CT is the most common diagnostic error in this situation.6 CT performed within 6 hours of headache onset is nearly 100% sensitive for this condition,7 but the sensitivity falls to 93% after the first 24 hours and to less than 60% after 5 days.8 In patients who have symptoms highly suggestive of aneurysmal subarachnoid hemorrhage but a normal CT, lumbar puncture is the next diagnostic step.
There are two alternatives to CT followed by lumbar puncture: ie, noncontrast CT followed by CT angiography,9,10 and magnetic resonance imaging followed by magnetic resonance angiography. In patients with suspicious clinical symptoms but negative CT results, CT followed by CT angiography can rule out aneurysmal subarachnoid hemorrhage with a 99% probability.9,10 However, CT followed by lumbar puncture remains the standard of care and carries a class I recommendation in the American Heart Association guidelines for ruling out subarachnoid hemorrhage.5
GRADING THE SEVERITY OF SUBARACHNOID HEMORRHAGE
Age, the thickness of the blood layer in the subarachnoid space, intraventricular hemorrhage and the findings of the neurologic examination at presentation are predictors of long-term outcomes in aneurysmal subarachnoid hemorrhage (Figure 1).
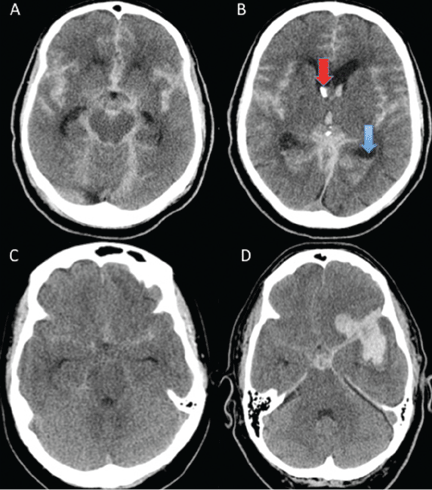
Different grading systems used in clinical practice are based on the findings on the initial neurologic examination and on the initial noncontrast CT (ie, the thickness of the blood, and whether intraventricular hemorrhage is present). Among the most widely used are those developed by Hunt and Hess12 and by the World Federation of Neurological Surgeons11 (WFNS), and the CT grading scales (Fisher13 or its modified version14) (Tables 1 and 2). With either the Hunt and Hess scale or the WFNS scale, the higher the score, the worse the patient’s probable outcome. Scores on both Fisher scales correlate with the risk of angiographic vasospasm. The higher the grade, the higher the risk of angiographic vasospasm.


The VASOGRADE score—a combination of the WFNS score and the modified Fisher scale—stratifies patients at risk of delayed cerebral ischemia, allowing for a tailored monitoring strategy.15 There are three variations:
- VASOGRADE green—Modified Fisher 1 or 2 and WFNS 1 or 2
- VASOGRADE yellow—Modified Fisher 3 or 4 and WFNS 1, 2, or 3
- VASOGRADE red—WFNS 4 or 5.
After the initial bleeding event, patients with aneurysmal subarachnoid hemorrhage are at high risk of delayed systemic and neurologic complications, with poor functional outcomes. Delayed cerebral ischemia holds the greatest risk of an unfavorable outcome and ultimately can lead to cerebral infarction, disability, and death.6,7
INITIAL MANAGEMENT
After aneurysmal subarachnoid hemorrhage is diagnosed, the initial management (Figure 2) includes appropriate medical prevention of rebleeding (which includes supportive care, blood pressure management, and, perhaps, the early use of a short course of an antifibrinolytic drug) and early transfer to a high-volume center for securing the aneurysm. The reported incidence of rebleeding varies from 5% to 22% in the first 72 hours. “Ultra-early” rebleeding (within 24 hours of hemorrhage) has been reported, with an incidence as high as 15% and a fatality rate around 70%. Patients with poor-grade aneurysmal subarachnoid hemorrhage, larger aneurysms, and “sentinel bleeds” are at higher risk of rebleeding.16
Outcomes are much better when patients are managed in a high-volume center, with a specialized neurointensive care unit17 and access to an interdisciplinary team.18 Regardless of the initial grade, patients with aneurysmal subarachnoid hemorrhage should be quickly transferred to a high-volume center, defined as one treating at least 35 cases per year, and the benefit is greater in centers treating more than 60 cases per year.19 The higher the caseload in any given hospital, the better the clinical outcomes in this population.20
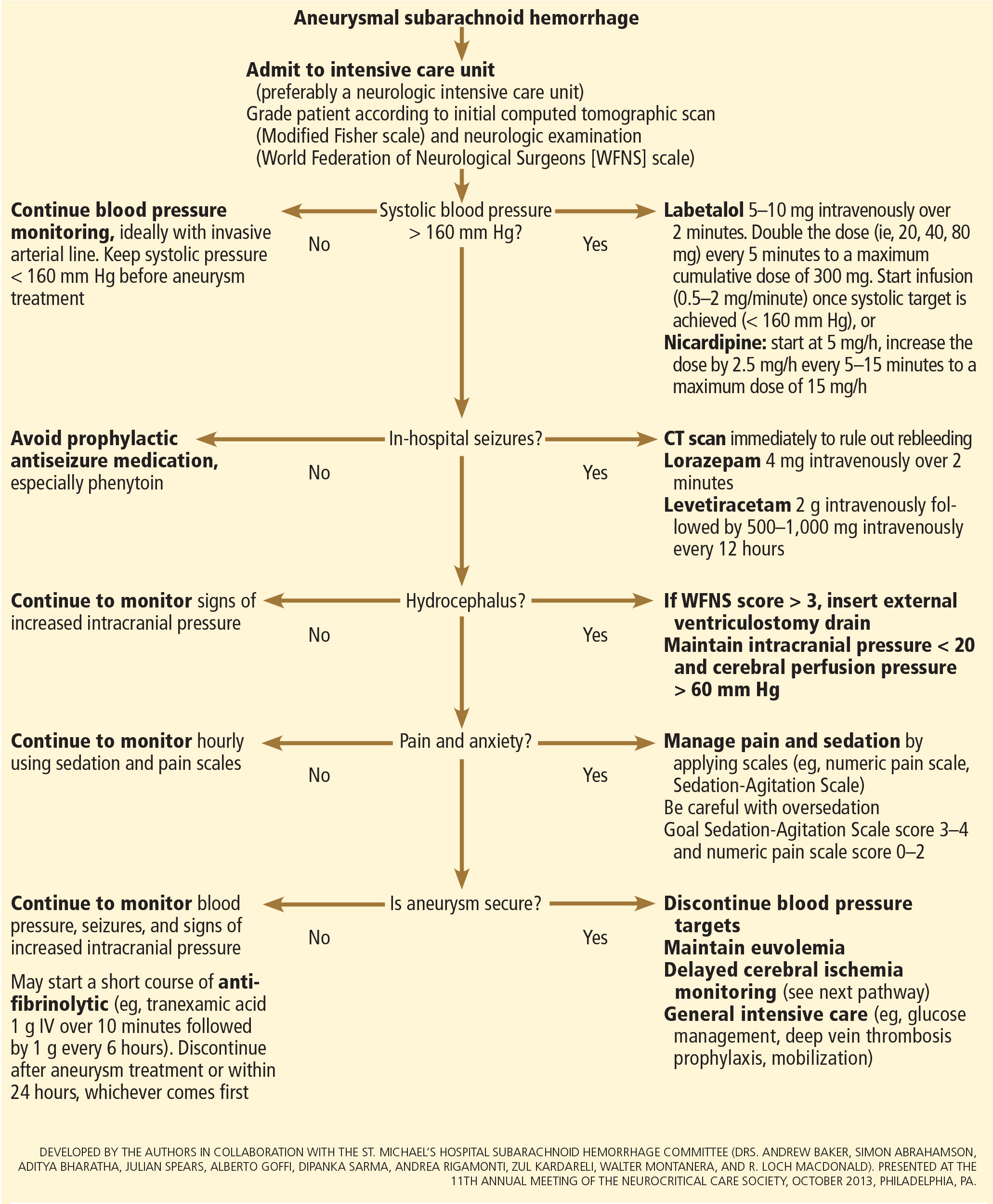
Treating cerebral aneurysm: Clipping or coiling
Early aneurysm repair is generally considered the standard of care and the best strategy to reduce the risk of rebleeding. Further, early treatment may be associated with a lower risk of delayed cerebral ischemia21 and better outcomes.22
Three randomized clinical trials have compared surgical clipping and endovascular repair (placement of small metal coils within the aneurysm to promote clotting).
The International Subarachnoid Aneurysm Trial23 showed a reduction of 23% in relative risk and of 7% in absolute risk in patients who underwent endovascular treatment compared with surgery. The survival benefit persisted at a mean of 9 years (range 6–14 years), but with a higher annual rate of aneurysm recurrence in the coiling group (2.9% vs 0.9%).24 Of note, this trial included only patients with aneurysms deemed suitable for both coiling and clipping, so that the exclusion rate was high. Most of the patients presented with good-grade (WFNS score 1–3), small aneurysms (< 5 mm) in the anterior circulation.
A single-center Finnish study25 found no differences in rates of recovery, disability, and death at 1 year, comparing surgery and endovascular treatment. Additionally, survival rates at a mean follow-up of 39 months were similar, with no late recurrences or aneurysmal bleeding.
Lastly, the Barrow Ruptured Aneurysm Trial26,27 found that patients assigned to endovascular treatment had better 1-year neurologic outcomes, defined as a modified Rankin score of 2 or less. Importantly, 37.7% of patients originally assigned to endovascular treatment crossed over to surgical treatment. The authors then performed intention-to-treat and as-treated analyses. Either way, patients treated by endovascular means had better neurologic outcomes at 1 year. However, no difference in the relative risk reduction in worse outcome was found on 3-year follow-up, and patients treated surgically had higher rates of aneurysm obliteration and required less aneurysm retreatment, both of which were statistically significant.
The question that remains is not whether to clip or whether to coil, but whom to clip and whom to coil.28 That question must be answered on a patient-to-patient basis and requires the expertise of an interventional neuroradiologist and a vascular neurosurgeon—one of the reasons these patients are best cared for in high-volume centers providing such expertise.
MEDICAL PREVENTION OF REBLEEDING
Blood pressure management
There are no systematic data on the optimal blood pressure before securing an aneurysm. Early studies of hemodynamic augmentation in cases of ruptured untreated aneurysm reported rebleeding when the systolic blood pressure was allowed to rise above 160 mm Hg.29,30 A recent study evaluating hypertensive intracerebral hemorrhage revealed better functional outcomes with intensive lowering of blood pressure (defined as systolic blood pressure < 140 mm Hg) but no significant reduction in the combined rate of death or severe disability.31 It is difficult to know if these results can be extrapolated to patients with aneurysmal subarachnoid hemorrhage. Current guidelines3,32 say that before the aneurysm is treated, the systolic pressure should be lower than 160 mm Hg.
There is no specific drug of choice, but a short-acting, titratable medication is preferable. Nicardipine is a very good option, and labetalol might be an appropriate alternative.33 Once the aneurysm is secured, all antihypertensive drugs should be held. Hypertension should not be treated unless the patient has clinical signs of a hypertensive crisis, such as flash pulmonary edema, myocardial infarction, or hypertensive encephalopathy.
Antifibrinolytic therapy
The role of antifibrinolytic therapy in aneurysmal subarachnoid hemorrhage is controversial and has been studied in 10 clinical trials. In a Swedish study,34 early use of tranexamic acid (1 g intravenously over 10 minutes followed by 1 g every 6 hours for a maximum of 24 hours) reduced the rebleeding rate substantially, from 10.8% to 2.4%, with an 80% reduction in the mortality rate from ultra-early rebleeding. However, a recent Cochrane review that included this study found no overall benefit.35
An ongoing multicenter randomized trial in the Netherlands will, we hope, answer this question in the near future.36 At present, some centers would consider a short course of tranexamic acid before aneurysm treatment.
DIAGNOSIS AND TREATMENT OF COMPLICATIONS
Medical complications are extremely common after aneurysmal subarachnoid hemorrhage. Between 75% and 100% of patients develop some type of systemic or further neurologic derangement, which in turn has a negative impact on the long-term outcome.37,38 In the first 72 hours, rebleeding is the most feared complication, and as mentioned previously, appropriate blood pressure management and early securing of the aneurysm minimize its risk.
NEUROLOGIC COMPLICATIONS
Hydrocephalus
Hydrocephalus is the most common early neurologic complication after aneurysmal subarachnoid hemorrhage, with an overall incidence of 50%.39 Many patients with poor-grade aneurysmal subarachnoid hemorrhage and patients whose condition deteriorates due to worsening of hydrocephalus require the insertion of an external ventricular drain (Figure 1).
Up to 30% of patients who have a poor-grade aneurysmal subarachnoid hemorrhage improve neurologically with cerebrospinal fluid drainage.40 An external ventricular drain can be safely placed, even before aneurysm treatment, and placement does not appear to increase the risk of rebleeding.39,41 After placement, rapid weaning from the drain (clamping within 24 hours of insertion) is safe, decreases length of stay in the intensive care unit and hospital, and may be more cost-effective than gradual weaning over 96 hours.42
Increased intracranial pressure
Intracranial hypertension is another potential early complication, and is frequently due to the development of hydrocephalus, cerebral edema, or rebleeding. The treatment of increased intracranial pressure does not differ from the approach used in managing severe traumatic brain injury, which includes elevating the head of the bed, sedation, analgesia, normoventilation, and cerebrospinal fluid drainage.
Hypertonic saline has been tested in several studies that were very small but nevertheless consistently showed control of intracranial pressure levels and improvement in cerebral blood flow measured by xenon CT.43–47 Two of these studies even showed better outcomes at discharge.43,44 However, the small number of patients prevents any meaningful conclusion regarding the use of hypertonic saline and functional outcomes.
Barbiturates, hypothermia, and decompressive craniectomy could be tried in refractory cases.48 Seule et al49 evaluated the role of therapeutic hypothermia with or without barbiturate coma in 100 patients with refractory intracranial hypertension. Only 13 patients received hypothermia by itself. At 1 year, 32 patients had achieved a good functional outcome (Glasgow Outcome Scale score 4 or 5). The remaining patients were severely disabled or had died. Of interest, the median duration of hypothermia was 7 days, and 93% of patients developed some medical complication such as electrolyte disorders (77%), pneumonia (52%), thrombocytopenia (47%), or septic shock syndrome (40%). Six patients died as a consequence of one of these complications.
Decompressive craniectomy can be life-saving in patients with refractory intracranial hypertension. However, most of these patients will die or remain severely disabled or comatose.50
Seizure prophylaxis is controversial
Seizures can occur at the onset of intracranial hemorrhage, perioperatively, or later (ie, after the first week). The incidence varied considerably in different reports, ranging from 4% to 26%.51 Seizures occurring perioperatively, ie, after hospital admission, are less frequent and are usually the manifestation of aneurysm rebleeding.24
Seizure prophylaxis remains controversial, especially because the use of phenytoin is associated with increased incidence of cerebral vasospasm, infarction, and worse cognitive outcomes after aneurysmal subarachnoid hemorrhage.52,53 Therefore, routine prophylactic use of phenytoin is not recommended in these patients,3 although the effect of other antiepileptic drugs is less studied and less clear. Patients may be considered for this therapy if they have multiple risk factors for seizures, such as intraparenchymal hematoma, advanced age (> 65), middle cerebral artery aneurysm, craniotomy for aneurysm clipping, and a short course (≤ 72 hours) of an antiepileptic drug other than phenytoin, especially while the aneurysm is unsecured.3
Levetiracetam may be an alternative to phenytoin, having better pharmacodynamic and kinetic profiles, minimal protein binding, and absence of hepatic metabolism, resulting in a very low risk of drug interaction and better tolerability.54,55 Because of these advantages, levetiracetam has become the drug of choice in several centers treating aneurysmal subarachnoid hemorrhage in the United States.
Addressing this question, a survey was sent to 25 high-volume aneurysmal subarachnoid hemorrhage academic centers in the United States. All 25 institutions answered the survey, and interestingly, levetiracetam was the first-line agent for 16 (94%) of the 17 responders that used prophylaxis, while only 1 used phenytoin as the agent of choice.56
A retrospective cohort study by Murphy-Human et al57 showed that a short course of levetiracetam (≤ 72 hours) was associated with higher rates of in-hospital seizures compared with an extended course of phenytoin (eg, entire hospital stay). However, the study did not address functional outcomes.57
Continuous electroencephalographic monitoring may be considered in comatose patients, in patients requiring controlled ventilation and sedation, or in patients with unexplained alteration in consciousness. In one series of patients with aneurysmal subarachnoid hemorrhage who received continuous monitoring, the incidence of nonconvulsive status epilepticus was 19%, with an associated mortality rate of 100%.58
Continuous quantitative electroencephalography is useful to monitor and to detect angiographic vasospasm and delayed cerebral ischemia. Relative alpha variability and the alpha-delta ratio decrease with ischemia, and this effect can precede angiographic vasospasm by 3 days.59,60
Delayed cerebral ischemia
Delayed cerebral ischemia is defined as the occurrence of focal neurologic impairment, or a decrease of at least 2 points on the Glasgow Coma Scale that lasts for at least 1 hour, is not apparent immediately after aneurysm occlusion, and not attributable to other causes (eg, hyponatremia, fever).61
Classically, neurologic deficits that occurred within 2 weeks of aneurysm rupture were ascribed to reduced cerebral blood flow caused by delayed large-vessel vasospasm causing cerebral ischemia.62 However, perfusion abnormalities have also been observed with either mild or no demonstrable vasospasm.63 Almost 70% of patients who survive the initial hemorrhage develop some degree of angiographic vasospasm. However, only 30% of those patients will experience symptoms.
In addition to vasospasm of large cerebral arteries, impaired autoregulation and early brain injury within the first 72 hours following subarachnoid hemorrhage may play important roles in the development of delayed cerebral ischemia.64 Therefore, the modern concept of delayed cerebral ischemia monitoring should focus on cerebral perfusion rather than vessel diameter measurements. This underscores the importance of comprehensive, standardized monitoring techniques that provide information not only on microvasculature, but also at the level of the microcirculation, with information on perfusion, oxygen utilization and extraction, and autoregulation.
Although transcranial Doppler has been the most commonly applied tool to monitor for angiographic vasospasm, it has a low sensitivity and negative predictive value.37 It is nevertheless a useful technique to monitor good-grade aneurysmal subarachnoid hemorrhage patients (WFNS score 1–3) combined with frequent neurologic examinations (Figure 3).
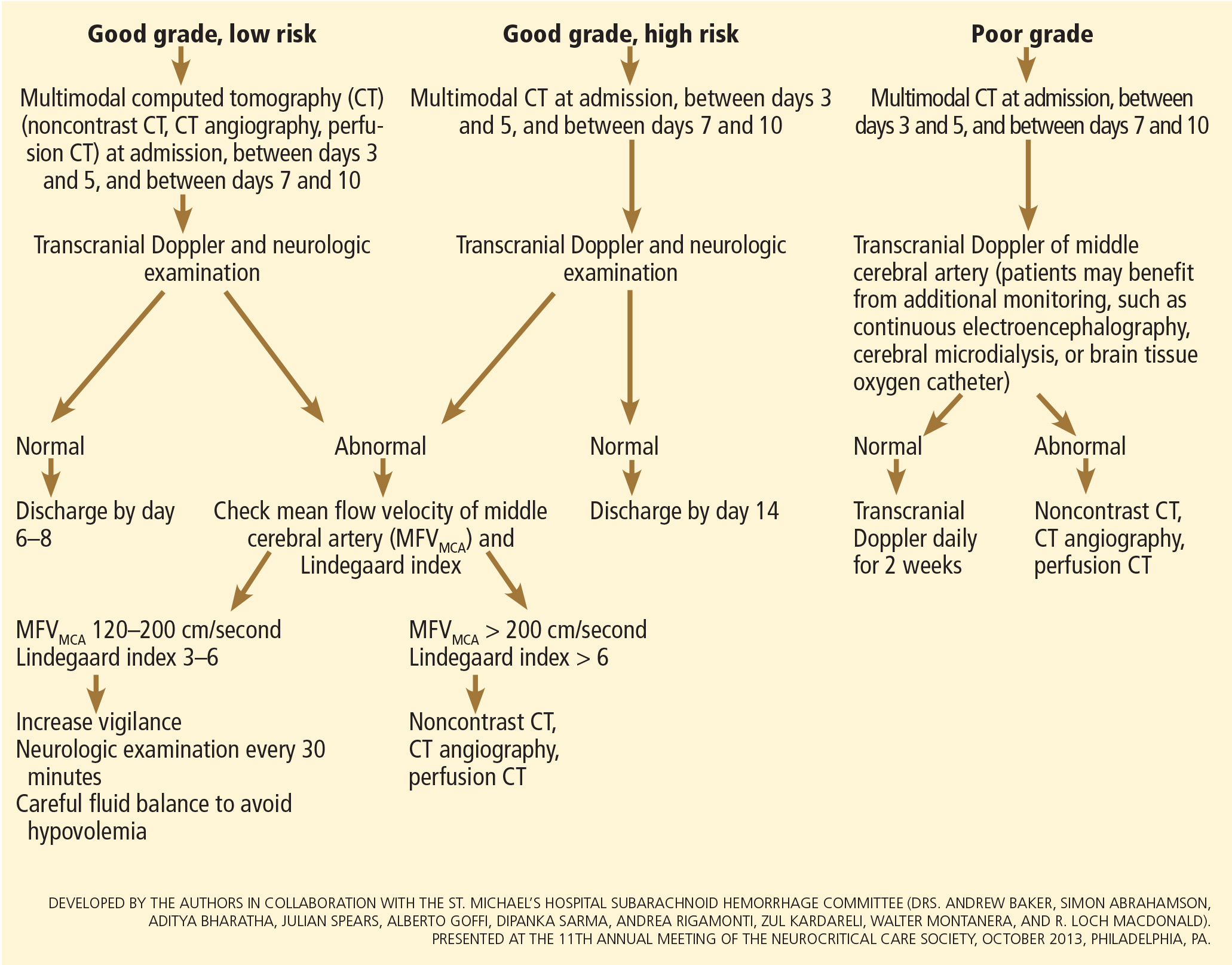
CT angiography is a good noninvasive alternative to digital subtraction angiography. However, it tends to overestimate the degree of vasoconstriction and does not provide information about perfusion and autoregulation.65 Nevertheless, CT angiography combined with a CT perfusion scan can add information about autoregulation and cerebral perfusion and has been shown to be more sensitive for the diagnosis of angiographic vasospasm than transcranial Doppler and digital subtraction angiography (Figure 4).
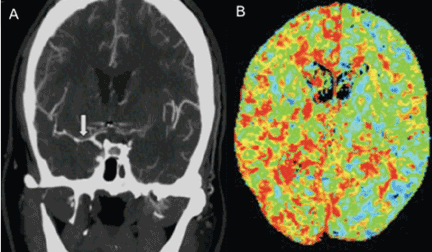
Patients with a poor clinical condition (WFNS score 4 or 5) or receiving continuous sedation constitute a challenge in monitoring for delayed neurologic deterioration. Neurologic examination is not sensitive enough in this setting to detect subtle changes. In these specific and challenging circumstances, multimodality neuromonitoring may be useful in the early detection of delayed cerebral ischemia and may help guide therapy.67
Several noninvasive and invasive techniques have been studied to monitor patients at risk of delayed cerebral ischemia after subarachnoid hemorrhage.66 These include continuous electroencephalography, brain tissue oxygenation monitoring (Ptio2), cerebral microdialysis, thermal diffusion flowmetry, and near-infrared spectroscopy. Of these techniques, Ptio2, cerebral microdialysis, and continuous electroencephalography (see discussion of seizure prophylaxis above) have been more extensively studied. However, most of the studies were observational and very small, limiting any recommendations for using these techniques in routine clinical practice.68
Ptio2 is measured by inserting an intraparenchymal oxygen-sensitive microelectrode, and microdialysis requires a microcatheter with a semipermeable membrane that allows small soluble substances to cross it into the dialysate. These substances, which include markers of ischemia (ie, glucose, lactate, and pyruvate), excitotoxins (ie, glutamate and aspartate), and membrane cell damage products (ie, glycerol), can be measured. Low Ptio2 values (< 15 mm Hg) and abnormal mycrodialysate findings (eg, glucose < 0.8 mmol/L, lactate-to-pyruvate ratio > 40) have both been associated with cerebral ischemic events and poor outcome.68
Preventing delayed cerebral ischemia
Oral nimodipine 60 mg every 4 hours for 21 days, started on admission, carries a class I, level of evidence A recommendation in the management of aneurysmal subarachnoid hemorrhage.3,32,69 It improves clinical outcome despite having no effect on the risk of angiographic vasospasm. The mechanism of improved outcome is unclear, but the effect may be a neuroprotective phenomenon limiting the extension of delayed cerebral ischemia.70
If hypotension occurs, the dose can be lowered to 30 mg every 2 hours. Whether to discontinue nimodipine in this situation is controversial. Of note, the clinical benefits of nimodipine have not been replicated with other calcium channel blockers (eg, nicardipine).71
Prophylactic hyperdynamic fluid therapy, known as “triple-H” (hypervolemia, hemodilution, and hypertension) was for years the mainstay of treatment in preventing delayed cerebral ischemia due to vasospasm. However, the clinical data supporting this intervention have been called into question, as analysis of two trials found that hypervolemia did not improve outcomes or reduce the incidence of delayed cerebral ischemia, and in fact increased the rate of complications.72,73 Based on these findings, current guidelines recommend maintaining euvolemia rather than prophylactic hypervolemia in patients with aneurysmal subarachnoid hemorrhage.3,32,69
TREATING DELAYED CEREBRAL ISCHEMIA
Hemodynamic augmentation
In patients with neurologic deterioration due to delayed cerebral ischemia, hemodynamic augmentation is the cornerstone of treatment. This is done according to a protocol, started early, involving specific physiologic goals, clinical improvement, and escalation to invasive therapies in a timely fashion in patients at high risk of further neurologic insult (Figure 5).
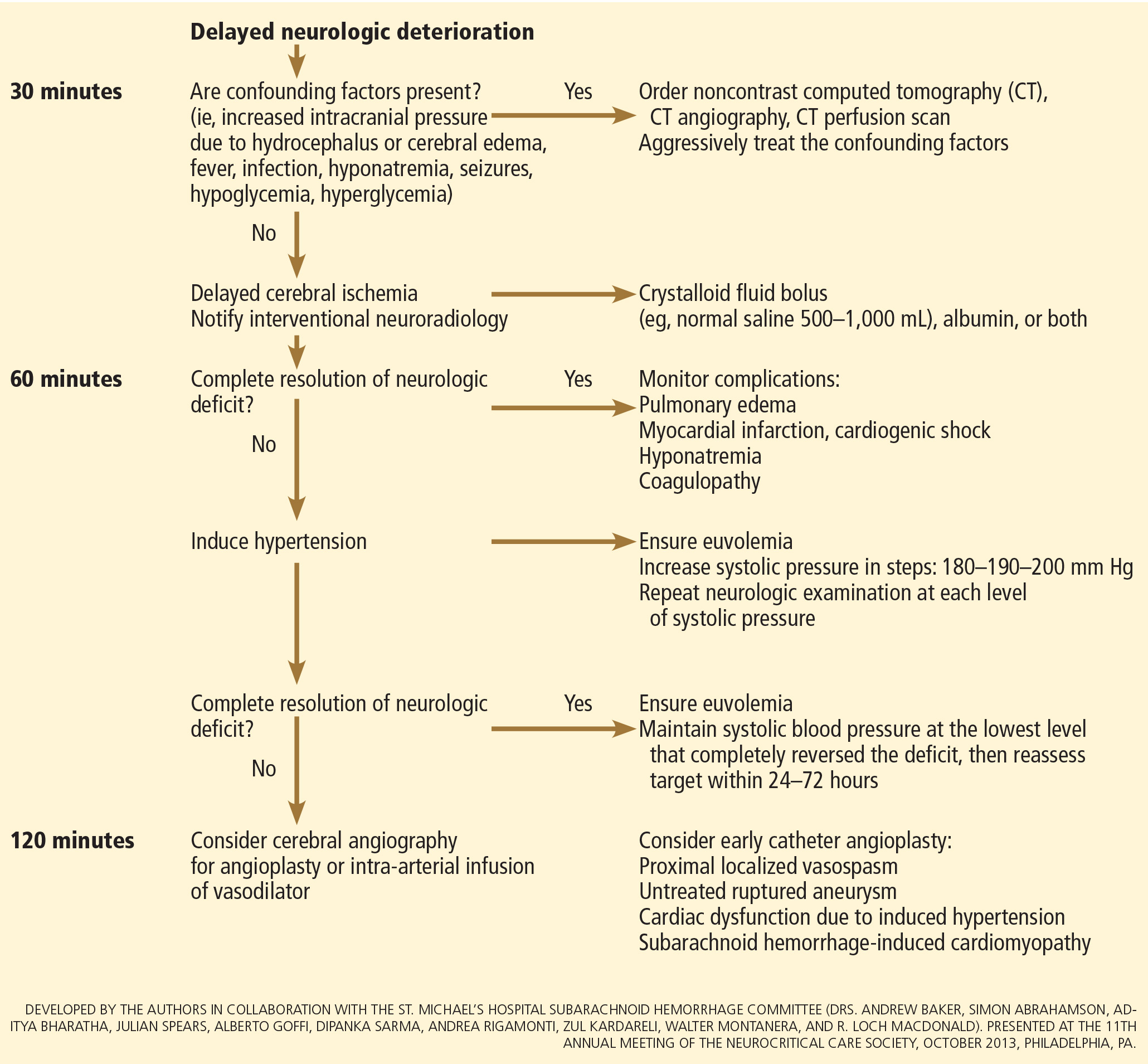
The physiologic goal is to increase the delivery of oxygen and glucose to the ischemic brain. Hypertension seems to be the most effective component of hemodynamic augmentation regardless of volume status, increasing cerebral blood flow and brain tissue oxygenation, with reversal of delayed cerebral ischemic symptoms in up to two-thirds of treated patients.74,75 However, this information comes from very small studies, with no randomized trials of induced hypertension available.
The effect of a normal saline fluid bolus in patients suspected of having delayed cerebral ischemia has been shown to increase cerebral blood flow in areas of cerebral ischemia.74 If volume augmentation fails to improve the neurologic status, the next step is to artificially induce hypertension using vasopressors. The blood pressure target should be based on clinical improvement. A stepwise approach is reasonable in this situation, and the lowest level of blood pressure at which there is a complete reversal of the new focal neurologic deficit should be maintained.3,29
Inotropic agents such as dobutamine or milrinone can be considered as alternatives in patients who have new neurologic deficits that are refractory to fluid boluses and vasopressors, or in a setting of subarachnoid hemorrhage-induced cardiomyopathy.76,77
Once the neurologic deficit is reversed by hemodynamic augmentation, the blood pressure should be maintained for 48 to 72 hours at the level that reversed the deficit completely, carefully reassessed thereafter, and the patient weaned slowly. Unruptured unsecured aneurysms should not prevent blood pressure augmentation in a setting of delayed cerebral ischemia if the culprit aneurysm is treated.3,32 If the ruptured aneurysm has not been secured, careful blood pressure augmentation can be attempted, keeping in mind that hypertension (> 160/95 mm Hg) is a risk factor for fatal aneurysm rupture.
Endovascular management of delayed cerebral ischemia
When medical augmentation fails to completely reverse the neurologic deficits, endovascular treatment can be considered. Although patients treated early in the course of delayed cerebral ischemia have better neurologic recovery, prophylactic endovascular treatment in asymptomatic patients, even if angiographic signs of spasm are present, does not improve clinical outcomes and carries the risk of fatal arterial rupture.78
SYSTEMIC COMPLICATIONS
Hyponatremia and hypovolemia
Aneurysmal subarachnoid hemorrhage is commonly associated with abnormalities of fluid balance and electrolyte derangements. Hyponatremia (serum sodium < 135 mmol/L) occurs in 30% to 50% of patients, while the rate of hypovolemia (decreased circulating blood volume) ranges from 17% to 30%.79 Both can negatively affect long-term outcomes.80,81
Decreased circulating blood volume is a well-described contributor to delayed cerebral ischemia and cerebral infarction after aneurysmal subarachnoid hemorrhage.80–82 Clinical variables such as heart rate, blood pressure, fluid balance, and serum sodium concentration are usually the cornerstones of intravascular volume status assessment. However, these variables correlate poorly with measured circulating blood volumes in those with aneurysmal subarachnoid hemorrhage.83,84
The mechanisms responsible for the development of hyponatremia and hypovolemia after aneurysmal subarachnoid hemorrhage are not completely understood. Several factors have been described and may contribute to the increased natriuresis and, hence, to a reduction in circulating blood volume: increased circulating natriuretic peptide concentrations,85–87 sympathetic nervous system hyperactivation,88 and hyperreninemic hypo-
aldosteronism syndrome.89,90
Lastly, the cerebral salt wasting syndrome, described in the 1950s,91 was thought to be a key mechanism in the development of hyponatremia and hypovolemia after aneurysmal subarachnoid hemorrhage. In contrast to the syndrome of inappropriate antidiuretic hormone, which is characterized by hyponatremia with a normal or slightly elevated intravascular volume, the characteristic feature of cerebral salt wasting syndrome is the development of hyponatremia in a setting of intravascular volume depletion.92 In critically ill neurologic and neurosurgical patients, this differential diagnosis is very difficult, especially in those with aneurysmal subarachnoid hemorrhage in whom the clinical assessment of fluid status is not reliable. These two syndromes might coexist and contribute to the development of hyponatremia after aneurysmal subarachnoid hemorrhage.92,93
Hoff et al83,84 prospectively compared the clinical assessment of fluid status by critical and intermediate care nurses and direct measurements of blood volume using pulse dye densitometry. The clinical assessment failed to accurately assess patients’ volume status. Using the same technique to measure circulating blood volume, this group showed that calculation of fluid balance does not provide adequate assessment of fluid status.83,84
Hemodynamic monitoring tools can help guide fluid replacement in this population. Mutoh et al94 randomized 160 patients within 24 hours of hemorrhage to receive early goal-directed fluid therapy (ie, preload volume and cardiac output monitored by transpulmonary thermodilution) vs standard therapy (ie, fluid balance or central venous pressure). Overall, no difference was found in the rates of delayed cerebral ischemia (33% vs 42%; P = .33) or favorable outcome (67% vs 57%; P = .22). However, in the subgroup of poor-grade patients (WFNS score 4 or 5), early goal-directed therapy was associated with a lower rate of delayed cerebral ischemia (5% vs 14%; P = .036) and with better functional outcomes at 3 months (52% vs 36%; P = .026).
Fluid restriction to treat hyponatremia in aneurysmal subarachnoid hemorrhage is no longer recommended because of the increased risk of cerebral infarction due to hypovolemic hypoperfusion.82
Prophylactic use of mineralocorticoids (eg, fludrocortisone, hydrocortisone) has been shown to limit natriuresis, hyponatremia, and the amount of fluid required to maintain euvolemia.95,96 Higher rates of hypokalemia and hyperglycemia, which can be easily treated, are the most common complications associated with this approach. Additionally, hypertonic saline (eg, 3% saline) can be used to correct hyponatremia in a setting of aneurysmal subarachnoid hemorrhage.79
Cardiac complications
Cardiac complications after subarachnoid hemorrhage are most likely related to sympathetic hyperactivity and catecholamine-induced myocyte dysfunction. The pathophysiology is complex, but cardiac complications have a significant negative impact on long-term outcome in these patients.97
Electrocardiographic changes and positive cardiac enzymes associated with aneurysmal subarachnoid hemorrhage have been extensively reported. More recently, data from studies of two-dimensional echocardiography have shown that subarachnoid hemorrhage can also be associated with significant wall-motion abnormalities and even overt cardiogenic shock.98–100
There is no specific curative therapy; the treatment is mainly supportive. Vasopressors and inotropes may be used for hemodynamic augmentation.
Pulmonary complications
Pulmonary complications occur in 20% to 30% of all aneurysmal subarachnoid hemorrhage patients and are associated with a higher risk of delayed cerebral ischemia and death. Common pulmonary complications in this population are mild acute respiratory distress syndrome (27%), hospital-acquired pneumonia (9%), cardiogenic pulmonary edema (8%), aspiration pneumonia (6%), neurogenic pulmonary edema (2%), and pulmonary embolism (1%).101–103
SUPPORTIVE CARE
Hyperthermia, hyperglycemia, and liberal use of transfusions have all been associated with longer stays in the intensive care unit and hospital, poorer neurologic outcomes, and higher mortality rates in patients with acute brain injury.104 Noninfectious fever is the most common systemic complication after subarachnoid hemorrhage.
Antipyretic drugs such as acetaminophen and ibuprofen are not very effective in reducing fever in the subarachnoid hemorrhage population, but should still be used as first-line therapy. The use of surface and intravascular devices can be considered when fevers do not respond to nonsteroidal anti-inflammatory drugs.
Although no prospective randomized trial has addressed the impact of induced normothermia on long-term outcome and mortality in aneurysmal subarachnoid hemorrhage patients, fever control has been shown to reduce cerebral metabolic distress, irrespective of intracranial pressure.105 Maintenance of normothermia (< 37.5°C) seems reasonable, especially in aneurysmal subarachnoid hemorrhage patients at risk of or with active delayed cerebral ischemia.106
Current guidelines3,32,69 strongly recommend avoiding hypoglycemia, defined as a serum glucose level less than 80 mg/dL, but suggest keeping the blood sugar level below 180 or 200 mg/dL.
At the moment, there is no clear threshold for transfusion in patients with aneurysmal subarachnoid hemorrhage. Current guidelines suggest keeping hemoglobin levels between 8 and 10 g/dL.3
Preventing venous thromboembolism
The incidence of venous thromboembolism after aneurysmal subarachnoid hemorrhage varies widely, from 1.5% to 18%.107 Active surveillance with venous Doppler ultrasonography has found asymptomatic deep vein thrombosis in up to 3.4% of poor-grade aneurysmal subarachnoid hemorrhage patients receiving pharmacologic thromboprophylaxis.108
In a retrospective study of 170 patients, our group showed that giving drugs to prevent venous thromboembolism (unfractionated heparin 5,000 IU subcutaneously every 12 hours or dalteparin 5,000 IU subcutaneously daily), starting within 24 hours of aneurysm treatment, could be safe.109 Fifty-eight percent of these patients had an external ventricular drain in place. One patient developed a major cerebral hemorrhagic complication and died while on unfractionated heparin; however, the patient was also on dual antiplatelet therapy with aspirin and clopidogrel.109
Current guidelines suggest that intermittent compression devices be applied in all patients before aneurysm treatment. Pharmacologic thromboprophylaxis with a heparinoid can be started 12 to 24 hours after aneurysm treatment.3,109
A TEAM APPROACH
Patients with subarachnoid hemorrhage need integrated care from different medical and nursing specialties. The best outcomes are achieved by systems that can focus as a team on the collective goal of quick intervention to secure the aneurysm, followed by measures to minimize secondary brain injury.
The modern concept of cerebral monitoring in a setting of subarachnoid hemorrhage should focus on brain perfusion rather than vascular diameter. Although the search continues for new diagnostic, prognostic, and therapeutic tools, there is no “silver bullet” that will help all patients. Instead, it is the systematic integration and application of many small advances that will ultimately lead to better outcomes.
ACKNOWLEDGMENT
This work was supported by research funding provided by the Bitove Foundation, which has been supportive of our clinical and research work for several years.
- van Gijn J, Rinkel GJ. Subarachnoid haemorrhage: diagnosis, causes and management. Brain 2001; 124:249–278.
- Go AS, Mozaffarian D, Roger VL, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2014 update: a report from the American Heart Association. Circulation 2014; 129:e28–e292.
- Diringer MN, Bleck TP, Claude Hemphill J 3rd, et al; Neurocritical Care Society. Critical care management of patients following aneurysmal subarachnoid hemorrhage: recommendations from the Neurocritical Care Society’s Multidisciplinary Consensus Conference. Neurocrit Care 2011; 15:211–240.
- Feigin VL, Rinkel GJ, Lawes CM, et al. Risk factors for subarachnoid hemorrhage: an updated systematic review of epidemiological studies. Stroke 2005; 36:2773–2780.
- Perry JJ, Stiell IG, Sivilotti ML, et al. Clinical decision rules to rule out subarachnoid hemorrhage for acute headache. JAMA 2013; 310:1248–1255.
- Kowalski RG, Claassen J, Kreiter KT, et al. Initial misdiagnosis and outcome after subarachnoid hemorrhage. JAMA 2004; 291:866–869.
- Perry JJ, Stiell IG, Sivilotti ML, et al. Sensitivity of computed tomography performed within six hours of onset of headache for diagnosis of subarachnoid haemorrhage: prospective cohort study. BMJ 2011; 343:d4277.
- van Gijn J, van Dongen KJ. The time course of aneurysmal haemorrhage on computed tomograms. Neuroradiology 1982; 23:153–156.
- McCormack RF, Hutson A. Can computed tomography angiography of the brain replace lumbar puncture in the evaluation of acute-onset headache after a negative noncontrast cranial computed tomography scan? Acad Emerg Med 2010; 17:444–451.
- Agid R, Andersson T, Almqvist H, et al. Negative CT angiography findings in patients with spontaneous subarachnoid hemorrhage: when is digital subtraction angiography still needed? AJNR Am J Neuroradiol 2010; 31:696–705.
- Teasdale GM, Drake CG, Hunt W, et al. A universal subarachnoid hemorrhage scale: report of a committee of the World Federation of Neurosurgical Societies. J Neurol Neurosurg Psychiatry 1988; 51:1457.
- Hunt WE, Hess RM. Surgical risk as related to time of intervention in the repair of intracranial aneurysms. J Neurosurg 1968; 28:14–20.
- Fisher CM, Kistler JP, Davis JM. Relation of cerebral vasospasm to subarachnoid hemorrhage visualized by computerized tomographic scanning. Neurosurgery 1980; 6:1–9.
- Frontera JA, Claassen J, Schmidt JM, et al. Prediction of symptomatic vasospasm after subarachnoid hemorrhage: the modified Fisher scale. Neurosurgery 2006; 59:21–27.
- de Oliveira Manoel AL, Turkel-Parrella D, Kouzmina E, et al. The VASOGRADE—a simple, reliable grading scale for aneurysmal subarachnoid hemorrhage. Neurology 2014; 82(suppl 10): P5.123.
- Naidech AM, Janjua N, Kreiter KT, et al. Predictors and impact of aneurysm rebleeding after subarachnoid hemorrhage. Arch Neurol 2005; 62:410–416.
- Rincon F, Mayer SA. Neurocritical care: a distinct discipline? Curr Opin Crit Care 2007; 13:115–121.
- Rabinstein AA, Lanzino G, Wijdicks EF. Multidisciplinary management and emerging therapeutic strategies in aneurysmal subarachnoid haemorrhage. Lancet Neurol 2010; 9:504–519.
- Vespa P, Diringer MN; Participants in the International Multi-Disciplinary Consensus Conference on the Critical Care Management of Subarachnoid Hemorrhage. High-volume centers. Neurocrit Care 2011; 15:369–372.
- McNeill L, English SW, Borg N, Matta BF, Menon DK. Effects of institutional caseload of subarachnoid hemorrhage on mortality: a secondary analysis of administrative data. Stroke 2013; 44:647–652.
- Dorhout Mees SM, Molyneux AJ, Kerr RS, Algra A, Rinkel GJ. Timing of aneurysm treatment after subarachnoid hemorrhage: relationship with delayed cerebral ischemia and poor outcome. Stroke 2012; 43:2126–2129.
- Laidlaw JD, Siu KH. Ultra-early surgery for aneurysmal subarachnoid hemorrhage: outcomes for a consecutive series of 391 patients not selected by grade or age. J Neurosurg 2002; 97:250–259.
- Molyneux A, Kerr R, Stratton I, et al; International Subarachnoid Aneurysm Trial (ISAT) Collaborative Group. International Subarachnoid Aneurysm Trial (ISAT) of neurosurgical clipping versus endovascular coiling in 2,143 patients with ruptured intracranial aneurysms: a randomised trial. Lancet 2002; 360:1267–1274.
- Molyneux AJ, Kerr RS, Yu LM, et al; International Subarachnoid Aneurysm Trial (ISAT) Collaborative Group. International subarachnoid aneurysm trial (ISAT) of neurosurgical clipping versus endovascular coiling in 2,143 patients with ruptured intracranial aneurysms: a randomised comparison of effects on survival, dependency, seizures, rebleeding, subgroups, and aneurysm occlusion. Lancet 2005; 366:809–817.
- Koivisto T, Vanninen R, Hurskainen H, Saari T, Hernesniemi J, Vapalahti M. Outcomes of early endovascular versus surgical treatment of ruptured cerebral aneurysms. A prospective randomized study. Stroke 2000; 31:2369–2377.
- McDougall CG, Spetzler RF, Zabramski JM, et al. The Barrow Ruptured Aneurysm Trial. J Neurosurg 2012; 116:135–144.
- Spetzler RF, McDougall CG, Albuquerque FC, et al. The Barrow Ruptured Aneurysm Trial: 3-year results. J Neurosurg 2013; 119:146–157.
- Connolly ES Jr, Meyers PM. Cerebral aneurysms: to clip or to coil? That is no longer the question. Nat Rev Neurol 2009; 5:412–413.
- Kassell NF, Peerless SJ, Durward QJ, Beck DW, Drake CG, Adams HP. Treatment of ischemic deficits from vasospasm with intravascular volume expansion and induced arterial hypertension. Neurosurgery 1982; 11:337–343.
- Otsubo H, Takemae T, Inoue T, Kobayashi S, Sugita K. Normovolaemic induced hypertension therapy for cerebral vasospasm after subarachnoid haemorrhage. Acta Neurochir (Wien) 1990; 103:18–26.
- Anderson CS, Heeley E, Huang Y, et al; INTERACT2 Investigators. Rapid blood-pressure lowering in patients with acute intracerebral hemorrhage. N Engl J Med 2013; 368:2355–2365.
- Connolly ES Jr, Rabinstein AA, Carhuapoma JR, et al; American Heart Association Stroke Council; Council on Cardiovascular Radiology and Intervention; Council on Cardiovascular Nursing; Council on Cardiovascular Surgery and Anesthesia; Council on Clinical Cardiology. Guidelines for the management of aneurysmal subarachnoid hemorrhage: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2012; 43:1711–1737.
- Ortega-Gutierrez S, Thomas J, Reccius A, et al. Effectiveness and safety of nicardipine and labetalol infusion for blood pressure management in patients with intracerebral and subarachnoid hemorrhage. Neurocrit Care 2013; 18:13–19.
- Hillman J, Fridriksson S, Nilsson O, Yu Z, Saveland H, Jakobsson KE. Immediate administration of tranexamic acid and reduced incidence of early rebleeding after aneurysmal subarachnoid hemorrhage: a prospective randomized study. J Neurosurg 2002; 97:771–778.
- Baharoglu MI, Germans MR, Rinkel GJ, et al. Antifibrinolytic therapy for aneurysmal subarachnoid haemorrhage. Cochrane Database Syst Rev 2013; 8:CD001245.
- Germans MR, Post R, Coert BA, Rinkel GJ, Vandertop WP, Verbaan D. Ultra-early tranexamic acid after subarachnoid hemorrhage (ULTRA): study protocol for a randomized controlled trial. Trials 2013; 14:143.
- Sloan MA, Alexandrov AV, Tegeler CH, et al; Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Assessment: transcranial Doppler ultrasonography: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology 2004; 62:1468–1481.
- Wartenberg KE, Schmidt JM, Claassen J, et al. Impact of medical complications on outcome after subarachnoid hemorrhage. Crit Care Med 2006; 34:617–623;
- Hellingman CA, van den Bergh WM, Beijer IS, et al. Risk of rebleeding after treatment of acute hydrocephalus in patients with aneurysmal subarachnoid hemorrhage. Stroke 2007; 38:96–99.
- Ransom ER, Mocco J, Komotar RJ, et al. External ventricular drainage response in poor grade aneurysmal subarachnoid hemorrhage: effect on preoperative grading and prognosis. Neurocrit Care 2007; 6:174–180.
- McIver JI, Friedman JA, Wijdicks EF, et al. Preoperative ventriculostomy and rebleeding after aneurysmal subarachnoid hemorrhage. J Neurosurg 2002; 97:1042–1044.
- Klopfenstein JD, Kim LJ, Feiz-Erfan I, et al. Comparison of rapid and gradual weaning from external ventricular drainage in patients with aneurysmal subarachnoid hemorrhage: a prospective randomized trial. J Neurosurg 2004; 100:225–229.
- Tseng MY, Al-Rawi PG, Czosnyka M, et al. Enhancement of cerebral blood flow using systemic hypertonic saline therapy improves outcome in patients with poor-grade spontaneous subarachnoid hemorrhage. J Neurosurg 2007; 107:274–282.
- Al-Rawi PG, Tseng MY, Richards HK, et al. Hypertonic saline in patients with poor-grade subarachnoid hemorrhage improves cerebral blood flow, brain tissue oxygen, and pH. Stroke 2010; 41:122–128.
- Tseng MY, Al-Rawi PG, Pickard JD, Rasulo FA, Kirkpatrick PJ. Effect of hypertonic saline on cerebral blood flow in poor-grade patients with subarachnoid hemorrhage. Stroke 2003; 34:1389–1396.
- Bentsen G, Breivik H, Lundar T, Stubhaug A. Hypertonic saline (7.2%) in 6% hydroxyethyl starch reduces intracranial pressure and improves hemodynamics in a placebo-controlled study involving stable patients with subarachnoid hemorrhage. Crit Care Med 2006; 34:2912–2917.
- Suarez JI, Qureshi AI, Parekh PD, et al. Administration of hypertonic (3%) sodium chloride/acetate in hyponatremic patients with symptomatic vasospasm following subarachnoid hemorrhage. J Neurosurg Anesthesiol 1999; 11:178–184.
- Stevens RD, Huff JS, Duckworth J, Papangelou A, Weingart SD, Smith WS. Emergency neurological life support: intracranial hypertension and herniation. Neurocrit Care 2012;17(suppl 1):S60–S65.
- Seule MA, Muroi C, Mink S, Yonekawa Y, Keller E. Therapeutic hypothermia in patients with aneurysmal subarachnoid hemorrhage, refractory intracranial hypertension, or cerebral vasospasm. Neurosurgery 2009; 64:86–93.
- Otani N, Takasato Y, Masaoka H, et al. Surgical outcome following decompressive craniectomy for poor-grade aneurysmal subarachnoid hemorrhage in patients with associated massive intracerebral or Sylvian hematomas. Cerebrovasc Dis 2008; 26:612–617.
- Lanzino G, D’Urso PI, Suarez J; Participants in the International Multi-Disciplinary Consensus Conference on the Critical Care Management of Subarachnoid Hemorrhage. Seizures and anticonvulsants after aneurysmal subarachnoid hemorrhage. Neurocrit Care 2011; 15:247–256.
- Naidech AM, Kreiter KT, Janjua N, et al. Phenytoin exposure is associated with functional and cognitive disability after subarachnoid hemorrhage. Stroke 2005; 36:583–587.
- Rosengart AJ, Huo JD, Tolentino J, et al. Outcome in patients with subarachnoid hemorrhage treated with antiepileptic drugs. J Neurosurg 2007; 107:253–260.
- Shah D, Husain AM. Utility of levetiracetam in patients with subarachnoid hemorrhage. Seizure 2009; 18:676–679.
- Patsalos PN. Pharmacokinetic profile of levetiracetam: toward ideal characteristics. Pharmacol Ther 2000; 85:77–85.
- Dewan MC, Mocco J. Current practice regarding seizure prophylaxis in aneurysmal subarachnoid hemorrhage across academic centers. J Neurointerv Surg 2014 Jan 28. doi: 10.1136/neurintsurg-2013-011075 [Epub ahead of print]
- Murphy-Human T, Welch E, Zipfel G, Diringer MN, Dhar R. Comparison of short-duration levetiracetam with extended-course phenytoin for seizure prophylaxis after subarachnoid hemorrhage. World Neurosurg 2011; 75:269–274.
- Dennis LJ, Claassen J, Hirsch LJ, Emerson RG, Connolly ES, Mayer SA. Nonconvulsive status epilepticus after subarachnoid hemorrhage. Neurosurgery 2002; 51:1136–1144.
- Vespa PM, Nuwer MR, Juhász C, et al. Early detection of vasospasm after acute subarachnoid hemorrhage using continuous EEG ICU monitoring. Electroencephalogr Clin Neurophysiol 1997; 103:607–615.
- Claassen J, Hirsch LJ, Kreiter KT, et al. Quantitative continuous EEG for detecting delayed cerebral ischemia in patients with poor-grade subarachnoid hemorrhage. Clin Neurophysiol 2004; 115:2699–2710.
- Vergouwen MD, Vermeulen M, van Gijn J, et al. Definition of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage as an outcome event in clinical trials and observational studies: proposal of a multidisciplinary research group. Stroke 2010; 41:2391–2395.
- Kelly PJ, Gorten RJ, Grossman RG, Eisenberg HM. Cerebral perfusion, vascular spasm, and outcome in patients with ruptured intracranial aneurysms. J Neurosurg 1977; 47:44–49.
- Aralasmak A, Akyuz M, Ozkaynak C, Sindel T, Tuncer R. CT angiography and perfusion imaging in patients with subarachnoid hemorrhage: correlation of vasospasm to perfusion abnormality. Neuroradiology 2009; 51:85–93.
- Sabri M, Lass E, Macdonald RL. Early brain injury: a common mechanism in subarachnoid hemorrhage and global cerebral ischemia. Stroke Res Treat 2013 Feb 28. doi: 10.1155/2013/394036 [Epub 2013 ahead of print]
- Yoon DY, Choi CS, Kim KH, Cho BM. Multidetector-row CT angiography of cerebral vasospasm after aneurysmal subarachnoid hemorrhage: comparison of volume-rendered images and digital subtraction angiography. AJNR Am J Neuroradiol 2006; 27:370–377.
- Wintermark M1, Ko NU, Smith WS, Liu S, Higashida RT, Dillon WP. Vasospasm after subarachnoid hemorrhage: utility of perfusion CT and CT angiography on diagnosis and management. AJNR Am J Neuroradiol 2006; 27:26–34.
- Helbok R, Madineni RC, Schmidt MJ, et al. Intracerebral monitoring of silent infarcts after subarachnoid hemorrhage. Neurocrit Care 2011; 14:162–167.
- Hänggi D; Participants in the International Multi-Disciplinary Consensus Conference on the Critical Care Management of Subarachnoid Hemorrhage. Monitoring and detection of vasospasm II: EEG and invasive monitoring. Neurocrit Care 2011; 15:318–323.
- Steiner T, Juvela S, Unterberg A, Jung C, Forsting M, Rinkel G; European Stroke Organization. European Stroke Organization guidelines for the management of intracranial aneurysms and subarachnoid haemorrhage. Cerebrovasc Dis 2013; 35:93–112.
- Pickard JD, Murray GD, Illingworth R, et al. Effect of oral nimodipine on cerebral infarction and outcome after subarachnoid haemorrhage: British aneurysm nimodipine trial. BMJ 1989; 298:636–642.
- Dorhout Mees SM, Rinkel GJ, Feigin VL, et al. Calcium antagonists for aneurysmal subarachnoid haemorrhage. Cochrane Database Syst Rev 2007; 3:CD000277.
- Lennihan L, Mayer SA, Fink ME, et al. Effect of hypervolemic therapy on cerebral blood flow after subarachnoid hemorrhage: a randomized controlled trial. Stroke 2000; 31:383–391.
- Egge A, Waterloo K, Sjøholm H, Solberg T, Ingebrigtsen T, Romner B. Prophylactic hyperdynamic postoperative fluid therapy after aneurysmal subarachnoid hemorrhage: a clinical, prospective, randomized, controlled study. Neurosurgery 2001; 49:593–606.
- Jost SC, Diringer MN, Zazulia AR, et al. Effect of normal saline bolus on cerebral blood flow in regions with low baseline flow in patients with vasospasm following subarachnoid hemorrhage. J Neurosurg 2005; 103:25–30.
- Muizelaar JP, Becker DP. Induced hypertension for the treatment of cerebral ischemia after subarachnoid hemorrhage. Direct effect on cerebral blood flow. Surg Neurol 1986; 25:317–325.
- Levy ML, Rabb CH, Zelman V, Giannotta SL. Cardiac performance enhancement from dobutamine in patients refractory to hypervolemic therapy for cerebral vasospasm. J Neurosurg 1993; 79:494–499.
- Lannes M, Teitelbaum J, del Pilar Cortés M, Cardoso M, Angle M. Milrinone and homeostasis to treat cerebral vasospasm associated with subarachnoid hemorrhage: the Montreal Neurological Hospital protocol. Neurocrit Care 2012; 16:354–362.
- Zwienenberg-Lee M, Hartman J, Rudisill N, et al; Balloon Prophylaxis for Aneurysmal Vasospasm (BPAV) Study Group. Effect of prophylactic transluminal balloon angioplasty on cerebral vasospasm and outcome in patients with Fisher grade III subarachnoid hemorrhage: results of a phase II multicenter, randomized, clinical trial. Stroke 2008; 39:1759–1765.
- Rabinstein AA, Bruder N. Management of hyponatremia and volume contraction. Neurocrit Care 2011; 15:354–360.
- Wijdicks EF, Vermeulen M, Hijdra A, van Gijn J. Hyponatremia and cerebral infarction in patients with ruptured intracranial aneurysms: is fluid restriction harmful? Ann Neurol 1985; 17:137–140.
- Hasan D, Wijdicks EF, Vermeulen M. Hyponatremia is associated with cerebral ischemia in patients with aneurysmal subarachnoid hemorrhage. Ann Neurol 1990; 27:106–108.
- Wijdicks EF, Vermeulen M, ten Haaf JA, Hijdra A, Bakker WH, van Gijn J. Volume depletion and natriuresis in patients with a ruptured intracranial aneurysm. Ann Neurol 1985; 18:211–216.
- Hoff RG, Rinkel GJ, Verweij BH, Algra A, Kalkman CJ. Nurses’ prediction of volume status after aneurysmal subarachnoid haemorrhage: a prospective cohort study. Crit Care 2008; 12:R153.
- Hoff RG, van Dijk GW, Algra A, Kalkman CJ, Rinkel GJ. Fluid balance and blood volume measurement after aneurysmal subarachnoid hemorrhage. Neurocrit Care 2008; 8:391–397.
- Berendes E, Walter M, Cullen P, et al. Secretion of brain natriuretic peptide in patients with aneurysmal subarachnoid haemorrhage. Lancet 1997; 349:245–249.
- Espiner EA, Leikis R, Ferch RD, et al. The neuro-cardio-endocrine response to acute subarachnoid haemorrhage. Clin Endocrinol (Oxf) 2002; 56:629–635.
- Isotani E, Suzuki R, Tomita K, et al. Alterations in plasma concentrations of natriuretic peptides and antidiuretic hormone after subarachnoid hemorrhage. Stroke 1994; 25:2198–2203.
- Benedict CR, Loach AB. Sympathetic nervous system activity in patients with subarachnoid hemorrhage. Stroke 1978; 9:237–244.
- Findling JW, Waters VO, Raff H. The dissociation of renin and aldosterone during critical illness. J Clin Endocrinol Metab 1987; 64:592–595.
- Solomon RA, Post KD, McMurtry JG 3rd. Depression of circulating blood volume in patients after subarachnoid hemorrhage: implications for the management of symptomatic vasospasm. Neurosurgery 1984; 15:354–361.
- Peters JP, Welt LG, Sims EA, Orloff J, Needham J. A salt-wasting syndrome associated with cerebral disease. Trans Assoc Am Physicians 1950; 63:57–64.
- Brimioulle S, Orellana-Jimenez C, Aminian A, Vincent JL. Hyponatremia in neurological patients: cerebral salt wasting versus inappropriate antidiuretic hormone secretion. Intensive Care Med 2008; 34:125–131.
- Singh S, Bohn D, Carlotti AP, Cusimano M, Rutka JT, Halperin ML. Cerebral salt wasting: truths, fallacies, theories, and challenges. Crit Care Med 2002; 30:2575–2579.
- Mutoh T, Kazumata K, Terasaka S, Taki Y, Suzuki A, Ishikawa T. Early intensive versus minimally invasive approach to postoperative hemodynamic management after subarachnoid hemorrhage. Stroke 2014; 45:1280–1284.
- Hasan D, Lindsay KW, Wijdicks EF, et al. Effect of fludrocortisone acetate in patients with subarachnoid hemorrhage. Stroke 1989; 20:1156–1161.
- Moro N, Katayama Y, Kojima J, Mori T, Kawamata T. Prophylactic management of excessive natriuresis with hydrocortisone for efficient hypervolemic therapy after subarachnoid hemorrhage. Stroke 2003; 34:2807–2811.
- Kilbourn KJ, Levy S, Staff I, Kureshi I, McCullough L. Clinical characteristics and outcomes of neurogenic stress cadiomyopathy in aneurysmal subarachnoid hemorrhage. Clin Neurol Neurosurg 2013; 115:909–914.
- Mayer SA, LiMandri G, Sherman D, et al. Electrocardiographic markers of abnormal left ventricular wall motion in acute subarachnoid hemorrhage. J Neurosurg 1995; 83:889–896.
- Deibert E, Barzilai B, Braverman AC, et al. Clinical significance of elevated troponin I levels in patients with nontraumatic subarachnoid hemorrhage. J Neurosurg 2003; 98:741–746.
- Banki N, Kopelnik A, Tung P, et al. Prospective analysis of prevalence, distribution, and rate of recovery of left ventricular systolic dysfunction in patients with subarachnoid hemorrhage. J Neurosurg 2006; 105:15–20.
- Kahn JM, Caldwell EC, Deem S, Newell DW, Heckbert SR, Rubenfeld GD. Acute lung injury in patients with subarachnoid hemorrhage: incidence, risk factors, and outcome. Crit Care Med 2006; 34:196–202.
- Kitamura Y, Nomura M, Shima H, et al. Acute lung injury associated with systemic inflammatory response syndrome following subarachnoid hemorrhage: a survey by the Shonan Neurosurgical Association. Neurol Med Chir (Tokyo) 2010; 50:456–460.
- Friedman JA, Pichelmann MA, Piepgras DG, et al. Pulmonary complications of aneurysmal subarachnoid hemorrhage. Neurosurgery 2003; 52:1025–1032.
- Oh HS, Jeong HS, Seo WS. Non-infectious hyperthermia in acute brain injury patients: relationships to mortality, blood pressure, intracranial pressure and cerebral perfusion pressure. Int J Nurs Pract 2012; 18:295–302.
- Oddo M, Frangos S, Milby A, et al. Induced normothermia attenuates cerebral metabolic distress in patients with aneurysmal subarachnoid hemorrhage and refractory fever. Stroke 2009; 40:1913–1916.
- Badjatia N, Fernandez L, Schmidt JM, et al. Impact of induced normothermia on outcome after subarachnoid hemorrhage: a case-control study. Neurosurgery 2010; 66:696-701.
- Serrone JC1, Wash EM, Hartings JA, Andaluz N, Zuccarello M. Venous thromboembolism in subarachnoid hemorrhage. World Neurosurg 2013; 80:859–863.
- Mack WJ, Ducruet AF, Hickman ZL, et al. Doppler ultrasonography screening of poor-grade subarachnoid hemorrhage patients increases the diagnosis of deep venous thrombosis. Neurol Res 2008; 30:889–892.
- de Oliveira Manoel AL, Turkel-Parrella D, Germans M, et al. Safety of early pharmacological thromboprophylaxis after subarachnoid hemorrhage. Can J Neurol Sci 2014; 41:554–561.
Aneurysmal subarachnoid hemorrhage is a devastating condition, with an estimated death rate of 30% during the initial episode.1,2 Approximately the same number of patients survive but leave the hospital with disabling neurologic deficits.3
However, better outcomes can be achieved by systems that are able to work as a team on the collective goal of quick intervention to secure the ruptured aneurysm, followed by the implementation of measures to minimize secondary brain injury. Although the search for new diagnostic, prognostic, and therapeutic modalities continues, it is clear that there exists no “silver bullet” that will help all patients. Instead, it is the systematic integration and application of small advances that will ultimately maximize the patient’s chances of survival and neurologic recovery.
This review focuses on the management of aneurysmal subarachnoid hemorrhage and its systemic and neurologic complications.
ANEURYSM IS THE MOST COMMON CAUSE OF SUBARACHNOID BLEEDING
Aneurysmal subarachnoid hemorrhage, ie, rupture of an intracranial aneurysm, flooding the subarachnoid space with blood, affects about 24,000 Americans each year.1,2 A ruptured aneurysm is the most common cause of subarachnoid hemorrhage, accounting for about 85% of cases. Less common causes include idiopathic benign perimesencephalic hemorrhage, arteriovenous malformation, dural arteriovenous fistula, and hemorrhagic mycotic aneurysm. These have their own natural history, pathophysiology, and specific treatment, and will not be addressed in this article.
Risk factors for aneurysmal subarachnoid hemorrhage include having a first-degree relative who had the disease, hypertension, smoking, and consuming more than 150 g of alcohol per week.4
CLINICAL PRESENTATION AND DIAGNOSIS
The key symptom of aneurysmal subarachnoid hemorrhage is the abrupt onset of severe headache that peaks in intensity over 1 hour,5 often described as “the worst headache of my life.” Headache is accompanied by brief loss of consciousness in 53% of cases (conversely, nearly half of patients maintain normal mental status), by nausea or vomiting in 77%, and by meningismus (neck pain or stiffness) in 35%.6
These clinical manifestations and risk factors have been incorporated into a decision rule:
Obtain brain imaging if the patient has acute headache reaching maximal intensity within 1 hour, associated with any of the following factors:
- Age 40 or older
- Neck pain or stiffness
- Witnessed loss of consciousness
- Onset during exertion
- “Thunderclap” headache (ie, instantly peaking pain)
- Limited neck flexion on examination.5
This decision rule has nearly 100% sensitivity for aneurysmal subarachnoid hemorrhage in clinical practice.5 All patients require brain imaging if they have a severe headache plus either abnormal neurologic findings (eg, a focal neurologic deficit) or a history of cerebral aneurysm.
Emergency physicians should have a low threshold for ordering noncontrast computed tomography (CT) of the head in patients with even mild symptoms suggesting aneurysmal subarachnoid hemorrhage. Failure to order CT is the most common diagnostic error in this situation.6 CT performed within 6 hours of headache onset is nearly 100% sensitive for this condition,7 but the sensitivity falls to 93% after the first 24 hours and to less than 60% after 5 days.8 In patients who have symptoms highly suggestive of aneurysmal subarachnoid hemorrhage but a normal CT, lumbar puncture is the next diagnostic step.
There are two alternatives to CT followed by lumbar puncture: ie, noncontrast CT followed by CT angiography,9,10 and magnetic resonance imaging followed by magnetic resonance angiography. In patients with suspicious clinical symptoms but negative CT results, CT followed by CT angiography can rule out aneurysmal subarachnoid hemorrhage with a 99% probability.9,10 However, CT followed by lumbar puncture remains the standard of care and carries a class I recommendation in the American Heart Association guidelines for ruling out subarachnoid hemorrhage.5
GRADING THE SEVERITY OF SUBARACHNOID HEMORRHAGE
Age, the thickness of the blood layer in the subarachnoid space, intraventricular hemorrhage and the findings of the neurologic examination at presentation are predictors of long-term outcomes in aneurysmal subarachnoid hemorrhage (Figure 1).
Different grading systems used in clinical practice are based on the findings on the initial neurologic examination and on the initial noncontrast CT (ie, the thickness of the blood, and whether intraventricular hemorrhage is present). Among the most widely used are those developed by Hunt and Hess12 and by the World Federation of Neurological Surgeons11 (WFNS), and the CT grading scales (Fisher13 or its modified version14) (Tables 1 and 2). With either the Hunt and Hess scale or the WFNS scale, the higher the score, the worse the patient’s probable outcome. Scores on both Fisher scales correlate with the risk of angiographic vasospasm. The higher the grade, the higher the risk of angiographic vasospasm.
The VASOGRADE score—a combination of the WFNS score and the modified Fisher scale—stratifies patients at risk of delayed cerebral ischemia, allowing for a tailored monitoring strategy.15 There are three variations:
- VASOGRADE green—Modified Fisher 1 or 2 and WFNS 1 or 2
- VASOGRADE yellow—Modified Fisher 3 or 4 and WFNS 1, 2, or 3
- VASOGRADE red—WFNS 4 or 5.
After the initial bleeding event, patients with aneurysmal subarachnoid hemorrhage are at high risk of delayed systemic and neurologic complications, with poor functional outcomes. Delayed cerebral ischemia holds the greatest risk of an unfavorable outcome and ultimately can lead to cerebral infarction, disability, and death.6,7
INITIAL MANAGEMENT
After aneurysmal subarachnoid hemorrhage is diagnosed, the initial management (Figure 2) includes appropriate medical prevention of rebleeding (which includes supportive care, blood pressure management, and, perhaps, the early use of a short course of an antifibrinolytic drug) and early transfer to a high-volume center for securing the aneurysm. The reported incidence of rebleeding varies from 5% to 22% in the first 72 hours. “Ultra-early” rebleeding (within 24 hours of hemorrhage) has been reported, with an incidence as high as 15% and a fatality rate around 70%. Patients with poor-grade aneurysmal subarachnoid hemorrhage, larger aneurysms, and “sentinel bleeds” are at higher risk of rebleeding.16
Outcomes are much better when patients are managed in a high-volume center, with a specialized neurointensive care unit17 and access to an interdisciplinary team.18 Regardless of the initial grade, patients with aneurysmal subarachnoid hemorrhage should be quickly transferred to a high-volume center, defined as one treating at least 35 cases per year, and the benefit is greater in centers treating more than 60 cases per year.19 The higher the caseload in any given hospital, the better the clinical outcomes in this population.20
Treating cerebral aneurysm: Clipping or coiling
Early aneurysm repair is generally considered the standard of care and the best strategy to reduce the risk of rebleeding. Further, early treatment may be associated with a lower risk of delayed cerebral ischemia21 and better outcomes.22
Three randomized clinical trials have compared surgical clipping and endovascular repair (placement of small metal coils within the aneurysm to promote clotting).
The International Subarachnoid Aneurysm Trial23 showed a reduction of 23% in relative risk and of 7% in absolute risk in patients who underwent endovascular treatment compared with surgery. The survival benefit persisted at a mean of 9 years (range 6–14 years), but with a higher annual rate of aneurysm recurrence in the coiling group (2.9% vs 0.9%).24 Of note, this trial included only patients with aneurysms deemed suitable for both coiling and clipping, so that the exclusion rate was high. Most of the patients presented with good-grade (WFNS score 1–3), small aneurysms (< 5 mm) in the anterior circulation.
A single-center Finnish study25 found no differences in rates of recovery, disability, and death at 1 year, comparing surgery and endovascular treatment. Additionally, survival rates at a mean follow-up of 39 months were similar, with no late recurrences or aneurysmal bleeding.
Lastly, the Barrow Ruptured Aneurysm Trial26,27 found that patients assigned to endovascular treatment had better 1-year neurologic outcomes, defined as a modified Rankin score of 2 or less. Importantly, 37.7% of patients originally assigned to endovascular treatment crossed over to surgical treatment. The authors then performed intention-to-treat and as-treated analyses. Either way, patients treated by endovascular means had better neurologic outcomes at 1 year. However, no difference in the relative risk reduction in worse outcome was found on 3-year follow-up, and patients treated surgically had higher rates of aneurysm obliteration and required less aneurysm retreatment, both of which were statistically significant.
The question that remains is not whether to clip or whether to coil, but whom to clip and whom to coil.28 That question must be answered on a patient-to-patient basis and requires the expertise of an interventional neuroradiologist and a vascular neurosurgeon—one of the reasons these patients are best cared for in high-volume centers providing such expertise.
MEDICAL PREVENTION OF REBLEEDING
Blood pressure management
There are no systematic data on the optimal blood pressure before securing an aneurysm. Early studies of hemodynamic augmentation in cases of ruptured untreated aneurysm reported rebleeding when the systolic blood pressure was allowed to rise above 160 mm Hg.29,30 A recent study evaluating hypertensive intracerebral hemorrhage revealed better functional outcomes with intensive lowering of blood pressure (defined as systolic blood pressure < 140 mm Hg) but no significant reduction in the combined rate of death or severe disability.31 It is difficult to know if these results can be extrapolated to patients with aneurysmal subarachnoid hemorrhage. Current guidelines3,32 say that before the aneurysm is treated, the systolic pressure should be lower than 160 mm Hg.
There is no specific drug of choice, but a short-acting, titratable medication is preferable. Nicardipine is a very good option, and labetalol might be an appropriate alternative.33 Once the aneurysm is secured, all antihypertensive drugs should be held. Hypertension should not be treated unless the patient has clinical signs of a hypertensive crisis, such as flash pulmonary edema, myocardial infarction, or hypertensive encephalopathy.
Antifibrinolytic therapy
The role of antifibrinolytic therapy in aneurysmal subarachnoid hemorrhage is controversial and has been studied in 10 clinical trials. In a Swedish study,34 early use of tranexamic acid (1 g intravenously over 10 minutes followed by 1 g every 6 hours for a maximum of 24 hours) reduced the rebleeding rate substantially, from 10.8% to 2.4%, with an 80% reduction in the mortality rate from ultra-early rebleeding. However, a recent Cochrane review that included this study found no overall benefit.35
An ongoing multicenter randomized trial in the Netherlands will, we hope, answer this question in the near future.36 At present, some centers would consider a short course of tranexamic acid before aneurysm treatment.
DIAGNOSIS AND TREATMENT OF COMPLICATIONS
Medical complications are extremely common after aneurysmal subarachnoid hemorrhage. Between 75% and 100% of patients develop some type of systemic or further neurologic derangement, which in turn has a negative impact on the long-term outcome.37,38 In the first 72 hours, rebleeding is the most feared complication, and as mentioned previously, appropriate blood pressure management and early securing of the aneurysm minimize its risk.
NEUROLOGIC COMPLICATIONS
Hydrocephalus
Hydrocephalus is the most common early neurologic complication after aneurysmal subarachnoid hemorrhage, with an overall incidence of 50%.39 Many patients with poor-grade aneurysmal subarachnoid hemorrhage and patients whose condition deteriorates due to worsening of hydrocephalus require the insertion of an external ventricular drain (Figure 1).
Up to 30% of patients who have a poor-grade aneurysmal subarachnoid hemorrhage improve neurologically with cerebrospinal fluid drainage.40 An external ventricular drain can be safely placed, even before aneurysm treatment, and placement does not appear to increase the risk of rebleeding.39,41 After placement, rapid weaning from the drain (clamping within 24 hours of insertion) is safe, decreases length of stay in the intensive care unit and hospital, and may be more cost-effective than gradual weaning over 96 hours.42
Increased intracranial pressure
Intracranial hypertension is another potential early complication, and is frequently due to the development of hydrocephalus, cerebral edema, or rebleeding. The treatment of increased intracranial pressure does not differ from the approach used in managing severe traumatic brain injury, which includes elevating the head of the bed, sedation, analgesia, normoventilation, and cerebrospinal fluid drainage.
Hypertonic saline has been tested in several studies that were very small but nevertheless consistently showed control of intracranial pressure levels and improvement in cerebral blood flow measured by xenon CT.43–47 Two of these studies even showed better outcomes at discharge.43,44 However, the small number of patients prevents any meaningful conclusion regarding the use of hypertonic saline and functional outcomes.
Barbiturates, hypothermia, and decompressive craniectomy could be tried in refractory cases.48 Seule et al49 evaluated the role of therapeutic hypothermia with or without barbiturate coma in 100 patients with refractory intracranial hypertension. Only 13 patients received hypothermia by itself. At 1 year, 32 patients had achieved a good functional outcome (Glasgow Outcome Scale score 4 or 5). The remaining patients were severely disabled or had died. Of interest, the median duration of hypothermia was 7 days, and 93% of patients developed some medical complication such as electrolyte disorders (77%), pneumonia (52%), thrombocytopenia (47%), or septic shock syndrome (40%). Six patients died as a consequence of one of these complications.
Decompressive craniectomy can be life-saving in patients with refractory intracranial hypertension. However, most of these patients will die or remain severely disabled or comatose.50
Seizure prophylaxis is controversial
Seizures can occur at the onset of intracranial hemorrhage, perioperatively, or later (ie, after the first week). The incidence varied considerably in different reports, ranging from 4% to 26%.51 Seizures occurring perioperatively, ie, after hospital admission, are less frequent and are usually the manifestation of aneurysm rebleeding.24
Seizure prophylaxis remains controversial, especially because the use of phenytoin is associated with increased incidence of cerebral vasospasm, infarction, and worse cognitive outcomes after aneurysmal subarachnoid hemorrhage.52,53 Therefore, routine prophylactic use of phenytoin is not recommended in these patients,3 although the effect of other antiepileptic drugs is less studied and less clear. Patients may be considered for this therapy if they have multiple risk factors for seizures, such as intraparenchymal hematoma, advanced age (> 65), middle cerebral artery aneurysm, craniotomy for aneurysm clipping, and a short course (≤ 72 hours) of an antiepileptic drug other than phenytoin, especially while the aneurysm is unsecured.3
Levetiracetam may be an alternative to phenytoin, having better pharmacodynamic and kinetic profiles, minimal protein binding, and absence of hepatic metabolism, resulting in a very low risk of drug interaction and better tolerability.54,55 Because of these advantages, levetiracetam has become the drug of choice in several centers treating aneurysmal subarachnoid hemorrhage in the United States.
Addressing this question, a survey was sent to 25 high-volume aneurysmal subarachnoid hemorrhage academic centers in the United States. All 25 institutions answered the survey, and interestingly, levetiracetam was the first-line agent for 16 (94%) of the 17 responders that used prophylaxis, while only 1 used phenytoin as the agent of choice.56
A retrospective cohort study by Murphy-Human et al57 showed that a short course of levetiracetam (≤ 72 hours) was associated with higher rates of in-hospital seizures compared with an extended course of phenytoin (eg, entire hospital stay). However, the study did not address functional outcomes.57
Continuous electroencephalographic monitoring may be considered in comatose patients, in patients requiring controlled ventilation and sedation, or in patients with unexplained alteration in consciousness. In one series of patients with aneurysmal subarachnoid hemorrhage who received continuous monitoring, the incidence of nonconvulsive status epilepticus was 19%, with an associated mortality rate of 100%.58
Continuous quantitative electroencephalography is useful to monitor and to detect angiographic vasospasm and delayed cerebral ischemia. Relative alpha variability and the alpha-delta ratio decrease with ischemia, and this effect can precede angiographic vasospasm by 3 days.59,60
Delayed cerebral ischemia
Delayed cerebral ischemia is defined as the occurrence of focal neurologic impairment, or a decrease of at least 2 points on the Glasgow Coma Scale that lasts for at least 1 hour, is not apparent immediately after aneurysm occlusion, and not attributable to other causes (eg, hyponatremia, fever).61
Classically, neurologic deficits that occurred within 2 weeks of aneurysm rupture were ascribed to reduced cerebral blood flow caused by delayed large-vessel vasospasm causing cerebral ischemia.62 However, perfusion abnormalities have also been observed with either mild or no demonstrable vasospasm.63 Almost 70% of patients who survive the initial hemorrhage develop some degree of angiographic vasospasm. However, only 30% of those patients will experience symptoms.
In addition to vasospasm of large cerebral arteries, impaired autoregulation and early brain injury within the first 72 hours following subarachnoid hemorrhage may play important roles in the development of delayed cerebral ischemia.64 Therefore, the modern concept of delayed cerebral ischemia monitoring should focus on cerebral perfusion rather than vessel diameter measurements. This underscores the importance of comprehensive, standardized monitoring techniques that provide information not only on microvasculature, but also at the level of the microcirculation, with information on perfusion, oxygen utilization and extraction, and autoregulation.
Although transcranial Doppler has been the most commonly applied tool to monitor for angiographic vasospasm, it has a low sensitivity and negative predictive value.37 It is nevertheless a useful technique to monitor good-grade aneurysmal subarachnoid hemorrhage patients (WFNS score 1–3) combined with frequent neurologic examinations (Figure 3).
CT angiography is a good noninvasive alternative to digital subtraction angiography. However, it tends to overestimate the degree of vasoconstriction and does not provide information about perfusion and autoregulation.65 Nevertheless, CT angiography combined with a CT perfusion scan can add information about autoregulation and cerebral perfusion and has been shown to be more sensitive for the diagnosis of angiographic vasospasm than transcranial Doppler and digital subtraction angiography (Figure 4).
Patients with a poor clinical condition (WFNS score 4 or 5) or receiving continuous sedation constitute a challenge in monitoring for delayed neurologic deterioration. Neurologic examination is not sensitive enough in this setting to detect subtle changes. In these specific and challenging circumstances, multimodality neuromonitoring may be useful in the early detection of delayed cerebral ischemia and may help guide therapy.67
Several noninvasive and invasive techniques have been studied to monitor patients at risk of delayed cerebral ischemia after subarachnoid hemorrhage.66 These include continuous electroencephalography, brain tissue oxygenation monitoring (Ptio2), cerebral microdialysis, thermal diffusion flowmetry, and near-infrared spectroscopy. Of these techniques, Ptio2, cerebral microdialysis, and continuous electroencephalography (see discussion of seizure prophylaxis above) have been more extensively studied. However, most of the studies were observational and very small, limiting any recommendations for using these techniques in routine clinical practice.68
Ptio2 is measured by inserting an intraparenchymal oxygen-sensitive microelectrode, and microdialysis requires a microcatheter with a semipermeable membrane that allows small soluble substances to cross it into the dialysate. These substances, which include markers of ischemia (ie, glucose, lactate, and pyruvate), excitotoxins (ie, glutamate and aspartate), and membrane cell damage products (ie, glycerol), can be measured. Low Ptio2 values (< 15 mm Hg) and abnormal mycrodialysate findings (eg, glucose < 0.8 mmol/L, lactate-to-pyruvate ratio > 40) have both been associated with cerebral ischemic events and poor outcome.68
Preventing delayed cerebral ischemia
Oral nimodipine 60 mg every 4 hours for 21 days, started on admission, carries a class I, level of evidence A recommendation in the management of aneurysmal subarachnoid hemorrhage.3,32,69 It improves clinical outcome despite having no effect on the risk of angiographic vasospasm. The mechanism of improved outcome is unclear, but the effect may be a neuroprotective phenomenon limiting the extension of delayed cerebral ischemia.70
If hypotension occurs, the dose can be lowered to 30 mg every 2 hours. Whether to discontinue nimodipine in this situation is controversial. Of note, the clinical benefits of nimodipine have not been replicated with other calcium channel blockers (eg, nicardipine).71
Prophylactic hyperdynamic fluid therapy, known as “triple-H” (hypervolemia, hemodilution, and hypertension) was for years the mainstay of treatment in preventing delayed cerebral ischemia due to vasospasm. However, the clinical data supporting this intervention have been called into question, as analysis of two trials found that hypervolemia did not improve outcomes or reduce the incidence of delayed cerebral ischemia, and in fact increased the rate of complications.72,73 Based on these findings, current guidelines recommend maintaining euvolemia rather than prophylactic hypervolemia in patients with aneurysmal subarachnoid hemorrhage.3,32,69
TREATING DELAYED CEREBRAL ISCHEMIA
Hemodynamic augmentation
In patients with neurologic deterioration due to delayed cerebral ischemia, hemodynamic augmentation is the cornerstone of treatment. This is done according to a protocol, started early, involving specific physiologic goals, clinical improvement, and escalation to invasive therapies in a timely fashion in patients at high risk of further neurologic insult (Figure 5).
The physiologic goal is to increase the delivery of oxygen and glucose to the ischemic brain. Hypertension seems to be the most effective component of hemodynamic augmentation regardless of volume status, increasing cerebral blood flow and brain tissue oxygenation, with reversal of delayed cerebral ischemic symptoms in up to two-thirds of treated patients.74,75 However, this information comes from very small studies, with no randomized trials of induced hypertension available.
The effect of a normal saline fluid bolus in patients suspected of having delayed cerebral ischemia has been shown to increase cerebral blood flow in areas of cerebral ischemia.74 If volume augmentation fails to improve the neurologic status, the next step is to artificially induce hypertension using vasopressors. The blood pressure target should be based on clinical improvement. A stepwise approach is reasonable in this situation, and the lowest level of blood pressure at which there is a complete reversal of the new focal neurologic deficit should be maintained.3,29
Inotropic agents such as dobutamine or milrinone can be considered as alternatives in patients who have new neurologic deficits that are refractory to fluid boluses and vasopressors, or in a setting of subarachnoid hemorrhage-induced cardiomyopathy.76,77
Once the neurologic deficit is reversed by hemodynamic augmentation, the blood pressure should be maintained for 48 to 72 hours at the level that reversed the deficit completely, carefully reassessed thereafter, and the patient weaned slowly. Unruptured unsecured aneurysms should not prevent blood pressure augmentation in a setting of delayed cerebral ischemia if the culprit aneurysm is treated.3,32 If the ruptured aneurysm has not been secured, careful blood pressure augmentation can be attempted, keeping in mind that hypertension (> 160/95 mm Hg) is a risk factor for fatal aneurysm rupture.
Endovascular management of delayed cerebral ischemia
When medical augmentation fails to completely reverse the neurologic deficits, endovascular treatment can be considered. Although patients treated early in the course of delayed cerebral ischemia have better neurologic recovery, prophylactic endovascular treatment in asymptomatic patients, even if angiographic signs of spasm are present, does not improve clinical outcomes and carries the risk of fatal arterial rupture.78
SYSTEMIC COMPLICATIONS
Hyponatremia and hypovolemia
Aneurysmal subarachnoid hemorrhage is commonly associated with abnormalities of fluid balance and electrolyte derangements. Hyponatremia (serum sodium < 135 mmol/L) occurs in 30% to 50% of patients, while the rate of hypovolemia (decreased circulating blood volume) ranges from 17% to 30%.79 Both can negatively affect long-term outcomes.80,81
Decreased circulating blood volume is a well-described contributor to delayed cerebral ischemia and cerebral infarction after aneurysmal subarachnoid hemorrhage.80–82 Clinical variables such as heart rate, blood pressure, fluid balance, and serum sodium concentration are usually the cornerstones of intravascular volume status assessment. However, these variables correlate poorly with measured circulating blood volumes in those with aneurysmal subarachnoid hemorrhage.83,84
The mechanisms responsible for the development of hyponatremia and hypovolemia after aneurysmal subarachnoid hemorrhage are not completely understood. Several factors have been described and may contribute to the increased natriuresis and, hence, to a reduction in circulating blood volume: increased circulating natriuretic peptide concentrations,85–87 sympathetic nervous system hyperactivation,88 and hyperreninemic hypo-
aldosteronism syndrome.89,90
Lastly, the cerebral salt wasting syndrome, described in the 1950s,91 was thought to be a key mechanism in the development of hyponatremia and hypovolemia after aneurysmal subarachnoid hemorrhage. In contrast to the syndrome of inappropriate antidiuretic hormone, which is characterized by hyponatremia with a normal or slightly elevated intravascular volume, the characteristic feature of cerebral salt wasting syndrome is the development of hyponatremia in a setting of intravascular volume depletion.92 In critically ill neurologic and neurosurgical patients, this differential diagnosis is very difficult, especially in those with aneurysmal subarachnoid hemorrhage in whom the clinical assessment of fluid status is not reliable. These two syndromes might coexist and contribute to the development of hyponatremia after aneurysmal subarachnoid hemorrhage.92,93
Hoff et al83,84 prospectively compared the clinical assessment of fluid status by critical and intermediate care nurses and direct measurements of blood volume using pulse dye densitometry. The clinical assessment failed to accurately assess patients’ volume status. Using the same technique to measure circulating blood volume, this group showed that calculation of fluid balance does not provide adequate assessment of fluid status.83,84
Hemodynamic monitoring tools can help guide fluid replacement in this population. Mutoh et al94 randomized 160 patients within 24 hours of hemorrhage to receive early goal-directed fluid therapy (ie, preload volume and cardiac output monitored by transpulmonary thermodilution) vs standard therapy (ie, fluid balance or central venous pressure). Overall, no difference was found in the rates of delayed cerebral ischemia (33% vs 42%; P = .33) or favorable outcome (67% vs 57%; P = .22). However, in the subgroup of poor-grade patients (WFNS score 4 or 5), early goal-directed therapy was associated with a lower rate of delayed cerebral ischemia (5% vs 14%; P = .036) and with better functional outcomes at 3 months (52% vs 36%; P = .026).
Fluid restriction to treat hyponatremia in aneurysmal subarachnoid hemorrhage is no longer recommended because of the increased risk of cerebral infarction due to hypovolemic hypoperfusion.82
Prophylactic use of mineralocorticoids (eg, fludrocortisone, hydrocortisone) has been shown to limit natriuresis, hyponatremia, and the amount of fluid required to maintain euvolemia.95,96 Higher rates of hypokalemia and hyperglycemia, which can be easily treated, are the most common complications associated with this approach. Additionally, hypertonic saline (eg, 3% saline) can be used to correct hyponatremia in a setting of aneurysmal subarachnoid hemorrhage.79
Cardiac complications
Cardiac complications after subarachnoid hemorrhage are most likely related to sympathetic hyperactivity and catecholamine-induced myocyte dysfunction. The pathophysiology is complex, but cardiac complications have a significant negative impact on long-term outcome in these patients.97
Electrocardiographic changes and positive cardiac enzymes associated with aneurysmal subarachnoid hemorrhage have been extensively reported. More recently, data from studies of two-dimensional echocardiography have shown that subarachnoid hemorrhage can also be associated with significant wall-motion abnormalities and even overt cardiogenic shock.98–100
There is no specific curative therapy; the treatment is mainly supportive. Vasopressors and inotropes may be used for hemodynamic augmentation.
Pulmonary complications
Pulmonary complications occur in 20% to 30% of all aneurysmal subarachnoid hemorrhage patients and are associated with a higher risk of delayed cerebral ischemia and death. Common pulmonary complications in this population are mild acute respiratory distress syndrome (27%), hospital-acquired pneumonia (9%), cardiogenic pulmonary edema (8%), aspiration pneumonia (6%), neurogenic pulmonary edema (2%), and pulmonary embolism (1%).101–103
SUPPORTIVE CARE
Hyperthermia, hyperglycemia, and liberal use of transfusions have all been associated with longer stays in the intensive care unit and hospital, poorer neurologic outcomes, and higher mortality rates in patients with acute brain injury.104 Noninfectious fever is the most common systemic complication after subarachnoid hemorrhage.
Antipyretic drugs such as acetaminophen and ibuprofen are not very effective in reducing fever in the subarachnoid hemorrhage population, but should still be used as first-line therapy. The use of surface and intravascular devices can be considered when fevers do not respond to nonsteroidal anti-inflammatory drugs.
Although no prospective randomized trial has addressed the impact of induced normothermia on long-term outcome and mortality in aneurysmal subarachnoid hemorrhage patients, fever control has been shown to reduce cerebral metabolic distress, irrespective of intracranial pressure.105 Maintenance of normothermia (< 37.5°C) seems reasonable, especially in aneurysmal subarachnoid hemorrhage patients at risk of or with active delayed cerebral ischemia.106
Current guidelines3,32,69 strongly recommend avoiding hypoglycemia, defined as a serum glucose level less than 80 mg/dL, but suggest keeping the blood sugar level below 180 or 200 mg/dL.
At the moment, there is no clear threshold for transfusion in patients with aneurysmal subarachnoid hemorrhage. Current guidelines suggest keeping hemoglobin levels between 8 and 10 g/dL.3
Preventing venous thromboembolism
The incidence of venous thromboembolism after aneurysmal subarachnoid hemorrhage varies widely, from 1.5% to 18%.107 Active surveillance with venous Doppler ultrasonography has found asymptomatic deep vein thrombosis in up to 3.4% of poor-grade aneurysmal subarachnoid hemorrhage patients receiving pharmacologic thromboprophylaxis.108
In a retrospective study of 170 patients, our group showed that giving drugs to prevent venous thromboembolism (unfractionated heparin 5,000 IU subcutaneously every 12 hours or dalteparin 5,000 IU subcutaneously daily), starting within 24 hours of aneurysm treatment, could be safe.109 Fifty-eight percent of these patients had an external ventricular drain in place. One patient developed a major cerebral hemorrhagic complication and died while on unfractionated heparin; however, the patient was also on dual antiplatelet therapy with aspirin and clopidogrel.109
Current guidelines suggest that intermittent compression devices be applied in all patients before aneurysm treatment. Pharmacologic thromboprophylaxis with a heparinoid can be started 12 to 24 hours after aneurysm treatment.3,109
A TEAM APPROACH
Patients with subarachnoid hemorrhage need integrated care from different medical and nursing specialties. The best outcomes are achieved by systems that can focus as a team on the collective goal of quick intervention to secure the aneurysm, followed by measures to minimize secondary brain injury.
The modern concept of cerebral monitoring in a setting of subarachnoid hemorrhage should focus on brain perfusion rather than vascular diameter. Although the search continues for new diagnostic, prognostic, and therapeutic tools, there is no “silver bullet” that will help all patients. Instead, it is the systematic integration and application of many small advances that will ultimately lead to better outcomes.
ACKNOWLEDGMENT
This work was supported by research funding provided by the Bitove Foundation, which has been supportive of our clinical and research work for several years.
Aneurysmal subarachnoid hemorrhage is a devastating condition, with an estimated death rate of 30% during the initial episode.1,2 Approximately the same number of patients survive but leave the hospital with disabling neurologic deficits.3
However, better outcomes can be achieved by systems that are able to work as a team on the collective goal of quick intervention to secure the ruptured aneurysm, followed by the implementation of measures to minimize secondary brain injury. Although the search for new diagnostic, prognostic, and therapeutic modalities continues, it is clear that there exists no “silver bullet” that will help all patients. Instead, it is the systematic integration and application of small advances that will ultimately maximize the patient’s chances of survival and neurologic recovery.
This review focuses on the management of aneurysmal subarachnoid hemorrhage and its systemic and neurologic complications.
ANEURYSM IS THE MOST COMMON CAUSE OF SUBARACHNOID BLEEDING
Aneurysmal subarachnoid hemorrhage, ie, rupture of an intracranial aneurysm, flooding the subarachnoid space with blood, affects about 24,000 Americans each year.1,2 A ruptured aneurysm is the most common cause of subarachnoid hemorrhage, accounting for about 85% of cases. Less common causes include idiopathic benign perimesencephalic hemorrhage, arteriovenous malformation, dural arteriovenous fistula, and hemorrhagic mycotic aneurysm. These have their own natural history, pathophysiology, and specific treatment, and will not be addressed in this article.
Risk factors for aneurysmal subarachnoid hemorrhage include having a first-degree relative who had the disease, hypertension, smoking, and consuming more than 150 g of alcohol per week.4
CLINICAL PRESENTATION AND DIAGNOSIS
The key symptom of aneurysmal subarachnoid hemorrhage is the abrupt onset of severe headache that peaks in intensity over 1 hour,5 often described as “the worst headache of my life.” Headache is accompanied by brief loss of consciousness in 53% of cases (conversely, nearly half of patients maintain normal mental status), by nausea or vomiting in 77%, and by meningismus (neck pain or stiffness) in 35%.6
These clinical manifestations and risk factors have been incorporated into a decision rule:
Obtain brain imaging if the patient has acute headache reaching maximal intensity within 1 hour, associated with any of the following factors:
- Age 40 or older
- Neck pain or stiffness
- Witnessed loss of consciousness
- Onset during exertion
- “Thunderclap” headache (ie, instantly peaking pain)
- Limited neck flexion on examination.5
This decision rule has nearly 100% sensitivity for aneurysmal subarachnoid hemorrhage in clinical practice.5 All patients require brain imaging if they have a severe headache plus either abnormal neurologic findings (eg, a focal neurologic deficit) or a history of cerebral aneurysm.
Emergency physicians should have a low threshold for ordering noncontrast computed tomography (CT) of the head in patients with even mild symptoms suggesting aneurysmal subarachnoid hemorrhage. Failure to order CT is the most common diagnostic error in this situation.6 CT performed within 6 hours of headache onset is nearly 100% sensitive for this condition,7 but the sensitivity falls to 93% after the first 24 hours and to less than 60% after 5 days.8 In patients who have symptoms highly suggestive of aneurysmal subarachnoid hemorrhage but a normal CT, lumbar puncture is the next diagnostic step.
There are two alternatives to CT followed by lumbar puncture: ie, noncontrast CT followed by CT angiography,9,10 and magnetic resonance imaging followed by magnetic resonance angiography. In patients with suspicious clinical symptoms but negative CT results, CT followed by CT angiography can rule out aneurysmal subarachnoid hemorrhage with a 99% probability.9,10 However, CT followed by lumbar puncture remains the standard of care and carries a class I recommendation in the American Heart Association guidelines for ruling out subarachnoid hemorrhage.5
GRADING THE SEVERITY OF SUBARACHNOID HEMORRHAGE
Age, the thickness of the blood layer in the subarachnoid space, intraventricular hemorrhage and the findings of the neurologic examination at presentation are predictors of long-term outcomes in aneurysmal subarachnoid hemorrhage (Figure 1).
Different grading systems used in clinical practice are based on the findings on the initial neurologic examination and on the initial noncontrast CT (ie, the thickness of the blood, and whether intraventricular hemorrhage is present). Among the most widely used are those developed by Hunt and Hess12 and by the World Federation of Neurological Surgeons11 (WFNS), and the CT grading scales (Fisher13 or its modified version14) (Tables 1 and 2). With either the Hunt and Hess scale or the WFNS scale, the higher the score, the worse the patient’s probable outcome. Scores on both Fisher scales correlate with the risk of angiographic vasospasm. The higher the grade, the higher the risk of angiographic vasospasm.
The VASOGRADE score—a combination of the WFNS score and the modified Fisher scale—stratifies patients at risk of delayed cerebral ischemia, allowing for a tailored monitoring strategy.15 There are three variations:
- VASOGRADE green—Modified Fisher 1 or 2 and WFNS 1 or 2
- VASOGRADE yellow—Modified Fisher 3 or 4 and WFNS 1, 2, or 3
- VASOGRADE red—WFNS 4 or 5.
After the initial bleeding event, patients with aneurysmal subarachnoid hemorrhage are at high risk of delayed systemic and neurologic complications, with poor functional outcomes. Delayed cerebral ischemia holds the greatest risk of an unfavorable outcome and ultimately can lead to cerebral infarction, disability, and death.6,7
INITIAL MANAGEMENT
After aneurysmal subarachnoid hemorrhage is diagnosed, the initial management (Figure 2) includes appropriate medical prevention of rebleeding (which includes supportive care, blood pressure management, and, perhaps, the early use of a short course of an antifibrinolytic drug) and early transfer to a high-volume center for securing the aneurysm. The reported incidence of rebleeding varies from 5% to 22% in the first 72 hours. “Ultra-early” rebleeding (within 24 hours of hemorrhage) has been reported, with an incidence as high as 15% and a fatality rate around 70%. Patients with poor-grade aneurysmal subarachnoid hemorrhage, larger aneurysms, and “sentinel bleeds” are at higher risk of rebleeding.16
Outcomes are much better when patients are managed in a high-volume center, with a specialized neurointensive care unit17 and access to an interdisciplinary team.18 Regardless of the initial grade, patients with aneurysmal subarachnoid hemorrhage should be quickly transferred to a high-volume center, defined as one treating at least 35 cases per year, and the benefit is greater in centers treating more than 60 cases per year.19 The higher the caseload in any given hospital, the better the clinical outcomes in this population.20
Treating cerebral aneurysm: Clipping or coiling
Early aneurysm repair is generally considered the standard of care and the best strategy to reduce the risk of rebleeding. Further, early treatment may be associated with a lower risk of delayed cerebral ischemia21 and better outcomes.22
Three randomized clinical trials have compared surgical clipping and endovascular repair (placement of small metal coils within the aneurysm to promote clotting).
The International Subarachnoid Aneurysm Trial23 showed a reduction of 23% in relative risk and of 7% in absolute risk in patients who underwent endovascular treatment compared with surgery. The survival benefit persisted at a mean of 9 years (range 6–14 years), but with a higher annual rate of aneurysm recurrence in the coiling group (2.9% vs 0.9%).24 Of note, this trial included only patients with aneurysms deemed suitable for both coiling and clipping, so that the exclusion rate was high. Most of the patients presented with good-grade (WFNS score 1–3), small aneurysms (< 5 mm) in the anterior circulation.
A single-center Finnish study25 found no differences in rates of recovery, disability, and death at 1 year, comparing surgery and endovascular treatment. Additionally, survival rates at a mean follow-up of 39 months were similar, with no late recurrences or aneurysmal bleeding.
Lastly, the Barrow Ruptured Aneurysm Trial26,27 found that patients assigned to endovascular treatment had better 1-year neurologic outcomes, defined as a modified Rankin score of 2 or less. Importantly, 37.7% of patients originally assigned to endovascular treatment crossed over to surgical treatment. The authors then performed intention-to-treat and as-treated analyses. Either way, patients treated by endovascular means had better neurologic outcomes at 1 year. However, no difference in the relative risk reduction in worse outcome was found on 3-year follow-up, and patients treated surgically had higher rates of aneurysm obliteration and required less aneurysm retreatment, both of which were statistically significant.
The question that remains is not whether to clip or whether to coil, but whom to clip and whom to coil.28 That question must be answered on a patient-to-patient basis and requires the expertise of an interventional neuroradiologist and a vascular neurosurgeon—one of the reasons these patients are best cared for in high-volume centers providing such expertise.
MEDICAL PREVENTION OF REBLEEDING
Blood pressure management
There are no systematic data on the optimal blood pressure before securing an aneurysm. Early studies of hemodynamic augmentation in cases of ruptured untreated aneurysm reported rebleeding when the systolic blood pressure was allowed to rise above 160 mm Hg.29,30 A recent study evaluating hypertensive intracerebral hemorrhage revealed better functional outcomes with intensive lowering of blood pressure (defined as systolic blood pressure < 140 mm Hg) but no significant reduction in the combined rate of death or severe disability.31 It is difficult to know if these results can be extrapolated to patients with aneurysmal subarachnoid hemorrhage. Current guidelines3,32 say that before the aneurysm is treated, the systolic pressure should be lower than 160 mm Hg.
There is no specific drug of choice, but a short-acting, titratable medication is preferable. Nicardipine is a very good option, and labetalol might be an appropriate alternative.33 Once the aneurysm is secured, all antihypertensive drugs should be held. Hypertension should not be treated unless the patient has clinical signs of a hypertensive crisis, such as flash pulmonary edema, myocardial infarction, or hypertensive encephalopathy.
Antifibrinolytic therapy
The role of antifibrinolytic therapy in aneurysmal subarachnoid hemorrhage is controversial and has been studied in 10 clinical trials. In a Swedish study,34 early use of tranexamic acid (1 g intravenously over 10 minutes followed by 1 g every 6 hours for a maximum of 24 hours) reduced the rebleeding rate substantially, from 10.8% to 2.4%, with an 80% reduction in the mortality rate from ultra-early rebleeding. However, a recent Cochrane review that included this study found no overall benefit.35
An ongoing multicenter randomized trial in the Netherlands will, we hope, answer this question in the near future.36 At present, some centers would consider a short course of tranexamic acid before aneurysm treatment.
DIAGNOSIS AND TREATMENT OF COMPLICATIONS
Medical complications are extremely common after aneurysmal subarachnoid hemorrhage. Between 75% and 100% of patients develop some type of systemic or further neurologic derangement, which in turn has a negative impact on the long-term outcome.37,38 In the first 72 hours, rebleeding is the most feared complication, and as mentioned previously, appropriate blood pressure management and early securing of the aneurysm minimize its risk.
NEUROLOGIC COMPLICATIONS
Hydrocephalus
Hydrocephalus is the most common early neurologic complication after aneurysmal subarachnoid hemorrhage, with an overall incidence of 50%.39 Many patients with poor-grade aneurysmal subarachnoid hemorrhage and patients whose condition deteriorates due to worsening of hydrocephalus require the insertion of an external ventricular drain (Figure 1).
Up to 30% of patients who have a poor-grade aneurysmal subarachnoid hemorrhage improve neurologically with cerebrospinal fluid drainage.40 An external ventricular drain can be safely placed, even before aneurysm treatment, and placement does not appear to increase the risk of rebleeding.39,41 After placement, rapid weaning from the drain (clamping within 24 hours of insertion) is safe, decreases length of stay in the intensive care unit and hospital, and may be more cost-effective than gradual weaning over 96 hours.42
Increased intracranial pressure
Intracranial hypertension is another potential early complication, and is frequently due to the development of hydrocephalus, cerebral edema, or rebleeding. The treatment of increased intracranial pressure does not differ from the approach used in managing severe traumatic brain injury, which includes elevating the head of the bed, sedation, analgesia, normoventilation, and cerebrospinal fluid drainage.
Hypertonic saline has been tested in several studies that were very small but nevertheless consistently showed control of intracranial pressure levels and improvement in cerebral blood flow measured by xenon CT.43–47 Two of these studies even showed better outcomes at discharge.43,44 However, the small number of patients prevents any meaningful conclusion regarding the use of hypertonic saline and functional outcomes.
Barbiturates, hypothermia, and decompressive craniectomy could be tried in refractory cases.48 Seule et al49 evaluated the role of therapeutic hypothermia with or without barbiturate coma in 100 patients with refractory intracranial hypertension. Only 13 patients received hypothermia by itself. At 1 year, 32 patients had achieved a good functional outcome (Glasgow Outcome Scale score 4 or 5). The remaining patients were severely disabled or had died. Of interest, the median duration of hypothermia was 7 days, and 93% of patients developed some medical complication such as electrolyte disorders (77%), pneumonia (52%), thrombocytopenia (47%), or septic shock syndrome (40%). Six patients died as a consequence of one of these complications.
Decompressive craniectomy can be life-saving in patients with refractory intracranial hypertension. However, most of these patients will die or remain severely disabled or comatose.50
Seizure prophylaxis is controversial
Seizures can occur at the onset of intracranial hemorrhage, perioperatively, or later (ie, after the first week). The incidence varied considerably in different reports, ranging from 4% to 26%.51 Seizures occurring perioperatively, ie, after hospital admission, are less frequent and are usually the manifestation of aneurysm rebleeding.24
Seizure prophylaxis remains controversial, especially because the use of phenytoin is associated with increased incidence of cerebral vasospasm, infarction, and worse cognitive outcomes after aneurysmal subarachnoid hemorrhage.52,53 Therefore, routine prophylactic use of phenytoin is not recommended in these patients,3 although the effect of other antiepileptic drugs is less studied and less clear. Patients may be considered for this therapy if they have multiple risk factors for seizures, such as intraparenchymal hematoma, advanced age (> 65), middle cerebral artery aneurysm, craniotomy for aneurysm clipping, and a short course (≤ 72 hours) of an antiepileptic drug other than phenytoin, especially while the aneurysm is unsecured.3
Levetiracetam may be an alternative to phenytoin, having better pharmacodynamic and kinetic profiles, minimal protein binding, and absence of hepatic metabolism, resulting in a very low risk of drug interaction and better tolerability.54,55 Because of these advantages, levetiracetam has become the drug of choice in several centers treating aneurysmal subarachnoid hemorrhage in the United States.
Addressing this question, a survey was sent to 25 high-volume aneurysmal subarachnoid hemorrhage academic centers in the United States. All 25 institutions answered the survey, and interestingly, levetiracetam was the first-line agent for 16 (94%) of the 17 responders that used prophylaxis, while only 1 used phenytoin as the agent of choice.56
A retrospective cohort study by Murphy-Human et al57 showed that a short course of levetiracetam (≤ 72 hours) was associated with higher rates of in-hospital seizures compared with an extended course of phenytoin (eg, entire hospital stay). However, the study did not address functional outcomes.57
Continuous electroencephalographic monitoring may be considered in comatose patients, in patients requiring controlled ventilation and sedation, or in patients with unexplained alteration in consciousness. In one series of patients with aneurysmal subarachnoid hemorrhage who received continuous monitoring, the incidence of nonconvulsive status epilepticus was 19%, with an associated mortality rate of 100%.58
Continuous quantitative electroencephalography is useful to monitor and to detect angiographic vasospasm and delayed cerebral ischemia. Relative alpha variability and the alpha-delta ratio decrease with ischemia, and this effect can precede angiographic vasospasm by 3 days.59,60
Delayed cerebral ischemia
Delayed cerebral ischemia is defined as the occurrence of focal neurologic impairment, or a decrease of at least 2 points on the Glasgow Coma Scale that lasts for at least 1 hour, is not apparent immediately after aneurysm occlusion, and not attributable to other causes (eg, hyponatremia, fever).61
Classically, neurologic deficits that occurred within 2 weeks of aneurysm rupture were ascribed to reduced cerebral blood flow caused by delayed large-vessel vasospasm causing cerebral ischemia.62 However, perfusion abnormalities have also been observed with either mild or no demonstrable vasospasm.63 Almost 70% of patients who survive the initial hemorrhage develop some degree of angiographic vasospasm. However, only 30% of those patients will experience symptoms.
In addition to vasospasm of large cerebral arteries, impaired autoregulation and early brain injury within the first 72 hours following subarachnoid hemorrhage may play important roles in the development of delayed cerebral ischemia.64 Therefore, the modern concept of delayed cerebral ischemia monitoring should focus on cerebral perfusion rather than vessel diameter measurements. This underscores the importance of comprehensive, standardized monitoring techniques that provide information not only on microvasculature, but also at the level of the microcirculation, with information on perfusion, oxygen utilization and extraction, and autoregulation.
Although transcranial Doppler has been the most commonly applied tool to monitor for angiographic vasospasm, it has a low sensitivity and negative predictive value.37 It is nevertheless a useful technique to monitor good-grade aneurysmal subarachnoid hemorrhage patients (WFNS score 1–3) combined with frequent neurologic examinations (Figure 3).
CT angiography is a good noninvasive alternative to digital subtraction angiography. However, it tends to overestimate the degree of vasoconstriction and does not provide information about perfusion and autoregulation.65 Nevertheless, CT angiography combined with a CT perfusion scan can add information about autoregulation and cerebral perfusion and has been shown to be more sensitive for the diagnosis of angiographic vasospasm than transcranial Doppler and digital subtraction angiography (Figure 4).
Patients with a poor clinical condition (WFNS score 4 or 5) or receiving continuous sedation constitute a challenge in monitoring for delayed neurologic deterioration. Neurologic examination is not sensitive enough in this setting to detect subtle changes. In these specific and challenging circumstances, multimodality neuromonitoring may be useful in the early detection of delayed cerebral ischemia and may help guide therapy.67
Several noninvasive and invasive techniques have been studied to monitor patients at risk of delayed cerebral ischemia after subarachnoid hemorrhage.66 These include continuous electroencephalography, brain tissue oxygenation monitoring (Ptio2), cerebral microdialysis, thermal diffusion flowmetry, and near-infrared spectroscopy. Of these techniques, Ptio2, cerebral microdialysis, and continuous electroencephalography (see discussion of seizure prophylaxis above) have been more extensively studied. However, most of the studies were observational and very small, limiting any recommendations for using these techniques in routine clinical practice.68
Ptio2 is measured by inserting an intraparenchymal oxygen-sensitive microelectrode, and microdialysis requires a microcatheter with a semipermeable membrane that allows small soluble substances to cross it into the dialysate. These substances, which include markers of ischemia (ie, glucose, lactate, and pyruvate), excitotoxins (ie, glutamate and aspartate), and membrane cell damage products (ie, glycerol), can be measured. Low Ptio2 values (< 15 mm Hg) and abnormal mycrodialysate findings (eg, glucose < 0.8 mmol/L, lactate-to-pyruvate ratio > 40) have both been associated with cerebral ischemic events and poor outcome.68
Preventing delayed cerebral ischemia
Oral nimodipine 60 mg every 4 hours for 21 days, started on admission, carries a class I, level of evidence A recommendation in the management of aneurysmal subarachnoid hemorrhage.3,32,69 It improves clinical outcome despite having no effect on the risk of angiographic vasospasm. The mechanism of improved outcome is unclear, but the effect may be a neuroprotective phenomenon limiting the extension of delayed cerebral ischemia.70
If hypotension occurs, the dose can be lowered to 30 mg every 2 hours. Whether to discontinue nimodipine in this situation is controversial. Of note, the clinical benefits of nimodipine have not been replicated with other calcium channel blockers (eg, nicardipine).71
Prophylactic hyperdynamic fluid therapy, known as “triple-H” (hypervolemia, hemodilution, and hypertension) was for years the mainstay of treatment in preventing delayed cerebral ischemia due to vasospasm. However, the clinical data supporting this intervention have been called into question, as analysis of two trials found that hypervolemia did not improve outcomes or reduce the incidence of delayed cerebral ischemia, and in fact increased the rate of complications.72,73 Based on these findings, current guidelines recommend maintaining euvolemia rather than prophylactic hypervolemia in patients with aneurysmal subarachnoid hemorrhage.3,32,69
TREATING DELAYED CEREBRAL ISCHEMIA
Hemodynamic augmentation
In patients with neurologic deterioration due to delayed cerebral ischemia, hemodynamic augmentation is the cornerstone of treatment. This is done according to a protocol, started early, involving specific physiologic goals, clinical improvement, and escalation to invasive therapies in a timely fashion in patients at high risk of further neurologic insult (Figure 5).
The physiologic goal is to increase the delivery of oxygen and glucose to the ischemic brain. Hypertension seems to be the most effective component of hemodynamic augmentation regardless of volume status, increasing cerebral blood flow and brain tissue oxygenation, with reversal of delayed cerebral ischemic symptoms in up to two-thirds of treated patients.74,75 However, this information comes from very small studies, with no randomized trials of induced hypertension available.
The effect of a normal saline fluid bolus in patients suspected of having delayed cerebral ischemia has been shown to increase cerebral blood flow in areas of cerebral ischemia.74 If volume augmentation fails to improve the neurologic status, the next step is to artificially induce hypertension using vasopressors. The blood pressure target should be based on clinical improvement. A stepwise approach is reasonable in this situation, and the lowest level of blood pressure at which there is a complete reversal of the new focal neurologic deficit should be maintained.3,29
Inotropic agents such as dobutamine or milrinone can be considered as alternatives in patients who have new neurologic deficits that are refractory to fluid boluses and vasopressors, or in a setting of subarachnoid hemorrhage-induced cardiomyopathy.76,77
Once the neurologic deficit is reversed by hemodynamic augmentation, the blood pressure should be maintained for 48 to 72 hours at the level that reversed the deficit completely, carefully reassessed thereafter, and the patient weaned slowly. Unruptured unsecured aneurysms should not prevent blood pressure augmentation in a setting of delayed cerebral ischemia if the culprit aneurysm is treated.3,32 If the ruptured aneurysm has not been secured, careful blood pressure augmentation can be attempted, keeping in mind that hypertension (> 160/95 mm Hg) is a risk factor for fatal aneurysm rupture.
Endovascular management of delayed cerebral ischemia
When medical augmentation fails to completely reverse the neurologic deficits, endovascular treatment can be considered. Although patients treated early in the course of delayed cerebral ischemia have better neurologic recovery, prophylactic endovascular treatment in asymptomatic patients, even if angiographic signs of spasm are present, does not improve clinical outcomes and carries the risk of fatal arterial rupture.78
SYSTEMIC COMPLICATIONS
Hyponatremia and hypovolemia
Aneurysmal subarachnoid hemorrhage is commonly associated with abnormalities of fluid balance and electrolyte derangements. Hyponatremia (serum sodium < 135 mmol/L) occurs in 30% to 50% of patients, while the rate of hypovolemia (decreased circulating blood volume) ranges from 17% to 30%.79 Both can negatively affect long-term outcomes.80,81
Decreased circulating blood volume is a well-described contributor to delayed cerebral ischemia and cerebral infarction after aneurysmal subarachnoid hemorrhage.80–82 Clinical variables such as heart rate, blood pressure, fluid balance, and serum sodium concentration are usually the cornerstones of intravascular volume status assessment. However, these variables correlate poorly with measured circulating blood volumes in those with aneurysmal subarachnoid hemorrhage.83,84
The mechanisms responsible for the development of hyponatremia and hypovolemia after aneurysmal subarachnoid hemorrhage are not completely understood. Several factors have been described and may contribute to the increased natriuresis and, hence, to a reduction in circulating blood volume: increased circulating natriuretic peptide concentrations,85–87 sympathetic nervous system hyperactivation,88 and hyperreninemic hypo-
aldosteronism syndrome.89,90
Lastly, the cerebral salt wasting syndrome, described in the 1950s,91 was thought to be a key mechanism in the development of hyponatremia and hypovolemia after aneurysmal subarachnoid hemorrhage. In contrast to the syndrome of inappropriate antidiuretic hormone, which is characterized by hyponatremia with a normal or slightly elevated intravascular volume, the characteristic feature of cerebral salt wasting syndrome is the development of hyponatremia in a setting of intravascular volume depletion.92 In critically ill neurologic and neurosurgical patients, this differential diagnosis is very difficult, especially in those with aneurysmal subarachnoid hemorrhage in whom the clinical assessment of fluid status is not reliable. These two syndromes might coexist and contribute to the development of hyponatremia after aneurysmal subarachnoid hemorrhage.92,93
Hoff et al83,84 prospectively compared the clinical assessment of fluid status by critical and intermediate care nurses and direct measurements of blood volume using pulse dye densitometry. The clinical assessment failed to accurately assess patients’ volume status. Using the same technique to measure circulating blood volume, this group showed that calculation of fluid balance does not provide adequate assessment of fluid status.83,84
Hemodynamic monitoring tools can help guide fluid replacement in this population. Mutoh et al94 randomized 160 patients within 24 hours of hemorrhage to receive early goal-directed fluid therapy (ie, preload volume and cardiac output monitored by transpulmonary thermodilution) vs standard therapy (ie, fluid balance or central venous pressure). Overall, no difference was found in the rates of delayed cerebral ischemia (33% vs 42%; P = .33) or favorable outcome (67% vs 57%; P = .22). However, in the subgroup of poor-grade patients (WFNS score 4 or 5), early goal-directed therapy was associated with a lower rate of delayed cerebral ischemia (5% vs 14%; P = .036) and with better functional outcomes at 3 months (52% vs 36%; P = .026).
Fluid restriction to treat hyponatremia in aneurysmal subarachnoid hemorrhage is no longer recommended because of the increased risk of cerebral infarction due to hypovolemic hypoperfusion.82
Prophylactic use of mineralocorticoids (eg, fludrocortisone, hydrocortisone) has been shown to limit natriuresis, hyponatremia, and the amount of fluid required to maintain euvolemia.95,96 Higher rates of hypokalemia and hyperglycemia, which can be easily treated, are the most common complications associated with this approach. Additionally, hypertonic saline (eg, 3% saline) can be used to correct hyponatremia in a setting of aneurysmal subarachnoid hemorrhage.79
Cardiac complications
Cardiac complications after subarachnoid hemorrhage are most likely related to sympathetic hyperactivity and catecholamine-induced myocyte dysfunction. The pathophysiology is complex, but cardiac complications have a significant negative impact on long-term outcome in these patients.97
Electrocardiographic changes and positive cardiac enzymes associated with aneurysmal subarachnoid hemorrhage have been extensively reported. More recently, data from studies of two-dimensional echocardiography have shown that subarachnoid hemorrhage can also be associated with significant wall-motion abnormalities and even overt cardiogenic shock.98–100
There is no specific curative therapy; the treatment is mainly supportive. Vasopressors and inotropes may be used for hemodynamic augmentation.
Pulmonary complications
Pulmonary complications occur in 20% to 30% of all aneurysmal subarachnoid hemorrhage patients and are associated with a higher risk of delayed cerebral ischemia and death. Common pulmonary complications in this population are mild acute respiratory distress syndrome (27%), hospital-acquired pneumonia (9%), cardiogenic pulmonary edema (8%), aspiration pneumonia (6%), neurogenic pulmonary edema (2%), and pulmonary embolism (1%).101–103
SUPPORTIVE CARE
Hyperthermia, hyperglycemia, and liberal use of transfusions have all been associated with longer stays in the intensive care unit and hospital, poorer neurologic outcomes, and higher mortality rates in patients with acute brain injury.104 Noninfectious fever is the most common systemic complication after subarachnoid hemorrhage.
Antipyretic drugs such as acetaminophen and ibuprofen are not very effective in reducing fever in the subarachnoid hemorrhage population, but should still be used as first-line therapy. The use of surface and intravascular devices can be considered when fevers do not respond to nonsteroidal anti-inflammatory drugs.
Although no prospective randomized trial has addressed the impact of induced normothermia on long-term outcome and mortality in aneurysmal subarachnoid hemorrhage patients, fever control has been shown to reduce cerebral metabolic distress, irrespective of intracranial pressure.105 Maintenance of normothermia (< 37.5°C) seems reasonable, especially in aneurysmal subarachnoid hemorrhage patients at risk of or with active delayed cerebral ischemia.106
Current guidelines3,32,69 strongly recommend avoiding hypoglycemia, defined as a serum glucose level less than 80 mg/dL, but suggest keeping the blood sugar level below 180 or 200 mg/dL.
At the moment, there is no clear threshold for transfusion in patients with aneurysmal subarachnoid hemorrhage. Current guidelines suggest keeping hemoglobin levels between 8 and 10 g/dL.3
Preventing venous thromboembolism
The incidence of venous thromboembolism after aneurysmal subarachnoid hemorrhage varies widely, from 1.5% to 18%.107 Active surveillance with venous Doppler ultrasonography has found asymptomatic deep vein thrombosis in up to 3.4% of poor-grade aneurysmal subarachnoid hemorrhage patients receiving pharmacologic thromboprophylaxis.108
In a retrospective study of 170 patients, our group showed that giving drugs to prevent venous thromboembolism (unfractionated heparin 5,000 IU subcutaneously every 12 hours or dalteparin 5,000 IU subcutaneously daily), starting within 24 hours of aneurysm treatment, could be safe.109 Fifty-eight percent of these patients had an external ventricular drain in place. One patient developed a major cerebral hemorrhagic complication and died while on unfractionated heparin; however, the patient was also on dual antiplatelet therapy with aspirin and clopidogrel.109
Current guidelines suggest that intermittent compression devices be applied in all patients before aneurysm treatment. Pharmacologic thromboprophylaxis with a heparinoid can be started 12 to 24 hours after aneurysm treatment.3,109
A TEAM APPROACH
Patients with subarachnoid hemorrhage need integrated care from different medical and nursing specialties. The best outcomes are achieved by systems that can focus as a team on the collective goal of quick intervention to secure the aneurysm, followed by measures to minimize secondary brain injury.
The modern concept of cerebral monitoring in a setting of subarachnoid hemorrhage should focus on brain perfusion rather than vascular diameter. Although the search continues for new diagnostic, prognostic, and therapeutic tools, there is no “silver bullet” that will help all patients. Instead, it is the systematic integration and application of many small advances that will ultimately lead to better outcomes.
ACKNOWLEDGMENT
This work was supported by research funding provided by the Bitove Foundation, which has been supportive of our clinical and research work for several years.
- van Gijn J, Rinkel GJ. Subarachnoid haemorrhage: diagnosis, causes and management. Brain 2001; 124:249–278.
- Go AS, Mozaffarian D, Roger VL, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2014 update: a report from the American Heart Association. Circulation 2014; 129:e28–e292.
- Diringer MN, Bleck TP, Claude Hemphill J 3rd, et al; Neurocritical Care Society. Critical care management of patients following aneurysmal subarachnoid hemorrhage: recommendations from the Neurocritical Care Society’s Multidisciplinary Consensus Conference. Neurocrit Care 2011; 15:211–240.
- Feigin VL, Rinkel GJ, Lawes CM, et al. Risk factors for subarachnoid hemorrhage: an updated systematic review of epidemiological studies. Stroke 2005; 36:2773–2780.
- Perry JJ, Stiell IG, Sivilotti ML, et al. Clinical decision rules to rule out subarachnoid hemorrhage for acute headache. JAMA 2013; 310:1248–1255.
- Kowalski RG, Claassen J, Kreiter KT, et al. Initial misdiagnosis and outcome after subarachnoid hemorrhage. JAMA 2004; 291:866–869.
- Perry JJ, Stiell IG, Sivilotti ML, et al. Sensitivity of computed tomography performed within six hours of onset of headache for diagnosis of subarachnoid haemorrhage: prospective cohort study. BMJ 2011; 343:d4277.
- van Gijn J, van Dongen KJ. The time course of aneurysmal haemorrhage on computed tomograms. Neuroradiology 1982; 23:153–156.
- McCormack RF, Hutson A. Can computed tomography angiography of the brain replace lumbar puncture in the evaluation of acute-onset headache after a negative noncontrast cranial computed tomography scan? Acad Emerg Med 2010; 17:444–451.
- Agid R, Andersson T, Almqvist H, et al. Negative CT angiography findings in patients with spontaneous subarachnoid hemorrhage: when is digital subtraction angiography still needed? AJNR Am J Neuroradiol 2010; 31:696–705.
- Teasdale GM, Drake CG, Hunt W, et al. A universal subarachnoid hemorrhage scale: report of a committee of the World Federation of Neurosurgical Societies. J Neurol Neurosurg Psychiatry 1988; 51:1457.
- Hunt WE, Hess RM. Surgical risk as related to time of intervention in the repair of intracranial aneurysms. J Neurosurg 1968; 28:14–20.
- Fisher CM, Kistler JP, Davis JM. Relation of cerebral vasospasm to subarachnoid hemorrhage visualized by computerized tomographic scanning. Neurosurgery 1980; 6:1–9.
- Frontera JA, Claassen J, Schmidt JM, et al. Prediction of symptomatic vasospasm after subarachnoid hemorrhage: the modified Fisher scale. Neurosurgery 2006; 59:21–27.
- de Oliveira Manoel AL, Turkel-Parrella D, Kouzmina E, et al. The VASOGRADE—a simple, reliable grading scale for aneurysmal subarachnoid hemorrhage. Neurology 2014; 82(suppl 10): P5.123.
- Naidech AM, Janjua N, Kreiter KT, et al. Predictors and impact of aneurysm rebleeding after subarachnoid hemorrhage. Arch Neurol 2005; 62:410–416.
- Rincon F, Mayer SA. Neurocritical care: a distinct discipline? Curr Opin Crit Care 2007; 13:115–121.
- Rabinstein AA, Lanzino G, Wijdicks EF. Multidisciplinary management and emerging therapeutic strategies in aneurysmal subarachnoid haemorrhage. Lancet Neurol 2010; 9:504–519.
- Vespa P, Diringer MN; Participants in the International Multi-Disciplinary Consensus Conference on the Critical Care Management of Subarachnoid Hemorrhage. High-volume centers. Neurocrit Care 2011; 15:369–372.
- McNeill L, English SW, Borg N, Matta BF, Menon DK. Effects of institutional caseload of subarachnoid hemorrhage on mortality: a secondary analysis of administrative data. Stroke 2013; 44:647–652.
- Dorhout Mees SM, Molyneux AJ, Kerr RS, Algra A, Rinkel GJ. Timing of aneurysm treatment after subarachnoid hemorrhage: relationship with delayed cerebral ischemia and poor outcome. Stroke 2012; 43:2126–2129.
- Laidlaw JD, Siu KH. Ultra-early surgery for aneurysmal subarachnoid hemorrhage: outcomes for a consecutive series of 391 patients not selected by grade or age. J Neurosurg 2002; 97:250–259.
- Molyneux A, Kerr R, Stratton I, et al; International Subarachnoid Aneurysm Trial (ISAT) Collaborative Group. International Subarachnoid Aneurysm Trial (ISAT) of neurosurgical clipping versus endovascular coiling in 2,143 patients with ruptured intracranial aneurysms: a randomised trial. Lancet 2002; 360:1267–1274.
- Molyneux AJ, Kerr RS, Yu LM, et al; International Subarachnoid Aneurysm Trial (ISAT) Collaborative Group. International subarachnoid aneurysm trial (ISAT) of neurosurgical clipping versus endovascular coiling in 2,143 patients with ruptured intracranial aneurysms: a randomised comparison of effects on survival, dependency, seizures, rebleeding, subgroups, and aneurysm occlusion. Lancet 2005; 366:809–817.
- Koivisto T, Vanninen R, Hurskainen H, Saari T, Hernesniemi J, Vapalahti M. Outcomes of early endovascular versus surgical treatment of ruptured cerebral aneurysms. A prospective randomized study. Stroke 2000; 31:2369–2377.
- McDougall CG, Spetzler RF, Zabramski JM, et al. The Barrow Ruptured Aneurysm Trial. J Neurosurg 2012; 116:135–144.
- Spetzler RF, McDougall CG, Albuquerque FC, et al. The Barrow Ruptured Aneurysm Trial: 3-year results. J Neurosurg 2013; 119:146–157.
- Connolly ES Jr, Meyers PM. Cerebral aneurysms: to clip or to coil? That is no longer the question. Nat Rev Neurol 2009; 5:412–413.
- Kassell NF, Peerless SJ, Durward QJ, Beck DW, Drake CG, Adams HP. Treatment of ischemic deficits from vasospasm with intravascular volume expansion and induced arterial hypertension. Neurosurgery 1982; 11:337–343.
- Otsubo H, Takemae T, Inoue T, Kobayashi S, Sugita K. Normovolaemic induced hypertension therapy for cerebral vasospasm after subarachnoid haemorrhage. Acta Neurochir (Wien) 1990; 103:18–26.
- Anderson CS, Heeley E, Huang Y, et al; INTERACT2 Investigators. Rapid blood-pressure lowering in patients with acute intracerebral hemorrhage. N Engl J Med 2013; 368:2355–2365.
- Connolly ES Jr, Rabinstein AA, Carhuapoma JR, et al; American Heart Association Stroke Council; Council on Cardiovascular Radiology and Intervention; Council on Cardiovascular Nursing; Council on Cardiovascular Surgery and Anesthesia; Council on Clinical Cardiology. Guidelines for the management of aneurysmal subarachnoid hemorrhage: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2012; 43:1711–1737.
- Ortega-Gutierrez S, Thomas J, Reccius A, et al. Effectiveness and safety of nicardipine and labetalol infusion for blood pressure management in patients with intracerebral and subarachnoid hemorrhage. Neurocrit Care 2013; 18:13–19.
- Hillman J, Fridriksson S, Nilsson O, Yu Z, Saveland H, Jakobsson KE. Immediate administration of tranexamic acid and reduced incidence of early rebleeding after aneurysmal subarachnoid hemorrhage: a prospective randomized study. J Neurosurg 2002; 97:771–778.
- Baharoglu MI, Germans MR, Rinkel GJ, et al. Antifibrinolytic therapy for aneurysmal subarachnoid haemorrhage. Cochrane Database Syst Rev 2013; 8:CD001245.
- Germans MR, Post R, Coert BA, Rinkel GJ, Vandertop WP, Verbaan D. Ultra-early tranexamic acid after subarachnoid hemorrhage (ULTRA): study protocol for a randomized controlled trial. Trials 2013; 14:143.
- Sloan MA, Alexandrov AV, Tegeler CH, et al; Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Assessment: transcranial Doppler ultrasonography: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology 2004; 62:1468–1481.
- Wartenberg KE, Schmidt JM, Claassen J, et al. Impact of medical complications on outcome after subarachnoid hemorrhage. Crit Care Med 2006; 34:617–623;
- Hellingman CA, van den Bergh WM, Beijer IS, et al. Risk of rebleeding after treatment of acute hydrocephalus in patients with aneurysmal subarachnoid hemorrhage. Stroke 2007; 38:96–99.
- Ransom ER, Mocco J, Komotar RJ, et al. External ventricular drainage response in poor grade aneurysmal subarachnoid hemorrhage: effect on preoperative grading and prognosis. Neurocrit Care 2007; 6:174–180.
- McIver JI, Friedman JA, Wijdicks EF, et al. Preoperative ventriculostomy and rebleeding after aneurysmal subarachnoid hemorrhage. J Neurosurg 2002; 97:1042–1044.
- Klopfenstein JD, Kim LJ, Feiz-Erfan I, et al. Comparison of rapid and gradual weaning from external ventricular drainage in patients with aneurysmal subarachnoid hemorrhage: a prospective randomized trial. J Neurosurg 2004; 100:225–229.
- Tseng MY, Al-Rawi PG, Czosnyka M, et al. Enhancement of cerebral blood flow using systemic hypertonic saline therapy improves outcome in patients with poor-grade spontaneous subarachnoid hemorrhage. J Neurosurg 2007; 107:274–282.
- Al-Rawi PG, Tseng MY, Richards HK, et al. Hypertonic saline in patients with poor-grade subarachnoid hemorrhage improves cerebral blood flow, brain tissue oxygen, and pH. Stroke 2010; 41:122–128.
- Tseng MY, Al-Rawi PG, Pickard JD, Rasulo FA, Kirkpatrick PJ. Effect of hypertonic saline on cerebral blood flow in poor-grade patients with subarachnoid hemorrhage. Stroke 2003; 34:1389–1396.
- Bentsen G, Breivik H, Lundar T, Stubhaug A. Hypertonic saline (7.2%) in 6% hydroxyethyl starch reduces intracranial pressure and improves hemodynamics in a placebo-controlled study involving stable patients with subarachnoid hemorrhage. Crit Care Med 2006; 34:2912–2917.
- Suarez JI, Qureshi AI, Parekh PD, et al. Administration of hypertonic (3%) sodium chloride/acetate in hyponatremic patients with symptomatic vasospasm following subarachnoid hemorrhage. J Neurosurg Anesthesiol 1999; 11:178–184.
- Stevens RD, Huff JS, Duckworth J, Papangelou A, Weingart SD, Smith WS. Emergency neurological life support: intracranial hypertension and herniation. Neurocrit Care 2012;17(suppl 1):S60–S65.
- Seule MA, Muroi C, Mink S, Yonekawa Y, Keller E. Therapeutic hypothermia in patients with aneurysmal subarachnoid hemorrhage, refractory intracranial hypertension, or cerebral vasospasm. Neurosurgery 2009; 64:86–93.
- Otani N, Takasato Y, Masaoka H, et al. Surgical outcome following decompressive craniectomy for poor-grade aneurysmal subarachnoid hemorrhage in patients with associated massive intracerebral or Sylvian hematomas. Cerebrovasc Dis 2008; 26:612–617.
- Lanzino G, D’Urso PI, Suarez J; Participants in the International Multi-Disciplinary Consensus Conference on the Critical Care Management of Subarachnoid Hemorrhage. Seizures and anticonvulsants after aneurysmal subarachnoid hemorrhage. Neurocrit Care 2011; 15:247–256.
- Naidech AM, Kreiter KT, Janjua N, et al. Phenytoin exposure is associated with functional and cognitive disability after subarachnoid hemorrhage. Stroke 2005; 36:583–587.
- Rosengart AJ, Huo JD, Tolentino J, et al. Outcome in patients with subarachnoid hemorrhage treated with antiepileptic drugs. J Neurosurg 2007; 107:253–260.
- Shah D, Husain AM. Utility of levetiracetam in patients with subarachnoid hemorrhage. Seizure 2009; 18:676–679.
- Patsalos PN. Pharmacokinetic profile of levetiracetam: toward ideal characteristics. Pharmacol Ther 2000; 85:77–85.
- Dewan MC, Mocco J. Current practice regarding seizure prophylaxis in aneurysmal subarachnoid hemorrhage across academic centers. J Neurointerv Surg 2014 Jan 28. doi: 10.1136/neurintsurg-2013-011075 [Epub ahead of print]
- Murphy-Human T, Welch E, Zipfel G, Diringer MN, Dhar R. Comparison of short-duration levetiracetam with extended-course phenytoin for seizure prophylaxis after subarachnoid hemorrhage. World Neurosurg 2011; 75:269–274.
- Dennis LJ, Claassen J, Hirsch LJ, Emerson RG, Connolly ES, Mayer SA. Nonconvulsive status epilepticus after subarachnoid hemorrhage. Neurosurgery 2002; 51:1136–1144.
- Vespa PM, Nuwer MR, Juhász C, et al. Early detection of vasospasm after acute subarachnoid hemorrhage using continuous EEG ICU monitoring. Electroencephalogr Clin Neurophysiol 1997; 103:607–615.
- Claassen J, Hirsch LJ, Kreiter KT, et al. Quantitative continuous EEG for detecting delayed cerebral ischemia in patients with poor-grade subarachnoid hemorrhage. Clin Neurophysiol 2004; 115:2699–2710.
- Vergouwen MD, Vermeulen M, van Gijn J, et al. Definition of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage as an outcome event in clinical trials and observational studies: proposal of a multidisciplinary research group. Stroke 2010; 41:2391–2395.
- Kelly PJ, Gorten RJ, Grossman RG, Eisenberg HM. Cerebral perfusion, vascular spasm, and outcome in patients with ruptured intracranial aneurysms. J Neurosurg 1977; 47:44–49.
- Aralasmak A, Akyuz M, Ozkaynak C, Sindel T, Tuncer R. CT angiography and perfusion imaging in patients with subarachnoid hemorrhage: correlation of vasospasm to perfusion abnormality. Neuroradiology 2009; 51:85–93.
- Sabri M, Lass E, Macdonald RL. Early brain injury: a common mechanism in subarachnoid hemorrhage and global cerebral ischemia. Stroke Res Treat 2013 Feb 28. doi: 10.1155/2013/394036 [Epub 2013 ahead of print]
- Yoon DY, Choi CS, Kim KH, Cho BM. Multidetector-row CT angiography of cerebral vasospasm after aneurysmal subarachnoid hemorrhage: comparison of volume-rendered images and digital subtraction angiography. AJNR Am J Neuroradiol 2006; 27:370–377.
- Wintermark M1, Ko NU, Smith WS, Liu S, Higashida RT, Dillon WP. Vasospasm after subarachnoid hemorrhage: utility of perfusion CT and CT angiography on diagnosis and management. AJNR Am J Neuroradiol 2006; 27:26–34.
- Helbok R, Madineni RC, Schmidt MJ, et al. Intracerebral monitoring of silent infarcts after subarachnoid hemorrhage. Neurocrit Care 2011; 14:162–167.
- Hänggi D; Participants in the International Multi-Disciplinary Consensus Conference on the Critical Care Management of Subarachnoid Hemorrhage. Monitoring and detection of vasospasm II: EEG and invasive monitoring. Neurocrit Care 2011; 15:318–323.
- Steiner T, Juvela S, Unterberg A, Jung C, Forsting M, Rinkel G; European Stroke Organization. European Stroke Organization guidelines for the management of intracranial aneurysms and subarachnoid haemorrhage. Cerebrovasc Dis 2013; 35:93–112.
- Pickard JD, Murray GD, Illingworth R, et al. Effect of oral nimodipine on cerebral infarction and outcome after subarachnoid haemorrhage: British aneurysm nimodipine trial. BMJ 1989; 298:636–642.
- Dorhout Mees SM, Rinkel GJ, Feigin VL, et al. Calcium antagonists for aneurysmal subarachnoid haemorrhage. Cochrane Database Syst Rev 2007; 3:CD000277.
- Lennihan L, Mayer SA, Fink ME, et al. Effect of hypervolemic therapy on cerebral blood flow after subarachnoid hemorrhage: a randomized controlled trial. Stroke 2000; 31:383–391.
- Egge A, Waterloo K, Sjøholm H, Solberg T, Ingebrigtsen T, Romner B. Prophylactic hyperdynamic postoperative fluid therapy after aneurysmal subarachnoid hemorrhage: a clinical, prospective, randomized, controlled study. Neurosurgery 2001; 49:593–606.
- Jost SC, Diringer MN, Zazulia AR, et al. Effect of normal saline bolus on cerebral blood flow in regions with low baseline flow in patients with vasospasm following subarachnoid hemorrhage. J Neurosurg 2005; 103:25–30.
- Muizelaar JP, Becker DP. Induced hypertension for the treatment of cerebral ischemia after subarachnoid hemorrhage. Direct effect on cerebral blood flow. Surg Neurol 1986; 25:317–325.
- Levy ML, Rabb CH, Zelman V, Giannotta SL. Cardiac performance enhancement from dobutamine in patients refractory to hypervolemic therapy for cerebral vasospasm. J Neurosurg 1993; 79:494–499.
- Lannes M, Teitelbaum J, del Pilar Cortés M, Cardoso M, Angle M. Milrinone and homeostasis to treat cerebral vasospasm associated with subarachnoid hemorrhage: the Montreal Neurological Hospital protocol. Neurocrit Care 2012; 16:354–362.
- Zwienenberg-Lee M, Hartman J, Rudisill N, et al; Balloon Prophylaxis for Aneurysmal Vasospasm (BPAV) Study Group. Effect of prophylactic transluminal balloon angioplasty on cerebral vasospasm and outcome in patients with Fisher grade III subarachnoid hemorrhage: results of a phase II multicenter, randomized, clinical trial. Stroke 2008; 39:1759–1765.
- Rabinstein AA, Bruder N. Management of hyponatremia and volume contraction. Neurocrit Care 2011; 15:354–360.
- Wijdicks EF, Vermeulen M, Hijdra A, van Gijn J. Hyponatremia and cerebral infarction in patients with ruptured intracranial aneurysms: is fluid restriction harmful? Ann Neurol 1985; 17:137–140.
- Hasan D, Wijdicks EF, Vermeulen M. Hyponatremia is associated with cerebral ischemia in patients with aneurysmal subarachnoid hemorrhage. Ann Neurol 1990; 27:106–108.
- Wijdicks EF, Vermeulen M, ten Haaf JA, Hijdra A, Bakker WH, van Gijn J. Volume depletion and natriuresis in patients with a ruptured intracranial aneurysm. Ann Neurol 1985; 18:211–216.
- Hoff RG, Rinkel GJ, Verweij BH, Algra A, Kalkman CJ. Nurses’ prediction of volume status after aneurysmal subarachnoid haemorrhage: a prospective cohort study. Crit Care 2008; 12:R153.
- Hoff RG, van Dijk GW, Algra A, Kalkman CJ, Rinkel GJ. Fluid balance and blood volume measurement after aneurysmal subarachnoid hemorrhage. Neurocrit Care 2008; 8:391–397.
- Berendes E, Walter M, Cullen P, et al. Secretion of brain natriuretic peptide in patients with aneurysmal subarachnoid haemorrhage. Lancet 1997; 349:245–249.
- Espiner EA, Leikis R, Ferch RD, et al. The neuro-cardio-endocrine response to acute subarachnoid haemorrhage. Clin Endocrinol (Oxf) 2002; 56:629–635.
- Isotani E, Suzuki R, Tomita K, et al. Alterations in plasma concentrations of natriuretic peptides and antidiuretic hormone after subarachnoid hemorrhage. Stroke 1994; 25:2198–2203.
- Benedict CR, Loach AB. Sympathetic nervous system activity in patients with subarachnoid hemorrhage. Stroke 1978; 9:237–244.
- Findling JW, Waters VO, Raff H. The dissociation of renin and aldosterone during critical illness. J Clin Endocrinol Metab 1987; 64:592–595.
- Solomon RA, Post KD, McMurtry JG 3rd. Depression of circulating blood volume in patients after subarachnoid hemorrhage: implications for the management of symptomatic vasospasm. Neurosurgery 1984; 15:354–361.
- Peters JP, Welt LG, Sims EA, Orloff J, Needham J. A salt-wasting syndrome associated with cerebral disease. Trans Assoc Am Physicians 1950; 63:57–64.
- Brimioulle S, Orellana-Jimenez C, Aminian A, Vincent JL. Hyponatremia in neurological patients: cerebral salt wasting versus inappropriate antidiuretic hormone secretion. Intensive Care Med 2008; 34:125–131.
- Singh S, Bohn D, Carlotti AP, Cusimano M, Rutka JT, Halperin ML. Cerebral salt wasting: truths, fallacies, theories, and challenges. Crit Care Med 2002; 30:2575–2579.
- Mutoh T, Kazumata K, Terasaka S, Taki Y, Suzuki A, Ishikawa T. Early intensive versus minimally invasive approach to postoperative hemodynamic management after subarachnoid hemorrhage. Stroke 2014; 45:1280–1284.
- Hasan D, Lindsay KW, Wijdicks EF, et al. Effect of fludrocortisone acetate in patients with subarachnoid hemorrhage. Stroke 1989; 20:1156–1161.
- Moro N, Katayama Y, Kojima J, Mori T, Kawamata T. Prophylactic management of excessive natriuresis with hydrocortisone for efficient hypervolemic therapy after subarachnoid hemorrhage. Stroke 2003; 34:2807–2811.
- Kilbourn KJ, Levy S, Staff I, Kureshi I, McCullough L. Clinical characteristics and outcomes of neurogenic stress cadiomyopathy in aneurysmal subarachnoid hemorrhage. Clin Neurol Neurosurg 2013; 115:909–914.
- Mayer SA, LiMandri G, Sherman D, et al. Electrocardiographic markers of abnormal left ventricular wall motion in acute subarachnoid hemorrhage. J Neurosurg 1995; 83:889–896.
- Deibert E, Barzilai B, Braverman AC, et al. Clinical significance of elevated troponin I levels in patients with nontraumatic subarachnoid hemorrhage. J Neurosurg 2003; 98:741–746.
- Banki N, Kopelnik A, Tung P, et al. Prospective analysis of prevalence, distribution, and rate of recovery of left ventricular systolic dysfunction in patients with subarachnoid hemorrhage. J Neurosurg 2006; 105:15–20.
- Kahn JM, Caldwell EC, Deem S, Newell DW, Heckbert SR, Rubenfeld GD. Acute lung injury in patients with subarachnoid hemorrhage: incidence, risk factors, and outcome. Crit Care Med 2006; 34:196–202.
- Kitamura Y, Nomura M, Shima H, et al. Acute lung injury associated with systemic inflammatory response syndrome following subarachnoid hemorrhage: a survey by the Shonan Neurosurgical Association. Neurol Med Chir (Tokyo) 2010; 50:456–460.
- Friedman JA, Pichelmann MA, Piepgras DG, et al. Pulmonary complications of aneurysmal subarachnoid hemorrhage. Neurosurgery 2003; 52:1025–1032.
- Oh HS, Jeong HS, Seo WS. Non-infectious hyperthermia in acute brain injury patients: relationships to mortality, blood pressure, intracranial pressure and cerebral perfusion pressure. Int J Nurs Pract 2012; 18:295–302.
- Oddo M, Frangos S, Milby A, et al. Induced normothermia attenuates cerebral metabolic distress in patients with aneurysmal subarachnoid hemorrhage and refractory fever. Stroke 2009; 40:1913–1916.
- Badjatia N, Fernandez L, Schmidt JM, et al. Impact of induced normothermia on outcome after subarachnoid hemorrhage: a case-control study. Neurosurgery 2010; 66:696-701.
- Serrone JC1, Wash EM, Hartings JA, Andaluz N, Zuccarello M. Venous thromboembolism in subarachnoid hemorrhage. World Neurosurg 2013; 80:859–863.
- Mack WJ, Ducruet AF, Hickman ZL, et al. Doppler ultrasonography screening of poor-grade subarachnoid hemorrhage patients increases the diagnosis of deep venous thrombosis. Neurol Res 2008; 30:889–892.
- de Oliveira Manoel AL, Turkel-Parrella D, Germans M, et al. Safety of early pharmacological thromboprophylaxis after subarachnoid hemorrhage. Can J Neurol Sci 2014; 41:554–561.
- van Gijn J, Rinkel GJ. Subarachnoid haemorrhage: diagnosis, causes and management. Brain 2001; 124:249–278.
- Go AS, Mozaffarian D, Roger VL, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2014 update: a report from the American Heart Association. Circulation 2014; 129:e28–e292.
- Diringer MN, Bleck TP, Claude Hemphill J 3rd, et al; Neurocritical Care Society. Critical care management of patients following aneurysmal subarachnoid hemorrhage: recommendations from the Neurocritical Care Society’s Multidisciplinary Consensus Conference. Neurocrit Care 2011; 15:211–240.
- Feigin VL, Rinkel GJ, Lawes CM, et al. Risk factors for subarachnoid hemorrhage: an updated systematic review of epidemiological studies. Stroke 2005; 36:2773–2780.
- Perry JJ, Stiell IG, Sivilotti ML, et al. Clinical decision rules to rule out subarachnoid hemorrhage for acute headache. JAMA 2013; 310:1248–1255.
- Kowalski RG, Claassen J, Kreiter KT, et al. Initial misdiagnosis and outcome after subarachnoid hemorrhage. JAMA 2004; 291:866–869.
- Perry JJ, Stiell IG, Sivilotti ML, et al. Sensitivity of computed tomography performed within six hours of onset of headache for diagnosis of subarachnoid haemorrhage: prospective cohort study. BMJ 2011; 343:d4277.
- van Gijn J, van Dongen KJ. The time course of aneurysmal haemorrhage on computed tomograms. Neuroradiology 1982; 23:153–156.
- McCormack RF, Hutson A. Can computed tomography angiography of the brain replace lumbar puncture in the evaluation of acute-onset headache after a negative noncontrast cranial computed tomography scan? Acad Emerg Med 2010; 17:444–451.
- Agid R, Andersson T, Almqvist H, et al. Negative CT angiography findings in patients with spontaneous subarachnoid hemorrhage: when is digital subtraction angiography still needed? AJNR Am J Neuroradiol 2010; 31:696–705.
- Teasdale GM, Drake CG, Hunt W, et al. A universal subarachnoid hemorrhage scale: report of a committee of the World Federation of Neurosurgical Societies. J Neurol Neurosurg Psychiatry 1988; 51:1457.
- Hunt WE, Hess RM. Surgical risk as related to time of intervention in the repair of intracranial aneurysms. J Neurosurg 1968; 28:14–20.
- Fisher CM, Kistler JP, Davis JM. Relation of cerebral vasospasm to subarachnoid hemorrhage visualized by computerized tomographic scanning. Neurosurgery 1980; 6:1–9.
- Frontera JA, Claassen J, Schmidt JM, et al. Prediction of symptomatic vasospasm after subarachnoid hemorrhage: the modified Fisher scale. Neurosurgery 2006; 59:21–27.
- de Oliveira Manoel AL, Turkel-Parrella D, Kouzmina E, et al. The VASOGRADE—a simple, reliable grading scale for aneurysmal subarachnoid hemorrhage. Neurology 2014; 82(suppl 10): P5.123.
- Naidech AM, Janjua N, Kreiter KT, et al. Predictors and impact of aneurysm rebleeding after subarachnoid hemorrhage. Arch Neurol 2005; 62:410–416.
- Rincon F, Mayer SA. Neurocritical care: a distinct discipline? Curr Opin Crit Care 2007; 13:115–121.
- Rabinstein AA, Lanzino G, Wijdicks EF. Multidisciplinary management and emerging therapeutic strategies in aneurysmal subarachnoid haemorrhage. Lancet Neurol 2010; 9:504–519.
- Vespa P, Diringer MN; Participants in the International Multi-Disciplinary Consensus Conference on the Critical Care Management of Subarachnoid Hemorrhage. High-volume centers. Neurocrit Care 2011; 15:369–372.
- McNeill L, English SW, Borg N, Matta BF, Menon DK. Effects of institutional caseload of subarachnoid hemorrhage on mortality: a secondary analysis of administrative data. Stroke 2013; 44:647–652.
- Dorhout Mees SM, Molyneux AJ, Kerr RS, Algra A, Rinkel GJ. Timing of aneurysm treatment after subarachnoid hemorrhage: relationship with delayed cerebral ischemia and poor outcome. Stroke 2012; 43:2126–2129.
- Laidlaw JD, Siu KH. Ultra-early surgery for aneurysmal subarachnoid hemorrhage: outcomes for a consecutive series of 391 patients not selected by grade or age. J Neurosurg 2002; 97:250–259.
- Molyneux A, Kerr R, Stratton I, et al; International Subarachnoid Aneurysm Trial (ISAT) Collaborative Group. International Subarachnoid Aneurysm Trial (ISAT) of neurosurgical clipping versus endovascular coiling in 2,143 patients with ruptured intracranial aneurysms: a randomised trial. Lancet 2002; 360:1267–1274.
- Molyneux AJ, Kerr RS, Yu LM, et al; International Subarachnoid Aneurysm Trial (ISAT) Collaborative Group. International subarachnoid aneurysm trial (ISAT) of neurosurgical clipping versus endovascular coiling in 2,143 patients with ruptured intracranial aneurysms: a randomised comparison of effects on survival, dependency, seizures, rebleeding, subgroups, and aneurysm occlusion. Lancet 2005; 366:809–817.
- Koivisto T, Vanninen R, Hurskainen H, Saari T, Hernesniemi J, Vapalahti M. Outcomes of early endovascular versus surgical treatment of ruptured cerebral aneurysms. A prospective randomized study. Stroke 2000; 31:2369–2377.
- McDougall CG, Spetzler RF, Zabramski JM, et al. The Barrow Ruptured Aneurysm Trial. J Neurosurg 2012; 116:135–144.
- Spetzler RF, McDougall CG, Albuquerque FC, et al. The Barrow Ruptured Aneurysm Trial: 3-year results. J Neurosurg 2013; 119:146–157.
- Connolly ES Jr, Meyers PM. Cerebral aneurysms: to clip or to coil? That is no longer the question. Nat Rev Neurol 2009; 5:412–413.
- Kassell NF, Peerless SJ, Durward QJ, Beck DW, Drake CG, Adams HP. Treatment of ischemic deficits from vasospasm with intravascular volume expansion and induced arterial hypertension. Neurosurgery 1982; 11:337–343.
- Otsubo H, Takemae T, Inoue T, Kobayashi S, Sugita K. Normovolaemic induced hypertension therapy for cerebral vasospasm after subarachnoid haemorrhage. Acta Neurochir (Wien) 1990; 103:18–26.
- Anderson CS, Heeley E, Huang Y, et al; INTERACT2 Investigators. Rapid blood-pressure lowering in patients with acute intracerebral hemorrhage. N Engl J Med 2013; 368:2355–2365.
- Connolly ES Jr, Rabinstein AA, Carhuapoma JR, et al; American Heart Association Stroke Council; Council on Cardiovascular Radiology and Intervention; Council on Cardiovascular Nursing; Council on Cardiovascular Surgery and Anesthesia; Council on Clinical Cardiology. Guidelines for the management of aneurysmal subarachnoid hemorrhage: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2012; 43:1711–1737.
- Ortega-Gutierrez S, Thomas J, Reccius A, et al. Effectiveness and safety of nicardipine and labetalol infusion for blood pressure management in patients with intracerebral and subarachnoid hemorrhage. Neurocrit Care 2013; 18:13–19.
- Hillman J, Fridriksson S, Nilsson O, Yu Z, Saveland H, Jakobsson KE. Immediate administration of tranexamic acid and reduced incidence of early rebleeding after aneurysmal subarachnoid hemorrhage: a prospective randomized study. J Neurosurg 2002; 97:771–778.
- Baharoglu MI, Germans MR, Rinkel GJ, et al. Antifibrinolytic therapy for aneurysmal subarachnoid haemorrhage. Cochrane Database Syst Rev 2013; 8:CD001245.
- Germans MR, Post R, Coert BA, Rinkel GJ, Vandertop WP, Verbaan D. Ultra-early tranexamic acid after subarachnoid hemorrhage (ULTRA): study protocol for a randomized controlled trial. Trials 2013; 14:143.
- Sloan MA, Alexandrov AV, Tegeler CH, et al; Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Assessment: transcranial Doppler ultrasonography: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology 2004; 62:1468–1481.
- Wartenberg KE, Schmidt JM, Claassen J, et al. Impact of medical complications on outcome after subarachnoid hemorrhage. Crit Care Med 2006; 34:617–623;
- Hellingman CA, van den Bergh WM, Beijer IS, et al. Risk of rebleeding after treatment of acute hydrocephalus in patients with aneurysmal subarachnoid hemorrhage. Stroke 2007; 38:96–99.
- Ransom ER, Mocco J, Komotar RJ, et al. External ventricular drainage response in poor grade aneurysmal subarachnoid hemorrhage: effect on preoperative grading and prognosis. Neurocrit Care 2007; 6:174–180.
- McIver JI, Friedman JA, Wijdicks EF, et al. Preoperative ventriculostomy and rebleeding after aneurysmal subarachnoid hemorrhage. J Neurosurg 2002; 97:1042–1044.
- Klopfenstein JD, Kim LJ, Feiz-Erfan I, et al. Comparison of rapid and gradual weaning from external ventricular drainage in patients with aneurysmal subarachnoid hemorrhage: a prospective randomized trial. J Neurosurg 2004; 100:225–229.
- Tseng MY, Al-Rawi PG, Czosnyka M, et al. Enhancement of cerebral blood flow using systemic hypertonic saline therapy improves outcome in patients with poor-grade spontaneous subarachnoid hemorrhage. J Neurosurg 2007; 107:274–282.
- Al-Rawi PG, Tseng MY, Richards HK, et al. Hypertonic saline in patients with poor-grade subarachnoid hemorrhage improves cerebral blood flow, brain tissue oxygen, and pH. Stroke 2010; 41:122–128.
- Tseng MY, Al-Rawi PG, Pickard JD, Rasulo FA, Kirkpatrick PJ. Effect of hypertonic saline on cerebral blood flow in poor-grade patients with subarachnoid hemorrhage. Stroke 2003; 34:1389–1396.
- Bentsen G, Breivik H, Lundar T, Stubhaug A. Hypertonic saline (7.2%) in 6% hydroxyethyl starch reduces intracranial pressure and improves hemodynamics in a placebo-controlled study involving stable patients with subarachnoid hemorrhage. Crit Care Med 2006; 34:2912–2917.
- Suarez JI, Qureshi AI, Parekh PD, et al. Administration of hypertonic (3%) sodium chloride/acetate in hyponatremic patients with symptomatic vasospasm following subarachnoid hemorrhage. J Neurosurg Anesthesiol 1999; 11:178–184.
- Stevens RD, Huff JS, Duckworth J, Papangelou A, Weingart SD, Smith WS. Emergency neurological life support: intracranial hypertension and herniation. Neurocrit Care 2012;17(suppl 1):S60–S65.
- Seule MA, Muroi C, Mink S, Yonekawa Y, Keller E. Therapeutic hypothermia in patients with aneurysmal subarachnoid hemorrhage, refractory intracranial hypertension, or cerebral vasospasm. Neurosurgery 2009; 64:86–93.
- Otani N, Takasato Y, Masaoka H, et al. Surgical outcome following decompressive craniectomy for poor-grade aneurysmal subarachnoid hemorrhage in patients with associated massive intracerebral or Sylvian hematomas. Cerebrovasc Dis 2008; 26:612–617.
- Lanzino G, D’Urso PI, Suarez J; Participants in the International Multi-Disciplinary Consensus Conference on the Critical Care Management of Subarachnoid Hemorrhage. Seizures and anticonvulsants after aneurysmal subarachnoid hemorrhage. Neurocrit Care 2011; 15:247–256.
- Naidech AM, Kreiter KT, Janjua N, et al. Phenytoin exposure is associated with functional and cognitive disability after subarachnoid hemorrhage. Stroke 2005; 36:583–587.
- Rosengart AJ, Huo JD, Tolentino J, et al. Outcome in patients with subarachnoid hemorrhage treated with antiepileptic drugs. J Neurosurg 2007; 107:253–260.
- Shah D, Husain AM. Utility of levetiracetam in patients with subarachnoid hemorrhage. Seizure 2009; 18:676–679.
- Patsalos PN. Pharmacokinetic profile of levetiracetam: toward ideal characteristics. Pharmacol Ther 2000; 85:77–85.
- Dewan MC, Mocco J. Current practice regarding seizure prophylaxis in aneurysmal subarachnoid hemorrhage across academic centers. J Neurointerv Surg 2014 Jan 28. doi: 10.1136/neurintsurg-2013-011075 [Epub ahead of print]
- Murphy-Human T, Welch E, Zipfel G, Diringer MN, Dhar R. Comparison of short-duration levetiracetam with extended-course phenytoin for seizure prophylaxis after subarachnoid hemorrhage. World Neurosurg 2011; 75:269–274.
- Dennis LJ, Claassen J, Hirsch LJ, Emerson RG, Connolly ES, Mayer SA. Nonconvulsive status epilepticus after subarachnoid hemorrhage. Neurosurgery 2002; 51:1136–1144.
- Vespa PM, Nuwer MR, Juhász C, et al. Early detection of vasospasm after acute subarachnoid hemorrhage using continuous EEG ICU monitoring. Electroencephalogr Clin Neurophysiol 1997; 103:607–615.
- Claassen J, Hirsch LJ, Kreiter KT, et al. Quantitative continuous EEG for detecting delayed cerebral ischemia in patients with poor-grade subarachnoid hemorrhage. Clin Neurophysiol 2004; 115:2699–2710.
- Vergouwen MD, Vermeulen M, van Gijn J, et al. Definition of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage as an outcome event in clinical trials and observational studies: proposal of a multidisciplinary research group. Stroke 2010; 41:2391–2395.
- Kelly PJ, Gorten RJ, Grossman RG, Eisenberg HM. Cerebral perfusion, vascular spasm, and outcome in patients with ruptured intracranial aneurysms. J Neurosurg 1977; 47:44–49.
- Aralasmak A, Akyuz M, Ozkaynak C, Sindel T, Tuncer R. CT angiography and perfusion imaging in patients with subarachnoid hemorrhage: correlation of vasospasm to perfusion abnormality. Neuroradiology 2009; 51:85–93.
- Sabri M, Lass E, Macdonald RL. Early brain injury: a common mechanism in subarachnoid hemorrhage and global cerebral ischemia. Stroke Res Treat 2013 Feb 28. doi: 10.1155/2013/394036 [Epub 2013 ahead of print]
- Yoon DY, Choi CS, Kim KH, Cho BM. Multidetector-row CT angiography of cerebral vasospasm after aneurysmal subarachnoid hemorrhage: comparison of volume-rendered images and digital subtraction angiography. AJNR Am J Neuroradiol 2006; 27:370–377.
- Wintermark M1, Ko NU, Smith WS, Liu S, Higashida RT, Dillon WP. Vasospasm after subarachnoid hemorrhage: utility of perfusion CT and CT angiography on diagnosis and management. AJNR Am J Neuroradiol 2006; 27:26–34.
- Helbok R, Madineni RC, Schmidt MJ, et al. Intracerebral monitoring of silent infarcts after subarachnoid hemorrhage. Neurocrit Care 2011; 14:162–167.
- Hänggi D; Participants in the International Multi-Disciplinary Consensus Conference on the Critical Care Management of Subarachnoid Hemorrhage. Monitoring and detection of vasospasm II: EEG and invasive monitoring. Neurocrit Care 2011; 15:318–323.
- Steiner T, Juvela S, Unterberg A, Jung C, Forsting M, Rinkel G; European Stroke Organization. European Stroke Organization guidelines for the management of intracranial aneurysms and subarachnoid haemorrhage. Cerebrovasc Dis 2013; 35:93–112.
- Pickard JD, Murray GD, Illingworth R, et al. Effect of oral nimodipine on cerebral infarction and outcome after subarachnoid haemorrhage: British aneurysm nimodipine trial. BMJ 1989; 298:636–642.
- Dorhout Mees SM, Rinkel GJ, Feigin VL, et al. Calcium antagonists for aneurysmal subarachnoid haemorrhage. Cochrane Database Syst Rev 2007; 3:CD000277.
- Lennihan L, Mayer SA, Fink ME, et al. Effect of hypervolemic therapy on cerebral blood flow after subarachnoid hemorrhage: a randomized controlled trial. Stroke 2000; 31:383–391.
- Egge A, Waterloo K, Sjøholm H, Solberg T, Ingebrigtsen T, Romner B. Prophylactic hyperdynamic postoperative fluid therapy after aneurysmal subarachnoid hemorrhage: a clinical, prospective, randomized, controlled study. Neurosurgery 2001; 49:593–606.
- Jost SC, Diringer MN, Zazulia AR, et al. Effect of normal saline bolus on cerebral blood flow in regions with low baseline flow in patients with vasospasm following subarachnoid hemorrhage. J Neurosurg 2005; 103:25–30.
- Muizelaar JP, Becker DP. Induced hypertension for the treatment of cerebral ischemia after subarachnoid hemorrhage. Direct effect on cerebral blood flow. Surg Neurol 1986; 25:317–325.
- Levy ML, Rabb CH, Zelman V, Giannotta SL. Cardiac performance enhancement from dobutamine in patients refractory to hypervolemic therapy for cerebral vasospasm. J Neurosurg 1993; 79:494–499.
- Lannes M, Teitelbaum J, del Pilar Cortés M, Cardoso M, Angle M. Milrinone and homeostasis to treat cerebral vasospasm associated with subarachnoid hemorrhage: the Montreal Neurological Hospital protocol. Neurocrit Care 2012; 16:354–362.
- Zwienenberg-Lee M, Hartman J, Rudisill N, et al; Balloon Prophylaxis for Aneurysmal Vasospasm (BPAV) Study Group. Effect of prophylactic transluminal balloon angioplasty on cerebral vasospasm and outcome in patients with Fisher grade III subarachnoid hemorrhage: results of a phase II multicenter, randomized, clinical trial. Stroke 2008; 39:1759–1765.
- Rabinstein AA, Bruder N. Management of hyponatremia and volume contraction. Neurocrit Care 2011; 15:354–360.
- Wijdicks EF, Vermeulen M, Hijdra A, van Gijn J. Hyponatremia and cerebral infarction in patients with ruptured intracranial aneurysms: is fluid restriction harmful? Ann Neurol 1985; 17:137–140.
- Hasan D, Wijdicks EF, Vermeulen M. Hyponatremia is associated with cerebral ischemia in patients with aneurysmal subarachnoid hemorrhage. Ann Neurol 1990; 27:106–108.
- Wijdicks EF, Vermeulen M, ten Haaf JA, Hijdra A, Bakker WH, van Gijn J. Volume depletion and natriuresis in patients with a ruptured intracranial aneurysm. Ann Neurol 1985; 18:211–216.
- Hoff RG, Rinkel GJ, Verweij BH, Algra A, Kalkman CJ. Nurses’ prediction of volume status after aneurysmal subarachnoid haemorrhage: a prospective cohort study. Crit Care 2008; 12:R153.
- Hoff RG, van Dijk GW, Algra A, Kalkman CJ, Rinkel GJ. Fluid balance and blood volume measurement after aneurysmal subarachnoid hemorrhage. Neurocrit Care 2008; 8:391–397.
- Berendes E, Walter M, Cullen P, et al. Secretion of brain natriuretic peptide in patients with aneurysmal subarachnoid haemorrhage. Lancet 1997; 349:245–249.
- Espiner EA, Leikis R, Ferch RD, et al. The neuro-cardio-endocrine response to acute subarachnoid haemorrhage. Clin Endocrinol (Oxf) 2002; 56:629–635.
- Isotani E, Suzuki R, Tomita K, et al. Alterations in plasma concentrations of natriuretic peptides and antidiuretic hormone after subarachnoid hemorrhage. Stroke 1994; 25:2198–2203.
- Benedict CR, Loach AB. Sympathetic nervous system activity in patients with subarachnoid hemorrhage. Stroke 1978; 9:237–244.
- Findling JW, Waters VO, Raff H. The dissociation of renin and aldosterone during critical illness. J Clin Endocrinol Metab 1987; 64:592–595.
- Solomon RA, Post KD, McMurtry JG 3rd. Depression of circulating blood volume in patients after subarachnoid hemorrhage: implications for the management of symptomatic vasospasm. Neurosurgery 1984; 15:354–361.
- Peters JP, Welt LG, Sims EA, Orloff J, Needham J. A salt-wasting syndrome associated with cerebral disease. Trans Assoc Am Physicians 1950; 63:57–64.
- Brimioulle S, Orellana-Jimenez C, Aminian A, Vincent JL. Hyponatremia in neurological patients: cerebral salt wasting versus inappropriate antidiuretic hormone secretion. Intensive Care Med 2008; 34:125–131.
- Singh S, Bohn D, Carlotti AP, Cusimano M, Rutka JT, Halperin ML. Cerebral salt wasting: truths, fallacies, theories, and challenges. Crit Care Med 2002; 30:2575–2579.
- Mutoh T, Kazumata K, Terasaka S, Taki Y, Suzuki A, Ishikawa T. Early intensive versus minimally invasive approach to postoperative hemodynamic management after subarachnoid hemorrhage. Stroke 2014; 45:1280–1284.
- Hasan D, Lindsay KW, Wijdicks EF, et al. Effect of fludrocortisone acetate in patients with subarachnoid hemorrhage. Stroke 1989; 20:1156–1161.
- Moro N, Katayama Y, Kojima J, Mori T, Kawamata T. Prophylactic management of excessive natriuresis with hydrocortisone for efficient hypervolemic therapy after subarachnoid hemorrhage. Stroke 2003; 34:2807–2811.
- Kilbourn KJ, Levy S, Staff I, Kureshi I, McCullough L. Clinical characteristics and outcomes of neurogenic stress cadiomyopathy in aneurysmal subarachnoid hemorrhage. Clin Neurol Neurosurg 2013; 115:909–914.
- Mayer SA, LiMandri G, Sherman D, et al. Electrocardiographic markers of abnormal left ventricular wall motion in acute subarachnoid hemorrhage. J Neurosurg 1995; 83:889–896.
- Deibert E, Barzilai B, Braverman AC, et al. Clinical significance of elevated troponin I levels in patients with nontraumatic subarachnoid hemorrhage. J Neurosurg 2003; 98:741–746.
- Banki N, Kopelnik A, Tung P, et al. Prospective analysis of prevalence, distribution, and rate of recovery of left ventricular systolic dysfunction in patients with subarachnoid hemorrhage. J Neurosurg 2006; 105:15–20.
- Kahn JM, Caldwell EC, Deem S, Newell DW, Heckbert SR, Rubenfeld GD. Acute lung injury in patients with subarachnoid hemorrhage: incidence, risk factors, and outcome. Crit Care Med 2006; 34:196–202.
- Kitamura Y, Nomura M, Shima H, et al. Acute lung injury associated with systemic inflammatory response syndrome following subarachnoid hemorrhage: a survey by the Shonan Neurosurgical Association. Neurol Med Chir (Tokyo) 2010; 50:456–460.
- Friedman JA, Pichelmann MA, Piepgras DG, et al. Pulmonary complications of aneurysmal subarachnoid hemorrhage. Neurosurgery 2003; 52:1025–1032.
- Oh HS, Jeong HS, Seo WS. Non-infectious hyperthermia in acute brain injury patients: relationships to mortality, blood pressure, intracranial pressure and cerebral perfusion pressure. Int J Nurs Pract 2012; 18:295–302.
- Oddo M, Frangos S, Milby A, et al. Induced normothermia attenuates cerebral metabolic distress in patients with aneurysmal subarachnoid hemorrhage and refractory fever. Stroke 2009; 40:1913–1916.
- Badjatia N, Fernandez L, Schmidt JM, et al. Impact of induced normothermia on outcome after subarachnoid hemorrhage: a case-control study. Neurosurgery 2010; 66:696-701.
- Serrone JC1, Wash EM, Hartings JA, Andaluz N, Zuccarello M. Venous thromboembolism in subarachnoid hemorrhage. World Neurosurg 2013; 80:859–863.
- Mack WJ, Ducruet AF, Hickman ZL, et al. Doppler ultrasonography screening of poor-grade subarachnoid hemorrhage patients increases the diagnosis of deep venous thrombosis. Neurol Res 2008; 30:889–892.
- de Oliveira Manoel AL, Turkel-Parrella D, Germans M, et al. Safety of early pharmacological thromboprophylaxis after subarachnoid hemorrhage. Can J Neurol Sci 2014; 41:554–561.
KEY POINTS
- The key symptom is the abrupt onset of severe headache, commonly described as “the worst headache of my life.
- Computed tomography without contrast should be done promptly when this condition is suspected.
- Outcomes are improved when patients are managed in a high-volume center with a specialized neurointensive care unit and access to an interdisciplinary team.
- Early aneurysm repair by surgical clipping or endovascular coiling is considered the standard of care and is the best strategy to reduce the risk of rebleeding.
- Medical and neurologic complications are extremely common and include hydrocephalus, increased intracranial pressure, seizures, delayed cerebral ischemia, hyponatremia, hypovolemia, and cardiac and pulmonary abnormalities.
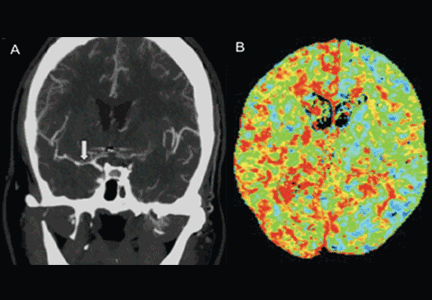
When the dissociation curve shifts to the left
A 48-year-old woman presented to the emergency department after 2 days of nonproductive cough, chest discomfort, worsening shortness of breath, and subjective fever. She had a history of systemic sclerosis. She was currently taking prednisone 20 mg daily and aspirin 81 mg daily.
Physical examination revealed tachypnea (28 breaths per minute), and bronchial breath sounds in the left lower chest posteriorly.
The initial laboratory workup revealed:
- Hemoglobin 106 g/L (reference range 115–155)
- Mean corpuscular volume 84 fL (80–100)
- White blood cell count 29.4 × 109/L (3.70–11.0), with 85% neutrophils
- Platelet count 180 × 109/L (150–350)
- Lactate dehydrogenase 312 U/L (100–220).
Chest radiography showed opacification of the lower lobe of the left lung.
She was admitted to the hospital and started treatment with intravenous azithromycin and ceftriaxone for presumed community-acquired pneumonia, based on the clinical presentation and findings on chest radiography. Because of her immunosuppression (due to chronic prednisone therapy) and her high lactate dehydrogenase level, Pneumocystis jirovecii pneumonia was suspected, and because she had a history of allergy to trimethoprim-sulfamethoxazole and pentamidine, she was started on dapsone.
During the next 24 hours, she developed worsening dyspnea, hypoxia, and cyanosis. She was placed on an air-entrainment mask, with a fraction of inspired oxygen of 0.5. Pulse oximetry showed an oxygen saturation of 85%, but arterial blood gas analysis indicated an oxyhemoglobin concentration of 95%.
THE ‘SATURATION GAP’
1. Which is most likely to have caused the discrepancy between the oxyhemoglobin concentration and the oxygen saturation by pulse oximetry in this patient?
- Methemoglobinemia
- Carbon monoxide poisoning
- Inappropriate placement of the pulse oximeter probe
- Pulmonary embolism
Methemoglobinemia is the most likely cause of the discrepancy between the oxyhemoglobin levels and the oxygen saturation by pulse oximetry, a phenomenon also known as the “saturation gap.” Other common causes are cyanide poisoning and carbon monoxide poisoning.
Carbon monoxide poisoning, however, does not explain our patient’s cyanosis. On the contrary, carbon monoxide poisoning can actually cause the patient’s lips and mucous membranes to appear unnaturally bright pink. Also, carbon monoxide poisoning raises the blood concentration of carboxyhemoglobin (which has a high affinity for oxygen), and this usually causes pulse oximetry to read inappropriately high, whereas in our patient it read low.
Incorrect placement of the pulse oximeter probe can result in an inaccurate measurement of oxygen saturation. Visualization of the waveform on the plethysmograph or the signal quality index can be used to assess adequate placement of the pulse oximeter probe. However, inadequate probe placement does not explain our patient’s dyspnea and cyanosis.
Pulmonary embolism can lead to hypoxia as a result of ventilation-perfusion mismatch. However, pulmonary embolism leading to low oxygen saturation on pulse oximetry will also lead to concomitantly low oxyhemoglobin levels as measured by arterial blood gas analysis, and this was not seen in our patient.
BACK TO OUR PATIENT
Because there was a discrepancy between our patient’s pulse oximetry reading and oxyhemoglobin concentration by arterial blood gas measurement, her methemoglobin level was checked and was found to be 30%, thus confirming the diagnosis of methemoglobinemia.
WHAT IS METHEMOGLOBINEMIA, AND WHAT CAUSES IT?
Oxygen is normally bound to iron in its ferrous (Fe2+) form in hemoglobin to form oxyhemoglobin. Oxidative stress in the body can cause iron to change from the ferrous to the ferric (Fe3+) state, forming methemoglobin. Methemoglobin is normally present in the blood in low levels (< 1% of the total hemoglobin), and ferric iron is reduced and recycled back to the ferrous form by NADH-cytochrome b5 reductase, an enzyme present in red blood cells. This protective mechanism maintains methemoglobin levels within safe limits. But increased production can lead to accumulation of methemoglobin, resulting in dyspnea and hypoxia and the condition referred to as methemoglobinemia.1
Increased levels of methemoglobin relative to normal hemoglobin cause tissue hypoxia by several mechanisms. Methemoglobin cannot efficiently carry oxygen; instead, it binds to water or to a hydroxide ion depending on the pH of the environment.2 Therefore, the hemoglobin molecule does not carry its usual load of oxygen, and hypoxia results from the reduced delivery of oxygen to tissues. In addition, an increased concentration of methemoglobin causes a leftward shift in the oxygen-hemoglobin dissociation curve, representing an increased affinity to bound oxygen in the remaining heme groups. The tightly bound oxygen is not adequately released at the tissue level, thus causing cellular hypoxia.
Methemoglobinemia is most often caused by exposure to an oxidizing chemical or drug that increases production of methemoglobin. In rare cases, it is caused by a congenital deficiency of NADH-cytochrome b5 reductase.3
2. Which of the following drugs can cause methemoglobinemia?
- Acetaminophen
- Dapsone
- Benzocaine
- Primaquine
All four of these drugs are common culprits for causing acquired methemoglobinemia; others include chloroquine, nitroglycerin, and sulfonamides.4–6
The increased production of methemoglobin caused by these drugs overwhelms the protective effect of reducing enzymes and can lead to an accumulation of methemoglobin. However, because of variability in cellular metabolism, not every person who takes these drugs develops dangerous levels of methemoglobin.

Dapsone and benzocaine are the most commonly encountered drugs known to cause methemoglobinemia (Table 1). Dapsone is an anti-inflammatory and antimicrobial agent most commonly used for treating lepromatous leprosy and dermatitis herpetiformis. It is also often prescribed for prophylaxis and treatment of P jirovecii pneumonia in immunosuppressed individuals.7 Benzocaine is a local anesthetic and was commonly used before procedures such as oral or dental surgery, transesophageal echocardiography, and endoscopy.8–10 Even low doses of benzocaine can lead to high levels of methemoglobinemia. However, the availability of other, safer anesthetics now limits the use of benzocaine in major US centers. In addition, the topical anesthetic Emla (lidocaine plus prilocaine) has been recently reported as a cause of methemoglobinemia in infants and children.11,12
Also, potentially fatal methemoglobinemia has been reported in patients with a deficiency of G-6-phosphate dehydrogenase (G6PD) who received rasburicase, a recombinant version of urate oxidase enzyme used to prevent and treat tumor lysis syndrome.13,14
Lastly, methemoglobinemia has been reported in patients with inflammatory bowel disease treated with mesalamine.
Although this adverse reaction is rare, clinicians should be aware of it, since these agents are commonly used in everyday medical practice.15
RECOGNIZING THE DANGER SIGNS
The clinical manifestations of methemoglobinemia are directly proportional to the percentage of methemoglobin in red blood cells. Cyanosis generally becomes apparent at concentrations around 15%, at which point the patient may still have no symptoms. Anxiety, lightheadedness, tachycardia, and dizziness manifest at levels of 20% to 30%. Fatigue, confusion, dizziness, tachypnea, and worsening tachycardia occur at levels of 30% to 50%. Levels of 50% to 70% cause coma, seizures, arrhythmias, and acidosis, and levels over 70% are considered lethal.16
While these levels provide a general guideline of symptomatology in an otherwise healthy person, it is important to remember that patients with underlying conditions such as anemia, lung disease (both of which our patient had), sepsis, thalassemia, G6PD deficiency, and sickle cell disease can manifest symptoms at lower concentrations of methemoglobin.1,17
Most patients who develop clinically significant levels of methemoglobin do so within the first few hours of starting one of the culprit drugs.
DIAGNOSIS: METHEMOGLOBINEMIA AND THE SATURATION GAP
In patients with methemoglobinemia, pulse oximetry gives lower values than arterial blood gas oxygen measurements. Regular pulse oximetry works by measuring light absorbance at two distinct wavelengths (660 and 940 nm) to calculate the ratio of oxyhemoglobin to deoxyhemoglobin. Methemoglobin absorbs light at both these wavelengths, thus lowering the pulse oximetry values.1
In contrast, oxygen saturation of arterial blood gas (oxyhemoglobin) is calculated indirectly from the concentration of dissolved oxygen in the blood and does not include oxygen bound to hemoglobin. Therefore, the measured arterial oxygen saturation is often normal in patients with methemoglobinemia since it relies only on inspired oxygen content and is independent of the methemoglobin concentration.18
Oxygen supplementation can raise the level of oxyhemoglobin, which is a measure of dissolved oxygen, but the oxygen saturation as measured by pulse oximetry remains largely unchanged—ie, the saturation gap. A difference of more than 5% between the oxygen saturation by pulse oximetry and blood gas analysis is abnormal. Patients with clinically significant methemoglobinemia usually have a saturation gap greater than 10%.
Several other unique features should raise suspicion of methemoglobinemia. It should be considered in a patient presenting with cyanosis out of proportion to the oxygen saturation and in a patient with low oxygen saturation and a normal chest radiograph. Other clues include blood that is chocolate-colored on gross examination, rather than the dark red of deoxygenated blood.
Co-oximetry measures oxygen saturation using different wavelengths of light to distinguish between fractions of oxyhemoglobin, deoxyhemoglobin, and methemoglobin, but it is not widely available.
THE NEXT STEP
3. What is the next step in the management of our patient?
- Discontinue the dapsone
- Start methylene blue
- Start hyperbaric oxygen
- Give sodium thiosulfate
- Discontinue dapsone and start methylene blue
The next step in her management should be to stop the dapsone and start an infusion of methylene blue. Hyperbaric oxygen is used in treating carbon monoxide poisoning, and sodium thiosulfate is used in treating cyanide toxicity. They would not be appropriate in this patient’s care.
MANAGEMENT OF ACQUIRED METHEMOGLOBINEMIA
The first, most critical step in managing acquired methemoglobinemia is to immediately discontinue the suspected offending agent. In most patients without a concomitant condition such as anemia or lung disease and with a methemoglobin level below 20%, discontinuing the offending agent may suffice. Patients with a level of 20% or greater and patients with cardiac and pulmonary disease, who develop symptoms at lower concentrations of methemoglobin, require infusion of methylene blue.
Methylene blue is converted to its reduced form, leukomethylene blue, by NADPH-methemoglobin reductase. As it is oxidized, leukomethylene blue reduces methemoglobin to hemoglobin. A dose of 1 mg/kg intravenously is given at first. The response is usually dramatic, with a reduction in methemoglobin levels and improvement in symptoms often within 30 to 60 minutes. If levels remain high, the dose can be repeated 1 hour later.19
A caveat: methylene blue therapy should be avoided in patients with complete G6PD deficiency. Methylene blue works through the enzyme NADPH-methemoglobin reductase, and since patients with G6PD deficiency lack this enzyme, methylene blue is ineffective. In fact, since it cannot be reduced, excessive methylene blue can oxidize hemoglobin to methemoglobin, further exacerbating the condition. In patients with partial G6PD deficiency, methylene blue is still recommended as a first-line treatment, but at a lower initial dose (0.3–0.5 mg/kg). However, in patients with significant hemolysis, an exchange transfusion is the only treatment option.
CASE CONCLUDED
Since dapsone was identified as the likely cause of methemoglobinemia in our patient, it was immediately discontinued. Because she was symptomatic, 70 mg of methylene blue was given intravenously. Over the next 60 minutes, her clinical condition improved significantly. A repeat methemoglobin measurement was 3%.
She was discharged home the next day on oral antibiotics to complete treatment for community-acquired pneumonia.
TAKE-HOME POINTS
- Consider methemoglobinemia in a patient with unexplained cyanosis.
- Pulse oximetry gives lower values than arterial blood gas oxygen measurements in patients with methemoglobinemia, and pulse oximetry readings do not improve with supplemental oxygen.
- A saturation gap greater than 5% strongly suggests methemoglobinemia.
- The diagnosis of methemoglobinemia is confirmed by measuring the methemoglobin concentration.
- Most healthy patients develop symptoms at methemoglobin levels of 20%, but patients with comorbidities can develop symptoms at lower levels.
- A number of drugs can cause methemoglobinemia, even at therapeutic dosages.
- Treatment is generally indicated in patients who have symptoms or in healthy patients who have a methemoglobin level of 20% or greater.
- Identifying and promptly discontinuing the causative agent and initiating methylene blue infusion (1 mg/kg over 5 minutes) is the preferred treatment.
- Cortazzo JA, Lichtman AD. Methemoglobinemia: a review and recommendations for management. J Cardiothorac Vasc Anesth 2014; 28:1055–1059.
- Margulies DR, Manookian CM. Methemoglobinemia as a cause of respiratory failure. J Trauma 2002; 52:796–797.
- Skold A, Cosco DL, Klein R. Methemoglobinemia: pathogenesis, diagnosis, and management. South Med J 2011; 104:757–761.
- Ash-Bernal R, Wise R, Wright SM. Acquired methemoglobinemia: a retrospective series of 138 cases at 2 teaching hospitals. Medicine (Baltimore) 2004; 83:265–273.
- Kanji HD, Mithani S, Boucher P, Dias VC, Yarema MC. Coma, metabolic acidosis, and methemoglobinemia in a patient with acetaminophen toxicity. J Popul Ther Clin Pharmacol 2013; 20:e207–e211.
- Kawasumi H, Tanaka E, Hoshi D, Kawaguchi Y, Yamanaka H. Methemoglobinemia induced by trimethoprim-sulfamethoxazole in a patient with systemic lupus erythematosus. Intern Med 2013; 52:1741–1743.
- Wieringa A, Bethlehem C, Hoogendoorn M, van der Maten J, van Roon EN. Very late recovery of dapsone-induced methemoglobinemia. Clin Toxicol (Phila) 2014; 52:80–81.
- Barclay JA, Ziemba SE, Ibrahim RB. Dapsone-induced methemoglobinemia: a primer for clinicians. Ann Pharmacother 2011; 45:1103–1115.
- Taleb M, Ashraf Z, Valavoor S, Tinkel J. Evaluation and management of acquired methemoglobinemia associated with topical benzocaine use. Am J Cardiovasc Drugs 2013; 13:325–330.
- Chowdhary S, Bukoye B, Bhansali AM, et al. Risk of topical anesthetic-induced methemoglobinemia: a 10-year retrospective case-control study. JAMA Intern Med 2013; 173:771–776.
- Larson A, Stidham T, Banerji S, Kaufman J. Seizures and methemoglobinemia in an infant after excessive EMLA application. Pediatr Emerg Care 2013; 29:377–379.
- Schmitt C, Matulic M, Kervégant M, et al. Methaemoglobinaemia in a child treated with Emla cream: circumstances and consequences of overdose [in French]. Ann Dermatol Venereol 2012; 139:824–827.
- Bucklin MH, Groth CM. Mortality following rasburicase-induced methemoglobinemia. Ann Pharmacother 2013; 47:1353–1358.
- Cheah CY, Lew TE, Seymour JF, Burbury K. Rasburicase causing severe oxidative hemolysis and methemoglobinemia in a patient with previously unrecognized glucose-6-phosphate dehydrogenase deficiency. Acta Haematol 2013; 130:254–259.
- Druez A, Rahier JF, Hébuterne X. Methaemoglobinaemia and renal failure following mesalazine for treatment of inflammatory bowel disease. J Crohns Colitis 2014; 8:900–901.
- Wright RO, Lewander WJ, Woolf AD. Methemoglobinemia: etiology, pharmacology, and clinical management. Ann Emerg Med 1999; 34:646–656.
- Groeper K, Katcher K, Tobias JD. Anesthetic management of a patient with methemoglobinemia. South Med J 2003; 96:504–509.
- Haymond S, Cariappa R, Eby CS, Scott MG. Laboratory assessment of oxygenation in methemoglobinemia. Clin Chem 2005; 51:434–444.
- Jang DH, Nelson LS, Hoffman RS. Methylene blue for distributive shock: a potential new use of an old antidote. J Med Toxicol 2013; 9:242–249.
A 48-year-old woman presented to the emergency department after 2 days of nonproductive cough, chest discomfort, worsening shortness of breath, and subjective fever. She had a history of systemic sclerosis. She was currently taking prednisone 20 mg daily and aspirin 81 mg daily.
Physical examination revealed tachypnea (28 breaths per minute), and bronchial breath sounds in the left lower chest posteriorly.
The initial laboratory workup revealed:
- Hemoglobin 106 g/L (reference range 115–155)
- Mean corpuscular volume 84 fL (80–100)
- White blood cell count 29.4 × 109/L (3.70–11.0), with 85% neutrophils
- Platelet count 180 × 109/L (150–350)
- Lactate dehydrogenase 312 U/L (100–220).
Chest radiography showed opacification of the lower lobe of the left lung.
She was admitted to the hospital and started treatment with intravenous azithromycin and ceftriaxone for presumed community-acquired pneumonia, based on the clinical presentation and findings on chest radiography. Because of her immunosuppression (due to chronic prednisone therapy) and her high lactate dehydrogenase level, Pneumocystis jirovecii pneumonia was suspected, and because she had a history of allergy to trimethoprim-sulfamethoxazole and pentamidine, she was started on dapsone.
During the next 24 hours, she developed worsening dyspnea, hypoxia, and cyanosis. She was placed on an air-entrainment mask, with a fraction of inspired oxygen of 0.5. Pulse oximetry showed an oxygen saturation of 85%, but arterial blood gas analysis indicated an oxyhemoglobin concentration of 95%.
THE ‘SATURATION GAP’
1. Which is most likely to have caused the discrepancy between the oxyhemoglobin concentration and the oxygen saturation by pulse oximetry in this patient?
- Methemoglobinemia
- Carbon monoxide poisoning
- Inappropriate placement of the pulse oximeter probe
- Pulmonary embolism
Methemoglobinemia is the most likely cause of the discrepancy between the oxyhemoglobin levels and the oxygen saturation by pulse oximetry, a phenomenon also known as the “saturation gap.” Other common causes are cyanide poisoning and carbon monoxide poisoning.
Carbon monoxide poisoning, however, does not explain our patient’s cyanosis. On the contrary, carbon monoxide poisoning can actually cause the patient’s lips and mucous membranes to appear unnaturally bright pink. Also, carbon monoxide poisoning raises the blood concentration of carboxyhemoglobin (which has a high affinity for oxygen), and this usually causes pulse oximetry to read inappropriately high, whereas in our patient it read low.
Incorrect placement of the pulse oximeter probe can result in an inaccurate measurement of oxygen saturation. Visualization of the waveform on the plethysmograph or the signal quality index can be used to assess adequate placement of the pulse oximeter probe. However, inadequate probe placement does not explain our patient’s dyspnea and cyanosis.
Pulmonary embolism can lead to hypoxia as a result of ventilation-perfusion mismatch. However, pulmonary embolism leading to low oxygen saturation on pulse oximetry will also lead to concomitantly low oxyhemoglobin levels as measured by arterial blood gas analysis, and this was not seen in our patient.
BACK TO OUR PATIENT
Because there was a discrepancy between our patient’s pulse oximetry reading and oxyhemoglobin concentration by arterial blood gas measurement, her methemoglobin level was checked and was found to be 30%, thus confirming the diagnosis of methemoglobinemia.
WHAT IS METHEMOGLOBINEMIA, AND WHAT CAUSES IT?
Oxygen is normally bound to iron in its ferrous (Fe2+) form in hemoglobin to form oxyhemoglobin. Oxidative stress in the body can cause iron to change from the ferrous to the ferric (Fe3+) state, forming methemoglobin. Methemoglobin is normally present in the blood in low levels (< 1% of the total hemoglobin), and ferric iron is reduced and recycled back to the ferrous form by NADH-cytochrome b5 reductase, an enzyme present in red blood cells. This protective mechanism maintains methemoglobin levels within safe limits. But increased production can lead to accumulation of methemoglobin, resulting in dyspnea and hypoxia and the condition referred to as methemoglobinemia.1
Increased levels of methemoglobin relative to normal hemoglobin cause tissue hypoxia by several mechanisms. Methemoglobin cannot efficiently carry oxygen; instead, it binds to water or to a hydroxide ion depending on the pH of the environment.2 Therefore, the hemoglobin molecule does not carry its usual load of oxygen, and hypoxia results from the reduced delivery of oxygen to tissues. In addition, an increased concentration of methemoglobin causes a leftward shift in the oxygen-hemoglobin dissociation curve, representing an increased affinity to bound oxygen in the remaining heme groups. The tightly bound oxygen is not adequately released at the tissue level, thus causing cellular hypoxia.
Methemoglobinemia is most often caused by exposure to an oxidizing chemical or drug that increases production of methemoglobin. In rare cases, it is caused by a congenital deficiency of NADH-cytochrome b5 reductase.3
2. Which of the following drugs can cause methemoglobinemia?
- Acetaminophen
- Dapsone
- Benzocaine
- Primaquine
All four of these drugs are common culprits for causing acquired methemoglobinemia; others include chloroquine, nitroglycerin, and sulfonamides.4–6
The increased production of methemoglobin caused by these drugs overwhelms the protective effect of reducing enzymes and can lead to an accumulation of methemoglobin. However, because of variability in cellular metabolism, not every person who takes these drugs develops dangerous levels of methemoglobin.
Dapsone and benzocaine are the most commonly encountered drugs known to cause methemoglobinemia (Table 1). Dapsone is an anti-inflammatory and antimicrobial agent most commonly used for treating lepromatous leprosy and dermatitis herpetiformis. It is also often prescribed for prophylaxis and treatment of P jirovecii pneumonia in immunosuppressed individuals.7 Benzocaine is a local anesthetic and was commonly used before procedures such as oral or dental surgery, transesophageal echocardiography, and endoscopy.8–10 Even low doses of benzocaine can lead to high levels of methemoglobinemia. However, the availability of other, safer anesthetics now limits the use of benzocaine in major US centers. In addition, the topical anesthetic Emla (lidocaine plus prilocaine) has been recently reported as a cause of methemoglobinemia in infants and children.11,12
Also, potentially fatal methemoglobinemia has been reported in patients with a deficiency of G-6-phosphate dehydrogenase (G6PD) who received rasburicase, a recombinant version of urate oxidase enzyme used to prevent and treat tumor lysis syndrome.13,14
Lastly, methemoglobinemia has been reported in patients with inflammatory bowel disease treated with mesalamine.
Although this adverse reaction is rare, clinicians should be aware of it, since these agents are commonly used in everyday medical practice.15
RECOGNIZING THE DANGER SIGNS
The clinical manifestations of methemoglobinemia are directly proportional to the percentage of methemoglobin in red blood cells. Cyanosis generally becomes apparent at concentrations around 15%, at which point the patient may still have no symptoms. Anxiety, lightheadedness, tachycardia, and dizziness manifest at levels of 20% to 30%. Fatigue, confusion, dizziness, tachypnea, and worsening tachycardia occur at levels of 30% to 50%. Levels of 50% to 70% cause coma, seizures, arrhythmias, and acidosis, and levels over 70% are considered lethal.16
While these levels provide a general guideline of symptomatology in an otherwise healthy person, it is important to remember that patients with underlying conditions such as anemia, lung disease (both of which our patient had), sepsis, thalassemia, G6PD deficiency, and sickle cell disease can manifest symptoms at lower concentrations of methemoglobin.1,17
Most patients who develop clinically significant levels of methemoglobin do so within the first few hours of starting one of the culprit drugs.
DIAGNOSIS: METHEMOGLOBINEMIA AND THE SATURATION GAP
In patients with methemoglobinemia, pulse oximetry gives lower values than arterial blood gas oxygen measurements. Regular pulse oximetry works by measuring light absorbance at two distinct wavelengths (660 and 940 nm) to calculate the ratio of oxyhemoglobin to deoxyhemoglobin. Methemoglobin absorbs light at both these wavelengths, thus lowering the pulse oximetry values.1
In contrast, oxygen saturation of arterial blood gas (oxyhemoglobin) is calculated indirectly from the concentration of dissolved oxygen in the blood and does not include oxygen bound to hemoglobin. Therefore, the measured arterial oxygen saturation is often normal in patients with methemoglobinemia since it relies only on inspired oxygen content and is independent of the methemoglobin concentration.18
Oxygen supplementation can raise the level of oxyhemoglobin, which is a measure of dissolved oxygen, but the oxygen saturation as measured by pulse oximetry remains largely unchanged—ie, the saturation gap. A difference of more than 5% between the oxygen saturation by pulse oximetry and blood gas analysis is abnormal. Patients with clinically significant methemoglobinemia usually have a saturation gap greater than 10%.
Several other unique features should raise suspicion of methemoglobinemia. It should be considered in a patient presenting with cyanosis out of proportion to the oxygen saturation and in a patient with low oxygen saturation and a normal chest radiograph. Other clues include blood that is chocolate-colored on gross examination, rather than the dark red of deoxygenated blood.
Co-oximetry measures oxygen saturation using different wavelengths of light to distinguish between fractions of oxyhemoglobin, deoxyhemoglobin, and methemoglobin, but it is not widely available.
THE NEXT STEP
3. What is the next step in the management of our patient?
- Discontinue the dapsone
- Start methylene blue
- Start hyperbaric oxygen
- Give sodium thiosulfate
- Discontinue dapsone and start methylene blue
The next step in her management should be to stop the dapsone and start an infusion of methylene blue. Hyperbaric oxygen is used in treating carbon monoxide poisoning, and sodium thiosulfate is used in treating cyanide toxicity. They would not be appropriate in this patient’s care.
MANAGEMENT OF ACQUIRED METHEMOGLOBINEMIA
The first, most critical step in managing acquired methemoglobinemia is to immediately discontinue the suspected offending agent. In most patients without a concomitant condition such as anemia or lung disease and with a methemoglobin level below 20%, discontinuing the offending agent may suffice. Patients with a level of 20% or greater and patients with cardiac and pulmonary disease, who develop symptoms at lower concentrations of methemoglobin, require infusion of methylene blue.
Methylene blue is converted to its reduced form, leukomethylene blue, by NADPH-methemoglobin reductase. As it is oxidized, leukomethylene blue reduces methemoglobin to hemoglobin. A dose of 1 mg/kg intravenously is given at first. The response is usually dramatic, with a reduction in methemoglobin levels and improvement in symptoms often within 30 to 60 minutes. If levels remain high, the dose can be repeated 1 hour later.19
A caveat: methylene blue therapy should be avoided in patients with complete G6PD deficiency. Methylene blue works through the enzyme NADPH-methemoglobin reductase, and since patients with G6PD deficiency lack this enzyme, methylene blue is ineffective. In fact, since it cannot be reduced, excessive methylene blue can oxidize hemoglobin to methemoglobin, further exacerbating the condition. In patients with partial G6PD deficiency, methylene blue is still recommended as a first-line treatment, but at a lower initial dose (0.3–0.5 mg/kg). However, in patients with significant hemolysis, an exchange transfusion is the only treatment option.
CASE CONCLUDED
Since dapsone was identified as the likely cause of methemoglobinemia in our patient, it was immediately discontinued. Because she was symptomatic, 70 mg of methylene blue was given intravenously. Over the next 60 minutes, her clinical condition improved significantly. A repeat methemoglobin measurement was 3%.
She was discharged home the next day on oral antibiotics to complete treatment for community-acquired pneumonia.
TAKE-HOME POINTS
- Consider methemoglobinemia in a patient with unexplained cyanosis.
- Pulse oximetry gives lower values than arterial blood gas oxygen measurements in patients with methemoglobinemia, and pulse oximetry readings do not improve with supplemental oxygen.
- A saturation gap greater than 5% strongly suggests methemoglobinemia.
- The diagnosis of methemoglobinemia is confirmed by measuring the methemoglobin concentration.
- Most healthy patients develop symptoms at methemoglobin levels of 20%, but patients with comorbidities can develop symptoms at lower levels.
- A number of drugs can cause methemoglobinemia, even at therapeutic dosages.
- Treatment is generally indicated in patients who have symptoms or in healthy patients who have a methemoglobin level of 20% or greater.
- Identifying and promptly discontinuing the causative agent and initiating methylene blue infusion (1 mg/kg over 5 minutes) is the preferred treatment.
A 48-year-old woman presented to the emergency department after 2 days of nonproductive cough, chest discomfort, worsening shortness of breath, and subjective fever. She had a history of systemic sclerosis. She was currently taking prednisone 20 mg daily and aspirin 81 mg daily.
Physical examination revealed tachypnea (28 breaths per minute), and bronchial breath sounds in the left lower chest posteriorly.
The initial laboratory workup revealed:
- Hemoglobin 106 g/L (reference range 115–155)
- Mean corpuscular volume 84 fL (80–100)
- White blood cell count 29.4 × 109/L (3.70–11.0), with 85% neutrophils
- Platelet count 180 × 109/L (150–350)
- Lactate dehydrogenase 312 U/L (100–220).
Chest radiography showed opacification of the lower lobe of the left lung.
She was admitted to the hospital and started treatment with intravenous azithromycin and ceftriaxone for presumed community-acquired pneumonia, based on the clinical presentation and findings on chest radiography. Because of her immunosuppression (due to chronic prednisone therapy) and her high lactate dehydrogenase level, Pneumocystis jirovecii pneumonia was suspected, and because she had a history of allergy to trimethoprim-sulfamethoxazole and pentamidine, she was started on dapsone.
During the next 24 hours, she developed worsening dyspnea, hypoxia, and cyanosis. She was placed on an air-entrainment mask, with a fraction of inspired oxygen of 0.5. Pulse oximetry showed an oxygen saturation of 85%, but arterial blood gas analysis indicated an oxyhemoglobin concentration of 95%.
THE ‘SATURATION GAP’
1. Which is most likely to have caused the discrepancy between the oxyhemoglobin concentration and the oxygen saturation by pulse oximetry in this patient?
- Methemoglobinemia
- Carbon monoxide poisoning
- Inappropriate placement of the pulse oximeter probe
- Pulmonary embolism
Methemoglobinemia is the most likely cause of the discrepancy between the oxyhemoglobin levels and the oxygen saturation by pulse oximetry, a phenomenon also known as the “saturation gap.” Other common causes are cyanide poisoning and carbon monoxide poisoning.
Carbon monoxide poisoning, however, does not explain our patient’s cyanosis. On the contrary, carbon monoxide poisoning can actually cause the patient’s lips and mucous membranes to appear unnaturally bright pink. Also, carbon monoxide poisoning raises the blood concentration of carboxyhemoglobin (which has a high affinity for oxygen), and this usually causes pulse oximetry to read inappropriately high, whereas in our patient it read low.
Incorrect placement of the pulse oximeter probe can result in an inaccurate measurement of oxygen saturation. Visualization of the waveform on the plethysmograph or the signal quality index can be used to assess adequate placement of the pulse oximeter probe. However, inadequate probe placement does not explain our patient’s dyspnea and cyanosis.
Pulmonary embolism can lead to hypoxia as a result of ventilation-perfusion mismatch. However, pulmonary embolism leading to low oxygen saturation on pulse oximetry will also lead to concomitantly low oxyhemoglobin levels as measured by arterial blood gas analysis, and this was not seen in our patient.
BACK TO OUR PATIENT
Because there was a discrepancy between our patient’s pulse oximetry reading and oxyhemoglobin concentration by arterial blood gas measurement, her methemoglobin level was checked and was found to be 30%, thus confirming the diagnosis of methemoglobinemia.
WHAT IS METHEMOGLOBINEMIA, AND WHAT CAUSES IT?
Oxygen is normally bound to iron in its ferrous (Fe2+) form in hemoglobin to form oxyhemoglobin. Oxidative stress in the body can cause iron to change from the ferrous to the ferric (Fe3+) state, forming methemoglobin. Methemoglobin is normally present in the blood in low levels (< 1% of the total hemoglobin), and ferric iron is reduced and recycled back to the ferrous form by NADH-cytochrome b5 reductase, an enzyme present in red blood cells. This protective mechanism maintains methemoglobin levels within safe limits. But increased production can lead to accumulation of methemoglobin, resulting in dyspnea and hypoxia and the condition referred to as methemoglobinemia.1
Increased levels of methemoglobin relative to normal hemoglobin cause tissue hypoxia by several mechanisms. Methemoglobin cannot efficiently carry oxygen; instead, it binds to water or to a hydroxide ion depending on the pH of the environment.2 Therefore, the hemoglobin molecule does not carry its usual load of oxygen, and hypoxia results from the reduced delivery of oxygen to tissues. In addition, an increased concentration of methemoglobin causes a leftward shift in the oxygen-hemoglobin dissociation curve, representing an increased affinity to bound oxygen in the remaining heme groups. The tightly bound oxygen is not adequately released at the tissue level, thus causing cellular hypoxia.
Methemoglobinemia is most often caused by exposure to an oxidizing chemical or drug that increases production of methemoglobin. In rare cases, it is caused by a congenital deficiency of NADH-cytochrome b5 reductase.3
2. Which of the following drugs can cause methemoglobinemia?
- Acetaminophen
- Dapsone
- Benzocaine
- Primaquine
All four of these drugs are common culprits for causing acquired methemoglobinemia; others include chloroquine, nitroglycerin, and sulfonamides.4–6
The increased production of methemoglobin caused by these drugs overwhelms the protective effect of reducing enzymes and can lead to an accumulation of methemoglobin. However, because of variability in cellular metabolism, not every person who takes these drugs develops dangerous levels of methemoglobin.
Dapsone and benzocaine are the most commonly encountered drugs known to cause methemoglobinemia (Table 1). Dapsone is an anti-inflammatory and antimicrobial agent most commonly used for treating lepromatous leprosy and dermatitis herpetiformis. It is also often prescribed for prophylaxis and treatment of P jirovecii pneumonia in immunosuppressed individuals.7 Benzocaine is a local anesthetic and was commonly used before procedures such as oral or dental surgery, transesophageal echocardiography, and endoscopy.8–10 Even low doses of benzocaine can lead to high levels of methemoglobinemia. However, the availability of other, safer anesthetics now limits the use of benzocaine in major US centers. In addition, the topical anesthetic Emla (lidocaine plus prilocaine) has been recently reported as a cause of methemoglobinemia in infants and children.11,12
Also, potentially fatal methemoglobinemia has been reported in patients with a deficiency of G-6-phosphate dehydrogenase (G6PD) who received rasburicase, a recombinant version of urate oxidase enzyme used to prevent and treat tumor lysis syndrome.13,14
Lastly, methemoglobinemia has been reported in patients with inflammatory bowel disease treated with mesalamine.
Although this adverse reaction is rare, clinicians should be aware of it, since these agents are commonly used in everyday medical practice.15
RECOGNIZING THE DANGER SIGNS
The clinical manifestations of methemoglobinemia are directly proportional to the percentage of methemoglobin in red blood cells. Cyanosis generally becomes apparent at concentrations around 15%, at which point the patient may still have no symptoms. Anxiety, lightheadedness, tachycardia, and dizziness manifest at levels of 20% to 30%. Fatigue, confusion, dizziness, tachypnea, and worsening tachycardia occur at levels of 30% to 50%. Levels of 50% to 70% cause coma, seizures, arrhythmias, and acidosis, and levels over 70% are considered lethal.16
While these levels provide a general guideline of symptomatology in an otherwise healthy person, it is important to remember that patients with underlying conditions such as anemia, lung disease (both of which our patient had), sepsis, thalassemia, G6PD deficiency, and sickle cell disease can manifest symptoms at lower concentrations of methemoglobin.1,17
Most patients who develop clinically significant levels of methemoglobin do so within the first few hours of starting one of the culprit drugs.
DIAGNOSIS: METHEMOGLOBINEMIA AND THE SATURATION GAP
In patients with methemoglobinemia, pulse oximetry gives lower values than arterial blood gas oxygen measurements. Regular pulse oximetry works by measuring light absorbance at two distinct wavelengths (660 and 940 nm) to calculate the ratio of oxyhemoglobin to deoxyhemoglobin. Methemoglobin absorbs light at both these wavelengths, thus lowering the pulse oximetry values.1
In contrast, oxygen saturation of arterial blood gas (oxyhemoglobin) is calculated indirectly from the concentration of dissolved oxygen in the blood and does not include oxygen bound to hemoglobin. Therefore, the measured arterial oxygen saturation is often normal in patients with methemoglobinemia since it relies only on inspired oxygen content and is independent of the methemoglobin concentration.18
Oxygen supplementation can raise the level of oxyhemoglobin, which is a measure of dissolved oxygen, but the oxygen saturation as measured by pulse oximetry remains largely unchanged—ie, the saturation gap. A difference of more than 5% between the oxygen saturation by pulse oximetry and blood gas analysis is abnormal. Patients with clinically significant methemoglobinemia usually have a saturation gap greater than 10%.
Several other unique features should raise suspicion of methemoglobinemia. It should be considered in a patient presenting with cyanosis out of proportion to the oxygen saturation and in a patient with low oxygen saturation and a normal chest radiograph. Other clues include blood that is chocolate-colored on gross examination, rather than the dark red of deoxygenated blood.
Co-oximetry measures oxygen saturation using different wavelengths of light to distinguish between fractions of oxyhemoglobin, deoxyhemoglobin, and methemoglobin, but it is not widely available.
THE NEXT STEP
3. What is the next step in the management of our patient?
- Discontinue the dapsone
- Start methylene blue
- Start hyperbaric oxygen
- Give sodium thiosulfate
- Discontinue dapsone and start methylene blue
The next step in her management should be to stop the dapsone and start an infusion of methylene blue. Hyperbaric oxygen is used in treating carbon monoxide poisoning, and sodium thiosulfate is used in treating cyanide toxicity. They would not be appropriate in this patient’s care.
MANAGEMENT OF ACQUIRED METHEMOGLOBINEMIA
The first, most critical step in managing acquired methemoglobinemia is to immediately discontinue the suspected offending agent. In most patients without a concomitant condition such as anemia or lung disease and with a methemoglobin level below 20%, discontinuing the offending agent may suffice. Patients with a level of 20% or greater and patients with cardiac and pulmonary disease, who develop symptoms at lower concentrations of methemoglobin, require infusion of methylene blue.
Methylene blue is converted to its reduced form, leukomethylene blue, by NADPH-methemoglobin reductase. As it is oxidized, leukomethylene blue reduces methemoglobin to hemoglobin. A dose of 1 mg/kg intravenously is given at first. The response is usually dramatic, with a reduction in methemoglobin levels and improvement in symptoms often within 30 to 60 minutes. If levels remain high, the dose can be repeated 1 hour later.19
A caveat: methylene blue therapy should be avoided in patients with complete G6PD deficiency. Methylene blue works through the enzyme NADPH-methemoglobin reductase, and since patients with G6PD deficiency lack this enzyme, methylene blue is ineffective. In fact, since it cannot be reduced, excessive methylene blue can oxidize hemoglobin to methemoglobin, further exacerbating the condition. In patients with partial G6PD deficiency, methylene blue is still recommended as a first-line treatment, but at a lower initial dose (0.3–0.5 mg/kg). However, in patients with significant hemolysis, an exchange transfusion is the only treatment option.
CASE CONCLUDED
Since dapsone was identified as the likely cause of methemoglobinemia in our patient, it was immediately discontinued. Because she was symptomatic, 70 mg of methylene blue was given intravenously. Over the next 60 minutes, her clinical condition improved significantly. A repeat methemoglobin measurement was 3%.
She was discharged home the next day on oral antibiotics to complete treatment for community-acquired pneumonia.
TAKE-HOME POINTS
- Consider methemoglobinemia in a patient with unexplained cyanosis.
- Pulse oximetry gives lower values than arterial blood gas oxygen measurements in patients with methemoglobinemia, and pulse oximetry readings do not improve with supplemental oxygen.
- A saturation gap greater than 5% strongly suggests methemoglobinemia.
- The diagnosis of methemoglobinemia is confirmed by measuring the methemoglobin concentration.
- Most healthy patients develop symptoms at methemoglobin levels of 20%, but patients with comorbidities can develop symptoms at lower levels.
- A number of drugs can cause methemoglobinemia, even at therapeutic dosages.
- Treatment is generally indicated in patients who have symptoms or in healthy patients who have a methemoglobin level of 20% or greater.
- Identifying and promptly discontinuing the causative agent and initiating methylene blue infusion (1 mg/kg over 5 minutes) is the preferred treatment.
- Cortazzo JA, Lichtman AD. Methemoglobinemia: a review and recommendations for management. J Cardiothorac Vasc Anesth 2014; 28:1055–1059.
- Margulies DR, Manookian CM. Methemoglobinemia as a cause of respiratory failure. J Trauma 2002; 52:796–797.
- Skold A, Cosco DL, Klein R. Methemoglobinemia: pathogenesis, diagnosis, and management. South Med J 2011; 104:757–761.
- Ash-Bernal R, Wise R, Wright SM. Acquired methemoglobinemia: a retrospective series of 138 cases at 2 teaching hospitals. Medicine (Baltimore) 2004; 83:265–273.
- Kanji HD, Mithani S, Boucher P, Dias VC, Yarema MC. Coma, metabolic acidosis, and methemoglobinemia in a patient with acetaminophen toxicity. J Popul Ther Clin Pharmacol 2013; 20:e207–e211.
- Kawasumi H, Tanaka E, Hoshi D, Kawaguchi Y, Yamanaka H. Methemoglobinemia induced by trimethoprim-sulfamethoxazole in a patient with systemic lupus erythematosus. Intern Med 2013; 52:1741–1743.
- Wieringa A, Bethlehem C, Hoogendoorn M, van der Maten J, van Roon EN. Very late recovery of dapsone-induced methemoglobinemia. Clin Toxicol (Phila) 2014; 52:80–81.
- Barclay JA, Ziemba SE, Ibrahim RB. Dapsone-induced methemoglobinemia: a primer for clinicians. Ann Pharmacother 2011; 45:1103–1115.
- Taleb M, Ashraf Z, Valavoor S, Tinkel J. Evaluation and management of acquired methemoglobinemia associated with topical benzocaine use. Am J Cardiovasc Drugs 2013; 13:325–330.
- Chowdhary S, Bukoye B, Bhansali AM, et al. Risk of topical anesthetic-induced methemoglobinemia: a 10-year retrospective case-control study. JAMA Intern Med 2013; 173:771–776.
- Larson A, Stidham T, Banerji S, Kaufman J. Seizures and methemoglobinemia in an infant after excessive EMLA application. Pediatr Emerg Care 2013; 29:377–379.
- Schmitt C, Matulic M, Kervégant M, et al. Methaemoglobinaemia in a child treated with Emla cream: circumstances and consequences of overdose [in French]. Ann Dermatol Venereol 2012; 139:824–827.
- Bucklin MH, Groth CM. Mortality following rasburicase-induced methemoglobinemia. Ann Pharmacother 2013; 47:1353–1358.
- Cheah CY, Lew TE, Seymour JF, Burbury K. Rasburicase causing severe oxidative hemolysis and methemoglobinemia in a patient with previously unrecognized glucose-6-phosphate dehydrogenase deficiency. Acta Haematol 2013; 130:254–259.
- Druez A, Rahier JF, Hébuterne X. Methaemoglobinaemia and renal failure following mesalazine for treatment of inflammatory bowel disease. J Crohns Colitis 2014; 8:900–901.
- Wright RO, Lewander WJ, Woolf AD. Methemoglobinemia: etiology, pharmacology, and clinical management. Ann Emerg Med 1999; 34:646–656.
- Groeper K, Katcher K, Tobias JD. Anesthetic management of a patient with methemoglobinemia. South Med J 2003; 96:504–509.
- Haymond S, Cariappa R, Eby CS, Scott MG. Laboratory assessment of oxygenation in methemoglobinemia. Clin Chem 2005; 51:434–444.
- Jang DH, Nelson LS, Hoffman RS. Methylene blue for distributive shock: a potential new use of an old antidote. J Med Toxicol 2013; 9:242–249.
- Cortazzo JA, Lichtman AD. Methemoglobinemia: a review and recommendations for management. J Cardiothorac Vasc Anesth 2014; 28:1055–1059.
- Margulies DR, Manookian CM. Methemoglobinemia as a cause of respiratory failure. J Trauma 2002; 52:796–797.
- Skold A, Cosco DL, Klein R. Methemoglobinemia: pathogenesis, diagnosis, and management. South Med J 2011; 104:757–761.
- Ash-Bernal R, Wise R, Wright SM. Acquired methemoglobinemia: a retrospective series of 138 cases at 2 teaching hospitals. Medicine (Baltimore) 2004; 83:265–273.
- Kanji HD, Mithani S, Boucher P, Dias VC, Yarema MC. Coma, metabolic acidosis, and methemoglobinemia in a patient with acetaminophen toxicity. J Popul Ther Clin Pharmacol 2013; 20:e207–e211.
- Kawasumi H, Tanaka E, Hoshi D, Kawaguchi Y, Yamanaka H. Methemoglobinemia induced by trimethoprim-sulfamethoxazole in a patient with systemic lupus erythematosus. Intern Med 2013; 52:1741–1743.
- Wieringa A, Bethlehem C, Hoogendoorn M, van der Maten J, van Roon EN. Very late recovery of dapsone-induced methemoglobinemia. Clin Toxicol (Phila) 2014; 52:80–81.
- Barclay JA, Ziemba SE, Ibrahim RB. Dapsone-induced methemoglobinemia: a primer for clinicians. Ann Pharmacother 2011; 45:1103–1115.
- Taleb M, Ashraf Z, Valavoor S, Tinkel J. Evaluation and management of acquired methemoglobinemia associated with topical benzocaine use. Am J Cardiovasc Drugs 2013; 13:325–330.
- Chowdhary S, Bukoye B, Bhansali AM, et al. Risk of topical anesthetic-induced methemoglobinemia: a 10-year retrospective case-control study. JAMA Intern Med 2013; 173:771–776.
- Larson A, Stidham T, Banerji S, Kaufman J. Seizures and methemoglobinemia in an infant after excessive EMLA application. Pediatr Emerg Care 2013; 29:377–379.
- Schmitt C, Matulic M, Kervégant M, et al. Methaemoglobinaemia in a child treated with Emla cream: circumstances and consequences of overdose [in French]. Ann Dermatol Venereol 2012; 139:824–827.
- Bucklin MH, Groth CM. Mortality following rasburicase-induced methemoglobinemia. Ann Pharmacother 2013; 47:1353–1358.
- Cheah CY, Lew TE, Seymour JF, Burbury K. Rasburicase causing severe oxidative hemolysis and methemoglobinemia in a patient with previously unrecognized glucose-6-phosphate dehydrogenase deficiency. Acta Haematol 2013; 130:254–259.
- Druez A, Rahier JF, Hébuterne X. Methaemoglobinaemia and renal failure following mesalazine for treatment of inflammatory bowel disease. J Crohns Colitis 2014; 8:900–901.
- Wright RO, Lewander WJ, Woolf AD. Methemoglobinemia: etiology, pharmacology, and clinical management. Ann Emerg Med 1999; 34:646–656.
- Groeper K, Katcher K, Tobias JD. Anesthetic management of a patient with methemoglobinemia. South Med J 2003; 96:504–509.
- Haymond S, Cariappa R, Eby CS, Scott MG. Laboratory assessment of oxygenation in methemoglobinemia. Clin Chem 2005; 51:434–444.
- Jang DH, Nelson LS, Hoffman RS. Methylene blue for distributive shock: a potential new use of an old antidote. J Med Toxicol 2013; 9:242–249.