User login
Thrown From Motorcycle
ANSWER
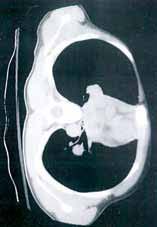
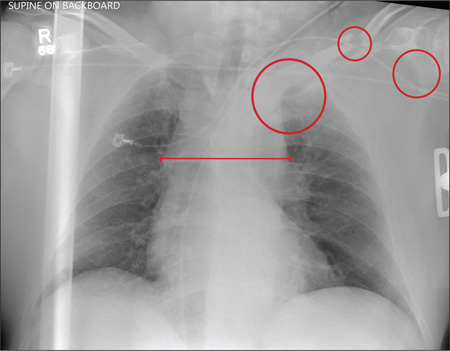
Several findings are evident from this radiograph. First, the quality is slightly diminished due to the patient’s size and artifact from the backboard. The patient’s mediastinum is somewhat widened, which is concerning for possible occult chest/vascular injury. There is some haziness within the left apical region suggestive of a hemothorax; no definite pneumothorax is seen. The left clavicle is fractured and displaced, and the left scapula is fractured as well.
ANSWER
Several findings are evident from this radiograph. First, the quality is slightly diminished due to the patient’s size and artifact from the backboard. The patient’s mediastinum is somewhat widened, which is concerning for possible occult chest/vascular injury. There is some haziness within the left apical region suggestive of a hemothorax; no definite pneumothorax is seen. The left clavicle is fractured and displaced, and the left scapula is fractured as well.
ANSWER
Several findings are evident from this radiograph. First, the quality is slightly diminished due to the patient’s size and artifact from the backboard. The patient’s mediastinum is somewhat widened, which is concerning for possible occult chest/vascular injury. There is some haziness within the left apical region suggestive of a hemothorax; no definite pneumothorax is seen. The left clavicle is fractured and displaced, and the left scapula is fractured as well.
A 57-year-old man is brought to your facility as a trauma code. He was riding a motorcycle on the highway, traveling approximately 45 to 50 mph, when the car in front of him abruptly stopped. He hit the car and was thrown from his bike. He believes he briefly lost consciousness but recalls emergency personnel tending to him. On arrival, he is awake and alert, complaining of pain in his neck, left arm, and left lower leg. Medical history is significant for borderline hypertension and a previous accident that resulted in an emergency laparotomy. Primary survey reveals stable vital signs: blood pressure of 157/100 mm Hg; heart rate, 110 beats/min; respiratory rate, 20 breaths/min; and O2 saturation, 98% with supplemental oxygen. Pupils are equal and reactive; there are slightly decreased breath sounds on the left side. Abdominal exam appears benign. There is decreased mobility and pain in the patient’s left upper and left lower extremities, although no obvious deformity is noted. Preliminary chest radiograph is obtained before the patient is sent for CT. What is your impression?
Capturing the Impact of Language Barriers on Asthma Management During an Emergency Department Visit
Study Overview
Objective. To compare rates of asthma action plan use in limited English proficiency (LEP) caregivers compared with English proficient (EP) caregivers.
Design. Cross-sectional survey.
Participants and setting. A convenience sample of 107 Latino caregivers of children with asthma at an urban academic emergency department (ED). Surveys in the preferred language of the patient (English or Spanish, with the translated version previously validated) were distributed at the time of the ED visit. Interpreters were utilized when requested.
Main outcome measure. Caregiver use of an asthma action plan.
Main results. 51 LEP caregivers and 56 EP caregivers completed the survey. Mothers completed the surveys 87% of the time and the average age of patients was 4 years. Among the EP caregivers, 64% reported using an asthma action plan, while only 39% of the LEP caregivers reported using one. The difference was statistally significant (P = 0.01). Through both correlations and regressions, English proficiency was the only variable (others included health insurance status and level of caregiver education) that showed a significant effect on asthma action plan use.
Conclusions. Children whose caregiver had LEP were significantly less likely to have and use an asthma action plan. Asthma education in the language of choice of the patient may help improve asthma care.
Commentary
With 20% of US households now speaking a language other than English at home [1], language barriers between providers and patients present multiple challenges to health services delivery and can significantly contribute to immigrant health disparities. Despite US laws and multiple federal agency policies requiring the use of interpreters during health care encounters, organizations continue to fall short of providing interpreter services and often lack adequate or equivalent materials for patient education. Too often, providers overestimate their language skills [2,3], use colleagues as ad hoc interpreters out of convenience [4], or rely on family members for interpretation [4]—a practice that is universally discouraged.
Recent research does suggest that the timing of interpreter use is critical. In planned encounters such as primary care visits, interpreters can and should be scheduled for visits when a language-concordant provider is not available. During hospitalizations, including ED visits, interpreters are most effective when used on admission, during patient teaching, and upon discharge, and the timing of these visits has been shown to affect length of stay and readmission rates [5,6].
This study magnifies the consequences of failing to provide language-concordant services to patients and their caregivers. It also helps to identify one of the sources of pediatric asthma health disparities in Latino populations. The emphasis on the role of the caregiver in action plan utilization is a unique aspect of this study and it is one of the first to examine the issue in this way. It highlights the importance of caregivers in health system transitions and illustrates how a language barrier can potentially impact transitions.
The authors’ explicit use of a power analysis to calculate their sample size is a strength of the study. Furthermore, the authors differentiated their respondents by country of origin, something that rarely occurs in studies of Latinos [7], and allows the reader to differentiate the impact of the intervention at a micro level within this population. The presentation of Spanish language quotes with their translations within the manuscript provides transparency for bilingual readers to verify the accuracy of the authors’ translation.
There are, however, a number of methodological issues that should be noted. The authors acknowledge that they did not account for asthma severity in the survey nor control for it in the analysis, did not assess health literacy, and did not differentiate their results based on country of origin. The latter point is important because the immigration experience and demographic profiles of Latinos differs significantly by country of origin and could factor in to action plan use. The translation process used for survey instrument translation also did not illustrate how it accounted for the well-established linguistic variation that occurs in the Spanish language. Additionally, US census data shows that the main countries of origin of Latinos in the service area of the study are Puerto Rico, Ecuador, and Mexico [1]. The survey itself had Ecuador as a write in and Dominican as a response option. The combination presented in the survey reflects the Latino demographic composition in the nearest large urban area. Thus, when collecting country of origin data on immigrant patients, country choices should reflect local demographics and not national trends for maximum precision.
Another concern is that Spanish language literacy was not assessed. Many Latino immigrants may have limited reading ability in Spanish. For Mexican immigrants in particular, Spanish may be a second language after their indigenous language. This is also true for some South American Latino immigrants from the Andean region. Many Latino immigrants come to the United States with less than an 8th grade education and likely come from educational systems of poor quality, which subsequently affects their Spanish language reading and writing skills [8]. Assessing education level based on US equivalents is not an accurate way to gauge literacy. Thus, assessing reading literacy in Spanish before surveying patients would have been a useful step that could have further refined the results. These factors will have implications for action plan utilization and implementation for any chronic disease.
Providers often think that language barriers are an obvious factor in health disparities and service delivery, but few studies have actually captured or quantified the effects of language barriers on health outcomes. Most studies only identify language barriers as an access issue. This study provides a good illustration of the impact of a language barrier on a known and effective intervention for pediatric asthma management. Practitioners can take the consequences illustrated in this study and easily extrapolate the contribution to health disparities on a broader scale.
Applications for Clinical Practice
Practitioners caring for patients in EDs where the patient or caregiver has a language barrier should make every effort to use appropriate interpreter services when patient teaching occurs. Assessing not only for health literacy but reading ability in the LEP patient or caregiver is also important, since it will affect dyad’s ability to implement self-care measures recommended in patient teaching sessions or action plan implementation. Asking the patient what their country of origin is, regardless of their legal status, will help practitioners refine patient teaching and the language they (and the interpreter when appropriate) use to illustrate what needs to be done to manage their condition.
—Allison Squires, PhD, RN
1. Ryan C. Language use in the United States : 2011. Migration Policy Institute: Washington, DC; 2013.
2. Diamond LC, Luft HS, Chung S, Jacobs EA. “Does this doctor speak my language?” Improving the characterization of physician non-English language skills. Health Serv Res 2012;47(1 Pt 2):556–69.
3. Jacobs EA. Patient centeredness in medical encounters requiring an interpreter. Am J Med 2000;109:515.
4. Hsieh E. Understanding medical interpreters: reconceptualizing bilingual health communication. Health Commun 2006;20:177–86.
5. Karliner LS, Kim SE, Meltzer DO, Auerbach AD. Influence of language barriers on outcomes of hospital care for general medicine inpatients. J Hosp Med 2010;5:276–82.
6. Lindholm M, Hargraves JL, Ferguson WJ, Reed G. Professional language interpretation and inpatient length of stay and readmission rates. J Gen Intern Med 2012;27:1294–9.
7. Gerchow L, Tagliaferro B, Squires A, et al. Latina food patterns in the United States: a qualitative metasynthesis. Nurs Res 2014;63:182–93.
8. Sudore RL, Landefeld CS, Pérez-Stable EJ, et al. Unraveling the relationship between literacy, language proficiency, and patient-physician communication. Patient Educ Couns 2009;75:398–402.
Study Overview
Objective. To compare rates of asthma action plan use in limited English proficiency (LEP) caregivers compared with English proficient (EP) caregivers.
Design. Cross-sectional survey.
Participants and setting. A convenience sample of 107 Latino caregivers of children with asthma at an urban academic emergency department (ED). Surveys in the preferred language of the patient (English or Spanish, with the translated version previously validated) were distributed at the time of the ED visit. Interpreters were utilized when requested.
Main outcome measure. Caregiver use of an asthma action plan.
Main results. 51 LEP caregivers and 56 EP caregivers completed the survey. Mothers completed the surveys 87% of the time and the average age of patients was 4 years. Among the EP caregivers, 64% reported using an asthma action plan, while only 39% of the LEP caregivers reported using one. The difference was statistally significant (P = 0.01). Through both correlations and regressions, English proficiency was the only variable (others included health insurance status and level of caregiver education) that showed a significant effect on asthma action plan use.
Conclusions. Children whose caregiver had LEP were significantly less likely to have and use an asthma action plan. Asthma education in the language of choice of the patient may help improve asthma care.
Commentary
With 20% of US households now speaking a language other than English at home [1], language barriers between providers and patients present multiple challenges to health services delivery and can significantly contribute to immigrant health disparities. Despite US laws and multiple federal agency policies requiring the use of interpreters during health care encounters, organizations continue to fall short of providing interpreter services and often lack adequate or equivalent materials for patient education. Too often, providers overestimate their language skills [2,3], use colleagues as ad hoc interpreters out of convenience [4], or rely on family members for interpretation [4]—a practice that is universally discouraged.
Recent research does suggest that the timing of interpreter use is critical. In planned encounters such as primary care visits, interpreters can and should be scheduled for visits when a language-concordant provider is not available. During hospitalizations, including ED visits, interpreters are most effective when used on admission, during patient teaching, and upon discharge, and the timing of these visits has been shown to affect length of stay and readmission rates [5,6].
This study magnifies the consequences of failing to provide language-concordant services to patients and their caregivers. It also helps to identify one of the sources of pediatric asthma health disparities in Latino populations. The emphasis on the role of the caregiver in action plan utilization is a unique aspect of this study and it is one of the first to examine the issue in this way. It highlights the importance of caregivers in health system transitions and illustrates how a language barrier can potentially impact transitions.
The authors’ explicit use of a power analysis to calculate their sample size is a strength of the study. Furthermore, the authors differentiated their respondents by country of origin, something that rarely occurs in studies of Latinos [7], and allows the reader to differentiate the impact of the intervention at a micro level within this population. The presentation of Spanish language quotes with their translations within the manuscript provides transparency for bilingual readers to verify the accuracy of the authors’ translation.
There are, however, a number of methodological issues that should be noted. The authors acknowledge that they did not account for asthma severity in the survey nor control for it in the analysis, did not assess health literacy, and did not differentiate their results based on country of origin. The latter point is important because the immigration experience and demographic profiles of Latinos differs significantly by country of origin and could factor in to action plan use. The translation process used for survey instrument translation also did not illustrate how it accounted for the well-established linguistic variation that occurs in the Spanish language. Additionally, US census data shows that the main countries of origin of Latinos in the service area of the study are Puerto Rico, Ecuador, and Mexico [1]. The survey itself had Ecuador as a write in and Dominican as a response option. The combination presented in the survey reflects the Latino demographic composition in the nearest large urban area. Thus, when collecting country of origin data on immigrant patients, country choices should reflect local demographics and not national trends for maximum precision.
Another concern is that Spanish language literacy was not assessed. Many Latino immigrants may have limited reading ability in Spanish. For Mexican immigrants in particular, Spanish may be a second language after their indigenous language. This is also true for some South American Latino immigrants from the Andean region. Many Latino immigrants come to the United States with less than an 8th grade education and likely come from educational systems of poor quality, which subsequently affects their Spanish language reading and writing skills [8]. Assessing education level based on US equivalents is not an accurate way to gauge literacy. Thus, assessing reading literacy in Spanish before surveying patients would have been a useful step that could have further refined the results. These factors will have implications for action plan utilization and implementation for any chronic disease.
Providers often think that language barriers are an obvious factor in health disparities and service delivery, but few studies have actually captured or quantified the effects of language barriers on health outcomes. Most studies only identify language barriers as an access issue. This study provides a good illustration of the impact of a language barrier on a known and effective intervention for pediatric asthma management. Practitioners can take the consequences illustrated in this study and easily extrapolate the contribution to health disparities on a broader scale.
Applications for Clinical Practice
Practitioners caring for patients in EDs where the patient or caregiver has a language barrier should make every effort to use appropriate interpreter services when patient teaching occurs. Assessing not only for health literacy but reading ability in the LEP patient or caregiver is also important, since it will affect dyad’s ability to implement self-care measures recommended in patient teaching sessions or action plan implementation. Asking the patient what their country of origin is, regardless of their legal status, will help practitioners refine patient teaching and the language they (and the interpreter when appropriate) use to illustrate what needs to be done to manage their condition.
—Allison Squires, PhD, RN
Study Overview
Objective. To compare rates of asthma action plan use in limited English proficiency (LEP) caregivers compared with English proficient (EP) caregivers.
Design. Cross-sectional survey.
Participants and setting. A convenience sample of 107 Latino caregivers of children with asthma at an urban academic emergency department (ED). Surveys in the preferred language of the patient (English or Spanish, with the translated version previously validated) were distributed at the time of the ED visit. Interpreters were utilized when requested.
Main outcome measure. Caregiver use of an asthma action plan.
Main results. 51 LEP caregivers and 56 EP caregivers completed the survey. Mothers completed the surveys 87% of the time and the average age of patients was 4 years. Among the EP caregivers, 64% reported using an asthma action plan, while only 39% of the LEP caregivers reported using one. The difference was statistally significant (P = 0.01). Through both correlations and regressions, English proficiency was the only variable (others included health insurance status and level of caregiver education) that showed a significant effect on asthma action plan use.
Conclusions. Children whose caregiver had LEP were significantly less likely to have and use an asthma action plan. Asthma education in the language of choice of the patient may help improve asthma care.
Commentary
With 20% of US households now speaking a language other than English at home [1], language barriers between providers and patients present multiple challenges to health services delivery and can significantly contribute to immigrant health disparities. Despite US laws and multiple federal agency policies requiring the use of interpreters during health care encounters, organizations continue to fall short of providing interpreter services and often lack adequate or equivalent materials for patient education. Too often, providers overestimate their language skills [2,3], use colleagues as ad hoc interpreters out of convenience [4], or rely on family members for interpretation [4]—a practice that is universally discouraged.
Recent research does suggest that the timing of interpreter use is critical. In planned encounters such as primary care visits, interpreters can and should be scheduled for visits when a language-concordant provider is not available. During hospitalizations, including ED visits, interpreters are most effective when used on admission, during patient teaching, and upon discharge, and the timing of these visits has been shown to affect length of stay and readmission rates [5,6].
This study magnifies the consequences of failing to provide language-concordant services to patients and their caregivers. It also helps to identify one of the sources of pediatric asthma health disparities in Latino populations. The emphasis on the role of the caregiver in action plan utilization is a unique aspect of this study and it is one of the first to examine the issue in this way. It highlights the importance of caregivers in health system transitions and illustrates how a language barrier can potentially impact transitions.
The authors’ explicit use of a power analysis to calculate their sample size is a strength of the study. Furthermore, the authors differentiated their respondents by country of origin, something that rarely occurs in studies of Latinos [7], and allows the reader to differentiate the impact of the intervention at a micro level within this population. The presentation of Spanish language quotes with their translations within the manuscript provides transparency for bilingual readers to verify the accuracy of the authors’ translation.
There are, however, a number of methodological issues that should be noted. The authors acknowledge that they did not account for asthma severity in the survey nor control for it in the analysis, did not assess health literacy, and did not differentiate their results based on country of origin. The latter point is important because the immigration experience and demographic profiles of Latinos differs significantly by country of origin and could factor in to action plan use. The translation process used for survey instrument translation also did not illustrate how it accounted for the well-established linguistic variation that occurs in the Spanish language. Additionally, US census data shows that the main countries of origin of Latinos in the service area of the study are Puerto Rico, Ecuador, and Mexico [1]. The survey itself had Ecuador as a write in and Dominican as a response option. The combination presented in the survey reflects the Latino demographic composition in the nearest large urban area. Thus, when collecting country of origin data on immigrant patients, country choices should reflect local demographics and not national trends for maximum precision.
Another concern is that Spanish language literacy was not assessed. Many Latino immigrants may have limited reading ability in Spanish. For Mexican immigrants in particular, Spanish may be a second language after their indigenous language. This is also true for some South American Latino immigrants from the Andean region. Many Latino immigrants come to the United States with less than an 8th grade education and likely come from educational systems of poor quality, which subsequently affects their Spanish language reading and writing skills [8]. Assessing education level based on US equivalents is not an accurate way to gauge literacy. Thus, assessing reading literacy in Spanish before surveying patients would have been a useful step that could have further refined the results. These factors will have implications for action plan utilization and implementation for any chronic disease.
Providers often think that language barriers are an obvious factor in health disparities and service delivery, but few studies have actually captured or quantified the effects of language barriers on health outcomes. Most studies only identify language barriers as an access issue. This study provides a good illustration of the impact of a language barrier on a known and effective intervention for pediatric asthma management. Practitioners can take the consequences illustrated in this study and easily extrapolate the contribution to health disparities on a broader scale.
Applications for Clinical Practice
Practitioners caring for patients in EDs where the patient or caregiver has a language barrier should make every effort to use appropriate interpreter services when patient teaching occurs. Assessing not only for health literacy but reading ability in the LEP patient or caregiver is also important, since it will affect dyad’s ability to implement self-care measures recommended in patient teaching sessions or action plan implementation. Asking the patient what their country of origin is, regardless of their legal status, will help practitioners refine patient teaching and the language they (and the interpreter when appropriate) use to illustrate what needs to be done to manage their condition.
—Allison Squires, PhD, RN
1. Ryan C. Language use in the United States : 2011. Migration Policy Institute: Washington, DC; 2013.
2. Diamond LC, Luft HS, Chung S, Jacobs EA. “Does this doctor speak my language?” Improving the characterization of physician non-English language skills. Health Serv Res 2012;47(1 Pt 2):556–69.
3. Jacobs EA. Patient centeredness in medical encounters requiring an interpreter. Am J Med 2000;109:515.
4. Hsieh E. Understanding medical interpreters: reconceptualizing bilingual health communication. Health Commun 2006;20:177–86.
5. Karliner LS, Kim SE, Meltzer DO, Auerbach AD. Influence of language barriers on outcomes of hospital care for general medicine inpatients. J Hosp Med 2010;5:276–82.
6. Lindholm M, Hargraves JL, Ferguson WJ, Reed G. Professional language interpretation and inpatient length of stay and readmission rates. J Gen Intern Med 2012;27:1294–9.
7. Gerchow L, Tagliaferro B, Squires A, et al. Latina food patterns in the United States: a qualitative metasynthesis. Nurs Res 2014;63:182–93.
8. Sudore RL, Landefeld CS, Pérez-Stable EJ, et al. Unraveling the relationship between literacy, language proficiency, and patient-physician communication. Patient Educ Couns 2009;75:398–402.
1. Ryan C. Language use in the United States : 2011. Migration Policy Institute: Washington, DC; 2013.
2. Diamond LC, Luft HS, Chung S, Jacobs EA. “Does this doctor speak my language?” Improving the characterization of physician non-English language skills. Health Serv Res 2012;47(1 Pt 2):556–69.
3. Jacobs EA. Patient centeredness in medical encounters requiring an interpreter. Am J Med 2000;109:515.
4. Hsieh E. Understanding medical interpreters: reconceptualizing bilingual health communication. Health Commun 2006;20:177–86.
5. Karliner LS, Kim SE, Meltzer DO, Auerbach AD. Influence of language barriers on outcomes of hospital care for general medicine inpatients. J Hosp Med 2010;5:276–82.
6. Lindholm M, Hargraves JL, Ferguson WJ, Reed G. Professional language interpretation and inpatient length of stay and readmission rates. J Gen Intern Med 2012;27:1294–9.
7. Gerchow L, Tagliaferro B, Squires A, et al. Latina food patterns in the United States: a qualitative metasynthesis. Nurs Res 2014;63:182–93.
8. Sudore RL, Landefeld CS, Pérez-Stable EJ, et al. Unraveling the relationship between literacy, language proficiency, and patient-physician communication. Patient Educ Couns 2009;75:398–402.
Case Studies in Toxicology: Death and Taxus
Case
A 50-year-old man ingests two handfuls of small, red berries that he picked from a shrub in front of his apartment building, with the belief that they would have medicinal value. Two hours later, he developed abdominal cramping and vomited multiple times, followed shortly thereafter by profuse diaphoresis, lethargy, and ataxia. His concerned family brought him to the ED where his vital signs on presentation were: blood pressure (BP), 78/43 mm Hg; heart rate (HR), 50 beats/minute; respiratory rate (RR), 12 breaths/minute; temperature (T), 97.8°F. With the exception of bradycardia, the patient’s cardiac, pulmonary, and abdominal examinations were normal. His skin was diaphoretic, and he had no focal motor or sensory deficits or tremor. Initial laboratory values were: hemoglobin, 12.6 g/dL; sodium, 137 mEq/L; potassium, 4.6 mEq/L; bicarbonate, 20 mEq/L; blood urea nitrogen, 17 mg/dL; creatinine, 2.2 mg/dL; glucose, 288 mg/dL. The patient’s troponin I level was slightly elevated at 0.06 ng/mL; electrocardiogram (ECG) results are shown in Figure 1.

Why do plant poisonings occur?
There is the general belief that what is natural is not only healthful but also safe. This is clearly not true: cyanide, uranium, and king cobras are all natural but hardly safe. While most plants chosen for their purported medicinal properties are generally harmless in most patients when taken in low doses, there are plants that are sufficiently poisonous to be consequential with even relatively small exposures. Some people, often unknowingly vulnerable due to genetic or other causes, are uniquely susceptible to even minute doses.
Humans probably learned about plant toxicity early on—most likely the hard way. To this day, however, the Internet is replete with traditional and avant-garde natural healing remedies involving the use of naturally-derived plant products. These numerous bioactive compounds are often sold in plant form or as extracts, the latter being more concerning given their more concentrated formulation.
Plant misidentification is a common cause of poisoning, whether the intended use is for food or medicine. For example, some mistake “deadly nightshade” (Atropa belladonna) berries, which are deep blue, for blueberries, or pokeweed roots for horseradish roots due to their similar appearances.1
Alternatively, even when a plant is correctly identified, patients may experience adverse effects if they exceed the “therapeutic dose” (eg, dysrhythmia from aconite roots used in traditional Chinese medicine) or if the plant is improperly prepared (eg, hypoglycemia from consuming unripe ackee fruit).2 In addition, a toxic plant such as Jimson weed (Datura stramonium) or coca leaf extract may be intentionally ingested for its psychoactive hallucinatory effects.2 Although rare in the United States, in certain parts of Asia, persons intent on self-harm may consume toxic plants.1
When ingested, what plants cause bradycardia and hypotension, and why do these effects occur?
The two broad classes of plant-derived toxins that can cause these findings are cardioactive steroids and sodium channel active agents.
Cardioactive Steroids
There are numerous botanical sources of cardioactive steroids (sometimes called cardiac glycosides) such as Digitalis lanata, from which digoxin is derived; and Digitalis purpurea, the source of digitoxin. Poisoning by Digitalis spp, squill, lily of the valley, oleander, yellow oleander, and Cerbera manghas are clinically similar. Cardioactive steroids act pharmacologically to block the sodium-potassium ATPase pump on the myocardial cell membrane. This in turn increases intracellular sodium, which subsequently inhibits the exchange of extracellular sodium for intracellular calcium, leading to inotropy. Clinical manifestations of toxicity include nausea, vomiting, hyperkalemia, bradycardia, cardiac dysrhythmias, and occasionally hypotension—some of which can be life-threatening.
Sodium Channel Active Agents
Several plant toxins affect the flow of sodium by blocking or activating the sodium channel. Both effects alter the rate and strength of cardiac contraction, causing cardiac dysrhythmias.
Aconite is often used in traditional Chinese medicine. In North America, it is mainly derived from Aconitinum napellus, commonly called monkshood, helmet flower, or wolfsbane. It effectively holds open the voltage-dependent sodium channel, increasing cellular excitability. By prolonging the sodium current influx, neuronal and cardiac repolarization eventually slow due to sodium overload, leading to bradycardia and hypotension, as well as neurological effects. Its cardiotoxicity resembles that caused by cardiac glycosides, though a history of paresthesias or muscle weakness may help to differentiate the two toxins.
Veratrum spp include false hellebore, Indian poke, and California hellebore. These plants are occasionally mistaken for leeks (ramps) and can cause vomiting, bradycardia, and hypotension by a mechanism of action similar to aconitine.

Grayanotoxins, a group of diterpenoid toxins found in death camas, azalea, Rhododendron spp, and mountain laurel, can become concentrated in honey made from these plants. Depending on the specific toxin, they variably open or close the sodium channel. In addition to causing bradycardia and hypotension, patients may exhibit mental status changes (“mad honey” poisoning) and seizures.2
Case Continuation
After rapid infusion of 1-liter of normal saline, the patient’s BP was 80/63 mm Hg and HR was 52 beats/minute. His wife arrived to the ED 30-minutes later with a plastic bag containing the red berries the patient had ingested. The emergency physician identified them as Taxus baccata, or more commonly, yew berries. The patient stated that he ingested both the red fleshy aril and chewed the hard central seed.
How is cardiotoxicity from yew berries treated?
Within hours of ingestion, toxicity progresses from nausea, abdominal pain, paresthesias, and ataxia, to bradycardia, cardiac conduction delays, wide-complex ventricular dysrhythmias and mental status changes.3 Although toxicity of Taxus has been known since antiquity, no antidote exists. Ventricular dysrhythmias causing hemodynamic instability should be electrically cardioverted, although there is no evidence to support the safety or efficacy of such therapy. Since the serum, and therefore cardiac concentration of taxine will be identical after cardioversion to its value prior, recurrent dysrhythmias are common.1 Sodium bicarbonate has been inconsistently effective in the treatment of wide-complex tachydysrhythmias,4 but its use seems counterintuitive for most cases. There may be merit to raising the sodium gradient on an already sodium overloaded myocyte, but short-term gain may lead to unintended consequences. Success with antidysrhythmics has been limited: although amiodarone is often used to treat wide-complex tachydysrhythmias, its efficacy in Taxus toxicity has been conflicting.4-6
There have been a few reported cases of yew alkaloid crossreactivity with digoxin assays, suggesting that digoxin-specific antibody fragments may bind taxine.7 There is no evidence, however, that cardioactive steroids are present in yew, and empiric use of antidigoxin Fab-fragments cannot be recommended. A single case report demonstrated that hemodialysis was ineffective in the removal of taxines, likely due to the toxin’s large volume of distribution.8 As a last resort, extracorporeal life support with membrane oxygenation is described favorably in two cases of yew berry poisoning refractory to conventional therapy.9,10
Case Conclusion
The patient’s ECGs showed a morphologically abnormal rhythm, possibly with a Brugada pattern, which are representative of the dysrhythmias caused by taxine’s inhibitory effects on the sodium and calcium channels. Despite an attempt at electrical cardioversion, the dysrhythmia persisted. He was given intravenous boluses of fluids and started on an amiodarone infusion. The patient’s BP gradually improved over the following 2 hours, and the dysrhythmia resolved with hemodynamic improvement. The amiodarone infusion was then discontinued, and he was admitted to the hospital for further testing. Echocardiography, electrophysiology studies, and cardiac catheterization were all normal. The absence of structural, dysrhythmogenic, and ischemic abnormalities supported the toxic etiology of his hemodynamic aberrations. He was discharged from the hospital 3 days later without report of sequelae.
Dr Nguyen is a medical toxicology fellow in the department of emergency medicine at New York University Langone Medical Center. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Bruneton J. Toxic Plants; Dangerous to Humans and Animals. Paris, France: Lavoisier Publishing; 1999:4-752.
- Palmer ME, Betz JM. Plants. In: Nelson LS, Lewin NA, Howland MA, Hoffman RS, Goldfrank LR, Flomenbaum NE. In: Goldfrank’s Toxicologic Emergencies. 9th ed. New York, NY: McGraw Hill; 2010:1537-1560.
- Nelson LS, Shih RD, Balick MJ. Handbook of Poisonous and Injurious Plants. 2nd ed. New York, NY: Springer/New York Botanical Garden; 2007:288-290.
- Pierog J, Kane B, Kane K, Donovan JW. Management of isolated yew berry toxicity with sodium bicarbonate: a case report in treatment efficacy. J Med Toxicol. 2009;5(2):84-89.
- Jones R, Jones J, Causer J, Ewins D, Goenka N, Joseph F. Yew tree poisoning: a near-fatal lesson from history. Clin Med. 2011;11(2):173-175.
- Willaert W, Claessens P, Vankelecom B, Vanderheyden M. Intoxication with Taxus baccata: cardiac arrhythmias following yew leaves ingestion. Pacing Clin Electrophysiol. 2002;25(4 Pt 1):511,512.
- Cummins RO, Haulman J, Quan L, Graves JR, Peterson D, Horan S. Near-fatal yew berry intoxication treated with external cardiac pacing and digoxin-specific FAB antibody fragments. Ann Emerg Med. 1990;19(1):38-43
- Dahlqvist M, Venzin R, König S, et al. Haemodialysis in Taxus baccata poisoning: a case report. QJM. 2012;105(4):359-361.
- Panzeri C, Bacis G, Ferri F, et al. Extracorporeal life support in severe Taxus baccata poisoning. Clin Toxicol. 2010;48(5):463-465.
- Soumagne N, Chauvet S, Chatellier D, Robert R, Charrière JM, Menu P. Treatment of yew leaf intoxication with extracorporeal circulation. Am J Emerg Med. 2011;29(3):354.e5-6.
Case
A 50-year-old man ingests two handfuls of small, red berries that he picked from a shrub in front of his apartment building, with the belief that they would have medicinal value. Two hours later, he developed abdominal cramping and vomited multiple times, followed shortly thereafter by profuse diaphoresis, lethargy, and ataxia. His concerned family brought him to the ED where his vital signs on presentation were: blood pressure (BP), 78/43 mm Hg; heart rate (HR), 50 beats/minute; respiratory rate (RR), 12 breaths/minute; temperature (T), 97.8°F. With the exception of bradycardia, the patient’s cardiac, pulmonary, and abdominal examinations were normal. His skin was diaphoretic, and he had no focal motor or sensory deficits or tremor. Initial laboratory values were: hemoglobin, 12.6 g/dL; sodium, 137 mEq/L; potassium, 4.6 mEq/L; bicarbonate, 20 mEq/L; blood urea nitrogen, 17 mg/dL; creatinine, 2.2 mg/dL; glucose, 288 mg/dL. The patient’s troponin I level was slightly elevated at 0.06 ng/mL; electrocardiogram (ECG) results are shown in Figure 1.
Why do plant poisonings occur?
There is the general belief that what is natural is not only healthful but also safe. This is clearly not true: cyanide, uranium, and king cobras are all natural but hardly safe. While most plants chosen for their purported medicinal properties are generally harmless in most patients when taken in low doses, there are plants that are sufficiently poisonous to be consequential with even relatively small exposures. Some people, often unknowingly vulnerable due to genetic or other causes, are uniquely susceptible to even minute doses.
Humans probably learned about plant toxicity early on—most likely the hard way. To this day, however, the Internet is replete with traditional and avant-garde natural healing remedies involving the use of naturally-derived plant products. These numerous bioactive compounds are often sold in plant form or as extracts, the latter being more concerning given their more concentrated formulation.
Plant misidentification is a common cause of poisoning, whether the intended use is for food or medicine. For example, some mistake “deadly nightshade” (Atropa belladonna) berries, which are deep blue, for blueberries, or pokeweed roots for horseradish roots due to their similar appearances.1
Alternatively, even when a plant is correctly identified, patients may experience adverse effects if they exceed the “therapeutic dose” (eg, dysrhythmia from aconite roots used in traditional Chinese medicine) or if the plant is improperly prepared (eg, hypoglycemia from consuming unripe ackee fruit).2 In addition, a toxic plant such as Jimson weed (Datura stramonium) or coca leaf extract may be intentionally ingested for its psychoactive hallucinatory effects.2 Although rare in the United States, in certain parts of Asia, persons intent on self-harm may consume toxic plants.1
When ingested, what plants cause bradycardia and hypotension, and why do these effects occur?
The two broad classes of plant-derived toxins that can cause these findings are cardioactive steroids and sodium channel active agents.
Cardioactive Steroids
There are numerous botanical sources of cardioactive steroids (sometimes called cardiac glycosides) such as Digitalis lanata, from which digoxin is derived; and Digitalis purpurea, the source of digitoxin. Poisoning by Digitalis spp, squill, lily of the valley, oleander, yellow oleander, and Cerbera manghas are clinically similar. Cardioactive steroids act pharmacologically to block the sodium-potassium ATPase pump on the myocardial cell membrane. This in turn increases intracellular sodium, which subsequently inhibits the exchange of extracellular sodium for intracellular calcium, leading to inotropy. Clinical manifestations of toxicity include nausea, vomiting, hyperkalemia, bradycardia, cardiac dysrhythmias, and occasionally hypotension—some of which can be life-threatening.
Sodium Channel Active Agents
Several plant toxins affect the flow of sodium by blocking or activating the sodium channel. Both effects alter the rate and strength of cardiac contraction, causing cardiac dysrhythmias.
Aconite is often used in traditional Chinese medicine. In North America, it is mainly derived from Aconitinum napellus, commonly called monkshood, helmet flower, or wolfsbane. It effectively holds open the voltage-dependent sodium channel, increasing cellular excitability. By prolonging the sodium current influx, neuronal and cardiac repolarization eventually slow due to sodium overload, leading to bradycardia and hypotension, as well as neurological effects. Its cardiotoxicity resembles that caused by cardiac glycosides, though a history of paresthesias or muscle weakness may help to differentiate the two toxins.
Veratrum spp include false hellebore, Indian poke, and California hellebore. These plants are occasionally mistaken for leeks (ramps) and can cause vomiting, bradycardia, and hypotension by a mechanism of action similar to aconitine.
Grayanotoxins, a group of diterpenoid toxins found in death camas, azalea, Rhododendron spp, and mountain laurel, can become concentrated in honey made from these plants. Depending on the specific toxin, they variably open or close the sodium channel. In addition to causing bradycardia and hypotension, patients may exhibit mental status changes (“mad honey” poisoning) and seizures.2
Case Continuation
After rapid infusion of 1-liter of normal saline, the patient’s BP was 80/63 mm Hg and HR was 52 beats/minute. His wife arrived to the ED 30-minutes later with a plastic bag containing the red berries the patient had ingested. The emergency physician identified them as Taxus baccata, or more commonly, yew berries. The patient stated that he ingested both the red fleshy aril and chewed the hard central seed.
How is cardiotoxicity from yew berries treated?
Within hours of ingestion, toxicity progresses from nausea, abdominal pain, paresthesias, and ataxia, to bradycardia, cardiac conduction delays, wide-complex ventricular dysrhythmias and mental status changes.3 Although toxicity of Taxus has been known since antiquity, no antidote exists. Ventricular dysrhythmias causing hemodynamic instability should be electrically cardioverted, although there is no evidence to support the safety or efficacy of such therapy. Since the serum, and therefore cardiac concentration of taxine will be identical after cardioversion to its value prior, recurrent dysrhythmias are common.1 Sodium bicarbonate has been inconsistently effective in the treatment of wide-complex tachydysrhythmias,4 but its use seems counterintuitive for most cases. There may be merit to raising the sodium gradient on an already sodium overloaded myocyte, but short-term gain may lead to unintended consequences. Success with antidysrhythmics has been limited: although amiodarone is often used to treat wide-complex tachydysrhythmias, its efficacy in Taxus toxicity has been conflicting.4-6
There have been a few reported cases of yew alkaloid crossreactivity with digoxin assays, suggesting that digoxin-specific antibody fragments may bind taxine.7 There is no evidence, however, that cardioactive steroids are present in yew, and empiric use of antidigoxin Fab-fragments cannot be recommended. A single case report demonstrated that hemodialysis was ineffective in the removal of taxines, likely due to the toxin’s large volume of distribution.8 As a last resort, extracorporeal life support with membrane oxygenation is described favorably in two cases of yew berry poisoning refractory to conventional therapy.9,10
Case Conclusion
The patient’s ECGs showed a morphologically abnormal rhythm, possibly with a Brugada pattern, which are representative of the dysrhythmias caused by taxine’s inhibitory effects on the sodium and calcium channels. Despite an attempt at electrical cardioversion, the dysrhythmia persisted. He was given intravenous boluses of fluids and started on an amiodarone infusion. The patient’s BP gradually improved over the following 2 hours, and the dysrhythmia resolved with hemodynamic improvement. The amiodarone infusion was then discontinued, and he was admitted to the hospital for further testing. Echocardiography, electrophysiology studies, and cardiac catheterization were all normal. The absence of structural, dysrhythmogenic, and ischemic abnormalities supported the toxic etiology of his hemodynamic aberrations. He was discharged from the hospital 3 days later without report of sequelae.
Dr Nguyen is a medical toxicology fellow in the department of emergency medicine at New York University Langone Medical Center. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
Case
A 50-year-old man ingests two handfuls of small, red berries that he picked from a shrub in front of his apartment building, with the belief that they would have medicinal value. Two hours later, he developed abdominal cramping and vomited multiple times, followed shortly thereafter by profuse diaphoresis, lethargy, and ataxia. His concerned family brought him to the ED where his vital signs on presentation were: blood pressure (BP), 78/43 mm Hg; heart rate (HR), 50 beats/minute; respiratory rate (RR), 12 breaths/minute; temperature (T), 97.8°F. With the exception of bradycardia, the patient’s cardiac, pulmonary, and abdominal examinations were normal. His skin was diaphoretic, and he had no focal motor or sensory deficits or tremor. Initial laboratory values were: hemoglobin, 12.6 g/dL; sodium, 137 mEq/L; potassium, 4.6 mEq/L; bicarbonate, 20 mEq/L; blood urea nitrogen, 17 mg/dL; creatinine, 2.2 mg/dL; glucose, 288 mg/dL. The patient’s troponin I level was slightly elevated at 0.06 ng/mL; electrocardiogram (ECG) results are shown in Figure 1.
Why do plant poisonings occur?
There is the general belief that what is natural is not only healthful but also safe. This is clearly not true: cyanide, uranium, and king cobras are all natural but hardly safe. While most plants chosen for their purported medicinal properties are generally harmless in most patients when taken in low doses, there are plants that are sufficiently poisonous to be consequential with even relatively small exposures. Some people, often unknowingly vulnerable due to genetic or other causes, are uniquely susceptible to even minute doses.
Humans probably learned about plant toxicity early on—most likely the hard way. To this day, however, the Internet is replete with traditional and avant-garde natural healing remedies involving the use of naturally-derived plant products. These numerous bioactive compounds are often sold in plant form or as extracts, the latter being more concerning given their more concentrated formulation.
Plant misidentification is a common cause of poisoning, whether the intended use is for food or medicine. For example, some mistake “deadly nightshade” (Atropa belladonna) berries, which are deep blue, for blueberries, or pokeweed roots for horseradish roots due to their similar appearances.1
Alternatively, even when a plant is correctly identified, patients may experience adverse effects if they exceed the “therapeutic dose” (eg, dysrhythmia from aconite roots used in traditional Chinese medicine) or if the plant is improperly prepared (eg, hypoglycemia from consuming unripe ackee fruit).2 In addition, a toxic plant such as Jimson weed (Datura stramonium) or coca leaf extract may be intentionally ingested for its psychoactive hallucinatory effects.2 Although rare in the United States, in certain parts of Asia, persons intent on self-harm may consume toxic plants.1
When ingested, what plants cause bradycardia and hypotension, and why do these effects occur?
The two broad classes of plant-derived toxins that can cause these findings are cardioactive steroids and sodium channel active agents.
Cardioactive Steroids
There are numerous botanical sources of cardioactive steroids (sometimes called cardiac glycosides) such as Digitalis lanata, from which digoxin is derived; and Digitalis purpurea, the source of digitoxin. Poisoning by Digitalis spp, squill, lily of the valley, oleander, yellow oleander, and Cerbera manghas are clinically similar. Cardioactive steroids act pharmacologically to block the sodium-potassium ATPase pump on the myocardial cell membrane. This in turn increases intracellular sodium, which subsequently inhibits the exchange of extracellular sodium for intracellular calcium, leading to inotropy. Clinical manifestations of toxicity include nausea, vomiting, hyperkalemia, bradycardia, cardiac dysrhythmias, and occasionally hypotension—some of which can be life-threatening.
Sodium Channel Active Agents
Several plant toxins affect the flow of sodium by blocking or activating the sodium channel. Both effects alter the rate and strength of cardiac contraction, causing cardiac dysrhythmias.
Aconite is often used in traditional Chinese medicine. In North America, it is mainly derived from Aconitinum napellus, commonly called monkshood, helmet flower, or wolfsbane. It effectively holds open the voltage-dependent sodium channel, increasing cellular excitability. By prolonging the sodium current influx, neuronal and cardiac repolarization eventually slow due to sodium overload, leading to bradycardia and hypotension, as well as neurological effects. Its cardiotoxicity resembles that caused by cardiac glycosides, though a history of paresthesias or muscle weakness may help to differentiate the two toxins.
Veratrum spp include false hellebore, Indian poke, and California hellebore. These plants are occasionally mistaken for leeks (ramps) and can cause vomiting, bradycardia, and hypotension by a mechanism of action similar to aconitine.
Grayanotoxins, a group of diterpenoid toxins found in death camas, azalea, Rhododendron spp, and mountain laurel, can become concentrated in honey made from these plants. Depending on the specific toxin, they variably open or close the sodium channel. In addition to causing bradycardia and hypotension, patients may exhibit mental status changes (“mad honey” poisoning) and seizures.2
Case Continuation
After rapid infusion of 1-liter of normal saline, the patient’s BP was 80/63 mm Hg and HR was 52 beats/minute. His wife arrived to the ED 30-minutes later with a plastic bag containing the red berries the patient had ingested. The emergency physician identified them as Taxus baccata, or more commonly, yew berries. The patient stated that he ingested both the red fleshy aril and chewed the hard central seed.
How is cardiotoxicity from yew berries treated?
Within hours of ingestion, toxicity progresses from nausea, abdominal pain, paresthesias, and ataxia, to bradycardia, cardiac conduction delays, wide-complex ventricular dysrhythmias and mental status changes.3 Although toxicity of Taxus has been known since antiquity, no antidote exists. Ventricular dysrhythmias causing hemodynamic instability should be electrically cardioverted, although there is no evidence to support the safety or efficacy of such therapy. Since the serum, and therefore cardiac concentration of taxine will be identical after cardioversion to its value prior, recurrent dysrhythmias are common.1 Sodium bicarbonate has been inconsistently effective in the treatment of wide-complex tachydysrhythmias,4 but its use seems counterintuitive for most cases. There may be merit to raising the sodium gradient on an already sodium overloaded myocyte, but short-term gain may lead to unintended consequences. Success with antidysrhythmics has been limited: although amiodarone is often used to treat wide-complex tachydysrhythmias, its efficacy in Taxus toxicity has been conflicting.4-6
There have been a few reported cases of yew alkaloid crossreactivity with digoxin assays, suggesting that digoxin-specific antibody fragments may bind taxine.7 There is no evidence, however, that cardioactive steroids are present in yew, and empiric use of antidigoxin Fab-fragments cannot be recommended. A single case report demonstrated that hemodialysis was ineffective in the removal of taxines, likely due to the toxin’s large volume of distribution.8 As a last resort, extracorporeal life support with membrane oxygenation is described favorably in two cases of yew berry poisoning refractory to conventional therapy.9,10
Case Conclusion
The patient’s ECGs showed a morphologically abnormal rhythm, possibly with a Brugada pattern, which are representative of the dysrhythmias caused by taxine’s inhibitory effects on the sodium and calcium channels. Despite an attempt at electrical cardioversion, the dysrhythmia persisted. He was given intravenous boluses of fluids and started on an amiodarone infusion. The patient’s BP gradually improved over the following 2 hours, and the dysrhythmia resolved with hemodynamic improvement. The amiodarone infusion was then discontinued, and he was admitted to the hospital for further testing. Echocardiography, electrophysiology studies, and cardiac catheterization were all normal. The absence of structural, dysrhythmogenic, and ischemic abnormalities supported the toxic etiology of his hemodynamic aberrations. He was discharged from the hospital 3 days later without report of sequelae.
Dr Nguyen is a medical toxicology fellow in the department of emergency medicine at New York University Langone Medical Center. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Bruneton J. Toxic Plants; Dangerous to Humans and Animals. Paris, France: Lavoisier Publishing; 1999:4-752.
- Palmer ME, Betz JM. Plants. In: Nelson LS, Lewin NA, Howland MA, Hoffman RS, Goldfrank LR, Flomenbaum NE. In: Goldfrank’s Toxicologic Emergencies. 9th ed. New York, NY: McGraw Hill; 2010:1537-1560.
- Nelson LS, Shih RD, Balick MJ. Handbook of Poisonous and Injurious Plants. 2nd ed. New York, NY: Springer/New York Botanical Garden; 2007:288-290.
- Pierog J, Kane B, Kane K, Donovan JW. Management of isolated yew berry toxicity with sodium bicarbonate: a case report in treatment efficacy. J Med Toxicol. 2009;5(2):84-89.
- Jones R, Jones J, Causer J, Ewins D, Goenka N, Joseph F. Yew tree poisoning: a near-fatal lesson from history. Clin Med. 2011;11(2):173-175.
- Willaert W, Claessens P, Vankelecom B, Vanderheyden M. Intoxication with Taxus baccata: cardiac arrhythmias following yew leaves ingestion. Pacing Clin Electrophysiol. 2002;25(4 Pt 1):511,512.
- Cummins RO, Haulman J, Quan L, Graves JR, Peterson D, Horan S. Near-fatal yew berry intoxication treated with external cardiac pacing and digoxin-specific FAB antibody fragments. Ann Emerg Med. 1990;19(1):38-43
- Dahlqvist M, Venzin R, König S, et al. Haemodialysis in Taxus baccata poisoning: a case report. QJM. 2012;105(4):359-361.
- Panzeri C, Bacis G, Ferri F, et al. Extracorporeal life support in severe Taxus baccata poisoning. Clin Toxicol. 2010;48(5):463-465.
- Soumagne N, Chauvet S, Chatellier D, Robert R, Charrière JM, Menu P. Treatment of yew leaf intoxication with extracorporeal circulation. Am J Emerg Med. 2011;29(3):354.e5-6.
- Bruneton J. Toxic Plants; Dangerous to Humans and Animals. Paris, France: Lavoisier Publishing; 1999:4-752.
- Palmer ME, Betz JM. Plants. In: Nelson LS, Lewin NA, Howland MA, Hoffman RS, Goldfrank LR, Flomenbaum NE. In: Goldfrank’s Toxicologic Emergencies. 9th ed. New York, NY: McGraw Hill; 2010:1537-1560.
- Nelson LS, Shih RD, Balick MJ. Handbook of Poisonous and Injurious Plants. 2nd ed. New York, NY: Springer/New York Botanical Garden; 2007:288-290.
- Pierog J, Kane B, Kane K, Donovan JW. Management of isolated yew berry toxicity with sodium bicarbonate: a case report in treatment efficacy. J Med Toxicol. 2009;5(2):84-89.
- Jones R, Jones J, Causer J, Ewins D, Goenka N, Joseph F. Yew tree poisoning: a near-fatal lesson from history. Clin Med. 2011;11(2):173-175.
- Willaert W, Claessens P, Vankelecom B, Vanderheyden M. Intoxication with Taxus baccata: cardiac arrhythmias following yew leaves ingestion. Pacing Clin Electrophysiol. 2002;25(4 Pt 1):511,512.
- Cummins RO, Haulman J, Quan L, Graves JR, Peterson D, Horan S. Near-fatal yew berry intoxication treated with external cardiac pacing and digoxin-specific FAB antibody fragments. Ann Emerg Med. 1990;19(1):38-43
- Dahlqvist M, Venzin R, König S, et al. Haemodialysis in Taxus baccata poisoning: a case report. QJM. 2012;105(4):359-361.
- Panzeri C, Bacis G, Ferri F, et al. Extracorporeal life support in severe Taxus baccata poisoning. Clin Toxicol. 2010;48(5):463-465.
- Soumagne N, Chauvet S, Chatellier D, Robert R, Charrière JM, Menu P. Treatment of yew leaf intoxication with extracorporeal circulation. Am J Emerg Med. 2011;29(3):354.e5-6.

Clipped by an Oncoming Car
ANSWER
The image shows a comminuted and depressed fracture of the lateral tibial plateau. It is depressed approximately 6 to 7 mm. The patient was admitted, and orthopedic consultation was obtained. The patient subsequently underwent an open reduction and internal fixation of the fracture.
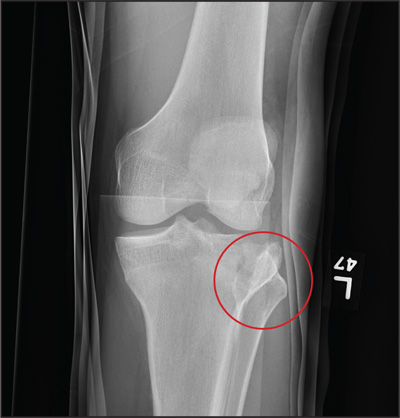
ANSWER
The image shows a comminuted and depressed fracture of the lateral tibial plateau. It is depressed approximately 6 to 7 mm. The patient was admitted, and orthopedic consultation was obtained. The patient subsequently underwent an open reduction and internal fixation of the fracture.
ANSWER
The image shows a comminuted and depressed fracture of the lateral tibial plateau. It is depressed approximately 6 to 7 mm. The patient was admitted, and orthopedic consultation was obtained. The patient subsequently underwent an open reduction and internal fixation of the fracture.
A 23-year-old man is brought in after being hit by a car. He was in the process of getting into his car when another vehicle coming from the opposite direction swerved into his lane. He tried to jump onto his hood to avoid the other car but was struck by the side mirror and landed on the ground. He is primarily complaining of left knee and lower leg pain. He denies any medical history. Primary survey appears to be stable except for scalp and facial lacerations. The patient is awake, alert, and oriented, and his vital signs are stable. His left lower extremity is in a splint immobilizer, placed by emergency medical personnel. There is a moderate amount of soft tissue swelling around the knee, which is exquisitely tender to palpation. The patient has limited flexion and extension of the knee due to pain. He is able to wiggle his toes, and distally in the leg and foot there appears to be no neurovascular compromise. Radiographs of the tibia are obtained. What is your impression?
Man, 26, With Sudden-Onset Right Lower Quadrant Pain
A 26-year-old man presented to the emergency department (ED) with a chief complaint of abdominal pain. After triage was complete, he was transported to an examination room, where the clinician obtained the history of presenting illness. The onset of pain was approximately 90 minutes prior to arrival at the ED and woke the patient from a “sound sleep.” He stated that the pain initially started as a “3 out of 10” but had progressed to a “12 out of 10,” and he described it as being in the right lower quadrant of his abdomen, with radiation to his right testicle. However, he was unsure where the pain started or if it was worse in either location. Nausea was the primary associated symptom, but he denied vomiting, diarrhea, fever, dysuria, or hematuria. Last, the patient denied history of trauma.
Medical history was noncontributory: He denied previous gastrointestinal diseases, and there was no history of renal stones, urinary tract infection, or any other genitourinary disease. He had no surgical history. The patient smoked less than a pack of cigarettes per day but denied alcohol or drug use.
Physical examination revealed a young man in moderate discomfort. Despite describing his pain as a “12 out of 10,” he had a blood pressure of 121/72 mm Hg; pulse, 59 beats/min; respiratory rate, 20 breaths/min; and temperature, 96.8°F. HEENT and cardiovascular, respiratory, musculoskeletal, and neurologic exam results were all within normal limits. Abdominal examination revealed a mildly tender right lower quadrant with deep palpation, but no rebound or guarding. Murphy sign was negative.
Because of the complaint of pain radiating to the testicles, a genitourinary examination was performed. The penis appeared unremarkable, with no lesions or discharge. There was no inguinal lymphadenopathy. The scrotum appeared appropriate in size and was also grossly unremarkable. The left testicle was nontender. However, palpation of the right testicle elicited moderate to severe pain. There was no visible swelling, and there were no palpable hernias or other masses. Cremasteric reflex was assessed bilaterally and deemed to be absent on the right side.
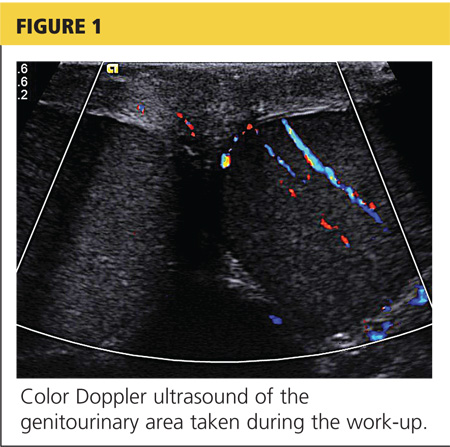
A workup was initiated that included a complete blood count, comprehensive metabolic panel, and urinalysis; the results of these tests were unremarkable. A differential diagnosis was formed, with emphasis on appendicitis and testicular torsion. Because of the specific nature and location of the pain, both ultrasound and CT of the abdomen/pelvis were considered. It was decided to order the ultrasound, with a plan to perform CT only if ultrasound was unremarkable. The patient was medicated for his pain and the ultrasound commenced. Halfway through the imaging, the clinician and attending physician were summoned to the examination room to review the image seen in Figure 1.
On the next page: Discussion and diagnosis >>
DISCUSSION
Testicular torsion may occur if the testicle twists or rotates on the spermatic cord. The twisting causes arterial ischemia and venous outflow obstruction, cutting off the testicle’s blood supply.1,2 Torsion may be extravaginal or intravaginal, depending on the extent of involvement of the surrounding structures.2
Extravaginal torsion is most commonly seen in neonates and occurs because the entire testicle may freely rotate prior to fixation to the scrotal wall via the tunica vaginalis.2Intravaginal torsion is more common in adolescents and often occurs as a result of a condition known as bell clapper deformity. This congenital abnormality enables the testicle to rotate within the tunica vaginalis and rest transversely in the scrotum instead of in a more vertical orientation.2,3 Torsion occurs if the testicle rotates 90° to 180°, with complete torsion occurring at 360° (torsion may extend to as much as 720°).2 Torsion may also occur as a result of trauma.1
Peak incidence of testicular torsion occurs at ages 13 to 14, but it can occur at any age; torsion affects approximately 1 in 4,000 males younger than 25.2-5 Ninety-five percent of all torsions are intravaginal.2 Torsion is the most common pathology for males who undergo surgical exploration for scrotal pain.3
The main goal in the diagnosis and treatment of torsion is testicular salvage. Torsion is considered a urologic emergency, making early diagnosis and treatment critical to prevent testicular loss. In fact, a review of the relevant literature reveals that the rate of testicular salvage is much higher if the diagnosis is made within 6 to 12 hours.1,2,5 Potential sequelae from delayed treatment include testicular infarction, loss of testicle, infertility problems, infections, cosmetic deformity, and increased risk for testicular malignancy.2
Because many men hesitate to seek medical attention for symptoms of testicular pain and swelling, the primary care clinician should openly discuss testicular disorders, especially with preadolescent males, during testicular examinations.6
Diagnosis
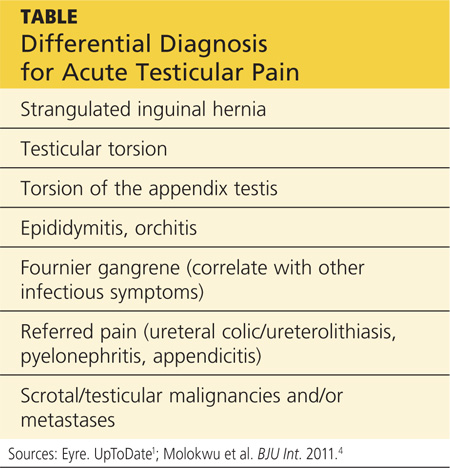
A testicular examination should be performed on any male presenting with a chief complaint of lower abdominal pain, back/flank pain, or any pain that radiates to the groin. The cremasteric reflex should be assessed because it can help differentiate among the causes of testicular pain.7 It is performed by gently stroking the upper inner thigh and observing for contraction of the ipsilateral testicle. One study found that, in cases of torsion, the absence of a cremasteric reflex had a sensitivity of 96% and a specificity of 88%.7 See the Table for the differential diagnosis for acute testicular pain.
While it is often possible to make the diagnosis of testicular torsion clinically, ultrasound with color Doppler is the diagnostic test of choice in cases for which the cause of acute scrotal pain is unclear.8 Ultrasound provides anatomic detail of the scrotum and its contents, and perfusion is assessed by adding the color Doppler images.8 It is important to note that, while the absence of blood flow is considered diagnostic for testicular torsion, the presence of flow does not necessarily exclude it.4
On the next page: Treatment >>
Treatment
Surgical exploration with intraoperative detorsion and orchiopexy (fixation of the testicle to the scrotal wall) is the mainstay of treatment for testicular torsion.1 Orchiopexy is often performed bilaterally in order to prevent future torsion of the unaffected testicle. In about 40% of males with the bell clapper deformity, the condition is present on both sides.2 Orchiectomy, the complete removal of the testicle, is necessary when the degree of torsion and subsequent ischemia have caused irreversible damage to the testicle.6 In one study in which 2,248 cases of torsion were reviewed, approximately 34% of males required orchiectomy.6
If surgery may be delayed, the clinician may attempt manual detorsion at the bedside. Despite the “open book” method described in many texts—which instructs the practitioner to rotate the testicle laterally—a review of the literature reveals that torsion takes place medially only 70% of the time.1,5 The clinician should always consider this when any attempts at manual detorsion are made and correlate his or her technique with physical examination and the patient’s response.5
Relief of pain and return of the testicle to its natural longitudinal lie are considered indicators of successful detorsion.1 Color Doppler ultrasound should be used to confirm the return of circulation. However, in one case review of pediatric patients who underwent surgical exploration after manual detorsion, some degree of residual torsion remained in 32%.5 Because of this risk, surgery is still indicated even in cases of successful bedside detorsion.5
On the next page: Case continuation >>
CASE CONTINUATION
The decision to perform bedside ultrasound was made because the diagnosis of testicular torsion is a surgical emergency, and the window of time to prevent complications can be extremely narrow. If the ultrasound had been normal, then a CT scan may have provided additional data on which to base the diagnosis.
The patient was given adequate parenteral pain medication. After color Doppler ultrasound confirmed the torsion, the testicle was laterally rotated approximately 360°. The patient reported alleviation of his symptoms. Color Doppler was again performed to confirm the return of hyperemic blood flow to the affected testicle (Figure 2). The urologist arrived shortly thereafter and the patient was taken to the operating room, where he underwent scrotal exploration and bilateral orchiopexy.
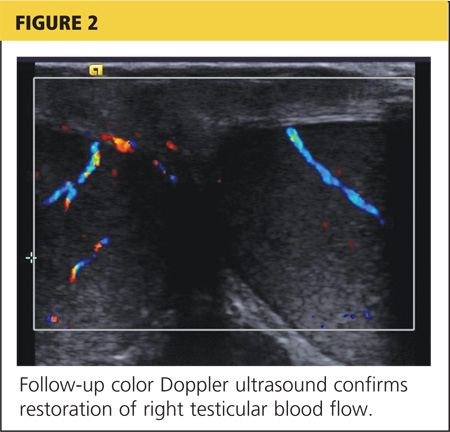
On the next page: Conclusion >>
CONCLUSION
A testicular examination should be performed on any male presenting with a chief complaint of lower abdominal pain, back/flank pain, or any pain that radiates to the groin. Testicular torsion is most commonly seen in infants and adolescents but can occur at any age. The condition is a surgical emergency and the goal is testicular salvage, which is most likely to occur before 12 hours have elapsed since the onset of symptoms. An important component of the physical examination is attempting to elicit the cremasteric reflex, which is likely to be absent in the presence of torsion.
The primary care provider’s goal is to rapidly diagnose testicular torsion, then refer the patient immediately to a urologist or ED. The skilled clinician may attempt manual detorsion, based on his/her expertise and comfort level; however, this procedure should never delay prompt surgical intervention.
REFERENCES
1. Eyre RC. Evaluation of the acute scrotum in adults. www.uptodate.com/contents/evaluation-of-the-acute-scrotum-in-adults. Accessed May 16, 2014.
2. Ogunyemi OI, Weiker M, Abel EJ. Testicular torsion. http://emedicine.medscape.com/article/2036003-overview. Accessed May 16, 2014.
3. Khan F, Muoka O, Watson GM. Bell clapper testis, torsion, and detorsion: a case report. Case Rep Urol. 2011;2011:631970.
4. Molokwu CN, Somani BK, Goodman CM. Outcomes of scrotal exploration for acute scrotal pain suspicious of testicular torsion: a consecutive case series of 173 patients. BJU Int. 2011;107(6):990-993.
5. Sessions AE, Rabinowitz R, Hulbert WC, et al. Testicular torsion: direction, degree, duration and disinformation. J Urol. 2003;169(2):663-665.
6. Mansbach JM, Forbes P, Peters C. Testicular torsion and risk factors for orchiectomy. Arch Pediatr Adolesc Med. 2005;159:1167-1171.
7. Schmitz D, Safranek S. How useful is a physical exam in diagnosing testicular torsion? J Fam Pract. 2009;58(8):433-434.
8. D’Andrea A, Coppolino F, Cesarano E, et al. US in the assessment of acute scrotum. Crit Ultrasound J. 2013;5(suppl 1):S8. www.criticalultrasound journal.com/content/5/S1/S8/. Accessed May 16, 2014.
A 26-year-old man presented to the emergency department (ED) with a chief complaint of abdominal pain. After triage was complete, he was transported to an examination room, where the clinician obtained the history of presenting illness. The onset of pain was approximately 90 minutes prior to arrival at the ED and woke the patient from a “sound sleep.” He stated that the pain initially started as a “3 out of 10” but had progressed to a “12 out of 10,” and he described it as being in the right lower quadrant of his abdomen, with radiation to his right testicle. However, he was unsure where the pain started or if it was worse in either location. Nausea was the primary associated symptom, but he denied vomiting, diarrhea, fever, dysuria, or hematuria. Last, the patient denied history of trauma.
Medical history was noncontributory: He denied previous gastrointestinal diseases, and there was no history of renal stones, urinary tract infection, or any other genitourinary disease. He had no surgical history. The patient smoked less than a pack of cigarettes per day but denied alcohol or drug use.
Physical examination revealed a young man in moderate discomfort. Despite describing his pain as a “12 out of 10,” he had a blood pressure of 121/72 mm Hg; pulse, 59 beats/min; respiratory rate, 20 breaths/min; and temperature, 96.8°F. HEENT and cardiovascular, respiratory, musculoskeletal, and neurologic exam results were all within normal limits. Abdominal examination revealed a mildly tender right lower quadrant with deep palpation, but no rebound or guarding. Murphy sign was negative.
Because of the complaint of pain radiating to the testicles, a genitourinary examination was performed. The penis appeared unremarkable, with no lesions or discharge. There was no inguinal lymphadenopathy. The scrotum appeared appropriate in size and was also grossly unremarkable. The left testicle was nontender. However, palpation of the right testicle elicited moderate to severe pain. There was no visible swelling, and there were no palpable hernias or other masses. Cremasteric reflex was assessed bilaterally and deemed to be absent on the right side.
A workup was initiated that included a complete blood count, comprehensive metabolic panel, and urinalysis; the results of these tests were unremarkable. A differential diagnosis was formed, with emphasis on appendicitis and testicular torsion. Because of the specific nature and location of the pain, both ultrasound and CT of the abdomen/pelvis were considered. It was decided to order the ultrasound, with a plan to perform CT only if ultrasound was unremarkable. The patient was medicated for his pain and the ultrasound commenced. Halfway through the imaging, the clinician and attending physician were summoned to the examination room to review the image seen in Figure 1.
On the next page: Discussion and diagnosis >>
DISCUSSION
Testicular torsion may occur if the testicle twists or rotates on the spermatic cord. The twisting causes arterial ischemia and venous outflow obstruction, cutting off the testicle’s blood supply.1,2 Torsion may be extravaginal or intravaginal, depending on the extent of involvement of the surrounding structures.2
Extravaginal torsion is most commonly seen in neonates and occurs because the entire testicle may freely rotate prior to fixation to the scrotal wall via the tunica vaginalis.2Intravaginal torsion is more common in adolescents and often occurs as a result of a condition known as bell clapper deformity. This congenital abnormality enables the testicle to rotate within the tunica vaginalis and rest transversely in the scrotum instead of in a more vertical orientation.2,3 Torsion occurs if the testicle rotates 90° to 180°, with complete torsion occurring at 360° (torsion may extend to as much as 720°).2 Torsion may also occur as a result of trauma.1
Peak incidence of testicular torsion occurs at ages 13 to 14, but it can occur at any age; torsion affects approximately 1 in 4,000 males younger than 25.2-5 Ninety-five percent of all torsions are intravaginal.2 Torsion is the most common pathology for males who undergo surgical exploration for scrotal pain.3
The main goal in the diagnosis and treatment of torsion is testicular salvage. Torsion is considered a urologic emergency, making early diagnosis and treatment critical to prevent testicular loss. In fact, a review of the relevant literature reveals that the rate of testicular salvage is much higher if the diagnosis is made within 6 to 12 hours.1,2,5 Potential sequelae from delayed treatment include testicular infarction, loss of testicle, infertility problems, infections, cosmetic deformity, and increased risk for testicular malignancy.2
Because many men hesitate to seek medical attention for symptoms of testicular pain and swelling, the primary care clinician should openly discuss testicular disorders, especially with preadolescent males, during testicular examinations.6
Diagnosis
A testicular examination should be performed on any male presenting with a chief complaint of lower abdominal pain, back/flank pain, or any pain that radiates to the groin. The cremasteric reflex should be assessed because it can help differentiate among the causes of testicular pain.7 It is performed by gently stroking the upper inner thigh and observing for contraction of the ipsilateral testicle. One study found that, in cases of torsion, the absence of a cremasteric reflex had a sensitivity of 96% and a specificity of 88%.7 See the Table for the differential diagnosis for acute testicular pain.
While it is often possible to make the diagnosis of testicular torsion clinically, ultrasound with color Doppler is the diagnostic test of choice in cases for which the cause of acute scrotal pain is unclear.8 Ultrasound provides anatomic detail of the scrotum and its contents, and perfusion is assessed by adding the color Doppler images.8 It is important to note that, while the absence of blood flow is considered diagnostic for testicular torsion, the presence of flow does not necessarily exclude it.4
On the next page: Treatment >>
Treatment
Surgical exploration with intraoperative detorsion and orchiopexy (fixation of the testicle to the scrotal wall) is the mainstay of treatment for testicular torsion.1 Orchiopexy is often performed bilaterally in order to prevent future torsion of the unaffected testicle. In about 40% of males with the bell clapper deformity, the condition is present on both sides.2 Orchiectomy, the complete removal of the testicle, is necessary when the degree of torsion and subsequent ischemia have caused irreversible damage to the testicle.6 In one study in which 2,248 cases of torsion were reviewed, approximately 34% of males required orchiectomy.6
If surgery may be delayed, the clinician may attempt manual detorsion at the bedside. Despite the “open book” method described in many texts—which instructs the practitioner to rotate the testicle laterally—a review of the literature reveals that torsion takes place medially only 70% of the time.1,5 The clinician should always consider this when any attempts at manual detorsion are made and correlate his or her technique with physical examination and the patient’s response.5
Relief of pain and return of the testicle to its natural longitudinal lie are considered indicators of successful detorsion.1 Color Doppler ultrasound should be used to confirm the return of circulation. However, in one case review of pediatric patients who underwent surgical exploration after manual detorsion, some degree of residual torsion remained in 32%.5 Because of this risk, surgery is still indicated even in cases of successful bedside detorsion.5
On the next page: Case continuation >>
CASE CONTINUATION
The decision to perform bedside ultrasound was made because the diagnosis of testicular torsion is a surgical emergency, and the window of time to prevent complications can be extremely narrow. If the ultrasound had been normal, then a CT scan may have provided additional data on which to base the diagnosis.
The patient was given adequate parenteral pain medication. After color Doppler ultrasound confirmed the torsion, the testicle was laterally rotated approximately 360°. The patient reported alleviation of his symptoms. Color Doppler was again performed to confirm the return of hyperemic blood flow to the affected testicle (Figure 2). The urologist arrived shortly thereafter and the patient was taken to the operating room, where he underwent scrotal exploration and bilateral orchiopexy.
On the next page: Conclusion >>
CONCLUSION
A testicular examination should be performed on any male presenting with a chief complaint of lower abdominal pain, back/flank pain, or any pain that radiates to the groin. Testicular torsion is most commonly seen in infants and adolescents but can occur at any age. The condition is a surgical emergency and the goal is testicular salvage, which is most likely to occur before 12 hours have elapsed since the onset of symptoms. An important component of the physical examination is attempting to elicit the cremasteric reflex, which is likely to be absent in the presence of torsion.
The primary care provider’s goal is to rapidly diagnose testicular torsion, then refer the patient immediately to a urologist or ED. The skilled clinician may attempt manual detorsion, based on his/her expertise and comfort level; however, this procedure should never delay prompt surgical intervention.
REFERENCES
1. Eyre RC. Evaluation of the acute scrotum in adults. www.uptodate.com/contents/evaluation-of-the-acute-scrotum-in-adults. Accessed May 16, 2014.
2. Ogunyemi OI, Weiker M, Abel EJ. Testicular torsion. http://emedicine.medscape.com/article/2036003-overview. Accessed May 16, 2014.
3. Khan F, Muoka O, Watson GM. Bell clapper testis, torsion, and detorsion: a case report. Case Rep Urol. 2011;2011:631970.
4. Molokwu CN, Somani BK, Goodman CM. Outcomes of scrotal exploration for acute scrotal pain suspicious of testicular torsion: a consecutive case series of 173 patients. BJU Int. 2011;107(6):990-993.
5. Sessions AE, Rabinowitz R, Hulbert WC, et al. Testicular torsion: direction, degree, duration and disinformation. J Urol. 2003;169(2):663-665.
6. Mansbach JM, Forbes P, Peters C. Testicular torsion and risk factors for orchiectomy. Arch Pediatr Adolesc Med. 2005;159:1167-1171.
7. Schmitz D, Safranek S. How useful is a physical exam in diagnosing testicular torsion? J Fam Pract. 2009;58(8):433-434.
8. D’Andrea A, Coppolino F, Cesarano E, et al. US in the assessment of acute scrotum. Crit Ultrasound J. 2013;5(suppl 1):S8. www.criticalultrasound journal.com/content/5/S1/S8/. Accessed May 16, 2014.
A 26-year-old man presented to the emergency department (ED) with a chief complaint of abdominal pain. After triage was complete, he was transported to an examination room, where the clinician obtained the history of presenting illness. The onset of pain was approximately 90 minutes prior to arrival at the ED and woke the patient from a “sound sleep.” He stated that the pain initially started as a “3 out of 10” but had progressed to a “12 out of 10,” and he described it as being in the right lower quadrant of his abdomen, with radiation to his right testicle. However, he was unsure where the pain started or if it was worse in either location. Nausea was the primary associated symptom, but he denied vomiting, diarrhea, fever, dysuria, or hematuria. Last, the patient denied history of trauma.
Medical history was noncontributory: He denied previous gastrointestinal diseases, and there was no history of renal stones, urinary tract infection, or any other genitourinary disease. He had no surgical history. The patient smoked less than a pack of cigarettes per day but denied alcohol or drug use.
Physical examination revealed a young man in moderate discomfort. Despite describing his pain as a “12 out of 10,” he had a blood pressure of 121/72 mm Hg; pulse, 59 beats/min; respiratory rate, 20 breaths/min; and temperature, 96.8°F. HEENT and cardiovascular, respiratory, musculoskeletal, and neurologic exam results were all within normal limits. Abdominal examination revealed a mildly tender right lower quadrant with deep palpation, but no rebound or guarding. Murphy sign was negative.
Because of the complaint of pain radiating to the testicles, a genitourinary examination was performed. The penis appeared unremarkable, with no lesions or discharge. There was no inguinal lymphadenopathy. The scrotum appeared appropriate in size and was also grossly unremarkable. The left testicle was nontender. However, palpation of the right testicle elicited moderate to severe pain. There was no visible swelling, and there were no palpable hernias or other masses. Cremasteric reflex was assessed bilaterally and deemed to be absent on the right side.
A workup was initiated that included a complete blood count, comprehensive metabolic panel, and urinalysis; the results of these tests were unremarkable. A differential diagnosis was formed, with emphasis on appendicitis and testicular torsion. Because of the specific nature and location of the pain, both ultrasound and CT of the abdomen/pelvis were considered. It was decided to order the ultrasound, with a plan to perform CT only if ultrasound was unremarkable. The patient was medicated for his pain and the ultrasound commenced. Halfway through the imaging, the clinician and attending physician were summoned to the examination room to review the image seen in Figure 1.
On the next page: Discussion and diagnosis >>
DISCUSSION
Testicular torsion may occur if the testicle twists or rotates on the spermatic cord. The twisting causes arterial ischemia and venous outflow obstruction, cutting off the testicle’s blood supply.1,2 Torsion may be extravaginal or intravaginal, depending on the extent of involvement of the surrounding structures.2
Extravaginal torsion is most commonly seen in neonates and occurs because the entire testicle may freely rotate prior to fixation to the scrotal wall via the tunica vaginalis.2Intravaginal torsion is more common in adolescents and often occurs as a result of a condition known as bell clapper deformity. This congenital abnormality enables the testicle to rotate within the tunica vaginalis and rest transversely in the scrotum instead of in a more vertical orientation.2,3 Torsion occurs if the testicle rotates 90° to 180°, with complete torsion occurring at 360° (torsion may extend to as much as 720°).2 Torsion may also occur as a result of trauma.1
Peak incidence of testicular torsion occurs at ages 13 to 14, but it can occur at any age; torsion affects approximately 1 in 4,000 males younger than 25.2-5 Ninety-five percent of all torsions are intravaginal.2 Torsion is the most common pathology for males who undergo surgical exploration for scrotal pain.3
The main goal in the diagnosis and treatment of torsion is testicular salvage. Torsion is considered a urologic emergency, making early diagnosis and treatment critical to prevent testicular loss. In fact, a review of the relevant literature reveals that the rate of testicular salvage is much higher if the diagnosis is made within 6 to 12 hours.1,2,5 Potential sequelae from delayed treatment include testicular infarction, loss of testicle, infertility problems, infections, cosmetic deformity, and increased risk for testicular malignancy.2
Because many men hesitate to seek medical attention for symptoms of testicular pain and swelling, the primary care clinician should openly discuss testicular disorders, especially with preadolescent males, during testicular examinations.6
Diagnosis
A testicular examination should be performed on any male presenting with a chief complaint of lower abdominal pain, back/flank pain, or any pain that radiates to the groin. The cremasteric reflex should be assessed because it can help differentiate among the causes of testicular pain.7 It is performed by gently stroking the upper inner thigh and observing for contraction of the ipsilateral testicle. One study found that, in cases of torsion, the absence of a cremasteric reflex had a sensitivity of 96% and a specificity of 88%.7 See the Table for the differential diagnosis for acute testicular pain.
While it is often possible to make the diagnosis of testicular torsion clinically, ultrasound with color Doppler is the diagnostic test of choice in cases for which the cause of acute scrotal pain is unclear.8 Ultrasound provides anatomic detail of the scrotum and its contents, and perfusion is assessed by adding the color Doppler images.8 It is important to note that, while the absence of blood flow is considered diagnostic for testicular torsion, the presence of flow does not necessarily exclude it.4
On the next page: Treatment >>
Treatment
Surgical exploration with intraoperative detorsion and orchiopexy (fixation of the testicle to the scrotal wall) is the mainstay of treatment for testicular torsion.1 Orchiopexy is often performed bilaterally in order to prevent future torsion of the unaffected testicle. In about 40% of males with the bell clapper deformity, the condition is present on both sides.2 Orchiectomy, the complete removal of the testicle, is necessary when the degree of torsion and subsequent ischemia have caused irreversible damage to the testicle.6 In one study in which 2,248 cases of torsion were reviewed, approximately 34% of males required orchiectomy.6
If surgery may be delayed, the clinician may attempt manual detorsion at the bedside. Despite the “open book” method described in many texts—which instructs the practitioner to rotate the testicle laterally—a review of the literature reveals that torsion takes place medially only 70% of the time.1,5 The clinician should always consider this when any attempts at manual detorsion are made and correlate his or her technique with physical examination and the patient’s response.5
Relief of pain and return of the testicle to its natural longitudinal lie are considered indicators of successful detorsion.1 Color Doppler ultrasound should be used to confirm the return of circulation. However, in one case review of pediatric patients who underwent surgical exploration after manual detorsion, some degree of residual torsion remained in 32%.5 Because of this risk, surgery is still indicated even in cases of successful bedside detorsion.5
On the next page: Case continuation >>
CASE CONTINUATION
The decision to perform bedside ultrasound was made because the diagnosis of testicular torsion is a surgical emergency, and the window of time to prevent complications can be extremely narrow. If the ultrasound had been normal, then a CT scan may have provided additional data on which to base the diagnosis.
The patient was given adequate parenteral pain medication. After color Doppler ultrasound confirmed the torsion, the testicle was laterally rotated approximately 360°. The patient reported alleviation of his symptoms. Color Doppler was again performed to confirm the return of hyperemic blood flow to the affected testicle (Figure 2). The urologist arrived shortly thereafter and the patient was taken to the operating room, where he underwent scrotal exploration and bilateral orchiopexy.
On the next page: Conclusion >>
CONCLUSION
A testicular examination should be performed on any male presenting with a chief complaint of lower abdominal pain, back/flank pain, or any pain that radiates to the groin. Testicular torsion is most commonly seen in infants and adolescents but can occur at any age. The condition is a surgical emergency and the goal is testicular salvage, which is most likely to occur before 12 hours have elapsed since the onset of symptoms. An important component of the physical examination is attempting to elicit the cremasteric reflex, which is likely to be absent in the presence of torsion.
The primary care provider’s goal is to rapidly diagnose testicular torsion, then refer the patient immediately to a urologist or ED. The skilled clinician may attempt manual detorsion, based on his/her expertise and comfort level; however, this procedure should never delay prompt surgical intervention.
REFERENCES
1. Eyre RC. Evaluation of the acute scrotum in adults. www.uptodate.com/contents/evaluation-of-the-acute-scrotum-in-adults. Accessed May 16, 2014.
2. Ogunyemi OI, Weiker M, Abel EJ. Testicular torsion. http://emedicine.medscape.com/article/2036003-overview. Accessed May 16, 2014.
3. Khan F, Muoka O, Watson GM. Bell clapper testis, torsion, and detorsion: a case report. Case Rep Urol. 2011;2011:631970.
4. Molokwu CN, Somani BK, Goodman CM. Outcomes of scrotal exploration for acute scrotal pain suspicious of testicular torsion: a consecutive case series of 173 patients. BJU Int. 2011;107(6):990-993.
5. Sessions AE, Rabinowitz R, Hulbert WC, et al. Testicular torsion: direction, degree, duration and disinformation. J Urol. 2003;169(2):663-665.
6. Mansbach JM, Forbes P, Peters C. Testicular torsion and risk factors for orchiectomy. Arch Pediatr Adolesc Med. 2005;159:1167-1171.
7. Schmitz D, Safranek S. How useful is a physical exam in diagnosing testicular torsion? J Fam Pract. 2009;58(8):433-434.
8. D’Andrea A, Coppolino F, Cesarano E, et al. US in the assessment of acute scrotum. Crit Ultrasound J. 2013;5(suppl 1):S8. www.criticalultrasound journal.com/content/5/S1/S8/. Accessed May 16, 2014.
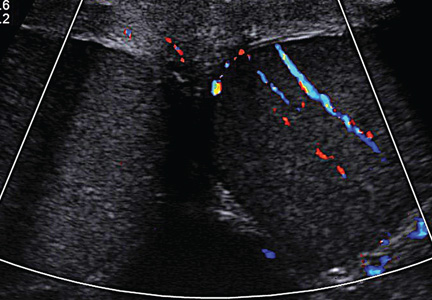
A 20-year-old woman with fatigue and palpitations
A 20-year-old woman presents to the emergency department with fatigue and the sudden onset of palpitations. She reports no history of significant illness or surgery. She says she is not currently taking prescription or over-the-counter medications. She does not smoke, drink alcohol, or use illicit drugs.
Her weight is 52 kg (115 lb), her height is 170 cm (67 in), and her body mass index (BMI) is 18 kg/m2. Vital signs: temperature 35.7°C (96.4°F), blood pressure 92/48 mm Hg, heart rate 73 bpm, respiratory rate 5 breaths per minute, and oxygen saturation 98% on room air.
She appears tired but is alert, conversant, and cooperative. Her skin is normal, and dentition is fair. Her pulse is regular, and respirations are slow. The abdomen is soft, non-tender, and flat. Strength is 4 on a scale of 5 in all extremities. Deep-tendon reflexes are 2+ and symmetric.
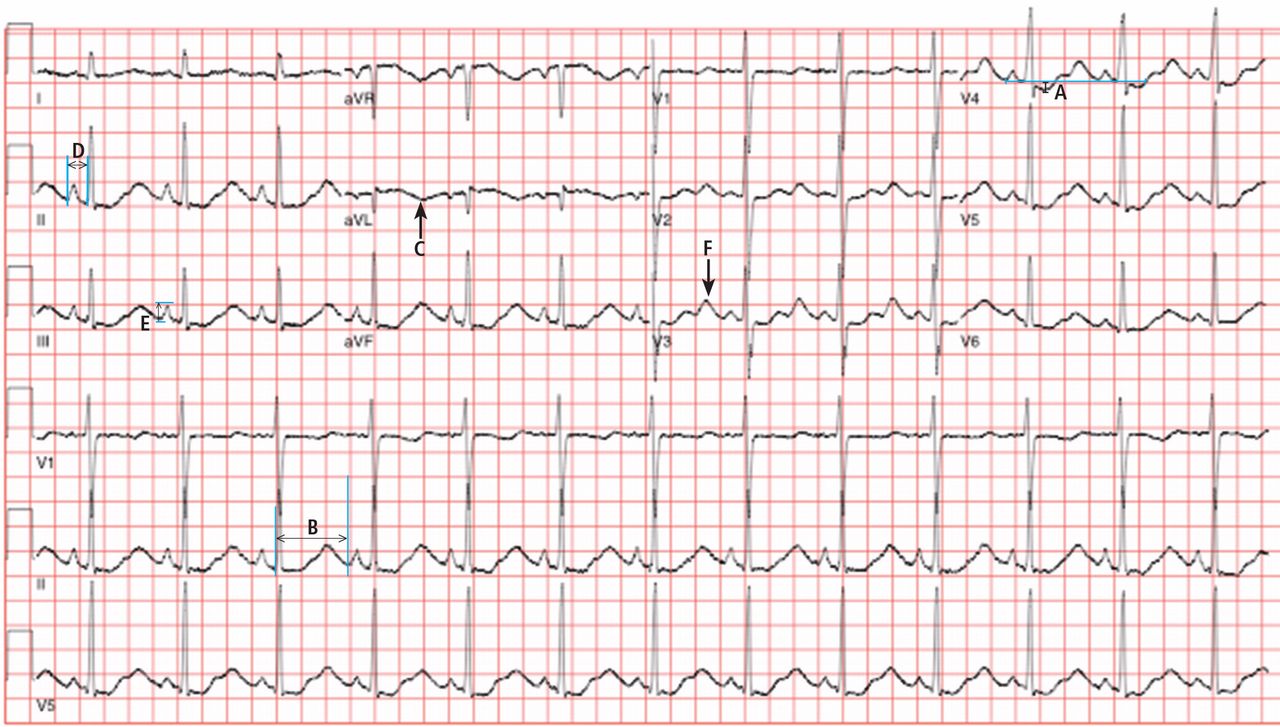
Electrocardiography (Figure 1) in the emergency department shows ST-segment depression, a prolonged corrected QT interval of 665 msec, T-wave inversion, PR prolongation, increased P-wave amplitude, and U waves.
1. Which electrolyte abnormality is associated with this electrocardiographic picture?
- Hypercalcemia
- Hyperkalemia
- Hypocalcemia
- Hypokalemia
Hypokalemia is the likely cause of these findings. The finding of U waves is considered significant when they are inverted, merged with the T wave, or have an amplitude greater than the T wave.1 U waves are best seen in the precordial leads. When severe, hypokalemia can lead to potentially fatal arrhythmias such as high-grade atrioventricular block, ventricular tachycardia, and ventricular fibrillation.2
Hyperkalemia is associated with peaked T waves, a prolonged PR interval, decreased P wave amplitude, and a widened QRS complex.2 When acute and severe, hyperkalemia is associated with ventricular arrhythmia.
Hypocalcemia is associated with a prolonged QT interval and ventricular dysrhythmia, but not U waves.2
Hypercalcemia is associated with bradydysrhythmia, as well as with a shortened QT interval.2
LABORATORY TESTING
Laboratory testing shows the following:
- Sodium 126 mmol/L (reference range 135–145)
- Potassium 1.5 mmol/L (3.5–5.1)
- Chloride 58 mmol/L (100–110)
- Bicarbonate 62 mmol/L (20–30)
- Blood urea nitrogen 16 mg/dL (7–18)
- Creatinine 0.8 mg/dL (0.5–1.0)
- Glucose 106 mg/dL (70–110)
- Ionized calcium 4.4 mg/dL (4.5–5.3)
- Magnesium 1.8 mg/dL (1.7–2.3)
- Phosphorus 4.1 mg/dL (2.5–4.5)
- Venous blood gases pH 7.56 (7.35–7.45), Pco2 69 mm Hg (35–45).
POTASSIUM HOMEOSTASIS
Ninety-eight percent of potassium is intracellular and only 2% is extracellular.3 The main cellular stores are myocytes and hepatocytes. Patients with decreased muscle mass may be at a higher risk of hypokalemia as a result of decreased skeletal muscle stores.4
The acute development of hypokalemia occurs from transcellular shifts. Alkalosis, insulin secretion, and beta-adrenergic stimulation promote the intracellular uptake of potassium. The major hormonal regulator of potassium excretion is aldosterone, which is stimulated by renal hypoperfusion and promotes potassium-ion secretion in the distal convoluted tubule.
Chronic hypokalemia develops in patients with ongoing renal or gastrointestinal potassium loss. If the cause of potassium loss is not elucidated by the history, the physical, and a review of medications, then one of two things is possible: either the patient has renal tubular disease affecting acid-base and potassium regulation, causing excessive mineralocorticoid secretion, which is associated with an abnormal response to aldosterone; or the patient is not being forthcoming in the history.
2. Which is the most likely cause of hypokalemia in this patient?
- Vomiting
- Liddle syndrome
- Bartter syndrome
- Gitelman syndrome
- Diuretic use
Her laboratory tests reveal hypokalemia and hyponatremic-hypochloremic metabolic alkalosis with compensatory respiratory acidosis. In metabolic alkalosis, the expected respiratory compensation is an increase of 0.7 mm Hg in Pco2 for each 1-mEq/L increase in bicarbonate. Therefore, the expected Pco2 is 67, close to the patient’s actual value of 69.
Protracted vomiting with loss of gastric acid juices could be a cause of the metabolic disturbances in this young woman, although she did not mention vomiting during the history.
Liddle syndrome, or pseudoaldosteronism, is a rare autosomal dominant disorder characterized by altered renal epithelial sodium channels, excessive sodium retention, and resultant hypertension. Hypokalemia and alkalosis are seen in Liddle syndrome, but the absence of hypertension in our patient makes Liddle syndrome unlikely.
Bartter syndrome is an inherited autosomal recessive disorder of the sodium-potassium-chloride cotransporter in the thick ascending loop of Henle, resulting in impaired reabsorption of chloride and sodium. Bartter syndrome mimics chronic loop-diuretic use and is associated with hypercalciuria. Bartter syndrome is possible in this patient; however, patients with Bartter syndrome are usually diagnosed in infancy or childhood and have evidence of growth impairment.
Gitelman syndrome is an autosomal recessive disorder of the thiazide-sensitive sodium-chloride cotransporter. Although Gitelman syndrome is more common than Bartter syndrome and presents at older ages, it is not usually associated with such profound metabolic alkalosis. Gitelman syndrome mimics chronic use of thiazide diuretics and is associated with hypocalciuria.
Diuretic use could also cause the metabolic disturbances described; however, the patient denied taking diuretics.
The most common cause of hypokalemia in clinical practice is diuretic use.4 In this young woman with unexplained hypokalemia, the most likely cause is either undisclosed self-induced vomiting or diuretic abuse. The degree of metabolic alkalosis suggests vomiting, since metabolic alkalosis this severe is usually seen only with protracted vomiting. Bartter and Gitelman syndromes are included in the differential diagnosis, but they are much less common than hypokalemia associated with diuretics or self-induced vomiting.5
3. Which test could help elucidate the cause of hypokalemia in this patient?
- Ratio of plasma aldosterone to rennin
- Urine chloride
- Ratio of urinary potassium to creatinine
- Urinary anion gap and urinary pH
APPROACH TO HYPOKALEMIA
Determining the cause of hypokalemia starts with a thorough history and physical examination. The history should focus on drugs such as diuretics and laxatives. Women should be asked about their menstrual history since irregular periods may suggest an eating disorder. The physical examination should focus on signs that suggest self-induced vomiting, such as dry skin, dental erosions, enlarged parotid glands, and calluses or scars on the knuckles.
Patients with an unclear cause of hypokalemia after a thorough history and physical examination can be categorized into one of three groups based on blood pressure and acid-base status:
- Hypokalemia, hypertension, metabolic alkalosis
- Hypokalemia, normal blood pressure, metabolic acidosis
- Hypokalemia, normal blood pressure, metabolic alkalosis.
Hypokalemia, hypertension, metabolic alkalosis
The blood pressure provides an important clue in the evaluation of hypokalemia. The combination of hypertension, hypokalemia, and alkalosis should raise concern for hyperaldosteronism or pseudoaldosteronism. Primary hyperaldosteronism from an adrenal adenoma (Conn syndrome) is characterized by a plasma aldosterone-renin ratio of greater than 20.6,7 In contrast, patients with secondary hyperaldosteronism due to renovascular disease have a plasma aldosterone-renin ratio of less than 10. Patients with pseudoaldosteronism have low aldosterone and renin levels and hypertension. Since our patient has a normal blood pressure, testing the plasma aldosterone and renin levels would not help determine the cause of her hypokalemia.
Hypokalemia, normal blood pressure, metabolic acidosis
Patients with normal blood pressure, hypokalemia, and normal plasma anion gap acidosis either have renal tubular acidosis or have lost potassium because of diarrhea or laxative abuse. In a patient who denies taking laxatives or denies a history of diarrhea, checking the urinary anion gap and urinary pH may help differentiate the cause of acidosis and hypokalemia.
The urinary anion gap, calculated by the equation sodium + potassium − chloride, is an indirect estimate of hydrogen excretion in the form of ammonium ion8; the normal value is 0 to 10 mEq/L. A negative value represents increased hydrogen excretion in response to systemic acidosis from gastrointestinal or renal loss of bicarbonate (proximal renal tubular acidosis). A urinary pH greater than 5.5 in the setting of systemic acidosis suggests impaired ability of the kidneys to acidify urine and raises the possibility of renal tubular acidosis.
This patient has metabolic alkalosis, so calculation of the urinary anion gap would not be helpful.
Hypokalemia, normal blood pressure, metabolic alkalosis
Patients such as ours, with normal blood pressure, hypokalemia, and alkalosis, have been vomiting, have used diuretics, or have an inherited renal tubulopathy such as Bartter or Gitelman syndrome. Usually, differentiating Bartter and Gitelman syndromes from chronic vomiting or diuretic use is done with the history and physical examination. However, in patients with a questionable history and a lack of findings on physical examination, checking the urinary chloride, potassium, calcium, and creatinine may be helpful.
A urinary potassium-creatinine ratio greater than 15 suggests renal loss, whereas a ratio less than 15 suggests extrarenal loss.9

Patients who are taking a diuretic or who have Bartter or Gitelman syndrome have a high urinary chloride concentration, ie, greater than 20 mmol/L, whereas patients with hypokalemia and alkalosis from chronic vomiting tend to have a concentration less than 10 mmol/L.10
Table 1 summarizes an approach to the evaluation of unexplained hypokalemia based on blood pressure and acid-base status.
A HIDDEN HISTORY
On further questioning, the patient admits to an 8-year history of daily self-induced vomiting in an attempt to lose weight, in addition to multiple hospitalizations for hypokalemia and a previous diagnosis of an eating disorder.
INITIAL MANAGEMENT OF HYPOKALEMIA
The initial management of hypokalemia should focus on life-threatening emergencies. While patients with potassium levels greater than 3 mmol/L are usually asymptomatic, those with levels below 3 mmol/L present with muscle weakness and rhabdomyolysis.4 An acute drop in serum potassium to less than 2 mmol/L is associated with respiratory muscle weakness and ventricular arrhythmias.4 If the patient has cardiac symptoms or hypoventilation due to respiratory muscle weakness, continuous monitoring in the intensive care unit and aggressive therapy are warranted.
4. Which potassium formulation is most appropriate for the treatment of hypokalemia in this patient?
- Potassium chloride
- Potassium phosphate
- Potassium acetate
Oral potassium is preferable in patients with a serum potassium above 2.5 mmol/L.4,11 Potassium phosphate should be used when supplementation with both potassium and phosphorus is needed. Potassium acetate should be reserved for patients with acidosis and hypokalemia. Otherwise, potassium chloride is typically preferred.4,12 It comes in liquid and tablet forms. Liquid forms have an unpleasant taste, whereas tablets are usually well tolerated. No more than 20 to 40 mEq of potassium chloride tablets should be given at a time, since higher doses are associated with gastrointestinal mucosal injury.12
Potassium chloride is particularly preferred in patients with metabolic alkalosis, since increased chloride intake and delivery to the distal tubule increases the expression of pendrin, a luminal chloride and bicarbonate exchanger in the cortical collecting duct.13 With metabolic alkalosis, increased excretion of bicarbonate occurs through up-regulation of pendrin. Potassium depletion down-regulates pendrin.13 Additionally, correction of metabolic alkalosis increases serum potassium by movement of potassium from the intracellular to the extracellular space.
Intravenous potassium should be reserved for patients with severe hypokalemia (< 2.5 mmol/L) or significant arrhythmias.11 Oral and intravenous potassium can safely be given simultaneously.11 The intravenous rate should not exceed more than 10 to 20 mEq of potassium chloride per hour unless the patient has a life-threatening arrhythmia, respiratory failure, or severe hypokalemia.14,15 In life-threatening situations, a femoral line should be placed, and potassium should be given as rapidly as 20 mEq over 15 to 20 minutes.14 Cannulation of the subclavian and internal jugular veins should be avoided in severe hypokalemia since mechanical irritation from guidewire placement can provoke ventricular arrhythmias.14
During intravenous administration of potassium, laboratory monitoring after every 20 mEq of potassium chloride is advised because of the possibility of rebound hyperkalemia. In patients with severe hypokalemia, avoidance of factors that can worsen intracellular shift of potassium is also important. Avoid dextrose-containing fluids to prevent insulin-induced shifting of potassium into cells. Restore intravascular volume to blunt hypovolemia-induced renin and aldosterone secretion. If a patient presents with severe hypokalemia and acidosis, correct the hypokalemia before the acidosis to avoid intracellular shift of potassium.
OUR PATIENT’S MANAGEMENT AND FOLLOW-UP PLAN
Given the severity of our patient’s hypokalemia and her complaint of palpitations, she was admitted to the hospital for monitoring. She required 180 mEq of intravenous potassium chloride and 140 mEq of oral potassium chloride during the first 24 hours in order to achieve a serum potassium level above 3 mmol/L. Electrocardiographic U waves resolved once the level was above 2 mmol/L, and ST depressions resolved once it was above 3 mmol/L. The QT interval normalized after 24 hours of hospitalization.
On discharge, she was prescribed oral potassium chloride 40 mEq daily and magnesium sulfate 400 mg twice daily, with plans for a followup visit with her outpatient therapy team, which includes a psychiatrist, a social worker, and her primary care provider. She declined a referral for inpatient therapy but agreed to a goal of decreasing the frequency of induced vomiting and outpatient visits. She was also educated on how and when to access emergency medical care.16
- Rautaharju PM, Surawicz B, Gettes LS, et al; American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; American College of Cardiology Foundation; Heart Rhythm Society. AHA/ACCF/HRS recommendations for the standardization and interpretation of the electrocardiogram: part IV: the ST segment, T and U waves, and the QT Interval: a scientific statement from the American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; the American College of Cardiology Foundation; and the Heart Rhythm Society. Endorsed by the International Society for Computerized Electrocardiology. J Am Coll Cardiol 2009; 53:982–991.
- Diercks DB, Shumaik GM, Harrigan RA, Brady WJ, Chan TC. Electrocardiographic manifestations: electrolyte abnormalities. J Emerg Med 2004; 27:153–160.
- Unwin RJ, Luft FC, Shirley DG. Pathophysiology and management of hypokalemia: a clinical perspective. Nat Rev Nephrol 2011; 7:75–84.
- Gennari FJ. Hypokalemia. N Engl J Med 1998; 339:451–458.
- Mehler PS. Clinical practice. Bulimia nervosa. N Engl J Med 2003; 349:875–881.
- Tzanela M, Effraimidis G, Vassiliadi D, et al. The aldosterone to renin ratio in the evaluation of patients with incidentally detected adrenal masses. Endocrine 2007; 32:136–142.
- Diederich S, Mai K, Bähr V, Helffrich S, Pfeiffer A, Perschel FH. The simultaneous measurement of plasma-aldosterone- and -renin-concentration allows rapid classification of all disorders of the renin-aldosterone system. Exp Clin Endocrinol Diabetes 2007; 115:433–438.
- Goldstein MB, Bear R, Richardson RM, Marsden PA, Halperin ML. The urine anion gap: a clinically useful index of ammonium excretion. Am J Med Sci 1986; 292:198–202.
- Groeneveld JH, Sijpkens YW, Lin SH, Davids MR, Halperin ML. An approach to the patient with severe hypokalaemia: the potassium quiz. QJM 2005; 98:305–316.
- Galla JH. Metabolic alkalosis. J Am Soc Nephrol 2000; 11:369–375.
- Asmar A, Mohandas R, Wingo CS. A physiologic-based approach to the treatment of a patient with hypokalemia. Am J Kidney Dis 2012; 60:492–497.
- Cohn JN, Kowey PR, Whelton PK, Prisant LM. New guidelines for potassium replacement in clinical practice: a contemporary review by the National Council on Potassium in Clinical Practice. Arch Intern Med 2000; 160:2429–2436.
- Luke RG, Galla JH. It is chloride depletion alkalosis, not contraction alkalosis. J Am Soc Nephrol 2012; 23:204–207.
- Kruse JA, Carlson RW. Rapid correction of hypokalemia using concentrated intravenous potassium chloride infusions. Arch Intern Med 1990; 150:613–617.
- Weiner ID, Wingo CS. Hypokalemia—consequences, causes, and correction. J Am Soc Nephrol 1997; 8:1179–1188.
- AED Medical Care Standards Task Force. Eating disorders: Critical Points for Early Recognition and Medical Risk Management in the Care of Individuals with Eating Disorders. AED Report 2012. www.aedweb.org/web/downloads/Guide-English.pdf. Accessed April 4, 2014.
A 20-year-old woman presents to the emergency department with fatigue and the sudden onset of palpitations. She reports no history of significant illness or surgery. She says she is not currently taking prescription or over-the-counter medications. She does not smoke, drink alcohol, or use illicit drugs.
Her weight is 52 kg (115 lb), her height is 170 cm (67 in), and her body mass index (BMI) is 18 kg/m2. Vital signs: temperature 35.7°C (96.4°F), blood pressure 92/48 mm Hg, heart rate 73 bpm, respiratory rate 5 breaths per minute, and oxygen saturation 98% on room air.
She appears tired but is alert, conversant, and cooperative. Her skin is normal, and dentition is fair. Her pulse is regular, and respirations are slow. The abdomen is soft, non-tender, and flat. Strength is 4 on a scale of 5 in all extremities. Deep-tendon reflexes are 2+ and symmetric.
Electrocardiography (Figure 1) in the emergency department shows ST-segment depression, a prolonged corrected QT interval of 665 msec, T-wave inversion, PR prolongation, increased P-wave amplitude, and U waves.
1. Which electrolyte abnormality is associated with this electrocardiographic picture?
- Hypercalcemia
- Hyperkalemia
- Hypocalcemia
- Hypokalemia
Hypokalemia is the likely cause of these findings. The finding of U waves is considered significant when they are inverted, merged with the T wave, or have an amplitude greater than the T wave.1 U waves are best seen in the precordial leads. When severe, hypokalemia can lead to potentially fatal arrhythmias such as high-grade atrioventricular block, ventricular tachycardia, and ventricular fibrillation.2
Hyperkalemia is associated with peaked T waves, a prolonged PR interval, decreased P wave amplitude, and a widened QRS complex.2 When acute and severe, hyperkalemia is associated with ventricular arrhythmia.
Hypocalcemia is associated with a prolonged QT interval and ventricular dysrhythmia, but not U waves.2
Hypercalcemia is associated with bradydysrhythmia, as well as with a shortened QT interval.2
LABORATORY TESTING
Laboratory testing shows the following:
- Sodium 126 mmol/L (reference range 135–145)
- Potassium 1.5 mmol/L (3.5–5.1)
- Chloride 58 mmol/L (100–110)
- Bicarbonate 62 mmol/L (20–30)
- Blood urea nitrogen 16 mg/dL (7–18)
- Creatinine 0.8 mg/dL (0.5–1.0)
- Glucose 106 mg/dL (70–110)
- Ionized calcium 4.4 mg/dL (4.5–5.3)
- Magnesium 1.8 mg/dL (1.7–2.3)
- Phosphorus 4.1 mg/dL (2.5–4.5)
- Venous blood gases pH 7.56 (7.35–7.45), Pco2 69 mm Hg (35–45).
POTASSIUM HOMEOSTASIS
Ninety-eight percent of potassium is intracellular and only 2% is extracellular.3 The main cellular stores are myocytes and hepatocytes. Patients with decreased muscle mass may be at a higher risk of hypokalemia as a result of decreased skeletal muscle stores.4
The acute development of hypokalemia occurs from transcellular shifts. Alkalosis, insulin secretion, and beta-adrenergic stimulation promote the intracellular uptake of potassium. The major hormonal regulator of potassium excretion is aldosterone, which is stimulated by renal hypoperfusion and promotes potassium-ion secretion in the distal convoluted tubule.
Chronic hypokalemia develops in patients with ongoing renal or gastrointestinal potassium loss. If the cause of potassium loss is not elucidated by the history, the physical, and a review of medications, then one of two things is possible: either the patient has renal tubular disease affecting acid-base and potassium regulation, causing excessive mineralocorticoid secretion, which is associated with an abnormal response to aldosterone; or the patient is not being forthcoming in the history.
2. Which is the most likely cause of hypokalemia in this patient?
- Vomiting
- Liddle syndrome
- Bartter syndrome
- Gitelman syndrome
- Diuretic use
Her laboratory tests reveal hypokalemia and hyponatremic-hypochloremic metabolic alkalosis with compensatory respiratory acidosis. In metabolic alkalosis, the expected respiratory compensation is an increase of 0.7 mm Hg in Pco2 for each 1-mEq/L increase in bicarbonate. Therefore, the expected Pco2 is 67, close to the patient’s actual value of 69.
Protracted vomiting with loss of gastric acid juices could be a cause of the metabolic disturbances in this young woman, although she did not mention vomiting during the history.
Liddle syndrome, or pseudoaldosteronism, is a rare autosomal dominant disorder characterized by altered renal epithelial sodium channels, excessive sodium retention, and resultant hypertension. Hypokalemia and alkalosis are seen in Liddle syndrome, but the absence of hypertension in our patient makes Liddle syndrome unlikely.
Bartter syndrome is an inherited autosomal recessive disorder of the sodium-potassium-chloride cotransporter in the thick ascending loop of Henle, resulting in impaired reabsorption of chloride and sodium. Bartter syndrome mimics chronic loop-diuretic use and is associated with hypercalciuria. Bartter syndrome is possible in this patient; however, patients with Bartter syndrome are usually diagnosed in infancy or childhood and have evidence of growth impairment.
Gitelman syndrome is an autosomal recessive disorder of the thiazide-sensitive sodium-chloride cotransporter. Although Gitelman syndrome is more common than Bartter syndrome and presents at older ages, it is not usually associated with such profound metabolic alkalosis. Gitelman syndrome mimics chronic use of thiazide diuretics and is associated with hypocalciuria.
Diuretic use could also cause the metabolic disturbances described; however, the patient denied taking diuretics.
The most common cause of hypokalemia in clinical practice is diuretic use.4 In this young woman with unexplained hypokalemia, the most likely cause is either undisclosed self-induced vomiting or diuretic abuse. The degree of metabolic alkalosis suggests vomiting, since metabolic alkalosis this severe is usually seen only with protracted vomiting. Bartter and Gitelman syndromes are included in the differential diagnosis, but they are much less common than hypokalemia associated with diuretics or self-induced vomiting.5
3. Which test could help elucidate the cause of hypokalemia in this patient?
- Ratio of plasma aldosterone to rennin
- Urine chloride
- Ratio of urinary potassium to creatinine
- Urinary anion gap and urinary pH
APPROACH TO HYPOKALEMIA
Determining the cause of hypokalemia starts with a thorough history and physical examination. The history should focus on drugs such as diuretics and laxatives. Women should be asked about their menstrual history since irregular periods may suggest an eating disorder. The physical examination should focus on signs that suggest self-induced vomiting, such as dry skin, dental erosions, enlarged parotid glands, and calluses or scars on the knuckles.
Patients with an unclear cause of hypokalemia after a thorough history and physical examination can be categorized into one of three groups based on blood pressure and acid-base status:
- Hypokalemia, hypertension, metabolic alkalosis
- Hypokalemia, normal blood pressure, metabolic acidosis
- Hypokalemia, normal blood pressure, metabolic alkalosis.
Hypokalemia, hypertension, metabolic alkalosis
The blood pressure provides an important clue in the evaluation of hypokalemia. The combination of hypertension, hypokalemia, and alkalosis should raise concern for hyperaldosteronism or pseudoaldosteronism. Primary hyperaldosteronism from an adrenal adenoma (Conn syndrome) is characterized by a plasma aldosterone-renin ratio of greater than 20.6,7 In contrast, patients with secondary hyperaldosteronism due to renovascular disease have a plasma aldosterone-renin ratio of less than 10. Patients with pseudoaldosteronism have low aldosterone and renin levels and hypertension. Since our patient has a normal blood pressure, testing the plasma aldosterone and renin levels would not help determine the cause of her hypokalemia.
Hypokalemia, normal blood pressure, metabolic acidosis
Patients with normal blood pressure, hypokalemia, and normal plasma anion gap acidosis either have renal tubular acidosis or have lost potassium because of diarrhea or laxative abuse. In a patient who denies taking laxatives or denies a history of diarrhea, checking the urinary anion gap and urinary pH may help differentiate the cause of acidosis and hypokalemia.
The urinary anion gap, calculated by the equation sodium + potassium − chloride, is an indirect estimate of hydrogen excretion in the form of ammonium ion8; the normal value is 0 to 10 mEq/L. A negative value represents increased hydrogen excretion in response to systemic acidosis from gastrointestinal or renal loss of bicarbonate (proximal renal tubular acidosis). A urinary pH greater than 5.5 in the setting of systemic acidosis suggests impaired ability of the kidneys to acidify urine and raises the possibility of renal tubular acidosis.
This patient has metabolic alkalosis, so calculation of the urinary anion gap would not be helpful.
Hypokalemia, normal blood pressure, metabolic alkalosis
Patients such as ours, with normal blood pressure, hypokalemia, and alkalosis, have been vomiting, have used diuretics, or have an inherited renal tubulopathy such as Bartter or Gitelman syndrome. Usually, differentiating Bartter and Gitelman syndromes from chronic vomiting or diuretic use is done with the history and physical examination. However, in patients with a questionable history and a lack of findings on physical examination, checking the urinary chloride, potassium, calcium, and creatinine may be helpful.
A urinary potassium-creatinine ratio greater than 15 suggests renal loss, whereas a ratio less than 15 suggests extrarenal loss.9
Patients who are taking a diuretic or who have Bartter or Gitelman syndrome have a high urinary chloride concentration, ie, greater than 20 mmol/L, whereas patients with hypokalemia and alkalosis from chronic vomiting tend to have a concentration less than 10 mmol/L.10
Table 1 summarizes an approach to the evaluation of unexplained hypokalemia based on blood pressure and acid-base status.
A HIDDEN HISTORY
On further questioning, the patient admits to an 8-year history of daily self-induced vomiting in an attempt to lose weight, in addition to multiple hospitalizations for hypokalemia and a previous diagnosis of an eating disorder.
INITIAL MANAGEMENT OF HYPOKALEMIA
The initial management of hypokalemia should focus on life-threatening emergencies. While patients with potassium levels greater than 3 mmol/L are usually asymptomatic, those with levels below 3 mmol/L present with muscle weakness and rhabdomyolysis.4 An acute drop in serum potassium to less than 2 mmol/L is associated with respiratory muscle weakness and ventricular arrhythmias.4 If the patient has cardiac symptoms or hypoventilation due to respiratory muscle weakness, continuous monitoring in the intensive care unit and aggressive therapy are warranted.
4. Which potassium formulation is most appropriate for the treatment of hypokalemia in this patient?
- Potassium chloride
- Potassium phosphate
- Potassium acetate
Oral potassium is preferable in patients with a serum potassium above 2.5 mmol/L.4,11 Potassium phosphate should be used when supplementation with both potassium and phosphorus is needed. Potassium acetate should be reserved for patients with acidosis and hypokalemia. Otherwise, potassium chloride is typically preferred.4,12 It comes in liquid and tablet forms. Liquid forms have an unpleasant taste, whereas tablets are usually well tolerated. No more than 20 to 40 mEq of potassium chloride tablets should be given at a time, since higher doses are associated with gastrointestinal mucosal injury.12
Potassium chloride is particularly preferred in patients with metabolic alkalosis, since increased chloride intake and delivery to the distal tubule increases the expression of pendrin, a luminal chloride and bicarbonate exchanger in the cortical collecting duct.13 With metabolic alkalosis, increased excretion of bicarbonate occurs through up-regulation of pendrin. Potassium depletion down-regulates pendrin.13 Additionally, correction of metabolic alkalosis increases serum potassium by movement of potassium from the intracellular to the extracellular space.
Intravenous potassium should be reserved for patients with severe hypokalemia (< 2.5 mmol/L) or significant arrhythmias.11 Oral and intravenous potassium can safely be given simultaneously.11 The intravenous rate should not exceed more than 10 to 20 mEq of potassium chloride per hour unless the patient has a life-threatening arrhythmia, respiratory failure, or severe hypokalemia.14,15 In life-threatening situations, a femoral line should be placed, and potassium should be given as rapidly as 20 mEq over 15 to 20 minutes.14 Cannulation of the subclavian and internal jugular veins should be avoided in severe hypokalemia since mechanical irritation from guidewire placement can provoke ventricular arrhythmias.14
During intravenous administration of potassium, laboratory monitoring after every 20 mEq of potassium chloride is advised because of the possibility of rebound hyperkalemia. In patients with severe hypokalemia, avoidance of factors that can worsen intracellular shift of potassium is also important. Avoid dextrose-containing fluids to prevent insulin-induced shifting of potassium into cells. Restore intravascular volume to blunt hypovolemia-induced renin and aldosterone secretion. If a patient presents with severe hypokalemia and acidosis, correct the hypokalemia before the acidosis to avoid intracellular shift of potassium.
OUR PATIENT’S MANAGEMENT AND FOLLOW-UP PLAN
Given the severity of our patient’s hypokalemia and her complaint of palpitations, she was admitted to the hospital for monitoring. She required 180 mEq of intravenous potassium chloride and 140 mEq of oral potassium chloride during the first 24 hours in order to achieve a serum potassium level above 3 mmol/L. Electrocardiographic U waves resolved once the level was above 2 mmol/L, and ST depressions resolved once it was above 3 mmol/L. The QT interval normalized after 24 hours of hospitalization.
On discharge, she was prescribed oral potassium chloride 40 mEq daily and magnesium sulfate 400 mg twice daily, with plans for a followup visit with her outpatient therapy team, which includes a psychiatrist, a social worker, and her primary care provider. She declined a referral for inpatient therapy but agreed to a goal of decreasing the frequency of induced vomiting and outpatient visits. She was also educated on how and when to access emergency medical care.16
A 20-year-old woman presents to the emergency department with fatigue and the sudden onset of palpitations. She reports no history of significant illness or surgery. She says she is not currently taking prescription or over-the-counter medications. She does not smoke, drink alcohol, or use illicit drugs.
Her weight is 52 kg (115 lb), her height is 170 cm (67 in), and her body mass index (BMI) is 18 kg/m2. Vital signs: temperature 35.7°C (96.4°F), blood pressure 92/48 mm Hg, heart rate 73 bpm, respiratory rate 5 breaths per minute, and oxygen saturation 98% on room air.
She appears tired but is alert, conversant, and cooperative. Her skin is normal, and dentition is fair. Her pulse is regular, and respirations are slow. The abdomen is soft, non-tender, and flat. Strength is 4 on a scale of 5 in all extremities. Deep-tendon reflexes are 2+ and symmetric.
Electrocardiography (Figure 1) in the emergency department shows ST-segment depression, a prolonged corrected QT interval of 665 msec, T-wave inversion, PR prolongation, increased P-wave amplitude, and U waves.
1. Which electrolyte abnormality is associated with this electrocardiographic picture?
- Hypercalcemia
- Hyperkalemia
- Hypocalcemia
- Hypokalemia
Hypokalemia is the likely cause of these findings. The finding of U waves is considered significant when they are inverted, merged with the T wave, or have an amplitude greater than the T wave.1 U waves are best seen in the precordial leads. When severe, hypokalemia can lead to potentially fatal arrhythmias such as high-grade atrioventricular block, ventricular tachycardia, and ventricular fibrillation.2
Hyperkalemia is associated with peaked T waves, a prolonged PR interval, decreased P wave amplitude, and a widened QRS complex.2 When acute and severe, hyperkalemia is associated with ventricular arrhythmia.
Hypocalcemia is associated with a prolonged QT interval and ventricular dysrhythmia, but not U waves.2
Hypercalcemia is associated with bradydysrhythmia, as well as with a shortened QT interval.2
LABORATORY TESTING
Laboratory testing shows the following:
- Sodium 126 mmol/L (reference range 135–145)
- Potassium 1.5 mmol/L (3.5–5.1)
- Chloride 58 mmol/L (100–110)
- Bicarbonate 62 mmol/L (20–30)
- Blood urea nitrogen 16 mg/dL (7–18)
- Creatinine 0.8 mg/dL (0.5–1.0)
- Glucose 106 mg/dL (70–110)
- Ionized calcium 4.4 mg/dL (4.5–5.3)
- Magnesium 1.8 mg/dL (1.7–2.3)
- Phosphorus 4.1 mg/dL (2.5–4.5)
- Venous blood gases pH 7.56 (7.35–7.45), Pco2 69 mm Hg (35–45).
POTASSIUM HOMEOSTASIS
Ninety-eight percent of potassium is intracellular and only 2% is extracellular.3 The main cellular stores are myocytes and hepatocytes. Patients with decreased muscle mass may be at a higher risk of hypokalemia as a result of decreased skeletal muscle stores.4
The acute development of hypokalemia occurs from transcellular shifts. Alkalosis, insulin secretion, and beta-adrenergic stimulation promote the intracellular uptake of potassium. The major hormonal regulator of potassium excretion is aldosterone, which is stimulated by renal hypoperfusion and promotes potassium-ion secretion in the distal convoluted tubule.
Chronic hypokalemia develops in patients with ongoing renal or gastrointestinal potassium loss. If the cause of potassium loss is not elucidated by the history, the physical, and a review of medications, then one of two things is possible: either the patient has renal tubular disease affecting acid-base and potassium regulation, causing excessive mineralocorticoid secretion, which is associated with an abnormal response to aldosterone; or the patient is not being forthcoming in the history.
2. Which is the most likely cause of hypokalemia in this patient?
- Vomiting
- Liddle syndrome
- Bartter syndrome
- Gitelman syndrome
- Diuretic use
Her laboratory tests reveal hypokalemia and hyponatremic-hypochloremic metabolic alkalosis with compensatory respiratory acidosis. In metabolic alkalosis, the expected respiratory compensation is an increase of 0.7 mm Hg in Pco2 for each 1-mEq/L increase in bicarbonate. Therefore, the expected Pco2 is 67, close to the patient’s actual value of 69.
Protracted vomiting with loss of gastric acid juices could be a cause of the metabolic disturbances in this young woman, although she did not mention vomiting during the history.
Liddle syndrome, or pseudoaldosteronism, is a rare autosomal dominant disorder characterized by altered renal epithelial sodium channels, excessive sodium retention, and resultant hypertension. Hypokalemia and alkalosis are seen in Liddle syndrome, but the absence of hypertension in our patient makes Liddle syndrome unlikely.
Bartter syndrome is an inherited autosomal recessive disorder of the sodium-potassium-chloride cotransporter in the thick ascending loop of Henle, resulting in impaired reabsorption of chloride and sodium. Bartter syndrome mimics chronic loop-diuretic use and is associated with hypercalciuria. Bartter syndrome is possible in this patient; however, patients with Bartter syndrome are usually diagnosed in infancy or childhood and have evidence of growth impairment.
Gitelman syndrome is an autosomal recessive disorder of the thiazide-sensitive sodium-chloride cotransporter. Although Gitelman syndrome is more common than Bartter syndrome and presents at older ages, it is not usually associated with such profound metabolic alkalosis. Gitelman syndrome mimics chronic use of thiazide diuretics and is associated with hypocalciuria.
Diuretic use could also cause the metabolic disturbances described; however, the patient denied taking diuretics.
The most common cause of hypokalemia in clinical practice is diuretic use.4 In this young woman with unexplained hypokalemia, the most likely cause is either undisclosed self-induced vomiting or diuretic abuse. The degree of metabolic alkalosis suggests vomiting, since metabolic alkalosis this severe is usually seen only with protracted vomiting. Bartter and Gitelman syndromes are included in the differential diagnosis, but they are much less common than hypokalemia associated with diuretics or self-induced vomiting.5
3. Which test could help elucidate the cause of hypokalemia in this patient?
- Ratio of plasma aldosterone to rennin
- Urine chloride
- Ratio of urinary potassium to creatinine
- Urinary anion gap and urinary pH
APPROACH TO HYPOKALEMIA
Determining the cause of hypokalemia starts with a thorough history and physical examination. The history should focus on drugs such as diuretics and laxatives. Women should be asked about their menstrual history since irregular periods may suggest an eating disorder. The physical examination should focus on signs that suggest self-induced vomiting, such as dry skin, dental erosions, enlarged parotid glands, and calluses or scars on the knuckles.
Patients with an unclear cause of hypokalemia after a thorough history and physical examination can be categorized into one of three groups based on blood pressure and acid-base status:
- Hypokalemia, hypertension, metabolic alkalosis
- Hypokalemia, normal blood pressure, metabolic acidosis
- Hypokalemia, normal blood pressure, metabolic alkalosis.
Hypokalemia, hypertension, metabolic alkalosis
The blood pressure provides an important clue in the evaluation of hypokalemia. The combination of hypertension, hypokalemia, and alkalosis should raise concern for hyperaldosteronism or pseudoaldosteronism. Primary hyperaldosteronism from an adrenal adenoma (Conn syndrome) is characterized by a plasma aldosterone-renin ratio of greater than 20.6,7 In contrast, patients with secondary hyperaldosteronism due to renovascular disease have a plasma aldosterone-renin ratio of less than 10. Patients with pseudoaldosteronism have low aldosterone and renin levels and hypertension. Since our patient has a normal blood pressure, testing the plasma aldosterone and renin levels would not help determine the cause of her hypokalemia.
Hypokalemia, normal blood pressure, metabolic acidosis
Patients with normal blood pressure, hypokalemia, and normal plasma anion gap acidosis either have renal tubular acidosis or have lost potassium because of diarrhea or laxative abuse. In a patient who denies taking laxatives or denies a history of diarrhea, checking the urinary anion gap and urinary pH may help differentiate the cause of acidosis and hypokalemia.
The urinary anion gap, calculated by the equation sodium + potassium − chloride, is an indirect estimate of hydrogen excretion in the form of ammonium ion8; the normal value is 0 to 10 mEq/L. A negative value represents increased hydrogen excretion in response to systemic acidosis from gastrointestinal or renal loss of bicarbonate (proximal renal tubular acidosis). A urinary pH greater than 5.5 in the setting of systemic acidosis suggests impaired ability of the kidneys to acidify urine and raises the possibility of renal tubular acidosis.
This patient has metabolic alkalosis, so calculation of the urinary anion gap would not be helpful.
Hypokalemia, normal blood pressure, metabolic alkalosis
Patients such as ours, with normal blood pressure, hypokalemia, and alkalosis, have been vomiting, have used diuretics, or have an inherited renal tubulopathy such as Bartter or Gitelman syndrome. Usually, differentiating Bartter and Gitelman syndromes from chronic vomiting or diuretic use is done with the history and physical examination. However, in patients with a questionable history and a lack of findings on physical examination, checking the urinary chloride, potassium, calcium, and creatinine may be helpful.
A urinary potassium-creatinine ratio greater than 15 suggests renal loss, whereas a ratio less than 15 suggests extrarenal loss.9
Patients who are taking a diuretic or who have Bartter or Gitelman syndrome have a high urinary chloride concentration, ie, greater than 20 mmol/L, whereas patients with hypokalemia and alkalosis from chronic vomiting tend to have a concentration less than 10 mmol/L.10
Table 1 summarizes an approach to the evaluation of unexplained hypokalemia based on blood pressure and acid-base status.
A HIDDEN HISTORY
On further questioning, the patient admits to an 8-year history of daily self-induced vomiting in an attempt to lose weight, in addition to multiple hospitalizations for hypokalemia and a previous diagnosis of an eating disorder.
INITIAL MANAGEMENT OF HYPOKALEMIA
The initial management of hypokalemia should focus on life-threatening emergencies. While patients with potassium levels greater than 3 mmol/L are usually asymptomatic, those with levels below 3 mmol/L present with muscle weakness and rhabdomyolysis.4 An acute drop in serum potassium to less than 2 mmol/L is associated with respiratory muscle weakness and ventricular arrhythmias.4 If the patient has cardiac symptoms or hypoventilation due to respiratory muscle weakness, continuous monitoring in the intensive care unit and aggressive therapy are warranted.
4. Which potassium formulation is most appropriate for the treatment of hypokalemia in this patient?
- Potassium chloride
- Potassium phosphate
- Potassium acetate
Oral potassium is preferable in patients with a serum potassium above 2.5 mmol/L.4,11 Potassium phosphate should be used when supplementation with both potassium and phosphorus is needed. Potassium acetate should be reserved for patients with acidosis and hypokalemia. Otherwise, potassium chloride is typically preferred.4,12 It comes in liquid and tablet forms. Liquid forms have an unpleasant taste, whereas tablets are usually well tolerated. No more than 20 to 40 mEq of potassium chloride tablets should be given at a time, since higher doses are associated with gastrointestinal mucosal injury.12
Potassium chloride is particularly preferred in patients with metabolic alkalosis, since increased chloride intake and delivery to the distal tubule increases the expression of pendrin, a luminal chloride and bicarbonate exchanger in the cortical collecting duct.13 With metabolic alkalosis, increased excretion of bicarbonate occurs through up-regulation of pendrin. Potassium depletion down-regulates pendrin.13 Additionally, correction of metabolic alkalosis increases serum potassium by movement of potassium from the intracellular to the extracellular space.
Intravenous potassium should be reserved for patients with severe hypokalemia (< 2.5 mmol/L) or significant arrhythmias.11 Oral and intravenous potassium can safely be given simultaneously.11 The intravenous rate should not exceed more than 10 to 20 mEq of potassium chloride per hour unless the patient has a life-threatening arrhythmia, respiratory failure, or severe hypokalemia.14,15 In life-threatening situations, a femoral line should be placed, and potassium should be given as rapidly as 20 mEq over 15 to 20 minutes.14 Cannulation of the subclavian and internal jugular veins should be avoided in severe hypokalemia since mechanical irritation from guidewire placement can provoke ventricular arrhythmias.14
During intravenous administration of potassium, laboratory monitoring after every 20 mEq of potassium chloride is advised because of the possibility of rebound hyperkalemia. In patients with severe hypokalemia, avoidance of factors that can worsen intracellular shift of potassium is also important. Avoid dextrose-containing fluids to prevent insulin-induced shifting of potassium into cells. Restore intravascular volume to blunt hypovolemia-induced renin and aldosterone secretion. If a patient presents with severe hypokalemia and acidosis, correct the hypokalemia before the acidosis to avoid intracellular shift of potassium.
OUR PATIENT’S MANAGEMENT AND FOLLOW-UP PLAN
Given the severity of our patient’s hypokalemia and her complaint of palpitations, she was admitted to the hospital for monitoring. She required 180 mEq of intravenous potassium chloride and 140 mEq of oral potassium chloride during the first 24 hours in order to achieve a serum potassium level above 3 mmol/L. Electrocardiographic U waves resolved once the level was above 2 mmol/L, and ST depressions resolved once it was above 3 mmol/L. The QT interval normalized after 24 hours of hospitalization.
On discharge, she was prescribed oral potassium chloride 40 mEq daily and magnesium sulfate 400 mg twice daily, with plans for a followup visit with her outpatient therapy team, which includes a psychiatrist, a social worker, and her primary care provider. She declined a referral for inpatient therapy but agreed to a goal of decreasing the frequency of induced vomiting and outpatient visits. She was also educated on how and when to access emergency medical care.16
- Rautaharju PM, Surawicz B, Gettes LS, et al; American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; American College of Cardiology Foundation; Heart Rhythm Society. AHA/ACCF/HRS recommendations for the standardization and interpretation of the electrocardiogram: part IV: the ST segment, T and U waves, and the QT Interval: a scientific statement from the American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; the American College of Cardiology Foundation; and the Heart Rhythm Society. Endorsed by the International Society for Computerized Electrocardiology. J Am Coll Cardiol 2009; 53:982–991.
- Diercks DB, Shumaik GM, Harrigan RA, Brady WJ, Chan TC. Electrocardiographic manifestations: electrolyte abnormalities. J Emerg Med 2004; 27:153–160.
- Unwin RJ, Luft FC, Shirley DG. Pathophysiology and management of hypokalemia: a clinical perspective. Nat Rev Nephrol 2011; 7:75–84.
- Gennari FJ. Hypokalemia. N Engl J Med 1998; 339:451–458.
- Mehler PS. Clinical practice. Bulimia nervosa. N Engl J Med 2003; 349:875–881.
- Tzanela M, Effraimidis G, Vassiliadi D, et al. The aldosterone to renin ratio in the evaluation of patients with incidentally detected adrenal masses. Endocrine 2007; 32:136–142.
- Diederich S, Mai K, Bähr V, Helffrich S, Pfeiffer A, Perschel FH. The simultaneous measurement of plasma-aldosterone- and -renin-concentration allows rapid classification of all disorders of the renin-aldosterone system. Exp Clin Endocrinol Diabetes 2007; 115:433–438.
- Goldstein MB, Bear R, Richardson RM, Marsden PA, Halperin ML. The urine anion gap: a clinically useful index of ammonium excretion. Am J Med Sci 1986; 292:198–202.
- Groeneveld JH, Sijpkens YW, Lin SH, Davids MR, Halperin ML. An approach to the patient with severe hypokalaemia: the potassium quiz. QJM 2005; 98:305–316.
- Galla JH. Metabolic alkalosis. J Am Soc Nephrol 2000; 11:369–375.
- Asmar A, Mohandas R, Wingo CS. A physiologic-based approach to the treatment of a patient with hypokalemia. Am J Kidney Dis 2012; 60:492–497.
- Cohn JN, Kowey PR, Whelton PK, Prisant LM. New guidelines for potassium replacement in clinical practice: a contemporary review by the National Council on Potassium in Clinical Practice. Arch Intern Med 2000; 160:2429–2436.
- Luke RG, Galla JH. It is chloride depletion alkalosis, not contraction alkalosis. J Am Soc Nephrol 2012; 23:204–207.
- Kruse JA, Carlson RW. Rapid correction of hypokalemia using concentrated intravenous potassium chloride infusions. Arch Intern Med 1990; 150:613–617.
- Weiner ID, Wingo CS. Hypokalemia—consequences, causes, and correction. J Am Soc Nephrol 1997; 8:1179–1188.
- AED Medical Care Standards Task Force. Eating disorders: Critical Points for Early Recognition and Medical Risk Management in the Care of Individuals with Eating Disorders. AED Report 2012. www.aedweb.org/web/downloads/Guide-English.pdf. Accessed April 4, 2014.
- Rautaharju PM, Surawicz B, Gettes LS, et al; American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; American College of Cardiology Foundation; Heart Rhythm Society. AHA/ACCF/HRS recommendations for the standardization and interpretation of the electrocardiogram: part IV: the ST segment, T and U waves, and the QT Interval: a scientific statement from the American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; the American College of Cardiology Foundation; and the Heart Rhythm Society. Endorsed by the International Society for Computerized Electrocardiology. J Am Coll Cardiol 2009; 53:982–991.
- Diercks DB, Shumaik GM, Harrigan RA, Brady WJ, Chan TC. Electrocardiographic manifestations: electrolyte abnormalities. J Emerg Med 2004; 27:153–160.
- Unwin RJ, Luft FC, Shirley DG. Pathophysiology and management of hypokalemia: a clinical perspective. Nat Rev Nephrol 2011; 7:75–84.
- Gennari FJ. Hypokalemia. N Engl J Med 1998; 339:451–458.
- Mehler PS. Clinical practice. Bulimia nervosa. N Engl J Med 2003; 349:875–881.
- Tzanela M, Effraimidis G, Vassiliadi D, et al. The aldosterone to renin ratio in the evaluation of patients with incidentally detected adrenal masses. Endocrine 2007; 32:136–142.
- Diederich S, Mai K, Bähr V, Helffrich S, Pfeiffer A, Perschel FH. The simultaneous measurement of plasma-aldosterone- and -renin-concentration allows rapid classification of all disorders of the renin-aldosterone system. Exp Clin Endocrinol Diabetes 2007; 115:433–438.
- Goldstein MB, Bear R, Richardson RM, Marsden PA, Halperin ML. The urine anion gap: a clinically useful index of ammonium excretion. Am J Med Sci 1986; 292:198–202.
- Groeneveld JH, Sijpkens YW, Lin SH, Davids MR, Halperin ML. An approach to the patient with severe hypokalaemia: the potassium quiz. QJM 2005; 98:305–316.
- Galla JH. Metabolic alkalosis. J Am Soc Nephrol 2000; 11:369–375.
- Asmar A, Mohandas R, Wingo CS. A physiologic-based approach to the treatment of a patient with hypokalemia. Am J Kidney Dis 2012; 60:492–497.
- Cohn JN, Kowey PR, Whelton PK, Prisant LM. New guidelines for potassium replacement in clinical practice: a contemporary review by the National Council on Potassium in Clinical Practice. Arch Intern Med 2000; 160:2429–2436.
- Luke RG, Galla JH. It is chloride depletion alkalosis, not contraction alkalosis. J Am Soc Nephrol 2012; 23:204–207.
- Kruse JA, Carlson RW. Rapid correction of hypokalemia using concentrated intravenous potassium chloride infusions. Arch Intern Med 1990; 150:613–617.
- Weiner ID, Wingo CS. Hypokalemia—consequences, causes, and correction. J Am Soc Nephrol 1997; 8:1179–1188.
- AED Medical Care Standards Task Force. Eating disorders: Critical Points for Early Recognition and Medical Risk Management in the Care of Individuals with Eating Disorders. AED Report 2012. www.aedweb.org/web/downloads/Guide-English.pdf. Accessed April 4, 2014.
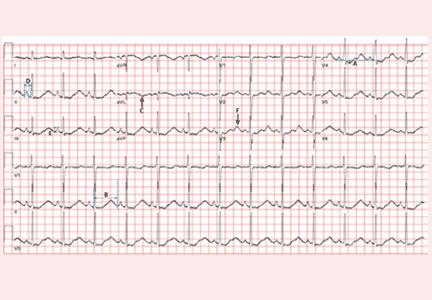
Inmate Falls From Top Bunk
ANSWER
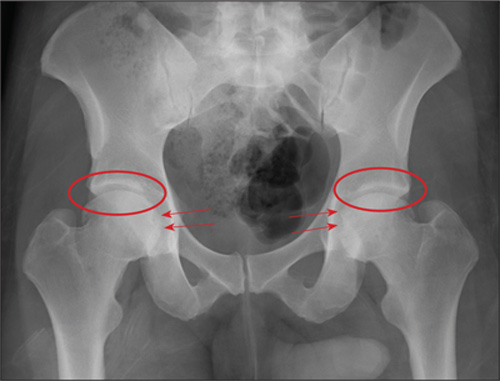
The radiograph demonstrates no acute osseous injury, such as fracture or dislocation. Of interest and note is increased sclerosis within both femoral heads, more so on the left versus the right side. Given the patient’s young age, such findings could be related to early avascular necrosis. His clinical symptoms certainly correlate. MRI or bone scan, as well as orthopedic evaluation, is warranted in such a case.
Fortunately, subsequent MRI of both hips did not show any avascular necrosis but rather osteoarthritic changes. The MRI of his spinal column was negative as well.
ANSWER
The radiograph demonstrates no acute osseous injury, such as fracture or dislocation. Of interest and note is increased sclerosis within both femoral heads, more so on the left versus the right side. Given the patient’s young age, such findings could be related to early avascular necrosis. His clinical symptoms certainly correlate. MRI or bone scan, as well as orthopedic evaluation, is warranted in such a case.
Fortunately, subsequent MRI of both hips did not show any avascular necrosis but rather osteoarthritic changes. The MRI of his spinal column was negative as well.
ANSWER
The radiograph demonstrates no acute osseous injury, such as fracture or dislocation. Of interest and note is increased sclerosis within both femoral heads, more so on the left versus the right side. Given the patient’s young age, such findings could be related to early avascular necrosis. His clinical symptoms certainly correlate. MRI or bone scan, as well as orthopedic evaluation, is warranted in such a case.
Fortunately, subsequent MRI of both hips did not show any avascular necrosis but rather osteoarthritic changes. The MRI of his spinal column was negative as well.
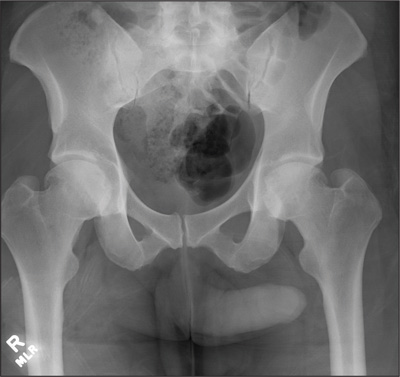
A 30-year-old man is transferred to your facility for evaluation of reported paraplegia after a fall. The patient is an inmate at a local prison. He states he was sleeping on the top bunk when he rolled over and fell off the bed, landing flat on his back on the concrete floor. He immediately started having severe back and hip pain and noticed that he could not move his legs. His primary complaint is severe bilateral hip pain. He was initially evaluated at an outside hospital, where CT of his head, cervical spine, and lumbar spine was negative for any acute pathology. He was sent to your facility for an MRI to rule out contusion or acute herniated disc. The patient denies any significant medical history, including back trauma. Currently, he reports no bowel/bladder issues or saddle anesthesia. On initial exam, he is awake, alert, and oriented, with normal vital signs. Musculoskeletal exam demonstrates a moderate amount of paraspinous tenderness and bilateral hip/pelvis tenderness. There is no instability detected, nor any leg shortening or rotation. He does have bilateral weakness in both lower extremities on the magnitude of 3-/5, although his exam seems limited due to the severity of his hip pain. Sensation is completely intact in both lower extremities. While the patient is awaiting his MRI, you order a portable pelvis radiograph, since none was performed at the outside facility. What is your impression?
Case Studies in Toxicology: A Patchwork of Problems in Parkinson Patients
Case
A 76-year-old man with Parkinson disease (PD) and hypertension presented to the ED with acute onset of severe tremulousness, blurred vision, salivation, lacrimation, diffuse muscle aches, and extremity weakness. His initial vital signs were: blood pressure, 175/74 mm Hg; heart rate, 62 beats/minute; respiratory rate, 16 breaths/minute; temperature, 37°C (98.6°F). Oxygen saturation was 100% on room air. On physical examination, the patient had excessive lacrimation and salivation, a coarse resting tremor, and 2/5 strength in both the upper and lower extremities. The remainder of the examination, including abdominal and pulmonary systems, was unremarkable compared with baseline findings.
How does the pathophysiology of PD explain how treatments are targeted?

What medications are used to treat PD? What are some associated complications?
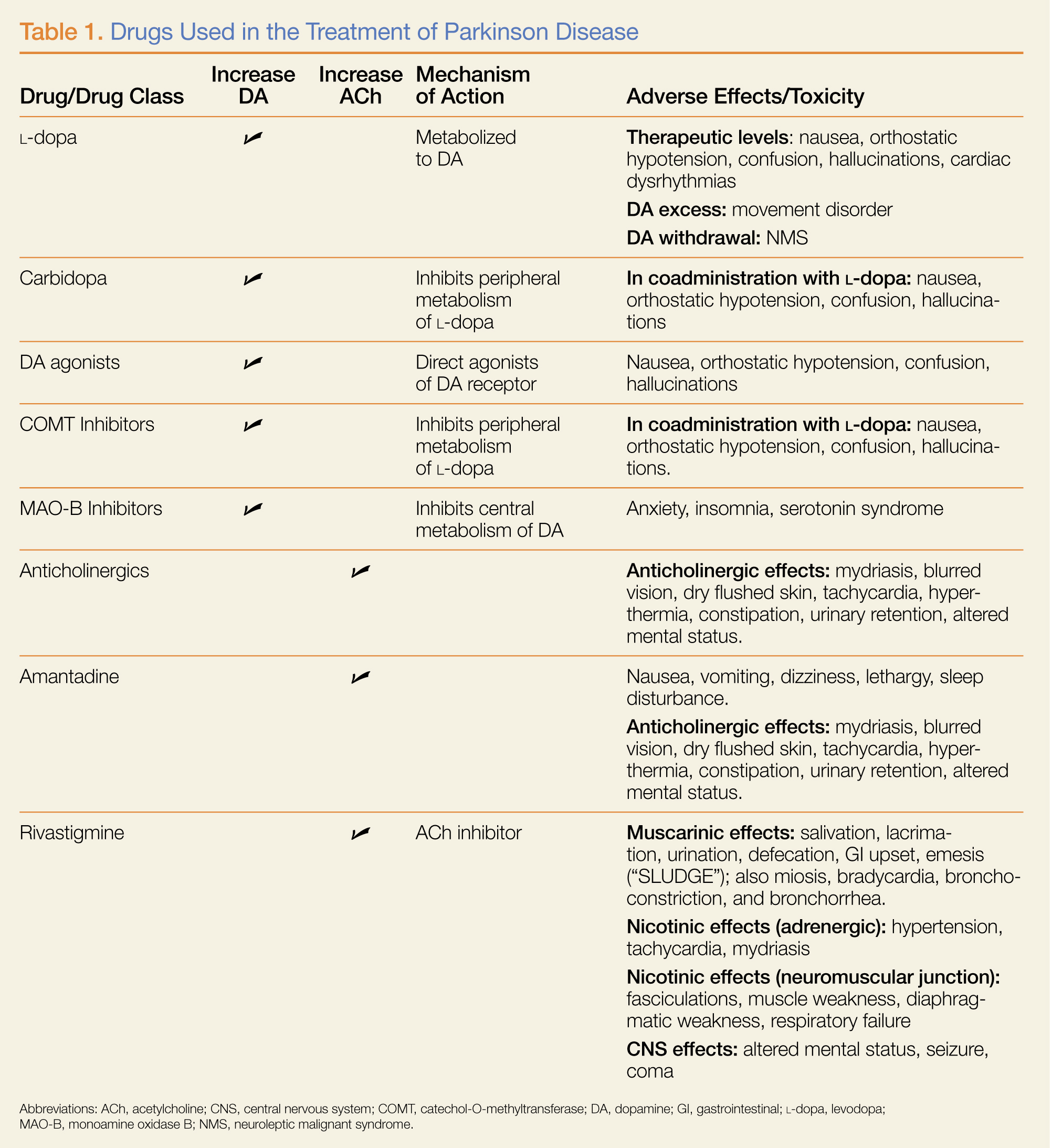
Dopamine Precursors and Agonists
(L-dopa) can be combined with the L-amino acid decarboxylase inhibitor carbidopa to prevent peripheral metabolism by this enzyme and thereby increase brain concentrations of DA following metabolism by DA decarboxylase in the central nervous system (CNS).1 Dopamine agonists, including bromocriptine, ropinirole, and pramipexole, do not depend on endogenous conversion to DA and have substantially longer durations of action, limiting the dose-related fluctuations in motor function common in some PD patients taking L-dopa.1 For these reasons, DA agonists have often replaced L-dopa as initial treatment, especially in younger patients. Catechol-O-methyltransferase inhibitors (tolcapone, entacapone) prevent peripheral breakdown of DA, allowing a higher fraction to reach the CNS.
With respect to side effects, all of the dopaminergic medications can cause nausea, hallucinations, confusion, and orthostatic hypotension.
Anticholinergic Drugs
Although the precise mechanism by which anticholinergic drugs improve PD is not fully understood, agents such as trihexyphenidyl, benztropine mesylate, and diphenhydramine hydrochloride were prescribed even before the discovery of L-dopa and continue to be used today.1 Adverse effects are a function of the antimuscarinic (anticholinergic) properties of the drugs and may include mydriasis and blurred vision, dry flushed skin, tachycardia, hyperthermia, constipation, urinary retention, and altered mental status.
Amantadine
In addition to the anticholinergics, amantadine is also used to treat PD. This antiviral agent alters DA release in the brain, produces anticholinergic effects, and blocks N-methyl-D-aspartate glutamate receptors.1 Common adverse drug effects include anticholinergic signs as well as nausea, vomiting, dizziness, lethargy, and sleep disturbance, all of which are usually mild and reversible.
Case Continuation
A review of the patient’s medication history revealed he has been taking L-dopa/carbidopa. In addition to L-dopa/carbidopa, he was recently prescribed transdermal rivastigmine patches (13.3 mg/24 h). At bedtime the evening prior to presentation, the patient applied more than 20 rivastigmine patches. Approximately 5 hours later, he awoke with the previously described findings whereupon his wife removed the patches and brought him to the ED.
What is rivastigmine and what is its role in PD
Rivastigmine is a carbamate-type cholinesterase inhibitor (CEI) indicated for the treatment of mild-to-moderate dementia associated with PD and Alzheimer disease.2 Tacrine, a medicinal noncarbamate CEI, is also prescribed for this use.2 Both drugs increase ACh concentrations in relevant brain regions and foster the formation of new memory.
Cholinesterase inhibitors are mechanistically analogous to the insecticidal carbamates (eg, aldicarb) and the organophosphates (OPs) (eg, malathion). They inhibit the metabolism of ACh by acetylcholinesterase (AChE) in the various cholinergic synapses, increasing the intrasynaptic concentration of ACh.
Additional AChEs include physostigmine, a carbamate commonly used in the ED to treat anticholinergic toxicity. Physostigmine raises the local synaptic concentration of ACh to compete for the muscarinic ACh receptor with drugs such as diphenhydramine or atropine. Other CEIs (eg, neostigmine, pyridostigmine, edrophonium) are used to raise intrasynaptic ACh concentrations and overcome antibody blockade of nicotinic ACh receptors at the neuromuscular junction in patients with myasthenia gravis.
What is the toxidrome associated with carbamate overdose
Carbamate toxicity, as manifested by the cholinergic toxidrome, largely resembles OP toxicity but with an important difference: Both OPs and carbamates function by binding to and inhibiting AChE; however, the carbamate-AChE bond undergoes spontaneous hydrolysis, thereby reactivating the enzyme. Consequently, the clinical effects of carbamate toxicity, though potentially severe, are self-limited and usually only last 24 hours or less.4
How should this patient be managed?
The general approach to a patient with medical carbamate toxicity is similar to that of a patient with OP poisoning. Dermal exposure, as is the case with this patient, should prompt skin decontamination to minimize ongoing exposure. Patch removal is necessary but is not sufficient to prevent ongoing absorption, since a depot of medication typically forms in the dermal tissue. In the presence of significant or life-threatening muscarinic effects (eg, bronchorrhea, bronchospasm, seizure), an antimuscarinic agent such as atropine is indicated. Various dosing schemes of atropine exist; at our institution, we recommend an initial dose of 1 to 3 mg intravenously (IV), with escalating doses every 5 minutes until reversal of bronchorrhea and bronchospasm occur.4 This is followed by initiation of an atropine infusion at a rate of 10% to 20% of the total loading dose per hour (to a maximum of 2 mg/h).4
Pralidoxime (2-PAM) and other oximes, accelerate the reactivation of carbamate-inhibited AChE and have effects at both the nicotinic and muscarinic synapses. Reactivation results in the enhanced metabolism of intrasynaptic ACh and decreased clinical cholinergic effects. Since atropine is only effective at muscarinic receptors, oximes were administered in this case to reverse neuromuscular weakness.
Although early administration of 2-PAM is indicated in the setting of significant OP poisoning (due to irreversible inhibition of AChE), its use for medical carbamate toxicity is controversial. Early animal studies of carbamate toxicity suggested that treatment with oximes worsened outcomes; however, this has not been demonstrated in more recent studies.5,6 Therefore, although 2-PAM may be beneficial in treating cases of clinically significant carbamate poisoning (which can be prolonged and severe), these benefits should be weighed against the potential risks.
Case Conclusion
Upon arrival to the ED, the patient’s skin was cleansed thoroughly. As he did not exhibit muscarinic findings of bradycardia, bronchoconstriction, or bronchorrhea, atropine was not indicated. He was treated conservatively with IV fluid hydration and admitted to the medicine floor. Since he continued to exhibit profound extremity weakness with no improvement 12 hours from the onset of symptoms, pralidoxime 1 g IV was administered over a 30-minute period. Shortly thereafter, patient’s motor strength improved from 2/5 to 4/5 in both upper and lower extremities. No complications were noted, and the patient‘s weakness and tremulousness continued to resolve. He was transferred to a skilled nursing facility on hospital day 6.
Dr Laskowski is a medical toxicology fellow in the department of emergency medicine at New York University Langone Medical Center. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Standaert DG, Roberson ED. Treatment of central nervous system degenerative disorders. In: Brunton LL, Chabner BA, Knollmann BC. Goodman & Gilman’s The Pharmacologic Basis of Therapeutics. 12th ed. New York, NY: McGraw-Hill; 2011:609-628
- Rösler M, Anand R, Cicin-Sain A, et al. Efficacy and safety of rivastigmine in patients with Alzheimer’s disease: international randomised controlled trial. BMJ. 1999;318(7184):633-638.
- Exelon Patch [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2013.
- Eddleston M, Clark RF. Insecticides: organic phosphorus compounds and carbamates. In: Nelson LS, Lewin NA, Howland MA, Hoffman RS, Goldfrank LR, Flomenbaum NE, eds. Goldfrank’s Toxicologic Emergencies. 9th ed. New York, NY: McGraw-Hill; 2011:1450-1466.
- Natoff IL, Reiff B. Effect of oximes on the acute toxicity of anticholinesterase carbamates. Toxicol Appl Pharmacol. 1973;25(4):569-575.
- Mercurio-Zappala M, Hack JB, Salvador A, Hoffman RS. Pralidoxime in carbaryl poisoning: an animal model. Hum Exp Toxicol. 2007;26(2)125-129.
Case
A 76-year-old man with Parkinson disease (PD) and hypertension presented to the ED with acute onset of severe tremulousness, blurred vision, salivation, lacrimation, diffuse muscle aches, and extremity weakness. His initial vital signs were: blood pressure, 175/74 mm Hg; heart rate, 62 beats/minute; respiratory rate, 16 breaths/minute; temperature, 37°C (98.6°F). Oxygen saturation was 100% on room air. On physical examination, the patient had excessive lacrimation and salivation, a coarse resting tremor, and 2/5 strength in both the upper and lower extremities. The remainder of the examination, including abdominal and pulmonary systems, was unremarkable compared with baseline findings.
How does the pathophysiology of PD explain how treatments are targeted?
What medications are used to treat PD? What are some associated complications?
Dopamine Precursors and Agonists
(L-dopa) can be combined with the L-amino acid decarboxylase inhibitor carbidopa to prevent peripheral metabolism by this enzyme and thereby increase brain concentrations of DA following metabolism by DA decarboxylase in the central nervous system (CNS).1 Dopamine agonists, including bromocriptine, ropinirole, and pramipexole, do not depend on endogenous conversion to DA and have substantially longer durations of action, limiting the dose-related fluctuations in motor function common in some PD patients taking L-dopa.1 For these reasons, DA agonists have often replaced L-dopa as initial treatment, especially in younger patients. Catechol-O-methyltransferase inhibitors (tolcapone, entacapone) prevent peripheral breakdown of DA, allowing a higher fraction to reach the CNS.
With respect to side effects, all of the dopaminergic medications can cause nausea, hallucinations, confusion, and orthostatic hypotension.
Anticholinergic Drugs
Although the precise mechanism by which anticholinergic drugs improve PD is not fully understood, agents such as trihexyphenidyl, benztropine mesylate, and diphenhydramine hydrochloride were prescribed even before the discovery of L-dopa and continue to be used today.1 Adverse effects are a function of the antimuscarinic (anticholinergic) properties of the drugs and may include mydriasis and blurred vision, dry flushed skin, tachycardia, hyperthermia, constipation, urinary retention, and altered mental status.
Amantadine
In addition to the anticholinergics, amantadine is also used to treat PD. This antiviral agent alters DA release in the brain, produces anticholinergic effects, and blocks N-methyl-D-aspartate glutamate receptors.1 Common adverse drug effects include anticholinergic signs as well as nausea, vomiting, dizziness, lethargy, and sleep disturbance, all of which are usually mild and reversible.
Case Continuation
A review of the patient’s medication history revealed he has been taking L-dopa/carbidopa. In addition to L-dopa/carbidopa, he was recently prescribed transdermal rivastigmine patches (13.3 mg/24 h). At bedtime the evening prior to presentation, the patient applied more than 20 rivastigmine patches. Approximately 5 hours later, he awoke with the previously described findings whereupon his wife removed the patches and brought him to the ED.
What is rivastigmine and what is its role in PD
Rivastigmine is a carbamate-type cholinesterase inhibitor (CEI) indicated for the treatment of mild-to-moderate dementia associated with PD and Alzheimer disease.2 Tacrine, a medicinal noncarbamate CEI, is also prescribed for this use.2 Both drugs increase ACh concentrations in relevant brain regions and foster the formation of new memory.
Cholinesterase inhibitors are mechanistically analogous to the insecticidal carbamates (eg, aldicarb) and the organophosphates (OPs) (eg, malathion). They inhibit the metabolism of ACh by acetylcholinesterase (AChE) in the various cholinergic synapses, increasing the intrasynaptic concentration of ACh.
Additional AChEs include physostigmine, a carbamate commonly used in the ED to treat anticholinergic toxicity. Physostigmine raises the local synaptic concentration of ACh to compete for the muscarinic ACh receptor with drugs such as diphenhydramine or atropine. Other CEIs (eg, neostigmine, pyridostigmine, edrophonium) are used to raise intrasynaptic ACh concentrations and overcome antibody blockade of nicotinic ACh receptors at the neuromuscular junction in patients with myasthenia gravis.
What is the toxidrome associated with carbamate overdose
Carbamate toxicity, as manifested by the cholinergic toxidrome, largely resembles OP toxicity but with an important difference: Both OPs and carbamates function by binding to and inhibiting AChE; however, the carbamate-AChE bond undergoes spontaneous hydrolysis, thereby reactivating the enzyme. Consequently, the clinical effects of carbamate toxicity, though potentially severe, are self-limited and usually only last 24 hours or less.4
How should this patient be managed?
The general approach to a patient with medical carbamate toxicity is similar to that of a patient with OP poisoning. Dermal exposure, as is the case with this patient, should prompt skin decontamination to minimize ongoing exposure. Patch removal is necessary but is not sufficient to prevent ongoing absorption, since a depot of medication typically forms in the dermal tissue. In the presence of significant or life-threatening muscarinic effects (eg, bronchorrhea, bronchospasm, seizure), an antimuscarinic agent such as atropine is indicated. Various dosing schemes of atropine exist; at our institution, we recommend an initial dose of 1 to 3 mg intravenously (IV), with escalating doses every 5 minutes until reversal of bronchorrhea and bronchospasm occur.4 This is followed by initiation of an atropine infusion at a rate of 10% to 20% of the total loading dose per hour (to a maximum of 2 mg/h).4
Pralidoxime (2-PAM) and other oximes, accelerate the reactivation of carbamate-inhibited AChE and have effects at both the nicotinic and muscarinic synapses. Reactivation results in the enhanced metabolism of intrasynaptic ACh and decreased clinical cholinergic effects. Since atropine is only effective at muscarinic receptors, oximes were administered in this case to reverse neuromuscular weakness.
Although early administration of 2-PAM is indicated in the setting of significant OP poisoning (due to irreversible inhibition of AChE), its use for medical carbamate toxicity is controversial. Early animal studies of carbamate toxicity suggested that treatment with oximes worsened outcomes; however, this has not been demonstrated in more recent studies.5,6 Therefore, although 2-PAM may be beneficial in treating cases of clinically significant carbamate poisoning (which can be prolonged and severe), these benefits should be weighed against the potential risks.
Case Conclusion
Upon arrival to the ED, the patient’s skin was cleansed thoroughly. As he did not exhibit muscarinic findings of bradycardia, bronchoconstriction, or bronchorrhea, atropine was not indicated. He was treated conservatively with IV fluid hydration and admitted to the medicine floor. Since he continued to exhibit profound extremity weakness with no improvement 12 hours from the onset of symptoms, pralidoxime 1 g IV was administered over a 30-minute period. Shortly thereafter, patient’s motor strength improved from 2/5 to 4/5 in both upper and lower extremities. No complications were noted, and the patient‘s weakness and tremulousness continued to resolve. He was transferred to a skilled nursing facility on hospital day 6.
Dr Laskowski is a medical toxicology fellow in the department of emergency medicine at New York University Langone Medical Center. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
Case
A 76-year-old man with Parkinson disease (PD) and hypertension presented to the ED with acute onset of severe tremulousness, blurred vision, salivation, lacrimation, diffuse muscle aches, and extremity weakness. His initial vital signs were: blood pressure, 175/74 mm Hg; heart rate, 62 beats/minute; respiratory rate, 16 breaths/minute; temperature, 37°C (98.6°F). Oxygen saturation was 100% on room air. On physical examination, the patient had excessive lacrimation and salivation, a coarse resting tremor, and 2/5 strength in both the upper and lower extremities. The remainder of the examination, including abdominal and pulmonary systems, was unremarkable compared with baseline findings.
How does the pathophysiology of PD explain how treatments are targeted?
What medications are used to treat PD? What are some associated complications?
Dopamine Precursors and Agonists
(L-dopa) can be combined with the L-amino acid decarboxylase inhibitor carbidopa to prevent peripheral metabolism by this enzyme and thereby increase brain concentrations of DA following metabolism by DA decarboxylase in the central nervous system (CNS).1 Dopamine agonists, including bromocriptine, ropinirole, and pramipexole, do not depend on endogenous conversion to DA and have substantially longer durations of action, limiting the dose-related fluctuations in motor function common in some PD patients taking L-dopa.1 For these reasons, DA agonists have often replaced L-dopa as initial treatment, especially in younger patients. Catechol-O-methyltransferase inhibitors (tolcapone, entacapone) prevent peripheral breakdown of DA, allowing a higher fraction to reach the CNS.
With respect to side effects, all of the dopaminergic medications can cause nausea, hallucinations, confusion, and orthostatic hypotension.
Anticholinergic Drugs
Although the precise mechanism by which anticholinergic drugs improve PD is not fully understood, agents such as trihexyphenidyl, benztropine mesylate, and diphenhydramine hydrochloride were prescribed even before the discovery of L-dopa and continue to be used today.1 Adverse effects are a function of the antimuscarinic (anticholinergic) properties of the drugs and may include mydriasis and blurred vision, dry flushed skin, tachycardia, hyperthermia, constipation, urinary retention, and altered mental status.
Amantadine
In addition to the anticholinergics, amantadine is also used to treat PD. This antiviral agent alters DA release in the brain, produces anticholinergic effects, and blocks N-methyl-D-aspartate glutamate receptors.1 Common adverse drug effects include anticholinergic signs as well as nausea, vomiting, dizziness, lethargy, and sleep disturbance, all of which are usually mild and reversible.
Case Continuation
A review of the patient’s medication history revealed he has been taking L-dopa/carbidopa. In addition to L-dopa/carbidopa, he was recently prescribed transdermal rivastigmine patches (13.3 mg/24 h). At bedtime the evening prior to presentation, the patient applied more than 20 rivastigmine patches. Approximately 5 hours later, he awoke with the previously described findings whereupon his wife removed the patches and brought him to the ED.
What is rivastigmine and what is its role in PD
Rivastigmine is a carbamate-type cholinesterase inhibitor (CEI) indicated for the treatment of mild-to-moderate dementia associated with PD and Alzheimer disease.2 Tacrine, a medicinal noncarbamate CEI, is also prescribed for this use.2 Both drugs increase ACh concentrations in relevant brain regions and foster the formation of new memory.
Cholinesterase inhibitors are mechanistically analogous to the insecticidal carbamates (eg, aldicarb) and the organophosphates (OPs) (eg, malathion). They inhibit the metabolism of ACh by acetylcholinesterase (AChE) in the various cholinergic synapses, increasing the intrasynaptic concentration of ACh.
Additional AChEs include physostigmine, a carbamate commonly used in the ED to treat anticholinergic toxicity. Physostigmine raises the local synaptic concentration of ACh to compete for the muscarinic ACh receptor with drugs such as diphenhydramine or atropine. Other CEIs (eg, neostigmine, pyridostigmine, edrophonium) are used to raise intrasynaptic ACh concentrations and overcome antibody blockade of nicotinic ACh receptors at the neuromuscular junction in patients with myasthenia gravis.
What is the toxidrome associated with carbamate overdose
Carbamate toxicity, as manifested by the cholinergic toxidrome, largely resembles OP toxicity but with an important difference: Both OPs and carbamates function by binding to and inhibiting AChE; however, the carbamate-AChE bond undergoes spontaneous hydrolysis, thereby reactivating the enzyme. Consequently, the clinical effects of carbamate toxicity, though potentially severe, are self-limited and usually only last 24 hours or less.4
How should this patient be managed?
The general approach to a patient with medical carbamate toxicity is similar to that of a patient with OP poisoning. Dermal exposure, as is the case with this patient, should prompt skin decontamination to minimize ongoing exposure. Patch removal is necessary but is not sufficient to prevent ongoing absorption, since a depot of medication typically forms in the dermal tissue. In the presence of significant or life-threatening muscarinic effects (eg, bronchorrhea, bronchospasm, seizure), an antimuscarinic agent such as atropine is indicated. Various dosing schemes of atropine exist; at our institution, we recommend an initial dose of 1 to 3 mg intravenously (IV), with escalating doses every 5 minutes until reversal of bronchorrhea and bronchospasm occur.4 This is followed by initiation of an atropine infusion at a rate of 10% to 20% of the total loading dose per hour (to a maximum of 2 mg/h).4
Pralidoxime (2-PAM) and other oximes, accelerate the reactivation of carbamate-inhibited AChE and have effects at both the nicotinic and muscarinic synapses. Reactivation results in the enhanced metabolism of intrasynaptic ACh and decreased clinical cholinergic effects. Since atropine is only effective at muscarinic receptors, oximes were administered in this case to reverse neuromuscular weakness.
Although early administration of 2-PAM is indicated in the setting of significant OP poisoning (due to irreversible inhibition of AChE), its use for medical carbamate toxicity is controversial. Early animal studies of carbamate toxicity suggested that treatment with oximes worsened outcomes; however, this has not been demonstrated in more recent studies.5,6 Therefore, although 2-PAM may be beneficial in treating cases of clinically significant carbamate poisoning (which can be prolonged and severe), these benefits should be weighed against the potential risks.
Case Conclusion
Upon arrival to the ED, the patient’s skin was cleansed thoroughly. As he did not exhibit muscarinic findings of bradycardia, bronchoconstriction, or bronchorrhea, atropine was not indicated. He was treated conservatively with IV fluid hydration and admitted to the medicine floor. Since he continued to exhibit profound extremity weakness with no improvement 12 hours from the onset of symptoms, pralidoxime 1 g IV was administered over a 30-minute period. Shortly thereafter, patient’s motor strength improved from 2/5 to 4/5 in both upper and lower extremities. No complications were noted, and the patient‘s weakness and tremulousness continued to resolve. He was transferred to a skilled nursing facility on hospital day 6.
Dr Laskowski is a medical toxicology fellow in the department of emergency medicine at New York University Langone Medical Center. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Standaert DG, Roberson ED. Treatment of central nervous system degenerative disorders. In: Brunton LL, Chabner BA, Knollmann BC. Goodman & Gilman’s The Pharmacologic Basis of Therapeutics. 12th ed. New York, NY: McGraw-Hill; 2011:609-628
- Rösler M, Anand R, Cicin-Sain A, et al. Efficacy and safety of rivastigmine in patients with Alzheimer’s disease: international randomised controlled trial. BMJ. 1999;318(7184):633-638.
- Exelon Patch [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2013.
- Eddleston M, Clark RF. Insecticides: organic phosphorus compounds and carbamates. In: Nelson LS, Lewin NA, Howland MA, Hoffman RS, Goldfrank LR, Flomenbaum NE, eds. Goldfrank’s Toxicologic Emergencies. 9th ed. New York, NY: McGraw-Hill; 2011:1450-1466.
- Natoff IL, Reiff B. Effect of oximes on the acute toxicity of anticholinesterase carbamates. Toxicol Appl Pharmacol. 1973;25(4):569-575.
- Mercurio-Zappala M, Hack JB, Salvador A, Hoffman RS. Pralidoxime in carbaryl poisoning: an animal model. Hum Exp Toxicol. 2007;26(2)125-129.
- Standaert DG, Roberson ED. Treatment of central nervous system degenerative disorders. In: Brunton LL, Chabner BA, Knollmann BC. Goodman & Gilman’s The Pharmacologic Basis of Therapeutics. 12th ed. New York, NY: McGraw-Hill; 2011:609-628
- Rösler M, Anand R, Cicin-Sain A, et al. Efficacy and safety of rivastigmine in patients with Alzheimer’s disease: international randomised controlled trial. BMJ. 1999;318(7184):633-638.
- Exelon Patch [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2013.
- Eddleston M, Clark RF. Insecticides: organic phosphorus compounds and carbamates. In: Nelson LS, Lewin NA, Howland MA, Hoffman RS, Goldfrank LR, Flomenbaum NE, eds. Goldfrank’s Toxicologic Emergencies. 9th ed. New York, NY: McGraw-Hill; 2011:1450-1466.
- Natoff IL, Reiff B. Effect of oximes on the acute toxicity of anticholinesterase carbamates. Toxicol Appl Pharmacol. 1973;25(4):569-575.
- Mercurio-Zappala M, Hack JB, Salvador A, Hoffman RS. Pralidoxime in carbaryl poisoning: an animal model. Hum Exp Toxicol. 2007;26(2)125-129.

Case Report: Nasal Septal Abscess
Case
A 28-year-old woman with history of bipolar disorder and methamphetamine abuse presented to the ED complaining of nasal swelling and pain. She was unable to provide any medical history regarding the onset of her symptoms or other details, which the ED team attributed to her underlying psychiatric disorder. She denied nasal trauma, insufflation, or insertion of foreign bodies into the nasal cavity. When the patient’s mother was contacted, she stated her daughter’s symptoms, which she believed were secondary to a domestic-violence-related injury, had been present and evolving over the past 2 weeks. She also related that the patient had been treated at another ED 4 days earlier and discharged with oral antibiotics.
On physical examination, the bilateral nares were entirely occluded by soft-tissue swelling, with fluctuance on palpation. The area was erythematous, and there were pustules scattered throughout the local region (Figure 1). There was no evidence of spreading cellulitis. During the examination, the patient had a labile level of alertness that fluctuated between somnolence and agitation; however, she was arousable and had satisfactory airway guarding. Patient’s vital signs remained stable throughout evaluation and treatment in the ED. On physical examination, her pupils were equal bilaterally, extraocular movements were intact, and no neurological deficits were detected. A complete blood cell count showed leukocytosis, with a white blood cell count of 18,240/uL and a predominance (88.2%) of neutrophils. All other laboratory values were within normal limits.
Computed tomography (CT) of the face revealed prominent soft-tissue swelling involving the inferior portion of the nose (Figure 2). In addition to swelling and obstruction of the bilateral nares, heterogeneity was also noted within the affected tissues and thought to represent a fluid component.
After the procedure, the patient was admitted to the hospital for observation on the medical psychiatric unit where she received additional IV antibiotic therapy as well as a psychiatric consultation. After a 24-hour observation period, she was discharged on a one-week regimen of oral clindamycin and instructions for outpatient follow-up with OMFS for septal repair. Cultures taken during exploration were positive for pan-sensitive Staphylococcus aureus. The working diagnosis at discharge was bilateral septal abscess from untreated bilateral septal hematoma due to an unreported facial trauma.
Discussion
Nasal septal abscess, a rare complication of a nasal septal hematoma, is defined as a collection of pus between the cartilaginous or bony nasal septum and its normally applied mucoperichondrium or mucoperiosteum. Patients most commonly present with fluctuant, tender, bilateral, or unilateral nasal obstruction as a result of anterior nasal septum swelling. Other symptoms include localized pain, swelling, fever, headache, or perinasal tenderness.1 The external portion of the nose is swollen, erythematous, and tender, and the anterior nasal cavities are occluded by a smooth, round, deep red or grey swelling.2 In a review of pediatric patients with nasal septal abscess, the most common complaint was nasal congestion (95%). Other significant complaints were nasal pain (50%), fever (50%), and headache (5%).3,4
Nasal septal abscess is most commonly caused by a hematoma. Although trauma is typically associated with this condition, it is not the sole cause. Other etiology includes nasal surgery, a furuncle of the nasal vestibule, sinusitis, or, in rare cases, infection from a dental extraction.3
Staphylococcus aureus is the most common pathogen. Streptococcus and other anaerobes are less common, and pediatric patients are more susceptible to Haemophilus influenza than adults. Although rare, Psuedomonas and Klebsiella have also been reported.3
When nasal septal abscess is suspected, prior to drainage, the diagnosis should be confirmed by CT of the face and include the paranasal sinuses. Computed tomography is an excellent imaging tool for abscess detection and is the community standard for evaluation. Magnetic resonance imaging is not usually utilized (especially in the acute or ED setting) as it is unlikely to affect or alter initial management. In radiographs, nasal septal abscess typically appears as fluid collection with thin rim enhancement in the cartilaginous nasal septum5 (Figure 2). These findings can be missed on brain CT alone.5
In patients presenting several days from a related trauma, distinguishing uncomplicated septal hematoma from nasal septal abscess can be very difficult—though nasal septal abscesses tend to be larger and more painful. In addition, there may be inflammation of the overlying mucosa, occasionally with exudates. In untreated cases, infection can extend into the cavernous sinus causing intracranial infections or cavernous sinus thrombosis. The most common complication of septal abscess is cartilage necrosis that can result in nasal structural collapse and “saddle-nose” deformity. Complications, including meningitis, can develop quickly (ie, within 3 to 4 days).6
The structural complications associated with septal abscess result from the avascular nature of the septal cartilage, which receives blood from the adherent mucoperichondrium. Hematoma and abscess can expand and obstruct the blood vessels that supply the nasal cartilage. Pressure of the hematoma on the septum causes progressive avascular necrosis.6
Patients with confirmed nasal septal abscess should obtain otolaryngology or OMFS consultation in the ED. Due to the high risk of complications and need for follow up, immediate drainage should also be directed by otolaryngology or OMFS. All patients should be discharged on oral broad-spectrum antibiotics, with a referral to an otolaryngologist or OMFS within 24 hours for evaluation and possible removal of nasal packs.7
Dr Yusuf is an academic chief resident, John Peter Smith Emergency Medicine Residency Program, Fort Worth, Texas. Dr Kirk is associate residency director and ultrasound director, department of emergency medicine, John Peter Smith Health System, Fort Worth, Texas.
- Huang PH, Chiang YC, Yang TH, Chao PZ, Lee FP. Nasal septal abscess. Otolaryngol Head Neck Surg. 2006;135(2):335,336.
- Shapiro RS. Nasal septal abscess. Can Med Assoc J. 1978;119(11):1321-1323.
- Lo SH, Wang PC. Nasal septal abscess as a complication of laser inferior turbinectomy. Chang Gung Med J. 2004;27(5):390-393.
- Canty PA, Berkowitz RG. Hematoma and abscess of the nasal septum in children. Arch Otolaryngol Head Neck Surg. 1996;122(12):1373-1376.
- Debnam JM, Gillenwater AM, Ginsberg LE. Nasal septal abscess in patients with immunosuppression. Am J Neuroradiol. 2007;28(10):1878,1879.
- Friedman M, Landsberg R, Chiampas G. Nasal septal hematoma evacuation. In: Reichman EF, Simon RR, eds. Emergency Medicine Procedures. New York, NY: McGraw-Hill; 2004. http://www.accessemergencymedicine.com/content.aspx?aID=45644. Accessed March 20, 2014.
- Summers SM, Bey T. Epistaxis, nasal fractures, and rhinosinusitis. In: Tintinalli JE, Stapczynski JS, Ma OJ, Cline DM, Cydulka RK, Meckler GD, eds. Tintinalli’s Emergency Medicine: A Comprehensive Study Guide. 7th ed. New York, NY: McGraw-Hill; 2011. http://www.accessemergencymedicine.com/content.aspx?aID=6388080. Accessed March 20, 2014.
Case
A 28-year-old woman with history of bipolar disorder and methamphetamine abuse presented to the ED complaining of nasal swelling and pain. She was unable to provide any medical history regarding the onset of her symptoms or other details, which the ED team attributed to her underlying psychiatric disorder. She denied nasal trauma, insufflation, or insertion of foreign bodies into the nasal cavity. When the patient’s mother was contacted, she stated her daughter’s symptoms, which she believed were secondary to a domestic-violence-related injury, had been present and evolving over the past 2 weeks. She also related that the patient had been treated at another ED 4 days earlier and discharged with oral antibiotics.
On physical examination, the bilateral nares were entirely occluded by soft-tissue swelling, with fluctuance on palpation. The area was erythematous, and there were pustules scattered throughout the local region (Figure 1). There was no evidence of spreading cellulitis. During the examination, the patient had a labile level of alertness that fluctuated between somnolence and agitation; however, she was arousable and had satisfactory airway guarding. Patient’s vital signs remained stable throughout evaluation and treatment in the ED. On physical examination, her pupils were equal bilaterally, extraocular movements were intact, and no neurological deficits were detected. A complete blood cell count showed leukocytosis, with a white blood cell count of 18,240/uL and a predominance (88.2%) of neutrophils. All other laboratory values were within normal limits.
Computed tomography (CT) of the face revealed prominent soft-tissue swelling involving the inferior portion of the nose (Figure 2). In addition to swelling and obstruction of the bilateral nares, heterogeneity was also noted within the affected tissues and thought to represent a fluid component.
After the procedure, the patient was admitted to the hospital for observation on the medical psychiatric unit where she received additional IV antibiotic therapy as well as a psychiatric consultation. After a 24-hour observation period, she was discharged on a one-week regimen of oral clindamycin and instructions for outpatient follow-up with OMFS for septal repair. Cultures taken during exploration were positive for pan-sensitive Staphylococcus aureus. The working diagnosis at discharge was bilateral septal abscess from untreated bilateral septal hematoma due to an unreported facial trauma.
Discussion
Nasal septal abscess, a rare complication of a nasal septal hematoma, is defined as a collection of pus between the cartilaginous or bony nasal septum and its normally applied mucoperichondrium or mucoperiosteum. Patients most commonly present with fluctuant, tender, bilateral, or unilateral nasal obstruction as a result of anterior nasal septum swelling. Other symptoms include localized pain, swelling, fever, headache, or perinasal tenderness.1 The external portion of the nose is swollen, erythematous, and tender, and the anterior nasal cavities are occluded by a smooth, round, deep red or grey swelling.2 In a review of pediatric patients with nasal septal abscess, the most common complaint was nasal congestion (95%). Other significant complaints were nasal pain (50%), fever (50%), and headache (5%).3,4
Nasal septal abscess is most commonly caused by a hematoma. Although trauma is typically associated with this condition, it is not the sole cause. Other etiology includes nasal surgery, a furuncle of the nasal vestibule, sinusitis, or, in rare cases, infection from a dental extraction.3
Staphylococcus aureus is the most common pathogen. Streptococcus and other anaerobes are less common, and pediatric patients are more susceptible to Haemophilus influenza than adults. Although rare, Psuedomonas and Klebsiella have also been reported.3
When nasal septal abscess is suspected, prior to drainage, the diagnosis should be confirmed by CT of the face and include the paranasal sinuses. Computed tomography is an excellent imaging tool for abscess detection and is the community standard for evaluation. Magnetic resonance imaging is not usually utilized (especially in the acute or ED setting) as it is unlikely to affect or alter initial management. In radiographs, nasal septal abscess typically appears as fluid collection with thin rim enhancement in the cartilaginous nasal septum5 (Figure 2). These findings can be missed on brain CT alone.5
In patients presenting several days from a related trauma, distinguishing uncomplicated septal hematoma from nasal septal abscess can be very difficult—though nasal septal abscesses tend to be larger and more painful. In addition, there may be inflammation of the overlying mucosa, occasionally with exudates. In untreated cases, infection can extend into the cavernous sinus causing intracranial infections or cavernous sinus thrombosis. The most common complication of septal abscess is cartilage necrosis that can result in nasal structural collapse and “saddle-nose” deformity. Complications, including meningitis, can develop quickly (ie, within 3 to 4 days).6
The structural complications associated with septal abscess result from the avascular nature of the septal cartilage, which receives blood from the adherent mucoperichondrium. Hematoma and abscess can expand and obstruct the blood vessels that supply the nasal cartilage. Pressure of the hematoma on the septum causes progressive avascular necrosis.6
Patients with confirmed nasal septal abscess should obtain otolaryngology or OMFS consultation in the ED. Due to the high risk of complications and need for follow up, immediate drainage should also be directed by otolaryngology or OMFS. All patients should be discharged on oral broad-spectrum antibiotics, with a referral to an otolaryngologist or OMFS within 24 hours for evaluation and possible removal of nasal packs.7
Dr Yusuf is an academic chief resident, John Peter Smith Emergency Medicine Residency Program, Fort Worth, Texas. Dr Kirk is associate residency director and ultrasound director, department of emergency medicine, John Peter Smith Health System, Fort Worth, Texas.
Case
A 28-year-old woman with history of bipolar disorder and methamphetamine abuse presented to the ED complaining of nasal swelling and pain. She was unable to provide any medical history regarding the onset of her symptoms or other details, which the ED team attributed to her underlying psychiatric disorder. She denied nasal trauma, insufflation, or insertion of foreign bodies into the nasal cavity. When the patient’s mother was contacted, she stated her daughter’s symptoms, which she believed were secondary to a domestic-violence-related injury, had been present and evolving over the past 2 weeks. She also related that the patient had been treated at another ED 4 days earlier and discharged with oral antibiotics.
On physical examination, the bilateral nares were entirely occluded by soft-tissue swelling, with fluctuance on palpation. The area was erythematous, and there were pustules scattered throughout the local region (Figure 1). There was no evidence of spreading cellulitis. During the examination, the patient had a labile level of alertness that fluctuated between somnolence and agitation; however, she was arousable and had satisfactory airway guarding. Patient’s vital signs remained stable throughout evaluation and treatment in the ED. On physical examination, her pupils were equal bilaterally, extraocular movements were intact, and no neurological deficits were detected. A complete blood cell count showed leukocytosis, with a white blood cell count of 18,240/uL and a predominance (88.2%) of neutrophils. All other laboratory values were within normal limits.
Computed tomography (CT) of the face revealed prominent soft-tissue swelling involving the inferior portion of the nose (Figure 2). In addition to swelling and obstruction of the bilateral nares, heterogeneity was also noted within the affected tissues and thought to represent a fluid component.
After the procedure, the patient was admitted to the hospital for observation on the medical psychiatric unit where she received additional IV antibiotic therapy as well as a psychiatric consultation. After a 24-hour observation period, she was discharged on a one-week regimen of oral clindamycin and instructions for outpatient follow-up with OMFS for septal repair. Cultures taken during exploration were positive for pan-sensitive Staphylococcus aureus. The working diagnosis at discharge was bilateral septal abscess from untreated bilateral septal hematoma due to an unreported facial trauma.
Discussion
Nasal septal abscess, a rare complication of a nasal septal hematoma, is defined as a collection of pus between the cartilaginous or bony nasal septum and its normally applied mucoperichondrium or mucoperiosteum. Patients most commonly present with fluctuant, tender, bilateral, or unilateral nasal obstruction as a result of anterior nasal septum swelling. Other symptoms include localized pain, swelling, fever, headache, or perinasal tenderness.1 The external portion of the nose is swollen, erythematous, and tender, and the anterior nasal cavities are occluded by a smooth, round, deep red or grey swelling.2 In a review of pediatric patients with nasal septal abscess, the most common complaint was nasal congestion (95%). Other significant complaints were nasal pain (50%), fever (50%), and headache (5%).3,4
Nasal septal abscess is most commonly caused by a hematoma. Although trauma is typically associated with this condition, it is not the sole cause. Other etiology includes nasal surgery, a furuncle of the nasal vestibule, sinusitis, or, in rare cases, infection from a dental extraction.3
Staphylococcus aureus is the most common pathogen. Streptococcus and other anaerobes are less common, and pediatric patients are more susceptible to Haemophilus influenza than adults. Although rare, Psuedomonas and Klebsiella have also been reported.3
When nasal septal abscess is suspected, prior to drainage, the diagnosis should be confirmed by CT of the face and include the paranasal sinuses. Computed tomography is an excellent imaging tool for abscess detection and is the community standard for evaluation. Magnetic resonance imaging is not usually utilized (especially in the acute or ED setting) as it is unlikely to affect or alter initial management. In radiographs, nasal septal abscess typically appears as fluid collection with thin rim enhancement in the cartilaginous nasal septum5 (Figure 2). These findings can be missed on brain CT alone.5
In patients presenting several days from a related trauma, distinguishing uncomplicated septal hematoma from nasal septal abscess can be very difficult—though nasal septal abscesses tend to be larger and more painful. In addition, there may be inflammation of the overlying mucosa, occasionally with exudates. In untreated cases, infection can extend into the cavernous sinus causing intracranial infections or cavernous sinus thrombosis. The most common complication of septal abscess is cartilage necrosis that can result in nasal structural collapse and “saddle-nose” deformity. Complications, including meningitis, can develop quickly (ie, within 3 to 4 days).6
The structural complications associated with septal abscess result from the avascular nature of the septal cartilage, which receives blood from the adherent mucoperichondrium. Hematoma and abscess can expand and obstruct the blood vessels that supply the nasal cartilage. Pressure of the hematoma on the septum causes progressive avascular necrosis.6
Patients with confirmed nasal septal abscess should obtain otolaryngology or OMFS consultation in the ED. Due to the high risk of complications and need for follow up, immediate drainage should also be directed by otolaryngology or OMFS. All patients should be discharged on oral broad-spectrum antibiotics, with a referral to an otolaryngologist or OMFS within 24 hours for evaluation and possible removal of nasal packs.7
Dr Yusuf is an academic chief resident, John Peter Smith Emergency Medicine Residency Program, Fort Worth, Texas. Dr Kirk is associate residency director and ultrasound director, department of emergency medicine, John Peter Smith Health System, Fort Worth, Texas.
- Huang PH, Chiang YC, Yang TH, Chao PZ, Lee FP. Nasal septal abscess. Otolaryngol Head Neck Surg. 2006;135(2):335,336.
- Shapiro RS. Nasal septal abscess. Can Med Assoc J. 1978;119(11):1321-1323.
- Lo SH, Wang PC. Nasal septal abscess as a complication of laser inferior turbinectomy. Chang Gung Med J. 2004;27(5):390-393.
- Canty PA, Berkowitz RG. Hematoma and abscess of the nasal septum in children. Arch Otolaryngol Head Neck Surg. 1996;122(12):1373-1376.
- Debnam JM, Gillenwater AM, Ginsberg LE. Nasal septal abscess in patients with immunosuppression. Am J Neuroradiol. 2007;28(10):1878,1879.
- Friedman M, Landsberg R, Chiampas G. Nasal septal hematoma evacuation. In: Reichman EF, Simon RR, eds. Emergency Medicine Procedures. New York, NY: McGraw-Hill; 2004. http://www.accessemergencymedicine.com/content.aspx?aID=45644. Accessed March 20, 2014.
- Summers SM, Bey T. Epistaxis, nasal fractures, and rhinosinusitis. In: Tintinalli JE, Stapczynski JS, Ma OJ, Cline DM, Cydulka RK, Meckler GD, eds. Tintinalli’s Emergency Medicine: A Comprehensive Study Guide. 7th ed. New York, NY: McGraw-Hill; 2011. http://www.accessemergencymedicine.com/content.aspx?aID=6388080. Accessed March 20, 2014.
- Huang PH, Chiang YC, Yang TH, Chao PZ, Lee FP. Nasal septal abscess. Otolaryngol Head Neck Surg. 2006;135(2):335,336.
- Shapiro RS. Nasal septal abscess. Can Med Assoc J. 1978;119(11):1321-1323.
- Lo SH, Wang PC. Nasal septal abscess as a complication of laser inferior turbinectomy. Chang Gung Med J. 2004;27(5):390-393.
- Canty PA, Berkowitz RG. Hematoma and abscess of the nasal septum in children. Arch Otolaryngol Head Neck Surg. 1996;122(12):1373-1376.
- Debnam JM, Gillenwater AM, Ginsberg LE. Nasal septal abscess in patients with immunosuppression. Am J Neuroradiol. 2007;28(10):1878,1879.
- Friedman M, Landsberg R, Chiampas G. Nasal septal hematoma evacuation. In: Reichman EF, Simon RR, eds. Emergency Medicine Procedures. New York, NY: McGraw-Hill; 2004. http://www.accessemergencymedicine.com/content.aspx?aID=45644. Accessed March 20, 2014.
- Summers SM, Bey T. Epistaxis, nasal fractures, and rhinosinusitis. In: Tintinalli JE, Stapczynski JS, Ma OJ, Cline DM, Cydulka RK, Meckler GD, eds. Tintinalli’s Emergency Medicine: A Comprehensive Study Guide. 7th ed. New York, NY: McGraw-Hill; 2011. http://www.accessemergencymedicine.com/content.aspx?aID=6388080. Accessed March 20, 2014.

Poland Syndrome: A Congenital Abnormality Mimicking a Traumatic Injury
Case
A 12-year-old boy presented to the ED via emergency medical services after he was struck by motor vehicle while skateboarding without a helmet or other safety equipment. He was thrown approximately 10 feet, but experienced no loss of consciousness, pain, or active bleeding at the site of the accident. Unaccompanied by family, he arrived to the ED fully immobilized on a long back board. His field vital signs were stable: blood pressure (BP), 100/65 mm Hg; heart rate (HR) 105 beats/minute; respiratory rate (RR), 22 breaths/minute; temperature, afebrile. Oxygen saturation was 100% on room air. The patient had an estimated Glasgow Coma Scale (GCS) of 14, with one point removed due to confusion.
Primary examination showed an intact airway with equal breath sounds bilaterally, and pulses were equal in all extremities with audible heart sounds. The patient was able to move all extremities, and showed no obvious deformities or bleeding. He was neurologically intact, with equal strength and sensation. He did, however, elicit some confusion during the examination, continuously stating it was “all his fault” and asking the medical staff where he was. This confusion persisted even after repeated reorientation. His vital signs remained stable, with slight tachycardia (BP, 105/67 mm hg; HR 100 beats/minute; RR, 17 breaths/minute; temperature, afebrile; pulse oxygen saturation, 99%). An abbreviated history revealed no allergies, medications, or past medical history. When questioned, the patient had no recollection of the accident or the last time he had eaten.
A secondary survey was significant for a small contusion/abrasion on the patient’s forehead but an otherwise normal head, ear, eyes, nose, and throat examination and no cervical c-spine tenderness. The patient denied any chest wall tenderness, but there was a dramatic palpable defect in the right chest wall, with profound asymmetry when compared to the left chest wall. No sharp, bony edges could be palpated, nor could any crepitance be felt. Breath sounds were reexamined and remained equal and nonlabored, and the patient continued to have a stable oxygen saturation of 99% on room air. The rest of the secondary survey was negative, and c-spine, pelvic, and portable chest X-rays were all negative for acute findings.

Due to the physical examination findings on the chest wall, a computed tomography (CT) scan of the chest was performed with contrast (Figure). The chest CT was normal, except for a lack of musculature over the right anterior chest wall. The patient’s mother arrived shortly after imaging studies, at which time he was reexamined. When interviewing his mother for further history, she stated that her son had been diagnosed with mild Poland Syndrome as a child, and that he has always had a chest deformity. All other studies, including a noncontrast CT of the brain, were normal. The child quickly improved during his 6-hour observation in the ED, and he was subsequently discharged home with the diagnosis of a concussion.
Discussion
Poland syndrome, also known as hand and ipsilateral thorax syndrome, is a rare congenital disorder with unknown etiology.1,2 The condition was first officially described in 1841 by Alfred Poland at Guy’s Hospital in London, though reports exist as early as 1826. Poland, a medical student, made the discovery while examining the cadaver of a hanged convict.
The occurrence of Poland syndrome is estimated to be from 1 in 25,000 to 1 in 75,000 to 100,000 by some reports,1-4 with a higher incidence in males than females (3:1 ratio) and 75% right-sided dominance.2 The syndrome is primarily described as unilateral, but there is one case report of suspected bilateral involvement.1 The components of the syndrome consist of aplasia of the sternal head of the pectoralis major muscle, hypoplasia of the pectoralis minor muscle, decreased development of breast and subcutaneous tissue, and a variety of ipsilateral hand abnormalities, including shortened carpels and phalanges, and syndactyly. The syndrome is quite variable, with different individuals eliciting combinations of the above components.
Poland syndrome was initially believed to be a nonfamilial disorder due to its sporadic nature, as illustrated by a case report of an isolated affected identical twin.3 However, enough cases of familial involvement have been reported that there is a proposed theory of an inheritable trait. Although over 250 patients with this syndrome have been described, there is no clear cause.2 The current theory of etiology is felt to be due to a lack of blood flow in the subclavian artery, or one of its branches, early in the development of the fetus, around the end of the sixth week of development. Individuals can have mild to severe manifestations, ranging as mild (eg, only pectoralis involvement), to severe (eg, rib hypoplasia, complete absence of ipsilateral hand, dextrocardia, lung herniation). Case reports of high functioning athletes with the disorder show that there is not necessarily functional impairment.
In addition to Poland syndrome, there are a number of congenital abnormalities that can also mimic traumatic chest injuries. Historically, surgeons have classified congenital wall deformities into one of five categories: Poland syndrome, pectus excavatum, pectus carinatum, sternal clefts, and generic skeletal and cartilage dysplasias (eg, absent ribs, rib torsion, vertebral anomalies).5-7 Of these categories, Poland syndrome, pectus excavatum, and some skeletal dysplasias cause anterior chest wall depression.5,6 Although these are examples of congenital thoracic wall abnormalities, one must also remember postoperative changes, which may also appear to be traumatic in origin. Examples of specific procedures are lumpectomy, mastectomy, rib resection, lung resection, or even cardiac surgery—all of which can alter the physical findings of the chest wall.
Conclusion
This report is an interesting case of an impaired patient presenting to the ED after a traumatic incident and unable to describe a past medical history of a congenital disorder. Although the patient was high functioning, as exemplified by his ability to complete normal adolescent activities such as skateboarding, he had a significant physical finding which appeared to correspond to the mechanism of his injury. He was initially thought to have a significant injury involving his chest wall, since secondary examination revealed a palpable defect. Although the patient was oxygenating well, and in no apparent distress, his altered mental status raised concerns about the accuracy of his report, with confusion and perseveration.
When a rare congenital abnormality imitates a traumatic condition, merely having the name of the condition—as we did when the family arrived—does not necessarily rule out the absence of a related deficit or injury. To better differentiate acute from preexisting physical deformities or deficits, one must gather and process multiple diagnostic clues. This is best accomplished by combining the presence or absence of symptoms (in this case, pain, dyspnea, or hemoptysis), physical examination findings (eg, ecchymosis, crepitance, flail segment), and supportive diagnostic tests (radiographs, CT, and echocardiograms). This approach will systematically eliminate or suggest acute traumatic diagnoses. With specific traumatic causes such as rib fracture, pneumothorax, or pulmonary contusion eliminated, one can expand the (nontraumatic) differential, keeping in mind the possibility of a congenital disorder.
Dr Martin is an emergency physician at Emergency Medical Associates of NY and NJ; and emergency medicine education director, Monmouth Medical Center, Long Branch, NJ.
Dr Martin reports no conflict of interest or financial arrangements.
- Fokin AA, Robicsek F. Poland syndrome revisited. Ann Thorac Surg. 2002;74(6):2218-2225
- Darian VB, Argenta LC, Pasyk KA. Familial Poland’s syndrome. Ann Plast Surg. 1989;23(6):531-537
- Stevens D, Fink B, Prevel C. Poland’s syndrome in one identical twin. J Pediatr Orthop. 2000;20(3):392-395.
- McGrath MH, Pomerantz J. Plastic surgery. In: Townsend CM Jr, Beauchamp RD, Evers BM, Mattox KL, eds. Sabiston Textbook of Surgery: The Biological Basis of Modern Surgical Practice. 19th ed. Philadelphia, PA: Elsevier Saunders; 2012:1935
- Spear SL, Pelletiere CV, Lee ES, Grotting JC. Anterior thoracic hypoplasia: a separate entity from Poland syndrome. Plast Reconstr Surg. 2004;113(1):
- Hodgkinson, DJ. Chest wall implants: their use for pectus excavatum, pectoralis muscle tears, Poland’s syndrome, and muscular insufficiency. Aesthetic Plast Surg. 1997;21(1):7-15.
- Hodgkinson, DJ. The management of anterior chest wall deformity in patients presenting for breast augmentation. Plast Reconstr Surg. 2002;109(5): 1714-1723.
Case
A 12-year-old boy presented to the ED via emergency medical services after he was struck by motor vehicle while skateboarding without a helmet or other safety equipment. He was thrown approximately 10 feet, but experienced no loss of consciousness, pain, or active bleeding at the site of the accident. Unaccompanied by family, he arrived to the ED fully immobilized on a long back board. His field vital signs were stable: blood pressure (BP), 100/65 mm Hg; heart rate (HR) 105 beats/minute; respiratory rate (RR), 22 breaths/minute; temperature, afebrile. Oxygen saturation was 100% on room air. The patient had an estimated Glasgow Coma Scale (GCS) of 14, with one point removed due to confusion.
Primary examination showed an intact airway with equal breath sounds bilaterally, and pulses were equal in all extremities with audible heart sounds. The patient was able to move all extremities, and showed no obvious deformities or bleeding. He was neurologically intact, with equal strength and sensation. He did, however, elicit some confusion during the examination, continuously stating it was “all his fault” and asking the medical staff where he was. This confusion persisted even after repeated reorientation. His vital signs remained stable, with slight tachycardia (BP, 105/67 mm hg; HR 100 beats/minute; RR, 17 breaths/minute; temperature, afebrile; pulse oxygen saturation, 99%). An abbreviated history revealed no allergies, medications, or past medical history. When questioned, the patient had no recollection of the accident or the last time he had eaten.
A secondary survey was significant for a small contusion/abrasion on the patient’s forehead but an otherwise normal head, ear, eyes, nose, and throat examination and no cervical c-spine tenderness. The patient denied any chest wall tenderness, but there was a dramatic palpable defect in the right chest wall, with profound asymmetry when compared to the left chest wall. No sharp, bony edges could be palpated, nor could any crepitance be felt. Breath sounds were reexamined and remained equal and nonlabored, and the patient continued to have a stable oxygen saturation of 99% on room air. The rest of the secondary survey was negative, and c-spine, pelvic, and portable chest X-rays were all negative for acute findings.
Due to the physical examination findings on the chest wall, a computed tomography (CT) scan of the chest was performed with contrast (Figure). The chest CT was normal, except for a lack of musculature over the right anterior chest wall. The patient’s mother arrived shortly after imaging studies, at which time he was reexamined. When interviewing his mother for further history, she stated that her son had been diagnosed with mild Poland Syndrome as a child, and that he has always had a chest deformity. All other studies, including a noncontrast CT of the brain, were normal. The child quickly improved during his 6-hour observation in the ED, and he was subsequently discharged home with the diagnosis of a concussion.
Discussion
Poland syndrome, also known as hand and ipsilateral thorax syndrome, is a rare congenital disorder with unknown etiology.1,2 The condition was first officially described in 1841 by Alfred Poland at Guy’s Hospital in London, though reports exist as early as 1826. Poland, a medical student, made the discovery while examining the cadaver of a hanged convict.
The occurrence of Poland syndrome is estimated to be from 1 in 25,000 to 1 in 75,000 to 100,000 by some reports,1-4 with a higher incidence in males than females (3:1 ratio) and 75% right-sided dominance.2 The syndrome is primarily described as unilateral, but there is one case report of suspected bilateral involvement.1 The components of the syndrome consist of aplasia of the sternal head of the pectoralis major muscle, hypoplasia of the pectoralis minor muscle, decreased development of breast and subcutaneous tissue, and a variety of ipsilateral hand abnormalities, including shortened carpels and phalanges, and syndactyly. The syndrome is quite variable, with different individuals eliciting combinations of the above components.
Poland syndrome was initially believed to be a nonfamilial disorder due to its sporadic nature, as illustrated by a case report of an isolated affected identical twin.3 However, enough cases of familial involvement have been reported that there is a proposed theory of an inheritable trait. Although over 250 patients with this syndrome have been described, there is no clear cause.2 The current theory of etiology is felt to be due to a lack of blood flow in the subclavian artery, or one of its branches, early in the development of the fetus, around the end of the sixth week of development. Individuals can have mild to severe manifestations, ranging as mild (eg, only pectoralis involvement), to severe (eg, rib hypoplasia, complete absence of ipsilateral hand, dextrocardia, lung herniation). Case reports of high functioning athletes with the disorder show that there is not necessarily functional impairment.
In addition to Poland syndrome, there are a number of congenital abnormalities that can also mimic traumatic chest injuries. Historically, surgeons have classified congenital wall deformities into one of five categories: Poland syndrome, pectus excavatum, pectus carinatum, sternal clefts, and generic skeletal and cartilage dysplasias (eg, absent ribs, rib torsion, vertebral anomalies).5-7 Of these categories, Poland syndrome, pectus excavatum, and some skeletal dysplasias cause anterior chest wall depression.5,6 Although these are examples of congenital thoracic wall abnormalities, one must also remember postoperative changes, which may also appear to be traumatic in origin. Examples of specific procedures are lumpectomy, mastectomy, rib resection, lung resection, or even cardiac surgery—all of which can alter the physical findings of the chest wall.
Conclusion
This report is an interesting case of an impaired patient presenting to the ED after a traumatic incident and unable to describe a past medical history of a congenital disorder. Although the patient was high functioning, as exemplified by his ability to complete normal adolescent activities such as skateboarding, he had a significant physical finding which appeared to correspond to the mechanism of his injury. He was initially thought to have a significant injury involving his chest wall, since secondary examination revealed a palpable defect. Although the patient was oxygenating well, and in no apparent distress, his altered mental status raised concerns about the accuracy of his report, with confusion and perseveration.
When a rare congenital abnormality imitates a traumatic condition, merely having the name of the condition—as we did when the family arrived—does not necessarily rule out the absence of a related deficit or injury. To better differentiate acute from preexisting physical deformities or deficits, one must gather and process multiple diagnostic clues. This is best accomplished by combining the presence or absence of symptoms (in this case, pain, dyspnea, or hemoptysis), physical examination findings (eg, ecchymosis, crepitance, flail segment), and supportive diagnostic tests (radiographs, CT, and echocardiograms). This approach will systematically eliminate or suggest acute traumatic diagnoses. With specific traumatic causes such as rib fracture, pneumothorax, or pulmonary contusion eliminated, one can expand the (nontraumatic) differential, keeping in mind the possibility of a congenital disorder.
Dr Martin is an emergency physician at Emergency Medical Associates of NY and NJ; and emergency medicine education director, Monmouth Medical Center, Long Branch, NJ.
Dr Martin reports no conflict of interest or financial arrangements.
Case
A 12-year-old boy presented to the ED via emergency medical services after he was struck by motor vehicle while skateboarding without a helmet or other safety equipment. He was thrown approximately 10 feet, but experienced no loss of consciousness, pain, or active bleeding at the site of the accident. Unaccompanied by family, he arrived to the ED fully immobilized on a long back board. His field vital signs were stable: blood pressure (BP), 100/65 mm Hg; heart rate (HR) 105 beats/minute; respiratory rate (RR), 22 breaths/minute; temperature, afebrile. Oxygen saturation was 100% on room air. The patient had an estimated Glasgow Coma Scale (GCS) of 14, with one point removed due to confusion.
Primary examination showed an intact airway with equal breath sounds bilaterally, and pulses were equal in all extremities with audible heart sounds. The patient was able to move all extremities, and showed no obvious deformities or bleeding. He was neurologically intact, with equal strength and sensation. He did, however, elicit some confusion during the examination, continuously stating it was “all his fault” and asking the medical staff where he was. This confusion persisted even after repeated reorientation. His vital signs remained stable, with slight tachycardia (BP, 105/67 mm hg; HR 100 beats/minute; RR, 17 breaths/minute; temperature, afebrile; pulse oxygen saturation, 99%). An abbreviated history revealed no allergies, medications, or past medical history. When questioned, the patient had no recollection of the accident or the last time he had eaten.
A secondary survey was significant for a small contusion/abrasion on the patient’s forehead but an otherwise normal head, ear, eyes, nose, and throat examination and no cervical c-spine tenderness. The patient denied any chest wall tenderness, but there was a dramatic palpable defect in the right chest wall, with profound asymmetry when compared to the left chest wall. No sharp, bony edges could be palpated, nor could any crepitance be felt. Breath sounds were reexamined and remained equal and nonlabored, and the patient continued to have a stable oxygen saturation of 99% on room air. The rest of the secondary survey was negative, and c-spine, pelvic, and portable chest X-rays were all negative for acute findings.
Due to the physical examination findings on the chest wall, a computed tomography (CT) scan of the chest was performed with contrast (Figure). The chest CT was normal, except for a lack of musculature over the right anterior chest wall. The patient’s mother arrived shortly after imaging studies, at which time he was reexamined. When interviewing his mother for further history, she stated that her son had been diagnosed with mild Poland Syndrome as a child, and that he has always had a chest deformity. All other studies, including a noncontrast CT of the brain, were normal. The child quickly improved during his 6-hour observation in the ED, and he was subsequently discharged home with the diagnosis of a concussion.
Discussion
Poland syndrome, also known as hand and ipsilateral thorax syndrome, is a rare congenital disorder with unknown etiology.1,2 The condition was first officially described in 1841 by Alfred Poland at Guy’s Hospital in London, though reports exist as early as 1826. Poland, a medical student, made the discovery while examining the cadaver of a hanged convict.
The occurrence of Poland syndrome is estimated to be from 1 in 25,000 to 1 in 75,000 to 100,000 by some reports,1-4 with a higher incidence in males than females (3:1 ratio) and 75% right-sided dominance.2 The syndrome is primarily described as unilateral, but there is one case report of suspected bilateral involvement.1 The components of the syndrome consist of aplasia of the sternal head of the pectoralis major muscle, hypoplasia of the pectoralis minor muscle, decreased development of breast and subcutaneous tissue, and a variety of ipsilateral hand abnormalities, including shortened carpels and phalanges, and syndactyly. The syndrome is quite variable, with different individuals eliciting combinations of the above components.
Poland syndrome was initially believed to be a nonfamilial disorder due to its sporadic nature, as illustrated by a case report of an isolated affected identical twin.3 However, enough cases of familial involvement have been reported that there is a proposed theory of an inheritable trait. Although over 250 patients with this syndrome have been described, there is no clear cause.2 The current theory of etiology is felt to be due to a lack of blood flow in the subclavian artery, or one of its branches, early in the development of the fetus, around the end of the sixth week of development. Individuals can have mild to severe manifestations, ranging as mild (eg, only pectoralis involvement), to severe (eg, rib hypoplasia, complete absence of ipsilateral hand, dextrocardia, lung herniation). Case reports of high functioning athletes with the disorder show that there is not necessarily functional impairment.
In addition to Poland syndrome, there are a number of congenital abnormalities that can also mimic traumatic chest injuries. Historically, surgeons have classified congenital wall deformities into one of five categories: Poland syndrome, pectus excavatum, pectus carinatum, sternal clefts, and generic skeletal and cartilage dysplasias (eg, absent ribs, rib torsion, vertebral anomalies).5-7 Of these categories, Poland syndrome, pectus excavatum, and some skeletal dysplasias cause anterior chest wall depression.5,6 Although these are examples of congenital thoracic wall abnormalities, one must also remember postoperative changes, which may also appear to be traumatic in origin. Examples of specific procedures are lumpectomy, mastectomy, rib resection, lung resection, or even cardiac surgery—all of which can alter the physical findings of the chest wall.
Conclusion
This report is an interesting case of an impaired patient presenting to the ED after a traumatic incident and unable to describe a past medical history of a congenital disorder. Although the patient was high functioning, as exemplified by his ability to complete normal adolescent activities such as skateboarding, he had a significant physical finding which appeared to correspond to the mechanism of his injury. He was initially thought to have a significant injury involving his chest wall, since secondary examination revealed a palpable defect. Although the patient was oxygenating well, and in no apparent distress, his altered mental status raised concerns about the accuracy of his report, with confusion and perseveration.
When a rare congenital abnormality imitates a traumatic condition, merely having the name of the condition—as we did when the family arrived—does not necessarily rule out the absence of a related deficit or injury. To better differentiate acute from preexisting physical deformities or deficits, one must gather and process multiple diagnostic clues. This is best accomplished by combining the presence or absence of symptoms (in this case, pain, dyspnea, or hemoptysis), physical examination findings (eg, ecchymosis, crepitance, flail segment), and supportive diagnostic tests (radiographs, CT, and echocardiograms). This approach will systematically eliminate or suggest acute traumatic diagnoses. With specific traumatic causes such as rib fracture, pneumothorax, or pulmonary contusion eliminated, one can expand the (nontraumatic) differential, keeping in mind the possibility of a congenital disorder.
Dr Martin is an emergency physician at Emergency Medical Associates of NY and NJ; and emergency medicine education director, Monmouth Medical Center, Long Branch, NJ.
Dr Martin reports no conflict of interest or financial arrangements.
- Fokin AA, Robicsek F. Poland syndrome revisited. Ann Thorac Surg. 2002;74(6):2218-2225
- Darian VB, Argenta LC, Pasyk KA. Familial Poland’s syndrome. Ann Plast Surg. 1989;23(6):531-537
- Stevens D, Fink B, Prevel C. Poland’s syndrome in one identical twin. J Pediatr Orthop. 2000;20(3):392-395.
- McGrath MH, Pomerantz J. Plastic surgery. In: Townsend CM Jr, Beauchamp RD, Evers BM, Mattox KL, eds. Sabiston Textbook of Surgery: The Biological Basis of Modern Surgical Practice. 19th ed. Philadelphia, PA: Elsevier Saunders; 2012:1935
- Spear SL, Pelletiere CV, Lee ES, Grotting JC. Anterior thoracic hypoplasia: a separate entity from Poland syndrome. Plast Reconstr Surg. 2004;113(1):
- Hodgkinson, DJ. Chest wall implants: their use for pectus excavatum, pectoralis muscle tears, Poland’s syndrome, and muscular insufficiency. Aesthetic Plast Surg. 1997;21(1):7-15.
- Hodgkinson, DJ. The management of anterior chest wall deformity in patients presenting for breast augmentation. Plast Reconstr Surg. 2002;109(5): 1714-1723.
- Fokin AA, Robicsek F. Poland syndrome revisited. Ann Thorac Surg. 2002;74(6):2218-2225
- Darian VB, Argenta LC, Pasyk KA. Familial Poland’s syndrome. Ann Plast Surg. 1989;23(6):531-537
- Stevens D, Fink B, Prevel C. Poland’s syndrome in one identical twin. J Pediatr Orthop. 2000;20(3):392-395.
- McGrath MH, Pomerantz J. Plastic surgery. In: Townsend CM Jr, Beauchamp RD, Evers BM, Mattox KL, eds. Sabiston Textbook of Surgery: The Biological Basis of Modern Surgical Practice. 19th ed. Philadelphia, PA: Elsevier Saunders; 2012:1935
- Spear SL, Pelletiere CV, Lee ES, Grotting JC. Anterior thoracic hypoplasia: a separate entity from Poland syndrome. Plast Reconstr Surg. 2004;113(1):
- Hodgkinson, DJ. Chest wall implants: their use for pectus excavatum, pectoralis muscle tears, Poland’s syndrome, and muscular insufficiency. Aesthetic Plast Surg. 1997;21(1):7-15.
- Hodgkinson, DJ. The management of anterior chest wall deformity in patients presenting for breast augmentation. Plast Reconstr Surg. 2002;109(5): 1714-1723.