User login
Canagliflozin
To the Editor: In a recent CCJM review of canagliflozin,1 this novel antihyperglycemic medication was noted to be associated with a dose-dependent increase in low-density lipoprotein (LDL) cholesterol, with an increase in LDL of 8.3 mg/dL (0.215 mmol/L) seen with the 300-mg/day dose of canagliflozin.
The Cholesterol Treatment Trialists’ (CTT) meta-analysis2 showed a significant 21% proportional reduction in major vascular events per 1.0 mmol/L reduction in LDL cholesterol in people with diabetes treated with statins over an average of 4.3 years. If we assume that raising LDL cholesterol by 1.0 mmol/L has the opposite effect, then patients taking 300 mg per day of canagliflozin would be expected to suffer an increase in major vascular events of about 4.5% over 4.3 years. Put another way, for every 22 diabetic patients treated with canagliflozin over 4.3 years, one additional major vascular event would be expected on the basis of the associated increase in LDL cholesterol.
The CTT data also showed a significant 9% decrease in all-cause mortality for every 1.0 mmol/L decrease in LDL cholesterol. Again, assuming that raising LDL has the opposite effect of lowering it, then we should expect an additional death for each 52 diabetic patients treated with 300 mg/day of canagliflozin per day for 4.3 years.
The hypotensive side effect of canagliflozin might tend to mitigate some of the above adverse effects, as might its antihyperglycemic effect. Still, it would seem prudent to use this novel agent only as a second- or third-line choice, particularly in diabetic patients who have already suffered a major vascular event.
- Vouyiouklis M. Canagliflozin: improving diabetes by making urine sweet. Cleve Clin J Med 2013; 80:683–687.
- Cholesterol Treatment Trialists’ (CTT) Collaborators; Kearney PM, Blackwell L, Collins R, et al. Efficacy of cholesterol-lowering therapy in 18,686 people with diabetes in 14 randomised trials of statins: a meta-analysis. Lancet 2008; 371:117–125.
To the Editor: In a recent CCJM review of canagliflozin,1 this novel antihyperglycemic medication was noted to be associated with a dose-dependent increase in low-density lipoprotein (LDL) cholesterol, with an increase in LDL of 8.3 mg/dL (0.215 mmol/L) seen with the 300-mg/day dose of canagliflozin.
The Cholesterol Treatment Trialists’ (CTT) meta-analysis2 showed a significant 21% proportional reduction in major vascular events per 1.0 mmol/L reduction in LDL cholesterol in people with diabetes treated with statins over an average of 4.3 years. If we assume that raising LDL cholesterol by 1.0 mmol/L has the opposite effect, then patients taking 300 mg per day of canagliflozin would be expected to suffer an increase in major vascular events of about 4.5% over 4.3 years. Put another way, for every 22 diabetic patients treated with canagliflozin over 4.3 years, one additional major vascular event would be expected on the basis of the associated increase in LDL cholesterol.
The CTT data also showed a significant 9% decrease in all-cause mortality for every 1.0 mmol/L decrease in LDL cholesterol. Again, assuming that raising LDL has the opposite effect of lowering it, then we should expect an additional death for each 52 diabetic patients treated with 300 mg/day of canagliflozin per day for 4.3 years.
The hypotensive side effect of canagliflozin might tend to mitigate some of the above adverse effects, as might its antihyperglycemic effect. Still, it would seem prudent to use this novel agent only as a second- or third-line choice, particularly in diabetic patients who have already suffered a major vascular event.
To the Editor: In a recent CCJM review of canagliflozin,1 this novel antihyperglycemic medication was noted to be associated with a dose-dependent increase in low-density lipoprotein (LDL) cholesterol, with an increase in LDL of 8.3 mg/dL (0.215 mmol/L) seen with the 300-mg/day dose of canagliflozin.
The Cholesterol Treatment Trialists’ (CTT) meta-analysis2 showed a significant 21% proportional reduction in major vascular events per 1.0 mmol/L reduction in LDL cholesterol in people with diabetes treated with statins over an average of 4.3 years. If we assume that raising LDL cholesterol by 1.0 mmol/L has the opposite effect, then patients taking 300 mg per day of canagliflozin would be expected to suffer an increase in major vascular events of about 4.5% over 4.3 years. Put another way, for every 22 diabetic patients treated with canagliflozin over 4.3 years, one additional major vascular event would be expected on the basis of the associated increase in LDL cholesterol.
The CTT data also showed a significant 9% decrease in all-cause mortality for every 1.0 mmol/L decrease in LDL cholesterol. Again, assuming that raising LDL has the opposite effect of lowering it, then we should expect an additional death for each 52 diabetic patients treated with 300 mg/day of canagliflozin per day for 4.3 years.
The hypotensive side effect of canagliflozin might tend to mitigate some of the above adverse effects, as might its antihyperglycemic effect. Still, it would seem prudent to use this novel agent only as a second- or third-line choice, particularly in diabetic patients who have already suffered a major vascular event.
- Vouyiouklis M. Canagliflozin: improving diabetes by making urine sweet. Cleve Clin J Med 2013; 80:683–687.
- Cholesterol Treatment Trialists’ (CTT) Collaborators; Kearney PM, Blackwell L, Collins R, et al. Efficacy of cholesterol-lowering therapy in 18,686 people with diabetes in 14 randomised trials of statins: a meta-analysis. Lancet 2008; 371:117–125.
- Vouyiouklis M. Canagliflozin: improving diabetes by making urine sweet. Cleve Clin J Med 2013; 80:683–687.
- Cholesterol Treatment Trialists’ (CTT) Collaborators; Kearney PM, Blackwell L, Collins R, et al. Efficacy of cholesterol-lowering therapy in 18,686 people with diabetes in 14 randomised trials of statins: a meta-analysis. Lancet 2008; 371:117–125.
In reply: Canagliflozin
In Reply: I would like to thank these readers very much for their response and comments.
Additional data provided from the study conducted by Lavalle-González et al evaluating the efficacy and safety of canagliflozin (100-mg and 300-mg doses) vs placebo and sitagliptin in patients with type 2 diabetes showed similar findings in weight and blood pressure reduction with slight LDL elevation with the studies mentioned in my article.1 At 52 weeks, as noted, canagliflozin 100 mg demonstrated noninferiority, and canagliflozin 300 mg showed a statistically significant superiority to sitagliptin in lowering hemoglobin A1c (a change of −0.73% with canagliflozin 100 mg, −0.88% with canagliflozin 300 mg, and −0.73% with sitagliptin), which may be considered in treatment decisions along with the other possible effects of this drug.1
The decision to use canagliflozin as second-or third-line therapy should be individualized after considering all of the patient’s risk factors as well as the potential benefit vs side effectsof this drug. Metformin remains my first-line choice in the management of type 2 diabetes. In my clinical practice, thus far, I have not used canagliflozin in patients with known coronary disease or a history of cardiovascular events. I have ensured that the LDL is certainly below goal before starting any patient on this drug, and I have followed the LDL closely, without hesitating to increase the statin drug to keep the LDL below goal. I agree that the slight increase of LDL is of concern, and certainly long-term studies are necessary to see whether there will be any increase in cardiovascular events from the use of canagliflozin.
- Lavalle-González FJ, Januszewicz A, Davidson J, et al. Efficacy and safety of canagliflozin compared with placebo and sitagliptin in patients with type 2 diabetes on background metformin monotherapy: a randomized trial. Diabetologia 2013; 56:2582–2592.
In Reply: I would like to thank these readers very much for their response and comments.
Additional data provided from the study conducted by Lavalle-González et al evaluating the efficacy and safety of canagliflozin (100-mg and 300-mg doses) vs placebo and sitagliptin in patients with type 2 diabetes showed similar findings in weight and blood pressure reduction with slight LDL elevation with the studies mentioned in my article.1 At 52 weeks, as noted, canagliflozin 100 mg demonstrated noninferiority, and canagliflozin 300 mg showed a statistically significant superiority to sitagliptin in lowering hemoglobin A1c (a change of −0.73% with canagliflozin 100 mg, −0.88% with canagliflozin 300 mg, and −0.73% with sitagliptin), which may be considered in treatment decisions along with the other possible effects of this drug.1
The decision to use canagliflozin as second-or third-line therapy should be individualized after considering all of the patient’s risk factors as well as the potential benefit vs side effectsof this drug. Metformin remains my first-line choice in the management of type 2 diabetes. In my clinical practice, thus far, I have not used canagliflozin in patients with known coronary disease or a history of cardiovascular events. I have ensured that the LDL is certainly below goal before starting any patient on this drug, and I have followed the LDL closely, without hesitating to increase the statin drug to keep the LDL below goal. I agree that the slight increase of LDL is of concern, and certainly long-term studies are necessary to see whether there will be any increase in cardiovascular events from the use of canagliflozin.
In Reply: I would like to thank these readers very much for their response and comments.
Additional data provided from the study conducted by Lavalle-González et al evaluating the efficacy and safety of canagliflozin (100-mg and 300-mg doses) vs placebo and sitagliptin in patients with type 2 diabetes showed similar findings in weight and blood pressure reduction with slight LDL elevation with the studies mentioned in my article.1 At 52 weeks, as noted, canagliflozin 100 mg demonstrated noninferiority, and canagliflozin 300 mg showed a statistically significant superiority to sitagliptin in lowering hemoglobin A1c (a change of −0.73% with canagliflozin 100 mg, −0.88% with canagliflozin 300 mg, and −0.73% with sitagliptin), which may be considered in treatment decisions along with the other possible effects of this drug.1
The decision to use canagliflozin as second-or third-line therapy should be individualized after considering all of the patient’s risk factors as well as the potential benefit vs side effectsof this drug. Metformin remains my first-line choice in the management of type 2 diabetes. In my clinical practice, thus far, I have not used canagliflozin in patients with known coronary disease or a history of cardiovascular events. I have ensured that the LDL is certainly below goal before starting any patient on this drug, and I have followed the LDL closely, without hesitating to increase the statin drug to keep the LDL below goal. I agree that the slight increase of LDL is of concern, and certainly long-term studies are necessary to see whether there will be any increase in cardiovascular events from the use of canagliflozin.
- Lavalle-González FJ, Januszewicz A, Davidson J, et al. Efficacy and safety of canagliflozin compared with placebo and sitagliptin in patients with type 2 diabetes on background metformin monotherapy: a randomized trial. Diabetologia 2013; 56:2582–2592.
- Lavalle-González FJ, Januszewicz A, Davidson J, et al. Efficacy and safety of canagliflozin compared with placebo and sitagliptin in patients with type 2 diabetes on background metformin monotherapy: a randomized trial. Diabetologia 2013; 56:2582–2592.
Anticoagulation and antiplatelet therapy in acute coronary syndromes
Antiplatelet and anticoagulant drugs are a cornerstone of the medical treatment of acute coronary syndrome (ACS), reducing the rates of both morbidity and death.1–4 However, reductions in ischemic events with these drugs have uniformly been accompanied by increases in bleeding complications, which reduce the net benefit.5 Thus, clinical research has been exploring ways to maximize the benefit while minimizing the risk.
Here, we review the guidelines and evidence supporting the use of antiplatelet and anticoagulant drugs in ACS.
ACUTE CORONARY SYNDROMES WITH OR WITHOUT ST ELEVATION

A key distinction when treating ACS is whether the electrocardiogram shows ST-segment elevation. In cases of non-ST-elevation ACS (ie, unstable angina or non-ST-elevation myocardial infarction), a second key question is whether the initial strategy will be invasive (with angiography performed urgently) or conservative (with angiography performed later). In ST-elevation myocardial infarction, another distinction is how perfusion is to be restored, ie, with primary percutaneous coronary intervention or with thrombolysis. All these questions affect the choice of antiplatelet and anticoagulant therapy.
Figure 1 and Figure 2 summarize the guidelines of the American College of Cardiology Foundation and American Heart Association.1,2,6,7
ANTIPLATELET THERAPY
Aspirin for all

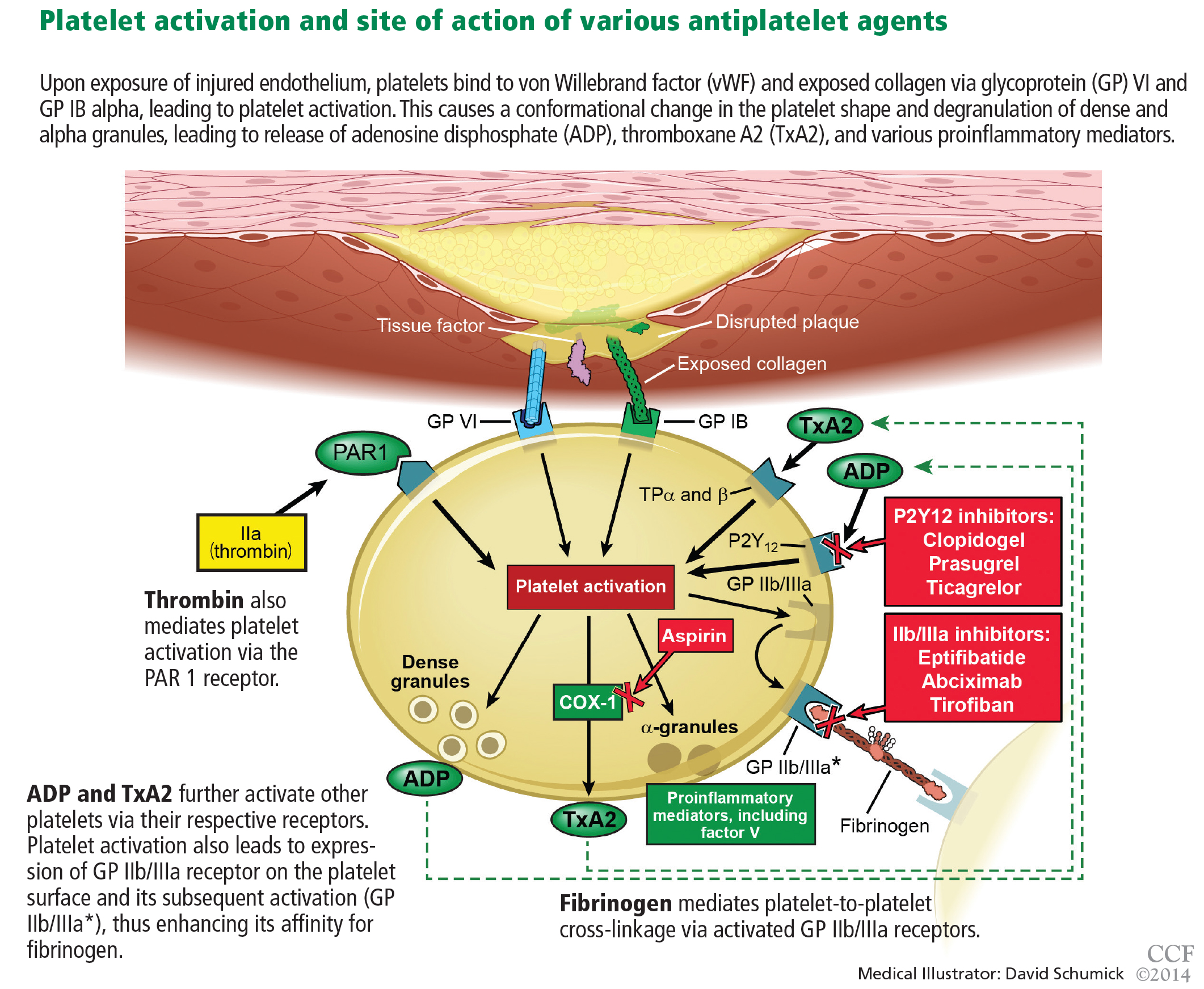
Aspirin irreversibly acetylates the enzyme cyclooxygenase-1, blocking intraplatelet formation of thromboxane A2 (Figure 3), a potent platelet aggregator and endothelial vasoconstrictor. Large clinical trials have confirmed that aspirin reduces morbidity and mortality rates by as much as 50% in patients with ACS.8
The ISIS-2 trial9 found that giving aspirin early in the emergency department significantly reduced the mortality rate.
The Antithrombotic Trialists’ Collaboration,10 in a meta-analysis of randomized controlled trials comparing different doses of aspirin in high-risk ACS patients, found no greater benefit for doses of aspirin higher than 162 mg per day when used long-term.
How to use. During an ACS, the patient should receive one dose of aspirin 325 mg (the standard high-dose pill in the United States). This dose should be chewed, as buccal absorption results in more rapid systemic effects.11
Thereafter, the patient should take 81 mg per day, continued indefinitely. The 81-mg dose also applies to patients who undergo a percutaneous coronary intervention with a drug-eluting stent.7 Previous recommendations called for higher doses, but studies have shown that higher doses pose a higher risk of bleeding without additional clinical benefit. The use of enteric-coated aspirin does not reduce this risk,12 and its delayed release may in fact cause aspirin “pseudoresistance.”13
The concurrent use of nonsteroidal anti-inflammatory drugs (NSAIDs) should be avoided, as NSAIDs reversibly bind to platelets, thus preventing aspirin from binding.14 As aspirin washes out of the body, NSAIDs may then become unbound from platelets, leaving platelets activated.
P2Y12 receptor inhibitors: Clopidogrel, prasugrel, ticagrelor
These agents bind to P2Y12 receptors on platelets to inhibit adenosine diphosphate-mediated platelet activation (Figure 3). Clopidogrel and prasugrel are irreversible prodrugs, whereas ticagrelor binds reversibly.
Clopidogrel, a prodrug
Clopidogrel has a half-life of 8 hours and a time to peak concentration of 4 hours. Eighty-five percent of a dose is inactivated by gut esterases. The remainder is metabolized primarily by the cytochrome P4502C19 enzyme system into its active metabolite.
How to use. The recommended dosage is a 600-mg bolus early in the course of ACS. This is associated with a lower rate of cardiovascular events than a 300-mg dose,2,15 although no trial has rigorously compared 300-mg vs 600-mg doses using major clinical end points. In patients presenting with ACS who cannot tolerate aspirin because of hypersensitivity or major gastrointestinal contraindication, clopidogrel is an alternative.1
The CURE trial16 randomized 12,526 patients with non-ST-elevation ACS to receive clopidogrel or placebo in addition to standard therapy. Clopidogrel was associated with a 20% lower rate of cardiovascular death, myocardial infarction, or stroke in both low- and high-risk patients regardless of whether an invasive or conservative strategy was pursued.

However, patients who underwent coronary artery bypass grafting (CABG) had a 53% higher risk of bleeding (an absolute risk of 3.3%) if they received clopidogrel within 5 days of the surgery. This has led to the practice in some centers of delaying giving clopidogrel until after the coronary anatomy has been defined. This deprives the patient of the anti-ischemic benefits conferred by giving clopidogrel early and remains a contentious issue, with most suggesting that the risk-benefit ratio still favors giving clopidogrel early, before angiography, unless there is a high likelihood that surgery will ultimately be required.17 Alternatively, one could consider using a shorter-acting intravenous glycoprotein IIb/IIIa inhibitor such as eptifibatide as a “bridge” until a definitive reperfusion strategy is chosen.
Effect of CYP2C19 variants. The CLOVIS-2 study18 assessed the effects of genetic variants on the clopidogrel concentration in 106 patients who had had a myocardial infarction. The study confirmed that patients who carry certain variants of the CYP2C19 gene attain lower plasma concentrations of clopidogrel after receiving this drug.19 This accounts for its delayed onset of action as well as its variability in response in patients who have reduced expression or inhibition of this enzyme system. Doubling the standard dose in patients who carry these variants does not appear to provide clinical benefit.20
Thus, the thought is emerging that one should consider using prasugrel or ticagrelor instead of clopidogrel in patients who have these polymorphisms, though this is yet to be backed by robust clinical evidence.
Possible interaction with proton pump inhibitors. Controversy exists about whether proton pump inhibitors inhibit clopidogrel’s action. Although the US Food and Drug Administration continues to warn against the concurrent use of omeprazole and clopidogrel,21 an analysis of the PLATO trial22 concluded that patients with ACS who were taking proton pump inhibitors were at higher risk of ischemic events regardless of whether they had been randomized to clopidogrel or ticagrelor (a drug that acts independently of the cytochrome P450 system). This observation suggests that patients on proton pump inhibitors are generally sicker and at higher risk of ischemic events regardless of the choice of antiplatelet therapy. The use of other gastroprotective agents did not appear to mitigate these risks.
Prasugrel: Faster metabolism to active drug
Prasugrel is an irreversible P2Y12 receptor antagonist (Figure 3) that is metabolized into its active metabolite faster and in a more predictable fashion than clopidogrel.23
The TRITON-TIMI 38 study24 included 13,608 ACS patients in whom an early invasive strategy was planned and who were pretreated with prasugrel or clopidogrel in addition to standard treatment. The rate of the primary efficacy end point of death, myocardial infarction, or stroke was 19% lower in the prasugrel group. In those who underwent percutaneous coronary intervention, the incidence of in-stent thrombosis was more than 50% lower in the prasugrel group regardless of whether bare metal stents or drug-eluting stents were used.
Greater platelet inhibition came at the price of a higher incidence of serious bleeding, particularly in the subgroups of patients who were over age 75, had a history of stroke or transient ischemic attack, or weighed less than 60 kg. Prasugrel is therefore contraindicated in patients with a history of transient ischemic attack or stroke. Some suggest that a 5-mg dose can be used with caution (rather than the usual 10-mg dose) in patients over age 75 years or those who have low body weight.
The TRILOGY-ACS trial25 compared prasugrel and clopidogrel in medically managed patients with high-risk non-ST-elevation ACS. It found no difference in the rates of the primary end points of cardiovascular death, myocardial infarction, or stroke at 1 year. In the prespecified subset of patients over age 75 years, the rate of bleeding end points was no higher with prasugrel 5 mg once daily than with clopidogrel.
Prasugrel’s half-life is 7 hours, and its peak antiplatelet effect is within 30 minutes after an oral dose, compared with 4 hours with clopidogrel. Therefore, if a patient with non-ST-elevation ACS is going to go to the catheterization laboratory soon, he or she should not receive prasugrel beforehand, and should receive it later only if the results of angiography indicate that CABG will not be needed urgently. This is an important consideration when using prasugrel, as the rate of surgery-related bleeding was four times higher than with clopidogrel. If possible, this drug should be withheld for at least 7 days before CABG.
Ticagrelor, a direct P2Y12 receptor inhibitor
Ticagrelor, a reversible direct inhibitor of the P2Y12 receptor, inhibits adenosine diphosphate-mediated activation and aggregation (Figure 3). It has a median time to peak concentration of 1.3 to 2 hours and a half-life of 9 hours.
The PLATO trial26 enrolled 18,624 patients with ACS who were given either ticagrelor or clopidogrel in addition to standard therapy. At 12 months, the composite primary end point of myocardial infarction, death, or stroke had occurred in 16% fewer patients receiving ticagrelor than in the clopidogrel group. Analyzed separately, there were 16% fewer myocardial infarctions, 21% fewer cardiovascular deaths, and 22% fewer deaths from any cause, regardless of whether an invasive or conservative strategy was used, and with or without prior clopidogrel use. Fewer cases of stent thrombosis occurred in the ticagrelor group, and the rate of major bleeding was the same.
In a prospectively defined subgroup analysis,27 ticagrelor was beneficial only in patients who received lower doses of aspirin (< 100 mg daily): the hazard ratio for the primary end point was 0.79 (95% confidence interval [CI] 0.71–0.88) in ticagrelor recipients who received low-dose aspirin and 1.45 (95% CI 1.01–2.09) in those who received high-dose aspirin.
Although this analysis is underpowered and controversial, the current evidence suggests that when used in combination with ticagrelor, the aspirin dose should be 81 mg.
Ticagrelor was also associated with a 19% higher incidence of non-CABG- or procedure-related major bleeding, more nonfatal and fatal intracranial bleeding, a higher incidence of dyspnea, and significantly more ventricular pauses.
Although ticagrelor carries no black-box warning about its use in patients with prior stroke or transient ischemic attack, the number of such patients in PLATO was small. Thus, caution should still be used in these patients.28
Ticagrelor should preferably be discontinued 5 days before CABG.
Glycoprotein IIb/IIIa inhibitors: Eptifibatide, tirofiban, abciximab
Glycoprotein IIb/IIIa inhibitors are intravenous agents that act by inhibiting fibrinogen-and von Willebrand factor-mediated platelet-to-platelet cross-linkage, the final pathway of platelet aggregation (Figure 3).
Use of these agents in ACS has been decreasing, as evidence supporting their use was largely established before the era of dual antiplatelet therapy.
A meta-analysis29 of 46,374 patients with non-ST-elevation ACS found that routinely adding a glycoprotein IIb/IIIa inhibitor “upstream” as a third agent in patients receiving dual antiplatelet therapy bought only a modest (11%) reduction in death or myocardial infarction at 30 days, at the price of a 23% increase in major bleeding and no decrease in the overall rate of death. Roughly 70% of the patients were receiving dual antiplatelet therapy before cardiac catheterization.
These agents can be considered in high-risk ACS patients, such as those with ST-segment changes or elevated troponin concentrations, and in diabetic patients, on the assumption that these patients likely have a high intracoronary thrombus burden and are at higher risk of microvascular embolization.6,30 They can also be considered at the time of primary percutaneous coronary intervention in selected patients receiving heparin.7
Eptifibatide
Eptifibatide is a small-molecule, short-acting glycoprotein IIb/IIIa inhibitor with a half-life of 2.5 hours. Its inhibition of platelet aggregation is reversible by stopping the drug infusion and is thought to be a result of dissociation of the drug from platelets.
The PURSUIT trial31 studied 10,948 patients presenting with non-ST-elevation ACS randomized to placebo, eptifibatide in a 180-μg/kg bolus followed by a 2.0-μg/kg/min infusion, or eptifibatide in a 180-μg/kg bolus followed by a 1.3-μg/kg/min infusion. Both eptifibatide groups had a 1.5% absolute reduction in the incidence of the primary end point of death or myocardial infarction, a benefit that was apparent at 96 hours and that persisted through 30 days. Bleeding was more common in the eptifibatide groups, but there was no increase in the rate of hemorrhagic stroke.
The ACUITY trial32 found that early use of eptifibatide or tirofiban had no effect on the primary outcome. (See the section below on bivalirudin for more information about the ACUITY trial.)
PARENTERAL ANTICOAGULANTS
Unfractionated heparin: A declining role
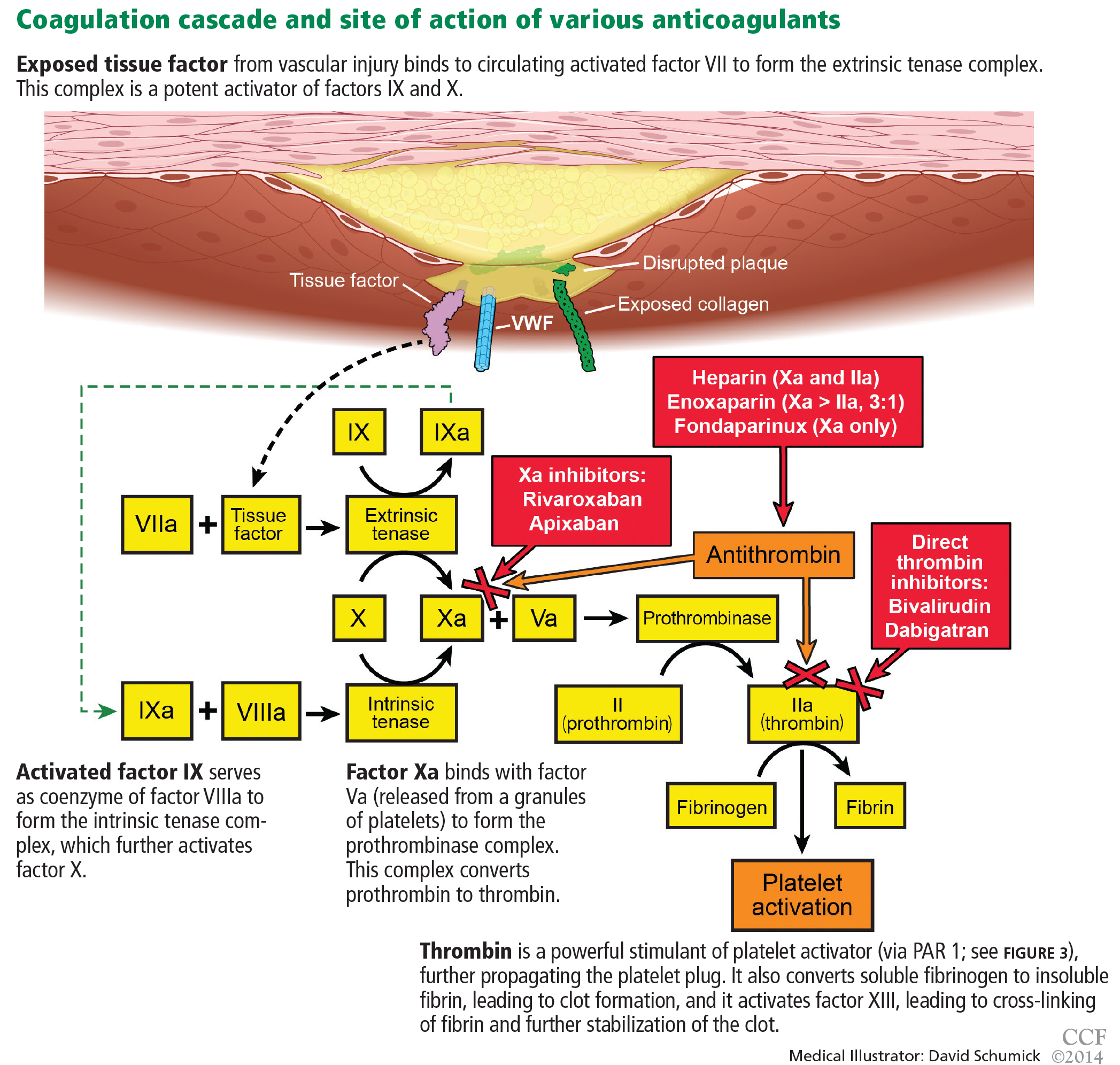
Heparin binds to antithrombin and induces a conformational change, causing rapid inhibition of factor IIa (thrombin), factor IXa, and factor Xa, thus preventing further thrombus propagation (Figure 4). An intravenous bolus of 60 units/kg produces a time to peak of 5 to 10 minutes and a half-life of 30 to 60 minutes.
Heparin can be reversed by giving protamine sulfate (1 mg per 100 units of heparin). For ACS, it is given in a bolus of 60 units/kg not exceeding 4,000 units, followed by an infusion of 12 units/kg/hour, with monitoring of the activated partial thromboplastin time every 6 hours with a goal value of 50 to 70 seconds or 1.5 to 2.5 times control.
Side effects include thrombocytopenia, heparin-induced thrombocytopenia (a distinct condition), and bleeding.
The use of unfractionated heparin was tested in ACS in the early 1990s. Oler et al33 performed a meta-analysis of six randomized trials and found a 33% lower rate of death in patients treated with heparin in addition to aspirin in ACS, as well less reported ischemic pain.
Advantages of unfractionated heparin are that it has stood the test of time, is inexpensive, and can be rapidly reversed. The disadvantages are that it can have serious side effects, including heparin-induced thrombocytopenia, and is more likely to cause bleeding than the newer intravenous anticoagulants discussed below. Thus, its position as the main anticoagulant in ACS is being challenged.
Bivalirudin, a direct thrombin inhibitor
Bivalirudin is a synthetic direct thrombin inhibitor of fluid-phase and clot-bound thrombin (Figure 4). It also inhibits platelets directly.
The ACUITY trial32 randomized 13,819 patients with moderate to high-risk ACS scheduled for invasive treatment into three treatment groups:
- Heparin (either unfractionated heparin or enoxaparin) plus a glycoprotein IIb/IIIa inhibitor (either eptifibatide, tirofiban, or abciximab)
- Bivalirudin plus a glycoprotein IIb/IIIa inhibitor
- Bivalirudin alone.
The bivalirudin-alone treatment was as sociated with noninferior rates of composite ischemia end points and significantly lower rates of major bleeding, adding up to a significant reduction in the net clinical outcome end point. An important caveat is that bivalirudin’s noninferiority was mostly in the group of patients already receiving a thienopyridine before angiography and percutaneous coronary intervention (RR 0.97 vs 1.27, P = .054). There was less major, nonmajor, minor, CABG-related, and non-CABG-related bleeding as well as need for transfusion in the bivalirudin-alone group, making bivalirudin monotherapy an attractive option in ACS patients with or without ST-segment elevation undergoing a percutaneous coronary intervention.1,31
The ISAR-REACT trial34 later compared bivalirudin alone vs unfractionated heparin and abciximab in patients with non-ST-elevation myocardial infarction undergoing percutaneous coronary intervention pretreated with aspirin and clopidogrel. The composite rate of ischemia was similar in the two treatment groups, with significantly lower rates of bleeding in the bivalirudin group.
HORIZONS-AMI35 randomized 3,602 patients with ST-elevation myocardial infarction receiving aspirin and clopidogrel either to unfractionated heparin and a glycoprotein IIb/IIIa inhibitor or to bivalirudin. As in the ACUITY trial, there was no difference in ischemic end points and a 40% to 45% lower rate of major bleeding end points in the bivalirudin group, translating into an overall lower rate of death.
Enoxaparin, a low-molecular weight heparin
Enoxaparin is a low-molecular-weight heparin that inhibits factor IIa and factor Xa via antithrombin, roughly in a ratio of 1:3 (Figure 4). It has a time to peak effect of 10 minutes when given intravenously36 and 3 to 5 hours when given subcutaneously.37 Its half-life is 4.5 hours, but it is longer in patients with renal dysfunction, requiring dose adjustments in this population.
Its anticoagulant effect is partially reversible. If it is to be reversed between 0 and 8 hours after dosing, the recommended reversal regimen is 1 mg of protamine sulfate for every 1 mg of enoxaparin used. At 8 to 12 hours, it is 0.5 mg of protamine for every 1 mg of enoxaparin. After 12 hours, no protamine is required.
Compared with unfractionated heparin, enoxaparin has less plasma protein binding and a more consistent anticoagulant effect. Its high bioavailability also allows for subcutaneous dosing. Its greater anti-Xa activity inhibits thrombin generation more effectively, and it causes lower rates of thrombocytopenia and heparin-induced thrombocytopenia.
de Lemos et al38 found that, in ACS patients in whom an early conservative approach of medical management was planned, enoxaparin was more efficacious than unfractionated heparin and caused a similar rate of bleeding.
Murphy et al,39 in a meta-analysis of 12 trials in 49,088 ACS patients, also found that enoxaparin had a net clinical benefit compared with unfractionated heparin in reducing rates of myocardial infarction and death despite more bleeding.
The ESSENCE trial40 compared enoxaparin vs unfractionated heparin in 3,171 patients with ACS. It found fewer ischemic events with enoxaparin in the early phase, more minor bleeding, but no increase in major bleeding.
The SYNERGY trial,41 in 10,027 patients with high-risk non-ST-elevation ACS undergoing percutaneous coronary intervention, compared subcutaneous enoxaparin with intravenous heparin. Enoxaparin was found to be noninferior to heparin but caused more bleeding, including major bleeding, drops in hemoglobin, and intracranial hemorrhage.
The EXTRACT-TIMI 25 trial.42 In patients with ST-elevation myocardial infarction, enoxaparin has been shown to be beneficial both in patients treated with fibrinolysis and in those who underwent primary percutaneous coronary intervention. The EXTRACT-TIMI 25 trial randomized 20,749 patients to receive either enoxaparin (an intravenous bolus and maintenance subcutaneous dosing based on renal function) or intravenous heparin in addition to thrombolysis within 6 hours of the diagnosis of ST-elevation myocardial infarction. Although the enoxaparin group had more bleeding end points, they had fewer primary and secondary efficacy end points, translating into an overall net clinical benefit in favor of enoxaparin.
The ATOLL trial43 examined the use of enoxaparin (0.5 mg/kg intravenously) or unfractionated heparin in 910 patients with ST-elevation myocardial infarction undergoing primary percutaneous coronary intervention (via the radial artery in 66% to 69%). Although there was a trend towards benefit in terms of the primary end point of death, myocardial infarction complications, procedure failure, and major bleeding favoring enoxaparin, it was not statistically significant (95% CI 0.68–1.01, P = .06).
However, there was a 37% to 42% lower rate of the secondary end point of death, recurrent myocardial infarction or ACS, or urgent target-vessel revascularization in the enoxaparin group, with a 40% reduction in death from any cause, death from a cardiac cause, or shock. The safety profiles of the two drugs were similar, and the net clinical benefit significantly favored enoxaparin.
Fondaparinux, a factor Xa inhibitor
Fondaparinux is a synthetic pentasaccharide that indirectly inhibits factor Xa through the action of antithrombin (Figure 4). After a 2.5-mg subcutaneous dose, it has a time to peak concentration of 2 hours and a half-life of 17 to 21 hours.
The OASIS-5 trial44 compared fondaparinux and enoxaparin in 20,078 patients treated for non-ST-elevation ACS. Although the rates of death, myocardial infarction, and refractory ischemia at 9 days were similar for both drugs, the fondaparinux group had a significantly (almost 50%) lower rate of bleeding at 30 days, translating into significantly fewer deaths at 30 days. However, patients receiving fondaparinux who underwent percutaneous coronary intervention had a threefold higher rate of catheter-related thrombosis.
The OASIS-6 trial45 compared fondaparinux vs usual care (placebo in those in whom unfractionated heparin was not indicated or unfractionated heparin for up to 48 hours followed by placebo for up to 8 days) in 12,092 patients with ST-elevation myocardial infarction. There was a 1.5% absolute risk reduction in death and reinfarction without an increase in bleeding at 30 days, with trends persisting 6 months into the study. However, fondaparinux was not superior to heparin in the 3% of patients who underwent primary percutaneous coronary intervention. As in OASIS-5, there was more catheter-related thrombosis in the fondaparinux group.
Although the use of supplemental unfractionated heparin appears to have mitigated this risk, fondaparinux remains a less-than-ideal option in the era of primary percutaneous coronary intervention for ST-elevation myocardial infarction and has therefore found limited use in this group of patients. It should, however, be considered in patients for whom a conservative strategy is planned, especially if bleeding risk is deemed to be high.
ORAL ANTICOAGULANTS
Oral anticoagulants provide ischemic benefit in selected patients with ACS—at the price of a higher risk of significant bleeding.
Warfarin
Warfarin was investigated after myocardial infarction in the WARIS II,46 CARS,47 and CHAMP48 trials.
WARIS II46 looked at the use of aspirin alone, warfarin alone, and aspirin and warfarin in combination. The rates of the primary end points of stroke, nonfatal infarction, and death were lower in the warfarin group.
CARS47 found no difference in the rate of the primary end point of fatal infarction, nonfatal ischemic stroke, or cardiovascular death with aspirin vs warfarin plus aspirin.
CHAMP48 saw similar trends, ie, no difference in the rate of death, recurrent myocardial infarction, or stroke with warfarin plus aspirin vs aspirin alone.
All three studies showed increases in major bleeding with warfarin use.
Putting these trials into context, the significant net clinical benefit of dual antiplatelet therapy in the current era compared with the significant bleeding and questionable conflicting evidence supporting benefit with warfarin has limited its use in ACS patients.
Rivaroxaban, an oral factor Xa inhibitor
Rivaroxaban is a novel oral direct reversible factor Xa inhibitor.
The ATLAS ACS 2-TIMI 51 trial49 found rivaroxaban 2.5 mg or 5 mg to yield a significantly lower rate of the primary outcome of cardiovascular death, myocardial infarction, ischemic stroke, and in-stent thrombosis compared with placebo, but significantly more major non-CABG bleeding and intracranial hemorrhage.
The dose used in this trial was much lower than the dose used in trials investigating the role of this drug in stroke prophylaxis in atrial fibrillation.
Apixaban, an oral factor Xa inhibitor
Apixaban is another direct factor Xa inhibitor.
The APPRAISE-2 trial50 compared apixaban 5 mg twice daily vs placebo in ACS. There was no difference in the rate of cardiovascular death, myocardial infarction, or stroke, but there was significantly more bleeding in the apixaban group, prompting early termination of this study.
Dabigatran, an oral thrombin inhibitor
Dabigatran is an oral direct thrombin inhibitor.
The RE-DEEM trial51 compared four doses of dabigatran (50, 75, 110, and 150 mg twice daily) and placebo in ACS patients. The dabigatran groups had more major and minor bleeding, and the higher the dose, the higher the incidence of bleeding. In addition, the rates of ischemic end points were no lower with dabigatran, although this trial was not powered to show differences in clinical events.
REDUCING THE RISK OF BLEEDING
In the treatment of ACS, the benefits of restoring perfusion by preventing further propagation of thrombus and platelet aggregation come at a significant price of higher bleeding risk. This in turn increases the risk of death through various mechanisms, including shock, worsening ischemia, discontinuation of antiplatelet and anticoagulation therapy causing stent thrombosis, and anemia leading to transfusion, which propagates the underlying inflammatory milieu.52
Giugliano and Braunwald53 provide practical suggestions to reduce this risk, advising physicians to:
- Avoid inappropriately high dosing, particularly in patients with renal insufficiency
- Preferentially use agents that cause less bleeding (eg, bivalirudin, fondaparinux) without compromising anti-ischemic efficacy
- Minimize the concomitant use of other drugs that cause bleeding (eg, NSAIDs)
- Use drugs that protect against bleeding (eg, proton pump inhibitors) in patients at high risk
- Prevent access-site bleeding by using the radial artery, smaller sheaths, and appropriate sheath and closure device management. Indeed, the use of radial interventions in ACS has been shown to reduce access-site-related bleeding, even in patients at high risk.54
The reduction in bleeding risk may provide future trials the opportunity to increase antithrombotic efficacy of different agents with goals of reducing ischemic end points.
- Wright RS, Anderson JL, Adams CD, et al; American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. 2011 ACCF/AHA focused update incorporated into the ACC/AHA 2007 Guidelines for the Management of Patients with Unstable Angina/Non-ST-Elevation Myocardial Infarction. J Am Coll Cardiol 2011; 57:e215–e367.
- Jneid H, Anderson JL, Wright RS, et al. 2012 ACCF/AHA focused update of the guideline for the management of patients with unstable angina/non-ST-elevation myocardial infarction (updating the 2007 guideline and replacing the 2011 focused update): a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2012; 60:645–681.
- CAPRIE Steering Committee. A randomised, blinded, trial of clopidogrel versus aspirin in patients at risk of ischaemic events (CAPRIE). CAPRIE Steering Committee. Lancet 1996; 348:1329–1339.
- Cohen M, Adams PC, Parry G, et al. Combination antithrombotic therapy in unstable rest angina and non-Q-wave infarction in nonprior aspirin users. Primary end points analysis from the ATACS trial. Antithrombotic Therapy in Acute Coronary Syndromes Research Group. Circulation 1994; 89:81–88.
- Moscucci M, Fox KA, Cannon CP, et al. Predictors of major bleeding in acute coronary syndromes: the Global Registry of Acute Coronary Events (GRACE). Eur Heart J 2003; 24:1815–1823.
- Levine GN, Bates ER, Blankenship JC, et al; American College of Cardiology Foundation; American Heart Association Task Force on Practice Guidelines; Society for Cardiovascular Angiography and Interventions. 2011 ACCF/AHA/SCAI Guideline for Percutaneous Coronary Intervention. A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines and the Society for Cardiovascular Angiography and Interventions. J Am Coll Cardiol 2011; 58:e44–e122.
- American College of Emergency Physicians; Society for Cardiovascular Angiography and Interventions; O’Gara PT, Kushner FG, Ascheim DD, et al. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013; 61:485–510.
- Lewis HD, Davis JW, Archibald DG, et al. Protective effects of aspirin against acute myocardial infarction and death in men with unstable angina. Results of a Veterans Administration Cooperative Study. N Engl J Med 1983; 309:396–403.
- ISIS-2 (Second International Study of Infarct Survival) Collaborative Group. Randomised trial of intravenous streptokinase, oral aspirin, both, or neither among 17,187 cases of suspected acute myocardial infarction: ISIS-2. Lancet 1988; 2:349–360.
- Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002; 324:71–86.
- Sweeny JM, Gorog DA, Fuster V. Antiplatelet drug ‘resistance’. Part 1: mechanisms and clinical measurements. Nat Rev Cardiol 2009; 6:273–282.
- Kelly JP, Kaufman DW, Jurgelon JM, Sheehan J, Koff RS, Shapiro S. Risk of aspirin-associated major upper-gastrointestinal bleeding with enteric-coated or buffered product. Lancet 1996; 348:1413–1416.
- Grosser T, Fries S, Lawson JA, Kapoor SC, Grant GR, FitzGerald GA. Drug resistance and pseudoresistance: an unintended consequence of enteric coating aspirin. Circulation 2013; 127:377–385.
- US Food and Drug Administration (FDA). Concomitant use of ibuprofen and aspirin: potential for attenuation of the anti-platelet effect of aspirin. http://www.fda.gov/downloads/drugs/drugsafety/postmarketdrugsafetyinformationforpatientsandproviders/ucm161282.pdf. Accessed November 30, 2013.
- Patti G, Colonna G, Pasceri V, Pepe LL, Montinaro A, Di Sciascio G. Randomized trial of high loading dose of clopidogrel for reduction of periprocedural myocardial infarction in patients undergoing coronary intervention: results from the ARMYDA-2 (Antiplatelet therapy for Reduction of MYocardial Damage during Angioplasty) study. Circulation 2005; 111:2099–2106.
- Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK; Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial Investigators. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med 2001; 345:494–502.
- Bavry AA, Lincoff AM. Is clopidogrel cardiovascular medicine’s double-edged sword? Circulation 2006; 113:1638–1640.
- Collet JP, Hulot JS, Anzaha G, et al; CLOVIS-2 Investigators. High doses of clopidogrel to overcome genetic resistance: the randomized crossover CLOVIS-2 (Clopidogrel and Response Variability Investigation Study 2). JACC Cardiovasc Interv 2011; 4:392–402.
- Hulot JS, Collet JP, Cayla G, et al. CYP2C19 but not PON1 genetic variants influence clopidogrel pharmacokinetics, pharmacodynamics, and clinical efficacy in post-myocardial infarction patients. Circ Cardiovasc Interv 2011; 4:422–428.
- Cuisset T, Quilici J, Cohen W, et al. Usefulness of high clopidogrel maintenance dose according to CYP2C19 genotypes in clopidogrel low responders undergoing coronary stenting for non ST elevation acute coronary syndrome. Am J Cardiol 2011; 108:760–765.
- US Food and Drug Administration (FDA). FDA reminder to avoid concomitant use of Plavix (clopidogrel) and omeprazole. http://www.fda.gov/Drugs/DrugSafety/ucm231161.htm. Accessed November 30, 2013.
- Goodman SG, Clare R, Pieper KS, et al; Platelet Inhibition and Patient Outcomes Trial Investigators. Association of proton pump inhibitor use on cardiovascular outcomes with clopidogrel and ticagrelor: insights from the platelet inhibition and patient outcomes trial. Circulation 2012; 125:978–986.
- Solomon S, Vacek JL. Reducing cardiac ischemic events in patients with ACS: prasugrel versus clopidogrel. Commentary. Postgrad Med 2010; 122:198–200.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Roe MT, Armstrong PW, Fox KA, et al; TRILOGY ACS Investigators. Prasugrel versus clopidogrel for acute coronary syndromes without revascularization. N Engl J Med 2012; 367:1297–1309.
- Wallentin L, Becker RC, Budaj A, et al; PLATO Investigators. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2009; 361:1045–1057.
- Mahaffey KW, Wojdyla DM, Carroll K, et al; PLATO Investigators. Ticagrelor compared with clopidogrel by geographic region in the Platelet Inhibition and Patient Outcomes (PLATO) trial. Circulation 2011; 124:544–554.
- Verheugt FW. Beware of novel antiplatelet therapy in acute coronary syndrome patients with previous stroke. Circulation 2012; 125:2821–2823.
- Tricoci P, Newby LK, Hasselblad V, et al. Upstream use of small-molecule glycoprotein iib/iiia inhibitors in patients with non-ST-segment elevation acute coronary syndromes: a systematic overview of randomized clinical trials. Circ Cardiovasc Qual Outcomes 2011; 4:448–458.
- Kastrati A, Mehilli J, Neumann FJ, et al; Intracoronary Stenting and Antithrombotic: Regimen Rapid Early Action for Coronary Treatment 2 (ISAR-REACT 2) Trial Investigators. Abciximab in patients with acute coronary syndromes undergoing percutaneous coronary intervention after clopidogrel pretreatment: the ISAR-REACT 2 randomized trial. JAMA 2006; 295:1531–1538.
- Inhibition of platelet glycoprotein IIb/IIIa with eptifibatide in patients with acute coronary syndromes. The PURSUIT Trial Investigators. Platelet Glycoprotein IIb/IIIa in Unstable Angina: Receptor Suppression Using Integrilin Therapy. N Engl J Med 1998; 339:436–443.
- Stone GW, McLaurin BT, Cox DA, et al; ACUITY Investigators. Bivalirudin for patients with acute coronary syndromes. N Engl J Med 2006; 355:2203–2216.
- Oler A, Whooley MA, Oler J, Grady D. Adding heparin to aspirin reduces the incidence of myocardial infarction and death in patients with unstable angina. A meta-analysis. JAMA 1996; 276:811–815.
- Kastrati A, Neumann FJ, Schulz S, et al; ISAR-REACT 4 Trial Investigators. Abciximab and heparin versus bivalirudin for non-ST-elevation myocardial infarction. N Engl J Med 2011; 365:1980–1989.
- Stone GW, Witzenbichler B, Guagliumi G, et al; HORIZONS-AMI Trial Investigators. Bivalirudin during primary PCI in acute myocardial infarction. N Engl J Med 2008; 358:2218–2230.
- Aslam MS, Sundberg S, Sabri MN, Cooke D, Lakier JB. Pharmacokinetics of intravenous/subcutaneous enoxaparin in patients with acute coronary syndrome undergoing percutaneous coronary interventions. Catheter Cardiovasc Interv 2002; 57:187–190.
- Sanofi-Aventis US. Lovenox (enoxaparin sodium injection) product information. http://www.lovenox.com/hcp/clinical-data.aspx. Accessed December 1, 2013.
- de Lemos JA, Blazing MA, Wiviott SD, et al. Enoxaparin versus unfractionated heparin in patients treated with tirofiban, aspirin and an early conservative initial management strategy: results from the A phase of the A-to-Z trial. Eur Heart J 2004; 25:1688–1694.
- Murphy SA, Gibson CM, Morrow DA, et al. Efficacy and safety of the low-molecular weight heparin enoxaparin compared with unfractionated heparin across the acute coronary syndrome spectrum: a meta-analysis. Eur Heart J 2007; 28:2077–2086.
- Cohen M, Demers C, Gurfinkel EP, et al. A comparison of low-molecular-weight heparin with unfractionated heparin for unstable coronary artery disease. Efficacy and Safety of Subcutaneous Enoxaparin in Non-Q-Wave Coronary Events Study Group. N Engl J Med 1997; 337:447–452.
- Ferguson JJ, Califf RM, Antman EM, et al; SYNERGY Trial Investigators. Enoxaparin vs unfractionated heparin in high-risk patients with non-ST-segment elevation acute coronary syndromes managed with an intended early invasive strategy: primary results of the SYNERGY randomized trial. JAMA 2004; 292:45–54.
- Antman EM, Morrow DA, McCabe CH, et al; ExTRACT-TIMI 25 Investigators. Enoxaparin versus unfractionated heparin with fibrinolysis for ST-elevation myocardial infarction. N Engl J Med 2006; 354:1477–1488.
- Montalescot G, Zeymer U, Silvain J, et al; ATOLL Investigators. Intravenous enoxaparin or unfractionated heparin in primary percutaneous coronary intervention for ST-elevation myocardial infarction: the international randomised open-label ATOLL trial. Lancet 2011; 378:693–703.
- Yusuf S, Mehta SR, Chrolavicius S, et al; Fifth Organization to Assess Strategies in Acute Ischemic Syndromes Investigators. Comparison of fondaparinux and enoxaparin in acute coronary syndromes. N Engl J Med 2006; 354:1464–1476.
- Yusuf S, Mehta SR, Chrolavicius S, et al; OASIS-6 Trial Group. Effects of fondaparinux on mortality and reinfarction in patients with acute ST-segment elevation myocardial infarction: the OASIS-6 randomized trial. JAMA 2006; 295:1519–1530.
- Hurlen M, Abdelnoor M, Smith P, Erikssen J, Arnesen H. Warfarin, aspirin, or both after myocardial infarction. N Engl J Med 2002; 347:969–974.
- Coumadin Aspirin Reinfarction Study (CARS) Investigators. Randomised double-blind trial of fixed low-dose warfarin with aspirin after myocardial infarction. Lancet 1997; 350:389–396.
- Fiore LD, Ezekowitz MD, Brophy MT, Lu D, Sacco J, Peduzzi P; Combination Hemotherapy and Mortality Prevention (CHAMP) Study Group. Department of Veterans Affairs Cooperative Studies Program Clinical Trial comparing combined warfarin and aspirin with aspirin alone in survivors of acute myocardial infarction: primary results of the CHAMP study. Circulation 2002; 105:557–563.
- Mega JL, Braunwald E, Wiviott SD, et al; ATLAS ACS 2–TIMI 51 Investigators. Rivaroxaban in patients with a recent acute coronary syndrome. N Engl J Med 2012; 366:9–19.
- Alexander JH, Lopes RD, James S, et al; APPRAISE-2 Investigators. Apixaban with antiplatelet therapy after acute coronary syndrome. N Engl J Med 2011; 365:699–708.
- Oldgren J, Budaj A, Granger CB, et al; RE-DEEM Investigators. Dabigatran vs placebo in patients with acute coronary syndromes on dual antiplatelet therapy: a randomized, double-blind, phase II trial. Eur Heart J 2011; 32:2781–2789.
- Steg PG, Huber K, Andreotti F, et al. Bleeding in acute coronary syndromes and percutaneous coronary interventions: position paper by the Working Group on Thrombosis of the European Society of Cardiology. Eur Heart J 2011; 32:1854–1864.
- Giugliano RP, Braunwald E. The year in non-ST-segment elevation acute coronary syndrome. J Am Coll Cardiol 2012; 60:2127–039.
- Rao SV, Ou FS, Wang TY, et al. Trends in the prevalence and outcomes of radial and femoral approaches to percutaneous coronary intervention: a report from the National Cardiovascular Data Registry. JACC Cardiovasc Interv 2008; 1:379–386.
Antiplatelet and anticoagulant drugs are a cornerstone of the medical treatment of acute coronary syndrome (ACS), reducing the rates of both morbidity and death.1–4 However, reductions in ischemic events with these drugs have uniformly been accompanied by increases in bleeding complications, which reduce the net benefit.5 Thus, clinical research has been exploring ways to maximize the benefit while minimizing the risk.
Here, we review the guidelines and evidence supporting the use of antiplatelet and anticoagulant drugs in ACS.
ACUTE CORONARY SYNDROMES WITH OR WITHOUT ST ELEVATION
A key distinction when treating ACS is whether the electrocardiogram shows ST-segment elevation. In cases of non-ST-elevation ACS (ie, unstable angina or non-ST-elevation myocardial infarction), a second key question is whether the initial strategy will be invasive (with angiography performed urgently) or conservative (with angiography performed later). In ST-elevation myocardial infarction, another distinction is how perfusion is to be restored, ie, with primary percutaneous coronary intervention or with thrombolysis. All these questions affect the choice of antiplatelet and anticoagulant therapy.
Figure 1 and Figure 2 summarize the guidelines of the American College of Cardiology Foundation and American Heart Association.1,2,6,7
ANTIPLATELET THERAPY
Aspirin for all
Aspirin irreversibly acetylates the enzyme cyclooxygenase-1, blocking intraplatelet formation of thromboxane A2 (Figure 3), a potent platelet aggregator and endothelial vasoconstrictor. Large clinical trials have confirmed that aspirin reduces morbidity and mortality rates by as much as 50% in patients with ACS.8
The ISIS-2 trial9 found that giving aspirin early in the emergency department significantly reduced the mortality rate.
The Antithrombotic Trialists’ Collaboration,10 in a meta-analysis of randomized controlled trials comparing different doses of aspirin in high-risk ACS patients, found no greater benefit for doses of aspirin higher than 162 mg per day when used long-term.
How to use. During an ACS, the patient should receive one dose of aspirin 325 mg (the standard high-dose pill in the United States). This dose should be chewed, as buccal absorption results in more rapid systemic effects.11
Thereafter, the patient should take 81 mg per day, continued indefinitely. The 81-mg dose also applies to patients who undergo a percutaneous coronary intervention with a drug-eluting stent.7 Previous recommendations called for higher doses, but studies have shown that higher doses pose a higher risk of bleeding without additional clinical benefit. The use of enteric-coated aspirin does not reduce this risk,12 and its delayed release may in fact cause aspirin “pseudoresistance.”13
The concurrent use of nonsteroidal anti-inflammatory drugs (NSAIDs) should be avoided, as NSAIDs reversibly bind to platelets, thus preventing aspirin from binding.14 As aspirin washes out of the body, NSAIDs may then become unbound from platelets, leaving platelets activated.
P2Y12 receptor inhibitors: Clopidogrel, prasugrel, ticagrelor
These agents bind to P2Y12 receptors on platelets to inhibit adenosine diphosphate-mediated platelet activation (Figure 3). Clopidogrel and prasugrel are irreversible prodrugs, whereas ticagrelor binds reversibly.
Clopidogrel, a prodrug
Clopidogrel has a half-life of 8 hours and a time to peak concentration of 4 hours. Eighty-five percent of a dose is inactivated by gut esterases. The remainder is metabolized primarily by the cytochrome P4502C19 enzyme system into its active metabolite.
How to use. The recommended dosage is a 600-mg bolus early in the course of ACS. This is associated with a lower rate of cardiovascular events than a 300-mg dose,2,15 although no trial has rigorously compared 300-mg vs 600-mg doses using major clinical end points. In patients presenting with ACS who cannot tolerate aspirin because of hypersensitivity or major gastrointestinal contraindication, clopidogrel is an alternative.1
The CURE trial16 randomized 12,526 patients with non-ST-elevation ACS to receive clopidogrel or placebo in addition to standard therapy. Clopidogrel was associated with a 20% lower rate of cardiovascular death, myocardial infarction, or stroke in both low- and high-risk patients regardless of whether an invasive or conservative strategy was pursued.
However, patients who underwent coronary artery bypass grafting (CABG) had a 53% higher risk of bleeding (an absolute risk of 3.3%) if they received clopidogrel within 5 days of the surgery. This has led to the practice in some centers of delaying giving clopidogrel until after the coronary anatomy has been defined. This deprives the patient of the anti-ischemic benefits conferred by giving clopidogrel early and remains a contentious issue, with most suggesting that the risk-benefit ratio still favors giving clopidogrel early, before angiography, unless there is a high likelihood that surgery will ultimately be required.17 Alternatively, one could consider using a shorter-acting intravenous glycoprotein IIb/IIIa inhibitor such as eptifibatide as a “bridge” until a definitive reperfusion strategy is chosen.
Effect of CYP2C19 variants. The CLOVIS-2 study18 assessed the effects of genetic variants on the clopidogrel concentration in 106 patients who had had a myocardial infarction. The study confirmed that patients who carry certain variants of the CYP2C19 gene attain lower plasma concentrations of clopidogrel after receiving this drug.19 This accounts for its delayed onset of action as well as its variability in response in patients who have reduced expression or inhibition of this enzyme system. Doubling the standard dose in patients who carry these variants does not appear to provide clinical benefit.20
Thus, the thought is emerging that one should consider using prasugrel or ticagrelor instead of clopidogrel in patients who have these polymorphisms, though this is yet to be backed by robust clinical evidence.
Possible interaction with proton pump inhibitors. Controversy exists about whether proton pump inhibitors inhibit clopidogrel’s action. Although the US Food and Drug Administration continues to warn against the concurrent use of omeprazole and clopidogrel,21 an analysis of the PLATO trial22 concluded that patients with ACS who were taking proton pump inhibitors were at higher risk of ischemic events regardless of whether they had been randomized to clopidogrel or ticagrelor (a drug that acts independently of the cytochrome P450 system). This observation suggests that patients on proton pump inhibitors are generally sicker and at higher risk of ischemic events regardless of the choice of antiplatelet therapy. The use of other gastroprotective agents did not appear to mitigate these risks.
Prasugrel: Faster metabolism to active drug
Prasugrel is an irreversible P2Y12 receptor antagonist (Figure 3) that is metabolized into its active metabolite faster and in a more predictable fashion than clopidogrel.23
The TRITON-TIMI 38 study24 included 13,608 ACS patients in whom an early invasive strategy was planned and who were pretreated with prasugrel or clopidogrel in addition to standard treatment. The rate of the primary efficacy end point of death, myocardial infarction, or stroke was 19% lower in the prasugrel group. In those who underwent percutaneous coronary intervention, the incidence of in-stent thrombosis was more than 50% lower in the prasugrel group regardless of whether bare metal stents or drug-eluting stents were used.
Greater platelet inhibition came at the price of a higher incidence of serious bleeding, particularly in the subgroups of patients who were over age 75, had a history of stroke or transient ischemic attack, or weighed less than 60 kg. Prasugrel is therefore contraindicated in patients with a history of transient ischemic attack or stroke. Some suggest that a 5-mg dose can be used with caution (rather than the usual 10-mg dose) in patients over age 75 years or those who have low body weight.
The TRILOGY-ACS trial25 compared prasugrel and clopidogrel in medically managed patients with high-risk non-ST-elevation ACS. It found no difference in the rates of the primary end points of cardiovascular death, myocardial infarction, or stroke at 1 year. In the prespecified subset of patients over age 75 years, the rate of bleeding end points was no higher with prasugrel 5 mg once daily than with clopidogrel.
Prasugrel’s half-life is 7 hours, and its peak antiplatelet effect is within 30 minutes after an oral dose, compared with 4 hours with clopidogrel. Therefore, if a patient with non-ST-elevation ACS is going to go to the catheterization laboratory soon, he or she should not receive prasugrel beforehand, and should receive it later only if the results of angiography indicate that CABG will not be needed urgently. This is an important consideration when using prasugrel, as the rate of surgery-related bleeding was four times higher than with clopidogrel. If possible, this drug should be withheld for at least 7 days before CABG.
Ticagrelor, a direct P2Y12 receptor inhibitor
Ticagrelor, a reversible direct inhibitor of the P2Y12 receptor, inhibits adenosine diphosphate-mediated activation and aggregation (Figure 3). It has a median time to peak concentration of 1.3 to 2 hours and a half-life of 9 hours.
The PLATO trial26 enrolled 18,624 patients with ACS who were given either ticagrelor or clopidogrel in addition to standard therapy. At 12 months, the composite primary end point of myocardial infarction, death, or stroke had occurred in 16% fewer patients receiving ticagrelor than in the clopidogrel group. Analyzed separately, there were 16% fewer myocardial infarctions, 21% fewer cardiovascular deaths, and 22% fewer deaths from any cause, regardless of whether an invasive or conservative strategy was used, and with or without prior clopidogrel use. Fewer cases of stent thrombosis occurred in the ticagrelor group, and the rate of major bleeding was the same.
In a prospectively defined subgroup analysis,27 ticagrelor was beneficial only in patients who received lower doses of aspirin (< 100 mg daily): the hazard ratio for the primary end point was 0.79 (95% confidence interval [CI] 0.71–0.88) in ticagrelor recipients who received low-dose aspirin and 1.45 (95% CI 1.01–2.09) in those who received high-dose aspirin.
Although this analysis is underpowered and controversial, the current evidence suggests that when used in combination with ticagrelor, the aspirin dose should be 81 mg.
Ticagrelor was also associated with a 19% higher incidence of non-CABG- or procedure-related major bleeding, more nonfatal and fatal intracranial bleeding, a higher incidence of dyspnea, and significantly more ventricular pauses.
Although ticagrelor carries no black-box warning about its use in patients with prior stroke or transient ischemic attack, the number of such patients in PLATO was small. Thus, caution should still be used in these patients.28
Ticagrelor should preferably be discontinued 5 days before CABG.
Glycoprotein IIb/IIIa inhibitors: Eptifibatide, tirofiban, abciximab
Glycoprotein IIb/IIIa inhibitors are intravenous agents that act by inhibiting fibrinogen-and von Willebrand factor-mediated platelet-to-platelet cross-linkage, the final pathway of platelet aggregation (Figure 3).
Use of these agents in ACS has been decreasing, as evidence supporting their use was largely established before the era of dual antiplatelet therapy.
A meta-analysis29 of 46,374 patients with non-ST-elevation ACS found that routinely adding a glycoprotein IIb/IIIa inhibitor “upstream” as a third agent in patients receiving dual antiplatelet therapy bought only a modest (11%) reduction in death or myocardial infarction at 30 days, at the price of a 23% increase in major bleeding and no decrease in the overall rate of death. Roughly 70% of the patients were receiving dual antiplatelet therapy before cardiac catheterization.
These agents can be considered in high-risk ACS patients, such as those with ST-segment changes or elevated troponin concentrations, and in diabetic patients, on the assumption that these patients likely have a high intracoronary thrombus burden and are at higher risk of microvascular embolization.6,30 They can also be considered at the time of primary percutaneous coronary intervention in selected patients receiving heparin.7
Eptifibatide
Eptifibatide is a small-molecule, short-acting glycoprotein IIb/IIIa inhibitor with a half-life of 2.5 hours. Its inhibition of platelet aggregation is reversible by stopping the drug infusion and is thought to be a result of dissociation of the drug from platelets.
The PURSUIT trial31 studied 10,948 patients presenting with non-ST-elevation ACS randomized to placebo, eptifibatide in a 180-μg/kg bolus followed by a 2.0-μg/kg/min infusion, or eptifibatide in a 180-μg/kg bolus followed by a 1.3-μg/kg/min infusion. Both eptifibatide groups had a 1.5% absolute reduction in the incidence of the primary end point of death or myocardial infarction, a benefit that was apparent at 96 hours and that persisted through 30 days. Bleeding was more common in the eptifibatide groups, but there was no increase in the rate of hemorrhagic stroke.
The ACUITY trial32 found that early use of eptifibatide or tirofiban had no effect on the primary outcome. (See the section below on bivalirudin for more information about the ACUITY trial.)
PARENTERAL ANTICOAGULANTS
Unfractionated heparin: A declining role
Heparin binds to antithrombin and induces a conformational change, causing rapid inhibition of factor IIa (thrombin), factor IXa, and factor Xa, thus preventing further thrombus propagation (Figure 4). An intravenous bolus of 60 units/kg produces a time to peak of 5 to 10 minutes and a half-life of 30 to 60 minutes.
Heparin can be reversed by giving protamine sulfate (1 mg per 100 units of heparin). For ACS, it is given in a bolus of 60 units/kg not exceeding 4,000 units, followed by an infusion of 12 units/kg/hour, with monitoring of the activated partial thromboplastin time every 6 hours with a goal value of 50 to 70 seconds or 1.5 to 2.5 times control.
Side effects include thrombocytopenia, heparin-induced thrombocytopenia (a distinct condition), and bleeding.
The use of unfractionated heparin was tested in ACS in the early 1990s. Oler et al33 performed a meta-analysis of six randomized trials and found a 33% lower rate of death in patients treated with heparin in addition to aspirin in ACS, as well less reported ischemic pain.
Advantages of unfractionated heparin are that it has stood the test of time, is inexpensive, and can be rapidly reversed. The disadvantages are that it can have serious side effects, including heparin-induced thrombocytopenia, and is more likely to cause bleeding than the newer intravenous anticoagulants discussed below. Thus, its position as the main anticoagulant in ACS is being challenged.
Bivalirudin, a direct thrombin inhibitor
Bivalirudin is a synthetic direct thrombin inhibitor of fluid-phase and clot-bound thrombin (Figure 4). It also inhibits platelets directly.
The ACUITY trial32 randomized 13,819 patients with moderate to high-risk ACS scheduled for invasive treatment into three treatment groups:
- Heparin (either unfractionated heparin or enoxaparin) plus a glycoprotein IIb/IIIa inhibitor (either eptifibatide, tirofiban, or abciximab)
- Bivalirudin plus a glycoprotein IIb/IIIa inhibitor
- Bivalirudin alone.
The bivalirudin-alone treatment was as sociated with noninferior rates of composite ischemia end points and significantly lower rates of major bleeding, adding up to a significant reduction in the net clinical outcome end point. An important caveat is that bivalirudin’s noninferiority was mostly in the group of patients already receiving a thienopyridine before angiography and percutaneous coronary intervention (RR 0.97 vs 1.27, P = .054). There was less major, nonmajor, minor, CABG-related, and non-CABG-related bleeding as well as need for transfusion in the bivalirudin-alone group, making bivalirudin monotherapy an attractive option in ACS patients with or without ST-segment elevation undergoing a percutaneous coronary intervention.1,31
The ISAR-REACT trial34 later compared bivalirudin alone vs unfractionated heparin and abciximab in patients with non-ST-elevation myocardial infarction undergoing percutaneous coronary intervention pretreated with aspirin and clopidogrel. The composite rate of ischemia was similar in the two treatment groups, with significantly lower rates of bleeding in the bivalirudin group.
HORIZONS-AMI35 randomized 3,602 patients with ST-elevation myocardial infarction receiving aspirin and clopidogrel either to unfractionated heparin and a glycoprotein IIb/IIIa inhibitor or to bivalirudin. As in the ACUITY trial, there was no difference in ischemic end points and a 40% to 45% lower rate of major bleeding end points in the bivalirudin group, translating into an overall lower rate of death.
Enoxaparin, a low-molecular weight heparin
Enoxaparin is a low-molecular-weight heparin that inhibits factor IIa and factor Xa via antithrombin, roughly in a ratio of 1:3 (Figure 4). It has a time to peak effect of 10 minutes when given intravenously36 and 3 to 5 hours when given subcutaneously.37 Its half-life is 4.5 hours, but it is longer in patients with renal dysfunction, requiring dose adjustments in this population.
Its anticoagulant effect is partially reversible. If it is to be reversed between 0 and 8 hours after dosing, the recommended reversal regimen is 1 mg of protamine sulfate for every 1 mg of enoxaparin used. At 8 to 12 hours, it is 0.5 mg of protamine for every 1 mg of enoxaparin. After 12 hours, no protamine is required.
Compared with unfractionated heparin, enoxaparin has less plasma protein binding and a more consistent anticoagulant effect. Its high bioavailability also allows for subcutaneous dosing. Its greater anti-Xa activity inhibits thrombin generation more effectively, and it causes lower rates of thrombocytopenia and heparin-induced thrombocytopenia.
de Lemos et al38 found that, in ACS patients in whom an early conservative approach of medical management was planned, enoxaparin was more efficacious than unfractionated heparin and caused a similar rate of bleeding.
Murphy et al,39 in a meta-analysis of 12 trials in 49,088 ACS patients, also found that enoxaparin had a net clinical benefit compared with unfractionated heparin in reducing rates of myocardial infarction and death despite more bleeding.
The ESSENCE trial40 compared enoxaparin vs unfractionated heparin in 3,171 patients with ACS. It found fewer ischemic events with enoxaparin in the early phase, more minor bleeding, but no increase in major bleeding.
The SYNERGY trial,41 in 10,027 patients with high-risk non-ST-elevation ACS undergoing percutaneous coronary intervention, compared subcutaneous enoxaparin with intravenous heparin. Enoxaparin was found to be noninferior to heparin but caused more bleeding, including major bleeding, drops in hemoglobin, and intracranial hemorrhage.
The EXTRACT-TIMI 25 trial.42 In patients with ST-elevation myocardial infarction, enoxaparin has been shown to be beneficial both in patients treated with fibrinolysis and in those who underwent primary percutaneous coronary intervention. The EXTRACT-TIMI 25 trial randomized 20,749 patients to receive either enoxaparin (an intravenous bolus and maintenance subcutaneous dosing based on renal function) or intravenous heparin in addition to thrombolysis within 6 hours of the diagnosis of ST-elevation myocardial infarction. Although the enoxaparin group had more bleeding end points, they had fewer primary and secondary efficacy end points, translating into an overall net clinical benefit in favor of enoxaparin.
The ATOLL trial43 examined the use of enoxaparin (0.5 mg/kg intravenously) or unfractionated heparin in 910 patients with ST-elevation myocardial infarction undergoing primary percutaneous coronary intervention (via the radial artery in 66% to 69%). Although there was a trend towards benefit in terms of the primary end point of death, myocardial infarction complications, procedure failure, and major bleeding favoring enoxaparin, it was not statistically significant (95% CI 0.68–1.01, P = .06).
However, there was a 37% to 42% lower rate of the secondary end point of death, recurrent myocardial infarction or ACS, or urgent target-vessel revascularization in the enoxaparin group, with a 40% reduction in death from any cause, death from a cardiac cause, or shock. The safety profiles of the two drugs were similar, and the net clinical benefit significantly favored enoxaparin.
Fondaparinux, a factor Xa inhibitor
Fondaparinux is a synthetic pentasaccharide that indirectly inhibits factor Xa through the action of antithrombin (Figure 4). After a 2.5-mg subcutaneous dose, it has a time to peak concentration of 2 hours and a half-life of 17 to 21 hours.
The OASIS-5 trial44 compared fondaparinux and enoxaparin in 20,078 patients treated for non-ST-elevation ACS. Although the rates of death, myocardial infarction, and refractory ischemia at 9 days were similar for both drugs, the fondaparinux group had a significantly (almost 50%) lower rate of bleeding at 30 days, translating into significantly fewer deaths at 30 days. However, patients receiving fondaparinux who underwent percutaneous coronary intervention had a threefold higher rate of catheter-related thrombosis.
The OASIS-6 trial45 compared fondaparinux vs usual care (placebo in those in whom unfractionated heparin was not indicated or unfractionated heparin for up to 48 hours followed by placebo for up to 8 days) in 12,092 patients with ST-elevation myocardial infarction. There was a 1.5% absolute risk reduction in death and reinfarction without an increase in bleeding at 30 days, with trends persisting 6 months into the study. However, fondaparinux was not superior to heparin in the 3% of patients who underwent primary percutaneous coronary intervention. As in OASIS-5, there was more catheter-related thrombosis in the fondaparinux group.
Although the use of supplemental unfractionated heparin appears to have mitigated this risk, fondaparinux remains a less-than-ideal option in the era of primary percutaneous coronary intervention for ST-elevation myocardial infarction and has therefore found limited use in this group of patients. It should, however, be considered in patients for whom a conservative strategy is planned, especially if bleeding risk is deemed to be high.
ORAL ANTICOAGULANTS
Oral anticoagulants provide ischemic benefit in selected patients with ACS—at the price of a higher risk of significant bleeding.
Warfarin
Warfarin was investigated after myocardial infarction in the WARIS II,46 CARS,47 and CHAMP48 trials.
WARIS II46 looked at the use of aspirin alone, warfarin alone, and aspirin and warfarin in combination. The rates of the primary end points of stroke, nonfatal infarction, and death were lower in the warfarin group.
CARS47 found no difference in the rate of the primary end point of fatal infarction, nonfatal ischemic stroke, or cardiovascular death with aspirin vs warfarin plus aspirin.
CHAMP48 saw similar trends, ie, no difference in the rate of death, recurrent myocardial infarction, or stroke with warfarin plus aspirin vs aspirin alone.
All three studies showed increases in major bleeding with warfarin use.
Putting these trials into context, the significant net clinical benefit of dual antiplatelet therapy in the current era compared with the significant bleeding and questionable conflicting evidence supporting benefit with warfarin has limited its use in ACS patients.
Rivaroxaban, an oral factor Xa inhibitor
Rivaroxaban is a novel oral direct reversible factor Xa inhibitor.
The ATLAS ACS 2-TIMI 51 trial49 found rivaroxaban 2.5 mg or 5 mg to yield a significantly lower rate of the primary outcome of cardiovascular death, myocardial infarction, ischemic stroke, and in-stent thrombosis compared with placebo, but significantly more major non-CABG bleeding and intracranial hemorrhage.
The dose used in this trial was much lower than the dose used in trials investigating the role of this drug in stroke prophylaxis in atrial fibrillation.
Apixaban, an oral factor Xa inhibitor
Apixaban is another direct factor Xa inhibitor.
The APPRAISE-2 trial50 compared apixaban 5 mg twice daily vs placebo in ACS. There was no difference in the rate of cardiovascular death, myocardial infarction, or stroke, but there was significantly more bleeding in the apixaban group, prompting early termination of this study.
Dabigatran, an oral thrombin inhibitor
Dabigatran is an oral direct thrombin inhibitor.
The RE-DEEM trial51 compared four doses of dabigatran (50, 75, 110, and 150 mg twice daily) and placebo in ACS patients. The dabigatran groups had more major and minor bleeding, and the higher the dose, the higher the incidence of bleeding. In addition, the rates of ischemic end points were no lower with dabigatran, although this trial was not powered to show differences in clinical events.
REDUCING THE RISK OF BLEEDING
In the treatment of ACS, the benefits of restoring perfusion by preventing further propagation of thrombus and platelet aggregation come at a significant price of higher bleeding risk. This in turn increases the risk of death through various mechanisms, including shock, worsening ischemia, discontinuation of antiplatelet and anticoagulation therapy causing stent thrombosis, and anemia leading to transfusion, which propagates the underlying inflammatory milieu.52
Giugliano and Braunwald53 provide practical suggestions to reduce this risk, advising physicians to:
- Avoid inappropriately high dosing, particularly in patients with renal insufficiency
- Preferentially use agents that cause less bleeding (eg, bivalirudin, fondaparinux) without compromising anti-ischemic efficacy
- Minimize the concomitant use of other drugs that cause bleeding (eg, NSAIDs)
- Use drugs that protect against bleeding (eg, proton pump inhibitors) in patients at high risk
- Prevent access-site bleeding by using the radial artery, smaller sheaths, and appropriate sheath and closure device management. Indeed, the use of radial interventions in ACS has been shown to reduce access-site-related bleeding, even in patients at high risk.54
The reduction in bleeding risk may provide future trials the opportunity to increase antithrombotic efficacy of different agents with goals of reducing ischemic end points.
Antiplatelet and anticoagulant drugs are a cornerstone of the medical treatment of acute coronary syndrome (ACS), reducing the rates of both morbidity and death.1–4 However, reductions in ischemic events with these drugs have uniformly been accompanied by increases in bleeding complications, which reduce the net benefit.5 Thus, clinical research has been exploring ways to maximize the benefit while minimizing the risk.
Here, we review the guidelines and evidence supporting the use of antiplatelet and anticoagulant drugs in ACS.
ACUTE CORONARY SYNDROMES WITH OR WITHOUT ST ELEVATION
A key distinction when treating ACS is whether the electrocardiogram shows ST-segment elevation. In cases of non-ST-elevation ACS (ie, unstable angina or non-ST-elevation myocardial infarction), a second key question is whether the initial strategy will be invasive (with angiography performed urgently) or conservative (with angiography performed later). In ST-elevation myocardial infarction, another distinction is how perfusion is to be restored, ie, with primary percutaneous coronary intervention or with thrombolysis. All these questions affect the choice of antiplatelet and anticoagulant therapy.
Figure 1 and Figure 2 summarize the guidelines of the American College of Cardiology Foundation and American Heart Association.1,2,6,7
ANTIPLATELET THERAPY
Aspirin for all
Aspirin irreversibly acetylates the enzyme cyclooxygenase-1, blocking intraplatelet formation of thromboxane A2 (Figure 3), a potent platelet aggregator and endothelial vasoconstrictor. Large clinical trials have confirmed that aspirin reduces morbidity and mortality rates by as much as 50% in patients with ACS.8
The ISIS-2 trial9 found that giving aspirin early in the emergency department significantly reduced the mortality rate.
The Antithrombotic Trialists’ Collaboration,10 in a meta-analysis of randomized controlled trials comparing different doses of aspirin in high-risk ACS patients, found no greater benefit for doses of aspirin higher than 162 mg per day when used long-term.
How to use. During an ACS, the patient should receive one dose of aspirin 325 mg (the standard high-dose pill in the United States). This dose should be chewed, as buccal absorption results in more rapid systemic effects.11
Thereafter, the patient should take 81 mg per day, continued indefinitely. The 81-mg dose also applies to patients who undergo a percutaneous coronary intervention with a drug-eluting stent.7 Previous recommendations called for higher doses, but studies have shown that higher doses pose a higher risk of bleeding without additional clinical benefit. The use of enteric-coated aspirin does not reduce this risk,12 and its delayed release may in fact cause aspirin “pseudoresistance.”13
The concurrent use of nonsteroidal anti-inflammatory drugs (NSAIDs) should be avoided, as NSAIDs reversibly bind to platelets, thus preventing aspirin from binding.14 As aspirin washes out of the body, NSAIDs may then become unbound from platelets, leaving platelets activated.
P2Y12 receptor inhibitors: Clopidogrel, prasugrel, ticagrelor
These agents bind to P2Y12 receptors on platelets to inhibit adenosine diphosphate-mediated platelet activation (Figure 3). Clopidogrel and prasugrel are irreversible prodrugs, whereas ticagrelor binds reversibly.
Clopidogrel, a prodrug
Clopidogrel has a half-life of 8 hours and a time to peak concentration of 4 hours. Eighty-five percent of a dose is inactivated by gut esterases. The remainder is metabolized primarily by the cytochrome P4502C19 enzyme system into its active metabolite.
How to use. The recommended dosage is a 600-mg bolus early in the course of ACS. This is associated with a lower rate of cardiovascular events than a 300-mg dose,2,15 although no trial has rigorously compared 300-mg vs 600-mg doses using major clinical end points. In patients presenting with ACS who cannot tolerate aspirin because of hypersensitivity or major gastrointestinal contraindication, clopidogrel is an alternative.1
The CURE trial16 randomized 12,526 patients with non-ST-elevation ACS to receive clopidogrel or placebo in addition to standard therapy. Clopidogrel was associated with a 20% lower rate of cardiovascular death, myocardial infarction, or stroke in both low- and high-risk patients regardless of whether an invasive or conservative strategy was pursued.
However, patients who underwent coronary artery bypass grafting (CABG) had a 53% higher risk of bleeding (an absolute risk of 3.3%) if they received clopidogrel within 5 days of the surgery. This has led to the practice in some centers of delaying giving clopidogrel until after the coronary anatomy has been defined. This deprives the patient of the anti-ischemic benefits conferred by giving clopidogrel early and remains a contentious issue, with most suggesting that the risk-benefit ratio still favors giving clopidogrel early, before angiography, unless there is a high likelihood that surgery will ultimately be required.17 Alternatively, one could consider using a shorter-acting intravenous glycoprotein IIb/IIIa inhibitor such as eptifibatide as a “bridge” until a definitive reperfusion strategy is chosen.
Effect of CYP2C19 variants. The CLOVIS-2 study18 assessed the effects of genetic variants on the clopidogrel concentration in 106 patients who had had a myocardial infarction. The study confirmed that patients who carry certain variants of the CYP2C19 gene attain lower plasma concentrations of clopidogrel after receiving this drug.19 This accounts for its delayed onset of action as well as its variability in response in patients who have reduced expression or inhibition of this enzyme system. Doubling the standard dose in patients who carry these variants does not appear to provide clinical benefit.20
Thus, the thought is emerging that one should consider using prasugrel or ticagrelor instead of clopidogrel in patients who have these polymorphisms, though this is yet to be backed by robust clinical evidence.
Possible interaction with proton pump inhibitors. Controversy exists about whether proton pump inhibitors inhibit clopidogrel’s action. Although the US Food and Drug Administration continues to warn against the concurrent use of omeprazole and clopidogrel,21 an analysis of the PLATO trial22 concluded that patients with ACS who were taking proton pump inhibitors were at higher risk of ischemic events regardless of whether they had been randomized to clopidogrel or ticagrelor (a drug that acts independently of the cytochrome P450 system). This observation suggests that patients on proton pump inhibitors are generally sicker and at higher risk of ischemic events regardless of the choice of antiplatelet therapy. The use of other gastroprotective agents did not appear to mitigate these risks.
Prasugrel: Faster metabolism to active drug
Prasugrel is an irreversible P2Y12 receptor antagonist (Figure 3) that is metabolized into its active metabolite faster and in a more predictable fashion than clopidogrel.23
The TRITON-TIMI 38 study24 included 13,608 ACS patients in whom an early invasive strategy was planned and who were pretreated with prasugrel or clopidogrel in addition to standard treatment. The rate of the primary efficacy end point of death, myocardial infarction, or stroke was 19% lower in the prasugrel group. In those who underwent percutaneous coronary intervention, the incidence of in-stent thrombosis was more than 50% lower in the prasugrel group regardless of whether bare metal stents or drug-eluting stents were used.
Greater platelet inhibition came at the price of a higher incidence of serious bleeding, particularly in the subgroups of patients who were over age 75, had a history of stroke or transient ischemic attack, or weighed less than 60 kg. Prasugrel is therefore contraindicated in patients with a history of transient ischemic attack or stroke. Some suggest that a 5-mg dose can be used with caution (rather than the usual 10-mg dose) in patients over age 75 years or those who have low body weight.
The TRILOGY-ACS trial25 compared prasugrel and clopidogrel in medically managed patients with high-risk non-ST-elevation ACS. It found no difference in the rates of the primary end points of cardiovascular death, myocardial infarction, or stroke at 1 year. In the prespecified subset of patients over age 75 years, the rate of bleeding end points was no higher with prasugrel 5 mg once daily than with clopidogrel.
Prasugrel’s half-life is 7 hours, and its peak antiplatelet effect is within 30 minutes after an oral dose, compared with 4 hours with clopidogrel. Therefore, if a patient with non-ST-elevation ACS is going to go to the catheterization laboratory soon, he or she should not receive prasugrel beforehand, and should receive it later only if the results of angiography indicate that CABG will not be needed urgently. This is an important consideration when using prasugrel, as the rate of surgery-related bleeding was four times higher than with clopidogrel. If possible, this drug should be withheld for at least 7 days before CABG.
Ticagrelor, a direct P2Y12 receptor inhibitor
Ticagrelor, a reversible direct inhibitor of the P2Y12 receptor, inhibits adenosine diphosphate-mediated activation and aggregation (Figure 3). It has a median time to peak concentration of 1.3 to 2 hours and a half-life of 9 hours.
The PLATO trial26 enrolled 18,624 patients with ACS who were given either ticagrelor or clopidogrel in addition to standard therapy. At 12 months, the composite primary end point of myocardial infarction, death, or stroke had occurred in 16% fewer patients receiving ticagrelor than in the clopidogrel group. Analyzed separately, there were 16% fewer myocardial infarctions, 21% fewer cardiovascular deaths, and 22% fewer deaths from any cause, regardless of whether an invasive or conservative strategy was used, and with or without prior clopidogrel use. Fewer cases of stent thrombosis occurred in the ticagrelor group, and the rate of major bleeding was the same.
In a prospectively defined subgroup analysis,27 ticagrelor was beneficial only in patients who received lower doses of aspirin (< 100 mg daily): the hazard ratio for the primary end point was 0.79 (95% confidence interval [CI] 0.71–0.88) in ticagrelor recipients who received low-dose aspirin and 1.45 (95% CI 1.01–2.09) in those who received high-dose aspirin.
Although this analysis is underpowered and controversial, the current evidence suggests that when used in combination with ticagrelor, the aspirin dose should be 81 mg.
Ticagrelor was also associated with a 19% higher incidence of non-CABG- or procedure-related major bleeding, more nonfatal and fatal intracranial bleeding, a higher incidence of dyspnea, and significantly more ventricular pauses.
Although ticagrelor carries no black-box warning about its use in patients with prior stroke or transient ischemic attack, the number of such patients in PLATO was small. Thus, caution should still be used in these patients.28
Ticagrelor should preferably be discontinued 5 days before CABG.
Glycoprotein IIb/IIIa inhibitors: Eptifibatide, tirofiban, abciximab
Glycoprotein IIb/IIIa inhibitors are intravenous agents that act by inhibiting fibrinogen-and von Willebrand factor-mediated platelet-to-platelet cross-linkage, the final pathway of platelet aggregation (Figure 3).
Use of these agents in ACS has been decreasing, as evidence supporting their use was largely established before the era of dual antiplatelet therapy.
A meta-analysis29 of 46,374 patients with non-ST-elevation ACS found that routinely adding a glycoprotein IIb/IIIa inhibitor “upstream” as a third agent in patients receiving dual antiplatelet therapy bought only a modest (11%) reduction in death or myocardial infarction at 30 days, at the price of a 23% increase in major bleeding and no decrease in the overall rate of death. Roughly 70% of the patients were receiving dual antiplatelet therapy before cardiac catheterization.
These agents can be considered in high-risk ACS patients, such as those with ST-segment changes or elevated troponin concentrations, and in diabetic patients, on the assumption that these patients likely have a high intracoronary thrombus burden and are at higher risk of microvascular embolization.6,30 They can also be considered at the time of primary percutaneous coronary intervention in selected patients receiving heparin.7
Eptifibatide
Eptifibatide is a small-molecule, short-acting glycoprotein IIb/IIIa inhibitor with a half-life of 2.5 hours. Its inhibition of platelet aggregation is reversible by stopping the drug infusion and is thought to be a result of dissociation of the drug from platelets.
The PURSUIT trial31 studied 10,948 patients presenting with non-ST-elevation ACS randomized to placebo, eptifibatide in a 180-μg/kg bolus followed by a 2.0-μg/kg/min infusion, or eptifibatide in a 180-μg/kg bolus followed by a 1.3-μg/kg/min infusion. Both eptifibatide groups had a 1.5% absolute reduction in the incidence of the primary end point of death or myocardial infarction, a benefit that was apparent at 96 hours and that persisted through 30 days. Bleeding was more common in the eptifibatide groups, but there was no increase in the rate of hemorrhagic stroke.
The ACUITY trial32 found that early use of eptifibatide or tirofiban had no effect on the primary outcome. (See the section below on bivalirudin for more information about the ACUITY trial.)
PARENTERAL ANTICOAGULANTS
Unfractionated heparin: A declining role
Heparin binds to antithrombin and induces a conformational change, causing rapid inhibition of factor IIa (thrombin), factor IXa, and factor Xa, thus preventing further thrombus propagation (Figure 4). An intravenous bolus of 60 units/kg produces a time to peak of 5 to 10 minutes and a half-life of 30 to 60 minutes.
Heparin can be reversed by giving protamine sulfate (1 mg per 100 units of heparin). For ACS, it is given in a bolus of 60 units/kg not exceeding 4,000 units, followed by an infusion of 12 units/kg/hour, with monitoring of the activated partial thromboplastin time every 6 hours with a goal value of 50 to 70 seconds or 1.5 to 2.5 times control.
Side effects include thrombocytopenia, heparin-induced thrombocytopenia (a distinct condition), and bleeding.
The use of unfractionated heparin was tested in ACS in the early 1990s. Oler et al33 performed a meta-analysis of six randomized trials and found a 33% lower rate of death in patients treated with heparin in addition to aspirin in ACS, as well less reported ischemic pain.
Advantages of unfractionated heparin are that it has stood the test of time, is inexpensive, and can be rapidly reversed. The disadvantages are that it can have serious side effects, including heparin-induced thrombocytopenia, and is more likely to cause bleeding than the newer intravenous anticoagulants discussed below. Thus, its position as the main anticoagulant in ACS is being challenged.
Bivalirudin, a direct thrombin inhibitor
Bivalirudin is a synthetic direct thrombin inhibitor of fluid-phase and clot-bound thrombin (Figure 4). It also inhibits platelets directly.
The ACUITY trial32 randomized 13,819 patients with moderate to high-risk ACS scheduled for invasive treatment into three treatment groups:
- Heparin (either unfractionated heparin or enoxaparin) plus a glycoprotein IIb/IIIa inhibitor (either eptifibatide, tirofiban, or abciximab)
- Bivalirudin plus a glycoprotein IIb/IIIa inhibitor
- Bivalirudin alone.
The bivalirudin-alone treatment was as sociated with noninferior rates of composite ischemia end points and significantly lower rates of major bleeding, adding up to a significant reduction in the net clinical outcome end point. An important caveat is that bivalirudin’s noninferiority was mostly in the group of patients already receiving a thienopyridine before angiography and percutaneous coronary intervention (RR 0.97 vs 1.27, P = .054). There was less major, nonmajor, minor, CABG-related, and non-CABG-related bleeding as well as need for transfusion in the bivalirudin-alone group, making bivalirudin monotherapy an attractive option in ACS patients with or without ST-segment elevation undergoing a percutaneous coronary intervention.1,31
The ISAR-REACT trial34 later compared bivalirudin alone vs unfractionated heparin and abciximab in patients with non-ST-elevation myocardial infarction undergoing percutaneous coronary intervention pretreated with aspirin and clopidogrel. The composite rate of ischemia was similar in the two treatment groups, with significantly lower rates of bleeding in the bivalirudin group.
HORIZONS-AMI35 randomized 3,602 patients with ST-elevation myocardial infarction receiving aspirin and clopidogrel either to unfractionated heparin and a glycoprotein IIb/IIIa inhibitor or to bivalirudin. As in the ACUITY trial, there was no difference in ischemic end points and a 40% to 45% lower rate of major bleeding end points in the bivalirudin group, translating into an overall lower rate of death.
Enoxaparin, a low-molecular weight heparin
Enoxaparin is a low-molecular-weight heparin that inhibits factor IIa and factor Xa via antithrombin, roughly in a ratio of 1:3 (Figure 4). It has a time to peak effect of 10 minutes when given intravenously36 and 3 to 5 hours when given subcutaneously.37 Its half-life is 4.5 hours, but it is longer in patients with renal dysfunction, requiring dose adjustments in this population.
Its anticoagulant effect is partially reversible. If it is to be reversed between 0 and 8 hours after dosing, the recommended reversal regimen is 1 mg of protamine sulfate for every 1 mg of enoxaparin used. At 8 to 12 hours, it is 0.5 mg of protamine for every 1 mg of enoxaparin. After 12 hours, no protamine is required.
Compared with unfractionated heparin, enoxaparin has less plasma protein binding and a more consistent anticoagulant effect. Its high bioavailability also allows for subcutaneous dosing. Its greater anti-Xa activity inhibits thrombin generation more effectively, and it causes lower rates of thrombocytopenia and heparin-induced thrombocytopenia.
de Lemos et al38 found that, in ACS patients in whom an early conservative approach of medical management was planned, enoxaparin was more efficacious than unfractionated heparin and caused a similar rate of bleeding.
Murphy et al,39 in a meta-analysis of 12 trials in 49,088 ACS patients, also found that enoxaparin had a net clinical benefit compared with unfractionated heparin in reducing rates of myocardial infarction and death despite more bleeding.
The ESSENCE trial40 compared enoxaparin vs unfractionated heparin in 3,171 patients with ACS. It found fewer ischemic events with enoxaparin in the early phase, more minor bleeding, but no increase in major bleeding.
The SYNERGY trial,41 in 10,027 patients with high-risk non-ST-elevation ACS undergoing percutaneous coronary intervention, compared subcutaneous enoxaparin with intravenous heparin. Enoxaparin was found to be noninferior to heparin but caused more bleeding, including major bleeding, drops in hemoglobin, and intracranial hemorrhage.
The EXTRACT-TIMI 25 trial.42 In patients with ST-elevation myocardial infarction, enoxaparin has been shown to be beneficial both in patients treated with fibrinolysis and in those who underwent primary percutaneous coronary intervention. The EXTRACT-TIMI 25 trial randomized 20,749 patients to receive either enoxaparin (an intravenous bolus and maintenance subcutaneous dosing based on renal function) or intravenous heparin in addition to thrombolysis within 6 hours of the diagnosis of ST-elevation myocardial infarction. Although the enoxaparin group had more bleeding end points, they had fewer primary and secondary efficacy end points, translating into an overall net clinical benefit in favor of enoxaparin.
The ATOLL trial43 examined the use of enoxaparin (0.5 mg/kg intravenously) or unfractionated heparin in 910 patients with ST-elevation myocardial infarction undergoing primary percutaneous coronary intervention (via the radial artery in 66% to 69%). Although there was a trend towards benefit in terms of the primary end point of death, myocardial infarction complications, procedure failure, and major bleeding favoring enoxaparin, it was not statistically significant (95% CI 0.68–1.01, P = .06).
However, there was a 37% to 42% lower rate of the secondary end point of death, recurrent myocardial infarction or ACS, or urgent target-vessel revascularization in the enoxaparin group, with a 40% reduction in death from any cause, death from a cardiac cause, or shock. The safety profiles of the two drugs were similar, and the net clinical benefit significantly favored enoxaparin.
Fondaparinux, a factor Xa inhibitor
Fondaparinux is a synthetic pentasaccharide that indirectly inhibits factor Xa through the action of antithrombin (Figure 4). After a 2.5-mg subcutaneous dose, it has a time to peak concentration of 2 hours and a half-life of 17 to 21 hours.
The OASIS-5 trial44 compared fondaparinux and enoxaparin in 20,078 patients treated for non-ST-elevation ACS. Although the rates of death, myocardial infarction, and refractory ischemia at 9 days were similar for both drugs, the fondaparinux group had a significantly (almost 50%) lower rate of bleeding at 30 days, translating into significantly fewer deaths at 30 days. However, patients receiving fondaparinux who underwent percutaneous coronary intervention had a threefold higher rate of catheter-related thrombosis.
The OASIS-6 trial45 compared fondaparinux vs usual care (placebo in those in whom unfractionated heparin was not indicated or unfractionated heparin for up to 48 hours followed by placebo for up to 8 days) in 12,092 patients with ST-elevation myocardial infarction. There was a 1.5% absolute risk reduction in death and reinfarction without an increase in bleeding at 30 days, with trends persisting 6 months into the study. However, fondaparinux was not superior to heparin in the 3% of patients who underwent primary percutaneous coronary intervention. As in OASIS-5, there was more catheter-related thrombosis in the fondaparinux group.
Although the use of supplemental unfractionated heparin appears to have mitigated this risk, fondaparinux remains a less-than-ideal option in the era of primary percutaneous coronary intervention for ST-elevation myocardial infarction and has therefore found limited use in this group of patients. It should, however, be considered in patients for whom a conservative strategy is planned, especially if bleeding risk is deemed to be high.
ORAL ANTICOAGULANTS
Oral anticoagulants provide ischemic benefit in selected patients with ACS—at the price of a higher risk of significant bleeding.
Warfarin
Warfarin was investigated after myocardial infarction in the WARIS II,46 CARS,47 and CHAMP48 trials.
WARIS II46 looked at the use of aspirin alone, warfarin alone, and aspirin and warfarin in combination. The rates of the primary end points of stroke, nonfatal infarction, and death were lower in the warfarin group.
CARS47 found no difference in the rate of the primary end point of fatal infarction, nonfatal ischemic stroke, or cardiovascular death with aspirin vs warfarin plus aspirin.
CHAMP48 saw similar trends, ie, no difference in the rate of death, recurrent myocardial infarction, or stroke with warfarin plus aspirin vs aspirin alone.
All three studies showed increases in major bleeding with warfarin use.
Putting these trials into context, the significant net clinical benefit of dual antiplatelet therapy in the current era compared with the significant bleeding and questionable conflicting evidence supporting benefit with warfarin has limited its use in ACS patients.
Rivaroxaban, an oral factor Xa inhibitor
Rivaroxaban is a novel oral direct reversible factor Xa inhibitor.
The ATLAS ACS 2-TIMI 51 trial49 found rivaroxaban 2.5 mg or 5 mg to yield a significantly lower rate of the primary outcome of cardiovascular death, myocardial infarction, ischemic stroke, and in-stent thrombosis compared with placebo, but significantly more major non-CABG bleeding and intracranial hemorrhage.
The dose used in this trial was much lower than the dose used in trials investigating the role of this drug in stroke prophylaxis in atrial fibrillation.
Apixaban, an oral factor Xa inhibitor
Apixaban is another direct factor Xa inhibitor.
The APPRAISE-2 trial50 compared apixaban 5 mg twice daily vs placebo in ACS. There was no difference in the rate of cardiovascular death, myocardial infarction, or stroke, but there was significantly more bleeding in the apixaban group, prompting early termination of this study.
Dabigatran, an oral thrombin inhibitor
Dabigatran is an oral direct thrombin inhibitor.
The RE-DEEM trial51 compared four doses of dabigatran (50, 75, 110, and 150 mg twice daily) and placebo in ACS patients. The dabigatran groups had more major and minor bleeding, and the higher the dose, the higher the incidence of bleeding. In addition, the rates of ischemic end points were no lower with dabigatran, although this trial was not powered to show differences in clinical events.
REDUCING THE RISK OF BLEEDING
In the treatment of ACS, the benefits of restoring perfusion by preventing further propagation of thrombus and platelet aggregation come at a significant price of higher bleeding risk. This in turn increases the risk of death through various mechanisms, including shock, worsening ischemia, discontinuation of antiplatelet and anticoagulation therapy causing stent thrombosis, and anemia leading to transfusion, which propagates the underlying inflammatory milieu.52
Giugliano and Braunwald53 provide practical suggestions to reduce this risk, advising physicians to:
- Avoid inappropriately high dosing, particularly in patients with renal insufficiency
- Preferentially use agents that cause less bleeding (eg, bivalirudin, fondaparinux) without compromising anti-ischemic efficacy
- Minimize the concomitant use of other drugs that cause bleeding (eg, NSAIDs)
- Use drugs that protect against bleeding (eg, proton pump inhibitors) in patients at high risk
- Prevent access-site bleeding by using the radial artery, smaller sheaths, and appropriate sheath and closure device management. Indeed, the use of radial interventions in ACS has been shown to reduce access-site-related bleeding, even in patients at high risk.54
The reduction in bleeding risk may provide future trials the opportunity to increase antithrombotic efficacy of different agents with goals of reducing ischemic end points.
- Wright RS, Anderson JL, Adams CD, et al; American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. 2011 ACCF/AHA focused update incorporated into the ACC/AHA 2007 Guidelines for the Management of Patients with Unstable Angina/Non-ST-Elevation Myocardial Infarction. J Am Coll Cardiol 2011; 57:e215–e367.
- Jneid H, Anderson JL, Wright RS, et al. 2012 ACCF/AHA focused update of the guideline for the management of patients with unstable angina/non-ST-elevation myocardial infarction (updating the 2007 guideline and replacing the 2011 focused update): a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2012; 60:645–681.
- CAPRIE Steering Committee. A randomised, blinded, trial of clopidogrel versus aspirin in patients at risk of ischaemic events (CAPRIE). CAPRIE Steering Committee. Lancet 1996; 348:1329–1339.
- Cohen M, Adams PC, Parry G, et al. Combination antithrombotic therapy in unstable rest angina and non-Q-wave infarction in nonprior aspirin users. Primary end points analysis from the ATACS trial. Antithrombotic Therapy in Acute Coronary Syndromes Research Group. Circulation 1994; 89:81–88.
- Moscucci M, Fox KA, Cannon CP, et al. Predictors of major bleeding in acute coronary syndromes: the Global Registry of Acute Coronary Events (GRACE). Eur Heart J 2003; 24:1815–1823.
- Levine GN, Bates ER, Blankenship JC, et al; American College of Cardiology Foundation; American Heart Association Task Force on Practice Guidelines; Society for Cardiovascular Angiography and Interventions. 2011 ACCF/AHA/SCAI Guideline for Percutaneous Coronary Intervention. A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines and the Society for Cardiovascular Angiography and Interventions. J Am Coll Cardiol 2011; 58:e44–e122.
- American College of Emergency Physicians; Society for Cardiovascular Angiography and Interventions; O’Gara PT, Kushner FG, Ascheim DD, et al. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013; 61:485–510.
- Lewis HD, Davis JW, Archibald DG, et al. Protective effects of aspirin against acute myocardial infarction and death in men with unstable angina. Results of a Veterans Administration Cooperative Study. N Engl J Med 1983; 309:396–403.
- ISIS-2 (Second International Study of Infarct Survival) Collaborative Group. Randomised trial of intravenous streptokinase, oral aspirin, both, or neither among 17,187 cases of suspected acute myocardial infarction: ISIS-2. Lancet 1988; 2:349–360.
- Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002; 324:71–86.
- Sweeny JM, Gorog DA, Fuster V. Antiplatelet drug ‘resistance’. Part 1: mechanisms and clinical measurements. Nat Rev Cardiol 2009; 6:273–282.
- Kelly JP, Kaufman DW, Jurgelon JM, Sheehan J, Koff RS, Shapiro S. Risk of aspirin-associated major upper-gastrointestinal bleeding with enteric-coated or buffered product. Lancet 1996; 348:1413–1416.
- Grosser T, Fries S, Lawson JA, Kapoor SC, Grant GR, FitzGerald GA. Drug resistance and pseudoresistance: an unintended consequence of enteric coating aspirin. Circulation 2013; 127:377–385.
- US Food and Drug Administration (FDA). Concomitant use of ibuprofen and aspirin: potential for attenuation of the anti-platelet effect of aspirin. http://www.fda.gov/downloads/drugs/drugsafety/postmarketdrugsafetyinformationforpatientsandproviders/ucm161282.pdf. Accessed November 30, 2013.
- Patti G, Colonna G, Pasceri V, Pepe LL, Montinaro A, Di Sciascio G. Randomized trial of high loading dose of clopidogrel for reduction of periprocedural myocardial infarction in patients undergoing coronary intervention: results from the ARMYDA-2 (Antiplatelet therapy for Reduction of MYocardial Damage during Angioplasty) study. Circulation 2005; 111:2099–2106.
- Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK; Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial Investigators. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med 2001; 345:494–502.
- Bavry AA, Lincoff AM. Is clopidogrel cardiovascular medicine’s double-edged sword? Circulation 2006; 113:1638–1640.
- Collet JP, Hulot JS, Anzaha G, et al; CLOVIS-2 Investigators. High doses of clopidogrel to overcome genetic resistance: the randomized crossover CLOVIS-2 (Clopidogrel and Response Variability Investigation Study 2). JACC Cardiovasc Interv 2011; 4:392–402.
- Hulot JS, Collet JP, Cayla G, et al. CYP2C19 but not PON1 genetic variants influence clopidogrel pharmacokinetics, pharmacodynamics, and clinical efficacy in post-myocardial infarction patients. Circ Cardiovasc Interv 2011; 4:422–428.
- Cuisset T, Quilici J, Cohen W, et al. Usefulness of high clopidogrel maintenance dose according to CYP2C19 genotypes in clopidogrel low responders undergoing coronary stenting for non ST elevation acute coronary syndrome. Am J Cardiol 2011; 108:760–765.
- US Food and Drug Administration (FDA). FDA reminder to avoid concomitant use of Plavix (clopidogrel) and omeprazole. http://www.fda.gov/Drugs/DrugSafety/ucm231161.htm. Accessed November 30, 2013.
- Goodman SG, Clare R, Pieper KS, et al; Platelet Inhibition and Patient Outcomes Trial Investigators. Association of proton pump inhibitor use on cardiovascular outcomes with clopidogrel and ticagrelor: insights from the platelet inhibition and patient outcomes trial. Circulation 2012; 125:978–986.
- Solomon S, Vacek JL. Reducing cardiac ischemic events in patients with ACS: prasugrel versus clopidogrel. Commentary. Postgrad Med 2010; 122:198–200.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Roe MT, Armstrong PW, Fox KA, et al; TRILOGY ACS Investigators. Prasugrel versus clopidogrel for acute coronary syndromes without revascularization. N Engl J Med 2012; 367:1297–1309.
- Wallentin L, Becker RC, Budaj A, et al; PLATO Investigators. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2009; 361:1045–1057.
- Mahaffey KW, Wojdyla DM, Carroll K, et al; PLATO Investigators. Ticagrelor compared with clopidogrel by geographic region in the Platelet Inhibition and Patient Outcomes (PLATO) trial. Circulation 2011; 124:544–554.
- Verheugt FW. Beware of novel antiplatelet therapy in acute coronary syndrome patients with previous stroke. Circulation 2012; 125:2821–2823.
- Tricoci P, Newby LK, Hasselblad V, et al. Upstream use of small-molecule glycoprotein iib/iiia inhibitors in patients with non-ST-segment elevation acute coronary syndromes: a systematic overview of randomized clinical trials. Circ Cardiovasc Qual Outcomes 2011; 4:448–458.
- Kastrati A, Mehilli J, Neumann FJ, et al; Intracoronary Stenting and Antithrombotic: Regimen Rapid Early Action for Coronary Treatment 2 (ISAR-REACT 2) Trial Investigators. Abciximab in patients with acute coronary syndromes undergoing percutaneous coronary intervention after clopidogrel pretreatment: the ISAR-REACT 2 randomized trial. JAMA 2006; 295:1531–1538.
- Inhibition of platelet glycoprotein IIb/IIIa with eptifibatide in patients with acute coronary syndromes. The PURSUIT Trial Investigators. Platelet Glycoprotein IIb/IIIa in Unstable Angina: Receptor Suppression Using Integrilin Therapy. N Engl J Med 1998; 339:436–443.
- Stone GW, McLaurin BT, Cox DA, et al; ACUITY Investigators. Bivalirudin for patients with acute coronary syndromes. N Engl J Med 2006; 355:2203–2216.
- Oler A, Whooley MA, Oler J, Grady D. Adding heparin to aspirin reduces the incidence of myocardial infarction and death in patients with unstable angina. A meta-analysis. JAMA 1996; 276:811–815.
- Kastrati A, Neumann FJ, Schulz S, et al; ISAR-REACT 4 Trial Investigators. Abciximab and heparin versus bivalirudin for non-ST-elevation myocardial infarction. N Engl J Med 2011; 365:1980–1989.
- Stone GW, Witzenbichler B, Guagliumi G, et al; HORIZONS-AMI Trial Investigators. Bivalirudin during primary PCI in acute myocardial infarction. N Engl J Med 2008; 358:2218–2230.
- Aslam MS, Sundberg S, Sabri MN, Cooke D, Lakier JB. Pharmacokinetics of intravenous/subcutaneous enoxaparin in patients with acute coronary syndrome undergoing percutaneous coronary interventions. Catheter Cardiovasc Interv 2002; 57:187–190.
- Sanofi-Aventis US. Lovenox (enoxaparin sodium injection) product information. http://www.lovenox.com/hcp/clinical-data.aspx. Accessed December 1, 2013.
- de Lemos JA, Blazing MA, Wiviott SD, et al. Enoxaparin versus unfractionated heparin in patients treated with tirofiban, aspirin and an early conservative initial management strategy: results from the A phase of the A-to-Z trial. Eur Heart J 2004; 25:1688–1694.
- Murphy SA, Gibson CM, Morrow DA, et al. Efficacy and safety of the low-molecular weight heparin enoxaparin compared with unfractionated heparin across the acute coronary syndrome spectrum: a meta-analysis. Eur Heart J 2007; 28:2077–2086.
- Cohen M, Demers C, Gurfinkel EP, et al. A comparison of low-molecular-weight heparin with unfractionated heparin for unstable coronary artery disease. Efficacy and Safety of Subcutaneous Enoxaparin in Non-Q-Wave Coronary Events Study Group. N Engl J Med 1997; 337:447–452.
- Ferguson JJ, Califf RM, Antman EM, et al; SYNERGY Trial Investigators. Enoxaparin vs unfractionated heparin in high-risk patients with non-ST-segment elevation acute coronary syndromes managed with an intended early invasive strategy: primary results of the SYNERGY randomized trial. JAMA 2004; 292:45–54.
- Antman EM, Morrow DA, McCabe CH, et al; ExTRACT-TIMI 25 Investigators. Enoxaparin versus unfractionated heparin with fibrinolysis for ST-elevation myocardial infarction. N Engl J Med 2006; 354:1477–1488.
- Montalescot G, Zeymer U, Silvain J, et al; ATOLL Investigators. Intravenous enoxaparin or unfractionated heparin in primary percutaneous coronary intervention for ST-elevation myocardial infarction: the international randomised open-label ATOLL trial. Lancet 2011; 378:693–703.
- Yusuf S, Mehta SR, Chrolavicius S, et al; Fifth Organization to Assess Strategies in Acute Ischemic Syndromes Investigators. Comparison of fondaparinux and enoxaparin in acute coronary syndromes. N Engl J Med 2006; 354:1464–1476.
- Yusuf S, Mehta SR, Chrolavicius S, et al; OASIS-6 Trial Group. Effects of fondaparinux on mortality and reinfarction in patients with acute ST-segment elevation myocardial infarction: the OASIS-6 randomized trial. JAMA 2006; 295:1519–1530.
- Hurlen M, Abdelnoor M, Smith P, Erikssen J, Arnesen H. Warfarin, aspirin, or both after myocardial infarction. N Engl J Med 2002; 347:969–974.
- Coumadin Aspirin Reinfarction Study (CARS) Investigators. Randomised double-blind trial of fixed low-dose warfarin with aspirin after myocardial infarction. Lancet 1997; 350:389–396.
- Fiore LD, Ezekowitz MD, Brophy MT, Lu D, Sacco J, Peduzzi P; Combination Hemotherapy and Mortality Prevention (CHAMP) Study Group. Department of Veterans Affairs Cooperative Studies Program Clinical Trial comparing combined warfarin and aspirin with aspirin alone in survivors of acute myocardial infarction: primary results of the CHAMP study. Circulation 2002; 105:557–563.
- Mega JL, Braunwald E, Wiviott SD, et al; ATLAS ACS 2–TIMI 51 Investigators. Rivaroxaban in patients with a recent acute coronary syndrome. N Engl J Med 2012; 366:9–19.
- Alexander JH, Lopes RD, James S, et al; APPRAISE-2 Investigators. Apixaban with antiplatelet therapy after acute coronary syndrome. N Engl J Med 2011; 365:699–708.
- Oldgren J, Budaj A, Granger CB, et al; RE-DEEM Investigators. Dabigatran vs placebo in patients with acute coronary syndromes on dual antiplatelet therapy: a randomized, double-blind, phase II trial. Eur Heart J 2011; 32:2781–2789.
- Steg PG, Huber K, Andreotti F, et al. Bleeding in acute coronary syndromes and percutaneous coronary interventions: position paper by the Working Group on Thrombosis of the European Society of Cardiology. Eur Heart J 2011; 32:1854–1864.
- Giugliano RP, Braunwald E. The year in non-ST-segment elevation acute coronary syndrome. J Am Coll Cardiol 2012; 60:2127–039.
- Rao SV, Ou FS, Wang TY, et al. Trends in the prevalence and outcomes of radial and femoral approaches to percutaneous coronary intervention: a report from the National Cardiovascular Data Registry. JACC Cardiovasc Interv 2008; 1:379–386.
- Wright RS, Anderson JL, Adams CD, et al; American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. 2011 ACCF/AHA focused update incorporated into the ACC/AHA 2007 Guidelines for the Management of Patients with Unstable Angina/Non-ST-Elevation Myocardial Infarction. J Am Coll Cardiol 2011; 57:e215–e367.
- Jneid H, Anderson JL, Wright RS, et al. 2012 ACCF/AHA focused update of the guideline for the management of patients with unstable angina/non-ST-elevation myocardial infarction (updating the 2007 guideline and replacing the 2011 focused update): a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2012; 60:645–681.
- CAPRIE Steering Committee. A randomised, blinded, trial of clopidogrel versus aspirin in patients at risk of ischaemic events (CAPRIE). CAPRIE Steering Committee. Lancet 1996; 348:1329–1339.
- Cohen M, Adams PC, Parry G, et al. Combination antithrombotic therapy in unstable rest angina and non-Q-wave infarction in nonprior aspirin users. Primary end points analysis from the ATACS trial. Antithrombotic Therapy in Acute Coronary Syndromes Research Group. Circulation 1994; 89:81–88.
- Moscucci M, Fox KA, Cannon CP, et al. Predictors of major bleeding in acute coronary syndromes: the Global Registry of Acute Coronary Events (GRACE). Eur Heart J 2003; 24:1815–1823.
- Levine GN, Bates ER, Blankenship JC, et al; American College of Cardiology Foundation; American Heart Association Task Force on Practice Guidelines; Society for Cardiovascular Angiography and Interventions. 2011 ACCF/AHA/SCAI Guideline for Percutaneous Coronary Intervention. A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines and the Society for Cardiovascular Angiography and Interventions. J Am Coll Cardiol 2011; 58:e44–e122.
- American College of Emergency Physicians; Society for Cardiovascular Angiography and Interventions; O’Gara PT, Kushner FG, Ascheim DD, et al. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013; 61:485–510.
- Lewis HD, Davis JW, Archibald DG, et al. Protective effects of aspirin against acute myocardial infarction and death in men with unstable angina. Results of a Veterans Administration Cooperative Study. N Engl J Med 1983; 309:396–403.
- ISIS-2 (Second International Study of Infarct Survival) Collaborative Group. Randomised trial of intravenous streptokinase, oral aspirin, both, or neither among 17,187 cases of suspected acute myocardial infarction: ISIS-2. Lancet 1988; 2:349–360.
- Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002; 324:71–86.
- Sweeny JM, Gorog DA, Fuster V. Antiplatelet drug ‘resistance’. Part 1: mechanisms and clinical measurements. Nat Rev Cardiol 2009; 6:273–282.
- Kelly JP, Kaufman DW, Jurgelon JM, Sheehan J, Koff RS, Shapiro S. Risk of aspirin-associated major upper-gastrointestinal bleeding with enteric-coated or buffered product. Lancet 1996; 348:1413–1416.
- Grosser T, Fries S, Lawson JA, Kapoor SC, Grant GR, FitzGerald GA. Drug resistance and pseudoresistance: an unintended consequence of enteric coating aspirin. Circulation 2013; 127:377–385.
- US Food and Drug Administration (FDA). Concomitant use of ibuprofen and aspirin: potential for attenuation of the anti-platelet effect of aspirin. http://www.fda.gov/downloads/drugs/drugsafety/postmarketdrugsafetyinformationforpatientsandproviders/ucm161282.pdf. Accessed November 30, 2013.
- Patti G, Colonna G, Pasceri V, Pepe LL, Montinaro A, Di Sciascio G. Randomized trial of high loading dose of clopidogrel for reduction of periprocedural myocardial infarction in patients undergoing coronary intervention: results from the ARMYDA-2 (Antiplatelet therapy for Reduction of MYocardial Damage during Angioplasty) study. Circulation 2005; 111:2099–2106.
- Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK; Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial Investigators. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med 2001; 345:494–502.
- Bavry AA, Lincoff AM. Is clopidogrel cardiovascular medicine’s double-edged sword? Circulation 2006; 113:1638–1640.
- Collet JP, Hulot JS, Anzaha G, et al; CLOVIS-2 Investigators. High doses of clopidogrel to overcome genetic resistance: the randomized crossover CLOVIS-2 (Clopidogrel and Response Variability Investigation Study 2). JACC Cardiovasc Interv 2011; 4:392–402.
- Hulot JS, Collet JP, Cayla G, et al. CYP2C19 but not PON1 genetic variants influence clopidogrel pharmacokinetics, pharmacodynamics, and clinical efficacy in post-myocardial infarction patients. Circ Cardiovasc Interv 2011; 4:422–428.
- Cuisset T, Quilici J, Cohen W, et al. Usefulness of high clopidogrel maintenance dose according to CYP2C19 genotypes in clopidogrel low responders undergoing coronary stenting for non ST elevation acute coronary syndrome. Am J Cardiol 2011; 108:760–765.
- US Food and Drug Administration (FDA). FDA reminder to avoid concomitant use of Plavix (clopidogrel) and omeprazole. http://www.fda.gov/Drugs/DrugSafety/ucm231161.htm. Accessed November 30, 2013.
- Goodman SG, Clare R, Pieper KS, et al; Platelet Inhibition and Patient Outcomes Trial Investigators. Association of proton pump inhibitor use on cardiovascular outcomes with clopidogrel and ticagrelor: insights from the platelet inhibition and patient outcomes trial. Circulation 2012; 125:978–986.
- Solomon S, Vacek JL. Reducing cardiac ischemic events in patients with ACS: prasugrel versus clopidogrel. Commentary. Postgrad Med 2010; 122:198–200.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Roe MT, Armstrong PW, Fox KA, et al; TRILOGY ACS Investigators. Prasugrel versus clopidogrel for acute coronary syndromes without revascularization. N Engl J Med 2012; 367:1297–1309.
- Wallentin L, Becker RC, Budaj A, et al; PLATO Investigators. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2009; 361:1045–1057.
- Mahaffey KW, Wojdyla DM, Carroll K, et al; PLATO Investigators. Ticagrelor compared with clopidogrel by geographic region in the Platelet Inhibition and Patient Outcomes (PLATO) trial. Circulation 2011; 124:544–554.
- Verheugt FW. Beware of novel antiplatelet therapy in acute coronary syndrome patients with previous stroke. Circulation 2012; 125:2821–2823.
- Tricoci P, Newby LK, Hasselblad V, et al. Upstream use of small-molecule glycoprotein iib/iiia inhibitors in patients with non-ST-segment elevation acute coronary syndromes: a systematic overview of randomized clinical trials. Circ Cardiovasc Qual Outcomes 2011; 4:448–458.
- Kastrati A, Mehilli J, Neumann FJ, et al; Intracoronary Stenting and Antithrombotic: Regimen Rapid Early Action for Coronary Treatment 2 (ISAR-REACT 2) Trial Investigators. Abciximab in patients with acute coronary syndromes undergoing percutaneous coronary intervention after clopidogrel pretreatment: the ISAR-REACT 2 randomized trial. JAMA 2006; 295:1531–1538.
- Inhibition of platelet glycoprotein IIb/IIIa with eptifibatide in patients with acute coronary syndromes. The PURSUIT Trial Investigators. Platelet Glycoprotein IIb/IIIa in Unstable Angina: Receptor Suppression Using Integrilin Therapy. N Engl J Med 1998; 339:436–443.
- Stone GW, McLaurin BT, Cox DA, et al; ACUITY Investigators. Bivalirudin for patients with acute coronary syndromes. N Engl J Med 2006; 355:2203–2216.
- Oler A, Whooley MA, Oler J, Grady D. Adding heparin to aspirin reduces the incidence of myocardial infarction and death in patients with unstable angina. A meta-analysis. JAMA 1996; 276:811–815.
- Kastrati A, Neumann FJ, Schulz S, et al; ISAR-REACT 4 Trial Investigators. Abciximab and heparin versus bivalirudin for non-ST-elevation myocardial infarction. N Engl J Med 2011; 365:1980–1989.
- Stone GW, Witzenbichler B, Guagliumi G, et al; HORIZONS-AMI Trial Investigators. Bivalirudin during primary PCI in acute myocardial infarction. N Engl J Med 2008; 358:2218–2230.
- Aslam MS, Sundberg S, Sabri MN, Cooke D, Lakier JB. Pharmacokinetics of intravenous/subcutaneous enoxaparin in patients with acute coronary syndrome undergoing percutaneous coronary interventions. Catheter Cardiovasc Interv 2002; 57:187–190.
- Sanofi-Aventis US. Lovenox (enoxaparin sodium injection) product information. http://www.lovenox.com/hcp/clinical-data.aspx. Accessed December 1, 2013.
- de Lemos JA, Blazing MA, Wiviott SD, et al. Enoxaparin versus unfractionated heparin in patients treated with tirofiban, aspirin and an early conservative initial management strategy: results from the A phase of the A-to-Z trial. Eur Heart J 2004; 25:1688–1694.
- Murphy SA, Gibson CM, Morrow DA, et al. Efficacy and safety of the low-molecular weight heparin enoxaparin compared with unfractionated heparin across the acute coronary syndrome spectrum: a meta-analysis. Eur Heart J 2007; 28:2077–2086.
- Cohen M, Demers C, Gurfinkel EP, et al. A comparison of low-molecular-weight heparin with unfractionated heparin for unstable coronary artery disease. Efficacy and Safety of Subcutaneous Enoxaparin in Non-Q-Wave Coronary Events Study Group. N Engl J Med 1997; 337:447–452.
- Ferguson JJ, Califf RM, Antman EM, et al; SYNERGY Trial Investigators. Enoxaparin vs unfractionated heparin in high-risk patients with non-ST-segment elevation acute coronary syndromes managed with an intended early invasive strategy: primary results of the SYNERGY randomized trial. JAMA 2004; 292:45–54.
- Antman EM, Morrow DA, McCabe CH, et al; ExTRACT-TIMI 25 Investigators. Enoxaparin versus unfractionated heparin with fibrinolysis for ST-elevation myocardial infarction. N Engl J Med 2006; 354:1477–1488.
- Montalescot G, Zeymer U, Silvain J, et al; ATOLL Investigators. Intravenous enoxaparin or unfractionated heparin in primary percutaneous coronary intervention for ST-elevation myocardial infarction: the international randomised open-label ATOLL trial. Lancet 2011; 378:693–703.
- Yusuf S, Mehta SR, Chrolavicius S, et al; Fifth Organization to Assess Strategies in Acute Ischemic Syndromes Investigators. Comparison of fondaparinux and enoxaparin in acute coronary syndromes. N Engl J Med 2006; 354:1464–1476.
- Yusuf S, Mehta SR, Chrolavicius S, et al; OASIS-6 Trial Group. Effects of fondaparinux on mortality and reinfarction in patients with acute ST-segment elevation myocardial infarction: the OASIS-6 randomized trial. JAMA 2006; 295:1519–1530.
- Hurlen M, Abdelnoor M, Smith P, Erikssen J, Arnesen H. Warfarin, aspirin, or both after myocardial infarction. N Engl J Med 2002; 347:969–974.
- Coumadin Aspirin Reinfarction Study (CARS) Investigators. Randomised double-blind trial of fixed low-dose warfarin with aspirin after myocardial infarction. Lancet 1997; 350:389–396.
- Fiore LD, Ezekowitz MD, Brophy MT, Lu D, Sacco J, Peduzzi P; Combination Hemotherapy and Mortality Prevention (CHAMP) Study Group. Department of Veterans Affairs Cooperative Studies Program Clinical Trial comparing combined warfarin and aspirin with aspirin alone in survivors of acute myocardial infarction: primary results of the CHAMP study. Circulation 2002; 105:557–563.
- Mega JL, Braunwald E, Wiviott SD, et al; ATLAS ACS 2–TIMI 51 Investigators. Rivaroxaban in patients with a recent acute coronary syndrome. N Engl J Med 2012; 366:9–19.
- Alexander JH, Lopes RD, James S, et al; APPRAISE-2 Investigators. Apixaban with antiplatelet therapy after acute coronary syndrome. N Engl J Med 2011; 365:699–708.
- Oldgren J, Budaj A, Granger CB, et al; RE-DEEM Investigators. Dabigatran vs placebo in patients with acute coronary syndromes on dual antiplatelet therapy: a randomized, double-blind, phase II trial. Eur Heart J 2011; 32:2781–2789.
- Steg PG, Huber K, Andreotti F, et al. Bleeding in acute coronary syndromes and percutaneous coronary interventions: position paper by the Working Group on Thrombosis of the European Society of Cardiology. Eur Heart J 2011; 32:1854–1864.
- Giugliano RP, Braunwald E. The year in non-ST-segment elevation acute coronary syndrome. J Am Coll Cardiol 2012; 60:2127–039.
- Rao SV, Ou FS, Wang TY, et al. Trends in the prevalence and outcomes of radial and femoral approaches to percutaneous coronary intervention: a report from the National Cardiovascular Data Registry. JACC Cardiovasc Interv 2008; 1:379–386.
KEY POINTS
- Although antiplatelet and anticoagulant drugs reduce the risk of ischemic events, including coronary death, they also increase the risk of bleeding, reducing their net benefit. But the risk of bleeding can be managed.
- All patients experiencing an ACS should receive a single dose of aspirin 325 mg and should be instructed to chew it; this should be followed by 81 mg daily.
- Patients who are not expected to undergo coronary artery bypass grafting on an urgent basis should also receive clopidogrel, prasugrel, or ticagrelor.
- Glycoprotein IIb/IIIa inhibitors are being used less now than in the past.
- The use of unfractionated heparin is being challenged by newer parenteral anticoagulants, ie, bivalirudin, enoxaparin, and fondaparinux.
- The role of oral anticoagulants (warfarin, rivaroxaban, apixaban, and dabigatran) in ACS is uncertain.
Should patients with gout avoid thiazides for hypertension?
The decision should be individualized, taking into consideration the degree to which the thiazide increases the serum urate level, whether this increase can be managed without overly complicating the patient’s hypouricemic therapy, and, most importantly, what effect switching to another drug will have on the control of the patient’s hypertension. No study has directly addressed this issue.
My practice in most patients, for reasons I explain below, is to use a thiazide if it helps to control the blood pressure and to adjust the dose of the hypouricemic therapy as needed to reduce the serum urate to the desired level.
THIAZIDES REMAIN IMPORTANT IN ANTIHYPERTENSIVE THERAPY
Many patients with gout also have hypertension, perhaps due in part to the same hyperuricemia that caused their gouty arthritis. It is well documented that thiazide diuretics can raise the serum urate level.1 In some studies2 (but not all3), patients using thiazides had a higher incidence of gouty arthritis. Thus, it is reasonable to ask if we should avoid thiazides in patients with coexistent gout and hypertension.
Many hypertensive patients fail to reach their target blood pressures (although with the “looser” recommendations in the latest guidelines,4 we may appear to be doing a better job). The reasons for failing to reach target pressures are complex and many: physicians may simply not be aggressive enough in pursuing a target blood pressure; patients cannot tolerate the drugs or cannot afford the drugs; and many patients need two or more antihypertensive drugs to achieve adequate control. Thiazides are cheap and effective5 and work synergistically with angiotensin-converting enzyme inhibitors and angiotensin receptor blockers.6

Thus, in many patients, avoiding or discontinuing a thiazide may inhibit our ability to control their hypertension, which is a key contributor to cardiovascular events and chronic kidney injury in patients with gout. Since other diuretics (eg, loop diuretics, which can lower blood pressure but often require split doses) also raise the serum urate level, switching to one of them will not eliminate concern over hyperuricemia.
Thiazides and serum urate
Thiazides slightly increase the serum urate level and in a dose-dependent manner. At the doses commonly used in treating hypertension (12.5 or 25 mg once a day), hydrochlorothiazide increases the serum urate level by 0.8 mg/dL or less in patients with normal renal function, as shown in a number of older hypertension treatment trials and in a recent prospective study.1 The effect of chlorthalidone is similar.
In patients with chronic gout treated with a xanthine oxidase inhibitor (allopurinol or febuxostat) to lower the serum urate to the American College of Rheumatology’s recommended target level7 of less than 6.0 mg/dL (or < 5 mg/dL in the British Rheumatology guidelines), this small elevation in serum urate is unlikely to negate the clinical efficacy of these drugs when dosing is optimized. Small studies have demonstrated a clinically insignificant pharmacodynamic interaction between thiazides and xanthine oxidase inhibitors.8,9 When I add a thiazide to a patient’s regimen, I do not usually need to increase the dose of allopurinol significantly to keep the serum urate level below the desired target.
Switch antihypertensive therapy
Occasionally, in a patient with chronic gout and mild hypertension who has a serum urate level marginally above the estimated precipitation threshold of 6.7 mg/dL, it is reasonable to simply switch the thiazide to another antihypertensive, such as losartan. Losartan is a weak uricosuric and can lower the serum urate level slightly, possibly making the addition of another hypouricemic agent unnecessary, while still controlling the blood pressure with a single pill. This decision must be individualized, taking into consideration the efficacy and cost of the alternative antihypertensive drug, as well as the potential but as yet unproven cardiovascular and renal benefits of lowering the serum urate with a more potent hypouricemic to a degree not likely to be attained with losartan alone.
Continue thiazide, adjust gout therapy
Discontinuing a thiazide or switching to another antihypertensive drug may increase the cost and decrease the efficacy of hypertensive therapy. Continuing thiazide therapy and, if necessary, adjusting hypouricemic therapy will not worsen the control of the serum urate level or gouty arthritis, and in most patients will not complicate the management of gout.
ASPIRIN AND HYPERURICEMIA
In answer to a separate but related question, aspirin in low doses for cardioprotection (81 mg daily) also need not be stopped in patients with hyperuricemia or gout in an effort to better control the serum urate level. Low-dose aspirin increases the serum urate level by about 0.3 mg/dL. Since patients with gout have a higher risk of having cardiovascular disease, metabolic syndrome, and chronic kidney disease, many will benefit from low-dose aspirin therapy.
- McAdams DeMarco MA, Maynard JW, Baer AN, et al. Diuretic use, increased serum urate levels, and risk of incident gout in a population-based study of adults with hypertension: the Atherosclerosis Risk in Communities cohort study. Arthritis Rheum 2012; 64:121–129.
- Choi HK, Soriano LC, Zhang Y, Rodríguez LA. Antihypertensive drugs and risk of incident gout among patients with hypertension: population based case-control study. BMJ 2012; 344:d8190.
- Hueskes BA, Roovers EA, Mantel-Teeuwisse AK, Janssens HJ, van de Lisdonk EH, Janssen M. Use of diuretics and the risk of gouty arthritis: a systematic review. Semin Arthritis Rheum 2012; 41:879–889.
- James PA, Oparil S, Carter BL, et al. 2014 Evidence-based guideline for the management of high blood pressure in adults. Report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 2013; doi: 10.1001/jama2013.284427
- Fuchs FD. Diuretics: still essential drugs for the management of hypertension. Expert Rev Cardiovasc Ther 2009; 7:591–598.
- Sood N, Reinhart KM, Baker WL. Combination therapy for the management of hypertension: a review of the evidence. Am J Health Syst Pharm 2010; 67:885–894.
- Khanna D, Fitzgerald JD, Khanna PP, et al; American College of Rheumatology. 2012 American College of Rheumatology guidelines for management of gout. Part 1: systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis Care Res (Hoboken) 2012; 64:1431–1446.
- Löffler W, Landthaler R, de Vries JX, et al. Interaction of allopurinol and hydrochlorothiazide during prolonged oral administration of both drugs in normal subjects. I. Uric acid kinetics. Clin Investig 1994; 72:1071–1075.
- Grabowski B, Khosravan R, Wu JT, Vernillet L, Lademacher C. Effect of hydrochlorothiazide on the pharmacokinetics and pharmacodynamics of febuxostat, a non-purine selective inhibitor of xanthine oxidase. Br J Clin Pharmacol 2010; 70:57–64.
The decision should be individualized, taking into consideration the degree to which the thiazide increases the serum urate level, whether this increase can be managed without overly complicating the patient’s hypouricemic therapy, and, most importantly, what effect switching to another drug will have on the control of the patient’s hypertension. No study has directly addressed this issue.
My practice in most patients, for reasons I explain below, is to use a thiazide if it helps to control the blood pressure and to adjust the dose of the hypouricemic therapy as needed to reduce the serum urate to the desired level.
THIAZIDES REMAIN IMPORTANT IN ANTIHYPERTENSIVE THERAPY
Many patients with gout also have hypertension, perhaps due in part to the same hyperuricemia that caused their gouty arthritis. It is well documented that thiazide diuretics can raise the serum urate level.1 In some studies2 (but not all3), patients using thiazides had a higher incidence of gouty arthritis. Thus, it is reasonable to ask if we should avoid thiazides in patients with coexistent gout and hypertension.
Many hypertensive patients fail to reach their target blood pressures (although with the “looser” recommendations in the latest guidelines,4 we may appear to be doing a better job). The reasons for failing to reach target pressures are complex and many: physicians may simply not be aggressive enough in pursuing a target blood pressure; patients cannot tolerate the drugs or cannot afford the drugs; and many patients need two or more antihypertensive drugs to achieve adequate control. Thiazides are cheap and effective5 and work synergistically with angiotensin-converting enzyme inhibitors and angiotensin receptor blockers.6
Thus, in many patients, avoiding or discontinuing a thiazide may inhibit our ability to control their hypertension, which is a key contributor to cardiovascular events and chronic kidney injury in patients with gout. Since other diuretics (eg, loop diuretics, which can lower blood pressure but often require split doses) also raise the serum urate level, switching to one of them will not eliminate concern over hyperuricemia.
Thiazides and serum urate
Thiazides slightly increase the serum urate level and in a dose-dependent manner. At the doses commonly used in treating hypertension (12.5 or 25 mg once a day), hydrochlorothiazide increases the serum urate level by 0.8 mg/dL or less in patients with normal renal function, as shown in a number of older hypertension treatment trials and in a recent prospective study.1 The effect of chlorthalidone is similar.
In patients with chronic gout treated with a xanthine oxidase inhibitor (allopurinol or febuxostat) to lower the serum urate to the American College of Rheumatology’s recommended target level7 of less than 6.0 mg/dL (or < 5 mg/dL in the British Rheumatology guidelines), this small elevation in serum urate is unlikely to negate the clinical efficacy of these drugs when dosing is optimized. Small studies have demonstrated a clinically insignificant pharmacodynamic interaction between thiazides and xanthine oxidase inhibitors.8,9 When I add a thiazide to a patient’s regimen, I do not usually need to increase the dose of allopurinol significantly to keep the serum urate level below the desired target.
Switch antihypertensive therapy
Occasionally, in a patient with chronic gout and mild hypertension who has a serum urate level marginally above the estimated precipitation threshold of 6.7 mg/dL, it is reasonable to simply switch the thiazide to another antihypertensive, such as losartan. Losartan is a weak uricosuric and can lower the serum urate level slightly, possibly making the addition of another hypouricemic agent unnecessary, while still controlling the blood pressure with a single pill. This decision must be individualized, taking into consideration the efficacy and cost of the alternative antihypertensive drug, as well as the potential but as yet unproven cardiovascular and renal benefits of lowering the serum urate with a more potent hypouricemic to a degree not likely to be attained with losartan alone.
Continue thiazide, adjust gout therapy
Discontinuing a thiazide or switching to another antihypertensive drug may increase the cost and decrease the efficacy of hypertensive therapy. Continuing thiazide therapy and, if necessary, adjusting hypouricemic therapy will not worsen the control of the serum urate level or gouty arthritis, and in most patients will not complicate the management of gout.
ASPIRIN AND HYPERURICEMIA
In answer to a separate but related question, aspirin in low doses for cardioprotection (81 mg daily) also need not be stopped in patients with hyperuricemia or gout in an effort to better control the serum urate level. Low-dose aspirin increases the serum urate level by about 0.3 mg/dL. Since patients with gout have a higher risk of having cardiovascular disease, metabolic syndrome, and chronic kidney disease, many will benefit from low-dose aspirin therapy.
The decision should be individualized, taking into consideration the degree to which the thiazide increases the serum urate level, whether this increase can be managed without overly complicating the patient’s hypouricemic therapy, and, most importantly, what effect switching to another drug will have on the control of the patient’s hypertension. No study has directly addressed this issue.
My practice in most patients, for reasons I explain below, is to use a thiazide if it helps to control the blood pressure and to adjust the dose of the hypouricemic therapy as needed to reduce the serum urate to the desired level.
THIAZIDES REMAIN IMPORTANT IN ANTIHYPERTENSIVE THERAPY
Many patients with gout also have hypertension, perhaps due in part to the same hyperuricemia that caused their gouty arthritis. It is well documented that thiazide diuretics can raise the serum urate level.1 In some studies2 (but not all3), patients using thiazides had a higher incidence of gouty arthritis. Thus, it is reasonable to ask if we should avoid thiazides in patients with coexistent gout and hypertension.
Many hypertensive patients fail to reach their target blood pressures (although with the “looser” recommendations in the latest guidelines,4 we may appear to be doing a better job). The reasons for failing to reach target pressures are complex and many: physicians may simply not be aggressive enough in pursuing a target blood pressure; patients cannot tolerate the drugs or cannot afford the drugs; and many patients need two or more antihypertensive drugs to achieve adequate control. Thiazides are cheap and effective5 and work synergistically with angiotensin-converting enzyme inhibitors and angiotensin receptor blockers.6
Thus, in many patients, avoiding or discontinuing a thiazide may inhibit our ability to control their hypertension, which is a key contributor to cardiovascular events and chronic kidney injury in patients with gout. Since other diuretics (eg, loop diuretics, which can lower blood pressure but often require split doses) also raise the serum urate level, switching to one of them will not eliminate concern over hyperuricemia.
Thiazides and serum urate
Thiazides slightly increase the serum urate level and in a dose-dependent manner. At the doses commonly used in treating hypertension (12.5 or 25 mg once a day), hydrochlorothiazide increases the serum urate level by 0.8 mg/dL or less in patients with normal renal function, as shown in a number of older hypertension treatment trials and in a recent prospective study.1 The effect of chlorthalidone is similar.
In patients with chronic gout treated with a xanthine oxidase inhibitor (allopurinol or febuxostat) to lower the serum urate to the American College of Rheumatology’s recommended target level7 of less than 6.0 mg/dL (or < 5 mg/dL in the British Rheumatology guidelines), this small elevation in serum urate is unlikely to negate the clinical efficacy of these drugs when dosing is optimized. Small studies have demonstrated a clinically insignificant pharmacodynamic interaction between thiazides and xanthine oxidase inhibitors.8,9 When I add a thiazide to a patient’s regimen, I do not usually need to increase the dose of allopurinol significantly to keep the serum urate level below the desired target.
Switch antihypertensive therapy
Occasionally, in a patient with chronic gout and mild hypertension who has a serum urate level marginally above the estimated precipitation threshold of 6.7 mg/dL, it is reasonable to simply switch the thiazide to another antihypertensive, such as losartan. Losartan is a weak uricosuric and can lower the serum urate level slightly, possibly making the addition of another hypouricemic agent unnecessary, while still controlling the blood pressure with a single pill. This decision must be individualized, taking into consideration the efficacy and cost of the alternative antihypertensive drug, as well as the potential but as yet unproven cardiovascular and renal benefits of lowering the serum urate with a more potent hypouricemic to a degree not likely to be attained with losartan alone.
Continue thiazide, adjust gout therapy
Discontinuing a thiazide or switching to another antihypertensive drug may increase the cost and decrease the efficacy of hypertensive therapy. Continuing thiazide therapy and, if necessary, adjusting hypouricemic therapy will not worsen the control of the serum urate level or gouty arthritis, and in most patients will not complicate the management of gout.
ASPIRIN AND HYPERURICEMIA
In answer to a separate but related question, aspirin in low doses for cardioprotection (81 mg daily) also need not be stopped in patients with hyperuricemia or gout in an effort to better control the serum urate level. Low-dose aspirin increases the serum urate level by about 0.3 mg/dL. Since patients with gout have a higher risk of having cardiovascular disease, metabolic syndrome, and chronic kidney disease, many will benefit from low-dose aspirin therapy.
- McAdams DeMarco MA, Maynard JW, Baer AN, et al. Diuretic use, increased serum urate levels, and risk of incident gout in a population-based study of adults with hypertension: the Atherosclerosis Risk in Communities cohort study. Arthritis Rheum 2012; 64:121–129.
- Choi HK, Soriano LC, Zhang Y, Rodríguez LA. Antihypertensive drugs and risk of incident gout among patients with hypertension: population based case-control study. BMJ 2012; 344:d8190.
- Hueskes BA, Roovers EA, Mantel-Teeuwisse AK, Janssens HJ, van de Lisdonk EH, Janssen M. Use of diuretics and the risk of gouty arthritis: a systematic review. Semin Arthritis Rheum 2012; 41:879–889.
- James PA, Oparil S, Carter BL, et al. 2014 Evidence-based guideline for the management of high blood pressure in adults. Report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 2013; doi: 10.1001/jama2013.284427
- Fuchs FD. Diuretics: still essential drugs for the management of hypertension. Expert Rev Cardiovasc Ther 2009; 7:591–598.
- Sood N, Reinhart KM, Baker WL. Combination therapy for the management of hypertension: a review of the evidence. Am J Health Syst Pharm 2010; 67:885–894.
- Khanna D, Fitzgerald JD, Khanna PP, et al; American College of Rheumatology. 2012 American College of Rheumatology guidelines for management of gout. Part 1: systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis Care Res (Hoboken) 2012; 64:1431–1446.
- Löffler W, Landthaler R, de Vries JX, et al. Interaction of allopurinol and hydrochlorothiazide during prolonged oral administration of both drugs in normal subjects. I. Uric acid kinetics. Clin Investig 1994; 72:1071–1075.
- Grabowski B, Khosravan R, Wu JT, Vernillet L, Lademacher C. Effect of hydrochlorothiazide on the pharmacokinetics and pharmacodynamics of febuxostat, a non-purine selective inhibitor of xanthine oxidase. Br J Clin Pharmacol 2010; 70:57–64.
- McAdams DeMarco MA, Maynard JW, Baer AN, et al. Diuretic use, increased serum urate levels, and risk of incident gout in a population-based study of adults with hypertension: the Atherosclerosis Risk in Communities cohort study. Arthritis Rheum 2012; 64:121–129.
- Choi HK, Soriano LC, Zhang Y, Rodríguez LA. Antihypertensive drugs and risk of incident gout among patients with hypertension: population based case-control study. BMJ 2012; 344:d8190.
- Hueskes BA, Roovers EA, Mantel-Teeuwisse AK, Janssens HJ, van de Lisdonk EH, Janssen M. Use of diuretics and the risk of gouty arthritis: a systematic review. Semin Arthritis Rheum 2012; 41:879–889.
- James PA, Oparil S, Carter BL, et al. 2014 Evidence-based guideline for the management of high blood pressure in adults. Report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 2013; doi: 10.1001/jama2013.284427
- Fuchs FD. Diuretics: still essential drugs for the management of hypertension. Expert Rev Cardiovasc Ther 2009; 7:591–598.
- Sood N, Reinhart KM, Baker WL. Combination therapy for the management of hypertension: a review of the evidence. Am J Health Syst Pharm 2010; 67:885–894.
- Khanna D, Fitzgerald JD, Khanna PP, et al; American College of Rheumatology. 2012 American College of Rheumatology guidelines for management of gout. Part 1: systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis Care Res (Hoboken) 2012; 64:1431–1446.
- Löffler W, Landthaler R, de Vries JX, et al. Interaction of allopurinol and hydrochlorothiazide during prolonged oral administration of both drugs in normal subjects. I. Uric acid kinetics. Clin Investig 1994; 72:1071–1075.
- Grabowski B, Khosravan R, Wu JT, Vernillet L, Lademacher C. Effect of hydrochlorothiazide on the pharmacokinetics and pharmacodynamics of febuxostat, a non-purine selective inhibitor of xanthine oxidase. Br J Clin Pharmacol 2010; 70:57–64.
The impact of anti-TNF therapy on the nonspecialist

About 15 years ago, the first anti-tumor necrosis factor (anti-TNF) drugs received approval for treating Crohn disease and rheumatoid arthritis, and a new era of pharmacotherapy was born. A few years before that, I was at a meeting discussing the potential benefits and pitfalls of these new biologic therapies, and I opined that no one would pay for them on an ongoing basis unless they were amazingly effective—which was unlikely, as the drugs only affected a single cytokine. And if they were effective, they would undoubtedly be associated with a host of opportunistic infections. Given my predictive skills, it is no surprise that Warren Buffett rarely calls to ask my opinion.
Clearly, anti-TNF drugs are effective and have raised the bar for how we define successful response to therapy. But recent studies in early rheumatoid arthritis indicate that they may not be much better than traditional combination therapy or monotherapy with methotrexate if the methotrexate and the other drugs are given and tolerated at full dose. This is clearly not the case for other inflammatory diseases.
Anti-TNF drugs and other biologics are now part of the arsenal of most medical specialists, so outpatient internists and hospitalists increasingly encounter patients taking these drugs. Since patients with systemic inflammatory disease have an increased prevalence of cardiovascular disease, cardiologists are also seeing more patients taking these drugs. Thus, the overview by Hadam et al in this issue of the Journal on the risks of biologic therapies is relevant to many readers.
Almost all prescriptions and requests for insurance approval for these drugs are written by subspecialists familiar with their risks. But patients may ask their primary care physicians about the tests and vaccines recommended for those about to start anti-TNF therapy. Before starting anti-TNF therapy, all patients should be tested for previous exposure to tuberculosis and should be treated for latent tuberculosis if appropriate. Blocking TNF leads to a breakdown of the protective granulomatous inflammatory response that contains the mycobacteria and, as with corticosteroid treatment, results in reactivation of the disease. Interestingly, the reactivation is quite often not in the lungs. And since anti-TNF therapy dramatically blunts the inflammatory response, as does corticosteroid therapy, reactivation may appear as nonspecific malaise or may be misinterpreted as a flare in the underlying disease, and thus it may go undiagnosed. Patients should also be screened for exposure to hepatitis B virus. Vaccines, particularly live vaccines, are generally given if possible before starting anti-TNF therapy, and all patients on chronic therapy should get annual influenza vaccines.
Despite initial concerns about a dramatically increased risk of routine and opportunistic infections in patients on anti-TNF therapy, this has not been observed. Even in the perioperative setting, the increased risk of infection is modest. What has struck me, however, is the way these drugs, like steroids, blunt and mask the signs of infection. I have seen deep soft-tissue, intra-abdominal, and native and prosthetic joint infections go unsuspected for days or even weeks in the absence of significant fever, elevation in acute-phase markers, or dramatic local findings. We must be extra vigilant.
There is a fear of malignancy arising or recurring in patients on anti-TNF therapy. This fear is certainly promoted by the required black-box warning about the risk of lymphoma and other malignancies that these drugs carry. The evidence of a significant increase in risk of malignancies other than hepatosplenic T-cell lymphoma in children and nonmelanoma skin cancers is not strong and is likely slanted by an increased risk of certain malignancies associated with the underlying rheumatic disease and other previous therapies. Nonetheless, I am reluctant to use these drugs in patients with a history of melanoma.
We still have much to learn about these drugs. Why are specific agents more effective in some diseases than others? For example, etanercept treats rheumatoid arthritis but not Crohn disease. Also, we still do not know how they can elicit reversible demyelinating disorders or autoantibodies with or without associated drug-induced lupus syndromes. Even odder is the occurrence of psoriasis induced by anti-TNF drugs, despite their being used to treat psoriasis.
My initial skepticism regarding anti-TNF drugs was unjustified. They are being tested and used successfully in an increasing number of diseases. But we all need to increase our familiarity with their unique risks and somehow find a way to deal with their unique cost.
About 15 years ago, the first anti-tumor necrosis factor (anti-TNF) drugs received approval for treating Crohn disease and rheumatoid arthritis, and a new era of pharmacotherapy was born. A few years before that, I was at a meeting discussing the potential benefits and pitfalls of these new biologic therapies, and I opined that no one would pay for them on an ongoing basis unless they were amazingly effective—which was unlikely, as the drugs only affected a single cytokine. And if they were effective, they would undoubtedly be associated with a host of opportunistic infections. Given my predictive skills, it is no surprise that Warren Buffett rarely calls to ask my opinion.
Clearly, anti-TNF drugs are effective and have raised the bar for how we define successful response to therapy. But recent studies in early rheumatoid arthritis indicate that they may not be much better than traditional combination therapy or monotherapy with methotrexate if the methotrexate and the other drugs are given and tolerated at full dose. This is clearly not the case for other inflammatory diseases.
Anti-TNF drugs and other biologics are now part of the arsenal of most medical specialists, so outpatient internists and hospitalists increasingly encounter patients taking these drugs. Since patients with systemic inflammatory disease have an increased prevalence of cardiovascular disease, cardiologists are also seeing more patients taking these drugs. Thus, the overview by Hadam et al in this issue of the Journal on the risks of biologic therapies is relevant to many readers.
Almost all prescriptions and requests for insurance approval for these drugs are written by subspecialists familiar with their risks. But patients may ask their primary care physicians about the tests and vaccines recommended for those about to start anti-TNF therapy. Before starting anti-TNF therapy, all patients should be tested for previous exposure to tuberculosis and should be treated for latent tuberculosis if appropriate. Blocking TNF leads to a breakdown of the protective granulomatous inflammatory response that contains the mycobacteria and, as with corticosteroid treatment, results in reactivation of the disease. Interestingly, the reactivation is quite often not in the lungs. And since anti-TNF therapy dramatically blunts the inflammatory response, as does corticosteroid therapy, reactivation may appear as nonspecific malaise or may be misinterpreted as a flare in the underlying disease, and thus it may go undiagnosed. Patients should also be screened for exposure to hepatitis B virus. Vaccines, particularly live vaccines, are generally given if possible before starting anti-TNF therapy, and all patients on chronic therapy should get annual influenza vaccines.
Despite initial concerns about a dramatically increased risk of routine and opportunistic infections in patients on anti-TNF therapy, this has not been observed. Even in the perioperative setting, the increased risk of infection is modest. What has struck me, however, is the way these drugs, like steroids, blunt and mask the signs of infection. I have seen deep soft-tissue, intra-abdominal, and native and prosthetic joint infections go unsuspected for days or even weeks in the absence of significant fever, elevation in acute-phase markers, or dramatic local findings. We must be extra vigilant.
There is a fear of malignancy arising or recurring in patients on anti-TNF therapy. This fear is certainly promoted by the required black-box warning about the risk of lymphoma and other malignancies that these drugs carry. The evidence of a significant increase in risk of malignancies other than hepatosplenic T-cell lymphoma in children and nonmelanoma skin cancers is not strong and is likely slanted by an increased risk of certain malignancies associated with the underlying rheumatic disease and other previous therapies. Nonetheless, I am reluctant to use these drugs in patients with a history of melanoma.
We still have much to learn about these drugs. Why are specific agents more effective in some diseases than others? For example, etanercept treats rheumatoid arthritis but not Crohn disease. Also, we still do not know how they can elicit reversible demyelinating disorders or autoantibodies with or without associated drug-induced lupus syndromes. Even odder is the occurrence of psoriasis induced by anti-TNF drugs, despite their being used to treat psoriasis.
My initial skepticism regarding anti-TNF drugs was unjustified. They are being tested and used successfully in an increasing number of diseases. But we all need to increase our familiarity with their unique risks and somehow find a way to deal with their unique cost.
About 15 years ago, the first anti-tumor necrosis factor (anti-TNF) drugs received approval for treating Crohn disease and rheumatoid arthritis, and a new era of pharmacotherapy was born. A few years before that, I was at a meeting discussing the potential benefits and pitfalls of these new biologic therapies, and I opined that no one would pay for them on an ongoing basis unless they were amazingly effective—which was unlikely, as the drugs only affected a single cytokine. And if they were effective, they would undoubtedly be associated with a host of opportunistic infections. Given my predictive skills, it is no surprise that Warren Buffett rarely calls to ask my opinion.
Clearly, anti-TNF drugs are effective and have raised the bar for how we define successful response to therapy. But recent studies in early rheumatoid arthritis indicate that they may not be much better than traditional combination therapy or monotherapy with methotrexate if the methotrexate and the other drugs are given and tolerated at full dose. This is clearly not the case for other inflammatory diseases.
Anti-TNF drugs and other biologics are now part of the arsenal of most medical specialists, so outpatient internists and hospitalists increasingly encounter patients taking these drugs. Since patients with systemic inflammatory disease have an increased prevalence of cardiovascular disease, cardiologists are also seeing more patients taking these drugs. Thus, the overview by Hadam et al in this issue of the Journal on the risks of biologic therapies is relevant to many readers.
Almost all prescriptions and requests for insurance approval for these drugs are written by subspecialists familiar with their risks. But patients may ask their primary care physicians about the tests and vaccines recommended for those about to start anti-TNF therapy. Before starting anti-TNF therapy, all patients should be tested for previous exposure to tuberculosis and should be treated for latent tuberculosis if appropriate. Blocking TNF leads to a breakdown of the protective granulomatous inflammatory response that contains the mycobacteria and, as with corticosteroid treatment, results in reactivation of the disease. Interestingly, the reactivation is quite often not in the lungs. And since anti-TNF therapy dramatically blunts the inflammatory response, as does corticosteroid therapy, reactivation may appear as nonspecific malaise or may be misinterpreted as a flare in the underlying disease, and thus it may go undiagnosed. Patients should also be screened for exposure to hepatitis B virus. Vaccines, particularly live vaccines, are generally given if possible before starting anti-TNF therapy, and all patients on chronic therapy should get annual influenza vaccines.
Despite initial concerns about a dramatically increased risk of routine and opportunistic infections in patients on anti-TNF therapy, this has not been observed. Even in the perioperative setting, the increased risk of infection is modest. What has struck me, however, is the way these drugs, like steroids, blunt and mask the signs of infection. I have seen deep soft-tissue, intra-abdominal, and native and prosthetic joint infections go unsuspected for days or even weeks in the absence of significant fever, elevation in acute-phase markers, or dramatic local findings. We must be extra vigilant.
There is a fear of malignancy arising or recurring in patients on anti-TNF therapy. This fear is certainly promoted by the required black-box warning about the risk of lymphoma and other malignancies that these drugs carry. The evidence of a significant increase in risk of malignancies other than hepatosplenic T-cell lymphoma in children and nonmelanoma skin cancers is not strong and is likely slanted by an increased risk of certain malignancies associated with the underlying rheumatic disease and other previous therapies. Nonetheless, I am reluctant to use these drugs in patients with a history of melanoma.
We still have much to learn about these drugs. Why are specific agents more effective in some diseases than others? For example, etanercept treats rheumatoid arthritis but not Crohn disease. Also, we still do not know how they can elicit reversible demyelinating disorders or autoantibodies with or without associated drug-induced lupus syndromes. Even odder is the occurrence of psoriasis induced by anti-TNF drugs, despite their being used to treat psoriasis.
My initial skepticism regarding anti-TNF drugs was unjustified. They are being tested and used successfully in an increasing number of diseases. But we all need to increase our familiarity with their unique risks and somehow find a way to deal with their unique cost.
Managing risks of TNF inhibitors: An update for the internist
Biologic agents such as those that block tumor necrosis factor (TNF) alpha have revolutionized the treatment of autoimmune diseases like rheumatoid arthritis and inflammatory bowel disease, dramatically improving disease control and quality of life. In addition, they have made true disease remission possible in some cases.
However, as with any new therapy, a variety of side effects must be considered.
WHAT ARE BIOLOGIC AGENTS?

Biologic agents are genetically engineered drugs manufactured or synthesized in vitro from molecules such as proteins, genes, and antibodies present in living organisms. By targeting specific molecular components of the inflammatory cascade, including cytokines, these drugs alter certain aspects of the body’s inflammatory response in autoimmune diseases. Because TNF inhibitors are the most widely used biologic agents in the United States, this review will focus on them.
TNF INHIBITORS
TNF inhibitors suppress the inflammatory cascade by inactivating TNF alpha, a cytokine that promotes inflammation in the intestine, synovial tissue, and other sites.1,2
Several TNF inhibitors are available. Etanercept and infliximab were the first two to receive US Food and Drug Administration (FDA) approval, and they are extensively used. Etanercept, a soluble TNF receptor given subcutaneously, was approved for treating rheumatoid arthritis in 1998. Infliximab, a chimeric monoclonal antibody (75% human and 25% mouse protein sequence) given intravenously, received FDA approval for treating Crohn disease in 1998 and for rheumatoid arthritis in 1999. Other anti-TNF agents with varying properties are also available.
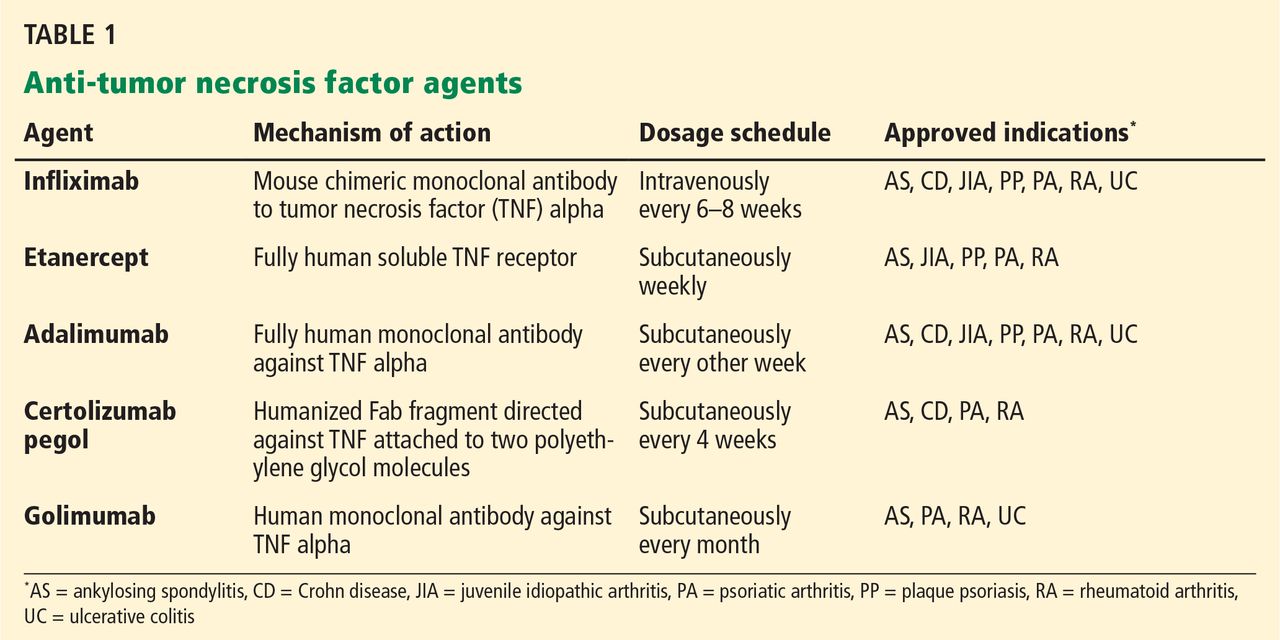
Table 1 lists anti-TNF agents approved for treating rheumatoid arthritis and inflammatory bowel disease. These are chronic, relapsing diseases that significantly and dramatically reduce quality of life, especially when they are poorly controlled.3,4 Untreated disease has been associated with malignancy, infection, pregnancy loss, and malnutrition.5–8 TNF inhibitors are aimed at patients who have moderate to severe disease or for whom previous treatments have failed, to help them achieve and maintain steroid-free remission. However, use of these agents is tempered by the risk of potentially serious side effects.
Special thought should also be given to the direct costs of the drugs (up to $30,000 per year, not counting the cost of their administration) and to the indirect costs such as time away from work to receive treatment. These are major considerations in some cases, and patients should be selected carefully for treatment with these drugs.
BEFORE STARTING THERAPY
Before starting anti-TNF therapy, several steps can reduce the risk of serious adverse events.
Take a focused history
The clinical history should include inquiries about previous bacterial, fungal, and tuberculosis infections or exposure; diabetes; and other immunocompromised states that increase the risk of acquiring potentially life-threatening infections.
Details of particular geographic areas of residence, occupational exposures, and social history should be sought. These include history of incarceration (which may put patients at risk of tuberculosis) and residence in the Ohio River valley or in the southwestern or midwestern United States (which may increase the risk of histoplasmosis, coccidioidomycosis, and other fungal infections).
Bring vaccinations up to date
Age-appropriate vaccinations should be discussed and given, ideally before starting therapy. These include influenza vaccine every year and tetanus boosters every 10 years for all and, as appropriate, varicella, human papillomavirus, and pneumococcal vaccinations. The US Centers for Disease Control and Prevention recommend an additional dose of the pneumococcal vaccine if more than 5 years have elapsed since the first one, and many clinicians opt to give it every 5 years.
In general, live-attenuated vaccines, including the intranasal influenza vaccine, are contraindicated in patients taking biologic agents.9 For patients at high risk of exposure or infection, it may be reasonable to hold the biologic agent for a period of time, vaccinate, and resume the biologic agent a month later.
Recent data suggest that the varicella zoster vaccine may be safely given to older patients with immune-mediated diseases such as rheumatoid arthritis and inflammatory bowel disease taking biologic agents.10 New guidelines from the American College of Rheumatology recommend age-appropriate vaccines for rheumatoid arthritis patients age 60 and older before biologic treatments are started. Case-by-case discussion with the subspecialist and the patient is recommended.
Screen for chronic infections
Tuberculosis screening with a purified protein derivative test or an interferon-gamma-release (Quantiferon) assay followed by chest radiography in patients with a positive test is mandatory before giving a TNF inhibitor.
Hepatitis B virus status should be determined before starting anti-TNF therapy.11
Hepatitis B vaccination has been recommended for patients with inflammatory bowel disease, but no clear recommendation exists for patients with rheumatic disease. Patients with inflammatory bowel disease tend to have low rates of response to hepatitis B vaccination12,13; possible reasons include their lack of an appropriate innate immune response to infectious agents, malnutrition, surgery, older age, and immunosuppressive drugs.14 An accelerated vaccination protocol with recombinant hepatitis B vaccine (Energix-B) in a double dose at 0, 1, and 2 months has been shown to improve response rates.15
Whenever possible, it may be better to vaccinate patients before starting immunosuppressive therapy, and to check postvaccination titers to ensure adequate response.
Perform an examination
A full physical examination with special attention to skin rashes should be performed. This may serve as a baseline to assist early detection of new rashes associated with anti-TNF therapy.

A baseline complete blood cell count and complete metabolic panel should be routinely obtained before starting therapy (and thereafter at the discretion of the physician). In conjunction with follow-up tests, they can help detect an unexpected decrease in white blood cell count or abnormal results on the liver panel.16 These baseline and follow-up tests are generally performed by the subspecialist, and the results are shared with the primary care physician.
Table 2 summarizes key information to be sought before starting a patient on a TNF inhibitor.
ADVERSE EFFECTS OF ANTI-TNF DRUGS
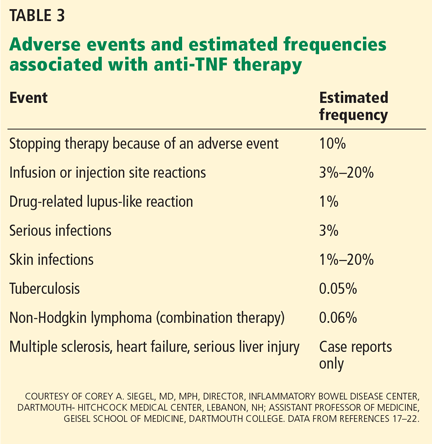
Infusion reactions, infections, cardiac arrhythmias, demyelinating disorders, skin infections, and malignancies have been reported with anti-TNF therapy. The relative frequencies of these adverse events are summarized in Table 3.17–22
NONINFECTIOUS COMPLICATIONS OF TNF INHIBITORS
Injection site reactions
When anti-TNF agents are given subcutaneously, injection site reactions are common (occurring in up to 40% of patients) and are considered minor.11 Reactions, including significant pain, typically occur within the first few months of therapy. They can last 2 to 5 days but rarely warrant stopping therapy. Treatment with ice and an antihistamine is almost always sufficient to control symptoms.
Infusion reactions with infliximab
Infliximab can cause both acute and delayed infusion reactions. Acute reactions can occur up to 24 hours after infusion but usually appear within 10 minutes of administration and are handled by the infusion suite staff. They range from the severe immunoglobulin E-mediated type I reaction, manifesting with hypotension, bronchospasm, and urticaria, to the milder anaphylactoid-type reaction, which constitutes the majority.23–25
While most primary care physicians will not encounter an acute reaction, family doctors and emergency room physicians may encounter delayed reactions, which can develop 1 to 14 days after infusion. These reactions usually resemble serum sickness and present with joint pain, fatigue, myalgia, and fever. But, unlike classic serum sickness, these reactions are generally not associated with a rash.
With nonspecific symptoms, the diagnosis may be easy to overlook. However, establishing this diagnosis is important because repeat therapy may result in a more severe reaction upon reexposure to the drug.
Once diagnosed, these reactions can be treated symptomatically with a combination of acetaminophen and diphenhydramine after discussion between the primary care physician and subspecialist.23,24
Autoimmune syndromes
Several studies have reported a small percentage of patients treated with anti-TNF agents who develop paradoxical autoimmune conditions. These range from asymptomatic immunologic alterations, including the formation of antinuclear antibodies and antibodies to double-stranded DNA, to life-threatening systemic autoimmune diseases.26,27
Autoimmune diseases associated with anti-TNF treatment include a lupus-like syndrome, vasculitides, and psoriatic skin lesions. These syndromes warrant stopping the inciting drug and, on occasion, giving corticosteroids. Most cases arise between 1 month and 1 year of starting treatment, and almost 75% resolve completely after the anti-TNF therapy is stopped.26
Interestingly, anti-TNF agents are approved for treating psoriasis and psoriatic arthritis, but psoriasis has paradoxically developed in patients being treated with these drugs for other autoimmune diseases. The FDA has reviewed 69 cases of new-onset psoriasis with anti-TNF therapy, including 17 pustular and 15 palmoplantar cases. The 12 most severe cases resulted in hospitalization, and symptoms resolved in most after treatment cessation.28
Fiorino et al29 counted 18 reported cases of psoriasis induced by anti-TNF therapy in patients with inflammatory bowel disease and concluded that it is rare. Harrison et al30 reported similar findings in patients with rheumatoid arthritis, with an increased incidence rate of 1.04 per 1,000 person-years. New-onset psoriasis was most common in patients treated with adalimumab. An example of the rash is seen in Figure 1.
CARDIOVASCULAR SIDE EFFECTS
Cardiovascular side effects of anti-TNF agents range from nonspecific and asymptomatic arrhythmias to worsening of heart failure.
Circulating levels of TNF are increased in patients with heart failure, and studies have evaluated the effects of TNF inhibition with infliximab on cardiac function and overall survival.31,32 The combined risk of death from any cause or hospitalization from heart failure was significantly higher in the infliximab groups, and the effects persisted for up to 5 months after stopping therapy.
Other studies22,33 have evaluated the effects of infliximab and etanercept on cardiac function and overall survival. Results showed possible exacerbation of heart failure with etanercept and increased risk of death with infliximab in patients with New York Heart Association (NYHA) class III or IV heart failure and left ventricular ejection fractions less than 35%.
Case reports have also described patients with worsening or new-onset heart failure on TNF inhibitors, including patients younger than 50 years and without identifiable cardiovascular risk factors.
Data analyses31,34,35 from large clinical registries have reported no significant increase in heart failure attributable to TNF inhibitors. However, we have concerns about the methodology of these analyses.
Currently, anti-TNF therapy is contraindicated in patients with NYHA class III or IV heart failure. Data are inconclusive for patients with class I or II heart failure. Baseline echocardiography and cardiology consultation can be considered, with close monitoring and avoidance of high doses of TNF inhibitors. If heart failure develops in a patient on anti-TNF therapy, the drug should be discontinued and the patient should be evaluated further.36
DEMYELINATING DISEASE, INCLUDING MULTIPLE SCLEROSIS
Anti-TNF agents have been associated with the onset or exacerbation of clinical symptoms and radiographic evidence of central nervous system demyelinating disorders, including multiple sclerosis.37–40 Mohan et al38 identified 19 cases of demyelinating events occurring after administration of anti-TNF agents in early 2001. In most cases, symptoms improved or resolved after therapy was stopped.
Optic neuritis,41,42 bilateral optic neuropathy,43 and aseptic meningitis44 have also been reported, but these have occurred only rarely.
How common are these effects? Postmarketing surveillance in patients with rheumatoid arthritis yields an estimated incidence of demyelinating disorders of 1 per 1,000 patient-years with adalimumab therapy.45 Complicating the assessment is an observed slight increase in risk of demyelinating conditions associated with inflammatory bowel disease.
Symptoms that should heighten the physician’s suspicion of this adverse effect include confusion, paresthesias, and ataxia. Patients on anti-TNF therapy who develop new visual symptoms should be checked for painless visual loss as a sign of early demyelinating disease.
Although data that conclusively link anti-TNF agents to multiple sclerosis are lacking, these drugs should not be initiated in patients who have a history of demyelinating disease, and treatment should be stopped promptly if the diagnosis is suspected.
MALIGNANCY
Whether anti-TNF therapy is directly linked to development of malignancies is difficult to determine. There are many confounding factors, including the risk of malignancy in underlying inflammatory disease and the concomitant use of other medications such as thiopurines, which have a known association with lymphoma.46
The incidence of lymphoma is twice as high in rheumatoid arthritis patients as in the general population.47 The risk is higher in those with more aggressive joint disease—the subset of rheumatoid arthritis patients who are more likely to be given anti-TNF agents.5
In patients with inflammatory bowel disease, the risk of cholangiocarcinoma is four times higher, and the risk of small-bowel adenocarcinoma is 16 to 18 times higher, but no increased risk of lymphoma has been identified in this population.48,49
Data from six clinical trials of infliximab, including a long-term study of its safety in Crohn disease, suggest it poses no increase in overall risk of malignancy.50–52 Similar results have been reported in patients with other rheumatic diseases.8 Information on this topic is constantly evolving, and studies range from case series to clinical trials to large patient registries.
The decision to use a TNF inhibitor should be based on the patient’s clinical picture and risk factors. Discussion of the risks and benefits of therapy with the patient should be clearly documented.
Non-Hodgkin lymphoma
Evidence about the risk of lymphoma with anti-TNF use is mixed, as up to two-thirds of patients on anti-TNF therapy have received concomitant nonbiologic immunosuppressive medications, making it difficult to determine the true risk from the biologic agents alone.53 Current evidence both supports9,53–56 and refutes7,55,57–60 the idea that anti-TNF agents increase lymphoma risk.
In patients with inflammatory bowel disease, several population-based studies have not shown a clear increase in lymphoma risk with anti-TNF use.56,59,60 Pedersen et al,61 in a meta-analysis of eight studies, confirmed these findings by showing no overall lymphoma risk in patients with inflammatory bowel disease.
However, a Canadian population-based study found a statistically significant increase in non-Hodgkin lymphoma in males with Crohn disease, with an incidence ratio of 3.63 (95% confidence interval [CI] 1.53–8.62).61 Additionally, Siegel et al62 found a significantly higher risk (6.1 cases per 10,000 patients) in patients treated with anti-TNF agents and thiopurines than in the general population (1.9 cases per 10,000 people). Although the difference was statistically significant, the overall risk is still very low.
Patients with rheumatoid arthritis seem to have a risk of lymphoma two to three times higher than in the general population. However, large population-based studies have not shown a statistically significant increase in the risk of lymphoma with anti-TNF therapy.63
Hepatosplenic T-cell lymphoma is a rare subtype of peripheral T-cell non-Hodgkin lymphoma; 25 cases have been reported in patients receiving anti-TNF therapy.64 Although the risk is extremely low (< 0.05%), physicians must carefully consider the risks and benefits of combination therapy, especially in young male patients with inflammatory bowel disease, since death is the usual outcome of this disease.65–67
Skin cancers
Wolfe and Michaud8 evaluated malignancy risk in rheumatoid arthritis patients being treated with biologic agents, including TNF inhibitors, using a large longitudinal database. These data were compared with those of the US Surveillance, Epidemiology, and End-Results (SEER) national database. No increase in the overall cancer rate was seen in rheumatoid arthritis patients (standardized incidence ratio [SIR] 1.0, 95% CI 1.0–1.1).
However, melanoma was more common in rheumatoid arthritis patients compared with SEER rates (SIR 1.7, 95% CI 1.3–2.3).8 In addition, biologic therapy was associated with a higher (but not statistically significant) risk of melanoma (odds ratio [OR] 2.3, 95% CI 0.9–5.4) and a higher risk of nonmelanoma skin cancer (OR 1.5, 95% CI 1.2–1.8), but not of other types of cancer.8
INFECTION
Patients on anti-TNF therapy are at a higher risk of infection, ranging from minor to life-threatening bacterial infections, and including the reactivation of granulomatous and fungal infections. More importantly, these agents are similar to steroids in blunting signs of infection, which may delay diagnosis and treatment.
The management of infection in patients on anti-TNF medications varies from case to case. In general, patients with a minor infection that does not require hospitalization or intravenous antibiotics can continue the biologic therapy while taking oral antibiotics. TNF inhibitors must be held in the event of a major infection.
Consultation with an infectious disease specialist is recommended, especially in complex cases.
Bacterial infections
An increased risk of minor bacterial infections such as urinary tract and respiratory infections has been well documented in several randomized control trials of anti-TNF agents, though other studies have shown no such increase in risk.33,51–59
The threshold for using antibiotics for a suspected bacterial infection is somewhat shifted in favor of treatment in patients on anti-TNF therapy. The reason is twofold: as previously noted, infections may be worse than they appear, because anti-TNF drugs can mask the signs and symptoms of a serious infection, and in patients on these drugs, an untreated bacterial infection may rapidly become life-threatening.
In general, broad-spectrum antibiotics are not warranted unless the source of infection is unclear or the patient is in danger of hemodynamic compromise.
Opportunistic infections
The association of anti-TNF agents with opportunistic infections could be viewed as an extension of their normal and intended therapeutic activity as potent immunosuppressive agents.68 Rheumatoid arthritis and inflammatory bowel disease are usually associated with conditions and situations that predispose patients to opportunistic infections, such as decreased immune response, malnutrition or malabsorption, surgeries, and concomitant immunosuppressive medications.7 Combination therapy with other immunosuppressive drugs and older age appear to markedly increase the risk of opportunistic infections, including mycobacterial and fungal infections, in patients with inflammatory bowel disease.7
Overall, opportunistic infections represent a measurable risk of anti-TNF therapy, and awareness and vigilance are important, especially in areas where opportunistic infections such as histoplasmosis and coccidiomycosis are endemic.50 Furthermore, physicians must be aware of the higher risk of opportunistic infections when multiple immunosuppressive drugs are used concurrently.
Granulomatous infections such as tuberculosis
Anti-TNF agents increase the risk of de novo granulomatous infections and of reactivating such infections. Granuloma formation and intracellular destruction of mycobacteria depend on TNF. TNF is important in maintaining the anatomic integrity of granulomas where these organisms have been sequestered, and blocking TNF leads to breakdown of granulomas and release of virulent organisms.69,70
TNF inhibitors increase the risk of reactivation of latent tuberculosis infection. The risk is greater with infliximab and adalimumab than with etanercept,71,72 and it has been described with certolizumab.74 Study results are varied thus far but show a risk of tuberculosis reactivation five to 30 times higher than in the general population, with tremendous variability in risk depending on background rates of previous exposure.
The absence of typical tuberculosis symptoms further complicates care in these cases. Fever, weight loss, and night sweats tend to be TNF-mediated and are therefore masked by anti-TNF agents, leading to atypical presentations. In addition, active tuberculosis infection associated with TNF inhibitors is more likely to involve extrapulmonary sites such as the skin and musculoskeletal system and to be disseminated at presentation.
A paradoxical worsening of tuberculosis symptoms may also be seen in patients with latent tuberculosis reactivation, especially after discontinuing anti-TNF therapy. This is thought to result from an immune reconstitution inflammatory syndrome.
The pretreatment evaluation should include a history of risk factors, a physical examination, and either a tuberculin skin test or an interferon-gamma-release assay. Interferon-gamma-release assays are particularly helpful in patients who have received bacille Calmette-Guérin vaccination. In patients who test positive or have been exposed, tuberculosis treatment should begin 4 weeks before starting anti-TNF therapy, though the optimal timing of antituberculosis agents is still controversial.74–77
If tuberculosis develops in a patient on anti-TNF therapy, he or she should receive antituberculosis drugs. Anti-TNF therapy should be stopped and should be resumed after 2 months only if no other treatment option is available.75
Invasive opportunistic fungal infections
Invasive opportunistic fungal infections have been reported with anti-TNF therapy, including histoplasmosis and coccidioidomycosis.78–80 Most patients who had histoplasmosis were treated with other immunosuppressive therapies and resided in or were raised near the Ohio or Mississippi River valleys, where this disease is endemic. Similarly, most cases of coccidioidomycosis were in endemic areas of Arizona, California, and Nevada, and patients on concomitant immunosuppressive therapy.78
Currently, there is no evidence to recommend obtaining Histoplasma capsulatum or Coccidioides immitis serologies before initiating anti-TNF therapy in patients in endemic areas.81 However, patients must be instructed to seek medical attention quickly for pulmonary or febrile illnesses.
Viral hepatitis infections
The data on hepatitis B and hepatitis C in patients on biologic therapies are mostly limited to case reports.
Hepatitis B. A small prospective study from Spain followed the liver biochemistry tests and hepatitis B status of 80 patients with Crohn disease treated with infliximab. Of three patients who were chronic hepatitis B carriers before starting infliximab, two experienced reactivation of hepatitis B after discontinuing infliximab, and one ultimately died. The third patient was treated with lamivudine concurrently with infliximab without clinical or biochemical changes during or after therapy.82
Similar findings were observed in two patients with rheumatoid arthritis on treatment with infliximab. One of the patients required liver transplantation, and both were treated with lamivudine, resulting in normalization of liver function test results.83,84
Recent reviews indicate that despite these findings, hepatitis B reactivation after anti-TNF withdrawal may not be common.85 There are limited data on hepatitis B reactivation and associated liver dysfunction in patients with inflammatory bowel disease treated with immunosuppressants. In a retrospective multicenter trial by Loras et al86 in patients with inflammatory bowel disease who had viral hepatitis, 36% of patients positive for hepatitis B surface antigen developed liver dysfunction, and six patients developed liver failure. In that study, treatment with more than two immunosuppressants was an independent predictor of hepatitis B reactivation.
Prolonged immunosuppression (longer than 3 months) has also been identified as an independent predictor of liver dysfunction (OR 3.06; 95% 95% CI 1.02–9.16).87
The European Association for the Study of the Liver and the American Association for the Study of Liver Diseases recommend starting lamivudine before chemotherapy or immunomodulator or immunosuppressive therapy in hepatitis B virus carriers and continuing preventive treatment for at least 6 months after stopping immunomodulating drugs. Lamivudine at a dose of 100 mg/day may reduce the risk of reactivation of hepatitis B.88–90 Tenofovir and entecavir may be useful alternatives in patients with hepatitis B who have never received nucleoside analogues. Hepatitis B reactivation did not occur in any of the 16 patients who received preventive entecavir treatment while receiving immunosuppressive treatments.89,91
In patients receiving immunosuppressive therapy, hepatitis B reactivation is associated with significant morbidity and mortality. Although risk factors for reactivation of hepatitis B virus infection have been identified, we recommend preventive treatment for all carriers positive for hepatitis B surface antigen. This should be done regardless of the number, type, and dosage of immunosuppressants and regardless of hepatitis B virus DNA levels.90
The frequency of hepatitis B and hepatitis C infection in patients with Crohn disease has been reported to be as high as 24%. The high incidence is thought to be secondary to multiple blood transfusions and surgeries.92 The use of biologic agents, including anti-TNF agents, in chronic hepatitis B virus- or hepatitis C virus-infected patients can lead to enhanced viral replication and hepatitis exacerbation.
Although active viral replication can occur during treatment with biologic agents, reactivation or exacerbation can also occur after the anti-TNF agent is stopped.82 This finding has prompted the recommendation that all candidates for biologic therapy be tested for hepatitis B immunization status, followed by immunization in nonimmune patients before starting anti-TNF therapy.93,94
Hepatitis C. There are no guidelines that adequately address the use of anti-TNF agents in patients with chronic hepatitis C infection.
Several small retrospective studies in rheumatoid arthritis patients with hepatitis C have shown that TNF inhibitors can be safely used without worsening liver function tests or changing the viral load.95–98 This is reassuring and provides the subspecialist with another treatment option, as other therapies such as disease-modifying antirheumatic drugs and steroids are known to aggravate viral hepatitis and increase the risk of viremia.96
Although small retrospective studies and one large randomized double-blind placebo-controlled trial have shown TNF inhibitors to be relatively safe in rheumatoid arthritis patients with hepatitis C, their use in these patients should be considered only with caution if they have evidence of hepatic synthetic dysfunction (eg, hypoalbuminemia, thrombocytopenia, increased international normalized ratio). The American College of Rheumatology recommends avoiding TNF inhibitors in Child-Pugh classes B and C.99
PREGNANCY
Physicians caring for patients with rheumatoid arthritis and inflammatory bowel disease must be aware of how these diseases affect fecundity and fertility and how the medications can affect conception and pregnancy. Many patients have difficulty conceiving while their autoimmune disease is active, and better disease control may improve fecundity and result in unanticipated pregnancy. Patients should be advised of the need for contraception if pregnancy is ill-advised or undesired.
Many patients seek advice about teratogenicity before conceiving, or seek guidance about rheumatoid arthritis and inflammatory bowel disease treatment while pregnant.
Several studies have reported a higher risk of adverse pregnancy outcomes in patients with rheumatoid arthritis and inflammatory bowel disease than in the general population.99–105 In inflammatory bowel disease, the odds of a premature delivery or having a low-birth-weight child are twice as high as in the normal population.100,103 Higher rates of cesarean delivery and stillbirth have also been reported. The main predisposing factor appears to be the disease activity at the time of conception, as active disease seems to be linked to adverse pregnancy outcomes.
Treatment with anti-TNF agents may rapidly achieve and maintain remission, raising the question of the safety of continued anti-TNF use during pregnancy. The FDA classifies anti-TNF agents as category B drugs, as animal studies have not demonstrated fetal risk, and no well-controlled prospective study has yet been conducted with pregnant women.
In an observational study, Schnitzler et al105 assessed the outcomes of 42 pregnancies in 35 patients with inflammatory bowel disease receiving either infliximab or adalimumab during pregnancy, compared with 56 pregnancies in 45 healthy patients without inflammatory bowel disease. There was no statistical difference in abortion rates between patients receiving anti-TNF agents and healthy women without inflammatory bowel disease (21% vs 14%, P = .4234). There was also no significant difference observed in birth weight, birth length, or cranial circumference of the children between the two groups. However, pregnancies with direct exposure to anti-TNF agents resulted in a higher frequency of premature delivery (25% vs 6%, P = .023).
Similar results were noted from the Crohn’s Therapy, Resource, Evaluation, and Assessment Tool, or TREAT, registry, as well as from a large systematic review by Vinet et al106 and case reports of rheumatoid arthritis and inflammatory bowel disease in women exposed to anti-TNF agents during pregnancy.
In addition, results from a recent systematic review of 38 studies of anti-TNF use and fetal risk, with a total of 437 women (189 on infliximab, 230 on adalimumab, 18 on certolizumab pegol), showed similar results.107 In pregnancies exposed to anti-TNF agents, the rates of congenital abnormalities (3.4%), fetal deaths (8.5%), and preterm births (2.7%) were similar to those in the general population.
For patients in disease remission on TNF inhibitors, it is reasonable to continue these agents during pregnancy after careful discussion with the patient. Fetal safety and infant immunization response after delivery are the primary concerns in these cases.
Both infliximab and adalimumab cross the placenta and remain detectable in the baby’s circulation 4 months (for adalimumab) to 6 months (for infliximab) after delivery. It is currently recommended that infliximab be stopped at 32 weeks of gestation for the remainder of the pregnancy and that adalimumab be stopped at 34 to 36 weeks, given a planned 40-week gestation.
Certolizumab does not cross the placenta in significant amounts and should be continued throughout pregnancy; drug levels in infants were shown to be less than 2 μg/mL, even when dosed the week of delivery.108–112
TAKE-HOME POINTS
- All health care providers of patients with rheumatoid arthritis and inflammatory bowel disease should be familiar with anti-TNF agents used in treating these diseases.
- The benefits of controlling the disease far outweigh the risks of therapy when used appropriately.
- Care of these patients should be multidisciplinary, with clear communication between primary care physician and specialist.
- Patient education and monitoring combined with prompt communication between primary care physician and specialist are key.
- Wijbrandts CA, Dijkgraaf MG, Kraan MC, et al. The clinical response to infliximab in rheumatoid arthritis is in part dependent on pretreatment tumour necrosis factor alpha expression in the synovium. Ann Rheum Dis 2008; 67:1139–1144.
- Sfikakis PP. The first decade of biologic TNF antagonists in clinical practice: lessons learned, unresolved issues and future directions. Curr Dir Autoimmun 2010; 11:180–210.
- Casellas F, Alcalá MJ, Prieto L, Miró JR, Malagelada JR. Assessment of the influence of disease activity on the quality of life of patients with inflammatory bowel disease using a short questionnaire. Am J Gastroenterol 2004; 99:457–461.
- Casellas F, López-Vivancos J, Badia X, Vilaseca J, Malagelada JR. Influence of inflammatory bowel disease on different dimensions of quality of life. Eur J Gastroenterol Hepatol 2001; 13:567–572.
- Baecklund E, Iliadou A, Askling J, et al. Association of chronic inflammation, not its treatment, with increased lymphoma risk in rheumatoid arthritis. Arthritis Rheum 2006; 54:692–701.
- Franklin J, Lunt M, Bunn D, Symmons D, Silman A. Incidence of lymphoma in a large primary care derived cohort of cases of inflammatory polyarthritis. Ann Rheum Dis 2006; 65:617–622.
- Toruner M, Loftus EV, Harmsen WS, et al. Risk factors for opportunistic infections in patients with inflammatory bowel disease. Gastroenterology 2008; 134:929–936.
- Wolfe F, Michaud K. Biologic treatment of rheumatoid arthritis and the risk of malignancy: analyses from a large US observational study. Arthritis Rheum 2007; 56:2886–2895.
- Prevention of pneumococcal disease: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 1997; 46:1–24.
- Zhang J, Xie F, Delzell E, et al. Association between vaccination for herpes zoster and risk of herpes zoster infection among older patients with selected immune-mediated diseases. JAMA 2012; 308:43–49.
- Moreland LW, Baumgartner SW, Schiff MH, et al. Treatment of rheumatoid arthritis with a recombinant human tumor necrosis factor receptor (p75)-Fc fusion protein. N Engl J Med 1997; 337:141–147.
- Altunöz ME, Senates E, Yesil A, Calhan T, Ovünç AO. Patients with inflammatory bowel disease have a lower response rate to HBV vaccination compared to controls. Dig Dis Sci 2012; 57:1039–1044.
- Gisbert JP, Villagrasa JR, Rodríguez-Nogueiras A, Chaparro M. Efficacy of hepatitis B vaccination and revaccination and factors impacting on response in patients with inflammatory bowel disease. Am J Gastroenterol 2012; 107:1460–1466.
- Carrera E, Manzano R, Garrido E. Efficacy of the vaccination in inflammatory bowel disease. World J Gastroenterol 2013; 19:1349–1353.
- Gisbert JP, Menchén L, García-Sánchez V, Marín I, Villagrasa JR, Chaparro M. Comparison of the effectiveness of two protocols for vaccination (standard and double dosage) against hepatitis B virus in patients with inflammatory bowel disease. Aliment Pharmacol Ther 2012; 35:1379–1385.
- Ferkolj I. How to improve the safety of biologic therapy in Crohn’s disease. J Physiol Pharmacol 2009; 60(suppl 7):67–70.
- Siegel CA. The risks of biologic therapy for inflammatory bowel disease. In:Bernstein ED, editor. The Inflammatory Bowel Disease Yearbook, volume 6. London, UK: Remedica, 2010:89–108.
- Remicade (infliximab) Package Insert. Horsham, PA: Janssen Biotech, Inc; 2013. http://www.remicade.com/shared/product/remicade/prescribing-information.pdf. Accessed January 2, 2014.
- Vermeire S, Noman M, Van Assche G, et al. Autoimmunity associated with anti-tumor necrosis factor alpha treatment in Crohn’s disease: a prospective cohort study. Gastroenterology 2003; 125:32–39.
- Cush JJ. Biological drug use: US perspectives on indications and monitoring. Ann Rheum Dis 2005; 64(suppl 4):iv18–iv23.
- TNF neutralization in MS: results of a randomized, placebo-controlled multicenter study. The Lenercept Multiple Sclerosis Study Group and The University of British Columbia MS/MRI Analysis Group. Neurology 1999; 53:457–465.
- Chung ES, Packer M, Lo KH, Fasanmade AA, Willerson JT; Anti-TNF Therapy Against Congestive Heart Failure Investigators. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: results of the anti-TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation 2003; 107:3133–3140.
- Cheifetz A, Mayer L. Monoclonal antibodies, immunogenicity, and associated infusion reactions. Mt Sinai J Med 2005; 72:250–256.
- Cheifetz A, Smedley M, Martin S, et al. The incidence and management of infusion reactions to infliximab: a large center experience. Am J Gastroenterol 2003; 98:1315–1324.
- Vultaggio A, Matucci A, Nencini F, et al. Anti-infliximab IgE and non-IgE antibodies and induction of infusion-related severe anaphylactic reactions. Allergy 2010; 65:657–661.
- Ramos-Casals M, Brito-Zerón P, Muñoz S, et al. Autoimmune diseases induced by TNF-targeted therapies: analysis of 233 cases. Medicine (Baltimore) 2007; 86:242–251.
- Stallmach A, Hagel S, Bruns T. Adverse effects of biologics used for treating IBD. Best Pract Res Clin Gastroenterol 2010; 24:167–182.
- Haagsma CJ, Blom HJ, van Riel PL, et al. Influence of sulphasalazine, methotrexate, and the combination of both on plasma homocysteine concentrations in patients with rheumatoid arthritis. Ann Rheum Dis 1999; 58:79–84.
- Fiorino G, Allez M, Malesci A, Danese S. Review article: anti TNF-alpha induced psoriasis in patients with inflammatory bowel disease. Aliment Pharmacol Ther 2009; 29:921–927.
- Harrison MJ, Dixon WG, Watson KD, et al; British Society for Rheumatology Biologics Register Control Centre Consortium; BSRBR. Rates of new-onset psoriasis in patients with rheumatoid arthritis receiving anti-tumour necrosis factor alpha therapy: results from the British Society for Rheumatology Biologics Register. Ann Rheum Dis 2009; 68:209–215.
- Marchesoni A, Zaccara E, Gorla R, et al. TNF-alpha antagonist survival rate in a cohort of rheumatoid arthritis patients observed under conditions of standard clinical practice. Ann N Y Acad Sci 2009; 1173:837–846.
- Tomas L, Lazurova I, Pundova L, et al. Acute and long-term effect of infliximab on humoral and echocardiographic parameters in patients with chronic inflammatory diseases. Clin Rheumatol 2013 32:61–66.
- Senel S, Cobankara V, Taskoylu O, et al. The safety and efficacy of etanercept on cardiac functions and lipid profile in patients with active rheumatoid arthritis. J Investig Med 2012; 60:62–65.
- Al-Aly Z, Pan H, Zeringue A, et al. Tumor necrosis factor-a blockade, cardiovascular outcomes, and survival in rheumatoid arthritis. Transl Res 2011; 157:10–18.
- Listing J, Strangfeld A, Kekow J, et al. Does tumor necrosis factor alpha inhibition promote or prevent heart failure in patients with rheumatoid arthritis? Arthritis Rheum 2008; 58:667–677.
- Wolfe F, Michaud K. Heart failure in rheumatoid arthritis: rates, predictors, and the effect of anti-tumor necrosis factor therapy. Am J Med 2004; 116:305–311.
- Enayati PJ, Papadakis KA. Association of anti-tumor necrosis factor therapy with the development of multiple sclerosis. J Clin Gastroenterol 2005; 39:303–306.
- Mohan N, Edwards ET, Cupps TR, et al. Demyelination occurring during anti-tumor necrosis factor alpha therapy for inflammatory arthritides. Arthritis Rheum 2001; 44:2862–2869.
- Sands BE, Anderson FH, Bernstein CN, et al. Infliximab maintenance therapy for fistulizing Crohn’s disease. N Engl J Med 2004; 350:876–885.
- Thomas CW, Weinshenker BG, Sandborn WJ. Demyelination during anti-tumor necrosis factor alpha therapy with infliximab for Crohn’s disease. Inflamm Bowel Dis 2004; 10:28–31.
- Foroozan R, Buono LM, Sergott RC, Savino PJ. Retrobulbar optic neuritis associated with infliximab. Arch Ophthalmol 2002; 120:985–987.
- Strong BY, Erny BC, Herzenberg H, Razzeca KJ. Retrobulbar optic neuritis associated with infliximab in a patient with Crohn disease. Ann Intern Med 2004; 140:W34.
- ten Tusscher MP, Jacobs PJ, Busch MJ, de Graaf L, Diemont WL. Bilateral anterior toxic optic neuropathy and the use of infliximab. BMJ 2003; 326:579.
- Hegde N, Gayomali C, Rich MW. Infliximab-induced headache and infliximab-induced meningitis: two ends of the same spectrum? South Med J 2005; 98:564–566.
- Schiff MH, Burmester GR, Kent JD, et al. Safety analyses of adalimumab (HUMIRA) in global clinical trials and US postmarketing surveillance of patients with rheumatoid arthritis. Ann Rheum Dis 2006; 65:889–894.
- Beaugerie L, Brousse N, Bouvier AM, et al; CESAME Study Group. Lymphoproliferative disorders in patients receiving thiopurines for inflammatory bowel disease: a prospective observational cohort study. Lancet 2009; 374:1617–1625.
- Ekström K, Hjalgrim H, Brandt L, et al. Risk of malignant lymphomas in patients with rheumatoid arthritis and in their first-degree relatives. Arthritis Rheum 2003; 48:963–970.
- Lakatos PL, Lakatos L. Risk for colorectal cancer in ulcerative colitis: changes, causes and management strategies. World J Gastroenterol 2008; 14:3937–3947.
- Persson PG, Karlén P, Bernell O, et al. Crohn’s disease and cancer: a population-based cohort study. Gastroenterology 1994; 107:1675–1679.
- de Silva S, Devlin S, Panaccione R. Optimizing the safety of biologic therapy for IBD. Nat Rev Gastroenterol Hepatol 2010; 7:93–101.
- Lichtenstein GR, Feagan BG, Cohen RD, et al. Serious infections and mortality in association with therapies for Crohn’s disease: TREAT registry. Clin Gastroenterol Hepatol 2006; 4:621–630.
- Peyrin-Biroulet L, Deltenre P, de Suray N, Branche J, Sandborn WJ, Colombel JF. Efficacy and safety of tumor necrosis factor antagonists in Crohn’s disease: meta-analysis of placebo-controlled trials. Clin Gastroenterol Hepatol 2008; 6:644–653.
- Hanauer SB, Feagan BG, Lichtenstein GR, et al; ACCENT I Study Group. Maintenance infliximab for Crohn’s disease: the ACCENT I randomised trial. Lancet 2002; 359:1541–1549.
- Colombel JF, Sandborn WJ, Rutgeerts P, et al. Adalimumab for maintenance of clinical response and remission in patients with Crohn’s disease: the CHARM trial. Gastroenterology 2007; 132:52–65.
- Sandborn WJ, Feagan BG, Stoinov S, et al; PRECISE 1 Study Investigators. Certolizumab pegol for the treatment of Crohn’s disease. N Engl J Med 2007; 357:228–238.
- Sandborn WJ, Hanauer SB, Rutgeerts P, et al. Adalimumab for maintenance treatment of Crohn’s disease: results of the CLASSIC II trial. Gut 2007; 56:1232–1239.
- Hanauer SB, Sandborn WJ, Rutgeerts P, et al. Human anti-tumor necrosis factor monoclonal antibody (adalimumab) in Crohn’s disease: the CLASSIC-I trial. Gastroenterology 2006; 130:323–333.
- Rutgeerts P, D’Haens G, Targan S, et al. Efficacy and safety of retreatment with anti-tumor necrosis factor antibody (infliximab) to maintain remission in Crohn’s disease. Gastroenterology 1999; 117:761–769.
- Rutgeerts P, Sandborn WJ, Feagan BG, et al. Infliximab for induction and maintenance therapy for ulcerative colitis. N Engl J Med 2005; 353:2462–2476.
- Pedersen N, Duricova D, Elkjaer M, Gamborg M, Munkholm P, Jess T. Risk of extra-intestinal cancer in inflammatory bowel disease: meta-analysis of population-based cohort studies. Am J Gastroenterol 2010; 105:1480–1487.
- Bernstein CN, Blanchard JF, Kliewer E, Wajda A. Cancer risk in patients with inflammatory bowel disease: a population-based study. Cancer 2001; 91:854–862.
- Siegel CA, Marden SM, Persing SN, Larson RJ, Sands BE. Risk of lymphoma associated with combination anti-tumor necrosis factor and immunomodulator therapy for the treatment of Crohn’s disease: a meta-analysis. Clin Gastroenterol Hepatol 2009; 7:874–881.
- Wolfe F, Michaud K. Lympyhoma in rheumatoid arthritis. The effect of methotrexate and anti-tumor necrosis factor therapy in 18,572 patients. Arthritis Rheum 2004; 50:1740–1751.
- Parakkal D, Sifuentes H, Semer R, Ehrenpreis ED. Hepatosplenic T-cell lymphoma in patients receiving TNF-a inhibitor therapy: expanding the groups at risk. Eur J Gastroenterol Hepatol 2011; 23:1150–1156.
- Rosh JR, Gross T, Mamula P, Griffiths A, Hyams J. Hepatosplenic T-cell lymphoma in adolescents and young adults with Crohn’s disease: a cautionary tale? Inflamm Bowel Dis 2007; 13:1024–1030.
- Shale M, Kanfer E, Panaccione R, Ghosh S. Hepatosplenic T cell lymphoma in inflammatory bowel disease. Gut 2008; 57:1639–1641.
- Thai A, Prindiville T. Hepatosplenic T-cell lymphoma and inflammatory bowel disease. J Crohns Colitis 2010; 4:511–522.
- Viget N, Vernier-Massouille G, Salmon-Ceron D, Yazdanpanah Y, Colombel JF. Opportunistic infections in patients with inflammatory bowel disease: prevention and diagnosis. Gut 2008; 57:549–558.
- Bekker LG, Freeman S, Murray PJ, Ryffel B, Kaplan G. TNF-alpha controls intracellular mycobacterial growth by both inducible nitric oxide synthase-dependent and inducible nitric oxide synthase-independent pathways. J Immunol 2001; 166:6728–6734.
- Roach DR, Bean AG, Demangel C, France MP, Briscoe H, Britton WJ. TNF regulates chemokine induction essential for cell recruitment, granuloma formation, and clearance of mycobacterial infection. J Immunol 2002; 168:4620–4627.
- Gómez-Reino JJ, Carmona L, Valverde VR, Mola EM, Montero MDBIOBADASER Group. Treatment of rheumatoid arthritis with tumor necrosis factor inhibitors may predispose to significant increase in tuberculosis risk: a multicenter active-surveillance report. Arthritis Rheum 2003; 48:2122–2127.
- Keane J, Gershon S, Wise RP, et al. Tuberculosis associated with infliximab, a tumor necrosis factor alpha-neutralizing agent. N Engl J Med 2001; 345:1098–1104.
- Smolen J, Landewé RB, Mease P, et al. Efficacy and safety of certolizumab pegol plus methotrexate in active rheumatoid arthritis: the RAPID 2 study. A randomised controlled trial. Ann Rheum Dis 2009; 68:797–804.
- Demkow U, Broniarek-Samson B, Filewska M, et al. Prevalence of latent tuberculosis infection in health care workers in Poland assessed by interferon-gamma whole blood and tuberculin skin tests. J Physiol Pharmacol 2008; 59(suppl 6):209–217.
- Pache I, Rogler G, Felley C. TNF-alpha blockers in inflammatory bowel diseases: practical consensus recommendations and a user’s guide. Swiss Med Wkly 2009; 139:278–287.
- Rahier JF, Ben-Horin S, Chowers Y, et al; European Crohn’s and Colitis Organisation (ECCO). European evidence-based consensus on the prevention, diagnosis and management of opportunistic infections in inflammatory bowel disease. J Crohns Colitis 2009; 3:47–91.
- Rahier JF, Yazdanpanah Y, Colombel JF, Travis S. The European (ECCO) consensus on infection in IBD: what does it change for the clinician? Gut 2009; 58:1313–1315.
- Bergstrom L, Yocum DE, Ampel NM, et al. Increased risk of coccidioidomycosis in patients treated with tumor necrosis factor alpha antagonists. Arthritis Rheum 2004; 50:1959–1966.
- Lee JH, Slifman NR, Gershon SK, et al. Life-threatening histoplasmosis complicating immunotherapy with tumor necrosis factor alpha antagonists infliximab and etanercept. Arthritis Rheum 2002; 46:2565–2570.
- Wood KL, Hage CA, Knox KS, et al. Histoplasmosis after treatment with anti-tumor necrosis factor-alpha therapy. Am J Respir Crit Care Med 2003; 167:1279–1282.
- Reddy JG, Loftus EV. Safety of infliximab and other biologic agents in the inflammatory bowel diseases. Gastroenterol Clin North Am 2006; 35:837–855.
- Esteve M, Saro C, González-Huix F, Suarez F, Forné M, Viver JM. Chronic hepatitis B reactivation following infliximab therapy in Crohn’s disease patients: need for primary prophylaxis. Gut 2004; 53:1363–1365.
- Michel M, Duvoux C, Hezode C, Cherqui D. Fulminant hepatitis after infliximab in a patient with hepatitis B virus treated for an adult onset Still’s disease. J Rheumatol 2003; 30:1624–1625.
- Ostuni P, Botsios C, Punzi L, Sfriso P, Todesco S. Hepatitis B reactivation in a chronic hepatitis B surface antigen carrier with rheumatoid arthritis treated with infliximab and low dose methotrexate. Ann Rheum Dis 2003; 62:686–687.
- Pérez-Alvarez R, Díaz-Lagares C, García-Hernández F, et al; BIOGEAS Study Group. Hepatitis B virus (HBV) reactivation in patients receiving tumor necrosis factor (TNF)-targeted therapy: analysis of 257 cases. Medicine (Baltimore) 2011; 90:359–371.
- Loras C, Gisbert JP, Mínguez M, et al; REPENTINA study; GETECCU (Grupo Español de Enfermedades de Crohn y Colitis Ulcerosa) Group. Liver dysfunction related to hepatitis B and C in patients with inflammatory bowel disease treated with immunosuppressive therapy. Gut 2010; 59:1340–1346.
- Park SH, Yang SK, Lim YS, et al. Clinical courses of chronic hepatitis B virus infection and inflammatory bowel disease in patients with both diseases. Inflamm Bowel Dis 2012; 18:2004–2010.
- Loomba R, Rowley A, Wesley R, et al. Systematic review: the effect of preventive lamivudine on hepatitis B reactivation during chemotherapy. Ann Intern Med 2008; 148:519–528.
- Lok AS, McMahon BJ. Chronic hepatitis B: update 2009. Hepatology 2009; 50:661–662.
- European Association For The Study Of The Liver. EASL Clinical Practice Guidelines: management of chronic hepatitis B. J Hepatol 2009; 50:227–242.
- Watanabe M, Shibuya A, Takada J, et al. Entecavir is an optional agent to prevent hepatitis B virus (HBV) reactivation: a review of 16 patients. Eur J Intern Med 2010; 21:333–337.
- Biancone L, Pavia M, Del Vecchio Blanco G, et al; Italian Group for the Study of the Colon and Rectum (GISC). Hepatitis B and C virus infection in Crohn’s disease. Inflamm Bowel Dis 2001; 7:287–294.
- Melmed GY. Vaccination strategies for patients with inflammatory bowel disease on immunomodulators and biologics. Inflamm Bowel Dis 2009; 15:1410–1416.
- Melmed GY, Ippoliti AF, Papadakis KA, et al. Patients with inflammatory bowel disease are at risk for vaccine-preventable illnesses. Am J Gastroenterol 2006; 101:1834–1840.
- Ferri C, Ferraccioli G, Ferrari D, et al. Safety of anti-tumor necrosis factor-alpha therapy in patients with rheumatoid arthritis and chronic hepatitis C virus infection. J Rheumatol 2008; 35:1944–1949.
- Mok MY, Ng WL, Yuen MF, Wong RW, Lau CS. Safety of disease modifying anti-rheumatic agents in rheumatoid arthritis patients with chronic viral hepatitis. Clin Exp Rheumatol 2000; 18:363–368.
- Vassilopoulos D, Calabrese LH. Risks of immunosuppressive therapies including biologic agents in patients with rheumatic diseases and coexisting chronic viral infections. Curr Opin Rheumatol 2007; 19:619–625.
- Vassilopoulos D, Apostolopoulou A, Hadziyannis E, et al. Long-term safety of anti-TNF treatment in patients with rheumatic diseases and chronic or resolved hepatitis B virus infection. Ann Rheum Dis 2010; 69:1352–1355.
- Saag KG, Teng GG, Patkar NM, et al; American College of Rheumatology. American College of Rheumatology 2008 recommendations for the use of nonbiologic and biologic disease-modifying antirheumatic drugs in rheumatoid arthritis. Arthritis Rheum 2008; 59:762–784.
- Cornish J, Tan E, Teare J, et al. A meta-analysis on the influence of inflammatory bowel disease on pregnancy. Gut 2007; 56:830–837.
- Dominitz JA, Young JC, Boyko EJ. Outcomes of infants born to mothers with inflammatory bowel disease: a population-based cohort study. Am J Gastroenterol 2002; 97:641–648.
- Kornfeld D, Cnattingius S, Ekbom A. Pregnancy outcomes in women with inflammatory bowel disease—a population-based cohort study. Am J Obstet Gynecol 1997; 177:942–946.
- Mahadevan U, Sandborn WJ, Li DK, Hakimian S, Kane S, Corley DA. Pregnancy outcomes in women with inflammatory bowel disease: a large community-based study from Northern California. Gastroenterology 2007; 133:1106–1112.
- Nguyen GC, Boudreau H, Harris ML, Maxwell CV. Outcomes of obstetric hospitalizations among women with inflammatory bowel disease in the United States. Clin Gastroenterol Hepatol 2009; 7:329–334.
- Schnitzler F, Fidder H, Ferrante M, et al. Outcome of pregnancy in women with inflammatory bowel disease treated with antitumor necrosis factor therapy. Inflamm Bowel Dis 2011; 17:1846–1854.
- Vinet E, Pineau C, Gordon C, Clarke AE, Bernatsky S. Biologic therapy and pregnancy outcomes in women with rheumatic diseases. Arthritis Rheum 2009; 61:587–592.
- Guidi L, Pugliese D, Armuzzi A. Update on the management of inflammatory bowel disease: specific role of adalimumab. Clin Exp Gastroenterol 2011; 4:163–172.
- Mahadevan U. Continuing immunomodulators and biologic medications in pregnant IBD patients – pro. IInflamm Bowel Dis 2007; 13:1439–1440.
- Mahadevan U. Gastrointestinal medications in pregnancy. Best Pract Res Clin Gastroenterol 2007; 21:849–877.
- Mahadevan U, Cucchiara S, Hyams JS, et al. The London Position Statement of the World Congress of Gastroenterology on Biological Therapy for IBD with the European Crohn’s and Colitis Organisation: pregnancy and pediatrics. Am J Gastroenterol 2011; 106:214–223.
- Mahadevan U, Kane S. Use of infliximab in pregnancy. Am J Gastroenterol 2010; 105:219–220.
- Vasiliauskas EA, Church JA, Silverman N, Barry M, Targan SR, Dubinsky MC. Case report: evidence for transplacental transfer of maternally administered infliximab to the newborn. Clin Gastroenterol Hepatol 2006; 4:1255–1258.
Biologic agents such as those that block tumor necrosis factor (TNF) alpha have revolutionized the treatment of autoimmune diseases like rheumatoid arthritis and inflammatory bowel disease, dramatically improving disease control and quality of life. In addition, they have made true disease remission possible in some cases.
However, as with any new therapy, a variety of side effects must be considered.
WHAT ARE BIOLOGIC AGENTS?
Biologic agents are genetically engineered drugs manufactured or synthesized in vitro from molecules such as proteins, genes, and antibodies present in living organisms. By targeting specific molecular components of the inflammatory cascade, including cytokines, these drugs alter certain aspects of the body’s inflammatory response in autoimmune diseases. Because TNF inhibitors are the most widely used biologic agents in the United States, this review will focus on them.
TNF INHIBITORS
TNF inhibitors suppress the inflammatory cascade by inactivating TNF alpha, a cytokine that promotes inflammation in the intestine, synovial tissue, and other sites.1,2
Several TNF inhibitors are available. Etanercept and infliximab were the first two to receive US Food and Drug Administration (FDA) approval, and they are extensively used. Etanercept, a soluble TNF receptor given subcutaneously, was approved for treating rheumatoid arthritis in 1998. Infliximab, a chimeric monoclonal antibody (75% human and 25% mouse protein sequence) given intravenously, received FDA approval for treating Crohn disease in 1998 and for rheumatoid arthritis in 1999. Other anti-TNF agents with varying properties are also available.
Table 1 lists anti-TNF agents approved for treating rheumatoid arthritis and inflammatory bowel disease. These are chronic, relapsing diseases that significantly and dramatically reduce quality of life, especially when they are poorly controlled.3,4 Untreated disease has been associated with malignancy, infection, pregnancy loss, and malnutrition.5–8 TNF inhibitors are aimed at patients who have moderate to severe disease or for whom previous treatments have failed, to help them achieve and maintain steroid-free remission. However, use of these agents is tempered by the risk of potentially serious side effects.
Special thought should also be given to the direct costs of the drugs (up to $30,000 per year, not counting the cost of their administration) and to the indirect costs such as time away from work to receive treatment. These are major considerations in some cases, and patients should be selected carefully for treatment with these drugs.
BEFORE STARTING THERAPY
Before starting anti-TNF therapy, several steps can reduce the risk of serious adverse events.
Take a focused history
The clinical history should include inquiries about previous bacterial, fungal, and tuberculosis infections or exposure; diabetes; and other immunocompromised states that increase the risk of acquiring potentially life-threatening infections.
Details of particular geographic areas of residence, occupational exposures, and social history should be sought. These include history of incarceration (which may put patients at risk of tuberculosis) and residence in the Ohio River valley or in the southwestern or midwestern United States (which may increase the risk of histoplasmosis, coccidioidomycosis, and other fungal infections).
Bring vaccinations up to date
Age-appropriate vaccinations should be discussed and given, ideally before starting therapy. These include influenza vaccine every year and tetanus boosters every 10 years for all and, as appropriate, varicella, human papillomavirus, and pneumococcal vaccinations. The US Centers for Disease Control and Prevention recommend an additional dose of the pneumococcal vaccine if more than 5 years have elapsed since the first one, and many clinicians opt to give it every 5 years.
In general, live-attenuated vaccines, including the intranasal influenza vaccine, are contraindicated in patients taking biologic agents.9 For patients at high risk of exposure or infection, it may be reasonable to hold the biologic agent for a period of time, vaccinate, and resume the biologic agent a month later.
Recent data suggest that the varicella zoster vaccine may be safely given to older patients with immune-mediated diseases such as rheumatoid arthritis and inflammatory bowel disease taking biologic agents.10 New guidelines from the American College of Rheumatology recommend age-appropriate vaccines for rheumatoid arthritis patients age 60 and older before biologic treatments are started. Case-by-case discussion with the subspecialist and the patient is recommended.
Screen for chronic infections
Tuberculosis screening with a purified protein derivative test or an interferon-gamma-release (Quantiferon) assay followed by chest radiography in patients with a positive test is mandatory before giving a TNF inhibitor.
Hepatitis B virus status should be determined before starting anti-TNF therapy.11
Hepatitis B vaccination has been recommended for patients with inflammatory bowel disease, but no clear recommendation exists for patients with rheumatic disease. Patients with inflammatory bowel disease tend to have low rates of response to hepatitis B vaccination12,13; possible reasons include their lack of an appropriate innate immune response to infectious agents, malnutrition, surgery, older age, and immunosuppressive drugs.14 An accelerated vaccination protocol with recombinant hepatitis B vaccine (Energix-B) in a double dose at 0, 1, and 2 months has been shown to improve response rates.15
Whenever possible, it may be better to vaccinate patients before starting immunosuppressive therapy, and to check postvaccination titers to ensure adequate response.
Perform an examination
A full physical examination with special attention to skin rashes should be performed. This may serve as a baseline to assist early detection of new rashes associated with anti-TNF therapy.
A baseline complete blood cell count and complete metabolic panel should be routinely obtained before starting therapy (and thereafter at the discretion of the physician). In conjunction with follow-up tests, they can help detect an unexpected decrease in white blood cell count or abnormal results on the liver panel.16 These baseline and follow-up tests are generally performed by the subspecialist, and the results are shared with the primary care physician.
Table 2 summarizes key information to be sought before starting a patient on a TNF inhibitor.
ADVERSE EFFECTS OF ANTI-TNF DRUGS
Infusion reactions, infections, cardiac arrhythmias, demyelinating disorders, skin infections, and malignancies have been reported with anti-TNF therapy. The relative frequencies of these adverse events are summarized in Table 3.17–22
NONINFECTIOUS COMPLICATIONS OF TNF INHIBITORS
Injection site reactions
When anti-TNF agents are given subcutaneously, injection site reactions are common (occurring in up to 40% of patients) and are considered minor.11 Reactions, including significant pain, typically occur within the first few months of therapy. They can last 2 to 5 days but rarely warrant stopping therapy. Treatment with ice and an antihistamine is almost always sufficient to control symptoms.
Infusion reactions with infliximab
Infliximab can cause both acute and delayed infusion reactions. Acute reactions can occur up to 24 hours after infusion but usually appear within 10 minutes of administration and are handled by the infusion suite staff. They range from the severe immunoglobulin E-mediated type I reaction, manifesting with hypotension, bronchospasm, and urticaria, to the milder anaphylactoid-type reaction, which constitutes the majority.23–25
While most primary care physicians will not encounter an acute reaction, family doctors and emergency room physicians may encounter delayed reactions, which can develop 1 to 14 days after infusion. These reactions usually resemble serum sickness and present with joint pain, fatigue, myalgia, and fever. But, unlike classic serum sickness, these reactions are generally not associated with a rash.
With nonspecific symptoms, the diagnosis may be easy to overlook. However, establishing this diagnosis is important because repeat therapy may result in a more severe reaction upon reexposure to the drug.
Once diagnosed, these reactions can be treated symptomatically with a combination of acetaminophen and diphenhydramine after discussion between the primary care physician and subspecialist.23,24
Autoimmune syndromes
Several studies have reported a small percentage of patients treated with anti-TNF agents who develop paradoxical autoimmune conditions. These range from asymptomatic immunologic alterations, including the formation of antinuclear antibodies and antibodies to double-stranded DNA, to life-threatening systemic autoimmune diseases.26,27
Autoimmune diseases associated with anti-TNF treatment include a lupus-like syndrome, vasculitides, and psoriatic skin lesions. These syndromes warrant stopping the inciting drug and, on occasion, giving corticosteroids. Most cases arise between 1 month and 1 year of starting treatment, and almost 75% resolve completely after the anti-TNF therapy is stopped.26
Interestingly, anti-TNF agents are approved for treating psoriasis and psoriatic arthritis, but psoriasis has paradoxically developed in patients being treated with these drugs for other autoimmune diseases. The FDA has reviewed 69 cases of new-onset psoriasis with anti-TNF therapy, including 17 pustular and 15 palmoplantar cases. The 12 most severe cases resulted in hospitalization, and symptoms resolved in most after treatment cessation.28
Fiorino et al29 counted 18 reported cases of psoriasis induced by anti-TNF therapy in patients with inflammatory bowel disease and concluded that it is rare. Harrison et al30 reported similar findings in patients with rheumatoid arthritis, with an increased incidence rate of 1.04 per 1,000 person-years. New-onset psoriasis was most common in patients treated with adalimumab. An example of the rash is seen in Figure 1.
CARDIOVASCULAR SIDE EFFECTS
Cardiovascular side effects of anti-TNF agents range from nonspecific and asymptomatic arrhythmias to worsening of heart failure.
Circulating levels of TNF are increased in patients with heart failure, and studies have evaluated the effects of TNF inhibition with infliximab on cardiac function and overall survival.31,32 The combined risk of death from any cause or hospitalization from heart failure was significantly higher in the infliximab groups, and the effects persisted for up to 5 months after stopping therapy.
Other studies22,33 have evaluated the effects of infliximab and etanercept on cardiac function and overall survival. Results showed possible exacerbation of heart failure with etanercept and increased risk of death with infliximab in patients with New York Heart Association (NYHA) class III or IV heart failure and left ventricular ejection fractions less than 35%.
Case reports have also described patients with worsening or new-onset heart failure on TNF inhibitors, including patients younger than 50 years and without identifiable cardiovascular risk factors.
Data analyses31,34,35 from large clinical registries have reported no significant increase in heart failure attributable to TNF inhibitors. However, we have concerns about the methodology of these analyses.
Currently, anti-TNF therapy is contraindicated in patients with NYHA class III or IV heart failure. Data are inconclusive for patients with class I or II heart failure. Baseline echocardiography and cardiology consultation can be considered, with close monitoring and avoidance of high doses of TNF inhibitors. If heart failure develops in a patient on anti-TNF therapy, the drug should be discontinued and the patient should be evaluated further.36
DEMYELINATING DISEASE, INCLUDING MULTIPLE SCLEROSIS
Anti-TNF agents have been associated with the onset or exacerbation of clinical symptoms and radiographic evidence of central nervous system demyelinating disorders, including multiple sclerosis.37–40 Mohan et al38 identified 19 cases of demyelinating events occurring after administration of anti-TNF agents in early 2001. In most cases, symptoms improved or resolved after therapy was stopped.
Optic neuritis,41,42 bilateral optic neuropathy,43 and aseptic meningitis44 have also been reported, but these have occurred only rarely.
How common are these effects? Postmarketing surveillance in patients with rheumatoid arthritis yields an estimated incidence of demyelinating disorders of 1 per 1,000 patient-years with adalimumab therapy.45 Complicating the assessment is an observed slight increase in risk of demyelinating conditions associated with inflammatory bowel disease.
Symptoms that should heighten the physician’s suspicion of this adverse effect include confusion, paresthesias, and ataxia. Patients on anti-TNF therapy who develop new visual symptoms should be checked for painless visual loss as a sign of early demyelinating disease.
Although data that conclusively link anti-TNF agents to multiple sclerosis are lacking, these drugs should not be initiated in patients who have a history of demyelinating disease, and treatment should be stopped promptly if the diagnosis is suspected.
MALIGNANCY
Whether anti-TNF therapy is directly linked to development of malignancies is difficult to determine. There are many confounding factors, including the risk of malignancy in underlying inflammatory disease and the concomitant use of other medications such as thiopurines, which have a known association with lymphoma.46
The incidence of lymphoma is twice as high in rheumatoid arthritis patients as in the general population.47 The risk is higher in those with more aggressive joint disease—the subset of rheumatoid arthritis patients who are more likely to be given anti-TNF agents.5
In patients with inflammatory bowel disease, the risk of cholangiocarcinoma is four times higher, and the risk of small-bowel adenocarcinoma is 16 to 18 times higher, but no increased risk of lymphoma has been identified in this population.48,49
Data from six clinical trials of infliximab, including a long-term study of its safety in Crohn disease, suggest it poses no increase in overall risk of malignancy.50–52 Similar results have been reported in patients with other rheumatic diseases.8 Information on this topic is constantly evolving, and studies range from case series to clinical trials to large patient registries.
The decision to use a TNF inhibitor should be based on the patient’s clinical picture and risk factors. Discussion of the risks and benefits of therapy with the patient should be clearly documented.
Non-Hodgkin lymphoma
Evidence about the risk of lymphoma with anti-TNF use is mixed, as up to two-thirds of patients on anti-TNF therapy have received concomitant nonbiologic immunosuppressive medications, making it difficult to determine the true risk from the biologic agents alone.53 Current evidence both supports9,53–56 and refutes7,55,57–60 the idea that anti-TNF agents increase lymphoma risk.
In patients with inflammatory bowel disease, several population-based studies have not shown a clear increase in lymphoma risk with anti-TNF use.56,59,60 Pedersen et al,61 in a meta-analysis of eight studies, confirmed these findings by showing no overall lymphoma risk in patients with inflammatory bowel disease.
However, a Canadian population-based study found a statistically significant increase in non-Hodgkin lymphoma in males with Crohn disease, with an incidence ratio of 3.63 (95% confidence interval [CI] 1.53–8.62).61 Additionally, Siegel et al62 found a significantly higher risk (6.1 cases per 10,000 patients) in patients treated with anti-TNF agents and thiopurines than in the general population (1.9 cases per 10,000 people). Although the difference was statistically significant, the overall risk is still very low.
Patients with rheumatoid arthritis seem to have a risk of lymphoma two to three times higher than in the general population. However, large population-based studies have not shown a statistically significant increase in the risk of lymphoma with anti-TNF therapy.63
Hepatosplenic T-cell lymphoma is a rare subtype of peripheral T-cell non-Hodgkin lymphoma; 25 cases have been reported in patients receiving anti-TNF therapy.64 Although the risk is extremely low (< 0.05%), physicians must carefully consider the risks and benefits of combination therapy, especially in young male patients with inflammatory bowel disease, since death is the usual outcome of this disease.65–67
Skin cancers
Wolfe and Michaud8 evaluated malignancy risk in rheumatoid arthritis patients being treated with biologic agents, including TNF inhibitors, using a large longitudinal database. These data were compared with those of the US Surveillance, Epidemiology, and End-Results (SEER) national database. No increase in the overall cancer rate was seen in rheumatoid arthritis patients (standardized incidence ratio [SIR] 1.0, 95% CI 1.0–1.1).
However, melanoma was more common in rheumatoid arthritis patients compared with SEER rates (SIR 1.7, 95% CI 1.3–2.3).8 In addition, biologic therapy was associated with a higher (but not statistically significant) risk of melanoma (odds ratio [OR] 2.3, 95% CI 0.9–5.4) and a higher risk of nonmelanoma skin cancer (OR 1.5, 95% CI 1.2–1.8), but not of other types of cancer.8
INFECTION
Patients on anti-TNF therapy are at a higher risk of infection, ranging from minor to life-threatening bacterial infections, and including the reactivation of granulomatous and fungal infections. More importantly, these agents are similar to steroids in blunting signs of infection, which may delay diagnosis and treatment.
The management of infection in patients on anti-TNF medications varies from case to case. In general, patients with a minor infection that does not require hospitalization or intravenous antibiotics can continue the biologic therapy while taking oral antibiotics. TNF inhibitors must be held in the event of a major infection.
Consultation with an infectious disease specialist is recommended, especially in complex cases.
Bacterial infections
An increased risk of minor bacterial infections such as urinary tract and respiratory infections has been well documented in several randomized control trials of anti-TNF agents, though other studies have shown no such increase in risk.33,51–59
The threshold for using antibiotics for a suspected bacterial infection is somewhat shifted in favor of treatment in patients on anti-TNF therapy. The reason is twofold: as previously noted, infections may be worse than they appear, because anti-TNF drugs can mask the signs and symptoms of a serious infection, and in patients on these drugs, an untreated bacterial infection may rapidly become life-threatening.
In general, broad-spectrum antibiotics are not warranted unless the source of infection is unclear or the patient is in danger of hemodynamic compromise.
Opportunistic infections
The association of anti-TNF agents with opportunistic infections could be viewed as an extension of their normal and intended therapeutic activity as potent immunosuppressive agents.68 Rheumatoid arthritis and inflammatory bowel disease are usually associated with conditions and situations that predispose patients to opportunistic infections, such as decreased immune response, malnutrition or malabsorption, surgeries, and concomitant immunosuppressive medications.7 Combination therapy with other immunosuppressive drugs and older age appear to markedly increase the risk of opportunistic infections, including mycobacterial and fungal infections, in patients with inflammatory bowel disease.7
Overall, opportunistic infections represent a measurable risk of anti-TNF therapy, and awareness and vigilance are important, especially in areas where opportunistic infections such as histoplasmosis and coccidiomycosis are endemic.50 Furthermore, physicians must be aware of the higher risk of opportunistic infections when multiple immunosuppressive drugs are used concurrently.
Granulomatous infections such as tuberculosis
Anti-TNF agents increase the risk of de novo granulomatous infections and of reactivating such infections. Granuloma formation and intracellular destruction of mycobacteria depend on TNF. TNF is important in maintaining the anatomic integrity of granulomas where these organisms have been sequestered, and blocking TNF leads to breakdown of granulomas and release of virulent organisms.69,70
TNF inhibitors increase the risk of reactivation of latent tuberculosis infection. The risk is greater with infliximab and adalimumab than with etanercept,71,72 and it has been described with certolizumab.74 Study results are varied thus far but show a risk of tuberculosis reactivation five to 30 times higher than in the general population, with tremendous variability in risk depending on background rates of previous exposure.
The absence of typical tuberculosis symptoms further complicates care in these cases. Fever, weight loss, and night sweats tend to be TNF-mediated and are therefore masked by anti-TNF agents, leading to atypical presentations. In addition, active tuberculosis infection associated with TNF inhibitors is more likely to involve extrapulmonary sites such as the skin and musculoskeletal system and to be disseminated at presentation.
A paradoxical worsening of tuberculosis symptoms may also be seen in patients with latent tuberculosis reactivation, especially after discontinuing anti-TNF therapy. This is thought to result from an immune reconstitution inflammatory syndrome.
The pretreatment evaluation should include a history of risk factors, a physical examination, and either a tuberculin skin test or an interferon-gamma-release assay. Interferon-gamma-release assays are particularly helpful in patients who have received bacille Calmette-Guérin vaccination. In patients who test positive or have been exposed, tuberculosis treatment should begin 4 weeks before starting anti-TNF therapy, though the optimal timing of antituberculosis agents is still controversial.74–77
If tuberculosis develops in a patient on anti-TNF therapy, he or she should receive antituberculosis drugs. Anti-TNF therapy should be stopped and should be resumed after 2 months only if no other treatment option is available.75
Invasive opportunistic fungal infections
Invasive opportunistic fungal infections have been reported with anti-TNF therapy, including histoplasmosis and coccidioidomycosis.78–80 Most patients who had histoplasmosis were treated with other immunosuppressive therapies and resided in or were raised near the Ohio or Mississippi River valleys, where this disease is endemic. Similarly, most cases of coccidioidomycosis were in endemic areas of Arizona, California, and Nevada, and patients on concomitant immunosuppressive therapy.78
Currently, there is no evidence to recommend obtaining Histoplasma capsulatum or Coccidioides immitis serologies before initiating anti-TNF therapy in patients in endemic areas.81 However, patients must be instructed to seek medical attention quickly for pulmonary or febrile illnesses.
Viral hepatitis infections
The data on hepatitis B and hepatitis C in patients on biologic therapies are mostly limited to case reports.
Hepatitis B. A small prospective study from Spain followed the liver biochemistry tests and hepatitis B status of 80 patients with Crohn disease treated with infliximab. Of three patients who were chronic hepatitis B carriers before starting infliximab, two experienced reactivation of hepatitis B after discontinuing infliximab, and one ultimately died. The third patient was treated with lamivudine concurrently with infliximab without clinical or biochemical changes during or after therapy.82
Similar findings were observed in two patients with rheumatoid arthritis on treatment with infliximab. One of the patients required liver transplantation, and both were treated with lamivudine, resulting in normalization of liver function test results.83,84
Recent reviews indicate that despite these findings, hepatitis B reactivation after anti-TNF withdrawal may not be common.85 There are limited data on hepatitis B reactivation and associated liver dysfunction in patients with inflammatory bowel disease treated with immunosuppressants. In a retrospective multicenter trial by Loras et al86 in patients with inflammatory bowel disease who had viral hepatitis, 36% of patients positive for hepatitis B surface antigen developed liver dysfunction, and six patients developed liver failure. In that study, treatment with more than two immunosuppressants was an independent predictor of hepatitis B reactivation.
Prolonged immunosuppression (longer than 3 months) has also been identified as an independent predictor of liver dysfunction (OR 3.06; 95% 95% CI 1.02–9.16).87
The European Association for the Study of the Liver and the American Association for the Study of Liver Diseases recommend starting lamivudine before chemotherapy or immunomodulator or immunosuppressive therapy in hepatitis B virus carriers and continuing preventive treatment for at least 6 months after stopping immunomodulating drugs. Lamivudine at a dose of 100 mg/day may reduce the risk of reactivation of hepatitis B.88–90 Tenofovir and entecavir may be useful alternatives in patients with hepatitis B who have never received nucleoside analogues. Hepatitis B reactivation did not occur in any of the 16 patients who received preventive entecavir treatment while receiving immunosuppressive treatments.89,91
In patients receiving immunosuppressive therapy, hepatitis B reactivation is associated with significant morbidity and mortality. Although risk factors for reactivation of hepatitis B virus infection have been identified, we recommend preventive treatment for all carriers positive for hepatitis B surface antigen. This should be done regardless of the number, type, and dosage of immunosuppressants and regardless of hepatitis B virus DNA levels.90
The frequency of hepatitis B and hepatitis C infection in patients with Crohn disease has been reported to be as high as 24%. The high incidence is thought to be secondary to multiple blood transfusions and surgeries.92 The use of biologic agents, including anti-TNF agents, in chronic hepatitis B virus- or hepatitis C virus-infected patients can lead to enhanced viral replication and hepatitis exacerbation.
Although active viral replication can occur during treatment with biologic agents, reactivation or exacerbation can also occur after the anti-TNF agent is stopped.82 This finding has prompted the recommendation that all candidates for biologic therapy be tested for hepatitis B immunization status, followed by immunization in nonimmune patients before starting anti-TNF therapy.93,94
Hepatitis C. There are no guidelines that adequately address the use of anti-TNF agents in patients with chronic hepatitis C infection.
Several small retrospective studies in rheumatoid arthritis patients with hepatitis C have shown that TNF inhibitors can be safely used without worsening liver function tests or changing the viral load.95–98 This is reassuring and provides the subspecialist with another treatment option, as other therapies such as disease-modifying antirheumatic drugs and steroids are known to aggravate viral hepatitis and increase the risk of viremia.96
Although small retrospective studies and one large randomized double-blind placebo-controlled trial have shown TNF inhibitors to be relatively safe in rheumatoid arthritis patients with hepatitis C, their use in these patients should be considered only with caution if they have evidence of hepatic synthetic dysfunction (eg, hypoalbuminemia, thrombocytopenia, increased international normalized ratio). The American College of Rheumatology recommends avoiding TNF inhibitors in Child-Pugh classes B and C.99
PREGNANCY
Physicians caring for patients with rheumatoid arthritis and inflammatory bowel disease must be aware of how these diseases affect fecundity and fertility and how the medications can affect conception and pregnancy. Many patients have difficulty conceiving while their autoimmune disease is active, and better disease control may improve fecundity and result in unanticipated pregnancy. Patients should be advised of the need for contraception if pregnancy is ill-advised or undesired.
Many patients seek advice about teratogenicity before conceiving, or seek guidance about rheumatoid arthritis and inflammatory bowel disease treatment while pregnant.
Several studies have reported a higher risk of adverse pregnancy outcomes in patients with rheumatoid arthritis and inflammatory bowel disease than in the general population.99–105 In inflammatory bowel disease, the odds of a premature delivery or having a low-birth-weight child are twice as high as in the normal population.100,103 Higher rates of cesarean delivery and stillbirth have also been reported. The main predisposing factor appears to be the disease activity at the time of conception, as active disease seems to be linked to adverse pregnancy outcomes.
Treatment with anti-TNF agents may rapidly achieve and maintain remission, raising the question of the safety of continued anti-TNF use during pregnancy. The FDA classifies anti-TNF agents as category B drugs, as animal studies have not demonstrated fetal risk, and no well-controlled prospective study has yet been conducted with pregnant women.
In an observational study, Schnitzler et al105 assessed the outcomes of 42 pregnancies in 35 patients with inflammatory bowel disease receiving either infliximab or adalimumab during pregnancy, compared with 56 pregnancies in 45 healthy patients without inflammatory bowel disease. There was no statistical difference in abortion rates between patients receiving anti-TNF agents and healthy women without inflammatory bowel disease (21% vs 14%, P = .4234). There was also no significant difference observed in birth weight, birth length, or cranial circumference of the children between the two groups. However, pregnancies with direct exposure to anti-TNF agents resulted in a higher frequency of premature delivery (25% vs 6%, P = .023).
Similar results were noted from the Crohn’s Therapy, Resource, Evaluation, and Assessment Tool, or TREAT, registry, as well as from a large systematic review by Vinet et al106 and case reports of rheumatoid arthritis and inflammatory bowel disease in women exposed to anti-TNF agents during pregnancy.
In addition, results from a recent systematic review of 38 studies of anti-TNF use and fetal risk, with a total of 437 women (189 on infliximab, 230 on adalimumab, 18 on certolizumab pegol), showed similar results.107 In pregnancies exposed to anti-TNF agents, the rates of congenital abnormalities (3.4%), fetal deaths (8.5%), and preterm births (2.7%) were similar to those in the general population.
For patients in disease remission on TNF inhibitors, it is reasonable to continue these agents during pregnancy after careful discussion with the patient. Fetal safety and infant immunization response after delivery are the primary concerns in these cases.
Both infliximab and adalimumab cross the placenta and remain detectable in the baby’s circulation 4 months (for adalimumab) to 6 months (for infliximab) after delivery. It is currently recommended that infliximab be stopped at 32 weeks of gestation for the remainder of the pregnancy and that adalimumab be stopped at 34 to 36 weeks, given a planned 40-week gestation.
Certolizumab does not cross the placenta in significant amounts and should be continued throughout pregnancy; drug levels in infants were shown to be less than 2 μg/mL, even when dosed the week of delivery.108–112
TAKE-HOME POINTS
- All health care providers of patients with rheumatoid arthritis and inflammatory bowel disease should be familiar with anti-TNF agents used in treating these diseases.
- The benefits of controlling the disease far outweigh the risks of therapy when used appropriately.
- Care of these patients should be multidisciplinary, with clear communication between primary care physician and specialist.
- Patient education and monitoring combined with prompt communication between primary care physician and specialist are key.
Biologic agents such as those that block tumor necrosis factor (TNF) alpha have revolutionized the treatment of autoimmune diseases like rheumatoid arthritis and inflammatory bowel disease, dramatically improving disease control and quality of life. In addition, they have made true disease remission possible in some cases.
However, as with any new therapy, a variety of side effects must be considered.
WHAT ARE BIOLOGIC AGENTS?
Biologic agents are genetically engineered drugs manufactured or synthesized in vitro from molecules such as proteins, genes, and antibodies present in living organisms. By targeting specific molecular components of the inflammatory cascade, including cytokines, these drugs alter certain aspects of the body’s inflammatory response in autoimmune diseases. Because TNF inhibitors are the most widely used biologic agents in the United States, this review will focus on them.
TNF INHIBITORS
TNF inhibitors suppress the inflammatory cascade by inactivating TNF alpha, a cytokine that promotes inflammation in the intestine, synovial tissue, and other sites.1,2
Several TNF inhibitors are available. Etanercept and infliximab were the first two to receive US Food and Drug Administration (FDA) approval, and they are extensively used. Etanercept, a soluble TNF receptor given subcutaneously, was approved for treating rheumatoid arthritis in 1998. Infliximab, a chimeric monoclonal antibody (75% human and 25% mouse protein sequence) given intravenously, received FDA approval for treating Crohn disease in 1998 and for rheumatoid arthritis in 1999. Other anti-TNF agents with varying properties are also available.
Table 1 lists anti-TNF agents approved for treating rheumatoid arthritis and inflammatory bowel disease. These are chronic, relapsing diseases that significantly and dramatically reduce quality of life, especially when they are poorly controlled.3,4 Untreated disease has been associated with malignancy, infection, pregnancy loss, and malnutrition.5–8 TNF inhibitors are aimed at patients who have moderate to severe disease or for whom previous treatments have failed, to help them achieve and maintain steroid-free remission. However, use of these agents is tempered by the risk of potentially serious side effects.
Special thought should also be given to the direct costs of the drugs (up to $30,000 per year, not counting the cost of their administration) and to the indirect costs such as time away from work to receive treatment. These are major considerations in some cases, and patients should be selected carefully for treatment with these drugs.
BEFORE STARTING THERAPY
Before starting anti-TNF therapy, several steps can reduce the risk of serious adverse events.
Take a focused history
The clinical history should include inquiries about previous bacterial, fungal, and tuberculosis infections or exposure; diabetes; and other immunocompromised states that increase the risk of acquiring potentially life-threatening infections.
Details of particular geographic areas of residence, occupational exposures, and social history should be sought. These include history of incarceration (which may put patients at risk of tuberculosis) and residence in the Ohio River valley or in the southwestern or midwestern United States (which may increase the risk of histoplasmosis, coccidioidomycosis, and other fungal infections).
Bring vaccinations up to date
Age-appropriate vaccinations should be discussed and given, ideally before starting therapy. These include influenza vaccine every year and tetanus boosters every 10 years for all and, as appropriate, varicella, human papillomavirus, and pneumococcal vaccinations. The US Centers for Disease Control and Prevention recommend an additional dose of the pneumococcal vaccine if more than 5 years have elapsed since the first one, and many clinicians opt to give it every 5 years.
In general, live-attenuated vaccines, including the intranasal influenza vaccine, are contraindicated in patients taking biologic agents.9 For patients at high risk of exposure or infection, it may be reasonable to hold the biologic agent for a period of time, vaccinate, and resume the biologic agent a month later.
Recent data suggest that the varicella zoster vaccine may be safely given to older patients with immune-mediated diseases such as rheumatoid arthritis and inflammatory bowel disease taking biologic agents.10 New guidelines from the American College of Rheumatology recommend age-appropriate vaccines for rheumatoid arthritis patients age 60 and older before biologic treatments are started. Case-by-case discussion with the subspecialist and the patient is recommended.
Screen for chronic infections
Tuberculosis screening with a purified protein derivative test or an interferon-gamma-release (Quantiferon) assay followed by chest radiography in patients with a positive test is mandatory before giving a TNF inhibitor.
Hepatitis B virus status should be determined before starting anti-TNF therapy.11
Hepatitis B vaccination has been recommended for patients with inflammatory bowel disease, but no clear recommendation exists for patients with rheumatic disease. Patients with inflammatory bowel disease tend to have low rates of response to hepatitis B vaccination12,13; possible reasons include their lack of an appropriate innate immune response to infectious agents, malnutrition, surgery, older age, and immunosuppressive drugs.14 An accelerated vaccination protocol with recombinant hepatitis B vaccine (Energix-B) in a double dose at 0, 1, and 2 months has been shown to improve response rates.15
Whenever possible, it may be better to vaccinate patients before starting immunosuppressive therapy, and to check postvaccination titers to ensure adequate response.
Perform an examination
A full physical examination with special attention to skin rashes should be performed. This may serve as a baseline to assist early detection of new rashes associated with anti-TNF therapy.
A baseline complete blood cell count and complete metabolic panel should be routinely obtained before starting therapy (and thereafter at the discretion of the physician). In conjunction with follow-up tests, they can help detect an unexpected decrease in white blood cell count or abnormal results on the liver panel.16 These baseline and follow-up tests are generally performed by the subspecialist, and the results are shared with the primary care physician.
Table 2 summarizes key information to be sought before starting a patient on a TNF inhibitor.
ADVERSE EFFECTS OF ANTI-TNF DRUGS
Infusion reactions, infections, cardiac arrhythmias, demyelinating disorders, skin infections, and malignancies have been reported with anti-TNF therapy. The relative frequencies of these adverse events are summarized in Table 3.17–22
NONINFECTIOUS COMPLICATIONS OF TNF INHIBITORS
Injection site reactions
When anti-TNF agents are given subcutaneously, injection site reactions are common (occurring in up to 40% of patients) and are considered minor.11 Reactions, including significant pain, typically occur within the first few months of therapy. They can last 2 to 5 days but rarely warrant stopping therapy. Treatment with ice and an antihistamine is almost always sufficient to control symptoms.
Infusion reactions with infliximab
Infliximab can cause both acute and delayed infusion reactions. Acute reactions can occur up to 24 hours after infusion but usually appear within 10 minutes of administration and are handled by the infusion suite staff. They range from the severe immunoglobulin E-mediated type I reaction, manifesting with hypotension, bronchospasm, and urticaria, to the milder anaphylactoid-type reaction, which constitutes the majority.23–25
While most primary care physicians will not encounter an acute reaction, family doctors and emergency room physicians may encounter delayed reactions, which can develop 1 to 14 days after infusion. These reactions usually resemble serum sickness and present with joint pain, fatigue, myalgia, and fever. But, unlike classic serum sickness, these reactions are generally not associated with a rash.
With nonspecific symptoms, the diagnosis may be easy to overlook. However, establishing this diagnosis is important because repeat therapy may result in a more severe reaction upon reexposure to the drug.
Once diagnosed, these reactions can be treated symptomatically with a combination of acetaminophen and diphenhydramine after discussion between the primary care physician and subspecialist.23,24
Autoimmune syndromes
Several studies have reported a small percentage of patients treated with anti-TNF agents who develop paradoxical autoimmune conditions. These range from asymptomatic immunologic alterations, including the formation of antinuclear antibodies and antibodies to double-stranded DNA, to life-threatening systemic autoimmune diseases.26,27
Autoimmune diseases associated with anti-TNF treatment include a lupus-like syndrome, vasculitides, and psoriatic skin lesions. These syndromes warrant stopping the inciting drug and, on occasion, giving corticosteroids. Most cases arise between 1 month and 1 year of starting treatment, and almost 75% resolve completely after the anti-TNF therapy is stopped.26
Interestingly, anti-TNF agents are approved for treating psoriasis and psoriatic arthritis, but psoriasis has paradoxically developed in patients being treated with these drugs for other autoimmune diseases. The FDA has reviewed 69 cases of new-onset psoriasis with anti-TNF therapy, including 17 pustular and 15 palmoplantar cases. The 12 most severe cases resulted in hospitalization, and symptoms resolved in most after treatment cessation.28
Fiorino et al29 counted 18 reported cases of psoriasis induced by anti-TNF therapy in patients with inflammatory bowel disease and concluded that it is rare. Harrison et al30 reported similar findings in patients with rheumatoid arthritis, with an increased incidence rate of 1.04 per 1,000 person-years. New-onset psoriasis was most common in patients treated with adalimumab. An example of the rash is seen in Figure 1.
CARDIOVASCULAR SIDE EFFECTS
Cardiovascular side effects of anti-TNF agents range from nonspecific and asymptomatic arrhythmias to worsening of heart failure.
Circulating levels of TNF are increased in patients with heart failure, and studies have evaluated the effects of TNF inhibition with infliximab on cardiac function and overall survival.31,32 The combined risk of death from any cause or hospitalization from heart failure was significantly higher in the infliximab groups, and the effects persisted for up to 5 months after stopping therapy.
Other studies22,33 have evaluated the effects of infliximab and etanercept on cardiac function and overall survival. Results showed possible exacerbation of heart failure with etanercept and increased risk of death with infliximab in patients with New York Heart Association (NYHA) class III or IV heart failure and left ventricular ejection fractions less than 35%.
Case reports have also described patients with worsening or new-onset heart failure on TNF inhibitors, including patients younger than 50 years and without identifiable cardiovascular risk factors.
Data analyses31,34,35 from large clinical registries have reported no significant increase in heart failure attributable to TNF inhibitors. However, we have concerns about the methodology of these analyses.
Currently, anti-TNF therapy is contraindicated in patients with NYHA class III or IV heart failure. Data are inconclusive for patients with class I or II heart failure. Baseline echocardiography and cardiology consultation can be considered, with close monitoring and avoidance of high doses of TNF inhibitors. If heart failure develops in a patient on anti-TNF therapy, the drug should be discontinued and the patient should be evaluated further.36
DEMYELINATING DISEASE, INCLUDING MULTIPLE SCLEROSIS
Anti-TNF agents have been associated with the onset or exacerbation of clinical symptoms and radiographic evidence of central nervous system demyelinating disorders, including multiple sclerosis.37–40 Mohan et al38 identified 19 cases of demyelinating events occurring after administration of anti-TNF agents in early 2001. In most cases, symptoms improved or resolved after therapy was stopped.
Optic neuritis,41,42 bilateral optic neuropathy,43 and aseptic meningitis44 have also been reported, but these have occurred only rarely.
How common are these effects? Postmarketing surveillance in patients with rheumatoid arthritis yields an estimated incidence of demyelinating disorders of 1 per 1,000 patient-years with adalimumab therapy.45 Complicating the assessment is an observed slight increase in risk of demyelinating conditions associated with inflammatory bowel disease.
Symptoms that should heighten the physician’s suspicion of this adverse effect include confusion, paresthesias, and ataxia. Patients on anti-TNF therapy who develop new visual symptoms should be checked for painless visual loss as a sign of early demyelinating disease.
Although data that conclusively link anti-TNF agents to multiple sclerosis are lacking, these drugs should not be initiated in patients who have a history of demyelinating disease, and treatment should be stopped promptly if the diagnosis is suspected.
MALIGNANCY
Whether anti-TNF therapy is directly linked to development of malignancies is difficult to determine. There are many confounding factors, including the risk of malignancy in underlying inflammatory disease and the concomitant use of other medications such as thiopurines, which have a known association with lymphoma.46
The incidence of lymphoma is twice as high in rheumatoid arthritis patients as in the general population.47 The risk is higher in those with more aggressive joint disease—the subset of rheumatoid arthritis patients who are more likely to be given anti-TNF agents.5
In patients with inflammatory bowel disease, the risk of cholangiocarcinoma is four times higher, and the risk of small-bowel adenocarcinoma is 16 to 18 times higher, but no increased risk of lymphoma has been identified in this population.48,49
Data from six clinical trials of infliximab, including a long-term study of its safety in Crohn disease, suggest it poses no increase in overall risk of malignancy.50–52 Similar results have been reported in patients with other rheumatic diseases.8 Information on this topic is constantly evolving, and studies range from case series to clinical trials to large patient registries.
The decision to use a TNF inhibitor should be based on the patient’s clinical picture and risk factors. Discussion of the risks and benefits of therapy with the patient should be clearly documented.
Non-Hodgkin lymphoma
Evidence about the risk of lymphoma with anti-TNF use is mixed, as up to two-thirds of patients on anti-TNF therapy have received concomitant nonbiologic immunosuppressive medications, making it difficult to determine the true risk from the biologic agents alone.53 Current evidence both supports9,53–56 and refutes7,55,57–60 the idea that anti-TNF agents increase lymphoma risk.
In patients with inflammatory bowel disease, several population-based studies have not shown a clear increase in lymphoma risk with anti-TNF use.56,59,60 Pedersen et al,61 in a meta-analysis of eight studies, confirmed these findings by showing no overall lymphoma risk in patients with inflammatory bowel disease.
However, a Canadian population-based study found a statistically significant increase in non-Hodgkin lymphoma in males with Crohn disease, with an incidence ratio of 3.63 (95% confidence interval [CI] 1.53–8.62).61 Additionally, Siegel et al62 found a significantly higher risk (6.1 cases per 10,000 patients) in patients treated with anti-TNF agents and thiopurines than in the general population (1.9 cases per 10,000 people). Although the difference was statistically significant, the overall risk is still very low.
Patients with rheumatoid arthritis seem to have a risk of lymphoma two to three times higher than in the general population. However, large population-based studies have not shown a statistically significant increase in the risk of lymphoma with anti-TNF therapy.63
Hepatosplenic T-cell lymphoma is a rare subtype of peripheral T-cell non-Hodgkin lymphoma; 25 cases have been reported in patients receiving anti-TNF therapy.64 Although the risk is extremely low (< 0.05%), physicians must carefully consider the risks and benefits of combination therapy, especially in young male patients with inflammatory bowel disease, since death is the usual outcome of this disease.65–67
Skin cancers
Wolfe and Michaud8 evaluated malignancy risk in rheumatoid arthritis patients being treated with biologic agents, including TNF inhibitors, using a large longitudinal database. These data were compared with those of the US Surveillance, Epidemiology, and End-Results (SEER) national database. No increase in the overall cancer rate was seen in rheumatoid arthritis patients (standardized incidence ratio [SIR] 1.0, 95% CI 1.0–1.1).
However, melanoma was more common in rheumatoid arthritis patients compared with SEER rates (SIR 1.7, 95% CI 1.3–2.3).8 In addition, biologic therapy was associated with a higher (but not statistically significant) risk of melanoma (odds ratio [OR] 2.3, 95% CI 0.9–5.4) and a higher risk of nonmelanoma skin cancer (OR 1.5, 95% CI 1.2–1.8), but not of other types of cancer.8
INFECTION
Patients on anti-TNF therapy are at a higher risk of infection, ranging from minor to life-threatening bacterial infections, and including the reactivation of granulomatous and fungal infections. More importantly, these agents are similar to steroids in blunting signs of infection, which may delay diagnosis and treatment.
The management of infection in patients on anti-TNF medications varies from case to case. In general, patients with a minor infection that does not require hospitalization or intravenous antibiotics can continue the biologic therapy while taking oral antibiotics. TNF inhibitors must be held in the event of a major infection.
Consultation with an infectious disease specialist is recommended, especially in complex cases.
Bacterial infections
An increased risk of minor bacterial infections such as urinary tract and respiratory infections has been well documented in several randomized control trials of anti-TNF agents, though other studies have shown no such increase in risk.33,51–59
The threshold for using antibiotics for a suspected bacterial infection is somewhat shifted in favor of treatment in patients on anti-TNF therapy. The reason is twofold: as previously noted, infections may be worse than they appear, because anti-TNF drugs can mask the signs and symptoms of a serious infection, and in patients on these drugs, an untreated bacterial infection may rapidly become life-threatening.
In general, broad-spectrum antibiotics are not warranted unless the source of infection is unclear or the patient is in danger of hemodynamic compromise.
Opportunistic infections
The association of anti-TNF agents with opportunistic infections could be viewed as an extension of their normal and intended therapeutic activity as potent immunosuppressive agents.68 Rheumatoid arthritis and inflammatory bowel disease are usually associated with conditions and situations that predispose patients to opportunistic infections, such as decreased immune response, malnutrition or malabsorption, surgeries, and concomitant immunosuppressive medications.7 Combination therapy with other immunosuppressive drugs and older age appear to markedly increase the risk of opportunistic infections, including mycobacterial and fungal infections, in patients with inflammatory bowel disease.7
Overall, opportunistic infections represent a measurable risk of anti-TNF therapy, and awareness and vigilance are important, especially in areas where opportunistic infections such as histoplasmosis and coccidiomycosis are endemic.50 Furthermore, physicians must be aware of the higher risk of opportunistic infections when multiple immunosuppressive drugs are used concurrently.
Granulomatous infections such as tuberculosis
Anti-TNF agents increase the risk of de novo granulomatous infections and of reactivating such infections. Granuloma formation and intracellular destruction of mycobacteria depend on TNF. TNF is important in maintaining the anatomic integrity of granulomas where these organisms have been sequestered, and blocking TNF leads to breakdown of granulomas and release of virulent organisms.69,70
TNF inhibitors increase the risk of reactivation of latent tuberculosis infection. The risk is greater with infliximab and adalimumab than with etanercept,71,72 and it has been described with certolizumab.74 Study results are varied thus far but show a risk of tuberculosis reactivation five to 30 times higher than in the general population, with tremendous variability in risk depending on background rates of previous exposure.
The absence of typical tuberculosis symptoms further complicates care in these cases. Fever, weight loss, and night sweats tend to be TNF-mediated and are therefore masked by anti-TNF agents, leading to atypical presentations. In addition, active tuberculosis infection associated with TNF inhibitors is more likely to involve extrapulmonary sites such as the skin and musculoskeletal system and to be disseminated at presentation.
A paradoxical worsening of tuberculosis symptoms may also be seen in patients with latent tuberculosis reactivation, especially after discontinuing anti-TNF therapy. This is thought to result from an immune reconstitution inflammatory syndrome.
The pretreatment evaluation should include a history of risk factors, a physical examination, and either a tuberculin skin test or an interferon-gamma-release assay. Interferon-gamma-release assays are particularly helpful in patients who have received bacille Calmette-Guérin vaccination. In patients who test positive or have been exposed, tuberculosis treatment should begin 4 weeks before starting anti-TNF therapy, though the optimal timing of antituberculosis agents is still controversial.74–77
If tuberculosis develops in a patient on anti-TNF therapy, he or she should receive antituberculosis drugs. Anti-TNF therapy should be stopped and should be resumed after 2 months only if no other treatment option is available.75
Invasive opportunistic fungal infections
Invasive opportunistic fungal infections have been reported with anti-TNF therapy, including histoplasmosis and coccidioidomycosis.78–80 Most patients who had histoplasmosis were treated with other immunosuppressive therapies and resided in or were raised near the Ohio or Mississippi River valleys, where this disease is endemic. Similarly, most cases of coccidioidomycosis were in endemic areas of Arizona, California, and Nevada, and patients on concomitant immunosuppressive therapy.78
Currently, there is no evidence to recommend obtaining Histoplasma capsulatum or Coccidioides immitis serologies before initiating anti-TNF therapy in patients in endemic areas.81 However, patients must be instructed to seek medical attention quickly for pulmonary or febrile illnesses.
Viral hepatitis infections
The data on hepatitis B and hepatitis C in patients on biologic therapies are mostly limited to case reports.
Hepatitis B. A small prospective study from Spain followed the liver biochemistry tests and hepatitis B status of 80 patients with Crohn disease treated with infliximab. Of three patients who were chronic hepatitis B carriers before starting infliximab, two experienced reactivation of hepatitis B after discontinuing infliximab, and one ultimately died. The third patient was treated with lamivudine concurrently with infliximab without clinical or biochemical changes during or after therapy.82
Similar findings were observed in two patients with rheumatoid arthritis on treatment with infliximab. One of the patients required liver transplantation, and both were treated with lamivudine, resulting in normalization of liver function test results.83,84
Recent reviews indicate that despite these findings, hepatitis B reactivation after anti-TNF withdrawal may not be common.85 There are limited data on hepatitis B reactivation and associated liver dysfunction in patients with inflammatory bowel disease treated with immunosuppressants. In a retrospective multicenter trial by Loras et al86 in patients with inflammatory bowel disease who had viral hepatitis, 36% of patients positive for hepatitis B surface antigen developed liver dysfunction, and six patients developed liver failure. In that study, treatment with more than two immunosuppressants was an independent predictor of hepatitis B reactivation.
Prolonged immunosuppression (longer than 3 months) has also been identified as an independent predictor of liver dysfunction (OR 3.06; 95% 95% CI 1.02–9.16).87
The European Association for the Study of the Liver and the American Association for the Study of Liver Diseases recommend starting lamivudine before chemotherapy or immunomodulator or immunosuppressive therapy in hepatitis B virus carriers and continuing preventive treatment for at least 6 months after stopping immunomodulating drugs. Lamivudine at a dose of 100 mg/day may reduce the risk of reactivation of hepatitis B.88–90 Tenofovir and entecavir may be useful alternatives in patients with hepatitis B who have never received nucleoside analogues. Hepatitis B reactivation did not occur in any of the 16 patients who received preventive entecavir treatment while receiving immunosuppressive treatments.89,91
In patients receiving immunosuppressive therapy, hepatitis B reactivation is associated with significant morbidity and mortality. Although risk factors for reactivation of hepatitis B virus infection have been identified, we recommend preventive treatment for all carriers positive for hepatitis B surface antigen. This should be done regardless of the number, type, and dosage of immunosuppressants and regardless of hepatitis B virus DNA levels.90
The frequency of hepatitis B and hepatitis C infection in patients with Crohn disease has been reported to be as high as 24%. The high incidence is thought to be secondary to multiple blood transfusions and surgeries.92 The use of biologic agents, including anti-TNF agents, in chronic hepatitis B virus- or hepatitis C virus-infected patients can lead to enhanced viral replication and hepatitis exacerbation.
Although active viral replication can occur during treatment with biologic agents, reactivation or exacerbation can also occur after the anti-TNF agent is stopped.82 This finding has prompted the recommendation that all candidates for biologic therapy be tested for hepatitis B immunization status, followed by immunization in nonimmune patients before starting anti-TNF therapy.93,94
Hepatitis C. There are no guidelines that adequately address the use of anti-TNF agents in patients with chronic hepatitis C infection.
Several small retrospective studies in rheumatoid arthritis patients with hepatitis C have shown that TNF inhibitors can be safely used without worsening liver function tests or changing the viral load.95–98 This is reassuring and provides the subspecialist with another treatment option, as other therapies such as disease-modifying antirheumatic drugs and steroids are known to aggravate viral hepatitis and increase the risk of viremia.96
Although small retrospective studies and one large randomized double-blind placebo-controlled trial have shown TNF inhibitors to be relatively safe in rheumatoid arthritis patients with hepatitis C, their use in these patients should be considered only with caution if they have evidence of hepatic synthetic dysfunction (eg, hypoalbuminemia, thrombocytopenia, increased international normalized ratio). The American College of Rheumatology recommends avoiding TNF inhibitors in Child-Pugh classes B and C.99
PREGNANCY
Physicians caring for patients with rheumatoid arthritis and inflammatory bowel disease must be aware of how these diseases affect fecundity and fertility and how the medications can affect conception and pregnancy. Many patients have difficulty conceiving while their autoimmune disease is active, and better disease control may improve fecundity and result in unanticipated pregnancy. Patients should be advised of the need for contraception if pregnancy is ill-advised or undesired.
Many patients seek advice about teratogenicity before conceiving, or seek guidance about rheumatoid arthritis and inflammatory bowel disease treatment while pregnant.
Several studies have reported a higher risk of adverse pregnancy outcomes in patients with rheumatoid arthritis and inflammatory bowel disease than in the general population.99–105 In inflammatory bowel disease, the odds of a premature delivery or having a low-birth-weight child are twice as high as in the normal population.100,103 Higher rates of cesarean delivery and stillbirth have also been reported. The main predisposing factor appears to be the disease activity at the time of conception, as active disease seems to be linked to adverse pregnancy outcomes.
Treatment with anti-TNF agents may rapidly achieve and maintain remission, raising the question of the safety of continued anti-TNF use during pregnancy. The FDA classifies anti-TNF agents as category B drugs, as animal studies have not demonstrated fetal risk, and no well-controlled prospective study has yet been conducted with pregnant women.
In an observational study, Schnitzler et al105 assessed the outcomes of 42 pregnancies in 35 patients with inflammatory bowel disease receiving either infliximab or adalimumab during pregnancy, compared with 56 pregnancies in 45 healthy patients without inflammatory bowel disease. There was no statistical difference in abortion rates between patients receiving anti-TNF agents and healthy women without inflammatory bowel disease (21% vs 14%, P = .4234). There was also no significant difference observed in birth weight, birth length, or cranial circumference of the children between the two groups. However, pregnancies with direct exposure to anti-TNF agents resulted in a higher frequency of premature delivery (25% vs 6%, P = .023).
Similar results were noted from the Crohn’s Therapy, Resource, Evaluation, and Assessment Tool, or TREAT, registry, as well as from a large systematic review by Vinet et al106 and case reports of rheumatoid arthritis and inflammatory bowel disease in women exposed to anti-TNF agents during pregnancy.
In addition, results from a recent systematic review of 38 studies of anti-TNF use and fetal risk, with a total of 437 women (189 on infliximab, 230 on adalimumab, 18 on certolizumab pegol), showed similar results.107 In pregnancies exposed to anti-TNF agents, the rates of congenital abnormalities (3.4%), fetal deaths (8.5%), and preterm births (2.7%) were similar to those in the general population.
For patients in disease remission on TNF inhibitors, it is reasonable to continue these agents during pregnancy after careful discussion with the patient. Fetal safety and infant immunization response after delivery are the primary concerns in these cases.
Both infliximab and adalimumab cross the placenta and remain detectable in the baby’s circulation 4 months (for adalimumab) to 6 months (for infliximab) after delivery. It is currently recommended that infliximab be stopped at 32 weeks of gestation for the remainder of the pregnancy and that adalimumab be stopped at 34 to 36 weeks, given a planned 40-week gestation.
Certolizumab does not cross the placenta in significant amounts and should be continued throughout pregnancy; drug levels in infants were shown to be less than 2 μg/mL, even when dosed the week of delivery.108–112
TAKE-HOME POINTS
- All health care providers of patients with rheumatoid arthritis and inflammatory bowel disease should be familiar with anti-TNF agents used in treating these diseases.
- The benefits of controlling the disease far outweigh the risks of therapy when used appropriately.
- Care of these patients should be multidisciplinary, with clear communication between primary care physician and specialist.
- Patient education and monitoring combined with prompt communication between primary care physician and specialist are key.
- Wijbrandts CA, Dijkgraaf MG, Kraan MC, et al. The clinical response to infliximab in rheumatoid arthritis is in part dependent on pretreatment tumour necrosis factor alpha expression in the synovium. Ann Rheum Dis 2008; 67:1139–1144.
- Sfikakis PP. The first decade of biologic TNF antagonists in clinical practice: lessons learned, unresolved issues and future directions. Curr Dir Autoimmun 2010; 11:180–210.
- Casellas F, Alcalá MJ, Prieto L, Miró JR, Malagelada JR. Assessment of the influence of disease activity on the quality of life of patients with inflammatory bowel disease using a short questionnaire. Am J Gastroenterol 2004; 99:457–461.
- Casellas F, López-Vivancos J, Badia X, Vilaseca J, Malagelada JR. Influence of inflammatory bowel disease on different dimensions of quality of life. Eur J Gastroenterol Hepatol 2001; 13:567–572.
- Baecklund E, Iliadou A, Askling J, et al. Association of chronic inflammation, not its treatment, with increased lymphoma risk in rheumatoid arthritis. Arthritis Rheum 2006; 54:692–701.
- Franklin J, Lunt M, Bunn D, Symmons D, Silman A. Incidence of lymphoma in a large primary care derived cohort of cases of inflammatory polyarthritis. Ann Rheum Dis 2006; 65:617–622.
- Toruner M, Loftus EV, Harmsen WS, et al. Risk factors for opportunistic infections in patients with inflammatory bowel disease. Gastroenterology 2008; 134:929–936.
- Wolfe F, Michaud K. Biologic treatment of rheumatoid arthritis and the risk of malignancy: analyses from a large US observational study. Arthritis Rheum 2007; 56:2886–2895.
- Prevention of pneumococcal disease: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 1997; 46:1–24.
- Zhang J, Xie F, Delzell E, et al. Association between vaccination for herpes zoster and risk of herpes zoster infection among older patients with selected immune-mediated diseases. JAMA 2012; 308:43–49.
- Moreland LW, Baumgartner SW, Schiff MH, et al. Treatment of rheumatoid arthritis with a recombinant human tumor necrosis factor receptor (p75)-Fc fusion protein. N Engl J Med 1997; 337:141–147.
- Altunöz ME, Senates E, Yesil A, Calhan T, Ovünç AO. Patients with inflammatory bowel disease have a lower response rate to HBV vaccination compared to controls. Dig Dis Sci 2012; 57:1039–1044.
- Gisbert JP, Villagrasa JR, Rodríguez-Nogueiras A, Chaparro M. Efficacy of hepatitis B vaccination and revaccination and factors impacting on response in patients with inflammatory bowel disease. Am J Gastroenterol 2012; 107:1460–1466.
- Carrera E, Manzano R, Garrido E. Efficacy of the vaccination in inflammatory bowel disease. World J Gastroenterol 2013; 19:1349–1353.
- Gisbert JP, Menchén L, García-Sánchez V, Marín I, Villagrasa JR, Chaparro M. Comparison of the effectiveness of two protocols for vaccination (standard and double dosage) against hepatitis B virus in patients with inflammatory bowel disease. Aliment Pharmacol Ther 2012; 35:1379–1385.
- Ferkolj I. How to improve the safety of biologic therapy in Crohn’s disease. J Physiol Pharmacol 2009; 60(suppl 7):67–70.
- Siegel CA. The risks of biologic therapy for inflammatory bowel disease. In:Bernstein ED, editor. The Inflammatory Bowel Disease Yearbook, volume 6. London, UK: Remedica, 2010:89–108.
- Remicade (infliximab) Package Insert. Horsham, PA: Janssen Biotech, Inc; 2013. http://www.remicade.com/shared/product/remicade/prescribing-information.pdf. Accessed January 2, 2014.
- Vermeire S, Noman M, Van Assche G, et al. Autoimmunity associated with anti-tumor necrosis factor alpha treatment in Crohn’s disease: a prospective cohort study. Gastroenterology 2003; 125:32–39.
- Cush JJ. Biological drug use: US perspectives on indications and monitoring. Ann Rheum Dis 2005; 64(suppl 4):iv18–iv23.
- TNF neutralization in MS: results of a randomized, placebo-controlled multicenter study. The Lenercept Multiple Sclerosis Study Group and The University of British Columbia MS/MRI Analysis Group. Neurology 1999; 53:457–465.
- Chung ES, Packer M, Lo KH, Fasanmade AA, Willerson JT; Anti-TNF Therapy Against Congestive Heart Failure Investigators. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: results of the anti-TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation 2003; 107:3133–3140.
- Cheifetz A, Mayer L. Monoclonal antibodies, immunogenicity, and associated infusion reactions. Mt Sinai J Med 2005; 72:250–256.
- Cheifetz A, Smedley M, Martin S, et al. The incidence and management of infusion reactions to infliximab: a large center experience. Am J Gastroenterol 2003; 98:1315–1324.
- Vultaggio A, Matucci A, Nencini F, et al. Anti-infliximab IgE and non-IgE antibodies and induction of infusion-related severe anaphylactic reactions. Allergy 2010; 65:657–661.
- Ramos-Casals M, Brito-Zerón P, Muñoz S, et al. Autoimmune diseases induced by TNF-targeted therapies: analysis of 233 cases. Medicine (Baltimore) 2007; 86:242–251.
- Stallmach A, Hagel S, Bruns T. Adverse effects of biologics used for treating IBD. Best Pract Res Clin Gastroenterol 2010; 24:167–182.
- Haagsma CJ, Blom HJ, van Riel PL, et al. Influence of sulphasalazine, methotrexate, and the combination of both on plasma homocysteine concentrations in patients with rheumatoid arthritis. Ann Rheum Dis 1999; 58:79–84.
- Fiorino G, Allez M, Malesci A, Danese S. Review article: anti TNF-alpha induced psoriasis in patients with inflammatory bowel disease. Aliment Pharmacol Ther 2009; 29:921–927.
- Harrison MJ, Dixon WG, Watson KD, et al; British Society for Rheumatology Biologics Register Control Centre Consortium; BSRBR. Rates of new-onset psoriasis in patients with rheumatoid arthritis receiving anti-tumour necrosis factor alpha therapy: results from the British Society for Rheumatology Biologics Register. Ann Rheum Dis 2009; 68:209–215.
- Marchesoni A, Zaccara E, Gorla R, et al. TNF-alpha antagonist survival rate in a cohort of rheumatoid arthritis patients observed under conditions of standard clinical practice. Ann N Y Acad Sci 2009; 1173:837–846.
- Tomas L, Lazurova I, Pundova L, et al. Acute and long-term effect of infliximab on humoral and echocardiographic parameters in patients with chronic inflammatory diseases. Clin Rheumatol 2013 32:61–66.
- Senel S, Cobankara V, Taskoylu O, et al. The safety and efficacy of etanercept on cardiac functions and lipid profile in patients with active rheumatoid arthritis. J Investig Med 2012; 60:62–65.
- Al-Aly Z, Pan H, Zeringue A, et al. Tumor necrosis factor-a blockade, cardiovascular outcomes, and survival in rheumatoid arthritis. Transl Res 2011; 157:10–18.
- Listing J, Strangfeld A, Kekow J, et al. Does tumor necrosis factor alpha inhibition promote or prevent heart failure in patients with rheumatoid arthritis? Arthritis Rheum 2008; 58:667–677.
- Wolfe F, Michaud K. Heart failure in rheumatoid arthritis: rates, predictors, and the effect of anti-tumor necrosis factor therapy. Am J Med 2004; 116:305–311.
- Enayati PJ, Papadakis KA. Association of anti-tumor necrosis factor therapy with the development of multiple sclerosis. J Clin Gastroenterol 2005; 39:303–306.
- Mohan N, Edwards ET, Cupps TR, et al. Demyelination occurring during anti-tumor necrosis factor alpha therapy for inflammatory arthritides. Arthritis Rheum 2001; 44:2862–2869.
- Sands BE, Anderson FH, Bernstein CN, et al. Infliximab maintenance therapy for fistulizing Crohn’s disease. N Engl J Med 2004; 350:876–885.
- Thomas CW, Weinshenker BG, Sandborn WJ. Demyelination during anti-tumor necrosis factor alpha therapy with infliximab for Crohn’s disease. Inflamm Bowel Dis 2004; 10:28–31.
- Foroozan R, Buono LM, Sergott RC, Savino PJ. Retrobulbar optic neuritis associated with infliximab. Arch Ophthalmol 2002; 120:985–987.
- Strong BY, Erny BC, Herzenberg H, Razzeca KJ. Retrobulbar optic neuritis associated with infliximab in a patient with Crohn disease. Ann Intern Med 2004; 140:W34.
- ten Tusscher MP, Jacobs PJ, Busch MJ, de Graaf L, Diemont WL. Bilateral anterior toxic optic neuropathy and the use of infliximab. BMJ 2003; 326:579.
- Hegde N, Gayomali C, Rich MW. Infliximab-induced headache and infliximab-induced meningitis: two ends of the same spectrum? South Med J 2005; 98:564–566.
- Schiff MH, Burmester GR, Kent JD, et al. Safety analyses of adalimumab (HUMIRA) in global clinical trials and US postmarketing surveillance of patients with rheumatoid arthritis. Ann Rheum Dis 2006; 65:889–894.
- Beaugerie L, Brousse N, Bouvier AM, et al; CESAME Study Group. Lymphoproliferative disorders in patients receiving thiopurines for inflammatory bowel disease: a prospective observational cohort study. Lancet 2009; 374:1617–1625.
- Ekström K, Hjalgrim H, Brandt L, et al. Risk of malignant lymphomas in patients with rheumatoid arthritis and in their first-degree relatives. Arthritis Rheum 2003; 48:963–970.
- Lakatos PL, Lakatos L. Risk for colorectal cancer in ulcerative colitis: changes, causes and management strategies. World J Gastroenterol 2008; 14:3937–3947.
- Persson PG, Karlén P, Bernell O, et al. Crohn’s disease and cancer: a population-based cohort study. Gastroenterology 1994; 107:1675–1679.
- de Silva S, Devlin S, Panaccione R. Optimizing the safety of biologic therapy for IBD. Nat Rev Gastroenterol Hepatol 2010; 7:93–101.
- Lichtenstein GR, Feagan BG, Cohen RD, et al. Serious infections and mortality in association with therapies for Crohn’s disease: TREAT registry. Clin Gastroenterol Hepatol 2006; 4:621–630.
- Peyrin-Biroulet L, Deltenre P, de Suray N, Branche J, Sandborn WJ, Colombel JF. Efficacy and safety of tumor necrosis factor antagonists in Crohn’s disease: meta-analysis of placebo-controlled trials. Clin Gastroenterol Hepatol 2008; 6:644–653.
- Hanauer SB, Feagan BG, Lichtenstein GR, et al; ACCENT I Study Group. Maintenance infliximab for Crohn’s disease: the ACCENT I randomised trial. Lancet 2002; 359:1541–1549.
- Colombel JF, Sandborn WJ, Rutgeerts P, et al. Adalimumab for maintenance of clinical response and remission in patients with Crohn’s disease: the CHARM trial. Gastroenterology 2007; 132:52–65.
- Sandborn WJ, Feagan BG, Stoinov S, et al; PRECISE 1 Study Investigators. Certolizumab pegol for the treatment of Crohn’s disease. N Engl J Med 2007; 357:228–238.
- Sandborn WJ, Hanauer SB, Rutgeerts P, et al. Adalimumab for maintenance treatment of Crohn’s disease: results of the CLASSIC II trial. Gut 2007; 56:1232–1239.
- Hanauer SB, Sandborn WJ, Rutgeerts P, et al. Human anti-tumor necrosis factor monoclonal antibody (adalimumab) in Crohn’s disease: the CLASSIC-I trial. Gastroenterology 2006; 130:323–333.
- Rutgeerts P, D’Haens G, Targan S, et al. Efficacy and safety of retreatment with anti-tumor necrosis factor antibody (infliximab) to maintain remission in Crohn’s disease. Gastroenterology 1999; 117:761–769.
- Rutgeerts P, Sandborn WJ, Feagan BG, et al. Infliximab for induction and maintenance therapy for ulcerative colitis. N Engl J Med 2005; 353:2462–2476.
- Pedersen N, Duricova D, Elkjaer M, Gamborg M, Munkholm P, Jess T. Risk of extra-intestinal cancer in inflammatory bowel disease: meta-analysis of population-based cohort studies. Am J Gastroenterol 2010; 105:1480–1487.
- Bernstein CN, Blanchard JF, Kliewer E, Wajda A. Cancer risk in patients with inflammatory bowel disease: a population-based study. Cancer 2001; 91:854–862.
- Siegel CA, Marden SM, Persing SN, Larson RJ, Sands BE. Risk of lymphoma associated with combination anti-tumor necrosis factor and immunomodulator therapy for the treatment of Crohn’s disease: a meta-analysis. Clin Gastroenterol Hepatol 2009; 7:874–881.
- Wolfe F, Michaud K. Lympyhoma in rheumatoid arthritis. The effect of methotrexate and anti-tumor necrosis factor therapy in 18,572 patients. Arthritis Rheum 2004; 50:1740–1751.
- Parakkal D, Sifuentes H, Semer R, Ehrenpreis ED. Hepatosplenic T-cell lymphoma in patients receiving TNF-a inhibitor therapy: expanding the groups at risk. Eur J Gastroenterol Hepatol 2011; 23:1150–1156.
- Rosh JR, Gross T, Mamula P, Griffiths A, Hyams J. Hepatosplenic T-cell lymphoma in adolescents and young adults with Crohn’s disease: a cautionary tale? Inflamm Bowel Dis 2007; 13:1024–1030.
- Shale M, Kanfer E, Panaccione R, Ghosh S. Hepatosplenic T cell lymphoma in inflammatory bowel disease. Gut 2008; 57:1639–1641.
- Thai A, Prindiville T. Hepatosplenic T-cell lymphoma and inflammatory bowel disease. J Crohns Colitis 2010; 4:511–522.
- Viget N, Vernier-Massouille G, Salmon-Ceron D, Yazdanpanah Y, Colombel JF. Opportunistic infections in patients with inflammatory bowel disease: prevention and diagnosis. Gut 2008; 57:549–558.
- Bekker LG, Freeman S, Murray PJ, Ryffel B, Kaplan G. TNF-alpha controls intracellular mycobacterial growth by both inducible nitric oxide synthase-dependent and inducible nitric oxide synthase-independent pathways. J Immunol 2001; 166:6728–6734.
- Roach DR, Bean AG, Demangel C, France MP, Briscoe H, Britton WJ. TNF regulates chemokine induction essential for cell recruitment, granuloma formation, and clearance of mycobacterial infection. J Immunol 2002; 168:4620–4627.
- Gómez-Reino JJ, Carmona L, Valverde VR, Mola EM, Montero MDBIOBADASER Group. Treatment of rheumatoid arthritis with tumor necrosis factor inhibitors may predispose to significant increase in tuberculosis risk: a multicenter active-surveillance report. Arthritis Rheum 2003; 48:2122–2127.
- Keane J, Gershon S, Wise RP, et al. Tuberculosis associated with infliximab, a tumor necrosis factor alpha-neutralizing agent. N Engl J Med 2001; 345:1098–1104.
- Smolen J, Landewé RB, Mease P, et al. Efficacy and safety of certolizumab pegol plus methotrexate in active rheumatoid arthritis: the RAPID 2 study. A randomised controlled trial. Ann Rheum Dis 2009; 68:797–804.
- Demkow U, Broniarek-Samson B, Filewska M, et al. Prevalence of latent tuberculosis infection in health care workers in Poland assessed by interferon-gamma whole blood and tuberculin skin tests. J Physiol Pharmacol 2008; 59(suppl 6):209–217.
- Pache I, Rogler G, Felley C. TNF-alpha blockers in inflammatory bowel diseases: practical consensus recommendations and a user’s guide. Swiss Med Wkly 2009; 139:278–287.
- Rahier JF, Ben-Horin S, Chowers Y, et al; European Crohn’s and Colitis Organisation (ECCO). European evidence-based consensus on the prevention, diagnosis and management of opportunistic infections in inflammatory bowel disease. J Crohns Colitis 2009; 3:47–91.
- Rahier JF, Yazdanpanah Y, Colombel JF, Travis S. The European (ECCO) consensus on infection in IBD: what does it change for the clinician? Gut 2009; 58:1313–1315.
- Bergstrom L, Yocum DE, Ampel NM, et al. Increased risk of coccidioidomycosis in patients treated with tumor necrosis factor alpha antagonists. Arthritis Rheum 2004; 50:1959–1966.
- Lee JH, Slifman NR, Gershon SK, et al. Life-threatening histoplasmosis complicating immunotherapy with tumor necrosis factor alpha antagonists infliximab and etanercept. Arthritis Rheum 2002; 46:2565–2570.
- Wood KL, Hage CA, Knox KS, et al. Histoplasmosis after treatment with anti-tumor necrosis factor-alpha therapy. Am J Respir Crit Care Med 2003; 167:1279–1282.
- Reddy JG, Loftus EV. Safety of infliximab and other biologic agents in the inflammatory bowel diseases. Gastroenterol Clin North Am 2006; 35:837–855.
- Esteve M, Saro C, González-Huix F, Suarez F, Forné M, Viver JM. Chronic hepatitis B reactivation following infliximab therapy in Crohn’s disease patients: need for primary prophylaxis. Gut 2004; 53:1363–1365.
- Michel M, Duvoux C, Hezode C, Cherqui D. Fulminant hepatitis after infliximab in a patient with hepatitis B virus treated for an adult onset Still’s disease. J Rheumatol 2003; 30:1624–1625.
- Ostuni P, Botsios C, Punzi L, Sfriso P, Todesco S. Hepatitis B reactivation in a chronic hepatitis B surface antigen carrier with rheumatoid arthritis treated with infliximab and low dose methotrexate. Ann Rheum Dis 2003; 62:686–687.
- Pérez-Alvarez R, Díaz-Lagares C, García-Hernández F, et al; BIOGEAS Study Group. Hepatitis B virus (HBV) reactivation in patients receiving tumor necrosis factor (TNF)-targeted therapy: analysis of 257 cases. Medicine (Baltimore) 2011; 90:359–371.
- Loras C, Gisbert JP, Mínguez M, et al; REPENTINA study; GETECCU (Grupo Español de Enfermedades de Crohn y Colitis Ulcerosa) Group. Liver dysfunction related to hepatitis B and C in patients with inflammatory bowel disease treated with immunosuppressive therapy. Gut 2010; 59:1340–1346.
- Park SH, Yang SK, Lim YS, et al. Clinical courses of chronic hepatitis B virus infection and inflammatory bowel disease in patients with both diseases. Inflamm Bowel Dis 2012; 18:2004–2010.
- Loomba R, Rowley A, Wesley R, et al. Systematic review: the effect of preventive lamivudine on hepatitis B reactivation during chemotherapy. Ann Intern Med 2008; 148:519–528.
- Lok AS, McMahon BJ. Chronic hepatitis B: update 2009. Hepatology 2009; 50:661–662.
- European Association For The Study Of The Liver. EASL Clinical Practice Guidelines: management of chronic hepatitis B. J Hepatol 2009; 50:227–242.
- Watanabe M, Shibuya A, Takada J, et al. Entecavir is an optional agent to prevent hepatitis B virus (HBV) reactivation: a review of 16 patients. Eur J Intern Med 2010; 21:333–337.
- Biancone L, Pavia M, Del Vecchio Blanco G, et al; Italian Group for the Study of the Colon and Rectum (GISC). Hepatitis B and C virus infection in Crohn’s disease. Inflamm Bowel Dis 2001; 7:287–294.
- Melmed GY. Vaccination strategies for patients with inflammatory bowel disease on immunomodulators and biologics. Inflamm Bowel Dis 2009; 15:1410–1416.
- Melmed GY, Ippoliti AF, Papadakis KA, et al. Patients with inflammatory bowel disease are at risk for vaccine-preventable illnesses. Am J Gastroenterol 2006; 101:1834–1840.
- Ferri C, Ferraccioli G, Ferrari D, et al. Safety of anti-tumor necrosis factor-alpha therapy in patients with rheumatoid arthritis and chronic hepatitis C virus infection. J Rheumatol 2008; 35:1944–1949.
- Mok MY, Ng WL, Yuen MF, Wong RW, Lau CS. Safety of disease modifying anti-rheumatic agents in rheumatoid arthritis patients with chronic viral hepatitis. Clin Exp Rheumatol 2000; 18:363–368.
- Vassilopoulos D, Calabrese LH. Risks of immunosuppressive therapies including biologic agents in patients with rheumatic diseases and coexisting chronic viral infections. Curr Opin Rheumatol 2007; 19:619–625.
- Vassilopoulos D, Apostolopoulou A, Hadziyannis E, et al. Long-term safety of anti-TNF treatment in patients with rheumatic diseases and chronic or resolved hepatitis B virus infection. Ann Rheum Dis 2010; 69:1352–1355.
- Saag KG, Teng GG, Patkar NM, et al; American College of Rheumatology. American College of Rheumatology 2008 recommendations for the use of nonbiologic and biologic disease-modifying antirheumatic drugs in rheumatoid arthritis. Arthritis Rheum 2008; 59:762–784.
- Cornish J, Tan E, Teare J, et al. A meta-analysis on the influence of inflammatory bowel disease on pregnancy. Gut 2007; 56:830–837.
- Dominitz JA, Young JC, Boyko EJ. Outcomes of infants born to mothers with inflammatory bowel disease: a population-based cohort study. Am J Gastroenterol 2002; 97:641–648.
- Kornfeld D, Cnattingius S, Ekbom A. Pregnancy outcomes in women with inflammatory bowel disease—a population-based cohort study. Am J Obstet Gynecol 1997; 177:942–946.
- Mahadevan U, Sandborn WJ, Li DK, Hakimian S, Kane S, Corley DA. Pregnancy outcomes in women with inflammatory bowel disease: a large community-based study from Northern California. Gastroenterology 2007; 133:1106–1112.
- Nguyen GC, Boudreau H, Harris ML, Maxwell CV. Outcomes of obstetric hospitalizations among women with inflammatory bowel disease in the United States. Clin Gastroenterol Hepatol 2009; 7:329–334.
- Schnitzler F, Fidder H, Ferrante M, et al. Outcome of pregnancy in women with inflammatory bowel disease treated with antitumor necrosis factor therapy. Inflamm Bowel Dis 2011; 17:1846–1854.
- Vinet E, Pineau C, Gordon C, Clarke AE, Bernatsky S. Biologic therapy and pregnancy outcomes in women with rheumatic diseases. Arthritis Rheum 2009; 61:587–592.
- Guidi L, Pugliese D, Armuzzi A. Update on the management of inflammatory bowel disease: specific role of adalimumab. Clin Exp Gastroenterol 2011; 4:163–172.
- Mahadevan U. Continuing immunomodulators and biologic medications in pregnant IBD patients – pro. IInflamm Bowel Dis 2007; 13:1439–1440.
- Mahadevan U. Gastrointestinal medications in pregnancy. Best Pract Res Clin Gastroenterol 2007; 21:849–877.
- Mahadevan U, Cucchiara S, Hyams JS, et al. The London Position Statement of the World Congress of Gastroenterology on Biological Therapy for IBD with the European Crohn’s and Colitis Organisation: pregnancy and pediatrics. Am J Gastroenterol 2011; 106:214–223.
- Mahadevan U, Kane S. Use of infliximab in pregnancy. Am J Gastroenterol 2010; 105:219–220.
- Vasiliauskas EA, Church JA, Silverman N, Barry M, Targan SR, Dubinsky MC. Case report: evidence for transplacental transfer of maternally administered infliximab to the newborn. Clin Gastroenterol Hepatol 2006; 4:1255–1258.
- Wijbrandts CA, Dijkgraaf MG, Kraan MC, et al. The clinical response to infliximab in rheumatoid arthritis is in part dependent on pretreatment tumour necrosis factor alpha expression in the synovium. Ann Rheum Dis 2008; 67:1139–1144.
- Sfikakis PP. The first decade of biologic TNF antagonists in clinical practice: lessons learned, unresolved issues and future directions. Curr Dir Autoimmun 2010; 11:180–210.
- Casellas F, Alcalá MJ, Prieto L, Miró JR, Malagelada JR. Assessment of the influence of disease activity on the quality of life of patients with inflammatory bowel disease using a short questionnaire. Am J Gastroenterol 2004; 99:457–461.
- Casellas F, López-Vivancos J, Badia X, Vilaseca J, Malagelada JR. Influence of inflammatory bowel disease on different dimensions of quality of life. Eur J Gastroenterol Hepatol 2001; 13:567–572.
- Baecklund E, Iliadou A, Askling J, et al. Association of chronic inflammation, not its treatment, with increased lymphoma risk in rheumatoid arthritis. Arthritis Rheum 2006; 54:692–701.
- Franklin J, Lunt M, Bunn D, Symmons D, Silman A. Incidence of lymphoma in a large primary care derived cohort of cases of inflammatory polyarthritis. Ann Rheum Dis 2006; 65:617–622.
- Toruner M, Loftus EV, Harmsen WS, et al. Risk factors for opportunistic infections in patients with inflammatory bowel disease. Gastroenterology 2008; 134:929–936.
- Wolfe F, Michaud K. Biologic treatment of rheumatoid arthritis and the risk of malignancy: analyses from a large US observational study. Arthritis Rheum 2007; 56:2886–2895.
- Prevention of pneumococcal disease: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 1997; 46:1–24.
- Zhang J, Xie F, Delzell E, et al. Association between vaccination for herpes zoster and risk of herpes zoster infection among older patients with selected immune-mediated diseases. JAMA 2012; 308:43–49.
- Moreland LW, Baumgartner SW, Schiff MH, et al. Treatment of rheumatoid arthritis with a recombinant human tumor necrosis factor receptor (p75)-Fc fusion protein. N Engl J Med 1997; 337:141–147.
- Altunöz ME, Senates E, Yesil A, Calhan T, Ovünç AO. Patients with inflammatory bowel disease have a lower response rate to HBV vaccination compared to controls. Dig Dis Sci 2012; 57:1039–1044.
- Gisbert JP, Villagrasa JR, Rodríguez-Nogueiras A, Chaparro M. Efficacy of hepatitis B vaccination and revaccination and factors impacting on response in patients with inflammatory bowel disease. Am J Gastroenterol 2012; 107:1460–1466.
- Carrera E, Manzano R, Garrido E. Efficacy of the vaccination in inflammatory bowel disease. World J Gastroenterol 2013; 19:1349–1353.
- Gisbert JP, Menchén L, García-Sánchez V, Marín I, Villagrasa JR, Chaparro M. Comparison of the effectiveness of two protocols for vaccination (standard and double dosage) against hepatitis B virus in patients with inflammatory bowel disease. Aliment Pharmacol Ther 2012; 35:1379–1385.
- Ferkolj I. How to improve the safety of biologic therapy in Crohn’s disease. J Physiol Pharmacol 2009; 60(suppl 7):67–70.
- Siegel CA. The risks of biologic therapy for inflammatory bowel disease. In:Bernstein ED, editor. The Inflammatory Bowel Disease Yearbook, volume 6. London, UK: Remedica, 2010:89–108.
- Remicade (infliximab) Package Insert. Horsham, PA: Janssen Biotech, Inc; 2013. http://www.remicade.com/shared/product/remicade/prescribing-information.pdf. Accessed January 2, 2014.
- Vermeire S, Noman M, Van Assche G, et al. Autoimmunity associated with anti-tumor necrosis factor alpha treatment in Crohn’s disease: a prospective cohort study. Gastroenterology 2003; 125:32–39.
- Cush JJ. Biological drug use: US perspectives on indications and monitoring. Ann Rheum Dis 2005; 64(suppl 4):iv18–iv23.
- TNF neutralization in MS: results of a randomized, placebo-controlled multicenter study. The Lenercept Multiple Sclerosis Study Group and The University of British Columbia MS/MRI Analysis Group. Neurology 1999; 53:457–465.
- Chung ES, Packer M, Lo KH, Fasanmade AA, Willerson JT; Anti-TNF Therapy Against Congestive Heart Failure Investigators. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: results of the anti-TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation 2003; 107:3133–3140.
- Cheifetz A, Mayer L. Monoclonal antibodies, immunogenicity, and associated infusion reactions. Mt Sinai J Med 2005; 72:250–256.
- Cheifetz A, Smedley M, Martin S, et al. The incidence and management of infusion reactions to infliximab: a large center experience. Am J Gastroenterol 2003; 98:1315–1324.
- Vultaggio A, Matucci A, Nencini F, et al. Anti-infliximab IgE and non-IgE antibodies and induction of infusion-related severe anaphylactic reactions. Allergy 2010; 65:657–661.
- Ramos-Casals M, Brito-Zerón P, Muñoz S, et al. Autoimmune diseases induced by TNF-targeted therapies: analysis of 233 cases. Medicine (Baltimore) 2007; 86:242–251.
- Stallmach A, Hagel S, Bruns T. Adverse effects of biologics used for treating IBD. Best Pract Res Clin Gastroenterol 2010; 24:167–182.
- Haagsma CJ, Blom HJ, van Riel PL, et al. Influence of sulphasalazine, methotrexate, and the combination of both on plasma homocysteine concentrations in patients with rheumatoid arthritis. Ann Rheum Dis 1999; 58:79–84.
- Fiorino G, Allez M, Malesci A, Danese S. Review article: anti TNF-alpha induced psoriasis in patients with inflammatory bowel disease. Aliment Pharmacol Ther 2009; 29:921–927.
- Harrison MJ, Dixon WG, Watson KD, et al; British Society for Rheumatology Biologics Register Control Centre Consortium; BSRBR. Rates of new-onset psoriasis in patients with rheumatoid arthritis receiving anti-tumour necrosis factor alpha therapy: results from the British Society for Rheumatology Biologics Register. Ann Rheum Dis 2009; 68:209–215.
- Marchesoni A, Zaccara E, Gorla R, et al. TNF-alpha antagonist survival rate in a cohort of rheumatoid arthritis patients observed under conditions of standard clinical practice. Ann N Y Acad Sci 2009; 1173:837–846.
- Tomas L, Lazurova I, Pundova L, et al. Acute and long-term effect of infliximab on humoral and echocardiographic parameters in patients with chronic inflammatory diseases. Clin Rheumatol 2013 32:61–66.
- Senel S, Cobankara V, Taskoylu O, et al. The safety and efficacy of etanercept on cardiac functions and lipid profile in patients with active rheumatoid arthritis. J Investig Med 2012; 60:62–65.
- Al-Aly Z, Pan H, Zeringue A, et al. Tumor necrosis factor-a blockade, cardiovascular outcomes, and survival in rheumatoid arthritis. Transl Res 2011; 157:10–18.
- Listing J, Strangfeld A, Kekow J, et al. Does tumor necrosis factor alpha inhibition promote or prevent heart failure in patients with rheumatoid arthritis? Arthritis Rheum 2008; 58:667–677.
- Wolfe F, Michaud K. Heart failure in rheumatoid arthritis: rates, predictors, and the effect of anti-tumor necrosis factor therapy. Am J Med 2004; 116:305–311.
- Enayati PJ, Papadakis KA. Association of anti-tumor necrosis factor therapy with the development of multiple sclerosis. J Clin Gastroenterol 2005; 39:303–306.
- Mohan N, Edwards ET, Cupps TR, et al. Demyelination occurring during anti-tumor necrosis factor alpha therapy for inflammatory arthritides. Arthritis Rheum 2001; 44:2862–2869.
- Sands BE, Anderson FH, Bernstein CN, et al. Infliximab maintenance therapy for fistulizing Crohn’s disease. N Engl J Med 2004; 350:876–885.
- Thomas CW, Weinshenker BG, Sandborn WJ. Demyelination during anti-tumor necrosis factor alpha therapy with infliximab for Crohn’s disease. Inflamm Bowel Dis 2004; 10:28–31.
- Foroozan R, Buono LM, Sergott RC, Savino PJ. Retrobulbar optic neuritis associated with infliximab. Arch Ophthalmol 2002; 120:985–987.
- Strong BY, Erny BC, Herzenberg H, Razzeca KJ. Retrobulbar optic neuritis associated with infliximab in a patient with Crohn disease. Ann Intern Med 2004; 140:W34.
- ten Tusscher MP, Jacobs PJ, Busch MJ, de Graaf L, Diemont WL. Bilateral anterior toxic optic neuropathy and the use of infliximab. BMJ 2003; 326:579.
- Hegde N, Gayomali C, Rich MW. Infliximab-induced headache and infliximab-induced meningitis: two ends of the same spectrum? South Med J 2005; 98:564–566.
- Schiff MH, Burmester GR, Kent JD, et al. Safety analyses of adalimumab (HUMIRA) in global clinical trials and US postmarketing surveillance of patients with rheumatoid arthritis. Ann Rheum Dis 2006; 65:889–894.
- Beaugerie L, Brousse N, Bouvier AM, et al; CESAME Study Group. Lymphoproliferative disorders in patients receiving thiopurines for inflammatory bowel disease: a prospective observational cohort study. Lancet 2009; 374:1617–1625.
- Ekström K, Hjalgrim H, Brandt L, et al. Risk of malignant lymphomas in patients with rheumatoid arthritis and in their first-degree relatives. Arthritis Rheum 2003; 48:963–970.
- Lakatos PL, Lakatos L. Risk for colorectal cancer in ulcerative colitis: changes, causes and management strategies. World J Gastroenterol 2008; 14:3937–3947.
- Persson PG, Karlén P, Bernell O, et al. Crohn’s disease and cancer: a population-based cohort study. Gastroenterology 1994; 107:1675–1679.
- de Silva S, Devlin S, Panaccione R. Optimizing the safety of biologic therapy for IBD. Nat Rev Gastroenterol Hepatol 2010; 7:93–101.
- Lichtenstein GR, Feagan BG, Cohen RD, et al. Serious infections and mortality in association with therapies for Crohn’s disease: TREAT registry. Clin Gastroenterol Hepatol 2006; 4:621–630.
- Peyrin-Biroulet L, Deltenre P, de Suray N, Branche J, Sandborn WJ, Colombel JF. Efficacy and safety of tumor necrosis factor antagonists in Crohn’s disease: meta-analysis of placebo-controlled trials. Clin Gastroenterol Hepatol 2008; 6:644–653.
- Hanauer SB, Feagan BG, Lichtenstein GR, et al; ACCENT I Study Group. Maintenance infliximab for Crohn’s disease: the ACCENT I randomised trial. Lancet 2002; 359:1541–1549.
- Colombel JF, Sandborn WJ, Rutgeerts P, et al. Adalimumab for maintenance of clinical response and remission in patients with Crohn’s disease: the CHARM trial. Gastroenterology 2007; 132:52–65.
- Sandborn WJ, Feagan BG, Stoinov S, et al; PRECISE 1 Study Investigators. Certolizumab pegol for the treatment of Crohn’s disease. N Engl J Med 2007; 357:228–238.
- Sandborn WJ, Hanauer SB, Rutgeerts P, et al. Adalimumab for maintenance treatment of Crohn’s disease: results of the CLASSIC II trial. Gut 2007; 56:1232–1239.
- Hanauer SB, Sandborn WJ, Rutgeerts P, et al. Human anti-tumor necrosis factor monoclonal antibody (adalimumab) in Crohn’s disease: the CLASSIC-I trial. Gastroenterology 2006; 130:323–333.
- Rutgeerts P, D’Haens G, Targan S, et al. Efficacy and safety of retreatment with anti-tumor necrosis factor antibody (infliximab) to maintain remission in Crohn’s disease. Gastroenterology 1999; 117:761–769.
- Rutgeerts P, Sandborn WJ, Feagan BG, et al. Infliximab for induction and maintenance therapy for ulcerative colitis. N Engl J Med 2005; 353:2462–2476.
- Pedersen N, Duricova D, Elkjaer M, Gamborg M, Munkholm P, Jess T. Risk of extra-intestinal cancer in inflammatory bowel disease: meta-analysis of population-based cohort studies. Am J Gastroenterol 2010; 105:1480–1487.
- Bernstein CN, Blanchard JF, Kliewer E, Wajda A. Cancer risk in patients with inflammatory bowel disease: a population-based study. Cancer 2001; 91:854–862.
- Siegel CA, Marden SM, Persing SN, Larson RJ, Sands BE. Risk of lymphoma associated with combination anti-tumor necrosis factor and immunomodulator therapy for the treatment of Crohn’s disease: a meta-analysis. Clin Gastroenterol Hepatol 2009; 7:874–881.
- Wolfe F, Michaud K. Lympyhoma in rheumatoid arthritis. The effect of methotrexate and anti-tumor necrosis factor therapy in 18,572 patients. Arthritis Rheum 2004; 50:1740–1751.
- Parakkal D, Sifuentes H, Semer R, Ehrenpreis ED. Hepatosplenic T-cell lymphoma in patients receiving TNF-a inhibitor therapy: expanding the groups at risk. Eur J Gastroenterol Hepatol 2011; 23:1150–1156.
- Rosh JR, Gross T, Mamula P, Griffiths A, Hyams J. Hepatosplenic T-cell lymphoma in adolescents and young adults with Crohn’s disease: a cautionary tale? Inflamm Bowel Dis 2007; 13:1024–1030.
- Shale M, Kanfer E, Panaccione R, Ghosh S. Hepatosplenic T cell lymphoma in inflammatory bowel disease. Gut 2008; 57:1639–1641.
- Thai A, Prindiville T. Hepatosplenic T-cell lymphoma and inflammatory bowel disease. J Crohns Colitis 2010; 4:511–522.
- Viget N, Vernier-Massouille G, Salmon-Ceron D, Yazdanpanah Y, Colombel JF. Opportunistic infections in patients with inflammatory bowel disease: prevention and diagnosis. Gut 2008; 57:549–558.
- Bekker LG, Freeman S, Murray PJ, Ryffel B, Kaplan G. TNF-alpha controls intracellular mycobacterial growth by both inducible nitric oxide synthase-dependent and inducible nitric oxide synthase-independent pathways. J Immunol 2001; 166:6728–6734.
- Roach DR, Bean AG, Demangel C, France MP, Briscoe H, Britton WJ. TNF regulates chemokine induction essential for cell recruitment, granuloma formation, and clearance of mycobacterial infection. J Immunol 2002; 168:4620–4627.
- Gómez-Reino JJ, Carmona L, Valverde VR, Mola EM, Montero MDBIOBADASER Group. Treatment of rheumatoid arthritis with tumor necrosis factor inhibitors may predispose to significant increase in tuberculosis risk: a multicenter active-surveillance report. Arthritis Rheum 2003; 48:2122–2127.
- Keane J, Gershon S, Wise RP, et al. Tuberculosis associated with infliximab, a tumor necrosis factor alpha-neutralizing agent. N Engl J Med 2001; 345:1098–1104.
- Smolen J, Landewé RB, Mease P, et al. Efficacy and safety of certolizumab pegol plus methotrexate in active rheumatoid arthritis: the RAPID 2 study. A randomised controlled trial. Ann Rheum Dis 2009; 68:797–804.
- Demkow U, Broniarek-Samson B, Filewska M, et al. Prevalence of latent tuberculosis infection in health care workers in Poland assessed by interferon-gamma whole blood and tuberculin skin tests. J Physiol Pharmacol 2008; 59(suppl 6):209–217.
- Pache I, Rogler G, Felley C. TNF-alpha blockers in inflammatory bowel diseases: practical consensus recommendations and a user’s guide. Swiss Med Wkly 2009; 139:278–287.
- Rahier JF, Ben-Horin S, Chowers Y, et al; European Crohn’s and Colitis Organisation (ECCO). European evidence-based consensus on the prevention, diagnosis and management of opportunistic infections in inflammatory bowel disease. J Crohns Colitis 2009; 3:47–91.
- Rahier JF, Yazdanpanah Y, Colombel JF, Travis S. The European (ECCO) consensus on infection in IBD: what does it change for the clinician? Gut 2009; 58:1313–1315.
- Bergstrom L, Yocum DE, Ampel NM, et al. Increased risk of coccidioidomycosis in patients treated with tumor necrosis factor alpha antagonists. Arthritis Rheum 2004; 50:1959–1966.
- Lee JH, Slifman NR, Gershon SK, et al. Life-threatening histoplasmosis complicating immunotherapy with tumor necrosis factor alpha antagonists infliximab and etanercept. Arthritis Rheum 2002; 46:2565–2570.
- Wood KL, Hage CA, Knox KS, et al. Histoplasmosis after treatment with anti-tumor necrosis factor-alpha therapy. Am J Respir Crit Care Med 2003; 167:1279–1282.
- Reddy JG, Loftus EV. Safety of infliximab and other biologic agents in the inflammatory bowel diseases. Gastroenterol Clin North Am 2006; 35:837–855.
- Esteve M, Saro C, González-Huix F, Suarez F, Forné M, Viver JM. Chronic hepatitis B reactivation following infliximab therapy in Crohn’s disease patients: need for primary prophylaxis. Gut 2004; 53:1363–1365.
- Michel M, Duvoux C, Hezode C, Cherqui D. Fulminant hepatitis after infliximab in a patient with hepatitis B virus treated for an adult onset Still’s disease. J Rheumatol 2003; 30:1624–1625.
- Ostuni P, Botsios C, Punzi L, Sfriso P, Todesco S. Hepatitis B reactivation in a chronic hepatitis B surface antigen carrier with rheumatoid arthritis treated with infliximab and low dose methotrexate. Ann Rheum Dis 2003; 62:686–687.
- Pérez-Alvarez R, Díaz-Lagares C, García-Hernández F, et al; BIOGEAS Study Group. Hepatitis B virus (HBV) reactivation in patients receiving tumor necrosis factor (TNF)-targeted therapy: analysis of 257 cases. Medicine (Baltimore) 2011; 90:359–371.
- Loras C, Gisbert JP, Mínguez M, et al; REPENTINA study; GETECCU (Grupo Español de Enfermedades de Crohn y Colitis Ulcerosa) Group. Liver dysfunction related to hepatitis B and C in patients with inflammatory bowel disease treated with immunosuppressive therapy. Gut 2010; 59:1340–1346.
- Park SH, Yang SK, Lim YS, et al. Clinical courses of chronic hepatitis B virus infection and inflammatory bowel disease in patients with both diseases. Inflamm Bowel Dis 2012; 18:2004–2010.
- Loomba R, Rowley A, Wesley R, et al. Systematic review: the effect of preventive lamivudine on hepatitis B reactivation during chemotherapy. Ann Intern Med 2008; 148:519–528.
- Lok AS, McMahon BJ. Chronic hepatitis B: update 2009. Hepatology 2009; 50:661–662.
- European Association For The Study Of The Liver. EASL Clinical Practice Guidelines: management of chronic hepatitis B. J Hepatol 2009; 50:227–242.
- Watanabe M, Shibuya A, Takada J, et al. Entecavir is an optional agent to prevent hepatitis B virus (HBV) reactivation: a review of 16 patients. Eur J Intern Med 2010; 21:333–337.
- Biancone L, Pavia M, Del Vecchio Blanco G, et al; Italian Group for the Study of the Colon and Rectum (GISC). Hepatitis B and C virus infection in Crohn’s disease. Inflamm Bowel Dis 2001; 7:287–294.
- Melmed GY. Vaccination strategies for patients with inflammatory bowel disease on immunomodulators and biologics. Inflamm Bowel Dis 2009; 15:1410–1416.
- Melmed GY, Ippoliti AF, Papadakis KA, et al. Patients with inflammatory bowel disease are at risk for vaccine-preventable illnesses. Am J Gastroenterol 2006; 101:1834–1840.
- Ferri C, Ferraccioli G, Ferrari D, et al. Safety of anti-tumor necrosis factor-alpha therapy in patients with rheumatoid arthritis and chronic hepatitis C virus infection. J Rheumatol 2008; 35:1944–1949.
- Mok MY, Ng WL, Yuen MF, Wong RW, Lau CS. Safety of disease modifying anti-rheumatic agents in rheumatoid arthritis patients with chronic viral hepatitis. Clin Exp Rheumatol 2000; 18:363–368.
- Vassilopoulos D, Calabrese LH. Risks of immunosuppressive therapies including biologic agents in patients with rheumatic diseases and coexisting chronic viral infections. Curr Opin Rheumatol 2007; 19:619–625.
- Vassilopoulos D, Apostolopoulou A, Hadziyannis E, et al. Long-term safety of anti-TNF treatment in patients with rheumatic diseases and chronic or resolved hepatitis B virus infection. Ann Rheum Dis 2010; 69:1352–1355.
- Saag KG, Teng GG, Patkar NM, et al; American College of Rheumatology. American College of Rheumatology 2008 recommendations for the use of nonbiologic and biologic disease-modifying antirheumatic drugs in rheumatoid arthritis. Arthritis Rheum 2008; 59:762–784.
- Cornish J, Tan E, Teare J, et al. A meta-analysis on the influence of inflammatory bowel disease on pregnancy. Gut 2007; 56:830–837.
- Dominitz JA, Young JC, Boyko EJ. Outcomes of infants born to mothers with inflammatory bowel disease: a population-based cohort study. Am J Gastroenterol 2002; 97:641–648.
- Kornfeld D, Cnattingius S, Ekbom A. Pregnancy outcomes in women with inflammatory bowel disease—a population-based cohort study. Am J Obstet Gynecol 1997; 177:942–946.
- Mahadevan U, Sandborn WJ, Li DK, Hakimian S, Kane S, Corley DA. Pregnancy outcomes in women with inflammatory bowel disease: a large community-based study from Northern California. Gastroenterology 2007; 133:1106–1112.
- Nguyen GC, Boudreau H, Harris ML, Maxwell CV. Outcomes of obstetric hospitalizations among women with inflammatory bowel disease in the United States. Clin Gastroenterol Hepatol 2009; 7:329–334.
- Schnitzler F, Fidder H, Ferrante M, et al. Outcome of pregnancy in women with inflammatory bowel disease treated with antitumor necrosis factor therapy. Inflamm Bowel Dis 2011; 17:1846–1854.
- Vinet E, Pineau C, Gordon C, Clarke AE, Bernatsky S. Biologic therapy and pregnancy outcomes in women with rheumatic diseases. Arthritis Rheum 2009; 61:587–592.
- Guidi L, Pugliese D, Armuzzi A. Update on the management of inflammatory bowel disease: specific role of adalimumab. Clin Exp Gastroenterol 2011; 4:163–172.
- Mahadevan U. Continuing immunomodulators and biologic medications in pregnant IBD patients – pro. IInflamm Bowel Dis 2007; 13:1439–1440.
- Mahadevan U. Gastrointestinal medications in pregnancy. Best Pract Res Clin Gastroenterol 2007; 21:849–877.
- Mahadevan U, Cucchiara S, Hyams JS, et al. The London Position Statement of the World Congress of Gastroenterology on Biological Therapy for IBD with the European Crohn’s and Colitis Organisation: pregnancy and pediatrics. Am J Gastroenterol 2011; 106:214–223.
- Mahadevan U, Kane S. Use of infliximab in pregnancy. Am J Gastroenterol 2010; 105:219–220.
- Vasiliauskas EA, Church JA, Silverman N, Barry M, Targan SR, Dubinsky MC. Case report: evidence for transplacental transfer of maternally administered infliximab to the newborn. Clin Gastroenterol Hepatol 2006; 4:1255–1258.
KEY POINTS
- Over the past 10 years, TNF inhibitors have substantially altered the management of autoimmune diseases such as rheumatoid arthritis and inflammatory bowel disease.
- Safety concerns include risks of infection, reactivation of latent infection (eg, fungal infection, granulomatous infection), malignancy, and autoimmune and neurologic effects.
- Before treating, take a complete history, including exposure to latent infections and geographic considerations, and bring patients’ immunizations up to date.
- Regular clinical and laboratory monitoring during treatment helps optimize therapy and minimize the risk of adverse effects.
- Physicians must be aware of atypical presentations of infection and understand how their treatment may differ in patients on biologic therapy.
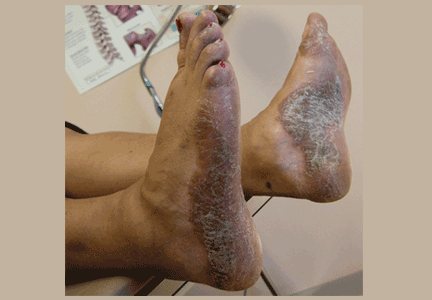
Do all hospitalized patients need stress ulcer prophylaxis?
No. Based on current evidence and guidelines, routine acid-suppressive therapy to prevent stress ulcers has no benefit in hospitalized patients outside the critical-care setting. Only critically ill patients who meet specific criteria, as described in the guidelines of the American Society of Health System Pharmacists, should receive acid-suppressive therapy.
Unfortunately, routine stress ulcer prophylaxis is common in US hospitals, unnecessarily putting patients at risk of complications and adding costs.
STRESS ULCER AND CRITICAL ILLNESS
Stress ulcers—ulcerations of the upper part of the gastrointestinal (GI) mucosa in the setting of acute disease—usually involve the fundus and body of the stomach. The stomach is lined with a glycoprotein mucous layer rich in bicarbonates, forming a physiologic barrier to protect the gastric wall from acid insult by neutralizing hydrogen ions. Disruption of this protective layer can occur in critically ill patients (eg, those with shock or sepsis) through overproduction of uremic toxins, increased reflux of bile salts, compromised blood flow, and increased stomach acidity through gastrin stimulation of parietal cells.
More than 75% of patients with major burns or cranial trauma develop endoscopic mucosal abnormalities within 72 hours of injury.1 In critically ill patients, the risk of ulcer-related overt bleeding is estimated to be 5% to 25%. Furthermore, 1% to 5% of stress ulcers can be deep enough to erode into the submucosa, causing clinically significant GI bleeding, defined as bleeding complicated by hemodynamic compromise or a drop in hemoglobin that requires a blood transfusion.2 In contrast, in inpatients who are not critically ill, the risk of overt bleeding from stress ulcers is less than 1%.3
ADDRESSING RISK
A multicenter prospective cohort study of 2,252 intensive care patients2 reported two main risk factors for significant bleeding caused by stress ulcers: mechanical ventilation for more than 48 hours and coagulopathy, defined as a platelet count below 50 × 109/L, an international normalized ratio greater than 1.5, or a partial thromboplastin time more than twice the control value.4 In hemodynamically stable patients receiving anticoagulation in a general medical or surgical ward, the risk of GI bleeding was low, and acid suppression failed to lower the rate of stress ulcer occurrence.3
Other risk factors include severe sepsis, shock, liver failure, kidney failure, burns over 35% of the total body surface, organ transplantation, cranial trauma, spinal cord trauma, history of peptic ulcer disease, and history of upper GI bleeding.3,5,6 Steroid therapy is not considered a risk factor for stress ulcers unless it is used in the presence of another risk factor such as use of aspirin or nonsteroidal antiinflammatory drugs (NSAIDs).2
INDICATIONS FOR PROPHYLAXIS
Prophylaxis with a proton pump inhibitor (PPI) is indicated in specific conditions—ie, peptic ulcer disease, gastroesophageal reflux disease, chronic NSAID therapy, and Zollinger-Ellison syndrome—and to eradicate Helicobacter pylori infection.7 But in the United States, stress ulcer prophylaxis is overused in general-care floors despite the lack of supporting evidence.
The American Society of Health System Pharmacists guidelines recommend it in the intensive care unit for patients with any of the following: coagulopathy, prolonged mechanical ventilation (more than 48 hours), GI ulcer or bleeding within the past year, sepsis, a stay longer than 1 week in the intensive care unit, occult GI bleeding for 6 or more days, and steroid therapy with more than 250 mg of hydrocortisone daily.8 Hemodynamically stable patients admitted to general-care floors should not receive stress ulcer prophylaxis, as it only negligibly decreases the rate of GI bleeding, from 0.33% to 0.22%.9
WHY ROUTINE ULCER PROPHYLAXIS IS NOT FOR ALL HOSPITALIZED PATIENTS
Although stress ulcer prophylaxis is often considered benign, its lack of proven benefit, additional cost, and risk of adverse effects, including interactions with foods and other drugs, preclude using it routinely for all hospitalized patients.10,11 Chronic use of PPIs has been associated with complications, as discussed below.
Infection
Acid suppression may impair the destruction of ingested microorganisms, resulting in overgrowth of bacteria.12 Overuse of PPIs may increase the risk of several infections:
- Diarrhea due to Clostridium difficile12
- Community-acquired pneumonia, from increased microaspiration of overgrown microorganisms into the lung.12
- Spontaneous bacterial peritonitis in patients with cirrhosis,13 although the mechanism is not clear. (Small-bowel bacterial overgrowth is the hypothesized cause.)
Bone fracture
PPIs lower gastric acidity, and this can inhibit intestinal calcium absorption. Furthermore, PPIs may directly inhibit bone resorption by osteoclasts.14
Reduction in clopidogrel efficacy
PPIs may reduce the efficacy of clopidogrel as a result of competitive inhibition of cytochrome CYP2C19, which is necessary to metabolize clopidogrel to its active forms. Therefore, concomitant use of clopidogrel with omeprazole, esomeprazole, or other CYP2C19 inhibitors is not recommended.15
Nutritional deficiencies
The overgrown microorganisms consume cobalamin in the stomach, resulting in vitamin B12 deficiency. Acid-suppressive therapy can also reduce the absorption of magnesium and iron.12
Unnecessary cost
Heidelbaugh and Inadomi16 reviewed the non-evidence-based use of stress ulcer prophylaxis in patients admitted to a large university hospital and estimated that it entailed a cost to the hospital of $111,791 over the course of a year.
WHICH ULCER PROPHYLAXIS SHOULD BE USED IN CRITICALLY ILL PATIENTS?
Studies have shown histamine-2 blockers to be superior to antacids and sucralfate in preventing stress ulcer and GI bleeding,8,15 but no study has compared PPIs with sucralfate and antacids.
When indicated, an oral PPI is preferred over an oral histamine-2 blocker for GI prophylaxis.17 This practice is considered cost-effective and is associated with lower rates of stress ulcer and GI bleeding. In intubated patients, however, an intravenous histamine-2 blocker is preferable to an intravenous PPI.3,8,11 Interestingly, no difference was reported between PPIs and histamine-2 blockers in terms of mortality rate or reduction in the incidence of nosocomial pneumonia.17
OUR RECOMMENDATION
Only critically ill patients who meet the specific criteria described here should receive stress ulcer prophylaxis. More effort is needed to educate residents, medical staff, and pharmacists about current guidelines. Computerized ordering templates and reminders to discontinue prophylaxis at discharge or step-down may decrease overall use, reduce costs, and limit potential side effects.18
- DePriest JL. Stress ulcer prophylaxis. Do critically ill patients need it? Postgrad Med 1995; 98:159–168.
- Cook DJ, Fuller HD, Guyatt GH, et al. Risk factors for gastrointestinal bleeding in critically ill patients. Canadian Critical Care Trials Group. N Engl J Med 1994; 330:377–381.
- Qadeer MA, Richter JE, Brotman DJ. Hospital-acquired gastrointestinal bleeding outside the critical care unit: risk factors, role of acid suppression, and endoscopy findings. J Hosp Med 2006; 1:13–20.
- Shuman RB, Schuster DP, Zuckerman GR. Prophylactic therapy for stress ulcer bleeding: a reappraisal. Ann Intern Med 1987; 106:562–567.
- Dellinger RP, Levy MM, Rhodes A, et al; Surviving Sepsis Campaign Guidelines Committee including the Pediatric Subgroup. Surviving sepsis campaign: international guidelines for management of severe sepsis and septic shock: 2012. Crit Care Med 2013; 41:580–637.
- Cook DJ, Reeve BK, Guyatt GH, et al. Stress ulcer prophylaxis in critically ill patients. Resolving discordant meta-analyses. JAMA 1996; 275:308–314.
- Kahrilas PJ, Shaheen NJ, Vaezi MF, et al; American Gastroenterological Association. American Gastroenterological Association Medical Position Statement on the management of gastroesophageal reflux disease. Gastroenterology 2008; 135:1383–1391.e1–1391.e5.
- Barkun AN, Bardou M, Pham CQ, Martel M. Proton pump inhibitors vs histamine 2 receptor antagonists for stress-related mucosal bleeding prophylaxis in critically ill patients: a meta-analysis. Am J Gastroenterol 2012; 107:507–520.
- Herzig SJ, Vaughn BP, Howell MD, Ngo LH, Marcantonio ER. Acid-suppressive medication use and the risk for nosocomial gastrointestinal tract bleeding. Arch Intern Med 2011; 171:991–997.
- Cook DJ. Stress ulcer prophylaxis: gastrointestinal bleeding and nosocomial pneumonia. Best evidence synthesis. Scand J Gastroenterol Suppl 1995; 210:48–52.
- Messori A, Trippoli S, Vaiani M, Gorini M, Corrado A. Bleeding and pneumonia in intensive care patients given ranitidine and sucralfate for prevention of stress ulcer: meta-analysis of randomised controlled trials. BMJ 2000; 321:1103–1106.
- Heidelbaugh JJ, Kim AH, Chang R, Walker PC. Overutilization of proton-pump inhibitors: what the clinician needs to know. Therap Adv Gastroenterol 2012; 5:219–232.
- Deshpande A, Pasupuleti V, Thota P, et al. Acid-suppressive therapy is associated with spontaneous bacterial peritonitis in cirrhotic patients: a meta-analysis. J Gastroenterol Hepatol 2013; 28:235–242.
- Farina C, Gagliardi S. Selective inhibition of osteoclast vacuolar H(+)- ATPase. Curr Pharm Des 2002; 8:2033–2048.
- ASHP Therapeutic Guidelines on Stress Ulcer Prophylaxis. ASHP Commission on Therapeutics and approved by the ASHP Board of Directors on November 14, 1998. Am J Health Syst Pharm 1999; 56:347–379.
- Heidelbaugh JJ, Inadomi JM. Magnitude and economic impact of inappropriate use of stress ulcer prophylaxis in non-ICU hospitalized patients. Am J Gastroenterol 2006; 101:2200–2205.
- Alhazzani W, Alenezi F, Jaeschke RZ, Moayyedi P, Cook DJ. Proton pump inhibitors versus histamine 2 receptor antagonists for stress ulcer prophylaxis in critically ill patients: a systematic review and meta-analysis. Crit Care Med 2013; 41:693–705.
- Liberman JD, Whelan CT. Brief report: reducing inappropriate usage of stress ulcer prophylaxis among internal medicine residents. A practice-based educational intervention. J Gen Intern Med 2006; 21:498–500.
No. Based on current evidence and guidelines, routine acid-suppressive therapy to prevent stress ulcers has no benefit in hospitalized patients outside the critical-care setting. Only critically ill patients who meet specific criteria, as described in the guidelines of the American Society of Health System Pharmacists, should receive acid-suppressive therapy.
Unfortunately, routine stress ulcer prophylaxis is common in US hospitals, unnecessarily putting patients at risk of complications and adding costs.
STRESS ULCER AND CRITICAL ILLNESS
Stress ulcers—ulcerations of the upper part of the gastrointestinal (GI) mucosa in the setting of acute disease—usually involve the fundus and body of the stomach. The stomach is lined with a glycoprotein mucous layer rich in bicarbonates, forming a physiologic barrier to protect the gastric wall from acid insult by neutralizing hydrogen ions. Disruption of this protective layer can occur in critically ill patients (eg, those with shock or sepsis) through overproduction of uremic toxins, increased reflux of bile salts, compromised blood flow, and increased stomach acidity through gastrin stimulation of parietal cells.
More than 75% of patients with major burns or cranial trauma develop endoscopic mucosal abnormalities within 72 hours of injury.1 In critically ill patients, the risk of ulcer-related overt bleeding is estimated to be 5% to 25%. Furthermore, 1% to 5% of stress ulcers can be deep enough to erode into the submucosa, causing clinically significant GI bleeding, defined as bleeding complicated by hemodynamic compromise or a drop in hemoglobin that requires a blood transfusion.2 In contrast, in inpatients who are not critically ill, the risk of overt bleeding from stress ulcers is less than 1%.3
ADDRESSING RISK
A multicenter prospective cohort study of 2,252 intensive care patients2 reported two main risk factors for significant bleeding caused by stress ulcers: mechanical ventilation for more than 48 hours and coagulopathy, defined as a platelet count below 50 × 109/L, an international normalized ratio greater than 1.5, or a partial thromboplastin time more than twice the control value.4 In hemodynamically stable patients receiving anticoagulation in a general medical or surgical ward, the risk of GI bleeding was low, and acid suppression failed to lower the rate of stress ulcer occurrence.3
Other risk factors include severe sepsis, shock, liver failure, kidney failure, burns over 35% of the total body surface, organ transplantation, cranial trauma, spinal cord trauma, history of peptic ulcer disease, and history of upper GI bleeding.3,5,6 Steroid therapy is not considered a risk factor for stress ulcers unless it is used in the presence of another risk factor such as use of aspirin or nonsteroidal antiinflammatory drugs (NSAIDs).2
INDICATIONS FOR PROPHYLAXIS
Prophylaxis with a proton pump inhibitor (PPI) is indicated in specific conditions—ie, peptic ulcer disease, gastroesophageal reflux disease, chronic NSAID therapy, and Zollinger-Ellison syndrome—and to eradicate Helicobacter pylori infection.7 But in the United States, stress ulcer prophylaxis is overused in general-care floors despite the lack of supporting evidence.
The American Society of Health System Pharmacists guidelines recommend it in the intensive care unit for patients with any of the following: coagulopathy, prolonged mechanical ventilation (more than 48 hours), GI ulcer or bleeding within the past year, sepsis, a stay longer than 1 week in the intensive care unit, occult GI bleeding for 6 or more days, and steroid therapy with more than 250 mg of hydrocortisone daily.8 Hemodynamically stable patients admitted to general-care floors should not receive stress ulcer prophylaxis, as it only negligibly decreases the rate of GI bleeding, from 0.33% to 0.22%.9
WHY ROUTINE ULCER PROPHYLAXIS IS NOT FOR ALL HOSPITALIZED PATIENTS
Although stress ulcer prophylaxis is often considered benign, its lack of proven benefit, additional cost, and risk of adverse effects, including interactions with foods and other drugs, preclude using it routinely for all hospitalized patients.10,11 Chronic use of PPIs has been associated with complications, as discussed below.
Infection
Acid suppression may impair the destruction of ingested microorganisms, resulting in overgrowth of bacteria.12 Overuse of PPIs may increase the risk of several infections:
- Diarrhea due to Clostridium difficile12
- Community-acquired pneumonia, from increased microaspiration of overgrown microorganisms into the lung.12
- Spontaneous bacterial peritonitis in patients with cirrhosis,13 although the mechanism is not clear. (Small-bowel bacterial overgrowth is the hypothesized cause.)
Bone fracture
PPIs lower gastric acidity, and this can inhibit intestinal calcium absorption. Furthermore, PPIs may directly inhibit bone resorption by osteoclasts.14
Reduction in clopidogrel efficacy
PPIs may reduce the efficacy of clopidogrel as a result of competitive inhibition of cytochrome CYP2C19, which is necessary to metabolize clopidogrel to its active forms. Therefore, concomitant use of clopidogrel with omeprazole, esomeprazole, or other CYP2C19 inhibitors is not recommended.15
Nutritional deficiencies
The overgrown microorganisms consume cobalamin in the stomach, resulting in vitamin B12 deficiency. Acid-suppressive therapy can also reduce the absorption of magnesium and iron.12
Unnecessary cost
Heidelbaugh and Inadomi16 reviewed the non-evidence-based use of stress ulcer prophylaxis in patients admitted to a large university hospital and estimated that it entailed a cost to the hospital of $111,791 over the course of a year.
WHICH ULCER PROPHYLAXIS SHOULD BE USED IN CRITICALLY ILL PATIENTS?
Studies have shown histamine-2 blockers to be superior to antacids and sucralfate in preventing stress ulcer and GI bleeding,8,15 but no study has compared PPIs with sucralfate and antacids.
When indicated, an oral PPI is preferred over an oral histamine-2 blocker for GI prophylaxis.17 This practice is considered cost-effective and is associated with lower rates of stress ulcer and GI bleeding. In intubated patients, however, an intravenous histamine-2 blocker is preferable to an intravenous PPI.3,8,11 Interestingly, no difference was reported between PPIs and histamine-2 blockers in terms of mortality rate or reduction in the incidence of nosocomial pneumonia.17
OUR RECOMMENDATION
Only critically ill patients who meet the specific criteria described here should receive stress ulcer prophylaxis. More effort is needed to educate residents, medical staff, and pharmacists about current guidelines. Computerized ordering templates and reminders to discontinue prophylaxis at discharge or step-down may decrease overall use, reduce costs, and limit potential side effects.18
No. Based on current evidence and guidelines, routine acid-suppressive therapy to prevent stress ulcers has no benefit in hospitalized patients outside the critical-care setting. Only critically ill patients who meet specific criteria, as described in the guidelines of the American Society of Health System Pharmacists, should receive acid-suppressive therapy.
Unfortunately, routine stress ulcer prophylaxis is common in US hospitals, unnecessarily putting patients at risk of complications and adding costs.
STRESS ULCER AND CRITICAL ILLNESS
Stress ulcers—ulcerations of the upper part of the gastrointestinal (GI) mucosa in the setting of acute disease—usually involve the fundus and body of the stomach. The stomach is lined with a glycoprotein mucous layer rich in bicarbonates, forming a physiologic barrier to protect the gastric wall from acid insult by neutralizing hydrogen ions. Disruption of this protective layer can occur in critically ill patients (eg, those with shock or sepsis) through overproduction of uremic toxins, increased reflux of bile salts, compromised blood flow, and increased stomach acidity through gastrin stimulation of parietal cells.
More than 75% of patients with major burns or cranial trauma develop endoscopic mucosal abnormalities within 72 hours of injury.1 In critically ill patients, the risk of ulcer-related overt bleeding is estimated to be 5% to 25%. Furthermore, 1% to 5% of stress ulcers can be deep enough to erode into the submucosa, causing clinically significant GI bleeding, defined as bleeding complicated by hemodynamic compromise or a drop in hemoglobin that requires a blood transfusion.2 In contrast, in inpatients who are not critically ill, the risk of overt bleeding from stress ulcers is less than 1%.3
ADDRESSING RISK
A multicenter prospective cohort study of 2,252 intensive care patients2 reported two main risk factors for significant bleeding caused by stress ulcers: mechanical ventilation for more than 48 hours and coagulopathy, defined as a platelet count below 50 × 109/L, an international normalized ratio greater than 1.5, or a partial thromboplastin time more than twice the control value.4 In hemodynamically stable patients receiving anticoagulation in a general medical or surgical ward, the risk of GI bleeding was low, and acid suppression failed to lower the rate of stress ulcer occurrence.3
Other risk factors include severe sepsis, shock, liver failure, kidney failure, burns over 35% of the total body surface, organ transplantation, cranial trauma, spinal cord trauma, history of peptic ulcer disease, and history of upper GI bleeding.3,5,6 Steroid therapy is not considered a risk factor for stress ulcers unless it is used in the presence of another risk factor such as use of aspirin or nonsteroidal antiinflammatory drugs (NSAIDs).2
INDICATIONS FOR PROPHYLAXIS
Prophylaxis with a proton pump inhibitor (PPI) is indicated in specific conditions—ie, peptic ulcer disease, gastroesophageal reflux disease, chronic NSAID therapy, and Zollinger-Ellison syndrome—and to eradicate Helicobacter pylori infection.7 But in the United States, stress ulcer prophylaxis is overused in general-care floors despite the lack of supporting evidence.
The American Society of Health System Pharmacists guidelines recommend it in the intensive care unit for patients with any of the following: coagulopathy, prolonged mechanical ventilation (more than 48 hours), GI ulcer or bleeding within the past year, sepsis, a stay longer than 1 week in the intensive care unit, occult GI bleeding for 6 or more days, and steroid therapy with more than 250 mg of hydrocortisone daily.8 Hemodynamically stable patients admitted to general-care floors should not receive stress ulcer prophylaxis, as it only negligibly decreases the rate of GI bleeding, from 0.33% to 0.22%.9
WHY ROUTINE ULCER PROPHYLAXIS IS NOT FOR ALL HOSPITALIZED PATIENTS
Although stress ulcer prophylaxis is often considered benign, its lack of proven benefit, additional cost, and risk of adverse effects, including interactions with foods and other drugs, preclude using it routinely for all hospitalized patients.10,11 Chronic use of PPIs has been associated with complications, as discussed below.
Infection
Acid suppression may impair the destruction of ingested microorganisms, resulting in overgrowth of bacteria.12 Overuse of PPIs may increase the risk of several infections:
- Diarrhea due to Clostridium difficile12
- Community-acquired pneumonia, from increased microaspiration of overgrown microorganisms into the lung.12
- Spontaneous bacterial peritonitis in patients with cirrhosis,13 although the mechanism is not clear. (Small-bowel bacterial overgrowth is the hypothesized cause.)
Bone fracture
PPIs lower gastric acidity, and this can inhibit intestinal calcium absorption. Furthermore, PPIs may directly inhibit bone resorption by osteoclasts.14
Reduction in clopidogrel efficacy
PPIs may reduce the efficacy of clopidogrel as a result of competitive inhibition of cytochrome CYP2C19, which is necessary to metabolize clopidogrel to its active forms. Therefore, concomitant use of clopidogrel with omeprazole, esomeprazole, or other CYP2C19 inhibitors is not recommended.15
Nutritional deficiencies
The overgrown microorganisms consume cobalamin in the stomach, resulting in vitamin B12 deficiency. Acid-suppressive therapy can also reduce the absorption of magnesium and iron.12
Unnecessary cost
Heidelbaugh and Inadomi16 reviewed the non-evidence-based use of stress ulcer prophylaxis in patients admitted to a large university hospital and estimated that it entailed a cost to the hospital of $111,791 over the course of a year.
WHICH ULCER PROPHYLAXIS SHOULD BE USED IN CRITICALLY ILL PATIENTS?
Studies have shown histamine-2 blockers to be superior to antacids and sucralfate in preventing stress ulcer and GI bleeding,8,15 but no study has compared PPIs with sucralfate and antacids.
When indicated, an oral PPI is preferred over an oral histamine-2 blocker for GI prophylaxis.17 This practice is considered cost-effective and is associated with lower rates of stress ulcer and GI bleeding. In intubated patients, however, an intravenous histamine-2 blocker is preferable to an intravenous PPI.3,8,11 Interestingly, no difference was reported between PPIs and histamine-2 blockers in terms of mortality rate or reduction in the incidence of nosocomial pneumonia.17
OUR RECOMMENDATION
Only critically ill patients who meet the specific criteria described here should receive stress ulcer prophylaxis. More effort is needed to educate residents, medical staff, and pharmacists about current guidelines. Computerized ordering templates and reminders to discontinue prophylaxis at discharge or step-down may decrease overall use, reduce costs, and limit potential side effects.18
- DePriest JL. Stress ulcer prophylaxis. Do critically ill patients need it? Postgrad Med 1995; 98:159–168.
- Cook DJ, Fuller HD, Guyatt GH, et al. Risk factors for gastrointestinal bleeding in critically ill patients. Canadian Critical Care Trials Group. N Engl J Med 1994; 330:377–381.
- Qadeer MA, Richter JE, Brotman DJ. Hospital-acquired gastrointestinal bleeding outside the critical care unit: risk factors, role of acid suppression, and endoscopy findings. J Hosp Med 2006; 1:13–20.
- Shuman RB, Schuster DP, Zuckerman GR. Prophylactic therapy for stress ulcer bleeding: a reappraisal. Ann Intern Med 1987; 106:562–567.
- Dellinger RP, Levy MM, Rhodes A, et al; Surviving Sepsis Campaign Guidelines Committee including the Pediatric Subgroup. Surviving sepsis campaign: international guidelines for management of severe sepsis and septic shock: 2012. Crit Care Med 2013; 41:580–637.
- Cook DJ, Reeve BK, Guyatt GH, et al. Stress ulcer prophylaxis in critically ill patients. Resolving discordant meta-analyses. JAMA 1996; 275:308–314.
- Kahrilas PJ, Shaheen NJ, Vaezi MF, et al; American Gastroenterological Association. American Gastroenterological Association Medical Position Statement on the management of gastroesophageal reflux disease. Gastroenterology 2008; 135:1383–1391.e1–1391.e5.
- Barkun AN, Bardou M, Pham CQ, Martel M. Proton pump inhibitors vs histamine 2 receptor antagonists for stress-related mucosal bleeding prophylaxis in critically ill patients: a meta-analysis. Am J Gastroenterol 2012; 107:507–520.
- Herzig SJ, Vaughn BP, Howell MD, Ngo LH, Marcantonio ER. Acid-suppressive medication use and the risk for nosocomial gastrointestinal tract bleeding. Arch Intern Med 2011; 171:991–997.
- Cook DJ. Stress ulcer prophylaxis: gastrointestinal bleeding and nosocomial pneumonia. Best evidence synthesis. Scand J Gastroenterol Suppl 1995; 210:48–52.
- Messori A, Trippoli S, Vaiani M, Gorini M, Corrado A. Bleeding and pneumonia in intensive care patients given ranitidine and sucralfate for prevention of stress ulcer: meta-analysis of randomised controlled trials. BMJ 2000; 321:1103–1106.
- Heidelbaugh JJ, Kim AH, Chang R, Walker PC. Overutilization of proton-pump inhibitors: what the clinician needs to know. Therap Adv Gastroenterol 2012; 5:219–232.
- Deshpande A, Pasupuleti V, Thota P, et al. Acid-suppressive therapy is associated with spontaneous bacterial peritonitis in cirrhotic patients: a meta-analysis. J Gastroenterol Hepatol 2013; 28:235–242.
- Farina C, Gagliardi S. Selective inhibition of osteoclast vacuolar H(+)- ATPase. Curr Pharm Des 2002; 8:2033–2048.
- ASHP Therapeutic Guidelines on Stress Ulcer Prophylaxis. ASHP Commission on Therapeutics and approved by the ASHP Board of Directors on November 14, 1998. Am J Health Syst Pharm 1999; 56:347–379.
- Heidelbaugh JJ, Inadomi JM. Magnitude and economic impact of inappropriate use of stress ulcer prophylaxis in non-ICU hospitalized patients. Am J Gastroenterol 2006; 101:2200–2205.
- Alhazzani W, Alenezi F, Jaeschke RZ, Moayyedi P, Cook DJ. Proton pump inhibitors versus histamine 2 receptor antagonists for stress ulcer prophylaxis in critically ill patients: a systematic review and meta-analysis. Crit Care Med 2013; 41:693–705.
- Liberman JD, Whelan CT. Brief report: reducing inappropriate usage of stress ulcer prophylaxis among internal medicine residents. A practice-based educational intervention. J Gen Intern Med 2006; 21:498–500.
- DePriest JL. Stress ulcer prophylaxis. Do critically ill patients need it? Postgrad Med 1995; 98:159–168.
- Cook DJ, Fuller HD, Guyatt GH, et al. Risk factors for gastrointestinal bleeding in critically ill patients. Canadian Critical Care Trials Group. N Engl J Med 1994; 330:377–381.
- Qadeer MA, Richter JE, Brotman DJ. Hospital-acquired gastrointestinal bleeding outside the critical care unit: risk factors, role of acid suppression, and endoscopy findings. J Hosp Med 2006; 1:13–20.
- Shuman RB, Schuster DP, Zuckerman GR. Prophylactic therapy for stress ulcer bleeding: a reappraisal. Ann Intern Med 1987; 106:562–567.
- Dellinger RP, Levy MM, Rhodes A, et al; Surviving Sepsis Campaign Guidelines Committee including the Pediatric Subgroup. Surviving sepsis campaign: international guidelines for management of severe sepsis and septic shock: 2012. Crit Care Med 2013; 41:580–637.
- Cook DJ, Reeve BK, Guyatt GH, et al. Stress ulcer prophylaxis in critically ill patients. Resolving discordant meta-analyses. JAMA 1996; 275:308–314.
- Kahrilas PJ, Shaheen NJ, Vaezi MF, et al; American Gastroenterological Association. American Gastroenterological Association Medical Position Statement on the management of gastroesophageal reflux disease. Gastroenterology 2008; 135:1383–1391.e1–1391.e5.
- Barkun AN, Bardou M, Pham CQ, Martel M. Proton pump inhibitors vs histamine 2 receptor antagonists for stress-related mucosal bleeding prophylaxis in critically ill patients: a meta-analysis. Am J Gastroenterol 2012; 107:507–520.
- Herzig SJ, Vaughn BP, Howell MD, Ngo LH, Marcantonio ER. Acid-suppressive medication use and the risk for nosocomial gastrointestinal tract bleeding. Arch Intern Med 2011; 171:991–997.
- Cook DJ. Stress ulcer prophylaxis: gastrointestinal bleeding and nosocomial pneumonia. Best evidence synthesis. Scand J Gastroenterol Suppl 1995; 210:48–52.
- Messori A, Trippoli S, Vaiani M, Gorini M, Corrado A. Bleeding and pneumonia in intensive care patients given ranitidine and sucralfate for prevention of stress ulcer: meta-analysis of randomised controlled trials. BMJ 2000; 321:1103–1106.
- Heidelbaugh JJ, Kim AH, Chang R, Walker PC. Overutilization of proton-pump inhibitors: what the clinician needs to know. Therap Adv Gastroenterol 2012; 5:219–232.
- Deshpande A, Pasupuleti V, Thota P, et al. Acid-suppressive therapy is associated with spontaneous bacterial peritonitis in cirrhotic patients: a meta-analysis. J Gastroenterol Hepatol 2013; 28:235–242.
- Farina C, Gagliardi S. Selective inhibition of osteoclast vacuolar H(+)- ATPase. Curr Pharm Des 2002; 8:2033–2048.
- ASHP Therapeutic Guidelines on Stress Ulcer Prophylaxis. ASHP Commission on Therapeutics and approved by the ASHP Board of Directors on November 14, 1998. Am J Health Syst Pharm 1999; 56:347–379.
- Heidelbaugh JJ, Inadomi JM. Magnitude and economic impact of inappropriate use of stress ulcer prophylaxis in non-ICU hospitalized patients. Am J Gastroenterol 2006; 101:2200–2205.
- Alhazzani W, Alenezi F, Jaeschke RZ, Moayyedi P, Cook DJ. Proton pump inhibitors versus histamine 2 receptor antagonists for stress ulcer prophylaxis in critically ill patients: a systematic review and meta-analysis. Crit Care Med 2013; 41:693–705.
- Liberman JD, Whelan CT. Brief report: reducing inappropriate usage of stress ulcer prophylaxis among internal medicine residents. A practice-based educational intervention. J Gen Intern Med 2006; 21:498–500.
Antiplatelet therapy to prevent recurrent stroke: Three good options
After a stroke, an important goal is to prevent another one.1,2 And for patients who have had an ischemic stroke or transient ischemic attack (TIA) due to atherosclerosis, an important part of secondary preventive therapy is a drug that inhibits platelets—ie, aspirin, extended-release dipyridamole, or clopidogrel. This has taken years to establish.
In the following pages, we discuss the antiplatelet agents that have been shown to be beneficial after stroke of atherosclerotic origin, and we briefly review the indications for surgery and stenting for the subset of patients whose strokes are caused by symptomatic carotid disease.
(Although managing modifiable risk factors such as smoking, hypertension, diabetes, and dyslipidemia is also important, we will not cover this topic here, nor will we talk about hemorrhagic stroke or stroke due to atrial fibrillation. Also not discussed here is cilostazol, which, although shown to be effective in preventing recurrent stroke when compared with placebo and aspirin,3,4 has not been approved for this use by the US Food and Drug Administration, as of this writing.)
HOW WE REVIEWED THE LITERATURE
We searched PubMed using the terms aspirin, acetylsalicylic acid, clopidogrel, and/or dipyridamole, in combination with stroke, cerebral ische(ae)mia, transient ische(ae)mic attacks, or retinal artery occlusion. We reviewed only clinical trials or meta-analyses of these drugs for either primary or secondary prevention of cerebrovascular disease.
As our aim was to review the topic and not to perform a meta-analysis, no cutoffs were used to exclude trials. The references in the selected papers were also reviewed to expand the articles. Finally, the references in the current American Heart Association and American Stroke Association secondary stroke prevention guideline were also reviewed.
For a summary of the trials included in our review, see the Data Supplement as an appendix to the online version of this article.
ASPIRIN: THE GOLD STANDARD
Prescribed by Hippocrates in the form of willow bark extract, aspirin has long been known for its antipyretic and anti-inflammatory properties. Its antiplatelet and antithrombotic properties, first described in 1967 by Weiss and Aledort,5 are mediated by irreversible inhibition of cyclooxygenase, leading to decreased thromboxane A2, a platelet-aggregation activator.
Fields et al,6,7 in 1977 and 1978, reported that in a controlled trial in patients with TIA or monocular blindness, fewer subsequent TIAs occurred in patients who received aspirin, although the difference was not statistically significant, with lower rates of events only in nonsurgical patients. Over the next 20 years, the results remained mixed.
The Danish Cooperative study8 (1983) found no significant difference in the rate of recurrent stroke with aspirin vs placebo.
AICLA.9 The Accidents Ischémiques Cérébraux Liés à l’Athérosclérose study of 1983 did find a difference. However, both the Danish Cooperative study and the AICLA were limited by lacking standardized computed tomographic imaging to rule out hemorrhagic stroke and by being relatively small.
The Swedish Cooperative Study10 (1987) found no statistical difference between high-dose aspirin and placebo in preventing recurrent vascular events (stroke, TIA, or myocardial infarction [MI]) 1 to 3 weeks after a stroke. However, it had several limitations: the aspirin group contained more patients with ischemic heart disease (who are more likely to die of cardiac causes), there were significantly more men in the aspirin group, and nearly one-fourth of the deaths were a result of the initial stroke, potentially masking the effect of aspirin in secondary prevention.
Later studies began to show a consistently favorable effect of aspirin.
Boysen et al11 in 1988 reported a nonsignificant trend toward fewer adverse events with aspirin.
UK-TIA.12 The United Kingdom Transient Ischaemic Attack trial in 1991 found a similar trend.
SALT.13 The Swedish Aspirin Low-dose Trial, also in 1991, showed a significant 18% lower rate of stroke or death in patients with recent TIA, minor stroke, or retinal occlusion treated with low-dose aspirin. The inclusion of patients with TIA helped broaden the population that might benefit. However, the study may have favored the aspirin group by having a run-in period in which patients were nonrandomly treated either with aspirin or with anticoagulation at the discretion of the patient’s physician and, if they suffered “several” TIAs, a stroke, retinal artery occlusion, or MI, were removed from the study.
ESPS-2.14 The second European Stroke Prevention Study in 1996 added to the evidence that aspirin prevents recurrent stroke. Patients with a history of TIA or stroke were randomized in double-blind fashion to four treatment groups: placebo, low-dose aspirin, dipyridamole, or aspirin plus dipyridamole. At 2 years, strokes had occurred in 18% fewer patients in the aspirin group than in the placebo group, and TIAs had occurred in 21.9% fewer. However, aspirin was associated with an absolute 0.5% increase in severe and fatal bleeding. The power of the study was limited because patients from one center were excluded because of “serious inconsistencies in patient case record forms and compliance assay determinations.” 14
Comment. The mixed results with aspirin in studies predating ESPS-2 were partly because the study populations were too small to show benefit.
ATT.15 The Antithrombotic Trialists’ Collaboration performed a meta-analysis that conclusively confirmed the benefit of aspirin after stroke or TIA. The investigators analyzed individual patient data pooled from randomized controlled trials published before 1997 that compared antiplatelet regimens (mostly aspirin) against placebo and against each other. The rates of vascular events were 10.7% with treatment vs 13.2% with placebo (P < .0001). Antiplatelet therapy was particularly effective in preventing ischemic stroke, with a 25% reduction in the rate of nonfatal stroke, and with an overall absolute benefit in stroke prevention across all high-risk patient groups. This translated to 25 fewer nonfatal strokes per 1,000 patients treated with antiplatelet therapy.
What is the optimal aspirin dose?
Studies of aspirin have used different daily doses—the earliest studies used large doses of 1,000 to 1,500 mg.6–10
Boysen et al11 in 1988 found a trend toward benefit (not statistically significant) with doses ranging from 50 mg to 100 mg.
In 1991, three separate studies found that higher doses of aspirin were no more effective than lower doses.
The UK-TIA trial12 compared aspirin 300 mg vs 1,200 mg and found a higher risk of gastrointestinal bleeding with the higher dose.
The SALT Collaborative Group13 found 75 mg to be effective.
The Dutch TIA trial16 compared 30 mg vs 283 mg; end point outcomes were similar but the rate of adverse events was higher with 283 mg.
ESPS-2 was able to show efficacy at a dose of only 50 mg.14
Taylor et al17 compared lower doses (81 or 325 mg) vs higher doses (650 or 1,300 mg) for patients undergoing carotid endarterectomy and found that the risk of adverse events was twice as high with the higher doses.
The ATT Collaboration15 found that efficacy was 40% lower with the highest dose of aspirin than with the lowest doses.
Algra and van Gijn18 performed a meta-analysis of all these studies and found no difference in risk reduction between low-dose and high-dose aspirin, with an overall relative risk reduction of 13% at any dose above 30 mg.
Campbell et al,19 in a 2007 review, found that doses greater than 300 mg conferred no benefit, and that rapid and maximum suppression of thromboxane A2 can be achieved by chewing or ingesting dissolved forms of aspirin 162 mg.
Conclusion. Aspirin doses higher than 81 mg (the US standard) confer no greater benefit and may even decrease the efficacy of aspirin. In an emergency, rapid suppression of thromboxane A2 can be achieved by chewing a minimum dose of 162 mg.
DIPYRIDAMOLE CAN BE ADDED TO ASPIRIN
In 1967, Weiss and Aledort5 found that aspirin’s antiplatelet effect could be blocked by adenosine diphosphate, which is released by activated platelet cells and is an essential part of thrombus formation. Adjacent platelets are then activated, leading to up-regulation of thromboxane A2 and glycoprotein IIb/IIIa receptors and resulting in a cascade of platelet activation and clot formation.20 Dipyridamole inhibits aggregation of platelets by inhibiting their ability to take up adenosine diphosphate.
Studies of dipyridamole
AICLA.9 Bousser et al9 randomized patients who suffered one or more cerebral or retinal infarctions to receive placebo, aspirin 1 g, or aspirin 1 g plus dipyridamole 225 mg. Aspirin was significantly better than placebo in preventing a recurrence of stroke. The event rate with aspirin plus dipyridamole was similar to the rate with aspirin alone, although on 2-by-2 analysis, the difference between placebo and aspirin plus dipyridamole did not reach statistical significance. However, the rate of carotid-origin stroke was 17% with aspirin alone and 6% with aspirin plus dipyridamole, a statistically significant difference.
Thus, this study confirmed the benefit of aspirin in preventing ischemic events but did not fully support the addition of dipyridamole, except in preventing stroke of carotid origin. The study had a number of limitations: the sample size was small, TIA was not included as an end point, computed tomography was not required for entry, and many patients were lost to follow-up, decreasing the statistical power of the trial.
The ESPS study21 was also a randomized controlled trial of aspirin plus dipyridamole vs placebo. But unlike AICLA, ESPS included patients with TIA.
ESPS found a 38.1% relative risk reduction in stroke with aspirin plus dipyridamole compared with placebo, and a 30.6% reduction in death from all causes. Interestingly, patients who had a TIA as the qualifying event had a lower end-point incidence and larger end-point reduction than those who had a stroke as the qualifying event. However, ESPS did not resolve the question of whether adding dipyridamole to aspirin affords any benefit over aspirin alone.
ESPS-214 hoped to answer this question. Patients were randomized to placebo, aspirin, dipyridamole, or aspirin plus dipyridamole. On 2 × 2 analysis, the dipyridamole group had a 16% lower rate of recurrent stroke than the placebo group, and patients on aspirin plus dipyridamole had a 37% lower rate. Aspirin plus dipyridamole yielded a 23.1% reduction compared with aspirin alone, and a 24.7% reduction compared with dipyridamole alone. Similar benefit was reported for the end point of TIA with combination therapy compared with either agent alone.
However, nearly 25% of patients had to withdraw because of side effects, particularly in the dipyridamole-alone and aspirin-dipyridamole groups, and, as mentioned above, verification of compliance in the aspirin group was an issue.14,22 Nevertheless, ESPS-2 clearly showed that aspirin plus dipyridamole was better than either drug alone in preventing recurrent stroke. It also showed the effectiveness of dipyridamole, which AICLA and ESPS could not do, because it had a larger study population, used a lower dose of aspirin, and perhaps because it used an extended-release form of dipyridamole.23
The ATT meta-analysis15 showed a clear benefit of antiplatelet therapy. However, much of this benefit was derived from aspirin therapy, with the addition of dipyridamole resulting in a nonsignificant 6% reduction of vascular events. Most of the patients on dipyridamole were from the ESPS-2 study. In effect, the ATT was a meta-analysis of aspirin, as aspirin studies dominated at that time.
A Cochrane review24 publsihed in 2003 attempted to rectify this by analyzing randomized controlled trials of dipyridamole vs placebo.24 Like the ATT meta-analysis, it did not bear out the benefits of dipyridamole: compared with placebo, there was no effect on the rate of vascular death, and only a minimal benefit in reduction of vascular events—and this latter point is only because of the inclusion of ESPS-2.
Directly comparing aspirin plus dipyridamole vs aspirin alone, the reviewers found no effect on the rate of vascular death, and with the exclusion of ESPS-2, no effect on vascular events.
The Cochrane review had the same limitation as the ATT meta-analysis, ie, dependence on a single trial (ESPS-2) to show benefit, and perhaps the fact that ESPS-2 was the only study that used an extended-release form of dipyridamole.
Leonardi-Bee et al25 performed a meta-analysis that overcame the limitation of ESPS-2 being the only study at the time with positive findings: they used pooled individual patient data from randomized trials and analyzed them en masse. Patients on aspirin plus dipyridamole had a 39% lower risk than with placebo and a 22% lower risk than with aspirin alone. Unlike the ATT and the Cochrane review, excluding ESPS-2 did not alter the statistically significant lower stroke rate with aspirin plus dipyridamole compared with controls. This meta-analysis helped to confirm ESPS-2’s finding of the additive effect of aspirin plus dipyridamole compared with aspirin and placebo control.
ESPRIT.26,27 The European/Australasian Stroke Prevention in Reversible Ischaemia Trial confirmed these findings. This randomized controlled trial compared aspirin plus dipyridamole against aspirin alone in patients with a TIA or minor ischemic stroke of arterial origin within the past 6 months. For the primary end point (death from all vascular causes, nonfatal stroke, nonfatal MI, nonfatal major bleeding complication), the hazard ratio was 0.80 favoring aspirin plus dipyridamole, with a number needed to treat of 104 over a mean of 3.5 years (absolute risk reduction of 1% per year). Importantly, twice as many patients taking aspirin plus dipyridamole discontinued the medication.
Caveats to interpreting this study are that it was not blinded, the aspirin doses varied (although the median aspirin dose—75 mg—was the same between the two groups), and not all patients received the extended-release form of dipyridamole.
Conclusions about dipyridamole
ESPS-2, ESPRIT, and the meta-analysis by Leonardi-Bee et al showed that aspirin plus dipyridamole is more effective than placebo or aspirin alone in secondary prevention of vascular events, including stroke. Also, extended-release dipyridamole appears to be more effective.
Unfortunately, many patients stop taking dipyridamole because of side effects (primarily headache).
Based on the results of ESPRIT, the absolute benefit of dipyridamole used alone may be small.
CLOPIDOGREL: SIMILAR TO ASPIRIN IN EFFICACY?
Like dipyridamole, clopidogrel targets adenosine diphosphate to prevent clot formation, blocking its ability to bind to its receptor on platelets. It is a thienopyridine and, unlike its sister drug ticlopidine, does not seem to be associated with the potentially serious side effects of neutropenia. However, a few cases of thrombotic thrombocytopenic purpura have been reported.28 The other drugs in this class have not been evaluated in clinical trials for secondary stroke prophylaxis.
Trials of clopidogrel
CAPRIE.29 The Clopidogrel Versus Aspirin in Patients at Risk of Ischaemic Events trial, in 1996, was one of the first to compare the clinical use of clopidogrel against aspirin. It was a randomized controlled noninferiority trial in patients over age 21 (inclusion criteria: ischemic stroke, MI, or peripheral arterial disease) randomized to aspirin 325 mg once daily or clopidogrel 75 mg once daily. Patients were followed for 1 to 3 years.
Patients on clopidogrel had a relative risk reduction of 8.7% in primary events (ischemic stroke, MI, or vascular death); patients on aspirin were at significantly higher risk of gastrointestinal hemorrhage. Patients with peripheral arterial disease as the qualifying event did particularly well on clopidogrel, with a significant relative risk reduction of 23.8%.
Limitations of the CAPRIE trial included its inability to measure the effect of treatment on individual outcomes, particularly stroke, and the fact that the relative risk reduction for patients with stroke as the qualifying event was not significant (P = .66). Another limitation was that it did not use TIA as an entry criterion or as part of the composite outcome. Also, the relative risk reduction had a wide confidence interval, and a large number of patients discontinued therapy for reasons other than the defined outcomes.
Nevertheless, the CAPRIE trial showed clopidogrel to be an effective antiplatelet prophylactic, particularly in patients with peripheral artery disease, but with no discernible difference from aspirin for those patients with MI or stroke as a qualifying event.
MATCH.30 The Management of Atherothrombosis With Clopidogrel in High-risk Patients trial hoped to better assess clopidogrel’s efficacy, particularly in patients with ischemic cerebral events. Cardiac studies leading up to MATCH suggested that adding a thienopyridine to aspirin might offer additive benefit in reducing the rate of vascular outcomes.15,31 MATCH randomized high-risk patients (inclusion criteria were ischemic stroke or TIA and a history of vascular disease) to clopidogrel or to aspirin plus clopidogrel.
There was a nonsignificant 6.4% relative risk reduction in the combined primary outcome of MI, ischemic stroke, vascular death, other vascular death, and re-hospitalization for acute ischemic events in the aspirin-plus-clopidogrel group compared with clopidogrel alone. However, this came at the cost of double the number of bleeding events in the combination group, mitigating most of the benefit of combination therapy.
An important caveat in interpreting the results of MATCH, as compared with the Clopidogrel in Unstable Angina to Prevent Recurrent Events (CURE) study, is that aspirin was being added to clopidogrel, not vice versa. CURE, which looked at the addition of clopidogrel to aspirin vs aspirin alone in cardiac patients, found a significant reduction of ischemic events taken as a group (relative risk 0.8), and a trend toward a lower rate of stroke (relative risk 0.86, but 95% confidence interval encompassing 1) for aspirin plus clopidogrel vs aspirin alone.31 However, patients in the CURE trial did not have high-risk vasculopathy per se but rather non-ST-elevation MI, perhaps skewing the benefit of combination therapy and lessening the risk of intracranial bleeding.
CHARISMA.32 The Clopidogrel for High Atherothrombotic Risk and Ischemic Stabilization, Management, and Avoidance trial, like the CURE trial, compared aspirin plus clopidogrel vs aspirin in patients with established cardiovascular, cerebrovascular, or peripheral arterial disease, or who were at high risk of events. As in the MATCH study, the findings for combination therapy were a nonsignificant relative risk of 0.93 for primary events (MI, stroke, or death from cardiovascular causes), and a significant reduction of secondary end points (primary end point event plus TIA or hospitalization for unstable angina) (relative risk 0.92, P = .04).
Importantly, combination therapy significantly increased the rate of bleeding events. In asymptomatic patients (those without documented vascular disease but with multiple atherothrombotic risk factors), there was actually harm with combined treatment. Conversely, for symptomatic patients (those with documented vascular disease), there was a negligible, but significant reduction in primary end points.
The result was that in patients with documented vascular disease, aspirin plus clopidogrel combination therapy provided little or no benefit over aspirin alone. For patients with elevated risk factors but no documented vascular burden, there may actually be harm from combination therapy.
PRoFESS.33 Logically following is the question of whether aspirin plus dipyridamole offers any benefit over clopidogrel as a stroke prophylactic. The Prevention Regimen for Effectively Avoiding Second Strokes trial hoped to answer this by comparing clopidogrel against aspirin plus dipyridamole, both with and without telmisartan, in patients with recent stroke.
The rate of recurrent stroke was similar in the two groups, but there were 25 fewer ischemic strokes in patients on aspirin plus dipyridamole, offset by an increase in hemorrhagic strokes. Rates of secondary outcomes of stroke, death, or MI were nearly identical between the groups. Early discontinuation of treatment was significantly more frequent in those patients taking aspirin plus dipyridamole, meaning better compliance for those taking clopidogrel.
Initially, patients were to be randomized to either aspirin plus dipyridamole or aspirin plus clopidogrel. However, after MATCH30 demonstrated a significantly higher bleeding risk with aspirin plus clopidogrel, patients were changed to clopidogrel alone. But despite this, the bleeding risk was still higher with aspirin plus dipyridamole.
During the trial, the entry criteria were expanded, allowing for the inclusion of younger patients and those with less recent strokes; but despite this change, the study remained underpowered to demonstrate its goal of noninferiority. Thus, it showed only a trend of noninferiority of clopidogrel vs aspirin plus dipyridamole.
What the clopidogrel trials tell us
Clopidogrel confers a benefit similar to that of aspirin (as shown in the CAPRIE study).29 Although aspirin plus dipyridamole confers greater benefit than aspirin alone (as shown in the ESPS-2,14 Leonardi-Bee,25 and ESPRIT26 studies), aspirin plus dipyridamole is not superior to clopidogrel, and may even be inferior.34
WARFARIN FOR ATRIAL FIBRILLATION ONLY
Warfarin acts by disrupting the coagulation cascade rather than acting at the site of platelet plug formation. In theory, warfarin should be as effective as the antiplatelet drugs in preventing clot formation, and so it was thought to possibly be effective in preventing stroke of arterial origin.
However, in at least three studies, warfarin increased the risk of death, MI, and hemorrhage, with perhaps a slight decrease in the risk of recurrent stroke in patients with suspected stroke or TIA.35–37 This should be differentiated from stroke originating from cardiac dysrhythmias, for which warfarin has clearly been shown to be beneficial.28
THREE GOOD MEDICAL OPTIONS FOR PREVENTING STROKE RECURRENCE
Antiplatelet therapy offers benefit in the primary and secondary prevention of stroke, with a 25% reduction in the rate of nonfatal stroke and a 17% reduction in the rate of death due to vascular causes.15
Aspirin is the best established
Aspirin is the best established, best tolerated, and least expensive of the three contemporary agents. Further, it is also the agent of choice for acute stroke care, to be given within 48 hours of a stroke to mitigate the risk of death and morbidity. The data for other agents in acute stroke management remain limited.38
Aspirin plus dipyridamole
Aspirin plus dipyridamole is slightly more efficacious than aspirin alone, and it is an alternative when aspirin is ineffective and when the patient can afford the additional cost. Aspirin plus dipyridamole offers up to a 22% relative risk reduction (but a small reduction in absolute risk) of stroke compared with aspirin alone, as demonstrated by ESPS-2,14 Leonardi-Bee et al,25 and ESPRIT.26
When is clopidogrel appropriate?
Up to one-third of patients may not tolerate aspirin plus dipyridamole because of side effects. Clopidogrel is an option for these patients. The CAPRIE study29 showed clopidogrel similar in efficacy to aspirin.
In contrast to aspirin plus dipyridamole, there is clearly no benefit to combining aspirin and clopidogrel for ischemic stroke prophylaxis. And data from PRoFESS33 suggested the combination was qualitatively inferior to aspirin plus dipyridamole. However, the PRoFESS trial was underpowered to fully bear this out.
Therefore, current guidelines consider all three agents as appropriate for secondary prevention of stroke. One is not preferred over another, and the selection should be based on individual patient characteristics and affordability.28
CAROTID SURGERY OR STENTING: BENEFITS AND LIMITATIONS
Atherosclerosis is the most common cause of stroke, and atherosclerosis of the common carotid bifurcation accounts for a small but significant percentage of all strokes.39–41
The degree of carotid stenosis and whether it is producing symptoms influence how it should be managed. For patients with symptomatic carotid stenosis of more than 70%, multicenter randomized trials have shown that surgery (ie, carotid endarterectomy) added to medical therapy decreases the rate of recurrent stroke by up to 17% and the rate of combined stroke and death by 10% to 12% over a 2- to 3-year follow-up period (level of evidence A).42–44 No study has proven the efficacy of surgery in patients with symptomatic stenosis of less than 50%.43,44
Similarly, in asymptomatic carotid disease, preventive surgery is a beneficial adjunct to medical therapy in certain patients. An approximate 6% reduction in the rate of stroke or death over 5 years has been shown in patients with moderate stenosis (> 60%), with men younger than age 75 and with greater than 70% stenosis deriving the most benefit.45–47
However, these robust, positive results with surgical intervention should not overshadow the importance of intensive and guided medical therapy, which has been shown to mitigate the risk of stroke.48,49
Is stenting as good as surgery? In the multicenter randomized Carotid Revascularization Endarterectomy vs Stenting Trial (CREST), stenting resulted in similar rates of stroke and MI in patients with symptomatic and asymptomatic disease.50 However, stenting carried a greater risk of perioperative stroke, and endarterectomy carried a greater risk of MI. Those under age 70 benefited more from stenting, and those over age 70 benefited more from endarterectomy.
But another fact to keep in mind is that the relationship between carotid narrowing and an ipsilateral stroke is not necessarily direct. Two follow-up studies in patients from the North American Symptomatic Carotid Endarterectomy Trial (NASCET) found that up to 45% of strokes that occurred after intervention in the distribution of the asymptomatic stenosed carotid artery were unrelated to the stenosis.51,52 Moreover, up to 20% of subsequent strokes in the distribution of the symptomatic artery were not of large-artery origin, increasing up to 35% for those with stenosis of less than 70%.51 Clearly, thorough screening of those with presumed symptomatic stenosis is needed to eliminate other possible causes.
- Zivin JA. Approach to cerebrovascular diseases. In:Goldman L, Schafer AI, editors. Goldman’s Cecil Medicine. 24th ed. Philadelphia, PA: Elsevier, 2012:2304–2309.
- Samsa GP, Bian J, Lipscomb J, Matchar DB. Epidemiology of recurrent cerebral infarction: a Medicare claims-based comparison of first and recurrent strokes on 2-year survival and cost. Stroke 1999; 30:338–349.
- Gotoh F, Tohgi H, Hirai S, et al. Cilostazol Stroke Prevention Study: a placebo-controlled double-blind trial for secondary prevention of cerebral infarction. J Stroke Cerebrovasc Dis 2000; 9:147–157.
- Shinohara Y, Katayama Y, Uchiyama S, et al; CSPS 2 group. Cilostazol for prevention of secondary stroke (CSPS 2): an aspirin-controlled, double-blind, randomised non-inferiority trial. Lancet Neurol 2010; 9:959–968.
- Weiss HJ, Aledort LM. Impaired platelet-connective-tissue reaction in man after aspirin ingestion. Lancet 1967; 2:495–497.
- Fields WS, Lemak NA, Frankowski RF, Hardy RJ. Controlled trial of aspirin in cerebral ischemia. Stroke 1977; 8:301–314.
- Fields WS, Lemak NA, Frankowski RF, Hardy RJ. Controlled trial of aspirin in cerebral ischemia. Part II: surgical group. Stroke 1978; 9:309–319.
- Sorensen PS, Pedersen H, Marquardsen J, et al. Acetylsalicylic acid in the prevention of stroke in patients with reversible cerebral ischemic attacks. A Danish cooperative study. Stroke 1983; 14:15–22.
- Bousser MG, Eschwege E, Haguenau M, et al. “AICLA” controlled trial of aspirin and dipyridamole in the secondary prevention of athero-thrombotic cerebral ischemia. Stroke 1983; 14:5–14.
- High-dose acetylsalicylic acid after cerebral infarction. A Swedish Cooperative Study. Stroke 1987; 18:325–334.
- Boysen G, Sørensen PS, Juhler M, et al. Danish very-low-dose aspirin after carotid endarterectomy trial. Stroke 1988; 19:1211–1215.
- Farrell B, Godwin J, Richards S, Warlow C. The United Kingdom transient ischaemic attack (UK-TIA) aspirin trial: final results. J Neurol Neurosurg Psychiatry 1991; 54:1044–1054.
- Swedish Aspirin Low-Dose Trial (SALT) of 75 mg aspirin as secondary prophylaxis after cerebrovascular ischaemic events. The SALT Collaborative Group. Lancet 1991; 338:1345–1349.
- Diener HC, Cunha L, Forbes C, Sivenius J, Smets P, Lowenthal A. European Stroke Prevention Study. 2. Dipyridamole and acetylsalicylic acid in the secondary prevention of stroke. J Neurol Sci 1996; 143:1–13.
- Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, MI, and stroke in high risk patients. BMJ 2002; 324:71–86.
- A comparison of two doses of aspirin (30 mg vs. 283 mg a day) in patients after a transient ischemic attack or minor ischemic stroke. The Dutch TIA Trial Study Group. N Engl J Med 1991; 325:1261–1266.
- Taylor DW, Barnett HJ, Haynes RB, et al. Low-dose and high-dose acetylsalicylic acid for patients undergoing carotid endarterectomy: a randomised controlled trial. ASA and Carotid Endarterectomy (ACE) Trial Collaborators. Lancet 1999; 353:2179–2184.
- Algra A, van Gijn J. Aspirin at any dose above 30 mg offers only modest protection after cerebral ischaemia. J Neurol Neurosurg Psychiatry 1996; 60:197–199.
- Campbell CL, Smyth S, Montalescot G, Steinhubl SR. Aspirin dose for the prevention of cardiovascular disease: a systematic review. JAMA 2007; 297:2018–2024.
- Weiss HJ, Aledort LM, Kochwa S. The effect of salicylates on the hemostatic properties of platelets in man. J Clin Invest 1968; 47:2169–2180.
- European Stroke Prevention Study. ESPS Group. Stroke 1990; 21:1122–1130.
- Davis SM, Donnan GA. Secondary prevention for stroke after CAPRIE and ESPS-2. Opinion 1. Cerebrovasc Dis 1998; 8:73–77.
- Diener HC. Dipyridamole trials in stroke prevention. Neurology 1998; 51(suppl 3):S17–S19.
- De Schryver EL, Algra A, van Gijn J. Cochrane review: dipyridamole for preventing major vascular events in patients with vascular disease. Stroke 2003; 34:2072–2080.
- Leonardi-Bee J, Bath PM, Bousser MG, et al; Dipyridamole in Stroke Collaboration (DISC). Dipyridamole for preventing recurrent ischemic stroke and other vascular events: a meta-analysis of individual patient data from randomized controlled trials. Stroke 2005; 36:162–168.
- ESPRIT Study Group; Halkes PH, van Gijn J, Kappelle LJ, Koudstaal PJ, Algra A. Aspirin plus dipyridamole versus aspirin alone after cerebral ischaemia of arterial origin (ESPRIT): randomised controlled trial. Lancet 2006; 367:1665–1673.
- Tirschwell D. Aspirin plus dipyridamole was more effective than aspirin alone for preventing vascular events after minor cerebral ischemia. ACP J Club 2006; 145:57.
- Furie KL, Kasner SE, Adams RJ, et al; American Heart Association Stroke Council, Council on Cardiovascular Nursing, Council on Clinical Cardiology, and Interdisciplinary Council on Quality of Care and Outcomes Research. Guidelines for the prevention of stroke in patients with stroke or transient ischemic attack: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2011; 42:227–276.
- CAPRIE Steering Committee. A randomised, blinded, trial of clopidogrel versus aspirin in patients at risk of ischaemic events (CAPRIE). CAPRIE Steering Committee. Lancet 1996; 348:1329–1339.
- Diener HC, Bogousslavsky J, Brass LM, et al; MATCH investigators. Aspirin and clopidogrel compared with clopidogrel alone after recent ischaemic stroke or transient ischaemic attack in high-risk patients (MATCH): randomised, double-blind, placebo-controlled trial. Lancet 2004; 364:331–337.
- Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK; Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial Investigators. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med 2001; 345:494–502.
- Bhatt DL, Fox KA, Hacke W, et al; CHARISMA Investigators. Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events. N Engl J Med 2006; 354:1706–1717.
- Sacco RL, Diener HC, Yusuf S, et al; PRoFESS Study Group. Aspirin and extended-release dipyridamole versus clopidogrel for recurrent stroke. N Engl J Med 2008; 359:1238–1251.
- Kent DM, Thaler DE. Stroke prevention—insights from incoherence. N Engl J Med 2008; 359:1287–1289.
- Chimowitz MI, Lynn MJ, Howlett-Smith H, et al; Warfarin-Aspirin Symptomatic Intracranial Disease Trial Investigators. Comparison of warfarin and aspirin for symptomatic intracranial arterial stenosis. N Engl J Med 2005; 352:1305–1316.
- ESPRIT Study Group; Halkes PH, van Gijn J, Kappelle LJ, Koudstaal PJ, Algra A. Medium intensity oral anticoagulants versus aspirin after cerebral ischaemia of arterial origin (ESPRIT): a randomised controlled trial. Lancet Neurol 2007; 6:115–124.
- Mohr JP, Thompson JL, Lazar RM, et al; Warfarin-Aspirin Recurrent Stroke Study Group. A comparison of warfarin and aspirin for the prevention of recurrent ischemic stroke. N Engl J Med 2001; 345:1444–1451.
- Jauch EC, Saver JL, Adams HP, et al; American Heart Association Stroke Council; Council on Cardiovascular Nursing; Council on Peripheral Vascular Disease; Council on Clinical Cardiology. Guidelines for the early management of patients with acute ischemic stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2013; 44:870–947.
- Kolominsky-Rabas PL, Weber M, Gefeller O, Neundoerfer B, Heuschmann PU. Epidemiology of ischemic stroke subtypes according to TOAST criteria: incidence, recurrence, and long-term survival in ischemic stroke subtypes: a population-based study. Stroke 2001; 32:2735–2740.
- Zivin JA. Ischemic cerebrovascular disease. In:Goldman L, Schafer AI, editors. Goldman’s Cecil Medicine 24th ed. Philadelphia, PA: Elsevier; 2012: chap 414. www.mdconsult.com. Accessed November 7, 2013.
- Smith WS, Johnston C, Easton D. Cerebrovascular diseases. In:Kasper DL, Braunwald E, Fauci AS, Hauser SL, Longo DL, Jameson JL, editors. Harrison’s Principles of Internal Medicine. 16th ed. New York, NY: McGraw Hill; 2005: chap 349. www.accessmedicine.com.
- Beneficial effect of carotid endarterectomy in symptomatic patients with high-grade carotid stenosis. North American Symptomatic Carotid Endarterectomy Trial Collaborators. N Engl J Med 1991; 325:445–453.
- Randomised trial of endarterectomy for recently symptomatic carotid stenosis: final results of the MRC European Carotid Surgery Trial (ECST). Lancet 1998; 351:1379–1387.
- Barnett HJ, Taylor DW, Eliasziw M, et al. Benefit of carotid endarterectomy in patients with symptomatic moderate or severe stenosis. North American Symptomatic Carotid Endarterectomy Trial Collaborators. N Engl J Med 1998; 339:1415–1425.
- Hobson RW, Weiss DG, Fields WS, et al. Efficacy of carotid endarterectomy for asymptomatic carotid stenosis. The Veterans Affairs Cooperative Study Group. N Engl J Med 1993; 328:221–227.
- Endarterectomy for asymptomatic carotid artery stenosis. Executive Committee for the Asymptomatic Carotid Atherosclerosis Study. JAMA 1995; 273:1421–1428.
- Halliday A, Mansfield A, Marro J, et al; MRC Asymptomatic Carotid Surgery Trial (ACST) Collaborative Group. Prevention of disabling and fatal strokes by successful carotid endarterectomy in patients without recent neurological symptoms: randomised controlled trial. Lancet 2004; 363:1491–1502.
- Marquardt L, Geraghty OC, Mehta Z, Rothwell PM. Low risk of ipsilateral stroke in patients with asymptomatic carotid stenosis on best medical treatment: a prospective, population-based study. Stroke 2010; 41:e11–e17.
- Spence JD, Coates V, Li H, et al. Effects of intensive medical therapy on microemboli and cardiovascular risk in asymptomatic carotid stenosis. Arch Neurol 2010; 67:180–186.
- Brott TG, Hobson RW, Howard G, et al; CREST Investigators. Stenting versus endarterectomy for treatment of carotid-artery stenosis. N Engl J Med 2010; 363:11–23.
- Barnett HJ, Gunton RW, Eliasziw M, et al. Causes and severity of ischemic stroke in patients with internal carotid artery stenosis. JAMA 2000; 283:1429–1436.
- Inzitari D, Eliasziw M, Gates P, et al. The causes and risk of stroke in patients with asymptomatic internal-carotid-artery stenosis. North American Symptomatic Carotid Endarterectomy Trial Collaborators. N Engl J Med 2000; 342:1693–1700.
After a stroke, an important goal is to prevent another one.1,2 And for patients who have had an ischemic stroke or transient ischemic attack (TIA) due to atherosclerosis, an important part of secondary preventive therapy is a drug that inhibits platelets—ie, aspirin, extended-release dipyridamole, or clopidogrel. This has taken years to establish.
In the following pages, we discuss the antiplatelet agents that have been shown to be beneficial after stroke of atherosclerotic origin, and we briefly review the indications for surgery and stenting for the subset of patients whose strokes are caused by symptomatic carotid disease.
(Although managing modifiable risk factors such as smoking, hypertension, diabetes, and dyslipidemia is also important, we will not cover this topic here, nor will we talk about hemorrhagic stroke or stroke due to atrial fibrillation. Also not discussed here is cilostazol, which, although shown to be effective in preventing recurrent stroke when compared with placebo and aspirin,3,4 has not been approved for this use by the US Food and Drug Administration, as of this writing.)
HOW WE REVIEWED THE LITERATURE
We searched PubMed using the terms aspirin, acetylsalicylic acid, clopidogrel, and/or dipyridamole, in combination with stroke, cerebral ische(ae)mia, transient ische(ae)mic attacks, or retinal artery occlusion. We reviewed only clinical trials or meta-analyses of these drugs for either primary or secondary prevention of cerebrovascular disease.
As our aim was to review the topic and not to perform a meta-analysis, no cutoffs were used to exclude trials. The references in the selected papers were also reviewed to expand the articles. Finally, the references in the current American Heart Association and American Stroke Association secondary stroke prevention guideline were also reviewed.
For a summary of the trials included in our review, see the Data Supplement as an appendix to the online version of this article.
ASPIRIN: THE GOLD STANDARD
Prescribed by Hippocrates in the form of willow bark extract, aspirin has long been known for its antipyretic and anti-inflammatory properties. Its antiplatelet and antithrombotic properties, first described in 1967 by Weiss and Aledort,5 are mediated by irreversible inhibition of cyclooxygenase, leading to decreased thromboxane A2, a platelet-aggregation activator.
Fields et al,6,7 in 1977 and 1978, reported that in a controlled trial in patients with TIA or monocular blindness, fewer subsequent TIAs occurred in patients who received aspirin, although the difference was not statistically significant, with lower rates of events only in nonsurgical patients. Over the next 20 years, the results remained mixed.
The Danish Cooperative study8 (1983) found no significant difference in the rate of recurrent stroke with aspirin vs placebo.
AICLA.9 The Accidents Ischémiques Cérébraux Liés à l’Athérosclérose study of 1983 did find a difference. However, both the Danish Cooperative study and the AICLA were limited by lacking standardized computed tomographic imaging to rule out hemorrhagic stroke and by being relatively small.
The Swedish Cooperative Study10 (1987) found no statistical difference between high-dose aspirin and placebo in preventing recurrent vascular events (stroke, TIA, or myocardial infarction [MI]) 1 to 3 weeks after a stroke. However, it had several limitations: the aspirin group contained more patients with ischemic heart disease (who are more likely to die of cardiac causes), there were significantly more men in the aspirin group, and nearly one-fourth of the deaths were a result of the initial stroke, potentially masking the effect of aspirin in secondary prevention.
Later studies began to show a consistently favorable effect of aspirin.
Boysen et al11 in 1988 reported a nonsignificant trend toward fewer adverse events with aspirin.
UK-TIA.12 The United Kingdom Transient Ischaemic Attack trial in 1991 found a similar trend.
SALT.13 The Swedish Aspirin Low-dose Trial, also in 1991, showed a significant 18% lower rate of stroke or death in patients with recent TIA, minor stroke, or retinal occlusion treated with low-dose aspirin. The inclusion of patients with TIA helped broaden the population that might benefit. However, the study may have favored the aspirin group by having a run-in period in which patients were nonrandomly treated either with aspirin or with anticoagulation at the discretion of the patient’s physician and, if they suffered “several” TIAs, a stroke, retinal artery occlusion, or MI, were removed from the study.
ESPS-2.14 The second European Stroke Prevention Study in 1996 added to the evidence that aspirin prevents recurrent stroke. Patients with a history of TIA or stroke were randomized in double-blind fashion to four treatment groups: placebo, low-dose aspirin, dipyridamole, or aspirin plus dipyridamole. At 2 years, strokes had occurred in 18% fewer patients in the aspirin group than in the placebo group, and TIAs had occurred in 21.9% fewer. However, aspirin was associated with an absolute 0.5% increase in severe and fatal bleeding. The power of the study was limited because patients from one center were excluded because of “serious inconsistencies in patient case record forms and compliance assay determinations.” 14
Comment. The mixed results with aspirin in studies predating ESPS-2 were partly because the study populations were too small to show benefit.
ATT.15 The Antithrombotic Trialists’ Collaboration performed a meta-analysis that conclusively confirmed the benefit of aspirin after stroke or TIA. The investigators analyzed individual patient data pooled from randomized controlled trials published before 1997 that compared antiplatelet regimens (mostly aspirin) against placebo and against each other. The rates of vascular events were 10.7% with treatment vs 13.2% with placebo (P < .0001). Antiplatelet therapy was particularly effective in preventing ischemic stroke, with a 25% reduction in the rate of nonfatal stroke, and with an overall absolute benefit in stroke prevention across all high-risk patient groups. This translated to 25 fewer nonfatal strokes per 1,000 patients treated with antiplatelet therapy.
What is the optimal aspirin dose?
Studies of aspirin have used different daily doses—the earliest studies used large doses of 1,000 to 1,500 mg.6–10
Boysen et al11 in 1988 found a trend toward benefit (not statistically significant) with doses ranging from 50 mg to 100 mg.
In 1991, three separate studies found that higher doses of aspirin were no more effective than lower doses.
The UK-TIA trial12 compared aspirin 300 mg vs 1,200 mg and found a higher risk of gastrointestinal bleeding with the higher dose.
The SALT Collaborative Group13 found 75 mg to be effective.
The Dutch TIA trial16 compared 30 mg vs 283 mg; end point outcomes were similar but the rate of adverse events was higher with 283 mg.
ESPS-2 was able to show efficacy at a dose of only 50 mg.14
Taylor et al17 compared lower doses (81 or 325 mg) vs higher doses (650 or 1,300 mg) for patients undergoing carotid endarterectomy and found that the risk of adverse events was twice as high with the higher doses.
The ATT Collaboration15 found that efficacy was 40% lower with the highest dose of aspirin than with the lowest doses.
Algra and van Gijn18 performed a meta-analysis of all these studies and found no difference in risk reduction between low-dose and high-dose aspirin, with an overall relative risk reduction of 13% at any dose above 30 mg.
Campbell et al,19 in a 2007 review, found that doses greater than 300 mg conferred no benefit, and that rapid and maximum suppression of thromboxane A2 can be achieved by chewing or ingesting dissolved forms of aspirin 162 mg.
Conclusion. Aspirin doses higher than 81 mg (the US standard) confer no greater benefit and may even decrease the efficacy of aspirin. In an emergency, rapid suppression of thromboxane A2 can be achieved by chewing a minimum dose of 162 mg.
DIPYRIDAMOLE CAN BE ADDED TO ASPIRIN
In 1967, Weiss and Aledort5 found that aspirin’s antiplatelet effect could be blocked by adenosine diphosphate, which is released by activated platelet cells and is an essential part of thrombus formation. Adjacent platelets are then activated, leading to up-regulation of thromboxane A2 and glycoprotein IIb/IIIa receptors and resulting in a cascade of platelet activation and clot formation.20 Dipyridamole inhibits aggregation of platelets by inhibiting their ability to take up adenosine diphosphate.
Studies of dipyridamole
AICLA.9 Bousser et al9 randomized patients who suffered one or more cerebral or retinal infarctions to receive placebo, aspirin 1 g, or aspirin 1 g plus dipyridamole 225 mg. Aspirin was significantly better than placebo in preventing a recurrence of stroke. The event rate with aspirin plus dipyridamole was similar to the rate with aspirin alone, although on 2-by-2 analysis, the difference between placebo and aspirin plus dipyridamole did not reach statistical significance. However, the rate of carotid-origin stroke was 17% with aspirin alone and 6% with aspirin plus dipyridamole, a statistically significant difference.
Thus, this study confirmed the benefit of aspirin in preventing ischemic events but did not fully support the addition of dipyridamole, except in preventing stroke of carotid origin. The study had a number of limitations: the sample size was small, TIA was not included as an end point, computed tomography was not required for entry, and many patients were lost to follow-up, decreasing the statistical power of the trial.
The ESPS study21 was also a randomized controlled trial of aspirin plus dipyridamole vs placebo. But unlike AICLA, ESPS included patients with TIA.
ESPS found a 38.1% relative risk reduction in stroke with aspirin plus dipyridamole compared with placebo, and a 30.6% reduction in death from all causes. Interestingly, patients who had a TIA as the qualifying event had a lower end-point incidence and larger end-point reduction than those who had a stroke as the qualifying event. However, ESPS did not resolve the question of whether adding dipyridamole to aspirin affords any benefit over aspirin alone.
ESPS-214 hoped to answer this question. Patients were randomized to placebo, aspirin, dipyridamole, or aspirin plus dipyridamole. On 2 × 2 analysis, the dipyridamole group had a 16% lower rate of recurrent stroke than the placebo group, and patients on aspirin plus dipyridamole had a 37% lower rate. Aspirin plus dipyridamole yielded a 23.1% reduction compared with aspirin alone, and a 24.7% reduction compared with dipyridamole alone. Similar benefit was reported for the end point of TIA with combination therapy compared with either agent alone.
However, nearly 25% of patients had to withdraw because of side effects, particularly in the dipyridamole-alone and aspirin-dipyridamole groups, and, as mentioned above, verification of compliance in the aspirin group was an issue.14,22 Nevertheless, ESPS-2 clearly showed that aspirin plus dipyridamole was better than either drug alone in preventing recurrent stroke. It also showed the effectiveness of dipyridamole, which AICLA and ESPS could not do, because it had a larger study population, used a lower dose of aspirin, and perhaps because it used an extended-release form of dipyridamole.23
The ATT meta-analysis15 showed a clear benefit of antiplatelet therapy. However, much of this benefit was derived from aspirin therapy, with the addition of dipyridamole resulting in a nonsignificant 6% reduction of vascular events. Most of the patients on dipyridamole were from the ESPS-2 study. In effect, the ATT was a meta-analysis of aspirin, as aspirin studies dominated at that time.
A Cochrane review24 publsihed in 2003 attempted to rectify this by analyzing randomized controlled trials of dipyridamole vs placebo.24 Like the ATT meta-analysis, it did not bear out the benefits of dipyridamole: compared with placebo, there was no effect on the rate of vascular death, and only a minimal benefit in reduction of vascular events—and this latter point is only because of the inclusion of ESPS-2.
Directly comparing aspirin plus dipyridamole vs aspirin alone, the reviewers found no effect on the rate of vascular death, and with the exclusion of ESPS-2, no effect on vascular events.
The Cochrane review had the same limitation as the ATT meta-analysis, ie, dependence on a single trial (ESPS-2) to show benefit, and perhaps the fact that ESPS-2 was the only study that used an extended-release form of dipyridamole.
Leonardi-Bee et al25 performed a meta-analysis that overcame the limitation of ESPS-2 being the only study at the time with positive findings: they used pooled individual patient data from randomized trials and analyzed them en masse. Patients on aspirin plus dipyridamole had a 39% lower risk than with placebo and a 22% lower risk than with aspirin alone. Unlike the ATT and the Cochrane review, excluding ESPS-2 did not alter the statistically significant lower stroke rate with aspirin plus dipyridamole compared with controls. This meta-analysis helped to confirm ESPS-2’s finding of the additive effect of aspirin plus dipyridamole compared with aspirin and placebo control.
ESPRIT.26,27 The European/Australasian Stroke Prevention in Reversible Ischaemia Trial confirmed these findings. This randomized controlled trial compared aspirin plus dipyridamole against aspirin alone in patients with a TIA or minor ischemic stroke of arterial origin within the past 6 months. For the primary end point (death from all vascular causes, nonfatal stroke, nonfatal MI, nonfatal major bleeding complication), the hazard ratio was 0.80 favoring aspirin plus dipyridamole, with a number needed to treat of 104 over a mean of 3.5 years (absolute risk reduction of 1% per year). Importantly, twice as many patients taking aspirin plus dipyridamole discontinued the medication.
Caveats to interpreting this study are that it was not blinded, the aspirin doses varied (although the median aspirin dose—75 mg—was the same between the two groups), and not all patients received the extended-release form of dipyridamole.
Conclusions about dipyridamole
ESPS-2, ESPRIT, and the meta-analysis by Leonardi-Bee et al showed that aspirin plus dipyridamole is more effective than placebo or aspirin alone in secondary prevention of vascular events, including stroke. Also, extended-release dipyridamole appears to be more effective.
Unfortunately, many patients stop taking dipyridamole because of side effects (primarily headache).
Based on the results of ESPRIT, the absolute benefit of dipyridamole used alone may be small.
CLOPIDOGREL: SIMILAR TO ASPIRIN IN EFFICACY?
Like dipyridamole, clopidogrel targets adenosine diphosphate to prevent clot formation, blocking its ability to bind to its receptor on platelets. It is a thienopyridine and, unlike its sister drug ticlopidine, does not seem to be associated with the potentially serious side effects of neutropenia. However, a few cases of thrombotic thrombocytopenic purpura have been reported.28 The other drugs in this class have not been evaluated in clinical trials for secondary stroke prophylaxis.
Trials of clopidogrel
CAPRIE.29 The Clopidogrel Versus Aspirin in Patients at Risk of Ischaemic Events trial, in 1996, was one of the first to compare the clinical use of clopidogrel against aspirin. It was a randomized controlled noninferiority trial in patients over age 21 (inclusion criteria: ischemic stroke, MI, or peripheral arterial disease) randomized to aspirin 325 mg once daily or clopidogrel 75 mg once daily. Patients were followed for 1 to 3 years.
Patients on clopidogrel had a relative risk reduction of 8.7% in primary events (ischemic stroke, MI, or vascular death); patients on aspirin were at significantly higher risk of gastrointestinal hemorrhage. Patients with peripheral arterial disease as the qualifying event did particularly well on clopidogrel, with a significant relative risk reduction of 23.8%.
Limitations of the CAPRIE trial included its inability to measure the effect of treatment on individual outcomes, particularly stroke, and the fact that the relative risk reduction for patients with stroke as the qualifying event was not significant (P = .66). Another limitation was that it did not use TIA as an entry criterion or as part of the composite outcome. Also, the relative risk reduction had a wide confidence interval, and a large number of patients discontinued therapy for reasons other than the defined outcomes.
Nevertheless, the CAPRIE trial showed clopidogrel to be an effective antiplatelet prophylactic, particularly in patients with peripheral artery disease, but with no discernible difference from aspirin for those patients with MI or stroke as a qualifying event.
MATCH.30 The Management of Atherothrombosis With Clopidogrel in High-risk Patients trial hoped to better assess clopidogrel’s efficacy, particularly in patients with ischemic cerebral events. Cardiac studies leading up to MATCH suggested that adding a thienopyridine to aspirin might offer additive benefit in reducing the rate of vascular outcomes.15,31 MATCH randomized high-risk patients (inclusion criteria were ischemic stroke or TIA and a history of vascular disease) to clopidogrel or to aspirin plus clopidogrel.
There was a nonsignificant 6.4% relative risk reduction in the combined primary outcome of MI, ischemic stroke, vascular death, other vascular death, and re-hospitalization for acute ischemic events in the aspirin-plus-clopidogrel group compared with clopidogrel alone. However, this came at the cost of double the number of bleeding events in the combination group, mitigating most of the benefit of combination therapy.
An important caveat in interpreting the results of MATCH, as compared with the Clopidogrel in Unstable Angina to Prevent Recurrent Events (CURE) study, is that aspirin was being added to clopidogrel, not vice versa. CURE, which looked at the addition of clopidogrel to aspirin vs aspirin alone in cardiac patients, found a significant reduction of ischemic events taken as a group (relative risk 0.8), and a trend toward a lower rate of stroke (relative risk 0.86, but 95% confidence interval encompassing 1) for aspirin plus clopidogrel vs aspirin alone.31 However, patients in the CURE trial did not have high-risk vasculopathy per se but rather non-ST-elevation MI, perhaps skewing the benefit of combination therapy and lessening the risk of intracranial bleeding.
CHARISMA.32 The Clopidogrel for High Atherothrombotic Risk and Ischemic Stabilization, Management, and Avoidance trial, like the CURE trial, compared aspirin plus clopidogrel vs aspirin in patients with established cardiovascular, cerebrovascular, or peripheral arterial disease, or who were at high risk of events. As in the MATCH study, the findings for combination therapy were a nonsignificant relative risk of 0.93 for primary events (MI, stroke, or death from cardiovascular causes), and a significant reduction of secondary end points (primary end point event plus TIA or hospitalization for unstable angina) (relative risk 0.92, P = .04).
Importantly, combination therapy significantly increased the rate of bleeding events. In asymptomatic patients (those without documented vascular disease but with multiple atherothrombotic risk factors), there was actually harm with combined treatment. Conversely, for symptomatic patients (those with documented vascular disease), there was a negligible, but significant reduction in primary end points.
The result was that in patients with documented vascular disease, aspirin plus clopidogrel combination therapy provided little or no benefit over aspirin alone. For patients with elevated risk factors but no documented vascular burden, there may actually be harm from combination therapy.
PRoFESS.33 Logically following is the question of whether aspirin plus dipyridamole offers any benefit over clopidogrel as a stroke prophylactic. The Prevention Regimen for Effectively Avoiding Second Strokes trial hoped to answer this by comparing clopidogrel against aspirin plus dipyridamole, both with and without telmisartan, in patients with recent stroke.
The rate of recurrent stroke was similar in the two groups, but there were 25 fewer ischemic strokes in patients on aspirin plus dipyridamole, offset by an increase in hemorrhagic strokes. Rates of secondary outcomes of stroke, death, or MI were nearly identical between the groups. Early discontinuation of treatment was significantly more frequent in those patients taking aspirin plus dipyridamole, meaning better compliance for those taking clopidogrel.
Initially, patients were to be randomized to either aspirin plus dipyridamole or aspirin plus clopidogrel. However, after MATCH30 demonstrated a significantly higher bleeding risk with aspirin plus clopidogrel, patients were changed to clopidogrel alone. But despite this, the bleeding risk was still higher with aspirin plus dipyridamole.
During the trial, the entry criteria were expanded, allowing for the inclusion of younger patients and those with less recent strokes; but despite this change, the study remained underpowered to demonstrate its goal of noninferiority. Thus, it showed only a trend of noninferiority of clopidogrel vs aspirin plus dipyridamole.
What the clopidogrel trials tell us
Clopidogrel confers a benefit similar to that of aspirin (as shown in the CAPRIE study).29 Although aspirin plus dipyridamole confers greater benefit than aspirin alone (as shown in the ESPS-2,14 Leonardi-Bee,25 and ESPRIT26 studies), aspirin plus dipyridamole is not superior to clopidogrel, and may even be inferior.34
WARFARIN FOR ATRIAL FIBRILLATION ONLY
Warfarin acts by disrupting the coagulation cascade rather than acting at the site of platelet plug formation. In theory, warfarin should be as effective as the antiplatelet drugs in preventing clot formation, and so it was thought to possibly be effective in preventing stroke of arterial origin.
However, in at least three studies, warfarin increased the risk of death, MI, and hemorrhage, with perhaps a slight decrease in the risk of recurrent stroke in patients with suspected stroke or TIA.35–37 This should be differentiated from stroke originating from cardiac dysrhythmias, for which warfarin has clearly been shown to be beneficial.28
THREE GOOD MEDICAL OPTIONS FOR PREVENTING STROKE RECURRENCE
Antiplatelet therapy offers benefit in the primary and secondary prevention of stroke, with a 25% reduction in the rate of nonfatal stroke and a 17% reduction in the rate of death due to vascular causes.15
Aspirin is the best established
Aspirin is the best established, best tolerated, and least expensive of the three contemporary agents. Further, it is also the agent of choice for acute stroke care, to be given within 48 hours of a stroke to mitigate the risk of death and morbidity. The data for other agents in acute stroke management remain limited.38
Aspirin plus dipyridamole
Aspirin plus dipyridamole is slightly more efficacious than aspirin alone, and it is an alternative when aspirin is ineffective and when the patient can afford the additional cost. Aspirin plus dipyridamole offers up to a 22% relative risk reduction (but a small reduction in absolute risk) of stroke compared with aspirin alone, as demonstrated by ESPS-2,14 Leonardi-Bee et al,25 and ESPRIT.26
When is clopidogrel appropriate?
Up to one-third of patients may not tolerate aspirin plus dipyridamole because of side effects. Clopidogrel is an option for these patients. The CAPRIE study29 showed clopidogrel similar in efficacy to aspirin.
In contrast to aspirin plus dipyridamole, there is clearly no benefit to combining aspirin and clopidogrel for ischemic stroke prophylaxis. And data from PRoFESS33 suggested the combination was qualitatively inferior to aspirin plus dipyridamole. However, the PRoFESS trial was underpowered to fully bear this out.
Therefore, current guidelines consider all three agents as appropriate for secondary prevention of stroke. One is not preferred over another, and the selection should be based on individual patient characteristics and affordability.28
CAROTID SURGERY OR STENTING: BENEFITS AND LIMITATIONS
Atherosclerosis is the most common cause of stroke, and atherosclerosis of the common carotid bifurcation accounts for a small but significant percentage of all strokes.39–41
The degree of carotid stenosis and whether it is producing symptoms influence how it should be managed. For patients with symptomatic carotid stenosis of more than 70%, multicenter randomized trials have shown that surgery (ie, carotid endarterectomy) added to medical therapy decreases the rate of recurrent stroke by up to 17% and the rate of combined stroke and death by 10% to 12% over a 2- to 3-year follow-up period (level of evidence A).42–44 No study has proven the efficacy of surgery in patients with symptomatic stenosis of less than 50%.43,44
Similarly, in asymptomatic carotid disease, preventive surgery is a beneficial adjunct to medical therapy in certain patients. An approximate 6% reduction in the rate of stroke or death over 5 years has been shown in patients with moderate stenosis (> 60%), with men younger than age 75 and with greater than 70% stenosis deriving the most benefit.45–47
However, these robust, positive results with surgical intervention should not overshadow the importance of intensive and guided medical therapy, which has been shown to mitigate the risk of stroke.48,49
Is stenting as good as surgery? In the multicenter randomized Carotid Revascularization Endarterectomy vs Stenting Trial (CREST), stenting resulted in similar rates of stroke and MI in patients with symptomatic and asymptomatic disease.50 However, stenting carried a greater risk of perioperative stroke, and endarterectomy carried a greater risk of MI. Those under age 70 benefited more from stenting, and those over age 70 benefited more from endarterectomy.
But another fact to keep in mind is that the relationship between carotid narrowing and an ipsilateral stroke is not necessarily direct. Two follow-up studies in patients from the North American Symptomatic Carotid Endarterectomy Trial (NASCET) found that up to 45% of strokes that occurred after intervention in the distribution of the asymptomatic stenosed carotid artery were unrelated to the stenosis.51,52 Moreover, up to 20% of subsequent strokes in the distribution of the symptomatic artery were not of large-artery origin, increasing up to 35% for those with stenosis of less than 70%.51 Clearly, thorough screening of those with presumed symptomatic stenosis is needed to eliminate other possible causes.
After a stroke, an important goal is to prevent another one.1,2 And for patients who have had an ischemic stroke or transient ischemic attack (TIA) due to atherosclerosis, an important part of secondary preventive therapy is a drug that inhibits platelets—ie, aspirin, extended-release dipyridamole, or clopidogrel. This has taken years to establish.
In the following pages, we discuss the antiplatelet agents that have been shown to be beneficial after stroke of atherosclerotic origin, and we briefly review the indications for surgery and stenting for the subset of patients whose strokes are caused by symptomatic carotid disease.
(Although managing modifiable risk factors such as smoking, hypertension, diabetes, and dyslipidemia is also important, we will not cover this topic here, nor will we talk about hemorrhagic stroke or stroke due to atrial fibrillation. Also not discussed here is cilostazol, which, although shown to be effective in preventing recurrent stroke when compared with placebo and aspirin,3,4 has not been approved for this use by the US Food and Drug Administration, as of this writing.)
HOW WE REVIEWED THE LITERATURE
We searched PubMed using the terms aspirin, acetylsalicylic acid, clopidogrel, and/or dipyridamole, in combination with stroke, cerebral ische(ae)mia, transient ische(ae)mic attacks, or retinal artery occlusion. We reviewed only clinical trials or meta-analyses of these drugs for either primary or secondary prevention of cerebrovascular disease.
As our aim was to review the topic and not to perform a meta-analysis, no cutoffs were used to exclude trials. The references in the selected papers were also reviewed to expand the articles. Finally, the references in the current American Heart Association and American Stroke Association secondary stroke prevention guideline were also reviewed.
For a summary of the trials included in our review, see the Data Supplement as an appendix to the online version of this article.
ASPIRIN: THE GOLD STANDARD
Prescribed by Hippocrates in the form of willow bark extract, aspirin has long been known for its antipyretic and anti-inflammatory properties. Its antiplatelet and antithrombotic properties, first described in 1967 by Weiss and Aledort,5 are mediated by irreversible inhibition of cyclooxygenase, leading to decreased thromboxane A2, a platelet-aggregation activator.
Fields et al,6,7 in 1977 and 1978, reported that in a controlled trial in patients with TIA or monocular blindness, fewer subsequent TIAs occurred in patients who received aspirin, although the difference was not statistically significant, with lower rates of events only in nonsurgical patients. Over the next 20 years, the results remained mixed.
The Danish Cooperative study8 (1983) found no significant difference in the rate of recurrent stroke with aspirin vs placebo.
AICLA.9 The Accidents Ischémiques Cérébraux Liés à l’Athérosclérose study of 1983 did find a difference. However, both the Danish Cooperative study and the AICLA were limited by lacking standardized computed tomographic imaging to rule out hemorrhagic stroke and by being relatively small.
The Swedish Cooperative Study10 (1987) found no statistical difference between high-dose aspirin and placebo in preventing recurrent vascular events (stroke, TIA, or myocardial infarction [MI]) 1 to 3 weeks after a stroke. However, it had several limitations: the aspirin group contained more patients with ischemic heart disease (who are more likely to die of cardiac causes), there were significantly more men in the aspirin group, and nearly one-fourth of the deaths were a result of the initial stroke, potentially masking the effect of aspirin in secondary prevention.
Later studies began to show a consistently favorable effect of aspirin.
Boysen et al11 in 1988 reported a nonsignificant trend toward fewer adverse events with aspirin.
UK-TIA.12 The United Kingdom Transient Ischaemic Attack trial in 1991 found a similar trend.
SALT.13 The Swedish Aspirin Low-dose Trial, also in 1991, showed a significant 18% lower rate of stroke or death in patients with recent TIA, minor stroke, or retinal occlusion treated with low-dose aspirin. The inclusion of patients with TIA helped broaden the population that might benefit. However, the study may have favored the aspirin group by having a run-in period in which patients were nonrandomly treated either with aspirin or with anticoagulation at the discretion of the patient’s physician and, if they suffered “several” TIAs, a stroke, retinal artery occlusion, or MI, were removed from the study.
ESPS-2.14 The second European Stroke Prevention Study in 1996 added to the evidence that aspirin prevents recurrent stroke. Patients with a history of TIA or stroke were randomized in double-blind fashion to four treatment groups: placebo, low-dose aspirin, dipyridamole, or aspirin plus dipyridamole. At 2 years, strokes had occurred in 18% fewer patients in the aspirin group than in the placebo group, and TIAs had occurred in 21.9% fewer. However, aspirin was associated with an absolute 0.5% increase in severe and fatal bleeding. The power of the study was limited because patients from one center were excluded because of “serious inconsistencies in patient case record forms and compliance assay determinations.” 14
Comment. The mixed results with aspirin in studies predating ESPS-2 were partly because the study populations were too small to show benefit.
ATT.15 The Antithrombotic Trialists’ Collaboration performed a meta-analysis that conclusively confirmed the benefit of aspirin after stroke or TIA. The investigators analyzed individual patient data pooled from randomized controlled trials published before 1997 that compared antiplatelet regimens (mostly aspirin) against placebo and against each other. The rates of vascular events were 10.7% with treatment vs 13.2% with placebo (P < .0001). Antiplatelet therapy was particularly effective in preventing ischemic stroke, with a 25% reduction in the rate of nonfatal stroke, and with an overall absolute benefit in stroke prevention across all high-risk patient groups. This translated to 25 fewer nonfatal strokes per 1,000 patients treated with antiplatelet therapy.
What is the optimal aspirin dose?
Studies of aspirin have used different daily doses—the earliest studies used large doses of 1,000 to 1,500 mg.6–10
Boysen et al11 in 1988 found a trend toward benefit (not statistically significant) with doses ranging from 50 mg to 100 mg.
In 1991, three separate studies found that higher doses of aspirin were no more effective than lower doses.
The UK-TIA trial12 compared aspirin 300 mg vs 1,200 mg and found a higher risk of gastrointestinal bleeding with the higher dose.
The SALT Collaborative Group13 found 75 mg to be effective.
The Dutch TIA trial16 compared 30 mg vs 283 mg; end point outcomes were similar but the rate of adverse events was higher with 283 mg.
ESPS-2 was able to show efficacy at a dose of only 50 mg.14
Taylor et al17 compared lower doses (81 or 325 mg) vs higher doses (650 or 1,300 mg) for patients undergoing carotid endarterectomy and found that the risk of adverse events was twice as high with the higher doses.
The ATT Collaboration15 found that efficacy was 40% lower with the highest dose of aspirin than with the lowest doses.
Algra and van Gijn18 performed a meta-analysis of all these studies and found no difference in risk reduction between low-dose and high-dose aspirin, with an overall relative risk reduction of 13% at any dose above 30 mg.
Campbell et al,19 in a 2007 review, found that doses greater than 300 mg conferred no benefit, and that rapid and maximum suppression of thromboxane A2 can be achieved by chewing or ingesting dissolved forms of aspirin 162 mg.
Conclusion. Aspirin doses higher than 81 mg (the US standard) confer no greater benefit and may even decrease the efficacy of aspirin. In an emergency, rapid suppression of thromboxane A2 can be achieved by chewing a minimum dose of 162 mg.
DIPYRIDAMOLE CAN BE ADDED TO ASPIRIN
In 1967, Weiss and Aledort5 found that aspirin’s antiplatelet effect could be blocked by adenosine diphosphate, which is released by activated platelet cells and is an essential part of thrombus formation. Adjacent platelets are then activated, leading to up-regulation of thromboxane A2 and glycoprotein IIb/IIIa receptors and resulting in a cascade of platelet activation and clot formation.20 Dipyridamole inhibits aggregation of platelets by inhibiting their ability to take up adenosine diphosphate.
Studies of dipyridamole
AICLA.9 Bousser et al9 randomized patients who suffered one or more cerebral or retinal infarctions to receive placebo, aspirin 1 g, or aspirin 1 g plus dipyridamole 225 mg. Aspirin was significantly better than placebo in preventing a recurrence of stroke. The event rate with aspirin plus dipyridamole was similar to the rate with aspirin alone, although on 2-by-2 analysis, the difference between placebo and aspirin plus dipyridamole did not reach statistical significance. However, the rate of carotid-origin stroke was 17% with aspirin alone and 6% with aspirin plus dipyridamole, a statistically significant difference.
Thus, this study confirmed the benefit of aspirin in preventing ischemic events but did not fully support the addition of dipyridamole, except in preventing stroke of carotid origin. The study had a number of limitations: the sample size was small, TIA was not included as an end point, computed tomography was not required for entry, and many patients were lost to follow-up, decreasing the statistical power of the trial.
The ESPS study21 was also a randomized controlled trial of aspirin plus dipyridamole vs placebo. But unlike AICLA, ESPS included patients with TIA.
ESPS found a 38.1% relative risk reduction in stroke with aspirin plus dipyridamole compared with placebo, and a 30.6% reduction in death from all causes. Interestingly, patients who had a TIA as the qualifying event had a lower end-point incidence and larger end-point reduction than those who had a stroke as the qualifying event. However, ESPS did not resolve the question of whether adding dipyridamole to aspirin affords any benefit over aspirin alone.
ESPS-214 hoped to answer this question. Patients were randomized to placebo, aspirin, dipyridamole, or aspirin plus dipyridamole. On 2 × 2 analysis, the dipyridamole group had a 16% lower rate of recurrent stroke than the placebo group, and patients on aspirin plus dipyridamole had a 37% lower rate. Aspirin plus dipyridamole yielded a 23.1% reduction compared with aspirin alone, and a 24.7% reduction compared with dipyridamole alone. Similar benefit was reported for the end point of TIA with combination therapy compared with either agent alone.
However, nearly 25% of patients had to withdraw because of side effects, particularly in the dipyridamole-alone and aspirin-dipyridamole groups, and, as mentioned above, verification of compliance in the aspirin group was an issue.14,22 Nevertheless, ESPS-2 clearly showed that aspirin plus dipyridamole was better than either drug alone in preventing recurrent stroke. It also showed the effectiveness of dipyridamole, which AICLA and ESPS could not do, because it had a larger study population, used a lower dose of aspirin, and perhaps because it used an extended-release form of dipyridamole.23
The ATT meta-analysis15 showed a clear benefit of antiplatelet therapy. However, much of this benefit was derived from aspirin therapy, with the addition of dipyridamole resulting in a nonsignificant 6% reduction of vascular events. Most of the patients on dipyridamole were from the ESPS-2 study. In effect, the ATT was a meta-analysis of aspirin, as aspirin studies dominated at that time.
A Cochrane review24 publsihed in 2003 attempted to rectify this by analyzing randomized controlled trials of dipyridamole vs placebo.24 Like the ATT meta-analysis, it did not bear out the benefits of dipyridamole: compared with placebo, there was no effect on the rate of vascular death, and only a minimal benefit in reduction of vascular events—and this latter point is only because of the inclusion of ESPS-2.
Directly comparing aspirin plus dipyridamole vs aspirin alone, the reviewers found no effect on the rate of vascular death, and with the exclusion of ESPS-2, no effect on vascular events.
The Cochrane review had the same limitation as the ATT meta-analysis, ie, dependence on a single trial (ESPS-2) to show benefit, and perhaps the fact that ESPS-2 was the only study that used an extended-release form of dipyridamole.
Leonardi-Bee et al25 performed a meta-analysis that overcame the limitation of ESPS-2 being the only study at the time with positive findings: they used pooled individual patient data from randomized trials and analyzed them en masse. Patients on aspirin plus dipyridamole had a 39% lower risk than with placebo and a 22% lower risk than with aspirin alone. Unlike the ATT and the Cochrane review, excluding ESPS-2 did not alter the statistically significant lower stroke rate with aspirin plus dipyridamole compared with controls. This meta-analysis helped to confirm ESPS-2’s finding of the additive effect of aspirin plus dipyridamole compared with aspirin and placebo control.
ESPRIT.26,27 The European/Australasian Stroke Prevention in Reversible Ischaemia Trial confirmed these findings. This randomized controlled trial compared aspirin plus dipyridamole against aspirin alone in patients with a TIA or minor ischemic stroke of arterial origin within the past 6 months. For the primary end point (death from all vascular causes, nonfatal stroke, nonfatal MI, nonfatal major bleeding complication), the hazard ratio was 0.80 favoring aspirin plus dipyridamole, with a number needed to treat of 104 over a mean of 3.5 years (absolute risk reduction of 1% per year). Importantly, twice as many patients taking aspirin plus dipyridamole discontinued the medication.
Caveats to interpreting this study are that it was not blinded, the aspirin doses varied (although the median aspirin dose—75 mg—was the same between the two groups), and not all patients received the extended-release form of dipyridamole.
Conclusions about dipyridamole
ESPS-2, ESPRIT, and the meta-analysis by Leonardi-Bee et al showed that aspirin plus dipyridamole is more effective than placebo or aspirin alone in secondary prevention of vascular events, including stroke. Also, extended-release dipyridamole appears to be more effective.
Unfortunately, many patients stop taking dipyridamole because of side effects (primarily headache).
Based on the results of ESPRIT, the absolute benefit of dipyridamole used alone may be small.
CLOPIDOGREL: SIMILAR TO ASPIRIN IN EFFICACY?
Like dipyridamole, clopidogrel targets adenosine diphosphate to prevent clot formation, blocking its ability to bind to its receptor on platelets. It is a thienopyridine and, unlike its sister drug ticlopidine, does not seem to be associated with the potentially serious side effects of neutropenia. However, a few cases of thrombotic thrombocytopenic purpura have been reported.28 The other drugs in this class have not been evaluated in clinical trials for secondary stroke prophylaxis.
Trials of clopidogrel
CAPRIE.29 The Clopidogrel Versus Aspirin in Patients at Risk of Ischaemic Events trial, in 1996, was one of the first to compare the clinical use of clopidogrel against aspirin. It was a randomized controlled noninferiority trial in patients over age 21 (inclusion criteria: ischemic stroke, MI, or peripheral arterial disease) randomized to aspirin 325 mg once daily or clopidogrel 75 mg once daily. Patients were followed for 1 to 3 years.
Patients on clopidogrel had a relative risk reduction of 8.7% in primary events (ischemic stroke, MI, or vascular death); patients on aspirin were at significantly higher risk of gastrointestinal hemorrhage. Patients with peripheral arterial disease as the qualifying event did particularly well on clopidogrel, with a significant relative risk reduction of 23.8%.
Limitations of the CAPRIE trial included its inability to measure the effect of treatment on individual outcomes, particularly stroke, and the fact that the relative risk reduction for patients with stroke as the qualifying event was not significant (P = .66). Another limitation was that it did not use TIA as an entry criterion or as part of the composite outcome. Also, the relative risk reduction had a wide confidence interval, and a large number of patients discontinued therapy for reasons other than the defined outcomes.
Nevertheless, the CAPRIE trial showed clopidogrel to be an effective antiplatelet prophylactic, particularly in patients with peripheral artery disease, but with no discernible difference from aspirin for those patients with MI or stroke as a qualifying event.
MATCH.30 The Management of Atherothrombosis With Clopidogrel in High-risk Patients trial hoped to better assess clopidogrel’s efficacy, particularly in patients with ischemic cerebral events. Cardiac studies leading up to MATCH suggested that adding a thienopyridine to aspirin might offer additive benefit in reducing the rate of vascular outcomes.15,31 MATCH randomized high-risk patients (inclusion criteria were ischemic stroke or TIA and a history of vascular disease) to clopidogrel or to aspirin plus clopidogrel.
There was a nonsignificant 6.4% relative risk reduction in the combined primary outcome of MI, ischemic stroke, vascular death, other vascular death, and re-hospitalization for acute ischemic events in the aspirin-plus-clopidogrel group compared with clopidogrel alone. However, this came at the cost of double the number of bleeding events in the combination group, mitigating most of the benefit of combination therapy.
An important caveat in interpreting the results of MATCH, as compared with the Clopidogrel in Unstable Angina to Prevent Recurrent Events (CURE) study, is that aspirin was being added to clopidogrel, not vice versa. CURE, which looked at the addition of clopidogrel to aspirin vs aspirin alone in cardiac patients, found a significant reduction of ischemic events taken as a group (relative risk 0.8), and a trend toward a lower rate of stroke (relative risk 0.86, but 95% confidence interval encompassing 1) for aspirin plus clopidogrel vs aspirin alone.31 However, patients in the CURE trial did not have high-risk vasculopathy per se but rather non-ST-elevation MI, perhaps skewing the benefit of combination therapy and lessening the risk of intracranial bleeding.
CHARISMA.32 The Clopidogrel for High Atherothrombotic Risk and Ischemic Stabilization, Management, and Avoidance trial, like the CURE trial, compared aspirin plus clopidogrel vs aspirin in patients with established cardiovascular, cerebrovascular, or peripheral arterial disease, or who were at high risk of events. As in the MATCH study, the findings for combination therapy were a nonsignificant relative risk of 0.93 for primary events (MI, stroke, or death from cardiovascular causes), and a significant reduction of secondary end points (primary end point event plus TIA or hospitalization for unstable angina) (relative risk 0.92, P = .04).
Importantly, combination therapy significantly increased the rate of bleeding events. In asymptomatic patients (those without documented vascular disease but with multiple atherothrombotic risk factors), there was actually harm with combined treatment. Conversely, for symptomatic patients (those with documented vascular disease), there was a negligible, but significant reduction in primary end points.
The result was that in patients with documented vascular disease, aspirin plus clopidogrel combination therapy provided little or no benefit over aspirin alone. For patients with elevated risk factors but no documented vascular burden, there may actually be harm from combination therapy.
PRoFESS.33 Logically following is the question of whether aspirin plus dipyridamole offers any benefit over clopidogrel as a stroke prophylactic. The Prevention Regimen for Effectively Avoiding Second Strokes trial hoped to answer this by comparing clopidogrel against aspirin plus dipyridamole, both with and without telmisartan, in patients with recent stroke.
The rate of recurrent stroke was similar in the two groups, but there were 25 fewer ischemic strokes in patients on aspirin plus dipyridamole, offset by an increase in hemorrhagic strokes. Rates of secondary outcomes of stroke, death, or MI were nearly identical between the groups. Early discontinuation of treatment was significantly more frequent in those patients taking aspirin plus dipyridamole, meaning better compliance for those taking clopidogrel.
Initially, patients were to be randomized to either aspirin plus dipyridamole or aspirin plus clopidogrel. However, after MATCH30 demonstrated a significantly higher bleeding risk with aspirin plus clopidogrel, patients were changed to clopidogrel alone. But despite this, the bleeding risk was still higher with aspirin plus dipyridamole.
During the trial, the entry criteria were expanded, allowing for the inclusion of younger patients and those with less recent strokes; but despite this change, the study remained underpowered to demonstrate its goal of noninferiority. Thus, it showed only a trend of noninferiority of clopidogrel vs aspirin plus dipyridamole.
What the clopidogrel trials tell us
Clopidogrel confers a benefit similar to that of aspirin (as shown in the CAPRIE study).29 Although aspirin plus dipyridamole confers greater benefit than aspirin alone (as shown in the ESPS-2,14 Leonardi-Bee,25 and ESPRIT26 studies), aspirin plus dipyridamole is not superior to clopidogrel, and may even be inferior.34
WARFARIN FOR ATRIAL FIBRILLATION ONLY
Warfarin acts by disrupting the coagulation cascade rather than acting at the site of platelet plug formation. In theory, warfarin should be as effective as the antiplatelet drugs in preventing clot formation, and so it was thought to possibly be effective in preventing stroke of arterial origin.
However, in at least three studies, warfarin increased the risk of death, MI, and hemorrhage, with perhaps a slight decrease in the risk of recurrent stroke in patients with suspected stroke or TIA.35–37 This should be differentiated from stroke originating from cardiac dysrhythmias, for which warfarin has clearly been shown to be beneficial.28
THREE GOOD MEDICAL OPTIONS FOR PREVENTING STROKE RECURRENCE
Antiplatelet therapy offers benefit in the primary and secondary prevention of stroke, with a 25% reduction in the rate of nonfatal stroke and a 17% reduction in the rate of death due to vascular causes.15
Aspirin is the best established
Aspirin is the best established, best tolerated, and least expensive of the three contemporary agents. Further, it is also the agent of choice for acute stroke care, to be given within 48 hours of a stroke to mitigate the risk of death and morbidity. The data for other agents in acute stroke management remain limited.38
Aspirin plus dipyridamole
Aspirin plus dipyridamole is slightly more efficacious than aspirin alone, and it is an alternative when aspirin is ineffective and when the patient can afford the additional cost. Aspirin plus dipyridamole offers up to a 22% relative risk reduction (but a small reduction in absolute risk) of stroke compared with aspirin alone, as demonstrated by ESPS-2,14 Leonardi-Bee et al,25 and ESPRIT.26
When is clopidogrel appropriate?
Up to one-third of patients may not tolerate aspirin plus dipyridamole because of side effects. Clopidogrel is an option for these patients. The CAPRIE study29 showed clopidogrel similar in efficacy to aspirin.
In contrast to aspirin plus dipyridamole, there is clearly no benefit to combining aspirin and clopidogrel for ischemic stroke prophylaxis. And data from PRoFESS33 suggested the combination was qualitatively inferior to aspirin plus dipyridamole. However, the PRoFESS trial was underpowered to fully bear this out.
Therefore, current guidelines consider all three agents as appropriate for secondary prevention of stroke. One is not preferred over another, and the selection should be based on individual patient characteristics and affordability.28
CAROTID SURGERY OR STENTING: BENEFITS AND LIMITATIONS
Atherosclerosis is the most common cause of stroke, and atherosclerosis of the common carotid bifurcation accounts for a small but significant percentage of all strokes.39–41
The degree of carotid stenosis and whether it is producing symptoms influence how it should be managed. For patients with symptomatic carotid stenosis of more than 70%, multicenter randomized trials have shown that surgery (ie, carotid endarterectomy) added to medical therapy decreases the rate of recurrent stroke by up to 17% and the rate of combined stroke and death by 10% to 12% over a 2- to 3-year follow-up period (level of evidence A).42–44 No study has proven the efficacy of surgery in patients with symptomatic stenosis of less than 50%.43,44
Similarly, in asymptomatic carotid disease, preventive surgery is a beneficial adjunct to medical therapy in certain patients. An approximate 6% reduction in the rate of stroke or death over 5 years has been shown in patients with moderate stenosis (> 60%), with men younger than age 75 and with greater than 70% stenosis deriving the most benefit.45–47
However, these robust, positive results with surgical intervention should not overshadow the importance of intensive and guided medical therapy, which has been shown to mitigate the risk of stroke.48,49
Is stenting as good as surgery? In the multicenter randomized Carotid Revascularization Endarterectomy vs Stenting Trial (CREST), stenting resulted in similar rates of stroke and MI in patients with symptomatic and asymptomatic disease.50 However, stenting carried a greater risk of perioperative stroke, and endarterectomy carried a greater risk of MI. Those under age 70 benefited more from stenting, and those over age 70 benefited more from endarterectomy.
But another fact to keep in mind is that the relationship between carotid narrowing and an ipsilateral stroke is not necessarily direct. Two follow-up studies in patients from the North American Symptomatic Carotid Endarterectomy Trial (NASCET) found that up to 45% of strokes that occurred after intervention in the distribution of the asymptomatic stenosed carotid artery were unrelated to the stenosis.51,52 Moreover, up to 20% of subsequent strokes in the distribution of the symptomatic artery were not of large-artery origin, increasing up to 35% for those with stenosis of less than 70%.51 Clearly, thorough screening of those with presumed symptomatic stenosis is needed to eliminate other possible causes.
- Zivin JA. Approach to cerebrovascular diseases. In:Goldman L, Schafer AI, editors. Goldman’s Cecil Medicine. 24th ed. Philadelphia, PA: Elsevier, 2012:2304–2309.
- Samsa GP, Bian J, Lipscomb J, Matchar DB. Epidemiology of recurrent cerebral infarction: a Medicare claims-based comparison of first and recurrent strokes on 2-year survival and cost. Stroke 1999; 30:338–349.
- Gotoh F, Tohgi H, Hirai S, et al. Cilostazol Stroke Prevention Study: a placebo-controlled double-blind trial for secondary prevention of cerebral infarction. J Stroke Cerebrovasc Dis 2000; 9:147–157.
- Shinohara Y, Katayama Y, Uchiyama S, et al; CSPS 2 group. Cilostazol for prevention of secondary stroke (CSPS 2): an aspirin-controlled, double-blind, randomised non-inferiority trial. Lancet Neurol 2010; 9:959–968.
- Weiss HJ, Aledort LM. Impaired platelet-connective-tissue reaction in man after aspirin ingestion. Lancet 1967; 2:495–497.
- Fields WS, Lemak NA, Frankowski RF, Hardy RJ. Controlled trial of aspirin in cerebral ischemia. Stroke 1977; 8:301–314.
- Fields WS, Lemak NA, Frankowski RF, Hardy RJ. Controlled trial of aspirin in cerebral ischemia. Part II: surgical group. Stroke 1978; 9:309–319.
- Sorensen PS, Pedersen H, Marquardsen J, et al. Acetylsalicylic acid in the prevention of stroke in patients with reversible cerebral ischemic attacks. A Danish cooperative study. Stroke 1983; 14:15–22.
- Bousser MG, Eschwege E, Haguenau M, et al. “AICLA” controlled trial of aspirin and dipyridamole in the secondary prevention of athero-thrombotic cerebral ischemia. Stroke 1983; 14:5–14.
- High-dose acetylsalicylic acid after cerebral infarction. A Swedish Cooperative Study. Stroke 1987; 18:325–334.
- Boysen G, Sørensen PS, Juhler M, et al. Danish very-low-dose aspirin after carotid endarterectomy trial. Stroke 1988; 19:1211–1215.
- Farrell B, Godwin J, Richards S, Warlow C. The United Kingdom transient ischaemic attack (UK-TIA) aspirin trial: final results. J Neurol Neurosurg Psychiatry 1991; 54:1044–1054.
- Swedish Aspirin Low-Dose Trial (SALT) of 75 mg aspirin as secondary prophylaxis after cerebrovascular ischaemic events. The SALT Collaborative Group. Lancet 1991; 338:1345–1349.
- Diener HC, Cunha L, Forbes C, Sivenius J, Smets P, Lowenthal A. European Stroke Prevention Study. 2. Dipyridamole and acetylsalicylic acid in the secondary prevention of stroke. J Neurol Sci 1996; 143:1–13.
- Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, MI, and stroke in high risk patients. BMJ 2002; 324:71–86.
- A comparison of two doses of aspirin (30 mg vs. 283 mg a day) in patients after a transient ischemic attack or minor ischemic stroke. The Dutch TIA Trial Study Group. N Engl J Med 1991; 325:1261–1266.
- Taylor DW, Barnett HJ, Haynes RB, et al. Low-dose and high-dose acetylsalicylic acid for patients undergoing carotid endarterectomy: a randomised controlled trial. ASA and Carotid Endarterectomy (ACE) Trial Collaborators. Lancet 1999; 353:2179–2184.
- Algra A, van Gijn J. Aspirin at any dose above 30 mg offers only modest protection after cerebral ischaemia. J Neurol Neurosurg Psychiatry 1996; 60:197–199.
- Campbell CL, Smyth S, Montalescot G, Steinhubl SR. Aspirin dose for the prevention of cardiovascular disease: a systematic review. JAMA 2007; 297:2018–2024.
- Weiss HJ, Aledort LM, Kochwa S. The effect of salicylates on the hemostatic properties of platelets in man. J Clin Invest 1968; 47:2169–2180.
- European Stroke Prevention Study. ESPS Group. Stroke 1990; 21:1122–1130.
- Davis SM, Donnan GA. Secondary prevention for stroke after CAPRIE and ESPS-2. Opinion 1. Cerebrovasc Dis 1998; 8:73–77.
- Diener HC. Dipyridamole trials in stroke prevention. Neurology 1998; 51(suppl 3):S17–S19.
- De Schryver EL, Algra A, van Gijn J. Cochrane review: dipyridamole for preventing major vascular events in patients with vascular disease. Stroke 2003; 34:2072–2080.
- Leonardi-Bee J, Bath PM, Bousser MG, et al; Dipyridamole in Stroke Collaboration (DISC). Dipyridamole for preventing recurrent ischemic stroke and other vascular events: a meta-analysis of individual patient data from randomized controlled trials. Stroke 2005; 36:162–168.
- ESPRIT Study Group; Halkes PH, van Gijn J, Kappelle LJ, Koudstaal PJ, Algra A. Aspirin plus dipyridamole versus aspirin alone after cerebral ischaemia of arterial origin (ESPRIT): randomised controlled trial. Lancet 2006; 367:1665–1673.
- Tirschwell D. Aspirin plus dipyridamole was more effective than aspirin alone for preventing vascular events after minor cerebral ischemia. ACP J Club 2006; 145:57.
- Furie KL, Kasner SE, Adams RJ, et al; American Heart Association Stroke Council, Council on Cardiovascular Nursing, Council on Clinical Cardiology, and Interdisciplinary Council on Quality of Care and Outcomes Research. Guidelines for the prevention of stroke in patients with stroke or transient ischemic attack: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2011; 42:227–276.
- CAPRIE Steering Committee. A randomised, blinded, trial of clopidogrel versus aspirin in patients at risk of ischaemic events (CAPRIE). CAPRIE Steering Committee. Lancet 1996; 348:1329–1339.
- Diener HC, Bogousslavsky J, Brass LM, et al; MATCH investigators. Aspirin and clopidogrel compared with clopidogrel alone after recent ischaemic stroke or transient ischaemic attack in high-risk patients (MATCH): randomised, double-blind, placebo-controlled trial. Lancet 2004; 364:331–337.
- Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK; Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial Investigators. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med 2001; 345:494–502.
- Bhatt DL, Fox KA, Hacke W, et al; CHARISMA Investigators. Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events. N Engl J Med 2006; 354:1706–1717.
- Sacco RL, Diener HC, Yusuf S, et al; PRoFESS Study Group. Aspirin and extended-release dipyridamole versus clopidogrel for recurrent stroke. N Engl J Med 2008; 359:1238–1251.
- Kent DM, Thaler DE. Stroke prevention—insights from incoherence. N Engl J Med 2008; 359:1287–1289.
- Chimowitz MI, Lynn MJ, Howlett-Smith H, et al; Warfarin-Aspirin Symptomatic Intracranial Disease Trial Investigators. Comparison of warfarin and aspirin for symptomatic intracranial arterial stenosis. N Engl J Med 2005; 352:1305–1316.
- ESPRIT Study Group; Halkes PH, van Gijn J, Kappelle LJ, Koudstaal PJ, Algra A. Medium intensity oral anticoagulants versus aspirin after cerebral ischaemia of arterial origin (ESPRIT): a randomised controlled trial. Lancet Neurol 2007; 6:115–124.
- Mohr JP, Thompson JL, Lazar RM, et al; Warfarin-Aspirin Recurrent Stroke Study Group. A comparison of warfarin and aspirin for the prevention of recurrent ischemic stroke. N Engl J Med 2001; 345:1444–1451.
- Jauch EC, Saver JL, Adams HP, et al; American Heart Association Stroke Council; Council on Cardiovascular Nursing; Council on Peripheral Vascular Disease; Council on Clinical Cardiology. Guidelines for the early management of patients with acute ischemic stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2013; 44:870–947.
- Kolominsky-Rabas PL, Weber M, Gefeller O, Neundoerfer B, Heuschmann PU. Epidemiology of ischemic stroke subtypes according to TOAST criteria: incidence, recurrence, and long-term survival in ischemic stroke subtypes: a population-based study. Stroke 2001; 32:2735–2740.
- Zivin JA. Ischemic cerebrovascular disease. In:Goldman L, Schafer AI, editors. Goldman’s Cecil Medicine 24th ed. Philadelphia, PA: Elsevier; 2012: chap 414. www.mdconsult.com. Accessed November 7, 2013.
- Smith WS, Johnston C, Easton D. Cerebrovascular diseases. In:Kasper DL, Braunwald E, Fauci AS, Hauser SL, Longo DL, Jameson JL, editors. Harrison’s Principles of Internal Medicine. 16th ed. New York, NY: McGraw Hill; 2005: chap 349. www.accessmedicine.com.
- Beneficial effect of carotid endarterectomy in symptomatic patients with high-grade carotid stenosis. North American Symptomatic Carotid Endarterectomy Trial Collaborators. N Engl J Med 1991; 325:445–453.
- Randomised trial of endarterectomy for recently symptomatic carotid stenosis: final results of the MRC European Carotid Surgery Trial (ECST). Lancet 1998; 351:1379–1387.
- Barnett HJ, Taylor DW, Eliasziw M, et al. Benefit of carotid endarterectomy in patients with symptomatic moderate or severe stenosis. North American Symptomatic Carotid Endarterectomy Trial Collaborators. N Engl J Med 1998; 339:1415–1425.
- Hobson RW, Weiss DG, Fields WS, et al. Efficacy of carotid endarterectomy for asymptomatic carotid stenosis. The Veterans Affairs Cooperative Study Group. N Engl J Med 1993; 328:221–227.
- Endarterectomy for asymptomatic carotid artery stenosis. Executive Committee for the Asymptomatic Carotid Atherosclerosis Study. JAMA 1995; 273:1421–1428.
- Halliday A, Mansfield A, Marro J, et al; MRC Asymptomatic Carotid Surgery Trial (ACST) Collaborative Group. Prevention of disabling and fatal strokes by successful carotid endarterectomy in patients without recent neurological symptoms: randomised controlled trial. Lancet 2004; 363:1491–1502.
- Marquardt L, Geraghty OC, Mehta Z, Rothwell PM. Low risk of ipsilateral stroke in patients with asymptomatic carotid stenosis on best medical treatment: a prospective, population-based study. Stroke 2010; 41:e11–e17.
- Spence JD, Coates V, Li H, et al. Effects of intensive medical therapy on microemboli and cardiovascular risk in asymptomatic carotid stenosis. Arch Neurol 2010; 67:180–186.
- Brott TG, Hobson RW, Howard G, et al; CREST Investigators. Stenting versus endarterectomy for treatment of carotid-artery stenosis. N Engl J Med 2010; 363:11–23.
- Barnett HJ, Gunton RW, Eliasziw M, et al. Causes and severity of ischemic stroke in patients with internal carotid artery stenosis. JAMA 2000; 283:1429–1436.
- Inzitari D, Eliasziw M, Gates P, et al. The causes and risk of stroke in patients with asymptomatic internal-carotid-artery stenosis. North American Symptomatic Carotid Endarterectomy Trial Collaborators. N Engl J Med 2000; 342:1693–1700.
- Zivin JA. Approach to cerebrovascular diseases. In:Goldman L, Schafer AI, editors. Goldman’s Cecil Medicine. 24th ed. Philadelphia, PA: Elsevier, 2012:2304–2309.
- Samsa GP, Bian J, Lipscomb J, Matchar DB. Epidemiology of recurrent cerebral infarction: a Medicare claims-based comparison of first and recurrent strokes on 2-year survival and cost. Stroke 1999; 30:338–349.
- Gotoh F, Tohgi H, Hirai S, et al. Cilostazol Stroke Prevention Study: a placebo-controlled double-blind trial for secondary prevention of cerebral infarction. J Stroke Cerebrovasc Dis 2000; 9:147–157.
- Shinohara Y, Katayama Y, Uchiyama S, et al; CSPS 2 group. Cilostazol for prevention of secondary stroke (CSPS 2): an aspirin-controlled, double-blind, randomised non-inferiority trial. Lancet Neurol 2010; 9:959–968.
- Weiss HJ, Aledort LM. Impaired platelet-connective-tissue reaction in man after aspirin ingestion. Lancet 1967; 2:495–497.
- Fields WS, Lemak NA, Frankowski RF, Hardy RJ. Controlled trial of aspirin in cerebral ischemia. Stroke 1977; 8:301–314.
- Fields WS, Lemak NA, Frankowski RF, Hardy RJ. Controlled trial of aspirin in cerebral ischemia. Part II: surgical group. Stroke 1978; 9:309–319.
- Sorensen PS, Pedersen H, Marquardsen J, et al. Acetylsalicylic acid in the prevention of stroke in patients with reversible cerebral ischemic attacks. A Danish cooperative study. Stroke 1983; 14:15–22.
- Bousser MG, Eschwege E, Haguenau M, et al. “AICLA” controlled trial of aspirin and dipyridamole in the secondary prevention of athero-thrombotic cerebral ischemia. Stroke 1983; 14:5–14.
- High-dose acetylsalicylic acid after cerebral infarction. A Swedish Cooperative Study. Stroke 1987; 18:325–334.
- Boysen G, Sørensen PS, Juhler M, et al. Danish very-low-dose aspirin after carotid endarterectomy trial. Stroke 1988; 19:1211–1215.
- Farrell B, Godwin J, Richards S, Warlow C. The United Kingdom transient ischaemic attack (UK-TIA) aspirin trial: final results. J Neurol Neurosurg Psychiatry 1991; 54:1044–1054.
- Swedish Aspirin Low-Dose Trial (SALT) of 75 mg aspirin as secondary prophylaxis after cerebrovascular ischaemic events. The SALT Collaborative Group. Lancet 1991; 338:1345–1349.
- Diener HC, Cunha L, Forbes C, Sivenius J, Smets P, Lowenthal A. European Stroke Prevention Study. 2. Dipyridamole and acetylsalicylic acid in the secondary prevention of stroke. J Neurol Sci 1996; 143:1–13.
- Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, MI, and stroke in high risk patients. BMJ 2002; 324:71–86.
- A comparison of two doses of aspirin (30 mg vs. 283 mg a day) in patients after a transient ischemic attack or minor ischemic stroke. The Dutch TIA Trial Study Group. N Engl J Med 1991; 325:1261–1266.
- Taylor DW, Barnett HJ, Haynes RB, et al. Low-dose and high-dose acetylsalicylic acid for patients undergoing carotid endarterectomy: a randomised controlled trial. ASA and Carotid Endarterectomy (ACE) Trial Collaborators. Lancet 1999; 353:2179–2184.
- Algra A, van Gijn J. Aspirin at any dose above 30 mg offers only modest protection after cerebral ischaemia. J Neurol Neurosurg Psychiatry 1996; 60:197–199.
- Campbell CL, Smyth S, Montalescot G, Steinhubl SR. Aspirin dose for the prevention of cardiovascular disease: a systematic review. JAMA 2007; 297:2018–2024.
- Weiss HJ, Aledort LM, Kochwa S. The effect of salicylates on the hemostatic properties of platelets in man. J Clin Invest 1968; 47:2169–2180.
- European Stroke Prevention Study. ESPS Group. Stroke 1990; 21:1122–1130.
- Davis SM, Donnan GA. Secondary prevention for stroke after CAPRIE and ESPS-2. Opinion 1. Cerebrovasc Dis 1998; 8:73–77.
- Diener HC. Dipyridamole trials in stroke prevention. Neurology 1998; 51(suppl 3):S17–S19.
- De Schryver EL, Algra A, van Gijn J. Cochrane review: dipyridamole for preventing major vascular events in patients with vascular disease. Stroke 2003; 34:2072–2080.
- Leonardi-Bee J, Bath PM, Bousser MG, et al; Dipyridamole in Stroke Collaboration (DISC). Dipyridamole for preventing recurrent ischemic stroke and other vascular events: a meta-analysis of individual patient data from randomized controlled trials. Stroke 2005; 36:162–168.
- ESPRIT Study Group; Halkes PH, van Gijn J, Kappelle LJ, Koudstaal PJ, Algra A. Aspirin plus dipyridamole versus aspirin alone after cerebral ischaemia of arterial origin (ESPRIT): randomised controlled trial. Lancet 2006; 367:1665–1673.
- Tirschwell D. Aspirin plus dipyridamole was more effective than aspirin alone for preventing vascular events after minor cerebral ischemia. ACP J Club 2006; 145:57.
- Furie KL, Kasner SE, Adams RJ, et al; American Heart Association Stroke Council, Council on Cardiovascular Nursing, Council on Clinical Cardiology, and Interdisciplinary Council on Quality of Care and Outcomes Research. Guidelines for the prevention of stroke in patients with stroke or transient ischemic attack: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2011; 42:227–276.
- CAPRIE Steering Committee. A randomised, blinded, trial of clopidogrel versus aspirin in patients at risk of ischaemic events (CAPRIE). CAPRIE Steering Committee. Lancet 1996; 348:1329–1339.
- Diener HC, Bogousslavsky J, Brass LM, et al; MATCH investigators. Aspirin and clopidogrel compared with clopidogrel alone after recent ischaemic stroke or transient ischaemic attack in high-risk patients (MATCH): randomised, double-blind, placebo-controlled trial. Lancet 2004; 364:331–337.
- Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK; Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial Investigators. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med 2001; 345:494–502.
- Bhatt DL, Fox KA, Hacke W, et al; CHARISMA Investigators. Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events. N Engl J Med 2006; 354:1706–1717.
- Sacco RL, Diener HC, Yusuf S, et al; PRoFESS Study Group. Aspirin and extended-release dipyridamole versus clopidogrel for recurrent stroke. N Engl J Med 2008; 359:1238–1251.
- Kent DM, Thaler DE. Stroke prevention—insights from incoherence. N Engl J Med 2008; 359:1287–1289.
- Chimowitz MI, Lynn MJ, Howlett-Smith H, et al; Warfarin-Aspirin Symptomatic Intracranial Disease Trial Investigators. Comparison of warfarin and aspirin for symptomatic intracranial arterial stenosis. N Engl J Med 2005; 352:1305–1316.
- ESPRIT Study Group; Halkes PH, van Gijn J, Kappelle LJ, Koudstaal PJ, Algra A. Medium intensity oral anticoagulants versus aspirin after cerebral ischaemia of arterial origin (ESPRIT): a randomised controlled trial. Lancet Neurol 2007; 6:115–124.
- Mohr JP, Thompson JL, Lazar RM, et al; Warfarin-Aspirin Recurrent Stroke Study Group. A comparison of warfarin and aspirin for the prevention of recurrent ischemic stroke. N Engl J Med 2001; 345:1444–1451.
- Jauch EC, Saver JL, Adams HP, et al; American Heart Association Stroke Council; Council on Cardiovascular Nursing; Council on Peripheral Vascular Disease; Council on Clinical Cardiology. Guidelines for the early management of patients with acute ischemic stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2013; 44:870–947.
- Kolominsky-Rabas PL, Weber M, Gefeller O, Neundoerfer B, Heuschmann PU. Epidemiology of ischemic stroke subtypes according to TOAST criteria: incidence, recurrence, and long-term survival in ischemic stroke subtypes: a population-based study. Stroke 2001; 32:2735–2740.
- Zivin JA. Ischemic cerebrovascular disease. In:Goldman L, Schafer AI, editors. Goldman’s Cecil Medicine 24th ed. Philadelphia, PA: Elsevier; 2012: chap 414. www.mdconsult.com. Accessed November 7, 2013.
- Smith WS, Johnston C, Easton D. Cerebrovascular diseases. In:Kasper DL, Braunwald E, Fauci AS, Hauser SL, Longo DL, Jameson JL, editors. Harrison’s Principles of Internal Medicine. 16th ed. New York, NY: McGraw Hill; 2005: chap 349. www.accessmedicine.com.
- Beneficial effect of carotid endarterectomy in symptomatic patients with high-grade carotid stenosis. North American Symptomatic Carotid Endarterectomy Trial Collaborators. N Engl J Med 1991; 325:445–453.
- Randomised trial of endarterectomy for recently symptomatic carotid stenosis: final results of the MRC European Carotid Surgery Trial (ECST). Lancet 1998; 351:1379–1387.
- Barnett HJ, Taylor DW, Eliasziw M, et al. Benefit of carotid endarterectomy in patients with symptomatic moderate or severe stenosis. North American Symptomatic Carotid Endarterectomy Trial Collaborators. N Engl J Med 1998; 339:1415–1425.
- Hobson RW, Weiss DG, Fields WS, et al. Efficacy of carotid endarterectomy for asymptomatic carotid stenosis. The Veterans Affairs Cooperative Study Group. N Engl J Med 1993; 328:221–227.
- Endarterectomy for asymptomatic carotid artery stenosis. Executive Committee for the Asymptomatic Carotid Atherosclerosis Study. JAMA 1995; 273:1421–1428.
- Halliday A, Mansfield A, Marro J, et al; MRC Asymptomatic Carotid Surgery Trial (ACST) Collaborative Group. Prevention of disabling and fatal strokes by successful carotid endarterectomy in patients without recent neurological symptoms: randomised controlled trial. Lancet 2004; 363:1491–1502.
- Marquardt L, Geraghty OC, Mehta Z, Rothwell PM. Low risk of ipsilateral stroke in patients with asymptomatic carotid stenosis on best medical treatment: a prospective, population-based study. Stroke 2010; 41:e11–e17.
- Spence JD, Coates V, Li H, et al. Effects of intensive medical therapy on microemboli and cardiovascular risk in asymptomatic carotid stenosis. Arch Neurol 2010; 67:180–186.
- Brott TG, Hobson RW, Howard G, et al; CREST Investigators. Stenting versus endarterectomy for treatment of carotid-artery stenosis. N Engl J Med 2010; 363:11–23.
- Barnett HJ, Gunton RW, Eliasziw M, et al. Causes and severity of ischemic stroke in patients with internal carotid artery stenosis. JAMA 2000; 283:1429–1436.
- Inzitari D, Eliasziw M, Gates P, et al. The causes and risk of stroke in patients with asymptomatic internal-carotid-artery stenosis. North American Symptomatic Carotid Endarterectomy Trial Collaborators. N Engl J Med 2000; 342:1693–1700.
KEY POINTS
- After a stroke, antiplatelet therapy lowers the rate of recurrent nonfatal stroke by about 25%.
- Aspirin is the most established, best tolerated, and least expensive of the three approved drugs.
- Adding dipyridamole to aspirin increases the efficacy, with a 22% reduction in relative risk, but only a 1% reduction in absolute risk.
- Clopidogrel is similar in efficacy to aspirin and to dipyridamole.
- All three agents are regarded as equal and appropriate for secondary prevention of stroke; the choice is based on individual patient characteristics.
- A small number of strokes result from atherosclerotic disease of the common carotid bifurcation, and patients with symptomatic carotid disease can be treated with the combination of surgery or stenting and drug therapy, or with drug therapy alone.

Can an ARB be given to patients who have had angioedema on an ACE inhibitor?
Current evidence suggests no absolute contraindication to angiotensin receptor blockers (ARBs) in patients who have had angioedema attributable to an angiotensin-converting enzyme (ACE) inhibitor. However, since ARBs can also cause angioedema, they should be prescribed with extreme caution after a thorough risk-benefit analysis and after educating the patient to watch for signs of angioedema while taking the drug.
A GROWING PROBLEM
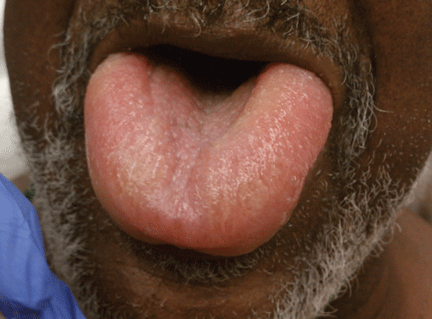
Angioedema is a potentially life-threatening swelling of the skin and subcutaneous tissues, often affecting the lips and tongue (Figure 1), and in some cases interfering with breathing and requiring tracheostomy.1 The incidence rate of angioedema in patients taking ACE inhibitors ranges from 0.1% to 0.7%.2–4 Although this rate may seem low, the widespread and growing use of ACE inhibitors and ARBs in patients with diabetes, diabetic nephropathy, and congestive heart failure5 makes angioedema fairly common in clinical practice.
ACE inhibitor-induced angioedema most commonly occurs within days of initiating therapy, but it also may occur weeks, months, or even years after the start of treatment.1 Patients who are over age 65, black, or female are at higher risk, as are renal transplant recipients taking mTOR inhibitors such as sirolimus. Diabetes appears to be associated with a lower risk.4,6,7 This adverse reaction to ACE inhibitors is thought to be a class side effect, and the future use of this class of drugs would be contraindicated.8,9
ACE inhibitors cause angioedema by direct interference with the degradation of bradykinin, thereby increasing bradykinin levels and potentiating its biologic effect, leading to increased vascular permeability, inflammation, and activation of nociceptors.8
EVIDENCE TO SUPPORT THE USE OF ARBs
ACE inhibitors and ARBs both block the renin-angiotensin-aldosterone pathway and confer similar advantages in patients with congestive heart failure, renal failure, and diabetes. But since ARBs directly inhibit the angiotensin receptor and do not interfere with bradykinin degradation, how they cause angioedema is unclear, and clinicians have questioned whether these agents might be used safely in patients who have had angioedema on an ACE inhibitor.
In a large meta-analysis of randomized clinical trials, Makani et al2 investigated the risk of angioedema with ARB use in 35,479 patients and compared this with other commonly used antihypertensive drugs. The weighted incidence of angioedema was 0.30% with an ACE inhibitor, 0.11% with an ARB, and 0.07% with placebo.2 In seven trials included in this study that compared ARBs with placebo, there was no significant difference in the risk of angioedema. Even in such a large study, the event rate was small, making definite conclusions difficult.
In a retrospective observational study of 4 million patients by Toh et al,3 patients on beta-blockers were used as a reference, and propensity scoring was used to estimate the hazard ratio of angioedema separately for drugs targeting the renin-angiotensin-aldosterone system, including ACE inhibitors and ARBs. The risk of angioedema, as measured by the cumulative incidence and incidence rate, was highest for ACE inhibitors and was similar between ARBs and beta-blockers. The risk of serious angioedema was three times higher with ACE inhibitors than with beta-blockers, and there was no higher risk of serious angioedema with ARBs than with beta-blockers.3
Looking specifically at the use of ARBs in patients who developed angioedema on an ACE inhibitor, Haymore et al10 performed a meta-analysis evaluating only three studies that showed the estimated risk of angioedema with an ARB was between 3.5% and 9.4% in patients with a history of ACE inhibitor-induced angioedema. Later, when the results of the Telmisartan Randomised Assessment Study in ACE Intolerant Subjects With Cardiovascular Disease trial11 were published, the previous meta-analysis was updated12: the risk of angioedema with an ARB was only 2.5% (95% confidence interval 0%–6.6%), and there was no statistically significant difference in the odds (odds ratio 1.1; 95% confidence interval 0.07–17) of angioedema between ARBs and placebo.10,12 Again, these results should be interpreted with caution, as only two patients in the ARB (telmisartan) group and three patients in the placebo group developed angioedema.
In another review, Beavers et al13 advised that the prescribing practitioner should carefully perform a risk-benefit analysis before substituting an ARB in patients with ACE inhibitor-induced angioedema. They concluded that an ARB could be considered in patients who are likely to have a large clinical benefit from an ARB, such as those with heart failure. They also suggested that angioedema related to ARBs was less severe and occurred earlier than with that linked to ACE inhibitors.
No large clinical trial has yet been specifically designed to address the use of ARBs in patients with a history of ACE inhibitor-induced angioedema. The package insert for the ARB losartan mentions that the risk of this adverse reaction might be higher in patients who have had angioedema on an ACE inhibitor. However, the issue of recurrent angioedema is not further addressed for this or other commonly used ARBs.
GENERAL RECOMMENDATIONS
The mechanisms of ARB-induced angioedema are yet unknown. However, studies have shown that the incidence of angioedema while on an ARB is low and is probably comparable to that of placebo.2,3,12–14 And since ARBs share many of the cardiac and renal protective effects of ACE inhibitors, ARBs may be beneficial for patients who discontinue an ACE inhibitor because of adverse effects including angioedema.9,15,16 Based on the discussion above, there is no clear evidence to suggest that ARBs are contraindicated in such patients, especially if there is a compelling indication for an ARB.
The National Kidney Foundation Kidney Disease Outcomes Quality Initiative (NKF KDOQI) guidelines on hypertension in chronic kidney disease recommend caution when substituting an ARB for an ACE inhibitor after angioedema.15 The joint guidelines of the American College of Cardiology and American Heart Association (ACC/AHA) for the diagnosis and management of heart failure in adults advise “extreme caution.”9,16
The risks and benefits of ARB therapy in this setting should be analyzed by the prescribing physician and discussed with the patient. The patient should be closely monitored for the recurrence of angioedema and should be given a clear plan of action should symptoms recur.
OUR ADVICE
In patients with ACE inhibitor-induced angioedema, we recommend the following:
- Determine that the patient truly has one of the evidence-based, compelling indications for an ARB. Carefully weigh the risks and benefits for the individual patient, and discuss the risk of angioedema based on age, race, sex, and medical history, and the availability of immediate medical care should angioedema occur.
- If there is an evidence-based indication for an ARB that outweighs the risk of angioedema, an ARB may be started with caution.
- Specifically discuss with the patient the possibility of recurrence of angioedema while on an ARB, and provide instructions on how to proceed if this should occur.
- Kaplan AP, Greaves MW. Angioedema. J Am Acad Dermatol 2005; 53:373–388.
- Makani H, Messerli FH, Romero J, et al. Meta-analysis of randomized trials of angioedema as an adverse event of renin-angiotensin system inhibitors. Am J Cardiol 2012; 110:383–391.
- Toh S, Reichman ME, Houstoun M, et al. Comparative risk for angioedema associated with the use of drugs that target the renin-angiotensin-aldosterone system. Arch Intern Med 2012; 172:1582–1589.
- Kostis JB, Kim HJ, Rusnak J, et al. Incidence and characteristics of angioedema associated with enalapril. Arch Intern Med 2005; 165:1637–1642.
- Taylor AA, Siragy H, Nesbitt S. Angiotensin receptor blockers: pharmacology, efficacy, and safety. J Clin Hypertens (Greenwich) 2011; 13:677–686.
- Duerr M, Glander P, Diekmann F, Dragun D, Neumayer HH, Budde K. Increased incidence of angioedema with ACE inhibitors in combination with mTOR inhibitors in kidney transplant recipients. Clin J Am Soc Nephrol 2010; 5:703–708.
- Byrd JB, Adam A, Brown NJ. Angiotensin-converting enzyme inhibitor-associated angioedema. Immunol Allergy Clin North Am 2006; 26:725–737.
- Inomata N. Recent advances in drug-induced angioedema. Allergol Int 2012; 61:545–557.
- Hunt SA, Abraham WT, Chin MH, et al; American College of Cardiology Foundation; American Heart Association. 2009 Focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines Developed in Collaboration With the International Society for Heart and Lung Transplantation. J Am Coll Cardiol 2009; 53:e1–e90.
- Haymore BR, Yoon J, Mikita CP, Klote MM, DeZee KJ. Risk of angioedema with angiotensin receptor blockers in patients with prior angioedema associated with angiotensin-converting enzyme inhibitors: a meta-analysis. Ann Allergy Asthma Immunol 2008; 101:495–499.
- Telmisartan Randomised Assessment Study in ACE Intolerant Subjects with Cardiovascular Disease (TRANSCEND) Investigators. Effects of the angiotensin-receptor blocker telmisartan on cardiovascular events in high-risk patients intolerant to angiotensin-converting enzyme inhibitors: a randomised controlled trial. Lancet 2008; 372:1174–1183.
- Haymore BR, DeZee KJ. Use of angiotensin receptor blockers after angioedema with an angiotensin-converting enzyme inhibitor. Ann Allergy Asthma Immunol 2009; 103:83–84.
- Beavers CJ, Dunn SP, Macaulay TE. The role of angiotensin receptor blockers in patients with angiotensin-converting enzyme inhibitor-induced angioedema. Ann Pharmacother 2011; 45:520–524.
- Caldeira D, David C, Sampaio C. Tolerability of angiotensin-receptor blockers in patients with intolerance to angiotensin-converting enzyme inhibitors: a systematic review and meta-analysis. Am J Cardiovasc Drugs 2012; 12:263–277.
- Kidney Disease Outcomes Quality Initiative (K/DOQI). K/DOQI clinical practice guidelines on hypertension and antihypertensive agents in chronic kidney disease. Am J Kidney Dis 2004; 43(suppl 1):S1–S290.
- Smith SC, Benjamin EJ, Bonow RO, et al. AHA/ACCF secondary prevention and risk reduction therapy for patients with coronary and other atherosclerotic vascular disease: 2011 update: a guideline from the American Heart Association and American College of Cardiology Foundation endorsed by the World Heart Federation and the Preventive Cardiovascular Nurses Association. J Am Coll Cardiol 2011; 58:2432–2446.
Current evidence suggests no absolute contraindication to angiotensin receptor blockers (ARBs) in patients who have had angioedema attributable to an angiotensin-converting enzyme (ACE) inhibitor. However, since ARBs can also cause angioedema, they should be prescribed with extreme caution after a thorough risk-benefit analysis and after educating the patient to watch for signs of angioedema while taking the drug.
A GROWING PROBLEM
Angioedema is a potentially life-threatening swelling of the skin and subcutaneous tissues, often affecting the lips and tongue (Figure 1), and in some cases interfering with breathing and requiring tracheostomy.1 The incidence rate of angioedema in patients taking ACE inhibitors ranges from 0.1% to 0.7%.2–4 Although this rate may seem low, the widespread and growing use of ACE inhibitors and ARBs in patients with diabetes, diabetic nephropathy, and congestive heart failure5 makes angioedema fairly common in clinical practice.
ACE inhibitor-induced angioedema most commonly occurs within days of initiating therapy, but it also may occur weeks, months, or even years after the start of treatment.1 Patients who are over age 65, black, or female are at higher risk, as are renal transplant recipients taking mTOR inhibitors such as sirolimus. Diabetes appears to be associated with a lower risk.4,6,7 This adverse reaction to ACE inhibitors is thought to be a class side effect, and the future use of this class of drugs would be contraindicated.8,9
ACE inhibitors cause angioedema by direct interference with the degradation of bradykinin, thereby increasing bradykinin levels and potentiating its biologic effect, leading to increased vascular permeability, inflammation, and activation of nociceptors.8
EVIDENCE TO SUPPORT THE USE OF ARBs
ACE inhibitors and ARBs both block the renin-angiotensin-aldosterone pathway and confer similar advantages in patients with congestive heart failure, renal failure, and diabetes. But since ARBs directly inhibit the angiotensin receptor and do not interfere with bradykinin degradation, how they cause angioedema is unclear, and clinicians have questioned whether these agents might be used safely in patients who have had angioedema on an ACE inhibitor.
In a large meta-analysis of randomized clinical trials, Makani et al2 investigated the risk of angioedema with ARB use in 35,479 patients and compared this with other commonly used antihypertensive drugs. The weighted incidence of angioedema was 0.30% with an ACE inhibitor, 0.11% with an ARB, and 0.07% with placebo.2 In seven trials included in this study that compared ARBs with placebo, there was no significant difference in the risk of angioedema. Even in such a large study, the event rate was small, making definite conclusions difficult.
In a retrospective observational study of 4 million patients by Toh et al,3 patients on beta-blockers were used as a reference, and propensity scoring was used to estimate the hazard ratio of angioedema separately for drugs targeting the renin-angiotensin-aldosterone system, including ACE inhibitors and ARBs. The risk of angioedema, as measured by the cumulative incidence and incidence rate, was highest for ACE inhibitors and was similar between ARBs and beta-blockers. The risk of serious angioedema was three times higher with ACE inhibitors than with beta-blockers, and there was no higher risk of serious angioedema with ARBs than with beta-blockers.3
Looking specifically at the use of ARBs in patients who developed angioedema on an ACE inhibitor, Haymore et al10 performed a meta-analysis evaluating only three studies that showed the estimated risk of angioedema with an ARB was between 3.5% and 9.4% in patients with a history of ACE inhibitor-induced angioedema. Later, when the results of the Telmisartan Randomised Assessment Study in ACE Intolerant Subjects With Cardiovascular Disease trial11 were published, the previous meta-analysis was updated12: the risk of angioedema with an ARB was only 2.5% (95% confidence interval 0%–6.6%), and there was no statistically significant difference in the odds (odds ratio 1.1; 95% confidence interval 0.07–17) of angioedema between ARBs and placebo.10,12 Again, these results should be interpreted with caution, as only two patients in the ARB (telmisartan) group and three patients in the placebo group developed angioedema.
In another review, Beavers et al13 advised that the prescribing practitioner should carefully perform a risk-benefit analysis before substituting an ARB in patients with ACE inhibitor-induced angioedema. They concluded that an ARB could be considered in patients who are likely to have a large clinical benefit from an ARB, such as those with heart failure. They also suggested that angioedema related to ARBs was less severe and occurred earlier than with that linked to ACE inhibitors.
No large clinical trial has yet been specifically designed to address the use of ARBs in patients with a history of ACE inhibitor-induced angioedema. The package insert for the ARB losartan mentions that the risk of this adverse reaction might be higher in patients who have had angioedema on an ACE inhibitor. However, the issue of recurrent angioedema is not further addressed for this or other commonly used ARBs.
GENERAL RECOMMENDATIONS
The mechanisms of ARB-induced angioedema are yet unknown. However, studies have shown that the incidence of angioedema while on an ARB is low and is probably comparable to that of placebo.2,3,12–14 And since ARBs share many of the cardiac and renal protective effects of ACE inhibitors, ARBs may be beneficial for patients who discontinue an ACE inhibitor because of adverse effects including angioedema.9,15,16 Based on the discussion above, there is no clear evidence to suggest that ARBs are contraindicated in such patients, especially if there is a compelling indication for an ARB.
The National Kidney Foundation Kidney Disease Outcomes Quality Initiative (NKF KDOQI) guidelines on hypertension in chronic kidney disease recommend caution when substituting an ARB for an ACE inhibitor after angioedema.15 The joint guidelines of the American College of Cardiology and American Heart Association (ACC/AHA) for the diagnosis and management of heart failure in adults advise “extreme caution.”9,16
The risks and benefits of ARB therapy in this setting should be analyzed by the prescribing physician and discussed with the patient. The patient should be closely monitored for the recurrence of angioedema and should be given a clear plan of action should symptoms recur.
OUR ADVICE
In patients with ACE inhibitor-induced angioedema, we recommend the following:
- Determine that the patient truly has one of the evidence-based, compelling indications for an ARB. Carefully weigh the risks and benefits for the individual patient, and discuss the risk of angioedema based on age, race, sex, and medical history, and the availability of immediate medical care should angioedema occur.
- If there is an evidence-based indication for an ARB that outweighs the risk of angioedema, an ARB may be started with caution.
- Specifically discuss with the patient the possibility of recurrence of angioedema while on an ARB, and provide instructions on how to proceed if this should occur.
Current evidence suggests no absolute contraindication to angiotensin receptor blockers (ARBs) in patients who have had angioedema attributable to an angiotensin-converting enzyme (ACE) inhibitor. However, since ARBs can also cause angioedema, they should be prescribed with extreme caution after a thorough risk-benefit analysis and after educating the patient to watch for signs of angioedema while taking the drug.
A GROWING PROBLEM
Angioedema is a potentially life-threatening swelling of the skin and subcutaneous tissues, often affecting the lips and tongue (Figure 1), and in some cases interfering with breathing and requiring tracheostomy.1 The incidence rate of angioedema in patients taking ACE inhibitors ranges from 0.1% to 0.7%.2–4 Although this rate may seem low, the widespread and growing use of ACE inhibitors and ARBs in patients with diabetes, diabetic nephropathy, and congestive heart failure5 makes angioedema fairly common in clinical practice.
ACE inhibitor-induced angioedema most commonly occurs within days of initiating therapy, but it also may occur weeks, months, or even years after the start of treatment.1 Patients who are over age 65, black, or female are at higher risk, as are renal transplant recipients taking mTOR inhibitors such as sirolimus. Diabetes appears to be associated with a lower risk.4,6,7 This adverse reaction to ACE inhibitors is thought to be a class side effect, and the future use of this class of drugs would be contraindicated.8,9
ACE inhibitors cause angioedema by direct interference with the degradation of bradykinin, thereby increasing bradykinin levels and potentiating its biologic effect, leading to increased vascular permeability, inflammation, and activation of nociceptors.8
EVIDENCE TO SUPPORT THE USE OF ARBs
ACE inhibitors and ARBs both block the renin-angiotensin-aldosterone pathway and confer similar advantages in patients with congestive heart failure, renal failure, and diabetes. But since ARBs directly inhibit the angiotensin receptor and do not interfere with bradykinin degradation, how they cause angioedema is unclear, and clinicians have questioned whether these agents might be used safely in patients who have had angioedema on an ACE inhibitor.
In a large meta-analysis of randomized clinical trials, Makani et al2 investigated the risk of angioedema with ARB use in 35,479 patients and compared this with other commonly used antihypertensive drugs. The weighted incidence of angioedema was 0.30% with an ACE inhibitor, 0.11% with an ARB, and 0.07% with placebo.2 In seven trials included in this study that compared ARBs with placebo, there was no significant difference in the risk of angioedema. Even in such a large study, the event rate was small, making definite conclusions difficult.
In a retrospective observational study of 4 million patients by Toh et al,3 patients on beta-blockers were used as a reference, and propensity scoring was used to estimate the hazard ratio of angioedema separately for drugs targeting the renin-angiotensin-aldosterone system, including ACE inhibitors and ARBs. The risk of angioedema, as measured by the cumulative incidence and incidence rate, was highest for ACE inhibitors and was similar between ARBs and beta-blockers. The risk of serious angioedema was three times higher with ACE inhibitors than with beta-blockers, and there was no higher risk of serious angioedema with ARBs than with beta-blockers.3
Looking specifically at the use of ARBs in patients who developed angioedema on an ACE inhibitor, Haymore et al10 performed a meta-analysis evaluating only three studies that showed the estimated risk of angioedema with an ARB was between 3.5% and 9.4% in patients with a history of ACE inhibitor-induced angioedema. Later, when the results of the Telmisartan Randomised Assessment Study in ACE Intolerant Subjects With Cardiovascular Disease trial11 were published, the previous meta-analysis was updated12: the risk of angioedema with an ARB was only 2.5% (95% confidence interval 0%–6.6%), and there was no statistically significant difference in the odds (odds ratio 1.1; 95% confidence interval 0.07–17) of angioedema between ARBs and placebo.10,12 Again, these results should be interpreted with caution, as only two patients in the ARB (telmisartan) group and three patients in the placebo group developed angioedema.
In another review, Beavers et al13 advised that the prescribing practitioner should carefully perform a risk-benefit analysis before substituting an ARB in patients with ACE inhibitor-induced angioedema. They concluded that an ARB could be considered in patients who are likely to have a large clinical benefit from an ARB, such as those with heart failure. They also suggested that angioedema related to ARBs was less severe and occurred earlier than with that linked to ACE inhibitors.
No large clinical trial has yet been specifically designed to address the use of ARBs in patients with a history of ACE inhibitor-induced angioedema. The package insert for the ARB losartan mentions that the risk of this adverse reaction might be higher in patients who have had angioedema on an ACE inhibitor. However, the issue of recurrent angioedema is not further addressed for this or other commonly used ARBs.
GENERAL RECOMMENDATIONS
The mechanisms of ARB-induced angioedema are yet unknown. However, studies have shown that the incidence of angioedema while on an ARB is low and is probably comparable to that of placebo.2,3,12–14 And since ARBs share many of the cardiac and renal protective effects of ACE inhibitors, ARBs may be beneficial for patients who discontinue an ACE inhibitor because of adverse effects including angioedema.9,15,16 Based on the discussion above, there is no clear evidence to suggest that ARBs are contraindicated in such patients, especially if there is a compelling indication for an ARB.
The National Kidney Foundation Kidney Disease Outcomes Quality Initiative (NKF KDOQI) guidelines on hypertension in chronic kidney disease recommend caution when substituting an ARB for an ACE inhibitor after angioedema.15 The joint guidelines of the American College of Cardiology and American Heart Association (ACC/AHA) for the diagnosis and management of heart failure in adults advise “extreme caution.”9,16
The risks and benefits of ARB therapy in this setting should be analyzed by the prescribing physician and discussed with the patient. The patient should be closely monitored for the recurrence of angioedema and should be given a clear plan of action should symptoms recur.
OUR ADVICE
In patients with ACE inhibitor-induced angioedema, we recommend the following:
- Determine that the patient truly has one of the evidence-based, compelling indications for an ARB. Carefully weigh the risks and benefits for the individual patient, and discuss the risk of angioedema based on age, race, sex, and medical history, and the availability of immediate medical care should angioedema occur.
- If there is an evidence-based indication for an ARB that outweighs the risk of angioedema, an ARB may be started with caution.
- Specifically discuss with the patient the possibility of recurrence of angioedema while on an ARB, and provide instructions on how to proceed if this should occur.
- Kaplan AP, Greaves MW. Angioedema. J Am Acad Dermatol 2005; 53:373–388.
- Makani H, Messerli FH, Romero J, et al. Meta-analysis of randomized trials of angioedema as an adverse event of renin-angiotensin system inhibitors. Am J Cardiol 2012; 110:383–391.
- Toh S, Reichman ME, Houstoun M, et al. Comparative risk for angioedema associated with the use of drugs that target the renin-angiotensin-aldosterone system. Arch Intern Med 2012; 172:1582–1589.
- Kostis JB, Kim HJ, Rusnak J, et al. Incidence and characteristics of angioedema associated with enalapril. Arch Intern Med 2005; 165:1637–1642.
- Taylor AA, Siragy H, Nesbitt S. Angiotensin receptor blockers: pharmacology, efficacy, and safety. J Clin Hypertens (Greenwich) 2011; 13:677–686.
- Duerr M, Glander P, Diekmann F, Dragun D, Neumayer HH, Budde K. Increased incidence of angioedema with ACE inhibitors in combination with mTOR inhibitors in kidney transplant recipients. Clin J Am Soc Nephrol 2010; 5:703–708.
- Byrd JB, Adam A, Brown NJ. Angiotensin-converting enzyme inhibitor-associated angioedema. Immunol Allergy Clin North Am 2006; 26:725–737.
- Inomata N. Recent advances in drug-induced angioedema. Allergol Int 2012; 61:545–557.
- Hunt SA, Abraham WT, Chin MH, et al; American College of Cardiology Foundation; American Heart Association. 2009 Focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines Developed in Collaboration With the International Society for Heart and Lung Transplantation. J Am Coll Cardiol 2009; 53:e1–e90.
- Haymore BR, Yoon J, Mikita CP, Klote MM, DeZee KJ. Risk of angioedema with angiotensin receptor blockers in patients with prior angioedema associated with angiotensin-converting enzyme inhibitors: a meta-analysis. Ann Allergy Asthma Immunol 2008; 101:495–499.
- Telmisartan Randomised Assessment Study in ACE Intolerant Subjects with Cardiovascular Disease (TRANSCEND) Investigators. Effects of the angiotensin-receptor blocker telmisartan on cardiovascular events in high-risk patients intolerant to angiotensin-converting enzyme inhibitors: a randomised controlled trial. Lancet 2008; 372:1174–1183.
- Haymore BR, DeZee KJ. Use of angiotensin receptor blockers after angioedema with an angiotensin-converting enzyme inhibitor. Ann Allergy Asthma Immunol 2009; 103:83–84.
- Beavers CJ, Dunn SP, Macaulay TE. The role of angiotensin receptor blockers in patients with angiotensin-converting enzyme inhibitor-induced angioedema. Ann Pharmacother 2011; 45:520–524.
- Caldeira D, David C, Sampaio C. Tolerability of angiotensin-receptor blockers in patients with intolerance to angiotensin-converting enzyme inhibitors: a systematic review and meta-analysis. Am J Cardiovasc Drugs 2012; 12:263–277.
- Kidney Disease Outcomes Quality Initiative (K/DOQI). K/DOQI clinical practice guidelines on hypertension and antihypertensive agents in chronic kidney disease. Am J Kidney Dis 2004; 43(suppl 1):S1–S290.
- Smith SC, Benjamin EJ, Bonow RO, et al. AHA/ACCF secondary prevention and risk reduction therapy for patients with coronary and other atherosclerotic vascular disease: 2011 update: a guideline from the American Heart Association and American College of Cardiology Foundation endorsed by the World Heart Federation and the Preventive Cardiovascular Nurses Association. J Am Coll Cardiol 2011; 58:2432–2446.
- Kaplan AP, Greaves MW. Angioedema. J Am Acad Dermatol 2005; 53:373–388.
- Makani H, Messerli FH, Romero J, et al. Meta-analysis of randomized trials of angioedema as an adverse event of renin-angiotensin system inhibitors. Am J Cardiol 2012; 110:383–391.
- Toh S, Reichman ME, Houstoun M, et al. Comparative risk for angioedema associated with the use of drugs that target the renin-angiotensin-aldosterone system. Arch Intern Med 2012; 172:1582–1589.
- Kostis JB, Kim HJ, Rusnak J, et al. Incidence and characteristics of angioedema associated with enalapril. Arch Intern Med 2005; 165:1637–1642.
- Taylor AA, Siragy H, Nesbitt S. Angiotensin receptor blockers: pharmacology, efficacy, and safety. J Clin Hypertens (Greenwich) 2011; 13:677–686.
- Duerr M, Glander P, Diekmann F, Dragun D, Neumayer HH, Budde K. Increased incidence of angioedema with ACE inhibitors in combination with mTOR inhibitors in kidney transplant recipients. Clin J Am Soc Nephrol 2010; 5:703–708.
- Byrd JB, Adam A, Brown NJ. Angiotensin-converting enzyme inhibitor-associated angioedema. Immunol Allergy Clin North Am 2006; 26:725–737.
- Inomata N. Recent advances in drug-induced angioedema. Allergol Int 2012; 61:545–557.
- Hunt SA, Abraham WT, Chin MH, et al; American College of Cardiology Foundation; American Heart Association. 2009 Focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines Developed in Collaboration With the International Society for Heart and Lung Transplantation. J Am Coll Cardiol 2009; 53:e1–e90.
- Haymore BR, Yoon J, Mikita CP, Klote MM, DeZee KJ. Risk of angioedema with angiotensin receptor blockers in patients with prior angioedema associated with angiotensin-converting enzyme inhibitors: a meta-analysis. Ann Allergy Asthma Immunol 2008; 101:495–499.
- Telmisartan Randomised Assessment Study in ACE Intolerant Subjects with Cardiovascular Disease (TRANSCEND) Investigators. Effects of the angiotensin-receptor blocker telmisartan on cardiovascular events in high-risk patients intolerant to angiotensin-converting enzyme inhibitors: a randomised controlled trial. Lancet 2008; 372:1174–1183.
- Haymore BR, DeZee KJ. Use of angiotensin receptor blockers after angioedema with an angiotensin-converting enzyme inhibitor. Ann Allergy Asthma Immunol 2009; 103:83–84.
- Beavers CJ, Dunn SP, Macaulay TE. The role of angiotensin receptor blockers in patients with angiotensin-converting enzyme inhibitor-induced angioedema. Ann Pharmacother 2011; 45:520–524.
- Caldeira D, David C, Sampaio C. Tolerability of angiotensin-receptor blockers in patients with intolerance to angiotensin-converting enzyme inhibitors: a systematic review and meta-analysis. Am J Cardiovasc Drugs 2012; 12:263–277.
- Kidney Disease Outcomes Quality Initiative (K/DOQI). K/DOQI clinical practice guidelines on hypertension and antihypertensive agents in chronic kidney disease. Am J Kidney Dis 2004; 43(suppl 1):S1–S290.
- Smith SC, Benjamin EJ, Bonow RO, et al. AHA/ACCF secondary prevention and risk reduction therapy for patients with coronary and other atherosclerotic vascular disease: 2011 update: a guideline from the American Heart Association and American College of Cardiology Foundation endorsed by the World Heart Federation and the Preventive Cardiovascular Nurses Association. J Am Coll Cardiol 2011; 58:2432–2446.
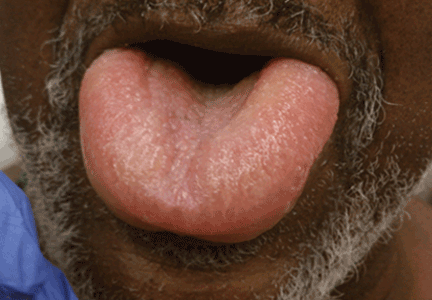
Myasthenia gravis: Newer therapies offer sustained improvement
Current therapies for myasthenia gravis can help most patients achieve sustained improvement. The overall prognosis has dramatically improved over the last 4 decades: the mortality rate used to be 75%; now it is 4.5%.1
Myasthenia gravis is the most common disorder of neuromuscular junction transmission and is also one of the best characterized autoimmune diseases. However, its symptoms—primarily weakness—vary from patient to patient, and in the same patient, by time of day and over longer time periods. The variation in symptoms can be very confusing to undiagnosed patients and puzzling to unsuspecting physicians. Such diagnostic uncertainty can give the patient additional frustration and emotional stress, which in turn exacerbate his or her condition.
In this review, we will give an overview of the pathogenesis, clinical manifestations, diagnosis, and treatment of myasthenia gravis.
TWO PEAKS IN INCIDENCE BY AGE

The annual incidence of myasthenia gravis is approximately 10 to 20 new cases per million, with a prevalence of about 150 to 200 per million.2
The age of onset has a bimodal distribution, with an early incidence peak in the second to third decade with a female predominance and a late peak in the 6th to the 8th decade with a male predominance.2
Myasthenia gravis is commonly associated with several other autoimmune disorders, including hypothyroidism, hyperthyroidism, systemic lupus erythematosus, rheumatoid arthritis, vitiligo, diabetes, and, more recently recognized, neuromyelitis optica.3
ANTIBODIES AGAINST AChR AND MuSK
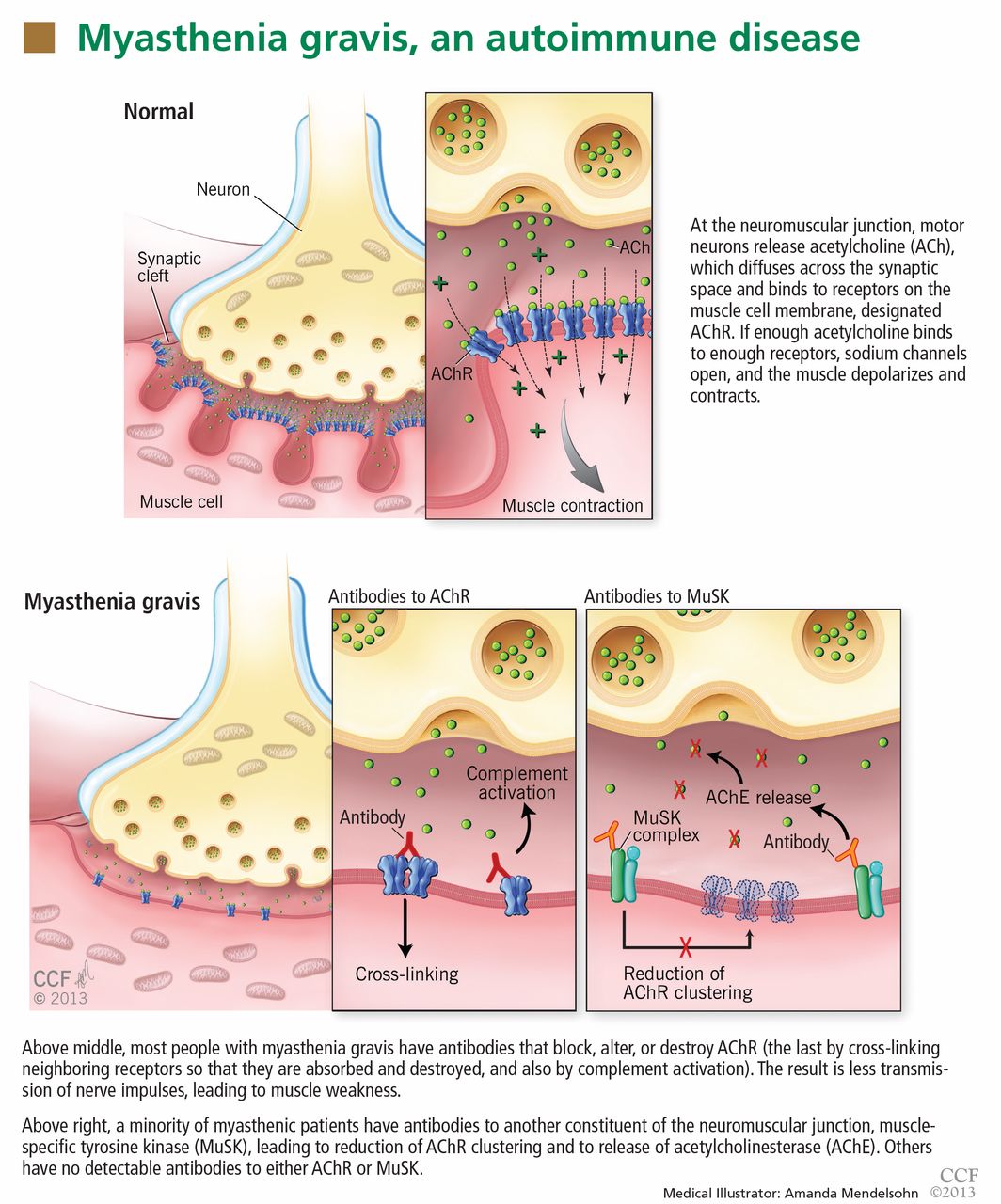
In most cases of myasthenia gravis the patient has autoimmune antibodies against constituents of the neuromuscular junction, specifically acetylcholine receptor (AChR) and muscle-specific tyrosine kinase (MuSK) (Figure 1).
AChR antibody-positive myasthenia gravis
When antibodies bind to AChR on the postsynaptic membrane, they cross-link neighboring AChR units, which are absorbed into the muscle fiber and are broken up.4 In addition, the complement system is activated to mediate further damage on the postsynaptic membrane.
AChR antibodies may come from germinal centers of the thymus, where clustered myoid cells express AChR on the plasma membrane surface.5 About 60% of AChR antibody-positive myasthenia gravis patients have an enlarged thymus, and 10% have a thymoma—a tumor of the epithelial cells of this organ. Conversely, about 15% of patients with a thymoma have clinical myasthenia gravis, and an additional 20% possess antibodies against AChR in the serum without myasthenic symptoms.5
MuSK antibody-positive myasthenia gravis
Like AChR, MuSK is a transmembrane component of the postsynaptic neuromuscular junction. During formation of the neuromuscular junction, MuSK is activated through the binding of agrin (a nerve-derived proteoglycan) to lipoprotein-related protein 4 (LRP4), after which complicated intracellular signaling promotes the assembly and stabilization of AChR.6
Unlike AChR antibodies, antibodies against MuSK do not activate the complement system, and complement fixation is not essential for clinical myasthenic symptoms to appear.7 Also, myasthenia gravis with MuSK antibodies is rarely associated with thymoma.8
The precise mechanism by which MuSK antibody impairs transmission at the neuromuscular junction has been a mystery until recently. Animal models, including MuSK-mutant mice and mice injected with MuSK protein or with purified immunoglobulin G from patients with this disease, have revealed a significant reduction of AChR clusters and destruction of neuromuscular junction structures.7,9–12
In addition, MuSK antibodies produce pre-synaptic dysfunction, manifesting as a reduction of acetylcholine content. This information is based on studies in mice and on in vitro electrophysiologic analyses of neuromuscular junctions from a patient with this disease.7,9–13
Finally, MuSK antibodies may indirectly affect the recycling of acetylcholine. After post-synaptic activation, acetylcholine is normally hydrolized by acetylcholinesterase, which is located in the synaptic cleft but anchored to MuSK on the postsynaptic membrane. MuSK antibodies block the binding of MuSK to acetylcholinesterase, possibly leading to less accumulation of acetylcholinesterase.14 This process may explain why patients with MuSK antibody-positive myasthenia gravis tend to respond poorly to acetylcholinesterase inhibitors (more about this below).
Seronegative myasthenia gravis
In a series of 562 consecutive patients with generalized weakness due to myasthenia gravis, 92% were positive for AChR antibody, 3% were positive for MuSK antibody, and 5% were seronegative (possessing neither antibody).15 In contrast, about 50% of patients with purely ocular myasthenia gravis (ie, with isolated weakness of the levator palpebrae superioris, orbicularis oculi, or oculomotor muscles) are seropositive for AChR antibody. Only a few ocular MuSK antibody-positive cases have been described, leaving the rest seronegative. Rarely, both antibodies can be detected in the same patient.16
In patients who are negative for AChR antibodies at the time of disease onset, sero-conversion may occur later during the course. Repeating serologic testing 6 to 12 months later may detect AChR antibodies in approximately 15% of patients who were initially seronegative.15,17
The clinical presentation, electrophysiologic findings, thymic pathologic findings, and treatment responses are similar in AChR antibody-positive and seronegative myasthenia gravis.17 Muscle biopsy study in seronegative cases demonstrates a loss of AChR as well.18
Based on these observations, it has been proposed that seronegative patients may have low-affinity antibodies that can bind to tightly clustered AChRs on the postsynaptic membrane but escape detection by routine radioimmunoassays in a solution phase. With a sensitive cell-based immunofluorescence assay, low-affinity antibodies to clustered AChRs were detected in 66% of patients with generalized myasthenia gravis and in 50% of those with ocular myasthenia gravis who were seronegative on standard assays.19,20 These low-affinity AChR antibodies can also activate complement in vitro, increasing the likelihood that they are pathogenic. However, assays to detect low-affinity AChR antibodies are not yet commercially available.
Within the past year, three research groups independently reported detecting antibodies to LRP4 in 2% to 50% of seronegative myasthenia gravis patients. This wide variation in the prevalence of LRP4 antibodies could be related to patient ethnicity and methods of detection.21–23 LRP4 is a receptor for agrin and is required for agrin-induced MuSK activation and AChR clustering. LRP antibodies can activate complement; therefore, it is plausible that LRP4 antibody binding leads to AChR loss on the postsynaptic membrane. However, additional study is needed to determine if LRP4 antibodies are truly pathogenic in myasthenia gravis.
A DISORDER OF FATIGABLE WEAKNESS

Myasthenia gravis is a disorder of fatigable weakness producing fluctuating symptoms. Symptoms related to the involvement of specific muscle groups are listed in Table 1. Muscle weakness is often worse later in the day or after exercise.
Ocular myasthenia gravis accounts for about 15% of all cases. Of patients initially presenting with ocular symptoms only, twothirds will ultimately develop generalized symptoms, most within the first 2 years.24 No factor has been identified that predicts conversion from an ocular to a generalized form.
Several clinical phenotypes of MuSK antibody-positive myasthenia gravis have been described. An oculobulbar form presents with diplopia, ptosis, dysarthria, and profound atrophy of the muscles of the tongue and face. A restricted myopathic form presents with prominent neck, shoulder, and respiratory weakness without ocular involvement. A third form is a combination of ocular and proximal limb weakness, indistinguishable from AChR antibody-positive disease.25
MuSK antibody-positive patients do not respond as well to acetylcholinesterase inhibitors as AChR antibody-positive patients do. In one study, nearly 70% of MuSK antibody-positive patients demonstrated no response, poor tolerance, or cholinergic hypersensitivity to these agents.25 Fortunately, most MuSK antibody-positive patients have a favorable response to immunosuppressive therapy—sometimes a dramatic improvement after plasmapheresis.8
DIAGNOSIS OF MYASTHENIA GRAVIS
The common differential diagnoses for myasthenia gravis are listed in Table 2.
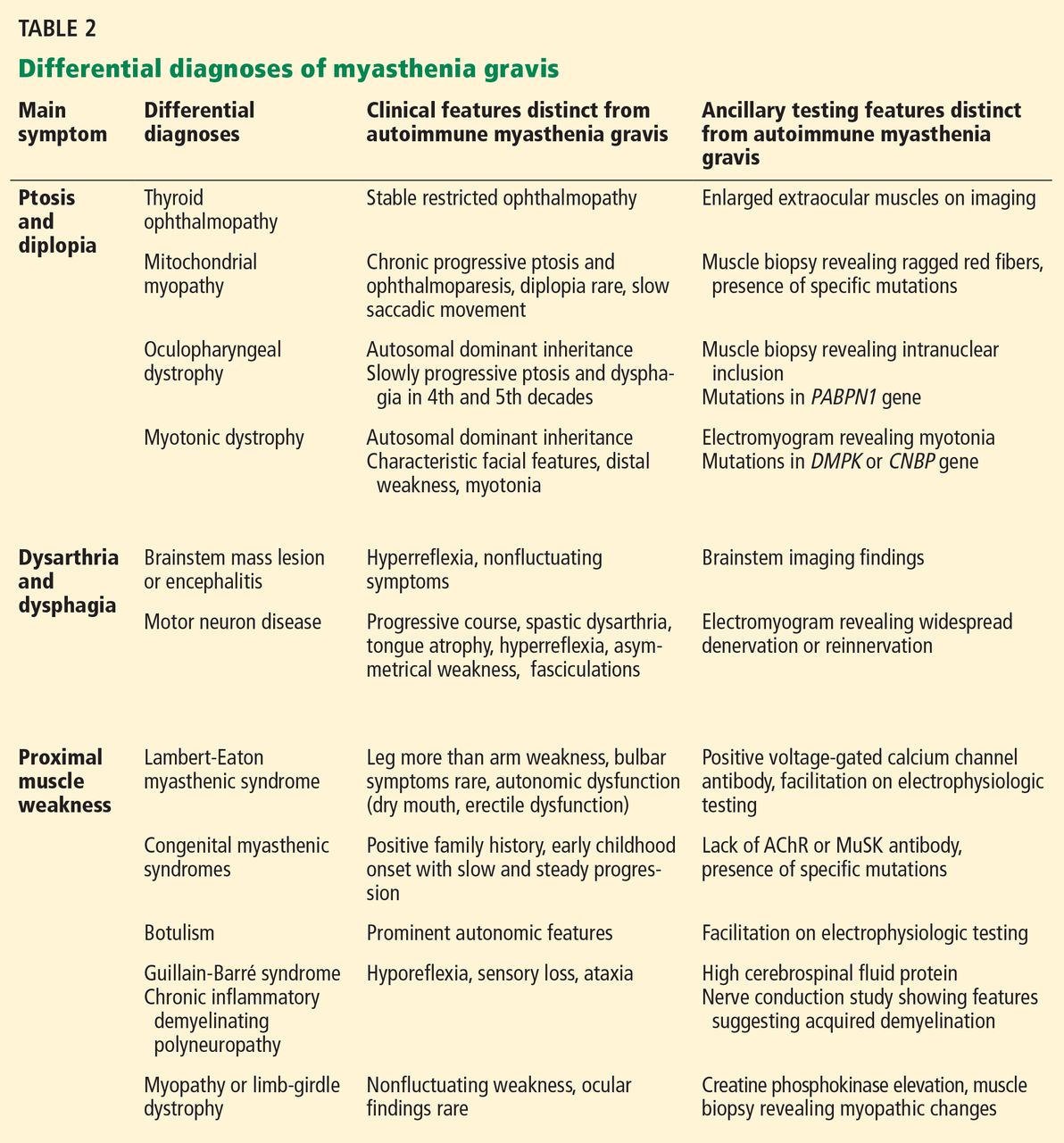
The essential feature of myasthenia gravis is fluctuating muscle weakness, often with fatigue. Many patients complain of weakness of specific muscle groups after their repeated usage. Pain is generally a less conspicuous symptom, and generalized fatigue without objective weakness is inconsistent with myasthenia gravis.
Signs of muscle weakness may include droopy eyelids, diplopia, inability to hold the head straight, difficulty swallowing or chewing, speech disturbances, difficulty breathing, and difficulty raising the arms or rising from the sitting position. A historical pattern of ptosis alternating from one eye to the other is fairly characteristic of myasthenia gravis.
The weakness of orbicularis oculi is easily identified on examination by prying open the eyes during forced eye closure. Limb weakness is usually more significant in the arms than in the legs. An often-neglected feature of myasthenia gravis is finger extensor weakness with a relative sparing of other distal hand muscles.2
The ice-pack test is performed by placing a small bag of ice over the ptotic eye for 2 to 5 minutes and assessing the degree of ptosis for any noticeable improvement. This test is not very helpful for assessing ocular motor weakness.
The edrophonium (Tensilon) test can be used for patients with ptosis or ophthalmoparesis. Edrophonium, a short-acting acetylcholinesterase inhibitor, is given intravenously while the patient is observed for objective improvement. The patient’s cardiovascular status should be monitored for arrhythmias and hypotension. Atropine should be immediately available in case severe bradycardia develops.
The ice-pack test and the edrophonium test can give false-negative and false-positive results, and the diagnosis of myasthenia gravis must be verified by other diagnostic tests.
Testing for antibodies
Testing for circulating AChR antibodies, MuSK antibodies, or both is the first step in the laboratory confirmation of myasthenia gravis.
There are three AChR antibody subtypes: binding, blocking, and modulating. Binding antibodies are present in 80% to 90% of patients with generalized myasthenia gravis and 50% of those with ocular myasthenia gravis. Testing for blocking and modulating AChR antibodies increases the sensitivity by less than 5% when added to testing for binding antibodies.
AChR antibody titers correlate poorly with disease severity between patients. However, in individual patients, antibody titers tend to go down in parallel with clinical improvement.
MuSK antibody is detected in nearly half of myasthenia gravis patients with generalized weakness who are negative for AChR antibody.
Electrophysiologic tests
Electrophysiologic tests can usually confirm the diagnosis of seronegative myasthenia gravis. They are also helpful in seropositive patients who have unusual clinical features or a poor response to treatment.
Repetitive nerve stimulation studies use a slow rate (2–5 Hz) of repetitive electrical stimulation. The study is positive if the motor response declines by more than 10%. However, a decremental response is not specific for myasthenia gravis, as it may be seen in other neuromuscular disorders such as motor neuron disease or Lambert-Eaton myasthenic syndrome.
This test is technically easier to do in distal muscles than in proximal muscles, but less sensitive. Therefore, proximal muscles such as the trapezius or facial muscles are usually also sampled to maximize the yield. To further maximize the sensitivity, muscles being tested should be warm, and acetylcholinesterase inhibitors should be withheld for 12 hours before.
Repetitive nerve stimulation studies in distal muscles are positive in approximately 75% of patients with generalized myasthenia gravis and in 30% with ocular myasthenia gravis.26
Single-fiber electromyography is more technically demanding than repetitive nerve stimulation and is less widely available. It is usually performed with a special needle electrode that can simultaneously identify action potentials arising from individual muscle fibers innervated by the same axon.
Variability in time of the second action potential relative to the first is called “jitter.” Abnormal jitter is seen in more than 95% of patients with generalized myasthenia gravis and in 85% to 90% of those with ocular myasthenia gravis.26,27 However, abnormal jitter can also be seen in other neuromuscular diseases such as motor neuron disease or in neuromuscular junctional disorders such as Lambert-Eaton myasthenic syndrome.
Imaging studies
Chest computed tomography or magnetic resonance imaging with contrast should be performed in all myasthenia gravis patients to look for a thymoma.
TREATMENT OF MYASTHENIA GRAVIS
Acetylcholinesterase inhibitors
As a reasonable first therapy in mild cases of myasthenia gravis, acetylcholinesterase inhibitors slow down the degradation of acetylcholine and prolong its effect in the neuromuscular junction, but they are not disease-modifying and their benefits are mild.
Pyridostigmine is the usual choice of acetylcholinesterase inhibitor. Its onset of action is rapid (15 to 30 minutes) and its action lasts for 3 to 4 hours. For most patients, the effective dosage range is 60 mg to 90 mg every 4 to 6 hours. A long-acting form is also available and can be given as a single nighttime dose.
Immunomodulating therapy
Patients who have moderate to severe symptoms require some form of immunomodulating therapy.
Plasmapheresis or intravenous immune globulin is reserved for patients with severe or rapidly worsening disease because their beneficial effects can be seen within the first week of treatment.
Longer-acting immunotherapies (corticosteroids, azathioprine, mycophenolate mofetil and others) have a slower onset of responses but provide sustained benefits. Which drug to use depends on factors such as comorbidity, side effects, and cost.
Drugs to avoid
A number of medications can exacerbate weakness in myasthenia gravis and should be avoided or used with caution. The list is long, but ones that deserve the most attention are penicillamine, interferons, procainamide, quinidine, and antibiotics, including quinolones and aminoglycosides. A more comprehensive list of medications that may exacerbate myasthenia gravis symptoms can be found in a review by Keesey.2
RAPID INDUCTION IMMUNOTHERAPIES : PLASMAPHERESIS, IMMUNE GLOBULIN
Both plasmapheresis and intravenous immune globulin act quickly over days, but in most patients their effects last only a few weeks. Both are used as rescue therapies for myasthenic crises, bridging therapy to slow-acting immunotherapeutic agents, or maintenance treatment for poorly controlled cases.
Several retrospective studies have confirmed the efficacy of plasmapheresis in more than 80% of patients with generalized symptoms.28,29
In a randomized trial in patients with generalized therapies, intravenous immune globulin improved muscle strength in the group of patients with severe symptoms.30 The effective dosage of intravenous immune globulin varies from 1 to 2 g/kg without observed difference between doses.31 Trials comparing the efficacy of intravenous immune globulin and plasmapheresis in acute and severe myasthenia gravis did not reveal a difference in efficacy.32,33 Intravenous immune globulin at a minimal dose of 0.4 g/kg every 3 months has been successfully used as a long-term maintenance monotherapy, and such a role could be expanded to more patients with further studies.34
The choice between plasmapheresis and intravenous immune globulin is often based on the ability of a patient to tolerate each treatment and on the availability of the plasmapheresis procedure. Intravenous immune globulin is easier to administer, is associated with fewer adverse events related to vascular access, and is therefore more appropriate than plasmapheresis in some centers.
CHRONIC MAINTENANCE IMMUNOMODULATING TREATMENT
Corticosteroids
Prednisone, the most commonly used agent, leads to remission or marked improvement in 70% to 80% of patients with ocular or generalized myasthenia gravis.35 It may also reduce the progression of ocular myasthenia gravis to the generalized form.36
The effective dose of prednisone depends on the severity and distribution of symptoms. Some patients may need up to 1.0 mg/kg/day (usually 50 to 80 mg per day). In patients with mild to moderate symptoms, a lower maximal dosage such as 20 to 40 mg per day can be sufficient.
Within 1 to 2 weeks after starting high-dose prednisone, up to 50% of patients may develop a transient deterioration, including possible precipitation of a myasthenic crisis.37 For this reason, high-dose prednisone is commonly started only in hospitalized patients who are also receiving plasmapheresis or intravenous immune globulin. Otherwise, an outpatient dose-escalation protocol can be used to achieve a target dose over several weeks.
Prednisone tapering can begin after the patient has been on the maximal dose for 1 to 2 months and significant improvement is evident. A monthly tapering of 5 to 10 mg is preferred, then more slowly after the daily dose reaches 30 mg. The usual maintenance dose averages about 5 mg daily.
Common side effects of prednisone include weight gain, cushingoid features, easy bruising, cataracts, glaucoma, hypertension, diabetes, dyslipidemia, and osteoporosis. Patients are advised to take supplemental calcium (1,500 mg per day) and vitamin D (400 to 800 IU per day). For those most at risk of osteoporosis, treatment with a bisphosphonate should be considered.
Other immunotherapeutic agents are often needed, either to replace the corticosteroid or to permit use of lower doses of it. Because of their delayed onset of action, starting such corticosteroid-sparing agents early in the course is often necessary. These agents are often initially combined with high-dose prednisone, with an eventual goal of weaning off prednisone entirely. This strategy offers the advantage of relatively rapid induction while avoiding the long-term adverse effects of corticosteroid treatment.
Azathioprine
Azathioprine doesn’t begin to show a beneficial effect in myasthenia gravis for 6 to 12 months, and it often reaches its maximal efficacy only after 1 to 2 years of treatment.38
In a study of 78 myasthenia gravis patients, 91% improved when treated with azathioprine alone or together with prednisone.39 In another study using azathioprine and prednisolone for generalized myasthenia gravis, nearly two-thirds of patients came off prednisolone while maintaining remission for 3 years.38
A typical maintenance dose is 2 to 3 mg/kg/day. Common side effects are nausea, vomiting, and malaise. Less frequent side effects include hematologic abnormalities, abnormal liver function, and pancreatitis. Monthly monitoring of complete blood cell counts and liver function tests is warranted for the first 6 months, then less often.
One in 300 people in the general population is homozygous for a mutant allele in the thiopurine methyltransferase (TPMT) gene. Patients with this genotype should not receive azathioprine because of the risk of life-threatening bone marrow suppression.40 A slightly increased risk of various forms of lymphoma has been documented.41
Mycophenolate mofetil
A well-tolerated medication with few side effects, mycophenolate mofetil is being used more in myasthenia gravis. The results of two recent randomized trials suggested that it is not effective in improving myasthenia gravis symptoms or sparing prednisone dosage when used for 90 days or 36 weeks.42,43 However, extensive clinical experience supports its longterm efficacy in myasthenia gravis.
In a retrospective study of 85 patients with generalized myasthenia gravis, mycophenolate at doses of 1 to 3 g daily improved symptoms in 73% and produced remission in 50%. Steroid dosage was reduced in 71% of patients.44
Another retrospective study, with 102 patients, verified a slow development of clinical benefit after months of mycophenolate therapy alone or in combination with prednisone. Approximately 50% of patients achieved a minimal manifestation status after 6 to 12 months of mycophenolate treatment. Eventually, at 24 months of treatment, 80% of patients had a desirable outcome of minimal clinical manifestation or better, 55% of patients were able to come off prednisone entirely, and 75% were taking less than 7.5 mg of prednisone per day.45
Common side effects of mycophenolate include nausea, diarrhea, and infections such as urinary tract infections and herpes reactivation. The complete blood cell count needs to be monitored frequently during the first 6 months of therapy. Leukopenia can occur but rarely necessitates stopping mycophenolate. Long-term safety data are lacking, but so far there has been no clearly increased risk of malignancy.
Mycophenolate exposure in pregnancy results in a high incidence of major fetal malformations. Therefore, its use in pregnant patients is discouraged, and women of child-bearing age should use effective contraception.46
Cyclosporine
A randomized trial in a small number of patients suggested that cyclosporine is fairly effective as monotherapy.47 Its onset of action in myasthenia gravis is faster than that of other corticosteroid-sparing agents, and clinical benefit can often be observed as early as 1 to 2 months. A dose of 5 mg/kg/day and a maintenance serum level of 100 to 150 ng/mL are generally recommended. However, renal, hepatic, and hematologic toxicities and interactions with other medications make cyclosporine a less attractive choice.
Methotrexate
A randomized trial evaluated the utility of methotrexate as a steroid-sparing agent compared with azathioprine.48 At 24 months, its steroid-sparing effect was similar to that of azathioprine, and the prednisone dosage had been reduced in more than 50% of patients.
Another phase II trial studying the efficacy of methotrexate in myasthenia gravis is under way.49
Rituximab
Rituximab is a monoclonal antibody against B-cell membrane marker CD20. A growing number of case series support its efficacy in patients with severe generalized myasthenia gravis refractory to multiple immunosuppressants.16,50 It seems particularly effective for MuSK antibody-positive disease, reducing MuSK antibody titers and having a treatment effect that lasts for years.
The standard dosage is 375 mg/m2 per week for 4 consecutive weeks. Peripheral B cells tend to be depleted within 2 weeks after the first infusion, while T-cell populations remain unchanged.50
A minimal infusion reaction such as flushing and chills can be seen with the first infusion. Patients may be more susceptible to certain infections such as reactivation of herpes zoster, but overall rituximab is well tolerated. Rare cases of progressive multifocal leukoencephalopathy have been reported in patients taking it, but none have occurred so far in myasthenia gravis treatment.
Cyclophosphamide
Cyclophosphamide is an alkylating agent that reduces proliferation of both B and T cells. It can be effective in myasthenia gravis, but potentially serious side effects limit its use. It should be reserved for the small percentage of cases that are refractory to other immunotherapies.
Thymectomy
Surgical treatment should be considered for patients with thymoma. If the tumor cannot be surgically resected, chemoradiotherapy can be considered for relief of myasthenic symptoms and for prevention of local invasion.
Thymomas recur in a minority of patients many years after the initial resection, sometimes without myasthenia gravis symptoms. A recurrence of symptoms does not necessarily indicate a recurrence of thymoma. The lack of correlation between myasthenia gravis symptoms and thymoma recurrence highlights the importance of radiologic follow-up in these patients.
For patients without thymoma, many experts believe that thymectomy is beneficial in patients under age 60 who have generalized myasthenia gravis. The likelihood of medication-free remission is about twice as high, and the likelihood of becoming asymptomatic is about one and a half times higher after thymectomy.51 However, it takes up to several years for the benefits of thymectomy to manifest, and thymectomy does not guarantee protection from developing AChR antibody-positive myasthenia gravis in the future.
The optimal timing of thymectomy is not well established; however, the procedure is usually recommended within the first 3 years of diagnosis.52 The response rates from thymectomy are similar for AChR antibody-positive and seronegative patients. In general, thymectomy for MuSK antibody-positive patients has not been effective, and its role in ocular myasthenia gravis is unclear.2,53
- Alshekhlee A, Miles JD, Katirji B, Preston DC, Kaminski HJ. Incidence and mortality rates of myasthenia gravis and myasthenic crisis in US hospitals. Neurology 2009; 72:1548–1554.
- Keesey JC. Clinical evaluation and management of myasthenia gravis. Muscle Nerve 2004; 29:484–505.
- Leite MI, Coutinho E, Lana-Peixoto M, et al. Myasthenia gravis and neuromyelitis optica spectrum disorder: a multicenter study of 16 patients. Neurology 2012; 78:1601–1607.
- Drachman DB, Angus CW, Adams RN, Michelson JD, Hoffman GJ. Myasthenic antibodies cross-link acetylcholine receptors to accelerate degradation. N Engl J Med 1978; 298:1116–1122.
- Fujii Y. The thymus, thymoma and myasthenia gravis. Surg Today 2013; 43:461–466.
- Evoli A, Lindstrom J. Myasthenia gravis with antibodies to MuSK: another step toward solving mystery? Neurology 2011; 77:1783–1784.
- Mori S, Kubo S, Akiyoshi T, et al. Antibodies against muscle-specific kinase impair both presynaptic and postsynaptic functions in a murine model of myasthenia gravis. Am J Pathol 2012; 180:798–810.
- Guptill JT, Sanders DB, Evoli A. Anti-MuSK antibody myasthenia gravis: clinical findings and response to treatment in two large cohorts. Muscle Nerve 2011; 44:36–40.
- Chevessier F, Girard E, Molgó J, et al. A mouse model for congenital myasthenic syndrome due to MuSK mutations reveals defects in structure and function of neuromuscular junctions. Hum Mol Genet 2008; 17:3577–3595.
- Richman DP, Nishi K, Morell SW, et al. Acute severe animal model of anti-muscle-specific kinase myasthenia: combined postsynaptic and presynaptic changes. Arch Neurol 2012; 69:453–460.
- Klooster R, Plomp JJ, Huijbers MG, et al. Muscle-specific kinase myasthenia gravis IgG4 autoantibodies cause severe neuromuscular junction dysfunction in mice. Brain 2012; 135:1081–1101.
- Viegas S, Jacobson L, Waters P, et al. Passive and active immunization models of MuSK-Ab positive myasthenia: electrophysiological evidence for pre and postsynaptic defects. Exp Neurol 2012; 234:506–512.
- Niks EH, Kuks JB, Wokke JH, et al. Pre- and postsynaptic neuromuscular junction abnormalities in musk myasthenia. Muscle Nerve 2010; 42:283–288.
- Kawakami Y, Ito M, Hirayama M, et al. Anti-MuSK autoantibodies block binding of collagen Q to MuSK. Neurology 2011; 77:1819–1826.
- Chan KH, Lachance DH, Harper CM, Lennon VA. Frequency of seronegativity in adult-acquired generalized myasthenia gravis. Muscle Nerve 2007; 36:651–658.
- Collongues N, Casez O, Lacour A, et al. Rituximab in refractory and non-refractory myasthenia: a retrospective multicenter study. Muscle Nerve 2012; 46:687–691.
- Sanders DB, Andrews PI, Howard JF, Massey JM. Seronegative myasthenia gravis. Neurology 1997; 48(suppl 5):40S–45S.
- Shiraishi H, Motomura M, Yoshimura T, et al. Acetylcholine receptors loss and postsynaptic damage in MuSK antibody-positive myasthenia gravis. Ann Neurol 2005; 57:289–293.
- Leite MI, Jacob S, Viegas S, et al. IgG1 antibodies to acetylcholine receptors in ‘seronegative’ myasthenia gravis. Brain 2008; 131:1940–1952.
- Jacob S, Viegas S, Leite MI, et al. Presence and pathogenic relevance of antibodies to clustered acetylcholine receptor in ocular and generalized myasthenia gravis. Arch Neurol 2012; 69:994–1001.
- Higuchi O, Hamuro J, Motomura M, Yamanashi Y. Autoantibodies to low-density lipoprotein receptor-related protein 4 in myasthenia gravis. Ann Neurol 2011; 69:418–422.
- Pevzner A, Schoser B, Peters K, et al. Anti-LRP4 autoantibodies in AChR- and MuSK-antibody-negative myasthenia gravis. J Neurol 2012; 259:427–435.
- Zhang B, Tzartos JS, Belimezi M, et al. Autoantibodies to lipoprotein-related protein 4 in patients with double-seronegative myasthenia gravis. Arch Neurol 2012; 69:445–451.
- Kupersmith MJ, Latkany R, Homel P. Development of generalized disease at 2 years in patients with ocular myasthenia gravis. Arch Neurol 2003; 60:243–248.
- Pasnoor M, Wolfe GI, Nations S, et al. Clinical findings in MuSK-antibody positive myasthenia gravis: a US experience. Muscle Nerve 2010; 41:370–374.
- Oh SJ, Kim DE, Kuruoglu R, Bradley RJ, Dwyer D. Diagnostic sensitivity of the laboratory tests in myasthenia gravis. Muscle Nerve 1992; 15:720–724.
- Sanders DB, Stålberg EV. AAEM minimonograph #25: single-fiber electromyography. Muscle Nerve 1996; 19:1069–1083.
- Lazo-Langner A, Espinosa-Poblano I, Tirado-Cárdenas N, et al. Therapeutic plasma exchange in Mexico: experience from a single institution. Am J Hematol 2002; 70:16–21.
- Carandina-Maffeis R, Nucci A, Marques JF, et al. Plasmapheresis in the treatment of myasthenia gravis: retrospective study of 26 patients. Arq Neuropsiquiatr 2004; 62:391–395.
- Zinman L, Ng E, Bril V. IV immunoglobulin in patients with myasthenia gravis: a randomized controlled trial. Neurology 2007; 68:837–841.
- Gajdos P, Tranchant C, Clair B, et al; Myasthenia Gravis Clinical Study Group. Treatment of myasthenia gravis exacerbation with intravenous immunoglobulin: a randomized double-blind clinical trial. Arch Neurol 2005; 62:1689–1693.
- Rønager J, Ravnborg M, Hermansen I, Vorstrup S. Immunoglobulin treatment versus plasma exchange in patients with chronic moderate to severe myasthenia gravis. Artif Organs 2001; 25:967–973.
- Barth D, Nabavi Nouri M, Ng E, Nwe P, Bril V. Comparison of IVIg and PLEX in patients with myasthenia gravis. Neurology 2011; 76:2017–2023.
- Wegner B, Ahmed I. Intravenous immunoglobulin monotherapy in long-term treatment of myasthenia gravis. Clin Neurol Neurosurg 2002; 105:3–8.
- Pascuzzi RM, Coslett HB, Johns TR. Long-term corticosteroid treatment of myasthenia gravis: report of 116 patients. Ann Neurol 1984; 15:291–298.
- Monsul NT, Patwa HS, Knorr AM, Lesser RL, Goldstein JM. The effect of prednisone on the progression from ocular to generalized myasthenia gravis. J Neurol Sci 2004; 217:131–133.
- Miller RG, Milner-Brown HS, Mirka A. Prednisone-induced worsening of neuromuscular function in myasthenia gravis. Neurology 1986; 36:729–732.
- Palace J, Newsom-Davis J, Lecky B. A randomized double-blind trial of prednisolone alone or with azathioprine in myasthenia gravis. Myasthenia Gravis Study Group. Neurology 1998; 50:1778–1783.
- Mertens HG, Hertel G, Reuther P, Ricker K. Effect of immunosuppressive drugs (azathioprine). Ann N Y Acad Sci 1981; 377:691–699.
- Relling MV, Gardner EE, Sandborn WJ, et al; Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing. Clin Pharmacol Ther 2011; 89:387–391.
- Finelli PF. Primary CNS lymphoma in myasthenic on long-term azathioprine. J Neurooncol 2005; 74:91–92.
- Sanders DB, Hart IK, Mantegazza R, et al. An international, phase III, randomized trial of mycophenolate mofetil in myasthenia gravis. Neurology 2008; 71:400–406.
- Muscle Study Group. A trial of mycophenolate mofetil with prednisone as initial immunotherapy in myasthenia gravis. Neurology 2008; 71:394–399.
- Meriggioli MN, Ciafaloni E, Al-Hayk KA, et al. Mycophenolate mofetil for myasthenia gravis: an analysis of efficacy, safety, and tolerability. Neurology 2003; 61:1438–1440.
- Hehir MK, Burns TM, Alpers J, Conaway MR, Sawa M, Sanders DB. Mycophenolate mofetil in AChR-antibody-positive myasthenia gravis: outcomes in 102 patients. Muscle Nerve 2010; 41:593–598.
- Merlob P, Stahl B, Klinger G. Tetrada of the possible mycophenolate mofetil embryopathy: a review. Reprod Toxicol 2009; 28:105–108.
- Tindall RS, Rollins JA, Phillips JT, Greenlee RG, Wells L, Belendiuk G. Preliminary results of a double-blind, randomized, placebo-controlled trial of cyclosporine in myasthenia gravis. N Engl J Med 1987; 316:719–724.
- Heckmann JM, Rawoot A, Bateman K, Renison R, Badri M. A single-blinded trial of methotrexate versus azathioprine as steroid-sparing agents in generalized myasthenia gravis. BMC Neurol 2011; 11:97.
- Pasnoor M, He J, Herbelin L, Dimachkie M, Barohn RJ; Muscle Study Group. Phase II trial of methotrexate in myasthenia gravis. Ann N Y Acad Sci 2012; 1275:23–28.
- Díaz-Manera J, Martínez-Hernández E, Querol L, et al. Long-lasting treatment effect of rituximab in MuSK myasthenia. Neurology 2012; 78:189–193.
- Gronseth GS, Barohn RJ. Practice parameter: thymectomy for autoimmune myasthenia gravis (an evidence-based review): Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2000; 55:7–15.
- Kumar V, Kaminski HJ. Treatment of myasthenia gravis. Curr Neurol Neurosci Rep 2011; 11:89–96.
- Pompeo E, Tacconi F, Massa R, Mineo D, Nahmias S, Mineo TC. Long-term outcome of thoracoscopic extended thymectomy for nonthymomatous myasthenia gravis. Eur J Cardiothorac Surg 2009; 36:164–169.
Current therapies for myasthenia gravis can help most patients achieve sustained improvement. The overall prognosis has dramatically improved over the last 4 decades: the mortality rate used to be 75%; now it is 4.5%.1
Myasthenia gravis is the most common disorder of neuromuscular junction transmission and is also one of the best characterized autoimmune diseases. However, its symptoms—primarily weakness—vary from patient to patient, and in the same patient, by time of day and over longer time periods. The variation in symptoms can be very confusing to undiagnosed patients and puzzling to unsuspecting physicians. Such diagnostic uncertainty can give the patient additional frustration and emotional stress, which in turn exacerbate his or her condition.
In this review, we will give an overview of the pathogenesis, clinical manifestations, diagnosis, and treatment of myasthenia gravis.
TWO PEAKS IN INCIDENCE BY AGE
The annual incidence of myasthenia gravis is approximately 10 to 20 new cases per million, with a prevalence of about 150 to 200 per million.2
The age of onset has a bimodal distribution, with an early incidence peak in the second to third decade with a female predominance and a late peak in the 6th to the 8th decade with a male predominance.2
Myasthenia gravis is commonly associated with several other autoimmune disorders, including hypothyroidism, hyperthyroidism, systemic lupus erythematosus, rheumatoid arthritis, vitiligo, diabetes, and, more recently recognized, neuromyelitis optica.3
ANTIBODIES AGAINST AChR AND MuSK
In most cases of myasthenia gravis the patient has autoimmune antibodies against constituents of the neuromuscular junction, specifically acetylcholine receptor (AChR) and muscle-specific tyrosine kinase (MuSK) (Figure 1).
AChR antibody-positive myasthenia gravis
When antibodies bind to AChR on the postsynaptic membrane, they cross-link neighboring AChR units, which are absorbed into the muscle fiber and are broken up.4 In addition, the complement system is activated to mediate further damage on the postsynaptic membrane.
AChR antibodies may come from germinal centers of the thymus, where clustered myoid cells express AChR on the plasma membrane surface.5 About 60% of AChR antibody-positive myasthenia gravis patients have an enlarged thymus, and 10% have a thymoma—a tumor of the epithelial cells of this organ. Conversely, about 15% of patients with a thymoma have clinical myasthenia gravis, and an additional 20% possess antibodies against AChR in the serum without myasthenic symptoms.5
MuSK antibody-positive myasthenia gravis
Like AChR, MuSK is a transmembrane component of the postsynaptic neuromuscular junction. During formation of the neuromuscular junction, MuSK is activated through the binding of agrin (a nerve-derived proteoglycan) to lipoprotein-related protein 4 (LRP4), after which complicated intracellular signaling promotes the assembly and stabilization of AChR.6
Unlike AChR antibodies, antibodies against MuSK do not activate the complement system, and complement fixation is not essential for clinical myasthenic symptoms to appear.7 Also, myasthenia gravis with MuSK antibodies is rarely associated with thymoma.8
The precise mechanism by which MuSK antibody impairs transmission at the neuromuscular junction has been a mystery until recently. Animal models, including MuSK-mutant mice and mice injected with MuSK protein or with purified immunoglobulin G from patients with this disease, have revealed a significant reduction of AChR clusters and destruction of neuromuscular junction structures.7,9–12
In addition, MuSK antibodies produce pre-synaptic dysfunction, manifesting as a reduction of acetylcholine content. This information is based on studies in mice and on in vitro electrophysiologic analyses of neuromuscular junctions from a patient with this disease.7,9–13
Finally, MuSK antibodies may indirectly affect the recycling of acetylcholine. After post-synaptic activation, acetylcholine is normally hydrolized by acetylcholinesterase, which is located in the synaptic cleft but anchored to MuSK on the postsynaptic membrane. MuSK antibodies block the binding of MuSK to acetylcholinesterase, possibly leading to less accumulation of acetylcholinesterase.14 This process may explain why patients with MuSK antibody-positive myasthenia gravis tend to respond poorly to acetylcholinesterase inhibitors (more about this below).
Seronegative myasthenia gravis
In a series of 562 consecutive patients with generalized weakness due to myasthenia gravis, 92% were positive for AChR antibody, 3% were positive for MuSK antibody, and 5% were seronegative (possessing neither antibody).15 In contrast, about 50% of patients with purely ocular myasthenia gravis (ie, with isolated weakness of the levator palpebrae superioris, orbicularis oculi, or oculomotor muscles) are seropositive for AChR antibody. Only a few ocular MuSK antibody-positive cases have been described, leaving the rest seronegative. Rarely, both antibodies can be detected in the same patient.16
In patients who are negative for AChR antibodies at the time of disease onset, sero-conversion may occur later during the course. Repeating serologic testing 6 to 12 months later may detect AChR antibodies in approximately 15% of patients who were initially seronegative.15,17
The clinical presentation, electrophysiologic findings, thymic pathologic findings, and treatment responses are similar in AChR antibody-positive and seronegative myasthenia gravis.17 Muscle biopsy study in seronegative cases demonstrates a loss of AChR as well.18
Based on these observations, it has been proposed that seronegative patients may have low-affinity antibodies that can bind to tightly clustered AChRs on the postsynaptic membrane but escape detection by routine radioimmunoassays in a solution phase. With a sensitive cell-based immunofluorescence assay, low-affinity antibodies to clustered AChRs were detected in 66% of patients with generalized myasthenia gravis and in 50% of those with ocular myasthenia gravis who were seronegative on standard assays.19,20 These low-affinity AChR antibodies can also activate complement in vitro, increasing the likelihood that they are pathogenic. However, assays to detect low-affinity AChR antibodies are not yet commercially available.
Within the past year, three research groups independently reported detecting antibodies to LRP4 in 2% to 50% of seronegative myasthenia gravis patients. This wide variation in the prevalence of LRP4 antibodies could be related to patient ethnicity and methods of detection.21–23 LRP4 is a receptor for agrin and is required for agrin-induced MuSK activation and AChR clustering. LRP antibodies can activate complement; therefore, it is plausible that LRP4 antibody binding leads to AChR loss on the postsynaptic membrane. However, additional study is needed to determine if LRP4 antibodies are truly pathogenic in myasthenia gravis.
A DISORDER OF FATIGABLE WEAKNESS
Myasthenia gravis is a disorder of fatigable weakness producing fluctuating symptoms. Symptoms related to the involvement of specific muscle groups are listed in Table 1. Muscle weakness is often worse later in the day or after exercise.
Ocular myasthenia gravis accounts for about 15% of all cases. Of patients initially presenting with ocular symptoms only, twothirds will ultimately develop generalized symptoms, most within the first 2 years.24 No factor has been identified that predicts conversion from an ocular to a generalized form.
Several clinical phenotypes of MuSK antibody-positive myasthenia gravis have been described. An oculobulbar form presents with diplopia, ptosis, dysarthria, and profound atrophy of the muscles of the tongue and face. A restricted myopathic form presents with prominent neck, shoulder, and respiratory weakness without ocular involvement. A third form is a combination of ocular and proximal limb weakness, indistinguishable from AChR antibody-positive disease.25
MuSK antibody-positive patients do not respond as well to acetylcholinesterase inhibitors as AChR antibody-positive patients do. In one study, nearly 70% of MuSK antibody-positive patients demonstrated no response, poor tolerance, or cholinergic hypersensitivity to these agents.25 Fortunately, most MuSK antibody-positive patients have a favorable response to immunosuppressive therapy—sometimes a dramatic improvement after plasmapheresis.8
DIAGNOSIS OF MYASTHENIA GRAVIS
The common differential diagnoses for myasthenia gravis are listed in Table 2.
The essential feature of myasthenia gravis is fluctuating muscle weakness, often with fatigue. Many patients complain of weakness of specific muscle groups after their repeated usage. Pain is generally a less conspicuous symptom, and generalized fatigue without objective weakness is inconsistent with myasthenia gravis.
Signs of muscle weakness may include droopy eyelids, diplopia, inability to hold the head straight, difficulty swallowing or chewing, speech disturbances, difficulty breathing, and difficulty raising the arms or rising from the sitting position. A historical pattern of ptosis alternating from one eye to the other is fairly characteristic of myasthenia gravis.
The weakness of orbicularis oculi is easily identified on examination by prying open the eyes during forced eye closure. Limb weakness is usually more significant in the arms than in the legs. An often-neglected feature of myasthenia gravis is finger extensor weakness with a relative sparing of other distal hand muscles.2
The ice-pack test is performed by placing a small bag of ice over the ptotic eye for 2 to 5 minutes and assessing the degree of ptosis for any noticeable improvement. This test is not very helpful for assessing ocular motor weakness.
The edrophonium (Tensilon) test can be used for patients with ptosis or ophthalmoparesis. Edrophonium, a short-acting acetylcholinesterase inhibitor, is given intravenously while the patient is observed for objective improvement. The patient’s cardiovascular status should be monitored for arrhythmias and hypotension. Atropine should be immediately available in case severe bradycardia develops.
The ice-pack test and the edrophonium test can give false-negative and false-positive results, and the diagnosis of myasthenia gravis must be verified by other diagnostic tests.
Testing for antibodies
Testing for circulating AChR antibodies, MuSK antibodies, or both is the first step in the laboratory confirmation of myasthenia gravis.
There are three AChR antibody subtypes: binding, blocking, and modulating. Binding antibodies are present in 80% to 90% of patients with generalized myasthenia gravis and 50% of those with ocular myasthenia gravis. Testing for blocking and modulating AChR antibodies increases the sensitivity by less than 5% when added to testing for binding antibodies.
AChR antibody titers correlate poorly with disease severity between patients. However, in individual patients, antibody titers tend to go down in parallel with clinical improvement.
MuSK antibody is detected in nearly half of myasthenia gravis patients with generalized weakness who are negative for AChR antibody.
Electrophysiologic tests
Electrophysiologic tests can usually confirm the diagnosis of seronegative myasthenia gravis. They are also helpful in seropositive patients who have unusual clinical features or a poor response to treatment.
Repetitive nerve stimulation studies use a slow rate (2–5 Hz) of repetitive electrical stimulation. The study is positive if the motor response declines by more than 10%. However, a decremental response is not specific for myasthenia gravis, as it may be seen in other neuromuscular disorders such as motor neuron disease or Lambert-Eaton myasthenic syndrome.
This test is technically easier to do in distal muscles than in proximal muscles, but less sensitive. Therefore, proximal muscles such as the trapezius or facial muscles are usually also sampled to maximize the yield. To further maximize the sensitivity, muscles being tested should be warm, and acetylcholinesterase inhibitors should be withheld for 12 hours before.
Repetitive nerve stimulation studies in distal muscles are positive in approximately 75% of patients with generalized myasthenia gravis and in 30% with ocular myasthenia gravis.26
Single-fiber electromyography is more technically demanding than repetitive nerve stimulation and is less widely available. It is usually performed with a special needle electrode that can simultaneously identify action potentials arising from individual muscle fibers innervated by the same axon.
Variability in time of the second action potential relative to the first is called “jitter.” Abnormal jitter is seen in more than 95% of patients with generalized myasthenia gravis and in 85% to 90% of those with ocular myasthenia gravis.26,27 However, abnormal jitter can also be seen in other neuromuscular diseases such as motor neuron disease or in neuromuscular junctional disorders such as Lambert-Eaton myasthenic syndrome.
Imaging studies
Chest computed tomography or magnetic resonance imaging with contrast should be performed in all myasthenia gravis patients to look for a thymoma.
TREATMENT OF MYASTHENIA GRAVIS
Acetylcholinesterase inhibitors
As a reasonable first therapy in mild cases of myasthenia gravis, acetylcholinesterase inhibitors slow down the degradation of acetylcholine and prolong its effect in the neuromuscular junction, but they are not disease-modifying and their benefits are mild.
Pyridostigmine is the usual choice of acetylcholinesterase inhibitor. Its onset of action is rapid (15 to 30 minutes) and its action lasts for 3 to 4 hours. For most patients, the effective dosage range is 60 mg to 90 mg every 4 to 6 hours. A long-acting form is also available and can be given as a single nighttime dose.
Immunomodulating therapy
Patients who have moderate to severe symptoms require some form of immunomodulating therapy.
Plasmapheresis or intravenous immune globulin is reserved for patients with severe or rapidly worsening disease because their beneficial effects can be seen within the first week of treatment.
Longer-acting immunotherapies (corticosteroids, azathioprine, mycophenolate mofetil and others) have a slower onset of responses but provide sustained benefits. Which drug to use depends on factors such as comorbidity, side effects, and cost.
Drugs to avoid
A number of medications can exacerbate weakness in myasthenia gravis and should be avoided or used with caution. The list is long, but ones that deserve the most attention are penicillamine, interferons, procainamide, quinidine, and antibiotics, including quinolones and aminoglycosides. A more comprehensive list of medications that may exacerbate myasthenia gravis symptoms can be found in a review by Keesey.2
RAPID INDUCTION IMMUNOTHERAPIES : PLASMAPHERESIS, IMMUNE GLOBULIN
Both plasmapheresis and intravenous immune globulin act quickly over days, but in most patients their effects last only a few weeks. Both are used as rescue therapies for myasthenic crises, bridging therapy to slow-acting immunotherapeutic agents, or maintenance treatment for poorly controlled cases.
Several retrospective studies have confirmed the efficacy of plasmapheresis in more than 80% of patients with generalized symptoms.28,29
In a randomized trial in patients with generalized therapies, intravenous immune globulin improved muscle strength in the group of patients with severe symptoms.30 The effective dosage of intravenous immune globulin varies from 1 to 2 g/kg without observed difference between doses.31 Trials comparing the efficacy of intravenous immune globulin and plasmapheresis in acute and severe myasthenia gravis did not reveal a difference in efficacy.32,33 Intravenous immune globulin at a minimal dose of 0.4 g/kg every 3 months has been successfully used as a long-term maintenance monotherapy, and such a role could be expanded to more patients with further studies.34
The choice between plasmapheresis and intravenous immune globulin is often based on the ability of a patient to tolerate each treatment and on the availability of the plasmapheresis procedure. Intravenous immune globulin is easier to administer, is associated with fewer adverse events related to vascular access, and is therefore more appropriate than plasmapheresis in some centers.
CHRONIC MAINTENANCE IMMUNOMODULATING TREATMENT
Corticosteroids
Prednisone, the most commonly used agent, leads to remission or marked improvement in 70% to 80% of patients with ocular or generalized myasthenia gravis.35 It may also reduce the progression of ocular myasthenia gravis to the generalized form.36
The effective dose of prednisone depends on the severity and distribution of symptoms. Some patients may need up to 1.0 mg/kg/day (usually 50 to 80 mg per day). In patients with mild to moderate symptoms, a lower maximal dosage such as 20 to 40 mg per day can be sufficient.
Within 1 to 2 weeks after starting high-dose prednisone, up to 50% of patients may develop a transient deterioration, including possible precipitation of a myasthenic crisis.37 For this reason, high-dose prednisone is commonly started only in hospitalized patients who are also receiving plasmapheresis or intravenous immune globulin. Otherwise, an outpatient dose-escalation protocol can be used to achieve a target dose over several weeks.
Prednisone tapering can begin after the patient has been on the maximal dose for 1 to 2 months and significant improvement is evident. A monthly tapering of 5 to 10 mg is preferred, then more slowly after the daily dose reaches 30 mg. The usual maintenance dose averages about 5 mg daily.
Common side effects of prednisone include weight gain, cushingoid features, easy bruising, cataracts, glaucoma, hypertension, diabetes, dyslipidemia, and osteoporosis. Patients are advised to take supplemental calcium (1,500 mg per day) and vitamin D (400 to 800 IU per day). For those most at risk of osteoporosis, treatment with a bisphosphonate should be considered.
Other immunotherapeutic agents are often needed, either to replace the corticosteroid or to permit use of lower doses of it. Because of their delayed onset of action, starting such corticosteroid-sparing agents early in the course is often necessary. These agents are often initially combined with high-dose prednisone, with an eventual goal of weaning off prednisone entirely. This strategy offers the advantage of relatively rapid induction while avoiding the long-term adverse effects of corticosteroid treatment.
Azathioprine
Azathioprine doesn’t begin to show a beneficial effect in myasthenia gravis for 6 to 12 months, and it often reaches its maximal efficacy only after 1 to 2 years of treatment.38
In a study of 78 myasthenia gravis patients, 91% improved when treated with azathioprine alone or together with prednisone.39 In another study using azathioprine and prednisolone for generalized myasthenia gravis, nearly two-thirds of patients came off prednisolone while maintaining remission for 3 years.38
A typical maintenance dose is 2 to 3 mg/kg/day. Common side effects are nausea, vomiting, and malaise. Less frequent side effects include hematologic abnormalities, abnormal liver function, and pancreatitis. Monthly monitoring of complete blood cell counts and liver function tests is warranted for the first 6 months, then less often.
One in 300 people in the general population is homozygous for a mutant allele in the thiopurine methyltransferase (TPMT) gene. Patients with this genotype should not receive azathioprine because of the risk of life-threatening bone marrow suppression.40 A slightly increased risk of various forms of lymphoma has been documented.41
Mycophenolate mofetil
A well-tolerated medication with few side effects, mycophenolate mofetil is being used more in myasthenia gravis. The results of two recent randomized trials suggested that it is not effective in improving myasthenia gravis symptoms or sparing prednisone dosage when used for 90 days or 36 weeks.42,43 However, extensive clinical experience supports its longterm efficacy in myasthenia gravis.
In a retrospective study of 85 patients with generalized myasthenia gravis, mycophenolate at doses of 1 to 3 g daily improved symptoms in 73% and produced remission in 50%. Steroid dosage was reduced in 71% of patients.44
Another retrospective study, with 102 patients, verified a slow development of clinical benefit after months of mycophenolate therapy alone or in combination with prednisone. Approximately 50% of patients achieved a minimal manifestation status after 6 to 12 months of mycophenolate treatment. Eventually, at 24 months of treatment, 80% of patients had a desirable outcome of minimal clinical manifestation or better, 55% of patients were able to come off prednisone entirely, and 75% were taking less than 7.5 mg of prednisone per day.45
Common side effects of mycophenolate include nausea, diarrhea, and infections such as urinary tract infections and herpes reactivation. The complete blood cell count needs to be monitored frequently during the first 6 months of therapy. Leukopenia can occur but rarely necessitates stopping mycophenolate. Long-term safety data are lacking, but so far there has been no clearly increased risk of malignancy.
Mycophenolate exposure in pregnancy results in a high incidence of major fetal malformations. Therefore, its use in pregnant patients is discouraged, and women of child-bearing age should use effective contraception.46
Cyclosporine
A randomized trial in a small number of patients suggested that cyclosporine is fairly effective as monotherapy.47 Its onset of action in myasthenia gravis is faster than that of other corticosteroid-sparing agents, and clinical benefit can often be observed as early as 1 to 2 months. A dose of 5 mg/kg/day and a maintenance serum level of 100 to 150 ng/mL are generally recommended. However, renal, hepatic, and hematologic toxicities and interactions with other medications make cyclosporine a less attractive choice.
Methotrexate
A randomized trial evaluated the utility of methotrexate as a steroid-sparing agent compared with azathioprine.48 At 24 months, its steroid-sparing effect was similar to that of azathioprine, and the prednisone dosage had been reduced in more than 50% of patients.
Another phase II trial studying the efficacy of methotrexate in myasthenia gravis is under way.49
Rituximab
Rituximab is a monoclonal antibody against B-cell membrane marker CD20. A growing number of case series support its efficacy in patients with severe generalized myasthenia gravis refractory to multiple immunosuppressants.16,50 It seems particularly effective for MuSK antibody-positive disease, reducing MuSK antibody titers and having a treatment effect that lasts for years.
The standard dosage is 375 mg/m2 per week for 4 consecutive weeks. Peripheral B cells tend to be depleted within 2 weeks after the first infusion, while T-cell populations remain unchanged.50
A minimal infusion reaction such as flushing and chills can be seen with the first infusion. Patients may be more susceptible to certain infections such as reactivation of herpes zoster, but overall rituximab is well tolerated. Rare cases of progressive multifocal leukoencephalopathy have been reported in patients taking it, but none have occurred so far in myasthenia gravis treatment.
Cyclophosphamide
Cyclophosphamide is an alkylating agent that reduces proliferation of both B and T cells. It can be effective in myasthenia gravis, but potentially serious side effects limit its use. It should be reserved for the small percentage of cases that are refractory to other immunotherapies.
Thymectomy
Surgical treatment should be considered for patients with thymoma. If the tumor cannot be surgically resected, chemoradiotherapy can be considered for relief of myasthenic symptoms and for prevention of local invasion.
Thymomas recur in a minority of patients many years after the initial resection, sometimes without myasthenia gravis symptoms. A recurrence of symptoms does not necessarily indicate a recurrence of thymoma. The lack of correlation between myasthenia gravis symptoms and thymoma recurrence highlights the importance of radiologic follow-up in these patients.
For patients without thymoma, many experts believe that thymectomy is beneficial in patients under age 60 who have generalized myasthenia gravis. The likelihood of medication-free remission is about twice as high, and the likelihood of becoming asymptomatic is about one and a half times higher after thymectomy.51 However, it takes up to several years for the benefits of thymectomy to manifest, and thymectomy does not guarantee protection from developing AChR antibody-positive myasthenia gravis in the future.
The optimal timing of thymectomy is not well established; however, the procedure is usually recommended within the first 3 years of diagnosis.52 The response rates from thymectomy are similar for AChR antibody-positive and seronegative patients. In general, thymectomy for MuSK antibody-positive patients has not been effective, and its role in ocular myasthenia gravis is unclear.2,53
Current therapies for myasthenia gravis can help most patients achieve sustained improvement. The overall prognosis has dramatically improved over the last 4 decades: the mortality rate used to be 75%; now it is 4.5%.1
Myasthenia gravis is the most common disorder of neuromuscular junction transmission and is also one of the best characterized autoimmune diseases. However, its symptoms—primarily weakness—vary from patient to patient, and in the same patient, by time of day and over longer time periods. The variation in symptoms can be very confusing to undiagnosed patients and puzzling to unsuspecting physicians. Such diagnostic uncertainty can give the patient additional frustration and emotional stress, which in turn exacerbate his or her condition.
In this review, we will give an overview of the pathogenesis, clinical manifestations, diagnosis, and treatment of myasthenia gravis.
TWO PEAKS IN INCIDENCE BY AGE
The annual incidence of myasthenia gravis is approximately 10 to 20 new cases per million, with a prevalence of about 150 to 200 per million.2
The age of onset has a bimodal distribution, with an early incidence peak in the second to third decade with a female predominance and a late peak in the 6th to the 8th decade with a male predominance.2
Myasthenia gravis is commonly associated with several other autoimmune disorders, including hypothyroidism, hyperthyroidism, systemic lupus erythematosus, rheumatoid arthritis, vitiligo, diabetes, and, more recently recognized, neuromyelitis optica.3
ANTIBODIES AGAINST AChR AND MuSK
In most cases of myasthenia gravis the patient has autoimmune antibodies against constituents of the neuromuscular junction, specifically acetylcholine receptor (AChR) and muscle-specific tyrosine kinase (MuSK) (Figure 1).
AChR antibody-positive myasthenia gravis
When antibodies bind to AChR on the postsynaptic membrane, they cross-link neighboring AChR units, which are absorbed into the muscle fiber and are broken up.4 In addition, the complement system is activated to mediate further damage on the postsynaptic membrane.
AChR antibodies may come from germinal centers of the thymus, where clustered myoid cells express AChR on the plasma membrane surface.5 About 60% of AChR antibody-positive myasthenia gravis patients have an enlarged thymus, and 10% have a thymoma—a tumor of the epithelial cells of this organ. Conversely, about 15% of patients with a thymoma have clinical myasthenia gravis, and an additional 20% possess antibodies against AChR in the serum without myasthenic symptoms.5
MuSK antibody-positive myasthenia gravis
Like AChR, MuSK is a transmembrane component of the postsynaptic neuromuscular junction. During formation of the neuromuscular junction, MuSK is activated through the binding of agrin (a nerve-derived proteoglycan) to lipoprotein-related protein 4 (LRP4), after which complicated intracellular signaling promotes the assembly and stabilization of AChR.6
Unlike AChR antibodies, antibodies against MuSK do not activate the complement system, and complement fixation is not essential for clinical myasthenic symptoms to appear.7 Also, myasthenia gravis with MuSK antibodies is rarely associated with thymoma.8
The precise mechanism by which MuSK antibody impairs transmission at the neuromuscular junction has been a mystery until recently. Animal models, including MuSK-mutant mice and mice injected with MuSK protein or with purified immunoglobulin G from patients with this disease, have revealed a significant reduction of AChR clusters and destruction of neuromuscular junction structures.7,9–12
In addition, MuSK antibodies produce pre-synaptic dysfunction, manifesting as a reduction of acetylcholine content. This information is based on studies in mice and on in vitro electrophysiologic analyses of neuromuscular junctions from a patient with this disease.7,9–13
Finally, MuSK antibodies may indirectly affect the recycling of acetylcholine. After post-synaptic activation, acetylcholine is normally hydrolized by acetylcholinesterase, which is located in the synaptic cleft but anchored to MuSK on the postsynaptic membrane. MuSK antibodies block the binding of MuSK to acetylcholinesterase, possibly leading to less accumulation of acetylcholinesterase.14 This process may explain why patients with MuSK antibody-positive myasthenia gravis tend to respond poorly to acetylcholinesterase inhibitors (more about this below).
Seronegative myasthenia gravis
In a series of 562 consecutive patients with generalized weakness due to myasthenia gravis, 92% were positive for AChR antibody, 3% were positive for MuSK antibody, and 5% were seronegative (possessing neither antibody).15 In contrast, about 50% of patients with purely ocular myasthenia gravis (ie, with isolated weakness of the levator palpebrae superioris, orbicularis oculi, or oculomotor muscles) are seropositive for AChR antibody. Only a few ocular MuSK antibody-positive cases have been described, leaving the rest seronegative. Rarely, both antibodies can be detected in the same patient.16
In patients who are negative for AChR antibodies at the time of disease onset, sero-conversion may occur later during the course. Repeating serologic testing 6 to 12 months later may detect AChR antibodies in approximately 15% of patients who were initially seronegative.15,17
The clinical presentation, electrophysiologic findings, thymic pathologic findings, and treatment responses are similar in AChR antibody-positive and seronegative myasthenia gravis.17 Muscle biopsy study in seronegative cases demonstrates a loss of AChR as well.18
Based on these observations, it has been proposed that seronegative patients may have low-affinity antibodies that can bind to tightly clustered AChRs on the postsynaptic membrane but escape detection by routine radioimmunoassays in a solution phase. With a sensitive cell-based immunofluorescence assay, low-affinity antibodies to clustered AChRs were detected in 66% of patients with generalized myasthenia gravis and in 50% of those with ocular myasthenia gravis who were seronegative on standard assays.19,20 These low-affinity AChR antibodies can also activate complement in vitro, increasing the likelihood that they are pathogenic. However, assays to detect low-affinity AChR antibodies are not yet commercially available.
Within the past year, three research groups independently reported detecting antibodies to LRP4 in 2% to 50% of seronegative myasthenia gravis patients. This wide variation in the prevalence of LRP4 antibodies could be related to patient ethnicity and methods of detection.21–23 LRP4 is a receptor for agrin and is required for agrin-induced MuSK activation and AChR clustering. LRP antibodies can activate complement; therefore, it is plausible that LRP4 antibody binding leads to AChR loss on the postsynaptic membrane. However, additional study is needed to determine if LRP4 antibodies are truly pathogenic in myasthenia gravis.
A DISORDER OF FATIGABLE WEAKNESS
Myasthenia gravis is a disorder of fatigable weakness producing fluctuating symptoms. Symptoms related to the involvement of specific muscle groups are listed in Table 1. Muscle weakness is often worse later in the day or after exercise.
Ocular myasthenia gravis accounts for about 15% of all cases. Of patients initially presenting with ocular symptoms only, twothirds will ultimately develop generalized symptoms, most within the first 2 years.24 No factor has been identified that predicts conversion from an ocular to a generalized form.
Several clinical phenotypes of MuSK antibody-positive myasthenia gravis have been described. An oculobulbar form presents with diplopia, ptosis, dysarthria, and profound atrophy of the muscles of the tongue and face. A restricted myopathic form presents with prominent neck, shoulder, and respiratory weakness without ocular involvement. A third form is a combination of ocular and proximal limb weakness, indistinguishable from AChR antibody-positive disease.25
MuSK antibody-positive patients do not respond as well to acetylcholinesterase inhibitors as AChR antibody-positive patients do. In one study, nearly 70% of MuSK antibody-positive patients demonstrated no response, poor tolerance, or cholinergic hypersensitivity to these agents.25 Fortunately, most MuSK antibody-positive patients have a favorable response to immunosuppressive therapy—sometimes a dramatic improvement after plasmapheresis.8
DIAGNOSIS OF MYASTHENIA GRAVIS
The common differential diagnoses for myasthenia gravis are listed in Table 2.
The essential feature of myasthenia gravis is fluctuating muscle weakness, often with fatigue. Many patients complain of weakness of specific muscle groups after their repeated usage. Pain is generally a less conspicuous symptom, and generalized fatigue without objective weakness is inconsistent with myasthenia gravis.
Signs of muscle weakness may include droopy eyelids, diplopia, inability to hold the head straight, difficulty swallowing or chewing, speech disturbances, difficulty breathing, and difficulty raising the arms or rising from the sitting position. A historical pattern of ptosis alternating from one eye to the other is fairly characteristic of myasthenia gravis.
The weakness of orbicularis oculi is easily identified on examination by prying open the eyes during forced eye closure. Limb weakness is usually more significant in the arms than in the legs. An often-neglected feature of myasthenia gravis is finger extensor weakness with a relative sparing of other distal hand muscles.2
The ice-pack test is performed by placing a small bag of ice over the ptotic eye for 2 to 5 minutes and assessing the degree of ptosis for any noticeable improvement. This test is not very helpful for assessing ocular motor weakness.
The edrophonium (Tensilon) test can be used for patients with ptosis or ophthalmoparesis. Edrophonium, a short-acting acetylcholinesterase inhibitor, is given intravenously while the patient is observed for objective improvement. The patient’s cardiovascular status should be monitored for arrhythmias and hypotension. Atropine should be immediately available in case severe bradycardia develops.
The ice-pack test and the edrophonium test can give false-negative and false-positive results, and the diagnosis of myasthenia gravis must be verified by other diagnostic tests.
Testing for antibodies
Testing for circulating AChR antibodies, MuSK antibodies, or both is the first step in the laboratory confirmation of myasthenia gravis.
There are three AChR antibody subtypes: binding, blocking, and modulating. Binding antibodies are present in 80% to 90% of patients with generalized myasthenia gravis and 50% of those with ocular myasthenia gravis. Testing for blocking and modulating AChR antibodies increases the sensitivity by less than 5% when added to testing for binding antibodies.
AChR antibody titers correlate poorly with disease severity between patients. However, in individual patients, antibody titers tend to go down in parallel with clinical improvement.
MuSK antibody is detected in nearly half of myasthenia gravis patients with generalized weakness who are negative for AChR antibody.
Electrophysiologic tests
Electrophysiologic tests can usually confirm the diagnosis of seronegative myasthenia gravis. They are also helpful in seropositive patients who have unusual clinical features or a poor response to treatment.
Repetitive nerve stimulation studies use a slow rate (2–5 Hz) of repetitive electrical stimulation. The study is positive if the motor response declines by more than 10%. However, a decremental response is not specific for myasthenia gravis, as it may be seen in other neuromuscular disorders such as motor neuron disease or Lambert-Eaton myasthenic syndrome.
This test is technically easier to do in distal muscles than in proximal muscles, but less sensitive. Therefore, proximal muscles such as the trapezius or facial muscles are usually also sampled to maximize the yield. To further maximize the sensitivity, muscles being tested should be warm, and acetylcholinesterase inhibitors should be withheld for 12 hours before.
Repetitive nerve stimulation studies in distal muscles are positive in approximately 75% of patients with generalized myasthenia gravis and in 30% with ocular myasthenia gravis.26
Single-fiber electromyography is more technically demanding than repetitive nerve stimulation and is less widely available. It is usually performed with a special needle electrode that can simultaneously identify action potentials arising from individual muscle fibers innervated by the same axon.
Variability in time of the second action potential relative to the first is called “jitter.” Abnormal jitter is seen in more than 95% of patients with generalized myasthenia gravis and in 85% to 90% of those with ocular myasthenia gravis.26,27 However, abnormal jitter can also be seen in other neuromuscular diseases such as motor neuron disease or in neuromuscular junctional disorders such as Lambert-Eaton myasthenic syndrome.
Imaging studies
Chest computed tomography or magnetic resonance imaging with contrast should be performed in all myasthenia gravis patients to look for a thymoma.
TREATMENT OF MYASTHENIA GRAVIS
Acetylcholinesterase inhibitors
As a reasonable first therapy in mild cases of myasthenia gravis, acetylcholinesterase inhibitors slow down the degradation of acetylcholine and prolong its effect in the neuromuscular junction, but they are not disease-modifying and their benefits are mild.
Pyridostigmine is the usual choice of acetylcholinesterase inhibitor. Its onset of action is rapid (15 to 30 minutes) and its action lasts for 3 to 4 hours. For most patients, the effective dosage range is 60 mg to 90 mg every 4 to 6 hours. A long-acting form is also available and can be given as a single nighttime dose.
Immunomodulating therapy
Patients who have moderate to severe symptoms require some form of immunomodulating therapy.
Plasmapheresis or intravenous immune globulin is reserved for patients with severe or rapidly worsening disease because their beneficial effects can be seen within the first week of treatment.
Longer-acting immunotherapies (corticosteroids, azathioprine, mycophenolate mofetil and others) have a slower onset of responses but provide sustained benefits. Which drug to use depends on factors such as comorbidity, side effects, and cost.
Drugs to avoid
A number of medications can exacerbate weakness in myasthenia gravis and should be avoided or used with caution. The list is long, but ones that deserve the most attention are penicillamine, interferons, procainamide, quinidine, and antibiotics, including quinolones and aminoglycosides. A more comprehensive list of medications that may exacerbate myasthenia gravis symptoms can be found in a review by Keesey.2
RAPID INDUCTION IMMUNOTHERAPIES : PLASMAPHERESIS, IMMUNE GLOBULIN
Both plasmapheresis and intravenous immune globulin act quickly over days, but in most patients their effects last only a few weeks. Both are used as rescue therapies for myasthenic crises, bridging therapy to slow-acting immunotherapeutic agents, or maintenance treatment for poorly controlled cases.
Several retrospective studies have confirmed the efficacy of plasmapheresis in more than 80% of patients with generalized symptoms.28,29
In a randomized trial in patients with generalized therapies, intravenous immune globulin improved muscle strength in the group of patients with severe symptoms.30 The effective dosage of intravenous immune globulin varies from 1 to 2 g/kg without observed difference between doses.31 Trials comparing the efficacy of intravenous immune globulin and plasmapheresis in acute and severe myasthenia gravis did not reveal a difference in efficacy.32,33 Intravenous immune globulin at a minimal dose of 0.4 g/kg every 3 months has been successfully used as a long-term maintenance monotherapy, and such a role could be expanded to more patients with further studies.34
The choice between plasmapheresis and intravenous immune globulin is often based on the ability of a patient to tolerate each treatment and on the availability of the plasmapheresis procedure. Intravenous immune globulin is easier to administer, is associated with fewer adverse events related to vascular access, and is therefore more appropriate than plasmapheresis in some centers.
CHRONIC MAINTENANCE IMMUNOMODULATING TREATMENT
Corticosteroids
Prednisone, the most commonly used agent, leads to remission or marked improvement in 70% to 80% of patients with ocular or generalized myasthenia gravis.35 It may also reduce the progression of ocular myasthenia gravis to the generalized form.36
The effective dose of prednisone depends on the severity and distribution of symptoms. Some patients may need up to 1.0 mg/kg/day (usually 50 to 80 mg per day). In patients with mild to moderate symptoms, a lower maximal dosage such as 20 to 40 mg per day can be sufficient.
Within 1 to 2 weeks after starting high-dose prednisone, up to 50% of patients may develop a transient deterioration, including possible precipitation of a myasthenic crisis.37 For this reason, high-dose prednisone is commonly started only in hospitalized patients who are also receiving plasmapheresis or intravenous immune globulin. Otherwise, an outpatient dose-escalation protocol can be used to achieve a target dose over several weeks.
Prednisone tapering can begin after the patient has been on the maximal dose for 1 to 2 months and significant improvement is evident. A monthly tapering of 5 to 10 mg is preferred, then more slowly after the daily dose reaches 30 mg. The usual maintenance dose averages about 5 mg daily.
Common side effects of prednisone include weight gain, cushingoid features, easy bruising, cataracts, glaucoma, hypertension, diabetes, dyslipidemia, and osteoporosis. Patients are advised to take supplemental calcium (1,500 mg per day) and vitamin D (400 to 800 IU per day). For those most at risk of osteoporosis, treatment with a bisphosphonate should be considered.
Other immunotherapeutic agents are often needed, either to replace the corticosteroid or to permit use of lower doses of it. Because of their delayed onset of action, starting such corticosteroid-sparing agents early in the course is often necessary. These agents are often initially combined with high-dose prednisone, with an eventual goal of weaning off prednisone entirely. This strategy offers the advantage of relatively rapid induction while avoiding the long-term adverse effects of corticosteroid treatment.
Azathioprine
Azathioprine doesn’t begin to show a beneficial effect in myasthenia gravis for 6 to 12 months, and it often reaches its maximal efficacy only after 1 to 2 years of treatment.38
In a study of 78 myasthenia gravis patients, 91% improved when treated with azathioprine alone or together with prednisone.39 In another study using azathioprine and prednisolone for generalized myasthenia gravis, nearly two-thirds of patients came off prednisolone while maintaining remission for 3 years.38
A typical maintenance dose is 2 to 3 mg/kg/day. Common side effects are nausea, vomiting, and malaise. Less frequent side effects include hematologic abnormalities, abnormal liver function, and pancreatitis. Monthly monitoring of complete blood cell counts and liver function tests is warranted for the first 6 months, then less often.
One in 300 people in the general population is homozygous for a mutant allele in the thiopurine methyltransferase (TPMT) gene. Patients with this genotype should not receive azathioprine because of the risk of life-threatening bone marrow suppression.40 A slightly increased risk of various forms of lymphoma has been documented.41
Mycophenolate mofetil
A well-tolerated medication with few side effects, mycophenolate mofetil is being used more in myasthenia gravis. The results of two recent randomized trials suggested that it is not effective in improving myasthenia gravis symptoms or sparing prednisone dosage when used for 90 days or 36 weeks.42,43 However, extensive clinical experience supports its longterm efficacy in myasthenia gravis.
In a retrospective study of 85 patients with generalized myasthenia gravis, mycophenolate at doses of 1 to 3 g daily improved symptoms in 73% and produced remission in 50%. Steroid dosage was reduced in 71% of patients.44
Another retrospective study, with 102 patients, verified a slow development of clinical benefit after months of mycophenolate therapy alone or in combination with prednisone. Approximately 50% of patients achieved a minimal manifestation status after 6 to 12 months of mycophenolate treatment. Eventually, at 24 months of treatment, 80% of patients had a desirable outcome of minimal clinical manifestation or better, 55% of patients were able to come off prednisone entirely, and 75% were taking less than 7.5 mg of prednisone per day.45
Common side effects of mycophenolate include nausea, diarrhea, and infections such as urinary tract infections and herpes reactivation. The complete blood cell count needs to be monitored frequently during the first 6 months of therapy. Leukopenia can occur but rarely necessitates stopping mycophenolate. Long-term safety data are lacking, but so far there has been no clearly increased risk of malignancy.
Mycophenolate exposure in pregnancy results in a high incidence of major fetal malformations. Therefore, its use in pregnant patients is discouraged, and women of child-bearing age should use effective contraception.46
Cyclosporine
A randomized trial in a small number of patients suggested that cyclosporine is fairly effective as monotherapy.47 Its onset of action in myasthenia gravis is faster than that of other corticosteroid-sparing agents, and clinical benefit can often be observed as early as 1 to 2 months. A dose of 5 mg/kg/day and a maintenance serum level of 100 to 150 ng/mL are generally recommended. However, renal, hepatic, and hematologic toxicities and interactions with other medications make cyclosporine a less attractive choice.
Methotrexate
A randomized trial evaluated the utility of methotrexate as a steroid-sparing agent compared with azathioprine.48 At 24 months, its steroid-sparing effect was similar to that of azathioprine, and the prednisone dosage had been reduced in more than 50% of patients.
Another phase II trial studying the efficacy of methotrexate in myasthenia gravis is under way.49
Rituximab
Rituximab is a monoclonal antibody against B-cell membrane marker CD20. A growing number of case series support its efficacy in patients with severe generalized myasthenia gravis refractory to multiple immunosuppressants.16,50 It seems particularly effective for MuSK antibody-positive disease, reducing MuSK antibody titers and having a treatment effect that lasts for years.
The standard dosage is 375 mg/m2 per week for 4 consecutive weeks. Peripheral B cells tend to be depleted within 2 weeks after the first infusion, while T-cell populations remain unchanged.50
A minimal infusion reaction such as flushing and chills can be seen with the first infusion. Patients may be more susceptible to certain infections such as reactivation of herpes zoster, but overall rituximab is well tolerated. Rare cases of progressive multifocal leukoencephalopathy have been reported in patients taking it, but none have occurred so far in myasthenia gravis treatment.
Cyclophosphamide
Cyclophosphamide is an alkylating agent that reduces proliferation of both B and T cells. It can be effective in myasthenia gravis, but potentially serious side effects limit its use. It should be reserved for the small percentage of cases that are refractory to other immunotherapies.
Thymectomy
Surgical treatment should be considered for patients with thymoma. If the tumor cannot be surgically resected, chemoradiotherapy can be considered for relief of myasthenic symptoms and for prevention of local invasion.
Thymomas recur in a minority of patients many years after the initial resection, sometimes without myasthenia gravis symptoms. A recurrence of symptoms does not necessarily indicate a recurrence of thymoma. The lack of correlation between myasthenia gravis symptoms and thymoma recurrence highlights the importance of radiologic follow-up in these patients.
For patients without thymoma, many experts believe that thymectomy is beneficial in patients under age 60 who have generalized myasthenia gravis. The likelihood of medication-free remission is about twice as high, and the likelihood of becoming asymptomatic is about one and a half times higher after thymectomy.51 However, it takes up to several years for the benefits of thymectomy to manifest, and thymectomy does not guarantee protection from developing AChR antibody-positive myasthenia gravis in the future.
The optimal timing of thymectomy is not well established; however, the procedure is usually recommended within the first 3 years of diagnosis.52 The response rates from thymectomy are similar for AChR antibody-positive and seronegative patients. In general, thymectomy for MuSK antibody-positive patients has not been effective, and its role in ocular myasthenia gravis is unclear.2,53
- Alshekhlee A, Miles JD, Katirji B, Preston DC, Kaminski HJ. Incidence and mortality rates of myasthenia gravis and myasthenic crisis in US hospitals. Neurology 2009; 72:1548–1554.
- Keesey JC. Clinical evaluation and management of myasthenia gravis. Muscle Nerve 2004; 29:484–505.
- Leite MI, Coutinho E, Lana-Peixoto M, et al. Myasthenia gravis and neuromyelitis optica spectrum disorder: a multicenter study of 16 patients. Neurology 2012; 78:1601–1607.
- Drachman DB, Angus CW, Adams RN, Michelson JD, Hoffman GJ. Myasthenic antibodies cross-link acetylcholine receptors to accelerate degradation. N Engl J Med 1978; 298:1116–1122.
- Fujii Y. The thymus, thymoma and myasthenia gravis. Surg Today 2013; 43:461–466.
- Evoli A, Lindstrom J. Myasthenia gravis with antibodies to MuSK: another step toward solving mystery? Neurology 2011; 77:1783–1784.
- Mori S, Kubo S, Akiyoshi T, et al. Antibodies against muscle-specific kinase impair both presynaptic and postsynaptic functions in a murine model of myasthenia gravis. Am J Pathol 2012; 180:798–810.
- Guptill JT, Sanders DB, Evoli A. Anti-MuSK antibody myasthenia gravis: clinical findings and response to treatment in two large cohorts. Muscle Nerve 2011; 44:36–40.
- Chevessier F, Girard E, Molgó J, et al. A mouse model for congenital myasthenic syndrome due to MuSK mutations reveals defects in structure and function of neuromuscular junctions. Hum Mol Genet 2008; 17:3577–3595.
- Richman DP, Nishi K, Morell SW, et al. Acute severe animal model of anti-muscle-specific kinase myasthenia: combined postsynaptic and presynaptic changes. Arch Neurol 2012; 69:453–460.
- Klooster R, Plomp JJ, Huijbers MG, et al. Muscle-specific kinase myasthenia gravis IgG4 autoantibodies cause severe neuromuscular junction dysfunction in mice. Brain 2012; 135:1081–1101.
- Viegas S, Jacobson L, Waters P, et al. Passive and active immunization models of MuSK-Ab positive myasthenia: electrophysiological evidence for pre and postsynaptic defects. Exp Neurol 2012; 234:506–512.
- Niks EH, Kuks JB, Wokke JH, et al. Pre- and postsynaptic neuromuscular junction abnormalities in musk myasthenia. Muscle Nerve 2010; 42:283–288.
- Kawakami Y, Ito M, Hirayama M, et al. Anti-MuSK autoantibodies block binding of collagen Q to MuSK. Neurology 2011; 77:1819–1826.
- Chan KH, Lachance DH, Harper CM, Lennon VA. Frequency of seronegativity in adult-acquired generalized myasthenia gravis. Muscle Nerve 2007; 36:651–658.
- Collongues N, Casez O, Lacour A, et al. Rituximab in refractory and non-refractory myasthenia: a retrospective multicenter study. Muscle Nerve 2012; 46:687–691.
- Sanders DB, Andrews PI, Howard JF, Massey JM. Seronegative myasthenia gravis. Neurology 1997; 48(suppl 5):40S–45S.
- Shiraishi H, Motomura M, Yoshimura T, et al. Acetylcholine receptors loss and postsynaptic damage in MuSK antibody-positive myasthenia gravis. Ann Neurol 2005; 57:289–293.
- Leite MI, Jacob S, Viegas S, et al. IgG1 antibodies to acetylcholine receptors in ‘seronegative’ myasthenia gravis. Brain 2008; 131:1940–1952.
- Jacob S, Viegas S, Leite MI, et al. Presence and pathogenic relevance of antibodies to clustered acetylcholine receptor in ocular and generalized myasthenia gravis. Arch Neurol 2012; 69:994–1001.
- Higuchi O, Hamuro J, Motomura M, Yamanashi Y. Autoantibodies to low-density lipoprotein receptor-related protein 4 in myasthenia gravis. Ann Neurol 2011; 69:418–422.
- Pevzner A, Schoser B, Peters K, et al. Anti-LRP4 autoantibodies in AChR- and MuSK-antibody-negative myasthenia gravis. J Neurol 2012; 259:427–435.
- Zhang B, Tzartos JS, Belimezi M, et al. Autoantibodies to lipoprotein-related protein 4 in patients with double-seronegative myasthenia gravis. Arch Neurol 2012; 69:445–451.
- Kupersmith MJ, Latkany R, Homel P. Development of generalized disease at 2 years in patients with ocular myasthenia gravis. Arch Neurol 2003; 60:243–248.
- Pasnoor M, Wolfe GI, Nations S, et al. Clinical findings in MuSK-antibody positive myasthenia gravis: a US experience. Muscle Nerve 2010; 41:370–374.
- Oh SJ, Kim DE, Kuruoglu R, Bradley RJ, Dwyer D. Diagnostic sensitivity of the laboratory tests in myasthenia gravis. Muscle Nerve 1992; 15:720–724.
- Sanders DB, Stålberg EV. AAEM minimonograph #25: single-fiber electromyography. Muscle Nerve 1996; 19:1069–1083.
- Lazo-Langner A, Espinosa-Poblano I, Tirado-Cárdenas N, et al. Therapeutic plasma exchange in Mexico: experience from a single institution. Am J Hematol 2002; 70:16–21.
- Carandina-Maffeis R, Nucci A, Marques JF, et al. Plasmapheresis in the treatment of myasthenia gravis: retrospective study of 26 patients. Arq Neuropsiquiatr 2004; 62:391–395.
- Zinman L, Ng E, Bril V. IV immunoglobulin in patients with myasthenia gravis: a randomized controlled trial. Neurology 2007; 68:837–841.
- Gajdos P, Tranchant C, Clair B, et al; Myasthenia Gravis Clinical Study Group. Treatment of myasthenia gravis exacerbation with intravenous immunoglobulin: a randomized double-blind clinical trial. Arch Neurol 2005; 62:1689–1693.
- Rønager J, Ravnborg M, Hermansen I, Vorstrup S. Immunoglobulin treatment versus plasma exchange in patients with chronic moderate to severe myasthenia gravis. Artif Organs 2001; 25:967–973.
- Barth D, Nabavi Nouri M, Ng E, Nwe P, Bril V. Comparison of IVIg and PLEX in patients with myasthenia gravis. Neurology 2011; 76:2017–2023.
- Wegner B, Ahmed I. Intravenous immunoglobulin monotherapy in long-term treatment of myasthenia gravis. Clin Neurol Neurosurg 2002; 105:3–8.
- Pascuzzi RM, Coslett HB, Johns TR. Long-term corticosteroid treatment of myasthenia gravis: report of 116 patients. Ann Neurol 1984; 15:291–298.
- Monsul NT, Patwa HS, Knorr AM, Lesser RL, Goldstein JM. The effect of prednisone on the progression from ocular to generalized myasthenia gravis. J Neurol Sci 2004; 217:131–133.
- Miller RG, Milner-Brown HS, Mirka A. Prednisone-induced worsening of neuromuscular function in myasthenia gravis. Neurology 1986; 36:729–732.
- Palace J, Newsom-Davis J, Lecky B. A randomized double-blind trial of prednisolone alone or with azathioprine in myasthenia gravis. Myasthenia Gravis Study Group. Neurology 1998; 50:1778–1783.
- Mertens HG, Hertel G, Reuther P, Ricker K. Effect of immunosuppressive drugs (azathioprine). Ann N Y Acad Sci 1981; 377:691–699.
- Relling MV, Gardner EE, Sandborn WJ, et al; Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing. Clin Pharmacol Ther 2011; 89:387–391.
- Finelli PF. Primary CNS lymphoma in myasthenic on long-term azathioprine. J Neurooncol 2005; 74:91–92.
- Sanders DB, Hart IK, Mantegazza R, et al. An international, phase III, randomized trial of mycophenolate mofetil in myasthenia gravis. Neurology 2008; 71:400–406.
- Muscle Study Group. A trial of mycophenolate mofetil with prednisone as initial immunotherapy in myasthenia gravis. Neurology 2008; 71:394–399.
- Meriggioli MN, Ciafaloni E, Al-Hayk KA, et al. Mycophenolate mofetil for myasthenia gravis: an analysis of efficacy, safety, and tolerability. Neurology 2003; 61:1438–1440.
- Hehir MK, Burns TM, Alpers J, Conaway MR, Sawa M, Sanders DB. Mycophenolate mofetil in AChR-antibody-positive myasthenia gravis: outcomes in 102 patients. Muscle Nerve 2010; 41:593–598.
- Merlob P, Stahl B, Klinger G. Tetrada of the possible mycophenolate mofetil embryopathy: a review. Reprod Toxicol 2009; 28:105–108.
- Tindall RS, Rollins JA, Phillips JT, Greenlee RG, Wells L, Belendiuk G. Preliminary results of a double-blind, randomized, placebo-controlled trial of cyclosporine in myasthenia gravis. N Engl J Med 1987; 316:719–724.
- Heckmann JM, Rawoot A, Bateman K, Renison R, Badri M. A single-blinded trial of methotrexate versus azathioprine as steroid-sparing agents in generalized myasthenia gravis. BMC Neurol 2011; 11:97.
- Pasnoor M, He J, Herbelin L, Dimachkie M, Barohn RJ; Muscle Study Group. Phase II trial of methotrexate in myasthenia gravis. Ann N Y Acad Sci 2012; 1275:23–28.
- Díaz-Manera J, Martínez-Hernández E, Querol L, et al. Long-lasting treatment effect of rituximab in MuSK myasthenia. Neurology 2012; 78:189–193.
- Gronseth GS, Barohn RJ. Practice parameter: thymectomy for autoimmune myasthenia gravis (an evidence-based review): Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2000; 55:7–15.
- Kumar V, Kaminski HJ. Treatment of myasthenia gravis. Curr Neurol Neurosci Rep 2011; 11:89–96.
- Pompeo E, Tacconi F, Massa R, Mineo D, Nahmias S, Mineo TC. Long-term outcome of thoracoscopic extended thymectomy for nonthymomatous myasthenia gravis. Eur J Cardiothorac Surg 2009; 36:164–169.
- Alshekhlee A, Miles JD, Katirji B, Preston DC, Kaminski HJ. Incidence and mortality rates of myasthenia gravis and myasthenic crisis in US hospitals. Neurology 2009; 72:1548–1554.
- Keesey JC. Clinical evaluation and management of myasthenia gravis. Muscle Nerve 2004; 29:484–505.
- Leite MI, Coutinho E, Lana-Peixoto M, et al. Myasthenia gravis and neuromyelitis optica spectrum disorder: a multicenter study of 16 patients. Neurology 2012; 78:1601–1607.
- Drachman DB, Angus CW, Adams RN, Michelson JD, Hoffman GJ. Myasthenic antibodies cross-link acetylcholine receptors to accelerate degradation. N Engl J Med 1978; 298:1116–1122.
- Fujii Y. The thymus, thymoma and myasthenia gravis. Surg Today 2013; 43:461–466.
- Evoli A, Lindstrom J. Myasthenia gravis with antibodies to MuSK: another step toward solving mystery? Neurology 2011; 77:1783–1784.
- Mori S, Kubo S, Akiyoshi T, et al. Antibodies against muscle-specific kinase impair both presynaptic and postsynaptic functions in a murine model of myasthenia gravis. Am J Pathol 2012; 180:798–810.
- Guptill JT, Sanders DB, Evoli A. Anti-MuSK antibody myasthenia gravis: clinical findings and response to treatment in two large cohorts. Muscle Nerve 2011; 44:36–40.
- Chevessier F, Girard E, Molgó J, et al. A mouse model for congenital myasthenic syndrome due to MuSK mutations reveals defects in structure and function of neuromuscular junctions. Hum Mol Genet 2008; 17:3577–3595.
- Richman DP, Nishi K, Morell SW, et al. Acute severe animal model of anti-muscle-specific kinase myasthenia: combined postsynaptic and presynaptic changes. Arch Neurol 2012; 69:453–460.
- Klooster R, Plomp JJ, Huijbers MG, et al. Muscle-specific kinase myasthenia gravis IgG4 autoantibodies cause severe neuromuscular junction dysfunction in mice. Brain 2012; 135:1081–1101.
- Viegas S, Jacobson L, Waters P, et al. Passive and active immunization models of MuSK-Ab positive myasthenia: electrophysiological evidence for pre and postsynaptic defects. Exp Neurol 2012; 234:506–512.
- Niks EH, Kuks JB, Wokke JH, et al. Pre- and postsynaptic neuromuscular junction abnormalities in musk myasthenia. Muscle Nerve 2010; 42:283–288.
- Kawakami Y, Ito M, Hirayama M, et al. Anti-MuSK autoantibodies block binding of collagen Q to MuSK. Neurology 2011; 77:1819–1826.
- Chan KH, Lachance DH, Harper CM, Lennon VA. Frequency of seronegativity in adult-acquired generalized myasthenia gravis. Muscle Nerve 2007; 36:651–658.
- Collongues N, Casez O, Lacour A, et al. Rituximab in refractory and non-refractory myasthenia: a retrospective multicenter study. Muscle Nerve 2012; 46:687–691.
- Sanders DB, Andrews PI, Howard JF, Massey JM. Seronegative myasthenia gravis. Neurology 1997; 48(suppl 5):40S–45S.
- Shiraishi H, Motomura M, Yoshimura T, et al. Acetylcholine receptors loss and postsynaptic damage in MuSK antibody-positive myasthenia gravis. Ann Neurol 2005; 57:289–293.
- Leite MI, Jacob S, Viegas S, et al. IgG1 antibodies to acetylcholine receptors in ‘seronegative’ myasthenia gravis. Brain 2008; 131:1940–1952.
- Jacob S, Viegas S, Leite MI, et al. Presence and pathogenic relevance of antibodies to clustered acetylcholine receptor in ocular and generalized myasthenia gravis. Arch Neurol 2012; 69:994–1001.
- Higuchi O, Hamuro J, Motomura M, Yamanashi Y. Autoantibodies to low-density lipoprotein receptor-related protein 4 in myasthenia gravis. Ann Neurol 2011; 69:418–422.
- Pevzner A, Schoser B, Peters K, et al. Anti-LRP4 autoantibodies in AChR- and MuSK-antibody-negative myasthenia gravis. J Neurol 2012; 259:427–435.
- Zhang B, Tzartos JS, Belimezi M, et al. Autoantibodies to lipoprotein-related protein 4 in patients with double-seronegative myasthenia gravis. Arch Neurol 2012; 69:445–451.
- Kupersmith MJ, Latkany R, Homel P. Development of generalized disease at 2 years in patients with ocular myasthenia gravis. Arch Neurol 2003; 60:243–248.
- Pasnoor M, Wolfe GI, Nations S, et al. Clinical findings in MuSK-antibody positive myasthenia gravis: a US experience. Muscle Nerve 2010; 41:370–374.
- Oh SJ, Kim DE, Kuruoglu R, Bradley RJ, Dwyer D. Diagnostic sensitivity of the laboratory tests in myasthenia gravis. Muscle Nerve 1992; 15:720–724.
- Sanders DB, Stålberg EV. AAEM minimonograph #25: single-fiber electromyography. Muscle Nerve 1996; 19:1069–1083.
- Lazo-Langner A, Espinosa-Poblano I, Tirado-Cárdenas N, et al. Therapeutic plasma exchange in Mexico: experience from a single institution. Am J Hematol 2002; 70:16–21.
- Carandina-Maffeis R, Nucci A, Marques JF, et al. Plasmapheresis in the treatment of myasthenia gravis: retrospective study of 26 patients. Arq Neuropsiquiatr 2004; 62:391–395.
- Zinman L, Ng E, Bril V. IV immunoglobulin in patients with myasthenia gravis: a randomized controlled trial. Neurology 2007; 68:837–841.
- Gajdos P, Tranchant C, Clair B, et al; Myasthenia Gravis Clinical Study Group. Treatment of myasthenia gravis exacerbation with intravenous immunoglobulin: a randomized double-blind clinical trial. Arch Neurol 2005; 62:1689–1693.
- Rønager J, Ravnborg M, Hermansen I, Vorstrup S. Immunoglobulin treatment versus plasma exchange in patients with chronic moderate to severe myasthenia gravis. Artif Organs 2001; 25:967–973.
- Barth D, Nabavi Nouri M, Ng E, Nwe P, Bril V. Comparison of IVIg and PLEX in patients with myasthenia gravis. Neurology 2011; 76:2017–2023.
- Wegner B, Ahmed I. Intravenous immunoglobulin monotherapy in long-term treatment of myasthenia gravis. Clin Neurol Neurosurg 2002; 105:3–8.
- Pascuzzi RM, Coslett HB, Johns TR. Long-term corticosteroid treatment of myasthenia gravis: report of 116 patients. Ann Neurol 1984; 15:291–298.
- Monsul NT, Patwa HS, Knorr AM, Lesser RL, Goldstein JM. The effect of prednisone on the progression from ocular to generalized myasthenia gravis. J Neurol Sci 2004; 217:131–133.
- Miller RG, Milner-Brown HS, Mirka A. Prednisone-induced worsening of neuromuscular function in myasthenia gravis. Neurology 1986; 36:729–732.
- Palace J, Newsom-Davis J, Lecky B. A randomized double-blind trial of prednisolone alone or with azathioprine in myasthenia gravis. Myasthenia Gravis Study Group. Neurology 1998; 50:1778–1783.
- Mertens HG, Hertel G, Reuther P, Ricker K. Effect of immunosuppressive drugs (azathioprine). Ann N Y Acad Sci 1981; 377:691–699.
- Relling MV, Gardner EE, Sandborn WJ, et al; Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing. Clin Pharmacol Ther 2011; 89:387–391.
- Finelli PF. Primary CNS lymphoma in myasthenic on long-term azathioprine. J Neurooncol 2005; 74:91–92.
- Sanders DB, Hart IK, Mantegazza R, et al. An international, phase III, randomized trial of mycophenolate mofetil in myasthenia gravis. Neurology 2008; 71:400–406.
- Muscle Study Group. A trial of mycophenolate mofetil with prednisone as initial immunotherapy in myasthenia gravis. Neurology 2008; 71:394–399.
- Meriggioli MN, Ciafaloni E, Al-Hayk KA, et al. Mycophenolate mofetil for myasthenia gravis: an analysis of efficacy, safety, and tolerability. Neurology 2003; 61:1438–1440.
- Hehir MK, Burns TM, Alpers J, Conaway MR, Sawa M, Sanders DB. Mycophenolate mofetil in AChR-antibody-positive myasthenia gravis: outcomes in 102 patients. Muscle Nerve 2010; 41:593–598.
- Merlob P, Stahl B, Klinger G. Tetrada of the possible mycophenolate mofetil embryopathy: a review. Reprod Toxicol 2009; 28:105–108.
- Tindall RS, Rollins JA, Phillips JT, Greenlee RG, Wells L, Belendiuk G. Preliminary results of a double-blind, randomized, placebo-controlled trial of cyclosporine in myasthenia gravis. N Engl J Med 1987; 316:719–724.
- Heckmann JM, Rawoot A, Bateman K, Renison R, Badri M. A single-blinded trial of methotrexate versus azathioprine as steroid-sparing agents in generalized myasthenia gravis. BMC Neurol 2011; 11:97.
- Pasnoor M, He J, Herbelin L, Dimachkie M, Barohn RJ; Muscle Study Group. Phase II trial of methotrexate in myasthenia gravis. Ann N Y Acad Sci 2012; 1275:23–28.
- Díaz-Manera J, Martínez-Hernández E, Querol L, et al. Long-lasting treatment effect of rituximab in MuSK myasthenia. Neurology 2012; 78:189–193.
- Gronseth GS, Barohn RJ. Practice parameter: thymectomy for autoimmune myasthenia gravis (an evidence-based review): Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2000; 55:7–15.
- Kumar V, Kaminski HJ. Treatment of myasthenia gravis. Curr Neurol Neurosci Rep 2011; 11:89–96.
- Pompeo E, Tacconi F, Massa R, Mineo D, Nahmias S, Mineo TC. Long-term outcome of thoracoscopic extended thymectomy for nonthymomatous myasthenia gravis. Eur J Cardiothorac Surg 2009; 36:164–169.
KEY POINTS
- In most cases of myasthenia gravis, the patient has antibodies against acetylcholine receptor (AChR) or musclespecific tyrosine kinase (MuSK).
- Myasthenia gravis is diagnosed by clinical signs, bedside tests (the ice-pack test or the edrophonium test), serologic tests for AChR antibodies or MuSK antibodies, and electrophysiologic tests.
- Acetylcholinesterase inhibitors are the first-step therapy, but patients who have moderate to severe symptoms require some form of immunomodulating therapy.
- A number of drugs can exacerbate weakness in myasthenia gravis and should be avoided or used with caution. These include penicillamine, interferons, procainamide, quinidine, and antibiotics such as quinolones and aminoglycosides.