User login
Dyspnea after treatment of recurrent urinary tract infection
A 71-year-old woman came to the hospital because of generalized weakness, fatigue, and exertional dyspnea.
She had a history of anemia, recurrent urinary tract infections, and hyperactive bladder. She had been taking nitrofurantoin for a urinary tract infection and phenazopyridine for dysuria, and she noticed that her urine was dark-colored.
She was of northern European descent. She was unaware of any family history of blood-related disorders. She had been admitted to the hospital 6 weeks earlier for symptomatic anemia after taking nitrofurantoin for a urinary tract infection. At that time, she received 2 units of packed red blood cells and then was discharged. Follow-up blood work done 2 weeks later—including a glucose-6 phosphate dehydrogenase (G6PD) assay—was normal.
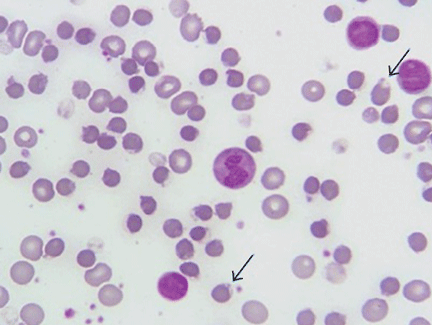
On physical examination, she was pale and weak. Her hemoglobin level was 5.5 g/dL (reference range 14.0–17.5), with normal white blood cell and platelet counts and an elevated reticulocyte count. A comprehensive metabolic panel showed elevated indirect bilirubin and lactate dehydrogenase levels. A direct Coombs test for autoimmune hemolytic anemia was negative, as was a haptoglobin assay to look for intravascular hemolytic anemia. G6PD levels were normal, yet a peripheral blood smear (Figure 1) showed features of G6PD deficiency.
What was the cause of her anemia?
GLUCOSE-6-PHOSPHATE DEHYDROGENASE DEFICIENCY
G6PD deficiency is an X-linked disorder1 that can present as hemolytic anemia. Symptoms of hemolysis can range from mild to severe on exposure to an inciting agent. Men are more commonly affected than women, and affected women are mostly heterozygous. The severity of hemolysis in heterozygous women depends on inactivation of the unaffected X chromosome in some cells.
When exposed to oxidizing agents, people with G6PD deficiency do not have enough nicotinamide adenine dinucleotide phosphate to protect red blood cells.2 This leads to oxidative denaturation of hemoglobin, formation of methemoglobin, and denaturation of globulin. These products are insoluble; they collect in red blood cells and are called Heinz bodies.3 When red blood cells containing Heinz bodies pass through the liver and spleen, the insoluble masses are taken up by macrophages, causing hemolysis and the formation of “bite cells”4 (so named because macrophages “bite” the Heinz bodies out of the red blood cells).
Patients with G6PD deficiency have all the clinical features of hemolytic anemia. On laboratory testing, the Coombs test is negative, the G6PD level is low, and the peripheral smear shows bite cells. The G6PD level is falsely normal or elevated during acute hemolysis because red blood cells deficient in G6PD are removed from circulation and replaced by young red blood cells. The G6PD level is also elevated after blood transfusion. Thus, the G6PD level should be tested 3 months after an acute event.
Hemolysis in G6PD is usually intermittent and self-limited. No treatment is needed except for avoidance of triggers and transfusion for symptomatic anemia. Of note, triggers include some of the drugs commonly used for urinary tract infections (sulfa drugs, nitrofurantoin, phenazopyridine) and antimalarials. Fava beans are also known to cause hemolytic crisis. A complete list of things to avoid can be found at www.g6pd.org/en/G6PDDeficiency/SafeUnsafe/DaEvitare_ISS-it.
There is no commercially available genetic testing kit for G6PD deficiency. Mutation analysis and G6PD gene sequencing are possible but are neither routinely done nor widely available.
BACK TO OUR PATIENT
Our patient’s hemolytic anemia was most likely drug-induced, secondary to a relative deficiency of G6PD. She had been taking nitrofurantoin and phenazopyridine; both of these are oxidizing agents and are known to cause acute hemolytic anemia in people with G6PD deficiency. The G6PD level can be normal after a recent blood transfusion and, as in our patient, during an acute episode of hemolysis.
Because of the strong suspicion of G6PD deficiency, both drugs were stopped when the patient was discharged from the hospital. She did not take either drug for 3 months. Her G6PD level was then retested and was found to be low, confirming the diagnosis. The patient was then advised not to take those drugs again. Since then, her hemoglobin level has remained stable and she has not needed any more blood transfusions.
- Mason PJ, Bautista JM, Gilsanz F. G6PD deficiency: the genotype-phenotype association. Blood Rev 2007; 21:267–283.
- Arese P, De Flora A. Pathophysiology of hemolysis in glucose-6-phosphate dehydrogenase deficiency. Semin Hematol 1990; 27:1–40.
- Jacob HS. Mechanisms of Heinz body formation and attachment to red cell membrane. Semin Hematol 1970; 7:341–354.
- Rifkind RA. Heinz body anemia: an ultrastructural study. II. Red cell sequestration and destruction. Blood 1965; 26:433–448.
A 71-year-old woman came to the hospital because of generalized weakness, fatigue, and exertional dyspnea.
She had a history of anemia, recurrent urinary tract infections, and hyperactive bladder. She had been taking nitrofurantoin for a urinary tract infection and phenazopyridine for dysuria, and she noticed that her urine was dark-colored.
She was of northern European descent. She was unaware of any family history of blood-related disorders. She had been admitted to the hospital 6 weeks earlier for symptomatic anemia after taking nitrofurantoin for a urinary tract infection. At that time, she received 2 units of packed red blood cells and then was discharged. Follow-up blood work done 2 weeks later—including a glucose-6 phosphate dehydrogenase (G6PD) assay—was normal.
On physical examination, she was pale and weak. Her hemoglobin level was 5.5 g/dL (reference range 14.0–17.5), with normal white blood cell and platelet counts and an elevated reticulocyte count. A comprehensive metabolic panel showed elevated indirect bilirubin and lactate dehydrogenase levels. A direct Coombs test for autoimmune hemolytic anemia was negative, as was a haptoglobin assay to look for intravascular hemolytic anemia. G6PD levels were normal, yet a peripheral blood smear (Figure 1) showed features of G6PD deficiency.
What was the cause of her anemia?
GLUCOSE-6-PHOSPHATE DEHYDROGENASE DEFICIENCY
G6PD deficiency is an X-linked disorder1 that can present as hemolytic anemia. Symptoms of hemolysis can range from mild to severe on exposure to an inciting agent. Men are more commonly affected than women, and affected women are mostly heterozygous. The severity of hemolysis in heterozygous women depends on inactivation of the unaffected X chromosome in some cells.
When exposed to oxidizing agents, people with G6PD deficiency do not have enough nicotinamide adenine dinucleotide phosphate to protect red blood cells.2 This leads to oxidative denaturation of hemoglobin, formation of methemoglobin, and denaturation of globulin. These products are insoluble; they collect in red blood cells and are called Heinz bodies.3 When red blood cells containing Heinz bodies pass through the liver and spleen, the insoluble masses are taken up by macrophages, causing hemolysis and the formation of “bite cells”4 (so named because macrophages “bite” the Heinz bodies out of the red blood cells).
Patients with G6PD deficiency have all the clinical features of hemolytic anemia. On laboratory testing, the Coombs test is negative, the G6PD level is low, and the peripheral smear shows bite cells. The G6PD level is falsely normal or elevated during acute hemolysis because red blood cells deficient in G6PD are removed from circulation and replaced by young red blood cells. The G6PD level is also elevated after blood transfusion. Thus, the G6PD level should be tested 3 months after an acute event.
Hemolysis in G6PD is usually intermittent and self-limited. No treatment is needed except for avoidance of triggers and transfusion for symptomatic anemia. Of note, triggers include some of the drugs commonly used for urinary tract infections (sulfa drugs, nitrofurantoin, phenazopyridine) and antimalarials. Fava beans are also known to cause hemolytic crisis. A complete list of things to avoid can be found at www.g6pd.org/en/G6PDDeficiency/SafeUnsafe/DaEvitare_ISS-it.
There is no commercially available genetic testing kit for G6PD deficiency. Mutation analysis and G6PD gene sequencing are possible but are neither routinely done nor widely available.
BACK TO OUR PATIENT
Our patient’s hemolytic anemia was most likely drug-induced, secondary to a relative deficiency of G6PD. She had been taking nitrofurantoin and phenazopyridine; both of these are oxidizing agents and are known to cause acute hemolytic anemia in people with G6PD deficiency. The G6PD level can be normal after a recent blood transfusion and, as in our patient, during an acute episode of hemolysis.
Because of the strong suspicion of G6PD deficiency, both drugs were stopped when the patient was discharged from the hospital. She did not take either drug for 3 months. Her G6PD level was then retested and was found to be low, confirming the diagnosis. The patient was then advised not to take those drugs again. Since then, her hemoglobin level has remained stable and she has not needed any more blood transfusions.
A 71-year-old woman came to the hospital because of generalized weakness, fatigue, and exertional dyspnea.
She had a history of anemia, recurrent urinary tract infections, and hyperactive bladder. She had been taking nitrofurantoin for a urinary tract infection and phenazopyridine for dysuria, and she noticed that her urine was dark-colored.
She was of northern European descent. She was unaware of any family history of blood-related disorders. She had been admitted to the hospital 6 weeks earlier for symptomatic anemia after taking nitrofurantoin for a urinary tract infection. At that time, she received 2 units of packed red blood cells and then was discharged. Follow-up blood work done 2 weeks later—including a glucose-6 phosphate dehydrogenase (G6PD) assay—was normal.
On physical examination, she was pale and weak. Her hemoglobin level was 5.5 g/dL (reference range 14.0–17.5), with normal white blood cell and platelet counts and an elevated reticulocyte count. A comprehensive metabolic panel showed elevated indirect bilirubin and lactate dehydrogenase levels. A direct Coombs test for autoimmune hemolytic anemia was negative, as was a haptoglobin assay to look for intravascular hemolytic anemia. G6PD levels were normal, yet a peripheral blood smear (Figure 1) showed features of G6PD deficiency.
What was the cause of her anemia?
GLUCOSE-6-PHOSPHATE DEHYDROGENASE DEFICIENCY
G6PD deficiency is an X-linked disorder1 that can present as hemolytic anemia. Symptoms of hemolysis can range from mild to severe on exposure to an inciting agent. Men are more commonly affected than women, and affected women are mostly heterozygous. The severity of hemolysis in heterozygous women depends on inactivation of the unaffected X chromosome in some cells.
When exposed to oxidizing agents, people with G6PD deficiency do not have enough nicotinamide adenine dinucleotide phosphate to protect red blood cells.2 This leads to oxidative denaturation of hemoglobin, formation of methemoglobin, and denaturation of globulin. These products are insoluble; they collect in red blood cells and are called Heinz bodies.3 When red blood cells containing Heinz bodies pass through the liver and spleen, the insoluble masses are taken up by macrophages, causing hemolysis and the formation of “bite cells”4 (so named because macrophages “bite” the Heinz bodies out of the red blood cells).
Patients with G6PD deficiency have all the clinical features of hemolytic anemia. On laboratory testing, the Coombs test is negative, the G6PD level is low, and the peripheral smear shows bite cells. The G6PD level is falsely normal or elevated during acute hemolysis because red blood cells deficient in G6PD are removed from circulation and replaced by young red blood cells. The G6PD level is also elevated after blood transfusion. Thus, the G6PD level should be tested 3 months after an acute event.
Hemolysis in G6PD is usually intermittent and self-limited. No treatment is needed except for avoidance of triggers and transfusion for symptomatic anemia. Of note, triggers include some of the drugs commonly used for urinary tract infections (sulfa drugs, nitrofurantoin, phenazopyridine) and antimalarials. Fava beans are also known to cause hemolytic crisis. A complete list of things to avoid can be found at www.g6pd.org/en/G6PDDeficiency/SafeUnsafe/DaEvitare_ISS-it.
There is no commercially available genetic testing kit for G6PD deficiency. Mutation analysis and G6PD gene sequencing are possible but are neither routinely done nor widely available.
BACK TO OUR PATIENT
Our patient’s hemolytic anemia was most likely drug-induced, secondary to a relative deficiency of G6PD. She had been taking nitrofurantoin and phenazopyridine; both of these are oxidizing agents and are known to cause acute hemolytic anemia in people with G6PD deficiency. The G6PD level can be normal after a recent blood transfusion and, as in our patient, during an acute episode of hemolysis.
Because of the strong suspicion of G6PD deficiency, both drugs were stopped when the patient was discharged from the hospital. She did not take either drug for 3 months. Her G6PD level was then retested and was found to be low, confirming the diagnosis. The patient was then advised not to take those drugs again. Since then, her hemoglobin level has remained stable and she has not needed any more blood transfusions.
- Mason PJ, Bautista JM, Gilsanz F. G6PD deficiency: the genotype-phenotype association. Blood Rev 2007; 21:267–283.
- Arese P, De Flora A. Pathophysiology of hemolysis in glucose-6-phosphate dehydrogenase deficiency. Semin Hematol 1990; 27:1–40.
- Jacob HS. Mechanisms of Heinz body formation and attachment to red cell membrane. Semin Hematol 1970; 7:341–354.
- Rifkind RA. Heinz body anemia: an ultrastructural study. II. Red cell sequestration and destruction. Blood 1965; 26:433–448.
- Mason PJ, Bautista JM, Gilsanz F. G6PD deficiency: the genotype-phenotype association. Blood Rev 2007; 21:267–283.
- Arese P, De Flora A. Pathophysiology of hemolysis in glucose-6-phosphate dehydrogenase deficiency. Semin Hematol 1990; 27:1–40.
- Jacob HS. Mechanisms of Heinz body formation and attachment to red cell membrane. Semin Hematol 1970; 7:341–354.
- Rifkind RA. Heinz body anemia: an ultrastructural study. II. Red cell sequestration and destruction. Blood 1965; 26:433–448.

Canagliflozin: Improving diabetes by making urine sweet
Glycosuria used to be a sign of uncontrolled diabetes and was something to be corrected, not a therapeutic mechanism. But now we have a new class of drugs that lower plasma glucose levels by increasing the renal excretion of glucose.
Here, we will review canagliflozin, the first in a new class of drugs for type 2 diabetes: how it works, who is a candidate for it, and what to watch out for.
THE NEED FOR NEW DIABETES DRUGS
Diabetes mellitus affects more than 25.8 million people in the United States—8.3% of the population—and this staggering number is rising.1 Among US residents age 65 and older, more than 10.9 million (26.9%) have diabetes.1 People with uncontrolled diabetes are at risk of microvascular complications such as retinopathy, nephropathy, and neuropathy, as well as cardiovascular disease. Diabetes is the leading cause of blindness, chronic kidney disease, and nontraumatic lower-limb amputation in the United States.1
Type 2 diabetes accounts for more than 90% of cases of diabetes in the United States, Europe, and Canada.2 It is characterized by insulin resistance, decreased beta-cell function, and progressive beta-cell decline.3
Current American Diabetes Association guidelines for the treatment of diabetes recommend a hemoglobin A1c target of less than 7.0%.4 Initial management includes lifestyle modifications such as changes in diet and an increase in exercise, as well as consideration of metformin treatment at the same time. If glucose levels remain uncontrolled despite these efforts, other drugs should be added.

A number of oral and injectable antihyperglycemic drugs are available to help achieve this goal, though none is without risk of adverse effects. Those available up to now include metformin, sulfonylureas, meglitinides, alpha-glucosidase inhibitors, thiazolidinediones, gliptins, glucagon-like peptide-1 agonists, amylin analogues, colesevelam, dopamine agonists, and insulin.5 Most of the available antihyperglycemics target the liver, pancreas, gut, and muscle to improve insulin sensitivity, reduce insulin resistance, or stimulate insulin secretion.
Despite the abundance of agents, type 2 diabetes remains uncontrolled in many patients. Only 57.1% of participants with previously diagnosed diabetes in the 2003–2006 National Health and Nutrition Examination Survey were at the hemoglobin A1c goal of less than 7.0%.6 Possible reasons for failure include adverse effects such as hypoglycemia, weight gain, and gastrointestinal symptoms resulting in discontinued use, nonadherence to the prescribed regimen, and failure to increase the dosage or to add additional agents, including insulin, to optimize glycemic control as beta-cell function declines over time.
HOW THE KIDNEYS HANDLE GLUCOSE
In the kidney, glucose is filtered in the glomerulus and then is reabsorbed in the proximal tubule. Normally, the filtered glucose is all reabsorbed unless the glucose load exceeds the kidney’s absorptive capacity. Membrane proteins called sodium-glucose cotransporters reabsorb glucose at the proximal tubule and return it into the peripheral circulation. Glucose enters the tubular epithelial cell with sodium by passive cotransport via the sodiumglucose cotransporters, and then exits on the other side via the glucose transporter GLUT in the basolateral membrane.
Two sodium-glucose transporters that act in the proximal tubule of the kidney have been identified: SGLT1 and SGLT2. SGLT2 reabsorbs most of the glucose in the early segment of the proximal tubule, while SLGT1 reabsorbs the remaining glucose at the distal end.7 SGLT2 is responsible for more than 90% of renal tubular reabsorption of glucose and is found only in the proximal tubule, whereas SGLT1 is found mainly in the gastrointestinal tract.8
Patients with type 2 diabetes have a higher capacity for glucose reabsorption in the proximal tubule as a result of the up-regulation of SGLT2.9
SGLT2 INHIBITORS AND TYPE 2 DIABETES
Drugs that inhibit SGLT2 block reabsorption of glucose in the proximal tubule, lowering the renal threshold for glucose and thereby increasing urinary glucose excretion and lowering the serum glucose level in patients with hyperglycemia. This mechanism of action is insulin-independent.
On March 29, 2013, canagliflozin became the first SGLT2 inhibitor to be approved in the United States for the treatment of type 2 diabetes.10 However, it is not the first of its class to be introduced.
Dapagliflozin was the first SGLT2 inhibitor approved in Europe and has been available there since November 2012. However, the US Food and Drug Administration withheld its approval in the United States in January 2012 because of concerns of a possible association with cancer, specifically breast and bladder cancers, as well as possible liver injury.10 Canagliflozin does not appear to share this risk.
Several other SGLT2 inhibitors may soon be available. Empagliflozin is in phase III trials, and the manufacturer has filed for approval in the United States. Ipragliflozin is awaiting approval in Japan.
CANAGLIFLOZIN: PHARMACOKINETICS AND THERAPEUTIC EFFICACY
Canagliflozin reaches its peak plasma concentration within 1 to 2 hours of oral administration.11 Its half-life is 10.6 hours with a 100-mg dose and 13.1 hours with a 300-mg dose. A steady state is typically achieved in 4 to 5 days.11
Canagliflozin lowers fasting plasma glucose and hemoglobin A1c levels in a dose-dependent manner.10,11 These effects are independent of age, sex, body mass index, and race.12 Postprandial glucose levels are also lowered.
Other potential benefits of canagliflozin include lowering of the systolic blood pressure and, especially important in obese people with type 2 diabetes, weight loss.12 Aside from metformin, which occasionally results in modest weight loss, other oral drugs used in treating type 2 diabetes are weight-neutral or can cause weight gain.
Trials of canagliflozin
Nine phase III trials of canagliflozin have enrolled 10,285 patients, in one of the largest clinical trial programs in type 2 diabetes to date.10 Several of these trials evaluated canagliflozin as monotherapy, whereas others assessed its effect as an add-on therapy in combination with another antihyperglycemic agent such as a sulfonylurea, metformin, pioglitazone, or insulin. There has not yet been a trial directly comparing canagliflozin with metformin.
Four of the placebo-controlled trials evaluated canagliflozin as monotherapy, canagliflozin added to metformin alone, canagliflozin added to metformin plus glimepiride, and canagliflozin added to metformin plus pioglitazone.
When canagliflozin was used as monotherapy, hemoglobin A1c levels at 26 weeks were an absolute 0.91% lower in the canagliflozin 100 mg/day group than in the placebo group, and an absolute 1.16% lower in the canagliflozin 300 mg/day group than in the placebo group (P < .001 for both).12 Patients lost 2.8% of their body weight with canagliflozin 100 mg and 3.3% with canagliflozin 300 mg, compared with 0.6% with placebo. Systolic blood pressure fell by a mean of 3.7 mm Hg with the 100-mg dose and by a mean of 5.4 mm Hg with the 300-mg dose compared with placebo (P < .001 for both dose groups).12
When canagliflozin was added to metformin, with glimepiride as the comparator drug, there was a 5.2% weight reduction with the 100-mg dose, a 5.7% reduction with 300 mg, and a 1% gain with glimepiride. Hemoglobin A1c fell about equally in the three groups.11
When canagliflozin was added to metformin and a sulfonylurea, with sitagliptin as the comparator third drug, the 300-mg canagliflozin dosage group had a 2.8% weight reduction.11
WHAT ARE THE ADVERSE EFFECTS?
Overall, canagliflozin seems to be well tolerated. The most common adverse effects reported in the clinical trials were genital yeast infections, urinary tract infections, and increased urination.
Genital yeast infections were more common in women than in men, occurring in 10.4% of women who received canagliflozin 100 mg and in 11.4% of women who received 300 mg, compared with only 3.2% in the placebo group.11
Urinary tract infections occurred in 5.9% of the 100-mg group and in 4.3% of the 300-mg group, compared with 4.0% of the placebo group.11
Postural hypotension. Lowering of blood pressure and symptoms of postural hypotension were also reported, and these may be attributed to the drug’s mild osmotic diuretic effect. The risk of adverse effects of volume depletion was dose-dependent; in patients over age 75, they occurred in 4.9% of those taking 100 mg and in 8.7% of those taking 300 mg, compared with 2.6% of those in the placebo or active-comparator groups.11 Therefore, one should exercise particular caution when starting this drug in the elderly or in patients taking diuretics or multiple antihypertensive drugs.
Hypoglycemia. When canagliflozin was used as monotherapy, the incidence of hypoglycemia over 26 weeks was similar to that with placebo, occurring in 3.6% of the 100-mg group, 3.0% of the 300-mg group, and 2.6% of the placebo group.12 Canagliflozin was associated with fewer episodes of hypoglycemia than were sulfonylureas, and the number of episodes was similar to that in patients taking gliptins. There was a higher overall incidence of hypoglycemia when canagliflozin was used in combination with a sulfonylurea or with insulin than when it was used as monotherapy.11
Hyperkalemia. Patients with moderate renal impairment or who are on potassiumsparing drugs or drugs that interfere with the renin-angiotensin-aldosterone system may be at higher risk of hyperkalemia, so close monitoring of potassium is recommended. There was also a dose-dependent increase in serum phosphate and magnesium levels, more notably in patients with moderate renal impairment within the first 3 weeks of starting the drug.11
Patients on canagliflozin who are also taking digoxin, ritonavir, phenytoin, phenobarbital, or rifampin should be closely monitored because of the risk of drug-drug interactions.11 Specifically, there was an increase in mean peak digoxin concentrations when used with canagliflozin 300 mg, and the use of phenytoin, phenobarbital, and ritonavir decreased the efficacy of canagliflozin.
WHAT ARE THE CARDIOVASCULAR RISKS OR LONG-TERM CONCERNS?
Dose-dependent increases in low-density lipoprotein cholesterol (LDL-C) may be seen with canagliflozin. Mean changes from baseline compared with placebo were 4.4 mg/dL (4.5%) with canagliflozin 100 mg and 8.3 mg/dL (8%) with canagliflozin 300 mg.11
There was also an increase in non-high-density lipoprotein cholesterol (non-HDL-C).12 Compared with placebo, mean non-HDL-C levels rose by 2.1 mg/dL (1.5%) with canagliflozin 100 mg and 5.1 mg/dL (3.6%) with 300 mg.11
In the 26-week canagliflozin monotherapy trial, archived blood samples in a small subgroup of patients (n = 349) were measured for apolipoprotein-B, which was found to increase by 1.2% with canagliflozin 100 mg and 3.5% with canagliflozin 300 mg, compared with 0.9% in the placebo group.12
Although small, the increase in LDL-C seen with this drug could be a concern, as diabetic patients are already at higher risk of cardiovascular events. The mechanism of this increase is not yet known, though it may be related to metabolic changes from urinary glucose excretion.12
The Canagliflozin Cardiovascular Assessment Study (CANVAS) is a randomized placebo-controlled trial in more than 4,000 patients with type 2 diabetes who have a history of or are at high risk of cardiovascular events. Currently under way, it is evaluating the occurrence of major adverse cardiovascular events (the primary end point) in patients randomized to receive canagliflozin 100 mg, canagliflozin 300 mg, or placebo once daily for up to 4 years. Secondary end points will be the drug’s effects on fasting plasma insulin and glucose, progression of albuminuria, body weight, blood pressure, HDL-C, LDL-C, bone mineral density, markers of bone turnover, and body composition.10 This trial will run for 9 years, to be completed in 2018.13
The CANVAS investigators have already reported that within the first month of treatment, 13 patients taking canagliflozin suffered a major cardiovascular event, including stroke (one of which was fatal) compared with just one patient taking placebo. These events were not seen after the first month. The hazard ratio for major adverse cardiovascular events within the first 30 days was 6.49, but this dropped to 0.89 after the first 30 days.10
Additional issues that should be addressed in long-term postmarketing studies include possible relationships with cancers and pancreatitis and the safety of the drug in pregnancy and in children with diabetes.10
WHO IS A CANDIDATE FOR THIS DRUG?
Canagliflozin is approved for use as monotherapy in addition to lifestyle modifications. It is also approved for use with other antihyperglycemic drugs, including metformin.
Obese patients with type 2 diabetes and normal kidney function may have the greatest benefit. Because of canagliflozin’s insulinin-dependent mechanism of action, patients with both early and late type 2 diabetes may benefit from its ability to lower hemoglobin A1c and blood glucose.14
Although it can be used in patients with moderate (but not severe) kidney disease, canagliflozin does not appear to be as effective in these patients, who had higher rates of adverse effects.11 It is not indicated for patients with type 1 diabetes, type 2 diabetes with ketonuria, or end-stage renal disease (estimated glomerular filtration rate < 45 mL/min or receiving dialysis).11 It also is not yet recommended for use in pregnant women or patients under age 18.
The recommended starting dose of canagliflozin is 100 mg once daily, taken with breakfast. This can be increased to 300 mg once daily if tolerated. However, patients with an estimated glomerular filtration rate of 45 to 60 mL/min should not exceed the 100- mg dose. No dose adjustment is required in patients with mild to moderate hepatic impairment. It is not recommended, however, in patients with severe hepatic impairment.11
Comment. Although canagliflozin is approved as monotherapy, metformin remains my choice for first-line oral therapy. Because canagliflozin is more expensive and its long-term affects are still relatively unknown, I prefer to use it as an adjunct, and believe it will be a useful addition, especially in obese patients who are seeking to lose weight.
WHAT IS THE COST OF THIS DRUG?
The suggested price is $10.53 per tablet (AmerisourceBergen), which is comparable to that of other newer drugs for type 2 diabetes.
THE BOTTOM LINE
The availability of canagliflozin as an additional oral antihyperglycemic option may prove helpful in managing patients with type 2 diabetes who experience adverse effects with other antihyperglycemic drugs.
As with any new drug, questions remain about the long-term risks of canagliflozin. However, it seems to be well tolerated, especially in patients with normal kidney function, and poses a low risk of hypoglycemia. The slight increase in LDL-C may prompt more aggressive lipid management. Whether blood pressure-lowering and weight loss will offset this increase in LDL-C is yet to be determined. Ongoing studies will help to further elucidate whether there is an increased risk of cardiovascular events.
Finally, canagliflozin distinguishes itself from other oral diabetes drugs by its added benefit of weight loss, an appealing side effect, especially in the growing population of obese individuals with type 2 diabetes mellitus.
- Centers for Disease Control and Prevention (CDC). Diabetes data and trends. www.cdc.gov/diabetes/statistics/. Accessed September 6, 2013.
- National Diabetes Information Clearinghouse (NDIC), National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). National diabetes statistics, 2011. www.diabetes.niddk.nih.gov/dm/pubs/statistics/. Accessed September 6, 2013.
- Campbell RK. Fate of the beta-cell in the pathophysiology of type 2 diabetes. J Am Pharm Assoc (2003). 2009; 49(suppl 1):S10–S15.
- American Diabetes Association. Executive summary: standards of medical care in diabetes—2012. Diabetes Care 2012; 35(suppl 1):S4–S10.
- Blonde L. Current antihyperglycemic treatment strategies for patients with type 2 diabetes mellitus. Cleve Clin J Med 2009; 76(suppl 5):S4–S11.
- Cheung BM, Ong KL, Cherny SS, Sham PC, Tso AW, Lam KS. Diabetes prevalence and therapeutic target achievement in the United States, 1999 to 2006. Am J Med 2009; 122:443–453.
- Vallon V. The proximal tubule in the pathophysiology of the diabetic kidney. Am J Physiol Regul Integr Comp Physiol 2011; 300:R1009–R1022.
- DeFronzo RA, Davidson JA, Del Prato S. The role of the kidneys in glucose homeostasis: a new path towards normalizing glycaemia. Diabetes Obes Metab 2012; 14:5–14.
- Pfister M, Whaley JM, Zhang L, List JF. Inhibition of SGLT2: a novel strategy for treatment of type 2 diabetes mellitus. Clin Pharmacol Ther 2011; 89:621–625.
- Food and Drug Administration (FDA). FDA Briefing Document. NDA 204042. Invokana (canagliflozin) Tablets. www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/EndocrinologicandMetabolicDrugsAdvisoryCommittee/UCM334550.pdf. Accessed September 6, 2013.
- INVOKANA (canagliflozin) tablets, for oral use. Prescribing Information. Janssen Pharmaceuticals, Inc. www.janssenpharmaceuticalsinc.com/assets/invokana_prescribing_info.pdf. Accessed September 6, 2013.
- Stenlöf K, Cefalu WT, Kim KA, et al. Efficacy and safety of canagliflozin monotherapy in subjects with type 2 diabetes mellitus inadequately controlled with diet and exercise. Diabetes Obes Metab 2013; 15:372–382.
- US National Institutes of Health. ClinicalTrials.gov. CANVAS—CA Nagliflozin cardio Vascular Assessment Study. http://clinicaltrials.gov/show/NCT01032629. Accessed September 6, 2013.
- Devineni D, Morrow L, Hompesch M, et al. Canagliflozin improves glycaemic control over 28 days in subjects with type 2 diabetes not optimally controlled on insulin. Diabetes Obes Metab 2012; 14:539–545.
Glycosuria used to be a sign of uncontrolled diabetes and was something to be corrected, not a therapeutic mechanism. But now we have a new class of drugs that lower plasma glucose levels by increasing the renal excretion of glucose.
Here, we will review canagliflozin, the first in a new class of drugs for type 2 diabetes: how it works, who is a candidate for it, and what to watch out for.
THE NEED FOR NEW DIABETES DRUGS
Diabetes mellitus affects more than 25.8 million people in the United States—8.3% of the population—and this staggering number is rising.1 Among US residents age 65 and older, more than 10.9 million (26.9%) have diabetes.1 People with uncontrolled diabetes are at risk of microvascular complications such as retinopathy, nephropathy, and neuropathy, as well as cardiovascular disease. Diabetes is the leading cause of blindness, chronic kidney disease, and nontraumatic lower-limb amputation in the United States.1
Type 2 diabetes accounts for more than 90% of cases of diabetes in the United States, Europe, and Canada.2 It is characterized by insulin resistance, decreased beta-cell function, and progressive beta-cell decline.3
Current American Diabetes Association guidelines for the treatment of diabetes recommend a hemoglobin A1c target of less than 7.0%.4 Initial management includes lifestyle modifications such as changes in diet and an increase in exercise, as well as consideration of metformin treatment at the same time. If glucose levels remain uncontrolled despite these efforts, other drugs should be added.
A number of oral and injectable antihyperglycemic drugs are available to help achieve this goal, though none is without risk of adverse effects. Those available up to now include metformin, sulfonylureas, meglitinides, alpha-glucosidase inhibitors, thiazolidinediones, gliptins, glucagon-like peptide-1 agonists, amylin analogues, colesevelam, dopamine agonists, and insulin.5 Most of the available antihyperglycemics target the liver, pancreas, gut, and muscle to improve insulin sensitivity, reduce insulin resistance, or stimulate insulin secretion.
Despite the abundance of agents, type 2 diabetes remains uncontrolled in many patients. Only 57.1% of participants with previously diagnosed diabetes in the 2003–2006 National Health and Nutrition Examination Survey were at the hemoglobin A1c goal of less than 7.0%.6 Possible reasons for failure include adverse effects such as hypoglycemia, weight gain, and gastrointestinal symptoms resulting in discontinued use, nonadherence to the prescribed regimen, and failure to increase the dosage or to add additional agents, including insulin, to optimize glycemic control as beta-cell function declines over time.
HOW THE KIDNEYS HANDLE GLUCOSE
In the kidney, glucose is filtered in the glomerulus and then is reabsorbed in the proximal tubule. Normally, the filtered glucose is all reabsorbed unless the glucose load exceeds the kidney’s absorptive capacity. Membrane proteins called sodium-glucose cotransporters reabsorb glucose at the proximal tubule and return it into the peripheral circulation. Glucose enters the tubular epithelial cell with sodium by passive cotransport via the sodiumglucose cotransporters, and then exits on the other side via the glucose transporter GLUT in the basolateral membrane.
Two sodium-glucose transporters that act in the proximal tubule of the kidney have been identified: SGLT1 and SGLT2. SGLT2 reabsorbs most of the glucose in the early segment of the proximal tubule, while SLGT1 reabsorbs the remaining glucose at the distal end.7 SGLT2 is responsible for more than 90% of renal tubular reabsorption of glucose and is found only in the proximal tubule, whereas SGLT1 is found mainly in the gastrointestinal tract.8
Patients with type 2 diabetes have a higher capacity for glucose reabsorption in the proximal tubule as a result of the up-regulation of SGLT2.9
SGLT2 INHIBITORS AND TYPE 2 DIABETES
Drugs that inhibit SGLT2 block reabsorption of glucose in the proximal tubule, lowering the renal threshold for glucose and thereby increasing urinary glucose excretion and lowering the serum glucose level in patients with hyperglycemia. This mechanism of action is insulin-independent.
On March 29, 2013, canagliflozin became the first SGLT2 inhibitor to be approved in the United States for the treatment of type 2 diabetes.10 However, it is not the first of its class to be introduced.
Dapagliflozin was the first SGLT2 inhibitor approved in Europe and has been available there since November 2012. However, the US Food and Drug Administration withheld its approval in the United States in January 2012 because of concerns of a possible association with cancer, specifically breast and bladder cancers, as well as possible liver injury.10 Canagliflozin does not appear to share this risk.
Several other SGLT2 inhibitors may soon be available. Empagliflozin is in phase III trials, and the manufacturer has filed for approval in the United States. Ipragliflozin is awaiting approval in Japan.
CANAGLIFLOZIN: PHARMACOKINETICS AND THERAPEUTIC EFFICACY
Canagliflozin reaches its peak plasma concentration within 1 to 2 hours of oral administration.11 Its half-life is 10.6 hours with a 100-mg dose and 13.1 hours with a 300-mg dose. A steady state is typically achieved in 4 to 5 days.11
Canagliflozin lowers fasting plasma glucose and hemoglobin A1c levels in a dose-dependent manner.10,11 These effects are independent of age, sex, body mass index, and race.12 Postprandial glucose levels are also lowered.
Other potential benefits of canagliflozin include lowering of the systolic blood pressure and, especially important in obese people with type 2 diabetes, weight loss.12 Aside from metformin, which occasionally results in modest weight loss, other oral drugs used in treating type 2 diabetes are weight-neutral or can cause weight gain.
Trials of canagliflozin
Nine phase III trials of canagliflozin have enrolled 10,285 patients, in one of the largest clinical trial programs in type 2 diabetes to date.10 Several of these trials evaluated canagliflozin as monotherapy, whereas others assessed its effect as an add-on therapy in combination with another antihyperglycemic agent such as a sulfonylurea, metformin, pioglitazone, or insulin. There has not yet been a trial directly comparing canagliflozin with metformin.
Four of the placebo-controlled trials evaluated canagliflozin as monotherapy, canagliflozin added to metformin alone, canagliflozin added to metformin plus glimepiride, and canagliflozin added to metformin plus pioglitazone.
When canagliflozin was used as monotherapy, hemoglobin A1c levels at 26 weeks were an absolute 0.91% lower in the canagliflozin 100 mg/day group than in the placebo group, and an absolute 1.16% lower in the canagliflozin 300 mg/day group than in the placebo group (P < .001 for both).12 Patients lost 2.8% of their body weight with canagliflozin 100 mg and 3.3% with canagliflozin 300 mg, compared with 0.6% with placebo. Systolic blood pressure fell by a mean of 3.7 mm Hg with the 100-mg dose and by a mean of 5.4 mm Hg with the 300-mg dose compared with placebo (P < .001 for both dose groups).12
When canagliflozin was added to metformin, with glimepiride as the comparator drug, there was a 5.2% weight reduction with the 100-mg dose, a 5.7% reduction with 300 mg, and a 1% gain with glimepiride. Hemoglobin A1c fell about equally in the three groups.11
When canagliflozin was added to metformin and a sulfonylurea, with sitagliptin as the comparator third drug, the 300-mg canagliflozin dosage group had a 2.8% weight reduction.11
WHAT ARE THE ADVERSE EFFECTS?
Overall, canagliflozin seems to be well tolerated. The most common adverse effects reported in the clinical trials were genital yeast infections, urinary tract infections, and increased urination.
Genital yeast infections were more common in women than in men, occurring in 10.4% of women who received canagliflozin 100 mg and in 11.4% of women who received 300 mg, compared with only 3.2% in the placebo group.11
Urinary tract infections occurred in 5.9% of the 100-mg group and in 4.3% of the 300-mg group, compared with 4.0% of the placebo group.11
Postural hypotension. Lowering of blood pressure and symptoms of postural hypotension were also reported, and these may be attributed to the drug’s mild osmotic diuretic effect. The risk of adverse effects of volume depletion was dose-dependent; in patients over age 75, they occurred in 4.9% of those taking 100 mg and in 8.7% of those taking 300 mg, compared with 2.6% of those in the placebo or active-comparator groups.11 Therefore, one should exercise particular caution when starting this drug in the elderly or in patients taking diuretics or multiple antihypertensive drugs.
Hypoglycemia. When canagliflozin was used as monotherapy, the incidence of hypoglycemia over 26 weeks was similar to that with placebo, occurring in 3.6% of the 100-mg group, 3.0% of the 300-mg group, and 2.6% of the placebo group.12 Canagliflozin was associated with fewer episodes of hypoglycemia than were sulfonylureas, and the number of episodes was similar to that in patients taking gliptins. There was a higher overall incidence of hypoglycemia when canagliflozin was used in combination with a sulfonylurea or with insulin than when it was used as monotherapy.11
Hyperkalemia. Patients with moderate renal impairment or who are on potassiumsparing drugs or drugs that interfere with the renin-angiotensin-aldosterone system may be at higher risk of hyperkalemia, so close monitoring of potassium is recommended. There was also a dose-dependent increase in serum phosphate and magnesium levels, more notably in patients with moderate renal impairment within the first 3 weeks of starting the drug.11
Patients on canagliflozin who are also taking digoxin, ritonavir, phenytoin, phenobarbital, or rifampin should be closely monitored because of the risk of drug-drug interactions.11 Specifically, there was an increase in mean peak digoxin concentrations when used with canagliflozin 300 mg, and the use of phenytoin, phenobarbital, and ritonavir decreased the efficacy of canagliflozin.
WHAT ARE THE CARDIOVASCULAR RISKS OR LONG-TERM CONCERNS?
Dose-dependent increases in low-density lipoprotein cholesterol (LDL-C) may be seen with canagliflozin. Mean changes from baseline compared with placebo were 4.4 mg/dL (4.5%) with canagliflozin 100 mg and 8.3 mg/dL (8%) with canagliflozin 300 mg.11
There was also an increase in non-high-density lipoprotein cholesterol (non-HDL-C).12 Compared with placebo, mean non-HDL-C levels rose by 2.1 mg/dL (1.5%) with canagliflozin 100 mg and 5.1 mg/dL (3.6%) with 300 mg.11
In the 26-week canagliflozin monotherapy trial, archived blood samples in a small subgroup of patients (n = 349) were measured for apolipoprotein-B, which was found to increase by 1.2% with canagliflozin 100 mg and 3.5% with canagliflozin 300 mg, compared with 0.9% in the placebo group.12
Although small, the increase in LDL-C seen with this drug could be a concern, as diabetic patients are already at higher risk of cardiovascular events. The mechanism of this increase is not yet known, though it may be related to metabolic changes from urinary glucose excretion.12
The Canagliflozin Cardiovascular Assessment Study (CANVAS) is a randomized placebo-controlled trial in more than 4,000 patients with type 2 diabetes who have a history of or are at high risk of cardiovascular events. Currently under way, it is evaluating the occurrence of major adverse cardiovascular events (the primary end point) in patients randomized to receive canagliflozin 100 mg, canagliflozin 300 mg, or placebo once daily for up to 4 years. Secondary end points will be the drug’s effects on fasting plasma insulin and glucose, progression of albuminuria, body weight, blood pressure, HDL-C, LDL-C, bone mineral density, markers of bone turnover, and body composition.10 This trial will run for 9 years, to be completed in 2018.13
The CANVAS investigators have already reported that within the first month of treatment, 13 patients taking canagliflozin suffered a major cardiovascular event, including stroke (one of which was fatal) compared with just one patient taking placebo. These events were not seen after the first month. The hazard ratio for major adverse cardiovascular events within the first 30 days was 6.49, but this dropped to 0.89 after the first 30 days.10
Additional issues that should be addressed in long-term postmarketing studies include possible relationships with cancers and pancreatitis and the safety of the drug in pregnancy and in children with diabetes.10
WHO IS A CANDIDATE FOR THIS DRUG?
Canagliflozin is approved for use as monotherapy in addition to lifestyle modifications. It is also approved for use with other antihyperglycemic drugs, including metformin.
Obese patients with type 2 diabetes and normal kidney function may have the greatest benefit. Because of canagliflozin’s insulinin-dependent mechanism of action, patients with both early and late type 2 diabetes may benefit from its ability to lower hemoglobin A1c and blood glucose.14
Although it can be used in patients with moderate (but not severe) kidney disease, canagliflozin does not appear to be as effective in these patients, who had higher rates of adverse effects.11 It is not indicated for patients with type 1 diabetes, type 2 diabetes with ketonuria, or end-stage renal disease (estimated glomerular filtration rate < 45 mL/min or receiving dialysis).11 It also is not yet recommended for use in pregnant women or patients under age 18.
The recommended starting dose of canagliflozin is 100 mg once daily, taken with breakfast. This can be increased to 300 mg once daily if tolerated. However, patients with an estimated glomerular filtration rate of 45 to 60 mL/min should not exceed the 100- mg dose. No dose adjustment is required in patients with mild to moderate hepatic impairment. It is not recommended, however, in patients with severe hepatic impairment.11
Comment. Although canagliflozin is approved as monotherapy, metformin remains my choice for first-line oral therapy. Because canagliflozin is more expensive and its long-term affects are still relatively unknown, I prefer to use it as an adjunct, and believe it will be a useful addition, especially in obese patients who are seeking to lose weight.
WHAT IS THE COST OF THIS DRUG?
The suggested price is $10.53 per tablet (AmerisourceBergen), which is comparable to that of other newer drugs for type 2 diabetes.
THE BOTTOM LINE
The availability of canagliflozin as an additional oral antihyperglycemic option may prove helpful in managing patients with type 2 diabetes who experience adverse effects with other antihyperglycemic drugs.
As with any new drug, questions remain about the long-term risks of canagliflozin. However, it seems to be well tolerated, especially in patients with normal kidney function, and poses a low risk of hypoglycemia. The slight increase in LDL-C may prompt more aggressive lipid management. Whether blood pressure-lowering and weight loss will offset this increase in LDL-C is yet to be determined. Ongoing studies will help to further elucidate whether there is an increased risk of cardiovascular events.
Finally, canagliflozin distinguishes itself from other oral diabetes drugs by its added benefit of weight loss, an appealing side effect, especially in the growing population of obese individuals with type 2 diabetes mellitus.
Glycosuria used to be a sign of uncontrolled diabetes and was something to be corrected, not a therapeutic mechanism. But now we have a new class of drugs that lower plasma glucose levels by increasing the renal excretion of glucose.
Here, we will review canagliflozin, the first in a new class of drugs for type 2 diabetes: how it works, who is a candidate for it, and what to watch out for.
THE NEED FOR NEW DIABETES DRUGS
Diabetes mellitus affects more than 25.8 million people in the United States—8.3% of the population—and this staggering number is rising.1 Among US residents age 65 and older, more than 10.9 million (26.9%) have diabetes.1 People with uncontrolled diabetes are at risk of microvascular complications such as retinopathy, nephropathy, and neuropathy, as well as cardiovascular disease. Diabetes is the leading cause of blindness, chronic kidney disease, and nontraumatic lower-limb amputation in the United States.1
Type 2 diabetes accounts for more than 90% of cases of diabetes in the United States, Europe, and Canada.2 It is characterized by insulin resistance, decreased beta-cell function, and progressive beta-cell decline.3
Current American Diabetes Association guidelines for the treatment of diabetes recommend a hemoglobin A1c target of less than 7.0%.4 Initial management includes lifestyle modifications such as changes in diet and an increase in exercise, as well as consideration of metformin treatment at the same time. If glucose levels remain uncontrolled despite these efforts, other drugs should be added.
A number of oral and injectable antihyperglycemic drugs are available to help achieve this goal, though none is without risk of adverse effects. Those available up to now include metformin, sulfonylureas, meglitinides, alpha-glucosidase inhibitors, thiazolidinediones, gliptins, glucagon-like peptide-1 agonists, amylin analogues, colesevelam, dopamine agonists, and insulin.5 Most of the available antihyperglycemics target the liver, pancreas, gut, and muscle to improve insulin sensitivity, reduce insulin resistance, or stimulate insulin secretion.
Despite the abundance of agents, type 2 diabetes remains uncontrolled in many patients. Only 57.1% of participants with previously diagnosed diabetes in the 2003–2006 National Health and Nutrition Examination Survey were at the hemoglobin A1c goal of less than 7.0%.6 Possible reasons for failure include adverse effects such as hypoglycemia, weight gain, and gastrointestinal symptoms resulting in discontinued use, nonadherence to the prescribed regimen, and failure to increase the dosage or to add additional agents, including insulin, to optimize glycemic control as beta-cell function declines over time.
HOW THE KIDNEYS HANDLE GLUCOSE
In the kidney, glucose is filtered in the glomerulus and then is reabsorbed in the proximal tubule. Normally, the filtered glucose is all reabsorbed unless the glucose load exceeds the kidney’s absorptive capacity. Membrane proteins called sodium-glucose cotransporters reabsorb glucose at the proximal tubule and return it into the peripheral circulation. Glucose enters the tubular epithelial cell with sodium by passive cotransport via the sodiumglucose cotransporters, and then exits on the other side via the glucose transporter GLUT in the basolateral membrane.
Two sodium-glucose transporters that act in the proximal tubule of the kidney have been identified: SGLT1 and SGLT2. SGLT2 reabsorbs most of the glucose in the early segment of the proximal tubule, while SLGT1 reabsorbs the remaining glucose at the distal end.7 SGLT2 is responsible for more than 90% of renal tubular reabsorption of glucose and is found only in the proximal tubule, whereas SGLT1 is found mainly in the gastrointestinal tract.8
Patients with type 2 diabetes have a higher capacity for glucose reabsorption in the proximal tubule as a result of the up-regulation of SGLT2.9
SGLT2 INHIBITORS AND TYPE 2 DIABETES
Drugs that inhibit SGLT2 block reabsorption of glucose in the proximal tubule, lowering the renal threshold for glucose and thereby increasing urinary glucose excretion and lowering the serum glucose level in patients with hyperglycemia. This mechanism of action is insulin-independent.
On March 29, 2013, canagliflozin became the first SGLT2 inhibitor to be approved in the United States for the treatment of type 2 diabetes.10 However, it is not the first of its class to be introduced.
Dapagliflozin was the first SGLT2 inhibitor approved in Europe and has been available there since November 2012. However, the US Food and Drug Administration withheld its approval in the United States in January 2012 because of concerns of a possible association with cancer, specifically breast and bladder cancers, as well as possible liver injury.10 Canagliflozin does not appear to share this risk.
Several other SGLT2 inhibitors may soon be available. Empagliflozin is in phase III trials, and the manufacturer has filed for approval in the United States. Ipragliflozin is awaiting approval in Japan.
CANAGLIFLOZIN: PHARMACOKINETICS AND THERAPEUTIC EFFICACY
Canagliflozin reaches its peak plasma concentration within 1 to 2 hours of oral administration.11 Its half-life is 10.6 hours with a 100-mg dose and 13.1 hours with a 300-mg dose. A steady state is typically achieved in 4 to 5 days.11
Canagliflozin lowers fasting plasma glucose and hemoglobin A1c levels in a dose-dependent manner.10,11 These effects are independent of age, sex, body mass index, and race.12 Postprandial glucose levels are also lowered.
Other potential benefits of canagliflozin include lowering of the systolic blood pressure and, especially important in obese people with type 2 diabetes, weight loss.12 Aside from metformin, which occasionally results in modest weight loss, other oral drugs used in treating type 2 diabetes are weight-neutral or can cause weight gain.
Trials of canagliflozin
Nine phase III trials of canagliflozin have enrolled 10,285 patients, in one of the largest clinical trial programs in type 2 diabetes to date.10 Several of these trials evaluated canagliflozin as monotherapy, whereas others assessed its effect as an add-on therapy in combination with another antihyperglycemic agent such as a sulfonylurea, metformin, pioglitazone, or insulin. There has not yet been a trial directly comparing canagliflozin with metformin.
Four of the placebo-controlled trials evaluated canagliflozin as monotherapy, canagliflozin added to metformin alone, canagliflozin added to metformin plus glimepiride, and canagliflozin added to metformin plus pioglitazone.
When canagliflozin was used as monotherapy, hemoglobin A1c levels at 26 weeks were an absolute 0.91% lower in the canagliflozin 100 mg/day group than in the placebo group, and an absolute 1.16% lower in the canagliflozin 300 mg/day group than in the placebo group (P < .001 for both).12 Patients lost 2.8% of their body weight with canagliflozin 100 mg and 3.3% with canagliflozin 300 mg, compared with 0.6% with placebo. Systolic blood pressure fell by a mean of 3.7 mm Hg with the 100-mg dose and by a mean of 5.4 mm Hg with the 300-mg dose compared with placebo (P < .001 for both dose groups).12
When canagliflozin was added to metformin, with glimepiride as the comparator drug, there was a 5.2% weight reduction with the 100-mg dose, a 5.7% reduction with 300 mg, and a 1% gain with glimepiride. Hemoglobin A1c fell about equally in the three groups.11
When canagliflozin was added to metformin and a sulfonylurea, with sitagliptin as the comparator third drug, the 300-mg canagliflozin dosage group had a 2.8% weight reduction.11
WHAT ARE THE ADVERSE EFFECTS?
Overall, canagliflozin seems to be well tolerated. The most common adverse effects reported in the clinical trials were genital yeast infections, urinary tract infections, and increased urination.
Genital yeast infections were more common in women than in men, occurring in 10.4% of women who received canagliflozin 100 mg and in 11.4% of women who received 300 mg, compared with only 3.2% in the placebo group.11
Urinary tract infections occurred in 5.9% of the 100-mg group and in 4.3% of the 300-mg group, compared with 4.0% of the placebo group.11
Postural hypotension. Lowering of blood pressure and symptoms of postural hypotension were also reported, and these may be attributed to the drug’s mild osmotic diuretic effect. The risk of adverse effects of volume depletion was dose-dependent; in patients over age 75, they occurred in 4.9% of those taking 100 mg and in 8.7% of those taking 300 mg, compared with 2.6% of those in the placebo or active-comparator groups.11 Therefore, one should exercise particular caution when starting this drug in the elderly or in patients taking diuretics or multiple antihypertensive drugs.
Hypoglycemia. When canagliflozin was used as monotherapy, the incidence of hypoglycemia over 26 weeks was similar to that with placebo, occurring in 3.6% of the 100-mg group, 3.0% of the 300-mg group, and 2.6% of the placebo group.12 Canagliflozin was associated with fewer episodes of hypoglycemia than were sulfonylureas, and the number of episodes was similar to that in patients taking gliptins. There was a higher overall incidence of hypoglycemia when canagliflozin was used in combination with a sulfonylurea or with insulin than when it was used as monotherapy.11
Hyperkalemia. Patients with moderate renal impairment or who are on potassiumsparing drugs or drugs that interfere with the renin-angiotensin-aldosterone system may be at higher risk of hyperkalemia, so close monitoring of potassium is recommended. There was also a dose-dependent increase in serum phosphate and magnesium levels, more notably in patients with moderate renal impairment within the first 3 weeks of starting the drug.11
Patients on canagliflozin who are also taking digoxin, ritonavir, phenytoin, phenobarbital, or rifampin should be closely monitored because of the risk of drug-drug interactions.11 Specifically, there was an increase in mean peak digoxin concentrations when used with canagliflozin 300 mg, and the use of phenytoin, phenobarbital, and ritonavir decreased the efficacy of canagliflozin.
WHAT ARE THE CARDIOVASCULAR RISKS OR LONG-TERM CONCERNS?
Dose-dependent increases in low-density lipoprotein cholesterol (LDL-C) may be seen with canagliflozin. Mean changes from baseline compared with placebo were 4.4 mg/dL (4.5%) with canagliflozin 100 mg and 8.3 mg/dL (8%) with canagliflozin 300 mg.11
There was also an increase in non-high-density lipoprotein cholesterol (non-HDL-C).12 Compared with placebo, mean non-HDL-C levels rose by 2.1 mg/dL (1.5%) with canagliflozin 100 mg and 5.1 mg/dL (3.6%) with 300 mg.11
In the 26-week canagliflozin monotherapy trial, archived blood samples in a small subgroup of patients (n = 349) were measured for apolipoprotein-B, which was found to increase by 1.2% with canagliflozin 100 mg and 3.5% with canagliflozin 300 mg, compared with 0.9% in the placebo group.12
Although small, the increase in LDL-C seen with this drug could be a concern, as diabetic patients are already at higher risk of cardiovascular events. The mechanism of this increase is not yet known, though it may be related to metabolic changes from urinary glucose excretion.12
The Canagliflozin Cardiovascular Assessment Study (CANVAS) is a randomized placebo-controlled trial in more than 4,000 patients with type 2 diabetes who have a history of or are at high risk of cardiovascular events. Currently under way, it is evaluating the occurrence of major adverse cardiovascular events (the primary end point) in patients randomized to receive canagliflozin 100 mg, canagliflozin 300 mg, or placebo once daily for up to 4 years. Secondary end points will be the drug’s effects on fasting plasma insulin and glucose, progression of albuminuria, body weight, blood pressure, HDL-C, LDL-C, bone mineral density, markers of bone turnover, and body composition.10 This trial will run for 9 years, to be completed in 2018.13
The CANVAS investigators have already reported that within the first month of treatment, 13 patients taking canagliflozin suffered a major cardiovascular event, including stroke (one of which was fatal) compared with just one patient taking placebo. These events were not seen after the first month. The hazard ratio for major adverse cardiovascular events within the first 30 days was 6.49, but this dropped to 0.89 after the first 30 days.10
Additional issues that should be addressed in long-term postmarketing studies include possible relationships with cancers and pancreatitis and the safety of the drug in pregnancy and in children with diabetes.10
WHO IS A CANDIDATE FOR THIS DRUG?
Canagliflozin is approved for use as monotherapy in addition to lifestyle modifications. It is also approved for use with other antihyperglycemic drugs, including metformin.
Obese patients with type 2 diabetes and normal kidney function may have the greatest benefit. Because of canagliflozin’s insulinin-dependent mechanism of action, patients with both early and late type 2 diabetes may benefit from its ability to lower hemoglobin A1c and blood glucose.14
Although it can be used in patients with moderate (but not severe) kidney disease, canagliflozin does not appear to be as effective in these patients, who had higher rates of adverse effects.11 It is not indicated for patients with type 1 diabetes, type 2 diabetes with ketonuria, or end-stage renal disease (estimated glomerular filtration rate < 45 mL/min or receiving dialysis).11 It also is not yet recommended for use in pregnant women or patients under age 18.
The recommended starting dose of canagliflozin is 100 mg once daily, taken with breakfast. This can be increased to 300 mg once daily if tolerated. However, patients with an estimated glomerular filtration rate of 45 to 60 mL/min should not exceed the 100- mg dose. No dose adjustment is required in patients with mild to moderate hepatic impairment. It is not recommended, however, in patients with severe hepatic impairment.11
Comment. Although canagliflozin is approved as monotherapy, metformin remains my choice for first-line oral therapy. Because canagliflozin is more expensive and its long-term affects are still relatively unknown, I prefer to use it as an adjunct, and believe it will be a useful addition, especially in obese patients who are seeking to lose weight.
WHAT IS THE COST OF THIS DRUG?
The suggested price is $10.53 per tablet (AmerisourceBergen), which is comparable to that of other newer drugs for type 2 diabetes.
THE BOTTOM LINE
The availability of canagliflozin as an additional oral antihyperglycemic option may prove helpful in managing patients with type 2 diabetes who experience adverse effects with other antihyperglycemic drugs.
As with any new drug, questions remain about the long-term risks of canagliflozin. However, it seems to be well tolerated, especially in patients with normal kidney function, and poses a low risk of hypoglycemia. The slight increase in LDL-C may prompt more aggressive lipid management. Whether blood pressure-lowering and weight loss will offset this increase in LDL-C is yet to be determined. Ongoing studies will help to further elucidate whether there is an increased risk of cardiovascular events.
Finally, canagliflozin distinguishes itself from other oral diabetes drugs by its added benefit of weight loss, an appealing side effect, especially in the growing population of obese individuals with type 2 diabetes mellitus.
- Centers for Disease Control and Prevention (CDC). Diabetes data and trends. www.cdc.gov/diabetes/statistics/. Accessed September 6, 2013.
- National Diabetes Information Clearinghouse (NDIC), National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). National diabetes statistics, 2011. www.diabetes.niddk.nih.gov/dm/pubs/statistics/. Accessed September 6, 2013.
- Campbell RK. Fate of the beta-cell in the pathophysiology of type 2 diabetes. J Am Pharm Assoc (2003). 2009; 49(suppl 1):S10–S15.
- American Diabetes Association. Executive summary: standards of medical care in diabetes—2012. Diabetes Care 2012; 35(suppl 1):S4–S10.
- Blonde L. Current antihyperglycemic treatment strategies for patients with type 2 diabetes mellitus. Cleve Clin J Med 2009; 76(suppl 5):S4–S11.
- Cheung BM, Ong KL, Cherny SS, Sham PC, Tso AW, Lam KS. Diabetes prevalence and therapeutic target achievement in the United States, 1999 to 2006. Am J Med 2009; 122:443–453.
- Vallon V. The proximal tubule in the pathophysiology of the diabetic kidney. Am J Physiol Regul Integr Comp Physiol 2011; 300:R1009–R1022.
- DeFronzo RA, Davidson JA, Del Prato S. The role of the kidneys in glucose homeostasis: a new path towards normalizing glycaemia. Diabetes Obes Metab 2012; 14:5–14.
- Pfister M, Whaley JM, Zhang L, List JF. Inhibition of SGLT2: a novel strategy for treatment of type 2 diabetes mellitus. Clin Pharmacol Ther 2011; 89:621–625.
- Food and Drug Administration (FDA). FDA Briefing Document. NDA 204042. Invokana (canagliflozin) Tablets. www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/EndocrinologicandMetabolicDrugsAdvisoryCommittee/UCM334550.pdf. Accessed September 6, 2013.
- INVOKANA (canagliflozin) tablets, for oral use. Prescribing Information. Janssen Pharmaceuticals, Inc. www.janssenpharmaceuticalsinc.com/assets/invokana_prescribing_info.pdf. Accessed September 6, 2013.
- Stenlöf K, Cefalu WT, Kim KA, et al. Efficacy and safety of canagliflozin monotherapy in subjects with type 2 diabetes mellitus inadequately controlled with diet and exercise. Diabetes Obes Metab 2013; 15:372–382.
- US National Institutes of Health. ClinicalTrials.gov. CANVAS—CA Nagliflozin cardio Vascular Assessment Study. http://clinicaltrials.gov/show/NCT01032629. Accessed September 6, 2013.
- Devineni D, Morrow L, Hompesch M, et al. Canagliflozin improves glycaemic control over 28 days in subjects with type 2 diabetes not optimally controlled on insulin. Diabetes Obes Metab 2012; 14:539–545.
- Centers for Disease Control and Prevention (CDC). Diabetes data and trends. www.cdc.gov/diabetes/statistics/. Accessed September 6, 2013.
- National Diabetes Information Clearinghouse (NDIC), National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). National diabetes statistics, 2011. www.diabetes.niddk.nih.gov/dm/pubs/statistics/. Accessed September 6, 2013.
- Campbell RK. Fate of the beta-cell in the pathophysiology of type 2 diabetes. J Am Pharm Assoc (2003). 2009; 49(suppl 1):S10–S15.
- American Diabetes Association. Executive summary: standards of medical care in diabetes—2012. Diabetes Care 2012; 35(suppl 1):S4–S10.
- Blonde L. Current antihyperglycemic treatment strategies for patients with type 2 diabetes mellitus. Cleve Clin J Med 2009; 76(suppl 5):S4–S11.
- Cheung BM, Ong KL, Cherny SS, Sham PC, Tso AW, Lam KS. Diabetes prevalence and therapeutic target achievement in the United States, 1999 to 2006. Am J Med 2009; 122:443–453.
- Vallon V. The proximal tubule in the pathophysiology of the diabetic kidney. Am J Physiol Regul Integr Comp Physiol 2011; 300:R1009–R1022.
- DeFronzo RA, Davidson JA, Del Prato S. The role of the kidneys in glucose homeostasis: a new path towards normalizing glycaemia. Diabetes Obes Metab 2012; 14:5–14.
- Pfister M, Whaley JM, Zhang L, List JF. Inhibition of SGLT2: a novel strategy for treatment of type 2 diabetes mellitus. Clin Pharmacol Ther 2011; 89:621–625.
- Food and Drug Administration (FDA). FDA Briefing Document. NDA 204042. Invokana (canagliflozin) Tablets. www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/EndocrinologicandMetabolicDrugsAdvisoryCommittee/UCM334550.pdf. Accessed September 6, 2013.
- INVOKANA (canagliflozin) tablets, for oral use. Prescribing Information. Janssen Pharmaceuticals, Inc. www.janssenpharmaceuticalsinc.com/assets/invokana_prescribing_info.pdf. Accessed September 6, 2013.
- Stenlöf K, Cefalu WT, Kim KA, et al. Efficacy and safety of canagliflozin monotherapy in subjects with type 2 diabetes mellitus inadequately controlled with diet and exercise. Diabetes Obes Metab 2013; 15:372–382.
- US National Institutes of Health. ClinicalTrials.gov. CANVAS—CA Nagliflozin cardio Vascular Assessment Study. http://clinicaltrials.gov/show/NCT01032629. Accessed September 6, 2013.
- Devineni D, Morrow L, Hompesch M, et al. Canagliflozin improves glycaemic control over 28 days in subjects with type 2 diabetes not optimally controlled on insulin. Diabetes Obes Metab 2012; 14:539–545.
KEY POINTS
- Type 2 diabetes is ubiquitous and, despite an abundance of agents, often remains uncontrolled.
- Canagliflozin and other drugs of its class cause glucose to be spilled in the urine by reducing the amount reabsorbed by the kidney.
- In clinical trials, canagliflozin lowered hemoglobin A1c levels by approximately 1 absolute percentage point.
- Beyond the adverse effects to be expected from the mechanism of action of the drug (ie, genital yeast infections, urinary tract infections, and hypotension caused by osmotic diuresis), canagliflozin seems to increase plasma levels of low-density lipoprotein cholesterol. This may be worrisome, as diabetic patients are already at increased risk of cardiovascular disease.
A practical approach to prescribing antidepressants
With the variety of drugs available for treating depression, choosing one can be daunting. Different agents have characteristics that may make them a better choice for different types of patients, but even so, treating any kind of mental illness often requires an element of trial and error.
Primary care providers are on the frontline of treating mental illness, often evaluating patients before they are seen by a psychiatrist. The purpose of this article is to provide insight into the art of prescribing antidepressants in the primary care setting. We will discuss common patient presentations, including depressed patients without other medical comorbidities as well as those with common comorbidities, with our recommendations for first-line treatment.
We hope our recommendations will help you to navigate the uncertainty more confidently, resulting in more efficient and tailored treatment for your patients.
BASELINE TESTING
When starting a patient on antidepressant drug therapy, we recommend obtaining a set of baseline laboratory tests to rule out underlying medical conditions that may be contributing to the patient’s depression or that may preclude the use of a given drug. (For example, elevation of liver enzymes may preclude the use of duloxetine.) Tests should include:
- A complete blood cell count
- A complete metabolic panel
- A thyroid-stimulating hormone level.
Electrocardiography may also be useful, as some antidepressants can prolong the QT interval or elevate the blood levels of other drugs with this effect.
GENERAL TREATMENT CONSIDERATIONS
There are several classes of antidepressants, and each class has a number of agents. Research has found little difference in efficacy among agents. So to simplify choosing which one to use, we recommend becoming comfortable with an agent from each class, ie:
- A selective serotonin reuptake inhibitor (SSRI)
- A selective serotonin-norepinephrine reuptake inhibitor (SNRI)
- A tricyclic antidepressant (TCA)
- A monoamine oxidase (MAO) inhibitor.
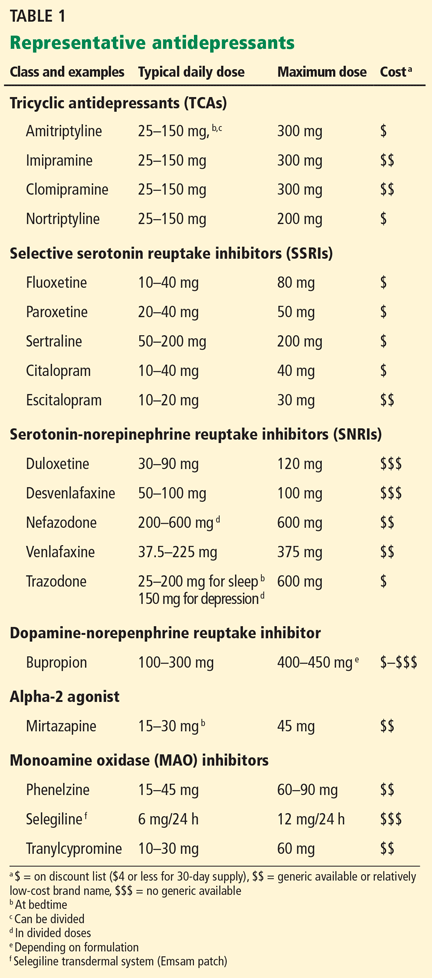
Each class includes generic agents, many of which are on the discount lists of retail pharmacies. Table 1 shows representative drugs from each class, with their relative costs.
Start low and go slow. In general, when starting an antidepressant, consider starting at half the normal dose, titrating upward as tolerated about every 14 days. This approach can minimize side effects. For example, if prescribing fluoxetine, start with 10 mg and titrate every 2 weeks based on tolerance and patient response. That said, each patient may respond differently, requiring perhaps a lower starting dose or a longer titration schedule.
Anticipate side effects. Most of the side effects of an antidepressant drug can be explained by its mechanism of action. Although side effects should certainly be considered when choosing an agent, patients can be reassured that most are transient and benign. A detailed discussion of side effects of antidepressant drugs is beyond the scope of this article, but a review by Khawam et al1 was published earlier in this journal.
Reassess. If after 4 to 6 weeks the patient has had little or no response, it is reasonable to switch agents. For a patient who was on an SSRI, the change can be to another SSRI or to an SNRI. However, if two SSRIs have already failed, then choose an SNRI. Agents are commonly cross-tapered during the switch to avoid abrupt cessation of one drug or the increased risk of adverse events such as cytochrome P450 interactions, serotonin syndrome, or hypertensive crisis (when switching to an MAO inhibitor).
Beware of interactions. All SSRIs and SNRIs are metabolized through the P450 system in the liver and therefore have the potential for drug-drug interactions. Care must be taken when giving these agents together with drugs whose metabolism can be altered by P450 inhibition. For TCAs, blood levels can be checked if there is concern about toxicity; however, dosing is not strictly based on this level. Great care should be taken if a TCA is given together with an SNRI or an SSRI, as the TCA blood level can become significantly elevated. This may result in QT interval prolongation, as mentioned earlier.
Refer. Referral to a psychiatrist is appropriate for patients for whom multiple classes have failed, for patients who have another psychiatric comorbidity (such as psychosis, hypomania, or mania), or for patients who may need hospitalization. Referral is also appropriate if the physician is concerned about suicide risk.
PATIENTS WITH MAJOR DEPRESSION ONLY
For a patient presenting with depression but no other significant medical comorbidity, the first-line therapy is often an SSRI. Several generic SSRIs are available, and some are on the discount lists at retail pharmacies.
Symptoms should start to improve in about 2 weeks, and the optimal response should be achieved in 4 to 6 weeks of treatment. If this does not occur, consider either adding an augmenting agent or switching to a different antidepressant.
PATIENTS WITH CHRONIC PAIN

Chronic pain and depression often go hand in hand and can potentiate each other. When considering an antidepressant in a patient who has both conditions, the SNRIs and TCAs are typically preferred. Some SNRIs, namely duloxetine and milnacipran, are approved for certain chronic pain conditions, such as fibromyalgia. SNRIs are frequently used off-label for other chronic pain conditions such as headache and neuropathic pain.2
TCAs such as amitriptyline, nortriptyline, and doxepin are also often used in patients with chronic pain. These agents, like the SNRIs, inhibit the reuptake of serotonin and norepinephrine and are used off-label for neuropathic pain,3,4 migraine, interstitial cystitis,5 and other pain conditions.6–9
For TCAs and SNRIs, the effective dose range for chronic pain overlaps that for depression. However, TCAs are often given at lower doses to patients without depression. We recommend starting at a low dose and slowly titrating upward to an effective dose. SNRIs are often preferred over TCAs because they do not have anticholinergic side effects and because an overdose is much less likely to be lethal.
PATIENTS WITH SEXUAL DYSFUNCTION
One of the more commonly reported side effects of antidepressants is sexual dysfunction, generally in the form of delayed orgasm or decreased libido.10 Typically, these complaints are attributed to SSRIs and SNRIs; however, TCAs and MAO inhibitors have also been associated wth sexual dysfunction.
Both erectile dysfunction and priapism have been linked to certain antidepressants. In particular, trazodone is a known cause of priapism. Even if using low doses for sleep, male patients should be made aware of this adverse effect.
Switching from one agent to another in the same class is not likely to improve sexual side effects. In particular, all the SSRIs are similar in their likelihood of causing sexual dysfunction. In a patient taking an SSRI who experiences this side effect, switching to bupropion11 or mirtazapine12 can be quite useful. Bupropion acts primarily on dopamine and norepinephrine, whereas mirtazapine acts on serotonin and norepinephrine but in a different manner from SSRIs and SNRIs.
Adjunctive treatment such as a cholinergic agonist, yohimbine (contraindicated with MAO inhibitors), a serotonergic agent (eg, buspirone), or a drug that acts on nitric oxide (eg, sildenafil, tadalafil) may have some utility but is often ineffective. Dose reduction, if possible, can be of value.
PATIENTS WITH ANXIETY
Many antidepressants are also approved for anxiety disorders, and still more are used off-label for this purpose. Anxiety and depression often occur together, so being able to treat both conditions with one drug can be quite useful.13 In general, the antidepressant effects are seen at lower doses of SSRIs and SNRIs, whereas more of the anxiolytic effects are seen at higher doses, particularly for obsessive-compulsive disorder.14
First-line treatment would be an SSRI or SNRI. Most anxiety disorders respond to either class, but there are some more-specific recommendations. SSRIs are best studied in panic disorder, generalized anxiety disorder, social anxiety disorder, posttraumatic stress disorder, and obsessive-compulsive disorder. Fluoxetine, citalopram, escitalopram, and sertraline15 can all be effective in both major depressive disorder and generalized anxiety disorder. Panic disorder also tends to respond well to SSRIs. SNRIs have been evaluated primarily in generalized anxiety disorder but may also be useful in many of the other conditions.
Additionally, mirtazapine (used off-label)12 and the TCAs16–18 can help treat anxiety. Clomipramine is used to treat obsessive-compulsive disorder.19 These drugs are especially useful for nighttime anxiety, as they can aid sleep. Of note, the anxiolytic effect of mirtazapine may be greater at higher doses.
MAO inhibitors often go unused because of the dietary and medication restrictions involved. However, very refractory cases of certain anxiety disorders may respond preferentially to these agents.
Bupropion tends to be more activating than other antidepressants, so is often avoided in anxious patients. However, some research suggests this is not always necessary.20 If the anxiety is secondary to depression, it will often improve significantly with this agent.
When starting or increasing the dose of an antidepressant, patients may experience increased anxiety or feel “jittery.” This feeling usually passes within the first week of treatment, and it is important to inform patients about this effect. “Start low and go slow” in patients with significant comorbid anxiety. Temporarily using a benzodiazepine such as clonazepam may make the transition more tolerable.
PATIENTS WITH CHRONIC FATIGUE SYNDROME OR FIBROMYALGIA
Increasing recognition of both chronic fatigue syndrome and fibromyalgia has led to more proactive treatment for these disorders. Depression can go hand in hand with these disorders, and certain antidepressants, namely the SNRIs, can be useful in this population.
More data exist for the treatment of fibromyalgia. Both duloxetine and milnacipran are approved by the US Food and Drug Administration (FDA) for the treatment of fibromyalgia.21 Venlafaxine is also used off-label for this purpose. SSRIs such as fluoxetine and citalopram have had mixed results.21–23 TCAs have been used with some success; however, their side effects and lethal potential are often limiting.21,24,25 A recent study in Spain also suggested there may be benefit from using MAO inhibitors for fibromyalgia, but data are quite limited.26
The data for treating chronic fatigue syndrome with SSRIs, SNRIs, or MAO inhibitors are conflicting.27–29 However, managing the co-existing depression may provide some relief in and of itself.
PATIENTS WITH FREQUENT INSOMNIA
Insomnia can be a symptom of depression, but it can also be a side effect of certain antidepressants. The SSRIs and SNRIs can disrupt sleep patterns in some patients by shortening the rapid-eye-movement (REM) stage.30,31
In patients with severe insomnia, it may be best to first recommend taking the antidepressant in the morning if they notice worsening sleep after initiating treatment. Patients can be told with any antidepressant, “If it makes you tired, take it at night, and if it wakes you up, take it in the morning.” Of note, a recent South African study suggested that escitalopram may be able to improve sleep.32
If that does not solve the problem, there are other options. For instance, mirtazapine, particularly in doses of 15 mg or 30 mg, aids depression and insomnia. At higher doses (45 mg), the sleep-aiding effect may be reduced. Low doses of TCAs, particularly doxepin, maprotiline (technically speaking, a tetracyclic antidepressant), amitriptyline, and nortriptyline can be effective sleep aids. These agents may be used as an adjunct to another antidepressant to enhance sleep and mood. However, the TCAs also shorten the REM stage of sleep.33
The previously mentioned drug interactions with SSRIs and SNRIs also need to be considered. Caution should be used when discontinuing these medications, as patients may experience rebound symptoms in the form of much more vivid dreams. MAO inhibitors may worsen insomnia because they suppress REM sleep.34
Trazodone is another agent that at lower doses (25–150 mg) can be an effective, nonaddicting sleep aid. When used as an antidepressant, it is generally prescribed at higher doses (300–400 mg), but its sedating effects can be quite limiting at these levels. It is important to remember the possibility of priapism in male patients.
GERIATRIC PATIENTS
Old age brings its own set of concerns when treating depression. Elderly patients are more susceptible to potential bradycardia caused by SSRIs. The TCAs have the more worrisome cardiac side effect of QTc prolongation. TCAs can slow cognitive function, whereas the SSRIs, bupropion, and the SNRIs tend not to affect cognition. Escitalopram and duloxetine have been suggested to be particularly effective in the elderly.35,36 A study from the Netherlands linked SSRIs with increased risk of falling in geriatric patients with dementia.37 Constipation, which could lead to ileus, is increased with TCAs and certain other agents (ie, paroxetine) in the geriatric population.
Mirtazapine is often very useful in elderly patients for many reasons: it treats both anxiety and depression, stimulates appetite and weight gain, can help with nausea, and is an effective sleep aid. Concerns about weight, appetite, and sleep are particularly common in the elderly, whereas younger patients can be less tolerant of drugs that make them gain weight and sleep more. Normal age-related changes to the sleep cycle contribute to decreased satisfaction with sleep as we age. In addition, depression often further impairs sleep. So, in the elderly, optimizing sleep is key. Research has also shown mirtazapine to be effective in patients with both Alzheimer dementia and depression.38
DIABETIC PATIENTS
One of the more worrisome side effects of psychiatric medications in diabetic patients is weight gain. Certain antidepressants have a greater propensity for weight gain and should likely be avoided as first-line treatments in this population.12 Typically, these agents include those that have more antihistamine action such as paroxetine and the TCAs. These agents also may lead to constipation, which could potentially worsen gastroparesis. Mirtazapine and the MAO inhibitors are also known to cause weight gain.
Bupropion and nefazodone are the most weight-neutral of all antidepressants. Nefazodone has fallen out of favor because of its potential to cause fulminant liver failure in rare cases. However, it remains a reasonable option for patients with comorbid anxiety and depression who have significant weight gain with other agents.
SSRIs and MAO inhibitors may improve or be neutral toward glucose metabolism, and some data suggest that SNRIs may impair this process.39
PATIENTS WITH CARDIAC CONDITIONS
Major depression often coexists with cardiac conditions. In particular, many patients develop depression after suffering a myocardial infarction, and increasingly they are being treated for it.40 Treatment in this situation is appropriate, since depression, if untreated, can increase the risk of recurrence of myocardial infarction.41
However, there are many concerns that accompany treating depression in cardiac patients. Therefore, a baseline electrocardiogram should be obtained before starting an antidepressant.
TCAs and tetracyclic agents have a tendency to prolong the QTc interval and potentiate ventricular arrhythmias,42 so it may be prudent to avoid these in patients at risk. These agents can also significantly increase the pulse rate. This tachycardia increases the risk of angina or myocardial infarction from the anticholinergic effects of these drugs.
In February 2013, the FDA issued a warning about possible arrhythmias with citalopram at doses greater than 40 mg in adult patients43; however, research has suggested citalopram is effective in treating depression in cardiac patients.44 Research has not shown an increase in efficacy at doses greater than 40 mg daily, so we recommend following the black-box warning.
TCAs and MAO inhibitors can also cause orthostatic hypotension. On the other hand, consuming large amounts of tyramine, in foods such as aged cheese, can precipitate a hypertensive crisis in patients taking MAO inhibitors.
Which antidepressants tend to be safer in cardiac patients? Sertraline has been shown to be safe in congestive heart failure and coronary artery disease,45–47 but the SSRIs are typically safe. Fluoxetine has shown efficacy in patients who have had a myocardial infarction.48 Mirtazapine has also been shown to be efficacious in cardiac patients.49 Nefazodone, mirtazapine, bupropion, SSRIs, and SNRIs have little or no tendency toward orthostatic hypotension.
- Khawam EA, Laurencic G, Malone DA. Side effects of antidepressants: an overview. Cleve Clin J Med 2006; 73:351–361.
- Ziegler D. Painful diabetic neuropathy: treatment and future aspects. Diabetes Metab Res Rev 2008; 24(suppl 1):S52–S57.
- Saarto T, Wiffen PJ. Antidepressants for neuropathic pain: a Cochrane review. J Neurol Neurosurg Psychiatry 2010; 81:1372–1373.
- Tanenberg RJ, Irving GA, Risser RC, et al. Duloxetine, pregabalin, and duloxetine plus gabapentin for diabetic peripheral neuropathic pain management in patients with inadequate pain response to gabapentin: an open-label, randomized, noninferiority comparison. Mayo Clin Proc 2011; 86:615–626.
- Hertle L, van Ophoven A. Long-term results of amitriptyline treatment for interstitial cystitis. Aktuelle Urol 2010; 41(suppl 1):S61–S65.
- Nguyen TM, Eslick GD. Systematic review: the treatment of noncardiac chest pain with antidepressants. Aliment Pharmacol Ther 2012; 35:493–500.
- Lee H, Kim JH, Min BH, et al. Efficacy of venlafaxine for symptomatic relief in young adult patients with functional chest pain: a randomized, double-blind, placebo-controlled, crossover trial. Am J Gastroenterol 2010; 105:1504–1512.
- Varia I, Logue E, O’Connor C, et al. Randomized trial of sertraline in patients with unexplained chest pain of noncardiac origin. Am Heart J 2000; 140:367–372.
- Doraiswamy PM, Varia I, Hellegers C, et al. A randomized controlled trial of paroxetine for noncardiac chest pain. Psychopharmacol Bull 2006; 39:15–24.
- Clayton AH. Understanding antidepressant mechanism of action and its effect on efficacy and safety. J Clin Psychiatry 2012; 73:e11.
- Gartlehner G, Hansen RA, Morgan LC, et al. Second-generation antidepressants in the pharmacologic treatment of adult depression: an update of the 2007 comparative effectiveness review (Internet). Rockville (MD): Agency for Healthcare Research and Quality (US); 2011 Dec. Comparative Effectiveness Reviews, No. 46. http://www.ncbi.nlm.nih.gov/books/NBK83442/. Accessed February 27, 2013.
- Watanabe N, Omori IM, Nakagawa A, et al. Mirtazapine versus other antidepressive agents for depression. Cochrane Database Syst Rev 2011; 12:CD006528.
- Hofmeijer-Sevink MK, Batelaan NM, van Megen HJ, et al. Clinical relevance of comorbidity in anxiety disorders: a report from the Netherlands Study of Depression and Anxiety (NESDA). J Affect Disord 2012; 137:106–112.
- Koen N, Stein DJ. Pharmacotherapy of anxiety disorders: a critical review. Dialogues Clin Neurosci 2011; 13:423–437.
- Sheehan DV, Kamijima K. An evidence-based review of the clinical use of sertraline in mood and anxiety disorders. Int Clin Psychopharmacol 2009; 24:43–60.
- Huh J, Goebert D, Takeshita J, Lu BY, Kang M. Treatment of generalized anxiety disorder: a comprehensive review of the literature for psychopharmacologic alternatives to newer antidepressants and benzodiazepines. Prim Care Companion CNS Disord 2011; 13: 4088/PCC.08r00709blu.
- Rickels K, Downing R, Schweizer E, Hassman H. Antidepressants for the treatment of generalized anxiety disorder. A placebo-controlled comparison of imipramine, trazodone, and diazepam. Arch Gen Psychiatry 1993; 50:884–895.
- Uher R, Maier W, Hauser J, et al. Differential efficacy of escitalopram and nortriptyline on dimensional measures of depression. Br J Psychiatry 2009; 194:252–259.
- Kellner M. Drug treatment of obsessive-compulsive disorder. Dialogues Clin Neurosci 2010; 12:187–197.
- Rush AJ, Trivedi MH, Carmody TJ, et al. Response in relation to baseline anxiety levels in major depressive disorder treated with bupropion sustained release or sertraline. Neuropsychopharmacology 2001; 25:131–138.
- Mease PJ, Dundon K, Sarzi-Puttini P. Pharmacotherapy of fibromyalgia. Best Pract Res Clin Rheumatol 2011; 25:285–297.
- Wolfe F, Cathey MA, Hawley DJ. A double-blind placebo controlled trial of fluoxetine in fibromyalgia. Scand J Rheumatol 1994; 23:255–259.
- Arnold LM, Hess EV, Hudson JI, Welge JA, Berno SE, Keck PE. A randomized, placebo-controlled, double-blind, flexible-dose study of fluoxetine in the treatment of women with fibromyalgia. Am J Med 2002; 112:191–197.
- Arnold LM, Keck PE, Welge JA. Antidepressant treatment of fibromyalgia. A meta-analysis and review. Psychosomatics 2000; 41:104–113.
- Goldenberg DL, Burckhardt C, Crofford L. Management of fibromyalgia syndrome. JAMA 2004; 292:2388–2395.
- Tort S, Urrútia G, Nishishinya MB, Walitt B. Monoamine oxidase inhibitors (MAOIs) for fibromyalgia syndrome. Cochrane Database Syst Rev 2012; 4:CD009807.
- Vercoulen JH, Swanink CM, Zitman FG, et al. Randomised, double-blind, placebo-controlled study of fluoxetine in chronic fatigue syndrome. Lancet 1996; 347:858–861.
- Natelson BH, Cheu J, Pareja J, Ellis SP, Policastro T, Findley TW. Randomized, double blind, controlled placebo-phase in trial of low dose phenelzine in the chronic fatigue syndrome. Psychopharmacology (Berl) 1996; 124:226–230.
- Reid S, Chalder T, Cleare A, Hotopf M, Wessely S. Chronic fatigue syndrome. BMJ 2000; 320:292–296.
- Kupfer DJ, Spiker DG, Coble PA, Neil JF, Ulrich R, Shaw DH. Sleep and treatment prediction in endogenous depression. Am J Psychiatry 1981; 138:429–434.
- Argyropoulos SV, Hicks JA, Nash JR, et al. Redistribution of slow wave activity of sleep during pharmacological treatment of depression with paroxetine but not with nefazodone. J Sleep Res 2009; 18:342–348.
- Stein DJ, Lopez AG. Effects of escitalopram on sleep problems in patients with major depression or generalized anxiety disorder. Adv Ther 2011; 28:1021–1037.
- Ehlers CL, Havstad JW, Kupfer DJ. Estimation of the time course of slow-wave sleep over the night in depressed patients: effects of clomipramine and clinical response. Biol Psychiatry 1996; 39:171–181.
- Landolt HP, Raimo EB, Schnierow BJ, Kelsoe JR, Rapaport MH, Gillin JC. Sleep and sleep electroencephalogram in depressed patients treated with phenelzine. Arch Gen Psychiatry 2001; 58:268–276.
- Chen YM, Huang XM, Thompson R, Zhao YB. Clinical features and efficacy of escitalopram treatment for geriatric depression. J Int Med Res 2011; 39:1946–1953.
- Dolder C, Nelson M, Stump A. Pharmacological and clinical profile of newer antidepressants: implications for the treatment of elderly patients. Drugs Aging 2010; 27:625–640.
- Sterke CS, Ziere G, van Beeck EF, Looman CW, van der Cammen TJ. Dose-response relationship between selective serotonin re-uptake inhibitors and injurious falls: a study in nursing home residents with dementia. Br J Clin Pharmacol 2012; 73:812–820.
- Raji MA, Brady SR. Mirtazapine for treatment of depression and comorbidities in Alzheimer disease. Ann Pharmacother 2001; 35:1024–1027.
- Hennings JM, Schaaf L, Fulda S. Glucose metabolism and antidepressant medication. Curr Pharm Des 2012; 18:5900–5919.
- Czarny MJ, Arthurs E, Coffie DF, et al. Prevalence of antidepressant prescription or use in patients with acute coronary syndrome: a systematic review. PLoS One 2011; 6:e27671.
- Zuidersma M, Ormel J, Conradi HJ, de Jonge P. An increase in depressive symptoms after myocardial infarction predicts new cardiac events irrespective of depressive symptoms before myocardial infarction. Psychol Med 2012; 42:683–693.
- van Noord C, Straus SM, Sturkenboom MC, et al. Psychotropic drugs associated with corrected QT interval prolongation. J Clin Psychopharmacol 2009; 29:9–15.
- US Food and Drug Administration (FDA). FDA Drug Safety Communication: abnormal heart rhythms associated with high doses of Celexa (citalopram hydrobromide). http://www.fda.gov/Drugs/DrugSafety/ucm269086.htm. Accessed August 25, 2013.
- Lespérance F, Frasure-Smith N, Koszycki D, et al; CREATE Investigators. Effects of citalopram and interpersonal psychotherapy on depression in patients with coronary artery disease: the Canadian Cardiac Randomized Evaluation of Antidepressant and Psychotherapy Efficacy (CREATE) trial. JAMA 2007; 297:367–379.
- O’Connor CM, Jiang W, Kuchibhatla M, et al; SADHART-CHF Investigators. Safety and efficacy of sertraline for depression in patients with heart failure: results of the SADHART-CHF (Sertraline Against Depression and Heart Disease in Chronic Heart Failure) trial. J Am Coll Cardiol 2010; 56:692–699.
- Glassman AH, O’Connor CM, Califf RM, et al; Sertraline Antidepressant Heart Attack Randomized Trial (SADHEART) Group. Sertraline treatment of major depression in patients with acute MI or unstable angina. JAMA 2002; 288:701–709.
- Swenson JR, O’Connor CM, Barton D, et al; Sertraline Antidepressant Heart Attack Randomized Trial (SADHART) Group. Influence of depression and effect of treatment with sertraline on quality of life after hospitalization for acute coronary syndrome. Am J Cardiol 2003; 92:1271–1276.
- Strik JJ, Honig A, Lousberg R, et al. Efficacy and safety of fluoxetine in the treatment of patients with major depression after first myocardial infarction: findings from a double-blind, placebo-controlled trial. Psychosom Med 2000; 62:783–789.
- Honig A, Kuyper AM, Schene AH, et al; MIND-IT investigators. Treatment of post-myocardial infarction depressive disorder: a randomized, placebo-controlled trial with mirtazapine. Psychosom Med 2007; 69:606–613.
With the variety of drugs available for treating depression, choosing one can be daunting. Different agents have characteristics that may make them a better choice for different types of patients, but even so, treating any kind of mental illness often requires an element of trial and error.
Primary care providers are on the frontline of treating mental illness, often evaluating patients before they are seen by a psychiatrist. The purpose of this article is to provide insight into the art of prescribing antidepressants in the primary care setting. We will discuss common patient presentations, including depressed patients without other medical comorbidities as well as those with common comorbidities, with our recommendations for first-line treatment.
We hope our recommendations will help you to navigate the uncertainty more confidently, resulting in more efficient and tailored treatment for your patients.
BASELINE TESTING
When starting a patient on antidepressant drug therapy, we recommend obtaining a set of baseline laboratory tests to rule out underlying medical conditions that may be contributing to the patient’s depression or that may preclude the use of a given drug. (For example, elevation of liver enzymes may preclude the use of duloxetine.) Tests should include:
- A complete blood cell count
- A complete metabolic panel
- A thyroid-stimulating hormone level.
Electrocardiography may also be useful, as some antidepressants can prolong the QT interval or elevate the blood levels of other drugs with this effect.
GENERAL TREATMENT CONSIDERATIONS
There are several classes of antidepressants, and each class has a number of agents. Research has found little difference in efficacy among agents. So to simplify choosing which one to use, we recommend becoming comfortable with an agent from each class, ie:
- A selective serotonin reuptake inhibitor (SSRI)
- A selective serotonin-norepinephrine reuptake inhibitor (SNRI)
- A tricyclic antidepressant (TCA)
- A monoamine oxidase (MAO) inhibitor.
Each class includes generic agents, many of which are on the discount lists of retail pharmacies. Table 1 shows representative drugs from each class, with their relative costs.
Start low and go slow. In general, when starting an antidepressant, consider starting at half the normal dose, titrating upward as tolerated about every 14 days. This approach can minimize side effects. For example, if prescribing fluoxetine, start with 10 mg and titrate every 2 weeks based on tolerance and patient response. That said, each patient may respond differently, requiring perhaps a lower starting dose or a longer titration schedule.
Anticipate side effects. Most of the side effects of an antidepressant drug can be explained by its mechanism of action. Although side effects should certainly be considered when choosing an agent, patients can be reassured that most are transient and benign. A detailed discussion of side effects of antidepressant drugs is beyond the scope of this article, but a review by Khawam et al1 was published earlier in this journal.
Reassess. If after 4 to 6 weeks the patient has had little or no response, it is reasonable to switch agents. For a patient who was on an SSRI, the change can be to another SSRI or to an SNRI. However, if two SSRIs have already failed, then choose an SNRI. Agents are commonly cross-tapered during the switch to avoid abrupt cessation of one drug or the increased risk of adverse events such as cytochrome P450 interactions, serotonin syndrome, or hypertensive crisis (when switching to an MAO inhibitor).
Beware of interactions. All SSRIs and SNRIs are metabolized through the P450 system in the liver and therefore have the potential for drug-drug interactions. Care must be taken when giving these agents together with drugs whose metabolism can be altered by P450 inhibition. For TCAs, blood levels can be checked if there is concern about toxicity; however, dosing is not strictly based on this level. Great care should be taken if a TCA is given together with an SNRI or an SSRI, as the TCA blood level can become significantly elevated. This may result in QT interval prolongation, as mentioned earlier.
Refer. Referral to a psychiatrist is appropriate for patients for whom multiple classes have failed, for patients who have another psychiatric comorbidity (such as psychosis, hypomania, or mania), or for patients who may need hospitalization. Referral is also appropriate if the physician is concerned about suicide risk.
PATIENTS WITH MAJOR DEPRESSION ONLY
For a patient presenting with depression but no other significant medical comorbidity, the first-line therapy is often an SSRI. Several generic SSRIs are available, and some are on the discount lists at retail pharmacies.
Symptoms should start to improve in about 2 weeks, and the optimal response should be achieved in 4 to 6 weeks of treatment. If this does not occur, consider either adding an augmenting agent or switching to a different antidepressant.
PATIENTS WITH CHRONIC PAIN
Chronic pain and depression often go hand in hand and can potentiate each other. When considering an antidepressant in a patient who has both conditions, the SNRIs and TCAs are typically preferred. Some SNRIs, namely duloxetine and milnacipran, are approved for certain chronic pain conditions, such as fibromyalgia. SNRIs are frequently used off-label for other chronic pain conditions such as headache and neuropathic pain.2
TCAs such as amitriptyline, nortriptyline, and doxepin are also often used in patients with chronic pain. These agents, like the SNRIs, inhibit the reuptake of serotonin and norepinephrine and are used off-label for neuropathic pain,3,4 migraine, interstitial cystitis,5 and other pain conditions.6–9
For TCAs and SNRIs, the effective dose range for chronic pain overlaps that for depression. However, TCAs are often given at lower doses to patients without depression. We recommend starting at a low dose and slowly titrating upward to an effective dose. SNRIs are often preferred over TCAs because they do not have anticholinergic side effects and because an overdose is much less likely to be lethal.
PATIENTS WITH SEXUAL DYSFUNCTION
One of the more commonly reported side effects of antidepressants is sexual dysfunction, generally in the form of delayed orgasm or decreased libido.10 Typically, these complaints are attributed to SSRIs and SNRIs; however, TCAs and MAO inhibitors have also been associated wth sexual dysfunction.
Both erectile dysfunction and priapism have been linked to certain antidepressants. In particular, trazodone is a known cause of priapism. Even if using low doses for sleep, male patients should be made aware of this adverse effect.
Switching from one agent to another in the same class is not likely to improve sexual side effects. In particular, all the SSRIs are similar in their likelihood of causing sexual dysfunction. In a patient taking an SSRI who experiences this side effect, switching to bupropion11 or mirtazapine12 can be quite useful. Bupropion acts primarily on dopamine and norepinephrine, whereas mirtazapine acts on serotonin and norepinephrine but in a different manner from SSRIs and SNRIs.
Adjunctive treatment such as a cholinergic agonist, yohimbine (contraindicated with MAO inhibitors), a serotonergic agent (eg, buspirone), or a drug that acts on nitric oxide (eg, sildenafil, tadalafil) may have some utility but is often ineffective. Dose reduction, if possible, can be of value.
PATIENTS WITH ANXIETY
Many antidepressants are also approved for anxiety disorders, and still more are used off-label for this purpose. Anxiety and depression often occur together, so being able to treat both conditions with one drug can be quite useful.13 In general, the antidepressant effects are seen at lower doses of SSRIs and SNRIs, whereas more of the anxiolytic effects are seen at higher doses, particularly for obsessive-compulsive disorder.14
First-line treatment would be an SSRI or SNRI. Most anxiety disorders respond to either class, but there are some more-specific recommendations. SSRIs are best studied in panic disorder, generalized anxiety disorder, social anxiety disorder, posttraumatic stress disorder, and obsessive-compulsive disorder. Fluoxetine, citalopram, escitalopram, and sertraline15 can all be effective in both major depressive disorder and generalized anxiety disorder. Panic disorder also tends to respond well to SSRIs. SNRIs have been evaluated primarily in generalized anxiety disorder but may also be useful in many of the other conditions.
Additionally, mirtazapine (used off-label)12 and the TCAs16–18 can help treat anxiety. Clomipramine is used to treat obsessive-compulsive disorder.19 These drugs are especially useful for nighttime anxiety, as they can aid sleep. Of note, the anxiolytic effect of mirtazapine may be greater at higher doses.
MAO inhibitors often go unused because of the dietary and medication restrictions involved. However, very refractory cases of certain anxiety disorders may respond preferentially to these agents.
Bupropion tends to be more activating than other antidepressants, so is often avoided in anxious patients. However, some research suggests this is not always necessary.20 If the anxiety is secondary to depression, it will often improve significantly with this agent.
When starting or increasing the dose of an antidepressant, patients may experience increased anxiety or feel “jittery.” This feeling usually passes within the first week of treatment, and it is important to inform patients about this effect. “Start low and go slow” in patients with significant comorbid anxiety. Temporarily using a benzodiazepine such as clonazepam may make the transition more tolerable.
PATIENTS WITH CHRONIC FATIGUE SYNDROME OR FIBROMYALGIA
Increasing recognition of both chronic fatigue syndrome and fibromyalgia has led to more proactive treatment for these disorders. Depression can go hand in hand with these disorders, and certain antidepressants, namely the SNRIs, can be useful in this population.
More data exist for the treatment of fibromyalgia. Both duloxetine and milnacipran are approved by the US Food and Drug Administration (FDA) for the treatment of fibromyalgia.21 Venlafaxine is also used off-label for this purpose. SSRIs such as fluoxetine and citalopram have had mixed results.21–23 TCAs have been used with some success; however, their side effects and lethal potential are often limiting.21,24,25 A recent study in Spain also suggested there may be benefit from using MAO inhibitors for fibromyalgia, but data are quite limited.26
The data for treating chronic fatigue syndrome with SSRIs, SNRIs, or MAO inhibitors are conflicting.27–29 However, managing the co-existing depression may provide some relief in and of itself.
PATIENTS WITH FREQUENT INSOMNIA
Insomnia can be a symptom of depression, but it can also be a side effect of certain antidepressants. The SSRIs and SNRIs can disrupt sleep patterns in some patients by shortening the rapid-eye-movement (REM) stage.30,31
In patients with severe insomnia, it may be best to first recommend taking the antidepressant in the morning if they notice worsening sleep after initiating treatment. Patients can be told with any antidepressant, “If it makes you tired, take it at night, and if it wakes you up, take it in the morning.” Of note, a recent South African study suggested that escitalopram may be able to improve sleep.32
If that does not solve the problem, there are other options. For instance, mirtazapine, particularly in doses of 15 mg or 30 mg, aids depression and insomnia. At higher doses (45 mg), the sleep-aiding effect may be reduced. Low doses of TCAs, particularly doxepin, maprotiline (technically speaking, a tetracyclic antidepressant), amitriptyline, and nortriptyline can be effective sleep aids. These agents may be used as an adjunct to another antidepressant to enhance sleep and mood. However, the TCAs also shorten the REM stage of sleep.33
The previously mentioned drug interactions with SSRIs and SNRIs also need to be considered. Caution should be used when discontinuing these medications, as patients may experience rebound symptoms in the form of much more vivid dreams. MAO inhibitors may worsen insomnia because they suppress REM sleep.34
Trazodone is another agent that at lower doses (25–150 mg) can be an effective, nonaddicting sleep aid. When used as an antidepressant, it is generally prescribed at higher doses (300–400 mg), but its sedating effects can be quite limiting at these levels. It is important to remember the possibility of priapism in male patients.
GERIATRIC PATIENTS
Old age brings its own set of concerns when treating depression. Elderly patients are more susceptible to potential bradycardia caused by SSRIs. The TCAs have the more worrisome cardiac side effect of QTc prolongation. TCAs can slow cognitive function, whereas the SSRIs, bupropion, and the SNRIs tend not to affect cognition. Escitalopram and duloxetine have been suggested to be particularly effective in the elderly.35,36 A study from the Netherlands linked SSRIs with increased risk of falling in geriatric patients with dementia.37 Constipation, which could lead to ileus, is increased with TCAs and certain other agents (ie, paroxetine) in the geriatric population.
Mirtazapine is often very useful in elderly patients for many reasons: it treats both anxiety and depression, stimulates appetite and weight gain, can help with nausea, and is an effective sleep aid. Concerns about weight, appetite, and sleep are particularly common in the elderly, whereas younger patients can be less tolerant of drugs that make them gain weight and sleep more. Normal age-related changes to the sleep cycle contribute to decreased satisfaction with sleep as we age. In addition, depression often further impairs sleep. So, in the elderly, optimizing sleep is key. Research has also shown mirtazapine to be effective in patients with both Alzheimer dementia and depression.38
DIABETIC PATIENTS
One of the more worrisome side effects of psychiatric medications in diabetic patients is weight gain. Certain antidepressants have a greater propensity for weight gain and should likely be avoided as first-line treatments in this population.12 Typically, these agents include those that have more antihistamine action such as paroxetine and the TCAs. These agents also may lead to constipation, which could potentially worsen gastroparesis. Mirtazapine and the MAO inhibitors are also known to cause weight gain.
Bupropion and nefazodone are the most weight-neutral of all antidepressants. Nefazodone has fallen out of favor because of its potential to cause fulminant liver failure in rare cases. However, it remains a reasonable option for patients with comorbid anxiety and depression who have significant weight gain with other agents.
SSRIs and MAO inhibitors may improve or be neutral toward glucose metabolism, and some data suggest that SNRIs may impair this process.39
PATIENTS WITH CARDIAC CONDITIONS
Major depression often coexists with cardiac conditions. In particular, many patients develop depression after suffering a myocardial infarction, and increasingly they are being treated for it.40 Treatment in this situation is appropriate, since depression, if untreated, can increase the risk of recurrence of myocardial infarction.41
However, there are many concerns that accompany treating depression in cardiac patients. Therefore, a baseline electrocardiogram should be obtained before starting an antidepressant.
TCAs and tetracyclic agents have a tendency to prolong the QTc interval and potentiate ventricular arrhythmias,42 so it may be prudent to avoid these in patients at risk. These agents can also significantly increase the pulse rate. This tachycardia increases the risk of angina or myocardial infarction from the anticholinergic effects of these drugs.
In February 2013, the FDA issued a warning about possible arrhythmias with citalopram at doses greater than 40 mg in adult patients43; however, research has suggested citalopram is effective in treating depression in cardiac patients.44 Research has not shown an increase in efficacy at doses greater than 40 mg daily, so we recommend following the black-box warning.
TCAs and MAO inhibitors can also cause orthostatic hypotension. On the other hand, consuming large amounts of tyramine, in foods such as aged cheese, can precipitate a hypertensive crisis in patients taking MAO inhibitors.
Which antidepressants tend to be safer in cardiac patients? Sertraline has been shown to be safe in congestive heart failure and coronary artery disease,45–47 but the SSRIs are typically safe. Fluoxetine has shown efficacy in patients who have had a myocardial infarction.48 Mirtazapine has also been shown to be efficacious in cardiac patients.49 Nefazodone, mirtazapine, bupropion, SSRIs, and SNRIs have little or no tendency toward orthostatic hypotension.
With the variety of drugs available for treating depression, choosing one can be daunting. Different agents have characteristics that may make them a better choice for different types of patients, but even so, treating any kind of mental illness often requires an element of trial and error.
Primary care providers are on the frontline of treating mental illness, often evaluating patients before they are seen by a psychiatrist. The purpose of this article is to provide insight into the art of prescribing antidepressants in the primary care setting. We will discuss common patient presentations, including depressed patients without other medical comorbidities as well as those with common comorbidities, with our recommendations for first-line treatment.
We hope our recommendations will help you to navigate the uncertainty more confidently, resulting in more efficient and tailored treatment for your patients.
BASELINE TESTING
When starting a patient on antidepressant drug therapy, we recommend obtaining a set of baseline laboratory tests to rule out underlying medical conditions that may be contributing to the patient’s depression or that may preclude the use of a given drug. (For example, elevation of liver enzymes may preclude the use of duloxetine.) Tests should include:
- A complete blood cell count
- A complete metabolic panel
- A thyroid-stimulating hormone level.
Electrocardiography may also be useful, as some antidepressants can prolong the QT interval or elevate the blood levels of other drugs with this effect.
GENERAL TREATMENT CONSIDERATIONS
There are several classes of antidepressants, and each class has a number of agents. Research has found little difference in efficacy among agents. So to simplify choosing which one to use, we recommend becoming comfortable with an agent from each class, ie:
- A selective serotonin reuptake inhibitor (SSRI)
- A selective serotonin-norepinephrine reuptake inhibitor (SNRI)
- A tricyclic antidepressant (TCA)
- A monoamine oxidase (MAO) inhibitor.
Each class includes generic agents, many of which are on the discount lists of retail pharmacies. Table 1 shows representative drugs from each class, with their relative costs.
Start low and go slow. In general, when starting an antidepressant, consider starting at half the normal dose, titrating upward as tolerated about every 14 days. This approach can minimize side effects. For example, if prescribing fluoxetine, start with 10 mg and titrate every 2 weeks based on tolerance and patient response. That said, each patient may respond differently, requiring perhaps a lower starting dose or a longer titration schedule.
Anticipate side effects. Most of the side effects of an antidepressant drug can be explained by its mechanism of action. Although side effects should certainly be considered when choosing an agent, patients can be reassured that most are transient and benign. A detailed discussion of side effects of antidepressant drugs is beyond the scope of this article, but a review by Khawam et al1 was published earlier in this journal.
Reassess. If after 4 to 6 weeks the patient has had little or no response, it is reasonable to switch agents. For a patient who was on an SSRI, the change can be to another SSRI or to an SNRI. However, if two SSRIs have already failed, then choose an SNRI. Agents are commonly cross-tapered during the switch to avoid abrupt cessation of one drug or the increased risk of adverse events such as cytochrome P450 interactions, serotonin syndrome, or hypertensive crisis (when switching to an MAO inhibitor).
Beware of interactions. All SSRIs and SNRIs are metabolized through the P450 system in the liver and therefore have the potential for drug-drug interactions. Care must be taken when giving these agents together with drugs whose metabolism can be altered by P450 inhibition. For TCAs, blood levels can be checked if there is concern about toxicity; however, dosing is not strictly based on this level. Great care should be taken if a TCA is given together with an SNRI or an SSRI, as the TCA blood level can become significantly elevated. This may result in QT interval prolongation, as mentioned earlier.
Refer. Referral to a psychiatrist is appropriate for patients for whom multiple classes have failed, for patients who have another psychiatric comorbidity (such as psychosis, hypomania, or mania), or for patients who may need hospitalization. Referral is also appropriate if the physician is concerned about suicide risk.
PATIENTS WITH MAJOR DEPRESSION ONLY
For a patient presenting with depression but no other significant medical comorbidity, the first-line therapy is often an SSRI. Several generic SSRIs are available, and some are on the discount lists at retail pharmacies.
Symptoms should start to improve in about 2 weeks, and the optimal response should be achieved in 4 to 6 weeks of treatment. If this does not occur, consider either adding an augmenting agent or switching to a different antidepressant.
PATIENTS WITH CHRONIC PAIN
Chronic pain and depression often go hand in hand and can potentiate each other. When considering an antidepressant in a patient who has both conditions, the SNRIs and TCAs are typically preferred. Some SNRIs, namely duloxetine and milnacipran, are approved for certain chronic pain conditions, such as fibromyalgia. SNRIs are frequently used off-label for other chronic pain conditions such as headache and neuropathic pain.2
TCAs such as amitriptyline, nortriptyline, and doxepin are also often used in patients with chronic pain. These agents, like the SNRIs, inhibit the reuptake of serotonin and norepinephrine and are used off-label for neuropathic pain,3,4 migraine, interstitial cystitis,5 and other pain conditions.6–9
For TCAs and SNRIs, the effective dose range for chronic pain overlaps that for depression. However, TCAs are often given at lower doses to patients without depression. We recommend starting at a low dose and slowly titrating upward to an effective dose. SNRIs are often preferred over TCAs because they do not have anticholinergic side effects and because an overdose is much less likely to be lethal.
PATIENTS WITH SEXUAL DYSFUNCTION
One of the more commonly reported side effects of antidepressants is sexual dysfunction, generally in the form of delayed orgasm or decreased libido.10 Typically, these complaints are attributed to SSRIs and SNRIs; however, TCAs and MAO inhibitors have also been associated wth sexual dysfunction.
Both erectile dysfunction and priapism have been linked to certain antidepressants. In particular, trazodone is a known cause of priapism. Even if using low doses for sleep, male patients should be made aware of this adverse effect.
Switching from one agent to another in the same class is not likely to improve sexual side effects. In particular, all the SSRIs are similar in their likelihood of causing sexual dysfunction. In a patient taking an SSRI who experiences this side effect, switching to bupropion11 or mirtazapine12 can be quite useful. Bupropion acts primarily on dopamine and norepinephrine, whereas mirtazapine acts on serotonin and norepinephrine but in a different manner from SSRIs and SNRIs.
Adjunctive treatment such as a cholinergic agonist, yohimbine (contraindicated with MAO inhibitors), a serotonergic agent (eg, buspirone), or a drug that acts on nitric oxide (eg, sildenafil, tadalafil) may have some utility but is often ineffective. Dose reduction, if possible, can be of value.
PATIENTS WITH ANXIETY
Many antidepressants are also approved for anxiety disorders, and still more are used off-label for this purpose. Anxiety and depression often occur together, so being able to treat both conditions with one drug can be quite useful.13 In general, the antidepressant effects are seen at lower doses of SSRIs and SNRIs, whereas more of the anxiolytic effects are seen at higher doses, particularly for obsessive-compulsive disorder.14
First-line treatment would be an SSRI or SNRI. Most anxiety disorders respond to either class, but there are some more-specific recommendations. SSRIs are best studied in panic disorder, generalized anxiety disorder, social anxiety disorder, posttraumatic stress disorder, and obsessive-compulsive disorder. Fluoxetine, citalopram, escitalopram, and sertraline15 can all be effective in both major depressive disorder and generalized anxiety disorder. Panic disorder also tends to respond well to SSRIs. SNRIs have been evaluated primarily in generalized anxiety disorder but may also be useful in many of the other conditions.
Additionally, mirtazapine (used off-label)12 and the TCAs16–18 can help treat anxiety. Clomipramine is used to treat obsessive-compulsive disorder.19 These drugs are especially useful for nighttime anxiety, as they can aid sleep. Of note, the anxiolytic effect of mirtazapine may be greater at higher doses.
MAO inhibitors often go unused because of the dietary and medication restrictions involved. However, very refractory cases of certain anxiety disorders may respond preferentially to these agents.
Bupropion tends to be more activating than other antidepressants, so is often avoided in anxious patients. However, some research suggests this is not always necessary.20 If the anxiety is secondary to depression, it will often improve significantly with this agent.
When starting or increasing the dose of an antidepressant, patients may experience increased anxiety or feel “jittery.” This feeling usually passes within the first week of treatment, and it is important to inform patients about this effect. “Start low and go slow” in patients with significant comorbid anxiety. Temporarily using a benzodiazepine such as clonazepam may make the transition more tolerable.
PATIENTS WITH CHRONIC FATIGUE SYNDROME OR FIBROMYALGIA
Increasing recognition of both chronic fatigue syndrome and fibromyalgia has led to more proactive treatment for these disorders. Depression can go hand in hand with these disorders, and certain antidepressants, namely the SNRIs, can be useful in this population.
More data exist for the treatment of fibromyalgia. Both duloxetine and milnacipran are approved by the US Food and Drug Administration (FDA) for the treatment of fibromyalgia.21 Venlafaxine is also used off-label for this purpose. SSRIs such as fluoxetine and citalopram have had mixed results.21–23 TCAs have been used with some success; however, their side effects and lethal potential are often limiting.21,24,25 A recent study in Spain also suggested there may be benefit from using MAO inhibitors for fibromyalgia, but data are quite limited.26
The data for treating chronic fatigue syndrome with SSRIs, SNRIs, or MAO inhibitors are conflicting.27–29 However, managing the co-existing depression may provide some relief in and of itself.
PATIENTS WITH FREQUENT INSOMNIA
Insomnia can be a symptom of depression, but it can also be a side effect of certain antidepressants. The SSRIs and SNRIs can disrupt sleep patterns in some patients by shortening the rapid-eye-movement (REM) stage.30,31
In patients with severe insomnia, it may be best to first recommend taking the antidepressant in the morning if they notice worsening sleep after initiating treatment. Patients can be told with any antidepressant, “If it makes you tired, take it at night, and if it wakes you up, take it in the morning.” Of note, a recent South African study suggested that escitalopram may be able to improve sleep.32
If that does not solve the problem, there are other options. For instance, mirtazapine, particularly in doses of 15 mg or 30 mg, aids depression and insomnia. At higher doses (45 mg), the sleep-aiding effect may be reduced. Low doses of TCAs, particularly doxepin, maprotiline (technically speaking, a tetracyclic antidepressant), amitriptyline, and nortriptyline can be effective sleep aids. These agents may be used as an adjunct to another antidepressant to enhance sleep and mood. However, the TCAs also shorten the REM stage of sleep.33
The previously mentioned drug interactions with SSRIs and SNRIs also need to be considered. Caution should be used when discontinuing these medications, as patients may experience rebound symptoms in the form of much more vivid dreams. MAO inhibitors may worsen insomnia because they suppress REM sleep.34
Trazodone is another agent that at lower doses (25–150 mg) can be an effective, nonaddicting sleep aid. When used as an antidepressant, it is generally prescribed at higher doses (300–400 mg), but its sedating effects can be quite limiting at these levels. It is important to remember the possibility of priapism in male patients.
GERIATRIC PATIENTS
Old age brings its own set of concerns when treating depression. Elderly patients are more susceptible to potential bradycardia caused by SSRIs. The TCAs have the more worrisome cardiac side effect of QTc prolongation. TCAs can slow cognitive function, whereas the SSRIs, bupropion, and the SNRIs tend not to affect cognition. Escitalopram and duloxetine have been suggested to be particularly effective in the elderly.35,36 A study from the Netherlands linked SSRIs with increased risk of falling in geriatric patients with dementia.37 Constipation, which could lead to ileus, is increased with TCAs and certain other agents (ie, paroxetine) in the geriatric population.
Mirtazapine is often very useful in elderly patients for many reasons: it treats both anxiety and depression, stimulates appetite and weight gain, can help with nausea, and is an effective sleep aid. Concerns about weight, appetite, and sleep are particularly common in the elderly, whereas younger patients can be less tolerant of drugs that make them gain weight and sleep more. Normal age-related changes to the sleep cycle contribute to decreased satisfaction with sleep as we age. In addition, depression often further impairs sleep. So, in the elderly, optimizing sleep is key. Research has also shown mirtazapine to be effective in patients with both Alzheimer dementia and depression.38
DIABETIC PATIENTS
One of the more worrisome side effects of psychiatric medications in diabetic patients is weight gain. Certain antidepressants have a greater propensity for weight gain and should likely be avoided as first-line treatments in this population.12 Typically, these agents include those that have more antihistamine action such as paroxetine and the TCAs. These agents also may lead to constipation, which could potentially worsen gastroparesis. Mirtazapine and the MAO inhibitors are also known to cause weight gain.
Bupropion and nefazodone are the most weight-neutral of all antidepressants. Nefazodone has fallen out of favor because of its potential to cause fulminant liver failure in rare cases. However, it remains a reasonable option for patients with comorbid anxiety and depression who have significant weight gain with other agents.
SSRIs and MAO inhibitors may improve or be neutral toward glucose metabolism, and some data suggest that SNRIs may impair this process.39
PATIENTS WITH CARDIAC CONDITIONS
Major depression often coexists with cardiac conditions. In particular, many patients develop depression after suffering a myocardial infarction, and increasingly they are being treated for it.40 Treatment in this situation is appropriate, since depression, if untreated, can increase the risk of recurrence of myocardial infarction.41
However, there are many concerns that accompany treating depression in cardiac patients. Therefore, a baseline electrocardiogram should be obtained before starting an antidepressant.
TCAs and tetracyclic agents have a tendency to prolong the QTc interval and potentiate ventricular arrhythmias,42 so it may be prudent to avoid these in patients at risk. These agents can also significantly increase the pulse rate. This tachycardia increases the risk of angina or myocardial infarction from the anticholinergic effects of these drugs.
In February 2013, the FDA issued a warning about possible arrhythmias with citalopram at doses greater than 40 mg in adult patients43; however, research has suggested citalopram is effective in treating depression in cardiac patients.44 Research has not shown an increase in efficacy at doses greater than 40 mg daily, so we recommend following the black-box warning.
TCAs and MAO inhibitors can also cause orthostatic hypotension. On the other hand, consuming large amounts of tyramine, in foods such as aged cheese, can precipitate a hypertensive crisis in patients taking MAO inhibitors.
Which antidepressants tend to be safer in cardiac patients? Sertraline has been shown to be safe in congestive heart failure and coronary artery disease,45–47 but the SSRIs are typically safe. Fluoxetine has shown efficacy in patients who have had a myocardial infarction.48 Mirtazapine has also been shown to be efficacious in cardiac patients.49 Nefazodone, mirtazapine, bupropion, SSRIs, and SNRIs have little or no tendency toward orthostatic hypotension.
- Khawam EA, Laurencic G, Malone DA. Side effects of antidepressants: an overview. Cleve Clin J Med 2006; 73:351–361.
- Ziegler D. Painful diabetic neuropathy: treatment and future aspects. Diabetes Metab Res Rev 2008; 24(suppl 1):S52–S57.
- Saarto T, Wiffen PJ. Antidepressants for neuropathic pain: a Cochrane review. J Neurol Neurosurg Psychiatry 2010; 81:1372–1373.
- Tanenberg RJ, Irving GA, Risser RC, et al. Duloxetine, pregabalin, and duloxetine plus gabapentin for diabetic peripheral neuropathic pain management in patients with inadequate pain response to gabapentin: an open-label, randomized, noninferiority comparison. Mayo Clin Proc 2011; 86:615–626.
- Hertle L, van Ophoven A. Long-term results of amitriptyline treatment for interstitial cystitis. Aktuelle Urol 2010; 41(suppl 1):S61–S65.
- Nguyen TM, Eslick GD. Systematic review: the treatment of noncardiac chest pain with antidepressants. Aliment Pharmacol Ther 2012; 35:493–500.
- Lee H, Kim JH, Min BH, et al. Efficacy of venlafaxine for symptomatic relief in young adult patients with functional chest pain: a randomized, double-blind, placebo-controlled, crossover trial. Am J Gastroenterol 2010; 105:1504–1512.
- Varia I, Logue E, O’Connor C, et al. Randomized trial of sertraline in patients with unexplained chest pain of noncardiac origin. Am Heart J 2000; 140:367–372.
- Doraiswamy PM, Varia I, Hellegers C, et al. A randomized controlled trial of paroxetine for noncardiac chest pain. Psychopharmacol Bull 2006; 39:15–24.
- Clayton AH. Understanding antidepressant mechanism of action and its effect on efficacy and safety. J Clin Psychiatry 2012; 73:e11.
- Gartlehner G, Hansen RA, Morgan LC, et al. Second-generation antidepressants in the pharmacologic treatment of adult depression: an update of the 2007 comparative effectiveness review (Internet). Rockville (MD): Agency for Healthcare Research and Quality (US); 2011 Dec. Comparative Effectiveness Reviews, No. 46. http://www.ncbi.nlm.nih.gov/books/NBK83442/. Accessed February 27, 2013.
- Watanabe N, Omori IM, Nakagawa A, et al. Mirtazapine versus other antidepressive agents for depression. Cochrane Database Syst Rev 2011; 12:CD006528.
- Hofmeijer-Sevink MK, Batelaan NM, van Megen HJ, et al. Clinical relevance of comorbidity in anxiety disorders: a report from the Netherlands Study of Depression and Anxiety (NESDA). J Affect Disord 2012; 137:106–112.
- Koen N, Stein DJ. Pharmacotherapy of anxiety disorders: a critical review. Dialogues Clin Neurosci 2011; 13:423–437.
- Sheehan DV, Kamijima K. An evidence-based review of the clinical use of sertraline in mood and anxiety disorders. Int Clin Psychopharmacol 2009; 24:43–60.
- Huh J, Goebert D, Takeshita J, Lu BY, Kang M. Treatment of generalized anxiety disorder: a comprehensive review of the literature for psychopharmacologic alternatives to newer antidepressants and benzodiazepines. Prim Care Companion CNS Disord 2011; 13: 4088/PCC.08r00709blu.
- Rickels K, Downing R, Schweizer E, Hassman H. Antidepressants for the treatment of generalized anxiety disorder. A placebo-controlled comparison of imipramine, trazodone, and diazepam. Arch Gen Psychiatry 1993; 50:884–895.
- Uher R, Maier W, Hauser J, et al. Differential efficacy of escitalopram and nortriptyline on dimensional measures of depression. Br J Psychiatry 2009; 194:252–259.
- Kellner M. Drug treatment of obsessive-compulsive disorder. Dialogues Clin Neurosci 2010; 12:187–197.
- Rush AJ, Trivedi MH, Carmody TJ, et al. Response in relation to baseline anxiety levels in major depressive disorder treated with bupropion sustained release or sertraline. Neuropsychopharmacology 2001; 25:131–138.
- Mease PJ, Dundon K, Sarzi-Puttini P. Pharmacotherapy of fibromyalgia. Best Pract Res Clin Rheumatol 2011; 25:285–297.
- Wolfe F, Cathey MA, Hawley DJ. A double-blind placebo controlled trial of fluoxetine in fibromyalgia. Scand J Rheumatol 1994; 23:255–259.
- Arnold LM, Hess EV, Hudson JI, Welge JA, Berno SE, Keck PE. A randomized, placebo-controlled, double-blind, flexible-dose study of fluoxetine in the treatment of women with fibromyalgia. Am J Med 2002; 112:191–197.
- Arnold LM, Keck PE, Welge JA. Antidepressant treatment of fibromyalgia. A meta-analysis and review. Psychosomatics 2000; 41:104–113.
- Goldenberg DL, Burckhardt C, Crofford L. Management of fibromyalgia syndrome. JAMA 2004; 292:2388–2395.
- Tort S, Urrútia G, Nishishinya MB, Walitt B. Monoamine oxidase inhibitors (MAOIs) for fibromyalgia syndrome. Cochrane Database Syst Rev 2012; 4:CD009807.
- Vercoulen JH, Swanink CM, Zitman FG, et al. Randomised, double-blind, placebo-controlled study of fluoxetine in chronic fatigue syndrome. Lancet 1996; 347:858–861.
- Natelson BH, Cheu J, Pareja J, Ellis SP, Policastro T, Findley TW. Randomized, double blind, controlled placebo-phase in trial of low dose phenelzine in the chronic fatigue syndrome. Psychopharmacology (Berl) 1996; 124:226–230.
- Reid S, Chalder T, Cleare A, Hotopf M, Wessely S. Chronic fatigue syndrome. BMJ 2000; 320:292–296.
- Kupfer DJ, Spiker DG, Coble PA, Neil JF, Ulrich R, Shaw DH. Sleep and treatment prediction in endogenous depression. Am J Psychiatry 1981; 138:429–434.
- Argyropoulos SV, Hicks JA, Nash JR, et al. Redistribution of slow wave activity of sleep during pharmacological treatment of depression with paroxetine but not with nefazodone. J Sleep Res 2009; 18:342–348.
- Stein DJ, Lopez AG. Effects of escitalopram on sleep problems in patients with major depression or generalized anxiety disorder. Adv Ther 2011; 28:1021–1037.
- Ehlers CL, Havstad JW, Kupfer DJ. Estimation of the time course of slow-wave sleep over the night in depressed patients: effects of clomipramine and clinical response. Biol Psychiatry 1996; 39:171–181.
- Landolt HP, Raimo EB, Schnierow BJ, Kelsoe JR, Rapaport MH, Gillin JC. Sleep and sleep electroencephalogram in depressed patients treated with phenelzine. Arch Gen Psychiatry 2001; 58:268–276.
- Chen YM, Huang XM, Thompson R, Zhao YB. Clinical features and efficacy of escitalopram treatment for geriatric depression. J Int Med Res 2011; 39:1946–1953.
- Dolder C, Nelson M, Stump A. Pharmacological and clinical profile of newer antidepressants: implications for the treatment of elderly patients. Drugs Aging 2010; 27:625–640.
- Sterke CS, Ziere G, van Beeck EF, Looman CW, van der Cammen TJ. Dose-response relationship between selective serotonin re-uptake inhibitors and injurious falls: a study in nursing home residents with dementia. Br J Clin Pharmacol 2012; 73:812–820.
- Raji MA, Brady SR. Mirtazapine for treatment of depression and comorbidities in Alzheimer disease. Ann Pharmacother 2001; 35:1024–1027.
- Hennings JM, Schaaf L, Fulda S. Glucose metabolism and antidepressant medication. Curr Pharm Des 2012; 18:5900–5919.
- Czarny MJ, Arthurs E, Coffie DF, et al. Prevalence of antidepressant prescription or use in patients with acute coronary syndrome: a systematic review. PLoS One 2011; 6:e27671.
- Zuidersma M, Ormel J, Conradi HJ, de Jonge P. An increase in depressive symptoms after myocardial infarction predicts new cardiac events irrespective of depressive symptoms before myocardial infarction. Psychol Med 2012; 42:683–693.
- van Noord C, Straus SM, Sturkenboom MC, et al. Psychotropic drugs associated with corrected QT interval prolongation. J Clin Psychopharmacol 2009; 29:9–15.
- US Food and Drug Administration (FDA). FDA Drug Safety Communication: abnormal heart rhythms associated with high doses of Celexa (citalopram hydrobromide). http://www.fda.gov/Drugs/DrugSafety/ucm269086.htm. Accessed August 25, 2013.
- Lespérance F, Frasure-Smith N, Koszycki D, et al; CREATE Investigators. Effects of citalopram and interpersonal psychotherapy on depression in patients with coronary artery disease: the Canadian Cardiac Randomized Evaluation of Antidepressant and Psychotherapy Efficacy (CREATE) trial. JAMA 2007; 297:367–379.
- O’Connor CM, Jiang W, Kuchibhatla M, et al; SADHART-CHF Investigators. Safety and efficacy of sertraline for depression in patients with heart failure: results of the SADHART-CHF (Sertraline Against Depression and Heart Disease in Chronic Heart Failure) trial. J Am Coll Cardiol 2010; 56:692–699.
- Glassman AH, O’Connor CM, Califf RM, et al; Sertraline Antidepressant Heart Attack Randomized Trial (SADHEART) Group. Sertraline treatment of major depression in patients with acute MI or unstable angina. JAMA 2002; 288:701–709.
- Swenson JR, O’Connor CM, Barton D, et al; Sertraline Antidepressant Heart Attack Randomized Trial (SADHART) Group. Influence of depression and effect of treatment with sertraline on quality of life after hospitalization for acute coronary syndrome. Am J Cardiol 2003; 92:1271–1276.
- Strik JJ, Honig A, Lousberg R, et al. Efficacy and safety of fluoxetine in the treatment of patients with major depression after first myocardial infarction: findings from a double-blind, placebo-controlled trial. Psychosom Med 2000; 62:783–789.
- Honig A, Kuyper AM, Schene AH, et al; MIND-IT investigators. Treatment of post-myocardial infarction depressive disorder: a randomized, placebo-controlled trial with mirtazapine. Psychosom Med 2007; 69:606–613.
- Khawam EA, Laurencic G, Malone DA. Side effects of antidepressants: an overview. Cleve Clin J Med 2006; 73:351–361.
- Ziegler D. Painful diabetic neuropathy: treatment and future aspects. Diabetes Metab Res Rev 2008; 24(suppl 1):S52–S57.
- Saarto T, Wiffen PJ. Antidepressants for neuropathic pain: a Cochrane review. J Neurol Neurosurg Psychiatry 2010; 81:1372–1373.
- Tanenberg RJ, Irving GA, Risser RC, et al. Duloxetine, pregabalin, and duloxetine plus gabapentin for diabetic peripheral neuropathic pain management in patients with inadequate pain response to gabapentin: an open-label, randomized, noninferiority comparison. Mayo Clin Proc 2011; 86:615–626.
- Hertle L, van Ophoven A. Long-term results of amitriptyline treatment for interstitial cystitis. Aktuelle Urol 2010; 41(suppl 1):S61–S65.
- Nguyen TM, Eslick GD. Systematic review: the treatment of noncardiac chest pain with antidepressants. Aliment Pharmacol Ther 2012; 35:493–500.
- Lee H, Kim JH, Min BH, et al. Efficacy of venlafaxine for symptomatic relief in young adult patients with functional chest pain: a randomized, double-blind, placebo-controlled, crossover trial. Am J Gastroenterol 2010; 105:1504–1512.
- Varia I, Logue E, O’Connor C, et al. Randomized trial of sertraline in patients with unexplained chest pain of noncardiac origin. Am Heart J 2000; 140:367–372.
- Doraiswamy PM, Varia I, Hellegers C, et al. A randomized controlled trial of paroxetine for noncardiac chest pain. Psychopharmacol Bull 2006; 39:15–24.
- Clayton AH. Understanding antidepressant mechanism of action and its effect on efficacy and safety. J Clin Psychiatry 2012; 73:e11.
- Gartlehner G, Hansen RA, Morgan LC, et al. Second-generation antidepressants in the pharmacologic treatment of adult depression: an update of the 2007 comparative effectiveness review (Internet). Rockville (MD): Agency for Healthcare Research and Quality (US); 2011 Dec. Comparative Effectiveness Reviews, No. 46. http://www.ncbi.nlm.nih.gov/books/NBK83442/. Accessed February 27, 2013.
- Watanabe N, Omori IM, Nakagawa A, et al. Mirtazapine versus other antidepressive agents for depression. Cochrane Database Syst Rev 2011; 12:CD006528.
- Hofmeijer-Sevink MK, Batelaan NM, van Megen HJ, et al. Clinical relevance of comorbidity in anxiety disorders: a report from the Netherlands Study of Depression and Anxiety (NESDA). J Affect Disord 2012; 137:106–112.
- Koen N, Stein DJ. Pharmacotherapy of anxiety disorders: a critical review. Dialogues Clin Neurosci 2011; 13:423–437.
- Sheehan DV, Kamijima K. An evidence-based review of the clinical use of sertraline in mood and anxiety disorders. Int Clin Psychopharmacol 2009; 24:43–60.
- Huh J, Goebert D, Takeshita J, Lu BY, Kang M. Treatment of generalized anxiety disorder: a comprehensive review of the literature for psychopharmacologic alternatives to newer antidepressants and benzodiazepines. Prim Care Companion CNS Disord 2011; 13: 4088/PCC.08r00709blu.
- Rickels K, Downing R, Schweizer E, Hassman H. Antidepressants for the treatment of generalized anxiety disorder. A placebo-controlled comparison of imipramine, trazodone, and diazepam. Arch Gen Psychiatry 1993; 50:884–895.
- Uher R, Maier W, Hauser J, et al. Differential efficacy of escitalopram and nortriptyline on dimensional measures of depression. Br J Psychiatry 2009; 194:252–259.
- Kellner M. Drug treatment of obsessive-compulsive disorder. Dialogues Clin Neurosci 2010; 12:187–197.
- Rush AJ, Trivedi MH, Carmody TJ, et al. Response in relation to baseline anxiety levels in major depressive disorder treated with bupropion sustained release or sertraline. Neuropsychopharmacology 2001; 25:131–138.
- Mease PJ, Dundon K, Sarzi-Puttini P. Pharmacotherapy of fibromyalgia. Best Pract Res Clin Rheumatol 2011; 25:285–297.
- Wolfe F, Cathey MA, Hawley DJ. A double-blind placebo controlled trial of fluoxetine in fibromyalgia. Scand J Rheumatol 1994; 23:255–259.
- Arnold LM, Hess EV, Hudson JI, Welge JA, Berno SE, Keck PE. A randomized, placebo-controlled, double-blind, flexible-dose study of fluoxetine in the treatment of women with fibromyalgia. Am J Med 2002; 112:191–197.
- Arnold LM, Keck PE, Welge JA. Antidepressant treatment of fibromyalgia. A meta-analysis and review. Psychosomatics 2000; 41:104–113.
- Goldenberg DL, Burckhardt C, Crofford L. Management of fibromyalgia syndrome. JAMA 2004; 292:2388–2395.
- Tort S, Urrútia G, Nishishinya MB, Walitt B. Monoamine oxidase inhibitors (MAOIs) for fibromyalgia syndrome. Cochrane Database Syst Rev 2012; 4:CD009807.
- Vercoulen JH, Swanink CM, Zitman FG, et al. Randomised, double-blind, placebo-controlled study of fluoxetine in chronic fatigue syndrome. Lancet 1996; 347:858–861.
- Natelson BH, Cheu J, Pareja J, Ellis SP, Policastro T, Findley TW. Randomized, double blind, controlled placebo-phase in trial of low dose phenelzine in the chronic fatigue syndrome. Psychopharmacology (Berl) 1996; 124:226–230.
- Reid S, Chalder T, Cleare A, Hotopf M, Wessely S. Chronic fatigue syndrome. BMJ 2000; 320:292–296.
- Kupfer DJ, Spiker DG, Coble PA, Neil JF, Ulrich R, Shaw DH. Sleep and treatment prediction in endogenous depression. Am J Psychiatry 1981; 138:429–434.
- Argyropoulos SV, Hicks JA, Nash JR, et al. Redistribution of slow wave activity of sleep during pharmacological treatment of depression with paroxetine but not with nefazodone. J Sleep Res 2009; 18:342–348.
- Stein DJ, Lopez AG. Effects of escitalopram on sleep problems in patients with major depression or generalized anxiety disorder. Adv Ther 2011; 28:1021–1037.
- Ehlers CL, Havstad JW, Kupfer DJ. Estimation of the time course of slow-wave sleep over the night in depressed patients: effects of clomipramine and clinical response. Biol Psychiatry 1996; 39:171–181.
- Landolt HP, Raimo EB, Schnierow BJ, Kelsoe JR, Rapaport MH, Gillin JC. Sleep and sleep electroencephalogram in depressed patients treated with phenelzine. Arch Gen Psychiatry 2001; 58:268–276.
- Chen YM, Huang XM, Thompson R, Zhao YB. Clinical features and efficacy of escitalopram treatment for geriatric depression. J Int Med Res 2011; 39:1946–1953.
- Dolder C, Nelson M, Stump A. Pharmacological and clinical profile of newer antidepressants: implications for the treatment of elderly patients. Drugs Aging 2010; 27:625–640.
- Sterke CS, Ziere G, van Beeck EF, Looman CW, van der Cammen TJ. Dose-response relationship between selective serotonin re-uptake inhibitors and injurious falls: a study in nursing home residents with dementia. Br J Clin Pharmacol 2012; 73:812–820.
- Raji MA, Brady SR. Mirtazapine for treatment of depression and comorbidities in Alzheimer disease. Ann Pharmacother 2001; 35:1024–1027.
- Hennings JM, Schaaf L, Fulda S. Glucose metabolism and antidepressant medication. Curr Pharm Des 2012; 18:5900–5919.
- Czarny MJ, Arthurs E, Coffie DF, et al. Prevalence of antidepressant prescription or use in patients with acute coronary syndrome: a systematic review. PLoS One 2011; 6:e27671.
- Zuidersma M, Ormel J, Conradi HJ, de Jonge P. An increase in depressive symptoms after myocardial infarction predicts new cardiac events irrespective of depressive symptoms before myocardial infarction. Psychol Med 2012; 42:683–693.
- van Noord C, Straus SM, Sturkenboom MC, et al. Psychotropic drugs associated with corrected QT interval prolongation. J Clin Psychopharmacol 2009; 29:9–15.
- US Food and Drug Administration (FDA). FDA Drug Safety Communication: abnormal heart rhythms associated with high doses of Celexa (citalopram hydrobromide). http://www.fda.gov/Drugs/DrugSafety/ucm269086.htm. Accessed August 25, 2013.
- Lespérance F, Frasure-Smith N, Koszycki D, et al; CREATE Investigators. Effects of citalopram and interpersonal psychotherapy on depression in patients with coronary artery disease: the Canadian Cardiac Randomized Evaluation of Antidepressant and Psychotherapy Efficacy (CREATE) trial. JAMA 2007; 297:367–379.
- O’Connor CM, Jiang W, Kuchibhatla M, et al; SADHART-CHF Investigators. Safety and efficacy of sertraline for depression in patients with heart failure: results of the SADHART-CHF (Sertraline Against Depression and Heart Disease in Chronic Heart Failure) trial. J Am Coll Cardiol 2010; 56:692–699.
- Glassman AH, O’Connor CM, Califf RM, et al; Sertraline Antidepressant Heart Attack Randomized Trial (SADHEART) Group. Sertraline treatment of major depression in patients with acute MI or unstable angina. JAMA 2002; 288:701–709.
- Swenson JR, O’Connor CM, Barton D, et al; Sertraline Antidepressant Heart Attack Randomized Trial (SADHART) Group. Influence of depression and effect of treatment with sertraline on quality of life after hospitalization for acute coronary syndrome. Am J Cardiol 2003; 92:1271–1276.
- Strik JJ, Honig A, Lousberg R, et al. Efficacy and safety of fluoxetine in the treatment of patients with major depression after first myocardial infarction: findings from a double-blind, placebo-controlled trial. Psychosom Med 2000; 62:783–789.
- Honig A, Kuyper AM, Schene AH, et al; MIND-IT investigators. Treatment of post-myocardial infarction depressive disorder: a randomized, placebo-controlled trial with mirtazapine. Psychosom Med 2007; 69:606–613.
KEY POINTS
- We suggest that clinicians become familiar with one drug from each class of antidepressants.
- Many antidepressants are also approved for conditions other than depression, and for patients who have both depression and one or more of these comcomitant conditions, these drugs can have a “two-for-one” benefit.
- Adverse effects of an antidepressant are usually predictable on the basis of the drug’s mechanism of action.
Which lower-extremity DVTs should be removed early?
Early thrombus removal for lower-extremity deep venous thrombosis (DVT) is at present only modestly supported by evidence and so remains controversial. It is largely aimed at preventing postthrombotic syndrome.
The decision to pursue early thrombus removal demands weighing the patient’s risk of postthrombotic syndrome against the risks and costs associated with thrombolysis and thrombectomy, such as bleeding complications. In the final analysis, this remains a subjective decision.
With these caveats in mind, the best candidate for early thrombus removal is a young patient with iliofemoral DVT with symptoms lasting fewer than 14 days.
POSTTHROMBOTIC SYNDROME IS COMMON
Anticoagulation with heparin and warfarin is the mainstay of DVT therapy. Indeed, the safety of this therapy and its effectiveness in reducing thrombus propagation and DVT recurrence are well established. Neither heparin nor warfarin, however, actively reduces the thrombus burden. Rather, both prevent the clot from propagating while it is, hopefully, gradually reabsorbed through endogenous mechanisms.
Up to 50% of DVT patients develop postthrombotic syndrome. A variety of mechanisms are involved, including persistent obstructive thrombosis and valvular injury.1 But much remains unknown about the etiology, and some patients develop the condition in the absence of abnormalities on objective testing.
Symptoms of postthrombotic syndrome can range from mild heaviness, edema, erythema, and cramping in the affected limb to debilitating pain with classic signs of venous hypertension (eg, venous ectasia and ulcers). It accounts for significant health care costs and has a detrimental effect on quality of life.1 Thus, there has been interest in early thrombus removal as initial therapy for DVT.
THROMBUS REMOVAL
Venous clots can be removed with open surgery or, more typically, with percutaneous catheter-based thrombolysis and thrombectomy devices that use high-velocity saline jets, ultrasonic energy, or wire oscillation to mechanically fragment the venous clot. All of these mechanisms help with drug delivery and pose a minimal risk of pulmonary embolism.
Evidence is weak
Patients with DVT of the iliac venous system or common femoral vein are at highest risk of postthrombotic syndrome. Therefore, the Society for Vascular Surgery and the American Venous Forum have issued a grade 2C (ie, weak) recommendation in favor of early thrombus removal in patients with a first-time episode of iliofemoral DVT with fewer than 14 days of symptoms.2 Moreover, patients must have a low risk of bleeding complications, be ambulatory, and have reasonable life expectancy.
The recommendation is buttressed by a Cochrane meta-analysis that included 101 patients.3 It concluded that there was a significant decrement in the development of postthrombotic syndrome with thrombolysis (but without mechanical thrombectomy) compared with standard therapy: the rate was 48% (29/61) with thrombolysis, and 65% (26/40) with standard therapy.3
More recently, the Catheter-Directed Thrombolysis Versus Standard Treatment for Acute Iliofemoral Deep Vein Thrombosis (CaVenT) study, a randomized prospective trial in 189 patients, demonstrated a lower rate of postthrombotic syndrome at 24 months and increased iliofemoral patency at 6 months with catheter-directed thrombolysis with alteplase (41.1% and 65.9%) vs anticoagulation with heparin and warfarin alone (55.6% and 47.4%).4
The Acute Venous Thrombosis: Thrombus Removal With Adjunctive Catheter-directed Thrombolysis (ATTRACT) trial is an ongoing prospective randomized multicenter trial of the effect of thrombolysis on postthrombotic syndrome that also hopes to clarify the relative benefits of different methods of pharmacomechanical clot removal.
While CaVenT has not been criticized extensively in the literature, other studies supporting early intervention for iliofemoral venous thrombosis generally have been noted to have a number of shortcomings, including a lack of randomization, and consequent bias, and the use of surrogate end points instead of a direct assessment of postthrombotic syndrome.
Reflecting the weakness of the evidence, the American College of Chest Physicians has issued a grade 2C recommendation against catheter-directed thrombolysis and against thrombectomy in favor of anticoagulant therapy.5
A subjective, case-by-case decision
The decision on standard vs interventional therapy must be made case by case. For example, thrombus removal may be more appropriate for a physically active young patient who is more likely to be impaired by postthrombotic syndrome, whereas standard warfarin therapy may be preferable for a sedentary patient. We are also more inclined to offer thrombus removal to patients who have worse symptoms.
Complicating the issue, many patients present with a mix of variables that support and oppose intervention—eg, a moderately active elderly patient with an unclear life expectancy and a history of gastrointestinal bleeding. At present, there is no way to quantitatively evaluate the risks and rewards of thrombus removal, and the final decision is essentially subjective.
Additional facts warranting consideration include the possibility that thrombolysis may require several days of therapy with daily venography for evaluation. Monitoring in the intensive care unit is normally required during the period of thrombolysis. Patients should be apprised of these elements of therapy beforehand; obviously, those who are unwilling to comply are not candidates.
Not a substitute for anticoagulation
It is important to recognize that thrombus removal is not a substitute for standard heparin-warfarin anticoagulation, which must also be prescribed.5 Thus, patients who cannot tolerate standard post-DVT anticoagulation should not undergo thrombus removal. Furthermore, the current evidence supports the use of standard anticoagulation over early thrombus removal of DVTs that are more distal in the lower extremity, such as those in the popliteal vein.5
PHLEGMASIA CERULEA DOLENS IS A SPECIAL CASE
Phlegmasia cerulea dolens—acute venous outflow obstruction associated with edema, cyanosis, and pain that in the worst cases may lead to shock, limb loss, and death—constitutes a special case. Although we lack robust supporting evidence, phlegmasia is a commonly accepted indication for early thrombus removal as a means of limb salvage.2,6
- Kahn SR. The post thrombotic syndrome. Thromb Res 2011; 127 (suppl 3):S89–S92.
- Meissner MH, Gloviczki P, Comerota AJ, et al; Society for Vascular Surgery; American Venous Forum. Early thrombus removal strategies for acute deep venous thrombosis: clinical practice guidelines of the Society for Vascular Surgery and the American Venous Forum. J Vasc Surg 2012; 55:1449–1462.
- Watson LI, Armon MP. Thrombolysis for acute deep vein thrombosis. Cochrane Database Syst Rev 2004; 4:CD002783.
- Enden T, Haig Y, Kløw NE, et al; CaVenT Study Group. Long-term outcome after additional catheter-directed thrombolysis versus standard treatment for acute iliofemoral deep vein thrombosis (the CaVenT study): a randomised controlled trial. Lancet 2012; 379:31–38.
- Kearon C, Akl EA, Comerota AJ, et al; American College of Chest Physicians. Antithrombotic therapy for VTE disease: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141 (suppl 2):e419S–e494S.
- Patterson BO, Hinchliffe R, Loftus IM, Thompson MM, Holt PJ. Indications for catheter-directed thrombolysis in the management of acute proximal deep venous thrombosis. Arterioscler Thromb Vasc Biol 2010; 30:669–674.
Early thrombus removal for lower-extremity deep venous thrombosis (DVT) is at present only modestly supported by evidence and so remains controversial. It is largely aimed at preventing postthrombotic syndrome.
The decision to pursue early thrombus removal demands weighing the patient’s risk of postthrombotic syndrome against the risks and costs associated with thrombolysis and thrombectomy, such as bleeding complications. In the final analysis, this remains a subjective decision.
With these caveats in mind, the best candidate for early thrombus removal is a young patient with iliofemoral DVT with symptoms lasting fewer than 14 days.
POSTTHROMBOTIC SYNDROME IS COMMON
Anticoagulation with heparin and warfarin is the mainstay of DVT therapy. Indeed, the safety of this therapy and its effectiveness in reducing thrombus propagation and DVT recurrence are well established. Neither heparin nor warfarin, however, actively reduces the thrombus burden. Rather, both prevent the clot from propagating while it is, hopefully, gradually reabsorbed through endogenous mechanisms.
Up to 50% of DVT patients develop postthrombotic syndrome. A variety of mechanisms are involved, including persistent obstructive thrombosis and valvular injury.1 But much remains unknown about the etiology, and some patients develop the condition in the absence of abnormalities on objective testing.
Symptoms of postthrombotic syndrome can range from mild heaviness, edema, erythema, and cramping in the affected limb to debilitating pain with classic signs of venous hypertension (eg, venous ectasia and ulcers). It accounts for significant health care costs and has a detrimental effect on quality of life.1 Thus, there has been interest in early thrombus removal as initial therapy for DVT.
THROMBUS REMOVAL
Venous clots can be removed with open surgery or, more typically, with percutaneous catheter-based thrombolysis and thrombectomy devices that use high-velocity saline jets, ultrasonic energy, or wire oscillation to mechanically fragment the venous clot. All of these mechanisms help with drug delivery and pose a minimal risk of pulmonary embolism.
Evidence is weak
Patients with DVT of the iliac venous system or common femoral vein are at highest risk of postthrombotic syndrome. Therefore, the Society for Vascular Surgery and the American Venous Forum have issued a grade 2C (ie, weak) recommendation in favor of early thrombus removal in patients with a first-time episode of iliofemoral DVT with fewer than 14 days of symptoms.2 Moreover, patients must have a low risk of bleeding complications, be ambulatory, and have reasonable life expectancy.
The recommendation is buttressed by a Cochrane meta-analysis that included 101 patients.3 It concluded that there was a significant decrement in the development of postthrombotic syndrome with thrombolysis (but without mechanical thrombectomy) compared with standard therapy: the rate was 48% (29/61) with thrombolysis, and 65% (26/40) with standard therapy.3
More recently, the Catheter-Directed Thrombolysis Versus Standard Treatment for Acute Iliofemoral Deep Vein Thrombosis (CaVenT) study, a randomized prospective trial in 189 patients, demonstrated a lower rate of postthrombotic syndrome at 24 months and increased iliofemoral patency at 6 months with catheter-directed thrombolysis with alteplase (41.1% and 65.9%) vs anticoagulation with heparin and warfarin alone (55.6% and 47.4%).4
The Acute Venous Thrombosis: Thrombus Removal With Adjunctive Catheter-directed Thrombolysis (ATTRACT) trial is an ongoing prospective randomized multicenter trial of the effect of thrombolysis on postthrombotic syndrome that also hopes to clarify the relative benefits of different methods of pharmacomechanical clot removal.
While CaVenT has not been criticized extensively in the literature, other studies supporting early intervention for iliofemoral venous thrombosis generally have been noted to have a number of shortcomings, including a lack of randomization, and consequent bias, and the use of surrogate end points instead of a direct assessment of postthrombotic syndrome.
Reflecting the weakness of the evidence, the American College of Chest Physicians has issued a grade 2C recommendation against catheter-directed thrombolysis and against thrombectomy in favor of anticoagulant therapy.5
A subjective, case-by-case decision
The decision on standard vs interventional therapy must be made case by case. For example, thrombus removal may be more appropriate for a physically active young patient who is more likely to be impaired by postthrombotic syndrome, whereas standard warfarin therapy may be preferable for a sedentary patient. We are also more inclined to offer thrombus removal to patients who have worse symptoms.
Complicating the issue, many patients present with a mix of variables that support and oppose intervention—eg, a moderately active elderly patient with an unclear life expectancy and a history of gastrointestinal bleeding. At present, there is no way to quantitatively evaluate the risks and rewards of thrombus removal, and the final decision is essentially subjective.
Additional facts warranting consideration include the possibility that thrombolysis may require several days of therapy with daily venography for evaluation. Monitoring in the intensive care unit is normally required during the period of thrombolysis. Patients should be apprised of these elements of therapy beforehand; obviously, those who are unwilling to comply are not candidates.
Not a substitute for anticoagulation
It is important to recognize that thrombus removal is not a substitute for standard heparin-warfarin anticoagulation, which must also be prescribed.5 Thus, patients who cannot tolerate standard post-DVT anticoagulation should not undergo thrombus removal. Furthermore, the current evidence supports the use of standard anticoagulation over early thrombus removal of DVTs that are more distal in the lower extremity, such as those in the popliteal vein.5
PHLEGMASIA CERULEA DOLENS IS A SPECIAL CASE
Phlegmasia cerulea dolens—acute venous outflow obstruction associated with edema, cyanosis, and pain that in the worst cases may lead to shock, limb loss, and death—constitutes a special case. Although we lack robust supporting evidence, phlegmasia is a commonly accepted indication for early thrombus removal as a means of limb salvage.2,6
Early thrombus removal for lower-extremity deep venous thrombosis (DVT) is at present only modestly supported by evidence and so remains controversial. It is largely aimed at preventing postthrombotic syndrome.
The decision to pursue early thrombus removal demands weighing the patient’s risk of postthrombotic syndrome against the risks and costs associated with thrombolysis and thrombectomy, such as bleeding complications. In the final analysis, this remains a subjective decision.
With these caveats in mind, the best candidate for early thrombus removal is a young patient with iliofemoral DVT with symptoms lasting fewer than 14 days.
POSTTHROMBOTIC SYNDROME IS COMMON
Anticoagulation with heparin and warfarin is the mainstay of DVT therapy. Indeed, the safety of this therapy and its effectiveness in reducing thrombus propagation and DVT recurrence are well established. Neither heparin nor warfarin, however, actively reduces the thrombus burden. Rather, both prevent the clot from propagating while it is, hopefully, gradually reabsorbed through endogenous mechanisms.
Up to 50% of DVT patients develop postthrombotic syndrome. A variety of mechanisms are involved, including persistent obstructive thrombosis and valvular injury.1 But much remains unknown about the etiology, and some patients develop the condition in the absence of abnormalities on objective testing.
Symptoms of postthrombotic syndrome can range from mild heaviness, edema, erythema, and cramping in the affected limb to debilitating pain with classic signs of venous hypertension (eg, venous ectasia and ulcers). It accounts for significant health care costs and has a detrimental effect on quality of life.1 Thus, there has been interest in early thrombus removal as initial therapy for DVT.
THROMBUS REMOVAL
Venous clots can be removed with open surgery or, more typically, with percutaneous catheter-based thrombolysis and thrombectomy devices that use high-velocity saline jets, ultrasonic energy, or wire oscillation to mechanically fragment the venous clot. All of these mechanisms help with drug delivery and pose a minimal risk of pulmonary embolism.
Evidence is weak
Patients with DVT of the iliac venous system or common femoral vein are at highest risk of postthrombotic syndrome. Therefore, the Society for Vascular Surgery and the American Venous Forum have issued a grade 2C (ie, weak) recommendation in favor of early thrombus removal in patients with a first-time episode of iliofemoral DVT with fewer than 14 days of symptoms.2 Moreover, patients must have a low risk of bleeding complications, be ambulatory, and have reasonable life expectancy.
The recommendation is buttressed by a Cochrane meta-analysis that included 101 patients.3 It concluded that there was a significant decrement in the development of postthrombotic syndrome with thrombolysis (but without mechanical thrombectomy) compared with standard therapy: the rate was 48% (29/61) with thrombolysis, and 65% (26/40) with standard therapy.3
More recently, the Catheter-Directed Thrombolysis Versus Standard Treatment for Acute Iliofemoral Deep Vein Thrombosis (CaVenT) study, a randomized prospective trial in 189 patients, demonstrated a lower rate of postthrombotic syndrome at 24 months and increased iliofemoral patency at 6 months with catheter-directed thrombolysis with alteplase (41.1% and 65.9%) vs anticoagulation with heparin and warfarin alone (55.6% and 47.4%).4
The Acute Venous Thrombosis: Thrombus Removal With Adjunctive Catheter-directed Thrombolysis (ATTRACT) trial is an ongoing prospective randomized multicenter trial of the effect of thrombolysis on postthrombotic syndrome that also hopes to clarify the relative benefits of different methods of pharmacomechanical clot removal.
While CaVenT has not been criticized extensively in the literature, other studies supporting early intervention for iliofemoral venous thrombosis generally have been noted to have a number of shortcomings, including a lack of randomization, and consequent bias, and the use of surrogate end points instead of a direct assessment of postthrombotic syndrome.
Reflecting the weakness of the evidence, the American College of Chest Physicians has issued a grade 2C recommendation against catheter-directed thrombolysis and against thrombectomy in favor of anticoagulant therapy.5
A subjective, case-by-case decision
The decision on standard vs interventional therapy must be made case by case. For example, thrombus removal may be more appropriate for a physically active young patient who is more likely to be impaired by postthrombotic syndrome, whereas standard warfarin therapy may be preferable for a sedentary patient. We are also more inclined to offer thrombus removal to patients who have worse symptoms.
Complicating the issue, many patients present with a mix of variables that support and oppose intervention—eg, a moderately active elderly patient with an unclear life expectancy and a history of gastrointestinal bleeding. At present, there is no way to quantitatively evaluate the risks and rewards of thrombus removal, and the final decision is essentially subjective.
Additional facts warranting consideration include the possibility that thrombolysis may require several days of therapy with daily venography for evaluation. Monitoring in the intensive care unit is normally required during the period of thrombolysis. Patients should be apprised of these elements of therapy beforehand; obviously, those who are unwilling to comply are not candidates.
Not a substitute for anticoagulation
It is important to recognize that thrombus removal is not a substitute for standard heparin-warfarin anticoagulation, which must also be prescribed.5 Thus, patients who cannot tolerate standard post-DVT anticoagulation should not undergo thrombus removal. Furthermore, the current evidence supports the use of standard anticoagulation over early thrombus removal of DVTs that are more distal in the lower extremity, such as those in the popliteal vein.5
PHLEGMASIA CERULEA DOLENS IS A SPECIAL CASE
Phlegmasia cerulea dolens—acute venous outflow obstruction associated with edema, cyanosis, and pain that in the worst cases may lead to shock, limb loss, and death—constitutes a special case. Although we lack robust supporting evidence, phlegmasia is a commonly accepted indication for early thrombus removal as a means of limb salvage.2,6
- Kahn SR. The post thrombotic syndrome. Thromb Res 2011; 127 (suppl 3):S89–S92.
- Meissner MH, Gloviczki P, Comerota AJ, et al; Society for Vascular Surgery; American Venous Forum. Early thrombus removal strategies for acute deep venous thrombosis: clinical practice guidelines of the Society for Vascular Surgery and the American Venous Forum. J Vasc Surg 2012; 55:1449–1462.
- Watson LI, Armon MP. Thrombolysis for acute deep vein thrombosis. Cochrane Database Syst Rev 2004; 4:CD002783.
- Enden T, Haig Y, Kløw NE, et al; CaVenT Study Group. Long-term outcome after additional catheter-directed thrombolysis versus standard treatment for acute iliofemoral deep vein thrombosis (the CaVenT study): a randomised controlled trial. Lancet 2012; 379:31–38.
- Kearon C, Akl EA, Comerota AJ, et al; American College of Chest Physicians. Antithrombotic therapy for VTE disease: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141 (suppl 2):e419S–e494S.
- Patterson BO, Hinchliffe R, Loftus IM, Thompson MM, Holt PJ. Indications for catheter-directed thrombolysis in the management of acute proximal deep venous thrombosis. Arterioscler Thromb Vasc Biol 2010; 30:669–674.
- Kahn SR. The post thrombotic syndrome. Thromb Res 2011; 127 (suppl 3):S89–S92.
- Meissner MH, Gloviczki P, Comerota AJ, et al; Society for Vascular Surgery; American Venous Forum. Early thrombus removal strategies for acute deep venous thrombosis: clinical practice guidelines of the Society for Vascular Surgery and the American Venous Forum. J Vasc Surg 2012; 55:1449–1462.
- Watson LI, Armon MP. Thrombolysis for acute deep vein thrombosis. Cochrane Database Syst Rev 2004; 4:CD002783.
- Enden T, Haig Y, Kløw NE, et al; CaVenT Study Group. Long-term outcome after additional catheter-directed thrombolysis versus standard treatment for acute iliofemoral deep vein thrombosis (the CaVenT study): a randomised controlled trial. Lancet 2012; 379:31–38.
- Kearon C, Akl EA, Comerota AJ, et al; American College of Chest Physicians. Antithrombotic therapy for VTE disease: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141 (suppl 2):e419S–e494S.
- Patterson BO, Hinchliffe R, Loftus IM, Thompson MM, Holt PJ. Indications for catheter-directed thrombolysis in the management of acute proximal deep venous thrombosis. Arterioscler Thromb Vasc Biol 2010; 30:669–674.
Azithromycin and risk of sudden cardiac death: Guilty as charged or falsely accused?
A March 2013 warning by the US Food and Drug Administration that azithromycin (Zithromax, Zmax, Z-pak) may increase the risk of sudden cardiac death does not mean we must abandon using it. We should, however, try to determine if our patients have cardiovascular risk factors for this extreme side effect and take appropriate precautions.
AZITHROMYCIN: THE SAFEST OF THE MACROLIDES?
Azithromycin, a broad-spectrum macrolide antibiotic, is used to treat or prevent a range of common bacterial infections, including upper and lower respiratory tract infections and certain sexually transmitted diseases.
In terms of overall toxicity, azithromycin has been considered the safest of the macrolides, as it neither undergoes CYP3A4 metabolism nor inhibits CYP3A4 to any clinically meaningful degree, and therefore does not interfere with the array of commonly used medications that undergo CYP3A4 metabolism.

Also, in vitro, azithromycin shows only limited blockade of the potassium channel hERG. This channel is critically involved in cardiomyocyte repolarization, and if it is blocked or otherwise malfunctioning, the result can be a prolonged QT interval, ventricular arrhythmias, and even sudden cardiac death.1–4 Therefore, lack of blockade, as reflected by a high inhibitory concentration (Table 1), boded well for the safety of azithromycin in terms of QT liability. However, we should be cautious in interpreting in vitro data.
With its broad antibiotic spectrum and perceived favorable safety profile, azithromycin has become one of the top 15 most prescribed drugs and the best-selling antibiotic in the United States, accounting for 55.4 million prescriptions in 2012, according to the IMS Institute for Healthcare Informatics.
THE FDA RECEIVES 203 REPORTS OF ADVERSE EVENTS IN 8 YEARS
However, beginning with a report of azithromycin-triggered torsades de pointes in 2001,5 a growing body of evidence, derived from postmarketing surveillance, has linked azithromycin to cardiac arrhythmias such as pronounced QT interval prolongation and associated torsades de pointes (which can progress to life-threatening ventricular fibrillation). Other, closely related macrolides such as clarithromycin and erythromycin are also linked to these effects.
Furthermore, in the 8-year period from 2004 to 2011, the US Food and Drug Administration (FDA) Adverse Event Reporting System (FAERS) received a total of 203 reports of azithromycin-associated QT prolongation, torsades de pointes, ventricular arrhythmia, or, in 65 cases, sudden cardiac death (Table 1).6
At face value, the number of FAERS reports appears to be similar between the various macrolide antibiotics. However, it is important to remember that these drugs differ substantially in the number of prescriptions written for them, with azithromycin being prescribed more often. Also, the FAERS numbers are subject to a number of well-known limitations such as confounding variables, uneven quality and completeness of reports, duplication, and underreporting. These limitations preclude the use of such adverse reporting databases in calculating and thereby comparing the true incidence of adverse events associated with the various macrolide antibiotics.6–9
RAY ET AL FIND A HIGHER RISK OF CARDIOVASCULAR DEATH
Despite these inherent flaws, initial postmarketing surveillance reports cast enough doubt on the long-standing notion that azithromycin is the safest macrolide antibiotic to prompt Ray et al10 to assess its safety in an observational, nonrandomized study of people enrolled in the Tennessee Medicaid program.

They found that, over the typical 5 days of therapy, people taking azithromycin had a rate of cardiovascular death 2.88 times higher than in people taking no antibiotic, and 2.49 times higher than in people taking amoxicillin (Table 2).
However, the absolute excess risk compared with amoxicillin varied considerably according to baseline risk score for cardiovascular disease, with 1 excess cardiovascular death per 4,100 in the highest-risk decile compared with 1 excess cardiovascular death per 100,000 in the lowest-risk decile.10,11
Moreover, the increase in deaths did not persist after the 5 days of therapy. This time-limited pattern directly correlated with expected peak azithromycin plasma levels during a standard 5-day course.
Ray et al used appropriate analytic methods to attempt to correct for any confounding bias intrinsic to the observational, nonrandomized study design. Nevertheless, the patients were Medicaid beneficiaries, who have a higher prevalence of comorbid conditions and higher mortality rates than the general population. Therefore, legitimate questions were raised about whether the results of the study could be generalized to populations with substantially lower baseline risk of cardiovascular disease and if differences in the baseline characteristics of the treatment groups were adequately controlled.12,13
THE FDA REVISES AZITHROMYCIN’S WARNINGS AND PRECAUTIONS
The striking observations by Ray et al,10 coupled with the concerns raised by postmarketing surveillance reports, compelled the FDA to review the labels of azithromycin and other macrolide antibiotics.
Ultimately, the FDA opted to revise the “warning and precautions” section of the azithromycin drug label to include a warning about the potential risk of fatal arrhythmias, specifically QT interval prolongation and torsades de pointes. In a March 2013 safety announcement, it also urged health care professionals to use caution when prescribing azithromycin to patients known to have risk factors for drug-related arrhythmias, including congenital long QT syndrome, acquired QT interval prolongation, hypokalemia, hypomagnesia, bradycardia, and concurrent use of other medications known to prolong the QT interval, specifically the class IA (eg, quinidine and procainamide) and class III (eg, amiodarone, sotalol, and dofetilide) antiarrhythmics.
SVANSTRÖM ET AL FIND NO INCREASED RISK
However, just when the medical community appeared ready to accept that azithromycin may not be as safe as we thought it was, a large prospective study by Svanström et al, published in early May 2013, found no increased risk of cardiovascular death associated with azithromycin (Table 2).14
The patients were a representative population of young to middle-aged Danish adults at low baseline risk of underlying cardiovascular disease.
Interestingly, Svanström et al were careful to point out that their study was only powered to rule out a moderate-to-high (> 55%) increase in the relative risk of cardiovascular death. Furthermore, profound differences existed in the baseline risk of death and cardiovascular risk factors between their patients and the Tennessee Medicaid patients studied by Ray et al.14 Therefore, the authors suggested that their study complements rather than contradicts the study by Ray et al. They attributed the differences in the findings to treatment-effect heterogeneity, in which the risk of azithromycin-associated cardiovascular mortality is largely limited to high-risk patients, namely those with multiple preexisting cardiovascular risk factors.14
ACC/AHA RECOMMENDATION: IDENTIFY THOSE AT RISK
Collectively, the data reviewed above provide compelling evidence that azithromycin is not completely free of the QT-prolonging and torsadogenic effects that have long been associated with other macrolide antibiotics. However, the findings from both the study by Ray et al and that of Svanström et al suggest that preexisting cardiovascular risk factors play a prominent role in determining the incidence of azithromycin-associated cardiovascular death in a given population (Table 2).10,14
These findings should prompt physicians to carefully reassess the risks and benefits of azithromycin use in their clinical practices. They also reinforce a recent call by the American Heart Association (AHA) and American College of Cardiology (ACC) to better identify, early on, patients at risk of drug-induced ventricular arrhythmias and sudden death and to subsequently improve how these patients are monitored when the use of QT-prolonging and torsadogenic drugs is medically necessary.15
AN ELECTRONIC MEDICAL RECORD FLAGS QTc ≥ 500 MS
On the heels of these AHA/ACC suggestions, our hospital has adopted an institution-wide QT alert system. Here, the electronic medical record system (Centricity EMR; GE Healthcare) uses a proprietary algorithm to detect and electronically alert ordering physicians when a patient has a prolonged QT interval, and gives information about the potential clinical significance of this electrocardiographic finding.16 Physicians also receive a warning when ordering QT-prolonging drugs in patients at risk.
This system is still in its infancy, but it has already confirmed that a prolonged QT interval (QTc ≥ 500 ms) is a powerful predictor of death from any cause and has demonstrated that mortality rates in those with prolonged QT intervals increase in a dose-dependent fashion with the patient’s number of modifiable risk factors (eg, electrolyte disturbances or QT-prolonging medications) and nonmodifiable risk factors (eg, genetic disposition, female sex, structural heart disease, diabetes mellitus).16 We have also found evidence that modifiable risk factors may have a more pronounced effect on mortality risk than non-modifiable risk factors.16
These findings suggest that information technology-based QT alert systems may one day provide physicians with an important tool to efficiently identify and possibly even modify the risk of cardiovascular death in patients at high risk, for example, by correcting electrolyte abnormalities or reducing the burden of QT-prolonging medications.
CONSIDER RISK OF QT PROLONGATION WHEN PRESCRIBING AZITHROMYCIN

For most institutions and clinical practices, such electronic QT alert systems are still years if not decades away. However, in light of the information summarized above, all physicians should begin considering risk factors for QT prolongation and torsades de pointes (summarized in Table 3) and weighing the risks and benefits of prescribing azithromycin vs alternative antibiotics with minimal QT liability. This should be relatively simple to do. Things to keep in mind:
- Although azithromycin may increase the relative risk of a cardiovascular event, for most otherwise-healthy patients, the absolute risk is miniscule.
- In a patient at risk (eg, with baseline QT prolongation or multiple risk factors for it), if azithromycin or another QT-prolonging antibiotic such as a macrolide or fluoroquinolone is medically necessary due to preferential bacterial susceptibility or patient allergies, every effort should be made to correct modifiable risk factors (eg, electrolyte abnormalities) and, if possible, to avoid polypharmacy with multiple QT-prolonging drugs.
- For patients who have multiple risk factors for QT prolongation in whom treatment with a known QT-prolonging medication is still deemed in the patient’s best interest, strong consideration should be given to inpatient administration and monitoring until the treatment has been completed.
With careful consideration of modifiable and nonmodifiable risk factors as well as a little extra caution when prescribing potential QT-prolonging medications such as azithromycin, the clinical benefit of these often-advantageous medications can be maximized and the incidence of these tragic but rare drug-induced sudden cardiac deaths can be reduced.
- Hopkins S. Clinical toleration and safety of azithromycin. Am J Med 1991; 91:40S–45S.
- Milberg P, Eckardt L, Bruns HJ, et al. Divergent proarrhythmic potential of macrolide antibiotics despite similar QT prolongation: fast phase 3 repolarization prevents early afterdepolarizations and torsade de pointes. J Pharmacol Exp Ther 2002; 303:218–225.
- Ioannidis JP, Contopoulos-Ioannidis DG, Chew P, Lau J. Meta-analysis of randomized controlled trials on the comparative efficacy and safety of azithromycin against other antibiotics for upper respiratory tract infections. J Antimicrob Chemother 2001; 48:677–689.
- Owens RC, Nolin TD. Antimicrobial-associated QT interval prolongation: pointes of interest. Clin Infect Dis 2006; 43:1603–1611.
- Arellano-Rodrigo E, García A, Mont L, Roqué M. Torsade de pointes and cardiorespiratory arrest induced by azithromycin in a patient with congenital long QT syndrome. (Article in Spanish.) Med Clin (Barc) 2001; 117:118–119.
- Raschi E, Poluzzi E, Koci A, Moretti U, Sturkenboom M, De Ponti F. Macrolides and torsadogenic risk: emerging issues from the fda pharmacovigilance database. J Pharmacovigilance 2013; 1:104.
- Shaffer D, Singer S, Korvick J, Honig P. Concomitant risk factors in reports of torsades de pointes associated with macrolide use: review of the United States Food and Drug Administration Adverse Event Reporting System. Clin Infect Dis 2002; 35:197–200.
- Stephenson WP, Hauben M. Data mining for signals in spontaneous reporting databases: proceed with caution. Pharmacoepidemiol Drug Saf 2007; 16:359–365.
- Bate A, Evans SJ. Quantitative signal detection using spontaneous ADR reporting. Pharmacoepidemiol Drug Saf 2009; 18:427–436.
- Ray WA, Murray KT, Hall K, Arbogast PG, Stein CM. Azithromycin and the risk of cardiovascular death. N Engl J Med 2012; 366:1881–1890.
- Mosholder AD, Mathew J, Alexander JJ, Smith H, Nambiar S. Cardiovascular risks with azithromycin and other antibacterial drugs. N Engl J Med 2013; 368:1665–1668.
- Louie R. Azithromycin and the risk of cardiovascular death. N Engl J Med 2012; 367:774–775.
- Koga T, Imaoka H. Azithromycin and the risk of cardiovascular death. N Engl J Med 2012; 367:774–775.
- Svanström H, Pasternak B, Hviid A. Use of azithromycin and death from cardiovascular causes. N Engl J Med 2013; 368:1704–1712.
- Drew BJ, Ackerman MJ, Funk M, et al; American Heart Association Acute Cardiac Care Committee of the Council on Clinical Cardiology, the Council on Cardiovascular Nursing, and the American College of Cardiology Foundation. Prevention of torsade de pointes in hospital settings: a scientific statement from the American Heart Association and the American College of Cardiology Foundation. Circulation 2010; 121:1047–1060.
- Haugaa KH, Bos JM, Tarrell RF, Morlan BW, Caraballo PJ, Ackerman MJ. Institution-wide QT alert system identifies patients with a high risk of mortality. Mayo Clin Proc 2013; 88:315–325.
A March 2013 warning by the US Food and Drug Administration that azithromycin (Zithromax, Zmax, Z-pak) may increase the risk of sudden cardiac death does not mean we must abandon using it. We should, however, try to determine if our patients have cardiovascular risk factors for this extreme side effect and take appropriate precautions.
AZITHROMYCIN: THE SAFEST OF THE MACROLIDES?
Azithromycin, a broad-spectrum macrolide antibiotic, is used to treat or prevent a range of common bacterial infections, including upper and lower respiratory tract infections and certain sexually transmitted diseases.
In terms of overall toxicity, azithromycin has been considered the safest of the macrolides, as it neither undergoes CYP3A4 metabolism nor inhibits CYP3A4 to any clinically meaningful degree, and therefore does not interfere with the array of commonly used medications that undergo CYP3A4 metabolism.
Also, in vitro, azithromycin shows only limited blockade of the potassium channel hERG. This channel is critically involved in cardiomyocyte repolarization, and if it is blocked or otherwise malfunctioning, the result can be a prolonged QT interval, ventricular arrhythmias, and even sudden cardiac death.1–4 Therefore, lack of blockade, as reflected by a high inhibitory concentration (Table 1), boded well for the safety of azithromycin in terms of QT liability. However, we should be cautious in interpreting in vitro data.
With its broad antibiotic spectrum and perceived favorable safety profile, azithromycin has become one of the top 15 most prescribed drugs and the best-selling antibiotic in the United States, accounting for 55.4 million prescriptions in 2012, according to the IMS Institute for Healthcare Informatics.
THE FDA RECEIVES 203 REPORTS OF ADVERSE EVENTS IN 8 YEARS
However, beginning with a report of azithromycin-triggered torsades de pointes in 2001,5 a growing body of evidence, derived from postmarketing surveillance, has linked azithromycin to cardiac arrhythmias such as pronounced QT interval prolongation and associated torsades de pointes (which can progress to life-threatening ventricular fibrillation). Other, closely related macrolides such as clarithromycin and erythromycin are also linked to these effects.
Furthermore, in the 8-year period from 2004 to 2011, the US Food and Drug Administration (FDA) Adverse Event Reporting System (FAERS) received a total of 203 reports of azithromycin-associated QT prolongation, torsades de pointes, ventricular arrhythmia, or, in 65 cases, sudden cardiac death (Table 1).6
At face value, the number of FAERS reports appears to be similar between the various macrolide antibiotics. However, it is important to remember that these drugs differ substantially in the number of prescriptions written for them, with azithromycin being prescribed more often. Also, the FAERS numbers are subject to a number of well-known limitations such as confounding variables, uneven quality and completeness of reports, duplication, and underreporting. These limitations preclude the use of such adverse reporting databases in calculating and thereby comparing the true incidence of adverse events associated with the various macrolide antibiotics.6–9
RAY ET AL FIND A HIGHER RISK OF CARDIOVASCULAR DEATH
Despite these inherent flaws, initial postmarketing surveillance reports cast enough doubt on the long-standing notion that azithromycin is the safest macrolide antibiotic to prompt Ray et al10 to assess its safety in an observational, nonrandomized study of people enrolled in the Tennessee Medicaid program.
They found that, over the typical 5 days of therapy, people taking azithromycin had a rate of cardiovascular death 2.88 times higher than in people taking no antibiotic, and 2.49 times higher than in people taking amoxicillin (Table 2).
However, the absolute excess risk compared with amoxicillin varied considerably according to baseline risk score for cardiovascular disease, with 1 excess cardiovascular death per 4,100 in the highest-risk decile compared with 1 excess cardiovascular death per 100,000 in the lowest-risk decile.10,11
Moreover, the increase in deaths did not persist after the 5 days of therapy. This time-limited pattern directly correlated with expected peak azithromycin plasma levels during a standard 5-day course.
Ray et al used appropriate analytic methods to attempt to correct for any confounding bias intrinsic to the observational, nonrandomized study design. Nevertheless, the patients were Medicaid beneficiaries, who have a higher prevalence of comorbid conditions and higher mortality rates than the general population. Therefore, legitimate questions were raised about whether the results of the study could be generalized to populations with substantially lower baseline risk of cardiovascular disease and if differences in the baseline characteristics of the treatment groups were adequately controlled.12,13
THE FDA REVISES AZITHROMYCIN’S WARNINGS AND PRECAUTIONS
The striking observations by Ray et al,10 coupled with the concerns raised by postmarketing surveillance reports, compelled the FDA to review the labels of azithromycin and other macrolide antibiotics.
Ultimately, the FDA opted to revise the “warning and precautions” section of the azithromycin drug label to include a warning about the potential risk of fatal arrhythmias, specifically QT interval prolongation and torsades de pointes. In a March 2013 safety announcement, it also urged health care professionals to use caution when prescribing azithromycin to patients known to have risk factors for drug-related arrhythmias, including congenital long QT syndrome, acquired QT interval prolongation, hypokalemia, hypomagnesia, bradycardia, and concurrent use of other medications known to prolong the QT interval, specifically the class IA (eg, quinidine and procainamide) and class III (eg, amiodarone, sotalol, and dofetilide) antiarrhythmics.
SVANSTRÖM ET AL FIND NO INCREASED RISK
However, just when the medical community appeared ready to accept that azithromycin may not be as safe as we thought it was, a large prospective study by Svanström et al, published in early May 2013, found no increased risk of cardiovascular death associated with azithromycin (Table 2).14
The patients were a representative population of young to middle-aged Danish adults at low baseline risk of underlying cardiovascular disease.
Interestingly, Svanström et al were careful to point out that their study was only powered to rule out a moderate-to-high (> 55%) increase in the relative risk of cardiovascular death. Furthermore, profound differences existed in the baseline risk of death and cardiovascular risk factors between their patients and the Tennessee Medicaid patients studied by Ray et al.14 Therefore, the authors suggested that their study complements rather than contradicts the study by Ray et al. They attributed the differences in the findings to treatment-effect heterogeneity, in which the risk of azithromycin-associated cardiovascular mortality is largely limited to high-risk patients, namely those with multiple preexisting cardiovascular risk factors.14
ACC/AHA RECOMMENDATION: IDENTIFY THOSE AT RISK
Collectively, the data reviewed above provide compelling evidence that azithromycin is not completely free of the QT-prolonging and torsadogenic effects that have long been associated with other macrolide antibiotics. However, the findings from both the study by Ray et al and that of Svanström et al suggest that preexisting cardiovascular risk factors play a prominent role in determining the incidence of azithromycin-associated cardiovascular death in a given population (Table 2).10,14
These findings should prompt physicians to carefully reassess the risks and benefits of azithromycin use in their clinical practices. They also reinforce a recent call by the American Heart Association (AHA) and American College of Cardiology (ACC) to better identify, early on, patients at risk of drug-induced ventricular arrhythmias and sudden death and to subsequently improve how these patients are monitored when the use of QT-prolonging and torsadogenic drugs is medically necessary.15
AN ELECTRONIC MEDICAL RECORD FLAGS QTc ≥ 500 MS
On the heels of these AHA/ACC suggestions, our hospital has adopted an institution-wide QT alert system. Here, the electronic medical record system (Centricity EMR; GE Healthcare) uses a proprietary algorithm to detect and electronically alert ordering physicians when a patient has a prolonged QT interval, and gives information about the potential clinical significance of this electrocardiographic finding.16 Physicians also receive a warning when ordering QT-prolonging drugs in patients at risk.
This system is still in its infancy, but it has already confirmed that a prolonged QT interval (QTc ≥ 500 ms) is a powerful predictor of death from any cause and has demonstrated that mortality rates in those with prolonged QT intervals increase in a dose-dependent fashion with the patient’s number of modifiable risk factors (eg, electrolyte disturbances or QT-prolonging medications) and nonmodifiable risk factors (eg, genetic disposition, female sex, structural heart disease, diabetes mellitus).16 We have also found evidence that modifiable risk factors may have a more pronounced effect on mortality risk than non-modifiable risk factors.16
These findings suggest that information technology-based QT alert systems may one day provide physicians with an important tool to efficiently identify and possibly even modify the risk of cardiovascular death in patients at high risk, for example, by correcting electrolyte abnormalities or reducing the burden of QT-prolonging medications.
CONSIDER RISK OF QT PROLONGATION WHEN PRESCRIBING AZITHROMYCIN
For most institutions and clinical practices, such electronic QT alert systems are still years if not decades away. However, in light of the information summarized above, all physicians should begin considering risk factors for QT prolongation and torsades de pointes (summarized in Table 3) and weighing the risks and benefits of prescribing azithromycin vs alternative antibiotics with minimal QT liability. This should be relatively simple to do. Things to keep in mind:
- Although azithromycin may increase the relative risk of a cardiovascular event, for most otherwise-healthy patients, the absolute risk is miniscule.
- In a patient at risk (eg, with baseline QT prolongation or multiple risk factors for it), if azithromycin or another QT-prolonging antibiotic such as a macrolide or fluoroquinolone is medically necessary due to preferential bacterial susceptibility or patient allergies, every effort should be made to correct modifiable risk factors (eg, electrolyte abnormalities) and, if possible, to avoid polypharmacy with multiple QT-prolonging drugs.
- For patients who have multiple risk factors for QT prolongation in whom treatment with a known QT-prolonging medication is still deemed in the patient’s best interest, strong consideration should be given to inpatient administration and monitoring until the treatment has been completed.
With careful consideration of modifiable and nonmodifiable risk factors as well as a little extra caution when prescribing potential QT-prolonging medications such as azithromycin, the clinical benefit of these often-advantageous medications can be maximized and the incidence of these tragic but rare drug-induced sudden cardiac deaths can be reduced.
A March 2013 warning by the US Food and Drug Administration that azithromycin (Zithromax, Zmax, Z-pak) may increase the risk of sudden cardiac death does not mean we must abandon using it. We should, however, try to determine if our patients have cardiovascular risk factors for this extreme side effect and take appropriate precautions.
AZITHROMYCIN: THE SAFEST OF THE MACROLIDES?
Azithromycin, a broad-spectrum macrolide antibiotic, is used to treat or prevent a range of common bacterial infections, including upper and lower respiratory tract infections and certain sexually transmitted diseases.
In terms of overall toxicity, azithromycin has been considered the safest of the macrolides, as it neither undergoes CYP3A4 metabolism nor inhibits CYP3A4 to any clinically meaningful degree, and therefore does not interfere with the array of commonly used medications that undergo CYP3A4 metabolism.
Also, in vitro, azithromycin shows only limited blockade of the potassium channel hERG. This channel is critically involved in cardiomyocyte repolarization, and if it is blocked or otherwise malfunctioning, the result can be a prolonged QT interval, ventricular arrhythmias, and even sudden cardiac death.1–4 Therefore, lack of blockade, as reflected by a high inhibitory concentration (Table 1), boded well for the safety of azithromycin in terms of QT liability. However, we should be cautious in interpreting in vitro data.
With its broad antibiotic spectrum and perceived favorable safety profile, azithromycin has become one of the top 15 most prescribed drugs and the best-selling antibiotic in the United States, accounting for 55.4 million prescriptions in 2012, according to the IMS Institute for Healthcare Informatics.
THE FDA RECEIVES 203 REPORTS OF ADVERSE EVENTS IN 8 YEARS
However, beginning with a report of azithromycin-triggered torsades de pointes in 2001,5 a growing body of evidence, derived from postmarketing surveillance, has linked azithromycin to cardiac arrhythmias such as pronounced QT interval prolongation and associated torsades de pointes (which can progress to life-threatening ventricular fibrillation). Other, closely related macrolides such as clarithromycin and erythromycin are also linked to these effects.
Furthermore, in the 8-year period from 2004 to 2011, the US Food and Drug Administration (FDA) Adverse Event Reporting System (FAERS) received a total of 203 reports of azithromycin-associated QT prolongation, torsades de pointes, ventricular arrhythmia, or, in 65 cases, sudden cardiac death (Table 1).6
At face value, the number of FAERS reports appears to be similar between the various macrolide antibiotics. However, it is important to remember that these drugs differ substantially in the number of prescriptions written for them, with azithromycin being prescribed more often. Also, the FAERS numbers are subject to a number of well-known limitations such as confounding variables, uneven quality and completeness of reports, duplication, and underreporting. These limitations preclude the use of such adverse reporting databases in calculating and thereby comparing the true incidence of adverse events associated with the various macrolide antibiotics.6–9
RAY ET AL FIND A HIGHER RISK OF CARDIOVASCULAR DEATH
Despite these inherent flaws, initial postmarketing surveillance reports cast enough doubt on the long-standing notion that azithromycin is the safest macrolide antibiotic to prompt Ray et al10 to assess its safety in an observational, nonrandomized study of people enrolled in the Tennessee Medicaid program.
They found that, over the typical 5 days of therapy, people taking azithromycin had a rate of cardiovascular death 2.88 times higher than in people taking no antibiotic, and 2.49 times higher than in people taking amoxicillin (Table 2).
However, the absolute excess risk compared with amoxicillin varied considerably according to baseline risk score for cardiovascular disease, with 1 excess cardiovascular death per 4,100 in the highest-risk decile compared with 1 excess cardiovascular death per 100,000 in the lowest-risk decile.10,11
Moreover, the increase in deaths did not persist after the 5 days of therapy. This time-limited pattern directly correlated with expected peak azithromycin plasma levels during a standard 5-day course.
Ray et al used appropriate analytic methods to attempt to correct for any confounding bias intrinsic to the observational, nonrandomized study design. Nevertheless, the patients were Medicaid beneficiaries, who have a higher prevalence of comorbid conditions and higher mortality rates than the general population. Therefore, legitimate questions were raised about whether the results of the study could be generalized to populations with substantially lower baseline risk of cardiovascular disease and if differences in the baseline characteristics of the treatment groups were adequately controlled.12,13
THE FDA REVISES AZITHROMYCIN’S WARNINGS AND PRECAUTIONS
The striking observations by Ray et al,10 coupled with the concerns raised by postmarketing surveillance reports, compelled the FDA to review the labels of azithromycin and other macrolide antibiotics.
Ultimately, the FDA opted to revise the “warning and precautions” section of the azithromycin drug label to include a warning about the potential risk of fatal arrhythmias, specifically QT interval prolongation and torsades de pointes. In a March 2013 safety announcement, it also urged health care professionals to use caution when prescribing azithromycin to patients known to have risk factors for drug-related arrhythmias, including congenital long QT syndrome, acquired QT interval prolongation, hypokalemia, hypomagnesia, bradycardia, and concurrent use of other medications known to prolong the QT interval, specifically the class IA (eg, quinidine and procainamide) and class III (eg, amiodarone, sotalol, and dofetilide) antiarrhythmics.
SVANSTRÖM ET AL FIND NO INCREASED RISK
However, just when the medical community appeared ready to accept that azithromycin may not be as safe as we thought it was, a large prospective study by Svanström et al, published in early May 2013, found no increased risk of cardiovascular death associated with azithromycin (Table 2).14
The patients were a representative population of young to middle-aged Danish adults at low baseline risk of underlying cardiovascular disease.
Interestingly, Svanström et al were careful to point out that their study was only powered to rule out a moderate-to-high (> 55%) increase in the relative risk of cardiovascular death. Furthermore, profound differences existed in the baseline risk of death and cardiovascular risk factors between their patients and the Tennessee Medicaid patients studied by Ray et al.14 Therefore, the authors suggested that their study complements rather than contradicts the study by Ray et al. They attributed the differences in the findings to treatment-effect heterogeneity, in which the risk of azithromycin-associated cardiovascular mortality is largely limited to high-risk patients, namely those with multiple preexisting cardiovascular risk factors.14
ACC/AHA RECOMMENDATION: IDENTIFY THOSE AT RISK
Collectively, the data reviewed above provide compelling evidence that azithromycin is not completely free of the QT-prolonging and torsadogenic effects that have long been associated with other macrolide antibiotics. However, the findings from both the study by Ray et al and that of Svanström et al suggest that preexisting cardiovascular risk factors play a prominent role in determining the incidence of azithromycin-associated cardiovascular death in a given population (Table 2).10,14
These findings should prompt physicians to carefully reassess the risks and benefits of azithromycin use in their clinical practices. They also reinforce a recent call by the American Heart Association (AHA) and American College of Cardiology (ACC) to better identify, early on, patients at risk of drug-induced ventricular arrhythmias and sudden death and to subsequently improve how these patients are monitored when the use of QT-prolonging and torsadogenic drugs is medically necessary.15
AN ELECTRONIC MEDICAL RECORD FLAGS QTc ≥ 500 MS
On the heels of these AHA/ACC suggestions, our hospital has adopted an institution-wide QT alert system. Here, the electronic medical record system (Centricity EMR; GE Healthcare) uses a proprietary algorithm to detect and electronically alert ordering physicians when a patient has a prolonged QT interval, and gives information about the potential clinical significance of this electrocardiographic finding.16 Physicians also receive a warning when ordering QT-prolonging drugs in patients at risk.
This system is still in its infancy, but it has already confirmed that a prolonged QT interval (QTc ≥ 500 ms) is a powerful predictor of death from any cause and has demonstrated that mortality rates in those with prolonged QT intervals increase in a dose-dependent fashion with the patient’s number of modifiable risk factors (eg, electrolyte disturbances or QT-prolonging medications) and nonmodifiable risk factors (eg, genetic disposition, female sex, structural heart disease, diabetes mellitus).16 We have also found evidence that modifiable risk factors may have a more pronounced effect on mortality risk than non-modifiable risk factors.16
These findings suggest that information technology-based QT alert systems may one day provide physicians with an important tool to efficiently identify and possibly even modify the risk of cardiovascular death in patients at high risk, for example, by correcting electrolyte abnormalities or reducing the burden of QT-prolonging medications.
CONSIDER RISK OF QT PROLONGATION WHEN PRESCRIBING AZITHROMYCIN
For most institutions and clinical practices, such electronic QT alert systems are still years if not decades away. However, in light of the information summarized above, all physicians should begin considering risk factors for QT prolongation and torsades de pointes (summarized in Table 3) and weighing the risks and benefits of prescribing azithromycin vs alternative antibiotics with minimal QT liability. This should be relatively simple to do. Things to keep in mind:
- Although azithromycin may increase the relative risk of a cardiovascular event, for most otherwise-healthy patients, the absolute risk is miniscule.
- In a patient at risk (eg, with baseline QT prolongation or multiple risk factors for it), if azithromycin or another QT-prolonging antibiotic such as a macrolide or fluoroquinolone is medically necessary due to preferential bacterial susceptibility or patient allergies, every effort should be made to correct modifiable risk factors (eg, electrolyte abnormalities) and, if possible, to avoid polypharmacy with multiple QT-prolonging drugs.
- For patients who have multiple risk factors for QT prolongation in whom treatment with a known QT-prolonging medication is still deemed in the patient’s best interest, strong consideration should be given to inpatient administration and monitoring until the treatment has been completed.
With careful consideration of modifiable and nonmodifiable risk factors as well as a little extra caution when prescribing potential QT-prolonging medications such as azithromycin, the clinical benefit of these often-advantageous medications can be maximized and the incidence of these tragic but rare drug-induced sudden cardiac deaths can be reduced.
- Hopkins S. Clinical toleration and safety of azithromycin. Am J Med 1991; 91:40S–45S.
- Milberg P, Eckardt L, Bruns HJ, et al. Divergent proarrhythmic potential of macrolide antibiotics despite similar QT prolongation: fast phase 3 repolarization prevents early afterdepolarizations and torsade de pointes. J Pharmacol Exp Ther 2002; 303:218–225.
- Ioannidis JP, Contopoulos-Ioannidis DG, Chew P, Lau J. Meta-analysis of randomized controlled trials on the comparative efficacy and safety of azithromycin against other antibiotics for upper respiratory tract infections. J Antimicrob Chemother 2001; 48:677–689.
- Owens RC, Nolin TD. Antimicrobial-associated QT interval prolongation: pointes of interest. Clin Infect Dis 2006; 43:1603–1611.
- Arellano-Rodrigo E, García A, Mont L, Roqué M. Torsade de pointes and cardiorespiratory arrest induced by azithromycin in a patient with congenital long QT syndrome. (Article in Spanish.) Med Clin (Barc) 2001; 117:118–119.
- Raschi E, Poluzzi E, Koci A, Moretti U, Sturkenboom M, De Ponti F. Macrolides and torsadogenic risk: emerging issues from the fda pharmacovigilance database. J Pharmacovigilance 2013; 1:104.
- Shaffer D, Singer S, Korvick J, Honig P. Concomitant risk factors in reports of torsades de pointes associated with macrolide use: review of the United States Food and Drug Administration Adverse Event Reporting System. Clin Infect Dis 2002; 35:197–200.
- Stephenson WP, Hauben M. Data mining for signals in spontaneous reporting databases: proceed with caution. Pharmacoepidemiol Drug Saf 2007; 16:359–365.
- Bate A, Evans SJ. Quantitative signal detection using spontaneous ADR reporting. Pharmacoepidemiol Drug Saf 2009; 18:427–436.
- Ray WA, Murray KT, Hall K, Arbogast PG, Stein CM. Azithromycin and the risk of cardiovascular death. N Engl J Med 2012; 366:1881–1890.
- Mosholder AD, Mathew J, Alexander JJ, Smith H, Nambiar S. Cardiovascular risks with azithromycin and other antibacterial drugs. N Engl J Med 2013; 368:1665–1668.
- Louie R. Azithromycin and the risk of cardiovascular death. N Engl J Med 2012; 367:774–775.
- Koga T, Imaoka H. Azithromycin and the risk of cardiovascular death. N Engl J Med 2012; 367:774–775.
- Svanström H, Pasternak B, Hviid A. Use of azithromycin and death from cardiovascular causes. N Engl J Med 2013; 368:1704–1712.
- Drew BJ, Ackerman MJ, Funk M, et al; American Heart Association Acute Cardiac Care Committee of the Council on Clinical Cardiology, the Council on Cardiovascular Nursing, and the American College of Cardiology Foundation. Prevention of torsade de pointes in hospital settings: a scientific statement from the American Heart Association and the American College of Cardiology Foundation. Circulation 2010; 121:1047–1060.
- Haugaa KH, Bos JM, Tarrell RF, Morlan BW, Caraballo PJ, Ackerman MJ. Institution-wide QT alert system identifies patients with a high risk of mortality. Mayo Clin Proc 2013; 88:315–325.
- Hopkins S. Clinical toleration and safety of azithromycin. Am J Med 1991; 91:40S–45S.
- Milberg P, Eckardt L, Bruns HJ, et al. Divergent proarrhythmic potential of macrolide antibiotics despite similar QT prolongation: fast phase 3 repolarization prevents early afterdepolarizations and torsade de pointes. J Pharmacol Exp Ther 2002; 303:218–225.
- Ioannidis JP, Contopoulos-Ioannidis DG, Chew P, Lau J. Meta-analysis of randomized controlled trials on the comparative efficacy and safety of azithromycin against other antibiotics for upper respiratory tract infections. J Antimicrob Chemother 2001; 48:677–689.
- Owens RC, Nolin TD. Antimicrobial-associated QT interval prolongation: pointes of interest. Clin Infect Dis 2006; 43:1603–1611.
- Arellano-Rodrigo E, García A, Mont L, Roqué M. Torsade de pointes and cardiorespiratory arrest induced by azithromycin in a patient with congenital long QT syndrome. (Article in Spanish.) Med Clin (Barc) 2001; 117:118–119.
- Raschi E, Poluzzi E, Koci A, Moretti U, Sturkenboom M, De Ponti F. Macrolides and torsadogenic risk: emerging issues from the fda pharmacovigilance database. J Pharmacovigilance 2013; 1:104.
- Shaffer D, Singer S, Korvick J, Honig P. Concomitant risk factors in reports of torsades de pointes associated with macrolide use: review of the United States Food and Drug Administration Adverse Event Reporting System. Clin Infect Dis 2002; 35:197–200.
- Stephenson WP, Hauben M. Data mining for signals in spontaneous reporting databases: proceed with caution. Pharmacoepidemiol Drug Saf 2007; 16:359–365.
- Bate A, Evans SJ. Quantitative signal detection using spontaneous ADR reporting. Pharmacoepidemiol Drug Saf 2009; 18:427–436.
- Ray WA, Murray KT, Hall K, Arbogast PG, Stein CM. Azithromycin and the risk of cardiovascular death. N Engl J Med 2012; 366:1881–1890.
- Mosholder AD, Mathew J, Alexander JJ, Smith H, Nambiar S. Cardiovascular risks with azithromycin and other antibacterial drugs. N Engl J Med 2013; 368:1665–1668.
- Louie R. Azithromycin and the risk of cardiovascular death. N Engl J Med 2012; 367:774–775.
- Koga T, Imaoka H. Azithromycin and the risk of cardiovascular death. N Engl J Med 2012; 367:774–775.
- Svanström H, Pasternak B, Hviid A. Use of azithromycin and death from cardiovascular causes. N Engl J Med 2013; 368:1704–1712.
- Drew BJ, Ackerman MJ, Funk M, et al; American Heart Association Acute Cardiac Care Committee of the Council on Clinical Cardiology, the Council on Cardiovascular Nursing, and the American College of Cardiology Foundation. Prevention of torsade de pointes in hospital settings: a scientific statement from the American Heart Association and the American College of Cardiology Foundation. Circulation 2010; 121:1047–1060.
- Haugaa KH, Bos JM, Tarrell RF, Morlan BW, Caraballo PJ, Ackerman MJ. Institution-wide QT alert system identifies patients with a high risk of mortality. Mayo Clin Proc 2013; 88:315–325.
Clinical applications of pharmacogenetics: Present and near future
“Change is the only constant.”
—Heraclitus (c 535–475 bce)
With the cost of health care rising and money to pay for it shrinking, there has never been a greater need to reduce waste.
Ineffective treatments and adverse drug effects account for much preventable morbidity and expense. New treatments, touted as more potent, are often introduced as replacements for traditional ones that are still effective in many patients, adding to costs and the potential for harm. For the pharmaceutical industry, the search for new “blockbuster” drugs seems to have hit a wall, at least in cardiovascular medicine.1 Advances often come at the cost of adverse effects, such as bleeding with triple antiplatelet therapy and diabetes with potent statin drugs.
The path to maximizing benefit and reducing harm now appears to lie in stratifying populations and appreciating patient individuality in response to treatment. For many decades we have known that patients vary widely in their response to drugs, owing to personal factors such as body surface area, age, environment, and genetics. And indeed, we treat our patients as individuals, for example by tailoring aminoglycoside dose to weight and renal function.
However, clinical trials typically give us an idea of the benefits only to the average patient. While subgroup analyses identify groups that may benefit more or less from treatment, the additional information they provide is not easily integrated into the clinical model of prescribing, in which one size fits all.
THE PROMISE OF PHARMACOGENETICS
The emerging field of pharmacogenetics promises to give clinicians the tools to make informed treatment decisions based on predictive genetic testing. This genetic testing aims to match treatment to an individual’s genetic profile. This often involves analyzing single-nucleotide polymorphisms in genes for enzymes that metabolize drugs, such as the cytochrome P450 enzymes, to predict efficacy or an adverse event with treatment.
Pharmacogenetics is playing an increasing role in clinical trials, particularly in the early stages of drug development, by helping to reduce the number of patients needed, prove efficacy, and identify subgroups in which alternative treatment can be targeted. At another level, a molecular understanding of disease is leading to truly targeted treatments based on genomics.
Over recent years, genetic testing has been increasingly used in clinical practice, thanks to a convergence of factors such as rapid, low-cost tests, a growing evidence base, and emerging interest among doctors and payers.
An advantage to using genetic testing as opposed to other types of laboratory testing, such as measuring the concentration of the drug in the blood during treatment, is that genetic tests can predict the response to treatment before the treatment is started. Moreover, with therapeutic drug monitoring after treatment has begun, there are sometimes no detectable measures of toxicity. For example, both carbamazepine and the antiviral drug abacavir can—fortunately only rarely—cause Stevens-Johnson syndrome. But before genetic markers were discovered, there was no method of estimating this risk apart from taking a family history.2,3 Considering the numbers of people involved, it was not feasible until recently to suggest genetic screening for patients starting on these drugs. However, the cost of genotyping and gene sequencing has been falling at a rate inversely faster than Moore’s law (an approximate annual doubling in computer power), and population genomics is becoming a reality.4
The US Food and Drug Administration (FDA) recognizes the current and future value of pharmacogenetics in drug safety and development. A number of approved pharmacogenetic biomarkers are listed on the FDA website (Table 1). Black box warnings have been mandated for a number of drugs on the basis of observational evidence.
The FDA also promotes rapid approval for novel drugs with pharmacogenetic “companion diagnostics.” A recent example of this was the approval of ivacaftor for cystic fibrosis patients who have the G551D mutation.5 Here, a molecular understanding of the condition led to the development of a targeted treatment. Although the cost of developing this drug was high, the path is now paved for similar advances. Oncologists are familiar with these advances with the emergence of new molecularly targeted treatments, eg, BRAF inhibitors in metastatic melanoma, imatinib in chronic myeloid leukemia, and gefitinib in non-small-cell lung cancer.
PHARMACOGENETICS IN CARDIOVASCULAR MEDICINE
Cardiovascular medicine also stands to benefit from rapid advances in pharmacogenetics.
While no treatment has been developed that targets the molecular basis of cardiovascular disease, a number of genomic biomarkers have emerged that identify patients at risk of adverse reactions or treatment failure. These include genetic tests to predict the maintenance dose and risk of bleeding with warfarin,6 the likelihood of myopathy and myositis with simvastatin,7 and the risk of recurrent thrombotic events with clopidogrel.8–10
Using pharmacogenetics in prescribing warfarin and its alternatives
The pharmacogenetics of warfarin has been extensively researched, but genotyping before prescribing this drug is not yet widely done.
In 2007, the FDA updated the labeling of warfarin to include information about the influence of two genes, VKORC1 and CYP2C9, on a patient’s response to this drug. In 2010 this was updated to add that testing for these genes could be used to predict the maintenance dose of the drug. Difficulties with algorithms used to integrate this into clinical practice have hindered adoption of this testing.
With the advent of new anticoagulants such as dabigatran, rivaroxaban, and apixaban, many have expected warfarin and its pharmacogenetics to become obsolete. However, the new agents cost considerably more. Further, they may not offer a very great advantage over warfarin: in the Randomized Evaluation of Long-term Anticoagulant Therapy (RE-LY) trial, the absolute risk reduction in intracranial hemorrhage with dabigatran vs warfarin was small.11 Therefore, dabigatran is probably not cost-effective in populations at low risk of bleeding.12 A cost-effectiveness analysis comparing warfarin with dabigatran in patients with uncomplicated atrial fibrillation has suggested that dabigatran is, however, cost-effective in patients at moderate risk.12
In the RE-LY trial, the international normalized ratios (INRs) of the patients in the warfarin group were in the therapeutic range only 64% of the time. The advantages of dabigatran over warfarin become less pronounced as warfarin control is tightened.13 Of note, pharmacogenetics and home monitoring of the INR have both been shown to lead to tighter control of the INR, with greater time within the therapeutic range.14,15
Moreover, genetic testing can help us reduce the number of bleeding events in patients taking warfarin.16 Patients who carry the CYP2C9*2 or CYP2C9*3 polymorphism metabolize S-warfarin slower and therefore have a threefold higher risk of hemorrhage after starting warfarin.17 We could speculate that patients carrying these variants may be better served by the newer anticoagulants, though this has not been tested in any clinical trial.
It is also worth appreciating that the conditions requiring anticoagulation, such as atrial fibrillation, also have a strong genetic basis. Variants in chromosomes 4q25, 1q21, and 16q22 have all been associated with atrial fibrillation.18 The risk of atrial fibrillation is five to six times higher in carriers of multiple variants within all of these loci.19 Genetic variants at 4q25 have been associated with the response to specific antiarrhythmic drug treatments,20 response to pulmonary vein isolation, 21,22 and direct-current cardioversion.23 One can imagine a future in which patients with palpitations, carrying multiple gene risk variants, will choose prolonged monitoring at home to confirm a diagnosis. They would then be provided with a personalized best management strategy, using their personal preferences, clinical data, and genetic profile to make a treatment decision.
Using pharmacogenetics in prescribing clopidogrel and its alternatives
The pharmacogenetics of clopidogrel is of particular interest, as it has the potential of establishing a rational basis for using newer antiplatelet drugs such as ticagrelor and prasugrel, which are considerably more expensive than generic clopidogrel.
Most of the people who do not respond to clopidogrel carry the common cytochrome P450 2C19 variants CYP2C19*2 or CYP2C19*3.9 These variants are present in particularly high frequency in Asians and African Americans, who often do not feature in large randomized trials.
Newer antiplatelet agents have failed to demonstrate consistent superiority to clopidogrel without a tradeoff of more bleeding. However, in the Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition With Prasugrel-Thrombolysis in Myocardial Infarction 39 (TRITON TIMI-38),24,25 patients with the *2 variant receiving prasugrel had lower cardiovascular event rates than *2 carriers receiving clopidogrel.
Similarly, the patients who benefit the most from ticagrelor are carriers of the 2C19 nonresponder variants. In a large study, clopidogrel responders who did not carry either 2C19 nonresponder genetic variants or ABCB1 variants had cardiovascular outcomes similar to those of patients receiving ticagrelor.26
Clinicians have been cautious in prescribing potent antiplatelet agents to all patients because of the risk of bleeding. One could assume that by reserving newer agents for clopidogrel nonresponders, the bleeding risk could be minimized and overall benefit could be preserved with this strategy.
Cost may also be contained. The cost-effectiveness of such an approach with prasugrel has been tested with computer modeling and appears favorable.27 On the other hand, a similar yet limited analysis did not find genotype-driven use of ticagrelor to be cost-effective.28 This was mostly due to fewer deaths in patients receiving ticagrelor. However, the cost estimate for genotype-guided therapy was overestimated, as heterozygotes in the model were treated with ticagrelor instead of a high dose of clopidogrel.
It now appears that heterozygotes, ie, patients with one copy of the nonresponder variant, can achieve similar platelet inhibition with clopidogrel 225 mg daily as noncarriers on 75 mg daily.29 Since genotype-guided antiplatelet therapy has not been tested in a randomized outcomes trial, this tailored strategy has not been widely accepted.
THE FUTURE
The barriers to adoption of pharmacogenetics are considerable. Clinicians need to be educated about it, reimbursement needs to be worked out, and the pharmaceutical industry needs to get behind it. Nevertheless, the future of pharmacogenetics is extremely promising.
Research networks are forming to support the use of pharmacogenetics in clinical practice. The Pharmgkb (www.pharmgkb.org) database serves as a hub for educating clinicians and researchers as well as curating data for reference. Vanderbilt University is piloting the BioVu project, in which DNA and genotype data on patients are being stored and matched to the electronic clinical records.30 These projects not only provide clinically useful information on the current state of the art of pharmacogenetics, they also aid in disseminating new information about genotype-phenotype relationships.
Analytical software that uses “natural language processing” is being applied to clinician-generated notes to derive new observations and associations between genetic variants and clinical phenotypes. Integrating this information in real-time decision-support modules in the electronic health record provides a feedback loop for a rapid assimilation of new knowledge. Similar innovative decision-support modules are being established by Cleveland Clinic’s Center for Personalized Healthcare.31
The rise of ‘omic’ sciences
Pharmacogenetics and pharmacogenomics are part of a larger set of “omic” sciences. The suffix “-omics” implies a larger, more holistic view and is being applied to a number of fields—for example, the study of proteins (proteomics) and the study of metabolites (metabolomics). Profiling proteins and metabolites delivers a deluge of information on a patient that can be clustered, using pattern-recognition software, into population subgroups. Patterns of multiple proteins or metabolites are extracted from this spectral data to identify disease or response to treatment (pharmacometabolomics).
Metabolomics has been shown to predict the response to statins,32 diagnose myocardial infarction,33 and reclassify cardiovascular risk status.34 In addition, whereas traditional laboratory chemistry is reductionist, using single biomarkers for single-disease diagnosis, omic technologies hold the potential to reveal information on a number of possible health or disease states. The identification of “healthy” profiles using these technologies can potentially provide positive feedback to patients undertaking lifestyle changes and treatment.
The instrument costs for proteomic and metabolomic profiling are relatively high. However, the ongoing running costs are minimal, estimated at as low as less than $13 per test, as there are no expensive reagents.33 High-volume testing therefore becomes very cost-effective.
Although omic science appears futuristic, proteomics and metabolomics are already used in many clinical laboratories to rapidly identify bacteria. These methods have already revolutionized the way laboratories identify microbes, since they are automated, reduce workload, and give very fast results.
The cost of genetic testing is falling
Critics of pharmacogenetics claim that the predictive value of genetic testing is poor, that evidence is lacking, and that the cost is too high. In all new technologies, the first iteration is coarse, but performance improves with use. The first major barrier is adoption. Projects like BioVu are establishing the infrastructure for a feedback loop to iteratively improve upon the status quo and provide the evidence base clinicians demand.
The cost of genetic testing is falling rapidly, with whole-genome sequencing and annotation now costing less than $5,000. The cost of a pharmacogenetic test can be as low as $100 using low-cost nanotechnology, and the test needs to be performed only once in a patient’s lifetime.27
As other related molecular technologies such as proteomics and metabolomics become available and are integrated with genomics, the predictive ability of this science will improve.
AWAY FROM ONE-SIZE-FITS-ALL MEDICINE
Over the last decade there has been a trend away from “one size fits all” to customized “markets of one” in everything from consumer products to education to medicine. Mass customizing, also known as personalization, has been embraced by the internet community as a means to increase efficiency and reduce cost. This occurs by eliminating waste in redundant work or production of ineffective products.
Personalization on the Internet has been enabled through the use of informatics, mathematics, and supercomputing. The same tools that have personalized the delivery of consumer products are also being applied to the field of pharmacogenetics. Applied in an evidence-based fashion, these new technologies should profoundly improve patient care now and in the future.
- Topol EJ. Past the wall in cardiovascular R&D. Nat Rev Drug Discov 2009; 8:259.
- Mallal S, Phillips E, Carosi G, et al; PREDICT-1 Study Team. HLA-B*5701 screening for hypersensitivity to abacavir. N Engl J Med 2008; 358:568–579.
- Hung SI, Chung WH, Jee SH, et al. Genetic susceptibility to carbamazepine-induced cutaneous adverse drug reactions. Pharmacogenet Genomics 2006; 16:297–306.
- Phimister EG, Feero WG, Guttmacher AE. Realizing genomic medicine. N Engl J Med 2012; 366:757–759.
- Van Goor F, Hadida S, Grootenhuis PD, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci USA 2009; 106:18825–18830.
- International Warfarin Pharmacogenetics Consortium; Klein TE, Altman RB, Eriksson N, et al. Estimation of the warfarin dose with clinical and pharmacogenetic data. N Engl J Med 2009; 360:753–764.
- SEARCH Collaborative Group; Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin-induced myopathy—a genomewide study. N Engl J Med 2008; 359:789–799.
- Simon T, Verstuyft C, Mary-Krause M, et al; French Registry of Acute ST-Elevation and Non-ST-Elevation Myocardial Infarction (FAST-MI) Investigators. Genetic determinants of response to clopidogrel and cardiovascular events. N Engl J Med 2009; 360:363–375.
- Mega JL, Close SL, Wiviott SD, et al. Cytochrome p-450 polymorphisms and response to clopidogrel. N Engl J Med 2009; 360:354–362
- Collet JP, Hulot JS, Pena A, et al. Cytochrome P450 2C19 polymorphism in young patients treated with clopidogrel after myocardial infarction: a cohort study. Lancet 2009; 373:309–317.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al; RE-LY Steering Committee and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med 2009; 361:1139–1151.
- Shah SV, Gage BF. Cost-effectiveness of dabigatran for stroke prophylaxis in atrial fibrillation. Circulation 2011; 123:2562–2570.
- Wallentin L, Yusuf S, Ezekowitz MD, et al; RE-LY investigators. Efficacy and safety of dabigatran compared with warfarin at different levels of international normalised ratio control for stroke prevention in atrial fibrillation: an analysis of the RE-LY trial. Lancet 2010; 376:975–983.
- Anderson JL, Horne BD, Stevens SM, et al; Couma-Gen Investigators. Randomized trial of genotype-guided versus standard warfarin dosing in patients initiating oral anticoagulation. Circulation 2007; 116:2563–2570.
- Matchar DB, Jacobson A, Dolor R, et al; THINRS Executive Committee and Site Investigators. Effect of home testing of international normalized ratio on clinical events. N Engl J Med 2010; 363:1608–1620.
- Epstein RS, Moyer TP, Aubert RE, et al. Warfarin genotyping reduces hospitalization rates results from the MM-WES (Medco-Mayo Warfarin Effectiveness study). J Am Coll Cardiol 2010; 55:2804–2812.
- Sanderson S, Emery J, Higgins J. CYP2C9 gene variants, drug dose, and bleeding risk in warfarin-treated patients: a HuGEnet systematic review and meta-analysis. Genet Med 2005; 7:97–104.
- Ellinor PT, Lunetta KL, Albert CM, et al. Meta-analysis identifies six new susceptibility loci for atrial fibrillation. Nat Genet 2012; 44:670–675.
- Lubitz SA, Sinner MF, Lunetta KL, et al. Independent susceptibility markers for atrial fibrillation on chromosome 4q25. Circulation 2010; 122:976–984.
- Parvez B, Vaglio J, Rowan S, et al. Symptomatic response to antiarrhythmic drug therapy is modulated by a common single nucleotide polymorphism in atrial fibrillation. J Am Coll Cardiol 2012; 60:539–545.
- Husser D, Adams V, Piorkowski C, Hindricks G, Bollmann A. Chromosome 4q25 variants and atrial fibrillation recurrence after catheter ablation. J Am Coll Cardiol 2010; 55:747–753.
- Benjamin Shoemaker M, Muhammad R, Parvez B, et al. Common atrial fibrillation risk alleles at 4q25 predict recurrence after catheter-based atrial fibrillation ablation.” Heart Rhythm 2012; Nov 23.pii: S1547-5271(12)01340–9. 10.1016/j.hrthm.2012.11.012. [Epub ahead of print]
- Parvez B, Benjamin Shoemaker M, Muhammad R, et al. Common genetic polymorphism at 4q25 locus predicts atrial fibrillation recurrence after successful cardioversion. Heart Rhythm 2013 Feb 18.pii: S1547-5271(13)00161–6. 10.1016/j.hrthm.2013.02.018. [Epub ahead of print]
- Mega JL, Close SL, Wiviott SD, et al. Genetic variants in ABCB1 and CYP2C19 and cardiovascular outcomes after treatment with clopidogrel and prasugrel in the TRITON-TIMI 38 trial: a pharmacogenetic analysis. Lancet 2010; 376:1312–1319.
- Mega JL, Close SL, Wiviott SD, et al. Cytochrome P450 genetic polymorphisms and the response to prasugrel: relationship to pharmacokinetic, pharmacodynamic, and clinical outcomes. Circulation 2009; 119:2553–2560.
- Wallentin L, James S, Storey RF, et al; PLATO investigators. Effect of CYP2C19 and ABCB1 single nucleotide polymorphisms on outcomes of treatment with ticagrelor versus clopidogrel for acute coronary syndromes: a genetic substudy of the PLATO trial. Lancet 2010; 376:1320–1328.
- Guzauskas GF, Hughes DA, Bradley SM, Veenstra DL. A risk-benefit assessment of prasugrel, clopidogrel, and genotype-guided therapy in patients undergoing percutaneous coronary intervention. Clin Pharmacol Ther 2012; 91:829–837.
- Crespin DJ, Federspiel JJ, Biddle AK, Jonas DE, Rossi JS. Ticagrelor versus genotype-driven antiplatelet therapy for secondary prevention after acute coronary syndrome: a cost-effectiveness analysis. Value Health 2011; 14:483–491.
- Mega JL, Hochholzer W, Frelinger AL, et al. Dosing clopidogrel based on CYP2C19 genotype and the effect on platelet reactivity in patients with stable cardiovascular disease. JAMA 2011; 306:2221–2228.
- Xu H, Jiang M, Oetjens M, et al. Facilitating pharmacogenetic studies using electronic health records and natural-language processing: a case study of warfarin. J Am Med Inform Assoc 2011; 18:387–391.
- Teng K, Eng C, Hess CA, et al. Building an innovative model for personalized healthcare. Cleve Clin J Med 2012; 79( suppl 1):S1–S9.
- Kaddurah-Daouk R, Baillie RA, Zhu H, et al. Enteric microbiome metabolites correlate with response to simvastatin treatment. PLoS One 2011; 6:e25482.
- Bodi V, Sanchis J, Morales JM, et al. Metabolomic profile of human myocardial ischemia by nuclear magnetic resonance spectroscopy of peripheral blood serum: a translational study based on transient coronary occlusion models. J Am Coll Cardiol 2012; 59:1629–1641.
- Shah SH, Sun JL, Stevens RD, et al. Baseline metabolomic profiles predict cardiovascular events in patients at risk for coronary artery disease. Am Heart J 2012; 163:844–850.e1.
“Change is the only constant.”
—Heraclitus (c 535–475 bce)
With the cost of health care rising and money to pay for it shrinking, there has never been a greater need to reduce waste.
Ineffective treatments and adverse drug effects account for much preventable morbidity and expense. New treatments, touted as more potent, are often introduced as replacements for traditional ones that are still effective in many patients, adding to costs and the potential for harm. For the pharmaceutical industry, the search for new “blockbuster” drugs seems to have hit a wall, at least in cardiovascular medicine.1 Advances often come at the cost of adverse effects, such as bleeding with triple antiplatelet therapy and diabetes with potent statin drugs.
The path to maximizing benefit and reducing harm now appears to lie in stratifying populations and appreciating patient individuality in response to treatment. For many decades we have known that patients vary widely in their response to drugs, owing to personal factors such as body surface area, age, environment, and genetics. And indeed, we treat our patients as individuals, for example by tailoring aminoglycoside dose to weight and renal function.
However, clinical trials typically give us an idea of the benefits only to the average patient. While subgroup analyses identify groups that may benefit more or less from treatment, the additional information they provide is not easily integrated into the clinical model of prescribing, in which one size fits all.
THE PROMISE OF PHARMACOGENETICS
The emerging field of pharmacogenetics promises to give clinicians the tools to make informed treatment decisions based on predictive genetic testing. This genetic testing aims to match treatment to an individual’s genetic profile. This often involves analyzing single-nucleotide polymorphisms in genes for enzymes that metabolize drugs, such as the cytochrome P450 enzymes, to predict efficacy or an adverse event with treatment.
Pharmacogenetics is playing an increasing role in clinical trials, particularly in the early stages of drug development, by helping to reduce the number of patients needed, prove efficacy, and identify subgroups in which alternative treatment can be targeted. At another level, a molecular understanding of disease is leading to truly targeted treatments based on genomics.
Over recent years, genetic testing has been increasingly used in clinical practice, thanks to a convergence of factors such as rapid, low-cost tests, a growing evidence base, and emerging interest among doctors and payers.
An advantage to using genetic testing as opposed to other types of laboratory testing, such as measuring the concentration of the drug in the blood during treatment, is that genetic tests can predict the response to treatment before the treatment is started. Moreover, with therapeutic drug monitoring after treatment has begun, there are sometimes no detectable measures of toxicity. For example, both carbamazepine and the antiviral drug abacavir can—fortunately only rarely—cause Stevens-Johnson syndrome. But before genetic markers were discovered, there was no method of estimating this risk apart from taking a family history.2,3 Considering the numbers of people involved, it was not feasible until recently to suggest genetic screening for patients starting on these drugs. However, the cost of genotyping and gene sequencing has been falling at a rate inversely faster than Moore’s law (an approximate annual doubling in computer power), and population genomics is becoming a reality.4
The US Food and Drug Administration (FDA) recognizes the current and future value of pharmacogenetics in drug safety and development. A number of approved pharmacogenetic biomarkers are listed on the FDA website (Table 1). Black box warnings have been mandated for a number of drugs on the basis of observational evidence.
The FDA also promotes rapid approval for novel drugs with pharmacogenetic “companion diagnostics.” A recent example of this was the approval of ivacaftor for cystic fibrosis patients who have the G551D mutation.5 Here, a molecular understanding of the condition led to the development of a targeted treatment. Although the cost of developing this drug was high, the path is now paved for similar advances. Oncologists are familiar with these advances with the emergence of new molecularly targeted treatments, eg, BRAF inhibitors in metastatic melanoma, imatinib in chronic myeloid leukemia, and gefitinib in non-small-cell lung cancer.
PHARMACOGENETICS IN CARDIOVASCULAR MEDICINE
Cardiovascular medicine also stands to benefit from rapid advances in pharmacogenetics.
While no treatment has been developed that targets the molecular basis of cardiovascular disease, a number of genomic biomarkers have emerged that identify patients at risk of adverse reactions or treatment failure. These include genetic tests to predict the maintenance dose and risk of bleeding with warfarin,6 the likelihood of myopathy and myositis with simvastatin,7 and the risk of recurrent thrombotic events with clopidogrel.8–10
Using pharmacogenetics in prescribing warfarin and its alternatives
The pharmacogenetics of warfarin has been extensively researched, but genotyping before prescribing this drug is not yet widely done.
In 2007, the FDA updated the labeling of warfarin to include information about the influence of two genes, VKORC1 and CYP2C9, on a patient’s response to this drug. In 2010 this was updated to add that testing for these genes could be used to predict the maintenance dose of the drug. Difficulties with algorithms used to integrate this into clinical practice have hindered adoption of this testing.
With the advent of new anticoagulants such as dabigatran, rivaroxaban, and apixaban, many have expected warfarin and its pharmacogenetics to become obsolete. However, the new agents cost considerably more. Further, they may not offer a very great advantage over warfarin: in the Randomized Evaluation of Long-term Anticoagulant Therapy (RE-LY) trial, the absolute risk reduction in intracranial hemorrhage with dabigatran vs warfarin was small.11 Therefore, dabigatran is probably not cost-effective in populations at low risk of bleeding.12 A cost-effectiveness analysis comparing warfarin with dabigatran in patients with uncomplicated atrial fibrillation has suggested that dabigatran is, however, cost-effective in patients at moderate risk.12
In the RE-LY trial, the international normalized ratios (INRs) of the patients in the warfarin group were in the therapeutic range only 64% of the time. The advantages of dabigatran over warfarin become less pronounced as warfarin control is tightened.13 Of note, pharmacogenetics and home monitoring of the INR have both been shown to lead to tighter control of the INR, with greater time within the therapeutic range.14,15
Moreover, genetic testing can help us reduce the number of bleeding events in patients taking warfarin.16 Patients who carry the CYP2C9*2 or CYP2C9*3 polymorphism metabolize S-warfarin slower and therefore have a threefold higher risk of hemorrhage after starting warfarin.17 We could speculate that patients carrying these variants may be better served by the newer anticoagulants, though this has not been tested in any clinical trial.
It is also worth appreciating that the conditions requiring anticoagulation, such as atrial fibrillation, also have a strong genetic basis. Variants in chromosomes 4q25, 1q21, and 16q22 have all been associated with atrial fibrillation.18 The risk of atrial fibrillation is five to six times higher in carriers of multiple variants within all of these loci.19 Genetic variants at 4q25 have been associated with the response to specific antiarrhythmic drug treatments,20 response to pulmonary vein isolation, 21,22 and direct-current cardioversion.23 One can imagine a future in which patients with palpitations, carrying multiple gene risk variants, will choose prolonged monitoring at home to confirm a diagnosis. They would then be provided with a personalized best management strategy, using their personal preferences, clinical data, and genetic profile to make a treatment decision.
Using pharmacogenetics in prescribing clopidogrel and its alternatives
The pharmacogenetics of clopidogrel is of particular interest, as it has the potential of establishing a rational basis for using newer antiplatelet drugs such as ticagrelor and prasugrel, which are considerably more expensive than generic clopidogrel.
Most of the people who do not respond to clopidogrel carry the common cytochrome P450 2C19 variants CYP2C19*2 or CYP2C19*3.9 These variants are present in particularly high frequency in Asians and African Americans, who often do not feature in large randomized trials.
Newer antiplatelet agents have failed to demonstrate consistent superiority to clopidogrel without a tradeoff of more bleeding. However, in the Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition With Prasugrel-Thrombolysis in Myocardial Infarction 39 (TRITON TIMI-38),24,25 patients with the *2 variant receiving prasugrel had lower cardiovascular event rates than *2 carriers receiving clopidogrel.
Similarly, the patients who benefit the most from ticagrelor are carriers of the 2C19 nonresponder variants. In a large study, clopidogrel responders who did not carry either 2C19 nonresponder genetic variants or ABCB1 variants had cardiovascular outcomes similar to those of patients receiving ticagrelor.26
Clinicians have been cautious in prescribing potent antiplatelet agents to all patients because of the risk of bleeding. One could assume that by reserving newer agents for clopidogrel nonresponders, the bleeding risk could be minimized and overall benefit could be preserved with this strategy.
Cost may also be contained. The cost-effectiveness of such an approach with prasugrel has been tested with computer modeling and appears favorable.27 On the other hand, a similar yet limited analysis did not find genotype-driven use of ticagrelor to be cost-effective.28 This was mostly due to fewer deaths in patients receiving ticagrelor. However, the cost estimate for genotype-guided therapy was overestimated, as heterozygotes in the model were treated with ticagrelor instead of a high dose of clopidogrel.
It now appears that heterozygotes, ie, patients with one copy of the nonresponder variant, can achieve similar platelet inhibition with clopidogrel 225 mg daily as noncarriers on 75 mg daily.29 Since genotype-guided antiplatelet therapy has not been tested in a randomized outcomes trial, this tailored strategy has not been widely accepted.
THE FUTURE
The barriers to adoption of pharmacogenetics are considerable. Clinicians need to be educated about it, reimbursement needs to be worked out, and the pharmaceutical industry needs to get behind it. Nevertheless, the future of pharmacogenetics is extremely promising.
Research networks are forming to support the use of pharmacogenetics in clinical practice. The Pharmgkb (www.pharmgkb.org) database serves as a hub for educating clinicians and researchers as well as curating data for reference. Vanderbilt University is piloting the BioVu project, in which DNA and genotype data on patients are being stored and matched to the electronic clinical records.30 These projects not only provide clinically useful information on the current state of the art of pharmacogenetics, they also aid in disseminating new information about genotype-phenotype relationships.
Analytical software that uses “natural language processing” is being applied to clinician-generated notes to derive new observations and associations between genetic variants and clinical phenotypes. Integrating this information in real-time decision-support modules in the electronic health record provides a feedback loop for a rapid assimilation of new knowledge. Similar innovative decision-support modules are being established by Cleveland Clinic’s Center for Personalized Healthcare.31
The rise of ‘omic’ sciences
Pharmacogenetics and pharmacogenomics are part of a larger set of “omic” sciences. The suffix “-omics” implies a larger, more holistic view and is being applied to a number of fields—for example, the study of proteins (proteomics) and the study of metabolites (metabolomics). Profiling proteins and metabolites delivers a deluge of information on a patient that can be clustered, using pattern-recognition software, into population subgroups. Patterns of multiple proteins or metabolites are extracted from this spectral data to identify disease or response to treatment (pharmacometabolomics).
Metabolomics has been shown to predict the response to statins,32 diagnose myocardial infarction,33 and reclassify cardiovascular risk status.34 In addition, whereas traditional laboratory chemistry is reductionist, using single biomarkers for single-disease diagnosis, omic technologies hold the potential to reveal information on a number of possible health or disease states. The identification of “healthy” profiles using these technologies can potentially provide positive feedback to patients undertaking lifestyle changes and treatment.
The instrument costs for proteomic and metabolomic profiling are relatively high. However, the ongoing running costs are minimal, estimated at as low as less than $13 per test, as there are no expensive reagents.33 High-volume testing therefore becomes very cost-effective.
Although omic science appears futuristic, proteomics and metabolomics are already used in many clinical laboratories to rapidly identify bacteria. These methods have already revolutionized the way laboratories identify microbes, since they are automated, reduce workload, and give very fast results.
The cost of genetic testing is falling
Critics of pharmacogenetics claim that the predictive value of genetic testing is poor, that evidence is lacking, and that the cost is too high. In all new technologies, the first iteration is coarse, but performance improves with use. The first major barrier is adoption. Projects like BioVu are establishing the infrastructure for a feedback loop to iteratively improve upon the status quo and provide the evidence base clinicians demand.
The cost of genetic testing is falling rapidly, with whole-genome sequencing and annotation now costing less than $5,000. The cost of a pharmacogenetic test can be as low as $100 using low-cost nanotechnology, and the test needs to be performed only once in a patient’s lifetime.27
As other related molecular technologies such as proteomics and metabolomics become available and are integrated with genomics, the predictive ability of this science will improve.
AWAY FROM ONE-SIZE-FITS-ALL MEDICINE
Over the last decade there has been a trend away from “one size fits all” to customized “markets of one” in everything from consumer products to education to medicine. Mass customizing, also known as personalization, has been embraced by the internet community as a means to increase efficiency and reduce cost. This occurs by eliminating waste in redundant work or production of ineffective products.
Personalization on the Internet has been enabled through the use of informatics, mathematics, and supercomputing. The same tools that have personalized the delivery of consumer products are also being applied to the field of pharmacogenetics. Applied in an evidence-based fashion, these new technologies should profoundly improve patient care now and in the future.
“Change is the only constant.”
—Heraclitus (c 535–475 bce)
With the cost of health care rising and money to pay for it shrinking, there has never been a greater need to reduce waste.
Ineffective treatments and adverse drug effects account for much preventable morbidity and expense. New treatments, touted as more potent, are often introduced as replacements for traditional ones that are still effective in many patients, adding to costs and the potential for harm. For the pharmaceutical industry, the search for new “blockbuster” drugs seems to have hit a wall, at least in cardiovascular medicine.1 Advances often come at the cost of adverse effects, such as bleeding with triple antiplatelet therapy and diabetes with potent statin drugs.
The path to maximizing benefit and reducing harm now appears to lie in stratifying populations and appreciating patient individuality in response to treatment. For many decades we have known that patients vary widely in their response to drugs, owing to personal factors such as body surface area, age, environment, and genetics. And indeed, we treat our patients as individuals, for example by tailoring aminoglycoside dose to weight and renal function.
However, clinical trials typically give us an idea of the benefits only to the average patient. While subgroup analyses identify groups that may benefit more or less from treatment, the additional information they provide is not easily integrated into the clinical model of prescribing, in which one size fits all.
THE PROMISE OF PHARMACOGENETICS
The emerging field of pharmacogenetics promises to give clinicians the tools to make informed treatment decisions based on predictive genetic testing. This genetic testing aims to match treatment to an individual’s genetic profile. This often involves analyzing single-nucleotide polymorphisms in genes for enzymes that metabolize drugs, such as the cytochrome P450 enzymes, to predict efficacy or an adverse event with treatment.
Pharmacogenetics is playing an increasing role in clinical trials, particularly in the early stages of drug development, by helping to reduce the number of patients needed, prove efficacy, and identify subgroups in which alternative treatment can be targeted. At another level, a molecular understanding of disease is leading to truly targeted treatments based on genomics.
Over recent years, genetic testing has been increasingly used in clinical practice, thanks to a convergence of factors such as rapid, low-cost tests, a growing evidence base, and emerging interest among doctors and payers.
An advantage to using genetic testing as opposed to other types of laboratory testing, such as measuring the concentration of the drug in the blood during treatment, is that genetic tests can predict the response to treatment before the treatment is started. Moreover, with therapeutic drug monitoring after treatment has begun, there are sometimes no detectable measures of toxicity. For example, both carbamazepine and the antiviral drug abacavir can—fortunately only rarely—cause Stevens-Johnson syndrome. But before genetic markers were discovered, there was no method of estimating this risk apart from taking a family history.2,3 Considering the numbers of people involved, it was not feasible until recently to suggest genetic screening for patients starting on these drugs. However, the cost of genotyping and gene sequencing has been falling at a rate inversely faster than Moore’s law (an approximate annual doubling in computer power), and population genomics is becoming a reality.4
The US Food and Drug Administration (FDA) recognizes the current and future value of pharmacogenetics in drug safety and development. A number of approved pharmacogenetic biomarkers are listed on the FDA website (Table 1). Black box warnings have been mandated for a number of drugs on the basis of observational evidence.
The FDA also promotes rapid approval for novel drugs with pharmacogenetic “companion diagnostics.” A recent example of this was the approval of ivacaftor for cystic fibrosis patients who have the G551D mutation.5 Here, a molecular understanding of the condition led to the development of a targeted treatment. Although the cost of developing this drug was high, the path is now paved for similar advances. Oncologists are familiar with these advances with the emergence of new molecularly targeted treatments, eg, BRAF inhibitors in metastatic melanoma, imatinib in chronic myeloid leukemia, and gefitinib in non-small-cell lung cancer.
PHARMACOGENETICS IN CARDIOVASCULAR MEDICINE
Cardiovascular medicine also stands to benefit from rapid advances in pharmacogenetics.
While no treatment has been developed that targets the molecular basis of cardiovascular disease, a number of genomic biomarkers have emerged that identify patients at risk of adverse reactions or treatment failure. These include genetic tests to predict the maintenance dose and risk of bleeding with warfarin,6 the likelihood of myopathy and myositis with simvastatin,7 and the risk of recurrent thrombotic events with clopidogrel.8–10
Using pharmacogenetics in prescribing warfarin and its alternatives
The pharmacogenetics of warfarin has been extensively researched, but genotyping before prescribing this drug is not yet widely done.
In 2007, the FDA updated the labeling of warfarin to include information about the influence of two genes, VKORC1 and CYP2C9, on a patient’s response to this drug. In 2010 this was updated to add that testing for these genes could be used to predict the maintenance dose of the drug. Difficulties with algorithms used to integrate this into clinical practice have hindered adoption of this testing.
With the advent of new anticoagulants such as dabigatran, rivaroxaban, and apixaban, many have expected warfarin and its pharmacogenetics to become obsolete. However, the new agents cost considerably more. Further, they may not offer a very great advantage over warfarin: in the Randomized Evaluation of Long-term Anticoagulant Therapy (RE-LY) trial, the absolute risk reduction in intracranial hemorrhage with dabigatran vs warfarin was small.11 Therefore, dabigatran is probably not cost-effective in populations at low risk of bleeding.12 A cost-effectiveness analysis comparing warfarin with dabigatran in patients with uncomplicated atrial fibrillation has suggested that dabigatran is, however, cost-effective in patients at moderate risk.12
In the RE-LY trial, the international normalized ratios (INRs) of the patients in the warfarin group were in the therapeutic range only 64% of the time. The advantages of dabigatran over warfarin become less pronounced as warfarin control is tightened.13 Of note, pharmacogenetics and home monitoring of the INR have both been shown to lead to tighter control of the INR, with greater time within the therapeutic range.14,15
Moreover, genetic testing can help us reduce the number of bleeding events in patients taking warfarin.16 Patients who carry the CYP2C9*2 or CYP2C9*3 polymorphism metabolize S-warfarin slower and therefore have a threefold higher risk of hemorrhage after starting warfarin.17 We could speculate that patients carrying these variants may be better served by the newer anticoagulants, though this has not been tested in any clinical trial.
It is also worth appreciating that the conditions requiring anticoagulation, such as atrial fibrillation, also have a strong genetic basis. Variants in chromosomes 4q25, 1q21, and 16q22 have all been associated with atrial fibrillation.18 The risk of atrial fibrillation is five to six times higher in carriers of multiple variants within all of these loci.19 Genetic variants at 4q25 have been associated with the response to specific antiarrhythmic drug treatments,20 response to pulmonary vein isolation, 21,22 and direct-current cardioversion.23 One can imagine a future in which patients with palpitations, carrying multiple gene risk variants, will choose prolonged monitoring at home to confirm a diagnosis. They would then be provided with a personalized best management strategy, using their personal preferences, clinical data, and genetic profile to make a treatment decision.
Using pharmacogenetics in prescribing clopidogrel and its alternatives
The pharmacogenetics of clopidogrel is of particular interest, as it has the potential of establishing a rational basis for using newer antiplatelet drugs such as ticagrelor and prasugrel, which are considerably more expensive than generic clopidogrel.
Most of the people who do not respond to clopidogrel carry the common cytochrome P450 2C19 variants CYP2C19*2 or CYP2C19*3.9 These variants are present in particularly high frequency in Asians and African Americans, who often do not feature in large randomized trials.
Newer antiplatelet agents have failed to demonstrate consistent superiority to clopidogrel without a tradeoff of more bleeding. However, in the Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition With Prasugrel-Thrombolysis in Myocardial Infarction 39 (TRITON TIMI-38),24,25 patients with the *2 variant receiving prasugrel had lower cardiovascular event rates than *2 carriers receiving clopidogrel.
Similarly, the patients who benefit the most from ticagrelor are carriers of the 2C19 nonresponder variants. In a large study, clopidogrel responders who did not carry either 2C19 nonresponder genetic variants or ABCB1 variants had cardiovascular outcomes similar to those of patients receiving ticagrelor.26
Clinicians have been cautious in prescribing potent antiplatelet agents to all patients because of the risk of bleeding. One could assume that by reserving newer agents for clopidogrel nonresponders, the bleeding risk could be minimized and overall benefit could be preserved with this strategy.
Cost may also be contained. The cost-effectiveness of such an approach with prasugrel has been tested with computer modeling and appears favorable.27 On the other hand, a similar yet limited analysis did not find genotype-driven use of ticagrelor to be cost-effective.28 This was mostly due to fewer deaths in patients receiving ticagrelor. However, the cost estimate for genotype-guided therapy was overestimated, as heterozygotes in the model were treated with ticagrelor instead of a high dose of clopidogrel.
It now appears that heterozygotes, ie, patients with one copy of the nonresponder variant, can achieve similar platelet inhibition with clopidogrel 225 mg daily as noncarriers on 75 mg daily.29 Since genotype-guided antiplatelet therapy has not been tested in a randomized outcomes trial, this tailored strategy has not been widely accepted.
THE FUTURE
The barriers to adoption of pharmacogenetics are considerable. Clinicians need to be educated about it, reimbursement needs to be worked out, and the pharmaceutical industry needs to get behind it. Nevertheless, the future of pharmacogenetics is extremely promising.
Research networks are forming to support the use of pharmacogenetics in clinical practice. The Pharmgkb (www.pharmgkb.org) database serves as a hub for educating clinicians and researchers as well as curating data for reference. Vanderbilt University is piloting the BioVu project, in which DNA and genotype data on patients are being stored and matched to the electronic clinical records.30 These projects not only provide clinically useful information on the current state of the art of pharmacogenetics, they also aid in disseminating new information about genotype-phenotype relationships.
Analytical software that uses “natural language processing” is being applied to clinician-generated notes to derive new observations and associations between genetic variants and clinical phenotypes. Integrating this information in real-time decision-support modules in the electronic health record provides a feedback loop for a rapid assimilation of new knowledge. Similar innovative decision-support modules are being established by Cleveland Clinic’s Center for Personalized Healthcare.31
The rise of ‘omic’ sciences
Pharmacogenetics and pharmacogenomics are part of a larger set of “omic” sciences. The suffix “-omics” implies a larger, more holistic view and is being applied to a number of fields—for example, the study of proteins (proteomics) and the study of metabolites (metabolomics). Profiling proteins and metabolites delivers a deluge of information on a patient that can be clustered, using pattern-recognition software, into population subgroups. Patterns of multiple proteins or metabolites are extracted from this spectral data to identify disease or response to treatment (pharmacometabolomics).
Metabolomics has been shown to predict the response to statins,32 diagnose myocardial infarction,33 and reclassify cardiovascular risk status.34 In addition, whereas traditional laboratory chemistry is reductionist, using single biomarkers for single-disease diagnosis, omic technologies hold the potential to reveal information on a number of possible health or disease states. The identification of “healthy” profiles using these technologies can potentially provide positive feedback to patients undertaking lifestyle changes and treatment.
The instrument costs for proteomic and metabolomic profiling are relatively high. However, the ongoing running costs are minimal, estimated at as low as less than $13 per test, as there are no expensive reagents.33 High-volume testing therefore becomes very cost-effective.
Although omic science appears futuristic, proteomics and metabolomics are already used in many clinical laboratories to rapidly identify bacteria. These methods have already revolutionized the way laboratories identify microbes, since they are automated, reduce workload, and give very fast results.
The cost of genetic testing is falling
Critics of pharmacogenetics claim that the predictive value of genetic testing is poor, that evidence is lacking, and that the cost is too high. In all new technologies, the first iteration is coarse, but performance improves with use. The first major barrier is adoption. Projects like BioVu are establishing the infrastructure for a feedback loop to iteratively improve upon the status quo and provide the evidence base clinicians demand.
The cost of genetic testing is falling rapidly, with whole-genome sequencing and annotation now costing less than $5,000. The cost of a pharmacogenetic test can be as low as $100 using low-cost nanotechnology, and the test needs to be performed only once in a patient’s lifetime.27
As other related molecular technologies such as proteomics and metabolomics become available and are integrated with genomics, the predictive ability of this science will improve.
AWAY FROM ONE-SIZE-FITS-ALL MEDICINE
Over the last decade there has been a trend away from “one size fits all” to customized “markets of one” in everything from consumer products to education to medicine. Mass customizing, also known as personalization, has been embraced by the internet community as a means to increase efficiency and reduce cost. This occurs by eliminating waste in redundant work or production of ineffective products.
Personalization on the Internet has been enabled through the use of informatics, mathematics, and supercomputing. The same tools that have personalized the delivery of consumer products are also being applied to the field of pharmacogenetics. Applied in an evidence-based fashion, these new technologies should profoundly improve patient care now and in the future.
- Topol EJ. Past the wall in cardiovascular R&D. Nat Rev Drug Discov 2009; 8:259.
- Mallal S, Phillips E, Carosi G, et al; PREDICT-1 Study Team. HLA-B*5701 screening for hypersensitivity to abacavir. N Engl J Med 2008; 358:568–579.
- Hung SI, Chung WH, Jee SH, et al. Genetic susceptibility to carbamazepine-induced cutaneous adverse drug reactions. Pharmacogenet Genomics 2006; 16:297–306.
- Phimister EG, Feero WG, Guttmacher AE. Realizing genomic medicine. N Engl J Med 2012; 366:757–759.
- Van Goor F, Hadida S, Grootenhuis PD, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci USA 2009; 106:18825–18830.
- International Warfarin Pharmacogenetics Consortium; Klein TE, Altman RB, Eriksson N, et al. Estimation of the warfarin dose with clinical and pharmacogenetic data. N Engl J Med 2009; 360:753–764.
- SEARCH Collaborative Group; Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin-induced myopathy—a genomewide study. N Engl J Med 2008; 359:789–799.
- Simon T, Verstuyft C, Mary-Krause M, et al; French Registry of Acute ST-Elevation and Non-ST-Elevation Myocardial Infarction (FAST-MI) Investigators. Genetic determinants of response to clopidogrel and cardiovascular events. N Engl J Med 2009; 360:363–375.
- Mega JL, Close SL, Wiviott SD, et al. Cytochrome p-450 polymorphisms and response to clopidogrel. N Engl J Med 2009; 360:354–362
- Collet JP, Hulot JS, Pena A, et al. Cytochrome P450 2C19 polymorphism in young patients treated with clopidogrel after myocardial infarction: a cohort study. Lancet 2009; 373:309–317.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al; RE-LY Steering Committee and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med 2009; 361:1139–1151.
- Shah SV, Gage BF. Cost-effectiveness of dabigatran for stroke prophylaxis in atrial fibrillation. Circulation 2011; 123:2562–2570.
- Wallentin L, Yusuf S, Ezekowitz MD, et al; RE-LY investigators. Efficacy and safety of dabigatran compared with warfarin at different levels of international normalised ratio control for stroke prevention in atrial fibrillation: an analysis of the RE-LY trial. Lancet 2010; 376:975–983.
- Anderson JL, Horne BD, Stevens SM, et al; Couma-Gen Investigators. Randomized trial of genotype-guided versus standard warfarin dosing in patients initiating oral anticoagulation. Circulation 2007; 116:2563–2570.
- Matchar DB, Jacobson A, Dolor R, et al; THINRS Executive Committee and Site Investigators. Effect of home testing of international normalized ratio on clinical events. N Engl J Med 2010; 363:1608–1620.
- Epstein RS, Moyer TP, Aubert RE, et al. Warfarin genotyping reduces hospitalization rates results from the MM-WES (Medco-Mayo Warfarin Effectiveness study). J Am Coll Cardiol 2010; 55:2804–2812.
- Sanderson S, Emery J, Higgins J. CYP2C9 gene variants, drug dose, and bleeding risk in warfarin-treated patients: a HuGEnet systematic review and meta-analysis. Genet Med 2005; 7:97–104.
- Ellinor PT, Lunetta KL, Albert CM, et al. Meta-analysis identifies six new susceptibility loci for atrial fibrillation. Nat Genet 2012; 44:670–675.
- Lubitz SA, Sinner MF, Lunetta KL, et al. Independent susceptibility markers for atrial fibrillation on chromosome 4q25. Circulation 2010; 122:976–984.
- Parvez B, Vaglio J, Rowan S, et al. Symptomatic response to antiarrhythmic drug therapy is modulated by a common single nucleotide polymorphism in atrial fibrillation. J Am Coll Cardiol 2012; 60:539–545.
- Husser D, Adams V, Piorkowski C, Hindricks G, Bollmann A. Chromosome 4q25 variants and atrial fibrillation recurrence after catheter ablation. J Am Coll Cardiol 2010; 55:747–753.
- Benjamin Shoemaker M, Muhammad R, Parvez B, et al. Common atrial fibrillation risk alleles at 4q25 predict recurrence after catheter-based atrial fibrillation ablation.” Heart Rhythm 2012; Nov 23.pii: S1547-5271(12)01340–9. 10.1016/j.hrthm.2012.11.012. [Epub ahead of print]
- Parvez B, Benjamin Shoemaker M, Muhammad R, et al. Common genetic polymorphism at 4q25 locus predicts atrial fibrillation recurrence after successful cardioversion. Heart Rhythm 2013 Feb 18.pii: S1547-5271(13)00161–6. 10.1016/j.hrthm.2013.02.018. [Epub ahead of print]
- Mega JL, Close SL, Wiviott SD, et al. Genetic variants in ABCB1 and CYP2C19 and cardiovascular outcomes after treatment with clopidogrel and prasugrel in the TRITON-TIMI 38 trial: a pharmacogenetic analysis. Lancet 2010; 376:1312–1319.
- Mega JL, Close SL, Wiviott SD, et al. Cytochrome P450 genetic polymorphisms and the response to prasugrel: relationship to pharmacokinetic, pharmacodynamic, and clinical outcomes. Circulation 2009; 119:2553–2560.
- Wallentin L, James S, Storey RF, et al; PLATO investigators. Effect of CYP2C19 and ABCB1 single nucleotide polymorphisms on outcomes of treatment with ticagrelor versus clopidogrel for acute coronary syndromes: a genetic substudy of the PLATO trial. Lancet 2010; 376:1320–1328.
- Guzauskas GF, Hughes DA, Bradley SM, Veenstra DL. A risk-benefit assessment of prasugrel, clopidogrel, and genotype-guided therapy in patients undergoing percutaneous coronary intervention. Clin Pharmacol Ther 2012; 91:829–837.
- Crespin DJ, Federspiel JJ, Biddle AK, Jonas DE, Rossi JS. Ticagrelor versus genotype-driven antiplatelet therapy for secondary prevention after acute coronary syndrome: a cost-effectiveness analysis. Value Health 2011; 14:483–491.
- Mega JL, Hochholzer W, Frelinger AL, et al. Dosing clopidogrel based on CYP2C19 genotype and the effect on platelet reactivity in patients with stable cardiovascular disease. JAMA 2011; 306:2221–2228.
- Xu H, Jiang M, Oetjens M, et al. Facilitating pharmacogenetic studies using electronic health records and natural-language processing: a case study of warfarin. J Am Med Inform Assoc 2011; 18:387–391.
- Teng K, Eng C, Hess CA, et al. Building an innovative model for personalized healthcare. Cleve Clin J Med 2012; 79( suppl 1):S1–S9.
- Kaddurah-Daouk R, Baillie RA, Zhu H, et al. Enteric microbiome metabolites correlate with response to simvastatin treatment. PLoS One 2011; 6:e25482.
- Bodi V, Sanchis J, Morales JM, et al. Metabolomic profile of human myocardial ischemia by nuclear magnetic resonance spectroscopy of peripheral blood serum: a translational study based on transient coronary occlusion models. J Am Coll Cardiol 2012; 59:1629–1641.
- Shah SH, Sun JL, Stevens RD, et al. Baseline metabolomic profiles predict cardiovascular events in patients at risk for coronary artery disease. Am Heart J 2012; 163:844–850.e1.
- Topol EJ. Past the wall in cardiovascular R&D. Nat Rev Drug Discov 2009; 8:259.
- Mallal S, Phillips E, Carosi G, et al; PREDICT-1 Study Team. HLA-B*5701 screening for hypersensitivity to abacavir. N Engl J Med 2008; 358:568–579.
- Hung SI, Chung WH, Jee SH, et al. Genetic susceptibility to carbamazepine-induced cutaneous adverse drug reactions. Pharmacogenet Genomics 2006; 16:297–306.
- Phimister EG, Feero WG, Guttmacher AE. Realizing genomic medicine. N Engl J Med 2012; 366:757–759.
- Van Goor F, Hadida S, Grootenhuis PD, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci USA 2009; 106:18825–18830.
- International Warfarin Pharmacogenetics Consortium; Klein TE, Altman RB, Eriksson N, et al. Estimation of the warfarin dose with clinical and pharmacogenetic data. N Engl J Med 2009; 360:753–764.
- SEARCH Collaborative Group; Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin-induced myopathy—a genomewide study. N Engl J Med 2008; 359:789–799.
- Simon T, Verstuyft C, Mary-Krause M, et al; French Registry of Acute ST-Elevation and Non-ST-Elevation Myocardial Infarction (FAST-MI) Investigators. Genetic determinants of response to clopidogrel and cardiovascular events. N Engl J Med 2009; 360:363–375.
- Mega JL, Close SL, Wiviott SD, et al. Cytochrome p-450 polymorphisms and response to clopidogrel. N Engl J Med 2009; 360:354–362
- Collet JP, Hulot JS, Pena A, et al. Cytochrome P450 2C19 polymorphism in young patients treated with clopidogrel after myocardial infarction: a cohort study. Lancet 2009; 373:309–317.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al; RE-LY Steering Committee and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med 2009; 361:1139–1151.
- Shah SV, Gage BF. Cost-effectiveness of dabigatran for stroke prophylaxis in atrial fibrillation. Circulation 2011; 123:2562–2570.
- Wallentin L, Yusuf S, Ezekowitz MD, et al; RE-LY investigators. Efficacy and safety of dabigatran compared with warfarin at different levels of international normalised ratio control for stroke prevention in atrial fibrillation: an analysis of the RE-LY trial. Lancet 2010; 376:975–983.
- Anderson JL, Horne BD, Stevens SM, et al; Couma-Gen Investigators. Randomized trial of genotype-guided versus standard warfarin dosing in patients initiating oral anticoagulation. Circulation 2007; 116:2563–2570.
- Matchar DB, Jacobson A, Dolor R, et al; THINRS Executive Committee and Site Investigators. Effect of home testing of international normalized ratio on clinical events. N Engl J Med 2010; 363:1608–1620.
- Epstein RS, Moyer TP, Aubert RE, et al. Warfarin genotyping reduces hospitalization rates results from the MM-WES (Medco-Mayo Warfarin Effectiveness study). J Am Coll Cardiol 2010; 55:2804–2812.
- Sanderson S, Emery J, Higgins J. CYP2C9 gene variants, drug dose, and bleeding risk in warfarin-treated patients: a HuGEnet systematic review and meta-analysis. Genet Med 2005; 7:97–104.
- Ellinor PT, Lunetta KL, Albert CM, et al. Meta-analysis identifies six new susceptibility loci for atrial fibrillation. Nat Genet 2012; 44:670–675.
- Lubitz SA, Sinner MF, Lunetta KL, et al. Independent susceptibility markers for atrial fibrillation on chromosome 4q25. Circulation 2010; 122:976–984.
- Parvez B, Vaglio J, Rowan S, et al. Symptomatic response to antiarrhythmic drug therapy is modulated by a common single nucleotide polymorphism in atrial fibrillation. J Am Coll Cardiol 2012; 60:539–545.
- Husser D, Adams V, Piorkowski C, Hindricks G, Bollmann A. Chromosome 4q25 variants and atrial fibrillation recurrence after catheter ablation. J Am Coll Cardiol 2010; 55:747–753.
- Benjamin Shoemaker M, Muhammad R, Parvez B, et al. Common atrial fibrillation risk alleles at 4q25 predict recurrence after catheter-based atrial fibrillation ablation.” Heart Rhythm 2012; Nov 23.pii: S1547-5271(12)01340–9. 10.1016/j.hrthm.2012.11.012. [Epub ahead of print]
- Parvez B, Benjamin Shoemaker M, Muhammad R, et al. Common genetic polymorphism at 4q25 locus predicts atrial fibrillation recurrence after successful cardioversion. Heart Rhythm 2013 Feb 18.pii: S1547-5271(13)00161–6. 10.1016/j.hrthm.2013.02.018. [Epub ahead of print]
- Mega JL, Close SL, Wiviott SD, et al. Genetic variants in ABCB1 and CYP2C19 and cardiovascular outcomes after treatment with clopidogrel and prasugrel in the TRITON-TIMI 38 trial: a pharmacogenetic analysis. Lancet 2010; 376:1312–1319.
- Mega JL, Close SL, Wiviott SD, et al. Cytochrome P450 genetic polymorphisms and the response to prasugrel: relationship to pharmacokinetic, pharmacodynamic, and clinical outcomes. Circulation 2009; 119:2553–2560.
- Wallentin L, James S, Storey RF, et al; PLATO investigators. Effect of CYP2C19 and ABCB1 single nucleotide polymorphisms on outcomes of treatment with ticagrelor versus clopidogrel for acute coronary syndromes: a genetic substudy of the PLATO trial. Lancet 2010; 376:1320–1328.
- Guzauskas GF, Hughes DA, Bradley SM, Veenstra DL. A risk-benefit assessment of prasugrel, clopidogrel, and genotype-guided therapy in patients undergoing percutaneous coronary intervention. Clin Pharmacol Ther 2012; 91:829–837.
- Crespin DJ, Federspiel JJ, Biddle AK, Jonas DE, Rossi JS. Ticagrelor versus genotype-driven antiplatelet therapy for secondary prevention after acute coronary syndrome: a cost-effectiveness analysis. Value Health 2011; 14:483–491.
- Mega JL, Hochholzer W, Frelinger AL, et al. Dosing clopidogrel based on CYP2C19 genotype and the effect on platelet reactivity in patients with stable cardiovascular disease. JAMA 2011; 306:2221–2228.
- Xu H, Jiang M, Oetjens M, et al. Facilitating pharmacogenetic studies using electronic health records and natural-language processing: a case study of warfarin. J Am Med Inform Assoc 2011; 18:387–391.
- Teng K, Eng C, Hess CA, et al. Building an innovative model for personalized healthcare. Cleve Clin J Med 2012; 79( suppl 1):S1–S9.
- Kaddurah-Daouk R, Baillie RA, Zhu H, et al. Enteric microbiome metabolites correlate with response to simvastatin treatment. PLoS One 2011; 6:e25482.
- Bodi V, Sanchis J, Morales JM, et al. Metabolomic profile of human myocardial ischemia by nuclear magnetic resonance spectroscopy of peripheral blood serum: a translational study based on transient coronary occlusion models. J Am Coll Cardiol 2012; 59:1629–1641.
- Shah SH, Sun JL, Stevens RD, et al. Baseline metabolomic profiles predict cardiovascular events in patients at risk for coronary artery disease. Am Heart J 2012; 163:844–850.e1.
Should we use pharmacogenetic testing when prescribing warfarin?
The answer is not clear. There is evidence in favor of pharmacogenetic testing, but not yet enough to strongly recommend it. However, we do believe that physicians should consider it when starting patients on warfarin therapy.
WARFARIN HAS A NARROW THERAPEUTIC WINDOW
Although newer drugs are available, warfarin is still the most commonly used oral anticoagulant for preventing and treating thromboembolism.1 It is highly effective but has a narrow therapeutic window and wide interindividual variability in dosage requirements, which poses challenges to achieving adequate anticoagulation.1–3 Inappropriate dosing contributes to a high rate of bleeding events and emergency room visits.4
Warfarin is monitored using the prothrombin time. Because the prothrombin time varies depending on the assay used, the standardized value called the international normalized ratio (INR) is more commonly used.
Clinical factors such as age, body size, and drug interactions affect warfarin dosage requirements and are important to consider,5 even though they account for only 15% to 20% of the variability in warfarin dose.6
Genetic factors also affect warfarin dosage requirements. The combination of genetic and clinical factors accounts for up to 47% of the dose variability.7
GENES THAT AFFECT WARFARIN
Several genes are known to influence warfarin’s pharmacokinetics and pharmacodynamics. Of these, the two most clinically relevant and well studied are CYP2C9 (which codes for cytochrome P450 2C9) and VKORC1 (which codes for vitamin K epoxide reductase).7 These genes are polymorphic, with some variants producing less-active enzymes that allow warfarin to be more active. Therefore, patients who carry these variants need lower doses of this drug (see below).
CYP2C9 variants
The CYP2C9 gene has several variants. Of these, CYP2C9*2 and CYP2C9*3 are associated with the lowest enzyme activity.
Patients with either of these variants require significantly lower warfarin doses to reach therapeutic levels than those with the wild-type gene (ie, CYP2C9*1). CYP2C9*2 reduces warfarin clearance by 40%, and the CYP2C9*3 variant reduces it by 75%.7 Having a *2 or *3 allele increases the risk of bleeding during warfarin therapy and the time needed to achieve a stable dose.8 Other variants associated with lower warfarin dose requirements are *5, *6, and *11.
The prevalence of these variants is significantly higher in people of European ancestry (roughly one-third) than in Asian people and African Americans,7 although no one has recommended not testing in these low-prevalence populations. Limdi et al9 reported that by including the *5, *6, and *11 variants in genetic testing (in addition to *2 and *3), they could identify more African Americans (9.7%) who carried at least one of these abnormal variants than reported previously. Differences among ethnic groups need to be taken into account when interpreting pharmacogenetic studies.
VKORC1 variants
Patients also need lower doses of warfarin if they carry the VKORC1 −1639G>A variant, and they spend more time with an INR above the therapeutic range and have higher overall INR values. However, having this variant does not appear to increase the risk of bleeding.
The −1639G>A variant is the most common variant of VKORC1. Rarer ones have also been described, but most commercially available tests do not detect them.
Racial differences exist in the prevalence rates of the various VKORC1 polymorphisms, with the most sensitive (low-dose) genotype predominating in Asians and the least sensitive (high-dose) genotype predominating in African Americans. Over 50% of people of European ancestry carry the intermediate-sensitivity genotype (typical dose).7
CURRENT RECOMMENDATIONS FOR OR AGAINST TESTING
FDA labeling
In 2007, the US Food and Drug Administration (FDA) required that the warfarin package insert carry information about initial dosing based on CYP2C9 and VKORC1 testing. This recommendation was revised in 2010 to include a table to help clinicians select an initial warfarin dose if CYP2C9 and VKORC1 genotype information is available. However, the FDA does not require pharmacogenetic testing, leaving the decision to the discretion of the clinician.7
American College of Chest Physicians
The American College of Chest Physicians recommends against routine pharmacogenetic testing (grade 1B) because of a lack of evidence that it improves clinical end points or that it is cost-effective.5
WHAT EVIDENCE SUPORTS GENETIC TESTING TO GUIDE WARFARIN THERAPY?
To date, no large randomized, controlled trial has been published that looked at clinical outcomes with warfarin dosing based on pharmacogenetic testing. However, several smaller studies have suggested it is beneficial.
One trial found that when dosing was informed by pharmacogenetic testing, patients had significantly more time in the therapeutic range, a lower percentage of INRs greater than 4 or less than 1.5, and fewer serious adverse events (death, myocardial infarction, stroke, thromboembolism, and clinically significant bleeding events).10 Patients whose dosage was determined using pharmacogenetic algorithms as opposed to traditional clinical algorithms maintained a therapeutic INR more consistently.11
In addition, compared with historical controls, patients whose physician used pharmacogenetic testing to guide warfarin dosing had a rate of hospitalization 31% lower and a rate of hospitalization specifically for bleeding or thromboembolism 28% lower during 6 months of follow-up.12,13
Several studies have attempted to assess the cost-effectiveness and utility of pharmacogenetic testing in warfarin therapy. As yet, the results have been inconclusive.14 Larger prospective trials are under way and are estimated to be completed in late 2013.15 These include:
- COAG (Clarification of Optimal Anticoagulation Through Genetics)
- GIFT (Genetics Informatics Trial of Warfarin to Prevent Venous Thrombosis)
- EU-PACT (European Pharmacogenetics of Anticoagulant Therapy-Warfarin).
We hope these studies will provide greater clarity on the clinical utility and cost-effectiveness of pharmacogenetic testing to guide warfarin dosing.
HOW SHOULD GENETIC INFORMATION BE USED TO GUIDE OR ALTER THERAPY?
Algorithms are available for estimating initial and maintenance warfarin doses based on genetic information (CYP2C9 and VKORC1), race or ethnicity, age, sex, body mass index, smoking status, and other medications taken. In addition, models incorporating the INR on day 4 and days 6 to 11 have been developed for dose refinement.15 The algorithms explain 30% to 60% of the variability of the data, with lower values for African Americans.7
A well-developed dosing model that includes traditional clinical factors and patient genetic status is publicly available online at www.warfarindosing.org.4
CPIC: A leader in applied pharmacogenetics
In late 2009, PharmGKB joined forces with the Pharmacogenomics Research Network of the National Institutes of Health to form the Clinical Pharmacogenetics Implementation Consortium (CPIC). This organization issues guidelines that are written by expert clinicians and scientists and then are peer-reviewed, published in leading journals, and simultaneously posted to the PharmGKB website along with supplemental information and updates.
CPIC’s goal is to review the current evidence and to address barriers to the adoption of pharmacogenetic testing into clinical practice. Its guidelines do not advise when or which pharmacogenetic tests should be ordered. Rather, they provide guidance on interpreting and applying such testing, should the test results be available.7
CPIC has guidelines on CYP2C9 and VKORC1 genotypes and warfarin dosing.8 If a patient’s genetic information is available, CPIC strongly recommends the use of pharmacogenetic algorithm-based dosing. If such an algorithm is not accessible, use of a genotype dosing table is recommended.8
Monitoring is still needed
Many factors can affect an individual’s response to warfarin above and beyond the above-noted clinical and genetic traits. These include diet, concomitant medications (both prescription and over-the-counter and herbal), and disease state. There may also be additional genetic polymorphisms not yet identified in various racial and ethnic groups that may affect dosing requirements. And as with all medications, patient compliance and dosing errors have a large potential to affect individual response. Therefore, clinicians should still be diligent about clinical monitoring.15
Most useful for initial dose
As with most pharmacogenetic information, the greatest benefit can be achieved when this information is used to guide the initial dose, although there is also some effect noted when this information is known and acted upon into the 2nd week of treatment.8
Patients on long-term warfarin treatment with stable doses and those unable to achieve stable dosing because of variable adherence or dietary vitamin K intake are less likely to benefit from genetic testing.
There are no published guidelines on the utility of pharmacogenetic testing if a patient is already on a stable dose of warfarin or has a known historical stable dose. There are also no published guidelines on changing the frequency of monitoring based on known genotype.
In children, the data are sparse at this time regarding the utility of pharmacogenetically informed dosing.
HOW DOES ONE ORDER TESTING, AND WHAT IS THE COST?
The FDA has approved four warfarin pharmacogenetic test kits. To be used in clinical decision-making, these tests must be done in a laboratory certified by the Clinical Laboratory Improvement Amendments (CLIA) program.
Testing typically costs a few hundred dollars and may take days for results to be returned if not available on site.15 At Cleveland Clinic, CYP2C9 and VKORC1 testing can be run in-house at a cost of about $700. Generally, many third-party payers do not reimburse for testing without a prior-approval process.
TO TEST OR NOT TO TEST
Pharmacogenetic testing is available and may help optimize warfarin dosing early in treatment, as well as help maintain therapeutic INRs more consistently. There is preliminary evidence that using this information to guide dosing improves clinical outcomes. Several large trials are under way to address additional questions of clinical utility, with results expected in the next year. There are also readily available decision-support tools to guide therapeutic dosing, and when pharmacogenetic test results are available, utilization of a warfarin dosing algorithm is recommended.
The largest barrier remaining appears to be cost (relative to perceived benefit), and until larger trials of clinical utility and cost-effectiveness are completed and analyzed, hurdles exist to obtaining coverage for such testing.
If it is readily available (and can be paid for by insurance companies or out-of-pocket) and test results can be obtained within 24 to 48 hours or before prescribing, pharmacogenetic testing can be a valuable tool to guide and manage warfarin dosing. Particularly for patients who want to be as proactive as possible, warfarin pharmacogenetic testing offers the ability to participate in this decision-making and to potentially reduce their risk of adverse drug events. And in view of the evidence and FDA recommendations, we propose that the discussion with our patients is not whether we should consider pharmacogenetic testing, but that we have considered pharmacogenetic testing, and why we have decided for or against it.
- Jacobs LG. Warfarin pharmacology, clinical management, and evaluation of hemorrhagic risk for the elderly. Clin Geriatr Med 2006; 22:17–32,vii–viii.
- Rieder MJ, Reiner AP, Gage BF, et al. Effect of VKORC1 haplotypes on transcriptional regulation and warfarin dose. N Engl J Med 2005; 352:2285–2293.
- Higashi MK, Veenstra DL, Kondo LM, et al. Association between CYP2C9 genetic variants and anticoagulation-related outcomes during warfarin therapy. JAMA 2002; 287:1690–1698.
- Shehab N, Sperling LS, Kegler SR, Budnitz DS. National estimates of emergency department visits for hemorrhage-related adverse events from clopidogrel plus aspirin and from warfarin. Arch Intern Med 2010; 170:1926–1933.
- Holbrook A, Schulman S, Witt DM, et al; American College of Chest Physicians. Evidence-based management of anticoagulant therapy: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e152S–e184S.
- Gage BF, Eby C, Johnson JA, et al. Use of pharmacogenetic and clinical factors to predict the therapeutic dose of warfarin. Clin Pharmacol Ther 2008; 84:326–331.
- Cavallari LH, Shin J, Perera MA. Role of pharmacogenomics in the management of traditional and novel oral anticoagulants. Pharmacotherapy 2011; 31:1192–1207.
- Johnson JA, Gong L, Whirl-Carillo M, et al; Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium guidelines for CYP2C9 and VKORC1 genotypes and warfarin dosing. Clin Pharmacol Ther 2011; 90:625–629.
- Limdi NA, McGwin G, Goldstein JA, et al. Influence of CYP2C9 and VKORC1 1173C/T genotype on the risk of hemorrhagic complications in African-American and European-American patients on warfarin. Clin Pharmacol Ther 2008; 83:312–321.
- Anderson JL, Horne BD, Stevens SM, et al. A randomized and clinical effectiveness trial comparing two pharmacogenetic algorithms and standard care for individualizing warfarin dosing (CoumaGen-II). Circulation 2012; 125:1997–2005.
- Yip VL, Pirmohamed M. Expanding role of pharmacogenomics in the management of cardiovascular disorders. Am J Cardiovasc Drugs 2013; 12 Apr; Epub ahead of print.
- Epstein RS, Moyer TP, Aubert RE, et al. Warfarin genotyping reduces hospitalization rates: results from the MM-WES (Medco-Mayo Warfarin Effectiveness Study). J Am Coll Cardiol 2010; 55:2804–2812.
- Wang L, McLeod HL, Weinshilboum RM. Genomics and drug response. N Engl J Med 2011; 364:1144–1153.
- Kitzmiller JP, Groen DK, Phelps MA, Sadee W. Pharmacogenomic testing: relevance in medical practice: why drugs work in some patients but not in others. Cleve Clin J Med 2011; 78:243–257.
- Carlquist JF, Anderson JL. Using pharmacogenetics in real time to guide warfarin initiation: a clinician update. Circulation 2011; 124:2554–2559.
The answer is not clear. There is evidence in favor of pharmacogenetic testing, but not yet enough to strongly recommend it. However, we do believe that physicians should consider it when starting patients on warfarin therapy.
WARFARIN HAS A NARROW THERAPEUTIC WINDOW
Although newer drugs are available, warfarin is still the most commonly used oral anticoagulant for preventing and treating thromboembolism.1 It is highly effective but has a narrow therapeutic window and wide interindividual variability in dosage requirements, which poses challenges to achieving adequate anticoagulation.1–3 Inappropriate dosing contributes to a high rate of bleeding events and emergency room visits.4
Warfarin is monitored using the prothrombin time. Because the prothrombin time varies depending on the assay used, the standardized value called the international normalized ratio (INR) is more commonly used.
Clinical factors such as age, body size, and drug interactions affect warfarin dosage requirements and are important to consider,5 even though they account for only 15% to 20% of the variability in warfarin dose.6
Genetic factors also affect warfarin dosage requirements. The combination of genetic and clinical factors accounts for up to 47% of the dose variability.7
GENES THAT AFFECT WARFARIN
Several genes are known to influence warfarin’s pharmacokinetics and pharmacodynamics. Of these, the two most clinically relevant and well studied are CYP2C9 (which codes for cytochrome P450 2C9) and VKORC1 (which codes for vitamin K epoxide reductase).7 These genes are polymorphic, with some variants producing less-active enzymes that allow warfarin to be more active. Therefore, patients who carry these variants need lower doses of this drug (see below).
CYP2C9 variants
The CYP2C9 gene has several variants. Of these, CYP2C9*2 and CYP2C9*3 are associated with the lowest enzyme activity.
Patients with either of these variants require significantly lower warfarin doses to reach therapeutic levels than those with the wild-type gene (ie, CYP2C9*1). CYP2C9*2 reduces warfarin clearance by 40%, and the CYP2C9*3 variant reduces it by 75%.7 Having a *2 or *3 allele increases the risk of bleeding during warfarin therapy and the time needed to achieve a stable dose.8 Other variants associated with lower warfarin dose requirements are *5, *6, and *11.
The prevalence of these variants is significantly higher in people of European ancestry (roughly one-third) than in Asian people and African Americans,7 although no one has recommended not testing in these low-prevalence populations. Limdi et al9 reported that by including the *5, *6, and *11 variants in genetic testing (in addition to *2 and *3), they could identify more African Americans (9.7%) who carried at least one of these abnormal variants than reported previously. Differences among ethnic groups need to be taken into account when interpreting pharmacogenetic studies.
VKORC1 variants
Patients also need lower doses of warfarin if they carry the VKORC1 −1639G>A variant, and they spend more time with an INR above the therapeutic range and have higher overall INR values. However, having this variant does not appear to increase the risk of bleeding.
The −1639G>A variant is the most common variant of VKORC1. Rarer ones have also been described, but most commercially available tests do not detect them.
Racial differences exist in the prevalence rates of the various VKORC1 polymorphisms, with the most sensitive (low-dose) genotype predominating in Asians and the least sensitive (high-dose) genotype predominating in African Americans. Over 50% of people of European ancestry carry the intermediate-sensitivity genotype (typical dose).7
CURRENT RECOMMENDATIONS FOR OR AGAINST TESTING
FDA labeling
In 2007, the US Food and Drug Administration (FDA) required that the warfarin package insert carry information about initial dosing based on CYP2C9 and VKORC1 testing. This recommendation was revised in 2010 to include a table to help clinicians select an initial warfarin dose if CYP2C9 and VKORC1 genotype information is available. However, the FDA does not require pharmacogenetic testing, leaving the decision to the discretion of the clinician.7
American College of Chest Physicians
The American College of Chest Physicians recommends against routine pharmacogenetic testing (grade 1B) because of a lack of evidence that it improves clinical end points or that it is cost-effective.5
WHAT EVIDENCE SUPORTS GENETIC TESTING TO GUIDE WARFARIN THERAPY?
To date, no large randomized, controlled trial has been published that looked at clinical outcomes with warfarin dosing based on pharmacogenetic testing. However, several smaller studies have suggested it is beneficial.
One trial found that when dosing was informed by pharmacogenetic testing, patients had significantly more time in the therapeutic range, a lower percentage of INRs greater than 4 or less than 1.5, and fewer serious adverse events (death, myocardial infarction, stroke, thromboembolism, and clinically significant bleeding events).10 Patients whose dosage was determined using pharmacogenetic algorithms as opposed to traditional clinical algorithms maintained a therapeutic INR more consistently.11
In addition, compared with historical controls, patients whose physician used pharmacogenetic testing to guide warfarin dosing had a rate of hospitalization 31% lower and a rate of hospitalization specifically for bleeding or thromboembolism 28% lower during 6 months of follow-up.12,13
Several studies have attempted to assess the cost-effectiveness and utility of pharmacogenetic testing in warfarin therapy. As yet, the results have been inconclusive.14 Larger prospective trials are under way and are estimated to be completed in late 2013.15 These include:
- COAG (Clarification of Optimal Anticoagulation Through Genetics)
- GIFT (Genetics Informatics Trial of Warfarin to Prevent Venous Thrombosis)
- EU-PACT (European Pharmacogenetics of Anticoagulant Therapy-Warfarin).
We hope these studies will provide greater clarity on the clinical utility and cost-effectiveness of pharmacogenetic testing to guide warfarin dosing.
HOW SHOULD GENETIC INFORMATION BE USED TO GUIDE OR ALTER THERAPY?
Algorithms are available for estimating initial and maintenance warfarin doses based on genetic information (CYP2C9 and VKORC1), race or ethnicity, age, sex, body mass index, smoking status, and other medications taken. In addition, models incorporating the INR on day 4 and days 6 to 11 have been developed for dose refinement.15 The algorithms explain 30% to 60% of the variability of the data, with lower values for African Americans.7
A well-developed dosing model that includes traditional clinical factors and patient genetic status is publicly available online at www.warfarindosing.org.4
CPIC: A leader in applied pharmacogenetics
In late 2009, PharmGKB joined forces with the Pharmacogenomics Research Network of the National Institutes of Health to form the Clinical Pharmacogenetics Implementation Consortium (CPIC). This organization issues guidelines that are written by expert clinicians and scientists and then are peer-reviewed, published in leading journals, and simultaneously posted to the PharmGKB website along with supplemental information and updates.
CPIC’s goal is to review the current evidence and to address barriers to the adoption of pharmacogenetic testing into clinical practice. Its guidelines do not advise when or which pharmacogenetic tests should be ordered. Rather, they provide guidance on interpreting and applying such testing, should the test results be available.7
CPIC has guidelines on CYP2C9 and VKORC1 genotypes and warfarin dosing.8 If a patient’s genetic information is available, CPIC strongly recommends the use of pharmacogenetic algorithm-based dosing. If such an algorithm is not accessible, use of a genotype dosing table is recommended.8
Monitoring is still needed
Many factors can affect an individual’s response to warfarin above and beyond the above-noted clinical and genetic traits. These include diet, concomitant medications (both prescription and over-the-counter and herbal), and disease state. There may also be additional genetic polymorphisms not yet identified in various racial and ethnic groups that may affect dosing requirements. And as with all medications, patient compliance and dosing errors have a large potential to affect individual response. Therefore, clinicians should still be diligent about clinical monitoring.15
Most useful for initial dose
As with most pharmacogenetic information, the greatest benefit can be achieved when this information is used to guide the initial dose, although there is also some effect noted when this information is known and acted upon into the 2nd week of treatment.8
Patients on long-term warfarin treatment with stable doses and those unable to achieve stable dosing because of variable adherence or dietary vitamin K intake are less likely to benefit from genetic testing.
There are no published guidelines on the utility of pharmacogenetic testing if a patient is already on a stable dose of warfarin or has a known historical stable dose. There are also no published guidelines on changing the frequency of monitoring based on known genotype.
In children, the data are sparse at this time regarding the utility of pharmacogenetically informed dosing.
HOW DOES ONE ORDER TESTING, AND WHAT IS THE COST?
The FDA has approved four warfarin pharmacogenetic test kits. To be used in clinical decision-making, these tests must be done in a laboratory certified by the Clinical Laboratory Improvement Amendments (CLIA) program.
Testing typically costs a few hundred dollars and may take days for results to be returned if not available on site.15 At Cleveland Clinic, CYP2C9 and VKORC1 testing can be run in-house at a cost of about $700. Generally, many third-party payers do not reimburse for testing without a prior-approval process.
TO TEST OR NOT TO TEST
Pharmacogenetic testing is available and may help optimize warfarin dosing early in treatment, as well as help maintain therapeutic INRs more consistently. There is preliminary evidence that using this information to guide dosing improves clinical outcomes. Several large trials are under way to address additional questions of clinical utility, with results expected in the next year. There are also readily available decision-support tools to guide therapeutic dosing, and when pharmacogenetic test results are available, utilization of a warfarin dosing algorithm is recommended.
The largest barrier remaining appears to be cost (relative to perceived benefit), and until larger trials of clinical utility and cost-effectiveness are completed and analyzed, hurdles exist to obtaining coverage for such testing.
If it is readily available (and can be paid for by insurance companies or out-of-pocket) and test results can be obtained within 24 to 48 hours or before prescribing, pharmacogenetic testing can be a valuable tool to guide and manage warfarin dosing. Particularly for patients who want to be as proactive as possible, warfarin pharmacogenetic testing offers the ability to participate in this decision-making and to potentially reduce their risk of adverse drug events. And in view of the evidence and FDA recommendations, we propose that the discussion with our patients is not whether we should consider pharmacogenetic testing, but that we have considered pharmacogenetic testing, and why we have decided for or against it.
The answer is not clear. There is evidence in favor of pharmacogenetic testing, but not yet enough to strongly recommend it. However, we do believe that physicians should consider it when starting patients on warfarin therapy.
WARFARIN HAS A NARROW THERAPEUTIC WINDOW
Although newer drugs are available, warfarin is still the most commonly used oral anticoagulant for preventing and treating thromboembolism.1 It is highly effective but has a narrow therapeutic window and wide interindividual variability in dosage requirements, which poses challenges to achieving adequate anticoagulation.1–3 Inappropriate dosing contributes to a high rate of bleeding events and emergency room visits.4
Warfarin is monitored using the prothrombin time. Because the prothrombin time varies depending on the assay used, the standardized value called the international normalized ratio (INR) is more commonly used.
Clinical factors such as age, body size, and drug interactions affect warfarin dosage requirements and are important to consider,5 even though they account for only 15% to 20% of the variability in warfarin dose.6
Genetic factors also affect warfarin dosage requirements. The combination of genetic and clinical factors accounts for up to 47% of the dose variability.7
GENES THAT AFFECT WARFARIN
Several genes are known to influence warfarin’s pharmacokinetics and pharmacodynamics. Of these, the two most clinically relevant and well studied are CYP2C9 (which codes for cytochrome P450 2C9) and VKORC1 (which codes for vitamin K epoxide reductase).7 These genes are polymorphic, with some variants producing less-active enzymes that allow warfarin to be more active. Therefore, patients who carry these variants need lower doses of this drug (see below).
CYP2C9 variants
The CYP2C9 gene has several variants. Of these, CYP2C9*2 and CYP2C9*3 are associated with the lowest enzyme activity.
Patients with either of these variants require significantly lower warfarin doses to reach therapeutic levels than those with the wild-type gene (ie, CYP2C9*1). CYP2C9*2 reduces warfarin clearance by 40%, and the CYP2C9*3 variant reduces it by 75%.7 Having a *2 or *3 allele increases the risk of bleeding during warfarin therapy and the time needed to achieve a stable dose.8 Other variants associated with lower warfarin dose requirements are *5, *6, and *11.
The prevalence of these variants is significantly higher in people of European ancestry (roughly one-third) than in Asian people and African Americans,7 although no one has recommended not testing in these low-prevalence populations. Limdi et al9 reported that by including the *5, *6, and *11 variants in genetic testing (in addition to *2 and *3), they could identify more African Americans (9.7%) who carried at least one of these abnormal variants than reported previously. Differences among ethnic groups need to be taken into account when interpreting pharmacogenetic studies.
VKORC1 variants
Patients also need lower doses of warfarin if they carry the VKORC1 −1639G>A variant, and they spend more time with an INR above the therapeutic range and have higher overall INR values. However, having this variant does not appear to increase the risk of bleeding.
The −1639G>A variant is the most common variant of VKORC1. Rarer ones have also been described, but most commercially available tests do not detect them.
Racial differences exist in the prevalence rates of the various VKORC1 polymorphisms, with the most sensitive (low-dose) genotype predominating in Asians and the least sensitive (high-dose) genotype predominating in African Americans. Over 50% of people of European ancestry carry the intermediate-sensitivity genotype (typical dose).7
CURRENT RECOMMENDATIONS FOR OR AGAINST TESTING
FDA labeling
In 2007, the US Food and Drug Administration (FDA) required that the warfarin package insert carry information about initial dosing based on CYP2C9 and VKORC1 testing. This recommendation was revised in 2010 to include a table to help clinicians select an initial warfarin dose if CYP2C9 and VKORC1 genotype information is available. However, the FDA does not require pharmacogenetic testing, leaving the decision to the discretion of the clinician.7
American College of Chest Physicians
The American College of Chest Physicians recommends against routine pharmacogenetic testing (grade 1B) because of a lack of evidence that it improves clinical end points or that it is cost-effective.5
WHAT EVIDENCE SUPORTS GENETIC TESTING TO GUIDE WARFARIN THERAPY?
To date, no large randomized, controlled trial has been published that looked at clinical outcomes with warfarin dosing based on pharmacogenetic testing. However, several smaller studies have suggested it is beneficial.
One trial found that when dosing was informed by pharmacogenetic testing, patients had significantly more time in the therapeutic range, a lower percentage of INRs greater than 4 or less than 1.5, and fewer serious adverse events (death, myocardial infarction, stroke, thromboembolism, and clinically significant bleeding events).10 Patients whose dosage was determined using pharmacogenetic algorithms as opposed to traditional clinical algorithms maintained a therapeutic INR more consistently.11
In addition, compared with historical controls, patients whose physician used pharmacogenetic testing to guide warfarin dosing had a rate of hospitalization 31% lower and a rate of hospitalization specifically for bleeding or thromboembolism 28% lower during 6 months of follow-up.12,13
Several studies have attempted to assess the cost-effectiveness and utility of pharmacogenetic testing in warfarin therapy. As yet, the results have been inconclusive.14 Larger prospective trials are under way and are estimated to be completed in late 2013.15 These include:
- COAG (Clarification of Optimal Anticoagulation Through Genetics)
- GIFT (Genetics Informatics Trial of Warfarin to Prevent Venous Thrombosis)
- EU-PACT (European Pharmacogenetics of Anticoagulant Therapy-Warfarin).
We hope these studies will provide greater clarity on the clinical utility and cost-effectiveness of pharmacogenetic testing to guide warfarin dosing.
HOW SHOULD GENETIC INFORMATION BE USED TO GUIDE OR ALTER THERAPY?
Algorithms are available for estimating initial and maintenance warfarin doses based on genetic information (CYP2C9 and VKORC1), race or ethnicity, age, sex, body mass index, smoking status, and other medications taken. In addition, models incorporating the INR on day 4 and days 6 to 11 have been developed for dose refinement.15 The algorithms explain 30% to 60% of the variability of the data, with lower values for African Americans.7
A well-developed dosing model that includes traditional clinical factors and patient genetic status is publicly available online at www.warfarindosing.org.4
CPIC: A leader in applied pharmacogenetics
In late 2009, PharmGKB joined forces with the Pharmacogenomics Research Network of the National Institutes of Health to form the Clinical Pharmacogenetics Implementation Consortium (CPIC). This organization issues guidelines that are written by expert clinicians and scientists and then are peer-reviewed, published in leading journals, and simultaneously posted to the PharmGKB website along with supplemental information and updates.
CPIC’s goal is to review the current evidence and to address barriers to the adoption of pharmacogenetic testing into clinical practice. Its guidelines do not advise when or which pharmacogenetic tests should be ordered. Rather, they provide guidance on interpreting and applying such testing, should the test results be available.7
CPIC has guidelines on CYP2C9 and VKORC1 genotypes and warfarin dosing.8 If a patient’s genetic information is available, CPIC strongly recommends the use of pharmacogenetic algorithm-based dosing. If such an algorithm is not accessible, use of a genotype dosing table is recommended.8
Monitoring is still needed
Many factors can affect an individual’s response to warfarin above and beyond the above-noted clinical and genetic traits. These include diet, concomitant medications (both prescription and over-the-counter and herbal), and disease state. There may also be additional genetic polymorphisms not yet identified in various racial and ethnic groups that may affect dosing requirements. And as with all medications, patient compliance and dosing errors have a large potential to affect individual response. Therefore, clinicians should still be diligent about clinical monitoring.15
Most useful for initial dose
As with most pharmacogenetic information, the greatest benefit can be achieved when this information is used to guide the initial dose, although there is also some effect noted when this information is known and acted upon into the 2nd week of treatment.8
Patients on long-term warfarin treatment with stable doses and those unable to achieve stable dosing because of variable adherence or dietary vitamin K intake are less likely to benefit from genetic testing.
There are no published guidelines on the utility of pharmacogenetic testing if a patient is already on a stable dose of warfarin or has a known historical stable dose. There are also no published guidelines on changing the frequency of monitoring based on known genotype.
In children, the data are sparse at this time regarding the utility of pharmacogenetically informed dosing.
HOW DOES ONE ORDER TESTING, AND WHAT IS THE COST?
The FDA has approved four warfarin pharmacogenetic test kits. To be used in clinical decision-making, these tests must be done in a laboratory certified by the Clinical Laboratory Improvement Amendments (CLIA) program.
Testing typically costs a few hundred dollars and may take days for results to be returned if not available on site.15 At Cleveland Clinic, CYP2C9 and VKORC1 testing can be run in-house at a cost of about $700. Generally, many third-party payers do not reimburse for testing without a prior-approval process.
TO TEST OR NOT TO TEST
Pharmacogenetic testing is available and may help optimize warfarin dosing early in treatment, as well as help maintain therapeutic INRs more consistently. There is preliminary evidence that using this information to guide dosing improves clinical outcomes. Several large trials are under way to address additional questions of clinical utility, with results expected in the next year. There are also readily available decision-support tools to guide therapeutic dosing, and when pharmacogenetic test results are available, utilization of a warfarin dosing algorithm is recommended.
The largest barrier remaining appears to be cost (relative to perceived benefit), and until larger trials of clinical utility and cost-effectiveness are completed and analyzed, hurdles exist to obtaining coverage for such testing.
If it is readily available (and can be paid for by insurance companies or out-of-pocket) and test results can be obtained within 24 to 48 hours or before prescribing, pharmacogenetic testing can be a valuable tool to guide and manage warfarin dosing. Particularly for patients who want to be as proactive as possible, warfarin pharmacogenetic testing offers the ability to participate in this decision-making and to potentially reduce their risk of adverse drug events. And in view of the evidence and FDA recommendations, we propose that the discussion with our patients is not whether we should consider pharmacogenetic testing, but that we have considered pharmacogenetic testing, and why we have decided for or against it.
- Jacobs LG. Warfarin pharmacology, clinical management, and evaluation of hemorrhagic risk for the elderly. Clin Geriatr Med 2006; 22:17–32,vii–viii.
- Rieder MJ, Reiner AP, Gage BF, et al. Effect of VKORC1 haplotypes on transcriptional regulation and warfarin dose. N Engl J Med 2005; 352:2285–2293.
- Higashi MK, Veenstra DL, Kondo LM, et al. Association between CYP2C9 genetic variants and anticoagulation-related outcomes during warfarin therapy. JAMA 2002; 287:1690–1698.
- Shehab N, Sperling LS, Kegler SR, Budnitz DS. National estimates of emergency department visits for hemorrhage-related adverse events from clopidogrel plus aspirin and from warfarin. Arch Intern Med 2010; 170:1926–1933.
- Holbrook A, Schulman S, Witt DM, et al; American College of Chest Physicians. Evidence-based management of anticoagulant therapy: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e152S–e184S.
- Gage BF, Eby C, Johnson JA, et al. Use of pharmacogenetic and clinical factors to predict the therapeutic dose of warfarin. Clin Pharmacol Ther 2008; 84:326–331.
- Cavallari LH, Shin J, Perera MA. Role of pharmacogenomics in the management of traditional and novel oral anticoagulants. Pharmacotherapy 2011; 31:1192–1207.
- Johnson JA, Gong L, Whirl-Carillo M, et al; Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium guidelines for CYP2C9 and VKORC1 genotypes and warfarin dosing. Clin Pharmacol Ther 2011; 90:625–629.
- Limdi NA, McGwin G, Goldstein JA, et al. Influence of CYP2C9 and VKORC1 1173C/T genotype on the risk of hemorrhagic complications in African-American and European-American patients on warfarin. Clin Pharmacol Ther 2008; 83:312–321.
- Anderson JL, Horne BD, Stevens SM, et al. A randomized and clinical effectiveness trial comparing two pharmacogenetic algorithms and standard care for individualizing warfarin dosing (CoumaGen-II). Circulation 2012; 125:1997–2005.
- Yip VL, Pirmohamed M. Expanding role of pharmacogenomics in the management of cardiovascular disorders. Am J Cardiovasc Drugs 2013; 12 Apr; Epub ahead of print.
- Epstein RS, Moyer TP, Aubert RE, et al. Warfarin genotyping reduces hospitalization rates: results from the MM-WES (Medco-Mayo Warfarin Effectiveness Study). J Am Coll Cardiol 2010; 55:2804–2812.
- Wang L, McLeod HL, Weinshilboum RM. Genomics and drug response. N Engl J Med 2011; 364:1144–1153.
- Kitzmiller JP, Groen DK, Phelps MA, Sadee W. Pharmacogenomic testing: relevance in medical practice: why drugs work in some patients but not in others. Cleve Clin J Med 2011; 78:243–257.
- Carlquist JF, Anderson JL. Using pharmacogenetics in real time to guide warfarin initiation: a clinician update. Circulation 2011; 124:2554–2559.
- Jacobs LG. Warfarin pharmacology, clinical management, and evaluation of hemorrhagic risk for the elderly. Clin Geriatr Med 2006; 22:17–32,vii–viii.
- Rieder MJ, Reiner AP, Gage BF, et al. Effect of VKORC1 haplotypes on transcriptional regulation and warfarin dose. N Engl J Med 2005; 352:2285–2293.
- Higashi MK, Veenstra DL, Kondo LM, et al. Association between CYP2C9 genetic variants and anticoagulation-related outcomes during warfarin therapy. JAMA 2002; 287:1690–1698.
- Shehab N, Sperling LS, Kegler SR, Budnitz DS. National estimates of emergency department visits for hemorrhage-related adverse events from clopidogrel plus aspirin and from warfarin. Arch Intern Med 2010; 170:1926–1933.
- Holbrook A, Schulman S, Witt DM, et al; American College of Chest Physicians. Evidence-based management of anticoagulant therapy: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e152S–e184S.
- Gage BF, Eby C, Johnson JA, et al. Use of pharmacogenetic and clinical factors to predict the therapeutic dose of warfarin. Clin Pharmacol Ther 2008; 84:326–331.
- Cavallari LH, Shin J, Perera MA. Role of pharmacogenomics in the management of traditional and novel oral anticoagulants. Pharmacotherapy 2011; 31:1192–1207.
- Johnson JA, Gong L, Whirl-Carillo M, et al; Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium guidelines for CYP2C9 and VKORC1 genotypes and warfarin dosing. Clin Pharmacol Ther 2011; 90:625–629.
- Limdi NA, McGwin G, Goldstein JA, et al. Influence of CYP2C9 and VKORC1 1173C/T genotype on the risk of hemorrhagic complications in African-American and European-American patients on warfarin. Clin Pharmacol Ther 2008; 83:312–321.
- Anderson JL, Horne BD, Stevens SM, et al. A randomized and clinical effectiveness trial comparing two pharmacogenetic algorithms and standard care for individualizing warfarin dosing (CoumaGen-II). Circulation 2012; 125:1997–2005.
- Yip VL, Pirmohamed M. Expanding role of pharmacogenomics in the management of cardiovascular disorders. Am J Cardiovasc Drugs 2013; 12 Apr; Epub ahead of print.
- Epstein RS, Moyer TP, Aubert RE, et al. Warfarin genotyping reduces hospitalization rates: results from the MM-WES (Medco-Mayo Warfarin Effectiveness Study). J Am Coll Cardiol 2010; 55:2804–2812.
- Wang L, McLeod HL, Weinshilboum RM. Genomics and drug response. N Engl J Med 2011; 364:1144–1153.
- Kitzmiller JP, Groen DK, Phelps MA, Sadee W. Pharmacogenomic testing: relevance in medical practice: why drugs work in some patients but not in others. Cleve Clin J Med 2011; 78:243–257.
- Carlquist JF, Anderson JL. Using pharmacogenetics in real time to guide warfarin initiation: a clinician update. Circulation 2011; 124:2554–2559.
The pipe and the plug: Is unblocking arteries enough?

It seems anachronistic that we still debate how best to fix the plumbing of clogged arteries. Our understanding of the pathogenesis of acute coronary syndromes has evolved in leaps and bounds since the first attempts at coronary revascularization. And yet, as Aggarwal et al discuss in their analysis of the FREEDOM trial,1 practical and technical questions about how best to open coronary blockages remain clinically relevant, even as we develop strategies to reverse the atherosclerotic processes that created those blockages.
Many acute coronary events arise not from coronary stenoses but from unstable, vulnerable plaques, which may be a distance away from the stable stenoses and thus undetectable. These unstable plaques, embedded within the remodeled arterial wall and without a protective fibrous cap, may rupture and cause an acute thrombotic occlusion. Statins, aspirin, and perhaps anti-inflammatory drugs (now including colchicine) decrease acute coronary events, likely by interfering with the chain of events initiated by plaque rupture.
So why should coronary artery bypass grafting (CABG) be superior to drug-eluting stents (with antiplatelet therapy) in some diabetic patients, as the FREEDOM trial1 found?
Stenting and balloon dilation repair discrete areas of critical narrowing presumed to be contributing to downstream myocardial ischemia. But areas of vulnerable, non-calcified plaque (with outward remodeling of the vessel wall but generally preserved lumen integrity) may be geographically separated from the identified stenosis and thus be left untreated by stenting. On the other hand, CABG may circumvent “silent” areas of nascent vulnerable plaque that, if left in place, might later rupture and cause acute syndromes or death.
This explanation is clearly hypothetical and one of many possibilities. But paying attention to the new biology of the atherosclerotic process should lead us all to be more aggressive in using treatments shown to reduce the progression of coronary artery disease and the occurrence of acute coronary syndromes. This is especially true in patients with diabetes who are known to have diffuse coronary involvement. So even as we more fully recognize the value of CABG in these patients, perhaps if we intervene earlier—with statins, hypertension control, improved diet, smoking cessation, prevention of chronic kidney disease, antiplatelet therapy, and anti-inflammatory therapy—we will not need it.
- Farkouh ME, Domanski M, Sleeper LA, et al. Strategies for multivessel revascularization in patients with diabetes. N Engl J Med 2012; 367:2375–2384.
It seems anachronistic that we still debate how best to fix the plumbing of clogged arteries. Our understanding of the pathogenesis of acute coronary syndromes has evolved in leaps and bounds since the first attempts at coronary revascularization. And yet, as Aggarwal et al discuss in their analysis of the FREEDOM trial,1 practical and technical questions about how best to open coronary blockages remain clinically relevant, even as we develop strategies to reverse the atherosclerotic processes that created those blockages.
Many acute coronary events arise not from coronary stenoses but from unstable, vulnerable plaques, which may be a distance away from the stable stenoses and thus undetectable. These unstable plaques, embedded within the remodeled arterial wall and without a protective fibrous cap, may rupture and cause an acute thrombotic occlusion. Statins, aspirin, and perhaps anti-inflammatory drugs (now including colchicine) decrease acute coronary events, likely by interfering with the chain of events initiated by plaque rupture.
So why should coronary artery bypass grafting (CABG) be superior to drug-eluting stents (with antiplatelet therapy) in some diabetic patients, as the FREEDOM trial1 found?
Stenting and balloon dilation repair discrete areas of critical narrowing presumed to be contributing to downstream myocardial ischemia. But areas of vulnerable, non-calcified plaque (with outward remodeling of the vessel wall but generally preserved lumen integrity) may be geographically separated from the identified stenosis and thus be left untreated by stenting. On the other hand, CABG may circumvent “silent” areas of nascent vulnerable plaque that, if left in place, might later rupture and cause acute syndromes or death.
This explanation is clearly hypothetical and one of many possibilities. But paying attention to the new biology of the atherosclerotic process should lead us all to be more aggressive in using treatments shown to reduce the progression of coronary artery disease and the occurrence of acute coronary syndromes. This is especially true in patients with diabetes who are known to have diffuse coronary involvement. So even as we more fully recognize the value of CABG in these patients, perhaps if we intervene earlier—with statins, hypertension control, improved diet, smoking cessation, prevention of chronic kidney disease, antiplatelet therapy, and anti-inflammatory therapy—we will not need it.
It seems anachronistic that we still debate how best to fix the plumbing of clogged arteries. Our understanding of the pathogenesis of acute coronary syndromes has evolved in leaps and bounds since the first attempts at coronary revascularization. And yet, as Aggarwal et al discuss in their analysis of the FREEDOM trial,1 practical and technical questions about how best to open coronary blockages remain clinically relevant, even as we develop strategies to reverse the atherosclerotic processes that created those blockages.
Many acute coronary events arise not from coronary stenoses but from unstable, vulnerable plaques, which may be a distance away from the stable stenoses and thus undetectable. These unstable plaques, embedded within the remodeled arterial wall and without a protective fibrous cap, may rupture and cause an acute thrombotic occlusion. Statins, aspirin, and perhaps anti-inflammatory drugs (now including colchicine) decrease acute coronary events, likely by interfering with the chain of events initiated by plaque rupture.
So why should coronary artery bypass grafting (CABG) be superior to drug-eluting stents (with antiplatelet therapy) in some diabetic patients, as the FREEDOM trial1 found?
Stenting and balloon dilation repair discrete areas of critical narrowing presumed to be contributing to downstream myocardial ischemia. But areas of vulnerable, non-calcified plaque (with outward remodeling of the vessel wall but generally preserved lumen integrity) may be geographically separated from the identified stenosis and thus be left untreated by stenting. On the other hand, CABG may circumvent “silent” areas of nascent vulnerable plaque that, if left in place, might later rupture and cause acute syndromes or death.
This explanation is clearly hypothetical and one of many possibilities. But paying attention to the new biology of the atherosclerotic process should lead us all to be more aggressive in using treatments shown to reduce the progression of coronary artery disease and the occurrence of acute coronary syndromes. This is especially true in patients with diabetes who are known to have diffuse coronary involvement. So even as we more fully recognize the value of CABG in these patients, perhaps if we intervene earlier—with statins, hypertension control, improved diet, smoking cessation, prevention of chronic kidney disease, antiplatelet therapy, and anti-inflammatory therapy—we will not need it.
- Farkouh ME, Domanski M, Sleeper LA, et al. Strategies for multivessel revascularization in patients with diabetes. N Engl J Med 2012; 367:2375–2384.
- Farkouh ME, Domanski M, Sleeper LA, et al. Strategies for multivessel revascularization in patients with diabetes. N Engl J Med 2012; 367:2375–2384.
Recurrent abdominal pain and vomiting
A 32-year-old man presents to the emergency department with excruciating abdominal pain associated with multiple episodes of vomiting for the past 2 days. He reports no fevers, headaches, diarrhea, constipation, hematochezia, melena, musculoskeletal symptoms, or weight loss. His abdominal pain is generalized and crampy. It does not radiate and has no precipitating factors. The pain is relieved only with intravenous narcotics.
He does not smoke, drink alcohol, or use illicit drugs. He has no known drug or food allergies. He says that his current condition causes him emotional stress that affects his performance at work.
About a year ago, after a complicated surgical procedure, he needed chronic high-dose narcotics. A few months later, he developed multiple bouts of abdominal pain and vomiting that required hospital visits. He now takes oral oxycodone 10–15 mg every 4–6 hours.
On admission, his vital signs are stable, but he is in excruciating pain. He is alert and oriented to person, place, and time. His sclera are anicteric, and the pupils are equal, round, and reactive to light. Lung and heart examinations are normal. The abdomen is soft and nondistended but tender in all four quadrants without guarding; the liver and spleen are not palpable, and no abdominal masses are detected. He has no skin rash, joint swelling or tenderness, or peripheral edema. The neurologic examination is normal. Computed tomography (CT) of the abdomen with contrast shows no signs of bowel obstruction, pancreatic calcifications or edema, cholecystitis, or hepatobiliary disease. Results of initial laboratory testing are shown in Table 1.
1. Based on the information available, which is the least likely cause of his symptoms?
- Acute pancreatitis
- Cyclic vomiting syndrome
- Acute intermittent porphyria
- Gastroparesis
Acute pancreatitis
Acute pancreatitis is the least likely cause of his symptoms. It is commonly caused by gallstones, alcohol, hypertriglyceridemia, and certain drugs.1 The associated abdominal pain is usually epigastric, radiates to the back, and is accompanied by nausea or vomiting, or both. The onset of pain is sudden and rapidly increases in severity within 30 minutes. CT shows enlargement of the pancreas with diffuse edema, heterogeneity of pancreatic parenchyma, peripancreatic stranding, and peripancreatic fluid collections.1 The diagnosis is based on two of the following three criteria: abdominal pain characteristic of acute pancreatitis; a serum amylase or lipase concentration three or more times the upper limit of normal; and characteristic findings of acute pancreatitis on CT.1
Cyclic vomiting syndrome
Cyclic vomiting syndrome is thought to be caused by episodic dysautonomia, mitochondrial DNA mutations, and hypothalamic emetic response oversensitivity,2–4 but the exact pathogenesis is unknown. The syndrome has been strongly linked to migraine and to the chronic excessive use of cannabinoids.5–9 The Rome III diagnostic criteria10 are the following: the vomiting episodes are stereotypical, ie, they are acute and last for less than 1 week; the patient has had three or more episodes in the previous year; and the patient has no nausea or vomiting between episodes. The patient must meet all three criteria. A history of migraine or a family history of migraine further supports the diagnosis.
Acute intermittent porphyria
Acute intermittent porphyria is characterized by neurovisceral symptoms such as convulsions, paresis, autonomic dysfunction, constipation, and diarrhea that result from the overproduction of porphyrin precursors and deficiency of porphobilinogen deaminase.11
Most patients have poorly localized, severe, steady abdominal pain that develops over hours to days and that may persist for days to weeks.11 Since the pain is neuropathic, abdominal tenderness is usually minimal during an acute attack. Other clues include signs of ileus, such as constipation, nausea, abdominal distention, or decreased bowel sounds; bladder dysfunction, eg, urinary retention, incontinence, or dysuria; reddish-brown urine; and sensory neuropathy of the chest, back, and extremities.11 Blistering skin lesions are usually not seen. The presence of porphobilinogen in the urine confirms the diagnosis.11
Gastroparesis
Gastroparesis is a result of discoordination between the sympathetic and parasympathetic nervous systems, neurons, and smooth muscles within the stomach, causing a decrease in gastric motility. Common causes are diabetes,12 scleroderma,13 and neurologic disorders.14 It can also be iatrogenic,15 resulting from visceral nerve injury and drug treatment with narcotics, calcium channel blockers, muscarinic cholinergic antagonists, or certain antidepressants. Symptoms are related to gastric stasis, ie, abdominal pain from gastric distention, bloating, vomiting, and early satiety.15 Abdominal pain may worsen after eating, and vomitus usually consists of recently ingested food. These patients may have abdominal distension or tenderness and succussion splash. After excluding possible mechanical obstruction, a gastric-emptying study may be necessary to make the diagnosis.15
CASE CONTINUED
A serum and urine drug screen in our patient is positive only for opioids. Urine measures of delta-aminolevulinic acid and porphobilinogen are normal. CT angiography of the abdomen shows no signs of mesenteric vascular occlusion. Esophagogastroduodenoscopy shows antral gastritis, but the esophagus and duodenum appear normal, and colonoscopy is normal as well. Histologic study of biopsy specimens obtained during endoscopy is unrevealing. A gastric-emptying study shows delayed emptying. The patient’s abdominal pain and vomiting persist with the initial dose of intravenous narcotic but resolve with escalating doses. When asked, the patient denies an excessive need for hot baths.
2. Which is the most likely diagnosis at this point?
- Narcotic bowel syndrome
- Opioid withdrawal
- Crohn disease
- Chronic pancreatitis
- Chronic mesenteric ischemia
- Cannabinoid hyperemesis
Narcotic bowel syndrome
Narcotic bowel syndrome is the most likely diagnosis. Grunkemeier et al16 described it as chronic or frequently recurring abdominal pain that is treated with narcotics, either chronically or acutely with high doses, and that includes all the following features16:
- The pain worsens or resolves incompletely with continued or increasing doses of narcotics
- The pain markedly worsens when the narcotic dose is decreased, and decreases when the drug is reinstituted (the “soarand-crash” effect)
- The frequency, duration, and intensity of the pain episodes gradually increase
- The nature of the pain and its intensity are not explained by a current or previous gastrointestinal diagnosis.16
This syndrome is common in patients who receive high doses of narcotics for postoperative pain or for other, nonmalignant causes of pain. Patients eventually become dependent on the drugs but are not aware that chronic use activates and facilitates areas in the brain that enhance the perception of pain.16 A study of a rat model of narcotic bowel syndrome17 showed that morphine-induced hyperalgesia depends on central sensitization involving the activation of spinal microglia. This eventually results in concomitant peripheral sensitization involving the colonic mucosal neuroimmune system, and also in central or peripheral activation of opioid kappa-receptors by dynorphin release.17
Patients tend to present with chronic or intermittent colicky abdominal pain that requires escalating doses of narcotics. Eventually, they develop tachyphylaxis and shortened pain-free periods and will require even higher doses of narcotics. This ultimately enhances the perception of pain and worsens opioid bowel symptoms, causing a vicious circle of pain and more narcotic use.16
Laboratory tests are usually normal, and imaging may show only ileus. Gastric emptying may be delayed in patients who have either narcotic bowel syndrome or gastroparesis, but since abdominal pain from narcotic bowel syndrome is a result of central and visceral hypersensitivity, these patients perceive more severe abdominal pain than patients with gastroparesis alone.
Opioid withdrawal
Symptoms of opioid withdrawal may appear as soon as 6 to 24 hours after cessation of the opioid in patients known to be dependent on opioids. These patients present with crampy abdominal pain with nausea.18 Other symptoms include agitation, rhinorrhea, lacrimation, excessive yawning, arthralgias, papillary dilation, and piloerection.18
Our patient did not have the typical signs of opioid withdrawal.
Crohn disease
Crohn disease is a multisystem disorder with specific clinical and pathologic features. It is characterized by focal, asymmetric, transmural, and occasionally granulomatous inflammation primarily affecting the gastrointestinal tract.19 Characteristic symptoms include abdominal pain, chronic diarrhea with or without rectal bleeding, and weight loss. Extraintestinal signs may include anemia and inflammatory changes in the eyes, skin, and joints. The diagnosis is based on endoscopic, radiographic, and pathologic findings.19
Our patient did not have diarrhea or signs of Crohn disease on CT, endoscopy, or histology.
Chronic pancreatitis
Chronic pancreatitis involves progressive inflammatory changes resulting in permanent structural damage to the pancreas and subsequent exocrine and endocrine dysfunction.20 Patients have epigastric abdominal pain that often radiates to the back20; it is associated with eating and is partly relieved with leaning forward. Symptoms of pancreatic insufficiency such as fat malabsorption (resulting in steatorrhea and fat-soluble vitamin deficiency) and diabetes are common. Calcifications within the pancreas on CT suggest chronic pancreatitis.20 Serum lipase and amylase levels may be normal or slightly elevated.20
Our patient’s abdominal pain was not typical of pancreatitis. He had no signs or symptoms of pancreatic insufficiency and no calcifications within the pancreas.
Chronic mesenteric ischemia
Chronic mesenteric ischemia (“intestinal angina”) is caused by a reduction in intestinal blood flow as a result of occlusion, vasospasm, or hypoperfusion of the mesenteric vasculature.21 It is commonly seen in patients who smoke or who have atherosclerotic vascular disease. These patients have chronic dull or crampy abdominal pain that usually occurs within 1 hour after eating.21 To avoid pain, patients avoid eating, resulting in weight loss.21 CT angiography with multi-detector CT is as effective as angiography (the gold standard) in depicting splanchnic arterial anatomy.22
Our patient is young and has no known risk factors for atherosclerosis such as smoking. His abdominal pain is more intermittent than chronic and is not associated with eating.
Cannabinoid hyperemesis
Cannabinoid hyperemesis should be considered in patients with long-term cannabis use presenting with cyclic vomiting, abdominal pain, compulsive use of hot showers, and improvement of symptoms with cannabis cessation.23 Although cannabinoids have been recognized for their antiemetic effects, long-term use may eventually cause autonomic instability and disturbances in the hypothalamic-pituitary-adrenal axis, resulting in cyclic vomiting and thermoregulatory impairment.23
Although our patient presented with multiple episodes of vomiting and abdominal pain, he denied using marijuana, he tested negative for tetrahydrocannabinol, and he did not associate any relief of his symptoms with hot baths.
CASE CONTINUED
Our patient receives intravenous hydration, antiemetics, and a narcotic in tapering intravenous doses, and his symptoms gradually improve. He is discharged from the hospital. However, a few weeks later he is readmitted with the same symptoms of abdominal pain and nausea.
3. What is the cornerstone of treatment for narcotic bowel syndrome?
- Establishing a therapeutic relationship
- Detoxification
- Supportive management with intravenous fluids, antiemetics, and stool-softeners
- Medical management with a short-acting narcotic, clonidine, lorazepam, and desipramine
MANAGEMENT OF NARCOTIC BOWEL SYNDROME
An effective therapeutic relationship with the patient is the cornerstone of treatment and should be established before starting detoxification.17 The physician must first learn to accept that the patient’s condition is real and must show genuine empathy as well as provide information about the pathophysiologic basis of the condition, the rationale for withholding narcotics, and the detrimental role narcotics play in the vicious circle of pain.
Detoxification involves gradually withdrawing the narcotic and substituting a nonnarcotic such as an antidepressant for pain control, as well as prescribing a drug such as a benzodiazepine or clonidine to prevent withdrawal symptoms and a laxative to prevent constipation.17,24 The physician must reassure the patient that he or she will not be abandoned in pain and that all medications will be continuously adjusted as needed to keep him or her comfortable throughout the detoxification process.17,24 The physician must continuously gauge the patient’s willingness to continue with treatment and must also be readily available to address the patient’s concerns in a timely manner.17,24 Involving family members and friends may provide additional support to the patient. Referral to a functional gastrointestinal motility program, a pain specialist, and a psychologist may also be considered.17,24 Follow-up care is essential, even after the withdrawal program has ended.17,24
BACK TO THE PATIENT
After successfully establishing a therapeutic relationship and discussing the treatment plan with our patient, we started him on the same dosage of narcotic that he had been receiving, calculated in intravenous morphine equivalents to achieve maximal comfort, and then decreased the dosage by 10% to 33% daily until he was completely off narcotics. An antidepressant and a benzodiazepine were given simultaneously with narcotic tapering. Oral clonidine (0.1–0.4 mg/day) was given after the narcotic dosage was reduced to about half, and polyethylene glycol was given as needed for constipation. The total duration of detoxification was 7 days.
The patient was referred to a psychologist for cognitive-behavioral and relaxation therapy, as well as for encouragement and support. At 6 months, he had had no recurrence of symptoms.
TAKE-HOME MESSAGE
In the United States, the number of patients taking a narcotic for nonmalignant pain is increasing, 25 and physicians should be more aware of complications such as narcotic bowel syndrome.
Narcotic bowel syndrome should be suspected in any patient with prolonged narcotic use presenting with multiple recurrent episodes of abdominal pain after other causes are ruled out.
Establishing a good therapeutic relationship with the patient is the cornerstone of successful treatment. Patients who understand their condition and are willing to be treated tend to have better outcomes.
Supportive treatment, symptom relief, and emotional support during detoxification increase compliance.
- Banks PA, Freeman ML; Practice Parameters Committee of the American College of Gastroenterology. Practice guidelines in acute pancreatitis. Am J Gastroenterol 2006; 101:2379–2400.
- Boles RG, Adams K, Ito M, Li BU. Maternal inheritance in cyclic vomiting syndrome with neuromuscular disease. Am J Med Genet A 2003; 120A:474–482.
- Wang Q, Ito M, Adams K, et al. Mitochondrial DNA control region sequence variation in migraine headache and cyclic vomiting syndrome. Am J Med Genet A 2004; 131:50–58.
- Taché Y. Cyclic vomiting syndrome: the corticotropinreleasing-factor hypothesis. Dig Dis Sci 1999; 44(suppl 8):79S–86S.
- Withers GD, Silburn SR, Forbes DA. Precipitants and aetiology of cyclic vomiting syndrome. Acta Paediatr 1998; 87:272–277.
- Whitney HB. Cyclic vomiting. A brief review of this affection as illustrated by a typical case. Arch Pediatr 1898; 15:839–845.
- Stickler GB. Relationship between cyclic vomiting syndrome and migraine. Clin Pediatr (Phila) 2005; 44:505–508.
- Li BU, Murray RD, Heitlinger LA, Robbins JL, Hayes JR. Is cyclic vomiting syndrome related to migraine? J Pediatr 1999; 134:567–572.
- Allen JH, de Moore GM, Heddle R, Twartz JC. Cannabinoid hyperemesis: cyclical hyperemesis in association with chronic cannabis abuse. Gut 2004; 53:1566–1570.
- Rome Foundation. Rome III disorders and diagnostic criteria. http://www.romecriteria.org/criteria/. Accessed February 27, 2013.
- Anderson KE, Bloomer JR, Bonkovsky HL, et al. Recommendations for the diagnosis and treatment of the acute porphyrias. Ann Intern Med 2005; 142:439–450.
- Camilleri M. Clinical practice. Diabetic gastroparesis. N Engl J Med 2007; 356:820–829.
- Maddern GJ, Horowitz M, Jamieson GG, Chatterton BE, Collins PJ, Roberts-Thomson P. Abnormalities of esophageal and gastric emptying in progressive systemic sclerosis. Gastroenterology 1984; 87:922–926.
- Jost WH. Gastrointestinal dysfunction in Parkinson’s disease. J Neurol Sci 2010; 289:69–73.
- Parkman HP, Hasler WL, Fisher RS; American Gastroenterological Association. American Gastroenterological Association technical review on the diagnosis and treatment of gastroparesis. Gastroenterology 2004; 127:1592–1622.
- Grunkemeier DM, Cassara JE, Dalton CB, Drossman DA. The narcotic bowel syndrome: clinical features, pathophysiology, and management. Clin Gastroenterol Hepatol 2007; 5:1126–1139.
- Agostini S, Eutamene H, Cartier C, et al. Evidence of central and peripheral sensitization in a rat model of narcotic bowel-like syndrome. Gastroenterology 2010; 139:553–563,563.e1–e5.
- Nicholls L, Bragaw L, Ruetsch C. Opioid dependence treatment and guidelines. J Manag Care Pharm 2010; 16(1 suppl B):S14–S21.
- Lichtenstein GR, Hanauer SB, Sandborn WJ; Practice Parameters Committee of American College of Gastroenterology. Management of Crohn’s disease in adults. Am J Gastroenterol 2009; 104:465–483.
- Steer ML, Waxman I, Freedman S. Chronic pancreatitis. N Engl J Med 1995; 332:1482–1490.
- American Gastroenterological Association Medical Position Statement: guidelines on intestinal ischemia. Gastroenterology 2000; 118:951–953.
- Savastano S, Teso S, Corrà S, Fantozzi O, Miotto D. Multislice CT angiography of the celiac and superior mesenteric arteries: comparison with arteriographic findings. Radiol Med 2002; 103:456–463.
- Simonetto DA, Oxentenko AS, Herman ML, Szostek JH. Cannabinoid hyperemesis: a case series of 98 patients. Mayo Clin Proc 2012; 87:114–119.
- Drossman DA, Morris CB, Edwards H, et al. Diagnosis, characterization, and 3-month outcome after detoxification of 39 patients with narcotic bowel syndrome. Am J Gastroenterol 2012; 107:1426–1440.
- Trescot AM, Boswell MV, Atluri SL, et al. Opioid guidelines in the management of chronic non-cancer pain. Pain Physician 2006; 9:1–39.
A 32-year-old man presents to the emergency department with excruciating abdominal pain associated with multiple episodes of vomiting for the past 2 days. He reports no fevers, headaches, diarrhea, constipation, hematochezia, melena, musculoskeletal symptoms, or weight loss. His abdominal pain is generalized and crampy. It does not radiate and has no precipitating factors. The pain is relieved only with intravenous narcotics.
He does not smoke, drink alcohol, or use illicit drugs. He has no known drug or food allergies. He says that his current condition causes him emotional stress that affects his performance at work.
About a year ago, after a complicated surgical procedure, he needed chronic high-dose narcotics. A few months later, he developed multiple bouts of abdominal pain and vomiting that required hospital visits. He now takes oral oxycodone 10–15 mg every 4–6 hours.
On admission, his vital signs are stable, but he is in excruciating pain. He is alert and oriented to person, place, and time. His sclera are anicteric, and the pupils are equal, round, and reactive to light. Lung and heart examinations are normal. The abdomen is soft and nondistended but tender in all four quadrants without guarding; the liver and spleen are not palpable, and no abdominal masses are detected. He has no skin rash, joint swelling or tenderness, or peripheral edema. The neurologic examination is normal. Computed tomography (CT) of the abdomen with contrast shows no signs of bowel obstruction, pancreatic calcifications or edema, cholecystitis, or hepatobiliary disease. Results of initial laboratory testing are shown in Table 1.
1. Based on the information available, which is the least likely cause of his symptoms?
- Acute pancreatitis
- Cyclic vomiting syndrome
- Acute intermittent porphyria
- Gastroparesis
Acute pancreatitis
Acute pancreatitis is the least likely cause of his symptoms. It is commonly caused by gallstones, alcohol, hypertriglyceridemia, and certain drugs.1 The associated abdominal pain is usually epigastric, radiates to the back, and is accompanied by nausea or vomiting, or both. The onset of pain is sudden and rapidly increases in severity within 30 minutes. CT shows enlargement of the pancreas with diffuse edema, heterogeneity of pancreatic parenchyma, peripancreatic stranding, and peripancreatic fluid collections.1 The diagnosis is based on two of the following three criteria: abdominal pain characteristic of acute pancreatitis; a serum amylase or lipase concentration three or more times the upper limit of normal; and characteristic findings of acute pancreatitis on CT.1
Cyclic vomiting syndrome
Cyclic vomiting syndrome is thought to be caused by episodic dysautonomia, mitochondrial DNA mutations, and hypothalamic emetic response oversensitivity,2–4 but the exact pathogenesis is unknown. The syndrome has been strongly linked to migraine and to the chronic excessive use of cannabinoids.5–9 The Rome III diagnostic criteria10 are the following: the vomiting episodes are stereotypical, ie, they are acute and last for less than 1 week; the patient has had three or more episodes in the previous year; and the patient has no nausea or vomiting between episodes. The patient must meet all three criteria. A history of migraine or a family history of migraine further supports the diagnosis.
Acute intermittent porphyria
Acute intermittent porphyria is characterized by neurovisceral symptoms such as convulsions, paresis, autonomic dysfunction, constipation, and diarrhea that result from the overproduction of porphyrin precursors and deficiency of porphobilinogen deaminase.11
Most patients have poorly localized, severe, steady abdominal pain that develops over hours to days and that may persist for days to weeks.11 Since the pain is neuropathic, abdominal tenderness is usually minimal during an acute attack. Other clues include signs of ileus, such as constipation, nausea, abdominal distention, or decreased bowel sounds; bladder dysfunction, eg, urinary retention, incontinence, or dysuria; reddish-brown urine; and sensory neuropathy of the chest, back, and extremities.11 Blistering skin lesions are usually not seen. The presence of porphobilinogen in the urine confirms the diagnosis.11
Gastroparesis
Gastroparesis is a result of discoordination between the sympathetic and parasympathetic nervous systems, neurons, and smooth muscles within the stomach, causing a decrease in gastric motility. Common causes are diabetes,12 scleroderma,13 and neurologic disorders.14 It can also be iatrogenic,15 resulting from visceral nerve injury and drug treatment with narcotics, calcium channel blockers, muscarinic cholinergic antagonists, or certain antidepressants. Symptoms are related to gastric stasis, ie, abdominal pain from gastric distention, bloating, vomiting, and early satiety.15 Abdominal pain may worsen after eating, and vomitus usually consists of recently ingested food. These patients may have abdominal distension or tenderness and succussion splash. After excluding possible mechanical obstruction, a gastric-emptying study may be necessary to make the diagnosis.15
CASE CONTINUED
A serum and urine drug screen in our patient is positive only for opioids. Urine measures of delta-aminolevulinic acid and porphobilinogen are normal. CT angiography of the abdomen shows no signs of mesenteric vascular occlusion. Esophagogastroduodenoscopy shows antral gastritis, but the esophagus and duodenum appear normal, and colonoscopy is normal as well. Histologic study of biopsy specimens obtained during endoscopy is unrevealing. A gastric-emptying study shows delayed emptying. The patient’s abdominal pain and vomiting persist with the initial dose of intravenous narcotic but resolve with escalating doses. When asked, the patient denies an excessive need for hot baths.
2. Which is the most likely diagnosis at this point?
- Narcotic bowel syndrome
- Opioid withdrawal
- Crohn disease
- Chronic pancreatitis
- Chronic mesenteric ischemia
- Cannabinoid hyperemesis
Narcotic bowel syndrome
Narcotic bowel syndrome is the most likely diagnosis. Grunkemeier et al16 described it as chronic or frequently recurring abdominal pain that is treated with narcotics, either chronically or acutely with high doses, and that includes all the following features16:
- The pain worsens or resolves incompletely with continued or increasing doses of narcotics
- The pain markedly worsens when the narcotic dose is decreased, and decreases when the drug is reinstituted (the “soarand-crash” effect)
- The frequency, duration, and intensity of the pain episodes gradually increase
- The nature of the pain and its intensity are not explained by a current or previous gastrointestinal diagnosis.16
This syndrome is common in patients who receive high doses of narcotics for postoperative pain or for other, nonmalignant causes of pain. Patients eventually become dependent on the drugs but are not aware that chronic use activates and facilitates areas in the brain that enhance the perception of pain.16 A study of a rat model of narcotic bowel syndrome17 showed that morphine-induced hyperalgesia depends on central sensitization involving the activation of spinal microglia. This eventually results in concomitant peripheral sensitization involving the colonic mucosal neuroimmune system, and also in central or peripheral activation of opioid kappa-receptors by dynorphin release.17
Patients tend to present with chronic or intermittent colicky abdominal pain that requires escalating doses of narcotics. Eventually, they develop tachyphylaxis and shortened pain-free periods and will require even higher doses of narcotics. This ultimately enhances the perception of pain and worsens opioid bowel symptoms, causing a vicious circle of pain and more narcotic use.16
Laboratory tests are usually normal, and imaging may show only ileus. Gastric emptying may be delayed in patients who have either narcotic bowel syndrome or gastroparesis, but since abdominal pain from narcotic bowel syndrome is a result of central and visceral hypersensitivity, these patients perceive more severe abdominal pain than patients with gastroparesis alone.
Opioid withdrawal
Symptoms of opioid withdrawal may appear as soon as 6 to 24 hours after cessation of the opioid in patients known to be dependent on opioids. These patients present with crampy abdominal pain with nausea.18 Other symptoms include agitation, rhinorrhea, lacrimation, excessive yawning, arthralgias, papillary dilation, and piloerection.18
Our patient did not have the typical signs of opioid withdrawal.
Crohn disease
Crohn disease is a multisystem disorder with specific clinical and pathologic features. It is characterized by focal, asymmetric, transmural, and occasionally granulomatous inflammation primarily affecting the gastrointestinal tract.19 Characteristic symptoms include abdominal pain, chronic diarrhea with or without rectal bleeding, and weight loss. Extraintestinal signs may include anemia and inflammatory changes in the eyes, skin, and joints. The diagnosis is based on endoscopic, radiographic, and pathologic findings.19
Our patient did not have diarrhea or signs of Crohn disease on CT, endoscopy, or histology.
Chronic pancreatitis
Chronic pancreatitis involves progressive inflammatory changes resulting in permanent structural damage to the pancreas and subsequent exocrine and endocrine dysfunction.20 Patients have epigastric abdominal pain that often radiates to the back20; it is associated with eating and is partly relieved with leaning forward. Symptoms of pancreatic insufficiency such as fat malabsorption (resulting in steatorrhea and fat-soluble vitamin deficiency) and diabetes are common. Calcifications within the pancreas on CT suggest chronic pancreatitis.20 Serum lipase and amylase levels may be normal or slightly elevated.20
Our patient’s abdominal pain was not typical of pancreatitis. He had no signs or symptoms of pancreatic insufficiency and no calcifications within the pancreas.
Chronic mesenteric ischemia
Chronic mesenteric ischemia (“intestinal angina”) is caused by a reduction in intestinal blood flow as a result of occlusion, vasospasm, or hypoperfusion of the mesenteric vasculature.21 It is commonly seen in patients who smoke or who have atherosclerotic vascular disease. These patients have chronic dull or crampy abdominal pain that usually occurs within 1 hour after eating.21 To avoid pain, patients avoid eating, resulting in weight loss.21 CT angiography with multi-detector CT is as effective as angiography (the gold standard) in depicting splanchnic arterial anatomy.22
Our patient is young and has no known risk factors for atherosclerosis such as smoking. His abdominal pain is more intermittent than chronic and is not associated with eating.
Cannabinoid hyperemesis
Cannabinoid hyperemesis should be considered in patients with long-term cannabis use presenting with cyclic vomiting, abdominal pain, compulsive use of hot showers, and improvement of symptoms with cannabis cessation.23 Although cannabinoids have been recognized for their antiemetic effects, long-term use may eventually cause autonomic instability and disturbances in the hypothalamic-pituitary-adrenal axis, resulting in cyclic vomiting and thermoregulatory impairment.23
Although our patient presented with multiple episodes of vomiting and abdominal pain, he denied using marijuana, he tested negative for tetrahydrocannabinol, and he did not associate any relief of his symptoms with hot baths.
CASE CONTINUED
Our patient receives intravenous hydration, antiemetics, and a narcotic in tapering intravenous doses, and his symptoms gradually improve. He is discharged from the hospital. However, a few weeks later he is readmitted with the same symptoms of abdominal pain and nausea.
3. What is the cornerstone of treatment for narcotic bowel syndrome?
- Establishing a therapeutic relationship
- Detoxification
- Supportive management with intravenous fluids, antiemetics, and stool-softeners
- Medical management with a short-acting narcotic, clonidine, lorazepam, and desipramine
MANAGEMENT OF NARCOTIC BOWEL SYNDROME
An effective therapeutic relationship with the patient is the cornerstone of treatment and should be established before starting detoxification.17 The physician must first learn to accept that the patient’s condition is real and must show genuine empathy as well as provide information about the pathophysiologic basis of the condition, the rationale for withholding narcotics, and the detrimental role narcotics play in the vicious circle of pain.
Detoxification involves gradually withdrawing the narcotic and substituting a nonnarcotic such as an antidepressant for pain control, as well as prescribing a drug such as a benzodiazepine or clonidine to prevent withdrawal symptoms and a laxative to prevent constipation.17,24 The physician must reassure the patient that he or she will not be abandoned in pain and that all medications will be continuously adjusted as needed to keep him or her comfortable throughout the detoxification process.17,24 The physician must continuously gauge the patient’s willingness to continue with treatment and must also be readily available to address the patient’s concerns in a timely manner.17,24 Involving family members and friends may provide additional support to the patient. Referral to a functional gastrointestinal motility program, a pain specialist, and a psychologist may also be considered.17,24 Follow-up care is essential, even after the withdrawal program has ended.17,24
BACK TO THE PATIENT
After successfully establishing a therapeutic relationship and discussing the treatment plan with our patient, we started him on the same dosage of narcotic that he had been receiving, calculated in intravenous morphine equivalents to achieve maximal comfort, and then decreased the dosage by 10% to 33% daily until he was completely off narcotics. An antidepressant and a benzodiazepine were given simultaneously with narcotic tapering. Oral clonidine (0.1–0.4 mg/day) was given after the narcotic dosage was reduced to about half, and polyethylene glycol was given as needed for constipation. The total duration of detoxification was 7 days.
The patient was referred to a psychologist for cognitive-behavioral and relaxation therapy, as well as for encouragement and support. At 6 months, he had had no recurrence of symptoms.
TAKE-HOME MESSAGE
In the United States, the number of patients taking a narcotic for nonmalignant pain is increasing, 25 and physicians should be more aware of complications such as narcotic bowel syndrome.
Narcotic bowel syndrome should be suspected in any patient with prolonged narcotic use presenting with multiple recurrent episodes of abdominal pain after other causes are ruled out.
Establishing a good therapeutic relationship with the patient is the cornerstone of successful treatment. Patients who understand their condition and are willing to be treated tend to have better outcomes.
Supportive treatment, symptom relief, and emotional support during detoxification increase compliance.
A 32-year-old man presents to the emergency department with excruciating abdominal pain associated with multiple episodes of vomiting for the past 2 days. He reports no fevers, headaches, diarrhea, constipation, hematochezia, melena, musculoskeletal symptoms, or weight loss. His abdominal pain is generalized and crampy. It does not radiate and has no precipitating factors. The pain is relieved only with intravenous narcotics.
He does not smoke, drink alcohol, or use illicit drugs. He has no known drug or food allergies. He says that his current condition causes him emotional stress that affects his performance at work.
About a year ago, after a complicated surgical procedure, he needed chronic high-dose narcotics. A few months later, he developed multiple bouts of abdominal pain and vomiting that required hospital visits. He now takes oral oxycodone 10–15 mg every 4–6 hours.
On admission, his vital signs are stable, but he is in excruciating pain. He is alert and oriented to person, place, and time. His sclera are anicteric, and the pupils are equal, round, and reactive to light. Lung and heart examinations are normal. The abdomen is soft and nondistended but tender in all four quadrants without guarding; the liver and spleen are not palpable, and no abdominal masses are detected. He has no skin rash, joint swelling or tenderness, or peripheral edema. The neurologic examination is normal. Computed tomography (CT) of the abdomen with contrast shows no signs of bowel obstruction, pancreatic calcifications or edema, cholecystitis, or hepatobiliary disease. Results of initial laboratory testing are shown in Table 1.
1. Based on the information available, which is the least likely cause of his symptoms?
- Acute pancreatitis
- Cyclic vomiting syndrome
- Acute intermittent porphyria
- Gastroparesis
Acute pancreatitis
Acute pancreatitis is the least likely cause of his symptoms. It is commonly caused by gallstones, alcohol, hypertriglyceridemia, and certain drugs.1 The associated abdominal pain is usually epigastric, radiates to the back, and is accompanied by nausea or vomiting, or both. The onset of pain is sudden and rapidly increases in severity within 30 minutes. CT shows enlargement of the pancreas with diffuse edema, heterogeneity of pancreatic parenchyma, peripancreatic stranding, and peripancreatic fluid collections.1 The diagnosis is based on two of the following three criteria: abdominal pain characteristic of acute pancreatitis; a serum amylase or lipase concentration three or more times the upper limit of normal; and characteristic findings of acute pancreatitis on CT.1
Cyclic vomiting syndrome
Cyclic vomiting syndrome is thought to be caused by episodic dysautonomia, mitochondrial DNA mutations, and hypothalamic emetic response oversensitivity,2–4 but the exact pathogenesis is unknown. The syndrome has been strongly linked to migraine and to the chronic excessive use of cannabinoids.5–9 The Rome III diagnostic criteria10 are the following: the vomiting episodes are stereotypical, ie, they are acute and last for less than 1 week; the patient has had three or more episodes in the previous year; and the patient has no nausea or vomiting between episodes. The patient must meet all three criteria. A history of migraine or a family history of migraine further supports the diagnosis.
Acute intermittent porphyria
Acute intermittent porphyria is characterized by neurovisceral symptoms such as convulsions, paresis, autonomic dysfunction, constipation, and diarrhea that result from the overproduction of porphyrin precursors and deficiency of porphobilinogen deaminase.11
Most patients have poorly localized, severe, steady abdominal pain that develops over hours to days and that may persist for days to weeks.11 Since the pain is neuropathic, abdominal tenderness is usually minimal during an acute attack. Other clues include signs of ileus, such as constipation, nausea, abdominal distention, or decreased bowel sounds; bladder dysfunction, eg, urinary retention, incontinence, or dysuria; reddish-brown urine; and sensory neuropathy of the chest, back, and extremities.11 Blistering skin lesions are usually not seen. The presence of porphobilinogen in the urine confirms the diagnosis.11
Gastroparesis
Gastroparesis is a result of discoordination between the sympathetic and parasympathetic nervous systems, neurons, and smooth muscles within the stomach, causing a decrease in gastric motility. Common causes are diabetes,12 scleroderma,13 and neurologic disorders.14 It can also be iatrogenic,15 resulting from visceral nerve injury and drug treatment with narcotics, calcium channel blockers, muscarinic cholinergic antagonists, or certain antidepressants. Symptoms are related to gastric stasis, ie, abdominal pain from gastric distention, bloating, vomiting, and early satiety.15 Abdominal pain may worsen after eating, and vomitus usually consists of recently ingested food. These patients may have abdominal distension or tenderness and succussion splash. After excluding possible mechanical obstruction, a gastric-emptying study may be necessary to make the diagnosis.15
CASE CONTINUED
A serum and urine drug screen in our patient is positive only for opioids. Urine measures of delta-aminolevulinic acid and porphobilinogen are normal. CT angiography of the abdomen shows no signs of mesenteric vascular occlusion. Esophagogastroduodenoscopy shows antral gastritis, but the esophagus and duodenum appear normal, and colonoscopy is normal as well. Histologic study of biopsy specimens obtained during endoscopy is unrevealing. A gastric-emptying study shows delayed emptying. The patient’s abdominal pain and vomiting persist with the initial dose of intravenous narcotic but resolve with escalating doses. When asked, the patient denies an excessive need for hot baths.
2. Which is the most likely diagnosis at this point?
- Narcotic bowel syndrome
- Opioid withdrawal
- Crohn disease
- Chronic pancreatitis
- Chronic mesenteric ischemia
- Cannabinoid hyperemesis
Narcotic bowel syndrome
Narcotic bowel syndrome is the most likely diagnosis. Grunkemeier et al16 described it as chronic or frequently recurring abdominal pain that is treated with narcotics, either chronically or acutely with high doses, and that includes all the following features16:
- The pain worsens or resolves incompletely with continued or increasing doses of narcotics
- The pain markedly worsens when the narcotic dose is decreased, and decreases when the drug is reinstituted (the “soarand-crash” effect)
- The frequency, duration, and intensity of the pain episodes gradually increase
- The nature of the pain and its intensity are not explained by a current or previous gastrointestinal diagnosis.16
This syndrome is common in patients who receive high doses of narcotics for postoperative pain or for other, nonmalignant causes of pain. Patients eventually become dependent on the drugs but are not aware that chronic use activates and facilitates areas in the brain that enhance the perception of pain.16 A study of a rat model of narcotic bowel syndrome17 showed that morphine-induced hyperalgesia depends on central sensitization involving the activation of spinal microglia. This eventually results in concomitant peripheral sensitization involving the colonic mucosal neuroimmune system, and also in central or peripheral activation of opioid kappa-receptors by dynorphin release.17
Patients tend to present with chronic or intermittent colicky abdominal pain that requires escalating doses of narcotics. Eventually, they develop tachyphylaxis and shortened pain-free periods and will require even higher doses of narcotics. This ultimately enhances the perception of pain and worsens opioid bowel symptoms, causing a vicious circle of pain and more narcotic use.16
Laboratory tests are usually normal, and imaging may show only ileus. Gastric emptying may be delayed in patients who have either narcotic bowel syndrome or gastroparesis, but since abdominal pain from narcotic bowel syndrome is a result of central and visceral hypersensitivity, these patients perceive more severe abdominal pain than patients with gastroparesis alone.
Opioid withdrawal
Symptoms of opioid withdrawal may appear as soon as 6 to 24 hours after cessation of the opioid in patients known to be dependent on opioids. These patients present with crampy abdominal pain with nausea.18 Other symptoms include agitation, rhinorrhea, lacrimation, excessive yawning, arthralgias, papillary dilation, and piloerection.18
Our patient did not have the typical signs of opioid withdrawal.
Crohn disease
Crohn disease is a multisystem disorder with specific clinical and pathologic features. It is characterized by focal, asymmetric, transmural, and occasionally granulomatous inflammation primarily affecting the gastrointestinal tract.19 Characteristic symptoms include abdominal pain, chronic diarrhea with or without rectal bleeding, and weight loss. Extraintestinal signs may include anemia and inflammatory changes in the eyes, skin, and joints. The diagnosis is based on endoscopic, radiographic, and pathologic findings.19
Our patient did not have diarrhea or signs of Crohn disease on CT, endoscopy, or histology.
Chronic pancreatitis
Chronic pancreatitis involves progressive inflammatory changes resulting in permanent structural damage to the pancreas and subsequent exocrine and endocrine dysfunction.20 Patients have epigastric abdominal pain that often radiates to the back20; it is associated with eating and is partly relieved with leaning forward. Symptoms of pancreatic insufficiency such as fat malabsorption (resulting in steatorrhea and fat-soluble vitamin deficiency) and diabetes are common. Calcifications within the pancreas on CT suggest chronic pancreatitis.20 Serum lipase and amylase levels may be normal or slightly elevated.20
Our patient’s abdominal pain was not typical of pancreatitis. He had no signs or symptoms of pancreatic insufficiency and no calcifications within the pancreas.
Chronic mesenteric ischemia
Chronic mesenteric ischemia (“intestinal angina”) is caused by a reduction in intestinal blood flow as a result of occlusion, vasospasm, or hypoperfusion of the mesenteric vasculature.21 It is commonly seen in patients who smoke or who have atherosclerotic vascular disease. These patients have chronic dull or crampy abdominal pain that usually occurs within 1 hour after eating.21 To avoid pain, patients avoid eating, resulting in weight loss.21 CT angiography with multi-detector CT is as effective as angiography (the gold standard) in depicting splanchnic arterial anatomy.22
Our patient is young and has no known risk factors for atherosclerosis such as smoking. His abdominal pain is more intermittent than chronic and is not associated with eating.
Cannabinoid hyperemesis
Cannabinoid hyperemesis should be considered in patients with long-term cannabis use presenting with cyclic vomiting, abdominal pain, compulsive use of hot showers, and improvement of symptoms with cannabis cessation.23 Although cannabinoids have been recognized for their antiemetic effects, long-term use may eventually cause autonomic instability and disturbances in the hypothalamic-pituitary-adrenal axis, resulting in cyclic vomiting and thermoregulatory impairment.23
Although our patient presented with multiple episodes of vomiting and abdominal pain, he denied using marijuana, he tested negative for tetrahydrocannabinol, and he did not associate any relief of his symptoms with hot baths.
CASE CONTINUED
Our patient receives intravenous hydration, antiemetics, and a narcotic in tapering intravenous doses, and his symptoms gradually improve. He is discharged from the hospital. However, a few weeks later he is readmitted with the same symptoms of abdominal pain and nausea.
3. What is the cornerstone of treatment for narcotic bowel syndrome?
- Establishing a therapeutic relationship
- Detoxification
- Supportive management with intravenous fluids, antiemetics, and stool-softeners
- Medical management with a short-acting narcotic, clonidine, lorazepam, and desipramine
MANAGEMENT OF NARCOTIC BOWEL SYNDROME
An effective therapeutic relationship with the patient is the cornerstone of treatment and should be established before starting detoxification.17 The physician must first learn to accept that the patient’s condition is real and must show genuine empathy as well as provide information about the pathophysiologic basis of the condition, the rationale for withholding narcotics, and the detrimental role narcotics play in the vicious circle of pain.
Detoxification involves gradually withdrawing the narcotic and substituting a nonnarcotic such as an antidepressant for pain control, as well as prescribing a drug such as a benzodiazepine or clonidine to prevent withdrawal symptoms and a laxative to prevent constipation.17,24 The physician must reassure the patient that he or she will not be abandoned in pain and that all medications will be continuously adjusted as needed to keep him or her comfortable throughout the detoxification process.17,24 The physician must continuously gauge the patient’s willingness to continue with treatment and must also be readily available to address the patient’s concerns in a timely manner.17,24 Involving family members and friends may provide additional support to the patient. Referral to a functional gastrointestinal motility program, a pain specialist, and a psychologist may also be considered.17,24 Follow-up care is essential, even after the withdrawal program has ended.17,24
BACK TO THE PATIENT
After successfully establishing a therapeutic relationship and discussing the treatment plan with our patient, we started him on the same dosage of narcotic that he had been receiving, calculated in intravenous morphine equivalents to achieve maximal comfort, and then decreased the dosage by 10% to 33% daily until he was completely off narcotics. An antidepressant and a benzodiazepine were given simultaneously with narcotic tapering. Oral clonidine (0.1–0.4 mg/day) was given after the narcotic dosage was reduced to about half, and polyethylene glycol was given as needed for constipation. The total duration of detoxification was 7 days.
The patient was referred to a psychologist for cognitive-behavioral and relaxation therapy, as well as for encouragement and support. At 6 months, he had had no recurrence of symptoms.
TAKE-HOME MESSAGE
In the United States, the number of patients taking a narcotic for nonmalignant pain is increasing, 25 and physicians should be more aware of complications such as narcotic bowel syndrome.
Narcotic bowel syndrome should be suspected in any patient with prolonged narcotic use presenting with multiple recurrent episodes of abdominal pain after other causes are ruled out.
Establishing a good therapeutic relationship with the patient is the cornerstone of successful treatment. Patients who understand their condition and are willing to be treated tend to have better outcomes.
Supportive treatment, symptom relief, and emotional support during detoxification increase compliance.
- Banks PA, Freeman ML; Practice Parameters Committee of the American College of Gastroenterology. Practice guidelines in acute pancreatitis. Am J Gastroenterol 2006; 101:2379–2400.
- Boles RG, Adams K, Ito M, Li BU. Maternal inheritance in cyclic vomiting syndrome with neuromuscular disease. Am J Med Genet A 2003; 120A:474–482.
- Wang Q, Ito M, Adams K, et al. Mitochondrial DNA control region sequence variation in migraine headache and cyclic vomiting syndrome. Am J Med Genet A 2004; 131:50–58.
- Taché Y. Cyclic vomiting syndrome: the corticotropinreleasing-factor hypothesis. Dig Dis Sci 1999; 44(suppl 8):79S–86S.
- Withers GD, Silburn SR, Forbes DA. Precipitants and aetiology of cyclic vomiting syndrome. Acta Paediatr 1998; 87:272–277.
- Whitney HB. Cyclic vomiting. A brief review of this affection as illustrated by a typical case. Arch Pediatr 1898; 15:839–845.
- Stickler GB. Relationship between cyclic vomiting syndrome and migraine. Clin Pediatr (Phila) 2005; 44:505–508.
- Li BU, Murray RD, Heitlinger LA, Robbins JL, Hayes JR. Is cyclic vomiting syndrome related to migraine? J Pediatr 1999; 134:567–572.
- Allen JH, de Moore GM, Heddle R, Twartz JC. Cannabinoid hyperemesis: cyclical hyperemesis in association with chronic cannabis abuse. Gut 2004; 53:1566–1570.
- Rome Foundation. Rome III disorders and diagnostic criteria. http://www.romecriteria.org/criteria/. Accessed February 27, 2013.
- Anderson KE, Bloomer JR, Bonkovsky HL, et al. Recommendations for the diagnosis and treatment of the acute porphyrias. Ann Intern Med 2005; 142:439–450.
- Camilleri M. Clinical practice. Diabetic gastroparesis. N Engl J Med 2007; 356:820–829.
- Maddern GJ, Horowitz M, Jamieson GG, Chatterton BE, Collins PJ, Roberts-Thomson P. Abnormalities of esophageal and gastric emptying in progressive systemic sclerosis. Gastroenterology 1984; 87:922–926.
- Jost WH. Gastrointestinal dysfunction in Parkinson’s disease. J Neurol Sci 2010; 289:69–73.
- Parkman HP, Hasler WL, Fisher RS; American Gastroenterological Association. American Gastroenterological Association technical review on the diagnosis and treatment of gastroparesis. Gastroenterology 2004; 127:1592–1622.
- Grunkemeier DM, Cassara JE, Dalton CB, Drossman DA. The narcotic bowel syndrome: clinical features, pathophysiology, and management. Clin Gastroenterol Hepatol 2007; 5:1126–1139.
- Agostini S, Eutamene H, Cartier C, et al. Evidence of central and peripheral sensitization in a rat model of narcotic bowel-like syndrome. Gastroenterology 2010; 139:553–563,563.e1–e5.
- Nicholls L, Bragaw L, Ruetsch C. Opioid dependence treatment and guidelines. J Manag Care Pharm 2010; 16(1 suppl B):S14–S21.
- Lichtenstein GR, Hanauer SB, Sandborn WJ; Practice Parameters Committee of American College of Gastroenterology. Management of Crohn’s disease in adults. Am J Gastroenterol 2009; 104:465–483.
- Steer ML, Waxman I, Freedman S. Chronic pancreatitis. N Engl J Med 1995; 332:1482–1490.
- American Gastroenterological Association Medical Position Statement: guidelines on intestinal ischemia. Gastroenterology 2000; 118:951–953.
- Savastano S, Teso S, Corrà S, Fantozzi O, Miotto D. Multislice CT angiography of the celiac and superior mesenteric arteries: comparison with arteriographic findings. Radiol Med 2002; 103:456–463.
- Simonetto DA, Oxentenko AS, Herman ML, Szostek JH. Cannabinoid hyperemesis: a case series of 98 patients. Mayo Clin Proc 2012; 87:114–119.
- Drossman DA, Morris CB, Edwards H, et al. Diagnosis, characterization, and 3-month outcome after detoxification of 39 patients with narcotic bowel syndrome. Am J Gastroenterol 2012; 107:1426–1440.
- Trescot AM, Boswell MV, Atluri SL, et al. Opioid guidelines in the management of chronic non-cancer pain. Pain Physician 2006; 9:1–39.
- Banks PA, Freeman ML; Practice Parameters Committee of the American College of Gastroenterology. Practice guidelines in acute pancreatitis. Am J Gastroenterol 2006; 101:2379–2400.
- Boles RG, Adams K, Ito M, Li BU. Maternal inheritance in cyclic vomiting syndrome with neuromuscular disease. Am J Med Genet A 2003; 120A:474–482.
- Wang Q, Ito M, Adams K, et al. Mitochondrial DNA control region sequence variation in migraine headache and cyclic vomiting syndrome. Am J Med Genet A 2004; 131:50–58.
- Taché Y. Cyclic vomiting syndrome: the corticotropinreleasing-factor hypothesis. Dig Dis Sci 1999; 44(suppl 8):79S–86S.
- Withers GD, Silburn SR, Forbes DA. Precipitants and aetiology of cyclic vomiting syndrome. Acta Paediatr 1998; 87:272–277.
- Whitney HB. Cyclic vomiting. A brief review of this affection as illustrated by a typical case. Arch Pediatr 1898; 15:839–845.
- Stickler GB. Relationship between cyclic vomiting syndrome and migraine. Clin Pediatr (Phila) 2005; 44:505–508.
- Li BU, Murray RD, Heitlinger LA, Robbins JL, Hayes JR. Is cyclic vomiting syndrome related to migraine? J Pediatr 1999; 134:567–572.
- Allen JH, de Moore GM, Heddle R, Twartz JC. Cannabinoid hyperemesis: cyclical hyperemesis in association with chronic cannabis abuse. Gut 2004; 53:1566–1570.
- Rome Foundation. Rome III disorders and diagnostic criteria. http://www.romecriteria.org/criteria/. Accessed February 27, 2013.
- Anderson KE, Bloomer JR, Bonkovsky HL, et al. Recommendations for the diagnosis and treatment of the acute porphyrias. Ann Intern Med 2005; 142:439–450.
- Camilleri M. Clinical practice. Diabetic gastroparesis. N Engl J Med 2007; 356:820–829.
- Maddern GJ, Horowitz M, Jamieson GG, Chatterton BE, Collins PJ, Roberts-Thomson P. Abnormalities of esophageal and gastric emptying in progressive systemic sclerosis. Gastroenterology 1984; 87:922–926.
- Jost WH. Gastrointestinal dysfunction in Parkinson’s disease. J Neurol Sci 2010; 289:69–73.
- Parkman HP, Hasler WL, Fisher RS; American Gastroenterological Association. American Gastroenterological Association technical review on the diagnosis and treatment of gastroparesis. Gastroenterology 2004; 127:1592–1622.
- Grunkemeier DM, Cassara JE, Dalton CB, Drossman DA. The narcotic bowel syndrome: clinical features, pathophysiology, and management. Clin Gastroenterol Hepatol 2007; 5:1126–1139.
- Agostini S, Eutamene H, Cartier C, et al. Evidence of central and peripheral sensitization in a rat model of narcotic bowel-like syndrome. Gastroenterology 2010; 139:553–563,563.e1–e5.
- Nicholls L, Bragaw L, Ruetsch C. Opioid dependence treatment and guidelines. J Manag Care Pharm 2010; 16(1 suppl B):S14–S21.
- Lichtenstein GR, Hanauer SB, Sandborn WJ; Practice Parameters Committee of American College of Gastroenterology. Management of Crohn’s disease in adults. Am J Gastroenterol 2009; 104:465–483.
- Steer ML, Waxman I, Freedman S. Chronic pancreatitis. N Engl J Med 1995; 332:1482–1490.
- American Gastroenterological Association Medical Position Statement: guidelines on intestinal ischemia. Gastroenterology 2000; 118:951–953.
- Savastano S, Teso S, Corrà S, Fantozzi O, Miotto D. Multislice CT angiography of the celiac and superior mesenteric arteries: comparison with arteriographic findings. Radiol Med 2002; 103:456–463.
- Simonetto DA, Oxentenko AS, Herman ML, Szostek JH. Cannabinoid hyperemesis: a case series of 98 patients. Mayo Clin Proc 2012; 87:114–119.
- Drossman DA, Morris CB, Edwards H, et al. Diagnosis, characterization, and 3-month outcome after detoxification of 39 patients with narcotic bowel syndrome. Am J Gastroenterol 2012; 107:1426–1440.
- Trescot AM, Boswell MV, Atluri SL, et al. Opioid guidelines in the management of chronic non-cancer pain. Pain Physician 2006; 9:1–39.
Paget disease of bone: Diagnosis and drug therapy
Paget disease of bone is a focal disorder of the aging skeleton that can be asymptomatic or can present with pain, bowing deformities, fractures, or nonspecific rheumatic complaints. Physicians often discover it in asymptomatic patients when serum alkaline phosphatase levels are elevated or as an incidental finding on radiography. Despite evidence of germline mutations and polymorphisms that predispose to Paget disease, the environmental determinants that permit disease expression in older people remain unknown.
A STRIKING GEOGRAPHIC DISTRIBUTION
Researchers have been studying the determinants and distribution of Paget disease ever since Sir James Paget first described it in 1877.1
Paget disease has a predilection for the axial skeleton, particularly the lumbosacral spine and pelvis, as well as the skull, femur, and tibia.2 Knowing this, investigators have used screening plain films of the abdomen (kidney-ureter-bladder views) to estimate its prevalence in different populations, as these images capture the lumbosacral spine, pelvis, and proximal femurs. Other means of assessing prevalence have included autopsy series, questionnaires, and screens for biochemical markers of bone turnover, such as elevated serum alkaline phosphatase from bone.3–6
Using these methods, Paget disease has been estimated to occur in 1% to 3% of people over age 55, and in as many as 8% of people over age 80 in certain countries.7
This disease has a striking geographic distribution, being frequent in Europe, Canada, the United States, Australia, New Zealand, and cities of South America, but rare in Scandinavia and Japan. It seems to be equally rare in other countries of the Far East and in India, Russia, and Africa, although its prevalence in these areas has not been thoroughly investigated.8
That it is an ancient disease has been corroborated by excavations in churchyards in Great Britain.9,10 It may be familial or sporadic, but its expression is delayed until late middle age in most persons, and it does not occur in children. For reasons unclear, the prevalence seems to be decreasing in many countries.11–13
GENETICS IS NOT THE WHOLE STORY
The variable prevalence of Paget disease in different geographic regions and its sometimes-familial expression suggest a genetic predisposition, environmental factor, or both.
Mutations in SQSTM1
In 2002, scientists investigating a cohort of French Canadian families found a mutation in the SQSTM1 gene that was present in almost 50% of people with familial Paget disease and in 16% of those with sporadic Paget disease.14 Hocking and his colleagues in the United Kingdom subsequently found the same mutation in 19% of cases of familial Paget disease and in 9% of sporadic cases.15
Further, investigators noted that the mutation was often present on a conserved haplotype, consistent with a stable genetic change occurring in the affected population.16 This observation of a “founder effect” dovetailed with the epidemiology of Paget disease,17 but only with this SQSTM1 mutation.

Throughout Europe, Australia, and the United States, comparable rates of the SQSTM1 mutation were reported in or around the ubiquitin-associated domain. Several specific mutations exist, the most common one being P392L, ie, a prolineto-leucine substitution at amino acid 392. Scientists have tried to correlate severity of disease with genotype, but the findings have been inconsistent.18–21
Investigations into the mechanism of disease have pointed to the role of p62, the product of SQSTM1, in signaling osteoclast activation via nuclear factor kappa B. Since this initial discovery, polymorphisms in the genes affecting osteoclast maturation, activation, and fusion pathways have been shown to predispose to Paget disease. Examples:
- TNFRSF11A, which codes for receptor activator of nuclear factor kappa B, or RANK
- TNFRSF11B, which codes for osteoprotegerin, or OPG
- CSF1, which codes for macrophage colony-stimulating factor 1, and
- OPTN, which codes for optineurin, a member of the nuclear factor kappa B-modulating protein family.
Clinicians interested in these details can read an excellent review of the pathogenesis of Paget disease.22
Other possible factors
Although there is good evidence that measles and canine distemper virus can infect osteoclasts and modify their phenotype, there is no good evidence that these infections by themselves cause Paget disease.23–25 It is, however, tempting to think of these RNA paramyxoviruses as precipitating factors; conceivably, an infectious agent might seed the ends of long bones, accounting for the fixed distribution of Paget disease and its late expression.
Epidemiologic studies from around the world have failed to identify conclusively any environmental exposure that predisposes to Paget disease, although a rural setting, trauma, infection, and milk ingestion have all been proposed.26–28 It is also possible that as bone ages and the marrow becomes less cellular and more fatty, these changes may permit the disease to develop.
The greatest risk factor for Paget disease is perhaps aging, followed by ancestry and a known family history of it. That genetics is not the whole story is evident by reports of people with SQSTM1 mutations who show no clinical evidence of Paget disease in their old age, and patients with Paget disease who have no SQSTM1 mutation.20,29
CLINICAL PRESENTATION
Most patients with Paget disease have no symptoms and come to medical attention because of an elevated serum alkaline phosphatase level or characteristic findings on radiographs ordered for other indications.11 Paget disease is the second most common disorder of aging bone after osteoporosis. Yet unlike osteoporosis, which presents as a systemic fragility of bone, the clinical manifestations of Paget disease depend on which bones are affected and how enlarged or misshapen they have become.
Common complications
As a consequence of this abnormal bone remodeling and overgrowth, many patients present with bone pain. Bone deformity, headache, and hearing loss may also occur (Figure 1), as well as fractures and nerve compression syndromes (eg, spinal stenosis, sciatica, cauda equina syndrome).

It is important to remember that “pagetic” bone may not be the source of pain, and that functional impairment caused by degenerative changes at affected sites is common (Figure 2).30,31
In a study from the New England Registry for Paget’s Disease,32 most patients knew fairly well which bones were affected and what complications resulted from this when deformity, fracture, or total joint replacement had occurred.32 Although Paget disease did affect their quality of life as measured by physical functioning on the Short Form-12 assessment, these impairments did not seem to affect their outlook, which was as good as or better than that in other people their age.
Metabolic complications

Metabolic complications of Paget disease are rare today but can occur in an elderly patient who has active, polyostotic (multibone) disease.33 The accelerated rate of bone remodeling and the increased vascularity of pagetic bone have been reported to lead to high-output heart failure. In theory, treatment should ease this by diminishing blood flow to pagetic bone and restoring bone turnover to more normal levels.34
Hypercalcemia can occur when patients with Paget disease are immobilized for any reason, and there is probably a higher incidence of renal stones in patients with Paget disease.35,36
Malignant complications
Osteosarcoma rarely arises in pagetic bone. Yet Paget disease may account for a significant number of cases of this cancer in the elderly.37 In these cases, osteosarcoma is presumed to be driven by a second genetic mutation, has a genetic signature distinct from that in osteosarcomas occurring in youth, and is quite resistant to treatment.38 In Scandinavia and Japan, where Paget disease is rare, the second peak of osteosarcoma that occurs with aging seems muted as well.39,40 These cancers present with pain, soft-tissue swelling, and variable elevations in serum alkaline phosphatase. Investigations to date suggest that pagetic lesions and osteosarcomas arising in pagetic bone are probably both driven to some extent by stromal cells overexpressing RANK ligand and may not represent defects intrinsic to the osteoclast.41
Giant-cell tumors of bone are also rare but can arise in pagetic bone. A cluster of cases was reported in Avellino and other towns of southern Italy.42 Again, the lesions occur in older individuals and in different sites than those seen in the benign giant-cell tumors recorded in patients without Paget disease.
Metastases from lymphomas, prostate cancer, and breast cancer certainly occur in bone, but rarely in pagetic sites.43 A recent case study noted that patients with prostate cancer who also had Paget disease had a later onset of metastasis to bone than patients without coincident Paget disease.44
A THOUGHTFUL ASSESSMENT
Evaluating a patient with Paget disease requires a thoughtful assessment of its musculoskeletal consequences in an aging skeleton. Pain in Paget disease is often multifactorial. In the elderly, end-stage degenerative disease of the spine, hip, and knees, mechanical instability, compression fractures of the spine, and neuropathies may compound the clinical picture. Therefore, a thorough evaluation is required to plan effective therapy.
Alkaline phosphatase and other markers
A screening serum alkaline phosphatase level is usually sufficient to measure bone turnover. Produced by osteoblasts, alkaline phosphatase is a marker of bone formation, but an imperfect one. Often it is elevated in active Paget disease—but not always.45 Many patients have normal serum alkaline phosphatase levels, particularly if they have monostotic (single-bone) disease. It is unclear why, in a disorder marked by accelerated bone remodeling, the biochemical markers are inconsistent measures of bone turnover.
Research into biochemical markers of Paget disease has had two aims: to identify the single best marker for baseline assessment of pagetic bone activity and to find out whether this measurement responds to therapy.46,47 Measures of bone formation such as bone-specific alkaline phosphatase, osteocalcin, and the procollagen type I peptides, and measures of bone resorption including the pyridinolines, hydroxyproline, and cross-linked collagens, have been analyzed as markers of bone remodeling and show no real advantage over the serum alkaline phosphatase level as reflections of bone turnover. As alkaline phosphatase measurement is inexpensive, available, and reliable, it should be used preferentially, with gamma-glutamyl transpeptidate or 5′ nucleotidase confirming the source as either liver or bone. Readers are directed to a recent review in which the utility of these markers is explored in more detail.48
Imaging studies

Bone scans can give us an idea of the extent, location, and general activity of the disease (Figure 3). Uptake is avid in affected bones, beginning in the subchondral region and spreading throughout the bone. Bone scans can be particularly useful in defining sites of active disease when the serum alkaline phosphatase level is normal.
Plain radiography of the affected bones outlines the anatomy of the problem and gives some insight into the cause of pain (Figure 3).
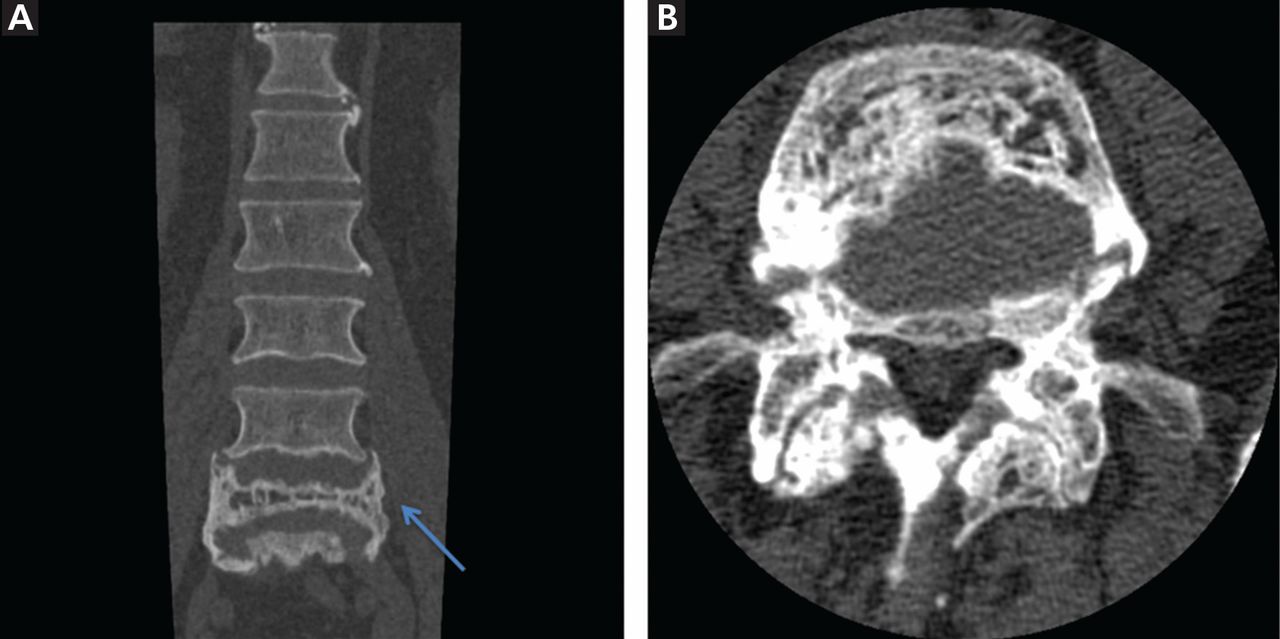
Computed tomography or magnetic resonance imaging may prove useful in cases of spinal stenosis, cauda equina syndrome, compression fractures, or suspected malignancy (Figure 4), but these studies are expensive and generally are not needed.
Radiographic features. Paget disease is presumed to be a disease of the osteoclast, and the earliest lesion is described as lytic. In my own experience, it is unusual to see a purely lytic lesion, although sometimes the disease presents in the skull in this way—osteoporosis circumscripta—or in the femur or tibia with an advancing edge of pure osteolysis.
More often, one sees evidence of both resorption by osteoclasts and formation by osteoblasts, reflecting the coupling of these two processes in this disease. Radiographic findings on plain films are usually definitive, showing enlargement of the affected bone, deformity, coarsened trabeculae, and thickened cortices with tunneling (Figure 5).49 In weightbearing bones, pseudofractures may stud the convex surface. These incongruities of bone may persist for years, heralding fracture only when there is focal pain (Figure 6).50
Biopsy is infrequently needed

If these diagnostic findings are not present, then biopsy is indicated. In the United Sates and Canada, where Paget disease is fairly common, biopsy is infrequently needed and is usually reserved for situations in which the differential diagnosis includes cancer, as when the cortex cannot be clearly visualized, the lesions are atypical in pattern or location, or there is a single sclerotic vertebral body on imaging.51
The other indication for biopsy is a “new” pagetic lesion. For reasons unknown, the pattern of skeletal involvement in Paget disease tends to be stable throughout the patient’s lifetime. This is another reason why a baseline bone scan is useful.
TREATMENT WITH BISPHOSPHONATES
Treatment of Paget disease today relies for the most part on the new generation of nitrogen-containing bisphosphonates. As a class, these are antiresorptive agents that inhibit osteoclasts; in this way they slow bone remodeling and enhance the deposition of normal lamellar bone. Their clinical efficacy in Paget disease, coupled with the observation that the earliest lesion in Paget disease is lytic, underscores the principle that Paget disease is a disorder of the osteoclast.
Oral bisphosphonates
Etidronate, approved in 1977, was the first bisphosphonate licensed to treat Paget disease, and it remains available for this indication in the United States. Used in 6-month regimens, it lowers the serum alkaline phosphatase level in some patients, but it has a narrow therapeutic margin. Drug-induced osteomalacia and worsening lytic lesions and fractures in weight-bearing bones are some of the complications.52 When the nitrogen-containing bisphosphonates were developed, they proved to be more potent antiresorptive agents that pose less risk of mineralization defects at prescribed doses.
Alendronate, approved in 1995, is an oral nitrogen-containing bisphosphonate that is effective in treating Paget disease.53 Alendronate is now available in the United States only through special programs (eg, the CVS ProCare Program); the paperwork required to secure this drug is onerous, so the drug is used infrequently. Studies in Paget disease showed that it normalizes the serum alkaline phosphatase level, improves the radiographic appearance, and eases pain in many patients.54 The dosage is 40 mg daily for 6 months.
Risedronate, approved in 1998, is another oral nitrogen-containing bisphosphonate and is comparable to alendronate in efficacy.55 The dosage is 30 mg daily for 2 months.
Tiludronate is another oral bisphosphonate with a different mechanism of action from the nitrogen-containing bisphosphonates.56 It is safe, often effective, but less potent than the newer agents.
The oral bisphosphonates are well tolerated, with few side effects other than gastrointestinal distress. As a class, they are poorly absorbed and so must be taken fasting with a full glass of water on rising, after which the patient should remain upright without food or drink for 30 to 60 minutes. This is a nuisance for elderly patients already on multiple medications and thus makes intravenous agents appealing.
Intravenous bisphosphonates
Pamidronate was approved in 1994. It is quite effective in many patients with Paget disease. There is no consensus around the world on dosing, with regimens ranging from 30 mg to 90 mg or more intravenously in divided doses given over 2 to 4 hours from once a day to once a week. In the United States, 30 mg is given over 4 hours on 3 consecutive days. Resistance to pamidronate has been described; the mechanism is unknown.
Zoledronic acid is a nitrogen-containing bisphosphonate. It is given as a single infusion over 15 minutes, and re-treatment may not be necessary for years. A randomized clinical trial in 2005 demonstrated the efficacy of zoledronic acid 5 mg by infusion compared with oral risedronate in the treatment of Paget disease.57 In observational extension studies lasting as long as 6.5 years, zoledronic acid has been shown to be superior to risedronate in terms of the proportion of patients experiencing a sustained clinical remission.58
While there are many bisphosphonates on the market, an infusion of 5 mg of zoledronic acid seems optimal in most patients who do not have a contraindication or an aversion to intravenous therapy. It tends to normalize the serum alkaline phosphatase level quickly and to leave more patients in sustained biochemical remission than do older bisphosphonates, as noted above. It also tends to be more effective in normalizing the serum alkaline phosphatase level when a patient has used other bisphosphonates in the past or has become resistant to them.
Bisphosphonates reduce bone turnover but do not correct deformities
In randomized clinical trials, bisphosphonates have been shown to restore bone remodeling to more normal levels, to ease pain from pagetic bone, to lower the serum alkaline phosphatase level, and to heal radiographic lesions, but these drugs have not been proven to prevent progression of deformity or to restore the structural integrity of bone (Figure 6).
The Paget’s Disease: Randomized Trial of Intensive Versus Symptomatic Management (PRISM), in 1,324 people with Paget disease in the United Kingdom, showed no difference in the incidence of fracture, orthopedic surgery, quality of life, or hearing thresholds over 2 to 5 years in patients treated with bisphosphonates vs those treated symptomatically, despite a significant difference in serum alkaline phosphatase in the two groups (P < .001).59
In the observational extension study of zoledronic acid described above,58 three of four fractures occurred in the group treated with zoledronic acid, echoing the findings of the PRISM study.
Adverse effects of bisphosphonates
The more potent the bisphosphonate is as an antiresorptive agent, the more it suppresses normal bone remodeling, which can lead to osteonecrosis of the jaw and to atypical femoral fractures.60,61 These complications are unusual in patients with Paget disease because the treatment is intermittent. Sometimes a single dose of zoledronic acid or one course of risedronate or alendronate will last for years.
All the nitrogen-containing bisphosphonates, particularly zoledronic acid, may provoke flulike symptoms of fever, arthralgias, and bone pain. This effect is self-limited, resolves in days, and does not tend to recur. Bone pain may be more sustained, but this also passes, and within weeks the antiresorptive process has abated and pagetic bone pain will ease. Atrial fibrillation is not an anticipated complication of treatment with a bisphosphonate.62 The risk of esophageal cancer is not confirmed at this time.63 Other rare complications of the bisphosphonates include iritis, acute renal failure, and allergy.
Bisphosphonates are not approved for use in patients with creatinine clearance less than 30 mL/min, or in pregnancy.
Other treatments
Calcitonin, an older agent, can still be useful in easing the pain of Paget disease, healing bone lesions, and reducing the metabolic activity of pagetic bone in patients who cannot receive bisphosphonates. It is given by injection in doses of 50 to 100 IU daily or every other day. Although unlikely to effect a sustained clinical remission, calcitonin remains a safe, well-tolerated, and well-studied medication in Paget disease and is approved for this indication.64,65
Denosumab has not been formally studied in Paget disease, but a recent case report indicated it was effective.66
A conservative strategy
Guidelines for treating Paget disease have been written at various times in many countries, including Italy (2007),67 the United Kingdom (2004),68 Japan (2006),69 and Canada (2007).70 Recommendations differ, in part because it is hard to ascertain whether long-term outcomes are improved by treatment, and in part because the prevalence of Paget disease is decreasing and its severity is lessening.11,12 Some guidelines are outdated, since they do not include the newer bisphosphonates.
If the natural history of untreated Paget disease involves the gradual evolution over more than 20 years of bowing deformities in the lower limbs, rigidity and overgrowth of the spine, and softening and enlargement of the skull, as described by Sir James Paget, then treatment should be initiated in hopes that it will modify the outcome. We have no lens to better focus this question on the effect of treatment on the natural history of the disease. We have the PRISM study, designed before zoledronic acid was approved and only 2 to 5 years in duration. And we have the epidemiologic data demonstrating that most patients have no symptoms during their lifetime.
We see the crippling bone disease described by Sir James Paget so infrequently today in the United States that we forget the profound morbidity that may attend the skeletal changes of Paget disease that were common in the early 20th century. Once the bones of the skull are overgrown, the limbs are bowed, and the degenerative joint disease is present, no medication can reverse these changes. Then, the integrity of the bone is lost, and the vulnerability to fracture, early osteoarthritis, nerve compression syndromes, and hearing loss persist. Understanding these consequences prompts the recommendation of early treatment in patients with Paget disease, in hopes of mitigating disease progression.
Patients with active Paget disease, documented either by an elevated serum alkaline phosphatase or by a bone scan, should be treated with a bisphosphonate if the disease is found in sites where remodeling of bone may lead to complications. Such sites include the skull, spine, and long bones of the lower extremity. Paget disease of bone in the pelvis tends to give little trouble (Figure 2) unless it is proximal to a joint, when pain and early arthritis may result. Treatment is safe and, I think, prudent to undertake in any person over age 55 with active disease. To prevent hypocalcemia during treatment, all patients should be repleted with vitamin D and maintained on calcium 1,200 mg daily through diet or supplements with meals.
Throughout the evaluation and treatment, it is important to remember that pain may not emanate from pagetic bone. If medication for Paget disease proves ineffective in the first few months, analgesics, bracing, walking aids, and operative management71 are adjunctive therapies to improve the functional status of these patients.
It is a remarkable clinical observation that treatment of Paget disease may rapidly reverse neurologic syndromes, resolve the erythema or warmth overlying active pagetic bone, and diminish the risk of bleeding with surgery. This response to therapy suggests that there is prompt inhibition and apoptosis of the osteoclasts, accompanied by diminished vascularity of bone. Whatever the mechanism, it is worth treating patients who have spinal stenosis, arthritis, and nerve compression syndromes with calcitonin or bisphosphonates before surgical intervention, whenever possible.34,72
- Paget J. On a form of chronic inflammation of bones (osteitis deformans). Med Chir Trans 1877; 60:37–64.9.
- Guyer PB, Chamberlain AT, Ackery DM, Rolfe EB. The anatomic distribution of osteitis deformans. Clin Orthop Relat Res 1981; 156:141–144.
- Tiegs RD, Lohse CM, Wollan PC, Melton LJ. Long-term trends in the incidence of Paget’s disease of bone. Bone 2000; 27:423–427.
- Altman RD, Bloch DA, Hochberg MC, Murphy WA. Prevalence of pelvic Paget’s disease of bone in the United States. J Bone Miner Res 2000; 15:461–465.
- Barker DJ. The epidemiology of Paget’s disease of bone. Br Med Bull 1984; 40:396–400.
- Detheridge FM, Guyer PB, Barker DJ. European distribution of Paget’s disease of bone. Br Med J (Clin Res Ed) 1982; 285:1005–1008.
- van Staa TP, Selby P, Leufkens HG, Lyles K, Sprafka JM, Cooper C. Incidence and natural history of Paget’s disease of bone in England and Wales. J Bone Miner Res 2002; 17:465–471.
- Barker DJ. The epidemiology of Paget’s disease. Metab Bone Dis Relat Res 1981; 3:231–233.
- Rogers J, Jeffrey DR, Watt I. Paget’s disease in an archeological population. J Bone Miner Res 2002; 17:1127–1134.
- Aaron JE, Rogers J, Kanis JA. Paleohistology of Paget’s disease in two medieval skeletons. Am J Phys Anthropol 1992; 89:325–331.
- Poór G, Donáth J, Fornet B, Cooper C. Epidemiology of Paget’s disease in Europe: the prevalence is decreasing. J Bone Miner Res 2006; 21:1545–1549.
- Cundy HR, Gamble G, Wattie D, Rutland M, Cundy T. Paget’s disease of bone in New Zealand: continued decline in disease severity. Calcif Tissue Int 2004; 75:358–364.
- Doyle T, Gunn J, Anderson G, Gill M, Cundy T. Paget’s disease in New Zealand: evidence for declining prevalence. Bone 2002; 31:616–619.
- Laurin N, Brown JP, Morissette J, Raymond V. Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am J Hum Genet 2002; 70:1582–1588.
- Hocking LJ, Lucas GJ, Daroszewska A, et al. Domain-specific mutations in sequestosome 1 (SQSTM1) cause familial and sporadic Paget’s disease. Hum Mol Genet 2002; 11:2735–2739.
- Lucas GJ, Hocking LJ, Daroszewska A, et al. Ubiquitin-associated domain mutations of SQSTM1 in Paget’s disease of bone: evidence for a founder effect in patients of British descent. J Bone Miner Res 2005; 20:227–231.
- Mays S. Archaeological skeletons support a northwest European origin for Paget’s disease of bone. J Bone Miner Res 2010; 25:1839–1841.
- Bolland MJ, Tong PC, Naot D, et al. Delayed development of Paget’s disease in offspring inheriting SQSTM1 mutations. J Bone Miner Res 2007; 22:411–415.
- Rea SL, Walsh JP, Ward L, et al. A novel mutation (K378X) in the sequestosome 1 gene associated with increased NF-kappaB signaling and Paget’s disease of bone with a severe phenotype. J Bone Miner Res 2006; 21:1136–1145.
- Morissette J, Laurin N, Brown JP. Sequestosome 1: mutation frequencies, haplotypes, and phenotypes in familial Paget’s disease of bone. J Bone Miner Res 2006; 21(suppl 2):P38–P44.
- Eekhoff EW, Karperien M, Houtsma D, et al. Familial Paget’s disease in The Netherlands: occurrence, identification of new mutations in the sequestosome 1 gene, and their clinical associations. Arthritis Rheum 2004; 50:1650–1654.
- Ralston SH, Layfield R. Pathogenesis of Paget disease of bone. Calcif Tissue Int 2012; 91:97–113.
- Kurihara N, Hiruma Y, Yamana K, et al. Contributions of the measles virus nucleocapsid gene and the SQSTM1/p62(P392L) mutation to Paget’s disease. Cell Metab 2011; 13:23–34.
- Kurihara N, Zhou H, Reddy SV, et al. Expression of measles virus nucleocapsid protein in osteoclasts induces Paget’s disease-like bone lesions in mice. J Bone Miner Res 2006; 21:446–455.
- Reddy SV, Singer FR, Roodman GD. Bone marrow mononuclear cells from patients with Paget’s disease contain measles virus nucleocapsid messenger ribonucleic acid that has mutations in a specific region of the sequence. J Clin Endocrinol Metab 1995; 80:2108–2111.
- Gennari L, Merlotti D, Martini G, Nuti R. Paget’s disease of bone in Italy. J Bone Miner Res 2006; 21(suppl 2):P14–P21.
- Seton M, Choi HK, Hansen MF, Sebaldt RJ, Cooper C. Analysis of environmental factors in familial versus sporadic Paget’s disease of bone—the New England Registry for Paget’s Disease of Bone. J Bone Miner Res 2003; 18:1519–1524.
- Siris ES. Extensive personal experience: Paget’s disease of bone. J Clin Endocrinol Metab 1995; 80:335–338.
- Lucas GJ, Daroszewska A, Ralston SH. Contribution of genetic factors to the pathogenesis of Paget’s disease of bone and related disorders. J Bone Miner Res 2006; 21(suppl 2):P31–P37.
- Seton M. Diagnosis, complications and treatment of Paget’s disease of bone. Aging Health 2009; 5:497–508.
- Siris E, Roodman GD. Paget’s Disease of Bone. 7th ed. Washington, DC: American Society for Bone and Mineral Research; 2008.
- Seton M, Moses AM, Bode RK, Schwartz C. Paget’s disease of bone: the skeletal distribution, complications and quality of life as perceived by patients. Bone 2011; 48:281–285.
- Seton M. Paget’s disease of bone. In:Hochberg MC, Silman AJ, Smolen JS, Weinblatt ME, Weisman MH, editors. Rheumatology. 5th ed. Philadelphia, PA: Mosby Elsevier; 2010:2021–2028.
- Douglas DL, Duckworth T, Kanis JA, Jefferson AA, Martin TJ, Russell RG. Spinal cord dysfunction in Paget’s disease of bone. Has medical treatment a vascular basis? J Bone Joint Surg Br 1981; 63B:495–503.
- Siris ES. Epidemiological aspects of Paget’s disease: family history and relationship to other medical conditions. Semin Arthritis Rheum 1994; 23:222–225.
- Kanis JA, Evanson JM, Russell RG. Paget’s disease of bone: diagnosis and management. Metab Bone Dis Relat Res 1981; 3:219–230.
- Mangham DC, Davie MW, Grimer RJ. Sarcoma arising in Paget’s disease of bone: declining incidence and increasing age at presentation. Bone 2009; 44:431–436.
- Hansen MF, Seton M, Merchant A. Osteosarcoma in Paget’s disease of bone. J Bone Miner Res 2006; 21(suppl 2):P58–P63.
- Price CH. The incidence of osteogenic sarcoma in South-West England and its relationship to Paget’s disease of bone. J Bone Joint Surg Br 1962; 44-B:366–376.
- Ishikawa Y, Tsukuma H, Miller RW. Low rates of Paget’s disease of bone and osteosarcoma in elderly Japanese. Lancet 1996; 347:1559.
- Sun SG, Lau YS, Itonaga I, Sabokbar A, Athanasou NA. Bone stromal cells in pagetic bone and Paget’s sarcoma express RANKL and support human osteoclast formation. J Pathol 2006; 209:114–120.
- Rendina D, Gennari L, De Filippo G, et al. Evidence for increased clinical severity of familial and sporadic Paget’s disease of bone in Campania, southern Italy. J Bone Miner Res 2006; 21:1828–1835.
- Fenton P, Resnick D. Metastases to bone affected by Paget’s disease. A report of three cases. Int Orthop 1991; 15:397–399.
- Tu SM, Som A, Tu B, Logothetis CJ, Lee MH, Yeung SC. Effect of Paget’s disease of bone (osteitis deformans) on the progression of prostate cancer bone metastasis. Br J Cancer 2012; 107:646–651.
- Eekhoff ME, van der Klift M, Kroon HM, et al. Paget’s disease of bone in The Netherlands: a population-based radiological and biochemical survey—the Rotterdam Study. J Bone Miner Res 2004; 19:566–570.
- Reid IR, Davidson JS, Wattie D, et al. Comparative responses of bone turnover markers to bisphosphonate therapy in Paget’s disease of bone. Bone 2004; 35:224–230.
- Alvarez L, Guañabens N, Peris P, et al. Usefulness of biochemical markers of bone turnover in assessing response to the treatment of Paget’s disease. Bone 2001; 29:447–452.
- Cundy T, Reid IR. Paget’s disease of bone. Clin Biochem 2012; 45:43–48.
- Cortis K, Micallef K, Mizzi A. Imaging Paget’s disease of bone—from head to toe. Clin Radiol 2011; 66:662–672.
- Redden JF, Dixon J, Vennart W, Hosking DJ. Management of fissure fractures in Paget’s disease. Int Orthop 1981; 5:103–106.
- Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 25-1993. A 67-year-old man with osteolytic lesions of T11 and T12. N Engl J Med 1993; 328:1836–1841.
- Evans RA, Dunstan CR, Hills E, Wong SY. Pathologic fracture due to severe osteomalacia following low-dose diphosphonate treatment of Paget’s disease of bone. Aust N Z J Med 1983; 13:277–279.
- Siris E, Weinstein RS, Altman R, et al. Comparative study of alendronate versus etidronate for the treatment of Paget’s disease of bone. J Clin Endocrinol Metab 1996; 81:961–967.
- Reid IR, Siris E. Alendronate in the treatment of Paget’s disease of bone. Int J Clin Pract Suppl 1999; 101:62–66.
- Miller PD, Brown JP, Siris ES, Hoseyni MS, Axelrod DW, Bekker PJ. A randomized, double-blind comparison of risedronate and etidronate in the treatment of Paget’s disease of bone. Paget’s Risedronate/Etidronate Study Group. Am J Med 1999; 106:513–520.
- Peris P, Alvarez L, Vidal S, Martínez MA, Monegal A, Guañabens N. Treatment with tiludronate has a similar effect to risedronate on Paget’s disease activity assessed by bone markers and bone scintigraphy. Clin Exp Rheumatol 2007; 25:206–210.
- Reid IR, Miller P, Lyles K, et al. Comparison of a single infusion of zoledronic acid with risedronate for Paget’s disease. N Engl J Med 2005; 353:898–908.
- Reid IR, Lyles K, Su G, et al. A single infusion of zoledronic acid produces sustained remissions in Paget disease: data to 6.5 years. J Bone Miner Res 2011; 26:2261–2270.
- Langston AL, Campbell MK, Fraser WD, MacLennan GS, Selby PL, Ralston SH; PRISM Trial Group. Randomized trial of intensive bisphosphonate treatment versus symptomatic management in Paget’s disease of bone. J Bone Miner Res 2010; 25:20–31.
- Abrahamsen B, Einhorn TA. Beyond a reasonable doubt? Bisphosphonates and atypical femur fractures. Bone 2012; 50:1196–1200.
- Seton M, Krane SM. Use of zoledronic acid in the treatment of Paget’s disease. Ther Clin Risk Manag 2007; 3:913–918.
- Sørensen HT, Christensen S, Mehnert F, et al. Use of bisphosphonates among women and risk of atrial fibrillation and flutter: Population based case-control study. BMJ 2008; 336:813–816.
- Dixon WG, Solomon DH. Bisphosphonates and esophageal cancer—a pathway through the confusion. Nat Rev Rheumatol 2011; 7:369–372.
- Singer FR, Krane SM. Paget’s disease of bone. In:Avioli LV, Krane SM, editors. Metabolic Bone Disease and Clinically Related Disorders. 2nd ed. Philadelphia, PA: W.B. Saunders Company; 1990:546–615.
- Kanis JA, Horn DB, Scott RD, Strong JA. Treatment of Paget’s disease of bone with synthetic salmon calcitonin. Br Med J 1974; 3:727–731.
- Schwarz P, Rasmussen AQ, Kvist TM, Andersen UB, Jørgensen NR. Paget’s disease of the bone after treatment with denosumab: a case report. Bone 2012; 50:1023–1025.
- Adami S, Bartolozzi P, Brandi ML, et al; Societa Italiana di Ortopedia e Traumatologia. [Italian guidelines for the diagnosis and treatment of Paget’s disease of bone.] Reumatismo 2007; 59:153–168. (Article in Italian.)
- Scarsbrok A, Brown M, Wilson D. UK guidelines on management of Paget’s disease of bone. Rheumatology (Oxford) 2004; 43:399–400.
- Takata S, Hashimoto J, Nakatsuka K, et a.l Guidelines for diagnosis and management of Paget’s disease of bone in Japan. J Bone Miner Metab 2006; 24:359–367.
- Josse RG, Hanley DA, Kendler D, Ste Marie L-G, Adachi JD, Brown J. Diagnosis and treatment of Paget’s disease of bone. Clin Invest Med 2007; 30:E210–E223.
- Kaplan FS. Paget’s disease of bone: orthopedic complications. Semin Arthritis Rheum 1994; 23:250–252.
- Kanis JA, Gray RE. Long-term follow-up observations on treatment in Paget’s disease of bone. Clin Orthop Relat Res 1987; 217:99–125.
Paget disease of bone is a focal disorder of the aging skeleton that can be asymptomatic or can present with pain, bowing deformities, fractures, or nonspecific rheumatic complaints. Physicians often discover it in asymptomatic patients when serum alkaline phosphatase levels are elevated or as an incidental finding on radiography. Despite evidence of germline mutations and polymorphisms that predispose to Paget disease, the environmental determinants that permit disease expression in older people remain unknown.
A STRIKING GEOGRAPHIC DISTRIBUTION
Researchers have been studying the determinants and distribution of Paget disease ever since Sir James Paget first described it in 1877.1
Paget disease has a predilection for the axial skeleton, particularly the lumbosacral spine and pelvis, as well as the skull, femur, and tibia.2 Knowing this, investigators have used screening plain films of the abdomen (kidney-ureter-bladder views) to estimate its prevalence in different populations, as these images capture the lumbosacral spine, pelvis, and proximal femurs. Other means of assessing prevalence have included autopsy series, questionnaires, and screens for biochemical markers of bone turnover, such as elevated serum alkaline phosphatase from bone.3–6
Using these methods, Paget disease has been estimated to occur in 1% to 3% of people over age 55, and in as many as 8% of people over age 80 in certain countries.7
This disease has a striking geographic distribution, being frequent in Europe, Canada, the United States, Australia, New Zealand, and cities of South America, but rare in Scandinavia and Japan. It seems to be equally rare in other countries of the Far East and in India, Russia, and Africa, although its prevalence in these areas has not been thoroughly investigated.8
That it is an ancient disease has been corroborated by excavations in churchyards in Great Britain.9,10 It may be familial or sporadic, but its expression is delayed until late middle age in most persons, and it does not occur in children. For reasons unclear, the prevalence seems to be decreasing in many countries.11–13
GENETICS IS NOT THE WHOLE STORY
The variable prevalence of Paget disease in different geographic regions and its sometimes-familial expression suggest a genetic predisposition, environmental factor, or both.
Mutations in SQSTM1
In 2002, scientists investigating a cohort of French Canadian families found a mutation in the SQSTM1 gene that was present in almost 50% of people with familial Paget disease and in 16% of those with sporadic Paget disease.14 Hocking and his colleagues in the United Kingdom subsequently found the same mutation in 19% of cases of familial Paget disease and in 9% of sporadic cases.15
Further, investigators noted that the mutation was often present on a conserved haplotype, consistent with a stable genetic change occurring in the affected population.16 This observation of a “founder effect” dovetailed with the epidemiology of Paget disease,17 but only with this SQSTM1 mutation.
Throughout Europe, Australia, and the United States, comparable rates of the SQSTM1 mutation were reported in or around the ubiquitin-associated domain. Several specific mutations exist, the most common one being P392L, ie, a prolineto-leucine substitution at amino acid 392. Scientists have tried to correlate severity of disease with genotype, but the findings have been inconsistent.18–21
Investigations into the mechanism of disease have pointed to the role of p62, the product of SQSTM1, in signaling osteoclast activation via nuclear factor kappa B. Since this initial discovery, polymorphisms in the genes affecting osteoclast maturation, activation, and fusion pathways have been shown to predispose to Paget disease. Examples:
- TNFRSF11A, which codes for receptor activator of nuclear factor kappa B, or RANK
- TNFRSF11B, which codes for osteoprotegerin, or OPG
- CSF1, which codes for macrophage colony-stimulating factor 1, and
- OPTN, which codes for optineurin, a member of the nuclear factor kappa B-modulating protein family.
Clinicians interested in these details can read an excellent review of the pathogenesis of Paget disease.22
Other possible factors
Although there is good evidence that measles and canine distemper virus can infect osteoclasts and modify their phenotype, there is no good evidence that these infections by themselves cause Paget disease.23–25 It is, however, tempting to think of these RNA paramyxoviruses as precipitating factors; conceivably, an infectious agent might seed the ends of long bones, accounting for the fixed distribution of Paget disease and its late expression.
Epidemiologic studies from around the world have failed to identify conclusively any environmental exposure that predisposes to Paget disease, although a rural setting, trauma, infection, and milk ingestion have all been proposed.26–28 It is also possible that as bone ages and the marrow becomes less cellular and more fatty, these changes may permit the disease to develop.
The greatest risk factor for Paget disease is perhaps aging, followed by ancestry and a known family history of it. That genetics is not the whole story is evident by reports of people with SQSTM1 mutations who show no clinical evidence of Paget disease in their old age, and patients with Paget disease who have no SQSTM1 mutation.20,29
CLINICAL PRESENTATION
Most patients with Paget disease have no symptoms and come to medical attention because of an elevated serum alkaline phosphatase level or characteristic findings on radiographs ordered for other indications.11 Paget disease is the second most common disorder of aging bone after osteoporosis. Yet unlike osteoporosis, which presents as a systemic fragility of bone, the clinical manifestations of Paget disease depend on which bones are affected and how enlarged or misshapen they have become.
Common complications
As a consequence of this abnormal bone remodeling and overgrowth, many patients present with bone pain. Bone deformity, headache, and hearing loss may also occur (Figure 1), as well as fractures and nerve compression syndromes (eg, spinal stenosis, sciatica, cauda equina syndrome).
It is important to remember that “pagetic” bone may not be the source of pain, and that functional impairment caused by degenerative changes at affected sites is common (Figure 2).30,31
In a study from the New England Registry for Paget’s Disease,32 most patients knew fairly well which bones were affected and what complications resulted from this when deformity, fracture, or total joint replacement had occurred.32 Although Paget disease did affect their quality of life as measured by physical functioning on the Short Form-12 assessment, these impairments did not seem to affect their outlook, which was as good as or better than that in other people their age.
Metabolic complications
Metabolic complications of Paget disease are rare today but can occur in an elderly patient who has active, polyostotic (multibone) disease.33 The accelerated rate of bone remodeling and the increased vascularity of pagetic bone have been reported to lead to high-output heart failure. In theory, treatment should ease this by diminishing blood flow to pagetic bone and restoring bone turnover to more normal levels.34
Hypercalcemia can occur when patients with Paget disease are immobilized for any reason, and there is probably a higher incidence of renal stones in patients with Paget disease.35,36
Malignant complications
Osteosarcoma rarely arises in pagetic bone. Yet Paget disease may account for a significant number of cases of this cancer in the elderly.37 In these cases, osteosarcoma is presumed to be driven by a second genetic mutation, has a genetic signature distinct from that in osteosarcomas occurring in youth, and is quite resistant to treatment.38 In Scandinavia and Japan, where Paget disease is rare, the second peak of osteosarcoma that occurs with aging seems muted as well.39,40 These cancers present with pain, soft-tissue swelling, and variable elevations in serum alkaline phosphatase. Investigations to date suggest that pagetic lesions and osteosarcomas arising in pagetic bone are probably both driven to some extent by stromal cells overexpressing RANK ligand and may not represent defects intrinsic to the osteoclast.41
Giant-cell tumors of bone are also rare but can arise in pagetic bone. A cluster of cases was reported in Avellino and other towns of southern Italy.42 Again, the lesions occur in older individuals and in different sites than those seen in the benign giant-cell tumors recorded in patients without Paget disease.
Metastases from lymphomas, prostate cancer, and breast cancer certainly occur in bone, but rarely in pagetic sites.43 A recent case study noted that patients with prostate cancer who also had Paget disease had a later onset of metastasis to bone than patients without coincident Paget disease.44
A THOUGHTFUL ASSESSMENT
Evaluating a patient with Paget disease requires a thoughtful assessment of its musculoskeletal consequences in an aging skeleton. Pain in Paget disease is often multifactorial. In the elderly, end-stage degenerative disease of the spine, hip, and knees, mechanical instability, compression fractures of the spine, and neuropathies may compound the clinical picture. Therefore, a thorough evaluation is required to plan effective therapy.
Alkaline phosphatase and other markers
A screening serum alkaline phosphatase level is usually sufficient to measure bone turnover. Produced by osteoblasts, alkaline phosphatase is a marker of bone formation, but an imperfect one. Often it is elevated in active Paget disease—but not always.45 Many patients have normal serum alkaline phosphatase levels, particularly if they have monostotic (single-bone) disease. It is unclear why, in a disorder marked by accelerated bone remodeling, the biochemical markers are inconsistent measures of bone turnover.
Research into biochemical markers of Paget disease has had two aims: to identify the single best marker for baseline assessment of pagetic bone activity and to find out whether this measurement responds to therapy.46,47 Measures of bone formation such as bone-specific alkaline phosphatase, osteocalcin, and the procollagen type I peptides, and measures of bone resorption including the pyridinolines, hydroxyproline, and cross-linked collagens, have been analyzed as markers of bone remodeling and show no real advantage over the serum alkaline phosphatase level as reflections of bone turnover. As alkaline phosphatase measurement is inexpensive, available, and reliable, it should be used preferentially, with gamma-glutamyl transpeptidate or 5′ nucleotidase confirming the source as either liver or bone. Readers are directed to a recent review in which the utility of these markers is explored in more detail.48
Imaging studies
Bone scans can give us an idea of the extent, location, and general activity of the disease (Figure 3). Uptake is avid in affected bones, beginning in the subchondral region and spreading throughout the bone. Bone scans can be particularly useful in defining sites of active disease when the serum alkaline phosphatase level is normal.
Plain radiography of the affected bones outlines the anatomy of the problem and gives some insight into the cause of pain (Figure 3).
Computed tomography or magnetic resonance imaging may prove useful in cases of spinal stenosis, cauda equina syndrome, compression fractures, or suspected malignancy (Figure 4), but these studies are expensive and generally are not needed.
Radiographic features. Paget disease is presumed to be a disease of the osteoclast, and the earliest lesion is described as lytic. In my own experience, it is unusual to see a purely lytic lesion, although sometimes the disease presents in the skull in this way—osteoporosis circumscripta—or in the femur or tibia with an advancing edge of pure osteolysis.
More often, one sees evidence of both resorption by osteoclasts and formation by osteoblasts, reflecting the coupling of these two processes in this disease. Radiographic findings on plain films are usually definitive, showing enlargement of the affected bone, deformity, coarsened trabeculae, and thickened cortices with tunneling (Figure 5).49 In weightbearing bones, pseudofractures may stud the convex surface. These incongruities of bone may persist for years, heralding fracture only when there is focal pain (Figure 6).50
Biopsy is infrequently needed
If these diagnostic findings are not present, then biopsy is indicated. In the United Sates and Canada, where Paget disease is fairly common, biopsy is infrequently needed and is usually reserved for situations in which the differential diagnosis includes cancer, as when the cortex cannot be clearly visualized, the lesions are atypical in pattern or location, or there is a single sclerotic vertebral body on imaging.51
The other indication for biopsy is a “new” pagetic lesion. For reasons unknown, the pattern of skeletal involvement in Paget disease tends to be stable throughout the patient’s lifetime. This is another reason why a baseline bone scan is useful.
TREATMENT WITH BISPHOSPHONATES
Treatment of Paget disease today relies for the most part on the new generation of nitrogen-containing bisphosphonates. As a class, these are antiresorptive agents that inhibit osteoclasts; in this way they slow bone remodeling and enhance the deposition of normal lamellar bone. Their clinical efficacy in Paget disease, coupled with the observation that the earliest lesion in Paget disease is lytic, underscores the principle that Paget disease is a disorder of the osteoclast.
Oral bisphosphonates
Etidronate, approved in 1977, was the first bisphosphonate licensed to treat Paget disease, and it remains available for this indication in the United States. Used in 6-month regimens, it lowers the serum alkaline phosphatase level in some patients, but it has a narrow therapeutic margin. Drug-induced osteomalacia and worsening lytic lesions and fractures in weight-bearing bones are some of the complications.52 When the nitrogen-containing bisphosphonates were developed, they proved to be more potent antiresorptive agents that pose less risk of mineralization defects at prescribed doses.
Alendronate, approved in 1995, is an oral nitrogen-containing bisphosphonate that is effective in treating Paget disease.53 Alendronate is now available in the United States only through special programs (eg, the CVS ProCare Program); the paperwork required to secure this drug is onerous, so the drug is used infrequently. Studies in Paget disease showed that it normalizes the serum alkaline phosphatase level, improves the radiographic appearance, and eases pain in many patients.54 The dosage is 40 mg daily for 6 months.
Risedronate, approved in 1998, is another oral nitrogen-containing bisphosphonate and is comparable to alendronate in efficacy.55 The dosage is 30 mg daily for 2 months.
Tiludronate is another oral bisphosphonate with a different mechanism of action from the nitrogen-containing bisphosphonates.56 It is safe, often effective, but less potent than the newer agents.
The oral bisphosphonates are well tolerated, with few side effects other than gastrointestinal distress. As a class, they are poorly absorbed and so must be taken fasting with a full glass of water on rising, after which the patient should remain upright without food or drink for 30 to 60 minutes. This is a nuisance for elderly patients already on multiple medications and thus makes intravenous agents appealing.
Intravenous bisphosphonates
Pamidronate was approved in 1994. It is quite effective in many patients with Paget disease. There is no consensus around the world on dosing, with regimens ranging from 30 mg to 90 mg or more intravenously in divided doses given over 2 to 4 hours from once a day to once a week. In the United States, 30 mg is given over 4 hours on 3 consecutive days. Resistance to pamidronate has been described; the mechanism is unknown.
Zoledronic acid is a nitrogen-containing bisphosphonate. It is given as a single infusion over 15 minutes, and re-treatment may not be necessary for years. A randomized clinical trial in 2005 demonstrated the efficacy of zoledronic acid 5 mg by infusion compared with oral risedronate in the treatment of Paget disease.57 In observational extension studies lasting as long as 6.5 years, zoledronic acid has been shown to be superior to risedronate in terms of the proportion of patients experiencing a sustained clinical remission.58
While there are many bisphosphonates on the market, an infusion of 5 mg of zoledronic acid seems optimal in most patients who do not have a contraindication or an aversion to intravenous therapy. It tends to normalize the serum alkaline phosphatase level quickly and to leave more patients in sustained biochemical remission than do older bisphosphonates, as noted above. It also tends to be more effective in normalizing the serum alkaline phosphatase level when a patient has used other bisphosphonates in the past or has become resistant to them.
Bisphosphonates reduce bone turnover but do not correct deformities
In randomized clinical trials, bisphosphonates have been shown to restore bone remodeling to more normal levels, to ease pain from pagetic bone, to lower the serum alkaline phosphatase level, and to heal radiographic lesions, but these drugs have not been proven to prevent progression of deformity or to restore the structural integrity of bone (Figure 6).
The Paget’s Disease: Randomized Trial of Intensive Versus Symptomatic Management (PRISM), in 1,324 people with Paget disease in the United Kingdom, showed no difference in the incidence of fracture, orthopedic surgery, quality of life, or hearing thresholds over 2 to 5 years in patients treated with bisphosphonates vs those treated symptomatically, despite a significant difference in serum alkaline phosphatase in the two groups (P < .001).59
In the observational extension study of zoledronic acid described above,58 three of four fractures occurred in the group treated with zoledronic acid, echoing the findings of the PRISM study.
Adverse effects of bisphosphonates
The more potent the bisphosphonate is as an antiresorptive agent, the more it suppresses normal bone remodeling, which can lead to osteonecrosis of the jaw and to atypical femoral fractures.60,61 These complications are unusual in patients with Paget disease because the treatment is intermittent. Sometimes a single dose of zoledronic acid or one course of risedronate or alendronate will last for years.
All the nitrogen-containing bisphosphonates, particularly zoledronic acid, may provoke flulike symptoms of fever, arthralgias, and bone pain. This effect is self-limited, resolves in days, and does not tend to recur. Bone pain may be more sustained, but this also passes, and within weeks the antiresorptive process has abated and pagetic bone pain will ease. Atrial fibrillation is not an anticipated complication of treatment with a bisphosphonate.62 The risk of esophageal cancer is not confirmed at this time.63 Other rare complications of the bisphosphonates include iritis, acute renal failure, and allergy.
Bisphosphonates are not approved for use in patients with creatinine clearance less than 30 mL/min, or in pregnancy.
Other treatments
Calcitonin, an older agent, can still be useful in easing the pain of Paget disease, healing bone lesions, and reducing the metabolic activity of pagetic bone in patients who cannot receive bisphosphonates. It is given by injection in doses of 50 to 100 IU daily or every other day. Although unlikely to effect a sustained clinical remission, calcitonin remains a safe, well-tolerated, and well-studied medication in Paget disease and is approved for this indication.64,65
Denosumab has not been formally studied in Paget disease, but a recent case report indicated it was effective.66
A conservative strategy
Guidelines for treating Paget disease have been written at various times in many countries, including Italy (2007),67 the United Kingdom (2004),68 Japan (2006),69 and Canada (2007).70 Recommendations differ, in part because it is hard to ascertain whether long-term outcomes are improved by treatment, and in part because the prevalence of Paget disease is decreasing and its severity is lessening.11,12 Some guidelines are outdated, since they do not include the newer bisphosphonates.
If the natural history of untreated Paget disease involves the gradual evolution over more than 20 years of bowing deformities in the lower limbs, rigidity and overgrowth of the spine, and softening and enlargement of the skull, as described by Sir James Paget, then treatment should be initiated in hopes that it will modify the outcome. We have no lens to better focus this question on the effect of treatment on the natural history of the disease. We have the PRISM study, designed before zoledronic acid was approved and only 2 to 5 years in duration. And we have the epidemiologic data demonstrating that most patients have no symptoms during their lifetime.
We see the crippling bone disease described by Sir James Paget so infrequently today in the United States that we forget the profound morbidity that may attend the skeletal changes of Paget disease that were common in the early 20th century. Once the bones of the skull are overgrown, the limbs are bowed, and the degenerative joint disease is present, no medication can reverse these changes. Then, the integrity of the bone is lost, and the vulnerability to fracture, early osteoarthritis, nerve compression syndromes, and hearing loss persist. Understanding these consequences prompts the recommendation of early treatment in patients with Paget disease, in hopes of mitigating disease progression.
Patients with active Paget disease, documented either by an elevated serum alkaline phosphatase or by a bone scan, should be treated with a bisphosphonate if the disease is found in sites where remodeling of bone may lead to complications. Such sites include the skull, spine, and long bones of the lower extremity. Paget disease of bone in the pelvis tends to give little trouble (Figure 2) unless it is proximal to a joint, when pain and early arthritis may result. Treatment is safe and, I think, prudent to undertake in any person over age 55 with active disease. To prevent hypocalcemia during treatment, all patients should be repleted with vitamin D and maintained on calcium 1,200 mg daily through diet or supplements with meals.
Throughout the evaluation and treatment, it is important to remember that pain may not emanate from pagetic bone. If medication for Paget disease proves ineffective in the first few months, analgesics, bracing, walking aids, and operative management71 are adjunctive therapies to improve the functional status of these patients.
It is a remarkable clinical observation that treatment of Paget disease may rapidly reverse neurologic syndromes, resolve the erythema or warmth overlying active pagetic bone, and diminish the risk of bleeding with surgery. This response to therapy suggests that there is prompt inhibition and apoptosis of the osteoclasts, accompanied by diminished vascularity of bone. Whatever the mechanism, it is worth treating patients who have spinal stenosis, arthritis, and nerve compression syndromes with calcitonin or bisphosphonates before surgical intervention, whenever possible.34,72
Paget disease of bone is a focal disorder of the aging skeleton that can be asymptomatic or can present with pain, bowing deformities, fractures, or nonspecific rheumatic complaints. Physicians often discover it in asymptomatic patients when serum alkaline phosphatase levels are elevated or as an incidental finding on radiography. Despite evidence of germline mutations and polymorphisms that predispose to Paget disease, the environmental determinants that permit disease expression in older people remain unknown.
A STRIKING GEOGRAPHIC DISTRIBUTION
Researchers have been studying the determinants and distribution of Paget disease ever since Sir James Paget first described it in 1877.1
Paget disease has a predilection for the axial skeleton, particularly the lumbosacral spine and pelvis, as well as the skull, femur, and tibia.2 Knowing this, investigators have used screening plain films of the abdomen (kidney-ureter-bladder views) to estimate its prevalence in different populations, as these images capture the lumbosacral spine, pelvis, and proximal femurs. Other means of assessing prevalence have included autopsy series, questionnaires, and screens for biochemical markers of bone turnover, such as elevated serum alkaline phosphatase from bone.3–6
Using these methods, Paget disease has been estimated to occur in 1% to 3% of people over age 55, and in as many as 8% of people over age 80 in certain countries.7
This disease has a striking geographic distribution, being frequent in Europe, Canada, the United States, Australia, New Zealand, and cities of South America, but rare in Scandinavia and Japan. It seems to be equally rare in other countries of the Far East and in India, Russia, and Africa, although its prevalence in these areas has not been thoroughly investigated.8
That it is an ancient disease has been corroborated by excavations in churchyards in Great Britain.9,10 It may be familial or sporadic, but its expression is delayed until late middle age in most persons, and it does not occur in children. For reasons unclear, the prevalence seems to be decreasing in many countries.11–13
GENETICS IS NOT THE WHOLE STORY
The variable prevalence of Paget disease in different geographic regions and its sometimes-familial expression suggest a genetic predisposition, environmental factor, or both.
Mutations in SQSTM1
In 2002, scientists investigating a cohort of French Canadian families found a mutation in the SQSTM1 gene that was present in almost 50% of people with familial Paget disease and in 16% of those with sporadic Paget disease.14 Hocking and his colleagues in the United Kingdom subsequently found the same mutation in 19% of cases of familial Paget disease and in 9% of sporadic cases.15
Further, investigators noted that the mutation was often present on a conserved haplotype, consistent with a stable genetic change occurring in the affected population.16 This observation of a “founder effect” dovetailed with the epidemiology of Paget disease,17 but only with this SQSTM1 mutation.
Throughout Europe, Australia, and the United States, comparable rates of the SQSTM1 mutation were reported in or around the ubiquitin-associated domain. Several specific mutations exist, the most common one being P392L, ie, a prolineto-leucine substitution at amino acid 392. Scientists have tried to correlate severity of disease with genotype, but the findings have been inconsistent.18–21
Investigations into the mechanism of disease have pointed to the role of p62, the product of SQSTM1, in signaling osteoclast activation via nuclear factor kappa B. Since this initial discovery, polymorphisms in the genes affecting osteoclast maturation, activation, and fusion pathways have been shown to predispose to Paget disease. Examples:
- TNFRSF11A, which codes for receptor activator of nuclear factor kappa B, or RANK
- TNFRSF11B, which codes for osteoprotegerin, or OPG
- CSF1, which codes for macrophage colony-stimulating factor 1, and
- OPTN, which codes for optineurin, a member of the nuclear factor kappa B-modulating protein family.
Clinicians interested in these details can read an excellent review of the pathogenesis of Paget disease.22
Other possible factors
Although there is good evidence that measles and canine distemper virus can infect osteoclasts and modify their phenotype, there is no good evidence that these infections by themselves cause Paget disease.23–25 It is, however, tempting to think of these RNA paramyxoviruses as precipitating factors; conceivably, an infectious agent might seed the ends of long bones, accounting for the fixed distribution of Paget disease and its late expression.
Epidemiologic studies from around the world have failed to identify conclusively any environmental exposure that predisposes to Paget disease, although a rural setting, trauma, infection, and milk ingestion have all been proposed.26–28 It is also possible that as bone ages and the marrow becomes less cellular and more fatty, these changes may permit the disease to develop.
The greatest risk factor for Paget disease is perhaps aging, followed by ancestry and a known family history of it. That genetics is not the whole story is evident by reports of people with SQSTM1 mutations who show no clinical evidence of Paget disease in their old age, and patients with Paget disease who have no SQSTM1 mutation.20,29
CLINICAL PRESENTATION
Most patients with Paget disease have no symptoms and come to medical attention because of an elevated serum alkaline phosphatase level or characteristic findings on radiographs ordered for other indications.11 Paget disease is the second most common disorder of aging bone after osteoporosis. Yet unlike osteoporosis, which presents as a systemic fragility of bone, the clinical manifestations of Paget disease depend on which bones are affected and how enlarged or misshapen they have become.
Common complications
As a consequence of this abnormal bone remodeling and overgrowth, many patients present with bone pain. Bone deformity, headache, and hearing loss may also occur (Figure 1), as well as fractures and nerve compression syndromes (eg, spinal stenosis, sciatica, cauda equina syndrome).
It is important to remember that “pagetic” bone may not be the source of pain, and that functional impairment caused by degenerative changes at affected sites is common (Figure 2).30,31
In a study from the New England Registry for Paget’s Disease,32 most patients knew fairly well which bones were affected and what complications resulted from this when deformity, fracture, or total joint replacement had occurred.32 Although Paget disease did affect their quality of life as measured by physical functioning on the Short Form-12 assessment, these impairments did not seem to affect their outlook, which was as good as or better than that in other people their age.
Metabolic complications
Metabolic complications of Paget disease are rare today but can occur in an elderly patient who has active, polyostotic (multibone) disease.33 The accelerated rate of bone remodeling and the increased vascularity of pagetic bone have been reported to lead to high-output heart failure. In theory, treatment should ease this by diminishing blood flow to pagetic bone and restoring bone turnover to more normal levels.34
Hypercalcemia can occur when patients with Paget disease are immobilized for any reason, and there is probably a higher incidence of renal stones in patients with Paget disease.35,36
Malignant complications
Osteosarcoma rarely arises in pagetic bone. Yet Paget disease may account for a significant number of cases of this cancer in the elderly.37 In these cases, osteosarcoma is presumed to be driven by a second genetic mutation, has a genetic signature distinct from that in osteosarcomas occurring in youth, and is quite resistant to treatment.38 In Scandinavia and Japan, where Paget disease is rare, the second peak of osteosarcoma that occurs with aging seems muted as well.39,40 These cancers present with pain, soft-tissue swelling, and variable elevations in serum alkaline phosphatase. Investigations to date suggest that pagetic lesions and osteosarcomas arising in pagetic bone are probably both driven to some extent by stromal cells overexpressing RANK ligand and may not represent defects intrinsic to the osteoclast.41
Giant-cell tumors of bone are also rare but can arise in pagetic bone. A cluster of cases was reported in Avellino and other towns of southern Italy.42 Again, the lesions occur in older individuals and in different sites than those seen in the benign giant-cell tumors recorded in patients without Paget disease.
Metastases from lymphomas, prostate cancer, and breast cancer certainly occur in bone, but rarely in pagetic sites.43 A recent case study noted that patients with prostate cancer who also had Paget disease had a later onset of metastasis to bone than patients without coincident Paget disease.44
A THOUGHTFUL ASSESSMENT
Evaluating a patient with Paget disease requires a thoughtful assessment of its musculoskeletal consequences in an aging skeleton. Pain in Paget disease is often multifactorial. In the elderly, end-stage degenerative disease of the spine, hip, and knees, mechanical instability, compression fractures of the spine, and neuropathies may compound the clinical picture. Therefore, a thorough evaluation is required to plan effective therapy.
Alkaline phosphatase and other markers
A screening serum alkaline phosphatase level is usually sufficient to measure bone turnover. Produced by osteoblasts, alkaline phosphatase is a marker of bone formation, but an imperfect one. Often it is elevated in active Paget disease—but not always.45 Many patients have normal serum alkaline phosphatase levels, particularly if they have monostotic (single-bone) disease. It is unclear why, in a disorder marked by accelerated bone remodeling, the biochemical markers are inconsistent measures of bone turnover.
Research into biochemical markers of Paget disease has had two aims: to identify the single best marker for baseline assessment of pagetic bone activity and to find out whether this measurement responds to therapy.46,47 Measures of bone formation such as bone-specific alkaline phosphatase, osteocalcin, and the procollagen type I peptides, and measures of bone resorption including the pyridinolines, hydroxyproline, and cross-linked collagens, have been analyzed as markers of bone remodeling and show no real advantage over the serum alkaline phosphatase level as reflections of bone turnover. As alkaline phosphatase measurement is inexpensive, available, and reliable, it should be used preferentially, with gamma-glutamyl transpeptidate or 5′ nucleotidase confirming the source as either liver or bone. Readers are directed to a recent review in which the utility of these markers is explored in more detail.48
Imaging studies
Bone scans can give us an idea of the extent, location, and general activity of the disease (Figure 3). Uptake is avid in affected bones, beginning in the subchondral region and spreading throughout the bone. Bone scans can be particularly useful in defining sites of active disease when the serum alkaline phosphatase level is normal.
Plain radiography of the affected bones outlines the anatomy of the problem and gives some insight into the cause of pain (Figure 3).
Computed tomography or magnetic resonance imaging may prove useful in cases of spinal stenosis, cauda equina syndrome, compression fractures, or suspected malignancy (Figure 4), but these studies are expensive and generally are not needed.
Radiographic features. Paget disease is presumed to be a disease of the osteoclast, and the earliest lesion is described as lytic. In my own experience, it is unusual to see a purely lytic lesion, although sometimes the disease presents in the skull in this way—osteoporosis circumscripta—or in the femur or tibia with an advancing edge of pure osteolysis.
More often, one sees evidence of both resorption by osteoclasts and formation by osteoblasts, reflecting the coupling of these two processes in this disease. Radiographic findings on plain films are usually definitive, showing enlargement of the affected bone, deformity, coarsened trabeculae, and thickened cortices with tunneling (Figure 5).49 In weightbearing bones, pseudofractures may stud the convex surface. These incongruities of bone may persist for years, heralding fracture only when there is focal pain (Figure 6).50
Biopsy is infrequently needed
If these diagnostic findings are not present, then biopsy is indicated. In the United Sates and Canada, where Paget disease is fairly common, biopsy is infrequently needed and is usually reserved for situations in which the differential diagnosis includes cancer, as when the cortex cannot be clearly visualized, the lesions are atypical in pattern or location, or there is a single sclerotic vertebral body on imaging.51
The other indication for biopsy is a “new” pagetic lesion. For reasons unknown, the pattern of skeletal involvement in Paget disease tends to be stable throughout the patient’s lifetime. This is another reason why a baseline bone scan is useful.
TREATMENT WITH BISPHOSPHONATES
Treatment of Paget disease today relies for the most part on the new generation of nitrogen-containing bisphosphonates. As a class, these are antiresorptive agents that inhibit osteoclasts; in this way they slow bone remodeling and enhance the deposition of normal lamellar bone. Their clinical efficacy in Paget disease, coupled with the observation that the earliest lesion in Paget disease is lytic, underscores the principle that Paget disease is a disorder of the osteoclast.
Oral bisphosphonates
Etidronate, approved in 1977, was the first bisphosphonate licensed to treat Paget disease, and it remains available for this indication in the United States. Used in 6-month regimens, it lowers the serum alkaline phosphatase level in some patients, but it has a narrow therapeutic margin. Drug-induced osteomalacia and worsening lytic lesions and fractures in weight-bearing bones are some of the complications.52 When the nitrogen-containing bisphosphonates were developed, they proved to be more potent antiresorptive agents that pose less risk of mineralization defects at prescribed doses.
Alendronate, approved in 1995, is an oral nitrogen-containing bisphosphonate that is effective in treating Paget disease.53 Alendronate is now available in the United States only through special programs (eg, the CVS ProCare Program); the paperwork required to secure this drug is onerous, so the drug is used infrequently. Studies in Paget disease showed that it normalizes the serum alkaline phosphatase level, improves the radiographic appearance, and eases pain in many patients.54 The dosage is 40 mg daily for 6 months.
Risedronate, approved in 1998, is another oral nitrogen-containing bisphosphonate and is comparable to alendronate in efficacy.55 The dosage is 30 mg daily for 2 months.
Tiludronate is another oral bisphosphonate with a different mechanism of action from the nitrogen-containing bisphosphonates.56 It is safe, often effective, but less potent than the newer agents.
The oral bisphosphonates are well tolerated, with few side effects other than gastrointestinal distress. As a class, they are poorly absorbed and so must be taken fasting with a full glass of water on rising, after which the patient should remain upright without food or drink for 30 to 60 minutes. This is a nuisance for elderly patients already on multiple medications and thus makes intravenous agents appealing.
Intravenous bisphosphonates
Pamidronate was approved in 1994. It is quite effective in many patients with Paget disease. There is no consensus around the world on dosing, with regimens ranging from 30 mg to 90 mg or more intravenously in divided doses given over 2 to 4 hours from once a day to once a week. In the United States, 30 mg is given over 4 hours on 3 consecutive days. Resistance to pamidronate has been described; the mechanism is unknown.
Zoledronic acid is a nitrogen-containing bisphosphonate. It is given as a single infusion over 15 minutes, and re-treatment may not be necessary for years. A randomized clinical trial in 2005 demonstrated the efficacy of zoledronic acid 5 mg by infusion compared with oral risedronate in the treatment of Paget disease.57 In observational extension studies lasting as long as 6.5 years, zoledronic acid has been shown to be superior to risedronate in terms of the proportion of patients experiencing a sustained clinical remission.58
While there are many bisphosphonates on the market, an infusion of 5 mg of zoledronic acid seems optimal in most patients who do not have a contraindication or an aversion to intravenous therapy. It tends to normalize the serum alkaline phosphatase level quickly and to leave more patients in sustained biochemical remission than do older bisphosphonates, as noted above. It also tends to be more effective in normalizing the serum alkaline phosphatase level when a patient has used other bisphosphonates in the past or has become resistant to them.
Bisphosphonates reduce bone turnover but do not correct deformities
In randomized clinical trials, bisphosphonates have been shown to restore bone remodeling to more normal levels, to ease pain from pagetic bone, to lower the serum alkaline phosphatase level, and to heal radiographic lesions, but these drugs have not been proven to prevent progression of deformity or to restore the structural integrity of bone (Figure 6).
The Paget’s Disease: Randomized Trial of Intensive Versus Symptomatic Management (PRISM), in 1,324 people with Paget disease in the United Kingdom, showed no difference in the incidence of fracture, orthopedic surgery, quality of life, or hearing thresholds over 2 to 5 years in patients treated with bisphosphonates vs those treated symptomatically, despite a significant difference in serum alkaline phosphatase in the two groups (P < .001).59
In the observational extension study of zoledronic acid described above,58 three of four fractures occurred in the group treated with zoledronic acid, echoing the findings of the PRISM study.
Adverse effects of bisphosphonates
The more potent the bisphosphonate is as an antiresorptive agent, the more it suppresses normal bone remodeling, which can lead to osteonecrosis of the jaw and to atypical femoral fractures.60,61 These complications are unusual in patients with Paget disease because the treatment is intermittent. Sometimes a single dose of zoledronic acid or one course of risedronate or alendronate will last for years.
All the nitrogen-containing bisphosphonates, particularly zoledronic acid, may provoke flulike symptoms of fever, arthralgias, and bone pain. This effect is self-limited, resolves in days, and does not tend to recur. Bone pain may be more sustained, but this also passes, and within weeks the antiresorptive process has abated and pagetic bone pain will ease. Atrial fibrillation is not an anticipated complication of treatment with a bisphosphonate.62 The risk of esophageal cancer is not confirmed at this time.63 Other rare complications of the bisphosphonates include iritis, acute renal failure, and allergy.
Bisphosphonates are not approved for use in patients with creatinine clearance less than 30 mL/min, or in pregnancy.
Other treatments
Calcitonin, an older agent, can still be useful in easing the pain of Paget disease, healing bone lesions, and reducing the metabolic activity of pagetic bone in patients who cannot receive bisphosphonates. It is given by injection in doses of 50 to 100 IU daily or every other day. Although unlikely to effect a sustained clinical remission, calcitonin remains a safe, well-tolerated, and well-studied medication in Paget disease and is approved for this indication.64,65
Denosumab has not been formally studied in Paget disease, but a recent case report indicated it was effective.66
A conservative strategy
Guidelines for treating Paget disease have been written at various times in many countries, including Italy (2007),67 the United Kingdom (2004),68 Japan (2006),69 and Canada (2007).70 Recommendations differ, in part because it is hard to ascertain whether long-term outcomes are improved by treatment, and in part because the prevalence of Paget disease is decreasing and its severity is lessening.11,12 Some guidelines are outdated, since they do not include the newer bisphosphonates.
If the natural history of untreated Paget disease involves the gradual evolution over more than 20 years of bowing deformities in the lower limbs, rigidity and overgrowth of the spine, and softening and enlargement of the skull, as described by Sir James Paget, then treatment should be initiated in hopes that it will modify the outcome. We have no lens to better focus this question on the effect of treatment on the natural history of the disease. We have the PRISM study, designed before zoledronic acid was approved and only 2 to 5 years in duration. And we have the epidemiologic data demonstrating that most patients have no symptoms during their lifetime.
We see the crippling bone disease described by Sir James Paget so infrequently today in the United States that we forget the profound morbidity that may attend the skeletal changes of Paget disease that were common in the early 20th century. Once the bones of the skull are overgrown, the limbs are bowed, and the degenerative joint disease is present, no medication can reverse these changes. Then, the integrity of the bone is lost, and the vulnerability to fracture, early osteoarthritis, nerve compression syndromes, and hearing loss persist. Understanding these consequences prompts the recommendation of early treatment in patients with Paget disease, in hopes of mitigating disease progression.
Patients with active Paget disease, documented either by an elevated serum alkaline phosphatase or by a bone scan, should be treated with a bisphosphonate if the disease is found in sites where remodeling of bone may lead to complications. Such sites include the skull, spine, and long bones of the lower extremity. Paget disease of bone in the pelvis tends to give little trouble (Figure 2) unless it is proximal to a joint, when pain and early arthritis may result. Treatment is safe and, I think, prudent to undertake in any person over age 55 with active disease. To prevent hypocalcemia during treatment, all patients should be repleted with vitamin D and maintained on calcium 1,200 mg daily through diet or supplements with meals.
Throughout the evaluation and treatment, it is important to remember that pain may not emanate from pagetic bone. If medication for Paget disease proves ineffective in the first few months, analgesics, bracing, walking aids, and operative management71 are adjunctive therapies to improve the functional status of these patients.
It is a remarkable clinical observation that treatment of Paget disease may rapidly reverse neurologic syndromes, resolve the erythema or warmth overlying active pagetic bone, and diminish the risk of bleeding with surgery. This response to therapy suggests that there is prompt inhibition and apoptosis of the osteoclasts, accompanied by diminished vascularity of bone. Whatever the mechanism, it is worth treating patients who have spinal stenosis, arthritis, and nerve compression syndromes with calcitonin or bisphosphonates before surgical intervention, whenever possible.34,72
- Paget J. On a form of chronic inflammation of bones (osteitis deformans). Med Chir Trans 1877; 60:37–64.9.
- Guyer PB, Chamberlain AT, Ackery DM, Rolfe EB. The anatomic distribution of osteitis deformans. Clin Orthop Relat Res 1981; 156:141–144.
- Tiegs RD, Lohse CM, Wollan PC, Melton LJ. Long-term trends in the incidence of Paget’s disease of bone. Bone 2000; 27:423–427.
- Altman RD, Bloch DA, Hochberg MC, Murphy WA. Prevalence of pelvic Paget’s disease of bone in the United States. J Bone Miner Res 2000; 15:461–465.
- Barker DJ. The epidemiology of Paget’s disease of bone. Br Med Bull 1984; 40:396–400.
- Detheridge FM, Guyer PB, Barker DJ. European distribution of Paget’s disease of bone. Br Med J (Clin Res Ed) 1982; 285:1005–1008.
- van Staa TP, Selby P, Leufkens HG, Lyles K, Sprafka JM, Cooper C. Incidence and natural history of Paget’s disease of bone in England and Wales. J Bone Miner Res 2002; 17:465–471.
- Barker DJ. The epidemiology of Paget’s disease. Metab Bone Dis Relat Res 1981; 3:231–233.
- Rogers J, Jeffrey DR, Watt I. Paget’s disease in an archeological population. J Bone Miner Res 2002; 17:1127–1134.
- Aaron JE, Rogers J, Kanis JA. Paleohistology of Paget’s disease in two medieval skeletons. Am J Phys Anthropol 1992; 89:325–331.
- Poór G, Donáth J, Fornet B, Cooper C. Epidemiology of Paget’s disease in Europe: the prevalence is decreasing. J Bone Miner Res 2006; 21:1545–1549.
- Cundy HR, Gamble G, Wattie D, Rutland M, Cundy T. Paget’s disease of bone in New Zealand: continued decline in disease severity. Calcif Tissue Int 2004; 75:358–364.
- Doyle T, Gunn J, Anderson G, Gill M, Cundy T. Paget’s disease in New Zealand: evidence for declining prevalence. Bone 2002; 31:616–619.
- Laurin N, Brown JP, Morissette J, Raymond V. Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am J Hum Genet 2002; 70:1582–1588.
- Hocking LJ, Lucas GJ, Daroszewska A, et al. Domain-specific mutations in sequestosome 1 (SQSTM1) cause familial and sporadic Paget’s disease. Hum Mol Genet 2002; 11:2735–2739.
- Lucas GJ, Hocking LJ, Daroszewska A, et al. Ubiquitin-associated domain mutations of SQSTM1 in Paget’s disease of bone: evidence for a founder effect in patients of British descent. J Bone Miner Res 2005; 20:227–231.
- Mays S. Archaeological skeletons support a northwest European origin for Paget’s disease of bone. J Bone Miner Res 2010; 25:1839–1841.
- Bolland MJ, Tong PC, Naot D, et al. Delayed development of Paget’s disease in offspring inheriting SQSTM1 mutations. J Bone Miner Res 2007; 22:411–415.
- Rea SL, Walsh JP, Ward L, et al. A novel mutation (K378X) in the sequestosome 1 gene associated with increased NF-kappaB signaling and Paget’s disease of bone with a severe phenotype. J Bone Miner Res 2006; 21:1136–1145.
- Morissette J, Laurin N, Brown JP. Sequestosome 1: mutation frequencies, haplotypes, and phenotypes in familial Paget’s disease of bone. J Bone Miner Res 2006; 21(suppl 2):P38–P44.
- Eekhoff EW, Karperien M, Houtsma D, et al. Familial Paget’s disease in The Netherlands: occurrence, identification of new mutations in the sequestosome 1 gene, and their clinical associations. Arthritis Rheum 2004; 50:1650–1654.
- Ralston SH, Layfield R. Pathogenesis of Paget disease of bone. Calcif Tissue Int 2012; 91:97–113.
- Kurihara N, Hiruma Y, Yamana K, et al. Contributions of the measles virus nucleocapsid gene and the SQSTM1/p62(P392L) mutation to Paget’s disease. Cell Metab 2011; 13:23–34.
- Kurihara N, Zhou H, Reddy SV, et al. Expression of measles virus nucleocapsid protein in osteoclasts induces Paget’s disease-like bone lesions in mice. J Bone Miner Res 2006; 21:446–455.
- Reddy SV, Singer FR, Roodman GD. Bone marrow mononuclear cells from patients with Paget’s disease contain measles virus nucleocapsid messenger ribonucleic acid that has mutations in a specific region of the sequence. J Clin Endocrinol Metab 1995; 80:2108–2111.
- Gennari L, Merlotti D, Martini G, Nuti R. Paget’s disease of bone in Italy. J Bone Miner Res 2006; 21(suppl 2):P14–P21.
- Seton M, Choi HK, Hansen MF, Sebaldt RJ, Cooper C. Analysis of environmental factors in familial versus sporadic Paget’s disease of bone—the New England Registry for Paget’s Disease of Bone. J Bone Miner Res 2003; 18:1519–1524.
- Siris ES. Extensive personal experience: Paget’s disease of bone. J Clin Endocrinol Metab 1995; 80:335–338.
- Lucas GJ, Daroszewska A, Ralston SH. Contribution of genetic factors to the pathogenesis of Paget’s disease of bone and related disorders. J Bone Miner Res 2006; 21(suppl 2):P31–P37.
- Seton M. Diagnosis, complications and treatment of Paget’s disease of bone. Aging Health 2009; 5:497–508.
- Siris E, Roodman GD. Paget’s Disease of Bone. 7th ed. Washington, DC: American Society for Bone and Mineral Research; 2008.
- Seton M, Moses AM, Bode RK, Schwartz C. Paget’s disease of bone: the skeletal distribution, complications and quality of life as perceived by patients. Bone 2011; 48:281–285.
- Seton M. Paget’s disease of bone. In:Hochberg MC, Silman AJ, Smolen JS, Weinblatt ME, Weisman MH, editors. Rheumatology. 5th ed. Philadelphia, PA: Mosby Elsevier; 2010:2021–2028.
- Douglas DL, Duckworth T, Kanis JA, Jefferson AA, Martin TJ, Russell RG. Spinal cord dysfunction in Paget’s disease of bone. Has medical treatment a vascular basis? J Bone Joint Surg Br 1981; 63B:495–503.
- Siris ES. Epidemiological aspects of Paget’s disease: family history and relationship to other medical conditions. Semin Arthritis Rheum 1994; 23:222–225.
- Kanis JA, Evanson JM, Russell RG. Paget’s disease of bone: diagnosis and management. Metab Bone Dis Relat Res 1981; 3:219–230.
- Mangham DC, Davie MW, Grimer RJ. Sarcoma arising in Paget’s disease of bone: declining incidence and increasing age at presentation. Bone 2009; 44:431–436.
- Hansen MF, Seton M, Merchant A. Osteosarcoma in Paget’s disease of bone. J Bone Miner Res 2006; 21(suppl 2):P58–P63.
- Price CH. The incidence of osteogenic sarcoma in South-West England and its relationship to Paget’s disease of bone. J Bone Joint Surg Br 1962; 44-B:366–376.
- Ishikawa Y, Tsukuma H, Miller RW. Low rates of Paget’s disease of bone and osteosarcoma in elderly Japanese. Lancet 1996; 347:1559.
- Sun SG, Lau YS, Itonaga I, Sabokbar A, Athanasou NA. Bone stromal cells in pagetic bone and Paget’s sarcoma express RANKL and support human osteoclast formation. J Pathol 2006; 209:114–120.
- Rendina D, Gennari L, De Filippo G, et al. Evidence for increased clinical severity of familial and sporadic Paget’s disease of bone in Campania, southern Italy. J Bone Miner Res 2006; 21:1828–1835.
- Fenton P, Resnick D. Metastases to bone affected by Paget’s disease. A report of three cases. Int Orthop 1991; 15:397–399.
- Tu SM, Som A, Tu B, Logothetis CJ, Lee MH, Yeung SC. Effect of Paget’s disease of bone (osteitis deformans) on the progression of prostate cancer bone metastasis. Br J Cancer 2012; 107:646–651.
- Eekhoff ME, van der Klift M, Kroon HM, et al. Paget’s disease of bone in The Netherlands: a population-based radiological and biochemical survey—the Rotterdam Study. J Bone Miner Res 2004; 19:566–570.
- Reid IR, Davidson JS, Wattie D, et al. Comparative responses of bone turnover markers to bisphosphonate therapy in Paget’s disease of bone. Bone 2004; 35:224–230.
- Alvarez L, Guañabens N, Peris P, et al. Usefulness of biochemical markers of bone turnover in assessing response to the treatment of Paget’s disease. Bone 2001; 29:447–452.
- Cundy T, Reid IR. Paget’s disease of bone. Clin Biochem 2012; 45:43–48.
- Cortis K, Micallef K, Mizzi A. Imaging Paget’s disease of bone—from head to toe. Clin Radiol 2011; 66:662–672.
- Redden JF, Dixon J, Vennart W, Hosking DJ. Management of fissure fractures in Paget’s disease. Int Orthop 1981; 5:103–106.
- Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 25-1993. A 67-year-old man with osteolytic lesions of T11 and T12. N Engl J Med 1993; 328:1836–1841.
- Evans RA, Dunstan CR, Hills E, Wong SY. Pathologic fracture due to severe osteomalacia following low-dose diphosphonate treatment of Paget’s disease of bone. Aust N Z J Med 1983; 13:277–279.
- Siris E, Weinstein RS, Altman R, et al. Comparative study of alendronate versus etidronate for the treatment of Paget’s disease of bone. J Clin Endocrinol Metab 1996; 81:961–967.
- Reid IR, Siris E. Alendronate in the treatment of Paget’s disease of bone. Int J Clin Pract Suppl 1999; 101:62–66.
- Miller PD, Brown JP, Siris ES, Hoseyni MS, Axelrod DW, Bekker PJ. A randomized, double-blind comparison of risedronate and etidronate in the treatment of Paget’s disease of bone. Paget’s Risedronate/Etidronate Study Group. Am J Med 1999; 106:513–520.
- Peris P, Alvarez L, Vidal S, Martínez MA, Monegal A, Guañabens N. Treatment with tiludronate has a similar effect to risedronate on Paget’s disease activity assessed by bone markers and bone scintigraphy. Clin Exp Rheumatol 2007; 25:206–210.
- Reid IR, Miller P, Lyles K, et al. Comparison of a single infusion of zoledronic acid with risedronate for Paget’s disease. N Engl J Med 2005; 353:898–908.
- Reid IR, Lyles K, Su G, et al. A single infusion of zoledronic acid produces sustained remissions in Paget disease: data to 6.5 years. J Bone Miner Res 2011; 26:2261–2270.
- Langston AL, Campbell MK, Fraser WD, MacLennan GS, Selby PL, Ralston SH; PRISM Trial Group. Randomized trial of intensive bisphosphonate treatment versus symptomatic management in Paget’s disease of bone. J Bone Miner Res 2010; 25:20–31.
- Abrahamsen B, Einhorn TA. Beyond a reasonable doubt? Bisphosphonates and atypical femur fractures. Bone 2012; 50:1196–1200.
- Seton M, Krane SM. Use of zoledronic acid in the treatment of Paget’s disease. Ther Clin Risk Manag 2007; 3:913–918.
- Sørensen HT, Christensen S, Mehnert F, et al. Use of bisphosphonates among women and risk of atrial fibrillation and flutter: Population based case-control study. BMJ 2008; 336:813–816.
- Dixon WG, Solomon DH. Bisphosphonates and esophageal cancer—a pathway through the confusion. Nat Rev Rheumatol 2011; 7:369–372.
- Singer FR, Krane SM. Paget’s disease of bone. In:Avioli LV, Krane SM, editors. Metabolic Bone Disease and Clinically Related Disorders. 2nd ed. Philadelphia, PA: W.B. Saunders Company; 1990:546–615.
- Kanis JA, Horn DB, Scott RD, Strong JA. Treatment of Paget’s disease of bone with synthetic salmon calcitonin. Br Med J 1974; 3:727–731.
- Schwarz P, Rasmussen AQ, Kvist TM, Andersen UB, Jørgensen NR. Paget’s disease of the bone after treatment with denosumab: a case report. Bone 2012; 50:1023–1025.
- Adami S, Bartolozzi P, Brandi ML, et al; Societa Italiana di Ortopedia e Traumatologia. [Italian guidelines for the diagnosis and treatment of Paget’s disease of bone.] Reumatismo 2007; 59:153–168. (Article in Italian.)
- Scarsbrok A, Brown M, Wilson D. UK guidelines on management of Paget’s disease of bone. Rheumatology (Oxford) 2004; 43:399–400.
- Takata S, Hashimoto J, Nakatsuka K, et a.l Guidelines for diagnosis and management of Paget’s disease of bone in Japan. J Bone Miner Metab 2006; 24:359–367.
- Josse RG, Hanley DA, Kendler D, Ste Marie L-G, Adachi JD, Brown J. Diagnosis and treatment of Paget’s disease of bone. Clin Invest Med 2007; 30:E210–E223.
- Kaplan FS. Paget’s disease of bone: orthopedic complications. Semin Arthritis Rheum 1994; 23:250–252.
- Kanis JA, Gray RE. Long-term follow-up observations on treatment in Paget’s disease of bone. Clin Orthop Relat Res 1987; 217:99–125.
- Paget J. On a form of chronic inflammation of bones (osteitis deformans). Med Chir Trans 1877; 60:37–64.9.
- Guyer PB, Chamberlain AT, Ackery DM, Rolfe EB. The anatomic distribution of osteitis deformans. Clin Orthop Relat Res 1981; 156:141–144.
- Tiegs RD, Lohse CM, Wollan PC, Melton LJ. Long-term trends in the incidence of Paget’s disease of bone. Bone 2000; 27:423–427.
- Altman RD, Bloch DA, Hochberg MC, Murphy WA. Prevalence of pelvic Paget’s disease of bone in the United States. J Bone Miner Res 2000; 15:461–465.
- Barker DJ. The epidemiology of Paget’s disease of bone. Br Med Bull 1984; 40:396–400.
- Detheridge FM, Guyer PB, Barker DJ. European distribution of Paget’s disease of bone. Br Med J (Clin Res Ed) 1982; 285:1005–1008.
- van Staa TP, Selby P, Leufkens HG, Lyles K, Sprafka JM, Cooper C. Incidence and natural history of Paget’s disease of bone in England and Wales. J Bone Miner Res 2002; 17:465–471.
- Barker DJ. The epidemiology of Paget’s disease. Metab Bone Dis Relat Res 1981; 3:231–233.
- Rogers J, Jeffrey DR, Watt I. Paget’s disease in an archeological population. J Bone Miner Res 2002; 17:1127–1134.
- Aaron JE, Rogers J, Kanis JA. Paleohistology of Paget’s disease in two medieval skeletons. Am J Phys Anthropol 1992; 89:325–331.
- Poór G, Donáth J, Fornet B, Cooper C. Epidemiology of Paget’s disease in Europe: the prevalence is decreasing. J Bone Miner Res 2006; 21:1545–1549.
- Cundy HR, Gamble G, Wattie D, Rutland M, Cundy T. Paget’s disease of bone in New Zealand: continued decline in disease severity. Calcif Tissue Int 2004; 75:358–364.
- Doyle T, Gunn J, Anderson G, Gill M, Cundy T. Paget’s disease in New Zealand: evidence for declining prevalence. Bone 2002; 31:616–619.
- Laurin N, Brown JP, Morissette J, Raymond V. Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am J Hum Genet 2002; 70:1582–1588.
- Hocking LJ, Lucas GJ, Daroszewska A, et al. Domain-specific mutations in sequestosome 1 (SQSTM1) cause familial and sporadic Paget’s disease. Hum Mol Genet 2002; 11:2735–2739.
- Lucas GJ, Hocking LJ, Daroszewska A, et al. Ubiquitin-associated domain mutations of SQSTM1 in Paget’s disease of bone: evidence for a founder effect in patients of British descent. J Bone Miner Res 2005; 20:227–231.
- Mays S. Archaeological skeletons support a northwest European origin for Paget’s disease of bone. J Bone Miner Res 2010; 25:1839–1841.
- Bolland MJ, Tong PC, Naot D, et al. Delayed development of Paget’s disease in offspring inheriting SQSTM1 mutations. J Bone Miner Res 2007; 22:411–415.
- Rea SL, Walsh JP, Ward L, et al. A novel mutation (K378X) in the sequestosome 1 gene associated with increased NF-kappaB signaling and Paget’s disease of bone with a severe phenotype. J Bone Miner Res 2006; 21:1136–1145.
- Morissette J, Laurin N, Brown JP. Sequestosome 1: mutation frequencies, haplotypes, and phenotypes in familial Paget’s disease of bone. J Bone Miner Res 2006; 21(suppl 2):P38–P44.
- Eekhoff EW, Karperien M, Houtsma D, et al. Familial Paget’s disease in The Netherlands: occurrence, identification of new mutations in the sequestosome 1 gene, and their clinical associations. Arthritis Rheum 2004; 50:1650–1654.
- Ralston SH, Layfield R. Pathogenesis of Paget disease of bone. Calcif Tissue Int 2012; 91:97–113.
- Kurihara N, Hiruma Y, Yamana K, et al. Contributions of the measles virus nucleocapsid gene and the SQSTM1/p62(P392L) mutation to Paget’s disease. Cell Metab 2011; 13:23–34.
- Kurihara N, Zhou H, Reddy SV, et al. Expression of measles virus nucleocapsid protein in osteoclasts induces Paget’s disease-like bone lesions in mice. J Bone Miner Res 2006; 21:446–455.
- Reddy SV, Singer FR, Roodman GD. Bone marrow mononuclear cells from patients with Paget’s disease contain measles virus nucleocapsid messenger ribonucleic acid that has mutations in a specific region of the sequence. J Clin Endocrinol Metab 1995; 80:2108–2111.
- Gennari L, Merlotti D, Martini G, Nuti R. Paget’s disease of bone in Italy. J Bone Miner Res 2006; 21(suppl 2):P14–P21.
- Seton M, Choi HK, Hansen MF, Sebaldt RJ, Cooper C. Analysis of environmental factors in familial versus sporadic Paget’s disease of bone—the New England Registry for Paget’s Disease of Bone. J Bone Miner Res 2003; 18:1519–1524.
- Siris ES. Extensive personal experience: Paget’s disease of bone. J Clin Endocrinol Metab 1995; 80:335–338.
- Lucas GJ, Daroszewska A, Ralston SH. Contribution of genetic factors to the pathogenesis of Paget’s disease of bone and related disorders. J Bone Miner Res 2006; 21(suppl 2):P31–P37.
- Seton M. Diagnosis, complications and treatment of Paget’s disease of bone. Aging Health 2009; 5:497–508.
- Siris E, Roodman GD. Paget’s Disease of Bone. 7th ed. Washington, DC: American Society for Bone and Mineral Research; 2008.
- Seton M, Moses AM, Bode RK, Schwartz C. Paget’s disease of bone: the skeletal distribution, complications and quality of life as perceived by patients. Bone 2011; 48:281–285.
- Seton M. Paget’s disease of bone. In:Hochberg MC, Silman AJ, Smolen JS, Weinblatt ME, Weisman MH, editors. Rheumatology. 5th ed. Philadelphia, PA: Mosby Elsevier; 2010:2021–2028.
- Douglas DL, Duckworth T, Kanis JA, Jefferson AA, Martin TJ, Russell RG. Spinal cord dysfunction in Paget’s disease of bone. Has medical treatment a vascular basis? J Bone Joint Surg Br 1981; 63B:495–503.
- Siris ES. Epidemiological aspects of Paget’s disease: family history and relationship to other medical conditions. Semin Arthritis Rheum 1994; 23:222–225.
- Kanis JA, Evanson JM, Russell RG. Paget’s disease of bone: diagnosis and management. Metab Bone Dis Relat Res 1981; 3:219–230.
- Mangham DC, Davie MW, Grimer RJ. Sarcoma arising in Paget’s disease of bone: declining incidence and increasing age at presentation. Bone 2009; 44:431–436.
- Hansen MF, Seton M, Merchant A. Osteosarcoma in Paget’s disease of bone. J Bone Miner Res 2006; 21(suppl 2):P58–P63.
- Price CH. The incidence of osteogenic sarcoma in South-West England and its relationship to Paget’s disease of bone. J Bone Joint Surg Br 1962; 44-B:366–376.
- Ishikawa Y, Tsukuma H, Miller RW. Low rates of Paget’s disease of bone and osteosarcoma in elderly Japanese. Lancet 1996; 347:1559.
- Sun SG, Lau YS, Itonaga I, Sabokbar A, Athanasou NA. Bone stromal cells in pagetic bone and Paget’s sarcoma express RANKL and support human osteoclast formation. J Pathol 2006; 209:114–120.
- Rendina D, Gennari L, De Filippo G, et al. Evidence for increased clinical severity of familial and sporadic Paget’s disease of bone in Campania, southern Italy. J Bone Miner Res 2006; 21:1828–1835.
- Fenton P, Resnick D. Metastases to bone affected by Paget’s disease. A report of three cases. Int Orthop 1991; 15:397–399.
- Tu SM, Som A, Tu B, Logothetis CJ, Lee MH, Yeung SC. Effect of Paget’s disease of bone (osteitis deformans) on the progression of prostate cancer bone metastasis. Br J Cancer 2012; 107:646–651.
- Eekhoff ME, van der Klift M, Kroon HM, et al. Paget’s disease of bone in The Netherlands: a population-based radiological and biochemical survey—the Rotterdam Study. J Bone Miner Res 2004; 19:566–570.
- Reid IR, Davidson JS, Wattie D, et al. Comparative responses of bone turnover markers to bisphosphonate therapy in Paget’s disease of bone. Bone 2004; 35:224–230.
- Alvarez L, Guañabens N, Peris P, et al. Usefulness of biochemical markers of bone turnover in assessing response to the treatment of Paget’s disease. Bone 2001; 29:447–452.
- Cundy T, Reid IR. Paget’s disease of bone. Clin Biochem 2012; 45:43–48.
- Cortis K, Micallef K, Mizzi A. Imaging Paget’s disease of bone—from head to toe. Clin Radiol 2011; 66:662–672.
- Redden JF, Dixon J, Vennart W, Hosking DJ. Management of fissure fractures in Paget’s disease. Int Orthop 1981; 5:103–106.
- Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 25-1993. A 67-year-old man with osteolytic lesions of T11 and T12. N Engl J Med 1993; 328:1836–1841.
- Evans RA, Dunstan CR, Hills E, Wong SY. Pathologic fracture due to severe osteomalacia following low-dose diphosphonate treatment of Paget’s disease of bone. Aust N Z J Med 1983; 13:277–279.
- Siris E, Weinstein RS, Altman R, et al. Comparative study of alendronate versus etidronate for the treatment of Paget’s disease of bone. J Clin Endocrinol Metab 1996; 81:961–967.
- Reid IR, Siris E. Alendronate in the treatment of Paget’s disease of bone. Int J Clin Pract Suppl 1999; 101:62–66.
- Miller PD, Brown JP, Siris ES, Hoseyni MS, Axelrod DW, Bekker PJ. A randomized, double-blind comparison of risedronate and etidronate in the treatment of Paget’s disease of bone. Paget’s Risedronate/Etidronate Study Group. Am J Med 1999; 106:513–520.
- Peris P, Alvarez L, Vidal S, Martínez MA, Monegal A, Guañabens N. Treatment with tiludronate has a similar effect to risedronate on Paget’s disease activity assessed by bone markers and bone scintigraphy. Clin Exp Rheumatol 2007; 25:206–210.
- Reid IR, Miller P, Lyles K, et al. Comparison of a single infusion of zoledronic acid with risedronate for Paget’s disease. N Engl J Med 2005; 353:898–908.
- Reid IR, Lyles K, Su G, et al. A single infusion of zoledronic acid produces sustained remissions in Paget disease: data to 6.5 years. J Bone Miner Res 2011; 26:2261–2270.
- Langston AL, Campbell MK, Fraser WD, MacLennan GS, Selby PL, Ralston SH; PRISM Trial Group. Randomized trial of intensive bisphosphonate treatment versus symptomatic management in Paget’s disease of bone. J Bone Miner Res 2010; 25:20–31.
- Abrahamsen B, Einhorn TA. Beyond a reasonable doubt? Bisphosphonates and atypical femur fractures. Bone 2012; 50:1196–1200.
- Seton M, Krane SM. Use of zoledronic acid in the treatment of Paget’s disease. Ther Clin Risk Manag 2007; 3:913–918.
- Sørensen HT, Christensen S, Mehnert F, et al. Use of bisphosphonates among women and risk of atrial fibrillation and flutter: Population based case-control study. BMJ 2008; 336:813–816.
- Dixon WG, Solomon DH. Bisphosphonates and esophageal cancer—a pathway through the confusion. Nat Rev Rheumatol 2011; 7:369–372.
- Singer FR, Krane SM. Paget’s disease of bone. In:Avioli LV, Krane SM, editors. Metabolic Bone Disease and Clinically Related Disorders. 2nd ed. Philadelphia, PA: W.B. Saunders Company; 1990:546–615.
- Kanis JA, Horn DB, Scott RD, Strong JA. Treatment of Paget’s disease of bone with synthetic salmon calcitonin. Br Med J 1974; 3:727–731.
- Schwarz P, Rasmussen AQ, Kvist TM, Andersen UB, Jørgensen NR. Paget’s disease of the bone after treatment with denosumab: a case report. Bone 2012; 50:1023–1025.
- Adami S, Bartolozzi P, Brandi ML, et al; Societa Italiana di Ortopedia e Traumatologia. [Italian guidelines for the diagnosis and treatment of Paget’s disease of bone.] Reumatismo 2007; 59:153–168. (Article in Italian.)
- Scarsbrok A, Brown M, Wilson D. UK guidelines on management of Paget’s disease of bone. Rheumatology (Oxford) 2004; 43:399–400.
- Takata S, Hashimoto J, Nakatsuka K, et a.l Guidelines for diagnosis and management of Paget’s disease of bone in Japan. J Bone Miner Metab 2006; 24:359–367.
- Josse RG, Hanley DA, Kendler D, Ste Marie L-G, Adachi JD, Brown J. Diagnosis and treatment of Paget’s disease of bone. Clin Invest Med 2007; 30:E210–E223.
- Kaplan FS. Paget’s disease of bone: orthopedic complications. Semin Arthritis Rheum 1994; 23:250–252.
- Kanis JA, Gray RE. Long-term follow-up observations on treatment in Paget’s disease of bone. Clin Orthop Relat Res 1987; 217:99–125.
KEY POINTS
- The variable prevalence of Paget disease in different geographic regions and its sometimes-familial expression suggest a genetic predisposition or an environmental factor, or both.
- Because Paget disease tends to occur in an aging skeleton, “pagetic” bone may not always be the source of pain. Rather, the pain may be from secondary degenerative changes of the spine or joints or from compression fractures.
- An elevated serum alkaline phosphatase level may signal Paget disease, but many patients have a normal serum alkaline phosphatase.
- Plain radiography of the affected bones outlines the anatomy of the problem and provides insight into the cause of pain.
- Treatment of Paget disease relies primarily on the new generation of nitrogen-containing bisphosphonates.
