User login
2015 Update on Parkinson disease
This has been a boom year for Parkinson disease, with the US Food and Drug Administration (FDA) approving two new therapies, and with others in the pipeline.
This article details clinical signs of Parkinson disease, discusses functional imaging, provides an update on current thinking on disease pathogenesis, and gives an overview of managing parkinsonian symptoms and dyskinesias.
DIAGNOSIS REMAINS CLINICAL
Although a better understanding of Parkinson disease has been gained in recent years, with the recognition of several premotor features and potential biomarkers, its diagnosis is still primarily based on clinical motor findings. The four cardinal motor features have the mnemonic TRAP:
- Tremor at rest can be subtle, involving just the thumb, best observed when the patient is sitting with the hand resting on the lap; or it can be obvious, involving the entire hand, arm, feet, lips, and chin.
- Rigidity can be felt rather than seen, by slowly passively rotating the patient’s wrist or elbow and feeling resistance. The right and left sides often differ.
- Akinesia or bradykinesia (slowness or lack of movement) can be observed by having the patient walk down a hallway. One may observe reduced arm swing and hesitation in initiating movement.
- Postural instability usually develops later rather than sooner in the disease progression. The patient may need to hold onto someone to maintain balance when getting up or walking.
At least two features must be present to make the diagnosis of parkinsonism. One feature must be tremor or rigidity.
Although the criteria for parkinsonism appear simple, the diagnosis of Parkinson disease is not always clear-cut. For example, shaking can be secondary to a dopamine receptor-blocking medication, to anxiety, or to essential tremor; rigidity and slowness may be due to arthritis; and postural instability can result from a neuropathy. Moreover, other neurodegenerative parkinsonian disorders may respond to levodopa (at least initially) and may present with levodopa-induced dyskinesias. Robust response to levodopa and the occurrence of dyskinesias are two additional features that strongly suggest the diagnosis of Parkinson disease.
Supporting parkinsonian features include stooped posture, masked facies, micrographia (small handwriting), drooling, speech changes (eg, hypophonia or soft speech, stuttering, slurring, monotonic speech), and a shuffling, festinating gait (quick short steps as if falling forward).
PARKINSON MIMICS
Parkinsonism is a broader term than Parkinson disease or idiopathic Parkinson disease. It is characterized by akinetic rigidity and impaired motor activity that leads to reduced function and falls; behavioral changes also may occur.
In the United States, Parkinson disease is the most common cause of parkinsonism. Other nonneurodegenerative causes are drug-induced parkinsonism (due to dopamine receptor antagonists such as antipsychotic or antiemetic drugs), stroke (in the basal ganglia or frontal lobe), and normal-pressure hydrocephalus (causing lower-body parkinsonism). Mimics of parkinsonism include essential tremor and psychogenic parkinsonism.
Parkinsonism can also be caused by Parkinson-plus disorders, ie, neurodegenerative conditions characterized by parkinsonism along with additional signs and symptoms, as listed below. Parkinson-plus disorders include progressive supranuclear palsy, multiple system atrophy, corticobasal degeneration, and Lewy body disease.
Clinical features that suggest a diagnosis other than Parkinson disease include1:
- Poor response to adequate dosages of levodopa
- Early onset of postural instability and falls
- Axial rigidity (eg, stiff neck) more than appendicular rigidity
- Early dementia
- Supranuclear gaze palsy
- Unusual movements besides tremor, eg, limb dystonia, myoclonus, limb levitation or alien limb syndrome
- Profound autonomic dysfunction
- Psychotic symptoms before taking levodopa or dopaminergic medication.
The precise diagnosis of Parkinson-plus disorders is not critical, as the treatment is generally the same for all of them: ie, levodopa (if it shows some efficacy and is well tolerated), with additional symptomatic treatment for features such as depression, cognitive impairment, and autonomic dysfunction, and supportive therapy including physical, occupational, speech, and swallowing therapy.
IMAGING MAY ASSIST IN THE DIAGNOSIS
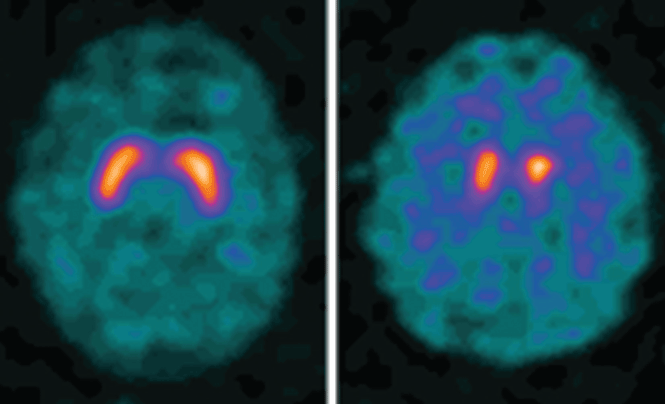
Dopamine transporter single-photon emission computed tomography (SPECT) is a functional imaging technique that supposedly reflects dopamine uptake by surviving presynaptic dopaminergic neurons in the striate bodies of the basal ganglia. Normal uptake shows distinct cashew-shaped enhancement bilaterally. In Parkinson disease, the enhanced areas are smaller and asymmetric, first with diminution of the tail (representing the putamen), then later involving the head (representing the caudate) along with the other striate bodies (Figure 1).

Dopamine transporter SPECT does not distinguish one neurodegenerative parkinsonian disorder from another. Therefore, it should not be used to distinguish Parkinson disease from other Parkinson-plus syndromes. But it does distinguish neurodegenerative parkinsonian disorders from nonneurodegenerative conditions and mimics, which have a normal result on dopamine transporter SPECT (Table 1).
SLOWING DISEASE PROGRESSION
Current treatments for Parkinson disease can significantly improve symptoms but, unfortunately, do not cure the disease or slow its progression. Testing whether agents modify the disease course is particularly difficult with Parkinson disease, because it affects individuals differently, has a wide spectrum of symptoms, has a long time course, and lacks definitive markers to monitor progression. Some agents have shown promise:
Caffeine. People who drink coffee are less likely to develop Parkinson disease, with the risk declining with the number of cups per day.2 For those who have the disease, drinking coffee is associated with reduced symptoms.
Exercise improves Parkinson disease and may prevent it, and some studies suggest that it can delay its progression.3 Exercise has been shown in an animal model to reduce the vulnerability of dopamine neurons to the toxic agent 6-hydroxydopamine.4 Functional magnetic resonance imaging studies have shown blood flow patterns before and after exercise that are similar to those seen in patients with and without Parkinson medication.3
Rasagiline, a monoamine oxidase B (MAO-B) inhibitor used for symptomatic treatment of Parkinson disease, had conflicting results in a neuroprotective clinical trial. Patients who received rasagiline 1 mg daily—but not those who received 2 mg daily—at the beginning of the trial had better Parkinson motor scores compared with patients who received rasagiline 9 months later.5
Inosine is a urate precursor that elevates urate levels in serum and the central nervous system. For unknown reasons, patients with Parkinson disease tend to have a low uric acid level, and higher levels are associated with milder disease. It is hoped that raising the uric acid level to a “pre-gout level” may slow the progression of Parkinson disease.
Isradipine, a calcium channel blocker, was found in an epidemiologic study of elderly patients to be associated with reduced likelihood of developing Parkinson disease.6 The drug is now undergoing clinical trials.
Smoking. Although cigarette smokers have long been recognized as having a very low risk of developing Parkinson disease, smoking is not recommended.
Agents found ineffective. Agents that have been tested and found ineffective in modifying the course of Parkinson disease include vitamin E, coenzyme Q10, riluzole, GPI-1485, pramipexole, cogane, CEP-1347, TCH-346, and creatine.
NOT JUST DOPAMINE—OR TREMORS
Dopamine deficiency is central to the current understanding of the pathogenesis of Parkinson disease and the focus of treatment efforts, but if dopamine deficiency were the only problem, replacing it should completely ameliorate all parkinsonian features. Other neurotransmitters also play roles: norepinephrine is implicated in orthostatic symptoms and apathy, acetylcholine in cognitive behaviors, glutamate in dyskinesias, and serotonin in depression, anxiety, and sleep abnormalities.
The most recognized area of involvement in the brain has traditionally been the substantia nigra in the midbrain. However, current thinking is that the disease starts lower in the caudal area of the brainstem (along with the olfactory tubercle), moves through the pons to the midbrain, then spreads across the cerebrum with extensive neocortical involvement.
Early premotor indicators are now recognized to occur 15 to 20 years before a tremor appears. The first signs are often hyposmia (diminished sense of smell, reflecting involvement of the olfactory tubercle) and constipation (reflecting involvement of the medulla and the vagus nucleus). With pons involvement, the patient can develop rapid eye movement sleep behavior disorder, depression, or anxiety. Only then does the disease spread to the midbrain and cause resting tremor, rigidity, and bradykinesia.7
Identifying the preclinical stages and starting disease-modifying treatments before the onset of motor symptoms may one day prove important, but at this point, the premotor symptoms (anosmia, constipation, depression) are too nonspecific to be useful, and such treatments have not yet been identified.
TREATMENT: LEVODOPA STILL PRIMARY
When to start drug treatment depends primarily on how much the symptoms bother the patient. Regardless of the clinician’s (or patient’s) belief in the benefits of delaying symptomatic treatment, it is universally considered necessary to start medication when gait problems develop because of the danger of a fall and resulting disability.
Carbidopa-levodopa combination therapy remains the most effective treatment; if it is not effective, another diagnosis may need to be considered. Carbidopa-levodopa improves tremor, rigidity, and bradykinesia, particularly in the early stages of Parkinson disease. It is well tolerated, has rapid onset, reduces the risk of death, and is the least expensive of the medications for Parkinson disease.
Immediate-release and continued-release formulations are available, as well as one that dissolves rapidly on the tongue and can be taken without water. An oral extended-release carbidopa-levodopa formulation (Rytary) was approved by the FDA in January 2015. Tablets are filled with drug-containing microbeads that dissolve at different rates to achieve therapeutic levodopa levels as quickly as the immediate-release formulation and maintain them for an extended time.8
The development of dyskinesias is the major psychological drawback of levodopa, occurring in 80% of patients after 5 to 10 years of treatment. Although many patients fear this side effect, most patients who develop it find it preferable to the rigidity and bradykinesia of Parkinson disease. In most cases, bothersome dyskinesias can be controlled by adjusting medications.9,10
Dopamine agonists include pramipexole, ropinirole, and rotigotine. They are available in generic form as three-times-daily dosing; once-daily dosing is also available, but not as a generic formulation. Dopamine agonists have the advantage of potentially improving depression and delaying the onset of dyskinesias.
However, dopamine agonists have a number of disadvantages compared with levodopa: they have a longer titration period, are less effective, and are less well tolerated, especially in the elderly. Side effects occur more frequently than with levodopa and include general and peripheral edema, hallucinations, nausea, lightheadedness, and sleepiness.11,12 These drugs are also associated with “sleep attacks” (sudden falling asleep while active, such as while driving or eating) and with compulsive and impulsive behaviors such as hypersexuality, buying, binge eating, and gambling. Although these behaviors occur in fewer than 10% of patients, they can be devastating, leading to marital, financial, and legal problems. A bothersome clinical state termed dopamine agonist withdrawal syndrome is characterized by anxiety, depression, jitteriness, and palpitations when dopamine agonists are tapered or discontinued because of a side effect.13
MAO-B inhibitors delay the breakdown of dopamine, allowing it to “stay” in the brain for a longer period of time. Rasagiline for early monotherapy has the advantages of once-daily dosing, no titration, and excellent tolerability, even in the elderly. Potential drug interactions should be considered when using this drug. Early warnings about interactions with tyramine-rich foods were lifted after trials showed that this was not a problem.14
Amantadine is an N-methyl-d-aspartate (NMDA) receptor antagonist often used in early Parkinson disease and for treatment of dyskinesias and fatigue. It is the only drug that is intrinsically antidyskinetic and also improves Parkinson symptoms.15 Side effects include leg swelling, livedo reticularis, and neuropsychiatric and anticholinergic effects.
Anticholinergic agents (eg, trihexyphenidyl) improve tremor but are not as useful for bradykinesia or rigidity, and often have anticholinergic effects such as mental dullness, dry mouth, dry eye, and urinary hesitancy, especially in the elderly, so they have a limited role in Parkinson treatment.
MOTOR COMPLICATIONS: FLUCTUATIONS AND DYSKINESIAS
Motor fluctuations are changes between the akinetic and mobile phases of Parkinson disease, or the off-periods and on-periods of drug treatment. A patient who is “off” is generally rigid and feels that the medication is not working. A patient who is “on” feels loose and mobile and that the medication is working. Variants of motor fluctuations include:
- End-of-dose deterioration
- Delayed onset of response (more than half an hour after taking medication)
- Drug-resistant offs—medication has become ineffective
- Random oscillation—on-off phenomenon
- Freezing—unpredictable inability to start or finish a movement.
Dyskinesias are abnormal involuntary movements such as writhing and twisting. They are associated with dopaminergic therapy at peak dose, when the drug starts to turn on or wear off (termed diphasic dyskinesias).16
The storage hypothesis provides a plausible explanation for the development of motor complications as the disease progresses. Although the half-life of levodopa is only 60 to 90 minutes, it is effective in early disease when given three times a day. It is believed that at this stage of the disease, enough dopaminergic neurons survive to “store” dopamine and release it as needed. As the disease progresses and dopaminergic neurons die, storage capacity diminishes, and the clinical effect slowly starts to approximate the pharmacokinetic profile of the drug. Upon taking the medication, the patient gets a surge of drug, causing dyskinesias, followed later by rigidity as the effect wears off since there are fewer surviving dopaminergic cells to store dopamine.
MANAGING DYSKINESIAS
Patients with dyskinesias should first be asked if they are bothered by them; not all patients are troubled by dyskinesias. If the movements only bother others (eg, family members), then education is often the only treatment needed. If the patient is uncomfortable, the following measures can be tried:
- Taking lower, more frequent doses of levodopa (however, risk of wearing off becomes a problem)
- Adding a dopamine agonist or MAO-B inhibitor while lowering the levodopa dose (however, MAO-B inhibitors pose a risk of side effects in elderly patients)
- Adding clozapine (periodic laboratory testing is required to monitor blood levels and liver and kidney function)
- Adding amantadine (however, this poses a risk of cognitive side effects).
Deep-brain-stimulation surgery is appropriate for select patients who are generally physically healthy, cognitively intact, and emotionally stable, with a strong family support system, but who are bothered by symptoms of parkinsonism (such as tremors), motor fluctuations, or dyskinesias.17
Infusion pump. In January 2015, the FDA approved a new system that continuously delivers levodopa-carbidopa in a 4:1 ratio in gel suspension for 16 hours directly into the small intestine, minimizing motor fluctuations. The patient changes the cartridge daily and turns it off at bedtime.
*Dr. Fernandez has received research support from AbbVie, Acadia, Auspex, Biotie Therapies, Civitas, Kyowa/ProStrakan, Michael J. Fox Foundation, Movement Disorders Society, NIH/NINDS, Parkinson Study Group, Rhythm, Synosia, and Teva. He also has received honoraria from Carling Communications, International Parkinson and Movement Disorders Society, The Ohio State University, and PRIME Education, Inc as a speaker in CME events. He has received honoraria from Biogen, GE Health Care, Lundbeck, Merz Pharmaceuticals, and Pfizer as a consultant. He has received royalty payments from Demos Publishing for serving as a book author/editor. Cleveland Clinic has contracts with AbbVie and Merz Pharmaceuticals for Dr. Fernandez’s role as a member of the Global Steering Committee for LCIG studies and as a consultant or speaker, and as Head Principal Investigator for the Xeomin Registry Study. Dr. Fernandez has received a stipend from International Parkinson and Movement Disorders Society for serving as medical editor of the Movement Disorders Society website.
- Wenning GK, Ben-Shlomo Y, Hughes A, Daniel SE, Lees A, Quinn NP. What clinical features are most useful to distinguish definite multiple system atrophy from Parkinson’s disease? J Neurol Neurosurg Psychiatry 2000; 68:434–440.
- Hernán MA, Takkouche B, Caamaño-Isoma F, et al. A meta-analysis of coffee drinking, cigarette smoking, and risk of Parkinson’s disease. Ann Neurol 2002; 52:276–84.
- Ridgel A, Thota A, Vitek JL, Alberts JL. Forced, not voluntary, exercise improves motor function in Parkinson’s disease patients. Neurorehabil Neural Repair 2009; 23:600–608.
- Smith AD, Zigmond MJ. Can the brain be protected through exercise? Lessons from an animal model of parkinsonism. Exp Neurol 2003; 184:31–39.
- Olanow CW, Rascol O, Hauser R, et al, for the ADAGIO Study Investigators. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N Engl J Med 2009; 361:1268–1278.
- Pasternak B, Svanström H, Nielsen NM, Fugger L, Melbye M, Hviid A. Use of calcium channel blockers and Parkinson’s disease. Am J Epidemiol 2012; 175:627-635.
- Braak H, Del Tredici K, Rüb U, de Vos RAI, Jansen Steur ENH, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 2003; 24:197–211.
- Hauser RA, Ellenbogen AL, Metman LV, et al. Crossover comparison of IPX066 and a standard levodopa formulation in advanced Parkinson’s disease. Mov Disord 2011; 26:2246–2252.
- Hely MA, Morris JG, Reid WG, Trafficante R. Sydney Multicenter Study of Parkinson’s disease: non-L-dopa-responsive problems dominate at 15 years. Mov Disord 2005; 20:190–199.
- Hung SW, Adeli GM, Arenovich T, Fox SH, Lang AE. Patient perception of dyskinesia in Parkinson’s disease. J Neurol Neurosurg Psychiatry 2010; 81:1112–1115.
- Rascol O, Brooks DJ, Korczyn AD, De Deyn PP, Clarke CE, Lang AE. A five-year study of the incidence of dyskinesia in patients with early Parkinson’s disease who were treated with ropinirole or levodopa. N Engl J Med 2000; 342:1484–1491.
- Parkinson Study Group. Pramipexole vs levodopa as initial treatment for Parkinson disease: a randomized controlled trial. Parkinson Study Group. JAMA 2000; 284:1931–1938.
- Nirenberg MJ. Dopamine agonist withdrawal syndrome: implications for patient care. Drugs Aging 2013; 30:587–592.
- Teva Neuroscience, Inc. Azilect prescribing information. https://www.azilect.com/Content/pdf/azi-40850-azilect-electronic-pi.pdf. Accessed June 29, 2015.
- Snow BJ, Macdonald L, Mcauley D, Wallis W. The effect of amantadine on levodopa-induced dyskinesias in Parkinson’s disease: a double-blind, placebo-controlled study. Clin Neuropharmacol 2000; 23:82–85.
- Adler CH, Ahlskog JE, eds. Parkinson’s Disease and Movement Disorders: Diagnosis and Treatment Guidelines for the Practicing Physician. Totowa, NJ: Humana Press; 2000.
- Machado A, Fernandez HH, Deogaonkar M. Deep brain stimulation: what can patients expect from it? Cleve Clin J Med 2012; 79:113–120.
This has been a boom year for Parkinson disease, with the US Food and Drug Administration (FDA) approving two new therapies, and with others in the pipeline.
This article details clinical signs of Parkinson disease, discusses functional imaging, provides an update on current thinking on disease pathogenesis, and gives an overview of managing parkinsonian symptoms and dyskinesias.
DIAGNOSIS REMAINS CLINICAL
Although a better understanding of Parkinson disease has been gained in recent years, with the recognition of several premotor features and potential biomarkers, its diagnosis is still primarily based on clinical motor findings. The four cardinal motor features have the mnemonic TRAP:
- Tremor at rest can be subtle, involving just the thumb, best observed when the patient is sitting with the hand resting on the lap; or it can be obvious, involving the entire hand, arm, feet, lips, and chin.
- Rigidity can be felt rather than seen, by slowly passively rotating the patient’s wrist or elbow and feeling resistance. The right and left sides often differ.
- Akinesia or bradykinesia (slowness or lack of movement) can be observed by having the patient walk down a hallway. One may observe reduced arm swing and hesitation in initiating movement.
- Postural instability usually develops later rather than sooner in the disease progression. The patient may need to hold onto someone to maintain balance when getting up or walking.
At least two features must be present to make the diagnosis of parkinsonism. One feature must be tremor or rigidity.
Although the criteria for parkinsonism appear simple, the diagnosis of Parkinson disease is not always clear-cut. For example, shaking can be secondary to a dopamine receptor-blocking medication, to anxiety, or to essential tremor; rigidity and slowness may be due to arthritis; and postural instability can result from a neuropathy. Moreover, other neurodegenerative parkinsonian disorders may respond to levodopa (at least initially) and may present with levodopa-induced dyskinesias. Robust response to levodopa and the occurrence of dyskinesias are two additional features that strongly suggest the diagnosis of Parkinson disease.
Supporting parkinsonian features include stooped posture, masked facies, micrographia (small handwriting), drooling, speech changes (eg, hypophonia or soft speech, stuttering, slurring, monotonic speech), and a shuffling, festinating gait (quick short steps as if falling forward).
PARKINSON MIMICS
Parkinsonism is a broader term than Parkinson disease or idiopathic Parkinson disease. It is characterized by akinetic rigidity and impaired motor activity that leads to reduced function and falls; behavioral changes also may occur.
In the United States, Parkinson disease is the most common cause of parkinsonism. Other nonneurodegenerative causes are drug-induced parkinsonism (due to dopamine receptor antagonists such as antipsychotic or antiemetic drugs), stroke (in the basal ganglia or frontal lobe), and normal-pressure hydrocephalus (causing lower-body parkinsonism). Mimics of parkinsonism include essential tremor and psychogenic parkinsonism.
Parkinsonism can also be caused by Parkinson-plus disorders, ie, neurodegenerative conditions characterized by parkinsonism along with additional signs and symptoms, as listed below. Parkinson-plus disorders include progressive supranuclear palsy, multiple system atrophy, corticobasal degeneration, and Lewy body disease.
Clinical features that suggest a diagnosis other than Parkinson disease include1:
- Poor response to adequate dosages of levodopa
- Early onset of postural instability and falls
- Axial rigidity (eg, stiff neck) more than appendicular rigidity
- Early dementia
- Supranuclear gaze palsy
- Unusual movements besides tremor, eg, limb dystonia, myoclonus, limb levitation or alien limb syndrome
- Profound autonomic dysfunction
- Psychotic symptoms before taking levodopa or dopaminergic medication.
The precise diagnosis of Parkinson-plus disorders is not critical, as the treatment is generally the same for all of them: ie, levodopa (if it shows some efficacy and is well tolerated), with additional symptomatic treatment for features such as depression, cognitive impairment, and autonomic dysfunction, and supportive therapy including physical, occupational, speech, and swallowing therapy.
IMAGING MAY ASSIST IN THE DIAGNOSIS
Dopamine transporter single-photon emission computed tomography (SPECT) is a functional imaging technique that supposedly reflects dopamine uptake by surviving presynaptic dopaminergic neurons in the striate bodies of the basal ganglia. Normal uptake shows distinct cashew-shaped enhancement bilaterally. In Parkinson disease, the enhanced areas are smaller and asymmetric, first with diminution of the tail (representing the putamen), then later involving the head (representing the caudate) along with the other striate bodies (Figure 1).
Dopamine transporter SPECT does not distinguish one neurodegenerative parkinsonian disorder from another. Therefore, it should not be used to distinguish Parkinson disease from other Parkinson-plus syndromes. But it does distinguish neurodegenerative parkinsonian disorders from nonneurodegenerative conditions and mimics, which have a normal result on dopamine transporter SPECT (Table 1).
SLOWING DISEASE PROGRESSION
Current treatments for Parkinson disease can significantly improve symptoms but, unfortunately, do not cure the disease or slow its progression. Testing whether agents modify the disease course is particularly difficult with Parkinson disease, because it affects individuals differently, has a wide spectrum of symptoms, has a long time course, and lacks definitive markers to monitor progression. Some agents have shown promise:
Caffeine. People who drink coffee are less likely to develop Parkinson disease, with the risk declining with the number of cups per day.2 For those who have the disease, drinking coffee is associated with reduced symptoms.
Exercise improves Parkinson disease and may prevent it, and some studies suggest that it can delay its progression.3 Exercise has been shown in an animal model to reduce the vulnerability of dopamine neurons to the toxic agent 6-hydroxydopamine.4 Functional magnetic resonance imaging studies have shown blood flow patterns before and after exercise that are similar to those seen in patients with and without Parkinson medication.3
Rasagiline, a monoamine oxidase B (MAO-B) inhibitor used for symptomatic treatment of Parkinson disease, had conflicting results in a neuroprotective clinical trial. Patients who received rasagiline 1 mg daily—but not those who received 2 mg daily—at the beginning of the trial had better Parkinson motor scores compared with patients who received rasagiline 9 months later.5
Inosine is a urate precursor that elevates urate levels in serum and the central nervous system. For unknown reasons, patients with Parkinson disease tend to have a low uric acid level, and higher levels are associated with milder disease. It is hoped that raising the uric acid level to a “pre-gout level” may slow the progression of Parkinson disease.
Isradipine, a calcium channel blocker, was found in an epidemiologic study of elderly patients to be associated with reduced likelihood of developing Parkinson disease.6 The drug is now undergoing clinical trials.
Smoking. Although cigarette smokers have long been recognized as having a very low risk of developing Parkinson disease, smoking is not recommended.
Agents found ineffective. Agents that have been tested and found ineffective in modifying the course of Parkinson disease include vitamin E, coenzyme Q10, riluzole, GPI-1485, pramipexole, cogane, CEP-1347, TCH-346, and creatine.
NOT JUST DOPAMINE—OR TREMORS
Dopamine deficiency is central to the current understanding of the pathogenesis of Parkinson disease and the focus of treatment efforts, but if dopamine deficiency were the only problem, replacing it should completely ameliorate all parkinsonian features. Other neurotransmitters also play roles: norepinephrine is implicated in orthostatic symptoms and apathy, acetylcholine in cognitive behaviors, glutamate in dyskinesias, and serotonin in depression, anxiety, and sleep abnormalities.
The most recognized area of involvement in the brain has traditionally been the substantia nigra in the midbrain. However, current thinking is that the disease starts lower in the caudal area of the brainstem (along with the olfactory tubercle), moves through the pons to the midbrain, then spreads across the cerebrum with extensive neocortical involvement.
Early premotor indicators are now recognized to occur 15 to 20 years before a tremor appears. The first signs are often hyposmia (diminished sense of smell, reflecting involvement of the olfactory tubercle) and constipation (reflecting involvement of the medulla and the vagus nucleus). With pons involvement, the patient can develop rapid eye movement sleep behavior disorder, depression, or anxiety. Only then does the disease spread to the midbrain and cause resting tremor, rigidity, and bradykinesia.7
Identifying the preclinical stages and starting disease-modifying treatments before the onset of motor symptoms may one day prove important, but at this point, the premotor symptoms (anosmia, constipation, depression) are too nonspecific to be useful, and such treatments have not yet been identified.
TREATMENT: LEVODOPA STILL PRIMARY
When to start drug treatment depends primarily on how much the symptoms bother the patient. Regardless of the clinician’s (or patient’s) belief in the benefits of delaying symptomatic treatment, it is universally considered necessary to start medication when gait problems develop because of the danger of a fall and resulting disability.
Carbidopa-levodopa combination therapy remains the most effective treatment; if it is not effective, another diagnosis may need to be considered. Carbidopa-levodopa improves tremor, rigidity, and bradykinesia, particularly in the early stages of Parkinson disease. It is well tolerated, has rapid onset, reduces the risk of death, and is the least expensive of the medications for Parkinson disease.
Immediate-release and continued-release formulations are available, as well as one that dissolves rapidly on the tongue and can be taken without water. An oral extended-release carbidopa-levodopa formulation (Rytary) was approved by the FDA in January 2015. Tablets are filled with drug-containing microbeads that dissolve at different rates to achieve therapeutic levodopa levels as quickly as the immediate-release formulation and maintain them for an extended time.8
The development of dyskinesias is the major psychological drawback of levodopa, occurring in 80% of patients after 5 to 10 years of treatment. Although many patients fear this side effect, most patients who develop it find it preferable to the rigidity and bradykinesia of Parkinson disease. In most cases, bothersome dyskinesias can be controlled by adjusting medications.9,10
Dopamine agonists include pramipexole, ropinirole, and rotigotine. They are available in generic form as three-times-daily dosing; once-daily dosing is also available, but not as a generic formulation. Dopamine agonists have the advantage of potentially improving depression and delaying the onset of dyskinesias.
However, dopamine agonists have a number of disadvantages compared with levodopa: they have a longer titration period, are less effective, and are less well tolerated, especially in the elderly. Side effects occur more frequently than with levodopa and include general and peripheral edema, hallucinations, nausea, lightheadedness, and sleepiness.11,12 These drugs are also associated with “sleep attacks” (sudden falling asleep while active, such as while driving or eating) and with compulsive and impulsive behaviors such as hypersexuality, buying, binge eating, and gambling. Although these behaviors occur in fewer than 10% of patients, they can be devastating, leading to marital, financial, and legal problems. A bothersome clinical state termed dopamine agonist withdrawal syndrome is characterized by anxiety, depression, jitteriness, and palpitations when dopamine agonists are tapered or discontinued because of a side effect.13
MAO-B inhibitors delay the breakdown of dopamine, allowing it to “stay” in the brain for a longer period of time. Rasagiline for early monotherapy has the advantages of once-daily dosing, no titration, and excellent tolerability, even in the elderly. Potential drug interactions should be considered when using this drug. Early warnings about interactions with tyramine-rich foods were lifted after trials showed that this was not a problem.14
Amantadine is an N-methyl-d-aspartate (NMDA) receptor antagonist often used in early Parkinson disease and for treatment of dyskinesias and fatigue. It is the only drug that is intrinsically antidyskinetic and also improves Parkinson symptoms.15 Side effects include leg swelling, livedo reticularis, and neuropsychiatric and anticholinergic effects.
Anticholinergic agents (eg, trihexyphenidyl) improve tremor but are not as useful for bradykinesia or rigidity, and often have anticholinergic effects such as mental dullness, dry mouth, dry eye, and urinary hesitancy, especially in the elderly, so they have a limited role in Parkinson treatment.
MOTOR COMPLICATIONS: FLUCTUATIONS AND DYSKINESIAS
Motor fluctuations are changes between the akinetic and mobile phases of Parkinson disease, or the off-periods and on-periods of drug treatment. A patient who is “off” is generally rigid and feels that the medication is not working. A patient who is “on” feels loose and mobile and that the medication is working. Variants of motor fluctuations include:
- End-of-dose deterioration
- Delayed onset of response (more than half an hour after taking medication)
- Drug-resistant offs—medication has become ineffective
- Random oscillation—on-off phenomenon
- Freezing—unpredictable inability to start or finish a movement.
Dyskinesias are abnormal involuntary movements such as writhing and twisting. They are associated with dopaminergic therapy at peak dose, when the drug starts to turn on or wear off (termed diphasic dyskinesias).16
The storage hypothesis provides a plausible explanation for the development of motor complications as the disease progresses. Although the half-life of levodopa is only 60 to 90 minutes, it is effective in early disease when given three times a day. It is believed that at this stage of the disease, enough dopaminergic neurons survive to “store” dopamine and release it as needed. As the disease progresses and dopaminergic neurons die, storage capacity diminishes, and the clinical effect slowly starts to approximate the pharmacokinetic profile of the drug. Upon taking the medication, the patient gets a surge of drug, causing dyskinesias, followed later by rigidity as the effect wears off since there are fewer surviving dopaminergic cells to store dopamine.
MANAGING DYSKINESIAS
Patients with dyskinesias should first be asked if they are bothered by them; not all patients are troubled by dyskinesias. If the movements only bother others (eg, family members), then education is often the only treatment needed. If the patient is uncomfortable, the following measures can be tried:
- Taking lower, more frequent doses of levodopa (however, risk of wearing off becomes a problem)
- Adding a dopamine agonist or MAO-B inhibitor while lowering the levodopa dose (however, MAO-B inhibitors pose a risk of side effects in elderly patients)
- Adding clozapine (periodic laboratory testing is required to monitor blood levels and liver and kidney function)
- Adding amantadine (however, this poses a risk of cognitive side effects).
Deep-brain-stimulation surgery is appropriate for select patients who are generally physically healthy, cognitively intact, and emotionally stable, with a strong family support system, but who are bothered by symptoms of parkinsonism (such as tremors), motor fluctuations, or dyskinesias.17
Infusion pump. In January 2015, the FDA approved a new system that continuously delivers levodopa-carbidopa in a 4:1 ratio in gel suspension for 16 hours directly into the small intestine, minimizing motor fluctuations. The patient changes the cartridge daily and turns it off at bedtime.
*Dr. Fernandez has received research support from AbbVie, Acadia, Auspex, Biotie Therapies, Civitas, Kyowa/ProStrakan, Michael J. Fox Foundation, Movement Disorders Society, NIH/NINDS, Parkinson Study Group, Rhythm, Synosia, and Teva. He also has received honoraria from Carling Communications, International Parkinson and Movement Disorders Society, The Ohio State University, and PRIME Education, Inc as a speaker in CME events. He has received honoraria from Biogen, GE Health Care, Lundbeck, Merz Pharmaceuticals, and Pfizer as a consultant. He has received royalty payments from Demos Publishing for serving as a book author/editor. Cleveland Clinic has contracts with AbbVie and Merz Pharmaceuticals for Dr. Fernandez’s role as a member of the Global Steering Committee for LCIG studies and as a consultant or speaker, and as Head Principal Investigator for the Xeomin Registry Study. Dr. Fernandez has received a stipend from International Parkinson and Movement Disorders Society for serving as medical editor of the Movement Disorders Society website.
This has been a boom year for Parkinson disease, with the US Food and Drug Administration (FDA) approving two new therapies, and with others in the pipeline.
This article details clinical signs of Parkinson disease, discusses functional imaging, provides an update on current thinking on disease pathogenesis, and gives an overview of managing parkinsonian symptoms and dyskinesias.
DIAGNOSIS REMAINS CLINICAL
Although a better understanding of Parkinson disease has been gained in recent years, with the recognition of several premotor features and potential biomarkers, its diagnosis is still primarily based on clinical motor findings. The four cardinal motor features have the mnemonic TRAP:
- Tremor at rest can be subtle, involving just the thumb, best observed when the patient is sitting with the hand resting on the lap; or it can be obvious, involving the entire hand, arm, feet, lips, and chin.
- Rigidity can be felt rather than seen, by slowly passively rotating the patient’s wrist or elbow and feeling resistance. The right and left sides often differ.
- Akinesia or bradykinesia (slowness or lack of movement) can be observed by having the patient walk down a hallway. One may observe reduced arm swing and hesitation in initiating movement.
- Postural instability usually develops later rather than sooner in the disease progression. The patient may need to hold onto someone to maintain balance when getting up or walking.
At least two features must be present to make the diagnosis of parkinsonism. One feature must be tremor or rigidity.
Although the criteria for parkinsonism appear simple, the diagnosis of Parkinson disease is not always clear-cut. For example, shaking can be secondary to a dopamine receptor-blocking medication, to anxiety, or to essential tremor; rigidity and slowness may be due to arthritis; and postural instability can result from a neuropathy. Moreover, other neurodegenerative parkinsonian disorders may respond to levodopa (at least initially) and may present with levodopa-induced dyskinesias. Robust response to levodopa and the occurrence of dyskinesias are two additional features that strongly suggest the diagnosis of Parkinson disease.
Supporting parkinsonian features include stooped posture, masked facies, micrographia (small handwriting), drooling, speech changes (eg, hypophonia or soft speech, stuttering, slurring, monotonic speech), and a shuffling, festinating gait (quick short steps as if falling forward).
PARKINSON MIMICS
Parkinsonism is a broader term than Parkinson disease or idiopathic Parkinson disease. It is characterized by akinetic rigidity and impaired motor activity that leads to reduced function and falls; behavioral changes also may occur.
In the United States, Parkinson disease is the most common cause of parkinsonism. Other nonneurodegenerative causes are drug-induced parkinsonism (due to dopamine receptor antagonists such as antipsychotic or antiemetic drugs), stroke (in the basal ganglia or frontal lobe), and normal-pressure hydrocephalus (causing lower-body parkinsonism). Mimics of parkinsonism include essential tremor and psychogenic parkinsonism.
Parkinsonism can also be caused by Parkinson-plus disorders, ie, neurodegenerative conditions characterized by parkinsonism along with additional signs and symptoms, as listed below. Parkinson-plus disorders include progressive supranuclear palsy, multiple system atrophy, corticobasal degeneration, and Lewy body disease.
Clinical features that suggest a diagnosis other than Parkinson disease include1:
- Poor response to adequate dosages of levodopa
- Early onset of postural instability and falls
- Axial rigidity (eg, stiff neck) more than appendicular rigidity
- Early dementia
- Supranuclear gaze palsy
- Unusual movements besides tremor, eg, limb dystonia, myoclonus, limb levitation or alien limb syndrome
- Profound autonomic dysfunction
- Psychotic symptoms before taking levodopa or dopaminergic medication.
The precise diagnosis of Parkinson-plus disorders is not critical, as the treatment is generally the same for all of them: ie, levodopa (if it shows some efficacy and is well tolerated), with additional symptomatic treatment for features such as depression, cognitive impairment, and autonomic dysfunction, and supportive therapy including physical, occupational, speech, and swallowing therapy.
IMAGING MAY ASSIST IN THE DIAGNOSIS
Dopamine transporter single-photon emission computed tomography (SPECT) is a functional imaging technique that supposedly reflects dopamine uptake by surviving presynaptic dopaminergic neurons in the striate bodies of the basal ganglia. Normal uptake shows distinct cashew-shaped enhancement bilaterally. In Parkinson disease, the enhanced areas are smaller and asymmetric, first with diminution of the tail (representing the putamen), then later involving the head (representing the caudate) along with the other striate bodies (Figure 1).
Dopamine transporter SPECT does not distinguish one neurodegenerative parkinsonian disorder from another. Therefore, it should not be used to distinguish Parkinson disease from other Parkinson-plus syndromes. But it does distinguish neurodegenerative parkinsonian disorders from nonneurodegenerative conditions and mimics, which have a normal result on dopamine transporter SPECT (Table 1).
SLOWING DISEASE PROGRESSION
Current treatments for Parkinson disease can significantly improve symptoms but, unfortunately, do not cure the disease or slow its progression. Testing whether agents modify the disease course is particularly difficult with Parkinson disease, because it affects individuals differently, has a wide spectrum of symptoms, has a long time course, and lacks definitive markers to monitor progression. Some agents have shown promise:
Caffeine. People who drink coffee are less likely to develop Parkinson disease, with the risk declining with the number of cups per day.2 For those who have the disease, drinking coffee is associated with reduced symptoms.
Exercise improves Parkinson disease and may prevent it, and some studies suggest that it can delay its progression.3 Exercise has been shown in an animal model to reduce the vulnerability of dopamine neurons to the toxic agent 6-hydroxydopamine.4 Functional magnetic resonance imaging studies have shown blood flow patterns before and after exercise that are similar to those seen in patients with and without Parkinson medication.3
Rasagiline, a monoamine oxidase B (MAO-B) inhibitor used for symptomatic treatment of Parkinson disease, had conflicting results in a neuroprotective clinical trial. Patients who received rasagiline 1 mg daily—but not those who received 2 mg daily—at the beginning of the trial had better Parkinson motor scores compared with patients who received rasagiline 9 months later.5
Inosine is a urate precursor that elevates urate levels in serum and the central nervous system. For unknown reasons, patients with Parkinson disease tend to have a low uric acid level, and higher levels are associated with milder disease. It is hoped that raising the uric acid level to a “pre-gout level” may slow the progression of Parkinson disease.
Isradipine, a calcium channel blocker, was found in an epidemiologic study of elderly patients to be associated with reduced likelihood of developing Parkinson disease.6 The drug is now undergoing clinical trials.
Smoking. Although cigarette smokers have long been recognized as having a very low risk of developing Parkinson disease, smoking is not recommended.
Agents found ineffective. Agents that have been tested and found ineffective in modifying the course of Parkinson disease include vitamin E, coenzyme Q10, riluzole, GPI-1485, pramipexole, cogane, CEP-1347, TCH-346, and creatine.
NOT JUST DOPAMINE—OR TREMORS
Dopamine deficiency is central to the current understanding of the pathogenesis of Parkinson disease and the focus of treatment efforts, but if dopamine deficiency were the only problem, replacing it should completely ameliorate all parkinsonian features. Other neurotransmitters also play roles: norepinephrine is implicated in orthostatic symptoms and apathy, acetylcholine in cognitive behaviors, glutamate in dyskinesias, and serotonin in depression, anxiety, and sleep abnormalities.
The most recognized area of involvement in the brain has traditionally been the substantia nigra in the midbrain. However, current thinking is that the disease starts lower in the caudal area of the brainstem (along with the olfactory tubercle), moves through the pons to the midbrain, then spreads across the cerebrum with extensive neocortical involvement.
Early premotor indicators are now recognized to occur 15 to 20 years before a tremor appears. The first signs are often hyposmia (diminished sense of smell, reflecting involvement of the olfactory tubercle) and constipation (reflecting involvement of the medulla and the vagus nucleus). With pons involvement, the patient can develop rapid eye movement sleep behavior disorder, depression, or anxiety. Only then does the disease spread to the midbrain and cause resting tremor, rigidity, and bradykinesia.7
Identifying the preclinical stages and starting disease-modifying treatments before the onset of motor symptoms may one day prove important, but at this point, the premotor symptoms (anosmia, constipation, depression) are too nonspecific to be useful, and such treatments have not yet been identified.
TREATMENT: LEVODOPA STILL PRIMARY
When to start drug treatment depends primarily on how much the symptoms bother the patient. Regardless of the clinician’s (or patient’s) belief in the benefits of delaying symptomatic treatment, it is universally considered necessary to start medication when gait problems develop because of the danger of a fall and resulting disability.
Carbidopa-levodopa combination therapy remains the most effective treatment; if it is not effective, another diagnosis may need to be considered. Carbidopa-levodopa improves tremor, rigidity, and bradykinesia, particularly in the early stages of Parkinson disease. It is well tolerated, has rapid onset, reduces the risk of death, and is the least expensive of the medications for Parkinson disease.
Immediate-release and continued-release formulations are available, as well as one that dissolves rapidly on the tongue and can be taken without water. An oral extended-release carbidopa-levodopa formulation (Rytary) was approved by the FDA in January 2015. Tablets are filled with drug-containing microbeads that dissolve at different rates to achieve therapeutic levodopa levels as quickly as the immediate-release formulation and maintain them for an extended time.8
The development of dyskinesias is the major psychological drawback of levodopa, occurring in 80% of patients after 5 to 10 years of treatment. Although many patients fear this side effect, most patients who develop it find it preferable to the rigidity and bradykinesia of Parkinson disease. In most cases, bothersome dyskinesias can be controlled by adjusting medications.9,10
Dopamine agonists include pramipexole, ropinirole, and rotigotine. They are available in generic form as three-times-daily dosing; once-daily dosing is also available, but not as a generic formulation. Dopamine agonists have the advantage of potentially improving depression and delaying the onset of dyskinesias.
However, dopamine agonists have a number of disadvantages compared with levodopa: they have a longer titration period, are less effective, and are less well tolerated, especially in the elderly. Side effects occur more frequently than with levodopa and include general and peripheral edema, hallucinations, nausea, lightheadedness, and sleepiness.11,12 These drugs are also associated with “sleep attacks” (sudden falling asleep while active, such as while driving or eating) and with compulsive and impulsive behaviors such as hypersexuality, buying, binge eating, and gambling. Although these behaviors occur in fewer than 10% of patients, they can be devastating, leading to marital, financial, and legal problems. A bothersome clinical state termed dopamine agonist withdrawal syndrome is characterized by anxiety, depression, jitteriness, and palpitations when dopamine agonists are tapered or discontinued because of a side effect.13
MAO-B inhibitors delay the breakdown of dopamine, allowing it to “stay” in the brain for a longer period of time. Rasagiline for early monotherapy has the advantages of once-daily dosing, no titration, and excellent tolerability, even in the elderly. Potential drug interactions should be considered when using this drug. Early warnings about interactions with tyramine-rich foods were lifted after trials showed that this was not a problem.14
Amantadine is an N-methyl-d-aspartate (NMDA) receptor antagonist often used in early Parkinson disease and for treatment of dyskinesias and fatigue. It is the only drug that is intrinsically antidyskinetic and also improves Parkinson symptoms.15 Side effects include leg swelling, livedo reticularis, and neuropsychiatric and anticholinergic effects.
Anticholinergic agents (eg, trihexyphenidyl) improve tremor but are not as useful for bradykinesia or rigidity, and often have anticholinergic effects such as mental dullness, dry mouth, dry eye, and urinary hesitancy, especially in the elderly, so they have a limited role in Parkinson treatment.
MOTOR COMPLICATIONS: FLUCTUATIONS AND DYSKINESIAS
Motor fluctuations are changes between the akinetic and mobile phases of Parkinson disease, or the off-periods and on-periods of drug treatment. A patient who is “off” is generally rigid and feels that the medication is not working. A patient who is “on” feels loose and mobile and that the medication is working. Variants of motor fluctuations include:
- End-of-dose deterioration
- Delayed onset of response (more than half an hour after taking medication)
- Drug-resistant offs—medication has become ineffective
- Random oscillation—on-off phenomenon
- Freezing—unpredictable inability to start or finish a movement.
Dyskinesias are abnormal involuntary movements such as writhing and twisting. They are associated with dopaminergic therapy at peak dose, when the drug starts to turn on or wear off (termed diphasic dyskinesias).16
The storage hypothesis provides a plausible explanation for the development of motor complications as the disease progresses. Although the half-life of levodopa is only 60 to 90 minutes, it is effective in early disease when given three times a day. It is believed that at this stage of the disease, enough dopaminergic neurons survive to “store” dopamine and release it as needed. As the disease progresses and dopaminergic neurons die, storage capacity diminishes, and the clinical effect slowly starts to approximate the pharmacokinetic profile of the drug. Upon taking the medication, the patient gets a surge of drug, causing dyskinesias, followed later by rigidity as the effect wears off since there are fewer surviving dopaminergic cells to store dopamine.
MANAGING DYSKINESIAS
Patients with dyskinesias should first be asked if they are bothered by them; not all patients are troubled by dyskinesias. If the movements only bother others (eg, family members), then education is often the only treatment needed. If the patient is uncomfortable, the following measures can be tried:
- Taking lower, more frequent doses of levodopa (however, risk of wearing off becomes a problem)
- Adding a dopamine agonist or MAO-B inhibitor while lowering the levodopa dose (however, MAO-B inhibitors pose a risk of side effects in elderly patients)
- Adding clozapine (periodic laboratory testing is required to monitor blood levels and liver and kidney function)
- Adding amantadine (however, this poses a risk of cognitive side effects).
Deep-brain-stimulation surgery is appropriate for select patients who are generally physically healthy, cognitively intact, and emotionally stable, with a strong family support system, but who are bothered by symptoms of parkinsonism (such as tremors), motor fluctuations, or dyskinesias.17
Infusion pump. In January 2015, the FDA approved a new system that continuously delivers levodopa-carbidopa in a 4:1 ratio in gel suspension for 16 hours directly into the small intestine, minimizing motor fluctuations. The patient changes the cartridge daily and turns it off at bedtime.
*Dr. Fernandez has received research support from AbbVie, Acadia, Auspex, Biotie Therapies, Civitas, Kyowa/ProStrakan, Michael J. Fox Foundation, Movement Disorders Society, NIH/NINDS, Parkinson Study Group, Rhythm, Synosia, and Teva. He also has received honoraria from Carling Communications, International Parkinson and Movement Disorders Society, The Ohio State University, and PRIME Education, Inc as a speaker in CME events. He has received honoraria from Biogen, GE Health Care, Lundbeck, Merz Pharmaceuticals, and Pfizer as a consultant. He has received royalty payments from Demos Publishing for serving as a book author/editor. Cleveland Clinic has contracts with AbbVie and Merz Pharmaceuticals for Dr. Fernandez’s role as a member of the Global Steering Committee for LCIG studies and as a consultant or speaker, and as Head Principal Investigator for the Xeomin Registry Study. Dr. Fernandez has received a stipend from International Parkinson and Movement Disorders Society for serving as medical editor of the Movement Disorders Society website.
- Wenning GK, Ben-Shlomo Y, Hughes A, Daniel SE, Lees A, Quinn NP. What clinical features are most useful to distinguish definite multiple system atrophy from Parkinson’s disease? J Neurol Neurosurg Psychiatry 2000; 68:434–440.
- Hernán MA, Takkouche B, Caamaño-Isoma F, et al. A meta-analysis of coffee drinking, cigarette smoking, and risk of Parkinson’s disease. Ann Neurol 2002; 52:276–84.
- Ridgel A, Thota A, Vitek JL, Alberts JL. Forced, not voluntary, exercise improves motor function in Parkinson’s disease patients. Neurorehabil Neural Repair 2009; 23:600–608.
- Smith AD, Zigmond MJ. Can the brain be protected through exercise? Lessons from an animal model of parkinsonism. Exp Neurol 2003; 184:31–39.
- Olanow CW, Rascol O, Hauser R, et al, for the ADAGIO Study Investigators. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N Engl J Med 2009; 361:1268–1278.
- Pasternak B, Svanström H, Nielsen NM, Fugger L, Melbye M, Hviid A. Use of calcium channel blockers and Parkinson’s disease. Am J Epidemiol 2012; 175:627-635.
- Braak H, Del Tredici K, Rüb U, de Vos RAI, Jansen Steur ENH, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 2003; 24:197–211.
- Hauser RA, Ellenbogen AL, Metman LV, et al. Crossover comparison of IPX066 and a standard levodopa formulation in advanced Parkinson’s disease. Mov Disord 2011; 26:2246–2252.
- Hely MA, Morris JG, Reid WG, Trafficante R. Sydney Multicenter Study of Parkinson’s disease: non-L-dopa-responsive problems dominate at 15 years. Mov Disord 2005; 20:190–199.
- Hung SW, Adeli GM, Arenovich T, Fox SH, Lang AE. Patient perception of dyskinesia in Parkinson’s disease. J Neurol Neurosurg Psychiatry 2010; 81:1112–1115.
- Rascol O, Brooks DJ, Korczyn AD, De Deyn PP, Clarke CE, Lang AE. A five-year study of the incidence of dyskinesia in patients with early Parkinson’s disease who were treated with ropinirole or levodopa. N Engl J Med 2000; 342:1484–1491.
- Parkinson Study Group. Pramipexole vs levodopa as initial treatment for Parkinson disease: a randomized controlled trial. Parkinson Study Group. JAMA 2000; 284:1931–1938.
- Nirenberg MJ. Dopamine agonist withdrawal syndrome: implications for patient care. Drugs Aging 2013; 30:587–592.
- Teva Neuroscience, Inc. Azilect prescribing information. https://www.azilect.com/Content/pdf/azi-40850-azilect-electronic-pi.pdf. Accessed June 29, 2015.
- Snow BJ, Macdonald L, Mcauley D, Wallis W. The effect of amantadine on levodopa-induced dyskinesias in Parkinson’s disease: a double-blind, placebo-controlled study. Clin Neuropharmacol 2000; 23:82–85.
- Adler CH, Ahlskog JE, eds. Parkinson’s Disease and Movement Disorders: Diagnosis and Treatment Guidelines for the Practicing Physician. Totowa, NJ: Humana Press; 2000.
- Machado A, Fernandez HH, Deogaonkar M. Deep brain stimulation: what can patients expect from it? Cleve Clin J Med 2012; 79:113–120.
- Wenning GK, Ben-Shlomo Y, Hughes A, Daniel SE, Lees A, Quinn NP. What clinical features are most useful to distinguish definite multiple system atrophy from Parkinson’s disease? J Neurol Neurosurg Psychiatry 2000; 68:434–440.
- Hernán MA, Takkouche B, Caamaño-Isoma F, et al. A meta-analysis of coffee drinking, cigarette smoking, and risk of Parkinson’s disease. Ann Neurol 2002; 52:276–84.
- Ridgel A, Thota A, Vitek JL, Alberts JL. Forced, not voluntary, exercise improves motor function in Parkinson’s disease patients. Neurorehabil Neural Repair 2009; 23:600–608.
- Smith AD, Zigmond MJ. Can the brain be protected through exercise? Lessons from an animal model of parkinsonism. Exp Neurol 2003; 184:31–39.
- Olanow CW, Rascol O, Hauser R, et al, for the ADAGIO Study Investigators. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N Engl J Med 2009; 361:1268–1278.
- Pasternak B, Svanström H, Nielsen NM, Fugger L, Melbye M, Hviid A. Use of calcium channel blockers and Parkinson’s disease. Am J Epidemiol 2012; 175:627-635.
- Braak H, Del Tredici K, Rüb U, de Vos RAI, Jansen Steur ENH, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 2003; 24:197–211.
- Hauser RA, Ellenbogen AL, Metman LV, et al. Crossover comparison of IPX066 and a standard levodopa formulation in advanced Parkinson’s disease. Mov Disord 2011; 26:2246–2252.
- Hely MA, Morris JG, Reid WG, Trafficante R. Sydney Multicenter Study of Parkinson’s disease: non-L-dopa-responsive problems dominate at 15 years. Mov Disord 2005; 20:190–199.
- Hung SW, Adeli GM, Arenovich T, Fox SH, Lang AE. Patient perception of dyskinesia in Parkinson’s disease. J Neurol Neurosurg Psychiatry 2010; 81:1112–1115.
- Rascol O, Brooks DJ, Korczyn AD, De Deyn PP, Clarke CE, Lang AE. A five-year study of the incidence of dyskinesia in patients with early Parkinson’s disease who were treated with ropinirole or levodopa. N Engl J Med 2000; 342:1484–1491.
- Parkinson Study Group. Pramipexole vs levodopa as initial treatment for Parkinson disease: a randomized controlled trial. Parkinson Study Group. JAMA 2000; 284:1931–1938.
- Nirenberg MJ. Dopamine agonist withdrawal syndrome: implications for patient care. Drugs Aging 2013; 30:587–592.
- Teva Neuroscience, Inc. Azilect prescribing information. https://www.azilect.com/Content/pdf/azi-40850-azilect-electronic-pi.pdf. Accessed June 29, 2015.
- Snow BJ, Macdonald L, Mcauley D, Wallis W. The effect of amantadine on levodopa-induced dyskinesias in Parkinson’s disease: a double-blind, placebo-controlled study. Clin Neuropharmacol 2000; 23:82–85.
- Adler CH, Ahlskog JE, eds. Parkinson’s Disease and Movement Disorders: Diagnosis and Treatment Guidelines for the Practicing Physician. Totowa, NJ: Humana Press; 2000.
- Machado A, Fernandez HH, Deogaonkar M. Deep brain stimulation: what can patients expect from it? Cleve Clin J Med 2012; 79:113–120.
KEY POINTS
- Parkinson disease is diagnosed by clinical signs with the mnemonic TRAP: Tremor at rest, Rigidity, Akinesia or bradykinesia, and Postural/gait instability.
- A dopamine transporter functional scan can distinguish neurodegenerative parkinsonian disorders from nonneurodegenerative etiologies such as drug-induced parkinsonism and vascular parkinsonism, and from mimics such as psychogenic parkinsonism and essential tremor.
- Coffee consumption and exercise may benefit patients with Parkinson disease.
- Carbidopa-levodopa combination therapy is still the most effective treatment, but most patients develop dyskinesia after 5 to 10 years of treatment.
- Dyskinesias can be managed by adjusting or changing medications, switching to the new levodopa infusion pump system, or with deep-brain-stimulation surgery.
Universal precautions to reduce stimulant misuse in treating adult ADHD
Children are not the only people affected by attention-deficit/hyperactivity disorder (ADHD). Characterized by high levels of inattention, overactivity, and impulsivity, ADHD affects 5% of school-aged children, but also 4% of adults.1–3 Adults with untreated ADHD are likely to develop serious psychosocial problems that manifest as unemployment, arrests, divorce, underachievement, and psychiatric comorbidities.4,5
However, many clinicians are reluctant to manage adults with ADHD, partly because of concerns about misuse of the stimulant drugs they must prescribe to treat it.
Here, we outline an approach whereby clinicians can diagnose and treat adult ADHD while taking “universal precautions” to discourage misuse of the medications involved.
RECOGNIZING ADHD IN ADULTS
ADHD is characterized by developmentally inappropriate levels of inattention, impulsiveness, and hyperactivity that arise in childhood and result in impairments that often persist.
The presentation of ADHD in adults may be influenced by the longevity of their ADHD, associated sequelae (eg, low self-esteem and interpersonal, educational, and occupational difficulties), and comorbid disorders.6 There are neither reliable biomarkers nor neuropsychological tests for diagnosis, and persons with ADHD typically have a complex presentation with at least one comorbidity.6,7
In patients diagnosed in childhood, difficulties with organization as well as initiating, maintaining, and completing tasks become more prominent in adulthood and hyperactivity tends to subside. Adult impulsivity may present as edginess, shopping sprees, quitting jobs, and risky behaviors.6
Overall, the clinical manifestations of ADHD in adolescents and adults include inattention, difficulties with task completion, disorganization, and executive dysfunction—all skills critical to managing the various roles of adult life.
OBSTACLES TO EFFECTIVE TREATMENT
In the past, ADHD treatment was routinely discontinued during adolescence, as it was unclear whether adults still had significant symptoms or benefited from treatment.8,9 Now, available ADHD guidelines suggest that children and adults who respond to pharmacotherapy should continue it for as long as it remains effective. In this context, there is increasing recognition of adult ADHD as a valid and treatable disorder.10
One of the challenges clinicians face is the reliability of adult recall of childhood ADHD. A controlled, prospective 16-year follow-up study found that of all adults retrospectively given a diagnosis of childhood ADHD, only 27% actually had the disorder.11 This study suggests that retrospective diagnoses of childhood ADHD made solely on the basis of self-reports are unlikely to be valid.
Another obstacle is that traditional medical education has seldom included training in adult ADHD.8,12 In a UK study, clinicians felt that they lacked training and knowledge to assess and manage adult ADHD patients.9
Even if adult ADHD is recognized, diagnosis is just the first step of care.13 These patients require ongoing management and follow-up assessments.
Although practice patterns vary, efforts to encourage doctors to provide adult ADHD care may be hindered by the fact that the gold standard of treatment is stimulant medication.4,10 Medications approved by the US Food and Drug Administration for adult ADHD include the stimulants lisdexamfetamine, osmotic-release methylphenidate, mixed amphetamine salts extended release, dexmethylphenidate extended release, and the nonstimulant atomoxetine.6 While stimulants are generally more efficacious for ADHD symptoms than nonstimulants, they are associated with misuse and diversion.14
UNIVERSAL PRECAUTIONS: A SIMPLIFIED APPROACH
The universal-precautions approach to prescribing stimulants aims to allay physician concerns and promote appropriate medication use to allow for proper management of this disorder.15 These precautions, to be applied to all adult ADHD patients for whom stimulants are being considered, include careful diagnosis and consideration of comorbidities, baseline risk stratification, informed consent processes, treatment agreements, periodic reassessments of treatment response, and meticulous documentation.
DIAGNOSIS
A frequently used screening assessment for adult ADHD is the ADHD Rating Scale (ADHD RS), which consists of two subscales for assessing hyperactivity/impulsivity and inattentiveness.16 ADHD can be classified into one of three subtypes based on symptoms: inattentive, hyperactive, or combined type. Symptoms must persist for at least 6 months for a diagnosis to be made. Other ADHD scales include the Conners Adult ADHD Rating Scales and the Brown Attention-Deficit Disorder Scales.4
High scores on screening scales must be interpreted within the clinical context. Clinicians need to ask about ADHD symptoms, establish their presence in various settings, and determine if these symptoms interfere with functioning. A diagnosis of adult ADHD also requires evidence of symptoms beginning in childhood.17 According to the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition, inattentive or hyperactive-impulsive symptoms must be present before age 12 in two or more settings and interfere with function and development.
Although self-reporting screening tools are helpful, these tests are not reliable for diagnostic purposes, and collateral information is also required.
Neuropsychological testing may detect impairments in persons with ADHD. The most consistently employed neuropsychological tests to evaluate ADHD include the Conners Continuous Performance Test, Stroop Color and Word Test, Trail-making Test, verbal fluency tests, Controlled Oral Word Association Test, and the Weschler Adult Intelligence Scale.6
COMORBIDITY
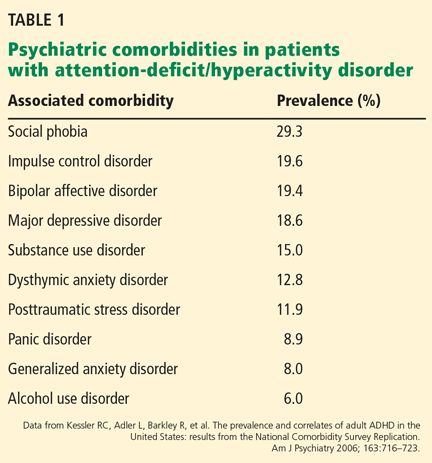
Epidemiologic studies suggest that adults with ADHD develop many psychiatric problems including anxiety, depression, and substance use disorders.7,16 Table 1 illustrates common comorbidities and their associated prevalence in the ADHD patient.7
Comorbid psychiatric disorders may affect the presentation of adult ADHD. For instance, adults with comorbid depression and ADHD are more likely to present with heightened irritability and difficulties concentrating on tasks than those with either condition alone.18 Similarly, antisocial personality disorder is more common in adults with ADHD.19 Such patients exhibit stable antisocial behavior (lying, stealing, and aggression) as well as medication misuse.5,14,19
While these comorbid disorders may obscure the ADHD diagnosis, their recognition is essential to effectively manage adult ADHD. In sum, a careful evaluation of the adult, including elucidating both ADHD and comorbid symptoms, functionality in several domains, and the degree of impairment, should precede initiating pharmacotherapy for adult ADHD.
BASELINE RISK STRATIFICATION: RISK FACTORS FOR STIMULANT MISUSE
After diagnosing ADHD, the prescriber must assess the risk for misuse of stimulant medications.20
One study revealed that nonmedical use of stimulant medications occurred in only 2% of the 4,300 people surveyed.21 Among the misusers, 66% had obtained medication from family or friends. Another 34% had stolen medication, and 20% had obtained prescriptions from a physician by falsely reporting symptoms. The study also assessed motivation for misuse. In this sample, 40% of misuse was to enhance performance, 34% was for recreation, and 23% was to stay awake.21
Other studies show that misuse of stimulant medications is common among youth in the United States, reporting that 18% of college students use some formulation of prescription stimulants.22

Still more research suggests that childhood conduct disorder or illicit drug use results in a higher risk of stimulant medication misuse.20 Additional risk factors for misuse include male sex, white ethnicity, upper-class background, Jewish or no religious affiliation, affiliation with a sorority or fraternity, off-campus housing, and a low grade-point average.23
Table 2 illustrates clinical interventions providers can use, once they have risk-stratified their patients, to monitor for stimulant misuse.
HOW SHOULD THESE RISK FACTORS AFFECT TREATMENT?
Although no formal scoring system exists to help clinicians risk-stratify these patients, the presence of multiple risk factors suggests the need for vigilance.14 Physicians should prescribe agents with less potential for abuse and monitor these patients more intensely.
Short-acting stimulant medications are the most likely to be abused, as phasic dopamine increase is more reinforcing than therapeutic dopamine release.24 Longer-acting stimulant medications are less likely to be abused, and they provide better symptom relief, as tonic dopamine release maintains a steady state and increases the therapeutic efficacy of these medications.25 For example, methylphenidate extended-release tablets have an osmotic oral controlled-release system and are less likely to be crushed for recreational inhalation.6,14
Lisdexamfetamine is a prodrug therapeutically inert until converted to d-amphetamine when lysine is cleaved from the molecule. This medication may be a good option for patients at high risk of misuse because it is tamper-resistant. However, it still may be subject to misuse for performance enhancement.26,27
The nonstimulant atomoxetine is also approved for ADHD, has no abuse potential, and may be particularly useful when anxiety, mood, and substance use disorders co-occur with ADHD.6 Rarely, atomoxetine can damage the liver, and liver function tests should be monitored if right upper quadrant pain develops.4,10
Other nonapproved agents such as bupropion and desipramine also have been used empirically and off-label for ADHD.4,10
Overall, treatment should be selected according to the risk of misuse of stimulant medication and the patient’s comorbidities.
INFORMED CONSENT
Informed consent may help patients appreciate the risks and benefits of the treatment options and develop realistic expectations about treatment.26 Patients are instructed to take their stimulant medications as prescribed and are informed of the risks of combining stimulants with other substances, particularly those that may interact with stimulants (eg, cocaine) and raise the risk of seizures and cardiovascular complications.
Stimulant medications lead to elevations in blood pressure and heart rate, although large-scale studies have shown no increase in the rate of serious cardiovascular events when these drugs are used appropriately.6 Less serious side effects associated with stimulant medications include insomnia, weight loss, decreased appetite, dry mouth, headache, and rarely, depression and anxiety.6
Patients need to be warned about diversion and abuse liability of stimulant medications, as well as alternative treatments.
The nonstimulant atomoxetine has no reinforcing properties but also raises the blood pressure and heart rate.6 As with stimulants, these elevations are generally minimal, time-limited, and of minor clinical significance.4,10 Frequent reasons to prescribe atomoxetine include poor tolerability of stimulants and a history of substance abuse. In addition, women with ADHD and high levels of emotional dysregulation or social anxiety appear to be particularly responsive to atomoxetine.6
Another consideration is cognitive behavioral therapy, which can augment the effects of pharmacotherapy.4 Cognitive behavioral therapy focuses on time management, prioritization, organization, problem-solving, motivation, and emotional regulation.4
Finally, patients also need to understand the possible consequences of nontreatment.5 Adults with untreated ADHD have high rates of academic and occupational difficulties, anti-social behaviors, and other forms of psychosocial adversity.4,5
Overall, this process should involve discussing risks and benefits of treatment options with the patient and promoting joint decision-making.
TREATMENT AGREEMENTS
Stimulant medications are classified by the US Drug Enforcement Administration as schedule II substances due to their abuse potential.20
It is important to inform patients of the addictive nature of the medication and to instruct them on how to store stimulants safely.27 Patients need to know that giving away or selling these medications is illegal.27
Treatment agreements establish rules for prescribing and are signed by the patient before initiating therapy.28,29 Patients are expected to attend all of their appointments, receive their prescriptions from one doctor, and obtain their medication from one pharmacy. These agreements may also require patients to submit to monitoring with random urine drug screens.29 Overall, they underscore the need for patients to follow a treatment plan in order to continue therapy with controlled substances.29
Manning27 recommends using agreements for high-risk college students prescribed stimulant medications. Red flags for misuse include signs of active substance use (eg, intoxication, a pattern of “lost” prescriptions, and doctor-shopping).27
The effectiveness in reducing risk of misuse in the adult ADHD population has not yet been investigated. Nonetheless, a method of communicating the seriousness of stimulant misuse to adult patients is essential to ADHD care.
STAYING ON TRACK
In the clinical setting, treatment response is measured not just by symptom reduction, but also by functional improvement. Thus, clinicians and patients must set functional goals whenever possible.27 Successful progress toward these goals justifies continuation of therapy, whereas lack of improvement signals the need to reconsider stimulant therapy.27
MONITORING AND DOCUMENTATION
Adults with ADHD present with varying levels of functional impairment and comorbidities, which may require different levels of monitoring.30 Not all patients with ADHD respond optimally to stimulant medications or tolerate them well.31,32 Hence, monitoring parameters for therapeutic change and adverse outcomes are important in that they guide the alteration or even discontinuation of pharmacotherapy.4,6,14
Documenting the decision-making process to continue stimulant medications under certain circumstances is also essential. Documentation should include discussion of goals and expectations, risks and benefits, and alternatives to stimulant use.
In adults, risk of stimulant medication misuse adds a new layer of complexity to monitoring.13,14 Adults may get multiple prescriptions from multiple providers, seek early refills, fill prescriptions at different pharmacies, or alter formulations. Many states track stimulant prescription use, and prescribers can use this information to determine if patients are refilling their prescriptions appropriately or obtaining stimulants from more than one provider.
Although these monitoring strategies are useful,6 it is prudent to structure the level of monitoring according to the patient’s risk of adverse events or medication misuse.14,27 Gourlay and Heit15 proposed the following “four-A” mnemonic for four domains to be explored at each visit in patients receiving pain medicine. This mnemonic can be applied to adult ADHD patients to more accurately monitor the patient throughout treatment.
THE ‘FOUR-A’ MNEMONIC
ADHD symptoms
Several ADHD scales can be used to track symptom changes over time.33 However, these self-report scales may be subject to positive illusory bias, a phenomenon observed in individuals with ADHD in which they tend to overrate their functioning,34 which may limit the accuracy of self-report scales.35
Activities of daily living
Since patients with ADHD tend to overrate their functioning in various aspects of living, collateral information should be gathered to corroborate patient self-reports whenever possible.
Adverse events
Blood pressure, heart rate, and weight should be assessed at baseline and monitored during stimulant treatment. Other symptoms to monitor include gastrointestinal distress, headache, aggression, depression, appetite, and sleeping habits.4,6 More intensive monitoring (eg, electrocardiography) may be indicated for those with hypertension and cardiovascular risk factors.
Aberrant behavior
Monitoring for misuse and diversion of stimulant medications is essential, as ADHD itself is a risk factor for addiction.20,21 Pill counts, prescription monitoring programs, urine drug screens, and collateral informants have all been proposed but not studied in monitoring for the misuse of stimulant medications.27 Before prescribing, it is prudent to check the prescription monitoring program, get a urine drug screen, and discuss any positive findings with the patient.36,37
Treatment agreements ensure that patients are aware of the consequences of misuse and allow the clinician to reference prior discussion when terminating treatment with stimulants.
LIVES CAN BE ENHANCED
ADHD is a common disorder that arises in childhood and can persist throughout life. Adults with untreated ADHD are at risk of severe impairments in various domains of functioning. Stimulant medications are an effective treatment but may be diverted into the street market. Using the universal-precautions model may reduce the risks of both nontreatment of ADHD and misuse of stimulants.
Accordingly, clinicians need to confirm the ADHD diagnosis, assess comorbidities, estimate risk of misuse, and provide informed consent prior to prescribing. Subsequent monitoring should involve the use of treatment agreements and evaluating treatment response, paying particular attention to ADHD symptom control but also to level of function, adverse effects, and aberrant behavior.
With these principles in mind, clinicians can address the risks of misuse and potentially enhance the lives of people who may have been suffering substantially due to lack of appropriate care.
- Polanczyk G, de Lima MS, Horta BL, Biederman J, Rohde LA. The worldwide prevalence of ADHD: a systematic review and metaregression analysis. Am J Psychiatry 2007; 164:942–948.
- Polanczyk GV, Wilcutt EG, Salum GA, Kieling C, Rohde LA. ADHD prevalence estimates across three decades: an updated systematic review and meta-regression analysis. Int J Epidemiol 2014; 43:434–442.
- Wilens TE. ADHD: Prevalence, diagnosis, and issues of comorbidity. CNS Spectr 2007; 12(suppl 6):1–5.
- Kooij SJ, Bejerot S, Blackwell A, et al. European consensus statement on diagnosis and treatment of adult ADHD: the European Network Adult ADHD. BMC Psychiatry 2010; 10:67.
- Shaw M, Hodgkins P, Caci H, et al. A systematic review and analysis of long-term outcomes in attention deficit hyperactivity disorder: effects of treatment and non-treatment. BMC Med 2012;10:99.
- Modesto-Lowe V, Meyer A, Soovajian V. A clinician’s guide to adult attention-deficit hyperactivity disorder. Conn Med 2012; 76:517–523.
- Kessler RC, Adler L, Barkley R, et al. The prevalence and correlates of adult ADHD in the United States: results from the National Comorbidity Survey Replication. Am J Psychiatry 2006; 163:716–723.
- Goodman DW, Surman CB, Scherer PB, Salinas GD, Brown JJ. Assessment of physician practices in adult attention-deficit/hyperactivity disorder. Prim Care Companion CNS Disord 2012; 14(4).
- Hall CL, Newell K, Taylor J, Sayal K, Swift KD, Hollis C. ‘Mind the gap’—mapping services for young people with ADHD transitioning from child to adult mental health services. BMC Psychiatry 2013; 13:186.
- National Institute for Health and Care Excellence. Attention deficit hyperactivity disorder: diagnosis and management of ADHD in children, young people and adults. The British Psychological Society and The Royal College of Psychiatrists: United Kingdom; 2009.
- Mannuzza S, Klein RG, Klein DF, Bessler A, Shrout P. Accuracy of adult recall of childhood attention deficit hyperactivity disorder. Am J Psychiatry 2002; 159:1882–1888.
- Wetzel MW. Medical student participation in an adult ADHD outpatient clinic: an ideal setting for education in outpatient psychiatry. Acad Psychiatry 2009; 33:80–81.
- Culpepper L, Mattingly G. Challenges in identifying and managing attention-deficit/hyperactivity disorder in adults in the primary care setting: a review of the literature. Prim Care Companion J Clin Psychiatry 2010; 12(6).
- Rabiner DL. Stimulant prescription cautions: addressing misuse, diversion and malingering. Curr Psychiatry Rep 2013; 15:375.
- Gourlay D, Heit H. Universal precautions: a matter of mutual trust and responsibility. Pain Med 2006; 7:210–211.
- Kessler RC, Adler L, Ames M, et al. The World Health Organization Adult ADHD Self-Report Scale (ASRS): a short screening scale for use in the general population. Psychol Med 2005; 35:245–256.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed. Arlington, VA: American Psychiatric Association; 2013.
- CADDRA Guidelines Steering Committee. Canadian ADHD practice guidelines: CADDRA 2008. http://www.naceonline.com/AdultADHDtoolkit/professionalresources/caddraguidelines.pdf. Accessed July 10, 2015.
- Mannuzza S, Klein RG, Bessler A, Malloy P, LaPadula M. Adult psychiatric status of hyperactive boys grown up. Am J Psychiatry 1998; 155:493–498.
- Kaye S, Darke S. The diversion and misuse of pharmaceutical stimulants: what do we know and why should we care? Addiction 2012; 107:467–477.
- Novak SP, Kroutil LA, Williams RL, Van Brunt DL. The nonmedical use of prescription ADHD medications: results from a national Internet panel. Subst Abuse Treat Prev Policy 2007; 2:32.
- Bavarian N, Flay BR, Ketcham P, et al. Using structural equation modeling to understand prescription stimulant misuse: a test of the Theory of Triadic Influence. Drug Alcohol Depend 2014; 138:193–201.
- McCabe SE, Teter CJ, Boyd CJ. Medical use, illicit use and diversion of prescription stimulant medication. J Psychoactive Drugs 2006; 38:43–56.
- Volkow ND. Stimulant medications: how to minimize their reinforcing effects? Am J Psychiatry 2006; 163:359–361.
- Kolar D, Keller A, Golfinopoulos M, Cumyn L, Syer C, Hechtman L. Treatment of adults with attention-deficit/hyperactivity disorder. Neuropsychiatr Dis Treat 2008; 4:107–121.
- Schachter D, Tharmalingam S, Kleinman I. Informed consent and stimulant medication: adolescents’ and parents’ ability to understand information about benefits and risks of stimulant medication for the treatment of attention-deficit/hyperactivity disorder. J Child Adolesc Psychopharmacol 2011; 21:139–148.
- Manning JS. Strategies for managing the risks associated with ADHD medications. J Clin Psychiatry 2013; 74:e19.
- Deep K. Use of narcotics contracts. Virtual Mentor 2013; 15:416–420.
- Cheatle MD, Savage SR. Informed consent in opioid therapy: a potential obligation and opportunity. J Pain Symptom Manage 2012; 44:105–116.
- Dias TG, Kieling C, Graeff-Martins AS, Moriyama TS, Rohde LA, Polanczyk GV. Developments and challenges in the diagnosis and treatment of ADHD. Rev Bras Psiquiatr 2013; 35(suppl 1):S40–S50.
- Mattingly GW, Weisler RH, Young J, et al. Clinical response and symptomatic remission in short- and long-term trials of lisdexamfetamine dimesylate in adults with attention-deficit/hyperactivity disorder. BMC Psychiatry 2013; 13:39.
- Contini V, Victor MM, Bertuzzi GP, et al. No significant association between genetic variants in 7 candidate genes and response to methylphenidate treatment in adult patients with ADHD. J Clin Psychopharmacol 2012; 32:820–823.
- Rösler M, Retz W, Thome J, Schneider M, Stieglitz RD, Falkai P. Psychopathological rating scales for diagnostic use in adults with attention-deficit/hyperactivity disorder (ADHD). Eur Arch Psychiatry Clin Neurosci 2006; 256(suppl 1):i3–i11.
- Prevatt F, Proctor B, Best L, Baker L, Van Walker J, Taylor NW. The positive illusory bias: does it explain self-evaluations in college students with ADHD? J Atten Disord 2012; 16:235–243.
- Jiang Y, Johnston C. The relationship between ADHD symptoms and competence as reported by both self and others. J Atten Disord 2012; 16:418–426.
- Darredeau C, Barrett SP, Jardin B, Pihl RO. Patterns and predictors of medication compliance, diversion, and misuse in adult prescribed methylphenidate users. Hum Psychopharmacol 2007; 22:529–536.
- Worley J. Prescription drug monitoring programs, a response to doctor shopping: purpose, effectiveness, and directions for future research. Issues Ment Health Nurs 2012; 33:319–328.
Children are not the only people affected by attention-deficit/hyperactivity disorder (ADHD). Characterized by high levels of inattention, overactivity, and impulsivity, ADHD affects 5% of school-aged children, but also 4% of adults.1–3 Adults with untreated ADHD are likely to develop serious psychosocial problems that manifest as unemployment, arrests, divorce, underachievement, and psychiatric comorbidities.4,5
However, many clinicians are reluctant to manage adults with ADHD, partly because of concerns about misuse of the stimulant drugs they must prescribe to treat it.
Here, we outline an approach whereby clinicians can diagnose and treat adult ADHD while taking “universal precautions” to discourage misuse of the medications involved.
RECOGNIZING ADHD IN ADULTS
ADHD is characterized by developmentally inappropriate levels of inattention, impulsiveness, and hyperactivity that arise in childhood and result in impairments that often persist.
The presentation of ADHD in adults may be influenced by the longevity of their ADHD, associated sequelae (eg, low self-esteem and interpersonal, educational, and occupational difficulties), and comorbid disorders.6 There are neither reliable biomarkers nor neuropsychological tests for diagnosis, and persons with ADHD typically have a complex presentation with at least one comorbidity.6,7
In patients diagnosed in childhood, difficulties with organization as well as initiating, maintaining, and completing tasks become more prominent in adulthood and hyperactivity tends to subside. Adult impulsivity may present as edginess, shopping sprees, quitting jobs, and risky behaviors.6
Overall, the clinical manifestations of ADHD in adolescents and adults include inattention, difficulties with task completion, disorganization, and executive dysfunction—all skills critical to managing the various roles of adult life.
OBSTACLES TO EFFECTIVE TREATMENT
In the past, ADHD treatment was routinely discontinued during adolescence, as it was unclear whether adults still had significant symptoms or benefited from treatment.8,9 Now, available ADHD guidelines suggest that children and adults who respond to pharmacotherapy should continue it for as long as it remains effective. In this context, there is increasing recognition of adult ADHD as a valid and treatable disorder.10
One of the challenges clinicians face is the reliability of adult recall of childhood ADHD. A controlled, prospective 16-year follow-up study found that of all adults retrospectively given a diagnosis of childhood ADHD, only 27% actually had the disorder.11 This study suggests that retrospective diagnoses of childhood ADHD made solely on the basis of self-reports are unlikely to be valid.
Another obstacle is that traditional medical education has seldom included training in adult ADHD.8,12 In a UK study, clinicians felt that they lacked training and knowledge to assess and manage adult ADHD patients.9
Even if adult ADHD is recognized, diagnosis is just the first step of care.13 These patients require ongoing management and follow-up assessments.
Although practice patterns vary, efforts to encourage doctors to provide adult ADHD care may be hindered by the fact that the gold standard of treatment is stimulant medication.4,10 Medications approved by the US Food and Drug Administration for adult ADHD include the stimulants lisdexamfetamine, osmotic-release methylphenidate, mixed amphetamine salts extended release, dexmethylphenidate extended release, and the nonstimulant atomoxetine.6 While stimulants are generally more efficacious for ADHD symptoms than nonstimulants, they are associated with misuse and diversion.14
UNIVERSAL PRECAUTIONS: A SIMPLIFIED APPROACH
The universal-precautions approach to prescribing stimulants aims to allay physician concerns and promote appropriate medication use to allow for proper management of this disorder.15 These precautions, to be applied to all adult ADHD patients for whom stimulants are being considered, include careful diagnosis and consideration of comorbidities, baseline risk stratification, informed consent processes, treatment agreements, periodic reassessments of treatment response, and meticulous documentation.
DIAGNOSIS
A frequently used screening assessment for adult ADHD is the ADHD Rating Scale (ADHD RS), which consists of two subscales for assessing hyperactivity/impulsivity and inattentiveness.16 ADHD can be classified into one of three subtypes based on symptoms: inattentive, hyperactive, or combined type. Symptoms must persist for at least 6 months for a diagnosis to be made. Other ADHD scales include the Conners Adult ADHD Rating Scales and the Brown Attention-Deficit Disorder Scales.4
High scores on screening scales must be interpreted within the clinical context. Clinicians need to ask about ADHD symptoms, establish their presence in various settings, and determine if these symptoms interfere with functioning. A diagnosis of adult ADHD also requires evidence of symptoms beginning in childhood.17 According to the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition, inattentive or hyperactive-impulsive symptoms must be present before age 12 in two or more settings and interfere with function and development.
Although self-reporting screening tools are helpful, these tests are not reliable for diagnostic purposes, and collateral information is also required.
Neuropsychological testing may detect impairments in persons with ADHD. The most consistently employed neuropsychological tests to evaluate ADHD include the Conners Continuous Performance Test, Stroop Color and Word Test, Trail-making Test, verbal fluency tests, Controlled Oral Word Association Test, and the Weschler Adult Intelligence Scale.6
COMORBIDITY
Epidemiologic studies suggest that adults with ADHD develop many psychiatric problems including anxiety, depression, and substance use disorders.7,16 Table 1 illustrates common comorbidities and their associated prevalence in the ADHD patient.7
Comorbid psychiatric disorders may affect the presentation of adult ADHD. For instance, adults with comorbid depression and ADHD are more likely to present with heightened irritability and difficulties concentrating on tasks than those with either condition alone.18 Similarly, antisocial personality disorder is more common in adults with ADHD.19 Such patients exhibit stable antisocial behavior (lying, stealing, and aggression) as well as medication misuse.5,14,19
While these comorbid disorders may obscure the ADHD diagnosis, their recognition is essential to effectively manage adult ADHD. In sum, a careful evaluation of the adult, including elucidating both ADHD and comorbid symptoms, functionality in several domains, and the degree of impairment, should precede initiating pharmacotherapy for adult ADHD.
BASELINE RISK STRATIFICATION: RISK FACTORS FOR STIMULANT MISUSE
After diagnosing ADHD, the prescriber must assess the risk for misuse of stimulant medications.20
One study revealed that nonmedical use of stimulant medications occurred in only 2% of the 4,300 people surveyed.21 Among the misusers, 66% had obtained medication from family or friends. Another 34% had stolen medication, and 20% had obtained prescriptions from a physician by falsely reporting symptoms. The study also assessed motivation for misuse. In this sample, 40% of misuse was to enhance performance, 34% was for recreation, and 23% was to stay awake.21
Other studies show that misuse of stimulant medications is common among youth in the United States, reporting that 18% of college students use some formulation of prescription stimulants.22
Still more research suggests that childhood conduct disorder or illicit drug use results in a higher risk of stimulant medication misuse.20 Additional risk factors for misuse include male sex, white ethnicity, upper-class background, Jewish or no religious affiliation, affiliation with a sorority or fraternity, off-campus housing, and a low grade-point average.23
Table 2 illustrates clinical interventions providers can use, once they have risk-stratified their patients, to monitor for stimulant misuse.
HOW SHOULD THESE RISK FACTORS AFFECT TREATMENT?
Although no formal scoring system exists to help clinicians risk-stratify these patients, the presence of multiple risk factors suggests the need for vigilance.14 Physicians should prescribe agents with less potential for abuse and monitor these patients more intensely.
Short-acting stimulant medications are the most likely to be abused, as phasic dopamine increase is more reinforcing than therapeutic dopamine release.24 Longer-acting stimulant medications are less likely to be abused, and they provide better symptom relief, as tonic dopamine release maintains a steady state and increases the therapeutic efficacy of these medications.25 For example, methylphenidate extended-release tablets have an osmotic oral controlled-release system and are less likely to be crushed for recreational inhalation.6,14
Lisdexamfetamine is a prodrug therapeutically inert until converted to d-amphetamine when lysine is cleaved from the molecule. This medication may be a good option for patients at high risk of misuse because it is tamper-resistant. However, it still may be subject to misuse for performance enhancement.26,27
The nonstimulant atomoxetine is also approved for ADHD, has no abuse potential, and may be particularly useful when anxiety, mood, and substance use disorders co-occur with ADHD.6 Rarely, atomoxetine can damage the liver, and liver function tests should be monitored if right upper quadrant pain develops.4,10
Other nonapproved agents such as bupropion and desipramine also have been used empirically and off-label for ADHD.4,10
Overall, treatment should be selected according to the risk of misuse of stimulant medication and the patient’s comorbidities.
INFORMED CONSENT
Informed consent may help patients appreciate the risks and benefits of the treatment options and develop realistic expectations about treatment.26 Patients are instructed to take their stimulant medications as prescribed and are informed of the risks of combining stimulants with other substances, particularly those that may interact with stimulants (eg, cocaine) and raise the risk of seizures and cardiovascular complications.
Stimulant medications lead to elevations in blood pressure and heart rate, although large-scale studies have shown no increase in the rate of serious cardiovascular events when these drugs are used appropriately.6 Less serious side effects associated with stimulant medications include insomnia, weight loss, decreased appetite, dry mouth, headache, and rarely, depression and anxiety.6
Patients need to be warned about diversion and abuse liability of stimulant medications, as well as alternative treatments.
The nonstimulant atomoxetine has no reinforcing properties but also raises the blood pressure and heart rate.6 As with stimulants, these elevations are generally minimal, time-limited, and of minor clinical significance.4,10 Frequent reasons to prescribe atomoxetine include poor tolerability of stimulants and a history of substance abuse. In addition, women with ADHD and high levels of emotional dysregulation or social anxiety appear to be particularly responsive to atomoxetine.6
Another consideration is cognitive behavioral therapy, which can augment the effects of pharmacotherapy.4 Cognitive behavioral therapy focuses on time management, prioritization, organization, problem-solving, motivation, and emotional regulation.4
Finally, patients also need to understand the possible consequences of nontreatment.5 Adults with untreated ADHD have high rates of academic and occupational difficulties, anti-social behaviors, and other forms of psychosocial adversity.4,5
Overall, this process should involve discussing risks and benefits of treatment options with the patient and promoting joint decision-making.
TREATMENT AGREEMENTS
Stimulant medications are classified by the US Drug Enforcement Administration as schedule II substances due to their abuse potential.20
It is important to inform patients of the addictive nature of the medication and to instruct them on how to store stimulants safely.27 Patients need to know that giving away or selling these medications is illegal.27
Treatment agreements establish rules for prescribing and are signed by the patient before initiating therapy.28,29 Patients are expected to attend all of their appointments, receive their prescriptions from one doctor, and obtain their medication from one pharmacy. These agreements may also require patients to submit to monitoring with random urine drug screens.29 Overall, they underscore the need for patients to follow a treatment plan in order to continue therapy with controlled substances.29
Manning27 recommends using agreements for high-risk college students prescribed stimulant medications. Red flags for misuse include signs of active substance use (eg, intoxication, a pattern of “lost” prescriptions, and doctor-shopping).27
The effectiveness in reducing risk of misuse in the adult ADHD population has not yet been investigated. Nonetheless, a method of communicating the seriousness of stimulant misuse to adult patients is essential to ADHD care.
STAYING ON TRACK
In the clinical setting, treatment response is measured not just by symptom reduction, but also by functional improvement. Thus, clinicians and patients must set functional goals whenever possible.27 Successful progress toward these goals justifies continuation of therapy, whereas lack of improvement signals the need to reconsider stimulant therapy.27
MONITORING AND DOCUMENTATION
Adults with ADHD present with varying levels of functional impairment and comorbidities, which may require different levels of monitoring.30 Not all patients with ADHD respond optimally to stimulant medications or tolerate them well.31,32 Hence, monitoring parameters for therapeutic change and adverse outcomes are important in that they guide the alteration or even discontinuation of pharmacotherapy.4,6,14
Documenting the decision-making process to continue stimulant medications under certain circumstances is also essential. Documentation should include discussion of goals and expectations, risks and benefits, and alternatives to stimulant use.
In adults, risk of stimulant medication misuse adds a new layer of complexity to monitoring.13,14 Adults may get multiple prescriptions from multiple providers, seek early refills, fill prescriptions at different pharmacies, or alter formulations. Many states track stimulant prescription use, and prescribers can use this information to determine if patients are refilling their prescriptions appropriately or obtaining stimulants from more than one provider.
Although these monitoring strategies are useful,6 it is prudent to structure the level of monitoring according to the patient’s risk of adverse events or medication misuse.14,27 Gourlay and Heit15 proposed the following “four-A” mnemonic for four domains to be explored at each visit in patients receiving pain medicine. This mnemonic can be applied to adult ADHD patients to more accurately monitor the patient throughout treatment.
THE ‘FOUR-A’ MNEMONIC
ADHD symptoms
Several ADHD scales can be used to track symptom changes over time.33 However, these self-report scales may be subject to positive illusory bias, a phenomenon observed in individuals with ADHD in which they tend to overrate their functioning,34 which may limit the accuracy of self-report scales.35
Activities of daily living
Since patients with ADHD tend to overrate their functioning in various aspects of living, collateral information should be gathered to corroborate patient self-reports whenever possible.
Adverse events
Blood pressure, heart rate, and weight should be assessed at baseline and monitored during stimulant treatment. Other symptoms to monitor include gastrointestinal distress, headache, aggression, depression, appetite, and sleeping habits.4,6 More intensive monitoring (eg, electrocardiography) may be indicated for those with hypertension and cardiovascular risk factors.
Aberrant behavior
Monitoring for misuse and diversion of stimulant medications is essential, as ADHD itself is a risk factor for addiction.20,21 Pill counts, prescription monitoring programs, urine drug screens, and collateral informants have all been proposed but not studied in monitoring for the misuse of stimulant medications.27 Before prescribing, it is prudent to check the prescription monitoring program, get a urine drug screen, and discuss any positive findings with the patient.36,37
Treatment agreements ensure that patients are aware of the consequences of misuse and allow the clinician to reference prior discussion when terminating treatment with stimulants.
LIVES CAN BE ENHANCED
ADHD is a common disorder that arises in childhood and can persist throughout life. Adults with untreated ADHD are at risk of severe impairments in various domains of functioning. Stimulant medications are an effective treatment but may be diverted into the street market. Using the universal-precautions model may reduce the risks of both nontreatment of ADHD and misuse of stimulants.
Accordingly, clinicians need to confirm the ADHD diagnosis, assess comorbidities, estimate risk of misuse, and provide informed consent prior to prescribing. Subsequent monitoring should involve the use of treatment agreements and evaluating treatment response, paying particular attention to ADHD symptom control but also to level of function, adverse effects, and aberrant behavior.
With these principles in mind, clinicians can address the risks of misuse and potentially enhance the lives of people who may have been suffering substantially due to lack of appropriate care.
Children are not the only people affected by attention-deficit/hyperactivity disorder (ADHD). Characterized by high levels of inattention, overactivity, and impulsivity, ADHD affects 5% of school-aged children, but also 4% of adults.1–3 Adults with untreated ADHD are likely to develop serious psychosocial problems that manifest as unemployment, arrests, divorce, underachievement, and psychiatric comorbidities.4,5
However, many clinicians are reluctant to manage adults with ADHD, partly because of concerns about misuse of the stimulant drugs they must prescribe to treat it.
Here, we outline an approach whereby clinicians can diagnose and treat adult ADHD while taking “universal precautions” to discourage misuse of the medications involved.
RECOGNIZING ADHD IN ADULTS
ADHD is characterized by developmentally inappropriate levels of inattention, impulsiveness, and hyperactivity that arise in childhood and result in impairments that often persist.
The presentation of ADHD in adults may be influenced by the longevity of their ADHD, associated sequelae (eg, low self-esteem and interpersonal, educational, and occupational difficulties), and comorbid disorders.6 There are neither reliable biomarkers nor neuropsychological tests for diagnosis, and persons with ADHD typically have a complex presentation with at least one comorbidity.6,7
In patients diagnosed in childhood, difficulties with organization as well as initiating, maintaining, and completing tasks become more prominent in adulthood and hyperactivity tends to subside. Adult impulsivity may present as edginess, shopping sprees, quitting jobs, and risky behaviors.6
Overall, the clinical manifestations of ADHD in adolescents and adults include inattention, difficulties with task completion, disorganization, and executive dysfunction—all skills critical to managing the various roles of adult life.
OBSTACLES TO EFFECTIVE TREATMENT
In the past, ADHD treatment was routinely discontinued during adolescence, as it was unclear whether adults still had significant symptoms or benefited from treatment.8,9 Now, available ADHD guidelines suggest that children and adults who respond to pharmacotherapy should continue it for as long as it remains effective. In this context, there is increasing recognition of adult ADHD as a valid and treatable disorder.10
One of the challenges clinicians face is the reliability of adult recall of childhood ADHD. A controlled, prospective 16-year follow-up study found that of all adults retrospectively given a diagnosis of childhood ADHD, only 27% actually had the disorder.11 This study suggests that retrospective diagnoses of childhood ADHD made solely on the basis of self-reports are unlikely to be valid.
Another obstacle is that traditional medical education has seldom included training in adult ADHD.8,12 In a UK study, clinicians felt that they lacked training and knowledge to assess and manage adult ADHD patients.9
Even if adult ADHD is recognized, diagnosis is just the first step of care.13 These patients require ongoing management and follow-up assessments.
Although practice patterns vary, efforts to encourage doctors to provide adult ADHD care may be hindered by the fact that the gold standard of treatment is stimulant medication.4,10 Medications approved by the US Food and Drug Administration for adult ADHD include the stimulants lisdexamfetamine, osmotic-release methylphenidate, mixed amphetamine salts extended release, dexmethylphenidate extended release, and the nonstimulant atomoxetine.6 While stimulants are generally more efficacious for ADHD symptoms than nonstimulants, they are associated with misuse and diversion.14
UNIVERSAL PRECAUTIONS: A SIMPLIFIED APPROACH
The universal-precautions approach to prescribing stimulants aims to allay physician concerns and promote appropriate medication use to allow for proper management of this disorder.15 These precautions, to be applied to all adult ADHD patients for whom stimulants are being considered, include careful diagnosis and consideration of comorbidities, baseline risk stratification, informed consent processes, treatment agreements, periodic reassessments of treatment response, and meticulous documentation.
DIAGNOSIS
A frequently used screening assessment for adult ADHD is the ADHD Rating Scale (ADHD RS), which consists of two subscales for assessing hyperactivity/impulsivity and inattentiveness.16 ADHD can be classified into one of three subtypes based on symptoms: inattentive, hyperactive, or combined type. Symptoms must persist for at least 6 months for a diagnosis to be made. Other ADHD scales include the Conners Adult ADHD Rating Scales and the Brown Attention-Deficit Disorder Scales.4
High scores on screening scales must be interpreted within the clinical context. Clinicians need to ask about ADHD symptoms, establish their presence in various settings, and determine if these symptoms interfere with functioning. A diagnosis of adult ADHD also requires evidence of symptoms beginning in childhood.17 According to the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition, inattentive or hyperactive-impulsive symptoms must be present before age 12 in two or more settings and interfere with function and development.
Although self-reporting screening tools are helpful, these tests are not reliable for diagnostic purposes, and collateral information is also required.
Neuropsychological testing may detect impairments in persons with ADHD. The most consistently employed neuropsychological tests to evaluate ADHD include the Conners Continuous Performance Test, Stroop Color and Word Test, Trail-making Test, verbal fluency tests, Controlled Oral Word Association Test, and the Weschler Adult Intelligence Scale.6
COMORBIDITY
Epidemiologic studies suggest that adults with ADHD develop many psychiatric problems including anxiety, depression, and substance use disorders.7,16 Table 1 illustrates common comorbidities and their associated prevalence in the ADHD patient.7
Comorbid psychiatric disorders may affect the presentation of adult ADHD. For instance, adults with comorbid depression and ADHD are more likely to present with heightened irritability and difficulties concentrating on tasks than those with either condition alone.18 Similarly, antisocial personality disorder is more common in adults with ADHD.19 Such patients exhibit stable antisocial behavior (lying, stealing, and aggression) as well as medication misuse.5,14,19
While these comorbid disorders may obscure the ADHD diagnosis, their recognition is essential to effectively manage adult ADHD. In sum, a careful evaluation of the adult, including elucidating both ADHD and comorbid symptoms, functionality in several domains, and the degree of impairment, should precede initiating pharmacotherapy for adult ADHD.
BASELINE RISK STRATIFICATION: RISK FACTORS FOR STIMULANT MISUSE
After diagnosing ADHD, the prescriber must assess the risk for misuse of stimulant medications.20
One study revealed that nonmedical use of stimulant medications occurred in only 2% of the 4,300 people surveyed.21 Among the misusers, 66% had obtained medication from family or friends. Another 34% had stolen medication, and 20% had obtained prescriptions from a physician by falsely reporting symptoms. The study also assessed motivation for misuse. In this sample, 40% of misuse was to enhance performance, 34% was for recreation, and 23% was to stay awake.21
Other studies show that misuse of stimulant medications is common among youth in the United States, reporting that 18% of college students use some formulation of prescription stimulants.22
Still more research suggests that childhood conduct disorder or illicit drug use results in a higher risk of stimulant medication misuse.20 Additional risk factors for misuse include male sex, white ethnicity, upper-class background, Jewish or no religious affiliation, affiliation with a sorority or fraternity, off-campus housing, and a low grade-point average.23
Table 2 illustrates clinical interventions providers can use, once they have risk-stratified their patients, to monitor for stimulant misuse.
HOW SHOULD THESE RISK FACTORS AFFECT TREATMENT?
Although no formal scoring system exists to help clinicians risk-stratify these patients, the presence of multiple risk factors suggests the need for vigilance.14 Physicians should prescribe agents with less potential for abuse and monitor these patients more intensely.
Short-acting stimulant medications are the most likely to be abused, as phasic dopamine increase is more reinforcing than therapeutic dopamine release.24 Longer-acting stimulant medications are less likely to be abused, and they provide better symptom relief, as tonic dopamine release maintains a steady state and increases the therapeutic efficacy of these medications.25 For example, methylphenidate extended-release tablets have an osmotic oral controlled-release system and are less likely to be crushed for recreational inhalation.6,14
Lisdexamfetamine is a prodrug therapeutically inert until converted to d-amphetamine when lysine is cleaved from the molecule. This medication may be a good option for patients at high risk of misuse because it is tamper-resistant. However, it still may be subject to misuse for performance enhancement.26,27
The nonstimulant atomoxetine is also approved for ADHD, has no abuse potential, and may be particularly useful when anxiety, mood, and substance use disorders co-occur with ADHD.6 Rarely, atomoxetine can damage the liver, and liver function tests should be monitored if right upper quadrant pain develops.4,10
Other nonapproved agents such as bupropion and desipramine also have been used empirically and off-label for ADHD.4,10
Overall, treatment should be selected according to the risk of misuse of stimulant medication and the patient’s comorbidities.
INFORMED CONSENT
Informed consent may help patients appreciate the risks and benefits of the treatment options and develop realistic expectations about treatment.26 Patients are instructed to take their stimulant medications as prescribed and are informed of the risks of combining stimulants with other substances, particularly those that may interact with stimulants (eg, cocaine) and raise the risk of seizures and cardiovascular complications.
Stimulant medications lead to elevations in blood pressure and heart rate, although large-scale studies have shown no increase in the rate of serious cardiovascular events when these drugs are used appropriately.6 Less serious side effects associated with stimulant medications include insomnia, weight loss, decreased appetite, dry mouth, headache, and rarely, depression and anxiety.6
Patients need to be warned about diversion and abuse liability of stimulant medications, as well as alternative treatments.
The nonstimulant atomoxetine has no reinforcing properties but also raises the blood pressure and heart rate.6 As with stimulants, these elevations are generally minimal, time-limited, and of minor clinical significance.4,10 Frequent reasons to prescribe atomoxetine include poor tolerability of stimulants and a history of substance abuse. In addition, women with ADHD and high levels of emotional dysregulation or social anxiety appear to be particularly responsive to atomoxetine.6
Another consideration is cognitive behavioral therapy, which can augment the effects of pharmacotherapy.4 Cognitive behavioral therapy focuses on time management, prioritization, organization, problem-solving, motivation, and emotional regulation.4
Finally, patients also need to understand the possible consequences of nontreatment.5 Adults with untreated ADHD have high rates of academic and occupational difficulties, anti-social behaviors, and other forms of psychosocial adversity.4,5
Overall, this process should involve discussing risks and benefits of treatment options with the patient and promoting joint decision-making.
TREATMENT AGREEMENTS
Stimulant medications are classified by the US Drug Enforcement Administration as schedule II substances due to their abuse potential.20
It is important to inform patients of the addictive nature of the medication and to instruct them on how to store stimulants safely.27 Patients need to know that giving away or selling these medications is illegal.27
Treatment agreements establish rules for prescribing and are signed by the patient before initiating therapy.28,29 Patients are expected to attend all of their appointments, receive their prescriptions from one doctor, and obtain their medication from one pharmacy. These agreements may also require patients to submit to monitoring with random urine drug screens.29 Overall, they underscore the need for patients to follow a treatment plan in order to continue therapy with controlled substances.29
Manning27 recommends using agreements for high-risk college students prescribed stimulant medications. Red flags for misuse include signs of active substance use (eg, intoxication, a pattern of “lost” prescriptions, and doctor-shopping).27
The effectiveness in reducing risk of misuse in the adult ADHD population has not yet been investigated. Nonetheless, a method of communicating the seriousness of stimulant misuse to adult patients is essential to ADHD care.
STAYING ON TRACK
In the clinical setting, treatment response is measured not just by symptom reduction, but also by functional improvement. Thus, clinicians and patients must set functional goals whenever possible.27 Successful progress toward these goals justifies continuation of therapy, whereas lack of improvement signals the need to reconsider stimulant therapy.27
MONITORING AND DOCUMENTATION
Adults with ADHD present with varying levels of functional impairment and comorbidities, which may require different levels of monitoring.30 Not all patients with ADHD respond optimally to stimulant medications or tolerate them well.31,32 Hence, monitoring parameters for therapeutic change and adverse outcomes are important in that they guide the alteration or even discontinuation of pharmacotherapy.4,6,14
Documenting the decision-making process to continue stimulant medications under certain circumstances is also essential. Documentation should include discussion of goals and expectations, risks and benefits, and alternatives to stimulant use.
In adults, risk of stimulant medication misuse adds a new layer of complexity to monitoring.13,14 Adults may get multiple prescriptions from multiple providers, seek early refills, fill prescriptions at different pharmacies, or alter formulations. Many states track stimulant prescription use, and prescribers can use this information to determine if patients are refilling their prescriptions appropriately or obtaining stimulants from more than one provider.
Although these monitoring strategies are useful,6 it is prudent to structure the level of monitoring according to the patient’s risk of adverse events or medication misuse.14,27 Gourlay and Heit15 proposed the following “four-A” mnemonic for four domains to be explored at each visit in patients receiving pain medicine. This mnemonic can be applied to adult ADHD patients to more accurately monitor the patient throughout treatment.
THE ‘FOUR-A’ MNEMONIC
ADHD symptoms
Several ADHD scales can be used to track symptom changes over time.33 However, these self-report scales may be subject to positive illusory bias, a phenomenon observed in individuals with ADHD in which they tend to overrate their functioning,34 which may limit the accuracy of self-report scales.35
Activities of daily living
Since patients with ADHD tend to overrate their functioning in various aspects of living, collateral information should be gathered to corroborate patient self-reports whenever possible.
Adverse events
Blood pressure, heart rate, and weight should be assessed at baseline and monitored during stimulant treatment. Other symptoms to monitor include gastrointestinal distress, headache, aggression, depression, appetite, and sleeping habits.4,6 More intensive monitoring (eg, electrocardiography) may be indicated for those with hypertension and cardiovascular risk factors.
Aberrant behavior
Monitoring for misuse and diversion of stimulant medications is essential, as ADHD itself is a risk factor for addiction.20,21 Pill counts, prescription monitoring programs, urine drug screens, and collateral informants have all been proposed but not studied in monitoring for the misuse of stimulant medications.27 Before prescribing, it is prudent to check the prescription monitoring program, get a urine drug screen, and discuss any positive findings with the patient.36,37
Treatment agreements ensure that patients are aware of the consequences of misuse and allow the clinician to reference prior discussion when terminating treatment with stimulants.
LIVES CAN BE ENHANCED
ADHD is a common disorder that arises in childhood and can persist throughout life. Adults with untreated ADHD are at risk of severe impairments in various domains of functioning. Stimulant medications are an effective treatment but may be diverted into the street market. Using the universal-precautions model may reduce the risks of both nontreatment of ADHD and misuse of stimulants.
Accordingly, clinicians need to confirm the ADHD diagnosis, assess comorbidities, estimate risk of misuse, and provide informed consent prior to prescribing. Subsequent monitoring should involve the use of treatment agreements and evaluating treatment response, paying particular attention to ADHD symptom control but also to level of function, adverse effects, and aberrant behavior.
With these principles in mind, clinicians can address the risks of misuse and potentially enhance the lives of people who may have been suffering substantially due to lack of appropriate care.
- Polanczyk G, de Lima MS, Horta BL, Biederman J, Rohde LA. The worldwide prevalence of ADHD: a systematic review and metaregression analysis. Am J Psychiatry 2007; 164:942–948.
- Polanczyk GV, Wilcutt EG, Salum GA, Kieling C, Rohde LA. ADHD prevalence estimates across three decades: an updated systematic review and meta-regression analysis. Int J Epidemiol 2014; 43:434–442.
- Wilens TE. ADHD: Prevalence, diagnosis, and issues of comorbidity. CNS Spectr 2007; 12(suppl 6):1–5.
- Kooij SJ, Bejerot S, Blackwell A, et al. European consensus statement on diagnosis and treatment of adult ADHD: the European Network Adult ADHD. BMC Psychiatry 2010; 10:67.
- Shaw M, Hodgkins P, Caci H, et al. A systematic review and analysis of long-term outcomes in attention deficit hyperactivity disorder: effects of treatment and non-treatment. BMC Med 2012;10:99.
- Modesto-Lowe V, Meyer A, Soovajian V. A clinician’s guide to adult attention-deficit hyperactivity disorder. Conn Med 2012; 76:517–523.
- Kessler RC, Adler L, Barkley R, et al. The prevalence and correlates of adult ADHD in the United States: results from the National Comorbidity Survey Replication. Am J Psychiatry 2006; 163:716–723.
- Goodman DW, Surman CB, Scherer PB, Salinas GD, Brown JJ. Assessment of physician practices in adult attention-deficit/hyperactivity disorder. Prim Care Companion CNS Disord 2012; 14(4).
- Hall CL, Newell K, Taylor J, Sayal K, Swift KD, Hollis C. ‘Mind the gap’—mapping services for young people with ADHD transitioning from child to adult mental health services. BMC Psychiatry 2013; 13:186.
- National Institute for Health and Care Excellence. Attention deficit hyperactivity disorder: diagnosis and management of ADHD in children, young people and adults. The British Psychological Society and The Royal College of Psychiatrists: United Kingdom; 2009.
- Mannuzza S, Klein RG, Klein DF, Bessler A, Shrout P. Accuracy of adult recall of childhood attention deficit hyperactivity disorder. Am J Psychiatry 2002; 159:1882–1888.
- Wetzel MW. Medical student participation in an adult ADHD outpatient clinic: an ideal setting for education in outpatient psychiatry. Acad Psychiatry 2009; 33:80–81.
- Culpepper L, Mattingly G. Challenges in identifying and managing attention-deficit/hyperactivity disorder in adults in the primary care setting: a review of the literature. Prim Care Companion J Clin Psychiatry 2010; 12(6).
- Rabiner DL. Stimulant prescription cautions: addressing misuse, diversion and malingering. Curr Psychiatry Rep 2013; 15:375.
- Gourlay D, Heit H. Universal precautions: a matter of mutual trust and responsibility. Pain Med 2006; 7:210–211.
- Kessler RC, Adler L, Ames M, et al. The World Health Organization Adult ADHD Self-Report Scale (ASRS): a short screening scale for use in the general population. Psychol Med 2005; 35:245–256.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed. Arlington, VA: American Psychiatric Association; 2013.
- CADDRA Guidelines Steering Committee. Canadian ADHD practice guidelines: CADDRA 2008. http://www.naceonline.com/AdultADHDtoolkit/professionalresources/caddraguidelines.pdf. Accessed July 10, 2015.
- Mannuzza S, Klein RG, Bessler A, Malloy P, LaPadula M. Adult psychiatric status of hyperactive boys grown up. Am J Psychiatry 1998; 155:493–498.
- Kaye S, Darke S. The diversion and misuse of pharmaceutical stimulants: what do we know and why should we care? Addiction 2012; 107:467–477.
- Novak SP, Kroutil LA, Williams RL, Van Brunt DL. The nonmedical use of prescription ADHD medications: results from a national Internet panel. Subst Abuse Treat Prev Policy 2007; 2:32.
- Bavarian N, Flay BR, Ketcham P, et al. Using structural equation modeling to understand prescription stimulant misuse: a test of the Theory of Triadic Influence. Drug Alcohol Depend 2014; 138:193–201.
- McCabe SE, Teter CJ, Boyd CJ. Medical use, illicit use and diversion of prescription stimulant medication. J Psychoactive Drugs 2006; 38:43–56.
- Volkow ND. Stimulant medications: how to minimize their reinforcing effects? Am J Psychiatry 2006; 163:359–361.
- Kolar D, Keller A, Golfinopoulos M, Cumyn L, Syer C, Hechtman L. Treatment of adults with attention-deficit/hyperactivity disorder. Neuropsychiatr Dis Treat 2008; 4:107–121.
- Schachter D, Tharmalingam S, Kleinman I. Informed consent and stimulant medication: adolescents’ and parents’ ability to understand information about benefits and risks of stimulant medication for the treatment of attention-deficit/hyperactivity disorder. J Child Adolesc Psychopharmacol 2011; 21:139–148.
- Manning JS. Strategies for managing the risks associated with ADHD medications. J Clin Psychiatry 2013; 74:e19.
- Deep K. Use of narcotics contracts. Virtual Mentor 2013; 15:416–420.
- Cheatle MD, Savage SR. Informed consent in opioid therapy: a potential obligation and opportunity. J Pain Symptom Manage 2012; 44:105–116.
- Dias TG, Kieling C, Graeff-Martins AS, Moriyama TS, Rohde LA, Polanczyk GV. Developments and challenges in the diagnosis and treatment of ADHD. Rev Bras Psiquiatr 2013; 35(suppl 1):S40–S50.
- Mattingly GW, Weisler RH, Young J, et al. Clinical response and symptomatic remission in short- and long-term trials of lisdexamfetamine dimesylate in adults with attention-deficit/hyperactivity disorder. BMC Psychiatry 2013; 13:39.
- Contini V, Victor MM, Bertuzzi GP, et al. No significant association between genetic variants in 7 candidate genes and response to methylphenidate treatment in adult patients with ADHD. J Clin Psychopharmacol 2012; 32:820–823.
- Rösler M, Retz W, Thome J, Schneider M, Stieglitz RD, Falkai P. Psychopathological rating scales for diagnostic use in adults with attention-deficit/hyperactivity disorder (ADHD). Eur Arch Psychiatry Clin Neurosci 2006; 256(suppl 1):i3–i11.
- Prevatt F, Proctor B, Best L, Baker L, Van Walker J, Taylor NW. The positive illusory bias: does it explain self-evaluations in college students with ADHD? J Atten Disord 2012; 16:235–243.
- Jiang Y, Johnston C. The relationship between ADHD symptoms and competence as reported by both self and others. J Atten Disord 2012; 16:418–426.
- Darredeau C, Barrett SP, Jardin B, Pihl RO. Patterns and predictors of medication compliance, diversion, and misuse in adult prescribed methylphenidate users. Hum Psychopharmacol 2007; 22:529–536.
- Worley J. Prescription drug monitoring programs, a response to doctor shopping: purpose, effectiveness, and directions for future research. Issues Ment Health Nurs 2012; 33:319–328.
- Polanczyk G, de Lima MS, Horta BL, Biederman J, Rohde LA. The worldwide prevalence of ADHD: a systematic review and metaregression analysis. Am J Psychiatry 2007; 164:942–948.
- Polanczyk GV, Wilcutt EG, Salum GA, Kieling C, Rohde LA. ADHD prevalence estimates across three decades: an updated systematic review and meta-regression analysis. Int J Epidemiol 2014; 43:434–442.
- Wilens TE. ADHD: Prevalence, diagnosis, and issues of comorbidity. CNS Spectr 2007; 12(suppl 6):1–5.
- Kooij SJ, Bejerot S, Blackwell A, et al. European consensus statement on diagnosis and treatment of adult ADHD: the European Network Adult ADHD. BMC Psychiatry 2010; 10:67.
- Shaw M, Hodgkins P, Caci H, et al. A systematic review and analysis of long-term outcomes in attention deficit hyperactivity disorder: effects of treatment and non-treatment. BMC Med 2012;10:99.
- Modesto-Lowe V, Meyer A, Soovajian V. A clinician’s guide to adult attention-deficit hyperactivity disorder. Conn Med 2012; 76:517–523.
- Kessler RC, Adler L, Barkley R, et al. The prevalence and correlates of adult ADHD in the United States: results from the National Comorbidity Survey Replication. Am J Psychiatry 2006; 163:716–723.
- Goodman DW, Surman CB, Scherer PB, Salinas GD, Brown JJ. Assessment of physician practices in adult attention-deficit/hyperactivity disorder. Prim Care Companion CNS Disord 2012; 14(4).
- Hall CL, Newell K, Taylor J, Sayal K, Swift KD, Hollis C. ‘Mind the gap’—mapping services for young people with ADHD transitioning from child to adult mental health services. BMC Psychiatry 2013; 13:186.
- National Institute for Health and Care Excellence. Attention deficit hyperactivity disorder: diagnosis and management of ADHD in children, young people and adults. The British Psychological Society and The Royal College of Psychiatrists: United Kingdom; 2009.
- Mannuzza S, Klein RG, Klein DF, Bessler A, Shrout P. Accuracy of adult recall of childhood attention deficit hyperactivity disorder. Am J Psychiatry 2002; 159:1882–1888.
- Wetzel MW. Medical student participation in an adult ADHD outpatient clinic: an ideal setting for education in outpatient psychiatry. Acad Psychiatry 2009; 33:80–81.
- Culpepper L, Mattingly G. Challenges in identifying and managing attention-deficit/hyperactivity disorder in adults in the primary care setting: a review of the literature. Prim Care Companion J Clin Psychiatry 2010; 12(6).
- Rabiner DL. Stimulant prescription cautions: addressing misuse, diversion and malingering. Curr Psychiatry Rep 2013; 15:375.
- Gourlay D, Heit H. Universal precautions: a matter of mutual trust and responsibility. Pain Med 2006; 7:210–211.
- Kessler RC, Adler L, Ames M, et al. The World Health Organization Adult ADHD Self-Report Scale (ASRS): a short screening scale for use in the general population. Psychol Med 2005; 35:245–256.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed. Arlington, VA: American Psychiatric Association; 2013.
- CADDRA Guidelines Steering Committee. Canadian ADHD practice guidelines: CADDRA 2008. http://www.naceonline.com/AdultADHDtoolkit/professionalresources/caddraguidelines.pdf. Accessed July 10, 2015.
- Mannuzza S, Klein RG, Bessler A, Malloy P, LaPadula M. Adult psychiatric status of hyperactive boys grown up. Am J Psychiatry 1998; 155:493–498.
- Kaye S, Darke S. The diversion and misuse of pharmaceutical stimulants: what do we know and why should we care? Addiction 2012; 107:467–477.
- Novak SP, Kroutil LA, Williams RL, Van Brunt DL. The nonmedical use of prescription ADHD medications: results from a national Internet panel. Subst Abuse Treat Prev Policy 2007; 2:32.
- Bavarian N, Flay BR, Ketcham P, et al. Using structural equation modeling to understand prescription stimulant misuse: a test of the Theory of Triadic Influence. Drug Alcohol Depend 2014; 138:193–201.
- McCabe SE, Teter CJ, Boyd CJ. Medical use, illicit use and diversion of prescription stimulant medication. J Psychoactive Drugs 2006; 38:43–56.
- Volkow ND. Stimulant medications: how to minimize their reinforcing effects? Am J Psychiatry 2006; 163:359–361.
- Kolar D, Keller A, Golfinopoulos M, Cumyn L, Syer C, Hechtman L. Treatment of adults with attention-deficit/hyperactivity disorder. Neuropsychiatr Dis Treat 2008; 4:107–121.
- Schachter D, Tharmalingam S, Kleinman I. Informed consent and stimulant medication: adolescents’ and parents’ ability to understand information about benefits and risks of stimulant medication for the treatment of attention-deficit/hyperactivity disorder. J Child Adolesc Psychopharmacol 2011; 21:139–148.
- Manning JS. Strategies for managing the risks associated with ADHD medications. J Clin Psychiatry 2013; 74:e19.
- Deep K. Use of narcotics contracts. Virtual Mentor 2013; 15:416–420.
- Cheatle MD, Savage SR. Informed consent in opioid therapy: a potential obligation and opportunity. J Pain Symptom Manage 2012; 44:105–116.
- Dias TG, Kieling C, Graeff-Martins AS, Moriyama TS, Rohde LA, Polanczyk GV. Developments and challenges in the diagnosis and treatment of ADHD. Rev Bras Psiquiatr 2013; 35(suppl 1):S40–S50.
- Mattingly GW, Weisler RH, Young J, et al. Clinical response and symptomatic remission in short- and long-term trials of lisdexamfetamine dimesylate in adults with attention-deficit/hyperactivity disorder. BMC Psychiatry 2013; 13:39.
- Contini V, Victor MM, Bertuzzi GP, et al. No significant association between genetic variants in 7 candidate genes and response to methylphenidate treatment in adult patients with ADHD. J Clin Psychopharmacol 2012; 32:820–823.
- Rösler M, Retz W, Thome J, Schneider M, Stieglitz RD, Falkai P. Psychopathological rating scales for diagnostic use in adults with attention-deficit/hyperactivity disorder (ADHD). Eur Arch Psychiatry Clin Neurosci 2006; 256(suppl 1):i3–i11.
- Prevatt F, Proctor B, Best L, Baker L, Van Walker J, Taylor NW. The positive illusory bias: does it explain self-evaluations in college students with ADHD? J Atten Disord 2012; 16:235–243.
- Jiang Y, Johnston C. The relationship between ADHD symptoms and competence as reported by both self and others. J Atten Disord 2012; 16:418–426.
- Darredeau C, Barrett SP, Jardin B, Pihl RO. Patterns and predictors of medication compliance, diversion, and misuse in adult prescribed methylphenidate users. Hum Psychopharmacol 2007; 22:529–536.
- Worley J. Prescription drug monitoring programs, a response to doctor shopping: purpose, effectiveness, and directions for future research. Issues Ment Health Nurs 2012; 33:319–328.
KEY POINTS
- Untreated adult ADHD is associated with negative outcomes that include unemployment, arrests, divorce, and psychiatric comorbidities.
- Available ADHD guidelines suggest that children and adults who respond to pharmacotherapy should continue it for as long as it remains effective. In this context, there is increasing recognition of adult ADHD as a valid and treatable disorder.
- Following the guidelines of universal precautions in the diagnosis and treatment of adult ADHD can alleviate clinicians’ concerns when diagnosing and treating this disorder.

Starting insulin in patients with type 2 diabetes: An individualized approach
Insulin therapy is one of the most effective tools clinicians can use to help patients reach their individualized hemoglobin A1c target. However, decisions about when and how to start insulin therapy have to be individualized to the needs and goals of each patient. Many insulin options are available, one of the most common being the addition of basal insulin to oral antidiabetic drugs. Although patients are often reluctant to start insulin, this reluctance can be overcome through patient education and hands-on training.
Here, we review hemoglobin A1c targets, factors that determine when to start insulin therapy, and the different regimens that can be used.
MOST PATIENTS EVENTUALLY NEED INSULIN
Type 2 diabetes mellitus is a chronic progressive disease associated with insulin resistance, beta-cell dysfunction, and decreased insulin secretion. Consequently, most patients eventually require insulin therapy to reduce the risk of long-term complications.

The efficacy of therapy can be assessed by measuring hemoglobin A1c, an important marker of the chronic hyperglycemic state. The hemoglobin A1c value can be reported as a ratio (%) standardized against the results of the Diabetes Control and Complications Trial,1 or as International Federation of Clinical Chemistry units (mmol/mol).2 Table 1 shows the relationship between hemoglobin A1c and average glucose values.3
WHAT IS AN APPROPRIATE HEMOGLOBIN A1c TARGET?
The short answer is, “It depends.”
Currently, the American Association of Clinical Endocrinologists (AACE) supports a hemoglobin A1c goal of less than 6.5% for otherwise healthy patients but states that the goal should be individualized for patients with concurrent illnesses or at risk of hypoglycemia.4
On the other hand, the American Diabetes Association (ADA) recommends a higher hemoglobin A1c target of less than 7% for most adults with type 2 diabetes mellitus.5 This value was shown to be associated with a reduction in the microvascular and macrovascular complications of diabetes.
Yet when three large trials6–8 recently compared intensive and standard glucose control regimens, tighter glucose control failed to improve cardiovascular outcomes. Moreover, in one of the trials,7 patients receiving intensive treatment had a higher rate of all-cause mortality. Details:
- Action in Diabetes and Vascular Disease (ADVANCE): 11,140 patients; average hemoglobin A1c levels 6.5% vs 7.3%6
- Action to Control Cardiovascular Risk in Diabetes (ACCORD): 10,251 patients; average hemoglobin A1c levels 6.4% vs 7.5%7
- Veterans Affairs Diabetes Trial (VADT): 1,791 patients; average hemoglobin A1c levels 6.9% vs 8.4%.8
Similarly, a 2013 Cochrane review9 that included 28 randomized controlled trials concluded that intensive control (in 18,717 patients) did not decrease all-cause and cardiovascular mortality rates compared with traditional glucose control (in 16,195 patients), and it increased the risk of hypoglycemia and serious adverse events.
As a result, the ADA5 states that a hemoglobin A1c target less than 6.5% is optional for patients with a long life expectancy, short duration of diabetes, low risk of hypoglycemia, and no significant cardiovascular disease. The ADA further defines a hemoglobin A1c goal of less than 8% for patients with a history of severe hypoglycemia, limited life expectancy, advanced microvascular or macrovascular complications, extensive comorbid conditions, and long-standing diabetes.
Therefore, the AACE and ADA are moving away from “one-size-fits-all” goals and toward individualizing their recommendations.
WHEN SHOULD INSULIN BE STARTED?
Physicians should consider the needs and preferences of each patient and individualize the treatment. The most recent recommendations from the ADA5 stress the importance of a patient-centered approach, with multiple factors taken into account. These include the patient’s attitude, expected compliance with treatment, risk of hypoglycemia, disease duration, life expectancy, and comorbidities, and the side effects of oral medications and insulin.
Compared with previous guidelines, there are fewer rules on how and when to start insulin therapy. But absolute and relative indications for insulin therapy should be considered in patients with the following:
Absolute indications for insulin
- Ketoacidosis or catabolic symptoms, including ketonuria
- Newly diagnosed type 2 diabetes with pronounced hyperglycemia (glucose ≥ 300 mg/dL or hemoglobin A1c ≥ 10.0%) with or without severe symptoms, including weight loss, polyuria, or polydipsia10
- Uncontrolled type 2 diabetes mellitus despite using one, two, or more oral antidiabetic drugs or glucagon-like peptide 1 (GLP-1) receptor agonists
- Gestational diabetes
- Preference for insulin.
Relative indications for insulin
- Hospitalized for surgery or acute illnesses
- Advanced renal or hepatic disease
- Inability to afford the cost or tolerate the side effects of oral antidiabetic drugs and GLP-1 receptor agonists.

Depending on the situation, blood glucose is measured fasting, before meals, or after meals after initiating or adjusting insulin regimens (Table 2).
WHAT ARE THE INSULIN REGIMENS?
Basal insulin
In the early stages of type 2 diabetes, metformin alone or in combination with another oral antidiabetic drug or with a GLP-1 receptor agonist is often used along with healthy eating, weight control, and increased physical activity.

When the target hemoglobin A1c cannot be achieved with one or two noninsulin drugs, the ADA suggests basal insulin be added to metformin or a two-medication regimen that includes metformin (Table 3). However, recent evidence suggests that combining a GLP-1 receptor agonist with basal insulin, in a regimen without metformin, is safe and improves glycemic control without hypoglycemia or weight gain.11
While a total daily dose of insulin of 0.1 to 0.2 units/kg could be initially used in patients with a hemoglobin A1c level less than 8%, a higher dose of 0.2 to 0.3 units/kg is required if the hemoglobin A1c level is between 8% and 10%. The dose can be titrated once or twice weekly if the fasting glucose is above the target level (usually < 130 mg/dL). If hypoglycemia develops (glucose < 70 mg/dL), the insulin dose should be reduced by 10% to 20%.10
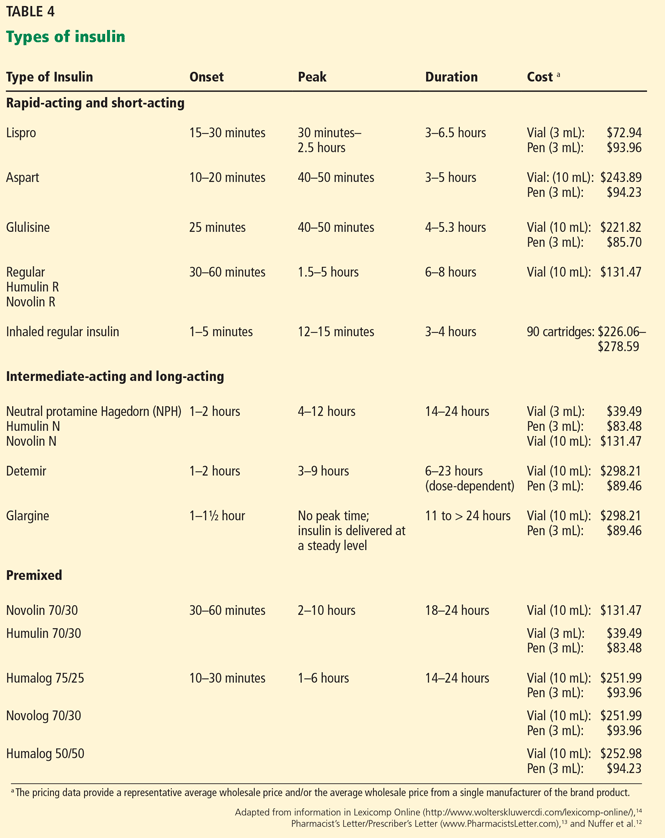
Available basal insulins include glargine, detemir, and neutral protamine Hagedorn (NPH) (Table 4).12–14 Because glargine and detemir offer better pharmacokinetic properties, less variability in response, and less risk of hypoglycemia, they are preferred over NPH. Glargine has a relatively constant plasma concentration over 24 hours, allowing once-daily dosing at any time during the day (Figure 1).15 The dose should be taken at the same time every day. Detemir and NPH are usually taken once or twice daily.

Patients treated once daily should take the dose with the evening meal or at bedtime. Patients who require a twice-daily regimen can take the first dose with breakfast and the second one with the evening meal, at bedtime, or 12 hours after the morning dose.
The randomized Treat-to-Target trial,16 in 756 patients, showed that both glargine and NPH, when added to oral therapy in patients with type 2 diabetes, achieve the target hemoglobin A1c, but NPH is associated with more episodes of nocturnal hypoglycemia. Similar results were found when NPH was compared with detemir insulin.17
A Cochrane review18 suggested that glargine and detemir are similar in efficacy and safety. However, detemir often needs to be injected twice daily, in a higher dose, and is associated with less weight gain. Furthermore, a meta-analysis of 46 randomized clinical trials19 showed that the weight increase at 1 year is less in patients treated with basal than with twice-daily or prandial regimens.
A noninterventional longitudinal study20 in 2,179 patients newly started on insulin showed that the mean weight increase at 1 year was 1.78 kg, and 24% of patients gained more than 5 kg. However, the factors independently associated with the weight gain were a higher hemoglobin A1c at baseline, a higher insulin dose at baseline and at 1 year, and a lower baseline body mass index, but not the type of insulin regimen.
Currently, a new class of ultralong-acting basal insulins is being studied. Insulins in this class are approved in other countries, but the US Food and Drug Administration requires additional data for approval. Ultralong-acting insulins are expected to reduce the risk of hypoglycemia, specifically the risk of nocturnal episodes. Also, given their longer duration of action and stable steady-state pharmacokinetics, they will offer flexibility in the dose timing.21
Basal-bolus regimens
Basal insulin often does not control postprandial hyperglycemia. The need for multiple doses of insulin (including one or more preprandial doses) is suggested by postprandial glucose values above target (usually > 180 mg/dL) or by a hemoglobin A1c above goal despite well-controlled fasting glucose levels. This usually becomes evident when the total daily dose of basal insulin exceeds 0.5 units/kg. Patients newly diagnosed with diabetes who have a hemoglobin A1c higher than 10% may also respond better to an initial basal-bolus regimen.
Available bolus insulins include lispro, aspart, glulisine, regular insulin, and the newly approved Technosphere inhaled regular insulin (Table 4).12–14 They can be taken before each meal, and the total bolus dose usually represents 50% of the total daily dose.22 Rapid-acting insulins have faster onset, shorter duration of action, and more predictable pharmacokinetics, which makes them preferable to regular insulin (Figure 1).15 Inhaled insulin is another option, but it is contraindicated in patients with chronic obstructive pulmonary disease or asthma because of the increased risk of acute bronchospasm.12
Alternatively, the transition to a basal-bolus regimen can be accomplished with a single dose of bolus insulin before the main meal, using a dose that represents approximately 10% of the total daily dose. Additional bolus doses can be added later based on the glycemic control. The adjustment of the preprandial insulin dose is done once or twice weekly, based on the postprandial glucose levels.10
Premixed combinations of long- and short-acting insulins in ratios of 50% to 50%, 70% to 30%, or 75% to 25% can be considered in patients who cannot adhere to a complex insulin regimen. A propensity-matched comparison of different insulin regimens (basal, premixed, mealtime plus basal, and mealtime) in patients with type 2 diabetes revealed that the hemoglobin A1c reduction was similar between the different groups.23 However, the number of hypoglycemic episodes was higher in the premixed insulin group, and the weight gain was less in the basal insulin group.
While premixed insulins require fewer injections, they do not provide dosing flexibility. In other words, dose adjustments for premixed insulins lead to increases in both basal and bolus amounts even though a dose adjustment is needed for only one insulin type. Thus, this is a common reason for increased hypoglycemic episodes.
Continuous subcutaneous insulin infusion
A meta-analysis showed that continuous subcutaneous insulin infusion (ie, use of an insulin pump) was similar to intensive therapy with multiple daily insulin injections in terms of glycemic control and hypoglycemia.24 Since both options can lead to similar glucose control, additional factors to consider when initiating insulin infusion include lifestyle and technical expertise. Some patients may or may not prefer having a pump attached for nearly all daily activities. Additionally, this type of therapy is complex and requires significant training to ensure efficacy and safety.25
WHAT IS THE COST OF INSULIN THERAPY?
A final factor to keep in mind when initiating insulin is cost (Table 4).12–14 Asking patients to check their prescription insurance formulary is important to ensure that an affordable option is selected. If patients do not have prescription insurance, medication assistance programs could be an option. However, if a patient is considering an insulin pump, insurance coverage is essential. Depending on the manufacturer, insulin pumps cost about $6,000 to $7,000, and the additional monthly supplies for the pump are also expensive.
If patients are engaged when considering and selecting insulin therapy, the likelihood of treatment success is greater.26–28
- The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med 1993; 329:977–986.
- Hanas R, John WG; International HbA1c Consensus Committee. 2013 Update on the worldwide standardization of the hemoglobin A1c measurement. Pediatr Diabetes 2014; 15:e1–e2.
- Nathan DM, Kuenen J, Borg R, Zheng H, Schoenfeld D, Heine RJ; A1c-Derived Average Glucose Study Group. Translating the A1C assay into estimated average glucose values. Diabetes Care 2008; 31:1473–1478.
- Garber AJ, Abrahamson MJ, Barzilay JI, et al; American Association of Clinical Endocrinologists. AACE comprehensive diabetes management algorithm 2013. Endocr Pract 2013; 19:327–336.
- American Diabetes Association. Standards of medical care in diabetes—2014. Diabetes Care 2014; 37(suppl 1):S14–S80.
- ADVANCE Collaborative Group; Patel A, MacMahon S, Chalmers J, et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med 2008; 358:2560–2572.
- Action to Control Cardiovascular Risk in Diabetes Study Group; Gerstein HC, Miller ME, Byington RP, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med 2008; 358:2545–2559.
- Duckworth W, Abraira C, Moritz T, et al; VADT Investigators. Glucose control and vascular complications in veterans with type 2 diabetes. N Engl J Med 2009; 360:129–139.
- Hemmingsen B, Lund SS, Gluud C, et al. Targeting intensive glycaemic control versus targeting conventional glycaemic control for type 2 diabetes mellitus. Cochrane Database Syst Rev 2013; 11:CD008143.
- Inzucchi SE, Bergenstal RM, Buse JB, et al; American Diabetes Association (ADA); European Association for the Study of Diabetes (EASD). Management of hyperglycemia in type 2 diabetes: a patient-centered approach: position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 2012; 35:1364–1379.
- Vora J, Bain SC, Damci T, et al. Incretin-based therapy in combination with basal insulin: a promising tactic for the treatment of type 2 diabetes. Diabetes Metab 2013; 39:6–15.
- Nuffer W, Trujillo JM, Ellis SL. Technosphere insulin (Afrezza): a new, inhaled prandial insulin. Ann Pharmacother 2015; 49:99–106.
- Pharmacist’s Letter/Prescriber’s Letter. Comparison of insulins and injectable diabetes meds. PL Detail-Document #281107 November 2012. www.PharmacistsLetter.com. Accessed July 2, 2015
- Lexicomp Online. www.wolterskluwercdi.com/lexicomp-online/. Accessed July 2, 2015.
- Hirsch IB. Insulin analogues. N Engl J Med 2005; 352:174-183.
- Riddle MC, Rosenstock J, Gerich J; Insulin Glargine 4002 Study Investigators. The treat-to-target trial: randomized addition of glargine or human NPH insulin to oral therapy of type 2 diabetic patients. Diabetes Care 2003; 26:3080–3086.
- Hermansen K, Davies M, Derezinski T, Martinez Ravn G, Clauson P, Home P. A 26-week, randomized, parallel, treat-to-target trial comparing insulin detemir with NPH insulin as add-on therapy to oral glucose-lowering drugs in insulin-naive people with type 2 diabetes. Diabetes Care 2006; 29:1269–1274.
- Swinnen SG, Simon AC, Holleman F, Hoekstra JB, Devries JH. Insulin detemir versus insulin glargine for type 2 diabetes mellitus. Cochrane Database Syst Rev 2011; 7:CD006383.
- Pontiroli AE, Miele L, Morabito A. Increase of body weight during the first year of intensive insulin treatment in type 2 diabetes: systematic review and meta-analysis. Diabetes Obes Metab 2011; 13:1008–1019.
- Balkau B, Home PD, Vincent M, Marre M, Freemantle N. Factors associated with weight gain in people with type 2 diabetes starting on insulin. Diabetes Care 2014; 37:2108–2113.
- Garber AJ. Will the next generation of basal insulins offer clinical advantages? Diabetes Obes Metab 2014; 16:483–491.
- Tamaki M, Shimizu T, Kanazawa A, et al. Effects of changes in basal/total daily insulin ratio in type 2 diabetes patients on intensive insulin therapy including insulin glargine (JUN-LAN Study 6). Diabetes Res Clin Pract 2008; 81:e1–e3.
- Freemantle N, Balkau B, Home PD. A propensity score matched comparison of different insulin regimens 1 year after beginning insulin in people with type 2 diabetes. Diabetes Obes Metab 2013; 15:1120–1127.
- Yeh HC, Brown TT, Maruthur N, et al. Comparative effectiveness and safety of methods of insulin delivery and glucose monitoring for diabetes mellitus: a systematic review and meta-analysis. Ann Intern Med 2012; 157:336–347.
- Schade DS, Valentine V. To pump or not to pump. Diabetes Care 2002; 25:2100–2102.
- Liu L, Lee MJ, Brateanu A. Improved A1C and lipid profile in patients referred to diabetes education programs in a wide health care network: a retrospective study. Diabetes Spectr 2014; 27:297–303.
- Funnell MM, Kruger DF, Spencer M. Self-management support for insulin therapy in type 2 diabetes. Diabetes Educ 2004; 30:274–280.
- Norris SL, Engelgau MM, Narayan KM. Effectiveness of self-management training in type 2 diabetes: a systematic review of randomized controlled trials. Diabetes Care 2001; 24:561–587.
Insulin therapy is one of the most effective tools clinicians can use to help patients reach their individualized hemoglobin A1c target. However, decisions about when and how to start insulin therapy have to be individualized to the needs and goals of each patient. Many insulin options are available, one of the most common being the addition of basal insulin to oral antidiabetic drugs. Although patients are often reluctant to start insulin, this reluctance can be overcome through patient education and hands-on training.
Here, we review hemoglobin A1c targets, factors that determine when to start insulin therapy, and the different regimens that can be used.
MOST PATIENTS EVENTUALLY NEED INSULIN
Type 2 diabetes mellitus is a chronic progressive disease associated with insulin resistance, beta-cell dysfunction, and decreased insulin secretion. Consequently, most patients eventually require insulin therapy to reduce the risk of long-term complications.
The efficacy of therapy can be assessed by measuring hemoglobin A1c, an important marker of the chronic hyperglycemic state. The hemoglobin A1c value can be reported as a ratio (%) standardized against the results of the Diabetes Control and Complications Trial,1 or as International Federation of Clinical Chemistry units (mmol/mol).2 Table 1 shows the relationship between hemoglobin A1c and average glucose values.3
WHAT IS AN APPROPRIATE HEMOGLOBIN A1c TARGET?
The short answer is, “It depends.”
Currently, the American Association of Clinical Endocrinologists (AACE) supports a hemoglobin A1c goal of less than 6.5% for otherwise healthy patients but states that the goal should be individualized for patients with concurrent illnesses or at risk of hypoglycemia.4
On the other hand, the American Diabetes Association (ADA) recommends a higher hemoglobin A1c target of less than 7% for most adults with type 2 diabetes mellitus.5 This value was shown to be associated with a reduction in the microvascular and macrovascular complications of diabetes.
Yet when three large trials6–8 recently compared intensive and standard glucose control regimens, tighter glucose control failed to improve cardiovascular outcomes. Moreover, in one of the trials,7 patients receiving intensive treatment had a higher rate of all-cause mortality. Details:
- Action in Diabetes and Vascular Disease (ADVANCE): 11,140 patients; average hemoglobin A1c levels 6.5% vs 7.3%6
- Action to Control Cardiovascular Risk in Diabetes (ACCORD): 10,251 patients; average hemoglobin A1c levels 6.4% vs 7.5%7
- Veterans Affairs Diabetes Trial (VADT): 1,791 patients; average hemoglobin A1c levels 6.9% vs 8.4%.8
Similarly, a 2013 Cochrane review9 that included 28 randomized controlled trials concluded that intensive control (in 18,717 patients) did not decrease all-cause and cardiovascular mortality rates compared with traditional glucose control (in 16,195 patients), and it increased the risk of hypoglycemia and serious adverse events.
As a result, the ADA5 states that a hemoglobin A1c target less than 6.5% is optional for patients with a long life expectancy, short duration of diabetes, low risk of hypoglycemia, and no significant cardiovascular disease. The ADA further defines a hemoglobin A1c goal of less than 8% for patients with a history of severe hypoglycemia, limited life expectancy, advanced microvascular or macrovascular complications, extensive comorbid conditions, and long-standing diabetes.
Therefore, the AACE and ADA are moving away from “one-size-fits-all” goals and toward individualizing their recommendations.
WHEN SHOULD INSULIN BE STARTED?
Physicians should consider the needs and preferences of each patient and individualize the treatment. The most recent recommendations from the ADA5 stress the importance of a patient-centered approach, with multiple factors taken into account. These include the patient’s attitude, expected compliance with treatment, risk of hypoglycemia, disease duration, life expectancy, and comorbidities, and the side effects of oral medications and insulin.
Compared with previous guidelines, there are fewer rules on how and when to start insulin therapy. But absolute and relative indications for insulin therapy should be considered in patients with the following:
Absolute indications for insulin
- Ketoacidosis or catabolic symptoms, including ketonuria
- Newly diagnosed type 2 diabetes with pronounced hyperglycemia (glucose ≥ 300 mg/dL or hemoglobin A1c ≥ 10.0%) with or without severe symptoms, including weight loss, polyuria, or polydipsia10
- Uncontrolled type 2 diabetes mellitus despite using one, two, or more oral antidiabetic drugs or glucagon-like peptide 1 (GLP-1) receptor agonists
- Gestational diabetes
- Preference for insulin.
Relative indications for insulin
- Hospitalized for surgery or acute illnesses
- Advanced renal or hepatic disease
- Inability to afford the cost or tolerate the side effects of oral antidiabetic drugs and GLP-1 receptor agonists.
Depending on the situation, blood glucose is measured fasting, before meals, or after meals after initiating or adjusting insulin regimens (Table 2).
WHAT ARE THE INSULIN REGIMENS?
Basal insulin
In the early stages of type 2 diabetes, metformin alone or in combination with another oral antidiabetic drug or with a GLP-1 receptor agonist is often used along with healthy eating, weight control, and increased physical activity.
When the target hemoglobin A1c cannot be achieved with one or two noninsulin drugs, the ADA suggests basal insulin be added to metformin or a two-medication regimen that includes metformin (Table 3). However, recent evidence suggests that combining a GLP-1 receptor agonist with basal insulin, in a regimen without metformin, is safe and improves glycemic control without hypoglycemia or weight gain.11
While a total daily dose of insulin of 0.1 to 0.2 units/kg could be initially used in patients with a hemoglobin A1c level less than 8%, a higher dose of 0.2 to 0.3 units/kg is required if the hemoglobin A1c level is between 8% and 10%. The dose can be titrated once or twice weekly if the fasting glucose is above the target level (usually < 130 mg/dL). If hypoglycemia develops (glucose < 70 mg/dL), the insulin dose should be reduced by 10% to 20%.10
Available basal insulins include glargine, detemir, and neutral protamine Hagedorn (NPH) (Table 4).12–14 Because glargine and detemir offer better pharmacokinetic properties, less variability in response, and less risk of hypoglycemia, they are preferred over NPH. Glargine has a relatively constant plasma concentration over 24 hours, allowing once-daily dosing at any time during the day (Figure 1).15 The dose should be taken at the same time every day. Detemir and NPH are usually taken once or twice daily.
Patients treated once daily should take the dose with the evening meal or at bedtime. Patients who require a twice-daily regimen can take the first dose with breakfast and the second one with the evening meal, at bedtime, or 12 hours after the morning dose.
The randomized Treat-to-Target trial,16 in 756 patients, showed that both glargine and NPH, when added to oral therapy in patients with type 2 diabetes, achieve the target hemoglobin A1c, but NPH is associated with more episodes of nocturnal hypoglycemia. Similar results were found when NPH was compared with detemir insulin.17
A Cochrane review18 suggested that glargine and detemir are similar in efficacy and safety. However, detemir often needs to be injected twice daily, in a higher dose, and is associated with less weight gain. Furthermore, a meta-analysis of 46 randomized clinical trials19 showed that the weight increase at 1 year is less in patients treated with basal than with twice-daily or prandial regimens.
A noninterventional longitudinal study20 in 2,179 patients newly started on insulin showed that the mean weight increase at 1 year was 1.78 kg, and 24% of patients gained more than 5 kg. However, the factors independently associated with the weight gain were a higher hemoglobin A1c at baseline, a higher insulin dose at baseline and at 1 year, and a lower baseline body mass index, but not the type of insulin regimen.
Currently, a new class of ultralong-acting basal insulins is being studied. Insulins in this class are approved in other countries, but the US Food and Drug Administration requires additional data for approval. Ultralong-acting insulins are expected to reduce the risk of hypoglycemia, specifically the risk of nocturnal episodes. Also, given their longer duration of action and stable steady-state pharmacokinetics, they will offer flexibility in the dose timing.21
Basal-bolus regimens
Basal insulin often does not control postprandial hyperglycemia. The need for multiple doses of insulin (including one or more preprandial doses) is suggested by postprandial glucose values above target (usually > 180 mg/dL) or by a hemoglobin A1c above goal despite well-controlled fasting glucose levels. This usually becomes evident when the total daily dose of basal insulin exceeds 0.5 units/kg. Patients newly diagnosed with diabetes who have a hemoglobin A1c higher than 10% may also respond better to an initial basal-bolus regimen.
Available bolus insulins include lispro, aspart, glulisine, regular insulin, and the newly approved Technosphere inhaled regular insulin (Table 4).12–14 They can be taken before each meal, and the total bolus dose usually represents 50% of the total daily dose.22 Rapid-acting insulins have faster onset, shorter duration of action, and more predictable pharmacokinetics, which makes them preferable to regular insulin (Figure 1).15 Inhaled insulin is another option, but it is contraindicated in patients with chronic obstructive pulmonary disease or asthma because of the increased risk of acute bronchospasm.12
Alternatively, the transition to a basal-bolus regimen can be accomplished with a single dose of bolus insulin before the main meal, using a dose that represents approximately 10% of the total daily dose. Additional bolus doses can be added later based on the glycemic control. The adjustment of the preprandial insulin dose is done once or twice weekly, based on the postprandial glucose levels.10
Premixed combinations of long- and short-acting insulins in ratios of 50% to 50%, 70% to 30%, or 75% to 25% can be considered in patients who cannot adhere to a complex insulin regimen. A propensity-matched comparison of different insulin regimens (basal, premixed, mealtime plus basal, and mealtime) in patients with type 2 diabetes revealed that the hemoglobin A1c reduction was similar between the different groups.23 However, the number of hypoglycemic episodes was higher in the premixed insulin group, and the weight gain was less in the basal insulin group.
While premixed insulins require fewer injections, they do not provide dosing flexibility. In other words, dose adjustments for premixed insulins lead to increases in both basal and bolus amounts even though a dose adjustment is needed for only one insulin type. Thus, this is a common reason for increased hypoglycemic episodes.
Continuous subcutaneous insulin infusion
A meta-analysis showed that continuous subcutaneous insulin infusion (ie, use of an insulin pump) was similar to intensive therapy with multiple daily insulin injections in terms of glycemic control and hypoglycemia.24 Since both options can lead to similar glucose control, additional factors to consider when initiating insulin infusion include lifestyle and technical expertise. Some patients may or may not prefer having a pump attached for nearly all daily activities. Additionally, this type of therapy is complex and requires significant training to ensure efficacy and safety.25
WHAT IS THE COST OF INSULIN THERAPY?
A final factor to keep in mind when initiating insulin is cost (Table 4).12–14 Asking patients to check their prescription insurance formulary is important to ensure that an affordable option is selected. If patients do not have prescription insurance, medication assistance programs could be an option. However, if a patient is considering an insulin pump, insurance coverage is essential. Depending on the manufacturer, insulin pumps cost about $6,000 to $7,000, and the additional monthly supplies for the pump are also expensive.
If patients are engaged when considering and selecting insulin therapy, the likelihood of treatment success is greater.26–28
Insulin therapy is one of the most effective tools clinicians can use to help patients reach their individualized hemoglobin A1c target. However, decisions about when and how to start insulin therapy have to be individualized to the needs and goals of each patient. Many insulin options are available, one of the most common being the addition of basal insulin to oral antidiabetic drugs. Although patients are often reluctant to start insulin, this reluctance can be overcome through patient education and hands-on training.
Here, we review hemoglobin A1c targets, factors that determine when to start insulin therapy, and the different regimens that can be used.
MOST PATIENTS EVENTUALLY NEED INSULIN
Type 2 diabetes mellitus is a chronic progressive disease associated with insulin resistance, beta-cell dysfunction, and decreased insulin secretion. Consequently, most patients eventually require insulin therapy to reduce the risk of long-term complications.
The efficacy of therapy can be assessed by measuring hemoglobin A1c, an important marker of the chronic hyperglycemic state. The hemoglobin A1c value can be reported as a ratio (%) standardized against the results of the Diabetes Control and Complications Trial,1 or as International Federation of Clinical Chemistry units (mmol/mol).2 Table 1 shows the relationship between hemoglobin A1c and average glucose values.3
WHAT IS AN APPROPRIATE HEMOGLOBIN A1c TARGET?
The short answer is, “It depends.”
Currently, the American Association of Clinical Endocrinologists (AACE) supports a hemoglobin A1c goal of less than 6.5% for otherwise healthy patients but states that the goal should be individualized for patients with concurrent illnesses or at risk of hypoglycemia.4
On the other hand, the American Diabetes Association (ADA) recommends a higher hemoglobin A1c target of less than 7% for most adults with type 2 diabetes mellitus.5 This value was shown to be associated with a reduction in the microvascular and macrovascular complications of diabetes.
Yet when three large trials6–8 recently compared intensive and standard glucose control regimens, tighter glucose control failed to improve cardiovascular outcomes. Moreover, in one of the trials,7 patients receiving intensive treatment had a higher rate of all-cause mortality. Details:
- Action in Diabetes and Vascular Disease (ADVANCE): 11,140 patients; average hemoglobin A1c levels 6.5% vs 7.3%6
- Action to Control Cardiovascular Risk in Diabetes (ACCORD): 10,251 patients; average hemoglobin A1c levels 6.4% vs 7.5%7
- Veterans Affairs Diabetes Trial (VADT): 1,791 patients; average hemoglobin A1c levels 6.9% vs 8.4%.8
Similarly, a 2013 Cochrane review9 that included 28 randomized controlled trials concluded that intensive control (in 18,717 patients) did not decrease all-cause and cardiovascular mortality rates compared with traditional glucose control (in 16,195 patients), and it increased the risk of hypoglycemia and serious adverse events.
As a result, the ADA5 states that a hemoglobin A1c target less than 6.5% is optional for patients with a long life expectancy, short duration of diabetes, low risk of hypoglycemia, and no significant cardiovascular disease. The ADA further defines a hemoglobin A1c goal of less than 8% for patients with a history of severe hypoglycemia, limited life expectancy, advanced microvascular or macrovascular complications, extensive comorbid conditions, and long-standing diabetes.
Therefore, the AACE and ADA are moving away from “one-size-fits-all” goals and toward individualizing their recommendations.
WHEN SHOULD INSULIN BE STARTED?
Physicians should consider the needs and preferences of each patient and individualize the treatment. The most recent recommendations from the ADA5 stress the importance of a patient-centered approach, with multiple factors taken into account. These include the patient’s attitude, expected compliance with treatment, risk of hypoglycemia, disease duration, life expectancy, and comorbidities, and the side effects of oral medications and insulin.
Compared with previous guidelines, there are fewer rules on how and when to start insulin therapy. But absolute and relative indications for insulin therapy should be considered in patients with the following:
Absolute indications for insulin
- Ketoacidosis or catabolic symptoms, including ketonuria
- Newly diagnosed type 2 diabetes with pronounced hyperglycemia (glucose ≥ 300 mg/dL or hemoglobin A1c ≥ 10.0%) with or without severe symptoms, including weight loss, polyuria, or polydipsia10
- Uncontrolled type 2 diabetes mellitus despite using one, two, or more oral antidiabetic drugs or glucagon-like peptide 1 (GLP-1) receptor agonists
- Gestational diabetes
- Preference for insulin.
Relative indications for insulin
- Hospitalized for surgery or acute illnesses
- Advanced renal or hepatic disease
- Inability to afford the cost or tolerate the side effects of oral antidiabetic drugs and GLP-1 receptor agonists.
Depending on the situation, blood glucose is measured fasting, before meals, or after meals after initiating or adjusting insulin regimens (Table 2).
WHAT ARE THE INSULIN REGIMENS?
Basal insulin
In the early stages of type 2 diabetes, metformin alone or in combination with another oral antidiabetic drug or with a GLP-1 receptor agonist is often used along with healthy eating, weight control, and increased physical activity.
When the target hemoglobin A1c cannot be achieved with one or two noninsulin drugs, the ADA suggests basal insulin be added to metformin or a two-medication regimen that includes metformin (Table 3). However, recent evidence suggests that combining a GLP-1 receptor agonist with basal insulin, in a regimen without metformin, is safe and improves glycemic control without hypoglycemia or weight gain.11
While a total daily dose of insulin of 0.1 to 0.2 units/kg could be initially used in patients with a hemoglobin A1c level less than 8%, a higher dose of 0.2 to 0.3 units/kg is required if the hemoglobin A1c level is between 8% and 10%. The dose can be titrated once or twice weekly if the fasting glucose is above the target level (usually < 130 mg/dL). If hypoglycemia develops (glucose < 70 mg/dL), the insulin dose should be reduced by 10% to 20%.10
Available basal insulins include glargine, detemir, and neutral protamine Hagedorn (NPH) (Table 4).12–14 Because glargine and detemir offer better pharmacokinetic properties, less variability in response, and less risk of hypoglycemia, they are preferred over NPH. Glargine has a relatively constant plasma concentration over 24 hours, allowing once-daily dosing at any time during the day (Figure 1).15 The dose should be taken at the same time every day. Detemir and NPH are usually taken once or twice daily.
Patients treated once daily should take the dose with the evening meal or at bedtime. Patients who require a twice-daily regimen can take the first dose with breakfast and the second one with the evening meal, at bedtime, or 12 hours after the morning dose.
The randomized Treat-to-Target trial,16 in 756 patients, showed that both glargine and NPH, when added to oral therapy in patients with type 2 diabetes, achieve the target hemoglobin A1c, but NPH is associated with more episodes of nocturnal hypoglycemia. Similar results were found when NPH was compared with detemir insulin.17
A Cochrane review18 suggested that glargine and detemir are similar in efficacy and safety. However, detemir often needs to be injected twice daily, in a higher dose, and is associated with less weight gain. Furthermore, a meta-analysis of 46 randomized clinical trials19 showed that the weight increase at 1 year is less in patients treated with basal than with twice-daily or prandial regimens.
A noninterventional longitudinal study20 in 2,179 patients newly started on insulin showed that the mean weight increase at 1 year was 1.78 kg, and 24% of patients gained more than 5 kg. However, the factors independently associated with the weight gain were a higher hemoglobin A1c at baseline, a higher insulin dose at baseline and at 1 year, and a lower baseline body mass index, but not the type of insulin regimen.
Currently, a new class of ultralong-acting basal insulins is being studied. Insulins in this class are approved in other countries, but the US Food and Drug Administration requires additional data for approval. Ultralong-acting insulins are expected to reduce the risk of hypoglycemia, specifically the risk of nocturnal episodes. Also, given their longer duration of action and stable steady-state pharmacokinetics, they will offer flexibility in the dose timing.21
Basal-bolus regimens
Basal insulin often does not control postprandial hyperglycemia. The need for multiple doses of insulin (including one or more preprandial doses) is suggested by postprandial glucose values above target (usually > 180 mg/dL) or by a hemoglobin A1c above goal despite well-controlled fasting glucose levels. This usually becomes evident when the total daily dose of basal insulin exceeds 0.5 units/kg. Patients newly diagnosed with diabetes who have a hemoglobin A1c higher than 10% may also respond better to an initial basal-bolus regimen.
Available bolus insulins include lispro, aspart, glulisine, regular insulin, and the newly approved Technosphere inhaled regular insulin (Table 4).12–14 They can be taken before each meal, and the total bolus dose usually represents 50% of the total daily dose.22 Rapid-acting insulins have faster onset, shorter duration of action, and more predictable pharmacokinetics, which makes them preferable to regular insulin (Figure 1).15 Inhaled insulin is another option, but it is contraindicated in patients with chronic obstructive pulmonary disease or asthma because of the increased risk of acute bronchospasm.12
Alternatively, the transition to a basal-bolus regimen can be accomplished with a single dose of bolus insulin before the main meal, using a dose that represents approximately 10% of the total daily dose. Additional bolus doses can be added later based on the glycemic control. The adjustment of the preprandial insulin dose is done once or twice weekly, based on the postprandial glucose levels.10
Premixed combinations of long- and short-acting insulins in ratios of 50% to 50%, 70% to 30%, or 75% to 25% can be considered in patients who cannot adhere to a complex insulin regimen. A propensity-matched comparison of different insulin regimens (basal, premixed, mealtime plus basal, and mealtime) in patients with type 2 diabetes revealed that the hemoglobin A1c reduction was similar between the different groups.23 However, the number of hypoglycemic episodes was higher in the premixed insulin group, and the weight gain was less in the basal insulin group.
While premixed insulins require fewer injections, they do not provide dosing flexibility. In other words, dose adjustments for premixed insulins lead to increases in both basal and bolus amounts even though a dose adjustment is needed for only one insulin type. Thus, this is a common reason for increased hypoglycemic episodes.
Continuous subcutaneous insulin infusion
A meta-analysis showed that continuous subcutaneous insulin infusion (ie, use of an insulin pump) was similar to intensive therapy with multiple daily insulin injections in terms of glycemic control and hypoglycemia.24 Since both options can lead to similar glucose control, additional factors to consider when initiating insulin infusion include lifestyle and technical expertise. Some patients may or may not prefer having a pump attached for nearly all daily activities. Additionally, this type of therapy is complex and requires significant training to ensure efficacy and safety.25
WHAT IS THE COST OF INSULIN THERAPY?
A final factor to keep in mind when initiating insulin is cost (Table 4).12–14 Asking patients to check their prescription insurance formulary is important to ensure that an affordable option is selected. If patients do not have prescription insurance, medication assistance programs could be an option. However, if a patient is considering an insulin pump, insurance coverage is essential. Depending on the manufacturer, insulin pumps cost about $6,000 to $7,000, and the additional monthly supplies for the pump are also expensive.
If patients are engaged when considering and selecting insulin therapy, the likelihood of treatment success is greater.26–28
- The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med 1993; 329:977–986.
- Hanas R, John WG; International HbA1c Consensus Committee. 2013 Update on the worldwide standardization of the hemoglobin A1c measurement. Pediatr Diabetes 2014; 15:e1–e2.
- Nathan DM, Kuenen J, Borg R, Zheng H, Schoenfeld D, Heine RJ; A1c-Derived Average Glucose Study Group. Translating the A1C assay into estimated average glucose values. Diabetes Care 2008; 31:1473–1478.
- Garber AJ, Abrahamson MJ, Barzilay JI, et al; American Association of Clinical Endocrinologists. AACE comprehensive diabetes management algorithm 2013. Endocr Pract 2013; 19:327–336.
- American Diabetes Association. Standards of medical care in diabetes—2014. Diabetes Care 2014; 37(suppl 1):S14–S80.
- ADVANCE Collaborative Group; Patel A, MacMahon S, Chalmers J, et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med 2008; 358:2560–2572.
- Action to Control Cardiovascular Risk in Diabetes Study Group; Gerstein HC, Miller ME, Byington RP, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med 2008; 358:2545–2559.
- Duckworth W, Abraira C, Moritz T, et al; VADT Investigators. Glucose control and vascular complications in veterans with type 2 diabetes. N Engl J Med 2009; 360:129–139.
- Hemmingsen B, Lund SS, Gluud C, et al. Targeting intensive glycaemic control versus targeting conventional glycaemic control for type 2 diabetes mellitus. Cochrane Database Syst Rev 2013; 11:CD008143.
- Inzucchi SE, Bergenstal RM, Buse JB, et al; American Diabetes Association (ADA); European Association for the Study of Diabetes (EASD). Management of hyperglycemia in type 2 diabetes: a patient-centered approach: position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 2012; 35:1364–1379.
- Vora J, Bain SC, Damci T, et al. Incretin-based therapy in combination with basal insulin: a promising tactic for the treatment of type 2 diabetes. Diabetes Metab 2013; 39:6–15.
- Nuffer W, Trujillo JM, Ellis SL. Technosphere insulin (Afrezza): a new, inhaled prandial insulin. Ann Pharmacother 2015; 49:99–106.
- Pharmacist’s Letter/Prescriber’s Letter. Comparison of insulins and injectable diabetes meds. PL Detail-Document #281107 November 2012. www.PharmacistsLetter.com. Accessed July 2, 2015
- Lexicomp Online. www.wolterskluwercdi.com/lexicomp-online/. Accessed July 2, 2015.
- Hirsch IB. Insulin analogues. N Engl J Med 2005; 352:174-183.
- Riddle MC, Rosenstock J, Gerich J; Insulin Glargine 4002 Study Investigators. The treat-to-target trial: randomized addition of glargine or human NPH insulin to oral therapy of type 2 diabetic patients. Diabetes Care 2003; 26:3080–3086.
- Hermansen K, Davies M, Derezinski T, Martinez Ravn G, Clauson P, Home P. A 26-week, randomized, parallel, treat-to-target trial comparing insulin detemir with NPH insulin as add-on therapy to oral glucose-lowering drugs in insulin-naive people with type 2 diabetes. Diabetes Care 2006; 29:1269–1274.
- Swinnen SG, Simon AC, Holleman F, Hoekstra JB, Devries JH. Insulin detemir versus insulin glargine for type 2 diabetes mellitus. Cochrane Database Syst Rev 2011; 7:CD006383.
- Pontiroli AE, Miele L, Morabito A. Increase of body weight during the first year of intensive insulin treatment in type 2 diabetes: systematic review and meta-analysis. Diabetes Obes Metab 2011; 13:1008–1019.
- Balkau B, Home PD, Vincent M, Marre M, Freemantle N. Factors associated with weight gain in people with type 2 diabetes starting on insulin. Diabetes Care 2014; 37:2108–2113.
- Garber AJ. Will the next generation of basal insulins offer clinical advantages? Diabetes Obes Metab 2014; 16:483–491.
- Tamaki M, Shimizu T, Kanazawa A, et al. Effects of changes in basal/total daily insulin ratio in type 2 diabetes patients on intensive insulin therapy including insulin glargine (JUN-LAN Study 6). Diabetes Res Clin Pract 2008; 81:e1–e3.
- Freemantle N, Balkau B, Home PD. A propensity score matched comparison of different insulin regimens 1 year after beginning insulin in people with type 2 diabetes. Diabetes Obes Metab 2013; 15:1120–1127.
- Yeh HC, Brown TT, Maruthur N, et al. Comparative effectiveness and safety of methods of insulin delivery and glucose monitoring for diabetes mellitus: a systematic review and meta-analysis. Ann Intern Med 2012; 157:336–347.
- Schade DS, Valentine V. To pump or not to pump. Diabetes Care 2002; 25:2100–2102.
- Liu L, Lee MJ, Brateanu A. Improved A1C and lipid profile in patients referred to diabetes education programs in a wide health care network: a retrospective study. Diabetes Spectr 2014; 27:297–303.
- Funnell MM, Kruger DF, Spencer M. Self-management support for insulin therapy in type 2 diabetes. Diabetes Educ 2004; 30:274–280.
- Norris SL, Engelgau MM, Narayan KM. Effectiveness of self-management training in type 2 diabetes: a systematic review of randomized controlled trials. Diabetes Care 2001; 24:561–587.
- The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med 1993; 329:977–986.
- Hanas R, John WG; International HbA1c Consensus Committee. 2013 Update on the worldwide standardization of the hemoglobin A1c measurement. Pediatr Diabetes 2014; 15:e1–e2.
- Nathan DM, Kuenen J, Borg R, Zheng H, Schoenfeld D, Heine RJ; A1c-Derived Average Glucose Study Group. Translating the A1C assay into estimated average glucose values. Diabetes Care 2008; 31:1473–1478.
- Garber AJ, Abrahamson MJ, Barzilay JI, et al; American Association of Clinical Endocrinologists. AACE comprehensive diabetes management algorithm 2013. Endocr Pract 2013; 19:327–336.
- American Diabetes Association. Standards of medical care in diabetes—2014. Diabetes Care 2014; 37(suppl 1):S14–S80.
- ADVANCE Collaborative Group; Patel A, MacMahon S, Chalmers J, et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med 2008; 358:2560–2572.
- Action to Control Cardiovascular Risk in Diabetes Study Group; Gerstein HC, Miller ME, Byington RP, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med 2008; 358:2545–2559.
- Duckworth W, Abraira C, Moritz T, et al; VADT Investigators. Glucose control and vascular complications in veterans with type 2 diabetes. N Engl J Med 2009; 360:129–139.
- Hemmingsen B, Lund SS, Gluud C, et al. Targeting intensive glycaemic control versus targeting conventional glycaemic control for type 2 diabetes mellitus. Cochrane Database Syst Rev 2013; 11:CD008143.
- Inzucchi SE, Bergenstal RM, Buse JB, et al; American Diabetes Association (ADA); European Association for the Study of Diabetes (EASD). Management of hyperglycemia in type 2 diabetes: a patient-centered approach: position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 2012; 35:1364–1379.
- Vora J, Bain SC, Damci T, et al. Incretin-based therapy in combination with basal insulin: a promising tactic for the treatment of type 2 diabetes. Diabetes Metab 2013; 39:6–15.
- Nuffer W, Trujillo JM, Ellis SL. Technosphere insulin (Afrezza): a new, inhaled prandial insulin. Ann Pharmacother 2015; 49:99–106.
- Pharmacist’s Letter/Prescriber’s Letter. Comparison of insulins and injectable diabetes meds. PL Detail-Document #281107 November 2012. www.PharmacistsLetter.com. Accessed July 2, 2015
- Lexicomp Online. www.wolterskluwercdi.com/lexicomp-online/. Accessed July 2, 2015.
- Hirsch IB. Insulin analogues. N Engl J Med 2005; 352:174-183.
- Riddle MC, Rosenstock J, Gerich J; Insulin Glargine 4002 Study Investigators. The treat-to-target trial: randomized addition of glargine or human NPH insulin to oral therapy of type 2 diabetic patients. Diabetes Care 2003; 26:3080–3086.
- Hermansen K, Davies M, Derezinski T, Martinez Ravn G, Clauson P, Home P. A 26-week, randomized, parallel, treat-to-target trial comparing insulin detemir with NPH insulin as add-on therapy to oral glucose-lowering drugs in insulin-naive people with type 2 diabetes. Diabetes Care 2006; 29:1269–1274.
- Swinnen SG, Simon AC, Holleman F, Hoekstra JB, Devries JH. Insulin detemir versus insulin glargine for type 2 diabetes mellitus. Cochrane Database Syst Rev 2011; 7:CD006383.
- Pontiroli AE, Miele L, Morabito A. Increase of body weight during the first year of intensive insulin treatment in type 2 diabetes: systematic review and meta-analysis. Diabetes Obes Metab 2011; 13:1008–1019.
- Balkau B, Home PD, Vincent M, Marre M, Freemantle N. Factors associated with weight gain in people with type 2 diabetes starting on insulin. Diabetes Care 2014; 37:2108–2113.
- Garber AJ. Will the next generation of basal insulins offer clinical advantages? Diabetes Obes Metab 2014; 16:483–491.
- Tamaki M, Shimizu T, Kanazawa A, et al. Effects of changes in basal/total daily insulin ratio in type 2 diabetes patients on intensive insulin therapy including insulin glargine (JUN-LAN Study 6). Diabetes Res Clin Pract 2008; 81:e1–e3.
- Freemantle N, Balkau B, Home PD. A propensity score matched comparison of different insulin regimens 1 year after beginning insulin in people with type 2 diabetes. Diabetes Obes Metab 2013; 15:1120–1127.
- Yeh HC, Brown TT, Maruthur N, et al. Comparative effectiveness and safety of methods of insulin delivery and glucose monitoring for diabetes mellitus: a systematic review and meta-analysis. Ann Intern Med 2012; 157:336–347.
- Schade DS, Valentine V. To pump or not to pump. Diabetes Care 2002; 25:2100–2102.
- Liu L, Lee MJ, Brateanu A. Improved A1C and lipid profile in patients referred to diabetes education programs in a wide health care network: a retrospective study. Diabetes Spectr 2014; 27:297–303.
- Funnell MM, Kruger DF, Spencer M. Self-management support for insulin therapy in type 2 diabetes. Diabetes Educ 2004; 30:274–280.
- Norris SL, Engelgau MM, Narayan KM. Effectiveness of self-management training in type 2 diabetes: a systematic review of randomized controlled trials. Diabetes Care 2001; 24:561–587.
KEY POINTS
- In deciding a patient’s hemoglobin A1c goal and whether it is time to start insulin therapy, one should take into account the patient’s age, life expectancy, concurrent illnesses, risk of hypoglycemia, and other factors.
- When the target hemoglobin A1c is not achieved with metformin or a two-drug regimen that includes metformin, the American Diabetes Association recommends adding a daily dose of basal insulin.
- Eventually, preprandial bolus doses may need to be added to the insulin regimen to control postprandial blood glucose levels and hemoglobin A1c.
Remembering that old dogs can still do tricks

More and more we are realizing that we need trials that use hard clinical end points to inform our clinical practice. Several things we used to do based on observational studies have fallen from grace after being evaluated in interventional trials. And faced with the US Food and Drug Administration’s mandate to demonstrate clinical impact, pharmaceutical companies can rarely count on using even well-accepted biomarkers instead of clinical outcomes when trying to bring new drugs to market.
This atmosphere often makes us a bit uncomfortable when prescribing older drugs that have passed the test of time and collective anecdotal experience but not rigorous clinical testing. In some cases this is good, and robust evaluation provides greater confidence in our choice of therapy: witness the demise of digoxin for heart failure.
Many older drugs have never been compared with newer drugs in well-designed trials using hard clinical outcomes and likely never will, owing to cost, marketing, and logistic reasons. But sometimes these trials are done, and the results are surprising. For instance, methotrexate in appropriate doses may actually be comparable to newer and far more expensive tumor necrosis factor inhibitors when used to treat rheumatoid arthritis.
Should we be willing to sometimes accept data on surrogate markers (eg, low-density lipoprotein cholesterol levels, blood pressure, hemoglobin A1c ) or even extensive clinical experience in the absence of hard outcome data when using older, tried-and-true drugs? Markers can mislead: consider the higher number of deaths recorded in the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial in the group receiving more aggressive control of their glucose levels.
So we should not be totally sanguine when using older drugs instead of newer ones. But some drugs may have slipped out of our mental formularies yet still have real value in niche or even common settings. Methyldopa remains an effective antihypertensive drug and may be especially useful in peripartum patients. Yet relatively few young physicians know the drug.
And so it may be with chlorthalidone. In this issue of the Journal, Cooney et al remind us not only that this drug is still around, but that it has proven efficacy and, compared with its more popular cousin hydrochlorothiazide, favorable pharmacokinetic properties such as longer action. Not to mention that it was a comparator drug in the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack (ALLHAT) trial.
In our current cost-saving environment, we should remember that some old dogs can still do good tricks.
More and more we are realizing that we need trials that use hard clinical end points to inform our clinical practice. Several things we used to do based on observational studies have fallen from grace after being evaluated in interventional trials. And faced with the US Food and Drug Administration’s mandate to demonstrate clinical impact, pharmaceutical companies can rarely count on using even well-accepted biomarkers instead of clinical outcomes when trying to bring new drugs to market.
This atmosphere often makes us a bit uncomfortable when prescribing older drugs that have passed the test of time and collective anecdotal experience but not rigorous clinical testing. In some cases this is good, and robust evaluation provides greater confidence in our choice of therapy: witness the demise of digoxin for heart failure.
Many older drugs have never been compared with newer drugs in well-designed trials using hard clinical outcomes and likely never will, owing to cost, marketing, and logistic reasons. But sometimes these trials are done, and the results are surprising. For instance, methotrexate in appropriate doses may actually be comparable to newer and far more expensive tumor necrosis factor inhibitors when used to treat rheumatoid arthritis.
Should we be willing to sometimes accept data on surrogate markers (eg, low-density lipoprotein cholesterol levels, blood pressure, hemoglobin A1c ) or even extensive clinical experience in the absence of hard outcome data when using older, tried-and-true drugs? Markers can mislead: consider the higher number of deaths recorded in the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial in the group receiving more aggressive control of their glucose levels.
So we should not be totally sanguine when using older drugs instead of newer ones. But some drugs may have slipped out of our mental formularies yet still have real value in niche or even common settings. Methyldopa remains an effective antihypertensive drug and may be especially useful in peripartum patients. Yet relatively few young physicians know the drug.
And so it may be with chlorthalidone. In this issue of the Journal, Cooney et al remind us not only that this drug is still around, but that it has proven efficacy and, compared with its more popular cousin hydrochlorothiazide, favorable pharmacokinetic properties such as longer action. Not to mention that it was a comparator drug in the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack (ALLHAT) trial.
In our current cost-saving environment, we should remember that some old dogs can still do good tricks.
More and more we are realizing that we need trials that use hard clinical end points to inform our clinical practice. Several things we used to do based on observational studies have fallen from grace after being evaluated in interventional trials. And faced with the US Food and Drug Administration’s mandate to demonstrate clinical impact, pharmaceutical companies can rarely count on using even well-accepted biomarkers instead of clinical outcomes when trying to bring new drugs to market.
This atmosphere often makes us a bit uncomfortable when prescribing older drugs that have passed the test of time and collective anecdotal experience but not rigorous clinical testing. In some cases this is good, and robust evaluation provides greater confidence in our choice of therapy: witness the demise of digoxin for heart failure.
Many older drugs have never been compared with newer drugs in well-designed trials using hard clinical outcomes and likely never will, owing to cost, marketing, and logistic reasons. But sometimes these trials are done, and the results are surprising. For instance, methotrexate in appropriate doses may actually be comparable to newer and far more expensive tumor necrosis factor inhibitors when used to treat rheumatoid arthritis.
Should we be willing to sometimes accept data on surrogate markers (eg, low-density lipoprotein cholesterol levels, blood pressure, hemoglobin A1c ) or even extensive clinical experience in the absence of hard outcome data when using older, tried-and-true drugs? Markers can mislead: consider the higher number of deaths recorded in the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial in the group receiving more aggressive control of their glucose levels.
So we should not be totally sanguine when using older drugs instead of newer ones. But some drugs may have slipped out of our mental formularies yet still have real value in niche or even common settings. Methyldopa remains an effective antihypertensive drug and may be especially useful in peripartum patients. Yet relatively few young physicians know the drug.
And so it may be with chlorthalidone. In this issue of the Journal, Cooney et al remind us not only that this drug is still around, but that it has proven efficacy and, compared with its more popular cousin hydrochlorothiazide, favorable pharmacokinetic properties such as longer action. Not to mention that it was a comparator drug in the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack (ALLHAT) trial.
In our current cost-saving environment, we should remember that some old dogs can still do good tricks.
Diuretics for hypertension: Hydrochlorothiazide or chlorthalidone?
The thiazide diuretic hydrochlorothiazide and the thiazidelike diuretic chlorthalidone are two old drugs that are still useful. Although similar, they differ in important ways still not fully appreciated more than a half century after they were introduced.
Most hypertension guidelines recommend thiazide diuretics as one of the classes of agents that can be used either as initial antihypertensive drug therapy or as part of combination therapy.1–3
In the United States, hydrochlorothiazide is used more often than chlorthalidone, but many clinical trials of antihypertensive therapy have used chlorthalidone.4,5 In recent years, particularly after the publication of the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT), interest in chlorthalidone has been increasing, and new data are now available comparing these two diuretics.6 While current US guidelines do not recommend one over the other, British guidelines prefer chlorthalidone.7
This review summarizes the data comparing the two drugs’ pharmacology, antihypertensive effect, and impact on clinical outcomes to help guide clinicians in choosing antihypertensive drug therapy.
PHARMACOLOGY AND MECHANISM OF ACTION
Many of the differences in effectiveness and adverse effects of hydrochlorothiazide and chlorthalidone are thought to be due to their different pharmacodynamic and pharmacokinetic effects.
Pharmacodynamic effects
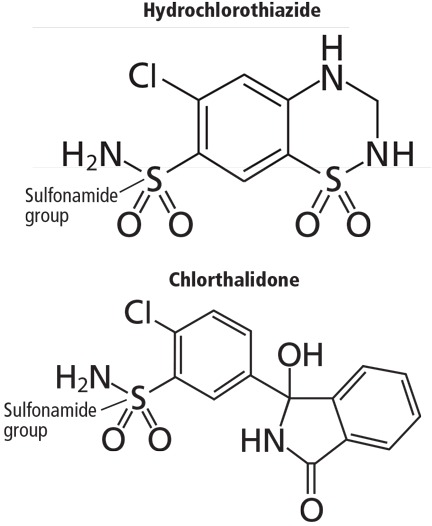
Hydrochlorothiazide and chlorthalidone differ significantly in chemical structure (Figure 1), but both contain a sulfonamide group that inhibits carbonic anhydrase activity, which may be associated with lower vascular contractility. Both drugs are concentrated in the kidney and secreted into the tubular lumen8; therefore, their therapeutic diuretic effects are often achieved with relatively low plasma concentrations.
Both drugs inhibit the sodium-chloride cotransporter in the luminal membrane of the distal convoluted tubule of the ascending loop of Henle, leading to a modest natriuresis and diuresis. The exact mechanism by which they lower blood pressure is not known: while the initial response is from diuresis and volume changes, long-term reduction in blood pressure is through uncertain mechanisms. In addition, chlorthalidone may have beneficial effects on endothelial function and oxidative stress.9,10
Both drugs also increase secretion of potassium and hydrogen ions and promote increased reabsorption of calcium through increased expression of a sodium-calcium exchange channel.8 Chlorthalidone may cause more inhibition of carbonic anhydrase than hydrochlorothiazide, which can lead to lower intracellular pH and cell volume. This effect may in part explain a pleiotropic effect of chlorthalidone, ie, inhibition of platelet function, which in turn may contribute to this drug’s beneficial effect on cardiovascular outcomes.9
Pharmacokinetic differences

Hydrochlorothiazide and chlorthalidone have important differences in their pharmacokinetic properties (Table 1).11
Hydrochlorothiazide has its onset of action in about 2 hours, and it reaches its peak in 4 to 6 hours. Though its duration of action is short—up to 12 hours—its pharmacodynamic response can be much longer than predicted by its kinetics, allowing once-daily dosing.8
Chlorthalidone has a longer duration of action than hydrochlorothiazide. This may be because it has a very high volume of distribution, since it is taken up into red blood cells and is bound to carbonic anhydrase.12 This may result in a “drug reservoir” that keeps drug levels higher for a longer time.13 Its long duration of action makes it a favorable choice for patients who have difficulty adhering to medication instructions. In addition, a missed dose is unlikely to have a “rebound” effect like that seen with some other antihypertensive agents. However, both chlorthalidone and hydrochlorothiazide are effective if taken once daily.
BLOOD PRESSURE-LOWERING
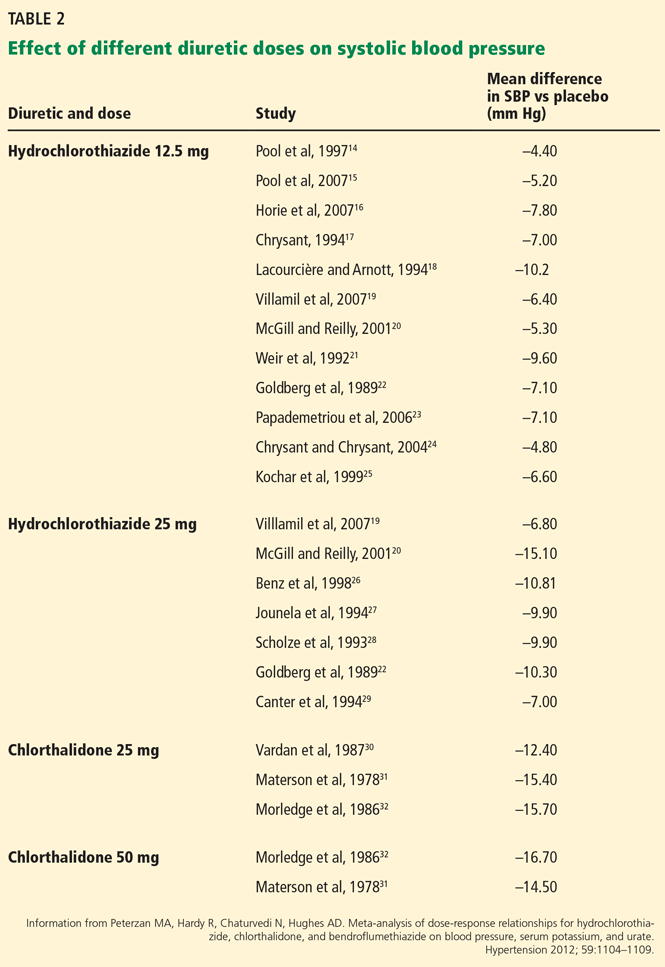
Both hydrochlorothiazide and chlorthalidone are effective antihypertensive agents. Table 2 summarizes findings from studies that evaluated their blood pressure-lowering effect at various doses.14–33 However, relatively few studies have directly compared these two agents’ effects on blood pressure.
Ernst et al,34 in a small study (but probably the best one to address this issue), compared chlorthalidone 12.5 mg/day (force-titrated to 25 mg/day) and hydrochlorothiazide 25 mg/day (force-titrated to 50 mg/day) in untreated hypertensive patients. After 8 weeks, ambulatory blood pressure monitoring indicated a greater reduction from baseline in systolic blood pressure with chlorthalidone 25 mg/day than with hydrochlorothiazide 50 mg/day (24-hour mean –12.4 vs –7.4 mm Hg, P = .05). Interestingly, the change in nighttime blood pressure was greater in the chlorthalidone group (–13.5 mm Hg) than in the hydrochlorothiazide group (–6.4 mm Hg; P = .009). These data suggest that at the doses studied, chlorthalidone is more effective than hydrochlorothiazide in lowering systolic blood pressure.
Bakris et al,35 using a different study design, compared the single-pill combination of azilsartan medoxomil and chlorthalidone vs coadministration of azilsartan medoxomil and hydrochlorothiazide in participants with stage 2 primary hypertension (≥ 160/100 mm Hg). Systolic blood pressure, as measured in the clinic, declined more with the chlorthalidone combination (–35.1 mm Hg) than with the hydrochlorothiazide combination (–29.5 mm Hg, mean difference –5.6 mm Hg, P < .001).
Meta-analyses also support the conclusion that chlorthalidone is more potent than hydrochlorothiazide in lowering blood pressure.35,36 Several studies have shown that chlorthalidone at the same dose is 1.5 to 2 times as potent as hydrochlorothiazide.33,36,37 Therefore, for clinical purposes, it is reasonable to consider chlorthalidone 12.5 mg daily as similar to 25 mg of hydrochlorothiazide daily.
ADVERSE EFFECTS
Electrolyte disturbances are a common adverse effect of thiazide diuretics.
Hypokalemia. All thiazide diuretics cause potassium wasting. The frequency of hypokalemia depends on the dose, frequency of administration, diet, and other pharmacologic agents used.
Two large clinical trials, the Systolic Hypertension in the Elderly Program and ALLHAT, found that chlorthalidone caused hypokalemia requiring therapy in about 6% to 8% of patients.38,39 Chlorthalidone therapy was associated with a lowering of serum potassium levels of 0.2 to 0.5 mmol/L.36 In ALLHAT, chlorthalidone was associated with a reduction in potassium levels of approximately 0.2 mmol/L after 4 years.38
All diuretics require monitoring of electrolytes, especially during the first 2 weeks of therapy. Once a steady state is reached, patients are not usually at risk of hypokalemia unless the dose is increased, extrarenal losses of potassium increase, or dietary potassium is reduced.
Other electrolyte changes. Thiazide and thiazide-like diuretics can cause other metabolic and endocrine abnormalities such as hypochloremic alkalosis, hyponatremia, and hypercalcemia.40,41 They can also cause photosensitivity and can precipitate gout.42
Observational studies have suggested that metabolic adverse effects such as hypokalemia and hyperuricemia are more common with chlorthalidone than with hydrochlorothiazide.43 It is prudent to monitor laboratory values periodically in patients on diuretic therapy.
DRUG INTERACTIONS
The drug interaction profiles of hydrochlorothiazide and chlorthalidone are also similar. The most common interactions are pharmacodynamic interactions resulting from potassium depletion caused by the diuretics.
Antiarrythymic drugs. Hypokalemia is a risk factor for arrhythmias such as torsades de pointes, and the risk is magnified with concomitant therapy with antiarrhythmic agents that prolong the QT interval independently of serum potassium concentration (eg, sotalol, dronedarone, ibutilide, propafenone). Therefore, combinations of drugs that can cause hypokalemia (eg, diuretics) and antiarrhythmic agents require vigilant monitoring of potassium and appropriate replenishment.44
Dofetilide is a class III antiarrhythmic agent and, like other antiarrhythmic drugs, carries a risk of QT prolongation and torsades de pointes, which is magnified by hypokalemia.45 In addition, dofetilide undergoes active tubular secretion in the kidney via the cation transport system, which is inhibited by hydrochlorothiazide.45 The resulting increase in plasma concentrations of dofetilide may magnify the risk of arrhythmias. Chlorthalidone has not been specifically studied in combination with dofetilide, but thiazide diuretics in general are thought to have a similar effect on tubular secretion and, therefore, should be considered similar to hydrochlorothiazide in this regard.
Digoxin. Similarly, digoxin toxicity may be enhanced in hypokalemia. As with antiarrhythmic agents, serum potassium should be carefully monitored and replenished appropriately when diuretics are used in combination with digoxin.
Lithium is reabsorbed in the proximal tubule along with sodium. Diuretics including hydrochlorothiazide and chlorthalidone that alter sodium reabsorption can also alter lithium absorption.46 When sodium reabsorption is decreased, lithium ion reabsorption is increased and may result in lithium toxicity. Although this combination is not contraindicated, monitoring of serum lithium concentrations and clinical signs and symptoms of lithium toxicity is recommended during initiation and dose adjustments of thiazide diuretics.
Nonsteroidal anti-inflammatory drugs can decrease the natriuretic, diuretic, and antihypertensive effects of both hydrochlorothiazide and chlorthalidone.47
Renin-angiotensin-aldosterone system antagonists, ie, angiotensin-converting enzyme inhibitors, angiotensin II receptor blockers, and the renin inhibitor aliskiren, have potentially beneficial interactions with hydrochlorothiazide and chlorthalidone, producing additive decreases in blood pressure when coadministered with these diuretics. These effects may be particularly potent early in concomitant therapy and allow use of lower doses of diuretics, typically 12.5 mg of hydrochlorothiazide in combination therapy.
LONG-TERM EFFECTS ON CARDIOVASCULAR EVENTS
The long-term goal in treating hypertension is to lower the risk of cardiovascular disease. Therefore, the clinician needs to consider the effect of antihypertensive drug therapy on long-term clinical outcomes.
Antihypertensive drug therapy based on thiazide diuretics has been shown to lower cardiovascular risk when compared with placebo.48 In addition, the effect of chlorthalidone-based antihypertensive therapy was similar to that of other antihypertensive drug classes in preventing most cardiovascular outcomes in ALLHAT.4
However, no study has directly compared hydrochlorothiazide and chlorthalidone with the primary outcome of reduction in long-term cardiovascular events. The data to date come from observational studies and meta-analyses. For example, in a retrospective analysis of the Multiple Risk Factor Intervention Trial, cardiovascular events were significantly fewer in those receiving chlorthalidone vs hydrochlorothiazide (P = .0016).43
In a systematic review and meta-analysis, chlorthalidone was associated with a 23% lower risk of heart failure and a 21% lower risk of all cardiovascular events.49
However, a Canadian observational study of 29,873 patients found no difference in the composite outcome of death or hospitalization for heart failure, stroke, or myocardial infarction between chlorthalidone recipients (3.2 events per 100 person-years) and hydrochlorothiazide recipients (3.4 events per 100 person-years; adjusted hazard ratio 0.93, 95% confidence interval 0.81–1.06).50
In summary, it is unclear whether chlorthalidone or hydrochlorothiazide is superior in preventing cardiovascular events.
SUMMARY
Thiazide and thiazidelike diuretics play an important role in managing hypertension in most patients. The eighth Joint National Committee guidelines do not recommend either hydrochlorothiazide or chlorthalidone over the other. The target dose recommendations are hydrochlorothiazide 25 to 50 mg or chlorthalidone 12.5 to 25 mg daily, with lower doses considered for the elderly.
There are important differences between hydrochlorothiazide and chlorthalidone in pharmacology, potency, and frequency of metabolic side effects. Clinicians should consider these factors to tailor the choice of thiazide diuretic therapy in hypertensive patients.
- James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 2014; 311:507–520.
- Dasgupta K, Quinn RR, Zarnke KB, et al; Canadian Hypertension Education Program. The 2014 Canadian Hypertension Education Program recommendations for blood pressure measurement, diagnosis, assessment of risk, prevention, and treatment of hypertension. Can J Cardiol 2014; 30:485–501.
- Mancia G, Fagard R, Narkiewicz K, et al. 2013 ESH/ESC guidelines for the management of arterial hypertension: the Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). Eur Heart J 2013; 34:2159–2219.
- ALLHAT Officers and Coordinators for the ALLHAT Collaborative Research Group; The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial. Major outcomes in high-risk hypertensive patients randomized to angiotensin-converting enzyme inhibitor or calcium channel blocker vs diuretic: the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). JAMA 2002; 288:2981–2997.
- Prevention of stroke by antihypertensive drug treatment in older persons with isolated systolic hypertension. Final results of the Systolic Hypertension in the Elderly Program (SHEP). SHEP Cooperative Research Group. JAMA 1991; 265:3255–3264.
- Roush GC, Kaur R, Ernst ME. Diuretics: a review and update. J Cardiovasc Pharmacol Ther 2014; 19:5–13.
- McCormack T, Krause T, O’Flynn N. Management of hypertension in adults in primary care: NICE guideline. Br J Gen Pract 2012; 62:163–164.
- Bhattacharaya M, Alper SL. Pharmacology of volume regulation. In: Golan DE, Tashjian AH Jr, Armstrong EJ, Armstrong AW, editors. Principles of Pharmacology: The pathophysiologic Basis of Drug Therapy. 3rd ed. Philadelphia, PA: Wolters Kluwer Health/Lippincott Williams & Wilkins; 2012:332–352.
- Woodman R, Brown C, Lockette W. Chlorthalidone decreases platelet aggregation and vascular permeability and promotes angiogenesis. Hypertension 2010; 56:463–470.
- Sato K, Dohi Y, Kojima M, Takase H, Suzuki S, Ito S. Antioxidative effects of thiazide diuretics in refractory hypertensive patients. A randomized crossover trial of chlortalidone and trichlormethiazide. Arzneimittelforschung 2010; 60:612–616.
- US National Library of Medicine. Dailymed. dailymed.nlm.nih.gov. Accessed May 14, 2015.
- Collste P, Garle M, Rawlins MD, Sjöqvist F. Interindividual differences in chlorthalidone concentration in plasma and red cells of man after single and multiple doses. Eur J Clin Pharmacol 1976; 9:319–325.
- Roush GC, Buddharaju V, Ernst ME, Holford TR. Chlorthalidone: mechanisms of action and effect on cardiovascular events. Curr Hypertens Rep 2013; 15:514–521.
- Pool JL, Cushman WC, Saini RK, Nwachuku CE, Battikha JP. Use of the factorial design and quadratic response surface models to evaluate the fosinopril and hydrochlorothiazide combination therapy in hypertension. Am J Hypertens 1997; 10:117–123.
- Pool JL, Glazer R, Weinberger M, Alvarado R, Huang J, Graff A. Comparison of valsartan/hydrochlorothiazide combination therapy at doses up to 320/25 mg versus monotherapy: a double-blind, placebo-controlled study followed by long-term combination therapy in hypertensive adults. Clin Ther 2007; 29:61–73.
- Horie Y, Higaki J, Takeuchi M. Design, statistical analysis and sample size calculation of dose response study of telmisartan and hydrochlorothiazide. Contemp Clin Trials 2007; 28:647–653.
- Chrysant SG. Antihypertensive effectiveness of low-dose lisinopril-hydrochlorothiazide combination. A large multicenter study. Lisinopril-Hydrochlorothiazide Group. Arch Intern Med 1994; 154:737–743.
- Lacourcière Y, Arnott W. Placebo-controlled comparison of the effects of nebivolol and low-dose hydrochlorothiazide as monotherapies and in combination on blood pressure and lipid profile in hypertensive patients. J Hum Hypertens 1994; 8:283–288.
- Villamil A, Chrysant SG, Calhoun D, et al. Renin inhibition with aliskiren provides additive antihypertensive efficacy when used in combination with hydrochlorothiazide. J Hypertens 2007; 25:217–226.
- McGill JB, Reilly PA. Telmisartan plus hydrochlorothiazide versus telmisartan or hydrochlorothiazide monotherapy in patients with mild to moderate hypertension: a multicenter, randomized, double-blind, placebo-controlled, parallel-group trial. Clin Ther 2001; 23:833–850.
- Weir MR, Weber MA, Punzi HA, Serfer HM, Rosenblatt S, Cady WJ. A dose escalation trial comparing the combination of diltiazem SR and hydrochlorothiazide with the monotherapies in patients with essential hypertension. J Hum Hypertens 1992; 6:133–138.
- Goldberg MR, Rockhold FW, Offen WW, Dornseif BE. Dose-effect and concentration-effect relationships of pinacidil and hydrochlorothiazide in hypertension. Clin Pharmacol Ther 1989; 46:208–218.
- Papademetriou V, Hainer JW, Sugg J, Munzer D; ATTACH Study Group. Factorial antihypertensive study of an extended-release metoprolol and hydrochlorothiazide combination. Am J Hypertens 2006; 19:1217–1225.
- Chrysant SG, Chrysant GS. Antihypertensive efficacy of olmesartan medoxomil alone and in combination with hydrochlorothiazide. Expert Opin Pharmacother 2004; 5:657–667.
- Kochar M, Guthrie R, Triscari J, Kassler-Taub K, Reeves RA. Matrix study of irbesartan with hydrochlorothiazide in mild-to-moderate hypertension. Am J Hypertens 1999; 12:797–805.
- Benz JR, Black HR, Graff A, Reed A, Fitzsimmons S, Shi Y. Valsartan and hydrochlorothiazide in patients with essential hypertension. A multiple dose, double-blind, placebo controlled trial comparing combination therapy with monotherapy. J Hum Hypertens 1998; 12:861–866.
- Jounela AJ, Lilja M, Lumme J, et al. Relation between low dose of hydrochlorothiazide, antihypertensive effect and adverse effects. Blood Press 1994; 3:231–235.
- Scholze J, Breitstadt A, Cairns V, et al. Short report: ramipril and hydrochlorothiazide combination therapy in hypertension: a clinical trial of factorial design. East Germany Collaborative Trial Group. J Hypertens 1993; 11:217–221.
- Canter D, Frank GJ, Knapp LE, Phelps M, Quade M, Texter M. Quinapril and hydrochlorothiazide combination for control of hypertension: assessment by factorial design. Quinapril Investigator Group. J Hum Hypertens 1994; 8:155–162.
- Vardan S, Mehrotra KG, Mookherjee S, Willsey GA, Gens JD, Green DE. Efficacy and reduced metabolic side effects of a 15-mg chlorthalidone formulation in the treatment of mild hypertension. A multicenter study. JAMA 1987; 258:484–488.
- Materson BJ, Oster JR, Michael UF, et al. Dose response to chlorthalidone in patients with mild hypertension. Efficacy of a lower dose. Clin Pharmacol Ther 1978; 24:192–198.
- Morledge JH, Ettinger B, Aranda J, et al. Isolated systolic hypertension in the elderly. A placebo-controlled, dose-response evaluation of chlorthalidone. J Am Geriatr Soc 1986; 34:199–206.
- Peterzan MA, Hardy R, Chaturvedi N, Hughes AD. Meta-analysis of dose-response relationships for hydrochlorothiazide, chlorthalidone, and bendroflumethiazide on blood pressure, serum potassium, and urate. Hypertension 2012; 59:1104–1109.
- Ernst ME, Carter BL, Goerdt CJ, et al. Comparative antihypertensive effects of hydrochlorothiazide and chlorthalidone on ambulatory and office blood pressure. Hypertension 2006; 47:352–358.
- Bakris GL, Sica D, White WB, et al. Antihypertensive efficacy of hydrochlorothiazide vs chlorthalidone combined with azilsartan medoxomil. Am J Med 2012; 25:1229.e1–1229.e10.
- Ernst ME, Carter BL, Zheng S, Grimm RH Jr. Meta-analysis of dose-response characteristics of hydrochlorothiazide and chlorthalidone: effects on systolic blood pressure and potassium. Am J Hypertens 2010; 23:440–446.
- Carter BL, Ernst ME, Cohen JD. Hydrochlorothiazide versus chlorthalidone: evidence supporting their interchangeability. Hypertension 2004; 43:4–9.
- Alderman MH, Piller LB, Ford CE, et al; Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial Collaborative Research Group. Clinical significance of incident hypokalemia and hyperkalemia in treated hypertensive patients in the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial. Hypertension 2012; 59:926–933.
- Franse LV, Pahor M, Di Bari M, Somes GW, Cushman WC, Applegate WB. Hypokalemia associated with diuretic use and cardiovascular events in the Systolic Hypertension in the Elderly Program. Hypertension 2000; 35:1025–1030.
- Egom EE, Chirico D, Clark AL. A review of thiazide-induced hyponatraemia. Clin Med 2011; 11:448–451.
- Palmer BF. Metabolic complications associated with use of diuretics. Semin Nephrol 2011; 31:542–552.
- Hueskes BA, Roovers EA, Mantel-Teeuwisse AK, Janssens HJ, van de Lisdonk EH, Janssen M. Use of diuretics and the risk of gouty arthritis: a systematic review. Semin Arthritis Rheum 2012; 41:879–889.
- Dorsch MP, Gillespie BW, Erickson SR, Bleske BE, Weder AB. Chlorthalidone reduces cardiovascular events compared with hydrochlorothiazide: a retrospective cohort analysis. Hypertension 2011; 57:689–694.
- Trinkley KE, Page RL 2nd, Lien H, Yamanouye K, Tisdale JE. QT interval prolongation and the risk of torsades de pointes: essentials for clinicians. Curr Med Res Opin 2013; 29:1719–1726.
- Crist LW, Dixon DL. Considerations for dofetilide use in the elderly. Consult Pharm 2014; 29:270–274.
- Handler J. Lithium and antihypertensive medication: a potentially dangerous interaction. J Clin Hypertens (Greenwich) 2009; 11:738–742.
- Pavlicević I, Kuzmanić M, Rumboldt M, Rumboldt Z. Interaction between antihypertensives and NSAIDs in primary care: a controlled trial. Can J Clin Pharmacol 2008; 15:e372–e382.
- Psaty BM, Smith NL, Siscovick DS, et al. Health outcomes associated with antihypertensive therapies used as first-line agents. A systematic review and meta-analysis. JAMA 1997; 277:739–745.
- Roush GC, Holford TR, Guddati AK. Chlorthalidone compared with hydrochlorothiazide in reducing cardiovascular events: systematic review and network meta-analyses. Hypertension 2012; 59:1110–1117.
- Dhalla IA, Gomes T, Yao Z, et al. Chlorthalidone versus hydrochlorothiazide for the treatment of hypertension in older adults: a population-based cohort study. Ann Intern Med 2013; 158:447–455.
The thiazide diuretic hydrochlorothiazide and the thiazidelike diuretic chlorthalidone are two old drugs that are still useful. Although similar, they differ in important ways still not fully appreciated more than a half century after they were introduced.
Most hypertension guidelines recommend thiazide diuretics as one of the classes of agents that can be used either as initial antihypertensive drug therapy or as part of combination therapy.1–3
In the United States, hydrochlorothiazide is used more often than chlorthalidone, but many clinical trials of antihypertensive therapy have used chlorthalidone.4,5 In recent years, particularly after the publication of the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT), interest in chlorthalidone has been increasing, and new data are now available comparing these two diuretics.6 While current US guidelines do not recommend one over the other, British guidelines prefer chlorthalidone.7
This review summarizes the data comparing the two drugs’ pharmacology, antihypertensive effect, and impact on clinical outcomes to help guide clinicians in choosing antihypertensive drug therapy.
PHARMACOLOGY AND MECHANISM OF ACTION
Many of the differences in effectiveness and adverse effects of hydrochlorothiazide and chlorthalidone are thought to be due to their different pharmacodynamic and pharmacokinetic effects.
Pharmacodynamic effects
Hydrochlorothiazide and chlorthalidone differ significantly in chemical structure (Figure 1), but both contain a sulfonamide group that inhibits carbonic anhydrase activity, which may be associated with lower vascular contractility. Both drugs are concentrated in the kidney and secreted into the tubular lumen8; therefore, their therapeutic diuretic effects are often achieved with relatively low plasma concentrations.
Both drugs inhibit the sodium-chloride cotransporter in the luminal membrane of the distal convoluted tubule of the ascending loop of Henle, leading to a modest natriuresis and diuresis. The exact mechanism by which they lower blood pressure is not known: while the initial response is from diuresis and volume changes, long-term reduction in blood pressure is through uncertain mechanisms. In addition, chlorthalidone may have beneficial effects on endothelial function and oxidative stress.9,10
Both drugs also increase secretion of potassium and hydrogen ions and promote increased reabsorption of calcium through increased expression of a sodium-calcium exchange channel.8 Chlorthalidone may cause more inhibition of carbonic anhydrase than hydrochlorothiazide, which can lead to lower intracellular pH and cell volume. This effect may in part explain a pleiotropic effect of chlorthalidone, ie, inhibition of platelet function, which in turn may contribute to this drug’s beneficial effect on cardiovascular outcomes.9
Pharmacokinetic differences
Hydrochlorothiazide and chlorthalidone have important differences in their pharmacokinetic properties (Table 1).11
Hydrochlorothiazide has its onset of action in about 2 hours, and it reaches its peak in 4 to 6 hours. Though its duration of action is short—up to 12 hours—its pharmacodynamic response can be much longer than predicted by its kinetics, allowing once-daily dosing.8
Chlorthalidone has a longer duration of action than hydrochlorothiazide. This may be because it has a very high volume of distribution, since it is taken up into red blood cells and is bound to carbonic anhydrase.12 This may result in a “drug reservoir” that keeps drug levels higher for a longer time.13 Its long duration of action makes it a favorable choice for patients who have difficulty adhering to medication instructions. In addition, a missed dose is unlikely to have a “rebound” effect like that seen with some other antihypertensive agents. However, both chlorthalidone and hydrochlorothiazide are effective if taken once daily.
BLOOD PRESSURE-LOWERING
Both hydrochlorothiazide and chlorthalidone are effective antihypertensive agents. Table 2 summarizes findings from studies that evaluated their blood pressure-lowering effect at various doses.14–33 However, relatively few studies have directly compared these two agents’ effects on blood pressure.
Ernst et al,34 in a small study (but probably the best one to address this issue), compared chlorthalidone 12.5 mg/day (force-titrated to 25 mg/day) and hydrochlorothiazide 25 mg/day (force-titrated to 50 mg/day) in untreated hypertensive patients. After 8 weeks, ambulatory blood pressure monitoring indicated a greater reduction from baseline in systolic blood pressure with chlorthalidone 25 mg/day than with hydrochlorothiazide 50 mg/day (24-hour mean –12.4 vs –7.4 mm Hg, P = .05). Interestingly, the change in nighttime blood pressure was greater in the chlorthalidone group (–13.5 mm Hg) than in the hydrochlorothiazide group (–6.4 mm Hg; P = .009). These data suggest that at the doses studied, chlorthalidone is more effective than hydrochlorothiazide in lowering systolic blood pressure.
Bakris et al,35 using a different study design, compared the single-pill combination of azilsartan medoxomil and chlorthalidone vs coadministration of azilsartan medoxomil and hydrochlorothiazide in participants with stage 2 primary hypertension (≥ 160/100 mm Hg). Systolic blood pressure, as measured in the clinic, declined more with the chlorthalidone combination (–35.1 mm Hg) than with the hydrochlorothiazide combination (–29.5 mm Hg, mean difference –5.6 mm Hg, P < .001).
Meta-analyses also support the conclusion that chlorthalidone is more potent than hydrochlorothiazide in lowering blood pressure.35,36 Several studies have shown that chlorthalidone at the same dose is 1.5 to 2 times as potent as hydrochlorothiazide.33,36,37 Therefore, for clinical purposes, it is reasonable to consider chlorthalidone 12.5 mg daily as similar to 25 mg of hydrochlorothiazide daily.
ADVERSE EFFECTS
Electrolyte disturbances are a common adverse effect of thiazide diuretics.
Hypokalemia. All thiazide diuretics cause potassium wasting. The frequency of hypokalemia depends on the dose, frequency of administration, diet, and other pharmacologic agents used.
Two large clinical trials, the Systolic Hypertension in the Elderly Program and ALLHAT, found that chlorthalidone caused hypokalemia requiring therapy in about 6% to 8% of patients.38,39 Chlorthalidone therapy was associated with a lowering of serum potassium levels of 0.2 to 0.5 mmol/L.36 In ALLHAT, chlorthalidone was associated with a reduction in potassium levels of approximately 0.2 mmol/L after 4 years.38
All diuretics require monitoring of electrolytes, especially during the first 2 weeks of therapy. Once a steady state is reached, patients are not usually at risk of hypokalemia unless the dose is increased, extrarenal losses of potassium increase, or dietary potassium is reduced.
Other electrolyte changes. Thiazide and thiazide-like diuretics can cause other metabolic and endocrine abnormalities such as hypochloremic alkalosis, hyponatremia, and hypercalcemia.40,41 They can also cause photosensitivity and can precipitate gout.42
Observational studies have suggested that metabolic adverse effects such as hypokalemia and hyperuricemia are more common with chlorthalidone than with hydrochlorothiazide.43 It is prudent to monitor laboratory values periodically in patients on diuretic therapy.
DRUG INTERACTIONS
The drug interaction profiles of hydrochlorothiazide and chlorthalidone are also similar. The most common interactions are pharmacodynamic interactions resulting from potassium depletion caused by the diuretics.
Antiarrythymic drugs. Hypokalemia is a risk factor for arrhythmias such as torsades de pointes, and the risk is magnified with concomitant therapy with antiarrhythmic agents that prolong the QT interval independently of serum potassium concentration (eg, sotalol, dronedarone, ibutilide, propafenone). Therefore, combinations of drugs that can cause hypokalemia (eg, diuretics) and antiarrhythmic agents require vigilant monitoring of potassium and appropriate replenishment.44
Dofetilide is a class III antiarrhythmic agent and, like other antiarrhythmic drugs, carries a risk of QT prolongation and torsades de pointes, which is magnified by hypokalemia.45 In addition, dofetilide undergoes active tubular secretion in the kidney via the cation transport system, which is inhibited by hydrochlorothiazide.45 The resulting increase in plasma concentrations of dofetilide may magnify the risk of arrhythmias. Chlorthalidone has not been specifically studied in combination with dofetilide, but thiazide diuretics in general are thought to have a similar effect on tubular secretion and, therefore, should be considered similar to hydrochlorothiazide in this regard.
Digoxin. Similarly, digoxin toxicity may be enhanced in hypokalemia. As with antiarrhythmic agents, serum potassium should be carefully monitored and replenished appropriately when diuretics are used in combination with digoxin.
Lithium is reabsorbed in the proximal tubule along with sodium. Diuretics including hydrochlorothiazide and chlorthalidone that alter sodium reabsorption can also alter lithium absorption.46 When sodium reabsorption is decreased, lithium ion reabsorption is increased and may result in lithium toxicity. Although this combination is not contraindicated, monitoring of serum lithium concentrations and clinical signs and symptoms of lithium toxicity is recommended during initiation and dose adjustments of thiazide diuretics.
Nonsteroidal anti-inflammatory drugs can decrease the natriuretic, diuretic, and antihypertensive effects of both hydrochlorothiazide and chlorthalidone.47
Renin-angiotensin-aldosterone system antagonists, ie, angiotensin-converting enzyme inhibitors, angiotensin II receptor blockers, and the renin inhibitor aliskiren, have potentially beneficial interactions with hydrochlorothiazide and chlorthalidone, producing additive decreases in blood pressure when coadministered with these diuretics. These effects may be particularly potent early in concomitant therapy and allow use of lower doses of diuretics, typically 12.5 mg of hydrochlorothiazide in combination therapy.
LONG-TERM EFFECTS ON CARDIOVASCULAR EVENTS
The long-term goal in treating hypertension is to lower the risk of cardiovascular disease. Therefore, the clinician needs to consider the effect of antihypertensive drug therapy on long-term clinical outcomes.
Antihypertensive drug therapy based on thiazide diuretics has been shown to lower cardiovascular risk when compared with placebo.48 In addition, the effect of chlorthalidone-based antihypertensive therapy was similar to that of other antihypertensive drug classes in preventing most cardiovascular outcomes in ALLHAT.4
However, no study has directly compared hydrochlorothiazide and chlorthalidone with the primary outcome of reduction in long-term cardiovascular events. The data to date come from observational studies and meta-analyses. For example, in a retrospective analysis of the Multiple Risk Factor Intervention Trial, cardiovascular events were significantly fewer in those receiving chlorthalidone vs hydrochlorothiazide (P = .0016).43
In a systematic review and meta-analysis, chlorthalidone was associated with a 23% lower risk of heart failure and a 21% lower risk of all cardiovascular events.49
However, a Canadian observational study of 29,873 patients found no difference in the composite outcome of death or hospitalization for heart failure, stroke, or myocardial infarction between chlorthalidone recipients (3.2 events per 100 person-years) and hydrochlorothiazide recipients (3.4 events per 100 person-years; adjusted hazard ratio 0.93, 95% confidence interval 0.81–1.06).50
In summary, it is unclear whether chlorthalidone or hydrochlorothiazide is superior in preventing cardiovascular events.
SUMMARY
Thiazide and thiazidelike diuretics play an important role in managing hypertension in most patients. The eighth Joint National Committee guidelines do not recommend either hydrochlorothiazide or chlorthalidone over the other. The target dose recommendations are hydrochlorothiazide 25 to 50 mg or chlorthalidone 12.5 to 25 mg daily, with lower doses considered for the elderly.
There are important differences between hydrochlorothiazide and chlorthalidone in pharmacology, potency, and frequency of metabolic side effects. Clinicians should consider these factors to tailor the choice of thiazide diuretic therapy in hypertensive patients.
The thiazide diuretic hydrochlorothiazide and the thiazidelike diuretic chlorthalidone are two old drugs that are still useful. Although similar, they differ in important ways still not fully appreciated more than a half century after they were introduced.
Most hypertension guidelines recommend thiazide diuretics as one of the classes of agents that can be used either as initial antihypertensive drug therapy or as part of combination therapy.1–3
In the United States, hydrochlorothiazide is used more often than chlorthalidone, but many clinical trials of antihypertensive therapy have used chlorthalidone.4,5 In recent years, particularly after the publication of the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT), interest in chlorthalidone has been increasing, and new data are now available comparing these two diuretics.6 While current US guidelines do not recommend one over the other, British guidelines prefer chlorthalidone.7
This review summarizes the data comparing the two drugs’ pharmacology, antihypertensive effect, and impact on clinical outcomes to help guide clinicians in choosing antihypertensive drug therapy.
PHARMACOLOGY AND MECHANISM OF ACTION
Many of the differences in effectiveness and adverse effects of hydrochlorothiazide and chlorthalidone are thought to be due to their different pharmacodynamic and pharmacokinetic effects.
Pharmacodynamic effects
Hydrochlorothiazide and chlorthalidone differ significantly in chemical structure (Figure 1), but both contain a sulfonamide group that inhibits carbonic anhydrase activity, which may be associated with lower vascular contractility. Both drugs are concentrated in the kidney and secreted into the tubular lumen8; therefore, their therapeutic diuretic effects are often achieved with relatively low plasma concentrations.
Both drugs inhibit the sodium-chloride cotransporter in the luminal membrane of the distal convoluted tubule of the ascending loop of Henle, leading to a modest natriuresis and diuresis. The exact mechanism by which they lower blood pressure is not known: while the initial response is from diuresis and volume changes, long-term reduction in blood pressure is through uncertain mechanisms. In addition, chlorthalidone may have beneficial effects on endothelial function and oxidative stress.9,10
Both drugs also increase secretion of potassium and hydrogen ions and promote increased reabsorption of calcium through increased expression of a sodium-calcium exchange channel.8 Chlorthalidone may cause more inhibition of carbonic anhydrase than hydrochlorothiazide, which can lead to lower intracellular pH and cell volume. This effect may in part explain a pleiotropic effect of chlorthalidone, ie, inhibition of platelet function, which in turn may contribute to this drug’s beneficial effect on cardiovascular outcomes.9
Pharmacokinetic differences
Hydrochlorothiazide and chlorthalidone have important differences in their pharmacokinetic properties (Table 1).11
Hydrochlorothiazide has its onset of action in about 2 hours, and it reaches its peak in 4 to 6 hours. Though its duration of action is short—up to 12 hours—its pharmacodynamic response can be much longer than predicted by its kinetics, allowing once-daily dosing.8
Chlorthalidone has a longer duration of action than hydrochlorothiazide. This may be because it has a very high volume of distribution, since it is taken up into red blood cells and is bound to carbonic anhydrase.12 This may result in a “drug reservoir” that keeps drug levels higher for a longer time.13 Its long duration of action makes it a favorable choice for patients who have difficulty adhering to medication instructions. In addition, a missed dose is unlikely to have a “rebound” effect like that seen with some other antihypertensive agents. However, both chlorthalidone and hydrochlorothiazide are effective if taken once daily.
BLOOD PRESSURE-LOWERING
Both hydrochlorothiazide and chlorthalidone are effective antihypertensive agents. Table 2 summarizes findings from studies that evaluated their blood pressure-lowering effect at various doses.14–33 However, relatively few studies have directly compared these two agents’ effects on blood pressure.
Ernst et al,34 in a small study (but probably the best one to address this issue), compared chlorthalidone 12.5 mg/day (force-titrated to 25 mg/day) and hydrochlorothiazide 25 mg/day (force-titrated to 50 mg/day) in untreated hypertensive patients. After 8 weeks, ambulatory blood pressure monitoring indicated a greater reduction from baseline in systolic blood pressure with chlorthalidone 25 mg/day than with hydrochlorothiazide 50 mg/day (24-hour mean –12.4 vs –7.4 mm Hg, P = .05). Interestingly, the change in nighttime blood pressure was greater in the chlorthalidone group (–13.5 mm Hg) than in the hydrochlorothiazide group (–6.4 mm Hg; P = .009). These data suggest that at the doses studied, chlorthalidone is more effective than hydrochlorothiazide in lowering systolic blood pressure.
Bakris et al,35 using a different study design, compared the single-pill combination of azilsartan medoxomil and chlorthalidone vs coadministration of azilsartan medoxomil and hydrochlorothiazide in participants with stage 2 primary hypertension (≥ 160/100 mm Hg). Systolic blood pressure, as measured in the clinic, declined more with the chlorthalidone combination (–35.1 mm Hg) than with the hydrochlorothiazide combination (–29.5 mm Hg, mean difference –5.6 mm Hg, P < .001).
Meta-analyses also support the conclusion that chlorthalidone is more potent than hydrochlorothiazide in lowering blood pressure.35,36 Several studies have shown that chlorthalidone at the same dose is 1.5 to 2 times as potent as hydrochlorothiazide.33,36,37 Therefore, for clinical purposes, it is reasonable to consider chlorthalidone 12.5 mg daily as similar to 25 mg of hydrochlorothiazide daily.
ADVERSE EFFECTS
Electrolyte disturbances are a common adverse effect of thiazide diuretics.
Hypokalemia. All thiazide diuretics cause potassium wasting. The frequency of hypokalemia depends on the dose, frequency of administration, diet, and other pharmacologic agents used.
Two large clinical trials, the Systolic Hypertension in the Elderly Program and ALLHAT, found that chlorthalidone caused hypokalemia requiring therapy in about 6% to 8% of patients.38,39 Chlorthalidone therapy was associated with a lowering of serum potassium levels of 0.2 to 0.5 mmol/L.36 In ALLHAT, chlorthalidone was associated with a reduction in potassium levels of approximately 0.2 mmol/L after 4 years.38
All diuretics require monitoring of electrolytes, especially during the first 2 weeks of therapy. Once a steady state is reached, patients are not usually at risk of hypokalemia unless the dose is increased, extrarenal losses of potassium increase, or dietary potassium is reduced.
Other electrolyte changes. Thiazide and thiazide-like diuretics can cause other metabolic and endocrine abnormalities such as hypochloremic alkalosis, hyponatremia, and hypercalcemia.40,41 They can also cause photosensitivity and can precipitate gout.42
Observational studies have suggested that metabolic adverse effects such as hypokalemia and hyperuricemia are more common with chlorthalidone than with hydrochlorothiazide.43 It is prudent to monitor laboratory values periodically in patients on diuretic therapy.
DRUG INTERACTIONS
The drug interaction profiles of hydrochlorothiazide and chlorthalidone are also similar. The most common interactions are pharmacodynamic interactions resulting from potassium depletion caused by the diuretics.
Antiarrythymic drugs. Hypokalemia is a risk factor for arrhythmias such as torsades de pointes, and the risk is magnified with concomitant therapy with antiarrhythmic agents that prolong the QT interval independently of serum potassium concentration (eg, sotalol, dronedarone, ibutilide, propafenone). Therefore, combinations of drugs that can cause hypokalemia (eg, diuretics) and antiarrhythmic agents require vigilant monitoring of potassium and appropriate replenishment.44
Dofetilide is a class III antiarrhythmic agent and, like other antiarrhythmic drugs, carries a risk of QT prolongation and torsades de pointes, which is magnified by hypokalemia.45 In addition, dofetilide undergoes active tubular secretion in the kidney via the cation transport system, which is inhibited by hydrochlorothiazide.45 The resulting increase in plasma concentrations of dofetilide may magnify the risk of arrhythmias. Chlorthalidone has not been specifically studied in combination with dofetilide, but thiazide diuretics in general are thought to have a similar effect on tubular secretion and, therefore, should be considered similar to hydrochlorothiazide in this regard.
Digoxin. Similarly, digoxin toxicity may be enhanced in hypokalemia. As with antiarrhythmic agents, serum potassium should be carefully monitored and replenished appropriately when diuretics are used in combination with digoxin.
Lithium is reabsorbed in the proximal tubule along with sodium. Diuretics including hydrochlorothiazide and chlorthalidone that alter sodium reabsorption can also alter lithium absorption.46 When sodium reabsorption is decreased, lithium ion reabsorption is increased and may result in lithium toxicity. Although this combination is not contraindicated, monitoring of serum lithium concentrations and clinical signs and symptoms of lithium toxicity is recommended during initiation and dose adjustments of thiazide diuretics.
Nonsteroidal anti-inflammatory drugs can decrease the natriuretic, diuretic, and antihypertensive effects of both hydrochlorothiazide and chlorthalidone.47
Renin-angiotensin-aldosterone system antagonists, ie, angiotensin-converting enzyme inhibitors, angiotensin II receptor blockers, and the renin inhibitor aliskiren, have potentially beneficial interactions with hydrochlorothiazide and chlorthalidone, producing additive decreases in blood pressure when coadministered with these diuretics. These effects may be particularly potent early in concomitant therapy and allow use of lower doses of diuretics, typically 12.5 mg of hydrochlorothiazide in combination therapy.
LONG-TERM EFFECTS ON CARDIOVASCULAR EVENTS
The long-term goal in treating hypertension is to lower the risk of cardiovascular disease. Therefore, the clinician needs to consider the effect of antihypertensive drug therapy on long-term clinical outcomes.
Antihypertensive drug therapy based on thiazide diuretics has been shown to lower cardiovascular risk when compared with placebo.48 In addition, the effect of chlorthalidone-based antihypertensive therapy was similar to that of other antihypertensive drug classes in preventing most cardiovascular outcomes in ALLHAT.4
However, no study has directly compared hydrochlorothiazide and chlorthalidone with the primary outcome of reduction in long-term cardiovascular events. The data to date come from observational studies and meta-analyses. For example, in a retrospective analysis of the Multiple Risk Factor Intervention Trial, cardiovascular events were significantly fewer in those receiving chlorthalidone vs hydrochlorothiazide (P = .0016).43
In a systematic review and meta-analysis, chlorthalidone was associated with a 23% lower risk of heart failure and a 21% lower risk of all cardiovascular events.49
However, a Canadian observational study of 29,873 patients found no difference in the composite outcome of death or hospitalization for heart failure, stroke, or myocardial infarction between chlorthalidone recipients (3.2 events per 100 person-years) and hydrochlorothiazide recipients (3.4 events per 100 person-years; adjusted hazard ratio 0.93, 95% confidence interval 0.81–1.06).50
In summary, it is unclear whether chlorthalidone or hydrochlorothiazide is superior in preventing cardiovascular events.
SUMMARY
Thiazide and thiazidelike diuretics play an important role in managing hypertension in most patients. The eighth Joint National Committee guidelines do not recommend either hydrochlorothiazide or chlorthalidone over the other. The target dose recommendations are hydrochlorothiazide 25 to 50 mg or chlorthalidone 12.5 to 25 mg daily, with lower doses considered for the elderly.
There are important differences between hydrochlorothiazide and chlorthalidone in pharmacology, potency, and frequency of metabolic side effects. Clinicians should consider these factors to tailor the choice of thiazide diuretic therapy in hypertensive patients.
- James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 2014; 311:507–520.
- Dasgupta K, Quinn RR, Zarnke KB, et al; Canadian Hypertension Education Program. The 2014 Canadian Hypertension Education Program recommendations for blood pressure measurement, diagnosis, assessment of risk, prevention, and treatment of hypertension. Can J Cardiol 2014; 30:485–501.
- Mancia G, Fagard R, Narkiewicz K, et al. 2013 ESH/ESC guidelines for the management of arterial hypertension: the Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). Eur Heart J 2013; 34:2159–2219.
- ALLHAT Officers and Coordinators for the ALLHAT Collaborative Research Group; The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial. Major outcomes in high-risk hypertensive patients randomized to angiotensin-converting enzyme inhibitor or calcium channel blocker vs diuretic: the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). JAMA 2002; 288:2981–2997.
- Prevention of stroke by antihypertensive drug treatment in older persons with isolated systolic hypertension. Final results of the Systolic Hypertension in the Elderly Program (SHEP). SHEP Cooperative Research Group. JAMA 1991; 265:3255–3264.
- Roush GC, Kaur R, Ernst ME. Diuretics: a review and update. J Cardiovasc Pharmacol Ther 2014; 19:5–13.
- McCormack T, Krause T, O’Flynn N. Management of hypertension in adults in primary care: NICE guideline. Br J Gen Pract 2012; 62:163–164.
- Bhattacharaya M, Alper SL. Pharmacology of volume regulation. In: Golan DE, Tashjian AH Jr, Armstrong EJ, Armstrong AW, editors. Principles of Pharmacology: The pathophysiologic Basis of Drug Therapy. 3rd ed. Philadelphia, PA: Wolters Kluwer Health/Lippincott Williams & Wilkins; 2012:332–352.
- Woodman R, Brown C, Lockette W. Chlorthalidone decreases platelet aggregation and vascular permeability and promotes angiogenesis. Hypertension 2010; 56:463–470.
- Sato K, Dohi Y, Kojima M, Takase H, Suzuki S, Ito S. Antioxidative effects of thiazide diuretics in refractory hypertensive patients. A randomized crossover trial of chlortalidone and trichlormethiazide. Arzneimittelforschung 2010; 60:612–616.
- US National Library of Medicine. Dailymed. dailymed.nlm.nih.gov. Accessed May 14, 2015.
- Collste P, Garle M, Rawlins MD, Sjöqvist F. Interindividual differences in chlorthalidone concentration in plasma and red cells of man after single and multiple doses. Eur J Clin Pharmacol 1976; 9:319–325.
- Roush GC, Buddharaju V, Ernst ME, Holford TR. Chlorthalidone: mechanisms of action and effect on cardiovascular events. Curr Hypertens Rep 2013; 15:514–521.
- Pool JL, Cushman WC, Saini RK, Nwachuku CE, Battikha JP. Use of the factorial design and quadratic response surface models to evaluate the fosinopril and hydrochlorothiazide combination therapy in hypertension. Am J Hypertens 1997; 10:117–123.
- Pool JL, Glazer R, Weinberger M, Alvarado R, Huang J, Graff A. Comparison of valsartan/hydrochlorothiazide combination therapy at doses up to 320/25 mg versus monotherapy: a double-blind, placebo-controlled study followed by long-term combination therapy in hypertensive adults. Clin Ther 2007; 29:61–73.
- Horie Y, Higaki J, Takeuchi M. Design, statistical analysis and sample size calculation of dose response study of telmisartan and hydrochlorothiazide. Contemp Clin Trials 2007; 28:647–653.
- Chrysant SG. Antihypertensive effectiveness of low-dose lisinopril-hydrochlorothiazide combination. A large multicenter study. Lisinopril-Hydrochlorothiazide Group. Arch Intern Med 1994; 154:737–743.
- Lacourcière Y, Arnott W. Placebo-controlled comparison of the effects of nebivolol and low-dose hydrochlorothiazide as monotherapies and in combination on blood pressure and lipid profile in hypertensive patients. J Hum Hypertens 1994; 8:283–288.
- Villamil A, Chrysant SG, Calhoun D, et al. Renin inhibition with aliskiren provides additive antihypertensive efficacy when used in combination with hydrochlorothiazide. J Hypertens 2007; 25:217–226.
- McGill JB, Reilly PA. Telmisartan plus hydrochlorothiazide versus telmisartan or hydrochlorothiazide monotherapy in patients with mild to moderate hypertension: a multicenter, randomized, double-blind, placebo-controlled, parallel-group trial. Clin Ther 2001; 23:833–850.
- Weir MR, Weber MA, Punzi HA, Serfer HM, Rosenblatt S, Cady WJ. A dose escalation trial comparing the combination of diltiazem SR and hydrochlorothiazide with the monotherapies in patients with essential hypertension. J Hum Hypertens 1992; 6:133–138.
- Goldberg MR, Rockhold FW, Offen WW, Dornseif BE. Dose-effect and concentration-effect relationships of pinacidil and hydrochlorothiazide in hypertension. Clin Pharmacol Ther 1989; 46:208–218.
- Papademetriou V, Hainer JW, Sugg J, Munzer D; ATTACH Study Group. Factorial antihypertensive study of an extended-release metoprolol and hydrochlorothiazide combination. Am J Hypertens 2006; 19:1217–1225.
- Chrysant SG, Chrysant GS. Antihypertensive efficacy of olmesartan medoxomil alone and in combination with hydrochlorothiazide. Expert Opin Pharmacother 2004; 5:657–667.
- Kochar M, Guthrie R, Triscari J, Kassler-Taub K, Reeves RA. Matrix study of irbesartan with hydrochlorothiazide in mild-to-moderate hypertension. Am J Hypertens 1999; 12:797–805.
- Benz JR, Black HR, Graff A, Reed A, Fitzsimmons S, Shi Y. Valsartan and hydrochlorothiazide in patients with essential hypertension. A multiple dose, double-blind, placebo controlled trial comparing combination therapy with monotherapy. J Hum Hypertens 1998; 12:861–866.
- Jounela AJ, Lilja M, Lumme J, et al. Relation between low dose of hydrochlorothiazide, antihypertensive effect and adverse effects. Blood Press 1994; 3:231–235.
- Scholze J, Breitstadt A, Cairns V, et al. Short report: ramipril and hydrochlorothiazide combination therapy in hypertension: a clinical trial of factorial design. East Germany Collaborative Trial Group. J Hypertens 1993; 11:217–221.
- Canter D, Frank GJ, Knapp LE, Phelps M, Quade M, Texter M. Quinapril and hydrochlorothiazide combination for control of hypertension: assessment by factorial design. Quinapril Investigator Group. J Hum Hypertens 1994; 8:155–162.
- Vardan S, Mehrotra KG, Mookherjee S, Willsey GA, Gens JD, Green DE. Efficacy and reduced metabolic side effects of a 15-mg chlorthalidone formulation in the treatment of mild hypertension. A multicenter study. JAMA 1987; 258:484–488.
- Materson BJ, Oster JR, Michael UF, et al. Dose response to chlorthalidone in patients with mild hypertension. Efficacy of a lower dose. Clin Pharmacol Ther 1978; 24:192–198.
- Morledge JH, Ettinger B, Aranda J, et al. Isolated systolic hypertension in the elderly. A placebo-controlled, dose-response evaluation of chlorthalidone. J Am Geriatr Soc 1986; 34:199–206.
- Peterzan MA, Hardy R, Chaturvedi N, Hughes AD. Meta-analysis of dose-response relationships for hydrochlorothiazide, chlorthalidone, and bendroflumethiazide on blood pressure, serum potassium, and urate. Hypertension 2012; 59:1104–1109.
- Ernst ME, Carter BL, Goerdt CJ, et al. Comparative antihypertensive effects of hydrochlorothiazide and chlorthalidone on ambulatory and office blood pressure. Hypertension 2006; 47:352–358.
- Bakris GL, Sica D, White WB, et al. Antihypertensive efficacy of hydrochlorothiazide vs chlorthalidone combined with azilsartan medoxomil. Am J Med 2012; 25:1229.e1–1229.e10.
- Ernst ME, Carter BL, Zheng S, Grimm RH Jr. Meta-analysis of dose-response characteristics of hydrochlorothiazide and chlorthalidone: effects on systolic blood pressure and potassium. Am J Hypertens 2010; 23:440–446.
- Carter BL, Ernst ME, Cohen JD. Hydrochlorothiazide versus chlorthalidone: evidence supporting their interchangeability. Hypertension 2004; 43:4–9.
- Alderman MH, Piller LB, Ford CE, et al; Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial Collaborative Research Group. Clinical significance of incident hypokalemia and hyperkalemia in treated hypertensive patients in the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial. Hypertension 2012; 59:926–933.
- Franse LV, Pahor M, Di Bari M, Somes GW, Cushman WC, Applegate WB. Hypokalemia associated with diuretic use and cardiovascular events in the Systolic Hypertension in the Elderly Program. Hypertension 2000; 35:1025–1030.
- Egom EE, Chirico D, Clark AL. A review of thiazide-induced hyponatraemia. Clin Med 2011; 11:448–451.
- Palmer BF. Metabolic complications associated with use of diuretics. Semin Nephrol 2011; 31:542–552.
- Hueskes BA, Roovers EA, Mantel-Teeuwisse AK, Janssens HJ, van de Lisdonk EH, Janssen M. Use of diuretics and the risk of gouty arthritis: a systematic review. Semin Arthritis Rheum 2012; 41:879–889.
- Dorsch MP, Gillespie BW, Erickson SR, Bleske BE, Weder AB. Chlorthalidone reduces cardiovascular events compared with hydrochlorothiazide: a retrospective cohort analysis. Hypertension 2011; 57:689–694.
- Trinkley KE, Page RL 2nd, Lien H, Yamanouye K, Tisdale JE. QT interval prolongation and the risk of torsades de pointes: essentials for clinicians. Curr Med Res Opin 2013; 29:1719–1726.
- Crist LW, Dixon DL. Considerations for dofetilide use in the elderly. Consult Pharm 2014; 29:270–274.
- Handler J. Lithium and antihypertensive medication: a potentially dangerous interaction. J Clin Hypertens (Greenwich) 2009; 11:738–742.
- Pavlicević I, Kuzmanić M, Rumboldt M, Rumboldt Z. Interaction between antihypertensives and NSAIDs in primary care: a controlled trial. Can J Clin Pharmacol 2008; 15:e372–e382.
- Psaty BM, Smith NL, Siscovick DS, et al. Health outcomes associated with antihypertensive therapies used as first-line agents. A systematic review and meta-analysis. JAMA 1997; 277:739–745.
- Roush GC, Holford TR, Guddati AK. Chlorthalidone compared with hydrochlorothiazide in reducing cardiovascular events: systematic review and network meta-analyses. Hypertension 2012; 59:1110–1117.
- Dhalla IA, Gomes T, Yao Z, et al. Chlorthalidone versus hydrochlorothiazide for the treatment of hypertension in older adults: a population-based cohort study. Ann Intern Med 2013; 158:447–455.
- James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 2014; 311:507–520.
- Dasgupta K, Quinn RR, Zarnke KB, et al; Canadian Hypertension Education Program. The 2014 Canadian Hypertension Education Program recommendations for blood pressure measurement, diagnosis, assessment of risk, prevention, and treatment of hypertension. Can J Cardiol 2014; 30:485–501.
- Mancia G, Fagard R, Narkiewicz K, et al. 2013 ESH/ESC guidelines for the management of arterial hypertension: the Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). Eur Heart J 2013; 34:2159–2219.
- ALLHAT Officers and Coordinators for the ALLHAT Collaborative Research Group; The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial. Major outcomes in high-risk hypertensive patients randomized to angiotensin-converting enzyme inhibitor or calcium channel blocker vs diuretic: the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). JAMA 2002; 288:2981–2997.
- Prevention of stroke by antihypertensive drug treatment in older persons with isolated systolic hypertension. Final results of the Systolic Hypertension in the Elderly Program (SHEP). SHEP Cooperative Research Group. JAMA 1991; 265:3255–3264.
- Roush GC, Kaur R, Ernst ME. Diuretics: a review and update. J Cardiovasc Pharmacol Ther 2014; 19:5–13.
- McCormack T, Krause T, O’Flynn N. Management of hypertension in adults in primary care: NICE guideline. Br J Gen Pract 2012; 62:163–164.
- Bhattacharaya M, Alper SL. Pharmacology of volume regulation. In: Golan DE, Tashjian AH Jr, Armstrong EJ, Armstrong AW, editors. Principles of Pharmacology: The pathophysiologic Basis of Drug Therapy. 3rd ed. Philadelphia, PA: Wolters Kluwer Health/Lippincott Williams & Wilkins; 2012:332–352.
- Woodman R, Brown C, Lockette W. Chlorthalidone decreases platelet aggregation and vascular permeability and promotes angiogenesis. Hypertension 2010; 56:463–470.
- Sato K, Dohi Y, Kojima M, Takase H, Suzuki S, Ito S. Antioxidative effects of thiazide diuretics in refractory hypertensive patients. A randomized crossover trial of chlortalidone and trichlormethiazide. Arzneimittelforschung 2010; 60:612–616.
- US National Library of Medicine. Dailymed. dailymed.nlm.nih.gov. Accessed May 14, 2015.
- Collste P, Garle M, Rawlins MD, Sjöqvist F. Interindividual differences in chlorthalidone concentration in plasma and red cells of man after single and multiple doses. Eur J Clin Pharmacol 1976; 9:319–325.
- Roush GC, Buddharaju V, Ernst ME, Holford TR. Chlorthalidone: mechanisms of action and effect on cardiovascular events. Curr Hypertens Rep 2013; 15:514–521.
- Pool JL, Cushman WC, Saini RK, Nwachuku CE, Battikha JP. Use of the factorial design and quadratic response surface models to evaluate the fosinopril and hydrochlorothiazide combination therapy in hypertension. Am J Hypertens 1997; 10:117–123.
- Pool JL, Glazer R, Weinberger M, Alvarado R, Huang J, Graff A. Comparison of valsartan/hydrochlorothiazide combination therapy at doses up to 320/25 mg versus monotherapy: a double-blind, placebo-controlled study followed by long-term combination therapy in hypertensive adults. Clin Ther 2007; 29:61–73.
- Horie Y, Higaki J, Takeuchi M. Design, statistical analysis and sample size calculation of dose response study of telmisartan and hydrochlorothiazide. Contemp Clin Trials 2007; 28:647–653.
- Chrysant SG. Antihypertensive effectiveness of low-dose lisinopril-hydrochlorothiazide combination. A large multicenter study. Lisinopril-Hydrochlorothiazide Group. Arch Intern Med 1994; 154:737–743.
- Lacourcière Y, Arnott W. Placebo-controlled comparison of the effects of nebivolol and low-dose hydrochlorothiazide as monotherapies and in combination on blood pressure and lipid profile in hypertensive patients. J Hum Hypertens 1994; 8:283–288.
- Villamil A, Chrysant SG, Calhoun D, et al. Renin inhibition with aliskiren provides additive antihypertensive efficacy when used in combination with hydrochlorothiazide. J Hypertens 2007; 25:217–226.
- McGill JB, Reilly PA. Telmisartan plus hydrochlorothiazide versus telmisartan or hydrochlorothiazide monotherapy in patients with mild to moderate hypertension: a multicenter, randomized, double-blind, placebo-controlled, parallel-group trial. Clin Ther 2001; 23:833–850.
- Weir MR, Weber MA, Punzi HA, Serfer HM, Rosenblatt S, Cady WJ. A dose escalation trial comparing the combination of diltiazem SR and hydrochlorothiazide with the monotherapies in patients with essential hypertension. J Hum Hypertens 1992; 6:133–138.
- Goldberg MR, Rockhold FW, Offen WW, Dornseif BE. Dose-effect and concentration-effect relationships of pinacidil and hydrochlorothiazide in hypertension. Clin Pharmacol Ther 1989; 46:208–218.
- Papademetriou V, Hainer JW, Sugg J, Munzer D; ATTACH Study Group. Factorial antihypertensive study of an extended-release metoprolol and hydrochlorothiazide combination. Am J Hypertens 2006; 19:1217–1225.
- Chrysant SG, Chrysant GS. Antihypertensive efficacy of olmesartan medoxomil alone and in combination with hydrochlorothiazide. Expert Opin Pharmacother 2004; 5:657–667.
- Kochar M, Guthrie R, Triscari J, Kassler-Taub K, Reeves RA. Matrix study of irbesartan with hydrochlorothiazide in mild-to-moderate hypertension. Am J Hypertens 1999; 12:797–805.
- Benz JR, Black HR, Graff A, Reed A, Fitzsimmons S, Shi Y. Valsartan and hydrochlorothiazide in patients with essential hypertension. A multiple dose, double-blind, placebo controlled trial comparing combination therapy with monotherapy. J Hum Hypertens 1998; 12:861–866.
- Jounela AJ, Lilja M, Lumme J, et al. Relation between low dose of hydrochlorothiazide, antihypertensive effect and adverse effects. Blood Press 1994; 3:231–235.
- Scholze J, Breitstadt A, Cairns V, et al. Short report: ramipril and hydrochlorothiazide combination therapy in hypertension: a clinical trial of factorial design. East Germany Collaborative Trial Group. J Hypertens 1993; 11:217–221.
- Canter D, Frank GJ, Knapp LE, Phelps M, Quade M, Texter M. Quinapril and hydrochlorothiazide combination for control of hypertension: assessment by factorial design. Quinapril Investigator Group. J Hum Hypertens 1994; 8:155–162.
- Vardan S, Mehrotra KG, Mookherjee S, Willsey GA, Gens JD, Green DE. Efficacy and reduced metabolic side effects of a 15-mg chlorthalidone formulation in the treatment of mild hypertension. A multicenter study. JAMA 1987; 258:484–488.
- Materson BJ, Oster JR, Michael UF, et al. Dose response to chlorthalidone in patients with mild hypertension. Efficacy of a lower dose. Clin Pharmacol Ther 1978; 24:192–198.
- Morledge JH, Ettinger B, Aranda J, et al. Isolated systolic hypertension in the elderly. A placebo-controlled, dose-response evaluation of chlorthalidone. J Am Geriatr Soc 1986; 34:199–206.
- Peterzan MA, Hardy R, Chaturvedi N, Hughes AD. Meta-analysis of dose-response relationships for hydrochlorothiazide, chlorthalidone, and bendroflumethiazide on blood pressure, serum potassium, and urate. Hypertension 2012; 59:1104–1109.
- Ernst ME, Carter BL, Goerdt CJ, et al. Comparative antihypertensive effects of hydrochlorothiazide and chlorthalidone on ambulatory and office blood pressure. Hypertension 2006; 47:352–358.
- Bakris GL, Sica D, White WB, et al. Antihypertensive efficacy of hydrochlorothiazide vs chlorthalidone combined with azilsartan medoxomil. Am J Med 2012; 25:1229.e1–1229.e10.
- Ernst ME, Carter BL, Zheng S, Grimm RH Jr. Meta-analysis of dose-response characteristics of hydrochlorothiazide and chlorthalidone: effects on systolic blood pressure and potassium. Am J Hypertens 2010; 23:440–446.
- Carter BL, Ernst ME, Cohen JD. Hydrochlorothiazide versus chlorthalidone: evidence supporting their interchangeability. Hypertension 2004; 43:4–9.
- Alderman MH, Piller LB, Ford CE, et al; Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial Collaborative Research Group. Clinical significance of incident hypokalemia and hyperkalemia in treated hypertensive patients in the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial. Hypertension 2012; 59:926–933.
- Franse LV, Pahor M, Di Bari M, Somes GW, Cushman WC, Applegate WB. Hypokalemia associated with diuretic use and cardiovascular events in the Systolic Hypertension in the Elderly Program. Hypertension 2000; 35:1025–1030.
- Egom EE, Chirico D, Clark AL. A review of thiazide-induced hyponatraemia. Clin Med 2011; 11:448–451.
- Palmer BF. Metabolic complications associated with use of diuretics. Semin Nephrol 2011; 31:542–552.
- Hueskes BA, Roovers EA, Mantel-Teeuwisse AK, Janssens HJ, van de Lisdonk EH, Janssen M. Use of diuretics and the risk of gouty arthritis: a systematic review. Semin Arthritis Rheum 2012; 41:879–889.
- Dorsch MP, Gillespie BW, Erickson SR, Bleske BE, Weder AB. Chlorthalidone reduces cardiovascular events compared with hydrochlorothiazide: a retrospective cohort analysis. Hypertension 2011; 57:689–694.
- Trinkley KE, Page RL 2nd, Lien H, Yamanouye K, Tisdale JE. QT interval prolongation and the risk of torsades de pointes: essentials for clinicians. Curr Med Res Opin 2013; 29:1719–1726.
- Crist LW, Dixon DL. Considerations for dofetilide use in the elderly. Consult Pharm 2014; 29:270–274.
- Handler J. Lithium and antihypertensive medication: a potentially dangerous interaction. J Clin Hypertens (Greenwich) 2009; 11:738–742.
- Pavlicević I, Kuzmanić M, Rumboldt M, Rumboldt Z. Interaction between antihypertensives and NSAIDs in primary care: a controlled trial. Can J Clin Pharmacol 2008; 15:e372–e382.
- Psaty BM, Smith NL, Siscovick DS, et al. Health outcomes associated with antihypertensive therapies used as first-line agents. A systematic review and meta-analysis. JAMA 1997; 277:739–745.
- Roush GC, Holford TR, Guddati AK. Chlorthalidone compared with hydrochlorothiazide in reducing cardiovascular events: systematic review and network meta-analyses. Hypertension 2012; 59:1110–1117.
- Dhalla IA, Gomes T, Yao Z, et al. Chlorthalidone versus hydrochlorothiazide for the treatment of hypertension in older adults: a population-based cohort study. Ann Intern Med 2013; 158:447–455.
KEY POINTS
- Chlorthalidone has a longer duration of action and a longer half-life than hydrochlorothiazide.
- Chlorthalidone may be more potent than hydrochlorothiazide in lowering blood pressure, but it also may be associated with more metabolic adverse effects, such as hypokalemia.
- No study has conclusively shown either drug to be better in preventing adverse clinical outcomes.
- These differences should be considered when making choices about thiazide diuretic therapy for hypertension.
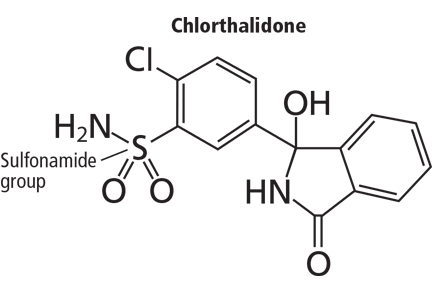
Urologic applications of botulinum toxin
Patients with loss of bladder control experience discomfort, embarrassment, personal care and health issues, and, often, significant pain, all with a decidedly negative impact on quality of life. Although some patients may find lifestyle modifications, drug therapy, and self-catheterization acceptable and effective, there is a clear need for more options.
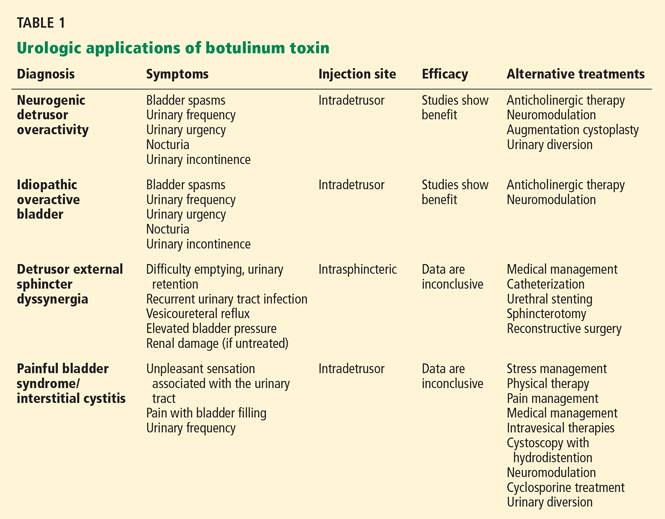
Botulinum toxin, or onabotulinumtoxinA, is currently approved by the US Food and Drug Administration (FDA) for neurogenic detrusor overactivity and overactive bladder refractory to drug therapy. Studies so far have shown botulinum toxin injection to be safe and effective for these conditions, and these results have led to interest in off-label uses, eg, for detrusor external sphincter dyssynergia (DESD), motor and sensory urgency, and painful bladder syndrome/interstitial cystitis (Table 1).
Although more data from clinical trials are needed, botulinum toxin injection offers patients a much-needed treatment option.
HOW BOTULINUM TOXIN WORKS
Seven serotypes identified
Discovered in 1897, botulinum toxin is a neurotoxin produced by the gram-positive, rod-shaped anaerobic bacterium Clostridium botulinum1 and is the most poisonous naturally occurring toxin known.2 Seven immunologically distinct antigenic serotypes have been identified (A, B, C1, D, E, F, and G),1 but only types A and B are available for clinical use.
Most research into potential therapeutic uses has focused on type A, which has the longest duration of action, a clinical advantage.3 Recently, work has been done to further characterize other serotypes and to isolate additional variants of botulinum toxin. For example, serotype E, the predominant serotype associated with foodborne botulism, is being studied in an effort to prevent future outbreaks.4
Our discussion focuses on clinical uses of the serotype A botulinum toxin preparation, which we will refer to simply as botulinum toxin.
Studies exploring how it works
Botulinum toxin exerts its effects by binding to peripheral cholinergic terminals, inhibiting release of acetylcholine at the neuromuscular junction. Flaccid paralysis ensues as a result.
Results of animal studies have shed additional light on the specific actions of botulinum toxin A:
- It may alter levels of nerve growth factor and transient receptor potential vanilloid 1 in rats, and this may provide an additional mechanism of reducing bladder detrusor overactivity.5
- In addition to blocking acetylcholine release from motor neurons, it inhibits the release of neurotransmitters involved in bladder sensory afferent pathways.6
- It inhibits the release of substance P and glutamate, neuropeptides involved in sensory and nociceptive pathways.6,7
- It promotes apoptosis in prostatic tissue; however, this effect has not been shown in the bladder.3
The time necessary to recover function after botulinum toxin paralysis depends on the subtype of botulinum toxin as well as on the type of nerve terminal. Chemodenervation lasts from 3 to 6 months when the toxin is injected into the neuromuscular junction of skeletal muscle, and considerably longer (up to 1 year) when injected into the autonomic neurons of smooth muscle.2,6
TREATMENT OF NEUROGENIC DETRUSOR OVERACTIVITY
Neurogenic detrusor overactivity involves involuntary contractions of the bladder resulting from spinal cord injury, multiple sclerosis, and other neurologic conditions. An estimated 273,000 people in the United States have a spinal cord injury, and 81% of them have urologic symptoms ranging from areflexia to overactivity.8 From 75% to 100% of patients with multiple sclerosis have urologic symptoms, and detrusor overactivity is the most common.9
Detrusor overactivity can cause urinary urgency, urinary frequency, and urgency incontinence, significantly affecting quality of life and leading to skin breakdown, sacral ulcerations, and challenges with personal care.
Anticholinergic drugs have been the mainstay of therapy. If drug therapy failed, the next option was reconstructive surgery, often augmentation cystoplasty. Thus, botulinum toxin injection is an important advance in treatment options.
Studies that showed effectiveness
Botulinum toxin for neurogenic detrusor overactivity was first studied by Schurch et al.10 In their study, 200 U or 300 U was injected into the trigone of 21 patients with spinal cord injury and urgency incontinence managed with intermittent self-catheterization.10 At 6 weeks after injection, 17 of the 19 patients seen at follow-up visits were completely continent. Urodynamic evaluation revealed significant increases in maximum cystometric capacity and in volume at first involuntary detrusor contraction, and a decrease in detrusor voiding pressure. Of the 11 patients available for follow-up at 16 and 36 weeks, improvements in measures of incontinence and urodynamic function persisted.
In addition, two small randomized controlled trials11,12 showed significant increases in cystometric bladder capacity, significant improvement in quality-of-life measures, and reduction in episodes of urgency incontinence.
In 2011 and 2012, two multicenter double-blind randomized controlled trials reported on patients with multiple sclerosis and spinal cord injury with neurogenic detrusor overactivity inadequately managed with drug therapy. The patients were randomized to botulinum toxin injection (200 U or 300 U) or placebo injection.13,14 The primary end point for both studies was the change from baseline in episodes of urinary incontinence per week at week 6. Secondary end points were maximum cystometric capacity, maximum detrusor pressure during first involuntary detrusor contraction, and score on the Incontinence Quality of Life scale.15
In both studies, the mean number of urinary incontinence episodes per week was 33 at baseline. At week 6, Cruz et al14 found that patients who received botulinum toxin injection had significantly fewer episodes per week (21.8 fewer with 200 U, 19.4 fewer with 300 U) than those in the placebo group, who had 13.2 fewer episodes per week (P < .01). Ginsberg et al13 reported decreases in the mean number of episodes of urinary incontinence of 21, 23, and 9 episodes per week in the 200 U, 300 U, and placebo groups, respectively (P < .001). The patients who received botulinum toxin had statistically significant improvements in maximum cystometric capacity, maximum detrusor pressure during first involuntary detrusor contraction, and Incontinence Quality of Life scores compared with placebo (P < .001). Thirty-eight percent of patients in the treatment group were fully continent.13,14
Safety and adverse effects
The most frequently reported adverse events were urinary tract infection (24% of patients)13,14 and urinary retention requiring initiation of clean intermittent catheterization. In the study by Cruz et al,14 these were reported in 30% with 200 U, 42% with 300 U, and 12% with placebo, while in the study by Ginsberg et al13 they were reported in 35% with 200 U, 42% with 300 U, and 10% with placebo.
In a study of long-term safety and efficacy of botulinum toxin injection in patients with neurogenic detrusor overactivity, Kennelly et al16 found that patients undergoing repeat injections had sustained reductions in episodes of incontinence and increases in the maximum cystometric capacity and quality of life scores, with no increase in adverse events over time.16
But is it cost-effective?
While botulinum toxin injection may be safe and effective for neurogenic detrusor overactivity, is it cost-effective?
Carlson et al17 used a Markov State Transition model to assess the cost of refractory neurogenic detrusor overactivity in patients receiving botulinum toxin vs best supportive care (incontinence pads, medications, intermittent self-catheterization).17 They found that the injections were more expensive than supportive care but were cost-effective when considering the reduction in episodes of incontinence, the reduced need for incontinence products, and improvement in measures of quality of life.
What the evidence indicates
Trials of botulinum toxin injection for neurogenic detrusor overactivity have shown that it improves continence, maximum cystometric capacity, detrusor pressures, and quality of life. The main adverse effects are urinary tract infection and urinary retention requiring intermittent self-catheterization.
Although many patients with this condition are already self-catheterizing, the physician must discuss this before botulinum toxin therapy to ensure that the patient or a family member is able to perform catheterization. Studies have shown that patients have an increase in urinary tract infections after botulinum injections. But in these studies, a urinary tract infection was defined as 100,000 colony-forming units or the presence of leukocytosis with or without symptoms. It is important to remember that patients on intermittent catheterization have bacteriuria and should be treated only for symptomatic, not asymptomatic, bacteriuria.
TREATMENT OF IDIOPATHIC OVERACTIVE BLADDER
Patients with idiopathic overactive bladder have urinary urgency accompanied by urgency incontinence, nocturia, or urinary frequency.18 The prevalence of this condition has been reported to range from 1.7% to 13.3% in men age 30 and older and 7% to 30.3% in women of similar ages. About one-third of women with overactive bladder also have detrusor overactivity.19 Overactive bladder presents a significant economic and medical burden on the healthcare system, as well as having a negative impact on quality of life.
The FDA approved botulinum toxin injection for treatment of idiopathic overactive bladder in January 2013.
Evidence of effectiveness
Two multicenter randomized controlled trials20,21 of botulinum toxin 100 U enrolled patients age 18 and older who had more than three episodes of urinary urgency incontinence in a 3-day period or more than eight micturitions per day inadequately managed by anticholinergic drug therapy. Primary end points were the change from baseline in the number of episodes of urinary incontinence per day and the proportion of patients with a positive response on the Treatment Benefit Scale22 at week 12. Secondary end points included episodes of urinary urgency incontinence, micturition, urgency, and nocturia, and scores on health-related quality of life questionnaires (Incontinence Quality of Life scale, King’s Health Questionnaire).
In both studies, patients receiving botulinum toxin had significantly fewer episodes of incontinence compared with placebo (−2.65 vs −0.87; P < .001 and −2.95 vs −1.03; P < .001).20,21 Reductions from baseline in all other symptoms of overactive bladder, a positive treatment response on the treatment benefit scale, and improvements in quality-of-life scores were also significantly greater with botulinum toxin injection than with placebo (P ≤ .01).
As in the studies of neurogenic detrusor overactivity, the most common adverse effects were urinary tract infection (occurring in 15.5%20 and 24.1%21 of patients) and urinary retention requiring self-catheterization (5.4%20 and 6.9%21).
The largest study to date of anticholinergic therapy vs botulinum toxin injection23 in women with urinary urgency incontinence, published in 2012, studied nearly 250 women who had five or more episodes of idiopathic urgency incontinence in a 3-day period. They were randomized either to daily oral therapy (solifenacin 5 mg with possible escalation to 10 mg and, if necessary, a subsequent switch to extended-release trospium 60 mg) plus one intradetrusor injection of saline, or to a daily oral placebo plus one injection of botulinum toxin 100 U.23
The dropout rate was low in both groups, with 93% of patients in both groups completing the 6-month protocol. Women experienced a mean reduction in urgency incontinence episodes of 3.4 per day (baseline 5) in the anticholinergic group vs 3.3 episodes in the botulinum toxin group (P = .81). However, more patients achieved complete resolution of urinary urgency incontinence in the botulinum toxin group than in the anticholinergic therapy group (27% vs 13%; P = .003). Quality of life improved in both groups without a significant difference between the groups. The botulinum toxin group had higher rates of initiation of self-catheterization (5% vs 0%, P = .01) and urinary tract infection (33% vs 13%, P < .001).23
Botulinum toxin as a third-line therapy
In May 2014, the American Urological Association updated its guidelines on idiopathic overactive bladder24 to include botulinum toxin injection as standard third-line therapy for patients in whom behavioral and medical management (ie, anticholinergics and beta-3-agonists) failed.
Interpreting the evidence to date
Overall, studies in idiopathic overactive bladder have shown a reduction in episodes of urgency incontinence and other symptoms, with some data also demonstrating a corresponding improvement in quality of life.
As in neurogenic detrusor overactivity, the main risks associated with botulinum toxin injection are urinary tract infection and the need to initiate self-catheterization. Although 94% of patients studied did not require self-catheterization after injection, the patient’s ability to perform self-catheterization should be determined before proceeding with botulinum toxin injections.
DETRUSOR EXTERNAL SPHINCTER DYSSYNERGIA
Botulinum toxin has been used not only to improve bladder storage but also to facilitate bladder emptying, as in patients with DESD, a lack of coordination between the bladder and the urinary sphincter. Normal voiding involves relaxation of the urinary sphincter and contraction of the bladder; in DESD the sphincter contracts and works against the bladder’s ability to empty. This leads not only to difficulty emptying the bladder but also to elevated bladder pressure, which can cause renal damage if untreated.
DESD can be seen after injury between the pontine micturition center, which coordinates activity between the bladder and the sphincter, and the caudal spinal cord. This can occur in spinal cord injury, multiple sclerosis, myelomeningocele, and transverse myelitis and can cause significant morbidity for the patient.
Treatment options include drug therapy, injection of botulinum toxin into the sphincter, clean intermittent catheterization, indwelling catheterization, urethral stenting, sphincterotomy, and reconstructive surgery such as urinary diversion.25
The goals of therapy are to avoid the need for clean intermittent catheterization in patients who have difficulty with manual dexterity, and to avoid the need for surgical procedures such as sphincterotomy and urinary diversion. The efficacy of urethral stenting is low, and medical management can be limited.26
In the first published report on botulinum toxin for DESD (in 1988),27 of 11 patients with spinal cord injury and DESD who received botulinum toxin injected into the external urethral sphincter, 10 showed signs of sphincter denervation on electromyography and reductions in urethral pressure profiles and postvoid residual volumes. Schurch et al28 and de Sèze et al29 also reported reductions in postvoid residual volume and maximal urethral pressures in patients with spinal cord injury and DESD.
In 2005, Gallien et al30 reported what is still the largest multicenter randomized controlled trial of botulinum toxin injection in DESD. Eighty-six patients with multiple sclerosis, DESD, and chronic urinary retention were randomized to receive either a single transperineal botulinum toxin injection of 100 U plus the alpha-1-blocker alfuzosin, or a placebo injection plus alfuzosin. Botulinum toxin treatment was associated with significantly increased voided volumes and reduced premicturition and maximal detrusor pressures, but no significant decrease in postvoid residual volume.30
More study needed
Despite these findings, a Cochrane Review concluded that, given the limited experience with intrasphincteric injection of botulinum toxin, data from larger randomized controlled trials are needed before making definitive recommendations.25 In the meantime, the clinician must weigh the low morbidity of the procedure against the limited options in the treatment of these patients.
OFF-LABEL UROLOGIC INDICATIONS
Botulinum toxin injection has been studied off-label for painful bladder syndrome/interstitial cystitis and for chronic prostatic pain. Patients with these conditions often describe pain with filling of the bladder, which leads to urinary frequency in an attempt to relieve the pain.
These pain syndromes can be difficult to treat and can have a devastating impact on quality of life. Treatment options include pain management, stress management, physical therapy, intravesical therapies, cystoscopy with hydrodistention, neuromodulation, cyclosporine, urinary diversion surgery, and botulinum toxin injection (an off-label use).31
In painful bladder syndrome/interstitial cystitis, botulinum toxin is thought to act on sensory afferent pathways, as well as to inhibit the release of substance P and glutamate, neuropeptides involved in sensory and nociceptive pathways.6 In animal studies,32 botulinum toxin was found to inhibit the afferent neural response by inhibiting mechanoreceptor-mediated release of adenosine triphosphate and by causing a decrease in calcitonin gene-related peptide, which helps regulate micturition and mediates painful bladder sensation.
Clinical studies to date in pelvic pain syndromes
Data from clinical studies of botulinum toxin injection for pelvic pain syndromes are limited. Zermann et al33 performed transurethral perisphincteric injection in 11 men with chronic prostatic pain, 9 of whom reported subjective pain relief, with an average decrease from 7.2 to 1.6 on a visual analogue scale. Postinjection urodynamic studies showed a decrease in functional urethral length, urethral closure pressure, and postvoid residual volume, and an increase in the peak and average flow rates.33
Abbott et al34 evaluated the effect of botulinum toxin injection into the levator ani in 12 women with chronic pelvic pain and pelvic floor hypertonicity. Pelvic floor manometry showed significant reduction in resting muscle pressures and improvements in dyspareunia and nonmenstrual pain. There were also improvements in quality of life and dyschezia, but these were not statistically significant.
Smith et al35 injected botulinum toxin into the detrusor of 13 women with refractory painful bladder syndrome and interstitial cystitis,35 and 9 women (69%) noted statistically significant improvements in the Interstitial Cystitis Symptom Index and Interstitial Cystitis Problem Index, daytime frequency, nocturia, pain, and urodynamic parameters (volume at first desire to void, and maximum cystometric capacity).
In a prospective randomized study of patients with refractory painful bladder syndrome and interstitial cystitis, Kuo and Chancellor36 compared suburothelial injection of 200 U or 100 U of botulinum toxin plus hydrodistention against hydrodistention alone.Patients who received botulinum toxin had increased bladder capacity and improved long-term pain relief, but no difference was noted between 200 U and 100 U, and more adverse effects were seen with the higher dose.36
Pinto et al37 treated 16 women with refractory painful bladder syndrome and interstitial cystitis with intratrigonal injections of botulinum toxin and reported improvements in pain scores, symptom scores, urinary frequency, and quality-of-life measures. The effect lasted 9.9 months (± 2.4 months) and persisted with successive injections.37
More study needed
Although these studies show that botulinum toxin injection for pelvic pain syndromes has the potential to improve pain, urinary frequency, bladder sensation, bladder capacity, and quality of life, larger randomized controlled trials are needed.
Again, the treatment options are limited for refractory painful bladder syndrome and interstitial cystitis. Patients may be desperate for relief from their symptoms. Practitioners must manage expectations and properly inform patients of the potential risks of treatments, especially with patients who will easily agree to further treatment with the smallest hope of relief.
INJECTION TECHNIQUES

For general points about the procedure to discuss with patients, see “What to tell patients.”
Cystoscopic detrusor injection
This procedure is usually done on an outpatient basis (eg, office, ambulatory surgery center). With the patient in the lithotomy position, 100 mL of 2% lidocaine is instilled into the bladder and is allowed 15 to 20 minutes to take effect. A flexible or rigid cystoscope can be used. Depending on the indication, the bladder is injected with 100 U to 300 U of botulinum toxin. The ideal depth of injection is 2 mm in the detrusor muscle, with each injection spaced about 1 cm apart. The recommended administration for 100 U is to inject 20 sites with 0.5 U per mL of saline and, for 200 U, to inject 30 sites with about 0.67 U per mL of saline.38 The location of the injections into the detrusor can vary, as long as adequate spacing is assured.
Injection sites vary. Proponents of injecting the trigone argue that as it is an area of greater nerve density, patients will have a better clinical response. Opponents argue that trigonal injection could result in distal ureteral paralysis and subsequent ureteral reflux. However, this theoretical concern has not been observed clinically.
Urethral injection (off-label use)
The urethra can be injected cystoscopically or periurethrally. Cystoscopic injection involves localization of the external sphincter using the rigid cystoscope and collagen needle; a total of 100 U is injected into the sphincter under direct vision, typically at the 3 o’clock and 9 o’clock positions.35 The periurethral technique is an option in women and involves a spinal needle with 100 U to 200 U of botulinum toxin injected into the external sphincter muscle at the 2 o’clock and 10 o’clock positions.
ADVERSE EFFECTS AND CONTRAINDICATIONS
Adverse effects are rare for urologic applications. The injections are localized, with little systemic absorption, and the doses are 1/1,000th of the theorized lethal dose in a 70-kg male.2 The maximum recommended dose for a 3-month period is 360 U.
Generalized muscle weakness has been reported in a paraplegic patient and in a tetraplegic patient after detrusor injections.2 Interestingly, both patients had return of bladder spasticity within 2 months, prompting speculation about diffusion of botulinum toxin through the bladder wall.2
Repeat injections can cause an immune response in up to 5% of patients.6 Patients undergoing repeat injections are at risk of forming neutralizing antibodies that can interfere with the efficacy of botulinum toxin.6 In a study by Schulte-Baukloh et al, all patients with antibodies to botulinum toxin had a history of recurrent urinary tract infection.39
Botulinum toxin injection is contraindicated in patients with preexisting neuromuscular disease, such as myasthenia gravis, Eaton-Lambert syndrome, and amyotrophic lateral sclerosis. It should also be avoided in patients who are breastfeeding, pregnant, or using agents that potentiate neuromuscular weakness, such as aminoglycosides.
Patients should be informed that some formulations of botulinum toxin include a stabilizer such as albumin derived from human blood, as this may be of religious or cultural significance.
- Leippold T, Reitz A, Schurch B. Botulinum toxin as a new therapy option for voiding disorders: current state of the art. Eur Urol 2003; 44:165–174.
- Sahai A, Khan M, Fowler CJ, Dasgupta P. Botulinum toxin for the treatment of lower urinary tract symptoms: a review. Neurourol Urodyn 2005; 24:2–12.
- Cruz F. Targets for botulinum toxin in the lower urinary tract. Neurourol Urodyn 2014; 33:31–38.
- Weedmark KA, Lambert DL, Mabon P, et al. Two novel toxin variants revealed by whole-genome sequencing of 175 Clostridium botulinum type E strains. Appl Environ Microbiol 2014; 80:6334–6345.
- Ha US, Park EY, Kim JC. Effect of botulinum toxin on expression of nerve growth factor and transient receptor potential vanilloid 1 in urothelium and detrusor muscle of rats with bladder outlet obstruction-induced detrusor overactivity. Urology 2011; 78:721.e1–721.e6
- Frenkl TL, Rackley RR. Injectable neuromodulatory agents: botulinum toxin therapy. Urol Clin North Am 2005; 32:89–99.
- Ikeda Y, Zabbarova IV, Birder LA, et al. Botulinum neurotoxin serotype A suppresses neurotransmitter release from afferent as well as efferent nerves in the urinary bladder. Eur Urol 2012; 62:1157–1164.
- Goldmark E, Niver B, Ginsberg DA. Neurogenic bladder: from diagnosis to management. Curr Urol Rep 2014; 15:448.
- Andersson KE. Current and future drugs for treatment of MS-associated bladder dysfunction. Ann Phys Rehabil Med 2014; 57:321–328.
- Schurch B, Stöhrer M, Kramer G, Schmid DM, Gaul G, Hauri D. Botulinum-A toxin for treating detrusor hyperreflexia in spinal cord injured patients: a new alternative to anticholinergic drugs? Preliminary results. J Urol 2000; 164:692–697.
- Schurch B, de Sèze M, Denys P, et al; Botox Detrusor Hyperreflexia Study Team. Botulinum toxin type a is a safe and effective treatment for neurogenic urinary incontinence: results of a single treatment, randomized, placebo controlled 6-month study. J Urol 2005; 174:196–200.
- Ehren I, Volz D, Farrelly E, et al. Efficacy and impact of botulinum toxin A on quality of life in patients with neurogenic detrusor overactivity: a randomised, placebo-controlled, double-blind study. Scand J Urol Nephrol 2007; 41:335–340.
- Ginsberg D, Gousse A, Keppenne V, et al. Phase 3 efficacy and tolerability study of onabotulinumtoxinA for urinary incontinence from neurogenic detrusor overactivity. J Urol 2012; 187:2131–2139.
- Cruz F, Herschorn S, Aliotta P, et al. Efficacy and safety of onabotulinumtoxinA in patients with urinary incontinence due to neurogenic detrusor overactivity: a randomised, double-blind, placebo-controlled trial. Eur Urol 2011; 60:742–750.
- Wagner TH, Patrick DL, Bavendam TG, Martin ML, Buesching DP. Quality of life of persons with urinary incontinence: development of a new measure. Urology 1996: 47:67–71.
- Kennelly M, Dmochowski R, Ethans K, et al. Long-term efficacy and safety of onabotulinumtoxinA in patients with urinary incontinence due to neurogenic detrusor overactivity: an interim analysis. Urology 2013; 81:491–497.
- Carlson JJ, Hansen RN, Dmochowski RR, Globe DR, Colayco DC, Sullivan SD. Estimating the cost-effectiveness of onabotulinumtoxinA for neurogenic detrusor overactivity in the United States. Clin Ther 2013; 35:414–424.
- Abrams P, Cardozo L, Fall M, et al; Standardisation Sub-Committee of the International Continence Society. The standardisation of terminology in lower urinary tract function: report from the standardisation sub-committee of the International Continence Society. Urology 2003; 61:37–49.
- Milsom I, Coyne KS, Nicholson S, Kvasz M, Chen CI, Wein AJ. Global prevalence and economic burden of urgency urinary incontinence: a systematic review. Eur Urol 2014; 65:79–95.
- Nitti VW, Dmochowski R, Herschorn S, et al; EMBARK Study Group. OnabotulinumtoxinA for the treatment of patients with overactive bladder and urinary incontinence: results of a phase 3, randomized, placebo controlled trial. J Urol 2013; 189:2186–2193.
- Chapple C, Sievert KD, MacDiarmid S, et al. OnabotulinumtoxinA 100 U significantly improves all idiopathic overactive bladder symptoms and quality of life in patients with overactive bladder and urinary incontinence: a randomised, double-blind, placebo-controlled trial. Eur Urol 2013; 64:249–256.
- Colman S, Chapple C, Nitti V, Haag-Molkenteller C, Hastedt C, Massow U. Validation of Treatment Benefit Scale for assessing subjective outcomes in treatment of overactive bladder. Urology 2008; 72:803–807.
- Visco AG, Brubaker L, Richter HE, et al; Pelvic Floor Disorders Network. Anticholinergic therapy vs onabotulinumtoxinA for urgency urinary incontinence. N Engl J Med 2012; 367:1803–1813.
- Gormley EA, Lightner DJ, Burgio KL, et al. Diagnosis and treatment of overactive bladder (non-neurogenic) in adults: AUA/SUFU Guideline. www.auanet.org/education/guidelines/overactive-bladder.cfm. Accessed June 11, 2015.
- Utomo E, Groen J, Blok BF. Surgical management of functional bladder outlet obstruction in adults with neurogenic bladder dysfunction. Cochrane Database Syst Rev 2014; 5:CD004927.
- Mahfouz W, Corcos J. Management of detrusor external sphincter dyssynergia in neurogenic bladder. Eur J Phys Rehabil Med 2011; 47:639–650.
- Dykstra DD, Sidi AA, Scott AB, Pagel JM, Goldish GD. Effects of botulinum A toxin on detrusor-sphincter dyssynergia in spinal cord injury patients. J Urol 1988; 139:919–922.
- Schurch B, Hauri D, Rodic B, Curt A, Meyer M, Rossier AB. Botulinum-A toxin as a treatment of detrusor-sphincter dyssynergia: a prospective study in 24 spinal cord injury patients. J Urol 1996; 155:1023–1029.
- de Sèze M, Petit H, Gallien, de Sèze MP, Joseph PA, Mazaux JM, Barat M. Botulinum a toxin and detrusor sphincter dyssynergia: a double-blind lidocaine-controlled study in 13 patients with spinal cord disease. Eur Urol 2002; 42:56–62.
- Gallien P, Reymann JM, Amarenco G, Nicolas B, de Sèze M, Bellissant E. Placebo controlled, randomised, double blind study of the effects of botulinum A toxin on detrusor sphincter dyssynergia in multiple sclerosis patients. J Neurol Neurosurg Psychiatry 2005; 76:1670–1676.
- Hanno PM, Burks DA, Clemens JQ, et al; Interstitial Cystitis Guidelines Panel of the American Urological Association Education and Research, Inc. AUA guideline for the diagnosis and treatment of interstitial cystitis/bladder pain syndrome. J Urol 2011; 185:2162–2170.
- Chuang YC, Yoshimura N, Huang CC, Chiang PH, Chancellor MB. Intravesical botulinum toxin a administration produces analgesia against acetic acid induced bladder pain responses in rats. J Urol 2004; 172:1529–1532.
- Zermann DH, Ishigooka M, Schubert J, Schmidt RA. Perisphincteric injection of botulinum toxin type A. A treatment option for patients with chronic prostatic pain? Eur Urol 2000; 38:393–399.
- Abbott JA, Jarvis SK, Lyons SD, Thomson A, Vancaille TG. Botulinum toxin type A for chronic pain and pelvic floor spasm in women: a randomized controlled trial. Obstet Gynecol 2006; 108:915–923.
- Smith CP, Radziszewski P, Borkowski A, Somogyi GT, Boone TB, Chancellor MB. Botulinum toxin A has antinociceptive effects in treating interstitial cystitis. Urology 2004; 64:871–875.
- Kuo HC, Chancellor MB. Comparison of intravesical botulinum toxin type A injections plus hydrodistention with hydrodistention alone for the treatment of refractory interstitial cystitis/painful bladder syndrome. BJU Int 2009: 104:657–661.
- Pinto R, Lopes T, Silva J, Silva C, Dinis P, Cruz F. Persistent therapeutic effect of repeated injections of onabotulinum toxin a in refractory bladder pain syndrome/interstitial cystitis. J Urol 2013; 189:548–553.
- Rovner E. Chapter 6: Practical aspects of administration of onabotulinumtoxinA. Neurourol Urodyn 2014; 33(suppl 3):S32–S37.
- Schulte-Baukloh H, Herholz J, Bigalke H, Miller K, Knispel HH. Results of a BoNT/A antibody study in children and adolescents after onabotulinumtoxin A (Botox®) detrusor injection. Urol Int 2011; 87:434–438.
Patients with loss of bladder control experience discomfort, embarrassment, personal care and health issues, and, often, significant pain, all with a decidedly negative impact on quality of life. Although some patients may find lifestyle modifications, drug therapy, and self-catheterization acceptable and effective, there is a clear need for more options.
Botulinum toxin, or onabotulinumtoxinA, is currently approved by the US Food and Drug Administration (FDA) for neurogenic detrusor overactivity and overactive bladder refractory to drug therapy. Studies so far have shown botulinum toxin injection to be safe and effective for these conditions, and these results have led to interest in off-label uses, eg, for detrusor external sphincter dyssynergia (DESD), motor and sensory urgency, and painful bladder syndrome/interstitial cystitis (Table 1).
Although more data from clinical trials are needed, botulinum toxin injection offers patients a much-needed treatment option.
HOW BOTULINUM TOXIN WORKS
Seven serotypes identified
Discovered in 1897, botulinum toxin is a neurotoxin produced by the gram-positive, rod-shaped anaerobic bacterium Clostridium botulinum1 and is the most poisonous naturally occurring toxin known.2 Seven immunologically distinct antigenic serotypes have been identified (A, B, C1, D, E, F, and G),1 but only types A and B are available for clinical use.
Most research into potential therapeutic uses has focused on type A, which has the longest duration of action, a clinical advantage.3 Recently, work has been done to further characterize other serotypes and to isolate additional variants of botulinum toxin. For example, serotype E, the predominant serotype associated with foodborne botulism, is being studied in an effort to prevent future outbreaks.4
Our discussion focuses on clinical uses of the serotype A botulinum toxin preparation, which we will refer to simply as botulinum toxin.
Studies exploring how it works
Botulinum toxin exerts its effects by binding to peripheral cholinergic terminals, inhibiting release of acetylcholine at the neuromuscular junction. Flaccid paralysis ensues as a result.
Results of animal studies have shed additional light on the specific actions of botulinum toxin A:
- It may alter levels of nerve growth factor and transient receptor potential vanilloid 1 in rats, and this may provide an additional mechanism of reducing bladder detrusor overactivity.5
- In addition to blocking acetylcholine release from motor neurons, it inhibits the release of neurotransmitters involved in bladder sensory afferent pathways.6
- It inhibits the release of substance P and glutamate, neuropeptides involved in sensory and nociceptive pathways.6,7
- It promotes apoptosis in prostatic tissue; however, this effect has not been shown in the bladder.3
The time necessary to recover function after botulinum toxin paralysis depends on the subtype of botulinum toxin as well as on the type of nerve terminal. Chemodenervation lasts from 3 to 6 months when the toxin is injected into the neuromuscular junction of skeletal muscle, and considerably longer (up to 1 year) when injected into the autonomic neurons of smooth muscle.2,6
TREATMENT OF NEUROGENIC DETRUSOR OVERACTIVITY
Neurogenic detrusor overactivity involves involuntary contractions of the bladder resulting from spinal cord injury, multiple sclerosis, and other neurologic conditions. An estimated 273,000 people in the United States have a spinal cord injury, and 81% of them have urologic symptoms ranging from areflexia to overactivity.8 From 75% to 100% of patients with multiple sclerosis have urologic symptoms, and detrusor overactivity is the most common.9
Detrusor overactivity can cause urinary urgency, urinary frequency, and urgency incontinence, significantly affecting quality of life and leading to skin breakdown, sacral ulcerations, and challenges with personal care.
Anticholinergic drugs have been the mainstay of therapy. If drug therapy failed, the next option was reconstructive surgery, often augmentation cystoplasty. Thus, botulinum toxin injection is an important advance in treatment options.
Studies that showed effectiveness
Botulinum toxin for neurogenic detrusor overactivity was first studied by Schurch et al.10 In their study, 200 U or 300 U was injected into the trigone of 21 patients with spinal cord injury and urgency incontinence managed with intermittent self-catheterization.10 At 6 weeks after injection, 17 of the 19 patients seen at follow-up visits were completely continent. Urodynamic evaluation revealed significant increases in maximum cystometric capacity and in volume at first involuntary detrusor contraction, and a decrease in detrusor voiding pressure. Of the 11 patients available for follow-up at 16 and 36 weeks, improvements in measures of incontinence and urodynamic function persisted.
In addition, two small randomized controlled trials11,12 showed significant increases in cystometric bladder capacity, significant improvement in quality-of-life measures, and reduction in episodes of urgency incontinence.
In 2011 and 2012, two multicenter double-blind randomized controlled trials reported on patients with multiple sclerosis and spinal cord injury with neurogenic detrusor overactivity inadequately managed with drug therapy. The patients were randomized to botulinum toxin injection (200 U or 300 U) or placebo injection.13,14 The primary end point for both studies was the change from baseline in episodes of urinary incontinence per week at week 6. Secondary end points were maximum cystometric capacity, maximum detrusor pressure during first involuntary detrusor contraction, and score on the Incontinence Quality of Life scale.15
In both studies, the mean number of urinary incontinence episodes per week was 33 at baseline. At week 6, Cruz et al14 found that patients who received botulinum toxin injection had significantly fewer episodes per week (21.8 fewer with 200 U, 19.4 fewer with 300 U) than those in the placebo group, who had 13.2 fewer episodes per week (P < .01). Ginsberg et al13 reported decreases in the mean number of episodes of urinary incontinence of 21, 23, and 9 episodes per week in the 200 U, 300 U, and placebo groups, respectively (P < .001). The patients who received botulinum toxin had statistically significant improvements in maximum cystometric capacity, maximum detrusor pressure during first involuntary detrusor contraction, and Incontinence Quality of Life scores compared with placebo (P < .001). Thirty-eight percent of patients in the treatment group were fully continent.13,14
Safety and adverse effects
The most frequently reported adverse events were urinary tract infection (24% of patients)13,14 and urinary retention requiring initiation of clean intermittent catheterization. In the study by Cruz et al,14 these were reported in 30% with 200 U, 42% with 300 U, and 12% with placebo, while in the study by Ginsberg et al13 they were reported in 35% with 200 U, 42% with 300 U, and 10% with placebo.
In a study of long-term safety and efficacy of botulinum toxin injection in patients with neurogenic detrusor overactivity, Kennelly et al16 found that patients undergoing repeat injections had sustained reductions in episodes of incontinence and increases in the maximum cystometric capacity and quality of life scores, with no increase in adverse events over time.16
But is it cost-effective?
While botulinum toxin injection may be safe and effective for neurogenic detrusor overactivity, is it cost-effective?
Carlson et al17 used a Markov State Transition model to assess the cost of refractory neurogenic detrusor overactivity in patients receiving botulinum toxin vs best supportive care (incontinence pads, medications, intermittent self-catheterization).17 They found that the injections were more expensive than supportive care but were cost-effective when considering the reduction in episodes of incontinence, the reduced need for incontinence products, and improvement in measures of quality of life.
What the evidence indicates
Trials of botulinum toxin injection for neurogenic detrusor overactivity have shown that it improves continence, maximum cystometric capacity, detrusor pressures, and quality of life. The main adverse effects are urinary tract infection and urinary retention requiring intermittent self-catheterization.
Although many patients with this condition are already self-catheterizing, the physician must discuss this before botulinum toxin therapy to ensure that the patient or a family member is able to perform catheterization. Studies have shown that patients have an increase in urinary tract infections after botulinum injections. But in these studies, a urinary tract infection was defined as 100,000 colony-forming units or the presence of leukocytosis with or without symptoms. It is important to remember that patients on intermittent catheterization have bacteriuria and should be treated only for symptomatic, not asymptomatic, bacteriuria.
TREATMENT OF IDIOPATHIC OVERACTIVE BLADDER
Patients with idiopathic overactive bladder have urinary urgency accompanied by urgency incontinence, nocturia, or urinary frequency.18 The prevalence of this condition has been reported to range from 1.7% to 13.3% in men age 30 and older and 7% to 30.3% in women of similar ages. About one-third of women with overactive bladder also have detrusor overactivity.19 Overactive bladder presents a significant economic and medical burden on the healthcare system, as well as having a negative impact on quality of life.
The FDA approved botulinum toxin injection for treatment of idiopathic overactive bladder in January 2013.
Evidence of effectiveness
Two multicenter randomized controlled trials20,21 of botulinum toxin 100 U enrolled patients age 18 and older who had more than three episodes of urinary urgency incontinence in a 3-day period or more than eight micturitions per day inadequately managed by anticholinergic drug therapy. Primary end points were the change from baseline in the number of episodes of urinary incontinence per day and the proportion of patients with a positive response on the Treatment Benefit Scale22 at week 12. Secondary end points included episodes of urinary urgency incontinence, micturition, urgency, and nocturia, and scores on health-related quality of life questionnaires (Incontinence Quality of Life scale, King’s Health Questionnaire).
In both studies, patients receiving botulinum toxin had significantly fewer episodes of incontinence compared with placebo (−2.65 vs −0.87; P < .001 and −2.95 vs −1.03; P < .001).20,21 Reductions from baseline in all other symptoms of overactive bladder, a positive treatment response on the treatment benefit scale, and improvements in quality-of-life scores were also significantly greater with botulinum toxin injection than with placebo (P ≤ .01).
As in the studies of neurogenic detrusor overactivity, the most common adverse effects were urinary tract infection (occurring in 15.5%20 and 24.1%21 of patients) and urinary retention requiring self-catheterization (5.4%20 and 6.9%21).
The largest study to date of anticholinergic therapy vs botulinum toxin injection23 in women with urinary urgency incontinence, published in 2012, studied nearly 250 women who had five or more episodes of idiopathic urgency incontinence in a 3-day period. They were randomized either to daily oral therapy (solifenacin 5 mg with possible escalation to 10 mg and, if necessary, a subsequent switch to extended-release trospium 60 mg) plus one intradetrusor injection of saline, or to a daily oral placebo plus one injection of botulinum toxin 100 U.23
The dropout rate was low in both groups, with 93% of patients in both groups completing the 6-month protocol. Women experienced a mean reduction in urgency incontinence episodes of 3.4 per day (baseline 5) in the anticholinergic group vs 3.3 episodes in the botulinum toxin group (P = .81). However, more patients achieved complete resolution of urinary urgency incontinence in the botulinum toxin group than in the anticholinergic therapy group (27% vs 13%; P = .003). Quality of life improved in both groups without a significant difference between the groups. The botulinum toxin group had higher rates of initiation of self-catheterization (5% vs 0%, P = .01) and urinary tract infection (33% vs 13%, P < .001).23
Botulinum toxin as a third-line therapy
In May 2014, the American Urological Association updated its guidelines on idiopathic overactive bladder24 to include botulinum toxin injection as standard third-line therapy for patients in whom behavioral and medical management (ie, anticholinergics and beta-3-agonists) failed.
Interpreting the evidence to date
Overall, studies in idiopathic overactive bladder have shown a reduction in episodes of urgency incontinence and other symptoms, with some data also demonstrating a corresponding improvement in quality of life.
As in neurogenic detrusor overactivity, the main risks associated with botulinum toxin injection are urinary tract infection and the need to initiate self-catheterization. Although 94% of patients studied did not require self-catheterization after injection, the patient’s ability to perform self-catheterization should be determined before proceeding with botulinum toxin injections.
DETRUSOR EXTERNAL SPHINCTER DYSSYNERGIA
Botulinum toxin has been used not only to improve bladder storage but also to facilitate bladder emptying, as in patients with DESD, a lack of coordination between the bladder and the urinary sphincter. Normal voiding involves relaxation of the urinary sphincter and contraction of the bladder; in DESD the sphincter contracts and works against the bladder’s ability to empty. This leads not only to difficulty emptying the bladder but also to elevated bladder pressure, which can cause renal damage if untreated.
DESD can be seen after injury between the pontine micturition center, which coordinates activity between the bladder and the sphincter, and the caudal spinal cord. This can occur in spinal cord injury, multiple sclerosis, myelomeningocele, and transverse myelitis and can cause significant morbidity for the patient.
Treatment options include drug therapy, injection of botulinum toxin into the sphincter, clean intermittent catheterization, indwelling catheterization, urethral stenting, sphincterotomy, and reconstructive surgery such as urinary diversion.25
The goals of therapy are to avoid the need for clean intermittent catheterization in patients who have difficulty with manual dexterity, and to avoid the need for surgical procedures such as sphincterotomy and urinary diversion. The efficacy of urethral stenting is low, and medical management can be limited.26
In the first published report on botulinum toxin for DESD (in 1988),27 of 11 patients with spinal cord injury and DESD who received botulinum toxin injected into the external urethral sphincter, 10 showed signs of sphincter denervation on electromyography and reductions in urethral pressure profiles and postvoid residual volumes. Schurch et al28 and de Sèze et al29 also reported reductions in postvoid residual volume and maximal urethral pressures in patients with spinal cord injury and DESD.
In 2005, Gallien et al30 reported what is still the largest multicenter randomized controlled trial of botulinum toxin injection in DESD. Eighty-six patients with multiple sclerosis, DESD, and chronic urinary retention were randomized to receive either a single transperineal botulinum toxin injection of 100 U plus the alpha-1-blocker alfuzosin, or a placebo injection plus alfuzosin. Botulinum toxin treatment was associated with significantly increased voided volumes and reduced premicturition and maximal detrusor pressures, but no significant decrease in postvoid residual volume.30
More study needed
Despite these findings, a Cochrane Review concluded that, given the limited experience with intrasphincteric injection of botulinum toxin, data from larger randomized controlled trials are needed before making definitive recommendations.25 In the meantime, the clinician must weigh the low morbidity of the procedure against the limited options in the treatment of these patients.
OFF-LABEL UROLOGIC INDICATIONS
Botulinum toxin injection has been studied off-label for painful bladder syndrome/interstitial cystitis and for chronic prostatic pain. Patients with these conditions often describe pain with filling of the bladder, which leads to urinary frequency in an attempt to relieve the pain.
These pain syndromes can be difficult to treat and can have a devastating impact on quality of life. Treatment options include pain management, stress management, physical therapy, intravesical therapies, cystoscopy with hydrodistention, neuromodulation, cyclosporine, urinary diversion surgery, and botulinum toxin injection (an off-label use).31
In painful bladder syndrome/interstitial cystitis, botulinum toxin is thought to act on sensory afferent pathways, as well as to inhibit the release of substance P and glutamate, neuropeptides involved in sensory and nociceptive pathways.6 In animal studies,32 botulinum toxin was found to inhibit the afferent neural response by inhibiting mechanoreceptor-mediated release of adenosine triphosphate and by causing a decrease in calcitonin gene-related peptide, which helps regulate micturition and mediates painful bladder sensation.
Clinical studies to date in pelvic pain syndromes
Data from clinical studies of botulinum toxin injection for pelvic pain syndromes are limited. Zermann et al33 performed transurethral perisphincteric injection in 11 men with chronic prostatic pain, 9 of whom reported subjective pain relief, with an average decrease from 7.2 to 1.6 on a visual analogue scale. Postinjection urodynamic studies showed a decrease in functional urethral length, urethral closure pressure, and postvoid residual volume, and an increase in the peak and average flow rates.33
Abbott et al34 evaluated the effect of botulinum toxin injection into the levator ani in 12 women with chronic pelvic pain and pelvic floor hypertonicity. Pelvic floor manometry showed significant reduction in resting muscle pressures and improvements in dyspareunia and nonmenstrual pain. There were also improvements in quality of life and dyschezia, but these were not statistically significant.
Smith et al35 injected botulinum toxin into the detrusor of 13 women with refractory painful bladder syndrome and interstitial cystitis,35 and 9 women (69%) noted statistically significant improvements in the Interstitial Cystitis Symptom Index and Interstitial Cystitis Problem Index, daytime frequency, nocturia, pain, and urodynamic parameters (volume at first desire to void, and maximum cystometric capacity).
In a prospective randomized study of patients with refractory painful bladder syndrome and interstitial cystitis, Kuo and Chancellor36 compared suburothelial injection of 200 U or 100 U of botulinum toxin plus hydrodistention against hydrodistention alone.Patients who received botulinum toxin had increased bladder capacity and improved long-term pain relief, but no difference was noted between 200 U and 100 U, and more adverse effects were seen with the higher dose.36
Pinto et al37 treated 16 women with refractory painful bladder syndrome and interstitial cystitis with intratrigonal injections of botulinum toxin and reported improvements in pain scores, symptom scores, urinary frequency, and quality-of-life measures. The effect lasted 9.9 months (± 2.4 months) and persisted with successive injections.37
More study needed
Although these studies show that botulinum toxin injection for pelvic pain syndromes has the potential to improve pain, urinary frequency, bladder sensation, bladder capacity, and quality of life, larger randomized controlled trials are needed.
Again, the treatment options are limited for refractory painful bladder syndrome and interstitial cystitis. Patients may be desperate for relief from their symptoms. Practitioners must manage expectations and properly inform patients of the potential risks of treatments, especially with patients who will easily agree to further treatment with the smallest hope of relief.
INJECTION TECHNIQUES
For general points about the procedure to discuss with patients, see “What to tell patients.”
Cystoscopic detrusor injection
This procedure is usually done on an outpatient basis (eg, office, ambulatory surgery center). With the patient in the lithotomy position, 100 mL of 2% lidocaine is instilled into the bladder and is allowed 15 to 20 minutes to take effect. A flexible or rigid cystoscope can be used. Depending on the indication, the bladder is injected with 100 U to 300 U of botulinum toxin. The ideal depth of injection is 2 mm in the detrusor muscle, with each injection spaced about 1 cm apart. The recommended administration for 100 U is to inject 20 sites with 0.5 U per mL of saline and, for 200 U, to inject 30 sites with about 0.67 U per mL of saline.38 The location of the injections into the detrusor can vary, as long as adequate spacing is assured.
Injection sites vary. Proponents of injecting the trigone argue that as it is an area of greater nerve density, patients will have a better clinical response. Opponents argue that trigonal injection could result in distal ureteral paralysis and subsequent ureteral reflux. However, this theoretical concern has not been observed clinically.
Urethral injection (off-label use)
The urethra can be injected cystoscopically or periurethrally. Cystoscopic injection involves localization of the external sphincter using the rigid cystoscope and collagen needle; a total of 100 U is injected into the sphincter under direct vision, typically at the 3 o’clock and 9 o’clock positions.35 The periurethral technique is an option in women and involves a spinal needle with 100 U to 200 U of botulinum toxin injected into the external sphincter muscle at the 2 o’clock and 10 o’clock positions.
ADVERSE EFFECTS AND CONTRAINDICATIONS
Adverse effects are rare for urologic applications. The injections are localized, with little systemic absorption, and the doses are 1/1,000th of the theorized lethal dose in a 70-kg male.2 The maximum recommended dose for a 3-month period is 360 U.
Generalized muscle weakness has been reported in a paraplegic patient and in a tetraplegic patient after detrusor injections.2 Interestingly, both patients had return of bladder spasticity within 2 months, prompting speculation about diffusion of botulinum toxin through the bladder wall.2
Repeat injections can cause an immune response in up to 5% of patients.6 Patients undergoing repeat injections are at risk of forming neutralizing antibodies that can interfere with the efficacy of botulinum toxin.6 In a study by Schulte-Baukloh et al, all patients with antibodies to botulinum toxin had a history of recurrent urinary tract infection.39
Botulinum toxin injection is contraindicated in patients with preexisting neuromuscular disease, such as myasthenia gravis, Eaton-Lambert syndrome, and amyotrophic lateral sclerosis. It should also be avoided in patients who are breastfeeding, pregnant, or using agents that potentiate neuromuscular weakness, such as aminoglycosides.
Patients should be informed that some formulations of botulinum toxin include a stabilizer such as albumin derived from human blood, as this may be of religious or cultural significance.
Patients with loss of bladder control experience discomfort, embarrassment, personal care and health issues, and, often, significant pain, all with a decidedly negative impact on quality of life. Although some patients may find lifestyle modifications, drug therapy, and self-catheterization acceptable and effective, there is a clear need for more options.
Botulinum toxin, or onabotulinumtoxinA, is currently approved by the US Food and Drug Administration (FDA) for neurogenic detrusor overactivity and overactive bladder refractory to drug therapy. Studies so far have shown botulinum toxin injection to be safe and effective for these conditions, and these results have led to interest in off-label uses, eg, for detrusor external sphincter dyssynergia (DESD), motor and sensory urgency, and painful bladder syndrome/interstitial cystitis (Table 1).
Although more data from clinical trials are needed, botulinum toxin injection offers patients a much-needed treatment option.
HOW BOTULINUM TOXIN WORKS
Seven serotypes identified
Discovered in 1897, botulinum toxin is a neurotoxin produced by the gram-positive, rod-shaped anaerobic bacterium Clostridium botulinum1 and is the most poisonous naturally occurring toxin known.2 Seven immunologically distinct antigenic serotypes have been identified (A, B, C1, D, E, F, and G),1 but only types A and B are available for clinical use.
Most research into potential therapeutic uses has focused on type A, which has the longest duration of action, a clinical advantage.3 Recently, work has been done to further characterize other serotypes and to isolate additional variants of botulinum toxin. For example, serotype E, the predominant serotype associated with foodborne botulism, is being studied in an effort to prevent future outbreaks.4
Our discussion focuses on clinical uses of the serotype A botulinum toxin preparation, which we will refer to simply as botulinum toxin.
Studies exploring how it works
Botulinum toxin exerts its effects by binding to peripheral cholinergic terminals, inhibiting release of acetylcholine at the neuromuscular junction. Flaccid paralysis ensues as a result.
Results of animal studies have shed additional light on the specific actions of botulinum toxin A:
- It may alter levels of nerve growth factor and transient receptor potential vanilloid 1 in rats, and this may provide an additional mechanism of reducing bladder detrusor overactivity.5
- In addition to blocking acetylcholine release from motor neurons, it inhibits the release of neurotransmitters involved in bladder sensory afferent pathways.6
- It inhibits the release of substance P and glutamate, neuropeptides involved in sensory and nociceptive pathways.6,7
- It promotes apoptosis in prostatic tissue; however, this effect has not been shown in the bladder.3
The time necessary to recover function after botulinum toxin paralysis depends on the subtype of botulinum toxin as well as on the type of nerve terminal. Chemodenervation lasts from 3 to 6 months when the toxin is injected into the neuromuscular junction of skeletal muscle, and considerably longer (up to 1 year) when injected into the autonomic neurons of smooth muscle.2,6
TREATMENT OF NEUROGENIC DETRUSOR OVERACTIVITY
Neurogenic detrusor overactivity involves involuntary contractions of the bladder resulting from spinal cord injury, multiple sclerosis, and other neurologic conditions. An estimated 273,000 people in the United States have a spinal cord injury, and 81% of them have urologic symptoms ranging from areflexia to overactivity.8 From 75% to 100% of patients with multiple sclerosis have urologic symptoms, and detrusor overactivity is the most common.9
Detrusor overactivity can cause urinary urgency, urinary frequency, and urgency incontinence, significantly affecting quality of life and leading to skin breakdown, sacral ulcerations, and challenges with personal care.
Anticholinergic drugs have been the mainstay of therapy. If drug therapy failed, the next option was reconstructive surgery, often augmentation cystoplasty. Thus, botulinum toxin injection is an important advance in treatment options.
Studies that showed effectiveness
Botulinum toxin for neurogenic detrusor overactivity was first studied by Schurch et al.10 In their study, 200 U or 300 U was injected into the trigone of 21 patients with spinal cord injury and urgency incontinence managed with intermittent self-catheterization.10 At 6 weeks after injection, 17 of the 19 patients seen at follow-up visits were completely continent. Urodynamic evaluation revealed significant increases in maximum cystometric capacity and in volume at first involuntary detrusor contraction, and a decrease in detrusor voiding pressure. Of the 11 patients available for follow-up at 16 and 36 weeks, improvements in measures of incontinence and urodynamic function persisted.
In addition, two small randomized controlled trials11,12 showed significant increases in cystometric bladder capacity, significant improvement in quality-of-life measures, and reduction in episodes of urgency incontinence.
In 2011 and 2012, two multicenter double-blind randomized controlled trials reported on patients with multiple sclerosis and spinal cord injury with neurogenic detrusor overactivity inadequately managed with drug therapy. The patients were randomized to botulinum toxin injection (200 U or 300 U) or placebo injection.13,14 The primary end point for both studies was the change from baseline in episodes of urinary incontinence per week at week 6. Secondary end points were maximum cystometric capacity, maximum detrusor pressure during first involuntary detrusor contraction, and score on the Incontinence Quality of Life scale.15
In both studies, the mean number of urinary incontinence episodes per week was 33 at baseline. At week 6, Cruz et al14 found that patients who received botulinum toxin injection had significantly fewer episodes per week (21.8 fewer with 200 U, 19.4 fewer with 300 U) than those in the placebo group, who had 13.2 fewer episodes per week (P < .01). Ginsberg et al13 reported decreases in the mean number of episodes of urinary incontinence of 21, 23, and 9 episodes per week in the 200 U, 300 U, and placebo groups, respectively (P < .001). The patients who received botulinum toxin had statistically significant improvements in maximum cystometric capacity, maximum detrusor pressure during first involuntary detrusor contraction, and Incontinence Quality of Life scores compared with placebo (P < .001). Thirty-eight percent of patients in the treatment group were fully continent.13,14
Safety and adverse effects
The most frequently reported adverse events were urinary tract infection (24% of patients)13,14 and urinary retention requiring initiation of clean intermittent catheterization. In the study by Cruz et al,14 these were reported in 30% with 200 U, 42% with 300 U, and 12% with placebo, while in the study by Ginsberg et al13 they were reported in 35% with 200 U, 42% with 300 U, and 10% with placebo.
In a study of long-term safety and efficacy of botulinum toxin injection in patients with neurogenic detrusor overactivity, Kennelly et al16 found that patients undergoing repeat injections had sustained reductions in episodes of incontinence and increases in the maximum cystometric capacity and quality of life scores, with no increase in adverse events over time.16
But is it cost-effective?
While botulinum toxin injection may be safe and effective for neurogenic detrusor overactivity, is it cost-effective?
Carlson et al17 used a Markov State Transition model to assess the cost of refractory neurogenic detrusor overactivity in patients receiving botulinum toxin vs best supportive care (incontinence pads, medications, intermittent self-catheterization).17 They found that the injections were more expensive than supportive care but were cost-effective when considering the reduction in episodes of incontinence, the reduced need for incontinence products, and improvement in measures of quality of life.
What the evidence indicates
Trials of botulinum toxin injection for neurogenic detrusor overactivity have shown that it improves continence, maximum cystometric capacity, detrusor pressures, and quality of life. The main adverse effects are urinary tract infection and urinary retention requiring intermittent self-catheterization.
Although many patients with this condition are already self-catheterizing, the physician must discuss this before botulinum toxin therapy to ensure that the patient or a family member is able to perform catheterization. Studies have shown that patients have an increase in urinary tract infections after botulinum injections. But in these studies, a urinary tract infection was defined as 100,000 colony-forming units or the presence of leukocytosis with or without symptoms. It is important to remember that patients on intermittent catheterization have bacteriuria and should be treated only for symptomatic, not asymptomatic, bacteriuria.
TREATMENT OF IDIOPATHIC OVERACTIVE BLADDER
Patients with idiopathic overactive bladder have urinary urgency accompanied by urgency incontinence, nocturia, or urinary frequency.18 The prevalence of this condition has been reported to range from 1.7% to 13.3% in men age 30 and older and 7% to 30.3% in women of similar ages. About one-third of women with overactive bladder also have detrusor overactivity.19 Overactive bladder presents a significant economic and medical burden on the healthcare system, as well as having a negative impact on quality of life.
The FDA approved botulinum toxin injection for treatment of idiopathic overactive bladder in January 2013.
Evidence of effectiveness
Two multicenter randomized controlled trials20,21 of botulinum toxin 100 U enrolled patients age 18 and older who had more than three episodes of urinary urgency incontinence in a 3-day period or more than eight micturitions per day inadequately managed by anticholinergic drug therapy. Primary end points were the change from baseline in the number of episodes of urinary incontinence per day and the proportion of patients with a positive response on the Treatment Benefit Scale22 at week 12. Secondary end points included episodes of urinary urgency incontinence, micturition, urgency, and nocturia, and scores on health-related quality of life questionnaires (Incontinence Quality of Life scale, King’s Health Questionnaire).
In both studies, patients receiving botulinum toxin had significantly fewer episodes of incontinence compared with placebo (−2.65 vs −0.87; P < .001 and −2.95 vs −1.03; P < .001).20,21 Reductions from baseline in all other symptoms of overactive bladder, a positive treatment response on the treatment benefit scale, and improvements in quality-of-life scores were also significantly greater with botulinum toxin injection than with placebo (P ≤ .01).
As in the studies of neurogenic detrusor overactivity, the most common adverse effects were urinary tract infection (occurring in 15.5%20 and 24.1%21 of patients) and urinary retention requiring self-catheterization (5.4%20 and 6.9%21).
The largest study to date of anticholinergic therapy vs botulinum toxin injection23 in women with urinary urgency incontinence, published in 2012, studied nearly 250 women who had five or more episodes of idiopathic urgency incontinence in a 3-day period. They were randomized either to daily oral therapy (solifenacin 5 mg with possible escalation to 10 mg and, if necessary, a subsequent switch to extended-release trospium 60 mg) plus one intradetrusor injection of saline, or to a daily oral placebo plus one injection of botulinum toxin 100 U.23
The dropout rate was low in both groups, with 93% of patients in both groups completing the 6-month protocol. Women experienced a mean reduction in urgency incontinence episodes of 3.4 per day (baseline 5) in the anticholinergic group vs 3.3 episodes in the botulinum toxin group (P = .81). However, more patients achieved complete resolution of urinary urgency incontinence in the botulinum toxin group than in the anticholinergic therapy group (27% vs 13%; P = .003). Quality of life improved in both groups without a significant difference between the groups. The botulinum toxin group had higher rates of initiation of self-catheterization (5% vs 0%, P = .01) and urinary tract infection (33% vs 13%, P < .001).23
Botulinum toxin as a third-line therapy
In May 2014, the American Urological Association updated its guidelines on idiopathic overactive bladder24 to include botulinum toxin injection as standard third-line therapy for patients in whom behavioral and medical management (ie, anticholinergics and beta-3-agonists) failed.
Interpreting the evidence to date
Overall, studies in idiopathic overactive bladder have shown a reduction in episodes of urgency incontinence and other symptoms, with some data also demonstrating a corresponding improvement in quality of life.
As in neurogenic detrusor overactivity, the main risks associated with botulinum toxin injection are urinary tract infection and the need to initiate self-catheterization. Although 94% of patients studied did not require self-catheterization after injection, the patient’s ability to perform self-catheterization should be determined before proceeding with botulinum toxin injections.
DETRUSOR EXTERNAL SPHINCTER DYSSYNERGIA
Botulinum toxin has been used not only to improve bladder storage but also to facilitate bladder emptying, as in patients with DESD, a lack of coordination between the bladder and the urinary sphincter. Normal voiding involves relaxation of the urinary sphincter and contraction of the bladder; in DESD the sphincter contracts and works against the bladder’s ability to empty. This leads not only to difficulty emptying the bladder but also to elevated bladder pressure, which can cause renal damage if untreated.
DESD can be seen after injury between the pontine micturition center, which coordinates activity between the bladder and the sphincter, and the caudal spinal cord. This can occur in spinal cord injury, multiple sclerosis, myelomeningocele, and transverse myelitis and can cause significant morbidity for the patient.
Treatment options include drug therapy, injection of botulinum toxin into the sphincter, clean intermittent catheterization, indwelling catheterization, urethral stenting, sphincterotomy, and reconstructive surgery such as urinary diversion.25
The goals of therapy are to avoid the need for clean intermittent catheterization in patients who have difficulty with manual dexterity, and to avoid the need for surgical procedures such as sphincterotomy and urinary diversion. The efficacy of urethral stenting is low, and medical management can be limited.26
In the first published report on botulinum toxin for DESD (in 1988),27 of 11 patients with spinal cord injury and DESD who received botulinum toxin injected into the external urethral sphincter, 10 showed signs of sphincter denervation on electromyography and reductions in urethral pressure profiles and postvoid residual volumes. Schurch et al28 and de Sèze et al29 also reported reductions in postvoid residual volume and maximal urethral pressures in patients with spinal cord injury and DESD.
In 2005, Gallien et al30 reported what is still the largest multicenter randomized controlled trial of botulinum toxin injection in DESD. Eighty-six patients with multiple sclerosis, DESD, and chronic urinary retention were randomized to receive either a single transperineal botulinum toxin injection of 100 U plus the alpha-1-blocker alfuzosin, or a placebo injection plus alfuzosin. Botulinum toxin treatment was associated with significantly increased voided volumes and reduced premicturition and maximal detrusor pressures, but no significant decrease in postvoid residual volume.30
More study needed
Despite these findings, a Cochrane Review concluded that, given the limited experience with intrasphincteric injection of botulinum toxin, data from larger randomized controlled trials are needed before making definitive recommendations.25 In the meantime, the clinician must weigh the low morbidity of the procedure against the limited options in the treatment of these patients.
OFF-LABEL UROLOGIC INDICATIONS
Botulinum toxin injection has been studied off-label for painful bladder syndrome/interstitial cystitis and for chronic prostatic pain. Patients with these conditions often describe pain with filling of the bladder, which leads to urinary frequency in an attempt to relieve the pain.
These pain syndromes can be difficult to treat and can have a devastating impact on quality of life. Treatment options include pain management, stress management, physical therapy, intravesical therapies, cystoscopy with hydrodistention, neuromodulation, cyclosporine, urinary diversion surgery, and botulinum toxin injection (an off-label use).31
In painful bladder syndrome/interstitial cystitis, botulinum toxin is thought to act on sensory afferent pathways, as well as to inhibit the release of substance P and glutamate, neuropeptides involved in sensory and nociceptive pathways.6 In animal studies,32 botulinum toxin was found to inhibit the afferent neural response by inhibiting mechanoreceptor-mediated release of adenosine triphosphate and by causing a decrease in calcitonin gene-related peptide, which helps regulate micturition and mediates painful bladder sensation.
Clinical studies to date in pelvic pain syndromes
Data from clinical studies of botulinum toxin injection for pelvic pain syndromes are limited. Zermann et al33 performed transurethral perisphincteric injection in 11 men with chronic prostatic pain, 9 of whom reported subjective pain relief, with an average decrease from 7.2 to 1.6 on a visual analogue scale. Postinjection urodynamic studies showed a decrease in functional urethral length, urethral closure pressure, and postvoid residual volume, and an increase in the peak and average flow rates.33
Abbott et al34 evaluated the effect of botulinum toxin injection into the levator ani in 12 women with chronic pelvic pain and pelvic floor hypertonicity. Pelvic floor manometry showed significant reduction in resting muscle pressures and improvements in dyspareunia and nonmenstrual pain. There were also improvements in quality of life and dyschezia, but these were not statistically significant.
Smith et al35 injected botulinum toxin into the detrusor of 13 women with refractory painful bladder syndrome and interstitial cystitis,35 and 9 women (69%) noted statistically significant improvements in the Interstitial Cystitis Symptom Index and Interstitial Cystitis Problem Index, daytime frequency, nocturia, pain, and urodynamic parameters (volume at first desire to void, and maximum cystometric capacity).
In a prospective randomized study of patients with refractory painful bladder syndrome and interstitial cystitis, Kuo and Chancellor36 compared suburothelial injection of 200 U or 100 U of botulinum toxin plus hydrodistention against hydrodistention alone.Patients who received botulinum toxin had increased bladder capacity and improved long-term pain relief, but no difference was noted between 200 U and 100 U, and more adverse effects were seen with the higher dose.36
Pinto et al37 treated 16 women with refractory painful bladder syndrome and interstitial cystitis with intratrigonal injections of botulinum toxin and reported improvements in pain scores, symptom scores, urinary frequency, and quality-of-life measures. The effect lasted 9.9 months (± 2.4 months) and persisted with successive injections.37
More study needed
Although these studies show that botulinum toxin injection for pelvic pain syndromes has the potential to improve pain, urinary frequency, bladder sensation, bladder capacity, and quality of life, larger randomized controlled trials are needed.
Again, the treatment options are limited for refractory painful bladder syndrome and interstitial cystitis. Patients may be desperate for relief from their symptoms. Practitioners must manage expectations and properly inform patients of the potential risks of treatments, especially with patients who will easily agree to further treatment with the smallest hope of relief.
INJECTION TECHNIQUES
For general points about the procedure to discuss with patients, see “What to tell patients.”
Cystoscopic detrusor injection
This procedure is usually done on an outpatient basis (eg, office, ambulatory surgery center). With the patient in the lithotomy position, 100 mL of 2% lidocaine is instilled into the bladder and is allowed 15 to 20 minutes to take effect. A flexible or rigid cystoscope can be used. Depending on the indication, the bladder is injected with 100 U to 300 U of botulinum toxin. The ideal depth of injection is 2 mm in the detrusor muscle, with each injection spaced about 1 cm apart. The recommended administration for 100 U is to inject 20 sites with 0.5 U per mL of saline and, for 200 U, to inject 30 sites with about 0.67 U per mL of saline.38 The location of the injections into the detrusor can vary, as long as adequate spacing is assured.
Injection sites vary. Proponents of injecting the trigone argue that as it is an area of greater nerve density, patients will have a better clinical response. Opponents argue that trigonal injection could result in distal ureteral paralysis and subsequent ureteral reflux. However, this theoretical concern has not been observed clinically.
Urethral injection (off-label use)
The urethra can be injected cystoscopically or periurethrally. Cystoscopic injection involves localization of the external sphincter using the rigid cystoscope and collagen needle; a total of 100 U is injected into the sphincter under direct vision, typically at the 3 o’clock and 9 o’clock positions.35 The periurethral technique is an option in women and involves a spinal needle with 100 U to 200 U of botulinum toxin injected into the external sphincter muscle at the 2 o’clock and 10 o’clock positions.
ADVERSE EFFECTS AND CONTRAINDICATIONS
Adverse effects are rare for urologic applications. The injections are localized, with little systemic absorption, and the doses are 1/1,000th of the theorized lethal dose in a 70-kg male.2 The maximum recommended dose for a 3-month period is 360 U.
Generalized muscle weakness has been reported in a paraplegic patient and in a tetraplegic patient after detrusor injections.2 Interestingly, both patients had return of bladder spasticity within 2 months, prompting speculation about diffusion of botulinum toxin through the bladder wall.2
Repeat injections can cause an immune response in up to 5% of patients.6 Patients undergoing repeat injections are at risk of forming neutralizing antibodies that can interfere with the efficacy of botulinum toxin.6 In a study by Schulte-Baukloh et al, all patients with antibodies to botulinum toxin had a history of recurrent urinary tract infection.39
Botulinum toxin injection is contraindicated in patients with preexisting neuromuscular disease, such as myasthenia gravis, Eaton-Lambert syndrome, and amyotrophic lateral sclerosis. It should also be avoided in patients who are breastfeeding, pregnant, or using agents that potentiate neuromuscular weakness, such as aminoglycosides.
Patients should be informed that some formulations of botulinum toxin include a stabilizer such as albumin derived from human blood, as this may be of religious or cultural significance.
- Leippold T, Reitz A, Schurch B. Botulinum toxin as a new therapy option for voiding disorders: current state of the art. Eur Urol 2003; 44:165–174.
- Sahai A, Khan M, Fowler CJ, Dasgupta P. Botulinum toxin for the treatment of lower urinary tract symptoms: a review. Neurourol Urodyn 2005; 24:2–12.
- Cruz F. Targets for botulinum toxin in the lower urinary tract. Neurourol Urodyn 2014; 33:31–38.
- Weedmark KA, Lambert DL, Mabon P, et al. Two novel toxin variants revealed by whole-genome sequencing of 175 Clostridium botulinum type E strains. Appl Environ Microbiol 2014; 80:6334–6345.
- Ha US, Park EY, Kim JC. Effect of botulinum toxin on expression of nerve growth factor and transient receptor potential vanilloid 1 in urothelium and detrusor muscle of rats with bladder outlet obstruction-induced detrusor overactivity. Urology 2011; 78:721.e1–721.e6
- Frenkl TL, Rackley RR. Injectable neuromodulatory agents: botulinum toxin therapy. Urol Clin North Am 2005; 32:89–99.
- Ikeda Y, Zabbarova IV, Birder LA, et al. Botulinum neurotoxin serotype A suppresses neurotransmitter release from afferent as well as efferent nerves in the urinary bladder. Eur Urol 2012; 62:1157–1164.
- Goldmark E, Niver B, Ginsberg DA. Neurogenic bladder: from diagnosis to management. Curr Urol Rep 2014; 15:448.
- Andersson KE. Current and future drugs for treatment of MS-associated bladder dysfunction. Ann Phys Rehabil Med 2014; 57:321–328.
- Schurch B, Stöhrer M, Kramer G, Schmid DM, Gaul G, Hauri D. Botulinum-A toxin for treating detrusor hyperreflexia in spinal cord injured patients: a new alternative to anticholinergic drugs? Preliminary results. J Urol 2000; 164:692–697.
- Schurch B, de Sèze M, Denys P, et al; Botox Detrusor Hyperreflexia Study Team. Botulinum toxin type a is a safe and effective treatment for neurogenic urinary incontinence: results of a single treatment, randomized, placebo controlled 6-month study. J Urol 2005; 174:196–200.
- Ehren I, Volz D, Farrelly E, et al. Efficacy and impact of botulinum toxin A on quality of life in patients with neurogenic detrusor overactivity: a randomised, placebo-controlled, double-blind study. Scand J Urol Nephrol 2007; 41:335–340.
- Ginsberg D, Gousse A, Keppenne V, et al. Phase 3 efficacy and tolerability study of onabotulinumtoxinA for urinary incontinence from neurogenic detrusor overactivity. J Urol 2012; 187:2131–2139.
- Cruz F, Herschorn S, Aliotta P, et al. Efficacy and safety of onabotulinumtoxinA in patients with urinary incontinence due to neurogenic detrusor overactivity: a randomised, double-blind, placebo-controlled trial. Eur Urol 2011; 60:742–750.
- Wagner TH, Patrick DL, Bavendam TG, Martin ML, Buesching DP. Quality of life of persons with urinary incontinence: development of a new measure. Urology 1996: 47:67–71.
- Kennelly M, Dmochowski R, Ethans K, et al. Long-term efficacy and safety of onabotulinumtoxinA in patients with urinary incontinence due to neurogenic detrusor overactivity: an interim analysis. Urology 2013; 81:491–497.
- Carlson JJ, Hansen RN, Dmochowski RR, Globe DR, Colayco DC, Sullivan SD. Estimating the cost-effectiveness of onabotulinumtoxinA for neurogenic detrusor overactivity in the United States. Clin Ther 2013; 35:414–424.
- Abrams P, Cardozo L, Fall M, et al; Standardisation Sub-Committee of the International Continence Society. The standardisation of terminology in lower urinary tract function: report from the standardisation sub-committee of the International Continence Society. Urology 2003; 61:37–49.
- Milsom I, Coyne KS, Nicholson S, Kvasz M, Chen CI, Wein AJ. Global prevalence and economic burden of urgency urinary incontinence: a systematic review. Eur Urol 2014; 65:79–95.
- Nitti VW, Dmochowski R, Herschorn S, et al; EMBARK Study Group. OnabotulinumtoxinA for the treatment of patients with overactive bladder and urinary incontinence: results of a phase 3, randomized, placebo controlled trial. J Urol 2013; 189:2186–2193.
- Chapple C, Sievert KD, MacDiarmid S, et al. OnabotulinumtoxinA 100 U significantly improves all idiopathic overactive bladder symptoms and quality of life in patients with overactive bladder and urinary incontinence: a randomised, double-blind, placebo-controlled trial. Eur Urol 2013; 64:249–256.
- Colman S, Chapple C, Nitti V, Haag-Molkenteller C, Hastedt C, Massow U. Validation of Treatment Benefit Scale for assessing subjective outcomes in treatment of overactive bladder. Urology 2008; 72:803–807.
- Visco AG, Brubaker L, Richter HE, et al; Pelvic Floor Disorders Network. Anticholinergic therapy vs onabotulinumtoxinA for urgency urinary incontinence. N Engl J Med 2012; 367:1803–1813.
- Gormley EA, Lightner DJ, Burgio KL, et al. Diagnosis and treatment of overactive bladder (non-neurogenic) in adults: AUA/SUFU Guideline. www.auanet.org/education/guidelines/overactive-bladder.cfm. Accessed June 11, 2015.
- Utomo E, Groen J, Blok BF. Surgical management of functional bladder outlet obstruction in adults with neurogenic bladder dysfunction. Cochrane Database Syst Rev 2014; 5:CD004927.
- Mahfouz W, Corcos J. Management of detrusor external sphincter dyssynergia in neurogenic bladder. Eur J Phys Rehabil Med 2011; 47:639–650.
- Dykstra DD, Sidi AA, Scott AB, Pagel JM, Goldish GD. Effects of botulinum A toxin on detrusor-sphincter dyssynergia in spinal cord injury patients. J Urol 1988; 139:919–922.
- Schurch B, Hauri D, Rodic B, Curt A, Meyer M, Rossier AB. Botulinum-A toxin as a treatment of detrusor-sphincter dyssynergia: a prospective study in 24 spinal cord injury patients. J Urol 1996; 155:1023–1029.
- de Sèze M, Petit H, Gallien, de Sèze MP, Joseph PA, Mazaux JM, Barat M. Botulinum a toxin and detrusor sphincter dyssynergia: a double-blind lidocaine-controlled study in 13 patients with spinal cord disease. Eur Urol 2002; 42:56–62.
- Gallien P, Reymann JM, Amarenco G, Nicolas B, de Sèze M, Bellissant E. Placebo controlled, randomised, double blind study of the effects of botulinum A toxin on detrusor sphincter dyssynergia in multiple sclerosis patients. J Neurol Neurosurg Psychiatry 2005; 76:1670–1676.
- Hanno PM, Burks DA, Clemens JQ, et al; Interstitial Cystitis Guidelines Panel of the American Urological Association Education and Research, Inc. AUA guideline for the diagnosis and treatment of interstitial cystitis/bladder pain syndrome. J Urol 2011; 185:2162–2170.
- Chuang YC, Yoshimura N, Huang CC, Chiang PH, Chancellor MB. Intravesical botulinum toxin a administration produces analgesia against acetic acid induced bladder pain responses in rats. J Urol 2004; 172:1529–1532.
- Zermann DH, Ishigooka M, Schubert J, Schmidt RA. Perisphincteric injection of botulinum toxin type A. A treatment option for patients with chronic prostatic pain? Eur Urol 2000; 38:393–399.
- Abbott JA, Jarvis SK, Lyons SD, Thomson A, Vancaille TG. Botulinum toxin type A for chronic pain and pelvic floor spasm in women: a randomized controlled trial. Obstet Gynecol 2006; 108:915–923.
- Smith CP, Radziszewski P, Borkowski A, Somogyi GT, Boone TB, Chancellor MB. Botulinum toxin A has antinociceptive effects in treating interstitial cystitis. Urology 2004; 64:871–875.
- Kuo HC, Chancellor MB. Comparison of intravesical botulinum toxin type A injections plus hydrodistention with hydrodistention alone for the treatment of refractory interstitial cystitis/painful bladder syndrome. BJU Int 2009: 104:657–661.
- Pinto R, Lopes T, Silva J, Silva C, Dinis P, Cruz F. Persistent therapeutic effect of repeated injections of onabotulinum toxin a in refractory bladder pain syndrome/interstitial cystitis. J Urol 2013; 189:548–553.
- Rovner E. Chapter 6: Practical aspects of administration of onabotulinumtoxinA. Neurourol Urodyn 2014; 33(suppl 3):S32–S37.
- Schulte-Baukloh H, Herholz J, Bigalke H, Miller K, Knispel HH. Results of a BoNT/A antibody study in children and adolescents after onabotulinumtoxin A (Botox®) detrusor injection. Urol Int 2011; 87:434–438.
- Leippold T, Reitz A, Schurch B. Botulinum toxin as a new therapy option for voiding disorders: current state of the art. Eur Urol 2003; 44:165–174.
- Sahai A, Khan M, Fowler CJ, Dasgupta P. Botulinum toxin for the treatment of lower urinary tract symptoms: a review. Neurourol Urodyn 2005; 24:2–12.
- Cruz F. Targets for botulinum toxin in the lower urinary tract. Neurourol Urodyn 2014; 33:31–38.
- Weedmark KA, Lambert DL, Mabon P, et al. Two novel toxin variants revealed by whole-genome sequencing of 175 Clostridium botulinum type E strains. Appl Environ Microbiol 2014; 80:6334–6345.
- Ha US, Park EY, Kim JC. Effect of botulinum toxin on expression of nerve growth factor and transient receptor potential vanilloid 1 in urothelium and detrusor muscle of rats with bladder outlet obstruction-induced detrusor overactivity. Urology 2011; 78:721.e1–721.e6
- Frenkl TL, Rackley RR. Injectable neuromodulatory agents: botulinum toxin therapy. Urol Clin North Am 2005; 32:89–99.
- Ikeda Y, Zabbarova IV, Birder LA, et al. Botulinum neurotoxin serotype A suppresses neurotransmitter release from afferent as well as efferent nerves in the urinary bladder. Eur Urol 2012; 62:1157–1164.
- Goldmark E, Niver B, Ginsberg DA. Neurogenic bladder: from diagnosis to management. Curr Urol Rep 2014; 15:448.
- Andersson KE. Current and future drugs for treatment of MS-associated bladder dysfunction. Ann Phys Rehabil Med 2014; 57:321–328.
- Schurch B, Stöhrer M, Kramer G, Schmid DM, Gaul G, Hauri D. Botulinum-A toxin for treating detrusor hyperreflexia in spinal cord injured patients: a new alternative to anticholinergic drugs? Preliminary results. J Urol 2000; 164:692–697.
- Schurch B, de Sèze M, Denys P, et al; Botox Detrusor Hyperreflexia Study Team. Botulinum toxin type a is a safe and effective treatment for neurogenic urinary incontinence: results of a single treatment, randomized, placebo controlled 6-month study. J Urol 2005; 174:196–200.
- Ehren I, Volz D, Farrelly E, et al. Efficacy and impact of botulinum toxin A on quality of life in patients with neurogenic detrusor overactivity: a randomised, placebo-controlled, double-blind study. Scand J Urol Nephrol 2007; 41:335–340.
- Ginsberg D, Gousse A, Keppenne V, et al. Phase 3 efficacy and tolerability study of onabotulinumtoxinA for urinary incontinence from neurogenic detrusor overactivity. J Urol 2012; 187:2131–2139.
- Cruz F, Herschorn S, Aliotta P, et al. Efficacy and safety of onabotulinumtoxinA in patients with urinary incontinence due to neurogenic detrusor overactivity: a randomised, double-blind, placebo-controlled trial. Eur Urol 2011; 60:742–750.
- Wagner TH, Patrick DL, Bavendam TG, Martin ML, Buesching DP. Quality of life of persons with urinary incontinence: development of a new measure. Urology 1996: 47:67–71.
- Kennelly M, Dmochowski R, Ethans K, et al. Long-term efficacy and safety of onabotulinumtoxinA in patients with urinary incontinence due to neurogenic detrusor overactivity: an interim analysis. Urology 2013; 81:491–497.
- Carlson JJ, Hansen RN, Dmochowski RR, Globe DR, Colayco DC, Sullivan SD. Estimating the cost-effectiveness of onabotulinumtoxinA for neurogenic detrusor overactivity in the United States. Clin Ther 2013; 35:414–424.
- Abrams P, Cardozo L, Fall M, et al; Standardisation Sub-Committee of the International Continence Society. The standardisation of terminology in lower urinary tract function: report from the standardisation sub-committee of the International Continence Society. Urology 2003; 61:37–49.
- Milsom I, Coyne KS, Nicholson S, Kvasz M, Chen CI, Wein AJ. Global prevalence and economic burden of urgency urinary incontinence: a systematic review. Eur Urol 2014; 65:79–95.
- Nitti VW, Dmochowski R, Herschorn S, et al; EMBARK Study Group. OnabotulinumtoxinA for the treatment of patients with overactive bladder and urinary incontinence: results of a phase 3, randomized, placebo controlled trial. J Urol 2013; 189:2186–2193.
- Chapple C, Sievert KD, MacDiarmid S, et al. OnabotulinumtoxinA 100 U significantly improves all idiopathic overactive bladder symptoms and quality of life in patients with overactive bladder and urinary incontinence: a randomised, double-blind, placebo-controlled trial. Eur Urol 2013; 64:249–256.
- Colman S, Chapple C, Nitti V, Haag-Molkenteller C, Hastedt C, Massow U. Validation of Treatment Benefit Scale for assessing subjective outcomes in treatment of overactive bladder. Urology 2008; 72:803–807.
- Visco AG, Brubaker L, Richter HE, et al; Pelvic Floor Disorders Network. Anticholinergic therapy vs onabotulinumtoxinA for urgency urinary incontinence. N Engl J Med 2012; 367:1803–1813.
- Gormley EA, Lightner DJ, Burgio KL, et al. Diagnosis and treatment of overactive bladder (non-neurogenic) in adults: AUA/SUFU Guideline. www.auanet.org/education/guidelines/overactive-bladder.cfm. Accessed June 11, 2015.
- Utomo E, Groen J, Blok BF. Surgical management of functional bladder outlet obstruction in adults with neurogenic bladder dysfunction. Cochrane Database Syst Rev 2014; 5:CD004927.
- Mahfouz W, Corcos J. Management of detrusor external sphincter dyssynergia in neurogenic bladder. Eur J Phys Rehabil Med 2011; 47:639–650.
- Dykstra DD, Sidi AA, Scott AB, Pagel JM, Goldish GD. Effects of botulinum A toxin on detrusor-sphincter dyssynergia in spinal cord injury patients. J Urol 1988; 139:919–922.
- Schurch B, Hauri D, Rodic B, Curt A, Meyer M, Rossier AB. Botulinum-A toxin as a treatment of detrusor-sphincter dyssynergia: a prospective study in 24 spinal cord injury patients. J Urol 1996; 155:1023–1029.
- de Sèze M, Petit H, Gallien, de Sèze MP, Joseph PA, Mazaux JM, Barat M. Botulinum a toxin and detrusor sphincter dyssynergia: a double-blind lidocaine-controlled study in 13 patients with spinal cord disease. Eur Urol 2002; 42:56–62.
- Gallien P, Reymann JM, Amarenco G, Nicolas B, de Sèze M, Bellissant E. Placebo controlled, randomised, double blind study of the effects of botulinum A toxin on detrusor sphincter dyssynergia in multiple sclerosis patients. J Neurol Neurosurg Psychiatry 2005; 76:1670–1676.
- Hanno PM, Burks DA, Clemens JQ, et al; Interstitial Cystitis Guidelines Panel of the American Urological Association Education and Research, Inc. AUA guideline for the diagnosis and treatment of interstitial cystitis/bladder pain syndrome. J Urol 2011; 185:2162–2170.
- Chuang YC, Yoshimura N, Huang CC, Chiang PH, Chancellor MB. Intravesical botulinum toxin a administration produces analgesia against acetic acid induced bladder pain responses in rats. J Urol 2004; 172:1529–1532.
- Zermann DH, Ishigooka M, Schubert J, Schmidt RA. Perisphincteric injection of botulinum toxin type A. A treatment option for patients with chronic prostatic pain? Eur Urol 2000; 38:393–399.
- Abbott JA, Jarvis SK, Lyons SD, Thomson A, Vancaille TG. Botulinum toxin type A for chronic pain and pelvic floor spasm in women: a randomized controlled trial. Obstet Gynecol 2006; 108:915–923.
- Smith CP, Radziszewski P, Borkowski A, Somogyi GT, Boone TB, Chancellor MB. Botulinum toxin A has antinociceptive effects in treating interstitial cystitis. Urology 2004; 64:871–875.
- Kuo HC, Chancellor MB. Comparison of intravesical botulinum toxin type A injections plus hydrodistention with hydrodistention alone for the treatment of refractory interstitial cystitis/painful bladder syndrome. BJU Int 2009: 104:657–661.
- Pinto R, Lopes T, Silva J, Silva C, Dinis P, Cruz F. Persistent therapeutic effect of repeated injections of onabotulinum toxin a in refractory bladder pain syndrome/interstitial cystitis. J Urol 2013; 189:548–553.
- Rovner E. Chapter 6: Practical aspects of administration of onabotulinumtoxinA. Neurourol Urodyn 2014; 33(suppl 3):S32–S37.
- Schulte-Baukloh H, Herholz J, Bigalke H, Miller K, Knispel HH. Results of a BoNT/A antibody study in children and adolescents after onabotulinumtoxin A (Botox®) detrusor injection. Urol Int 2011; 87:434–438.
KEY POINTS
- Anticholinergic drugs have been the first-line therapy for neurogenic detrusor overactivity. If drug therapy failed, the next option was reconstructive surgery such as cystoplasty. Botulinum toxin injection may be an option in select patients.
- Urinary tract infection and urinary retention requiring intermittent self-catheterization are the most common adverse events of botulinum toxin injection in trials of patients with neurogenic detrusor overactivity or idiopathic overactive bladder.
- Small studies have shown that botulinum toxin injection for painful bladder syndrome/interstitial cystitis can improve pain, urinary frequency, and quality of life. But larger randomized controlled trials are needed.
Ceftaroline fosamil: A super-cephalosporin?
Ceftaroline fosamil (Teflaro), introduced to the US market in October 2010, is the first beta-lactam agent with clinically useful activity against methicillin-resistant Staphylococcus aureus (MRSA). Currently, it is approved by the US Food and Drug Administration (FDA) to treat acute bacterial skin and skin-structure infections and community-acquired bacterial pneumonia caused by susceptible microorganisms.
In an era of increasing drug resistance and limited numbers of antimicrobials in the drug-production pipeline, ceftaroline is a step forward in fulfilling the Infectious Diseases Society of America’s “10 × ’20 Initiative” to increase support for drug research and manufacturing, with the goal of producing 10 new antimicrobial drugs by the year 2020.1 Ceftaroline was the first of several antibiotics to receive FDA approval in response to this initiative. It was followed by dalbavancin (May 2014), tedizolid phosphate (June 2014), oritavancin (August 2014), ceftolozane-tazobactam (December 2014), and ceftazidime-avibactam (February 2015). These antibiotic agents are aimed at treating infections caused by drug-resistant gram-positive and gram-negative microorganisms. It is important to understand and optimize the use of these new antibiotic agents in order to decrease the risk of emerging antibiotic resistance and superinfections (eg, Clostridium difficile infection) caused by antibiotic overuse or misuse.
This article provides an overview of ceftaroline’s mechanisms of action and resistance, spectrum of activity, pharmacokinetic properties, adverse effects, and current place in therapy.
AN ERA OF MULTIDRUG-RESISTANT MICROORGANISMS
Increasing rates of antimicrobial resistance threaten the efficacy of antimicrobial drugs in the daily practice of medicine. The World Health Organization has labeled antimicrobial resistance one of the three greatest threats to human health. Global efforts are under way to stimulate development of new antimicrobial agents and to decrease rates of antimicrobial resistance.
Staphylococcus aureus: A threat, even with vancomycin
Between 1998 and 2005, S aureus was one of the most common inpatient and outpatient isolates reported by clinical laboratories throughout the United States.2
Treatment of S aureus infection is complicated by a variety of resistance mechanisms that have evolved over time. In fact, the first resistant isolate of S aureus emerged not long after penicillin’s debut into clinical practice, and now the majority of strains are resistant to penicillin.
Methicillin was designed to overcome this beta-lactamase resistance and became the treatment of choice for penicillin-resistant S aureus isolates. However, MRSA isolates soon emerged because of the organism’s acquisition of penicillin-binding protein PBP2a via the mecA gene, leading to decreased binding affinity of methicillin.3
Since then, several agents active against MRSA (vancomycin, daptomycin, linezolid, tigecycline) have been introduced and continue to be widely used. While vancomycin is considered the first-line option for a variety of MRSA infections, its use has been threatened because of the emergence of vancomycin-intermediate-resistant S aureus (VISA), S aureus strains displaying vancomycin heteroresistance (hVISA), and vancomycin-resistant S aureus (VRSA) strains.4
VISA and hVISA isolates emerged through sequential mutations that lead to autolytic activity and cell-wall thickening. In contrast, the mechanism of resistance in VRSA is by acquisition of the vanA resistance gene, which alters the binding site of vancomycin from d-alanine-d-alanine to d-alanine-d-lactate.5
Streptococcus pneumoniae resistance: A continuing problem
The prevalence of drug resistance in S pneumoniae has risen since the late 1990s. A 2013 report from the SENTRY Antimicrobial Surveillance Program stated that almost 20% of S pneumoniae isolates were resistant to amoxicillin-clavulanate, and similar trends have been observed for penicillin (14.8%) and ceftriaxone (11.7%).6
S pneumoniae resistance is acquired through modifications of the penicillin-binding proteins, namely PBP1a, PBP2b, PBP2x, and, less frequently, PBP2a. These modifications lead to decreased binding affinity for most beta-lactams.7
Clinical impact of multidrug-resistant S aureus and S pneumoniae
In 2011, the US Centers for Disease Control and Prevention reported an estimated 80,000 severe MRSA infections and 11,000 MRSA-related deaths in the United States.8 In the same report, drug-resistant S pneumoniae was estimated to be responsible for almost 1.2 million illnesses and 7,000 deaths per year, leading to upwards of $96 million in related medical costs.
While invasive drug-resistant S pneumoniae infections usually affect patients at the extremes of age (under age 5 and over age 65), they have had a serious impact on patients of all ages.8
In light of the increasing prevalence of multidrug-resistant organisms, newer antimicrobial agents with novel mechanisms of action are needed.
CEFTAROLINE: A BETA-LACTAM WITH ANTI-MRSA ACTIVITY
The cephalosporins, a class of beta-lactam antibiotics, were originally derived from the fungus Cephalosporium (now called Acremonium). There are now many agents in this class, each containing a nucleus consisting of a beta-lactam ring fused to a six-member dihydrothiazine ring, and two side chains that can be modified to affect antibacterial activity and pharmacokinetic properties.
Cephalosporins are typically categorized into “generations.” With some exceptions, the first- and second-generation agents have good activity against gram-positive microorganisms, including methicillin-susceptible S aureus—but not against MRSA. The third- and fourth-generation cephalosporins have better gram-negative activity, with many agents having activity against the gram-negative bacterium Pseudomonas aeruginosa.
Enterococcal isolates are intrinsically resistant to cephalosporins. Additionally, cephalosporins are not active against anaerobic bacteria, except for a subset of structurally unique second-generation cephalosporins, ie, cefotetan and cefoxitin.
Ceftaroline was synthesized with specific manipulations of the side chains to provide enhanced activity against MRSA and multidrug-resistant S pneumoniae isolates, making it the first available beta-lactam with this ability.
Mechanism of action
Ceftaroline binds to penicillin-binding proteins, inhibiting transpeptidation. This interaction blocks the final stage of peptidoglycan synthesis and inhibits bacterial cell wall formation, ultimately leading to cellular autolysis and microorganism death. Ceftaroline binds with high affinity to PBP2a and PBP2x, expanding its activity to encompass MRSA and penicillin-resistant S pneumoniae isolates.9
Spectrum of activity
Ceftaroline has in vitro activity against many gram-positive and gram-negative bacteria,10–13 including (Table 1):
- Methicillin-susceptible and methicillin-resistant staphylococci
- VISA, VRSA, and hVISA
- Daptomycin-nonsusceptible S aureus
- Streptococcal species, including penicillin-resistant S pneumoniae
- Enterobacteriaceae, including Klebsiella pneumoniae, Klebsiella oxytoca, Escherichia coli, Citrobacter koseri, Citrobacter freundii, Enterobacter cloacae, Enterobacter aerogenes, Moraxella catarrhalis, Morganella morganii, and Proteus mirabilis.
Of note, ceftaroline is not active against Pseudomonas species, Enterococcus species, or Bacteroides fragilis. In addition, it is not active against the “atypical” respiratory pathogens Mycoplasma pneumoniae, Chlamydophila pneumoniae, and Legionella pneumophila.
Ceftaroline resistance
Gram-negative organisms appear to develop resistance to ceftaroline at rates similar to those observed with the other oxyimino-cephalosporins (eg, ceftriaxone). Ceftaroline is inactive against gram-negative organisms producing extended-spectrum beta-lactamases, including K pneumoniae carbapenemase and metallo-beta-lactamases.14 In addition, it induces the expression of AmpC beta-lactamases.
Although currently uncommon, resistance to ceftaroline has also been reported in S aureus strains.15 The mechanism of resistance is decreased binding affinity for PBP2a due to amino acid substitutions on the nonpenicillin-binding domains.15
Pharmacokinetic profile
An understanding of pharmacokinetics is key in optimizing the dose of antimicrobials so that the drugs are used most effectively and pathogens do not develop resistance to them.

Ceftaroline fosamil is a prodrug that, upon intravenous administration, is rapidly converted by phosphatase enzymes to its active moiety, ceftaroline. Its pharmacokinetic profile is summarized in Table 2.16,17 Its volume of distribution is similar to that of the fourth-generation cephalosporin cefepime.
Ceftaroline is then hydrolyzed into its inactive metabolite, ceftaroline M-1. It undergoes little hepatic metabolism and lacks properties to make it a substrate, inhibitor, or inducer of the CYP450 enzyme system and therefore is not likely to cause notable CYP450-related drug-drug interactions.
Like most other beta-lactams, ceftaroline is primarily excreted by the kidneys. Furthermore, an estimated 21% of a dose is eliminated with each intermittent hemodialysis session. Therefore, renal and intermittent hemodialysis dose adjustments are necessary. The estimated elimination half-life is 2.6 hours, necessitating dosing two to three times daily, depending on the indication and infectious inoculum.
Ceftaroline dosing
Ceftaroline is available only in a parenteral preparation and is typically given at a dose of 600 mg every 12 hours.10 The intravenous infusion is given over 1 hour.
The current stability data require reconstituted ceftaroline to be used within 6 hours at room temperature and within 24 hours if refrigerated.10
Ceftaroline requires dosing adjustments for patients with renal insufficiency. Per the manufacturer, renal dosing adjustments are based on the creatinine clearance rate, as estimated by the Cockroft-Gault formula:
- Creatinine clearance > 50 mL/min: no dosage adjustment necessary
- Creatinine clearance > 30 to ≤ 50 mL/min: give 400 mg every 12 hours
- Creatinine clearance ≥ 15 to ≤ 30 mL/min: give 300 mg every 12 hours
- Creatinine clearance < 15 mL/min or on intermittent dialysis: give 200 mg every 12 hours.
Ongoing clinical trials are investigating a higher-dosing strategy of 600 mg every 8 hours for patients with community-acquired bacterial pneumonia at risk of MRSA bacteremia.18
CLINICAL TRIALS LEADING TO CEFTAROLINE’S APPROVAL
Ceftaroline was approved for the treatment of community-acquired bacterial pneumonia and acute bacterial skin and skin-structure infections due to susceptible pathogens on the basis of phase 3 comparator trials.
Community-acquired bacterial pneumonia: The FOCUS 1 and 2 trials
The efficacy and safety of ceftaroline in the treatment of community-acquired bacterial pneumonia was studied in two randomized, double-blind, noninferiority trials, known as Ceftaroline Community-acquired Pneumonia vs Ceftriaxone (FOCUS) 1 and FOCUS 2.19,20
Patients were adults and not critically ill, as was reflected by their being in Pneumonia Outcomes Research Team (PORT) risk class III or IV (with class V indicating the highest risk of death). Therefore, the results may not be completely applicable to critically ill patients or those not admitted to the hospital. Of note, patients were excluded from the trials if they had infections known or thought to be due to MRSA or to atypical organisms.21 Baseline characteristics and patient demographics were similar between study groups in both trials.
A bacterial pathogen was identified in 26.1% of the patients included in the modified intent-to-treat analysis of the pooled data of the trials; the most common pathogens were S pneumoniae, methicillin-sensitive S aureus, Haemophilus influenzae, K pneumoniae, and E coli.21
Treatment. Patients received either ceftaroline 600 mg every 12 hours (or a lower dose based on renal function) or ceftriaxone 1 g every 24 hours. In addition, in the FOCUS 1 trial, patients in both treatment groups received clarithromycin 500 mg every 12 hours for the first day.19

Results. In both trials and in the integrated analysis, ceftaroline was noninferior to ceftriaxone (Table 3).22 In the integrated analysis of both trials, compared with the ceftriaxone group, the ceftaroline group had a higher clinical cure rate among patients classified as PORT risk class III (86.8% vs 79.2%, weighted treatment difference 12.6%, 95% confidence interval [CI] 1.3–13.8) and among patients who had not received prior antibiotic treatment (85.5% vs 74.9%, weighted treatment difference 11.2%, 95% CI 4.5–18.0).21
Acute bacterial skin and skin-structure infections: The CANVAS 1 and 2 trials
The efficacy and safety of ceftaroline in the treatment of complicated acute bacterial skin and skin-structure infections was studied in two randomized, double-blind trials: Ceftaroline Versus Vancomycin in Skin and Skin Structure Infections (CANVAS) 1 and CANVAS 2.23,24
Patients. Adult patients with a diagnosis of community-acquired skin and skin-structure infections warranting at least 5 days of intravenous antimicrobial therapy were included in the trials. Important protocol exclusions were patients with diabetic foot ulcers, decubitus ulcers, burns, ulcers associated with peripheral vascular disease accompanied by osteomyelitis, and suspected P aeruginosa infections.25 This limits the external validity of ceftaroline use in the aforementioned excluded patient populations.
Patients in each treatment group of the trials had similar demographic characteristics. The most common infections were cellulitis, major abscess requiring surgical intervention, wound infection, and infected ulcer. Bacteremia was present in 4.2% of patients in the ceftaroline group and in 3.8% of patients in the vancomycin-aztreonam group. The most common pathogen was S aureus. Methicillin resistance was present in 40% of the ceftaroline group and 34% of the control group.
Treatment. Patients received either ceftaroline 600 mg every 12 hours or the combination of vancomycin 1 g plus aztreonam 1 g given 12 hours, for 5 to 14 days.

Results. As assessed at a “test-of-cure” visit 8 to 15 days after the last dose of study medication, the efficacy of ceftaroline was similar to that of vancomycin-aztreonam, meeting the set noninferiority goal (Table 4).25 Moreover, if assessed on day 2 or 3 (a new end point recommended by the FDA), the rate of cessation of erythema spread and absence of fever was higher in the ceftaroline group than in the vancomycin-aztreonam group.26 However, this end point was not in the original trial protocol.
CEFTAROLINE FOR OTHER INDICATIONS
As noted, ceftaroline has been approved for treating community-acquired bacterial pneumonia and acute bacterial skin and skin-structure infections. In addition, it has been used in several studies in animals, and case reports of non-FDA approved indications including endocarditis and osteomyelitis have been published. Clinical trials are evaluating its use in pediatric patients, as well as for community-acquired bacterial pneumonia with risk for MRSA and for MRSA bacteremia.
Endocarditis
Animal studies have demonstrated ceftaroline to have bactericidal activity against MRSA and hVISA in endocarditis.27
A few case series have been published describing ceftaroline’s use as salvage therapy for persistent MRSA bacteremia and endocarditis. For example, Ho et al28 reported using it in three patients who had endocarditis as a source of their persistent bacteremia. All three patients had resolution of their MRSA bloodstream infection following ceftaroline therapy. The dosage was 600 mg every 8 hours, which is higher than in the manufacturer’s prescribing information.
Lin et al29 reported using ceftaroline in five patients with either possible or probable endocarditis. Three of the five patients had clinical cure as defined by resolution or improvement of all signs and symptoms of infection, and not requiring further antimicrobial therapy.29
More data from clinical trials would be beneficial in defining ceftaroline’s role in treating endocarditis caused by susceptible microorganisms.
Osteomyelitis
In animal studies of osteomyelitis, ceftaroline exhibited activity against MRSA in infected bone and joint fluid. Compared with vancomycin and linezolid, ceftaroline was associated with more significant decreases in bacterial load in the infected joint fluid, bone marrow, and bone.30
Lin et al29 gave ceftaroline to two patients with bone and joint infections, both of whom had received other therapies that had failed. The doses of ceftaroline were higher than those recommended in the prescribing information; clinical cure was noted in both cases following the switch.
These data come from case series, and more study of ceftaroline’s role in the treatment of osteomyelitis infections is warranted.
Meningitis
The use of ceftaroline in meningitis has been studied in rabbits. While ceftaroline penetrated into the cerebrospinal fluid in only negligible amounts in healthy rabbits (3% penetration), its penetration improved to 15% in animals with inflamed meninges. Ceftaroline cerebrospinal fluid levels in inflamed meninges were sufficient to provide bactericidal activity against penicillin-sensitive and resistant S pneumoniae strains as well as K pneumoniae and E coli strains.31,32
REPORTED ADVERSE EFFECTS OF CEFTAROLINE
Overall, ceftaroline was well tolerated in clinical trials, and its safety profile was similar to those of the comparator agents (ceftriaxone and vancomycin-aztreonam).
As with the other cephalosporins, hypersensitivity reactions have been reported with ceftaroline. In the clinical trials, 3% of patients developed a rash with ceftaroline.33,34 Patients with a history of beta-lactam allergy were excluded from the trials, so the rate of cross-reactivity with penicillins and with other cephalosporins is unknown.
In the phase 3 clinical trials, gastrointestinal side effects including diarrhea (5%), nausea (4%), and vomiting (2%) were reported with ceftaroline. C difficile-associated diarrhea has also been reported.33
As with other cephalosporins, ceftaroline can cause a false-positive result on the Coombs test. Approximately 11% of ceftaroline-treated patients in phase 3 clinical trials had a positive Coombs test, but hemolytic anemia did not occur in any patients.33,34
Discontinuation of ceftaroline due to an adverse reaction was reported in 2.7% of patients receiving the drug during phase 3 trials, compared with 3.7% with comparator agents.
WHEN SHOULD CEFTAROLINE BE USED IN DAILY PRACTICE?
Ceftaroline has been shown to be at least as effective as ceftriaxone in treating community-acquired bacterial pneumonia, and at least as effective as vancomycin-aztreonam in treating acute bacterial skin and skin-structure infections. The 2014 Infectious Diseases Society of America’s guidelines for the diagnosis and management of skin and soft-tissue infections recommend ceftaroline as an option for empiric therapy for purulent skin and soft-tissue infections.35
The guidelines on community-acquired pneumonia have not been updated since 2007, which was before ceftaroline was approved. However, these guidelines are currently undergoing revision and may provide insight on ceftaroline’s place in the treatment of community-acquired bacterial pneumonia.36
Currently, ceftaroline’s routine use for these indications should be balanced by its higher cost ($150 for a 600-mg dose) compared with ceftriaxone ($5 for a 1-g dose) or vancomycin ($25 for a 1-g dose). The drug’s in vitro activity against drug-resistant pneumococci and S aureus, including MRSA, hVISA, and VISA may help fill an unmet need or provide a safer and more tolerable alternative to currently available therapies.
However, ceftaroline’s lack of activity against P aeruginosa and carbapenem-resistant Enterobacteriaceae does not meet the public health threat needs stemming from these multidrug-resistant microorganisms. Ongoing clinical trials in patients with more serious MRSA infections will provide important information about ceftaroline’s role as an anti-MRSA agent.
While the discovery of antimicrobials has had one of the greatest impacts on medicine, continued antibiotic use is threatened by the emergence of drug-resistant pathogens. Therefore, it is as important as ever to be good stewards of our currently available antimicrobials. Developing usage and dosing criteria for antimicrobials based on available data and literature is a step forward in optimizing the use of antibiotics—a precious medical resource.
- Infectious Diseases Society of America. The 10 x ‘20 Initiative: pursuing a global commitment to develop 10 new antibacterial drugs by 2020. Clin Infect Dis 2010; 50:1081–1083.
- Styers D, Sheehan DJ, Hogan P, Sahm DF. Laboratory-based surveillance of current antimicrobial resistance patterns and trends among Staphylococcus aureus: 2005 status in the United States. Ann Clin Microbiol Antimicrob 2006; 5:2.
- Farrell DJ, Castanheira M, Mendes RE, Sader HS, Jones RN. In vitro activity of ceftaroline against multidrug-resistant Staphylococcus aureus and Streptococcus pneumoniae: a review of published studies and the AWARE Surveillance Program (2008-2010). Clin Infect Dis 2012; 55(suppl 3):S206–S214.
- Holmes NE, Johnson PD, Howden BP. Relationship between vancomycin-resistant Staphylococcus aureus, vancomycin-intermediate S. aureus, high vancomycin MIC, and outcome in serious S. aureus infections. J Clin Microbiol 2012; 50:2548–2552.
- Lowy FD. Antimicrobial resistance: the example of Staphylococcus aureus. J Clin Invest 2003; 111:1265–1273.
- Jones RN, Sader HS, Mendes RE, Flamm RK. Update on antimicrobial susceptibility trends among Streptococcus pneumoniae in the United States: report of ceftaroline activity from the SENTRY Antimicrobial Surveillance Program (1998-2011). Diag Microbiol Infect Dis 2013; 75:107–109.
- Zapun A, Contreras-Martel C, Vernet T. Penicillin-binding proteins and beta-lactam resistance. FEMS Microbiol Rev 2008; 32:361–385.
- Centers for Disease Control and Prevention (CDC). Antibiotic resistance threats in the United States 2013. cdc.gov/drugresistance/threat-report-2013/pdf/ar-threats-2013-508.pdf. Accessed June 1, 2015.
- Moisan H, Pruneau M, Malouin F. Binding of ceftaroline to penicillin-binding proteins of Staphylococcus aureus and Streptococcus pneumoniae. J Antimicrob Chemother 2010; 65:713–716.
- Forest Laboratories, Inc. Teflaro® (ceftaroline fosamil): prescribing information. www.frx.com/pi/teflaro_pi.pdf. Accessed June 1, 2015.
- Richter SS, Heilmann KP, Dohrn CL, et al. Activity of ceftaroline and epidemiologic trends in Staphylococcus aureus isolates collected from 43 medical centers in the United States in 2009. Antimicrob Agents Chemother 2011; 55:4154–4160.
- Ge Y, Biek D, Talbot GH, Sahm DF. In vitro profiling of ceftaroline against a collection of recent bacterial clinical isolates from across the United States. Antimicrob Agents Chemother 2008; 52:3398–3407.
- Saravolatz L, Pawlak J, Johnson L. In vitro activity of ceftaroline against community-associated methicillin-resistant, vancomycin-intermediate, vancomycin-resistant, and daptomycin-nonsusceptible Staphylococcus aureus isolates. Antimicrob Agents Chemother 2010; 54:3027–3030.
- Mushtaq S, Livermore DM. AmpC induction by ceftaroline. J Antimicrob Chemother 2010; 65:586–588.
- Mendes RE, Tsakris A, Sader HS, et al. Characterization of methicillin-resistant Staphylococcus aureus displaying increased MICs of ceftaroline. J Antimicrob Chemother 2012; 67:1321–1324.
- Lodise TP, Low DE. Ceftaroline fosamil in the treatment of community-acquired bacterial pneumonia and acute bacterial skin and skin structure infections. Drugs 2012; 72:1473–1493.
- Riccobene TA, Su SF, Rank D. Single- and multiple-dose study to determine the safety, tolerability, and pharmacokinetics of ceftaroline fosamil in combination with avibactam in healthy subjects. Antimicrob Agents Chemother 2013; 57:1496–1504.
- US National Institutes of Health. ClinicalTrials.gov. Evaluation of ceftaroline fosamil versus a comparator in adult subjects with community-acquired bacterial pneumonia (CABP) with risk for methicillin-resistant Staphylococcus aureus. http://clinicaltrials.gov/ct2/show/NCT01645735. Accessed June 1, 2015.
- File TM Jr, Low DE, Eckburg PB, et al; FOCUS 1 investigators. FOCUS 1: a randomized, double-blinded, multicentre, phase III trial of the efficacy and safety of ceftaroline fosamil versus ceftriaxone in community-acquired pneumonia. J Antimicrob Chemother 2011; 66(suppl 3):iii19–iii32.
- Low DE, File TM Jr, Eckburg PB, et al; FOCUS 2 investigators. FOCUS 2: a randomized, double-blinded, multicentre, phase III trial of the efficacy and safety of ceftaroline fosamil versus ceftriaxone in community-acquired pneumonia. J Antimicrob Chemother 2011; 66(suppl 3):iii33–iii44.
- File TM Jr, Low DE, Eckburg PB, et al. Integrated analysis of FOCUS 1 and FOCUS 2: randomized, doubled-blinded, multicenter phase 3 trials of the efficacy and safety of ceftaroline fosamil versus ceftriaxone in patients with community-acquired pneumonia. Clin Infect Dis 2010; 51:1395–1405.
- Food and Drug Administration (FDA). Ceftaroline fosamil for the treatment of community-acquired bacterial pneumonia and complicated skin and skin structure infections. www.fda.gov/downloads/advisorycommittees/committeesmeetingmaterials/drugs/anti-infectivedrugsadvisorycommittee/ucm224656.pdf. Accessed June 1, 2015.
- Corey GR, Wilcox MH, Talbot GH, Thye D, Friedland D, Baculik T; CANVAS 1 investigators. CANVAS 1: the first phase III, randomized, double-blind study evaluating ceftaroline fosamil for the treatment of patients with complicated skin and skin structure infections. J Antimicrob Chemother 2010; 65(suppl 4):iv41–iv51.
- Wilcox MH, Corey GR, Talbot GH, Thye D, Friedland D, Baculik T; CANVAS 2 investigators. CANVAS 2: the second phase III, randomized, double-blind study evaluating ceftaroline fosamil for the treatment of patients with complicated skin and skin structure infections. J Antimicrob Chemother 2010; 65(suppl 4):iv53-iv65.
- Corey GR, Wilcox M, Talbot GH, et al. Integrated analysis of CANVAS 1 and 2: phase 3, multicenter, randomized, double-blind studies to evaluate the safety and efficacy of ceftaroline versus vancomycin plus aztreonam in complicated skin and skin-structure infection. Clin Infect Dis 2010; 51:641–650.
- Friedland HD, O’Neal T, Biek D, et al. CANVAS 1 and 2: analysis of clinical response at day 3 in two phase 3 trials of ceftaroline fosamil versus vancomycin plus aztreonam in treatment of acute bacterial skin and skin structure infections. Antimicrob Agents Chemother 2012; 56:2231–2236.
- Jacqueline C, Caillon J, Le Mabecque V, et al. In vivo efficacy of ceftaroline (PPI-0903), a new broad-spectrum cephalosporin, compared with linezolid and vancomycin against methicillin-resistant and vancomycin-intermediate Staphylococcus aureus in a rabbit endocarditis model. Antimicrob Agents Chemother 2007; 51:3397–3400.
- Ho TT, Cadena J, Childs LM, Gonzalez-Velez M, Lewis JS 2nd. Methicillin-resistant Staphylococcus aureus bacteraemia and endocarditis treated with ceftaroline salvage therapy. J Antimicrob Chemother 2012; 67:1267–1270.
- Lin JC, Aung G, Thomas A, Jahng M, Johns S, Fierer J. The use of ceftaroline fosamil in methicillin-resistant Staphylococcus aureus endocarditis and deep-seated MRSA infections: a retrospective case series of 10 patients. J Infect Chemother 2013; 19:42–49.
- Jacqueline C, Amador G, Caillon J, et al. Efficacy of the new cephalosporin ceftaroline in the treatment of experimental methicillin-resistant Staphylococcus aureus acute osteomyelitis. J Antimicrob Chemother 2010; 65:1749–1752.
- Stucki A, Acosta F, Cottagnoud M, Cottagnoud P. Efficacy of ceftaroline fosamil against Escherichia coli and Klebsiella pneumoniae strains in a rabbit meningitis model. Antimicrob Agents Chemother 2013; 57:5808–5810.
- Cottagnoud P, Cottagnoud M, Acosta F, Stucki A. Efficacy of ceftaroline fosamil against penicillin-sensitive and -resistant Streptococcus pneumoniae in an experimental rabbit meningitis model. Antimicrob Agents Chemother 2013; 57:4653–4655.
- Corrado ML. Integrated safety summary of CANVAS 1 and 2 trials: phase III, randomized, double-blind studies evaluating ceftaroline fosamil for the treatment of patients with complicated skin and skin structure infections. J Antimicrob Chemother 2010; 65(suppl 4):iv67–iv71.
- Rank DR, Friedland HD, Laudano JB. Integrated safety summary of FOCUS 1 and FOCUS 2 trials: phase III randomized, double-blind studies evaluating ceftaroline fosamil for the treatment of patients with community-acquired pneumonia. J Antimicrob Chemother 2011; 66(suppl 3):iii53–iii59.
- Stevens DL, Bisno AL, Chambers HF, et al. Practice guidelines for the diagnosis and management of skin and soft tissue infections: 2014 update by the Infectious Diseases Society of America. Clin Infect Dis 2014; 59:147–159.
- Mandell LA, Wunderink RG, Anzueto A, et al; Infectious Diseases Society of America; American Thoracic Society. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis 2007; 44:S27–S72.
Ceftaroline fosamil (Teflaro), introduced to the US market in October 2010, is the first beta-lactam agent with clinically useful activity against methicillin-resistant Staphylococcus aureus (MRSA). Currently, it is approved by the US Food and Drug Administration (FDA) to treat acute bacterial skin and skin-structure infections and community-acquired bacterial pneumonia caused by susceptible microorganisms.
In an era of increasing drug resistance and limited numbers of antimicrobials in the drug-production pipeline, ceftaroline is a step forward in fulfilling the Infectious Diseases Society of America’s “10 × ’20 Initiative” to increase support for drug research and manufacturing, with the goal of producing 10 new antimicrobial drugs by the year 2020.1 Ceftaroline was the first of several antibiotics to receive FDA approval in response to this initiative. It was followed by dalbavancin (May 2014), tedizolid phosphate (June 2014), oritavancin (August 2014), ceftolozane-tazobactam (December 2014), and ceftazidime-avibactam (February 2015). These antibiotic agents are aimed at treating infections caused by drug-resistant gram-positive and gram-negative microorganisms. It is important to understand and optimize the use of these new antibiotic agents in order to decrease the risk of emerging antibiotic resistance and superinfections (eg, Clostridium difficile infection) caused by antibiotic overuse or misuse.
This article provides an overview of ceftaroline’s mechanisms of action and resistance, spectrum of activity, pharmacokinetic properties, adverse effects, and current place in therapy.
AN ERA OF MULTIDRUG-RESISTANT MICROORGANISMS
Increasing rates of antimicrobial resistance threaten the efficacy of antimicrobial drugs in the daily practice of medicine. The World Health Organization has labeled antimicrobial resistance one of the three greatest threats to human health. Global efforts are under way to stimulate development of new antimicrobial agents and to decrease rates of antimicrobial resistance.
Staphylococcus aureus: A threat, even with vancomycin
Between 1998 and 2005, S aureus was one of the most common inpatient and outpatient isolates reported by clinical laboratories throughout the United States.2
Treatment of S aureus infection is complicated by a variety of resistance mechanisms that have evolved over time. In fact, the first resistant isolate of S aureus emerged not long after penicillin’s debut into clinical practice, and now the majority of strains are resistant to penicillin.
Methicillin was designed to overcome this beta-lactamase resistance and became the treatment of choice for penicillin-resistant S aureus isolates. However, MRSA isolates soon emerged because of the organism’s acquisition of penicillin-binding protein PBP2a via the mecA gene, leading to decreased binding affinity of methicillin.3
Since then, several agents active against MRSA (vancomycin, daptomycin, linezolid, tigecycline) have been introduced and continue to be widely used. While vancomycin is considered the first-line option for a variety of MRSA infections, its use has been threatened because of the emergence of vancomycin-intermediate-resistant S aureus (VISA), S aureus strains displaying vancomycin heteroresistance (hVISA), and vancomycin-resistant S aureus (VRSA) strains.4
VISA and hVISA isolates emerged through sequential mutations that lead to autolytic activity and cell-wall thickening. In contrast, the mechanism of resistance in VRSA is by acquisition of the vanA resistance gene, which alters the binding site of vancomycin from d-alanine-d-alanine to d-alanine-d-lactate.5
Streptococcus pneumoniae resistance: A continuing problem
The prevalence of drug resistance in S pneumoniae has risen since the late 1990s. A 2013 report from the SENTRY Antimicrobial Surveillance Program stated that almost 20% of S pneumoniae isolates were resistant to amoxicillin-clavulanate, and similar trends have been observed for penicillin (14.8%) and ceftriaxone (11.7%).6
S pneumoniae resistance is acquired through modifications of the penicillin-binding proteins, namely PBP1a, PBP2b, PBP2x, and, less frequently, PBP2a. These modifications lead to decreased binding affinity for most beta-lactams.7
Clinical impact of multidrug-resistant S aureus and S pneumoniae
In 2011, the US Centers for Disease Control and Prevention reported an estimated 80,000 severe MRSA infections and 11,000 MRSA-related deaths in the United States.8 In the same report, drug-resistant S pneumoniae was estimated to be responsible for almost 1.2 million illnesses and 7,000 deaths per year, leading to upwards of $96 million in related medical costs.
While invasive drug-resistant S pneumoniae infections usually affect patients at the extremes of age (under age 5 and over age 65), they have had a serious impact on patients of all ages.8
In light of the increasing prevalence of multidrug-resistant organisms, newer antimicrobial agents with novel mechanisms of action are needed.
CEFTAROLINE: A BETA-LACTAM WITH ANTI-MRSA ACTIVITY
The cephalosporins, a class of beta-lactam antibiotics, were originally derived from the fungus Cephalosporium (now called Acremonium). There are now many agents in this class, each containing a nucleus consisting of a beta-lactam ring fused to a six-member dihydrothiazine ring, and two side chains that can be modified to affect antibacterial activity and pharmacokinetic properties.
Cephalosporins are typically categorized into “generations.” With some exceptions, the first- and second-generation agents have good activity against gram-positive microorganisms, including methicillin-susceptible S aureus—but not against MRSA. The third- and fourth-generation cephalosporins have better gram-negative activity, with many agents having activity against the gram-negative bacterium Pseudomonas aeruginosa.
Enterococcal isolates are intrinsically resistant to cephalosporins. Additionally, cephalosporins are not active against anaerobic bacteria, except for a subset of structurally unique second-generation cephalosporins, ie, cefotetan and cefoxitin.
Ceftaroline was synthesized with specific manipulations of the side chains to provide enhanced activity against MRSA and multidrug-resistant S pneumoniae isolates, making it the first available beta-lactam with this ability.
Mechanism of action
Ceftaroline binds to penicillin-binding proteins, inhibiting transpeptidation. This interaction blocks the final stage of peptidoglycan synthesis and inhibits bacterial cell wall formation, ultimately leading to cellular autolysis and microorganism death. Ceftaroline binds with high affinity to PBP2a and PBP2x, expanding its activity to encompass MRSA and penicillin-resistant S pneumoniae isolates.9
Spectrum of activity
Ceftaroline has in vitro activity against many gram-positive and gram-negative bacteria,10–13 including (Table 1):
- Methicillin-susceptible and methicillin-resistant staphylococci
- VISA, VRSA, and hVISA
- Daptomycin-nonsusceptible S aureus
- Streptococcal species, including penicillin-resistant S pneumoniae
- Enterobacteriaceae, including Klebsiella pneumoniae, Klebsiella oxytoca, Escherichia coli, Citrobacter koseri, Citrobacter freundii, Enterobacter cloacae, Enterobacter aerogenes, Moraxella catarrhalis, Morganella morganii, and Proteus mirabilis.
Of note, ceftaroline is not active against Pseudomonas species, Enterococcus species, or Bacteroides fragilis. In addition, it is not active against the “atypical” respiratory pathogens Mycoplasma pneumoniae, Chlamydophila pneumoniae, and Legionella pneumophila.
Ceftaroline resistance
Gram-negative organisms appear to develop resistance to ceftaroline at rates similar to those observed with the other oxyimino-cephalosporins (eg, ceftriaxone). Ceftaroline is inactive against gram-negative organisms producing extended-spectrum beta-lactamases, including K pneumoniae carbapenemase and metallo-beta-lactamases.14 In addition, it induces the expression of AmpC beta-lactamases.
Although currently uncommon, resistance to ceftaroline has also been reported in S aureus strains.15 The mechanism of resistance is decreased binding affinity for PBP2a due to amino acid substitutions on the nonpenicillin-binding domains.15
Pharmacokinetic profile
An understanding of pharmacokinetics is key in optimizing the dose of antimicrobials so that the drugs are used most effectively and pathogens do not develop resistance to them.
Ceftaroline fosamil is a prodrug that, upon intravenous administration, is rapidly converted by phosphatase enzymes to its active moiety, ceftaroline. Its pharmacokinetic profile is summarized in Table 2.16,17 Its volume of distribution is similar to that of the fourth-generation cephalosporin cefepime.
Ceftaroline is then hydrolyzed into its inactive metabolite, ceftaroline M-1. It undergoes little hepatic metabolism and lacks properties to make it a substrate, inhibitor, or inducer of the CYP450 enzyme system and therefore is not likely to cause notable CYP450-related drug-drug interactions.
Like most other beta-lactams, ceftaroline is primarily excreted by the kidneys. Furthermore, an estimated 21% of a dose is eliminated with each intermittent hemodialysis session. Therefore, renal and intermittent hemodialysis dose adjustments are necessary. The estimated elimination half-life is 2.6 hours, necessitating dosing two to three times daily, depending on the indication and infectious inoculum.
Ceftaroline dosing
Ceftaroline is available only in a parenteral preparation and is typically given at a dose of 600 mg every 12 hours.10 The intravenous infusion is given over 1 hour.
The current stability data require reconstituted ceftaroline to be used within 6 hours at room temperature and within 24 hours if refrigerated.10
Ceftaroline requires dosing adjustments for patients with renal insufficiency. Per the manufacturer, renal dosing adjustments are based on the creatinine clearance rate, as estimated by the Cockroft-Gault formula:
- Creatinine clearance > 50 mL/min: no dosage adjustment necessary
- Creatinine clearance > 30 to ≤ 50 mL/min: give 400 mg every 12 hours
- Creatinine clearance ≥ 15 to ≤ 30 mL/min: give 300 mg every 12 hours
- Creatinine clearance < 15 mL/min or on intermittent dialysis: give 200 mg every 12 hours.
Ongoing clinical trials are investigating a higher-dosing strategy of 600 mg every 8 hours for patients with community-acquired bacterial pneumonia at risk of MRSA bacteremia.18
CLINICAL TRIALS LEADING TO CEFTAROLINE’S APPROVAL
Ceftaroline was approved for the treatment of community-acquired bacterial pneumonia and acute bacterial skin and skin-structure infections due to susceptible pathogens on the basis of phase 3 comparator trials.
Community-acquired bacterial pneumonia: The FOCUS 1 and 2 trials
The efficacy and safety of ceftaroline in the treatment of community-acquired bacterial pneumonia was studied in two randomized, double-blind, noninferiority trials, known as Ceftaroline Community-acquired Pneumonia vs Ceftriaxone (FOCUS) 1 and FOCUS 2.19,20
Patients were adults and not critically ill, as was reflected by their being in Pneumonia Outcomes Research Team (PORT) risk class III or IV (with class V indicating the highest risk of death). Therefore, the results may not be completely applicable to critically ill patients or those not admitted to the hospital. Of note, patients were excluded from the trials if they had infections known or thought to be due to MRSA or to atypical organisms.21 Baseline characteristics and patient demographics were similar between study groups in both trials.
A bacterial pathogen was identified in 26.1% of the patients included in the modified intent-to-treat analysis of the pooled data of the trials; the most common pathogens were S pneumoniae, methicillin-sensitive S aureus, Haemophilus influenzae, K pneumoniae, and E coli.21
Treatment. Patients received either ceftaroline 600 mg every 12 hours (or a lower dose based on renal function) or ceftriaxone 1 g every 24 hours. In addition, in the FOCUS 1 trial, patients in both treatment groups received clarithromycin 500 mg every 12 hours for the first day.19
Results. In both trials and in the integrated analysis, ceftaroline was noninferior to ceftriaxone (Table 3).22 In the integrated analysis of both trials, compared with the ceftriaxone group, the ceftaroline group had a higher clinical cure rate among patients classified as PORT risk class III (86.8% vs 79.2%, weighted treatment difference 12.6%, 95% confidence interval [CI] 1.3–13.8) and among patients who had not received prior antibiotic treatment (85.5% vs 74.9%, weighted treatment difference 11.2%, 95% CI 4.5–18.0).21
Acute bacterial skin and skin-structure infections: The CANVAS 1 and 2 trials
The efficacy and safety of ceftaroline in the treatment of complicated acute bacterial skin and skin-structure infections was studied in two randomized, double-blind trials: Ceftaroline Versus Vancomycin in Skin and Skin Structure Infections (CANVAS) 1 and CANVAS 2.23,24
Patients. Adult patients with a diagnosis of community-acquired skin and skin-structure infections warranting at least 5 days of intravenous antimicrobial therapy were included in the trials. Important protocol exclusions were patients with diabetic foot ulcers, decubitus ulcers, burns, ulcers associated with peripheral vascular disease accompanied by osteomyelitis, and suspected P aeruginosa infections.25 This limits the external validity of ceftaroline use in the aforementioned excluded patient populations.
Patients in each treatment group of the trials had similar demographic characteristics. The most common infections were cellulitis, major abscess requiring surgical intervention, wound infection, and infected ulcer. Bacteremia was present in 4.2% of patients in the ceftaroline group and in 3.8% of patients in the vancomycin-aztreonam group. The most common pathogen was S aureus. Methicillin resistance was present in 40% of the ceftaroline group and 34% of the control group.
Treatment. Patients received either ceftaroline 600 mg every 12 hours or the combination of vancomycin 1 g plus aztreonam 1 g given 12 hours, for 5 to 14 days.
Results. As assessed at a “test-of-cure” visit 8 to 15 days after the last dose of study medication, the efficacy of ceftaroline was similar to that of vancomycin-aztreonam, meeting the set noninferiority goal (Table 4).25 Moreover, if assessed on day 2 or 3 (a new end point recommended by the FDA), the rate of cessation of erythema spread and absence of fever was higher in the ceftaroline group than in the vancomycin-aztreonam group.26 However, this end point was not in the original trial protocol.
CEFTAROLINE FOR OTHER INDICATIONS
As noted, ceftaroline has been approved for treating community-acquired bacterial pneumonia and acute bacterial skin and skin-structure infections. In addition, it has been used in several studies in animals, and case reports of non-FDA approved indications including endocarditis and osteomyelitis have been published. Clinical trials are evaluating its use in pediatric patients, as well as for community-acquired bacterial pneumonia with risk for MRSA and for MRSA bacteremia.
Endocarditis
Animal studies have demonstrated ceftaroline to have bactericidal activity against MRSA and hVISA in endocarditis.27
A few case series have been published describing ceftaroline’s use as salvage therapy for persistent MRSA bacteremia and endocarditis. For example, Ho et al28 reported using it in three patients who had endocarditis as a source of their persistent bacteremia. All three patients had resolution of their MRSA bloodstream infection following ceftaroline therapy. The dosage was 600 mg every 8 hours, which is higher than in the manufacturer’s prescribing information.
Lin et al29 reported using ceftaroline in five patients with either possible or probable endocarditis. Three of the five patients had clinical cure as defined by resolution or improvement of all signs and symptoms of infection, and not requiring further antimicrobial therapy.29
More data from clinical trials would be beneficial in defining ceftaroline’s role in treating endocarditis caused by susceptible microorganisms.
Osteomyelitis
In animal studies of osteomyelitis, ceftaroline exhibited activity against MRSA in infected bone and joint fluid. Compared with vancomycin and linezolid, ceftaroline was associated with more significant decreases in bacterial load in the infected joint fluid, bone marrow, and bone.30
Lin et al29 gave ceftaroline to two patients with bone and joint infections, both of whom had received other therapies that had failed. The doses of ceftaroline were higher than those recommended in the prescribing information; clinical cure was noted in both cases following the switch.
These data come from case series, and more study of ceftaroline’s role in the treatment of osteomyelitis infections is warranted.
Meningitis
The use of ceftaroline in meningitis has been studied in rabbits. While ceftaroline penetrated into the cerebrospinal fluid in only negligible amounts in healthy rabbits (3% penetration), its penetration improved to 15% in animals with inflamed meninges. Ceftaroline cerebrospinal fluid levels in inflamed meninges were sufficient to provide bactericidal activity against penicillin-sensitive and resistant S pneumoniae strains as well as K pneumoniae and E coli strains.31,32
REPORTED ADVERSE EFFECTS OF CEFTAROLINE
Overall, ceftaroline was well tolerated in clinical trials, and its safety profile was similar to those of the comparator agents (ceftriaxone and vancomycin-aztreonam).
As with the other cephalosporins, hypersensitivity reactions have been reported with ceftaroline. In the clinical trials, 3% of patients developed a rash with ceftaroline.33,34 Patients with a history of beta-lactam allergy were excluded from the trials, so the rate of cross-reactivity with penicillins and with other cephalosporins is unknown.
In the phase 3 clinical trials, gastrointestinal side effects including diarrhea (5%), nausea (4%), and vomiting (2%) were reported with ceftaroline. C difficile-associated diarrhea has also been reported.33
As with other cephalosporins, ceftaroline can cause a false-positive result on the Coombs test. Approximately 11% of ceftaroline-treated patients in phase 3 clinical trials had a positive Coombs test, but hemolytic anemia did not occur in any patients.33,34
Discontinuation of ceftaroline due to an adverse reaction was reported in 2.7% of patients receiving the drug during phase 3 trials, compared with 3.7% with comparator agents.
WHEN SHOULD CEFTAROLINE BE USED IN DAILY PRACTICE?
Ceftaroline has been shown to be at least as effective as ceftriaxone in treating community-acquired bacterial pneumonia, and at least as effective as vancomycin-aztreonam in treating acute bacterial skin and skin-structure infections. The 2014 Infectious Diseases Society of America’s guidelines for the diagnosis and management of skin and soft-tissue infections recommend ceftaroline as an option for empiric therapy for purulent skin and soft-tissue infections.35
The guidelines on community-acquired pneumonia have not been updated since 2007, which was before ceftaroline was approved. However, these guidelines are currently undergoing revision and may provide insight on ceftaroline’s place in the treatment of community-acquired bacterial pneumonia.36
Currently, ceftaroline’s routine use for these indications should be balanced by its higher cost ($150 for a 600-mg dose) compared with ceftriaxone ($5 for a 1-g dose) or vancomycin ($25 for a 1-g dose). The drug’s in vitro activity against drug-resistant pneumococci and S aureus, including MRSA, hVISA, and VISA may help fill an unmet need or provide a safer and more tolerable alternative to currently available therapies.
However, ceftaroline’s lack of activity against P aeruginosa and carbapenem-resistant Enterobacteriaceae does not meet the public health threat needs stemming from these multidrug-resistant microorganisms. Ongoing clinical trials in patients with more serious MRSA infections will provide important information about ceftaroline’s role as an anti-MRSA agent.
While the discovery of antimicrobials has had one of the greatest impacts on medicine, continued antibiotic use is threatened by the emergence of drug-resistant pathogens. Therefore, it is as important as ever to be good stewards of our currently available antimicrobials. Developing usage and dosing criteria for antimicrobials based on available data and literature is a step forward in optimizing the use of antibiotics—a precious medical resource.
Ceftaroline fosamil (Teflaro), introduced to the US market in October 2010, is the first beta-lactam agent with clinically useful activity against methicillin-resistant Staphylococcus aureus (MRSA). Currently, it is approved by the US Food and Drug Administration (FDA) to treat acute bacterial skin and skin-structure infections and community-acquired bacterial pneumonia caused by susceptible microorganisms.
In an era of increasing drug resistance and limited numbers of antimicrobials in the drug-production pipeline, ceftaroline is a step forward in fulfilling the Infectious Diseases Society of America’s “10 × ’20 Initiative” to increase support for drug research and manufacturing, with the goal of producing 10 new antimicrobial drugs by the year 2020.1 Ceftaroline was the first of several antibiotics to receive FDA approval in response to this initiative. It was followed by dalbavancin (May 2014), tedizolid phosphate (June 2014), oritavancin (August 2014), ceftolozane-tazobactam (December 2014), and ceftazidime-avibactam (February 2015). These antibiotic agents are aimed at treating infections caused by drug-resistant gram-positive and gram-negative microorganisms. It is important to understand and optimize the use of these new antibiotic agents in order to decrease the risk of emerging antibiotic resistance and superinfections (eg, Clostridium difficile infection) caused by antibiotic overuse or misuse.
This article provides an overview of ceftaroline’s mechanisms of action and resistance, spectrum of activity, pharmacokinetic properties, adverse effects, and current place in therapy.
AN ERA OF MULTIDRUG-RESISTANT MICROORGANISMS
Increasing rates of antimicrobial resistance threaten the efficacy of antimicrobial drugs in the daily practice of medicine. The World Health Organization has labeled antimicrobial resistance one of the three greatest threats to human health. Global efforts are under way to stimulate development of new antimicrobial agents and to decrease rates of antimicrobial resistance.
Staphylococcus aureus: A threat, even with vancomycin
Between 1998 and 2005, S aureus was one of the most common inpatient and outpatient isolates reported by clinical laboratories throughout the United States.2
Treatment of S aureus infection is complicated by a variety of resistance mechanisms that have evolved over time. In fact, the first resistant isolate of S aureus emerged not long after penicillin’s debut into clinical practice, and now the majority of strains are resistant to penicillin.
Methicillin was designed to overcome this beta-lactamase resistance and became the treatment of choice for penicillin-resistant S aureus isolates. However, MRSA isolates soon emerged because of the organism’s acquisition of penicillin-binding protein PBP2a via the mecA gene, leading to decreased binding affinity of methicillin.3
Since then, several agents active against MRSA (vancomycin, daptomycin, linezolid, tigecycline) have been introduced and continue to be widely used. While vancomycin is considered the first-line option for a variety of MRSA infections, its use has been threatened because of the emergence of vancomycin-intermediate-resistant S aureus (VISA), S aureus strains displaying vancomycin heteroresistance (hVISA), and vancomycin-resistant S aureus (VRSA) strains.4
VISA and hVISA isolates emerged through sequential mutations that lead to autolytic activity and cell-wall thickening. In contrast, the mechanism of resistance in VRSA is by acquisition of the vanA resistance gene, which alters the binding site of vancomycin from d-alanine-d-alanine to d-alanine-d-lactate.5
Streptococcus pneumoniae resistance: A continuing problem
The prevalence of drug resistance in S pneumoniae has risen since the late 1990s. A 2013 report from the SENTRY Antimicrobial Surveillance Program stated that almost 20% of S pneumoniae isolates were resistant to amoxicillin-clavulanate, and similar trends have been observed for penicillin (14.8%) and ceftriaxone (11.7%).6
S pneumoniae resistance is acquired through modifications of the penicillin-binding proteins, namely PBP1a, PBP2b, PBP2x, and, less frequently, PBP2a. These modifications lead to decreased binding affinity for most beta-lactams.7
Clinical impact of multidrug-resistant S aureus and S pneumoniae
In 2011, the US Centers for Disease Control and Prevention reported an estimated 80,000 severe MRSA infections and 11,000 MRSA-related deaths in the United States.8 In the same report, drug-resistant S pneumoniae was estimated to be responsible for almost 1.2 million illnesses and 7,000 deaths per year, leading to upwards of $96 million in related medical costs.
While invasive drug-resistant S pneumoniae infections usually affect patients at the extremes of age (under age 5 and over age 65), they have had a serious impact on patients of all ages.8
In light of the increasing prevalence of multidrug-resistant organisms, newer antimicrobial agents with novel mechanisms of action are needed.
CEFTAROLINE: A BETA-LACTAM WITH ANTI-MRSA ACTIVITY
The cephalosporins, a class of beta-lactam antibiotics, were originally derived from the fungus Cephalosporium (now called Acremonium). There are now many agents in this class, each containing a nucleus consisting of a beta-lactam ring fused to a six-member dihydrothiazine ring, and two side chains that can be modified to affect antibacterial activity and pharmacokinetic properties.
Cephalosporins are typically categorized into “generations.” With some exceptions, the first- and second-generation agents have good activity against gram-positive microorganisms, including methicillin-susceptible S aureus—but not against MRSA. The third- and fourth-generation cephalosporins have better gram-negative activity, with many agents having activity against the gram-negative bacterium Pseudomonas aeruginosa.
Enterococcal isolates are intrinsically resistant to cephalosporins. Additionally, cephalosporins are not active against anaerobic bacteria, except for a subset of structurally unique second-generation cephalosporins, ie, cefotetan and cefoxitin.
Ceftaroline was synthesized with specific manipulations of the side chains to provide enhanced activity against MRSA and multidrug-resistant S pneumoniae isolates, making it the first available beta-lactam with this ability.
Mechanism of action
Ceftaroline binds to penicillin-binding proteins, inhibiting transpeptidation. This interaction blocks the final stage of peptidoglycan synthesis and inhibits bacterial cell wall formation, ultimately leading to cellular autolysis and microorganism death. Ceftaroline binds with high affinity to PBP2a and PBP2x, expanding its activity to encompass MRSA and penicillin-resistant S pneumoniae isolates.9
Spectrum of activity
Ceftaroline has in vitro activity against many gram-positive and gram-negative bacteria,10–13 including (Table 1):
- Methicillin-susceptible and methicillin-resistant staphylococci
- VISA, VRSA, and hVISA
- Daptomycin-nonsusceptible S aureus
- Streptococcal species, including penicillin-resistant S pneumoniae
- Enterobacteriaceae, including Klebsiella pneumoniae, Klebsiella oxytoca, Escherichia coli, Citrobacter koseri, Citrobacter freundii, Enterobacter cloacae, Enterobacter aerogenes, Moraxella catarrhalis, Morganella morganii, and Proteus mirabilis.
Of note, ceftaroline is not active against Pseudomonas species, Enterococcus species, or Bacteroides fragilis. In addition, it is not active against the “atypical” respiratory pathogens Mycoplasma pneumoniae, Chlamydophila pneumoniae, and Legionella pneumophila.
Ceftaroline resistance
Gram-negative organisms appear to develop resistance to ceftaroline at rates similar to those observed with the other oxyimino-cephalosporins (eg, ceftriaxone). Ceftaroline is inactive against gram-negative organisms producing extended-spectrum beta-lactamases, including K pneumoniae carbapenemase and metallo-beta-lactamases.14 In addition, it induces the expression of AmpC beta-lactamases.
Although currently uncommon, resistance to ceftaroline has also been reported in S aureus strains.15 The mechanism of resistance is decreased binding affinity for PBP2a due to amino acid substitutions on the nonpenicillin-binding domains.15
Pharmacokinetic profile
An understanding of pharmacokinetics is key in optimizing the dose of antimicrobials so that the drugs are used most effectively and pathogens do not develop resistance to them.
Ceftaroline fosamil is a prodrug that, upon intravenous administration, is rapidly converted by phosphatase enzymes to its active moiety, ceftaroline. Its pharmacokinetic profile is summarized in Table 2.16,17 Its volume of distribution is similar to that of the fourth-generation cephalosporin cefepime.
Ceftaroline is then hydrolyzed into its inactive metabolite, ceftaroline M-1. It undergoes little hepatic metabolism and lacks properties to make it a substrate, inhibitor, or inducer of the CYP450 enzyme system and therefore is not likely to cause notable CYP450-related drug-drug interactions.
Like most other beta-lactams, ceftaroline is primarily excreted by the kidneys. Furthermore, an estimated 21% of a dose is eliminated with each intermittent hemodialysis session. Therefore, renal and intermittent hemodialysis dose adjustments are necessary. The estimated elimination half-life is 2.6 hours, necessitating dosing two to three times daily, depending on the indication and infectious inoculum.
Ceftaroline dosing
Ceftaroline is available only in a parenteral preparation and is typically given at a dose of 600 mg every 12 hours.10 The intravenous infusion is given over 1 hour.
The current stability data require reconstituted ceftaroline to be used within 6 hours at room temperature and within 24 hours if refrigerated.10
Ceftaroline requires dosing adjustments for patients with renal insufficiency. Per the manufacturer, renal dosing adjustments are based on the creatinine clearance rate, as estimated by the Cockroft-Gault formula:
- Creatinine clearance > 50 mL/min: no dosage adjustment necessary
- Creatinine clearance > 30 to ≤ 50 mL/min: give 400 mg every 12 hours
- Creatinine clearance ≥ 15 to ≤ 30 mL/min: give 300 mg every 12 hours
- Creatinine clearance < 15 mL/min or on intermittent dialysis: give 200 mg every 12 hours.
Ongoing clinical trials are investigating a higher-dosing strategy of 600 mg every 8 hours for patients with community-acquired bacterial pneumonia at risk of MRSA bacteremia.18
CLINICAL TRIALS LEADING TO CEFTAROLINE’S APPROVAL
Ceftaroline was approved for the treatment of community-acquired bacterial pneumonia and acute bacterial skin and skin-structure infections due to susceptible pathogens on the basis of phase 3 comparator trials.
Community-acquired bacterial pneumonia: The FOCUS 1 and 2 trials
The efficacy and safety of ceftaroline in the treatment of community-acquired bacterial pneumonia was studied in two randomized, double-blind, noninferiority trials, known as Ceftaroline Community-acquired Pneumonia vs Ceftriaxone (FOCUS) 1 and FOCUS 2.19,20
Patients were adults and not critically ill, as was reflected by their being in Pneumonia Outcomes Research Team (PORT) risk class III or IV (with class V indicating the highest risk of death). Therefore, the results may not be completely applicable to critically ill patients or those not admitted to the hospital. Of note, patients were excluded from the trials if they had infections known or thought to be due to MRSA or to atypical organisms.21 Baseline characteristics and patient demographics were similar between study groups in both trials.
A bacterial pathogen was identified in 26.1% of the patients included in the modified intent-to-treat analysis of the pooled data of the trials; the most common pathogens were S pneumoniae, methicillin-sensitive S aureus, Haemophilus influenzae, K pneumoniae, and E coli.21
Treatment. Patients received either ceftaroline 600 mg every 12 hours (or a lower dose based on renal function) or ceftriaxone 1 g every 24 hours. In addition, in the FOCUS 1 trial, patients in both treatment groups received clarithromycin 500 mg every 12 hours for the first day.19
Results. In both trials and in the integrated analysis, ceftaroline was noninferior to ceftriaxone (Table 3).22 In the integrated analysis of both trials, compared with the ceftriaxone group, the ceftaroline group had a higher clinical cure rate among patients classified as PORT risk class III (86.8% vs 79.2%, weighted treatment difference 12.6%, 95% confidence interval [CI] 1.3–13.8) and among patients who had not received prior antibiotic treatment (85.5% vs 74.9%, weighted treatment difference 11.2%, 95% CI 4.5–18.0).21
Acute bacterial skin and skin-structure infections: The CANVAS 1 and 2 trials
The efficacy and safety of ceftaroline in the treatment of complicated acute bacterial skin and skin-structure infections was studied in two randomized, double-blind trials: Ceftaroline Versus Vancomycin in Skin and Skin Structure Infections (CANVAS) 1 and CANVAS 2.23,24
Patients. Adult patients with a diagnosis of community-acquired skin and skin-structure infections warranting at least 5 days of intravenous antimicrobial therapy were included in the trials. Important protocol exclusions were patients with diabetic foot ulcers, decubitus ulcers, burns, ulcers associated with peripheral vascular disease accompanied by osteomyelitis, and suspected P aeruginosa infections.25 This limits the external validity of ceftaroline use in the aforementioned excluded patient populations.
Patients in each treatment group of the trials had similar demographic characteristics. The most common infections were cellulitis, major abscess requiring surgical intervention, wound infection, and infected ulcer. Bacteremia was present in 4.2% of patients in the ceftaroline group and in 3.8% of patients in the vancomycin-aztreonam group. The most common pathogen was S aureus. Methicillin resistance was present in 40% of the ceftaroline group and 34% of the control group.
Treatment. Patients received either ceftaroline 600 mg every 12 hours or the combination of vancomycin 1 g plus aztreonam 1 g given 12 hours, for 5 to 14 days.
Results. As assessed at a “test-of-cure” visit 8 to 15 days after the last dose of study medication, the efficacy of ceftaroline was similar to that of vancomycin-aztreonam, meeting the set noninferiority goal (Table 4).25 Moreover, if assessed on day 2 or 3 (a new end point recommended by the FDA), the rate of cessation of erythema spread and absence of fever was higher in the ceftaroline group than in the vancomycin-aztreonam group.26 However, this end point was not in the original trial protocol.
CEFTAROLINE FOR OTHER INDICATIONS
As noted, ceftaroline has been approved for treating community-acquired bacterial pneumonia and acute bacterial skin and skin-structure infections. In addition, it has been used in several studies in animals, and case reports of non-FDA approved indications including endocarditis and osteomyelitis have been published. Clinical trials are evaluating its use in pediatric patients, as well as for community-acquired bacterial pneumonia with risk for MRSA and for MRSA bacteremia.
Endocarditis
Animal studies have demonstrated ceftaroline to have bactericidal activity against MRSA and hVISA in endocarditis.27
A few case series have been published describing ceftaroline’s use as salvage therapy for persistent MRSA bacteremia and endocarditis. For example, Ho et al28 reported using it in three patients who had endocarditis as a source of their persistent bacteremia. All three patients had resolution of their MRSA bloodstream infection following ceftaroline therapy. The dosage was 600 mg every 8 hours, which is higher than in the manufacturer’s prescribing information.
Lin et al29 reported using ceftaroline in five patients with either possible or probable endocarditis. Three of the five patients had clinical cure as defined by resolution or improvement of all signs and symptoms of infection, and not requiring further antimicrobial therapy.29
More data from clinical trials would be beneficial in defining ceftaroline’s role in treating endocarditis caused by susceptible microorganisms.
Osteomyelitis
In animal studies of osteomyelitis, ceftaroline exhibited activity against MRSA in infected bone and joint fluid. Compared with vancomycin and linezolid, ceftaroline was associated with more significant decreases in bacterial load in the infected joint fluid, bone marrow, and bone.30
Lin et al29 gave ceftaroline to two patients with bone and joint infections, both of whom had received other therapies that had failed. The doses of ceftaroline were higher than those recommended in the prescribing information; clinical cure was noted in both cases following the switch.
These data come from case series, and more study of ceftaroline’s role in the treatment of osteomyelitis infections is warranted.
Meningitis
The use of ceftaroline in meningitis has been studied in rabbits. While ceftaroline penetrated into the cerebrospinal fluid in only negligible amounts in healthy rabbits (3% penetration), its penetration improved to 15% in animals with inflamed meninges. Ceftaroline cerebrospinal fluid levels in inflamed meninges were sufficient to provide bactericidal activity against penicillin-sensitive and resistant S pneumoniae strains as well as K pneumoniae and E coli strains.31,32
REPORTED ADVERSE EFFECTS OF CEFTAROLINE
Overall, ceftaroline was well tolerated in clinical trials, and its safety profile was similar to those of the comparator agents (ceftriaxone and vancomycin-aztreonam).
As with the other cephalosporins, hypersensitivity reactions have been reported with ceftaroline. In the clinical trials, 3% of patients developed a rash with ceftaroline.33,34 Patients with a history of beta-lactam allergy were excluded from the trials, so the rate of cross-reactivity with penicillins and with other cephalosporins is unknown.
In the phase 3 clinical trials, gastrointestinal side effects including diarrhea (5%), nausea (4%), and vomiting (2%) were reported with ceftaroline. C difficile-associated diarrhea has also been reported.33
As with other cephalosporins, ceftaroline can cause a false-positive result on the Coombs test. Approximately 11% of ceftaroline-treated patients in phase 3 clinical trials had a positive Coombs test, but hemolytic anemia did not occur in any patients.33,34
Discontinuation of ceftaroline due to an adverse reaction was reported in 2.7% of patients receiving the drug during phase 3 trials, compared with 3.7% with comparator agents.
WHEN SHOULD CEFTAROLINE BE USED IN DAILY PRACTICE?
Ceftaroline has been shown to be at least as effective as ceftriaxone in treating community-acquired bacterial pneumonia, and at least as effective as vancomycin-aztreonam in treating acute bacterial skin and skin-structure infections. The 2014 Infectious Diseases Society of America’s guidelines for the diagnosis and management of skin and soft-tissue infections recommend ceftaroline as an option for empiric therapy for purulent skin and soft-tissue infections.35
The guidelines on community-acquired pneumonia have not been updated since 2007, which was before ceftaroline was approved. However, these guidelines are currently undergoing revision and may provide insight on ceftaroline’s place in the treatment of community-acquired bacterial pneumonia.36
Currently, ceftaroline’s routine use for these indications should be balanced by its higher cost ($150 for a 600-mg dose) compared with ceftriaxone ($5 for a 1-g dose) or vancomycin ($25 for a 1-g dose). The drug’s in vitro activity against drug-resistant pneumococci and S aureus, including MRSA, hVISA, and VISA may help fill an unmet need or provide a safer and more tolerable alternative to currently available therapies.
However, ceftaroline’s lack of activity against P aeruginosa and carbapenem-resistant Enterobacteriaceae does not meet the public health threat needs stemming from these multidrug-resistant microorganisms. Ongoing clinical trials in patients with more serious MRSA infections will provide important information about ceftaroline’s role as an anti-MRSA agent.
While the discovery of antimicrobials has had one of the greatest impacts on medicine, continued antibiotic use is threatened by the emergence of drug-resistant pathogens. Therefore, it is as important as ever to be good stewards of our currently available antimicrobials. Developing usage and dosing criteria for antimicrobials based on available data and literature is a step forward in optimizing the use of antibiotics—a precious medical resource.
- Infectious Diseases Society of America. The 10 x ‘20 Initiative: pursuing a global commitment to develop 10 new antibacterial drugs by 2020. Clin Infect Dis 2010; 50:1081–1083.
- Styers D, Sheehan DJ, Hogan P, Sahm DF. Laboratory-based surveillance of current antimicrobial resistance patterns and trends among Staphylococcus aureus: 2005 status in the United States. Ann Clin Microbiol Antimicrob 2006; 5:2.
- Farrell DJ, Castanheira M, Mendes RE, Sader HS, Jones RN. In vitro activity of ceftaroline against multidrug-resistant Staphylococcus aureus and Streptococcus pneumoniae: a review of published studies and the AWARE Surveillance Program (2008-2010). Clin Infect Dis 2012; 55(suppl 3):S206–S214.
- Holmes NE, Johnson PD, Howden BP. Relationship between vancomycin-resistant Staphylococcus aureus, vancomycin-intermediate S. aureus, high vancomycin MIC, and outcome in serious S. aureus infections. J Clin Microbiol 2012; 50:2548–2552.
- Lowy FD. Antimicrobial resistance: the example of Staphylococcus aureus. J Clin Invest 2003; 111:1265–1273.
- Jones RN, Sader HS, Mendes RE, Flamm RK. Update on antimicrobial susceptibility trends among Streptococcus pneumoniae in the United States: report of ceftaroline activity from the SENTRY Antimicrobial Surveillance Program (1998-2011). Diag Microbiol Infect Dis 2013; 75:107–109.
- Zapun A, Contreras-Martel C, Vernet T. Penicillin-binding proteins and beta-lactam resistance. FEMS Microbiol Rev 2008; 32:361–385.
- Centers for Disease Control and Prevention (CDC). Antibiotic resistance threats in the United States 2013. cdc.gov/drugresistance/threat-report-2013/pdf/ar-threats-2013-508.pdf. Accessed June 1, 2015.
- Moisan H, Pruneau M, Malouin F. Binding of ceftaroline to penicillin-binding proteins of Staphylococcus aureus and Streptococcus pneumoniae. J Antimicrob Chemother 2010; 65:713–716.
- Forest Laboratories, Inc. Teflaro® (ceftaroline fosamil): prescribing information. www.frx.com/pi/teflaro_pi.pdf. Accessed June 1, 2015.
- Richter SS, Heilmann KP, Dohrn CL, et al. Activity of ceftaroline and epidemiologic trends in Staphylococcus aureus isolates collected from 43 medical centers in the United States in 2009. Antimicrob Agents Chemother 2011; 55:4154–4160.
- Ge Y, Biek D, Talbot GH, Sahm DF. In vitro profiling of ceftaroline against a collection of recent bacterial clinical isolates from across the United States. Antimicrob Agents Chemother 2008; 52:3398–3407.
- Saravolatz L, Pawlak J, Johnson L. In vitro activity of ceftaroline against community-associated methicillin-resistant, vancomycin-intermediate, vancomycin-resistant, and daptomycin-nonsusceptible Staphylococcus aureus isolates. Antimicrob Agents Chemother 2010; 54:3027–3030.
- Mushtaq S, Livermore DM. AmpC induction by ceftaroline. J Antimicrob Chemother 2010; 65:586–588.
- Mendes RE, Tsakris A, Sader HS, et al. Characterization of methicillin-resistant Staphylococcus aureus displaying increased MICs of ceftaroline. J Antimicrob Chemother 2012; 67:1321–1324.
- Lodise TP, Low DE. Ceftaroline fosamil in the treatment of community-acquired bacterial pneumonia and acute bacterial skin and skin structure infections. Drugs 2012; 72:1473–1493.
- Riccobene TA, Su SF, Rank D. Single- and multiple-dose study to determine the safety, tolerability, and pharmacokinetics of ceftaroline fosamil in combination with avibactam in healthy subjects. Antimicrob Agents Chemother 2013; 57:1496–1504.
- US National Institutes of Health. ClinicalTrials.gov. Evaluation of ceftaroline fosamil versus a comparator in adult subjects with community-acquired bacterial pneumonia (CABP) with risk for methicillin-resistant Staphylococcus aureus. http://clinicaltrials.gov/ct2/show/NCT01645735. Accessed June 1, 2015.
- File TM Jr, Low DE, Eckburg PB, et al; FOCUS 1 investigators. FOCUS 1: a randomized, double-blinded, multicentre, phase III trial of the efficacy and safety of ceftaroline fosamil versus ceftriaxone in community-acquired pneumonia. J Antimicrob Chemother 2011; 66(suppl 3):iii19–iii32.
- Low DE, File TM Jr, Eckburg PB, et al; FOCUS 2 investigators. FOCUS 2: a randomized, double-blinded, multicentre, phase III trial of the efficacy and safety of ceftaroline fosamil versus ceftriaxone in community-acquired pneumonia. J Antimicrob Chemother 2011; 66(suppl 3):iii33–iii44.
- File TM Jr, Low DE, Eckburg PB, et al. Integrated analysis of FOCUS 1 and FOCUS 2: randomized, doubled-blinded, multicenter phase 3 trials of the efficacy and safety of ceftaroline fosamil versus ceftriaxone in patients with community-acquired pneumonia. Clin Infect Dis 2010; 51:1395–1405.
- Food and Drug Administration (FDA). Ceftaroline fosamil for the treatment of community-acquired bacterial pneumonia and complicated skin and skin structure infections. www.fda.gov/downloads/advisorycommittees/committeesmeetingmaterials/drugs/anti-infectivedrugsadvisorycommittee/ucm224656.pdf. Accessed June 1, 2015.
- Corey GR, Wilcox MH, Talbot GH, Thye D, Friedland D, Baculik T; CANVAS 1 investigators. CANVAS 1: the first phase III, randomized, double-blind study evaluating ceftaroline fosamil for the treatment of patients with complicated skin and skin structure infections. J Antimicrob Chemother 2010; 65(suppl 4):iv41–iv51.
- Wilcox MH, Corey GR, Talbot GH, Thye D, Friedland D, Baculik T; CANVAS 2 investigators. CANVAS 2: the second phase III, randomized, double-blind study evaluating ceftaroline fosamil for the treatment of patients with complicated skin and skin structure infections. J Antimicrob Chemother 2010; 65(suppl 4):iv53-iv65.
- Corey GR, Wilcox M, Talbot GH, et al. Integrated analysis of CANVAS 1 and 2: phase 3, multicenter, randomized, double-blind studies to evaluate the safety and efficacy of ceftaroline versus vancomycin plus aztreonam in complicated skin and skin-structure infection. Clin Infect Dis 2010; 51:641–650.
- Friedland HD, O’Neal T, Biek D, et al. CANVAS 1 and 2: analysis of clinical response at day 3 in two phase 3 trials of ceftaroline fosamil versus vancomycin plus aztreonam in treatment of acute bacterial skin and skin structure infections. Antimicrob Agents Chemother 2012; 56:2231–2236.
- Jacqueline C, Caillon J, Le Mabecque V, et al. In vivo efficacy of ceftaroline (PPI-0903), a new broad-spectrum cephalosporin, compared with linezolid and vancomycin against methicillin-resistant and vancomycin-intermediate Staphylococcus aureus in a rabbit endocarditis model. Antimicrob Agents Chemother 2007; 51:3397–3400.
- Ho TT, Cadena J, Childs LM, Gonzalez-Velez M, Lewis JS 2nd. Methicillin-resistant Staphylococcus aureus bacteraemia and endocarditis treated with ceftaroline salvage therapy. J Antimicrob Chemother 2012; 67:1267–1270.
- Lin JC, Aung G, Thomas A, Jahng M, Johns S, Fierer J. The use of ceftaroline fosamil in methicillin-resistant Staphylococcus aureus endocarditis and deep-seated MRSA infections: a retrospective case series of 10 patients. J Infect Chemother 2013; 19:42–49.
- Jacqueline C, Amador G, Caillon J, et al. Efficacy of the new cephalosporin ceftaroline in the treatment of experimental methicillin-resistant Staphylococcus aureus acute osteomyelitis. J Antimicrob Chemother 2010; 65:1749–1752.
- Stucki A, Acosta F, Cottagnoud M, Cottagnoud P. Efficacy of ceftaroline fosamil against Escherichia coli and Klebsiella pneumoniae strains in a rabbit meningitis model. Antimicrob Agents Chemother 2013; 57:5808–5810.
- Cottagnoud P, Cottagnoud M, Acosta F, Stucki A. Efficacy of ceftaroline fosamil against penicillin-sensitive and -resistant Streptococcus pneumoniae in an experimental rabbit meningitis model. Antimicrob Agents Chemother 2013; 57:4653–4655.
- Corrado ML. Integrated safety summary of CANVAS 1 and 2 trials: phase III, randomized, double-blind studies evaluating ceftaroline fosamil for the treatment of patients with complicated skin and skin structure infections. J Antimicrob Chemother 2010; 65(suppl 4):iv67–iv71.
- Rank DR, Friedland HD, Laudano JB. Integrated safety summary of FOCUS 1 and FOCUS 2 trials: phase III randomized, double-blind studies evaluating ceftaroline fosamil for the treatment of patients with community-acquired pneumonia. J Antimicrob Chemother 2011; 66(suppl 3):iii53–iii59.
- Stevens DL, Bisno AL, Chambers HF, et al. Practice guidelines for the diagnosis and management of skin and soft tissue infections: 2014 update by the Infectious Diseases Society of America. Clin Infect Dis 2014; 59:147–159.
- Mandell LA, Wunderink RG, Anzueto A, et al; Infectious Diseases Society of America; American Thoracic Society. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis 2007; 44:S27–S72.
- Infectious Diseases Society of America. The 10 x ‘20 Initiative: pursuing a global commitment to develop 10 new antibacterial drugs by 2020. Clin Infect Dis 2010; 50:1081–1083.
- Styers D, Sheehan DJ, Hogan P, Sahm DF. Laboratory-based surveillance of current antimicrobial resistance patterns and trends among Staphylococcus aureus: 2005 status in the United States. Ann Clin Microbiol Antimicrob 2006; 5:2.
- Farrell DJ, Castanheira M, Mendes RE, Sader HS, Jones RN. In vitro activity of ceftaroline against multidrug-resistant Staphylococcus aureus and Streptococcus pneumoniae: a review of published studies and the AWARE Surveillance Program (2008-2010). Clin Infect Dis 2012; 55(suppl 3):S206–S214.
- Holmes NE, Johnson PD, Howden BP. Relationship between vancomycin-resistant Staphylococcus aureus, vancomycin-intermediate S. aureus, high vancomycin MIC, and outcome in serious S. aureus infections. J Clin Microbiol 2012; 50:2548–2552.
- Lowy FD. Antimicrobial resistance: the example of Staphylococcus aureus. J Clin Invest 2003; 111:1265–1273.
- Jones RN, Sader HS, Mendes RE, Flamm RK. Update on antimicrobial susceptibility trends among Streptococcus pneumoniae in the United States: report of ceftaroline activity from the SENTRY Antimicrobial Surveillance Program (1998-2011). Diag Microbiol Infect Dis 2013; 75:107–109.
- Zapun A, Contreras-Martel C, Vernet T. Penicillin-binding proteins and beta-lactam resistance. FEMS Microbiol Rev 2008; 32:361–385.
- Centers for Disease Control and Prevention (CDC). Antibiotic resistance threats in the United States 2013. cdc.gov/drugresistance/threat-report-2013/pdf/ar-threats-2013-508.pdf. Accessed June 1, 2015.
- Moisan H, Pruneau M, Malouin F. Binding of ceftaroline to penicillin-binding proteins of Staphylococcus aureus and Streptococcus pneumoniae. J Antimicrob Chemother 2010; 65:713–716.
- Forest Laboratories, Inc. Teflaro® (ceftaroline fosamil): prescribing information. www.frx.com/pi/teflaro_pi.pdf. Accessed June 1, 2015.
- Richter SS, Heilmann KP, Dohrn CL, et al. Activity of ceftaroline and epidemiologic trends in Staphylococcus aureus isolates collected from 43 medical centers in the United States in 2009. Antimicrob Agents Chemother 2011; 55:4154–4160.
- Ge Y, Biek D, Talbot GH, Sahm DF. In vitro profiling of ceftaroline against a collection of recent bacterial clinical isolates from across the United States. Antimicrob Agents Chemother 2008; 52:3398–3407.
- Saravolatz L, Pawlak J, Johnson L. In vitro activity of ceftaroline against community-associated methicillin-resistant, vancomycin-intermediate, vancomycin-resistant, and daptomycin-nonsusceptible Staphylococcus aureus isolates. Antimicrob Agents Chemother 2010; 54:3027–3030.
- Mushtaq S, Livermore DM. AmpC induction by ceftaroline. J Antimicrob Chemother 2010; 65:586–588.
- Mendes RE, Tsakris A, Sader HS, et al. Characterization of methicillin-resistant Staphylococcus aureus displaying increased MICs of ceftaroline. J Antimicrob Chemother 2012; 67:1321–1324.
- Lodise TP, Low DE. Ceftaroline fosamil in the treatment of community-acquired bacterial pneumonia and acute bacterial skin and skin structure infections. Drugs 2012; 72:1473–1493.
- Riccobene TA, Su SF, Rank D. Single- and multiple-dose study to determine the safety, tolerability, and pharmacokinetics of ceftaroline fosamil in combination with avibactam in healthy subjects. Antimicrob Agents Chemother 2013; 57:1496–1504.
- US National Institutes of Health. ClinicalTrials.gov. Evaluation of ceftaroline fosamil versus a comparator in adult subjects with community-acquired bacterial pneumonia (CABP) with risk for methicillin-resistant Staphylococcus aureus. http://clinicaltrials.gov/ct2/show/NCT01645735. Accessed June 1, 2015.
- File TM Jr, Low DE, Eckburg PB, et al; FOCUS 1 investigators. FOCUS 1: a randomized, double-blinded, multicentre, phase III trial of the efficacy and safety of ceftaroline fosamil versus ceftriaxone in community-acquired pneumonia. J Antimicrob Chemother 2011; 66(suppl 3):iii19–iii32.
- Low DE, File TM Jr, Eckburg PB, et al; FOCUS 2 investigators. FOCUS 2: a randomized, double-blinded, multicentre, phase III trial of the efficacy and safety of ceftaroline fosamil versus ceftriaxone in community-acquired pneumonia. J Antimicrob Chemother 2011; 66(suppl 3):iii33–iii44.
- File TM Jr, Low DE, Eckburg PB, et al. Integrated analysis of FOCUS 1 and FOCUS 2: randomized, doubled-blinded, multicenter phase 3 trials of the efficacy and safety of ceftaroline fosamil versus ceftriaxone in patients with community-acquired pneumonia. Clin Infect Dis 2010; 51:1395–1405.
- Food and Drug Administration (FDA). Ceftaroline fosamil for the treatment of community-acquired bacterial pneumonia and complicated skin and skin structure infections. www.fda.gov/downloads/advisorycommittees/committeesmeetingmaterials/drugs/anti-infectivedrugsadvisorycommittee/ucm224656.pdf. Accessed June 1, 2015.
- Corey GR, Wilcox MH, Talbot GH, Thye D, Friedland D, Baculik T; CANVAS 1 investigators. CANVAS 1: the first phase III, randomized, double-blind study evaluating ceftaroline fosamil for the treatment of patients with complicated skin and skin structure infections. J Antimicrob Chemother 2010; 65(suppl 4):iv41–iv51.
- Wilcox MH, Corey GR, Talbot GH, Thye D, Friedland D, Baculik T; CANVAS 2 investigators. CANVAS 2: the second phase III, randomized, double-blind study evaluating ceftaroline fosamil for the treatment of patients with complicated skin and skin structure infections. J Antimicrob Chemother 2010; 65(suppl 4):iv53-iv65.
- Corey GR, Wilcox M, Talbot GH, et al. Integrated analysis of CANVAS 1 and 2: phase 3, multicenter, randomized, double-blind studies to evaluate the safety and efficacy of ceftaroline versus vancomycin plus aztreonam in complicated skin and skin-structure infection. Clin Infect Dis 2010; 51:641–650.
- Friedland HD, O’Neal T, Biek D, et al. CANVAS 1 and 2: analysis of clinical response at day 3 in two phase 3 trials of ceftaroline fosamil versus vancomycin plus aztreonam in treatment of acute bacterial skin and skin structure infections. Antimicrob Agents Chemother 2012; 56:2231–2236.
- Jacqueline C, Caillon J, Le Mabecque V, et al. In vivo efficacy of ceftaroline (PPI-0903), a new broad-spectrum cephalosporin, compared with linezolid and vancomycin against methicillin-resistant and vancomycin-intermediate Staphylococcus aureus in a rabbit endocarditis model. Antimicrob Agents Chemother 2007; 51:3397–3400.
- Ho TT, Cadena J, Childs LM, Gonzalez-Velez M, Lewis JS 2nd. Methicillin-resistant Staphylococcus aureus bacteraemia and endocarditis treated with ceftaroline salvage therapy. J Antimicrob Chemother 2012; 67:1267–1270.
- Lin JC, Aung G, Thomas A, Jahng M, Johns S, Fierer J. The use of ceftaroline fosamil in methicillin-resistant Staphylococcus aureus endocarditis and deep-seated MRSA infections: a retrospective case series of 10 patients. J Infect Chemother 2013; 19:42–49.
- Jacqueline C, Amador G, Caillon J, et al. Efficacy of the new cephalosporin ceftaroline in the treatment of experimental methicillin-resistant Staphylococcus aureus acute osteomyelitis. J Antimicrob Chemother 2010; 65:1749–1752.
- Stucki A, Acosta F, Cottagnoud M, Cottagnoud P. Efficacy of ceftaroline fosamil against Escherichia coli and Klebsiella pneumoniae strains in a rabbit meningitis model. Antimicrob Agents Chemother 2013; 57:5808–5810.
- Cottagnoud P, Cottagnoud M, Acosta F, Stucki A. Efficacy of ceftaroline fosamil against penicillin-sensitive and -resistant Streptococcus pneumoniae in an experimental rabbit meningitis model. Antimicrob Agents Chemother 2013; 57:4653–4655.
- Corrado ML. Integrated safety summary of CANVAS 1 and 2 trials: phase III, randomized, double-blind studies evaluating ceftaroline fosamil for the treatment of patients with complicated skin and skin structure infections. J Antimicrob Chemother 2010; 65(suppl 4):iv67–iv71.
- Rank DR, Friedland HD, Laudano JB. Integrated safety summary of FOCUS 1 and FOCUS 2 trials: phase III randomized, double-blind studies evaluating ceftaroline fosamil for the treatment of patients with community-acquired pneumonia. J Antimicrob Chemother 2011; 66(suppl 3):iii53–iii59.
- Stevens DL, Bisno AL, Chambers HF, et al. Practice guidelines for the diagnosis and management of skin and soft tissue infections: 2014 update by the Infectious Diseases Society of America. Clin Infect Dis 2014; 59:147–159.
- Mandell LA, Wunderink RG, Anzueto A, et al; Infectious Diseases Society of America; American Thoracic Society. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis 2007; 44:S27–S72.
KEY POINTS
- Resistance of S aureus and S pneumoniae to multiple antimicrobial drugs is on the rise, and new agents are urgently needed.
- Ceftaroline’s molecular structure was designed to provide enhanced activity against MRSA and multidrug-resistant S pneumoniae.
- In clinical trials leading to its approval, ceftaroline was found to be at least as effective as ceftriaxone in treating community-acquired pneumonia and at least as effective as vancomycin plus aztreonam in treating acute bacterial skin and skin-structure infections.
- The routine use of ceftaroline for these indications should be balanced by its higher cost compared with ceftriaxone or vancomycin. Ongoing studies should shed more light on its role in treatment.
Electrocardiographic changes in amitriptyline overdose
A 49-year-old woman with a history of depression, bipolar disorder, and chronic back pain was brought to the emergency department unresponsive after having taken an unknown quantity of amitriptyline tablets.
On arrival, she was comatose, with a score of 3 (the lowest possible score) on the 15-point Glasgow Coma Scale. Her blood pressure was 65/22 mm Hg, heart rate 121 beats per minute, respiratory rate 14 per minute, and oxygen saturation 88% on room air. The rest of the initial physical examination was normal.
She was immediately intubated, put on mechanical ventilation, and given an infusion of a 1-L bolus of normal saline and 50 mmol (1 mmol/kg) of sodium bicarbonate. Norepinephrine infusion was started. Gastric lavage was not done.
Results of initial laboratory testing showed a serum potassium of 2.9 mmol/L (reference range 3.5–5.0) and a serum magnesium of 1.6 mmol/L (1.7–2.6), which were corrected with infusion of 60 mmol of potassium chloride and 2 g of magnesium sulfate. The serum amitriptyline measurement was ordered at the time of her presentation to the emergency department.
Arterial blood gas analysis showed:
- pH 7.15 (normal range 7.35–7.45)
- Paco2 66 mm Hg (34–46)
- Pao2 229 mm Hg (85–95)
- Bicarbonate 22 mmol/L (22–26).
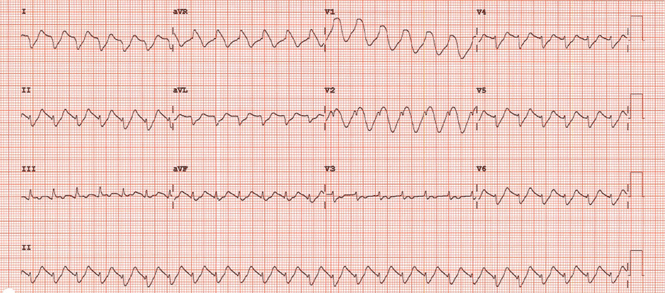
The initial electrocardiogram (ECG) (Figure 1) showed regular wide-complex tachycardia with no definite right or left bundle branch block morphology, no discernible P waves, a QRS duration of 198 msec, right axis deviation, and no Brugada criteria to suggest ventricular tachycardia.
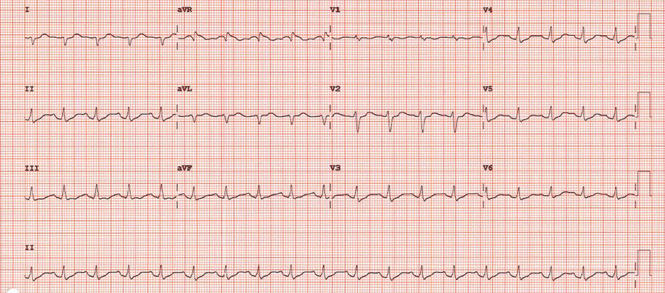
She remained hypotensive, with regular wide-complex tachycardia on the ECG. She was given an additional 1-L bolus of normal saline and 100 mmol (2 mmol/kg) of sodium bicarbonate, and within 1 minute the wide-complex tachycardia resolved to narrow-complex sinus tachycardia (Figure 2). At this point, an infusion of 150 mmol/L of sodium bicarbonate in dextrose 5% in water was started, with serial ECGs to monitor the QRS duration and serial arterial blood gas monitoring to maintain the pH between 7.45 and 7.55.
TRANSFER TO THE ICU
She was then transferred to the intensive care unit (ICU), where she remained for 2 weeks. While in the ICU, she had a single recurrence of wide-complex tachycardia that resolved immediately with an infusion of 100 mmol of sodium bicarbonate. A urine toxicology screen was negative, and the serum amitriptyline measurement, returned from the laboratory 48 hours after her initial presentation, was 594 ng/mL (reference range 100–250 ng/mL). She was eventually weaned off the norepinephrine infusion after 20 hours, the sodium bicarbonate infusion was discontinued after 4 days, and she was taken off mechanical ventilation after 10 days. Also during her ICU stay, she had seizures on day 3 and developed aspiration pneumonia.
From the ICU, she was transferred to a regular floor, where she stayed for another week and then was transferred to a rehabilitation center. This patient was known to have clinical depression and to have attempted suicide once before. She had recently been under additional psychosocial stresses, which likely prompted this second attempt.
She reportedly had no neurologic or cardiovascular sequelae after her discharge from the hospital.
AMITRIPTYLINE OVERDOSE
Amitriptyline causes a relatively high number of fatal overdoses, at 34 per 1 million prescriptions.1 Death is usually from hypotension and ventricular arrhythmia caused by blockage of cardiac fast sodium channels leading to disturbances of cardiac conduction such as wide-complex tachycardia.
Other manifestations of amitriptyline overdose include seizures, sedation, and anticholinergic toxicity from variable blockade of gamma-aminobutyric acid receptors, histamine 1 receptors, and alpha receptors.2
Of the various changes on ECG described with amitriptyline overdose, sinus tachycardia is the most common. A QRS duration greater than 100 msec, right to extreme-right axis deviation with negative QRS complexes in leads I and aVL, and an R-wave amplitude greater than 3 mm in lead aVR are indications for sodium bicarbonate infusion, especially in hemodynamically unstable patients.3 Sodium bicarbonate increases the serum concentration of sodium and thereby overcomes the sodium channel blockade. It also alkalinizes the serum, favoring an electrically neutral form of amitriptyline that binds less to receptors and binds more to alpha-1-acid glycoprotein, decreasing the fraction of free drug available for toxicity.4
In patients with amitriptyline overdose, wide-complex tachycardia and hypotension refractory to sodium bicarbonate infusion can be treated with lidocaine, magnesium sulfate, direct-current cardioversion, and lipid resuscitation.5,6 Treatment with class IA, IC, and III antiarrhythmics is contraindicated, as they block sodium channels and thus can worsen conduction disturbances.
- Henry JA, Alexander CA, Sener EK. Relative mortality from overdose of antidepressants. BMJ 1995; 310:221–224.
- Shannon M, Merola J, Lovejoy FH Jr. Hypotension in severe tricyclic antidepressant overdose. Am J Emerg Med 1988; 6:439–442.
- Liebelt EL, Francis PD, Woolf AD. ECG lead aVR versus QRS interval in predicting seizures and arrhythmias in acute tricyclic antidepressant toxicity. Ann Emerg Med 1995; 26:195–201.
- Sayniuk BI, Jhamandas V. Mechanism of reversal of toxic effects of amitriptyline on cardiac Purkinje fibres by sodium bicarbonate. J Pharmacol Exp Ther 1984; 231:387.
- Kiberd MB, Minor SF. Lipid therapy for the treatment of a refractory amitriptyline overdose. CJEM 2012; 14:193–197.
- Harvey M, Cave G. Case report: successful lipid resuscitation in multidrug overdose with predominant tricyclic antidepressant toxidrome. Int J Emerg Med 2012; 5:8.
A 49-year-old woman with a history of depression, bipolar disorder, and chronic back pain was brought to the emergency department unresponsive after having taken an unknown quantity of amitriptyline tablets.
On arrival, she was comatose, with a score of 3 (the lowest possible score) on the 15-point Glasgow Coma Scale. Her blood pressure was 65/22 mm Hg, heart rate 121 beats per minute, respiratory rate 14 per minute, and oxygen saturation 88% on room air. The rest of the initial physical examination was normal.
She was immediately intubated, put on mechanical ventilation, and given an infusion of a 1-L bolus of normal saline and 50 mmol (1 mmol/kg) of sodium bicarbonate. Norepinephrine infusion was started. Gastric lavage was not done.
Results of initial laboratory testing showed a serum potassium of 2.9 mmol/L (reference range 3.5–5.0) and a serum magnesium of 1.6 mmol/L (1.7–2.6), which were corrected with infusion of 60 mmol of potassium chloride and 2 g of magnesium sulfate. The serum amitriptyline measurement was ordered at the time of her presentation to the emergency department.
Arterial blood gas analysis showed:
- pH 7.15 (normal range 7.35–7.45)
- Paco2 66 mm Hg (34–46)
- Pao2 229 mm Hg (85–95)
- Bicarbonate 22 mmol/L (22–26).
The initial electrocardiogram (ECG) (Figure 1) showed regular wide-complex tachycardia with no definite right or left bundle branch block morphology, no discernible P waves, a QRS duration of 198 msec, right axis deviation, and no Brugada criteria to suggest ventricular tachycardia.
She remained hypotensive, with regular wide-complex tachycardia on the ECG. She was given an additional 1-L bolus of normal saline and 100 mmol (2 mmol/kg) of sodium bicarbonate, and within 1 minute the wide-complex tachycardia resolved to narrow-complex sinus tachycardia (Figure 2). At this point, an infusion of 150 mmol/L of sodium bicarbonate in dextrose 5% in water was started, with serial ECGs to monitor the QRS duration and serial arterial blood gas monitoring to maintain the pH between 7.45 and 7.55.
TRANSFER TO THE ICU
She was then transferred to the intensive care unit (ICU), where she remained for 2 weeks. While in the ICU, she had a single recurrence of wide-complex tachycardia that resolved immediately with an infusion of 100 mmol of sodium bicarbonate. A urine toxicology screen was negative, and the serum amitriptyline measurement, returned from the laboratory 48 hours after her initial presentation, was 594 ng/mL (reference range 100–250 ng/mL). She was eventually weaned off the norepinephrine infusion after 20 hours, the sodium bicarbonate infusion was discontinued after 4 days, and she was taken off mechanical ventilation after 10 days. Also during her ICU stay, she had seizures on day 3 and developed aspiration pneumonia.
From the ICU, she was transferred to a regular floor, where she stayed for another week and then was transferred to a rehabilitation center. This patient was known to have clinical depression and to have attempted suicide once before. She had recently been under additional psychosocial stresses, which likely prompted this second attempt.
She reportedly had no neurologic or cardiovascular sequelae after her discharge from the hospital.
AMITRIPTYLINE OVERDOSE
Amitriptyline causes a relatively high number of fatal overdoses, at 34 per 1 million prescriptions.1 Death is usually from hypotension and ventricular arrhythmia caused by blockage of cardiac fast sodium channels leading to disturbances of cardiac conduction such as wide-complex tachycardia.
Other manifestations of amitriptyline overdose include seizures, sedation, and anticholinergic toxicity from variable blockade of gamma-aminobutyric acid receptors, histamine 1 receptors, and alpha receptors.2
Of the various changes on ECG described with amitriptyline overdose, sinus tachycardia is the most common. A QRS duration greater than 100 msec, right to extreme-right axis deviation with negative QRS complexes in leads I and aVL, and an R-wave amplitude greater than 3 mm in lead aVR are indications for sodium bicarbonate infusion, especially in hemodynamically unstable patients.3 Sodium bicarbonate increases the serum concentration of sodium and thereby overcomes the sodium channel blockade. It also alkalinizes the serum, favoring an electrically neutral form of amitriptyline that binds less to receptors and binds more to alpha-1-acid glycoprotein, decreasing the fraction of free drug available for toxicity.4
In patients with amitriptyline overdose, wide-complex tachycardia and hypotension refractory to sodium bicarbonate infusion can be treated with lidocaine, magnesium sulfate, direct-current cardioversion, and lipid resuscitation.5,6 Treatment with class IA, IC, and III antiarrhythmics is contraindicated, as they block sodium channels and thus can worsen conduction disturbances.
A 49-year-old woman with a history of depression, bipolar disorder, and chronic back pain was brought to the emergency department unresponsive after having taken an unknown quantity of amitriptyline tablets.
On arrival, she was comatose, with a score of 3 (the lowest possible score) on the 15-point Glasgow Coma Scale. Her blood pressure was 65/22 mm Hg, heart rate 121 beats per minute, respiratory rate 14 per minute, and oxygen saturation 88% on room air. The rest of the initial physical examination was normal.
She was immediately intubated, put on mechanical ventilation, and given an infusion of a 1-L bolus of normal saline and 50 mmol (1 mmol/kg) of sodium bicarbonate. Norepinephrine infusion was started. Gastric lavage was not done.
Results of initial laboratory testing showed a serum potassium of 2.9 mmol/L (reference range 3.5–5.0) and a serum magnesium of 1.6 mmol/L (1.7–2.6), which were corrected with infusion of 60 mmol of potassium chloride and 2 g of magnesium sulfate. The serum amitriptyline measurement was ordered at the time of her presentation to the emergency department.
Arterial blood gas analysis showed:
- pH 7.15 (normal range 7.35–7.45)
- Paco2 66 mm Hg (34–46)
- Pao2 229 mm Hg (85–95)
- Bicarbonate 22 mmol/L (22–26).
The initial electrocardiogram (ECG) (Figure 1) showed regular wide-complex tachycardia with no definite right or left bundle branch block morphology, no discernible P waves, a QRS duration of 198 msec, right axis deviation, and no Brugada criteria to suggest ventricular tachycardia.
She remained hypotensive, with regular wide-complex tachycardia on the ECG. She was given an additional 1-L bolus of normal saline and 100 mmol (2 mmol/kg) of sodium bicarbonate, and within 1 minute the wide-complex tachycardia resolved to narrow-complex sinus tachycardia (Figure 2). At this point, an infusion of 150 mmol/L of sodium bicarbonate in dextrose 5% in water was started, with serial ECGs to monitor the QRS duration and serial arterial blood gas monitoring to maintain the pH between 7.45 and 7.55.
TRANSFER TO THE ICU
She was then transferred to the intensive care unit (ICU), where she remained for 2 weeks. While in the ICU, she had a single recurrence of wide-complex tachycardia that resolved immediately with an infusion of 100 mmol of sodium bicarbonate. A urine toxicology screen was negative, and the serum amitriptyline measurement, returned from the laboratory 48 hours after her initial presentation, was 594 ng/mL (reference range 100–250 ng/mL). She was eventually weaned off the norepinephrine infusion after 20 hours, the sodium bicarbonate infusion was discontinued after 4 days, and she was taken off mechanical ventilation after 10 days. Also during her ICU stay, she had seizures on day 3 and developed aspiration pneumonia.
From the ICU, she was transferred to a regular floor, where she stayed for another week and then was transferred to a rehabilitation center. This patient was known to have clinical depression and to have attempted suicide once before. She had recently been under additional psychosocial stresses, which likely prompted this second attempt.
She reportedly had no neurologic or cardiovascular sequelae after her discharge from the hospital.
AMITRIPTYLINE OVERDOSE
Amitriptyline causes a relatively high number of fatal overdoses, at 34 per 1 million prescriptions.1 Death is usually from hypotension and ventricular arrhythmia caused by blockage of cardiac fast sodium channels leading to disturbances of cardiac conduction such as wide-complex tachycardia.
Other manifestations of amitriptyline overdose include seizures, sedation, and anticholinergic toxicity from variable blockade of gamma-aminobutyric acid receptors, histamine 1 receptors, and alpha receptors.2
Of the various changes on ECG described with amitriptyline overdose, sinus tachycardia is the most common. A QRS duration greater than 100 msec, right to extreme-right axis deviation with negative QRS complexes in leads I and aVL, and an R-wave amplitude greater than 3 mm in lead aVR are indications for sodium bicarbonate infusion, especially in hemodynamically unstable patients.3 Sodium bicarbonate increases the serum concentration of sodium and thereby overcomes the sodium channel blockade. It also alkalinizes the serum, favoring an electrically neutral form of amitriptyline that binds less to receptors and binds more to alpha-1-acid glycoprotein, decreasing the fraction of free drug available for toxicity.4
In patients with amitriptyline overdose, wide-complex tachycardia and hypotension refractory to sodium bicarbonate infusion can be treated with lidocaine, magnesium sulfate, direct-current cardioversion, and lipid resuscitation.5,6 Treatment with class IA, IC, and III antiarrhythmics is contraindicated, as they block sodium channels and thus can worsen conduction disturbances.
- Henry JA, Alexander CA, Sener EK. Relative mortality from overdose of antidepressants. BMJ 1995; 310:221–224.
- Shannon M, Merola J, Lovejoy FH Jr. Hypotension in severe tricyclic antidepressant overdose. Am J Emerg Med 1988; 6:439–442.
- Liebelt EL, Francis PD, Woolf AD. ECG lead aVR versus QRS interval in predicting seizures and arrhythmias in acute tricyclic antidepressant toxicity. Ann Emerg Med 1995; 26:195–201.
- Sayniuk BI, Jhamandas V. Mechanism of reversal of toxic effects of amitriptyline on cardiac Purkinje fibres by sodium bicarbonate. J Pharmacol Exp Ther 1984; 231:387.
- Kiberd MB, Minor SF. Lipid therapy for the treatment of a refractory amitriptyline overdose. CJEM 2012; 14:193–197.
- Harvey M, Cave G. Case report: successful lipid resuscitation in multidrug overdose with predominant tricyclic antidepressant toxidrome. Int J Emerg Med 2012; 5:8.
- Henry JA, Alexander CA, Sener EK. Relative mortality from overdose of antidepressants. BMJ 1995; 310:221–224.
- Shannon M, Merola J, Lovejoy FH Jr. Hypotension in severe tricyclic antidepressant overdose. Am J Emerg Med 1988; 6:439–442.
- Liebelt EL, Francis PD, Woolf AD. ECG lead aVR versus QRS interval in predicting seizures and arrhythmias in acute tricyclic antidepressant toxicity. Ann Emerg Med 1995; 26:195–201.
- Sayniuk BI, Jhamandas V. Mechanism of reversal of toxic effects of amitriptyline on cardiac Purkinje fibres by sodium bicarbonate. J Pharmacol Exp Ther 1984; 231:387.
- Kiberd MB, Minor SF. Lipid therapy for the treatment of a refractory amitriptyline overdose. CJEM 2012; 14:193–197.
- Harvey M, Cave G. Case report: successful lipid resuscitation in multidrug overdose with predominant tricyclic antidepressant toxidrome. Int J Emerg Med 2012; 5:8.
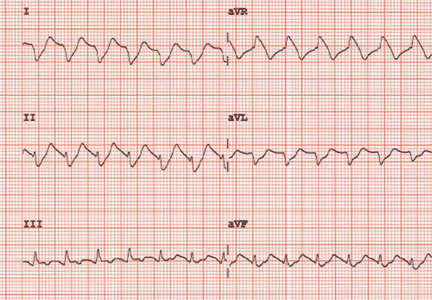
The cohabitation of art and genomic science
The art of medicine includes picking the right drug for the right patient, especially when we can choose between different classes of efficacious therapies. But, in view of our growing understanding of the human genome, can science replace art?
That question is part of the promise of pharmacogenetics, the study of how inter-individual genetic differences influence a patient’s response to a specific drug. A patient’s genome dictates the expression of specific enzymes that metabolize a drug with various efficiencies: variant alleles may result in slightly different proteins that express different enzymatic activity, ie, different substrate affinities for a drug resulting in more or less efficient metabolism. Genomic differences may also dictate whether a specific biochemical pathway is dominant in generating a specific pathophysiologic response, in which case drugs that affect that pathway may be strikingly effective. This may partly explain the various responses to different antihypertensive drugs.
Another less well-understood example of pharmacogenetics is the link between specific HLA haplotypes and a dramatic increase in allergic reactions to specific medications, such as the link between HLA-B*57:01 and abacavir hypersensitivity.
In this issue of the Journal, DiPiero et al discuss thiopurine methyltransferase (TPMT), an enzyme responsible for the degradation of azathioprine, and how knowing the genetically determined relative activity of this enzyme should influence our initial dosing of this and related drugs. Patients with certain variant alleles of TPMT degrade azathioprine more slowly, and these patients are at higher risk of myelosuppressive toxicity from the drug when it is given at the full weight-based dose. The TPMT test is expensive but not prohibitively so, and it would seem that genomic testing is a reasonable clinical and cost-effective option.
As in the abacavir scenario noted above, genomic-based dosing of azathioprine makes scientific sense and offers proof of principle for the validity of pharmacogenomics. But is it truly a clinical game-changer?
The answer depends in part on how the prescribing physician doses the drug, which depends in part on what disease is being treated, how fast the drug needs to be at full dose, and whether there are equally effective alternatives. Recommendations have been offered that state if TPMT activity is normal, we can start at the usual maintenance dose of 1.5 to 2 mg/kg/day (or occasionally more). But if the patient is heterozygous for the wild-type gene and thus is a slower drug metabolizer, then initial dosing “should” be reduced to 25 to 50 mg/day, with close observation of the white blood cell count as the dose is slowly increased to the target. The very rare patient who is homozygous for a non–wild-type allele should not be given the drug.
My usual practice has been to start patients on 50 mg or less daily and slowly titrate up, asking them how they are tolerating the drug and watching the white count—notably, the same approach to be taken if I had done genotyping before starting the drug and had found the patient to be heterozygous for the TPMT gene.
Interestingly, one pragmatic clinical trial tested whether genotyping patients before starting azathioprine—with subsequent suggested dosing of the drug based on the genotype as above—was safer and cheaper than letting physicians dose as they chose.1 It turned out that physicians participating in this study still dosed their patients conservatively. Even knowing that they might be able to give full doses from the start in patients with normal TPMT activity, many chose not to. I assume that many of those physicians felt as I do that there was no urgency in reaching the presumed-to-be-effective full weight-based therapeutic dose. (We don’t have a good clinical marker of azathioprine’s efficacy). At 4 months, the maintenance dose was about the same in all groups.
We have robust evidence to support the role of pharmacogenetics in informing the dosing of several medications, more than just the ones I have mentioned here. And in the right settings, we should use pharmacogenetic testing to limit toxicity and perhaps enhance efficacy in our drug selection. As the field moves rapidly forward, we will have many opportunities to improve clinical care by using our patients’ genomic information.
But like it or bemoan it, even when we have science in the house, the art of medicine still plays a role in our clinical decisions.
- Thompson AJ, Newman WG, Elliott RA, Roberts SA, Tricker K, Payne K. The cost-effectiveness of a pharmacogenetic test: a trial-based evaluation of TPMT genotyping for azathioprine. Value Health 2014; 17:22–33.
The art of medicine includes picking the right drug for the right patient, especially when we can choose between different classes of efficacious therapies. But, in view of our growing understanding of the human genome, can science replace art?
That question is part of the promise of pharmacogenetics, the study of how inter-individual genetic differences influence a patient’s response to a specific drug. A patient’s genome dictates the expression of specific enzymes that metabolize a drug with various efficiencies: variant alleles may result in slightly different proteins that express different enzymatic activity, ie, different substrate affinities for a drug resulting in more or less efficient metabolism. Genomic differences may also dictate whether a specific biochemical pathway is dominant in generating a specific pathophysiologic response, in which case drugs that affect that pathway may be strikingly effective. This may partly explain the various responses to different antihypertensive drugs.
Another less well-understood example of pharmacogenetics is the link between specific HLA haplotypes and a dramatic increase in allergic reactions to specific medications, such as the link between HLA-B*57:01 and abacavir hypersensitivity.
In this issue of the Journal, DiPiero et al discuss thiopurine methyltransferase (TPMT), an enzyme responsible for the degradation of azathioprine, and how knowing the genetically determined relative activity of this enzyme should influence our initial dosing of this and related drugs. Patients with certain variant alleles of TPMT degrade azathioprine more slowly, and these patients are at higher risk of myelosuppressive toxicity from the drug when it is given at the full weight-based dose. The TPMT test is expensive but not prohibitively so, and it would seem that genomic testing is a reasonable clinical and cost-effective option.
As in the abacavir scenario noted above, genomic-based dosing of azathioprine makes scientific sense and offers proof of principle for the validity of pharmacogenomics. But is it truly a clinical game-changer?
The answer depends in part on how the prescribing physician doses the drug, which depends in part on what disease is being treated, how fast the drug needs to be at full dose, and whether there are equally effective alternatives. Recommendations have been offered that state if TPMT activity is normal, we can start at the usual maintenance dose of 1.5 to 2 mg/kg/day (or occasionally more). But if the patient is heterozygous for the wild-type gene and thus is a slower drug metabolizer, then initial dosing “should” be reduced to 25 to 50 mg/day, with close observation of the white blood cell count as the dose is slowly increased to the target. The very rare patient who is homozygous for a non–wild-type allele should not be given the drug.
My usual practice has been to start patients on 50 mg or less daily and slowly titrate up, asking them how they are tolerating the drug and watching the white count—notably, the same approach to be taken if I had done genotyping before starting the drug and had found the patient to be heterozygous for the TPMT gene.
Interestingly, one pragmatic clinical trial tested whether genotyping patients before starting azathioprine—with subsequent suggested dosing of the drug based on the genotype as above—was safer and cheaper than letting physicians dose as they chose.1 It turned out that physicians participating in this study still dosed their patients conservatively. Even knowing that they might be able to give full doses from the start in patients with normal TPMT activity, many chose not to. I assume that many of those physicians felt as I do that there was no urgency in reaching the presumed-to-be-effective full weight-based therapeutic dose. (We don’t have a good clinical marker of azathioprine’s efficacy). At 4 months, the maintenance dose was about the same in all groups.
We have robust evidence to support the role of pharmacogenetics in informing the dosing of several medications, more than just the ones I have mentioned here. And in the right settings, we should use pharmacogenetic testing to limit toxicity and perhaps enhance efficacy in our drug selection. As the field moves rapidly forward, we will have many opportunities to improve clinical care by using our patients’ genomic information.
But like it or bemoan it, even when we have science in the house, the art of medicine still plays a role in our clinical decisions.
The art of medicine includes picking the right drug for the right patient, especially when we can choose between different classes of efficacious therapies. But, in view of our growing understanding of the human genome, can science replace art?
That question is part of the promise of pharmacogenetics, the study of how inter-individual genetic differences influence a patient’s response to a specific drug. A patient’s genome dictates the expression of specific enzymes that metabolize a drug with various efficiencies: variant alleles may result in slightly different proteins that express different enzymatic activity, ie, different substrate affinities for a drug resulting in more or less efficient metabolism. Genomic differences may also dictate whether a specific biochemical pathway is dominant in generating a specific pathophysiologic response, in which case drugs that affect that pathway may be strikingly effective. This may partly explain the various responses to different antihypertensive drugs.
Another less well-understood example of pharmacogenetics is the link between specific HLA haplotypes and a dramatic increase in allergic reactions to specific medications, such as the link between HLA-B*57:01 and abacavir hypersensitivity.
In this issue of the Journal, DiPiero et al discuss thiopurine methyltransferase (TPMT), an enzyme responsible for the degradation of azathioprine, and how knowing the genetically determined relative activity of this enzyme should influence our initial dosing of this and related drugs. Patients with certain variant alleles of TPMT degrade azathioprine more slowly, and these patients are at higher risk of myelosuppressive toxicity from the drug when it is given at the full weight-based dose. The TPMT test is expensive but not prohibitively so, and it would seem that genomic testing is a reasonable clinical and cost-effective option.
As in the abacavir scenario noted above, genomic-based dosing of azathioprine makes scientific sense and offers proof of principle for the validity of pharmacogenomics. But is it truly a clinical game-changer?
The answer depends in part on how the prescribing physician doses the drug, which depends in part on what disease is being treated, how fast the drug needs to be at full dose, and whether there are equally effective alternatives. Recommendations have been offered that state if TPMT activity is normal, we can start at the usual maintenance dose of 1.5 to 2 mg/kg/day (or occasionally more). But if the patient is heterozygous for the wild-type gene and thus is a slower drug metabolizer, then initial dosing “should” be reduced to 25 to 50 mg/day, with close observation of the white blood cell count as the dose is slowly increased to the target. The very rare patient who is homozygous for a non–wild-type allele should not be given the drug.
My usual practice has been to start patients on 50 mg or less daily and slowly titrate up, asking them how they are tolerating the drug and watching the white count—notably, the same approach to be taken if I had done genotyping before starting the drug and had found the patient to be heterozygous for the TPMT gene.
Interestingly, one pragmatic clinical trial tested whether genotyping patients before starting azathioprine—with subsequent suggested dosing of the drug based on the genotype as above—was safer and cheaper than letting physicians dose as they chose.1 It turned out that physicians participating in this study still dosed their patients conservatively. Even knowing that they might be able to give full doses from the start in patients with normal TPMT activity, many chose not to. I assume that many of those physicians felt as I do that there was no urgency in reaching the presumed-to-be-effective full weight-based therapeutic dose. (We don’t have a good clinical marker of azathioprine’s efficacy). At 4 months, the maintenance dose was about the same in all groups.
We have robust evidence to support the role of pharmacogenetics in informing the dosing of several medications, more than just the ones I have mentioned here. And in the right settings, we should use pharmacogenetic testing to limit toxicity and perhaps enhance efficacy in our drug selection. As the field moves rapidly forward, we will have many opportunities to improve clinical care by using our patients’ genomic information.
But like it or bemoan it, even when we have science in the house, the art of medicine still plays a role in our clinical decisions.
- Thompson AJ, Newman WG, Elliott RA, Roberts SA, Tricker K, Payne K. The cost-effectiveness of a pharmacogenetic test: a trial-based evaluation of TPMT genotyping for azathioprine. Value Health 2014; 17:22–33.
- Thompson AJ, Newman WG, Elliott RA, Roberts SA, Tricker K, Payne K. The cost-effectiveness of a pharmacogenetic test: a trial-based evaluation of TPMT genotyping for azathioprine. Value Health 2014; 17:22–33.
Should thiopurine methyltransferase (TPMT) activity be determined before prescribing azathioprine, mercaptopurine, or thioguanine?
The thiopurines azathioprine, mercaptopurine, and thioguanine are prodrugs that are converted to active thioguanine nucleotide metabolites or methylated by thiopurine methyltransferase (TPMT) to compounds with less pharmacologic activity. In the absence of TPMT activity, patients are likely to have higher concentrations of thioguanine nucleotides, which can pose an increased risk of severe life-threatening myelosuppression. Determining TPMT activity, either directly by phenotyping or indirectly by determining the specific genetic allele (different alleles have different enzymatic activity), can help identify patients at greater risk of severe myelosuppression. Therefore, we recommend that TPMT testing be strongly considered before initiating therapy with a thiopurine.
THIOPURINES AND TPMT
Azathioprine, mercaptopurine, and thioguanine are used for treating autoimmune and inflammatory diseases1–3 and certain types of cancer such as leukemias and lymphomas.1,4–6 Typically, azathioprine is used to treat nonmalignant conditions, thioguanine is used to treat malignancies, and mercaptopurine can be used to treat both malignant and nonmalignant conditions.
Although the exact mechanism of action of these drugs has not been completely elucidated, the active thioguanine nucleotide metabolites are thought to be incorporated into the DNA of leukocytes, resulting in DNA damage that subsequently leads to cell death and myelosuppression.7–9
Variants of the TPMT gene may alter the activity of the TPMT enzyme, resulting in individual variability in thiopurine metabolism. Compared with people with normal (high) TPMT activity, those with intermediate or low TPMT activity metabolize the drugs more slowly, and are likely to have higher thioguanine nucleotide concentrations and therefore an increased risk of myelosuppression.
One of the earliest correlations between TPMT activity and thiopurine-induced myelosuppression was described in a pediatric patient with acute lymphocytic leukemia.10 After being prescribed a conventional mercaptopurine dosage (75 mg/m2 daily), the patient developed severe myelosuppression and was observed to have a thioguanine nucleotide metabolite concentration seven times the observed population median. TPMT phenotyping demonstrated that the patient had low TPMT activity. Reducing the mercaptopurine dose by approximately 90% resulted in normalization of thioguanine nucleotide metabolite concentrations, and the myelosuppression subsequently resolved.
Approximately 10% of the population has intermediate TPMT activity and 0.3% has low or absent TPMT activity, though these percentages vary depending on ancestry.1 Research has demonstrated that approximately 30% to 60% of those with intermediate TPMT activity cannot tolerate a full thiopurine dose (eg, azathioprine 2–3 mg/kg/day or mercaptopurine 1.5 mg/kg/day).1 Almost all patients with low TPMT activity will develop life-threating myelosuppression if prescribed a full thiopurine dose.1
SHOULD TPMT ACTIVITY BE DETERMINED FOR EVERY PATIENT PRESCRIBED A THIOPURINE?
Although determining TPMT activity in thiopurine-naïve patients will assist clinicians in selecting a thiopurine starting dose or in deciding if an alternative agent is warranted, there are instances when a clinician may elect to not perform a TPMT genotype or phenotype test. For example, determining TPMT activity is not recommended for patients who previously tolerated thiopurine therapy at full steady-state doses.
The required starting dose of a thiopurine can influence the decision on whether or not to test for TPMT activity. TPMT genotyping or phenotyping may be of most benefit for patients requiring immediate full doses of a thiopurine.11 Ideally, TPMT activity should be determined before prescribing immediate full doses of a thiopurine. This could be achieved by preemptively ordering a TPMT test in patients likely to require immunosuppression—for example, in patients diagnosed with inflammatory or autoimmune diseases. If therapy cannot be delayed and TPMT activity is unknown, ordering a TPMT test at the time of prescribing a full thiopurine dose is still of benefit. Depending on the clinical laboratory utilized for testing, TPMT phenotype results are usually reported in 3 to 5 days, and TPMT genotype results are usually reported in 5 to 7 days. Because most patients will not reach steady-state concentrations for 2 to 6 weeks, clinicians could initiate immediate full doses of a thiopurine and modify therapy based on TPMT test results before accumulation of thioguanine nucleotide metabolites occurs. Caution should be used with this approach, particularly in situations where the clinical laboratory may not return results in a timely manner.
For patients who are candidates for an initial low dose of a thiopurine, clinicians may choose to slowly titrate doses based on response and tolerability instead of determining TPMT activity.11 Depending on the starting dose and how slowly titration occurs, initiating a thiopurine at a low dose and titrating based on response can be a feasible approach for patients with intermediate TPMT activity. Because drastic thiopurine dose reductions of approximately 10-fold are required for patients with low TPMT activity, which is a much smaller dosage than most clinicians will initially prescribe, the starting dosage will likely not be low enough to prevent myelosuppression in patients with low TPMT activity.1,10
Determining TPMT activity can help clinicians establish an appropriate titration schedule. Patients with normal TPMT activity will usually reach thiopurine steady-state concentrations in 2 weeks, and the dosage can be titrated based on response.1 Alterations in TPMT activity influence the pharmacokinetic parameters of thiopurines, and the time to reach steady-state is extended to 4 or 6 weeks for those with intermediate or low TPMT activity.1 Increasing the thiopurine dosage before reaching steady state can lead to the prescribing of doses that will not be tolerated, resulting in myelosuppression.
Factors to consider when deciding if TPMT activity should be assessed include the disease state being treated and corresponding starting dose, the need for immediate full doses, and previous documented tolerance of thiopurines at steady-state doses. As with many aspects of medicine that have multiple options, coupled with an increase in patient access to healthcare information, the decision to test for TPMT activity may include shared decision-making between patients and providers. Although TPMT genotyping or phenotyping can help identify those at greatest risk of severe myelosuppression, such assays do not replace routine monitoring for myelosuppression, hepatotoxicity, or pancreatitis that may be caused by thiopurines.
WHAT TESTS ARE AVAILABLE TO DETERMINE TPMT ACTIVITY?
Patients with intermediate or low TPMT activity can be identified by either genotyping or phenotyping. There are considerations, though, that clinicians should be aware of before selecting a particular test.
TPMT genotyping
![]()
Four TPMT alleles, TPMT*2, *3A, *3B, and *3C, account for over 90% of inactivating polymorphisms.12 Therefore, most reference laboratories only analyze for those genetic variants. Based on the reported test result, a predicted phenotype (eg, normal, intermediate, or low TPMT activity) can be assigned. Table 1 lists the predicted phenotypes for select genotyping results.
TPMT phenotyping
Phenotyping quantitates TPMT enzyme activity in erythrocytes, and based on the result, patients are classified as having normal, intermediate, or low TPMT activity. Because internal standards and other testing conditions may differ between reference laboratories, test results must be interpreted in the context of the laboratory that performed the assay.
Which test is right for my patient?
In most cases, either the genotype or the phenotype test provides sufficient information to guide thiopurine therapy. There are certain circumstances, though, in which the genotype or phenotype test is less informative.
TPMT genotyping, when performed using a blood specimen, is not recommended in those with a history of allogeneic bone marrow transplantation, as the result would reflect the donor’s genotype, not the patient’s. In such instances, monitoring of white blood cell counts and thiopurine metabolites may be more beneficial.
TPMT phenotyping may be inaccurate if performed within 30 to 90 days of an erythrocyte transfusion, as the test result may be influenced by donor erythrocytes. If a patient is receiving erythrocyte transfusions, TPMT genotyping is preferable to phenotyping.
Test cost may also be a consideration when determining if the genotype or phenotype test is best for your patient. Costs vary by laboratory, but phenotyping is generally less expensive than genotyping. The cost of genotyping, though, continues to decrease.13 The approximate commercial cost is $200 for phenotyping and $450 for genotyping, but laboratory fees may be substantially higher. Several insurance plans, including Medicare, cover TPMT testing, but reimbursement and copayments vary, depending on the patient’s specific plan.
There are conflicting data as to whether determining TPMT status is11,14–18 or is not19 cost-effective. Multiple studies suggest that the cost of genotyping a sufficient number of patients to identify a single individual at high risk of myelosuppression is cheaper than the costs associated with treating an adverse event. Additional cost-benefit studies are needed, particularly studies that consider how bundled payments and outcomes-based reimbursement influence cost-effectiveness.
MODIFYING THIOPURINE THERAPY BASED ON TPMT ACTIVITY
![]()
There is a strong correlation between TPMT activity and tolerated thiopurine doses, with those having intermediate or low TPMT activity requiring lower doses.10,20–23 Adjusting mercaptopurine doses based on TPMT activity to prevent hematopoietic toxicity has been successfully demonstrated in pediatric patients with acute lymphoblastic leukemia.24 Furthermore, reducing initial thiopurine doses to avoid myelosuppression and titrating based on response has been shown to not compromise outcomes.1,25,26 The Clinical Pharmacogenetic Implementation Consortium (CPIC) has developed an evidence-based guideline on how to adjust thiopurine doses based on TPMT activity,1 summarized in Table 2. These dosing recommendations are classified as “strong.”
Patients with normal TPMT activity should be prescribed the usual thiopurine starting dose as indicated by disease-specific guidelines.
For those with intermediate TPMT activity, the CPIC guideline recommends reducing the initial targeted full dose of azathioprine and mercaptopurine by 30% to 70% and reducing the targeted full dose of thioguanine by 30% to 50%. The percentage of dose reduction depends on the targeted full dose. Siegel and Sands27 suggested that for those who are diagnosed with inflammatory bowel disease and have intermediate TPMT activity, azathioprine should be initiated at a low dose and titrated to 1.25 mg/kg and mercaptopurine should be initiated at a low dose and titrated to 0.75 mg/kg. Based on these titration goals, if the targeted full dose for mercaptopurine is 1 mg/kg, then a dose reduction of approximately 30% would be more appropriate. If the targeted full dose is 1.5 mg/kg, a dose reduction of approximately 50% would be more appropriate. Thiopurine doses should be titrated based on response and disease-specific guidelines, allowing 2 to 4 weeks to reach steady state before dose titration.
For those with low TPMT activity, alternative therapy should be considered for nonmalignant conditions because of the risk of severe myelosuppression. For malignancy, or if a thiopurine is warranted for a nonmalignant condition, consider a 90% dose reduction and give the drug three times per week instead of daily. For example, acute lymphoblastic leukemia patients with low TPMT activity can be started on mercaptopurine 10 mg/m2 three times per week instead of the usual starting dose.10 Thiopurine doses should be titrated based on response and disease-specific guidelines, allowing 4 to 6 weeks to reach steady state before dose titration.
RECOMMENDATIONS
Individuals with intermediate or low TPMT activity have an increased risk of myelosuppression. Because of the elevated risk for morbidity and death, especially for patients with low TPMT activity, multiple guidelines and regulatory agencies recommend TPMT genotyping or phenotyping if a thiopurine is prescribed.25,28–32 Although additional cost-benefit analysis studies are needed, evidence suggests testing for TPMT activity may be cheaper than the costs associated with treating myelosuppression.
In view of treatment guidelines, the recommendations of regulatory agencies, cost-benefit analyses, and the availability of gene-based dosing recommendations, we consider the benefits of testing for TPMT activity to greatly outweigh any associated risks. Therefore, we recommend that TPMT testing be strongly considered before initiating therapy with a thiopurine.
- Relling MV, Gardner EE, Sandborn WJ, et al; Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing. Clin Pharmacol Ther 2011; 89:387–391.
- Ansari A, Arenas M, Greenfield SM, et al. Prospective evaluation of the pharmacogenetics of azathioprine in the treatment of inflammatory bowel disease. Aliment Pharmacol Ther 2008; 28:973–983.
- Beswick L, Friedman AB, Sparrow MP. The role of thiopurine metabolite monitoring in inflammatory bowel disease. Expert Rev Gastroenterol Hepatol 2014; 8:383–392.
- Gervasini G, Vagace JM. Impact of genetic polymorphisms on chemotherapy toxicity in childhood acute lymphoblastic leukemia. Front Genet 2012; 3:249.
- Levinsen M, Rotevatn EØ, Rosthøj S, et al; Nordic Society of Paediatric Haematology, Oncology. Pharmacogenetically based dosing of thiopurines in childhood acute lymphoblastic leukemia: influence on cure rates and risk of second cancer. Pediatr Blood Cancer 2014; 61:797–802.
- Adam de Beaumais T, Jacqz-Aigrain E. Pharmacogenetic determinants of mercaptopurine disposition in children with acute lymphoblastic leukemia. Eur J Clin Pharmacol 2012; 68:1233–1242.
- Derijks LJ, Gilissen LP, Hooymans PM, Hommes DW. Review article: thiopurines in inflammatory bowel disease. Aliment Pharmacol Ther 2006; 24:715–729.
- Fairchild CR, Maybaum J, Kennedy KA. Concurrent unilateral chromatid damage and DNA strand breakage in response to 6-thioguanine treatment. Biochem Pharmacol 1986; 35:3533–3541.
- Karran P. Thiopurines, DNA damage, DNA repair and therapy-related cancer. Br Med Bull 2006; 79–80:153–170.
- Evans WE, Horner M, Chu YQ, Kalwinsky D, Roberts WM. Altered mercaptopurine metabolism, toxic effects, and dosage requirement in a thiopurine methyltransferase-deficient child with acute lymphocytic leukemia. J Pediatr 1991; 119:985–989.
- Gardiner SJ, Gearry RB, Barclay ML, Begg EJ. Two cases of thiopurine methyltransferase (TPMT) deficiency—a lucky save and a near miss with azathioprine. Br J Clin Pharmacol 2006; 62:473–476.
- Relling MV, Gardner EE, Sandborn WJ, et al. Clinical pharmacogenetics implementation consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing: 2013 update. Clin Pharmacol Ther 2013; 93:324–325.
- Altman RB. Pharmacogenomics: “noninferiority” is sufficient for initial implementation. Clin Pharmacol Ther 2011; 89:348–350.
- van den Akker-van Marle ME, Gurwitz D, Detmar SB, et al. Cost-effectiveness of pharmacogenomics in clinical practice: a case study of thiopurine methyltransferase genotyping in acute lymphoblastic leukemia in Europe. Pharmacogenomics 2006; 7:783–792.
- Clunie GP, Lennard L. Relevance of thiopurine methyltransferase status in rheumatology patients receiving azathioprine. Rheumatology (Oxford) 2004; 43:13–18.
- Dubinsky MC, Reyes E, Ofman J, Chiou CF, Wade S, Sandborn WJ. A cost-effectiveness analysis of alternative disease management strategies in patients with Crohn’s disease treated with azathioprine or 6-mercaptopurine. Am J Gastroenterol 2005; 100:2239–2247.
- Winter J, Walker A, Shapiro D, Gaffney D, Spooner RJ, Mills PR. Cost-effectiveness of thiopurine methyltransferase genotype screening in patients about to commence azathioprine therapy for treatment of inflammatory bowel disease. Aliment Pharmacol Ther 2004; 20:593–599.
- Marra CA, Esdaile JM, Anis AH. Practical pharmacogenetics: the cost effectiveness of screening for thiopurine s-methyltransferase polymorphisms in patients with rheumatological conditions treated with azathioprine. J Rheumatol 2002; 29:2507–2512.
- Donnan JR, Ungar WJ, Mathews M, Hancock-Howard RL, Rahman P. A cost effectiveness analysis of thiopurine methyltransferase testing for guiding 6-mercaptopurine dosing in children with acute lymphoblastic leukemia. Pediatr Blood Cancer 2011; 57:231–239.
- Lennard L, Gibson BE, Nicole T, Lilleyman JS. Congenital thiopurine methyltransferase deficiency and 6-mercaptopurine toxicity during treatment for acute lymphoblastic leukaemia. Arch Dis Child 1993; 69:577–579.
- Hindorf U, Lindqvist M, Hildebrand H, Fagerberg U, Almer S. Adverse events leading to modification of therapy in a large cohort of patients with inflammatory bowel disease. Aliment Pharmacol Ther 2006; 24:331–342.
- Relling MV, Hancock ML, Rivera GK, et al. Mercaptopurine therapy intolerance and heterozygosity at the thiopurine S-methyltransferase gene locus. J Natl Cancer Inst 1999; 91:2001–2008.
- Relling MV, Hancock ML, Boyett JM, Pui CH, Evans WE. Prognostic importance of 6-mercaptopurine dose intensity in acute lymphoblastic leukemia. Blood 1999; 93:2817–2823.
- Pui CH, Pei D, Sandlund JT, et al. Long-term results of St Jude Total Therapy Studies 11, 12, 13A, 13B, and 14 for childhood acute lymphoblastic leukemia. Leukemia 2010; 24:371–382.
- Ford LT, Berg JD. Thiopurine S-methyltransferase (TPMT) assessment prior to starting thiopurine drug treatment; a pharmacogenomic test whose time has come. J Clin Pathol 2010; 63:288–295.
- Schmiegelow K, Forestier E, Hellebostad M, et al; Nordic Society of Paediatric Haematology and Oncology. Long-term results of NOPHO ALL-92 and ALL-2000 studies of childhood acute lymphoblastic leukemia. Leukemia 2010; 24:345–354.
- Siegel CA, Sands BE. Review article: practical management of inflammatory bowel disease patients taking immunomodulators. Aliment Pharmacol Ther 2005; 22:1–16.
- Mayberry JF, Lobo A, Ford AC, Thomas A. NICE clinical guideline (CG152): the management of Crohn’s disease in adults, children and young people. Aliment Pharmacol Ther 2013; 37:195–203
- Mowat C, Cole A, Windsor A, et al; IBD Section of the British Society of Gastroenterology. Guidelines for the management of inflammatory bowel disease in adults. Gut 2011; 60:571–607.
- Turner D, Levine A, Escher JC, et al; European Crohn’s and Colitis Organization; European Society for Paediatric Gastroenterology, Hepatology, and Nutrition. Management of pediatric ulcerative colitis: joint ECCO and ESPGHAN evidence-based consensus guidelines. J Pediatr Gastroenterol Nutr 2012; 55:340–361.
- Bernstein CN, Fried M, Krabshuis JH, et al. World Gastroenterology Organization Practice Guidelines for the diagnosis and management of IBD in 2010. Inflamm Bowel Dis 2010; 16:112–124.
- Becquemont L, Alfirevic A, Amstutz U, et al. Practical recommendations for pharmacogenomics-based prescription: 2010 ESF-UB Conference on Pharmacogenetics and Pharmacogenomics. Pharmacogenomics 2011; 12:113–124.
The thiopurines azathioprine, mercaptopurine, and thioguanine are prodrugs that are converted to active thioguanine nucleotide metabolites or methylated by thiopurine methyltransferase (TPMT) to compounds with less pharmacologic activity. In the absence of TPMT activity, patients are likely to have higher concentrations of thioguanine nucleotides, which can pose an increased risk of severe life-threatening myelosuppression. Determining TPMT activity, either directly by phenotyping or indirectly by determining the specific genetic allele (different alleles have different enzymatic activity), can help identify patients at greater risk of severe myelosuppression. Therefore, we recommend that TPMT testing be strongly considered before initiating therapy with a thiopurine.
THIOPURINES AND TPMT
Azathioprine, mercaptopurine, and thioguanine are used for treating autoimmune and inflammatory diseases1–3 and certain types of cancer such as leukemias and lymphomas.1,4–6 Typically, azathioprine is used to treat nonmalignant conditions, thioguanine is used to treat malignancies, and mercaptopurine can be used to treat both malignant and nonmalignant conditions.
Although the exact mechanism of action of these drugs has not been completely elucidated, the active thioguanine nucleotide metabolites are thought to be incorporated into the DNA of leukocytes, resulting in DNA damage that subsequently leads to cell death and myelosuppression.7–9
Variants of the TPMT gene may alter the activity of the TPMT enzyme, resulting in individual variability in thiopurine metabolism. Compared with people with normal (high) TPMT activity, those with intermediate or low TPMT activity metabolize the drugs more slowly, and are likely to have higher thioguanine nucleotide concentrations and therefore an increased risk of myelosuppression.
One of the earliest correlations between TPMT activity and thiopurine-induced myelosuppression was described in a pediatric patient with acute lymphocytic leukemia.10 After being prescribed a conventional mercaptopurine dosage (75 mg/m2 daily), the patient developed severe myelosuppression and was observed to have a thioguanine nucleotide metabolite concentration seven times the observed population median. TPMT phenotyping demonstrated that the patient had low TPMT activity. Reducing the mercaptopurine dose by approximately 90% resulted in normalization of thioguanine nucleotide metabolite concentrations, and the myelosuppression subsequently resolved.
Approximately 10% of the population has intermediate TPMT activity and 0.3% has low or absent TPMT activity, though these percentages vary depending on ancestry.1 Research has demonstrated that approximately 30% to 60% of those with intermediate TPMT activity cannot tolerate a full thiopurine dose (eg, azathioprine 2–3 mg/kg/day or mercaptopurine 1.5 mg/kg/day).1 Almost all patients with low TPMT activity will develop life-threating myelosuppression if prescribed a full thiopurine dose.1
SHOULD TPMT ACTIVITY BE DETERMINED FOR EVERY PATIENT PRESCRIBED A THIOPURINE?
Although determining TPMT activity in thiopurine-naïve patients will assist clinicians in selecting a thiopurine starting dose or in deciding if an alternative agent is warranted, there are instances when a clinician may elect to not perform a TPMT genotype or phenotype test. For example, determining TPMT activity is not recommended for patients who previously tolerated thiopurine therapy at full steady-state doses.
The required starting dose of a thiopurine can influence the decision on whether or not to test for TPMT activity. TPMT genotyping or phenotyping may be of most benefit for patients requiring immediate full doses of a thiopurine.11 Ideally, TPMT activity should be determined before prescribing immediate full doses of a thiopurine. This could be achieved by preemptively ordering a TPMT test in patients likely to require immunosuppression—for example, in patients diagnosed with inflammatory or autoimmune diseases. If therapy cannot be delayed and TPMT activity is unknown, ordering a TPMT test at the time of prescribing a full thiopurine dose is still of benefit. Depending on the clinical laboratory utilized for testing, TPMT phenotype results are usually reported in 3 to 5 days, and TPMT genotype results are usually reported in 5 to 7 days. Because most patients will not reach steady-state concentrations for 2 to 6 weeks, clinicians could initiate immediate full doses of a thiopurine and modify therapy based on TPMT test results before accumulation of thioguanine nucleotide metabolites occurs. Caution should be used with this approach, particularly in situations where the clinical laboratory may not return results in a timely manner.
For patients who are candidates for an initial low dose of a thiopurine, clinicians may choose to slowly titrate doses based on response and tolerability instead of determining TPMT activity.11 Depending on the starting dose and how slowly titration occurs, initiating a thiopurine at a low dose and titrating based on response can be a feasible approach for patients with intermediate TPMT activity. Because drastic thiopurine dose reductions of approximately 10-fold are required for patients with low TPMT activity, which is a much smaller dosage than most clinicians will initially prescribe, the starting dosage will likely not be low enough to prevent myelosuppression in patients with low TPMT activity.1,10
Determining TPMT activity can help clinicians establish an appropriate titration schedule. Patients with normal TPMT activity will usually reach thiopurine steady-state concentrations in 2 weeks, and the dosage can be titrated based on response.1 Alterations in TPMT activity influence the pharmacokinetic parameters of thiopurines, and the time to reach steady-state is extended to 4 or 6 weeks for those with intermediate or low TPMT activity.1 Increasing the thiopurine dosage before reaching steady state can lead to the prescribing of doses that will not be tolerated, resulting in myelosuppression.
Factors to consider when deciding if TPMT activity should be assessed include the disease state being treated and corresponding starting dose, the need for immediate full doses, and previous documented tolerance of thiopurines at steady-state doses. As with many aspects of medicine that have multiple options, coupled with an increase in patient access to healthcare information, the decision to test for TPMT activity may include shared decision-making between patients and providers. Although TPMT genotyping or phenotyping can help identify those at greatest risk of severe myelosuppression, such assays do not replace routine monitoring for myelosuppression, hepatotoxicity, or pancreatitis that may be caused by thiopurines.
WHAT TESTS ARE AVAILABLE TO DETERMINE TPMT ACTIVITY?
Patients with intermediate or low TPMT activity can be identified by either genotyping or phenotyping. There are considerations, though, that clinicians should be aware of before selecting a particular test.
TPMT genotyping
![]()
Four TPMT alleles, TPMT*2, *3A, *3B, and *3C, account for over 90% of inactivating polymorphisms.12 Therefore, most reference laboratories only analyze for those genetic variants. Based on the reported test result, a predicted phenotype (eg, normal, intermediate, or low TPMT activity) can be assigned. Table 1 lists the predicted phenotypes for select genotyping results.
TPMT phenotyping
Phenotyping quantitates TPMT enzyme activity in erythrocytes, and based on the result, patients are classified as having normal, intermediate, or low TPMT activity. Because internal standards and other testing conditions may differ between reference laboratories, test results must be interpreted in the context of the laboratory that performed the assay.
Which test is right for my patient?
In most cases, either the genotype or the phenotype test provides sufficient information to guide thiopurine therapy. There are certain circumstances, though, in which the genotype or phenotype test is less informative.
TPMT genotyping, when performed using a blood specimen, is not recommended in those with a history of allogeneic bone marrow transplantation, as the result would reflect the donor’s genotype, not the patient’s. In such instances, monitoring of white blood cell counts and thiopurine metabolites may be more beneficial.
TPMT phenotyping may be inaccurate if performed within 30 to 90 days of an erythrocyte transfusion, as the test result may be influenced by donor erythrocytes. If a patient is receiving erythrocyte transfusions, TPMT genotyping is preferable to phenotyping.
Test cost may also be a consideration when determining if the genotype or phenotype test is best for your patient. Costs vary by laboratory, but phenotyping is generally less expensive than genotyping. The cost of genotyping, though, continues to decrease.13 The approximate commercial cost is $200 for phenotyping and $450 for genotyping, but laboratory fees may be substantially higher. Several insurance plans, including Medicare, cover TPMT testing, but reimbursement and copayments vary, depending on the patient’s specific plan.
There are conflicting data as to whether determining TPMT status is11,14–18 or is not19 cost-effective. Multiple studies suggest that the cost of genotyping a sufficient number of patients to identify a single individual at high risk of myelosuppression is cheaper than the costs associated with treating an adverse event. Additional cost-benefit studies are needed, particularly studies that consider how bundled payments and outcomes-based reimbursement influence cost-effectiveness.
MODIFYING THIOPURINE THERAPY BASED ON TPMT ACTIVITY
![]()
There is a strong correlation between TPMT activity and tolerated thiopurine doses, with those having intermediate or low TPMT activity requiring lower doses.10,20–23 Adjusting mercaptopurine doses based on TPMT activity to prevent hematopoietic toxicity has been successfully demonstrated in pediatric patients with acute lymphoblastic leukemia.24 Furthermore, reducing initial thiopurine doses to avoid myelosuppression and titrating based on response has been shown to not compromise outcomes.1,25,26 The Clinical Pharmacogenetic Implementation Consortium (CPIC) has developed an evidence-based guideline on how to adjust thiopurine doses based on TPMT activity,1 summarized in Table 2. These dosing recommendations are classified as “strong.”
Patients with normal TPMT activity should be prescribed the usual thiopurine starting dose as indicated by disease-specific guidelines.
For those with intermediate TPMT activity, the CPIC guideline recommends reducing the initial targeted full dose of azathioprine and mercaptopurine by 30% to 70% and reducing the targeted full dose of thioguanine by 30% to 50%. The percentage of dose reduction depends on the targeted full dose. Siegel and Sands27 suggested that for those who are diagnosed with inflammatory bowel disease and have intermediate TPMT activity, azathioprine should be initiated at a low dose and titrated to 1.25 mg/kg and mercaptopurine should be initiated at a low dose and titrated to 0.75 mg/kg. Based on these titration goals, if the targeted full dose for mercaptopurine is 1 mg/kg, then a dose reduction of approximately 30% would be more appropriate. If the targeted full dose is 1.5 mg/kg, a dose reduction of approximately 50% would be more appropriate. Thiopurine doses should be titrated based on response and disease-specific guidelines, allowing 2 to 4 weeks to reach steady state before dose titration.
For those with low TPMT activity, alternative therapy should be considered for nonmalignant conditions because of the risk of severe myelosuppression. For malignancy, or if a thiopurine is warranted for a nonmalignant condition, consider a 90% dose reduction and give the drug three times per week instead of daily. For example, acute lymphoblastic leukemia patients with low TPMT activity can be started on mercaptopurine 10 mg/m2 three times per week instead of the usual starting dose.10 Thiopurine doses should be titrated based on response and disease-specific guidelines, allowing 4 to 6 weeks to reach steady state before dose titration.
RECOMMENDATIONS
Individuals with intermediate or low TPMT activity have an increased risk of myelosuppression. Because of the elevated risk for morbidity and death, especially for patients with low TPMT activity, multiple guidelines and regulatory agencies recommend TPMT genotyping or phenotyping if a thiopurine is prescribed.25,28–32 Although additional cost-benefit analysis studies are needed, evidence suggests testing for TPMT activity may be cheaper than the costs associated with treating myelosuppression.
In view of treatment guidelines, the recommendations of regulatory agencies, cost-benefit analyses, and the availability of gene-based dosing recommendations, we consider the benefits of testing for TPMT activity to greatly outweigh any associated risks. Therefore, we recommend that TPMT testing be strongly considered before initiating therapy with a thiopurine.
The thiopurines azathioprine, mercaptopurine, and thioguanine are prodrugs that are converted to active thioguanine nucleotide metabolites or methylated by thiopurine methyltransferase (TPMT) to compounds with less pharmacologic activity. In the absence of TPMT activity, patients are likely to have higher concentrations of thioguanine nucleotides, which can pose an increased risk of severe life-threatening myelosuppression. Determining TPMT activity, either directly by phenotyping or indirectly by determining the specific genetic allele (different alleles have different enzymatic activity), can help identify patients at greater risk of severe myelosuppression. Therefore, we recommend that TPMT testing be strongly considered before initiating therapy with a thiopurine.
THIOPURINES AND TPMT
Azathioprine, mercaptopurine, and thioguanine are used for treating autoimmune and inflammatory diseases1–3 and certain types of cancer such as leukemias and lymphomas.1,4–6 Typically, azathioprine is used to treat nonmalignant conditions, thioguanine is used to treat malignancies, and mercaptopurine can be used to treat both malignant and nonmalignant conditions.
Although the exact mechanism of action of these drugs has not been completely elucidated, the active thioguanine nucleotide metabolites are thought to be incorporated into the DNA of leukocytes, resulting in DNA damage that subsequently leads to cell death and myelosuppression.7–9
Variants of the TPMT gene may alter the activity of the TPMT enzyme, resulting in individual variability in thiopurine metabolism. Compared with people with normal (high) TPMT activity, those with intermediate or low TPMT activity metabolize the drugs more slowly, and are likely to have higher thioguanine nucleotide concentrations and therefore an increased risk of myelosuppression.
One of the earliest correlations between TPMT activity and thiopurine-induced myelosuppression was described in a pediatric patient with acute lymphocytic leukemia.10 After being prescribed a conventional mercaptopurine dosage (75 mg/m2 daily), the patient developed severe myelosuppression and was observed to have a thioguanine nucleotide metabolite concentration seven times the observed population median. TPMT phenotyping demonstrated that the patient had low TPMT activity. Reducing the mercaptopurine dose by approximately 90% resulted in normalization of thioguanine nucleotide metabolite concentrations, and the myelosuppression subsequently resolved.
Approximately 10% of the population has intermediate TPMT activity and 0.3% has low or absent TPMT activity, though these percentages vary depending on ancestry.1 Research has demonstrated that approximately 30% to 60% of those with intermediate TPMT activity cannot tolerate a full thiopurine dose (eg, azathioprine 2–3 mg/kg/day or mercaptopurine 1.5 mg/kg/day).1 Almost all patients with low TPMT activity will develop life-threating myelosuppression if prescribed a full thiopurine dose.1
SHOULD TPMT ACTIVITY BE DETERMINED FOR EVERY PATIENT PRESCRIBED A THIOPURINE?
Although determining TPMT activity in thiopurine-naïve patients will assist clinicians in selecting a thiopurine starting dose or in deciding if an alternative agent is warranted, there are instances when a clinician may elect to not perform a TPMT genotype or phenotype test. For example, determining TPMT activity is not recommended for patients who previously tolerated thiopurine therapy at full steady-state doses.
The required starting dose of a thiopurine can influence the decision on whether or not to test for TPMT activity. TPMT genotyping or phenotyping may be of most benefit for patients requiring immediate full doses of a thiopurine.11 Ideally, TPMT activity should be determined before prescribing immediate full doses of a thiopurine. This could be achieved by preemptively ordering a TPMT test in patients likely to require immunosuppression—for example, in patients diagnosed with inflammatory or autoimmune diseases. If therapy cannot be delayed and TPMT activity is unknown, ordering a TPMT test at the time of prescribing a full thiopurine dose is still of benefit. Depending on the clinical laboratory utilized for testing, TPMT phenotype results are usually reported in 3 to 5 days, and TPMT genotype results are usually reported in 5 to 7 days. Because most patients will not reach steady-state concentrations for 2 to 6 weeks, clinicians could initiate immediate full doses of a thiopurine and modify therapy based on TPMT test results before accumulation of thioguanine nucleotide metabolites occurs. Caution should be used with this approach, particularly in situations where the clinical laboratory may not return results in a timely manner.
For patients who are candidates for an initial low dose of a thiopurine, clinicians may choose to slowly titrate doses based on response and tolerability instead of determining TPMT activity.11 Depending on the starting dose and how slowly titration occurs, initiating a thiopurine at a low dose and titrating based on response can be a feasible approach for patients with intermediate TPMT activity. Because drastic thiopurine dose reductions of approximately 10-fold are required for patients with low TPMT activity, which is a much smaller dosage than most clinicians will initially prescribe, the starting dosage will likely not be low enough to prevent myelosuppression in patients with low TPMT activity.1,10
Determining TPMT activity can help clinicians establish an appropriate titration schedule. Patients with normal TPMT activity will usually reach thiopurine steady-state concentrations in 2 weeks, and the dosage can be titrated based on response.1 Alterations in TPMT activity influence the pharmacokinetic parameters of thiopurines, and the time to reach steady-state is extended to 4 or 6 weeks for those with intermediate or low TPMT activity.1 Increasing the thiopurine dosage before reaching steady state can lead to the prescribing of doses that will not be tolerated, resulting in myelosuppression.
Factors to consider when deciding if TPMT activity should be assessed include the disease state being treated and corresponding starting dose, the need for immediate full doses, and previous documented tolerance of thiopurines at steady-state doses. As with many aspects of medicine that have multiple options, coupled with an increase in patient access to healthcare information, the decision to test for TPMT activity may include shared decision-making between patients and providers. Although TPMT genotyping or phenotyping can help identify those at greatest risk of severe myelosuppression, such assays do not replace routine monitoring for myelosuppression, hepatotoxicity, or pancreatitis that may be caused by thiopurines.
WHAT TESTS ARE AVAILABLE TO DETERMINE TPMT ACTIVITY?
Patients with intermediate or low TPMT activity can be identified by either genotyping or phenotyping. There are considerations, though, that clinicians should be aware of before selecting a particular test.
TPMT genotyping
![]()
Four TPMT alleles, TPMT*2, *3A, *3B, and *3C, account for over 90% of inactivating polymorphisms.12 Therefore, most reference laboratories only analyze for those genetic variants. Based on the reported test result, a predicted phenotype (eg, normal, intermediate, or low TPMT activity) can be assigned. Table 1 lists the predicted phenotypes for select genotyping results.
TPMT phenotyping
Phenotyping quantitates TPMT enzyme activity in erythrocytes, and based on the result, patients are classified as having normal, intermediate, or low TPMT activity. Because internal standards and other testing conditions may differ between reference laboratories, test results must be interpreted in the context of the laboratory that performed the assay.
Which test is right for my patient?
In most cases, either the genotype or the phenotype test provides sufficient information to guide thiopurine therapy. There are certain circumstances, though, in which the genotype or phenotype test is less informative.
TPMT genotyping, when performed using a blood specimen, is not recommended in those with a history of allogeneic bone marrow transplantation, as the result would reflect the donor’s genotype, not the patient’s. In such instances, monitoring of white blood cell counts and thiopurine metabolites may be more beneficial.
TPMT phenotyping may be inaccurate if performed within 30 to 90 days of an erythrocyte transfusion, as the test result may be influenced by donor erythrocytes. If a patient is receiving erythrocyte transfusions, TPMT genotyping is preferable to phenotyping.
Test cost may also be a consideration when determining if the genotype or phenotype test is best for your patient. Costs vary by laboratory, but phenotyping is generally less expensive than genotyping. The cost of genotyping, though, continues to decrease.13 The approximate commercial cost is $200 for phenotyping and $450 for genotyping, but laboratory fees may be substantially higher. Several insurance plans, including Medicare, cover TPMT testing, but reimbursement and copayments vary, depending on the patient’s specific plan.
There are conflicting data as to whether determining TPMT status is11,14–18 or is not19 cost-effective. Multiple studies suggest that the cost of genotyping a sufficient number of patients to identify a single individual at high risk of myelosuppression is cheaper than the costs associated with treating an adverse event. Additional cost-benefit studies are needed, particularly studies that consider how bundled payments and outcomes-based reimbursement influence cost-effectiveness.
MODIFYING THIOPURINE THERAPY BASED ON TPMT ACTIVITY
![]()
There is a strong correlation between TPMT activity and tolerated thiopurine doses, with those having intermediate or low TPMT activity requiring lower doses.10,20–23 Adjusting mercaptopurine doses based on TPMT activity to prevent hematopoietic toxicity has been successfully demonstrated in pediatric patients with acute lymphoblastic leukemia.24 Furthermore, reducing initial thiopurine doses to avoid myelosuppression and titrating based on response has been shown to not compromise outcomes.1,25,26 The Clinical Pharmacogenetic Implementation Consortium (CPIC) has developed an evidence-based guideline on how to adjust thiopurine doses based on TPMT activity,1 summarized in Table 2. These dosing recommendations are classified as “strong.”
Patients with normal TPMT activity should be prescribed the usual thiopurine starting dose as indicated by disease-specific guidelines.
For those with intermediate TPMT activity, the CPIC guideline recommends reducing the initial targeted full dose of azathioprine and mercaptopurine by 30% to 70% and reducing the targeted full dose of thioguanine by 30% to 50%. The percentage of dose reduction depends on the targeted full dose. Siegel and Sands27 suggested that for those who are diagnosed with inflammatory bowel disease and have intermediate TPMT activity, azathioprine should be initiated at a low dose and titrated to 1.25 mg/kg and mercaptopurine should be initiated at a low dose and titrated to 0.75 mg/kg. Based on these titration goals, if the targeted full dose for mercaptopurine is 1 mg/kg, then a dose reduction of approximately 30% would be more appropriate. If the targeted full dose is 1.5 mg/kg, a dose reduction of approximately 50% would be more appropriate. Thiopurine doses should be titrated based on response and disease-specific guidelines, allowing 2 to 4 weeks to reach steady state before dose titration.
For those with low TPMT activity, alternative therapy should be considered for nonmalignant conditions because of the risk of severe myelosuppression. For malignancy, or if a thiopurine is warranted for a nonmalignant condition, consider a 90% dose reduction and give the drug three times per week instead of daily. For example, acute lymphoblastic leukemia patients with low TPMT activity can be started on mercaptopurine 10 mg/m2 three times per week instead of the usual starting dose.10 Thiopurine doses should be titrated based on response and disease-specific guidelines, allowing 4 to 6 weeks to reach steady state before dose titration.
RECOMMENDATIONS
Individuals with intermediate or low TPMT activity have an increased risk of myelosuppression. Because of the elevated risk for morbidity and death, especially for patients with low TPMT activity, multiple guidelines and regulatory agencies recommend TPMT genotyping or phenotyping if a thiopurine is prescribed.25,28–32 Although additional cost-benefit analysis studies are needed, evidence suggests testing for TPMT activity may be cheaper than the costs associated with treating myelosuppression.
In view of treatment guidelines, the recommendations of regulatory agencies, cost-benefit analyses, and the availability of gene-based dosing recommendations, we consider the benefits of testing for TPMT activity to greatly outweigh any associated risks. Therefore, we recommend that TPMT testing be strongly considered before initiating therapy with a thiopurine.
- Relling MV, Gardner EE, Sandborn WJ, et al; Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing. Clin Pharmacol Ther 2011; 89:387–391.
- Ansari A, Arenas M, Greenfield SM, et al. Prospective evaluation of the pharmacogenetics of azathioprine in the treatment of inflammatory bowel disease. Aliment Pharmacol Ther 2008; 28:973–983.
- Beswick L, Friedman AB, Sparrow MP. The role of thiopurine metabolite monitoring in inflammatory bowel disease. Expert Rev Gastroenterol Hepatol 2014; 8:383–392.
- Gervasini G, Vagace JM. Impact of genetic polymorphisms on chemotherapy toxicity in childhood acute lymphoblastic leukemia. Front Genet 2012; 3:249.
- Levinsen M, Rotevatn EØ, Rosthøj S, et al; Nordic Society of Paediatric Haematology, Oncology. Pharmacogenetically based dosing of thiopurines in childhood acute lymphoblastic leukemia: influence on cure rates and risk of second cancer. Pediatr Blood Cancer 2014; 61:797–802.
- Adam de Beaumais T, Jacqz-Aigrain E. Pharmacogenetic determinants of mercaptopurine disposition in children with acute lymphoblastic leukemia. Eur J Clin Pharmacol 2012; 68:1233–1242.
- Derijks LJ, Gilissen LP, Hooymans PM, Hommes DW. Review article: thiopurines in inflammatory bowel disease. Aliment Pharmacol Ther 2006; 24:715–729.
- Fairchild CR, Maybaum J, Kennedy KA. Concurrent unilateral chromatid damage and DNA strand breakage in response to 6-thioguanine treatment. Biochem Pharmacol 1986; 35:3533–3541.
- Karran P. Thiopurines, DNA damage, DNA repair and therapy-related cancer. Br Med Bull 2006; 79–80:153–170.
- Evans WE, Horner M, Chu YQ, Kalwinsky D, Roberts WM. Altered mercaptopurine metabolism, toxic effects, and dosage requirement in a thiopurine methyltransferase-deficient child with acute lymphocytic leukemia. J Pediatr 1991; 119:985–989.
- Gardiner SJ, Gearry RB, Barclay ML, Begg EJ. Two cases of thiopurine methyltransferase (TPMT) deficiency—a lucky save and a near miss with azathioprine. Br J Clin Pharmacol 2006; 62:473–476.
- Relling MV, Gardner EE, Sandborn WJ, et al. Clinical pharmacogenetics implementation consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing: 2013 update. Clin Pharmacol Ther 2013; 93:324–325.
- Altman RB. Pharmacogenomics: “noninferiority” is sufficient for initial implementation. Clin Pharmacol Ther 2011; 89:348–350.
- van den Akker-van Marle ME, Gurwitz D, Detmar SB, et al. Cost-effectiveness of pharmacogenomics in clinical practice: a case study of thiopurine methyltransferase genotyping in acute lymphoblastic leukemia in Europe. Pharmacogenomics 2006; 7:783–792.
- Clunie GP, Lennard L. Relevance of thiopurine methyltransferase status in rheumatology patients receiving azathioprine. Rheumatology (Oxford) 2004; 43:13–18.
- Dubinsky MC, Reyes E, Ofman J, Chiou CF, Wade S, Sandborn WJ. A cost-effectiveness analysis of alternative disease management strategies in patients with Crohn’s disease treated with azathioprine or 6-mercaptopurine. Am J Gastroenterol 2005; 100:2239–2247.
- Winter J, Walker A, Shapiro D, Gaffney D, Spooner RJ, Mills PR. Cost-effectiveness of thiopurine methyltransferase genotype screening in patients about to commence azathioprine therapy for treatment of inflammatory bowel disease. Aliment Pharmacol Ther 2004; 20:593–599.
- Marra CA, Esdaile JM, Anis AH. Practical pharmacogenetics: the cost effectiveness of screening for thiopurine s-methyltransferase polymorphisms in patients with rheumatological conditions treated with azathioprine. J Rheumatol 2002; 29:2507–2512.
- Donnan JR, Ungar WJ, Mathews M, Hancock-Howard RL, Rahman P. A cost effectiveness analysis of thiopurine methyltransferase testing for guiding 6-mercaptopurine dosing in children with acute lymphoblastic leukemia. Pediatr Blood Cancer 2011; 57:231–239.
- Lennard L, Gibson BE, Nicole T, Lilleyman JS. Congenital thiopurine methyltransferase deficiency and 6-mercaptopurine toxicity during treatment for acute lymphoblastic leukaemia. Arch Dis Child 1993; 69:577–579.
- Hindorf U, Lindqvist M, Hildebrand H, Fagerberg U, Almer S. Adverse events leading to modification of therapy in a large cohort of patients with inflammatory bowel disease. Aliment Pharmacol Ther 2006; 24:331–342.
- Relling MV, Hancock ML, Rivera GK, et al. Mercaptopurine therapy intolerance and heterozygosity at the thiopurine S-methyltransferase gene locus. J Natl Cancer Inst 1999; 91:2001–2008.
- Relling MV, Hancock ML, Boyett JM, Pui CH, Evans WE. Prognostic importance of 6-mercaptopurine dose intensity in acute lymphoblastic leukemia. Blood 1999; 93:2817–2823.
- Pui CH, Pei D, Sandlund JT, et al. Long-term results of St Jude Total Therapy Studies 11, 12, 13A, 13B, and 14 for childhood acute lymphoblastic leukemia. Leukemia 2010; 24:371–382.
- Ford LT, Berg JD. Thiopurine S-methyltransferase (TPMT) assessment prior to starting thiopurine drug treatment; a pharmacogenomic test whose time has come. J Clin Pathol 2010; 63:288–295.
- Schmiegelow K, Forestier E, Hellebostad M, et al; Nordic Society of Paediatric Haematology and Oncology. Long-term results of NOPHO ALL-92 and ALL-2000 studies of childhood acute lymphoblastic leukemia. Leukemia 2010; 24:345–354.
- Siegel CA, Sands BE. Review article: practical management of inflammatory bowel disease patients taking immunomodulators. Aliment Pharmacol Ther 2005; 22:1–16.
- Mayberry JF, Lobo A, Ford AC, Thomas A. NICE clinical guideline (CG152): the management of Crohn’s disease in adults, children and young people. Aliment Pharmacol Ther 2013; 37:195–203
- Mowat C, Cole A, Windsor A, et al; IBD Section of the British Society of Gastroenterology. Guidelines for the management of inflammatory bowel disease in adults. Gut 2011; 60:571–607.
- Turner D, Levine A, Escher JC, et al; European Crohn’s and Colitis Organization; European Society for Paediatric Gastroenterology, Hepatology, and Nutrition. Management of pediatric ulcerative colitis: joint ECCO and ESPGHAN evidence-based consensus guidelines. J Pediatr Gastroenterol Nutr 2012; 55:340–361.
- Bernstein CN, Fried M, Krabshuis JH, et al. World Gastroenterology Organization Practice Guidelines for the diagnosis and management of IBD in 2010. Inflamm Bowel Dis 2010; 16:112–124.
- Becquemont L, Alfirevic A, Amstutz U, et al. Practical recommendations for pharmacogenomics-based prescription: 2010 ESF-UB Conference on Pharmacogenetics and Pharmacogenomics. Pharmacogenomics 2011; 12:113–124.
- Relling MV, Gardner EE, Sandborn WJ, et al; Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing. Clin Pharmacol Ther 2011; 89:387–391.
- Ansari A, Arenas M, Greenfield SM, et al. Prospective evaluation of the pharmacogenetics of azathioprine in the treatment of inflammatory bowel disease. Aliment Pharmacol Ther 2008; 28:973–983.
- Beswick L, Friedman AB, Sparrow MP. The role of thiopurine metabolite monitoring in inflammatory bowel disease. Expert Rev Gastroenterol Hepatol 2014; 8:383–392.
- Gervasini G, Vagace JM. Impact of genetic polymorphisms on chemotherapy toxicity in childhood acute lymphoblastic leukemia. Front Genet 2012; 3:249.
- Levinsen M, Rotevatn EØ, Rosthøj S, et al; Nordic Society of Paediatric Haematology, Oncology. Pharmacogenetically based dosing of thiopurines in childhood acute lymphoblastic leukemia: influence on cure rates and risk of second cancer. Pediatr Blood Cancer 2014; 61:797–802.
- Adam de Beaumais T, Jacqz-Aigrain E. Pharmacogenetic determinants of mercaptopurine disposition in children with acute lymphoblastic leukemia. Eur J Clin Pharmacol 2012; 68:1233–1242.
- Derijks LJ, Gilissen LP, Hooymans PM, Hommes DW. Review article: thiopurines in inflammatory bowel disease. Aliment Pharmacol Ther 2006; 24:715–729.
- Fairchild CR, Maybaum J, Kennedy KA. Concurrent unilateral chromatid damage and DNA strand breakage in response to 6-thioguanine treatment. Biochem Pharmacol 1986; 35:3533–3541.
- Karran P. Thiopurines, DNA damage, DNA repair and therapy-related cancer. Br Med Bull 2006; 79–80:153–170.
- Evans WE, Horner M, Chu YQ, Kalwinsky D, Roberts WM. Altered mercaptopurine metabolism, toxic effects, and dosage requirement in a thiopurine methyltransferase-deficient child with acute lymphocytic leukemia. J Pediatr 1991; 119:985–989.
- Gardiner SJ, Gearry RB, Barclay ML, Begg EJ. Two cases of thiopurine methyltransferase (TPMT) deficiency—a lucky save and a near miss with azathioprine. Br J Clin Pharmacol 2006; 62:473–476.
- Relling MV, Gardner EE, Sandborn WJ, et al. Clinical pharmacogenetics implementation consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing: 2013 update. Clin Pharmacol Ther 2013; 93:324–325.
- Altman RB. Pharmacogenomics: “noninferiority” is sufficient for initial implementation. Clin Pharmacol Ther 2011; 89:348–350.
- van den Akker-van Marle ME, Gurwitz D, Detmar SB, et al. Cost-effectiveness of pharmacogenomics in clinical practice: a case study of thiopurine methyltransferase genotyping in acute lymphoblastic leukemia in Europe. Pharmacogenomics 2006; 7:783–792.
- Clunie GP, Lennard L. Relevance of thiopurine methyltransferase status in rheumatology patients receiving azathioprine. Rheumatology (Oxford) 2004; 43:13–18.
- Dubinsky MC, Reyes E, Ofman J, Chiou CF, Wade S, Sandborn WJ. A cost-effectiveness analysis of alternative disease management strategies in patients with Crohn’s disease treated with azathioprine or 6-mercaptopurine. Am J Gastroenterol 2005; 100:2239–2247.
- Winter J, Walker A, Shapiro D, Gaffney D, Spooner RJ, Mills PR. Cost-effectiveness of thiopurine methyltransferase genotype screening in patients about to commence azathioprine therapy for treatment of inflammatory bowel disease. Aliment Pharmacol Ther 2004; 20:593–599.
- Marra CA, Esdaile JM, Anis AH. Practical pharmacogenetics: the cost effectiveness of screening for thiopurine s-methyltransferase polymorphisms in patients with rheumatological conditions treated with azathioprine. J Rheumatol 2002; 29:2507–2512.
- Donnan JR, Ungar WJ, Mathews M, Hancock-Howard RL, Rahman P. A cost effectiveness analysis of thiopurine methyltransferase testing for guiding 6-mercaptopurine dosing in children with acute lymphoblastic leukemia. Pediatr Blood Cancer 2011; 57:231–239.
- Lennard L, Gibson BE, Nicole T, Lilleyman JS. Congenital thiopurine methyltransferase deficiency and 6-mercaptopurine toxicity during treatment for acute lymphoblastic leukaemia. Arch Dis Child 1993; 69:577–579.
- Hindorf U, Lindqvist M, Hildebrand H, Fagerberg U, Almer S. Adverse events leading to modification of therapy in a large cohort of patients with inflammatory bowel disease. Aliment Pharmacol Ther 2006; 24:331–342.
- Relling MV, Hancock ML, Rivera GK, et al. Mercaptopurine therapy intolerance and heterozygosity at the thiopurine S-methyltransferase gene locus. J Natl Cancer Inst 1999; 91:2001–2008.
- Relling MV, Hancock ML, Boyett JM, Pui CH, Evans WE. Prognostic importance of 6-mercaptopurine dose intensity in acute lymphoblastic leukemia. Blood 1999; 93:2817–2823.
- Pui CH, Pei D, Sandlund JT, et al. Long-term results of St Jude Total Therapy Studies 11, 12, 13A, 13B, and 14 for childhood acute lymphoblastic leukemia. Leukemia 2010; 24:371–382.
- Ford LT, Berg JD. Thiopurine S-methyltransferase (TPMT) assessment prior to starting thiopurine drug treatment; a pharmacogenomic test whose time has come. J Clin Pathol 2010; 63:288–295.
- Schmiegelow K, Forestier E, Hellebostad M, et al; Nordic Society of Paediatric Haematology and Oncology. Long-term results of NOPHO ALL-92 and ALL-2000 studies of childhood acute lymphoblastic leukemia. Leukemia 2010; 24:345–354.
- Siegel CA, Sands BE. Review article: practical management of inflammatory bowel disease patients taking immunomodulators. Aliment Pharmacol Ther 2005; 22:1–16.
- Mayberry JF, Lobo A, Ford AC, Thomas A. NICE clinical guideline (CG152): the management of Crohn’s disease in adults, children and young people. Aliment Pharmacol Ther 2013; 37:195–203
- Mowat C, Cole A, Windsor A, et al; IBD Section of the British Society of Gastroenterology. Guidelines for the management of inflammatory bowel disease in adults. Gut 2011; 60:571–607.
- Turner D, Levine A, Escher JC, et al; European Crohn’s and Colitis Organization; European Society for Paediatric Gastroenterology, Hepatology, and Nutrition. Management of pediatric ulcerative colitis: joint ECCO and ESPGHAN evidence-based consensus guidelines. J Pediatr Gastroenterol Nutr 2012; 55:340–361.
- Bernstein CN, Fried M, Krabshuis JH, et al. World Gastroenterology Organization Practice Guidelines for the diagnosis and management of IBD in 2010. Inflamm Bowel Dis 2010; 16:112–124.
- Becquemont L, Alfirevic A, Amstutz U, et al. Practical recommendations for pharmacogenomics-based prescription: 2010 ESF-UB Conference on Pharmacogenetics and Pharmacogenomics. Pharmacogenomics 2011; 12:113–124.