User login
Noncosmetic uses of botulinum toxin in otolaryngology
Botulinum toxin is commonly used to treat movement disorders of the head and neck. It was first used to treat focal eye dystonia (blepharospasm) and laryngeal dystonia (spasmodic dysphonia) and is now also used for other head and neck dystonias, movement disorders, and muscle spasticity or contraction.
This article reviews the use of botulinum toxin for primary disorders of the laryngopharynx—adductor and abductor spasmodic dysphonias, laryngopharyngeal tremor, and cricopharyngeus muscle dysfunction—and its efficacy and side effects for the different conditions.
ABNORMAL MUSCLE MOVEMENT
Dystonia is abnormal muscle movement characterized by repetitive involuntary contractions. Dystonic contractions are described as either sustained (tonic) or spasmodic (clonic) and are typically induced by conscious action to move the muscle group.1,2 Dystonia can be categorized according to the amount of muscle involvement: generalized (widespread muscle activity), segmental (involving neighboring groups of muscles), or focal (involving only one or a few local small muscles).3 Activity may be associated with gross posturing and disfigurement, depending on the size and location of the muscle contractions, although the muscle action is usually normal during rest.
The cause of dystonia has been the focus of much debate and investigation. Some types of dystonia have strong family inheritance patterns, but most are sporadic, possibly brought on by trauma or infection. In most cases, dystonia is idiopathic, although it may be associated with other muscle group dystonias, tremor, neurologic injury or insults, other neurologic diseases and neurodegenerative disorders, or tardive syndromes.1 Because of the relationship with other neurologic diseases, consultation with a neurologist should be considered.
Treatment of the muscle contractions of the various dystonias includes drug therapy and physical, occupational, and voice therapy. Botulinum toxin is a principal treatment for head and neck dystonias and works by blocking muscular contractions.4 It has the advantages of having few side effects and predictable results for many conditions, although repeat injections are usually required to achieve a sustained effect.
LARYNGEAL DYSTONIAS CAUSE VOICE ABNORMALITIES
The most common laryngeal dystonia is spasmodic dysphonia, a focal dystonia of the larynx. It is subdivided into two types according to whether spasm of the vocal folds occurs during adduction or abduction.
Adductor spasmodic dysphonia accounts for 80% to 90% of cases. It is characterized by irregular speech with pitch breaks and a strained or strangulated voice. It was formerly treated by resection of the nerve to the vocal folds, but results were neither consistent nor persistent. Currently, the primary treatment is injection of botulinum toxin, which has a high success rate,5 with patients reporting about a 90% return of normal function.
Abductor spasmodic dysphonia accounts for 10% to 20% of cases.6 Patients have a breathy quality to the voice with a short duration of vocalization due to excessive loss of air on phonation. This is especially noticeable when the patient speaks words that begin with a voiceless consonant followed by a vowel (eg, pat, puppy). Response to botulinum toxin injection is more variable,6 possibly because of the pathophysiology of the disorder or because of the technical challenges of administering the injection.
Fewer than 1% of patients have both abductor and adductor components, and their treatment can be particularly challenging.
Adductor spasmodic dysphonia: Treatment usually successful
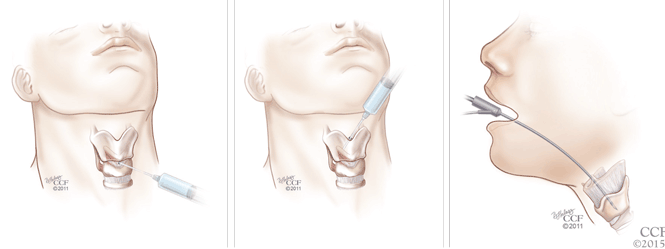
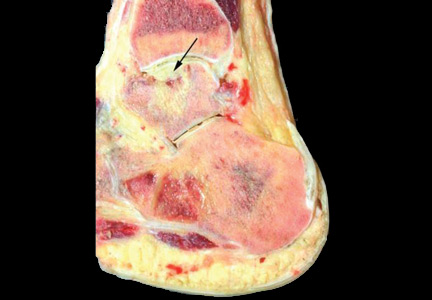
Botulinum toxin can be injected for adductor spasmodic dysphonia via a number of approaches, the most common being through the cricothyroid membrane (Figure 1). Injections can be made into one or both vocal folds and can be performed under guidance with laryngeal electromyography or with a flexible laryngoscope to visualize the larynx.
Patients typically experience breathiness beginning 1 or 2 days after the injection, and this effect may last for up to 2 weeks. During that time, the patient may be more susceptible to aspiration of thin liquids and so is instructed to drink cautiously. Treatment benefits typically last for 3 to 6 months. As the botulinum toxin wears off, the patient notices a gradual increase in vocal straining and effort.
Dosages of botulinum toxin for subsequent treatments are adjusted by balancing the period of benefit with postinjection breathiness. The desire of the patient should be paramount. Some are willing to tolerate more side effects to avoid frequent injections, so they can be given a larger dose. Others cannot tolerate the breathiness but are willing to accept more frequent injections, so they should be given a smaller dose. In rare cases, patients have significant breathiness from even small doses; they may be helped by injecting into only one vocal fold or, alternatively, into a false vocal fold, allowing diffusion of the toxin down to the muscle of the true vocal fold.
Abductor spasmodic dysphonia: Treatment more challenging
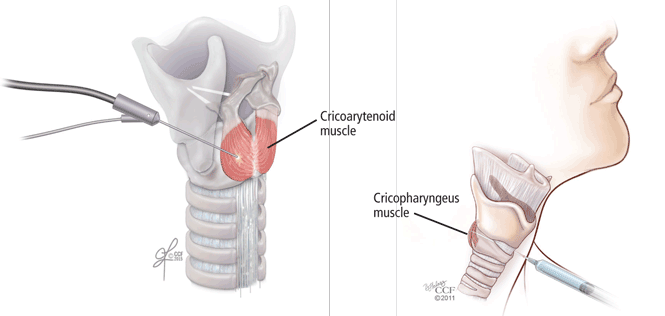
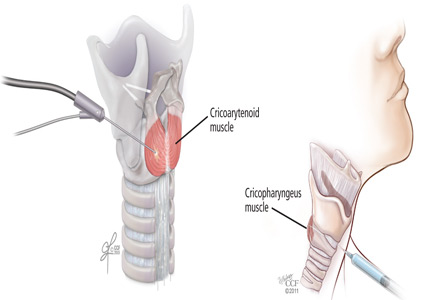
The success of botulinum toxin treatment for abductor spasmodic dysphonia is more variable than for the adductor type. The injections are made into the posterior cricoarytenoid muscle (Figure 2); because this muscle cannot be directly visualized, this procedure requires guidance with laryngeal electromyography. Most patients note improvement, and about 20% have a good response.6 Most require a second injection about 1 month later, often on the other side. Bilateral injections at one sitting may compromise the airway, and vocal fold motion should be evaluated at the time of the contralateral injection to assess airway patency. Interest has increased in simultaneous bilateral injections with lower doses of botulinum toxin, and this approach has been shown to be safe.7
ESSENTIAL TREMOR OF THE VOICE
Essential tremor is an action tremor that can occur with voluntary movement. It can occur anywhere in the body, often the head or hand, but the voice can also be affected. About half of cases are hereditary. Essential tremor of the voice causes a rhythmic oscillation of pitch and intensity.
Consultation with a neurologist is recommended to evaluate the cause, although voice tremor is often idiopathic and occurs in about 30% of patients with essential tremor in the arms or legs, as well as in about 30% of patients with spasmodic dysphonia. Extremity tremor can usually be successfully managed medically, but this is not true for voice tremor.
Botulinum toxin injection is the mainstay of treatment for essential tremor of the voice, although its success is marginal. About two-thirds of patients have some degree of improvement from traditional botulinum toxin injections in the true vocal fold.8
The results of treatment are likely to be inconsistent because tremor tends to involve several different muscles used in voice production, commonly in the soft palate, tongue base, pharyngeal walls, strap muscles, false vocal folds, and true vocal folds. A location-oriented tremor scoring system9 can help identify the involved muscles to guide injections. Treatment is less likely to be successful in patients with multiple sites of voice tremor. Injection into the false vocal fold, true vocal fold, and interarytenoid muscle10 can safely be performed; injections into the palate, tongue base, and strap muscles are to be avoided because of the high risk of postinjection aspiration.
Patients who have good results can have repeat treatments as needed. The dosage of botulinum toxin is adjusted according to response, side effects (eg, breathy voice, dysphagia), and patient preference.
CRICOPHARYNGEUS MUSCLE DYSFUNCTION: TROUBLE SWALLOWING
Dysfunction of the cricopharyngeus muscle causes difficulty swallowing, especially swallowing solid foods. It can be attributed to a mechanical stricture or to hyperfunction (spasm).
Mechanical stricture at the esophageal inlet frequently occurs in patients who have had a total laryngectomy for advanced laryngeal cancer. Fibrosis tends to be worse in patients who have also undergone radiation therapy.
Stricture can be treated with botulinum toxin injections and dilation. Conservative treatment is preferred to surgical myotomy for patients with complex postlaryngectomy anatomy and scarring from radiation therapy.
Cricopharyngeus muscle spasm or hyperfunction can be an important cause of dysphagia, especially in the elderly. Patients should be evaluated with barium esophagography or a modified barium swallow. The finding of a cricopharyngeal “bar” provides evidence of contraction of the muscle that impedes the passage of food.
Botulinum toxin injections for cricopharyngeus muscle dysfunction (Figure 2) can be effective in some cases, especially if the toxin is injected bilaterally. However, because the cricopharyngeus muscle plays an important role in preventing esophageal reflux into the laryngopharynx, botulinum toxin injection in patients with substantial hiatal hernia or laryngopharyngeal reflux disease should only be done with caution. In addition, treatment of reflux disease should be considered in any patient undergoing botulinum toxin injection for cricopharyngeus muscle dysfunction.
Most patients require repeat injections when the toxin wears off, although occasionally one or two injections provide long-term or permanent relief. Dosages are adjusted for the patient’s age, the presence of other swallowing problems, and reflux. Patients may experience increased difficulty swallowing for 1 or 2 weeks after the procedure and so should be counseled to eat slowly and carefully.
- Cultrara A, Chitkara A, Blitzer A. Botulinum toxin injections for the treatment of oromandibular dystonia. Oper Tech Otolaryngol Head Neck Surg 2004; 15:97–102.
- Fahn S. The varied clinical expressions of dystonia. Neurol Clin 1984; 2:541–554.
- Fahn S. Concept and classification of dystonia. Adv Neurol 1988; 50:1–8.
- Benninger MS, Knott PD. Techniques of botulinum toxin injections in the head and neck. San Diego, CA: Plural Publishing, Inc; 2012.
- Benninger MS, Gardner G, Grywalski C. Outcomes of botulinum toxin treatment for spasmodic dysphonia. Arch Otolaryngol Head Neck Surg 2001; 127:1083–1085.
- Blitzer A, Brin MF, Stewart CF. Botulinum toxin management of spasmodic dysphonia (laryngeal dystonia): a 12-year experience in more than 900 patients. Laryngoscope 1998; 108:1435–1441.
- Klein AM, Stong BC, Wise J, DelGaudio JM, Hapner ER, Johns MM 3rd. Vocal outcome measures after bilateral posterior cricoarytenoid muscle botulinum toxin injections for abductor spasmodic dysphonia. Otolaryngol Head Neck Surg 2008; 139:421–423.
- Hertegård S, Granqvist S, Lindestad PA. Botulinum toxin injections for essential voice tremor. Ann Otol Rhinol Laryngol 2000; 109:204–209.
- Bové M, Daamen N, Rosen C, Wang CC, Sulica L, Gartner-Schmidt J. Development and validation of the vocal tremor scoring system. Laryngoscope 2006; 116:1662–1667.
- Kendall KA, Leonard RJ. Interarytenoid muscle Botox injection for treatment of adductor spasmodic dysphonia with vocal tremor. J Voice 2001; 25:114–119.
Botulinum toxin is commonly used to treat movement disorders of the head and neck. It was first used to treat focal eye dystonia (blepharospasm) and laryngeal dystonia (spasmodic dysphonia) and is now also used for other head and neck dystonias, movement disorders, and muscle spasticity or contraction.
This article reviews the use of botulinum toxin for primary disorders of the laryngopharynx—adductor and abductor spasmodic dysphonias, laryngopharyngeal tremor, and cricopharyngeus muscle dysfunction—and its efficacy and side effects for the different conditions.
ABNORMAL MUSCLE MOVEMENT
Dystonia is abnormal muscle movement characterized by repetitive involuntary contractions. Dystonic contractions are described as either sustained (tonic) or spasmodic (clonic) and are typically induced by conscious action to move the muscle group.1,2 Dystonia can be categorized according to the amount of muscle involvement: generalized (widespread muscle activity), segmental (involving neighboring groups of muscles), or focal (involving only one or a few local small muscles).3 Activity may be associated with gross posturing and disfigurement, depending on the size and location of the muscle contractions, although the muscle action is usually normal during rest.
The cause of dystonia has been the focus of much debate and investigation. Some types of dystonia have strong family inheritance patterns, but most are sporadic, possibly brought on by trauma or infection. In most cases, dystonia is idiopathic, although it may be associated with other muscle group dystonias, tremor, neurologic injury or insults, other neurologic diseases and neurodegenerative disorders, or tardive syndromes.1 Because of the relationship with other neurologic diseases, consultation with a neurologist should be considered.
Treatment of the muscle contractions of the various dystonias includes drug therapy and physical, occupational, and voice therapy. Botulinum toxin is a principal treatment for head and neck dystonias and works by blocking muscular contractions.4 It has the advantages of having few side effects and predictable results for many conditions, although repeat injections are usually required to achieve a sustained effect.
LARYNGEAL DYSTONIAS CAUSE VOICE ABNORMALITIES
The most common laryngeal dystonia is spasmodic dysphonia, a focal dystonia of the larynx. It is subdivided into two types according to whether spasm of the vocal folds occurs during adduction or abduction.
Adductor spasmodic dysphonia accounts for 80% to 90% of cases. It is characterized by irregular speech with pitch breaks and a strained or strangulated voice. It was formerly treated by resection of the nerve to the vocal folds, but results were neither consistent nor persistent. Currently, the primary treatment is injection of botulinum toxin, which has a high success rate,5 with patients reporting about a 90% return of normal function.
Abductor spasmodic dysphonia accounts for 10% to 20% of cases.6 Patients have a breathy quality to the voice with a short duration of vocalization due to excessive loss of air on phonation. This is especially noticeable when the patient speaks words that begin with a voiceless consonant followed by a vowel (eg, pat, puppy). Response to botulinum toxin injection is more variable,6 possibly because of the pathophysiology of the disorder or because of the technical challenges of administering the injection.
Fewer than 1% of patients have both abductor and adductor components, and their treatment can be particularly challenging.
Adductor spasmodic dysphonia: Treatment usually successful
Botulinum toxin can be injected for adductor spasmodic dysphonia via a number of approaches, the most common being through the cricothyroid membrane (Figure 1). Injections can be made into one or both vocal folds and can be performed under guidance with laryngeal electromyography or with a flexible laryngoscope to visualize the larynx.
Patients typically experience breathiness beginning 1 or 2 days after the injection, and this effect may last for up to 2 weeks. During that time, the patient may be more susceptible to aspiration of thin liquids and so is instructed to drink cautiously. Treatment benefits typically last for 3 to 6 months. As the botulinum toxin wears off, the patient notices a gradual increase in vocal straining and effort.
Dosages of botulinum toxin for subsequent treatments are adjusted by balancing the period of benefit with postinjection breathiness. The desire of the patient should be paramount. Some are willing to tolerate more side effects to avoid frequent injections, so they can be given a larger dose. Others cannot tolerate the breathiness but are willing to accept more frequent injections, so they should be given a smaller dose. In rare cases, patients have significant breathiness from even small doses; they may be helped by injecting into only one vocal fold or, alternatively, into a false vocal fold, allowing diffusion of the toxin down to the muscle of the true vocal fold.
Abductor spasmodic dysphonia: Treatment more challenging
The success of botulinum toxin treatment for abductor spasmodic dysphonia is more variable than for the adductor type. The injections are made into the posterior cricoarytenoid muscle (Figure 2); because this muscle cannot be directly visualized, this procedure requires guidance with laryngeal electromyography. Most patients note improvement, and about 20% have a good response.6 Most require a second injection about 1 month later, often on the other side. Bilateral injections at one sitting may compromise the airway, and vocal fold motion should be evaluated at the time of the contralateral injection to assess airway patency. Interest has increased in simultaneous bilateral injections with lower doses of botulinum toxin, and this approach has been shown to be safe.7
ESSENTIAL TREMOR OF THE VOICE
Essential tremor is an action tremor that can occur with voluntary movement. It can occur anywhere in the body, often the head or hand, but the voice can also be affected. About half of cases are hereditary. Essential tremor of the voice causes a rhythmic oscillation of pitch and intensity.
Consultation with a neurologist is recommended to evaluate the cause, although voice tremor is often idiopathic and occurs in about 30% of patients with essential tremor in the arms or legs, as well as in about 30% of patients with spasmodic dysphonia. Extremity tremor can usually be successfully managed medically, but this is not true for voice tremor.
Botulinum toxin injection is the mainstay of treatment for essential tremor of the voice, although its success is marginal. About two-thirds of patients have some degree of improvement from traditional botulinum toxin injections in the true vocal fold.8
The results of treatment are likely to be inconsistent because tremor tends to involve several different muscles used in voice production, commonly in the soft palate, tongue base, pharyngeal walls, strap muscles, false vocal folds, and true vocal folds. A location-oriented tremor scoring system9 can help identify the involved muscles to guide injections. Treatment is less likely to be successful in patients with multiple sites of voice tremor. Injection into the false vocal fold, true vocal fold, and interarytenoid muscle10 can safely be performed; injections into the palate, tongue base, and strap muscles are to be avoided because of the high risk of postinjection aspiration.
Patients who have good results can have repeat treatments as needed. The dosage of botulinum toxin is adjusted according to response, side effects (eg, breathy voice, dysphagia), and patient preference.
CRICOPHARYNGEUS MUSCLE DYSFUNCTION: TROUBLE SWALLOWING
Dysfunction of the cricopharyngeus muscle causes difficulty swallowing, especially swallowing solid foods. It can be attributed to a mechanical stricture or to hyperfunction (spasm).
Mechanical stricture at the esophageal inlet frequently occurs in patients who have had a total laryngectomy for advanced laryngeal cancer. Fibrosis tends to be worse in patients who have also undergone radiation therapy.
Stricture can be treated with botulinum toxin injections and dilation. Conservative treatment is preferred to surgical myotomy for patients with complex postlaryngectomy anatomy and scarring from radiation therapy.
Cricopharyngeus muscle spasm or hyperfunction can be an important cause of dysphagia, especially in the elderly. Patients should be evaluated with barium esophagography or a modified barium swallow. The finding of a cricopharyngeal “bar” provides evidence of contraction of the muscle that impedes the passage of food.
Botulinum toxin injections for cricopharyngeus muscle dysfunction (Figure 2) can be effective in some cases, especially if the toxin is injected bilaterally. However, because the cricopharyngeus muscle plays an important role in preventing esophageal reflux into the laryngopharynx, botulinum toxin injection in patients with substantial hiatal hernia or laryngopharyngeal reflux disease should only be done with caution. In addition, treatment of reflux disease should be considered in any patient undergoing botulinum toxin injection for cricopharyngeus muscle dysfunction.
Most patients require repeat injections when the toxin wears off, although occasionally one or two injections provide long-term or permanent relief. Dosages are adjusted for the patient’s age, the presence of other swallowing problems, and reflux. Patients may experience increased difficulty swallowing for 1 or 2 weeks after the procedure and so should be counseled to eat slowly and carefully.
Botulinum toxin is commonly used to treat movement disorders of the head and neck. It was first used to treat focal eye dystonia (blepharospasm) and laryngeal dystonia (spasmodic dysphonia) and is now also used for other head and neck dystonias, movement disorders, and muscle spasticity or contraction.
This article reviews the use of botulinum toxin for primary disorders of the laryngopharynx—adductor and abductor spasmodic dysphonias, laryngopharyngeal tremor, and cricopharyngeus muscle dysfunction—and its efficacy and side effects for the different conditions.
ABNORMAL MUSCLE MOVEMENT
Dystonia is abnormal muscle movement characterized by repetitive involuntary contractions. Dystonic contractions are described as either sustained (tonic) or spasmodic (clonic) and are typically induced by conscious action to move the muscle group.1,2 Dystonia can be categorized according to the amount of muscle involvement: generalized (widespread muscle activity), segmental (involving neighboring groups of muscles), or focal (involving only one or a few local small muscles).3 Activity may be associated with gross posturing and disfigurement, depending on the size and location of the muscle contractions, although the muscle action is usually normal during rest.
The cause of dystonia has been the focus of much debate and investigation. Some types of dystonia have strong family inheritance patterns, but most are sporadic, possibly brought on by trauma or infection. In most cases, dystonia is idiopathic, although it may be associated with other muscle group dystonias, tremor, neurologic injury or insults, other neurologic diseases and neurodegenerative disorders, or tardive syndromes.1 Because of the relationship with other neurologic diseases, consultation with a neurologist should be considered.
Treatment of the muscle contractions of the various dystonias includes drug therapy and physical, occupational, and voice therapy. Botulinum toxin is a principal treatment for head and neck dystonias and works by blocking muscular contractions.4 It has the advantages of having few side effects and predictable results for many conditions, although repeat injections are usually required to achieve a sustained effect.
LARYNGEAL DYSTONIAS CAUSE VOICE ABNORMALITIES
The most common laryngeal dystonia is spasmodic dysphonia, a focal dystonia of the larynx. It is subdivided into two types according to whether spasm of the vocal folds occurs during adduction or abduction.
Adductor spasmodic dysphonia accounts for 80% to 90% of cases. It is characterized by irregular speech with pitch breaks and a strained or strangulated voice. It was formerly treated by resection of the nerve to the vocal folds, but results were neither consistent nor persistent. Currently, the primary treatment is injection of botulinum toxin, which has a high success rate,5 with patients reporting about a 90% return of normal function.
Abductor spasmodic dysphonia accounts for 10% to 20% of cases.6 Patients have a breathy quality to the voice with a short duration of vocalization due to excessive loss of air on phonation. This is especially noticeable when the patient speaks words that begin with a voiceless consonant followed by a vowel (eg, pat, puppy). Response to botulinum toxin injection is more variable,6 possibly because of the pathophysiology of the disorder or because of the technical challenges of administering the injection.
Fewer than 1% of patients have both abductor and adductor components, and their treatment can be particularly challenging.
Adductor spasmodic dysphonia: Treatment usually successful
Botulinum toxin can be injected for adductor spasmodic dysphonia via a number of approaches, the most common being through the cricothyroid membrane (Figure 1). Injections can be made into one or both vocal folds and can be performed under guidance with laryngeal electromyography or with a flexible laryngoscope to visualize the larynx.
Patients typically experience breathiness beginning 1 or 2 days after the injection, and this effect may last for up to 2 weeks. During that time, the patient may be more susceptible to aspiration of thin liquids and so is instructed to drink cautiously. Treatment benefits typically last for 3 to 6 months. As the botulinum toxin wears off, the patient notices a gradual increase in vocal straining and effort.
Dosages of botulinum toxin for subsequent treatments are adjusted by balancing the period of benefit with postinjection breathiness. The desire of the patient should be paramount. Some are willing to tolerate more side effects to avoid frequent injections, so they can be given a larger dose. Others cannot tolerate the breathiness but are willing to accept more frequent injections, so they should be given a smaller dose. In rare cases, patients have significant breathiness from even small doses; they may be helped by injecting into only one vocal fold or, alternatively, into a false vocal fold, allowing diffusion of the toxin down to the muscle of the true vocal fold.
Abductor spasmodic dysphonia: Treatment more challenging
The success of botulinum toxin treatment for abductor spasmodic dysphonia is more variable than for the adductor type. The injections are made into the posterior cricoarytenoid muscle (Figure 2); because this muscle cannot be directly visualized, this procedure requires guidance with laryngeal electromyography. Most patients note improvement, and about 20% have a good response.6 Most require a second injection about 1 month later, often on the other side. Bilateral injections at one sitting may compromise the airway, and vocal fold motion should be evaluated at the time of the contralateral injection to assess airway patency. Interest has increased in simultaneous bilateral injections with lower doses of botulinum toxin, and this approach has been shown to be safe.7
ESSENTIAL TREMOR OF THE VOICE
Essential tremor is an action tremor that can occur with voluntary movement. It can occur anywhere in the body, often the head or hand, but the voice can also be affected. About half of cases are hereditary. Essential tremor of the voice causes a rhythmic oscillation of pitch and intensity.
Consultation with a neurologist is recommended to evaluate the cause, although voice tremor is often idiopathic and occurs in about 30% of patients with essential tremor in the arms or legs, as well as in about 30% of patients with spasmodic dysphonia. Extremity tremor can usually be successfully managed medically, but this is not true for voice tremor.
Botulinum toxin injection is the mainstay of treatment for essential tremor of the voice, although its success is marginal. About two-thirds of patients have some degree of improvement from traditional botulinum toxin injections in the true vocal fold.8
The results of treatment are likely to be inconsistent because tremor tends to involve several different muscles used in voice production, commonly in the soft palate, tongue base, pharyngeal walls, strap muscles, false vocal folds, and true vocal folds. A location-oriented tremor scoring system9 can help identify the involved muscles to guide injections. Treatment is less likely to be successful in patients with multiple sites of voice tremor. Injection into the false vocal fold, true vocal fold, and interarytenoid muscle10 can safely be performed; injections into the palate, tongue base, and strap muscles are to be avoided because of the high risk of postinjection aspiration.
Patients who have good results can have repeat treatments as needed. The dosage of botulinum toxin is adjusted according to response, side effects (eg, breathy voice, dysphagia), and patient preference.
CRICOPHARYNGEUS MUSCLE DYSFUNCTION: TROUBLE SWALLOWING
Dysfunction of the cricopharyngeus muscle causes difficulty swallowing, especially swallowing solid foods. It can be attributed to a mechanical stricture or to hyperfunction (spasm).
Mechanical stricture at the esophageal inlet frequently occurs in patients who have had a total laryngectomy for advanced laryngeal cancer. Fibrosis tends to be worse in patients who have also undergone radiation therapy.
Stricture can be treated with botulinum toxin injections and dilation. Conservative treatment is preferred to surgical myotomy for patients with complex postlaryngectomy anatomy and scarring from radiation therapy.
Cricopharyngeus muscle spasm or hyperfunction can be an important cause of dysphagia, especially in the elderly. Patients should be evaluated with barium esophagography or a modified barium swallow. The finding of a cricopharyngeal “bar” provides evidence of contraction of the muscle that impedes the passage of food.
Botulinum toxin injections for cricopharyngeus muscle dysfunction (Figure 2) can be effective in some cases, especially if the toxin is injected bilaterally. However, because the cricopharyngeus muscle plays an important role in preventing esophageal reflux into the laryngopharynx, botulinum toxin injection in patients with substantial hiatal hernia or laryngopharyngeal reflux disease should only be done with caution. In addition, treatment of reflux disease should be considered in any patient undergoing botulinum toxin injection for cricopharyngeus muscle dysfunction.
Most patients require repeat injections when the toxin wears off, although occasionally one or two injections provide long-term or permanent relief. Dosages are adjusted for the patient’s age, the presence of other swallowing problems, and reflux. Patients may experience increased difficulty swallowing for 1 or 2 weeks after the procedure and so should be counseled to eat slowly and carefully.
- Cultrara A, Chitkara A, Blitzer A. Botulinum toxin injections for the treatment of oromandibular dystonia. Oper Tech Otolaryngol Head Neck Surg 2004; 15:97–102.
- Fahn S. The varied clinical expressions of dystonia. Neurol Clin 1984; 2:541–554.
- Fahn S. Concept and classification of dystonia. Adv Neurol 1988; 50:1–8.
- Benninger MS, Knott PD. Techniques of botulinum toxin injections in the head and neck. San Diego, CA: Plural Publishing, Inc; 2012.
- Benninger MS, Gardner G, Grywalski C. Outcomes of botulinum toxin treatment for spasmodic dysphonia. Arch Otolaryngol Head Neck Surg 2001; 127:1083–1085.
- Blitzer A, Brin MF, Stewart CF. Botulinum toxin management of spasmodic dysphonia (laryngeal dystonia): a 12-year experience in more than 900 patients. Laryngoscope 1998; 108:1435–1441.
- Klein AM, Stong BC, Wise J, DelGaudio JM, Hapner ER, Johns MM 3rd. Vocal outcome measures after bilateral posterior cricoarytenoid muscle botulinum toxin injections for abductor spasmodic dysphonia. Otolaryngol Head Neck Surg 2008; 139:421–423.
- Hertegård S, Granqvist S, Lindestad PA. Botulinum toxin injections for essential voice tremor. Ann Otol Rhinol Laryngol 2000; 109:204–209.
- Bové M, Daamen N, Rosen C, Wang CC, Sulica L, Gartner-Schmidt J. Development and validation of the vocal tremor scoring system. Laryngoscope 2006; 116:1662–1667.
- Kendall KA, Leonard RJ. Interarytenoid muscle Botox injection for treatment of adductor spasmodic dysphonia with vocal tremor. J Voice 2001; 25:114–119.
- Cultrara A, Chitkara A, Blitzer A. Botulinum toxin injections for the treatment of oromandibular dystonia. Oper Tech Otolaryngol Head Neck Surg 2004; 15:97–102.
- Fahn S. The varied clinical expressions of dystonia. Neurol Clin 1984; 2:541–554.
- Fahn S. Concept and classification of dystonia. Adv Neurol 1988; 50:1–8.
- Benninger MS, Knott PD. Techniques of botulinum toxin injections in the head and neck. San Diego, CA: Plural Publishing, Inc; 2012.
- Benninger MS, Gardner G, Grywalski C. Outcomes of botulinum toxin treatment for spasmodic dysphonia. Arch Otolaryngol Head Neck Surg 2001; 127:1083–1085.
- Blitzer A, Brin MF, Stewart CF. Botulinum toxin management of spasmodic dysphonia (laryngeal dystonia): a 12-year experience in more than 900 patients. Laryngoscope 1998; 108:1435–1441.
- Klein AM, Stong BC, Wise J, DelGaudio JM, Hapner ER, Johns MM 3rd. Vocal outcome measures after bilateral posterior cricoarytenoid muscle botulinum toxin injections for abductor spasmodic dysphonia. Otolaryngol Head Neck Surg 2008; 139:421–423.
- Hertegård S, Granqvist S, Lindestad PA. Botulinum toxin injections for essential voice tremor. Ann Otol Rhinol Laryngol 2000; 109:204–209.
- Bové M, Daamen N, Rosen C, Wang CC, Sulica L, Gartner-Schmidt J. Development and validation of the vocal tremor scoring system. Laryngoscope 2006; 116:1662–1667.
- Kendall KA, Leonard RJ. Interarytenoid muscle Botox injection for treatment of adductor spasmodic dysphonia with vocal tremor. J Voice 2001; 25:114–119.
KEY POINTS
- Botulinum toxin can be injected with a variety of approaches directly into the affected muscle exhibiting abnormal contractions.
- Depending on the muscles involved, side effects may include breathiness or difficulty swallowing for a period soon after injection.
- Injections can be repeated as needed as the toxin wears off.
- Some conditions are more amenable to treatment than others. Benefit can be enhanced by altering the dosage or injection site.
Women’s health 2015: An update for the internist
Women's health encompasses a broad range of issues unique to the female patient, with a scope that has expanded beyond reproductive health. Providers who care for women must develop cross-disciplinary competencies and understand the complex role of sex and gender on disease expression and treatment outcomes. Staying current with the literature in this rapidly changing field can be challenging for the busy clinician.
This article reviews recent advances in the treatment of depression in pregnancy, nonhormonal therapies for menopausal symptoms, and heart failure therapy in women, highlighting notable studies published in 2014 and early 2015.
TREATMENT OF DEPRESSION IN PREGNANCY
A 32-year-old woman with well-controlled but recurrent depression presents to the clinic for preconception counseling. Her depression has been successfully managed with a selective serotonin reuptake inhibitor (SSRI). She and her husband would like to try to conceive soon, but she is worried that continuing on her current SSRI may harm her baby. How should you advise her?
Concern for teratogenic effects of SSRIs
Depression is common during pregnancy: 11.8% to 13.5% of pregnant women report symptoms of depression,1 and 7.5% of pregnant women take an antidepressant.2
SSRI use during pregnancy has drawn attention because of mixed reports of teratogenic effects on the newborn, such as omphalocele, congenital heart defects, and craniosynostosis.3 Previous observational studies have specifically linked paroxetine to small but significant increases in right ventricular outflow tract obstruction4,5 and have linked sertraline to ventricular septal defects.6
However, reports of associations of congenital malformations and SSRI use in pregnancy in observational studies have been questioned, with concern that these studies had low statistical power, self-reported data leading to recall bias, and limited assessment for confounding factors.3,7
Recent studies refute risk of cardiac malformations
Several newer studies have been published that further examine the association between SSRI use in pregnancy and congenital heart defects, and their findings suggest that once adjusted for confounding variables, SSRI use in pregnancy may not be associated with cardiac malformations.
Huybrechts et al,8 in a large study published in 2014, extracted data on 950,000 pregnant women from the Medicaid database over a 7-year period and examined it for SSRI use during the first 90 days of pregnancy. Though SSRI use was associated with cardiac malformations when unadjusted for confounding variables (unadjusted relative risk 1.25, 95% confidence interval [CI] 1.13–1.38), once the cohort was restricted to women with a diagnosis of only depression and was adjusted based on propensity scoring, the association was no longer statistically significant (adjusted relative risk 1.06, 95% CI 0.93–1.22).
Additionally, there was no association between sertraline and ventricular septal defects (63 cases in 14,040 women exposed to sertraline, adjusted relative risk 1.04, 95% CI 0.76–1.41), or between paroxetine and right ventricular outflow tract obstruction (93 cases in 11,126 women exposed to paroxetine, adjusted relative risk 1.07, 95% CI 0.59–1.93).8
Furu et al7 conducted a sibling-matched case-control comparison published in 2015, in which more than 2 million live births from five Nordic countries were examined in the full cohort study and 2,288 births in the sibling-matched case-control cohort. SSRI or venlafaxine use in the first 90 days of pregnancy was examined. There was a slightly higher rate of cardiac defects in infants born to SSRI or venlafaxine recipients in the cohort study (adjusted odds ratio 1.15, 95% CI 1.05–1.26). However, in the sibling-controlled analyses, neither an SSRI nor venlafaxine was associated with heart defects (adjusted odds ratio 0.92, 95% CI 0.72–1.17), leading the authors to conclude that there might be familial factors or other lifestyle factors that were not taken into consideration and that could have confounded the cohort results.
Bérard et al9 examined antidepressant use in the first trimester of pregnancy in a cohort of women in Canada and concluded that sertraline was associated with congenital atrial and ventricular defects (risk ratio 1.34; 95% CI 1.02–1.76).9 However, this association should be interpreted with caution, as the Canadian cohort was notably smaller than those in other studies we have discussed, with only 18,493 pregnancies in the total cohort, and this conclusion was drawn from 9 cases of ventricular or atrial septal defects in babies of 366 women exposed to sertraline.
Although at first glance SSRIs may appear to be associated with congenital heart defects, these recent studies are reassuring and suggest that the association may actually not be significant. As with any statistical analysis, thoughtful study design, adequate statistical power, and adjustment for confounding factors must be considered before drawing conclusions.
SSRIs, offspring psychiatric outcomes, and miscarriage rates
Clements et al10 studied a cohort extracted from Partners Healthcare consisting of newborns with autism spectrum disorder, newborns with attention-deficit hyperactivity disorder (ADHD), and healthy matched controls and found that SSRI use during pregnancy was not associated with offspring autism spectrum disorder (adjusted odds ratio 1.10, 95% CI 0.7–1.70). However, they did find an increased risk of ADHD with SSRI use during pregnancy (adjusted odds ratio 1.81, 95% CI 1.22–2.70).
Andersen et al11 examined more than 1 million pregnancies in Denmark and found no difference in risk of miscarriage between women who used an SSRI during pregnancy (adjusted hazard ratio 1.27) and women who discontinued their SSRI at least 3 months before pregnancy (adjusted hazard ratio 1.24, P = .47). The authors concluded that because of the similar rate of miscarriage in both groups, there was no association between SSRI use and miscarriage, and that the small increased risk of miscarriage in both groups could have been attributable to a confounding factor that was not measured.
Should our patient continue her SSRI through pregnancy?
Our patient has recurrent depression, and her risk of relapse with antidepressant cessation is high. Though previous, less well-done studies suggested a small risk of congenital heart defects, recent larger high-quality studies provide significant reassurance that SSRI use in pregnancy is not strongly associated with cardiac malformations. Recent studies also show no association with miscarriage or autism spectrum disorder, though there may be risk of offspring ADHD.
She can be counseled that she may continue on her SSRI during pregnancy and can be reassured that the risk to her baby is small compared with her risk of recurrent or postpartum depression.
NONHORMONAL TREATMENT FOR VASOMOTOR SYMPTOMS OF MENOPAUSE
You see a patient who is struggling with symptoms of menopause. She tells you she has terrible hot flashes day and night, and she would like to try drug therapy. She does not want hormone replacement therapy because she is worried about the risk of adverse events. Are there safe and effective nonhormonal pharmacologic treatments for her vasomotor symptoms?
Paroxetine 7.5 mg is approved for vasomotor symptoms of menopause
As many as 75% of menopausal women in the United States experience vasomotor symptoms related to menopause, or hot flashes and night sweats.12 These symptoms can disrupt sleep and negatively affect quality of life. Though previously thought to occur during a short and self-limited time period, a recently published large observational study reported the median duration of vasomotor symptoms was 7.4 years, and in African American women in the cohort the median duration of vasomotor symptoms was 10.1 years—an entire decade of life.13
In 2013, the US Food and Drug Administration (FDA) approved paroxetine 7.5 mg daily for treating moderate to severe hot flashes associated with menopause. It is the only approved nonhormonal treatment for vasomotor symptoms; the only other approved treatments are estrogen therapy for women who have had a hysterectomy and combination estrogen-progesterone therapy for women who have not had a hysterectomy.
Further studies of paroxetine for menopausal symptoms
Since its approval, further studies have been published supporting the use of paroxetine 7.5 mg in treating symptoms of menopause. In addition to reducing hot flashes, this treatment also improves sleep disturbance in women with menopause.14
Pinkerton et al,14 in a pooled analysis of the data from the phase 3 clinical trials of paroxetine 7.5 mg per day, found that participants in groups assigned to paroxetine reported a 62% reduction in nighttime awakenings due to hot flashes compared with a 43% reduction in the placebo group (P < .001). Those who took paroxetine also reported a statistically significantly greater increase in duration of sleep than those who took placebo (37 minutes in the treatment group vs 27 minutes in the placebo group, P = .03).
Some patients are hesitant to take an SSRI because of concerns about adverse effects when used for psychiatric conditions. However, the dose of paroxetine that was studied and approved for vasomotor symptoms is lower than doses used for psychiatric indications and does not appear to be associated with these adverse effects.
Portman et al15 in 2014 examined the effect of paroxetine 7.5 mg vs placebo on weight gain and sexual function in women with vasomotor symptoms of menopause and found no significant increase in weight or decrease in sexual function at 24 weeks of use. Participants were weighed during study visits, and those in the paroxetine group gained on average 0.48% from baseline at 24 weeks, compared with 0.09% in the placebo group (P = .29).
Sexual dysfunction was assessed using the Arizona Sexual Experience Scale, which has been validated in psychiatric patients using antidepressants, and there was no significant difference in symptoms such as sex drive, sexual arousal, vaginal lubrication, or ability to achieve orgasm between the treatment group and placebo group.15
Of note, paroxetine is a potent inhibitor of the cytochrome P-450 CYP2D6 enzyme, and concurrent use of paroxetine with tamoxifen decreases tamoxifen activity.12,16 Since women with a history of breast cancer who cannot use estrogen for hot flashes may be seeking nonhormonal treatment for their vasomotor symptoms, providers should perform careful medication reconciliation and be aware that concomitant use of paroxetine and tamoxifen is not recommended.
Other antidepressants show promise but are not approved for menopausal symptoms
In addition to paroxetine, other nonhormonal drugs have been studied for treating hot flashes, but they have been unable to secure FDA approval for this indication. One of these is the serotonin-norepinephrine reuptake inhibitor venlafaxine, and a 2014 study17 confirmed its efficacy in treating menopausal vasomotor symptoms.
Joffe et al17 performed a three-armed trial comparing venlafaxine 75 mg/day, estradiol 0.5 mg/day, and placebo and found that both of the active treatments were better than placebo at reducing vasomotor symptoms. Compared with each other, estradiol 0.5 mg/day reduced hot flash frequency by an additional 0.6 events per day compared with venlafaxine 75 mg/day (P = .09). Though this difference was statistically significant, the authors pointed out that the clinical significance of such a small absolute difference is questionable. Additionally, providers should be aware that venlafaxine has little or no effect on the metabolism of tamoxifen.16
Shams et al,18 in a meta-analysis published in 2014, concluded that SSRIs as a class are more effective than placebo in treating hot flashes, supporting their widespread off-label use for this purpose. Their analysis examined the results of 11 studies, which included more than 2,000 patients in total, and found that compared with placebo, SSRI use was associated with a significant decrease in hot flashes (mean difference –0.93 events per day, 95% CI –1.49 to –0.37). A mixed treatment comparison analysis was also performed to try to model performance of individual SSRIs based on the pooled data, and the model suggests that escitalopram may be the most efficacious SSRI at reducing hot flash severity.
These studies support the effectiveness of SSRIs18 and venlafaxine17 in reducing hot flashes compared with placebo, though providers should be aware that they are still not FDA-approved for this indication.
Nonhormonal therapy for our patient
We would recommend paroxetine 7.5 mg nightly to this patient, as it is an FDA-approved nonhormonal medication that has been shown to help patients with vasomotor symptoms of menopause as well as sleep disturbance, without sexual side effects or weight gain. If the patient cannot tolerate paroxetine, off-label use of another SSRI or venlafaxine is supported by the recent literature.
HEART DISEASE IN WOMEN: CARDIAC RESYNCHRONIZATION THERAPY
A 68-year-old woman with a history of nonischemic cardiomyopathy presents for routine follow-up in your office. Despite maximal medical therapy on a beta-blocker, an angiotensin II receptor blocker, and a diuretic, she has New York Heart Association (NYHA) class III symptoms. Her most recent studies showed an ejection fraction of 30% by echocardiography and left bundle-branch block on electrocardiography, with a QRS duration of 140 ms. She recently saw her cardiologist, who recommended cardiac resynchronization therapy, and she wants your opinion as to whether or not to proceed with this recommendation. How should you counsel her?
Which patients are candidates for cardiac resynchronization therapy?
Heart disease continues to be the number one cause of death in the United States for both men and women, and almost the same number of women and men die from heart disease every year.19 Though coronary artery disease accounts for most cases of cardiovascular disease in the United States, heart failure is a significant and growing contributor. Approximately 6.6 million adults had heart failure in 2010 in the United States, and an additional 3 million are projected to have heart failure by 2030.20 The burden of disease on our health system is high, with about 1 million hospitalizations and more than 3 million outpatient office visits attributable to heart failure yearly.20
Patients with heart failure may have symptoms of dyspnea, fatigue, orthopnea, and peripheral edema; laboratory and radiologic findings of pulmonary edema, renal insufficiency, and hyponatremia; and electrocardiographic findings of atrial fibrillation or prolonged QRS.21 Intraventricular conduction delay (QRS duration > 120 ms) is associated with dyssynchronous ventricular contraction and impaired pump function and is present in almost one-third of patients who have advanced heart failure.21
Cardiac resynchronization therapy, or biventricular pacing, can improve symptoms and pump function and has been shown to decrease rates of hospitalization and death in these patients.22 According to the joint 2012 guidelines of the American College of Cardiology Foundation, American Heart Association, and Heart Rhythm Society,22 it is indicated for patients with an ejection fraction of 35% or less, left bundle-branch block with QRS duration of 150 ms or more, and NYHA class II to IV symptoms who are in sinus rhythm (class I recommendation, level of evidence A).
Studies of cardiac resynchronization therapy in women
Recently published studies have suggested that women may derive greater benefit than men from cardiac resynchronization therapy.
Zusterzeel et al23 (2014) evaluated sex-specific data from the National Cardiovascular Data Registry, which contains data on all biventricular pacemaker and implantable cardioverter-defibrillator implantations from 80% of US hospitals.23 Of the 21,152 patients who had left bundle-branch block and received cardiac resynchronization therapy, women derived greater benefit in terms of death than men did, with a 21% lower risk of death than men (adjusted hazard ratio 0.79, 95% CI 0.74–0.84, P < .001). This study was also notable in that 36% of the patients were women, whereas in most earlier studies of cardiac resynchronization therapy women accounted for only 22% to 30% of the study population.22
Goldenberg et al24 (2014) performed a follow-up analysis of the Multicenter Automatic Defibrillator Implantation Trial With Cardiac Resynchronization Therapy. Subgroup analysis showed that although both men and women had a lower risk of death if they received cardiac resynchronization therapy compared with an implantable cardioverter-defibrillator only, the magnitude of benefit may be greater for women (hazard ratio 0.48, 95% CI 0.25–0.91, P = .03) than for men (hazard ratio 0.69, 95% CI 0.50–0.95, P = .02).
In addition to deriving greater mortality benefit, women may actually benefit from cardiac resynchronization therapy at shorter QRS durations than what is currently recommended. Women have a shorter baseline QRS than men, and a smaller left ventricular cavity.25 In an FDA meta-analysis published in August 2014, pooled data from more than 4,000 patients in three studies suggested that women with left bundle-branch block benefited from cardiac resynchronization therapy more than men with left bundle-branch block.26 Neither men nor women with left bundle-branch block benefited from it if their QRS duration was less than 130 ms, and both sexes benefited from it if they had left bundle-branch block and a QRS duration longer than 150 ms. However, women who received it who had left bundle-branch block and a QRS duration of 130 to 149 ms had a significant 76% reduction in the primary composite outcome of a heart failure event or death (hazard ratio 0.24, 95% CI 0.11–0.53, P < .001), while men in the same group did not derive significant benefit (hazard ratio 0.85, 95% CI 0.60–1.21, P = .38).
Despite the increasing evidence that there are sex-specific differences in the benefit from cardiac resynchronization therapy, what we know is limited by the low rates of female enrollment in most of the studies of this treatment. In a systematic review published in 2015, Herz et al27 found that 90% of the 183 studies they reviewed enrolled 35% women or less, and half of the studies enrolled less than 23% women. Furthermore, only 20 of the 183 studies reported baseline characteristics by sex.
Recognizing this lack of adequate data, in August 2014 the FDA issued an official guidance statement outlining its expectations regarding sex-specific patient recruitment, data analysis, and data reporting in future medical device studies.28 Hopefully, with this support for sex-specific research by the FDA, future studies will be able to identify therapeutic outcome differences that may exist between male and female patients.
Should our patient receive cardiac resynchronization therapy?
Regarding our patient with heart failure, the above studies suggest she will likely have a lower risk of death if she receives cardiac resynchronization therapy, even though her QRS interval is shorter than 150 ms. Providers who are aware of the emerging data regarding sex differences and treatment response can be powerful advocates for their patients, even in subspecialty areas, as highlighted by this case. We recommend counseling this patient to proceed with cardiac resynchronization therapy.
- Evans J, Heron J, Francomb H, Oke S, Golding J. Cohort study of depressed mood during pregnancy and after childbirth. BMJ 2001; 323:257–260.
- Mitchell AA, Gilboa SM, Werler MM, Kelley KE, Louik C, Hernández-Díaz S; National Birth Defects Prevention Study. Medication use during pregnancy, with particular focus on prescription drugs: 1976–2008. Am J Obstet Gynecol 2011; 205:51.e1–e8.
- Greene MF. Teratogenicity of SSRIs—serious concern or much ado about little? N Engl J Med 2007; 356:2732–2733.
- Louik C, Lin AE, Werler MM, Hernández-Díaz S, Mitchell AA. First-trimester use of selective serotonin-reuptake inhibitors and the risk of birth defects. N Engl J Med 2007; 356:2675–2683.
- Alwan S, Reefhuis J, Rasmussen SA, Olney RS, Friedman JM; National Birth Defects Prevention Study. Use of selective serotonin-reuptake inhibitors in pregnancy and the risk of birth defects. N Engl J Med 2007; 356:2684–2692.
- Pedersen LH, Henriksen TB, Vestergaard M, Olsen J, Bech BH. Selective serotonin reuptake inhibitors in pregnancy and congenital malformations: population based cohort study. BMJ 2009; 339:b3569.
- Furu K, Kieler H, Haglund B, et al. Selective serotonin reuptake inhibitors and venlafaxine in early pregnancy and risk of birth defects: population based cohort study and sibling design. BMJ 2015; 350:h1798.
- Huybrechts KF, Palmsten K, Avorn J, et al. Antidepressant use in pregnancy and the risk of cardiac defects. N Engl J Med 2014; 370:2397–2407.
- Bérard A, Zhao J-P, Sheehy O. Sertraline use during pregnancy and the risk of major malformations. Am J Obstet Gynecol 2015; 212:795.e1–795.e12.
- Clements CC, Castro VM, Blumenthal SR, et al. Prenatal antidepressant exposure is associated with risk for attention-deficit hyperactivity disorder but not autism spectrum disorder in a large health system. Mol Psychiatry 2015; 20:727–734.
- Andersen JT, Andersen NL, Horwitz H, Poulsen HE, Jimenez-Solem E. Exposure to selective serotonin reuptake inhibitors in early pregnancy and the risk of miscarriage. Obstet Gynecol 2014; 124:655–661.
- Orleans RJ, Li L, Kim M-J, et al. FDA approval of paroxetine for menopausal hot flushes. N Engl J Med 2014; 370:1777–1779.
- Avis NE, Crawford SL, Greendale G, et al; Study of Women’s Health Across the Nation. Duration of menopausal vasomotor symptoms over the menopause transition. JAMA Intern Med 2015; 175:531–539.
- Pinkerton JV, Joffe H, Kazempour K, Mekonnen H, Bhaskar S, Lippman J. Low-dose paroxetine (7.5 mg) improves sleep in women with vasomotor symptoms associated with menopause. Menopause 2015; 22:50–58.
- Portman DJ, Kaunitz AM, Kazempour K, Mekonnen H, Bhaskar S, Lippman J. Effects of low-dose paroxetine 7.5 mg on weight and sexual function during treatment of vasomotor symptoms associated with menopause. Menopause 2014; 21:1082–1090.
- Desmarais JE, Looper KJ. Interactions between tamoxifen and antidepressants via cytochrome P450 2D6. J Clin Psychiatry 2009; 70:1688–1697.
- Joffe H, Guthrie KA, LaCroix AZ, et al. Low-dose estradiol and the serotonin-norepinephrine reuptake inhibitor venlafaxine for vasomotor symptoms: a randomized clinical trial. JAMA Intern Med 2014; 174:1058–1066.
- Shams T, Firwana B, Habib F, et al. SSRIs for hot flashes: a systematic review and meta-analysis of randomized trials. J Gen Intern Med 2014; 29:204–213.
- Kochanek KD, Xu J, Murphy SL, Minino AM, Kung H-C. Deaths: final data for 2009. Nat Vital Stat Rep 2012; 60(3):1–117.
- Roger VL, Go AS, Lloyd-Jones DM, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—-2012 update: a report from the American Heart Association. Circulation 2012; 125:e2–e220.
- McMurray JJV. Clinical practice. Systolic heart failure. N Engl J Med 2010; 362:228–238.
- Tracy CM, Epstein AE, Darbar D, et al. 2012 ACCF/AHA/HRS focused update incorporated into the ACCF/AHA/HRS 2008 guidelines for device-based therapy of cardiac rhythm abnormalities: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol 2013; 61:e6–e75.
- Zusterzeel R, Curtis JP, Canos DA, et al. Sex-specific mortality risk by QRS morphology and duration in patients receiving CRT. J Am Coll Cardiol 2014; 64:887–894.
- Goldenberg I, Kutyifa V, Klein HU, et al. Survival with cardiac-resynchronization therapy in mild heart failure. N Engl J Med 2014; 370:1694–1701.
- Dec GW. Leaning toward a better understanding of CRT in women. J Am Coll Cardiol 2014; 64:895–897.
- Zusterzeel R, Selzman KA, Sanders WE, et al. Cardiac resynchronization therapy in women: US Food and Drug Administration meta-analysis of patient-level data. JAMA Intern Med 2014; 174:1340–1348.
- Herz ND, Engeda J, Zusterzeel R, et al. Sex differences in device therapy for heart failure: utilization, outcomes, and adverse events. J Women’s Health 2015; 24:261–271.
- U.S. Department of Health and Human Services, Food and Drug Administration. Evaluation of sex-specific data in medical device clinical studies: guidance for industry and Food and Drug Administration staff. 2014; 1–30. www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/UCM283707.pdf. Accessed October 1, 2015.
Women's health encompasses a broad range of issues unique to the female patient, with a scope that has expanded beyond reproductive health. Providers who care for women must develop cross-disciplinary competencies and understand the complex role of sex and gender on disease expression and treatment outcomes. Staying current with the literature in this rapidly changing field can be challenging for the busy clinician.
This article reviews recent advances in the treatment of depression in pregnancy, nonhormonal therapies for menopausal symptoms, and heart failure therapy in women, highlighting notable studies published in 2014 and early 2015.
TREATMENT OF DEPRESSION IN PREGNANCY
A 32-year-old woman with well-controlled but recurrent depression presents to the clinic for preconception counseling. Her depression has been successfully managed with a selective serotonin reuptake inhibitor (SSRI). She and her husband would like to try to conceive soon, but she is worried that continuing on her current SSRI may harm her baby. How should you advise her?
Concern for teratogenic effects of SSRIs
Depression is common during pregnancy: 11.8% to 13.5% of pregnant women report symptoms of depression,1 and 7.5% of pregnant women take an antidepressant.2
SSRI use during pregnancy has drawn attention because of mixed reports of teratogenic effects on the newborn, such as omphalocele, congenital heart defects, and craniosynostosis.3 Previous observational studies have specifically linked paroxetine to small but significant increases in right ventricular outflow tract obstruction4,5 and have linked sertraline to ventricular septal defects.6
However, reports of associations of congenital malformations and SSRI use in pregnancy in observational studies have been questioned, with concern that these studies had low statistical power, self-reported data leading to recall bias, and limited assessment for confounding factors.3,7
Recent studies refute risk of cardiac malformations
Several newer studies have been published that further examine the association between SSRI use in pregnancy and congenital heart defects, and their findings suggest that once adjusted for confounding variables, SSRI use in pregnancy may not be associated with cardiac malformations.
Huybrechts et al,8 in a large study published in 2014, extracted data on 950,000 pregnant women from the Medicaid database over a 7-year period and examined it for SSRI use during the first 90 days of pregnancy. Though SSRI use was associated with cardiac malformations when unadjusted for confounding variables (unadjusted relative risk 1.25, 95% confidence interval [CI] 1.13–1.38), once the cohort was restricted to women with a diagnosis of only depression and was adjusted based on propensity scoring, the association was no longer statistically significant (adjusted relative risk 1.06, 95% CI 0.93–1.22).
Additionally, there was no association between sertraline and ventricular septal defects (63 cases in 14,040 women exposed to sertraline, adjusted relative risk 1.04, 95% CI 0.76–1.41), or between paroxetine and right ventricular outflow tract obstruction (93 cases in 11,126 women exposed to paroxetine, adjusted relative risk 1.07, 95% CI 0.59–1.93).8
Furu et al7 conducted a sibling-matched case-control comparison published in 2015, in which more than 2 million live births from five Nordic countries were examined in the full cohort study and 2,288 births in the sibling-matched case-control cohort. SSRI or venlafaxine use in the first 90 days of pregnancy was examined. There was a slightly higher rate of cardiac defects in infants born to SSRI or venlafaxine recipients in the cohort study (adjusted odds ratio 1.15, 95% CI 1.05–1.26). However, in the sibling-controlled analyses, neither an SSRI nor venlafaxine was associated with heart defects (adjusted odds ratio 0.92, 95% CI 0.72–1.17), leading the authors to conclude that there might be familial factors or other lifestyle factors that were not taken into consideration and that could have confounded the cohort results.
Bérard et al9 examined antidepressant use in the first trimester of pregnancy in a cohort of women in Canada and concluded that sertraline was associated with congenital atrial and ventricular defects (risk ratio 1.34; 95% CI 1.02–1.76).9 However, this association should be interpreted with caution, as the Canadian cohort was notably smaller than those in other studies we have discussed, with only 18,493 pregnancies in the total cohort, and this conclusion was drawn from 9 cases of ventricular or atrial septal defects in babies of 366 women exposed to sertraline.
Although at first glance SSRIs may appear to be associated with congenital heart defects, these recent studies are reassuring and suggest that the association may actually not be significant. As with any statistical analysis, thoughtful study design, adequate statistical power, and adjustment for confounding factors must be considered before drawing conclusions.
SSRIs, offspring psychiatric outcomes, and miscarriage rates
Clements et al10 studied a cohort extracted from Partners Healthcare consisting of newborns with autism spectrum disorder, newborns with attention-deficit hyperactivity disorder (ADHD), and healthy matched controls and found that SSRI use during pregnancy was not associated with offspring autism spectrum disorder (adjusted odds ratio 1.10, 95% CI 0.7–1.70). However, they did find an increased risk of ADHD with SSRI use during pregnancy (adjusted odds ratio 1.81, 95% CI 1.22–2.70).
Andersen et al11 examined more than 1 million pregnancies in Denmark and found no difference in risk of miscarriage between women who used an SSRI during pregnancy (adjusted hazard ratio 1.27) and women who discontinued their SSRI at least 3 months before pregnancy (adjusted hazard ratio 1.24, P = .47). The authors concluded that because of the similar rate of miscarriage in both groups, there was no association between SSRI use and miscarriage, and that the small increased risk of miscarriage in both groups could have been attributable to a confounding factor that was not measured.
Should our patient continue her SSRI through pregnancy?
Our patient has recurrent depression, and her risk of relapse with antidepressant cessation is high. Though previous, less well-done studies suggested a small risk of congenital heart defects, recent larger high-quality studies provide significant reassurance that SSRI use in pregnancy is not strongly associated with cardiac malformations. Recent studies also show no association with miscarriage or autism spectrum disorder, though there may be risk of offspring ADHD.
She can be counseled that she may continue on her SSRI during pregnancy and can be reassured that the risk to her baby is small compared with her risk of recurrent or postpartum depression.
NONHORMONAL TREATMENT FOR VASOMOTOR SYMPTOMS OF MENOPAUSE
You see a patient who is struggling with symptoms of menopause. She tells you she has terrible hot flashes day and night, and she would like to try drug therapy. She does not want hormone replacement therapy because she is worried about the risk of adverse events. Are there safe and effective nonhormonal pharmacologic treatments for her vasomotor symptoms?
Paroxetine 7.5 mg is approved for vasomotor symptoms of menopause
As many as 75% of menopausal women in the United States experience vasomotor symptoms related to menopause, or hot flashes and night sweats.12 These symptoms can disrupt sleep and negatively affect quality of life. Though previously thought to occur during a short and self-limited time period, a recently published large observational study reported the median duration of vasomotor symptoms was 7.4 years, and in African American women in the cohort the median duration of vasomotor symptoms was 10.1 years—an entire decade of life.13
In 2013, the US Food and Drug Administration (FDA) approved paroxetine 7.5 mg daily for treating moderate to severe hot flashes associated with menopause. It is the only approved nonhormonal treatment for vasomotor symptoms; the only other approved treatments are estrogen therapy for women who have had a hysterectomy and combination estrogen-progesterone therapy for women who have not had a hysterectomy.
Further studies of paroxetine for menopausal symptoms
Since its approval, further studies have been published supporting the use of paroxetine 7.5 mg in treating symptoms of menopause. In addition to reducing hot flashes, this treatment also improves sleep disturbance in women with menopause.14
Pinkerton et al,14 in a pooled analysis of the data from the phase 3 clinical trials of paroxetine 7.5 mg per day, found that participants in groups assigned to paroxetine reported a 62% reduction in nighttime awakenings due to hot flashes compared with a 43% reduction in the placebo group (P < .001). Those who took paroxetine also reported a statistically significantly greater increase in duration of sleep than those who took placebo (37 minutes in the treatment group vs 27 minutes in the placebo group, P = .03).
Some patients are hesitant to take an SSRI because of concerns about adverse effects when used for psychiatric conditions. However, the dose of paroxetine that was studied and approved for vasomotor symptoms is lower than doses used for psychiatric indications and does not appear to be associated with these adverse effects.
Portman et al15 in 2014 examined the effect of paroxetine 7.5 mg vs placebo on weight gain and sexual function in women with vasomotor symptoms of menopause and found no significant increase in weight or decrease in sexual function at 24 weeks of use. Participants were weighed during study visits, and those in the paroxetine group gained on average 0.48% from baseline at 24 weeks, compared with 0.09% in the placebo group (P = .29).
Sexual dysfunction was assessed using the Arizona Sexual Experience Scale, which has been validated in psychiatric patients using antidepressants, and there was no significant difference in symptoms such as sex drive, sexual arousal, vaginal lubrication, or ability to achieve orgasm between the treatment group and placebo group.15
Of note, paroxetine is a potent inhibitor of the cytochrome P-450 CYP2D6 enzyme, and concurrent use of paroxetine with tamoxifen decreases tamoxifen activity.12,16 Since women with a history of breast cancer who cannot use estrogen for hot flashes may be seeking nonhormonal treatment for their vasomotor symptoms, providers should perform careful medication reconciliation and be aware that concomitant use of paroxetine and tamoxifen is not recommended.
Other antidepressants show promise but are not approved for menopausal symptoms
In addition to paroxetine, other nonhormonal drugs have been studied for treating hot flashes, but they have been unable to secure FDA approval for this indication. One of these is the serotonin-norepinephrine reuptake inhibitor venlafaxine, and a 2014 study17 confirmed its efficacy in treating menopausal vasomotor symptoms.
Joffe et al17 performed a three-armed trial comparing venlafaxine 75 mg/day, estradiol 0.5 mg/day, and placebo and found that both of the active treatments were better than placebo at reducing vasomotor symptoms. Compared with each other, estradiol 0.5 mg/day reduced hot flash frequency by an additional 0.6 events per day compared with venlafaxine 75 mg/day (P = .09). Though this difference was statistically significant, the authors pointed out that the clinical significance of such a small absolute difference is questionable. Additionally, providers should be aware that venlafaxine has little or no effect on the metabolism of tamoxifen.16
Shams et al,18 in a meta-analysis published in 2014, concluded that SSRIs as a class are more effective than placebo in treating hot flashes, supporting their widespread off-label use for this purpose. Their analysis examined the results of 11 studies, which included more than 2,000 patients in total, and found that compared with placebo, SSRI use was associated with a significant decrease in hot flashes (mean difference –0.93 events per day, 95% CI –1.49 to –0.37). A mixed treatment comparison analysis was also performed to try to model performance of individual SSRIs based on the pooled data, and the model suggests that escitalopram may be the most efficacious SSRI at reducing hot flash severity.
These studies support the effectiveness of SSRIs18 and venlafaxine17 in reducing hot flashes compared with placebo, though providers should be aware that they are still not FDA-approved for this indication.
Nonhormonal therapy for our patient
We would recommend paroxetine 7.5 mg nightly to this patient, as it is an FDA-approved nonhormonal medication that has been shown to help patients with vasomotor symptoms of menopause as well as sleep disturbance, without sexual side effects or weight gain. If the patient cannot tolerate paroxetine, off-label use of another SSRI or venlafaxine is supported by the recent literature.
HEART DISEASE IN WOMEN: CARDIAC RESYNCHRONIZATION THERAPY
A 68-year-old woman with a history of nonischemic cardiomyopathy presents for routine follow-up in your office. Despite maximal medical therapy on a beta-blocker, an angiotensin II receptor blocker, and a diuretic, she has New York Heart Association (NYHA) class III symptoms. Her most recent studies showed an ejection fraction of 30% by echocardiography and left bundle-branch block on electrocardiography, with a QRS duration of 140 ms. She recently saw her cardiologist, who recommended cardiac resynchronization therapy, and she wants your opinion as to whether or not to proceed with this recommendation. How should you counsel her?
Which patients are candidates for cardiac resynchronization therapy?
Heart disease continues to be the number one cause of death in the United States for both men and women, and almost the same number of women and men die from heart disease every year.19 Though coronary artery disease accounts for most cases of cardiovascular disease in the United States, heart failure is a significant and growing contributor. Approximately 6.6 million adults had heart failure in 2010 in the United States, and an additional 3 million are projected to have heart failure by 2030.20 The burden of disease on our health system is high, with about 1 million hospitalizations and more than 3 million outpatient office visits attributable to heart failure yearly.20
Patients with heart failure may have symptoms of dyspnea, fatigue, orthopnea, and peripheral edema; laboratory and radiologic findings of pulmonary edema, renal insufficiency, and hyponatremia; and electrocardiographic findings of atrial fibrillation or prolonged QRS.21 Intraventricular conduction delay (QRS duration > 120 ms) is associated with dyssynchronous ventricular contraction and impaired pump function and is present in almost one-third of patients who have advanced heart failure.21
Cardiac resynchronization therapy, or biventricular pacing, can improve symptoms and pump function and has been shown to decrease rates of hospitalization and death in these patients.22 According to the joint 2012 guidelines of the American College of Cardiology Foundation, American Heart Association, and Heart Rhythm Society,22 it is indicated for patients with an ejection fraction of 35% or less, left bundle-branch block with QRS duration of 150 ms or more, and NYHA class II to IV symptoms who are in sinus rhythm (class I recommendation, level of evidence A).
Studies of cardiac resynchronization therapy in women
Recently published studies have suggested that women may derive greater benefit than men from cardiac resynchronization therapy.
Zusterzeel et al23 (2014) evaluated sex-specific data from the National Cardiovascular Data Registry, which contains data on all biventricular pacemaker and implantable cardioverter-defibrillator implantations from 80% of US hospitals.23 Of the 21,152 patients who had left bundle-branch block and received cardiac resynchronization therapy, women derived greater benefit in terms of death than men did, with a 21% lower risk of death than men (adjusted hazard ratio 0.79, 95% CI 0.74–0.84, P < .001). This study was also notable in that 36% of the patients were women, whereas in most earlier studies of cardiac resynchronization therapy women accounted for only 22% to 30% of the study population.22
Goldenberg et al24 (2014) performed a follow-up analysis of the Multicenter Automatic Defibrillator Implantation Trial With Cardiac Resynchronization Therapy. Subgroup analysis showed that although both men and women had a lower risk of death if they received cardiac resynchronization therapy compared with an implantable cardioverter-defibrillator only, the magnitude of benefit may be greater for women (hazard ratio 0.48, 95% CI 0.25–0.91, P = .03) than for men (hazard ratio 0.69, 95% CI 0.50–0.95, P = .02).
In addition to deriving greater mortality benefit, women may actually benefit from cardiac resynchronization therapy at shorter QRS durations than what is currently recommended. Women have a shorter baseline QRS than men, and a smaller left ventricular cavity.25 In an FDA meta-analysis published in August 2014, pooled data from more than 4,000 patients in three studies suggested that women with left bundle-branch block benefited from cardiac resynchronization therapy more than men with left bundle-branch block.26 Neither men nor women with left bundle-branch block benefited from it if their QRS duration was less than 130 ms, and both sexes benefited from it if they had left bundle-branch block and a QRS duration longer than 150 ms. However, women who received it who had left bundle-branch block and a QRS duration of 130 to 149 ms had a significant 76% reduction in the primary composite outcome of a heart failure event or death (hazard ratio 0.24, 95% CI 0.11–0.53, P < .001), while men in the same group did not derive significant benefit (hazard ratio 0.85, 95% CI 0.60–1.21, P = .38).
Despite the increasing evidence that there are sex-specific differences in the benefit from cardiac resynchronization therapy, what we know is limited by the low rates of female enrollment in most of the studies of this treatment. In a systematic review published in 2015, Herz et al27 found that 90% of the 183 studies they reviewed enrolled 35% women or less, and half of the studies enrolled less than 23% women. Furthermore, only 20 of the 183 studies reported baseline characteristics by sex.
Recognizing this lack of adequate data, in August 2014 the FDA issued an official guidance statement outlining its expectations regarding sex-specific patient recruitment, data analysis, and data reporting in future medical device studies.28 Hopefully, with this support for sex-specific research by the FDA, future studies will be able to identify therapeutic outcome differences that may exist between male and female patients.
Should our patient receive cardiac resynchronization therapy?
Regarding our patient with heart failure, the above studies suggest she will likely have a lower risk of death if she receives cardiac resynchronization therapy, even though her QRS interval is shorter than 150 ms. Providers who are aware of the emerging data regarding sex differences and treatment response can be powerful advocates for their patients, even in subspecialty areas, as highlighted by this case. We recommend counseling this patient to proceed with cardiac resynchronization therapy.
Women's health encompasses a broad range of issues unique to the female patient, with a scope that has expanded beyond reproductive health. Providers who care for women must develop cross-disciplinary competencies and understand the complex role of sex and gender on disease expression and treatment outcomes. Staying current with the literature in this rapidly changing field can be challenging for the busy clinician.
This article reviews recent advances in the treatment of depression in pregnancy, nonhormonal therapies for menopausal symptoms, and heart failure therapy in women, highlighting notable studies published in 2014 and early 2015.
TREATMENT OF DEPRESSION IN PREGNANCY
A 32-year-old woman with well-controlled but recurrent depression presents to the clinic for preconception counseling. Her depression has been successfully managed with a selective serotonin reuptake inhibitor (SSRI). She and her husband would like to try to conceive soon, but she is worried that continuing on her current SSRI may harm her baby. How should you advise her?
Concern for teratogenic effects of SSRIs
Depression is common during pregnancy: 11.8% to 13.5% of pregnant women report symptoms of depression,1 and 7.5% of pregnant women take an antidepressant.2
SSRI use during pregnancy has drawn attention because of mixed reports of teratogenic effects on the newborn, such as omphalocele, congenital heart defects, and craniosynostosis.3 Previous observational studies have specifically linked paroxetine to small but significant increases in right ventricular outflow tract obstruction4,5 and have linked sertraline to ventricular septal defects.6
However, reports of associations of congenital malformations and SSRI use in pregnancy in observational studies have been questioned, with concern that these studies had low statistical power, self-reported data leading to recall bias, and limited assessment for confounding factors.3,7
Recent studies refute risk of cardiac malformations
Several newer studies have been published that further examine the association between SSRI use in pregnancy and congenital heart defects, and their findings suggest that once adjusted for confounding variables, SSRI use in pregnancy may not be associated with cardiac malformations.
Huybrechts et al,8 in a large study published in 2014, extracted data on 950,000 pregnant women from the Medicaid database over a 7-year period and examined it for SSRI use during the first 90 days of pregnancy. Though SSRI use was associated with cardiac malformations when unadjusted for confounding variables (unadjusted relative risk 1.25, 95% confidence interval [CI] 1.13–1.38), once the cohort was restricted to women with a diagnosis of only depression and was adjusted based on propensity scoring, the association was no longer statistically significant (adjusted relative risk 1.06, 95% CI 0.93–1.22).
Additionally, there was no association between sertraline and ventricular septal defects (63 cases in 14,040 women exposed to sertraline, adjusted relative risk 1.04, 95% CI 0.76–1.41), or between paroxetine and right ventricular outflow tract obstruction (93 cases in 11,126 women exposed to paroxetine, adjusted relative risk 1.07, 95% CI 0.59–1.93).8
Furu et al7 conducted a sibling-matched case-control comparison published in 2015, in which more than 2 million live births from five Nordic countries were examined in the full cohort study and 2,288 births in the sibling-matched case-control cohort. SSRI or venlafaxine use in the first 90 days of pregnancy was examined. There was a slightly higher rate of cardiac defects in infants born to SSRI or venlafaxine recipients in the cohort study (adjusted odds ratio 1.15, 95% CI 1.05–1.26). However, in the sibling-controlled analyses, neither an SSRI nor venlafaxine was associated with heart defects (adjusted odds ratio 0.92, 95% CI 0.72–1.17), leading the authors to conclude that there might be familial factors or other lifestyle factors that were not taken into consideration and that could have confounded the cohort results.
Bérard et al9 examined antidepressant use in the first trimester of pregnancy in a cohort of women in Canada and concluded that sertraline was associated with congenital atrial and ventricular defects (risk ratio 1.34; 95% CI 1.02–1.76).9 However, this association should be interpreted with caution, as the Canadian cohort was notably smaller than those in other studies we have discussed, with only 18,493 pregnancies in the total cohort, and this conclusion was drawn from 9 cases of ventricular or atrial septal defects in babies of 366 women exposed to sertraline.
Although at first glance SSRIs may appear to be associated with congenital heart defects, these recent studies are reassuring and suggest that the association may actually not be significant. As with any statistical analysis, thoughtful study design, adequate statistical power, and adjustment for confounding factors must be considered before drawing conclusions.
SSRIs, offspring psychiatric outcomes, and miscarriage rates
Clements et al10 studied a cohort extracted from Partners Healthcare consisting of newborns with autism spectrum disorder, newborns with attention-deficit hyperactivity disorder (ADHD), and healthy matched controls and found that SSRI use during pregnancy was not associated with offspring autism spectrum disorder (adjusted odds ratio 1.10, 95% CI 0.7–1.70). However, they did find an increased risk of ADHD with SSRI use during pregnancy (adjusted odds ratio 1.81, 95% CI 1.22–2.70).
Andersen et al11 examined more than 1 million pregnancies in Denmark and found no difference in risk of miscarriage between women who used an SSRI during pregnancy (adjusted hazard ratio 1.27) and women who discontinued their SSRI at least 3 months before pregnancy (adjusted hazard ratio 1.24, P = .47). The authors concluded that because of the similar rate of miscarriage in both groups, there was no association between SSRI use and miscarriage, and that the small increased risk of miscarriage in both groups could have been attributable to a confounding factor that was not measured.
Should our patient continue her SSRI through pregnancy?
Our patient has recurrent depression, and her risk of relapse with antidepressant cessation is high. Though previous, less well-done studies suggested a small risk of congenital heart defects, recent larger high-quality studies provide significant reassurance that SSRI use in pregnancy is not strongly associated with cardiac malformations. Recent studies also show no association with miscarriage or autism spectrum disorder, though there may be risk of offspring ADHD.
She can be counseled that she may continue on her SSRI during pregnancy and can be reassured that the risk to her baby is small compared with her risk of recurrent or postpartum depression.
NONHORMONAL TREATMENT FOR VASOMOTOR SYMPTOMS OF MENOPAUSE
You see a patient who is struggling with symptoms of menopause. She tells you she has terrible hot flashes day and night, and she would like to try drug therapy. She does not want hormone replacement therapy because she is worried about the risk of adverse events. Are there safe and effective nonhormonal pharmacologic treatments for her vasomotor symptoms?
Paroxetine 7.5 mg is approved for vasomotor symptoms of menopause
As many as 75% of menopausal women in the United States experience vasomotor symptoms related to menopause, or hot flashes and night sweats.12 These symptoms can disrupt sleep and negatively affect quality of life. Though previously thought to occur during a short and self-limited time period, a recently published large observational study reported the median duration of vasomotor symptoms was 7.4 years, and in African American women in the cohort the median duration of vasomotor symptoms was 10.1 years—an entire decade of life.13
In 2013, the US Food and Drug Administration (FDA) approved paroxetine 7.5 mg daily for treating moderate to severe hot flashes associated with menopause. It is the only approved nonhormonal treatment for vasomotor symptoms; the only other approved treatments are estrogen therapy for women who have had a hysterectomy and combination estrogen-progesterone therapy for women who have not had a hysterectomy.
Further studies of paroxetine for menopausal symptoms
Since its approval, further studies have been published supporting the use of paroxetine 7.5 mg in treating symptoms of menopause. In addition to reducing hot flashes, this treatment also improves sleep disturbance in women with menopause.14
Pinkerton et al,14 in a pooled analysis of the data from the phase 3 clinical trials of paroxetine 7.5 mg per day, found that participants in groups assigned to paroxetine reported a 62% reduction in nighttime awakenings due to hot flashes compared with a 43% reduction in the placebo group (P < .001). Those who took paroxetine also reported a statistically significantly greater increase in duration of sleep than those who took placebo (37 minutes in the treatment group vs 27 minutes in the placebo group, P = .03).
Some patients are hesitant to take an SSRI because of concerns about adverse effects when used for psychiatric conditions. However, the dose of paroxetine that was studied and approved for vasomotor symptoms is lower than doses used for psychiatric indications and does not appear to be associated with these adverse effects.
Portman et al15 in 2014 examined the effect of paroxetine 7.5 mg vs placebo on weight gain and sexual function in women with vasomotor symptoms of menopause and found no significant increase in weight or decrease in sexual function at 24 weeks of use. Participants were weighed during study visits, and those in the paroxetine group gained on average 0.48% from baseline at 24 weeks, compared with 0.09% in the placebo group (P = .29).
Sexual dysfunction was assessed using the Arizona Sexual Experience Scale, which has been validated in psychiatric patients using antidepressants, and there was no significant difference in symptoms such as sex drive, sexual arousal, vaginal lubrication, or ability to achieve orgasm between the treatment group and placebo group.15
Of note, paroxetine is a potent inhibitor of the cytochrome P-450 CYP2D6 enzyme, and concurrent use of paroxetine with tamoxifen decreases tamoxifen activity.12,16 Since women with a history of breast cancer who cannot use estrogen for hot flashes may be seeking nonhormonal treatment for their vasomotor symptoms, providers should perform careful medication reconciliation and be aware that concomitant use of paroxetine and tamoxifen is not recommended.
Other antidepressants show promise but are not approved for menopausal symptoms
In addition to paroxetine, other nonhormonal drugs have been studied for treating hot flashes, but they have been unable to secure FDA approval for this indication. One of these is the serotonin-norepinephrine reuptake inhibitor venlafaxine, and a 2014 study17 confirmed its efficacy in treating menopausal vasomotor symptoms.
Joffe et al17 performed a three-armed trial comparing venlafaxine 75 mg/day, estradiol 0.5 mg/day, and placebo and found that both of the active treatments were better than placebo at reducing vasomotor symptoms. Compared with each other, estradiol 0.5 mg/day reduced hot flash frequency by an additional 0.6 events per day compared with venlafaxine 75 mg/day (P = .09). Though this difference was statistically significant, the authors pointed out that the clinical significance of such a small absolute difference is questionable. Additionally, providers should be aware that venlafaxine has little or no effect on the metabolism of tamoxifen.16
Shams et al,18 in a meta-analysis published in 2014, concluded that SSRIs as a class are more effective than placebo in treating hot flashes, supporting their widespread off-label use for this purpose. Their analysis examined the results of 11 studies, which included more than 2,000 patients in total, and found that compared with placebo, SSRI use was associated with a significant decrease in hot flashes (mean difference –0.93 events per day, 95% CI –1.49 to –0.37). A mixed treatment comparison analysis was also performed to try to model performance of individual SSRIs based on the pooled data, and the model suggests that escitalopram may be the most efficacious SSRI at reducing hot flash severity.
These studies support the effectiveness of SSRIs18 and venlafaxine17 in reducing hot flashes compared with placebo, though providers should be aware that they are still not FDA-approved for this indication.
Nonhormonal therapy for our patient
We would recommend paroxetine 7.5 mg nightly to this patient, as it is an FDA-approved nonhormonal medication that has been shown to help patients with vasomotor symptoms of menopause as well as sleep disturbance, without sexual side effects or weight gain. If the patient cannot tolerate paroxetine, off-label use of another SSRI or venlafaxine is supported by the recent literature.
HEART DISEASE IN WOMEN: CARDIAC RESYNCHRONIZATION THERAPY
A 68-year-old woman with a history of nonischemic cardiomyopathy presents for routine follow-up in your office. Despite maximal medical therapy on a beta-blocker, an angiotensin II receptor blocker, and a diuretic, she has New York Heart Association (NYHA) class III symptoms. Her most recent studies showed an ejection fraction of 30% by echocardiography and left bundle-branch block on electrocardiography, with a QRS duration of 140 ms. She recently saw her cardiologist, who recommended cardiac resynchronization therapy, and she wants your opinion as to whether or not to proceed with this recommendation. How should you counsel her?
Which patients are candidates for cardiac resynchronization therapy?
Heart disease continues to be the number one cause of death in the United States for both men and women, and almost the same number of women and men die from heart disease every year.19 Though coronary artery disease accounts for most cases of cardiovascular disease in the United States, heart failure is a significant and growing contributor. Approximately 6.6 million adults had heart failure in 2010 in the United States, and an additional 3 million are projected to have heart failure by 2030.20 The burden of disease on our health system is high, with about 1 million hospitalizations and more than 3 million outpatient office visits attributable to heart failure yearly.20
Patients with heart failure may have symptoms of dyspnea, fatigue, orthopnea, and peripheral edema; laboratory and radiologic findings of pulmonary edema, renal insufficiency, and hyponatremia; and electrocardiographic findings of atrial fibrillation or prolonged QRS.21 Intraventricular conduction delay (QRS duration > 120 ms) is associated with dyssynchronous ventricular contraction and impaired pump function and is present in almost one-third of patients who have advanced heart failure.21
Cardiac resynchronization therapy, or biventricular pacing, can improve symptoms and pump function and has been shown to decrease rates of hospitalization and death in these patients.22 According to the joint 2012 guidelines of the American College of Cardiology Foundation, American Heart Association, and Heart Rhythm Society,22 it is indicated for patients with an ejection fraction of 35% or less, left bundle-branch block with QRS duration of 150 ms or more, and NYHA class II to IV symptoms who are in sinus rhythm (class I recommendation, level of evidence A).
Studies of cardiac resynchronization therapy in women
Recently published studies have suggested that women may derive greater benefit than men from cardiac resynchronization therapy.
Zusterzeel et al23 (2014) evaluated sex-specific data from the National Cardiovascular Data Registry, which contains data on all biventricular pacemaker and implantable cardioverter-defibrillator implantations from 80% of US hospitals.23 Of the 21,152 patients who had left bundle-branch block and received cardiac resynchronization therapy, women derived greater benefit in terms of death than men did, with a 21% lower risk of death than men (adjusted hazard ratio 0.79, 95% CI 0.74–0.84, P < .001). This study was also notable in that 36% of the patients were women, whereas in most earlier studies of cardiac resynchronization therapy women accounted for only 22% to 30% of the study population.22
Goldenberg et al24 (2014) performed a follow-up analysis of the Multicenter Automatic Defibrillator Implantation Trial With Cardiac Resynchronization Therapy. Subgroup analysis showed that although both men and women had a lower risk of death if they received cardiac resynchronization therapy compared with an implantable cardioverter-defibrillator only, the magnitude of benefit may be greater for women (hazard ratio 0.48, 95% CI 0.25–0.91, P = .03) than for men (hazard ratio 0.69, 95% CI 0.50–0.95, P = .02).
In addition to deriving greater mortality benefit, women may actually benefit from cardiac resynchronization therapy at shorter QRS durations than what is currently recommended. Women have a shorter baseline QRS than men, and a smaller left ventricular cavity.25 In an FDA meta-analysis published in August 2014, pooled data from more than 4,000 patients in three studies suggested that women with left bundle-branch block benefited from cardiac resynchronization therapy more than men with left bundle-branch block.26 Neither men nor women with left bundle-branch block benefited from it if their QRS duration was less than 130 ms, and both sexes benefited from it if they had left bundle-branch block and a QRS duration longer than 150 ms. However, women who received it who had left bundle-branch block and a QRS duration of 130 to 149 ms had a significant 76% reduction in the primary composite outcome of a heart failure event or death (hazard ratio 0.24, 95% CI 0.11–0.53, P < .001), while men in the same group did not derive significant benefit (hazard ratio 0.85, 95% CI 0.60–1.21, P = .38).
Despite the increasing evidence that there are sex-specific differences in the benefit from cardiac resynchronization therapy, what we know is limited by the low rates of female enrollment in most of the studies of this treatment. In a systematic review published in 2015, Herz et al27 found that 90% of the 183 studies they reviewed enrolled 35% women or less, and half of the studies enrolled less than 23% women. Furthermore, only 20 of the 183 studies reported baseline characteristics by sex.
Recognizing this lack of adequate data, in August 2014 the FDA issued an official guidance statement outlining its expectations regarding sex-specific patient recruitment, data analysis, and data reporting in future medical device studies.28 Hopefully, with this support for sex-specific research by the FDA, future studies will be able to identify therapeutic outcome differences that may exist between male and female patients.
Should our patient receive cardiac resynchronization therapy?
Regarding our patient with heart failure, the above studies suggest she will likely have a lower risk of death if she receives cardiac resynchronization therapy, even though her QRS interval is shorter than 150 ms. Providers who are aware of the emerging data regarding sex differences and treatment response can be powerful advocates for their patients, even in subspecialty areas, as highlighted by this case. We recommend counseling this patient to proceed with cardiac resynchronization therapy.
- Evans J, Heron J, Francomb H, Oke S, Golding J. Cohort study of depressed mood during pregnancy and after childbirth. BMJ 2001; 323:257–260.
- Mitchell AA, Gilboa SM, Werler MM, Kelley KE, Louik C, Hernández-Díaz S; National Birth Defects Prevention Study. Medication use during pregnancy, with particular focus on prescription drugs: 1976–2008. Am J Obstet Gynecol 2011; 205:51.e1–e8.
- Greene MF. Teratogenicity of SSRIs—serious concern or much ado about little? N Engl J Med 2007; 356:2732–2733.
- Louik C, Lin AE, Werler MM, Hernández-Díaz S, Mitchell AA. First-trimester use of selective serotonin-reuptake inhibitors and the risk of birth defects. N Engl J Med 2007; 356:2675–2683.
- Alwan S, Reefhuis J, Rasmussen SA, Olney RS, Friedman JM; National Birth Defects Prevention Study. Use of selective serotonin-reuptake inhibitors in pregnancy and the risk of birth defects. N Engl J Med 2007; 356:2684–2692.
- Pedersen LH, Henriksen TB, Vestergaard M, Olsen J, Bech BH. Selective serotonin reuptake inhibitors in pregnancy and congenital malformations: population based cohort study. BMJ 2009; 339:b3569.
- Furu K, Kieler H, Haglund B, et al. Selective serotonin reuptake inhibitors and venlafaxine in early pregnancy and risk of birth defects: population based cohort study and sibling design. BMJ 2015; 350:h1798.
- Huybrechts KF, Palmsten K, Avorn J, et al. Antidepressant use in pregnancy and the risk of cardiac defects. N Engl J Med 2014; 370:2397–2407.
- Bérard A, Zhao J-P, Sheehy O. Sertraline use during pregnancy and the risk of major malformations. Am J Obstet Gynecol 2015; 212:795.e1–795.e12.
- Clements CC, Castro VM, Blumenthal SR, et al. Prenatal antidepressant exposure is associated with risk for attention-deficit hyperactivity disorder but not autism spectrum disorder in a large health system. Mol Psychiatry 2015; 20:727–734.
- Andersen JT, Andersen NL, Horwitz H, Poulsen HE, Jimenez-Solem E. Exposure to selective serotonin reuptake inhibitors in early pregnancy and the risk of miscarriage. Obstet Gynecol 2014; 124:655–661.
- Orleans RJ, Li L, Kim M-J, et al. FDA approval of paroxetine for menopausal hot flushes. N Engl J Med 2014; 370:1777–1779.
- Avis NE, Crawford SL, Greendale G, et al; Study of Women’s Health Across the Nation. Duration of menopausal vasomotor symptoms over the menopause transition. JAMA Intern Med 2015; 175:531–539.
- Pinkerton JV, Joffe H, Kazempour K, Mekonnen H, Bhaskar S, Lippman J. Low-dose paroxetine (7.5 mg) improves sleep in women with vasomotor symptoms associated with menopause. Menopause 2015; 22:50–58.
- Portman DJ, Kaunitz AM, Kazempour K, Mekonnen H, Bhaskar S, Lippman J. Effects of low-dose paroxetine 7.5 mg on weight and sexual function during treatment of vasomotor symptoms associated with menopause. Menopause 2014; 21:1082–1090.
- Desmarais JE, Looper KJ. Interactions between tamoxifen and antidepressants via cytochrome P450 2D6. J Clin Psychiatry 2009; 70:1688–1697.
- Joffe H, Guthrie KA, LaCroix AZ, et al. Low-dose estradiol and the serotonin-norepinephrine reuptake inhibitor venlafaxine for vasomotor symptoms: a randomized clinical trial. JAMA Intern Med 2014; 174:1058–1066.
- Shams T, Firwana B, Habib F, et al. SSRIs for hot flashes: a systematic review and meta-analysis of randomized trials. J Gen Intern Med 2014; 29:204–213.
- Kochanek KD, Xu J, Murphy SL, Minino AM, Kung H-C. Deaths: final data for 2009. Nat Vital Stat Rep 2012; 60(3):1–117.
- Roger VL, Go AS, Lloyd-Jones DM, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—-2012 update: a report from the American Heart Association. Circulation 2012; 125:e2–e220.
- McMurray JJV. Clinical practice. Systolic heart failure. N Engl J Med 2010; 362:228–238.
- Tracy CM, Epstein AE, Darbar D, et al. 2012 ACCF/AHA/HRS focused update incorporated into the ACCF/AHA/HRS 2008 guidelines for device-based therapy of cardiac rhythm abnormalities: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol 2013; 61:e6–e75.
- Zusterzeel R, Curtis JP, Canos DA, et al. Sex-specific mortality risk by QRS morphology and duration in patients receiving CRT. J Am Coll Cardiol 2014; 64:887–894.
- Goldenberg I, Kutyifa V, Klein HU, et al. Survival with cardiac-resynchronization therapy in mild heart failure. N Engl J Med 2014; 370:1694–1701.
- Dec GW. Leaning toward a better understanding of CRT in women. J Am Coll Cardiol 2014; 64:895–897.
- Zusterzeel R, Selzman KA, Sanders WE, et al. Cardiac resynchronization therapy in women: US Food and Drug Administration meta-analysis of patient-level data. JAMA Intern Med 2014; 174:1340–1348.
- Herz ND, Engeda J, Zusterzeel R, et al. Sex differences in device therapy for heart failure: utilization, outcomes, and adverse events. J Women’s Health 2015; 24:261–271.
- U.S. Department of Health and Human Services, Food and Drug Administration. Evaluation of sex-specific data in medical device clinical studies: guidance for industry and Food and Drug Administration staff. 2014; 1–30. www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/UCM283707.pdf. Accessed October 1, 2015.
- Evans J, Heron J, Francomb H, Oke S, Golding J. Cohort study of depressed mood during pregnancy and after childbirth. BMJ 2001; 323:257–260.
- Mitchell AA, Gilboa SM, Werler MM, Kelley KE, Louik C, Hernández-Díaz S; National Birth Defects Prevention Study. Medication use during pregnancy, with particular focus on prescription drugs: 1976–2008. Am J Obstet Gynecol 2011; 205:51.e1–e8.
- Greene MF. Teratogenicity of SSRIs—serious concern or much ado about little? N Engl J Med 2007; 356:2732–2733.
- Louik C, Lin AE, Werler MM, Hernández-Díaz S, Mitchell AA. First-trimester use of selective serotonin-reuptake inhibitors and the risk of birth defects. N Engl J Med 2007; 356:2675–2683.
- Alwan S, Reefhuis J, Rasmussen SA, Olney RS, Friedman JM; National Birth Defects Prevention Study. Use of selective serotonin-reuptake inhibitors in pregnancy and the risk of birth defects. N Engl J Med 2007; 356:2684–2692.
- Pedersen LH, Henriksen TB, Vestergaard M, Olsen J, Bech BH. Selective serotonin reuptake inhibitors in pregnancy and congenital malformations: population based cohort study. BMJ 2009; 339:b3569.
- Furu K, Kieler H, Haglund B, et al. Selective serotonin reuptake inhibitors and venlafaxine in early pregnancy and risk of birth defects: population based cohort study and sibling design. BMJ 2015; 350:h1798.
- Huybrechts KF, Palmsten K, Avorn J, et al. Antidepressant use in pregnancy and the risk of cardiac defects. N Engl J Med 2014; 370:2397–2407.
- Bérard A, Zhao J-P, Sheehy O. Sertraline use during pregnancy and the risk of major malformations. Am J Obstet Gynecol 2015; 212:795.e1–795.e12.
- Clements CC, Castro VM, Blumenthal SR, et al. Prenatal antidepressant exposure is associated with risk for attention-deficit hyperactivity disorder but not autism spectrum disorder in a large health system. Mol Psychiatry 2015; 20:727–734.
- Andersen JT, Andersen NL, Horwitz H, Poulsen HE, Jimenez-Solem E. Exposure to selective serotonin reuptake inhibitors in early pregnancy and the risk of miscarriage. Obstet Gynecol 2014; 124:655–661.
- Orleans RJ, Li L, Kim M-J, et al. FDA approval of paroxetine for menopausal hot flushes. N Engl J Med 2014; 370:1777–1779.
- Avis NE, Crawford SL, Greendale G, et al; Study of Women’s Health Across the Nation. Duration of menopausal vasomotor symptoms over the menopause transition. JAMA Intern Med 2015; 175:531–539.
- Pinkerton JV, Joffe H, Kazempour K, Mekonnen H, Bhaskar S, Lippman J. Low-dose paroxetine (7.5 mg) improves sleep in women with vasomotor symptoms associated with menopause. Menopause 2015; 22:50–58.
- Portman DJ, Kaunitz AM, Kazempour K, Mekonnen H, Bhaskar S, Lippman J. Effects of low-dose paroxetine 7.5 mg on weight and sexual function during treatment of vasomotor symptoms associated with menopause. Menopause 2014; 21:1082–1090.
- Desmarais JE, Looper KJ. Interactions between tamoxifen and antidepressants via cytochrome P450 2D6. J Clin Psychiatry 2009; 70:1688–1697.
- Joffe H, Guthrie KA, LaCroix AZ, et al. Low-dose estradiol and the serotonin-norepinephrine reuptake inhibitor venlafaxine for vasomotor symptoms: a randomized clinical trial. JAMA Intern Med 2014; 174:1058–1066.
- Shams T, Firwana B, Habib F, et al. SSRIs for hot flashes: a systematic review and meta-analysis of randomized trials. J Gen Intern Med 2014; 29:204–213.
- Kochanek KD, Xu J, Murphy SL, Minino AM, Kung H-C. Deaths: final data for 2009. Nat Vital Stat Rep 2012; 60(3):1–117.
- Roger VL, Go AS, Lloyd-Jones DM, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—-2012 update: a report from the American Heart Association. Circulation 2012; 125:e2–e220.
- McMurray JJV. Clinical practice. Systolic heart failure. N Engl J Med 2010; 362:228–238.
- Tracy CM, Epstein AE, Darbar D, et al. 2012 ACCF/AHA/HRS focused update incorporated into the ACCF/AHA/HRS 2008 guidelines for device-based therapy of cardiac rhythm abnormalities: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol 2013; 61:e6–e75.
- Zusterzeel R, Curtis JP, Canos DA, et al. Sex-specific mortality risk by QRS morphology and duration in patients receiving CRT. J Am Coll Cardiol 2014; 64:887–894.
- Goldenberg I, Kutyifa V, Klein HU, et al. Survival with cardiac-resynchronization therapy in mild heart failure. N Engl J Med 2014; 370:1694–1701.
- Dec GW. Leaning toward a better understanding of CRT in women. J Am Coll Cardiol 2014; 64:895–897.
- Zusterzeel R, Selzman KA, Sanders WE, et al. Cardiac resynchronization therapy in women: US Food and Drug Administration meta-analysis of patient-level data. JAMA Intern Med 2014; 174:1340–1348.
- Herz ND, Engeda J, Zusterzeel R, et al. Sex differences in device therapy for heart failure: utilization, outcomes, and adverse events. J Women’s Health 2015; 24:261–271.
- U.S. Department of Health and Human Services, Food and Drug Administration. Evaluation of sex-specific data in medical device clinical studies: guidance for industry and Food and Drug Administration staff. 2014; 1–30. www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/UCM283707.pdf. Accessed October 1, 2015.
KEY POINTS
- Earlier trials had raised concerns about possible teratogenic effects of selective serotonin reuptake inhibitors, but more recent trials have found no strong association between these drugs and congenital heart defects, and no association with miscarriage or autism spectrum disorder, though there may be a risk of attention deficit hyperactivity disorder in offspring.
- Paroxetine is approved for treating vasomotor symptoms of menopause, but in a lower dose (7.5 mg) than those used for depression and other psychiatric indications. Clinical trials have also shown good results with other antidepressants for treating hot flashes, but the drugs are not yet approved for this indication.
- Women with heart failure and left bundle-branch block can decrease their risk of death with cardiac resynchronization therapy more than men with the same condition. Moreover, women may benefit from this therapy even if their QRS duration is somewhat shorter than the established cutoff, ie, if it is in the range of 130 to 149 ms.

Should all patients with significant proteinuria take a renin-angiotensin inhibitor?
Most patients with proteinuria benefit from a renin-angiotensin-aldosterone system (RAAS) inhibitor. Exceptions due to adverse effects are discussed below.
WHY RAAS INHIBITORS?
RAAS inhibitors—particularly angiotensin-converting enzyme (ACE) inhibitors and angiotensin II receptor blockers (ARBs)—reduce proteinuria and slow the progression of chronic kidney disease by improving glomerular hemodynamics, restoring the altered glomerular barrier function, and limiting the nonhemodynamic effects of angiotensin II and aldosterone, such as fibrosis and vascular endothelial dysfunction.1 Studies have shown that these protective effects are, at least in part, independent of the reduction in systemic blood pressure.2,3
EVIDENCE FOR USING RAAS INHIBITORS IN PATIENTS WITH PROTEINURIA
In nondiabetic kidney disease, there is strong evidence from the REIN and AASK trials that treatment with ACE inhibitors results in slower decline in glomerular filtration rate (GFR), and this risk reduction is more pronounced in patients with a higher degree of proteinuria.4–6
In type 1 diabetes, treatment with an ACE inhibitor in patients with overt proteinuria was associated with a 50% decrease in the risk of the combined end point of death, dialysis, or renal transplant.7 Patients with moderately increased albuminuria who were treated with an ACE inhibitor also had a reduced incidence of progression to overt proteinuria.8 Angiotensin inhibition may be beneficial even in normotensive patients with type 1 diabetes and persistent moderately increased albuminuria.9,10

In type 2 diabetes, the IDNT and RENAAL trials showed that treatment with an ARB in patients with overt nephropathy was associated with a statistically significant decrease (20% in IDNT, 16% in RENAAL) in the risk of the combined end point of death, end-stage renal disease, or doubling of serum creatinine.11,12 While there are more data for ARBs than for ACE inhibitors in type 2 diabetes, the DETAIL study showed that an ACE inhibitor was at least as effective as an ARB in providing long-term renal protection in type 2 diabetes and moderately increased albuminuria.13
Data are limited on the role of angiotensin inhibition in normotensive patients with type 2 diabetes and persistent moderately increased albuminuria, but consensus opinion suggests treatment with an ACE inhibitor or ARB in these patients if there are no contraindications.
LIMITATIONS
Adverse effects of ACE inhibitors and ARBs include cough (more with ACE inhibitors), angioedema (more with ACE inhibitors), and hyperkalemia.
The use of ARBs in patients with a history of ACE inhibitor-related angioedema has been previously discussed in this Journal.14 Guidelines advocate caution when prescribing ARBs for patients who will benefit from RAAS inhibition and have had ACE inhibitor-related angioedema.15
RAAS inhibitor therapy can cause a modest rise in creatinine due to reduction in intraglomerular pressure. An elevation in creatinine of up to 30% that stabilizes in the first 2 months is not necessarily a reason to discontinue therapy. However, a continued rise in creatinine should prompt evaluation for excessive fall in blood pressure (especially with volume depletion from concomitant diuretic use), possible bilateral renal artery stenosis, or both. There is no level of GFR or serum creatinine at which an ACE inhibitor or ARB is absolutely contraindicated, and this decision should be made on an individual basis in conjunction with a nephrologist.
Risks for hyperkalemia should always be kept in mind at lower GFR levels. It would be prudent to check serum creatinine and potassium levels within the first week or two after starting or intensifying RAAS inhibition in these patients.
CAUTION
Combination therapy with an ACE inhibitor and an ARB was hypothesized to provide more complete RAAS blockade, with the hope of better clinical outcomes. However, this strategy has been questioned with results from three studies—ONTARGET, ALTITUDE, and the VA NEPHRON-D study—all of which showed worse renal outcomes, hypertension, and hyperkalemia with use of dual RAAS blockade.16–20 The combined evidence so far suggests that dual RAAS blockade should not be routinely prescribed.
RAAS INHIBITION IN PRACTICE
RAAS inhibition should be instituted and continued in patients with proteinuria who are able to tolerate the therapy and do not experience adverse effects as discussed above. Although there is no specific consensus guideline on the frequency of assessment of albumin excretion after diagnosis of albuminuria and institution of RAAS inhibition and blood pressure control in patients with diabetes, periodic surveillance at least once a year is reasonable to assess response to therapy and possible disease progression.21 If there is significant proteinuria or possibility of nondiabetic kidney disease, the patient should be referred to a nephrologist.
- Taal MW, Brenner BM. Renoprotective benefits of RAS inhibition: from ACEI to angiotensin II antagonists. Kidney Int 2000; 57:1803–1817.
- Atkins RC, Briganti EM, Lewis JB, et al. Proteinuria reduction and progression to renal failure in patients with type 2 diabetes mellitus and overt nephropathy. Am J Kidney Dis 2005; 45:281–287.
- de Zeeuw D, Remuzzi G, Parving HH, et al. Proteinuria, a target for renoprotection in patients with type 2 diabetic nephropathy: lessons from RENAAL. Kidney Int 2004; 65:2309–2320.
- Randomised placebo-controlled trial of effect of ramipril on decline in glomerular filtration rate and risk of terminal renal failure in proteinuric, non-diabetic nephropathy. The GISEN Group (Gruppo Italiano di Studi Epidemiologici in Nefrologia). Lancet 1997; 349:1857–1863.
- Ruggenenti P, Perna A, Gherardi G, et al. Renoprotective properties of ACE-inhibition in non-diabetic nephropathies with non-nephrotic proteinuria. Lancet 1999; 354:359–364.
- Agodoa LY, Appel L, Bakris GL, et al; African American Study of Kidney Disease and Hypertension (AASK) Study Group. Effect of ramipril vs amlodipine on renal outcomes in hypertensive nephrosclerosis: a randomized controlled trial. JAMA 2001; 285:2719–2728.
- Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med 1993; 329:1456–1462.
- Viberti G, Mogensen CE, Groop LC, Pauls JF. Effect of captopril on progression to clinical proteinuria in patients with insulin-dependent diabetes mellitus and microalbuminuria. European Microalbuminuria Captopril Study Group. JAMA 1994; 271:275–279.
- ACE Inhibitors in Diabetic Nephropathy Trialist Group. Should all patients with type 1 diabetes mellitus and microalbuminuria receive angiotensin-converting enzyme inhibitors? A meta-analysis of individual patient data. Ann Intern Med 2001; 134:370–379.
- Randomised placebo-controlled trial of lisinopril in normotensive patients with insulin-dependent diabetes and normoalbuminuria or microalbuminuria. The EUCLID Study Group. Lancet 1997; 349:1787–1792.
- Lewis EJ, Hunsicker LG, Clarke WR, et al; Collaborative Study Group. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med 2001; 345:851–860.
- Brenner BM, Copper ME, de Zeeuw D, et al; RENAAL study investigators. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 2001; 345:861–869.
- Barnett AH, Bain SC, Bouter P, et al; Diabetics Exposed to Telmisartan and Enalapril Study Group. Angiotensin-receptor blockade versus converting-enzyme inhibition in type 2 diabetes and nephropathy. N Engl J Med 2004; 351:1952–1961.
- Sharma P, Nagarajan V. Q: Can an ARB be given to patients who have had angioedema on an ACE inhibitor? Cleve Clin J Med 2013; 80:755–757.
- Kidney Disease Outcomes Quality Initiative (K/DOQI).K/DOQI clinical practice guidelines on hypertension and antihypertensive agents in chronic kidney disease. Am J Kidney Dis 2004; 43(suppl 1):S1–S290.
- ONTARGET Investigators; Yusuf S, Teo KK, Pogue J, et al. Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med 2008; 358:1547–1559.
- Mann JF, Schmieder RE, McQueen M, et al; ONTARGET investigators. Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): a multicentre, randomised, double-blind, controlled trial. Lancet 2008; 372:547–553.
- Mann JF, Anderson C, Gao P, et al; ONTARGET Investigators. Dual inhibition of the renin-angiotensin system in high-risk diabetes and risk for stroke and other outcomes: results of the ONTARGET trial. J Hypertens 2013; 31:414–421.
- Parving HH, Brenner BM, McMurray JJ, et al; ALTITUDE Investigators. Cardiorenal end points in a trial of aliskiren for type 2 diabetes. N Engl J Med 2012; 367:2204–2213.
- Fried LF, Emanuele N, Zhang JH, et al; VA NEPHRON-D Investigators. Combined angiotensin inhibition for the treatment of diabetic nephropathy. N Engl J Med 2013; 369:1892–1903.
- American Diabetes Association. Microvascular complications and foot care. Sec. 9. In: Standards of Medical Care in Diabetes—2015. Diabetes Care 2015;38(suppl 1):S58–S66.
Most patients with proteinuria benefit from a renin-angiotensin-aldosterone system (RAAS) inhibitor. Exceptions due to adverse effects are discussed below.
WHY RAAS INHIBITORS?
RAAS inhibitors—particularly angiotensin-converting enzyme (ACE) inhibitors and angiotensin II receptor blockers (ARBs)—reduce proteinuria and slow the progression of chronic kidney disease by improving glomerular hemodynamics, restoring the altered glomerular barrier function, and limiting the nonhemodynamic effects of angiotensin II and aldosterone, such as fibrosis and vascular endothelial dysfunction.1 Studies have shown that these protective effects are, at least in part, independent of the reduction in systemic blood pressure.2,3
EVIDENCE FOR USING RAAS INHIBITORS IN PATIENTS WITH PROTEINURIA
In nondiabetic kidney disease, there is strong evidence from the REIN and AASK trials that treatment with ACE inhibitors results in slower decline in glomerular filtration rate (GFR), and this risk reduction is more pronounced in patients with a higher degree of proteinuria.4–6
In type 1 diabetes, treatment with an ACE inhibitor in patients with overt proteinuria was associated with a 50% decrease in the risk of the combined end point of death, dialysis, or renal transplant.7 Patients with moderately increased albuminuria who were treated with an ACE inhibitor also had a reduced incidence of progression to overt proteinuria.8 Angiotensin inhibition may be beneficial even in normotensive patients with type 1 diabetes and persistent moderately increased albuminuria.9,10
In type 2 diabetes, the IDNT and RENAAL trials showed that treatment with an ARB in patients with overt nephropathy was associated with a statistically significant decrease (20% in IDNT, 16% in RENAAL) in the risk of the combined end point of death, end-stage renal disease, or doubling of serum creatinine.11,12 While there are more data for ARBs than for ACE inhibitors in type 2 diabetes, the DETAIL study showed that an ACE inhibitor was at least as effective as an ARB in providing long-term renal protection in type 2 diabetes and moderately increased albuminuria.13
Data are limited on the role of angiotensin inhibition in normotensive patients with type 2 diabetes and persistent moderately increased albuminuria, but consensus opinion suggests treatment with an ACE inhibitor or ARB in these patients if there are no contraindications.
LIMITATIONS
Adverse effects of ACE inhibitors and ARBs include cough (more with ACE inhibitors), angioedema (more with ACE inhibitors), and hyperkalemia.
The use of ARBs in patients with a history of ACE inhibitor-related angioedema has been previously discussed in this Journal.14 Guidelines advocate caution when prescribing ARBs for patients who will benefit from RAAS inhibition and have had ACE inhibitor-related angioedema.15
RAAS inhibitor therapy can cause a modest rise in creatinine due to reduction in intraglomerular pressure. An elevation in creatinine of up to 30% that stabilizes in the first 2 months is not necessarily a reason to discontinue therapy. However, a continued rise in creatinine should prompt evaluation for excessive fall in blood pressure (especially with volume depletion from concomitant diuretic use), possible bilateral renal artery stenosis, or both. There is no level of GFR or serum creatinine at which an ACE inhibitor or ARB is absolutely contraindicated, and this decision should be made on an individual basis in conjunction with a nephrologist.
Risks for hyperkalemia should always be kept in mind at lower GFR levels. It would be prudent to check serum creatinine and potassium levels within the first week or two after starting or intensifying RAAS inhibition in these patients.
CAUTION
Combination therapy with an ACE inhibitor and an ARB was hypothesized to provide more complete RAAS blockade, with the hope of better clinical outcomes. However, this strategy has been questioned with results from three studies—ONTARGET, ALTITUDE, and the VA NEPHRON-D study—all of which showed worse renal outcomes, hypertension, and hyperkalemia with use of dual RAAS blockade.16–20 The combined evidence so far suggests that dual RAAS blockade should not be routinely prescribed.
RAAS INHIBITION IN PRACTICE
RAAS inhibition should be instituted and continued in patients with proteinuria who are able to tolerate the therapy and do not experience adverse effects as discussed above. Although there is no specific consensus guideline on the frequency of assessment of albumin excretion after diagnosis of albuminuria and institution of RAAS inhibition and blood pressure control in patients with diabetes, periodic surveillance at least once a year is reasonable to assess response to therapy and possible disease progression.21 If there is significant proteinuria or possibility of nondiabetic kidney disease, the patient should be referred to a nephrologist.
Most patients with proteinuria benefit from a renin-angiotensin-aldosterone system (RAAS) inhibitor. Exceptions due to adverse effects are discussed below.
WHY RAAS INHIBITORS?
RAAS inhibitors—particularly angiotensin-converting enzyme (ACE) inhibitors and angiotensin II receptor blockers (ARBs)—reduce proteinuria and slow the progression of chronic kidney disease by improving glomerular hemodynamics, restoring the altered glomerular barrier function, and limiting the nonhemodynamic effects of angiotensin II and aldosterone, such as fibrosis and vascular endothelial dysfunction.1 Studies have shown that these protective effects are, at least in part, independent of the reduction in systemic blood pressure.2,3
EVIDENCE FOR USING RAAS INHIBITORS IN PATIENTS WITH PROTEINURIA
In nondiabetic kidney disease, there is strong evidence from the REIN and AASK trials that treatment with ACE inhibitors results in slower decline in glomerular filtration rate (GFR), and this risk reduction is more pronounced in patients with a higher degree of proteinuria.4–6
In type 1 diabetes, treatment with an ACE inhibitor in patients with overt proteinuria was associated with a 50% decrease in the risk of the combined end point of death, dialysis, or renal transplant.7 Patients with moderately increased albuminuria who were treated with an ACE inhibitor also had a reduced incidence of progression to overt proteinuria.8 Angiotensin inhibition may be beneficial even in normotensive patients with type 1 diabetes and persistent moderately increased albuminuria.9,10
In type 2 diabetes, the IDNT and RENAAL trials showed that treatment with an ARB in patients with overt nephropathy was associated with a statistically significant decrease (20% in IDNT, 16% in RENAAL) in the risk of the combined end point of death, end-stage renal disease, or doubling of serum creatinine.11,12 While there are more data for ARBs than for ACE inhibitors in type 2 diabetes, the DETAIL study showed that an ACE inhibitor was at least as effective as an ARB in providing long-term renal protection in type 2 diabetes and moderately increased albuminuria.13
Data are limited on the role of angiotensin inhibition in normotensive patients with type 2 diabetes and persistent moderately increased albuminuria, but consensus opinion suggests treatment with an ACE inhibitor or ARB in these patients if there are no contraindications.
LIMITATIONS
Adverse effects of ACE inhibitors and ARBs include cough (more with ACE inhibitors), angioedema (more with ACE inhibitors), and hyperkalemia.
The use of ARBs in patients with a history of ACE inhibitor-related angioedema has been previously discussed in this Journal.14 Guidelines advocate caution when prescribing ARBs for patients who will benefit from RAAS inhibition and have had ACE inhibitor-related angioedema.15
RAAS inhibitor therapy can cause a modest rise in creatinine due to reduction in intraglomerular pressure. An elevation in creatinine of up to 30% that stabilizes in the first 2 months is not necessarily a reason to discontinue therapy. However, a continued rise in creatinine should prompt evaluation for excessive fall in blood pressure (especially with volume depletion from concomitant diuretic use), possible bilateral renal artery stenosis, or both. There is no level of GFR or serum creatinine at which an ACE inhibitor or ARB is absolutely contraindicated, and this decision should be made on an individual basis in conjunction with a nephrologist.
Risks for hyperkalemia should always be kept in mind at lower GFR levels. It would be prudent to check serum creatinine and potassium levels within the first week or two after starting or intensifying RAAS inhibition in these patients.
CAUTION
Combination therapy with an ACE inhibitor and an ARB was hypothesized to provide more complete RAAS blockade, with the hope of better clinical outcomes. However, this strategy has been questioned with results from three studies—ONTARGET, ALTITUDE, and the VA NEPHRON-D study—all of which showed worse renal outcomes, hypertension, and hyperkalemia with use of dual RAAS blockade.16–20 The combined evidence so far suggests that dual RAAS blockade should not be routinely prescribed.
RAAS INHIBITION IN PRACTICE
RAAS inhibition should be instituted and continued in patients with proteinuria who are able to tolerate the therapy and do not experience adverse effects as discussed above. Although there is no specific consensus guideline on the frequency of assessment of albumin excretion after diagnosis of albuminuria and institution of RAAS inhibition and blood pressure control in patients with diabetes, periodic surveillance at least once a year is reasonable to assess response to therapy and possible disease progression.21 If there is significant proteinuria or possibility of nondiabetic kidney disease, the patient should be referred to a nephrologist.
- Taal MW, Brenner BM. Renoprotective benefits of RAS inhibition: from ACEI to angiotensin II antagonists. Kidney Int 2000; 57:1803–1817.
- Atkins RC, Briganti EM, Lewis JB, et al. Proteinuria reduction and progression to renal failure in patients with type 2 diabetes mellitus and overt nephropathy. Am J Kidney Dis 2005; 45:281–287.
- de Zeeuw D, Remuzzi G, Parving HH, et al. Proteinuria, a target for renoprotection in patients with type 2 diabetic nephropathy: lessons from RENAAL. Kidney Int 2004; 65:2309–2320.
- Randomised placebo-controlled trial of effect of ramipril on decline in glomerular filtration rate and risk of terminal renal failure in proteinuric, non-diabetic nephropathy. The GISEN Group (Gruppo Italiano di Studi Epidemiologici in Nefrologia). Lancet 1997; 349:1857–1863.
- Ruggenenti P, Perna A, Gherardi G, et al. Renoprotective properties of ACE-inhibition in non-diabetic nephropathies with non-nephrotic proteinuria. Lancet 1999; 354:359–364.
- Agodoa LY, Appel L, Bakris GL, et al; African American Study of Kidney Disease and Hypertension (AASK) Study Group. Effect of ramipril vs amlodipine on renal outcomes in hypertensive nephrosclerosis: a randomized controlled trial. JAMA 2001; 285:2719–2728.
- Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med 1993; 329:1456–1462.
- Viberti G, Mogensen CE, Groop LC, Pauls JF. Effect of captopril on progression to clinical proteinuria in patients with insulin-dependent diabetes mellitus and microalbuminuria. European Microalbuminuria Captopril Study Group. JAMA 1994; 271:275–279.
- ACE Inhibitors in Diabetic Nephropathy Trialist Group. Should all patients with type 1 diabetes mellitus and microalbuminuria receive angiotensin-converting enzyme inhibitors? A meta-analysis of individual patient data. Ann Intern Med 2001; 134:370–379.
- Randomised placebo-controlled trial of lisinopril in normotensive patients with insulin-dependent diabetes and normoalbuminuria or microalbuminuria. The EUCLID Study Group. Lancet 1997; 349:1787–1792.
- Lewis EJ, Hunsicker LG, Clarke WR, et al; Collaborative Study Group. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med 2001; 345:851–860.
- Brenner BM, Copper ME, de Zeeuw D, et al; RENAAL study investigators. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 2001; 345:861–869.
- Barnett AH, Bain SC, Bouter P, et al; Diabetics Exposed to Telmisartan and Enalapril Study Group. Angiotensin-receptor blockade versus converting-enzyme inhibition in type 2 diabetes and nephropathy. N Engl J Med 2004; 351:1952–1961.
- Sharma P, Nagarajan V. Q: Can an ARB be given to patients who have had angioedema on an ACE inhibitor? Cleve Clin J Med 2013; 80:755–757.
- Kidney Disease Outcomes Quality Initiative (K/DOQI).K/DOQI clinical practice guidelines on hypertension and antihypertensive agents in chronic kidney disease. Am J Kidney Dis 2004; 43(suppl 1):S1–S290.
- ONTARGET Investigators; Yusuf S, Teo KK, Pogue J, et al. Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med 2008; 358:1547–1559.
- Mann JF, Schmieder RE, McQueen M, et al; ONTARGET investigators. Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): a multicentre, randomised, double-blind, controlled trial. Lancet 2008; 372:547–553.
- Mann JF, Anderson C, Gao P, et al; ONTARGET Investigators. Dual inhibition of the renin-angiotensin system in high-risk diabetes and risk for stroke and other outcomes: results of the ONTARGET trial. J Hypertens 2013; 31:414–421.
- Parving HH, Brenner BM, McMurray JJ, et al; ALTITUDE Investigators. Cardiorenal end points in a trial of aliskiren for type 2 diabetes. N Engl J Med 2012; 367:2204–2213.
- Fried LF, Emanuele N, Zhang JH, et al; VA NEPHRON-D Investigators. Combined angiotensin inhibition for the treatment of diabetic nephropathy. N Engl J Med 2013; 369:1892–1903.
- American Diabetes Association. Microvascular complications and foot care. Sec. 9. In: Standards of Medical Care in Diabetes—2015. Diabetes Care 2015;38(suppl 1):S58–S66.
- Taal MW, Brenner BM. Renoprotective benefits of RAS inhibition: from ACEI to angiotensin II antagonists. Kidney Int 2000; 57:1803–1817.
- Atkins RC, Briganti EM, Lewis JB, et al. Proteinuria reduction and progression to renal failure in patients with type 2 diabetes mellitus and overt nephropathy. Am J Kidney Dis 2005; 45:281–287.
- de Zeeuw D, Remuzzi G, Parving HH, et al. Proteinuria, a target for renoprotection in patients with type 2 diabetic nephropathy: lessons from RENAAL. Kidney Int 2004; 65:2309–2320.
- Randomised placebo-controlled trial of effect of ramipril on decline in glomerular filtration rate and risk of terminal renal failure in proteinuric, non-diabetic nephropathy. The GISEN Group (Gruppo Italiano di Studi Epidemiologici in Nefrologia). Lancet 1997; 349:1857–1863.
- Ruggenenti P, Perna A, Gherardi G, et al. Renoprotective properties of ACE-inhibition in non-diabetic nephropathies with non-nephrotic proteinuria. Lancet 1999; 354:359–364.
- Agodoa LY, Appel L, Bakris GL, et al; African American Study of Kidney Disease and Hypertension (AASK) Study Group. Effect of ramipril vs amlodipine on renal outcomes in hypertensive nephrosclerosis: a randomized controlled trial. JAMA 2001; 285:2719–2728.
- Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med 1993; 329:1456–1462.
- Viberti G, Mogensen CE, Groop LC, Pauls JF. Effect of captopril on progression to clinical proteinuria in patients with insulin-dependent diabetes mellitus and microalbuminuria. European Microalbuminuria Captopril Study Group. JAMA 1994; 271:275–279.
- ACE Inhibitors in Diabetic Nephropathy Trialist Group. Should all patients with type 1 diabetes mellitus and microalbuminuria receive angiotensin-converting enzyme inhibitors? A meta-analysis of individual patient data. Ann Intern Med 2001; 134:370–379.
- Randomised placebo-controlled trial of lisinopril in normotensive patients with insulin-dependent diabetes and normoalbuminuria or microalbuminuria. The EUCLID Study Group. Lancet 1997; 349:1787–1792.
- Lewis EJ, Hunsicker LG, Clarke WR, et al; Collaborative Study Group. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med 2001; 345:851–860.
- Brenner BM, Copper ME, de Zeeuw D, et al; RENAAL study investigators. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 2001; 345:861–869.
- Barnett AH, Bain SC, Bouter P, et al; Diabetics Exposed to Telmisartan and Enalapril Study Group. Angiotensin-receptor blockade versus converting-enzyme inhibition in type 2 diabetes and nephropathy. N Engl J Med 2004; 351:1952–1961.
- Sharma P, Nagarajan V. Q: Can an ARB be given to patients who have had angioedema on an ACE inhibitor? Cleve Clin J Med 2013; 80:755–757.
- Kidney Disease Outcomes Quality Initiative (K/DOQI).K/DOQI clinical practice guidelines on hypertension and antihypertensive agents in chronic kidney disease. Am J Kidney Dis 2004; 43(suppl 1):S1–S290.
- ONTARGET Investigators; Yusuf S, Teo KK, Pogue J, et al. Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med 2008; 358:1547–1559.
- Mann JF, Schmieder RE, McQueen M, et al; ONTARGET investigators. Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): a multicentre, randomised, double-blind, controlled trial. Lancet 2008; 372:547–553.
- Mann JF, Anderson C, Gao P, et al; ONTARGET Investigators. Dual inhibition of the renin-angiotensin system in high-risk diabetes and risk for stroke and other outcomes: results of the ONTARGET trial. J Hypertens 2013; 31:414–421.
- Parving HH, Brenner BM, McMurray JJ, et al; ALTITUDE Investigators. Cardiorenal end points in a trial of aliskiren for type 2 diabetes. N Engl J Med 2012; 367:2204–2213.
- Fried LF, Emanuele N, Zhang JH, et al; VA NEPHRON-D Investigators. Combined angiotensin inhibition for the treatment of diabetic nephropathy. N Engl J Med 2013; 369:1892–1903.
- American Diabetes Association. Microvascular complications and foot care. Sec. 9. In: Standards of Medical Care in Diabetes—2015. Diabetes Care 2015;38(suppl 1):S58–S66.
Starting insulin therapy
To the Editor: I would like to add two points to the excellent review on starting insulin in patients with type 2 diabetes by Brateanu et al in the August 2015 issue.1
First, in my practice, I review glucose patterns and recommend that mealtime insulin be started early after basal insulin is started and not simply wait for the next hemoglobin A1c result. In my experience, basal insulin is often mindlessly up-titrated, month after month, to fix a high fasting glucose. During the first 2 to 3 weeks of basal insulin titration, I ask patients to test before breakfast, dinner, and bedtime, not just fasting. In so doing, I detect, in most patients, significant bedtime hyperglycemia arising from dinner, usually their largest meal. Then I prescribe dinnertime rapid-acting insulin to correct the bedtime hyperglycemia, and this in turn ameliorates the fasting hyperglycemia. Additional mealtime doses can be added if necessary.2
After all, why should we ignore hyperglycemia occurring at other times and focus only on fasting glucose? With blood glucose pattern review, we can detect those glucose elevations that need to be targeted regardless of when they occur. It has been repeatedly shown that up to almost 50% of patients will fail to reach a hemoglobin A1c below 7%, even after months of up-titration of basal insulin.3,4 Most patients will benefit by starting mealtime rapid-acting insulin early on.
And second, when adjusting mealtime rapid-acting injected insulin, there is no need to measure postprandial glucose in most patients with type 2 diabetes. A rigorous clinical trial5 showed that testing before the next meal or, in the case of dinner, before bedtime worked as well as or better than postprandial testing. By implementing the above steps, I think we all can provide better, more individualized therapy for our patients.
- Brateanu A, Russo-Alvarez G, Nielsen C. Starting insulin in patients with type 2 diabetes: an individualized approach. Cleve Clin J Med 2015; 82:513–519.
- Rodbard HW, Visco VE, Andersen H, Hiort LC, Shu DHW. Treatment intensification with stepwise addition of prandial insulin aspart boluses compared with full basal-bolus therapy (FullSTEP Study): a randomized, treat-to-target clinical trial. Lancet Diabetes Endocrinol 2014; 2:30–37.
- Riddle MC, Rosenstock J, Gerich J; Insulin Glargine 4002 Study Investigators. The Treat-to-Target Trial: randomized addition of glargine or human NPH insulin to oral therapy of type 2 diabetic patients. Diabetes Care 2003; 26:3080–3086.
- Rosenstock J, Davies M, Home PD, Larsen J, Koenen C, Schernthaner G. A randomized 52-week treat-to-target trial comparing insulin detemir with insulin glargine when administered as add-on to glucose-lowering drugs in insulin-naive people with type 2 diabetes. Diabetologia 2008; 51:408–416.
- Meneghini L, Mersebach H, Kumar S, Svendsen AL, Hermansen K. Comparison of 2 intensification regimens with rapid-acting insulin aspart in type 2 diabetes mellitus inadequately controlled by once-daily insulin detemir and oral antidiabetes drugs: the Step-Wise Randomized Study. Endocr Pract 2011; 17:727–736.
To the Editor: I would like to add two points to the excellent review on starting insulin in patients with type 2 diabetes by Brateanu et al in the August 2015 issue.1
First, in my practice, I review glucose patterns and recommend that mealtime insulin be started early after basal insulin is started and not simply wait for the next hemoglobin A1c result. In my experience, basal insulin is often mindlessly up-titrated, month after month, to fix a high fasting glucose. During the first 2 to 3 weeks of basal insulin titration, I ask patients to test before breakfast, dinner, and bedtime, not just fasting. In so doing, I detect, in most patients, significant bedtime hyperglycemia arising from dinner, usually their largest meal. Then I prescribe dinnertime rapid-acting insulin to correct the bedtime hyperglycemia, and this in turn ameliorates the fasting hyperglycemia. Additional mealtime doses can be added if necessary.2
After all, why should we ignore hyperglycemia occurring at other times and focus only on fasting glucose? With blood glucose pattern review, we can detect those glucose elevations that need to be targeted regardless of when they occur. It has been repeatedly shown that up to almost 50% of patients will fail to reach a hemoglobin A1c below 7%, even after months of up-titration of basal insulin.3,4 Most patients will benefit by starting mealtime rapid-acting insulin early on.
And second, when adjusting mealtime rapid-acting injected insulin, there is no need to measure postprandial glucose in most patients with type 2 diabetes. A rigorous clinical trial5 showed that testing before the next meal or, in the case of dinner, before bedtime worked as well as or better than postprandial testing. By implementing the above steps, I think we all can provide better, more individualized therapy for our patients.
To the Editor: I would like to add two points to the excellent review on starting insulin in patients with type 2 diabetes by Brateanu et al in the August 2015 issue.1
First, in my practice, I review glucose patterns and recommend that mealtime insulin be started early after basal insulin is started and not simply wait for the next hemoglobin A1c result. In my experience, basal insulin is often mindlessly up-titrated, month after month, to fix a high fasting glucose. During the first 2 to 3 weeks of basal insulin titration, I ask patients to test before breakfast, dinner, and bedtime, not just fasting. In so doing, I detect, in most patients, significant bedtime hyperglycemia arising from dinner, usually their largest meal. Then I prescribe dinnertime rapid-acting insulin to correct the bedtime hyperglycemia, and this in turn ameliorates the fasting hyperglycemia. Additional mealtime doses can be added if necessary.2
After all, why should we ignore hyperglycemia occurring at other times and focus only on fasting glucose? With blood glucose pattern review, we can detect those glucose elevations that need to be targeted regardless of when they occur. It has been repeatedly shown that up to almost 50% of patients will fail to reach a hemoglobin A1c below 7%, even after months of up-titration of basal insulin.3,4 Most patients will benefit by starting mealtime rapid-acting insulin early on.
And second, when adjusting mealtime rapid-acting injected insulin, there is no need to measure postprandial glucose in most patients with type 2 diabetes. A rigorous clinical trial5 showed that testing before the next meal or, in the case of dinner, before bedtime worked as well as or better than postprandial testing. By implementing the above steps, I think we all can provide better, more individualized therapy for our patients.
- Brateanu A, Russo-Alvarez G, Nielsen C. Starting insulin in patients with type 2 diabetes: an individualized approach. Cleve Clin J Med 2015; 82:513–519.
- Rodbard HW, Visco VE, Andersen H, Hiort LC, Shu DHW. Treatment intensification with stepwise addition of prandial insulin aspart boluses compared with full basal-bolus therapy (FullSTEP Study): a randomized, treat-to-target clinical trial. Lancet Diabetes Endocrinol 2014; 2:30–37.
- Riddle MC, Rosenstock J, Gerich J; Insulin Glargine 4002 Study Investigators. The Treat-to-Target Trial: randomized addition of glargine or human NPH insulin to oral therapy of type 2 diabetic patients. Diabetes Care 2003; 26:3080–3086.
- Rosenstock J, Davies M, Home PD, Larsen J, Koenen C, Schernthaner G. A randomized 52-week treat-to-target trial comparing insulin detemir with insulin glargine when administered as add-on to glucose-lowering drugs in insulin-naive people with type 2 diabetes. Diabetologia 2008; 51:408–416.
- Meneghini L, Mersebach H, Kumar S, Svendsen AL, Hermansen K. Comparison of 2 intensification regimens with rapid-acting insulin aspart in type 2 diabetes mellitus inadequately controlled by once-daily insulin detemir and oral antidiabetes drugs: the Step-Wise Randomized Study. Endocr Pract 2011; 17:727–736.
- Brateanu A, Russo-Alvarez G, Nielsen C. Starting insulin in patients with type 2 diabetes: an individualized approach. Cleve Clin J Med 2015; 82:513–519.
- Rodbard HW, Visco VE, Andersen H, Hiort LC, Shu DHW. Treatment intensification with stepwise addition of prandial insulin aspart boluses compared with full basal-bolus therapy (FullSTEP Study): a randomized, treat-to-target clinical trial. Lancet Diabetes Endocrinol 2014; 2:30–37.
- Riddle MC, Rosenstock J, Gerich J; Insulin Glargine 4002 Study Investigators. The Treat-to-Target Trial: randomized addition of glargine or human NPH insulin to oral therapy of type 2 diabetic patients. Diabetes Care 2003; 26:3080–3086.
- Rosenstock J, Davies M, Home PD, Larsen J, Koenen C, Schernthaner G. A randomized 52-week treat-to-target trial comparing insulin detemir with insulin glargine when administered as add-on to glucose-lowering drugs in insulin-naive people with type 2 diabetes. Diabetologia 2008; 51:408–416.
- Meneghini L, Mersebach H, Kumar S, Svendsen AL, Hermansen K. Comparison of 2 intensification regimens with rapid-acting insulin aspart in type 2 diabetes mellitus inadequately controlled by once-daily insulin detemir and oral antidiabetes drugs: the Step-Wise Randomized Study. Endocr Pract 2011; 17:727–736.
In reply: Starting insulin therapy
In Reply: We thank Dr. Weiss for his insightful comments and for the opportunity to clarify a number of points from our article.
We agree that controlling the fasting glucose should not take months. As mentioned in our article, adjusting the basal insulin dose should be done with 2 to 4 units every 2 to 3 days in order to reach the fasting glycemic goal. Applying this approach and systematically titrating the NPH, glargine, or detemir insulin will smoothly decrease the fasting glucose within 12 weeks, as described in the 24-week1 and 52-week2 treat-to-target trials in which basal insulin was added to the oral therapy in patients with type 2 diabetes.
When basal insulin is no longer sufficient to reach a target hemoglobin A1c, a glucagon-like peptide-1 receptor agonist or prandial insulin can be used. The basal-bolus or twice-daily premixed insulin analogues can also be considered as the initial therapy, depending on the patient, disease, and drug characteristics.3 We agree that once a prandial insulin regimen is initiated, the dose titration can be done based on preprandial or postprandial blood glucose measurements, as shown in Table 2 in our article. However, adding the prandial insulin without first optimizing the basal therapy was considered a limitation of the Orals Plus Apidra and Lantus (OPAL) study,4 which investigated the addition of one prandial insulin injection to basal glargine insulin.5 As a consequence, the subsequent studies investigating the effects of initiating and titrating the preprandial rapid-acting insulin (as a single dose or using a stepwise approach) in patients inadequately controlled with once-daily basal insulin and oral antidiabetic drugs had run-in periods of 12 to 14 weeks, in order to optimize the basal insulin dosage and achieve target fasting blood glucose levels of 110 mg/dL or less. This approach had the additional benefit of achieving a target hemoglobin A1c level of less than 7% in a significant number of patients (up to 37%),6 before starting the preprandial insulin.6–8
Regardless of the regimen selected, titration of the insulin doses can only be achieved with understanding the pharmacodynamic characteristics of each type of insulin used.9
- Riddle MC, Rosenstock J, Gerich J; Insulin Glargine 4002 Study Investigators. The Treat-to-Target Trial: randomized addition of glargine or human NPH insulin to oral therapy of type 2 diabetic patients. Diabetes Care 2003; 26:3080–3086.
- Rosenstock J, Davies M, Home PD, Larsen J, Koenen C, Schernthaner G. A randomised, 52-week, treat-to-target trial comparing insulin detemir with insulin glargine when administered as add-on to glucose-lowering drugs in insulin-naive people with type 2 diabetes. Diabetologia 2008; 51:408–416.
- Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of hyperglycaemia in type 2 diabetes, 2015: a patient-centered approach. Update to a position statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetologia 2015; 58:429–442.
- Owens DR. Stepwise intensification of insulin therapy in type 2 diabetes management—exploring the concept of the basal-plus approach in clinical practice. Diabet Med 2013; 30:276–288.
- Lankisch MR, Ferlinz KC, Leahy JL, Scherbaum WA; Orals Plus Apidra and Lantus (OPAL) Study Group. Introducing a simplified approach to insulin therapy in type 2 diabetes: a comparison of two single-dose regimens of insulin glulisine plus insulin glargine and oral antidiabetic drugs. Diabetes Obes Metab 2008; 10:1178–1185.
- Davidson MB, Raskin P, Tanenberg RJ, Vlajnic A, Hollander P. A stepwise approach to insulin therapy in patients with type 2 diabetes mellitus and basal insulin treatment failure. Endocr Pract 2011; 17:395–403.
- Meneghini L, Mersebach H, Kumar S, Svendsen AL, Hermansen K. Comparison of 2 intensification regimens with rapid-acting insulin aspart in type 2 diabetes mellitus inadequately controlled by once-daily insulin detemir and oral antidiabetes drugs: the Step-Wise Randomized Study. Endocrine Practice 2011; 17:727–736.
- Owens DR, Luzio SD, Sert-Langeron C, Riddle MC. Effects of initiation and titration of a single pre-prandial dose of insulin glulisine while continuing titrated insulin glargine in type 2 diabetes: a 6-month ‘proof-of-concept’ study. Diabetes Obes Metab 2011; 13:1020–1027.
- American Diabetes Association. 7. Approaches to glycemic treatment. Diabetes Care 2015; 38(suppl):S41–S48.
In Reply: We thank Dr. Weiss for his insightful comments and for the opportunity to clarify a number of points from our article.
We agree that controlling the fasting glucose should not take months. As mentioned in our article, adjusting the basal insulin dose should be done with 2 to 4 units every 2 to 3 days in order to reach the fasting glycemic goal. Applying this approach and systematically titrating the NPH, glargine, or detemir insulin will smoothly decrease the fasting glucose within 12 weeks, as described in the 24-week1 and 52-week2 treat-to-target trials in which basal insulin was added to the oral therapy in patients with type 2 diabetes.
When basal insulin is no longer sufficient to reach a target hemoglobin A1c, a glucagon-like peptide-1 receptor agonist or prandial insulin can be used. The basal-bolus or twice-daily premixed insulin analogues can also be considered as the initial therapy, depending on the patient, disease, and drug characteristics.3 We agree that once a prandial insulin regimen is initiated, the dose titration can be done based on preprandial or postprandial blood glucose measurements, as shown in Table 2 in our article. However, adding the prandial insulin without first optimizing the basal therapy was considered a limitation of the Orals Plus Apidra and Lantus (OPAL) study,4 which investigated the addition of one prandial insulin injection to basal glargine insulin.5 As a consequence, the subsequent studies investigating the effects of initiating and titrating the preprandial rapid-acting insulin (as a single dose or using a stepwise approach) in patients inadequately controlled with once-daily basal insulin and oral antidiabetic drugs had run-in periods of 12 to 14 weeks, in order to optimize the basal insulin dosage and achieve target fasting blood glucose levels of 110 mg/dL or less. This approach had the additional benefit of achieving a target hemoglobin A1c level of less than 7% in a significant number of patients (up to 37%),6 before starting the preprandial insulin.6–8
Regardless of the regimen selected, titration of the insulin doses can only be achieved with understanding the pharmacodynamic characteristics of each type of insulin used.9
In Reply: We thank Dr. Weiss for his insightful comments and for the opportunity to clarify a number of points from our article.
We agree that controlling the fasting glucose should not take months. As mentioned in our article, adjusting the basal insulin dose should be done with 2 to 4 units every 2 to 3 days in order to reach the fasting glycemic goal. Applying this approach and systematically titrating the NPH, glargine, or detemir insulin will smoothly decrease the fasting glucose within 12 weeks, as described in the 24-week1 and 52-week2 treat-to-target trials in which basal insulin was added to the oral therapy in patients with type 2 diabetes.
When basal insulin is no longer sufficient to reach a target hemoglobin A1c, a glucagon-like peptide-1 receptor agonist or prandial insulin can be used. The basal-bolus or twice-daily premixed insulin analogues can also be considered as the initial therapy, depending on the patient, disease, and drug characteristics.3 We agree that once a prandial insulin regimen is initiated, the dose titration can be done based on preprandial or postprandial blood glucose measurements, as shown in Table 2 in our article. However, adding the prandial insulin without first optimizing the basal therapy was considered a limitation of the Orals Plus Apidra and Lantus (OPAL) study,4 which investigated the addition of one prandial insulin injection to basal glargine insulin.5 As a consequence, the subsequent studies investigating the effects of initiating and titrating the preprandial rapid-acting insulin (as a single dose or using a stepwise approach) in patients inadequately controlled with once-daily basal insulin and oral antidiabetic drugs had run-in periods of 12 to 14 weeks, in order to optimize the basal insulin dosage and achieve target fasting blood glucose levels of 110 mg/dL or less. This approach had the additional benefit of achieving a target hemoglobin A1c level of less than 7% in a significant number of patients (up to 37%),6 before starting the preprandial insulin.6–8
Regardless of the regimen selected, titration of the insulin doses can only be achieved with understanding the pharmacodynamic characteristics of each type of insulin used.9
- Riddle MC, Rosenstock J, Gerich J; Insulin Glargine 4002 Study Investigators. The Treat-to-Target Trial: randomized addition of glargine or human NPH insulin to oral therapy of type 2 diabetic patients. Diabetes Care 2003; 26:3080–3086.
- Rosenstock J, Davies M, Home PD, Larsen J, Koenen C, Schernthaner G. A randomised, 52-week, treat-to-target trial comparing insulin detemir with insulin glargine when administered as add-on to glucose-lowering drugs in insulin-naive people with type 2 diabetes. Diabetologia 2008; 51:408–416.
- Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of hyperglycaemia in type 2 diabetes, 2015: a patient-centered approach. Update to a position statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetologia 2015; 58:429–442.
- Owens DR. Stepwise intensification of insulin therapy in type 2 diabetes management—exploring the concept of the basal-plus approach in clinical practice. Diabet Med 2013; 30:276–288.
- Lankisch MR, Ferlinz KC, Leahy JL, Scherbaum WA; Orals Plus Apidra and Lantus (OPAL) Study Group. Introducing a simplified approach to insulin therapy in type 2 diabetes: a comparison of two single-dose regimens of insulin glulisine plus insulin glargine and oral antidiabetic drugs. Diabetes Obes Metab 2008; 10:1178–1185.
- Davidson MB, Raskin P, Tanenberg RJ, Vlajnic A, Hollander P. A stepwise approach to insulin therapy in patients with type 2 diabetes mellitus and basal insulin treatment failure. Endocr Pract 2011; 17:395–403.
- Meneghini L, Mersebach H, Kumar S, Svendsen AL, Hermansen K. Comparison of 2 intensification regimens with rapid-acting insulin aspart in type 2 diabetes mellitus inadequately controlled by once-daily insulin detemir and oral antidiabetes drugs: the Step-Wise Randomized Study. Endocrine Practice 2011; 17:727–736.
- Owens DR, Luzio SD, Sert-Langeron C, Riddle MC. Effects of initiation and titration of a single pre-prandial dose of insulin glulisine while continuing titrated insulin glargine in type 2 diabetes: a 6-month ‘proof-of-concept’ study. Diabetes Obes Metab 2011; 13:1020–1027.
- American Diabetes Association. 7. Approaches to glycemic treatment. Diabetes Care 2015; 38(suppl):S41–S48.
- Riddle MC, Rosenstock J, Gerich J; Insulin Glargine 4002 Study Investigators. The Treat-to-Target Trial: randomized addition of glargine or human NPH insulin to oral therapy of type 2 diabetic patients. Diabetes Care 2003; 26:3080–3086.
- Rosenstock J, Davies M, Home PD, Larsen J, Koenen C, Schernthaner G. A randomised, 52-week, treat-to-target trial comparing insulin detemir with insulin glargine when administered as add-on to glucose-lowering drugs in insulin-naive people with type 2 diabetes. Diabetologia 2008; 51:408–416.
- Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of hyperglycaemia in type 2 diabetes, 2015: a patient-centered approach. Update to a position statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetologia 2015; 58:429–442.
- Owens DR. Stepwise intensification of insulin therapy in type 2 diabetes management—exploring the concept of the basal-plus approach in clinical practice. Diabet Med 2013; 30:276–288.
- Lankisch MR, Ferlinz KC, Leahy JL, Scherbaum WA; Orals Plus Apidra and Lantus (OPAL) Study Group. Introducing a simplified approach to insulin therapy in type 2 diabetes: a comparison of two single-dose regimens of insulin glulisine plus insulin glargine and oral antidiabetic drugs. Diabetes Obes Metab 2008; 10:1178–1185.
- Davidson MB, Raskin P, Tanenberg RJ, Vlajnic A, Hollander P. A stepwise approach to insulin therapy in patients with type 2 diabetes mellitus and basal insulin treatment failure. Endocr Pract 2011; 17:395–403.
- Meneghini L, Mersebach H, Kumar S, Svendsen AL, Hermansen K. Comparison of 2 intensification regimens with rapid-acting insulin aspart in type 2 diabetes mellitus inadequately controlled by once-daily insulin detemir and oral antidiabetes drugs: the Step-Wise Randomized Study. Endocrine Practice 2011; 17:727–736.
- Owens DR, Luzio SD, Sert-Langeron C, Riddle MC. Effects of initiation and titration of a single pre-prandial dose of insulin glulisine while continuing titrated insulin glargine in type 2 diabetes: a 6-month ‘proof-of-concept’ study. Diabetes Obes Metab 2011; 13:1020–1027.
- American Diabetes Association. 7. Approaches to glycemic treatment. Diabetes Care 2015; 38(suppl):S41–S48.
Sacubitril-valsartan and the evolution of heart failure care
Three decades ago, the only drugs we had for treating chronic heart failure were digitalis and loop diuretics. The mortality rate was very high, and heart transplantation was a newly developing treatment that could help only a very few patients.
The early 80s heralded new hope for patients with heart failure, with the introduction of angiotensin-converting enzyme (ACE) inhibitors1–5 and, later, beta-blockers. Beta-blockers were considered contraindicated in heart failure until new trials provided evidence of dramatic benefit such as better quality of life and longer survival.6–8 ACE inhibitors, along with beta-blockers, quickly became the standard of care for all patients with systolic heart failure.
The implantable cardioverter-defibrillator (ICD) required numerous clinical trials in ischemic and nonischemic cardiomyopathy to define its role.9,10 Cardiac resynchronization therapy did not arrive until 15 years ago and is now indicated in a specific niche of patients with left bundle branch block.11,12 Mineralocorticoid antagonists required three pivotal clinical trials before their important role in the treatment of systolic heart failure was defined.13–16
And in the current decade, the roles of ACE inhibitors, angiotensin II receptor blockers (ARBs), beta-blockers, mineralocorticoid antagonists, ICDs, and cardiac resynchronization therapy have been further defined, as reflected in the latest guidelines for the treatment of systolic heart failure.17
Guideline-directed medical therapy for systolic heart failure with the agents and devices mentioned above improves quality of life and extends survival. It was therefore hard to imagine that any new additive therapy could offer significant incremental improvement. However, more than 5 years ago, in an ambitious effort, the largest global clinical trial ever performed in chronic heart failure was launched with a novel agent.18
THE PARADIGM-HF TRIAL
In this issue of the Journal, Sabe et al19 describe the results of the Prospective Comparison of ARNI With ACEI to Determine Impact on Global Mortality and Morbidity in Heart Failure (PARADIGM-HF) trial of the novel combination drug sacubitril-valsartan, designated LCZ696 during its development and now available as Entresto.20
The mean age of the 8,442 patients in PARADIGM-HF was 64, and 78% were men. Despite guideline-directed medical therapy (93% of the patients were receiving a beta-blocker, and 60% were receiving a mineralocorticoid receptor antagonist), patients had persistent symptoms and signs of heart failure, diminished health-related quality of life, reduced ejection fraction (mean 29%), and elevated n-terminal pro-B-type natriuretic peptide levels (median 1,608 pg/mL, interquartile range 886–3,221).
The investigators reported a remarkable 20% reduction in the primary outcome of death from cardiovascular causes or hospitalization for heart failure in the patients who received sacubitril-valsartan compared with enalapril.20
Sacubitril-valsartan was reviewed under a US Food and Drug Administration (FDA) program that provides expedited review of drugs that are intended to treat a serious disease or condition and that may provide a significant improvement over available therapy. It was also granted a fast-track designation, which supports FDA efforts to facilitate the development and expedite the review of drugs to treat serious and life-threatening conditions and fill an unmet medical need. The FDA approved sacubitril-valsartan on July 7, 2015, for use in place of an ACE inhibitor or ARB in patients with New York Heart Association class II, III, or IV heart failure with reduced ejection fraction.21
WHAT WE STILL NEED TO KNOW
The results of PARADIGM-HF are generalizable, and sacubitril-valsartan was well tolerated in patients whose blood pressure was acceptable and who were able to tolerate ACE inhibitors in target doses. More than 90% of patients were receiving a beta-blocker. The dosing of enalapril (target 10 mg twice a day) is the guideline-directed target dose, and ACE inhibition is considered the gold standard for heart failure with reduced ejection fraction. Sacubitril-valsartan vs enalapril was a very appropriate comparison.
Far fewer PARADIGM-HF patients outside the United States had an ICD than those in the United States, which is a common finding in global clinical trials. However, Desai et al reported that sacubitril-valsartan reduced rates of cardiovascular mortality both from worsening heart failure and from sudden cardiac death, independent of whether the patient had an ICD.22
Sacubitril-valsartan is taken twice a day, but most heart failure patients already take medications at several times during the day, so this should not pose a problem.
More information is needed on the use of this new drug in patients with New York Heart Association class IV symptoms, as only 60 patients with class IV symptoms were included in the PARADIGM-HF trial. Also, the efficacy of the drug in patients unable to tolerate a full dose will need to be analyzed.
PARADIGM-HF was conducted in stable, nonhospitalized patients with chronic heart failure; the use of the drug in new-onset heart failure and its initiation in hospitalized patients will require further study. In addition, the PARAGON-HF trial23 will examine the efficacy of sacubitril-valsartan in patients with heart failure and an ejection fraction of 45% or higher.
Sacubitril-valsartan ushers in a new era in heart failure treatment for patients with reduced ejection fraction and will certainly prompt quick revision of heart failure guidelines.
- Captopril Multicenter Research Group. A placebo-controlled trial of captopril in refractory chronic congestive heart failure. J Am Coll Cardiol 1983; 2:755–763.
- Effects of enalapril on mortality in severe congestive heart failure. Results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS). The CONSENSUS Trial Study Group. N Engl J Med 1987; 316:1429–1435.
- The SOLVD Investigators. Effect of enalapril on survival in patients with reduced left ventricular ejection fraction and congestive heart failure. N Engl J Med 1991; 325:293–302.
- Cohn JN, Johnson G, Ziesche S, et al. A comparison of enalapril with hydralazine-isosorbide dinitrate in the treatment of chronic congestive heart failure. N Engl J Med 1991; 325:303–310.
- Pfeffer MA, Braunwald E, Moyé LA, et al. Effect of captopril on mortality and morbidity in patients with left ventricular dysfunction after myocardial infarction. Results of the survival and ventricular enlargement trial. The SAVE Investigators. N Engl J Med 1992; 327:669–677.
- Packer M, Coats AJ, Fowler MB, et al. Effect of carvedilol on survival in severe chronic heart failure. N Engl J Med 2001; 344:1651–1658.
- Effect of metoprolol CR/XL in chronic heart failure: Metoprolol CR/XL Randomised Intervention Trial in Congestive Heart Failure (MERIT-HF). Lancet 1999; 353:2001–2007.
- Brophy JM, Joseph L, Rouleau JL. Beta-blockers in congestive heart failure. A Bayesian meta-analysis. Ann Intern Med 2001; 134:550–560.
- Buxton AE, Lee KL, Fisher JD, et al. A randomized study of the prevention of sudden death in patients with coronary artery disease. Multicenter Unsustained Tachycardia Trial Investigators. N Engl J Med 1999; 341:1882–1890.
- Moss AJ, Zareba W, Hall WJ, et al. Prophylactic implantation of a defibrillator in patients with myocardial infarction and reduced ejection fraction. N Engl J Med 2002; 346:877–883.
- Abraham WT, Fisher WG, Smith AL, et al; MIRACLE Study Group. Multicenter InSync Randomized Clinical Evaluation. Cardiac resynchronization in chronic heart failure. N Engl J Med 2002; 346:1845–1853.
- McAlister FA, Ezekowitz J, Hooton N, et al. Cardiac resynchronization therapy for patients with left ventricular systolic dysfunction: a systematic review. JAMA 2007; 297:2502–2514.
- Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med 1999; 341:709–717.
- Pitt B, Remme W, Zannad F, et al; Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study Investigators. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med 2003; 348:1309–1321.
- Pitt B, White H, Nicolau J, et al; EPHESUS Investigators. Eplerenone reduces mortality 30 days after randomization following acute myocardial infarction in patients with left ventricular systolic dysfunction and heart failure. J Am Coll Cardiol 2005; 46:425–431.
- Zannad F, McMurray JJ, Krum H, et al; EMPHASIS-HF Study Group. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med 2011; 364:11–21.
- Yancy CW, Jessup M, Bozkurt B, et al; American College of Cardiology Foundation; American Heart Association Task Force on Practice Guidelines. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013; 62:e147–e239.
- McMurray JJ, Packer M, Desai AS, et al; PARADIGM-HF Committees and Investigators. Dual angiotensin receptor and neprilysin inhibition as an alternative to angiotensin-converting enzyme inhibition in patients with chronic systolic heart failure: rationale for and design of the Prospective comparison of ARNI with ACEI to Determine Impact on Global Mortality and morbidity in Heart Failure trial (PARADIGM-HF). Eur J Heart Fail 2013; 15:1062–1073.
- Sabe IA, Jacob MS, Taylor DO. A new class of drugs for systolic heart failure: The PARADIGM-HF study. Cleve Clin J Med 2015; 82:693–701.
- McMurray JJ, Packer M, Desai AS, Gong J, et al; PARADIGM-HF Investigators and Committees. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N Engl J Med 2014; 371:993–1004.
- US Food and Drug Administration. FDA approves new drug to treat heart failure. www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm453845.htm. Accessed September 2, 2015.
- Desai AS, McMurray JJ, Packer M, et al. Effect of the angiotensin-receptor-neprilysin inhibitor LCZ696 compared with enalapril on mode of death in heart failure patients. Eur Heart J 2015; 36:1990–1997.
- ClinicalTrials.gov. Efficacy and Safety of LCZ696 Compared to Valsartan, on Morbidity and Mortality in Heart Failure Patients With Preserved Ejection Fraction (PARAGON-HF). https://clinicaltrials.gov/ct2/show/NCT01920711. Accessed September 2, 2015.
Three decades ago, the only drugs we had for treating chronic heart failure were digitalis and loop diuretics. The mortality rate was very high, and heart transplantation was a newly developing treatment that could help only a very few patients.
The early 80s heralded new hope for patients with heart failure, with the introduction of angiotensin-converting enzyme (ACE) inhibitors1–5 and, later, beta-blockers. Beta-blockers were considered contraindicated in heart failure until new trials provided evidence of dramatic benefit such as better quality of life and longer survival.6–8 ACE inhibitors, along with beta-blockers, quickly became the standard of care for all patients with systolic heart failure.
The implantable cardioverter-defibrillator (ICD) required numerous clinical trials in ischemic and nonischemic cardiomyopathy to define its role.9,10 Cardiac resynchronization therapy did not arrive until 15 years ago and is now indicated in a specific niche of patients with left bundle branch block.11,12 Mineralocorticoid antagonists required three pivotal clinical trials before their important role in the treatment of systolic heart failure was defined.13–16
And in the current decade, the roles of ACE inhibitors, angiotensin II receptor blockers (ARBs), beta-blockers, mineralocorticoid antagonists, ICDs, and cardiac resynchronization therapy have been further defined, as reflected in the latest guidelines for the treatment of systolic heart failure.17
Guideline-directed medical therapy for systolic heart failure with the agents and devices mentioned above improves quality of life and extends survival. It was therefore hard to imagine that any new additive therapy could offer significant incremental improvement. However, more than 5 years ago, in an ambitious effort, the largest global clinical trial ever performed in chronic heart failure was launched with a novel agent.18
THE PARADIGM-HF TRIAL
In this issue of the Journal, Sabe et al19 describe the results of the Prospective Comparison of ARNI With ACEI to Determine Impact on Global Mortality and Morbidity in Heart Failure (PARADIGM-HF) trial of the novel combination drug sacubitril-valsartan, designated LCZ696 during its development and now available as Entresto.20
The mean age of the 8,442 patients in PARADIGM-HF was 64, and 78% were men. Despite guideline-directed medical therapy (93% of the patients were receiving a beta-blocker, and 60% were receiving a mineralocorticoid receptor antagonist), patients had persistent symptoms and signs of heart failure, diminished health-related quality of life, reduced ejection fraction (mean 29%), and elevated n-terminal pro-B-type natriuretic peptide levels (median 1,608 pg/mL, interquartile range 886–3,221).
The investigators reported a remarkable 20% reduction in the primary outcome of death from cardiovascular causes or hospitalization for heart failure in the patients who received sacubitril-valsartan compared with enalapril.20
Sacubitril-valsartan was reviewed under a US Food and Drug Administration (FDA) program that provides expedited review of drugs that are intended to treat a serious disease or condition and that may provide a significant improvement over available therapy. It was also granted a fast-track designation, which supports FDA efforts to facilitate the development and expedite the review of drugs to treat serious and life-threatening conditions and fill an unmet medical need. The FDA approved sacubitril-valsartan on July 7, 2015, for use in place of an ACE inhibitor or ARB in patients with New York Heart Association class II, III, or IV heart failure with reduced ejection fraction.21
WHAT WE STILL NEED TO KNOW
The results of PARADIGM-HF are generalizable, and sacubitril-valsartan was well tolerated in patients whose blood pressure was acceptable and who were able to tolerate ACE inhibitors in target doses. More than 90% of patients were receiving a beta-blocker. The dosing of enalapril (target 10 mg twice a day) is the guideline-directed target dose, and ACE inhibition is considered the gold standard for heart failure with reduced ejection fraction. Sacubitril-valsartan vs enalapril was a very appropriate comparison.
Far fewer PARADIGM-HF patients outside the United States had an ICD than those in the United States, which is a common finding in global clinical trials. However, Desai et al reported that sacubitril-valsartan reduced rates of cardiovascular mortality both from worsening heart failure and from sudden cardiac death, independent of whether the patient had an ICD.22
Sacubitril-valsartan is taken twice a day, but most heart failure patients already take medications at several times during the day, so this should not pose a problem.
More information is needed on the use of this new drug in patients with New York Heart Association class IV symptoms, as only 60 patients with class IV symptoms were included in the PARADIGM-HF trial. Also, the efficacy of the drug in patients unable to tolerate a full dose will need to be analyzed.
PARADIGM-HF was conducted in stable, nonhospitalized patients with chronic heart failure; the use of the drug in new-onset heart failure and its initiation in hospitalized patients will require further study. In addition, the PARAGON-HF trial23 will examine the efficacy of sacubitril-valsartan in patients with heart failure and an ejection fraction of 45% or higher.
Sacubitril-valsartan ushers in a new era in heart failure treatment for patients with reduced ejection fraction and will certainly prompt quick revision of heart failure guidelines.
Three decades ago, the only drugs we had for treating chronic heart failure were digitalis and loop diuretics. The mortality rate was very high, and heart transplantation was a newly developing treatment that could help only a very few patients.
The early 80s heralded new hope for patients with heart failure, with the introduction of angiotensin-converting enzyme (ACE) inhibitors1–5 and, later, beta-blockers. Beta-blockers were considered contraindicated in heart failure until new trials provided evidence of dramatic benefit such as better quality of life and longer survival.6–8 ACE inhibitors, along with beta-blockers, quickly became the standard of care for all patients with systolic heart failure.
The implantable cardioverter-defibrillator (ICD) required numerous clinical trials in ischemic and nonischemic cardiomyopathy to define its role.9,10 Cardiac resynchronization therapy did not arrive until 15 years ago and is now indicated in a specific niche of patients with left bundle branch block.11,12 Mineralocorticoid antagonists required three pivotal clinical trials before their important role in the treatment of systolic heart failure was defined.13–16
And in the current decade, the roles of ACE inhibitors, angiotensin II receptor blockers (ARBs), beta-blockers, mineralocorticoid antagonists, ICDs, and cardiac resynchronization therapy have been further defined, as reflected in the latest guidelines for the treatment of systolic heart failure.17
Guideline-directed medical therapy for systolic heart failure with the agents and devices mentioned above improves quality of life and extends survival. It was therefore hard to imagine that any new additive therapy could offer significant incremental improvement. However, more than 5 years ago, in an ambitious effort, the largest global clinical trial ever performed in chronic heart failure was launched with a novel agent.18
THE PARADIGM-HF TRIAL
In this issue of the Journal, Sabe et al19 describe the results of the Prospective Comparison of ARNI With ACEI to Determine Impact on Global Mortality and Morbidity in Heart Failure (PARADIGM-HF) trial of the novel combination drug sacubitril-valsartan, designated LCZ696 during its development and now available as Entresto.20
The mean age of the 8,442 patients in PARADIGM-HF was 64, and 78% were men. Despite guideline-directed medical therapy (93% of the patients were receiving a beta-blocker, and 60% were receiving a mineralocorticoid receptor antagonist), patients had persistent symptoms and signs of heart failure, diminished health-related quality of life, reduced ejection fraction (mean 29%), and elevated n-terminal pro-B-type natriuretic peptide levels (median 1,608 pg/mL, interquartile range 886–3,221).
The investigators reported a remarkable 20% reduction in the primary outcome of death from cardiovascular causes or hospitalization for heart failure in the patients who received sacubitril-valsartan compared with enalapril.20
Sacubitril-valsartan was reviewed under a US Food and Drug Administration (FDA) program that provides expedited review of drugs that are intended to treat a serious disease or condition and that may provide a significant improvement over available therapy. It was also granted a fast-track designation, which supports FDA efforts to facilitate the development and expedite the review of drugs to treat serious and life-threatening conditions and fill an unmet medical need. The FDA approved sacubitril-valsartan on July 7, 2015, for use in place of an ACE inhibitor or ARB in patients with New York Heart Association class II, III, or IV heart failure with reduced ejection fraction.21
WHAT WE STILL NEED TO KNOW
The results of PARADIGM-HF are generalizable, and sacubitril-valsartan was well tolerated in patients whose blood pressure was acceptable and who were able to tolerate ACE inhibitors in target doses. More than 90% of patients were receiving a beta-blocker. The dosing of enalapril (target 10 mg twice a day) is the guideline-directed target dose, and ACE inhibition is considered the gold standard for heart failure with reduced ejection fraction. Sacubitril-valsartan vs enalapril was a very appropriate comparison.
Far fewer PARADIGM-HF patients outside the United States had an ICD than those in the United States, which is a common finding in global clinical trials. However, Desai et al reported that sacubitril-valsartan reduced rates of cardiovascular mortality both from worsening heart failure and from sudden cardiac death, independent of whether the patient had an ICD.22
Sacubitril-valsartan is taken twice a day, but most heart failure patients already take medications at several times during the day, so this should not pose a problem.
More information is needed on the use of this new drug in patients with New York Heart Association class IV symptoms, as only 60 patients with class IV symptoms were included in the PARADIGM-HF trial. Also, the efficacy of the drug in patients unable to tolerate a full dose will need to be analyzed.
PARADIGM-HF was conducted in stable, nonhospitalized patients with chronic heart failure; the use of the drug in new-onset heart failure and its initiation in hospitalized patients will require further study. In addition, the PARAGON-HF trial23 will examine the efficacy of sacubitril-valsartan in patients with heart failure and an ejection fraction of 45% or higher.
Sacubitril-valsartan ushers in a new era in heart failure treatment for patients with reduced ejection fraction and will certainly prompt quick revision of heart failure guidelines.
- Captopril Multicenter Research Group. A placebo-controlled trial of captopril in refractory chronic congestive heart failure. J Am Coll Cardiol 1983; 2:755–763.
- Effects of enalapril on mortality in severe congestive heart failure. Results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS). The CONSENSUS Trial Study Group. N Engl J Med 1987; 316:1429–1435.
- The SOLVD Investigators. Effect of enalapril on survival in patients with reduced left ventricular ejection fraction and congestive heart failure. N Engl J Med 1991; 325:293–302.
- Cohn JN, Johnson G, Ziesche S, et al. A comparison of enalapril with hydralazine-isosorbide dinitrate in the treatment of chronic congestive heart failure. N Engl J Med 1991; 325:303–310.
- Pfeffer MA, Braunwald E, Moyé LA, et al. Effect of captopril on mortality and morbidity in patients with left ventricular dysfunction after myocardial infarction. Results of the survival and ventricular enlargement trial. The SAVE Investigators. N Engl J Med 1992; 327:669–677.
- Packer M, Coats AJ, Fowler MB, et al. Effect of carvedilol on survival in severe chronic heart failure. N Engl J Med 2001; 344:1651–1658.
- Effect of metoprolol CR/XL in chronic heart failure: Metoprolol CR/XL Randomised Intervention Trial in Congestive Heart Failure (MERIT-HF). Lancet 1999; 353:2001–2007.
- Brophy JM, Joseph L, Rouleau JL. Beta-blockers in congestive heart failure. A Bayesian meta-analysis. Ann Intern Med 2001; 134:550–560.
- Buxton AE, Lee KL, Fisher JD, et al. A randomized study of the prevention of sudden death in patients with coronary artery disease. Multicenter Unsustained Tachycardia Trial Investigators. N Engl J Med 1999; 341:1882–1890.
- Moss AJ, Zareba W, Hall WJ, et al. Prophylactic implantation of a defibrillator in patients with myocardial infarction and reduced ejection fraction. N Engl J Med 2002; 346:877–883.
- Abraham WT, Fisher WG, Smith AL, et al; MIRACLE Study Group. Multicenter InSync Randomized Clinical Evaluation. Cardiac resynchronization in chronic heart failure. N Engl J Med 2002; 346:1845–1853.
- McAlister FA, Ezekowitz J, Hooton N, et al. Cardiac resynchronization therapy for patients with left ventricular systolic dysfunction: a systematic review. JAMA 2007; 297:2502–2514.
- Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med 1999; 341:709–717.
- Pitt B, Remme W, Zannad F, et al; Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study Investigators. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med 2003; 348:1309–1321.
- Pitt B, White H, Nicolau J, et al; EPHESUS Investigators. Eplerenone reduces mortality 30 days after randomization following acute myocardial infarction in patients with left ventricular systolic dysfunction and heart failure. J Am Coll Cardiol 2005; 46:425–431.
- Zannad F, McMurray JJ, Krum H, et al; EMPHASIS-HF Study Group. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med 2011; 364:11–21.
- Yancy CW, Jessup M, Bozkurt B, et al; American College of Cardiology Foundation; American Heart Association Task Force on Practice Guidelines. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013; 62:e147–e239.
- McMurray JJ, Packer M, Desai AS, et al; PARADIGM-HF Committees and Investigators. Dual angiotensin receptor and neprilysin inhibition as an alternative to angiotensin-converting enzyme inhibition in patients with chronic systolic heart failure: rationale for and design of the Prospective comparison of ARNI with ACEI to Determine Impact on Global Mortality and morbidity in Heart Failure trial (PARADIGM-HF). Eur J Heart Fail 2013; 15:1062–1073.
- Sabe IA, Jacob MS, Taylor DO. A new class of drugs for systolic heart failure: The PARADIGM-HF study. Cleve Clin J Med 2015; 82:693–701.
- McMurray JJ, Packer M, Desai AS, Gong J, et al; PARADIGM-HF Investigators and Committees. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N Engl J Med 2014; 371:993–1004.
- US Food and Drug Administration. FDA approves new drug to treat heart failure. www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm453845.htm. Accessed September 2, 2015.
- Desai AS, McMurray JJ, Packer M, et al. Effect of the angiotensin-receptor-neprilysin inhibitor LCZ696 compared with enalapril on mode of death in heart failure patients. Eur Heart J 2015; 36:1990–1997.
- ClinicalTrials.gov. Efficacy and Safety of LCZ696 Compared to Valsartan, on Morbidity and Mortality in Heart Failure Patients With Preserved Ejection Fraction (PARAGON-HF). https://clinicaltrials.gov/ct2/show/NCT01920711. Accessed September 2, 2015.
- Captopril Multicenter Research Group. A placebo-controlled trial of captopril in refractory chronic congestive heart failure. J Am Coll Cardiol 1983; 2:755–763.
- Effects of enalapril on mortality in severe congestive heart failure. Results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS). The CONSENSUS Trial Study Group. N Engl J Med 1987; 316:1429–1435.
- The SOLVD Investigators. Effect of enalapril on survival in patients with reduced left ventricular ejection fraction and congestive heart failure. N Engl J Med 1991; 325:293–302.
- Cohn JN, Johnson G, Ziesche S, et al. A comparison of enalapril with hydralazine-isosorbide dinitrate in the treatment of chronic congestive heart failure. N Engl J Med 1991; 325:303–310.
- Pfeffer MA, Braunwald E, Moyé LA, et al. Effect of captopril on mortality and morbidity in patients with left ventricular dysfunction after myocardial infarction. Results of the survival and ventricular enlargement trial. The SAVE Investigators. N Engl J Med 1992; 327:669–677.
- Packer M, Coats AJ, Fowler MB, et al. Effect of carvedilol on survival in severe chronic heart failure. N Engl J Med 2001; 344:1651–1658.
- Effect of metoprolol CR/XL in chronic heart failure: Metoprolol CR/XL Randomised Intervention Trial in Congestive Heart Failure (MERIT-HF). Lancet 1999; 353:2001–2007.
- Brophy JM, Joseph L, Rouleau JL. Beta-blockers in congestive heart failure. A Bayesian meta-analysis. Ann Intern Med 2001; 134:550–560.
- Buxton AE, Lee KL, Fisher JD, et al. A randomized study of the prevention of sudden death in patients with coronary artery disease. Multicenter Unsustained Tachycardia Trial Investigators. N Engl J Med 1999; 341:1882–1890.
- Moss AJ, Zareba W, Hall WJ, et al. Prophylactic implantation of a defibrillator in patients with myocardial infarction and reduced ejection fraction. N Engl J Med 2002; 346:877–883.
- Abraham WT, Fisher WG, Smith AL, et al; MIRACLE Study Group. Multicenter InSync Randomized Clinical Evaluation. Cardiac resynchronization in chronic heart failure. N Engl J Med 2002; 346:1845–1853.
- McAlister FA, Ezekowitz J, Hooton N, et al. Cardiac resynchronization therapy for patients with left ventricular systolic dysfunction: a systematic review. JAMA 2007; 297:2502–2514.
- Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med 1999; 341:709–717.
- Pitt B, Remme W, Zannad F, et al; Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study Investigators. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med 2003; 348:1309–1321.
- Pitt B, White H, Nicolau J, et al; EPHESUS Investigators. Eplerenone reduces mortality 30 days after randomization following acute myocardial infarction in patients with left ventricular systolic dysfunction and heart failure. J Am Coll Cardiol 2005; 46:425–431.
- Zannad F, McMurray JJ, Krum H, et al; EMPHASIS-HF Study Group. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med 2011; 364:11–21.
- Yancy CW, Jessup M, Bozkurt B, et al; American College of Cardiology Foundation; American Heart Association Task Force on Practice Guidelines. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013; 62:e147–e239.
- McMurray JJ, Packer M, Desai AS, et al; PARADIGM-HF Committees and Investigators. Dual angiotensin receptor and neprilysin inhibition as an alternative to angiotensin-converting enzyme inhibition in patients with chronic systolic heart failure: rationale for and design of the Prospective comparison of ARNI with ACEI to Determine Impact on Global Mortality and morbidity in Heart Failure trial (PARADIGM-HF). Eur J Heart Fail 2013; 15:1062–1073.
- Sabe IA, Jacob MS, Taylor DO. A new class of drugs for systolic heart failure: The PARADIGM-HF study. Cleve Clin J Med 2015; 82:693–701.
- McMurray JJ, Packer M, Desai AS, Gong J, et al; PARADIGM-HF Investigators and Committees. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N Engl J Med 2014; 371:993–1004.
- US Food and Drug Administration. FDA approves new drug to treat heart failure. www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm453845.htm. Accessed September 2, 2015.
- Desai AS, McMurray JJ, Packer M, et al. Effect of the angiotensin-receptor-neprilysin inhibitor LCZ696 compared with enalapril on mode of death in heart failure patients. Eur Heart J 2015; 36:1990–1997.
- ClinicalTrials.gov. Efficacy and Safety of LCZ696 Compared to Valsartan, on Morbidity and Mortality in Heart Failure Patients With Preserved Ejection Fraction (PARAGON-HF). https://clinicaltrials.gov/ct2/show/NCT01920711. Accessed September 2, 2015.
A new class of drugs for systolic heart failure: The PARADIGM-HF study
In a large phase trial, a combination drug that contains the angiotensin II receptor blocker (ARB) valsartan and the neprilysin inhibitor sacubitril was found to be superior to the angiotensin-converting enzyme (ACE) inhibitor enalapril in terms of important end points, including death and hospitalization for heart failure, in patients with heart failure with reduced ejection fraction.1
Recently approved by the US Food and Drug Administration, this combination drug, marketed under the brand name Entresto, represents a new drug class, angiotensin receptor-neprilysin inhibitors, or ARNIs.
This article is an overview of the Prospective Comparison of ARNI With ACEI to Determine Impact on Global Mortality and Morbidity in Heart Failure (PARADIGM-HF) trial1 and the implications it may have on the care of patients with chronic heart failure.
NEED FOR NEW HEART FAILURE DRUGS

Heart failure is a major public health problem, and the care of patients with heart failure is challenging.
Almost 6 million US adults have heart failure, and the prevalence is projected to increase in the next few decades as the population continues to age.2 Furthermore, the total healthcare cost for heart failure patients was almost $31 billion in 2012 and is projected to rise to $70 billion by 2030.2
The care of patients with severely decompensated heart failure has changed dramatically in the last few decades with advances in heart transplantation and mechanical support devices. But day-to-day management of patients with chronic mildly to moderately symptomatic heart failure continues to pose a clinical challenge.
The drugs currently available for these patients include beta-blockers, ACE inhibitors, ARBs, aldosterone antagonists, digoxin, diuretics, and vasodilators. But even with these drugs, the death and readmission rates of patients with heart failure with reduced ejection fraction remain high. More than 50% of patients with heart failure die within 5 years of diagnosis,3 and 25% of patients hospitalized with heart failure are readmitted within 30 days of discharge.2 Furthermore, death rates are higher in those patients who have a history of heart failure hospitalization.4
Although heart failure with preserved ejection fraction encompasses an important group of heart failure patients with high morbidity, the focus of this article will be on patients with heart failure with reduced ejection fraction.
Available drugs to date
The cornerstone drugs that lower the odds of death in patients with heart failure with reduced ejection fraction are ACE inhibitors, ARBs, beta-blockers, and mineralocorticoid antagonists.
ACE inhibitors were the first class of drugs shown to reduce the death rate in patients with heart failure with reduced ejection fraction. The landmark CONSENSUS trial,5 published in 1987, found that the death rate in patients who received enalapril was 27% lower than in those receiving placebo, an effect driven entirely by a reduction in progressive heart failure. Similarly, the SOLVD trial,6 published in 1991, showed a 26% reduction in heart failure hospitalization and a 16% lower rate of death with enalapril compared with placebo, an effect driven predominantly by a decrease in the progression of heart failure.
ARBs have also been shown to decrease the rate of death, although not by as much as ACE inhibitors. In the CHARM trial,7 compared with placebo, candesartan significantly decreased the risk of death from any cause, of death from cardiovascular causes, and of hospitalization related to heart failure.7
Beta-blockers. The MERIT-HF trial,8 published in 1999, was stopped early because fewer patients were dying in the group receiving metoprolol succinate than in the group receiving placebo (relative risk 0.66). Similarly, in 2001, the COPERNICUS trial9 reported a 34% reduction in deaths in patients receiving carvedilol in addition to an ACE inhibitor compared with those receiving an ACE inhibitor alone.
Mineralocorticoid receptor antagonists were found to be beneficial when added to standard therapy for chronic symptomatic heart failure in the RALES10 and EMPHASIS-HF11 trials.
Vasodilators (specifically, the combination of isosorbide dinitrate and hydralazine) were found to have benefit in terms of mortality when added to standard therapy in African American patients in the A-HeEFT trial.12
WHY INHIBIT BOTH ANGIOTENSIN AND NEPRILYSIN?
The renin-angiotensin-aldosterone system is a major focus in treating heart failure, as overactivity of this system plays a key role in the pathophysiology of this disease. Therefore, essential drugs for heart failure patients include those that inhibit overactivity of this system such as ACE inhibitors, ARBs, and aldosterone antagonists.
The natriuretic peptide system is another important pathway that can be targeted in patients with heart failure. Natriuretic peptides are key molecules that counteract heart failure, as they contribute to diuresis and vasodilation and protect against vascular remodeling.13 An increased understanding of the importance of this system in slowing the progression of heart failure has motivated evaluation of drugs such as nesiritide in patients with symptomatic heart failure. However, these drugs can cause hypotension and have limited bioavailability.14
Neprilysin is an endopeptidase—an endogenous enzyme that degrades vasoactive peptides such as bradykinin and natriuretic peptides.14 Drugs that inhibit neprilysin increase the levels of these peptides and thus counteract neurohormonal stimuli that lead to cardiac remodeling, sodium retention, and vasoconstriction.15
However, neprilysin also hydrolyzes angiotensin I to angiotensin (1–7), an inhibitor of angiotensin II. Thus, inhibition of neprilysin alone could lead to increased activity of angiotensin II and so have an overall neutral effect. To be beneficial, neprilysin inhibition needs to be combined with renin-angiotensin system inhibition. Furthermore, the benefit of renin-angiotensin-aldosterone system blockade may be amplified by up-regulation of the endogenous natriuretic peptide system.15
Omapatrilat, the most studied combination neprilysin inhibitor and ACE inhibitor, improved cardiac function and decreased cardiac mass in animal experiments.15 In addition, this drug showed promise in terms of blood pressure, heart failure readmissions, death, and preservation of renal function when compared with ACE inhibitors in patients with heart failure.15–17 But in clinical trials this drug posed a greater risk of hypotension, dizziness, and, its major shortcoming, an unacceptably high incidence of angioedema compared with ACE inhibitors.15,16,18 This higher risk of angioedema is thought to be from inhibition of three enzymes that break down bradykinin: ACE, neprilysin, and aminopeptidase P.19
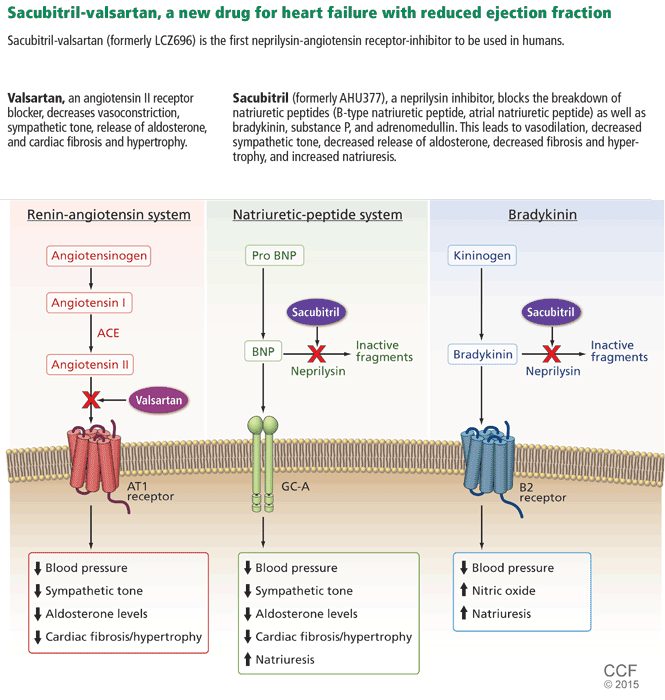
ARNIs contain an angiotensin receptor blocker rather than an ACE inhibitor, and thus in theory they may be associated with a lower risk of angioedema.19 Sacubitril-valsartan, the first drug of this class, contains its two constitutive drugs in a one-to-one molecular ratio (Figure 1).
PARADIGM-HF investigated the benefit of this drug in patients with systolic heart failure.1
STUDY DESIGN AND OBJECTIVES
PARADIGM-HF was a double-blind, randomized controlled trial comparing sacubitril-valsartan and enalapril in patients with chronic systolic heart failure. As such, it was the first trial in recent years to study a new drug in comparison with a well-established heart failure drug rather than as an add-on strategy.1
Inclusion criteria
To be included in the PARADIGM-HF trial, patients had to have:
- A left ventricular ejection fraction of 40% or less (later changed to ≤ 35%)
- New York Heart Association class II, III, or IV symptoms
- A B-type natriuretic peptide (BNP) level of at least 150 pg/mL or an N-terminal proBNP (NT-proBNP) level of at least 600 pg/mL; for patients hospitalized for heart failure within the previous 12 months, the cut points were lower (BNP ≥ 100 pg/mL or NT-proBNP ≥ 400 pg/mL).
End points
The primary end point was the composite of cardiovascular death or first hospitalization for heart failure. Other outcomes assessed were time to death from any cause, the change from baseline in the Kansas City Cardiomyopathy Questionnaire (KCCQ) score at 8 months, time to new-onset atrial fibrillation, and the time to decline in renal function (defined as end-stage renal disease or a decrease in estimated glomerular filtration rate of at least 50% from randomization). All end points were blindly adjudicated by a clinical end points committee.
Two run-in periods
The study enrolled 10,521 patients from 1,043 centers in 47 countries, who entered the initial run-in period consisting of 2 weeks of treatment with enalapril at the study dosage (10 mg twice daily) in order to ensure no unacceptable side effects. At this point, 1,102 patients exited the study, leaving 9,419 who entered the second run-in period.
The second run-in period consisted of 4 weeks of treatment with sacubitril-valsartan, initially at half the study regimen (100 mg twice daily) and eventually at the full study dosage (200 mg twice daily). During the second run-in period, 977 participants left the study, leaving a total of 8,442 patients who underwent randomization. Forty-three patients were then excluded (6 because of invalid randomization and 37 because of four sites that closed because of major violations of good clinical practice).
Of those randomized, 4,187 patients were assigned to the sacubitril-valsartan treatment group and 4,212 were assigned to the enalapril group. The investigators used an intention-to-treat analysis for this study.
Most patients had NYHA class II symptoms
The randomized patients had a mean age of 64 years, 75% were men, 66% were white, and 58% were from Europe (only 7% were from North America). The mean left ventricular ejection fraction was about 30%, and 60% of the study participants had an ischemic cause for their cardiomyopathy. Although one of the inclusion criteria was New York Heart Association class II, III, or IV symptoms, about 5% of the patients had class I symptoms. Seventy percent had class II symptoms, 24% had class III, and less than 1% had class IV symptoms.
At the time of randomization, 78% of the patients were taking an ACE inhibitor and 93% were taking a beta-blocker, but only a little more than half were taking a mineralocorticoid antagonist and only about 15% had an implantable cardioverter-defibrillator.
STUDY OUTCOMES
This study was designed to detect a 15% lower risk of cardiovascular death in the sacubitril-valsartan group. It was projected to continue for at least 34 months but was stopped early because of an overwhelming benefit of the new drug at a median follow-up of 27 months.
Major findings
The primary composite outcome (cardiovascular death or first hospitalization for heart failure)1 occurred in 21.8% of the patients in the sacubitril-valsartan group vs 26.5% of patients in the enalapril group (hazard ratio [HR] 0.80, 95% confidence interval [CI] 0.73–0.87, P < .001). The number of patients who needed to be treated to prevent one occurrence of the primary composite outcome (100/absolute risk reduction) was only 21. The benefit was strong and consistent across both of the individual components of the composite outcome:
- Cardiovascular death 13.3% vs 16.5%, HR 0.80 (95% CI 0.71–0.89), P < .001
- First hospitalization for worsening heart failure 12.8% vs 15.6%, HR 0.79 (95% CI 0.71–0.89), P < .001.
Secondary outcomes. The sacubitril-valsartan group had a significantly lower rate of death from any cause (17.0% vs 19.8%, HR 0.84, 95% CI 0.76–0.93, P < .001) and a lower mean decrease in KCCQ clinical summary scores at 8 months (2.99 points vs 4.63 points, mean difference 1.64, 95% CI 0.63–2.65, P = .001). The KCCQ score measures subjective symptoms and physical limitations caused by heart failure; possible scores range from 0 to 100, with a higher score indicating better functional status. Notably, sacubitril-valsartan did not increase the KCCQ score in these patients; rather, sacubitril-valsartan recipients had a lower decrease in their scores than those in the enalapril group.
The incidence of new-onset atrial fibrillation was the same in both groups (3.1% and 3.1%).1 A decline in renal function (defined as end-stage renal disease, a decrease of 50% or more in estimated glomerular filtration rate from the value at randomization or a decrease in the estimated glomerular filtration rate of more than 30 mL/min/1.73 m2 to less than 60 mL/min/1.73 m2) occurred in 2.2% of the valsartan-sacubitril group and 2.6% of the enalapril group (P = .28).
The effects of the study drug on the primary composite outcome and on death from a cardiovascular cause were similar in all prespecified subgroups except for NYHA class: the reduction in the risk of the composite outcome was lower in sacubitril-valsartan recipients with NYHA I or II symptoms than in those with NYHA III or IV symptoms (P for interaction .03). However, there were no differences in the other prespecified subgroups, defined by age, sex, race, region, estimated glomerular filtration rate, diabetes, systolic blood pressure, ejection fraction, atrial fibrillation, NT-proBNP, hypertension, previous use of an ACE inhibitor, previous use of an aldosterone antagonist, previous heart failure hospitalization, and time since diagnosis of heart failure.
SAFETY: ANGIOEDEMA, HYPOTENSION, AND RENAL DYSFUNCTION
Angioedema
As noted above, the combination of neprilysin inhibitors and ACE inhibitors has been associated with an increased risk of angioedema. That was an important consideration before starting this study, which used a combination of a neprilysin inhibitor and an ARB in an attempt to avoid this serious side effect.
As it happened, there was no increased risk of significant angioedema with sacubitril-valsartan use compared with enalapril. Rates were similar to those in other studies, which showed a less than 1% risk of angioedema caused by ACE inhibitors.20,21 Only 19 patients (0.5%) in the sacubitril-valsartan group and 10 patients (0.2%) in the enalapril group experienced any angioedema. Of these, just three patients in the sacubitril-valsartan group and one patient in the enalapril group experienced angioedema that required hospitalization (P = .31). None of these patients had airway compromise due to angioedema.
Hypotension, cough, renal dysfunction, hyperkalemia
Other safety issues that were assessed included hypotension, worsening renal function, increase in potassium levels, and cough. Patients in the sacubitril-valsartan group were more likely to have symptomatic hypotension than patients in the enalapril group (14.0% vs 9.2%, P < .001); however, the authors noted that this was a rare cause of drug discontinuation.
Patients in the sacubitril-valsartan group were less likely to develop cough (11.3% vs 14.3%, P < .001), a serum creatinine level of 2.5 mg/dL or more (3.3% vs 4.5%, P = .007), or a serum potassium level of more than 6.0 mmol/L (11.3% vs 14.3%, P = .007).1
During the two run-in periods combined, 12% of the patients in the study withdrew because of adverse events, including cough, renal dysfunction, hyperkalemia, and symptomatic hypotension. During the enalapril run-in period, 591 patients (5.6%) withdrew due to adverse events, and 547 patients (5.8%) withdrew due to these events during the sacubitril-valsartan run-in period. After adjusting for the shorter time on enalapril, there was a higher rate of withdrawal because of adverse events from enalapril than from sacubitril-valsartan.
LOWER RISK OF CLINICAL PROGRESSION
In a separate paper,22 the PARADIGM-HF investigators reported that, among the survivors in the study, those who received sacubitril-valsartan fared better in terms of a number of markers of progression of heart failure, with lower rates of:
- Intensification of medical treatment for heart failure
- Emergency department visits for worsening heart failure
- Hospitalization for worsening heart failure
- Need for intensive care
- Need for intravenous inotropic agents
- Need for cardiac devices or heart transplants
- Worsening symptom scores
- Elevation of biomarkers of myocardial injury.
QUESTIONS AND CONCERNS
Although this study, which was funded by the manufacturer, showed consistent benefit for sacubitril-valsartan over enalapril, questions remain.
Are the findings generalizable?
Despite the study’s rigorous run-in period, 12% of patients dropped out because of adverse events, and thus the patients who completed the study may not be representative of the general population of heart failure patients. The authors included this double-level wash-out to ensure patient tolerance of both drugs. But in everyday practice, a significant number of patients may be unable to tolerate one of these drugs.
Moreover, after adjusting for the difference in the run-in periods, patients actually withdrew more often during the enalapril run-in period than during the sacubitril-valsartan run-in period. However, there may be overlap in tolerability in these two drugs, which both affect the renin-angiotensin-aldosterone system. Thus, the enalapril run-in period may have contributed to the lower tolerability of this drug compared with sacubitril-valsartan.
Were patients receiving the best possible therapy?
Another important point when considering how we treat heart failure patients in the United States is how few patients in this study were using cardiac implantable electronic devices. Only 15% of the patients in this study had an implantable cardioverter-defibrillator despite a mean left ventricular ejection fraction less than 30%. This likely reflects differences in practice internationally; however, based on American College of Cardiology, American Heart Association, and Heart Rhythm Society guidelines, these patients would have a class I indication for an implantable cardioverter-defibrillator for primary prevention of sudden cardiac death.23
Therefore, based on these recommendations, the patients in this study were not necessarily on optimal medical and device therapy and furthermore may not be representative of heart failure patients in the United States.
Was enalapril 10 mg twice a day a fair comparison?
Another concern about the results of this study relates to the dosages used in the two treatment groups. The sacubitril-valsartan formulation included full-dose valsartan, whereas the ACE inhibitor group received enalapril at less than a full dose.
Although the authors explained that the dose of enalapril chosen for the study was based on the one used in previous studies that showed survival benefit, this raises the question of whether the significant difference in outcomes in this trial was due to a greater inhibition of the renin-angiotensin-aldosterone system related to a higher dose of drug in the sacubitril-valsartan group.
What about black patients taking hydralazine-isosorbide?
Only about 5% of patients in the PARADIGM-HF trial were black. Based on the A-HeFT study results, black patients can be prescribed an ACE inhibitor as well as hydralazine and isosorbide dinitrate as tolerated to decrease the risk of death. Does sacubitril-valsartan offer benefit to these patients compared with a regimen of an ACE inhibitor, hydralazine, and isosorbide dinitrate?
Another concern is that the incidence of angioedema observed with ACE inhibitors and omapatrilat is higher in black patients.15,21 Thus, it would be prudent to investigate whether the risk of angioedema with sacubitril-valsartan would be higher if more black patients are studied.
IMPLICATIONS AND CONSIDERATIONS
In this study, sacubitril-valsartan showed impressive and consistent results, with an almost 20% decrease in the composite end point of heart failure hospitalization or cardiovascular death and a similar decrease in the composite outcomes with a very low number needed to treat (21 patients). It did not show a decrease in the incidence of new-onset atrial fibrillation; however, only 80 cases of atrial fibrillation were reported, so there may have been a lack of statistical power to detect a difference.
To avoid angioedema, wait at least 36 hours after stopping an ACE inhibitor. Sacubitril-valsartan was not associated with an increased risk of severe angioedema, and no patients experienced life-threatening angioedema. In the trial, the sacubitril-valsartan run-in period was started at least 24 hours after enalapril was stopped, and thus the authors recommended at least a 1-day washout period after discontinuing an ACE inhibitor to avoid angioedema in patients starting sacubitril-valsartan.
Hypotension is a concern. Although there was actually a decreased risk of renal dysfunction, hyperkalemia, and cough compared with enalapril, there was a significantly increased rate of symptomatic hypotension in the sacubitril-valsartan group, which raises the question of patient tolerance and physician comfort when prescribing and titrating this drug in clinical practice. This side effect will be an important consideration when attempting to titrate the drug to target doses.
Start treatment early. This trial largely consisted of patients with NYHA class II or III symptoms, with about 70% of patients with class II symptoms. Since this drug showed benefit in patients with mildly to moderately symptomatic heart failure, clinicians who are considering prescribing this drug should not wait until the patient is closer to end-stage disease. Patients with mildly symptomatic heart failure may be followed by a general cardiologist, internist, or both, and thus it is important to emphasize to the entire medical community the need to start this medication early on.

How much will it cost? Cost is a concern that could heavily weigh on the decision to prescribe this drug. Generic ACE inhibitors are relatively inexpensive, and it may difficult to switch from an affordable generic drug to a new drug that is likely to be much more expensive. Arguably, this drug may be cost-effective in the long run owing to a large decrease in heart failure readmissions. We await further analyses to evaluate this issue.
Will patients take a twice-a-day drug as prescribed? Most patients who are prescribed an ACE inhibitor take it just once a day, and switching from a daily to a twice-daily drug may present a challenge for some.
What about other outcomes? Based on this study, it is unclear what effect sacubitril-valsartan has on the incidence of fatal arrhythmias, sudden cardiac death, and pump failure. Furthermore, the effect on quality of life is still uncertain. Quality of life is an integral component in the evaluation of heart failure patients, and in this study the changes in KCCQ scores were not impressive. We hope to see further evaluations of this drug’s impact on quality of life of patients with heart failure. Furthermore, it would be interesting to study if this drug has any long-term effects on the need for advanced therapies such as left ventricular assist devices and orthotopic heart transplant.
What about patients with heart failure with preserved ejection fraction? This study included only patients with heart failure with reduced ejection fraction. However PARAMOUNT, a phase 2 study that evaluated the benefit of sacubitril-valsartan in patients with heart failure with preserved ejection fraction, has shown encouraging results.24 We look forward to further investigation of this agent in patients with heart failure with preserved ejection fraction.
Sacubitril-valsartan, the first ARNI to be studied in humans, has a dual action in that it enhances the activity of the natriuretic peptide system and inhibits that of the renin-angiotensin-aldosterone system. It is the first drug in over a decade to show mortality benefit in patients with chronic systolic heart failure when compared with an already well-established heart failure medication. It appears to decrease rates of mortality and heart failure hospitalization without increasing the risk of severe angioedema in patients with mild or moderate chronic systolic heart failure. Symptomatic hypotension and high cost may pose the largest barriers to the use of this new drug. And we have yet to see how the clinical community and patients with heart failure will respond to it.
- McMurray JJ, Packer M, Desai AS, et al; PARADIGM-HF Investigators and Committees. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N Engl J Med 2014; 371:993–1004.
- Dunlay SM, Pereira NL, Kushwaha SS. Contemporary strategies in the diagnosis and management of heart failure. Mayo Clin Proc 2014; 89:662–676.
- Go AS, Mozaffarian D, Roger VL, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2013 update: a report from the American Heart Association. Circulation 2013; 127:e6–e245.
- Bello NA, Claggett B, Desai AS, et al. Influence of previous heart failure hospitalization on cardiovascular events in patients with reduced and preserved ejection fraction. Circ Heart Fail 2014; 7:590–595.
- Effects of enalapril on mortality in severe congestive heart failure. Results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS). The CONSENSUS Trial Study Group. N Engl J Med 1987; 316:1429–1435.
- Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congestive heart failure. The SOLVD Investigators. N Engl J Med 1991; 325:293–302.
- Pfeffer MA, Swedberg K, Granger CB, et al; CHARM Investigators and Committees. Effects of candesartan on mortality and morbidity in patients with chronic heart failure: the CHARM-Overall programme. Lancet 2003; 362:759–766.
- Effect of metoprolol CR/XL in chronic heart failure: Metoprolol CR/XL Randomised Intervention Trial in Congestive Heart Failure (MERIT-HF). Lancet 1999; 353:2001–2007.
- Packer M, Coats AJ, Fowler MB, et al; Carvedilol Prospective Randomized Cumulative Survival Study Group. Effect of carvedilol on survival in severe chronic heart failure. N Engl J Med 2001; 344:1651–1658.
- Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med 1999; 341:709–717.
- Zannad F, McMurray JJ, Krum H, et al; EMPHASIS-HF Study Group. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med 2011; 364:11–21.
- Taylor AL, Ziesche S, Yancy C, et al; African-American Heart Failure Trial Investigators. Combination of isosorbide dinitrate and hydralazine in blacks with heart failure. N Engl J Med 2004; 351:2049–2057.
- Schreiner GF, Protter AA. B-type natriuretic peptide for the treatment of congestive heart failure. Curr Opin Pharmacol 2002; 2:142–147.
- von Lueder TG, Sangaralingham SJ, Wang BH, et al. Renin-angiotensin blockade combined with natriuretic peptide system augmentation: novel therapeutic concepts to combat heart failure. Circ Heart Fail 2013; 6:594–605.
- Abassi Z, Karram T, Ellaham S, Winaver J, Hoffman A. Implications of the natriuretic peptide system in the pathogenesis of heart failure: diagnostic and therapeutic importance. Pharmacol Ther 2004; 102:223–241.
- Packer M, Califf RM, Konstam MA, et al. Comparison of omapatrilat and enalapril in patients with chronic heart failure: the Omapatrilat Versus Enalapril Randomized Trial of Utility in Reducing Events (OVERTURE). Circulation 2002; 106:920–926.
- Rouleau JL, Pfeffer MA, Stewart DJ, et al. Comparison of vasopeptidase inhibitor, omapatrilat, and lisinopril on exercise tolerance and morbidity in patients with heart failure: IMPRESS randomised trial. Lancet 2000; 356:615–620.
- Kostis JB, Packer M, Black HR, Schmieder R, Henry D, Levy E. Omapatrilat and enalapril in patients with hypertension: the Omapatrilat Cardiovascular Treatment vs. Enalapril (OCTAVE) trial. Am J Hypertens 2004; 17:103–111.
- McMurray JJ, Packer M, Desai AS, et al; PARADIGM-HF Committees and Investigators. Dual angiotensin receptor and neprilysin inhibition as an alternative to angiotensin-converting enzyme inhibition in patients with chronic systolic heart failure: rationale for and design of the Prospective comparison of ARNI with ACEI to Determine Impact on Global Mortality and morbidity in Heart Failure trial (PARADIGM-HF). Eur J Heart Fail 2013; 15:1062–1073.
- Toh S, Reichman ME, Houstoun M, et al. Comparative risk for angioedema associated with the use of drugs that target the renin-angiotensin-aldosterone system. Arch Intern Med 2012; 172:1582–1589.
- Kostis JB, Kim HJ, Rusnak J, et al. Incidence and characteristics of angioedema associated with enalapril. Arch Intern Med 2005; 165:1637–1642.
- Packer M, McMurray JJV, Desai AS, et al; on behalf of the PARADIGM-HF Investigators and Coordinators. Angiotensin receptor neprilysin inhibition compared with enalapril on the risk of clinical progression in surviving patients with heart failure. Circulation 2015; 131:54–61.
- Epstein AE, Dimarco JP, Ellenbogen KA, et al; American College of Cardiology/American Heart Association Task Force on Practice; American Association for Thoracic Surgery; Society of Thoracic Surgeons. ACC/AHA/HRS 2008 guidelines for Device-Based Therapy of Cardiac Rhythm Abnormalities: executive summary. Heart Rhythm 2008; 5:934–955.
- Solomon SD, Zile M, Pieske B, et al; Prospective comparison of ARNI with ARB on Management Of Heart Failure with Preserved Ejection Fraction (PARAMOUNT) Investigators. The angiotensin receptor neprilysin inhibitor LCZ696 in heart failure with preserved ejection fraction: a phase 2 double-blind randomised controlled trial. Lancet 2012; 380:1387–1395.
In a large phase trial, a combination drug that contains the angiotensin II receptor blocker (ARB) valsartan and the neprilysin inhibitor sacubitril was found to be superior to the angiotensin-converting enzyme (ACE) inhibitor enalapril in terms of important end points, including death and hospitalization for heart failure, in patients with heart failure with reduced ejection fraction.1
Recently approved by the US Food and Drug Administration, this combination drug, marketed under the brand name Entresto, represents a new drug class, angiotensin receptor-neprilysin inhibitors, or ARNIs.
This article is an overview of the Prospective Comparison of ARNI With ACEI to Determine Impact on Global Mortality and Morbidity in Heart Failure (PARADIGM-HF) trial1 and the implications it may have on the care of patients with chronic heart failure.
NEED FOR NEW HEART FAILURE DRUGS
Heart failure is a major public health problem, and the care of patients with heart failure is challenging.
Almost 6 million US adults have heart failure, and the prevalence is projected to increase in the next few decades as the population continues to age.2 Furthermore, the total healthcare cost for heart failure patients was almost $31 billion in 2012 and is projected to rise to $70 billion by 2030.2
The care of patients with severely decompensated heart failure has changed dramatically in the last few decades with advances in heart transplantation and mechanical support devices. But day-to-day management of patients with chronic mildly to moderately symptomatic heart failure continues to pose a clinical challenge.
The drugs currently available for these patients include beta-blockers, ACE inhibitors, ARBs, aldosterone antagonists, digoxin, diuretics, and vasodilators. But even with these drugs, the death and readmission rates of patients with heart failure with reduced ejection fraction remain high. More than 50% of patients with heart failure die within 5 years of diagnosis,3 and 25% of patients hospitalized with heart failure are readmitted within 30 days of discharge.2 Furthermore, death rates are higher in those patients who have a history of heart failure hospitalization.4
Although heart failure with preserved ejection fraction encompasses an important group of heart failure patients with high morbidity, the focus of this article will be on patients with heart failure with reduced ejection fraction.
Available drugs to date
The cornerstone drugs that lower the odds of death in patients with heart failure with reduced ejection fraction are ACE inhibitors, ARBs, beta-blockers, and mineralocorticoid antagonists.
ACE inhibitors were the first class of drugs shown to reduce the death rate in patients with heart failure with reduced ejection fraction. The landmark CONSENSUS trial,5 published in 1987, found that the death rate in patients who received enalapril was 27% lower than in those receiving placebo, an effect driven entirely by a reduction in progressive heart failure. Similarly, the SOLVD trial,6 published in 1991, showed a 26% reduction in heart failure hospitalization and a 16% lower rate of death with enalapril compared with placebo, an effect driven predominantly by a decrease in the progression of heart failure.
ARBs have also been shown to decrease the rate of death, although not by as much as ACE inhibitors. In the CHARM trial,7 compared with placebo, candesartan significantly decreased the risk of death from any cause, of death from cardiovascular causes, and of hospitalization related to heart failure.7
Beta-blockers. The MERIT-HF trial,8 published in 1999, was stopped early because fewer patients were dying in the group receiving metoprolol succinate than in the group receiving placebo (relative risk 0.66). Similarly, in 2001, the COPERNICUS trial9 reported a 34% reduction in deaths in patients receiving carvedilol in addition to an ACE inhibitor compared with those receiving an ACE inhibitor alone.
Mineralocorticoid receptor antagonists were found to be beneficial when added to standard therapy for chronic symptomatic heart failure in the RALES10 and EMPHASIS-HF11 trials.
Vasodilators (specifically, the combination of isosorbide dinitrate and hydralazine) were found to have benefit in terms of mortality when added to standard therapy in African American patients in the A-HeEFT trial.12
WHY INHIBIT BOTH ANGIOTENSIN AND NEPRILYSIN?
The renin-angiotensin-aldosterone system is a major focus in treating heart failure, as overactivity of this system plays a key role in the pathophysiology of this disease. Therefore, essential drugs for heart failure patients include those that inhibit overactivity of this system such as ACE inhibitors, ARBs, and aldosterone antagonists.
The natriuretic peptide system is another important pathway that can be targeted in patients with heart failure. Natriuretic peptides are key molecules that counteract heart failure, as they contribute to diuresis and vasodilation and protect against vascular remodeling.13 An increased understanding of the importance of this system in slowing the progression of heart failure has motivated evaluation of drugs such as nesiritide in patients with symptomatic heart failure. However, these drugs can cause hypotension and have limited bioavailability.14
Neprilysin is an endopeptidase—an endogenous enzyme that degrades vasoactive peptides such as bradykinin and natriuretic peptides.14 Drugs that inhibit neprilysin increase the levels of these peptides and thus counteract neurohormonal stimuli that lead to cardiac remodeling, sodium retention, and vasoconstriction.15
However, neprilysin also hydrolyzes angiotensin I to angiotensin (1–7), an inhibitor of angiotensin II. Thus, inhibition of neprilysin alone could lead to increased activity of angiotensin II and so have an overall neutral effect. To be beneficial, neprilysin inhibition needs to be combined with renin-angiotensin system inhibition. Furthermore, the benefit of renin-angiotensin-aldosterone system blockade may be amplified by up-regulation of the endogenous natriuretic peptide system.15
Omapatrilat, the most studied combination neprilysin inhibitor and ACE inhibitor, improved cardiac function and decreased cardiac mass in animal experiments.15 In addition, this drug showed promise in terms of blood pressure, heart failure readmissions, death, and preservation of renal function when compared with ACE inhibitors in patients with heart failure.15–17 But in clinical trials this drug posed a greater risk of hypotension, dizziness, and, its major shortcoming, an unacceptably high incidence of angioedema compared with ACE inhibitors.15,16,18 This higher risk of angioedema is thought to be from inhibition of three enzymes that break down bradykinin: ACE, neprilysin, and aminopeptidase P.19
ARNIs contain an angiotensin receptor blocker rather than an ACE inhibitor, and thus in theory they may be associated with a lower risk of angioedema.19 Sacubitril-valsartan, the first drug of this class, contains its two constitutive drugs in a one-to-one molecular ratio (Figure 1).
PARADIGM-HF investigated the benefit of this drug in patients with systolic heart failure.1
STUDY DESIGN AND OBJECTIVES
PARADIGM-HF was a double-blind, randomized controlled trial comparing sacubitril-valsartan and enalapril in patients with chronic systolic heart failure. As such, it was the first trial in recent years to study a new drug in comparison with a well-established heart failure drug rather than as an add-on strategy.1
Inclusion criteria
To be included in the PARADIGM-HF trial, patients had to have:
- A left ventricular ejection fraction of 40% or less (later changed to ≤ 35%)
- New York Heart Association class II, III, or IV symptoms
- A B-type natriuretic peptide (BNP) level of at least 150 pg/mL or an N-terminal proBNP (NT-proBNP) level of at least 600 pg/mL; for patients hospitalized for heart failure within the previous 12 months, the cut points were lower (BNP ≥ 100 pg/mL or NT-proBNP ≥ 400 pg/mL).
End points
The primary end point was the composite of cardiovascular death or first hospitalization for heart failure. Other outcomes assessed were time to death from any cause, the change from baseline in the Kansas City Cardiomyopathy Questionnaire (KCCQ) score at 8 months, time to new-onset atrial fibrillation, and the time to decline in renal function (defined as end-stage renal disease or a decrease in estimated glomerular filtration rate of at least 50% from randomization). All end points were blindly adjudicated by a clinical end points committee.
Two run-in periods
The study enrolled 10,521 patients from 1,043 centers in 47 countries, who entered the initial run-in period consisting of 2 weeks of treatment with enalapril at the study dosage (10 mg twice daily) in order to ensure no unacceptable side effects. At this point, 1,102 patients exited the study, leaving 9,419 who entered the second run-in period.
The second run-in period consisted of 4 weeks of treatment with sacubitril-valsartan, initially at half the study regimen (100 mg twice daily) and eventually at the full study dosage (200 mg twice daily). During the second run-in period, 977 participants left the study, leaving a total of 8,442 patients who underwent randomization. Forty-three patients were then excluded (6 because of invalid randomization and 37 because of four sites that closed because of major violations of good clinical practice).
Of those randomized, 4,187 patients were assigned to the sacubitril-valsartan treatment group and 4,212 were assigned to the enalapril group. The investigators used an intention-to-treat analysis for this study.
Most patients had NYHA class II symptoms
The randomized patients had a mean age of 64 years, 75% were men, 66% were white, and 58% were from Europe (only 7% were from North America). The mean left ventricular ejection fraction was about 30%, and 60% of the study participants had an ischemic cause for their cardiomyopathy. Although one of the inclusion criteria was New York Heart Association class II, III, or IV symptoms, about 5% of the patients had class I symptoms. Seventy percent had class II symptoms, 24% had class III, and less than 1% had class IV symptoms.
At the time of randomization, 78% of the patients were taking an ACE inhibitor and 93% were taking a beta-blocker, but only a little more than half were taking a mineralocorticoid antagonist and only about 15% had an implantable cardioverter-defibrillator.
STUDY OUTCOMES
This study was designed to detect a 15% lower risk of cardiovascular death in the sacubitril-valsartan group. It was projected to continue for at least 34 months but was stopped early because of an overwhelming benefit of the new drug at a median follow-up of 27 months.
Major findings
The primary composite outcome (cardiovascular death or first hospitalization for heart failure)1 occurred in 21.8% of the patients in the sacubitril-valsartan group vs 26.5% of patients in the enalapril group (hazard ratio [HR] 0.80, 95% confidence interval [CI] 0.73–0.87, P < .001). The number of patients who needed to be treated to prevent one occurrence of the primary composite outcome (100/absolute risk reduction) was only 21. The benefit was strong and consistent across both of the individual components of the composite outcome:
- Cardiovascular death 13.3% vs 16.5%, HR 0.80 (95% CI 0.71–0.89), P < .001
- First hospitalization for worsening heart failure 12.8% vs 15.6%, HR 0.79 (95% CI 0.71–0.89), P < .001.
Secondary outcomes. The sacubitril-valsartan group had a significantly lower rate of death from any cause (17.0% vs 19.8%, HR 0.84, 95% CI 0.76–0.93, P < .001) and a lower mean decrease in KCCQ clinical summary scores at 8 months (2.99 points vs 4.63 points, mean difference 1.64, 95% CI 0.63–2.65, P = .001). The KCCQ score measures subjective symptoms and physical limitations caused by heart failure; possible scores range from 0 to 100, with a higher score indicating better functional status. Notably, sacubitril-valsartan did not increase the KCCQ score in these patients; rather, sacubitril-valsartan recipients had a lower decrease in their scores than those in the enalapril group.
The incidence of new-onset atrial fibrillation was the same in both groups (3.1% and 3.1%).1 A decline in renal function (defined as end-stage renal disease, a decrease of 50% or more in estimated glomerular filtration rate from the value at randomization or a decrease in the estimated glomerular filtration rate of more than 30 mL/min/1.73 m2 to less than 60 mL/min/1.73 m2) occurred in 2.2% of the valsartan-sacubitril group and 2.6% of the enalapril group (P = .28).
The effects of the study drug on the primary composite outcome and on death from a cardiovascular cause were similar in all prespecified subgroups except for NYHA class: the reduction in the risk of the composite outcome was lower in sacubitril-valsartan recipients with NYHA I or II symptoms than in those with NYHA III or IV symptoms (P for interaction .03). However, there were no differences in the other prespecified subgroups, defined by age, sex, race, region, estimated glomerular filtration rate, diabetes, systolic blood pressure, ejection fraction, atrial fibrillation, NT-proBNP, hypertension, previous use of an ACE inhibitor, previous use of an aldosterone antagonist, previous heart failure hospitalization, and time since diagnosis of heart failure.
SAFETY: ANGIOEDEMA, HYPOTENSION, AND RENAL DYSFUNCTION
Angioedema
As noted above, the combination of neprilysin inhibitors and ACE inhibitors has been associated with an increased risk of angioedema. That was an important consideration before starting this study, which used a combination of a neprilysin inhibitor and an ARB in an attempt to avoid this serious side effect.
As it happened, there was no increased risk of significant angioedema with sacubitril-valsartan use compared with enalapril. Rates were similar to those in other studies, which showed a less than 1% risk of angioedema caused by ACE inhibitors.20,21 Only 19 patients (0.5%) in the sacubitril-valsartan group and 10 patients (0.2%) in the enalapril group experienced any angioedema. Of these, just three patients in the sacubitril-valsartan group and one patient in the enalapril group experienced angioedema that required hospitalization (P = .31). None of these patients had airway compromise due to angioedema.
Hypotension, cough, renal dysfunction, hyperkalemia
Other safety issues that were assessed included hypotension, worsening renal function, increase in potassium levels, and cough. Patients in the sacubitril-valsartan group were more likely to have symptomatic hypotension than patients in the enalapril group (14.0% vs 9.2%, P < .001); however, the authors noted that this was a rare cause of drug discontinuation.
Patients in the sacubitril-valsartan group were less likely to develop cough (11.3% vs 14.3%, P < .001), a serum creatinine level of 2.5 mg/dL or more (3.3% vs 4.5%, P = .007), or a serum potassium level of more than 6.0 mmol/L (11.3% vs 14.3%, P = .007).1
During the two run-in periods combined, 12% of the patients in the study withdrew because of adverse events, including cough, renal dysfunction, hyperkalemia, and symptomatic hypotension. During the enalapril run-in period, 591 patients (5.6%) withdrew due to adverse events, and 547 patients (5.8%) withdrew due to these events during the sacubitril-valsartan run-in period. After adjusting for the shorter time on enalapril, there was a higher rate of withdrawal because of adverse events from enalapril than from sacubitril-valsartan.
LOWER RISK OF CLINICAL PROGRESSION
In a separate paper,22 the PARADIGM-HF investigators reported that, among the survivors in the study, those who received sacubitril-valsartan fared better in terms of a number of markers of progression of heart failure, with lower rates of:
- Intensification of medical treatment for heart failure
- Emergency department visits for worsening heart failure
- Hospitalization for worsening heart failure
- Need for intensive care
- Need for intravenous inotropic agents
- Need for cardiac devices or heart transplants
- Worsening symptom scores
- Elevation of biomarkers of myocardial injury.
QUESTIONS AND CONCERNS
Although this study, which was funded by the manufacturer, showed consistent benefit for sacubitril-valsartan over enalapril, questions remain.
Are the findings generalizable?
Despite the study’s rigorous run-in period, 12% of patients dropped out because of adverse events, and thus the patients who completed the study may not be representative of the general population of heart failure patients. The authors included this double-level wash-out to ensure patient tolerance of both drugs. But in everyday practice, a significant number of patients may be unable to tolerate one of these drugs.
Moreover, after adjusting for the difference in the run-in periods, patients actually withdrew more often during the enalapril run-in period than during the sacubitril-valsartan run-in period. However, there may be overlap in tolerability in these two drugs, which both affect the renin-angiotensin-aldosterone system. Thus, the enalapril run-in period may have contributed to the lower tolerability of this drug compared with sacubitril-valsartan.
Were patients receiving the best possible therapy?
Another important point when considering how we treat heart failure patients in the United States is how few patients in this study were using cardiac implantable electronic devices. Only 15% of the patients in this study had an implantable cardioverter-defibrillator despite a mean left ventricular ejection fraction less than 30%. This likely reflects differences in practice internationally; however, based on American College of Cardiology, American Heart Association, and Heart Rhythm Society guidelines, these patients would have a class I indication for an implantable cardioverter-defibrillator for primary prevention of sudden cardiac death.23
Therefore, based on these recommendations, the patients in this study were not necessarily on optimal medical and device therapy and furthermore may not be representative of heart failure patients in the United States.
Was enalapril 10 mg twice a day a fair comparison?
Another concern about the results of this study relates to the dosages used in the two treatment groups. The sacubitril-valsartan formulation included full-dose valsartan, whereas the ACE inhibitor group received enalapril at less than a full dose.
Although the authors explained that the dose of enalapril chosen for the study was based on the one used in previous studies that showed survival benefit, this raises the question of whether the significant difference in outcomes in this trial was due to a greater inhibition of the renin-angiotensin-aldosterone system related to a higher dose of drug in the sacubitril-valsartan group.
What about black patients taking hydralazine-isosorbide?
Only about 5% of patients in the PARADIGM-HF trial were black. Based on the A-HeFT study results, black patients can be prescribed an ACE inhibitor as well as hydralazine and isosorbide dinitrate as tolerated to decrease the risk of death. Does sacubitril-valsartan offer benefit to these patients compared with a regimen of an ACE inhibitor, hydralazine, and isosorbide dinitrate?
Another concern is that the incidence of angioedema observed with ACE inhibitors and omapatrilat is higher in black patients.15,21 Thus, it would be prudent to investigate whether the risk of angioedema with sacubitril-valsartan would be higher if more black patients are studied.
IMPLICATIONS AND CONSIDERATIONS
In this study, sacubitril-valsartan showed impressive and consistent results, with an almost 20% decrease in the composite end point of heart failure hospitalization or cardiovascular death and a similar decrease in the composite outcomes with a very low number needed to treat (21 patients). It did not show a decrease in the incidence of new-onset atrial fibrillation; however, only 80 cases of atrial fibrillation were reported, so there may have been a lack of statistical power to detect a difference.
To avoid angioedema, wait at least 36 hours after stopping an ACE inhibitor. Sacubitril-valsartan was not associated with an increased risk of severe angioedema, and no patients experienced life-threatening angioedema. In the trial, the sacubitril-valsartan run-in period was started at least 24 hours after enalapril was stopped, and thus the authors recommended at least a 1-day washout period after discontinuing an ACE inhibitor to avoid angioedema in patients starting sacubitril-valsartan.
Hypotension is a concern. Although there was actually a decreased risk of renal dysfunction, hyperkalemia, and cough compared with enalapril, there was a significantly increased rate of symptomatic hypotension in the sacubitril-valsartan group, which raises the question of patient tolerance and physician comfort when prescribing and titrating this drug in clinical practice. This side effect will be an important consideration when attempting to titrate the drug to target doses.
Start treatment early. This trial largely consisted of patients with NYHA class II or III symptoms, with about 70% of patients with class II symptoms. Since this drug showed benefit in patients with mildly to moderately symptomatic heart failure, clinicians who are considering prescribing this drug should not wait until the patient is closer to end-stage disease. Patients with mildly symptomatic heart failure may be followed by a general cardiologist, internist, or both, and thus it is important to emphasize to the entire medical community the need to start this medication early on.
How much will it cost? Cost is a concern that could heavily weigh on the decision to prescribe this drug. Generic ACE inhibitors are relatively inexpensive, and it may difficult to switch from an affordable generic drug to a new drug that is likely to be much more expensive. Arguably, this drug may be cost-effective in the long run owing to a large decrease in heart failure readmissions. We await further analyses to evaluate this issue.
Will patients take a twice-a-day drug as prescribed? Most patients who are prescribed an ACE inhibitor take it just once a day, and switching from a daily to a twice-daily drug may present a challenge for some.
What about other outcomes? Based on this study, it is unclear what effect sacubitril-valsartan has on the incidence of fatal arrhythmias, sudden cardiac death, and pump failure. Furthermore, the effect on quality of life is still uncertain. Quality of life is an integral component in the evaluation of heart failure patients, and in this study the changes in KCCQ scores were not impressive. We hope to see further evaluations of this drug’s impact on quality of life of patients with heart failure. Furthermore, it would be interesting to study if this drug has any long-term effects on the need for advanced therapies such as left ventricular assist devices and orthotopic heart transplant.
What about patients with heart failure with preserved ejection fraction? This study included only patients with heart failure with reduced ejection fraction. However PARAMOUNT, a phase 2 study that evaluated the benefit of sacubitril-valsartan in patients with heart failure with preserved ejection fraction, has shown encouraging results.24 We look forward to further investigation of this agent in patients with heart failure with preserved ejection fraction.
Sacubitril-valsartan, the first ARNI to be studied in humans, has a dual action in that it enhances the activity of the natriuretic peptide system and inhibits that of the renin-angiotensin-aldosterone system. It is the first drug in over a decade to show mortality benefit in patients with chronic systolic heart failure when compared with an already well-established heart failure medication. It appears to decrease rates of mortality and heart failure hospitalization without increasing the risk of severe angioedema in patients with mild or moderate chronic systolic heart failure. Symptomatic hypotension and high cost may pose the largest barriers to the use of this new drug. And we have yet to see how the clinical community and patients with heart failure will respond to it.
In a large phase trial, a combination drug that contains the angiotensin II receptor blocker (ARB) valsartan and the neprilysin inhibitor sacubitril was found to be superior to the angiotensin-converting enzyme (ACE) inhibitor enalapril in terms of important end points, including death and hospitalization for heart failure, in patients with heart failure with reduced ejection fraction.1
Recently approved by the US Food and Drug Administration, this combination drug, marketed under the brand name Entresto, represents a new drug class, angiotensin receptor-neprilysin inhibitors, or ARNIs.
This article is an overview of the Prospective Comparison of ARNI With ACEI to Determine Impact on Global Mortality and Morbidity in Heart Failure (PARADIGM-HF) trial1 and the implications it may have on the care of patients with chronic heart failure.
NEED FOR NEW HEART FAILURE DRUGS
Heart failure is a major public health problem, and the care of patients with heart failure is challenging.
Almost 6 million US adults have heart failure, and the prevalence is projected to increase in the next few decades as the population continues to age.2 Furthermore, the total healthcare cost for heart failure patients was almost $31 billion in 2012 and is projected to rise to $70 billion by 2030.2
The care of patients with severely decompensated heart failure has changed dramatically in the last few decades with advances in heart transplantation and mechanical support devices. But day-to-day management of patients with chronic mildly to moderately symptomatic heart failure continues to pose a clinical challenge.
The drugs currently available for these patients include beta-blockers, ACE inhibitors, ARBs, aldosterone antagonists, digoxin, diuretics, and vasodilators. But even with these drugs, the death and readmission rates of patients with heart failure with reduced ejection fraction remain high. More than 50% of patients with heart failure die within 5 years of diagnosis,3 and 25% of patients hospitalized with heart failure are readmitted within 30 days of discharge.2 Furthermore, death rates are higher in those patients who have a history of heart failure hospitalization.4
Although heart failure with preserved ejection fraction encompasses an important group of heart failure patients with high morbidity, the focus of this article will be on patients with heart failure with reduced ejection fraction.
Available drugs to date
The cornerstone drugs that lower the odds of death in patients with heart failure with reduced ejection fraction are ACE inhibitors, ARBs, beta-blockers, and mineralocorticoid antagonists.
ACE inhibitors were the first class of drugs shown to reduce the death rate in patients with heart failure with reduced ejection fraction. The landmark CONSENSUS trial,5 published in 1987, found that the death rate in patients who received enalapril was 27% lower than in those receiving placebo, an effect driven entirely by a reduction in progressive heart failure. Similarly, the SOLVD trial,6 published in 1991, showed a 26% reduction in heart failure hospitalization and a 16% lower rate of death with enalapril compared with placebo, an effect driven predominantly by a decrease in the progression of heart failure.
ARBs have also been shown to decrease the rate of death, although not by as much as ACE inhibitors. In the CHARM trial,7 compared with placebo, candesartan significantly decreased the risk of death from any cause, of death from cardiovascular causes, and of hospitalization related to heart failure.7
Beta-blockers. The MERIT-HF trial,8 published in 1999, was stopped early because fewer patients were dying in the group receiving metoprolol succinate than in the group receiving placebo (relative risk 0.66). Similarly, in 2001, the COPERNICUS trial9 reported a 34% reduction in deaths in patients receiving carvedilol in addition to an ACE inhibitor compared with those receiving an ACE inhibitor alone.
Mineralocorticoid receptor antagonists were found to be beneficial when added to standard therapy for chronic symptomatic heart failure in the RALES10 and EMPHASIS-HF11 trials.
Vasodilators (specifically, the combination of isosorbide dinitrate and hydralazine) were found to have benefit in terms of mortality when added to standard therapy in African American patients in the A-HeEFT trial.12
WHY INHIBIT BOTH ANGIOTENSIN AND NEPRILYSIN?
The renin-angiotensin-aldosterone system is a major focus in treating heart failure, as overactivity of this system plays a key role in the pathophysiology of this disease. Therefore, essential drugs for heart failure patients include those that inhibit overactivity of this system such as ACE inhibitors, ARBs, and aldosterone antagonists.
The natriuretic peptide system is another important pathway that can be targeted in patients with heart failure. Natriuretic peptides are key molecules that counteract heart failure, as they contribute to diuresis and vasodilation and protect against vascular remodeling.13 An increased understanding of the importance of this system in slowing the progression of heart failure has motivated evaluation of drugs such as nesiritide in patients with symptomatic heart failure. However, these drugs can cause hypotension and have limited bioavailability.14
Neprilysin is an endopeptidase—an endogenous enzyme that degrades vasoactive peptides such as bradykinin and natriuretic peptides.14 Drugs that inhibit neprilysin increase the levels of these peptides and thus counteract neurohormonal stimuli that lead to cardiac remodeling, sodium retention, and vasoconstriction.15
However, neprilysin also hydrolyzes angiotensin I to angiotensin (1–7), an inhibitor of angiotensin II. Thus, inhibition of neprilysin alone could lead to increased activity of angiotensin II and so have an overall neutral effect. To be beneficial, neprilysin inhibition needs to be combined with renin-angiotensin system inhibition. Furthermore, the benefit of renin-angiotensin-aldosterone system blockade may be amplified by up-regulation of the endogenous natriuretic peptide system.15
Omapatrilat, the most studied combination neprilysin inhibitor and ACE inhibitor, improved cardiac function and decreased cardiac mass in animal experiments.15 In addition, this drug showed promise in terms of blood pressure, heart failure readmissions, death, and preservation of renal function when compared with ACE inhibitors in patients with heart failure.15–17 But in clinical trials this drug posed a greater risk of hypotension, dizziness, and, its major shortcoming, an unacceptably high incidence of angioedema compared with ACE inhibitors.15,16,18 This higher risk of angioedema is thought to be from inhibition of three enzymes that break down bradykinin: ACE, neprilysin, and aminopeptidase P.19
ARNIs contain an angiotensin receptor blocker rather than an ACE inhibitor, and thus in theory they may be associated with a lower risk of angioedema.19 Sacubitril-valsartan, the first drug of this class, contains its two constitutive drugs in a one-to-one molecular ratio (Figure 1).
PARADIGM-HF investigated the benefit of this drug in patients with systolic heart failure.1
STUDY DESIGN AND OBJECTIVES
PARADIGM-HF was a double-blind, randomized controlled trial comparing sacubitril-valsartan and enalapril in patients with chronic systolic heart failure. As such, it was the first trial in recent years to study a new drug in comparison with a well-established heart failure drug rather than as an add-on strategy.1
Inclusion criteria
To be included in the PARADIGM-HF trial, patients had to have:
- A left ventricular ejection fraction of 40% or less (later changed to ≤ 35%)
- New York Heart Association class II, III, or IV symptoms
- A B-type natriuretic peptide (BNP) level of at least 150 pg/mL or an N-terminal proBNP (NT-proBNP) level of at least 600 pg/mL; for patients hospitalized for heart failure within the previous 12 months, the cut points were lower (BNP ≥ 100 pg/mL or NT-proBNP ≥ 400 pg/mL).
End points
The primary end point was the composite of cardiovascular death or first hospitalization for heart failure. Other outcomes assessed were time to death from any cause, the change from baseline in the Kansas City Cardiomyopathy Questionnaire (KCCQ) score at 8 months, time to new-onset atrial fibrillation, and the time to decline in renal function (defined as end-stage renal disease or a decrease in estimated glomerular filtration rate of at least 50% from randomization). All end points were blindly adjudicated by a clinical end points committee.
Two run-in periods
The study enrolled 10,521 patients from 1,043 centers in 47 countries, who entered the initial run-in period consisting of 2 weeks of treatment with enalapril at the study dosage (10 mg twice daily) in order to ensure no unacceptable side effects. At this point, 1,102 patients exited the study, leaving 9,419 who entered the second run-in period.
The second run-in period consisted of 4 weeks of treatment with sacubitril-valsartan, initially at half the study regimen (100 mg twice daily) and eventually at the full study dosage (200 mg twice daily). During the second run-in period, 977 participants left the study, leaving a total of 8,442 patients who underwent randomization. Forty-three patients were then excluded (6 because of invalid randomization and 37 because of four sites that closed because of major violations of good clinical practice).
Of those randomized, 4,187 patients were assigned to the sacubitril-valsartan treatment group and 4,212 were assigned to the enalapril group. The investigators used an intention-to-treat analysis for this study.
Most patients had NYHA class II symptoms
The randomized patients had a mean age of 64 years, 75% were men, 66% were white, and 58% were from Europe (only 7% were from North America). The mean left ventricular ejection fraction was about 30%, and 60% of the study participants had an ischemic cause for their cardiomyopathy. Although one of the inclusion criteria was New York Heart Association class II, III, or IV symptoms, about 5% of the patients had class I symptoms. Seventy percent had class II symptoms, 24% had class III, and less than 1% had class IV symptoms.
At the time of randomization, 78% of the patients were taking an ACE inhibitor and 93% were taking a beta-blocker, but only a little more than half were taking a mineralocorticoid antagonist and only about 15% had an implantable cardioverter-defibrillator.
STUDY OUTCOMES
This study was designed to detect a 15% lower risk of cardiovascular death in the sacubitril-valsartan group. It was projected to continue for at least 34 months but was stopped early because of an overwhelming benefit of the new drug at a median follow-up of 27 months.
Major findings
The primary composite outcome (cardiovascular death or first hospitalization for heart failure)1 occurred in 21.8% of the patients in the sacubitril-valsartan group vs 26.5% of patients in the enalapril group (hazard ratio [HR] 0.80, 95% confidence interval [CI] 0.73–0.87, P < .001). The number of patients who needed to be treated to prevent one occurrence of the primary composite outcome (100/absolute risk reduction) was only 21. The benefit was strong and consistent across both of the individual components of the composite outcome:
- Cardiovascular death 13.3% vs 16.5%, HR 0.80 (95% CI 0.71–0.89), P < .001
- First hospitalization for worsening heart failure 12.8% vs 15.6%, HR 0.79 (95% CI 0.71–0.89), P < .001.
Secondary outcomes. The sacubitril-valsartan group had a significantly lower rate of death from any cause (17.0% vs 19.8%, HR 0.84, 95% CI 0.76–0.93, P < .001) and a lower mean decrease in KCCQ clinical summary scores at 8 months (2.99 points vs 4.63 points, mean difference 1.64, 95% CI 0.63–2.65, P = .001). The KCCQ score measures subjective symptoms and physical limitations caused by heart failure; possible scores range from 0 to 100, with a higher score indicating better functional status. Notably, sacubitril-valsartan did not increase the KCCQ score in these patients; rather, sacubitril-valsartan recipients had a lower decrease in their scores than those in the enalapril group.
The incidence of new-onset atrial fibrillation was the same in both groups (3.1% and 3.1%).1 A decline in renal function (defined as end-stage renal disease, a decrease of 50% or more in estimated glomerular filtration rate from the value at randomization or a decrease in the estimated glomerular filtration rate of more than 30 mL/min/1.73 m2 to less than 60 mL/min/1.73 m2) occurred in 2.2% of the valsartan-sacubitril group and 2.6% of the enalapril group (P = .28).
The effects of the study drug on the primary composite outcome and on death from a cardiovascular cause were similar in all prespecified subgroups except for NYHA class: the reduction in the risk of the composite outcome was lower in sacubitril-valsartan recipients with NYHA I or II symptoms than in those with NYHA III or IV symptoms (P for interaction .03). However, there were no differences in the other prespecified subgroups, defined by age, sex, race, region, estimated glomerular filtration rate, diabetes, systolic blood pressure, ejection fraction, atrial fibrillation, NT-proBNP, hypertension, previous use of an ACE inhibitor, previous use of an aldosterone antagonist, previous heart failure hospitalization, and time since diagnosis of heart failure.
SAFETY: ANGIOEDEMA, HYPOTENSION, AND RENAL DYSFUNCTION
Angioedema
As noted above, the combination of neprilysin inhibitors and ACE inhibitors has been associated with an increased risk of angioedema. That was an important consideration before starting this study, which used a combination of a neprilysin inhibitor and an ARB in an attempt to avoid this serious side effect.
As it happened, there was no increased risk of significant angioedema with sacubitril-valsartan use compared with enalapril. Rates were similar to those in other studies, which showed a less than 1% risk of angioedema caused by ACE inhibitors.20,21 Only 19 patients (0.5%) in the sacubitril-valsartan group and 10 patients (0.2%) in the enalapril group experienced any angioedema. Of these, just three patients in the sacubitril-valsartan group and one patient in the enalapril group experienced angioedema that required hospitalization (P = .31). None of these patients had airway compromise due to angioedema.
Hypotension, cough, renal dysfunction, hyperkalemia
Other safety issues that were assessed included hypotension, worsening renal function, increase in potassium levels, and cough. Patients in the sacubitril-valsartan group were more likely to have symptomatic hypotension than patients in the enalapril group (14.0% vs 9.2%, P < .001); however, the authors noted that this was a rare cause of drug discontinuation.
Patients in the sacubitril-valsartan group were less likely to develop cough (11.3% vs 14.3%, P < .001), a serum creatinine level of 2.5 mg/dL or more (3.3% vs 4.5%, P = .007), or a serum potassium level of more than 6.0 mmol/L (11.3% vs 14.3%, P = .007).1
During the two run-in periods combined, 12% of the patients in the study withdrew because of adverse events, including cough, renal dysfunction, hyperkalemia, and symptomatic hypotension. During the enalapril run-in period, 591 patients (5.6%) withdrew due to adverse events, and 547 patients (5.8%) withdrew due to these events during the sacubitril-valsartan run-in period. After adjusting for the shorter time on enalapril, there was a higher rate of withdrawal because of adverse events from enalapril than from sacubitril-valsartan.
LOWER RISK OF CLINICAL PROGRESSION
In a separate paper,22 the PARADIGM-HF investigators reported that, among the survivors in the study, those who received sacubitril-valsartan fared better in terms of a number of markers of progression of heart failure, with lower rates of:
- Intensification of medical treatment for heart failure
- Emergency department visits for worsening heart failure
- Hospitalization for worsening heart failure
- Need for intensive care
- Need for intravenous inotropic agents
- Need for cardiac devices or heart transplants
- Worsening symptom scores
- Elevation of biomarkers of myocardial injury.
QUESTIONS AND CONCERNS
Although this study, which was funded by the manufacturer, showed consistent benefit for sacubitril-valsartan over enalapril, questions remain.
Are the findings generalizable?
Despite the study’s rigorous run-in period, 12% of patients dropped out because of adverse events, and thus the patients who completed the study may not be representative of the general population of heart failure patients. The authors included this double-level wash-out to ensure patient tolerance of both drugs. But in everyday practice, a significant number of patients may be unable to tolerate one of these drugs.
Moreover, after adjusting for the difference in the run-in periods, patients actually withdrew more often during the enalapril run-in period than during the sacubitril-valsartan run-in period. However, there may be overlap in tolerability in these two drugs, which both affect the renin-angiotensin-aldosterone system. Thus, the enalapril run-in period may have contributed to the lower tolerability of this drug compared with sacubitril-valsartan.
Were patients receiving the best possible therapy?
Another important point when considering how we treat heart failure patients in the United States is how few patients in this study were using cardiac implantable electronic devices. Only 15% of the patients in this study had an implantable cardioverter-defibrillator despite a mean left ventricular ejection fraction less than 30%. This likely reflects differences in practice internationally; however, based on American College of Cardiology, American Heart Association, and Heart Rhythm Society guidelines, these patients would have a class I indication for an implantable cardioverter-defibrillator for primary prevention of sudden cardiac death.23
Therefore, based on these recommendations, the patients in this study were not necessarily on optimal medical and device therapy and furthermore may not be representative of heart failure patients in the United States.
Was enalapril 10 mg twice a day a fair comparison?
Another concern about the results of this study relates to the dosages used in the two treatment groups. The sacubitril-valsartan formulation included full-dose valsartan, whereas the ACE inhibitor group received enalapril at less than a full dose.
Although the authors explained that the dose of enalapril chosen for the study was based on the one used in previous studies that showed survival benefit, this raises the question of whether the significant difference in outcomes in this trial was due to a greater inhibition of the renin-angiotensin-aldosterone system related to a higher dose of drug in the sacubitril-valsartan group.
What about black patients taking hydralazine-isosorbide?
Only about 5% of patients in the PARADIGM-HF trial were black. Based on the A-HeFT study results, black patients can be prescribed an ACE inhibitor as well as hydralazine and isosorbide dinitrate as tolerated to decrease the risk of death. Does sacubitril-valsartan offer benefit to these patients compared with a regimen of an ACE inhibitor, hydralazine, and isosorbide dinitrate?
Another concern is that the incidence of angioedema observed with ACE inhibitors and omapatrilat is higher in black patients.15,21 Thus, it would be prudent to investigate whether the risk of angioedema with sacubitril-valsartan would be higher if more black patients are studied.
IMPLICATIONS AND CONSIDERATIONS
In this study, sacubitril-valsartan showed impressive and consistent results, with an almost 20% decrease in the composite end point of heart failure hospitalization or cardiovascular death and a similar decrease in the composite outcomes with a very low number needed to treat (21 patients). It did not show a decrease in the incidence of new-onset atrial fibrillation; however, only 80 cases of atrial fibrillation were reported, so there may have been a lack of statistical power to detect a difference.
To avoid angioedema, wait at least 36 hours after stopping an ACE inhibitor. Sacubitril-valsartan was not associated with an increased risk of severe angioedema, and no patients experienced life-threatening angioedema. In the trial, the sacubitril-valsartan run-in period was started at least 24 hours after enalapril was stopped, and thus the authors recommended at least a 1-day washout period after discontinuing an ACE inhibitor to avoid angioedema in patients starting sacubitril-valsartan.
Hypotension is a concern. Although there was actually a decreased risk of renal dysfunction, hyperkalemia, and cough compared with enalapril, there was a significantly increased rate of symptomatic hypotension in the sacubitril-valsartan group, which raises the question of patient tolerance and physician comfort when prescribing and titrating this drug in clinical practice. This side effect will be an important consideration when attempting to titrate the drug to target doses.
Start treatment early. This trial largely consisted of patients with NYHA class II or III symptoms, with about 70% of patients with class II symptoms. Since this drug showed benefit in patients with mildly to moderately symptomatic heart failure, clinicians who are considering prescribing this drug should not wait until the patient is closer to end-stage disease. Patients with mildly symptomatic heart failure may be followed by a general cardiologist, internist, or both, and thus it is important to emphasize to the entire medical community the need to start this medication early on.
How much will it cost? Cost is a concern that could heavily weigh on the decision to prescribe this drug. Generic ACE inhibitors are relatively inexpensive, and it may difficult to switch from an affordable generic drug to a new drug that is likely to be much more expensive. Arguably, this drug may be cost-effective in the long run owing to a large decrease in heart failure readmissions. We await further analyses to evaluate this issue.
Will patients take a twice-a-day drug as prescribed? Most patients who are prescribed an ACE inhibitor take it just once a day, and switching from a daily to a twice-daily drug may present a challenge for some.
What about other outcomes? Based on this study, it is unclear what effect sacubitril-valsartan has on the incidence of fatal arrhythmias, sudden cardiac death, and pump failure. Furthermore, the effect on quality of life is still uncertain. Quality of life is an integral component in the evaluation of heart failure patients, and in this study the changes in KCCQ scores were not impressive. We hope to see further evaluations of this drug’s impact on quality of life of patients with heart failure. Furthermore, it would be interesting to study if this drug has any long-term effects on the need for advanced therapies such as left ventricular assist devices and orthotopic heart transplant.
What about patients with heart failure with preserved ejection fraction? This study included only patients with heart failure with reduced ejection fraction. However PARAMOUNT, a phase 2 study that evaluated the benefit of sacubitril-valsartan in patients with heart failure with preserved ejection fraction, has shown encouraging results.24 We look forward to further investigation of this agent in patients with heart failure with preserved ejection fraction.
Sacubitril-valsartan, the first ARNI to be studied in humans, has a dual action in that it enhances the activity of the natriuretic peptide system and inhibits that of the renin-angiotensin-aldosterone system. It is the first drug in over a decade to show mortality benefit in patients with chronic systolic heart failure when compared with an already well-established heart failure medication. It appears to decrease rates of mortality and heart failure hospitalization without increasing the risk of severe angioedema in patients with mild or moderate chronic systolic heart failure. Symptomatic hypotension and high cost may pose the largest barriers to the use of this new drug. And we have yet to see how the clinical community and patients with heart failure will respond to it.
- McMurray JJ, Packer M, Desai AS, et al; PARADIGM-HF Investigators and Committees. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N Engl J Med 2014; 371:993–1004.
- Dunlay SM, Pereira NL, Kushwaha SS. Contemporary strategies in the diagnosis and management of heart failure. Mayo Clin Proc 2014; 89:662–676.
- Go AS, Mozaffarian D, Roger VL, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2013 update: a report from the American Heart Association. Circulation 2013; 127:e6–e245.
- Bello NA, Claggett B, Desai AS, et al. Influence of previous heart failure hospitalization on cardiovascular events in patients with reduced and preserved ejection fraction. Circ Heart Fail 2014; 7:590–595.
- Effects of enalapril on mortality in severe congestive heart failure. Results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS). The CONSENSUS Trial Study Group. N Engl J Med 1987; 316:1429–1435.
- Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congestive heart failure. The SOLVD Investigators. N Engl J Med 1991; 325:293–302.
- Pfeffer MA, Swedberg K, Granger CB, et al; CHARM Investigators and Committees. Effects of candesartan on mortality and morbidity in patients with chronic heart failure: the CHARM-Overall programme. Lancet 2003; 362:759–766.
- Effect of metoprolol CR/XL in chronic heart failure: Metoprolol CR/XL Randomised Intervention Trial in Congestive Heart Failure (MERIT-HF). Lancet 1999; 353:2001–2007.
- Packer M, Coats AJ, Fowler MB, et al; Carvedilol Prospective Randomized Cumulative Survival Study Group. Effect of carvedilol on survival in severe chronic heart failure. N Engl J Med 2001; 344:1651–1658.
- Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med 1999; 341:709–717.
- Zannad F, McMurray JJ, Krum H, et al; EMPHASIS-HF Study Group. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med 2011; 364:11–21.
- Taylor AL, Ziesche S, Yancy C, et al; African-American Heart Failure Trial Investigators. Combination of isosorbide dinitrate and hydralazine in blacks with heart failure. N Engl J Med 2004; 351:2049–2057.
- Schreiner GF, Protter AA. B-type natriuretic peptide for the treatment of congestive heart failure. Curr Opin Pharmacol 2002; 2:142–147.
- von Lueder TG, Sangaralingham SJ, Wang BH, et al. Renin-angiotensin blockade combined with natriuretic peptide system augmentation: novel therapeutic concepts to combat heart failure. Circ Heart Fail 2013; 6:594–605.
- Abassi Z, Karram T, Ellaham S, Winaver J, Hoffman A. Implications of the natriuretic peptide system in the pathogenesis of heart failure: diagnostic and therapeutic importance. Pharmacol Ther 2004; 102:223–241.
- Packer M, Califf RM, Konstam MA, et al. Comparison of omapatrilat and enalapril in patients with chronic heart failure: the Omapatrilat Versus Enalapril Randomized Trial of Utility in Reducing Events (OVERTURE). Circulation 2002; 106:920–926.
- Rouleau JL, Pfeffer MA, Stewart DJ, et al. Comparison of vasopeptidase inhibitor, omapatrilat, and lisinopril on exercise tolerance and morbidity in patients with heart failure: IMPRESS randomised trial. Lancet 2000; 356:615–620.
- Kostis JB, Packer M, Black HR, Schmieder R, Henry D, Levy E. Omapatrilat and enalapril in patients with hypertension: the Omapatrilat Cardiovascular Treatment vs. Enalapril (OCTAVE) trial. Am J Hypertens 2004; 17:103–111.
- McMurray JJ, Packer M, Desai AS, et al; PARADIGM-HF Committees and Investigators. Dual angiotensin receptor and neprilysin inhibition as an alternative to angiotensin-converting enzyme inhibition in patients with chronic systolic heart failure: rationale for and design of the Prospective comparison of ARNI with ACEI to Determine Impact on Global Mortality and morbidity in Heart Failure trial (PARADIGM-HF). Eur J Heart Fail 2013; 15:1062–1073.
- Toh S, Reichman ME, Houstoun M, et al. Comparative risk for angioedema associated with the use of drugs that target the renin-angiotensin-aldosterone system. Arch Intern Med 2012; 172:1582–1589.
- Kostis JB, Kim HJ, Rusnak J, et al. Incidence and characteristics of angioedema associated with enalapril. Arch Intern Med 2005; 165:1637–1642.
- Packer M, McMurray JJV, Desai AS, et al; on behalf of the PARADIGM-HF Investigators and Coordinators. Angiotensin receptor neprilysin inhibition compared with enalapril on the risk of clinical progression in surviving patients with heart failure. Circulation 2015; 131:54–61.
- Epstein AE, Dimarco JP, Ellenbogen KA, et al; American College of Cardiology/American Heart Association Task Force on Practice; American Association for Thoracic Surgery; Society of Thoracic Surgeons. ACC/AHA/HRS 2008 guidelines for Device-Based Therapy of Cardiac Rhythm Abnormalities: executive summary. Heart Rhythm 2008; 5:934–955.
- Solomon SD, Zile M, Pieske B, et al; Prospective comparison of ARNI with ARB on Management Of Heart Failure with Preserved Ejection Fraction (PARAMOUNT) Investigators. The angiotensin receptor neprilysin inhibitor LCZ696 in heart failure with preserved ejection fraction: a phase 2 double-blind randomised controlled trial. Lancet 2012; 380:1387–1395.
- McMurray JJ, Packer M, Desai AS, et al; PARADIGM-HF Investigators and Committees. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N Engl J Med 2014; 371:993–1004.
- Dunlay SM, Pereira NL, Kushwaha SS. Contemporary strategies in the diagnosis and management of heart failure. Mayo Clin Proc 2014; 89:662–676.
- Go AS, Mozaffarian D, Roger VL, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2013 update: a report from the American Heart Association. Circulation 2013; 127:e6–e245.
- Bello NA, Claggett B, Desai AS, et al. Influence of previous heart failure hospitalization on cardiovascular events in patients with reduced and preserved ejection fraction. Circ Heart Fail 2014; 7:590–595.
- Effects of enalapril on mortality in severe congestive heart failure. Results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS). The CONSENSUS Trial Study Group. N Engl J Med 1987; 316:1429–1435.
- Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congestive heart failure. The SOLVD Investigators. N Engl J Med 1991; 325:293–302.
- Pfeffer MA, Swedberg K, Granger CB, et al; CHARM Investigators and Committees. Effects of candesartan on mortality and morbidity in patients with chronic heart failure: the CHARM-Overall programme. Lancet 2003; 362:759–766.
- Effect of metoprolol CR/XL in chronic heart failure: Metoprolol CR/XL Randomised Intervention Trial in Congestive Heart Failure (MERIT-HF). Lancet 1999; 353:2001–2007.
- Packer M, Coats AJ, Fowler MB, et al; Carvedilol Prospective Randomized Cumulative Survival Study Group. Effect of carvedilol on survival in severe chronic heart failure. N Engl J Med 2001; 344:1651–1658.
- Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med 1999; 341:709–717.
- Zannad F, McMurray JJ, Krum H, et al; EMPHASIS-HF Study Group. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med 2011; 364:11–21.
- Taylor AL, Ziesche S, Yancy C, et al; African-American Heart Failure Trial Investigators. Combination of isosorbide dinitrate and hydralazine in blacks with heart failure. N Engl J Med 2004; 351:2049–2057.
- Schreiner GF, Protter AA. B-type natriuretic peptide for the treatment of congestive heart failure. Curr Opin Pharmacol 2002; 2:142–147.
- von Lueder TG, Sangaralingham SJ, Wang BH, et al. Renin-angiotensin blockade combined with natriuretic peptide system augmentation: novel therapeutic concepts to combat heart failure. Circ Heart Fail 2013; 6:594–605.
- Abassi Z, Karram T, Ellaham S, Winaver J, Hoffman A. Implications of the natriuretic peptide system in the pathogenesis of heart failure: diagnostic and therapeutic importance. Pharmacol Ther 2004; 102:223–241.
- Packer M, Califf RM, Konstam MA, et al. Comparison of omapatrilat and enalapril in patients with chronic heart failure: the Omapatrilat Versus Enalapril Randomized Trial of Utility in Reducing Events (OVERTURE). Circulation 2002; 106:920–926.
- Rouleau JL, Pfeffer MA, Stewart DJ, et al. Comparison of vasopeptidase inhibitor, omapatrilat, and lisinopril on exercise tolerance and morbidity in patients with heart failure: IMPRESS randomised trial. Lancet 2000; 356:615–620.
- Kostis JB, Packer M, Black HR, Schmieder R, Henry D, Levy E. Omapatrilat and enalapril in patients with hypertension: the Omapatrilat Cardiovascular Treatment vs. Enalapril (OCTAVE) trial. Am J Hypertens 2004; 17:103–111.
- McMurray JJ, Packer M, Desai AS, et al; PARADIGM-HF Committees and Investigators. Dual angiotensin receptor and neprilysin inhibition as an alternative to angiotensin-converting enzyme inhibition in patients with chronic systolic heart failure: rationale for and design of the Prospective comparison of ARNI with ACEI to Determine Impact on Global Mortality and morbidity in Heart Failure trial (PARADIGM-HF). Eur J Heart Fail 2013; 15:1062–1073.
- Toh S, Reichman ME, Houstoun M, et al. Comparative risk for angioedema associated with the use of drugs that target the renin-angiotensin-aldosterone system. Arch Intern Med 2012; 172:1582–1589.
- Kostis JB, Kim HJ, Rusnak J, et al. Incidence and characteristics of angioedema associated with enalapril. Arch Intern Med 2005; 165:1637–1642.
- Packer M, McMurray JJV, Desai AS, et al; on behalf of the PARADIGM-HF Investigators and Coordinators. Angiotensin receptor neprilysin inhibition compared with enalapril on the risk of clinical progression in surviving patients with heart failure. Circulation 2015; 131:54–61.
- Epstein AE, Dimarco JP, Ellenbogen KA, et al; American College of Cardiology/American Heart Association Task Force on Practice; American Association for Thoracic Surgery; Society of Thoracic Surgeons. ACC/AHA/HRS 2008 guidelines for Device-Based Therapy of Cardiac Rhythm Abnormalities: executive summary. Heart Rhythm 2008; 5:934–955.
- Solomon SD, Zile M, Pieske B, et al; Prospective comparison of ARNI with ARB on Management Of Heart Failure with Preserved Ejection Fraction (PARAMOUNT) Investigators. The angiotensin receptor neprilysin inhibitor LCZ696 in heart failure with preserved ejection fraction: a phase 2 double-blind randomised controlled trial. Lancet 2012; 380:1387–1395.
KEY POINTS
- Neprilysin is an endogenous enzyme that degrades vasoactive peptides such as bradykinin and natriuretic peptides. Inhibition of neprilysin raises the levels of these peptides, leading to less cardiac remodeling, less sodium retention, and less vasoconstriction.
- Neprilysin inhibition must be combined with inhibition of the renin-angiotensin-aldosterone system, optimally with an angiotensin II receptor blocker.
- PARADIGM-HF showed a 20% reduction in the primary outcome of death from cardiovascular causes or hospitalization for heart failure with sacubitril-valsartan 200 mg twice daily vs enalapril 10 mg twice daily at a median follow-up of 27 months.
- The ultimate role of combined neprilysin and angiotensin receptor inhibitors remains to be determined.
Are breast and pelvic exams necessary when prescribing hormonal contraception?
No. According to 2013 guidelines of the US Centers for Disease Control and Prevention (CDC),1 there is little evidence of benefit for many of the tests commonly mandated by healthcare providers before prescribing hormonal contraception (pill, ring, patch). These tests include breast and pelvic examinations, screening for cervical and sexually transmitted infections, laboratory testing, and mammography.
Only a medical history and blood pressure measurement are needed before prescribing estrogen-containing contraceptives. Patients who have elevated blood pressure but have not been previously diagnosed with hypertension should be preferentially offered other forms of contraception to avoid an additional risk of stroke or myocardial infarction, such as progestin-only products and intrauterine devices (IUDs). Women with blood pressures between 140/90 and 160/100 mm Hg may use estrogen-containing contraceptives only if other options are not appropriate. The CDC guidelines further state that if a patient is unable to come to the office for blood pressure assessment, then a community reading reported by the patient may be used to guide decision-making.
IS A PELVIC EXAMINATION NEEDED?
A pelvic examination (cervical inspection and bimanual examination) will not affect decisions related to prescribing contraceptives, except when prescribing female barrier methods (diaphragm, cervical cap) or IUDs.
Based on a systematic review of the literature between 1946 and 2014, the American College of Physicians now recommends against a screening pelvic examination in asymptomatic, nonpregnant, adult women when a Papanicolaou test is not otherwise indicated.2
The American College of Obstetricians and Gynecologists (ACOG) acknowledges that no current scientific evidence supports or refutes the need for an annual pelvic examination for an asymptomatic, low-risk patient. But ACOG supports pelvic examinations as a way to establish open communication with patients about sexual health and reproduction.3 ACOG also recommends an annual health visit for all women. Whether or not a pelvic examination is performed, women should be counseled annually about birth control and offered contraception.
Patients should also be encouraged to keep their preventive care up-to-date, including cervical cancer screening with a Papanicolaou test or a human papillomavirus test (or both) at appropriate intervals, especially if the patient has cervical abnormalities requiring follow-up. However, falling behind on preventive care should not be a barrier to obtaining contraception.
IMPROVING ADHERENCE, DECREASING UNINTENDED PREGNANCY
One goal of the CDC’s 2013 guidelines was to remove unnecessary barriers to women’s access to contraceptives. In the United States, half of all pregnancies are unintended, and almost half of unintended pregnancies lead to abortion.4 Only half of women who have had an abortion used any contraceptive method within the last month.5 This suggests high levels of unprotected and underprotected sex.
For most patients, several national societies now recommend long-acting reversible contraceptive (LARC) methods, which include IUDs and progestin-only arm implants, because they have lower failure rates in a real-world setting.1,6,7 LARC methods offer the advantage of the patient’s not having to remember to take, apply, or insert the contraceptive (ie, they are worry-free), and of not having to rely on a yearly appointment for refills.
The Contraceptive CHOICE Project8 was a large prospective cohort study that assessed the impact of offering contraception free of charge in St. Louis, Missouri. Most of the 9,256 women who participated selected a LARC method.8 Those taking combined hormonal contraceptives (ie, birth control pill, patch, or ring) had a higher contraceptive failure rate than those using LARC methods (4.55 vs 0.27 per 100 participant-years; hazard ratio after adjustment for age, education, and unintended pregnancy history, 21.8; 95% confidence interval 13.7–34.9). The rate of unintended pregnancy in those under age 21 using combined hormonal contraceptives was almost twice as high as in older participants. Subsequent analyses showed that the abortion rates in the St. Louis region decreased to less than a quarter of the national average after the start of this project.9
Given that the failure rate with combined hormonal contraceptives averages 9% per year,1 it is of the utmost importance that providers not limit access to patients’ prescriptions by requesting unnecessary visits and tests. If oral contraception is selected, women who are dispensed a full year’s supply of pill packs are more likely to continue with their contraceptive in the long term.10
THE PATIENT WITH A COMPLEX MEDICAL HISTORY
Limiting a woman’s contraceptive choices can increase her odds of experiencing an unintended pregnancy, which is associated with a far greater risk of adverse events than any contraceptive.11 Thus, the CDC developed separate guidelines in 2010 to help determine all available options for the patient with medical comorbidities and with a concerning family history (ie, breast cancer, venous thromboembolism).12 It can be helpful to consult the 2010 CDC medical eligibility criteria before offering contraception to these patients. Compared with the 2013 guidelines, which provide practical advice on how to use each contraceptive, the 2010 guidelines give guidance on when it is appropriate to prescribe each contraceptive—eg, which contraceptives are preferred based on a patient’s risk factors, medical history, and medication use. In addition to a two-page color summary chart of the 2010 medical eligibility criteria on the CDC website (https://www.cdc.gov/reproductivehealth/unintendedpregnancy/pdf/legal_summary-chart_english_final_tag508.pdf), a free mobile app is also available to guide decision-making.13

Pregnancy should be ruled out before initiating any contraceptive. This can be done through a detailed history. The six-item checklist in Table 1 has a 99.8% negative predictive value, so healthcare providers may be confident that a woman is not pregnant if pregnancy is excluded based on this history.14 A pregnancy test is needed in those who test positive on the checklist if they wish to start a LARC method such as an IUD or a progestin-only arm implant. However, because the test has a high false-positive rate, initiation of shorter-acting methods such as combined hormonal contraceptives should not be delayed on the basis of a positive checklist screen alone.1
Emergency contraception taken orally should be offered without an office visit, as its short duration of use allows women with traditional contraindications to hormonal contraceptives to safely use this birth control method.1,12 Because all emergency contraceptives must be used within 5 days of intercourse (the earlier the better), unnecessary office visits delay access and effectiveness.
Although a levonorgestrel-based emergency contraceptive is available over the counter, ulipristal acetate is more effective, especially in women who are overweight.15 A copper IUD placed within 5 days of intercourse is the most effective form of emergency contraception15 but requires an office visit. This method is an option for most women but should be strongly considered for women at highest risk of pregnancy (previous unintended pregnancy, intercourse at midcycle, obesity).
In summary, most women may safely begin their hormonal contraceptive with a detailed medical history alone, without additional office visits, examinations, or screening tests.
- Division of Reproductive Health, National Center for Chronic Disease Prevention and Health Promotion, Centers for Disease Control and Prevention (CDC). US selected practice recommendations for contraceptive use, 2013: adapted from the World Health Organization selected practice recommendations for contraceptive use, 2nd edition. MMWR Recomm Rep 2013; 62:1–60.
- Qaseem A, Humphrey LL, Harris R, et al; Clinical Guidelines Committee of the American College of Physicians. Screening pelvic examination in adult women: a clinical practice guideline from the American College of Physicians. Ann Intern Med 2014; 161:67–72.
- American Congress of Obstetricians and Gynecologists. ACOG practice advisory on annual pelvic examination recommendations; 2014. www.acog.org/About-ACOG/News-Room/Practice-Advisories/ACOG-Practice-Advisory-on-Annual-Pelvic-Examination-Recommendations. Accessed September 8, 2015.
- Finer LB, Zolna MR. Unintended pregnancy in the United States: incidence and disparities, 2006. Contraception 2011; 84:478–485.
- Jones RK, Darroch JE, Henshaw SK. Contraceptive use among US women having abortions in 2000-2001. Perspect Sex Reprod Health 2002; 34:294–303.
- Committee on Health Care for Underserved Women. Committee opinion no. 615: access to contraception. Obstet Gynecol 2015; 125:250–255.
- Committee on Adolescent Health Care. Committee opinion no. 598: the initial reproductive health visit. Obstet Gynecol 2014; 123:1143–1147.
- Winner B, Peipert JF, Zhao Q, et al. Effectiveness of long-acting reversible contraception. N Engl J Med 2012; 366:1998–2007.
- Secura GM, Madden T, McNicholas C, et al. Provision of no-cost, long-acting contraception and teenage pregnancy. N Engl J Med 2014; 371:1316–1323.
- Committee on Gynecologic Practice, American College of Obstetricians and Gynecologists. Over-the-counter access to oral contraceptives. Committee opinion no 544. Obstet Gynecol 2012; 120:1527–1531.
- Committee on Gynecologic Practice. ACOG committee opinion number 540: risk of venous thromboembolism among users of drospirenone-containing oral contraceptive pills. Obstet Gynecol 2012; 120:1239–1242.
- Centers for Disease Control and Prevention (CDC). US medical eligibility criteria for contraceptive use, 2010. MMWR Recomm Rep 2010; 59:1–86.
- Centers for Disease Control and Prevention (CDC). United States medical eligibility criteria (US MEC) for contraceptive use, 2010. www.cdc.gov/reproductivehealth/unintendedpregnancy/usmec.htm. Accessed September 8, 2015.
- Min J, Buckel C, Secura GM, Peipert JF, Madden T. Performance of a checklist to exclude pregnancy at the time of contraceptive initiation among women with a negative urine pregnancy test. Contraception 2015; 91:80–84.
- Batur P. Emergency contraception: separating fact from fiction. Cleve Clin J Med 2012; 79:771–776.
No. According to 2013 guidelines of the US Centers for Disease Control and Prevention (CDC),1 there is little evidence of benefit for many of the tests commonly mandated by healthcare providers before prescribing hormonal contraception (pill, ring, patch). These tests include breast and pelvic examinations, screening for cervical and sexually transmitted infections, laboratory testing, and mammography.
Only a medical history and blood pressure measurement are needed before prescribing estrogen-containing contraceptives. Patients who have elevated blood pressure but have not been previously diagnosed with hypertension should be preferentially offered other forms of contraception to avoid an additional risk of stroke or myocardial infarction, such as progestin-only products and intrauterine devices (IUDs). Women with blood pressures between 140/90 and 160/100 mm Hg may use estrogen-containing contraceptives only if other options are not appropriate. The CDC guidelines further state that if a patient is unable to come to the office for blood pressure assessment, then a community reading reported by the patient may be used to guide decision-making.
IS A PELVIC EXAMINATION NEEDED?
A pelvic examination (cervical inspection and bimanual examination) will not affect decisions related to prescribing contraceptives, except when prescribing female barrier methods (diaphragm, cervical cap) or IUDs.
Based on a systematic review of the literature between 1946 and 2014, the American College of Physicians now recommends against a screening pelvic examination in asymptomatic, nonpregnant, adult women when a Papanicolaou test is not otherwise indicated.2
The American College of Obstetricians and Gynecologists (ACOG) acknowledges that no current scientific evidence supports or refutes the need for an annual pelvic examination for an asymptomatic, low-risk patient. But ACOG supports pelvic examinations as a way to establish open communication with patients about sexual health and reproduction.3 ACOG also recommends an annual health visit for all women. Whether or not a pelvic examination is performed, women should be counseled annually about birth control and offered contraception.
Patients should also be encouraged to keep their preventive care up-to-date, including cervical cancer screening with a Papanicolaou test or a human papillomavirus test (or both) at appropriate intervals, especially if the patient has cervical abnormalities requiring follow-up. However, falling behind on preventive care should not be a barrier to obtaining contraception.
IMPROVING ADHERENCE, DECREASING UNINTENDED PREGNANCY
One goal of the CDC’s 2013 guidelines was to remove unnecessary barriers to women’s access to contraceptives. In the United States, half of all pregnancies are unintended, and almost half of unintended pregnancies lead to abortion.4 Only half of women who have had an abortion used any contraceptive method within the last month.5 This suggests high levels of unprotected and underprotected sex.
For most patients, several national societies now recommend long-acting reversible contraceptive (LARC) methods, which include IUDs and progestin-only arm implants, because they have lower failure rates in a real-world setting.1,6,7 LARC methods offer the advantage of the patient’s not having to remember to take, apply, or insert the contraceptive (ie, they are worry-free), and of not having to rely on a yearly appointment for refills.
The Contraceptive CHOICE Project8 was a large prospective cohort study that assessed the impact of offering contraception free of charge in St. Louis, Missouri. Most of the 9,256 women who participated selected a LARC method.8 Those taking combined hormonal contraceptives (ie, birth control pill, patch, or ring) had a higher contraceptive failure rate than those using LARC methods (4.55 vs 0.27 per 100 participant-years; hazard ratio after adjustment for age, education, and unintended pregnancy history, 21.8; 95% confidence interval 13.7–34.9). The rate of unintended pregnancy in those under age 21 using combined hormonal contraceptives was almost twice as high as in older participants. Subsequent analyses showed that the abortion rates in the St. Louis region decreased to less than a quarter of the national average after the start of this project.9
Given that the failure rate with combined hormonal contraceptives averages 9% per year,1 it is of the utmost importance that providers not limit access to patients’ prescriptions by requesting unnecessary visits and tests. If oral contraception is selected, women who are dispensed a full year’s supply of pill packs are more likely to continue with their contraceptive in the long term.10
THE PATIENT WITH A COMPLEX MEDICAL HISTORY
Limiting a woman’s contraceptive choices can increase her odds of experiencing an unintended pregnancy, which is associated with a far greater risk of adverse events than any contraceptive.11 Thus, the CDC developed separate guidelines in 2010 to help determine all available options for the patient with medical comorbidities and with a concerning family history (ie, breast cancer, venous thromboembolism).12 It can be helpful to consult the 2010 CDC medical eligibility criteria before offering contraception to these patients. Compared with the 2013 guidelines, which provide practical advice on how to use each contraceptive, the 2010 guidelines give guidance on when it is appropriate to prescribe each contraceptive—eg, which contraceptives are preferred based on a patient’s risk factors, medical history, and medication use. In addition to a two-page color summary chart of the 2010 medical eligibility criteria on the CDC website (https://www.cdc.gov/reproductivehealth/unintendedpregnancy/pdf/legal_summary-chart_english_final_tag508.pdf), a free mobile app is also available to guide decision-making.13
Pregnancy should be ruled out before initiating any contraceptive. This can be done through a detailed history. The six-item checklist in Table 1 has a 99.8% negative predictive value, so healthcare providers may be confident that a woman is not pregnant if pregnancy is excluded based on this history.14 A pregnancy test is needed in those who test positive on the checklist if they wish to start a LARC method such as an IUD or a progestin-only arm implant. However, because the test has a high false-positive rate, initiation of shorter-acting methods such as combined hormonal contraceptives should not be delayed on the basis of a positive checklist screen alone.1
Emergency contraception taken orally should be offered without an office visit, as its short duration of use allows women with traditional contraindications to hormonal contraceptives to safely use this birth control method.1,12 Because all emergency contraceptives must be used within 5 days of intercourse (the earlier the better), unnecessary office visits delay access and effectiveness.
Although a levonorgestrel-based emergency contraceptive is available over the counter, ulipristal acetate is more effective, especially in women who are overweight.15 A copper IUD placed within 5 days of intercourse is the most effective form of emergency contraception15 but requires an office visit. This method is an option for most women but should be strongly considered for women at highest risk of pregnancy (previous unintended pregnancy, intercourse at midcycle, obesity).
In summary, most women may safely begin their hormonal contraceptive with a detailed medical history alone, without additional office visits, examinations, or screening tests.
No. According to 2013 guidelines of the US Centers for Disease Control and Prevention (CDC),1 there is little evidence of benefit for many of the tests commonly mandated by healthcare providers before prescribing hormonal contraception (pill, ring, patch). These tests include breast and pelvic examinations, screening for cervical and sexually transmitted infections, laboratory testing, and mammography.
Only a medical history and blood pressure measurement are needed before prescribing estrogen-containing contraceptives. Patients who have elevated blood pressure but have not been previously diagnosed with hypertension should be preferentially offered other forms of contraception to avoid an additional risk of stroke or myocardial infarction, such as progestin-only products and intrauterine devices (IUDs). Women with blood pressures between 140/90 and 160/100 mm Hg may use estrogen-containing contraceptives only if other options are not appropriate. The CDC guidelines further state that if a patient is unable to come to the office for blood pressure assessment, then a community reading reported by the patient may be used to guide decision-making.
IS A PELVIC EXAMINATION NEEDED?
A pelvic examination (cervical inspection and bimanual examination) will not affect decisions related to prescribing contraceptives, except when prescribing female barrier methods (diaphragm, cervical cap) or IUDs.
Based on a systematic review of the literature between 1946 and 2014, the American College of Physicians now recommends against a screening pelvic examination in asymptomatic, nonpregnant, adult women when a Papanicolaou test is not otherwise indicated.2
The American College of Obstetricians and Gynecologists (ACOG) acknowledges that no current scientific evidence supports or refutes the need for an annual pelvic examination for an asymptomatic, low-risk patient. But ACOG supports pelvic examinations as a way to establish open communication with patients about sexual health and reproduction.3 ACOG also recommends an annual health visit for all women. Whether or not a pelvic examination is performed, women should be counseled annually about birth control and offered contraception.
Patients should also be encouraged to keep their preventive care up-to-date, including cervical cancer screening with a Papanicolaou test or a human papillomavirus test (or both) at appropriate intervals, especially if the patient has cervical abnormalities requiring follow-up. However, falling behind on preventive care should not be a barrier to obtaining contraception.
IMPROVING ADHERENCE, DECREASING UNINTENDED PREGNANCY
One goal of the CDC’s 2013 guidelines was to remove unnecessary barriers to women’s access to contraceptives. In the United States, half of all pregnancies are unintended, and almost half of unintended pregnancies lead to abortion.4 Only half of women who have had an abortion used any contraceptive method within the last month.5 This suggests high levels of unprotected and underprotected sex.
For most patients, several national societies now recommend long-acting reversible contraceptive (LARC) methods, which include IUDs and progestin-only arm implants, because they have lower failure rates in a real-world setting.1,6,7 LARC methods offer the advantage of the patient’s not having to remember to take, apply, or insert the contraceptive (ie, they are worry-free), and of not having to rely on a yearly appointment for refills.
The Contraceptive CHOICE Project8 was a large prospective cohort study that assessed the impact of offering contraception free of charge in St. Louis, Missouri. Most of the 9,256 women who participated selected a LARC method.8 Those taking combined hormonal contraceptives (ie, birth control pill, patch, or ring) had a higher contraceptive failure rate than those using LARC methods (4.55 vs 0.27 per 100 participant-years; hazard ratio after adjustment for age, education, and unintended pregnancy history, 21.8; 95% confidence interval 13.7–34.9). The rate of unintended pregnancy in those under age 21 using combined hormonal contraceptives was almost twice as high as in older participants. Subsequent analyses showed that the abortion rates in the St. Louis region decreased to less than a quarter of the national average after the start of this project.9
Given that the failure rate with combined hormonal contraceptives averages 9% per year,1 it is of the utmost importance that providers not limit access to patients’ prescriptions by requesting unnecessary visits and tests. If oral contraception is selected, women who are dispensed a full year’s supply of pill packs are more likely to continue with their contraceptive in the long term.10
THE PATIENT WITH A COMPLEX MEDICAL HISTORY
Limiting a woman’s contraceptive choices can increase her odds of experiencing an unintended pregnancy, which is associated with a far greater risk of adverse events than any contraceptive.11 Thus, the CDC developed separate guidelines in 2010 to help determine all available options for the patient with medical comorbidities and with a concerning family history (ie, breast cancer, venous thromboembolism).12 It can be helpful to consult the 2010 CDC medical eligibility criteria before offering contraception to these patients. Compared with the 2013 guidelines, which provide practical advice on how to use each contraceptive, the 2010 guidelines give guidance on when it is appropriate to prescribe each contraceptive—eg, which contraceptives are preferred based on a patient’s risk factors, medical history, and medication use. In addition to a two-page color summary chart of the 2010 medical eligibility criteria on the CDC website (https://www.cdc.gov/reproductivehealth/unintendedpregnancy/pdf/legal_summary-chart_english_final_tag508.pdf), a free mobile app is also available to guide decision-making.13
Pregnancy should be ruled out before initiating any contraceptive. This can be done through a detailed history. The six-item checklist in Table 1 has a 99.8% negative predictive value, so healthcare providers may be confident that a woman is not pregnant if pregnancy is excluded based on this history.14 A pregnancy test is needed in those who test positive on the checklist if they wish to start a LARC method such as an IUD or a progestin-only arm implant. However, because the test has a high false-positive rate, initiation of shorter-acting methods such as combined hormonal contraceptives should not be delayed on the basis of a positive checklist screen alone.1
Emergency contraception taken orally should be offered without an office visit, as its short duration of use allows women with traditional contraindications to hormonal contraceptives to safely use this birth control method.1,12 Because all emergency contraceptives must be used within 5 days of intercourse (the earlier the better), unnecessary office visits delay access and effectiveness.
Although a levonorgestrel-based emergency contraceptive is available over the counter, ulipristal acetate is more effective, especially in women who are overweight.15 A copper IUD placed within 5 days of intercourse is the most effective form of emergency contraception15 but requires an office visit. This method is an option for most women but should be strongly considered for women at highest risk of pregnancy (previous unintended pregnancy, intercourse at midcycle, obesity).
In summary, most women may safely begin their hormonal contraceptive with a detailed medical history alone, without additional office visits, examinations, or screening tests.
- Division of Reproductive Health, National Center for Chronic Disease Prevention and Health Promotion, Centers for Disease Control and Prevention (CDC). US selected practice recommendations for contraceptive use, 2013: adapted from the World Health Organization selected practice recommendations for contraceptive use, 2nd edition. MMWR Recomm Rep 2013; 62:1–60.
- Qaseem A, Humphrey LL, Harris R, et al; Clinical Guidelines Committee of the American College of Physicians. Screening pelvic examination in adult women: a clinical practice guideline from the American College of Physicians. Ann Intern Med 2014; 161:67–72.
- American Congress of Obstetricians and Gynecologists. ACOG practice advisory on annual pelvic examination recommendations; 2014. www.acog.org/About-ACOG/News-Room/Practice-Advisories/ACOG-Practice-Advisory-on-Annual-Pelvic-Examination-Recommendations. Accessed September 8, 2015.
- Finer LB, Zolna MR. Unintended pregnancy in the United States: incidence and disparities, 2006. Contraception 2011; 84:478–485.
- Jones RK, Darroch JE, Henshaw SK. Contraceptive use among US women having abortions in 2000-2001. Perspect Sex Reprod Health 2002; 34:294–303.
- Committee on Health Care for Underserved Women. Committee opinion no. 615: access to contraception. Obstet Gynecol 2015; 125:250–255.
- Committee on Adolescent Health Care. Committee opinion no. 598: the initial reproductive health visit. Obstet Gynecol 2014; 123:1143–1147.
- Winner B, Peipert JF, Zhao Q, et al. Effectiveness of long-acting reversible contraception. N Engl J Med 2012; 366:1998–2007.
- Secura GM, Madden T, McNicholas C, et al. Provision of no-cost, long-acting contraception and teenage pregnancy. N Engl J Med 2014; 371:1316–1323.
- Committee on Gynecologic Practice, American College of Obstetricians and Gynecologists. Over-the-counter access to oral contraceptives. Committee opinion no 544. Obstet Gynecol 2012; 120:1527–1531.
- Committee on Gynecologic Practice. ACOG committee opinion number 540: risk of venous thromboembolism among users of drospirenone-containing oral contraceptive pills. Obstet Gynecol 2012; 120:1239–1242.
- Centers for Disease Control and Prevention (CDC). US medical eligibility criteria for contraceptive use, 2010. MMWR Recomm Rep 2010; 59:1–86.
- Centers for Disease Control and Prevention (CDC). United States medical eligibility criteria (US MEC) for contraceptive use, 2010. www.cdc.gov/reproductivehealth/unintendedpregnancy/usmec.htm. Accessed September 8, 2015.
- Min J, Buckel C, Secura GM, Peipert JF, Madden T. Performance of a checklist to exclude pregnancy at the time of contraceptive initiation among women with a negative urine pregnancy test. Contraception 2015; 91:80–84.
- Batur P. Emergency contraception: separating fact from fiction. Cleve Clin J Med 2012; 79:771–776.
- Division of Reproductive Health, National Center for Chronic Disease Prevention and Health Promotion, Centers for Disease Control and Prevention (CDC). US selected practice recommendations for contraceptive use, 2013: adapted from the World Health Organization selected practice recommendations for contraceptive use, 2nd edition. MMWR Recomm Rep 2013; 62:1–60.
- Qaseem A, Humphrey LL, Harris R, et al; Clinical Guidelines Committee of the American College of Physicians. Screening pelvic examination in adult women: a clinical practice guideline from the American College of Physicians. Ann Intern Med 2014; 161:67–72.
- American Congress of Obstetricians and Gynecologists. ACOG practice advisory on annual pelvic examination recommendations; 2014. www.acog.org/About-ACOG/News-Room/Practice-Advisories/ACOG-Practice-Advisory-on-Annual-Pelvic-Examination-Recommendations. Accessed September 8, 2015.
- Finer LB, Zolna MR. Unintended pregnancy in the United States: incidence and disparities, 2006. Contraception 2011; 84:478–485.
- Jones RK, Darroch JE, Henshaw SK. Contraceptive use among US women having abortions in 2000-2001. Perspect Sex Reprod Health 2002; 34:294–303.
- Committee on Health Care for Underserved Women. Committee opinion no. 615: access to contraception. Obstet Gynecol 2015; 125:250–255.
- Committee on Adolescent Health Care. Committee opinion no. 598: the initial reproductive health visit. Obstet Gynecol 2014; 123:1143–1147.
- Winner B, Peipert JF, Zhao Q, et al. Effectiveness of long-acting reversible contraception. N Engl J Med 2012; 366:1998–2007.
- Secura GM, Madden T, McNicholas C, et al. Provision of no-cost, long-acting contraception and teenage pregnancy. N Engl J Med 2014; 371:1316–1323.
- Committee on Gynecologic Practice, American College of Obstetricians and Gynecologists. Over-the-counter access to oral contraceptives. Committee opinion no 544. Obstet Gynecol 2012; 120:1527–1531.
- Committee on Gynecologic Practice. ACOG committee opinion number 540: risk of venous thromboembolism among users of drospirenone-containing oral contraceptive pills. Obstet Gynecol 2012; 120:1239–1242.
- Centers for Disease Control and Prevention (CDC). US medical eligibility criteria for contraceptive use, 2010. MMWR Recomm Rep 2010; 59:1–86.
- Centers for Disease Control and Prevention (CDC). United States medical eligibility criteria (US MEC) for contraceptive use, 2010. www.cdc.gov/reproductivehealth/unintendedpregnancy/usmec.htm. Accessed September 8, 2015.
- Min J, Buckel C, Secura GM, Peipert JF, Madden T. Performance of a checklist to exclude pregnancy at the time of contraceptive initiation among women with a negative urine pregnancy test. Contraception 2015; 91:80–84.
- Batur P. Emergency contraception: separating fact from fiction. Cleve Clin J Med 2012; 79:771–776.
Reflux redux

Symptoms compatible with gastroesophageal reflux disease (GERD) are incredibly prevalent. The typical ones are common, and the atypical ones are so often attributed to GERD that they too are extremely common. It seems that few patients in my clinic are not taking a proton pump inhibitor (PPI).
Drs. Alzubaidi and Gabbard, in their review of GERD in this issue, note that up to 40% of people experience symptoms of GERD at least once monthly. Since these symptoms can be intermittent, diagnosis poses a problem when the diagnostic algorithm includes a trial of a PPI. It is sometimes unclear whether PPI therapy relieved the symptoms or whether the symptoms abated for other reasons. I suspect that many patients remain on PPI therapy longer than needed (and often longer than initially intended) because of a false sense of improvement and continued need. When patients are diagnosed on clinical grounds, we need to intermittently reassess the continued need for PPI therapy. The authors discuss and place in reasonable perspective a few of the potential complications of chronic PPI use, but not the effects on absorption of iron, calcium, and micronutrients, or PPI-associated gastric polyposis. These can be clinically significant in some patients.
I believe that some atypical symptoms such as cough and hoarseness are overly attributed to GERD, so that PPI therapy is started, continued, and escalated due to premature closure of the diagnosis. I believe that the diagnosis should be reassessed at least once with observed withdrawal of PPI therapy in patients who did not have a firm physiologic diagnosis. Asking the patient to keep a symptom diary may help.
Lack of a significant response to PPI therapy should cast doubt on the diagnosis of GERD and warrant exploration for an alternative cause of the symptoms (eg, eosinophilic esophagitis, bile reflux, sinus disease, dysmotility). The possibility that the patient was not given an optimal trial of a PPI must also be considered: eg, the dose may have been inadequate, the timing of administration may have been suboptimal (not preprandial), or the patient may have been taking over-the-counter NSAIDs.
GERD is so prevalent in the general population that we must train ourselves to consider the possibility that, even if totally relieved by PPI therapy, the symptoms might be associated with aggravating comorbid conditions such as obstructive sleep apnea, Raynaud phenomenon, drugs that can decrease the tone of the lower esophageal sphincter, or even scleroderma.
Finally, in patients who have had a less-than-total response to full-dose PPI therapy and have had other diagnoses excluded, we shouldn’t forget the value of adding appropriately timed histamine 2 receptor antagonist therapy (and asking the patient about use of medications that can exacerbate symptoms).
Even the diseases we deal with every day sometimes warrant a second look.
Symptoms compatible with gastroesophageal reflux disease (GERD) are incredibly prevalent. The typical ones are common, and the atypical ones are so often attributed to GERD that they too are extremely common. It seems that few patients in my clinic are not taking a proton pump inhibitor (PPI).
Drs. Alzubaidi and Gabbard, in their review of GERD in this issue, note that up to 40% of people experience symptoms of GERD at least once monthly. Since these symptoms can be intermittent, diagnosis poses a problem when the diagnostic algorithm includes a trial of a PPI. It is sometimes unclear whether PPI therapy relieved the symptoms or whether the symptoms abated for other reasons. I suspect that many patients remain on PPI therapy longer than needed (and often longer than initially intended) because of a false sense of improvement and continued need. When patients are diagnosed on clinical grounds, we need to intermittently reassess the continued need for PPI therapy. The authors discuss and place in reasonable perspective a few of the potential complications of chronic PPI use, but not the effects on absorption of iron, calcium, and micronutrients, or PPI-associated gastric polyposis. These can be clinically significant in some patients.
I believe that some atypical symptoms such as cough and hoarseness are overly attributed to GERD, so that PPI therapy is started, continued, and escalated due to premature closure of the diagnosis. I believe that the diagnosis should be reassessed at least once with observed withdrawal of PPI therapy in patients who did not have a firm physiologic diagnosis. Asking the patient to keep a symptom diary may help.
Lack of a significant response to PPI therapy should cast doubt on the diagnosis of GERD and warrant exploration for an alternative cause of the symptoms (eg, eosinophilic esophagitis, bile reflux, sinus disease, dysmotility). The possibility that the patient was not given an optimal trial of a PPI must also be considered: eg, the dose may have been inadequate, the timing of administration may have been suboptimal (not preprandial), or the patient may have been taking over-the-counter NSAIDs.
GERD is so prevalent in the general population that we must train ourselves to consider the possibility that, even if totally relieved by PPI therapy, the symptoms might be associated with aggravating comorbid conditions such as obstructive sleep apnea, Raynaud phenomenon, drugs that can decrease the tone of the lower esophageal sphincter, or even scleroderma.
Finally, in patients who have had a less-than-total response to full-dose PPI therapy and have had other diagnoses excluded, we shouldn’t forget the value of adding appropriately timed histamine 2 receptor antagonist therapy (and asking the patient about use of medications that can exacerbate symptoms).
Even the diseases we deal with every day sometimes warrant a second look.
Symptoms compatible with gastroesophageal reflux disease (GERD) are incredibly prevalent. The typical ones are common, and the atypical ones are so often attributed to GERD that they too are extremely common. It seems that few patients in my clinic are not taking a proton pump inhibitor (PPI).
Drs. Alzubaidi and Gabbard, in their review of GERD in this issue, note that up to 40% of people experience symptoms of GERD at least once monthly. Since these symptoms can be intermittent, diagnosis poses a problem when the diagnostic algorithm includes a trial of a PPI. It is sometimes unclear whether PPI therapy relieved the symptoms or whether the symptoms abated for other reasons. I suspect that many patients remain on PPI therapy longer than needed (and often longer than initially intended) because of a false sense of improvement and continued need. When patients are diagnosed on clinical grounds, we need to intermittently reassess the continued need for PPI therapy. The authors discuss and place in reasonable perspective a few of the potential complications of chronic PPI use, but not the effects on absorption of iron, calcium, and micronutrients, or PPI-associated gastric polyposis. These can be clinically significant in some patients.
I believe that some atypical symptoms such as cough and hoarseness are overly attributed to GERD, so that PPI therapy is started, continued, and escalated due to premature closure of the diagnosis. I believe that the diagnosis should be reassessed at least once with observed withdrawal of PPI therapy in patients who did not have a firm physiologic diagnosis. Asking the patient to keep a symptom diary may help.
Lack of a significant response to PPI therapy should cast doubt on the diagnosis of GERD and warrant exploration for an alternative cause of the symptoms (eg, eosinophilic esophagitis, bile reflux, sinus disease, dysmotility). The possibility that the patient was not given an optimal trial of a PPI must also be considered: eg, the dose may have been inadequate, the timing of administration may have been suboptimal (not preprandial), or the patient may have been taking over-the-counter NSAIDs.
GERD is so prevalent in the general population that we must train ourselves to consider the possibility that, even if totally relieved by PPI therapy, the symptoms might be associated with aggravating comorbid conditions such as obstructive sleep apnea, Raynaud phenomenon, drugs that can decrease the tone of the lower esophageal sphincter, or even scleroderma.
Finally, in patients who have had a less-than-total response to full-dose PPI therapy and have had other diagnoses excluded, we shouldn’t forget the value of adding appropriately timed histamine 2 receptor antagonist therapy (and asking the patient about use of medications that can exacerbate symptoms).
Even the diseases we deal with every day sometimes warrant a second look.
Ankle pain in a young woman with Gaucher disease
A 20-year-old woman with Gaucher disease presents with pain in her right ankle and in her back. She has had the ankle pain for the past 12 months and the back pain for the past 2 years. She describes the ankle pain as stabbing and moderately severe. It is constant, present both at rest and during physical activity, but aggravated by walking and twisting movements. She has noticed grinding and clicking sounds as she moves her ankle. The ankle pain has worsened over the past several months.
She says her back pain is similar to her ankle pain but less severe. She also reports generalized mild aches and bone pain. No other joints are involved. She has no history of fever, chills, or trauma.
A COMPLICATED MEDICAL HISTORY
Her Gaucher disease was diagnosed at age 4 when she presented with failure to thrive and with thrombocytopenia and splenomegaly. She and was found to have an N370S/IVS2+1 mutation of the GBA gene. She underwent removal of 90% of her spleen at the time of diagnosis and was on enzyme replacement therapy with imiglucerase until 3 years ago, when the treatment was stopped because the drug had become unavailable (because of a temporary closure of the manufacturing facility), and because she had developed neutralizing antibodies to it. Despite a dosage as high as 120 U/kg every 2 weeks (the recommended range is 2.5 U/kg three times a week up to 60 U/kg every 2 weeks), her anemia and thrombocytopenia worsened to the point that she became dependent on transfusion of red blood cells and platelets. She has also taken glucocorticoids at various times in the past as a premedication before enzyme replacement therapy.
About 3 years ago, she developed dryness of the skin, pruritus, shiny skin, hardening of the skin, and decreased oral aperture, which was diagnosed as scleroderma.
During the past 5 years, she has had multiple episodes of pale coloration of her skin on exposure to cold, suggestive of Raynaud phenomenon. And for the past 5 months, she has noticed a burning sensation in her throat and retrosternal pain, suggestive of gastroesophageal reflux disease.
She is a college student, with no history of smoking or use of alcohol or recreational drugs. She is sexually active, with no history of sexually transmitted disease, and she uses condoms and oral contraceptives for contraception.
Her father and mother are both carriers of Gaucher disease. She is not of Ashkenazi Jewish descent.
FINDINGS ON PHYSICAL EXAMINATION
On physical examination, her temperature, blood pressure, pulse, and respiratory rate are within normal limits. She has extensive tattooing on her upper chest to hide scarring from previous cannulation ports. The right ankle joint is moderately swollen but shows no other signs of inflammation; its range of motion is limited by severe pain. She has tenderness of the spinous processes and paraspinal area, in addition to multiple tender points in the thoracolumbar area. Palpation of the right hip reveals tenderness of the groin and trochanteric bursa.
No lymphadenopathy, hepatomegaly, splenomegaly, or abdominal masses are noted. Neurologic examination is essentially nonfocal.
Her current medications include omeprazole, ergocalciferol, calcium carbonate, gabapentin, citalopram, and celecoxib. She also takes a multivitamin daily.
1. Which is the most likely underlying cause of her ankle pain?
- Rheumatoid arthritis
- Gaucher disease
- Septic arthritis
- Avascular necrosis secondary to steroid use
Rheumatoid arthritis varies in its presentation. It is usually insidious in onset, migratory, and intermittent, with polyarticular or even monoarticular involvement, and it presents with pain, stiffness, and swelling of the joint.1 Most often affected are the metacarpophalangeal, proximal interphalangeal, wrist, and metatarsophalangeal joints. Involvement of large joints of the upper and lower limbs is also common.2 This is not the most likely cause of this patient’s symptoms, based on the history and the current presentation.
Gaucher disease is a lipidosis caused by accumulation of cellular glycolipids, especially glucocerebrosides, due to deficiency of the enzyme beta-glucosidase. Clinical manifestations include hepatomegaly, splenomegaly, and bone marrow disease presenting as anemia, thrombocytopenia, or skeletal disease.3 Skeletal involvement in Gaucher disease includes bone pain, bone infarcts, and lytic lesions.
Whether splenectomy predisposes the patient to bone manifestations is controversial. Some believe that splenectomy decreases the total body reservoir for the storage of glycolipids and predisposes to their deposition in bone, which in turn results in cortical thinning, impaired remodeling, and decreased intraosseous blood flow, leading to osteonecrosis and fractures.4 This is more common in patients with type 1 Gaucher disease who have undergone splenectomy. (Types 2 and 3 are much rarer, occurring mainly in children; central nervous system involvement is a key feature. A discussion of these types is beyond the focus of this paper.) However, some studies suggest that the increase in bone manifestations after splenectomy may be simply because of severe disease.5 It should be noted that, since the advent of enzyme replacement therapy for Gaucher disease, splenectomy is now rarely performed.6
Anemia is also considered an independent risk factor for the development of avascular necrosis in type 1 Gaucher disease.7 Osteonecrosis due to Gaucher disease is relatively common in the femur, tibia, and humerus and uncommon in the ankle joints.8
Septic arthritis is unlikely in this patient in the absence of fever or signs of inflammation of the joint. Her long-standing history of ankle pain would also be unusual for infection, but a superimposed infectious process should always be suspected in an arthritic joint.
Avascular necrosis secondary to steroid use. Glucocorticoids are notorious for their adverse effects on bone. They induce osteocyte apoptosis and a decrease in bone remodeling, potentially predisposing to osteonecrosis.9 There is a high incidence of osteoporosis, osteonecrosis, and fracture risk with glucocorticoid therapy, and the incidence is dose-dependent. Discontinuation of the drug only partially restores fracture risk to baseline levels.10,11
A meta-analysis of cohort studies with a total sample size of about 42,000 reported an increased risk of fracture at all ages with the use of glucocorticoids.12 Because the minimum dosage and duration of therapy to prevent glucocorticoid-induced osteoporosis are not known, the only recommendation is to keep the dosage as low as possible.13
Glucocorticoid therapy is the most common cause of nontraumatic avascular necrosis. The risk of osteonecrosis in patients on long-term glucocorticoid therapy may be as high as 40%.14 The risk is increased with prolonged treatment and with high doses, but it can also occur with short-term exposure to high doses. The increased risk has been shown to persist for as long as 2 years after the drugs are discontinued.15 Glucocorticoid-induced bone disease commonly affects the hip and vertebrae.
At this stage of the workup, we cannot completely rule out glucocorticoid use as the cause. However, after considering this patient’s presentation and the key features of the other diagnoses, her ankle pain and back pain are more likely caused by her preexisting Gaucher disease.
CONTINUED EVALUATION
Initial laboratory tests (Table 1) reveal severe anemia and thrombocytopenia. Bone marrow biopsy of the iliac crest done as part of the workup for these conditions shows extensive bone marrow space replacement by histiocytic infiltrate, consistent with Gaucher disease. No other marrow process is observed.
Radiography of the ankle (Figure 1) shows a subtle lucency in the talar dome with minimal subarticular collapse seen on the lateral view, suggestive of avascular necrosis and diffuse osteopenia. Joint spaces are maintained.
Magnetic resonance imaging (MRI) of the ankle shows numerous bone infarcts with an approximately 15-mm region of mild articular surface collapse in the central and lateral aspect of the talar dome.
MRI of the back shows extensive abnormal bone marrow signal intensity throughout the spine, compatible with a marrow replacement process. Patchy nonexpansile T2/stir hyperintensity with serpiginous enhancement within the T9, T11, T12, L2, and L3 vertebral bodies as well as throughout the entire sacrum is consistent with bone infarct.
2. Based on the results of radiographic studies, which is most likely the immediate cause of her ankle pain?
- Talar avascular necrosis secondary to rheumatoid arthritis
- Talar avascular necrosis secondary to Gaucher disease
- Trauma-induced fracture of the talus
- Plantar fasciitis
Of the bones of the feet, the talus is unique. It is the second largest of the tarsal bones and does not have muscular or tendinous attachments. Sixty percent of the talus bone is covered by articular cartilage,16 so only a limited area is available for penetration of blood vessels. Also, small nutrient vessels and variations of intraosseous anastomoses with a lack of collateral circulation predispose the talus to osteonecrosis when the vascular supply is compromised.16
Radiographic evidence of avascular necrosis is the presence of bone that is more radiopaque than normal bone; this is necrotic bone surrounded by osteopenic bone. Avascular necrosis causes hyperemia and resorption of bone. The resorption does not take place in necrotic bone because of the lack of a vascular supply, and so it appears radiopaque, whereas the bone surrounding the necrotic bone becomes osteopenic and radiolucent.
The sclerotic rim of a bone infarct is also enhanced by an attempted healing process in which new bone forms on the surface of necrotic trabeculae, a process known as “creeping substitution.” This gives a typical sclerotic picture of the talus.
MRI is the most sensitive technique for detecting osteonecrosis. A characteristic radiographic pattern is seen with osteonecrosis of the talus starting with talar dome opacity, followed by deformity and, in severe cases, articular collapse and bone fragmentation.17
The radiograph in our patient’s case is not consistent with features of rheumatoid arthritis or traumatic fracture of the talus. In plantar fasciitis, radiographs are used to rule out other pathologies of the foot, and the only finding may be a bone spur seen at the site of pain. The bone spur is not the cause of pain in plantar fasciitis but may be a result of the plantar fasciitis itself.
Therefore, avascular necrosis secondary to Gaucher disease is most likely the immediate cause of her ankle pain.
THE COURSE OF TREATMENT
The patient is started on enzyme replacement therapy with taliglucerase alfa (see discussion of enzyme replacement below). For the ankle pain, conservative management is prescribed, with application of a splint and a boot.
After 4 months of conservative management, radiography (Figure 2) and magnetic resonance imaging (Figure 3) show progressive deterioration of the talus body, and her ankle pain has worsened. A 6-week trial of an ankle brace also proves futile. Her pain continues to worsen and is not controllable with high doses of pain medication. She requests below-the-knee amputation.
Given the complexity of this patient’s medical condition, fusion of the ankle and hindfoot—which in some patients is preferable to amputation—is not considered because of her extensive bone involvement and ongoing thrombocytopenia, which would impede healing after the procedure. Below-the-knee amputation is performed without complications.
Study of the specimen after amputation reveals talar bone necrosis and bone marrow infiltration by foamy macrophages, consistent with Gaucher disease (Figures 4–6).
GAUCHER DISEASE
Pharmacologic treatments, effective only for type 1 Gaucher disease, target hepatosplenomegaly, cytopenia, and bone manifestations. Two approaches are enzyme replacement therapy—ie, to replace the defective enzyme—and substrate reduction therapy—ie, to reduce the production and thus the accumulation of glucocerebroside. Enzyme replacement is the first choice of therapy; substrate reduction is reserved for patients unable to tolerate enzyme replacement therapy.
Enzyme replacement
Current drugs for enzyme replacement therapy are imiglucerase, taliglucerase alfa, and velaglucerase alfa. The drugs are given by intravenous infusion over 1 to 2 hours in an outpatient clinic or office every 2 weeks.
These drugs are extremely expensive. Currently, the estimated cost of therapy for 1 year would be $432,978 for imiglucerase, $324,870 for taliglucerase alfa, and $368,550 for velaglucerase alfa. (The estimated costs are for 1 year of treatment for a 70-kg patient at 60 U/kg every 2 weeks.)18 Taliglucerase alfa is less expensive than the other two because it is plant-derived and thus can be more readily produced on a large scale.19
Substrate reduction
Current drugs for substrate reduction therapy are eliglustat and miglustat. They are given orally. Eliglustat is the first oral drug approved as a first-line treatment for Gaucher disease.20 Miglustat is approved only for mild to moderate disease when enzyme replacement fails or is not tolerated.
Patients can develop antibodies to any of the enzyme replacement drugs. It is not known whether this antibody response differs among the three drugs.21
Avascular necrosis of bone can occur in many clinical settings especially after a fracture, particularly of the head of the femur, which leads to interruption of blood supply to the area. Patients with sickle cell disease, those on corticosteroids or bisphosphonates (the latter causing osteonecrosis of the jaw), and those who have pancreatitis or human immunodeficiency virus infection are more prone to this bone complication.
In Gaucher disease, osteonecrosis is associated with splenectomy and severe disease and tends to occur at a younger age than in patients with other diagnoses.8 The plasma chitotriosidase activity and pulmonary and activation-regulated chemokines (PARC/CCL18), which are 10 to 40 times higher than normal in symptomatic patients with Gaucher disease, can be used as a biomarker of disease activity.8 Only plasma chitotriosidase is clinically available and used on a routine basis.
Bone involvement is seen in approximately 75% of the patients with type 1 Gaucher disease,22 and osteonecrosis is a severe form of bone involvement. Monitoring of patients for bone involvement is recommended. Enzyme replacement therapy for Gaucher disease needs to be started even if visceral disease is absent if the patient has evidence of bone involvement in the form of avascular necrosis.7 Prospective studies have shown that enzyme replacement therapy reduces the incidence of osteonecrosis.23
FOLLOW-UP MANAGEMENT OF OUR PATIENT
Avascular necrosis in Gaucher disease more typically involves the hips and shoulders. In the case of our patient, the talus was the most affected bone. Other contributing factors may have been the use of steroids as a premedication (often unnecessary) for her enzyme replacement therapy, as well as the coexistent scleroderma.24
The decision to switch from imiglucerase, to which she developed antibodies, to taliglucerase was made in the hope that the antibodies would not cross-react. After she started taliglucerase, her complete blood count values improved steadily. She did not require transfusions for more than 1 year. Her platelet count rose to 90 × 109/L, and her hemoglobin to 12 g/dL.
A multidisciplinary approach with regular monitoring and appropriate initiation of therapy is necessary to prevent disastrous complications in patients with Gaucher disease.
- Lee DM, Weinblatt ME. Rheumatoid arthritis. Lancet 2001; 358:903–911.
- Fleming A, Crown JM, Corbett M. Early rheumatoid disease. I. Onset. Ann Rheum Dis 1976; 35:357–360.
- Grabowski GA, Andria G, Baldellou A, et al. Pediatric non-neuronopathic Gaucher disease: presentation, diagnosis, and assessment. Consensus statements. Eur J Pediatr 2004; 163:58–66.
- Rodrigue SW, Rosenthal DI, Barton NW, Zurakowski D, Mankin HJ. Risk factors for osteonecrosis in patients with type 1 Gaucher’s disease. Clin Orthop Relat Res 1999; May (362):201–207.
- Lee RE. The pathology of Gaucher disease. Prog Clin Biol Res 1982; 95:177–217.
- Cox TM, Aerts JM, Belmatoug N, et al. Management of non-neuronopathic Gaucher disease with special reference to pregnancy, splenectomy, bisphosphonate therapy, use of biomarkers and bone disease monitoring. J Inherit Metab Dis 2008; 31:319–336.
- Khan A, Hangartner T, Weinreb NJ, Taylor JS, Mistry PK. Risk factors for fractures and avascular osteonecrosis in type 1 Gaucher disease: a study from the International Collaborative Gaucher Group (ICGG) Gaucher Registry. J Bone Miner Res 2012; 27:1839–1848.
- Deegan PB, Pavlova E, Tindall J, et al. Osseous manifestations of adult Gaucher disease in the era of enzyme replacement therapy. Medicine (Baltimore) 2011; 90:52–60.
- Weinstein RS. Glucocorticoid-induced osteonecrosis. Endocrine 2012; 41:183–190.
- Compston J. Management of glucocorticoid-induced osteoporosis. Nat Rev Rheumatol 2010; 6:82–88.
- Van Staa TP, Laan RF, Barton IP, Cohen S, Reid DM, Cooper C. Bone density threshold and other predictors of vertebral fracture in patients receiving oral glucocorticoid therapy. Arthritis Rheum 2003; 48:3224–3229.
- Kanis JA, Johnell O, Oden A, et al. The risk and burden of vertebral fractures in Sweden. Osteoporos Int 2004; 15:20–26.
- Seguro LP, Rosario C, Shoenfeld Y. Long-term complications of past glucocorticoid use. Autoimmun Rev 2013; 12:629–632.
- Weinstein RS. Glucocorticoid-induced osteoporosis and osteonecrosis. Endocrinol Metab Clin North Am 2012; 41:595–611.
- Cooper C, Steinbuch M, Stevenson R, Miday R, Watts NB. The epidemiology of osteonecrosis: findings from the GPRD and THIN databases in the UK. Osteoporos Int 2010; 21:569–577.
- Mulfinger GL, Trueta J. The blood supply of the talus. J Bone Joint Surg Br 1970; 52:160–167.
- Pearce DH, Mongiardi CN, Fornasier VL, Daniels TR. Avascular necrosis of the talus: a pictoral essay. Radiographics 2005; 25:399–410.
- In brief: Taliglucerase (Elelyso) for Gaucher disease. Med Lett Drugs Ther 2012 Jul 9; 54(1394):56.
- Hollak CE. An evidence-based review of the potential benefits of taliglucerase alfa in the treatment of patients with Gaucher disease. Core Evid 2012; 7:15–20.
- Poole RM. Eliglustat: first global approval. Drugs 2014; 74:1829–1836.
- Bennett LL, Mohan D. Gaucher disease and its treatment options. Ann Pharmacother 2013; 47:1182–1193.
- Germain DP. Gaucher’s disease: a paradigm for interventional genetics. Clin Genet 2004; 65:77–86.
- Sims KB, Pastores GM, Weinreb NJ, et al. Improvement of bone disease by imiglucerase (Cerezyme) therapy in patients with skeletal manifestations of type 1 Gaucher disease: results of a 48-month longitudinal cohort study. Clin Genet 2008; 73:430–440.
- Rennie C, Britton J, Prouse P. Bilateral avascular necrosis of the lunate in a patient with severe Raynaud’s phenomenon and scleroderma. J Clin Rheumatol 1999; 5:165–168.
A 20-year-old woman with Gaucher disease presents with pain in her right ankle and in her back. She has had the ankle pain for the past 12 months and the back pain for the past 2 years. She describes the ankle pain as stabbing and moderately severe. It is constant, present both at rest and during physical activity, but aggravated by walking and twisting movements. She has noticed grinding and clicking sounds as she moves her ankle. The ankle pain has worsened over the past several months.
She says her back pain is similar to her ankle pain but less severe. She also reports generalized mild aches and bone pain. No other joints are involved. She has no history of fever, chills, or trauma.
A COMPLICATED MEDICAL HISTORY
Her Gaucher disease was diagnosed at age 4 when she presented with failure to thrive and with thrombocytopenia and splenomegaly. She and was found to have an N370S/IVS2+1 mutation of the GBA gene. She underwent removal of 90% of her spleen at the time of diagnosis and was on enzyme replacement therapy with imiglucerase until 3 years ago, when the treatment was stopped because the drug had become unavailable (because of a temporary closure of the manufacturing facility), and because she had developed neutralizing antibodies to it. Despite a dosage as high as 120 U/kg every 2 weeks (the recommended range is 2.5 U/kg three times a week up to 60 U/kg every 2 weeks), her anemia and thrombocytopenia worsened to the point that she became dependent on transfusion of red blood cells and platelets. She has also taken glucocorticoids at various times in the past as a premedication before enzyme replacement therapy.
About 3 years ago, she developed dryness of the skin, pruritus, shiny skin, hardening of the skin, and decreased oral aperture, which was diagnosed as scleroderma.
During the past 5 years, she has had multiple episodes of pale coloration of her skin on exposure to cold, suggestive of Raynaud phenomenon. And for the past 5 months, she has noticed a burning sensation in her throat and retrosternal pain, suggestive of gastroesophageal reflux disease.
She is a college student, with no history of smoking or use of alcohol or recreational drugs. She is sexually active, with no history of sexually transmitted disease, and she uses condoms and oral contraceptives for contraception.
Her father and mother are both carriers of Gaucher disease. She is not of Ashkenazi Jewish descent.
FINDINGS ON PHYSICAL EXAMINATION
On physical examination, her temperature, blood pressure, pulse, and respiratory rate are within normal limits. She has extensive tattooing on her upper chest to hide scarring from previous cannulation ports. The right ankle joint is moderately swollen but shows no other signs of inflammation; its range of motion is limited by severe pain. She has tenderness of the spinous processes and paraspinal area, in addition to multiple tender points in the thoracolumbar area. Palpation of the right hip reveals tenderness of the groin and trochanteric bursa.
No lymphadenopathy, hepatomegaly, splenomegaly, or abdominal masses are noted. Neurologic examination is essentially nonfocal.
Her current medications include omeprazole, ergocalciferol, calcium carbonate, gabapentin, citalopram, and celecoxib. She also takes a multivitamin daily.
1. Which is the most likely underlying cause of her ankle pain?
- Rheumatoid arthritis
- Gaucher disease
- Septic arthritis
- Avascular necrosis secondary to steroid use
Rheumatoid arthritis varies in its presentation. It is usually insidious in onset, migratory, and intermittent, with polyarticular or even monoarticular involvement, and it presents with pain, stiffness, and swelling of the joint.1 Most often affected are the metacarpophalangeal, proximal interphalangeal, wrist, and metatarsophalangeal joints. Involvement of large joints of the upper and lower limbs is also common.2 This is not the most likely cause of this patient’s symptoms, based on the history and the current presentation.
Gaucher disease is a lipidosis caused by accumulation of cellular glycolipids, especially glucocerebrosides, due to deficiency of the enzyme beta-glucosidase. Clinical manifestations include hepatomegaly, splenomegaly, and bone marrow disease presenting as anemia, thrombocytopenia, or skeletal disease.3 Skeletal involvement in Gaucher disease includes bone pain, bone infarcts, and lytic lesions.
Whether splenectomy predisposes the patient to bone manifestations is controversial. Some believe that splenectomy decreases the total body reservoir for the storage of glycolipids and predisposes to their deposition in bone, which in turn results in cortical thinning, impaired remodeling, and decreased intraosseous blood flow, leading to osteonecrosis and fractures.4 This is more common in patients with type 1 Gaucher disease who have undergone splenectomy. (Types 2 and 3 are much rarer, occurring mainly in children; central nervous system involvement is a key feature. A discussion of these types is beyond the focus of this paper.) However, some studies suggest that the increase in bone manifestations after splenectomy may be simply because of severe disease.5 It should be noted that, since the advent of enzyme replacement therapy for Gaucher disease, splenectomy is now rarely performed.6
Anemia is also considered an independent risk factor for the development of avascular necrosis in type 1 Gaucher disease.7 Osteonecrosis due to Gaucher disease is relatively common in the femur, tibia, and humerus and uncommon in the ankle joints.8
Septic arthritis is unlikely in this patient in the absence of fever or signs of inflammation of the joint. Her long-standing history of ankle pain would also be unusual for infection, but a superimposed infectious process should always be suspected in an arthritic joint.
Avascular necrosis secondary to steroid use. Glucocorticoids are notorious for their adverse effects on bone. They induce osteocyte apoptosis and a decrease in bone remodeling, potentially predisposing to osteonecrosis.9 There is a high incidence of osteoporosis, osteonecrosis, and fracture risk with glucocorticoid therapy, and the incidence is dose-dependent. Discontinuation of the drug only partially restores fracture risk to baseline levels.10,11
A meta-analysis of cohort studies with a total sample size of about 42,000 reported an increased risk of fracture at all ages with the use of glucocorticoids.12 Because the minimum dosage and duration of therapy to prevent glucocorticoid-induced osteoporosis are not known, the only recommendation is to keep the dosage as low as possible.13
Glucocorticoid therapy is the most common cause of nontraumatic avascular necrosis. The risk of osteonecrosis in patients on long-term glucocorticoid therapy may be as high as 40%.14 The risk is increased with prolonged treatment and with high doses, but it can also occur with short-term exposure to high doses. The increased risk has been shown to persist for as long as 2 years after the drugs are discontinued.15 Glucocorticoid-induced bone disease commonly affects the hip and vertebrae.
At this stage of the workup, we cannot completely rule out glucocorticoid use as the cause. However, after considering this patient’s presentation and the key features of the other diagnoses, her ankle pain and back pain are more likely caused by her preexisting Gaucher disease.
CONTINUED EVALUATION
Initial laboratory tests (Table 1) reveal severe anemia and thrombocytopenia. Bone marrow biopsy of the iliac crest done as part of the workup for these conditions shows extensive bone marrow space replacement by histiocytic infiltrate, consistent with Gaucher disease. No other marrow process is observed.
Radiography of the ankle (Figure 1) shows a subtle lucency in the talar dome with minimal subarticular collapse seen on the lateral view, suggestive of avascular necrosis and diffuse osteopenia. Joint spaces are maintained.
Magnetic resonance imaging (MRI) of the ankle shows numerous bone infarcts with an approximately 15-mm region of mild articular surface collapse in the central and lateral aspect of the talar dome.
MRI of the back shows extensive abnormal bone marrow signal intensity throughout the spine, compatible with a marrow replacement process. Patchy nonexpansile T2/stir hyperintensity with serpiginous enhancement within the T9, T11, T12, L2, and L3 vertebral bodies as well as throughout the entire sacrum is consistent with bone infarct.
2. Based on the results of radiographic studies, which is most likely the immediate cause of her ankle pain?
- Talar avascular necrosis secondary to rheumatoid arthritis
- Talar avascular necrosis secondary to Gaucher disease
- Trauma-induced fracture of the talus
- Plantar fasciitis
Of the bones of the feet, the talus is unique. It is the second largest of the tarsal bones and does not have muscular or tendinous attachments. Sixty percent of the talus bone is covered by articular cartilage,16 so only a limited area is available for penetration of blood vessels. Also, small nutrient vessels and variations of intraosseous anastomoses with a lack of collateral circulation predispose the talus to osteonecrosis when the vascular supply is compromised.16
Radiographic evidence of avascular necrosis is the presence of bone that is more radiopaque than normal bone; this is necrotic bone surrounded by osteopenic bone. Avascular necrosis causes hyperemia and resorption of bone. The resorption does not take place in necrotic bone because of the lack of a vascular supply, and so it appears radiopaque, whereas the bone surrounding the necrotic bone becomes osteopenic and radiolucent.
The sclerotic rim of a bone infarct is also enhanced by an attempted healing process in which new bone forms on the surface of necrotic trabeculae, a process known as “creeping substitution.” This gives a typical sclerotic picture of the talus.
MRI is the most sensitive technique for detecting osteonecrosis. A characteristic radiographic pattern is seen with osteonecrosis of the talus starting with talar dome opacity, followed by deformity and, in severe cases, articular collapse and bone fragmentation.17
The radiograph in our patient’s case is not consistent with features of rheumatoid arthritis or traumatic fracture of the talus. In plantar fasciitis, radiographs are used to rule out other pathologies of the foot, and the only finding may be a bone spur seen at the site of pain. The bone spur is not the cause of pain in plantar fasciitis but may be a result of the plantar fasciitis itself.
Therefore, avascular necrosis secondary to Gaucher disease is most likely the immediate cause of her ankle pain.
THE COURSE OF TREATMENT
The patient is started on enzyme replacement therapy with taliglucerase alfa (see discussion of enzyme replacement below). For the ankle pain, conservative management is prescribed, with application of a splint and a boot.
After 4 months of conservative management, radiography (Figure 2) and magnetic resonance imaging (Figure 3) show progressive deterioration of the talus body, and her ankle pain has worsened. A 6-week trial of an ankle brace also proves futile. Her pain continues to worsen and is not controllable with high doses of pain medication. She requests below-the-knee amputation.
Given the complexity of this patient’s medical condition, fusion of the ankle and hindfoot—which in some patients is preferable to amputation—is not considered because of her extensive bone involvement and ongoing thrombocytopenia, which would impede healing after the procedure. Below-the-knee amputation is performed without complications.
Study of the specimen after amputation reveals talar bone necrosis and bone marrow infiltration by foamy macrophages, consistent with Gaucher disease (Figures 4–6).
GAUCHER DISEASE
Pharmacologic treatments, effective only for type 1 Gaucher disease, target hepatosplenomegaly, cytopenia, and bone manifestations. Two approaches are enzyme replacement therapy—ie, to replace the defective enzyme—and substrate reduction therapy—ie, to reduce the production and thus the accumulation of glucocerebroside. Enzyme replacement is the first choice of therapy; substrate reduction is reserved for patients unable to tolerate enzyme replacement therapy.
Enzyme replacement
Current drugs for enzyme replacement therapy are imiglucerase, taliglucerase alfa, and velaglucerase alfa. The drugs are given by intravenous infusion over 1 to 2 hours in an outpatient clinic or office every 2 weeks.
These drugs are extremely expensive. Currently, the estimated cost of therapy for 1 year would be $432,978 for imiglucerase, $324,870 for taliglucerase alfa, and $368,550 for velaglucerase alfa. (The estimated costs are for 1 year of treatment for a 70-kg patient at 60 U/kg every 2 weeks.)18 Taliglucerase alfa is less expensive than the other two because it is plant-derived and thus can be more readily produced on a large scale.19
Substrate reduction
Current drugs for substrate reduction therapy are eliglustat and miglustat. They are given orally. Eliglustat is the first oral drug approved as a first-line treatment for Gaucher disease.20 Miglustat is approved only for mild to moderate disease when enzyme replacement fails or is not tolerated.
Patients can develop antibodies to any of the enzyme replacement drugs. It is not known whether this antibody response differs among the three drugs.21
Avascular necrosis of bone can occur in many clinical settings especially after a fracture, particularly of the head of the femur, which leads to interruption of blood supply to the area. Patients with sickle cell disease, those on corticosteroids or bisphosphonates (the latter causing osteonecrosis of the jaw), and those who have pancreatitis or human immunodeficiency virus infection are more prone to this bone complication.
In Gaucher disease, osteonecrosis is associated with splenectomy and severe disease and tends to occur at a younger age than in patients with other diagnoses.8 The plasma chitotriosidase activity and pulmonary and activation-regulated chemokines (PARC/CCL18), which are 10 to 40 times higher than normal in symptomatic patients with Gaucher disease, can be used as a biomarker of disease activity.8 Only plasma chitotriosidase is clinically available and used on a routine basis.
Bone involvement is seen in approximately 75% of the patients with type 1 Gaucher disease,22 and osteonecrosis is a severe form of bone involvement. Monitoring of patients for bone involvement is recommended. Enzyme replacement therapy for Gaucher disease needs to be started even if visceral disease is absent if the patient has evidence of bone involvement in the form of avascular necrosis.7 Prospective studies have shown that enzyme replacement therapy reduces the incidence of osteonecrosis.23
FOLLOW-UP MANAGEMENT OF OUR PATIENT
Avascular necrosis in Gaucher disease more typically involves the hips and shoulders. In the case of our patient, the talus was the most affected bone. Other contributing factors may have been the use of steroids as a premedication (often unnecessary) for her enzyme replacement therapy, as well as the coexistent scleroderma.24
The decision to switch from imiglucerase, to which she developed antibodies, to taliglucerase was made in the hope that the antibodies would not cross-react. After she started taliglucerase, her complete blood count values improved steadily. She did not require transfusions for more than 1 year. Her platelet count rose to 90 × 109/L, and her hemoglobin to 12 g/dL.
A multidisciplinary approach with regular monitoring and appropriate initiation of therapy is necessary to prevent disastrous complications in patients with Gaucher disease.
A 20-year-old woman with Gaucher disease presents with pain in her right ankle and in her back. She has had the ankle pain for the past 12 months and the back pain for the past 2 years. She describes the ankle pain as stabbing and moderately severe. It is constant, present both at rest and during physical activity, but aggravated by walking and twisting movements. She has noticed grinding and clicking sounds as she moves her ankle. The ankle pain has worsened over the past several months.
She says her back pain is similar to her ankle pain but less severe. She also reports generalized mild aches and bone pain. No other joints are involved. She has no history of fever, chills, or trauma.
A COMPLICATED MEDICAL HISTORY
Her Gaucher disease was diagnosed at age 4 when she presented with failure to thrive and with thrombocytopenia and splenomegaly. She and was found to have an N370S/IVS2+1 mutation of the GBA gene. She underwent removal of 90% of her spleen at the time of diagnosis and was on enzyme replacement therapy with imiglucerase until 3 years ago, when the treatment was stopped because the drug had become unavailable (because of a temporary closure of the manufacturing facility), and because she had developed neutralizing antibodies to it. Despite a dosage as high as 120 U/kg every 2 weeks (the recommended range is 2.5 U/kg three times a week up to 60 U/kg every 2 weeks), her anemia and thrombocytopenia worsened to the point that she became dependent on transfusion of red blood cells and platelets. She has also taken glucocorticoids at various times in the past as a premedication before enzyme replacement therapy.
About 3 years ago, she developed dryness of the skin, pruritus, shiny skin, hardening of the skin, and decreased oral aperture, which was diagnosed as scleroderma.
During the past 5 years, she has had multiple episodes of pale coloration of her skin on exposure to cold, suggestive of Raynaud phenomenon. And for the past 5 months, she has noticed a burning sensation in her throat and retrosternal pain, suggestive of gastroesophageal reflux disease.
She is a college student, with no history of smoking or use of alcohol or recreational drugs. She is sexually active, with no history of sexually transmitted disease, and she uses condoms and oral contraceptives for contraception.
Her father and mother are both carriers of Gaucher disease. She is not of Ashkenazi Jewish descent.
FINDINGS ON PHYSICAL EXAMINATION
On physical examination, her temperature, blood pressure, pulse, and respiratory rate are within normal limits. She has extensive tattooing on her upper chest to hide scarring from previous cannulation ports. The right ankle joint is moderately swollen but shows no other signs of inflammation; its range of motion is limited by severe pain. She has tenderness of the spinous processes and paraspinal area, in addition to multiple tender points in the thoracolumbar area. Palpation of the right hip reveals tenderness of the groin and trochanteric bursa.
No lymphadenopathy, hepatomegaly, splenomegaly, or abdominal masses are noted. Neurologic examination is essentially nonfocal.
Her current medications include omeprazole, ergocalciferol, calcium carbonate, gabapentin, citalopram, and celecoxib. She also takes a multivitamin daily.
1. Which is the most likely underlying cause of her ankle pain?
- Rheumatoid arthritis
- Gaucher disease
- Septic arthritis
- Avascular necrosis secondary to steroid use
Rheumatoid arthritis varies in its presentation. It is usually insidious in onset, migratory, and intermittent, with polyarticular or even monoarticular involvement, and it presents with pain, stiffness, and swelling of the joint.1 Most often affected are the metacarpophalangeal, proximal interphalangeal, wrist, and metatarsophalangeal joints. Involvement of large joints of the upper and lower limbs is also common.2 This is not the most likely cause of this patient’s symptoms, based on the history and the current presentation.
Gaucher disease is a lipidosis caused by accumulation of cellular glycolipids, especially glucocerebrosides, due to deficiency of the enzyme beta-glucosidase. Clinical manifestations include hepatomegaly, splenomegaly, and bone marrow disease presenting as anemia, thrombocytopenia, or skeletal disease.3 Skeletal involvement in Gaucher disease includes bone pain, bone infarcts, and lytic lesions.
Whether splenectomy predisposes the patient to bone manifestations is controversial. Some believe that splenectomy decreases the total body reservoir for the storage of glycolipids and predisposes to their deposition in bone, which in turn results in cortical thinning, impaired remodeling, and decreased intraosseous blood flow, leading to osteonecrosis and fractures.4 This is more common in patients with type 1 Gaucher disease who have undergone splenectomy. (Types 2 and 3 are much rarer, occurring mainly in children; central nervous system involvement is a key feature. A discussion of these types is beyond the focus of this paper.) However, some studies suggest that the increase in bone manifestations after splenectomy may be simply because of severe disease.5 It should be noted that, since the advent of enzyme replacement therapy for Gaucher disease, splenectomy is now rarely performed.6
Anemia is also considered an independent risk factor for the development of avascular necrosis in type 1 Gaucher disease.7 Osteonecrosis due to Gaucher disease is relatively common in the femur, tibia, and humerus and uncommon in the ankle joints.8
Septic arthritis is unlikely in this patient in the absence of fever or signs of inflammation of the joint. Her long-standing history of ankle pain would also be unusual for infection, but a superimposed infectious process should always be suspected in an arthritic joint.
Avascular necrosis secondary to steroid use. Glucocorticoids are notorious for their adverse effects on bone. They induce osteocyte apoptosis and a decrease in bone remodeling, potentially predisposing to osteonecrosis.9 There is a high incidence of osteoporosis, osteonecrosis, and fracture risk with glucocorticoid therapy, and the incidence is dose-dependent. Discontinuation of the drug only partially restores fracture risk to baseline levels.10,11
A meta-analysis of cohort studies with a total sample size of about 42,000 reported an increased risk of fracture at all ages with the use of glucocorticoids.12 Because the minimum dosage and duration of therapy to prevent glucocorticoid-induced osteoporosis are not known, the only recommendation is to keep the dosage as low as possible.13
Glucocorticoid therapy is the most common cause of nontraumatic avascular necrosis. The risk of osteonecrosis in patients on long-term glucocorticoid therapy may be as high as 40%.14 The risk is increased with prolonged treatment and with high doses, but it can also occur with short-term exposure to high doses. The increased risk has been shown to persist for as long as 2 years after the drugs are discontinued.15 Glucocorticoid-induced bone disease commonly affects the hip and vertebrae.
At this stage of the workup, we cannot completely rule out glucocorticoid use as the cause. However, after considering this patient’s presentation and the key features of the other diagnoses, her ankle pain and back pain are more likely caused by her preexisting Gaucher disease.
CONTINUED EVALUATION
Initial laboratory tests (Table 1) reveal severe anemia and thrombocytopenia. Bone marrow biopsy of the iliac crest done as part of the workup for these conditions shows extensive bone marrow space replacement by histiocytic infiltrate, consistent with Gaucher disease. No other marrow process is observed.
Radiography of the ankle (Figure 1) shows a subtle lucency in the talar dome with minimal subarticular collapse seen on the lateral view, suggestive of avascular necrosis and diffuse osteopenia. Joint spaces are maintained.
Magnetic resonance imaging (MRI) of the ankle shows numerous bone infarcts with an approximately 15-mm region of mild articular surface collapse in the central and lateral aspect of the talar dome.
MRI of the back shows extensive abnormal bone marrow signal intensity throughout the spine, compatible with a marrow replacement process. Patchy nonexpansile T2/stir hyperintensity with serpiginous enhancement within the T9, T11, T12, L2, and L3 vertebral bodies as well as throughout the entire sacrum is consistent with bone infarct.
2. Based on the results of radiographic studies, which is most likely the immediate cause of her ankle pain?
- Talar avascular necrosis secondary to rheumatoid arthritis
- Talar avascular necrosis secondary to Gaucher disease
- Trauma-induced fracture of the talus
- Plantar fasciitis
Of the bones of the feet, the talus is unique. It is the second largest of the tarsal bones and does not have muscular or tendinous attachments. Sixty percent of the talus bone is covered by articular cartilage,16 so only a limited area is available for penetration of blood vessels. Also, small nutrient vessels and variations of intraosseous anastomoses with a lack of collateral circulation predispose the talus to osteonecrosis when the vascular supply is compromised.16
Radiographic evidence of avascular necrosis is the presence of bone that is more radiopaque than normal bone; this is necrotic bone surrounded by osteopenic bone. Avascular necrosis causes hyperemia and resorption of bone. The resorption does not take place in necrotic bone because of the lack of a vascular supply, and so it appears radiopaque, whereas the bone surrounding the necrotic bone becomes osteopenic and radiolucent.
The sclerotic rim of a bone infarct is also enhanced by an attempted healing process in which new bone forms on the surface of necrotic trabeculae, a process known as “creeping substitution.” This gives a typical sclerotic picture of the talus.
MRI is the most sensitive technique for detecting osteonecrosis. A characteristic radiographic pattern is seen with osteonecrosis of the talus starting with talar dome opacity, followed by deformity and, in severe cases, articular collapse and bone fragmentation.17
The radiograph in our patient’s case is not consistent with features of rheumatoid arthritis or traumatic fracture of the talus. In plantar fasciitis, radiographs are used to rule out other pathologies of the foot, and the only finding may be a bone spur seen at the site of pain. The bone spur is not the cause of pain in plantar fasciitis but may be a result of the plantar fasciitis itself.
Therefore, avascular necrosis secondary to Gaucher disease is most likely the immediate cause of her ankle pain.
THE COURSE OF TREATMENT
The patient is started on enzyme replacement therapy with taliglucerase alfa (see discussion of enzyme replacement below). For the ankle pain, conservative management is prescribed, with application of a splint and a boot.
After 4 months of conservative management, radiography (Figure 2) and magnetic resonance imaging (Figure 3) show progressive deterioration of the talus body, and her ankle pain has worsened. A 6-week trial of an ankle brace also proves futile. Her pain continues to worsen and is not controllable with high doses of pain medication. She requests below-the-knee amputation.
Given the complexity of this patient’s medical condition, fusion of the ankle and hindfoot—which in some patients is preferable to amputation—is not considered because of her extensive bone involvement and ongoing thrombocytopenia, which would impede healing after the procedure. Below-the-knee amputation is performed without complications.
Study of the specimen after amputation reveals talar bone necrosis and bone marrow infiltration by foamy macrophages, consistent with Gaucher disease (Figures 4–6).
GAUCHER DISEASE
Pharmacologic treatments, effective only for type 1 Gaucher disease, target hepatosplenomegaly, cytopenia, and bone manifestations. Two approaches are enzyme replacement therapy—ie, to replace the defective enzyme—and substrate reduction therapy—ie, to reduce the production and thus the accumulation of glucocerebroside. Enzyme replacement is the first choice of therapy; substrate reduction is reserved for patients unable to tolerate enzyme replacement therapy.
Enzyme replacement
Current drugs for enzyme replacement therapy are imiglucerase, taliglucerase alfa, and velaglucerase alfa. The drugs are given by intravenous infusion over 1 to 2 hours in an outpatient clinic or office every 2 weeks.
These drugs are extremely expensive. Currently, the estimated cost of therapy for 1 year would be $432,978 for imiglucerase, $324,870 for taliglucerase alfa, and $368,550 for velaglucerase alfa. (The estimated costs are for 1 year of treatment for a 70-kg patient at 60 U/kg every 2 weeks.)18 Taliglucerase alfa is less expensive than the other two because it is plant-derived and thus can be more readily produced on a large scale.19
Substrate reduction
Current drugs for substrate reduction therapy are eliglustat and miglustat. They are given orally. Eliglustat is the first oral drug approved as a first-line treatment for Gaucher disease.20 Miglustat is approved only for mild to moderate disease when enzyme replacement fails or is not tolerated.
Patients can develop antibodies to any of the enzyme replacement drugs. It is not known whether this antibody response differs among the three drugs.21
Avascular necrosis of bone can occur in many clinical settings especially after a fracture, particularly of the head of the femur, which leads to interruption of blood supply to the area. Patients with sickle cell disease, those on corticosteroids or bisphosphonates (the latter causing osteonecrosis of the jaw), and those who have pancreatitis or human immunodeficiency virus infection are more prone to this bone complication.
In Gaucher disease, osteonecrosis is associated with splenectomy and severe disease and tends to occur at a younger age than in patients with other diagnoses.8 The plasma chitotriosidase activity and pulmonary and activation-regulated chemokines (PARC/CCL18), which are 10 to 40 times higher than normal in symptomatic patients with Gaucher disease, can be used as a biomarker of disease activity.8 Only plasma chitotriosidase is clinically available and used on a routine basis.
Bone involvement is seen in approximately 75% of the patients with type 1 Gaucher disease,22 and osteonecrosis is a severe form of bone involvement. Monitoring of patients for bone involvement is recommended. Enzyme replacement therapy for Gaucher disease needs to be started even if visceral disease is absent if the patient has evidence of bone involvement in the form of avascular necrosis.7 Prospective studies have shown that enzyme replacement therapy reduces the incidence of osteonecrosis.23
FOLLOW-UP MANAGEMENT OF OUR PATIENT
Avascular necrosis in Gaucher disease more typically involves the hips and shoulders. In the case of our patient, the talus was the most affected bone. Other contributing factors may have been the use of steroids as a premedication (often unnecessary) for her enzyme replacement therapy, as well as the coexistent scleroderma.24
The decision to switch from imiglucerase, to which she developed antibodies, to taliglucerase was made in the hope that the antibodies would not cross-react. After she started taliglucerase, her complete blood count values improved steadily. She did not require transfusions for more than 1 year. Her platelet count rose to 90 × 109/L, and her hemoglobin to 12 g/dL.
A multidisciplinary approach with regular monitoring and appropriate initiation of therapy is necessary to prevent disastrous complications in patients with Gaucher disease.
- Lee DM, Weinblatt ME. Rheumatoid arthritis. Lancet 2001; 358:903–911.
- Fleming A, Crown JM, Corbett M. Early rheumatoid disease. I. Onset. Ann Rheum Dis 1976; 35:357–360.
- Grabowski GA, Andria G, Baldellou A, et al. Pediatric non-neuronopathic Gaucher disease: presentation, diagnosis, and assessment. Consensus statements. Eur J Pediatr 2004; 163:58–66.
- Rodrigue SW, Rosenthal DI, Barton NW, Zurakowski D, Mankin HJ. Risk factors for osteonecrosis in patients with type 1 Gaucher’s disease. Clin Orthop Relat Res 1999; May (362):201–207.
- Lee RE. The pathology of Gaucher disease. Prog Clin Biol Res 1982; 95:177–217.
- Cox TM, Aerts JM, Belmatoug N, et al. Management of non-neuronopathic Gaucher disease with special reference to pregnancy, splenectomy, bisphosphonate therapy, use of biomarkers and bone disease monitoring. J Inherit Metab Dis 2008; 31:319–336.
- Khan A, Hangartner T, Weinreb NJ, Taylor JS, Mistry PK. Risk factors for fractures and avascular osteonecrosis in type 1 Gaucher disease: a study from the International Collaborative Gaucher Group (ICGG) Gaucher Registry. J Bone Miner Res 2012; 27:1839–1848.
- Deegan PB, Pavlova E, Tindall J, et al. Osseous manifestations of adult Gaucher disease in the era of enzyme replacement therapy. Medicine (Baltimore) 2011; 90:52–60.
- Weinstein RS. Glucocorticoid-induced osteonecrosis. Endocrine 2012; 41:183–190.
- Compston J. Management of glucocorticoid-induced osteoporosis. Nat Rev Rheumatol 2010; 6:82–88.
- Van Staa TP, Laan RF, Barton IP, Cohen S, Reid DM, Cooper C. Bone density threshold and other predictors of vertebral fracture in patients receiving oral glucocorticoid therapy. Arthritis Rheum 2003; 48:3224–3229.
- Kanis JA, Johnell O, Oden A, et al. The risk and burden of vertebral fractures in Sweden. Osteoporos Int 2004; 15:20–26.
- Seguro LP, Rosario C, Shoenfeld Y. Long-term complications of past glucocorticoid use. Autoimmun Rev 2013; 12:629–632.
- Weinstein RS. Glucocorticoid-induced osteoporosis and osteonecrosis. Endocrinol Metab Clin North Am 2012; 41:595–611.
- Cooper C, Steinbuch M, Stevenson R, Miday R, Watts NB. The epidemiology of osteonecrosis: findings from the GPRD and THIN databases in the UK. Osteoporos Int 2010; 21:569–577.
- Mulfinger GL, Trueta J. The blood supply of the talus. J Bone Joint Surg Br 1970; 52:160–167.
- Pearce DH, Mongiardi CN, Fornasier VL, Daniels TR. Avascular necrosis of the talus: a pictoral essay. Radiographics 2005; 25:399–410.
- In brief: Taliglucerase (Elelyso) for Gaucher disease. Med Lett Drugs Ther 2012 Jul 9; 54(1394):56.
- Hollak CE. An evidence-based review of the potential benefits of taliglucerase alfa in the treatment of patients with Gaucher disease. Core Evid 2012; 7:15–20.
- Poole RM. Eliglustat: first global approval. Drugs 2014; 74:1829–1836.
- Bennett LL, Mohan D. Gaucher disease and its treatment options. Ann Pharmacother 2013; 47:1182–1193.
- Germain DP. Gaucher’s disease: a paradigm for interventional genetics. Clin Genet 2004; 65:77–86.
- Sims KB, Pastores GM, Weinreb NJ, et al. Improvement of bone disease by imiglucerase (Cerezyme) therapy in patients with skeletal manifestations of type 1 Gaucher disease: results of a 48-month longitudinal cohort study. Clin Genet 2008; 73:430–440.
- Rennie C, Britton J, Prouse P. Bilateral avascular necrosis of the lunate in a patient with severe Raynaud’s phenomenon and scleroderma. J Clin Rheumatol 1999; 5:165–168.
- Lee DM, Weinblatt ME. Rheumatoid arthritis. Lancet 2001; 358:903–911.
- Fleming A, Crown JM, Corbett M. Early rheumatoid disease. I. Onset. Ann Rheum Dis 1976; 35:357–360.
- Grabowski GA, Andria G, Baldellou A, et al. Pediatric non-neuronopathic Gaucher disease: presentation, diagnosis, and assessment. Consensus statements. Eur J Pediatr 2004; 163:58–66.
- Rodrigue SW, Rosenthal DI, Barton NW, Zurakowski D, Mankin HJ. Risk factors for osteonecrosis in patients with type 1 Gaucher’s disease. Clin Orthop Relat Res 1999; May (362):201–207.
- Lee RE. The pathology of Gaucher disease. Prog Clin Biol Res 1982; 95:177–217.
- Cox TM, Aerts JM, Belmatoug N, et al. Management of non-neuronopathic Gaucher disease with special reference to pregnancy, splenectomy, bisphosphonate therapy, use of biomarkers and bone disease monitoring. J Inherit Metab Dis 2008; 31:319–336.
- Khan A, Hangartner T, Weinreb NJ, Taylor JS, Mistry PK. Risk factors for fractures and avascular osteonecrosis in type 1 Gaucher disease: a study from the International Collaborative Gaucher Group (ICGG) Gaucher Registry. J Bone Miner Res 2012; 27:1839–1848.
- Deegan PB, Pavlova E, Tindall J, et al. Osseous manifestations of adult Gaucher disease in the era of enzyme replacement therapy. Medicine (Baltimore) 2011; 90:52–60.
- Weinstein RS. Glucocorticoid-induced osteonecrosis. Endocrine 2012; 41:183–190.
- Compston J. Management of glucocorticoid-induced osteoporosis. Nat Rev Rheumatol 2010; 6:82–88.
- Van Staa TP, Laan RF, Barton IP, Cohen S, Reid DM, Cooper C. Bone density threshold and other predictors of vertebral fracture in patients receiving oral glucocorticoid therapy. Arthritis Rheum 2003; 48:3224–3229.
- Kanis JA, Johnell O, Oden A, et al. The risk and burden of vertebral fractures in Sweden. Osteoporos Int 2004; 15:20–26.
- Seguro LP, Rosario C, Shoenfeld Y. Long-term complications of past glucocorticoid use. Autoimmun Rev 2013; 12:629–632.
- Weinstein RS. Glucocorticoid-induced osteoporosis and osteonecrosis. Endocrinol Metab Clin North Am 2012; 41:595–611.
- Cooper C, Steinbuch M, Stevenson R, Miday R, Watts NB. The epidemiology of osteonecrosis: findings from the GPRD and THIN databases in the UK. Osteoporos Int 2010; 21:569–577.
- Mulfinger GL, Trueta J. The blood supply of the talus. J Bone Joint Surg Br 1970; 52:160–167.
- Pearce DH, Mongiardi CN, Fornasier VL, Daniels TR. Avascular necrosis of the talus: a pictoral essay. Radiographics 2005; 25:399–410.
- In brief: Taliglucerase (Elelyso) for Gaucher disease. Med Lett Drugs Ther 2012 Jul 9; 54(1394):56.
- Hollak CE. An evidence-based review of the potential benefits of taliglucerase alfa in the treatment of patients with Gaucher disease. Core Evid 2012; 7:15–20.
- Poole RM. Eliglustat: first global approval. Drugs 2014; 74:1829–1836.
- Bennett LL, Mohan D. Gaucher disease and its treatment options. Ann Pharmacother 2013; 47:1182–1193.
- Germain DP. Gaucher’s disease: a paradigm for interventional genetics. Clin Genet 2004; 65:77–86.
- Sims KB, Pastores GM, Weinreb NJ, et al. Improvement of bone disease by imiglucerase (Cerezyme) therapy in patients with skeletal manifestations of type 1 Gaucher disease: results of a 48-month longitudinal cohort study. Clin Genet 2008; 73:430–440.
- Rennie C, Britton J, Prouse P. Bilateral avascular necrosis of the lunate in a patient with severe Raynaud’s phenomenon and scleroderma. J Clin Rheumatol 1999; 5:165–168.