User login
Improving medication safety during hospital-based transitions of care
Any time patients enter or leave the hospital, they risk being harmed by errors in their medications.1 Adverse events from medication errors during transitions of care are common but often preventable. One key approach is to systematically review every patient’s medication list on admission and discharge and resolve any discrepancies. These transitions are also an opportunity to address other medication-related problems, such as adherence, drug interactions, and clinical appropriateness.
This article summarizes the types and prevalence of medication problems that occur during hospital-based transitions of care, and suggests strategies to decrease the risk of medication errors, focusing on medication reconciliation and related interventions that clinicians can use at the bedside to improve medication safety.
DEFINING TERMS
A medication discrepancy is any variance noted in a patient’s documented medication regimen across different medication lists or sites of care. While some differences reflect intentional and clinically appropriate changes to the regimen, others are unintentional and reflect inaccurate or incomplete information. These unintentional discrepancies are medication errors.
Depending on the clinical circumstances and medications involved, such errors may lead to an adverse drug event (ADE), defined as actual harm or injury resulting from a medication. Sometimes a medication error does not cause harm immediately but could if left uncorrected; this is called a potential ADE.
An important goal during transitions of care is to reduce unintentional medication discrepancies, thereby reducing potential and actual ADEs.
ERRORS ARISE AT DISCHARGE—AND EVEN MORE AT ADMISSION
Hospital discharge is a widely recognized transition in which patient harm occurs. As many as 70% of patients may have an unintentional medication discrepancy at hospital discharge, with many of those discrepancies having potential for harm.2 Indeed, during the first few weeks after discharge, 50% of patients have a clinically important medication error,3 and 20% experience an adverse event, most commonly an ADE.4 ADEs are associated with excess health care utilization,5–7 and many are preventable through strategies such as medication reconciliation.5,8
Importantly, more errors arise at hospital admission than at other times.9,10
Errors in medication histories are the most common source of discrepancies, affecting up to two-thirds of admitted patients.11,12 More than one-quarter of hospital prescribing errors can be attributed to incomplete medication histories at the time of admission,13 and nearly three times as many clinically important medication discrepancies are related to history-taking errors on admission rather than reconciliation errors at discharge.9
Most discrepancies occurring at the time of admission have the potential to cause harm, particularly if the errors persist beyond discharge.14 Therefore, taking a complete and accurate medication history on hospital admission is critical to ensuring safe care transitions.
MEDICATION RECONCILIATION: BARRIERS AND FACILITATORS
Medication reconciliation is a strategy for reducing medication discrepancies in patients moving across care settings. Simply put, it is the process by which a patient’s medication list is obtained, compared, and clarified across different sites of care.15 It has consistently been shown to decrease medication errors compared with usual care,16 and it is strongly supported by national and international organizations.17–21

In clinical practice, many physicians and institutions have found medication reconciliation difficult to implement, owing to barriers at the level of the patient, provider, and system (Table 1). In response to these challenges, two initiatives have synthesized best practices and offer toolkits that hospitals and clinicians can use: the Medications at Transitions and Clinical Handoffs (MATCH) program22 and the Multi-Center Medication Reconciliation Quality Improvement Study (MARQUIS).23
Lack of resources is a widely acknowledged challenge. Thus, the MARQUIS investigators23 suggested focusing on the admission history, where most errors occur, and applying the most resource-intensive interventions in patients at highest risk of ADEs, ie, those who are elderly, have multiple comorbid conditions, or take numerous medications.16
Although the risk of an ADE increases with the number of medications a patient takes,4 the exact number of drugs that defines high risk has not been well established. Targeting patients who take 10 or more maintenance medications is a reasonable initial approach,24 but institutions should tailor risk stratification to their patient populations and available resources. Patients taking high-risk medications such as anticoagulants and insulin could also be prioritized for review, since these medications are more likely to cause serious patient harm when used without appropriate clinical oversight.7
Using pharmacy staff to perform medication history-taking, reconciliation, and patient counseling has been shown to produce favorable patient outcomes, particularly for higher-risk patients.16,23
The MARQUIS investigators found they could boost the chances of success by sharing stories of patient harm to foster “buy-in” among frontline staff, providing formal training to clinicians on how to take a medication history, and obtaining the support of nursing leaders to champion improvement efforts.23
Additionally, patients should be empowered to maintain an accurate medication list. We address strategies for improving patient engagement and adherence in a later section.
BEST PRACTICES FOR IMPROVING MEDICATION SAFETY
Medication reconciliation is one of several measures necessary to optimize medication safety during transitions of care. It typically includes the following actions:
- Interview the patient or caregiver to determine the list of medications the patient is currently taking (or supposed to be taking); consult other sources if needed.
- List medications that are being ordered during the clinical encounter.
- Compare these two lists, making note of medications that are stopped, changed, or newly prescribed.
- Resolve any discrepancies.
- Communicate the reconciled list to the patient, appropriate caregivers, and providers of follow-up care.

At a rudimentary level, medication reconciliation encompasses medication list management along the continuum of care. However, we recommend leveraging medication reconciliation as an opportunity to further enhance medication safety by reviewing the appropriateness of each medication, seizing opportunities to streamline or simplify the regimen, assessing patient and caregiver understanding of medication instructions and potential ADEs, and delivering appropriate counseling to enhance medication use. Table 2 outlines our framework for medication management during hospital-based transitions.
STEP 1: OBTAIN A COMPLETE PREADMISSION MEDICATION LIST
The “best-possible medication history” is obtained in a systematic process of interviewing the patient or caregiver plus reviewing at least one other reliable source.23 The resulting list should include all medications the patient is taking (prescription and nonprescription), doses, directions for use, formulations if applicable, indications, start and stop dates, and medication allergies and reactions.
Review existing information. Before eliciting a history from the patient, review his or her recorded medical history and existing medication lists (eg, prior discharge summaries, records from other facilities, records from outpatient visits, pharmacy fill data). This will provide context about the regimen and help identify issues and questions that can be addressed during the history-taking.

Ask open-ended questions. Instead of just asking the patient to confirm the accuracy of the existing medication list, we recommend actively obtaining the full medication list from the patient or caregiver. The conversation should begin with an open-ended question such as, “What medications do you take at home?” This approach will also allow the clinician to gauge the patient’s level of understanding of each medication’s indication and dosing instructions. Using a series of prompts such as those recommended in Table 3 will elicit a best-possible medical history, while verifying all of the medications on the existing list.
Clarify discrepancies. Resolve differences between the existing medication lists and the patient’s or caregiver’s report during the preadmission interview. Examples include errors of omission (a medication is missing), errors of commission (an additional medication is present), and discrepancies in the strength, formulation, dosing instructions, and indications for the drugs. If necessary, other sources of information should be consulted, such as the patient’s medication bottles, pharmacy or pharmacies, primary care physician, and a family member or caregiver.
Assess adherence. The extent to which patients take their medications as directed is an important component of the history, but is often left out. Medication nonadherence rates in the United States are 40% to 70%,25 contributing to poor patient outcomes and imposing extraordinary costs on the health care system.26
Asking open-ended, nonjudgmental questions at the time of hospital admission will help to uncover medication-taking behaviors as well as barriers to adherence (Table 3). The patient’s responses should be taken into account when determining the treatment plan.
Document your findings. After completing the medication history and clarifying discrepancies, document the preadmission list in the medical record. All members of the health care team should have access to view and update the same list, as new information about the preadmission medications may be uncovered after the initial history.
Make clinical decisions. Complete the admission medication reconciliation by deciding whether each medication on the list should be continued, changed, held, or discontinued on the basis of the patient’s clinical condition. Well-designed information technology applications enable the provider to document each action and the rationale for it, as well as carry that information into the order-entry system. Medications marked as held or discontinued on admission should be revisited as the patient’s clinical condition changes and at discharge.
STEP 2: AVOID RECONCILIATION ERRORS
Reconciliation errors reflect discrepancies between the medication history and the medications that are ordered after admission.
Reconciliation errors are less common than medication history errors, accounting for approximately one-third of potentially harmful medication errors in hospitalized medical patients.9 These include errors of omission (a medication is omitted from the orders), errors of commission (a medication is prescribed with no indication for continuation), and therapeutic duplication.
Preventing errors of omission
Medications are often held at transition points for appropriate clinical reasons. Examples include holding anticoagulants and antiplatelet agents in patients who have gastrointestinal bleeding or an upcoming procedure, antihypertensives in patients with hemodynamic instability, and other chronic medications in patients with an acute illness.
Poor documentation and communication of these decisions can lead to a failure to resume the medications—an error of omission—at hospital discharge.
Hospitalized patients are at risk of unintentional discontinuation of their chronic medications, including antiplatelet drugs, anticoagulants, statins, and thyroid replacement, particularly if admitted to the intensive care unit.12 These errors can be minimized by a standardized medication reconciliation process at each transition and clear documentation of the medication plan.
Communication among providers can be improved if the admitting clinician documents clearly whether each preadmission medication is being continued, changed, or stopped, along with the reason for doing so, and makes this information available throughout the hospital stay. Upon transfer to another unit and at discharge, the physician should review each; preadmission medication that was held and the patient’s current clinical status and, based on that information, decide whether medications that were held should be resumed. If a medication will be restarted later, specific instructions should be documented and communicated to the patient and the physicians who are taking over his or her care.
Preventing errors of commission
Failure to perform a complete reconciliation at each transition of care and match each medication with an appropriate indication can lead to errors of commission.
One study showed that 44% of patients were prescribed at least one unnecessary drug at hospital discharge, one-fourth of which were started during the hospitalization.27 Commonly prescribed unnecessarily were gastrointestinal agents, central nervous system drugs, nutrients, and supplements.
It is critical to assess each medication’s ongoing need, appropriateness, and risk-benefit ratio at every transition. Medications no longer indicated should be discontinued in order to simplify the regimen, avoid unnecessary drug exposure, and prevent ADEs.
For example, proton pump inhibitors or histamine 2 receptor blockers are often started in the hospital for stress ulcer prophylaxis. One-third of patients are then discharged home on the medication, and 6 months later half of those patients are still taking the unnecessary drug.28 This situation can be avoided by limiting use of these medications to appropriate circumstances, clearly marking the indication as stress ulcer prophylaxis (as opposed to an ongoing condition that will require continuing it after discharge), and discontinuing the agent when appropriate.
All drugs, even common and seemingly benign ones, carry some risk and should be discontinued when no longer needed. Thus, medications added during the hospitalization to control acute symptoms should also be reviewed at each transition to prevent inappropriate continuation when symptoms have resolved.
One study, for example, found that many patients were discharged with inappropriate prescriptions for atypical antipsychotics after receiving them in the intensive care unit, likely for delirium.29 Documenting that an acute issue such as delirium has resolved should prompt the discontinuation of therapy.
Preventing therapeutic duplication
Therapeutic duplication occurs in about 8% of discharges.1 These errors often result from formulary substitutions or altering the dosage form in the acute setting. For example, patients who receive a prescription for the substituted agent at discharge and also resume their prehospitalization medications end up with duplicate therapy.
Therapeutic substitution is common at the time of admission to the hospital as a result of formulary restrictions. Drug classes that are frequently substituted include statins, antihypertensives, urinary antispasmodics, and proton pump inhibitors. Physicians should be familiar with the preferred agents on the hospital formulary and make careful note when a substitution occurs. Furthermore, hospital systems should be developed to remind the physician to switch back to the outpatient medication at discharge.
Similar problems occur when home medications are replaced with different dosage forms with different pharmacokinetic properties. For example, a long-acting medication may be temporarily replaced with an intravenous solution or immediate-release tablet for several reasons, including nothing-by-mouth status, unstable clinical condition, need for titration, and need to crush the tablet to give the drug per tube. The differing formulations must be reconciled throughout the patient’s hospital course and at discharge to avoid therapeutic duplication and serious medication errors. Deliberate changes to the dosage form should be clearly communicated in the discharge medication list so that patients and other clinicians are aware.
Hospital systems should also have the capability to identify duplications in the medication list and to warn prescribers of these errors. The ability to group medications by drug class or sort the medication list alphabetically by generic name can help uncover duplication errors.
STEP 3: REVIEW THE LIST IN VIEW OF THE CLINICAL PICTURE
Transitions of care should prompt providers to review the medication list for possible drug-disease interactions, confirm compliance with evidence-based guidelines, and evaluate the risks and benefits of each medication in the context of the patient’s age and acute and chronic medical issues. This is also an opportunity to screen the full list for potentially inappropriate medications and high-alert drugs such as insulin or anticoagulants, which are more likely than other drugs to cause severe harm when used in error.
Acute kidney injury. New drug-disease interactions can arise during a hospitalization and can affect dosing and the choice of drug. The onset of acute kidney injury, for example, often necessitates adjusting or discontinuing nephrotoxic and renally excreted medications. ADEs or potential ADEs have been reported in 43% of hospitalized patients with acute kidney injury.30
Because acute kidney injury is often transient, medications may need to be held or adjusted several times until renal function stabilizes. This can be challenging across the continuum of care and requires close monitoring of the serum creatinine level and associated drug doses and levels, if applicable. Well-designed clinical decision support tools can integrate laboratory data and alert the prescriber to a clinically important increase or decrease in serum creatinine that may warrant a change in therapy. Modifications to the regimen and a plan for timely follow-up of the serum creatinine level should be clearly documented in the discharge plan.
Liver disease. Similar attention should be given to drugs that are hepatically metabolized if a patient has acute or chronic liver impairment.
Geriatric patients, particularly those who present with altered mental status, falls, or urinary retention, should have their medication list reviewed for potentially inappropriate medications, which are drugs that pose increased risk of poor outcomes in older adults.31,32 Patients and providers may have been willing to accept the risk of medications such as anticholinergics or sedative-hypnotics when the drugs were initiated, but circumstances can change over time, especially in this patient population. Hospitalization is a prime opportunity to screen for medications that meet the Beers criteria31 for agents to avoid or use with caution in older adults.
As-needed medications. Medications prescribed on an as-needed basis in the hospital should be reviewed for continuation or discontinuation at discharge. How often the medication was given can inform this decision.
For example, if as-needed opioids were used frequently, failure to develop a plan of care for pain can lead to persistent symptoms and, possibly, to readmission.33,34 Similar scenarios occur with use of as-needed blood pressure medications, laxatives, and correction-dose insulin.
If an as-needed medication was used consistently during hospitalization, the physician should consider whether a regularly scheduled medication is needed. Conversely, if the medication was not used during the inpatient admission, it can likely be discontinued.
STEP 4: PREPARE THE PATIENT AND FOLLOW-UP PROVIDER
Once a clinician has performed medication reconciliation, including obtaining a best-possible medical history and carefully reviewing the medication list and orders for errors and clinical appropriateness, the next steps are to ensure the patient understands what he or she needs to do and to confirm that suitable follow-up plans are in place. These measures should be taken at all transitions of care but are critically important at hospital discharge.
Preparing the patient and caregiver
An accurate, reconciled medication list should be given to the patient, caregiver, or both, and should be reviewed before discharge.17

Approximately one-third of Americans have low health literacy skills, so medication lists and associated materials should be easy to understand.35 Medication lists should be written in plain language and formatted for optimal readability (Table 4), clearly stating which medications to continue, change, hold temporarily, and stop.
Patients recall and comprehend about half of the information provided during a medical encounter.36 Thus, medication teaching should focus on key points including changes or additions to the regimen, specific instructions for follow-up and monitoring, and how to handle common and serious side effects.
To confirm patient understanding, clinicians should use “teach-back,” ie, provide the patient with information and then ask him or her to repeat back key points.37,38 The patient and family should also be encouraged to ask questions before discharge.

If not already addressed during the hospital stay, barriers to medication adherence and ability to obtain the medications should be attended to at this time (Table 5). Also, the plan to pick up the medications should be verified with the patient and caregiver. Verify that there is transportation to a particular pharmacy that is open at the time of discharge, and that the patient can afford the medications.
Ensuring appropriate follow-up
Studies have shown that timely in-home or telephone follow-up after discharge can decrease adverse events and postdischarge health care utilization.39,40 Telephone follow-up that includes thorough medication reconciliation can help detect and resolve medication issues early after discharge and can close gaps related to monitoring and follow-up.

Medication reconciliation by telephone can be time-consuming. Depending on the number of medications that need to be reviewed, calls can take between 10 and 60 minutes. Postdischarge phone calls should be performed by clinical personnel who are able to identify medication-related problems. A pharmacist should be an available resource to assist with complex regimens, to help resolve medication discrepancies, and to address patient concerns. Table 6 provides tips for conducting follow-up phone calls.
Resolving discrepancies identified during follow-up calls can be difficult, as changes to the medication regimen are often not communicated effectively to other members of the care team. Physicians should document the complete medication list and plan in the discharge summary, and there should be a method for the caller to record updates to the medication list in the medical record so that they are apparent at the outpatient follow-up visit.
An additional challenge is that it is frequently unclear which physician “owns” which medications. Therefore, designating a contact person for each medication until follow-up can be very valuable. At a minimum, a “physician owner” for high-alert medications such as insulin, anticoagulants, and diuretics should be identified to provide close follow-up, titration, and monitoring.
There should also be a plan for the patient to obtain refills of essential long-term medications, such as antiplatelet agents following stent placement.
SUMMARY AND RECOMMENDATIONS
Medication-related problems during hospital admission and discharge are common and range from minor discrepancies in the medication list to errors in history-taking, prescribing, and reconciliation that can lead to potential or actual patient harm. Putting systems in place to facilitate medication reconciliation can decrease the occurrence of medication discrepancies and ADEs, thereby improving patient safety during these critical transitions between care settings and providers. Institutional medication reconciliation programs should focus resources on the admission history-taking step, target the highest-risk patients for the most intensive interventions, and involve pharmacy personnel when possible.
On an individual level, clinicians can incorporate additional interventions into their workflows to optimize medication safety for hospitalized patients. Using a structured approach to obtain a complete and accurate medication list at the time of hospital admission will help providers identify medication-related problems and prevent the propagation of errors throughout the hospital stay and at discharge. Focusing additional time and effort on a comprehensive review of the medication list for errors of omission and commission, patient-specific needs, and high-alert drugs will further decrease the risk of medication errors. Finally, providing discharge counseling targeting patient barriers to adherence and ensuring a proper handover of medication information and rationale for medication changes to outpatient providers will improve the chances of a safe transition.
- Coleman EA, Smith JD, Raha D, Min SJ. Posthospital medication discrepancies: prevalence and contributing factors. Arch Intern Med 2005; 165:1842–1847.
- Wong JD, Bajcar JM, Wong GG, et al. Medication reconciliation at hospital discharge: evaluating discrepancies. Ann Pharmacother 2008; 42:1373–1379.
- Kripalani S, Roumie CL, Dalal AK, et al; PILL-CVD (Pharmacist Intervention for Low Literacy in Cardiovascular Disease) Study Group. Effect of a pharmacist intervention on clinically important medication errors after hospital discharge: a randomized trial. Ann Intern Med 2012; 157:1-10.
- Forster AJ, Murff HJ, Peterson JF, Gandhi TK, Bates DW. The incidence and severity of adverse events affecting patients after discharge from the hospital. Ann Intern Med 2003; 138:161–167.
- Forster AJ, Murff HJ, Peterson JF, Gandhi TK, Bates DW. Adverse drug events occurring following hospital discharge. J Gen Intern Med 2005; 20:317–323.
- Johnson JA, Bootman JL. Drug-related morbidity and mortality. A cost-of-illness model. Arch Intern Med 1995; 155:1949–1956.
- Budnitz DS, Lovegrove MC, Shehab N, Richards CL. Emergency hospitalizations for adverse drug events in older Americans. N Engl J Med 2011; 365:2002–2012.
- Bates DW, Boyle DL, Vander Vliet MB, Schneider J, Leape L. Relationship between medication errors and adverse drug events. J Gen Intern Med 1995;10:199–205.
- Pippins JR, Gandhi TK, Hamann C, et al. Classifying and predicting errors of inpatient medication reconciliation. J Gen Intern Med 2008; 23:1414–1422.
- Lau HS, Florax C, Porsius AJ, De Boer A.The completeness of medication histories in hospital medical records of patients admitted to general internal medicine wards. Br J Clin Pharmacol 2000; 49:597–603.
- Tam VC, Knowles SR, Cornish PL, Fine N, Marchesano R, Etchells EE. Frequency, type and clinical importance of medication history errors at admission to hospital: a systematic review. CMAJ 2005; 173:510–515.
- Bell CM, Brener SS, Gunraj N, et al. Association of ICU or hospital admission with unintentional discontinuation of medications for chronic diseases. JAMA 2011; 306:840–847.
- Dobranski S, Hammond I, Khan G, Holdsworth H. The nature of hospital prescribing errors. Br J Clin Governance 2002; 7:187–193.
- Gleason KM, Groszek JM, Sullivan C, Rooney D, Barnard C, Noskin GA. Reconciliation of discrepancies in medication histories and admission orders of newly hospitalized patients. Am J Health Syst Pharm 2004; 61:1689–1695.
- Rozich JD, Resar KR. Medication safety: one organization’s approach to the challenge. J Clin Outcomes Manage 2001; 8:27–34.
- Mueller SK, Sponsler KC, Kripalani S, Schnipper JL. Hospital-based medication reconciliation practices: a systematic review. Arch Intern Med 2012; 172:1057–1069.
- Joint Commission. Using medication reconciliation to prevent errors. Sentinel Event Alert 2006, Issue 35. www.jointcommission.org/assets/1/18/SEA_35.pdf. Accessed March 31, 2015.
- Greenwald JL, Halasyamani L, Greene J, et al. Making inpatient medication reconciliation patient centered, clinically relevant and implementable: a consensus statement on key principles and necessary first steps. J Hosp Med 2010; 5:477–485.
- Berwick DM, Calkins DR, McCannon CJ, Hackbarth AD. The 100,000 lives campaign: setting a goal and a deadline for improving health care quality. JAMA 2006; 295:324–327.
- McCannon CJ, Hackbarth AD, Griffin FA. Miles to go: an introduction to the 5 Million Lives Campaign. Jt Comm J Qual Patient Saf 2007; 33:477–484.
- Leotsakos A, Caisley L, Karga M, Kelly E, O’Leary D, Timmons K. High 5s: addressing excellence in patient safety. World Hosp Health Serv 2009; 45:19–22.
- Gleason KM, Brake H, Agramonte V, Perfetti C. Medications at Transitions and Clinical Handoffs (MATCH) Toolkit for Medication Reconciliation. www.ahrq.gov/professionals/quality-patient-safety/patient-safety-resources/resources/match/match.pdf. Accessed March 31, 2015.
- Mueller SK, Kripalani S, Stein J, et al. A toolkit to disseminate best practices in inpatient medication reconciliation: multi-center medication reconciliation quality improvement study (MARQUIS). Jt Comm J Qual Patient Saf 2013; 39:371–382.
- Pal A, Babbott S, Wilkinson ST. Can the targeted use of a discharge pharmacist significantly decrease 30-day readmissions? Hosp Pharm 2013; 48:380–388.
- Claxton AJ, Cramer J, Pierce C. A systematic review of the associations between dose regimens and medication compliance. Clin Ther 2001; 23:1296–1310.
- Osterberg L, Blaschke T. Adherence to medication. N Engl J Med 2005; 353:487–497.
- Hajjar ER, Hanlon JT, Sloane RJ, et al. Unnecessary drug use in frail older people at hospital discharge. J Am Geriatr Soc 2005; 53:1518–1523.
- Zink DA, Pohlman M, Barnes M, Cannon ME. Long-term use of acid suppression started inappropriately during hospitalization. Aliment Pharmacol Ther 2005; 21:1203–1209.
- Morandi A, Vasilevskis E, Pandharipande PP, et al. Inappropriate medication prescriptions in elderly adults surviving an intensive care unit hospitalization. J Am Geriatr Soc 2013; 61:1128–1134.
- Cox ZL, McCoy AB, Matheny ME, et al. Adverse drug events during AKI and its recovery. Clin J Am Soc Nephrol 2013; 8:1070–1078.
- American Geriatrics Society 2012 Beers Criteria Update Expert Panel. American Geriatrics Society updated Beers Criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc 2012; 60:616–631.
- Gallagher P, O’Mahony D. STOPP (Screening Tool of Older Persons’ potentially inappropriate Prescriptions): application to acutely ill elderly patients and comparison with Beers’ criteria. Age Ageing 2008; 37:673–679.
- Tjia J, Bonner A, Briesacher BA, McGee S, Terrill E, Miller K. Medication discrepancies upon hospital to skilled nursing facility transitions. J Gen Intern Med 2009; 24:630–635.
- Boockvar K, Fishman E, Kyriacou CK, Monias A, Gavi S, Cortes T. Adverse events due to discontinuations in drug use and dose changes in patients transferred between acute and long-term care facilities. Arch Intern Med 2004; 164:545–550.
- Kutner M, Greenberg E, Jin Y, Paulsen C. The Health Literacy of America’s Adults: Results From the 2003 National Assessment of Adult Literacy. http://nces.ed.gov/pubs2006/2006483_1.pdf. Accessed March 31, 2015.
- Crane JA. Patient comprehension of doctor-patient communication on discharge from the emergency department. J Emerg Med 1997; 15:1–7.
- DeWalt DA, Callahan LF, Hawk VH, et al. Health Literacy Universal Precautions Toolkit. www.ahrq.gov/qual/literacy/healthliteracytoolkit.pdf. Accessed March 31, 2015.
- Schillinger D, Piette J, Grumbach K, et al. Closing the loop: physician communication with diabetic patients who have low health literacy. Arch Intern Med 2003; 163:83–90.
- Coleman EA, Parry C, Chalmers S, Min SJ. The care transitions intervention: results of a randomized controlled trial. Arch Intern Med 2006; 166:1822–1828.
- Jack BW, Chetty VK, Anthony D, et al. A reengineered hospital discharge program to decrease rehospitalization: a randomized trial. Ann Intern Med 2009; 150:178–187.
Any time patients enter or leave the hospital, they risk being harmed by errors in their medications.1 Adverse events from medication errors during transitions of care are common but often preventable. One key approach is to systematically review every patient’s medication list on admission and discharge and resolve any discrepancies. These transitions are also an opportunity to address other medication-related problems, such as adherence, drug interactions, and clinical appropriateness.
This article summarizes the types and prevalence of medication problems that occur during hospital-based transitions of care, and suggests strategies to decrease the risk of medication errors, focusing on medication reconciliation and related interventions that clinicians can use at the bedside to improve medication safety.
DEFINING TERMS
A medication discrepancy is any variance noted in a patient’s documented medication regimen across different medication lists or sites of care. While some differences reflect intentional and clinically appropriate changes to the regimen, others are unintentional and reflect inaccurate or incomplete information. These unintentional discrepancies are medication errors.
Depending on the clinical circumstances and medications involved, such errors may lead to an adverse drug event (ADE), defined as actual harm or injury resulting from a medication. Sometimes a medication error does not cause harm immediately but could if left uncorrected; this is called a potential ADE.
An important goal during transitions of care is to reduce unintentional medication discrepancies, thereby reducing potential and actual ADEs.
ERRORS ARISE AT DISCHARGE—AND EVEN MORE AT ADMISSION
Hospital discharge is a widely recognized transition in which patient harm occurs. As many as 70% of patients may have an unintentional medication discrepancy at hospital discharge, with many of those discrepancies having potential for harm.2 Indeed, during the first few weeks after discharge, 50% of patients have a clinically important medication error,3 and 20% experience an adverse event, most commonly an ADE.4 ADEs are associated with excess health care utilization,5–7 and many are preventable through strategies such as medication reconciliation.5,8
Importantly, more errors arise at hospital admission than at other times.9,10
Errors in medication histories are the most common source of discrepancies, affecting up to two-thirds of admitted patients.11,12 More than one-quarter of hospital prescribing errors can be attributed to incomplete medication histories at the time of admission,13 and nearly three times as many clinically important medication discrepancies are related to history-taking errors on admission rather than reconciliation errors at discharge.9
Most discrepancies occurring at the time of admission have the potential to cause harm, particularly if the errors persist beyond discharge.14 Therefore, taking a complete and accurate medication history on hospital admission is critical to ensuring safe care transitions.
MEDICATION RECONCILIATION: BARRIERS AND FACILITATORS
Medication reconciliation is a strategy for reducing medication discrepancies in patients moving across care settings. Simply put, it is the process by which a patient’s medication list is obtained, compared, and clarified across different sites of care.15 It has consistently been shown to decrease medication errors compared with usual care,16 and it is strongly supported by national and international organizations.17–21
In clinical practice, many physicians and institutions have found medication reconciliation difficult to implement, owing to barriers at the level of the patient, provider, and system (Table 1). In response to these challenges, two initiatives have synthesized best practices and offer toolkits that hospitals and clinicians can use: the Medications at Transitions and Clinical Handoffs (MATCH) program22 and the Multi-Center Medication Reconciliation Quality Improvement Study (MARQUIS).23
Lack of resources is a widely acknowledged challenge. Thus, the MARQUIS investigators23 suggested focusing on the admission history, where most errors occur, and applying the most resource-intensive interventions in patients at highest risk of ADEs, ie, those who are elderly, have multiple comorbid conditions, or take numerous medications.16
Although the risk of an ADE increases with the number of medications a patient takes,4 the exact number of drugs that defines high risk has not been well established. Targeting patients who take 10 or more maintenance medications is a reasonable initial approach,24 but institutions should tailor risk stratification to their patient populations and available resources. Patients taking high-risk medications such as anticoagulants and insulin could also be prioritized for review, since these medications are more likely to cause serious patient harm when used without appropriate clinical oversight.7
Using pharmacy staff to perform medication history-taking, reconciliation, and patient counseling has been shown to produce favorable patient outcomes, particularly for higher-risk patients.16,23
The MARQUIS investigators found they could boost the chances of success by sharing stories of patient harm to foster “buy-in” among frontline staff, providing formal training to clinicians on how to take a medication history, and obtaining the support of nursing leaders to champion improvement efforts.23
Additionally, patients should be empowered to maintain an accurate medication list. We address strategies for improving patient engagement and adherence in a later section.
BEST PRACTICES FOR IMPROVING MEDICATION SAFETY
Medication reconciliation is one of several measures necessary to optimize medication safety during transitions of care. It typically includes the following actions:
- Interview the patient or caregiver to determine the list of medications the patient is currently taking (or supposed to be taking); consult other sources if needed.
- List medications that are being ordered during the clinical encounter.
- Compare these two lists, making note of medications that are stopped, changed, or newly prescribed.
- Resolve any discrepancies.
- Communicate the reconciled list to the patient, appropriate caregivers, and providers of follow-up care.
At a rudimentary level, medication reconciliation encompasses medication list management along the continuum of care. However, we recommend leveraging medication reconciliation as an opportunity to further enhance medication safety by reviewing the appropriateness of each medication, seizing opportunities to streamline or simplify the regimen, assessing patient and caregiver understanding of medication instructions and potential ADEs, and delivering appropriate counseling to enhance medication use. Table 2 outlines our framework for medication management during hospital-based transitions.
STEP 1: OBTAIN A COMPLETE PREADMISSION MEDICATION LIST
The “best-possible medication history” is obtained in a systematic process of interviewing the patient or caregiver plus reviewing at least one other reliable source.23 The resulting list should include all medications the patient is taking (prescription and nonprescription), doses, directions for use, formulations if applicable, indications, start and stop dates, and medication allergies and reactions.
Review existing information. Before eliciting a history from the patient, review his or her recorded medical history and existing medication lists (eg, prior discharge summaries, records from other facilities, records from outpatient visits, pharmacy fill data). This will provide context about the regimen and help identify issues and questions that can be addressed during the history-taking.
Ask open-ended questions. Instead of just asking the patient to confirm the accuracy of the existing medication list, we recommend actively obtaining the full medication list from the patient or caregiver. The conversation should begin with an open-ended question such as, “What medications do you take at home?” This approach will also allow the clinician to gauge the patient’s level of understanding of each medication’s indication and dosing instructions. Using a series of prompts such as those recommended in Table 3 will elicit a best-possible medical history, while verifying all of the medications on the existing list.
Clarify discrepancies. Resolve differences between the existing medication lists and the patient’s or caregiver’s report during the preadmission interview. Examples include errors of omission (a medication is missing), errors of commission (an additional medication is present), and discrepancies in the strength, formulation, dosing instructions, and indications for the drugs. If necessary, other sources of information should be consulted, such as the patient’s medication bottles, pharmacy or pharmacies, primary care physician, and a family member or caregiver.
Assess adherence. The extent to which patients take their medications as directed is an important component of the history, but is often left out. Medication nonadherence rates in the United States are 40% to 70%,25 contributing to poor patient outcomes and imposing extraordinary costs on the health care system.26
Asking open-ended, nonjudgmental questions at the time of hospital admission will help to uncover medication-taking behaviors as well as barriers to adherence (Table 3). The patient’s responses should be taken into account when determining the treatment plan.
Document your findings. After completing the medication history and clarifying discrepancies, document the preadmission list in the medical record. All members of the health care team should have access to view and update the same list, as new information about the preadmission medications may be uncovered after the initial history.
Make clinical decisions. Complete the admission medication reconciliation by deciding whether each medication on the list should be continued, changed, held, or discontinued on the basis of the patient’s clinical condition. Well-designed information technology applications enable the provider to document each action and the rationale for it, as well as carry that information into the order-entry system. Medications marked as held or discontinued on admission should be revisited as the patient’s clinical condition changes and at discharge.
STEP 2: AVOID RECONCILIATION ERRORS
Reconciliation errors reflect discrepancies between the medication history and the medications that are ordered after admission.
Reconciliation errors are less common than medication history errors, accounting for approximately one-third of potentially harmful medication errors in hospitalized medical patients.9 These include errors of omission (a medication is omitted from the orders), errors of commission (a medication is prescribed with no indication for continuation), and therapeutic duplication.
Preventing errors of omission
Medications are often held at transition points for appropriate clinical reasons. Examples include holding anticoagulants and antiplatelet agents in patients who have gastrointestinal bleeding or an upcoming procedure, antihypertensives in patients with hemodynamic instability, and other chronic medications in patients with an acute illness.
Poor documentation and communication of these decisions can lead to a failure to resume the medications—an error of omission—at hospital discharge.
Hospitalized patients are at risk of unintentional discontinuation of their chronic medications, including antiplatelet drugs, anticoagulants, statins, and thyroid replacement, particularly if admitted to the intensive care unit.12 These errors can be minimized by a standardized medication reconciliation process at each transition and clear documentation of the medication plan.
Communication among providers can be improved if the admitting clinician documents clearly whether each preadmission medication is being continued, changed, or stopped, along with the reason for doing so, and makes this information available throughout the hospital stay. Upon transfer to another unit and at discharge, the physician should review each; preadmission medication that was held and the patient’s current clinical status and, based on that information, decide whether medications that were held should be resumed. If a medication will be restarted later, specific instructions should be documented and communicated to the patient and the physicians who are taking over his or her care.
Preventing errors of commission
Failure to perform a complete reconciliation at each transition of care and match each medication with an appropriate indication can lead to errors of commission.
One study showed that 44% of patients were prescribed at least one unnecessary drug at hospital discharge, one-fourth of which were started during the hospitalization.27 Commonly prescribed unnecessarily were gastrointestinal agents, central nervous system drugs, nutrients, and supplements.
It is critical to assess each medication’s ongoing need, appropriateness, and risk-benefit ratio at every transition. Medications no longer indicated should be discontinued in order to simplify the regimen, avoid unnecessary drug exposure, and prevent ADEs.
For example, proton pump inhibitors or histamine 2 receptor blockers are often started in the hospital for stress ulcer prophylaxis. One-third of patients are then discharged home on the medication, and 6 months later half of those patients are still taking the unnecessary drug.28 This situation can be avoided by limiting use of these medications to appropriate circumstances, clearly marking the indication as stress ulcer prophylaxis (as opposed to an ongoing condition that will require continuing it after discharge), and discontinuing the agent when appropriate.
All drugs, even common and seemingly benign ones, carry some risk and should be discontinued when no longer needed. Thus, medications added during the hospitalization to control acute symptoms should also be reviewed at each transition to prevent inappropriate continuation when symptoms have resolved.
One study, for example, found that many patients were discharged with inappropriate prescriptions for atypical antipsychotics after receiving them in the intensive care unit, likely for delirium.29 Documenting that an acute issue such as delirium has resolved should prompt the discontinuation of therapy.
Preventing therapeutic duplication
Therapeutic duplication occurs in about 8% of discharges.1 These errors often result from formulary substitutions or altering the dosage form in the acute setting. For example, patients who receive a prescription for the substituted agent at discharge and also resume their prehospitalization medications end up with duplicate therapy.
Therapeutic substitution is common at the time of admission to the hospital as a result of formulary restrictions. Drug classes that are frequently substituted include statins, antihypertensives, urinary antispasmodics, and proton pump inhibitors. Physicians should be familiar with the preferred agents on the hospital formulary and make careful note when a substitution occurs. Furthermore, hospital systems should be developed to remind the physician to switch back to the outpatient medication at discharge.
Similar problems occur when home medications are replaced with different dosage forms with different pharmacokinetic properties. For example, a long-acting medication may be temporarily replaced with an intravenous solution or immediate-release tablet for several reasons, including nothing-by-mouth status, unstable clinical condition, need for titration, and need to crush the tablet to give the drug per tube. The differing formulations must be reconciled throughout the patient’s hospital course and at discharge to avoid therapeutic duplication and serious medication errors. Deliberate changes to the dosage form should be clearly communicated in the discharge medication list so that patients and other clinicians are aware.
Hospital systems should also have the capability to identify duplications in the medication list and to warn prescribers of these errors. The ability to group medications by drug class or sort the medication list alphabetically by generic name can help uncover duplication errors.
STEP 3: REVIEW THE LIST IN VIEW OF THE CLINICAL PICTURE
Transitions of care should prompt providers to review the medication list for possible drug-disease interactions, confirm compliance with evidence-based guidelines, and evaluate the risks and benefits of each medication in the context of the patient’s age and acute and chronic medical issues. This is also an opportunity to screen the full list for potentially inappropriate medications and high-alert drugs such as insulin or anticoagulants, which are more likely than other drugs to cause severe harm when used in error.
Acute kidney injury. New drug-disease interactions can arise during a hospitalization and can affect dosing and the choice of drug. The onset of acute kidney injury, for example, often necessitates adjusting or discontinuing nephrotoxic and renally excreted medications. ADEs or potential ADEs have been reported in 43% of hospitalized patients with acute kidney injury.30
Because acute kidney injury is often transient, medications may need to be held or adjusted several times until renal function stabilizes. This can be challenging across the continuum of care and requires close monitoring of the serum creatinine level and associated drug doses and levels, if applicable. Well-designed clinical decision support tools can integrate laboratory data and alert the prescriber to a clinically important increase or decrease in serum creatinine that may warrant a change in therapy. Modifications to the regimen and a plan for timely follow-up of the serum creatinine level should be clearly documented in the discharge plan.
Liver disease. Similar attention should be given to drugs that are hepatically metabolized if a patient has acute or chronic liver impairment.
Geriatric patients, particularly those who present with altered mental status, falls, or urinary retention, should have their medication list reviewed for potentially inappropriate medications, which are drugs that pose increased risk of poor outcomes in older adults.31,32 Patients and providers may have been willing to accept the risk of medications such as anticholinergics or sedative-hypnotics when the drugs were initiated, but circumstances can change over time, especially in this patient population. Hospitalization is a prime opportunity to screen for medications that meet the Beers criteria31 for agents to avoid or use with caution in older adults.
As-needed medications. Medications prescribed on an as-needed basis in the hospital should be reviewed for continuation or discontinuation at discharge. How often the medication was given can inform this decision.
For example, if as-needed opioids were used frequently, failure to develop a plan of care for pain can lead to persistent symptoms and, possibly, to readmission.33,34 Similar scenarios occur with use of as-needed blood pressure medications, laxatives, and correction-dose insulin.
If an as-needed medication was used consistently during hospitalization, the physician should consider whether a regularly scheduled medication is needed. Conversely, if the medication was not used during the inpatient admission, it can likely be discontinued.
STEP 4: PREPARE THE PATIENT AND FOLLOW-UP PROVIDER
Once a clinician has performed medication reconciliation, including obtaining a best-possible medical history and carefully reviewing the medication list and orders for errors and clinical appropriateness, the next steps are to ensure the patient understands what he or she needs to do and to confirm that suitable follow-up plans are in place. These measures should be taken at all transitions of care but are critically important at hospital discharge.
Preparing the patient and caregiver
An accurate, reconciled medication list should be given to the patient, caregiver, or both, and should be reviewed before discharge.17
Approximately one-third of Americans have low health literacy skills, so medication lists and associated materials should be easy to understand.35 Medication lists should be written in plain language and formatted for optimal readability (Table 4), clearly stating which medications to continue, change, hold temporarily, and stop.
Patients recall and comprehend about half of the information provided during a medical encounter.36 Thus, medication teaching should focus on key points including changes or additions to the regimen, specific instructions for follow-up and monitoring, and how to handle common and serious side effects.
To confirm patient understanding, clinicians should use “teach-back,” ie, provide the patient with information and then ask him or her to repeat back key points.37,38 The patient and family should also be encouraged to ask questions before discharge.
If not already addressed during the hospital stay, barriers to medication adherence and ability to obtain the medications should be attended to at this time (Table 5). Also, the plan to pick up the medications should be verified with the patient and caregiver. Verify that there is transportation to a particular pharmacy that is open at the time of discharge, and that the patient can afford the medications.
Ensuring appropriate follow-up
Studies have shown that timely in-home or telephone follow-up after discharge can decrease adverse events and postdischarge health care utilization.39,40 Telephone follow-up that includes thorough medication reconciliation can help detect and resolve medication issues early after discharge and can close gaps related to monitoring and follow-up.
Medication reconciliation by telephone can be time-consuming. Depending on the number of medications that need to be reviewed, calls can take between 10 and 60 minutes. Postdischarge phone calls should be performed by clinical personnel who are able to identify medication-related problems. A pharmacist should be an available resource to assist with complex regimens, to help resolve medication discrepancies, and to address patient concerns. Table 6 provides tips for conducting follow-up phone calls.
Resolving discrepancies identified during follow-up calls can be difficult, as changes to the medication regimen are often not communicated effectively to other members of the care team. Physicians should document the complete medication list and plan in the discharge summary, and there should be a method for the caller to record updates to the medication list in the medical record so that they are apparent at the outpatient follow-up visit.
An additional challenge is that it is frequently unclear which physician “owns” which medications. Therefore, designating a contact person for each medication until follow-up can be very valuable. At a minimum, a “physician owner” for high-alert medications such as insulin, anticoagulants, and diuretics should be identified to provide close follow-up, titration, and monitoring.
There should also be a plan for the patient to obtain refills of essential long-term medications, such as antiplatelet agents following stent placement.
SUMMARY AND RECOMMENDATIONS
Medication-related problems during hospital admission and discharge are common and range from minor discrepancies in the medication list to errors in history-taking, prescribing, and reconciliation that can lead to potential or actual patient harm. Putting systems in place to facilitate medication reconciliation can decrease the occurrence of medication discrepancies and ADEs, thereby improving patient safety during these critical transitions between care settings and providers. Institutional medication reconciliation programs should focus resources on the admission history-taking step, target the highest-risk patients for the most intensive interventions, and involve pharmacy personnel when possible.
On an individual level, clinicians can incorporate additional interventions into their workflows to optimize medication safety for hospitalized patients. Using a structured approach to obtain a complete and accurate medication list at the time of hospital admission will help providers identify medication-related problems and prevent the propagation of errors throughout the hospital stay and at discharge. Focusing additional time and effort on a comprehensive review of the medication list for errors of omission and commission, patient-specific needs, and high-alert drugs will further decrease the risk of medication errors. Finally, providing discharge counseling targeting patient barriers to adherence and ensuring a proper handover of medication information and rationale for medication changes to outpatient providers will improve the chances of a safe transition.
Any time patients enter or leave the hospital, they risk being harmed by errors in their medications.1 Adverse events from medication errors during transitions of care are common but often preventable. One key approach is to systematically review every patient’s medication list on admission and discharge and resolve any discrepancies. These transitions are also an opportunity to address other medication-related problems, such as adherence, drug interactions, and clinical appropriateness.
This article summarizes the types and prevalence of medication problems that occur during hospital-based transitions of care, and suggests strategies to decrease the risk of medication errors, focusing on medication reconciliation and related interventions that clinicians can use at the bedside to improve medication safety.
DEFINING TERMS
A medication discrepancy is any variance noted in a patient’s documented medication regimen across different medication lists or sites of care. While some differences reflect intentional and clinically appropriate changes to the regimen, others are unintentional and reflect inaccurate or incomplete information. These unintentional discrepancies are medication errors.
Depending on the clinical circumstances and medications involved, such errors may lead to an adverse drug event (ADE), defined as actual harm or injury resulting from a medication. Sometimes a medication error does not cause harm immediately but could if left uncorrected; this is called a potential ADE.
An important goal during transitions of care is to reduce unintentional medication discrepancies, thereby reducing potential and actual ADEs.
ERRORS ARISE AT DISCHARGE—AND EVEN MORE AT ADMISSION
Hospital discharge is a widely recognized transition in which patient harm occurs. As many as 70% of patients may have an unintentional medication discrepancy at hospital discharge, with many of those discrepancies having potential for harm.2 Indeed, during the first few weeks after discharge, 50% of patients have a clinically important medication error,3 and 20% experience an adverse event, most commonly an ADE.4 ADEs are associated with excess health care utilization,5–7 and many are preventable through strategies such as medication reconciliation.5,8
Importantly, more errors arise at hospital admission than at other times.9,10
Errors in medication histories are the most common source of discrepancies, affecting up to two-thirds of admitted patients.11,12 More than one-quarter of hospital prescribing errors can be attributed to incomplete medication histories at the time of admission,13 and nearly three times as many clinically important medication discrepancies are related to history-taking errors on admission rather than reconciliation errors at discharge.9
Most discrepancies occurring at the time of admission have the potential to cause harm, particularly if the errors persist beyond discharge.14 Therefore, taking a complete and accurate medication history on hospital admission is critical to ensuring safe care transitions.
MEDICATION RECONCILIATION: BARRIERS AND FACILITATORS
Medication reconciliation is a strategy for reducing medication discrepancies in patients moving across care settings. Simply put, it is the process by which a patient’s medication list is obtained, compared, and clarified across different sites of care.15 It has consistently been shown to decrease medication errors compared with usual care,16 and it is strongly supported by national and international organizations.17–21
In clinical practice, many physicians and institutions have found medication reconciliation difficult to implement, owing to barriers at the level of the patient, provider, and system (Table 1). In response to these challenges, two initiatives have synthesized best practices and offer toolkits that hospitals and clinicians can use: the Medications at Transitions and Clinical Handoffs (MATCH) program22 and the Multi-Center Medication Reconciliation Quality Improvement Study (MARQUIS).23
Lack of resources is a widely acknowledged challenge. Thus, the MARQUIS investigators23 suggested focusing on the admission history, where most errors occur, and applying the most resource-intensive interventions in patients at highest risk of ADEs, ie, those who are elderly, have multiple comorbid conditions, or take numerous medications.16
Although the risk of an ADE increases with the number of medications a patient takes,4 the exact number of drugs that defines high risk has not been well established. Targeting patients who take 10 or more maintenance medications is a reasonable initial approach,24 but institutions should tailor risk stratification to their patient populations and available resources. Patients taking high-risk medications such as anticoagulants and insulin could also be prioritized for review, since these medications are more likely to cause serious patient harm when used without appropriate clinical oversight.7
Using pharmacy staff to perform medication history-taking, reconciliation, and patient counseling has been shown to produce favorable patient outcomes, particularly for higher-risk patients.16,23
The MARQUIS investigators found they could boost the chances of success by sharing stories of patient harm to foster “buy-in” among frontline staff, providing formal training to clinicians on how to take a medication history, and obtaining the support of nursing leaders to champion improvement efforts.23
Additionally, patients should be empowered to maintain an accurate medication list. We address strategies for improving patient engagement and adherence in a later section.
BEST PRACTICES FOR IMPROVING MEDICATION SAFETY
Medication reconciliation is one of several measures necessary to optimize medication safety during transitions of care. It typically includes the following actions:
- Interview the patient or caregiver to determine the list of medications the patient is currently taking (or supposed to be taking); consult other sources if needed.
- List medications that are being ordered during the clinical encounter.
- Compare these two lists, making note of medications that are stopped, changed, or newly prescribed.
- Resolve any discrepancies.
- Communicate the reconciled list to the patient, appropriate caregivers, and providers of follow-up care.
At a rudimentary level, medication reconciliation encompasses medication list management along the continuum of care. However, we recommend leveraging medication reconciliation as an opportunity to further enhance medication safety by reviewing the appropriateness of each medication, seizing opportunities to streamline or simplify the regimen, assessing patient and caregiver understanding of medication instructions and potential ADEs, and delivering appropriate counseling to enhance medication use. Table 2 outlines our framework for medication management during hospital-based transitions.
STEP 1: OBTAIN A COMPLETE PREADMISSION MEDICATION LIST
The “best-possible medication history” is obtained in a systematic process of interviewing the patient or caregiver plus reviewing at least one other reliable source.23 The resulting list should include all medications the patient is taking (prescription and nonprescription), doses, directions for use, formulations if applicable, indications, start and stop dates, and medication allergies and reactions.
Review existing information. Before eliciting a history from the patient, review his or her recorded medical history and existing medication lists (eg, prior discharge summaries, records from other facilities, records from outpatient visits, pharmacy fill data). This will provide context about the regimen and help identify issues and questions that can be addressed during the history-taking.
Ask open-ended questions. Instead of just asking the patient to confirm the accuracy of the existing medication list, we recommend actively obtaining the full medication list from the patient or caregiver. The conversation should begin with an open-ended question such as, “What medications do you take at home?” This approach will also allow the clinician to gauge the patient’s level of understanding of each medication’s indication and dosing instructions. Using a series of prompts such as those recommended in Table 3 will elicit a best-possible medical history, while verifying all of the medications on the existing list.
Clarify discrepancies. Resolve differences between the existing medication lists and the patient’s or caregiver’s report during the preadmission interview. Examples include errors of omission (a medication is missing), errors of commission (an additional medication is present), and discrepancies in the strength, formulation, dosing instructions, and indications for the drugs. If necessary, other sources of information should be consulted, such as the patient’s medication bottles, pharmacy or pharmacies, primary care physician, and a family member or caregiver.
Assess adherence. The extent to which patients take their medications as directed is an important component of the history, but is often left out. Medication nonadherence rates in the United States are 40% to 70%,25 contributing to poor patient outcomes and imposing extraordinary costs on the health care system.26
Asking open-ended, nonjudgmental questions at the time of hospital admission will help to uncover medication-taking behaviors as well as barriers to adherence (Table 3). The patient’s responses should be taken into account when determining the treatment plan.
Document your findings. After completing the medication history and clarifying discrepancies, document the preadmission list in the medical record. All members of the health care team should have access to view and update the same list, as new information about the preadmission medications may be uncovered after the initial history.
Make clinical decisions. Complete the admission medication reconciliation by deciding whether each medication on the list should be continued, changed, held, or discontinued on the basis of the patient’s clinical condition. Well-designed information technology applications enable the provider to document each action and the rationale for it, as well as carry that information into the order-entry system. Medications marked as held or discontinued on admission should be revisited as the patient’s clinical condition changes and at discharge.
STEP 2: AVOID RECONCILIATION ERRORS
Reconciliation errors reflect discrepancies between the medication history and the medications that are ordered after admission.
Reconciliation errors are less common than medication history errors, accounting for approximately one-third of potentially harmful medication errors in hospitalized medical patients.9 These include errors of omission (a medication is omitted from the orders), errors of commission (a medication is prescribed with no indication for continuation), and therapeutic duplication.
Preventing errors of omission
Medications are often held at transition points for appropriate clinical reasons. Examples include holding anticoagulants and antiplatelet agents in patients who have gastrointestinal bleeding or an upcoming procedure, antihypertensives in patients with hemodynamic instability, and other chronic medications in patients with an acute illness.
Poor documentation and communication of these decisions can lead to a failure to resume the medications—an error of omission—at hospital discharge.
Hospitalized patients are at risk of unintentional discontinuation of their chronic medications, including antiplatelet drugs, anticoagulants, statins, and thyroid replacement, particularly if admitted to the intensive care unit.12 These errors can be minimized by a standardized medication reconciliation process at each transition and clear documentation of the medication plan.
Communication among providers can be improved if the admitting clinician documents clearly whether each preadmission medication is being continued, changed, or stopped, along with the reason for doing so, and makes this information available throughout the hospital stay. Upon transfer to another unit and at discharge, the physician should review each; preadmission medication that was held and the patient’s current clinical status and, based on that information, decide whether medications that were held should be resumed. If a medication will be restarted later, specific instructions should be documented and communicated to the patient and the physicians who are taking over his or her care.
Preventing errors of commission
Failure to perform a complete reconciliation at each transition of care and match each medication with an appropriate indication can lead to errors of commission.
One study showed that 44% of patients were prescribed at least one unnecessary drug at hospital discharge, one-fourth of which were started during the hospitalization.27 Commonly prescribed unnecessarily were gastrointestinal agents, central nervous system drugs, nutrients, and supplements.
It is critical to assess each medication’s ongoing need, appropriateness, and risk-benefit ratio at every transition. Medications no longer indicated should be discontinued in order to simplify the regimen, avoid unnecessary drug exposure, and prevent ADEs.
For example, proton pump inhibitors or histamine 2 receptor blockers are often started in the hospital for stress ulcer prophylaxis. One-third of patients are then discharged home on the medication, and 6 months later half of those patients are still taking the unnecessary drug.28 This situation can be avoided by limiting use of these medications to appropriate circumstances, clearly marking the indication as stress ulcer prophylaxis (as opposed to an ongoing condition that will require continuing it after discharge), and discontinuing the agent when appropriate.
All drugs, even common and seemingly benign ones, carry some risk and should be discontinued when no longer needed. Thus, medications added during the hospitalization to control acute symptoms should also be reviewed at each transition to prevent inappropriate continuation when symptoms have resolved.
One study, for example, found that many patients were discharged with inappropriate prescriptions for atypical antipsychotics after receiving them in the intensive care unit, likely for delirium.29 Documenting that an acute issue such as delirium has resolved should prompt the discontinuation of therapy.
Preventing therapeutic duplication
Therapeutic duplication occurs in about 8% of discharges.1 These errors often result from formulary substitutions or altering the dosage form in the acute setting. For example, patients who receive a prescription for the substituted agent at discharge and also resume their prehospitalization medications end up with duplicate therapy.
Therapeutic substitution is common at the time of admission to the hospital as a result of formulary restrictions. Drug classes that are frequently substituted include statins, antihypertensives, urinary antispasmodics, and proton pump inhibitors. Physicians should be familiar with the preferred agents on the hospital formulary and make careful note when a substitution occurs. Furthermore, hospital systems should be developed to remind the physician to switch back to the outpatient medication at discharge.
Similar problems occur when home medications are replaced with different dosage forms with different pharmacokinetic properties. For example, a long-acting medication may be temporarily replaced with an intravenous solution or immediate-release tablet for several reasons, including nothing-by-mouth status, unstable clinical condition, need for titration, and need to crush the tablet to give the drug per tube. The differing formulations must be reconciled throughout the patient’s hospital course and at discharge to avoid therapeutic duplication and serious medication errors. Deliberate changes to the dosage form should be clearly communicated in the discharge medication list so that patients and other clinicians are aware.
Hospital systems should also have the capability to identify duplications in the medication list and to warn prescribers of these errors. The ability to group medications by drug class or sort the medication list alphabetically by generic name can help uncover duplication errors.
STEP 3: REVIEW THE LIST IN VIEW OF THE CLINICAL PICTURE
Transitions of care should prompt providers to review the medication list for possible drug-disease interactions, confirm compliance with evidence-based guidelines, and evaluate the risks and benefits of each medication in the context of the patient’s age and acute and chronic medical issues. This is also an opportunity to screen the full list for potentially inappropriate medications and high-alert drugs such as insulin or anticoagulants, which are more likely than other drugs to cause severe harm when used in error.
Acute kidney injury. New drug-disease interactions can arise during a hospitalization and can affect dosing and the choice of drug. The onset of acute kidney injury, for example, often necessitates adjusting or discontinuing nephrotoxic and renally excreted medications. ADEs or potential ADEs have been reported in 43% of hospitalized patients with acute kidney injury.30
Because acute kidney injury is often transient, medications may need to be held or adjusted several times until renal function stabilizes. This can be challenging across the continuum of care and requires close monitoring of the serum creatinine level and associated drug doses and levels, if applicable. Well-designed clinical decision support tools can integrate laboratory data and alert the prescriber to a clinically important increase or decrease in serum creatinine that may warrant a change in therapy. Modifications to the regimen and a plan for timely follow-up of the serum creatinine level should be clearly documented in the discharge plan.
Liver disease. Similar attention should be given to drugs that are hepatically metabolized if a patient has acute or chronic liver impairment.
Geriatric patients, particularly those who present with altered mental status, falls, or urinary retention, should have their medication list reviewed for potentially inappropriate medications, which are drugs that pose increased risk of poor outcomes in older adults.31,32 Patients and providers may have been willing to accept the risk of medications such as anticholinergics or sedative-hypnotics when the drugs were initiated, but circumstances can change over time, especially in this patient population. Hospitalization is a prime opportunity to screen for medications that meet the Beers criteria31 for agents to avoid or use with caution in older adults.
As-needed medications. Medications prescribed on an as-needed basis in the hospital should be reviewed for continuation or discontinuation at discharge. How often the medication was given can inform this decision.
For example, if as-needed opioids were used frequently, failure to develop a plan of care for pain can lead to persistent symptoms and, possibly, to readmission.33,34 Similar scenarios occur with use of as-needed blood pressure medications, laxatives, and correction-dose insulin.
If an as-needed medication was used consistently during hospitalization, the physician should consider whether a regularly scheduled medication is needed. Conversely, if the medication was not used during the inpatient admission, it can likely be discontinued.
STEP 4: PREPARE THE PATIENT AND FOLLOW-UP PROVIDER
Once a clinician has performed medication reconciliation, including obtaining a best-possible medical history and carefully reviewing the medication list and orders for errors and clinical appropriateness, the next steps are to ensure the patient understands what he or she needs to do and to confirm that suitable follow-up plans are in place. These measures should be taken at all transitions of care but are critically important at hospital discharge.
Preparing the patient and caregiver
An accurate, reconciled medication list should be given to the patient, caregiver, or both, and should be reviewed before discharge.17
Approximately one-third of Americans have low health literacy skills, so medication lists and associated materials should be easy to understand.35 Medication lists should be written in plain language and formatted for optimal readability (Table 4), clearly stating which medications to continue, change, hold temporarily, and stop.
Patients recall and comprehend about half of the information provided during a medical encounter.36 Thus, medication teaching should focus on key points including changes or additions to the regimen, specific instructions for follow-up and monitoring, and how to handle common and serious side effects.
To confirm patient understanding, clinicians should use “teach-back,” ie, provide the patient with information and then ask him or her to repeat back key points.37,38 The patient and family should also be encouraged to ask questions before discharge.
If not already addressed during the hospital stay, barriers to medication adherence and ability to obtain the medications should be attended to at this time (Table 5). Also, the plan to pick up the medications should be verified with the patient and caregiver. Verify that there is transportation to a particular pharmacy that is open at the time of discharge, and that the patient can afford the medications.
Ensuring appropriate follow-up
Studies have shown that timely in-home or telephone follow-up after discharge can decrease adverse events and postdischarge health care utilization.39,40 Telephone follow-up that includes thorough medication reconciliation can help detect and resolve medication issues early after discharge and can close gaps related to monitoring and follow-up.
Medication reconciliation by telephone can be time-consuming. Depending on the number of medications that need to be reviewed, calls can take between 10 and 60 minutes. Postdischarge phone calls should be performed by clinical personnel who are able to identify medication-related problems. A pharmacist should be an available resource to assist with complex regimens, to help resolve medication discrepancies, and to address patient concerns. Table 6 provides tips for conducting follow-up phone calls.
Resolving discrepancies identified during follow-up calls can be difficult, as changes to the medication regimen are often not communicated effectively to other members of the care team. Physicians should document the complete medication list and plan in the discharge summary, and there should be a method for the caller to record updates to the medication list in the medical record so that they are apparent at the outpatient follow-up visit.
An additional challenge is that it is frequently unclear which physician “owns” which medications. Therefore, designating a contact person for each medication until follow-up can be very valuable. At a minimum, a “physician owner” for high-alert medications such as insulin, anticoagulants, and diuretics should be identified to provide close follow-up, titration, and monitoring.
There should also be a plan for the patient to obtain refills of essential long-term medications, such as antiplatelet agents following stent placement.
SUMMARY AND RECOMMENDATIONS
Medication-related problems during hospital admission and discharge are common and range from minor discrepancies in the medication list to errors in history-taking, prescribing, and reconciliation that can lead to potential or actual patient harm. Putting systems in place to facilitate medication reconciliation can decrease the occurrence of medication discrepancies and ADEs, thereby improving patient safety during these critical transitions between care settings and providers. Institutional medication reconciliation programs should focus resources on the admission history-taking step, target the highest-risk patients for the most intensive interventions, and involve pharmacy personnel when possible.
On an individual level, clinicians can incorporate additional interventions into their workflows to optimize medication safety for hospitalized patients. Using a structured approach to obtain a complete and accurate medication list at the time of hospital admission will help providers identify medication-related problems and prevent the propagation of errors throughout the hospital stay and at discharge. Focusing additional time and effort on a comprehensive review of the medication list for errors of omission and commission, patient-specific needs, and high-alert drugs will further decrease the risk of medication errors. Finally, providing discharge counseling targeting patient barriers to adherence and ensuring a proper handover of medication information and rationale for medication changes to outpatient providers will improve the chances of a safe transition.
- Coleman EA, Smith JD, Raha D, Min SJ. Posthospital medication discrepancies: prevalence and contributing factors. Arch Intern Med 2005; 165:1842–1847.
- Wong JD, Bajcar JM, Wong GG, et al. Medication reconciliation at hospital discharge: evaluating discrepancies. Ann Pharmacother 2008; 42:1373–1379.
- Kripalani S, Roumie CL, Dalal AK, et al; PILL-CVD (Pharmacist Intervention for Low Literacy in Cardiovascular Disease) Study Group. Effect of a pharmacist intervention on clinically important medication errors after hospital discharge: a randomized trial. Ann Intern Med 2012; 157:1-10.
- Forster AJ, Murff HJ, Peterson JF, Gandhi TK, Bates DW. The incidence and severity of adverse events affecting patients after discharge from the hospital. Ann Intern Med 2003; 138:161–167.
- Forster AJ, Murff HJ, Peterson JF, Gandhi TK, Bates DW. Adverse drug events occurring following hospital discharge. J Gen Intern Med 2005; 20:317–323.
- Johnson JA, Bootman JL. Drug-related morbidity and mortality. A cost-of-illness model. Arch Intern Med 1995; 155:1949–1956.
- Budnitz DS, Lovegrove MC, Shehab N, Richards CL. Emergency hospitalizations for adverse drug events in older Americans. N Engl J Med 2011; 365:2002–2012.
- Bates DW, Boyle DL, Vander Vliet MB, Schneider J, Leape L. Relationship between medication errors and adverse drug events. J Gen Intern Med 1995;10:199–205.
- Pippins JR, Gandhi TK, Hamann C, et al. Classifying and predicting errors of inpatient medication reconciliation. J Gen Intern Med 2008; 23:1414–1422.
- Lau HS, Florax C, Porsius AJ, De Boer A.The completeness of medication histories in hospital medical records of patients admitted to general internal medicine wards. Br J Clin Pharmacol 2000; 49:597–603.
- Tam VC, Knowles SR, Cornish PL, Fine N, Marchesano R, Etchells EE. Frequency, type and clinical importance of medication history errors at admission to hospital: a systematic review. CMAJ 2005; 173:510–515.
- Bell CM, Brener SS, Gunraj N, et al. Association of ICU or hospital admission with unintentional discontinuation of medications for chronic diseases. JAMA 2011; 306:840–847.
- Dobranski S, Hammond I, Khan G, Holdsworth H. The nature of hospital prescribing errors. Br J Clin Governance 2002; 7:187–193.
- Gleason KM, Groszek JM, Sullivan C, Rooney D, Barnard C, Noskin GA. Reconciliation of discrepancies in medication histories and admission orders of newly hospitalized patients. Am J Health Syst Pharm 2004; 61:1689–1695.
- Rozich JD, Resar KR. Medication safety: one organization’s approach to the challenge. J Clin Outcomes Manage 2001; 8:27–34.
- Mueller SK, Sponsler KC, Kripalani S, Schnipper JL. Hospital-based medication reconciliation practices: a systematic review. Arch Intern Med 2012; 172:1057–1069.
- Joint Commission. Using medication reconciliation to prevent errors. Sentinel Event Alert 2006, Issue 35. www.jointcommission.org/assets/1/18/SEA_35.pdf. Accessed March 31, 2015.
- Greenwald JL, Halasyamani L, Greene J, et al. Making inpatient medication reconciliation patient centered, clinically relevant and implementable: a consensus statement on key principles and necessary first steps. J Hosp Med 2010; 5:477–485.
- Berwick DM, Calkins DR, McCannon CJ, Hackbarth AD. The 100,000 lives campaign: setting a goal and a deadline for improving health care quality. JAMA 2006; 295:324–327.
- McCannon CJ, Hackbarth AD, Griffin FA. Miles to go: an introduction to the 5 Million Lives Campaign. Jt Comm J Qual Patient Saf 2007; 33:477–484.
- Leotsakos A, Caisley L, Karga M, Kelly E, O’Leary D, Timmons K. High 5s: addressing excellence in patient safety. World Hosp Health Serv 2009; 45:19–22.
- Gleason KM, Brake H, Agramonte V, Perfetti C. Medications at Transitions and Clinical Handoffs (MATCH) Toolkit for Medication Reconciliation. www.ahrq.gov/professionals/quality-patient-safety/patient-safety-resources/resources/match/match.pdf. Accessed March 31, 2015.
- Mueller SK, Kripalani S, Stein J, et al. A toolkit to disseminate best practices in inpatient medication reconciliation: multi-center medication reconciliation quality improvement study (MARQUIS). Jt Comm J Qual Patient Saf 2013; 39:371–382.
- Pal A, Babbott S, Wilkinson ST. Can the targeted use of a discharge pharmacist significantly decrease 30-day readmissions? Hosp Pharm 2013; 48:380–388.
- Claxton AJ, Cramer J, Pierce C. A systematic review of the associations between dose regimens and medication compliance. Clin Ther 2001; 23:1296–1310.
- Osterberg L, Blaschke T. Adherence to medication. N Engl J Med 2005; 353:487–497.
- Hajjar ER, Hanlon JT, Sloane RJ, et al. Unnecessary drug use in frail older people at hospital discharge. J Am Geriatr Soc 2005; 53:1518–1523.
- Zink DA, Pohlman M, Barnes M, Cannon ME. Long-term use of acid suppression started inappropriately during hospitalization. Aliment Pharmacol Ther 2005; 21:1203–1209.
- Morandi A, Vasilevskis E, Pandharipande PP, et al. Inappropriate medication prescriptions in elderly adults surviving an intensive care unit hospitalization. J Am Geriatr Soc 2013; 61:1128–1134.
- Cox ZL, McCoy AB, Matheny ME, et al. Adverse drug events during AKI and its recovery. Clin J Am Soc Nephrol 2013; 8:1070–1078.
- American Geriatrics Society 2012 Beers Criteria Update Expert Panel. American Geriatrics Society updated Beers Criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc 2012; 60:616–631.
- Gallagher P, O’Mahony D. STOPP (Screening Tool of Older Persons’ potentially inappropriate Prescriptions): application to acutely ill elderly patients and comparison with Beers’ criteria. Age Ageing 2008; 37:673–679.
- Tjia J, Bonner A, Briesacher BA, McGee S, Terrill E, Miller K. Medication discrepancies upon hospital to skilled nursing facility transitions. J Gen Intern Med 2009; 24:630–635.
- Boockvar K, Fishman E, Kyriacou CK, Monias A, Gavi S, Cortes T. Adverse events due to discontinuations in drug use and dose changes in patients transferred between acute and long-term care facilities. Arch Intern Med 2004; 164:545–550.
- Kutner M, Greenberg E, Jin Y, Paulsen C. The Health Literacy of America’s Adults: Results From the 2003 National Assessment of Adult Literacy. http://nces.ed.gov/pubs2006/2006483_1.pdf. Accessed March 31, 2015.
- Crane JA. Patient comprehension of doctor-patient communication on discharge from the emergency department. J Emerg Med 1997; 15:1–7.
- DeWalt DA, Callahan LF, Hawk VH, et al. Health Literacy Universal Precautions Toolkit. www.ahrq.gov/qual/literacy/healthliteracytoolkit.pdf. Accessed March 31, 2015.
- Schillinger D, Piette J, Grumbach K, et al. Closing the loop: physician communication with diabetic patients who have low health literacy. Arch Intern Med 2003; 163:83–90.
- Coleman EA, Parry C, Chalmers S, Min SJ. The care transitions intervention: results of a randomized controlled trial. Arch Intern Med 2006; 166:1822–1828.
- Jack BW, Chetty VK, Anthony D, et al. A reengineered hospital discharge program to decrease rehospitalization: a randomized trial. Ann Intern Med 2009; 150:178–187.
- Coleman EA, Smith JD, Raha D, Min SJ. Posthospital medication discrepancies: prevalence and contributing factors. Arch Intern Med 2005; 165:1842–1847.
- Wong JD, Bajcar JM, Wong GG, et al. Medication reconciliation at hospital discharge: evaluating discrepancies. Ann Pharmacother 2008; 42:1373–1379.
- Kripalani S, Roumie CL, Dalal AK, et al; PILL-CVD (Pharmacist Intervention for Low Literacy in Cardiovascular Disease) Study Group. Effect of a pharmacist intervention on clinically important medication errors after hospital discharge: a randomized trial. Ann Intern Med 2012; 157:1-10.
- Forster AJ, Murff HJ, Peterson JF, Gandhi TK, Bates DW. The incidence and severity of adverse events affecting patients after discharge from the hospital. Ann Intern Med 2003; 138:161–167.
- Forster AJ, Murff HJ, Peterson JF, Gandhi TK, Bates DW. Adverse drug events occurring following hospital discharge. J Gen Intern Med 2005; 20:317–323.
- Johnson JA, Bootman JL. Drug-related morbidity and mortality. A cost-of-illness model. Arch Intern Med 1995; 155:1949–1956.
- Budnitz DS, Lovegrove MC, Shehab N, Richards CL. Emergency hospitalizations for adverse drug events in older Americans. N Engl J Med 2011; 365:2002–2012.
- Bates DW, Boyle DL, Vander Vliet MB, Schneider J, Leape L. Relationship between medication errors and adverse drug events. J Gen Intern Med 1995;10:199–205.
- Pippins JR, Gandhi TK, Hamann C, et al. Classifying and predicting errors of inpatient medication reconciliation. J Gen Intern Med 2008; 23:1414–1422.
- Lau HS, Florax C, Porsius AJ, De Boer A.The completeness of medication histories in hospital medical records of patients admitted to general internal medicine wards. Br J Clin Pharmacol 2000; 49:597–603.
- Tam VC, Knowles SR, Cornish PL, Fine N, Marchesano R, Etchells EE. Frequency, type and clinical importance of medication history errors at admission to hospital: a systematic review. CMAJ 2005; 173:510–515.
- Bell CM, Brener SS, Gunraj N, et al. Association of ICU or hospital admission with unintentional discontinuation of medications for chronic diseases. JAMA 2011; 306:840–847.
- Dobranski S, Hammond I, Khan G, Holdsworth H. The nature of hospital prescribing errors. Br J Clin Governance 2002; 7:187–193.
- Gleason KM, Groszek JM, Sullivan C, Rooney D, Barnard C, Noskin GA. Reconciliation of discrepancies in medication histories and admission orders of newly hospitalized patients. Am J Health Syst Pharm 2004; 61:1689–1695.
- Rozich JD, Resar KR. Medication safety: one organization’s approach to the challenge. J Clin Outcomes Manage 2001; 8:27–34.
- Mueller SK, Sponsler KC, Kripalani S, Schnipper JL. Hospital-based medication reconciliation practices: a systematic review. Arch Intern Med 2012; 172:1057–1069.
- Joint Commission. Using medication reconciliation to prevent errors. Sentinel Event Alert 2006, Issue 35. www.jointcommission.org/assets/1/18/SEA_35.pdf. Accessed March 31, 2015.
- Greenwald JL, Halasyamani L, Greene J, et al. Making inpatient medication reconciliation patient centered, clinically relevant and implementable: a consensus statement on key principles and necessary first steps. J Hosp Med 2010; 5:477–485.
- Berwick DM, Calkins DR, McCannon CJ, Hackbarth AD. The 100,000 lives campaign: setting a goal and a deadline for improving health care quality. JAMA 2006; 295:324–327.
- McCannon CJ, Hackbarth AD, Griffin FA. Miles to go: an introduction to the 5 Million Lives Campaign. Jt Comm J Qual Patient Saf 2007; 33:477–484.
- Leotsakos A, Caisley L, Karga M, Kelly E, O’Leary D, Timmons K. High 5s: addressing excellence in patient safety. World Hosp Health Serv 2009; 45:19–22.
- Gleason KM, Brake H, Agramonte V, Perfetti C. Medications at Transitions and Clinical Handoffs (MATCH) Toolkit for Medication Reconciliation. www.ahrq.gov/professionals/quality-patient-safety/patient-safety-resources/resources/match/match.pdf. Accessed March 31, 2015.
- Mueller SK, Kripalani S, Stein J, et al. A toolkit to disseminate best practices in inpatient medication reconciliation: multi-center medication reconciliation quality improvement study (MARQUIS). Jt Comm J Qual Patient Saf 2013; 39:371–382.
- Pal A, Babbott S, Wilkinson ST. Can the targeted use of a discharge pharmacist significantly decrease 30-day readmissions? Hosp Pharm 2013; 48:380–388.
- Claxton AJ, Cramer J, Pierce C. A systematic review of the associations between dose regimens and medication compliance. Clin Ther 2001; 23:1296–1310.
- Osterberg L, Blaschke T. Adherence to medication. N Engl J Med 2005; 353:487–497.
- Hajjar ER, Hanlon JT, Sloane RJ, et al. Unnecessary drug use in frail older people at hospital discharge. J Am Geriatr Soc 2005; 53:1518–1523.
- Zink DA, Pohlman M, Barnes M, Cannon ME. Long-term use of acid suppression started inappropriately during hospitalization. Aliment Pharmacol Ther 2005; 21:1203–1209.
- Morandi A, Vasilevskis E, Pandharipande PP, et al. Inappropriate medication prescriptions in elderly adults surviving an intensive care unit hospitalization. J Am Geriatr Soc 2013; 61:1128–1134.
- Cox ZL, McCoy AB, Matheny ME, et al. Adverse drug events during AKI and its recovery. Clin J Am Soc Nephrol 2013; 8:1070–1078.
- American Geriatrics Society 2012 Beers Criteria Update Expert Panel. American Geriatrics Society updated Beers Criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc 2012; 60:616–631.
- Gallagher P, O’Mahony D. STOPP (Screening Tool of Older Persons’ potentially inappropriate Prescriptions): application to acutely ill elderly patients and comparison with Beers’ criteria. Age Ageing 2008; 37:673–679.
- Tjia J, Bonner A, Briesacher BA, McGee S, Terrill E, Miller K. Medication discrepancies upon hospital to skilled nursing facility transitions. J Gen Intern Med 2009; 24:630–635.
- Boockvar K, Fishman E, Kyriacou CK, Monias A, Gavi S, Cortes T. Adverse events due to discontinuations in drug use and dose changes in patients transferred between acute and long-term care facilities. Arch Intern Med 2004; 164:545–550.
- Kutner M, Greenberg E, Jin Y, Paulsen C. The Health Literacy of America’s Adults: Results From the 2003 National Assessment of Adult Literacy. http://nces.ed.gov/pubs2006/2006483_1.pdf. Accessed March 31, 2015.
- Crane JA. Patient comprehension of doctor-patient communication on discharge from the emergency department. J Emerg Med 1997; 15:1–7.
- DeWalt DA, Callahan LF, Hawk VH, et al. Health Literacy Universal Precautions Toolkit. www.ahrq.gov/qual/literacy/healthliteracytoolkit.pdf. Accessed March 31, 2015.
- Schillinger D, Piette J, Grumbach K, et al. Closing the loop: physician communication with diabetic patients who have low health literacy. Arch Intern Med 2003; 163:83–90.
- Coleman EA, Parry C, Chalmers S, Min SJ. The care transitions intervention: results of a randomized controlled trial. Arch Intern Med 2006; 166:1822–1828.
- Jack BW, Chetty VK, Anthony D, et al. A reengineered hospital discharge program to decrease rehospitalization: a randomized trial. Ann Intern Med 2009; 150:178–187.
KEY POINTS
- Institutional medication reconciliation programs should include taking a best-possible medication history at admission, intervening when patients are at high risk, and involving pharmacy staff when possible.
- Clinicians can incorporate additional interventions into their workflows to optimize medication safety for hospitalized patients.
- Reviewing the medication list for errors of omission and commission, patient-specific needs, and “high-alert” drugs further decreases the risk of medication errors.
- At discharge, patients should receive counseling to ensure understanding of medications and follow-up plans. Hospital physicians should communicate with outpatient providers about medications and rationales for medication changes.

Dermatology update: The dawn of targeted treatment
New targeted therapies are changing the way patients with advanced dermatologic diseases are treated. Innovative molecular biology techniques developed as far back as the 1970s have engendered tremendous insight into the cellular and molecular pathogenesis of numerous diseases. Novel medications based on these insights are now bearing fruit, as directed biologic therapies that are revolutionizing clinical practice are increasingly becoming available.
This article reviews advances in targeted therapies for advanced basal cell carcinoma, psoriasis, and metastatic melanoma.
TARGETED THERAPY FOR BASAL CELL CARCINOMA
Case 1. A 56-year-old man presents with a progressively enlarging leg ulcer. Although it has been treated empirically for years as a venous stasis ulcer, biopsy reveals that it is basal cell carcinoma. Imaging shows muscle and tendon invasion, making surgical intervention short of amputation challenging (Figure 1). What are his options?
Basal cell carcinoma is the most common cancer in humans, accounting for 25% of all cancers and more than 2 million cases in the United States every year. In most cases, surgical excision is curative, but a subset of patients have inoperable, locally advanced, or metastatic disease that drastically limits treatment options. The median survival in metastatic basal cell carcinoma is 24 months, and conventional chemotherapy has not been shown to improve the prognosis.1,2
In addition to the burden of sporadic basal cell carcinoma, patients with the rare autosomal-dominant genetic disorder basal cell nevus syndrome (Gorlin syndrome) develop multiple basal cell lesions over their lifetime. The syndrome may also involve abnormalities of the skeletal system, genitourinary tract, and central nervous system, including development of medulloblastoma.
In Gorlin syndrome, basal cell carcinomas occur often and early; about half of white patients with the syndrome develop their first lesions by age 21, and 90% by age 35. The lesions occur in multiple numbers and can develop anywhere on the body, including on non–sun-exposed areas. Patients who have Gorlin syndrome need meticulous monitoring every 2 to 3 months so that basal cell lesions can be recognized early and treated before they become locally advanced. Keeping up with the numerous medical appointments and invasive treatments can be physically and mentally taxing for patients.
Specific pathway and mutations identified
In 1996, Gorlin syndrome was found to be caused by mutations of the human homolog of the PATCHED gene, which codes for a receptor in the “hedgehog” pathway.3 Two years later, the same mutations were found to be involved in many sporadic basal cell carcinomas, and we now believe that at least 85% of basal cell carcinomas involve abnormal activation of hedgehog pathway signaling.4,5
Vismodegib developed as targeted therapy
In 2009, Robarge et al6 described a potent inhibitor of the hedgehog pathway that was later optimized for potency and desirable pharmacologic traits, resulting in the drug vismodegib.7,8
Two phase 2 multicenter clinical trials9,10 of vismodegib were published in 2012. In the first, which was not randomized,9 33 patients with metastatic basal cell carcinoma and 63 patients with locally advanced disease were treated with vismodegib. Of those with metastatic disease, 30% achieved an objective response. Of those with locally advanced disease, 43% achieved an objective response and 21% achieved a complete response.
In the second trial,10 patients with Gorlin syndrome were randomized to either vismodegib (26 patients) or placebo (16 patients). After 8 months, the vismodegib group had developed significantly fewer new surgically eligible tumors (2 vs 29 per year), their tumors were smaller (change from baseline of the sum of the longest diameters –65% vs –11%), and they needed fewer surgeries (mean 0.31 vs 4.4 per patient). No tumors progressed in the treatment group. Results in some patients were dramatic, with complete healing of large ulcerative tumors. The trial was ended early in view of significant efficacy in the treatment group.
Based on these trials, the US Food and Drug Administration (FDA) approved vismodegib for treating metastatic and locally advanced basal cell carcinoma.
Resistance and adverse effects common
Unfortunately, vismodegib has significant drawbacks. About 20% of patients develop resistance, with tumors recurring after several months of therapy.11 Adverse effects most commonly reported were muscle spasms (68%), alopecia (63%), taste distortion (51%), weight loss (46%), and fatigue (36%). Although these effects were considered mild or moderate, they tended to persist, and almost every patient developed at least one. In the nonrandomized trial,9 more than 25% of patients discontinued treatment because of adverse effects, and more than half of patients did the same in the basal cell nevus syndrome trial.10
New uses may reduce shortcomings
Studies are under way to determine how best to use vismodegib.
One possibility is to use the drug for a few months to shrink tumors to the point that they become eligible for surgery. This is especially important for high-risk tumors, such as those near the eye or other vital structures. In 11 patients, Ally et al12 found that the surgical defect area was reduced by 27% from baseline after 4 months of treatment with vismodegib, allowing for curative surgery in some.
Another option is to combine vismodegib with other agents—either new ones on the horizon or existing nonspecific medications. For example, the antifungal itraconazole has been shown to inhibit hedgehog signaling and perhaps could be combined with vismodegib to increase response and reduce resistance.
Finally, a topical or intralesional form of vismodegib would be useful not only to reduce systemic toxicity, but also to increase efficacy when combined with other topical or systemic medications.
TARGETED THERAPY FOR PSORIASIS VULGARIS
Case 2. A 28-year-old woman presents with worsening psoriasis. About 35% of her body surface is involved, including the palms and soles, making it difficult for her to perform activities of daily living (Figure 2). What are her options?
Psoriasis is a chronic immune-mediated disease that affects up to 3% of people worldwide. In its moderate to severe forms, we recognize psoriasis as a systemic inflammatory disease that may adversely affect organ systems other than the skin. Commonly associated comorbid diseases include inflammatory (psoriatic) arthritis, cardiovascular disease, malignancies (eg, lymphoma), and inflammatory bowel disease. In addition, patients are well known to have significantly impaired quality of life because of low self-esteem, stigmatization affecting their social and work relationships, and, in up to 60%, clinical depression.13,14 The onset of psoriatic arthritis, particularly of erosive disease, is an important decision point for starting aggressive treatment, as joint destruction is irreversible.
Early targeted therapy aimed at TNF alpha, IL-12, and IL-23
Histologically, psoriasis involves thickening of the epidermis caused by hyperproliferation of keratinocytes. Based on this, prior to the 1980s, the dominant hypothesis concerning its pathogenesis was that it was caused by an inherent defect of keratinocytes. In the 1980s and 1990s, however, molecular research revealed that psoriasis was an immune-mediated disease caused by immunologic dysregulation predominantly involving T-helper 1 (Th-1) cells, with the inflammatory cytokines tumor necrosis factor (TNF) alpha, interferon gamma, interleukin (IL) 12, and IL-23 playing prominent roles.15 These findings led to the development and FDA approval of the first effective, targeted, psoriasis treatments, TNF-alpha inhibitors and the IL-12/23 inhibitor ustekinumab.
Etanercept, the first TNF-alpha inhibitor to become available, was approved in 2004 for moderate to severe psoriasis. In 2008, the IL-12/23 inhibitor ustekinumab was approved for the same indication. These drugs are efficacious, are generally safe, and have revolutionized the treatment of psoriasis and psoriatic arthritis, and they are now prescribed on a daily basis.16,17
In the clinical trials that led to approval of these drugs, the main outcome measure was the Psoriasis Area and Severity Index (PASI), a clinical scoring tool that assesses clinical aspects of psoriatic disease including body surface area involvement, degree of thickness, erythema, and scaling of psoriatic plaques. PASI scores range from 0 (no psoriasis) to 72 (most severe psoriasis). Achieving “PASI 75” indicates at least 75% improvement from the baseline score and represents the most common primary outcome measure in clinical trials assessing efficacy of new treatments. Up to 80% of patients who received currently available TNF-alpha inhibitors and ustekinumab in pivotal clinical trials achieved PASI 75 when assessed at 12 to 16 weeks after starting treatment. A moderate percentage of patients (19%–57%, depending on the trial) achieved 90% improvement (PASI 90), and a minority (up to 18%) achieved PASI 100, indicating complete clearing of their psoriasis.18–22
Newly developed therapies target IL-17A
In the mid-2000s, Th-17 cells were discovered, a new lineage of T cells distinct from Th-1 and Th-2 cells. Th-17 cells are characterized by their production of IL-17, a pro-inflammatory cytokine with six family members (IL-17A through IL-17F). Over the next few years, experiments revealed that Th-17 cells and IL-17A play key roles in psoriasis immunologic dysregulation.15 These findings led to a paradigm shift in hypotheses concerning psoriasis pathogenesis, with Th-17 cells and IL-17 replacing Th-1 cells and associated cytokines as dominant mediators of tissue damage.
Additionally, these findings led to new ideas for treatment. Three monoclonal antibodies that target IL-17 inhibition are currently under investigation. Secukinumab and ixekizumab bind to IL-17A and inhibit it from downstream signaling, whereas brodalumab binds to the IL-17A receptor, blocking all six IL-17 cytokines (IL-17A to IL-17F).23
Clinical trials of IL-17 inhibitors show excellent skin improvement
Secukinumab. In 2014, the results of two phase 3 trials of secukinumab were published.24
In the Efficacy and Safety of Subcutaneous Secukinumab for Moderate to Severe Chronic Plaque-type Psoriasis for up to 1 Year trial,24 patients were given either secukinumab 300 mg or 150 mg subcutaneously at defined time points; 82% and 72%, respectively, attained PASI 75 at 12 weeks.
Similar results were seen in the Safety and Efficacy of Secukinumab Compared to Etanercept in Subjects With Moderate to Severe, Chronic Plaque-Type Psoriasis study,24 in which PASI 75 was achieved by 77% of patients receiving secukinumab 300 mg, 67% of those receiving secukinumab 150 mg, and only 44% of those receiving etanercept 50 mg twice weekly at 12 weeks. Rates of infection with secukinumab and etanercept were similar.
The most striking results of these trials were that more than half of patients receiving the 300-mg dose achieved at least 90% improvement in their PASI score (PASI 90) by week 12, and in more than a quarter of patients the psoriasis completely cleared (PASI 100). These results were dramatically better than for etanercept (PASI 90 21%; PASI 100 4%).
Additionally, secukinumab worked fast. The median time to PASI 50 with secukinumab 300 mg was less than half that seen with etanercept (3 weeks vs 7 weeks).
Ixekizumab. In 2012, a phase 2 trial evaluated subcutaneous injections of ixekizumab in dosages ranging from 10 to 150 mg at defined intervals for 16 weeks.25 Of those receiving the highest dosage, 82% attained PASI 75 at 12 weeks, on par with what is noted in patients receiving TNF-alpha inhibitors and IL-12/23 inhibitors. Remarkably, however, almost three-quarters of patients (71%) achieved PASI 90, and 39% achieved PASI 100. Improvement in psoriasis was apparent as early as 1 week after injection.
Brodalumab. A 2012 phase 2 trial of various dosages of the IL-17 receptor inhibitor brodalumab26 also showed excellent PASI 75 achievement with the highest dosage (82%). Astonishingly, though, PASI 90 was achieved by 75% of patients, and PASI 100 by 62%.
Overall, although the percentages of patients achieving PASI 75 with the new IL-17 inhibitor drugs are comparable to those seen with TNF-alpha inhibitors and IL-12/23 inhibitors, the extraordinarily high percentages of patients who achieved PASI 90 and PASI 100 are unprecedented.18–22
Arthritis improvement not shown
Where the IL-17 inhibitors eventually settle within algorithms of psoriasis treatment largely depends on their efficacy in treating psoriatic arthritis compared with TNF-alpha inhibitors and IL-12/23 inhibitors. Joint inflammation is typically evaluated with the American College of Rheumatology (ACR) scoring tool, which in simple terms can be thought of as analogous to the PASI scoring tool for the skin. Although the ACR scoring tool was developed to assess joint inflammation in clinical trials for patients with rheumatoid arthritis, it is commonly used to assess improvement of psoriatic arthritis in clinical trials. The ACR tool involves assessing and scoring the number of swollen and tender joints, but also incorporates serologic assessment of acute-phase reactants (erythrocyte sedimentation rate or C-reactive protein level), patient and physician global assessment, pain, and function. ACR 20 implies roughly a 20% improvement in these criteria, whereas ACR 50 indicates 50% improvement, and so on.
Two phase 2 trials of IL-17 inhibitors for psoriatic arthritis have been published, one with secukinumab27 and one with brodalumab.28 Neither had impressive improvement in the ACR score vs TNF inhibitors—39% for ACR 20 at week 12 and less than 10% for ACR 70. Clinical trial design may have played a role, and phase 3 trials are under way for all three IL-17 inhibitors.
Adverse effects of IL-17 inhibitors
For the most part, adverse effects reported with the IL-17 inhibitors have been mild and similar to those reported with available biologic treatments for psoriasis. Adverse effects most commonly reported have been nasopharyngitis, upper respiratory infection, arthralgia, and mild injection-site reactions. In the future, attention will be paid to the rate of infections known to be associated with IL-17, mainly localized infections with Staphylococcus aureus and Candida species. Some patients have developed Candida esophagitis, but this appears to resolve with discontinuation of the drugs. Neutropenia has occurred, but very few patients have developed grade 3 (500–1,000 cells/mm3) or worse. All adverse effects were reversible with discontinuation of treatment.
Approval of secukinumab, and current studies of IL-17 inhibitors
On January 21, 2015, secukinumab was approved by the FDA for treatment of moderate to severe psoriasis vulgaris in adult patients and is now available by prescription.
More trials of IL-17 inhibitors for the treatment of psoriasis and psoriatic arthritis are under way and are at various phases at the time of this writing.23
TARGETED THERAPY FOR ADVANCED MELANOMA
Case 3. A 58-year-old man presents with an irregular pigmented lesion on his back. Biopsy shows malignant melanoma with an intense, chronic inflammatory infiltrate surrounding the tumor (Figure 3). The tumor was surgically excised with standard margins. Two years later, the patient developed multiple pigmented lesions on the face and complained of headache. Magnetic resonance imaging of the brain revealed multiple enhancing lesions consistent with metastatic melanoma (Figure 3). What are this patient’s options?
Melanoma is the fifth most common cancer in humans, with about 132,000 new cases diagnosed worldwide each year and 48,000 deaths from advanced disease. Its incidence has risen rapidly over the last few decades. Advanced disease has a poor prognosis, with the median overall survival less than 1 year and 5-year survival less than 10%.
Despite decades of research, a paucity of FDA-approved medications were available to treat advanced melanoma until recently. The alkylating agent dacarbazine was approved in 1975, interferon alpha in 1995, and high-dose IL-2 in 1998. Although some patients respond, studies have not shown significant improvement in survival with any of these medications.29–31
In 2002, Davies et al32 found that 50% to 65% of metastatic cutaneous melanomas have a mutation in the BRAF gene. Interestingly, 80% of these patients share a single specific mutation: substitution of glutamic acid for valine in codon 600 (BRAF V600E). The second most common mutation is a single substitution of a lysine for that same valine (BRAF V600K). Additionally, NRAS is mutated in about 20% of melanomas. These discoveries implicated a mitogen-activated protein kinase (MAPK) pathway (Figure 4) as playing a critical role in metastatic melanoma for a large percentage of patients.29
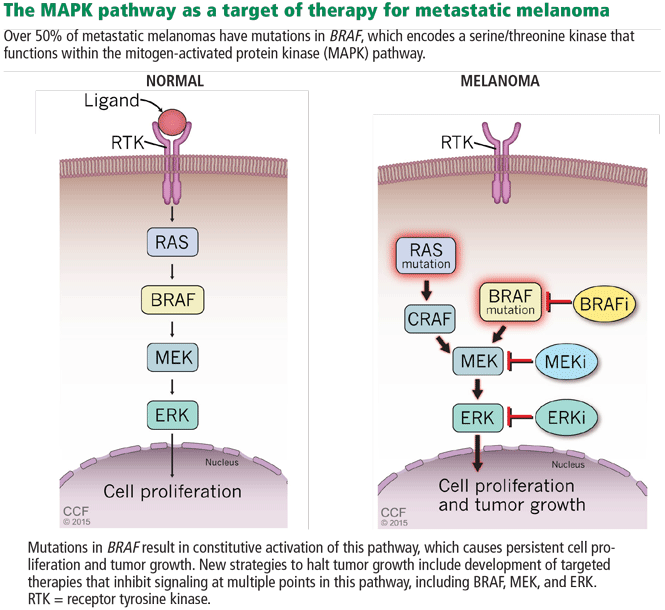
Based on this knowledge, several targeted therapies for melanoma have been developed, and some have been approved.
BRAF inhibitors—first success against melanoma
Vemurafenib. In 2010, Flaherty et al33 reported on a phase 1 and phase 2 clinical trial of vemurafenib (960 mg orally twice daily), a potent inhibitor of BRAF with the V600E mutation. They demonstrated a clinical benefit in 80% of patients with stage IV BRAF-mutant melanoma, an unprecedented response that opened the door to changes in the treatment of metastatic melanoma.
The phase 3 BRAF Inhibitor in Melanoma (BRIM)-3 clinical trial,34 published in 2011, randomized 675 previously untreated patients with advanced melanoma to either vemurafenib 960 mg orally twice daily or dacarbazine, the standard of care. The trial was terminated early when an interim analysis showed a significant overall advantage for vemurafenib (median progression-free survival 5.3 months vs 1.6 months for dacarbazine). Based on these results, vemurafenib was FDA-approved in August 2011 for use in patients with BRAF-mutant melanoma.
Dabrafenib. In a phase 3 clinical trial in 2012, Hauschild et al35 randomized 250 patients with BRAF (V600E)-mutated melanoma in a 3:1 ratio to receive either dabrafenib, a more potent second-generation BRAF inhibitor, or dacarbazine. Half of patients responded to dabrafenib, with a significantly improved progression-free survival rate (5.1 vs 2.7 months respectively), leading to FDA approval for its use in BRAF-mutant melanoma in May 2013.
Adverse effects common to vemurafenib and dabrafenib include rash, fatigue, fever, and joint pain. In addition, up to 25% of patients develop multiple secondary cutaneous squamous cell carcinomas and keratoacanthomas, usually within the first few months of therapy, which are believed to be caused by paradoxical activation of the MAPK pathway.
A more important problem with these medications is the development of resistance. Tumors typically progress again after a median progression-free survival of 6 to 7 months.
MEK inhibitors—another line of defense
Inhibitors of MEK—a serine-threonine kinase that is part of the same MAPK pathway involving BRAF—have been developed as well.
Trametinib. In 2012, trametinib, an allosteric MEK inhibitor, was used in an open-label phase 3 trial in 322 patients with advanced melanoma. Progression-free survival was 4.8 months for trametinib-treated patients compared with 1.5 months for the standard chemotherapy group (dacarbazine or paclitaxel).36 These results led to FDA approval of trametinib in May 2013 for treating BRAF-mutant melanoma.29
Cobimetinib is a second MEK inhibitor being evaluated alone and in combination with other targeted treatments for advanced melanoma.
Both MEK inhibitors have adverse effects similar to those seen with the BRAF inhibitors, including diarrhea, rash, fatigue, and edema. They also tend to cause asymptomatic elevated creatine kinase and transient retinopathy, reduced ejection fraction, and ventricular dysfunction. Unlike BRAF inhibitors, they are not associated with development of secondary cutaneous squamous cell carcinomas or keratoacanthomas. However, as with BRAF inhibitors, resistance is a problem with MEK inhibitors, with most patients relapsing less than a year after starting therapy.
Combination therapy improves outcomes
Possible mechanisms underlying resistance to these medications are being studied. A number of important factors appear to drive resistance, including expression of truncated BRAF proteins that do not bind the BRAF inhibitors and still activate downstream signaling, and amplification of BRAF to such a degree that it overwhelms the medications. This has led to the idea of combining BRAF inhibitors and MEK inhibitors to block the MAPK pathway at two points, potentially increasing the response and decreasing resistance.
Two trials have evaluated combinations of BRAF and MEK inhibitors in patients with advanced melanoma. Larkin et al37 conducted a phase 3 study evaluating combined vemurafenib (a BRAF inhibitor) and cobimetinib (a MEK inhibitor) vs combined vemurafenib and placebo. Survival with the combination therapy was 9.9 months vs 6.2 months with the single treatment.
The incidence of serious adverse effects was not significantly increased with the combination therapy, and keratoacanthomas, cutaneous squamous cell carcinomas, alopecia, and arthralgias were reduced compared with the vemurafenib and placebo group.
Another trial38 evaluating combined dabrafenib (a BRAF inhibitor) and trametinib (a MEK inhibitor) vs combined dabrafenib and placebo had similar findings: increased survival in the combined therapy group (9.3 months vs 8.8 months) and lower rates of squamous cell carcinoma (2% vs 9%).
In January 2014, the FDA approved the combination of BRAF and MEK inhibitors for the treatment of BRAF-mutant metastatic melanoma based on improved survival and generally reduced adverse effects.
IMMUNOTHERAPIES FOR NON-BRAF MELANOMA
Although BRAF and MEK inhibitors represent tremendous advances, their use is limited to the approximately 50% to 65% of patients with advanced melanoma who have BRAF V600 mutations. For others, only the traditional standard medications have been available until recently.
Two of those standard FDA-approved medications, interferon alpha-2b and IL-2, represent immunotherapies. Interferon alpha-2b up-regulates antigen presentation and increases antigen recognition by T cells. Overall, about 20% of patients in clinical trials have achieved responses.
IL-2 is a cytokine that increases T-cell proliferation and maturation into effector T cells. High-dose IL-2 has produced responses in 15% of patients, with a durable complete response in a small proportion.
Though success with these medications was modest, the fact that some patients responded to them indicates that immunotherapy could be a viable strategy for treating metastatic melanoma.30 This is underscored by the fact that some patients can mount an adaptive immune response specifically directed against antigenic proteins expressed in their tumors, resulting in expansion of cytotoxic T cells and control or even elimination of the malignancy.30
Tumors manipulate host immune checkpoints
Molecular biology has provided tremendous insight into tumor immunology over the past several decades, and we now recognize that a hallmark of cancer is escape from immune control.
Cancer cells contain a multitude of mutations that produce proteins that should be recognized by the immune system as foreign but in most individuals are not. This is because T-cell activity is down-regulated in cancer due to cancer cells’ ability to manipulate the host’s normal immunologic inhibitory pathways critical for maintaining self-tolerance.
In general, T-cell activation is initiated when an antigen-presenting cell presents an antigen to a T cell in a major histocompatibility complex-restricted manner. To prevent T cells from being activated by self-antigens and initiating autoimmunity, the interaction between antigen-presenting cells and T cells is regulated by checkpoints (Figure 5). First, for an antigen-presenting cell/T-cell interaction to result in T-cell activation, the T-cell receptor CD28 must bind CD80 on the antigen-presenting cell to drive a “positive” signal. Early in the interaction, the T-cell receptor CTLA-4 is up-regulated and competes with CD28 for binding of CD80. If CTLA-4, and not CD28, binds CD80, a “negative” signal is sent to the T cell and down-regulates it, making the interaction unproductive. Importantly, it is the CTLA-4:CD80 interaction that appears to be crucial for the ability of tumors to dampen T-cell responses to cancer cells.
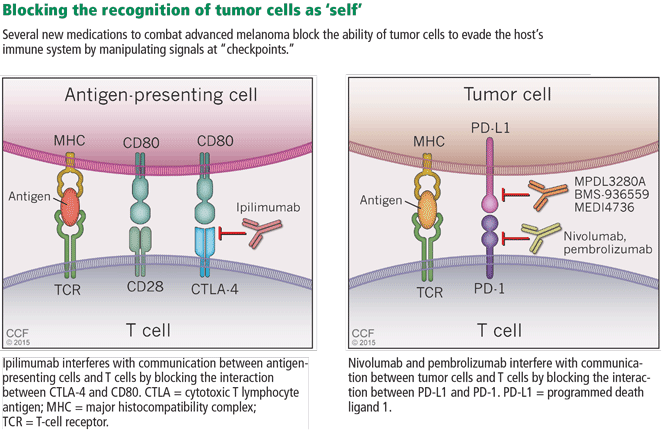
Ipilimumab is a fully humanized monoclonal antibody that binds to CTLA-4, blocking its ability to bind to CD80 and thereby enhancing T-cell activation. In a phase 3 trial, Hodi et al39 evaluated its use in treating advanced melanoma, with some enrolled patients having failed IL-2 treatment. Patients receiving ipilimumab with or without a glycoprotein-100 peptide vaccine (gp100) had an overall survival benefit of 10.1 months compared with 6.4 months for patients treated with gp100 alone. At 24 months, the survival rate with ipilimumab alone was 23.5%, almost double that of patients receiving gp100 alone.
Ipilimumab received FDA approval for treatment of metastatic melanoma in March 2011. This, and the BRAF inhibitors, were the first drugs approved by the FDA for the treatment of advanced melanoma in more than a decade.
Common adverse effects of ipilimumab include fatigue, diarrhea, rash, and pruritus. As expected, given its mechanism of action, up to about 25% of patients experience severe autoimmune-related events that may variably manifest as colitis, rash, hepatitis, neuritis, hypothyroidism, hypopituitarism, and hypophysitis. Another problem with this medication is that a subset of patients do not respond.
Cancer cells disguised as normal cells
Cancer cells can also manipulate another immunologic checkpoint to evade attack by the host immune system (Figure 5). Cytotoxic T cells may recognize antigens on tumor cells and become activated and primed to directly destroy them. However, tumor cells, like normal cells express the programmed death ligands RTK-L1 and PD-L2. These ligands function to bind to the PD-1 receptor on activated T cells to indicate they are “self” and inhibit the cytotoxic T cells from destroying them.
Evasion of immune system attack by manipulating checkpoints involving CTLA-4 and PD-1 helps explain why malignancies can seemingly be associated with brisk inflammatory responses, such as the tumor in Case 3, yet progress and eventually metastasize (Figure 3).
Two medications—nivolumab and pembrolizumab—have been developed in an attempt to disrupt the ability of tumor cells to trick the immune system into accepting them as “self” by manipulating the PD-L1/PD-L2: PD-1 interaction. Both drugs are monoclonal antibodies that bind to PD-1 and, thus, effectively block the ability of PD-L1 or PD-L2 on tumor cells to bind these ligands and signal to activated T cells that they are “self.” This blocking allows T cells to then carry out their killing of tumor cells they initially recognize as foreign.
Nivolumab. In 2014, a phase 3 trial40 compared nivolumab and dacarbazine in patients with untreated advanced melanoma without a BRAF mutation. Objective response rates were 40.0% in the nivolumab group vs 13.9% in the dacarbazine group. This trial was stopped early because of significantly better survival rates in patients taking nivolumab compared with standard chemotherapy.
Interestingly, only 35% of patients who responded to nivolumab had evidence of PD-L1 expression on the surface of their tumor cells as assessed by immunohistochemical assay. Regardless of PD-L1 status, nivolumab-treated patients had improved overall survival compared with those treated with dacarbazine. The response rate with nivolumab was only slightly better in the subgroup of patients whose tumors expressed PD-L1 than in the subgroup without PD-L1.
On December 22, 2014, the FDA granted accelerated approval to nivolumab for the treatment of patients with unresectable or metastatic melanoma and disease progression following ipilimumab treatment and, if BRAF V600 mutation-positive, a BRAF inhibitor.
Pembrolizumab. Also in 2014, an open-label, randomized, phase 1b trial of pembrolizumab treatment at two different dosage schedules was conducted in patients with advanced melanoma that had become refractory either to ipilimumab or a BRAF inhibitor.41 Treatment with pembrolizumab had an objective response rate of 26% at both doses.
In September 2014, the FDA granted accelerated approval for the use of pembrolizumab to treat patients with unresectable or metastatic melanoma and disease progression following treatment with ipilimumab or a BRAF inhibitor.
Adverse effects of PD-1 inhibitors are similar to those seen with ipilimumab, the most common (occurring in at least 20%) being fatigue, cough, nausea, pruritus, rash, decreased appetite, constipation, muscle pain, and diarrhea. Serious effects from pembrolizumab (occurring in at least 2%) were kidney failure, dyspnea, pneumonia, and cellulitis. As seen with ipilimumab, clinically significant autoimmune adverse reactions occur with PD-1 inhibitors, including pneumonitis, colitis, hypophysitis, nephritis, and hepatitis.
Combination therapy under investigation
A phase 1 trial using combination therapy with both immune checkpoint inhibitors—nivolumab (anti-PD-1) and ipilimumab (anti-CTLA-4)—in patients with treatment-resistant metastatic melanoma was published in 2013.42 More than half of patients achieved objective responses, with tumor regression of at least 80% in those who had a response. Tumor response was evident in all subgroups of patients studied—those with pretreatment elevated lactate dehydrogenase levels (one of the strongest prognostic factors in metastatic melanoma), metastases to distant sites, and bulky, multifocal tumor burden. Based on these results, a phase 3 trial is now under way looking at the combination of these two medications vs either one alone.
In summary, targeted treatments are changing the paradigm of how common dermatologic conditions associated with significant morbidity and mortality are treated. Although implementation of the above treatments into everyday clinical practice is exciting, future studies surrounding each are needed to address unanswered issues, such as the optimal dosing and treatment schedules in terms of both disease response and inhibition of resistance, optimal patient/disease characteristics for use, and optimal drug treatment combinations. In the meantime, basic research still utilizing classic molecular biology techniques to uncover pathogenic disease mechanisms in even more detail is ongoing and hopefully will lead to development of even better targeted treatments or even cures for these diseases.
- Lyons TG, O’Kane GM, Kelly CM. Efficacy and safety of vismodegib: a new therapeutic agent in the treatment of basal cell carcinoma. Expert Opin Drug Saf 2014; 13:1125–1132.
- McCusker M, Basset-Sequin N, Dummer R, et al. Metastatic basal cell carcinoma: prognosis dependent on anatomic site and spread of disease. Eur J Cancer 2014; 50:774–783.
- Hahn H, Wicking C, Zaphiropoulous PG, et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 1996; 85:841–851.
- Aszterbaum M, Rothman A, Johnson RL, et al. Identification of mutations in the human PATCHED gene in sporadic basal cell carcinomas and in patients with the basal cell nevus syndrome. J Invest Dermatol 1998; 110:885–888.
- Ingham PW, Placzek M. Orchestrating ontogenesis: variations on a theme by sonic hedgehog. Nat Rev Genet 2006; 7:841–850.
- Robarge KD, Brunton SA, Castanedo GM, et al. GDC-0449-a potent inhibitor of the hedgehog pathway. Bioorg Med Chem Lett 2009; 19:5576–5581.
- Proctor AE, Thompson LA, O’Bryant CL. Vismodegib: an inhibitor of the Hedgehog signaling pathway in the treatment of basal cell carcinoma. Ann Pharmacother 2014; 48:99–106.
- Dessinioti C, Plaka M, Stratigos AJ. Vismodegib for the treatment of basal cell carcinoma: results and implications of the ERIVANCE BCC trial. Future Oncol 2014; 10:927–936.
- Sekulic A, Migden MR, Oro AE, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med 2012; 366:2171–2179.
- Tang JY, Mackay-Wiggan JM, Aszterbaum M, et al. Inhibiting the hedgehog pathway in patients with basal-cell nevus syndrome. N Engl J Med 2012; 366:2180–2188.
- Brinkhuizen T, Reinders MG, van Geel M, et al. Acquired resistance to the Hedgehog pathway inhibitor vismodegib due to smoothened mutations in treatment of locally advanced basal cell carcinoma. J Am Acad Dermatol 2014; 71:1005–1008.
- Ally MS, Aasi S, Wysong A, et al. An investigator-initiated open-label clinical trial of vismodegib as a neoadjuvant to surgery for high-risk basal cell carcinoma. J Am Acad Dermatol 2014; 71:904–911.
- Rapp SR, Feldman SR, Exum ML, Fleischer AB Jr, Reboussin DM. Psoriasis causes as much disability as other major medical diseases. J Am Acad Dermatol 1999; 41:401–407.
- Gelfand JM, Niemann AL, Shin DB, Wang X, Margolis DJ, Troxel AB. Risk of myocardial infarction in patients with psoriasis. JAMA 2006; 296:1735–1741.
- Lynde CW, Poulin Y, Vender R, Bourcier M, Khalil S. Interleukin 17A: toward a new understanding of psoriasis pathogenesis. J Am Acad Dermatol 2014; 71:141–150.
- Tracey D, Klareskog L, Sasso EH, Salfeld JG, Tak PP. Tumor necrosis factor antagonist mechanisms of action: a comprehensive review. Pharmacol Ther 2008; 117:244–279.
- Nestle FL, Kaplan DH, Barker J. Psoriasis. N Engl J Med 2009; 361:496–509.
- Mentor A, Tyring SK, Gordon K, et al. Adalimumab therapy for moderate to severe psoriasis: a randomized, controlled phase III trial. J Am Acad Dermatol 2007; 58:106–115.
- Leonardi CL, Powers JL, Matheson RT, et al. Etanercept as monotherapy in patients with psoriasis. N Engl J Med 2003; 349:2014–2022.
- Reich K, Nestle FO, Papp K, et al. Infliximab induction and maintenance therapy for moderate-to-severe psoriasis: a phase III, multicentre, double-blind trial. Lancet 2005; 366:1367–1374.
- Leonardi CL, Kimball AB, Papp KA, et al. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 76-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 1). Lancet 2008; 371:1665–1674.
- Papp KA, Langley RG, Lebwohl M, et al; PHOENIX 2 study investigators. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 52-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 2). Lancet 2008; 371:1675–1684.
- Leonardi CL, Gordon KB. New and emerging therapies in psoriasis. Semin Cut Med Surg 2014; 33(suppl 2):S37–S41.
- Langley RG, Elewski BE, Lebwohl, et al for the ERASURE and FIXTURE Study Groups. Secukinumab in plaque psorisis—results of two phase 3 trials. N Engl J Med 2014; 371:326–338.
- Leonardi C, Matheson R, Zachariae C. Anti-interleukin-17 monoclonal antibody ixekizumab in chronic plaque psoriasis. N Engl J Med 2012; 366:1190–1199.
- Papp KA, Leonardi C, Menter A, et al. Brodalumab, an anti-interleukin-17-receptor antibody for psoriasis. N Engl J Med 2012; 366:1181–1189.
- McInnes IB, Sieper J, Braun J, et al. Efficacy and safety of secukinumab, a fully human anti-interleukin-17A monoclonal antibody, in patients with moderate-to-severe psoriatic arthritis: a 24-week, randomised, double-blind, placebo-controlled, phase II proof-of-concept trial. Ann Rheum Dis 2014; 73:349–356.
- Mease PJ, Genovese MC, Greenwald MW, et al. Brodalumab, an anti-IL17RA monoclonal antibody, in psoriatic arthritis. N Engl J Med 2014; 370:2295–2306.
- Girotti MR, Saturno G, Lorigan P, Marais R. No longer an untreatable disease: how targeted and immunotherapies have changed the management of melanoma patients. Molec Oncol 2014, 8:1140–1158.
- Saranga-Perry V, Ambe C, Zager JS, Kudchadkar RR. Recent developments in the medical and surgical treatment of melanoma. CA Canc J Clin 2014; 64:171–185.
- Shah DJ, Dronca RS. Latest advances in chemotherapeutic, targeted, and immune approaches in the treatment of metastatic melanoma. Mayo Clin Proc 2014; 89:504–519.
- Davies H, Ignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature 2002; 417:949–954.
- Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med 2010; 363:809–819.
- Chapman PB, Hauschild A, Robert C. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011; 364:2507–2516.
- Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012; 380:358–365.
- Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med 2012; 367:107–114.
- Larkin J, Ascierto PA, Dreno B, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med 2014; 371:1867–1876.
- Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Eng J Med 2014; 371:1877–1888.
- Hodi FS, O’Day SJ, McDermott DF, Weber RW. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010; 363:711–723.
- Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015; 372:320–330.
- Robert C, Ribas A, Wolchok JD, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet 2014; 384:1109–1117.
- Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med 2013; 369:122–133.
New targeted therapies are changing the way patients with advanced dermatologic diseases are treated. Innovative molecular biology techniques developed as far back as the 1970s have engendered tremendous insight into the cellular and molecular pathogenesis of numerous diseases. Novel medications based on these insights are now bearing fruit, as directed biologic therapies that are revolutionizing clinical practice are increasingly becoming available.
This article reviews advances in targeted therapies for advanced basal cell carcinoma, psoriasis, and metastatic melanoma.
TARGETED THERAPY FOR BASAL CELL CARCINOMA
Case 1. A 56-year-old man presents with a progressively enlarging leg ulcer. Although it has been treated empirically for years as a venous stasis ulcer, biopsy reveals that it is basal cell carcinoma. Imaging shows muscle and tendon invasion, making surgical intervention short of amputation challenging (Figure 1). What are his options?
Basal cell carcinoma is the most common cancer in humans, accounting for 25% of all cancers and more than 2 million cases in the United States every year. In most cases, surgical excision is curative, but a subset of patients have inoperable, locally advanced, or metastatic disease that drastically limits treatment options. The median survival in metastatic basal cell carcinoma is 24 months, and conventional chemotherapy has not been shown to improve the prognosis.1,2
In addition to the burden of sporadic basal cell carcinoma, patients with the rare autosomal-dominant genetic disorder basal cell nevus syndrome (Gorlin syndrome) develop multiple basal cell lesions over their lifetime. The syndrome may also involve abnormalities of the skeletal system, genitourinary tract, and central nervous system, including development of medulloblastoma.
In Gorlin syndrome, basal cell carcinomas occur often and early; about half of white patients with the syndrome develop their first lesions by age 21, and 90% by age 35. The lesions occur in multiple numbers and can develop anywhere on the body, including on non–sun-exposed areas. Patients who have Gorlin syndrome need meticulous monitoring every 2 to 3 months so that basal cell lesions can be recognized early and treated before they become locally advanced. Keeping up with the numerous medical appointments and invasive treatments can be physically and mentally taxing for patients.
Specific pathway and mutations identified
In 1996, Gorlin syndrome was found to be caused by mutations of the human homolog of the PATCHED gene, which codes for a receptor in the “hedgehog” pathway.3 Two years later, the same mutations were found to be involved in many sporadic basal cell carcinomas, and we now believe that at least 85% of basal cell carcinomas involve abnormal activation of hedgehog pathway signaling.4,5
Vismodegib developed as targeted therapy
In 2009, Robarge et al6 described a potent inhibitor of the hedgehog pathway that was later optimized for potency and desirable pharmacologic traits, resulting in the drug vismodegib.7,8
Two phase 2 multicenter clinical trials9,10 of vismodegib were published in 2012. In the first, which was not randomized,9 33 patients with metastatic basal cell carcinoma and 63 patients with locally advanced disease were treated with vismodegib. Of those with metastatic disease, 30% achieved an objective response. Of those with locally advanced disease, 43% achieved an objective response and 21% achieved a complete response.
In the second trial,10 patients with Gorlin syndrome were randomized to either vismodegib (26 patients) or placebo (16 patients). After 8 months, the vismodegib group had developed significantly fewer new surgically eligible tumors (2 vs 29 per year), their tumors were smaller (change from baseline of the sum of the longest diameters –65% vs –11%), and they needed fewer surgeries (mean 0.31 vs 4.4 per patient). No tumors progressed in the treatment group. Results in some patients were dramatic, with complete healing of large ulcerative tumors. The trial was ended early in view of significant efficacy in the treatment group.
Based on these trials, the US Food and Drug Administration (FDA) approved vismodegib for treating metastatic and locally advanced basal cell carcinoma.
Resistance and adverse effects common
Unfortunately, vismodegib has significant drawbacks. About 20% of patients develop resistance, with tumors recurring after several months of therapy.11 Adverse effects most commonly reported were muscle spasms (68%), alopecia (63%), taste distortion (51%), weight loss (46%), and fatigue (36%). Although these effects were considered mild or moderate, they tended to persist, and almost every patient developed at least one. In the nonrandomized trial,9 more than 25% of patients discontinued treatment because of adverse effects, and more than half of patients did the same in the basal cell nevus syndrome trial.10
New uses may reduce shortcomings
Studies are under way to determine how best to use vismodegib.
One possibility is to use the drug for a few months to shrink tumors to the point that they become eligible for surgery. This is especially important for high-risk tumors, such as those near the eye or other vital structures. In 11 patients, Ally et al12 found that the surgical defect area was reduced by 27% from baseline after 4 months of treatment with vismodegib, allowing for curative surgery in some.
Another option is to combine vismodegib with other agents—either new ones on the horizon or existing nonspecific medications. For example, the antifungal itraconazole has been shown to inhibit hedgehog signaling and perhaps could be combined with vismodegib to increase response and reduce resistance.
Finally, a topical or intralesional form of vismodegib would be useful not only to reduce systemic toxicity, but also to increase efficacy when combined with other topical or systemic medications.
TARGETED THERAPY FOR PSORIASIS VULGARIS
Case 2. A 28-year-old woman presents with worsening psoriasis. About 35% of her body surface is involved, including the palms and soles, making it difficult for her to perform activities of daily living (Figure 2). What are her options?
Psoriasis is a chronic immune-mediated disease that affects up to 3% of people worldwide. In its moderate to severe forms, we recognize psoriasis as a systemic inflammatory disease that may adversely affect organ systems other than the skin. Commonly associated comorbid diseases include inflammatory (psoriatic) arthritis, cardiovascular disease, malignancies (eg, lymphoma), and inflammatory bowel disease. In addition, patients are well known to have significantly impaired quality of life because of low self-esteem, stigmatization affecting their social and work relationships, and, in up to 60%, clinical depression.13,14 The onset of psoriatic arthritis, particularly of erosive disease, is an important decision point for starting aggressive treatment, as joint destruction is irreversible.
Early targeted therapy aimed at TNF alpha, IL-12, and IL-23
Histologically, psoriasis involves thickening of the epidermis caused by hyperproliferation of keratinocytes. Based on this, prior to the 1980s, the dominant hypothesis concerning its pathogenesis was that it was caused by an inherent defect of keratinocytes. In the 1980s and 1990s, however, molecular research revealed that psoriasis was an immune-mediated disease caused by immunologic dysregulation predominantly involving T-helper 1 (Th-1) cells, with the inflammatory cytokines tumor necrosis factor (TNF) alpha, interferon gamma, interleukin (IL) 12, and IL-23 playing prominent roles.15 These findings led to the development and FDA approval of the first effective, targeted, psoriasis treatments, TNF-alpha inhibitors and the IL-12/23 inhibitor ustekinumab.
Etanercept, the first TNF-alpha inhibitor to become available, was approved in 2004 for moderate to severe psoriasis. In 2008, the IL-12/23 inhibitor ustekinumab was approved for the same indication. These drugs are efficacious, are generally safe, and have revolutionized the treatment of psoriasis and psoriatic arthritis, and they are now prescribed on a daily basis.16,17
In the clinical trials that led to approval of these drugs, the main outcome measure was the Psoriasis Area and Severity Index (PASI), a clinical scoring tool that assesses clinical aspects of psoriatic disease including body surface area involvement, degree of thickness, erythema, and scaling of psoriatic plaques. PASI scores range from 0 (no psoriasis) to 72 (most severe psoriasis). Achieving “PASI 75” indicates at least 75% improvement from the baseline score and represents the most common primary outcome measure in clinical trials assessing efficacy of new treatments. Up to 80% of patients who received currently available TNF-alpha inhibitors and ustekinumab in pivotal clinical trials achieved PASI 75 when assessed at 12 to 16 weeks after starting treatment. A moderate percentage of patients (19%–57%, depending on the trial) achieved 90% improvement (PASI 90), and a minority (up to 18%) achieved PASI 100, indicating complete clearing of their psoriasis.18–22
Newly developed therapies target IL-17A
In the mid-2000s, Th-17 cells were discovered, a new lineage of T cells distinct from Th-1 and Th-2 cells. Th-17 cells are characterized by their production of IL-17, a pro-inflammatory cytokine with six family members (IL-17A through IL-17F). Over the next few years, experiments revealed that Th-17 cells and IL-17A play key roles in psoriasis immunologic dysregulation.15 These findings led to a paradigm shift in hypotheses concerning psoriasis pathogenesis, with Th-17 cells and IL-17 replacing Th-1 cells and associated cytokines as dominant mediators of tissue damage.
Additionally, these findings led to new ideas for treatment. Three monoclonal antibodies that target IL-17 inhibition are currently under investigation. Secukinumab and ixekizumab bind to IL-17A and inhibit it from downstream signaling, whereas brodalumab binds to the IL-17A receptor, blocking all six IL-17 cytokines (IL-17A to IL-17F).23
Clinical trials of IL-17 inhibitors show excellent skin improvement
Secukinumab. In 2014, the results of two phase 3 trials of secukinumab were published.24
In the Efficacy and Safety of Subcutaneous Secukinumab for Moderate to Severe Chronic Plaque-type Psoriasis for up to 1 Year trial,24 patients were given either secukinumab 300 mg or 150 mg subcutaneously at defined time points; 82% and 72%, respectively, attained PASI 75 at 12 weeks.
Similar results were seen in the Safety and Efficacy of Secukinumab Compared to Etanercept in Subjects With Moderate to Severe, Chronic Plaque-Type Psoriasis study,24 in which PASI 75 was achieved by 77% of patients receiving secukinumab 300 mg, 67% of those receiving secukinumab 150 mg, and only 44% of those receiving etanercept 50 mg twice weekly at 12 weeks. Rates of infection with secukinumab and etanercept were similar.
The most striking results of these trials were that more than half of patients receiving the 300-mg dose achieved at least 90% improvement in their PASI score (PASI 90) by week 12, and in more than a quarter of patients the psoriasis completely cleared (PASI 100). These results were dramatically better than for etanercept (PASI 90 21%; PASI 100 4%).
Additionally, secukinumab worked fast. The median time to PASI 50 with secukinumab 300 mg was less than half that seen with etanercept (3 weeks vs 7 weeks).
Ixekizumab. In 2012, a phase 2 trial evaluated subcutaneous injections of ixekizumab in dosages ranging from 10 to 150 mg at defined intervals for 16 weeks.25 Of those receiving the highest dosage, 82% attained PASI 75 at 12 weeks, on par with what is noted in patients receiving TNF-alpha inhibitors and IL-12/23 inhibitors. Remarkably, however, almost three-quarters of patients (71%) achieved PASI 90, and 39% achieved PASI 100. Improvement in psoriasis was apparent as early as 1 week after injection.
Brodalumab. A 2012 phase 2 trial of various dosages of the IL-17 receptor inhibitor brodalumab26 also showed excellent PASI 75 achievement with the highest dosage (82%). Astonishingly, though, PASI 90 was achieved by 75% of patients, and PASI 100 by 62%.
Overall, although the percentages of patients achieving PASI 75 with the new IL-17 inhibitor drugs are comparable to those seen with TNF-alpha inhibitors and IL-12/23 inhibitors, the extraordinarily high percentages of patients who achieved PASI 90 and PASI 100 are unprecedented.18–22
Arthritis improvement not shown
Where the IL-17 inhibitors eventually settle within algorithms of psoriasis treatment largely depends on their efficacy in treating psoriatic arthritis compared with TNF-alpha inhibitors and IL-12/23 inhibitors. Joint inflammation is typically evaluated with the American College of Rheumatology (ACR) scoring tool, which in simple terms can be thought of as analogous to the PASI scoring tool for the skin. Although the ACR scoring tool was developed to assess joint inflammation in clinical trials for patients with rheumatoid arthritis, it is commonly used to assess improvement of psoriatic arthritis in clinical trials. The ACR tool involves assessing and scoring the number of swollen and tender joints, but also incorporates serologic assessment of acute-phase reactants (erythrocyte sedimentation rate or C-reactive protein level), patient and physician global assessment, pain, and function. ACR 20 implies roughly a 20% improvement in these criteria, whereas ACR 50 indicates 50% improvement, and so on.
Two phase 2 trials of IL-17 inhibitors for psoriatic arthritis have been published, one with secukinumab27 and one with brodalumab.28 Neither had impressive improvement in the ACR score vs TNF inhibitors—39% for ACR 20 at week 12 and less than 10% for ACR 70. Clinical trial design may have played a role, and phase 3 trials are under way for all three IL-17 inhibitors.
Adverse effects of IL-17 inhibitors
For the most part, adverse effects reported with the IL-17 inhibitors have been mild and similar to those reported with available biologic treatments for psoriasis. Adverse effects most commonly reported have been nasopharyngitis, upper respiratory infection, arthralgia, and mild injection-site reactions. In the future, attention will be paid to the rate of infections known to be associated with IL-17, mainly localized infections with Staphylococcus aureus and Candida species. Some patients have developed Candida esophagitis, but this appears to resolve with discontinuation of the drugs. Neutropenia has occurred, but very few patients have developed grade 3 (500–1,000 cells/mm3) or worse. All adverse effects were reversible with discontinuation of treatment.
Approval of secukinumab, and current studies of IL-17 inhibitors
On January 21, 2015, secukinumab was approved by the FDA for treatment of moderate to severe psoriasis vulgaris in adult patients and is now available by prescription.
More trials of IL-17 inhibitors for the treatment of psoriasis and psoriatic arthritis are under way and are at various phases at the time of this writing.23
TARGETED THERAPY FOR ADVANCED MELANOMA
Case 3. A 58-year-old man presents with an irregular pigmented lesion on his back. Biopsy shows malignant melanoma with an intense, chronic inflammatory infiltrate surrounding the tumor (Figure 3). The tumor was surgically excised with standard margins. Two years later, the patient developed multiple pigmented lesions on the face and complained of headache. Magnetic resonance imaging of the brain revealed multiple enhancing lesions consistent with metastatic melanoma (Figure 3). What are this patient’s options?
Melanoma is the fifth most common cancer in humans, with about 132,000 new cases diagnosed worldwide each year and 48,000 deaths from advanced disease. Its incidence has risen rapidly over the last few decades. Advanced disease has a poor prognosis, with the median overall survival less than 1 year and 5-year survival less than 10%.
Despite decades of research, a paucity of FDA-approved medications were available to treat advanced melanoma until recently. The alkylating agent dacarbazine was approved in 1975, interferon alpha in 1995, and high-dose IL-2 in 1998. Although some patients respond, studies have not shown significant improvement in survival with any of these medications.29–31
In 2002, Davies et al32 found that 50% to 65% of metastatic cutaneous melanomas have a mutation in the BRAF gene. Interestingly, 80% of these patients share a single specific mutation: substitution of glutamic acid for valine in codon 600 (BRAF V600E). The second most common mutation is a single substitution of a lysine for that same valine (BRAF V600K). Additionally, NRAS is mutated in about 20% of melanomas. These discoveries implicated a mitogen-activated protein kinase (MAPK) pathway (Figure 4) as playing a critical role in metastatic melanoma for a large percentage of patients.29
Based on this knowledge, several targeted therapies for melanoma have been developed, and some have been approved.
BRAF inhibitors—first success against melanoma
Vemurafenib. In 2010, Flaherty et al33 reported on a phase 1 and phase 2 clinical trial of vemurafenib (960 mg orally twice daily), a potent inhibitor of BRAF with the V600E mutation. They demonstrated a clinical benefit in 80% of patients with stage IV BRAF-mutant melanoma, an unprecedented response that opened the door to changes in the treatment of metastatic melanoma.
The phase 3 BRAF Inhibitor in Melanoma (BRIM)-3 clinical trial,34 published in 2011, randomized 675 previously untreated patients with advanced melanoma to either vemurafenib 960 mg orally twice daily or dacarbazine, the standard of care. The trial was terminated early when an interim analysis showed a significant overall advantage for vemurafenib (median progression-free survival 5.3 months vs 1.6 months for dacarbazine). Based on these results, vemurafenib was FDA-approved in August 2011 for use in patients with BRAF-mutant melanoma.
Dabrafenib. In a phase 3 clinical trial in 2012, Hauschild et al35 randomized 250 patients with BRAF (V600E)-mutated melanoma in a 3:1 ratio to receive either dabrafenib, a more potent second-generation BRAF inhibitor, or dacarbazine. Half of patients responded to dabrafenib, with a significantly improved progression-free survival rate (5.1 vs 2.7 months respectively), leading to FDA approval for its use in BRAF-mutant melanoma in May 2013.
Adverse effects common to vemurafenib and dabrafenib include rash, fatigue, fever, and joint pain. In addition, up to 25% of patients develop multiple secondary cutaneous squamous cell carcinomas and keratoacanthomas, usually within the first few months of therapy, which are believed to be caused by paradoxical activation of the MAPK pathway.
A more important problem with these medications is the development of resistance. Tumors typically progress again after a median progression-free survival of 6 to 7 months.
MEK inhibitors—another line of defense
Inhibitors of MEK—a serine-threonine kinase that is part of the same MAPK pathway involving BRAF—have been developed as well.
Trametinib. In 2012, trametinib, an allosteric MEK inhibitor, was used in an open-label phase 3 trial in 322 patients with advanced melanoma. Progression-free survival was 4.8 months for trametinib-treated patients compared with 1.5 months for the standard chemotherapy group (dacarbazine or paclitaxel).36 These results led to FDA approval of trametinib in May 2013 for treating BRAF-mutant melanoma.29
Cobimetinib is a second MEK inhibitor being evaluated alone and in combination with other targeted treatments for advanced melanoma.
Both MEK inhibitors have adverse effects similar to those seen with the BRAF inhibitors, including diarrhea, rash, fatigue, and edema. They also tend to cause asymptomatic elevated creatine kinase and transient retinopathy, reduced ejection fraction, and ventricular dysfunction. Unlike BRAF inhibitors, they are not associated with development of secondary cutaneous squamous cell carcinomas or keratoacanthomas. However, as with BRAF inhibitors, resistance is a problem with MEK inhibitors, with most patients relapsing less than a year after starting therapy.
Combination therapy improves outcomes
Possible mechanisms underlying resistance to these medications are being studied. A number of important factors appear to drive resistance, including expression of truncated BRAF proteins that do not bind the BRAF inhibitors and still activate downstream signaling, and amplification of BRAF to such a degree that it overwhelms the medications. This has led to the idea of combining BRAF inhibitors and MEK inhibitors to block the MAPK pathway at two points, potentially increasing the response and decreasing resistance.
Two trials have evaluated combinations of BRAF and MEK inhibitors in patients with advanced melanoma. Larkin et al37 conducted a phase 3 study evaluating combined vemurafenib (a BRAF inhibitor) and cobimetinib (a MEK inhibitor) vs combined vemurafenib and placebo. Survival with the combination therapy was 9.9 months vs 6.2 months with the single treatment.
The incidence of serious adverse effects was not significantly increased with the combination therapy, and keratoacanthomas, cutaneous squamous cell carcinomas, alopecia, and arthralgias were reduced compared with the vemurafenib and placebo group.
Another trial38 evaluating combined dabrafenib (a BRAF inhibitor) and trametinib (a MEK inhibitor) vs combined dabrafenib and placebo had similar findings: increased survival in the combined therapy group (9.3 months vs 8.8 months) and lower rates of squamous cell carcinoma (2% vs 9%).
In January 2014, the FDA approved the combination of BRAF and MEK inhibitors for the treatment of BRAF-mutant metastatic melanoma based on improved survival and generally reduced adverse effects.
IMMUNOTHERAPIES FOR NON-BRAF MELANOMA
Although BRAF and MEK inhibitors represent tremendous advances, their use is limited to the approximately 50% to 65% of patients with advanced melanoma who have BRAF V600 mutations. For others, only the traditional standard medications have been available until recently.
Two of those standard FDA-approved medications, interferon alpha-2b and IL-2, represent immunotherapies. Interferon alpha-2b up-regulates antigen presentation and increases antigen recognition by T cells. Overall, about 20% of patients in clinical trials have achieved responses.
IL-2 is a cytokine that increases T-cell proliferation and maturation into effector T cells. High-dose IL-2 has produced responses in 15% of patients, with a durable complete response in a small proportion.
Though success with these medications was modest, the fact that some patients responded to them indicates that immunotherapy could be a viable strategy for treating metastatic melanoma.30 This is underscored by the fact that some patients can mount an adaptive immune response specifically directed against antigenic proteins expressed in their tumors, resulting in expansion of cytotoxic T cells and control or even elimination of the malignancy.30
Tumors manipulate host immune checkpoints
Molecular biology has provided tremendous insight into tumor immunology over the past several decades, and we now recognize that a hallmark of cancer is escape from immune control.
Cancer cells contain a multitude of mutations that produce proteins that should be recognized by the immune system as foreign but in most individuals are not. This is because T-cell activity is down-regulated in cancer due to cancer cells’ ability to manipulate the host’s normal immunologic inhibitory pathways critical for maintaining self-tolerance.
In general, T-cell activation is initiated when an antigen-presenting cell presents an antigen to a T cell in a major histocompatibility complex-restricted manner. To prevent T cells from being activated by self-antigens and initiating autoimmunity, the interaction between antigen-presenting cells and T cells is regulated by checkpoints (Figure 5). First, for an antigen-presenting cell/T-cell interaction to result in T-cell activation, the T-cell receptor CD28 must bind CD80 on the antigen-presenting cell to drive a “positive” signal. Early in the interaction, the T-cell receptor CTLA-4 is up-regulated and competes with CD28 for binding of CD80. If CTLA-4, and not CD28, binds CD80, a “negative” signal is sent to the T cell and down-regulates it, making the interaction unproductive. Importantly, it is the CTLA-4:CD80 interaction that appears to be crucial for the ability of tumors to dampen T-cell responses to cancer cells.
Ipilimumab is a fully humanized monoclonal antibody that binds to CTLA-4, blocking its ability to bind to CD80 and thereby enhancing T-cell activation. In a phase 3 trial, Hodi et al39 evaluated its use in treating advanced melanoma, with some enrolled patients having failed IL-2 treatment. Patients receiving ipilimumab with or without a glycoprotein-100 peptide vaccine (gp100) had an overall survival benefit of 10.1 months compared with 6.4 months for patients treated with gp100 alone. At 24 months, the survival rate with ipilimumab alone was 23.5%, almost double that of patients receiving gp100 alone.
Ipilimumab received FDA approval for treatment of metastatic melanoma in March 2011. This, and the BRAF inhibitors, were the first drugs approved by the FDA for the treatment of advanced melanoma in more than a decade.
Common adverse effects of ipilimumab include fatigue, diarrhea, rash, and pruritus. As expected, given its mechanism of action, up to about 25% of patients experience severe autoimmune-related events that may variably manifest as colitis, rash, hepatitis, neuritis, hypothyroidism, hypopituitarism, and hypophysitis. Another problem with this medication is that a subset of patients do not respond.
Cancer cells disguised as normal cells
Cancer cells can also manipulate another immunologic checkpoint to evade attack by the host immune system (Figure 5). Cytotoxic T cells may recognize antigens on tumor cells and become activated and primed to directly destroy them. However, tumor cells, like normal cells express the programmed death ligands RTK-L1 and PD-L2. These ligands function to bind to the PD-1 receptor on activated T cells to indicate they are “self” and inhibit the cytotoxic T cells from destroying them.
Evasion of immune system attack by manipulating checkpoints involving CTLA-4 and PD-1 helps explain why malignancies can seemingly be associated with brisk inflammatory responses, such as the tumor in Case 3, yet progress and eventually metastasize (Figure 3).
Two medications—nivolumab and pembrolizumab—have been developed in an attempt to disrupt the ability of tumor cells to trick the immune system into accepting them as “self” by manipulating the PD-L1/PD-L2: PD-1 interaction. Both drugs are monoclonal antibodies that bind to PD-1 and, thus, effectively block the ability of PD-L1 or PD-L2 on tumor cells to bind these ligands and signal to activated T cells that they are “self.” This blocking allows T cells to then carry out their killing of tumor cells they initially recognize as foreign.
Nivolumab. In 2014, a phase 3 trial40 compared nivolumab and dacarbazine in patients with untreated advanced melanoma without a BRAF mutation. Objective response rates were 40.0% in the nivolumab group vs 13.9% in the dacarbazine group. This trial was stopped early because of significantly better survival rates in patients taking nivolumab compared with standard chemotherapy.
Interestingly, only 35% of patients who responded to nivolumab had evidence of PD-L1 expression on the surface of their tumor cells as assessed by immunohistochemical assay. Regardless of PD-L1 status, nivolumab-treated patients had improved overall survival compared with those treated with dacarbazine. The response rate with nivolumab was only slightly better in the subgroup of patients whose tumors expressed PD-L1 than in the subgroup without PD-L1.
On December 22, 2014, the FDA granted accelerated approval to nivolumab for the treatment of patients with unresectable or metastatic melanoma and disease progression following ipilimumab treatment and, if BRAF V600 mutation-positive, a BRAF inhibitor.
Pembrolizumab. Also in 2014, an open-label, randomized, phase 1b trial of pembrolizumab treatment at two different dosage schedules was conducted in patients with advanced melanoma that had become refractory either to ipilimumab or a BRAF inhibitor.41 Treatment with pembrolizumab had an objective response rate of 26% at both doses.
In September 2014, the FDA granted accelerated approval for the use of pembrolizumab to treat patients with unresectable or metastatic melanoma and disease progression following treatment with ipilimumab or a BRAF inhibitor.
Adverse effects of PD-1 inhibitors are similar to those seen with ipilimumab, the most common (occurring in at least 20%) being fatigue, cough, nausea, pruritus, rash, decreased appetite, constipation, muscle pain, and diarrhea. Serious effects from pembrolizumab (occurring in at least 2%) were kidney failure, dyspnea, pneumonia, and cellulitis. As seen with ipilimumab, clinically significant autoimmune adverse reactions occur with PD-1 inhibitors, including pneumonitis, colitis, hypophysitis, nephritis, and hepatitis.
Combination therapy under investigation
A phase 1 trial using combination therapy with both immune checkpoint inhibitors—nivolumab (anti-PD-1) and ipilimumab (anti-CTLA-4)—in patients with treatment-resistant metastatic melanoma was published in 2013.42 More than half of patients achieved objective responses, with tumor regression of at least 80% in those who had a response. Tumor response was evident in all subgroups of patients studied—those with pretreatment elevated lactate dehydrogenase levels (one of the strongest prognostic factors in metastatic melanoma), metastases to distant sites, and bulky, multifocal tumor burden. Based on these results, a phase 3 trial is now under way looking at the combination of these two medications vs either one alone.
In summary, targeted treatments are changing the paradigm of how common dermatologic conditions associated with significant morbidity and mortality are treated. Although implementation of the above treatments into everyday clinical practice is exciting, future studies surrounding each are needed to address unanswered issues, such as the optimal dosing and treatment schedules in terms of both disease response and inhibition of resistance, optimal patient/disease characteristics for use, and optimal drug treatment combinations. In the meantime, basic research still utilizing classic molecular biology techniques to uncover pathogenic disease mechanisms in even more detail is ongoing and hopefully will lead to development of even better targeted treatments or even cures for these diseases.
New targeted therapies are changing the way patients with advanced dermatologic diseases are treated. Innovative molecular biology techniques developed as far back as the 1970s have engendered tremendous insight into the cellular and molecular pathogenesis of numerous diseases. Novel medications based on these insights are now bearing fruit, as directed biologic therapies that are revolutionizing clinical practice are increasingly becoming available.
This article reviews advances in targeted therapies for advanced basal cell carcinoma, psoriasis, and metastatic melanoma.
TARGETED THERAPY FOR BASAL CELL CARCINOMA
Case 1. A 56-year-old man presents with a progressively enlarging leg ulcer. Although it has been treated empirically for years as a venous stasis ulcer, biopsy reveals that it is basal cell carcinoma. Imaging shows muscle and tendon invasion, making surgical intervention short of amputation challenging (Figure 1). What are his options?
Basal cell carcinoma is the most common cancer in humans, accounting for 25% of all cancers and more than 2 million cases in the United States every year. In most cases, surgical excision is curative, but a subset of patients have inoperable, locally advanced, or metastatic disease that drastically limits treatment options. The median survival in metastatic basal cell carcinoma is 24 months, and conventional chemotherapy has not been shown to improve the prognosis.1,2
In addition to the burden of sporadic basal cell carcinoma, patients with the rare autosomal-dominant genetic disorder basal cell nevus syndrome (Gorlin syndrome) develop multiple basal cell lesions over their lifetime. The syndrome may also involve abnormalities of the skeletal system, genitourinary tract, and central nervous system, including development of medulloblastoma.
In Gorlin syndrome, basal cell carcinomas occur often and early; about half of white patients with the syndrome develop their first lesions by age 21, and 90% by age 35. The lesions occur in multiple numbers and can develop anywhere on the body, including on non–sun-exposed areas. Patients who have Gorlin syndrome need meticulous monitoring every 2 to 3 months so that basal cell lesions can be recognized early and treated before they become locally advanced. Keeping up with the numerous medical appointments and invasive treatments can be physically and mentally taxing for patients.
Specific pathway and mutations identified
In 1996, Gorlin syndrome was found to be caused by mutations of the human homolog of the PATCHED gene, which codes for a receptor in the “hedgehog” pathway.3 Two years later, the same mutations were found to be involved in many sporadic basal cell carcinomas, and we now believe that at least 85% of basal cell carcinomas involve abnormal activation of hedgehog pathway signaling.4,5
Vismodegib developed as targeted therapy
In 2009, Robarge et al6 described a potent inhibitor of the hedgehog pathway that was later optimized for potency and desirable pharmacologic traits, resulting in the drug vismodegib.7,8
Two phase 2 multicenter clinical trials9,10 of vismodegib were published in 2012. In the first, which was not randomized,9 33 patients with metastatic basal cell carcinoma and 63 patients with locally advanced disease were treated with vismodegib. Of those with metastatic disease, 30% achieved an objective response. Of those with locally advanced disease, 43% achieved an objective response and 21% achieved a complete response.
In the second trial,10 patients with Gorlin syndrome were randomized to either vismodegib (26 patients) or placebo (16 patients). After 8 months, the vismodegib group had developed significantly fewer new surgically eligible tumors (2 vs 29 per year), their tumors were smaller (change from baseline of the sum of the longest diameters –65% vs –11%), and they needed fewer surgeries (mean 0.31 vs 4.4 per patient). No tumors progressed in the treatment group. Results in some patients were dramatic, with complete healing of large ulcerative tumors. The trial was ended early in view of significant efficacy in the treatment group.
Based on these trials, the US Food and Drug Administration (FDA) approved vismodegib for treating metastatic and locally advanced basal cell carcinoma.
Resistance and adverse effects common
Unfortunately, vismodegib has significant drawbacks. About 20% of patients develop resistance, with tumors recurring after several months of therapy.11 Adverse effects most commonly reported were muscle spasms (68%), alopecia (63%), taste distortion (51%), weight loss (46%), and fatigue (36%). Although these effects were considered mild or moderate, they tended to persist, and almost every patient developed at least one. In the nonrandomized trial,9 more than 25% of patients discontinued treatment because of adverse effects, and more than half of patients did the same in the basal cell nevus syndrome trial.10
New uses may reduce shortcomings
Studies are under way to determine how best to use vismodegib.
One possibility is to use the drug for a few months to shrink tumors to the point that they become eligible for surgery. This is especially important for high-risk tumors, such as those near the eye or other vital structures. In 11 patients, Ally et al12 found that the surgical defect area was reduced by 27% from baseline after 4 months of treatment with vismodegib, allowing for curative surgery in some.
Another option is to combine vismodegib with other agents—either new ones on the horizon or existing nonspecific medications. For example, the antifungal itraconazole has been shown to inhibit hedgehog signaling and perhaps could be combined with vismodegib to increase response and reduce resistance.
Finally, a topical or intralesional form of vismodegib would be useful not only to reduce systemic toxicity, but also to increase efficacy when combined with other topical or systemic medications.
TARGETED THERAPY FOR PSORIASIS VULGARIS
Case 2. A 28-year-old woman presents with worsening psoriasis. About 35% of her body surface is involved, including the palms and soles, making it difficult for her to perform activities of daily living (Figure 2). What are her options?
Psoriasis is a chronic immune-mediated disease that affects up to 3% of people worldwide. In its moderate to severe forms, we recognize psoriasis as a systemic inflammatory disease that may adversely affect organ systems other than the skin. Commonly associated comorbid diseases include inflammatory (psoriatic) arthritis, cardiovascular disease, malignancies (eg, lymphoma), and inflammatory bowel disease. In addition, patients are well known to have significantly impaired quality of life because of low self-esteem, stigmatization affecting their social and work relationships, and, in up to 60%, clinical depression.13,14 The onset of psoriatic arthritis, particularly of erosive disease, is an important decision point for starting aggressive treatment, as joint destruction is irreversible.
Early targeted therapy aimed at TNF alpha, IL-12, and IL-23
Histologically, psoriasis involves thickening of the epidermis caused by hyperproliferation of keratinocytes. Based on this, prior to the 1980s, the dominant hypothesis concerning its pathogenesis was that it was caused by an inherent defect of keratinocytes. In the 1980s and 1990s, however, molecular research revealed that psoriasis was an immune-mediated disease caused by immunologic dysregulation predominantly involving T-helper 1 (Th-1) cells, with the inflammatory cytokines tumor necrosis factor (TNF) alpha, interferon gamma, interleukin (IL) 12, and IL-23 playing prominent roles.15 These findings led to the development and FDA approval of the first effective, targeted, psoriasis treatments, TNF-alpha inhibitors and the IL-12/23 inhibitor ustekinumab.
Etanercept, the first TNF-alpha inhibitor to become available, was approved in 2004 for moderate to severe psoriasis. In 2008, the IL-12/23 inhibitor ustekinumab was approved for the same indication. These drugs are efficacious, are generally safe, and have revolutionized the treatment of psoriasis and psoriatic arthritis, and they are now prescribed on a daily basis.16,17
In the clinical trials that led to approval of these drugs, the main outcome measure was the Psoriasis Area and Severity Index (PASI), a clinical scoring tool that assesses clinical aspects of psoriatic disease including body surface area involvement, degree of thickness, erythema, and scaling of psoriatic plaques. PASI scores range from 0 (no psoriasis) to 72 (most severe psoriasis). Achieving “PASI 75” indicates at least 75% improvement from the baseline score and represents the most common primary outcome measure in clinical trials assessing efficacy of new treatments. Up to 80% of patients who received currently available TNF-alpha inhibitors and ustekinumab in pivotal clinical trials achieved PASI 75 when assessed at 12 to 16 weeks after starting treatment. A moderate percentage of patients (19%–57%, depending on the trial) achieved 90% improvement (PASI 90), and a minority (up to 18%) achieved PASI 100, indicating complete clearing of their psoriasis.18–22
Newly developed therapies target IL-17A
In the mid-2000s, Th-17 cells were discovered, a new lineage of T cells distinct from Th-1 and Th-2 cells. Th-17 cells are characterized by their production of IL-17, a pro-inflammatory cytokine with six family members (IL-17A through IL-17F). Over the next few years, experiments revealed that Th-17 cells and IL-17A play key roles in psoriasis immunologic dysregulation.15 These findings led to a paradigm shift in hypotheses concerning psoriasis pathogenesis, with Th-17 cells and IL-17 replacing Th-1 cells and associated cytokines as dominant mediators of tissue damage.
Additionally, these findings led to new ideas for treatment. Three monoclonal antibodies that target IL-17 inhibition are currently under investigation. Secukinumab and ixekizumab bind to IL-17A and inhibit it from downstream signaling, whereas brodalumab binds to the IL-17A receptor, blocking all six IL-17 cytokines (IL-17A to IL-17F).23
Clinical trials of IL-17 inhibitors show excellent skin improvement
Secukinumab. In 2014, the results of two phase 3 trials of secukinumab were published.24
In the Efficacy and Safety of Subcutaneous Secukinumab for Moderate to Severe Chronic Plaque-type Psoriasis for up to 1 Year trial,24 patients were given either secukinumab 300 mg or 150 mg subcutaneously at defined time points; 82% and 72%, respectively, attained PASI 75 at 12 weeks.
Similar results were seen in the Safety and Efficacy of Secukinumab Compared to Etanercept in Subjects With Moderate to Severe, Chronic Plaque-Type Psoriasis study,24 in which PASI 75 was achieved by 77% of patients receiving secukinumab 300 mg, 67% of those receiving secukinumab 150 mg, and only 44% of those receiving etanercept 50 mg twice weekly at 12 weeks. Rates of infection with secukinumab and etanercept were similar.
The most striking results of these trials were that more than half of patients receiving the 300-mg dose achieved at least 90% improvement in their PASI score (PASI 90) by week 12, and in more than a quarter of patients the psoriasis completely cleared (PASI 100). These results were dramatically better than for etanercept (PASI 90 21%; PASI 100 4%).
Additionally, secukinumab worked fast. The median time to PASI 50 with secukinumab 300 mg was less than half that seen with etanercept (3 weeks vs 7 weeks).
Ixekizumab. In 2012, a phase 2 trial evaluated subcutaneous injections of ixekizumab in dosages ranging from 10 to 150 mg at defined intervals for 16 weeks.25 Of those receiving the highest dosage, 82% attained PASI 75 at 12 weeks, on par with what is noted in patients receiving TNF-alpha inhibitors and IL-12/23 inhibitors. Remarkably, however, almost three-quarters of patients (71%) achieved PASI 90, and 39% achieved PASI 100. Improvement in psoriasis was apparent as early as 1 week after injection.
Brodalumab. A 2012 phase 2 trial of various dosages of the IL-17 receptor inhibitor brodalumab26 also showed excellent PASI 75 achievement with the highest dosage (82%). Astonishingly, though, PASI 90 was achieved by 75% of patients, and PASI 100 by 62%.
Overall, although the percentages of patients achieving PASI 75 with the new IL-17 inhibitor drugs are comparable to those seen with TNF-alpha inhibitors and IL-12/23 inhibitors, the extraordinarily high percentages of patients who achieved PASI 90 and PASI 100 are unprecedented.18–22
Arthritis improvement not shown
Where the IL-17 inhibitors eventually settle within algorithms of psoriasis treatment largely depends on their efficacy in treating psoriatic arthritis compared with TNF-alpha inhibitors and IL-12/23 inhibitors. Joint inflammation is typically evaluated with the American College of Rheumatology (ACR) scoring tool, which in simple terms can be thought of as analogous to the PASI scoring tool for the skin. Although the ACR scoring tool was developed to assess joint inflammation in clinical trials for patients with rheumatoid arthritis, it is commonly used to assess improvement of psoriatic arthritis in clinical trials. The ACR tool involves assessing and scoring the number of swollen and tender joints, but also incorporates serologic assessment of acute-phase reactants (erythrocyte sedimentation rate or C-reactive protein level), patient and physician global assessment, pain, and function. ACR 20 implies roughly a 20% improvement in these criteria, whereas ACR 50 indicates 50% improvement, and so on.
Two phase 2 trials of IL-17 inhibitors for psoriatic arthritis have been published, one with secukinumab27 and one with brodalumab.28 Neither had impressive improvement in the ACR score vs TNF inhibitors—39% for ACR 20 at week 12 and less than 10% for ACR 70. Clinical trial design may have played a role, and phase 3 trials are under way for all three IL-17 inhibitors.
Adverse effects of IL-17 inhibitors
For the most part, adverse effects reported with the IL-17 inhibitors have been mild and similar to those reported with available biologic treatments for psoriasis. Adverse effects most commonly reported have been nasopharyngitis, upper respiratory infection, arthralgia, and mild injection-site reactions. In the future, attention will be paid to the rate of infections known to be associated with IL-17, mainly localized infections with Staphylococcus aureus and Candida species. Some patients have developed Candida esophagitis, but this appears to resolve with discontinuation of the drugs. Neutropenia has occurred, but very few patients have developed grade 3 (500–1,000 cells/mm3) or worse. All adverse effects were reversible with discontinuation of treatment.
Approval of secukinumab, and current studies of IL-17 inhibitors
On January 21, 2015, secukinumab was approved by the FDA for treatment of moderate to severe psoriasis vulgaris in adult patients and is now available by prescription.
More trials of IL-17 inhibitors for the treatment of psoriasis and psoriatic arthritis are under way and are at various phases at the time of this writing.23
TARGETED THERAPY FOR ADVANCED MELANOMA
Case 3. A 58-year-old man presents with an irregular pigmented lesion on his back. Biopsy shows malignant melanoma with an intense, chronic inflammatory infiltrate surrounding the tumor (Figure 3). The tumor was surgically excised with standard margins. Two years later, the patient developed multiple pigmented lesions on the face and complained of headache. Magnetic resonance imaging of the brain revealed multiple enhancing lesions consistent with metastatic melanoma (Figure 3). What are this patient’s options?
Melanoma is the fifth most common cancer in humans, with about 132,000 new cases diagnosed worldwide each year and 48,000 deaths from advanced disease. Its incidence has risen rapidly over the last few decades. Advanced disease has a poor prognosis, with the median overall survival less than 1 year and 5-year survival less than 10%.
Despite decades of research, a paucity of FDA-approved medications were available to treat advanced melanoma until recently. The alkylating agent dacarbazine was approved in 1975, interferon alpha in 1995, and high-dose IL-2 in 1998. Although some patients respond, studies have not shown significant improvement in survival with any of these medications.29–31
In 2002, Davies et al32 found that 50% to 65% of metastatic cutaneous melanomas have a mutation in the BRAF gene. Interestingly, 80% of these patients share a single specific mutation: substitution of glutamic acid for valine in codon 600 (BRAF V600E). The second most common mutation is a single substitution of a lysine for that same valine (BRAF V600K). Additionally, NRAS is mutated in about 20% of melanomas. These discoveries implicated a mitogen-activated protein kinase (MAPK) pathway (Figure 4) as playing a critical role in metastatic melanoma for a large percentage of patients.29
Based on this knowledge, several targeted therapies for melanoma have been developed, and some have been approved.
BRAF inhibitors—first success against melanoma
Vemurafenib. In 2010, Flaherty et al33 reported on a phase 1 and phase 2 clinical trial of vemurafenib (960 mg orally twice daily), a potent inhibitor of BRAF with the V600E mutation. They demonstrated a clinical benefit in 80% of patients with stage IV BRAF-mutant melanoma, an unprecedented response that opened the door to changes in the treatment of metastatic melanoma.
The phase 3 BRAF Inhibitor in Melanoma (BRIM)-3 clinical trial,34 published in 2011, randomized 675 previously untreated patients with advanced melanoma to either vemurafenib 960 mg orally twice daily or dacarbazine, the standard of care. The trial was terminated early when an interim analysis showed a significant overall advantage for vemurafenib (median progression-free survival 5.3 months vs 1.6 months for dacarbazine). Based on these results, vemurafenib was FDA-approved in August 2011 for use in patients with BRAF-mutant melanoma.
Dabrafenib. In a phase 3 clinical trial in 2012, Hauschild et al35 randomized 250 patients with BRAF (V600E)-mutated melanoma in a 3:1 ratio to receive either dabrafenib, a more potent second-generation BRAF inhibitor, or dacarbazine. Half of patients responded to dabrafenib, with a significantly improved progression-free survival rate (5.1 vs 2.7 months respectively), leading to FDA approval for its use in BRAF-mutant melanoma in May 2013.
Adverse effects common to vemurafenib and dabrafenib include rash, fatigue, fever, and joint pain. In addition, up to 25% of patients develop multiple secondary cutaneous squamous cell carcinomas and keratoacanthomas, usually within the first few months of therapy, which are believed to be caused by paradoxical activation of the MAPK pathway.
A more important problem with these medications is the development of resistance. Tumors typically progress again after a median progression-free survival of 6 to 7 months.
MEK inhibitors—another line of defense
Inhibitors of MEK—a serine-threonine kinase that is part of the same MAPK pathway involving BRAF—have been developed as well.
Trametinib. In 2012, trametinib, an allosteric MEK inhibitor, was used in an open-label phase 3 trial in 322 patients with advanced melanoma. Progression-free survival was 4.8 months for trametinib-treated patients compared with 1.5 months for the standard chemotherapy group (dacarbazine or paclitaxel).36 These results led to FDA approval of trametinib in May 2013 for treating BRAF-mutant melanoma.29
Cobimetinib is a second MEK inhibitor being evaluated alone and in combination with other targeted treatments for advanced melanoma.
Both MEK inhibitors have adverse effects similar to those seen with the BRAF inhibitors, including diarrhea, rash, fatigue, and edema. They also tend to cause asymptomatic elevated creatine kinase and transient retinopathy, reduced ejection fraction, and ventricular dysfunction. Unlike BRAF inhibitors, they are not associated with development of secondary cutaneous squamous cell carcinomas or keratoacanthomas. However, as with BRAF inhibitors, resistance is a problem with MEK inhibitors, with most patients relapsing less than a year after starting therapy.
Combination therapy improves outcomes
Possible mechanisms underlying resistance to these medications are being studied. A number of important factors appear to drive resistance, including expression of truncated BRAF proteins that do not bind the BRAF inhibitors and still activate downstream signaling, and amplification of BRAF to such a degree that it overwhelms the medications. This has led to the idea of combining BRAF inhibitors and MEK inhibitors to block the MAPK pathway at two points, potentially increasing the response and decreasing resistance.
Two trials have evaluated combinations of BRAF and MEK inhibitors in patients with advanced melanoma. Larkin et al37 conducted a phase 3 study evaluating combined vemurafenib (a BRAF inhibitor) and cobimetinib (a MEK inhibitor) vs combined vemurafenib and placebo. Survival with the combination therapy was 9.9 months vs 6.2 months with the single treatment.
The incidence of serious adverse effects was not significantly increased with the combination therapy, and keratoacanthomas, cutaneous squamous cell carcinomas, alopecia, and arthralgias were reduced compared with the vemurafenib and placebo group.
Another trial38 evaluating combined dabrafenib (a BRAF inhibitor) and trametinib (a MEK inhibitor) vs combined dabrafenib and placebo had similar findings: increased survival in the combined therapy group (9.3 months vs 8.8 months) and lower rates of squamous cell carcinoma (2% vs 9%).
In January 2014, the FDA approved the combination of BRAF and MEK inhibitors for the treatment of BRAF-mutant metastatic melanoma based on improved survival and generally reduced adverse effects.
IMMUNOTHERAPIES FOR NON-BRAF MELANOMA
Although BRAF and MEK inhibitors represent tremendous advances, their use is limited to the approximately 50% to 65% of patients with advanced melanoma who have BRAF V600 mutations. For others, only the traditional standard medications have been available until recently.
Two of those standard FDA-approved medications, interferon alpha-2b and IL-2, represent immunotherapies. Interferon alpha-2b up-regulates antigen presentation and increases antigen recognition by T cells. Overall, about 20% of patients in clinical trials have achieved responses.
IL-2 is a cytokine that increases T-cell proliferation and maturation into effector T cells. High-dose IL-2 has produced responses in 15% of patients, with a durable complete response in a small proportion.
Though success with these medications was modest, the fact that some patients responded to them indicates that immunotherapy could be a viable strategy for treating metastatic melanoma.30 This is underscored by the fact that some patients can mount an adaptive immune response specifically directed against antigenic proteins expressed in their tumors, resulting in expansion of cytotoxic T cells and control or even elimination of the malignancy.30
Tumors manipulate host immune checkpoints
Molecular biology has provided tremendous insight into tumor immunology over the past several decades, and we now recognize that a hallmark of cancer is escape from immune control.
Cancer cells contain a multitude of mutations that produce proteins that should be recognized by the immune system as foreign but in most individuals are not. This is because T-cell activity is down-regulated in cancer due to cancer cells’ ability to manipulate the host’s normal immunologic inhibitory pathways critical for maintaining self-tolerance.
In general, T-cell activation is initiated when an antigen-presenting cell presents an antigen to a T cell in a major histocompatibility complex-restricted manner. To prevent T cells from being activated by self-antigens and initiating autoimmunity, the interaction between antigen-presenting cells and T cells is regulated by checkpoints (Figure 5). First, for an antigen-presenting cell/T-cell interaction to result in T-cell activation, the T-cell receptor CD28 must bind CD80 on the antigen-presenting cell to drive a “positive” signal. Early in the interaction, the T-cell receptor CTLA-4 is up-regulated and competes with CD28 for binding of CD80. If CTLA-4, and not CD28, binds CD80, a “negative” signal is sent to the T cell and down-regulates it, making the interaction unproductive. Importantly, it is the CTLA-4:CD80 interaction that appears to be crucial for the ability of tumors to dampen T-cell responses to cancer cells.
Ipilimumab is a fully humanized monoclonal antibody that binds to CTLA-4, blocking its ability to bind to CD80 and thereby enhancing T-cell activation. In a phase 3 trial, Hodi et al39 evaluated its use in treating advanced melanoma, with some enrolled patients having failed IL-2 treatment. Patients receiving ipilimumab with or without a glycoprotein-100 peptide vaccine (gp100) had an overall survival benefit of 10.1 months compared with 6.4 months for patients treated with gp100 alone. At 24 months, the survival rate with ipilimumab alone was 23.5%, almost double that of patients receiving gp100 alone.
Ipilimumab received FDA approval for treatment of metastatic melanoma in March 2011. This, and the BRAF inhibitors, were the first drugs approved by the FDA for the treatment of advanced melanoma in more than a decade.
Common adverse effects of ipilimumab include fatigue, diarrhea, rash, and pruritus. As expected, given its mechanism of action, up to about 25% of patients experience severe autoimmune-related events that may variably manifest as colitis, rash, hepatitis, neuritis, hypothyroidism, hypopituitarism, and hypophysitis. Another problem with this medication is that a subset of patients do not respond.
Cancer cells disguised as normal cells
Cancer cells can also manipulate another immunologic checkpoint to evade attack by the host immune system (Figure 5). Cytotoxic T cells may recognize antigens on tumor cells and become activated and primed to directly destroy them. However, tumor cells, like normal cells express the programmed death ligands RTK-L1 and PD-L2. These ligands function to bind to the PD-1 receptor on activated T cells to indicate they are “self” and inhibit the cytotoxic T cells from destroying them.
Evasion of immune system attack by manipulating checkpoints involving CTLA-4 and PD-1 helps explain why malignancies can seemingly be associated with brisk inflammatory responses, such as the tumor in Case 3, yet progress and eventually metastasize (Figure 3).
Two medications—nivolumab and pembrolizumab—have been developed in an attempt to disrupt the ability of tumor cells to trick the immune system into accepting them as “self” by manipulating the PD-L1/PD-L2: PD-1 interaction. Both drugs are monoclonal antibodies that bind to PD-1 and, thus, effectively block the ability of PD-L1 or PD-L2 on tumor cells to bind these ligands and signal to activated T cells that they are “self.” This blocking allows T cells to then carry out their killing of tumor cells they initially recognize as foreign.
Nivolumab. In 2014, a phase 3 trial40 compared nivolumab and dacarbazine in patients with untreated advanced melanoma without a BRAF mutation. Objective response rates were 40.0% in the nivolumab group vs 13.9% in the dacarbazine group. This trial was stopped early because of significantly better survival rates in patients taking nivolumab compared with standard chemotherapy.
Interestingly, only 35% of patients who responded to nivolumab had evidence of PD-L1 expression on the surface of their tumor cells as assessed by immunohistochemical assay. Regardless of PD-L1 status, nivolumab-treated patients had improved overall survival compared with those treated with dacarbazine. The response rate with nivolumab was only slightly better in the subgroup of patients whose tumors expressed PD-L1 than in the subgroup without PD-L1.
On December 22, 2014, the FDA granted accelerated approval to nivolumab for the treatment of patients with unresectable or metastatic melanoma and disease progression following ipilimumab treatment and, if BRAF V600 mutation-positive, a BRAF inhibitor.
Pembrolizumab. Also in 2014, an open-label, randomized, phase 1b trial of pembrolizumab treatment at two different dosage schedules was conducted in patients with advanced melanoma that had become refractory either to ipilimumab or a BRAF inhibitor.41 Treatment with pembrolizumab had an objective response rate of 26% at both doses.
In September 2014, the FDA granted accelerated approval for the use of pembrolizumab to treat patients with unresectable or metastatic melanoma and disease progression following treatment with ipilimumab or a BRAF inhibitor.
Adverse effects of PD-1 inhibitors are similar to those seen with ipilimumab, the most common (occurring in at least 20%) being fatigue, cough, nausea, pruritus, rash, decreased appetite, constipation, muscle pain, and diarrhea. Serious effects from pembrolizumab (occurring in at least 2%) were kidney failure, dyspnea, pneumonia, and cellulitis. As seen with ipilimumab, clinically significant autoimmune adverse reactions occur with PD-1 inhibitors, including pneumonitis, colitis, hypophysitis, nephritis, and hepatitis.
Combination therapy under investigation
A phase 1 trial using combination therapy with both immune checkpoint inhibitors—nivolumab (anti-PD-1) and ipilimumab (anti-CTLA-4)—in patients with treatment-resistant metastatic melanoma was published in 2013.42 More than half of patients achieved objective responses, with tumor regression of at least 80% in those who had a response. Tumor response was evident in all subgroups of patients studied—those with pretreatment elevated lactate dehydrogenase levels (one of the strongest prognostic factors in metastatic melanoma), metastases to distant sites, and bulky, multifocal tumor burden. Based on these results, a phase 3 trial is now under way looking at the combination of these two medications vs either one alone.
In summary, targeted treatments are changing the paradigm of how common dermatologic conditions associated with significant morbidity and mortality are treated. Although implementation of the above treatments into everyday clinical practice is exciting, future studies surrounding each are needed to address unanswered issues, such as the optimal dosing and treatment schedules in terms of both disease response and inhibition of resistance, optimal patient/disease characteristics for use, and optimal drug treatment combinations. In the meantime, basic research still utilizing classic molecular biology techniques to uncover pathogenic disease mechanisms in even more detail is ongoing and hopefully will lead to development of even better targeted treatments or even cures for these diseases.
- Lyons TG, O’Kane GM, Kelly CM. Efficacy and safety of vismodegib: a new therapeutic agent in the treatment of basal cell carcinoma. Expert Opin Drug Saf 2014; 13:1125–1132.
- McCusker M, Basset-Sequin N, Dummer R, et al. Metastatic basal cell carcinoma: prognosis dependent on anatomic site and spread of disease. Eur J Cancer 2014; 50:774–783.
- Hahn H, Wicking C, Zaphiropoulous PG, et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 1996; 85:841–851.
- Aszterbaum M, Rothman A, Johnson RL, et al. Identification of mutations in the human PATCHED gene in sporadic basal cell carcinomas and in patients with the basal cell nevus syndrome. J Invest Dermatol 1998; 110:885–888.
- Ingham PW, Placzek M. Orchestrating ontogenesis: variations on a theme by sonic hedgehog. Nat Rev Genet 2006; 7:841–850.
- Robarge KD, Brunton SA, Castanedo GM, et al. GDC-0449-a potent inhibitor of the hedgehog pathway. Bioorg Med Chem Lett 2009; 19:5576–5581.
- Proctor AE, Thompson LA, O’Bryant CL. Vismodegib: an inhibitor of the Hedgehog signaling pathway in the treatment of basal cell carcinoma. Ann Pharmacother 2014; 48:99–106.
- Dessinioti C, Plaka M, Stratigos AJ. Vismodegib for the treatment of basal cell carcinoma: results and implications of the ERIVANCE BCC trial. Future Oncol 2014; 10:927–936.
- Sekulic A, Migden MR, Oro AE, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med 2012; 366:2171–2179.
- Tang JY, Mackay-Wiggan JM, Aszterbaum M, et al. Inhibiting the hedgehog pathway in patients with basal-cell nevus syndrome. N Engl J Med 2012; 366:2180–2188.
- Brinkhuizen T, Reinders MG, van Geel M, et al. Acquired resistance to the Hedgehog pathway inhibitor vismodegib due to smoothened mutations in treatment of locally advanced basal cell carcinoma. J Am Acad Dermatol 2014; 71:1005–1008.
- Ally MS, Aasi S, Wysong A, et al. An investigator-initiated open-label clinical trial of vismodegib as a neoadjuvant to surgery for high-risk basal cell carcinoma. J Am Acad Dermatol 2014; 71:904–911.
- Rapp SR, Feldman SR, Exum ML, Fleischer AB Jr, Reboussin DM. Psoriasis causes as much disability as other major medical diseases. J Am Acad Dermatol 1999; 41:401–407.
- Gelfand JM, Niemann AL, Shin DB, Wang X, Margolis DJ, Troxel AB. Risk of myocardial infarction in patients with psoriasis. JAMA 2006; 296:1735–1741.
- Lynde CW, Poulin Y, Vender R, Bourcier M, Khalil S. Interleukin 17A: toward a new understanding of psoriasis pathogenesis. J Am Acad Dermatol 2014; 71:141–150.
- Tracey D, Klareskog L, Sasso EH, Salfeld JG, Tak PP. Tumor necrosis factor antagonist mechanisms of action: a comprehensive review. Pharmacol Ther 2008; 117:244–279.
- Nestle FL, Kaplan DH, Barker J. Psoriasis. N Engl J Med 2009; 361:496–509.
- Mentor A, Tyring SK, Gordon K, et al. Adalimumab therapy for moderate to severe psoriasis: a randomized, controlled phase III trial. J Am Acad Dermatol 2007; 58:106–115.
- Leonardi CL, Powers JL, Matheson RT, et al. Etanercept as monotherapy in patients with psoriasis. N Engl J Med 2003; 349:2014–2022.
- Reich K, Nestle FO, Papp K, et al. Infliximab induction and maintenance therapy for moderate-to-severe psoriasis: a phase III, multicentre, double-blind trial. Lancet 2005; 366:1367–1374.
- Leonardi CL, Kimball AB, Papp KA, et al. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 76-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 1). Lancet 2008; 371:1665–1674.
- Papp KA, Langley RG, Lebwohl M, et al; PHOENIX 2 study investigators. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 52-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 2). Lancet 2008; 371:1675–1684.
- Leonardi CL, Gordon KB. New and emerging therapies in psoriasis. Semin Cut Med Surg 2014; 33(suppl 2):S37–S41.
- Langley RG, Elewski BE, Lebwohl, et al for the ERASURE and FIXTURE Study Groups. Secukinumab in plaque psorisis—results of two phase 3 trials. N Engl J Med 2014; 371:326–338.
- Leonardi C, Matheson R, Zachariae C. Anti-interleukin-17 monoclonal antibody ixekizumab in chronic plaque psoriasis. N Engl J Med 2012; 366:1190–1199.
- Papp KA, Leonardi C, Menter A, et al. Brodalumab, an anti-interleukin-17-receptor antibody for psoriasis. N Engl J Med 2012; 366:1181–1189.
- McInnes IB, Sieper J, Braun J, et al. Efficacy and safety of secukinumab, a fully human anti-interleukin-17A monoclonal antibody, in patients with moderate-to-severe psoriatic arthritis: a 24-week, randomised, double-blind, placebo-controlled, phase II proof-of-concept trial. Ann Rheum Dis 2014; 73:349–356.
- Mease PJ, Genovese MC, Greenwald MW, et al. Brodalumab, an anti-IL17RA monoclonal antibody, in psoriatic arthritis. N Engl J Med 2014; 370:2295–2306.
- Girotti MR, Saturno G, Lorigan P, Marais R. No longer an untreatable disease: how targeted and immunotherapies have changed the management of melanoma patients. Molec Oncol 2014, 8:1140–1158.
- Saranga-Perry V, Ambe C, Zager JS, Kudchadkar RR. Recent developments in the medical and surgical treatment of melanoma. CA Canc J Clin 2014; 64:171–185.
- Shah DJ, Dronca RS. Latest advances in chemotherapeutic, targeted, and immune approaches in the treatment of metastatic melanoma. Mayo Clin Proc 2014; 89:504–519.
- Davies H, Ignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature 2002; 417:949–954.
- Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med 2010; 363:809–819.
- Chapman PB, Hauschild A, Robert C. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011; 364:2507–2516.
- Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012; 380:358–365.
- Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med 2012; 367:107–114.
- Larkin J, Ascierto PA, Dreno B, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med 2014; 371:1867–1876.
- Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Eng J Med 2014; 371:1877–1888.
- Hodi FS, O’Day SJ, McDermott DF, Weber RW. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010; 363:711–723.
- Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015; 372:320–330.
- Robert C, Ribas A, Wolchok JD, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet 2014; 384:1109–1117.
- Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med 2013; 369:122–133.
- Lyons TG, O’Kane GM, Kelly CM. Efficacy and safety of vismodegib: a new therapeutic agent in the treatment of basal cell carcinoma. Expert Opin Drug Saf 2014; 13:1125–1132.
- McCusker M, Basset-Sequin N, Dummer R, et al. Metastatic basal cell carcinoma: prognosis dependent on anatomic site and spread of disease. Eur J Cancer 2014; 50:774–783.
- Hahn H, Wicking C, Zaphiropoulous PG, et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 1996; 85:841–851.
- Aszterbaum M, Rothman A, Johnson RL, et al. Identification of mutations in the human PATCHED gene in sporadic basal cell carcinomas and in patients with the basal cell nevus syndrome. J Invest Dermatol 1998; 110:885–888.
- Ingham PW, Placzek M. Orchestrating ontogenesis: variations on a theme by sonic hedgehog. Nat Rev Genet 2006; 7:841–850.
- Robarge KD, Brunton SA, Castanedo GM, et al. GDC-0449-a potent inhibitor of the hedgehog pathway. Bioorg Med Chem Lett 2009; 19:5576–5581.
- Proctor AE, Thompson LA, O’Bryant CL. Vismodegib: an inhibitor of the Hedgehog signaling pathway in the treatment of basal cell carcinoma. Ann Pharmacother 2014; 48:99–106.
- Dessinioti C, Plaka M, Stratigos AJ. Vismodegib for the treatment of basal cell carcinoma: results and implications of the ERIVANCE BCC trial. Future Oncol 2014; 10:927–936.
- Sekulic A, Migden MR, Oro AE, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med 2012; 366:2171–2179.
- Tang JY, Mackay-Wiggan JM, Aszterbaum M, et al. Inhibiting the hedgehog pathway in patients with basal-cell nevus syndrome. N Engl J Med 2012; 366:2180–2188.
- Brinkhuizen T, Reinders MG, van Geel M, et al. Acquired resistance to the Hedgehog pathway inhibitor vismodegib due to smoothened mutations in treatment of locally advanced basal cell carcinoma. J Am Acad Dermatol 2014; 71:1005–1008.
- Ally MS, Aasi S, Wysong A, et al. An investigator-initiated open-label clinical trial of vismodegib as a neoadjuvant to surgery for high-risk basal cell carcinoma. J Am Acad Dermatol 2014; 71:904–911.
- Rapp SR, Feldman SR, Exum ML, Fleischer AB Jr, Reboussin DM. Psoriasis causes as much disability as other major medical diseases. J Am Acad Dermatol 1999; 41:401–407.
- Gelfand JM, Niemann AL, Shin DB, Wang X, Margolis DJ, Troxel AB. Risk of myocardial infarction in patients with psoriasis. JAMA 2006; 296:1735–1741.
- Lynde CW, Poulin Y, Vender R, Bourcier M, Khalil S. Interleukin 17A: toward a new understanding of psoriasis pathogenesis. J Am Acad Dermatol 2014; 71:141–150.
- Tracey D, Klareskog L, Sasso EH, Salfeld JG, Tak PP. Tumor necrosis factor antagonist mechanisms of action: a comprehensive review. Pharmacol Ther 2008; 117:244–279.
- Nestle FL, Kaplan DH, Barker J. Psoriasis. N Engl J Med 2009; 361:496–509.
- Mentor A, Tyring SK, Gordon K, et al. Adalimumab therapy for moderate to severe psoriasis: a randomized, controlled phase III trial. J Am Acad Dermatol 2007; 58:106–115.
- Leonardi CL, Powers JL, Matheson RT, et al. Etanercept as monotherapy in patients with psoriasis. N Engl J Med 2003; 349:2014–2022.
- Reich K, Nestle FO, Papp K, et al. Infliximab induction and maintenance therapy for moderate-to-severe psoriasis: a phase III, multicentre, double-blind trial. Lancet 2005; 366:1367–1374.
- Leonardi CL, Kimball AB, Papp KA, et al. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 76-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 1). Lancet 2008; 371:1665–1674.
- Papp KA, Langley RG, Lebwohl M, et al; PHOENIX 2 study investigators. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 52-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 2). Lancet 2008; 371:1675–1684.
- Leonardi CL, Gordon KB. New and emerging therapies in psoriasis. Semin Cut Med Surg 2014; 33(suppl 2):S37–S41.
- Langley RG, Elewski BE, Lebwohl, et al for the ERASURE and FIXTURE Study Groups. Secukinumab in plaque psorisis—results of two phase 3 trials. N Engl J Med 2014; 371:326–338.
- Leonardi C, Matheson R, Zachariae C. Anti-interleukin-17 monoclonal antibody ixekizumab in chronic plaque psoriasis. N Engl J Med 2012; 366:1190–1199.
- Papp KA, Leonardi C, Menter A, et al. Brodalumab, an anti-interleukin-17-receptor antibody for psoriasis. N Engl J Med 2012; 366:1181–1189.
- McInnes IB, Sieper J, Braun J, et al. Efficacy and safety of secukinumab, a fully human anti-interleukin-17A monoclonal antibody, in patients with moderate-to-severe psoriatic arthritis: a 24-week, randomised, double-blind, placebo-controlled, phase II proof-of-concept trial. Ann Rheum Dis 2014; 73:349–356.
- Mease PJ, Genovese MC, Greenwald MW, et al. Brodalumab, an anti-IL17RA monoclonal antibody, in psoriatic arthritis. N Engl J Med 2014; 370:2295–2306.
- Girotti MR, Saturno G, Lorigan P, Marais R. No longer an untreatable disease: how targeted and immunotherapies have changed the management of melanoma patients. Molec Oncol 2014, 8:1140–1158.
- Saranga-Perry V, Ambe C, Zager JS, Kudchadkar RR. Recent developments in the medical and surgical treatment of melanoma. CA Canc J Clin 2014; 64:171–185.
- Shah DJ, Dronca RS. Latest advances in chemotherapeutic, targeted, and immune approaches in the treatment of metastatic melanoma. Mayo Clin Proc 2014; 89:504–519.
- Davies H, Ignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature 2002; 417:949–954.
- Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med 2010; 363:809–819.
- Chapman PB, Hauschild A, Robert C. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011; 364:2507–2516.
- Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012; 380:358–365.
- Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med 2012; 367:107–114.
- Larkin J, Ascierto PA, Dreno B, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med 2014; 371:1867–1876.
- Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Eng J Med 2014; 371:1877–1888.
- Hodi FS, O’Day SJ, McDermott DF, Weber RW. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010; 363:711–723.
- Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015; 372:320–330.
- Robert C, Ribas A, Wolchok JD, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet 2014; 384:1109–1117.
- Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med 2013; 369:122–133.
KEY POINTS
- Vismodegib, an inhibitor of the “hedgehog” pathway, dramatically shrinks basal cell carcinomas, but resistance and adverse effects remain troublesome. Using it to shrink tumors to operable size may be its best future role.
- Th-17 cells and interleukin 17 are now thought to play central roles in the pathogenesis of psoriasis. Clinical trials of new drugs that block interleukin 17 show striking improvement in skin manifestations with few side effects. Benefits in psoriatic arthritis have not yet been shown.
- About half of patients with melanoma harbor BRAF mutations, and new treatments that target this pathway have improved survival rates. For melanoma not involving BRAF mutations, a better understanding of how tumors evade immune control has led to improved immunotherapies. These targeted medications mark the first major advancements in metastatic melanoma treatment in decades.
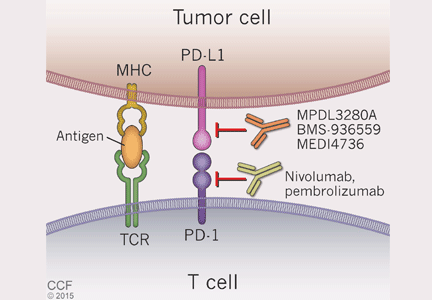
Penicillin allergy: A practical guide for clinicians
Most patients who report that they are allergic to penicillin can ultimately receive penicillin or a penicillin-type antibiotic again after an appropriate evaluation and, possibly, treatment. This course of action decreases the need for broad-spectrum antibiotics,1–4 reduces health care costs, and prevents the development of multidrug-resistant pathogens.5
About 10% of the general population say that they are allergic to penicillin.1,6,7 Although the prevalence of life-threatening anaphylactic reactions to penicillin has been estimated to be between 0.02% and 0.04%,6 the most common reaction is a cutaneous eruption. Since anaphylactic reactions are mediated by immunoglobulin E (IgE), evaluation of patients with a history of penicillin allergy by penicillin skin testing is recommended to rule out IgE-mediated reactions.
This review outlines a practical approach to evaluating a suspected IgE-mediated reaction to penicillin, with key points in the history and diagnostic testing. We also review subsequent management and cross-reactivity with other beta-lactam-containing antibiotics.
EVALUATING ALLERGIC PATIENTS
Evaluation of patients with a history of penicillin allergy can be improved with an understanding of the classification of drug reactions, risk factors for allergy, and the pathophysiology of penicillin allergy.
Classification of drug reactions
Adverse drug reactions include all unintended pharmacologic effects of a drug and can be classified as predictable (type A) or unpredictable (type B). Predictable reactions are dose-dependent, are related to the known pharmacologic actions of the medication, and occur in otherwise healthy individuals. Unpredictable reactions are further classified into drug intolerance, drug idiosyncrasy, drug allergy, and pseudoallergic reactions.8,9

Penicillin allergy can manifest as any hypersensitivity reaction of the Gell and Coombs classification (Table 1).9 Type I (immediate) and type IV (delayed) reactions are the most common types of reactions that occur with antibiotics and should be classified based on the onset of symptoms as immediate (within 1 hour) or delayed (days or weeks).8
Risk factors for IgE-mediated reaction
Risk factors for a hypersensitivity reaction include frequent or repetitive courses of penicillin10 and high-dose parenteral (rather than oral) administration.
Age and atopy are not risk factors for penicillin allergy.7 However, atopy increases the risk of a more severe anaphylactic reaction to penicillin, and anaphylactic reactions are most commonly reported between the ages of 20 and 49.6
Pathophysiology of penicillin allergy
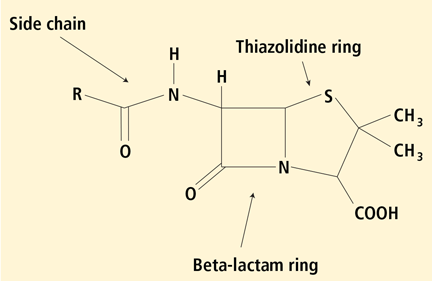
All penicillins share a common core ring structure (beta-lactam and thiazolidine rings) but differ in their side chains (R group) (Figure 1).
Under physiologic conditions, the core ring structure is metabolized into major (penicilloyl) and minor (penicillin itself, penicilloate and penilloate) antigenic determinants that may trigger an immediate IgE-dependent response.9 In the United States, commercial forms of antigenic determinates for skin testing exist in the form of penicillin G (minor determinant) and penicilloyl-polylysine, better known as Prepen (major determinant).
Immediate-type reactions to similar antibiotics such as aminopenicillins and cephalosporins may be caused by IgE antibodies against the R-group side chain rather than the core penicillin major and minor determinants.11
Questions to ask patients who have a history of penicillin allergy
Patients should be questioned closely about previous and current reactions to penicillin and should undergo skin-prick and intradermal testing, followed by graded-dose challenge or drug tolerance desensitization (Figure 2).
Questions to ask patients who have a history of penicillin allergy (Table 2)9,12 include the following:

Do you remember the details of the reaction? These include the route of administration, the time between the dose of penicillin and the appearance of symptoms, and how the reaction was managed.
Immediate reactions (ie, IgE-mediated, or Gell and Coombs type I) usually occur within the first hour after the first dose of the antibiotic, although they occasionally take up to 2 hours to occur, especially if the medication is taken orally and is taken with food. Symptoms consistent with IgE-mediated reactions include urticaria (most common), pruritus, angioedema, laryngeal edema, wheezing, shortness of breath, presyncope or syncope, hypotension, and cardiorespiratory collapse.
In contrast, symptoms of a non–IgE-mediated reaction are delayed in onset, occurring after days of treatment. They include nonpruritic maculopapular eruptions, hemolytic anemia, serum sickness, Stevens-Johnson syndrome, drug rash with eosinophilia and systemic symptoms, acute interstitial nephritis, and toxic epidermal necrolysis.9
If the patient has had severe non–IgE-mediated reactions to penicillin (eg, Stevens-Johnson syndrome, toxic epidermal necrolysis, acute interstitial nephritis, hemolytic anemia, or serum sickness) in the past, skin testing, graded-dose challenge, and desensitization are contraindicated.
How many years ago did the reaction occur? Most patients lose their sensitivity to penicillin over time.7,13–15 Nearly 50% of patients with IgE-mediated penicillin allergy lose their sensitivity within 5 years of the reaction,15 increasing to 80% or more by 10 years.13
How was the reaction managed? What was the outcome? Use of and positive response to epinephrine and histamine 1 receptor antagonists (antihistamines) with resolution or significant improvement of symptoms within a few hours may indicate an IgE-mediated reaction.
What was the indication for penicillin? Many cutaneous reactions are a result of an underlying viral or bacterial infection. For example, up to 90% of patients with Epstein-Barr virus infection develop a maculopapular rash when given penicillin.16
Have you tolerated other forms of penicillin since the reaction? Sometimes the patient has already tolerated other beta-lactams such as aminopenicillins, cephalosporins, and semisynthetic penicillins (piperacillin-tazobactam). Patients who tolerate other beta-lactams without adverse reactions are not allergic to beta-lactams.
Diagnostic tests
Skin testing. The only validated test for diagnosing IgE-mediated reactions caused by penicillin is the immediate hypersensitivity skin test,9 which should be performed by a board-certified allergist. The test consists of skin-prick and intradermal testing with the major determinant (penicilloyl-polylysine), the minor determinant (penicillin G), a negative control (normal saline), and a positive control (histamine). Minor-determinant mix is not commercially available in the United States.
Results of skin-prick testing are read 15 minutes after application. A positive response is a wheal at least 3 mm larger in diameter (with equivalent erythema) than the negative control done simultaneously. Intradermal testing is only done after a negative skin-prick test. If the allergic reaction was severe (ie, anaphylaxis), skin testing should be done at least 4 to 6 weeks after the reaction.
A history of severe non–IgE-mediated reaction to penicillin is a contraindication to skin-prick testing for penicillin allergy. The positive predictive value of penicillin skin testing is 50%, and the negative predictive value is 97%.3,7,9,13
Commercial in vitro testing (serum-specific IgE assays) for IgE-mediated hypersensitivity to penicillin is inferior to skin testing in terms of the negative predictive value and is not a suitable substitute for penicillin skin testing.
MANAGING PENICILLIN ALLERGY
If skin testing is positive, use another antibiotic, or refer for desensitization
If penicillin skin testing is positive (Figure 2), use another antibiotic that is equally efficacious. Patients who absolutely need a beta-lactam may undergo drug desensitization, performed by a board-certified allergist.
During desensitization, patients receive progressively higher doses of the drug every 15 to 20 minutes subcutaneously or intravenously, or every 20 to 30 minutes orally, until a full therapeutic dose is tolerated. Most protocols begin with a dose ranging from 1/10,000 to 1/1,000 of the final dose, depending on the severity of the allergic reaction.9,17
Using modern protocols, the success rate for tolerance induction is extremely high (75% to 100% in patients with cystic fibrosis, a group with a high rate of drug allergy18–20).
Drug desensitization is contraindicated in patients with non–IgE-mediated reactions.
If skin testing is negative, refer for graded-dose challenge
If skin testing is negative (Figure 2), graded-dose challenge is recommended. This procedure must be done by a board-certified allergist. If the original reaction was life-threatening, graded-dose challenge may entail giving 1/100 of the therapeutic dose. Then, if no reaction occurs during a brief observation period (usually 30 minutes), a full dose is given. However, many patients can start with 1/10 or even a full dose of the drug, especially if the original reaction was limited to the skin and the penicillin skin test is negative.
Graded-dose challenge is contraindicated if the original reaction was a severe non–IgE-mediated reaction.
UNDERSTANDING CROSS-REACTIVITY OF PENICILLIN

Penicillin is the only antibiotic for which skin testing is reliable and validated. If a drug that cross-reacts with penicillin is needed, it is important to know the rate of cross-reactivity (Table 3). The rate of cross-reactivity between penicillin and aminopenicillins (amoxicillin and ampicillin) is less than 1.3% in the United States.10,21 However, the cross-reactivity rate among aminopenicillins and cephalosporins is between 10% to 40%. For that reason, patients with prior reactions to aminopenicillins should avoid cephalosporins that share identical R-chain side groups with aminopenicillins.9,22
The rate of cross-reactivity between penicillin and cephalosporins was reported as 10% 40 years ago.23,24 But this was with early, first-generation cephalosporins that may have been contaminated with penicillin. The cross-reactivity rate with cephalosporins today is 3%.25 In general, first- and second-generation cephalosporins cause more allergic reactions than third- and fourth-generation cephalosporins.26
Patients with a history of penicillin allergy who require a cephalosporin should still undergo penicillin skin testing. Skin testing with cephalosporins has not been validated. However, skin testing with nonirritating concentrations of cephalosporins9 may be done to elucidate IgE reactions.
In a study by Romano et al,27 110 patients who had positive results on penicillin skin testing completed graded-dose challenge with the carbapenem antibiotic imipenem. The rate of cross-reactivity between penicillin and imipenem was less than 1%.
Monobactam antibiotics do not cross-react with other beta-lactams, except ceftazidime with aztreonam. This is probably because of similarities in their chemical structure.
- Park M, Markus P, Matesic D, Li JT. Safety and effectiveness of a preoperative allergy clinic in decreasing vancomycin use in patients with a history of penicillin allergy. Ann Allergy Asthma Immunol 2006; 97:681–687.
- Arroliga ME, Radojicic C, Gordon SM, et al. A prospective observational study of the effect of penicillin skin testing on antibiotic use in the intensive care unit. Infect Control Hosp Epidemiol 2003; 24:347–350.
- del Real GA, Rose ME, Ramirez-Atamoros MT, et al. Penicillin skin testing in patients with a history of beta-lactam allergy. Ann Allergy Asthma Immunol 2007; 98:355–359.
- Nadarajah K, Green GR, Naglak M. Clinical outcomes of penicillin skin testing. Ann Allergy Asthma Immunol 2005; 95:541–545.
- Harris AD, Sauberman L, Kabbash L, Greineder DK, Samore MH. Penicillin skin testing: a way to optimize antibiotic utilization. Am J Med 1999; 107:166–168.
- Idsoe O, Guthe T, Willcox RR, de Weck AL. Nature and extent of penicillin side-reactions, with particular reference to fatalities from anaphylactic shock. Bull World Health Organ 1968; 38:159–188.
- Gadde J, Spence M, Wheeler B, Adkinson NF Jr. Clinical experience with penicillin skin testing in a large inner-city STD clinic. JAMA 1993; 270:2456–2463.
- Johansson SG, Bieber T, Dahl R, et al. Revised nomenclature for allergy for global use: report of the Nomenclature Review Committee of the World Allergy Organization, October 2003. J Allergy Clin Immunol 2004; 113:832–836.
- Joint Task Force on Practice Parameters; American Academy of Allergy, Asthma and Immunology; American College of Allergy, Asthma and Immunology; Joint Council of Allergy, Asthma and Immunology. Drug allergy: an updated practice parameter. Ann Allergy Asthma Immunol 2010; 105:259–273.
- Park MA, Matesic D, Markus PJ, Li JT. Female sex as a risk factor for penicillin allergy. Ann Allergy Asthma Immunol 2007; 99:54–58.
- Moreno E, Macias E, Davila I, Laffond E, Ruiz A, Lorente F. Hypersensitivity reactions to cephalosporins. Expert Opin Drug Saf 2008; 7:295–304.
- Salkind AR, Cuddy PG, Foxworth JW. The rational clinical examination. Is this patient allergic to penicillin? An evidence-based analysis of the likelihood of penicillin allergy. JAMA 2001; 285:2498–2505.
- Sullivan TJ, Wedner HJ, Shatz GS, Yecies LD, Parker CW. Skin testing to detect penicillin allergy. J Allergy Clin Immunol 1981; 68:171–180.
- Macy E, Schatz M, Lin C, Poon KY. The falling rate of positive penicillin skin tests from 1995 to 2007. Perm J 2009; 13:12–18.
- Blanca M, Torres MJ, Garcia JJ, et al. Natural evolution of skin test sensitivity in patients allergic to beta-lactam antibiotics. J Allergy Clin Immunol 1999; 103:918–924.
- Patel BM. Skin rash with infectious mononucleosis and ampicillin. Pediatrics 1967; 40:910–911.
- Liu A, Fanning L, Chong H, et al. Desensitization regimens for drug allergy: state of the art in the 21st century. Clin Exp Allergy 2011; 41:1679–1688.
- Burrows JA, Toon M, Bell SC. Antibiotic desensitization in adults with cystic fibrosis. Respirology 2003; 8:359–364.
- Turvey SE, Cronin B, Arnold AD, Dioun AF. Antibiotic desensitization for the allergic patient: 5 years of experience and practice. Ann Allergy Asthma Immunol 2004; 92:426–432.
- Legere HJ 3rd, Palis RI, Rodriguez Bouza T, Uluer AZ, Castells MC. A safe protocol for rapid desensitization in patients with cystic fibrosis and antibiotic hypersensitivity. J Cyst Fibros 2009; 8:418–424.
- Lin E, Saxon A, Riedl M. Penicillin allergy: value of including amoxicillin as a determinant in penicillin skin testing. Int Arch Allergy Immunol 2010; 152:313–318.
- Dickson SD, Salazar KC. Diagnosis and management of immediate hypersensitivity reactions to cephalosporins. Clin Rev Allergy Immunol 2013; 45:131–142.
- Dash CH. Penicillin allergy and the cephalosporins. J Antimicrob Chemother 1975; 1(suppl 3):107–118.
- Petz LD. Immunologic cross-reactivity between penicillins and cephalosporins: a review. J Infect Dis 1978; 137(suppl):S74–S79.
- American Academy of Allergy Asthma & Immunology. Cephalosporin administration to patients with a history of penicillin allergy. www.aaaai.org/Aaaai/media/MediaLibrary/PDF%20Documents/Practice%20and%20Parameters/Cephalosporin-administration-2009.pdf. Accessed April 2, 2015.
- Fonacier L, Hirschberg R, Gerson S. Adverse drug reactions to cephalosporins in hospitalized patients with a history of penicillin allergy. Allergy Asthma Proc 2005; 26:135–141.
- Romano A, Viola M, Gueant-Rodriguez RM, Gaeta F, Pettinato R, Gueant JL. Imipenem in patients with immediate hypersensitivity to penicillins. N Engl J Med 2006; 354:2835–2837.
Most patients who report that they are allergic to penicillin can ultimately receive penicillin or a penicillin-type antibiotic again after an appropriate evaluation and, possibly, treatment. This course of action decreases the need for broad-spectrum antibiotics,1–4 reduces health care costs, and prevents the development of multidrug-resistant pathogens.5
About 10% of the general population say that they are allergic to penicillin.1,6,7 Although the prevalence of life-threatening anaphylactic reactions to penicillin has been estimated to be between 0.02% and 0.04%,6 the most common reaction is a cutaneous eruption. Since anaphylactic reactions are mediated by immunoglobulin E (IgE), evaluation of patients with a history of penicillin allergy by penicillin skin testing is recommended to rule out IgE-mediated reactions.
This review outlines a practical approach to evaluating a suspected IgE-mediated reaction to penicillin, with key points in the history and diagnostic testing. We also review subsequent management and cross-reactivity with other beta-lactam-containing antibiotics.
EVALUATING ALLERGIC PATIENTS
Evaluation of patients with a history of penicillin allergy can be improved with an understanding of the classification of drug reactions, risk factors for allergy, and the pathophysiology of penicillin allergy.
Classification of drug reactions
Adverse drug reactions include all unintended pharmacologic effects of a drug and can be classified as predictable (type A) or unpredictable (type B). Predictable reactions are dose-dependent, are related to the known pharmacologic actions of the medication, and occur in otherwise healthy individuals. Unpredictable reactions are further classified into drug intolerance, drug idiosyncrasy, drug allergy, and pseudoallergic reactions.8,9
Penicillin allergy can manifest as any hypersensitivity reaction of the Gell and Coombs classification (Table 1).9 Type I (immediate) and type IV (delayed) reactions are the most common types of reactions that occur with antibiotics and should be classified based on the onset of symptoms as immediate (within 1 hour) or delayed (days or weeks).8
Risk factors for IgE-mediated reaction
Risk factors for a hypersensitivity reaction include frequent or repetitive courses of penicillin10 and high-dose parenteral (rather than oral) administration.
Age and atopy are not risk factors for penicillin allergy.7 However, atopy increases the risk of a more severe anaphylactic reaction to penicillin, and anaphylactic reactions are most commonly reported between the ages of 20 and 49.6
Pathophysiology of penicillin allergy
All penicillins share a common core ring structure (beta-lactam and thiazolidine rings) but differ in their side chains (R group) (Figure 1).
Under physiologic conditions, the core ring structure is metabolized into major (penicilloyl) and minor (penicillin itself, penicilloate and penilloate) antigenic determinants that may trigger an immediate IgE-dependent response.9 In the United States, commercial forms of antigenic determinates for skin testing exist in the form of penicillin G (minor determinant) and penicilloyl-polylysine, better known as Prepen (major determinant).
Immediate-type reactions to similar antibiotics such as aminopenicillins and cephalosporins may be caused by IgE antibodies against the R-group side chain rather than the core penicillin major and minor determinants.11
Questions to ask patients who have a history of penicillin allergy
Patients should be questioned closely about previous and current reactions to penicillin and should undergo skin-prick and intradermal testing, followed by graded-dose challenge or drug tolerance desensitization (Figure 2).
Questions to ask patients who have a history of penicillin allergy (Table 2)9,12 include the following:
Do you remember the details of the reaction? These include the route of administration, the time between the dose of penicillin and the appearance of symptoms, and how the reaction was managed.
Immediate reactions (ie, IgE-mediated, or Gell and Coombs type I) usually occur within the first hour after the first dose of the antibiotic, although they occasionally take up to 2 hours to occur, especially if the medication is taken orally and is taken with food. Symptoms consistent with IgE-mediated reactions include urticaria (most common), pruritus, angioedema, laryngeal edema, wheezing, shortness of breath, presyncope or syncope, hypotension, and cardiorespiratory collapse.
In contrast, symptoms of a non–IgE-mediated reaction are delayed in onset, occurring after days of treatment. They include nonpruritic maculopapular eruptions, hemolytic anemia, serum sickness, Stevens-Johnson syndrome, drug rash with eosinophilia and systemic symptoms, acute interstitial nephritis, and toxic epidermal necrolysis.9
If the patient has had severe non–IgE-mediated reactions to penicillin (eg, Stevens-Johnson syndrome, toxic epidermal necrolysis, acute interstitial nephritis, hemolytic anemia, or serum sickness) in the past, skin testing, graded-dose challenge, and desensitization are contraindicated.
How many years ago did the reaction occur? Most patients lose their sensitivity to penicillin over time.7,13–15 Nearly 50% of patients with IgE-mediated penicillin allergy lose their sensitivity within 5 years of the reaction,15 increasing to 80% or more by 10 years.13
How was the reaction managed? What was the outcome? Use of and positive response to epinephrine and histamine 1 receptor antagonists (antihistamines) with resolution or significant improvement of symptoms within a few hours may indicate an IgE-mediated reaction.
What was the indication for penicillin? Many cutaneous reactions are a result of an underlying viral or bacterial infection. For example, up to 90% of patients with Epstein-Barr virus infection develop a maculopapular rash when given penicillin.16
Have you tolerated other forms of penicillin since the reaction? Sometimes the patient has already tolerated other beta-lactams such as aminopenicillins, cephalosporins, and semisynthetic penicillins (piperacillin-tazobactam). Patients who tolerate other beta-lactams without adverse reactions are not allergic to beta-lactams.
Diagnostic tests
Skin testing. The only validated test for diagnosing IgE-mediated reactions caused by penicillin is the immediate hypersensitivity skin test,9 which should be performed by a board-certified allergist. The test consists of skin-prick and intradermal testing with the major determinant (penicilloyl-polylysine), the minor determinant (penicillin G), a negative control (normal saline), and a positive control (histamine). Minor-determinant mix is not commercially available in the United States.
Results of skin-prick testing are read 15 minutes after application. A positive response is a wheal at least 3 mm larger in diameter (with equivalent erythema) than the negative control done simultaneously. Intradermal testing is only done after a negative skin-prick test. If the allergic reaction was severe (ie, anaphylaxis), skin testing should be done at least 4 to 6 weeks after the reaction.
A history of severe non–IgE-mediated reaction to penicillin is a contraindication to skin-prick testing for penicillin allergy. The positive predictive value of penicillin skin testing is 50%, and the negative predictive value is 97%.3,7,9,13
Commercial in vitro testing (serum-specific IgE assays) for IgE-mediated hypersensitivity to penicillin is inferior to skin testing in terms of the negative predictive value and is not a suitable substitute for penicillin skin testing.
MANAGING PENICILLIN ALLERGY
If skin testing is positive, use another antibiotic, or refer for desensitization
If penicillin skin testing is positive (Figure 2), use another antibiotic that is equally efficacious. Patients who absolutely need a beta-lactam may undergo drug desensitization, performed by a board-certified allergist.
During desensitization, patients receive progressively higher doses of the drug every 15 to 20 minutes subcutaneously or intravenously, or every 20 to 30 minutes orally, until a full therapeutic dose is tolerated. Most protocols begin with a dose ranging from 1/10,000 to 1/1,000 of the final dose, depending on the severity of the allergic reaction.9,17
Using modern protocols, the success rate for tolerance induction is extremely high (75% to 100% in patients with cystic fibrosis, a group with a high rate of drug allergy18–20).
Drug desensitization is contraindicated in patients with non–IgE-mediated reactions.
If skin testing is negative, refer for graded-dose challenge
If skin testing is negative (Figure 2), graded-dose challenge is recommended. This procedure must be done by a board-certified allergist. If the original reaction was life-threatening, graded-dose challenge may entail giving 1/100 of the therapeutic dose. Then, if no reaction occurs during a brief observation period (usually 30 minutes), a full dose is given. However, many patients can start with 1/10 or even a full dose of the drug, especially if the original reaction was limited to the skin and the penicillin skin test is negative.
Graded-dose challenge is contraindicated if the original reaction was a severe non–IgE-mediated reaction.
UNDERSTANDING CROSS-REACTIVITY OF PENICILLIN
Penicillin is the only antibiotic for which skin testing is reliable and validated. If a drug that cross-reacts with penicillin is needed, it is important to know the rate of cross-reactivity (Table 3). The rate of cross-reactivity between penicillin and aminopenicillins (amoxicillin and ampicillin) is less than 1.3% in the United States.10,21 However, the cross-reactivity rate among aminopenicillins and cephalosporins is between 10% to 40%. For that reason, patients with prior reactions to aminopenicillins should avoid cephalosporins that share identical R-chain side groups with aminopenicillins.9,22
The rate of cross-reactivity between penicillin and cephalosporins was reported as 10% 40 years ago.23,24 But this was with early, first-generation cephalosporins that may have been contaminated with penicillin. The cross-reactivity rate with cephalosporins today is 3%.25 In general, first- and second-generation cephalosporins cause more allergic reactions than third- and fourth-generation cephalosporins.26
Patients with a history of penicillin allergy who require a cephalosporin should still undergo penicillin skin testing. Skin testing with cephalosporins has not been validated. However, skin testing with nonirritating concentrations of cephalosporins9 may be done to elucidate IgE reactions.
In a study by Romano et al,27 110 patients who had positive results on penicillin skin testing completed graded-dose challenge with the carbapenem antibiotic imipenem. The rate of cross-reactivity between penicillin and imipenem was less than 1%.
Monobactam antibiotics do not cross-react with other beta-lactams, except ceftazidime with aztreonam. This is probably because of similarities in their chemical structure.
Most patients who report that they are allergic to penicillin can ultimately receive penicillin or a penicillin-type antibiotic again after an appropriate evaluation and, possibly, treatment. This course of action decreases the need for broad-spectrum antibiotics,1–4 reduces health care costs, and prevents the development of multidrug-resistant pathogens.5
About 10% of the general population say that they are allergic to penicillin.1,6,7 Although the prevalence of life-threatening anaphylactic reactions to penicillin has been estimated to be between 0.02% and 0.04%,6 the most common reaction is a cutaneous eruption. Since anaphylactic reactions are mediated by immunoglobulin E (IgE), evaluation of patients with a history of penicillin allergy by penicillin skin testing is recommended to rule out IgE-mediated reactions.
This review outlines a practical approach to evaluating a suspected IgE-mediated reaction to penicillin, with key points in the history and diagnostic testing. We also review subsequent management and cross-reactivity with other beta-lactam-containing antibiotics.
EVALUATING ALLERGIC PATIENTS
Evaluation of patients with a history of penicillin allergy can be improved with an understanding of the classification of drug reactions, risk factors for allergy, and the pathophysiology of penicillin allergy.
Classification of drug reactions
Adverse drug reactions include all unintended pharmacologic effects of a drug and can be classified as predictable (type A) or unpredictable (type B). Predictable reactions are dose-dependent, are related to the known pharmacologic actions of the medication, and occur in otherwise healthy individuals. Unpredictable reactions are further classified into drug intolerance, drug idiosyncrasy, drug allergy, and pseudoallergic reactions.8,9
Penicillin allergy can manifest as any hypersensitivity reaction of the Gell and Coombs classification (Table 1).9 Type I (immediate) and type IV (delayed) reactions are the most common types of reactions that occur with antibiotics and should be classified based on the onset of symptoms as immediate (within 1 hour) or delayed (days or weeks).8
Risk factors for IgE-mediated reaction
Risk factors for a hypersensitivity reaction include frequent or repetitive courses of penicillin10 and high-dose parenteral (rather than oral) administration.
Age and atopy are not risk factors for penicillin allergy.7 However, atopy increases the risk of a more severe anaphylactic reaction to penicillin, and anaphylactic reactions are most commonly reported between the ages of 20 and 49.6
Pathophysiology of penicillin allergy
All penicillins share a common core ring structure (beta-lactam and thiazolidine rings) but differ in their side chains (R group) (Figure 1).
Under physiologic conditions, the core ring structure is metabolized into major (penicilloyl) and minor (penicillin itself, penicilloate and penilloate) antigenic determinants that may trigger an immediate IgE-dependent response.9 In the United States, commercial forms of antigenic determinates for skin testing exist in the form of penicillin G (minor determinant) and penicilloyl-polylysine, better known as Prepen (major determinant).
Immediate-type reactions to similar antibiotics such as aminopenicillins and cephalosporins may be caused by IgE antibodies against the R-group side chain rather than the core penicillin major and minor determinants.11
Questions to ask patients who have a history of penicillin allergy
Patients should be questioned closely about previous and current reactions to penicillin and should undergo skin-prick and intradermal testing, followed by graded-dose challenge or drug tolerance desensitization (Figure 2).
Questions to ask patients who have a history of penicillin allergy (Table 2)9,12 include the following:
Do you remember the details of the reaction? These include the route of administration, the time between the dose of penicillin and the appearance of symptoms, and how the reaction was managed.
Immediate reactions (ie, IgE-mediated, or Gell and Coombs type I) usually occur within the first hour after the first dose of the antibiotic, although they occasionally take up to 2 hours to occur, especially if the medication is taken orally and is taken with food. Symptoms consistent with IgE-mediated reactions include urticaria (most common), pruritus, angioedema, laryngeal edema, wheezing, shortness of breath, presyncope or syncope, hypotension, and cardiorespiratory collapse.
In contrast, symptoms of a non–IgE-mediated reaction are delayed in onset, occurring after days of treatment. They include nonpruritic maculopapular eruptions, hemolytic anemia, serum sickness, Stevens-Johnson syndrome, drug rash with eosinophilia and systemic symptoms, acute interstitial nephritis, and toxic epidermal necrolysis.9
If the patient has had severe non–IgE-mediated reactions to penicillin (eg, Stevens-Johnson syndrome, toxic epidermal necrolysis, acute interstitial nephritis, hemolytic anemia, or serum sickness) in the past, skin testing, graded-dose challenge, and desensitization are contraindicated.
How many years ago did the reaction occur? Most patients lose their sensitivity to penicillin over time.7,13–15 Nearly 50% of patients with IgE-mediated penicillin allergy lose their sensitivity within 5 years of the reaction,15 increasing to 80% or more by 10 years.13
How was the reaction managed? What was the outcome? Use of and positive response to epinephrine and histamine 1 receptor antagonists (antihistamines) with resolution or significant improvement of symptoms within a few hours may indicate an IgE-mediated reaction.
What was the indication for penicillin? Many cutaneous reactions are a result of an underlying viral or bacterial infection. For example, up to 90% of patients with Epstein-Barr virus infection develop a maculopapular rash when given penicillin.16
Have you tolerated other forms of penicillin since the reaction? Sometimes the patient has already tolerated other beta-lactams such as aminopenicillins, cephalosporins, and semisynthetic penicillins (piperacillin-tazobactam). Patients who tolerate other beta-lactams without adverse reactions are not allergic to beta-lactams.
Diagnostic tests
Skin testing. The only validated test for diagnosing IgE-mediated reactions caused by penicillin is the immediate hypersensitivity skin test,9 which should be performed by a board-certified allergist. The test consists of skin-prick and intradermal testing with the major determinant (penicilloyl-polylysine), the minor determinant (penicillin G), a negative control (normal saline), and a positive control (histamine). Minor-determinant mix is not commercially available in the United States.
Results of skin-prick testing are read 15 minutes after application. A positive response is a wheal at least 3 mm larger in diameter (with equivalent erythema) than the negative control done simultaneously. Intradermal testing is only done after a negative skin-prick test. If the allergic reaction was severe (ie, anaphylaxis), skin testing should be done at least 4 to 6 weeks after the reaction.
A history of severe non–IgE-mediated reaction to penicillin is a contraindication to skin-prick testing for penicillin allergy. The positive predictive value of penicillin skin testing is 50%, and the negative predictive value is 97%.3,7,9,13
Commercial in vitro testing (serum-specific IgE assays) for IgE-mediated hypersensitivity to penicillin is inferior to skin testing in terms of the negative predictive value and is not a suitable substitute for penicillin skin testing.
MANAGING PENICILLIN ALLERGY
If skin testing is positive, use another antibiotic, or refer for desensitization
If penicillin skin testing is positive (Figure 2), use another antibiotic that is equally efficacious. Patients who absolutely need a beta-lactam may undergo drug desensitization, performed by a board-certified allergist.
During desensitization, patients receive progressively higher doses of the drug every 15 to 20 minutes subcutaneously or intravenously, or every 20 to 30 minutes orally, until a full therapeutic dose is tolerated. Most protocols begin with a dose ranging from 1/10,000 to 1/1,000 of the final dose, depending on the severity of the allergic reaction.9,17
Using modern protocols, the success rate for tolerance induction is extremely high (75% to 100% in patients with cystic fibrosis, a group with a high rate of drug allergy18–20).
Drug desensitization is contraindicated in patients with non–IgE-mediated reactions.
If skin testing is negative, refer for graded-dose challenge
If skin testing is negative (Figure 2), graded-dose challenge is recommended. This procedure must be done by a board-certified allergist. If the original reaction was life-threatening, graded-dose challenge may entail giving 1/100 of the therapeutic dose. Then, if no reaction occurs during a brief observation period (usually 30 minutes), a full dose is given. However, many patients can start with 1/10 or even a full dose of the drug, especially if the original reaction was limited to the skin and the penicillin skin test is negative.
Graded-dose challenge is contraindicated if the original reaction was a severe non–IgE-mediated reaction.
UNDERSTANDING CROSS-REACTIVITY OF PENICILLIN
Penicillin is the only antibiotic for which skin testing is reliable and validated. If a drug that cross-reacts with penicillin is needed, it is important to know the rate of cross-reactivity (Table 3). The rate of cross-reactivity between penicillin and aminopenicillins (amoxicillin and ampicillin) is less than 1.3% in the United States.10,21 However, the cross-reactivity rate among aminopenicillins and cephalosporins is between 10% to 40%. For that reason, patients with prior reactions to aminopenicillins should avoid cephalosporins that share identical R-chain side groups with aminopenicillins.9,22
The rate of cross-reactivity between penicillin and cephalosporins was reported as 10% 40 years ago.23,24 But this was with early, first-generation cephalosporins that may have been contaminated with penicillin. The cross-reactivity rate with cephalosporins today is 3%.25 In general, first- and second-generation cephalosporins cause more allergic reactions than third- and fourth-generation cephalosporins.26
Patients with a history of penicillin allergy who require a cephalosporin should still undergo penicillin skin testing. Skin testing with cephalosporins has not been validated. However, skin testing with nonirritating concentrations of cephalosporins9 may be done to elucidate IgE reactions.
In a study by Romano et al,27 110 patients who had positive results on penicillin skin testing completed graded-dose challenge with the carbapenem antibiotic imipenem. The rate of cross-reactivity between penicillin and imipenem was less than 1%.
Monobactam antibiotics do not cross-react with other beta-lactams, except ceftazidime with aztreonam. This is probably because of similarities in their chemical structure.
- Park M, Markus P, Matesic D, Li JT. Safety and effectiveness of a preoperative allergy clinic in decreasing vancomycin use in patients with a history of penicillin allergy. Ann Allergy Asthma Immunol 2006; 97:681–687.
- Arroliga ME, Radojicic C, Gordon SM, et al. A prospective observational study of the effect of penicillin skin testing on antibiotic use in the intensive care unit. Infect Control Hosp Epidemiol 2003; 24:347–350.
- del Real GA, Rose ME, Ramirez-Atamoros MT, et al. Penicillin skin testing in patients with a history of beta-lactam allergy. Ann Allergy Asthma Immunol 2007; 98:355–359.
- Nadarajah K, Green GR, Naglak M. Clinical outcomes of penicillin skin testing. Ann Allergy Asthma Immunol 2005; 95:541–545.
- Harris AD, Sauberman L, Kabbash L, Greineder DK, Samore MH. Penicillin skin testing: a way to optimize antibiotic utilization. Am J Med 1999; 107:166–168.
- Idsoe O, Guthe T, Willcox RR, de Weck AL. Nature and extent of penicillin side-reactions, with particular reference to fatalities from anaphylactic shock. Bull World Health Organ 1968; 38:159–188.
- Gadde J, Spence M, Wheeler B, Adkinson NF Jr. Clinical experience with penicillin skin testing in a large inner-city STD clinic. JAMA 1993; 270:2456–2463.
- Johansson SG, Bieber T, Dahl R, et al. Revised nomenclature for allergy for global use: report of the Nomenclature Review Committee of the World Allergy Organization, October 2003. J Allergy Clin Immunol 2004; 113:832–836.
- Joint Task Force on Practice Parameters; American Academy of Allergy, Asthma and Immunology; American College of Allergy, Asthma and Immunology; Joint Council of Allergy, Asthma and Immunology. Drug allergy: an updated practice parameter. Ann Allergy Asthma Immunol 2010; 105:259–273.
- Park MA, Matesic D, Markus PJ, Li JT. Female sex as a risk factor for penicillin allergy. Ann Allergy Asthma Immunol 2007; 99:54–58.
- Moreno E, Macias E, Davila I, Laffond E, Ruiz A, Lorente F. Hypersensitivity reactions to cephalosporins. Expert Opin Drug Saf 2008; 7:295–304.
- Salkind AR, Cuddy PG, Foxworth JW. The rational clinical examination. Is this patient allergic to penicillin? An evidence-based analysis of the likelihood of penicillin allergy. JAMA 2001; 285:2498–2505.
- Sullivan TJ, Wedner HJ, Shatz GS, Yecies LD, Parker CW. Skin testing to detect penicillin allergy. J Allergy Clin Immunol 1981; 68:171–180.
- Macy E, Schatz M, Lin C, Poon KY. The falling rate of positive penicillin skin tests from 1995 to 2007. Perm J 2009; 13:12–18.
- Blanca M, Torres MJ, Garcia JJ, et al. Natural evolution of skin test sensitivity in patients allergic to beta-lactam antibiotics. J Allergy Clin Immunol 1999; 103:918–924.
- Patel BM. Skin rash with infectious mononucleosis and ampicillin. Pediatrics 1967; 40:910–911.
- Liu A, Fanning L, Chong H, et al. Desensitization regimens for drug allergy: state of the art in the 21st century. Clin Exp Allergy 2011; 41:1679–1688.
- Burrows JA, Toon M, Bell SC. Antibiotic desensitization in adults with cystic fibrosis. Respirology 2003; 8:359–364.
- Turvey SE, Cronin B, Arnold AD, Dioun AF. Antibiotic desensitization for the allergic patient: 5 years of experience and practice. Ann Allergy Asthma Immunol 2004; 92:426–432.
- Legere HJ 3rd, Palis RI, Rodriguez Bouza T, Uluer AZ, Castells MC. A safe protocol for rapid desensitization in patients with cystic fibrosis and antibiotic hypersensitivity. J Cyst Fibros 2009; 8:418–424.
- Lin E, Saxon A, Riedl M. Penicillin allergy: value of including amoxicillin as a determinant in penicillin skin testing. Int Arch Allergy Immunol 2010; 152:313–318.
- Dickson SD, Salazar KC. Diagnosis and management of immediate hypersensitivity reactions to cephalosporins. Clin Rev Allergy Immunol 2013; 45:131–142.
- Dash CH. Penicillin allergy and the cephalosporins. J Antimicrob Chemother 1975; 1(suppl 3):107–118.
- Petz LD. Immunologic cross-reactivity between penicillins and cephalosporins: a review. J Infect Dis 1978; 137(suppl):S74–S79.
- American Academy of Allergy Asthma & Immunology. Cephalosporin administration to patients with a history of penicillin allergy. www.aaaai.org/Aaaai/media/MediaLibrary/PDF%20Documents/Practice%20and%20Parameters/Cephalosporin-administration-2009.pdf. Accessed April 2, 2015.
- Fonacier L, Hirschberg R, Gerson S. Adverse drug reactions to cephalosporins in hospitalized patients with a history of penicillin allergy. Allergy Asthma Proc 2005; 26:135–141.
- Romano A, Viola M, Gueant-Rodriguez RM, Gaeta F, Pettinato R, Gueant JL. Imipenem in patients with immediate hypersensitivity to penicillins. N Engl J Med 2006; 354:2835–2837.
- Park M, Markus P, Matesic D, Li JT. Safety and effectiveness of a preoperative allergy clinic in decreasing vancomycin use in patients with a history of penicillin allergy. Ann Allergy Asthma Immunol 2006; 97:681–687.
- Arroliga ME, Radojicic C, Gordon SM, et al. A prospective observational study of the effect of penicillin skin testing on antibiotic use in the intensive care unit. Infect Control Hosp Epidemiol 2003; 24:347–350.
- del Real GA, Rose ME, Ramirez-Atamoros MT, et al. Penicillin skin testing in patients with a history of beta-lactam allergy. Ann Allergy Asthma Immunol 2007; 98:355–359.
- Nadarajah K, Green GR, Naglak M. Clinical outcomes of penicillin skin testing. Ann Allergy Asthma Immunol 2005; 95:541–545.
- Harris AD, Sauberman L, Kabbash L, Greineder DK, Samore MH. Penicillin skin testing: a way to optimize antibiotic utilization. Am J Med 1999; 107:166–168.
- Idsoe O, Guthe T, Willcox RR, de Weck AL. Nature and extent of penicillin side-reactions, with particular reference to fatalities from anaphylactic shock. Bull World Health Organ 1968; 38:159–188.
- Gadde J, Spence M, Wheeler B, Adkinson NF Jr. Clinical experience with penicillin skin testing in a large inner-city STD clinic. JAMA 1993; 270:2456–2463.
- Johansson SG, Bieber T, Dahl R, et al. Revised nomenclature for allergy for global use: report of the Nomenclature Review Committee of the World Allergy Organization, October 2003. J Allergy Clin Immunol 2004; 113:832–836.
- Joint Task Force on Practice Parameters; American Academy of Allergy, Asthma and Immunology; American College of Allergy, Asthma and Immunology; Joint Council of Allergy, Asthma and Immunology. Drug allergy: an updated practice parameter. Ann Allergy Asthma Immunol 2010; 105:259–273.
- Park MA, Matesic D, Markus PJ, Li JT. Female sex as a risk factor for penicillin allergy. Ann Allergy Asthma Immunol 2007; 99:54–58.
- Moreno E, Macias E, Davila I, Laffond E, Ruiz A, Lorente F. Hypersensitivity reactions to cephalosporins. Expert Opin Drug Saf 2008; 7:295–304.
- Salkind AR, Cuddy PG, Foxworth JW. The rational clinical examination. Is this patient allergic to penicillin? An evidence-based analysis of the likelihood of penicillin allergy. JAMA 2001; 285:2498–2505.
- Sullivan TJ, Wedner HJ, Shatz GS, Yecies LD, Parker CW. Skin testing to detect penicillin allergy. J Allergy Clin Immunol 1981; 68:171–180.
- Macy E, Schatz M, Lin C, Poon KY. The falling rate of positive penicillin skin tests from 1995 to 2007. Perm J 2009; 13:12–18.
- Blanca M, Torres MJ, Garcia JJ, et al. Natural evolution of skin test sensitivity in patients allergic to beta-lactam antibiotics. J Allergy Clin Immunol 1999; 103:918–924.
- Patel BM. Skin rash with infectious mononucleosis and ampicillin. Pediatrics 1967; 40:910–911.
- Liu A, Fanning L, Chong H, et al. Desensitization regimens for drug allergy: state of the art in the 21st century. Clin Exp Allergy 2011; 41:1679–1688.
- Burrows JA, Toon M, Bell SC. Antibiotic desensitization in adults with cystic fibrosis. Respirology 2003; 8:359–364.
- Turvey SE, Cronin B, Arnold AD, Dioun AF. Antibiotic desensitization for the allergic patient: 5 years of experience and practice. Ann Allergy Asthma Immunol 2004; 92:426–432.
- Legere HJ 3rd, Palis RI, Rodriguez Bouza T, Uluer AZ, Castells MC. A safe protocol for rapid desensitization in patients with cystic fibrosis and antibiotic hypersensitivity. J Cyst Fibros 2009; 8:418–424.
- Lin E, Saxon A, Riedl M. Penicillin allergy: value of including amoxicillin as a determinant in penicillin skin testing. Int Arch Allergy Immunol 2010; 152:313–318.
- Dickson SD, Salazar KC. Diagnosis and management of immediate hypersensitivity reactions to cephalosporins. Clin Rev Allergy Immunol 2013; 45:131–142.
- Dash CH. Penicillin allergy and the cephalosporins. J Antimicrob Chemother 1975; 1(suppl 3):107–118.
- Petz LD. Immunologic cross-reactivity between penicillins and cephalosporins: a review. J Infect Dis 1978; 137(suppl):S74–S79.
- American Academy of Allergy Asthma & Immunology. Cephalosporin administration to patients with a history of penicillin allergy. www.aaaai.org/Aaaai/media/MediaLibrary/PDF%20Documents/Practice%20and%20Parameters/Cephalosporin-administration-2009.pdf. Accessed April 2, 2015.
- Fonacier L, Hirschberg R, Gerson S. Adverse drug reactions to cephalosporins in hospitalized patients with a history of penicillin allergy. Allergy Asthma Proc 2005; 26:135–141.
- Romano A, Viola M, Gueant-Rodriguez RM, Gaeta F, Pettinato R, Gueant JL. Imipenem in patients with immediate hypersensitivity to penicillins. N Engl J Med 2006; 354:2835–2837.
KEY POINTS
- The prevalence of reported penicillin allergy is 10% in the general population. However, more than 90% of these patients are found not to be allergic to penicillin after skin testing.
- In patients found to have penicillin allergy, the frequency of positive results on skin testing decreases by 10% per year of avoidance. Therefore, 80% to 100% of patients are expected to test negative for penicillin allergy by 10 years after their reaction.
- Skin testing for penicillin allergy is only useful for type 1 IgE-mediated reactions. However, in properly selected patients, the negative predictive value of penicillin skin testing is nearly 97%.
- The rate of cross-reactivity between penicillin and cephalosporins is approximately 3%.
Clinical utility of warfarin pharmacogenomics
To the Editor: We previously addressed whether VKORC1 and CYP2C9 pharmacogenomic testing should be considered when prescribing warfarin.1 Our recommendation, based on available evidence at that time, was that physicians should consider pharmacogenomic testing for any patient who is started on warfarin therapy.
Since the publication of this recommendation, two major trials, COAG (Clarification of Optimal Anticoagulation Through Genetics)2 and EU-PACT (European Pharmacogenetics of Anticoagulant Therapy-Warfarin),3 were published along with commentaries debating the clinical utility of warfarin pharmacogenomics.4–15 Based on these publications, we would like to update our recommendations for pharmacogenomic testing for warfarin therapy.
COAG compared the efficacy of a clinical algorithm or a clinical algorithm plus VKORC1 and CYP2C9 genotyping to guide warfarin dosage. At the end of 4 weeks, the mean percentage of time within the therapeutic international normalized ratio (INR) range was 45.4% for those in the clinical algorithm arm and 45.2% for those in the genotyping arm (95% confidence interval [CI] –3.4 to 3.1, P = .91). For both treatment groups, clinical data that included body surface area, age, target INR, concomitantly prescribed drugs, and smoking status were used to predict warfarin dose, with the genotyping arm including VKORC1 and CYP2C9. Although VKORC1 and CYP2C9 genotyping offered no additional benefit, caution should be used when extrapolating this conclusion to clinical settings in which warfarin therapy is initiated using a standardized starting dose (eg, 5 mg daily) instead of a clinical dosing algorithm.
Of interest, in the COAG trial, among black patients, the mean percentage of time in the therapeutic INR range was significantly less for those in the genotype-guided arm than for those in the clinically guided arm—ie, 35.2% vs 43.5% (95% CI –15.0 to –2.0, P = .01). The percentage of time with therapeutic INR has been identified as a surrogate marker for poor outcomes such as death, stroke, or major hemorrhage, with those with a lower percentage of time in therapeutic INR being at greater risk of an adverse event.16 Wan et al17 demonstrated that a 6.9% improvement of time in therapeutic INR decreased the risk of major hemorrhage by one event per 100 patient-years.17 Therefore, black patients in the COAG genotyping arm may have been at greater risk for an adverse event because of a lower observed percentage of time within the therapeutic INR range.
In the COAG trial, genotyping was done for only one VKORC1 variant and for two CYP2C9 alleles (CYP2C9*2, and CYP2C9*3). Other genetic variants are of clinical importance for warfarin dosing in black patients, and the lack of genotyping for these additional variants may explain why black patients in the genotyping arm performed worse.5,7,11 In particular, CYP2C9*8 may be an important predictor of warfarin dose in black patients.18
EU-PACT compared the efficacy of standardized warfarin dosing and that of a clinical algorithm.3 Patients in the standardized dosing arm were prescribed warfarin 10 mg on the first day of treatment (5 mg for those over age 75), and 5 mg on days 2 and 3, with subsequent dosing adjustments based on INR. Patients in the genotyping arm were prescribed warfarin based on an algorithm that incorporated clinical data that included body surface area, age, and concomitantly prescribed drugs, as well as VKORC1 and CYP2C9 genotypes. At the end of 12 weeks, the mean percentage of time in the therapeutic INR range was 60.3% for those in the standardized-dosing arm and 67.4% for those in the genotyping arm (95% CI 3.3 to 10.6, P < .001).2 The approximate 7% improvement in percentage of time in the therapeutic INR range may predict a lower risk of hemorrhage for those in the genotyping arm.17 Although patients in the genotyping arm had a higher percentage of time in the therapeutic INR range, it is unclear whether genotyping alone is superior to standardized dosing because the dosing algorithm used both clinical data and genotype data.
There are substantial differences between the COAG and EU-PACT trials, including dosing schemes, racial diversity, and trial length, and these differences could have contributed to the conflicting results. Based on these two trials, a possible conclusion is that genotype-guided warfarin dosing may be superior to standardized dosing, but may be no better than utilizing a clinical algorithm in white patients. For black patients, additional studies are needed to determine which genetic variants are of importance for guiding warfarin dosing.
We would like to update the recommendations we made in our previously published article,1 to state that genotyping for CYP2C9 and VKORC1 may be of clinical utility in white patients depending on whether standardized dosing or a clinical algorithm is used to initiate warfarin therapy. Routine genotyping in black patients is not recommended until further studies clarify which genetic variants are of importance for guiding warfarin dosing.
The ongoing Genetics Informatics Trial of Warfarin to Prevent Venous Thrombosis may bring much needed clarity to the clinical utility of warfarin pharmacogenomics. We hope to publish a more detailed update of our 2013 article after completion of that trial.
- Rouse M, Cristiani C, Teng KA. Should we use pharmacogenetic testing when prescribing warfarin? Cleve Clin J Med 2013; 80:483–486.
- Kimmel SE, French B, Kasner SE, et al; COAG Investigators. A pharmacogenetic versus a clinical algorithm for warfarin dosing. N Engl J Med 2013; 369:2283–2293.
- Pirmohamed M, Burnside G, Eriksson N, et al; EU-PACT Group. A randomized trial of genotype-guided dosing of warfarin. N Engl J Med 2013; 369:2294–2303.
- Cavallari LH, Kittles RA, Perera MA. Genotype-guided dosing of vitamin K antagonists. N Engl J Med 2014; 370:1763.
- Cavallari LH, Nutescu EA. Warfarin pharmacogenetics: to genotype or not to genotype, that is the question. Clin Pharmacol Ther 2014; 96:22–24.
- Daneshjou R, Klein TE, Altman RB. Genotype-guided dosing of vitamin K antagonists. N Engl J Med 2014; 370:1762–1763.
- Hernandez W, Gamazon ER, Aquino-Michaels K, et al. Ethnicity-specific pharmacogenetics: the case of warfarin in African Americans. Pharmacogenomics J 2014; 14:223–228.
- Kimmel SE, French B, Geller NL; COAG Investigators. Genotype-guided dosing of vitamin K antagonists. N Engl J Med 2014; 370:1763–1764.
- Koller EA, Roche JC, Rollins JA. Genotype-guided dosing of vitamin K antagonists. N Engl J Med 2014; 370:1761.
- Pereira NL, Rihal CS, Weinshilboum RM. Genotype-guided dosing of vitamin K antagonists. N Engl J Med 2014; 370:1762.
- Perera MA, Cavallari LH, Johnson JA. Warfarin pharmacogenetics: an illustration of the importance of studies in minority populations. Clin Pharmacol Ther 2014; 95:242–244.
- Pirmohamed M, Wadelius M, Kamali F; EU-PACT Group. Genotype-guided dosing of vitamin K antagonists. N Engl J Med 2014; 370:1764–1765.
- Schwarz UI, Kim RB, Tirona RG. Genotype-guided dosing of vitamin K antagonists. N Engl J Med 2014; 370:1761–1762.
- Scott SA, Lubitz SA. Warfarin pharmacogenetic trials: is there a future for pharmacogenetic-guided dosing? Pharmacogenomics 2014; 15:719–722.
- Zineh I, Pacanowski M, Woodcock J. Pharmacogenetics and coumarin dosing—recalibrating expectations. N Engl J Med 2013; 369:2273–2275.
- Hylek EM. Vitamin K antagonists and time in the therapeutic range: implications, challenges, and strategies for improvement. J Thromb Thrombolysis 2013; 35:333–335.
- Wan Y, Heneghan C, Perera R, et al. Anticoagulation control and prediction of adverse events in patients with atrial fibrillation: a systematic review. Circ Cardiovasc Qual Outcomes 2008;1:84-91.
- Nagai R, Ohara M, Cavallari LH, et al. Factors influencing pharmacokinetics of warfarin in African-Americans: implications for pharmacogenetic dosing algorithms. Pharmacogenomics 2015;16:217–225.
To the Editor: We previously addressed whether VKORC1 and CYP2C9 pharmacogenomic testing should be considered when prescribing warfarin.1 Our recommendation, based on available evidence at that time, was that physicians should consider pharmacogenomic testing for any patient who is started on warfarin therapy.
Since the publication of this recommendation, two major trials, COAG (Clarification of Optimal Anticoagulation Through Genetics)2 and EU-PACT (European Pharmacogenetics of Anticoagulant Therapy-Warfarin),3 were published along with commentaries debating the clinical utility of warfarin pharmacogenomics.4–15 Based on these publications, we would like to update our recommendations for pharmacogenomic testing for warfarin therapy.
COAG compared the efficacy of a clinical algorithm or a clinical algorithm plus VKORC1 and CYP2C9 genotyping to guide warfarin dosage. At the end of 4 weeks, the mean percentage of time within the therapeutic international normalized ratio (INR) range was 45.4% for those in the clinical algorithm arm and 45.2% for those in the genotyping arm (95% confidence interval [CI] –3.4 to 3.1, P = .91). For both treatment groups, clinical data that included body surface area, age, target INR, concomitantly prescribed drugs, and smoking status were used to predict warfarin dose, with the genotyping arm including VKORC1 and CYP2C9. Although VKORC1 and CYP2C9 genotyping offered no additional benefit, caution should be used when extrapolating this conclusion to clinical settings in which warfarin therapy is initiated using a standardized starting dose (eg, 5 mg daily) instead of a clinical dosing algorithm.
Of interest, in the COAG trial, among black patients, the mean percentage of time in the therapeutic INR range was significantly less for those in the genotype-guided arm than for those in the clinically guided arm—ie, 35.2% vs 43.5% (95% CI –15.0 to –2.0, P = .01). The percentage of time with therapeutic INR has been identified as a surrogate marker for poor outcomes such as death, stroke, or major hemorrhage, with those with a lower percentage of time in therapeutic INR being at greater risk of an adverse event.16 Wan et al17 demonstrated that a 6.9% improvement of time in therapeutic INR decreased the risk of major hemorrhage by one event per 100 patient-years.17 Therefore, black patients in the COAG genotyping arm may have been at greater risk for an adverse event because of a lower observed percentage of time within the therapeutic INR range.
In the COAG trial, genotyping was done for only one VKORC1 variant and for two CYP2C9 alleles (CYP2C9*2, and CYP2C9*3). Other genetic variants are of clinical importance for warfarin dosing in black patients, and the lack of genotyping for these additional variants may explain why black patients in the genotyping arm performed worse.5,7,11 In particular, CYP2C9*8 may be an important predictor of warfarin dose in black patients.18
EU-PACT compared the efficacy of standardized warfarin dosing and that of a clinical algorithm.3 Patients in the standardized dosing arm were prescribed warfarin 10 mg on the first day of treatment (5 mg for those over age 75), and 5 mg on days 2 and 3, with subsequent dosing adjustments based on INR. Patients in the genotyping arm were prescribed warfarin based on an algorithm that incorporated clinical data that included body surface area, age, and concomitantly prescribed drugs, as well as VKORC1 and CYP2C9 genotypes. At the end of 12 weeks, the mean percentage of time in the therapeutic INR range was 60.3% for those in the standardized-dosing arm and 67.4% for those in the genotyping arm (95% CI 3.3 to 10.6, P < .001).2 The approximate 7% improvement in percentage of time in the therapeutic INR range may predict a lower risk of hemorrhage for those in the genotyping arm.17 Although patients in the genotyping arm had a higher percentage of time in the therapeutic INR range, it is unclear whether genotyping alone is superior to standardized dosing because the dosing algorithm used both clinical data and genotype data.
There are substantial differences between the COAG and EU-PACT trials, including dosing schemes, racial diversity, and trial length, and these differences could have contributed to the conflicting results. Based on these two trials, a possible conclusion is that genotype-guided warfarin dosing may be superior to standardized dosing, but may be no better than utilizing a clinical algorithm in white patients. For black patients, additional studies are needed to determine which genetic variants are of importance for guiding warfarin dosing.
We would like to update the recommendations we made in our previously published article,1 to state that genotyping for CYP2C9 and VKORC1 may be of clinical utility in white patients depending on whether standardized dosing or a clinical algorithm is used to initiate warfarin therapy. Routine genotyping in black patients is not recommended until further studies clarify which genetic variants are of importance for guiding warfarin dosing.
The ongoing Genetics Informatics Trial of Warfarin to Prevent Venous Thrombosis may bring much needed clarity to the clinical utility of warfarin pharmacogenomics. We hope to publish a more detailed update of our 2013 article after completion of that trial.
To the Editor: We previously addressed whether VKORC1 and CYP2C9 pharmacogenomic testing should be considered when prescribing warfarin.1 Our recommendation, based on available evidence at that time, was that physicians should consider pharmacogenomic testing for any patient who is started on warfarin therapy.
Since the publication of this recommendation, two major trials, COAG (Clarification of Optimal Anticoagulation Through Genetics)2 and EU-PACT (European Pharmacogenetics of Anticoagulant Therapy-Warfarin),3 were published along with commentaries debating the clinical utility of warfarin pharmacogenomics.4–15 Based on these publications, we would like to update our recommendations for pharmacogenomic testing for warfarin therapy.
COAG compared the efficacy of a clinical algorithm or a clinical algorithm plus VKORC1 and CYP2C9 genotyping to guide warfarin dosage. At the end of 4 weeks, the mean percentage of time within the therapeutic international normalized ratio (INR) range was 45.4% for those in the clinical algorithm arm and 45.2% for those in the genotyping arm (95% confidence interval [CI] –3.4 to 3.1, P = .91). For both treatment groups, clinical data that included body surface area, age, target INR, concomitantly prescribed drugs, and smoking status were used to predict warfarin dose, with the genotyping arm including VKORC1 and CYP2C9. Although VKORC1 and CYP2C9 genotyping offered no additional benefit, caution should be used when extrapolating this conclusion to clinical settings in which warfarin therapy is initiated using a standardized starting dose (eg, 5 mg daily) instead of a clinical dosing algorithm.
Of interest, in the COAG trial, among black patients, the mean percentage of time in the therapeutic INR range was significantly less for those in the genotype-guided arm than for those in the clinically guided arm—ie, 35.2% vs 43.5% (95% CI –15.0 to –2.0, P = .01). The percentage of time with therapeutic INR has been identified as a surrogate marker for poor outcomes such as death, stroke, or major hemorrhage, with those with a lower percentage of time in therapeutic INR being at greater risk of an adverse event.16 Wan et al17 demonstrated that a 6.9% improvement of time in therapeutic INR decreased the risk of major hemorrhage by one event per 100 patient-years.17 Therefore, black patients in the COAG genotyping arm may have been at greater risk for an adverse event because of a lower observed percentage of time within the therapeutic INR range.
In the COAG trial, genotyping was done for only one VKORC1 variant and for two CYP2C9 alleles (CYP2C9*2, and CYP2C9*3). Other genetic variants are of clinical importance for warfarin dosing in black patients, and the lack of genotyping for these additional variants may explain why black patients in the genotyping arm performed worse.5,7,11 In particular, CYP2C9*8 may be an important predictor of warfarin dose in black patients.18
EU-PACT compared the efficacy of standardized warfarin dosing and that of a clinical algorithm.3 Patients in the standardized dosing arm were prescribed warfarin 10 mg on the first day of treatment (5 mg for those over age 75), and 5 mg on days 2 and 3, with subsequent dosing adjustments based on INR. Patients in the genotyping arm were prescribed warfarin based on an algorithm that incorporated clinical data that included body surface area, age, and concomitantly prescribed drugs, as well as VKORC1 and CYP2C9 genotypes. At the end of 12 weeks, the mean percentage of time in the therapeutic INR range was 60.3% for those in the standardized-dosing arm and 67.4% for those in the genotyping arm (95% CI 3.3 to 10.6, P < .001).2 The approximate 7% improvement in percentage of time in the therapeutic INR range may predict a lower risk of hemorrhage for those in the genotyping arm.17 Although patients in the genotyping arm had a higher percentage of time in the therapeutic INR range, it is unclear whether genotyping alone is superior to standardized dosing because the dosing algorithm used both clinical data and genotype data.
There are substantial differences between the COAG and EU-PACT trials, including dosing schemes, racial diversity, and trial length, and these differences could have contributed to the conflicting results. Based on these two trials, a possible conclusion is that genotype-guided warfarin dosing may be superior to standardized dosing, but may be no better than utilizing a clinical algorithm in white patients. For black patients, additional studies are needed to determine which genetic variants are of importance for guiding warfarin dosing.
We would like to update the recommendations we made in our previously published article,1 to state that genotyping for CYP2C9 and VKORC1 may be of clinical utility in white patients depending on whether standardized dosing or a clinical algorithm is used to initiate warfarin therapy. Routine genotyping in black patients is not recommended until further studies clarify which genetic variants are of importance for guiding warfarin dosing.
The ongoing Genetics Informatics Trial of Warfarin to Prevent Venous Thrombosis may bring much needed clarity to the clinical utility of warfarin pharmacogenomics. We hope to publish a more detailed update of our 2013 article after completion of that trial.
- Rouse M, Cristiani C, Teng KA. Should we use pharmacogenetic testing when prescribing warfarin? Cleve Clin J Med 2013; 80:483–486.
- Kimmel SE, French B, Kasner SE, et al; COAG Investigators. A pharmacogenetic versus a clinical algorithm for warfarin dosing. N Engl J Med 2013; 369:2283–2293.
- Pirmohamed M, Burnside G, Eriksson N, et al; EU-PACT Group. A randomized trial of genotype-guided dosing of warfarin. N Engl J Med 2013; 369:2294–2303.
- Cavallari LH, Kittles RA, Perera MA. Genotype-guided dosing of vitamin K antagonists. N Engl J Med 2014; 370:1763.
- Cavallari LH, Nutescu EA. Warfarin pharmacogenetics: to genotype or not to genotype, that is the question. Clin Pharmacol Ther 2014; 96:22–24.
- Daneshjou R, Klein TE, Altman RB. Genotype-guided dosing of vitamin K antagonists. N Engl J Med 2014; 370:1762–1763.
- Hernandez W, Gamazon ER, Aquino-Michaels K, et al. Ethnicity-specific pharmacogenetics: the case of warfarin in African Americans. Pharmacogenomics J 2014; 14:223–228.
- Kimmel SE, French B, Geller NL; COAG Investigators. Genotype-guided dosing of vitamin K antagonists. N Engl J Med 2014; 370:1763–1764.
- Koller EA, Roche JC, Rollins JA. Genotype-guided dosing of vitamin K antagonists. N Engl J Med 2014; 370:1761.
- Pereira NL, Rihal CS, Weinshilboum RM. Genotype-guided dosing of vitamin K antagonists. N Engl J Med 2014; 370:1762.
- Perera MA, Cavallari LH, Johnson JA. Warfarin pharmacogenetics: an illustration of the importance of studies in minority populations. Clin Pharmacol Ther 2014; 95:242–244.
- Pirmohamed M, Wadelius M, Kamali F; EU-PACT Group. Genotype-guided dosing of vitamin K antagonists. N Engl J Med 2014; 370:1764–1765.
- Schwarz UI, Kim RB, Tirona RG. Genotype-guided dosing of vitamin K antagonists. N Engl J Med 2014; 370:1761–1762.
- Scott SA, Lubitz SA. Warfarin pharmacogenetic trials: is there a future for pharmacogenetic-guided dosing? Pharmacogenomics 2014; 15:719–722.
- Zineh I, Pacanowski M, Woodcock J. Pharmacogenetics and coumarin dosing—recalibrating expectations. N Engl J Med 2013; 369:2273–2275.
- Hylek EM. Vitamin K antagonists and time in the therapeutic range: implications, challenges, and strategies for improvement. J Thromb Thrombolysis 2013; 35:333–335.
- Wan Y, Heneghan C, Perera R, et al. Anticoagulation control and prediction of adverse events in patients with atrial fibrillation: a systematic review. Circ Cardiovasc Qual Outcomes 2008;1:84-91.
- Nagai R, Ohara M, Cavallari LH, et al. Factors influencing pharmacokinetics of warfarin in African-Americans: implications for pharmacogenetic dosing algorithms. Pharmacogenomics 2015;16:217–225.
- Rouse M, Cristiani C, Teng KA. Should we use pharmacogenetic testing when prescribing warfarin? Cleve Clin J Med 2013; 80:483–486.
- Kimmel SE, French B, Kasner SE, et al; COAG Investigators. A pharmacogenetic versus a clinical algorithm for warfarin dosing. N Engl J Med 2013; 369:2283–2293.
- Pirmohamed M, Burnside G, Eriksson N, et al; EU-PACT Group. A randomized trial of genotype-guided dosing of warfarin. N Engl J Med 2013; 369:2294–2303.
- Cavallari LH, Kittles RA, Perera MA. Genotype-guided dosing of vitamin K antagonists. N Engl J Med 2014; 370:1763.
- Cavallari LH, Nutescu EA. Warfarin pharmacogenetics: to genotype or not to genotype, that is the question. Clin Pharmacol Ther 2014; 96:22–24.
- Daneshjou R, Klein TE, Altman RB. Genotype-guided dosing of vitamin K antagonists. N Engl J Med 2014; 370:1762–1763.
- Hernandez W, Gamazon ER, Aquino-Michaels K, et al. Ethnicity-specific pharmacogenetics: the case of warfarin in African Americans. Pharmacogenomics J 2014; 14:223–228.
- Kimmel SE, French B, Geller NL; COAG Investigators. Genotype-guided dosing of vitamin K antagonists. N Engl J Med 2014; 370:1763–1764.
- Koller EA, Roche JC, Rollins JA. Genotype-guided dosing of vitamin K antagonists. N Engl J Med 2014; 370:1761.
- Pereira NL, Rihal CS, Weinshilboum RM. Genotype-guided dosing of vitamin K antagonists. N Engl J Med 2014; 370:1762.
- Perera MA, Cavallari LH, Johnson JA. Warfarin pharmacogenetics: an illustration of the importance of studies in minority populations. Clin Pharmacol Ther 2014; 95:242–244.
- Pirmohamed M, Wadelius M, Kamali F; EU-PACT Group. Genotype-guided dosing of vitamin K antagonists. N Engl J Med 2014; 370:1764–1765.
- Schwarz UI, Kim RB, Tirona RG. Genotype-guided dosing of vitamin K antagonists. N Engl J Med 2014; 370:1761–1762.
- Scott SA, Lubitz SA. Warfarin pharmacogenetic trials: is there a future for pharmacogenetic-guided dosing? Pharmacogenomics 2014; 15:719–722.
- Zineh I, Pacanowski M, Woodcock J. Pharmacogenetics and coumarin dosing—recalibrating expectations. N Engl J Med 2013; 369:2273–2275.
- Hylek EM. Vitamin K antagonists and time in the therapeutic range: implications, challenges, and strategies for improvement. J Thromb Thrombolysis 2013; 35:333–335.
- Wan Y, Heneghan C, Perera R, et al. Anticoagulation control and prediction of adverse events in patients with atrial fibrillation: a systematic review. Circ Cardiovasc Qual Outcomes 2008;1:84-91.
- Nagai R, Ohara M, Cavallari LH, et al. Factors influencing pharmacokinetics of warfarin in African-Americans: implications for pharmacogenetic dosing algorithms. Pharmacogenomics 2015;16:217–225.
Optimizing Inpatient Pharmacotherapy Using a Single Clinical Policy Streamlining Pharmacy Protocols
From the Ernest Mario School of Pharmacy, Rutgers, The State University of New Jersey, Piscataway, NJ.
Abstract
- Objectives: To describe the implementation of broadly scoped clinical pharmacy protocols positioned as a singular policy in a community hospital. These protocols were designed to expand the established benefits demonstrated using narrower, traditional protocols.
- Methods: A retrospective chart review of protocol interventions in the first year of the policy’s implementation was conducted to evaluate prescriber acceptance of protocol interventions. Interventions were identified from required email notifications. The frequency of use of each protocol was assessed, including evaluation of novel characteristics of specific protocols. Pharmacist utilization patterns were assessed for job classification, shift, and practice setting (ie, centralized or decentralized).
- Results: In the 1-year assessment period, 145 interventions were reported and 144 were accepted by the prescribing physicians. Interventions involved orders from hospitalists and intensivists most frequently, with the renal dosing and dose formulations protocols being the most commonly utilized. Staff pharmacists used the policy more frequently than clinical pharmacists, primarily during day shift from decentralized locations on the patient care units.
- Conclusions: The implementation of broadly scoped clinical pharmacy protocols for items our pharmacists routinely contact physicians about (and our physicians deemed were within the practice of pharmacy) instituted a cultural shift that expanded the elements considered to be part of routine pharmacy practice. As a result, pharmacists more seamlessly applied their expertise as pharmacotherapy specialists to optimize pharmacotherapy, which streamlined workflow for both pharmacists and physicians. This expanded the proven benefits of allowing professionals to work to their fullest extent, as established in the literature.
Allowing pharmacists to apply their expertise has been associated with improved outcomes in both pharmacotherapy quality (eg, reduction in mortality and length of stay [1]) and savings in health care dollars. Studies of focused protocols, including intravenous-to-oral (IV-to-PO) switch [2–20], renal dosing [21], stress ulcer prophylaxis [22] and anticoagulation management [1,23,24] demonstrate these benefits in a multitude of practice areas. While such protocols have become commonplace in the acute care setting [25–28], most continue to be singularly focused and impose patient population restrictions that preclude comprehensive patient evaluation. Many are administered as a task within the pharmacist workflow using a patient list generated by the limited protocol criteria, which are often restricted to agent or patient characteristics.
Better outcomes are associated with permitting professionals such as pharmacists to work to the fullest extent of their scope and expertise [29–31]. In specific cases, studies evaluating pharmacists’ impact within a multi-disciplinary health care team have demonstrated improved outcomes in regard to both patient care and cost [29–31]. Recognizing this, accountable care organizations (ACOs) have developed practice models that are based on this benefit. Each team member is expected to robustly apply their training and expertise to achieve the best outcomes [32,33]. As health care moves toward a more integrative approach, it is paramount that pharmacists utilize the full scope of the skills in which they are trained.
This report describes the development, implementation, and outcomes of a singular policy outlining comprehensively scoped protocols allowing acute care hospital pharmacists within Princeton HealthCare System to optimize pharmacotherapy during the course of their usual clinical practice.
Methods
Setting
The University Medical Center of Princeton at Plainsboro (UMCPP), part of the Princeton HealthCare System, is a 230-bed community acute care hospital located in central New Jersey. The hospital facility relocated in May 2012 from its previous location in Princeton to a new state-of-the-art facility in Plainsboro. As an affiliate of the Robert Wood Johnson Medical School and the Ernest Mario School of Pharmacy at Rutgers, The State University of New Jersey (ie, Rutgers), it is an academic teaching hospital with a mixed model for providing patient care. UMCPP employs both faculty physicians leading academic teams alongside hospitalists and private attendings.
Pharmacy services are provided on facility 24 hours a day, 365 days a year. The department of pharmacy services provides a full scope medication services from a centralized location with 3 full-time day pharmacists and 1 oncology satellite pharmacist. During weekdays, decentralized pharmacists provide medication review, patient education, and medication reconciliation on 2 to 3 inpatient care units. Centralized support decreases to 2 pharmacists in the evening and 1 overnight. Clinical pharmacists, both hospital-based and Rutgers faculty, work in conjunction with the staff pharmacists to ensure appropriate management of patients throughout different levels of care.
Program Overview and Implementation
To enhance protocols allowing pharmacists to more holistically and robustly optimize pharmacotherapy, UMCPP implemented the Clinical Pharmacy Services policy in February 2012. The policy outlined 8 protocols through which registered pharmacists within the acute care hospital could implement outlined medication order adjustments for adults of inpatient status. Pediatric patients or those treated outside of the acute care hospital (eg, in the psychiatric hospital, surgical center or outpatient facilities) were excluded. While the hospital had existing traditional programs such as IV-to-PO conversions, the programs were restricted to specific agents or conditions. As such, pharmacists were assigned to review queues in the clinical computer system to which orders for the agents outlined by the specific program would flow. Review would occur at set intervals and focus on that detail of the patient’s care as opposed to broadly encompassing an evaluation of the patient’s comprehensive pharmacotherapy. The goal of the new policy was to better utilize the pharmacists’ expertise by broadening these assessments to all applicable agents, refine workflow (by allowing protocol management instead of requiring individual prescriber calls for each issue) and integrate holistic refinement of pharmacotherapy regimens during the usual course of the pharmacist’s clinical care.
In the state of New Jersey, the Pharmacy Practice Act (updated on 14 January 2004) formally recognizes pharmacists as health care professionals and permits for collaborative practice in the community setting [34]. However, pharmacist management by protocol in the acute care hospital setting is defined separately, requiring only medical approvals within the system [35]. In accordance, the policy and associated protocols were approved by the institution’s multidisciplinary pharmacy and therapeutics (P&T) and medical executive committee processes.
To ensure appropriate oversight, the policy required that the pharmacist making changes submit notification of protocol intervention to the patient’s attending physician, the physician who generated the original order (if other than the attending) and a designated clinical pharmacist (for auditing purposes). All notifications were made via email within the clinical computer system in “interrupt” status to ensure active recognition by the prescriber(s).
Program Evaluation
An evaluation of the first year’s interventions was conducted to validate the program, describe its utility, and provide a basis for re-evaluation and continued evolution. The aim was to evaluate the institution’s experience with the program, focusing on both specific physician and pharmacist elements. One of the primary goals was to evaluate which physician’s orders were associated with interventions as well as the rate of physician acceptance of protocol interventions, as their acceptance clearly validates the pharmacist’s ability to appropriately apply the protocols in patient-specific contexts.
To evaluate the pharmacist’s experience, trends in pharmacist utilization were captured, including which pharmacist by job classification (ie, staff or clinical pharmacist) implemented interventions, during which shift, and in what operational capacity (ie, centralized or decentralized) the pharmacist was practicing. Lastly, the study sought to characterize the frequency to which each protocol was applied. Based on the existing experiences described in the literature as well as with consideration of institutional culture and operation, we hypothesized that all pharmacists would apply protocols with equal efficacy with more interventions likely generated by staff pharmacists due to their role in primary order review and that the types of interventions would vary based on shift and location.
A retrospective review of cases throughout the first year of the policy’s implementation was conducted, including interventions made between 1 February 2012 and 31 January 2013. Cases were identified through the required email notification of the auditing clinical pharmacist. The patient’s electronic medical record for that defined visit was reviewed. To assess pharmacist utilization patterns, data captured included the agent involved in the intervention, date, day of week and shift, whether the pharmacist was centralized or decentralized, and whether that pharmacist was classified as staff or clinical. Decentralized pharmacists were defined as a pharmacist working on the patient care unit with direct access to other practitioners and patients, rather than those performing their functions from within the confines of the pharmacy department.
Prescribers were described both by status (ie, attending or resident/training) and specialty. Physician acceptance was assessed through evaluation of order trends as the electronic medical record allows for all changes to an order to be audited and tracked; a review of progress notes to capture any commentary or rationale regarding interventions or the surrounding circumstances; as well as a review of any associated laboratory or diagnostic reports and nursing notes. If the order was not altered by the physician within 24 hours (ie, the time frame in which orders must be reviewed by the prescriber per institutional standards) of the pharmacist’s protocol change it was deemed accepted by the physician. Changes made within 24 hours for clinical reasons unrelated to the protocol change as verified by documentation in the progress notes were considered as accepted. These included, for example, the discontinuation of empiric antibiotics that had been dose adjusted by the pharmacist for patients in whom infection had been ruled out or a change from the adjusted agent to one of another class (such as might occur during de-escalation of antibiotic therapy). Interventions were excluded if there were insufficient patient and/or intervention details to allow complete assessment.
For protocol evaluation, details concerning the nature of the adjustment were collected. For formulation changes, agents were classified by their bioavailability. Renal dose adjustments were classified by the patient’s estimated creatinine clearance range since interventions were not restricted to ranges or agents. Stress ulcer prophylaxis adjustments were classified as those involving initiation, changes or discontinuation of therapy. For parenteral product adjustments, the initial and final base solution and/or the change in concentration was captured. Pain management order adjustments were classified as those involving the same agent with overlapping indications or those with oral and intravenous orders for the same pain scale range. When laboratory tests were ordered, the type of test was captured.
The study was approved by the institutional review boards of Princeton HealthCare System and Rutgers.
Results
There were 145 interventions occurring between 1 February 2012 and 31 January 2013, with 144 (99.3%) of those being accepted by the prescriber. The 1 intervention that was not accepted involved an IV to oral conversion of levothyroxine. The pharmacist performed the conversion appropriately as the patient was tolerating other oral medications. However, on the day of the change, the patient refused all oral medications despite having the ability to accept them and, as a result, all medications were converted back to parenteral formulations.
Pharmacist Evaluation
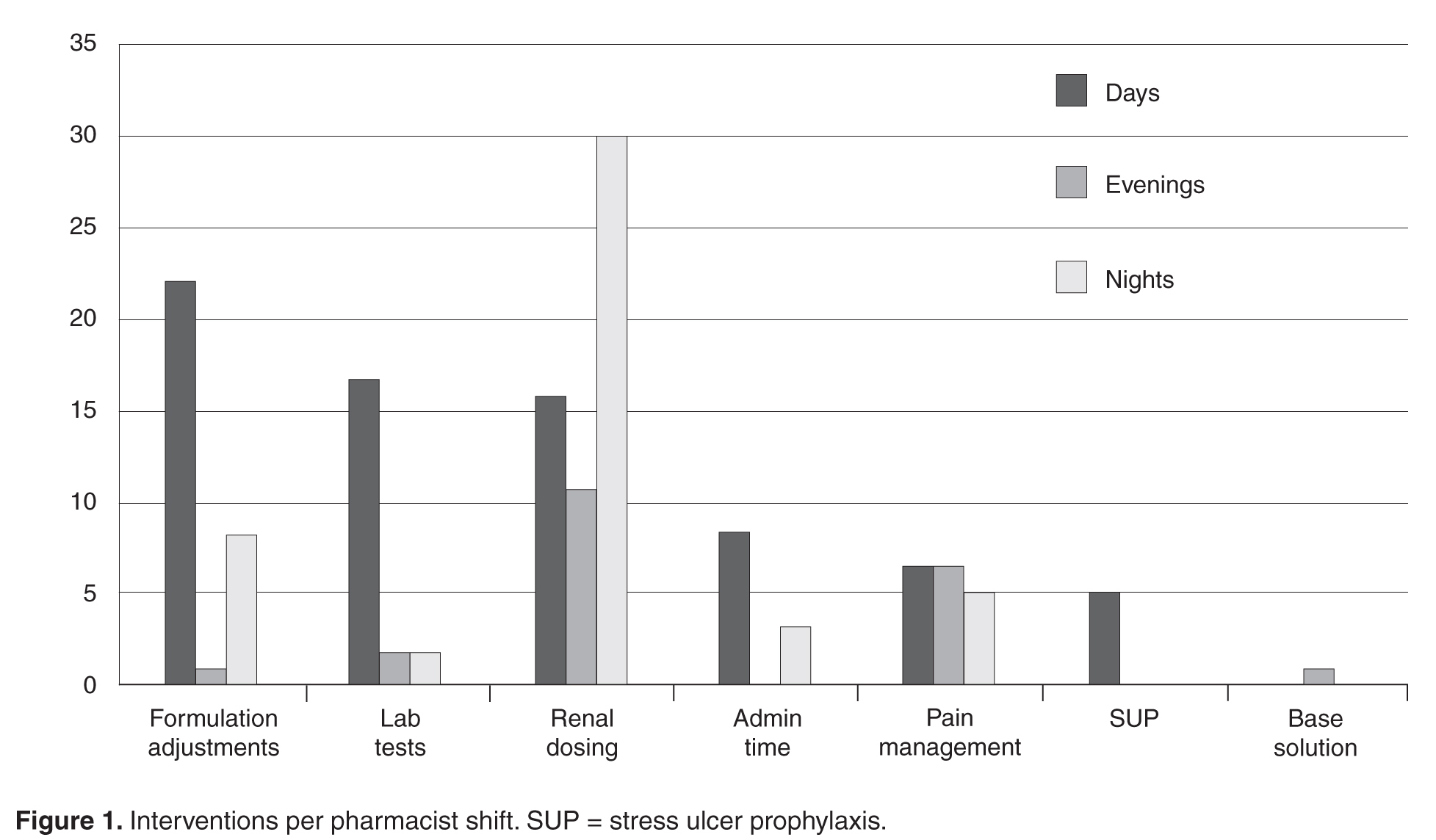
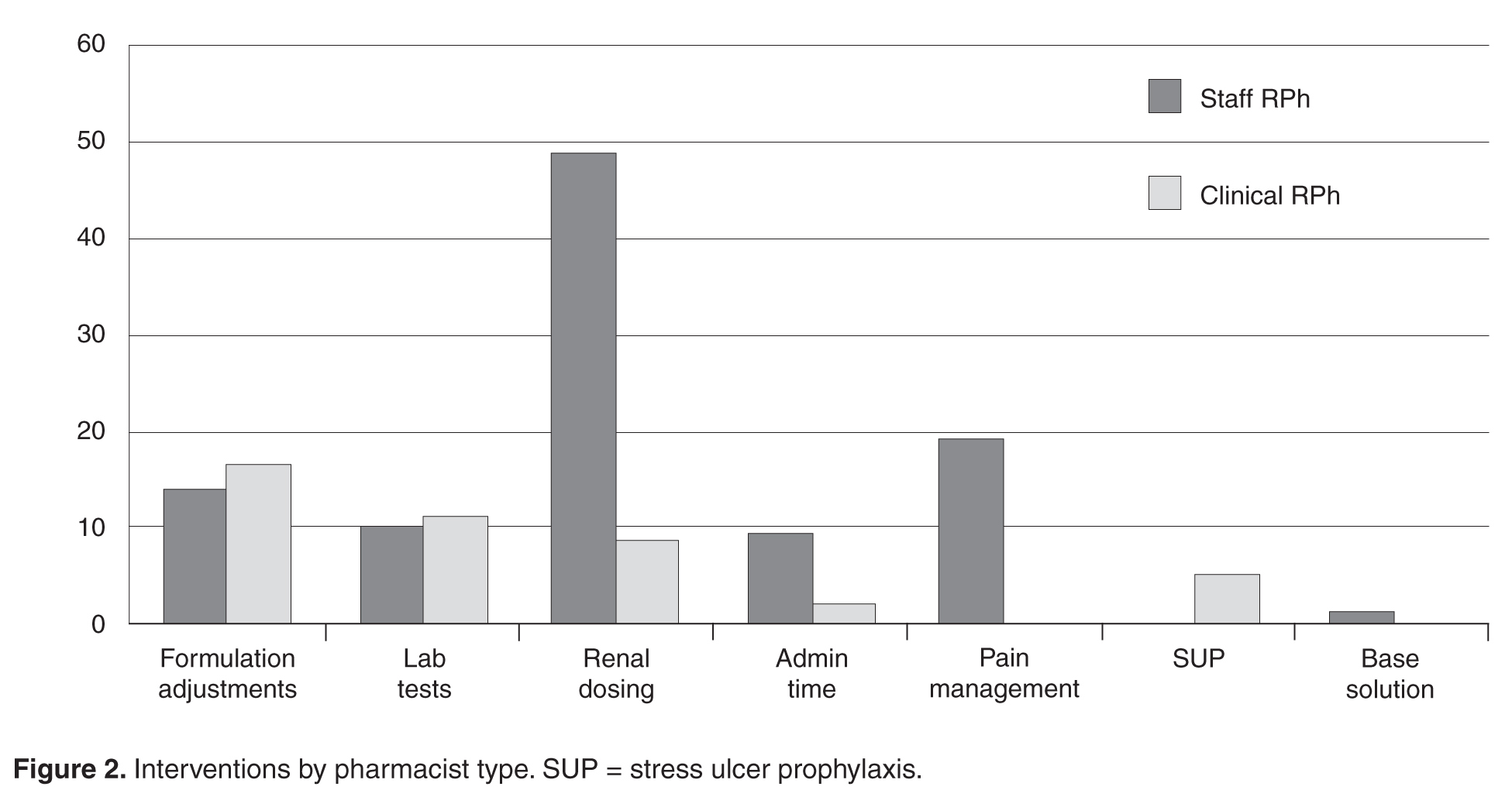
Prescriber Evaluation
An evaluation of prescribers revealed that the primary physician groups (ie, order generators) involved were hospitalists (n = 32) and critical care attendings (n = 24) at 22% and 17% of all orders, respectively. The remaining 89 interventions were distributed across other attending types (including general medicine physicians, specialty physicians and surgeons) and trainees (residents and fellows) with no more than eight orders for any individual physician category.
Protocol Evaluation
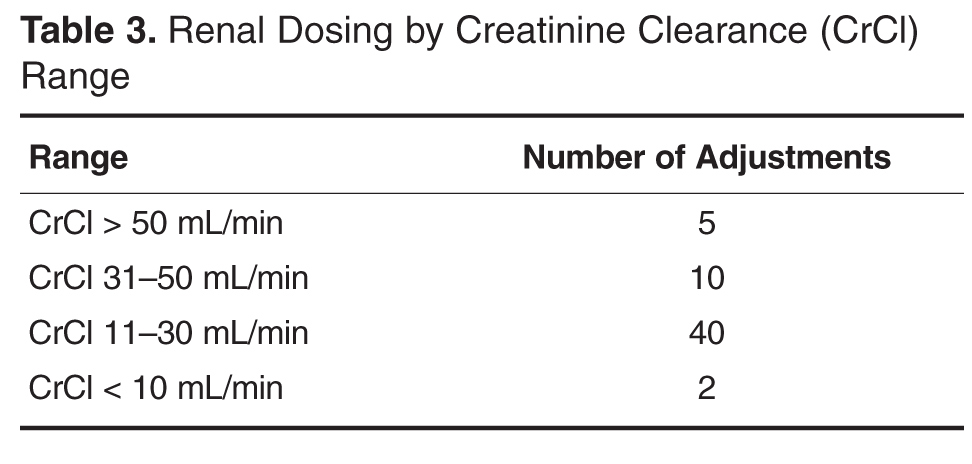
The total number of laboratory tests ordered accounted for 14% (n = 21) of all interventions. Studies related to the management of anti-infective agents and blood formation, coagulation, and thrombosis agents consisted of the majority of the lab tests ordered; INR/PTT and vancomycin levels were the most commonly ordered. Thirteen percent (n = 19) of all interventions include pain management adjustments with an even distribution between pain medications.
Several protocols were less frequently used, specifically the stress ulcer prophylaxis protocol (representing 3% of all interventions or n = 5), base solution changes (< 1% of all interventions or n = 1), and adjustment of administration time (7.6% of all interventions, n = 11). Of the time adjustments, more than 50% (n = 6) involved furosemide.
Discussion
While the literature has many studies describing pharmacists improving outcomes through successful provision of clinical programs by protocol in the acute care hospital setting, the majority of studies are limited to single or focused protocols [2–24,27,37,38]. This approach fails to recognize or limits application of a pharmacist’s expertise in pharmacotherapy, as intervention is permitted only on defined agents under specific circumstances. This is the only report we are aware of that addresses a broader approach in permitting pharmacists to optimize pharmaco-therapy during the course of their usual practice through a single policy. As better outcomes are associated with allowing professionals to work to the fullest extent of their expertise, a broad range of protocols identified as pharmacy clinical services were selected and integrated into a singular policy that would be the foundation for instituting cultural change in regard to the elements considered to be routine pharmacy practice. Thus, the protocols applied here did not specify agents that could be adjusted for renal function or classes for which formulation conversion were permissible. This is also the case for dose formulation adjustments, where the protocol allowed for the pharmacist to apply their expertise beyond 1:1 conversions using standardized drug information references (Table 1 and Table 2). As such, the protocols allowed for the full application of the pharmacist’s expertise as a pharmacotherapy consultant within these intervention categories to assure that therapies are optimized. Additionally, eliminating phone calls streamlined the workflow for both the pharmacist and physicians, thus minimizing interruptions that distract from the other functions in which they are engaged.
During the approval process, physicians inquired whether all pharmacists were equally capable of making the clinical judgments involved with the protocols as described and, thusly, whether protocol management should be limited to clinical pharmacists who have less traditional dispensing roles and more experience and time at the bedside. During those discussions we contended that the nature of these protocols were fundamental and applicable to all practicing pharmacists and, if limited, would result in missed opportunities as the clinical pharmacists are focused in specialized areas during weekdays only at UMCPP. For example, a single, centralized night-shift pharmacist could make routine dose or formulation adjustments without the need to awaken a physician as the UMCPP electronic medical record makes available all progress notes, laboratory results, and diagnostics crucial to clinical decision making. All pharmacists, regardless of job title, meet the same requirements for licensure. Post-doctoral residency or fellowship training and advanced certifications in specialty areas of practice exist among both groups as well. The study results support the validity of this argument. The majority of interventions were successfully performed by staff pharmacists with involvement from all shifts, including a third that occurred overnight. This is important because, like at most hospitals, the UMCPP staffing ratio decreases throughout the course of the day presenting changing workflow challenges throughout different shifts.
Several limitations of this study should be noted. Due to its retrospective nature, it is likely that not all interventions were captured. Some decentralized pharmacists reported not emailing interventions as they had verbally communicated the adjustments prior to having the opportunity to send the email. Four interventions could not be assessed as the email notification did not contain all the required patient identifiers or intervention information to permit for appropriate evaluation. The hospital also moved to a newly built facility in the fourth month of protocol implementation, which required significant changes in drug distribution methods, and this could have contributed to the small sample size of interventions. The move temporarily shifted departmental resources to support operational needs.
Another important factor is the voluntary nature of the policy; while it was within the pharmacist’s professional judgment to apply the protocols, pharmacists were encouraged to contact prescribers if there was any ambiguity. Therefore, while one might have expected more resident physicians to be involved with orders that were adjusted, the UMCPP practice philosophy supports contacting training physicians about changes so that they may learn from the discussion to support developing stronger prescribing habits. Future development should therefore support more universal protocol application to all eligible patients to optimize the benefits described here. Lastly, data measuring the clinical outcomes and time savings or increased productivity secondary to the elimination of physician phone calls was not directly measured. We thus sought to first demonstrate to the physician base that pharmacists could successfully apply a variety of protocols that were broader than those formally studied with equal accuracy. With that effectiveness established, future studies should explore if broader protocol application produces a greater optimization of outcomes.
After the study was completed, a survey was conducted of the pharmacists to assess perceptions and guide further policy development. We received a 63.6% response rate (14 of 22 possible respondents) with a strong majority of the respondents expressing a favorable perception of the protocols. A few respondents indicated some protocols were infrequently utilized and there was limited familiarity with others. We anticipate this is largely based on various shift and unit assignments that would make some protocols more applicable than others to the populations serviced. One of the survey questions polled the respondents on the necessity of the email notification to the prescriber given that this practice is of a higher level of notification than other established hospital protocols which only requires a notation of the change within the medication order. Seventy-one percent (n = 10) of respondents favored removing the email notification, citing primarily that it would be consistent with physician comments regarding the existing notifications. Pharmacists also identified further areas of protocol development including electrocardiogram ordering for QTc monitoring, implementation of a standardized vancomycin dosing protocol, discontinuation of duplicate orders, product substitution for nonformulary items and addition of a protocol for pharmacists to order over-the-counter or nonprescription products as they would in a community setting. This input will shape the revision of the policy and its protocols.
Conclusion
Consistent with the published literature, pharmacists effectively performed pharmacotherapy interventions in a multitude of practice categories for adult inpatients of an acute care community-teaching hospital using a single, comprehensive clinical policy. Providing these broadly scoped protocols in a singular policy allowed pharmacists to increase the autonomy with which they applied their pharmacotherapy expertise during the course of their routine, prospective care and expanded the established benefit of allowing professionals to work to their fullest extent. Pharmacist protocol intervention was met with a high physician acceptance rate.
Acknowledgment: We thank all the pharmacists at UMCPP for supporting our efforts to refine pharmacy practice for our patients.
Corresponding author: Liza Barbarello Andrews, PharmD, BSPharm, BCPS, Rutgers, The State University of New Jersey, 160 Frelinghuysen Rd, Piscataway, NJ 08854, [email protected].
Financial disclosures: None.
1. Bond CA, Raehl CL. Pharmacist provided anticoagulation management in United States hospitals: death rates, length of stay, Medicare charges, bleeding complications and transfusions. Pharmacother 2004;24:953–63.
2. Yen YH, Chen HY, Wuan-Jin L, et al. Clinical and economic impact of a pharmacist-managed iv-to-po conversion service for levofloxacin in Taiwan. Int J Clin Pharmacol Ther 2012;50:136–41.
3. Buyle F, Vogelaers D, PelemanR, et al. Implementation of guidelines for sequential therapy with fluoroquinolones in a Belgian hospital. Pharm World Sci 2010;32:404–10.
4. Davis SL, Delgado G, McKinnon PS. Pharmacoeconomic consideration associated with the use of intravenous-to-oral moxifloxacin for community-acquired pneumonia Clin Infect Dis 2005;41Supp2;5:136–43.
5. Ho BP, Lau TT, Balen RM, et al. The impact of a pharmacist-managed dosage form conversion service on ciprofloxacin usage at a major Canadian teaching hospital: a pre- and post-intervention study. BMC Health Serv Res 2005;5:48.
6. Kuti JL, Le TN, Nightingale CH, et al. Pharmacoeconomics of a pharmacist-managed program for automatically converting levofloxacin route from iv to oral. Am J Health Sys Pharm 2002;59:2209–15.
7. Cohen SM, Lipsett PA, Buchman TG, et al. Comparison of intravenous/oral ciprofloxacin plus metronidazole versus piperacillin/tazobactam in the treatment of complicated intraabdominal infections. Ann Surg 2000;232:254–62.
8. Plouffe J, Schwartz DB, Kolokathis A, et al. Clinical efficacy of intravenous followed by oral azithromycin monotherapy in hospitalized patients with community-acquired pneumonia. Antimicrob Agents Chemother 2000;44:1796–802.
9. Wetzstein GA. Intravenous to oral (IV:PO) anti-infective combination therapy. Cancer Control 2000;7:170–6.
10. Paladino JA. Pharmacoeconomics of antimicrobial therapy. Am J Healthsys Pharm 1999;56(Supp3):S25–8.
11. Ahkee S, Smith S, et al. Early switch from intravenous to oral antibiotics in hospitalized patient with infections: a six-month prospective study. Pharmacotherapy 1997;17:569–75.
12. Przybylski KG, Rybak MJ, Martin PR, et al. A pharmacist-initiated program of intravenous to oral antibiotic conversion. Pharmacotherapy 1997;17:271–6.
13. Jewesson P. Cost-effectiveness and value of an IV switch. Pharmacoeconomics 1994;5(Supp2):20–6.
14. Ramirez J. Advances in antibiotic use: switch therapy. Curr Ther Res 1994;55(suppA):30–3.
15. Shepard MF. Making the switch from IV to PO. Am J Healthsys Pharm 1994;50:2510.
16. Chessin LN. When to switch from IV to oral antibiotics. Patient Care 1993;15:113–25.
17. Cohen MR. Important news! IV route not needed to justify hospitalization for antibiotics. Hospital Pharmacy 1993;28:946.
18. Schentag JJ. Changes in antimicrobial agent usage resulting from interactions among clinical pharmacy, infectious disease division and the microbiology laboratory. Diagnos Microbiol Infect Dis 1993;16:255–64.
19. Fighetto L. Intravenous to oral stepdown program: four years’ experience in a large teaching hospital. Ann Pharmacother 1992;26:1447–51.
20. Powers T. Clinical and economic effect of ciprofloxacin as an alternative to injectable antimicrobial therapy. Am J Healthsys Pharm 1990;47:1781–4.
21. Ament PW, McGuire WM. Setting up an automatic pharmacist-initiated pharmacokinetic dosing service. Hosp Formul 1993;28:589–92.
22. Mousavi M, Dashti-Khavidaki S, Khalili H, et al. Impact of clinical pharmacy services on stress ulcer prophylaxis prescribing and related cost in patients with renal insufficiency. Int J Pharm Pract 2012;Nov 9.
23. Damaske DL, Baird RW. Development and implementation of a pharmacist managed inpatient warfarin protocol. Proc (Baylor Univ Med Cent) 2005;18:397–400.
24. Radley AS, Hall J, Farrow M, et al. Evaluation of anticoagulation control in a pharmacist operated anticoagulation clinic. J Clin Pathol 1995;48:545–47.
25. Pharmacy and Therapeutics Committee. Medication policies and protocols of Nebraska Methodist Hospital, Methodist Women’s Hospital.
26. University of Kentucky Pharmacy Services. Therapeutic interchanges [Internet]. Available at www.hosp.uky.edu/pharmacy/interchange.asp
27. Bayshore Community Hospital Department of Pharmacy. Conversion of intravenous azithromycin (Zithromax), ceftriaxone (Rocephin), ciprofloxacin (Cipro), fluconazole (Diflucan), lansoprazole (Prevacid), levofloxacin (Levaquin), linezolid (Zyvox), metronidazole (Flagyl), moxifloxacin (Avelox), potassium chloride (KC), or ranitidine (Zantac) to oral medication [Internet]. [cited 2013 May 9]. Available at www.ashp.org/s_ashp/docs/files/R-IVtoPOConvPol-2.pdf.
28. Medication Education Safety Approval Committee, Massachusetts General Hospital. Automatic intravenous to oral protocol [Internet]. MESAC memo; 2005 July [cited 2013 March 9]. Available at www2.massgeneral.org/pharmacy/mesac/mesac_memo3.pdf.
29. Hanson RL, Habibi M, Khamo N, et al. Integrated clinical and specialty pharmacy practice model for management of patients with multiple sclerosis. Am J Health Syst Pharm 2014;71:463–9.
30. Making pharmacists part of the multidisciplinary team. Hosp Case Manag 2014;22:13–6.
31. Preslaski CR, Lat I, MacLaren R, et al. Pharmacist contributions as members of the multidisciplinary ICU team. Chest 2013;144:1687–95.
32. Ripley TL, Adamson PB, Hennebry TA, et al. Collaborative practice model between cardiologists and clinical pharmacist for management of patients with cardiovascular disease in an outpatient clinic. Ann Pharmacother 2014;48:412–9.
33. Smith M, Bates DW, Bodenheimer TS. Pharmacists belong in accountable care organizations and integrated care teams. Health Aff (Millwood) 2013;32:1963–70.
34. Ukens C. New Jersey rewrites its state pharmacy practice act. Drug Topics 2004;148:41.
35. New Jersey Board of Pharmacy Laws. Statute 45:14-64. Inapplicability relative to collaborative drug therapy management in hospital. [Internet] July 2011 [cited 11 September 14]. Available from: www.njconsumeraffairs.gov/laws/pharmlaws.pdf.
36. ASHP Commission on Therapeutics. ASHP therapeutic guidelines on stress ulcer prophylaxis. Am J Health Syst Pharm 1999;56:347–79.
37. Bond CA, Raehl CL. Clinical pharmacy services, pharmacy staffing, and hospital mortality rates. Pharmacotherapy 2007;27:481–93.
38. Bond CA, Raehl CL, Franke T. Interrelationships among mortality rates, drug costs, total cost of care, and length of stay in United States hospitals: summary and recommendations for clinical pharmacy services and staffing. Pharmacotherapy 2001;21:129–41.
From the Ernest Mario School of Pharmacy, Rutgers, The State University of New Jersey, Piscataway, NJ.
Abstract
- Objectives: To describe the implementation of broadly scoped clinical pharmacy protocols positioned as a singular policy in a community hospital. These protocols were designed to expand the established benefits demonstrated using narrower, traditional protocols.
- Methods: A retrospective chart review of protocol interventions in the first year of the policy’s implementation was conducted to evaluate prescriber acceptance of protocol interventions. Interventions were identified from required email notifications. The frequency of use of each protocol was assessed, including evaluation of novel characteristics of specific protocols. Pharmacist utilization patterns were assessed for job classification, shift, and practice setting (ie, centralized or decentralized).
- Results: In the 1-year assessment period, 145 interventions were reported and 144 were accepted by the prescribing physicians. Interventions involved orders from hospitalists and intensivists most frequently, with the renal dosing and dose formulations protocols being the most commonly utilized. Staff pharmacists used the policy more frequently than clinical pharmacists, primarily during day shift from decentralized locations on the patient care units.
- Conclusions: The implementation of broadly scoped clinical pharmacy protocols for items our pharmacists routinely contact physicians about (and our physicians deemed were within the practice of pharmacy) instituted a cultural shift that expanded the elements considered to be part of routine pharmacy practice. As a result, pharmacists more seamlessly applied their expertise as pharmacotherapy specialists to optimize pharmacotherapy, which streamlined workflow for both pharmacists and physicians. This expanded the proven benefits of allowing professionals to work to their fullest extent, as established in the literature.
Allowing pharmacists to apply their expertise has been associated with improved outcomes in both pharmacotherapy quality (eg, reduction in mortality and length of stay [1]) and savings in health care dollars. Studies of focused protocols, including intravenous-to-oral (IV-to-PO) switch [2–20], renal dosing [21], stress ulcer prophylaxis [22] and anticoagulation management [1,23,24] demonstrate these benefits in a multitude of practice areas. While such protocols have become commonplace in the acute care setting [25–28], most continue to be singularly focused and impose patient population restrictions that preclude comprehensive patient evaluation. Many are administered as a task within the pharmacist workflow using a patient list generated by the limited protocol criteria, which are often restricted to agent or patient characteristics.
Better outcomes are associated with permitting professionals such as pharmacists to work to the fullest extent of their scope and expertise [29–31]. In specific cases, studies evaluating pharmacists’ impact within a multi-disciplinary health care team have demonstrated improved outcomes in regard to both patient care and cost [29–31]. Recognizing this, accountable care organizations (ACOs) have developed practice models that are based on this benefit. Each team member is expected to robustly apply their training and expertise to achieve the best outcomes [32,33]. As health care moves toward a more integrative approach, it is paramount that pharmacists utilize the full scope of the skills in which they are trained.
This report describes the development, implementation, and outcomes of a singular policy outlining comprehensively scoped protocols allowing acute care hospital pharmacists within Princeton HealthCare System to optimize pharmacotherapy during the course of their usual clinical practice.
Methods
Setting
The University Medical Center of Princeton at Plainsboro (UMCPP), part of the Princeton HealthCare System, is a 230-bed community acute care hospital located in central New Jersey. The hospital facility relocated in May 2012 from its previous location in Princeton to a new state-of-the-art facility in Plainsboro. As an affiliate of the Robert Wood Johnson Medical School and the Ernest Mario School of Pharmacy at Rutgers, The State University of New Jersey (ie, Rutgers), it is an academic teaching hospital with a mixed model for providing patient care. UMCPP employs both faculty physicians leading academic teams alongside hospitalists and private attendings.
Pharmacy services are provided on facility 24 hours a day, 365 days a year. The department of pharmacy services provides a full scope medication services from a centralized location with 3 full-time day pharmacists and 1 oncology satellite pharmacist. During weekdays, decentralized pharmacists provide medication review, patient education, and medication reconciliation on 2 to 3 inpatient care units. Centralized support decreases to 2 pharmacists in the evening and 1 overnight. Clinical pharmacists, both hospital-based and Rutgers faculty, work in conjunction with the staff pharmacists to ensure appropriate management of patients throughout different levels of care.
Program Overview and Implementation
To enhance protocols allowing pharmacists to more holistically and robustly optimize pharmacotherapy, UMCPP implemented the Clinical Pharmacy Services policy in February 2012. The policy outlined 8 protocols through which registered pharmacists within the acute care hospital could implement outlined medication order adjustments for adults of inpatient status. Pediatric patients or those treated outside of the acute care hospital (eg, in the psychiatric hospital, surgical center or outpatient facilities) were excluded. While the hospital had existing traditional programs such as IV-to-PO conversions, the programs were restricted to specific agents or conditions. As such, pharmacists were assigned to review queues in the clinical computer system to which orders for the agents outlined by the specific program would flow. Review would occur at set intervals and focus on that detail of the patient’s care as opposed to broadly encompassing an evaluation of the patient’s comprehensive pharmacotherapy. The goal of the new policy was to better utilize the pharmacists’ expertise by broadening these assessments to all applicable agents, refine workflow (by allowing protocol management instead of requiring individual prescriber calls for each issue) and integrate holistic refinement of pharmacotherapy regimens during the usual course of the pharmacist’s clinical care.
In the state of New Jersey, the Pharmacy Practice Act (updated on 14 January 2004) formally recognizes pharmacists as health care professionals and permits for collaborative practice in the community setting [34]. However, pharmacist management by protocol in the acute care hospital setting is defined separately, requiring only medical approvals within the system [35]. In accordance, the policy and associated protocols were approved by the institution’s multidisciplinary pharmacy and therapeutics (P&T) and medical executive committee processes.
To ensure appropriate oversight, the policy required that the pharmacist making changes submit notification of protocol intervention to the patient’s attending physician, the physician who generated the original order (if other than the attending) and a designated clinical pharmacist (for auditing purposes). All notifications were made via email within the clinical computer system in “interrupt” status to ensure active recognition by the prescriber(s).
Program Evaluation
An evaluation of the first year’s interventions was conducted to validate the program, describe its utility, and provide a basis for re-evaluation and continued evolution. The aim was to evaluate the institution’s experience with the program, focusing on both specific physician and pharmacist elements. One of the primary goals was to evaluate which physician’s orders were associated with interventions as well as the rate of physician acceptance of protocol interventions, as their acceptance clearly validates the pharmacist’s ability to appropriately apply the protocols in patient-specific contexts.
To evaluate the pharmacist’s experience, trends in pharmacist utilization were captured, including which pharmacist by job classification (ie, staff or clinical pharmacist) implemented interventions, during which shift, and in what operational capacity (ie, centralized or decentralized) the pharmacist was practicing. Lastly, the study sought to characterize the frequency to which each protocol was applied. Based on the existing experiences described in the literature as well as with consideration of institutional culture and operation, we hypothesized that all pharmacists would apply protocols with equal efficacy with more interventions likely generated by staff pharmacists due to their role in primary order review and that the types of interventions would vary based on shift and location.
A retrospective review of cases throughout the first year of the policy’s implementation was conducted, including interventions made between 1 February 2012 and 31 January 2013. Cases were identified through the required email notification of the auditing clinical pharmacist. The patient’s electronic medical record for that defined visit was reviewed. To assess pharmacist utilization patterns, data captured included the agent involved in the intervention, date, day of week and shift, whether the pharmacist was centralized or decentralized, and whether that pharmacist was classified as staff or clinical. Decentralized pharmacists were defined as a pharmacist working on the patient care unit with direct access to other practitioners and patients, rather than those performing their functions from within the confines of the pharmacy department.
Prescribers were described both by status (ie, attending or resident/training) and specialty. Physician acceptance was assessed through evaluation of order trends as the electronic medical record allows for all changes to an order to be audited and tracked; a review of progress notes to capture any commentary or rationale regarding interventions or the surrounding circumstances; as well as a review of any associated laboratory or diagnostic reports and nursing notes. If the order was not altered by the physician within 24 hours (ie, the time frame in which orders must be reviewed by the prescriber per institutional standards) of the pharmacist’s protocol change it was deemed accepted by the physician. Changes made within 24 hours for clinical reasons unrelated to the protocol change as verified by documentation in the progress notes were considered as accepted. These included, for example, the discontinuation of empiric antibiotics that had been dose adjusted by the pharmacist for patients in whom infection had been ruled out or a change from the adjusted agent to one of another class (such as might occur during de-escalation of antibiotic therapy). Interventions were excluded if there were insufficient patient and/or intervention details to allow complete assessment.
For protocol evaluation, details concerning the nature of the adjustment were collected. For formulation changes, agents were classified by their bioavailability. Renal dose adjustments were classified by the patient’s estimated creatinine clearance range since interventions were not restricted to ranges or agents. Stress ulcer prophylaxis adjustments were classified as those involving initiation, changes or discontinuation of therapy. For parenteral product adjustments, the initial and final base solution and/or the change in concentration was captured. Pain management order adjustments were classified as those involving the same agent with overlapping indications or those with oral and intravenous orders for the same pain scale range. When laboratory tests were ordered, the type of test was captured.
The study was approved by the institutional review boards of Princeton HealthCare System and Rutgers.
Results
There were 145 interventions occurring between 1 February 2012 and 31 January 2013, with 144 (99.3%) of those being accepted by the prescriber. The 1 intervention that was not accepted involved an IV to oral conversion of levothyroxine. The pharmacist performed the conversion appropriately as the patient was tolerating other oral medications. However, on the day of the change, the patient refused all oral medications despite having the ability to accept them and, as a result, all medications were converted back to parenteral formulations.
Pharmacist Evaluation
Prescriber Evaluation
An evaluation of prescribers revealed that the primary physician groups (ie, order generators) involved were hospitalists (n = 32) and critical care attendings (n = 24) at 22% and 17% of all orders, respectively. The remaining 89 interventions were distributed across other attending types (including general medicine physicians, specialty physicians and surgeons) and trainees (residents and fellows) with no more than eight orders for any individual physician category.
Protocol Evaluation
The total number of laboratory tests ordered accounted for 14% (n = 21) of all interventions. Studies related to the management of anti-infective agents and blood formation, coagulation, and thrombosis agents consisted of the majority of the lab tests ordered; INR/PTT and vancomycin levels were the most commonly ordered. Thirteen percent (n = 19) of all interventions include pain management adjustments with an even distribution between pain medications.
Several protocols were less frequently used, specifically the stress ulcer prophylaxis protocol (representing 3% of all interventions or n = 5), base solution changes (< 1% of all interventions or n = 1), and adjustment of administration time (7.6% of all interventions, n = 11). Of the time adjustments, more than 50% (n = 6) involved furosemide.
Discussion
While the literature has many studies describing pharmacists improving outcomes through successful provision of clinical programs by protocol in the acute care hospital setting, the majority of studies are limited to single or focused protocols [2–24,27,37,38]. This approach fails to recognize or limits application of a pharmacist’s expertise in pharmacotherapy, as intervention is permitted only on defined agents under specific circumstances. This is the only report we are aware of that addresses a broader approach in permitting pharmacists to optimize pharmaco-therapy during the course of their usual practice through a single policy. As better outcomes are associated with allowing professionals to work to the fullest extent of their expertise, a broad range of protocols identified as pharmacy clinical services were selected and integrated into a singular policy that would be the foundation for instituting cultural change in regard to the elements considered to be routine pharmacy practice. Thus, the protocols applied here did not specify agents that could be adjusted for renal function or classes for which formulation conversion were permissible. This is also the case for dose formulation adjustments, where the protocol allowed for the pharmacist to apply their expertise beyond 1:1 conversions using standardized drug information references (Table 1 and Table 2). As such, the protocols allowed for the full application of the pharmacist’s expertise as a pharmacotherapy consultant within these intervention categories to assure that therapies are optimized. Additionally, eliminating phone calls streamlined the workflow for both the pharmacist and physicians, thus minimizing interruptions that distract from the other functions in which they are engaged.
During the approval process, physicians inquired whether all pharmacists were equally capable of making the clinical judgments involved with the protocols as described and, thusly, whether protocol management should be limited to clinical pharmacists who have less traditional dispensing roles and more experience and time at the bedside. During those discussions we contended that the nature of these protocols were fundamental and applicable to all practicing pharmacists and, if limited, would result in missed opportunities as the clinical pharmacists are focused in specialized areas during weekdays only at UMCPP. For example, a single, centralized night-shift pharmacist could make routine dose or formulation adjustments without the need to awaken a physician as the UMCPP electronic medical record makes available all progress notes, laboratory results, and diagnostics crucial to clinical decision making. All pharmacists, regardless of job title, meet the same requirements for licensure. Post-doctoral residency or fellowship training and advanced certifications in specialty areas of practice exist among both groups as well. The study results support the validity of this argument. The majority of interventions were successfully performed by staff pharmacists with involvement from all shifts, including a third that occurred overnight. This is important because, like at most hospitals, the UMCPP staffing ratio decreases throughout the course of the day presenting changing workflow challenges throughout different shifts.
Several limitations of this study should be noted. Due to its retrospective nature, it is likely that not all interventions were captured. Some decentralized pharmacists reported not emailing interventions as they had verbally communicated the adjustments prior to having the opportunity to send the email. Four interventions could not be assessed as the email notification did not contain all the required patient identifiers or intervention information to permit for appropriate evaluation. The hospital also moved to a newly built facility in the fourth month of protocol implementation, which required significant changes in drug distribution methods, and this could have contributed to the small sample size of interventions. The move temporarily shifted departmental resources to support operational needs.
Another important factor is the voluntary nature of the policy; while it was within the pharmacist’s professional judgment to apply the protocols, pharmacists were encouraged to contact prescribers if there was any ambiguity. Therefore, while one might have expected more resident physicians to be involved with orders that were adjusted, the UMCPP practice philosophy supports contacting training physicians about changes so that they may learn from the discussion to support developing stronger prescribing habits. Future development should therefore support more universal protocol application to all eligible patients to optimize the benefits described here. Lastly, data measuring the clinical outcomes and time savings or increased productivity secondary to the elimination of physician phone calls was not directly measured. We thus sought to first demonstrate to the physician base that pharmacists could successfully apply a variety of protocols that were broader than those formally studied with equal accuracy. With that effectiveness established, future studies should explore if broader protocol application produces a greater optimization of outcomes.
After the study was completed, a survey was conducted of the pharmacists to assess perceptions and guide further policy development. We received a 63.6% response rate (14 of 22 possible respondents) with a strong majority of the respondents expressing a favorable perception of the protocols. A few respondents indicated some protocols were infrequently utilized and there was limited familiarity with others. We anticipate this is largely based on various shift and unit assignments that would make some protocols more applicable than others to the populations serviced. One of the survey questions polled the respondents on the necessity of the email notification to the prescriber given that this practice is of a higher level of notification than other established hospital protocols which only requires a notation of the change within the medication order. Seventy-one percent (n = 10) of respondents favored removing the email notification, citing primarily that it would be consistent with physician comments regarding the existing notifications. Pharmacists also identified further areas of protocol development including electrocardiogram ordering for QTc monitoring, implementation of a standardized vancomycin dosing protocol, discontinuation of duplicate orders, product substitution for nonformulary items and addition of a protocol for pharmacists to order over-the-counter or nonprescription products as they would in a community setting. This input will shape the revision of the policy and its protocols.
Conclusion
Consistent with the published literature, pharmacists effectively performed pharmacotherapy interventions in a multitude of practice categories for adult inpatients of an acute care community-teaching hospital using a single, comprehensive clinical policy. Providing these broadly scoped protocols in a singular policy allowed pharmacists to increase the autonomy with which they applied their pharmacotherapy expertise during the course of their routine, prospective care and expanded the established benefit of allowing professionals to work to their fullest extent. Pharmacist protocol intervention was met with a high physician acceptance rate.
Acknowledgment: We thank all the pharmacists at UMCPP for supporting our efforts to refine pharmacy practice for our patients.
Corresponding author: Liza Barbarello Andrews, PharmD, BSPharm, BCPS, Rutgers, The State University of New Jersey, 160 Frelinghuysen Rd, Piscataway, NJ 08854, [email protected].
Financial disclosures: None.
From the Ernest Mario School of Pharmacy, Rutgers, The State University of New Jersey, Piscataway, NJ.
Abstract
- Objectives: To describe the implementation of broadly scoped clinical pharmacy protocols positioned as a singular policy in a community hospital. These protocols were designed to expand the established benefits demonstrated using narrower, traditional protocols.
- Methods: A retrospective chart review of protocol interventions in the first year of the policy’s implementation was conducted to evaluate prescriber acceptance of protocol interventions. Interventions were identified from required email notifications. The frequency of use of each protocol was assessed, including evaluation of novel characteristics of specific protocols. Pharmacist utilization patterns were assessed for job classification, shift, and practice setting (ie, centralized or decentralized).
- Results: In the 1-year assessment period, 145 interventions were reported and 144 were accepted by the prescribing physicians. Interventions involved orders from hospitalists and intensivists most frequently, with the renal dosing and dose formulations protocols being the most commonly utilized. Staff pharmacists used the policy more frequently than clinical pharmacists, primarily during day shift from decentralized locations on the patient care units.
- Conclusions: The implementation of broadly scoped clinical pharmacy protocols for items our pharmacists routinely contact physicians about (and our physicians deemed were within the practice of pharmacy) instituted a cultural shift that expanded the elements considered to be part of routine pharmacy practice. As a result, pharmacists more seamlessly applied their expertise as pharmacotherapy specialists to optimize pharmacotherapy, which streamlined workflow for both pharmacists and physicians. This expanded the proven benefits of allowing professionals to work to their fullest extent, as established in the literature.
Allowing pharmacists to apply their expertise has been associated with improved outcomes in both pharmacotherapy quality (eg, reduction in mortality and length of stay [1]) and savings in health care dollars. Studies of focused protocols, including intravenous-to-oral (IV-to-PO) switch [2–20], renal dosing [21], stress ulcer prophylaxis [22] and anticoagulation management [1,23,24] demonstrate these benefits in a multitude of practice areas. While such protocols have become commonplace in the acute care setting [25–28], most continue to be singularly focused and impose patient population restrictions that preclude comprehensive patient evaluation. Many are administered as a task within the pharmacist workflow using a patient list generated by the limited protocol criteria, which are often restricted to agent or patient characteristics.
Better outcomes are associated with permitting professionals such as pharmacists to work to the fullest extent of their scope and expertise [29–31]. In specific cases, studies evaluating pharmacists’ impact within a multi-disciplinary health care team have demonstrated improved outcomes in regard to both patient care and cost [29–31]. Recognizing this, accountable care organizations (ACOs) have developed practice models that are based on this benefit. Each team member is expected to robustly apply their training and expertise to achieve the best outcomes [32,33]. As health care moves toward a more integrative approach, it is paramount that pharmacists utilize the full scope of the skills in which they are trained.
This report describes the development, implementation, and outcomes of a singular policy outlining comprehensively scoped protocols allowing acute care hospital pharmacists within Princeton HealthCare System to optimize pharmacotherapy during the course of their usual clinical practice.
Methods
Setting
The University Medical Center of Princeton at Plainsboro (UMCPP), part of the Princeton HealthCare System, is a 230-bed community acute care hospital located in central New Jersey. The hospital facility relocated in May 2012 from its previous location in Princeton to a new state-of-the-art facility in Plainsboro. As an affiliate of the Robert Wood Johnson Medical School and the Ernest Mario School of Pharmacy at Rutgers, The State University of New Jersey (ie, Rutgers), it is an academic teaching hospital with a mixed model for providing patient care. UMCPP employs both faculty physicians leading academic teams alongside hospitalists and private attendings.
Pharmacy services are provided on facility 24 hours a day, 365 days a year. The department of pharmacy services provides a full scope medication services from a centralized location with 3 full-time day pharmacists and 1 oncology satellite pharmacist. During weekdays, decentralized pharmacists provide medication review, patient education, and medication reconciliation on 2 to 3 inpatient care units. Centralized support decreases to 2 pharmacists in the evening and 1 overnight. Clinical pharmacists, both hospital-based and Rutgers faculty, work in conjunction with the staff pharmacists to ensure appropriate management of patients throughout different levels of care.
Program Overview and Implementation
To enhance protocols allowing pharmacists to more holistically and robustly optimize pharmacotherapy, UMCPP implemented the Clinical Pharmacy Services policy in February 2012. The policy outlined 8 protocols through which registered pharmacists within the acute care hospital could implement outlined medication order adjustments for adults of inpatient status. Pediatric patients or those treated outside of the acute care hospital (eg, in the psychiatric hospital, surgical center or outpatient facilities) were excluded. While the hospital had existing traditional programs such as IV-to-PO conversions, the programs were restricted to specific agents or conditions. As such, pharmacists were assigned to review queues in the clinical computer system to which orders for the agents outlined by the specific program would flow. Review would occur at set intervals and focus on that detail of the patient’s care as opposed to broadly encompassing an evaluation of the patient’s comprehensive pharmacotherapy. The goal of the new policy was to better utilize the pharmacists’ expertise by broadening these assessments to all applicable agents, refine workflow (by allowing protocol management instead of requiring individual prescriber calls for each issue) and integrate holistic refinement of pharmacotherapy regimens during the usual course of the pharmacist’s clinical care.
In the state of New Jersey, the Pharmacy Practice Act (updated on 14 January 2004) formally recognizes pharmacists as health care professionals and permits for collaborative practice in the community setting [34]. However, pharmacist management by protocol in the acute care hospital setting is defined separately, requiring only medical approvals within the system [35]. In accordance, the policy and associated protocols were approved by the institution’s multidisciplinary pharmacy and therapeutics (P&T) and medical executive committee processes.
To ensure appropriate oversight, the policy required that the pharmacist making changes submit notification of protocol intervention to the patient’s attending physician, the physician who generated the original order (if other than the attending) and a designated clinical pharmacist (for auditing purposes). All notifications were made via email within the clinical computer system in “interrupt” status to ensure active recognition by the prescriber(s).
Program Evaluation
An evaluation of the first year’s interventions was conducted to validate the program, describe its utility, and provide a basis for re-evaluation and continued evolution. The aim was to evaluate the institution’s experience with the program, focusing on both specific physician and pharmacist elements. One of the primary goals was to evaluate which physician’s orders were associated with interventions as well as the rate of physician acceptance of protocol interventions, as their acceptance clearly validates the pharmacist’s ability to appropriately apply the protocols in patient-specific contexts.
To evaluate the pharmacist’s experience, trends in pharmacist utilization were captured, including which pharmacist by job classification (ie, staff or clinical pharmacist) implemented interventions, during which shift, and in what operational capacity (ie, centralized or decentralized) the pharmacist was practicing. Lastly, the study sought to characterize the frequency to which each protocol was applied. Based on the existing experiences described in the literature as well as with consideration of institutional culture and operation, we hypothesized that all pharmacists would apply protocols with equal efficacy with more interventions likely generated by staff pharmacists due to their role in primary order review and that the types of interventions would vary based on shift and location.
A retrospective review of cases throughout the first year of the policy’s implementation was conducted, including interventions made between 1 February 2012 and 31 January 2013. Cases were identified through the required email notification of the auditing clinical pharmacist. The patient’s electronic medical record for that defined visit was reviewed. To assess pharmacist utilization patterns, data captured included the agent involved in the intervention, date, day of week and shift, whether the pharmacist was centralized or decentralized, and whether that pharmacist was classified as staff or clinical. Decentralized pharmacists were defined as a pharmacist working on the patient care unit with direct access to other practitioners and patients, rather than those performing their functions from within the confines of the pharmacy department.
Prescribers were described both by status (ie, attending or resident/training) and specialty. Physician acceptance was assessed through evaluation of order trends as the electronic medical record allows for all changes to an order to be audited and tracked; a review of progress notes to capture any commentary or rationale regarding interventions or the surrounding circumstances; as well as a review of any associated laboratory or diagnostic reports and nursing notes. If the order was not altered by the physician within 24 hours (ie, the time frame in which orders must be reviewed by the prescriber per institutional standards) of the pharmacist’s protocol change it was deemed accepted by the physician. Changes made within 24 hours for clinical reasons unrelated to the protocol change as verified by documentation in the progress notes were considered as accepted. These included, for example, the discontinuation of empiric antibiotics that had been dose adjusted by the pharmacist for patients in whom infection had been ruled out or a change from the adjusted agent to one of another class (such as might occur during de-escalation of antibiotic therapy). Interventions were excluded if there were insufficient patient and/or intervention details to allow complete assessment.
For protocol evaluation, details concerning the nature of the adjustment were collected. For formulation changes, agents were classified by their bioavailability. Renal dose adjustments were classified by the patient’s estimated creatinine clearance range since interventions were not restricted to ranges or agents. Stress ulcer prophylaxis adjustments were classified as those involving initiation, changes or discontinuation of therapy. For parenteral product adjustments, the initial and final base solution and/or the change in concentration was captured. Pain management order adjustments were classified as those involving the same agent with overlapping indications or those with oral and intravenous orders for the same pain scale range. When laboratory tests were ordered, the type of test was captured.
The study was approved by the institutional review boards of Princeton HealthCare System and Rutgers.
Results
There were 145 interventions occurring between 1 February 2012 and 31 January 2013, with 144 (99.3%) of those being accepted by the prescriber. The 1 intervention that was not accepted involved an IV to oral conversion of levothyroxine. The pharmacist performed the conversion appropriately as the patient was tolerating other oral medications. However, on the day of the change, the patient refused all oral medications despite having the ability to accept them and, as a result, all medications were converted back to parenteral formulations.
Pharmacist Evaluation
Prescriber Evaluation
An evaluation of prescribers revealed that the primary physician groups (ie, order generators) involved were hospitalists (n = 32) and critical care attendings (n = 24) at 22% and 17% of all orders, respectively. The remaining 89 interventions were distributed across other attending types (including general medicine physicians, specialty physicians and surgeons) and trainees (residents and fellows) with no more than eight orders for any individual physician category.
Protocol Evaluation
The total number of laboratory tests ordered accounted for 14% (n = 21) of all interventions. Studies related to the management of anti-infective agents and blood formation, coagulation, and thrombosis agents consisted of the majority of the lab tests ordered; INR/PTT and vancomycin levels were the most commonly ordered. Thirteen percent (n = 19) of all interventions include pain management adjustments with an even distribution between pain medications.
Several protocols were less frequently used, specifically the stress ulcer prophylaxis protocol (representing 3% of all interventions or n = 5), base solution changes (< 1% of all interventions or n = 1), and adjustment of administration time (7.6% of all interventions, n = 11). Of the time adjustments, more than 50% (n = 6) involved furosemide.
Discussion
While the literature has many studies describing pharmacists improving outcomes through successful provision of clinical programs by protocol in the acute care hospital setting, the majority of studies are limited to single or focused protocols [2–24,27,37,38]. This approach fails to recognize or limits application of a pharmacist’s expertise in pharmacotherapy, as intervention is permitted only on defined agents under specific circumstances. This is the only report we are aware of that addresses a broader approach in permitting pharmacists to optimize pharmaco-therapy during the course of their usual practice through a single policy. As better outcomes are associated with allowing professionals to work to the fullest extent of their expertise, a broad range of protocols identified as pharmacy clinical services were selected and integrated into a singular policy that would be the foundation for instituting cultural change in regard to the elements considered to be routine pharmacy practice. Thus, the protocols applied here did not specify agents that could be adjusted for renal function or classes for which formulation conversion were permissible. This is also the case for dose formulation adjustments, where the protocol allowed for the pharmacist to apply their expertise beyond 1:1 conversions using standardized drug information references (Table 1 and Table 2). As such, the protocols allowed for the full application of the pharmacist’s expertise as a pharmacotherapy consultant within these intervention categories to assure that therapies are optimized. Additionally, eliminating phone calls streamlined the workflow for both the pharmacist and physicians, thus minimizing interruptions that distract from the other functions in which they are engaged.
During the approval process, physicians inquired whether all pharmacists were equally capable of making the clinical judgments involved with the protocols as described and, thusly, whether protocol management should be limited to clinical pharmacists who have less traditional dispensing roles and more experience and time at the bedside. During those discussions we contended that the nature of these protocols were fundamental and applicable to all practicing pharmacists and, if limited, would result in missed opportunities as the clinical pharmacists are focused in specialized areas during weekdays only at UMCPP. For example, a single, centralized night-shift pharmacist could make routine dose or formulation adjustments without the need to awaken a physician as the UMCPP electronic medical record makes available all progress notes, laboratory results, and diagnostics crucial to clinical decision making. All pharmacists, regardless of job title, meet the same requirements for licensure. Post-doctoral residency or fellowship training and advanced certifications in specialty areas of practice exist among both groups as well. The study results support the validity of this argument. The majority of interventions were successfully performed by staff pharmacists with involvement from all shifts, including a third that occurred overnight. This is important because, like at most hospitals, the UMCPP staffing ratio decreases throughout the course of the day presenting changing workflow challenges throughout different shifts.
Several limitations of this study should be noted. Due to its retrospective nature, it is likely that not all interventions were captured. Some decentralized pharmacists reported not emailing interventions as they had verbally communicated the adjustments prior to having the opportunity to send the email. Four interventions could not be assessed as the email notification did not contain all the required patient identifiers or intervention information to permit for appropriate evaluation. The hospital also moved to a newly built facility in the fourth month of protocol implementation, which required significant changes in drug distribution methods, and this could have contributed to the small sample size of interventions. The move temporarily shifted departmental resources to support operational needs.
Another important factor is the voluntary nature of the policy; while it was within the pharmacist’s professional judgment to apply the protocols, pharmacists were encouraged to contact prescribers if there was any ambiguity. Therefore, while one might have expected more resident physicians to be involved with orders that were adjusted, the UMCPP practice philosophy supports contacting training physicians about changes so that they may learn from the discussion to support developing stronger prescribing habits. Future development should therefore support more universal protocol application to all eligible patients to optimize the benefits described here. Lastly, data measuring the clinical outcomes and time savings or increased productivity secondary to the elimination of physician phone calls was not directly measured. We thus sought to first demonstrate to the physician base that pharmacists could successfully apply a variety of protocols that were broader than those formally studied with equal accuracy. With that effectiveness established, future studies should explore if broader protocol application produces a greater optimization of outcomes.
After the study was completed, a survey was conducted of the pharmacists to assess perceptions and guide further policy development. We received a 63.6% response rate (14 of 22 possible respondents) with a strong majority of the respondents expressing a favorable perception of the protocols. A few respondents indicated some protocols were infrequently utilized and there was limited familiarity with others. We anticipate this is largely based on various shift and unit assignments that would make some protocols more applicable than others to the populations serviced. One of the survey questions polled the respondents on the necessity of the email notification to the prescriber given that this practice is of a higher level of notification than other established hospital protocols which only requires a notation of the change within the medication order. Seventy-one percent (n = 10) of respondents favored removing the email notification, citing primarily that it would be consistent with physician comments regarding the existing notifications. Pharmacists also identified further areas of protocol development including electrocardiogram ordering for QTc monitoring, implementation of a standardized vancomycin dosing protocol, discontinuation of duplicate orders, product substitution for nonformulary items and addition of a protocol for pharmacists to order over-the-counter or nonprescription products as they would in a community setting. This input will shape the revision of the policy and its protocols.
Conclusion
Consistent with the published literature, pharmacists effectively performed pharmacotherapy interventions in a multitude of practice categories for adult inpatients of an acute care community-teaching hospital using a single, comprehensive clinical policy. Providing these broadly scoped protocols in a singular policy allowed pharmacists to increase the autonomy with which they applied their pharmacotherapy expertise during the course of their routine, prospective care and expanded the established benefit of allowing professionals to work to their fullest extent. Pharmacist protocol intervention was met with a high physician acceptance rate.
Acknowledgment: We thank all the pharmacists at UMCPP for supporting our efforts to refine pharmacy practice for our patients.
Corresponding author: Liza Barbarello Andrews, PharmD, BSPharm, BCPS, Rutgers, The State University of New Jersey, 160 Frelinghuysen Rd, Piscataway, NJ 08854, [email protected].
Financial disclosures: None.
1. Bond CA, Raehl CL. Pharmacist provided anticoagulation management in United States hospitals: death rates, length of stay, Medicare charges, bleeding complications and transfusions. Pharmacother 2004;24:953–63.
2. Yen YH, Chen HY, Wuan-Jin L, et al. Clinical and economic impact of a pharmacist-managed iv-to-po conversion service for levofloxacin in Taiwan. Int J Clin Pharmacol Ther 2012;50:136–41.
3. Buyle F, Vogelaers D, PelemanR, et al. Implementation of guidelines for sequential therapy with fluoroquinolones in a Belgian hospital. Pharm World Sci 2010;32:404–10.
4. Davis SL, Delgado G, McKinnon PS. Pharmacoeconomic consideration associated with the use of intravenous-to-oral moxifloxacin for community-acquired pneumonia Clin Infect Dis 2005;41Supp2;5:136–43.
5. Ho BP, Lau TT, Balen RM, et al. The impact of a pharmacist-managed dosage form conversion service on ciprofloxacin usage at a major Canadian teaching hospital: a pre- and post-intervention study. BMC Health Serv Res 2005;5:48.
6. Kuti JL, Le TN, Nightingale CH, et al. Pharmacoeconomics of a pharmacist-managed program for automatically converting levofloxacin route from iv to oral. Am J Health Sys Pharm 2002;59:2209–15.
7. Cohen SM, Lipsett PA, Buchman TG, et al. Comparison of intravenous/oral ciprofloxacin plus metronidazole versus piperacillin/tazobactam in the treatment of complicated intraabdominal infections. Ann Surg 2000;232:254–62.
8. Plouffe J, Schwartz DB, Kolokathis A, et al. Clinical efficacy of intravenous followed by oral azithromycin monotherapy in hospitalized patients with community-acquired pneumonia. Antimicrob Agents Chemother 2000;44:1796–802.
9. Wetzstein GA. Intravenous to oral (IV:PO) anti-infective combination therapy. Cancer Control 2000;7:170–6.
10. Paladino JA. Pharmacoeconomics of antimicrobial therapy. Am J Healthsys Pharm 1999;56(Supp3):S25–8.
11. Ahkee S, Smith S, et al. Early switch from intravenous to oral antibiotics in hospitalized patient with infections: a six-month prospective study. Pharmacotherapy 1997;17:569–75.
12. Przybylski KG, Rybak MJ, Martin PR, et al. A pharmacist-initiated program of intravenous to oral antibiotic conversion. Pharmacotherapy 1997;17:271–6.
13. Jewesson P. Cost-effectiveness and value of an IV switch. Pharmacoeconomics 1994;5(Supp2):20–6.
14. Ramirez J. Advances in antibiotic use: switch therapy. Curr Ther Res 1994;55(suppA):30–3.
15. Shepard MF. Making the switch from IV to PO. Am J Healthsys Pharm 1994;50:2510.
16. Chessin LN. When to switch from IV to oral antibiotics. Patient Care 1993;15:113–25.
17. Cohen MR. Important news! IV route not needed to justify hospitalization for antibiotics. Hospital Pharmacy 1993;28:946.
18. Schentag JJ. Changes in antimicrobial agent usage resulting from interactions among clinical pharmacy, infectious disease division and the microbiology laboratory. Diagnos Microbiol Infect Dis 1993;16:255–64.
19. Fighetto L. Intravenous to oral stepdown program: four years’ experience in a large teaching hospital. Ann Pharmacother 1992;26:1447–51.
20. Powers T. Clinical and economic effect of ciprofloxacin as an alternative to injectable antimicrobial therapy. Am J Healthsys Pharm 1990;47:1781–4.
21. Ament PW, McGuire WM. Setting up an automatic pharmacist-initiated pharmacokinetic dosing service. Hosp Formul 1993;28:589–92.
22. Mousavi M, Dashti-Khavidaki S, Khalili H, et al. Impact of clinical pharmacy services on stress ulcer prophylaxis prescribing and related cost in patients with renal insufficiency. Int J Pharm Pract 2012;Nov 9.
23. Damaske DL, Baird RW. Development and implementation of a pharmacist managed inpatient warfarin protocol. Proc (Baylor Univ Med Cent) 2005;18:397–400.
24. Radley AS, Hall J, Farrow M, et al. Evaluation of anticoagulation control in a pharmacist operated anticoagulation clinic. J Clin Pathol 1995;48:545–47.
25. Pharmacy and Therapeutics Committee. Medication policies and protocols of Nebraska Methodist Hospital, Methodist Women’s Hospital.
26. University of Kentucky Pharmacy Services. Therapeutic interchanges [Internet]. Available at www.hosp.uky.edu/pharmacy/interchange.asp
27. Bayshore Community Hospital Department of Pharmacy. Conversion of intravenous azithromycin (Zithromax), ceftriaxone (Rocephin), ciprofloxacin (Cipro), fluconazole (Diflucan), lansoprazole (Prevacid), levofloxacin (Levaquin), linezolid (Zyvox), metronidazole (Flagyl), moxifloxacin (Avelox), potassium chloride (KC), or ranitidine (Zantac) to oral medication [Internet]. [cited 2013 May 9]. Available at www.ashp.org/s_ashp/docs/files/R-IVtoPOConvPol-2.pdf.
28. Medication Education Safety Approval Committee, Massachusetts General Hospital. Automatic intravenous to oral protocol [Internet]. MESAC memo; 2005 July [cited 2013 March 9]. Available at www2.massgeneral.org/pharmacy/mesac/mesac_memo3.pdf.
29. Hanson RL, Habibi M, Khamo N, et al. Integrated clinical and specialty pharmacy practice model for management of patients with multiple sclerosis. Am J Health Syst Pharm 2014;71:463–9.
30. Making pharmacists part of the multidisciplinary team. Hosp Case Manag 2014;22:13–6.
31. Preslaski CR, Lat I, MacLaren R, et al. Pharmacist contributions as members of the multidisciplinary ICU team. Chest 2013;144:1687–95.
32. Ripley TL, Adamson PB, Hennebry TA, et al. Collaborative practice model between cardiologists and clinical pharmacist for management of patients with cardiovascular disease in an outpatient clinic. Ann Pharmacother 2014;48:412–9.
33. Smith M, Bates DW, Bodenheimer TS. Pharmacists belong in accountable care organizations and integrated care teams. Health Aff (Millwood) 2013;32:1963–70.
34. Ukens C. New Jersey rewrites its state pharmacy practice act. Drug Topics 2004;148:41.
35. New Jersey Board of Pharmacy Laws. Statute 45:14-64. Inapplicability relative to collaborative drug therapy management in hospital. [Internet] July 2011 [cited 11 September 14]. Available from: www.njconsumeraffairs.gov/laws/pharmlaws.pdf.
36. ASHP Commission on Therapeutics. ASHP therapeutic guidelines on stress ulcer prophylaxis. Am J Health Syst Pharm 1999;56:347–79.
37. Bond CA, Raehl CL. Clinical pharmacy services, pharmacy staffing, and hospital mortality rates. Pharmacotherapy 2007;27:481–93.
38. Bond CA, Raehl CL, Franke T. Interrelationships among mortality rates, drug costs, total cost of care, and length of stay in United States hospitals: summary and recommendations for clinical pharmacy services and staffing. Pharmacotherapy 2001;21:129–41.
1. Bond CA, Raehl CL. Pharmacist provided anticoagulation management in United States hospitals: death rates, length of stay, Medicare charges, bleeding complications and transfusions. Pharmacother 2004;24:953–63.
2. Yen YH, Chen HY, Wuan-Jin L, et al. Clinical and economic impact of a pharmacist-managed iv-to-po conversion service for levofloxacin in Taiwan. Int J Clin Pharmacol Ther 2012;50:136–41.
3. Buyle F, Vogelaers D, PelemanR, et al. Implementation of guidelines for sequential therapy with fluoroquinolones in a Belgian hospital. Pharm World Sci 2010;32:404–10.
4. Davis SL, Delgado G, McKinnon PS. Pharmacoeconomic consideration associated with the use of intravenous-to-oral moxifloxacin for community-acquired pneumonia Clin Infect Dis 2005;41Supp2;5:136–43.
5. Ho BP, Lau TT, Balen RM, et al. The impact of a pharmacist-managed dosage form conversion service on ciprofloxacin usage at a major Canadian teaching hospital: a pre- and post-intervention study. BMC Health Serv Res 2005;5:48.
6. Kuti JL, Le TN, Nightingale CH, et al. Pharmacoeconomics of a pharmacist-managed program for automatically converting levofloxacin route from iv to oral. Am J Health Sys Pharm 2002;59:2209–15.
7. Cohen SM, Lipsett PA, Buchman TG, et al. Comparison of intravenous/oral ciprofloxacin plus metronidazole versus piperacillin/tazobactam in the treatment of complicated intraabdominal infections. Ann Surg 2000;232:254–62.
8. Plouffe J, Schwartz DB, Kolokathis A, et al. Clinical efficacy of intravenous followed by oral azithromycin monotherapy in hospitalized patients with community-acquired pneumonia. Antimicrob Agents Chemother 2000;44:1796–802.
9. Wetzstein GA. Intravenous to oral (IV:PO) anti-infective combination therapy. Cancer Control 2000;7:170–6.
10. Paladino JA. Pharmacoeconomics of antimicrobial therapy. Am J Healthsys Pharm 1999;56(Supp3):S25–8.
11. Ahkee S, Smith S, et al. Early switch from intravenous to oral antibiotics in hospitalized patient with infections: a six-month prospective study. Pharmacotherapy 1997;17:569–75.
12. Przybylski KG, Rybak MJ, Martin PR, et al. A pharmacist-initiated program of intravenous to oral antibiotic conversion. Pharmacotherapy 1997;17:271–6.
13. Jewesson P. Cost-effectiveness and value of an IV switch. Pharmacoeconomics 1994;5(Supp2):20–6.
14. Ramirez J. Advances in antibiotic use: switch therapy. Curr Ther Res 1994;55(suppA):30–3.
15. Shepard MF. Making the switch from IV to PO. Am J Healthsys Pharm 1994;50:2510.
16. Chessin LN. When to switch from IV to oral antibiotics. Patient Care 1993;15:113–25.
17. Cohen MR. Important news! IV route not needed to justify hospitalization for antibiotics. Hospital Pharmacy 1993;28:946.
18. Schentag JJ. Changes in antimicrobial agent usage resulting from interactions among clinical pharmacy, infectious disease division and the microbiology laboratory. Diagnos Microbiol Infect Dis 1993;16:255–64.
19. Fighetto L. Intravenous to oral stepdown program: four years’ experience in a large teaching hospital. Ann Pharmacother 1992;26:1447–51.
20. Powers T. Clinical and economic effect of ciprofloxacin as an alternative to injectable antimicrobial therapy. Am J Healthsys Pharm 1990;47:1781–4.
21. Ament PW, McGuire WM. Setting up an automatic pharmacist-initiated pharmacokinetic dosing service. Hosp Formul 1993;28:589–92.
22. Mousavi M, Dashti-Khavidaki S, Khalili H, et al. Impact of clinical pharmacy services on stress ulcer prophylaxis prescribing and related cost in patients with renal insufficiency. Int J Pharm Pract 2012;Nov 9.
23. Damaske DL, Baird RW. Development and implementation of a pharmacist managed inpatient warfarin protocol. Proc (Baylor Univ Med Cent) 2005;18:397–400.
24. Radley AS, Hall J, Farrow M, et al. Evaluation of anticoagulation control in a pharmacist operated anticoagulation clinic. J Clin Pathol 1995;48:545–47.
25. Pharmacy and Therapeutics Committee. Medication policies and protocols of Nebraska Methodist Hospital, Methodist Women’s Hospital.
26. University of Kentucky Pharmacy Services. Therapeutic interchanges [Internet]. Available at www.hosp.uky.edu/pharmacy/interchange.asp
27. Bayshore Community Hospital Department of Pharmacy. Conversion of intravenous azithromycin (Zithromax), ceftriaxone (Rocephin), ciprofloxacin (Cipro), fluconazole (Diflucan), lansoprazole (Prevacid), levofloxacin (Levaquin), linezolid (Zyvox), metronidazole (Flagyl), moxifloxacin (Avelox), potassium chloride (KC), or ranitidine (Zantac) to oral medication [Internet]. [cited 2013 May 9]. Available at www.ashp.org/s_ashp/docs/files/R-IVtoPOConvPol-2.pdf.
28. Medication Education Safety Approval Committee, Massachusetts General Hospital. Automatic intravenous to oral protocol [Internet]. MESAC memo; 2005 July [cited 2013 March 9]. Available at www2.massgeneral.org/pharmacy/mesac/mesac_memo3.pdf.
29. Hanson RL, Habibi M, Khamo N, et al. Integrated clinical and specialty pharmacy practice model for management of patients with multiple sclerosis. Am J Health Syst Pharm 2014;71:463–9.
30. Making pharmacists part of the multidisciplinary team. Hosp Case Manag 2014;22:13–6.
31. Preslaski CR, Lat I, MacLaren R, et al. Pharmacist contributions as members of the multidisciplinary ICU team. Chest 2013;144:1687–95.
32. Ripley TL, Adamson PB, Hennebry TA, et al. Collaborative practice model between cardiologists and clinical pharmacist for management of patients with cardiovascular disease in an outpatient clinic. Ann Pharmacother 2014;48:412–9.
33. Smith M, Bates DW, Bodenheimer TS. Pharmacists belong in accountable care organizations and integrated care teams. Health Aff (Millwood) 2013;32:1963–70.
34. Ukens C. New Jersey rewrites its state pharmacy practice act. Drug Topics 2004;148:41.
35. New Jersey Board of Pharmacy Laws. Statute 45:14-64. Inapplicability relative to collaborative drug therapy management in hospital. [Internet] July 2011 [cited 11 September 14]. Available from: www.njconsumeraffairs.gov/laws/pharmlaws.pdf.
36. ASHP Commission on Therapeutics. ASHP therapeutic guidelines on stress ulcer prophylaxis. Am J Health Syst Pharm 1999;56:347–79.
37. Bond CA, Raehl CL. Clinical pharmacy services, pharmacy staffing, and hospital mortality rates. Pharmacotherapy 2007;27:481–93.
38. Bond CA, Raehl CL, Franke T. Interrelationships among mortality rates, drug costs, total cost of care, and length of stay in United States hospitals: summary and recommendations for clinical pharmacy services and staffing. Pharmacotherapy 2001;21:129–41.
A 57-year-old woman with abdominal pain
A 57-year-old woman presented to the emergency department with left lower quadrant pain, which had started 1 week earlier and was constant, dull, aching, and nonradiating. There were no aggravating or alleviating factors. The pain was associated with low-grade fever and nausea. She reported no vomiting, no change in bowel habits, and no hematemesis, hematochezia, or melena. She did not have urinary urgency, frequency, or dysuria. She had no cardiac, respiratory, or neurologic symptoms.
Her medical history included hypothyroidism, type 2 diabetes mellitus, diverticulosis, and chronic obstructive pulmonary disease. Her medications included metformin, insulin, levothyroxine, and inhaled tiotropium. She had no allergies. She had never undergone surgery, including cesarean delivery. She was postmenopausal. She had two children, both of whom had been born vaginally at full term. She denied using alcohol, tobacco, and illicit drugs. Her family history was noncontributory.
On examination, she was not in acute distress. Her temperature was 36.7°C (98.1°F), blood pressure 130/90 mm Hg, heart rate 86 beats per minute and regular, respiratory rate 16 breaths per minute, and oxygen saturation 98% on ambient air. Examination of her head and neck was unremarkable. Cardiopulmonary examination was normal. Abdominal examination revealed normal bowel sounds, mild tenderness in the left lower quadrant with localized guarding, and rebound tenderness. Neurologic examination was unremarkable.
Initial laboratory data showed mild leukocytosis. Computed tomography with contrast of the abdomen and pelvis suggested acute diverticulitis.
ATRIAL FIBRILLATION, AND THEN A TURN FOR THE WORSE
The patient was admitted with an initial diagnosis of acute diverticulitis. She was started on antibiotics, hydration, and pain medications, and her abdominal pain gradually improved.
On the third hospital day, she suddenly experienced shortness of breath and palpitations. At the time of admission her electrocardiogram had been normal, but it now showed atrial fibrillation with a rapid ventricular response. She also developed elevated troponin levels, which were thought to represent type 2 non-ST-elevation myocardial infarction.
She was started on aspirin, clopidogrel, and anticoagulation with heparin bridged with warfarin for the new-onset atrial fibrillation. Her heart rate was controlled with metoprolol, and her shortness of breath improved. An echocardiogram was normal.
On the seventh hospital day, she developed severe right-sided lower abdominal pain and bruising. Her blood pressure was 90/60 mm Hg, heart rate 110 beats per minute and irregularly irregular, respiratory rate 22 breaths per minute, and oxygen saturation 97% on room air. Her abdomen was diffusely tender with a palpable mass in the right lower quadrant and hypoactive bowel sounds. Ecchymosis was noted (Figure 1).
DIFFERENTIAL DIAGNOSIS
1. What is the likely cause of her decompensation?
- Acute mesenteric ischemia
- Perforation of the gastrointestinal tract
- Rectus sheath hematoma
- Abdominal compartment syndrome due to acute pancreatitis
Acute mesenteric ischemia
Signs and symptoms of acute mesenteric ischemia can be vague. Moreover, when it leads to bowel necrosis the mortality rate is high, ranging from 30% to 65%.1 Hence, one should suspect it and try to diagnose it early.
Most patients with this condition have comorbidities; risk factors include atherosclerotic disease, cardiac conditions (congestive heart failure, recent myocardial infarction, and atrial fibrillation), systemic illness, and inherited or acquired hypercoagulable states.2
The four major causes are:
- Acute thromboembolic occlusion of the superior mesenteric artery (the most common site of occlusion because of the acute angle of origin from the aorta)
- Acute thrombosis of the mesenteric vein
- Acute thrombosis of the mesenteric artery
- Nonocclusive disease affecting the mesenteric vessels2
Nonocclusive disease is seen in conditions in which the mesenteric vessels are already compromised due to background stenosis owing to atherosclerosis. Also, conditions such as septic and cardiogenic shock can compromise these arteries, leading to ischemia, which, if it persists, can lead to bowel infarction. Ischemic colitis falls under this category. It commonly involves the descending and sigmoid colon.3
The initial symptom of ischemia may be abdominal pain that is brought on by eating large meals (“postprandial intestinal angina.”2 When the ischemia worsens to infarction, patients may have a diffusely tender abdomen and constant pain that does not vary with palpation. Surprisingly, patients do not exhibit peritoneal signs early on. This gives rise to the description of “pain out of proportion to the physical findings” traditionally associated with acute mesenteric ischemia.2
Diagnosis. Supportive laboratory data include marked leukocytosis, elevated hematocrit due to hemoconcentration, metabolic acidosis, and elevated lactate.4 Newer markers such as serum alpha-glutathione S-transferase (alpha-GST) and intestinal fatty acid-binding protein (I-FABP) may be used to support the diagnosis.
Elevated alpha-GST has 72% sensitivity and 77% specificity in the diagnosis of acute mesenteric ischemia.5 The caveat is that it cannot reliably differentiate ischemia from infarction. Its sensitivity can be improved to 97% to 100% by using the white blood cell count and lactate levels in combination.5
An I-FABP level higher than 100 ng/mL has 100% sensitivity for diagnosing mesenteric infarction but only 25% sensitivity for bowel strangulation.6
Early use of abdominal computed tomography with contrast can aid in recognizing this diagnosis.7 Thus, it should be ordered in suspected cases, even in patients who have elevated creatinine levels (which would normally preclude the use of contrast), since early diagnosis followed by endovascular therapy is associated with survival benefit, and the risk of contrast-induced nephropathy appears to be small.8 Computed tomography helps to determine the state of mesenteric vessels and bowel perfusion before ischemia progresses to infarction. It also helps to rule out other common diagnoses. Findings that suggest acute mesenteric ischemia include segmental bowel wall thickening, intestinal pneumatosis with gas in the portal vein, bowel dilation, mesenteric stranding, portomesenteric thrombosis, and solid-organ infarction.9
Treatment. If superior mesenteric artery occlusion is diagnosed on computed tomography, the next step is to determine if there is peritonitis.10 In patients who have evidence of peritonitis, exploratory laparotomy is performed. For emboli in such patients, open embolectomy followed by on-table angiography is carried out in combination with damage-control surgery. For patients with peritonitis and acute thrombosis, stenting along with damage-control surgery is preferred.10
On the other hand, if there is no peritonitis, the thrombosis may be amenable to endovascular intervention. For patients with acute embolic occlusion with no contraindications to thrombolysis, aspiration embolectomy in combination with local catheter-directed thrombolysis with recombinant tissue plasminogen activator can be performed. This can be combined with endovascular mechanical embolectomy for more complete management.10 Patients with contraindications to thrombolysis can be treated either with aspiration and mechanical embolectomy or with open embolectomy with angiography.10
During laparotomy, the surgeon carefully inspects the bowel for signs of necrosis. Signs that bowel is still viable include pink color, bleeding from cut surfaces, good peristalsis, and visible pulsations in the arterial arcade of the mesentery.
Acute mesenteric artery thrombosis arising from chronic atherosclerotic disease can be treated with stenting of the stenotic lesion.10 Patients with this condition would also benefit from aggressive management of atherosclerotic disease with statins along with antiplatelet agents.10
Mesenteric vein thrombosis requires prompt institution of anticoagulation. However, in advanced cases leading to bowel infarction, exploratory laparotomy with resection of the necrotic bowel may be required. Anticoagulation should be continued for at least 6 months, and further therapy should be determined by the underlying precipitating condition.10
Critically ill patients who develop mesenteric ischemia secondary to persistent hypotension usually respond to adequate volume resuscitation, cessation of vasopressors, and overall improvement in their hemodynamic status. These patients must be closely monitored for development of gangrene of the bowel because they may be intubated and not able to complain. Any sudden deterioration in their condition should prompt physicians to consider bowel necrosis developing in these patients. Elevation of lactate levels out of proportion to the degree of hypotension may be corroborative evidence.4
Our patient had risk factors for acute mesenteric ischemia that included atrial fibrillation and recent non-ST-elevation myocardial infarction. She could have had arterial emboli due to atrial fibrillation, in situ superior mesenteric arterial thrombosis, or splanchnic arterial vasoconstriction due to the myocardial infarction associated with transient hypotension.
Arguing against this diagnosis, although she had a grossly distended abdomen, abdominal bruising usually is not seen. Also, a palpable mass in the right lower quadrant is uncommon except when acute mesenteric ischemia occurs due to segmental intestinal strangulation, as with strangulated hernia or volvulus. She also had therapeutic international normalized ratio (INR) levels constantly while on anticoagulation. Nevertheless, acute mesenteric ischemia should be strongly considered in the initial differential diagnosis of this patient’s acute decompensation.
Perforation of the gastrointestinal tract
Diverticulitis is the acute inflammation of one or more diverticula, which are small pouches created by herniation of the mucosa into the wall of the colon. The condition is caused by microscopic or macroscopic perforation of the diverticula. Microscopic perforation is usually self-limited (uncomplicated diverticulitis) and responds to conservative treatment, whereas macroscopic perforation can be associated with fecal or purulent peritonitis, abscess, enteric fistula, bowel obstruction, and stricture (complicated diverticulitis), in which case surgery may be necessary.
Patients with peritonitis due to free perforation present with generalized tenderness with rebound tenderness and guarding on abdominal examination. The abdomen may be distended and tympanic to percussion, with diminished or absent bowel sounds. Patients may have hemodynamic compromise.
Plain upright abdominal radiographs may show free air under the diaphragm. Computed tomography may show oral contrast outside the lumen and detect even small amounts of free intraperitoneal air (more clearly seen on a lung window setting).
Our patient initially presented with acute diverticulitis. She later developed diffuse abdominal tenderness with hypoactive bowel sounds. Bowel perforation is certainly a possibility at this stage, though it is usually not associated with abdominal bruising. She would need additional imaging to rule out this complication.
Other differential diagnoses to be considered in this patient with right lower-quadrant pain include acute appendicitis, incarcerated inguinal hernia, volvulus (particularly cecal volvulus), small-bowel obstruction, pyelonephritis, and gynecologic causes such as adnexal torsion, ruptured ovarian cyst, and tubo-ovarian abscess. Computed tomography helps to differentiate most of these causes.
Rectus sheath hematoma
Rectus sheath hematoma is relatively uncommon and often not considered in the initial differential diagnosis of an acute abdomen. This gives it the rightful term “a great masquerader.” It usually results from bleeding into the rectus sheath from damage to the superior (more common) or inferior epigastric arteries and occasionally from a direct tear of the rectus abdominis muscle. Predisposing factors include anticoagulant therapy (most common), advanced age, hypertension, previous abdominal surgery, trauma, paroxysmal coughing, medication injections, pregnancy, blood dyscrasias, severe vomiting, violent physical activity, and leukemia.11
Clinical manifestations include acute abdominal pain, often associated with fever, nausea, and vomiting. Physical examination may reveal signs of hypovolemic shock, a palpable nonpulsatile abdominal mass, and signs of local peritoneal irritation. The Carnett sign11 (tenderness within the abdominal wall that persists and does not improve with raising the head) and the Fothergill sign11 (a tender abdominal mass that does not cross the midline and remains palpable with tensing of the rectus sheath) may be elicited.
Computed tomography is more sensitive than abdominal ultrasonography in differentiating rectus sheath hematoma from an intra-abdominal pathology.11 In addition, computed tomography also helps to determine if the bleeding is active or not, which has therapeutic implications.
In our patient, rectus sheath hematoma is a possibility because of her ongoing anticoagulation, findings of localized abdominal bruising, and palpable right lower-quadrant mass, and it is high on the list of differential diagnoses. Rectus sheath hematoma should be considered in the differential diagnosis of lower abdominal pain particularly in elderly women who are on anticoagulation and in whom the onset of pain coincides with a paroxysm of cough.12 Women are twice as likely as men to develop rectus sheath hematoma, owing to their different muscle mass.13 In addition, anterior abdominal wall muscles are stretched during pregnancy.13
Abdominal compartment syndrome
Abdominal compartment syndrome has been classically associated with surgical patients. However, it is being increasingly recognized in critically ill medical patients, in whom detecting and treating it early may result in significant reduction in rates of morbidity and death.14
Abdominal compartment syndrome is of three types: primary, secondary, and recurrent. Primary abdominal compartment syndrome refers to the classic surgical patients with evidence of direct injury to the abdominal or pelvic organs through major trauma or extensive abdominal surgeries. Secondary abdominal compartment syndrome refers to its development in critically ill intensive care patients in whom the pathology does not directly involve the abdominal or pelvic organs.
Various medical conditions can culminate in abdominal compartment syndrome and result in multiorgan failure. Recurrent abdominal compartment syndrome refers to its development after management of either primary or secondary intra-abdominal hypertension or abdominal compartment syndrome.15 Clinicians thus must be aware of secondary and recurrent abdominal compartment syndrome occurring in critically ill patients.
The normal intra-abdominal pressure is around 5 to 7 mm Hg, even in most critically ill patients. Persistent elevation, ie, higher than 12 mm Hg, is referred to as intra-abdominal hypertension.16–18 Intra-abdominal hypertension is subdivided into four grades:
- Grade I: 12–15 mm Hg
- Grade II: 16–20 mm Hg
- Grade III: 21–25 mm Hg
- Grade IV: > 25 mm Hg.
The World Society of the Abdominal Compartment Syndrome (WSACS) defines abdominal compartment syndrome as pressure higher than 20 mm Hg along with organ damage.18 It may or may not be associated with an abdominal perfusion pressure less than 60 mm Hg.18
Risk factors associated with abdominal compartment syndrome include conditions causing decreased gut motility (gastroparesis, ileus, and colonic pseudo-obstruction), intra-abdominal or retroperitoneal masses or abscesses, ascites, hemoperitoneum, acute pancreatitis, third-spacing due to massive fluid resuscitation with transfusions, peritoneal dialysis, and shock.18,19
Physical examination has a sensitivity of only 40% to 60% in detecting intra-abdominal hypertension.20 The gold-standard method of measuring the intra-abdominal pressure is the modified Kron technique,18 using a Foley catheter in the bladder connected to a pressure transducer. With the patient in the supine position, the transducer is zeroed at the mid-axillary line at the level of the iliac crest, and 25 mL of normal saline is instilled into the bladder and maintained for 30 to 60 seconds to let the detrusor muscle relax.15 Pressure tracings are then recorded at the end of expiration. Factors that are known to affect the transbladder pressure include patient position, respiratory movement, and body mass index, and should be taken into account when reading the pressure recordings.15,21 Other techniques that can be used include intragastric, intra-inferior vena cava, and intrarectal approaches.15
The WSACS recommends that any patient admitted to a critical care unit or in whom new organ failure develops should be screened for risk factors for intra-abdominal hypertension and abdominal compartment syndrome. If a patient has at least two of the risk factors suggested by WSACS, a baseline intra-abdominal pressure measurement should be obtained. Patients at risk for intra-abdominal hypertension should have the intra-abdominal pressure measured every 4 to 6 hours. However, in the face of hemodynamic instability and worsening multiorgan failure, the pressure may need to be measured hourly.18
Clinicians managing patients in the intensive care unit should think of intra-abdominal pressure alongside blood pressure, urine output, and mental status when evaluating hemodynamic status. Clinical manifestations of abdominal compartment syndrome reflect the underlying organ dysfunction and include hypotension, refractory shock, decreased urine output, intracranial hypertension, progressive hypoxemia and hypercarbia, elevated pulmonary peak pressures, and worsening of metabolic acidosis.22
Treatment. The standard treatment for primary abdominal compartment syndrome is surgical decompression. According to WSACS guidelines, insertion of a percutaneous drainage catheter should be advocated in patients with gross ascites and in whom decompressive surgery is not feasible. A damage-control resuscitation strategy used for patients undergoing damage-control laparotomy has been found to increase the 30-day survival rate.23 A damage-control resuscitation strategy consists of increasing the use of plasma and platelet transfusions over packed red cell transfusions, limiting the use of crystalloid solutions in early fluid resuscitation, and allowing for permissive hypotension.
Secondary abdominal compartment syndrome is treated conservatively in most cases, since patients with this condition are very poor surgical candidates owing to their comorbidities.18 However, in patients with progressive organ dysfunction in whom medical management has failed, surgical decompression should be considered.18 Medical management of secondary abdominal compartment syndrome depends on the underlying etiology. Strategies include nasogastric or colonic decompression, use of prokinetic agents, paracentesis in cases with gross ascites, and maintaining a cumulative negative fluid balance. The WSACS does not recommend routine use of diuretics, albumin infusion, or renal replacement strategies. Pain should be adequately controlled to improve abdominal wall compliance.18,24 Neuromuscular blockade agents may be used to aid this process. Neostigmine may be used to treat colonic pseudo-obstruction when other conservative methods fail. Use of enteral nutrition should be minimized.18
Our patient might have abdominal compartment syndrome, but a definitive diagnosis cannot be made at this point without measuring the intra-abdominal pressure.
WHICH IMAGING TEST WOULD BE BEST?
2. Which imaging test would be best for establishing the diagnosis in this patient?
- Plain abdominal radiography
- Abdominal ultrasonography
- Computed tomography of the abdomen and pelvis with contrast
- Magnetic resonance imaging of the abdomen and pelvis
Plain abdominal radiography
Plain abdominal radiography can help to determine if there is free gas under the diaphragm (due to bowel perforation), obstructed bowel, sentinel loop, volvulus, or fecoliths causing the abdominal pain. It cannot diagnose rectus sheath hematoma or acute mesenteric ischemia.
Abdominal ultrasonography
Abdominal ultrasonography can be used as the first diagnostic test, as it is widely available, safe, effective, and tolerable. It does not expose the patient to radiation or intravenous contrast agents. It helps to diagnose rectus sheath hematoma and helps to follow its maturation and resolution once a diagnosis is made. It can provide a rapid assessment of the size, location, extent, and physical characteristics of the mass.
Rectus sheath hematoma appears spindle-shaped on sagittal sections and ovoid on coronal sections. It often appears sonolucent in the early stages and sonodense in the late stage, but the appearance may be heterogeneous depending on the combined presence of clot and fresh blood. These findings are sufficient to make the diagnosis.
Abdominal ultrasonography has 85% to 96% sensitivity in diagnosing rectus sheath hematoma.25 It can help diagnose other causes of the abdominal pain, such as renal stones and cholecystitis. It is the preferred imaging test in pediatric patients, pregnant patients, and those with renal insufficiency.
Abdominal computed tomography
Abdominal computed tomography has a sensitivity and specificity of 100% for diagnosing acute rectus sheath hematoma with a duration of less than 5 days.25 It not only helps to determine the precise location and extent, but also helps to determine if there is active extravasation. Even in patients with renal insufficiency, noncontrast computed tomography will help to confirm the diagnosis, although it may not show extravasation or it may miss certain abdominal pathologies because of the lack of contrast.
Acute rectus sheath hematoma appears as a hyperdense mass posterior to the rectus abdominis muscle with ipsilateral anterolateral muscular enlargement. Chronic rectus sheath hematoma appears isodense or hypodense relative to the surrounding muscle. Above the arcuate line, rectus sheath hematoma has a spindle shape; below the arcuate line, it is typically spherical.
In 1996, Berná et al26 classified rectus sheath hematoma into three grades based on findings of computed tomography:
- Grade I is intramuscular and unilateral
- Grade II may involve bilateral rectus muscles without extension into the prevesicular space
- Grade III extends into the peritoneum and prevesicular space
Magnetic resonance imaging
Magnetic resonance imaging is useful to differentiate chronic rectus sheath hematoma (greater than 5-day duration) from an anterior abdominal wall mass. Chronic rectus sheath hematoma will have high signal intensity on both T1- and T2-weighted images up to 10 months after the onset of the hematoma.27
Back to our patient
Since our patient’s symptoms are acute and of less than 5 days’ duration, computed tomography of the abdomen and pelvis would be the best diagnostic test, with therapeutic implications if there is ongoing extravasation.
Computed tomography of the abdomen with contrast showed a new hematoma, measuring 25 by 14 by 13.5 cm, in the right rectus sheath (Figure 2), with no other findings. The hematoma was grade I, since it was intramuscular and unilateral without extension elsewhere.
Laboratory workup showed that the patient’s hematocrit was falling, from 34% to 24%, and her INR was elevated at 2.5. She was resuscitated with fluids, blood transfusion, and fresh-frozen plasma. Anticoagulation was withheld. In spite of resuscitation, her hematocrit kept falling, though she remained hemodynamically stable.
THE WAY FORWARD
3. At this point, what would be the best approach to management in this patient?
- Serial clinical examinations and frequent monitoring of the complete blood cell count
- Urgent surgical consult for exploratory laparotomy with evaluation of the hematoma and ligation of the bleeding vessel
- Repeat computed tomographic angiography to identify a possible bleeding vessel; consideration of radiographically guided arterial embolization
- Measuring the intra-abdominal pressure using the intrabladder pressure for abdominal compartment syndrome and consideration of surgical drainage
The key clinical concern in a patient with a rectus sheath hematoma who is hemodynamically stable is whether the hematoma is expanding. This patient responded to initial resuscitation, but her falling hematocrit was evidence of ongoing bleeding leading to an expanding rectus sheath hematoma. Thus, serial clinical examinations and frequent monitoring of the complete blood cell count would not be enough, as it could miss fatal ongoing bleeding.
Radiographically guided embolization with Gelfoam, thrombin, or coils should be attempted first, as this is less invasive than exploratory laparotomy.28 It can achieve hemostasis, decrease the size of the hematoma, limit the need for blood products, and prevent rupture into the abdomen. If this is unsuccessful, the next step is ligation of the bleeding vessel.29
Surgical treatment includes evacuation of the hematoma, repair of the rectus sheath, ligation of bleeding vessels, and abdominal wall closure. Surgical evacuation or guided drainage of a rectus sheath hematoma on its own is not normally indicated and may indeed cause persistent bleeding by diminishing a potential tamponade effect. However, it may become necessary if the hematoma is very large or infected, if it causes marked respiratory impairment, or if abdominal compartment syndrome is suspected.
Abdominal compartment syndrome is very rare but is associated with a 50% mortality rate.30 It should be suspected in patients with oliguria, low cardiac output, changes in minute ventilation, and altered splanchnic blood flow. The diagnosis is confirmed with indwelling catheter manometry of the bladder to measure intra-abdominal pressure. Intra-abominal pressure above 25 mm Hg should be treated with decompressive laparotomy.30 However, the clinical suspicion of abdominal compartment syndrome was low in this patient.
The best choice at this point would be urgent computed tomographic angiography to identify a bleeding vessel, along with consideration of radiographically guided arterial embolization.
TREATMENT IS USUALLY CONSERVATIVE
Treatment of rectus sheath hematoma is conservative in most hemodynamically stable patients, with embolization or surgical intervention reserved for unstable patients or those in whom the hematoma is expanding.
Knowledge of the grading system of Berná et al26 helps to assess the patient’s risk and to anticipate potential complications. Grade I hematomas are mild and do not necessitate admission. Patients with grade II hematoma can be admitted to the floor for 24 to 48 hours for observation. Grade III usually occurs in patients receiving anticoagulant therapy and frequently requires blood products. These patients have a prolonged hospital stay and more complications, including hypovolemic shock, myonecrosis, acute coronary syndrome, arrhythmias, acute renal failure, small-bowel infarction, and abdominal compartment syndrome—all of which increases the risk of morbidity and death. Thus, patients who are on anticoagulation who develop grade III rectus sheath hematoma should be admitted to the hospital, preferably to the intensive care unit, to ensure that the hematoma is not expanding and to plan reinstitution of anticoagulation as appropriate.
In most cases, rectus sheath hematomas resolve within 1 to 3 months. Resolution of large hematomas may be hastened with the use of pulsed ultrasound.31 However, this treatment should be used only after the acute phase is over, when there is evidence of an organized thrombus and coagulation measures have returned to the target range. This helps to reduce the risk of bleeding and to prevent symptoms from worsening.31
OUR PATIENT’S COURSE
Our patient underwent urgent computed tomographic angiography, which showed a modest increase in the size of the rectus sheath hematoma. However, no definitive blush of contrast was seen to suggest active arterial bleeding. Her hematocrit stabilized, and she remained hemodynamically stable without requiring additional intervention. Most likely her bleeding was self-contained. She had normal intra-abdominal pressure on serial monitoring. She was later transferred to acute inpatient rehabilitation in view of deconditioning and is currently doing well. The hematoma persisted, decreasing only slightly in size over the next 3 weeks.
- Kougias P, Lau D, El Sayed HF, Zhou W, Huynh TT, Lin PH. Determinants of mortality and treatment outcome following surgical interventions for acute mesenteric ischemia. J Vasc Surg 2007; 46:467–474.
- Sise MJ. Acute mesenteric ischemia. Surg Clin North Am 2014; 94:165–181.
- Scharff JR, Longo WE, Vartanian SM, Jacobs DL, Bahadursingh AN, Kaminski DL. Ischemic colitis: spectrum of disease and outcome. Surgery 2003; 134:624–629.
- Lange H, Jäckel R. Usefulness of plasma lactate concentration in the diagnosis of acute abdominal disease. Eur J Surg 1994; 160:381–384.
- Gearhart SL, Delaney CP, Senagore AJ, et al. Prospective assessment of the predictive value of alpha-glutathione S-transferase for intestinal ischemia. Am Surg 2003; 69:324–329.
- Kanda T, Fujii H, Tani T, et al. Intestinal fatty acid-binding protein is a useful diagnostic marker for mesenteric infarction in humans. Gastroenterology 1996; 110:339–343.
- Menke J. Diagnostic accuracy of multidetector CT in acute mesenteric ischemia: systematic review and meta-analysis. Radiology 2010; 256:93–101.
- Acosta S, Björnsson S, Ekberg O, Resch T. CT angiography followed by endovascular intervention for acute superior mesenteric artery occlusion does not increase risk of contrast-induced renal failure. Eur J Vasc Endovasc Surg 2010; 39:726–730.
- Clark RA. Computed tomography of bowel infarction. J Comput Assist Tomogr 1987; 11:757–762.
- Acosta S, Björck M. Modern treatment of acute mesenteric ischaemia. Br J Surg 2014; 101:e100–e108.
- Smithson A, Ruiz J, Perello R, Valverde M, Ramos J, Garzo L. Diagnostic and management of spontaneous rectus sheath hematoma. Eur J Intern Med 2013; 24:579–582.
- Moreno Gallego A, Aguayo JL, Flores B, et al. Ultrasonography and computed tomography reduce unnecessary surgery in abdominal rectus sheath haematoma. Br J Surg 1997; 84:1295–1297.
- Dubinsky IL. Hematoma of the rectus abdominis muscle: case report and review of the literature. J Emerg Med 1997; 15:165–167.
- Yi M, Yao G, Bai Y. The monitoring of intra-abdominal pressure in critically ill patients. (In Chinese.) Zhonghua Wei Zhong Bing Ji Jiu Yi Xue 2014; 26:175–178.
- Hunt L, Frost SA, Hillman K, Newton PJ, Davidson PM. Management of intra-abdominal hypertension and abdominal compartment syndrome: a review. J Trauma Manag Outcomes 2014; 8:2.
- Malbrain ML, Cheatham ML, Kirkpatrick A, et al. Results from the International Conference of Experts on Intra-abdominal Hypertension and Abdominal Compartment Syndrome. I. Definitions. Intensive Care Med 2006; 32:1722–1732.
- Malbrain ML, Chiumello D, Cesana BM, et al; WAKE-Up! Investigators. A systematic review and individual patient data meta-analysis on intra-abdominal hypertension in critically ill patients: the wake-up project. World initiative on Abdominal Hypertension Epidemiology, a Unifying Project (WAKE-Up!). Minerva Anestesiol 2014; 80:293–306.
- Kirkpatrick AW, Roberts DJ, De Waele J, et al; Pediatric Guidelines Sub-Committee for the World Society of the Abdominal Compartment Syndrome. Intra-abdominal hypertension and the abdominal compartment syndrome: updated consensus definitions and clinical practice guidelines from the World Society of the Abdominal Compartment Syndrome. Intensive Care Med 2013; 39:1190–1206.
- Holodinsky JK, Roberts DJ, Ball CG, et al. Risk factors for intra-abdominal hypertension and abdominal compartment syndrome among adult intensive care unit patients: a systematic review and meta-analysis. Crit Care 2013; 17:R249.
- Sugrue M, Bauman A, Jones F, et al. Clinical examination is an inaccurate predictor of intraabdominal pressure. World J Surg 2002; 26:1428–1431.
- Cheatham ML, De Waele JJ, De Laet I, et al; World Society of the Abdominal Compartment Syndrome (WSACS) Clinical Trials Working Group. The impact of body position on intra-abdominal pressure measurement: a multicenter analysis. Crit Care Med 2009; 37:2187–2190.
- Ortiz-Diaz E, Lan CK. Intra-abdominal hypertension in medical critically ill patients: a narrative review. Shock 2014; 41:175–180.
- Cotton BA, Reddy N, Hatch QM, et al. Damage control resuscitation is associated with a reduction in resuscitation volumes and improvement in survival in 390 damage control laparotomy patients. Ann Surg 2011; 254:598–605.
- An G, West MA. Abdominal compartment syndrome: a concise clinical review. Crit Care Med 2008; 36:1304–1310.
- Tolcher MC, Nitsche JF, Arendt KW, Rose CH. Spontaneous rectus sheath hematoma pregnancy: case report and review of the literature. Obstet Gynecol Surv 2010; 65:517–522.
- Berná JD, Garcia-Medina V, Guirao J, Garcia-Medina J. Rectus sheath hematoma: diagnostic classification by CT. Abdom Imaging 1996; 21:62–64.
- Unger EC, Glazer HS, Lee JK, Ling D. MRI of extracranial hematomas: preliminary observations. AJR Am J Roentgenol 1986; 146:403–407.
- Rimola J, Perendreu J, Falcó J, Fortuño JR, Massuet A, Branera J. Percutaneous arterial embolization in the management of rectus sheath hematoma. AJR Am J Roentgenol 2007; 188:W497–W502.
- Titone C, Lipsius M, Krakauer JS. “Spontaneous” hematoma of the rectus abdominis muscle: critical review of 50 cases with emphasis on early diagnosis and treatment. Surgery 1972; 72:568–572.
- Osinbowale O, Bartholomew JR. Rectus sheath hematoma. Vasc Med 2008; 13:275–279.
- Berná-Serna JD, Sánchez-Garre J, Madrigal M, Zuazu I, Berná-Mestre JD. Ultrasound therapy in rectus sheath hematoma. Phys Ther 2005; 85:352–357.
A 57-year-old woman presented to the emergency department with left lower quadrant pain, which had started 1 week earlier and was constant, dull, aching, and nonradiating. There were no aggravating or alleviating factors. The pain was associated with low-grade fever and nausea. She reported no vomiting, no change in bowel habits, and no hematemesis, hematochezia, or melena. She did not have urinary urgency, frequency, or dysuria. She had no cardiac, respiratory, or neurologic symptoms.
Her medical history included hypothyroidism, type 2 diabetes mellitus, diverticulosis, and chronic obstructive pulmonary disease. Her medications included metformin, insulin, levothyroxine, and inhaled tiotropium. She had no allergies. She had never undergone surgery, including cesarean delivery. She was postmenopausal. She had two children, both of whom had been born vaginally at full term. She denied using alcohol, tobacco, and illicit drugs. Her family history was noncontributory.
On examination, she was not in acute distress. Her temperature was 36.7°C (98.1°F), blood pressure 130/90 mm Hg, heart rate 86 beats per minute and regular, respiratory rate 16 breaths per minute, and oxygen saturation 98% on ambient air. Examination of her head and neck was unremarkable. Cardiopulmonary examination was normal. Abdominal examination revealed normal bowel sounds, mild tenderness in the left lower quadrant with localized guarding, and rebound tenderness. Neurologic examination was unremarkable.
Initial laboratory data showed mild leukocytosis. Computed tomography with contrast of the abdomen and pelvis suggested acute diverticulitis.
ATRIAL FIBRILLATION, AND THEN A TURN FOR THE WORSE
The patient was admitted with an initial diagnosis of acute diverticulitis. She was started on antibiotics, hydration, and pain medications, and her abdominal pain gradually improved.
On the third hospital day, she suddenly experienced shortness of breath and palpitations. At the time of admission her electrocardiogram had been normal, but it now showed atrial fibrillation with a rapid ventricular response. She also developed elevated troponin levels, which were thought to represent type 2 non-ST-elevation myocardial infarction.
She was started on aspirin, clopidogrel, and anticoagulation with heparin bridged with warfarin for the new-onset atrial fibrillation. Her heart rate was controlled with metoprolol, and her shortness of breath improved. An echocardiogram was normal.
On the seventh hospital day, she developed severe right-sided lower abdominal pain and bruising. Her blood pressure was 90/60 mm Hg, heart rate 110 beats per minute and irregularly irregular, respiratory rate 22 breaths per minute, and oxygen saturation 97% on room air. Her abdomen was diffusely tender with a palpable mass in the right lower quadrant and hypoactive bowel sounds. Ecchymosis was noted (Figure 1).
DIFFERENTIAL DIAGNOSIS
1. What is the likely cause of her decompensation?
- Acute mesenteric ischemia
- Perforation of the gastrointestinal tract
- Rectus sheath hematoma
- Abdominal compartment syndrome due to acute pancreatitis
Acute mesenteric ischemia
Signs and symptoms of acute mesenteric ischemia can be vague. Moreover, when it leads to bowel necrosis the mortality rate is high, ranging from 30% to 65%.1 Hence, one should suspect it and try to diagnose it early.
Most patients with this condition have comorbidities; risk factors include atherosclerotic disease, cardiac conditions (congestive heart failure, recent myocardial infarction, and atrial fibrillation), systemic illness, and inherited or acquired hypercoagulable states.2
The four major causes are:
- Acute thromboembolic occlusion of the superior mesenteric artery (the most common site of occlusion because of the acute angle of origin from the aorta)
- Acute thrombosis of the mesenteric vein
- Acute thrombosis of the mesenteric artery
- Nonocclusive disease affecting the mesenteric vessels2
Nonocclusive disease is seen in conditions in which the mesenteric vessels are already compromised due to background stenosis owing to atherosclerosis. Also, conditions such as septic and cardiogenic shock can compromise these arteries, leading to ischemia, which, if it persists, can lead to bowel infarction. Ischemic colitis falls under this category. It commonly involves the descending and sigmoid colon.3
The initial symptom of ischemia may be abdominal pain that is brought on by eating large meals (“postprandial intestinal angina.”2 When the ischemia worsens to infarction, patients may have a diffusely tender abdomen and constant pain that does not vary with palpation. Surprisingly, patients do not exhibit peritoneal signs early on. This gives rise to the description of “pain out of proportion to the physical findings” traditionally associated with acute mesenteric ischemia.2
Diagnosis. Supportive laboratory data include marked leukocytosis, elevated hematocrit due to hemoconcentration, metabolic acidosis, and elevated lactate.4 Newer markers such as serum alpha-glutathione S-transferase (alpha-GST) and intestinal fatty acid-binding protein (I-FABP) may be used to support the diagnosis.
Elevated alpha-GST has 72% sensitivity and 77% specificity in the diagnosis of acute mesenteric ischemia.5 The caveat is that it cannot reliably differentiate ischemia from infarction. Its sensitivity can be improved to 97% to 100% by using the white blood cell count and lactate levels in combination.5
An I-FABP level higher than 100 ng/mL has 100% sensitivity for diagnosing mesenteric infarction but only 25% sensitivity for bowel strangulation.6
Early use of abdominal computed tomography with contrast can aid in recognizing this diagnosis.7 Thus, it should be ordered in suspected cases, even in patients who have elevated creatinine levels (which would normally preclude the use of contrast), since early diagnosis followed by endovascular therapy is associated with survival benefit, and the risk of contrast-induced nephropathy appears to be small.8 Computed tomography helps to determine the state of mesenteric vessels and bowel perfusion before ischemia progresses to infarction. It also helps to rule out other common diagnoses. Findings that suggest acute mesenteric ischemia include segmental bowel wall thickening, intestinal pneumatosis with gas in the portal vein, bowel dilation, mesenteric stranding, portomesenteric thrombosis, and solid-organ infarction.9
Treatment. If superior mesenteric artery occlusion is diagnosed on computed tomography, the next step is to determine if there is peritonitis.10 In patients who have evidence of peritonitis, exploratory laparotomy is performed. For emboli in such patients, open embolectomy followed by on-table angiography is carried out in combination with damage-control surgery. For patients with peritonitis and acute thrombosis, stenting along with damage-control surgery is preferred.10
On the other hand, if there is no peritonitis, the thrombosis may be amenable to endovascular intervention. For patients with acute embolic occlusion with no contraindications to thrombolysis, aspiration embolectomy in combination with local catheter-directed thrombolysis with recombinant tissue plasminogen activator can be performed. This can be combined with endovascular mechanical embolectomy for more complete management.10 Patients with contraindications to thrombolysis can be treated either with aspiration and mechanical embolectomy or with open embolectomy with angiography.10
During laparotomy, the surgeon carefully inspects the bowel for signs of necrosis. Signs that bowel is still viable include pink color, bleeding from cut surfaces, good peristalsis, and visible pulsations in the arterial arcade of the mesentery.
Acute mesenteric artery thrombosis arising from chronic atherosclerotic disease can be treated with stenting of the stenotic lesion.10 Patients with this condition would also benefit from aggressive management of atherosclerotic disease with statins along with antiplatelet agents.10
Mesenteric vein thrombosis requires prompt institution of anticoagulation. However, in advanced cases leading to bowel infarction, exploratory laparotomy with resection of the necrotic bowel may be required. Anticoagulation should be continued for at least 6 months, and further therapy should be determined by the underlying precipitating condition.10
Critically ill patients who develop mesenteric ischemia secondary to persistent hypotension usually respond to adequate volume resuscitation, cessation of vasopressors, and overall improvement in their hemodynamic status. These patients must be closely monitored for development of gangrene of the bowel because they may be intubated and not able to complain. Any sudden deterioration in their condition should prompt physicians to consider bowel necrosis developing in these patients. Elevation of lactate levels out of proportion to the degree of hypotension may be corroborative evidence.4
Our patient had risk factors for acute mesenteric ischemia that included atrial fibrillation and recent non-ST-elevation myocardial infarction. She could have had arterial emboli due to atrial fibrillation, in situ superior mesenteric arterial thrombosis, or splanchnic arterial vasoconstriction due to the myocardial infarction associated with transient hypotension.
Arguing against this diagnosis, although she had a grossly distended abdomen, abdominal bruising usually is not seen. Also, a palpable mass in the right lower quadrant is uncommon except when acute mesenteric ischemia occurs due to segmental intestinal strangulation, as with strangulated hernia or volvulus. She also had therapeutic international normalized ratio (INR) levels constantly while on anticoagulation. Nevertheless, acute mesenteric ischemia should be strongly considered in the initial differential diagnosis of this patient’s acute decompensation.
Perforation of the gastrointestinal tract
Diverticulitis is the acute inflammation of one or more diverticula, which are small pouches created by herniation of the mucosa into the wall of the colon. The condition is caused by microscopic or macroscopic perforation of the diverticula. Microscopic perforation is usually self-limited (uncomplicated diverticulitis) and responds to conservative treatment, whereas macroscopic perforation can be associated with fecal or purulent peritonitis, abscess, enteric fistula, bowel obstruction, and stricture (complicated diverticulitis), in which case surgery may be necessary.
Patients with peritonitis due to free perforation present with generalized tenderness with rebound tenderness and guarding on abdominal examination. The abdomen may be distended and tympanic to percussion, with diminished or absent bowel sounds. Patients may have hemodynamic compromise.
Plain upright abdominal radiographs may show free air under the diaphragm. Computed tomography may show oral contrast outside the lumen and detect even small amounts of free intraperitoneal air (more clearly seen on a lung window setting).
Our patient initially presented with acute diverticulitis. She later developed diffuse abdominal tenderness with hypoactive bowel sounds. Bowel perforation is certainly a possibility at this stage, though it is usually not associated with abdominal bruising. She would need additional imaging to rule out this complication.
Other differential diagnoses to be considered in this patient with right lower-quadrant pain include acute appendicitis, incarcerated inguinal hernia, volvulus (particularly cecal volvulus), small-bowel obstruction, pyelonephritis, and gynecologic causes such as adnexal torsion, ruptured ovarian cyst, and tubo-ovarian abscess. Computed tomography helps to differentiate most of these causes.
Rectus sheath hematoma
Rectus sheath hematoma is relatively uncommon and often not considered in the initial differential diagnosis of an acute abdomen. This gives it the rightful term “a great masquerader.” It usually results from bleeding into the rectus sheath from damage to the superior (more common) or inferior epigastric arteries and occasionally from a direct tear of the rectus abdominis muscle. Predisposing factors include anticoagulant therapy (most common), advanced age, hypertension, previous abdominal surgery, trauma, paroxysmal coughing, medication injections, pregnancy, blood dyscrasias, severe vomiting, violent physical activity, and leukemia.11
Clinical manifestations include acute abdominal pain, often associated with fever, nausea, and vomiting. Physical examination may reveal signs of hypovolemic shock, a palpable nonpulsatile abdominal mass, and signs of local peritoneal irritation. The Carnett sign11 (tenderness within the abdominal wall that persists and does not improve with raising the head) and the Fothergill sign11 (a tender abdominal mass that does not cross the midline and remains palpable with tensing of the rectus sheath) may be elicited.
Computed tomography is more sensitive than abdominal ultrasonography in differentiating rectus sheath hematoma from an intra-abdominal pathology.11 In addition, computed tomography also helps to determine if the bleeding is active or not, which has therapeutic implications.
In our patient, rectus sheath hematoma is a possibility because of her ongoing anticoagulation, findings of localized abdominal bruising, and palpable right lower-quadrant mass, and it is high on the list of differential diagnoses. Rectus sheath hematoma should be considered in the differential diagnosis of lower abdominal pain particularly in elderly women who are on anticoagulation and in whom the onset of pain coincides with a paroxysm of cough.12 Women are twice as likely as men to develop rectus sheath hematoma, owing to their different muscle mass.13 In addition, anterior abdominal wall muscles are stretched during pregnancy.13
Abdominal compartment syndrome
Abdominal compartment syndrome has been classically associated with surgical patients. However, it is being increasingly recognized in critically ill medical patients, in whom detecting and treating it early may result in significant reduction in rates of morbidity and death.14
Abdominal compartment syndrome is of three types: primary, secondary, and recurrent. Primary abdominal compartment syndrome refers to the classic surgical patients with evidence of direct injury to the abdominal or pelvic organs through major trauma or extensive abdominal surgeries. Secondary abdominal compartment syndrome refers to its development in critically ill intensive care patients in whom the pathology does not directly involve the abdominal or pelvic organs.
Various medical conditions can culminate in abdominal compartment syndrome and result in multiorgan failure. Recurrent abdominal compartment syndrome refers to its development after management of either primary or secondary intra-abdominal hypertension or abdominal compartment syndrome.15 Clinicians thus must be aware of secondary and recurrent abdominal compartment syndrome occurring in critically ill patients.
The normal intra-abdominal pressure is around 5 to 7 mm Hg, even in most critically ill patients. Persistent elevation, ie, higher than 12 mm Hg, is referred to as intra-abdominal hypertension.16–18 Intra-abdominal hypertension is subdivided into four grades:
- Grade I: 12–15 mm Hg
- Grade II: 16–20 mm Hg
- Grade III: 21–25 mm Hg
- Grade IV: > 25 mm Hg.
The World Society of the Abdominal Compartment Syndrome (WSACS) defines abdominal compartment syndrome as pressure higher than 20 mm Hg along with organ damage.18 It may or may not be associated with an abdominal perfusion pressure less than 60 mm Hg.18
Risk factors associated with abdominal compartment syndrome include conditions causing decreased gut motility (gastroparesis, ileus, and colonic pseudo-obstruction), intra-abdominal or retroperitoneal masses or abscesses, ascites, hemoperitoneum, acute pancreatitis, third-spacing due to massive fluid resuscitation with transfusions, peritoneal dialysis, and shock.18,19
Physical examination has a sensitivity of only 40% to 60% in detecting intra-abdominal hypertension.20 The gold-standard method of measuring the intra-abdominal pressure is the modified Kron technique,18 using a Foley catheter in the bladder connected to a pressure transducer. With the patient in the supine position, the transducer is zeroed at the mid-axillary line at the level of the iliac crest, and 25 mL of normal saline is instilled into the bladder and maintained for 30 to 60 seconds to let the detrusor muscle relax.15 Pressure tracings are then recorded at the end of expiration. Factors that are known to affect the transbladder pressure include patient position, respiratory movement, and body mass index, and should be taken into account when reading the pressure recordings.15,21 Other techniques that can be used include intragastric, intra-inferior vena cava, and intrarectal approaches.15
The WSACS recommends that any patient admitted to a critical care unit or in whom new organ failure develops should be screened for risk factors for intra-abdominal hypertension and abdominal compartment syndrome. If a patient has at least two of the risk factors suggested by WSACS, a baseline intra-abdominal pressure measurement should be obtained. Patients at risk for intra-abdominal hypertension should have the intra-abdominal pressure measured every 4 to 6 hours. However, in the face of hemodynamic instability and worsening multiorgan failure, the pressure may need to be measured hourly.18
Clinicians managing patients in the intensive care unit should think of intra-abdominal pressure alongside blood pressure, urine output, and mental status when evaluating hemodynamic status. Clinical manifestations of abdominal compartment syndrome reflect the underlying organ dysfunction and include hypotension, refractory shock, decreased urine output, intracranial hypertension, progressive hypoxemia and hypercarbia, elevated pulmonary peak pressures, and worsening of metabolic acidosis.22
Treatment. The standard treatment for primary abdominal compartment syndrome is surgical decompression. According to WSACS guidelines, insertion of a percutaneous drainage catheter should be advocated in patients with gross ascites and in whom decompressive surgery is not feasible. A damage-control resuscitation strategy used for patients undergoing damage-control laparotomy has been found to increase the 30-day survival rate.23 A damage-control resuscitation strategy consists of increasing the use of plasma and platelet transfusions over packed red cell transfusions, limiting the use of crystalloid solutions in early fluid resuscitation, and allowing for permissive hypotension.
Secondary abdominal compartment syndrome is treated conservatively in most cases, since patients with this condition are very poor surgical candidates owing to their comorbidities.18 However, in patients with progressive organ dysfunction in whom medical management has failed, surgical decompression should be considered.18 Medical management of secondary abdominal compartment syndrome depends on the underlying etiology. Strategies include nasogastric or colonic decompression, use of prokinetic agents, paracentesis in cases with gross ascites, and maintaining a cumulative negative fluid balance. The WSACS does not recommend routine use of diuretics, albumin infusion, or renal replacement strategies. Pain should be adequately controlled to improve abdominal wall compliance.18,24 Neuromuscular blockade agents may be used to aid this process. Neostigmine may be used to treat colonic pseudo-obstruction when other conservative methods fail. Use of enteral nutrition should be minimized.18
Our patient might have abdominal compartment syndrome, but a definitive diagnosis cannot be made at this point without measuring the intra-abdominal pressure.
WHICH IMAGING TEST WOULD BE BEST?
2. Which imaging test would be best for establishing the diagnosis in this patient?
- Plain abdominal radiography
- Abdominal ultrasonography
- Computed tomography of the abdomen and pelvis with contrast
- Magnetic resonance imaging of the abdomen and pelvis
Plain abdominal radiography
Plain abdominal radiography can help to determine if there is free gas under the diaphragm (due to bowel perforation), obstructed bowel, sentinel loop, volvulus, or fecoliths causing the abdominal pain. It cannot diagnose rectus sheath hematoma or acute mesenteric ischemia.
Abdominal ultrasonography
Abdominal ultrasonography can be used as the first diagnostic test, as it is widely available, safe, effective, and tolerable. It does not expose the patient to radiation or intravenous contrast agents. It helps to diagnose rectus sheath hematoma and helps to follow its maturation and resolution once a diagnosis is made. It can provide a rapid assessment of the size, location, extent, and physical characteristics of the mass.
Rectus sheath hematoma appears spindle-shaped on sagittal sections and ovoid on coronal sections. It often appears sonolucent in the early stages and sonodense in the late stage, but the appearance may be heterogeneous depending on the combined presence of clot and fresh blood. These findings are sufficient to make the diagnosis.
Abdominal ultrasonography has 85% to 96% sensitivity in diagnosing rectus sheath hematoma.25 It can help diagnose other causes of the abdominal pain, such as renal stones and cholecystitis. It is the preferred imaging test in pediatric patients, pregnant patients, and those with renal insufficiency.
Abdominal computed tomography
Abdominal computed tomography has a sensitivity and specificity of 100% for diagnosing acute rectus sheath hematoma with a duration of less than 5 days.25 It not only helps to determine the precise location and extent, but also helps to determine if there is active extravasation. Even in patients with renal insufficiency, noncontrast computed tomography will help to confirm the diagnosis, although it may not show extravasation or it may miss certain abdominal pathologies because of the lack of contrast.
Acute rectus sheath hematoma appears as a hyperdense mass posterior to the rectus abdominis muscle with ipsilateral anterolateral muscular enlargement. Chronic rectus sheath hematoma appears isodense or hypodense relative to the surrounding muscle. Above the arcuate line, rectus sheath hematoma has a spindle shape; below the arcuate line, it is typically spherical.
In 1996, Berná et al26 classified rectus sheath hematoma into three grades based on findings of computed tomography:
- Grade I is intramuscular and unilateral
- Grade II may involve bilateral rectus muscles without extension into the prevesicular space
- Grade III extends into the peritoneum and prevesicular space
Magnetic resonance imaging
Magnetic resonance imaging is useful to differentiate chronic rectus sheath hematoma (greater than 5-day duration) from an anterior abdominal wall mass. Chronic rectus sheath hematoma will have high signal intensity on both T1- and T2-weighted images up to 10 months after the onset of the hematoma.27
Back to our patient
Since our patient’s symptoms are acute and of less than 5 days’ duration, computed tomography of the abdomen and pelvis would be the best diagnostic test, with therapeutic implications if there is ongoing extravasation.
Computed tomography of the abdomen with contrast showed a new hematoma, measuring 25 by 14 by 13.5 cm, in the right rectus sheath (Figure 2), with no other findings. The hematoma was grade I, since it was intramuscular and unilateral without extension elsewhere.
Laboratory workup showed that the patient’s hematocrit was falling, from 34% to 24%, and her INR was elevated at 2.5. She was resuscitated with fluids, blood transfusion, and fresh-frozen plasma. Anticoagulation was withheld. In spite of resuscitation, her hematocrit kept falling, though she remained hemodynamically stable.
THE WAY FORWARD
3. At this point, what would be the best approach to management in this patient?
- Serial clinical examinations and frequent monitoring of the complete blood cell count
- Urgent surgical consult for exploratory laparotomy with evaluation of the hematoma and ligation of the bleeding vessel
- Repeat computed tomographic angiography to identify a possible bleeding vessel; consideration of radiographically guided arterial embolization
- Measuring the intra-abdominal pressure using the intrabladder pressure for abdominal compartment syndrome and consideration of surgical drainage
The key clinical concern in a patient with a rectus sheath hematoma who is hemodynamically stable is whether the hematoma is expanding. This patient responded to initial resuscitation, but her falling hematocrit was evidence of ongoing bleeding leading to an expanding rectus sheath hematoma. Thus, serial clinical examinations and frequent monitoring of the complete blood cell count would not be enough, as it could miss fatal ongoing bleeding.
Radiographically guided embolization with Gelfoam, thrombin, or coils should be attempted first, as this is less invasive than exploratory laparotomy.28 It can achieve hemostasis, decrease the size of the hematoma, limit the need for blood products, and prevent rupture into the abdomen. If this is unsuccessful, the next step is ligation of the bleeding vessel.29
Surgical treatment includes evacuation of the hematoma, repair of the rectus sheath, ligation of bleeding vessels, and abdominal wall closure. Surgical evacuation or guided drainage of a rectus sheath hematoma on its own is not normally indicated and may indeed cause persistent bleeding by diminishing a potential tamponade effect. However, it may become necessary if the hematoma is very large or infected, if it causes marked respiratory impairment, or if abdominal compartment syndrome is suspected.
Abdominal compartment syndrome is very rare but is associated with a 50% mortality rate.30 It should be suspected in patients with oliguria, low cardiac output, changes in minute ventilation, and altered splanchnic blood flow. The diagnosis is confirmed with indwelling catheter manometry of the bladder to measure intra-abdominal pressure. Intra-abominal pressure above 25 mm Hg should be treated with decompressive laparotomy.30 However, the clinical suspicion of abdominal compartment syndrome was low in this patient.
The best choice at this point would be urgent computed tomographic angiography to identify a bleeding vessel, along with consideration of radiographically guided arterial embolization.
TREATMENT IS USUALLY CONSERVATIVE
Treatment of rectus sheath hematoma is conservative in most hemodynamically stable patients, with embolization or surgical intervention reserved for unstable patients or those in whom the hematoma is expanding.
Knowledge of the grading system of Berná et al26 helps to assess the patient’s risk and to anticipate potential complications. Grade I hematomas are mild and do not necessitate admission. Patients with grade II hematoma can be admitted to the floor for 24 to 48 hours for observation. Grade III usually occurs in patients receiving anticoagulant therapy and frequently requires blood products. These patients have a prolonged hospital stay and more complications, including hypovolemic shock, myonecrosis, acute coronary syndrome, arrhythmias, acute renal failure, small-bowel infarction, and abdominal compartment syndrome—all of which increases the risk of morbidity and death. Thus, patients who are on anticoagulation who develop grade III rectus sheath hematoma should be admitted to the hospital, preferably to the intensive care unit, to ensure that the hematoma is not expanding and to plan reinstitution of anticoagulation as appropriate.
In most cases, rectus sheath hematomas resolve within 1 to 3 months. Resolution of large hematomas may be hastened with the use of pulsed ultrasound.31 However, this treatment should be used only after the acute phase is over, when there is evidence of an organized thrombus and coagulation measures have returned to the target range. This helps to reduce the risk of bleeding and to prevent symptoms from worsening.31
OUR PATIENT’S COURSE
Our patient underwent urgent computed tomographic angiography, which showed a modest increase in the size of the rectus sheath hematoma. However, no definitive blush of contrast was seen to suggest active arterial bleeding. Her hematocrit stabilized, and she remained hemodynamically stable without requiring additional intervention. Most likely her bleeding was self-contained. She had normal intra-abdominal pressure on serial monitoring. She was later transferred to acute inpatient rehabilitation in view of deconditioning and is currently doing well. The hematoma persisted, decreasing only slightly in size over the next 3 weeks.
A 57-year-old woman presented to the emergency department with left lower quadrant pain, which had started 1 week earlier and was constant, dull, aching, and nonradiating. There were no aggravating or alleviating factors. The pain was associated with low-grade fever and nausea. She reported no vomiting, no change in bowel habits, and no hematemesis, hematochezia, or melena. She did not have urinary urgency, frequency, or dysuria. She had no cardiac, respiratory, or neurologic symptoms.
Her medical history included hypothyroidism, type 2 diabetes mellitus, diverticulosis, and chronic obstructive pulmonary disease. Her medications included metformin, insulin, levothyroxine, and inhaled tiotropium. She had no allergies. She had never undergone surgery, including cesarean delivery. She was postmenopausal. She had two children, both of whom had been born vaginally at full term. She denied using alcohol, tobacco, and illicit drugs. Her family history was noncontributory.
On examination, she was not in acute distress. Her temperature was 36.7°C (98.1°F), blood pressure 130/90 mm Hg, heart rate 86 beats per minute and regular, respiratory rate 16 breaths per minute, and oxygen saturation 98% on ambient air. Examination of her head and neck was unremarkable. Cardiopulmonary examination was normal. Abdominal examination revealed normal bowel sounds, mild tenderness in the left lower quadrant with localized guarding, and rebound tenderness. Neurologic examination was unremarkable.
Initial laboratory data showed mild leukocytosis. Computed tomography with contrast of the abdomen and pelvis suggested acute diverticulitis.
ATRIAL FIBRILLATION, AND THEN A TURN FOR THE WORSE
The patient was admitted with an initial diagnosis of acute diverticulitis. She was started on antibiotics, hydration, and pain medications, and her abdominal pain gradually improved.
On the third hospital day, she suddenly experienced shortness of breath and palpitations. At the time of admission her electrocardiogram had been normal, but it now showed atrial fibrillation with a rapid ventricular response. She also developed elevated troponin levels, which were thought to represent type 2 non-ST-elevation myocardial infarction.
She was started on aspirin, clopidogrel, and anticoagulation with heparin bridged with warfarin for the new-onset atrial fibrillation. Her heart rate was controlled with metoprolol, and her shortness of breath improved. An echocardiogram was normal.
On the seventh hospital day, she developed severe right-sided lower abdominal pain and bruising. Her blood pressure was 90/60 mm Hg, heart rate 110 beats per minute and irregularly irregular, respiratory rate 22 breaths per minute, and oxygen saturation 97% on room air. Her abdomen was diffusely tender with a palpable mass in the right lower quadrant and hypoactive bowel sounds. Ecchymosis was noted (Figure 1).
DIFFERENTIAL DIAGNOSIS
1. What is the likely cause of her decompensation?
- Acute mesenteric ischemia
- Perforation of the gastrointestinal tract
- Rectus sheath hematoma
- Abdominal compartment syndrome due to acute pancreatitis
Acute mesenteric ischemia
Signs and symptoms of acute mesenteric ischemia can be vague. Moreover, when it leads to bowel necrosis the mortality rate is high, ranging from 30% to 65%.1 Hence, one should suspect it and try to diagnose it early.
Most patients with this condition have comorbidities; risk factors include atherosclerotic disease, cardiac conditions (congestive heart failure, recent myocardial infarction, and atrial fibrillation), systemic illness, and inherited or acquired hypercoagulable states.2
The four major causes are:
- Acute thromboembolic occlusion of the superior mesenteric artery (the most common site of occlusion because of the acute angle of origin from the aorta)
- Acute thrombosis of the mesenteric vein
- Acute thrombosis of the mesenteric artery
- Nonocclusive disease affecting the mesenteric vessels2
Nonocclusive disease is seen in conditions in which the mesenteric vessels are already compromised due to background stenosis owing to atherosclerosis. Also, conditions such as septic and cardiogenic shock can compromise these arteries, leading to ischemia, which, if it persists, can lead to bowel infarction. Ischemic colitis falls under this category. It commonly involves the descending and sigmoid colon.3
The initial symptom of ischemia may be abdominal pain that is brought on by eating large meals (“postprandial intestinal angina.”2 When the ischemia worsens to infarction, patients may have a diffusely tender abdomen and constant pain that does not vary with palpation. Surprisingly, patients do not exhibit peritoneal signs early on. This gives rise to the description of “pain out of proportion to the physical findings” traditionally associated with acute mesenteric ischemia.2
Diagnosis. Supportive laboratory data include marked leukocytosis, elevated hematocrit due to hemoconcentration, metabolic acidosis, and elevated lactate.4 Newer markers such as serum alpha-glutathione S-transferase (alpha-GST) and intestinal fatty acid-binding protein (I-FABP) may be used to support the diagnosis.
Elevated alpha-GST has 72% sensitivity and 77% specificity in the diagnosis of acute mesenteric ischemia.5 The caveat is that it cannot reliably differentiate ischemia from infarction. Its sensitivity can be improved to 97% to 100% by using the white blood cell count and lactate levels in combination.5
An I-FABP level higher than 100 ng/mL has 100% sensitivity for diagnosing mesenteric infarction but only 25% sensitivity for bowel strangulation.6
Early use of abdominal computed tomography with contrast can aid in recognizing this diagnosis.7 Thus, it should be ordered in suspected cases, even in patients who have elevated creatinine levels (which would normally preclude the use of contrast), since early diagnosis followed by endovascular therapy is associated with survival benefit, and the risk of contrast-induced nephropathy appears to be small.8 Computed tomography helps to determine the state of mesenteric vessels and bowel perfusion before ischemia progresses to infarction. It also helps to rule out other common diagnoses. Findings that suggest acute mesenteric ischemia include segmental bowel wall thickening, intestinal pneumatosis with gas in the portal vein, bowel dilation, mesenteric stranding, portomesenteric thrombosis, and solid-organ infarction.9
Treatment. If superior mesenteric artery occlusion is diagnosed on computed tomography, the next step is to determine if there is peritonitis.10 In patients who have evidence of peritonitis, exploratory laparotomy is performed. For emboli in such patients, open embolectomy followed by on-table angiography is carried out in combination with damage-control surgery. For patients with peritonitis and acute thrombosis, stenting along with damage-control surgery is preferred.10
On the other hand, if there is no peritonitis, the thrombosis may be amenable to endovascular intervention. For patients with acute embolic occlusion with no contraindications to thrombolysis, aspiration embolectomy in combination with local catheter-directed thrombolysis with recombinant tissue plasminogen activator can be performed. This can be combined with endovascular mechanical embolectomy for more complete management.10 Patients with contraindications to thrombolysis can be treated either with aspiration and mechanical embolectomy or with open embolectomy with angiography.10
During laparotomy, the surgeon carefully inspects the bowel for signs of necrosis. Signs that bowel is still viable include pink color, bleeding from cut surfaces, good peristalsis, and visible pulsations in the arterial arcade of the mesentery.
Acute mesenteric artery thrombosis arising from chronic atherosclerotic disease can be treated with stenting of the stenotic lesion.10 Patients with this condition would also benefit from aggressive management of atherosclerotic disease with statins along with antiplatelet agents.10
Mesenteric vein thrombosis requires prompt institution of anticoagulation. However, in advanced cases leading to bowel infarction, exploratory laparotomy with resection of the necrotic bowel may be required. Anticoagulation should be continued for at least 6 months, and further therapy should be determined by the underlying precipitating condition.10
Critically ill patients who develop mesenteric ischemia secondary to persistent hypotension usually respond to adequate volume resuscitation, cessation of vasopressors, and overall improvement in their hemodynamic status. These patients must be closely monitored for development of gangrene of the bowel because they may be intubated and not able to complain. Any sudden deterioration in their condition should prompt physicians to consider bowel necrosis developing in these patients. Elevation of lactate levels out of proportion to the degree of hypotension may be corroborative evidence.4
Our patient had risk factors for acute mesenteric ischemia that included atrial fibrillation and recent non-ST-elevation myocardial infarction. She could have had arterial emboli due to atrial fibrillation, in situ superior mesenteric arterial thrombosis, or splanchnic arterial vasoconstriction due to the myocardial infarction associated with transient hypotension.
Arguing against this diagnosis, although she had a grossly distended abdomen, abdominal bruising usually is not seen. Also, a palpable mass in the right lower quadrant is uncommon except when acute mesenteric ischemia occurs due to segmental intestinal strangulation, as with strangulated hernia or volvulus. She also had therapeutic international normalized ratio (INR) levels constantly while on anticoagulation. Nevertheless, acute mesenteric ischemia should be strongly considered in the initial differential diagnosis of this patient’s acute decompensation.
Perforation of the gastrointestinal tract
Diverticulitis is the acute inflammation of one or more diverticula, which are small pouches created by herniation of the mucosa into the wall of the colon. The condition is caused by microscopic or macroscopic perforation of the diverticula. Microscopic perforation is usually self-limited (uncomplicated diverticulitis) and responds to conservative treatment, whereas macroscopic perforation can be associated with fecal or purulent peritonitis, abscess, enteric fistula, bowel obstruction, and stricture (complicated diverticulitis), in which case surgery may be necessary.
Patients with peritonitis due to free perforation present with generalized tenderness with rebound tenderness and guarding on abdominal examination. The abdomen may be distended and tympanic to percussion, with diminished or absent bowel sounds. Patients may have hemodynamic compromise.
Plain upright abdominal radiographs may show free air under the diaphragm. Computed tomography may show oral contrast outside the lumen and detect even small amounts of free intraperitoneal air (more clearly seen on a lung window setting).
Our patient initially presented with acute diverticulitis. She later developed diffuse abdominal tenderness with hypoactive bowel sounds. Bowel perforation is certainly a possibility at this stage, though it is usually not associated with abdominal bruising. She would need additional imaging to rule out this complication.
Other differential diagnoses to be considered in this patient with right lower-quadrant pain include acute appendicitis, incarcerated inguinal hernia, volvulus (particularly cecal volvulus), small-bowel obstruction, pyelonephritis, and gynecologic causes such as adnexal torsion, ruptured ovarian cyst, and tubo-ovarian abscess. Computed tomography helps to differentiate most of these causes.
Rectus sheath hematoma
Rectus sheath hematoma is relatively uncommon and often not considered in the initial differential diagnosis of an acute abdomen. This gives it the rightful term “a great masquerader.” It usually results from bleeding into the rectus sheath from damage to the superior (more common) or inferior epigastric arteries and occasionally from a direct tear of the rectus abdominis muscle. Predisposing factors include anticoagulant therapy (most common), advanced age, hypertension, previous abdominal surgery, trauma, paroxysmal coughing, medication injections, pregnancy, blood dyscrasias, severe vomiting, violent physical activity, and leukemia.11
Clinical manifestations include acute abdominal pain, often associated with fever, nausea, and vomiting. Physical examination may reveal signs of hypovolemic shock, a palpable nonpulsatile abdominal mass, and signs of local peritoneal irritation. The Carnett sign11 (tenderness within the abdominal wall that persists and does not improve with raising the head) and the Fothergill sign11 (a tender abdominal mass that does not cross the midline and remains palpable with tensing of the rectus sheath) may be elicited.
Computed tomography is more sensitive than abdominal ultrasonography in differentiating rectus sheath hematoma from an intra-abdominal pathology.11 In addition, computed tomography also helps to determine if the bleeding is active or not, which has therapeutic implications.
In our patient, rectus sheath hematoma is a possibility because of her ongoing anticoagulation, findings of localized abdominal bruising, and palpable right lower-quadrant mass, and it is high on the list of differential diagnoses. Rectus sheath hematoma should be considered in the differential diagnosis of lower abdominal pain particularly in elderly women who are on anticoagulation and in whom the onset of pain coincides with a paroxysm of cough.12 Women are twice as likely as men to develop rectus sheath hematoma, owing to their different muscle mass.13 In addition, anterior abdominal wall muscles are stretched during pregnancy.13
Abdominal compartment syndrome
Abdominal compartment syndrome has been classically associated with surgical patients. However, it is being increasingly recognized in critically ill medical patients, in whom detecting and treating it early may result in significant reduction in rates of morbidity and death.14
Abdominal compartment syndrome is of three types: primary, secondary, and recurrent. Primary abdominal compartment syndrome refers to the classic surgical patients with evidence of direct injury to the abdominal or pelvic organs through major trauma or extensive abdominal surgeries. Secondary abdominal compartment syndrome refers to its development in critically ill intensive care patients in whom the pathology does not directly involve the abdominal or pelvic organs.
Various medical conditions can culminate in abdominal compartment syndrome and result in multiorgan failure. Recurrent abdominal compartment syndrome refers to its development after management of either primary or secondary intra-abdominal hypertension or abdominal compartment syndrome.15 Clinicians thus must be aware of secondary and recurrent abdominal compartment syndrome occurring in critically ill patients.
The normal intra-abdominal pressure is around 5 to 7 mm Hg, even in most critically ill patients. Persistent elevation, ie, higher than 12 mm Hg, is referred to as intra-abdominal hypertension.16–18 Intra-abdominal hypertension is subdivided into four grades:
- Grade I: 12–15 mm Hg
- Grade II: 16–20 mm Hg
- Grade III: 21–25 mm Hg
- Grade IV: > 25 mm Hg.
The World Society of the Abdominal Compartment Syndrome (WSACS) defines abdominal compartment syndrome as pressure higher than 20 mm Hg along with organ damage.18 It may or may not be associated with an abdominal perfusion pressure less than 60 mm Hg.18
Risk factors associated with abdominal compartment syndrome include conditions causing decreased gut motility (gastroparesis, ileus, and colonic pseudo-obstruction), intra-abdominal or retroperitoneal masses or abscesses, ascites, hemoperitoneum, acute pancreatitis, third-spacing due to massive fluid resuscitation with transfusions, peritoneal dialysis, and shock.18,19
Physical examination has a sensitivity of only 40% to 60% in detecting intra-abdominal hypertension.20 The gold-standard method of measuring the intra-abdominal pressure is the modified Kron technique,18 using a Foley catheter in the bladder connected to a pressure transducer. With the patient in the supine position, the transducer is zeroed at the mid-axillary line at the level of the iliac crest, and 25 mL of normal saline is instilled into the bladder and maintained for 30 to 60 seconds to let the detrusor muscle relax.15 Pressure tracings are then recorded at the end of expiration. Factors that are known to affect the transbladder pressure include patient position, respiratory movement, and body mass index, and should be taken into account when reading the pressure recordings.15,21 Other techniques that can be used include intragastric, intra-inferior vena cava, and intrarectal approaches.15
The WSACS recommends that any patient admitted to a critical care unit or in whom new organ failure develops should be screened for risk factors for intra-abdominal hypertension and abdominal compartment syndrome. If a patient has at least two of the risk factors suggested by WSACS, a baseline intra-abdominal pressure measurement should be obtained. Patients at risk for intra-abdominal hypertension should have the intra-abdominal pressure measured every 4 to 6 hours. However, in the face of hemodynamic instability and worsening multiorgan failure, the pressure may need to be measured hourly.18
Clinicians managing patients in the intensive care unit should think of intra-abdominal pressure alongside blood pressure, urine output, and mental status when evaluating hemodynamic status. Clinical manifestations of abdominal compartment syndrome reflect the underlying organ dysfunction and include hypotension, refractory shock, decreased urine output, intracranial hypertension, progressive hypoxemia and hypercarbia, elevated pulmonary peak pressures, and worsening of metabolic acidosis.22
Treatment. The standard treatment for primary abdominal compartment syndrome is surgical decompression. According to WSACS guidelines, insertion of a percutaneous drainage catheter should be advocated in patients with gross ascites and in whom decompressive surgery is not feasible. A damage-control resuscitation strategy used for patients undergoing damage-control laparotomy has been found to increase the 30-day survival rate.23 A damage-control resuscitation strategy consists of increasing the use of plasma and platelet transfusions over packed red cell transfusions, limiting the use of crystalloid solutions in early fluid resuscitation, and allowing for permissive hypotension.
Secondary abdominal compartment syndrome is treated conservatively in most cases, since patients with this condition are very poor surgical candidates owing to their comorbidities.18 However, in patients with progressive organ dysfunction in whom medical management has failed, surgical decompression should be considered.18 Medical management of secondary abdominal compartment syndrome depends on the underlying etiology. Strategies include nasogastric or colonic decompression, use of prokinetic agents, paracentesis in cases with gross ascites, and maintaining a cumulative negative fluid balance. The WSACS does not recommend routine use of diuretics, albumin infusion, or renal replacement strategies. Pain should be adequately controlled to improve abdominal wall compliance.18,24 Neuromuscular blockade agents may be used to aid this process. Neostigmine may be used to treat colonic pseudo-obstruction when other conservative methods fail. Use of enteral nutrition should be minimized.18
Our patient might have abdominal compartment syndrome, but a definitive diagnosis cannot be made at this point without measuring the intra-abdominal pressure.
WHICH IMAGING TEST WOULD BE BEST?
2. Which imaging test would be best for establishing the diagnosis in this patient?
- Plain abdominal radiography
- Abdominal ultrasonography
- Computed tomography of the abdomen and pelvis with contrast
- Magnetic resonance imaging of the abdomen and pelvis
Plain abdominal radiography
Plain abdominal radiography can help to determine if there is free gas under the diaphragm (due to bowel perforation), obstructed bowel, sentinel loop, volvulus, or fecoliths causing the abdominal pain. It cannot diagnose rectus sheath hematoma or acute mesenteric ischemia.
Abdominal ultrasonography
Abdominal ultrasonography can be used as the first diagnostic test, as it is widely available, safe, effective, and tolerable. It does not expose the patient to radiation or intravenous contrast agents. It helps to diagnose rectus sheath hematoma and helps to follow its maturation and resolution once a diagnosis is made. It can provide a rapid assessment of the size, location, extent, and physical characteristics of the mass.
Rectus sheath hematoma appears spindle-shaped on sagittal sections and ovoid on coronal sections. It often appears sonolucent in the early stages and sonodense in the late stage, but the appearance may be heterogeneous depending on the combined presence of clot and fresh blood. These findings are sufficient to make the diagnosis.
Abdominal ultrasonography has 85% to 96% sensitivity in diagnosing rectus sheath hematoma.25 It can help diagnose other causes of the abdominal pain, such as renal stones and cholecystitis. It is the preferred imaging test in pediatric patients, pregnant patients, and those with renal insufficiency.
Abdominal computed tomography
Abdominal computed tomography has a sensitivity and specificity of 100% for diagnosing acute rectus sheath hematoma with a duration of less than 5 days.25 It not only helps to determine the precise location and extent, but also helps to determine if there is active extravasation. Even in patients with renal insufficiency, noncontrast computed tomography will help to confirm the diagnosis, although it may not show extravasation or it may miss certain abdominal pathologies because of the lack of contrast.
Acute rectus sheath hematoma appears as a hyperdense mass posterior to the rectus abdominis muscle with ipsilateral anterolateral muscular enlargement. Chronic rectus sheath hematoma appears isodense or hypodense relative to the surrounding muscle. Above the arcuate line, rectus sheath hematoma has a spindle shape; below the arcuate line, it is typically spherical.
In 1996, Berná et al26 classified rectus sheath hematoma into three grades based on findings of computed tomography:
- Grade I is intramuscular and unilateral
- Grade II may involve bilateral rectus muscles without extension into the prevesicular space
- Grade III extends into the peritoneum and prevesicular space
Magnetic resonance imaging
Magnetic resonance imaging is useful to differentiate chronic rectus sheath hematoma (greater than 5-day duration) from an anterior abdominal wall mass. Chronic rectus sheath hematoma will have high signal intensity on both T1- and T2-weighted images up to 10 months after the onset of the hematoma.27
Back to our patient
Since our patient’s symptoms are acute and of less than 5 days’ duration, computed tomography of the abdomen and pelvis would be the best diagnostic test, with therapeutic implications if there is ongoing extravasation.
Computed tomography of the abdomen with contrast showed a new hematoma, measuring 25 by 14 by 13.5 cm, in the right rectus sheath (Figure 2), with no other findings. The hematoma was grade I, since it was intramuscular and unilateral without extension elsewhere.
Laboratory workup showed that the patient’s hematocrit was falling, from 34% to 24%, and her INR was elevated at 2.5. She was resuscitated with fluids, blood transfusion, and fresh-frozen plasma. Anticoagulation was withheld. In spite of resuscitation, her hematocrit kept falling, though she remained hemodynamically stable.
THE WAY FORWARD
3. At this point, what would be the best approach to management in this patient?
- Serial clinical examinations and frequent monitoring of the complete blood cell count
- Urgent surgical consult for exploratory laparotomy with evaluation of the hematoma and ligation of the bleeding vessel
- Repeat computed tomographic angiography to identify a possible bleeding vessel; consideration of radiographically guided arterial embolization
- Measuring the intra-abdominal pressure using the intrabladder pressure for abdominal compartment syndrome and consideration of surgical drainage
The key clinical concern in a patient with a rectus sheath hematoma who is hemodynamically stable is whether the hematoma is expanding. This patient responded to initial resuscitation, but her falling hematocrit was evidence of ongoing bleeding leading to an expanding rectus sheath hematoma. Thus, serial clinical examinations and frequent monitoring of the complete blood cell count would not be enough, as it could miss fatal ongoing bleeding.
Radiographically guided embolization with Gelfoam, thrombin, or coils should be attempted first, as this is less invasive than exploratory laparotomy.28 It can achieve hemostasis, decrease the size of the hematoma, limit the need for blood products, and prevent rupture into the abdomen. If this is unsuccessful, the next step is ligation of the bleeding vessel.29
Surgical treatment includes evacuation of the hematoma, repair of the rectus sheath, ligation of bleeding vessels, and abdominal wall closure. Surgical evacuation or guided drainage of a rectus sheath hematoma on its own is not normally indicated and may indeed cause persistent bleeding by diminishing a potential tamponade effect. However, it may become necessary if the hematoma is very large or infected, if it causes marked respiratory impairment, or if abdominal compartment syndrome is suspected.
Abdominal compartment syndrome is very rare but is associated with a 50% mortality rate.30 It should be suspected in patients with oliguria, low cardiac output, changes in minute ventilation, and altered splanchnic blood flow. The diagnosis is confirmed with indwelling catheter manometry of the bladder to measure intra-abdominal pressure. Intra-abominal pressure above 25 mm Hg should be treated with decompressive laparotomy.30 However, the clinical suspicion of abdominal compartment syndrome was low in this patient.
The best choice at this point would be urgent computed tomographic angiography to identify a bleeding vessel, along with consideration of radiographically guided arterial embolization.
TREATMENT IS USUALLY CONSERVATIVE
Treatment of rectus sheath hematoma is conservative in most hemodynamically stable patients, with embolization or surgical intervention reserved for unstable patients or those in whom the hematoma is expanding.
Knowledge of the grading system of Berná et al26 helps to assess the patient’s risk and to anticipate potential complications. Grade I hematomas are mild and do not necessitate admission. Patients with grade II hematoma can be admitted to the floor for 24 to 48 hours for observation. Grade III usually occurs in patients receiving anticoagulant therapy and frequently requires blood products. These patients have a prolonged hospital stay and more complications, including hypovolemic shock, myonecrosis, acute coronary syndrome, arrhythmias, acute renal failure, small-bowel infarction, and abdominal compartment syndrome—all of which increases the risk of morbidity and death. Thus, patients who are on anticoagulation who develop grade III rectus sheath hematoma should be admitted to the hospital, preferably to the intensive care unit, to ensure that the hematoma is not expanding and to plan reinstitution of anticoagulation as appropriate.
In most cases, rectus sheath hematomas resolve within 1 to 3 months. Resolution of large hematomas may be hastened with the use of pulsed ultrasound.31 However, this treatment should be used only after the acute phase is over, when there is evidence of an organized thrombus and coagulation measures have returned to the target range. This helps to reduce the risk of bleeding and to prevent symptoms from worsening.31
OUR PATIENT’S COURSE
Our patient underwent urgent computed tomographic angiography, which showed a modest increase in the size of the rectus sheath hematoma. However, no definitive blush of contrast was seen to suggest active arterial bleeding. Her hematocrit stabilized, and she remained hemodynamically stable without requiring additional intervention. Most likely her bleeding was self-contained. She had normal intra-abdominal pressure on serial monitoring. She was later transferred to acute inpatient rehabilitation in view of deconditioning and is currently doing well. The hematoma persisted, decreasing only slightly in size over the next 3 weeks.
- Kougias P, Lau D, El Sayed HF, Zhou W, Huynh TT, Lin PH. Determinants of mortality and treatment outcome following surgical interventions for acute mesenteric ischemia. J Vasc Surg 2007; 46:467–474.
- Sise MJ. Acute mesenteric ischemia. Surg Clin North Am 2014; 94:165–181.
- Scharff JR, Longo WE, Vartanian SM, Jacobs DL, Bahadursingh AN, Kaminski DL. Ischemic colitis: spectrum of disease and outcome. Surgery 2003; 134:624–629.
- Lange H, Jäckel R. Usefulness of plasma lactate concentration in the diagnosis of acute abdominal disease. Eur J Surg 1994; 160:381–384.
- Gearhart SL, Delaney CP, Senagore AJ, et al. Prospective assessment of the predictive value of alpha-glutathione S-transferase for intestinal ischemia. Am Surg 2003; 69:324–329.
- Kanda T, Fujii H, Tani T, et al. Intestinal fatty acid-binding protein is a useful diagnostic marker for mesenteric infarction in humans. Gastroenterology 1996; 110:339–343.
- Menke J. Diagnostic accuracy of multidetector CT in acute mesenteric ischemia: systematic review and meta-analysis. Radiology 2010; 256:93–101.
- Acosta S, Björnsson S, Ekberg O, Resch T. CT angiography followed by endovascular intervention for acute superior mesenteric artery occlusion does not increase risk of contrast-induced renal failure. Eur J Vasc Endovasc Surg 2010; 39:726–730.
- Clark RA. Computed tomography of bowel infarction. J Comput Assist Tomogr 1987; 11:757–762.
- Acosta S, Björck M. Modern treatment of acute mesenteric ischaemia. Br J Surg 2014; 101:e100–e108.
- Smithson A, Ruiz J, Perello R, Valverde M, Ramos J, Garzo L. Diagnostic and management of spontaneous rectus sheath hematoma. Eur J Intern Med 2013; 24:579–582.
- Moreno Gallego A, Aguayo JL, Flores B, et al. Ultrasonography and computed tomography reduce unnecessary surgery in abdominal rectus sheath haematoma. Br J Surg 1997; 84:1295–1297.
- Dubinsky IL. Hematoma of the rectus abdominis muscle: case report and review of the literature. J Emerg Med 1997; 15:165–167.
- Yi M, Yao G, Bai Y. The monitoring of intra-abdominal pressure in critically ill patients. (In Chinese.) Zhonghua Wei Zhong Bing Ji Jiu Yi Xue 2014; 26:175–178.
- Hunt L, Frost SA, Hillman K, Newton PJ, Davidson PM. Management of intra-abdominal hypertension and abdominal compartment syndrome: a review. J Trauma Manag Outcomes 2014; 8:2.
- Malbrain ML, Cheatham ML, Kirkpatrick A, et al. Results from the International Conference of Experts on Intra-abdominal Hypertension and Abdominal Compartment Syndrome. I. Definitions. Intensive Care Med 2006; 32:1722–1732.
- Malbrain ML, Chiumello D, Cesana BM, et al; WAKE-Up! Investigators. A systematic review and individual patient data meta-analysis on intra-abdominal hypertension in critically ill patients: the wake-up project. World initiative on Abdominal Hypertension Epidemiology, a Unifying Project (WAKE-Up!). Minerva Anestesiol 2014; 80:293–306.
- Kirkpatrick AW, Roberts DJ, De Waele J, et al; Pediatric Guidelines Sub-Committee for the World Society of the Abdominal Compartment Syndrome. Intra-abdominal hypertension and the abdominal compartment syndrome: updated consensus definitions and clinical practice guidelines from the World Society of the Abdominal Compartment Syndrome. Intensive Care Med 2013; 39:1190–1206.
- Holodinsky JK, Roberts DJ, Ball CG, et al. Risk factors for intra-abdominal hypertension and abdominal compartment syndrome among adult intensive care unit patients: a systematic review and meta-analysis. Crit Care 2013; 17:R249.
- Sugrue M, Bauman A, Jones F, et al. Clinical examination is an inaccurate predictor of intraabdominal pressure. World J Surg 2002; 26:1428–1431.
- Cheatham ML, De Waele JJ, De Laet I, et al; World Society of the Abdominal Compartment Syndrome (WSACS) Clinical Trials Working Group. The impact of body position on intra-abdominal pressure measurement: a multicenter analysis. Crit Care Med 2009; 37:2187–2190.
- Ortiz-Diaz E, Lan CK. Intra-abdominal hypertension in medical critically ill patients: a narrative review. Shock 2014; 41:175–180.
- Cotton BA, Reddy N, Hatch QM, et al. Damage control resuscitation is associated with a reduction in resuscitation volumes and improvement in survival in 390 damage control laparotomy patients. Ann Surg 2011; 254:598–605.
- An G, West MA. Abdominal compartment syndrome: a concise clinical review. Crit Care Med 2008; 36:1304–1310.
- Tolcher MC, Nitsche JF, Arendt KW, Rose CH. Spontaneous rectus sheath hematoma pregnancy: case report and review of the literature. Obstet Gynecol Surv 2010; 65:517–522.
- Berná JD, Garcia-Medina V, Guirao J, Garcia-Medina J. Rectus sheath hematoma: diagnostic classification by CT. Abdom Imaging 1996; 21:62–64.
- Unger EC, Glazer HS, Lee JK, Ling D. MRI of extracranial hematomas: preliminary observations. AJR Am J Roentgenol 1986; 146:403–407.
- Rimola J, Perendreu J, Falcó J, Fortuño JR, Massuet A, Branera J. Percutaneous arterial embolization in the management of rectus sheath hematoma. AJR Am J Roentgenol 2007; 188:W497–W502.
- Titone C, Lipsius M, Krakauer JS. “Spontaneous” hematoma of the rectus abdominis muscle: critical review of 50 cases with emphasis on early diagnosis and treatment. Surgery 1972; 72:568–572.
- Osinbowale O, Bartholomew JR. Rectus sheath hematoma. Vasc Med 2008; 13:275–279.
- Berná-Serna JD, Sánchez-Garre J, Madrigal M, Zuazu I, Berná-Mestre JD. Ultrasound therapy in rectus sheath hematoma. Phys Ther 2005; 85:352–357.
- Kougias P, Lau D, El Sayed HF, Zhou W, Huynh TT, Lin PH. Determinants of mortality and treatment outcome following surgical interventions for acute mesenteric ischemia. J Vasc Surg 2007; 46:467–474.
- Sise MJ. Acute mesenteric ischemia. Surg Clin North Am 2014; 94:165–181.
- Scharff JR, Longo WE, Vartanian SM, Jacobs DL, Bahadursingh AN, Kaminski DL. Ischemic colitis: spectrum of disease and outcome. Surgery 2003; 134:624–629.
- Lange H, Jäckel R. Usefulness of plasma lactate concentration in the diagnosis of acute abdominal disease. Eur J Surg 1994; 160:381–384.
- Gearhart SL, Delaney CP, Senagore AJ, et al. Prospective assessment of the predictive value of alpha-glutathione S-transferase for intestinal ischemia. Am Surg 2003; 69:324–329.
- Kanda T, Fujii H, Tani T, et al. Intestinal fatty acid-binding protein is a useful diagnostic marker for mesenteric infarction in humans. Gastroenterology 1996; 110:339–343.
- Menke J. Diagnostic accuracy of multidetector CT in acute mesenteric ischemia: systematic review and meta-analysis. Radiology 2010; 256:93–101.
- Acosta S, Björnsson S, Ekberg O, Resch T. CT angiography followed by endovascular intervention for acute superior mesenteric artery occlusion does not increase risk of contrast-induced renal failure. Eur J Vasc Endovasc Surg 2010; 39:726–730.
- Clark RA. Computed tomography of bowel infarction. J Comput Assist Tomogr 1987; 11:757–762.
- Acosta S, Björck M. Modern treatment of acute mesenteric ischaemia. Br J Surg 2014; 101:e100–e108.
- Smithson A, Ruiz J, Perello R, Valverde M, Ramos J, Garzo L. Diagnostic and management of spontaneous rectus sheath hematoma. Eur J Intern Med 2013; 24:579–582.
- Moreno Gallego A, Aguayo JL, Flores B, et al. Ultrasonography and computed tomography reduce unnecessary surgery in abdominal rectus sheath haematoma. Br J Surg 1997; 84:1295–1297.
- Dubinsky IL. Hematoma of the rectus abdominis muscle: case report and review of the literature. J Emerg Med 1997; 15:165–167.
- Yi M, Yao G, Bai Y. The monitoring of intra-abdominal pressure in critically ill patients. (In Chinese.) Zhonghua Wei Zhong Bing Ji Jiu Yi Xue 2014; 26:175–178.
- Hunt L, Frost SA, Hillman K, Newton PJ, Davidson PM. Management of intra-abdominal hypertension and abdominal compartment syndrome: a review. J Trauma Manag Outcomes 2014; 8:2.
- Malbrain ML, Cheatham ML, Kirkpatrick A, et al. Results from the International Conference of Experts on Intra-abdominal Hypertension and Abdominal Compartment Syndrome. I. Definitions. Intensive Care Med 2006; 32:1722–1732.
- Malbrain ML, Chiumello D, Cesana BM, et al; WAKE-Up! Investigators. A systematic review and individual patient data meta-analysis on intra-abdominal hypertension in critically ill patients: the wake-up project. World initiative on Abdominal Hypertension Epidemiology, a Unifying Project (WAKE-Up!). Minerva Anestesiol 2014; 80:293–306.
- Kirkpatrick AW, Roberts DJ, De Waele J, et al; Pediatric Guidelines Sub-Committee for the World Society of the Abdominal Compartment Syndrome. Intra-abdominal hypertension and the abdominal compartment syndrome: updated consensus definitions and clinical practice guidelines from the World Society of the Abdominal Compartment Syndrome. Intensive Care Med 2013; 39:1190–1206.
- Holodinsky JK, Roberts DJ, Ball CG, et al. Risk factors for intra-abdominal hypertension and abdominal compartment syndrome among adult intensive care unit patients: a systematic review and meta-analysis. Crit Care 2013; 17:R249.
- Sugrue M, Bauman A, Jones F, et al. Clinical examination is an inaccurate predictor of intraabdominal pressure. World J Surg 2002; 26:1428–1431.
- Cheatham ML, De Waele JJ, De Laet I, et al; World Society of the Abdominal Compartment Syndrome (WSACS) Clinical Trials Working Group. The impact of body position on intra-abdominal pressure measurement: a multicenter analysis. Crit Care Med 2009; 37:2187–2190.
- Ortiz-Diaz E, Lan CK. Intra-abdominal hypertension in medical critically ill patients: a narrative review. Shock 2014; 41:175–180.
- Cotton BA, Reddy N, Hatch QM, et al. Damage control resuscitation is associated with a reduction in resuscitation volumes and improvement in survival in 390 damage control laparotomy patients. Ann Surg 2011; 254:598–605.
- An G, West MA. Abdominal compartment syndrome: a concise clinical review. Crit Care Med 2008; 36:1304–1310.
- Tolcher MC, Nitsche JF, Arendt KW, Rose CH. Spontaneous rectus sheath hematoma pregnancy: case report and review of the literature. Obstet Gynecol Surv 2010; 65:517–522.
- Berná JD, Garcia-Medina V, Guirao J, Garcia-Medina J. Rectus sheath hematoma: diagnostic classification by CT. Abdom Imaging 1996; 21:62–64.
- Unger EC, Glazer HS, Lee JK, Ling D. MRI of extracranial hematomas: preliminary observations. AJR Am J Roentgenol 1986; 146:403–407.
- Rimola J, Perendreu J, Falcó J, Fortuño JR, Massuet A, Branera J. Percutaneous arterial embolization in the management of rectus sheath hematoma. AJR Am J Roentgenol 2007; 188:W497–W502.
- Titone C, Lipsius M, Krakauer JS. “Spontaneous” hematoma of the rectus abdominis muscle: critical review of 50 cases with emphasis on early diagnosis and treatment. Surgery 1972; 72:568–572.
- Osinbowale O, Bartholomew JR. Rectus sheath hematoma. Vasc Med 2008; 13:275–279.
- Berná-Serna JD, Sánchez-Garre J, Madrigal M, Zuazu I, Berná-Mestre JD. Ultrasound therapy in rectus sheath hematoma. Phys Ther 2005; 85:352–357.
When the dissociation curve shifts to the left
A 48-year-old woman presented to the emergency department after 2 days of nonproductive cough, chest discomfort, worsening shortness of breath, and subjective fever. She had a history of systemic sclerosis. She was currently taking prednisone 20 mg daily and aspirin 81 mg daily.
Physical examination revealed tachypnea (28 breaths per minute), and bronchial breath sounds in the left lower chest posteriorly.
The initial laboratory workup revealed:
- Hemoglobin 106 g/L (reference range 115–155)
- Mean corpuscular volume 84 fL (80–100)
- White blood cell count 29.4 × 109/L (3.70–11.0), with 85% neutrophils
- Platelet count 180 × 109/L (150–350)
- Lactate dehydrogenase 312 U/L (100–220).
Chest radiography showed opacification of the lower lobe of the left lung.
She was admitted to the hospital and started treatment with intravenous azithromycin and ceftriaxone for presumed community-acquired pneumonia, based on the clinical presentation and findings on chest radiography. Because of her immunosuppression (due to chronic prednisone therapy) and her high lactate dehydrogenase level, Pneumocystis jirovecii pneumonia was suspected, and because she had a history of allergy to trimethoprim-sulfamethoxazole and pentamidine, she was started on dapsone.
During the next 24 hours, she developed worsening dyspnea, hypoxia, and cyanosis. She was placed on an air-entrainment mask, with a fraction of inspired oxygen of 0.5. Pulse oximetry showed an oxygen saturation of 85%, but arterial blood gas analysis indicated an oxyhemoglobin concentration of 95%.
THE ‘SATURATION GAP’
1. Which is most likely to have caused the discrepancy between the oxyhemoglobin concentration and the oxygen saturation by pulse oximetry in this patient?
- Methemoglobinemia
- Carbon monoxide poisoning
- Inappropriate placement of the pulse oximeter probe
- Pulmonary embolism
Methemoglobinemia is the most likely cause of the discrepancy between the oxyhemoglobin levels and the oxygen saturation by pulse oximetry, a phenomenon also known as the “saturation gap.” Other common causes are cyanide poisoning and carbon monoxide poisoning.
Carbon monoxide poisoning, however, does not explain our patient’s cyanosis. On the contrary, carbon monoxide poisoning can actually cause the patient’s lips and mucous membranes to appear unnaturally bright pink. Also, carbon monoxide poisoning raises the blood concentration of carboxyhemoglobin (which has a high affinity for oxygen), and this usually causes pulse oximetry to read inappropriately high, whereas in our patient it read low.
Incorrect placement of the pulse oximeter probe can result in an inaccurate measurement of oxygen saturation. Visualization of the waveform on the plethysmograph or the signal quality index can be used to assess adequate placement of the pulse oximeter probe. However, inadequate probe placement does not explain our patient’s dyspnea and cyanosis.
Pulmonary embolism can lead to hypoxia as a result of ventilation-perfusion mismatch. However, pulmonary embolism leading to low oxygen saturation on pulse oximetry will also lead to concomitantly low oxyhemoglobin levels as measured by arterial blood gas analysis, and this was not seen in our patient.
BACK TO OUR PATIENT
Because there was a discrepancy between our patient’s pulse oximetry reading and oxyhemoglobin concentration by arterial blood gas measurement, her methemoglobin level was checked and was found to be 30%, thus confirming the diagnosis of methemoglobinemia.
WHAT IS METHEMOGLOBINEMIA, AND WHAT CAUSES IT?
Oxygen is normally bound to iron in its ferrous (Fe2+) form in hemoglobin to form oxyhemoglobin. Oxidative stress in the body can cause iron to change from the ferrous to the ferric (Fe3+) state, forming methemoglobin. Methemoglobin is normally present in the blood in low levels (< 1% of the total hemoglobin), and ferric iron is reduced and recycled back to the ferrous form by NADH-cytochrome b5 reductase, an enzyme present in red blood cells. This protective mechanism maintains methemoglobin levels within safe limits. But increased production can lead to accumulation of methemoglobin, resulting in dyspnea and hypoxia and the condition referred to as methemoglobinemia.1
Increased levels of methemoglobin relative to normal hemoglobin cause tissue hypoxia by several mechanisms. Methemoglobin cannot efficiently carry oxygen; instead, it binds to water or to a hydroxide ion depending on the pH of the environment.2 Therefore, the hemoglobin molecule does not carry its usual load of oxygen, and hypoxia results from the reduced delivery of oxygen to tissues. In addition, an increased concentration of methemoglobin causes a leftward shift in the oxygen-hemoglobin dissociation curve, representing an increased affinity to bound oxygen in the remaining heme groups. The tightly bound oxygen is not adequately released at the tissue level, thus causing cellular hypoxia.
Methemoglobinemia is most often caused by exposure to an oxidizing chemical or drug that increases production of methemoglobin. In rare cases, it is caused by a congenital deficiency of NADH-cytochrome b5 reductase.3
2. Which of the following drugs can cause methemoglobinemia?
- Acetaminophen
- Dapsone
- Benzocaine
- Primaquine
All four of these drugs are common culprits for causing acquired methemoglobinemia; others include chloroquine, nitroglycerin, and sulfonamides.4–6
The increased production of methemoglobin caused by these drugs overwhelms the protective effect of reducing enzymes and can lead to an accumulation of methemoglobin. However, because of variability in cellular metabolism, not every person who takes these drugs develops dangerous levels of methemoglobin.

Dapsone and benzocaine are the most commonly encountered drugs known to cause methemoglobinemia (Table 1). Dapsone is an anti-inflammatory and antimicrobial agent most commonly used for treating lepromatous leprosy and dermatitis herpetiformis. It is also often prescribed for prophylaxis and treatment of P jirovecii pneumonia in immunosuppressed individuals.7 Benzocaine is a local anesthetic and was commonly used before procedures such as oral or dental surgery, transesophageal echocardiography, and endoscopy.8–10 Even low doses of benzocaine can lead to high levels of methemoglobinemia. However, the availability of other, safer anesthetics now limits the use of benzocaine in major US centers. In addition, the topical anesthetic Emla (lidocaine plus prilocaine) has been recently reported as a cause of methemoglobinemia in infants and children.11,12
Also, potentially fatal methemoglobinemia has been reported in patients with a deficiency of G-6-phosphate dehydrogenase (G6PD) who received rasburicase, a recombinant version of urate oxidase enzyme used to prevent and treat tumor lysis syndrome.13,14
Lastly, methemoglobinemia has been reported in patients with inflammatory bowel disease treated with mesalamine.
Although this adverse reaction is rare, clinicians should be aware of it, since these agents are commonly used in everyday medical practice.15
RECOGNIZING THE DANGER SIGNS
The clinical manifestations of methemoglobinemia are directly proportional to the percentage of methemoglobin in red blood cells. Cyanosis generally becomes apparent at concentrations around 15%, at which point the patient may still have no symptoms. Anxiety, lightheadedness, tachycardia, and dizziness manifest at levels of 20% to 30%. Fatigue, confusion, dizziness, tachypnea, and worsening tachycardia occur at levels of 30% to 50%. Levels of 50% to 70% cause coma, seizures, arrhythmias, and acidosis, and levels over 70% are considered lethal.16
While these levels provide a general guideline of symptomatology in an otherwise healthy person, it is important to remember that patients with underlying conditions such as anemia, lung disease (both of which our patient had), sepsis, thalassemia, G6PD deficiency, and sickle cell disease can manifest symptoms at lower concentrations of methemoglobin.1,17
Most patients who develop clinically significant levels of methemoglobin do so within the first few hours of starting one of the culprit drugs.
DIAGNOSIS: METHEMOGLOBINEMIA AND THE SATURATION GAP
In patients with methemoglobinemia, pulse oximetry gives lower values than arterial blood gas oxygen measurements. Regular pulse oximetry works by measuring light absorbance at two distinct wavelengths (660 and 940 nm) to calculate the ratio of oxyhemoglobin to deoxyhemoglobin. Methemoglobin absorbs light at both these wavelengths, thus lowering the pulse oximetry values.1
In contrast, oxygen saturation of arterial blood gas (oxyhemoglobin) is calculated indirectly from the concentration of dissolved oxygen in the blood and does not include oxygen bound to hemoglobin. Therefore, the measured arterial oxygen saturation is often normal in patients with methemoglobinemia since it relies only on inspired oxygen content and is independent of the methemoglobin concentration.18
Oxygen supplementation can raise the level of oxyhemoglobin, which is a measure of dissolved oxygen, but the oxygen saturation as measured by pulse oximetry remains largely unchanged—ie, the saturation gap. A difference of more than 5% between the oxygen saturation by pulse oximetry and blood gas analysis is abnormal. Patients with clinically significant methemoglobinemia usually have a saturation gap greater than 10%.
Several other unique features should raise suspicion of methemoglobinemia. It should be considered in a patient presenting with cyanosis out of proportion to the oxygen saturation and in a patient with low oxygen saturation and a normal chest radiograph. Other clues include blood that is chocolate-colored on gross examination, rather than the dark red of deoxygenated blood.
Co-oximetry measures oxygen saturation using different wavelengths of light to distinguish between fractions of oxyhemoglobin, deoxyhemoglobin, and methemoglobin, but it is not widely available.
THE NEXT STEP
3. What is the next step in the management of our patient?
- Discontinue the dapsone
- Start methylene blue
- Start hyperbaric oxygen
- Give sodium thiosulfate
- Discontinue dapsone and start methylene blue
The next step in her management should be to stop the dapsone and start an infusion of methylene blue. Hyperbaric oxygen is used in treating carbon monoxide poisoning, and sodium thiosulfate is used in treating cyanide toxicity. They would not be appropriate in this patient’s care.
MANAGEMENT OF ACQUIRED METHEMOGLOBINEMIA
The first, most critical step in managing acquired methemoglobinemia is to immediately discontinue the suspected offending agent. In most patients without a concomitant condition such as anemia or lung disease and with a methemoglobin level below 20%, discontinuing the offending agent may suffice. Patients with a level of 20% or greater and patients with cardiac and pulmonary disease, who develop symptoms at lower concentrations of methemoglobin, require infusion of methylene blue.
Methylene blue is converted to its reduced form, leukomethylene blue, by NADPH-methemoglobin reductase. As it is oxidized, leukomethylene blue reduces methemoglobin to hemoglobin. A dose of 1 mg/kg intravenously is given at first. The response is usually dramatic, with a reduction in methemoglobin levels and improvement in symptoms often within 30 to 60 minutes. If levels remain high, the dose can be repeated 1 hour later.19
A caveat: methylene blue therapy should be avoided in patients with complete G6PD deficiency. Methylene blue works through the enzyme NADPH-methemoglobin reductase, and since patients with G6PD deficiency lack this enzyme, methylene blue is ineffective. In fact, since it cannot be reduced, excessive methylene blue can oxidize hemoglobin to methemoglobin, further exacerbating the condition. In patients with partial G6PD deficiency, methylene blue is still recommended as a first-line treatment, but at a lower initial dose (0.3–0.5 mg/kg). However, in patients with significant hemolysis, an exchange transfusion is the only treatment option.
CASE CONCLUDED
Since dapsone was identified as the likely cause of methemoglobinemia in our patient, it was immediately discontinued. Because she was symptomatic, 70 mg of methylene blue was given intravenously. Over the next 60 minutes, her clinical condition improved significantly. A repeat methemoglobin measurement was 3%.
She was discharged home the next day on oral antibiotics to complete treatment for community-acquired pneumonia.
TAKE-HOME POINTS
- Consider methemoglobinemia in a patient with unexplained cyanosis.
- Pulse oximetry gives lower values than arterial blood gas oxygen measurements in patients with methemoglobinemia, and pulse oximetry readings do not improve with supplemental oxygen.
- A saturation gap greater than 5% strongly suggests methemoglobinemia.
- The diagnosis of methemoglobinemia is confirmed by measuring the methemoglobin concentration.
- Most healthy patients develop symptoms at methemoglobin levels of 20%, but patients with comorbidities can develop symptoms at lower levels.
- A number of drugs can cause methemoglobinemia, even at therapeutic dosages.
- Treatment is generally indicated in patients who have symptoms or in healthy patients who have a methemoglobin level of 20% or greater.
- Identifying and promptly discontinuing the causative agent and initiating methylene blue infusion (1 mg/kg over 5 minutes) is the preferred treatment.
- Cortazzo JA, Lichtman AD. Methemoglobinemia: a review and recommendations for management. J Cardiothorac Vasc Anesth 2014; 28:1055–1059.
- Margulies DR, Manookian CM. Methemoglobinemia as a cause of respiratory failure. J Trauma 2002; 52:796–797.
- Skold A, Cosco DL, Klein R. Methemoglobinemia: pathogenesis, diagnosis, and management. South Med J 2011; 104:757–761.
- Ash-Bernal R, Wise R, Wright SM. Acquired methemoglobinemia: a retrospective series of 138 cases at 2 teaching hospitals. Medicine (Baltimore) 2004; 83:265–273.
- Kanji HD, Mithani S, Boucher P, Dias VC, Yarema MC. Coma, metabolic acidosis, and methemoglobinemia in a patient with acetaminophen toxicity. J Popul Ther Clin Pharmacol 2013; 20:e207–e211.
- Kawasumi H, Tanaka E, Hoshi D, Kawaguchi Y, Yamanaka H. Methemoglobinemia induced by trimethoprim-sulfamethoxazole in a patient with systemic lupus erythematosus. Intern Med 2013; 52:1741–1743.
- Wieringa A, Bethlehem C, Hoogendoorn M, van der Maten J, van Roon EN. Very late recovery of dapsone-induced methemoglobinemia. Clin Toxicol (Phila) 2014; 52:80–81.
- Barclay JA, Ziemba SE, Ibrahim RB. Dapsone-induced methemoglobinemia: a primer for clinicians. Ann Pharmacother 2011; 45:1103–1115.
- Taleb M, Ashraf Z, Valavoor S, Tinkel J. Evaluation and management of acquired methemoglobinemia associated with topical benzocaine use. Am J Cardiovasc Drugs 2013; 13:325–330.
- Chowdhary S, Bukoye B, Bhansali AM, et al. Risk of topical anesthetic-induced methemoglobinemia: a 10-year retrospective case-control study. JAMA Intern Med 2013; 173:771–776.
- Larson A, Stidham T, Banerji S, Kaufman J. Seizures and methemoglobinemia in an infant after excessive EMLA application. Pediatr Emerg Care 2013; 29:377–379.
- Schmitt C, Matulic M, Kervégant M, et al. Methaemoglobinaemia in a child treated with Emla cream: circumstances and consequences of overdose [in French]. Ann Dermatol Venereol 2012; 139:824–827.
- Bucklin MH, Groth CM. Mortality following rasburicase-induced methemoglobinemia. Ann Pharmacother 2013; 47:1353–1358.
- Cheah CY, Lew TE, Seymour JF, Burbury K. Rasburicase causing severe oxidative hemolysis and methemoglobinemia in a patient with previously unrecognized glucose-6-phosphate dehydrogenase deficiency. Acta Haematol 2013; 130:254–259.
- Druez A, Rahier JF, Hébuterne X. Methaemoglobinaemia and renal failure following mesalazine for treatment of inflammatory bowel disease. J Crohns Colitis 2014; 8:900–901.
- Wright RO, Lewander WJ, Woolf AD. Methemoglobinemia: etiology, pharmacology, and clinical management. Ann Emerg Med 1999; 34:646–656.
- Groeper K, Katcher K, Tobias JD. Anesthetic management of a patient with methemoglobinemia. South Med J 2003; 96:504–509.
- Haymond S, Cariappa R, Eby CS, Scott MG. Laboratory assessment of oxygenation in methemoglobinemia. Clin Chem 2005; 51:434–444.
- Jang DH, Nelson LS, Hoffman RS. Methylene blue for distributive shock: a potential new use of an old antidote. J Med Toxicol 2013; 9:242–249.
A 48-year-old woman presented to the emergency department after 2 days of nonproductive cough, chest discomfort, worsening shortness of breath, and subjective fever. She had a history of systemic sclerosis. She was currently taking prednisone 20 mg daily and aspirin 81 mg daily.
Physical examination revealed tachypnea (28 breaths per minute), and bronchial breath sounds in the left lower chest posteriorly.
The initial laboratory workup revealed:
- Hemoglobin 106 g/L (reference range 115–155)
- Mean corpuscular volume 84 fL (80–100)
- White blood cell count 29.4 × 109/L (3.70–11.0), with 85% neutrophils
- Platelet count 180 × 109/L (150–350)
- Lactate dehydrogenase 312 U/L (100–220).
Chest radiography showed opacification of the lower lobe of the left lung.
She was admitted to the hospital and started treatment with intravenous azithromycin and ceftriaxone for presumed community-acquired pneumonia, based on the clinical presentation and findings on chest radiography. Because of her immunosuppression (due to chronic prednisone therapy) and her high lactate dehydrogenase level, Pneumocystis jirovecii pneumonia was suspected, and because she had a history of allergy to trimethoprim-sulfamethoxazole and pentamidine, she was started on dapsone.
During the next 24 hours, she developed worsening dyspnea, hypoxia, and cyanosis. She was placed on an air-entrainment mask, with a fraction of inspired oxygen of 0.5. Pulse oximetry showed an oxygen saturation of 85%, but arterial blood gas analysis indicated an oxyhemoglobin concentration of 95%.
THE ‘SATURATION GAP’
1. Which is most likely to have caused the discrepancy between the oxyhemoglobin concentration and the oxygen saturation by pulse oximetry in this patient?
- Methemoglobinemia
- Carbon monoxide poisoning
- Inappropriate placement of the pulse oximeter probe
- Pulmonary embolism
Methemoglobinemia is the most likely cause of the discrepancy between the oxyhemoglobin levels and the oxygen saturation by pulse oximetry, a phenomenon also known as the “saturation gap.” Other common causes are cyanide poisoning and carbon monoxide poisoning.
Carbon monoxide poisoning, however, does not explain our patient’s cyanosis. On the contrary, carbon monoxide poisoning can actually cause the patient’s lips and mucous membranes to appear unnaturally bright pink. Also, carbon monoxide poisoning raises the blood concentration of carboxyhemoglobin (which has a high affinity for oxygen), and this usually causes pulse oximetry to read inappropriately high, whereas in our patient it read low.
Incorrect placement of the pulse oximeter probe can result in an inaccurate measurement of oxygen saturation. Visualization of the waveform on the plethysmograph or the signal quality index can be used to assess adequate placement of the pulse oximeter probe. However, inadequate probe placement does not explain our patient’s dyspnea and cyanosis.
Pulmonary embolism can lead to hypoxia as a result of ventilation-perfusion mismatch. However, pulmonary embolism leading to low oxygen saturation on pulse oximetry will also lead to concomitantly low oxyhemoglobin levels as measured by arterial blood gas analysis, and this was not seen in our patient.
BACK TO OUR PATIENT
Because there was a discrepancy between our patient’s pulse oximetry reading and oxyhemoglobin concentration by arterial blood gas measurement, her methemoglobin level was checked and was found to be 30%, thus confirming the diagnosis of methemoglobinemia.
WHAT IS METHEMOGLOBINEMIA, AND WHAT CAUSES IT?
Oxygen is normally bound to iron in its ferrous (Fe2+) form in hemoglobin to form oxyhemoglobin. Oxidative stress in the body can cause iron to change from the ferrous to the ferric (Fe3+) state, forming methemoglobin. Methemoglobin is normally present in the blood in low levels (< 1% of the total hemoglobin), and ferric iron is reduced and recycled back to the ferrous form by NADH-cytochrome b5 reductase, an enzyme present in red blood cells. This protective mechanism maintains methemoglobin levels within safe limits. But increased production can lead to accumulation of methemoglobin, resulting in dyspnea and hypoxia and the condition referred to as methemoglobinemia.1
Increased levels of methemoglobin relative to normal hemoglobin cause tissue hypoxia by several mechanisms. Methemoglobin cannot efficiently carry oxygen; instead, it binds to water or to a hydroxide ion depending on the pH of the environment.2 Therefore, the hemoglobin molecule does not carry its usual load of oxygen, and hypoxia results from the reduced delivery of oxygen to tissues. In addition, an increased concentration of methemoglobin causes a leftward shift in the oxygen-hemoglobin dissociation curve, representing an increased affinity to bound oxygen in the remaining heme groups. The tightly bound oxygen is not adequately released at the tissue level, thus causing cellular hypoxia.
Methemoglobinemia is most often caused by exposure to an oxidizing chemical or drug that increases production of methemoglobin. In rare cases, it is caused by a congenital deficiency of NADH-cytochrome b5 reductase.3
2. Which of the following drugs can cause methemoglobinemia?
- Acetaminophen
- Dapsone
- Benzocaine
- Primaquine
All four of these drugs are common culprits for causing acquired methemoglobinemia; others include chloroquine, nitroglycerin, and sulfonamides.4–6
The increased production of methemoglobin caused by these drugs overwhelms the protective effect of reducing enzymes and can lead to an accumulation of methemoglobin. However, because of variability in cellular metabolism, not every person who takes these drugs develops dangerous levels of methemoglobin.
Dapsone and benzocaine are the most commonly encountered drugs known to cause methemoglobinemia (Table 1). Dapsone is an anti-inflammatory and antimicrobial agent most commonly used for treating lepromatous leprosy and dermatitis herpetiformis. It is also often prescribed for prophylaxis and treatment of P jirovecii pneumonia in immunosuppressed individuals.7 Benzocaine is a local anesthetic and was commonly used before procedures such as oral or dental surgery, transesophageal echocardiography, and endoscopy.8–10 Even low doses of benzocaine can lead to high levels of methemoglobinemia. However, the availability of other, safer anesthetics now limits the use of benzocaine in major US centers. In addition, the topical anesthetic Emla (lidocaine plus prilocaine) has been recently reported as a cause of methemoglobinemia in infants and children.11,12
Also, potentially fatal methemoglobinemia has been reported in patients with a deficiency of G-6-phosphate dehydrogenase (G6PD) who received rasburicase, a recombinant version of urate oxidase enzyme used to prevent and treat tumor lysis syndrome.13,14
Lastly, methemoglobinemia has been reported in patients with inflammatory bowel disease treated with mesalamine.
Although this adverse reaction is rare, clinicians should be aware of it, since these agents are commonly used in everyday medical practice.15
RECOGNIZING THE DANGER SIGNS
The clinical manifestations of methemoglobinemia are directly proportional to the percentage of methemoglobin in red blood cells. Cyanosis generally becomes apparent at concentrations around 15%, at which point the patient may still have no symptoms. Anxiety, lightheadedness, tachycardia, and dizziness manifest at levels of 20% to 30%. Fatigue, confusion, dizziness, tachypnea, and worsening tachycardia occur at levels of 30% to 50%. Levels of 50% to 70% cause coma, seizures, arrhythmias, and acidosis, and levels over 70% are considered lethal.16
While these levels provide a general guideline of symptomatology in an otherwise healthy person, it is important to remember that patients with underlying conditions such as anemia, lung disease (both of which our patient had), sepsis, thalassemia, G6PD deficiency, and sickle cell disease can manifest symptoms at lower concentrations of methemoglobin.1,17
Most patients who develop clinically significant levels of methemoglobin do so within the first few hours of starting one of the culprit drugs.
DIAGNOSIS: METHEMOGLOBINEMIA AND THE SATURATION GAP
In patients with methemoglobinemia, pulse oximetry gives lower values than arterial blood gas oxygen measurements. Regular pulse oximetry works by measuring light absorbance at two distinct wavelengths (660 and 940 nm) to calculate the ratio of oxyhemoglobin to deoxyhemoglobin. Methemoglobin absorbs light at both these wavelengths, thus lowering the pulse oximetry values.1
In contrast, oxygen saturation of arterial blood gas (oxyhemoglobin) is calculated indirectly from the concentration of dissolved oxygen in the blood and does not include oxygen bound to hemoglobin. Therefore, the measured arterial oxygen saturation is often normal in patients with methemoglobinemia since it relies only on inspired oxygen content and is independent of the methemoglobin concentration.18
Oxygen supplementation can raise the level of oxyhemoglobin, which is a measure of dissolved oxygen, but the oxygen saturation as measured by pulse oximetry remains largely unchanged—ie, the saturation gap. A difference of more than 5% between the oxygen saturation by pulse oximetry and blood gas analysis is abnormal. Patients with clinically significant methemoglobinemia usually have a saturation gap greater than 10%.
Several other unique features should raise suspicion of methemoglobinemia. It should be considered in a patient presenting with cyanosis out of proportion to the oxygen saturation and in a patient with low oxygen saturation and a normal chest radiograph. Other clues include blood that is chocolate-colored on gross examination, rather than the dark red of deoxygenated blood.
Co-oximetry measures oxygen saturation using different wavelengths of light to distinguish between fractions of oxyhemoglobin, deoxyhemoglobin, and methemoglobin, but it is not widely available.
THE NEXT STEP
3. What is the next step in the management of our patient?
- Discontinue the dapsone
- Start methylene blue
- Start hyperbaric oxygen
- Give sodium thiosulfate
- Discontinue dapsone and start methylene blue
The next step in her management should be to stop the dapsone and start an infusion of methylene blue. Hyperbaric oxygen is used in treating carbon monoxide poisoning, and sodium thiosulfate is used in treating cyanide toxicity. They would not be appropriate in this patient’s care.
MANAGEMENT OF ACQUIRED METHEMOGLOBINEMIA
The first, most critical step in managing acquired methemoglobinemia is to immediately discontinue the suspected offending agent. In most patients without a concomitant condition such as anemia or lung disease and with a methemoglobin level below 20%, discontinuing the offending agent may suffice. Patients with a level of 20% or greater and patients with cardiac and pulmonary disease, who develop symptoms at lower concentrations of methemoglobin, require infusion of methylene blue.
Methylene blue is converted to its reduced form, leukomethylene blue, by NADPH-methemoglobin reductase. As it is oxidized, leukomethylene blue reduces methemoglobin to hemoglobin. A dose of 1 mg/kg intravenously is given at first. The response is usually dramatic, with a reduction in methemoglobin levels and improvement in symptoms often within 30 to 60 minutes. If levels remain high, the dose can be repeated 1 hour later.19
A caveat: methylene blue therapy should be avoided in patients with complete G6PD deficiency. Methylene blue works through the enzyme NADPH-methemoglobin reductase, and since patients with G6PD deficiency lack this enzyme, methylene blue is ineffective. In fact, since it cannot be reduced, excessive methylene blue can oxidize hemoglobin to methemoglobin, further exacerbating the condition. In patients with partial G6PD deficiency, methylene blue is still recommended as a first-line treatment, but at a lower initial dose (0.3–0.5 mg/kg). However, in patients with significant hemolysis, an exchange transfusion is the only treatment option.
CASE CONCLUDED
Since dapsone was identified as the likely cause of methemoglobinemia in our patient, it was immediately discontinued. Because she was symptomatic, 70 mg of methylene blue was given intravenously. Over the next 60 minutes, her clinical condition improved significantly. A repeat methemoglobin measurement was 3%.
She was discharged home the next day on oral antibiotics to complete treatment for community-acquired pneumonia.
TAKE-HOME POINTS
- Consider methemoglobinemia in a patient with unexplained cyanosis.
- Pulse oximetry gives lower values than arterial blood gas oxygen measurements in patients with methemoglobinemia, and pulse oximetry readings do not improve with supplemental oxygen.
- A saturation gap greater than 5% strongly suggests methemoglobinemia.
- The diagnosis of methemoglobinemia is confirmed by measuring the methemoglobin concentration.
- Most healthy patients develop symptoms at methemoglobin levels of 20%, but patients with comorbidities can develop symptoms at lower levels.
- A number of drugs can cause methemoglobinemia, even at therapeutic dosages.
- Treatment is generally indicated in patients who have symptoms or in healthy patients who have a methemoglobin level of 20% or greater.
- Identifying and promptly discontinuing the causative agent and initiating methylene blue infusion (1 mg/kg over 5 minutes) is the preferred treatment.
A 48-year-old woman presented to the emergency department after 2 days of nonproductive cough, chest discomfort, worsening shortness of breath, and subjective fever. She had a history of systemic sclerosis. She was currently taking prednisone 20 mg daily and aspirin 81 mg daily.
Physical examination revealed tachypnea (28 breaths per minute), and bronchial breath sounds in the left lower chest posteriorly.
The initial laboratory workup revealed:
- Hemoglobin 106 g/L (reference range 115–155)
- Mean corpuscular volume 84 fL (80–100)
- White blood cell count 29.4 × 109/L (3.70–11.0), with 85% neutrophils
- Platelet count 180 × 109/L (150–350)
- Lactate dehydrogenase 312 U/L (100–220).
Chest radiography showed opacification of the lower lobe of the left lung.
She was admitted to the hospital and started treatment with intravenous azithromycin and ceftriaxone for presumed community-acquired pneumonia, based on the clinical presentation and findings on chest radiography. Because of her immunosuppression (due to chronic prednisone therapy) and her high lactate dehydrogenase level, Pneumocystis jirovecii pneumonia was suspected, and because she had a history of allergy to trimethoprim-sulfamethoxazole and pentamidine, she was started on dapsone.
During the next 24 hours, she developed worsening dyspnea, hypoxia, and cyanosis. She was placed on an air-entrainment mask, with a fraction of inspired oxygen of 0.5. Pulse oximetry showed an oxygen saturation of 85%, but arterial blood gas analysis indicated an oxyhemoglobin concentration of 95%.
THE ‘SATURATION GAP’
1. Which is most likely to have caused the discrepancy between the oxyhemoglobin concentration and the oxygen saturation by pulse oximetry in this patient?
- Methemoglobinemia
- Carbon monoxide poisoning
- Inappropriate placement of the pulse oximeter probe
- Pulmonary embolism
Methemoglobinemia is the most likely cause of the discrepancy between the oxyhemoglobin levels and the oxygen saturation by pulse oximetry, a phenomenon also known as the “saturation gap.” Other common causes are cyanide poisoning and carbon monoxide poisoning.
Carbon monoxide poisoning, however, does not explain our patient’s cyanosis. On the contrary, carbon monoxide poisoning can actually cause the patient’s lips and mucous membranes to appear unnaturally bright pink. Also, carbon monoxide poisoning raises the blood concentration of carboxyhemoglobin (which has a high affinity for oxygen), and this usually causes pulse oximetry to read inappropriately high, whereas in our patient it read low.
Incorrect placement of the pulse oximeter probe can result in an inaccurate measurement of oxygen saturation. Visualization of the waveform on the plethysmograph or the signal quality index can be used to assess adequate placement of the pulse oximeter probe. However, inadequate probe placement does not explain our patient’s dyspnea and cyanosis.
Pulmonary embolism can lead to hypoxia as a result of ventilation-perfusion mismatch. However, pulmonary embolism leading to low oxygen saturation on pulse oximetry will also lead to concomitantly low oxyhemoglobin levels as measured by arterial blood gas analysis, and this was not seen in our patient.
BACK TO OUR PATIENT
Because there was a discrepancy between our patient’s pulse oximetry reading and oxyhemoglobin concentration by arterial blood gas measurement, her methemoglobin level was checked and was found to be 30%, thus confirming the diagnosis of methemoglobinemia.
WHAT IS METHEMOGLOBINEMIA, AND WHAT CAUSES IT?
Oxygen is normally bound to iron in its ferrous (Fe2+) form in hemoglobin to form oxyhemoglobin. Oxidative stress in the body can cause iron to change from the ferrous to the ferric (Fe3+) state, forming methemoglobin. Methemoglobin is normally present in the blood in low levels (< 1% of the total hemoglobin), and ferric iron is reduced and recycled back to the ferrous form by NADH-cytochrome b5 reductase, an enzyme present in red blood cells. This protective mechanism maintains methemoglobin levels within safe limits. But increased production can lead to accumulation of methemoglobin, resulting in dyspnea and hypoxia and the condition referred to as methemoglobinemia.1
Increased levels of methemoglobin relative to normal hemoglobin cause tissue hypoxia by several mechanisms. Methemoglobin cannot efficiently carry oxygen; instead, it binds to water or to a hydroxide ion depending on the pH of the environment.2 Therefore, the hemoglobin molecule does not carry its usual load of oxygen, and hypoxia results from the reduced delivery of oxygen to tissues. In addition, an increased concentration of methemoglobin causes a leftward shift in the oxygen-hemoglobin dissociation curve, representing an increased affinity to bound oxygen in the remaining heme groups. The tightly bound oxygen is not adequately released at the tissue level, thus causing cellular hypoxia.
Methemoglobinemia is most often caused by exposure to an oxidizing chemical or drug that increases production of methemoglobin. In rare cases, it is caused by a congenital deficiency of NADH-cytochrome b5 reductase.3
2. Which of the following drugs can cause methemoglobinemia?
- Acetaminophen
- Dapsone
- Benzocaine
- Primaquine
All four of these drugs are common culprits for causing acquired methemoglobinemia; others include chloroquine, nitroglycerin, and sulfonamides.4–6
The increased production of methemoglobin caused by these drugs overwhelms the protective effect of reducing enzymes and can lead to an accumulation of methemoglobin. However, because of variability in cellular metabolism, not every person who takes these drugs develops dangerous levels of methemoglobin.
Dapsone and benzocaine are the most commonly encountered drugs known to cause methemoglobinemia (Table 1). Dapsone is an anti-inflammatory and antimicrobial agent most commonly used for treating lepromatous leprosy and dermatitis herpetiformis. It is also often prescribed for prophylaxis and treatment of P jirovecii pneumonia in immunosuppressed individuals.7 Benzocaine is a local anesthetic and was commonly used before procedures such as oral or dental surgery, transesophageal echocardiography, and endoscopy.8–10 Even low doses of benzocaine can lead to high levels of methemoglobinemia. However, the availability of other, safer anesthetics now limits the use of benzocaine in major US centers. In addition, the topical anesthetic Emla (lidocaine plus prilocaine) has been recently reported as a cause of methemoglobinemia in infants and children.11,12
Also, potentially fatal methemoglobinemia has been reported in patients with a deficiency of G-6-phosphate dehydrogenase (G6PD) who received rasburicase, a recombinant version of urate oxidase enzyme used to prevent and treat tumor lysis syndrome.13,14
Lastly, methemoglobinemia has been reported in patients with inflammatory bowel disease treated with mesalamine.
Although this adverse reaction is rare, clinicians should be aware of it, since these agents are commonly used in everyday medical practice.15
RECOGNIZING THE DANGER SIGNS
The clinical manifestations of methemoglobinemia are directly proportional to the percentage of methemoglobin in red blood cells. Cyanosis generally becomes apparent at concentrations around 15%, at which point the patient may still have no symptoms. Anxiety, lightheadedness, tachycardia, and dizziness manifest at levels of 20% to 30%. Fatigue, confusion, dizziness, tachypnea, and worsening tachycardia occur at levels of 30% to 50%. Levels of 50% to 70% cause coma, seizures, arrhythmias, and acidosis, and levels over 70% are considered lethal.16
While these levels provide a general guideline of symptomatology in an otherwise healthy person, it is important to remember that patients with underlying conditions such as anemia, lung disease (both of which our patient had), sepsis, thalassemia, G6PD deficiency, and sickle cell disease can manifest symptoms at lower concentrations of methemoglobin.1,17
Most patients who develop clinically significant levels of methemoglobin do so within the first few hours of starting one of the culprit drugs.
DIAGNOSIS: METHEMOGLOBINEMIA AND THE SATURATION GAP
In patients with methemoglobinemia, pulse oximetry gives lower values than arterial blood gas oxygen measurements. Regular pulse oximetry works by measuring light absorbance at two distinct wavelengths (660 and 940 nm) to calculate the ratio of oxyhemoglobin to deoxyhemoglobin. Methemoglobin absorbs light at both these wavelengths, thus lowering the pulse oximetry values.1
In contrast, oxygen saturation of arterial blood gas (oxyhemoglobin) is calculated indirectly from the concentration of dissolved oxygen in the blood and does not include oxygen bound to hemoglobin. Therefore, the measured arterial oxygen saturation is often normal in patients with methemoglobinemia since it relies only on inspired oxygen content and is independent of the methemoglobin concentration.18
Oxygen supplementation can raise the level of oxyhemoglobin, which is a measure of dissolved oxygen, but the oxygen saturation as measured by pulse oximetry remains largely unchanged—ie, the saturation gap. A difference of more than 5% between the oxygen saturation by pulse oximetry and blood gas analysis is abnormal. Patients with clinically significant methemoglobinemia usually have a saturation gap greater than 10%.
Several other unique features should raise suspicion of methemoglobinemia. It should be considered in a patient presenting with cyanosis out of proportion to the oxygen saturation and in a patient with low oxygen saturation and a normal chest radiograph. Other clues include blood that is chocolate-colored on gross examination, rather than the dark red of deoxygenated blood.
Co-oximetry measures oxygen saturation using different wavelengths of light to distinguish between fractions of oxyhemoglobin, deoxyhemoglobin, and methemoglobin, but it is not widely available.
THE NEXT STEP
3. What is the next step in the management of our patient?
- Discontinue the dapsone
- Start methylene blue
- Start hyperbaric oxygen
- Give sodium thiosulfate
- Discontinue dapsone and start methylene blue
The next step in her management should be to stop the dapsone and start an infusion of methylene blue. Hyperbaric oxygen is used in treating carbon monoxide poisoning, and sodium thiosulfate is used in treating cyanide toxicity. They would not be appropriate in this patient’s care.
MANAGEMENT OF ACQUIRED METHEMOGLOBINEMIA
The first, most critical step in managing acquired methemoglobinemia is to immediately discontinue the suspected offending agent. In most patients without a concomitant condition such as anemia or lung disease and with a methemoglobin level below 20%, discontinuing the offending agent may suffice. Patients with a level of 20% or greater and patients with cardiac and pulmonary disease, who develop symptoms at lower concentrations of methemoglobin, require infusion of methylene blue.
Methylene blue is converted to its reduced form, leukomethylene blue, by NADPH-methemoglobin reductase. As it is oxidized, leukomethylene blue reduces methemoglobin to hemoglobin. A dose of 1 mg/kg intravenously is given at first. The response is usually dramatic, with a reduction in methemoglobin levels and improvement in symptoms often within 30 to 60 minutes. If levels remain high, the dose can be repeated 1 hour later.19
A caveat: methylene blue therapy should be avoided in patients with complete G6PD deficiency. Methylene blue works through the enzyme NADPH-methemoglobin reductase, and since patients with G6PD deficiency lack this enzyme, methylene blue is ineffective. In fact, since it cannot be reduced, excessive methylene blue can oxidize hemoglobin to methemoglobin, further exacerbating the condition. In patients with partial G6PD deficiency, methylene blue is still recommended as a first-line treatment, but at a lower initial dose (0.3–0.5 mg/kg). However, in patients with significant hemolysis, an exchange transfusion is the only treatment option.
CASE CONCLUDED
Since dapsone was identified as the likely cause of methemoglobinemia in our patient, it was immediately discontinued. Because she was symptomatic, 70 mg of methylene blue was given intravenously. Over the next 60 minutes, her clinical condition improved significantly. A repeat methemoglobin measurement was 3%.
She was discharged home the next day on oral antibiotics to complete treatment for community-acquired pneumonia.
TAKE-HOME POINTS
- Consider methemoglobinemia in a patient with unexplained cyanosis.
- Pulse oximetry gives lower values than arterial blood gas oxygen measurements in patients with methemoglobinemia, and pulse oximetry readings do not improve with supplemental oxygen.
- A saturation gap greater than 5% strongly suggests methemoglobinemia.
- The diagnosis of methemoglobinemia is confirmed by measuring the methemoglobin concentration.
- Most healthy patients develop symptoms at methemoglobin levels of 20%, but patients with comorbidities can develop symptoms at lower levels.
- A number of drugs can cause methemoglobinemia, even at therapeutic dosages.
- Treatment is generally indicated in patients who have symptoms or in healthy patients who have a methemoglobin level of 20% or greater.
- Identifying and promptly discontinuing the causative agent and initiating methylene blue infusion (1 mg/kg over 5 minutes) is the preferred treatment.
- Cortazzo JA, Lichtman AD. Methemoglobinemia: a review and recommendations for management. J Cardiothorac Vasc Anesth 2014; 28:1055–1059.
- Margulies DR, Manookian CM. Methemoglobinemia as a cause of respiratory failure. J Trauma 2002; 52:796–797.
- Skold A, Cosco DL, Klein R. Methemoglobinemia: pathogenesis, diagnosis, and management. South Med J 2011; 104:757–761.
- Ash-Bernal R, Wise R, Wright SM. Acquired methemoglobinemia: a retrospective series of 138 cases at 2 teaching hospitals. Medicine (Baltimore) 2004; 83:265–273.
- Kanji HD, Mithani S, Boucher P, Dias VC, Yarema MC. Coma, metabolic acidosis, and methemoglobinemia in a patient with acetaminophen toxicity. J Popul Ther Clin Pharmacol 2013; 20:e207–e211.
- Kawasumi H, Tanaka E, Hoshi D, Kawaguchi Y, Yamanaka H. Methemoglobinemia induced by trimethoprim-sulfamethoxazole in a patient with systemic lupus erythematosus. Intern Med 2013; 52:1741–1743.
- Wieringa A, Bethlehem C, Hoogendoorn M, van der Maten J, van Roon EN. Very late recovery of dapsone-induced methemoglobinemia. Clin Toxicol (Phila) 2014; 52:80–81.
- Barclay JA, Ziemba SE, Ibrahim RB. Dapsone-induced methemoglobinemia: a primer for clinicians. Ann Pharmacother 2011; 45:1103–1115.
- Taleb M, Ashraf Z, Valavoor S, Tinkel J. Evaluation and management of acquired methemoglobinemia associated with topical benzocaine use. Am J Cardiovasc Drugs 2013; 13:325–330.
- Chowdhary S, Bukoye B, Bhansali AM, et al. Risk of topical anesthetic-induced methemoglobinemia: a 10-year retrospective case-control study. JAMA Intern Med 2013; 173:771–776.
- Larson A, Stidham T, Banerji S, Kaufman J. Seizures and methemoglobinemia in an infant after excessive EMLA application. Pediatr Emerg Care 2013; 29:377–379.
- Schmitt C, Matulic M, Kervégant M, et al. Methaemoglobinaemia in a child treated with Emla cream: circumstances and consequences of overdose [in French]. Ann Dermatol Venereol 2012; 139:824–827.
- Bucklin MH, Groth CM. Mortality following rasburicase-induced methemoglobinemia. Ann Pharmacother 2013; 47:1353–1358.
- Cheah CY, Lew TE, Seymour JF, Burbury K. Rasburicase causing severe oxidative hemolysis and methemoglobinemia in a patient with previously unrecognized glucose-6-phosphate dehydrogenase deficiency. Acta Haematol 2013; 130:254–259.
- Druez A, Rahier JF, Hébuterne X. Methaemoglobinaemia and renal failure following mesalazine for treatment of inflammatory bowel disease. J Crohns Colitis 2014; 8:900–901.
- Wright RO, Lewander WJ, Woolf AD. Methemoglobinemia: etiology, pharmacology, and clinical management. Ann Emerg Med 1999; 34:646–656.
- Groeper K, Katcher K, Tobias JD. Anesthetic management of a patient with methemoglobinemia. South Med J 2003; 96:504–509.
- Haymond S, Cariappa R, Eby CS, Scott MG. Laboratory assessment of oxygenation in methemoglobinemia. Clin Chem 2005; 51:434–444.
- Jang DH, Nelson LS, Hoffman RS. Methylene blue for distributive shock: a potential new use of an old antidote. J Med Toxicol 2013; 9:242–249.
- Cortazzo JA, Lichtman AD. Methemoglobinemia: a review and recommendations for management. J Cardiothorac Vasc Anesth 2014; 28:1055–1059.
- Margulies DR, Manookian CM. Methemoglobinemia as a cause of respiratory failure. J Trauma 2002; 52:796–797.
- Skold A, Cosco DL, Klein R. Methemoglobinemia: pathogenesis, diagnosis, and management. South Med J 2011; 104:757–761.
- Ash-Bernal R, Wise R, Wright SM. Acquired methemoglobinemia: a retrospective series of 138 cases at 2 teaching hospitals. Medicine (Baltimore) 2004; 83:265–273.
- Kanji HD, Mithani S, Boucher P, Dias VC, Yarema MC. Coma, metabolic acidosis, and methemoglobinemia in a patient with acetaminophen toxicity. J Popul Ther Clin Pharmacol 2013; 20:e207–e211.
- Kawasumi H, Tanaka E, Hoshi D, Kawaguchi Y, Yamanaka H. Methemoglobinemia induced by trimethoprim-sulfamethoxazole in a patient with systemic lupus erythematosus. Intern Med 2013; 52:1741–1743.
- Wieringa A, Bethlehem C, Hoogendoorn M, van der Maten J, van Roon EN. Very late recovery of dapsone-induced methemoglobinemia. Clin Toxicol (Phila) 2014; 52:80–81.
- Barclay JA, Ziemba SE, Ibrahim RB. Dapsone-induced methemoglobinemia: a primer for clinicians. Ann Pharmacother 2011; 45:1103–1115.
- Taleb M, Ashraf Z, Valavoor S, Tinkel J. Evaluation and management of acquired methemoglobinemia associated with topical benzocaine use. Am J Cardiovasc Drugs 2013; 13:325–330.
- Chowdhary S, Bukoye B, Bhansali AM, et al. Risk of topical anesthetic-induced methemoglobinemia: a 10-year retrospective case-control study. JAMA Intern Med 2013; 173:771–776.
- Larson A, Stidham T, Banerji S, Kaufman J. Seizures and methemoglobinemia in an infant after excessive EMLA application. Pediatr Emerg Care 2013; 29:377–379.
- Schmitt C, Matulic M, Kervégant M, et al. Methaemoglobinaemia in a child treated with Emla cream: circumstances and consequences of overdose [in French]. Ann Dermatol Venereol 2012; 139:824–827.
- Bucklin MH, Groth CM. Mortality following rasburicase-induced methemoglobinemia. Ann Pharmacother 2013; 47:1353–1358.
- Cheah CY, Lew TE, Seymour JF, Burbury K. Rasburicase causing severe oxidative hemolysis and methemoglobinemia in a patient with previously unrecognized glucose-6-phosphate dehydrogenase deficiency. Acta Haematol 2013; 130:254–259.
- Druez A, Rahier JF, Hébuterne X. Methaemoglobinaemia and renal failure following mesalazine for treatment of inflammatory bowel disease. J Crohns Colitis 2014; 8:900–901.
- Wright RO, Lewander WJ, Woolf AD. Methemoglobinemia: etiology, pharmacology, and clinical management. Ann Emerg Med 1999; 34:646–656.
- Groeper K, Katcher K, Tobias JD. Anesthetic management of a patient with methemoglobinemia. South Med J 2003; 96:504–509.
- Haymond S, Cariappa R, Eby CS, Scott MG. Laboratory assessment of oxygenation in methemoglobinemia. Clin Chem 2005; 51:434–444.
- Jang DH, Nelson LS, Hoffman RS. Methylene blue for distributive shock: a potential new use of an old antidote. J Med Toxicol 2013; 9:242–249.
Should patients stop taking aspirin for primary prevention?
In view of current evidence, we do not recommend routinely using aspirin for primary prevention of cardiovascular disease, even in patients with diabetes mellitus. The decision must be individualized on the basis of the patient’s risks of cardiovascular disease and bleeding, especially the risk of serious bleeding events such as gastrointestinal and intracranial hemorrhage.
For example, patients with a family history of myocardial infarction at an early age and patients who smoke or have multiple cardiovascular risk factors may be most likely to benefit, whereas those with risk factors for gastrointestinal bleeding such as dyspepsia or ulcer would not be good candidates. Of note, current recommendations are mixed and confusing and will need to be reevaluated as new trial data become available.
TRIALS THAT SET THE STAGE FOR CURRENT PRACTICE
Routine use of aspirin for primary prevention of cardiovascular disease remains controversial.1,2 Aspirin’s safety and efficacy for this indication was studied in six major trials (Table 1).3–8 In the late 1980s, the first two primary prevention trials of aspirin enrolled healthy male physicians who had minimal cardiovascular risk factors3,4:
The British Doctors’ Trial3 observed no significant differences between aspirin (300–500 mg/day) and no aspirin in the rates of the primary end point of cardiovascular death or in the individual secondary end points of nonfatal myocardial infarction, nonfatal stroke, or bleeding.3
The Physicians’ Health Study4 found no differences in the rates of cardiovascular mortality or ischemic stroke between aspirin (325 mg every other day) and placebo. The rate of nonfatal myocardial infarction was significantly lower with aspirin than with placebo, but with a higher risk of bleeding. Relative risks and 95% confidence intervals with aspirin vs placebo:
- Nonfatal myocardial infarction
0.59 (0.47–0.74), P < .00001 - Bleeding
1.32 (1.25–1.40), P < .00001 - Blood transfusions
1.71 (1.09–2.69), P = .02 - Hemorrhagic stroke
2.14 (0.96–4.77), P = .06.
A subgroup analysis revealed that the benefit of aspirin for myocardial infarction in the Physicians’ Health Study was predominantly in those age 50 and older.4 This finding established the common clinical practice of routinely using aspirin for primary prevention in men age 50 and older.1
Later, aspirin for primary prevention was studied in four trials,5–8 three of which enrolled patients at higher cardiovascular risk5–7:
The Thrombosis Prevention Trial5 was conducted in men in the highest quintile of cardiovascular risk. The aspirin dosage was 75 mg/day.
The Hypertension Optimal Treatment6 trial included men and women ages 50 to 80 with hypertension. Aspirin dosage: 75 mg/day.
The Primary Prevention Project7 involved men and women age 50 and older with at least one risk factor for cardiovascular disease.1,5–7 The aspirin dosage was 100 mg/day.
In these trials (Table 1), aspirin significantly lowered the rate of ischemic events compared with placebo or control: nonfatal myocardial infarction in the Thrombosis Prevention Trial; myocardial infarction and major adverse cardiac event (ie, cardiovascular death, myocardial infarction, or stroke) in the Hypertension Optimal Treatment trial; and cardiovascular mortality and major cardiovascular events (cardiovascular death, nonfatal myocardial infarction, nonfatal stroke, angina pectoris, transient ischemic attack, peripheral artery disease, or revascularization procedures) in the Primary Prevention Project. However, aspirin’s benefit in each trial was largely offset by a higher rate of various bleeding end points.5–7
The Women’s Health Study
A subgroup analysis of the Hypertension Optimal Treatment trial suggested that sex may influence the efficacy of aspirin—specifically, aspirin did not prevent nonfatal myocardial infarction in women.9 Given the paucity of female participants in the previous primary prevention trials, the Women’s Health Study8 was designed to determine the efficacy and safety of aspirin (100 mg every other day) in women age 45 and older with very few cardiovascular risk factors.8
Aspirin did not significantly reduce the rate of the primary end point of cardiovascular death, myocardial infarction, or stroke, though a significant effect was observed in the subgroup of women age 65 and older. Although overall the Women’s Health Study found no benefit in the rate of myocardial infarction, there was a significant reduction in the rate of ischemic stroke (which needs to be interpreted cautiously in an overall neutral trial) and a nonsignificant increase in the rate of hemorrhagic stroke. As in other trials, rates of bleeding, including gastrointestinal bleeding, were higher with aspirin.
A meta-analysis of six trials of aspirin for primary prevention
In 2009, the Antithrombotic Trialists’ Collaboration10 published a meta-analysis of six trials of aspirin for primary prevention. In this analysis, aspirin did not reduce the rate of cardiovascular death, but it did reduce the yearly risk of:
- Death from coronary heart disease or nonfatal myocardial infarction
(0.28% vs 0.34%, P < .0001) - Nonfatal myocardial infarction
(0.18% vs 0.23%, P < .0001) - Ischemic stroke
(0.11% vs 0.12%, P = .05).10
Despite aspirin’s apparent efficacy, the absolute yearly risk for major extracranial bleeding and hemorrhagic stroke was also significantly increased with aspirin use by 0.3% and 0.1%, respectively. The efficacy of aspirin for preventing all serious vascular events (vascular death, myocardial infarction, or stroke) was similar in men and women.10 The authors concluded that the net benefit of aspirin did not outweigh the increased risks of bleeding.
WHAT ABOUT PATIENTS WITH DIABETES?
When considering whether to prescribe aspirin for primary prevention, the individual patient’s risks of cardiovascular disease and bleeding must be carefully assessed. Those at highest risk of cardiovascular disease and at low risk of bleeding may still benefit, but current evidence does not clearly support this strategy.
For example, diabetes mellitus has traditionally been considered a coronary heart disease equivalent, and aspirin was routinely prescribed as “secondary prevention.”11 In the six trials of aspirin for primary prevention, the prevalence of diabetic patients ranged from 1% to 17%, the efficacy of aspirin in this subgroup was inconsistent among the trials, and aspirin did not confer a net clinical benefit according to the 2009 Antithrombotic Trialists’ Collaboration meta-analysis.1,3–8,10
Additionally, two trials of aspirin for primary prevention in diabetes12,13 failed to demonstrate significant efficacy for aspirin compared with no aspirin, either in Japanese patients with type 2 diabetes and no history of cardiovascular disease12 or in patients with asymptomatic peripheral artery disease.13
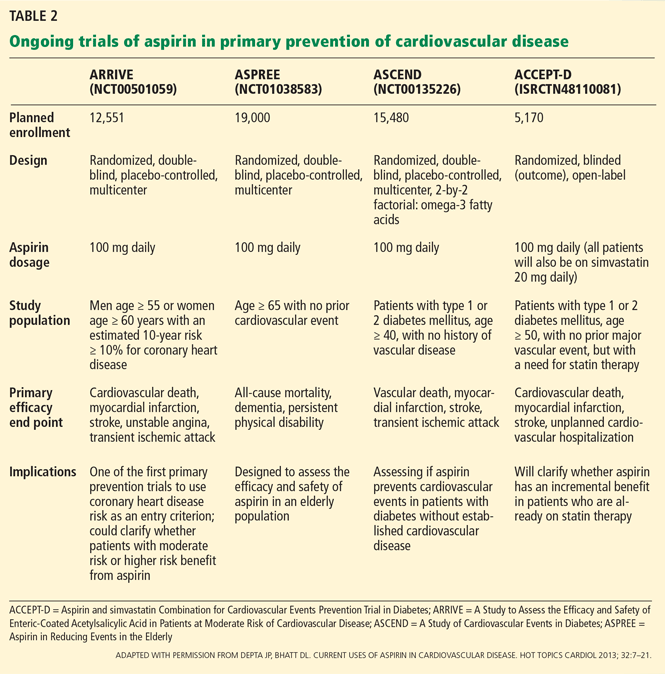
Thus, the current evidence for aspirin for primary prevention in diabetes does not demonstrate a net clinical benefit, but ongoing trials (Table 2) may provide evidence for the use of aspirin in this important subgroup.
An important finding from the 2009 Antithrombotic Trialists’ Collaboration was that traditional risk factors for cardiovascular disease also increase the risk of major bleeding, thus making it difficult to determine who will receive the maximum net clinical benefit.10 Additionally, many of the aspirin primary prevention trials predated the widespread use of statins and the current lower prevalence of smoking, which may further limit the generalizability of the positive signals seen in earlier trials.
THE DATA ARE MIXED, BUT ONE MESSAGE IS CLEAR

Based on the current available evidence, the US Food and Drug Administration recently issued a Consumer Update that does not support aspirin for primary prevention and warns patients about the risk of serious bleeding complications.14 Moreover, current guidelines and consensus panels (Table 3) for aspirin in primary prevention differ from one another,15–21 making it challenging for clinicians to determine which patients would benefit. One message is clear in the most current clinical guidelines, namely, that routine use of aspirin for primary prevention is not recommended.15–21 Several ongoing trials may resolve this important clinical dilemma.
- Depta JP, Bhatt DL. Current uses of aspirin in cardiovascular disease. Hot Topics Cardiol 2013; 32:7–21.
- Nemerovski CW, Salinitri FD, Morbitzer KA, Moser LR. Aspirin for primary prevention of cardiovascular disease events. Pharmacotherapy 2012; 32:1020–1035.
- Peto R, Gray R, Collins R, et al. Randomised trial of prophylactic daily aspirin in British male doctors. Br Med J (Clin Res Ed) 1988; 296:313–316.
- Final report on the aspirin component of the ongoing Physicians’ Health Study. Steering Committee of the Physicians’ Health Study Research Group. N Engl J Med 1989; 321:129–135.
- Thrombosis prevention trial: randomised trial of low-intensity oral anticoagulation with warfarin and low-dose aspirin in the primary prevention of ischaemic heart disease in men at increased risk. The Medical Research Council’s General Practice Research Framework. Lancet 1998; 351:233–241.
- Hansson L, Zanchetti A, Carruthers SG, et al. Effects of intensive blood-pressure lowering and low-dose aspirin in patients with hypertension: principal results of the Hypertension Optimal Treatment (HOT) randomised trial. HOT Study Group. Lancet 1998; 351:1755–1762.
- de Gaetano G; Collaborative Group of the Primary Prevention Project. Low-dose aspirin and vitamin E in people at cardiovascular risk: a randomised trial in general practice. Collaborative Group of the Primary Prevention Project. Lancet 2001; 357:89–95.
- Ridker PM, Cook NR, Lee IM, et al. A randomized trial of low-dose aspirin in the primary prevention of cardiovascular disease in women. N Engl J Med 2005; 352:1293–1304.
- Kjeldsen SE, Kolloch RE, Leonetti G, et al. Influence of gender and age on preventing cardiovascular disease by antihypertensive treatment and acetylsalicylic acid. The HOT study. Hypertension Optimal Treatment. J Hypertens 2000; 18:629–642.
- Antithrombotic Trialists’ (ATT) Collaboration; Baigent C, Blackwell L, Collins R, et al. Aspirin in the primary and secondary prevention of vascular disease: collaborative meta-analysis of individual participant data from randomised trials. Lancet 2009; 373:1849–1860.
- Buse JB, Ginsberg HN, Bakris GL, et al; American Heart Association; American Diabetes Association. Primary prevention of cardiovascular diseases in people with diabetes mellitus: a scientific statement from the American Heart Association and the American Diabetes Association. Circulation 2007; 115:114–126.
- Ogawa H, Nakayama M, Morimoto T, et al; Japanese Primary Prevention of Atherosclerosis With Aspirin for Diabetes (JPAD) Trial Investigators. Low-dose aspirin for primary prevention of atherosclerotic events in patients with type 2 diabetes: a randomized controlled trial. JAMA 2008; 300:2134–2141.
- Belch J, MacCuish A, Campbell I, et al; Prevention of Progression of Arterial Disease and Diabetes Study Group; Diabetes Registry Group; Royal College of Physicians Edinburgh. The prevention of progression of arterial disease and diabetes (POPADAD) trial: factorial randomised placebo controlled trial of aspirin and antioxidants in patients with diabetes and asymptomatic peripheral arterial disease. BMJ 2008; 337:a1840.
- US Food and Drug Administration (FDA). Use of aspirin for primary prevention of heart attack and stroke. http://www.fda.gov/drugs/resourcesforyou/consumers/ucm390574.htm. Accessed January 9, 2015.
- Vandvik PO, Lincoff AM, Gore JM, et al; American College of Chest Physicians. Primary and secondary prevention of cardiovascular disease: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e637S–e668S.
- American Diabetes Association. Standards of medical care in diabetes—2013. Diabetes Care 2013; 36(suppl 1):S11–S66.
- Pearson TA, Blair SN, Daniels SR, et al. AHA Guidelines for Primary Prevention of Cardiovascular Disease and Stroke: 2002 Update: Consensus Panel Guide to Comprehensive Risk Reduction for Adult Patients Without Coronary or Other Atherosclerotic Vascular Diseases. American Heart Association Science Advisory and Coordinating Committee. Circulation 2002; 106:388–391.
- Mosca L, Benjamin EJ, Berra K, et al. Effectiveness-based Guidelines for the Prevention of Cardiovascular Disease in Women—2011 Update: a Guideline from the American Heart Association. Circulation 2011; 123:1243–1262.
- Bell AD, Roussin A, Cartier R, et al; Canadian Cardiovascular Society. The use of antiplatelet therapy in the outpatient setting: Canadian Cardiovascular Society Guidelines. Can J Cardiol 2011; 27(suppl A):S1–S59.
- Perk J, De Backer G, Gohlke H, et al; European Association for Cardiovascular Prevention & Rehabilitation (EACPR); ESC Committee for Practice Guidelines (CPG). European guidelines on cardiovascular disease prevention in clinical practice (version 2012). The Fifth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of nine societies and by invited experts). Eur Heart J 2012; 33:1635–1701.
- US Preventive Services Task Force. Aspirin for the prevention of cardiovascular disease: US Preventive Services Task Force recommendation statement. Ann Intern Med 2009; 150:396–404.
In view of current evidence, we do not recommend routinely using aspirin for primary prevention of cardiovascular disease, even in patients with diabetes mellitus. The decision must be individualized on the basis of the patient’s risks of cardiovascular disease and bleeding, especially the risk of serious bleeding events such as gastrointestinal and intracranial hemorrhage.
For example, patients with a family history of myocardial infarction at an early age and patients who smoke or have multiple cardiovascular risk factors may be most likely to benefit, whereas those with risk factors for gastrointestinal bleeding such as dyspepsia or ulcer would not be good candidates. Of note, current recommendations are mixed and confusing and will need to be reevaluated as new trial data become available.
TRIALS THAT SET THE STAGE FOR CURRENT PRACTICE
Routine use of aspirin for primary prevention of cardiovascular disease remains controversial.1,2 Aspirin’s safety and efficacy for this indication was studied in six major trials (Table 1).3–8 In the late 1980s, the first two primary prevention trials of aspirin enrolled healthy male physicians who had minimal cardiovascular risk factors3,4:
The British Doctors’ Trial3 observed no significant differences between aspirin (300–500 mg/day) and no aspirin in the rates of the primary end point of cardiovascular death or in the individual secondary end points of nonfatal myocardial infarction, nonfatal stroke, or bleeding.3
The Physicians’ Health Study4 found no differences in the rates of cardiovascular mortality or ischemic stroke between aspirin (325 mg every other day) and placebo. The rate of nonfatal myocardial infarction was significantly lower with aspirin than with placebo, but with a higher risk of bleeding. Relative risks and 95% confidence intervals with aspirin vs placebo:
- Nonfatal myocardial infarction
0.59 (0.47–0.74), P < .00001 - Bleeding
1.32 (1.25–1.40), P < .00001 - Blood transfusions
1.71 (1.09–2.69), P = .02 - Hemorrhagic stroke
2.14 (0.96–4.77), P = .06.
A subgroup analysis revealed that the benefit of aspirin for myocardial infarction in the Physicians’ Health Study was predominantly in those age 50 and older.4 This finding established the common clinical practice of routinely using aspirin for primary prevention in men age 50 and older.1
Later, aspirin for primary prevention was studied in four trials,5–8 three of which enrolled patients at higher cardiovascular risk5–7:
The Thrombosis Prevention Trial5 was conducted in men in the highest quintile of cardiovascular risk. The aspirin dosage was 75 mg/day.
The Hypertension Optimal Treatment6 trial included men and women ages 50 to 80 with hypertension. Aspirin dosage: 75 mg/day.
The Primary Prevention Project7 involved men and women age 50 and older with at least one risk factor for cardiovascular disease.1,5–7 The aspirin dosage was 100 mg/day.
In these trials (Table 1), aspirin significantly lowered the rate of ischemic events compared with placebo or control: nonfatal myocardial infarction in the Thrombosis Prevention Trial; myocardial infarction and major adverse cardiac event (ie, cardiovascular death, myocardial infarction, or stroke) in the Hypertension Optimal Treatment trial; and cardiovascular mortality and major cardiovascular events (cardiovascular death, nonfatal myocardial infarction, nonfatal stroke, angina pectoris, transient ischemic attack, peripheral artery disease, or revascularization procedures) in the Primary Prevention Project. However, aspirin’s benefit in each trial was largely offset by a higher rate of various bleeding end points.5–7
The Women’s Health Study
A subgroup analysis of the Hypertension Optimal Treatment trial suggested that sex may influence the efficacy of aspirin—specifically, aspirin did not prevent nonfatal myocardial infarction in women.9 Given the paucity of female participants in the previous primary prevention trials, the Women’s Health Study8 was designed to determine the efficacy and safety of aspirin (100 mg every other day) in women age 45 and older with very few cardiovascular risk factors.8
Aspirin did not significantly reduce the rate of the primary end point of cardiovascular death, myocardial infarction, or stroke, though a significant effect was observed in the subgroup of women age 65 and older. Although overall the Women’s Health Study found no benefit in the rate of myocardial infarction, there was a significant reduction in the rate of ischemic stroke (which needs to be interpreted cautiously in an overall neutral trial) and a nonsignificant increase in the rate of hemorrhagic stroke. As in other trials, rates of bleeding, including gastrointestinal bleeding, were higher with aspirin.
A meta-analysis of six trials of aspirin for primary prevention
In 2009, the Antithrombotic Trialists’ Collaboration10 published a meta-analysis of six trials of aspirin for primary prevention. In this analysis, aspirin did not reduce the rate of cardiovascular death, but it did reduce the yearly risk of:
- Death from coronary heart disease or nonfatal myocardial infarction
(0.28% vs 0.34%, P < .0001) - Nonfatal myocardial infarction
(0.18% vs 0.23%, P < .0001) - Ischemic stroke
(0.11% vs 0.12%, P = .05).10
Despite aspirin’s apparent efficacy, the absolute yearly risk for major extracranial bleeding and hemorrhagic stroke was also significantly increased with aspirin use by 0.3% and 0.1%, respectively. The efficacy of aspirin for preventing all serious vascular events (vascular death, myocardial infarction, or stroke) was similar in men and women.10 The authors concluded that the net benefit of aspirin did not outweigh the increased risks of bleeding.
WHAT ABOUT PATIENTS WITH DIABETES?
When considering whether to prescribe aspirin for primary prevention, the individual patient’s risks of cardiovascular disease and bleeding must be carefully assessed. Those at highest risk of cardiovascular disease and at low risk of bleeding may still benefit, but current evidence does not clearly support this strategy.
For example, diabetes mellitus has traditionally been considered a coronary heart disease equivalent, and aspirin was routinely prescribed as “secondary prevention.”11 In the six trials of aspirin for primary prevention, the prevalence of diabetic patients ranged from 1% to 17%, the efficacy of aspirin in this subgroup was inconsistent among the trials, and aspirin did not confer a net clinical benefit according to the 2009 Antithrombotic Trialists’ Collaboration meta-analysis.1,3–8,10
Additionally, two trials of aspirin for primary prevention in diabetes12,13 failed to demonstrate significant efficacy for aspirin compared with no aspirin, either in Japanese patients with type 2 diabetes and no history of cardiovascular disease12 or in patients with asymptomatic peripheral artery disease.13
Thus, the current evidence for aspirin for primary prevention in diabetes does not demonstrate a net clinical benefit, but ongoing trials (Table 2) may provide evidence for the use of aspirin in this important subgroup.
An important finding from the 2009 Antithrombotic Trialists’ Collaboration was that traditional risk factors for cardiovascular disease also increase the risk of major bleeding, thus making it difficult to determine who will receive the maximum net clinical benefit.10 Additionally, many of the aspirin primary prevention trials predated the widespread use of statins and the current lower prevalence of smoking, which may further limit the generalizability of the positive signals seen in earlier trials.
THE DATA ARE MIXED, BUT ONE MESSAGE IS CLEAR
Based on the current available evidence, the US Food and Drug Administration recently issued a Consumer Update that does not support aspirin for primary prevention and warns patients about the risk of serious bleeding complications.14 Moreover, current guidelines and consensus panels (Table 3) for aspirin in primary prevention differ from one another,15–21 making it challenging for clinicians to determine which patients would benefit. One message is clear in the most current clinical guidelines, namely, that routine use of aspirin for primary prevention is not recommended.15–21 Several ongoing trials may resolve this important clinical dilemma.
In view of current evidence, we do not recommend routinely using aspirin for primary prevention of cardiovascular disease, even in patients with diabetes mellitus. The decision must be individualized on the basis of the patient’s risks of cardiovascular disease and bleeding, especially the risk of serious bleeding events such as gastrointestinal and intracranial hemorrhage.
For example, patients with a family history of myocardial infarction at an early age and patients who smoke or have multiple cardiovascular risk factors may be most likely to benefit, whereas those with risk factors for gastrointestinal bleeding such as dyspepsia or ulcer would not be good candidates. Of note, current recommendations are mixed and confusing and will need to be reevaluated as new trial data become available.
TRIALS THAT SET THE STAGE FOR CURRENT PRACTICE
Routine use of aspirin for primary prevention of cardiovascular disease remains controversial.1,2 Aspirin’s safety and efficacy for this indication was studied in six major trials (Table 1).3–8 In the late 1980s, the first two primary prevention trials of aspirin enrolled healthy male physicians who had minimal cardiovascular risk factors3,4:
The British Doctors’ Trial3 observed no significant differences between aspirin (300–500 mg/day) and no aspirin in the rates of the primary end point of cardiovascular death or in the individual secondary end points of nonfatal myocardial infarction, nonfatal stroke, or bleeding.3
The Physicians’ Health Study4 found no differences in the rates of cardiovascular mortality or ischemic stroke between aspirin (325 mg every other day) and placebo. The rate of nonfatal myocardial infarction was significantly lower with aspirin than with placebo, but with a higher risk of bleeding. Relative risks and 95% confidence intervals with aspirin vs placebo:
- Nonfatal myocardial infarction
0.59 (0.47–0.74), P < .00001 - Bleeding
1.32 (1.25–1.40), P < .00001 - Blood transfusions
1.71 (1.09–2.69), P = .02 - Hemorrhagic stroke
2.14 (0.96–4.77), P = .06.
A subgroup analysis revealed that the benefit of aspirin for myocardial infarction in the Physicians’ Health Study was predominantly in those age 50 and older.4 This finding established the common clinical practice of routinely using aspirin for primary prevention in men age 50 and older.1
Later, aspirin for primary prevention was studied in four trials,5–8 three of which enrolled patients at higher cardiovascular risk5–7:
The Thrombosis Prevention Trial5 was conducted in men in the highest quintile of cardiovascular risk. The aspirin dosage was 75 mg/day.
The Hypertension Optimal Treatment6 trial included men and women ages 50 to 80 with hypertension. Aspirin dosage: 75 mg/day.
The Primary Prevention Project7 involved men and women age 50 and older with at least one risk factor for cardiovascular disease.1,5–7 The aspirin dosage was 100 mg/day.
In these trials (Table 1), aspirin significantly lowered the rate of ischemic events compared with placebo or control: nonfatal myocardial infarction in the Thrombosis Prevention Trial; myocardial infarction and major adverse cardiac event (ie, cardiovascular death, myocardial infarction, or stroke) in the Hypertension Optimal Treatment trial; and cardiovascular mortality and major cardiovascular events (cardiovascular death, nonfatal myocardial infarction, nonfatal stroke, angina pectoris, transient ischemic attack, peripheral artery disease, or revascularization procedures) in the Primary Prevention Project. However, aspirin’s benefit in each trial was largely offset by a higher rate of various bleeding end points.5–7
The Women’s Health Study
A subgroup analysis of the Hypertension Optimal Treatment trial suggested that sex may influence the efficacy of aspirin—specifically, aspirin did not prevent nonfatal myocardial infarction in women.9 Given the paucity of female participants in the previous primary prevention trials, the Women’s Health Study8 was designed to determine the efficacy and safety of aspirin (100 mg every other day) in women age 45 and older with very few cardiovascular risk factors.8
Aspirin did not significantly reduce the rate of the primary end point of cardiovascular death, myocardial infarction, or stroke, though a significant effect was observed in the subgroup of women age 65 and older. Although overall the Women’s Health Study found no benefit in the rate of myocardial infarction, there was a significant reduction in the rate of ischemic stroke (which needs to be interpreted cautiously in an overall neutral trial) and a nonsignificant increase in the rate of hemorrhagic stroke. As in other trials, rates of bleeding, including gastrointestinal bleeding, were higher with aspirin.
A meta-analysis of six trials of aspirin for primary prevention
In 2009, the Antithrombotic Trialists’ Collaboration10 published a meta-analysis of six trials of aspirin for primary prevention. In this analysis, aspirin did not reduce the rate of cardiovascular death, but it did reduce the yearly risk of:
- Death from coronary heart disease or nonfatal myocardial infarction
(0.28% vs 0.34%, P < .0001) - Nonfatal myocardial infarction
(0.18% vs 0.23%, P < .0001) - Ischemic stroke
(0.11% vs 0.12%, P = .05).10
Despite aspirin’s apparent efficacy, the absolute yearly risk for major extracranial bleeding and hemorrhagic stroke was also significantly increased with aspirin use by 0.3% and 0.1%, respectively. The efficacy of aspirin for preventing all serious vascular events (vascular death, myocardial infarction, or stroke) was similar in men and women.10 The authors concluded that the net benefit of aspirin did not outweigh the increased risks of bleeding.
WHAT ABOUT PATIENTS WITH DIABETES?
When considering whether to prescribe aspirin for primary prevention, the individual patient’s risks of cardiovascular disease and bleeding must be carefully assessed. Those at highest risk of cardiovascular disease and at low risk of bleeding may still benefit, but current evidence does not clearly support this strategy.
For example, diabetes mellitus has traditionally been considered a coronary heart disease equivalent, and aspirin was routinely prescribed as “secondary prevention.”11 In the six trials of aspirin for primary prevention, the prevalence of diabetic patients ranged from 1% to 17%, the efficacy of aspirin in this subgroup was inconsistent among the trials, and aspirin did not confer a net clinical benefit according to the 2009 Antithrombotic Trialists’ Collaboration meta-analysis.1,3–8,10
Additionally, two trials of aspirin for primary prevention in diabetes12,13 failed to demonstrate significant efficacy for aspirin compared with no aspirin, either in Japanese patients with type 2 diabetes and no history of cardiovascular disease12 or in patients with asymptomatic peripheral artery disease.13
Thus, the current evidence for aspirin for primary prevention in diabetes does not demonstrate a net clinical benefit, but ongoing trials (Table 2) may provide evidence for the use of aspirin in this important subgroup.
An important finding from the 2009 Antithrombotic Trialists’ Collaboration was that traditional risk factors for cardiovascular disease also increase the risk of major bleeding, thus making it difficult to determine who will receive the maximum net clinical benefit.10 Additionally, many of the aspirin primary prevention trials predated the widespread use of statins and the current lower prevalence of smoking, which may further limit the generalizability of the positive signals seen in earlier trials.
THE DATA ARE MIXED, BUT ONE MESSAGE IS CLEAR
Based on the current available evidence, the US Food and Drug Administration recently issued a Consumer Update that does not support aspirin for primary prevention and warns patients about the risk of serious bleeding complications.14 Moreover, current guidelines and consensus panels (Table 3) for aspirin in primary prevention differ from one another,15–21 making it challenging for clinicians to determine which patients would benefit. One message is clear in the most current clinical guidelines, namely, that routine use of aspirin for primary prevention is not recommended.15–21 Several ongoing trials may resolve this important clinical dilemma.
- Depta JP, Bhatt DL. Current uses of aspirin in cardiovascular disease. Hot Topics Cardiol 2013; 32:7–21.
- Nemerovski CW, Salinitri FD, Morbitzer KA, Moser LR. Aspirin for primary prevention of cardiovascular disease events. Pharmacotherapy 2012; 32:1020–1035.
- Peto R, Gray R, Collins R, et al. Randomised trial of prophylactic daily aspirin in British male doctors. Br Med J (Clin Res Ed) 1988; 296:313–316.
- Final report on the aspirin component of the ongoing Physicians’ Health Study. Steering Committee of the Physicians’ Health Study Research Group. N Engl J Med 1989; 321:129–135.
- Thrombosis prevention trial: randomised trial of low-intensity oral anticoagulation with warfarin and low-dose aspirin in the primary prevention of ischaemic heart disease in men at increased risk. The Medical Research Council’s General Practice Research Framework. Lancet 1998; 351:233–241.
- Hansson L, Zanchetti A, Carruthers SG, et al. Effects of intensive blood-pressure lowering and low-dose aspirin in patients with hypertension: principal results of the Hypertension Optimal Treatment (HOT) randomised trial. HOT Study Group. Lancet 1998; 351:1755–1762.
- de Gaetano G; Collaborative Group of the Primary Prevention Project. Low-dose aspirin and vitamin E in people at cardiovascular risk: a randomised trial in general practice. Collaborative Group of the Primary Prevention Project. Lancet 2001; 357:89–95.
- Ridker PM, Cook NR, Lee IM, et al. A randomized trial of low-dose aspirin in the primary prevention of cardiovascular disease in women. N Engl J Med 2005; 352:1293–1304.
- Kjeldsen SE, Kolloch RE, Leonetti G, et al. Influence of gender and age on preventing cardiovascular disease by antihypertensive treatment and acetylsalicylic acid. The HOT study. Hypertension Optimal Treatment. J Hypertens 2000; 18:629–642.
- Antithrombotic Trialists’ (ATT) Collaboration; Baigent C, Blackwell L, Collins R, et al. Aspirin in the primary and secondary prevention of vascular disease: collaborative meta-analysis of individual participant data from randomised trials. Lancet 2009; 373:1849–1860.
- Buse JB, Ginsberg HN, Bakris GL, et al; American Heart Association; American Diabetes Association. Primary prevention of cardiovascular diseases in people with diabetes mellitus: a scientific statement from the American Heart Association and the American Diabetes Association. Circulation 2007; 115:114–126.
- Ogawa H, Nakayama M, Morimoto T, et al; Japanese Primary Prevention of Atherosclerosis With Aspirin for Diabetes (JPAD) Trial Investigators. Low-dose aspirin for primary prevention of atherosclerotic events in patients with type 2 diabetes: a randomized controlled trial. JAMA 2008; 300:2134–2141.
- Belch J, MacCuish A, Campbell I, et al; Prevention of Progression of Arterial Disease and Diabetes Study Group; Diabetes Registry Group; Royal College of Physicians Edinburgh. The prevention of progression of arterial disease and diabetes (POPADAD) trial: factorial randomised placebo controlled trial of aspirin and antioxidants in patients with diabetes and asymptomatic peripheral arterial disease. BMJ 2008; 337:a1840.
- US Food and Drug Administration (FDA). Use of aspirin for primary prevention of heart attack and stroke. http://www.fda.gov/drugs/resourcesforyou/consumers/ucm390574.htm. Accessed January 9, 2015.
- Vandvik PO, Lincoff AM, Gore JM, et al; American College of Chest Physicians. Primary and secondary prevention of cardiovascular disease: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e637S–e668S.
- American Diabetes Association. Standards of medical care in diabetes—2013. Diabetes Care 2013; 36(suppl 1):S11–S66.
- Pearson TA, Blair SN, Daniels SR, et al. AHA Guidelines for Primary Prevention of Cardiovascular Disease and Stroke: 2002 Update: Consensus Panel Guide to Comprehensive Risk Reduction for Adult Patients Without Coronary or Other Atherosclerotic Vascular Diseases. American Heart Association Science Advisory and Coordinating Committee. Circulation 2002; 106:388–391.
- Mosca L, Benjamin EJ, Berra K, et al. Effectiveness-based Guidelines for the Prevention of Cardiovascular Disease in Women—2011 Update: a Guideline from the American Heart Association. Circulation 2011; 123:1243–1262.
- Bell AD, Roussin A, Cartier R, et al; Canadian Cardiovascular Society. The use of antiplatelet therapy in the outpatient setting: Canadian Cardiovascular Society Guidelines. Can J Cardiol 2011; 27(suppl A):S1–S59.
- Perk J, De Backer G, Gohlke H, et al; European Association for Cardiovascular Prevention & Rehabilitation (EACPR); ESC Committee for Practice Guidelines (CPG). European guidelines on cardiovascular disease prevention in clinical practice (version 2012). The Fifth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of nine societies and by invited experts). Eur Heart J 2012; 33:1635–1701.
- US Preventive Services Task Force. Aspirin for the prevention of cardiovascular disease: US Preventive Services Task Force recommendation statement. Ann Intern Med 2009; 150:396–404.
- Depta JP, Bhatt DL. Current uses of aspirin in cardiovascular disease. Hot Topics Cardiol 2013; 32:7–21.
- Nemerovski CW, Salinitri FD, Morbitzer KA, Moser LR. Aspirin for primary prevention of cardiovascular disease events. Pharmacotherapy 2012; 32:1020–1035.
- Peto R, Gray R, Collins R, et al. Randomised trial of prophylactic daily aspirin in British male doctors. Br Med J (Clin Res Ed) 1988; 296:313–316.
- Final report on the aspirin component of the ongoing Physicians’ Health Study. Steering Committee of the Physicians’ Health Study Research Group. N Engl J Med 1989; 321:129–135.
- Thrombosis prevention trial: randomised trial of low-intensity oral anticoagulation with warfarin and low-dose aspirin in the primary prevention of ischaemic heart disease in men at increased risk. The Medical Research Council’s General Practice Research Framework. Lancet 1998; 351:233–241.
- Hansson L, Zanchetti A, Carruthers SG, et al. Effects of intensive blood-pressure lowering and low-dose aspirin in patients with hypertension: principal results of the Hypertension Optimal Treatment (HOT) randomised trial. HOT Study Group. Lancet 1998; 351:1755–1762.
- de Gaetano G; Collaborative Group of the Primary Prevention Project. Low-dose aspirin and vitamin E in people at cardiovascular risk: a randomised trial in general practice. Collaborative Group of the Primary Prevention Project. Lancet 2001; 357:89–95.
- Ridker PM, Cook NR, Lee IM, et al. A randomized trial of low-dose aspirin in the primary prevention of cardiovascular disease in women. N Engl J Med 2005; 352:1293–1304.
- Kjeldsen SE, Kolloch RE, Leonetti G, et al. Influence of gender and age on preventing cardiovascular disease by antihypertensive treatment and acetylsalicylic acid. The HOT study. Hypertension Optimal Treatment. J Hypertens 2000; 18:629–642.
- Antithrombotic Trialists’ (ATT) Collaboration; Baigent C, Blackwell L, Collins R, et al. Aspirin in the primary and secondary prevention of vascular disease: collaborative meta-analysis of individual participant data from randomised trials. Lancet 2009; 373:1849–1860.
- Buse JB, Ginsberg HN, Bakris GL, et al; American Heart Association; American Diabetes Association. Primary prevention of cardiovascular diseases in people with diabetes mellitus: a scientific statement from the American Heart Association and the American Diabetes Association. Circulation 2007; 115:114–126.
- Ogawa H, Nakayama M, Morimoto T, et al; Japanese Primary Prevention of Atherosclerosis With Aspirin for Diabetes (JPAD) Trial Investigators. Low-dose aspirin for primary prevention of atherosclerotic events in patients with type 2 diabetes: a randomized controlled trial. JAMA 2008; 300:2134–2141.
- Belch J, MacCuish A, Campbell I, et al; Prevention of Progression of Arterial Disease and Diabetes Study Group; Diabetes Registry Group; Royal College of Physicians Edinburgh. The prevention of progression of arterial disease and diabetes (POPADAD) trial: factorial randomised placebo controlled trial of aspirin and antioxidants in patients with diabetes and asymptomatic peripheral arterial disease. BMJ 2008; 337:a1840.
- US Food and Drug Administration (FDA). Use of aspirin for primary prevention of heart attack and stroke. http://www.fda.gov/drugs/resourcesforyou/consumers/ucm390574.htm. Accessed January 9, 2015.
- Vandvik PO, Lincoff AM, Gore JM, et al; American College of Chest Physicians. Primary and secondary prevention of cardiovascular disease: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e637S–e668S.
- American Diabetes Association. Standards of medical care in diabetes—2013. Diabetes Care 2013; 36(suppl 1):S11–S66.
- Pearson TA, Blair SN, Daniels SR, et al. AHA Guidelines for Primary Prevention of Cardiovascular Disease and Stroke: 2002 Update: Consensus Panel Guide to Comprehensive Risk Reduction for Adult Patients Without Coronary or Other Atherosclerotic Vascular Diseases. American Heart Association Science Advisory and Coordinating Committee. Circulation 2002; 106:388–391.
- Mosca L, Benjamin EJ, Berra K, et al. Effectiveness-based Guidelines for the Prevention of Cardiovascular Disease in Women—2011 Update: a Guideline from the American Heart Association. Circulation 2011; 123:1243–1262.
- Bell AD, Roussin A, Cartier R, et al; Canadian Cardiovascular Society. The use of antiplatelet therapy in the outpatient setting: Canadian Cardiovascular Society Guidelines. Can J Cardiol 2011; 27(suppl A):S1–S59.
- Perk J, De Backer G, Gohlke H, et al; European Association for Cardiovascular Prevention & Rehabilitation (EACPR); ESC Committee for Practice Guidelines (CPG). European guidelines on cardiovascular disease prevention in clinical practice (version 2012). The Fifth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of nine societies and by invited experts). Eur Heart J 2012; 33:1635–1701.
- US Preventive Services Task Force. Aspirin for the prevention of cardiovascular disease: US Preventive Services Task Force recommendation statement. Ann Intern Med 2009; 150:396–404.

Denosumab: A novel antiresorptive drug for osteoporosis
A 68-year-old white woman presents with mid- thoracic back pain. Plain radiographs reveal a compression fracture of the 10th thoracic vertebra. She is diagnosed with osteoporosis on the basis of dual energy x-ray absorptiometry (DXA) scans that show T scores of –2.9 in her lumbar spine and –2.6 in her left femoral neck. Her 10-year probability of fracture is estimated as 23% for major osteoporotic fracture and 5.9% for hip fracture (based on the World Health Organization’s absolute fracture risk assessment tool, adapted for the United States, and available at www.shef.ac.uk/FRAX).
After excluding common secondary causes of osteoporosis, her physician recommends a bisphosphonate to reduce her risk of fracture, but she develops upper-gastrointestinal adverse effects with both alendronate and risedronate despite correctly following the instructions for oral administration.
What should her physician consider next?
OSTEOPOROSIS IS A MAJOR PROBLEM
Osteoporosis is a systemic skeletal disease characterized by low bone mass and microarchitectural deterioration of bone tissue, predisposing to an increased risk of fragility fractures, particularly of the spine, hip, and wrist.
It is a major public health problem, affecting 200 million people throughout the world, with 9 million osteoporotic fractures reported in the year 2000.1 The incidence of hip fracture alone is predicted to rise to 2.6 million by the year 2025, and to 4.5 million by the year 2050.2 In the United States, the total burden was estimated to be about 2 million incident fractures in the year 2005, projected to rise by another 50% by the year 2025,3 primarily because of the aging of the population. Population studies have indeed suggested that about 40% of white women and 13% of white men over the age of 50 are at risk of sustaining an osteoporotic fracture during the remainder of their lifetime.4
The consequences of osteoporotic fractures can be devastating. Hip fractures are associated with a risk of death ranging from 8.4% to 36% during the first year after fracture.5 One-fifth of patients who sustain a hip fracture require long-term nursing home care, and more than half of the survivors do not regain their previous level of independence.
Patients with vertebral fractures are also at increased risk of death, although the results of some studies suggest that this could be the result of comorbid factors.6–9 Vertebral fractures can result in chronic back pain, loss of height from spinal deformity, reduced mobility, loss of self-esteem, and in severe cases, respiratory and digestive problems because of contact between the lower ribs and pelvis.
A person with one vertebral compression fracture is five times more likely to have another vertebral fracture,10 and a person with two or more compression fractures is 12 times more likely.11
The costs of treating osteoporotic fractures are greater than those of treating myocardial infarction or stroke12,13; they include not only direct costs incurred in treating the fracture, but also indirect societal costs owing to the long-term morbidity associated with the fracture. In the United States, the total cost of treating osteoporotic fractures was estimated at $19 billion in the year 2005.3 By 2025, the annual costs are projected to rise by almost 50%.3
A NEED FOR MORE OPTIONS
Until fairly recently, bisphosphonates were the only drugs of first choice, but adherence to oral bisphosphonate therapy is generally poor (< 50% at 1 year),14 most commonly because of dyspepsia,15 and poor adherence has been shown to be associated with increased fracture risk.16,17 Hence the need for additional therapeutic options.
In this review, we discuss denosumab, an antiresorptive drug approved by the US Food and Drug Administration (FDA) in 2010. First, we discuss its mechanism of action, efficacy, and safety, and then we offer recommendations for its use in clinical practice.
WHAT IS DENOSUMAB AND HOW DOES IT WORK?
Bone remodeling is a dynamic process involving a balance between bone resorption by osteoclasts on the one hand and new bone formation by osteoblasts on the other. A net gain in bone occurs when the activity of osteoblasts exceeds that of osteoclasts, and bone loss occurs when there is increased osteoclast activity or reduced osteoblast activity, or both. The activities of osteoblasts and osteoclasts are tightly coupled because of the opposing effects of two sets of proteins, namely, receptor activator of nuclear factor kappa b ligand (RANKL) and osteoprotegerin.
Both RANKL and osteoprotegerin are produced by osteoblasts. RANKL binds to its receptor (RANK) on preosteoclasts and osteoclasts and induces their differentiation and activation, respectively. Osteoprotegerin is the decoy receptor and natural antagonist for RANKL. By binding with RANKL, it blocks its interaction with RANK.18 In healthy individuals, a fine balance between RANKL and osteoprotegerin ensures that bone remodeling is regulated.
In postmenopausal women, estrogen deficiency leads to an imbalance between RANKL and osteoprotegerin (increased RANKL and reduced osteoprotegerin), resulting in net bone loss. This imbalance is also a feature of rheumatoid arthritis, myeloma bone disease, and osteolytic metastatic bone disease; it also occurs in those receiving androgen deprivation therapy for prostate cancer or aromatase inhibitors for breast cancer.
Denosumab is a fully human monoclonal antibody that targets RANKL.19 By binding to RANKL, this drug prevents the maturation and differentiation of preosteoclasts and promotes apoptosis of osteoclasts. Bone resorption is therefore slowed. It was parenteral osteoprotegerin that was initially developed by denosumab’s manufacturer,20 but this approach failed because neutralizing antibodies developed to osteoprotegerin, rendering it ineffective. Development of neutralizing antibodies has thus far not been a problem with denosumab.
Denosumab, with its property of RANKL inhibition, has also been used to prevent skeletal events in patients with bone metastases from solid tumors and to treat unresectable giant cell tumors of the bone (both FDA-approved indications) and hypercalcemia of malignancy. There is limited clinical experience in Paget disease of the bone as well.21–23 These other potential uses of denosumab are beyond the scope of this review.
HOW WELL DOES DENOSUMAB WORK FOR OSTEOPOROSIS?
Several phase 2 and phase 3 randomized controlled trials have evaluated the efficacy of denosumab, but only one, the Fracture Reduction Evaluation of Denosumab in Osteoporosis Every 6 Months (FREEDOM) trial, included fracture reduction as the primary outcome measure. The rest evaluated changes in bone mineral density or in markers of bone turnover, or both.
FREEDOM was a double-blind, randomized controlled trial in 7,808 postmenopausal women with T scores between –2.5 and –4.0 at the lumbar spine or hip.24 Twenty-four percent of the patients had vertebral fractures at baseline. Patients were randomized to receive either denosumab 60 mg (n = 3,902) or placebo (n = 3,906) every 6 months for up to 36 months. All patients also received adequate calcium and vitamin D supplementation.
At 36 months, compared with those who were randomized to receive placebo, those who were randomized to denosumab had lower incidence rates of:
- New vertebral fracture
(2.3% vs 7.2%, risk ratio 0.32,
95% CI 0.26–0.41, P < .001) - Nonvertebral fracture
(6.5% vs 8.0%, risk ratio 0.80,
95% CI 0.67–0.95, P = .01) - Hip fracture
(0.7% vs 1.2%, risk ratio 0.60,
95% CI 0.37–0.97, P = .04).
Increases in bone mineral density at the lumbar spine and hip, and decreases in bone turnover markers were also significantly greater in the denosumab group. The number needed to treat to prevent one new fracture over 3 years was 21 for vertebral fracture, 67 for nonvertebral fracture, and 200 for hip fracture, reflecting the relatively low event rate in the study.
In an open-label extension of the FREEDOM trial, the fracture incidence rates among participants who continued to receive denosumab for an additional 5 years remained low, and still below those projected for a “virtual placebo cohort” (total duration of exposure of 8 years). The rates among participants who switched from placebo to denosumab were similar to those of the denosumab group from the parent trial.25,26
A subgroup analysis of the FREEDOM trial suggested that denosumab reduced the risk of new vertebral fractures irrespective of age, body mass index, femoral neck bone mineral density, prevalent vertebral fractures, or prior nonvertebral fractures (risk ratio 0.32; 95% CI 0.26–0.41, P < .001), whereas the risk of nonvertebral fractures was only reduced in those women with body mass indices less than 25 kg/m2, femoral neck bone mineral density T scores less than –2.5, and in those without a prevalent vertebral fracture.27
A post hoc analysis revealed that denosumab significantly reduced the risk of new vertebral and hip fractures even in subgroups of women at higher risk of fracture.28 At 10% fracture probability (as estimated by the FRAX risk calculator), denosumab reduced the fracture risk by 11% (P = .629), whereas at 30% probability (moderate to high risk), the reduction was 50% (P = .001).29
Other phase 2 and phase 3 trials, in postmenopausal women with low bone mineral density, demonstrated that compared with placebo, denosumab significantly increased bone mineral density at all skeletal sites, increased volumetric bone mineral density at the distal radius, improved hip structural analysis parameters, and reduced bone turnover markers.30–33 Increases in bone mineral density and reductions in bone turnover markers with denosumab have been shown in men as well.34
In a randomized controlled trial,35 improvement in bone mineral density was better in those who received the combination of denosumab and teriparatide than in those who received either drug on its own.
Denosumab has also been shown to reduce the incidence of new vertebral fractures and improve bone mineral density in men receiving androgen-deprivation therapy for nonmetastatic prostate cancer,36 and to improve bone mineral density in women with metastatic breast cancer and low bone mass who were receiving adjuvant aromatase inhibitor therapy.37
HOW DOES DENOSUMAB COMPARE WITH OTHER OSTEOPOROSIS DRUGS?
A double-blind randomized controlled trial in postmenopausal women with low bone mass demonstrated that denosumab was superior to alendronate in improving bone mineral density at all skeletal sites (3.5% vs 2.6% for total hip bone mineral density, P < .0001).38
Another double-blind trial demonstrated that in patients previously treated with alendronate, switching to denosumab resulted in significantly greater increases in bone mineral density at all skeletal sites compared with continuing with alendronate (P < .0001).39
Denosumab has also been shown to be superior to alendronate in improving cortical bone mineral density, as measured by quantitative computed tomography.40
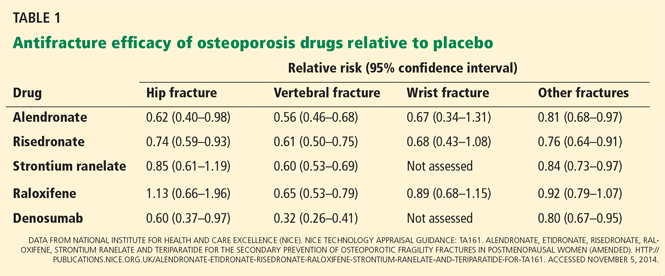
No trial has directly compared the efficacy of denosumab with other osteoporosis drugs in reducing fracture risk, but a systematic literature review of multiple databases,41 comparing the antifracture efficacy of nine osteoporosis drugs, concluded that teriparatide, zoledronic acid, and denosumab had the highest probabilities of being most efficacious for nonvertebral and vertebral fractures, with the greatest effect sizes. Indirect comparisons of the relative risk of fracture with denosumab (based on the results of FREEDOM), alendronate, risedronate, raloxifene, and strontium (based on a meta-analysis of randomized controlled trials) are presented in Table 1.42
A 2-year randomized, open-label, crossover study43 randomized patients to receive either denosumab followed by alendronate or alendronate followed by denosumab over successive 12-month periods. The results suggested that postmenopausal women with osteoporosis were more adherent, compliant, and persistent with denosumab therapy (a subcutaneous injection every 6 months) than with alendronate therapy in the form of oral tablets, self-administered weekly (7.5% nonadherence vs 36.5% at the end of 2 years). After receiving both treatments, women reported greater satisfaction with denosumab, with 92.4% preferring it over oral alendronate. Bone mineral density remained stable when patients were switched from denosumab to alendronate, but improved further when they were switched from alendronate to denosumab.
HOW SAFE IS DENOSUMAB?
The most frequent adverse events with denosumab reported in the long-term extension of one phase 2 study were upper respiratory tract infections (13.5%), arthralgia (11.5%), and back pain (9.0%).30
Increased risk of infection, cancer, and dermatologic reactions has been a concern, as RANKL and RANK are expressed by a wide variety of cells, including T lymphocytes, B cells, and dendritic cells.44 However, there were no significant differences in the overall incidences of adverse events between patients who received denosumab and those who received placebo or alendronate in any of the phase 2, phase 3, or extension studies.
In the FREEDOM trial,24 there was no significant difference between the two groups in the overall incidence of infection (52.9% with denosumab vs 54.4% with placebo, P = .17), or serious infection (4.1% with denosumab vs 3.4% with placebo, P = .14), although the incidence of “serious” cellulitis requiring hospitalization was higher in the denosumab group (0.3% vs < 0.1%, P = .002). There were more serious infections involving the gastrointestinal system, urinary tract, and ear and cases of endocarditis in the denosumab group, but the number of events was small, and there was no relationship with the timing of administration or duration of exposure to denosumab.45 Eczema was more common in the denosumab than in the placebo group (3.0% vs 1.7%, P < .001), but the extension data from the first 3 years did not provide any evidence for an increased risk of cellulitis or eczema with denosumab.26
Although randomized controlled trials reported more cases of neoplasms in the denosumab than in the placebo groups, meta-analyses have failed to detect a statistically significant difference (risk ratio 1.11, 95% CI 0.91–1.36).46 The overall incidence of adverse and serious adverse events reported in the 8-year extension of FREEDOM were consistent with data reported in the previous extension studies.25
In the FREEDOM extension trial, four events in the long-term group (n = 2,343), and two in the crossover group (n = 2,207) were adjudicated as being consistent with osteonecrosis of the jaw.26 One mid-shaft fracture in the crossover group was adjudicated as an atypical femoral fracture. There were, however, no reports of osteonecrosis of the jaw or atypical femoral fracture in the long-term phase 2 trial after 8 years of follow-up.30 By September 2013, postmarketing safety surveillance data for denosumab (estimated exposure of 1.2 million patient-years) had recorded four cases of atypical femoral fracture. All four patients had previously been on bisphosphonates. There were also 32 reports of osteonecrosis of the jaw.47
Denosumab’s manufacturer aims to communicate the risks of treatment to health care professionals and patients. Information is available online at www.proliahcp.com/risk-evaluation-mitigation-strategy/.
WHAT ARE THE PRECAUTIONS?
Several precautions need to be taken when considering treatment with denosumab.
Antiresorptives can aggravate hypocalcemia by inhibiting bone turnover. Serum calcium should therefore be checked and preexisting hypocalcemia should be corrected before starting denosumab.48
Denosumab is contraindicated in women who are pregnant or are planning to become pregnant, as fetal loss and teratogenicity have been reported in animal experiments. (Denosumab is unlikely to be used in premenopausal women, as it is not approved for use in this group.)
There are no data on excretion of denosumab in human milk, so it should not be given to nursing mothers.
Renal impairment is not a contraindication, and no dose adjustment is necessary (even for patients on renal replacement therapy), as denosumab, being an antibody, is eliminated through the reticuloendothelial system.49,50 However, in practice, any antiresorptive agent should be used with caution in patients with severe renal impairment because of the possible presence of adynamic bone disease. Further reduction of bone turnover would be detrimental in such patients. Also, severe hypocalcemia has been reported in patients with a creatinine clearance rate less than 30 mL/min and in those receiving dialysis.51,52 Postmarketing surveillance data have reported eight cases of severe symptomatic hypocalcemia, of which seven were in patients with chronic kidney disease.47
The manufacturer suggests that patients receive a dental examination with appropriate preventive dentistry before starting denosumab to reduce the incidence of osteonecrosis of the jaw, despite the lack of evidence in support of this strategy. The American Dental Association recommends regular dental visits and maintenance of good oral hygiene for patients already established on antiresorptive therapy.53,54
SHOULD PATIENTS ON DENOSUMAB BE OFFERED A DRUG HOLIDAY?
A drug holiday (temporary discontinuation of the drug after a certain duration of treatment) has been proposed for patients receiving bisphosphonates because of the risk of atypical femoral fracture and osteonecrosis of the jaw (although small) consequent to long-term continuous suppression of bone turnover.55 The antifracture efficacy of bisphosphonates is likely to persist for an unknown length of time after discontinuation because of their long skeletal half-life, while the risks gradually diminish.
By contrast, denosumab targets RANKL in the extracellular fluid and does not become embedded within the bone tissue.56 Pharmacokinetic studies have shown that denosumab has a rapid offset of action, with a half-life of only 26 days and biological activity lasting only 6 months.57 The results of a phase 2 extension study suggest that bone mineral density starts to decline and bone turnover markers start to rise within 12 months of discontinuing denosumab.58
Although fracture risk did not increase in those who were randomized to stopping the treatment and bone mineral density increased further when treatment was restarted, a drug holiday cannot presently be recommended for patients receiving denosumab because of the lack of supportive data.
HOW COST-EFFECTIVE IS DENOSUMAB?
The wholesale acquisition cost is $825 per 60-mg prefilled syringe of denosumab, although this may vary depending on where the drug is obtained. This does not include physician-related service costs associated with administration of denosumab.
Cost-effectiveness analyses conducted in the United States, the United Kingdom, and Sweden have all concluded that denosumab would offer a cost-effective alternative to other osteoporosis medications for primary prevention and secondary prevention of fractures.59–61
The Swedish study also incorporated adherence in the cost-effectiveness model and showed that denosumab was a cost-effective alternative to oral bisphosphonates, particularly for patients who were not expected to adhere well to oral treatments.61
WHICH OSTEOPOROSIS PATIENTS ARE CANDIDATES FOR DENOSUMAB?
The FDA has approved denosumab for the treatment of postmenopausal women and men at high risk of fracture (defined as having a history of osteoporotic fracture or multiple risk factors for fracture), or in those who cannot tolerate other osteoporosis medications or for whom other medications have failed.
Denosumab is also approved for men at high risk of fracture receiving androgen deprivation therapy for nonmetastatic prostate cancer, and for women at high risk of fracture receiving adjuvant aromatase inhibitor therapy for breast cancer.
WHAT DO THE GUIDELINES RECOMMEND?
The National Osteoporosis Foundation guidelines recommend pharmacologic treatment for patients with hip or vertebral fractures (clinical or asymptomatic); T scores lower than –2.5 at the femoral neck, total hip, or lumbar spine; and those with a 10-year probability of hip fracture of more than 3% or of a major osteoporotic fracture more than 20% based on the US-adapted FRAX calculator.62 The American College of Endocrinology guidelines have proposed similar thresholds for pharmacologic treatment, and they recommend alendronate, risedronate, zoledronate, and denosumab as first-line agents.63
The 2010 Osteoporosis Canada guidelines recommend denosumab, alendronate, risedronate, and zoledronate as first-line therapies for preventing hip, nonvertebral, and vertebral fractures in postmenopausal women (grade A recommendation).64 The National Institute of Health and Clinical Excellence in England and Wales, on the other hand, recommends denosumab only for patients who are unable to take a bisphosphonate.65
PRACTICAL PRESCRIBING TIPS
The patient described at the beginning of this article has already sustained a vertebral compression fracture, and her DXA scan shows T scores in the osteoporotic range. She is therefore at increased risk of another fragility fracture (with a fivefold higher risk of another vertebral fracture). Pharmacologic therapy should be considered. In addition, she should be encouraged to adhere to lifestyle measures such as a healthy diet and regular weight-bearing exercise, her risk of falling should be assessed, and adequate calcium and vitamin D supplementation should be given.
Secondary causes of osteoporosis are present in about 30% of women and 55% of men who have vertebral fractures.66 A complete blood count, erythrocyte sedimentation rate, bone biochemistry, 25-hydroxyvitamin D, thyroid-stimulating hormone, and renal and liver function tests should be requested in all patients. Further tests should be considered depending on the clinical evaluation and results of initial investigations.
Because this patient cannot tolerate oral bisphosphonates, she could be offered the option of annual intravenous zoledronic acid infusions or 6-monthly subcutaneous denosumab injections. In clinical trials, gastrointestinal adverse effects were noted with intravenous bisphosphonates as well, but the adverse effects reported were no different than those with placebo. The potential advantages with denosumab include better bone mineral density gains, adherence and patient satisfaction compared with oral bisphosphonates, convenient twice-yearly administration, safety in patients with renal impairment, and absence of gastrointestinal effects.
Raloxifene, a selective estrogen receptor modulator, has estrogen-like action on the bone and antiestrogen actions on the breast and uterus. Unlike standard hormone replacement therapy, raloxifene can therefore increase bone mineral density without increasing the risk of breast and endometrial cancers. However, it has only been shown to reduce the risk of vertebral fracture, not hip fracture. Hence, it would be a more appropriate choice for younger postmenopausal women. Moreover, it may cause troublesome menopausal symptoms.
Teriparatide, the recombinant parathyroid hormone, is an anabolic agent. It is very expensive, and because of this, guidelines in several countries restrict its use to women with severe osteoporosis and multiple fractures who fail to respond to standard treatments. It cannot be used for longer than 2 years because of its association with osteosarcoma in rats.
If our patient prefers denosumab, therapy should be initiated after appropriate counseling (see precautions above). The dose is 60 mg, given subcutaneously, once every 6 months.
Monitoring
There is no consensus regarding the optimal frequency for monitoring patients on treatment, owing to the lack of prospective trial data. The National Osteoporosis Foundation recommends repeating the bone mineral density measurements about 2 years after starting therapy, and about every 2 years thereafter.62 Some studies suggest that changes in bone mineral density correlate with reduction in fracture risk.67,68 A change in bone mineral density is considered significant when it is greater than the range of error of the densitometer (also known as the least significant change).69 If the bone mineral density is stable or improving, therapy could be continued, but if it is declining and the decline is greater than the least significant change, a change in therapy should be considered if no secondary causes for bone loss are evident (but see What are the areas of uncertainty? below).
The National Osteoporosis Foundation also recommends measuring a bone turnover marker at baseline and then 3 to 6 months later, as its suppression predicts greater bone mineral density responses and fracture risk reduction.70 If there is a decrease of more than 30% in serum carboxy-terminal collagen crosslinks (CTX) or more than 50% in urinary N-telopeptide (NTX),71 the patient can be reassured that the next bone mineral density measurement will be stable or improved. In patients on oral bisphosphonates, measurement of bone turnover markers also provides evidence of compliance.
Clinical trials suggest that a numerical increase in bone mineral density can be expected in most patients on treatment, though this depends on the measurement site and the length of time between examinations. In one phase 3 trial of denosumab in postmenopausal women, only 5% of the participants had unchanged or diminished bone mineral density at the lumbar spine, and 8% at the hip, after 36 months of treatment.72 However, the CTX levels fell to below the lower limit of the reference interval as early as 1 month after commencing treatment in all denosumab-treated patients.68
Hence, bone turnover markers may be a more sensitive indicator of treatment effect than bone mineral density, but this would ultimately need to be evaluated against fracture rates in a real-world setting.
WHAT ARE THE AREAS OF UNCERTAINTY?
There are currently no guidelines for long-term management of patients on denosumab, and also no data to suggest whether patients should be switched to a weaker antiresorptive drug after a certain number of years in order to reduce the possible risk of atypical femoral fracture or osteonecrosis of the jaw.
No head-to-head trials have directly compared the antifracture efficacy of denosumab with that of other standard osteoporosis therapies. The antifracture efficacy and safety of combination therapies involving denosumab are also uncertain. For adherent patients who have a suboptimal response, there is no evidence to guide the further course of action. The International Osteoporosis Foundation guidelines suggest replacing a stronger antiresorptive with an anabolic agent, but acknowledge that this is only based on expert opinion.71
The very-long-term effects (beyond 8 years) of continuous denosumab administration on increasing the risk of atypical femoral fracture, osteonecrosis of the jaw, malignancy, or infection or the duration after which risks would start to outweigh benefits is not known. However, postmarketing safety data continue to be collected through the voluntary Post-marketing Active Safety Surveillance Program (for prespecified adverse events) in addition to the FDA’s MedWatch program.
CASE PROGRESSION
The patient described in the vignette is presented with two options—zoledronate and denosumab. She chooses denosumab. Her renal function and serum calcium are checked and are found to be satisfactory. She undergoes a dental examination, which is also satisfactory. She is counseled about the possible increased risk of infection, and then she is started on 60 mg of denosumab subcutaneously, once every 6 months.
When reviewed after 2 years, she reports no further fractures. Her bone mineral density remains stable compared with the values obtained before starting treatment. She reports no adverse effects and is happy to continue with denosumab.
- Johnell O, Kanis JA. An estimate of the worldwide prevalence and disability associated with osteoporotic fractures. Osteoporos Int 2006; 17:1726–1733.
- Gullberg B, Johnell O, Kanis JA. World-wide projections for hip fracture. Osteoporos Int 1997; 7:407–413.
- Burge R, Dawson-Hughes B, Solomon DH, Wong JB, King A, Tosteson A. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005–2025. J Bone Miner Res 2007; 22:465–475.
- Melton LJ 3rd, Chrischilles EA, Cooper C, Lane AW, Riggs BL. Perspective. How many women have osteoporosis? J Bone Miner Res 1992; 7:1005–1010.
- Abrahamsen B, van Staa T, Ariely R, Olson M, Cooper C. Excess mortality following hip fracture: a systematic epidemiological review. Osteoporos Int 2009; 20:1633–1650.
- Jalava T, Sarna S, Pylkkänen L, et al. Association between vertebral fracture and increased mortality in osteoporotic patients. J Bone Miner Res 2003; 18:1254–1260.
- Ismail AA, O’Neill TW, Cooper C, et al. Mortality associated with vertebral deformity in men and women: results from the European Prospective Osteoporosis Study (EPOS). Osteoporos Int 1998; 8:291–297.
- Ensrud KE, Thompson DE, Cauley JA, et al. Prevalent vertebral deformities predict mortality and hospitalization in older women with low bone mass. Fracture Intervention Trial Research Group. J Am Geriatr Soc 2000; 48:241–249.
- Kado DM, Browner WS, Palermo L, Nevitt MC, Genant HK, Cummings SR. Vertebral fractures and mortality in older women: a prospective study. Study of Osteoporotic Fractures Research Group. Arch Intern Med 1999; 159:1215–1220.
- Lindsay R, Silverman SL, Cooper C, et al. Risk of new vertebral fracture in the year following a fracture. JAMA 2001; 285:320–323.
- Ross PD, Davis JW, Epstein RS, Wasnich RD. Pre-existing fractures and bone mass predict vertebral fracture incidence in women. Ann Intern Med 1991; 114:919–923.
- Piscitelli P, Iolascon G, Argentiero A, et al. Incidence and costs of hip fractures vs strokes and acute myocardial infarction in Italy: comparative analysis based on national hospitalization records. Clin Interv Aging 2012; 7:575–583.
- Johnell O, Kanis JA, Jonsson B, Oden A, Johansson H, De Laet C. The burden of hospitalised fractures in Sweden. Osteoporos Int 2005; 16:222–228.
- Confavreux CB, Canoui-Poitrine F, Schott AM, Ambrosi V, Tainturier V, Chapurlat RD. Persistence at 1 year of oral antiosteoporotic drugs: a prospective study in a comprehensive health insurance database. Eur J Endocrinol 2012; 166:735–741.
- Biswas PN, Wilton LV, Shakir SA. Pharmacovigilance study of alendronate in England. Osteoporos Int 2003; 14:507–514.
- Landfeldt E, Ström O, Robbins S, Borgström F. Adherence to treatment of primary osteoporosis and its association to fractures—the Swedish Adherence Register Analysis (SARA). Osteoporos Int 2012; 23:433–443.
- Sampalis JS, Adachi JD, Rampakakis E, Vaillancourt J, Karellis A, Kindundu C. Long-term impact of adherence to oral bisphosphonates on osteoporotic fracture incidence. J Bone Miner Res 2012; 27:202–210.
- Schwarz EM, Ritchlin CT. Clinical development of anti-RANKL therapy. Arthritis Res Ther 2007; 9(suppl 1):S7.
- Hanley DA, Adachi JD, Bell A, Brown V. Denosumab: mechanism of action and clinical outcomes. Int J Clin Pract 2012; 66:1139–1146.
- Bekker PJ, Holloway D, Nakanishi A, Arrighi M, Leese PT, Dunstan CR. The effect of a single dose of osteoprotegerin in postmenopausal women. J Bone Miner Res 2001; 16:348–360.
- Rizzoli R, Body JJ, Brandi ML, et al; International Osteoporosis Foundation Committee of Scientific Advisors Working Group on Cancer-Induced Bone Disease. Cancer-associated bone disease. Osteoporos Int 2013; 24:2929–2953.
- Schwarz P, Rasmussen AQ, Kvist TM, Andersen UB, Jørgensen NR. Paget’s disease of the bone after treatment with denosumab: a case report. Bone 2012; 50:1023–1025.
- Hu MI, Glezerman IG, Leboulleux S, et al. Denosumab for treatment of hypercalcemia of malignancy. J Clin Endocrinol Metab 2014; Jun 10 [Epub ahead of print].
- Cummings SR, San Martin J, McClung MR, et al; FREEDOM Trial. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med 2009; 361:756–765.
- Papapoulos S, Lippuner K, Roux C, et al. Eight years of denosumab treatment in postmenopausal women with osteoporosis: results from the first five years of the FREEDOM extension [abstract]. Presented at the 2013 annual meeting of the American Society for Bone and Mineral Research, Baltimore, MD, October 4–7, 2013.
- Bone HG, Chapurlat R, Brandi ML, et al. The effect of three or six years of denosumab exposure in women with postmenopausal osteoporosis: results from the FREEDOM extension. J Clin Endocrinol Metab 2013; 98:4483–4492.
- McClung MR, Boonen S, Törring O, et al. Effect of denosumab treatment on the risk of fractures in subgroups of women with postmenopausal osteoporosis. J Bone Miner Res 2012; 27:211–218.
- Boonen S, Adachi JD, Man Z, et al. Treatment with denosumab reduces the incidence of new vertebral and hip fractures in postmenopausal women at high risk. J Clin Endocrinol Metab 2011; 96:1727–1736.
- McCloskey EV, Johansson H, Oden A, et al. Denosumab reduces the risk of osteoporotic fractures in postmenopausal women, particularly in those with moderate to high fracture risk as assessed with FRAX. J Bone Miner Res 2012; 27:1480–1486.
- McClung MR, Lewiecki EM, Geller ML, et al. Effect of denosumab on bone mineral density and biochemical markers of bone turnover: 8-year results of a phase 2 clinical trial. Osteoporos Int 2013; 24:227–235.
- McClung MR, Lewiecki EM, Cohen SB, et al; AMG 162 Bone Loss Study Group. Denosumab in postmenopausal women with low bone mineral density. N Engl J Med 2006; 354:821–831.
- Bone HG, Bolognese MA, Yuen CK, et al. Effects of denosumab on bone mineral density and bone turnover in postmenopausal women. J Clin Endocrinol Metab 2008; 93:2149–2157.
- Genant HK, Engelke K, Hanley DA, et al. Denosumab improves density and strength parameters as measured by QCT of the radius in postmenopausal women with low bone mineral density. Bone 2010; 47:131–139.
- Orwoll E, Teglbjærg CS, Langdahl BL, et al. A randomized, placebo-controlled study of the effects of denosumab for the treatment of men with low bone mineral density. J Clin Endocrinol Metab 2012; 97:3161–3169.
- Tsai JN, Uihlein AV, Lee H, et al. Teriparatide and denosumab, alone or combined, in women with postmenopausal osteoporosis: the DATA study randomised trial. Lancet 2013; 382:50–56.
- Smith MR, Egerdie B, Hernández Toriz N, et al; Denosumab HALT Prostate Cancer Study Group. Denosumab in men receiving androgen-deprivation therapy for prostate cancer. N Engl J Med 2009; 361:745–755.
- Ellis GK, Bone HG, Chlebowski R, et al. Randomized trial of denosumab in patients receiving adjuvant aromatase inhibitors for nonmetastatic breast cancer. J Clin Oncol 2008; 26:4875–4882.
- Brown JP, Prince RL, Deal C, et al. Comparison of the effect of denosumab and alendronate on BMD and biochemical markers of bone turnover in postmenopausal women with low bone mass: a randomized, blinded, phase 3 trial. J Bone Miner Res 2009; 24:153–161.
- Kendler DL, Roux C, Benhamou CL, et al. Effects of denosumab on bone mineral density and bone turnover in postmenopausal women transitioning from alendronate therapy. J Bone Miner Res 2010; 25:72–81.
- Seeman E, Delmas PD, Hanley DA, et al. Microarchitectural deterioration of cortical and trabecular bone: differing effects of denosumab and alendronate. J Bone Miner Res 2010; 25:1886–1894.
- Hopkins RB, Goeree R, Pullenayegum E, et al. The relative efficacy of nine osteoporosis medications for reducing the rate of fractures in post-menopausal women. BMC Musculoskelet Disord 2011; 12:209.
- National Institute for Health and Care Excellence (NICE). NICE technology appraisal guidance: TA161. Alendronate, etidronate, risedronate, raloxifene, strontium ranelate and teriparatide for the secondary prevention of osteoporotic fragility fractures in postmenopausal women (amended). http://publications.nice.org.uk/alendronate-etidronate-risedronate-raloxifene-strontium-ranelate-and-teriparatide-for-ta161. Accessed January 9, 2015.
- Freemantle N, Satram-Hoang S, Tang ET, et al; DAPS Investigators. Final results of the DAPS (Denosumab Adherence Preference Satisfaction) study: a 24-month, randomized, crossover comparison with alendronate in postmenopausal women. Osteoporos Int 2012; 23:317–326.
- Lewiecki EM. Safety and tolerability of denosumab for the treatment of postmenopausal osteoporosis. Drug Healthc Patient Saf 2011; 3:79–91.
- Watts NB, Roux C, Modlin JF, et al. Infections in postmenopausal women with osteoporosis treated with denosumab or placebo: coincidence or causal association? Osteoporos Int 2012; 23:327–337.
- von Keyserlingk C, Hopkins R, Anastasilakis A, et al. Clinical efficacy and safety of denosumab in postmenopausal women with low bone mineral density and osteoporosis: a meta-analysis. Semin Arthritis Rheum 2011; 41:178–186.
- Geller M, Wagman RB, Ho PR, et al. Early findings from Prolia postmarketing safety surveillance for atypical femoral fracture, osteonecrosis of the jaw, severe symptomatic hypocalcemia, and anaphylaxis (abstract). Osteoporos Int 2014; 25(suppl 2). OC40; www.wco-iof-esceo.org/sites/ecceo14/docs/wco14-abstractbook.pdf. Accessed January 9, 2015.
- McCormick BB, Davis J, Burns KD. Severe hypocalcemia following denosumab injection in a hemodialysis patient. Am J Kidney Dis 2012; 60:626–628.
- Jamal SA, Ljunggren O, Stehman-Breen C, et al. Effects of denosumab on fracture and bone mineral density by level of kidney function. J Bone Miner Res 2011; 26:1829–1835.
- Block GA, Bone HG, Fang L, Lee E, Padhi D. A single-dose study of denosumab in patients with various degrees of renal impairment. J Bone Miner Res 2012; 27:1471–1479.
- Ungprasert P, Cheungpasitporn W, Srivali N, Kittanamongkolchai W, Bischof EF. Life-threatening hypocalcemia associated with denosumab in a patient with moderate renal insufficiency. Am J Emerg Med 2013; 31:756.e1–e2.
- McCormick BB, Davis J, Burns KD. Severe hypocalcemia following denosumab injection in a hemodialysis patient. Am J Kidney Dis 2012; 60:626–628.
- Rachner TD, Platzbecker U, Felsenberg D, Hofbauer LC. Osteonecrosis of the jaw after osteoporosis therapy with denosumab following long-term bisphosphonate therapy. Mayo Clin Proc 2013; 88:418–419.
- Epstein MS, Ephros HD, Epstein JB. Review of current literature and implications of RANKL inhibitors for oral health care providers. Oral Surg Oral Med Oral Pathol Oral Radiol 2013; 116:e437–e442.
- McClung M, Harris ST, Miller PD, et al. Bisphosphonate therapy for osteoporosis: benefits, risks, and drug holiday. Am J Med 2013; 126:13–20.
- Baron R, Ferrari S, Russell RG. Denosumab and bisphosphonates: different mechanisms of action and effects. Bone 2011; 48:677–692.
- Bekker PJ, Holloway DL, Rasmussen AS, et al. A single-dose placebo-controlled study of AMG 162, a fully human monoclonal antibody to RANKL, in postmenopausal women. J Bone Miner Res 2004; 19:1059–1066.
- Miller PD, Bolognese MA, Lewiecki EM, et al; Amg Bone Loss Study Group. Effect of denosumab on bone density and turnover in postmenopausal women with low bone mass after long-term continued, discontinued, and restarting of therapy: a randomized blinded phase 2 clinical trial. Bone 2008; 43:222–229.
- Parthan A, Kruse M, Yurgin N, Huang J, Viswanathan HN, Taylor D. Cost effectiveness of denosumab versus oral bisphosphonates for postmenopausal osteoporosis in the US. Appl Health Econ Health Policy 2013; 11:485–497.
- Scotland G, Waugh N, Royle P, McNamee P, Henderson R, Hollick R. Denosumab for the prevention of osteoporotic fractures in post-menopausal women: a NICE single technology appraisal. Pharmacoeconomics 2011; 29:951–961.
- Jönsson B, Ström O, Eisman JA, et al. Cost-effectiveness of denosumab for the treatment of postmenopausal osteoporosis. Osteoporos Int 2011; 22:967–982.
- Clinician’s guide to prevention and treatment of osteoporosis. Washington DC: National Osteoporosis Foundation, 2013.
- Watts NB, Bilezikian JP, Camacho PM, et al; AACE Osteoporosis Task Force. American Association of Clinical Endocrinologists Medical Guidelines for Clinical Practice for the diagnosis and treatment of postmenopausal osteoporosis. Endocr Pract 2010;16(suppl 3):1–37.
- Papaioannou A, Morin S, Cheung AM, et al; Scientific Advisory Council of Osteoporosis Canada. 2010 clinical practice guidelines for the diagnosis and management of osteoporosis in Canada: summary. CMAJ 2010; 182:1864–1673.
- National Institute for Health and Care Excellence (NICE). NICE technology appraisal: TA204. Denosumab for the prevention of osteoporotic fractures in postmenopausal women. http://guidance.nice.org.uk/TA204. Accessed January 9, 2015.
- Premaor MO, Compston JE. Testing for secondary causes of osteoporosis. BMJ 2010; 341:c6959.
- Hochberg MC, Ross PD, Black D, et al. Larger increases in bone mineral density during alendronate therapy are associated with a lower risk of new vertebral fractures in women with postmenopausal osteoporosis. Fracture Intervention Trial Research Group. Arthritis Rheum 1999; 42:1246–1254.
- Eastell R, Vrijens B, Cahall DL, Ringe JD, Garnero P, Watts NB. Bone turnover markers and bone mineral density response with risedronate therapy: relationship with fracture risk and patient adherence. J Bone Miner Res 2011; 26:1662–1669.
- Diez-Perez A, Adachi JD, Agnusdei D, et al; IOF CSA Inadequate Responders Working Group. Treatment failure in osteoporosis. Osteoporos Int 2012; 23:2769–2774.
- Eastell R, Barton I, Hannon RA, Chines A, Garnero P, Delmas PD. Relationship of early changes in bone resorption to the reduction in fracture risk with risedronate. J Bone Miner Res 2003; 18:1051–1056.
- Rosen HN, Moses AC, Garber J, Ross DS, Lee SL, Greenspan SL. Utility of biochemical markers of bone turnover in the follow-up of patients treated with bisphosphonates. Calcif Tissue Int 1998; 63:363–368.
- Bolognese MA, Teglbjærg CS, Zanchetta JR, et al. Denosumab significantly increases DXA BMD at both trabecular and cortical sites: results from the FREEDOM study. J Clin Densitom 2013; 16:147–153.
A 68-year-old white woman presents with mid- thoracic back pain. Plain radiographs reveal a compression fracture of the 10th thoracic vertebra. She is diagnosed with osteoporosis on the basis of dual energy x-ray absorptiometry (DXA) scans that show T scores of –2.9 in her lumbar spine and –2.6 in her left femoral neck. Her 10-year probability of fracture is estimated as 23% for major osteoporotic fracture and 5.9% for hip fracture (based on the World Health Organization’s absolute fracture risk assessment tool, adapted for the United States, and available at www.shef.ac.uk/FRAX).
After excluding common secondary causes of osteoporosis, her physician recommends a bisphosphonate to reduce her risk of fracture, but she develops upper-gastrointestinal adverse effects with both alendronate and risedronate despite correctly following the instructions for oral administration.
What should her physician consider next?
OSTEOPOROSIS IS A MAJOR PROBLEM
Osteoporosis is a systemic skeletal disease characterized by low bone mass and microarchitectural deterioration of bone tissue, predisposing to an increased risk of fragility fractures, particularly of the spine, hip, and wrist.
It is a major public health problem, affecting 200 million people throughout the world, with 9 million osteoporotic fractures reported in the year 2000.1 The incidence of hip fracture alone is predicted to rise to 2.6 million by the year 2025, and to 4.5 million by the year 2050.2 In the United States, the total burden was estimated to be about 2 million incident fractures in the year 2005, projected to rise by another 50% by the year 2025,3 primarily because of the aging of the population. Population studies have indeed suggested that about 40% of white women and 13% of white men over the age of 50 are at risk of sustaining an osteoporotic fracture during the remainder of their lifetime.4
The consequences of osteoporotic fractures can be devastating. Hip fractures are associated with a risk of death ranging from 8.4% to 36% during the first year after fracture.5 One-fifth of patients who sustain a hip fracture require long-term nursing home care, and more than half of the survivors do not regain their previous level of independence.
Patients with vertebral fractures are also at increased risk of death, although the results of some studies suggest that this could be the result of comorbid factors.6–9 Vertebral fractures can result in chronic back pain, loss of height from spinal deformity, reduced mobility, loss of self-esteem, and in severe cases, respiratory and digestive problems because of contact between the lower ribs and pelvis.
A person with one vertebral compression fracture is five times more likely to have another vertebral fracture,10 and a person with two or more compression fractures is 12 times more likely.11
The costs of treating osteoporotic fractures are greater than those of treating myocardial infarction or stroke12,13; they include not only direct costs incurred in treating the fracture, but also indirect societal costs owing to the long-term morbidity associated with the fracture. In the United States, the total cost of treating osteoporotic fractures was estimated at $19 billion in the year 2005.3 By 2025, the annual costs are projected to rise by almost 50%.3
A NEED FOR MORE OPTIONS
Until fairly recently, bisphosphonates were the only drugs of first choice, but adherence to oral bisphosphonate therapy is generally poor (< 50% at 1 year),14 most commonly because of dyspepsia,15 and poor adherence has been shown to be associated with increased fracture risk.16,17 Hence the need for additional therapeutic options.
In this review, we discuss denosumab, an antiresorptive drug approved by the US Food and Drug Administration (FDA) in 2010. First, we discuss its mechanism of action, efficacy, and safety, and then we offer recommendations for its use in clinical practice.
WHAT IS DENOSUMAB AND HOW DOES IT WORK?
Bone remodeling is a dynamic process involving a balance between bone resorption by osteoclasts on the one hand and new bone formation by osteoblasts on the other. A net gain in bone occurs when the activity of osteoblasts exceeds that of osteoclasts, and bone loss occurs when there is increased osteoclast activity or reduced osteoblast activity, or both. The activities of osteoblasts and osteoclasts are tightly coupled because of the opposing effects of two sets of proteins, namely, receptor activator of nuclear factor kappa b ligand (RANKL) and osteoprotegerin.
Both RANKL and osteoprotegerin are produced by osteoblasts. RANKL binds to its receptor (RANK) on preosteoclasts and osteoclasts and induces their differentiation and activation, respectively. Osteoprotegerin is the decoy receptor and natural antagonist for RANKL. By binding with RANKL, it blocks its interaction with RANK.18 In healthy individuals, a fine balance between RANKL and osteoprotegerin ensures that bone remodeling is regulated.
In postmenopausal women, estrogen deficiency leads to an imbalance between RANKL and osteoprotegerin (increased RANKL and reduced osteoprotegerin), resulting in net bone loss. This imbalance is also a feature of rheumatoid arthritis, myeloma bone disease, and osteolytic metastatic bone disease; it also occurs in those receiving androgen deprivation therapy for prostate cancer or aromatase inhibitors for breast cancer.
Denosumab is a fully human monoclonal antibody that targets RANKL.19 By binding to RANKL, this drug prevents the maturation and differentiation of preosteoclasts and promotes apoptosis of osteoclasts. Bone resorption is therefore slowed. It was parenteral osteoprotegerin that was initially developed by denosumab’s manufacturer,20 but this approach failed because neutralizing antibodies developed to osteoprotegerin, rendering it ineffective. Development of neutralizing antibodies has thus far not been a problem with denosumab.
Denosumab, with its property of RANKL inhibition, has also been used to prevent skeletal events in patients with bone metastases from solid tumors and to treat unresectable giant cell tumors of the bone (both FDA-approved indications) and hypercalcemia of malignancy. There is limited clinical experience in Paget disease of the bone as well.21–23 These other potential uses of denosumab are beyond the scope of this review.
HOW WELL DOES DENOSUMAB WORK FOR OSTEOPOROSIS?
Several phase 2 and phase 3 randomized controlled trials have evaluated the efficacy of denosumab, but only one, the Fracture Reduction Evaluation of Denosumab in Osteoporosis Every 6 Months (FREEDOM) trial, included fracture reduction as the primary outcome measure. The rest evaluated changes in bone mineral density or in markers of bone turnover, or both.
FREEDOM was a double-blind, randomized controlled trial in 7,808 postmenopausal women with T scores between –2.5 and –4.0 at the lumbar spine or hip.24 Twenty-four percent of the patients had vertebral fractures at baseline. Patients were randomized to receive either denosumab 60 mg (n = 3,902) or placebo (n = 3,906) every 6 months for up to 36 months. All patients also received adequate calcium and vitamin D supplementation.
At 36 months, compared with those who were randomized to receive placebo, those who were randomized to denosumab had lower incidence rates of:
- New vertebral fracture
(2.3% vs 7.2%, risk ratio 0.32,
95% CI 0.26–0.41, P < .001) - Nonvertebral fracture
(6.5% vs 8.0%, risk ratio 0.80,
95% CI 0.67–0.95, P = .01) - Hip fracture
(0.7% vs 1.2%, risk ratio 0.60,
95% CI 0.37–0.97, P = .04).
Increases in bone mineral density at the lumbar spine and hip, and decreases in bone turnover markers were also significantly greater in the denosumab group. The number needed to treat to prevent one new fracture over 3 years was 21 for vertebral fracture, 67 for nonvertebral fracture, and 200 for hip fracture, reflecting the relatively low event rate in the study.
In an open-label extension of the FREEDOM trial, the fracture incidence rates among participants who continued to receive denosumab for an additional 5 years remained low, and still below those projected for a “virtual placebo cohort” (total duration of exposure of 8 years). The rates among participants who switched from placebo to denosumab were similar to those of the denosumab group from the parent trial.25,26
A subgroup analysis of the FREEDOM trial suggested that denosumab reduced the risk of new vertebral fractures irrespective of age, body mass index, femoral neck bone mineral density, prevalent vertebral fractures, or prior nonvertebral fractures (risk ratio 0.32; 95% CI 0.26–0.41, P < .001), whereas the risk of nonvertebral fractures was only reduced in those women with body mass indices less than 25 kg/m2, femoral neck bone mineral density T scores less than –2.5, and in those without a prevalent vertebral fracture.27
A post hoc analysis revealed that denosumab significantly reduced the risk of new vertebral and hip fractures even in subgroups of women at higher risk of fracture.28 At 10% fracture probability (as estimated by the FRAX risk calculator), denosumab reduced the fracture risk by 11% (P = .629), whereas at 30% probability (moderate to high risk), the reduction was 50% (P = .001).29
Other phase 2 and phase 3 trials, in postmenopausal women with low bone mineral density, demonstrated that compared with placebo, denosumab significantly increased bone mineral density at all skeletal sites, increased volumetric bone mineral density at the distal radius, improved hip structural analysis parameters, and reduced bone turnover markers.30–33 Increases in bone mineral density and reductions in bone turnover markers with denosumab have been shown in men as well.34
In a randomized controlled trial,35 improvement in bone mineral density was better in those who received the combination of denosumab and teriparatide than in those who received either drug on its own.
Denosumab has also been shown to reduce the incidence of new vertebral fractures and improve bone mineral density in men receiving androgen-deprivation therapy for nonmetastatic prostate cancer,36 and to improve bone mineral density in women with metastatic breast cancer and low bone mass who were receiving adjuvant aromatase inhibitor therapy.37
HOW DOES DENOSUMAB COMPARE WITH OTHER OSTEOPOROSIS DRUGS?
A double-blind randomized controlled trial in postmenopausal women with low bone mass demonstrated that denosumab was superior to alendronate in improving bone mineral density at all skeletal sites (3.5% vs 2.6% for total hip bone mineral density, P < .0001).38
Another double-blind trial demonstrated that in patients previously treated with alendronate, switching to denosumab resulted in significantly greater increases in bone mineral density at all skeletal sites compared with continuing with alendronate (P < .0001).39
Denosumab has also been shown to be superior to alendronate in improving cortical bone mineral density, as measured by quantitative computed tomography.40
No trial has directly compared the efficacy of denosumab with other osteoporosis drugs in reducing fracture risk, but a systematic literature review of multiple databases,41 comparing the antifracture efficacy of nine osteoporosis drugs, concluded that teriparatide, zoledronic acid, and denosumab had the highest probabilities of being most efficacious for nonvertebral and vertebral fractures, with the greatest effect sizes. Indirect comparisons of the relative risk of fracture with denosumab (based on the results of FREEDOM), alendronate, risedronate, raloxifene, and strontium (based on a meta-analysis of randomized controlled trials) are presented in Table 1.42
A 2-year randomized, open-label, crossover study43 randomized patients to receive either denosumab followed by alendronate or alendronate followed by denosumab over successive 12-month periods. The results suggested that postmenopausal women with osteoporosis were more adherent, compliant, and persistent with denosumab therapy (a subcutaneous injection every 6 months) than with alendronate therapy in the form of oral tablets, self-administered weekly (7.5% nonadherence vs 36.5% at the end of 2 years). After receiving both treatments, women reported greater satisfaction with denosumab, with 92.4% preferring it over oral alendronate. Bone mineral density remained stable when patients were switched from denosumab to alendronate, but improved further when they were switched from alendronate to denosumab.
HOW SAFE IS DENOSUMAB?
The most frequent adverse events with denosumab reported in the long-term extension of one phase 2 study were upper respiratory tract infections (13.5%), arthralgia (11.5%), and back pain (9.0%).30
Increased risk of infection, cancer, and dermatologic reactions has been a concern, as RANKL and RANK are expressed by a wide variety of cells, including T lymphocytes, B cells, and dendritic cells.44 However, there were no significant differences in the overall incidences of adverse events between patients who received denosumab and those who received placebo or alendronate in any of the phase 2, phase 3, or extension studies.
In the FREEDOM trial,24 there was no significant difference between the two groups in the overall incidence of infection (52.9% with denosumab vs 54.4% with placebo, P = .17), or serious infection (4.1% with denosumab vs 3.4% with placebo, P = .14), although the incidence of “serious” cellulitis requiring hospitalization was higher in the denosumab group (0.3% vs < 0.1%, P = .002). There were more serious infections involving the gastrointestinal system, urinary tract, and ear and cases of endocarditis in the denosumab group, but the number of events was small, and there was no relationship with the timing of administration or duration of exposure to denosumab.45 Eczema was more common in the denosumab than in the placebo group (3.0% vs 1.7%, P < .001), but the extension data from the first 3 years did not provide any evidence for an increased risk of cellulitis or eczema with denosumab.26
Although randomized controlled trials reported more cases of neoplasms in the denosumab than in the placebo groups, meta-analyses have failed to detect a statistically significant difference (risk ratio 1.11, 95% CI 0.91–1.36).46 The overall incidence of adverse and serious adverse events reported in the 8-year extension of FREEDOM were consistent with data reported in the previous extension studies.25
In the FREEDOM extension trial, four events in the long-term group (n = 2,343), and two in the crossover group (n = 2,207) were adjudicated as being consistent with osteonecrosis of the jaw.26 One mid-shaft fracture in the crossover group was adjudicated as an atypical femoral fracture. There were, however, no reports of osteonecrosis of the jaw or atypical femoral fracture in the long-term phase 2 trial after 8 years of follow-up.30 By September 2013, postmarketing safety surveillance data for denosumab (estimated exposure of 1.2 million patient-years) had recorded four cases of atypical femoral fracture. All four patients had previously been on bisphosphonates. There were also 32 reports of osteonecrosis of the jaw.47
Denosumab’s manufacturer aims to communicate the risks of treatment to health care professionals and patients. Information is available online at www.proliahcp.com/risk-evaluation-mitigation-strategy/.
WHAT ARE THE PRECAUTIONS?
Several precautions need to be taken when considering treatment with denosumab.
Antiresorptives can aggravate hypocalcemia by inhibiting bone turnover. Serum calcium should therefore be checked and preexisting hypocalcemia should be corrected before starting denosumab.48
Denosumab is contraindicated in women who are pregnant or are planning to become pregnant, as fetal loss and teratogenicity have been reported in animal experiments. (Denosumab is unlikely to be used in premenopausal women, as it is not approved for use in this group.)
There are no data on excretion of denosumab in human milk, so it should not be given to nursing mothers.
Renal impairment is not a contraindication, and no dose adjustment is necessary (even for patients on renal replacement therapy), as denosumab, being an antibody, is eliminated through the reticuloendothelial system.49,50 However, in practice, any antiresorptive agent should be used with caution in patients with severe renal impairment because of the possible presence of adynamic bone disease. Further reduction of bone turnover would be detrimental in such patients. Also, severe hypocalcemia has been reported in patients with a creatinine clearance rate less than 30 mL/min and in those receiving dialysis.51,52 Postmarketing surveillance data have reported eight cases of severe symptomatic hypocalcemia, of which seven were in patients with chronic kidney disease.47
The manufacturer suggests that patients receive a dental examination with appropriate preventive dentistry before starting denosumab to reduce the incidence of osteonecrosis of the jaw, despite the lack of evidence in support of this strategy. The American Dental Association recommends regular dental visits and maintenance of good oral hygiene for patients already established on antiresorptive therapy.53,54
SHOULD PATIENTS ON DENOSUMAB BE OFFERED A DRUG HOLIDAY?
A drug holiday (temporary discontinuation of the drug after a certain duration of treatment) has been proposed for patients receiving bisphosphonates because of the risk of atypical femoral fracture and osteonecrosis of the jaw (although small) consequent to long-term continuous suppression of bone turnover.55 The antifracture efficacy of bisphosphonates is likely to persist for an unknown length of time after discontinuation because of their long skeletal half-life, while the risks gradually diminish.
By contrast, denosumab targets RANKL in the extracellular fluid and does not become embedded within the bone tissue.56 Pharmacokinetic studies have shown that denosumab has a rapid offset of action, with a half-life of only 26 days and biological activity lasting only 6 months.57 The results of a phase 2 extension study suggest that bone mineral density starts to decline and bone turnover markers start to rise within 12 months of discontinuing denosumab.58
Although fracture risk did not increase in those who were randomized to stopping the treatment and bone mineral density increased further when treatment was restarted, a drug holiday cannot presently be recommended for patients receiving denosumab because of the lack of supportive data.
HOW COST-EFFECTIVE IS DENOSUMAB?
The wholesale acquisition cost is $825 per 60-mg prefilled syringe of denosumab, although this may vary depending on where the drug is obtained. This does not include physician-related service costs associated with administration of denosumab.
Cost-effectiveness analyses conducted in the United States, the United Kingdom, and Sweden have all concluded that denosumab would offer a cost-effective alternative to other osteoporosis medications for primary prevention and secondary prevention of fractures.59–61
The Swedish study also incorporated adherence in the cost-effectiveness model and showed that denosumab was a cost-effective alternative to oral bisphosphonates, particularly for patients who were not expected to adhere well to oral treatments.61
WHICH OSTEOPOROSIS PATIENTS ARE CANDIDATES FOR DENOSUMAB?
The FDA has approved denosumab for the treatment of postmenopausal women and men at high risk of fracture (defined as having a history of osteoporotic fracture or multiple risk factors for fracture), or in those who cannot tolerate other osteoporosis medications or for whom other medications have failed.
Denosumab is also approved for men at high risk of fracture receiving androgen deprivation therapy for nonmetastatic prostate cancer, and for women at high risk of fracture receiving adjuvant aromatase inhibitor therapy for breast cancer.
WHAT DO THE GUIDELINES RECOMMEND?
The National Osteoporosis Foundation guidelines recommend pharmacologic treatment for patients with hip or vertebral fractures (clinical or asymptomatic); T scores lower than –2.5 at the femoral neck, total hip, or lumbar spine; and those with a 10-year probability of hip fracture of more than 3% or of a major osteoporotic fracture more than 20% based on the US-adapted FRAX calculator.62 The American College of Endocrinology guidelines have proposed similar thresholds for pharmacologic treatment, and they recommend alendronate, risedronate, zoledronate, and denosumab as first-line agents.63
The 2010 Osteoporosis Canada guidelines recommend denosumab, alendronate, risedronate, and zoledronate as first-line therapies for preventing hip, nonvertebral, and vertebral fractures in postmenopausal women (grade A recommendation).64 The National Institute of Health and Clinical Excellence in England and Wales, on the other hand, recommends denosumab only for patients who are unable to take a bisphosphonate.65
PRACTICAL PRESCRIBING TIPS
The patient described at the beginning of this article has already sustained a vertebral compression fracture, and her DXA scan shows T scores in the osteoporotic range. She is therefore at increased risk of another fragility fracture (with a fivefold higher risk of another vertebral fracture). Pharmacologic therapy should be considered. In addition, she should be encouraged to adhere to lifestyle measures such as a healthy diet and regular weight-bearing exercise, her risk of falling should be assessed, and adequate calcium and vitamin D supplementation should be given.
Secondary causes of osteoporosis are present in about 30% of women and 55% of men who have vertebral fractures.66 A complete blood count, erythrocyte sedimentation rate, bone biochemistry, 25-hydroxyvitamin D, thyroid-stimulating hormone, and renal and liver function tests should be requested in all patients. Further tests should be considered depending on the clinical evaluation and results of initial investigations.
Because this patient cannot tolerate oral bisphosphonates, she could be offered the option of annual intravenous zoledronic acid infusions or 6-monthly subcutaneous denosumab injections. In clinical trials, gastrointestinal adverse effects were noted with intravenous bisphosphonates as well, but the adverse effects reported were no different than those with placebo. The potential advantages with denosumab include better bone mineral density gains, adherence and patient satisfaction compared with oral bisphosphonates, convenient twice-yearly administration, safety in patients with renal impairment, and absence of gastrointestinal effects.
Raloxifene, a selective estrogen receptor modulator, has estrogen-like action on the bone and antiestrogen actions on the breast and uterus. Unlike standard hormone replacement therapy, raloxifene can therefore increase bone mineral density without increasing the risk of breast and endometrial cancers. However, it has only been shown to reduce the risk of vertebral fracture, not hip fracture. Hence, it would be a more appropriate choice for younger postmenopausal women. Moreover, it may cause troublesome menopausal symptoms.
Teriparatide, the recombinant parathyroid hormone, is an anabolic agent. It is very expensive, and because of this, guidelines in several countries restrict its use to women with severe osteoporosis and multiple fractures who fail to respond to standard treatments. It cannot be used for longer than 2 years because of its association with osteosarcoma in rats.
If our patient prefers denosumab, therapy should be initiated after appropriate counseling (see precautions above). The dose is 60 mg, given subcutaneously, once every 6 months.
Monitoring
There is no consensus regarding the optimal frequency for monitoring patients on treatment, owing to the lack of prospective trial data. The National Osteoporosis Foundation recommends repeating the bone mineral density measurements about 2 years after starting therapy, and about every 2 years thereafter.62 Some studies suggest that changes in bone mineral density correlate with reduction in fracture risk.67,68 A change in bone mineral density is considered significant when it is greater than the range of error of the densitometer (also known as the least significant change).69 If the bone mineral density is stable or improving, therapy could be continued, but if it is declining and the decline is greater than the least significant change, a change in therapy should be considered if no secondary causes for bone loss are evident (but see What are the areas of uncertainty? below).
The National Osteoporosis Foundation also recommends measuring a bone turnover marker at baseline and then 3 to 6 months later, as its suppression predicts greater bone mineral density responses and fracture risk reduction.70 If there is a decrease of more than 30% in serum carboxy-terminal collagen crosslinks (CTX) or more than 50% in urinary N-telopeptide (NTX),71 the patient can be reassured that the next bone mineral density measurement will be stable or improved. In patients on oral bisphosphonates, measurement of bone turnover markers also provides evidence of compliance.
Clinical trials suggest that a numerical increase in bone mineral density can be expected in most patients on treatment, though this depends on the measurement site and the length of time between examinations. In one phase 3 trial of denosumab in postmenopausal women, only 5% of the participants had unchanged or diminished bone mineral density at the lumbar spine, and 8% at the hip, after 36 months of treatment.72 However, the CTX levels fell to below the lower limit of the reference interval as early as 1 month after commencing treatment in all denosumab-treated patients.68
Hence, bone turnover markers may be a more sensitive indicator of treatment effect than bone mineral density, but this would ultimately need to be evaluated against fracture rates in a real-world setting.
WHAT ARE THE AREAS OF UNCERTAINTY?
There are currently no guidelines for long-term management of patients on denosumab, and also no data to suggest whether patients should be switched to a weaker antiresorptive drug after a certain number of years in order to reduce the possible risk of atypical femoral fracture or osteonecrosis of the jaw.
No head-to-head trials have directly compared the antifracture efficacy of denosumab with that of other standard osteoporosis therapies. The antifracture efficacy and safety of combination therapies involving denosumab are also uncertain. For adherent patients who have a suboptimal response, there is no evidence to guide the further course of action. The International Osteoporosis Foundation guidelines suggest replacing a stronger antiresorptive with an anabolic agent, but acknowledge that this is only based on expert opinion.71
The very-long-term effects (beyond 8 years) of continuous denosumab administration on increasing the risk of atypical femoral fracture, osteonecrosis of the jaw, malignancy, or infection or the duration after which risks would start to outweigh benefits is not known. However, postmarketing safety data continue to be collected through the voluntary Post-marketing Active Safety Surveillance Program (for prespecified adverse events) in addition to the FDA’s MedWatch program.
CASE PROGRESSION
The patient described in the vignette is presented with two options—zoledronate and denosumab. She chooses denosumab. Her renal function and serum calcium are checked and are found to be satisfactory. She undergoes a dental examination, which is also satisfactory. She is counseled about the possible increased risk of infection, and then she is started on 60 mg of denosumab subcutaneously, once every 6 months.
When reviewed after 2 years, she reports no further fractures. Her bone mineral density remains stable compared with the values obtained before starting treatment. She reports no adverse effects and is happy to continue with denosumab.
A 68-year-old white woman presents with mid- thoracic back pain. Plain radiographs reveal a compression fracture of the 10th thoracic vertebra. She is diagnosed with osteoporosis on the basis of dual energy x-ray absorptiometry (DXA) scans that show T scores of –2.9 in her lumbar spine and –2.6 in her left femoral neck. Her 10-year probability of fracture is estimated as 23% for major osteoporotic fracture and 5.9% for hip fracture (based on the World Health Organization’s absolute fracture risk assessment tool, adapted for the United States, and available at www.shef.ac.uk/FRAX).
After excluding common secondary causes of osteoporosis, her physician recommends a bisphosphonate to reduce her risk of fracture, but she develops upper-gastrointestinal adverse effects with both alendronate and risedronate despite correctly following the instructions for oral administration.
What should her physician consider next?
OSTEOPOROSIS IS A MAJOR PROBLEM
Osteoporosis is a systemic skeletal disease characterized by low bone mass and microarchitectural deterioration of bone tissue, predisposing to an increased risk of fragility fractures, particularly of the spine, hip, and wrist.
It is a major public health problem, affecting 200 million people throughout the world, with 9 million osteoporotic fractures reported in the year 2000.1 The incidence of hip fracture alone is predicted to rise to 2.6 million by the year 2025, and to 4.5 million by the year 2050.2 In the United States, the total burden was estimated to be about 2 million incident fractures in the year 2005, projected to rise by another 50% by the year 2025,3 primarily because of the aging of the population. Population studies have indeed suggested that about 40% of white women and 13% of white men over the age of 50 are at risk of sustaining an osteoporotic fracture during the remainder of their lifetime.4
The consequences of osteoporotic fractures can be devastating. Hip fractures are associated with a risk of death ranging from 8.4% to 36% during the first year after fracture.5 One-fifth of patients who sustain a hip fracture require long-term nursing home care, and more than half of the survivors do not regain their previous level of independence.
Patients with vertebral fractures are also at increased risk of death, although the results of some studies suggest that this could be the result of comorbid factors.6–9 Vertebral fractures can result in chronic back pain, loss of height from spinal deformity, reduced mobility, loss of self-esteem, and in severe cases, respiratory and digestive problems because of contact between the lower ribs and pelvis.
A person with one vertebral compression fracture is five times more likely to have another vertebral fracture,10 and a person with two or more compression fractures is 12 times more likely.11
The costs of treating osteoporotic fractures are greater than those of treating myocardial infarction or stroke12,13; they include not only direct costs incurred in treating the fracture, but also indirect societal costs owing to the long-term morbidity associated with the fracture. In the United States, the total cost of treating osteoporotic fractures was estimated at $19 billion in the year 2005.3 By 2025, the annual costs are projected to rise by almost 50%.3
A NEED FOR MORE OPTIONS
Until fairly recently, bisphosphonates were the only drugs of first choice, but adherence to oral bisphosphonate therapy is generally poor (< 50% at 1 year),14 most commonly because of dyspepsia,15 and poor adherence has been shown to be associated with increased fracture risk.16,17 Hence the need for additional therapeutic options.
In this review, we discuss denosumab, an antiresorptive drug approved by the US Food and Drug Administration (FDA) in 2010. First, we discuss its mechanism of action, efficacy, and safety, and then we offer recommendations for its use in clinical practice.
WHAT IS DENOSUMAB AND HOW DOES IT WORK?
Bone remodeling is a dynamic process involving a balance between bone resorption by osteoclasts on the one hand and new bone formation by osteoblasts on the other. A net gain in bone occurs when the activity of osteoblasts exceeds that of osteoclasts, and bone loss occurs when there is increased osteoclast activity or reduced osteoblast activity, or both. The activities of osteoblasts and osteoclasts are tightly coupled because of the opposing effects of two sets of proteins, namely, receptor activator of nuclear factor kappa b ligand (RANKL) and osteoprotegerin.
Both RANKL and osteoprotegerin are produced by osteoblasts. RANKL binds to its receptor (RANK) on preosteoclasts and osteoclasts and induces their differentiation and activation, respectively. Osteoprotegerin is the decoy receptor and natural antagonist for RANKL. By binding with RANKL, it blocks its interaction with RANK.18 In healthy individuals, a fine balance between RANKL and osteoprotegerin ensures that bone remodeling is regulated.
In postmenopausal women, estrogen deficiency leads to an imbalance between RANKL and osteoprotegerin (increased RANKL and reduced osteoprotegerin), resulting in net bone loss. This imbalance is also a feature of rheumatoid arthritis, myeloma bone disease, and osteolytic metastatic bone disease; it also occurs in those receiving androgen deprivation therapy for prostate cancer or aromatase inhibitors for breast cancer.
Denosumab is a fully human monoclonal antibody that targets RANKL.19 By binding to RANKL, this drug prevents the maturation and differentiation of preosteoclasts and promotes apoptosis of osteoclasts. Bone resorption is therefore slowed. It was parenteral osteoprotegerin that was initially developed by denosumab’s manufacturer,20 but this approach failed because neutralizing antibodies developed to osteoprotegerin, rendering it ineffective. Development of neutralizing antibodies has thus far not been a problem with denosumab.
Denosumab, with its property of RANKL inhibition, has also been used to prevent skeletal events in patients with bone metastases from solid tumors and to treat unresectable giant cell tumors of the bone (both FDA-approved indications) and hypercalcemia of malignancy. There is limited clinical experience in Paget disease of the bone as well.21–23 These other potential uses of denosumab are beyond the scope of this review.
HOW WELL DOES DENOSUMAB WORK FOR OSTEOPOROSIS?
Several phase 2 and phase 3 randomized controlled trials have evaluated the efficacy of denosumab, but only one, the Fracture Reduction Evaluation of Denosumab in Osteoporosis Every 6 Months (FREEDOM) trial, included fracture reduction as the primary outcome measure. The rest evaluated changes in bone mineral density or in markers of bone turnover, or both.
FREEDOM was a double-blind, randomized controlled trial in 7,808 postmenopausal women with T scores between –2.5 and –4.0 at the lumbar spine or hip.24 Twenty-four percent of the patients had vertebral fractures at baseline. Patients were randomized to receive either denosumab 60 mg (n = 3,902) or placebo (n = 3,906) every 6 months for up to 36 months. All patients also received adequate calcium and vitamin D supplementation.
At 36 months, compared with those who were randomized to receive placebo, those who were randomized to denosumab had lower incidence rates of:
- New vertebral fracture
(2.3% vs 7.2%, risk ratio 0.32,
95% CI 0.26–0.41, P < .001) - Nonvertebral fracture
(6.5% vs 8.0%, risk ratio 0.80,
95% CI 0.67–0.95, P = .01) - Hip fracture
(0.7% vs 1.2%, risk ratio 0.60,
95% CI 0.37–0.97, P = .04).
Increases in bone mineral density at the lumbar spine and hip, and decreases in bone turnover markers were also significantly greater in the denosumab group. The number needed to treat to prevent one new fracture over 3 years was 21 for vertebral fracture, 67 for nonvertebral fracture, and 200 for hip fracture, reflecting the relatively low event rate in the study.
In an open-label extension of the FREEDOM trial, the fracture incidence rates among participants who continued to receive denosumab for an additional 5 years remained low, and still below those projected for a “virtual placebo cohort” (total duration of exposure of 8 years). The rates among participants who switched from placebo to denosumab were similar to those of the denosumab group from the parent trial.25,26
A subgroup analysis of the FREEDOM trial suggested that denosumab reduced the risk of new vertebral fractures irrespective of age, body mass index, femoral neck bone mineral density, prevalent vertebral fractures, or prior nonvertebral fractures (risk ratio 0.32; 95% CI 0.26–0.41, P < .001), whereas the risk of nonvertebral fractures was only reduced in those women with body mass indices less than 25 kg/m2, femoral neck bone mineral density T scores less than –2.5, and in those without a prevalent vertebral fracture.27
A post hoc analysis revealed that denosumab significantly reduced the risk of new vertebral and hip fractures even in subgroups of women at higher risk of fracture.28 At 10% fracture probability (as estimated by the FRAX risk calculator), denosumab reduced the fracture risk by 11% (P = .629), whereas at 30% probability (moderate to high risk), the reduction was 50% (P = .001).29
Other phase 2 and phase 3 trials, in postmenopausal women with low bone mineral density, demonstrated that compared with placebo, denosumab significantly increased bone mineral density at all skeletal sites, increased volumetric bone mineral density at the distal radius, improved hip structural analysis parameters, and reduced bone turnover markers.30–33 Increases in bone mineral density and reductions in bone turnover markers with denosumab have been shown in men as well.34
In a randomized controlled trial,35 improvement in bone mineral density was better in those who received the combination of denosumab and teriparatide than in those who received either drug on its own.
Denosumab has also been shown to reduce the incidence of new vertebral fractures and improve bone mineral density in men receiving androgen-deprivation therapy for nonmetastatic prostate cancer,36 and to improve bone mineral density in women with metastatic breast cancer and low bone mass who were receiving adjuvant aromatase inhibitor therapy.37
HOW DOES DENOSUMAB COMPARE WITH OTHER OSTEOPOROSIS DRUGS?
A double-blind randomized controlled trial in postmenopausal women with low bone mass demonstrated that denosumab was superior to alendronate in improving bone mineral density at all skeletal sites (3.5% vs 2.6% for total hip bone mineral density, P < .0001).38
Another double-blind trial demonstrated that in patients previously treated with alendronate, switching to denosumab resulted in significantly greater increases in bone mineral density at all skeletal sites compared with continuing with alendronate (P < .0001).39
Denosumab has also been shown to be superior to alendronate in improving cortical bone mineral density, as measured by quantitative computed tomography.40
No trial has directly compared the efficacy of denosumab with other osteoporosis drugs in reducing fracture risk, but a systematic literature review of multiple databases,41 comparing the antifracture efficacy of nine osteoporosis drugs, concluded that teriparatide, zoledronic acid, and denosumab had the highest probabilities of being most efficacious for nonvertebral and vertebral fractures, with the greatest effect sizes. Indirect comparisons of the relative risk of fracture with denosumab (based on the results of FREEDOM), alendronate, risedronate, raloxifene, and strontium (based on a meta-analysis of randomized controlled trials) are presented in Table 1.42
A 2-year randomized, open-label, crossover study43 randomized patients to receive either denosumab followed by alendronate or alendronate followed by denosumab over successive 12-month periods. The results suggested that postmenopausal women with osteoporosis were more adherent, compliant, and persistent with denosumab therapy (a subcutaneous injection every 6 months) than with alendronate therapy in the form of oral tablets, self-administered weekly (7.5% nonadherence vs 36.5% at the end of 2 years). After receiving both treatments, women reported greater satisfaction with denosumab, with 92.4% preferring it over oral alendronate. Bone mineral density remained stable when patients were switched from denosumab to alendronate, but improved further when they were switched from alendronate to denosumab.
HOW SAFE IS DENOSUMAB?
The most frequent adverse events with denosumab reported in the long-term extension of one phase 2 study were upper respiratory tract infections (13.5%), arthralgia (11.5%), and back pain (9.0%).30
Increased risk of infection, cancer, and dermatologic reactions has been a concern, as RANKL and RANK are expressed by a wide variety of cells, including T lymphocytes, B cells, and dendritic cells.44 However, there were no significant differences in the overall incidences of adverse events between patients who received denosumab and those who received placebo or alendronate in any of the phase 2, phase 3, or extension studies.
In the FREEDOM trial,24 there was no significant difference between the two groups in the overall incidence of infection (52.9% with denosumab vs 54.4% with placebo, P = .17), or serious infection (4.1% with denosumab vs 3.4% with placebo, P = .14), although the incidence of “serious” cellulitis requiring hospitalization was higher in the denosumab group (0.3% vs < 0.1%, P = .002). There were more serious infections involving the gastrointestinal system, urinary tract, and ear and cases of endocarditis in the denosumab group, but the number of events was small, and there was no relationship with the timing of administration or duration of exposure to denosumab.45 Eczema was more common in the denosumab than in the placebo group (3.0% vs 1.7%, P < .001), but the extension data from the first 3 years did not provide any evidence for an increased risk of cellulitis or eczema with denosumab.26
Although randomized controlled trials reported more cases of neoplasms in the denosumab than in the placebo groups, meta-analyses have failed to detect a statistically significant difference (risk ratio 1.11, 95% CI 0.91–1.36).46 The overall incidence of adverse and serious adverse events reported in the 8-year extension of FREEDOM were consistent with data reported in the previous extension studies.25
In the FREEDOM extension trial, four events in the long-term group (n = 2,343), and two in the crossover group (n = 2,207) were adjudicated as being consistent with osteonecrosis of the jaw.26 One mid-shaft fracture in the crossover group was adjudicated as an atypical femoral fracture. There were, however, no reports of osteonecrosis of the jaw or atypical femoral fracture in the long-term phase 2 trial after 8 years of follow-up.30 By September 2013, postmarketing safety surveillance data for denosumab (estimated exposure of 1.2 million patient-years) had recorded four cases of atypical femoral fracture. All four patients had previously been on bisphosphonates. There were also 32 reports of osteonecrosis of the jaw.47
Denosumab’s manufacturer aims to communicate the risks of treatment to health care professionals and patients. Information is available online at www.proliahcp.com/risk-evaluation-mitigation-strategy/.
WHAT ARE THE PRECAUTIONS?
Several precautions need to be taken when considering treatment with denosumab.
Antiresorptives can aggravate hypocalcemia by inhibiting bone turnover. Serum calcium should therefore be checked and preexisting hypocalcemia should be corrected before starting denosumab.48
Denosumab is contraindicated in women who are pregnant or are planning to become pregnant, as fetal loss and teratogenicity have been reported in animal experiments. (Denosumab is unlikely to be used in premenopausal women, as it is not approved for use in this group.)
There are no data on excretion of denosumab in human milk, so it should not be given to nursing mothers.
Renal impairment is not a contraindication, and no dose adjustment is necessary (even for patients on renal replacement therapy), as denosumab, being an antibody, is eliminated through the reticuloendothelial system.49,50 However, in practice, any antiresorptive agent should be used with caution in patients with severe renal impairment because of the possible presence of adynamic bone disease. Further reduction of bone turnover would be detrimental in such patients. Also, severe hypocalcemia has been reported in patients with a creatinine clearance rate less than 30 mL/min and in those receiving dialysis.51,52 Postmarketing surveillance data have reported eight cases of severe symptomatic hypocalcemia, of which seven were in patients with chronic kidney disease.47
The manufacturer suggests that patients receive a dental examination with appropriate preventive dentistry before starting denosumab to reduce the incidence of osteonecrosis of the jaw, despite the lack of evidence in support of this strategy. The American Dental Association recommends regular dental visits and maintenance of good oral hygiene for patients already established on antiresorptive therapy.53,54
SHOULD PATIENTS ON DENOSUMAB BE OFFERED A DRUG HOLIDAY?
A drug holiday (temporary discontinuation of the drug after a certain duration of treatment) has been proposed for patients receiving bisphosphonates because of the risk of atypical femoral fracture and osteonecrosis of the jaw (although small) consequent to long-term continuous suppression of bone turnover.55 The antifracture efficacy of bisphosphonates is likely to persist for an unknown length of time after discontinuation because of their long skeletal half-life, while the risks gradually diminish.
By contrast, denosumab targets RANKL in the extracellular fluid and does not become embedded within the bone tissue.56 Pharmacokinetic studies have shown that denosumab has a rapid offset of action, with a half-life of only 26 days and biological activity lasting only 6 months.57 The results of a phase 2 extension study suggest that bone mineral density starts to decline and bone turnover markers start to rise within 12 months of discontinuing denosumab.58
Although fracture risk did not increase in those who were randomized to stopping the treatment and bone mineral density increased further when treatment was restarted, a drug holiday cannot presently be recommended for patients receiving denosumab because of the lack of supportive data.
HOW COST-EFFECTIVE IS DENOSUMAB?
The wholesale acquisition cost is $825 per 60-mg prefilled syringe of denosumab, although this may vary depending on where the drug is obtained. This does not include physician-related service costs associated with administration of denosumab.
Cost-effectiveness analyses conducted in the United States, the United Kingdom, and Sweden have all concluded that denosumab would offer a cost-effective alternative to other osteoporosis medications for primary prevention and secondary prevention of fractures.59–61
The Swedish study also incorporated adherence in the cost-effectiveness model and showed that denosumab was a cost-effective alternative to oral bisphosphonates, particularly for patients who were not expected to adhere well to oral treatments.61
WHICH OSTEOPOROSIS PATIENTS ARE CANDIDATES FOR DENOSUMAB?
The FDA has approved denosumab for the treatment of postmenopausal women and men at high risk of fracture (defined as having a history of osteoporotic fracture or multiple risk factors for fracture), or in those who cannot tolerate other osteoporosis medications or for whom other medications have failed.
Denosumab is also approved for men at high risk of fracture receiving androgen deprivation therapy for nonmetastatic prostate cancer, and for women at high risk of fracture receiving adjuvant aromatase inhibitor therapy for breast cancer.
WHAT DO THE GUIDELINES RECOMMEND?
The National Osteoporosis Foundation guidelines recommend pharmacologic treatment for patients with hip or vertebral fractures (clinical or asymptomatic); T scores lower than –2.5 at the femoral neck, total hip, or lumbar spine; and those with a 10-year probability of hip fracture of more than 3% or of a major osteoporotic fracture more than 20% based on the US-adapted FRAX calculator.62 The American College of Endocrinology guidelines have proposed similar thresholds for pharmacologic treatment, and they recommend alendronate, risedronate, zoledronate, and denosumab as first-line agents.63
The 2010 Osteoporosis Canada guidelines recommend denosumab, alendronate, risedronate, and zoledronate as first-line therapies for preventing hip, nonvertebral, and vertebral fractures in postmenopausal women (grade A recommendation).64 The National Institute of Health and Clinical Excellence in England and Wales, on the other hand, recommends denosumab only for patients who are unable to take a bisphosphonate.65
PRACTICAL PRESCRIBING TIPS
The patient described at the beginning of this article has already sustained a vertebral compression fracture, and her DXA scan shows T scores in the osteoporotic range. She is therefore at increased risk of another fragility fracture (with a fivefold higher risk of another vertebral fracture). Pharmacologic therapy should be considered. In addition, she should be encouraged to adhere to lifestyle measures such as a healthy diet and regular weight-bearing exercise, her risk of falling should be assessed, and adequate calcium and vitamin D supplementation should be given.
Secondary causes of osteoporosis are present in about 30% of women and 55% of men who have vertebral fractures.66 A complete blood count, erythrocyte sedimentation rate, bone biochemistry, 25-hydroxyvitamin D, thyroid-stimulating hormone, and renal and liver function tests should be requested in all patients. Further tests should be considered depending on the clinical evaluation and results of initial investigations.
Because this patient cannot tolerate oral bisphosphonates, she could be offered the option of annual intravenous zoledronic acid infusions or 6-monthly subcutaneous denosumab injections. In clinical trials, gastrointestinal adverse effects were noted with intravenous bisphosphonates as well, but the adverse effects reported were no different than those with placebo. The potential advantages with denosumab include better bone mineral density gains, adherence and patient satisfaction compared with oral bisphosphonates, convenient twice-yearly administration, safety in patients with renal impairment, and absence of gastrointestinal effects.
Raloxifene, a selective estrogen receptor modulator, has estrogen-like action on the bone and antiestrogen actions on the breast and uterus. Unlike standard hormone replacement therapy, raloxifene can therefore increase bone mineral density without increasing the risk of breast and endometrial cancers. However, it has only been shown to reduce the risk of vertebral fracture, not hip fracture. Hence, it would be a more appropriate choice for younger postmenopausal women. Moreover, it may cause troublesome menopausal symptoms.
Teriparatide, the recombinant parathyroid hormone, is an anabolic agent. It is very expensive, and because of this, guidelines in several countries restrict its use to women with severe osteoporosis and multiple fractures who fail to respond to standard treatments. It cannot be used for longer than 2 years because of its association with osteosarcoma in rats.
If our patient prefers denosumab, therapy should be initiated after appropriate counseling (see precautions above). The dose is 60 mg, given subcutaneously, once every 6 months.
Monitoring
There is no consensus regarding the optimal frequency for monitoring patients on treatment, owing to the lack of prospective trial data. The National Osteoporosis Foundation recommends repeating the bone mineral density measurements about 2 years after starting therapy, and about every 2 years thereafter.62 Some studies suggest that changes in bone mineral density correlate with reduction in fracture risk.67,68 A change in bone mineral density is considered significant when it is greater than the range of error of the densitometer (also known as the least significant change).69 If the bone mineral density is stable or improving, therapy could be continued, but if it is declining and the decline is greater than the least significant change, a change in therapy should be considered if no secondary causes for bone loss are evident (but see What are the areas of uncertainty? below).
The National Osteoporosis Foundation also recommends measuring a bone turnover marker at baseline and then 3 to 6 months later, as its suppression predicts greater bone mineral density responses and fracture risk reduction.70 If there is a decrease of more than 30% in serum carboxy-terminal collagen crosslinks (CTX) or more than 50% in urinary N-telopeptide (NTX),71 the patient can be reassured that the next bone mineral density measurement will be stable or improved. In patients on oral bisphosphonates, measurement of bone turnover markers also provides evidence of compliance.
Clinical trials suggest that a numerical increase in bone mineral density can be expected in most patients on treatment, though this depends on the measurement site and the length of time between examinations. In one phase 3 trial of denosumab in postmenopausal women, only 5% of the participants had unchanged or diminished bone mineral density at the lumbar spine, and 8% at the hip, after 36 months of treatment.72 However, the CTX levels fell to below the lower limit of the reference interval as early as 1 month after commencing treatment in all denosumab-treated patients.68
Hence, bone turnover markers may be a more sensitive indicator of treatment effect than bone mineral density, but this would ultimately need to be evaluated against fracture rates in a real-world setting.
WHAT ARE THE AREAS OF UNCERTAINTY?
There are currently no guidelines for long-term management of patients on denosumab, and also no data to suggest whether patients should be switched to a weaker antiresorptive drug after a certain number of years in order to reduce the possible risk of atypical femoral fracture or osteonecrosis of the jaw.
No head-to-head trials have directly compared the antifracture efficacy of denosumab with that of other standard osteoporosis therapies. The antifracture efficacy and safety of combination therapies involving denosumab are also uncertain. For adherent patients who have a suboptimal response, there is no evidence to guide the further course of action. The International Osteoporosis Foundation guidelines suggest replacing a stronger antiresorptive with an anabolic agent, but acknowledge that this is only based on expert opinion.71
The very-long-term effects (beyond 8 years) of continuous denosumab administration on increasing the risk of atypical femoral fracture, osteonecrosis of the jaw, malignancy, or infection or the duration after which risks would start to outweigh benefits is not known. However, postmarketing safety data continue to be collected through the voluntary Post-marketing Active Safety Surveillance Program (for prespecified adverse events) in addition to the FDA’s MedWatch program.
CASE PROGRESSION
The patient described in the vignette is presented with two options—zoledronate and denosumab. She chooses denosumab. Her renal function and serum calcium are checked and are found to be satisfactory. She undergoes a dental examination, which is also satisfactory. She is counseled about the possible increased risk of infection, and then she is started on 60 mg of denosumab subcutaneously, once every 6 months.
When reviewed after 2 years, she reports no further fractures. Her bone mineral density remains stable compared with the values obtained before starting treatment. She reports no adverse effects and is happy to continue with denosumab.
- Johnell O, Kanis JA. An estimate of the worldwide prevalence and disability associated with osteoporotic fractures. Osteoporos Int 2006; 17:1726–1733.
- Gullberg B, Johnell O, Kanis JA. World-wide projections for hip fracture. Osteoporos Int 1997; 7:407–413.
- Burge R, Dawson-Hughes B, Solomon DH, Wong JB, King A, Tosteson A. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005–2025. J Bone Miner Res 2007; 22:465–475.
- Melton LJ 3rd, Chrischilles EA, Cooper C, Lane AW, Riggs BL. Perspective. How many women have osteoporosis? J Bone Miner Res 1992; 7:1005–1010.
- Abrahamsen B, van Staa T, Ariely R, Olson M, Cooper C. Excess mortality following hip fracture: a systematic epidemiological review. Osteoporos Int 2009; 20:1633–1650.
- Jalava T, Sarna S, Pylkkänen L, et al. Association between vertebral fracture and increased mortality in osteoporotic patients. J Bone Miner Res 2003; 18:1254–1260.
- Ismail AA, O’Neill TW, Cooper C, et al. Mortality associated with vertebral deformity in men and women: results from the European Prospective Osteoporosis Study (EPOS). Osteoporos Int 1998; 8:291–297.
- Ensrud KE, Thompson DE, Cauley JA, et al. Prevalent vertebral deformities predict mortality and hospitalization in older women with low bone mass. Fracture Intervention Trial Research Group. J Am Geriatr Soc 2000; 48:241–249.
- Kado DM, Browner WS, Palermo L, Nevitt MC, Genant HK, Cummings SR. Vertebral fractures and mortality in older women: a prospective study. Study of Osteoporotic Fractures Research Group. Arch Intern Med 1999; 159:1215–1220.
- Lindsay R, Silverman SL, Cooper C, et al. Risk of new vertebral fracture in the year following a fracture. JAMA 2001; 285:320–323.
- Ross PD, Davis JW, Epstein RS, Wasnich RD. Pre-existing fractures and bone mass predict vertebral fracture incidence in women. Ann Intern Med 1991; 114:919–923.
- Piscitelli P, Iolascon G, Argentiero A, et al. Incidence and costs of hip fractures vs strokes and acute myocardial infarction in Italy: comparative analysis based on national hospitalization records. Clin Interv Aging 2012; 7:575–583.
- Johnell O, Kanis JA, Jonsson B, Oden A, Johansson H, De Laet C. The burden of hospitalised fractures in Sweden. Osteoporos Int 2005; 16:222–228.
- Confavreux CB, Canoui-Poitrine F, Schott AM, Ambrosi V, Tainturier V, Chapurlat RD. Persistence at 1 year of oral antiosteoporotic drugs: a prospective study in a comprehensive health insurance database. Eur J Endocrinol 2012; 166:735–741.
- Biswas PN, Wilton LV, Shakir SA. Pharmacovigilance study of alendronate in England. Osteoporos Int 2003; 14:507–514.
- Landfeldt E, Ström O, Robbins S, Borgström F. Adherence to treatment of primary osteoporosis and its association to fractures—the Swedish Adherence Register Analysis (SARA). Osteoporos Int 2012; 23:433–443.
- Sampalis JS, Adachi JD, Rampakakis E, Vaillancourt J, Karellis A, Kindundu C. Long-term impact of adherence to oral bisphosphonates on osteoporotic fracture incidence. J Bone Miner Res 2012; 27:202–210.
- Schwarz EM, Ritchlin CT. Clinical development of anti-RANKL therapy. Arthritis Res Ther 2007; 9(suppl 1):S7.
- Hanley DA, Adachi JD, Bell A, Brown V. Denosumab: mechanism of action and clinical outcomes. Int J Clin Pract 2012; 66:1139–1146.
- Bekker PJ, Holloway D, Nakanishi A, Arrighi M, Leese PT, Dunstan CR. The effect of a single dose of osteoprotegerin in postmenopausal women. J Bone Miner Res 2001; 16:348–360.
- Rizzoli R, Body JJ, Brandi ML, et al; International Osteoporosis Foundation Committee of Scientific Advisors Working Group on Cancer-Induced Bone Disease. Cancer-associated bone disease. Osteoporos Int 2013; 24:2929–2953.
- Schwarz P, Rasmussen AQ, Kvist TM, Andersen UB, Jørgensen NR. Paget’s disease of the bone after treatment with denosumab: a case report. Bone 2012; 50:1023–1025.
- Hu MI, Glezerman IG, Leboulleux S, et al. Denosumab for treatment of hypercalcemia of malignancy. J Clin Endocrinol Metab 2014; Jun 10 [Epub ahead of print].
- Cummings SR, San Martin J, McClung MR, et al; FREEDOM Trial. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med 2009; 361:756–765.
- Papapoulos S, Lippuner K, Roux C, et al. Eight years of denosumab treatment in postmenopausal women with osteoporosis: results from the first five years of the FREEDOM extension [abstract]. Presented at the 2013 annual meeting of the American Society for Bone and Mineral Research, Baltimore, MD, October 4–7, 2013.
- Bone HG, Chapurlat R, Brandi ML, et al. The effect of three or six years of denosumab exposure in women with postmenopausal osteoporosis: results from the FREEDOM extension. J Clin Endocrinol Metab 2013; 98:4483–4492.
- McClung MR, Boonen S, Törring O, et al. Effect of denosumab treatment on the risk of fractures in subgroups of women with postmenopausal osteoporosis. J Bone Miner Res 2012; 27:211–218.
- Boonen S, Adachi JD, Man Z, et al. Treatment with denosumab reduces the incidence of new vertebral and hip fractures in postmenopausal women at high risk. J Clin Endocrinol Metab 2011; 96:1727–1736.
- McCloskey EV, Johansson H, Oden A, et al. Denosumab reduces the risk of osteoporotic fractures in postmenopausal women, particularly in those with moderate to high fracture risk as assessed with FRAX. J Bone Miner Res 2012; 27:1480–1486.
- McClung MR, Lewiecki EM, Geller ML, et al. Effect of denosumab on bone mineral density and biochemical markers of bone turnover: 8-year results of a phase 2 clinical trial. Osteoporos Int 2013; 24:227–235.
- McClung MR, Lewiecki EM, Cohen SB, et al; AMG 162 Bone Loss Study Group. Denosumab in postmenopausal women with low bone mineral density. N Engl J Med 2006; 354:821–831.
- Bone HG, Bolognese MA, Yuen CK, et al. Effects of denosumab on bone mineral density and bone turnover in postmenopausal women. J Clin Endocrinol Metab 2008; 93:2149–2157.
- Genant HK, Engelke K, Hanley DA, et al. Denosumab improves density and strength parameters as measured by QCT of the radius in postmenopausal women with low bone mineral density. Bone 2010; 47:131–139.
- Orwoll E, Teglbjærg CS, Langdahl BL, et al. A randomized, placebo-controlled study of the effects of denosumab for the treatment of men with low bone mineral density. J Clin Endocrinol Metab 2012; 97:3161–3169.
- Tsai JN, Uihlein AV, Lee H, et al. Teriparatide and denosumab, alone or combined, in women with postmenopausal osteoporosis: the DATA study randomised trial. Lancet 2013; 382:50–56.
- Smith MR, Egerdie B, Hernández Toriz N, et al; Denosumab HALT Prostate Cancer Study Group. Denosumab in men receiving androgen-deprivation therapy for prostate cancer. N Engl J Med 2009; 361:745–755.
- Ellis GK, Bone HG, Chlebowski R, et al. Randomized trial of denosumab in patients receiving adjuvant aromatase inhibitors for nonmetastatic breast cancer. J Clin Oncol 2008; 26:4875–4882.
- Brown JP, Prince RL, Deal C, et al. Comparison of the effect of denosumab and alendronate on BMD and biochemical markers of bone turnover in postmenopausal women with low bone mass: a randomized, blinded, phase 3 trial. J Bone Miner Res 2009; 24:153–161.
- Kendler DL, Roux C, Benhamou CL, et al. Effects of denosumab on bone mineral density and bone turnover in postmenopausal women transitioning from alendronate therapy. J Bone Miner Res 2010; 25:72–81.
- Seeman E, Delmas PD, Hanley DA, et al. Microarchitectural deterioration of cortical and trabecular bone: differing effects of denosumab and alendronate. J Bone Miner Res 2010; 25:1886–1894.
- Hopkins RB, Goeree R, Pullenayegum E, et al. The relative efficacy of nine osteoporosis medications for reducing the rate of fractures in post-menopausal women. BMC Musculoskelet Disord 2011; 12:209.
- National Institute for Health and Care Excellence (NICE). NICE technology appraisal guidance: TA161. Alendronate, etidronate, risedronate, raloxifene, strontium ranelate and teriparatide for the secondary prevention of osteoporotic fragility fractures in postmenopausal women (amended). http://publications.nice.org.uk/alendronate-etidronate-risedronate-raloxifene-strontium-ranelate-and-teriparatide-for-ta161. Accessed January 9, 2015.
- Freemantle N, Satram-Hoang S, Tang ET, et al; DAPS Investigators. Final results of the DAPS (Denosumab Adherence Preference Satisfaction) study: a 24-month, randomized, crossover comparison with alendronate in postmenopausal women. Osteoporos Int 2012; 23:317–326.
- Lewiecki EM. Safety and tolerability of denosumab for the treatment of postmenopausal osteoporosis. Drug Healthc Patient Saf 2011; 3:79–91.
- Watts NB, Roux C, Modlin JF, et al. Infections in postmenopausal women with osteoporosis treated with denosumab or placebo: coincidence or causal association? Osteoporos Int 2012; 23:327–337.
- von Keyserlingk C, Hopkins R, Anastasilakis A, et al. Clinical efficacy and safety of denosumab in postmenopausal women with low bone mineral density and osteoporosis: a meta-analysis. Semin Arthritis Rheum 2011; 41:178–186.
- Geller M, Wagman RB, Ho PR, et al. Early findings from Prolia postmarketing safety surveillance for atypical femoral fracture, osteonecrosis of the jaw, severe symptomatic hypocalcemia, and anaphylaxis (abstract). Osteoporos Int 2014; 25(suppl 2). OC40; www.wco-iof-esceo.org/sites/ecceo14/docs/wco14-abstractbook.pdf. Accessed January 9, 2015.
- McCormick BB, Davis J, Burns KD. Severe hypocalcemia following denosumab injection in a hemodialysis patient. Am J Kidney Dis 2012; 60:626–628.
- Jamal SA, Ljunggren O, Stehman-Breen C, et al. Effects of denosumab on fracture and bone mineral density by level of kidney function. J Bone Miner Res 2011; 26:1829–1835.
- Block GA, Bone HG, Fang L, Lee E, Padhi D. A single-dose study of denosumab in patients with various degrees of renal impairment. J Bone Miner Res 2012; 27:1471–1479.
- Ungprasert P, Cheungpasitporn W, Srivali N, Kittanamongkolchai W, Bischof EF. Life-threatening hypocalcemia associated with denosumab in a patient with moderate renal insufficiency. Am J Emerg Med 2013; 31:756.e1–e2.
- McCormick BB, Davis J, Burns KD. Severe hypocalcemia following denosumab injection in a hemodialysis patient. Am J Kidney Dis 2012; 60:626–628.
- Rachner TD, Platzbecker U, Felsenberg D, Hofbauer LC. Osteonecrosis of the jaw after osteoporosis therapy with denosumab following long-term bisphosphonate therapy. Mayo Clin Proc 2013; 88:418–419.
- Epstein MS, Ephros HD, Epstein JB. Review of current literature and implications of RANKL inhibitors for oral health care providers. Oral Surg Oral Med Oral Pathol Oral Radiol 2013; 116:e437–e442.
- McClung M, Harris ST, Miller PD, et al. Bisphosphonate therapy for osteoporosis: benefits, risks, and drug holiday. Am J Med 2013; 126:13–20.
- Baron R, Ferrari S, Russell RG. Denosumab and bisphosphonates: different mechanisms of action and effects. Bone 2011; 48:677–692.
- Bekker PJ, Holloway DL, Rasmussen AS, et al. A single-dose placebo-controlled study of AMG 162, a fully human monoclonal antibody to RANKL, in postmenopausal women. J Bone Miner Res 2004; 19:1059–1066.
- Miller PD, Bolognese MA, Lewiecki EM, et al; Amg Bone Loss Study Group. Effect of denosumab on bone density and turnover in postmenopausal women with low bone mass after long-term continued, discontinued, and restarting of therapy: a randomized blinded phase 2 clinical trial. Bone 2008; 43:222–229.
- Parthan A, Kruse M, Yurgin N, Huang J, Viswanathan HN, Taylor D. Cost effectiveness of denosumab versus oral bisphosphonates for postmenopausal osteoporosis in the US. Appl Health Econ Health Policy 2013; 11:485–497.
- Scotland G, Waugh N, Royle P, McNamee P, Henderson R, Hollick R. Denosumab for the prevention of osteoporotic fractures in post-menopausal women: a NICE single technology appraisal. Pharmacoeconomics 2011; 29:951–961.
- Jönsson B, Ström O, Eisman JA, et al. Cost-effectiveness of denosumab for the treatment of postmenopausal osteoporosis. Osteoporos Int 2011; 22:967–982.
- Clinician’s guide to prevention and treatment of osteoporosis. Washington DC: National Osteoporosis Foundation, 2013.
- Watts NB, Bilezikian JP, Camacho PM, et al; AACE Osteoporosis Task Force. American Association of Clinical Endocrinologists Medical Guidelines for Clinical Practice for the diagnosis and treatment of postmenopausal osteoporosis. Endocr Pract 2010;16(suppl 3):1–37.
- Papaioannou A, Morin S, Cheung AM, et al; Scientific Advisory Council of Osteoporosis Canada. 2010 clinical practice guidelines for the diagnosis and management of osteoporosis in Canada: summary. CMAJ 2010; 182:1864–1673.
- National Institute for Health and Care Excellence (NICE). NICE technology appraisal: TA204. Denosumab for the prevention of osteoporotic fractures in postmenopausal women. http://guidance.nice.org.uk/TA204. Accessed January 9, 2015.
- Premaor MO, Compston JE. Testing for secondary causes of osteoporosis. BMJ 2010; 341:c6959.
- Hochberg MC, Ross PD, Black D, et al. Larger increases in bone mineral density during alendronate therapy are associated with a lower risk of new vertebral fractures in women with postmenopausal osteoporosis. Fracture Intervention Trial Research Group. Arthritis Rheum 1999; 42:1246–1254.
- Eastell R, Vrijens B, Cahall DL, Ringe JD, Garnero P, Watts NB. Bone turnover markers and bone mineral density response with risedronate therapy: relationship with fracture risk and patient adherence. J Bone Miner Res 2011; 26:1662–1669.
- Diez-Perez A, Adachi JD, Agnusdei D, et al; IOF CSA Inadequate Responders Working Group. Treatment failure in osteoporosis. Osteoporos Int 2012; 23:2769–2774.
- Eastell R, Barton I, Hannon RA, Chines A, Garnero P, Delmas PD. Relationship of early changes in bone resorption to the reduction in fracture risk with risedronate. J Bone Miner Res 2003; 18:1051–1056.
- Rosen HN, Moses AC, Garber J, Ross DS, Lee SL, Greenspan SL. Utility of biochemical markers of bone turnover in the follow-up of patients treated with bisphosphonates. Calcif Tissue Int 1998; 63:363–368.
- Bolognese MA, Teglbjærg CS, Zanchetta JR, et al. Denosumab significantly increases DXA BMD at both trabecular and cortical sites: results from the FREEDOM study. J Clin Densitom 2013; 16:147–153.
- Johnell O, Kanis JA. An estimate of the worldwide prevalence and disability associated with osteoporotic fractures. Osteoporos Int 2006; 17:1726–1733.
- Gullberg B, Johnell O, Kanis JA. World-wide projections for hip fracture. Osteoporos Int 1997; 7:407–413.
- Burge R, Dawson-Hughes B, Solomon DH, Wong JB, King A, Tosteson A. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005–2025. J Bone Miner Res 2007; 22:465–475.
- Melton LJ 3rd, Chrischilles EA, Cooper C, Lane AW, Riggs BL. Perspective. How many women have osteoporosis? J Bone Miner Res 1992; 7:1005–1010.
- Abrahamsen B, van Staa T, Ariely R, Olson M, Cooper C. Excess mortality following hip fracture: a systematic epidemiological review. Osteoporos Int 2009; 20:1633–1650.
- Jalava T, Sarna S, Pylkkänen L, et al. Association between vertebral fracture and increased mortality in osteoporotic patients. J Bone Miner Res 2003; 18:1254–1260.
- Ismail AA, O’Neill TW, Cooper C, et al. Mortality associated with vertebral deformity in men and women: results from the European Prospective Osteoporosis Study (EPOS). Osteoporos Int 1998; 8:291–297.
- Ensrud KE, Thompson DE, Cauley JA, et al. Prevalent vertebral deformities predict mortality and hospitalization in older women with low bone mass. Fracture Intervention Trial Research Group. J Am Geriatr Soc 2000; 48:241–249.
- Kado DM, Browner WS, Palermo L, Nevitt MC, Genant HK, Cummings SR. Vertebral fractures and mortality in older women: a prospective study. Study of Osteoporotic Fractures Research Group. Arch Intern Med 1999; 159:1215–1220.
- Lindsay R, Silverman SL, Cooper C, et al. Risk of new vertebral fracture in the year following a fracture. JAMA 2001; 285:320–323.
- Ross PD, Davis JW, Epstein RS, Wasnich RD. Pre-existing fractures and bone mass predict vertebral fracture incidence in women. Ann Intern Med 1991; 114:919–923.
- Piscitelli P, Iolascon G, Argentiero A, et al. Incidence and costs of hip fractures vs strokes and acute myocardial infarction in Italy: comparative analysis based on national hospitalization records. Clin Interv Aging 2012; 7:575–583.
- Johnell O, Kanis JA, Jonsson B, Oden A, Johansson H, De Laet C. The burden of hospitalised fractures in Sweden. Osteoporos Int 2005; 16:222–228.
- Confavreux CB, Canoui-Poitrine F, Schott AM, Ambrosi V, Tainturier V, Chapurlat RD. Persistence at 1 year of oral antiosteoporotic drugs: a prospective study in a comprehensive health insurance database. Eur J Endocrinol 2012; 166:735–741.
- Biswas PN, Wilton LV, Shakir SA. Pharmacovigilance study of alendronate in England. Osteoporos Int 2003; 14:507–514.
- Landfeldt E, Ström O, Robbins S, Borgström F. Adherence to treatment of primary osteoporosis and its association to fractures—the Swedish Adherence Register Analysis (SARA). Osteoporos Int 2012; 23:433–443.
- Sampalis JS, Adachi JD, Rampakakis E, Vaillancourt J, Karellis A, Kindundu C. Long-term impact of adherence to oral bisphosphonates on osteoporotic fracture incidence. J Bone Miner Res 2012; 27:202–210.
- Schwarz EM, Ritchlin CT. Clinical development of anti-RANKL therapy. Arthritis Res Ther 2007; 9(suppl 1):S7.
- Hanley DA, Adachi JD, Bell A, Brown V. Denosumab: mechanism of action and clinical outcomes. Int J Clin Pract 2012; 66:1139–1146.
- Bekker PJ, Holloway D, Nakanishi A, Arrighi M, Leese PT, Dunstan CR. The effect of a single dose of osteoprotegerin in postmenopausal women. J Bone Miner Res 2001; 16:348–360.
- Rizzoli R, Body JJ, Brandi ML, et al; International Osteoporosis Foundation Committee of Scientific Advisors Working Group on Cancer-Induced Bone Disease. Cancer-associated bone disease. Osteoporos Int 2013; 24:2929–2953.
- Schwarz P, Rasmussen AQ, Kvist TM, Andersen UB, Jørgensen NR. Paget’s disease of the bone after treatment with denosumab: a case report. Bone 2012; 50:1023–1025.
- Hu MI, Glezerman IG, Leboulleux S, et al. Denosumab for treatment of hypercalcemia of malignancy. J Clin Endocrinol Metab 2014; Jun 10 [Epub ahead of print].
- Cummings SR, San Martin J, McClung MR, et al; FREEDOM Trial. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med 2009; 361:756–765.
- Papapoulos S, Lippuner K, Roux C, et al. Eight years of denosumab treatment in postmenopausal women with osteoporosis: results from the first five years of the FREEDOM extension [abstract]. Presented at the 2013 annual meeting of the American Society for Bone and Mineral Research, Baltimore, MD, October 4–7, 2013.
- Bone HG, Chapurlat R, Brandi ML, et al. The effect of three or six years of denosumab exposure in women with postmenopausal osteoporosis: results from the FREEDOM extension. J Clin Endocrinol Metab 2013; 98:4483–4492.
- McClung MR, Boonen S, Törring O, et al. Effect of denosumab treatment on the risk of fractures in subgroups of women with postmenopausal osteoporosis. J Bone Miner Res 2012; 27:211–218.
- Boonen S, Adachi JD, Man Z, et al. Treatment with denosumab reduces the incidence of new vertebral and hip fractures in postmenopausal women at high risk. J Clin Endocrinol Metab 2011; 96:1727–1736.
- McCloskey EV, Johansson H, Oden A, et al. Denosumab reduces the risk of osteoporotic fractures in postmenopausal women, particularly in those with moderate to high fracture risk as assessed with FRAX. J Bone Miner Res 2012; 27:1480–1486.
- McClung MR, Lewiecki EM, Geller ML, et al. Effect of denosumab on bone mineral density and biochemical markers of bone turnover: 8-year results of a phase 2 clinical trial. Osteoporos Int 2013; 24:227–235.
- McClung MR, Lewiecki EM, Cohen SB, et al; AMG 162 Bone Loss Study Group. Denosumab in postmenopausal women with low bone mineral density. N Engl J Med 2006; 354:821–831.
- Bone HG, Bolognese MA, Yuen CK, et al. Effects of denosumab on bone mineral density and bone turnover in postmenopausal women. J Clin Endocrinol Metab 2008; 93:2149–2157.
- Genant HK, Engelke K, Hanley DA, et al. Denosumab improves density and strength parameters as measured by QCT of the radius in postmenopausal women with low bone mineral density. Bone 2010; 47:131–139.
- Orwoll E, Teglbjærg CS, Langdahl BL, et al. A randomized, placebo-controlled study of the effects of denosumab for the treatment of men with low bone mineral density. J Clin Endocrinol Metab 2012; 97:3161–3169.
- Tsai JN, Uihlein AV, Lee H, et al. Teriparatide and denosumab, alone or combined, in women with postmenopausal osteoporosis: the DATA study randomised trial. Lancet 2013; 382:50–56.
- Smith MR, Egerdie B, Hernández Toriz N, et al; Denosumab HALT Prostate Cancer Study Group. Denosumab in men receiving androgen-deprivation therapy for prostate cancer. N Engl J Med 2009; 361:745–755.
- Ellis GK, Bone HG, Chlebowski R, et al. Randomized trial of denosumab in patients receiving adjuvant aromatase inhibitors for nonmetastatic breast cancer. J Clin Oncol 2008; 26:4875–4882.
- Brown JP, Prince RL, Deal C, et al. Comparison of the effect of denosumab and alendronate on BMD and biochemical markers of bone turnover in postmenopausal women with low bone mass: a randomized, blinded, phase 3 trial. J Bone Miner Res 2009; 24:153–161.
- Kendler DL, Roux C, Benhamou CL, et al. Effects of denosumab on bone mineral density and bone turnover in postmenopausal women transitioning from alendronate therapy. J Bone Miner Res 2010; 25:72–81.
- Seeman E, Delmas PD, Hanley DA, et al. Microarchitectural deterioration of cortical and trabecular bone: differing effects of denosumab and alendronate. J Bone Miner Res 2010; 25:1886–1894.
- Hopkins RB, Goeree R, Pullenayegum E, et al. The relative efficacy of nine osteoporosis medications for reducing the rate of fractures in post-menopausal women. BMC Musculoskelet Disord 2011; 12:209.
- National Institute for Health and Care Excellence (NICE). NICE technology appraisal guidance: TA161. Alendronate, etidronate, risedronate, raloxifene, strontium ranelate and teriparatide for the secondary prevention of osteoporotic fragility fractures in postmenopausal women (amended). http://publications.nice.org.uk/alendronate-etidronate-risedronate-raloxifene-strontium-ranelate-and-teriparatide-for-ta161. Accessed January 9, 2015.
- Freemantle N, Satram-Hoang S, Tang ET, et al; DAPS Investigators. Final results of the DAPS (Denosumab Adherence Preference Satisfaction) study: a 24-month, randomized, crossover comparison with alendronate in postmenopausal women. Osteoporos Int 2012; 23:317–326.
- Lewiecki EM. Safety and tolerability of denosumab for the treatment of postmenopausal osteoporosis. Drug Healthc Patient Saf 2011; 3:79–91.
- Watts NB, Roux C, Modlin JF, et al. Infections in postmenopausal women with osteoporosis treated with denosumab or placebo: coincidence or causal association? Osteoporos Int 2012; 23:327–337.
- von Keyserlingk C, Hopkins R, Anastasilakis A, et al. Clinical efficacy and safety of denosumab in postmenopausal women with low bone mineral density and osteoporosis: a meta-analysis. Semin Arthritis Rheum 2011; 41:178–186.
- Geller M, Wagman RB, Ho PR, et al. Early findings from Prolia postmarketing safety surveillance for atypical femoral fracture, osteonecrosis of the jaw, severe symptomatic hypocalcemia, and anaphylaxis (abstract). Osteoporos Int 2014; 25(suppl 2). OC40; www.wco-iof-esceo.org/sites/ecceo14/docs/wco14-abstractbook.pdf. Accessed January 9, 2015.
- McCormick BB, Davis J, Burns KD. Severe hypocalcemia following denosumab injection in a hemodialysis patient. Am J Kidney Dis 2012; 60:626–628.
- Jamal SA, Ljunggren O, Stehman-Breen C, et al. Effects of denosumab on fracture and bone mineral density by level of kidney function. J Bone Miner Res 2011; 26:1829–1835.
- Block GA, Bone HG, Fang L, Lee E, Padhi D. A single-dose study of denosumab in patients with various degrees of renal impairment. J Bone Miner Res 2012; 27:1471–1479.
- Ungprasert P, Cheungpasitporn W, Srivali N, Kittanamongkolchai W, Bischof EF. Life-threatening hypocalcemia associated with denosumab in a patient with moderate renal insufficiency. Am J Emerg Med 2013; 31:756.e1–e2.
- McCormick BB, Davis J, Burns KD. Severe hypocalcemia following denosumab injection in a hemodialysis patient. Am J Kidney Dis 2012; 60:626–628.
- Rachner TD, Platzbecker U, Felsenberg D, Hofbauer LC. Osteonecrosis of the jaw after osteoporosis therapy with denosumab following long-term bisphosphonate therapy. Mayo Clin Proc 2013; 88:418–419.
- Epstein MS, Ephros HD, Epstein JB. Review of current literature and implications of RANKL inhibitors for oral health care providers. Oral Surg Oral Med Oral Pathol Oral Radiol 2013; 116:e437–e442.
- McClung M, Harris ST, Miller PD, et al. Bisphosphonate therapy for osteoporosis: benefits, risks, and drug holiday. Am J Med 2013; 126:13–20.
- Baron R, Ferrari S, Russell RG. Denosumab and bisphosphonates: different mechanisms of action and effects. Bone 2011; 48:677–692.
- Bekker PJ, Holloway DL, Rasmussen AS, et al. A single-dose placebo-controlled study of AMG 162, a fully human monoclonal antibody to RANKL, in postmenopausal women. J Bone Miner Res 2004; 19:1059–1066.
- Miller PD, Bolognese MA, Lewiecki EM, et al; Amg Bone Loss Study Group. Effect of denosumab on bone density and turnover in postmenopausal women with low bone mass after long-term continued, discontinued, and restarting of therapy: a randomized blinded phase 2 clinical trial. Bone 2008; 43:222–229.
- Parthan A, Kruse M, Yurgin N, Huang J, Viswanathan HN, Taylor D. Cost effectiveness of denosumab versus oral bisphosphonates for postmenopausal osteoporosis in the US. Appl Health Econ Health Policy 2013; 11:485–497.
- Scotland G, Waugh N, Royle P, McNamee P, Henderson R, Hollick R. Denosumab for the prevention of osteoporotic fractures in post-menopausal women: a NICE single technology appraisal. Pharmacoeconomics 2011; 29:951–961.
- Jönsson B, Ström O, Eisman JA, et al. Cost-effectiveness of denosumab for the treatment of postmenopausal osteoporosis. Osteoporos Int 2011; 22:967–982.
- Clinician’s guide to prevention and treatment of osteoporosis. Washington DC: National Osteoporosis Foundation, 2013.
- Watts NB, Bilezikian JP, Camacho PM, et al; AACE Osteoporosis Task Force. American Association of Clinical Endocrinologists Medical Guidelines for Clinical Practice for the diagnosis and treatment of postmenopausal osteoporosis. Endocr Pract 2010;16(suppl 3):1–37.
- Papaioannou A, Morin S, Cheung AM, et al; Scientific Advisory Council of Osteoporosis Canada. 2010 clinical practice guidelines for the diagnosis and management of osteoporosis in Canada: summary. CMAJ 2010; 182:1864–1673.
- National Institute for Health and Care Excellence (NICE). NICE technology appraisal: TA204. Denosumab for the prevention of osteoporotic fractures in postmenopausal women. http://guidance.nice.org.uk/TA204. Accessed January 9, 2015.
- Premaor MO, Compston JE. Testing for secondary causes of osteoporosis. BMJ 2010; 341:c6959.
- Hochberg MC, Ross PD, Black D, et al. Larger increases in bone mineral density during alendronate therapy are associated with a lower risk of new vertebral fractures in women with postmenopausal osteoporosis. Fracture Intervention Trial Research Group. Arthritis Rheum 1999; 42:1246–1254.
- Eastell R, Vrijens B, Cahall DL, Ringe JD, Garnero P, Watts NB. Bone turnover markers and bone mineral density response with risedronate therapy: relationship with fracture risk and patient adherence. J Bone Miner Res 2011; 26:1662–1669.
- Diez-Perez A, Adachi JD, Agnusdei D, et al; IOF CSA Inadequate Responders Working Group. Treatment failure in osteoporosis. Osteoporos Int 2012; 23:2769–2774.
- Eastell R, Barton I, Hannon RA, Chines A, Garnero P, Delmas PD. Relationship of early changes in bone resorption to the reduction in fracture risk with risedronate. J Bone Miner Res 2003; 18:1051–1056.
- Rosen HN, Moses AC, Garber J, Ross DS, Lee SL, Greenspan SL. Utility of biochemical markers of bone turnover in the follow-up of patients treated with bisphosphonates. Calcif Tissue Int 1998; 63:363–368.
- Bolognese MA, Teglbjærg CS, Zanchetta JR, et al. Denosumab significantly increases DXA BMD at both trabecular and cortical sites: results from the FREEDOM study. J Clin Densitom 2013; 16:147–153.
KEY POINTS
- Denosumab is a fully human monoclonal antibody that targets the receptor activator of nuclear factor kappa b ligand, a key mediator of osteoclastic bone resorption.
- Commpared with placebo, denosumab has been shown to significantly reduce the risk of vertebral, nonvertebral, and hip fractures in postmenopausal women with osteoporosis.
- Patients taking denosumab are more adherent, compliant, and persistent with therapy than those taking alendronate. Denosumab is also superior to alendronate in improving bone mineral density at all skeletal sites.
- Denosumab is safe, with safety data now available for up to 8 years of exposure.
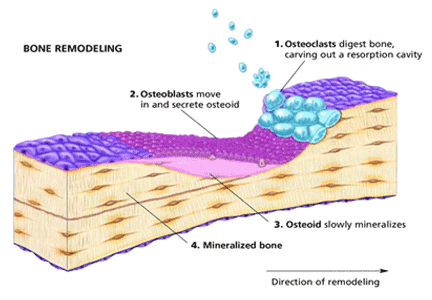
Genetics and hepatitis C: It’s good to be ‘CC’
What a difference a single nucleotide can make! The human genome contains more than 3 billion base pairs. Yet having a different nucleotide in only one pair can make a big difference in how we respond to a disease or its treatment.
Specifically, in hepatitis C virus infection, people born with the nucleotide cytosine (C) at location rs12979860 in both alleles of the gene that codes for interleukin 28B (the IL28B CC genotype) can count themselves luckier than those born with thymine (T) in this location in one of their alleles (the CT genotype) or both of their alleles (the TT genotype). Those with the CC genotype are more likely to clear the virus spontaneously, and even if the infection persists, it is less likely to progress to liver cancer and more likely to respond to treatment with interferon.
Here, we review the IL28B polymorphism and its implications in treating hepatitis C.
GENETIC POLYMORPHISM AND HUMAN DISEASE
Of the 3 billion base pairs of nucleotides, fewer than 1% differ between individuals, but this 1% is responsible for the diversity of human beings. Differences in genetic sequences among individuals are called genetic polymorphisms. A single-nucleotide polymorphism is a DNA sequence variation that occurs in a single nucleotide in the genome. For example, two sequenced DNA fragments from different individuals, AAGCCTA and AAGCTTA, contain a difference in a single nucleotide.
Genetic variations such as these underlie some of the differences in our susceptibility to disease, the severity of illness we develop, and our response to treatments. Therefore, identifying genetic polymorphisms may shed light on biologic pathways involved in diseases and may uncover new targets for therapy.1
Genome-wide association studies have looked at hundreds of thousands of single-nucleotide polymorphisms to try to identify most of the common genetic differences among people and relate them to common chronic diseases such as coronary artery disease,2 type 2 diabetes,3 stroke,4 breast cancer,5 rheumatoid arthritis,6 Alzheimer disease,7 and, more recently, hepatitis C virus infection.8
HEPATITIS C VIRUS: A MAJOR CAUSE OF LIVER DISEASE
Hepatitis C virus infection is a major cause of chronic liver disease and hepatocellular carcinoma and has become the most common indication for liver transplantation in the United States.9
This virus has six distinct genotypes throughout the world, with multiple subtypes in each genotype. (A genotype is a classification of a virus based on its RNA.9) In this review, we will focus on genotype 1; hence, “hepatitis C virus” will refer to hepatitis C virus genotype 1.
Our knowledge of the biology, pathogenesis, and treatment of hepatitis C has been advancing. Originally, fewer than 50% of patients responded to therapy with the combination of pegylated interferon and ribavirin,10,11 but since 2011 the response rate has increased to approximately 70% with the approval of the protease inhibitors telaprevir and boceprevir, used in combination with pegylated interferon and ribavirin.12–15
Unfortunately, interferon-based treatment is often complicated by side effects such as fatigue, influenza-like symptoms, hematologic abnormalities, and neuropsychiatric symptoms. An accurate way to predict response would help patients make informed decisions about antiviral treatment, taking into account the risk and possible benefit for individual patients.
GENETIC POLYMORPHISM AND HEPATITIS C VIRUS INFECTION
Genome-wide association studies have identified single-nucleotide polymorphisms in the IL28B gene that are associated with differences in response to hepatitis C treatment.8
Studying 565,759 polymorphisms in 1,137 patients, researchers at Duke University identified a single-nucleotide polymorphism at location rs12979860 in IL28B (Figure 1) that was strongly associated with response to combination therapy with pegylated interferon and ribavirin.8 The chance of cure with this standard treatment is twice as high in patients who are homozygous for cytosine in this location (the CC genotype) than in those who are heterozygous (CT) or homozygous for thymine in this location (the TT genotype) (Table 1).

Adding one of the new protease inhibitors, telaprevir or boceprevir, to the standard hepatitis C treatment substantially improves the cure rates in all three IL28B genotypes, but especially in people with CT or TT, in whom the response rate almost triples with the addition of one of these drugs. Those with the CC genotype (who are more likely to be cured with pegylated interferon and ribavirin alone) also achieve an increase (although minimal) in cure rates when a protease inhibitor is included in the regimen (TABLE 1).13–15 Thus, it remains unclear if adding a protease inhibitor to pegylated interferon plus ribavirin in patients with the IL28B CC genotype translates into added effectiveness worth the additional cost of the protease inhibitor in previously untreated patients.
Additionally, the effect of the IL28B genotype on telaprevir-based triple therapy has been disputed in more recent studies. In a subgroup analysis of the results of a trial that evaluated telaprevir in the treatment of hepatitis C, researchers found that sustained virologic response rates were significantly higher in the telaprevir group, and this was similar across the different IL28B polymorphisms.16
The favorable IL28B CC genotype is associated with higher rates of rapid virologic response to antiviral therapy.13–15 Of note, almost all patients who achieve a rapid virologic response do well, with a high rate of sustained virologic response even after a shorter duration of therapy (24 vs 48 weeks). Therefore, in addition to predicting response to interferon before starting treatment, the IL28B CC genotype may also identify patients who need only a shorter duration of therapy.
Interestingly, the C allele is much more frequent in white than in African American populations, an important observation that explains the racial difference in response to hepatitis C therapy.8
Two other research groups, from Asia and Australia, performed independent genome-wide association studies that identified different single-nucleotide polymorphisms (eg, rs8099917) in the same IL28B gene as predictors of response to treatment in patients with hepatitis C virus infection.17,18 These findings may be explained by linkage disequilibrium, which means that these single-nucleotide polymorphisms are found more frequently together in the same patient due to their proximity to each other. In this review, we will focus on the rs12979860 polymorphism; hence “IL28B genotype” will refer to the single-nucleotide polymorphism at rs12979860, unless otherwise specified.
The favorable CC genotype is less common in African Americans than in patients of other ethnicities.19 Moreover, although IL28B CC is associated with a better response rate to interferon-based antiviral therapy across all ethnicities, those of African American descent with the CC genotype are less likely to achieve a sustained virologic response than white or Hispanic Americans.8
BIOLOGIC ASSOCIATION: IL28B POLYMORPHISM AND HEPATITIS C
The interferon lambda family consists of three cytokines:
- Interleukin 29 (interferon lambda 1)
- Interleukin 28A (interferon lambda 2)
- Interleukin 28B (interferon lambda 3).
Production of these three molecules can be triggered by viral infection, and they induce antiviral activity through both innate and adaptive immune pathways. They signal through the IL10R-IL28R receptor complex.20–22 This receptor activates the JAK-STAT (Janus kinase-signal transducer and activator of transcription) pathway, which regulates a large number of interferon-stimulated genes, primarily through the interferon-stimulated response element (Figure 2).
A 2013 study found that interferon-stimulated gene expression levels in patients with normal livers were highest in those with the CC genotype, intermediate with CT, and lowest with TT. Interestingly, this pattern was reversed in those with hepatitis C virus infection, indicating a relationship between the IL28B genotype and gene expression before infection.23
The mechanism underlying the association between the IL28B polymorphism and response to hepatitis C treatment is not well understood. The unfavorable TT genotype seems to lead to continuous activation of a subset of interferon-stimulated genes in the presence of intracellular hepatitis C viral RNA. But this level of expression is not sufficient to eliminate the virus from the cells. Instead, it might lead to up-regulation of interferon-inhibitory molecules that suppress JAK-STAT signaling, thereby reducing sensitivity to interferon signaling. Therefore, the hepatocyte not only cannot clear the virus by itself, but also cannot induce strong interferon-stimulated gene expression when interferon is given during therapy.20–22
The recently identified ss469425590 polymorphism, which is located in close proximity to rs12979860 in the IL28B gene, is particularly interesting, as it suggests a possible molecular mechanism. The delta G frameshift variant creates a novel gene called IFNL4, which is transiently activated in response to hepatitis C virus infection.24IFNL4 stimulates STAT1 and STAT2 phosphorylation and induces the expression of interferon-stimulated genes. Increased interferon-stimulated gene expression has been shown to be associated with decreased response to pegylated interferon-ribavirin treatment. These observations suggest that the ss469425590 delta G allele is responsible for the increased activation of interferon-stimulated genes and the lower sustained virologic response rate observed in patients who receive pegylated interferon-ribavirin treatment. It is possible that the activation of interferon-stimulated genes in patients with the ss469425590 delta G/delta G genotype reduces interferon-stimulated gene responsiveness to interferon alpha, which normally activates interferon-stimulated genes and inhibits hepatitis C progression.24
IL28B POLYMORPHISM AND ACUTE HEPATITIS C VIRUS INFECTION
From 70% to 80% of acute hepatitis C virus infections persist and become chronic, while 20% to 30% spontaneously resolve. Epidemiologic, viral, and host factors have been associated with the differences in viral clearance or persistence, and studies have found that a strong host immune response against the virus favors viral clearance. Thus, variation in the genes involved in the immune response may contribute to one’s ability to clear the virus. Consistent with these observations, recent studies have shown that the polymorphism in the IL28 gene region encoding interferon lambda 3 strongly predicts spontaneous resolution of acute hepatitis C virus infection. People who have the IL28B CC genotype are three times more likely to spontaneously clear the virus than those with the CT or TT genotype (Figure 3).24
IL28B POLYMORPHISM AND THE NATURAL HISTORY OF HEPATITIS C
In people in whom hepatitis C virus infection persists, up to 20% develop progressive liver fibrosis and eventually cirrhosis over 10 to 20 years.19,25,26 The speed at which fibrosis develops in these patients is variable and unpredictable.25 The relationship between IL28B polymorphisms and hepatic fibrosis in patients with chronic hepatitis C virus infection has not been clearly established, although a study indicated that in patients with a known date of infection, the IL28B genotype is not associated with progression of hepatic fibrosis.27 Obstacles in this field of study are that it is difficult to determine accurately when the patient contracted the virus, and that serial liver biopsies are needed to investigate the progression of hepatic fibrosis.
Patients with chronic hepatitis C virus infection are also at higher risk of hepatocellular carcinoma compared with the general population.28 An analysis of explanted livers of patients with hepatitis C found that the prevalence of hepatocellular carcinoma in those with the unfavorable TT genotype was significantly higher than with the other genotypes.29 Similarly, an earlier study demonstrated that patients with hepatitis C-associated hepatocellular carcinoma carried the T allele more frequently.30 As with other aspects of IL28B associations with hepatitis C, these findings indicate that the C allele confers a certain degree of protection.
An important implication of these relationships is that they may eventually help identify patients at greater risk, who therefore need earlier intervention.
IL28B POLYMORPHISM AND LIVER TRANSPLANTATION
Hepatitis C virus infection always recurs after liver transplantation, with serious consequences that include cirrhosis and liver failure. Recurrent hepatitis C virus infection has become an important reason for repeat transplantation in the United States.
Results of treatment with pegylated interferon and ribavirin for recurrent hepatitis C after liver transplantation have been disappointing, with response rates lower than 30% and significant side effects.31 Identifying the factors that predict the response to therapy allows for better selection of treatment candidates.
Similar to the way the IL28B genotype predicts response to antiviral therapy in the nontransplant setting, the IL28B genotypes of both the recipient and the donor are strongly and independently associated with response to interferon-based treatment in patients with hepatitis C after liver transplantation. The IL28B CC genotype in either the recipient or the donor is associated with a higher rate of response to pegylated interferon and ribavirin combination therapy after liver transplantation.30,32 For example, the response rate to therapy after liver transplantation reaches 86% in CC-donor and CC-recipient livers, compared with 0% in TT-donor and TT-recipient livers.
Additionally, the IL28B genotype of the recipient may determine the severity of histologic recurrence of hepatitis C, as indicated by progressive hepatic fibrosis. A recipient IL28B TT genotype is associated with more severe histologic recurrence of hepatitis C.33
These data suggest that CC donor livers might be preferentially allocated to patients with hepatitis C virus infection.
IL28B AND OTHER FACTORS IN HEPATITIS C VIRUS INFECTION
Although it is tempting to think that the IL28B polymorphism is the sole predictor of response to antiviral therapy, it is but one of several known factors in the virus and the host.
While IL28B polymorphisms are the most important predictor of sustained virologic response with an interferon-based regimen, a rapid virologic response (undetectable viral load at 4 weeks) had superior predictive value and specificity in one study.34 In fact, for patients with chronic hepatitis C infection who achieved a rapid virologic response with pegylated interferon and ribavirin, the IL28B polymorphism had no effect on the rate of sustained virologic response. However, it did predict a sustained virologic response in the group who did not achieve rapid virologic response.
In a study of patients with acute hepatitis C infection,35 jaundice and the IL28 rs12979860 CC genotype both predicted spontaneous clearance. The best predictor of viral persistence was the combination of the CT or TT genotype plus the absence of jaundice, which had a predictive value of 98%.
IL28B AND THE FUTURE OF HEPATITIS C VIRUS THERAPY
New oral agents were recently approved for treating hepatitis C. As of November 2014, these included simeprevir, sofosbuvir, and ledipasvir.
Simeprevir is a second-generation NS3/4A protease inhibitor approved for use in combination with pegylated interferon and ribavirin. A recent phase 3 trial evaluating simeprevir in patients who had relapsed after prior therapy found sustained virologic response rates to be higher with simeprevir than with placebo, irrespective of IL28B status.36 This finding was similar to that of a trial of telaprevir.16
Sofosbuvir is a nucleotide analogue NS5B polymerase inhibitor that becomes incorporated into the growing RNA, inducing a chain termination event.37 In phase 3 trials,38,39 researchers found an initial rapid decrease in viral load for patients treated with this agent regardless of IL28B status.
In the NEUTRINO trial (Sofosbuvir With Peginterferon Alfa 2a and Ribavirin for 12 Weeks in Treatment-Naive Subjects With Chronic Genotype 1, 4, 5, or 6 HCV Infection),38 which used sofusbuvir in combination with interferon and ribavirin, the rate of sustained virologic response was higher in those with the favorable CC genotype (98%) than with a non-CC genotype (87%).
In COSMOS (A Study of TMC435 in Combination With PSI-7977 [GS7977] in Chronic Hepatitis C Genotype 1-Infected Prior Null Responders to Peginterferon/Ribavirin Therapy or HCV Treatment-Naive Patients),39 which used a combination of simeprevir, sofosbuvir, and ribavirin, the rate of sustained virologic response was higher in those with the CC genotype (100%) than with the TT genotype (83%; Table 1).
These new medications have radically changed the landscape of hepatitis C therapy and have also unlocked the potential for developing completely interferon-free regimens.
Other new interferon-free regimens such as ledipasvir, daclatasvir, and asunaprevir promise high rates of sustained virologic response, which makes the utility of testing for IL28B polymorphisms to predict sustained virologic response very much diminished (Table 1).40,41 However, these new drugs are expected to be expensive, and IL28B polymorphisms may be used to identify candidates who are more likely to respond to pegylated interferon and ribavirin, particularly in resource-poor settings and in developing countries. Additionally, patients who have contraindications to these newer therapies will still likely need an interferon-based regimen, and thus the IL28B polymorphism will still be important in predicting treatment response and prognosis.
IL28B WILL STILL BE RELEVANT IN THE INTERFERON-FREE AGE
The IL28B polymorphism is a strong predictor of spontaneous clearance of hepatitis C virus and responsiveness to interferon-based therapy, and testing for it has demonstrated a great potential to improve patient care. IL28B testing has become available for clinical use and may optimize the outcome of hepatitis C treatment by helping us to select the best treatment for individual patients and minimizing the duration of therapy and the side effects associated with interferon-based antiviral medications.
As newer therapies have shifted toward interferon-free regimens that offer very high sustained virologic response rates, the usefulness of IL28B polymorphism as a clinical test to predict the response rate to antiviral therapy is minimized substantially. It may remain clinically relevant in resource-poor settings and in developing countries, especially in light of the potentially prohibitive costs of the newer regimens, and for patients in whom these treatments are contraindicated. This does not minimize the lesson we learned from the discovery of the IL28B gene and the impact on our understanding of the pathogenesis of hepatitis C virus infection.
- Attia J, Ioannidis JP, Thakkinstian A, et al. How to use an article about genetic association: A: background concepts. JAMA 2009; 301:74–81.
- Samani NJ, Erdmann J, Hall AS, et al; WTCCC and the Cardiogenics Consortium. Genomewide association analysis of coronary artery disease. N Engl J Med 2007; 357:443–453.
- Zeggini E, Weedon MN, Lindgren CM, et al; Wellcome Trust Case Control Consortium (WTCCC). Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science 2007; 316:1336–1341.
- Matarín M, Brown WM, Scholz S, et al. A genome-wide genotyping study in patients with ischaemic stroke: initial analysis and data release. Lancet Neurol 2007; 6:414–420.
- Easton DF, Pooley KA, Dunning AM, et al; AOCS Management Group. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 2007; 447:1087–1093.
- Plenge RM, Seielstad M, Padyukov L, et al. TRAF1-C5 as a risk locus for rheumatoid arthritis—a genomewide study. N Engl J Med 2007; 357:1199–1209.
- Coon KD, Myers AJ, Craig DW, et al. A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer’s disease. J Clin Psychiatry 2007; 68:613–618.
- Ge D, Fellay J, Thompson AJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance (letter). Nature 2009; 461:399–401.
- Ali A, Zein NN. Hepatitis C infection: a systemic disease with extrahepatic manifestations. Cleve Clin J Med 2005; 72:1005-1019.
- Hanouneh IA, Feldstein AE, Lopez R, et al. Clinical significance of metabolic syndrome in the setting of chronic hepatitis C virus infection. Clin Gastroenterol Hepatol 2008; 6:584–589.
- Elgouhari HM, Zein CO, Hanouneh I, Feldstein AE, Zein NN. Diabetes mellitus is associated with impaired response to antiviral therapy in chronic hepatitis C infection. Dig Dis Sci 2009; 54:2699–2705.
- Alkhouri N, Zein NN. Protease inhibitors: silver bullets for chronic hepatitis C infection? Cleve Clin J Med 2012; 79:213–222.
- McHutchison JG, Everson GT, Gordon SC, et al; PROVE1 Study Team. Telaprevir with peginterferon and ribavirin for chronic HCV genotype 1 infection. N Engl J Med 2009; 360:1827–1838.
- Jacobson IM, McHutchison JG, Dusheiko G, et al; ADVANCE Study Team. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med 2011; 364:2405–2416.
- Jacobson IM, Catlett I, Marcellin P, et al. Telaprevir substantially improved SVR rates across all IL28B genotypes in the ADVANCE trial. J Hepatol 2011; 54(suppl 1):S542–S543.
- Pol S, Aerssens J, Zeuzem S, et al. Limited impact of IL28B genotype on response rates in telaprevir-treated patients with prior treatment failure. J Hepatol 2013; 58:883–889.
- Suppiah V, Moldovan M, Ahlenstiel G, et al. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat Genet 2009; 41:1100–1104.
- Tanaka Y, Nishida N, Sugiyama M, et al. Genome-wide association of IL28B with response to pegylated interferon-alpha and ribavirin therapy for chronic hepatitis C. Nat Genet 2009; 41:1105–1109.
- Thomas DL, Thio CL, Martin MP, et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature 2009; 461:798–801.
- Rehermann B. Hepatitis C virus versus innate and adaptive immune responses: a tale of coevolution and coexistence. J Clin Invest 2009; 119:1745–1754.
- Marcello T, Grakoui A, Barba-Spaeth G, et al. Interferons alpha and lambda inhibit hepatitis C virus replication with distinct signal transduction and gene regulation kinetics. Gastroenterology 2006; 131:1887–1898.
- Doyle SE, Schreckhise H, Khuu-Duong K, et al. Interleukin-29 uses a type 1 interferon-like program to promote antiviral responses in human hepatocytes. Hepatology 2006; 44:896–906.
- Raglow Z, Thoma-Perry C, Gilroy R, Wan YJ. IL28B genotype and the expression of ISGs in normal liver. Liver Int 2013; 33:991–998.
- Prokunina-Olsson L, Muchmore B, Tang W, et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat Genet 2013; 45:164–171.
- Hanouneh IA, Zein NN, Askar M, Lopez R, John B. Interleukin-28B polymorphisms are associated with fibrosing cholestatic hepatitis in recurrent hepatitis C after liver transplantation. Clin Transplant 2012; 26:E335–E336.
- Poynard T, Bedossa P, Opolon P. Natural history of liver fibrosis progression in patients with chronic hepatitis C. The OBSVIRC, METAVIR, CLINIVIR, and DOSVIRC groups. Lancet 1997; 349:825–832.
- Thomas DL, Astemborski J, Rai RM, et al. The natural history of hepatitis C virus infection: host, viral, and environmental factors. JAMA 2000; 284:450–456.
- Bochud PY, Cai T, Overbeck K, et al; Swiss Hepatitis C Cohort Study Group. Genotype 3 is associated with accelerated fibrosis progression in chronic hepatitis C. J Hepatol 2009; 51:655–666.
- Marabita F, Aghemo A, De Nicola S, et al. Genetic variation in the interleukin-28B gene is not associated with fibrosis progression in patients with chronic hepatitis C and known date of infection. Hepatology 2011; 54:1127–1134.
- Fabris C, Falleti E, Cussigh A, et al. IL-28B rs12979860 C/T allele distribution in patients with liver cirrhosis: role in the course of chronic viral hepatitis and the development of HCC. J Hepatol 2011; 54:716–722.
- Eurich D, Boas-Knoop S, Bahra M, et al. Role of IL28B polymorphism in the development of hepatitis C virus-induced hepatocellular carcinoma, graft fibrosis, and posttransplant antiviral therapy. Transplantation 2012; 93:644–649.
- Hanouneh IA, Miller C, Aucejo F, Lopez R, Quinn MK, Zein NN. Recurrent hepatitis C after liver transplantation: on-treatment prediction of response to peginterferon/ribavirin therapy. Liver Transpl 2008; 14:53–58.
- Charlton MR, Thompson A, Veldt BJ, et al. Interleukin-28B polymorphisms are associated with histological recurrence and treatment response following liver transplantation in patients with hepatitis C virus infection. Hepatology 2011; 53:317–324.
- Thompson AJ, Muir AJ, Sulkowski MS, et al. Interleukin-28B polymorphism improves viral kinetics and is the strongest pretreatment predictor of sustained virologic response in genotype 1 hepatitis C virus. Gastroenterology 2010; 139:120–129.e18.
- Beinhardt S, Payer BA, Datz C, et al. A diagnostic score for the prediction of spontaneous resolution of acute hepatitis C virus infection. J Hepatol 2013; 59:972–977.
- Forns X, Lawitz E, Zeuzem S, et al. Simeprevir with peginterferon and ribavirin leads to high rates of SVR in patients with HCV genotype 1 who relapsed after previous therapy: a phase 3 trial. Gastroenterology 2014; 146:1669–1679.e3.
- Sofia MJ, Bao D, Chang W, et al. Discovery of a ß-d-2’-deoxy-2’-ß-fluoro-2’-ß-C-methyluridine nucleotide prodrug (PSI-7977) for the treatment of hepatitis C virus. J Med Chem 2010; 53:7202–7218.
- Lawitz E, Mangia A, Wyles D, et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med 2013; 368:1878–1887.
- Sulkowski MS, Jacobson IM, Ghalib R, et al. Once-daily simeprevir (TMC435) plus sofosbuvir (GS-7977) with or without ribavirin in HCV genotype 1 prior null responders with metavir F0-2: COSMOS study subgroup analysis. 49th EASL, April 2014, London. Oral abstract O7. www.natap.org/2014/EASL/EASL_46.htm. Accesed January 9, 2015.
- Lok AS, Gardiner DF, Lawitz E, et al. Preliminary study of two antiviral agents for hepatitis C genotype 1. N Engl J Med 2012; 366:216–224.
- Afdhal N, Zeuzem S, Kwo P, et al; ION-1 Investigators. Ledipasvir and sofosbuvir for untreated HCV genotype 1 infection. N Engl J Med 2014; 370:1889–1898.
What a difference a single nucleotide can make! The human genome contains more than 3 billion base pairs. Yet having a different nucleotide in only one pair can make a big difference in how we respond to a disease or its treatment.
Specifically, in hepatitis C virus infection, people born with the nucleotide cytosine (C) at location rs12979860 in both alleles of the gene that codes for interleukin 28B (the IL28B CC genotype) can count themselves luckier than those born with thymine (T) in this location in one of their alleles (the CT genotype) or both of their alleles (the TT genotype). Those with the CC genotype are more likely to clear the virus spontaneously, and even if the infection persists, it is less likely to progress to liver cancer and more likely to respond to treatment with interferon.
Here, we review the IL28B polymorphism and its implications in treating hepatitis C.
GENETIC POLYMORPHISM AND HUMAN DISEASE
Of the 3 billion base pairs of nucleotides, fewer than 1% differ between individuals, but this 1% is responsible for the diversity of human beings. Differences in genetic sequences among individuals are called genetic polymorphisms. A single-nucleotide polymorphism is a DNA sequence variation that occurs in a single nucleotide in the genome. For example, two sequenced DNA fragments from different individuals, AAGCCTA and AAGCTTA, contain a difference in a single nucleotide.
Genetic variations such as these underlie some of the differences in our susceptibility to disease, the severity of illness we develop, and our response to treatments. Therefore, identifying genetic polymorphisms may shed light on biologic pathways involved in diseases and may uncover new targets for therapy.1
Genome-wide association studies have looked at hundreds of thousands of single-nucleotide polymorphisms to try to identify most of the common genetic differences among people and relate them to common chronic diseases such as coronary artery disease,2 type 2 diabetes,3 stroke,4 breast cancer,5 rheumatoid arthritis,6 Alzheimer disease,7 and, more recently, hepatitis C virus infection.8
HEPATITIS C VIRUS: A MAJOR CAUSE OF LIVER DISEASE
Hepatitis C virus infection is a major cause of chronic liver disease and hepatocellular carcinoma and has become the most common indication for liver transplantation in the United States.9
This virus has six distinct genotypes throughout the world, with multiple subtypes in each genotype. (A genotype is a classification of a virus based on its RNA.9) In this review, we will focus on genotype 1; hence, “hepatitis C virus” will refer to hepatitis C virus genotype 1.
Our knowledge of the biology, pathogenesis, and treatment of hepatitis C has been advancing. Originally, fewer than 50% of patients responded to therapy with the combination of pegylated interferon and ribavirin,10,11 but since 2011 the response rate has increased to approximately 70% with the approval of the protease inhibitors telaprevir and boceprevir, used in combination with pegylated interferon and ribavirin.12–15
Unfortunately, interferon-based treatment is often complicated by side effects such as fatigue, influenza-like symptoms, hematologic abnormalities, and neuropsychiatric symptoms. An accurate way to predict response would help patients make informed decisions about antiviral treatment, taking into account the risk and possible benefit for individual patients.
GENETIC POLYMORPHISM AND HEPATITIS C VIRUS INFECTION
Genome-wide association studies have identified single-nucleotide polymorphisms in the IL28B gene that are associated with differences in response to hepatitis C treatment.8
Studying 565,759 polymorphisms in 1,137 patients, researchers at Duke University identified a single-nucleotide polymorphism at location rs12979860 in IL28B (Figure 1) that was strongly associated with response to combination therapy with pegylated interferon and ribavirin.8 The chance of cure with this standard treatment is twice as high in patients who are homozygous for cytosine in this location (the CC genotype) than in those who are heterozygous (CT) or homozygous for thymine in this location (the TT genotype) (Table 1).
Adding one of the new protease inhibitors, telaprevir or boceprevir, to the standard hepatitis C treatment substantially improves the cure rates in all three IL28B genotypes, but especially in people with CT or TT, in whom the response rate almost triples with the addition of one of these drugs. Those with the CC genotype (who are more likely to be cured with pegylated interferon and ribavirin alone) also achieve an increase (although minimal) in cure rates when a protease inhibitor is included in the regimen (TABLE 1).13–15 Thus, it remains unclear if adding a protease inhibitor to pegylated interferon plus ribavirin in patients with the IL28B CC genotype translates into added effectiveness worth the additional cost of the protease inhibitor in previously untreated patients.
Additionally, the effect of the IL28B genotype on telaprevir-based triple therapy has been disputed in more recent studies. In a subgroup analysis of the results of a trial that evaluated telaprevir in the treatment of hepatitis C, researchers found that sustained virologic response rates were significantly higher in the telaprevir group, and this was similar across the different IL28B polymorphisms.16
The favorable IL28B CC genotype is associated with higher rates of rapid virologic response to antiviral therapy.13–15 Of note, almost all patients who achieve a rapid virologic response do well, with a high rate of sustained virologic response even after a shorter duration of therapy (24 vs 48 weeks). Therefore, in addition to predicting response to interferon before starting treatment, the IL28B CC genotype may also identify patients who need only a shorter duration of therapy.
Interestingly, the C allele is much more frequent in white than in African American populations, an important observation that explains the racial difference in response to hepatitis C therapy.8
Two other research groups, from Asia and Australia, performed independent genome-wide association studies that identified different single-nucleotide polymorphisms (eg, rs8099917) in the same IL28B gene as predictors of response to treatment in patients with hepatitis C virus infection.17,18 These findings may be explained by linkage disequilibrium, which means that these single-nucleotide polymorphisms are found more frequently together in the same patient due to their proximity to each other. In this review, we will focus on the rs12979860 polymorphism; hence “IL28B genotype” will refer to the single-nucleotide polymorphism at rs12979860, unless otherwise specified.
The favorable CC genotype is less common in African Americans than in patients of other ethnicities.19 Moreover, although IL28B CC is associated with a better response rate to interferon-based antiviral therapy across all ethnicities, those of African American descent with the CC genotype are less likely to achieve a sustained virologic response than white or Hispanic Americans.8
BIOLOGIC ASSOCIATION: IL28B POLYMORPHISM AND HEPATITIS C
The interferon lambda family consists of three cytokines:
- Interleukin 29 (interferon lambda 1)
- Interleukin 28A (interferon lambda 2)
- Interleukin 28B (interferon lambda 3).
Production of these three molecules can be triggered by viral infection, and they induce antiviral activity through both innate and adaptive immune pathways. They signal through the IL10R-IL28R receptor complex.20–22 This receptor activates the JAK-STAT (Janus kinase-signal transducer and activator of transcription) pathway, which regulates a large number of interferon-stimulated genes, primarily through the interferon-stimulated response element (Figure 2).
A 2013 study found that interferon-stimulated gene expression levels in patients with normal livers were highest in those with the CC genotype, intermediate with CT, and lowest with TT. Interestingly, this pattern was reversed in those with hepatitis C virus infection, indicating a relationship between the IL28B genotype and gene expression before infection.23
The mechanism underlying the association between the IL28B polymorphism and response to hepatitis C treatment is not well understood. The unfavorable TT genotype seems to lead to continuous activation of a subset of interferon-stimulated genes in the presence of intracellular hepatitis C viral RNA. But this level of expression is not sufficient to eliminate the virus from the cells. Instead, it might lead to up-regulation of interferon-inhibitory molecules that suppress JAK-STAT signaling, thereby reducing sensitivity to interferon signaling. Therefore, the hepatocyte not only cannot clear the virus by itself, but also cannot induce strong interferon-stimulated gene expression when interferon is given during therapy.20–22
The recently identified ss469425590 polymorphism, which is located in close proximity to rs12979860 in the IL28B gene, is particularly interesting, as it suggests a possible molecular mechanism. The delta G frameshift variant creates a novel gene called IFNL4, which is transiently activated in response to hepatitis C virus infection.24IFNL4 stimulates STAT1 and STAT2 phosphorylation and induces the expression of interferon-stimulated genes. Increased interferon-stimulated gene expression has been shown to be associated with decreased response to pegylated interferon-ribavirin treatment. These observations suggest that the ss469425590 delta G allele is responsible for the increased activation of interferon-stimulated genes and the lower sustained virologic response rate observed in patients who receive pegylated interferon-ribavirin treatment. It is possible that the activation of interferon-stimulated genes in patients with the ss469425590 delta G/delta G genotype reduces interferon-stimulated gene responsiveness to interferon alpha, which normally activates interferon-stimulated genes and inhibits hepatitis C progression.24
IL28B POLYMORPHISM AND ACUTE HEPATITIS C VIRUS INFECTION
From 70% to 80% of acute hepatitis C virus infections persist and become chronic, while 20% to 30% spontaneously resolve. Epidemiologic, viral, and host factors have been associated with the differences in viral clearance or persistence, and studies have found that a strong host immune response against the virus favors viral clearance. Thus, variation in the genes involved in the immune response may contribute to one’s ability to clear the virus. Consistent with these observations, recent studies have shown that the polymorphism in the IL28 gene region encoding interferon lambda 3 strongly predicts spontaneous resolution of acute hepatitis C virus infection. People who have the IL28B CC genotype are three times more likely to spontaneously clear the virus than those with the CT or TT genotype (Figure 3).24
IL28B POLYMORPHISM AND THE NATURAL HISTORY OF HEPATITIS C
In people in whom hepatitis C virus infection persists, up to 20% develop progressive liver fibrosis and eventually cirrhosis over 10 to 20 years.19,25,26 The speed at which fibrosis develops in these patients is variable and unpredictable.25 The relationship between IL28B polymorphisms and hepatic fibrosis in patients with chronic hepatitis C virus infection has not been clearly established, although a study indicated that in patients with a known date of infection, the IL28B genotype is not associated with progression of hepatic fibrosis.27 Obstacles in this field of study are that it is difficult to determine accurately when the patient contracted the virus, and that serial liver biopsies are needed to investigate the progression of hepatic fibrosis.
Patients with chronic hepatitis C virus infection are also at higher risk of hepatocellular carcinoma compared with the general population.28 An analysis of explanted livers of patients with hepatitis C found that the prevalence of hepatocellular carcinoma in those with the unfavorable TT genotype was significantly higher than with the other genotypes.29 Similarly, an earlier study demonstrated that patients with hepatitis C-associated hepatocellular carcinoma carried the T allele more frequently.30 As with other aspects of IL28B associations with hepatitis C, these findings indicate that the C allele confers a certain degree of protection.
An important implication of these relationships is that they may eventually help identify patients at greater risk, who therefore need earlier intervention.
IL28B POLYMORPHISM AND LIVER TRANSPLANTATION
Hepatitis C virus infection always recurs after liver transplantation, with serious consequences that include cirrhosis and liver failure. Recurrent hepatitis C virus infection has become an important reason for repeat transplantation in the United States.
Results of treatment with pegylated interferon and ribavirin for recurrent hepatitis C after liver transplantation have been disappointing, with response rates lower than 30% and significant side effects.31 Identifying the factors that predict the response to therapy allows for better selection of treatment candidates.
Similar to the way the IL28B genotype predicts response to antiviral therapy in the nontransplant setting, the IL28B genotypes of both the recipient and the donor are strongly and independently associated with response to interferon-based treatment in patients with hepatitis C after liver transplantation. The IL28B CC genotype in either the recipient or the donor is associated with a higher rate of response to pegylated interferon and ribavirin combination therapy after liver transplantation.30,32 For example, the response rate to therapy after liver transplantation reaches 86% in CC-donor and CC-recipient livers, compared with 0% in TT-donor and TT-recipient livers.
Additionally, the IL28B genotype of the recipient may determine the severity of histologic recurrence of hepatitis C, as indicated by progressive hepatic fibrosis. A recipient IL28B TT genotype is associated with more severe histologic recurrence of hepatitis C.33
These data suggest that CC donor livers might be preferentially allocated to patients with hepatitis C virus infection.
IL28B AND OTHER FACTORS IN HEPATITIS C VIRUS INFECTION
Although it is tempting to think that the IL28B polymorphism is the sole predictor of response to antiviral therapy, it is but one of several known factors in the virus and the host.
While IL28B polymorphisms are the most important predictor of sustained virologic response with an interferon-based regimen, a rapid virologic response (undetectable viral load at 4 weeks) had superior predictive value and specificity in one study.34 In fact, for patients with chronic hepatitis C infection who achieved a rapid virologic response with pegylated interferon and ribavirin, the IL28B polymorphism had no effect on the rate of sustained virologic response. However, it did predict a sustained virologic response in the group who did not achieve rapid virologic response.
In a study of patients with acute hepatitis C infection,35 jaundice and the IL28 rs12979860 CC genotype both predicted spontaneous clearance. The best predictor of viral persistence was the combination of the CT or TT genotype plus the absence of jaundice, which had a predictive value of 98%.
IL28B AND THE FUTURE OF HEPATITIS C VIRUS THERAPY
New oral agents were recently approved for treating hepatitis C. As of November 2014, these included simeprevir, sofosbuvir, and ledipasvir.
Simeprevir is a second-generation NS3/4A protease inhibitor approved for use in combination with pegylated interferon and ribavirin. A recent phase 3 trial evaluating simeprevir in patients who had relapsed after prior therapy found sustained virologic response rates to be higher with simeprevir than with placebo, irrespective of IL28B status.36 This finding was similar to that of a trial of telaprevir.16
Sofosbuvir is a nucleotide analogue NS5B polymerase inhibitor that becomes incorporated into the growing RNA, inducing a chain termination event.37 In phase 3 trials,38,39 researchers found an initial rapid decrease in viral load for patients treated with this agent regardless of IL28B status.
In the NEUTRINO trial (Sofosbuvir With Peginterferon Alfa 2a and Ribavirin for 12 Weeks in Treatment-Naive Subjects With Chronic Genotype 1, 4, 5, or 6 HCV Infection),38 which used sofusbuvir in combination with interferon and ribavirin, the rate of sustained virologic response was higher in those with the favorable CC genotype (98%) than with a non-CC genotype (87%).
In COSMOS (A Study of TMC435 in Combination With PSI-7977 [GS7977] in Chronic Hepatitis C Genotype 1-Infected Prior Null Responders to Peginterferon/Ribavirin Therapy or HCV Treatment-Naive Patients),39 which used a combination of simeprevir, sofosbuvir, and ribavirin, the rate of sustained virologic response was higher in those with the CC genotype (100%) than with the TT genotype (83%; Table 1).
These new medications have radically changed the landscape of hepatitis C therapy and have also unlocked the potential for developing completely interferon-free regimens.
Other new interferon-free regimens such as ledipasvir, daclatasvir, and asunaprevir promise high rates of sustained virologic response, which makes the utility of testing for IL28B polymorphisms to predict sustained virologic response very much diminished (Table 1).40,41 However, these new drugs are expected to be expensive, and IL28B polymorphisms may be used to identify candidates who are more likely to respond to pegylated interferon and ribavirin, particularly in resource-poor settings and in developing countries. Additionally, patients who have contraindications to these newer therapies will still likely need an interferon-based regimen, and thus the IL28B polymorphism will still be important in predicting treatment response and prognosis.
IL28B WILL STILL BE RELEVANT IN THE INTERFERON-FREE AGE
The IL28B polymorphism is a strong predictor of spontaneous clearance of hepatitis C virus and responsiveness to interferon-based therapy, and testing for it has demonstrated a great potential to improve patient care. IL28B testing has become available for clinical use and may optimize the outcome of hepatitis C treatment by helping us to select the best treatment for individual patients and minimizing the duration of therapy and the side effects associated with interferon-based antiviral medications.
As newer therapies have shifted toward interferon-free regimens that offer very high sustained virologic response rates, the usefulness of IL28B polymorphism as a clinical test to predict the response rate to antiviral therapy is minimized substantially. It may remain clinically relevant in resource-poor settings and in developing countries, especially in light of the potentially prohibitive costs of the newer regimens, and for patients in whom these treatments are contraindicated. This does not minimize the lesson we learned from the discovery of the IL28B gene and the impact on our understanding of the pathogenesis of hepatitis C virus infection.
What a difference a single nucleotide can make! The human genome contains more than 3 billion base pairs. Yet having a different nucleotide in only one pair can make a big difference in how we respond to a disease or its treatment.
Specifically, in hepatitis C virus infection, people born with the nucleotide cytosine (C) at location rs12979860 in both alleles of the gene that codes for interleukin 28B (the IL28B CC genotype) can count themselves luckier than those born with thymine (T) in this location in one of their alleles (the CT genotype) or both of their alleles (the TT genotype). Those with the CC genotype are more likely to clear the virus spontaneously, and even if the infection persists, it is less likely to progress to liver cancer and more likely to respond to treatment with interferon.
Here, we review the IL28B polymorphism and its implications in treating hepatitis C.
GENETIC POLYMORPHISM AND HUMAN DISEASE
Of the 3 billion base pairs of nucleotides, fewer than 1% differ between individuals, but this 1% is responsible for the diversity of human beings. Differences in genetic sequences among individuals are called genetic polymorphisms. A single-nucleotide polymorphism is a DNA sequence variation that occurs in a single nucleotide in the genome. For example, two sequenced DNA fragments from different individuals, AAGCCTA and AAGCTTA, contain a difference in a single nucleotide.
Genetic variations such as these underlie some of the differences in our susceptibility to disease, the severity of illness we develop, and our response to treatments. Therefore, identifying genetic polymorphisms may shed light on biologic pathways involved in diseases and may uncover new targets for therapy.1
Genome-wide association studies have looked at hundreds of thousands of single-nucleotide polymorphisms to try to identify most of the common genetic differences among people and relate them to common chronic diseases such as coronary artery disease,2 type 2 diabetes,3 stroke,4 breast cancer,5 rheumatoid arthritis,6 Alzheimer disease,7 and, more recently, hepatitis C virus infection.8
HEPATITIS C VIRUS: A MAJOR CAUSE OF LIVER DISEASE
Hepatitis C virus infection is a major cause of chronic liver disease and hepatocellular carcinoma and has become the most common indication for liver transplantation in the United States.9
This virus has six distinct genotypes throughout the world, with multiple subtypes in each genotype. (A genotype is a classification of a virus based on its RNA.9) In this review, we will focus on genotype 1; hence, “hepatitis C virus” will refer to hepatitis C virus genotype 1.
Our knowledge of the biology, pathogenesis, and treatment of hepatitis C has been advancing. Originally, fewer than 50% of patients responded to therapy with the combination of pegylated interferon and ribavirin,10,11 but since 2011 the response rate has increased to approximately 70% with the approval of the protease inhibitors telaprevir and boceprevir, used in combination with pegylated interferon and ribavirin.12–15
Unfortunately, interferon-based treatment is often complicated by side effects such as fatigue, influenza-like symptoms, hematologic abnormalities, and neuropsychiatric symptoms. An accurate way to predict response would help patients make informed decisions about antiviral treatment, taking into account the risk and possible benefit for individual patients.
GENETIC POLYMORPHISM AND HEPATITIS C VIRUS INFECTION
Genome-wide association studies have identified single-nucleotide polymorphisms in the IL28B gene that are associated with differences in response to hepatitis C treatment.8
Studying 565,759 polymorphisms in 1,137 patients, researchers at Duke University identified a single-nucleotide polymorphism at location rs12979860 in IL28B (Figure 1) that was strongly associated with response to combination therapy with pegylated interferon and ribavirin.8 The chance of cure with this standard treatment is twice as high in patients who are homozygous for cytosine in this location (the CC genotype) than in those who are heterozygous (CT) or homozygous for thymine in this location (the TT genotype) (Table 1).
Adding one of the new protease inhibitors, telaprevir or boceprevir, to the standard hepatitis C treatment substantially improves the cure rates in all three IL28B genotypes, but especially in people with CT or TT, in whom the response rate almost triples with the addition of one of these drugs. Those with the CC genotype (who are more likely to be cured with pegylated interferon and ribavirin alone) also achieve an increase (although minimal) in cure rates when a protease inhibitor is included in the regimen (TABLE 1).13–15 Thus, it remains unclear if adding a protease inhibitor to pegylated interferon plus ribavirin in patients with the IL28B CC genotype translates into added effectiveness worth the additional cost of the protease inhibitor in previously untreated patients.
Additionally, the effect of the IL28B genotype on telaprevir-based triple therapy has been disputed in more recent studies. In a subgroup analysis of the results of a trial that evaluated telaprevir in the treatment of hepatitis C, researchers found that sustained virologic response rates were significantly higher in the telaprevir group, and this was similar across the different IL28B polymorphisms.16
The favorable IL28B CC genotype is associated with higher rates of rapid virologic response to antiviral therapy.13–15 Of note, almost all patients who achieve a rapid virologic response do well, with a high rate of sustained virologic response even after a shorter duration of therapy (24 vs 48 weeks). Therefore, in addition to predicting response to interferon before starting treatment, the IL28B CC genotype may also identify patients who need only a shorter duration of therapy.
Interestingly, the C allele is much more frequent in white than in African American populations, an important observation that explains the racial difference in response to hepatitis C therapy.8
Two other research groups, from Asia and Australia, performed independent genome-wide association studies that identified different single-nucleotide polymorphisms (eg, rs8099917) in the same IL28B gene as predictors of response to treatment in patients with hepatitis C virus infection.17,18 These findings may be explained by linkage disequilibrium, which means that these single-nucleotide polymorphisms are found more frequently together in the same patient due to their proximity to each other. In this review, we will focus on the rs12979860 polymorphism; hence “IL28B genotype” will refer to the single-nucleotide polymorphism at rs12979860, unless otherwise specified.
The favorable CC genotype is less common in African Americans than in patients of other ethnicities.19 Moreover, although IL28B CC is associated with a better response rate to interferon-based antiviral therapy across all ethnicities, those of African American descent with the CC genotype are less likely to achieve a sustained virologic response than white or Hispanic Americans.8
BIOLOGIC ASSOCIATION: IL28B POLYMORPHISM AND HEPATITIS C
The interferon lambda family consists of three cytokines:
- Interleukin 29 (interferon lambda 1)
- Interleukin 28A (interferon lambda 2)
- Interleukin 28B (interferon lambda 3).
Production of these three molecules can be triggered by viral infection, and they induce antiviral activity through both innate and adaptive immune pathways. They signal through the IL10R-IL28R receptor complex.20–22 This receptor activates the JAK-STAT (Janus kinase-signal transducer and activator of transcription) pathway, which regulates a large number of interferon-stimulated genes, primarily through the interferon-stimulated response element (Figure 2).
A 2013 study found that interferon-stimulated gene expression levels in patients with normal livers were highest in those with the CC genotype, intermediate with CT, and lowest with TT. Interestingly, this pattern was reversed in those with hepatitis C virus infection, indicating a relationship between the IL28B genotype and gene expression before infection.23
The mechanism underlying the association between the IL28B polymorphism and response to hepatitis C treatment is not well understood. The unfavorable TT genotype seems to lead to continuous activation of a subset of interferon-stimulated genes in the presence of intracellular hepatitis C viral RNA. But this level of expression is not sufficient to eliminate the virus from the cells. Instead, it might lead to up-regulation of interferon-inhibitory molecules that suppress JAK-STAT signaling, thereby reducing sensitivity to interferon signaling. Therefore, the hepatocyte not only cannot clear the virus by itself, but also cannot induce strong interferon-stimulated gene expression when interferon is given during therapy.20–22
The recently identified ss469425590 polymorphism, which is located in close proximity to rs12979860 in the IL28B gene, is particularly interesting, as it suggests a possible molecular mechanism. The delta G frameshift variant creates a novel gene called IFNL4, which is transiently activated in response to hepatitis C virus infection.24IFNL4 stimulates STAT1 and STAT2 phosphorylation and induces the expression of interferon-stimulated genes. Increased interferon-stimulated gene expression has been shown to be associated with decreased response to pegylated interferon-ribavirin treatment. These observations suggest that the ss469425590 delta G allele is responsible for the increased activation of interferon-stimulated genes and the lower sustained virologic response rate observed in patients who receive pegylated interferon-ribavirin treatment. It is possible that the activation of interferon-stimulated genes in patients with the ss469425590 delta G/delta G genotype reduces interferon-stimulated gene responsiveness to interferon alpha, which normally activates interferon-stimulated genes and inhibits hepatitis C progression.24
IL28B POLYMORPHISM AND ACUTE HEPATITIS C VIRUS INFECTION
From 70% to 80% of acute hepatitis C virus infections persist and become chronic, while 20% to 30% spontaneously resolve. Epidemiologic, viral, and host factors have been associated with the differences in viral clearance or persistence, and studies have found that a strong host immune response against the virus favors viral clearance. Thus, variation in the genes involved in the immune response may contribute to one’s ability to clear the virus. Consistent with these observations, recent studies have shown that the polymorphism in the IL28 gene region encoding interferon lambda 3 strongly predicts spontaneous resolution of acute hepatitis C virus infection. People who have the IL28B CC genotype are three times more likely to spontaneously clear the virus than those with the CT or TT genotype (Figure 3).24
IL28B POLYMORPHISM AND THE NATURAL HISTORY OF HEPATITIS C
In people in whom hepatitis C virus infection persists, up to 20% develop progressive liver fibrosis and eventually cirrhosis over 10 to 20 years.19,25,26 The speed at which fibrosis develops in these patients is variable and unpredictable.25 The relationship between IL28B polymorphisms and hepatic fibrosis in patients with chronic hepatitis C virus infection has not been clearly established, although a study indicated that in patients with a known date of infection, the IL28B genotype is not associated with progression of hepatic fibrosis.27 Obstacles in this field of study are that it is difficult to determine accurately when the patient contracted the virus, and that serial liver biopsies are needed to investigate the progression of hepatic fibrosis.
Patients with chronic hepatitis C virus infection are also at higher risk of hepatocellular carcinoma compared with the general population.28 An analysis of explanted livers of patients with hepatitis C found that the prevalence of hepatocellular carcinoma in those with the unfavorable TT genotype was significantly higher than with the other genotypes.29 Similarly, an earlier study demonstrated that patients with hepatitis C-associated hepatocellular carcinoma carried the T allele more frequently.30 As with other aspects of IL28B associations with hepatitis C, these findings indicate that the C allele confers a certain degree of protection.
An important implication of these relationships is that they may eventually help identify patients at greater risk, who therefore need earlier intervention.
IL28B POLYMORPHISM AND LIVER TRANSPLANTATION
Hepatitis C virus infection always recurs after liver transplantation, with serious consequences that include cirrhosis and liver failure. Recurrent hepatitis C virus infection has become an important reason for repeat transplantation in the United States.
Results of treatment with pegylated interferon and ribavirin for recurrent hepatitis C after liver transplantation have been disappointing, with response rates lower than 30% and significant side effects.31 Identifying the factors that predict the response to therapy allows for better selection of treatment candidates.
Similar to the way the IL28B genotype predicts response to antiviral therapy in the nontransplant setting, the IL28B genotypes of both the recipient and the donor are strongly and independently associated with response to interferon-based treatment in patients with hepatitis C after liver transplantation. The IL28B CC genotype in either the recipient or the donor is associated with a higher rate of response to pegylated interferon and ribavirin combination therapy after liver transplantation.30,32 For example, the response rate to therapy after liver transplantation reaches 86% in CC-donor and CC-recipient livers, compared with 0% in TT-donor and TT-recipient livers.
Additionally, the IL28B genotype of the recipient may determine the severity of histologic recurrence of hepatitis C, as indicated by progressive hepatic fibrosis. A recipient IL28B TT genotype is associated with more severe histologic recurrence of hepatitis C.33
These data suggest that CC donor livers might be preferentially allocated to patients with hepatitis C virus infection.
IL28B AND OTHER FACTORS IN HEPATITIS C VIRUS INFECTION
Although it is tempting to think that the IL28B polymorphism is the sole predictor of response to antiviral therapy, it is but one of several known factors in the virus and the host.
While IL28B polymorphisms are the most important predictor of sustained virologic response with an interferon-based regimen, a rapid virologic response (undetectable viral load at 4 weeks) had superior predictive value and specificity in one study.34 In fact, for patients with chronic hepatitis C infection who achieved a rapid virologic response with pegylated interferon and ribavirin, the IL28B polymorphism had no effect on the rate of sustained virologic response. However, it did predict a sustained virologic response in the group who did not achieve rapid virologic response.
In a study of patients with acute hepatitis C infection,35 jaundice and the IL28 rs12979860 CC genotype both predicted spontaneous clearance. The best predictor of viral persistence was the combination of the CT or TT genotype plus the absence of jaundice, which had a predictive value of 98%.
IL28B AND THE FUTURE OF HEPATITIS C VIRUS THERAPY
New oral agents were recently approved for treating hepatitis C. As of November 2014, these included simeprevir, sofosbuvir, and ledipasvir.
Simeprevir is a second-generation NS3/4A protease inhibitor approved for use in combination with pegylated interferon and ribavirin. A recent phase 3 trial evaluating simeprevir in patients who had relapsed after prior therapy found sustained virologic response rates to be higher with simeprevir than with placebo, irrespective of IL28B status.36 This finding was similar to that of a trial of telaprevir.16
Sofosbuvir is a nucleotide analogue NS5B polymerase inhibitor that becomes incorporated into the growing RNA, inducing a chain termination event.37 In phase 3 trials,38,39 researchers found an initial rapid decrease in viral load for patients treated with this agent regardless of IL28B status.
In the NEUTRINO trial (Sofosbuvir With Peginterferon Alfa 2a and Ribavirin for 12 Weeks in Treatment-Naive Subjects With Chronic Genotype 1, 4, 5, or 6 HCV Infection),38 which used sofusbuvir in combination with interferon and ribavirin, the rate of sustained virologic response was higher in those with the favorable CC genotype (98%) than with a non-CC genotype (87%).
In COSMOS (A Study of TMC435 in Combination With PSI-7977 [GS7977] in Chronic Hepatitis C Genotype 1-Infected Prior Null Responders to Peginterferon/Ribavirin Therapy or HCV Treatment-Naive Patients),39 which used a combination of simeprevir, sofosbuvir, and ribavirin, the rate of sustained virologic response was higher in those with the CC genotype (100%) than with the TT genotype (83%; Table 1).
These new medications have radically changed the landscape of hepatitis C therapy and have also unlocked the potential for developing completely interferon-free regimens.
Other new interferon-free regimens such as ledipasvir, daclatasvir, and asunaprevir promise high rates of sustained virologic response, which makes the utility of testing for IL28B polymorphisms to predict sustained virologic response very much diminished (Table 1).40,41 However, these new drugs are expected to be expensive, and IL28B polymorphisms may be used to identify candidates who are more likely to respond to pegylated interferon and ribavirin, particularly in resource-poor settings and in developing countries. Additionally, patients who have contraindications to these newer therapies will still likely need an interferon-based regimen, and thus the IL28B polymorphism will still be important in predicting treatment response and prognosis.
IL28B WILL STILL BE RELEVANT IN THE INTERFERON-FREE AGE
The IL28B polymorphism is a strong predictor of spontaneous clearance of hepatitis C virus and responsiveness to interferon-based therapy, and testing for it has demonstrated a great potential to improve patient care. IL28B testing has become available for clinical use and may optimize the outcome of hepatitis C treatment by helping us to select the best treatment for individual patients and minimizing the duration of therapy and the side effects associated with interferon-based antiviral medications.
As newer therapies have shifted toward interferon-free regimens that offer very high sustained virologic response rates, the usefulness of IL28B polymorphism as a clinical test to predict the response rate to antiviral therapy is minimized substantially. It may remain clinically relevant in resource-poor settings and in developing countries, especially in light of the potentially prohibitive costs of the newer regimens, and for patients in whom these treatments are contraindicated. This does not minimize the lesson we learned from the discovery of the IL28B gene and the impact on our understanding of the pathogenesis of hepatitis C virus infection.
- Attia J, Ioannidis JP, Thakkinstian A, et al. How to use an article about genetic association: A: background concepts. JAMA 2009; 301:74–81.
- Samani NJ, Erdmann J, Hall AS, et al; WTCCC and the Cardiogenics Consortium. Genomewide association analysis of coronary artery disease. N Engl J Med 2007; 357:443–453.
- Zeggini E, Weedon MN, Lindgren CM, et al; Wellcome Trust Case Control Consortium (WTCCC). Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science 2007; 316:1336–1341.
- Matarín M, Brown WM, Scholz S, et al. A genome-wide genotyping study in patients with ischaemic stroke: initial analysis and data release. Lancet Neurol 2007; 6:414–420.
- Easton DF, Pooley KA, Dunning AM, et al; AOCS Management Group. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 2007; 447:1087–1093.
- Plenge RM, Seielstad M, Padyukov L, et al. TRAF1-C5 as a risk locus for rheumatoid arthritis—a genomewide study. N Engl J Med 2007; 357:1199–1209.
- Coon KD, Myers AJ, Craig DW, et al. A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer’s disease. J Clin Psychiatry 2007; 68:613–618.
- Ge D, Fellay J, Thompson AJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance (letter). Nature 2009; 461:399–401.
- Ali A, Zein NN. Hepatitis C infection: a systemic disease with extrahepatic manifestations. Cleve Clin J Med 2005; 72:1005-1019.
- Hanouneh IA, Feldstein AE, Lopez R, et al. Clinical significance of metabolic syndrome in the setting of chronic hepatitis C virus infection. Clin Gastroenterol Hepatol 2008; 6:584–589.
- Elgouhari HM, Zein CO, Hanouneh I, Feldstein AE, Zein NN. Diabetes mellitus is associated with impaired response to antiviral therapy in chronic hepatitis C infection. Dig Dis Sci 2009; 54:2699–2705.
- Alkhouri N, Zein NN. Protease inhibitors: silver bullets for chronic hepatitis C infection? Cleve Clin J Med 2012; 79:213–222.
- McHutchison JG, Everson GT, Gordon SC, et al; PROVE1 Study Team. Telaprevir with peginterferon and ribavirin for chronic HCV genotype 1 infection. N Engl J Med 2009; 360:1827–1838.
- Jacobson IM, McHutchison JG, Dusheiko G, et al; ADVANCE Study Team. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med 2011; 364:2405–2416.
- Jacobson IM, Catlett I, Marcellin P, et al. Telaprevir substantially improved SVR rates across all IL28B genotypes in the ADVANCE trial. J Hepatol 2011; 54(suppl 1):S542–S543.
- Pol S, Aerssens J, Zeuzem S, et al. Limited impact of IL28B genotype on response rates in telaprevir-treated patients with prior treatment failure. J Hepatol 2013; 58:883–889.
- Suppiah V, Moldovan M, Ahlenstiel G, et al. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat Genet 2009; 41:1100–1104.
- Tanaka Y, Nishida N, Sugiyama M, et al. Genome-wide association of IL28B with response to pegylated interferon-alpha and ribavirin therapy for chronic hepatitis C. Nat Genet 2009; 41:1105–1109.
- Thomas DL, Thio CL, Martin MP, et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature 2009; 461:798–801.
- Rehermann B. Hepatitis C virus versus innate and adaptive immune responses: a tale of coevolution and coexistence. J Clin Invest 2009; 119:1745–1754.
- Marcello T, Grakoui A, Barba-Spaeth G, et al. Interferons alpha and lambda inhibit hepatitis C virus replication with distinct signal transduction and gene regulation kinetics. Gastroenterology 2006; 131:1887–1898.
- Doyle SE, Schreckhise H, Khuu-Duong K, et al. Interleukin-29 uses a type 1 interferon-like program to promote antiviral responses in human hepatocytes. Hepatology 2006; 44:896–906.
- Raglow Z, Thoma-Perry C, Gilroy R, Wan YJ. IL28B genotype and the expression of ISGs in normal liver. Liver Int 2013; 33:991–998.
- Prokunina-Olsson L, Muchmore B, Tang W, et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat Genet 2013; 45:164–171.
- Hanouneh IA, Zein NN, Askar M, Lopez R, John B. Interleukin-28B polymorphisms are associated with fibrosing cholestatic hepatitis in recurrent hepatitis C after liver transplantation. Clin Transplant 2012; 26:E335–E336.
- Poynard T, Bedossa P, Opolon P. Natural history of liver fibrosis progression in patients with chronic hepatitis C. The OBSVIRC, METAVIR, CLINIVIR, and DOSVIRC groups. Lancet 1997; 349:825–832.
- Thomas DL, Astemborski J, Rai RM, et al. The natural history of hepatitis C virus infection: host, viral, and environmental factors. JAMA 2000; 284:450–456.
- Bochud PY, Cai T, Overbeck K, et al; Swiss Hepatitis C Cohort Study Group. Genotype 3 is associated with accelerated fibrosis progression in chronic hepatitis C. J Hepatol 2009; 51:655–666.
- Marabita F, Aghemo A, De Nicola S, et al. Genetic variation in the interleukin-28B gene is not associated with fibrosis progression in patients with chronic hepatitis C and known date of infection. Hepatology 2011; 54:1127–1134.
- Fabris C, Falleti E, Cussigh A, et al. IL-28B rs12979860 C/T allele distribution in patients with liver cirrhosis: role in the course of chronic viral hepatitis and the development of HCC. J Hepatol 2011; 54:716–722.
- Eurich D, Boas-Knoop S, Bahra M, et al. Role of IL28B polymorphism in the development of hepatitis C virus-induced hepatocellular carcinoma, graft fibrosis, and posttransplant antiviral therapy. Transplantation 2012; 93:644–649.
- Hanouneh IA, Miller C, Aucejo F, Lopez R, Quinn MK, Zein NN. Recurrent hepatitis C after liver transplantation: on-treatment prediction of response to peginterferon/ribavirin therapy. Liver Transpl 2008; 14:53–58.
- Charlton MR, Thompson A, Veldt BJ, et al. Interleukin-28B polymorphisms are associated with histological recurrence and treatment response following liver transplantation in patients with hepatitis C virus infection. Hepatology 2011; 53:317–324.
- Thompson AJ, Muir AJ, Sulkowski MS, et al. Interleukin-28B polymorphism improves viral kinetics and is the strongest pretreatment predictor of sustained virologic response in genotype 1 hepatitis C virus. Gastroenterology 2010; 139:120–129.e18.
- Beinhardt S, Payer BA, Datz C, et al. A diagnostic score for the prediction of spontaneous resolution of acute hepatitis C virus infection. J Hepatol 2013; 59:972–977.
- Forns X, Lawitz E, Zeuzem S, et al. Simeprevir with peginterferon and ribavirin leads to high rates of SVR in patients with HCV genotype 1 who relapsed after previous therapy: a phase 3 trial. Gastroenterology 2014; 146:1669–1679.e3.
- Sofia MJ, Bao D, Chang W, et al. Discovery of a ß-d-2’-deoxy-2’-ß-fluoro-2’-ß-C-methyluridine nucleotide prodrug (PSI-7977) for the treatment of hepatitis C virus. J Med Chem 2010; 53:7202–7218.
- Lawitz E, Mangia A, Wyles D, et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med 2013; 368:1878–1887.
- Sulkowski MS, Jacobson IM, Ghalib R, et al. Once-daily simeprevir (TMC435) plus sofosbuvir (GS-7977) with or without ribavirin in HCV genotype 1 prior null responders with metavir F0-2: COSMOS study subgroup analysis. 49th EASL, April 2014, London. Oral abstract O7. www.natap.org/2014/EASL/EASL_46.htm. Accesed January 9, 2015.
- Lok AS, Gardiner DF, Lawitz E, et al. Preliminary study of two antiviral agents for hepatitis C genotype 1. N Engl J Med 2012; 366:216–224.
- Afdhal N, Zeuzem S, Kwo P, et al; ION-1 Investigators. Ledipasvir and sofosbuvir for untreated HCV genotype 1 infection. N Engl J Med 2014; 370:1889–1898.
- Attia J, Ioannidis JP, Thakkinstian A, et al. How to use an article about genetic association: A: background concepts. JAMA 2009; 301:74–81.
- Samani NJ, Erdmann J, Hall AS, et al; WTCCC and the Cardiogenics Consortium. Genomewide association analysis of coronary artery disease. N Engl J Med 2007; 357:443–453.
- Zeggini E, Weedon MN, Lindgren CM, et al; Wellcome Trust Case Control Consortium (WTCCC). Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science 2007; 316:1336–1341.
- Matarín M, Brown WM, Scholz S, et al. A genome-wide genotyping study in patients with ischaemic stroke: initial analysis and data release. Lancet Neurol 2007; 6:414–420.
- Easton DF, Pooley KA, Dunning AM, et al; AOCS Management Group. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 2007; 447:1087–1093.
- Plenge RM, Seielstad M, Padyukov L, et al. TRAF1-C5 as a risk locus for rheumatoid arthritis—a genomewide study. N Engl J Med 2007; 357:1199–1209.
- Coon KD, Myers AJ, Craig DW, et al. A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer’s disease. J Clin Psychiatry 2007; 68:613–618.
- Ge D, Fellay J, Thompson AJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance (letter). Nature 2009; 461:399–401.
- Ali A, Zein NN. Hepatitis C infection: a systemic disease with extrahepatic manifestations. Cleve Clin J Med 2005; 72:1005-1019.
- Hanouneh IA, Feldstein AE, Lopez R, et al. Clinical significance of metabolic syndrome in the setting of chronic hepatitis C virus infection. Clin Gastroenterol Hepatol 2008; 6:584–589.
- Elgouhari HM, Zein CO, Hanouneh I, Feldstein AE, Zein NN. Diabetes mellitus is associated with impaired response to antiviral therapy in chronic hepatitis C infection. Dig Dis Sci 2009; 54:2699–2705.
- Alkhouri N, Zein NN. Protease inhibitors: silver bullets for chronic hepatitis C infection? Cleve Clin J Med 2012; 79:213–222.
- McHutchison JG, Everson GT, Gordon SC, et al; PROVE1 Study Team. Telaprevir with peginterferon and ribavirin for chronic HCV genotype 1 infection. N Engl J Med 2009; 360:1827–1838.
- Jacobson IM, McHutchison JG, Dusheiko G, et al; ADVANCE Study Team. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med 2011; 364:2405–2416.
- Jacobson IM, Catlett I, Marcellin P, et al. Telaprevir substantially improved SVR rates across all IL28B genotypes in the ADVANCE trial. J Hepatol 2011; 54(suppl 1):S542–S543.
- Pol S, Aerssens J, Zeuzem S, et al. Limited impact of IL28B genotype on response rates in telaprevir-treated patients with prior treatment failure. J Hepatol 2013; 58:883–889.
- Suppiah V, Moldovan M, Ahlenstiel G, et al. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat Genet 2009; 41:1100–1104.
- Tanaka Y, Nishida N, Sugiyama M, et al. Genome-wide association of IL28B with response to pegylated interferon-alpha and ribavirin therapy for chronic hepatitis C. Nat Genet 2009; 41:1105–1109.
- Thomas DL, Thio CL, Martin MP, et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature 2009; 461:798–801.
- Rehermann B. Hepatitis C virus versus innate and adaptive immune responses: a tale of coevolution and coexistence. J Clin Invest 2009; 119:1745–1754.
- Marcello T, Grakoui A, Barba-Spaeth G, et al. Interferons alpha and lambda inhibit hepatitis C virus replication with distinct signal transduction and gene regulation kinetics. Gastroenterology 2006; 131:1887–1898.
- Doyle SE, Schreckhise H, Khuu-Duong K, et al. Interleukin-29 uses a type 1 interferon-like program to promote antiviral responses in human hepatocytes. Hepatology 2006; 44:896–906.
- Raglow Z, Thoma-Perry C, Gilroy R, Wan YJ. IL28B genotype and the expression of ISGs in normal liver. Liver Int 2013; 33:991–998.
- Prokunina-Olsson L, Muchmore B, Tang W, et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat Genet 2013; 45:164–171.
- Hanouneh IA, Zein NN, Askar M, Lopez R, John B. Interleukin-28B polymorphisms are associated with fibrosing cholestatic hepatitis in recurrent hepatitis C after liver transplantation. Clin Transplant 2012; 26:E335–E336.
- Poynard T, Bedossa P, Opolon P. Natural history of liver fibrosis progression in patients with chronic hepatitis C. The OBSVIRC, METAVIR, CLINIVIR, and DOSVIRC groups. Lancet 1997; 349:825–832.
- Thomas DL, Astemborski J, Rai RM, et al. The natural history of hepatitis C virus infection: host, viral, and environmental factors. JAMA 2000; 284:450–456.
- Bochud PY, Cai T, Overbeck K, et al; Swiss Hepatitis C Cohort Study Group. Genotype 3 is associated with accelerated fibrosis progression in chronic hepatitis C. J Hepatol 2009; 51:655–666.
- Marabita F, Aghemo A, De Nicola S, et al. Genetic variation in the interleukin-28B gene is not associated with fibrosis progression in patients with chronic hepatitis C and known date of infection. Hepatology 2011; 54:1127–1134.
- Fabris C, Falleti E, Cussigh A, et al. IL-28B rs12979860 C/T allele distribution in patients with liver cirrhosis: role in the course of chronic viral hepatitis and the development of HCC. J Hepatol 2011; 54:716–722.
- Eurich D, Boas-Knoop S, Bahra M, et al. Role of IL28B polymorphism in the development of hepatitis C virus-induced hepatocellular carcinoma, graft fibrosis, and posttransplant antiviral therapy. Transplantation 2012; 93:644–649.
- Hanouneh IA, Miller C, Aucejo F, Lopez R, Quinn MK, Zein NN. Recurrent hepatitis C after liver transplantation: on-treatment prediction of response to peginterferon/ribavirin therapy. Liver Transpl 2008; 14:53–58.
- Charlton MR, Thompson A, Veldt BJ, et al. Interleukin-28B polymorphisms are associated with histological recurrence and treatment response following liver transplantation in patients with hepatitis C virus infection. Hepatology 2011; 53:317–324.
- Thompson AJ, Muir AJ, Sulkowski MS, et al. Interleukin-28B polymorphism improves viral kinetics and is the strongest pretreatment predictor of sustained virologic response in genotype 1 hepatitis C virus. Gastroenterology 2010; 139:120–129.e18.
- Beinhardt S, Payer BA, Datz C, et al. A diagnostic score for the prediction of spontaneous resolution of acute hepatitis C virus infection. J Hepatol 2013; 59:972–977.
- Forns X, Lawitz E, Zeuzem S, et al. Simeprevir with peginterferon and ribavirin leads to high rates of SVR in patients with HCV genotype 1 who relapsed after previous therapy: a phase 3 trial. Gastroenterology 2014; 146:1669–1679.e3.
- Sofia MJ, Bao D, Chang W, et al. Discovery of a ß-d-2’-deoxy-2’-ß-fluoro-2’-ß-C-methyluridine nucleotide prodrug (PSI-7977) for the treatment of hepatitis C virus. J Med Chem 2010; 53:7202–7218.
- Lawitz E, Mangia A, Wyles D, et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med 2013; 368:1878–1887.
- Sulkowski MS, Jacobson IM, Ghalib R, et al. Once-daily simeprevir (TMC435) plus sofosbuvir (GS-7977) with or without ribavirin in HCV genotype 1 prior null responders with metavir F0-2: COSMOS study subgroup analysis. 49th EASL, April 2014, London. Oral abstract O7. www.natap.org/2014/EASL/EASL_46.htm. Accesed January 9, 2015.
- Lok AS, Gardiner DF, Lawitz E, et al. Preliminary study of two antiviral agents for hepatitis C genotype 1. N Engl J Med 2012; 366:216–224.
- Afdhal N, Zeuzem S, Kwo P, et al; ION-1 Investigators. Ledipasvir and sofosbuvir for untreated HCV genotype 1 infection. N Engl J Med 2014; 370:1889–1898.
KEY POINTS
- In IL28B, the rs12979860 location can be occupied by either cytosine (C) or thymine (T). The CC genotype is more favorable than the CT or TT genotype.
- Testing for the IL28B polymorphism is currently available and allows for better outcomes through proper selection of treatment, particularly with interferon-based treatment.
- Although newer therapies have shifted toward regimens that do not use interferon, the IL28B polymorphism remains clinically significant, especially in light of the potentially prohibitive costs of the newer regimens, and for patients in whom these treatments are contraindicated.