User login
Temporal Triangular Alopecia Acquired in Adulthood
To the Editor:
Temporal triangular alopecia (TTA), a condition first described by Sabouraud1 in 1905, is a circumscribed nonscarring form of alopecia. Also referred to as congenital triangular alopecia, TTA presents as a triangular or lancet-shaped area of hair loss involving the frontotemporal hairline. Temporal triangular alopecia is characterized histologically by a normal number of miniaturized hair follicles without notable inflammation.2 Although the majority of cases arise between birth and 9 years of age,3,4 rare cases of adult-onset TTA also have been reported.5,6 Adult-onset cases can cause notable diagnostic confusion and inappropriate treatment, as reported in our patient.
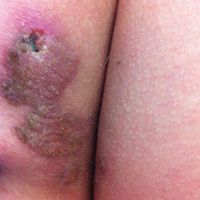
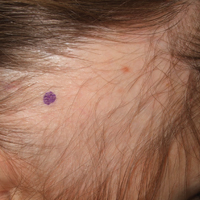
A 25-year-old woman with a history of Hashimoto thyroiditis presented with hair loss affecting the right temporal scalp of 3 years' duration that was first noticed by her husband. The lesion was an asymptomatic, 6×8-cm, roughly lancet-shaped patch of alopecia located on the right temporal scalp, bordering on the frontal hairline (Figure 1). Centrally, the patch appeared almost hairless with a few retained terminal hairs. The frontal hairline was thinned but still present. There was no scaling or erythema, and fine vellus hairs and a few isolated terminal hairs covered the area. The corresponding skin on the contralateral temporal scalp showed normal hair density. The patient insisted that she had normal hair at the affected area until 22 years of age, and she denied a history of trauma or tight hairstyles. Initially diagnosed with alopecia areata by her primary care provider, the patient was treated with topical corticosteroids for 6 months without benefit. She was subsequently referred to a dermatologist who again offered a diagnosis of alopecia areata and treated the lesions with 2 intralesional corticosteroid injections without benefit. No biopsies of the affected area were performed, and the patient was given a trial of topical minoxidil.
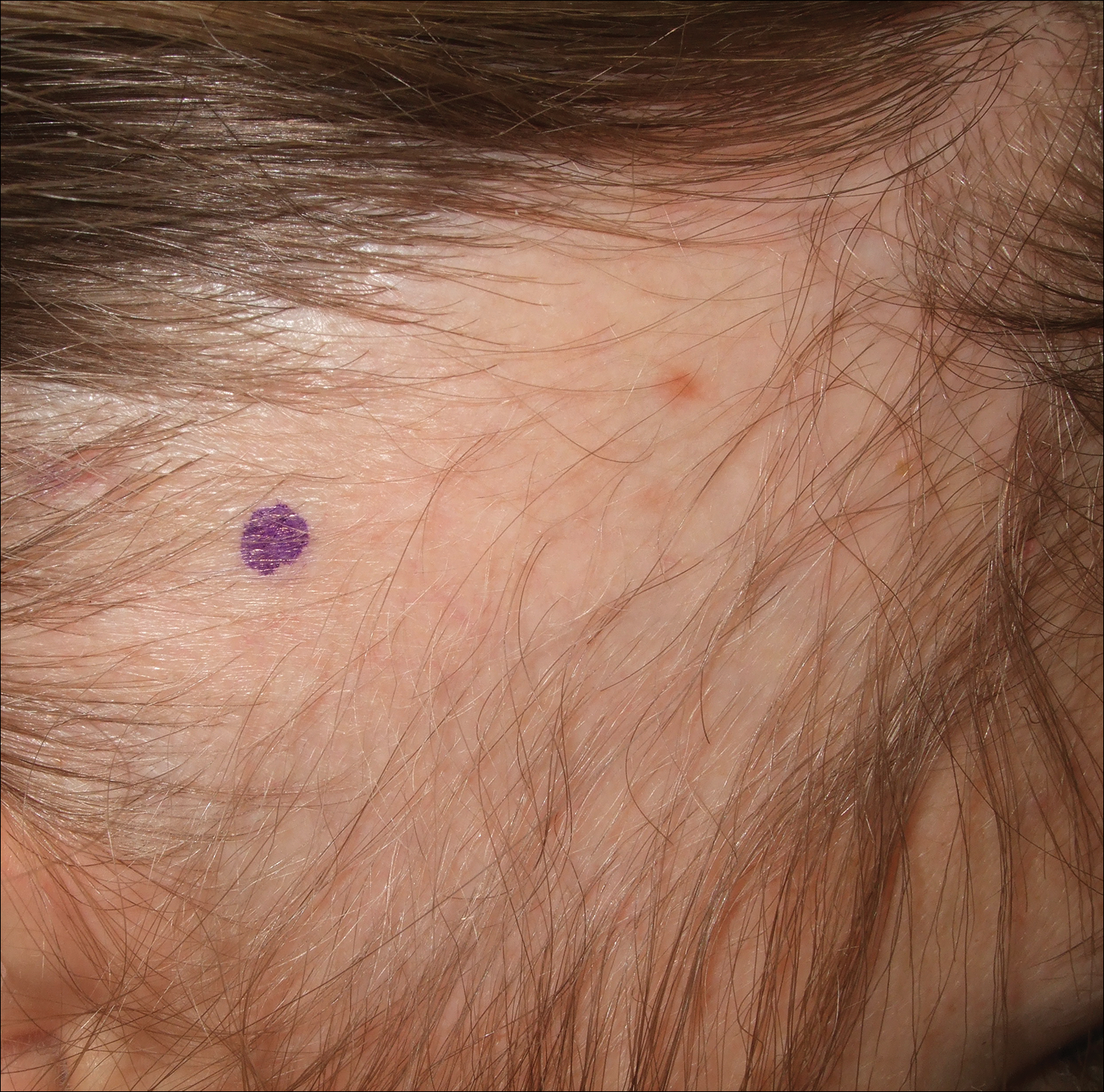
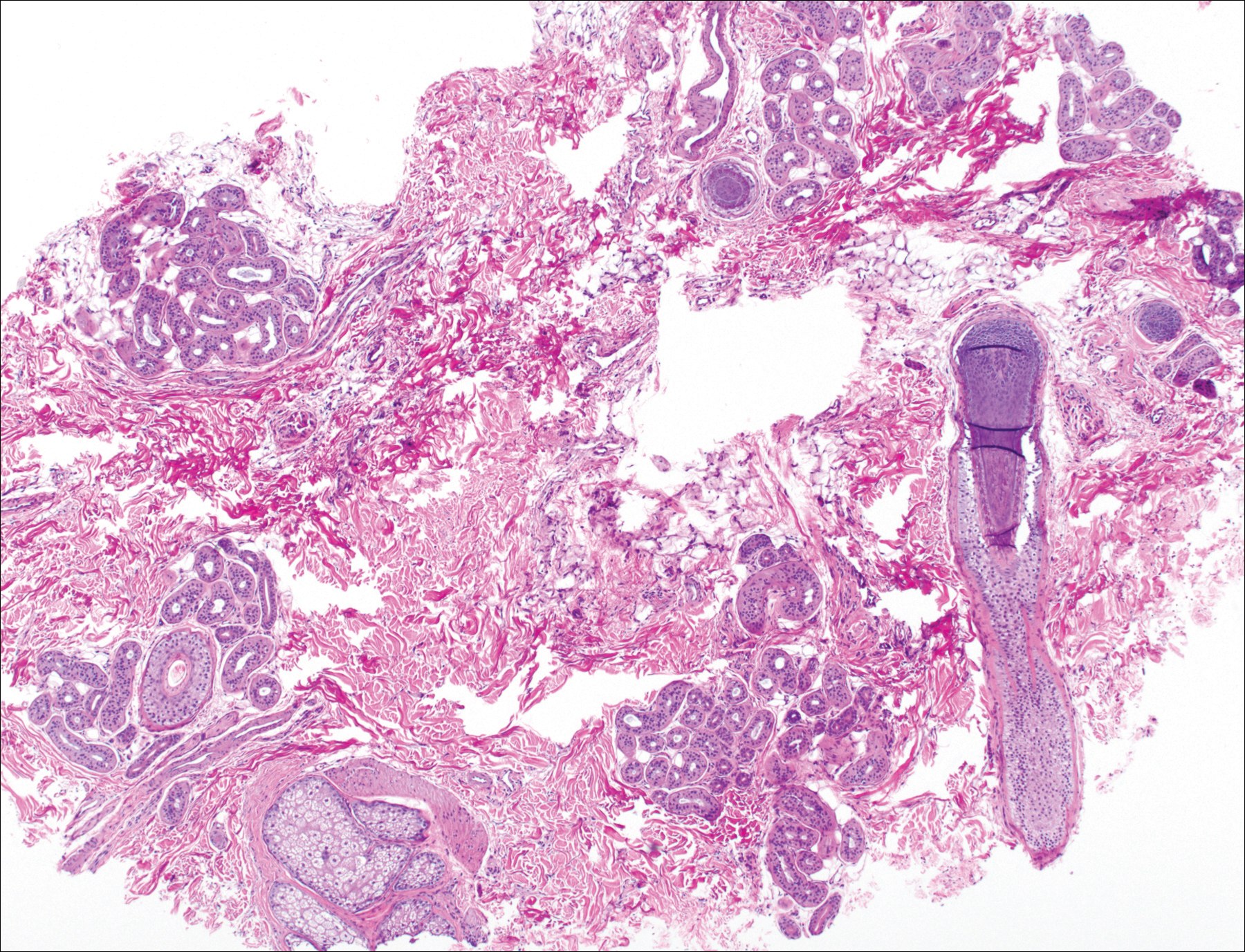
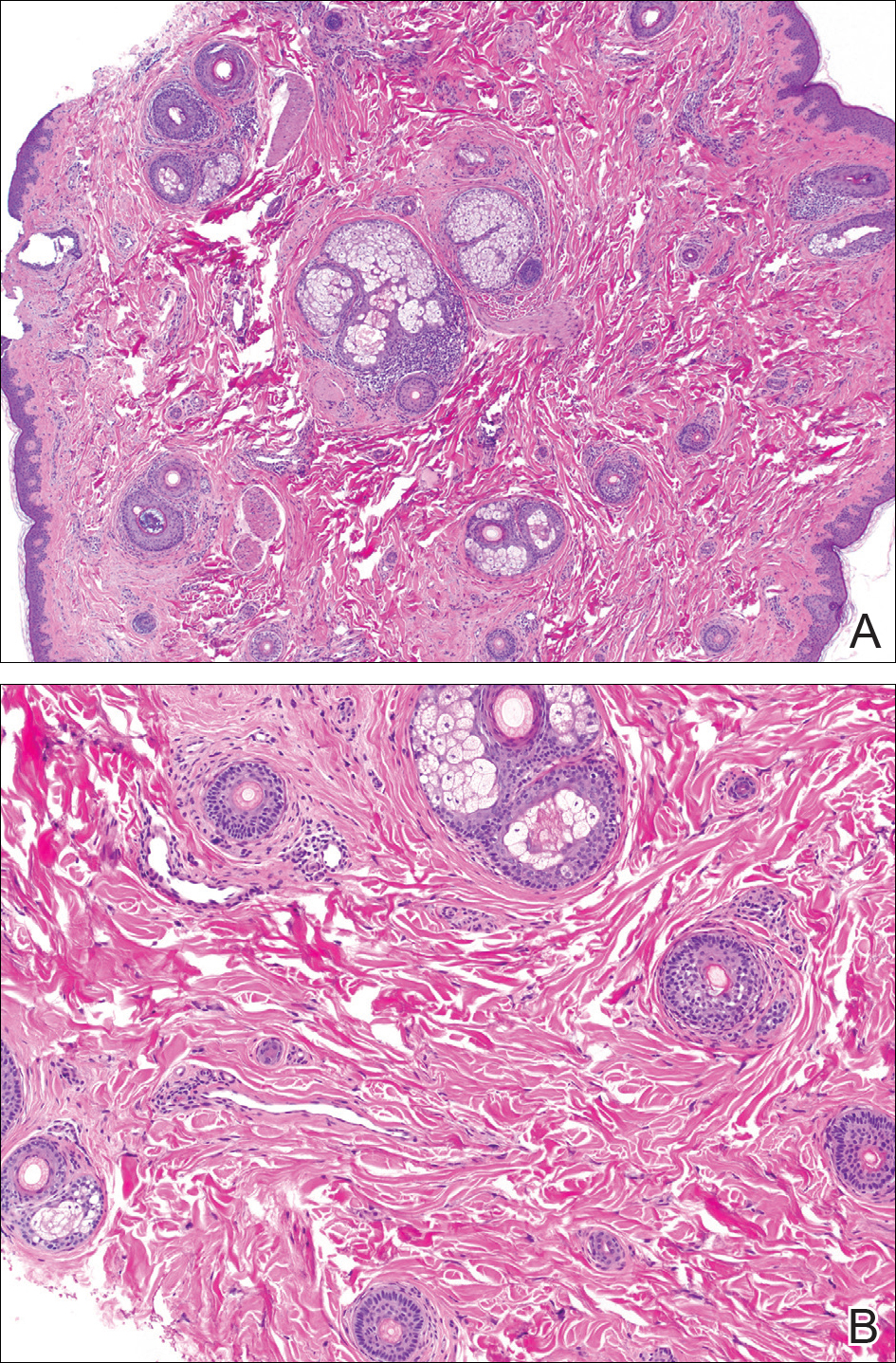
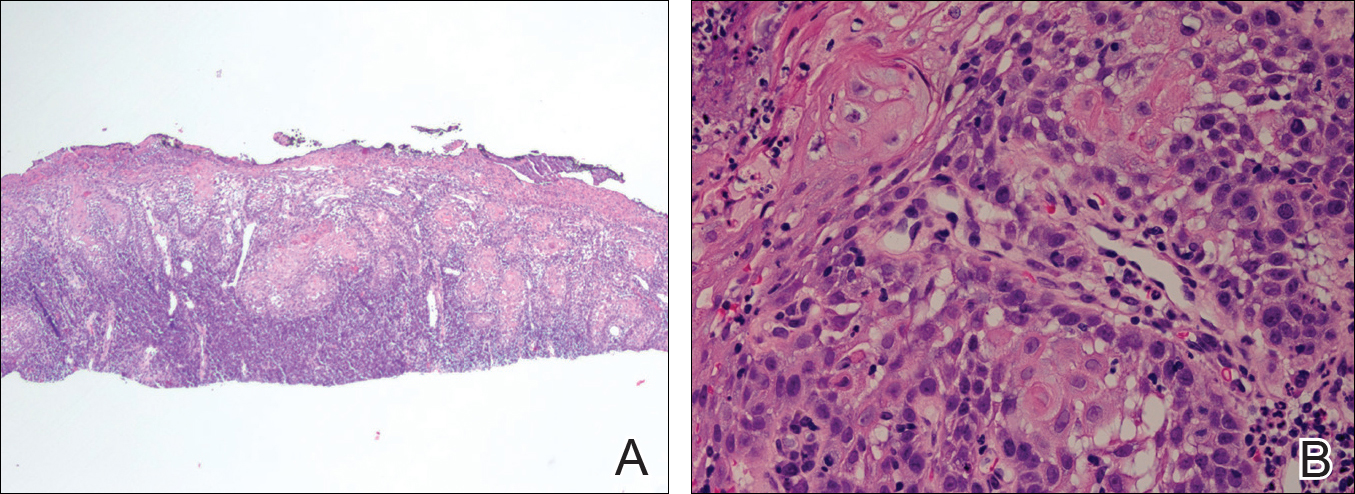
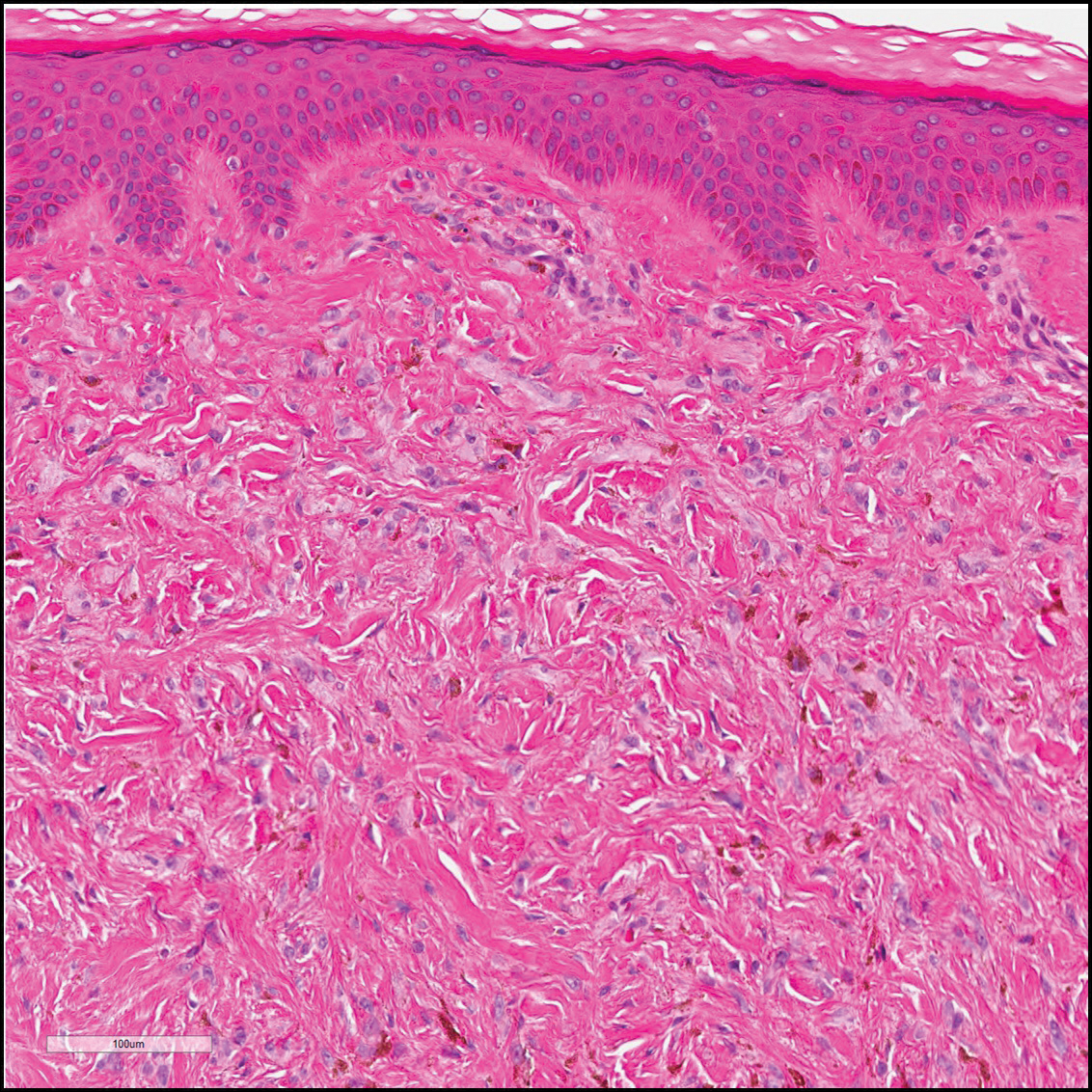
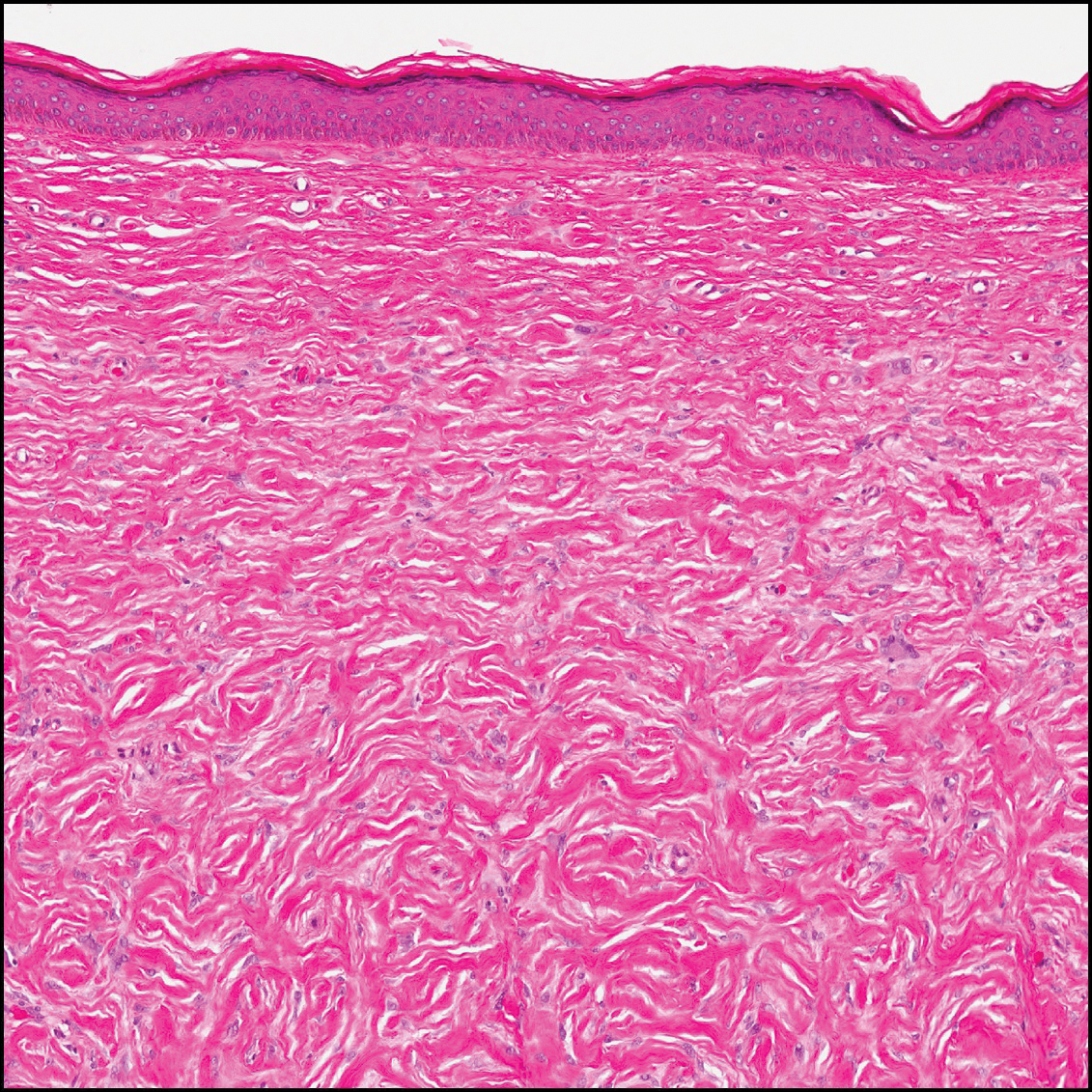
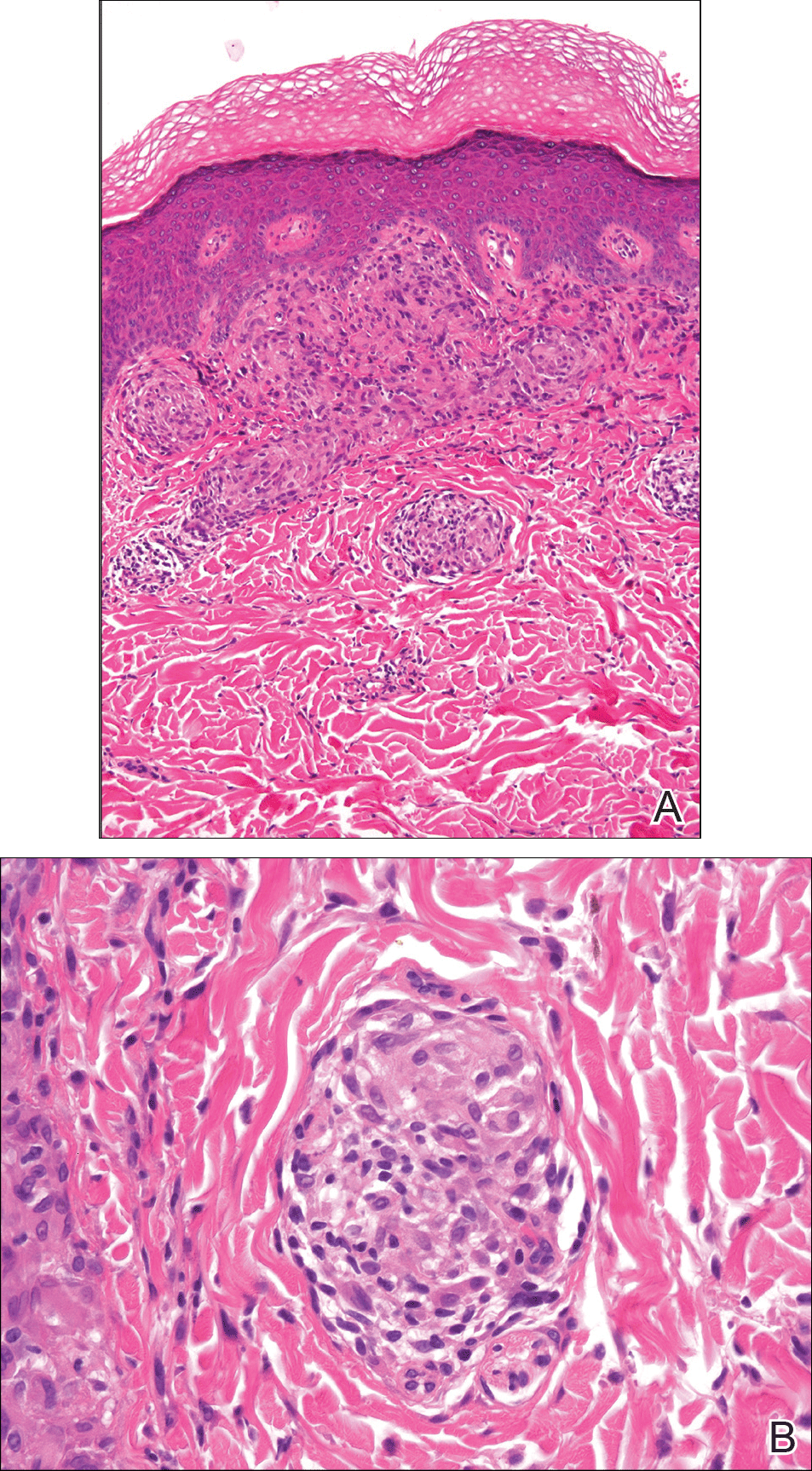
The patient consulted a new primary care provider and was diagnosed with scarring alopecia. She was referred to our dermatology department for further treatment. An initial biopsy at the edge of the affected area was interpreted as normal, but after failing additional intralesional corticosteroid injections, she was referred to our hair clinic where another biopsy was performed in the central portion of the lesion. A 4-mm diameter punch biopsy specimen revealed a normal epidermis and dermis; however, in the lower dermis only a single terminal follicle was seen (Figure 2). Sections through the upper dermis (Figure 3) showed that the total number of hairs was normal or nearly normal with at least 22 follicles, but most were vellus and indeterminate hairs with only a single terminal hair. The dermal architecture was otherwise normal. Given the clinical and histologic findings, a diagnosis of TTA was made. Subsequent to the diagnosis, the patient did not pursue any additional treatment options and preferred to style her hair so that the area of TTA remained covered.
The differential diagnosis in adults presenting with a patch of localized alopecia includes alopecia areata, trichotillomania, pressure-induced alopecia, traction alopecia, lichen planopilaris, discoid lupus erythematosus, and rarely TTA. Temporal triangular alopecia is a fairly common, if underreported, nonscarring form of alopecia that mainly affects young children. A PubMed search of articles indexed for MEDLINE using the terms temporal triangular alopecia or congenital triangular alopecia or triangular alopecia documented only 76 cases of TTA including our own, with the majority of patients diagnosed before 9 years of age. Only 2 cases of adult-onset TTA have been reported,5,6 possibly leading to misdiagnosis of adult patients who present with similar areas of hair loss. As with some prior cases of TTA,5,7 our patient was misdiagnosed with alopecia areata and scarring alopecia, both treated unsuccessfully before a diagnosis of TTA was considered. Clues to the diagnosis included the location, the lack of change in size and shape, the lack of response to intralesional corticosteroids, and the presence of numerous vellus hairs on the surface. A biopsy of the visibly hairless zone was confirmatory. The normal or nearly normal number of miniaturized hairs in specimens of TTA suggest that topical minoxidil therapy (eg, 5% solution twice daily for at least 6 months) might be useful, but the authors have tried it on a few other patients with clinically typical TTA without discernible benefit. When lesions are small, excision provides a fast and permanent solution to the problem, albeit with the usual risks of minor surgery.
- Sabouraud RJA. Manuel Élémentaire de Dermatologie Topographique Régionale. Paris, France: Masson & Cie; 1905:197.
- Trakimas C, Sperling LC, Skelton HG 3rd, et al. Clinical and histologic findings in temporal triangular alopecia. J Am Acad Dermatol. 1994;31:205-209.
- Yamazaki M, Irisawa R, Tsuboi R. Temporal triangular alopecia and a review of 52 past cases. J Dermatol. 2010;37:360-362.
- Sarifakioglu E, Yilmaz AE, Gorpelioglu C, et al. Prevalence of scalp disorders and hair loss in children. Cutis. 2012;90:225-229.
- Trakimas CA, Sperling LC. Temporal triangular alopecia acquired in adulthood. J Am Acad Dermatol. 1999;40:842-844.
- Akan IM, Yildirim S, Avci G, et al. Bilateral temporal triangular alopecia acquired in adulthood. Plast Reconstr Surg. 2001;107:1616-1617.
- Gupta LK, Khare AK, Garg A, et al. Congenital triangular alopecia--a close mimicker of alopecia areata. Int J Trichology. 2011;3:40-41.
To the Editor:
Temporal triangular alopecia (TTA), a condition first described by Sabouraud1 in 1905, is a circumscribed nonscarring form of alopecia. Also referred to as congenital triangular alopecia, TTA presents as a triangular or lancet-shaped area of hair loss involving the frontotemporal hairline. Temporal triangular alopecia is characterized histologically by a normal number of miniaturized hair follicles without notable inflammation.2 Although the majority of cases arise between birth and 9 years of age,3,4 rare cases of adult-onset TTA also have been reported.5,6 Adult-onset cases can cause notable diagnostic confusion and inappropriate treatment, as reported in our patient.
A 25-year-old woman with a history of Hashimoto thyroiditis presented with hair loss affecting the right temporal scalp of 3 years' duration that was first noticed by her husband. The lesion was an asymptomatic, 6×8-cm, roughly lancet-shaped patch of alopecia located on the right temporal scalp, bordering on the frontal hairline (Figure 1). Centrally, the patch appeared almost hairless with a few retained terminal hairs. The frontal hairline was thinned but still present. There was no scaling or erythema, and fine vellus hairs and a few isolated terminal hairs covered the area. The corresponding skin on the contralateral temporal scalp showed normal hair density. The patient insisted that she had normal hair at the affected area until 22 years of age, and she denied a history of trauma or tight hairstyles. Initially diagnosed with alopecia areata by her primary care provider, the patient was treated with topical corticosteroids for 6 months without benefit. She was subsequently referred to a dermatologist who again offered a diagnosis of alopecia areata and treated the lesions with 2 intralesional corticosteroid injections without benefit. No biopsies of the affected area were performed, and the patient was given a trial of topical minoxidil.
The patient consulted a new primary care provider and was diagnosed with scarring alopecia. She was referred to our dermatology department for further treatment. An initial biopsy at the edge of the affected area was interpreted as normal, but after failing additional intralesional corticosteroid injections, she was referred to our hair clinic where another biopsy was performed in the central portion of the lesion. A 4-mm diameter punch biopsy specimen revealed a normal epidermis and dermis; however, in the lower dermis only a single terminal follicle was seen (Figure 2). Sections through the upper dermis (Figure 3) showed that the total number of hairs was normal or nearly normal with at least 22 follicles, but most were vellus and indeterminate hairs with only a single terminal hair. The dermal architecture was otherwise normal. Given the clinical and histologic findings, a diagnosis of TTA was made. Subsequent to the diagnosis, the patient did not pursue any additional treatment options and preferred to style her hair so that the area of TTA remained covered.
The differential diagnosis in adults presenting with a patch of localized alopecia includes alopecia areata, trichotillomania, pressure-induced alopecia, traction alopecia, lichen planopilaris, discoid lupus erythematosus, and rarely TTA. Temporal triangular alopecia is a fairly common, if underreported, nonscarring form of alopecia that mainly affects young children. A PubMed search of articles indexed for MEDLINE using the terms temporal triangular alopecia or congenital triangular alopecia or triangular alopecia documented only 76 cases of TTA including our own, with the majority of patients diagnosed before 9 years of age. Only 2 cases of adult-onset TTA have been reported,5,6 possibly leading to misdiagnosis of adult patients who present with similar areas of hair loss. As with some prior cases of TTA,5,7 our patient was misdiagnosed with alopecia areata and scarring alopecia, both treated unsuccessfully before a diagnosis of TTA was considered. Clues to the diagnosis included the location, the lack of change in size and shape, the lack of response to intralesional corticosteroids, and the presence of numerous vellus hairs on the surface. A biopsy of the visibly hairless zone was confirmatory. The normal or nearly normal number of miniaturized hairs in specimens of TTA suggest that topical minoxidil therapy (eg, 5% solution twice daily for at least 6 months) might be useful, but the authors have tried it on a few other patients with clinically typical TTA without discernible benefit. When lesions are small, excision provides a fast and permanent solution to the problem, albeit with the usual risks of minor surgery.
To the Editor:
Temporal triangular alopecia (TTA), a condition first described by Sabouraud1 in 1905, is a circumscribed nonscarring form of alopecia. Also referred to as congenital triangular alopecia, TTA presents as a triangular or lancet-shaped area of hair loss involving the frontotemporal hairline. Temporal triangular alopecia is characterized histologically by a normal number of miniaturized hair follicles without notable inflammation.2 Although the majority of cases arise between birth and 9 years of age,3,4 rare cases of adult-onset TTA also have been reported.5,6 Adult-onset cases can cause notable diagnostic confusion and inappropriate treatment, as reported in our patient.
A 25-year-old woman with a history of Hashimoto thyroiditis presented with hair loss affecting the right temporal scalp of 3 years' duration that was first noticed by her husband. The lesion was an asymptomatic, 6×8-cm, roughly lancet-shaped patch of alopecia located on the right temporal scalp, bordering on the frontal hairline (Figure 1). Centrally, the patch appeared almost hairless with a few retained terminal hairs. The frontal hairline was thinned but still present. There was no scaling or erythema, and fine vellus hairs and a few isolated terminal hairs covered the area. The corresponding skin on the contralateral temporal scalp showed normal hair density. The patient insisted that she had normal hair at the affected area until 22 years of age, and she denied a history of trauma or tight hairstyles. Initially diagnosed with alopecia areata by her primary care provider, the patient was treated with topical corticosteroids for 6 months without benefit. She was subsequently referred to a dermatologist who again offered a diagnosis of alopecia areata and treated the lesions with 2 intralesional corticosteroid injections without benefit. No biopsies of the affected area were performed, and the patient was given a trial of topical minoxidil.
The patient consulted a new primary care provider and was diagnosed with scarring alopecia. She was referred to our dermatology department for further treatment. An initial biopsy at the edge of the affected area was interpreted as normal, but after failing additional intralesional corticosteroid injections, she was referred to our hair clinic where another biopsy was performed in the central portion of the lesion. A 4-mm diameter punch biopsy specimen revealed a normal epidermis and dermis; however, in the lower dermis only a single terminal follicle was seen (Figure 2). Sections through the upper dermis (Figure 3) showed that the total number of hairs was normal or nearly normal with at least 22 follicles, but most were vellus and indeterminate hairs with only a single terminal hair. The dermal architecture was otherwise normal. Given the clinical and histologic findings, a diagnosis of TTA was made. Subsequent to the diagnosis, the patient did not pursue any additional treatment options and preferred to style her hair so that the area of TTA remained covered.
The differential diagnosis in adults presenting with a patch of localized alopecia includes alopecia areata, trichotillomania, pressure-induced alopecia, traction alopecia, lichen planopilaris, discoid lupus erythematosus, and rarely TTA. Temporal triangular alopecia is a fairly common, if underreported, nonscarring form of alopecia that mainly affects young children. A PubMed search of articles indexed for MEDLINE using the terms temporal triangular alopecia or congenital triangular alopecia or triangular alopecia documented only 76 cases of TTA including our own, with the majority of patients diagnosed before 9 years of age. Only 2 cases of adult-onset TTA have been reported,5,6 possibly leading to misdiagnosis of adult patients who present with similar areas of hair loss. As with some prior cases of TTA,5,7 our patient was misdiagnosed with alopecia areata and scarring alopecia, both treated unsuccessfully before a diagnosis of TTA was considered. Clues to the diagnosis included the location, the lack of change in size and shape, the lack of response to intralesional corticosteroids, and the presence of numerous vellus hairs on the surface. A biopsy of the visibly hairless zone was confirmatory. The normal or nearly normal number of miniaturized hairs in specimens of TTA suggest that topical minoxidil therapy (eg, 5% solution twice daily for at least 6 months) might be useful, but the authors have tried it on a few other patients with clinically typical TTA without discernible benefit. When lesions are small, excision provides a fast and permanent solution to the problem, albeit with the usual risks of minor surgery.
- Sabouraud RJA. Manuel Élémentaire de Dermatologie Topographique Régionale. Paris, France: Masson & Cie; 1905:197.
- Trakimas C, Sperling LC, Skelton HG 3rd, et al. Clinical and histologic findings in temporal triangular alopecia. J Am Acad Dermatol. 1994;31:205-209.
- Yamazaki M, Irisawa R, Tsuboi R. Temporal triangular alopecia and a review of 52 past cases. J Dermatol. 2010;37:360-362.
- Sarifakioglu E, Yilmaz AE, Gorpelioglu C, et al. Prevalence of scalp disorders and hair loss in children. Cutis. 2012;90:225-229.
- Trakimas CA, Sperling LC. Temporal triangular alopecia acquired in adulthood. J Am Acad Dermatol. 1999;40:842-844.
- Akan IM, Yildirim S, Avci G, et al. Bilateral temporal triangular alopecia acquired in adulthood. Plast Reconstr Surg. 2001;107:1616-1617.
- Gupta LK, Khare AK, Garg A, et al. Congenital triangular alopecia--a close mimicker of alopecia areata. Int J Trichology. 2011;3:40-41.
- Sabouraud RJA. Manuel Élémentaire de Dermatologie Topographique Régionale. Paris, France: Masson & Cie; 1905:197.
- Trakimas C, Sperling LC, Skelton HG 3rd, et al. Clinical and histologic findings in temporal triangular alopecia. J Am Acad Dermatol. 1994;31:205-209.
- Yamazaki M, Irisawa R, Tsuboi R. Temporal triangular alopecia and a review of 52 past cases. J Dermatol. 2010;37:360-362.
- Sarifakioglu E, Yilmaz AE, Gorpelioglu C, et al. Prevalence of scalp disorders and hair loss in children. Cutis. 2012;90:225-229.
- Trakimas CA, Sperling LC. Temporal triangular alopecia acquired in adulthood. J Am Acad Dermatol. 1999;40:842-844.
- Akan IM, Yildirim S, Avci G, et al. Bilateral temporal triangular alopecia acquired in adulthood. Plast Reconstr Surg. 2001;107:1616-1617.
- Gupta LK, Khare AK, Garg A, et al. Congenital triangular alopecia--a close mimicker of alopecia areata. Int J Trichology. 2011;3:40-41.
Practice Points
- Temporal triangular alopecia (TTA) in adults often is confused with alopecia areata.
- An acquired, persistent, unchanging, circumscribed hairless spot in an adult that does not respond to intralesional corticosteroids may represent TTA.
- Hair miniaturization without peribulbar inflammation is consistent with a diagnosis of TTA.
Eroded Plaque on the Lower Lip
The Diagnosis: Squamous Cell Carcinoma
The initial clinical presentation suggested a diagnosis of herpes simplex labialis. The patient reported no response to topical acyclovir, and because the plaque persisted, a biopsy was performed. Pathology demonstrated squamous cell carcinoma (SCC) that was moderately well differentiated and invasive (Figure).
Approximately 38% of all oral SCCs in the United States occur on the lower lip and typically are solar-related cancers developing within the epidermis.1 Oral lesions initially may be asymptomatic and may not be of concern to the patient; however, it is important to recognize SCC early, as invasive lesions have the potential to metastasize. Some factors that increase the chance for the development of metastases include tumor size larger than 2 cm; location on the ear, lip, or other sites on the head and neck; and history of prior unsuccessful treatment.2 Any solitary ulcer, lump, wound, or lesion that will not heal and persists for more than 3 weeks should be regarded as cancer until proven otherwise. Although few oral SCCs are detected by clinicians at an early stage, diagnostic aids such as vital staining and molecular markers in tissues and saliva may be implemented.3 Toluidine blue is a simple, fast, and inexpensive technique that stains the nuclear material of malignant lesions, but not normal mucosa, and may be a worthwhile diagnostic adjunct to clinical inspection.4
Our patient presented with a lesion that clinically looked herpetic, though he reported no prodromal signs of tingling, burning, or pain before the occurrence of the lesion. Due to the persistence of the lesion and lack of response to treatment, a biopsy was indicated. The differential diagnoses include aphthous ulcers, which may occasionally extend on to the vermilion border of the lip and exhibit nondiagnostic histology.5 Bullous oral lichen planus is the least common variant of oral lichen planus, is unlikely to present as a solitary lesion, and is rarely seen on the lips. Histologically, the lesion demonstrated lichenoid inflammation.6 Solitary keratoacanthoma, though histologically similar to SCC, typically presents as a rapidly growing crateriform nodule without erosion or ulceration.7 The differential diagnoses are summarized in the Table.

The patient underwent wide excision with repair by mucosal advancement flap. He continues to be regularly seen in the clinic for monitoring of other skin cancers and is doing well. Clinicians encountering any wound or ulcer that does not show signs of healing should be wary of underlying malignancy and be prompted to perform a biopsy.
- Fehrenbach MJ. Extraoral and intraoral clinical assessment. In: Darby ML, Walsh MM, eds. Dental Hygiene: Theory and Practice. 4th ed. St Louis, MO: Elsevier; 2014:214-233.
- Hawrot A, Alam M, Ratner D. Squamous cell carcinoma. Curr Probl Dermatol. 2003;15:91-133.
- Scully C, Bagan J. Oral squamous cell carcinoma overview. Oral Oncol. 2009;45:301-308.
- Chhabra N, Chhabra S, Sapra N. Diagnostic modalities for squamous cell carcinoma: an extensive review of literature considering toluidine blue as a useful adjunct. J Oral Maxillofac Surg. 2015;14:188-200.
- Porter SR, Scully C, Pedersen A. Recurrent aphthous stomatitis. Crit Rev Oral Biol Med. 2003;9:1499-1505.
- Bricker SL. Oral lichen planus: a review. Semin Dermatol. 1994;13:87-90.
- Cabrijan L, Lipozencic´ J, Batinac T, et al. Differences between keratoacanthoma and squamous cell carcinoma using TGF-alpha. Coll Antropol. 2013;37:147-150.
- Douglas GD, Couch RB. A prospective study of chronic herpes simplex virus infection and recurrent herpes labialis in humans. J Immunol. 1970;104:289-295.
- Alam M, Ratner D. Cutaneous squamous-cell carcinoma. N Engl J Med. 2001;344:976-983.
- van Tuyll van Serooskerken AM, van Marion AM, de Zwart-Storm E, et al. Lichen planus with bullous manifestation on the lip. Int J Dermatol. 2007;46(suppl 3):25-26.
- Messadi DV, Younai F. Apthous ulcers. Dermatol Ther. 2010;23:281-290.
- Ko CJ. Keratoacanthoma: facts and controversies. Clin Dermatol. 2010;28:254-261.
The Diagnosis: Squamous Cell Carcinoma
The initial clinical presentation suggested a diagnosis of herpes simplex labialis. The patient reported no response to topical acyclovir, and because the plaque persisted, a biopsy was performed. Pathology demonstrated squamous cell carcinoma (SCC) that was moderately well differentiated and invasive (Figure).
Approximately 38% of all oral SCCs in the United States occur on the lower lip and typically are solar-related cancers developing within the epidermis.1 Oral lesions initially may be asymptomatic and may not be of concern to the patient; however, it is important to recognize SCC early, as invasive lesions have the potential to metastasize. Some factors that increase the chance for the development of metastases include tumor size larger than 2 cm; location on the ear, lip, or other sites on the head and neck; and history of prior unsuccessful treatment.2 Any solitary ulcer, lump, wound, or lesion that will not heal and persists for more than 3 weeks should be regarded as cancer until proven otherwise. Although few oral SCCs are detected by clinicians at an early stage, diagnostic aids such as vital staining and molecular markers in tissues and saliva may be implemented.3 Toluidine blue is a simple, fast, and inexpensive technique that stains the nuclear material of malignant lesions, but not normal mucosa, and may be a worthwhile diagnostic adjunct to clinical inspection.4
Our patient presented with a lesion that clinically looked herpetic, though he reported no prodromal signs of tingling, burning, or pain before the occurrence of the lesion. Due to the persistence of the lesion and lack of response to treatment, a biopsy was indicated. The differential diagnoses include aphthous ulcers, which may occasionally extend on to the vermilion border of the lip and exhibit nondiagnostic histology.5 Bullous oral lichen planus is the least common variant of oral lichen planus, is unlikely to present as a solitary lesion, and is rarely seen on the lips. Histologically, the lesion demonstrated lichenoid inflammation.6 Solitary keratoacanthoma, though histologically similar to SCC, typically presents as a rapidly growing crateriform nodule without erosion or ulceration.7 The differential diagnoses are summarized in the Table.
The patient underwent wide excision with repair by mucosal advancement flap. He continues to be regularly seen in the clinic for monitoring of other skin cancers and is doing well. Clinicians encountering any wound or ulcer that does not show signs of healing should be wary of underlying malignancy and be prompted to perform a biopsy.
The Diagnosis: Squamous Cell Carcinoma
The initial clinical presentation suggested a diagnosis of herpes simplex labialis. The patient reported no response to topical acyclovir, and because the plaque persisted, a biopsy was performed. Pathology demonstrated squamous cell carcinoma (SCC) that was moderately well differentiated and invasive (Figure).
Approximately 38% of all oral SCCs in the United States occur on the lower lip and typically are solar-related cancers developing within the epidermis.1 Oral lesions initially may be asymptomatic and may not be of concern to the patient; however, it is important to recognize SCC early, as invasive lesions have the potential to metastasize. Some factors that increase the chance for the development of metastases include tumor size larger than 2 cm; location on the ear, lip, or other sites on the head and neck; and history of prior unsuccessful treatment.2 Any solitary ulcer, lump, wound, or lesion that will not heal and persists for more than 3 weeks should be regarded as cancer until proven otherwise. Although few oral SCCs are detected by clinicians at an early stage, diagnostic aids such as vital staining and molecular markers in tissues and saliva may be implemented.3 Toluidine blue is a simple, fast, and inexpensive technique that stains the nuclear material of malignant lesions, but not normal mucosa, and may be a worthwhile diagnostic adjunct to clinical inspection.4
Our patient presented with a lesion that clinically looked herpetic, though he reported no prodromal signs of tingling, burning, or pain before the occurrence of the lesion. Due to the persistence of the lesion and lack of response to treatment, a biopsy was indicated. The differential diagnoses include aphthous ulcers, which may occasionally extend on to the vermilion border of the lip and exhibit nondiagnostic histology.5 Bullous oral lichen planus is the least common variant of oral lichen planus, is unlikely to present as a solitary lesion, and is rarely seen on the lips. Histologically, the lesion demonstrated lichenoid inflammation.6 Solitary keratoacanthoma, though histologically similar to SCC, typically presents as a rapidly growing crateriform nodule without erosion or ulceration.7 The differential diagnoses are summarized in the Table.
The patient underwent wide excision with repair by mucosal advancement flap. He continues to be regularly seen in the clinic for monitoring of other skin cancers and is doing well. Clinicians encountering any wound or ulcer that does not show signs of healing should be wary of underlying malignancy and be prompted to perform a biopsy.
- Fehrenbach MJ. Extraoral and intraoral clinical assessment. In: Darby ML, Walsh MM, eds. Dental Hygiene: Theory and Practice. 4th ed. St Louis, MO: Elsevier; 2014:214-233.
- Hawrot A, Alam M, Ratner D. Squamous cell carcinoma. Curr Probl Dermatol. 2003;15:91-133.
- Scully C, Bagan J. Oral squamous cell carcinoma overview. Oral Oncol. 2009;45:301-308.
- Chhabra N, Chhabra S, Sapra N. Diagnostic modalities for squamous cell carcinoma: an extensive review of literature considering toluidine blue as a useful adjunct. J Oral Maxillofac Surg. 2015;14:188-200.
- Porter SR, Scully C, Pedersen A. Recurrent aphthous stomatitis. Crit Rev Oral Biol Med. 2003;9:1499-1505.
- Bricker SL. Oral lichen planus: a review. Semin Dermatol. 1994;13:87-90.
- Cabrijan L, Lipozencic´ J, Batinac T, et al. Differences between keratoacanthoma and squamous cell carcinoma using TGF-alpha. Coll Antropol. 2013;37:147-150.
- Douglas GD, Couch RB. A prospective study of chronic herpes simplex virus infection and recurrent herpes labialis in humans. J Immunol. 1970;104:289-295.
- Alam M, Ratner D. Cutaneous squamous-cell carcinoma. N Engl J Med. 2001;344:976-983.
- van Tuyll van Serooskerken AM, van Marion AM, de Zwart-Storm E, et al. Lichen planus with bullous manifestation on the lip. Int J Dermatol. 2007;46(suppl 3):25-26.
- Messadi DV, Younai F. Apthous ulcers. Dermatol Ther. 2010;23:281-290.
- Ko CJ. Keratoacanthoma: facts and controversies. Clin Dermatol. 2010;28:254-261.
- Fehrenbach MJ. Extraoral and intraoral clinical assessment. In: Darby ML, Walsh MM, eds. Dental Hygiene: Theory and Practice. 4th ed. St Louis, MO: Elsevier; 2014:214-233.
- Hawrot A, Alam M, Ratner D. Squamous cell carcinoma. Curr Probl Dermatol. 2003;15:91-133.
- Scully C, Bagan J. Oral squamous cell carcinoma overview. Oral Oncol. 2009;45:301-308.
- Chhabra N, Chhabra S, Sapra N. Diagnostic modalities for squamous cell carcinoma: an extensive review of literature considering toluidine blue as a useful adjunct. J Oral Maxillofac Surg. 2015;14:188-200.
- Porter SR, Scully C, Pedersen A. Recurrent aphthous stomatitis. Crit Rev Oral Biol Med. 2003;9:1499-1505.
- Bricker SL. Oral lichen planus: a review. Semin Dermatol. 1994;13:87-90.
- Cabrijan L, Lipozencic´ J, Batinac T, et al. Differences between keratoacanthoma and squamous cell carcinoma using TGF-alpha. Coll Antropol. 2013;37:147-150.
- Douglas GD, Couch RB. A prospective study of chronic herpes simplex virus infection and recurrent herpes labialis in humans. J Immunol. 1970;104:289-295.
- Alam M, Ratner D. Cutaneous squamous-cell carcinoma. N Engl J Med. 2001;344:976-983.
- van Tuyll van Serooskerken AM, van Marion AM, de Zwart-Storm E, et al. Lichen planus with bullous manifestation on the lip. Int J Dermatol. 2007;46(suppl 3):25-26.
- Messadi DV, Younai F. Apthous ulcers. Dermatol Ther. 2010;23:281-290.
- Ko CJ. Keratoacanthoma: facts and controversies. Clin Dermatol. 2010;28:254-261.
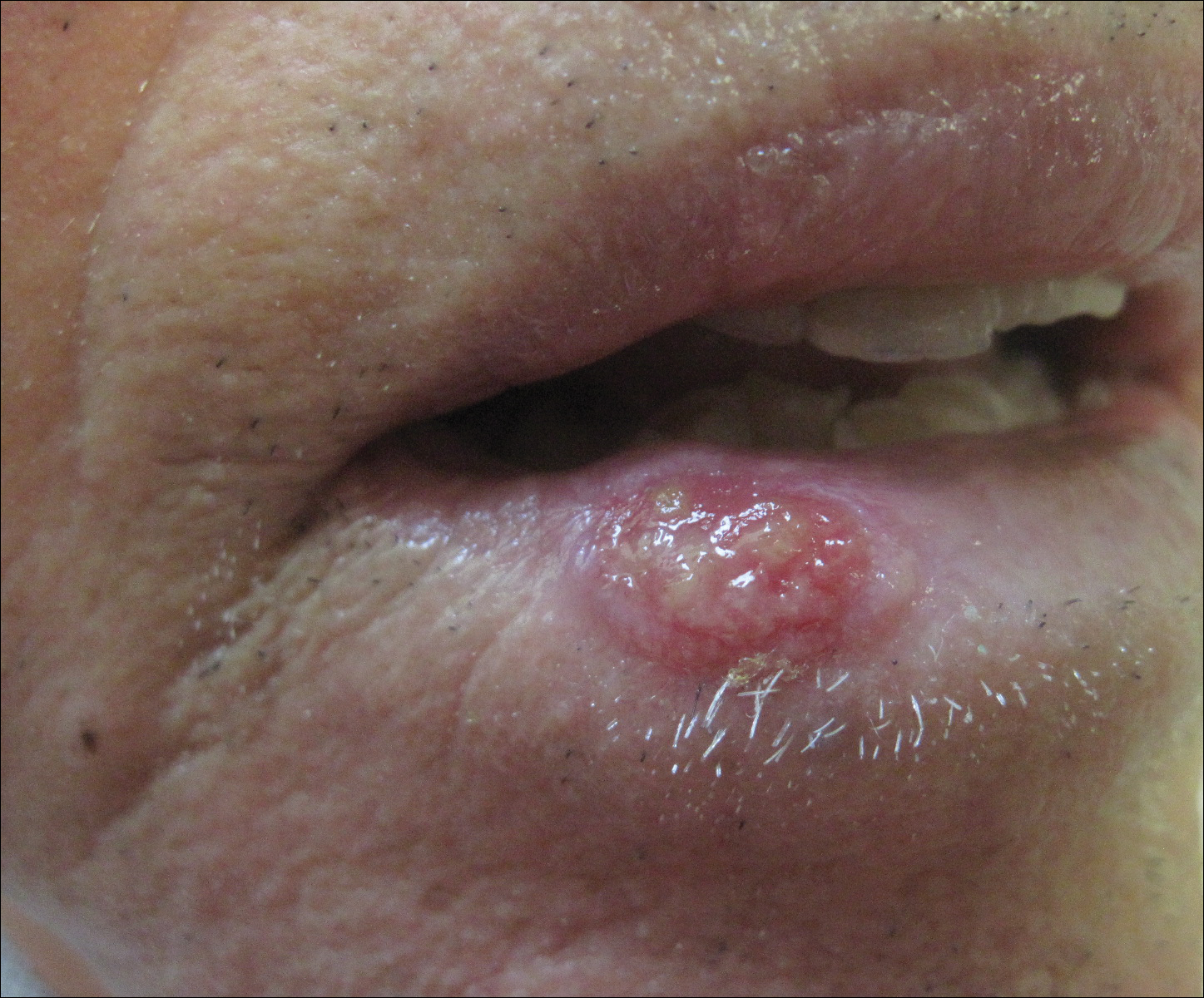
An 83-year-old man presented with a new-onset 1.2-cm eroded plaque on the vermilion border of the right lower lip that reportedly developed 2 weeks prior and was increasing in size. The plaque was moist and was composed of confluent glistening papules. Medical history was notable for the presence of both basal cell and squamous cell carcinomas.
Large Hyperpigmented Nodule on the Leg
The Diagnosis: Dermatofibroma
Dermatofibroma (DF) is a commonly encountered lesion. Although usually a straightforward clinical diagnosis, histopathological diagnosis is sometimes required. Conventional histologic findings of DF are hyperkeratosis, induction of the epidermis with acanthosis, and basal layer hyperpigmentation.1,2 Within the dermis there usually is proliferation of fibroblasts, histiocytes, and blood vessels that sometimes spares the overlying papillary dermis. Nomenclature of specific variants may be assigned based on the predominant component (eg, nodular subepidermal fibrosis, histiocytoma, sclerosing hemangioma) or histologic findings (eg, fibrocollagenous, sclerotic, cellular, histiocytic, lipidized, angiomatous, aneurysmal, clear cell, monster cell, myxoid, keloidal, palisading, osteoclastic, epithelioid).3-5 Of the histologic variants, fibrocollagenous is most common, but knowledge of other variants is important for accurate diagnosis, especially to exclude malignancy.
The sclerosing hemangioma variant of DF may pre-sent a diagnostic dilemma. In addition to typical features of DF, pseudovascular spaces, abundant hemosiderin, and reactive-appearing spindled cells are histologically demonstrated. The marked sclerosis and pigment deposition may mimic a blue nevus, and the dilated pseudovascular spaces may be reminiscent of a vascular neoplasm such as angiosarcoma or Kaposi sarcoma. However, the presence of characteristic features such as peripheral collagen trapping and overlying epidermal hyperplasia provide important clues for correct diagnosis.
Angiosarcomas (Figure 1) are malignant neoplasms with vascular differentiation. Cutaneous angiosarcomas present as purple plaques or nodules on the head and/or neck in elderly individuals as well as in patients with chronic lymphedema or prior radiation exposure.6-9 They are aggressive neoplasms with high rates of recurrence and metastases. Microscopically, the tumor is composed of anastomosing vascular channels lined by atypical endothelial cells with a multilayered appearance. There is frequent red blood cell extravasation, and substantial hemosiderin deposition may be noted in long-standing lesions. Neoplastic cells are positive for vascular markers (CD34, CD31, ETS-related gene transcription factor). Notably, cases associated with radiation exposure and chronic lymphedema are positive for MYC.10

Blue nevi (Figure 2) are benign melanocytic tumors that occur most frequently in children but may pre-sent in any age group. Clinical presentation is a blue to black, slightly raised papule that may be found on any site of the body. Biopsy typically shows a wedge-shaped infiltrate of spindled melanocytes with elongated dendritic processes in a sclerotic collagenous stroma. There frequently is a striking population of heavily pigmented melanophages. The melanocytes are positive for melanoma antigen recognized by T cells (MART-1)/melan-A, S-100, and transcription factor SOX-10. In contrast to other benign nevi, human melanoma black-45 will be positive in the dermal component.
Dermatofibrosarcoma protuberans (Figure 3) is a dermal-based tumor of intermediate malignant potential with a high rate of local recurrence and potential for sarcomatous transformation. Dermatofibrosarcoma protuberans most commonly presents in young adults as firm, pink to brown plaques and can occur on any site of the body. Histologically, they show a dermal proliferation of spindled cells that infiltrate in a storiform fashion into the subcutaneous adipose tissue,11 which imparts a honeycomb or Swiss cheese pattern. The tumor characteristically demonstrates positive staining for CD34. Loss of CD34 staining, increased mitoses, nuclear atypia, and fascicular growth are features suggestive of sarcomatous transformation.11,12 Dermatofibrosarcoma protuberans is associated with chromosomal abnormalities of chromosomes 17 and 22, resulting in COL1A1 (collagen type 1 alpha 1 chain) and PDGF-β (platelet-derived growth factor subunit B) gene fusion.13
Sclerotic fibromas (also known as storiform collagenomas)(Figure 4) may represent regressed DFs and are frequently associated with prior trauma to the affected area.14,15 They usually appear as flesh-colored papules or nodules on the face and trunk. The presence of multiple sclerotic fibromas is associated with Cowden syndrome.16,17 Histologically, the lesions present as well-demarcated, nonencapsulated, dermal nodules composed of a storiform or whorled arrangement of collagen with spindled fibroblasts. The sclerotic collagen bundles often are separated by small clefts imparting a plywoodlike pattern.16
The differential diagnosis for DF expands once atypical clinical and histopathological findings are present. In this case, the nodule was much larger and darker than the usual appearance of DF (3-10 mm).2,4 Given the lesion's nodularity, the clinical dimple sign on lateral compression could not be seen. On biopsy, the predominance of blood vessels and sclerosis further complicated the diagnostic picture. In unusual cases such as this one, correlation of clinical history, histology, and immunophenotype is ever important.
- Zeidi M, North JP. Sebaceous induction in dermatofibroma: a common feature of dermatofibromas on the shoulder. J Cutan Pathol. 2015;42:400-405.
- Şenel E, Yuyucu Karabulut Y, Doğruer S¸enel S. Clinical, histopathological, dermatoscopic and digital microscopic features of dermatofibroma: a retrospective analysis of 200 lesions. J Eur Acad Dermatol Venereol. 2015;29:1958-1966.
- Vilanova JR, Flint A. The morphological variations of fibrous histiocytomas. J Cutan Pathol. 1974;1:155-164.
- Han TY, Chang HS, Lee JH, et al. A clinical and histopathological study of 122 cases of dermatofibroma (benign fibrous histiocytoma)[published online May 27, 2011]. Ann Dermatol. 2011;23:185-192.
- Alves JVP, Matos DM, Barreiros HF, et al. Variants of dermatofibroma--a histopathological study. An Bras Dermatol. 2014;89:472-477.
- Rosai J, Sumner HW, Major MC, et al. Angiosarcoma of the skin: a clinicopathologic and fine structural study. Hum Pathol. 1976;7:83-109.
- Haustein UF. Angiosarcoma of the face and scalp. Int J Dermatol. 1991;30:851-856.
- Stewart FW, Treves N. Lymphangiosarcoma in postmastectomy lymphedema: a report of six cases in elephantiasis chirurgica. Cancer. 1948;1:64-81.
- Goette DK, Detlefs RL. Postirradiation angiosarcoma. J Am Acad Dermatol. 1985;12(5 pt 2):922-926.
- Manner J, Radlwimmer B, Hohenberger P, et al. MYC high level gene amplification is a distinctive feature of angiosarcomas after irradiation or chronic lymphedema. Am J Pathol. 2010;176:34-39.
- Voth H, Landsberg J, Hinz T, et al. Management of dermatofibrosarcoma protuberans with fibrosarcomatous transformation: an evidence-based review of the literature. J Eur Acad Dermatol Venereol. 2011;25:1385-1391.
- Goldblum JR. CD34 positivity in fibrosarcomas which arise in dermatofibrosarcoma protuberans. Arch Pathol Lab Med. 1995;119:238-241.
- Patel KU, Szabo SS, Hernandez VS, et al. Dermatofibrosarcoma protuberans COL1A1-PDGFB fusion is identified in virtually all dermatofibrosarcoma protuberans cases when investigated by newly developed multiplex reverse transcription polymerase chain reaction and fluorescence in situ hybridization assays. Hum Pathol. 2008;39:184-193.
- Sohn IB, Hwang SM, Lee SH, et al. Dermatofibroma with sclerotic areas resembling a sclerotic fibroma of the skin. J Cutan Pathol. 2002;29:44-47.
- Pujol RM, de Castro F, Schroeter AL, et al. Solitary sclerotic fibroma of the skin: a sclerotic dermatofibroma? Am J Dermatopathol. 1996;18:620-624.
- Requena L, Gutiérrez J, Sánchez Yus E. Multiple sclerotic fibromas of the skin: a cutaneous marker of Cowden's disease. J Cutan Pathol. 1992;19:346-351.
- Weary PE, Gorlin RJ, Gentry WC Jr, et al. Multiple hamartoma syndrome (Cowden's disease). Arch Dermatol. 1972;106:682-690.
The Diagnosis: Dermatofibroma
Dermatofibroma (DF) is a commonly encountered lesion. Although usually a straightforward clinical diagnosis, histopathological diagnosis is sometimes required. Conventional histologic findings of DF are hyperkeratosis, induction of the epidermis with acanthosis, and basal layer hyperpigmentation.1,2 Within the dermis there usually is proliferation of fibroblasts, histiocytes, and blood vessels that sometimes spares the overlying papillary dermis. Nomenclature of specific variants may be assigned based on the predominant component (eg, nodular subepidermal fibrosis, histiocytoma, sclerosing hemangioma) or histologic findings (eg, fibrocollagenous, sclerotic, cellular, histiocytic, lipidized, angiomatous, aneurysmal, clear cell, monster cell, myxoid, keloidal, palisading, osteoclastic, epithelioid).3-5 Of the histologic variants, fibrocollagenous is most common, but knowledge of other variants is important for accurate diagnosis, especially to exclude malignancy.
The sclerosing hemangioma variant of DF may pre-sent a diagnostic dilemma. In addition to typical features of DF, pseudovascular spaces, abundant hemosiderin, and reactive-appearing spindled cells are histologically demonstrated. The marked sclerosis and pigment deposition may mimic a blue nevus, and the dilated pseudovascular spaces may be reminiscent of a vascular neoplasm such as angiosarcoma or Kaposi sarcoma. However, the presence of characteristic features such as peripheral collagen trapping and overlying epidermal hyperplasia provide important clues for correct diagnosis.
Angiosarcomas (Figure 1) are malignant neoplasms with vascular differentiation. Cutaneous angiosarcomas present as purple plaques or nodules on the head and/or neck in elderly individuals as well as in patients with chronic lymphedema or prior radiation exposure.6-9 They are aggressive neoplasms with high rates of recurrence and metastases. Microscopically, the tumor is composed of anastomosing vascular channels lined by atypical endothelial cells with a multilayered appearance. There is frequent red blood cell extravasation, and substantial hemosiderin deposition may be noted in long-standing lesions. Neoplastic cells are positive for vascular markers (CD34, CD31, ETS-related gene transcription factor). Notably, cases associated with radiation exposure and chronic lymphedema are positive for MYC.10
Blue nevi (Figure 2) are benign melanocytic tumors that occur most frequently in children but may pre-sent in any age group. Clinical presentation is a blue to black, slightly raised papule that may be found on any site of the body. Biopsy typically shows a wedge-shaped infiltrate of spindled melanocytes with elongated dendritic processes in a sclerotic collagenous stroma. There frequently is a striking population of heavily pigmented melanophages. The melanocytes are positive for melanoma antigen recognized by T cells (MART-1)/melan-A, S-100, and transcription factor SOX-10. In contrast to other benign nevi, human melanoma black-45 will be positive in the dermal component.
Dermatofibrosarcoma protuberans (Figure 3) is a dermal-based tumor of intermediate malignant potential with a high rate of local recurrence and potential for sarcomatous transformation. Dermatofibrosarcoma protuberans most commonly presents in young adults as firm, pink to brown plaques and can occur on any site of the body. Histologically, they show a dermal proliferation of spindled cells that infiltrate in a storiform fashion into the subcutaneous adipose tissue,11 which imparts a honeycomb or Swiss cheese pattern. The tumor characteristically demonstrates positive staining for CD34. Loss of CD34 staining, increased mitoses, nuclear atypia, and fascicular growth are features suggestive of sarcomatous transformation.11,12 Dermatofibrosarcoma protuberans is associated with chromosomal abnormalities of chromosomes 17 and 22, resulting in COL1A1 (collagen type 1 alpha 1 chain) and PDGF-β (platelet-derived growth factor subunit B) gene fusion.13
Sclerotic fibromas (also known as storiform collagenomas)(Figure 4) may represent regressed DFs and are frequently associated with prior trauma to the affected area.14,15 They usually appear as flesh-colored papules or nodules on the face and trunk. The presence of multiple sclerotic fibromas is associated with Cowden syndrome.16,17 Histologically, the lesions present as well-demarcated, nonencapsulated, dermal nodules composed of a storiform or whorled arrangement of collagen with spindled fibroblasts. The sclerotic collagen bundles often are separated by small clefts imparting a plywoodlike pattern.16
The differential diagnosis for DF expands once atypical clinical and histopathological findings are present. In this case, the nodule was much larger and darker than the usual appearance of DF (3-10 mm).2,4 Given the lesion's nodularity, the clinical dimple sign on lateral compression could not be seen. On biopsy, the predominance of blood vessels and sclerosis further complicated the diagnostic picture. In unusual cases such as this one, correlation of clinical history, histology, and immunophenotype is ever important.
The Diagnosis: Dermatofibroma
Dermatofibroma (DF) is a commonly encountered lesion. Although usually a straightforward clinical diagnosis, histopathological diagnosis is sometimes required. Conventional histologic findings of DF are hyperkeratosis, induction of the epidermis with acanthosis, and basal layer hyperpigmentation.1,2 Within the dermis there usually is proliferation of fibroblasts, histiocytes, and blood vessels that sometimes spares the overlying papillary dermis. Nomenclature of specific variants may be assigned based on the predominant component (eg, nodular subepidermal fibrosis, histiocytoma, sclerosing hemangioma) or histologic findings (eg, fibrocollagenous, sclerotic, cellular, histiocytic, lipidized, angiomatous, aneurysmal, clear cell, monster cell, myxoid, keloidal, palisading, osteoclastic, epithelioid).3-5 Of the histologic variants, fibrocollagenous is most common, but knowledge of other variants is important for accurate diagnosis, especially to exclude malignancy.
The sclerosing hemangioma variant of DF may pre-sent a diagnostic dilemma. In addition to typical features of DF, pseudovascular spaces, abundant hemosiderin, and reactive-appearing spindled cells are histologically demonstrated. The marked sclerosis and pigment deposition may mimic a blue nevus, and the dilated pseudovascular spaces may be reminiscent of a vascular neoplasm such as angiosarcoma or Kaposi sarcoma. However, the presence of characteristic features such as peripheral collagen trapping and overlying epidermal hyperplasia provide important clues for correct diagnosis.
Angiosarcomas (Figure 1) are malignant neoplasms with vascular differentiation. Cutaneous angiosarcomas present as purple plaques or nodules on the head and/or neck in elderly individuals as well as in patients with chronic lymphedema or prior radiation exposure.6-9 They are aggressive neoplasms with high rates of recurrence and metastases. Microscopically, the tumor is composed of anastomosing vascular channels lined by atypical endothelial cells with a multilayered appearance. There is frequent red blood cell extravasation, and substantial hemosiderin deposition may be noted in long-standing lesions. Neoplastic cells are positive for vascular markers (CD34, CD31, ETS-related gene transcription factor). Notably, cases associated with radiation exposure and chronic lymphedema are positive for MYC.10
Blue nevi (Figure 2) are benign melanocytic tumors that occur most frequently in children but may pre-sent in any age group. Clinical presentation is a blue to black, slightly raised papule that may be found on any site of the body. Biopsy typically shows a wedge-shaped infiltrate of spindled melanocytes with elongated dendritic processes in a sclerotic collagenous stroma. There frequently is a striking population of heavily pigmented melanophages. The melanocytes are positive for melanoma antigen recognized by T cells (MART-1)/melan-A, S-100, and transcription factor SOX-10. In contrast to other benign nevi, human melanoma black-45 will be positive in the dermal component.
Dermatofibrosarcoma protuberans (Figure 3) is a dermal-based tumor of intermediate malignant potential with a high rate of local recurrence and potential for sarcomatous transformation. Dermatofibrosarcoma protuberans most commonly presents in young adults as firm, pink to brown plaques and can occur on any site of the body. Histologically, they show a dermal proliferation of spindled cells that infiltrate in a storiform fashion into the subcutaneous adipose tissue,11 which imparts a honeycomb or Swiss cheese pattern. The tumor characteristically demonstrates positive staining for CD34. Loss of CD34 staining, increased mitoses, nuclear atypia, and fascicular growth are features suggestive of sarcomatous transformation.11,12 Dermatofibrosarcoma protuberans is associated with chromosomal abnormalities of chromosomes 17 and 22, resulting in COL1A1 (collagen type 1 alpha 1 chain) and PDGF-β (platelet-derived growth factor subunit B) gene fusion.13
Sclerotic fibromas (also known as storiform collagenomas)(Figure 4) may represent regressed DFs and are frequently associated with prior trauma to the affected area.14,15 They usually appear as flesh-colored papules or nodules on the face and trunk. The presence of multiple sclerotic fibromas is associated with Cowden syndrome.16,17 Histologically, the lesions present as well-demarcated, nonencapsulated, dermal nodules composed of a storiform or whorled arrangement of collagen with spindled fibroblasts. The sclerotic collagen bundles often are separated by small clefts imparting a plywoodlike pattern.16
The differential diagnosis for DF expands once atypical clinical and histopathological findings are present. In this case, the nodule was much larger and darker than the usual appearance of DF (3-10 mm).2,4 Given the lesion's nodularity, the clinical dimple sign on lateral compression could not be seen. On biopsy, the predominance of blood vessels and sclerosis further complicated the diagnostic picture. In unusual cases such as this one, correlation of clinical history, histology, and immunophenotype is ever important.
- Zeidi M, North JP. Sebaceous induction in dermatofibroma: a common feature of dermatofibromas on the shoulder. J Cutan Pathol. 2015;42:400-405.
- Şenel E, Yuyucu Karabulut Y, Doğruer S¸enel S. Clinical, histopathological, dermatoscopic and digital microscopic features of dermatofibroma: a retrospective analysis of 200 lesions. J Eur Acad Dermatol Venereol. 2015;29:1958-1966.
- Vilanova JR, Flint A. The morphological variations of fibrous histiocytomas. J Cutan Pathol. 1974;1:155-164.
- Han TY, Chang HS, Lee JH, et al. A clinical and histopathological study of 122 cases of dermatofibroma (benign fibrous histiocytoma)[published online May 27, 2011]. Ann Dermatol. 2011;23:185-192.
- Alves JVP, Matos DM, Barreiros HF, et al. Variants of dermatofibroma--a histopathological study. An Bras Dermatol. 2014;89:472-477.
- Rosai J, Sumner HW, Major MC, et al. Angiosarcoma of the skin: a clinicopathologic and fine structural study. Hum Pathol. 1976;7:83-109.
- Haustein UF. Angiosarcoma of the face and scalp. Int J Dermatol. 1991;30:851-856.
- Stewart FW, Treves N. Lymphangiosarcoma in postmastectomy lymphedema: a report of six cases in elephantiasis chirurgica. Cancer. 1948;1:64-81.
- Goette DK, Detlefs RL. Postirradiation angiosarcoma. J Am Acad Dermatol. 1985;12(5 pt 2):922-926.
- Manner J, Radlwimmer B, Hohenberger P, et al. MYC high level gene amplification is a distinctive feature of angiosarcomas after irradiation or chronic lymphedema. Am J Pathol. 2010;176:34-39.
- Voth H, Landsberg J, Hinz T, et al. Management of dermatofibrosarcoma protuberans with fibrosarcomatous transformation: an evidence-based review of the literature. J Eur Acad Dermatol Venereol. 2011;25:1385-1391.
- Goldblum JR. CD34 positivity in fibrosarcomas which arise in dermatofibrosarcoma protuberans. Arch Pathol Lab Med. 1995;119:238-241.
- Patel KU, Szabo SS, Hernandez VS, et al. Dermatofibrosarcoma protuberans COL1A1-PDGFB fusion is identified in virtually all dermatofibrosarcoma protuberans cases when investigated by newly developed multiplex reverse transcription polymerase chain reaction and fluorescence in situ hybridization assays. Hum Pathol. 2008;39:184-193.
- Sohn IB, Hwang SM, Lee SH, et al. Dermatofibroma with sclerotic areas resembling a sclerotic fibroma of the skin. J Cutan Pathol. 2002;29:44-47.
- Pujol RM, de Castro F, Schroeter AL, et al. Solitary sclerotic fibroma of the skin: a sclerotic dermatofibroma? Am J Dermatopathol. 1996;18:620-624.
- Requena L, Gutiérrez J, Sánchez Yus E. Multiple sclerotic fibromas of the skin: a cutaneous marker of Cowden's disease. J Cutan Pathol. 1992;19:346-351.
- Weary PE, Gorlin RJ, Gentry WC Jr, et al. Multiple hamartoma syndrome (Cowden's disease). Arch Dermatol. 1972;106:682-690.
- Zeidi M, North JP. Sebaceous induction in dermatofibroma: a common feature of dermatofibromas on the shoulder. J Cutan Pathol. 2015;42:400-405.
- Şenel E, Yuyucu Karabulut Y, Doğruer S¸enel S. Clinical, histopathological, dermatoscopic and digital microscopic features of dermatofibroma: a retrospective analysis of 200 lesions. J Eur Acad Dermatol Venereol. 2015;29:1958-1966.
- Vilanova JR, Flint A. The morphological variations of fibrous histiocytomas. J Cutan Pathol. 1974;1:155-164.
- Han TY, Chang HS, Lee JH, et al. A clinical and histopathological study of 122 cases of dermatofibroma (benign fibrous histiocytoma)[published online May 27, 2011]. Ann Dermatol. 2011;23:185-192.
- Alves JVP, Matos DM, Barreiros HF, et al. Variants of dermatofibroma--a histopathological study. An Bras Dermatol. 2014;89:472-477.
- Rosai J, Sumner HW, Major MC, et al. Angiosarcoma of the skin: a clinicopathologic and fine structural study. Hum Pathol. 1976;7:83-109.
- Haustein UF. Angiosarcoma of the face and scalp. Int J Dermatol. 1991;30:851-856.
- Stewart FW, Treves N. Lymphangiosarcoma in postmastectomy lymphedema: a report of six cases in elephantiasis chirurgica. Cancer. 1948;1:64-81.
- Goette DK, Detlefs RL. Postirradiation angiosarcoma. J Am Acad Dermatol. 1985;12(5 pt 2):922-926.
- Manner J, Radlwimmer B, Hohenberger P, et al. MYC high level gene amplification is a distinctive feature of angiosarcomas after irradiation or chronic lymphedema. Am J Pathol. 2010;176:34-39.
- Voth H, Landsberg J, Hinz T, et al. Management of dermatofibrosarcoma protuberans with fibrosarcomatous transformation: an evidence-based review of the literature. J Eur Acad Dermatol Venereol. 2011;25:1385-1391.
- Goldblum JR. CD34 positivity in fibrosarcomas which arise in dermatofibrosarcoma protuberans. Arch Pathol Lab Med. 1995;119:238-241.
- Patel KU, Szabo SS, Hernandez VS, et al. Dermatofibrosarcoma protuberans COL1A1-PDGFB fusion is identified in virtually all dermatofibrosarcoma protuberans cases when investigated by newly developed multiplex reverse transcription polymerase chain reaction and fluorescence in situ hybridization assays. Hum Pathol. 2008;39:184-193.
- Sohn IB, Hwang SM, Lee SH, et al. Dermatofibroma with sclerotic areas resembling a sclerotic fibroma of the skin. J Cutan Pathol. 2002;29:44-47.
- Pujol RM, de Castro F, Schroeter AL, et al. Solitary sclerotic fibroma of the skin: a sclerotic dermatofibroma? Am J Dermatopathol. 1996;18:620-624.
- Requena L, Gutiérrez J, Sánchez Yus E. Multiple sclerotic fibromas of the skin: a cutaneous marker of Cowden's disease. J Cutan Pathol. 1992;19:346-351.
- Weary PE, Gorlin RJ, Gentry WC Jr, et al. Multiple hamartoma syndrome (Cowden's disease). Arch Dermatol. 1972;106:682-690.

A 61-year-old woman presented with a 2.5-cm hyperpigmented exophytic nodule on the anterior aspect of the left shin of approximately 2 years' duration. The patient initially noticed a small lesion following a bee sting, but it subsequently grew over the ensuing 2 years. A shave biopsy was obtained.
Product News: 07 2017
Glytone Sunscreen Lotion Broad Spectrum SPF 40
Pierre Fabre Dermo-Cosmetique USA introduces Glytone Sunscreen Lotion Broad Spectrum SPF 40, a mineral-based formula for face and body with micronized zinc oxide, octinoxate, and octisalate. The lightweight formula is water resistant for up to 40 minutes and contains hyaluronic acid to nourish the skin and help boost natural moisture levels to visibly reduce the appearance of fine lines and wrinkles. For more information, visit www.glytone-usa.com.
proactivMD
The Proactiv Company launches the proactivMD Essentials System, a 3-step acne regimen that has been reformulated to include
If you would like your product included in Product News, please email a press release to the Editorial Office at [email protected].
Glytone Sunscreen Lotion Broad Spectrum SPF 40
Pierre Fabre Dermo-Cosmetique USA introduces Glytone Sunscreen Lotion Broad Spectrum SPF 40, a mineral-based formula for face and body with micronized zinc oxide, octinoxate, and octisalate. The lightweight formula is water resistant for up to 40 minutes and contains hyaluronic acid to nourish the skin and help boost natural moisture levels to visibly reduce the appearance of fine lines and wrinkles. For more information, visit www.glytone-usa.com.
proactivMD
The Proactiv Company launches the proactivMD Essentials System, a 3-step acne regimen that has been reformulated to include
If you would like your product included in Product News, please email a press release to the Editorial Office at [email protected].
Glytone Sunscreen Lotion Broad Spectrum SPF 40
Pierre Fabre Dermo-Cosmetique USA introduces Glytone Sunscreen Lotion Broad Spectrum SPF 40, a mineral-based formula for face and body with micronized zinc oxide, octinoxate, and octisalate. The lightweight formula is water resistant for up to 40 minutes and contains hyaluronic acid to nourish the skin and help boost natural moisture levels to visibly reduce the appearance of fine lines and wrinkles. For more information, visit www.glytone-usa.com.
proactivMD
The Proactiv Company launches the proactivMD Essentials System, a 3-step acne regimen that has been reformulated to include
If you would like your product included in Product News, please email a press release to the Editorial Office at [email protected].

Metastatic Crohn Disease: A Review of Dermatologic Manifestations and Treatment
Almost half of Crohn disease (CD) patients experience a dermatologic manifestation of the disease. A rare entity, metastatic CD (MCD) presents a diagnostic challenge without a high index of suspicion. Its etiology is not well defined; however, it appears to be an autoimmune response to gut antigens. Herein, we review the etiology/epidemiology, diagnostic criteria, and treatment for this uncommon condition.
Epidemiology and Clinical Characteristics of MCD
Metastatic CD was first described by Parks et al1 in 1965 and refers to a diverse collection of macroscopic dermatologic manifestations in tissue not contiguous with the gastrointestinal (GI) tract. To be classified as MCD, the tissue must demonstrate characteristic histopathologic findings, which invariably include noncaseating granulomas.
Crohn disease may affect any part of the GI tract from the mouth to anus, with a multitude of associated cutaneous manifestations having been described. The terminal ileum is the most commonly affected portion of the GI tract in CD, but the large intestine also may be involved in 55% to 80% of cases.2 The incidence of non-MCD-associated anal lesions seems to correlate with intestinal involvement in that as few as 25% of patients with ileal-localized CD have anal lesions compared to nearly 80% of patients with large intestinal involvement.3
It has been estimated that 18% to 44% of patients with CD have some form of cutaneous manifestation,4 with MCD being a rare subcategory. As few as 100 cases have been described from 1965 to the present.5 The presence of MCD does not correlate well with severity of intestinal CD, and although a majority of MCD cases present after at least 6 months of GI symptoms,6 there are instances in which MCD presents without prior or existing evidence of intestinal CD.7
With regard to MCD, the term metastatic is sometimes supplanted in the literature by cutaneous to avoid any implication of cancer; however, due to a myriad of dermatologic manifestations, both terms can cause confusion. The categorization of the various types of cutaneous findings in CD is well summarized in a review by Palamaras et al8 with the following classifications: (1) granulomatous by direct extension (oral or perianal), (2) MCD lesions (genital and nongenital), (3) immune-related lesions, and (4) lesions from nutritional deficiencies. Of the cutaneous manifestations relating to CD, MCD is the least common cutaneous categorical manifestation and is further divided into subcategories of genital and nongenital lesions.8
The nongenital distribution of MCD is the more common variety in adults and particularly seems to affect the legs and plantar surfaces (38%), the trunk and abdomen (24%), and the face (15%).5,9 These nongenital MCD manifestations are most commonly described as nodules, ulcerations, or erythematous to purple plaques, and less commonly described as abscesses, pustules, or papules.
The sequence of cutaneous symptoms of MCD relative to intestinal disease depends to some degree on patient age. In adults diagnosed with MCD, it has been noted that a GI flare is expected 2 months to 4 years after diagnosis; however, in children the subsequent GI flare has been noted to vary more widely from 9 months to 14 years following presentation of MCD.8 Furthermore, roughly 50% of children diagnosed with MCD present concomitantly with their first symptoms of a GI flare, whereas 70% of adults with MCD had been previously diagnosed with intestinal CD.8 In one review of 80 reported cases of MCD, 20% (16/80) had no symptoms of intestinal disease at the time of MCD diagnosis, and the majority of the asymptomatic cases were in children; interestingly, the majority of these same children were diagnosed with CD months to years later.9
Both the location and characteristics of cutaneous findings in MCD correlate with age.9 Metastatic CD has been identified in all age groups; however, lymphedema is more common in children/young adults, while nodules, ulceration, and fistulating disease are more often seen in adults.10 Affected children and adolescents with MCD range from 5 to 17 years of age, with a mean age at disease onset of 11.1 years and equal incidence in males and females.8 Adults with MCD range from 18 to 78 years of age, with a mean age at presentation of 38.4 years.8,11
Concerning anatomic location of disease, adults with MCD most commonly have nodules with or without plaques on the arms and legs and less commonly in the genital area.8 In contrast, children with MCD are more prone to genital lesions, with up to 85% of cases including some degree of genital erythematous or nonerythematous swelling with or without induration.8 Genitourinary complications of CD as a broad category, however, are estimated to occur in only 5% to 20% of intestinal CD cases in both children and adults.12
There have been conflicting reports regarding gender predilection in MCD. Based on a review by Samitz et al13 of 200 cases of CD over an 18-year period, 22% of patients with CD were found to have cutaneous manifestations--presumably not MCD but rather perianal, perineal, vulvar fistulae, fissures, or abscesses--with a male to female preponderance of almost 2 to 1. A more recent review of the literature by Palamaras et al8 in 2008 reported that contiguous non-MCD affects adult females and children more often than adult males, with 63% adult cases being female. This review seems to be more congruent with other reports in the literature implicating that females are twice as commonly affected by MCD than males.9,14
Pathophysiology
The etiology of MCD has not been well defined. One proposed mechanism of the distal tissue involvement of MCD is through passage of antigens to the skin with subsequent granulomatous response at the level of the dermis.10 Another proposed mechanism suggests antibody sensitization to gut antigens, possibly bacterial antigens, that then coincidentally cross-react with analogous skin antigens.8,14 Burgdorf11 supported this notion in a 1981 report in which it was suggested that the granulomatous reaction was related to deposition of immune complexes in the skin. Slater et al15 and Tatnall et al16 offered a variation of Burgdorf's notion, suggesting that it was sensitized T cells to circulating antigens that were the initiators of granuloma formation in the periphery.
An examination of MCD tissue in 1990 by Shum and Guenther17 under electron microscopy and immunofluorescence provided evidence against prior studies that purported to have identified immune complexes as the causative agents of MCD. In this study, the authors found no evidence of immune complexes in the dermis of MCD lesions. In addition, an attempt to react serum antibodies of a patient with MCD, which were postulated to have IgG, IgM, and IgA antibodies to specific gut antigens, yielded no response when reacted with the tongue, ileum, and colon tissue from a rat. As a culminant finding, the authors also noted MCD dermis tissue with granulomas without vasculitis, suggesting a T-cell mediated type IV hypersensitivity response with a secondary vasculitis from T-cell origin lymphokines and T-cell mediated monocyte activation.17
Research implicating other immunologic entities involved in the pathophysiology of CD such as β-2 integrin,18 CD14+ monocytes,19 and the role of the DNA repair gene MLH1 (mutL homolog 1)20 have been considered but without a clearly definitive role in the manifestations of MCD.
The utility of metronidazole in the treatment of MCD has been suggested as evidence that certain bacteria in the gut may either serve as the causative antigen or may induce its formation21; however, the causative antigen has yet to be identified, and whether it travels distally to the skin or merely resembles a similar antigen normally present in the dermis has not yet been determined. Some research has used in situ polymerase chain reaction techniques to attempt to detect similar microbial pathogens in both the vasculature of active bowel lesions and in the skin, but to date, bacterial RNA noted to be present in the gut vasculature adjacent to CD lesions has not been detected in skin lesions.22
Diagnosis
Physical Findings
Overall, it is estimated that roughly 56% of all MCD cases affect the external genitalia.23 The classic appearance of MCD includes well-demarcated ulcerations in the areas of intertriginous skin folds with or without diffuse edema and tenderness to palpation.23 Although MCD has been historically noted as having a predilection for moist skin folds, there are numerous case reports of MCD all over the body, including the face,7,24-29 retroauricular areas,30 arms and legs,16,17,31-34 lower abdomen,3,5 under the breasts,1 perineum,35 external genitalia,1,9,36-40 and even the lungs41 and bladder.42
As a dermatologic disease, MCD has been referred to as yet another great imitator, both on the macroscopic and microscopic levels.8 As such, more common causes of genital edema should be considered first and investigated based on the patient's history, physical examination, skin biopsy, lymphangiogram, ultrasound, and cystogram.43 Ultrasonography and color Doppler sonography have been shown to be helpful in patients with genital involvement. This modality can evaluate not only the presence of normal testes but also intratesticular and scrotal wall fluid, especially when the physical examination reveals swelling that makes testicle palpation more difficult.6 Clinically, the correct diagnosis of MCD often is made through suspicion of inflammatory bowel disease based on classic symptoms and/or physical findings including abdominal pain, weight loss, bloody stool, diarrhea, perianal skin tags, and anal fissures or fistulas. Any of these GI findings should prompt an intestinal biopsy to rule out any histologic evidence of CD.
Metastatic CD affecting the vulva often presents with vulvar pain and pruritus and may clinically mimic a more benign disease such as balanitis plasmacellularis, also referred to as Zoon vulvitis.23 Similar to MCD on any given body surface, there is dramatic variation in the macroscopic presentation of vulvar MCD, with physical examination findings ranging from bilateral diffuse, edematous, deeply macerated, red, ulcerated lesions over the vulva with lymphadenopathy to findings of bilateral vulvar pain with yellow drainage from the labia majora.23 There have been cases of vulvar MCD that include exquisite vulvar pain but without structural abnormalities including normal uterus, cervix, adnexa, rectovaginal septum, and rectum. In these more nebulous cases of vulvar MCD, the diagnosis often is discovered incidentally when nonspecific diagnostic imaging suggests underlying CD.23
Beyond the case-by-case variations on physical examination, the great difficulty in diagnosis, particularly in children, occurs in the absence of any GI symptoms and therefore no logical consideration of underlying CD. Consequently, there have been cases of children presenting with irritation of the vulva who were eventually diagnosed with MCD only after erroneous treatment of contact dermatitis, candidiasis, and even consideration of sexual abuse.37 Because it is so rare and obscure among practicing clinicians, the diagnosis of MCD often is considered only after irritation or swelling of the external genitalia has not responded to standard therapies. If and when the diagnosis of MCD is considered in children, it has been suggested to screen patients for anorectal stricture, as case studies have found the condition to be relatively common in this subpopulation.44
In the less common case of adults with genitourinary symptoms that suggest possible MCD, the differential diagnosis for penile or vaginal ulcers should include contact and irritant dermatitis, chronic infectious lesions (eg, hidradenitis suppurativa, actinomycosis, tuberculosis),45 sexually transmitted ulcerative diseases (eg, chancroid, lymphogranuloma venereum, herpes genitalia, granuloma inguinale),46 drug reactions, and even extramammary Paget disease.47
Histologic Findings
Because MCD has so much macroscopic variation and can present anywhere on the surface of the body, formal diagnosis relies on microscopy. As an added measure of difficulty in diagnosis, one random biopsy of a suspicious segment of tissue may not contain the expected histologic findings; therefore, clinical suspicion may warrant a second biopsy.10 There have been reported cases of an adult patient without history of CD presenting with a lesion that resembled a more common pathology, such as a genital wart, and the correct diagnosis of MCD with pseudocondylomatous morphology was made only after intestinal manifestations prompted the clinician to consider such an unusual diagnosis.48
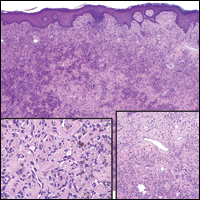
From a histopathologic perspective, MCD is characterized by discrete, noncaseating, sarcoidlike granulomas with abundant multinucleated giant cells (Langhans giant cells) in the superficial dermis (papillary), deep dermis (reticular), and adipose tissue (Figure).8,17 In the presence of concomitant intestinal disease, the granulomas of both the intestinal and dermal tissues should share the same microscopic characteristics.8 In addition, copious neutrophils and granulomas surrounding the microvasculature have been described,34 as well as general lymphocyte and plasma cell infiltrate.45 Some histologic samples have included collagen degeneration termed necrobiosis in the middle dermal layer as another variable finding in MCD.14,34
On microscopy, it has been reported that use of Verhoeff-van Gieson staining may be helpful to highlight the presence of neutrophil obstruction within the dermal vasculature, particularly the arterial lumen, as well as to aid in highlighting swelling of the endothelium with fragmentation of the internal elastic lamina.17 Although not part of the routine diagnosis, electron microscopy of MCD tissue samples have confirmed hypertrophy of the endothelial cells composing the capillaries with resulting extravasation of fibrin, red blood cells, lymphocytes, and epithelioid histiocytes.17 Observation of tissue under direct immunofluorescence has been less helpful, as it has shown only nonspecific fibrinogen deposition within the dermis and dermal vessels.17
In an article on treatment of MCD, Escher et al43 reinforced that the macroscopic findings of MCD are diverse, and the microscopic findings characteristic of MCD also can be mimicked by other etiologies such as sarcoidosis, tuberculosis, fungal infections, lymphogranuloma venereum, leishmaniasis, and connective tissue disorders.43 As such, the workup to rule out infectious, anatomic, and autoimmune etiologies should be diverse. Often, the workup for MCD will include special stains such as Ziehl-Neelsen stain to rule out Mycobacterium tuberculosis and acid-fast bacilli and Fite stain to consider atypical mycobacteria. Other tests such as tissue culture, chest radiograph, tuberculin skin test (Mantoux test), IFN-γ release assay, or polarized light microscopy may rule out infectious etiologies.9,49 Serologic testing might include VDRL test, Treponema pallidum hemagglutination assay, hepatitis B, hepatitis C, and human immunodeficiency virus.5
Crohn disease is characterized histologically by sarcoidlike noncaseating granulomas, and as such, it is important to differentiate MCD from sarcoidosis prior to histologic analysis. Sarcoidosis also can be considered much less likely with a normal chest radiograph and in the absence of increased serum calcium and angiotensin-converting enzyme levels.7 The differentiation of sarcoidosis from MCD on the microscopic scale is subtle but is sometimes facilitated in the presence of an ulcerated epidermis or lymphocytic/eosinophilic infiltrate and edema within the dermis, all suggestive of MCD.14
Metastatic CD also should be differentiated from erythema nodosum and pyoderma gangrenosum, which are among the most common cutaneous findings associated with CD.14 Pyoderma gangrenosum can be distinguished histologically by identifying copious neutrophilic infiltrate with pseudoepitheliomatous hyperplasia.50
Treatment
Because MCD is relatively rare, there are no known randomized trials suggesting a particular medical or surgical treatment. In a review of perineal MCD from 2007, the 40-year-old recommendation by Moutain3 opting for surgical debridement versus medical management still resonates, particularly for perineal disease, as an effective measure in all but the mildest of presentations.51 However, recent case reports also suggest that the tumor necrosis factor α (TNF-α) inhibitors such as infliximab and adalimumab should be considered prior to surgery even with severe perineal MCD.51 Moreover, even if medical management with TNF-α inhibitors or some combination of immunosuppressants and antibiotics does not eradicate the disease, it often helps reduce the size of the ulcers prior to surgery.52 With a limited understanding of MCD, one might think that removal of the affected bowel would eliminate cutaneous disease, but it has been shown that this strategy is not effective.53,54
The composition and location of the particular lesion affects the trajectory of treatment. For example, MCD manifesting as local ulcers and plaques has been described as responding well to topical and intralesional steroids.10,55,56 In the case of penile swelling and/or phimosis, circumcision has been helpful to improve the patient's ability to void as well as to attain and maintain erection.10 In the case of scrotal swelling secondary to MCD, early treatment (ie, within 4 to 6 months) with oral steroids and/or metronidazole is likely beneficial to prevent refractory edematous organization of the tissue.57
As a general rule, an effective treatment will include a combination of an immunosuppressant, antibiotic therapy, and sometimes surgery. The most commonly used immunosuppressant agents include topical or intralesional steroids, infliximab,43,58 cyclosporine A,59,60 dapsone, minocycline, thalidomide, methotrexate, mycophenolate mofetil, sulfasalazine, azathioprine, tacrolimus, and 6-mercaptopurine.4 Steroids have been the conventional treatment of extraintestinal manifestations of CD61; however, perineal CD has been poorly controlled with systemic steroids.62 If steroids are found not to be effective, sometimes agents such as dapsone or thalidomide are considered. One case report noted stabilization of MCD penile ulcers with oral thalidomide 300 mg once daily, oral minocycline 100 mg once daily, and topical tacrolimus 0.3% with benzocaine twice daily with continuation of prednisolone and methotrexate as parts of previously unsuccessful regimen.52
Metronidazole is perhaps the most commonly used antibiotic, having been a component of many successful regimens.4,63 For example, a 27-year-old patient with MCD presenting as a nonhealing ulcerative lesion in the subcoronal area of the penis and scrotum was treated successfully with a 6-month course of mesalamine, prednisone, and metronidazole.45 Another case report of vulvar MCD reported initial success with intravenous methylprednisolone, ciprofloxacin, and metronidazole.23 The primary limitation of metronidazole is that subsequent tapering of the dose seems to result in recurrence of disease.64 Consequently, patients must remain on the antibiotic for an indeterminate course, with dosages ranging from 5 mg/kg daily in adolescents65 to 1000 to 1500 mg daily in adults.66
Of the various immunosuppressants available, infliximab has been listed in numerous reports as a successful agent in both the induction and maintenance of extraintestinal manifestations of CD including MCD.67-71 Infliximab has been reported to be effective in the treatment of penile and scrotal edema secondary to MCD that did not respond to other immunosuppressants including oral prednisolone, azathioprine, and cyclosporine.43 Infliximab may be a good option to help heal draining fistulas, particularly in combination with an antibiotic such as metronidazole and ciprofloxacin, which helps to prevent abscess formation during healing.72 The response to infliximab has been dramatic, with resolution of cutaneous lesions after just 6 weeks in some cases.73 The dosing regimen of infliximab has been suggested at 5 mg/kg administered at 0, 2, and 6 weeks, with subsequent maintenance infusions every 10 weeks,70 or at 0, 4, and 12 weeks, with subsequent infusions every 8 weeks.43
Adalimumab may be considered as an alternative to infliximab and is potentially less allergenic as a fully humanized monoclonal antibody to TNF-α, which also has been used successfully to both induce and maintain remission of moderate to severe CD.42,74,75 Proposed dosing of adalimumab includes a loading dose of 160 mg subcutaneously on day 1, followed by an 80-mg dose 2 weeks later and a 40-mg maintenance dose every other week indefinitely.48 Of note, adalimumab has been noted in the literature to have many potential side effects, including one particular case in which severe headaches were attributed to its use.59 As a consequence of the headaches, the patient was switched from adalimumab to cyclosporine and responded well with no subsequent flare-ups on follow-up.
In summary, treatment of MCD depends on cutaneous location, severity, physician experience with certain antibiotics or immunosuppressants, availability of medication, and patient disposition. It seems reasonable to attempt medical management with one or more medical regimens before committing to surgical intervention. Furthermore, even with debridement, curettage, skin graft, or other surgical strategy, the patient is likely to require some period of immunosuppression to provide long-lasting remission.
Conclusion
Patients with inflammatory bowel disease often develop dermatologic sequelae, with MCD being a rare but serious process. Patients may present with a wide array of physical concerns and symptoms, many resembling other disease processes. As such, education and a high index of suspicion are needed for proper diagnosis and treatment.
- Parks AG, Morson BC, Pegum JS. Crohn's disease with cutaneous involvement. Proc R Soc Med. 1965;58:241-242.
- Friedman S, Blumber RS. Inflammatory bowel disease. In: Kasper DL, Braunwald E, Fauci AS, et al, eds. Harrison's Principles of Internal Medicine. 16th ed. New York, NY: McGraw-Hill; 2005:1778-1784.
- Moutain JC. Cutaneous ulceration in Crohn's disease. Gut. 1970;11:18-26.
- Lester LU, Rapini RP. Dermatologic manifestations of colonic disorders. Curr Opin Gastroenterol. 2008;25:66-73.
- Teixeira M, Machado S, Lago P, et al. Cutaneous Crohn's disease. Int J Dermatol. 2006;45:1074-1076.
- Simoneaux SF, Ball TI, Atkinson GO Jr. Scrotal swelling: unusual first presentation of Crohn's disease. Pediatr Radiol. 1995;25:375-376.
- Albuquerque A, Magro F, Rodrigues S, et al. Metastatic cutaneous Crohn's disease of the face: a case report and review of literature. Eur J Gastroenterol Hepatol. 2011;23:954-956.
- Palamaras I, El-Jabbour J, Pietropaolo N, et al. Metastatic Crohn's disease: a review. J Eur Acad Dermatol Venereol. 2008;22:1033-1043.
- Ploysangam T, Heubi JE, Eisen D, et al. Cutaneous Crohn's disease in children. J Am Acad Dermatol. 1997;36:697-704.
- Vint R, Husain E, Hassain F, et al. Metastatic Crohn's disease of the penis: two cases. Int Urol Nephrol. 2012;44:45-49.
- Burgdorf W. Cutaneous manifestations of Crohn's disease. J Am Acad Dermatol. 1981;5:689-695.
- Resnick MI, Kursh ED. Extrinsic obstruction of the ureter. In: Walsh PC, Retik AB, Stamey TA, et al, eds. Campbell's Urology. 7th ed. Philadelphia, PA: WB Saunders; 1998:400-402.
- Samitz MH, Dana AS Jr, Rosenberg P. Cutaneous vasculitis in association with Crohn's disease--review of statistics of skin complications. Cutis. 1970;6:51-56.
- Emanuel PO, Phelps RG. Metastatic Crohn's disease: a histo-pathologic study of 12 cases. J Cutan Pathol. 2008;35:457-461.
- Slater DN, Waller PC, Reilly G. Cutaneous granulomatous vasculitis: presenting features of Crohn's disease. J R Soc Med. 1985;78:589-590.
- Tatnall FM, Dodd HJ, Sarkany I. Crohn's disease with metastatic cutaneous involvement and granulomatous cheilitis. J R Soc Med. 1987;80:49-51.
- Shum DT, Guenther L. Metastatic Crohn's disease: case report and review of literature. Arch Dermatol. 1990;126:645-648.
- Bernstein CN, Sargent M, Gallatin WM. Beta2 integrin/ICAM expression in Crohn's disease. Clin Immunol Immunopathol. 1998;86:147-160.
- Grimm MC, Pavli P, Van de Pol E, et al. Evidence for a CD-14+ population of monocytes in inflammatory bowel disease mucosa--implications for pathogenesis. Clin Exp Immunol. 1995;100:291-297.
- Pokorny RM, Hofmeister A, Galandiuk S, et al. Crohn's disease and ulcerative colitis are associated with the DNA repair gene MLH1. Ann Surg. 1997;225:718-723; discussion 723-725.
- Ursing B, Kamme C. Metronidazole for Crohn's disease. Lancet. 1975;1:775-777.
- Crowson AN, Nuovo GJ, Mihm MC Jr, et al. Cutaneous manifestations of Crohn's disease, its spectrum, and pathogenesis: intracellular consensus bacterial 16S rRNA is associated with the gastrointestinal but not the cutaneous manifestations of Crohn's disease. Hum Pathol. 2003;34:1185.
- Leu S, Sun PK, Collyer J, et al. Clinical spectrum of vulva metastatic Crohn's disease. Dig Dis Sci. 2009;54:1565-1571.
- Chen W, Blume-Peytavi U, Goerdt S, et al. Metastatic Crohn's disease of the face. J Am Acad Dermatol. 1996;35:986-988.
- Ogram AE, Sobanko JF, Nigra TP. Metastatic cutaneous Crohn disease of the face: a case report and review of the literature. Cutis. 2010;85:25-27.
- Graham D, Jager D, Borum M. Metastatic Crohn's disease of the face. Dig Dis Sci. 2006;51:2062-2063.
- Biancone L, Geboes K, Spagnoli LG, et al. Metastatic Crohn's disease of the forehead. Inflamm Bowel Dis. 2002;8:101-105.
- Kolansky G, Green CK, Dubin H. Metastatic Crohn's disease of the face: an uncommon presentation. Arch Dermatol. 1993;129:1348-1349.
- Mahadevan U, Sandborn WJ. Infliximab for the treatment of orofacial Crohn's disease. Inflamm Bowel Dis. 2001;7:38-42.
- McCallum DI, Gray WM. Metastatic Crohn's disease. Br J Dermatol. 1976;95:551-554.
- Lieberman TR, Greene JF Jr. Transient subcutaneous granulomatosis of the upper extremities in Crohn's disease. Am J Gastroenterol. 1979;72:89-91.
- Kafity AA, Pellegrini AE, Fromkes JJ. Metastatic Crohn's disease: a rare cutaneous manifestation. J Clin Gastroenterol. 1993;17:300-303.
- Marotta PJ, Reynolds RP. Metastatic Crohn's disease. Am J Gastroenterol. 1996;91:373-375.
- Hackzell-Bradley M, Hedblad MA, Stephansson EA. Metastatic Crohn's disease. report of 3 cases with special reference to histopathologic findings. Arch Dermatol. 1996;132:928-932.
- van Dulleman HM, de Jong E, Slors F, et al. Treatment of therapy resistant perineal metastatic Crohn's disease after proctectomy using anti-tumor necrosis factor chimeric monoclonal antibody, cA2: report of two cases. Dis Colon Rectum. 1998;41:98-102.
- Lavery HA, Pinkerton JH, Sloan J. Crohn's disease of the vulva--two further cases. Br J Dermatol. 1985;113:359-363.
- Lally MR, Orenstein SR, Cohen BA. Crohn's disease of the vulva in an 8-year-old girl. Pediatr Dermatol. 1988;5:103-106.
- Tuffnell D, Buchan PC. Crohn's disease of the vulva in childhood. Br J Clin Pract. 1991;45:159-160.
- Schrodt BJ, Callen JP. Metastatic Crohn's disease presenting as chronic perivulvar and perirectal ulcerations in an adolescent patient. Pediatrics. 1999;103:500-502.
- Slaney G, Muller S, Clay J, et al. Crohn's disease involving the penis. Gut. 1986;27:329-333.
- Calder CJ, Lacy D, Raafat F, et al. Crohn's disease with pulmonary involvement in a 3 year old boy. Gut. 1993;34:1636-1638.
- Saha S, Fichera A, Bales G, et al. Metastatic Crohn's disease of the bladder. Inflamm Bowel Dis. 2008;14:140-142.
- Escher JC, Stoof TJ, van Deventer SJ, et al. Successful treatment of metastatic Crohn disease with infliximab. J Pediatr Gastroenterol Nutr. 2002;34:420-423.
- Saadah OI, Oliver MR, Bines JE, et al. Anorectal strictures and genital Crohn's disease: an unusual clinical association. J Pediatr Gastroenterol Nutr. 2003;36:403-406.
- Martinez-Salamanca JI, Jara J, Miralles P, et al. Metastatic Crohn's disease: penile and scrotal involvement. Scand J Urol Nephrol. 2004;38:436-437.
- Podolsky DK. Inflammatory bowel disease. N Engl J Med. 2002;347:417-429.
- Acker SM, Sahn EE, Rogers HC, et al. Genital cutaneous Crohn disease. Am J Dermatopathol. 2000;22:443-446.
- Lestre S, Ramos J, Joao A, et al. Cutaneous Crohn's disease presenting as genital warts: successful treatment with adalimumab. Eur J Dermatol. 2010;20:504-505.
- Yu JT, Chong LY, Lee KC. Metastatic Crohn's disease in a Chinese girl. Hong Kong Med J. 2006;12:467-469.
- Wilson-Jones E, Winkelmann RK. Superficial granulomatous pyoderma: a localized vegetative form of pyoderma gangrenosum. J Am Acad Dermatol. 1988;18:511-521.
- Moyes LH, Glen P, Pickford IR. Perineal metastatic Crohn's disease: a case report and review of the literature. Ann R Coll Surg Engl. 2007;89:W1-W3.
- Rajpara SM, Siddha SK, Ormerod AD, et al. Cutaneous penile and perianal Crohn's disease treated with a combination of medical and surgical interventions. Australas J Dermatol. 2008;49:21-24.
- Cockburn AG, Krolikowski J, Balogh K, et al. Crohn disease of penile and scrotal skin. Urology. 1980;15:596-598.
- Guest GD, Fink RL. Metastatic Crohn's disease: case report of an unusual variant and review of the literature. Dis Colon Rectum. 2000;43:1764-1766.
- Sangueza OP, Davis LS, Gourdin FW. Metastatic Crohn disease. South Med J. 1997;90:897-900.
- Chiba M, Iizuka M, Horie Y, et al. Metastatic Crohn's disease involving the penis. J Gastroenterol. 1997;32:817-821.
- Poon KS, Gilks CB, Masterson JS. Metastatic Crohn's disease involving the genitalia. J Urol. 2002;167:2541-2542.
- Shanahan F. Anti-TNF therapy for Crohn's disease: a perspective (infliximab is not the drug we have been waiting for). Inflamm Bowel Dis. 2000;6:137-139.
- Carranza DC, Young L. Successful treatment of metastatic Crohn's disease with cyclosporine. J Drugs Dermatol. 2008;7:789-791.
- Bardazzi F, Guidetti MS, Passarini B, et al. Cyclosporine A in metastatic Crohn's disease. Acta Derm Venereol. 1995;75:324-325.
- Faubion WA Jr, Loftus EV Jr, Harmsen WS, et al. The natural history of corticosteroid therapy for inflammatory bowel disease: a population-based study. Gastroenterology. 2001;121:255-260.
- Gelbmann CM, Rogler G, Gross V, et al. Prior bowel resections, perianal disease, and a high initial Crohn's disease activity index are associated with corticosteroid resistance in active Crohn's disease. Am J Gastroenterol. 2002;97:1438-1445.
- Thukral C, Travassos WJ, Peppercorn MA. The role of antibiotics in inflammatory bowel disease. Curr Treat Options Gastroenterol. 2005;8:223-228.
- Brandt LJ, Berstein LH, Boley SJ, et al. Metronidazole therapy for perineal Crohn's disease: a follow-up study. Gastroenterology. 1982;83:383-387.
- Lehrnbecher T, Kontny HU, Jeschke R. Metastatic Crohn's disease in a 9-year-old boy. J Pediatr Gastroenterol Nutr. 1999;28:321-323.
- Abide JM. Metastatic Crohn disease: clearance with metronidazole. J Am Acad Dermatol. 2011;64:448-449.
- Rispo A, Scarpa R, Di Girolamo E, et al. Infliximab in the treatment of extra-intestinal manifestations of Crohn's disease. Scand J Rheumatol. 2005;34:387-391.
- Kaufman I, Caspi D, Yeshurun D, et al. The effect of infliximab on extraintestinal manifestations of Crohn's disease. Rheumatol Int. 2005;25:406-410.
- Konrad A, Seibold F. Response of cutaneous Crohn's disease to infliximab and methotrexate. Dig Liver Dis. 2003;35:351-356.
- Miller AM, Elliott PR, Fink R, et al. Rapid response of severe refractory metastatic Crohn's disease to infliximab. J Gastroenterol Hepatol. 2001;16:940-942.
- Chuah JH, Kim DS, Allen C, et al. Metastatic Crohn's disease of the ear. Int J Otolaryngol. 2009;2009:871567.
- Present DH, Rutgeerts P, Targan S, et al. Infliximab for the treatment of fistulas in patients with Crohn's disease. N Engl J Med. 1999;340:1398-1405.
- Petrolati A, Altavilla N, Cipolla R, et al. Cutaneous metastatic Crohn's disease responsive to infliximab. Am J Gastroenterol. 2009;104:1058.
- Hanauer SB, Sandborn WJ, Rutgeerts P, et al. Human anti-tumor necrosis factor monoclonal antibody (adalimumab) in Crohn's disease: the CLASSIC-I trial. Gastroenterology. 2006;130:323-333.
- Cury DB, Moss A, Elias G, et al. Adalimumab for cutaneous metastatic Crohn's disease. Inflamm Bowel Dis. 2010;16:723-724.
Almost half of Crohn disease (CD) patients experience a dermatologic manifestation of the disease. A rare entity, metastatic CD (MCD) presents a diagnostic challenge without a high index of suspicion. Its etiology is not well defined; however, it appears to be an autoimmune response to gut antigens. Herein, we review the etiology/epidemiology, diagnostic criteria, and treatment for this uncommon condition.
Epidemiology and Clinical Characteristics of MCD
Metastatic CD was first described by Parks et al1 in 1965 and refers to a diverse collection of macroscopic dermatologic manifestations in tissue not contiguous with the gastrointestinal (GI) tract. To be classified as MCD, the tissue must demonstrate characteristic histopathologic findings, which invariably include noncaseating granulomas.
Crohn disease may affect any part of the GI tract from the mouth to anus, with a multitude of associated cutaneous manifestations having been described. The terminal ileum is the most commonly affected portion of the GI tract in CD, but the large intestine also may be involved in 55% to 80% of cases.2 The incidence of non-MCD-associated anal lesions seems to correlate with intestinal involvement in that as few as 25% of patients with ileal-localized CD have anal lesions compared to nearly 80% of patients with large intestinal involvement.3
It has been estimated that 18% to 44% of patients with CD have some form of cutaneous manifestation,4 with MCD being a rare subcategory. As few as 100 cases have been described from 1965 to the present.5 The presence of MCD does not correlate well with severity of intestinal CD, and although a majority of MCD cases present after at least 6 months of GI symptoms,6 there are instances in which MCD presents without prior or existing evidence of intestinal CD.7
With regard to MCD, the term metastatic is sometimes supplanted in the literature by cutaneous to avoid any implication of cancer; however, due to a myriad of dermatologic manifestations, both terms can cause confusion. The categorization of the various types of cutaneous findings in CD is well summarized in a review by Palamaras et al8 with the following classifications: (1) granulomatous by direct extension (oral or perianal), (2) MCD lesions (genital and nongenital), (3) immune-related lesions, and (4) lesions from nutritional deficiencies. Of the cutaneous manifestations relating to CD, MCD is the least common cutaneous categorical manifestation and is further divided into subcategories of genital and nongenital lesions.8
The nongenital distribution of MCD is the more common variety in adults and particularly seems to affect the legs and plantar surfaces (38%), the trunk and abdomen (24%), and the face (15%).5,9 These nongenital MCD manifestations are most commonly described as nodules, ulcerations, or erythematous to purple plaques, and less commonly described as abscesses, pustules, or papules.
The sequence of cutaneous symptoms of MCD relative to intestinal disease depends to some degree on patient age. In adults diagnosed with MCD, it has been noted that a GI flare is expected 2 months to 4 years after diagnosis; however, in children the subsequent GI flare has been noted to vary more widely from 9 months to 14 years following presentation of MCD.8 Furthermore, roughly 50% of children diagnosed with MCD present concomitantly with their first symptoms of a GI flare, whereas 70% of adults with MCD had been previously diagnosed with intestinal CD.8 In one review of 80 reported cases of MCD, 20% (16/80) had no symptoms of intestinal disease at the time of MCD diagnosis, and the majority of the asymptomatic cases were in children; interestingly, the majority of these same children were diagnosed with CD months to years later.9
Both the location and characteristics of cutaneous findings in MCD correlate with age.9 Metastatic CD has been identified in all age groups; however, lymphedema is more common in children/young adults, while nodules, ulceration, and fistulating disease are more often seen in adults.10 Affected children and adolescents with MCD range from 5 to 17 years of age, with a mean age at disease onset of 11.1 years and equal incidence in males and females.8 Adults with MCD range from 18 to 78 years of age, with a mean age at presentation of 38.4 years.8,11
Concerning anatomic location of disease, adults with MCD most commonly have nodules with or without plaques on the arms and legs and less commonly in the genital area.8 In contrast, children with MCD are more prone to genital lesions, with up to 85% of cases including some degree of genital erythematous or nonerythematous swelling with or without induration.8 Genitourinary complications of CD as a broad category, however, are estimated to occur in only 5% to 20% of intestinal CD cases in both children and adults.12
There have been conflicting reports regarding gender predilection in MCD. Based on a review by Samitz et al13 of 200 cases of CD over an 18-year period, 22% of patients with CD were found to have cutaneous manifestations--presumably not MCD but rather perianal, perineal, vulvar fistulae, fissures, or abscesses--with a male to female preponderance of almost 2 to 1. A more recent review of the literature by Palamaras et al8 in 2008 reported that contiguous non-MCD affects adult females and children more often than adult males, with 63% adult cases being female. This review seems to be more congruent with other reports in the literature implicating that females are twice as commonly affected by MCD than males.9,14
Pathophysiology
The etiology of MCD has not been well defined. One proposed mechanism of the distal tissue involvement of MCD is through passage of antigens to the skin with subsequent granulomatous response at the level of the dermis.10 Another proposed mechanism suggests antibody sensitization to gut antigens, possibly bacterial antigens, that then coincidentally cross-react with analogous skin antigens.8,14 Burgdorf11 supported this notion in a 1981 report in which it was suggested that the granulomatous reaction was related to deposition of immune complexes in the skin. Slater et al15 and Tatnall et al16 offered a variation of Burgdorf's notion, suggesting that it was sensitized T cells to circulating antigens that were the initiators of granuloma formation in the periphery.
An examination of MCD tissue in 1990 by Shum and Guenther17 under electron microscopy and immunofluorescence provided evidence against prior studies that purported to have identified immune complexes as the causative agents of MCD. In this study, the authors found no evidence of immune complexes in the dermis of MCD lesions. In addition, an attempt to react serum antibodies of a patient with MCD, which were postulated to have IgG, IgM, and IgA antibodies to specific gut antigens, yielded no response when reacted with the tongue, ileum, and colon tissue from a rat. As a culminant finding, the authors also noted MCD dermis tissue with granulomas without vasculitis, suggesting a T-cell mediated type IV hypersensitivity response with a secondary vasculitis from T-cell origin lymphokines and T-cell mediated monocyte activation.17
Research implicating other immunologic entities involved in the pathophysiology of CD such as β-2 integrin,18 CD14+ monocytes,19 and the role of the DNA repair gene MLH1 (mutL homolog 1)20 have been considered but without a clearly definitive role in the manifestations of MCD.
The utility of metronidazole in the treatment of MCD has been suggested as evidence that certain bacteria in the gut may either serve as the causative antigen or may induce its formation21; however, the causative antigen has yet to be identified, and whether it travels distally to the skin or merely resembles a similar antigen normally present in the dermis has not yet been determined. Some research has used in situ polymerase chain reaction techniques to attempt to detect similar microbial pathogens in both the vasculature of active bowel lesions and in the skin, but to date, bacterial RNA noted to be present in the gut vasculature adjacent to CD lesions has not been detected in skin lesions.22
Diagnosis
Physical Findings
Overall, it is estimated that roughly 56% of all MCD cases affect the external genitalia.23 The classic appearance of MCD includes well-demarcated ulcerations in the areas of intertriginous skin folds with or without diffuse edema and tenderness to palpation.23 Although MCD has been historically noted as having a predilection for moist skin folds, there are numerous case reports of MCD all over the body, including the face,7,24-29 retroauricular areas,30 arms and legs,16,17,31-34 lower abdomen,3,5 under the breasts,1 perineum,35 external genitalia,1,9,36-40 and even the lungs41 and bladder.42
As a dermatologic disease, MCD has been referred to as yet another great imitator, both on the macroscopic and microscopic levels.8 As such, more common causes of genital edema should be considered first and investigated based on the patient's history, physical examination, skin biopsy, lymphangiogram, ultrasound, and cystogram.43 Ultrasonography and color Doppler sonography have been shown to be helpful in patients with genital involvement. This modality can evaluate not only the presence of normal testes but also intratesticular and scrotal wall fluid, especially when the physical examination reveals swelling that makes testicle palpation more difficult.6 Clinically, the correct diagnosis of MCD often is made through suspicion of inflammatory bowel disease based on classic symptoms and/or physical findings including abdominal pain, weight loss, bloody stool, diarrhea, perianal skin tags, and anal fissures or fistulas. Any of these GI findings should prompt an intestinal biopsy to rule out any histologic evidence of CD.
Metastatic CD affecting the vulva often presents with vulvar pain and pruritus and may clinically mimic a more benign disease such as balanitis plasmacellularis, also referred to as Zoon vulvitis.23 Similar to MCD on any given body surface, there is dramatic variation in the macroscopic presentation of vulvar MCD, with physical examination findings ranging from bilateral diffuse, edematous, deeply macerated, red, ulcerated lesions over the vulva with lymphadenopathy to findings of bilateral vulvar pain with yellow drainage from the labia majora.23 There have been cases of vulvar MCD that include exquisite vulvar pain but without structural abnormalities including normal uterus, cervix, adnexa, rectovaginal septum, and rectum. In these more nebulous cases of vulvar MCD, the diagnosis often is discovered incidentally when nonspecific diagnostic imaging suggests underlying CD.23
Beyond the case-by-case variations on physical examination, the great difficulty in diagnosis, particularly in children, occurs in the absence of any GI symptoms and therefore no logical consideration of underlying CD. Consequently, there have been cases of children presenting with irritation of the vulva who were eventually diagnosed with MCD only after erroneous treatment of contact dermatitis, candidiasis, and even consideration of sexual abuse.37 Because it is so rare and obscure among practicing clinicians, the diagnosis of MCD often is considered only after irritation or swelling of the external genitalia has not responded to standard therapies. If and when the diagnosis of MCD is considered in children, it has been suggested to screen patients for anorectal stricture, as case studies have found the condition to be relatively common in this subpopulation.44
In the less common case of adults with genitourinary symptoms that suggest possible MCD, the differential diagnosis for penile or vaginal ulcers should include contact and irritant dermatitis, chronic infectious lesions (eg, hidradenitis suppurativa, actinomycosis, tuberculosis),45 sexually transmitted ulcerative diseases (eg, chancroid, lymphogranuloma venereum, herpes genitalia, granuloma inguinale),46 drug reactions, and even extramammary Paget disease.47
Histologic Findings
Because MCD has so much macroscopic variation and can present anywhere on the surface of the body, formal diagnosis relies on microscopy. As an added measure of difficulty in diagnosis, one random biopsy of a suspicious segment of tissue may not contain the expected histologic findings; therefore, clinical suspicion may warrant a second biopsy.10 There have been reported cases of an adult patient without history of CD presenting with a lesion that resembled a more common pathology, such as a genital wart, and the correct diagnosis of MCD with pseudocondylomatous morphology was made only after intestinal manifestations prompted the clinician to consider such an unusual diagnosis.48
From a histopathologic perspective, MCD is characterized by discrete, noncaseating, sarcoidlike granulomas with abundant multinucleated giant cells (Langhans giant cells) in the superficial dermis (papillary), deep dermis (reticular), and adipose tissue (Figure).8,17 In the presence of concomitant intestinal disease, the granulomas of both the intestinal and dermal tissues should share the same microscopic characteristics.8 In addition, copious neutrophils and granulomas surrounding the microvasculature have been described,34 as well as general lymphocyte and plasma cell infiltrate.45 Some histologic samples have included collagen degeneration termed necrobiosis in the middle dermal layer as another variable finding in MCD.14,34
On microscopy, it has been reported that use of Verhoeff-van Gieson staining may be helpful to highlight the presence of neutrophil obstruction within the dermal vasculature, particularly the arterial lumen, as well as to aid in highlighting swelling of the endothelium with fragmentation of the internal elastic lamina.17 Although not part of the routine diagnosis, electron microscopy of MCD tissue samples have confirmed hypertrophy of the endothelial cells composing the capillaries with resulting extravasation of fibrin, red blood cells, lymphocytes, and epithelioid histiocytes.17 Observation of tissue under direct immunofluorescence has been less helpful, as it has shown only nonspecific fibrinogen deposition within the dermis and dermal vessels.17
In an article on treatment of MCD, Escher et al43 reinforced that the macroscopic findings of MCD are diverse, and the microscopic findings characteristic of MCD also can be mimicked by other etiologies such as sarcoidosis, tuberculosis, fungal infections, lymphogranuloma venereum, leishmaniasis, and connective tissue disorders.43 As such, the workup to rule out infectious, anatomic, and autoimmune etiologies should be diverse. Often, the workup for MCD will include special stains such as Ziehl-Neelsen stain to rule out Mycobacterium tuberculosis and acid-fast bacilli and Fite stain to consider atypical mycobacteria. Other tests such as tissue culture, chest radiograph, tuberculin skin test (Mantoux test), IFN-γ release assay, or polarized light microscopy may rule out infectious etiologies.9,49 Serologic testing might include VDRL test, Treponema pallidum hemagglutination assay, hepatitis B, hepatitis C, and human immunodeficiency virus.5
Crohn disease is characterized histologically by sarcoidlike noncaseating granulomas, and as such, it is important to differentiate MCD from sarcoidosis prior to histologic analysis. Sarcoidosis also can be considered much less likely with a normal chest radiograph and in the absence of increased serum calcium and angiotensin-converting enzyme levels.7 The differentiation of sarcoidosis from MCD on the microscopic scale is subtle but is sometimes facilitated in the presence of an ulcerated epidermis or lymphocytic/eosinophilic infiltrate and edema within the dermis, all suggestive of MCD.14
Metastatic CD also should be differentiated from erythema nodosum and pyoderma gangrenosum, which are among the most common cutaneous findings associated with CD.14 Pyoderma gangrenosum can be distinguished histologically by identifying copious neutrophilic infiltrate with pseudoepitheliomatous hyperplasia.50
Treatment
Because MCD is relatively rare, there are no known randomized trials suggesting a particular medical or surgical treatment. In a review of perineal MCD from 2007, the 40-year-old recommendation by Moutain3 opting for surgical debridement versus medical management still resonates, particularly for perineal disease, as an effective measure in all but the mildest of presentations.51 However, recent case reports also suggest that the tumor necrosis factor α (TNF-α) inhibitors such as infliximab and adalimumab should be considered prior to surgery even with severe perineal MCD.51 Moreover, even if medical management with TNF-α inhibitors or some combination of immunosuppressants and antibiotics does not eradicate the disease, it often helps reduce the size of the ulcers prior to surgery.52 With a limited understanding of MCD, one might think that removal of the affected bowel would eliminate cutaneous disease, but it has been shown that this strategy is not effective.53,54
The composition and location of the particular lesion affects the trajectory of treatment. For example, MCD manifesting as local ulcers and plaques has been described as responding well to topical and intralesional steroids.10,55,56 In the case of penile swelling and/or phimosis, circumcision has been helpful to improve the patient's ability to void as well as to attain and maintain erection.10 In the case of scrotal swelling secondary to MCD, early treatment (ie, within 4 to 6 months) with oral steroids and/or metronidazole is likely beneficial to prevent refractory edematous organization of the tissue.57
As a general rule, an effective treatment will include a combination of an immunosuppressant, antibiotic therapy, and sometimes surgery. The most commonly used immunosuppressant agents include topical or intralesional steroids, infliximab,43,58 cyclosporine A,59,60 dapsone, minocycline, thalidomide, methotrexate, mycophenolate mofetil, sulfasalazine, azathioprine, tacrolimus, and 6-mercaptopurine.4 Steroids have been the conventional treatment of extraintestinal manifestations of CD61; however, perineal CD has been poorly controlled with systemic steroids.62 If steroids are found not to be effective, sometimes agents such as dapsone or thalidomide are considered. One case report noted stabilization of MCD penile ulcers with oral thalidomide 300 mg once daily, oral minocycline 100 mg once daily, and topical tacrolimus 0.3% with benzocaine twice daily with continuation of prednisolone and methotrexate as parts of previously unsuccessful regimen.52
Metronidazole is perhaps the most commonly used antibiotic, having been a component of many successful regimens.4,63 For example, a 27-year-old patient with MCD presenting as a nonhealing ulcerative lesion in the subcoronal area of the penis and scrotum was treated successfully with a 6-month course of mesalamine, prednisone, and metronidazole.45 Another case report of vulvar MCD reported initial success with intravenous methylprednisolone, ciprofloxacin, and metronidazole.23 The primary limitation of metronidazole is that subsequent tapering of the dose seems to result in recurrence of disease.64 Consequently, patients must remain on the antibiotic for an indeterminate course, with dosages ranging from 5 mg/kg daily in adolescents65 to 1000 to 1500 mg daily in adults.66
Of the various immunosuppressants available, infliximab has been listed in numerous reports as a successful agent in both the induction and maintenance of extraintestinal manifestations of CD including MCD.67-71 Infliximab has been reported to be effective in the treatment of penile and scrotal edema secondary to MCD that did not respond to other immunosuppressants including oral prednisolone, azathioprine, and cyclosporine.43 Infliximab may be a good option to help heal draining fistulas, particularly in combination with an antibiotic such as metronidazole and ciprofloxacin, which helps to prevent abscess formation during healing.72 The response to infliximab has been dramatic, with resolution of cutaneous lesions after just 6 weeks in some cases.73 The dosing regimen of infliximab has been suggested at 5 mg/kg administered at 0, 2, and 6 weeks, with subsequent maintenance infusions every 10 weeks,70 or at 0, 4, and 12 weeks, with subsequent infusions every 8 weeks.43
Adalimumab may be considered as an alternative to infliximab and is potentially less allergenic as a fully humanized monoclonal antibody to TNF-α, which also has been used successfully to both induce and maintain remission of moderate to severe CD.42,74,75 Proposed dosing of adalimumab includes a loading dose of 160 mg subcutaneously on day 1, followed by an 80-mg dose 2 weeks later and a 40-mg maintenance dose every other week indefinitely.48 Of note, adalimumab has been noted in the literature to have many potential side effects, including one particular case in which severe headaches were attributed to its use.59 As a consequence of the headaches, the patient was switched from adalimumab to cyclosporine and responded well with no subsequent flare-ups on follow-up.
In summary, treatment of MCD depends on cutaneous location, severity, physician experience with certain antibiotics or immunosuppressants, availability of medication, and patient disposition. It seems reasonable to attempt medical management with one or more medical regimens before committing to surgical intervention. Furthermore, even with debridement, curettage, skin graft, or other surgical strategy, the patient is likely to require some period of immunosuppression to provide long-lasting remission.
Conclusion
Patients with inflammatory bowel disease often develop dermatologic sequelae, with MCD being a rare but serious process. Patients may present with a wide array of physical concerns and symptoms, many resembling other disease processes. As such, education and a high index of suspicion are needed for proper diagnosis and treatment.
Almost half of Crohn disease (CD) patients experience a dermatologic manifestation of the disease. A rare entity, metastatic CD (MCD) presents a diagnostic challenge without a high index of suspicion. Its etiology is not well defined; however, it appears to be an autoimmune response to gut antigens. Herein, we review the etiology/epidemiology, diagnostic criteria, and treatment for this uncommon condition.
Epidemiology and Clinical Characteristics of MCD
Metastatic CD was first described by Parks et al1 in 1965 and refers to a diverse collection of macroscopic dermatologic manifestations in tissue not contiguous with the gastrointestinal (GI) tract. To be classified as MCD, the tissue must demonstrate characteristic histopathologic findings, which invariably include noncaseating granulomas.
Crohn disease may affect any part of the GI tract from the mouth to anus, with a multitude of associated cutaneous manifestations having been described. The terminal ileum is the most commonly affected portion of the GI tract in CD, but the large intestine also may be involved in 55% to 80% of cases.2 The incidence of non-MCD-associated anal lesions seems to correlate with intestinal involvement in that as few as 25% of patients with ileal-localized CD have anal lesions compared to nearly 80% of patients with large intestinal involvement.3
It has been estimated that 18% to 44% of patients with CD have some form of cutaneous manifestation,4 with MCD being a rare subcategory. As few as 100 cases have been described from 1965 to the present.5 The presence of MCD does not correlate well with severity of intestinal CD, and although a majority of MCD cases present after at least 6 months of GI symptoms,6 there are instances in which MCD presents without prior or existing evidence of intestinal CD.7
With regard to MCD, the term metastatic is sometimes supplanted in the literature by cutaneous to avoid any implication of cancer; however, due to a myriad of dermatologic manifestations, both terms can cause confusion. The categorization of the various types of cutaneous findings in CD is well summarized in a review by Palamaras et al8 with the following classifications: (1) granulomatous by direct extension (oral or perianal), (2) MCD lesions (genital and nongenital), (3) immune-related lesions, and (4) lesions from nutritional deficiencies. Of the cutaneous manifestations relating to CD, MCD is the least common cutaneous categorical manifestation and is further divided into subcategories of genital and nongenital lesions.8
The nongenital distribution of MCD is the more common variety in adults and particularly seems to affect the legs and plantar surfaces (38%), the trunk and abdomen (24%), and the face (15%).5,9 These nongenital MCD manifestations are most commonly described as nodules, ulcerations, or erythematous to purple plaques, and less commonly described as abscesses, pustules, or papules.
The sequence of cutaneous symptoms of MCD relative to intestinal disease depends to some degree on patient age. In adults diagnosed with MCD, it has been noted that a GI flare is expected 2 months to 4 years after diagnosis; however, in children the subsequent GI flare has been noted to vary more widely from 9 months to 14 years following presentation of MCD.8 Furthermore, roughly 50% of children diagnosed with MCD present concomitantly with their first symptoms of a GI flare, whereas 70% of adults with MCD had been previously diagnosed with intestinal CD.8 In one review of 80 reported cases of MCD, 20% (16/80) had no symptoms of intestinal disease at the time of MCD diagnosis, and the majority of the asymptomatic cases were in children; interestingly, the majority of these same children were diagnosed with CD months to years later.9
Both the location and characteristics of cutaneous findings in MCD correlate with age.9 Metastatic CD has been identified in all age groups; however, lymphedema is more common in children/young adults, while nodules, ulceration, and fistulating disease are more often seen in adults.10 Affected children and adolescents with MCD range from 5 to 17 years of age, with a mean age at disease onset of 11.1 years and equal incidence in males and females.8 Adults with MCD range from 18 to 78 years of age, with a mean age at presentation of 38.4 years.8,11
Concerning anatomic location of disease, adults with MCD most commonly have nodules with or without plaques on the arms and legs and less commonly in the genital area.8 In contrast, children with MCD are more prone to genital lesions, with up to 85% of cases including some degree of genital erythematous or nonerythematous swelling with or without induration.8 Genitourinary complications of CD as a broad category, however, are estimated to occur in only 5% to 20% of intestinal CD cases in both children and adults.12
There have been conflicting reports regarding gender predilection in MCD. Based on a review by Samitz et al13 of 200 cases of CD over an 18-year period, 22% of patients with CD were found to have cutaneous manifestations--presumably not MCD but rather perianal, perineal, vulvar fistulae, fissures, or abscesses--with a male to female preponderance of almost 2 to 1. A more recent review of the literature by Palamaras et al8 in 2008 reported that contiguous non-MCD affects adult females and children more often than adult males, with 63% adult cases being female. This review seems to be more congruent with other reports in the literature implicating that females are twice as commonly affected by MCD than males.9,14
Pathophysiology
The etiology of MCD has not been well defined. One proposed mechanism of the distal tissue involvement of MCD is through passage of antigens to the skin with subsequent granulomatous response at the level of the dermis.10 Another proposed mechanism suggests antibody sensitization to gut antigens, possibly bacterial antigens, that then coincidentally cross-react with analogous skin antigens.8,14 Burgdorf11 supported this notion in a 1981 report in which it was suggested that the granulomatous reaction was related to deposition of immune complexes in the skin. Slater et al15 and Tatnall et al16 offered a variation of Burgdorf's notion, suggesting that it was sensitized T cells to circulating antigens that were the initiators of granuloma formation in the periphery.
An examination of MCD tissue in 1990 by Shum and Guenther17 under electron microscopy and immunofluorescence provided evidence against prior studies that purported to have identified immune complexes as the causative agents of MCD. In this study, the authors found no evidence of immune complexes in the dermis of MCD lesions. In addition, an attempt to react serum antibodies of a patient with MCD, which were postulated to have IgG, IgM, and IgA antibodies to specific gut antigens, yielded no response when reacted with the tongue, ileum, and colon tissue from a rat. As a culminant finding, the authors also noted MCD dermis tissue with granulomas without vasculitis, suggesting a T-cell mediated type IV hypersensitivity response with a secondary vasculitis from T-cell origin lymphokines and T-cell mediated monocyte activation.17
Research implicating other immunologic entities involved in the pathophysiology of CD such as β-2 integrin,18 CD14+ monocytes,19 and the role of the DNA repair gene MLH1 (mutL homolog 1)20 have been considered but without a clearly definitive role in the manifestations of MCD.
The utility of metronidazole in the treatment of MCD has been suggested as evidence that certain bacteria in the gut may either serve as the causative antigen or may induce its formation21; however, the causative antigen has yet to be identified, and whether it travels distally to the skin or merely resembles a similar antigen normally present in the dermis has not yet been determined. Some research has used in situ polymerase chain reaction techniques to attempt to detect similar microbial pathogens in both the vasculature of active bowel lesions and in the skin, but to date, bacterial RNA noted to be present in the gut vasculature adjacent to CD lesions has not been detected in skin lesions.22
Diagnosis
Physical Findings
Overall, it is estimated that roughly 56% of all MCD cases affect the external genitalia.23 The classic appearance of MCD includes well-demarcated ulcerations in the areas of intertriginous skin folds with or without diffuse edema and tenderness to palpation.23 Although MCD has been historically noted as having a predilection for moist skin folds, there are numerous case reports of MCD all over the body, including the face,7,24-29 retroauricular areas,30 arms and legs,16,17,31-34 lower abdomen,3,5 under the breasts,1 perineum,35 external genitalia,1,9,36-40 and even the lungs41 and bladder.42
As a dermatologic disease, MCD has been referred to as yet another great imitator, both on the macroscopic and microscopic levels.8 As such, more common causes of genital edema should be considered first and investigated based on the patient's history, physical examination, skin biopsy, lymphangiogram, ultrasound, and cystogram.43 Ultrasonography and color Doppler sonography have been shown to be helpful in patients with genital involvement. This modality can evaluate not only the presence of normal testes but also intratesticular and scrotal wall fluid, especially when the physical examination reveals swelling that makes testicle palpation more difficult.6 Clinically, the correct diagnosis of MCD often is made through suspicion of inflammatory bowel disease based on classic symptoms and/or physical findings including abdominal pain, weight loss, bloody stool, diarrhea, perianal skin tags, and anal fissures or fistulas. Any of these GI findings should prompt an intestinal biopsy to rule out any histologic evidence of CD.
Metastatic CD affecting the vulva often presents with vulvar pain and pruritus and may clinically mimic a more benign disease such as balanitis plasmacellularis, also referred to as Zoon vulvitis.23 Similar to MCD on any given body surface, there is dramatic variation in the macroscopic presentation of vulvar MCD, with physical examination findings ranging from bilateral diffuse, edematous, deeply macerated, red, ulcerated lesions over the vulva with lymphadenopathy to findings of bilateral vulvar pain with yellow drainage from the labia majora.23 There have been cases of vulvar MCD that include exquisite vulvar pain but without structural abnormalities including normal uterus, cervix, adnexa, rectovaginal septum, and rectum. In these more nebulous cases of vulvar MCD, the diagnosis often is discovered incidentally when nonspecific diagnostic imaging suggests underlying CD.23
Beyond the case-by-case variations on physical examination, the great difficulty in diagnosis, particularly in children, occurs in the absence of any GI symptoms and therefore no logical consideration of underlying CD. Consequently, there have been cases of children presenting with irritation of the vulva who were eventually diagnosed with MCD only after erroneous treatment of contact dermatitis, candidiasis, and even consideration of sexual abuse.37 Because it is so rare and obscure among practicing clinicians, the diagnosis of MCD often is considered only after irritation or swelling of the external genitalia has not responded to standard therapies. If and when the diagnosis of MCD is considered in children, it has been suggested to screen patients for anorectal stricture, as case studies have found the condition to be relatively common in this subpopulation.44
In the less common case of adults with genitourinary symptoms that suggest possible MCD, the differential diagnosis for penile or vaginal ulcers should include contact and irritant dermatitis, chronic infectious lesions (eg, hidradenitis suppurativa, actinomycosis, tuberculosis),45 sexually transmitted ulcerative diseases (eg, chancroid, lymphogranuloma venereum, herpes genitalia, granuloma inguinale),46 drug reactions, and even extramammary Paget disease.47
Histologic Findings
Because MCD has so much macroscopic variation and can present anywhere on the surface of the body, formal diagnosis relies on microscopy. As an added measure of difficulty in diagnosis, one random biopsy of a suspicious segment of tissue may not contain the expected histologic findings; therefore, clinical suspicion may warrant a second biopsy.10 There have been reported cases of an adult patient without history of CD presenting with a lesion that resembled a more common pathology, such as a genital wart, and the correct diagnosis of MCD with pseudocondylomatous morphology was made only after intestinal manifestations prompted the clinician to consider such an unusual diagnosis.48
From a histopathologic perspective, MCD is characterized by discrete, noncaseating, sarcoidlike granulomas with abundant multinucleated giant cells (Langhans giant cells) in the superficial dermis (papillary), deep dermis (reticular), and adipose tissue (Figure).8,17 In the presence of concomitant intestinal disease, the granulomas of both the intestinal and dermal tissues should share the same microscopic characteristics.8 In addition, copious neutrophils and granulomas surrounding the microvasculature have been described,34 as well as general lymphocyte and plasma cell infiltrate.45 Some histologic samples have included collagen degeneration termed necrobiosis in the middle dermal layer as another variable finding in MCD.14,34
On microscopy, it has been reported that use of Verhoeff-van Gieson staining may be helpful to highlight the presence of neutrophil obstruction within the dermal vasculature, particularly the arterial lumen, as well as to aid in highlighting swelling of the endothelium with fragmentation of the internal elastic lamina.17 Although not part of the routine diagnosis, electron microscopy of MCD tissue samples have confirmed hypertrophy of the endothelial cells composing the capillaries with resulting extravasation of fibrin, red blood cells, lymphocytes, and epithelioid histiocytes.17 Observation of tissue under direct immunofluorescence has been less helpful, as it has shown only nonspecific fibrinogen deposition within the dermis and dermal vessels.17
In an article on treatment of MCD, Escher et al43 reinforced that the macroscopic findings of MCD are diverse, and the microscopic findings characteristic of MCD also can be mimicked by other etiologies such as sarcoidosis, tuberculosis, fungal infections, lymphogranuloma venereum, leishmaniasis, and connective tissue disorders.43 As such, the workup to rule out infectious, anatomic, and autoimmune etiologies should be diverse. Often, the workup for MCD will include special stains such as Ziehl-Neelsen stain to rule out Mycobacterium tuberculosis and acid-fast bacilli and Fite stain to consider atypical mycobacteria. Other tests such as tissue culture, chest radiograph, tuberculin skin test (Mantoux test), IFN-γ release assay, or polarized light microscopy may rule out infectious etiologies.9,49 Serologic testing might include VDRL test, Treponema pallidum hemagglutination assay, hepatitis B, hepatitis C, and human immunodeficiency virus.5
Crohn disease is characterized histologically by sarcoidlike noncaseating granulomas, and as such, it is important to differentiate MCD from sarcoidosis prior to histologic analysis. Sarcoidosis also can be considered much less likely with a normal chest radiograph and in the absence of increased serum calcium and angiotensin-converting enzyme levels.7 The differentiation of sarcoidosis from MCD on the microscopic scale is subtle but is sometimes facilitated in the presence of an ulcerated epidermis or lymphocytic/eosinophilic infiltrate and edema within the dermis, all suggestive of MCD.14
Metastatic CD also should be differentiated from erythema nodosum and pyoderma gangrenosum, which are among the most common cutaneous findings associated with CD.14 Pyoderma gangrenosum can be distinguished histologically by identifying copious neutrophilic infiltrate with pseudoepitheliomatous hyperplasia.50
Treatment
Because MCD is relatively rare, there are no known randomized trials suggesting a particular medical or surgical treatment. In a review of perineal MCD from 2007, the 40-year-old recommendation by Moutain3 opting for surgical debridement versus medical management still resonates, particularly for perineal disease, as an effective measure in all but the mildest of presentations.51 However, recent case reports also suggest that the tumor necrosis factor α (TNF-α) inhibitors such as infliximab and adalimumab should be considered prior to surgery even with severe perineal MCD.51 Moreover, even if medical management with TNF-α inhibitors or some combination of immunosuppressants and antibiotics does not eradicate the disease, it often helps reduce the size of the ulcers prior to surgery.52 With a limited understanding of MCD, one might think that removal of the affected bowel would eliminate cutaneous disease, but it has been shown that this strategy is not effective.53,54
The composition and location of the particular lesion affects the trajectory of treatment. For example, MCD manifesting as local ulcers and plaques has been described as responding well to topical and intralesional steroids.10,55,56 In the case of penile swelling and/or phimosis, circumcision has been helpful to improve the patient's ability to void as well as to attain and maintain erection.10 In the case of scrotal swelling secondary to MCD, early treatment (ie, within 4 to 6 months) with oral steroids and/or metronidazole is likely beneficial to prevent refractory edematous organization of the tissue.57
As a general rule, an effective treatment will include a combination of an immunosuppressant, antibiotic therapy, and sometimes surgery. The most commonly used immunosuppressant agents include topical or intralesional steroids, infliximab,43,58 cyclosporine A,59,60 dapsone, minocycline, thalidomide, methotrexate, mycophenolate mofetil, sulfasalazine, azathioprine, tacrolimus, and 6-mercaptopurine.4 Steroids have been the conventional treatment of extraintestinal manifestations of CD61; however, perineal CD has been poorly controlled with systemic steroids.62 If steroids are found not to be effective, sometimes agents such as dapsone or thalidomide are considered. One case report noted stabilization of MCD penile ulcers with oral thalidomide 300 mg once daily, oral minocycline 100 mg once daily, and topical tacrolimus 0.3% with benzocaine twice daily with continuation of prednisolone and methotrexate as parts of previously unsuccessful regimen.52
Metronidazole is perhaps the most commonly used antibiotic, having been a component of many successful regimens.4,63 For example, a 27-year-old patient with MCD presenting as a nonhealing ulcerative lesion in the subcoronal area of the penis and scrotum was treated successfully with a 6-month course of mesalamine, prednisone, and metronidazole.45 Another case report of vulvar MCD reported initial success with intravenous methylprednisolone, ciprofloxacin, and metronidazole.23 The primary limitation of metronidazole is that subsequent tapering of the dose seems to result in recurrence of disease.64 Consequently, patients must remain on the antibiotic for an indeterminate course, with dosages ranging from 5 mg/kg daily in adolescents65 to 1000 to 1500 mg daily in adults.66
Of the various immunosuppressants available, infliximab has been listed in numerous reports as a successful agent in both the induction and maintenance of extraintestinal manifestations of CD including MCD.67-71 Infliximab has been reported to be effective in the treatment of penile and scrotal edema secondary to MCD that did not respond to other immunosuppressants including oral prednisolone, azathioprine, and cyclosporine.43 Infliximab may be a good option to help heal draining fistulas, particularly in combination with an antibiotic such as metronidazole and ciprofloxacin, which helps to prevent abscess formation during healing.72 The response to infliximab has been dramatic, with resolution of cutaneous lesions after just 6 weeks in some cases.73 The dosing regimen of infliximab has been suggested at 5 mg/kg administered at 0, 2, and 6 weeks, with subsequent maintenance infusions every 10 weeks,70 or at 0, 4, and 12 weeks, with subsequent infusions every 8 weeks.43
Adalimumab may be considered as an alternative to infliximab and is potentially less allergenic as a fully humanized monoclonal antibody to TNF-α, which also has been used successfully to both induce and maintain remission of moderate to severe CD.42,74,75 Proposed dosing of adalimumab includes a loading dose of 160 mg subcutaneously on day 1, followed by an 80-mg dose 2 weeks later and a 40-mg maintenance dose every other week indefinitely.48 Of note, adalimumab has been noted in the literature to have many potential side effects, including one particular case in which severe headaches were attributed to its use.59 As a consequence of the headaches, the patient was switched from adalimumab to cyclosporine and responded well with no subsequent flare-ups on follow-up.
In summary, treatment of MCD depends on cutaneous location, severity, physician experience with certain antibiotics or immunosuppressants, availability of medication, and patient disposition. It seems reasonable to attempt medical management with one or more medical regimens before committing to surgical intervention. Furthermore, even with debridement, curettage, skin graft, or other surgical strategy, the patient is likely to require some period of immunosuppression to provide long-lasting remission.
Conclusion
Patients with inflammatory bowel disease often develop dermatologic sequelae, with MCD being a rare but serious process. Patients may present with a wide array of physical concerns and symptoms, many resembling other disease processes. As such, education and a high index of suspicion are needed for proper diagnosis and treatment.
- Parks AG, Morson BC, Pegum JS. Crohn's disease with cutaneous involvement. Proc R Soc Med. 1965;58:241-242.
- Friedman S, Blumber RS. Inflammatory bowel disease. In: Kasper DL, Braunwald E, Fauci AS, et al, eds. Harrison's Principles of Internal Medicine. 16th ed. New York, NY: McGraw-Hill; 2005:1778-1784.
- Moutain JC. Cutaneous ulceration in Crohn's disease. Gut. 1970;11:18-26.
- Lester LU, Rapini RP. Dermatologic manifestations of colonic disorders. Curr Opin Gastroenterol. 2008;25:66-73.
- Teixeira M, Machado S, Lago P, et al. Cutaneous Crohn's disease. Int J Dermatol. 2006;45:1074-1076.
- Simoneaux SF, Ball TI, Atkinson GO Jr. Scrotal swelling: unusual first presentation of Crohn's disease. Pediatr Radiol. 1995;25:375-376.
- Albuquerque A, Magro F, Rodrigues S, et al. Metastatic cutaneous Crohn's disease of the face: a case report and review of literature. Eur J Gastroenterol Hepatol. 2011;23:954-956.
- Palamaras I, El-Jabbour J, Pietropaolo N, et al. Metastatic Crohn's disease: a review. J Eur Acad Dermatol Venereol. 2008;22:1033-1043.
- Ploysangam T, Heubi JE, Eisen D, et al. Cutaneous Crohn's disease in children. J Am Acad Dermatol. 1997;36:697-704.
- Vint R, Husain E, Hassain F, et al. Metastatic Crohn's disease of the penis: two cases. Int Urol Nephrol. 2012;44:45-49.
- Burgdorf W. Cutaneous manifestations of Crohn's disease. J Am Acad Dermatol. 1981;5:689-695.
- Resnick MI, Kursh ED. Extrinsic obstruction of the ureter. In: Walsh PC, Retik AB, Stamey TA, et al, eds. Campbell's Urology. 7th ed. Philadelphia, PA: WB Saunders; 1998:400-402.
- Samitz MH, Dana AS Jr, Rosenberg P. Cutaneous vasculitis in association with Crohn's disease--review of statistics of skin complications. Cutis. 1970;6:51-56.
- Emanuel PO, Phelps RG. Metastatic Crohn's disease: a histo-pathologic study of 12 cases. J Cutan Pathol. 2008;35:457-461.
- Slater DN, Waller PC, Reilly G. Cutaneous granulomatous vasculitis: presenting features of Crohn's disease. J R Soc Med. 1985;78:589-590.
- Tatnall FM, Dodd HJ, Sarkany I. Crohn's disease with metastatic cutaneous involvement and granulomatous cheilitis. J R Soc Med. 1987;80:49-51.
- Shum DT, Guenther L. Metastatic Crohn's disease: case report and review of literature. Arch Dermatol. 1990;126:645-648.
- Bernstein CN, Sargent M, Gallatin WM. Beta2 integrin/ICAM expression in Crohn's disease. Clin Immunol Immunopathol. 1998;86:147-160.
- Grimm MC, Pavli P, Van de Pol E, et al. Evidence for a CD-14+ population of monocytes in inflammatory bowel disease mucosa--implications for pathogenesis. Clin Exp Immunol. 1995;100:291-297.
- Pokorny RM, Hofmeister A, Galandiuk S, et al. Crohn's disease and ulcerative colitis are associated with the DNA repair gene MLH1. Ann Surg. 1997;225:718-723; discussion 723-725.
- Ursing B, Kamme C. Metronidazole for Crohn's disease. Lancet. 1975;1:775-777.
- Crowson AN, Nuovo GJ, Mihm MC Jr, et al. Cutaneous manifestations of Crohn's disease, its spectrum, and pathogenesis: intracellular consensus bacterial 16S rRNA is associated with the gastrointestinal but not the cutaneous manifestations of Crohn's disease. Hum Pathol. 2003;34:1185.
- Leu S, Sun PK, Collyer J, et al. Clinical spectrum of vulva metastatic Crohn's disease. Dig Dis Sci. 2009;54:1565-1571.
- Chen W, Blume-Peytavi U, Goerdt S, et al. Metastatic Crohn's disease of the face. J Am Acad Dermatol. 1996;35:986-988.
- Ogram AE, Sobanko JF, Nigra TP. Metastatic cutaneous Crohn disease of the face: a case report and review of the literature. Cutis. 2010;85:25-27.
- Graham D, Jager D, Borum M. Metastatic Crohn's disease of the face. Dig Dis Sci. 2006;51:2062-2063.
- Biancone L, Geboes K, Spagnoli LG, et al. Metastatic Crohn's disease of the forehead. Inflamm Bowel Dis. 2002;8:101-105.
- Kolansky G, Green CK, Dubin H. Metastatic Crohn's disease of the face: an uncommon presentation. Arch Dermatol. 1993;129:1348-1349.
- Mahadevan U, Sandborn WJ. Infliximab for the treatment of orofacial Crohn's disease. Inflamm Bowel Dis. 2001;7:38-42.
- McCallum DI, Gray WM. Metastatic Crohn's disease. Br J Dermatol. 1976;95:551-554.
- Lieberman TR, Greene JF Jr. Transient subcutaneous granulomatosis of the upper extremities in Crohn's disease. Am J Gastroenterol. 1979;72:89-91.
- Kafity AA, Pellegrini AE, Fromkes JJ. Metastatic Crohn's disease: a rare cutaneous manifestation. J Clin Gastroenterol. 1993;17:300-303.
- Marotta PJ, Reynolds RP. Metastatic Crohn's disease. Am J Gastroenterol. 1996;91:373-375.
- Hackzell-Bradley M, Hedblad MA, Stephansson EA. Metastatic Crohn's disease. report of 3 cases with special reference to histopathologic findings. Arch Dermatol. 1996;132:928-932.
- van Dulleman HM, de Jong E, Slors F, et al. Treatment of therapy resistant perineal metastatic Crohn's disease after proctectomy using anti-tumor necrosis factor chimeric monoclonal antibody, cA2: report of two cases. Dis Colon Rectum. 1998;41:98-102.
- Lavery HA, Pinkerton JH, Sloan J. Crohn's disease of the vulva--two further cases. Br J Dermatol. 1985;113:359-363.
- Lally MR, Orenstein SR, Cohen BA. Crohn's disease of the vulva in an 8-year-old girl. Pediatr Dermatol. 1988;5:103-106.
- Tuffnell D, Buchan PC. Crohn's disease of the vulva in childhood. Br J Clin Pract. 1991;45:159-160.
- Schrodt BJ, Callen JP. Metastatic Crohn's disease presenting as chronic perivulvar and perirectal ulcerations in an adolescent patient. Pediatrics. 1999;103:500-502.
- Slaney G, Muller S, Clay J, et al. Crohn's disease involving the penis. Gut. 1986;27:329-333.
- Calder CJ, Lacy D, Raafat F, et al. Crohn's disease with pulmonary involvement in a 3 year old boy. Gut. 1993;34:1636-1638.
- Saha S, Fichera A, Bales G, et al. Metastatic Crohn's disease of the bladder. Inflamm Bowel Dis. 2008;14:140-142.
- Escher JC, Stoof TJ, van Deventer SJ, et al. Successful treatment of metastatic Crohn disease with infliximab. J Pediatr Gastroenterol Nutr. 2002;34:420-423.
- Saadah OI, Oliver MR, Bines JE, et al. Anorectal strictures and genital Crohn's disease: an unusual clinical association. J Pediatr Gastroenterol Nutr. 2003;36:403-406.
- Martinez-Salamanca JI, Jara J, Miralles P, et al. Metastatic Crohn's disease: penile and scrotal involvement. Scand J Urol Nephrol. 2004;38:436-437.
- Podolsky DK. Inflammatory bowel disease. N Engl J Med. 2002;347:417-429.
- Acker SM, Sahn EE, Rogers HC, et al. Genital cutaneous Crohn disease. Am J Dermatopathol. 2000;22:443-446.
- Lestre S, Ramos J, Joao A, et al. Cutaneous Crohn's disease presenting as genital warts: successful treatment with adalimumab. Eur J Dermatol. 2010;20:504-505.
- Yu JT, Chong LY, Lee KC. Metastatic Crohn's disease in a Chinese girl. Hong Kong Med J. 2006;12:467-469.
- Wilson-Jones E, Winkelmann RK. Superficial granulomatous pyoderma: a localized vegetative form of pyoderma gangrenosum. J Am Acad Dermatol. 1988;18:511-521.
- Moyes LH, Glen P, Pickford IR. Perineal metastatic Crohn's disease: a case report and review of the literature. Ann R Coll Surg Engl. 2007;89:W1-W3.
- Rajpara SM, Siddha SK, Ormerod AD, et al. Cutaneous penile and perianal Crohn's disease treated with a combination of medical and surgical interventions. Australas J Dermatol. 2008;49:21-24.
- Cockburn AG, Krolikowski J, Balogh K, et al. Crohn disease of penile and scrotal skin. Urology. 1980;15:596-598.
- Guest GD, Fink RL. Metastatic Crohn's disease: case report of an unusual variant and review of the literature. Dis Colon Rectum. 2000;43:1764-1766.
- Sangueza OP, Davis LS, Gourdin FW. Metastatic Crohn disease. South Med J. 1997;90:897-900.
- Chiba M, Iizuka M, Horie Y, et al. Metastatic Crohn's disease involving the penis. J Gastroenterol. 1997;32:817-821.
- Poon KS, Gilks CB, Masterson JS. Metastatic Crohn's disease involving the genitalia. J Urol. 2002;167:2541-2542.
- Shanahan F. Anti-TNF therapy for Crohn's disease: a perspective (infliximab is not the drug we have been waiting for). Inflamm Bowel Dis. 2000;6:137-139.
- Carranza DC, Young L. Successful treatment of metastatic Crohn's disease with cyclosporine. J Drugs Dermatol. 2008;7:789-791.
- Bardazzi F, Guidetti MS, Passarini B, et al. Cyclosporine A in metastatic Crohn's disease. Acta Derm Venereol. 1995;75:324-325.
- Faubion WA Jr, Loftus EV Jr, Harmsen WS, et al. The natural history of corticosteroid therapy for inflammatory bowel disease: a population-based study. Gastroenterology. 2001;121:255-260.
- Gelbmann CM, Rogler G, Gross V, et al. Prior bowel resections, perianal disease, and a high initial Crohn's disease activity index are associated with corticosteroid resistance in active Crohn's disease. Am J Gastroenterol. 2002;97:1438-1445.
- Thukral C, Travassos WJ, Peppercorn MA. The role of antibiotics in inflammatory bowel disease. Curr Treat Options Gastroenterol. 2005;8:223-228.
- Brandt LJ, Berstein LH, Boley SJ, et al. Metronidazole therapy for perineal Crohn's disease: a follow-up study. Gastroenterology. 1982;83:383-387.
- Lehrnbecher T, Kontny HU, Jeschke R. Metastatic Crohn's disease in a 9-year-old boy. J Pediatr Gastroenterol Nutr. 1999;28:321-323.
- Abide JM. Metastatic Crohn disease: clearance with metronidazole. J Am Acad Dermatol. 2011;64:448-449.
- Rispo A, Scarpa R, Di Girolamo E, et al. Infliximab in the treatment of extra-intestinal manifestations of Crohn's disease. Scand J Rheumatol. 2005;34:387-391.
- Kaufman I, Caspi D, Yeshurun D, et al. The effect of infliximab on extraintestinal manifestations of Crohn's disease. Rheumatol Int. 2005;25:406-410.
- Konrad A, Seibold F. Response of cutaneous Crohn's disease to infliximab and methotrexate. Dig Liver Dis. 2003;35:351-356.
- Miller AM, Elliott PR, Fink R, et al. Rapid response of severe refractory metastatic Crohn's disease to infliximab. J Gastroenterol Hepatol. 2001;16:940-942.
- Chuah JH, Kim DS, Allen C, et al. Metastatic Crohn's disease of the ear. Int J Otolaryngol. 2009;2009:871567.
- Present DH, Rutgeerts P, Targan S, et al. Infliximab for the treatment of fistulas in patients with Crohn's disease. N Engl J Med. 1999;340:1398-1405.
- Petrolati A, Altavilla N, Cipolla R, et al. Cutaneous metastatic Crohn's disease responsive to infliximab. Am J Gastroenterol. 2009;104:1058.
- Hanauer SB, Sandborn WJ, Rutgeerts P, et al. Human anti-tumor necrosis factor monoclonal antibody (adalimumab) in Crohn's disease: the CLASSIC-I trial. Gastroenterology. 2006;130:323-333.
- Cury DB, Moss A, Elias G, et al. Adalimumab for cutaneous metastatic Crohn's disease. Inflamm Bowel Dis. 2010;16:723-724.
- Parks AG, Morson BC, Pegum JS. Crohn's disease with cutaneous involvement. Proc R Soc Med. 1965;58:241-242.
- Friedman S, Blumber RS. Inflammatory bowel disease. In: Kasper DL, Braunwald E, Fauci AS, et al, eds. Harrison's Principles of Internal Medicine. 16th ed. New York, NY: McGraw-Hill; 2005:1778-1784.
- Moutain JC. Cutaneous ulceration in Crohn's disease. Gut. 1970;11:18-26.
- Lester LU, Rapini RP. Dermatologic manifestations of colonic disorders. Curr Opin Gastroenterol. 2008;25:66-73.
- Teixeira M, Machado S, Lago P, et al. Cutaneous Crohn's disease. Int J Dermatol. 2006;45:1074-1076.
- Simoneaux SF, Ball TI, Atkinson GO Jr. Scrotal swelling: unusual first presentation of Crohn's disease. Pediatr Radiol. 1995;25:375-376.
- Albuquerque A, Magro F, Rodrigues S, et al. Metastatic cutaneous Crohn's disease of the face: a case report and review of literature. Eur J Gastroenterol Hepatol. 2011;23:954-956.
- Palamaras I, El-Jabbour J, Pietropaolo N, et al. Metastatic Crohn's disease: a review. J Eur Acad Dermatol Venereol. 2008;22:1033-1043.
- Ploysangam T, Heubi JE, Eisen D, et al. Cutaneous Crohn's disease in children. J Am Acad Dermatol. 1997;36:697-704.
- Vint R, Husain E, Hassain F, et al. Metastatic Crohn's disease of the penis: two cases. Int Urol Nephrol. 2012;44:45-49.
- Burgdorf W. Cutaneous manifestations of Crohn's disease. J Am Acad Dermatol. 1981;5:689-695.
- Resnick MI, Kursh ED. Extrinsic obstruction of the ureter. In: Walsh PC, Retik AB, Stamey TA, et al, eds. Campbell's Urology. 7th ed. Philadelphia, PA: WB Saunders; 1998:400-402.
- Samitz MH, Dana AS Jr, Rosenberg P. Cutaneous vasculitis in association with Crohn's disease--review of statistics of skin complications. Cutis. 1970;6:51-56.
- Emanuel PO, Phelps RG. Metastatic Crohn's disease: a histo-pathologic study of 12 cases. J Cutan Pathol. 2008;35:457-461.
- Slater DN, Waller PC, Reilly G. Cutaneous granulomatous vasculitis: presenting features of Crohn's disease. J R Soc Med. 1985;78:589-590.
- Tatnall FM, Dodd HJ, Sarkany I. Crohn's disease with metastatic cutaneous involvement and granulomatous cheilitis. J R Soc Med. 1987;80:49-51.
- Shum DT, Guenther L. Metastatic Crohn's disease: case report and review of literature. Arch Dermatol. 1990;126:645-648.
- Bernstein CN, Sargent M, Gallatin WM. Beta2 integrin/ICAM expression in Crohn's disease. Clin Immunol Immunopathol. 1998;86:147-160.
- Grimm MC, Pavli P, Van de Pol E, et al. Evidence for a CD-14+ population of monocytes in inflammatory bowel disease mucosa--implications for pathogenesis. Clin Exp Immunol. 1995;100:291-297.
- Pokorny RM, Hofmeister A, Galandiuk S, et al. Crohn's disease and ulcerative colitis are associated with the DNA repair gene MLH1. Ann Surg. 1997;225:718-723; discussion 723-725.
- Ursing B, Kamme C. Metronidazole for Crohn's disease. Lancet. 1975;1:775-777.
- Crowson AN, Nuovo GJ, Mihm MC Jr, et al. Cutaneous manifestations of Crohn's disease, its spectrum, and pathogenesis: intracellular consensus bacterial 16S rRNA is associated with the gastrointestinal but not the cutaneous manifestations of Crohn's disease. Hum Pathol. 2003;34:1185.
- Leu S, Sun PK, Collyer J, et al. Clinical spectrum of vulva metastatic Crohn's disease. Dig Dis Sci. 2009;54:1565-1571.
- Chen W, Blume-Peytavi U, Goerdt S, et al. Metastatic Crohn's disease of the face. J Am Acad Dermatol. 1996;35:986-988.
- Ogram AE, Sobanko JF, Nigra TP. Metastatic cutaneous Crohn disease of the face: a case report and review of the literature. Cutis. 2010;85:25-27.
- Graham D, Jager D, Borum M. Metastatic Crohn's disease of the face. Dig Dis Sci. 2006;51:2062-2063.
- Biancone L, Geboes K, Spagnoli LG, et al. Metastatic Crohn's disease of the forehead. Inflamm Bowel Dis. 2002;8:101-105.
- Kolansky G, Green CK, Dubin H. Metastatic Crohn's disease of the face: an uncommon presentation. Arch Dermatol. 1993;129:1348-1349.
- Mahadevan U, Sandborn WJ. Infliximab for the treatment of orofacial Crohn's disease. Inflamm Bowel Dis. 2001;7:38-42.
- McCallum DI, Gray WM. Metastatic Crohn's disease. Br J Dermatol. 1976;95:551-554.
- Lieberman TR, Greene JF Jr. Transient subcutaneous granulomatosis of the upper extremities in Crohn's disease. Am J Gastroenterol. 1979;72:89-91.
- Kafity AA, Pellegrini AE, Fromkes JJ. Metastatic Crohn's disease: a rare cutaneous manifestation. J Clin Gastroenterol. 1993;17:300-303.
- Marotta PJ, Reynolds RP. Metastatic Crohn's disease. Am J Gastroenterol. 1996;91:373-375.
- Hackzell-Bradley M, Hedblad MA, Stephansson EA. Metastatic Crohn's disease. report of 3 cases with special reference to histopathologic findings. Arch Dermatol. 1996;132:928-932.
- van Dulleman HM, de Jong E, Slors F, et al. Treatment of therapy resistant perineal metastatic Crohn's disease after proctectomy using anti-tumor necrosis factor chimeric monoclonal antibody, cA2: report of two cases. Dis Colon Rectum. 1998;41:98-102.
- Lavery HA, Pinkerton JH, Sloan J. Crohn's disease of the vulva--two further cases. Br J Dermatol. 1985;113:359-363.
- Lally MR, Orenstein SR, Cohen BA. Crohn's disease of the vulva in an 8-year-old girl. Pediatr Dermatol. 1988;5:103-106.
- Tuffnell D, Buchan PC. Crohn's disease of the vulva in childhood. Br J Clin Pract. 1991;45:159-160.
- Schrodt BJ, Callen JP. Metastatic Crohn's disease presenting as chronic perivulvar and perirectal ulcerations in an adolescent patient. Pediatrics. 1999;103:500-502.
- Slaney G, Muller S, Clay J, et al. Crohn's disease involving the penis. Gut. 1986;27:329-333.
- Calder CJ, Lacy D, Raafat F, et al. Crohn's disease with pulmonary involvement in a 3 year old boy. Gut. 1993;34:1636-1638.
- Saha S, Fichera A, Bales G, et al. Metastatic Crohn's disease of the bladder. Inflamm Bowel Dis. 2008;14:140-142.
- Escher JC, Stoof TJ, van Deventer SJ, et al. Successful treatment of metastatic Crohn disease with infliximab. J Pediatr Gastroenterol Nutr. 2002;34:420-423.
- Saadah OI, Oliver MR, Bines JE, et al. Anorectal strictures and genital Crohn's disease: an unusual clinical association. J Pediatr Gastroenterol Nutr. 2003;36:403-406.
- Martinez-Salamanca JI, Jara J, Miralles P, et al. Metastatic Crohn's disease: penile and scrotal involvement. Scand J Urol Nephrol. 2004;38:436-437.
- Podolsky DK. Inflammatory bowel disease. N Engl J Med. 2002;347:417-429.
- Acker SM, Sahn EE, Rogers HC, et al. Genital cutaneous Crohn disease. Am J Dermatopathol. 2000;22:443-446.
- Lestre S, Ramos J, Joao A, et al. Cutaneous Crohn's disease presenting as genital warts: successful treatment with adalimumab. Eur J Dermatol. 2010;20:504-505.
- Yu JT, Chong LY, Lee KC. Metastatic Crohn's disease in a Chinese girl. Hong Kong Med J. 2006;12:467-469.
- Wilson-Jones E, Winkelmann RK. Superficial granulomatous pyoderma: a localized vegetative form of pyoderma gangrenosum. J Am Acad Dermatol. 1988;18:511-521.
- Moyes LH, Glen P, Pickford IR. Perineal metastatic Crohn's disease: a case report and review of the literature. Ann R Coll Surg Engl. 2007;89:W1-W3.
- Rajpara SM, Siddha SK, Ormerod AD, et al. Cutaneous penile and perianal Crohn's disease treated with a combination of medical and surgical interventions. Australas J Dermatol. 2008;49:21-24.
- Cockburn AG, Krolikowski J, Balogh K, et al. Crohn disease of penile and scrotal skin. Urology. 1980;15:596-598.
- Guest GD, Fink RL. Metastatic Crohn's disease: case report of an unusual variant and review of the literature. Dis Colon Rectum. 2000;43:1764-1766.
- Sangueza OP, Davis LS, Gourdin FW. Metastatic Crohn disease. South Med J. 1997;90:897-900.
- Chiba M, Iizuka M, Horie Y, et al. Metastatic Crohn's disease involving the penis. J Gastroenterol. 1997;32:817-821.
- Poon KS, Gilks CB, Masterson JS. Metastatic Crohn's disease involving the genitalia. J Urol. 2002;167:2541-2542.
- Shanahan F. Anti-TNF therapy for Crohn's disease: a perspective (infliximab is not the drug we have been waiting for). Inflamm Bowel Dis. 2000;6:137-139.
- Carranza DC, Young L. Successful treatment of metastatic Crohn's disease with cyclosporine. J Drugs Dermatol. 2008;7:789-791.
- Bardazzi F, Guidetti MS, Passarini B, et al. Cyclosporine A in metastatic Crohn's disease. Acta Derm Venereol. 1995;75:324-325.
- Faubion WA Jr, Loftus EV Jr, Harmsen WS, et al. The natural history of corticosteroid therapy for inflammatory bowel disease: a population-based study. Gastroenterology. 2001;121:255-260.
- Gelbmann CM, Rogler G, Gross V, et al. Prior bowel resections, perianal disease, and a high initial Crohn's disease activity index are associated with corticosteroid resistance in active Crohn's disease. Am J Gastroenterol. 2002;97:1438-1445.
- Thukral C, Travassos WJ, Peppercorn MA. The role of antibiotics in inflammatory bowel disease. Curr Treat Options Gastroenterol. 2005;8:223-228.
- Brandt LJ, Berstein LH, Boley SJ, et al. Metronidazole therapy for perineal Crohn's disease: a follow-up study. Gastroenterology. 1982;83:383-387.
- Lehrnbecher T, Kontny HU, Jeschke R. Metastatic Crohn's disease in a 9-year-old boy. J Pediatr Gastroenterol Nutr. 1999;28:321-323.
- Abide JM. Metastatic Crohn disease: clearance with metronidazole. J Am Acad Dermatol. 2011;64:448-449.
- Rispo A, Scarpa R, Di Girolamo E, et al. Infliximab in the treatment of extra-intestinal manifestations of Crohn's disease. Scand J Rheumatol. 2005;34:387-391.
- Kaufman I, Caspi D, Yeshurun D, et al. The effect of infliximab on extraintestinal manifestations of Crohn's disease. Rheumatol Int. 2005;25:406-410.
- Konrad A, Seibold F. Response of cutaneous Crohn's disease to infliximab and methotrexate. Dig Liver Dis. 2003;35:351-356.
- Miller AM, Elliott PR, Fink R, et al. Rapid response of severe refractory metastatic Crohn's disease to infliximab. J Gastroenterol Hepatol. 2001;16:940-942.
- Chuah JH, Kim DS, Allen C, et al. Metastatic Crohn's disease of the ear. Int J Otolaryngol. 2009;2009:871567.
- Present DH, Rutgeerts P, Targan S, et al. Infliximab for the treatment of fistulas in patients with Crohn's disease. N Engl J Med. 1999;340:1398-1405.
- Petrolati A, Altavilla N, Cipolla R, et al. Cutaneous metastatic Crohn's disease responsive to infliximab. Am J Gastroenterol. 2009;104:1058.
- Hanauer SB, Sandborn WJ, Rutgeerts P, et al. Human anti-tumor necrosis factor monoclonal antibody (adalimumab) in Crohn's disease: the CLASSIC-I trial. Gastroenterology. 2006;130:323-333.
- Cury DB, Moss A, Elias G, et al. Adalimumab for cutaneous metastatic Crohn's disease. Inflamm Bowel Dis. 2010;16:723-724.
Practice Points
- Almost half of patients with Crohn disease develop a dermatologic manifestation of the disease.
- The etiology of metastatic Crohn disease is unknown and diagnosis requires a high index of suspicion with exclusion of other processes.
Small skin abscesses: Add antibiotics to drainage
For patients who have a single, small skin abscess, the addition of oral antibiotics to standard incision and drainage of the lesion improves cure rates and decreases recurrence rates, according to a study published online June 28 in the New England Journal of Medicine.
The results of this multicenter prospective randomized double-blind placebo-controlled trial, taken together with those of another recent large study, “call into question the perception – largely based on expert opinion or smaller, underpowered, and lower-quality noninferiority trials – that cure rates do not improve with the addition of systemic antibiotic treatment after incision and drainage,” said Robert S. Daum, MD, professor of pediatrics at the University of Chicago, and his associates.
The trial involved 786 patients of all ages (64% were adults and 36% were children; mean age was 25.5 years) who had a single, uncomplicated skin abscess of 5 cm or smaller and were treated at the University of Chicago; San Francisco General Hospital; Harbor-UCLA Medical Center; Vanderbilt University Medical Center, Nashville; Washington University, St. Louis; or Emory University, Atlanta. A total of 266 patients were assigned to receive oral clindamycin, 263 to receive oral trimethoprim–sulfamethoxazole (TMP-SMX), and 257 to receive matching placebo for 10 days after the lesions were incised and drained.
At follow-up 7-10 days following the conclusion of treatment, the rates of clinical cure were 83.1% with clindamycin and 81.7% with TMP-SMX, both significantly greater than the 68.9% cure rate with placebo (P less than .001 for both comparisons). Similarly, at 1-month follow-up, 78.6% of the clindamycin group and 73.0% of the TMP-SMX group “remained cured,” compared with 62.6% of the placebo group (N Engl J Med. 2017 June 28. doi: 10.1056/NEJMoa1607033).
Among those with cultures positive for Staphylococcus aureus, cure rates 7-10 days after treatment ended were 83.5% and 83.2% in the clindamycin and TMP-SMX groups, respectively, significantly higher than in the placebo group (63.8%). Among those positive for methicillin-resistant S. aureus, cure rates were 81.7% and 84.6% in the clindamycin and TMP-SMX groups, respectively, significantly higher than in the placebo group (62.9%).
The rate of treatment-associated adverse events was higher with clindamycin (21.9%) than with TMP-SMX (11.1%) or with placebo (12.5%). The most common adverse events were diarrhea and nausea, which were mild to moderate in severity and resolved with sequelae. There were no cases of Clostridium difficile–associated diarrhea or severe allergic reactions. One patient had a hypersensitivity reaction that was considered to be related to TMP-SMX, which involved fever, rash, thrombocytopenia, and hepatitis and which resolved without sequelae.
The National Institute of Allergy and Infectious Diseases and the National Center for Research Resources supported the study. Dr. Daum reported ties to Pfizer, Dynavax, Theravance, and Merck, and his associates reported ties to numerous industry sources.
For patients who have a single, small skin abscess, the addition of oral antibiotics to standard incision and drainage of the lesion improves cure rates and decreases recurrence rates, according to a study published online June 28 in the New England Journal of Medicine.
The results of this multicenter prospective randomized double-blind placebo-controlled trial, taken together with those of another recent large study, “call into question the perception – largely based on expert opinion or smaller, underpowered, and lower-quality noninferiority trials – that cure rates do not improve with the addition of systemic antibiotic treatment after incision and drainage,” said Robert S. Daum, MD, professor of pediatrics at the University of Chicago, and his associates.
The trial involved 786 patients of all ages (64% were adults and 36% were children; mean age was 25.5 years) who had a single, uncomplicated skin abscess of 5 cm or smaller and were treated at the University of Chicago; San Francisco General Hospital; Harbor-UCLA Medical Center; Vanderbilt University Medical Center, Nashville; Washington University, St. Louis; or Emory University, Atlanta. A total of 266 patients were assigned to receive oral clindamycin, 263 to receive oral trimethoprim–sulfamethoxazole (TMP-SMX), and 257 to receive matching placebo for 10 days after the lesions were incised and drained.
At follow-up 7-10 days following the conclusion of treatment, the rates of clinical cure were 83.1% with clindamycin and 81.7% with TMP-SMX, both significantly greater than the 68.9% cure rate with placebo (P less than .001 for both comparisons). Similarly, at 1-month follow-up, 78.6% of the clindamycin group and 73.0% of the TMP-SMX group “remained cured,” compared with 62.6% of the placebo group (N Engl J Med. 2017 June 28. doi: 10.1056/NEJMoa1607033).
Among those with cultures positive for Staphylococcus aureus, cure rates 7-10 days after treatment ended were 83.5% and 83.2% in the clindamycin and TMP-SMX groups, respectively, significantly higher than in the placebo group (63.8%). Among those positive for methicillin-resistant S. aureus, cure rates were 81.7% and 84.6% in the clindamycin and TMP-SMX groups, respectively, significantly higher than in the placebo group (62.9%).
The rate of treatment-associated adverse events was higher with clindamycin (21.9%) than with TMP-SMX (11.1%) or with placebo (12.5%). The most common adverse events were diarrhea and nausea, which were mild to moderate in severity and resolved with sequelae. There were no cases of Clostridium difficile–associated diarrhea or severe allergic reactions. One patient had a hypersensitivity reaction that was considered to be related to TMP-SMX, which involved fever, rash, thrombocytopenia, and hepatitis and which resolved without sequelae.
The National Institute of Allergy and Infectious Diseases and the National Center for Research Resources supported the study. Dr. Daum reported ties to Pfizer, Dynavax, Theravance, and Merck, and his associates reported ties to numerous industry sources.
For patients who have a single, small skin abscess, the addition of oral antibiotics to standard incision and drainage of the lesion improves cure rates and decreases recurrence rates, according to a study published online June 28 in the New England Journal of Medicine.
The results of this multicenter prospective randomized double-blind placebo-controlled trial, taken together with those of another recent large study, “call into question the perception – largely based on expert opinion or smaller, underpowered, and lower-quality noninferiority trials – that cure rates do not improve with the addition of systemic antibiotic treatment after incision and drainage,” said Robert S. Daum, MD, professor of pediatrics at the University of Chicago, and his associates.
The trial involved 786 patients of all ages (64% were adults and 36% were children; mean age was 25.5 years) who had a single, uncomplicated skin abscess of 5 cm or smaller and were treated at the University of Chicago; San Francisco General Hospital; Harbor-UCLA Medical Center; Vanderbilt University Medical Center, Nashville; Washington University, St. Louis; or Emory University, Atlanta. A total of 266 patients were assigned to receive oral clindamycin, 263 to receive oral trimethoprim–sulfamethoxazole (TMP-SMX), and 257 to receive matching placebo for 10 days after the lesions were incised and drained.
At follow-up 7-10 days following the conclusion of treatment, the rates of clinical cure were 83.1% with clindamycin and 81.7% with TMP-SMX, both significantly greater than the 68.9% cure rate with placebo (P less than .001 for both comparisons). Similarly, at 1-month follow-up, 78.6% of the clindamycin group and 73.0% of the TMP-SMX group “remained cured,” compared with 62.6% of the placebo group (N Engl J Med. 2017 June 28. doi: 10.1056/NEJMoa1607033).
Among those with cultures positive for Staphylococcus aureus, cure rates 7-10 days after treatment ended were 83.5% and 83.2% in the clindamycin and TMP-SMX groups, respectively, significantly higher than in the placebo group (63.8%). Among those positive for methicillin-resistant S. aureus, cure rates were 81.7% and 84.6% in the clindamycin and TMP-SMX groups, respectively, significantly higher than in the placebo group (62.9%).
The rate of treatment-associated adverse events was higher with clindamycin (21.9%) than with TMP-SMX (11.1%) or with placebo (12.5%). The most common adverse events were diarrhea and nausea, which were mild to moderate in severity and resolved with sequelae. There were no cases of Clostridium difficile–associated diarrhea or severe allergic reactions. One patient had a hypersensitivity reaction that was considered to be related to TMP-SMX, which involved fever, rash, thrombocytopenia, and hepatitis and which resolved without sequelae.
The National Institute of Allergy and Infectious Diseases and the National Center for Research Resources supported the study. Dr. Daum reported ties to Pfizer, Dynavax, Theravance, and Merck, and his associates reported ties to numerous industry sources.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: For patients who have a single, small skin abscess, the addition of oral antibiotics to standard incision and drainage of the lesion improves cure rates and decreases recurrence rates.
Major finding: At follow-up 7-10 days following the completion of treatment, clinical cure rates were 83.1% with clindamycin and 81.7% with TMP-SMX, both significantly greater than the 68.9% cure rate with placebo.
Data source: A multicenter prospective randomized double-blind placebo-controlled trial involving 786 adults and children who had single, small skin abscesses.
Disclosures: The National Institute of Allergy and Infectious Diseases and the National Center for Research Resources supported the study. Dr. Daum reported ties to Pfizer, Dynavax, Theravance, and Merck, and his associates reported ties to numerous industry sources.
Recurring Yellowish Papules and Plaques on the Back
The Diagnosis: Nevus Lipomatosus Cutaneous Superficialis
A punch biopsy was obtained from a skin lesion, which showed orthokeratosis, irregular acanthosis, papillomatosis, intense edema in the upper dermis, and mature fat lobules that dissected collagen fibers in the reticular dermis (Figure). Classical-type nevus lipomatosus cutaneous superficialis (NLCS) was diagnosed based on these clinical and histopathological findings. The patient was referred to the plastic surgery clinic for total excision of all lesions.
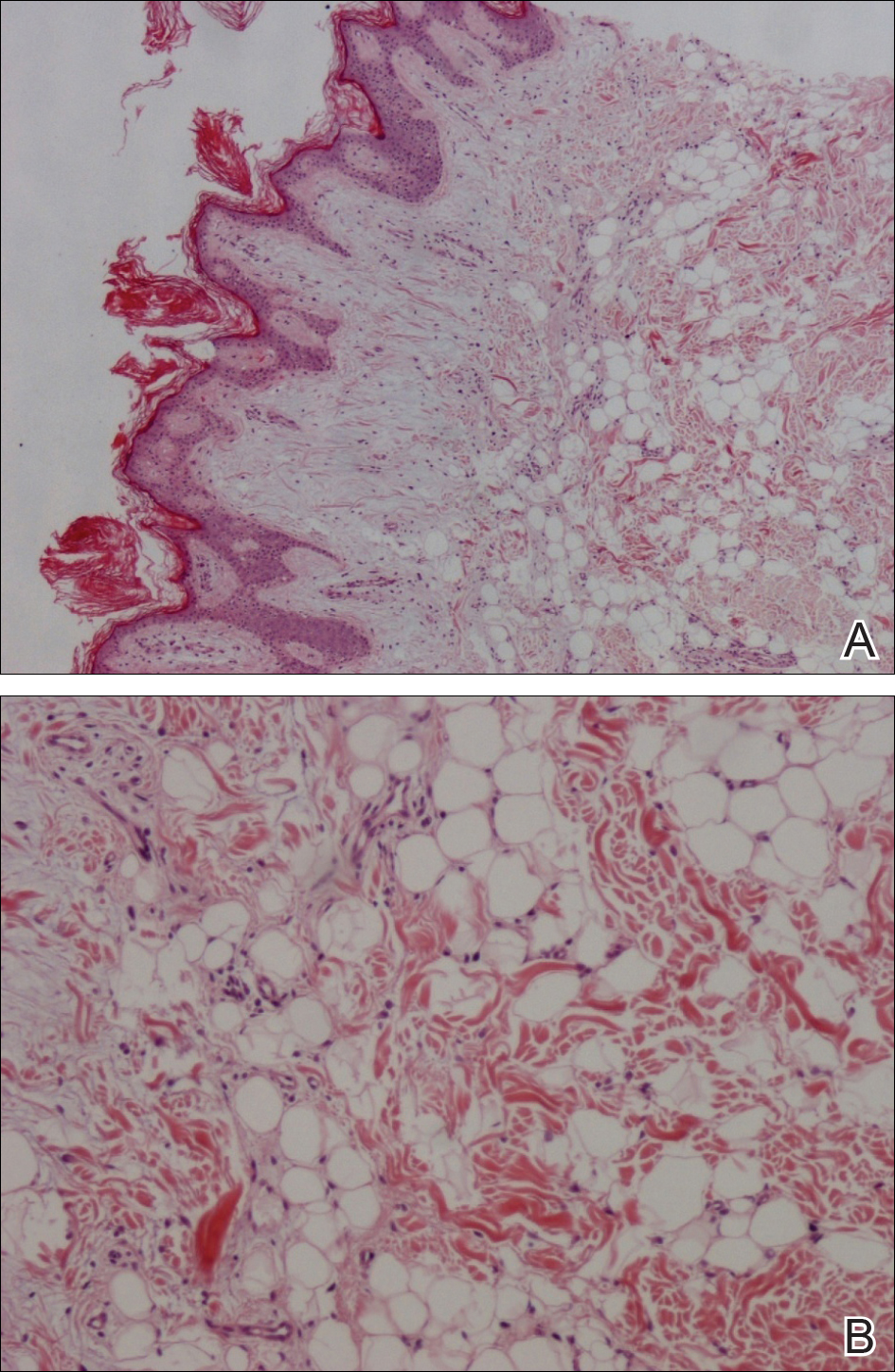
Nevus lipomatosus cutaneous superficialis is a rare hamartoma characterized by ectopic deposition of mature adipose tissue in the dermis.1 It was first described by Hoffmann and Zurhelle2 in 1921. Clinically, NLCS is classified into 2 subtypes: classical (multiple) and solitary. Classical-type NLCS is characterized by multiple pedunculated or sessile, soft, cerebriform, yellowish papules and nodules, especially in the pelvic area. Solitary-type NLCS presents as a sessile papule or nodule with no predilection for localization. Although the classical form of NLCS generally occurs in the first 2 decades of life, the solitary form usually appears in adulthood.3 Nevus lipomatosus cutaneous superficialis has no gender predilection and there is no genetic or congenital defect association.1,4
The pathogenesis of NLCS still is unknown, but some theories have been proposed, such as the development of adipose metaplasia secondary to degeneration of connective tissue, the formation of a true nevus resulting from heterotopic development of adipose tissue, and the development of mature adipocytes from pericytes in dermal vessels.1,5
Histopathology of NLCS shows clusters of ectopic mature adipose tissue in varying rates (10%-50%) between collagen bundles in the dermis. Characteristically, there is no connection between the ectopic mature adipose tissue and the subcutaneous adipose tissue.3 The differential diagnosis of NLCS includes neurofibroma, lymphangioma, sebaceous nevus, fibroepithelial polyps, leiomyoma, and lipomas.1,6
Treatment of NLCS generally involves basic surgical excision; however, patients treated with CO2 laser also have been reported in the literature.5 Because of the growth tendency and the large size of the classical form of NLCS, recurrence may occur, as in our case. In such cases, gradual surgical excision is recommended.5 We present this case to indicate that undesirable surgical results or relapse may occur in untreated patients because of lesion growth and delayed diagnosis.
- Goucha S, Khaled A, Zéglaoui F, et al. Nevus lipomatosus cutaneous superficialis: report of eight cases. Dermatol Ther (Heidelb). 2011;1:25-30.
- Hoffmann E, Zurhelle E. Ubereinen nevus lipomatodes cutaneous superficialis der linkenglutaalgegend. Arch Dermatol Syph. 1921;130:327-333.
- Patil SB, Narchal S, Paricharak M, et al. Nevus lipomatosus cutaneous superficialis: a rare case report. Iran J Med Sci. 2014;39:304-307.
- Bancalari E, Martínez-Sánchez D, Tardío JC. Nevus lipomatosus superficialis with a folliculosebaceous component: report of 2 cases. Patholog Res Int. 2011;2011:105973.
- Kim YJ, Choi JH, Kim H, et al. Recurrence of nevus lipomatosus cutaneous superficialis after CO(2) laser treatment [published online November 14, 2012]. Arch Plast Surg. 2012;39:671-673.
- Wollina U. Photoletter to the editor - nevus lipomatosus superficialis (Hoffmann-Zurhelle). three new cases including one with ulceration and one with ipsilateral gluteal hypertrophy. J Dermatol Case Rep. 2013;7:71-73.
The Diagnosis: Nevus Lipomatosus Cutaneous Superficialis
A punch biopsy was obtained from a skin lesion, which showed orthokeratosis, irregular acanthosis, papillomatosis, intense edema in the upper dermis, and mature fat lobules that dissected collagen fibers in the reticular dermis (Figure). Classical-type nevus lipomatosus cutaneous superficialis (NLCS) was diagnosed based on these clinical and histopathological findings. The patient was referred to the plastic surgery clinic for total excision of all lesions.
Nevus lipomatosus cutaneous superficialis is a rare hamartoma characterized by ectopic deposition of mature adipose tissue in the dermis.1 It was first described by Hoffmann and Zurhelle2 in 1921. Clinically, NLCS is classified into 2 subtypes: classical (multiple) and solitary. Classical-type NLCS is characterized by multiple pedunculated or sessile, soft, cerebriform, yellowish papules and nodules, especially in the pelvic area. Solitary-type NLCS presents as a sessile papule or nodule with no predilection for localization. Although the classical form of NLCS generally occurs in the first 2 decades of life, the solitary form usually appears in adulthood.3 Nevus lipomatosus cutaneous superficialis has no gender predilection and there is no genetic or congenital defect association.1,4
The pathogenesis of NLCS still is unknown, but some theories have been proposed, such as the development of adipose metaplasia secondary to degeneration of connective tissue, the formation of a true nevus resulting from heterotopic development of adipose tissue, and the development of mature adipocytes from pericytes in dermal vessels.1,5
Histopathology of NLCS shows clusters of ectopic mature adipose tissue in varying rates (10%-50%) between collagen bundles in the dermis. Characteristically, there is no connection between the ectopic mature adipose tissue and the subcutaneous adipose tissue.3 The differential diagnosis of NLCS includes neurofibroma, lymphangioma, sebaceous nevus, fibroepithelial polyps, leiomyoma, and lipomas.1,6
Treatment of NLCS generally involves basic surgical excision; however, patients treated with CO2 laser also have been reported in the literature.5 Because of the growth tendency and the large size of the classical form of NLCS, recurrence may occur, as in our case. In such cases, gradual surgical excision is recommended.5 We present this case to indicate that undesirable surgical results or relapse may occur in untreated patients because of lesion growth and delayed diagnosis.
The Diagnosis: Nevus Lipomatosus Cutaneous Superficialis
A punch biopsy was obtained from a skin lesion, which showed orthokeratosis, irregular acanthosis, papillomatosis, intense edema in the upper dermis, and mature fat lobules that dissected collagen fibers in the reticular dermis (Figure). Classical-type nevus lipomatosus cutaneous superficialis (NLCS) was diagnosed based on these clinical and histopathological findings. The patient was referred to the plastic surgery clinic for total excision of all lesions.
Nevus lipomatosus cutaneous superficialis is a rare hamartoma characterized by ectopic deposition of mature adipose tissue in the dermis.1 It was first described by Hoffmann and Zurhelle2 in 1921. Clinically, NLCS is classified into 2 subtypes: classical (multiple) and solitary. Classical-type NLCS is characterized by multiple pedunculated or sessile, soft, cerebriform, yellowish papules and nodules, especially in the pelvic area. Solitary-type NLCS presents as a sessile papule or nodule with no predilection for localization. Although the classical form of NLCS generally occurs in the first 2 decades of life, the solitary form usually appears in adulthood.3 Nevus lipomatosus cutaneous superficialis has no gender predilection and there is no genetic or congenital defect association.1,4
The pathogenesis of NLCS still is unknown, but some theories have been proposed, such as the development of adipose metaplasia secondary to degeneration of connective tissue, the formation of a true nevus resulting from heterotopic development of adipose tissue, and the development of mature adipocytes from pericytes in dermal vessels.1,5
Histopathology of NLCS shows clusters of ectopic mature adipose tissue in varying rates (10%-50%) between collagen bundles in the dermis. Characteristically, there is no connection between the ectopic mature adipose tissue and the subcutaneous adipose tissue.3 The differential diagnosis of NLCS includes neurofibroma, lymphangioma, sebaceous nevus, fibroepithelial polyps, leiomyoma, and lipomas.1,6
Treatment of NLCS generally involves basic surgical excision; however, patients treated with CO2 laser also have been reported in the literature.5 Because of the growth tendency and the large size of the classical form of NLCS, recurrence may occur, as in our case. In such cases, gradual surgical excision is recommended.5 We present this case to indicate that undesirable surgical results or relapse may occur in untreated patients because of lesion growth and delayed diagnosis.
- Goucha S, Khaled A, Zéglaoui F, et al. Nevus lipomatosus cutaneous superficialis: report of eight cases. Dermatol Ther (Heidelb). 2011;1:25-30.
- Hoffmann E, Zurhelle E. Ubereinen nevus lipomatodes cutaneous superficialis der linkenglutaalgegend. Arch Dermatol Syph. 1921;130:327-333.
- Patil SB, Narchal S, Paricharak M, et al. Nevus lipomatosus cutaneous superficialis: a rare case report. Iran J Med Sci. 2014;39:304-307.
- Bancalari E, Martínez-Sánchez D, Tardío JC. Nevus lipomatosus superficialis with a folliculosebaceous component: report of 2 cases. Patholog Res Int. 2011;2011:105973.
- Kim YJ, Choi JH, Kim H, et al. Recurrence of nevus lipomatosus cutaneous superficialis after CO(2) laser treatment [published online November 14, 2012]. Arch Plast Surg. 2012;39:671-673.
- Wollina U. Photoletter to the editor - nevus lipomatosus superficialis (Hoffmann-Zurhelle). three new cases including one with ulceration and one with ipsilateral gluteal hypertrophy. J Dermatol Case Rep. 2013;7:71-73.
- Goucha S, Khaled A, Zéglaoui F, et al. Nevus lipomatosus cutaneous superficialis: report of eight cases. Dermatol Ther (Heidelb). 2011;1:25-30.
- Hoffmann E, Zurhelle E. Ubereinen nevus lipomatodes cutaneous superficialis der linkenglutaalgegend. Arch Dermatol Syph. 1921;130:327-333.
- Patil SB, Narchal S, Paricharak M, et al. Nevus lipomatosus cutaneous superficialis: a rare case report. Iran J Med Sci. 2014;39:304-307.
- Bancalari E, Martínez-Sánchez D, Tardío JC. Nevus lipomatosus superficialis with a folliculosebaceous component: report of 2 cases. Patholog Res Int. 2011;2011:105973.
- Kim YJ, Choi JH, Kim H, et al. Recurrence of nevus lipomatosus cutaneous superficialis after CO(2) laser treatment [published online November 14, 2012]. Arch Plast Surg. 2012;39:671-673.
- Wollina U. Photoletter to the editor - nevus lipomatosus superficialis (Hoffmann-Zurhelle). three new cases including one with ulceration and one with ipsilateral gluteal hypertrophy. J Dermatol Case Rep. 2013;7:71-73.
A 36-year-old man presented with a group of partially erythematous, yellowish papules and plaques ranging from 5 to 20 mm in diameter on the right side of the upper back of 20 years' duration. They were surgically excised 8 years prior but recurred and spread. The lesions occasionally were painful and tender with redness and discharge.
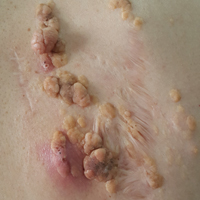
Desmoplastic trichoepithelioma may co-occur with BCC
SYDNEY, AUSTRALIA – Watchful waiting may not be the safest approach for managing patients with desmoplastic trichoepithelioma, according to a speaker at the annual meeting of the Australasian College of Dermatologists, who described five cases of the benign tumor combined with basal cell carcinoma.
Desmoplastic trichoepithelioma (DTE), a rare benign tumor that typically presents as a small, slow-growing, asymptomatic, skin-colored lesion on the face, with a depressed nonulcerated center and often raised edges, is managed with watchful waiting or local excision. While its key histopathologic features are narrow cords or strands of basaloid cells, numerous small keratin-filled cysts, and a surrounding desmoplastic core, DTE can be confused with morpheaform basal cell carcinoma (BCC), Tristan Blake, MD, dermatology registrar at Royal Brisbane and Womens’ Hospital, Brisbane, Australia, said at the meeting.

“At this stage, there’s no way to confidently say, looking at the slides, if those cases were desmoplastic trichoepithelioma arising in basal cell carcinoma or vice versa, or if they were a single tumor with divergent differentiation, or an occlusion of two separate tumors,” he said.
Dr. Blake added that this was the first time, to his knowledge, that such a combination had been reported, and that the finding had the potential to change the way DTE is managed.
“How can you now confidently elect to leave or watch the desmoplastic trichoepithelioma patients you have, knowing that not an insignificant portion might also harbor BCC or develop BCC in the future?” he said. This dilemma is made more acute by the fact that DTEs are typically found in younger patients and on the face, he added.
Two dermatopathologists involved in the retrospective review of cases reported that histochemistry was not particularly useful in differentiating DTE from other tumors, he noted.
Patients in the study were also interviewed about their tumors and reported no symptoms; when asked how long the lesions had been there prior to diagnosis, those who could recall said the lesions had likely been present for decades.
In an interview, Dr. Blake said that the discovery of coexisting DTE and BCC was a surprise, and cast doubt on the practice of watchful waiting.
No conflicts of interest were declared.
SYDNEY, AUSTRALIA – Watchful waiting may not be the safest approach for managing patients with desmoplastic trichoepithelioma, according to a speaker at the annual meeting of the Australasian College of Dermatologists, who described five cases of the benign tumor combined with basal cell carcinoma.
Desmoplastic trichoepithelioma (DTE), a rare benign tumor that typically presents as a small, slow-growing, asymptomatic, skin-colored lesion on the face, with a depressed nonulcerated center and often raised edges, is managed with watchful waiting or local excision. While its key histopathologic features are narrow cords or strands of basaloid cells, numerous small keratin-filled cysts, and a surrounding desmoplastic core, DTE can be confused with morpheaform basal cell carcinoma (BCC), Tristan Blake, MD, dermatology registrar at Royal Brisbane and Womens’ Hospital, Brisbane, Australia, said at the meeting.
“At this stage, there’s no way to confidently say, looking at the slides, if those cases were desmoplastic trichoepithelioma arising in basal cell carcinoma or vice versa, or if they were a single tumor with divergent differentiation, or an occlusion of two separate tumors,” he said.
Dr. Blake added that this was the first time, to his knowledge, that such a combination had been reported, and that the finding had the potential to change the way DTE is managed.
“How can you now confidently elect to leave or watch the desmoplastic trichoepithelioma patients you have, knowing that not an insignificant portion might also harbor BCC or develop BCC in the future?” he said. This dilemma is made more acute by the fact that DTEs are typically found in younger patients and on the face, he added.
Two dermatopathologists involved in the retrospective review of cases reported that histochemistry was not particularly useful in differentiating DTE from other tumors, he noted.
Patients in the study were also interviewed about their tumors and reported no symptoms; when asked how long the lesions had been there prior to diagnosis, those who could recall said the lesions had likely been present for decades.
In an interview, Dr. Blake said that the discovery of coexisting DTE and BCC was a surprise, and cast doubt on the practice of watchful waiting.
No conflicts of interest were declared.
SYDNEY, AUSTRALIA – Watchful waiting may not be the safest approach for managing patients with desmoplastic trichoepithelioma, according to a speaker at the annual meeting of the Australasian College of Dermatologists, who described five cases of the benign tumor combined with basal cell carcinoma.
Desmoplastic trichoepithelioma (DTE), a rare benign tumor that typically presents as a small, slow-growing, asymptomatic, skin-colored lesion on the face, with a depressed nonulcerated center and often raised edges, is managed with watchful waiting or local excision. While its key histopathologic features are narrow cords or strands of basaloid cells, numerous small keratin-filled cysts, and a surrounding desmoplastic core, DTE can be confused with morpheaform basal cell carcinoma (BCC), Tristan Blake, MD, dermatology registrar at Royal Brisbane and Womens’ Hospital, Brisbane, Australia, said at the meeting.
“At this stage, there’s no way to confidently say, looking at the slides, if those cases were desmoplastic trichoepithelioma arising in basal cell carcinoma or vice versa, or if they were a single tumor with divergent differentiation, or an occlusion of two separate tumors,” he said.
Dr. Blake added that this was the first time, to his knowledge, that such a combination had been reported, and that the finding had the potential to change the way DTE is managed.
“How can you now confidently elect to leave or watch the desmoplastic trichoepithelioma patients you have, knowing that not an insignificant portion might also harbor BCC or develop BCC in the future?” he said. This dilemma is made more acute by the fact that DTEs are typically found in younger patients and on the face, he added.
Two dermatopathologists involved in the retrospective review of cases reported that histochemistry was not particularly useful in differentiating DTE from other tumors, he noted.
Patients in the study were also interviewed about their tumors and reported no symptoms; when asked how long the lesions had been there prior to diagnosis, those who could recall said the lesions had likely been present for decades.
In an interview, Dr. Blake said that the discovery of coexisting DTE and BCC was a surprise, and cast doubt on the practice of watchful waiting.
No conflicts of interest were declared.
AT ACDASM 2017
Key clinical point: Watchful waiting may no longer be the obvious choice for desmoplastic trichoepithelioma, with evidence that the benign tumor may co-occur with basal cell carcinoma.
Major finding: Researchers reported five cases in which both DTE and BCC were identified in the same pathology specimen.
Data source: A retrospective review of 27 patients with DTE, which included reexamination of specimens.
Disclosures: No conflicts of interest were declared.
Perifollicular Papules on the Trunk
The Diagnosis: Disseminate and Recurrent Infundibulofolliculitis
A punch biopsy of a representative lesion on the trunk was performed. Histopathologic examination revealed a chronic lymphohistiocytic proliferation, focal spongiosis, and lymphocytic exocytosis primarily involving the isthmus of the hair follicle (Figure 1). At the follicular opening there was associated parakeratosis of the adjacent epidermis (Figure 2). Given these clinical and histopathological findings, a diagnosis of disseminate and recurrent infundibulofolliculitis (DRIF) was made.
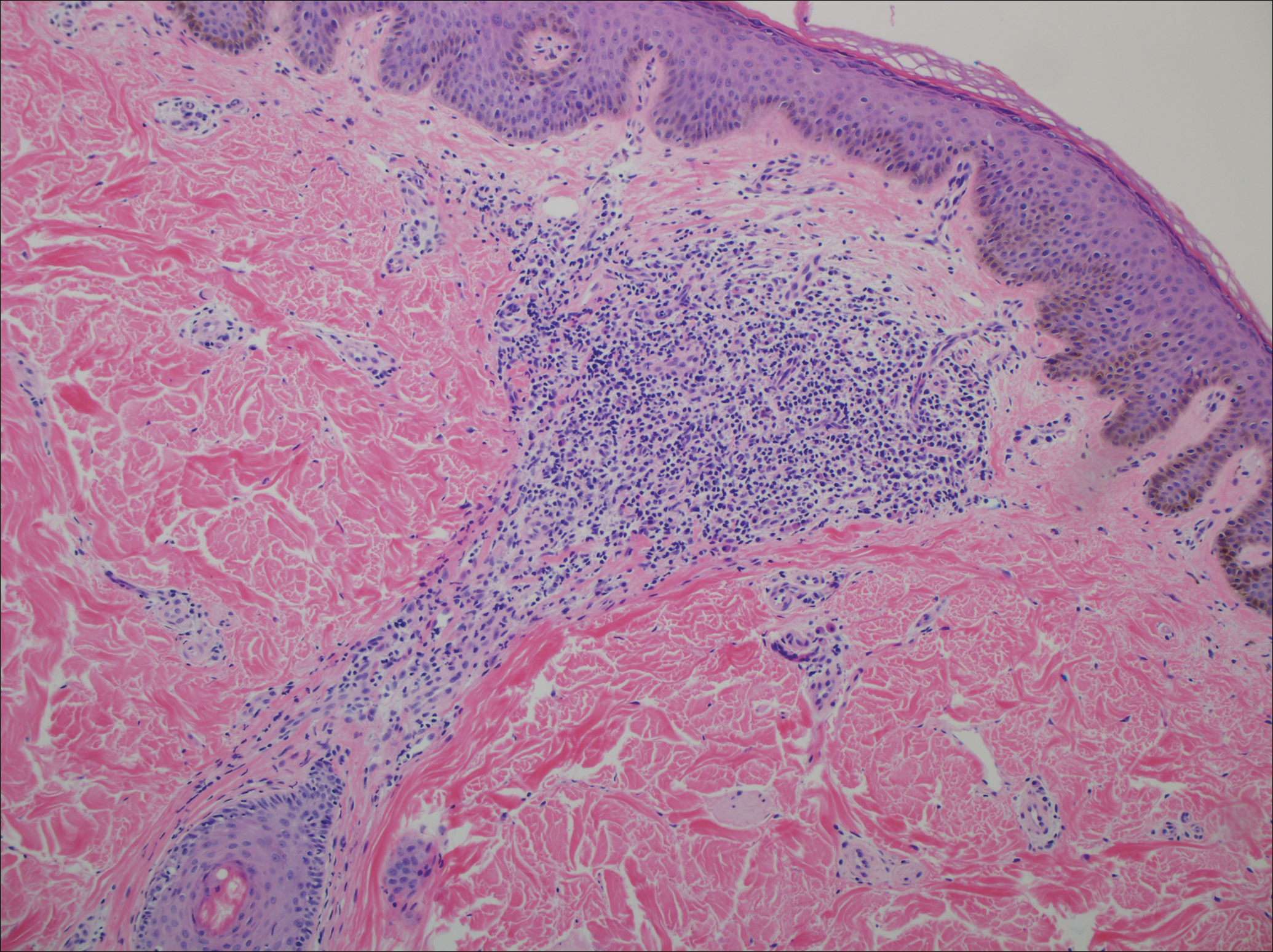
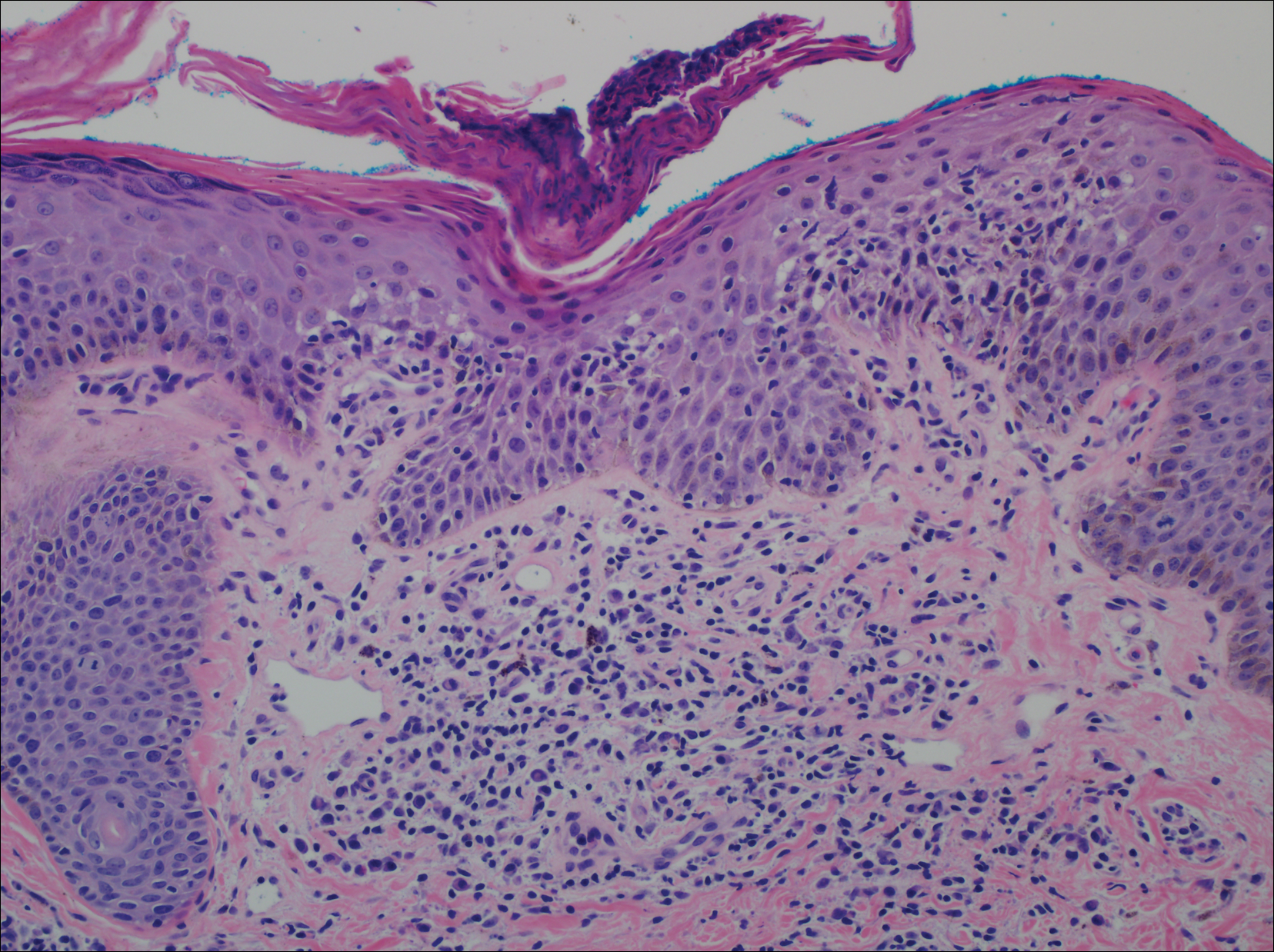
Disseminate and recurrent infundibulofolliculitis was first described by Hitch and Lund1 in 1968 in a healthy 27-year-old black man as a widespread recurrent follicular eruption. Disseminate and recurrent infundibulofolliculitis usually affects young adult males with darkly pigmented skin.2,3 It has less commonly been described in children, females, and white individuals.3,4 Associations with atopy, systemic diseases, or medications are unknown.3-6 The onset usually is sudden and the disease course may be characterized by intermittent recurrences. Pruritus usually is reported but may be mild.5
Histopathology is characterized by spongiosis centered on the infundibulum of the hair follicle and a primarily lymphocytic inflammatory infiltrate. Neutrophils also may be identified.3 Disseminate and recurrent infundibulofolliculitis can be differentiated histologically from clinically similar entities such as keratosis pilaris, which has a keratin plug filling the infundibulum; lichen nitidus, which is characterized by a clawlike downgrowth of the rete ridges surrounding a central foci of inflammation; or folliculitis, which is characterized by perifollicular suppurative inflammation.
Treatment of DRIF is anecdotal and limited to case reports. Vitamin A alone or in combination with vitamin E has been reported to lead to some improvement.5 Tetracycline-class antibiotics, keratolytics, antihistamines, and topical retinoids have not been successful, and mixed results have been seen with topical steroids.5-7 There is a reported case of improvement with a 3-week regimen of psoralen plus UVA followed by twice-weekly maintenance.8 Promising results in the treatment of DRIF have been shown with oral isotretinoin once daily.3-5 Finally, DRIF may resolve independently6; therefore, treatment of DRIF should be addressed on a case-by-case basis.
- Hitch JM, Lund HZ. Disseminate and recurrent infundibulo-folliculitis: report of a case. Arch Dermatol. 1968;97:432-435.
- Hitch JM, Lund HZ. Disseminate and recurrent infundibulo-folliculitis. Arch Dermatol. 1972;105:580-583.
- Calka O, Metin A, Ozen S. A case of disseminated and recurrent infundibulofolliculitis responsive to treatment with systemic isotretinoin. J Dermatol. 2002;29:431-434.
- Aroni K, Grapsa A, Agapitos E. Disseminate and recurrent infundibulofolliculitis: response to isotretinoin. J Drugs Dermatol. 2004;3:434-435.
- Aroni K, Aivaliotis M, Davaris P. Disseminated and recurrent infundibular folliculitis (D.R.I.F.): report of a case successfully treated with isotretinoin. J Dermatol. 1998;25:51-53.
- Owen WR, Wood C. Disseminate and recurrent infundibulofolliculitis. Arch Dermatol. 1979;115:174-175.
- Hinds GA, Heald PW. A case of disseminate and recurrent infundibulofolliculitis responsive to treatment with topical steroids. Dermatol Online J. 2008;14:11.
- Goihman-Yahr M. Disseminate and recurrent infundibulofolliculitis: response to psoralen plus UVA therapy. Int J Dermatol. 1999;38:75-78.
The Diagnosis: Disseminate and Recurrent Infundibulofolliculitis
A punch biopsy of a representative lesion on the trunk was performed. Histopathologic examination revealed a chronic lymphohistiocytic proliferation, focal spongiosis, and lymphocytic exocytosis primarily involving the isthmus of the hair follicle (Figure 1). At the follicular opening there was associated parakeratosis of the adjacent epidermis (Figure 2). Given these clinical and histopathological findings, a diagnosis of disseminate and recurrent infundibulofolliculitis (DRIF) was made.
Disseminate and recurrent infundibulofolliculitis was first described by Hitch and Lund1 in 1968 in a healthy 27-year-old black man as a widespread recurrent follicular eruption. Disseminate and recurrent infundibulofolliculitis usually affects young adult males with darkly pigmented skin.2,3 It has less commonly been described in children, females, and white individuals.3,4 Associations with atopy, systemic diseases, or medications are unknown.3-6 The onset usually is sudden and the disease course may be characterized by intermittent recurrences. Pruritus usually is reported but may be mild.5
Histopathology is characterized by spongiosis centered on the infundibulum of the hair follicle and a primarily lymphocytic inflammatory infiltrate. Neutrophils also may be identified.3 Disseminate and recurrent infundibulofolliculitis can be differentiated histologically from clinically similar entities such as keratosis pilaris, which has a keratin plug filling the infundibulum; lichen nitidus, which is characterized by a clawlike downgrowth of the rete ridges surrounding a central foci of inflammation; or folliculitis, which is characterized by perifollicular suppurative inflammation.
Treatment of DRIF is anecdotal and limited to case reports. Vitamin A alone or in combination with vitamin E has been reported to lead to some improvement.5 Tetracycline-class antibiotics, keratolytics, antihistamines, and topical retinoids have not been successful, and mixed results have been seen with topical steroids.5-7 There is a reported case of improvement with a 3-week regimen of psoralen plus UVA followed by twice-weekly maintenance.8 Promising results in the treatment of DRIF have been shown with oral isotretinoin once daily.3-5 Finally, DRIF may resolve independently6; therefore, treatment of DRIF should be addressed on a case-by-case basis.
The Diagnosis: Disseminate and Recurrent Infundibulofolliculitis
A punch biopsy of a representative lesion on the trunk was performed. Histopathologic examination revealed a chronic lymphohistiocytic proliferation, focal spongiosis, and lymphocytic exocytosis primarily involving the isthmus of the hair follicle (Figure 1). At the follicular opening there was associated parakeratosis of the adjacent epidermis (Figure 2). Given these clinical and histopathological findings, a diagnosis of disseminate and recurrent infundibulofolliculitis (DRIF) was made.
Disseminate and recurrent infundibulofolliculitis was first described by Hitch and Lund1 in 1968 in a healthy 27-year-old black man as a widespread recurrent follicular eruption. Disseminate and recurrent infundibulofolliculitis usually affects young adult males with darkly pigmented skin.2,3 It has less commonly been described in children, females, and white individuals.3,4 Associations with atopy, systemic diseases, or medications are unknown.3-6 The onset usually is sudden and the disease course may be characterized by intermittent recurrences. Pruritus usually is reported but may be mild.5
Histopathology is characterized by spongiosis centered on the infundibulum of the hair follicle and a primarily lymphocytic inflammatory infiltrate. Neutrophils also may be identified.3 Disseminate and recurrent infundibulofolliculitis can be differentiated histologically from clinically similar entities such as keratosis pilaris, which has a keratin plug filling the infundibulum; lichen nitidus, which is characterized by a clawlike downgrowth of the rete ridges surrounding a central foci of inflammation; or folliculitis, which is characterized by perifollicular suppurative inflammation.
Treatment of DRIF is anecdotal and limited to case reports. Vitamin A alone or in combination with vitamin E has been reported to lead to some improvement.5 Tetracycline-class antibiotics, keratolytics, antihistamines, and topical retinoids have not been successful, and mixed results have been seen with topical steroids.5-7 There is a reported case of improvement with a 3-week regimen of psoralen plus UVA followed by twice-weekly maintenance.8 Promising results in the treatment of DRIF have been shown with oral isotretinoin once daily.3-5 Finally, DRIF may resolve independently6; therefore, treatment of DRIF should be addressed on a case-by-case basis.
- Hitch JM, Lund HZ. Disseminate and recurrent infundibulo-folliculitis: report of a case. Arch Dermatol. 1968;97:432-435.
- Hitch JM, Lund HZ. Disseminate and recurrent infundibulo-folliculitis. Arch Dermatol. 1972;105:580-583.
- Calka O, Metin A, Ozen S. A case of disseminated and recurrent infundibulofolliculitis responsive to treatment with systemic isotretinoin. J Dermatol. 2002;29:431-434.
- Aroni K, Grapsa A, Agapitos E. Disseminate and recurrent infundibulofolliculitis: response to isotretinoin. J Drugs Dermatol. 2004;3:434-435.
- Aroni K, Aivaliotis M, Davaris P. Disseminated and recurrent infundibular folliculitis (D.R.I.F.): report of a case successfully treated with isotretinoin. J Dermatol. 1998;25:51-53.
- Owen WR, Wood C. Disseminate and recurrent infundibulofolliculitis. Arch Dermatol. 1979;115:174-175.
- Hinds GA, Heald PW. A case of disseminate and recurrent infundibulofolliculitis responsive to treatment with topical steroids. Dermatol Online J. 2008;14:11.
- Goihman-Yahr M. Disseminate and recurrent infundibulofolliculitis: response to psoralen plus UVA therapy. Int J Dermatol. 1999;38:75-78.
- Hitch JM, Lund HZ. Disseminate and recurrent infundibulo-folliculitis: report of a case. Arch Dermatol. 1968;97:432-435.
- Hitch JM, Lund HZ. Disseminate and recurrent infundibulo-folliculitis. Arch Dermatol. 1972;105:580-583.
- Calka O, Metin A, Ozen S. A case of disseminated and recurrent infundibulofolliculitis responsive to treatment with systemic isotretinoin. J Dermatol. 2002;29:431-434.
- Aroni K, Grapsa A, Agapitos E. Disseminate and recurrent infundibulofolliculitis: response to isotretinoin. J Drugs Dermatol. 2004;3:434-435.
- Aroni K, Aivaliotis M, Davaris P. Disseminated and recurrent infundibular folliculitis (D.R.I.F.): report of a case successfully treated with isotretinoin. J Dermatol. 1998;25:51-53.
- Owen WR, Wood C. Disseminate and recurrent infundibulofolliculitis. Arch Dermatol. 1979;115:174-175.
- Hinds GA, Heald PW. A case of disseminate and recurrent infundibulofolliculitis responsive to treatment with topical steroids. Dermatol Online J. 2008;14:11.
- Goihman-Yahr M. Disseminate and recurrent infundibulofolliculitis: response to psoralen plus UVA therapy. Int J Dermatol. 1999;38:75-78.
A 40-year-old black man presented with numerous perifollicular flesh-colored papules on the back, chest, abdomen, and proximal aspect of the arms of 6 years' duration. He described these lesions as persistent, nonpainful, and nonpruritic. He previously was treated with an unknown cream without any benefit. These lesions were cosmetically bothersome.
Unilateral Verrucous Porokeratosis of the Gluteal Cleft
To the Editor:
Verrucous porokeratosis of the gluteal cleft is a rare skin condition that has distinct clinical and histologic features. A review of 5 cases described a characteristic clinical presentation of a butterfly-shaped bilateral gluteal cleft lesion on most patients.1 We present an unusual case of verrucous porokeratosis presenting as a unilateral single lesion in the gluteal area that emulated seborrheic keratosis with histology consistent with verrucous porokeratosis. This case adds to the variable presentation of this unusual disease.
A 40-year-old man who presented to the dermatology clinic for a follow-up on a basal cell carcinoma of the temple region was concerned about a lesion on the left buttock of 1 year’s duration. Physical examination revealed a unilateral hyperkeratotic plaque that clinically resembled seborrheic keratosis (Figure 1). Biopsy revealed hyperkeratosis with numerous columns of parakeratosis, psoriasiform epidermal hyperplasia (Figures 2A and 2B), dyskeratotic keratinocytes (Figure 2C), pigment incontinence, and mild superficial chronic inflammation consistent with verrucous porokeratosis. The patient was treated with urea lotion but ultimately was lost to follow-up.
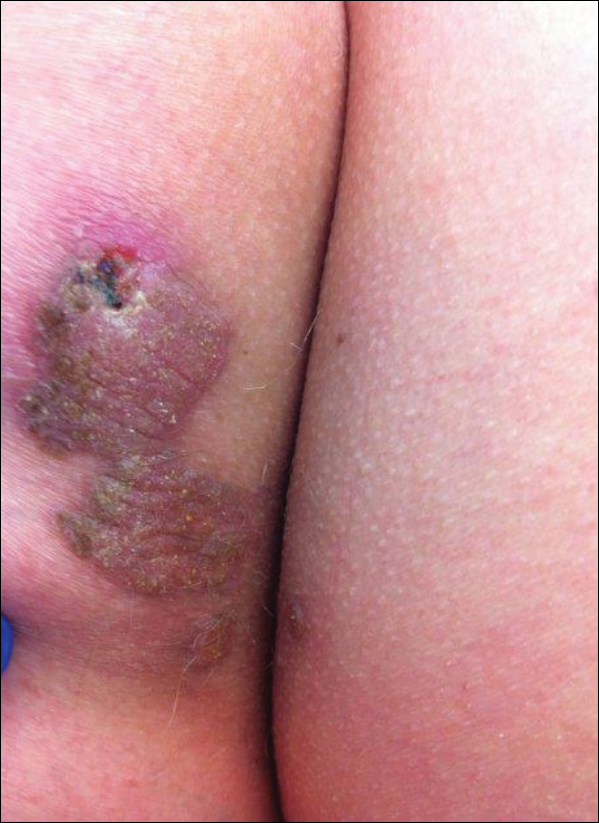
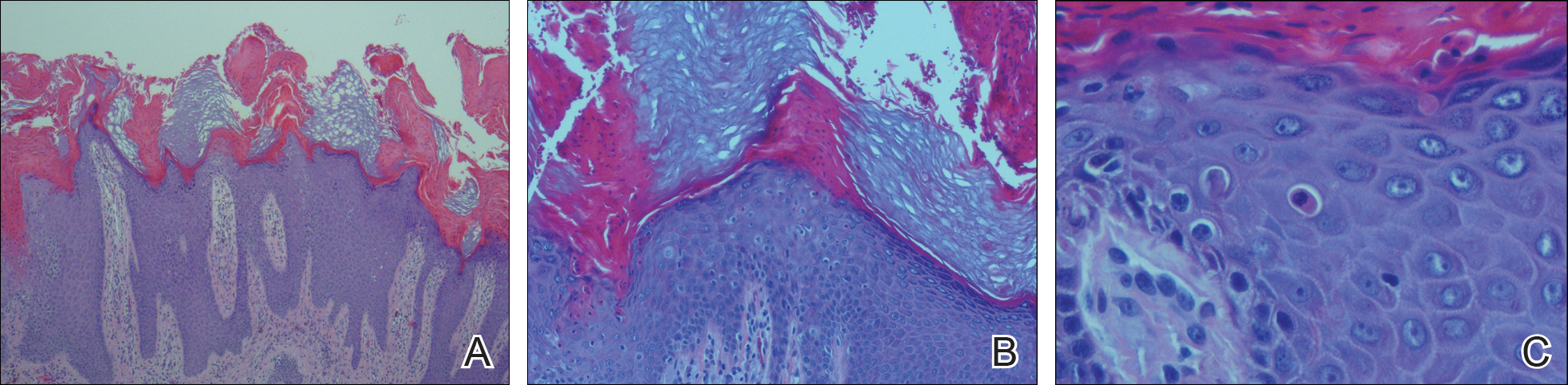
We present a unique case of unilateral verrucous porokeratosis of the gluteal cleft. The clinical differential diagnosis included seborrheic keratosis, condyloma acuminata, and inflammatory linear verrucous epidermal nevus. Histopathology was consistent with verrucous porokeratosis. Porokeratosis is a heterogeneous group of keratinization disorders containing several described variants such as classici porokeratosis of Mibelli, disseminated superficial porokeratosis, porokeratosis palmaris et plantaris disseminata, linear porokeratosis, and punctuate porokeratosis.1,2 Most patients present clinically with plaquelike bilateral (butterfly) lesions with threadlike (ridge) borders, though some patients initially have a unilateral lesion that subsequently develops into a bilateral lesion.1 The clinical course is slow growing, but it can potentially give rise to malignancies such as squamous cell carcinoma.3 Histologically, numerous columns of parakeratosis overlying epidermal cells with attenuated granular layer are observed with the concentric cornoid lamellae considered unique to the verrucous variant.1 Although our patient had only a single unilateral lesion on the gluteal cleft, the histology was consistent with verrucous porokeratosis. Our case adds to the growing clinical presentations of this unusual disease.
- Takiguchi R, White K, Clifton W, et al. Verrucous porokeratosis of the gluteal cleft (porokeratosis stychotropica): a rare disorder easily misdiagnosed. J Cutan Pathol. 2010;37:802-807.
- McGuigan K, Shurman D, Campanelli C, et al. Porokeratosis ptychotropica: a clinically distinct variant of porokeratosis. J Am Acad Dermatol. 2009;60:501-503.
- Malek J, Chedraoui A, Kibbi AG, et al. Genitogluteal porokeratosis: 10 years to make the diagnosis! Am J Dermatopathol. 2009;31:604-606.
To the Editor:
Verrucous porokeratosis of the gluteal cleft is a rare skin condition that has distinct clinical and histologic features. A review of 5 cases described a characteristic clinical presentation of a butterfly-shaped bilateral gluteal cleft lesion on most patients.1 We present an unusual case of verrucous porokeratosis presenting as a unilateral single lesion in the gluteal area that emulated seborrheic keratosis with histology consistent with verrucous porokeratosis. This case adds to the variable presentation of this unusual disease.
A 40-year-old man who presented to the dermatology clinic for a follow-up on a basal cell carcinoma of the temple region was concerned about a lesion on the left buttock of 1 year’s duration. Physical examination revealed a unilateral hyperkeratotic plaque that clinically resembled seborrheic keratosis (Figure 1). Biopsy revealed hyperkeratosis with numerous columns of parakeratosis, psoriasiform epidermal hyperplasia (Figures 2A and 2B), dyskeratotic keratinocytes (Figure 2C), pigment incontinence, and mild superficial chronic inflammation consistent with verrucous porokeratosis. The patient was treated with urea lotion but ultimately was lost to follow-up.
We present a unique case of unilateral verrucous porokeratosis of the gluteal cleft. The clinical differential diagnosis included seborrheic keratosis, condyloma acuminata, and inflammatory linear verrucous epidermal nevus. Histopathology was consistent with verrucous porokeratosis. Porokeratosis is a heterogeneous group of keratinization disorders containing several described variants such as classici porokeratosis of Mibelli, disseminated superficial porokeratosis, porokeratosis palmaris et plantaris disseminata, linear porokeratosis, and punctuate porokeratosis.1,2 Most patients present clinically with plaquelike bilateral (butterfly) lesions with threadlike (ridge) borders, though some patients initially have a unilateral lesion that subsequently develops into a bilateral lesion.1 The clinical course is slow growing, but it can potentially give rise to malignancies such as squamous cell carcinoma.3 Histologically, numerous columns of parakeratosis overlying epidermal cells with attenuated granular layer are observed with the concentric cornoid lamellae considered unique to the verrucous variant.1 Although our patient had only a single unilateral lesion on the gluteal cleft, the histology was consistent with verrucous porokeratosis. Our case adds to the growing clinical presentations of this unusual disease.
To the Editor:
Verrucous porokeratosis of the gluteal cleft is a rare skin condition that has distinct clinical and histologic features. A review of 5 cases described a characteristic clinical presentation of a butterfly-shaped bilateral gluteal cleft lesion on most patients.1 We present an unusual case of verrucous porokeratosis presenting as a unilateral single lesion in the gluteal area that emulated seborrheic keratosis with histology consistent with verrucous porokeratosis. This case adds to the variable presentation of this unusual disease.
A 40-year-old man who presented to the dermatology clinic for a follow-up on a basal cell carcinoma of the temple region was concerned about a lesion on the left buttock of 1 year’s duration. Physical examination revealed a unilateral hyperkeratotic plaque that clinically resembled seborrheic keratosis (Figure 1). Biopsy revealed hyperkeratosis with numerous columns of parakeratosis, psoriasiform epidermal hyperplasia (Figures 2A and 2B), dyskeratotic keratinocytes (Figure 2C), pigment incontinence, and mild superficial chronic inflammation consistent with verrucous porokeratosis. The patient was treated with urea lotion but ultimately was lost to follow-up.
We present a unique case of unilateral verrucous porokeratosis of the gluteal cleft. The clinical differential diagnosis included seborrheic keratosis, condyloma acuminata, and inflammatory linear verrucous epidermal nevus. Histopathology was consistent with verrucous porokeratosis. Porokeratosis is a heterogeneous group of keratinization disorders containing several described variants such as classici porokeratosis of Mibelli, disseminated superficial porokeratosis, porokeratosis palmaris et plantaris disseminata, linear porokeratosis, and punctuate porokeratosis.1,2 Most patients present clinically with plaquelike bilateral (butterfly) lesions with threadlike (ridge) borders, though some patients initially have a unilateral lesion that subsequently develops into a bilateral lesion.1 The clinical course is slow growing, but it can potentially give rise to malignancies such as squamous cell carcinoma.3 Histologically, numerous columns of parakeratosis overlying epidermal cells with attenuated granular layer are observed with the concentric cornoid lamellae considered unique to the verrucous variant.1 Although our patient had only a single unilateral lesion on the gluteal cleft, the histology was consistent with verrucous porokeratosis. Our case adds to the growing clinical presentations of this unusual disease.
- Takiguchi R, White K, Clifton W, et al. Verrucous porokeratosis of the gluteal cleft (porokeratosis stychotropica): a rare disorder easily misdiagnosed. J Cutan Pathol. 2010;37:802-807.
- McGuigan K, Shurman D, Campanelli C, et al. Porokeratosis ptychotropica: a clinically distinct variant of porokeratosis. J Am Acad Dermatol. 2009;60:501-503.
- Malek J, Chedraoui A, Kibbi AG, et al. Genitogluteal porokeratosis: 10 years to make the diagnosis! Am J Dermatopathol. 2009;31:604-606.
- Takiguchi R, White K, Clifton W, et al. Verrucous porokeratosis of the gluteal cleft (porokeratosis stychotropica): a rare disorder easily misdiagnosed. J Cutan Pathol. 2010;37:802-807.
- McGuigan K, Shurman D, Campanelli C, et al. Porokeratosis ptychotropica: a clinically distinct variant of porokeratosis. J Am Acad Dermatol. 2009;60:501-503.
- Malek J, Chedraoui A, Kibbi AG, et al. Genitogluteal porokeratosis: 10 years to make the diagnosis! Am J Dermatopathol. 2009;31:604-606.
Practice Point
- Porokeratosis of the gluteal cleft typically is bilateral but may be unilateral.