User login
BEST PRACTICES IN: Helping Patients Eat More Seafood
A supplement to Family Practice News. This supplement was sponsored by National Fisheries Institute.

Click here to download PDF
Topics
- Introduction
- Survey Reveals Knowledge Gap That Inhibits Patient Dialogue
- Barriers to Eating More Seafood
- Conclusions
Faculty/Faculty Disclosure
Jeffrey D. Fisher, MD
Cardiology and Internal Medicine
Weill Cornell Medical College
New York, New York
William Goodnight, MD
Maternal and Fetal Medicine University of North Carolina
Chapel Hill Chapel Hill, North Carolina
Laura Jana, MD
Pediatrician, Health Communicator
Omaha, Nebraska
John La Puma, MD
C.H.E.F. Clinic
Santa Barbara, California
Drs Goodnight, Jana and La Puma have nothing to disclose. Dr Fisher is a consultant to, and has received funding for, clinical grants from National Fisheries Institute.
Copyright © 2012 Elsevier Inc.
A supplement to Family Practice News. This supplement was sponsored by National Fisheries Institute.
Click here to download PDF
Topics
- Introduction
- Survey Reveals Knowledge Gap That Inhibits Patient Dialogue
- Barriers to Eating More Seafood
- Conclusions
Faculty/Faculty Disclosure
Jeffrey D. Fisher, MD
Cardiology and Internal Medicine
Weill Cornell Medical College
New York, New York
William Goodnight, MD
Maternal and Fetal Medicine University of North Carolina
Chapel Hill Chapel Hill, North Carolina
Laura Jana, MD
Pediatrician, Health Communicator
Omaha, Nebraska
John La Puma, MD
C.H.E.F. Clinic
Santa Barbara, California
Drs Goodnight, Jana and La Puma have nothing to disclose. Dr Fisher is a consultant to, and has received funding for, clinical grants from National Fisheries Institute.
Copyright © 2012 Elsevier Inc.
A supplement to Family Practice News. This supplement was sponsored by National Fisheries Institute.
Click here to download PDF
Topics
- Introduction
- Survey Reveals Knowledge Gap That Inhibits Patient Dialogue
- Barriers to Eating More Seafood
- Conclusions
Faculty/Faculty Disclosure
Jeffrey D. Fisher, MD
Cardiology and Internal Medicine
Weill Cornell Medical College
New York, New York
William Goodnight, MD
Maternal and Fetal Medicine University of North Carolina
Chapel Hill Chapel Hill, North Carolina
Laura Jana, MD
Pediatrician, Health Communicator
Omaha, Nebraska
John La Puma, MD
C.H.E.F. Clinic
Santa Barbara, California
Drs Goodnight, Jana and La Puma have nothing to disclose. Dr Fisher is a consultant to, and has received funding for, clinical grants from National Fisheries Institute.
Copyright © 2012 Elsevier Inc.

Addressing Key Questions with Statin Therapy
Managing the Multiple Symptoms of Benign Prostatic Hyperplasia — CME
Managing Type 2 Diabetes in Men
Meeting New Challenges with Antiplatelet Therapy in Primary Care
Dr. Toth has disclosed that he is on the speakers’ bureaus and is a consultant for Abbott, AstraZeneca, Kowa, Lilly, and Merck. He is on the speakers’ bureaus for Boehringer-Ingelheim and GlaxoSmithKline and is a consultant for Genentech and Genzyme.
Statins have become an important therapeutic option for managing cardiovascular (CV) risk, yet many questions remain regarding their use. This article addresses some of these questions in the primary care management of patients and highlights the impact of long-term statin therapy on CV end points. Because pitavastatin has recently become available in the United States, more detailed information about this agent is also presented.
LEARNING OBJECTIVES
After reviewing this activity on statin therapy, the reader will be able to:
- Describe the long-term benefits of statin therapy.
- Compare the efficacy and safety of pitavastatin with other statins.
- Select and modify statin therapy based upon individual patient factors.
TARGET AUDIENCE
Family physicians and clinicians who wish to gain increased knowledge and greater competency regarding statin therapy in the primary care management of patients with dyslipidemia.
ACKNOWLEDGEMENT
Dr. Toth was paid an honorarium by and received editorial assistance from the Primary Care Education Consortium in the development of this activity.
DISCLOSURES
As a continuing medical education provider accredited by the Accreditation Council for Continuing Medical Education (ACCME), it is the policy of the Primary Care Education Consortium (PCEC) to require any individual in a position to influence educational content to disclose the existence of any financial interest or other personal relationship with the manufacturer(s) of any commercial product(s).
Dr. Toth has disclosed that he is on the speakers’ bureaus and is a consultant for Abbott, AstraZeneca, Kowa, Lilly, and Merck. He is on the speakers’ bureaus for Boehringer-Ingelheim and GlaxoSmithKline and is a consultant for Genentech and Genzyme.
The medical accuracy and continuing medical education (CME) reviewer for this activity, Dr. Ron Pollack, has no real or apparent conflicts of interest to report.
PRIMARY CARE EDUCATION CONSORTIUM STAFF
Dr. Brunton has disclosed that he is on the advisory boards and speakers’ bureaus for Boehringer Ingelheim, Eli Lilly, Kowa, Novo Nordisk, Inc, and Teva Pharmaceuticals, and is on the advisory boards for Abbott and Sunovion.
Other PCEC staff has provided financial disclosure and have no conflicts of interest to resolve related to this activity.
CONFLICTS OF INTEREST
When individuals in a position to control content have reported financial relationships with one or more commercial interests, the Primary Care Education Consortium works with them to resolve such conflicts to ensure that the content presented is free of commercial bias. The content of this activity was vetted by the following mechanisms and modified as required to meet this standard:
- Content peer-review by an external topic expert
- Content peer-review by an external CME reviewer
- Content validation by internal Primary Care Education Consortium clinical editorial staff
OFF-LABEL DISCLOSURE
In accordance with ACCME guidelines, the faculty author has been asked to disclose discussion on unlabeled or unapproved uses of drugs or devices during the course of the activity.
SPONSORSHIP
This activity is sponsored by the Primary Care Education Consortium.
ACCREDITATION
This journal-based CME activity, Addressing Key Questions with Statin Therapy, has been reviewed and is acceptable for up to 1.0 prescribed credit by the American Academy of Family Physicians. AAFP accreditation begins June 1, 2012. Term of approval is for one year from this date with option for yearly renewal.
Physicians should claim only the credit commensurate with the extent of their participation in the activity.
MEDIUM
Text publication in the form of a journal article.
METHOD OF PHYSICIAN PARTICIPATION
To receive CME credit, please read the journal article, and upon completion go to: www.pceconsortium.org/menshealthSTATIN to complete the online evaluation to receive your certification of completion.
SUPPORT
This activity was supported by an educational grant from Kowa Pharmaceuticals America, Inc. and Lilly USA, LLC.
Recent Clinical Evidence
Findings from clinical trials continue to add to our understanding of the safety and efficacy of statin therapy; for example, extended follow-up studies from 2 landmark trials show lasting benefit and no evidence of emerging hazards. An analysis of the Heart Protection Study demonstrated that participants randomized to simvastatin 40 mg during the initial 5-year trial had maintained the vascular event reduction of 23% (95% confidence interval [CI], 19-28; P < .0001) at the 6-year follow-up.1 Similarly, 8 years after the close of the 3-year lipid-lowering arm of the Anglo-Scandinavian Cardiac Outcomes Trial (ASCOT), primary prevention patients originally randomized to atorvastatin had maintained a 14% reduction in all-cause mortality (95% CI, 0.76-0.98; P = .02) and a 15% lower rate of non-CV death (95% CI, 0.73-0.99; P = .03) compared with placebo.2 Cancer incidence among those receiving a statin versus those receiving a placebo was similar in both trials. Collectively, these data provide reassurance for the long-term continuation of statin therapy.
Results from a meta-analysis involving 34,272 participants without coronary heart disease from 14 randomized controlled trials (16 trial arms) comparing statins to placebo demonstrated significant reductions in all major events with statins, including a reduction of 16% in all-cause mortality (95% CI, 0.73-0.96), 30% in combined fatal and nonfatal CV disease end points (95% CI, 0.61-0.79), and 34% in revascularization rates (95% CI, 0.53-0.83).3 The meta-analysis found no evidence of significant harm caused by a statin or negative effects on patient quality of life.
Pitavastatin
Pitavastatin was approved in the United States in 2009, although it has been available in Japan since 2003. Pitava-statin is a synthetic lipophilic statin with an 11-hour half-life. Following oral ingestion, it enters the enterohepatic circulation without the formation of active metabolites. Pitavastatin is principally metabolized by the cytochrome-P450 (CYP) 2C9 isoenzyme and avoids the major CYP3A4 pathway; thus CYP-mediated drug interactions are greatly reduced.4
Several 12-week dose comparative studies with pitavastatin have been conducted. The first study randomized subjects (N = 857) to 1 of 4 groups: pitavastatin 2 or 4 mg/d or simvastatin 20 or 40 mg/d.5 Pitavastatin 2 mg demonstrated significantly greater reductions in low- density lipoprotein cholesterol (LDL-C; 39% vs 35%; P = .014) and greater reductions in non–high-density lipoprotein cholesterol (non–HDL-C) than did simvastatin 20 mg/d. Pitavastatin 4 mg/d and simvastatin 40 mg/d each reduced LDL-C by about 44%. Pitavastatin 4 mg/d has also been compared to atorvastatin 20 mg/d in 418 subjects.6 After 12 weeks, pitavastatin 4 mg/d and atorvastatin 20 mg/d produced similar reductions in LDL-C (~42%). No differences between groups were noted for other parameters, including HDL-C and non–HDL-C.
Long-term extension studies have evaluated the safety and efficacy of pitavastatin. Patients randomized to pitava-statin, atorvastatin, or simvastatin for 12 weeks received open-label pitavastatin 4 mg/d for up to 52 weeks (N = 1353).7 Notable findings included maintenance of LDL-C reductions from the end of the 12-week trial to 52 weeks with all 3 treatments. HDL-C levels continued to increase during follow up, rising 14.3% from baseline. Another long-term study compared pitavastatin 4 mg/d and atorvastatin 20 or 40 mg/d (N = 212).6 Both statins produced similar reductions in LDL-C and improvements in other major lipoproteins; however, atorvastatin significantly increased fasting blood glucose from baseline (7.2%; P < .05), whereas pitavastatin showed a nonsignificant increase of 2.1%.
The Japanese LIVALO Effectiveness and Safety (LIVES) Study (N = 20,000) evaluated the effects of pitavastatin 1 to 4 mg daily in clinical practice.8 Among patients with abnormal baseline values, treatment with pitavastatin was associated with a 29% reduction in LDL-C and a 23% reduction in triglycerides after 2 years. There was a 5.9% overall increase in HDL-C and a 24.6% increase among those with baseline HDL-C values <40 mg/dL. Pitavastatin was also associated with an improvement in glycosylated hemoglobin (A1C) values among those with diabetes mellitus (DM). Concomitant antidiabetic therapy was continued during the study. These findings suggest that pitavastatin does not worsen glycemic parameters. A 5-year extension of the LIVES study (N = 6582) demonstrated that long-term treatment with pitavastatin maintained the LDL-C reductions observed in the 2-year trial.8 Furthermore, HDL-C levels continued to climb, with an overall 29% increase among those with baseline values < 40 mg/dL. Patients who achieved both LDL-C and HDL-C targets experienced the greatest reductions in CV and cerebrovascular risk.
Finally, the Japan Assessment of Pitavastatin and Ator-vastatin in Acute Coronary Syndrome (JAPAN-ACS) study was a prospective, open-label trial that investigated the effects of pitavastatin 4 mg/d and atorvastatin 20 mg/d on coronary plaque volume (PV) among patients with acute coronary syndrome (N = 252) undergoing intravascular ultrasound.9 After 8 to 12 months of treatment, the mean change in PV was – 16.9 ± 13.9% and – 18.1 ± 14.2% in the pitavastatin and atorvastatin groups, respectively. Each statin produced significant but equivalent regression of PV.
Other key findings from additional pitavastatin clinical trials are found in TABLE 1 .10-17
table 1
Key findings from pitavastatin clinical trials
| Statins | Population | Findings/Comments |
|---|---|---|
| Dose Comparative Studies | ||
| Pitavastatin 4 mg vs Simvastatin 40 mg15 | Dyslipidemic adults with ≥2 CV risk factors (N = 355) | Each statin: LDL-C ↓ by 44% at 12 weeks >80% reached LDL-C goal |
| Pitavastatin 2 mg, 4 mg17 | Dyslipidemic adults age ≥65 years (N = 545) | LDL-C ↓ by 43%, HDL-C ↑ by 9.6% at 60 weeks Only 17% required uptitration to 4 mg 89%-94% achieved LDL-C goals |
| Pitavastatin 4 mg vs Simvastatin 40-80 mg16 | Dyslipidemic adults with ≥ 2 CV risk factors (N=178) | Each statin: LDL-C ↓ by ~42% at 44 weeks Discontinuation (5.8% vs 10.5%), myalgia (4.1% vs 12.3%) for pitavastatin vs simvastatin, respectively |
| Other Clinical Trials | ||
| Pitavastatin 2 mg vs Atorvastatin 10 mg vs Rosuvastatin 2.5 mg10 | Dyslipidemic adults with CV risk factors (N=302) | All agents: LDL-C ↓ by 40%-45% at 16 weeks Atorvastatin and rosuvastatin: A1C ↑ |
| Pitavastatin 2 mg vs Rosuvastatin 2.5 mg11 | Dyslipidemic adults with type 2 DM (N = 90) | Both agents: Inflammation ↓, lipids improved, no adverse effects on glycemic control Rosuvastatin: Greater LDL-C ↓, hsCRP vs pitavastatin |
| Pitavastatin 2.3 mg vs Atorvastatin 11.3 mg vs Pravastatin 10.3 mg vs No statin13 | Previous PCI (N = 743) | Each statin: Major coronary events ↓ LDL-C and HDL-C: Predicted coronary events Pitavastatin and atorvastatin: Greater LDL-C ↓ vs pravastatin Only pitavastatin: Significant HDL-C ↑ vs no statin |
| Pitavastatin 2 mg vs Atorvastatin 10 mg12 | ACS patients who underwent emergency PCI and IVUS (N = 160) | Fibrofatty composition, PV: Significant ↓ with pitavastatin |
| Pitavastatin 2 mg14 | Adults with acute MI (N = 1039) | 71% achieved LDL-C goal at 12 months Pitavastatin: Favorable effects on biomarkers maintained at 12 months |
| A1C, glycosylated hemoglobin; ACS, acute coronary syndrome; CV, cardiovascular; DM, diabetes mellitus; HDL-C, high-density lipoprotein cholesterol; hsCRP, high-sensitivity C-reactive protein; IVUS, intravascular ultrasound; LDL-C, low-density lipoprotein cholesterol; MI, myocardial infarction; PCI, percutaneous coronary intervention; PV, plaque volume. | ||
Key Questions
The following are common questions asked by family physicians when considering statin therapy to treat patients with dyslipidemia.
What are the key lipoprotein differences among available statins?
Nearly all statins are able to provide the minimal 30% to 40% LDL-C reduction as suggested by the National Cholesterol Education Program Adult Treatment Panel III for high-risk patients ( TABLE 2 ).18-22 If greater reductions are required, higher doses of more potent agents, such as atorvastatin and rosuvastatin, may be needed.
Statins also provide moderate increases in HDL-C, with subtle differences observed among the agents. Atorvastatin and fluvastatin usually provide the smallest increases in HDL-C (up to ~6%), whereas simvastatin, pitavastatin, and rosuvastatin produce more robust increases (~5% to 10%).20,21,23 The effect of statins on non–HDL-C is similar to their effect on LDL-C.22 Non–HDL-C is a secondary target of therapy in patients with triglyceride levels ≥200 mg/dL. Non–HDL-C includes all atherogenic particles (ie, LDL-C and triglyceride-rich lipoproteins) and is calculated as the difference between total cholesterol and HDL-C. The non–HDL-C goal is 30 mg/dL higher than the LDL-C goal. Clinical investigation continues to demonstrate that non–HDL-C is a valuable predictor of CV risk. An analysis of statin-treated patients indicated that compared with LDL-C and apolipoprotein B, non–HDL-C has a greater strength of association for risk of future CV events.24
table 2
Range of Low-Density Lipoprotein Cholesterol (LDL-C)–lowering among statins18-21
| LDL-C Range (↓) | Atorvastatin | Fluvastatin | Lovastatin | Pitavastatin | Pravastatin | Rosuvastatin | Simvastatin |
|---|---|---|---|---|---|---|---|
| 20%-25% | — | 20 mg | — | — | — | — | — |
| 25%-30% | — | 40 mg | — | — | 10 mg | — | — |
| 30%-35% | — | 80 mg | 20 mg | 1 mg | 20 mg | — | 10 mg |
| 35%-40% | 10 mg | — | 40 mg | 2 mg | 40 mg | — | 20 mg |
| 40%-45% | 20 mg | — | 80 mg | 4 mg | 80 mg | 5 mg | 40 mg |
| 45%-50% | 40 mg | — | — | — | — | 10 mg | — |
| 50%-60% | 80 mg | — | — | — | — | 20 mg | — |
| >60% | — | — | — | — | — | 40 mg | — |
Is diabetes really a consequence of statin therapy? If so, do differences exist among the statins?
The US Food and Drug Administration (FDA) recently added warnings to all statin labeling indicating that statins can raise blood glucose and A1C levels.25 These effects appear to be modest and dose dependent. This concern initially emerged in the Justification for the Use of statins in Prevention: an Intervention Trial Evaluating Rosuva-statin (JUPITER) study when statin users experienced a 25% higher incidence of new onset DM compared to those receiving placebo.26 The short-term effects of various atorvastatin doses on glycemic indices further support these findings.27 Compared to placebo, all atorvastatin doses significantly increased A1C and fasting plasma insulin levels after 8 weeks (all, P < .01). Additionally, a meta-analysis of 5 major statin trials involving 32,752 patients demonstrated that patients receiving intensive-dose statin therapy had a 12% higher risk of developing DM than patients receiving moderate-dose statin therapy.28
The association between statin therapy and DM is considered a class effect; differences among the statins are controversial. In an analysis of 13 major randomized controlled trials, pravastatin produced a nonsignificant 3% increase in new onset DM, whereas rosuvastatin was associated with an 18% increase.28 A 16-week, head-to-head comparison showed that pitavastatin had no effect on A1C, while modest increases were seen with low-dose atorvastatin and rosuvastatin.10 In another study, atorvastatin but not pitavastatin produced significant (P < .03) increases in glycoalbumin and A1C (P < .01), whereas fasting glucose and insulin levels tended to decrease with pitavastatin.29 However, findings from the meta-analysis showed that the individual studies lacked sufficient specific data to detect heterogeneity between statins.30
Overall, statins are associated with modest increases in glycemic indices and new onset DM. This association appears to be greater with high-dose therapy; however, additional trials are needed to fully understand possible differences among statins.
Which drug interactions are clinically important?
As statin pharmacokinetic data have accumulated, critical drug interactions have become more apparent. The major concern is increased statin exposure secondary to limited metabolism, resulting in more dose-dependent AEs, such as muscle injury. CYP3A4 isoenzyme involvement is common in clinically significant interactions. Lovastatin, simvastatin, and to a lesser extent, atorvastatin are all substrates for CYP3A4.31 The FDA recently updated labeling for simvastatin and lovastatin to provide information on contraindications and dose limitations with concomitant agents [www.fda.gov/Drugs/DrugSafety/ucm293877.htm].18,25
Statins have differing effects on warfarin metabolism, with most agents increasing the international normalized ratio (INR). Conversely, atorvastatin and pitavastatin have shown no significant effect on prothrombin time when added to chronic warfarin therapy.23,32 Despite this, appropriate INR monitoring is suggested when any statin is added to warfarin treatment.
Another recent FDA advisory focusing on human immunodeficiency virus and hepatitis C virus protease inhibitors further emphasizes the importance of statin interactions.33 The advisory provides specific dose limitations and contraindications for 7 statins. Similar to other potent CYP3A4 inhibitors, protease inhibitors can increase lovastatin and simvastatin levels by 13- to 20-fold. No information is available for fluvastatin, while no dose limitations are needed for pitavastatin or pravastatin.33
Mechanisms implicating statins in other drug interactions include inhibition of CYP2C9, glucuronidation, and organic anion transporting polypeptide (OATP).31 Concomitant treatment with gemfibrozil and a statin produces a significant interaction, as this combination inhibits CYP2C9 and glucuronidation, resulting in marked increases in statin exposure. Similarly, the coadministration of a statin with cyclosporine is clinically relevant. Cyclosporine blocks another key step in statin metabolism, OATP, resulting in elevated concentrations of nearly all statins. The concomitant use of cyclosporine with lovastatin, simvastatin, or pitavastatin is contraindicated, whereas most other agents require dose limitations.18,23,25,31
Do statins possess a dose-dependent threshold for adverse events?
A general dose-dependent threshold for AEs has been observed with statin therapy. This upper limit is more apparent with certain statins and primarily manifests as myotoxicity or increased hepatic transaminase levels. High-dose simvastatin has shown the most evidence regarding increased myopathy. In the Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH) trial, 53 patients (0.9%) in the simvastatin 80-mg group experienced myopathy, including 7 cases (0.1%) of rhabdomyolysis, over a mean of 6.7 years of follow-up.34 By comparison, there were 2 reports of myopathy (0.03%) in the 20-mg group. Similarly, in Phase Z of the A to Z trial, 9 reports (0.4%) of myopathy, including 3 cases of rhabdomyolysis (0.13%), were reported with simvastatin 80 mg over a median of 2 years of follow-up, compared to none with lower doses.35 Lower rates of myopathy and rhabdomyolysis (0.0%-0.3% and 0.0%-0.1%, respectively) were found with atorvastatin 80 mg, fluvastatin 80 mg, and rosuvastatin 40 mg in major trials.36 These data prompted the FDA to publish an advisory on simvastatin dose limitations, including restricting the 80-mg dose.18 A threshold also has been observed with other statins, as an approximate 3-fold higher incidence of creatine kinase (CK) and hepatic transaminase elevations occur when titrating from moderate to maximal doses.37
Should ethnicity be a factor in selecting a statin?
While no specific recommendations presently exist regarding the selection of statin therapy based on ethnicity, rosuvastatin doses, including the 5-mg starting dose, should be reduced in patients of Asian ancestry because of a 2-fold increase in pharmacokinetic parameters compared to whites.38 Otherwise, the few studies evaluating individual agents among various ethnic groups generally suggest similar effects on pharmacokinetic parameters, lipid changes, and CV outcomes.
One study compared pharmacokinetic parameters of pitavastatin between healthy Caucasian and Japanese men.39 Pitavastatin demonstrated pharmacokinetic bioequivalence between the 2 groups with no clinically relevant differences. A substudy of ASCOT assessed the lipid effects of atorvastatin among whites, blacks, and South Asians.40 No significant differences were observed in the reductions in total cholesterol, LDL-C, or triglycerides. Lastly, outcomes were evaluated among different ethnicities in the JUPITER study.41 Similar reductions in major CV events were noted for whites versus non-whites with Hispanics and blacks experiencing comparable risk reductions.
How should statin-associated myalgia be managed?
Approximately 11% of patients receiving moderate- to high-dose statin therapy experience muscle symptoms.42 This common AE can greatly affect therapy by reducing quality of life and adherence and limiting treatment outcomes. A step-wise approach can be implemented to minimize the risk of myotoxicity.
The first step is to avoid critical drug interactions that increase statin exposure. The statins most susceptible to interactions are those metabolized by CYP3A4—simvastatin, lovastatin, and atorvastatin. Medications commonly used that inhibit CYP3A4 include macrolide antibiotics and azole antifungals.42
Second, establishing a firm diagnosis of statin- associated myalgia is critical. This is often challenging given that many comorbid conditions (eg, arthritis) are associated with muscle symptoms. Ruling out other possible contributors, such as thyroid dysfunction, electrolyte abnormalities, and recent muscle injury, also should be considered. Temporary discontinuation of the statin to determine if symptoms improve is suggested. Monitoring the CK level is prudent in symptomatic patients to gauge potential myotoxicity and determine if therapy should be discontinued. The National Lipid Association recommends stopping statin therapy when signs and symptoms of rhabdomyolysis are present, including CK >10,000 IU/L or >10 times the upper limit of normal with elevated serum creatinine or requiring intravenous hydration.42
Other steps include switching to a different statin, reducing the statin dose, or using intermittent dosing (eg, every other day or twice weekly) with an extended half-life statin (eg, atorvastatin or rosuvastatin).42 Lastly, a bile acid resin or the cholesterol absorption inhibitor ezetimibe can be used. These classes produce only moderate reductions in LDL-C (~20%) but are unlikely to cause muscle symptoms.
Continue to complete the online evaluation and receive your certification of completion.
1. Bulbulia R, Bowman L, Wallendszus K, et al. for Heart Protection Study Collaborative Group. Effects on 11-year mortality and morbidity of lowering LDL cholesterol with simvastatin for about 5 years in 20,536 high-risk individuals: a randomised controlled trial. Lancet. 2011;378(9808):2013-2020.
2. Sever PS, Chang CL, Gupta AK, Whitehouse A, Poulter NR. for ASCOT Investigators. The Anglo-Scandinavian Cardiac Outcomes Trial: 11-year mortality follow-up of the lipid-lowering arm in the U.K. Eur Heart J. 2011;32(20):2525-2532.
3. Taylor F, Ward K, Moore TH, et al. Statins for the primary prevention of cardiovascular disease. Cochrane Database Syst Rev. 2011;(1):CD004816.-
4. Kawai Y, Sato-Ishida R, Motoyama A, Kajinami K. Place of pitavastatin in the statin armamentarium: promising evidence for a role in diabetes mellitus. Drug Des Devel Ther. 2011;5:283-297.
5. Ose L, Budinski D, Hounslow N, Arneson V. Comparison of pitavastatin with simvastatin in primary hypercholesterolaemia or combined dyslipidaemia. Curr Med Res Opin. 2009;25(11):2755-2764.
6. Gumprecht J, Gosho M, Budinski D, Hounslow N. Comparative long-term efficacy and tolerability of pitavastatin 4 mg and atorvastatin 20-40 mg in patients with type 2 diabetes mellitus and combined (mixed) dyslipidaemia. Diabetes Obes Metab. 2011;13(11):1047-1055.
7. Ose L, Budinski D, Hounslow N, Arneson V. Long-term treatment with pitavastatin is effective and well tolerated by patients with primary hypercholesterolemia or combined dyslipidemia. Atherosclerosis. 2010;210(1):202-208.
8. Teramoto T. Pitavastatin: clinical effects from the LIVES Study. Atheroscler Suppl. 2011;12(3):285-288.
9. Hiro T, Kimura T, Morimoto T, et al. for JAPAN-ACS Investigators. Effect of intensive statin therapy on regression of coronary atherosclerosis in patients with acute coronary syndrome: a multicenter randomized trial evaluated by volumetric intravascular ultrasound using pitavastatin versus atorvastatin (JAPAN-ACS [Japan assessment of pitavastatin and atorvastatin in acute coronary syndrome] study). J Am Coll Cardiol. 2009;54(4):293-302.
10. Saku K, Zhang B, Noda K. and PATROL Trial Investigators. Randomized head-to-head comparison of pitavastatin, atorvastatin, and rosuvastatin for safety and efficacy (quantity and quality of LDL): the PATROL trial. Circ J. 2011;75(6):1493-1505.
11. Yanagi K, Monden T, Ikeda S, Matsumura M, Kasai K. A crossover study of rosuvastatin and pitavastatin in patients with type 2 diabetes. Adv Ther. 2011;28(2):160-171.
12. Toi T, Taguchi I, Yoneda S, et al. Early effect of lipid-lowering therapy with pitavastatin on regression of coronary atherosclerotic plaque. Comparison with atorvastatin. Circ J. 2009;73(8):1466-1472.
13. Maruyama T, Takada M, Nishibori Y, et al. Comparison of preventive effect on cardiovascular events with different statins: the CIRCLE study. Circ J. 2011;75(8):1951-1959.
14. Suh SY, Rha SW, Ahn TH, et al. for LAMIS Investigators. Long-term safety and efficacy of pitavastatin in patients with acute myocardial infarction (from the Livalo Acute Myocardial Infarction Study [LAMIS]). Am J Cardiol. 2011;108(11):1530-1535.
15. Eriksson M, Budinski D, Hounslow N. Comparative efficacy of pitavastatin and simvastatin in high-risk patients: a randomized controlled trial. Adv Ther. 2011;28(9):811-823.
16. Eriksson M, Budinski D, Hounslow N. Long-term efficacy of pitavastatin versus simvastatin. Adv Ther. 2011;28(9):799-810.
17. Stender S, Budinski D, Hounslow N. Pitavastatin demonstrates long-term efficacy, safety and tolerability in elderly patients with primary hypercholesterolaemia or combined (mixed) dyslipidaemia [published online ahead of print January 23, 2012]. Eur J Prev Cardiol. 2012;doi:10.1177/2047487312437326.
18. US Food and Drug Administration. FDA drug safety communication: New restrictions, contraindications, and dose limitations for Zocor (simvastatin) to reduce the risk of muscle injury. http://www.fda.gov/drugs/drugsafety/ucm256581.htm. Published 2011. Accessed May 18, 2012.
19. Betteridge J. Pitavastatin—results from phase III & IV. Atheroscler Suppl. 2010;11(3):8-14.
20. Jones PH, Davidson MH, Stein EA, et al. for STELLAR Study Group. Comparison of the efficacy and safety of rosuvastatin versus atorvastatin, simvastatin, and pravastatin across doses (STELLAR* Trial). Am J Cardiol. 2003;92(2):152-160.
21. Jones P, Kafonek S, Laurora I, Hunninghake D. Comparative dose efficacy study of atorvastatin versus simvastatin, pravastatin, lovastatin, and fluvastatin in patients with hypercholesterolemia (the CURVES study). Am J Cardiol. 1998;81(5):582-587.
22. Grundy SM, Cleeman JI, Merz CN, et al. National Heart, Lung, and Blood Institute; American College of Cardiology Foundation; American Heart Association. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation. 2004;110(2):227-239.
23. Livalo [package insert]. Montgomery, AL: Kowa Pharmaceuticals America, Inc.; 2012.
24. Boekholdt SM, Arsenault BJ, Mora S, et al. Association of LDL cholesterol, non-HDL cholesterol, and apolipoprotein B levels with risk of cardiovascular events among patients treated with statins: a meta-analysis. JAMA. 2012;307(12):1302-1309.
25. US Food and Drug Administration. FDA drug safety communication: Important safety label changes to cholesterol-lowering statin drugs. http://www.fda.gov/Drugs/DrugSafety/ucm293101.htm. Published 2012. Accessed May 18, 2012.
26. Ridker PM, Danielson E, Fonseca FA, et al. JUPITER Study Group. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359(21):2195-2207.
27. Koh KK, Quon MJ, Han SH, Lee Y, Kim SJ, Shin EK. Atorvastatin causes insulin resistance and increases ambient glycemia in hypercholesterolemic patients. J Am Coll Cardiol. 2010;55(12):1209-1216.
28. Preiss D, Seshasai SR, Welsh P, et al. Risk of incident diabetes with intensive-dose compared with moderate-dose statin therapy: a meta-analysis. JAMA. 2011;305(24):2556-2564.
29. Yokote K, Saito Y. CHIBA study investigators. Influence of statins on glucose tolerance in patients with type 2 diabetes mellitus: subanalysis of the collaborative study on hypercholesterolemia drug intervention and their benefits for atherosclerosis prevention (CHIBA study). J Atheroscler Thromb. 2009;16(3):297-298.
30. Preiss D, Sattar N. Statins and the risk of new-onset diabetes: a review of recent evidence. Curr Opin Lipidol. 2011;22(6):460-466.
31. Bottorff MB. Statin safety and drug interactions: clinical implications. Am J Cardiol. 2006;97(8A):27C-31C.
32. Andrus MR. Oral anticoagulant drug interactions with statins: case report of fluvastatin and review of the literature. Pharmacotherapy. 2004;24(2):285-290.
33. US Food and Drug Administration. FDA drug safety communication: Interactions beteween certain HIV or hepatitis C drugs and cholesterol-lowering statin drugs can increase the risk of muscle injury. http://www.fda.gov/Drugs/DrugSafety/ucm293877.htm. Published 2012. Accessed May 18, 2012.
34. Armitage J, Bowman L, Wallendszus K, et al. for Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH) Collaborative Group. Intensive lowering of LDL cholesterol with 80 mg versus 20 mg simvastatin daily in 12,064 survivors of myocardial infarction: a double-blind randomised trial. Lancet. 2010;376(9753):1658-1669.
35. de Lemos JA, Blazing MA, Wiviott SD, et al. for A to Z Investigators. Early intensive vs a delayed conservative simvastatin strategy in patients with acute coronary syndromes: phase Z of the A to Z trial. JAMA. 2004;292(11):1307-1316.
36. Backes JM, Howard PA, Ruisinger JF, Moriarty PM. Does simvastatin cause more myotoxicity compared with other statins? Ann Pharmacother. 2009;43(12):2012-2020.
37. Jacobson TA. Statin safety: lessons from new drug applications for marketed statins. Am J Cardiol. 2006;97(8A):44C-51C.
38. Toth PP, Dayspring TD. Drug safety evaluation of rosuvastatin. Expert Opin Drug Saf. 2011;10(6):969-986.
39. Warrington S, Nagakawa S, Hounslow N. Comparison of the pharmacokinetics of pitavastatin by formulation and ethnic group: an open-label, single-dose, two-way crossover pharmacokinetic study in healthy Caucasian and Japanese men. Clin Drug Investig. 2011;31(10):735-743.
40. Chapman N, Chang CL, Caulfield M, et al. Ethnic variations in lipid-lowering in response to a statin (EVIREST): a substudy of the Anglo-Scandinavian Cardiac Outcomes Trial (ASCOT). Ethn Dis. 2011;21(2):150-157.
41. Albert MA, Glynn RJ, Fonseca FA, et al. Race, ethnicity, and the efficacy of rosuvastatin in primary prevention: the Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER) trial. Am Heart J. 2011;162(1):106-114.
42. Jacobson TA. Toward “pain-free” statin prescribing: clinical algorithm for diagnosis and management of myalgia. Mayo Clin Proc. 2008;83(6):687-700.
![]()
Peter P. Toth, MD, PhD
Professor of Clinical Family and Community Medicine, University of Illinois College of Medicine, Peoria, IL, Director of Preventative Cardiology, CGH Medical Center, Sterling IL
![]()
Peter P. Toth, MD, PhD
Professor of Clinical Family and Community Medicine, University of Illinois College of Medicine, Peoria, IL, Director of Preventative Cardiology, CGH Medical Center, Sterling IL
![]()
Peter P. Toth, MD, PhD
Professor of Clinical Family and Community Medicine, University of Illinois College of Medicine, Peoria, IL, Director of Preventative Cardiology, CGH Medical Center, Sterling IL
Managing the Multiple Symptoms of Benign Prostatic Hyperplasia — CME
Managing Type 2 Diabetes in Men
Meeting New Challenges with Antiplatelet Therapy in Primary Care
Dr. Toth has disclosed that he is on the speakers’ bureaus and is a consultant for Abbott, AstraZeneca, Kowa, Lilly, and Merck. He is on the speakers’ bureaus for Boehringer-Ingelheim and GlaxoSmithKline and is a consultant for Genentech and Genzyme.
Statins have become an important therapeutic option for managing cardiovascular (CV) risk, yet many questions remain regarding their use. This article addresses some of these questions in the primary care management of patients and highlights the impact of long-term statin therapy on CV end points. Because pitavastatin has recently become available in the United States, more detailed information about this agent is also presented.
LEARNING OBJECTIVES
After reviewing this activity on statin therapy, the reader will be able to:
- Describe the long-term benefits of statin therapy.
- Compare the efficacy and safety of pitavastatin with other statins.
- Select and modify statin therapy based upon individual patient factors.
TARGET AUDIENCE
Family physicians and clinicians who wish to gain increased knowledge and greater competency regarding statin therapy in the primary care management of patients with dyslipidemia.
ACKNOWLEDGEMENT
Dr. Toth was paid an honorarium by and received editorial assistance from the Primary Care Education Consortium in the development of this activity.
DISCLOSURES
As a continuing medical education provider accredited by the Accreditation Council for Continuing Medical Education (ACCME), it is the policy of the Primary Care Education Consortium (PCEC) to require any individual in a position to influence educational content to disclose the existence of any financial interest or other personal relationship with the manufacturer(s) of any commercial product(s).
Dr. Toth has disclosed that he is on the speakers’ bureaus and is a consultant for Abbott, AstraZeneca, Kowa, Lilly, and Merck. He is on the speakers’ bureaus for Boehringer-Ingelheim and GlaxoSmithKline and is a consultant for Genentech and Genzyme.
The medical accuracy and continuing medical education (CME) reviewer for this activity, Dr. Ron Pollack, has no real or apparent conflicts of interest to report.
PRIMARY CARE EDUCATION CONSORTIUM STAFF
Dr. Brunton has disclosed that he is on the advisory boards and speakers’ bureaus for Boehringer Ingelheim, Eli Lilly, Kowa, Novo Nordisk, Inc, and Teva Pharmaceuticals, and is on the advisory boards for Abbott and Sunovion.
Other PCEC staff has provided financial disclosure and have no conflicts of interest to resolve related to this activity.
CONFLICTS OF INTEREST
When individuals in a position to control content have reported financial relationships with one or more commercial interests, the Primary Care Education Consortium works with them to resolve such conflicts to ensure that the content presented is free of commercial bias. The content of this activity was vetted by the following mechanisms and modified as required to meet this standard:
- Content peer-review by an external topic expert
- Content peer-review by an external CME reviewer
- Content validation by internal Primary Care Education Consortium clinical editorial staff
OFF-LABEL DISCLOSURE
In accordance with ACCME guidelines, the faculty author has been asked to disclose discussion on unlabeled or unapproved uses of drugs or devices during the course of the activity.
SPONSORSHIP
This activity is sponsored by the Primary Care Education Consortium.
ACCREDITATION
This journal-based CME activity, Addressing Key Questions with Statin Therapy, has been reviewed and is acceptable for up to 1.0 prescribed credit by the American Academy of Family Physicians. AAFP accreditation begins June 1, 2012. Term of approval is for one year from this date with option for yearly renewal.
Physicians should claim only the credit commensurate with the extent of their participation in the activity.
MEDIUM
Text publication in the form of a journal article.
METHOD OF PHYSICIAN PARTICIPATION
To receive CME credit, please read the journal article, and upon completion go to: www.pceconsortium.org/menshealthSTATIN to complete the online evaluation to receive your certification of completion.
SUPPORT
This activity was supported by an educational grant from Kowa Pharmaceuticals America, Inc. and Lilly USA, LLC.
Recent Clinical Evidence
Findings from clinical trials continue to add to our understanding of the safety and efficacy of statin therapy; for example, extended follow-up studies from 2 landmark trials show lasting benefit and no evidence of emerging hazards. An analysis of the Heart Protection Study demonstrated that participants randomized to simvastatin 40 mg during the initial 5-year trial had maintained the vascular event reduction of 23% (95% confidence interval [CI], 19-28; P < .0001) at the 6-year follow-up.1 Similarly, 8 years after the close of the 3-year lipid-lowering arm of the Anglo-Scandinavian Cardiac Outcomes Trial (ASCOT), primary prevention patients originally randomized to atorvastatin had maintained a 14% reduction in all-cause mortality (95% CI, 0.76-0.98; P = .02) and a 15% lower rate of non-CV death (95% CI, 0.73-0.99; P = .03) compared with placebo.2 Cancer incidence among those receiving a statin versus those receiving a placebo was similar in both trials. Collectively, these data provide reassurance for the long-term continuation of statin therapy.
Results from a meta-analysis involving 34,272 participants without coronary heart disease from 14 randomized controlled trials (16 trial arms) comparing statins to placebo demonstrated significant reductions in all major events with statins, including a reduction of 16% in all-cause mortality (95% CI, 0.73-0.96), 30% in combined fatal and nonfatal CV disease end points (95% CI, 0.61-0.79), and 34% in revascularization rates (95% CI, 0.53-0.83).3 The meta-analysis found no evidence of significant harm caused by a statin or negative effects on patient quality of life.
Pitavastatin
Pitavastatin was approved in the United States in 2009, although it has been available in Japan since 2003. Pitava-statin is a synthetic lipophilic statin with an 11-hour half-life. Following oral ingestion, it enters the enterohepatic circulation without the formation of active metabolites. Pitavastatin is principally metabolized by the cytochrome-P450 (CYP) 2C9 isoenzyme and avoids the major CYP3A4 pathway; thus CYP-mediated drug interactions are greatly reduced.4
Several 12-week dose comparative studies with pitavastatin have been conducted. The first study randomized subjects (N = 857) to 1 of 4 groups: pitavastatin 2 or 4 mg/d or simvastatin 20 or 40 mg/d.5 Pitavastatin 2 mg demonstrated significantly greater reductions in low- density lipoprotein cholesterol (LDL-C; 39% vs 35%; P = .014) and greater reductions in non–high-density lipoprotein cholesterol (non–HDL-C) than did simvastatin 20 mg/d. Pitavastatin 4 mg/d and simvastatin 40 mg/d each reduced LDL-C by about 44%. Pitavastatin 4 mg/d has also been compared to atorvastatin 20 mg/d in 418 subjects.6 After 12 weeks, pitavastatin 4 mg/d and atorvastatin 20 mg/d produced similar reductions in LDL-C (~42%). No differences between groups were noted for other parameters, including HDL-C and non–HDL-C.
Long-term extension studies have evaluated the safety and efficacy of pitavastatin. Patients randomized to pitava-statin, atorvastatin, or simvastatin for 12 weeks received open-label pitavastatin 4 mg/d for up to 52 weeks (N = 1353).7 Notable findings included maintenance of LDL-C reductions from the end of the 12-week trial to 52 weeks with all 3 treatments. HDL-C levels continued to increase during follow up, rising 14.3% from baseline. Another long-term study compared pitavastatin 4 mg/d and atorvastatin 20 or 40 mg/d (N = 212).6 Both statins produced similar reductions in LDL-C and improvements in other major lipoproteins; however, atorvastatin significantly increased fasting blood glucose from baseline (7.2%; P < .05), whereas pitavastatin showed a nonsignificant increase of 2.1%.
The Japanese LIVALO Effectiveness and Safety (LIVES) Study (N = 20,000) evaluated the effects of pitavastatin 1 to 4 mg daily in clinical practice.8 Among patients with abnormal baseline values, treatment with pitavastatin was associated with a 29% reduction in LDL-C and a 23% reduction in triglycerides after 2 years. There was a 5.9% overall increase in HDL-C and a 24.6% increase among those with baseline HDL-C values <40 mg/dL. Pitavastatin was also associated with an improvement in glycosylated hemoglobin (A1C) values among those with diabetes mellitus (DM). Concomitant antidiabetic therapy was continued during the study. These findings suggest that pitavastatin does not worsen glycemic parameters. A 5-year extension of the LIVES study (N = 6582) demonstrated that long-term treatment with pitavastatin maintained the LDL-C reductions observed in the 2-year trial.8 Furthermore, HDL-C levels continued to climb, with an overall 29% increase among those with baseline values < 40 mg/dL. Patients who achieved both LDL-C and HDL-C targets experienced the greatest reductions in CV and cerebrovascular risk.
Finally, the Japan Assessment of Pitavastatin and Ator-vastatin in Acute Coronary Syndrome (JAPAN-ACS) study was a prospective, open-label trial that investigated the effects of pitavastatin 4 mg/d and atorvastatin 20 mg/d on coronary plaque volume (PV) among patients with acute coronary syndrome (N = 252) undergoing intravascular ultrasound.9 After 8 to 12 months of treatment, the mean change in PV was – 16.9 ± 13.9% and – 18.1 ± 14.2% in the pitavastatin and atorvastatin groups, respectively. Each statin produced significant but equivalent regression of PV.
Other key findings from additional pitavastatin clinical trials are found in TABLE 1 .10-17
table 1
Key findings from pitavastatin clinical trials
| Statins | Population | Findings/Comments |
|---|---|---|
| Dose Comparative Studies | ||
| Pitavastatin 4 mg vs Simvastatin 40 mg15 | Dyslipidemic adults with ≥2 CV risk factors (N = 355) | Each statin: LDL-C ↓ by 44% at 12 weeks >80% reached LDL-C goal |
| Pitavastatin 2 mg, 4 mg17 | Dyslipidemic adults age ≥65 years (N = 545) | LDL-C ↓ by 43%, HDL-C ↑ by 9.6% at 60 weeks Only 17% required uptitration to 4 mg 89%-94% achieved LDL-C goals |
| Pitavastatin 4 mg vs Simvastatin 40-80 mg16 | Dyslipidemic adults with ≥ 2 CV risk factors (N=178) | Each statin: LDL-C ↓ by ~42% at 44 weeks Discontinuation (5.8% vs 10.5%), myalgia (4.1% vs 12.3%) for pitavastatin vs simvastatin, respectively |
| Other Clinical Trials | ||
| Pitavastatin 2 mg vs Atorvastatin 10 mg vs Rosuvastatin 2.5 mg10 | Dyslipidemic adults with CV risk factors (N=302) | All agents: LDL-C ↓ by 40%-45% at 16 weeks Atorvastatin and rosuvastatin: A1C ↑ |
| Pitavastatin 2 mg vs Rosuvastatin 2.5 mg11 | Dyslipidemic adults with type 2 DM (N = 90) | Both agents: Inflammation ↓, lipids improved, no adverse effects on glycemic control Rosuvastatin: Greater LDL-C ↓, hsCRP vs pitavastatin |
| Pitavastatin 2.3 mg vs Atorvastatin 11.3 mg vs Pravastatin 10.3 mg vs No statin13 | Previous PCI (N = 743) | Each statin: Major coronary events ↓ LDL-C and HDL-C: Predicted coronary events Pitavastatin and atorvastatin: Greater LDL-C ↓ vs pravastatin Only pitavastatin: Significant HDL-C ↑ vs no statin |
| Pitavastatin 2 mg vs Atorvastatin 10 mg12 | ACS patients who underwent emergency PCI and IVUS (N = 160) | Fibrofatty composition, PV: Significant ↓ with pitavastatin |
| Pitavastatin 2 mg14 | Adults with acute MI (N = 1039) | 71% achieved LDL-C goal at 12 months Pitavastatin: Favorable effects on biomarkers maintained at 12 months |
| A1C, glycosylated hemoglobin; ACS, acute coronary syndrome; CV, cardiovascular; DM, diabetes mellitus; HDL-C, high-density lipoprotein cholesterol; hsCRP, high-sensitivity C-reactive protein; IVUS, intravascular ultrasound; LDL-C, low-density lipoprotein cholesterol; MI, myocardial infarction; PCI, percutaneous coronary intervention; PV, plaque volume. | ||
Key Questions
The following are common questions asked by family physicians when considering statin therapy to treat patients with dyslipidemia.
What are the key lipoprotein differences among available statins?
Nearly all statins are able to provide the minimal 30% to 40% LDL-C reduction as suggested by the National Cholesterol Education Program Adult Treatment Panel III for high-risk patients ( TABLE 2 ).18-22 If greater reductions are required, higher doses of more potent agents, such as atorvastatin and rosuvastatin, may be needed.
Statins also provide moderate increases in HDL-C, with subtle differences observed among the agents. Atorvastatin and fluvastatin usually provide the smallest increases in HDL-C (up to ~6%), whereas simvastatin, pitavastatin, and rosuvastatin produce more robust increases (~5% to 10%).20,21,23 The effect of statins on non–HDL-C is similar to their effect on LDL-C.22 Non–HDL-C is a secondary target of therapy in patients with triglyceride levels ≥200 mg/dL. Non–HDL-C includes all atherogenic particles (ie, LDL-C and triglyceride-rich lipoproteins) and is calculated as the difference between total cholesterol and HDL-C. The non–HDL-C goal is 30 mg/dL higher than the LDL-C goal. Clinical investigation continues to demonstrate that non–HDL-C is a valuable predictor of CV risk. An analysis of statin-treated patients indicated that compared with LDL-C and apolipoprotein B, non–HDL-C has a greater strength of association for risk of future CV events.24
table 2
Range of Low-Density Lipoprotein Cholesterol (LDL-C)–lowering among statins18-21
| LDL-C Range (↓) | Atorvastatin | Fluvastatin | Lovastatin | Pitavastatin | Pravastatin | Rosuvastatin | Simvastatin |
|---|---|---|---|---|---|---|---|
| 20%-25% | — | 20 mg | — | — | — | — | — |
| 25%-30% | — | 40 mg | — | — | 10 mg | — | — |
| 30%-35% | — | 80 mg | 20 mg | 1 mg | 20 mg | — | 10 mg |
| 35%-40% | 10 mg | — | 40 mg | 2 mg | 40 mg | — | 20 mg |
| 40%-45% | 20 mg | — | 80 mg | 4 mg | 80 mg | 5 mg | 40 mg |
| 45%-50% | 40 mg | — | — | — | — | 10 mg | — |
| 50%-60% | 80 mg | — | — | — | — | 20 mg | — |
| >60% | — | — | — | — | — | 40 mg | — |
Is diabetes really a consequence of statin therapy? If so, do differences exist among the statins?
The US Food and Drug Administration (FDA) recently added warnings to all statin labeling indicating that statins can raise blood glucose and A1C levels.25 These effects appear to be modest and dose dependent. This concern initially emerged in the Justification for the Use of statins in Prevention: an Intervention Trial Evaluating Rosuva-statin (JUPITER) study when statin users experienced a 25% higher incidence of new onset DM compared to those receiving placebo.26 The short-term effects of various atorvastatin doses on glycemic indices further support these findings.27 Compared to placebo, all atorvastatin doses significantly increased A1C and fasting plasma insulin levels after 8 weeks (all, P < .01). Additionally, a meta-analysis of 5 major statin trials involving 32,752 patients demonstrated that patients receiving intensive-dose statin therapy had a 12% higher risk of developing DM than patients receiving moderate-dose statin therapy.28
The association between statin therapy and DM is considered a class effect; differences among the statins are controversial. In an analysis of 13 major randomized controlled trials, pravastatin produced a nonsignificant 3% increase in new onset DM, whereas rosuvastatin was associated with an 18% increase.28 A 16-week, head-to-head comparison showed that pitavastatin had no effect on A1C, while modest increases were seen with low-dose atorvastatin and rosuvastatin.10 In another study, atorvastatin but not pitavastatin produced significant (P < .03) increases in glycoalbumin and A1C (P < .01), whereas fasting glucose and insulin levels tended to decrease with pitavastatin.29 However, findings from the meta-analysis showed that the individual studies lacked sufficient specific data to detect heterogeneity between statins.30
Overall, statins are associated with modest increases in glycemic indices and new onset DM. This association appears to be greater with high-dose therapy; however, additional trials are needed to fully understand possible differences among statins.
Which drug interactions are clinically important?
As statin pharmacokinetic data have accumulated, critical drug interactions have become more apparent. The major concern is increased statin exposure secondary to limited metabolism, resulting in more dose-dependent AEs, such as muscle injury. CYP3A4 isoenzyme involvement is common in clinically significant interactions. Lovastatin, simvastatin, and to a lesser extent, atorvastatin are all substrates for CYP3A4.31 The FDA recently updated labeling for simvastatin and lovastatin to provide information on contraindications and dose limitations with concomitant agents [www.fda.gov/Drugs/DrugSafety/ucm293877.htm].18,25
Statins have differing effects on warfarin metabolism, with most agents increasing the international normalized ratio (INR). Conversely, atorvastatin and pitavastatin have shown no significant effect on prothrombin time when added to chronic warfarin therapy.23,32 Despite this, appropriate INR monitoring is suggested when any statin is added to warfarin treatment.
Another recent FDA advisory focusing on human immunodeficiency virus and hepatitis C virus protease inhibitors further emphasizes the importance of statin interactions.33 The advisory provides specific dose limitations and contraindications for 7 statins. Similar to other potent CYP3A4 inhibitors, protease inhibitors can increase lovastatin and simvastatin levels by 13- to 20-fold. No information is available for fluvastatin, while no dose limitations are needed for pitavastatin or pravastatin.33
Mechanisms implicating statins in other drug interactions include inhibition of CYP2C9, glucuronidation, and organic anion transporting polypeptide (OATP).31 Concomitant treatment with gemfibrozil and a statin produces a significant interaction, as this combination inhibits CYP2C9 and glucuronidation, resulting in marked increases in statin exposure. Similarly, the coadministration of a statin with cyclosporine is clinically relevant. Cyclosporine blocks another key step in statin metabolism, OATP, resulting in elevated concentrations of nearly all statins. The concomitant use of cyclosporine with lovastatin, simvastatin, or pitavastatin is contraindicated, whereas most other agents require dose limitations.18,23,25,31
Do statins possess a dose-dependent threshold for adverse events?
A general dose-dependent threshold for AEs has been observed with statin therapy. This upper limit is more apparent with certain statins and primarily manifests as myotoxicity or increased hepatic transaminase levels. High-dose simvastatin has shown the most evidence regarding increased myopathy. In the Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH) trial, 53 patients (0.9%) in the simvastatin 80-mg group experienced myopathy, including 7 cases (0.1%) of rhabdomyolysis, over a mean of 6.7 years of follow-up.34 By comparison, there were 2 reports of myopathy (0.03%) in the 20-mg group. Similarly, in Phase Z of the A to Z trial, 9 reports (0.4%) of myopathy, including 3 cases of rhabdomyolysis (0.13%), were reported with simvastatin 80 mg over a median of 2 years of follow-up, compared to none with lower doses.35 Lower rates of myopathy and rhabdomyolysis (0.0%-0.3% and 0.0%-0.1%, respectively) were found with atorvastatin 80 mg, fluvastatin 80 mg, and rosuvastatin 40 mg in major trials.36 These data prompted the FDA to publish an advisory on simvastatin dose limitations, including restricting the 80-mg dose.18 A threshold also has been observed with other statins, as an approximate 3-fold higher incidence of creatine kinase (CK) and hepatic transaminase elevations occur when titrating from moderate to maximal doses.37
Should ethnicity be a factor in selecting a statin?
While no specific recommendations presently exist regarding the selection of statin therapy based on ethnicity, rosuvastatin doses, including the 5-mg starting dose, should be reduced in patients of Asian ancestry because of a 2-fold increase in pharmacokinetic parameters compared to whites.38 Otherwise, the few studies evaluating individual agents among various ethnic groups generally suggest similar effects on pharmacokinetic parameters, lipid changes, and CV outcomes.
One study compared pharmacokinetic parameters of pitavastatin between healthy Caucasian and Japanese men.39 Pitavastatin demonstrated pharmacokinetic bioequivalence between the 2 groups with no clinically relevant differences. A substudy of ASCOT assessed the lipid effects of atorvastatin among whites, blacks, and South Asians.40 No significant differences were observed in the reductions in total cholesterol, LDL-C, or triglycerides. Lastly, outcomes were evaluated among different ethnicities in the JUPITER study.41 Similar reductions in major CV events were noted for whites versus non-whites with Hispanics and blacks experiencing comparable risk reductions.
How should statin-associated myalgia be managed?
Approximately 11% of patients receiving moderate- to high-dose statin therapy experience muscle symptoms.42 This common AE can greatly affect therapy by reducing quality of life and adherence and limiting treatment outcomes. A step-wise approach can be implemented to minimize the risk of myotoxicity.
The first step is to avoid critical drug interactions that increase statin exposure. The statins most susceptible to interactions are those metabolized by CYP3A4—simvastatin, lovastatin, and atorvastatin. Medications commonly used that inhibit CYP3A4 include macrolide antibiotics and azole antifungals.42
Second, establishing a firm diagnosis of statin- associated myalgia is critical. This is often challenging given that many comorbid conditions (eg, arthritis) are associated with muscle symptoms. Ruling out other possible contributors, such as thyroid dysfunction, electrolyte abnormalities, and recent muscle injury, also should be considered. Temporary discontinuation of the statin to determine if symptoms improve is suggested. Monitoring the CK level is prudent in symptomatic patients to gauge potential myotoxicity and determine if therapy should be discontinued. The National Lipid Association recommends stopping statin therapy when signs and symptoms of rhabdomyolysis are present, including CK >10,000 IU/L or >10 times the upper limit of normal with elevated serum creatinine or requiring intravenous hydration.42
Other steps include switching to a different statin, reducing the statin dose, or using intermittent dosing (eg, every other day or twice weekly) with an extended half-life statin (eg, atorvastatin or rosuvastatin).42 Lastly, a bile acid resin or the cholesterol absorption inhibitor ezetimibe can be used. These classes produce only moderate reductions in LDL-C (~20%) but are unlikely to cause muscle symptoms.
Continue to complete the online evaluation and receive your certification of completion.
Managing the Multiple Symptoms of Benign Prostatic Hyperplasia — CME
Managing Type 2 Diabetes in Men
Meeting New Challenges with Antiplatelet Therapy in Primary Care
Dr. Toth has disclosed that he is on the speakers’ bureaus and is a consultant for Abbott, AstraZeneca, Kowa, Lilly, and Merck. He is on the speakers’ bureaus for Boehringer-Ingelheim and GlaxoSmithKline and is a consultant for Genentech and Genzyme.
Statins have become an important therapeutic option for managing cardiovascular (CV) risk, yet many questions remain regarding their use. This article addresses some of these questions in the primary care management of patients and highlights the impact of long-term statin therapy on CV end points. Because pitavastatin has recently become available in the United States, more detailed information about this agent is also presented.
LEARNING OBJECTIVES
After reviewing this activity on statin therapy, the reader will be able to:
- Describe the long-term benefits of statin therapy.
- Compare the efficacy and safety of pitavastatin with other statins.
- Select and modify statin therapy based upon individual patient factors.
TARGET AUDIENCE
Family physicians and clinicians who wish to gain increased knowledge and greater competency regarding statin therapy in the primary care management of patients with dyslipidemia.
ACKNOWLEDGEMENT
Dr. Toth was paid an honorarium by and received editorial assistance from the Primary Care Education Consortium in the development of this activity.
DISCLOSURES
As a continuing medical education provider accredited by the Accreditation Council for Continuing Medical Education (ACCME), it is the policy of the Primary Care Education Consortium (PCEC) to require any individual in a position to influence educational content to disclose the existence of any financial interest or other personal relationship with the manufacturer(s) of any commercial product(s).
Dr. Toth has disclosed that he is on the speakers’ bureaus and is a consultant for Abbott, AstraZeneca, Kowa, Lilly, and Merck. He is on the speakers’ bureaus for Boehringer-Ingelheim and GlaxoSmithKline and is a consultant for Genentech and Genzyme.
The medical accuracy and continuing medical education (CME) reviewer for this activity, Dr. Ron Pollack, has no real or apparent conflicts of interest to report.
PRIMARY CARE EDUCATION CONSORTIUM STAFF
Dr. Brunton has disclosed that he is on the advisory boards and speakers’ bureaus for Boehringer Ingelheim, Eli Lilly, Kowa, Novo Nordisk, Inc, and Teva Pharmaceuticals, and is on the advisory boards for Abbott and Sunovion.
Other PCEC staff has provided financial disclosure and have no conflicts of interest to resolve related to this activity.
CONFLICTS OF INTEREST
When individuals in a position to control content have reported financial relationships with one or more commercial interests, the Primary Care Education Consortium works with them to resolve such conflicts to ensure that the content presented is free of commercial bias. The content of this activity was vetted by the following mechanisms and modified as required to meet this standard:
- Content peer-review by an external topic expert
- Content peer-review by an external CME reviewer
- Content validation by internal Primary Care Education Consortium clinical editorial staff
OFF-LABEL DISCLOSURE
In accordance with ACCME guidelines, the faculty author has been asked to disclose discussion on unlabeled or unapproved uses of drugs or devices during the course of the activity.
SPONSORSHIP
This activity is sponsored by the Primary Care Education Consortium.
ACCREDITATION
This journal-based CME activity, Addressing Key Questions with Statin Therapy, has been reviewed and is acceptable for up to 1.0 prescribed credit by the American Academy of Family Physicians. AAFP accreditation begins June 1, 2012. Term of approval is for one year from this date with option for yearly renewal.
Physicians should claim only the credit commensurate with the extent of their participation in the activity.
MEDIUM
Text publication in the form of a journal article.
METHOD OF PHYSICIAN PARTICIPATION
To receive CME credit, please read the journal article, and upon completion go to: www.pceconsortium.org/menshealthSTATIN to complete the online evaluation to receive your certification of completion.
SUPPORT
This activity was supported by an educational grant from Kowa Pharmaceuticals America, Inc. and Lilly USA, LLC.
Recent Clinical Evidence
Findings from clinical trials continue to add to our understanding of the safety and efficacy of statin therapy; for example, extended follow-up studies from 2 landmark trials show lasting benefit and no evidence of emerging hazards. An analysis of the Heart Protection Study demonstrated that participants randomized to simvastatin 40 mg during the initial 5-year trial had maintained the vascular event reduction of 23% (95% confidence interval [CI], 19-28; P < .0001) at the 6-year follow-up.1 Similarly, 8 years after the close of the 3-year lipid-lowering arm of the Anglo-Scandinavian Cardiac Outcomes Trial (ASCOT), primary prevention patients originally randomized to atorvastatin had maintained a 14% reduction in all-cause mortality (95% CI, 0.76-0.98; P = .02) and a 15% lower rate of non-CV death (95% CI, 0.73-0.99; P = .03) compared with placebo.2 Cancer incidence among those receiving a statin versus those receiving a placebo was similar in both trials. Collectively, these data provide reassurance for the long-term continuation of statin therapy.
Results from a meta-analysis involving 34,272 participants without coronary heart disease from 14 randomized controlled trials (16 trial arms) comparing statins to placebo demonstrated significant reductions in all major events with statins, including a reduction of 16% in all-cause mortality (95% CI, 0.73-0.96), 30% in combined fatal and nonfatal CV disease end points (95% CI, 0.61-0.79), and 34% in revascularization rates (95% CI, 0.53-0.83).3 The meta-analysis found no evidence of significant harm caused by a statin or negative effects on patient quality of life.
Pitavastatin
Pitavastatin was approved in the United States in 2009, although it has been available in Japan since 2003. Pitava-statin is a synthetic lipophilic statin with an 11-hour half-life. Following oral ingestion, it enters the enterohepatic circulation without the formation of active metabolites. Pitavastatin is principally metabolized by the cytochrome-P450 (CYP) 2C9 isoenzyme and avoids the major CYP3A4 pathway; thus CYP-mediated drug interactions are greatly reduced.4
Several 12-week dose comparative studies with pitavastatin have been conducted. The first study randomized subjects (N = 857) to 1 of 4 groups: pitavastatin 2 or 4 mg/d or simvastatin 20 or 40 mg/d.5 Pitavastatin 2 mg demonstrated significantly greater reductions in low- density lipoprotein cholesterol (LDL-C; 39% vs 35%; P = .014) and greater reductions in non–high-density lipoprotein cholesterol (non–HDL-C) than did simvastatin 20 mg/d. Pitavastatin 4 mg/d and simvastatin 40 mg/d each reduced LDL-C by about 44%. Pitavastatin 4 mg/d has also been compared to atorvastatin 20 mg/d in 418 subjects.6 After 12 weeks, pitavastatin 4 mg/d and atorvastatin 20 mg/d produced similar reductions in LDL-C (~42%). No differences between groups were noted for other parameters, including HDL-C and non–HDL-C.
Long-term extension studies have evaluated the safety and efficacy of pitavastatin. Patients randomized to pitava-statin, atorvastatin, or simvastatin for 12 weeks received open-label pitavastatin 4 mg/d for up to 52 weeks (N = 1353).7 Notable findings included maintenance of LDL-C reductions from the end of the 12-week trial to 52 weeks with all 3 treatments. HDL-C levels continued to increase during follow up, rising 14.3% from baseline. Another long-term study compared pitavastatin 4 mg/d and atorvastatin 20 or 40 mg/d (N = 212).6 Both statins produced similar reductions in LDL-C and improvements in other major lipoproteins; however, atorvastatin significantly increased fasting blood glucose from baseline (7.2%; P < .05), whereas pitavastatin showed a nonsignificant increase of 2.1%.
The Japanese LIVALO Effectiveness and Safety (LIVES) Study (N = 20,000) evaluated the effects of pitavastatin 1 to 4 mg daily in clinical practice.8 Among patients with abnormal baseline values, treatment with pitavastatin was associated with a 29% reduction in LDL-C and a 23% reduction in triglycerides after 2 years. There was a 5.9% overall increase in HDL-C and a 24.6% increase among those with baseline HDL-C values <40 mg/dL. Pitavastatin was also associated with an improvement in glycosylated hemoglobin (A1C) values among those with diabetes mellitus (DM). Concomitant antidiabetic therapy was continued during the study. These findings suggest that pitavastatin does not worsen glycemic parameters. A 5-year extension of the LIVES study (N = 6582) demonstrated that long-term treatment with pitavastatin maintained the LDL-C reductions observed in the 2-year trial.8 Furthermore, HDL-C levels continued to climb, with an overall 29% increase among those with baseline values < 40 mg/dL. Patients who achieved both LDL-C and HDL-C targets experienced the greatest reductions in CV and cerebrovascular risk.
Finally, the Japan Assessment of Pitavastatin and Ator-vastatin in Acute Coronary Syndrome (JAPAN-ACS) study was a prospective, open-label trial that investigated the effects of pitavastatin 4 mg/d and atorvastatin 20 mg/d on coronary plaque volume (PV) among patients with acute coronary syndrome (N = 252) undergoing intravascular ultrasound.9 After 8 to 12 months of treatment, the mean change in PV was – 16.9 ± 13.9% and – 18.1 ± 14.2% in the pitavastatin and atorvastatin groups, respectively. Each statin produced significant but equivalent regression of PV.
Other key findings from additional pitavastatin clinical trials are found in TABLE 1 .10-17
table 1
Key findings from pitavastatin clinical trials
| Statins | Population | Findings/Comments |
|---|---|---|
| Dose Comparative Studies | ||
| Pitavastatin 4 mg vs Simvastatin 40 mg15 | Dyslipidemic adults with ≥2 CV risk factors (N = 355) | Each statin: LDL-C ↓ by 44% at 12 weeks >80% reached LDL-C goal |
| Pitavastatin 2 mg, 4 mg17 | Dyslipidemic adults age ≥65 years (N = 545) | LDL-C ↓ by 43%, HDL-C ↑ by 9.6% at 60 weeks Only 17% required uptitration to 4 mg 89%-94% achieved LDL-C goals |
| Pitavastatin 4 mg vs Simvastatin 40-80 mg16 | Dyslipidemic adults with ≥ 2 CV risk factors (N=178) | Each statin: LDL-C ↓ by ~42% at 44 weeks Discontinuation (5.8% vs 10.5%), myalgia (4.1% vs 12.3%) for pitavastatin vs simvastatin, respectively |
| Other Clinical Trials | ||
| Pitavastatin 2 mg vs Atorvastatin 10 mg vs Rosuvastatin 2.5 mg10 | Dyslipidemic adults with CV risk factors (N=302) | All agents: LDL-C ↓ by 40%-45% at 16 weeks Atorvastatin and rosuvastatin: A1C ↑ |
| Pitavastatin 2 mg vs Rosuvastatin 2.5 mg11 | Dyslipidemic adults with type 2 DM (N = 90) | Both agents: Inflammation ↓, lipids improved, no adverse effects on glycemic control Rosuvastatin: Greater LDL-C ↓, hsCRP vs pitavastatin |
| Pitavastatin 2.3 mg vs Atorvastatin 11.3 mg vs Pravastatin 10.3 mg vs No statin13 | Previous PCI (N = 743) | Each statin: Major coronary events ↓ LDL-C and HDL-C: Predicted coronary events Pitavastatin and atorvastatin: Greater LDL-C ↓ vs pravastatin Only pitavastatin: Significant HDL-C ↑ vs no statin |
| Pitavastatin 2 mg vs Atorvastatin 10 mg12 | ACS patients who underwent emergency PCI and IVUS (N = 160) | Fibrofatty composition, PV: Significant ↓ with pitavastatin |
| Pitavastatin 2 mg14 | Adults with acute MI (N = 1039) | 71% achieved LDL-C goal at 12 months Pitavastatin: Favorable effects on biomarkers maintained at 12 months |
| A1C, glycosylated hemoglobin; ACS, acute coronary syndrome; CV, cardiovascular; DM, diabetes mellitus; HDL-C, high-density lipoprotein cholesterol; hsCRP, high-sensitivity C-reactive protein; IVUS, intravascular ultrasound; LDL-C, low-density lipoprotein cholesterol; MI, myocardial infarction; PCI, percutaneous coronary intervention; PV, plaque volume. | ||
Key Questions
The following are common questions asked by family physicians when considering statin therapy to treat patients with dyslipidemia.
What are the key lipoprotein differences among available statins?
Nearly all statins are able to provide the minimal 30% to 40% LDL-C reduction as suggested by the National Cholesterol Education Program Adult Treatment Panel III for high-risk patients ( TABLE 2 ).18-22 If greater reductions are required, higher doses of more potent agents, such as atorvastatin and rosuvastatin, may be needed.
Statins also provide moderate increases in HDL-C, with subtle differences observed among the agents. Atorvastatin and fluvastatin usually provide the smallest increases in HDL-C (up to ~6%), whereas simvastatin, pitavastatin, and rosuvastatin produce more robust increases (~5% to 10%).20,21,23 The effect of statins on non–HDL-C is similar to their effect on LDL-C.22 Non–HDL-C is a secondary target of therapy in patients with triglyceride levels ≥200 mg/dL. Non–HDL-C includes all atherogenic particles (ie, LDL-C and triglyceride-rich lipoproteins) and is calculated as the difference between total cholesterol and HDL-C. The non–HDL-C goal is 30 mg/dL higher than the LDL-C goal. Clinical investigation continues to demonstrate that non–HDL-C is a valuable predictor of CV risk. An analysis of statin-treated patients indicated that compared with LDL-C and apolipoprotein B, non–HDL-C has a greater strength of association for risk of future CV events.24
table 2
Range of Low-Density Lipoprotein Cholesterol (LDL-C)–lowering among statins18-21
| LDL-C Range (↓) | Atorvastatin | Fluvastatin | Lovastatin | Pitavastatin | Pravastatin | Rosuvastatin | Simvastatin |
|---|---|---|---|---|---|---|---|
| 20%-25% | — | 20 mg | — | — | — | — | — |
| 25%-30% | — | 40 mg | — | — | 10 mg | — | — |
| 30%-35% | — | 80 mg | 20 mg | 1 mg | 20 mg | — | 10 mg |
| 35%-40% | 10 mg | — | 40 mg | 2 mg | 40 mg | — | 20 mg |
| 40%-45% | 20 mg | — | 80 mg | 4 mg | 80 mg | 5 mg | 40 mg |
| 45%-50% | 40 mg | — | — | — | — | 10 mg | — |
| 50%-60% | 80 mg | — | — | — | — | 20 mg | — |
| >60% | — | — | — | — | — | 40 mg | — |
Is diabetes really a consequence of statin therapy? If so, do differences exist among the statins?
The US Food and Drug Administration (FDA) recently added warnings to all statin labeling indicating that statins can raise blood glucose and A1C levels.25 These effects appear to be modest and dose dependent. This concern initially emerged in the Justification for the Use of statins in Prevention: an Intervention Trial Evaluating Rosuva-statin (JUPITER) study when statin users experienced a 25% higher incidence of new onset DM compared to those receiving placebo.26 The short-term effects of various atorvastatin doses on glycemic indices further support these findings.27 Compared to placebo, all atorvastatin doses significantly increased A1C and fasting plasma insulin levels after 8 weeks (all, P < .01). Additionally, a meta-analysis of 5 major statin trials involving 32,752 patients demonstrated that patients receiving intensive-dose statin therapy had a 12% higher risk of developing DM than patients receiving moderate-dose statin therapy.28
The association between statin therapy and DM is considered a class effect; differences among the statins are controversial. In an analysis of 13 major randomized controlled trials, pravastatin produced a nonsignificant 3% increase in new onset DM, whereas rosuvastatin was associated with an 18% increase.28 A 16-week, head-to-head comparison showed that pitavastatin had no effect on A1C, while modest increases were seen with low-dose atorvastatin and rosuvastatin.10 In another study, atorvastatin but not pitavastatin produced significant (P < .03) increases in glycoalbumin and A1C (P < .01), whereas fasting glucose and insulin levels tended to decrease with pitavastatin.29 However, findings from the meta-analysis showed that the individual studies lacked sufficient specific data to detect heterogeneity between statins.30
Overall, statins are associated with modest increases in glycemic indices and new onset DM. This association appears to be greater with high-dose therapy; however, additional trials are needed to fully understand possible differences among statins.
Which drug interactions are clinically important?
As statin pharmacokinetic data have accumulated, critical drug interactions have become more apparent. The major concern is increased statin exposure secondary to limited metabolism, resulting in more dose-dependent AEs, such as muscle injury. CYP3A4 isoenzyme involvement is common in clinically significant interactions. Lovastatin, simvastatin, and to a lesser extent, atorvastatin are all substrates for CYP3A4.31 The FDA recently updated labeling for simvastatin and lovastatin to provide information on contraindications and dose limitations with concomitant agents [www.fda.gov/Drugs/DrugSafety/ucm293877.htm].18,25
Statins have differing effects on warfarin metabolism, with most agents increasing the international normalized ratio (INR). Conversely, atorvastatin and pitavastatin have shown no significant effect on prothrombin time when added to chronic warfarin therapy.23,32 Despite this, appropriate INR monitoring is suggested when any statin is added to warfarin treatment.
Another recent FDA advisory focusing on human immunodeficiency virus and hepatitis C virus protease inhibitors further emphasizes the importance of statin interactions.33 The advisory provides specific dose limitations and contraindications for 7 statins. Similar to other potent CYP3A4 inhibitors, protease inhibitors can increase lovastatin and simvastatin levels by 13- to 20-fold. No information is available for fluvastatin, while no dose limitations are needed for pitavastatin or pravastatin.33
Mechanisms implicating statins in other drug interactions include inhibition of CYP2C9, glucuronidation, and organic anion transporting polypeptide (OATP).31 Concomitant treatment with gemfibrozil and a statin produces a significant interaction, as this combination inhibits CYP2C9 and glucuronidation, resulting in marked increases in statin exposure. Similarly, the coadministration of a statin with cyclosporine is clinically relevant. Cyclosporine blocks another key step in statin metabolism, OATP, resulting in elevated concentrations of nearly all statins. The concomitant use of cyclosporine with lovastatin, simvastatin, or pitavastatin is contraindicated, whereas most other agents require dose limitations.18,23,25,31
Do statins possess a dose-dependent threshold for adverse events?
A general dose-dependent threshold for AEs has been observed with statin therapy. This upper limit is more apparent with certain statins and primarily manifests as myotoxicity or increased hepatic transaminase levels. High-dose simvastatin has shown the most evidence regarding increased myopathy. In the Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH) trial, 53 patients (0.9%) in the simvastatin 80-mg group experienced myopathy, including 7 cases (0.1%) of rhabdomyolysis, over a mean of 6.7 years of follow-up.34 By comparison, there were 2 reports of myopathy (0.03%) in the 20-mg group. Similarly, in Phase Z of the A to Z trial, 9 reports (0.4%) of myopathy, including 3 cases of rhabdomyolysis (0.13%), were reported with simvastatin 80 mg over a median of 2 years of follow-up, compared to none with lower doses.35 Lower rates of myopathy and rhabdomyolysis (0.0%-0.3% and 0.0%-0.1%, respectively) were found with atorvastatin 80 mg, fluvastatin 80 mg, and rosuvastatin 40 mg in major trials.36 These data prompted the FDA to publish an advisory on simvastatin dose limitations, including restricting the 80-mg dose.18 A threshold also has been observed with other statins, as an approximate 3-fold higher incidence of creatine kinase (CK) and hepatic transaminase elevations occur when titrating from moderate to maximal doses.37
Should ethnicity be a factor in selecting a statin?
While no specific recommendations presently exist regarding the selection of statin therapy based on ethnicity, rosuvastatin doses, including the 5-mg starting dose, should be reduced in patients of Asian ancestry because of a 2-fold increase in pharmacokinetic parameters compared to whites.38 Otherwise, the few studies evaluating individual agents among various ethnic groups generally suggest similar effects on pharmacokinetic parameters, lipid changes, and CV outcomes.
One study compared pharmacokinetic parameters of pitavastatin between healthy Caucasian and Japanese men.39 Pitavastatin demonstrated pharmacokinetic bioequivalence between the 2 groups with no clinically relevant differences. A substudy of ASCOT assessed the lipid effects of atorvastatin among whites, blacks, and South Asians.40 No significant differences were observed in the reductions in total cholesterol, LDL-C, or triglycerides. Lastly, outcomes were evaluated among different ethnicities in the JUPITER study.41 Similar reductions in major CV events were noted for whites versus non-whites with Hispanics and blacks experiencing comparable risk reductions.
How should statin-associated myalgia be managed?
Approximately 11% of patients receiving moderate- to high-dose statin therapy experience muscle symptoms.42 This common AE can greatly affect therapy by reducing quality of life and adherence and limiting treatment outcomes. A step-wise approach can be implemented to minimize the risk of myotoxicity.
The first step is to avoid critical drug interactions that increase statin exposure. The statins most susceptible to interactions are those metabolized by CYP3A4—simvastatin, lovastatin, and atorvastatin. Medications commonly used that inhibit CYP3A4 include macrolide antibiotics and azole antifungals.42
Second, establishing a firm diagnosis of statin- associated myalgia is critical. This is often challenging given that many comorbid conditions (eg, arthritis) are associated with muscle symptoms. Ruling out other possible contributors, such as thyroid dysfunction, electrolyte abnormalities, and recent muscle injury, also should be considered. Temporary discontinuation of the statin to determine if symptoms improve is suggested. Monitoring the CK level is prudent in symptomatic patients to gauge potential myotoxicity and determine if therapy should be discontinued. The National Lipid Association recommends stopping statin therapy when signs and symptoms of rhabdomyolysis are present, including CK >10,000 IU/L or >10 times the upper limit of normal with elevated serum creatinine or requiring intravenous hydration.42
Other steps include switching to a different statin, reducing the statin dose, or using intermittent dosing (eg, every other day or twice weekly) with an extended half-life statin (eg, atorvastatin or rosuvastatin).42 Lastly, a bile acid resin or the cholesterol absorption inhibitor ezetimibe can be used. These classes produce only moderate reductions in LDL-C (~20%) but are unlikely to cause muscle symptoms.
Continue to complete the online evaluation and receive your certification of completion.
1. Bulbulia R, Bowman L, Wallendszus K, et al. for Heart Protection Study Collaborative Group. Effects on 11-year mortality and morbidity of lowering LDL cholesterol with simvastatin for about 5 years in 20,536 high-risk individuals: a randomised controlled trial. Lancet. 2011;378(9808):2013-2020.
2. Sever PS, Chang CL, Gupta AK, Whitehouse A, Poulter NR. for ASCOT Investigators. The Anglo-Scandinavian Cardiac Outcomes Trial: 11-year mortality follow-up of the lipid-lowering arm in the U.K. Eur Heart J. 2011;32(20):2525-2532.
3. Taylor F, Ward K, Moore TH, et al. Statins for the primary prevention of cardiovascular disease. Cochrane Database Syst Rev. 2011;(1):CD004816.-
4. Kawai Y, Sato-Ishida R, Motoyama A, Kajinami K. Place of pitavastatin in the statin armamentarium: promising evidence for a role in diabetes mellitus. Drug Des Devel Ther. 2011;5:283-297.
5. Ose L, Budinski D, Hounslow N, Arneson V. Comparison of pitavastatin with simvastatin in primary hypercholesterolaemia or combined dyslipidaemia. Curr Med Res Opin. 2009;25(11):2755-2764.
6. Gumprecht J, Gosho M, Budinski D, Hounslow N. Comparative long-term efficacy and tolerability of pitavastatin 4 mg and atorvastatin 20-40 mg in patients with type 2 diabetes mellitus and combined (mixed) dyslipidaemia. Diabetes Obes Metab. 2011;13(11):1047-1055.
7. Ose L, Budinski D, Hounslow N, Arneson V. Long-term treatment with pitavastatin is effective and well tolerated by patients with primary hypercholesterolemia or combined dyslipidemia. Atherosclerosis. 2010;210(1):202-208.
8. Teramoto T. Pitavastatin: clinical effects from the LIVES Study. Atheroscler Suppl. 2011;12(3):285-288.
9. Hiro T, Kimura T, Morimoto T, et al. for JAPAN-ACS Investigators. Effect of intensive statin therapy on regression of coronary atherosclerosis in patients with acute coronary syndrome: a multicenter randomized trial evaluated by volumetric intravascular ultrasound using pitavastatin versus atorvastatin (JAPAN-ACS [Japan assessment of pitavastatin and atorvastatin in acute coronary syndrome] study). J Am Coll Cardiol. 2009;54(4):293-302.
10. Saku K, Zhang B, Noda K. and PATROL Trial Investigators. Randomized head-to-head comparison of pitavastatin, atorvastatin, and rosuvastatin for safety and efficacy (quantity and quality of LDL): the PATROL trial. Circ J. 2011;75(6):1493-1505.
11. Yanagi K, Monden T, Ikeda S, Matsumura M, Kasai K. A crossover study of rosuvastatin and pitavastatin in patients with type 2 diabetes. Adv Ther. 2011;28(2):160-171.
12. Toi T, Taguchi I, Yoneda S, et al. Early effect of lipid-lowering therapy with pitavastatin on regression of coronary atherosclerotic plaque. Comparison with atorvastatin. Circ J. 2009;73(8):1466-1472.
13. Maruyama T, Takada M, Nishibori Y, et al. Comparison of preventive effect on cardiovascular events with different statins: the CIRCLE study. Circ J. 2011;75(8):1951-1959.
14. Suh SY, Rha SW, Ahn TH, et al. for LAMIS Investigators. Long-term safety and efficacy of pitavastatin in patients with acute myocardial infarction (from the Livalo Acute Myocardial Infarction Study [LAMIS]). Am J Cardiol. 2011;108(11):1530-1535.
15. Eriksson M, Budinski D, Hounslow N. Comparative efficacy of pitavastatin and simvastatin in high-risk patients: a randomized controlled trial. Adv Ther. 2011;28(9):811-823.
16. Eriksson M, Budinski D, Hounslow N. Long-term efficacy of pitavastatin versus simvastatin. Adv Ther. 2011;28(9):799-810.
17. Stender S, Budinski D, Hounslow N. Pitavastatin demonstrates long-term efficacy, safety and tolerability in elderly patients with primary hypercholesterolaemia or combined (mixed) dyslipidaemia [published online ahead of print January 23, 2012]. Eur J Prev Cardiol. 2012;doi:10.1177/2047487312437326.
18. US Food and Drug Administration. FDA drug safety communication: New restrictions, contraindications, and dose limitations for Zocor (simvastatin) to reduce the risk of muscle injury. http://www.fda.gov/drugs/drugsafety/ucm256581.htm. Published 2011. Accessed May 18, 2012.
19. Betteridge J. Pitavastatin—results from phase III & IV. Atheroscler Suppl. 2010;11(3):8-14.
20. Jones PH, Davidson MH, Stein EA, et al. for STELLAR Study Group. Comparison of the efficacy and safety of rosuvastatin versus atorvastatin, simvastatin, and pravastatin across doses (STELLAR* Trial). Am J Cardiol. 2003;92(2):152-160.
21. Jones P, Kafonek S, Laurora I, Hunninghake D. Comparative dose efficacy study of atorvastatin versus simvastatin, pravastatin, lovastatin, and fluvastatin in patients with hypercholesterolemia (the CURVES study). Am J Cardiol. 1998;81(5):582-587.
22. Grundy SM, Cleeman JI, Merz CN, et al. National Heart, Lung, and Blood Institute; American College of Cardiology Foundation; American Heart Association. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation. 2004;110(2):227-239.
23. Livalo [package insert]. Montgomery, AL: Kowa Pharmaceuticals America, Inc.; 2012.
24. Boekholdt SM, Arsenault BJ, Mora S, et al. Association of LDL cholesterol, non-HDL cholesterol, and apolipoprotein B levels with risk of cardiovascular events among patients treated with statins: a meta-analysis. JAMA. 2012;307(12):1302-1309.
25. US Food and Drug Administration. FDA drug safety communication: Important safety label changes to cholesterol-lowering statin drugs. http://www.fda.gov/Drugs/DrugSafety/ucm293101.htm. Published 2012. Accessed May 18, 2012.
26. Ridker PM, Danielson E, Fonseca FA, et al. JUPITER Study Group. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359(21):2195-2207.
27. Koh KK, Quon MJ, Han SH, Lee Y, Kim SJ, Shin EK. Atorvastatin causes insulin resistance and increases ambient glycemia in hypercholesterolemic patients. J Am Coll Cardiol. 2010;55(12):1209-1216.
28. Preiss D, Seshasai SR, Welsh P, et al. Risk of incident diabetes with intensive-dose compared with moderate-dose statin therapy: a meta-analysis. JAMA. 2011;305(24):2556-2564.
29. Yokote K, Saito Y. CHIBA study investigators. Influence of statins on glucose tolerance in patients with type 2 diabetes mellitus: subanalysis of the collaborative study on hypercholesterolemia drug intervention and their benefits for atherosclerosis prevention (CHIBA study). J Atheroscler Thromb. 2009;16(3):297-298.
30. Preiss D, Sattar N. Statins and the risk of new-onset diabetes: a review of recent evidence. Curr Opin Lipidol. 2011;22(6):460-466.
31. Bottorff MB. Statin safety and drug interactions: clinical implications. Am J Cardiol. 2006;97(8A):27C-31C.
32. Andrus MR. Oral anticoagulant drug interactions with statins: case report of fluvastatin and review of the literature. Pharmacotherapy. 2004;24(2):285-290.
33. US Food and Drug Administration. FDA drug safety communication: Interactions beteween certain HIV or hepatitis C drugs and cholesterol-lowering statin drugs can increase the risk of muscle injury. http://www.fda.gov/Drugs/DrugSafety/ucm293877.htm. Published 2012. Accessed May 18, 2012.
34. Armitage J, Bowman L, Wallendszus K, et al. for Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH) Collaborative Group. Intensive lowering of LDL cholesterol with 80 mg versus 20 mg simvastatin daily in 12,064 survivors of myocardial infarction: a double-blind randomised trial. Lancet. 2010;376(9753):1658-1669.
35. de Lemos JA, Blazing MA, Wiviott SD, et al. for A to Z Investigators. Early intensive vs a delayed conservative simvastatin strategy in patients with acute coronary syndromes: phase Z of the A to Z trial. JAMA. 2004;292(11):1307-1316.
36. Backes JM, Howard PA, Ruisinger JF, Moriarty PM. Does simvastatin cause more myotoxicity compared with other statins? Ann Pharmacother. 2009;43(12):2012-2020.
37. Jacobson TA. Statin safety: lessons from new drug applications for marketed statins. Am J Cardiol. 2006;97(8A):44C-51C.
38. Toth PP, Dayspring TD. Drug safety evaluation of rosuvastatin. Expert Opin Drug Saf. 2011;10(6):969-986.
39. Warrington S, Nagakawa S, Hounslow N. Comparison of the pharmacokinetics of pitavastatin by formulation and ethnic group: an open-label, single-dose, two-way crossover pharmacokinetic study in healthy Caucasian and Japanese men. Clin Drug Investig. 2011;31(10):735-743.
40. Chapman N, Chang CL, Caulfield M, et al. Ethnic variations in lipid-lowering in response to a statin (EVIREST): a substudy of the Anglo-Scandinavian Cardiac Outcomes Trial (ASCOT). Ethn Dis. 2011;21(2):150-157.
41. Albert MA, Glynn RJ, Fonseca FA, et al. Race, ethnicity, and the efficacy of rosuvastatin in primary prevention: the Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER) trial. Am Heart J. 2011;162(1):106-114.
42. Jacobson TA. Toward “pain-free” statin prescribing: clinical algorithm for diagnosis and management of myalgia. Mayo Clin Proc. 2008;83(6):687-700.
1. Bulbulia R, Bowman L, Wallendszus K, et al. for Heart Protection Study Collaborative Group. Effects on 11-year mortality and morbidity of lowering LDL cholesterol with simvastatin for about 5 years in 20,536 high-risk individuals: a randomised controlled trial. Lancet. 2011;378(9808):2013-2020.
2. Sever PS, Chang CL, Gupta AK, Whitehouse A, Poulter NR. for ASCOT Investigators. The Anglo-Scandinavian Cardiac Outcomes Trial: 11-year mortality follow-up of the lipid-lowering arm in the U.K. Eur Heart J. 2011;32(20):2525-2532.
3. Taylor F, Ward K, Moore TH, et al. Statins for the primary prevention of cardiovascular disease. Cochrane Database Syst Rev. 2011;(1):CD004816.-
4. Kawai Y, Sato-Ishida R, Motoyama A, Kajinami K. Place of pitavastatin in the statin armamentarium: promising evidence for a role in diabetes mellitus. Drug Des Devel Ther. 2011;5:283-297.
5. Ose L, Budinski D, Hounslow N, Arneson V. Comparison of pitavastatin with simvastatin in primary hypercholesterolaemia or combined dyslipidaemia. Curr Med Res Opin. 2009;25(11):2755-2764.
6. Gumprecht J, Gosho M, Budinski D, Hounslow N. Comparative long-term efficacy and tolerability of pitavastatin 4 mg and atorvastatin 20-40 mg in patients with type 2 diabetes mellitus and combined (mixed) dyslipidaemia. Diabetes Obes Metab. 2011;13(11):1047-1055.
7. Ose L, Budinski D, Hounslow N, Arneson V. Long-term treatment with pitavastatin is effective and well tolerated by patients with primary hypercholesterolemia or combined dyslipidemia. Atherosclerosis. 2010;210(1):202-208.
8. Teramoto T. Pitavastatin: clinical effects from the LIVES Study. Atheroscler Suppl. 2011;12(3):285-288.
9. Hiro T, Kimura T, Morimoto T, et al. for JAPAN-ACS Investigators. Effect of intensive statin therapy on regression of coronary atherosclerosis in patients with acute coronary syndrome: a multicenter randomized trial evaluated by volumetric intravascular ultrasound using pitavastatin versus atorvastatin (JAPAN-ACS [Japan assessment of pitavastatin and atorvastatin in acute coronary syndrome] study). J Am Coll Cardiol. 2009;54(4):293-302.
10. Saku K, Zhang B, Noda K. and PATROL Trial Investigators. Randomized head-to-head comparison of pitavastatin, atorvastatin, and rosuvastatin for safety and efficacy (quantity and quality of LDL): the PATROL trial. Circ J. 2011;75(6):1493-1505.
11. Yanagi K, Monden T, Ikeda S, Matsumura M, Kasai K. A crossover study of rosuvastatin and pitavastatin in patients with type 2 diabetes. Adv Ther. 2011;28(2):160-171.
12. Toi T, Taguchi I, Yoneda S, et al. Early effect of lipid-lowering therapy with pitavastatin on regression of coronary atherosclerotic plaque. Comparison with atorvastatin. Circ J. 2009;73(8):1466-1472.
13. Maruyama T, Takada M, Nishibori Y, et al. Comparison of preventive effect on cardiovascular events with different statins: the CIRCLE study. Circ J. 2011;75(8):1951-1959.
14. Suh SY, Rha SW, Ahn TH, et al. for LAMIS Investigators. Long-term safety and efficacy of pitavastatin in patients with acute myocardial infarction (from the Livalo Acute Myocardial Infarction Study [LAMIS]). Am J Cardiol. 2011;108(11):1530-1535.
15. Eriksson M, Budinski D, Hounslow N. Comparative efficacy of pitavastatin and simvastatin in high-risk patients: a randomized controlled trial. Adv Ther. 2011;28(9):811-823.
16. Eriksson M, Budinski D, Hounslow N. Long-term efficacy of pitavastatin versus simvastatin. Adv Ther. 2011;28(9):799-810.
17. Stender S, Budinski D, Hounslow N. Pitavastatin demonstrates long-term efficacy, safety and tolerability in elderly patients with primary hypercholesterolaemia or combined (mixed) dyslipidaemia [published online ahead of print January 23, 2012]. Eur J Prev Cardiol. 2012;doi:10.1177/2047487312437326.
18. US Food and Drug Administration. FDA drug safety communication: New restrictions, contraindications, and dose limitations for Zocor (simvastatin) to reduce the risk of muscle injury. http://www.fda.gov/drugs/drugsafety/ucm256581.htm. Published 2011. Accessed May 18, 2012.
19. Betteridge J. Pitavastatin—results from phase III & IV. Atheroscler Suppl. 2010;11(3):8-14.
20. Jones PH, Davidson MH, Stein EA, et al. for STELLAR Study Group. Comparison of the efficacy and safety of rosuvastatin versus atorvastatin, simvastatin, and pravastatin across doses (STELLAR* Trial). Am J Cardiol. 2003;92(2):152-160.
21. Jones P, Kafonek S, Laurora I, Hunninghake D. Comparative dose efficacy study of atorvastatin versus simvastatin, pravastatin, lovastatin, and fluvastatin in patients with hypercholesterolemia (the CURVES study). Am J Cardiol. 1998;81(5):582-587.
22. Grundy SM, Cleeman JI, Merz CN, et al. National Heart, Lung, and Blood Institute; American College of Cardiology Foundation; American Heart Association. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation. 2004;110(2):227-239.
23. Livalo [package insert]. Montgomery, AL: Kowa Pharmaceuticals America, Inc.; 2012.
24. Boekholdt SM, Arsenault BJ, Mora S, et al. Association of LDL cholesterol, non-HDL cholesterol, and apolipoprotein B levels with risk of cardiovascular events among patients treated with statins: a meta-analysis. JAMA. 2012;307(12):1302-1309.
25. US Food and Drug Administration. FDA drug safety communication: Important safety label changes to cholesterol-lowering statin drugs. http://www.fda.gov/Drugs/DrugSafety/ucm293101.htm. Published 2012. Accessed May 18, 2012.
26. Ridker PM, Danielson E, Fonseca FA, et al. JUPITER Study Group. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359(21):2195-2207.
27. Koh KK, Quon MJ, Han SH, Lee Y, Kim SJ, Shin EK. Atorvastatin causes insulin resistance and increases ambient glycemia in hypercholesterolemic patients. J Am Coll Cardiol. 2010;55(12):1209-1216.
28. Preiss D, Seshasai SR, Welsh P, et al. Risk of incident diabetes with intensive-dose compared with moderate-dose statin therapy: a meta-analysis. JAMA. 2011;305(24):2556-2564.
29. Yokote K, Saito Y. CHIBA study investigators. Influence of statins on glucose tolerance in patients with type 2 diabetes mellitus: subanalysis of the collaborative study on hypercholesterolemia drug intervention and their benefits for atherosclerosis prevention (CHIBA study). J Atheroscler Thromb. 2009;16(3):297-298.
30. Preiss D, Sattar N. Statins and the risk of new-onset diabetes: a review of recent evidence. Curr Opin Lipidol. 2011;22(6):460-466.
31. Bottorff MB. Statin safety and drug interactions: clinical implications. Am J Cardiol. 2006;97(8A):27C-31C.
32. Andrus MR. Oral anticoagulant drug interactions with statins: case report of fluvastatin and review of the literature. Pharmacotherapy. 2004;24(2):285-290.
33. US Food and Drug Administration. FDA drug safety communication: Interactions beteween certain HIV or hepatitis C drugs and cholesterol-lowering statin drugs can increase the risk of muscle injury. http://www.fda.gov/Drugs/DrugSafety/ucm293877.htm. Published 2012. Accessed May 18, 2012.
34. Armitage J, Bowman L, Wallendszus K, et al. for Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH) Collaborative Group. Intensive lowering of LDL cholesterol with 80 mg versus 20 mg simvastatin daily in 12,064 survivors of myocardial infarction: a double-blind randomised trial. Lancet. 2010;376(9753):1658-1669.
35. de Lemos JA, Blazing MA, Wiviott SD, et al. for A to Z Investigators. Early intensive vs a delayed conservative simvastatin strategy in patients with acute coronary syndromes: phase Z of the A to Z trial. JAMA. 2004;292(11):1307-1316.
36. Backes JM, Howard PA, Ruisinger JF, Moriarty PM. Does simvastatin cause more myotoxicity compared with other statins? Ann Pharmacother. 2009;43(12):2012-2020.
37. Jacobson TA. Statin safety: lessons from new drug applications for marketed statins. Am J Cardiol. 2006;97(8A):44C-51C.
38. Toth PP, Dayspring TD. Drug safety evaluation of rosuvastatin. Expert Opin Drug Saf. 2011;10(6):969-986.
39. Warrington S, Nagakawa S, Hounslow N. Comparison of the pharmacokinetics of pitavastatin by formulation and ethnic group: an open-label, single-dose, two-way crossover pharmacokinetic study in healthy Caucasian and Japanese men. Clin Drug Investig. 2011;31(10):735-743.
40. Chapman N, Chang CL, Caulfield M, et al. Ethnic variations in lipid-lowering in response to a statin (EVIREST): a substudy of the Anglo-Scandinavian Cardiac Outcomes Trial (ASCOT). Ethn Dis. 2011;21(2):150-157.
41. Albert MA, Glynn RJ, Fonseca FA, et al. Race, ethnicity, and the efficacy of rosuvastatin in primary prevention: the Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER) trial. Am Heart J. 2011;162(1):106-114.
42. Jacobson TA. Toward “pain-free” statin prescribing: clinical algorithm for diagnosis and management of myalgia. Mayo Clin Proc. 2008;83(6):687-700.
Coronary Heart Disease in Men
Managing the Multiple Symptoms of Benign Prostatic Hyperplasia — CME
Managing Type 2 Diabetes in Men
Meeting New Challenges with Antiplatelet Therapy in Primary Care
Dr. Cobble has disclosed that he is on the advisory boards and speakers’ bureaus for AstraZeneca and Bristol-Myers Squibb and is on the speakers’ bureaus for Eli Lilly, Forest, and Kowa.
SUPPORT
This program is sponsored by the PCEC and is supported by funding from AstraZeneca.
The death rate from coronary heart disease (CHD) declined by 59% from 1950 to 1999 in the United States, yet CHD remains a major cause of morbidity and mortality, resulting in an estimated 1.5 million heart attacks in 2011.1 Better recognition and treatment of the 9 modifiable risk factors for CHD identified by the INTERHEART study ( FIGURE 1 ), as well as changes in lifestyle practices, undoubtedly contributed to the decline in CHD mortality, but further improvement is possible.2 Estimates derived from the Second National Health and Nutrition Examination Survey (NHANES II) baseline data and 17-year mortality follow-up data indicate that 45% of CHD deaths in men and 64% in women could be avoided by eliminating 3 major risk factors: elevated total cholesterol (≥240 mg/dL), hypertension, and smoking.3
The evidence indicates that these 3 risk factors are not well controlled. Data from the National Cholesterol Education Program (NCEP) Evaluation ProjecT Utilizing Novel E-Technology (NEPTUNE) II survey and the Lipid Treatment Assessment Project 2 (L-TAP 2), as well as more recent evidence, indicate that many patients do not achieve low-density lipoprotein cholesterol (LDL-C) and triglyceride targets.4-10 Similarly, although there has been significant improvement in blood pressure (BP) control over the past 2 decades, BP is controlled in only half of all hypertensive patients.11,12 Finally, the sharp declines in the prevalence of cigarette smoking seen in the past have slowed in recent years, such that approximately 20% of US adults still smoke cigarettes.13
These trends are a concern since a greater risk factor burden in middle age is associated with poorer quality of life and higher medical costs, as well as a higher incidence of cardiovascular events in older age.1 A recent meta-analysis of 18 cohort studies involving 257,384 adults showed a higher incidence of cardiovascular events in later life with an increasing number of risk factors.14 For example, adults 55 years of age with an optimal risk factor profile (ie, total cholesterol <180 mg/dL, BP <120/80 mm Hg, nonsmoker, nondiabetic) had much lower risks of death from cardiovascular disease (CVD) through the age of 80 years than those with 2 or more risk factors (4.7% vs 29.6% among men, 6.4% vs 20.5% among women). This translates into a relative risk (RR) of cardiovascular death of 6 times for men and 3 times for women without optimal risk profiles. Similar trends were observed for risk of fatal CHD/nonfatal myocardial infarction (MI) (3.6% vs 37.5% among men, <1% vs 18.3% among women). These findings point to the critical importance of modifying multiple risk factors early in adulthood, well in advance of symptoms. However, the Study to Help Improve Early Evaluation and Management of Risk Factors Leading to Diabetes (SHIELD) showed that about half of patients with CHD are not diagnosed until symptoms become apparent, and fewer than one quarter are diagnosed as a result of screening.15
This review focuses on patient assessment and treatment strategies to modify abnormal lipid levels and high BP for primary prevention. Addressing other modifiable risk factors is also important, especially since risk factors such as abdominal obesity impact other risk factors ( FIGURE 1 ). An emphasis is placed on strategies in men, since the prevalence of CHD among patients aged 45 years and older is higher in men than in women ( FIGURE 2 ).16 Furthermore, men experience a first cardiovascular event a decade earlier than women, and a more serious CHD event, such as MI or sudden death, 2 decades earlier.1
FIGURE 1
Modifiable risk factors for myocardial infarction (MI)2

ApoB/ApoA-I, apolipoprotein B/apolipoprotein A-I.
FIGURE 2
Prevalence of heart disease by age and gender16
Assessment
The assessment of CHD risk in men need not be complicated and should be made practical so that it is applied consistently. A family and personal medical history and physical examination combined with laboratory determination of lipid levels and glycosylated hemoglobin can help assess modifiable risk factors. The assessment of CHD risk can be facilitated by using 1 of 2 risk calculators. The Framingham Risk Score [www.framinghamheartstudy.org/risk/gencardio.html] is widely used but may underestimate risk, especially in younger persons or those who appear to be healthy but may have other risk factors for CHD.17-19 The Reynolds Risk Score [www.reynoldsriskscore.org/] includes other risk factors, such as parental history of MI before age 60 years, low levels of apolipoprotein A (apoA), high levels of apolipoprotein B (apoB), and increased levels of high-sensitivity C-reactive protein (hs-CRP).19 The Reynolds Risk Score has been validated in healthy, nondiabetic men.20
The relevance of apolipoprotein levels, particularly apoB, to cardiovascular risk is increasingly appreciated.21 ApoB concentration represents the sum of atherogenic particles found on all atherogenic lipoproteins, including very-low-density lipoprotein, intermediate-density lipoprotein, low-density lipoprotein, and lipoprotein(a) cholesterol, whereas apoA represents the sum of antiatherogenic particles found on high-density lipoprotein cholesterol (HDL-C), the antiatherogenic lipoprotein.22 The ratio of apoB/apoA-I has, in fact, been shown to be a good predictor of cardiovascular events in young men without hypertension and diabetes but with chest pain.23 High-sensitivity C-reactive protein is a sensitive marker of acute inflammation and is associated with coronary risk.24 Measuring hs-CRP is a recommended option to determine enhanced absolute risk in people with an intermediate 10-year CHD risk of 10% to 20%.25
There remains some uncertainty regarding which lipid levels should be measured when screening for cardiovascular risk. The National Cholesterol Education Program Adult Treatment Panel III (NCEP ATP III) advises that total cholesterol, LDL-C, HDL-C, and triglycerides be measured.26 More recent results from The Emerging Risk Factors Collaboration suggest that a more simplified approach may be reasonable.27 Review of data from 68 long-term prospective studies involving 302,430 people without initial vascular disease and 2.79 million person-years of follow-up showed that lipid assessment of vascular risk could be accomplished by measuring either total cholesterol and HDL-C levels or apolipoprotein levels; measuring the triglyceride level was of no added benefit in assessing vascular risk. In addition, fasting and nonfasting lipid levels were found to be of similar value in assessing risk. Other evidence shows that the combination of a triglyceride level ≥178 mg/dL and waist circumference ≥35.4 inches—the hypertriglyceridemic waist phenotype—is as discriminatory a screening tool as the NCEP ATP III guidelines to identify individuals at increased cardiometabolic risk.28 The use of more comprehensive lipoprotein and apolipoprotein testing, as well as noninvasive imaging, may have value in future cardiovascular risk assessment.
Treatment
The main goal of treatment in persons with 1 or more modifiable risk factors is to prevent an incident or primary cardiovascular event. Treatment strategies to achieve this goal in men and women are the same. Prevention of recurrent or secondary events will not be addressed here.
Lipids
Numerous clinical trials, such as the Air Force/Texas Coronary Atherosclerosis Prevention Study (AFCAPS/TexCAPS),29 Anglo-Scandinavian Cardiac Outcomes Trial-Lipid Lowering Arm (ASCOT-LLA),30 and West of Scotland Coronary Prevention Study (WOSCOPS),31 definitively established the benefit of cardiovascular risk reduction with lipid-lowering treatment, particularly LDL-C-lowering treatment. Low-density lipoprotein cholesterol is the principal lipid target in most patients, with the treatment goal based on the presence of additional risk factors.32 Discussion of treatments for low HDL-C and elevated triglyceride levels is beyond the scope of this review but is expected to be included in the NCEP ATP IV guidelines scheduled for release later in 2012.
The Justification for the Use of Statins in Prevention: An Intervention Trial Evaluating Rosuvastatin (JUPITER) also established significant benefits of statin therapy in primary prevention, compared with placebo, in persons with normal or modestly elevated LDL-C (<130 mg/dL) and elevated hs-CRP (≥2 mg/L).33 Rates of the primary end point (MI, stroke, arterial revascularization, hospitalization for unstable angina, or cardiovascular death) were 0.77 and 1.36 per 100 person-years of follow-up in the rosuvastatin and placebo groups, respectively (hazard ratio [HR], 0.56; 95% CI, 0.46-0.69; P < .00001). Further analysis showed that patients who achieved LDL-C <70 mg/dL had a 55% lower rate of vascular events compared with placebo.34
Results from large primary prevention clinical trials such as JUPITER have led to recommendations over the past decade or so for progressively lower LDL-C goals. A meta-analysis of 25 large clinical trials involving 155,613 subjects showed that for every 25 mg/dL reduction in LDL-C, the RR for several cardiovascular outcomes was reduced: vascular mortality, 0.89; major vascular events, 0.86; major coronary events, 0.84; and stroke, 0.90. Put differently, there was a 20% reduction in major coronary events for every 39 mg/dL LDL-C reduction.35
Recent trials support the benefits of intensive high-dose statin therapy in greatly reducing lipid levels, with associated benefits in terms of cardiovascular events. A meta-analysis of 7 trials involving 50,972 high-risk patients with a mean follow-up of 3.1 years showed significant reductions in the risk for cardiovascular events with intensive statin therapy. Those who achieved LDL-C <82 mg/dL with intensive statin therapy had lower cardiovascular risks compared with those with LDL-C ≥82 mg/dL: stroke, odds ratio (OR): 0.80; major coronary events, OR: 0.74; and CVD or CHD death, OR: 0.84.36 Significantly higher liver enzyme abnormalities were observed in patients treated with high-dose statin therapy. [See also Addressing Key Questions with Statin Therapy in this supplement.] The benefits of intensive statin therapy on the progression of coronary atherosclerosis have also been investigated. The Study of Coronary Atheroma by Intravascular Ultrasound: Effect of Rosuvastatin versus Atorvastatin (SATURN) by Nicholls et al37 included patients (N = 1039) with documented coronary vessel stenosis of at least 20% and a target vessel for imaging with less than 50% obstruction. Patients received either atorvastatin 80 mg daily or rosu-vastatin 40 mg daily for 104 weeks. In the rosuvastatin group, end-of-study LDL-C levels were lower (62.6 vs 70.2 mg/dL; P < .001) and HDL-C levels higher (50.4 vs 48.6 mg/dL; P = .01) compared with the atorvastatin group, respectively. The percent atheroma volume decreased by 1.22% with rosuvastatin and 0.99% with atorvastatin (P = .17). The normalized total atheroma volume decreased 6.39 mm3 with rosuvastatin and 4.42 mm3 with atorvastatin (P = .01). Atheroma regression was induced in the majority of patients in both groups.
Further support for treating with statin doses higher than those recommended for initial therapy comes from a prospective trial involving 1337 consecutive patients followed over a median of 33 months.10 Although 83% of these patients were on statin therapy, only 51% had an LDL-C <100 mg/dL, and only 15% of the very high-risk patients (n = 941) had an LDL-C <70 mg/dL. The use of intensive statin therapy was associated with a 12-fold higher possibility of achieving an LDL-C <70 mg/dL. Very high-risk patients who achieved an LDL-C <70 mg/dL had a significantly lower risk of all cardiovascular events (HR, 0.34; P = .003).
Blood pressure
As with dyslipidemia, the cardiovascular benefits of lowering elevated BP are well established. While the usual BP goal is <140/90 mm Hg, in those with hypertension and concomitant diabetes or renal disease, the goal is <130/80 mm Hg.38 It is not clear how best to achieve these goals, but therapy must be individualized based on patient comorbidities and drug side effects as recommended in current guidelines.38-40 With these guidelines as a basis, a simplified ABCD approach can be considered in selecting initial antihypertensive therapy ( FIGURE 3 ).
FIGURE 3
ABCD approach to initial antihypertensive therapy38-40
ACE-I, angiotensin converting enzyme inhibitor; ARB, angiotensin receptor blocker; MI, myocardial infarction. Monotherapy, however, does not result in BP control in most patients. As shown by the Antihypertensive Lipid-Lowering Treatment to Prevent Heart Attacks Trial (ALLHAT), BP control typically requires at least 2 different classes of drugs, with 3 or more drugs required in about 1 in 6 patients within 3 years and 1 in 4 patients within 5 years. A higher percentage of patients with diabetes mellitus or kidney impairment (creatinine ≥1.5 mg/dL) require 3 or more antihypertensive drugs after 5 years (33% and 40%, respectively).41
Several meta-analyses have been conducted recently to assess the magnitude of BP (systolic/diastolic) lowering in the different classes of antihypertensive drugs. While these meta-analyses have important limitations, such as differences in study design and the lack of a clear description of outcomes, some general impressions can be made. In 1 meta-analysis, thiazide diuretics were found to lower BP by 6/3 and 8/4 mm Hg at doses of 1 and 2 times the recommended starting dose, respectively. A BP-lowering effect of 6/3 mm Hg was observed with starting doses of loop diuretics.42 Another meta-analysis failed to find a statistically or clinically significant BP-lowering effect with potassium-sparing diuretics at low doses.43 For spironolactone, a review of 5 crossover studies found a reduction in BP of 21/7 mm Hg. In this review, daily doses of 25 to 100 mg were found to provide the best balance between BP reduction and safety and tolerability.44
Several meta-analyses of angiotensin receptor blockers (ARBs) have found BP reductions to be similar among the various ARB drugs. Generally, at maximum recommended doses, a BP reduction of 8/5 mm Hg is observed with these drugs, except for losartan, which produces a smaller BP reduction.45-49 Heran et al45 found a BP reduction of 12/7 mm Hg among the ARBs 1 to 12 hours after the dose was taken. When cost per quality-adjusted life-year gained was considered, 1 meta-analysis found that the slightly greater BP reduction with candesartan compared with losartan was not cost-effective.46 However, other benefits of candesartan compared with losartan therapy (eg, lower risk for cardiovascular disease, heart failure, dysrhythmias, and peripheral artery disease) should be considered.50 Adverse events were generally found to be similar among the ARBs.
No differences in BP lowering were observed among 92 trials of 14 different angiotensin-converting enzyme inhibitors. As a class, these drugs were found to produce a reduction in BP of 8/5 mm Hg.51
Because of the modest BP-lowering effects of each of the antihypertensive drugs currently available, consideration should be given to starting antihypertensive therapy with 2 agents for patients with stage 2 hypertension (ie, BP ≥160/100 mm Hg).
Summary
Elimination of key risk factors such as dyslipidemia and hypertension is important for reducing cardiovascular events later in life. A medical history, physical examination, and laboratory determination of lipid and glycosylated hemoglobin levels provide a good assessment of cardiovascular risk. A statin is first-line therapy for reducing LDL-C, which is the primary lipid target in most patients. High-dose statin therapy may be required to reach desired target levels. The choice of initial antihypertensive therapy is based on patient comorbidities and drug side effects; however, most patients require combination antihypertensive therapy to reach goal. The combination of this multifactorial risk approach along with smoking cessation and modification of other risk factors should complement current and future cardiovascular care for men.
1. Roger VL, Go AS, Lloyd-Jones DM, et al. Heart disease and stroke statistics—2012 update: a report from the American Heart Association. Circulation. 2012;125(1):e2-e220.
2. Yusuf S, Hawken S, Ounpuu S, et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control study. Lancet. 2004;364(9438):937-952.
3. Mensah GA, Brown DW, Croft JB, Greenlund KJ. Major coronary risk factors and death from coronary heart disease: baseline and follow-up mortality data from the Second National Health and Nutrition Examination Survey (NHANES II). Am J Prev Med. 2005;29(5 suppl 1):68-74.
4. Davidson MH, Maki KC, Pearson TA, et al. Results of the National Cholesterol Education (NCEP) Program Evaluation ProjecT Utilizing Novel E-Technology (NEPTUNE) II survey and implications for treatment under the recent NCEP Writing Group recommendations. Am J Cardiol. 2005;96(4):556-563.
5. Waters DD, Brotons C, Chiang CW, et al. Lipid treatment assessment project 2: a multinational survey to evaluate the proportion of patients achieving low-density lipoprotein cholesterol goals. Circulation. 2009;120(1):28-34.
6. Vande Griend JP, Saseen JJ. Low-density lipoprotein cholesterol goal attainment in high-risk family medicine patients. J Clin Lipidol. 2009;3(3):195-200.
7. Barham AH, Goff DC, Jr, Chen H, et al. Appropriateness of cholesterol management in primary care by sex and level of cardiovascular risk. Prev Cardiol. 2009;12(2):95-101.
8. Kitkungvan D, Lynn Fillipon NM, Dani SS, Downey BC. Low-density lipoprotein cholesterol target achievement in patients at high risk for coronary heart disease. J Clin Lipidol. 2010;4(4):293-297.
9. Leiter LA, Lundman P, da Silva PM, et al. Persistent lipid abnormalities in statin- treated patients with diabetes mellitus in Europe and Canada: results of the Dyslipidaemia International Study. Diabet Med. 2011;28(11):1343-1351.
10. Rallidis LS, Kotakos C, Sourides V, et al. Attainment of optional low-density lipoprotein cholesterol goal of less than 70 mg/dl and impact on prognosis of very high risk stable coronary patients: a 3-year follow-up. Expert Opin Pharmacother. 2011;12(10):1481-1489.
11. Centers for Disease Control and Prevention. High blood pressure facts. http://www.cdc.gov/bloodpressure/facts.htm. Published 2012. Accessed May 2, 2012.
12. Egan BM, Zhao Y, Axon RN. US trends in prevalence, awareness, treatment, and control of hypertension, 1988-2008. JAMA. 2010;303(20):2043-2050.
13. McClave A, Rock V, Thorne S, Malarcher A. State-specific prevalence of cigarette smoking and smokeless tobacco use among adults—United States, 2009. MMWR Morb Mortal Wkly Rep. 2010;59(43):1400-1406.
14. Berry JD, Dyer A, Cai X, et al. Lifetime risks of cardiovascular disease. N Engl J Med. 2012;366(4):321-329.
15. Lewis SJ, Fox KM, Grandy S. Shield Study Group. Self-reported diagnosis of heart disease: results from the SHIELD study. Int J Clin Pract. 2009;63(5):726-734.
16. National Center for Health Statistics. Health, United States, 2010: with special feature on death and dying. http://www.cdc.gov/nchs/data/hus/hus10.pdf. Published 2011. Accessed May 2, 2012.
17. Hemann BA, Bimson WF, Taylor AJ. The Framingham Risk Score: an appraisal of its benefits and limitations. Am Heart Hosp J. 2007;5(2):91-96.
18. Karim R, Hodis HN, Detrano R, Liu CR, Liu CH, Mack WJ. Relation of Framingham risk score to subclinical atherosclerosis evaluated across three arterial sites. Am J Cardiol. 2008;102(7):825-830.
19. Ridker PM, Buring JE, Rifai N, Cook NR. Development and validation of improved algorithms for the assessment of global cardiovascular risk in women: the Reynolds Risk Score. JAMA. 2007;297(6):611-619.
20. Ridker PM, Paynter NP, Rifai N, Gaziano JM, Cook NR. C-reactive protein and parental history improve global cardiovascular risk prediction: the Reynolds Risk Score for men. Circulation. 2008;118(22):2243-2251.
21. Brunzell JD, Davidson M, Furberg CD, et al. Lipoprotein management in patients with cardiometabolic risk: consensus conference report from the American Diabetes Association and the American College of Cardiology Foundation. J Am Coll Cardiol. 2008;51(15):1512-1524.
22. Fruchart JC, Sacks FM, Hermans MP, et al. The Residual Risk Reduction Initiative: a call to action to reduce residual vascular risk in dyslipidaemic patient. Diab Vasc Dis Res. 2008;5(4):319-335.
23. Koz C, Baysan O, Hasimi A, et al. Conventional and non-conventional coronary risk factors in male premature coronary artery disease patients already having a low Framingham risk score. Acta Cardiol. 2008;63(5):623-628.
24. Koenig W, Khuseyinova N. Biomarkers of atherosclerotic plaque instability and rupture. Arterioscler Thromb Vasc Biol. 2007;27(1):15-26.
25. Pearson TA, Mensah GA, Alexander RW, et al. Markers of inflammation and cardiovascular disease: application to clinical and public health practice: a statement for healthcare professionals from the Centers for Disease Control and Prevention and the American Heart Association. Circulation. 2003;107(3):499-511.
26. Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation. 2002;106(25):3143-3421.
27. Di Angelantonio E, Sarwar N, Perry P, et al. Emerging Risk Factors Collaboration. Major lipids, apolipoproteins, and risk of vascular disease. JAMA. 2009;302(18):1993-2000.
28. Blackburn P, Lemieux I, Alméras N, et al. The hypertriglyceridemic waist phenotype versus the National Cholesterol Education Program-Adult Treatment Panel III and International Diabetes Federation clinical criteria to identify high-risk men with an altered cardiometabolic risk profile. Metabolism. 2009;58(8):1123-1130.
29. Downs JR, Clearfield M, Weis S, et al. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA. 1998;279(20):1615-1622.
30. Sever PS, Dahlöf B, Poulter NR, et al. Prevention of coronary and stroke events with atorvastatin in hypertensive patients who have average or lower-than-average cholesterol concentrations, in the Anglo-Scandinavian Cardiac Outcomes Trial—Lipid Lowering Arm (ASCOT-LLA): a multicentre randomised controlled trial. Lancet. 2003;361(9364):1149-1158.
31. Shepherd J, Cobbe SM, Ford I, et al. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. West of Scotland Coronary Prevention Study Group. N Engl J Med. 1995;333(20):1301-1307.
32. Grundy SM, Cleeman JI, Merz CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation. 2004;110(2):227-239.
33. Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359(21):2195-2207.
34. Ridker PM, Danielson E, Fonseca FA, et al. Reduction in C-reactive protein and LDL cholesterol and cardiovascular event rates after initiation of rosuvastatin: a prospective study of the JUPITER trial. Lancet. 2009;373(9670):1175-1182.
35. Delahoy PJ, Magliano DJ, Webb K, Grobler M, Liew D. The relationship between reduction in low-density lipoprotein cholesterol by statins and reduction in risk of cardiovascular outcomes: an updated meta-analysis. Clin Ther. 2009;31(2):236-244.
36. Chan DK, O’Rourke F, Shen Q, Mak JC, Hung WT. Meta-analysis of the cardiovascular benefits of intensive lipid lowering with statins. Acta Neurol Scand. 2011;124(3):188-195.
37. Nicholls SJ, Ballantyne CM, Barter PJ, et al. Effect of two intensive statin regimens on progression of coronary disease. N Engl J Med. 2011;365(22):2078-2087.
38. Chobanian AV, Bakris GL, Black HR, et al. The seventh report of the Joint National Committee on prevention, detection, evaluation, and treatment of high blood pressure: the JNC 7 report. JAMA. 2003;289(19):2560-2572.
39. Whitworth JA. World Health Organization, International Society of Hypertension Writing Group. 2003 World Health Organization (WHO)/International Society of Hypertension (ISH) statement on management of hypertension. J Hypertens. 2003;21(11):1983-1992.
40. National Clinical Guideline Centre. Hypertension: the clinical management of primary hypertension in adults. http://www.nice.org.uk/nicemedia/live/12167/54727/54727.pdf. Published 2011. Accessed May 2, 2012.
41. Cushman WC, Ford CE, Cutler JA, et al. Success and predictors of blood pressure control in diverse North American settings: the antihypertensive and lipid-lowering treatment to prevent heart attack trial (ALLHAT). J Clin Hypertens (Greenwich). 2002;4(6):393-404.
42. Chen JM, Heran BS, Wright JM. Blood pressure lowering efficacy of diuretics as second-line therapy for primary hypertension. Cochrane Database Syst Rev. 2009;(4):CD007187.-
43. Heran BS, Chen JM, Wang JJ, Wright JM. Blood pressure lowering efficacy of potassium-sparing diuretics (that block the epithelial sodium channel) for primary hypertension. Cochrane Database Syst Rev. 2010;(1):CD008167.-
44. Batterink J, Stabler SN, Tejani AM, Fowkes CT. Spironolactone for hypertension. Cochrane Database Syst Rev. 2010;(8):CD008169.-
45. Heran BS, Wong MM, Heran IK, Wright JM. Blood pressure lowering efficacy of angiotensin receptor blockers for primary hypertension. Cochrane Database Syst Rev. 2008;(4):CD003822.-
46. Grosso AM, Bodalia PN, Macallister RJ, Hingorani AD, Moon JC, Scott MA. Comparative clinical-and cost-effectiveness of candesartan and losartan in the management of hypertension and heart failure: a systematic review, meta-and cost-utility analysis. Int J Clin Pract. 2011;65(3):253-263.
47. Nixon RM, Müller E, Lowy A, Falvey H. Valsartan vs. other angiotensin II receptor blockers in the treatment of hypertension: a meta-analytical approach. Int J Clin Pract. 2009;63(5):766-775.
48. Zhenfeng Z, Huilan S, Junya J, Dong L, Shan L. A systematic review and meta-analysis of candesartan and losartan in the management of essential hypertension. J Renin Angiotensin Aldosterone Syst. 2011;12(3):365-374.
49. Zheng Z, Lin S, Shi H. A systematic review and meta-analysis of telmisartan versus valsartan in the management of essential hypertension. J Clin Hypertens (Greenwich). 2010;12(6):414-421.
50. Kjeldsen SE, Stålhammar J, Hasvole P, Bodegard J, Olsson U, Russell D. Effects of losartan vs candesartan in reducing cardiovascular events in the primary treatment of hypertension. J Hum Hypertens. 2010;24(4):263-273.
51. Heran BS, Wong MM, Heran IK, Wright JM. Blood pressure lowering efficacy of angiotensin converting enzyme (ACE) inhibitors for primary hypertension. Cochrane Database Syst Rev. 2008;(4):CD003823.-
Managing the Multiple Symptoms of Benign Prostatic Hyperplasia — CME
Managing Type 2 Diabetes in Men
Meeting New Challenges with Antiplatelet Therapy in Primary Care
Dr. Cobble has disclosed that he is on the advisory boards and speakers’ bureaus for AstraZeneca and Bristol-Myers Squibb and is on the speakers’ bureaus for Eli Lilly, Forest, and Kowa.
SUPPORT
This program is sponsored by the PCEC and is supported by funding from AstraZeneca.
The death rate from coronary heart disease (CHD) declined by 59% from 1950 to 1999 in the United States, yet CHD remains a major cause of morbidity and mortality, resulting in an estimated 1.5 million heart attacks in 2011.1 Better recognition and treatment of the 9 modifiable risk factors for CHD identified by the INTERHEART study ( FIGURE 1 ), as well as changes in lifestyle practices, undoubtedly contributed to the decline in CHD mortality, but further improvement is possible.2 Estimates derived from the Second National Health and Nutrition Examination Survey (NHANES II) baseline data and 17-year mortality follow-up data indicate that 45% of CHD deaths in men and 64% in women could be avoided by eliminating 3 major risk factors: elevated total cholesterol (≥240 mg/dL), hypertension, and smoking.3
The evidence indicates that these 3 risk factors are not well controlled. Data from the National Cholesterol Education Program (NCEP) Evaluation ProjecT Utilizing Novel E-Technology (NEPTUNE) II survey and the Lipid Treatment Assessment Project 2 (L-TAP 2), as well as more recent evidence, indicate that many patients do not achieve low-density lipoprotein cholesterol (LDL-C) and triglyceride targets.4-10 Similarly, although there has been significant improvement in blood pressure (BP) control over the past 2 decades, BP is controlled in only half of all hypertensive patients.11,12 Finally, the sharp declines in the prevalence of cigarette smoking seen in the past have slowed in recent years, such that approximately 20% of US adults still smoke cigarettes.13
These trends are a concern since a greater risk factor burden in middle age is associated with poorer quality of life and higher medical costs, as well as a higher incidence of cardiovascular events in older age.1 A recent meta-analysis of 18 cohort studies involving 257,384 adults showed a higher incidence of cardiovascular events in later life with an increasing number of risk factors.14 For example, adults 55 years of age with an optimal risk factor profile (ie, total cholesterol <180 mg/dL, BP <120/80 mm Hg, nonsmoker, nondiabetic) had much lower risks of death from cardiovascular disease (CVD) through the age of 80 years than those with 2 or more risk factors (4.7% vs 29.6% among men, 6.4% vs 20.5% among women). This translates into a relative risk (RR) of cardiovascular death of 6 times for men and 3 times for women without optimal risk profiles. Similar trends were observed for risk of fatal CHD/nonfatal myocardial infarction (MI) (3.6% vs 37.5% among men, <1% vs 18.3% among women). These findings point to the critical importance of modifying multiple risk factors early in adulthood, well in advance of symptoms. However, the Study to Help Improve Early Evaluation and Management of Risk Factors Leading to Diabetes (SHIELD) showed that about half of patients with CHD are not diagnosed until symptoms become apparent, and fewer than one quarter are diagnosed as a result of screening.15
This review focuses on patient assessment and treatment strategies to modify abnormal lipid levels and high BP for primary prevention. Addressing other modifiable risk factors is also important, especially since risk factors such as abdominal obesity impact other risk factors ( FIGURE 1 ). An emphasis is placed on strategies in men, since the prevalence of CHD among patients aged 45 years and older is higher in men than in women ( FIGURE 2 ).16 Furthermore, men experience a first cardiovascular event a decade earlier than women, and a more serious CHD event, such as MI or sudden death, 2 decades earlier.1
FIGURE 1
Modifiable risk factors for myocardial infarction (MI)2
ApoB/ApoA-I, apolipoprotein B/apolipoprotein A-I.
FIGURE 2
Prevalence of heart disease by age and gender16
Assessment
The assessment of CHD risk in men need not be complicated and should be made practical so that it is applied consistently. A family and personal medical history and physical examination combined with laboratory determination of lipid levels and glycosylated hemoglobin can help assess modifiable risk factors. The assessment of CHD risk can be facilitated by using 1 of 2 risk calculators. The Framingham Risk Score [www.framinghamheartstudy.org/risk/gencardio.html] is widely used but may underestimate risk, especially in younger persons or those who appear to be healthy but may have other risk factors for CHD.17-19 The Reynolds Risk Score [www.reynoldsriskscore.org/] includes other risk factors, such as parental history of MI before age 60 years, low levels of apolipoprotein A (apoA), high levels of apolipoprotein B (apoB), and increased levels of high-sensitivity C-reactive protein (hs-CRP).19 The Reynolds Risk Score has been validated in healthy, nondiabetic men.20
The relevance of apolipoprotein levels, particularly apoB, to cardiovascular risk is increasingly appreciated.21 ApoB concentration represents the sum of atherogenic particles found on all atherogenic lipoproteins, including very-low-density lipoprotein, intermediate-density lipoprotein, low-density lipoprotein, and lipoprotein(a) cholesterol, whereas apoA represents the sum of antiatherogenic particles found on high-density lipoprotein cholesterol (HDL-C), the antiatherogenic lipoprotein.22 The ratio of apoB/apoA-I has, in fact, been shown to be a good predictor of cardiovascular events in young men without hypertension and diabetes but with chest pain.23 High-sensitivity C-reactive protein is a sensitive marker of acute inflammation and is associated with coronary risk.24 Measuring hs-CRP is a recommended option to determine enhanced absolute risk in people with an intermediate 10-year CHD risk of 10% to 20%.25
There remains some uncertainty regarding which lipid levels should be measured when screening for cardiovascular risk. The National Cholesterol Education Program Adult Treatment Panel III (NCEP ATP III) advises that total cholesterol, LDL-C, HDL-C, and triglycerides be measured.26 More recent results from The Emerging Risk Factors Collaboration suggest that a more simplified approach may be reasonable.27 Review of data from 68 long-term prospective studies involving 302,430 people without initial vascular disease and 2.79 million person-years of follow-up showed that lipid assessment of vascular risk could be accomplished by measuring either total cholesterol and HDL-C levels or apolipoprotein levels; measuring the triglyceride level was of no added benefit in assessing vascular risk. In addition, fasting and nonfasting lipid levels were found to be of similar value in assessing risk. Other evidence shows that the combination of a triglyceride level ≥178 mg/dL and waist circumference ≥35.4 inches—the hypertriglyceridemic waist phenotype—is as discriminatory a screening tool as the NCEP ATP III guidelines to identify individuals at increased cardiometabolic risk.28 The use of more comprehensive lipoprotein and apolipoprotein testing, as well as noninvasive imaging, may have value in future cardiovascular risk assessment.
Treatment
The main goal of treatment in persons with 1 or more modifiable risk factors is to prevent an incident or primary cardiovascular event. Treatment strategies to achieve this goal in men and women are the same. Prevention of recurrent or secondary events will not be addressed here.
Lipids
Numerous clinical trials, such as the Air Force/Texas Coronary Atherosclerosis Prevention Study (AFCAPS/TexCAPS),29 Anglo-Scandinavian Cardiac Outcomes Trial-Lipid Lowering Arm (ASCOT-LLA),30 and West of Scotland Coronary Prevention Study (WOSCOPS),31 definitively established the benefit of cardiovascular risk reduction with lipid-lowering treatment, particularly LDL-C-lowering treatment. Low-density lipoprotein cholesterol is the principal lipid target in most patients, with the treatment goal based on the presence of additional risk factors.32 Discussion of treatments for low HDL-C and elevated triglyceride levels is beyond the scope of this review but is expected to be included in the NCEP ATP IV guidelines scheduled for release later in 2012.
The Justification for the Use of Statins in Prevention: An Intervention Trial Evaluating Rosuvastatin (JUPITER) also established significant benefits of statin therapy in primary prevention, compared with placebo, in persons with normal or modestly elevated LDL-C (<130 mg/dL) and elevated hs-CRP (≥2 mg/L).33 Rates of the primary end point (MI, stroke, arterial revascularization, hospitalization for unstable angina, or cardiovascular death) were 0.77 and 1.36 per 100 person-years of follow-up in the rosuvastatin and placebo groups, respectively (hazard ratio [HR], 0.56; 95% CI, 0.46-0.69; P < .00001). Further analysis showed that patients who achieved LDL-C <70 mg/dL had a 55% lower rate of vascular events compared with placebo.34
Results from large primary prevention clinical trials such as JUPITER have led to recommendations over the past decade or so for progressively lower LDL-C goals. A meta-analysis of 25 large clinical trials involving 155,613 subjects showed that for every 25 mg/dL reduction in LDL-C, the RR for several cardiovascular outcomes was reduced: vascular mortality, 0.89; major vascular events, 0.86; major coronary events, 0.84; and stroke, 0.90. Put differently, there was a 20% reduction in major coronary events for every 39 mg/dL LDL-C reduction.35
Recent trials support the benefits of intensive high-dose statin therapy in greatly reducing lipid levels, with associated benefits in terms of cardiovascular events. A meta-analysis of 7 trials involving 50,972 high-risk patients with a mean follow-up of 3.1 years showed significant reductions in the risk for cardiovascular events with intensive statin therapy. Those who achieved LDL-C <82 mg/dL with intensive statin therapy had lower cardiovascular risks compared with those with LDL-C ≥82 mg/dL: stroke, odds ratio (OR): 0.80; major coronary events, OR: 0.74; and CVD or CHD death, OR: 0.84.36 Significantly higher liver enzyme abnormalities were observed in patients treated with high-dose statin therapy. [See also Addressing Key Questions with Statin Therapy in this supplement.] The benefits of intensive statin therapy on the progression of coronary atherosclerosis have also been investigated. The Study of Coronary Atheroma by Intravascular Ultrasound: Effect of Rosuvastatin versus Atorvastatin (SATURN) by Nicholls et al37 included patients (N = 1039) with documented coronary vessel stenosis of at least 20% and a target vessel for imaging with less than 50% obstruction. Patients received either atorvastatin 80 mg daily or rosu-vastatin 40 mg daily for 104 weeks. In the rosuvastatin group, end-of-study LDL-C levels were lower (62.6 vs 70.2 mg/dL; P < .001) and HDL-C levels higher (50.4 vs 48.6 mg/dL; P = .01) compared with the atorvastatin group, respectively. The percent atheroma volume decreased by 1.22% with rosuvastatin and 0.99% with atorvastatin (P = .17). The normalized total atheroma volume decreased 6.39 mm3 with rosuvastatin and 4.42 mm3 with atorvastatin (P = .01). Atheroma regression was induced in the majority of patients in both groups.
Further support for treating with statin doses higher than those recommended for initial therapy comes from a prospective trial involving 1337 consecutive patients followed over a median of 33 months.10 Although 83% of these patients were on statin therapy, only 51% had an LDL-C <100 mg/dL, and only 15% of the very high-risk patients (n = 941) had an LDL-C <70 mg/dL. The use of intensive statin therapy was associated with a 12-fold higher possibility of achieving an LDL-C <70 mg/dL. Very high-risk patients who achieved an LDL-C <70 mg/dL had a significantly lower risk of all cardiovascular events (HR, 0.34; P = .003).
Blood pressure
As with dyslipidemia, the cardiovascular benefits of lowering elevated BP are well established. While the usual BP goal is <140/90 mm Hg, in those with hypertension and concomitant diabetes or renal disease, the goal is <130/80 mm Hg.38 It is not clear how best to achieve these goals, but therapy must be individualized based on patient comorbidities and drug side effects as recommended in current guidelines.38-40 With these guidelines as a basis, a simplified ABCD approach can be considered in selecting initial antihypertensive therapy ( FIGURE 3 ).
FIGURE 3
ABCD approach to initial antihypertensive therapy38-40
ACE-I, angiotensin converting enzyme inhibitor; ARB, angiotensin receptor blocker; MI, myocardial infarction. Monotherapy, however, does not result in BP control in most patients. As shown by the Antihypertensive Lipid-Lowering Treatment to Prevent Heart Attacks Trial (ALLHAT), BP control typically requires at least 2 different classes of drugs, with 3 or more drugs required in about 1 in 6 patients within 3 years and 1 in 4 patients within 5 years. A higher percentage of patients with diabetes mellitus or kidney impairment (creatinine ≥1.5 mg/dL) require 3 or more antihypertensive drugs after 5 years (33% and 40%, respectively).41
Several meta-analyses have been conducted recently to assess the magnitude of BP (systolic/diastolic) lowering in the different classes of antihypertensive drugs. While these meta-analyses have important limitations, such as differences in study design and the lack of a clear description of outcomes, some general impressions can be made. In 1 meta-analysis, thiazide diuretics were found to lower BP by 6/3 and 8/4 mm Hg at doses of 1 and 2 times the recommended starting dose, respectively. A BP-lowering effect of 6/3 mm Hg was observed with starting doses of loop diuretics.42 Another meta-analysis failed to find a statistically or clinically significant BP-lowering effect with potassium-sparing diuretics at low doses.43 For spironolactone, a review of 5 crossover studies found a reduction in BP of 21/7 mm Hg. In this review, daily doses of 25 to 100 mg were found to provide the best balance between BP reduction and safety and tolerability.44
Several meta-analyses of angiotensin receptor blockers (ARBs) have found BP reductions to be similar among the various ARB drugs. Generally, at maximum recommended doses, a BP reduction of 8/5 mm Hg is observed with these drugs, except for losartan, which produces a smaller BP reduction.45-49 Heran et al45 found a BP reduction of 12/7 mm Hg among the ARBs 1 to 12 hours after the dose was taken. When cost per quality-adjusted life-year gained was considered, 1 meta-analysis found that the slightly greater BP reduction with candesartan compared with losartan was not cost-effective.46 However, other benefits of candesartan compared with losartan therapy (eg, lower risk for cardiovascular disease, heart failure, dysrhythmias, and peripheral artery disease) should be considered.50 Adverse events were generally found to be similar among the ARBs.
No differences in BP lowering were observed among 92 trials of 14 different angiotensin-converting enzyme inhibitors. As a class, these drugs were found to produce a reduction in BP of 8/5 mm Hg.51
Because of the modest BP-lowering effects of each of the antihypertensive drugs currently available, consideration should be given to starting antihypertensive therapy with 2 agents for patients with stage 2 hypertension (ie, BP ≥160/100 mm Hg).
Summary
Elimination of key risk factors such as dyslipidemia and hypertension is important for reducing cardiovascular events later in life. A medical history, physical examination, and laboratory determination of lipid and glycosylated hemoglobin levels provide a good assessment of cardiovascular risk. A statin is first-line therapy for reducing LDL-C, which is the primary lipid target in most patients. High-dose statin therapy may be required to reach desired target levels. The choice of initial antihypertensive therapy is based on patient comorbidities and drug side effects; however, most patients require combination antihypertensive therapy to reach goal. The combination of this multifactorial risk approach along with smoking cessation and modification of other risk factors should complement current and future cardiovascular care for men.
Managing the Multiple Symptoms of Benign Prostatic Hyperplasia — CME
Managing Type 2 Diabetes in Men
Meeting New Challenges with Antiplatelet Therapy in Primary Care
Dr. Cobble has disclosed that he is on the advisory boards and speakers’ bureaus for AstraZeneca and Bristol-Myers Squibb and is on the speakers’ bureaus for Eli Lilly, Forest, and Kowa.
SUPPORT
This program is sponsored by the PCEC and is supported by funding from AstraZeneca.
The death rate from coronary heart disease (CHD) declined by 59% from 1950 to 1999 in the United States, yet CHD remains a major cause of morbidity and mortality, resulting in an estimated 1.5 million heart attacks in 2011.1 Better recognition and treatment of the 9 modifiable risk factors for CHD identified by the INTERHEART study ( FIGURE 1 ), as well as changes in lifestyle practices, undoubtedly contributed to the decline in CHD mortality, but further improvement is possible.2 Estimates derived from the Second National Health and Nutrition Examination Survey (NHANES II) baseline data and 17-year mortality follow-up data indicate that 45% of CHD deaths in men and 64% in women could be avoided by eliminating 3 major risk factors: elevated total cholesterol (≥240 mg/dL), hypertension, and smoking.3
The evidence indicates that these 3 risk factors are not well controlled. Data from the National Cholesterol Education Program (NCEP) Evaluation ProjecT Utilizing Novel E-Technology (NEPTUNE) II survey and the Lipid Treatment Assessment Project 2 (L-TAP 2), as well as more recent evidence, indicate that many patients do not achieve low-density lipoprotein cholesterol (LDL-C) and triglyceride targets.4-10 Similarly, although there has been significant improvement in blood pressure (BP) control over the past 2 decades, BP is controlled in only half of all hypertensive patients.11,12 Finally, the sharp declines in the prevalence of cigarette smoking seen in the past have slowed in recent years, such that approximately 20% of US adults still smoke cigarettes.13
These trends are a concern since a greater risk factor burden in middle age is associated with poorer quality of life and higher medical costs, as well as a higher incidence of cardiovascular events in older age.1 A recent meta-analysis of 18 cohort studies involving 257,384 adults showed a higher incidence of cardiovascular events in later life with an increasing number of risk factors.14 For example, adults 55 years of age with an optimal risk factor profile (ie, total cholesterol <180 mg/dL, BP <120/80 mm Hg, nonsmoker, nondiabetic) had much lower risks of death from cardiovascular disease (CVD) through the age of 80 years than those with 2 or more risk factors (4.7% vs 29.6% among men, 6.4% vs 20.5% among women). This translates into a relative risk (RR) of cardiovascular death of 6 times for men and 3 times for women without optimal risk profiles. Similar trends were observed for risk of fatal CHD/nonfatal myocardial infarction (MI) (3.6% vs 37.5% among men, <1% vs 18.3% among women). These findings point to the critical importance of modifying multiple risk factors early in adulthood, well in advance of symptoms. However, the Study to Help Improve Early Evaluation and Management of Risk Factors Leading to Diabetes (SHIELD) showed that about half of patients with CHD are not diagnosed until symptoms become apparent, and fewer than one quarter are diagnosed as a result of screening.15
This review focuses on patient assessment and treatment strategies to modify abnormal lipid levels and high BP for primary prevention. Addressing other modifiable risk factors is also important, especially since risk factors such as abdominal obesity impact other risk factors ( FIGURE 1 ). An emphasis is placed on strategies in men, since the prevalence of CHD among patients aged 45 years and older is higher in men than in women ( FIGURE 2 ).16 Furthermore, men experience a first cardiovascular event a decade earlier than women, and a more serious CHD event, such as MI or sudden death, 2 decades earlier.1
FIGURE 1
Modifiable risk factors for myocardial infarction (MI)2
ApoB/ApoA-I, apolipoprotein B/apolipoprotein A-I.
FIGURE 2
Prevalence of heart disease by age and gender16
Assessment
The assessment of CHD risk in men need not be complicated and should be made practical so that it is applied consistently. A family and personal medical history and physical examination combined with laboratory determination of lipid levels and glycosylated hemoglobin can help assess modifiable risk factors. The assessment of CHD risk can be facilitated by using 1 of 2 risk calculators. The Framingham Risk Score [www.framinghamheartstudy.org/risk/gencardio.html] is widely used but may underestimate risk, especially in younger persons or those who appear to be healthy but may have other risk factors for CHD.17-19 The Reynolds Risk Score [www.reynoldsriskscore.org/] includes other risk factors, such as parental history of MI before age 60 years, low levels of apolipoprotein A (apoA), high levels of apolipoprotein B (apoB), and increased levels of high-sensitivity C-reactive protein (hs-CRP).19 The Reynolds Risk Score has been validated in healthy, nondiabetic men.20
The relevance of apolipoprotein levels, particularly apoB, to cardiovascular risk is increasingly appreciated.21 ApoB concentration represents the sum of atherogenic particles found on all atherogenic lipoproteins, including very-low-density lipoprotein, intermediate-density lipoprotein, low-density lipoprotein, and lipoprotein(a) cholesterol, whereas apoA represents the sum of antiatherogenic particles found on high-density lipoprotein cholesterol (HDL-C), the antiatherogenic lipoprotein.22 The ratio of apoB/apoA-I has, in fact, been shown to be a good predictor of cardiovascular events in young men without hypertension and diabetes but with chest pain.23 High-sensitivity C-reactive protein is a sensitive marker of acute inflammation and is associated with coronary risk.24 Measuring hs-CRP is a recommended option to determine enhanced absolute risk in people with an intermediate 10-year CHD risk of 10% to 20%.25
There remains some uncertainty regarding which lipid levels should be measured when screening for cardiovascular risk. The National Cholesterol Education Program Adult Treatment Panel III (NCEP ATP III) advises that total cholesterol, LDL-C, HDL-C, and triglycerides be measured.26 More recent results from The Emerging Risk Factors Collaboration suggest that a more simplified approach may be reasonable.27 Review of data from 68 long-term prospective studies involving 302,430 people without initial vascular disease and 2.79 million person-years of follow-up showed that lipid assessment of vascular risk could be accomplished by measuring either total cholesterol and HDL-C levels or apolipoprotein levels; measuring the triglyceride level was of no added benefit in assessing vascular risk. In addition, fasting and nonfasting lipid levels were found to be of similar value in assessing risk. Other evidence shows that the combination of a triglyceride level ≥178 mg/dL and waist circumference ≥35.4 inches—the hypertriglyceridemic waist phenotype—is as discriminatory a screening tool as the NCEP ATP III guidelines to identify individuals at increased cardiometabolic risk.28 The use of more comprehensive lipoprotein and apolipoprotein testing, as well as noninvasive imaging, may have value in future cardiovascular risk assessment.
Treatment
The main goal of treatment in persons with 1 or more modifiable risk factors is to prevent an incident or primary cardiovascular event. Treatment strategies to achieve this goal in men and women are the same. Prevention of recurrent or secondary events will not be addressed here.
Lipids
Numerous clinical trials, such as the Air Force/Texas Coronary Atherosclerosis Prevention Study (AFCAPS/TexCAPS),29 Anglo-Scandinavian Cardiac Outcomes Trial-Lipid Lowering Arm (ASCOT-LLA),30 and West of Scotland Coronary Prevention Study (WOSCOPS),31 definitively established the benefit of cardiovascular risk reduction with lipid-lowering treatment, particularly LDL-C-lowering treatment. Low-density lipoprotein cholesterol is the principal lipid target in most patients, with the treatment goal based on the presence of additional risk factors.32 Discussion of treatments for low HDL-C and elevated triglyceride levels is beyond the scope of this review but is expected to be included in the NCEP ATP IV guidelines scheduled for release later in 2012.
The Justification for the Use of Statins in Prevention: An Intervention Trial Evaluating Rosuvastatin (JUPITER) also established significant benefits of statin therapy in primary prevention, compared with placebo, in persons with normal or modestly elevated LDL-C (<130 mg/dL) and elevated hs-CRP (≥2 mg/L).33 Rates of the primary end point (MI, stroke, arterial revascularization, hospitalization for unstable angina, or cardiovascular death) were 0.77 and 1.36 per 100 person-years of follow-up in the rosuvastatin and placebo groups, respectively (hazard ratio [HR], 0.56; 95% CI, 0.46-0.69; P < .00001). Further analysis showed that patients who achieved LDL-C <70 mg/dL had a 55% lower rate of vascular events compared with placebo.34
Results from large primary prevention clinical trials such as JUPITER have led to recommendations over the past decade or so for progressively lower LDL-C goals. A meta-analysis of 25 large clinical trials involving 155,613 subjects showed that for every 25 mg/dL reduction in LDL-C, the RR for several cardiovascular outcomes was reduced: vascular mortality, 0.89; major vascular events, 0.86; major coronary events, 0.84; and stroke, 0.90. Put differently, there was a 20% reduction in major coronary events for every 39 mg/dL LDL-C reduction.35
Recent trials support the benefits of intensive high-dose statin therapy in greatly reducing lipid levels, with associated benefits in terms of cardiovascular events. A meta-analysis of 7 trials involving 50,972 high-risk patients with a mean follow-up of 3.1 years showed significant reductions in the risk for cardiovascular events with intensive statin therapy. Those who achieved LDL-C <82 mg/dL with intensive statin therapy had lower cardiovascular risks compared with those with LDL-C ≥82 mg/dL: stroke, odds ratio (OR): 0.80; major coronary events, OR: 0.74; and CVD or CHD death, OR: 0.84.36 Significantly higher liver enzyme abnormalities were observed in patients treated with high-dose statin therapy. [See also Addressing Key Questions with Statin Therapy in this supplement.] The benefits of intensive statin therapy on the progression of coronary atherosclerosis have also been investigated. The Study of Coronary Atheroma by Intravascular Ultrasound: Effect of Rosuvastatin versus Atorvastatin (SATURN) by Nicholls et al37 included patients (N = 1039) with documented coronary vessel stenosis of at least 20% and a target vessel for imaging with less than 50% obstruction. Patients received either atorvastatin 80 mg daily or rosu-vastatin 40 mg daily for 104 weeks. In the rosuvastatin group, end-of-study LDL-C levels were lower (62.6 vs 70.2 mg/dL; P < .001) and HDL-C levels higher (50.4 vs 48.6 mg/dL; P = .01) compared with the atorvastatin group, respectively. The percent atheroma volume decreased by 1.22% with rosuvastatin and 0.99% with atorvastatin (P = .17). The normalized total atheroma volume decreased 6.39 mm3 with rosuvastatin and 4.42 mm3 with atorvastatin (P = .01). Atheroma regression was induced in the majority of patients in both groups.
Further support for treating with statin doses higher than those recommended for initial therapy comes from a prospective trial involving 1337 consecutive patients followed over a median of 33 months.10 Although 83% of these patients were on statin therapy, only 51% had an LDL-C <100 mg/dL, and only 15% of the very high-risk patients (n = 941) had an LDL-C <70 mg/dL. The use of intensive statin therapy was associated with a 12-fold higher possibility of achieving an LDL-C <70 mg/dL. Very high-risk patients who achieved an LDL-C <70 mg/dL had a significantly lower risk of all cardiovascular events (HR, 0.34; P = .003).
Blood pressure
As with dyslipidemia, the cardiovascular benefits of lowering elevated BP are well established. While the usual BP goal is <140/90 mm Hg, in those with hypertension and concomitant diabetes or renal disease, the goal is <130/80 mm Hg.38 It is not clear how best to achieve these goals, but therapy must be individualized based on patient comorbidities and drug side effects as recommended in current guidelines.38-40 With these guidelines as a basis, a simplified ABCD approach can be considered in selecting initial antihypertensive therapy ( FIGURE 3 ).
FIGURE 3
ABCD approach to initial antihypertensive therapy38-40
ACE-I, angiotensin converting enzyme inhibitor; ARB, angiotensin receptor blocker; MI, myocardial infarction. Monotherapy, however, does not result in BP control in most patients. As shown by the Antihypertensive Lipid-Lowering Treatment to Prevent Heart Attacks Trial (ALLHAT), BP control typically requires at least 2 different classes of drugs, with 3 or more drugs required in about 1 in 6 patients within 3 years and 1 in 4 patients within 5 years. A higher percentage of patients with diabetes mellitus or kidney impairment (creatinine ≥1.5 mg/dL) require 3 or more antihypertensive drugs after 5 years (33% and 40%, respectively).41
Several meta-analyses have been conducted recently to assess the magnitude of BP (systolic/diastolic) lowering in the different classes of antihypertensive drugs. While these meta-analyses have important limitations, such as differences in study design and the lack of a clear description of outcomes, some general impressions can be made. In 1 meta-analysis, thiazide diuretics were found to lower BP by 6/3 and 8/4 mm Hg at doses of 1 and 2 times the recommended starting dose, respectively. A BP-lowering effect of 6/3 mm Hg was observed with starting doses of loop diuretics.42 Another meta-analysis failed to find a statistically or clinically significant BP-lowering effect with potassium-sparing diuretics at low doses.43 For spironolactone, a review of 5 crossover studies found a reduction in BP of 21/7 mm Hg. In this review, daily doses of 25 to 100 mg were found to provide the best balance between BP reduction and safety and tolerability.44
Several meta-analyses of angiotensin receptor blockers (ARBs) have found BP reductions to be similar among the various ARB drugs. Generally, at maximum recommended doses, a BP reduction of 8/5 mm Hg is observed with these drugs, except for losartan, which produces a smaller BP reduction.45-49 Heran et al45 found a BP reduction of 12/7 mm Hg among the ARBs 1 to 12 hours after the dose was taken. When cost per quality-adjusted life-year gained was considered, 1 meta-analysis found that the slightly greater BP reduction with candesartan compared with losartan was not cost-effective.46 However, other benefits of candesartan compared with losartan therapy (eg, lower risk for cardiovascular disease, heart failure, dysrhythmias, and peripheral artery disease) should be considered.50 Adverse events were generally found to be similar among the ARBs.
No differences in BP lowering were observed among 92 trials of 14 different angiotensin-converting enzyme inhibitors. As a class, these drugs were found to produce a reduction in BP of 8/5 mm Hg.51
Because of the modest BP-lowering effects of each of the antihypertensive drugs currently available, consideration should be given to starting antihypertensive therapy with 2 agents for patients with stage 2 hypertension (ie, BP ≥160/100 mm Hg).
Summary
Elimination of key risk factors such as dyslipidemia and hypertension is important for reducing cardiovascular events later in life. A medical history, physical examination, and laboratory determination of lipid and glycosylated hemoglobin levels provide a good assessment of cardiovascular risk. A statin is first-line therapy for reducing LDL-C, which is the primary lipid target in most patients. High-dose statin therapy may be required to reach desired target levels. The choice of initial antihypertensive therapy is based on patient comorbidities and drug side effects; however, most patients require combination antihypertensive therapy to reach goal. The combination of this multifactorial risk approach along with smoking cessation and modification of other risk factors should complement current and future cardiovascular care for men.
1. Roger VL, Go AS, Lloyd-Jones DM, et al. Heart disease and stroke statistics—2012 update: a report from the American Heart Association. Circulation. 2012;125(1):e2-e220.
2. Yusuf S, Hawken S, Ounpuu S, et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control study. Lancet. 2004;364(9438):937-952.
3. Mensah GA, Brown DW, Croft JB, Greenlund KJ. Major coronary risk factors and death from coronary heart disease: baseline and follow-up mortality data from the Second National Health and Nutrition Examination Survey (NHANES II). Am J Prev Med. 2005;29(5 suppl 1):68-74.
4. Davidson MH, Maki KC, Pearson TA, et al. Results of the National Cholesterol Education (NCEP) Program Evaluation ProjecT Utilizing Novel E-Technology (NEPTUNE) II survey and implications for treatment under the recent NCEP Writing Group recommendations. Am J Cardiol. 2005;96(4):556-563.
5. Waters DD, Brotons C, Chiang CW, et al. Lipid treatment assessment project 2: a multinational survey to evaluate the proportion of patients achieving low-density lipoprotein cholesterol goals. Circulation. 2009;120(1):28-34.
6. Vande Griend JP, Saseen JJ. Low-density lipoprotein cholesterol goal attainment in high-risk family medicine patients. J Clin Lipidol. 2009;3(3):195-200.
7. Barham AH, Goff DC, Jr, Chen H, et al. Appropriateness of cholesterol management in primary care by sex and level of cardiovascular risk. Prev Cardiol. 2009;12(2):95-101.
8. Kitkungvan D, Lynn Fillipon NM, Dani SS, Downey BC. Low-density lipoprotein cholesterol target achievement in patients at high risk for coronary heart disease. J Clin Lipidol. 2010;4(4):293-297.
9. Leiter LA, Lundman P, da Silva PM, et al. Persistent lipid abnormalities in statin- treated patients with diabetes mellitus in Europe and Canada: results of the Dyslipidaemia International Study. Diabet Med. 2011;28(11):1343-1351.
10. Rallidis LS, Kotakos C, Sourides V, et al. Attainment of optional low-density lipoprotein cholesterol goal of less than 70 mg/dl and impact on prognosis of very high risk stable coronary patients: a 3-year follow-up. Expert Opin Pharmacother. 2011;12(10):1481-1489.
11. Centers for Disease Control and Prevention. High blood pressure facts. http://www.cdc.gov/bloodpressure/facts.htm. Published 2012. Accessed May 2, 2012.
12. Egan BM, Zhao Y, Axon RN. US trends in prevalence, awareness, treatment, and control of hypertension, 1988-2008. JAMA. 2010;303(20):2043-2050.
13. McClave A, Rock V, Thorne S, Malarcher A. State-specific prevalence of cigarette smoking and smokeless tobacco use among adults—United States, 2009. MMWR Morb Mortal Wkly Rep. 2010;59(43):1400-1406.
14. Berry JD, Dyer A, Cai X, et al. Lifetime risks of cardiovascular disease. N Engl J Med. 2012;366(4):321-329.
15. Lewis SJ, Fox KM, Grandy S. Shield Study Group. Self-reported diagnosis of heart disease: results from the SHIELD study. Int J Clin Pract. 2009;63(5):726-734.
16. National Center for Health Statistics. Health, United States, 2010: with special feature on death and dying. http://www.cdc.gov/nchs/data/hus/hus10.pdf. Published 2011. Accessed May 2, 2012.
17. Hemann BA, Bimson WF, Taylor AJ. The Framingham Risk Score: an appraisal of its benefits and limitations. Am Heart Hosp J. 2007;5(2):91-96.
18. Karim R, Hodis HN, Detrano R, Liu CR, Liu CH, Mack WJ. Relation of Framingham risk score to subclinical atherosclerosis evaluated across three arterial sites. Am J Cardiol. 2008;102(7):825-830.
19. Ridker PM, Buring JE, Rifai N, Cook NR. Development and validation of improved algorithms for the assessment of global cardiovascular risk in women: the Reynolds Risk Score. JAMA. 2007;297(6):611-619.
20. Ridker PM, Paynter NP, Rifai N, Gaziano JM, Cook NR. C-reactive protein and parental history improve global cardiovascular risk prediction: the Reynolds Risk Score for men. Circulation. 2008;118(22):2243-2251.
21. Brunzell JD, Davidson M, Furberg CD, et al. Lipoprotein management in patients with cardiometabolic risk: consensus conference report from the American Diabetes Association and the American College of Cardiology Foundation. J Am Coll Cardiol. 2008;51(15):1512-1524.
22. Fruchart JC, Sacks FM, Hermans MP, et al. The Residual Risk Reduction Initiative: a call to action to reduce residual vascular risk in dyslipidaemic patient. Diab Vasc Dis Res. 2008;5(4):319-335.
23. Koz C, Baysan O, Hasimi A, et al. Conventional and non-conventional coronary risk factors in male premature coronary artery disease patients already having a low Framingham risk score. Acta Cardiol. 2008;63(5):623-628.
24. Koenig W, Khuseyinova N. Biomarkers of atherosclerotic plaque instability and rupture. Arterioscler Thromb Vasc Biol. 2007;27(1):15-26.
25. Pearson TA, Mensah GA, Alexander RW, et al. Markers of inflammation and cardiovascular disease: application to clinical and public health practice: a statement for healthcare professionals from the Centers for Disease Control and Prevention and the American Heart Association. Circulation. 2003;107(3):499-511.
26. Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation. 2002;106(25):3143-3421.
27. Di Angelantonio E, Sarwar N, Perry P, et al. Emerging Risk Factors Collaboration. Major lipids, apolipoproteins, and risk of vascular disease. JAMA. 2009;302(18):1993-2000.
28. Blackburn P, Lemieux I, Alméras N, et al. The hypertriglyceridemic waist phenotype versus the National Cholesterol Education Program-Adult Treatment Panel III and International Diabetes Federation clinical criteria to identify high-risk men with an altered cardiometabolic risk profile. Metabolism. 2009;58(8):1123-1130.
29. Downs JR, Clearfield M, Weis S, et al. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA. 1998;279(20):1615-1622.
30. Sever PS, Dahlöf B, Poulter NR, et al. Prevention of coronary and stroke events with atorvastatin in hypertensive patients who have average or lower-than-average cholesterol concentrations, in the Anglo-Scandinavian Cardiac Outcomes Trial—Lipid Lowering Arm (ASCOT-LLA): a multicentre randomised controlled trial. Lancet. 2003;361(9364):1149-1158.
31. Shepherd J, Cobbe SM, Ford I, et al. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. West of Scotland Coronary Prevention Study Group. N Engl J Med. 1995;333(20):1301-1307.
32. Grundy SM, Cleeman JI, Merz CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation. 2004;110(2):227-239.
33. Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359(21):2195-2207.
34. Ridker PM, Danielson E, Fonseca FA, et al. Reduction in C-reactive protein and LDL cholesterol and cardiovascular event rates after initiation of rosuvastatin: a prospective study of the JUPITER trial. Lancet. 2009;373(9670):1175-1182.
35. Delahoy PJ, Magliano DJ, Webb K, Grobler M, Liew D. The relationship between reduction in low-density lipoprotein cholesterol by statins and reduction in risk of cardiovascular outcomes: an updated meta-analysis. Clin Ther. 2009;31(2):236-244.
36. Chan DK, O’Rourke F, Shen Q, Mak JC, Hung WT. Meta-analysis of the cardiovascular benefits of intensive lipid lowering with statins. Acta Neurol Scand. 2011;124(3):188-195.
37. Nicholls SJ, Ballantyne CM, Barter PJ, et al. Effect of two intensive statin regimens on progression of coronary disease. N Engl J Med. 2011;365(22):2078-2087.
38. Chobanian AV, Bakris GL, Black HR, et al. The seventh report of the Joint National Committee on prevention, detection, evaluation, and treatment of high blood pressure: the JNC 7 report. JAMA. 2003;289(19):2560-2572.
39. Whitworth JA. World Health Organization, International Society of Hypertension Writing Group. 2003 World Health Organization (WHO)/International Society of Hypertension (ISH) statement on management of hypertension. J Hypertens. 2003;21(11):1983-1992.
40. National Clinical Guideline Centre. Hypertension: the clinical management of primary hypertension in adults. http://www.nice.org.uk/nicemedia/live/12167/54727/54727.pdf. Published 2011. Accessed May 2, 2012.
41. Cushman WC, Ford CE, Cutler JA, et al. Success and predictors of blood pressure control in diverse North American settings: the antihypertensive and lipid-lowering treatment to prevent heart attack trial (ALLHAT). J Clin Hypertens (Greenwich). 2002;4(6):393-404.
42. Chen JM, Heran BS, Wright JM. Blood pressure lowering efficacy of diuretics as second-line therapy for primary hypertension. Cochrane Database Syst Rev. 2009;(4):CD007187.-
43. Heran BS, Chen JM, Wang JJ, Wright JM. Blood pressure lowering efficacy of potassium-sparing diuretics (that block the epithelial sodium channel) for primary hypertension. Cochrane Database Syst Rev. 2010;(1):CD008167.-
44. Batterink J, Stabler SN, Tejani AM, Fowkes CT. Spironolactone for hypertension. Cochrane Database Syst Rev. 2010;(8):CD008169.-
45. Heran BS, Wong MM, Heran IK, Wright JM. Blood pressure lowering efficacy of angiotensin receptor blockers for primary hypertension. Cochrane Database Syst Rev. 2008;(4):CD003822.-
46. Grosso AM, Bodalia PN, Macallister RJ, Hingorani AD, Moon JC, Scott MA. Comparative clinical-and cost-effectiveness of candesartan and losartan in the management of hypertension and heart failure: a systematic review, meta-and cost-utility analysis. Int J Clin Pract. 2011;65(3):253-263.
47. Nixon RM, Müller E, Lowy A, Falvey H. Valsartan vs. other angiotensin II receptor blockers in the treatment of hypertension: a meta-analytical approach. Int J Clin Pract. 2009;63(5):766-775.
48. Zhenfeng Z, Huilan S, Junya J, Dong L, Shan L. A systematic review and meta-analysis of candesartan and losartan in the management of essential hypertension. J Renin Angiotensin Aldosterone Syst. 2011;12(3):365-374.
49. Zheng Z, Lin S, Shi H. A systematic review and meta-analysis of telmisartan versus valsartan in the management of essential hypertension. J Clin Hypertens (Greenwich). 2010;12(6):414-421.
50. Kjeldsen SE, Stålhammar J, Hasvole P, Bodegard J, Olsson U, Russell D. Effects of losartan vs candesartan in reducing cardiovascular events in the primary treatment of hypertension. J Hum Hypertens. 2010;24(4):263-273.
51. Heran BS, Wong MM, Heran IK, Wright JM. Blood pressure lowering efficacy of angiotensin converting enzyme (ACE) inhibitors for primary hypertension. Cochrane Database Syst Rev. 2008;(4):CD003823.-
1. Roger VL, Go AS, Lloyd-Jones DM, et al. Heart disease and stroke statistics—2012 update: a report from the American Heart Association. Circulation. 2012;125(1):e2-e220.
2. Yusuf S, Hawken S, Ounpuu S, et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control study. Lancet. 2004;364(9438):937-952.
3. Mensah GA, Brown DW, Croft JB, Greenlund KJ. Major coronary risk factors and death from coronary heart disease: baseline and follow-up mortality data from the Second National Health and Nutrition Examination Survey (NHANES II). Am J Prev Med. 2005;29(5 suppl 1):68-74.
4. Davidson MH, Maki KC, Pearson TA, et al. Results of the National Cholesterol Education (NCEP) Program Evaluation ProjecT Utilizing Novel E-Technology (NEPTUNE) II survey and implications for treatment under the recent NCEP Writing Group recommendations. Am J Cardiol. 2005;96(4):556-563.
5. Waters DD, Brotons C, Chiang CW, et al. Lipid treatment assessment project 2: a multinational survey to evaluate the proportion of patients achieving low-density lipoprotein cholesterol goals. Circulation. 2009;120(1):28-34.
6. Vande Griend JP, Saseen JJ. Low-density lipoprotein cholesterol goal attainment in high-risk family medicine patients. J Clin Lipidol. 2009;3(3):195-200.
7. Barham AH, Goff DC, Jr, Chen H, et al. Appropriateness of cholesterol management in primary care by sex and level of cardiovascular risk. Prev Cardiol. 2009;12(2):95-101.
8. Kitkungvan D, Lynn Fillipon NM, Dani SS, Downey BC. Low-density lipoprotein cholesterol target achievement in patients at high risk for coronary heart disease. J Clin Lipidol. 2010;4(4):293-297.
9. Leiter LA, Lundman P, da Silva PM, et al. Persistent lipid abnormalities in statin- treated patients with diabetes mellitus in Europe and Canada: results of the Dyslipidaemia International Study. Diabet Med. 2011;28(11):1343-1351.
10. Rallidis LS, Kotakos C, Sourides V, et al. Attainment of optional low-density lipoprotein cholesterol goal of less than 70 mg/dl and impact on prognosis of very high risk stable coronary patients: a 3-year follow-up. Expert Opin Pharmacother. 2011;12(10):1481-1489.
11. Centers for Disease Control and Prevention. High blood pressure facts. http://www.cdc.gov/bloodpressure/facts.htm. Published 2012. Accessed May 2, 2012.
12. Egan BM, Zhao Y, Axon RN. US trends in prevalence, awareness, treatment, and control of hypertension, 1988-2008. JAMA. 2010;303(20):2043-2050.
13. McClave A, Rock V, Thorne S, Malarcher A. State-specific prevalence of cigarette smoking and smokeless tobacco use among adults—United States, 2009. MMWR Morb Mortal Wkly Rep. 2010;59(43):1400-1406.
14. Berry JD, Dyer A, Cai X, et al. Lifetime risks of cardiovascular disease. N Engl J Med. 2012;366(4):321-329.
15. Lewis SJ, Fox KM, Grandy S. Shield Study Group. Self-reported diagnosis of heart disease: results from the SHIELD study. Int J Clin Pract. 2009;63(5):726-734.
16. National Center for Health Statistics. Health, United States, 2010: with special feature on death and dying. http://www.cdc.gov/nchs/data/hus/hus10.pdf. Published 2011. Accessed May 2, 2012.
17. Hemann BA, Bimson WF, Taylor AJ. The Framingham Risk Score: an appraisal of its benefits and limitations. Am Heart Hosp J. 2007;5(2):91-96.
18. Karim R, Hodis HN, Detrano R, Liu CR, Liu CH, Mack WJ. Relation of Framingham risk score to subclinical atherosclerosis evaluated across three arterial sites. Am J Cardiol. 2008;102(7):825-830.
19. Ridker PM, Buring JE, Rifai N, Cook NR. Development and validation of improved algorithms for the assessment of global cardiovascular risk in women: the Reynolds Risk Score. JAMA. 2007;297(6):611-619.
20. Ridker PM, Paynter NP, Rifai N, Gaziano JM, Cook NR. C-reactive protein and parental history improve global cardiovascular risk prediction: the Reynolds Risk Score for men. Circulation. 2008;118(22):2243-2251.
21. Brunzell JD, Davidson M, Furberg CD, et al. Lipoprotein management in patients with cardiometabolic risk: consensus conference report from the American Diabetes Association and the American College of Cardiology Foundation. J Am Coll Cardiol. 2008;51(15):1512-1524.
22. Fruchart JC, Sacks FM, Hermans MP, et al. The Residual Risk Reduction Initiative: a call to action to reduce residual vascular risk in dyslipidaemic patient. Diab Vasc Dis Res. 2008;5(4):319-335.
23. Koz C, Baysan O, Hasimi A, et al. Conventional and non-conventional coronary risk factors in male premature coronary artery disease patients already having a low Framingham risk score. Acta Cardiol. 2008;63(5):623-628.
24. Koenig W, Khuseyinova N. Biomarkers of atherosclerotic plaque instability and rupture. Arterioscler Thromb Vasc Biol. 2007;27(1):15-26.
25. Pearson TA, Mensah GA, Alexander RW, et al. Markers of inflammation and cardiovascular disease: application to clinical and public health practice: a statement for healthcare professionals from the Centers for Disease Control and Prevention and the American Heart Association. Circulation. 2003;107(3):499-511.
26. Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation. 2002;106(25):3143-3421.
27. Di Angelantonio E, Sarwar N, Perry P, et al. Emerging Risk Factors Collaboration. Major lipids, apolipoproteins, and risk of vascular disease. JAMA. 2009;302(18):1993-2000.
28. Blackburn P, Lemieux I, Alméras N, et al. The hypertriglyceridemic waist phenotype versus the National Cholesterol Education Program-Adult Treatment Panel III and International Diabetes Federation clinical criteria to identify high-risk men with an altered cardiometabolic risk profile. Metabolism. 2009;58(8):1123-1130.
29. Downs JR, Clearfield M, Weis S, et al. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA. 1998;279(20):1615-1622.
30. Sever PS, Dahlöf B, Poulter NR, et al. Prevention of coronary and stroke events with atorvastatin in hypertensive patients who have average or lower-than-average cholesterol concentrations, in the Anglo-Scandinavian Cardiac Outcomes Trial—Lipid Lowering Arm (ASCOT-LLA): a multicentre randomised controlled trial. Lancet. 2003;361(9364):1149-1158.
31. Shepherd J, Cobbe SM, Ford I, et al. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. West of Scotland Coronary Prevention Study Group. N Engl J Med. 1995;333(20):1301-1307.
32. Grundy SM, Cleeman JI, Merz CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation. 2004;110(2):227-239.
33. Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359(21):2195-2207.
34. Ridker PM, Danielson E, Fonseca FA, et al. Reduction in C-reactive protein and LDL cholesterol and cardiovascular event rates after initiation of rosuvastatin: a prospective study of the JUPITER trial. Lancet. 2009;373(9670):1175-1182.
35. Delahoy PJ, Magliano DJ, Webb K, Grobler M, Liew D. The relationship between reduction in low-density lipoprotein cholesterol by statins and reduction in risk of cardiovascular outcomes: an updated meta-analysis. Clin Ther. 2009;31(2):236-244.
36. Chan DK, O’Rourke F, Shen Q, Mak JC, Hung WT. Meta-analysis of the cardiovascular benefits of intensive lipid lowering with statins. Acta Neurol Scand. 2011;124(3):188-195.
37. Nicholls SJ, Ballantyne CM, Barter PJ, et al. Effect of two intensive statin regimens on progression of coronary disease. N Engl J Med. 2011;365(22):2078-2087.
38. Chobanian AV, Bakris GL, Black HR, et al. The seventh report of the Joint National Committee on prevention, detection, evaluation, and treatment of high blood pressure: the JNC 7 report. JAMA. 2003;289(19):2560-2572.
39. Whitworth JA. World Health Organization, International Society of Hypertension Writing Group. 2003 World Health Organization (WHO)/International Society of Hypertension (ISH) statement on management of hypertension. J Hypertens. 2003;21(11):1983-1992.
40. National Clinical Guideline Centre. Hypertension: the clinical management of primary hypertension in adults. http://www.nice.org.uk/nicemedia/live/12167/54727/54727.pdf. Published 2011. Accessed May 2, 2012.
41. Cushman WC, Ford CE, Cutler JA, et al. Success and predictors of blood pressure control in diverse North American settings: the antihypertensive and lipid-lowering treatment to prevent heart attack trial (ALLHAT). J Clin Hypertens (Greenwich). 2002;4(6):393-404.
42. Chen JM, Heran BS, Wright JM. Blood pressure lowering efficacy of diuretics as second-line therapy for primary hypertension. Cochrane Database Syst Rev. 2009;(4):CD007187.-
43. Heran BS, Chen JM, Wang JJ, Wright JM. Blood pressure lowering efficacy of potassium-sparing diuretics (that block the epithelial sodium channel) for primary hypertension. Cochrane Database Syst Rev. 2010;(1):CD008167.-
44. Batterink J, Stabler SN, Tejani AM, Fowkes CT. Spironolactone for hypertension. Cochrane Database Syst Rev. 2010;(8):CD008169.-
45. Heran BS, Wong MM, Heran IK, Wright JM. Blood pressure lowering efficacy of angiotensin receptor blockers for primary hypertension. Cochrane Database Syst Rev. 2008;(4):CD003822.-
46. Grosso AM, Bodalia PN, Macallister RJ, Hingorani AD, Moon JC, Scott MA. Comparative clinical-and cost-effectiveness of candesartan and losartan in the management of hypertension and heart failure: a systematic review, meta-and cost-utility analysis. Int J Clin Pract. 2011;65(3):253-263.
47. Nixon RM, Müller E, Lowy A, Falvey H. Valsartan vs. other angiotensin II receptor blockers in the treatment of hypertension: a meta-analytical approach. Int J Clin Pract. 2009;63(5):766-775.
48. Zhenfeng Z, Huilan S, Junya J, Dong L, Shan L. A systematic review and meta-analysis of candesartan and losartan in the management of essential hypertension. J Renin Angiotensin Aldosterone Syst. 2011;12(3):365-374.
49. Zheng Z, Lin S, Shi H. A systematic review and meta-analysis of telmisartan versus valsartan in the management of essential hypertension. J Clin Hypertens (Greenwich). 2010;12(6):414-421.
50. Kjeldsen SE, Stålhammar J, Hasvole P, Bodegard J, Olsson U, Russell D. Effects of losartan vs candesartan in reducing cardiovascular events in the primary treatment of hypertension. J Hum Hypertens. 2010;24(4):263-273.
51. Heran BS, Wong MM, Heran IK, Wright JM. Blood pressure lowering efficacy of angiotensin converting enzyme (ACE) inhibitors for primary hypertension. Cochrane Database Syst Rev. 2008;(4):CD003823.-
Meeting New Challenges with Antiplatelet Therapy in Primary Care
Managing the Multiple Symptoms of Benign Prostatic Hyperplasia — CME
Managing Type 2 Diabetes in Men
Meeting New Challenges with Antiplatelet Therapy in Primary Care
Dr. Kuritzky has nothing to disclose.
Dr. Díez has nothing to disclose.
SUPPORT
This program is sponsored by the PCEC and supported by funding from AstraZeneca. Dr. Kuritzky received no financial support for this article.
Introduction
The importance of acute coronary syndrome (ACS) (ie, patients with ST-segment elevation myocardial infarction [MI] [STEMI], non-ST segment elevation MI [NSTEMI], or unstable angina) in primary care is highlighted by its prevalence. Acute coronary syndrome was the primary or secondary discharge diagnosis in 1.19 million hospitalizations in the United States in 2009, a slight majority of which were in men.1 Platelet activation plays a central role in the pathophysiology of ACS. Despite well established benefits of antiplatelet therapy in both primary and secondary prevention of ACS, adverse events—particularly bleeding—require ongoing vigilance.2 Among the several classes of antiplatelet agents currently available, the thromboxane A2 inhibitor (ie, aspirin) and P2Y12 inhibitors (ie, clopidogrel, prasugrel, and ticagrelor) are those most commonly used; ticlopidine is not commonly used due to nausea/vomiting and bone marrow toxicity.3
Antiplatelet Agents
It is well established that hemostasis is protected by multilayered, overlapping, and sometimes redundant pathways. Even though currently available antiplatelet agents are highly efficacious in inhibiting 1 or more phases of platelet activity pertinent to coagulation (eg, activation, adhesion, and aggregation), because of the multiple backup pathways involved, no single antiplatelet agent is anticipated to totally eliminate platelet activity. In addition, every combination of antiplatelet agents—though potentially more efficacious because of multipathway activity—is also laden with greater bleeding risk. The 3 primary pathways of platelet activation for which pharmacologic antagonists have been developed are the thromboxane, adenosine diphosphonate (ADP)-P2Y12, and ADP-A2 pathways. While dual antiplatelet therapy with aspirin and clopidogrel may be the current standard of care, the focus of this review is on the ADP-P2Y12 inhibitors as the two newest agents, prasugrel and ticagrelor, are less familiar to family physicians. The second section addresses questions often encountered by family physicians when caring for patients who have recently experienced ACS.
P2Y12 Inhibitors
Two groups of agents exert their antiplatelet effects by inhibiting the platelet P2Y12 receptor: (1) thienopyridines (ie, ticlopidine, clopidogrel, and prasugrel) and (2) the cyclopentyltriazolopyrimidines (ie, ticagrelor). Both groups inhibit ADP-dependent platelet function but at different sites on the platelet P2Y12 receptor. Thienopyridine activity is mediated via short-lived active metabolites formed in the liver. Platelet exposure to the active metabolite of prasugrel is about 10-fold higher than to the active metabolite of clopidogrel, resulting in a higher level and less individual variation of platelet inhibition with prasugrel. Hepatic metabolism of clopidogrel makes it subject to genetic, as well as drug-induced, variation in activity; prasugrel is not affected by these same limitations. Recovery of platelet function following withdrawal of thienopyridine therapy occurs over 7 to 8 days as a function of platelet turnover.2,3 This slow recovery of platelet function has important implications when any surgical intervention is needed.
In contrast to the thienopyridines, ticagrelor does not require metabolic activation by the liver. Ticagrelor and its active metabolite display approximately equipotent antiplatelet activity and are direct P2Y12 inhibitors. Ticargrelor non-competitively antagonizes ADP-induced receptor activation. Ticagrelor is rapidly absorbed reaching its peak plasma concentration in 1.5 to 3 hours, thereby providing a rapid antiplatelet effect. Twice-daily administration is required because of its rapid offset of platelet inhibition.2,4,5
Prasugrel
Prasugrel is indicated by the US Food and Drug Administration (FDA) for reduction of thrombotic cardiovascular (CV) events (including stent thrombosis) in patients with ACS who are to be managed with percutaneous coronary intervention (PCI) as follows: (1) unstable angina or NSTEMI or (2) STEMI when managed with primary or delayed PCI.6
The efficacy and safety of prasugrel have been investigated in several clinical trials. The Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition with Prasugrel-Thrombolysis in Myocardial Infarction (TRITON-TIMI) 38 is the largest and has many planned sub-analyses ( TABLE 1 ).7-9 TRITON TIMI 38 involved patients with moderate- to high-risk ACS scheduled for PCI
(N = 13,608).7 Patients were randomized to prasugrel 60 mg as a loading dose followed by 10 mg daily or clopidogrel 300 mg as a loading dose followed by 75 mg daily for 6 to 15 months. Aspirin 75 to 162 mg once daily was recommended, but was left up to the physician. The primary efficacy end point was a composite of CV death, nonfatal MI, or nonfatal stroke.
Findings from TRITON TIMI 38 show that, compared with clopidogrel, prasugrel was associated with significantly reduced rates of ischemic events, including nonfatal MI and stent thrombosis. The benefit with prasugrel was primarily due to a significant reduction in the rate of MI compared with clopidogrel. However, patients treated with prasugrel experienced a higher rate of major bleeding, including fatal and life-threatening bleeding. Prasugrel was found to be more effective than clopidogrel in preventing ischemic events without excess bleeding in patients with STEMI undergoing secondary PCI (treated between 12 hours and 14 days after symptom onset). In patients with ACS undergoing PCI without stent implantation, ischemic events occurred at similar rates in patients treated with prasugrel or clopidogrel; however, bleeding was more common with prasugrel.
Not all patients benefited from prasugrel therapy. Compared with clopidogrel, patients with previous stroke/transient ischemic attack (TIA) had net harm from prasugrel. In addition, no net benefit from prasugrel compared with clopidogrel was observed in patients age ≥75 years or body weight <60 kg. The results of TRITON TIMI 38 contributed to the boxed warnings regarding bleeding risk recommending that prasugrel not be used in patients age ≥75 years, in patients with active pathological bleeding or a history of TIA or stroke, or patients likely to undergo coronary artery bypass graft (CABG) surgery. In addition, patients with body weight < 60 kg are also at increased risk for bleeding.6
TABLE 1
Prasugrel: TRITON-TIMI 38 and subanalyses
| TRITON-TIMI 38 Cohort7 | TRITON-TIMI 38 Selected Subanalyses8,9 | ||
|---|---|---|---|
| Treatment | Pr 60 mg LD, then 10 mg QD or Cl 300 mg LD, then 75 mg QD plus Aspirin 75-162 mg QD for 6-15 mos (median 14.5 mos) | ||
| Population | Moderate/High-risk ACS scheduled for PCI (N = 13,608) | PCI for STEMI (N = 3534) | PCI without ST elevation (N = 569) |
| Efficacy Outcomes | Primary end point (CV death, nonfatal MI, or nonfatal stroke):
(P = .31) Nonfatal MI: Cl 9.5% vs Pr 7.3% (P < .001) Nonfatal stroke: Cl 1.0% vs Pr 1.0% (P = .93) Urgent target-vessel revascularization: Cl 3.7% vs Pr 2.5% (P < .001) Stent thrombosis: Cl 2.4% vs Pr 1.1% (P < .001) | Primary end point (CV death, nonfatal MI, or nonfatal stroke):
| Primary end point (CV death, nonfatal MI, or nonfatal stroke): Cl 17.1% vs Pr 14.2% (P = .27) Urgent target-vessel revascularization: Cl 8.2% vs Pr 3.6% (P = .04) |
| Safety Outcomes | Non-CABG TIMI major bleeding: Cl 1.8% vs Pr 2.4% (P = .03) Fatal bleeding: Cl 0.1% vs Pr 0.4% (P = .002) Life-threatening bleeding: Cl 0.9% vs Pr 1.4% (P = .01) Non-fatal bleeding: Cl 0.9% vs Pr 1.1% (P = .23) | TIMI major bleedinga unrelated to CABG:
| TIMI major bleedinga unrelated to CABG: Cl 0% vs Pr 2.1% (P = .03) |
| Key Findings | Prasugrel was associated with significantly reduced rates of ischemic events, including nonfatal MI and stent thrombosis, but with an increased risk of major bleeding, including fatal and life-threatening bleeding. Compared to clopidogrel, patients with previous stroke/TIA had net harm from prasugrel; patients with age ≥ 75 y had no net benefit from prasugrel; patients with BW < 60 kg had no net benefit from prasugrel. | Net clinical outcome All-cause death, MI, stroke, TIMI major bleeding unrelated to CABG:
| In patients with ACS undergoing PCI without stent implantation, ischemic events occurred at similar rates in patients treated with prasugrel or clopidogrel; however, bleeding was more common with prasugrel. |
| ACS, acute coronary syndrome; BW, body weight; CABG, coronary artery bypass graft; Cl, clopidogrel; CV, cardiovascular; LD, loading dose; MI, myocardial infarction; PCI, percutaneous coronary intervention; Pr, prasugrel; QD, once daily; STEMI, ST-segment elevation in myocardial infarction; TIA, transient ischemic attack; TIMI, thrombolysis in myocardial infarction. aTIMI major bleed (intracranial bleed or intrapericardial bleed with cardiac tamponade or a decline of 5.0 g/dL or more in hemoglobin after adjusting for red blood cell transfusions). | |||
Ticagrelor
Ticagrelor is the most recent antiplatelet agent to be approved by the US FDA. Ticagrelor is indicated to reduce the rate of thrombotic CV events in patients with ACS (eg, unstable angina, NSTEMI, or STEMI).10
The efficacy and safety of ticagrelor has been assessed in the Study of Platelet Inhibition and Patient Outcomes (PLATO) and several planned sub-analyses ( TABLE 2 ).11-16 PLATO was a 12-month, multicenter, double-blind, randomized trial that involved patients with ACS with or without ST-segment elevation (N = 18,624).11 Patients were randomized to ticagrelor 180 mg loading dose then 90 mg twice daily or clopidogrel 300 to 600 mg loading dose then 75 mg once daily for 12 months. The primary efficacy end point was a composite of death from vascular causes, MI, or stroke.
The results of PLATO and sub-analyses show that in patients with ACS and compared with clopidogrel, ticagrelor significantly reduced the primary efficacy end point with a similar rate of major bleeding
( TABLE 1 ). These safety results contributed to the boxed warnings regarding bleeding risk that ticagrelor not be used in patients with active pathological bleeding or a history of intracranial hemorrhage, or in patients planned to undergo urgent CABG surgery. In addition, maintenance aspirin therapy at a dose above 100 mg reduces the effectiveness of ticagrelor and should be avoided.10
Consistent with the general PLATO population, in patients intended for non-invasive management, ticagrelor significantly reduced the rate of death from vascular causes, MI, or stroke compared with clopidogrel with a similar rate of major bleeding. In patients with ACS and ST elevation or left bundle branch block planned for PCI, ticagrelor reduced CV and all-cause death, MI, stent thrombosis, and improved survival compared with clopidogrel, with a similar rate of major bleeding. Ticagrelor, compared with clopidogrel, reduced all-cause and CV death without excess risk of CABG-related bleeding in patients with ACS undergoing CABG within 7 days of the last dose of clopidogrel or ticagrelor. Finally, in ACS with chronic kidney disease (estimated creatinine clearance < 60 mL/minute), ticagrelor compared with clopidogrel significantly reduced ischemic end points and mortality without a significant increase in major bleeding and with a similar rate of non–CABG-related bleeding.
TABLE 2
Ticagrelor: PLATO and subanalyses
| PLATO Cohort9,12 | PLATO Selected Subanalyses13-16 | ||||
|---|---|---|---|---|---|
| Treatment | Ti 180 mg LD, then 90 mg BID or Cl 300-600 mg LD then 75 mg QD plus Aspirin 75-325 mg QD for 12 months | ||||
| Population | ACS with/without ST elevation (N = 18,624) | ACS planned for non-invasive management (N = 5216) | ACS with ST elevation or left bundle branch block planned for PCI (N = 7544) | ACS with/without ST elevation managed with CABG (N = 1261) | ACS with/without ST elevation but with chronic kidney disease (eCrCl < 60 mL/min) (n = 3237) |
| Efficacy Outcomes | Primary end point (death from vascular causes, MI, or stroke): Cl 11.7% vs Ti 9.8% (P < .001) Death from any cause, MI, or stroke: Cl 12.3% vs Ti 10.2% (P < .001) Death from any cause, MI, stroke, severe recurrent ischemia, recurrent ischemia, TIA, or other arterial thrombotic event: Cl 16.7% vs Ti 14.6% (P < .001) Death from nonvascular causes: Cl 0.8% vs Ti 0.5% (P = .08) | Primary end point (death from vascular causes, MI, or stroke): Cl 14.3% vs. Ti 12.0% (P = .045) CV death: Cl 7.2% vs Ti 5.5% (P = .019) | Primary end point (death from vascular causes, MI, or stroke): Cl 10.8% vs Ti 9.4% (P = .07) CV death, MI (excluding silent): Cl 10.2% vs Ti 8.4% (P = .01) All cause death, MI (excluding silent), stroke: Cl 11.3% vs Ti 9.8% (P = .05) CV death, total MI, stroke, severe recurrent cardiac ischemia, recurrent cardiac ischemia, TIA, arterial thrombotic events: Cl 15.0% vs Ti 13.3% (P = .03) MI (excluding silent): Cl 5.8% vs Ti 4.7% (P = .03) Stroke: Cl 1.0% vs Ti 1.7% (P = .02) All-cause mortality: Cl 6.1% vs Ti 5.0% (P = .05) Definite, probable, or possible stent thrombosis: Cl 4.3% vs Ti 3.3% (P = .04) | Primary end point (death from vascular causes, MI, or stroke): Cl 13.1% vs Ti 10.6% (P = .29) All-cause death: Cl 9.7% vs Ti 4.7% (P < .01) CV death: Cl 7.9% vs Ti 4.1% (P < .01) Non-CV death: Cl 2.0% vs Ti 0.7% (P = .07) Stroke: Cl 2.1% vs Ti 2.1% (P = .70) | Primary end point (death from vascular causes, MI, or stroke): Cl 22.0% vs Ti 17.3% All-cause death: Cl 14.0% vs Ti 10.0% |
| Safety Outcomes | TIMI major bleedinga: Cl 7.7% vs Ti 7.9% (P = .57) TIMI major bleedinga unrelated to CABG: Cl 2.2% vs Ti 2.8% (P = .03) PLATO major bleedingb: Cl 11.2% vs Ti 11.6% (P = .43) PLATO major bleedingb unrelated to CABG: Cl 3.8% vs Ti 4.5% (P = .03) Dyspnea requiring discontinuation: Cl 0.1% vs Ti 0.9% (P < .001) | PLATO major bleedingb: Cl 10.3% vs Ti 11.9% (P = .079) Life-threatening/fatal bleeding: Cl 5.6% vs Ti 5.5% (P= . 911) Major/Minor bleeding unrelated to CABG: Cl 6.7% vs Ti 8.3% (P = .0182) | PLATO major bleeding: Cl 9.2% vs Ti 9.0% (P = .76) TIMI major bleeding: Cl 6.4% vs Ti 6.1% (P = .66) PLATO non-procedure-related major/ minor bleeding: Cl 3.7% vs Ti 5.1% (P = .02) PLATO minor bleeding: Cl 3.8% vs Ti 4.9% (P = .05) Dyspnea requiring discontinuation: Cl 0.1% vs Ti 0.5% (P = .0004) | Major/Life-threatening CABG-related bleeding causing death within 7 d after CABG: Cl 3.0% vs Ti 1.3% (P = .052) Major CABG bleeding: Cl 80.1% vs Ti 81.2% (P = .669) TIMI major CABG bleeding: Cl 57.6% vs Ti 59.3% (P = .53) | PLATO major bleeding: Cl 14.3% vs Ti 15.1% PLATO fatal major bleeding: Cl 0.77% vs Ti 0.34% PLATO non-CABG major bleeding: Cl 7.3% vs Ti 8.5% Dyspnea: Cl 11.5% vs Ti 16.4% |
| Key Findings | Ticagrelor significantly reduced the rate of CV death, MI, or stroke compared to clopidogrel with a similar rate of major bleeding; ticagrelor led to increased major bleeding unrelated to CABG. Fatal bleeding was low and did not differ between groups. | Consistent with the general PLATO population, ticagrelor significantly reduced the rate of CV death, MI, or stroke compared to clopidogrel with a similar rate of major bleeding. | Consistent with the general PLATO population, compared with clopidogrel, ticagrelor reduced CV and all-cause death, MI, stent thrombosis and improved survival without increasing major bleeding. Ticagrelor resulted in a higher rate of stroke. | Ticagrelor compared with clopidogrel reduced all-cause and CV death without excess risk of CABG-related bleeding in patients with ACS undergoing CABG within 7 days of the last dose of clopidogrel or ticagrelor. | In ACS with CKD, ticagrelor compared with clopidogrel significantly reduced ischemic end points and mortality without a significant increase in major bleeding and with a similar rate of non-procedure- related bleeding. |
| ACS, acute coronary syndrome; BID, twice daily; CABG, coronary artery bypass graft; Cl, clopidogrel; CKD, chronic kidney disease; CV, cardiovascular; eCrCL, estimated creatinine clearance; LD, loading dose; MI, myocardial infarction; PCI, percutaneous coronary intervention; QD, once daily; Ti, ticagrelor; TIA, transient ischemic attack; TIMI, thrombolysis in myocardial infarction. aTIMI major bleed (intracranial bleed or intrapericardial bleed with cardiac tamponade or a decline of 5.0 g/dL or more in hemoglobin after adjusting for red blood cell transfusions). bPLATO major bleed (fatal bleeding, intrapericardial bleeding with cardiac tamponade, intracranial bleeding, severe hypotension, or hypovolemic shock due to bleeding and requiring either vasopressors or surgical intervention, a decline in hemoglobin of 5.0 g/dL or more after adjusting for red blood cell transfusions, or the need for transfusion of 4 or more units of packed red blood cells) | |||||
Common Questions Regarding Antiplatelet Therapy in Primary Care
The preceding discussion confirms that many patients with ACS benefit from antiplatelet therapy. However, the use of antiplatelet agents in primary care can be challenging. The following are some of the evolving issues and questions regarding antiplatelet therapy faced by family physicians.
If a patient has experienced gastrointestinal bleeding while taking low-dose aspirin in the past and has an acute coronary syndrome, what course of action should be taken?
Dual antiplatelet therapy is still recommended in this setting, but therapy with a proton pump inhibitor (PPI) for gastrointestinal (GI) protection is recommended.2,3,17 For patients at low risk of upper GI bleeding, routine PPI prophylaxis is not recommended. Currently available data do not demonstrate the prophylactic superiority of one PPI over another, but do show that PPI therapy is more effective in decreasing GI bleeding associated with aspirin and is, therefore, preferred over a histamine H2 receptor antagonist.17 For instance, high-dose famotidine has been shown to be less effective than pantoprazole in patients with aspirin-related peptic ulcers/erosions.18
Can a proton pump inhibitor be used for gastrointestinal protection in conjunction with clopidogrel?
Yes, although the evidence is conflicting about whether specific PPIs should be avoided because of reduced clinical efficacy of clopidogrel. The results of a meta-analysis of 23 studies demonstrated a clinically significant interaction that reduces the antiplatelet effectiveness of clopidogrel when combined with some PPIs.19 The results of 4 prospective, crossover pharmacokinetic studies in healthy subjects (N = 282) also suggest an interaction between clopidogrel and omeprazole but not between clopidogrel and pantoprazole.20 A subanalysis of PLATO showed that the use of a PPI was independently associated with a higher rate of CV events in patients with ACS treated with clopidogrel or ticagrelor.21 The observed effect with both agents, as well as a higher rate of major bleeding among PPI vs non-PPI users suggests that PPI use may be more of a marker for rather than a cause of higher rates of CV events. In fact, data from the Clopidogrel and the Optimization of Gastrointestinal Events Trial (COGENT) found that in patients treated with clopidogrel and aspirin, the addition of omeprazole reduced the rate of a GI event, compared with placebo at 180 days (1.1% vs. 2.9%, respectively;
P < .001).22 Overt upper GI bleeding occurred less frequently in the omeprazole group (hazard ratio, 0.13; 95% confidence interval, 0.03 to 0.56; P = .001). A CV event was observed in 4.9% of patients treated with omeprazole and 5.7% of placebo patients (P = .96). While limited, these prospective data do not suggest a detriment to clopidogrel efficacy when used in combination with a PPI. The dose of PPI to use for GI protection is not well-established; the following drugs and doses have been used: omeprazole 20 to 40 mg once daily; esomeprazole 20 mg once or twice daily; pantoprazole 20 mg once daily; or lansoprazole 30 mg once daily.18,23-28
Should I avoid starting clopidogrel in patients with acute coronary syndrome because of concerns about “poor metabolizers”?
Clopidogrel is a prodrug, requiring CYP450 metabolism to its active metabolite. Because of genetic CYP450 variations, as many as one-third of patients lack fully active CYP450 pathways, resulting in reduced (or even absent) conversion from the parent drug to the active metabolite, with a corresponding diminution of antiplatelet effects.3,29 Recent recommendations about dealing with these genetic polymorphisms include direct measurement of CYP450 pathway status and selection of alternative pharmacologic agents which are not dependent upon similar CYP pathway activation. There are, unfortunately, no prospective clinical trials based upon CYP2C19 genotyping confirming that patient selection based upon genotyping is associated with improved outcomes.
In terms of alternative antiplatelet therapy in clopidogrel nonresponders, the Response to Ticagrelor in Clopidogrel Nonresponders and Responders (RESPOND) study shows ticagrelor to be beneficial, at least as measured in vitro.30 Following laboratory assessment of patients’ responsiveness to clopidogrel, both responders and nonresponders were randomized to clopidogrel or ticagrelor. After 14 days, all clopidogrel nonresponders and half of the responders switched treatment. The antiplatelet effects of ticagrelor were similar whether the patient was a clopidogrel responder or not. The Prasugrel in Comparison to Clopidogrel for Inhibition of Platelet Activation and Aggregation-Thrombolysis in Myocardial Infarction 44 (PRINCIPLE-TIMI 44) showed higher inhibition of platelet aggregation (IPA) with prasugrel 60 mg compared with clopidogrel 600 mg 6 hours after initiation.31 Following crossover, IPA was higher in subjects receiving prasugrel 10 mg/d compared with clopidogrel 150 mg/d (61% vs 46%, respectively; P < .0001). While not measuring clopidogrel responsiveness, this suggests that prasugrel might be effective in clopidogrel nonresponders. Not all patients treated with prasugrel achieve optimal inhibition of platelet reactivity. In patients who underwent successful PCI for ACS (N = 301) 25.2% were observed to have high on-treatment platelet reactivity following a 60 mg loading dose of prasugrel.32 Such patients had a significantly higher risk for a major adverse cardiovascular event after PCI. The clinical trials which demonstrate improved clinical outcomes when clopidogrel is compared with other antiplatelet agents suggest that the above-mentioned in vitro metrics are clinically relevant.
I’ve heard a lot about testing platelet aggregability. Should I be considering that for my patients?
Not at the present time. One prospective study evaluated the capability of platelet function tests to predict clinical outcome in patients taking clopidogrel undergoing elective stent implantation.33 On-treatment platelet reactivity was measured using: light transmittance aggregometry, VerifyNow P2Y12, Plateletworks, and the IMPACT-R and the platelet function analysis system (PFA-100) (with the Dade PFA collagen/ADP cartridge and Innovance PFA P2Y). After 1 year of follow-up, only the light transmittance aggregometry, VerifyNow, Plateletworks, and Innovance PFA P2Y tests were significantly associated with patient outcome, but had only modest predictive accuracy. Also, none of the tests studied provided accurate prognostic information to identify patients at higher risk of bleeding following stent implantation.
How concerning are the findings on ticagrelor and dyspnea?
The occurrence of dyspnea associated with ticagrelor was observed during its clinical development. While the mechanism is not known, dyspnea is a transient phenomenon, and there is no suggestion that ticagrelor is associated with an increased incidence of heart failure.
The incidence and characterization of dyspnea has been investigated in subanalyses of 2 large clinical trials of ticagrelor. Prospective analysis of the ONSET/OFFSET study (N = 123) showed that dyspnea was experienced by more patients treated with ticagrelor than clopidogrel or placebo over 6 weeks (38.6% vs 9.3% vs 8.3%, respectively; P < .001).34 Episodes of dyspnea were generally mild, lasted <24 hours, and easily tolerated. Moderate dyspnea that led to study discontinuation occurred in 3 patients (5.3%) treated with ticagrelor. Dyspnea occurred within the first 24 hours in 8 of 22 patients (36.4%) and within the first week in 17 of 22 patients (77.3%) of the ticagrelor-treated patients who experienced dyspnea. Dyspnea persisted through the study follow-up (10 days after the 6 week study) in 3 of 22 patients (13.6%) treated with ticagrelor. Dyspnea was not associated with any significant adverse change in cardiac or pulmonary function tests.34
In a subanalysis of the PLATO study to investigate the occurrence of dyspnea (N = 18,421), dyspnea occurred in 14.5% of patients treated with ticagrelor and 8.7% of patients treated with clopidogrel.35 Severe dyspnea occurred in 0.4% and 0.3% of patients, respectively. Dyspnea had no impact on the composite end point after excluding dyspnea that occurred after the secondary end point of MI. The mechanism whereby ticagrelor induces dyspnea is not certain, but may be mediated via an adenosine-related mechanism.36
Conclusion
Aspirin and clopidogrel have been the predominant antiplatelet agents used in the management of patients with ACS, yet their use can be challenging. Differences in the clinical pharmacology of prasugrel and ticagrelor provide the opportunity to address some of these challenges and better enable antiplatelet therapy to be individualized.
1. Roger VL, Go AS, Lloyd-Jones DM, et al. Heart disease and stroke statistics—2012 update: a report from the American Heart Association. Circulation. 2012;125(1):e2-e220.
2. Patrono C, Andreotti F, Arnesen H, et al. Antiplatelet agents for the treatment and prevention of atherothrombosis. Eur Heart J. 2011;32(23):2922-2932.
3. Eikelboom JW, Hirsh J, Spencer FA, Baglin TP, Weitz JI. Antiplatelet Drugs: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141 (2 Suppl):e89S-e119S.
4. Abergel E, Nikolsky E. Ticagrelor: an investigational oral antiplatelet treatment for reduction of major adverse cardiac events in patients with acute coronary syndrome. Vasc Health Risk Manag. 2010;6:963-977.
5. Gurbel PA, Bliden KP, Butler K, et al. Randomized double-blind assessment of the ONSET and OFFSET of the antiplatelet effects of ticagrelor versus clopidogrel in patients with stable coronary artery disease: the ONSET/OFFSET study. Circulation. 2009;120(25):2577-2585.
6. Effient [package insert]. Indianapolis, IN: Eli Lilly and Co.; 2011.
7. Wiviott SD, Braunwald E, McCabe CH, et al. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2007;357(20):2001-2015.
8. Montalescot G, Wiviott SD, Braunwald E, et al. Prasugrel compared with clopidogrel in patients undergoing percutaneous coronary intervention for ST-elevation myocardial infarction (TRITON-TIMI 38): double-blind, randomised controlled trial. Lancet. 2009;373(9665):723-731.
9. Pride YB, Wiviott SD, Buros JL, et al. Effect of prasugrel versus clopidogrel on outcomes among patients with acute coronary syndrome undergoing percutaneous coronary intervention without stent implantation: a TRial to assess Improvement in Therapeutic Outcomes by optimizing platelet inhibitioN with prasugrel (TRITON)-Thrombolysis in Myocardial Infarction (TIMI) 38 substudy. Am Heart J. 2009;158(3):e21-e26.
10. Brilinta [package insert]. Wilmington, DE: AstraZeneca; 2011.
11. Wallentin L, Becker RC, Budaj A, et al. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2009;361(11):1045-1057.
12. Becker RC, Bassand JP, Budaj A, et al. Bleeding complications with the P2Y12 receptor antagonists clopidogrel and ticagrelor in the PLATelet inhibition and patient Outcomes (PLATO) trial. Eur Heart J. 2011;32(23):2933-2944.
13. James SK, Roe MT, Cannon CP, et al. Ticagrelor versus clopidogrel in patients with acute coronary syndromes intended for non-invasive management: substudy from prospective randomised PLATelet inhibition and patient Outcomes (PLATO) trial. BMJ. 2011;342:d3527-
14. Steg PG, James S, Harrington RA, et al. Ticagrelor versus clopidogrel in patients with ST-elevation acute coronary syndromes intended for reperfusion with primary percutaneous coronary intervention: A Platelet Inhibition and Patient Outcomes (PLATO) trial subgroup analysis. Circulation. 2010;122(21):2131-2141.
15. Held C, Asenblad N, Bassand JP, et al. Ticagrelor versus clopidogrel in patients with acute coronary syndromes undergoing coronary artery bypass surgery: results from the PLATO (Platelet Inhibition and Patient Outcomes) trial. J Am Coll Cardiol. 2011;57(6):672-684.
16. James S, Budaj A, Aylward P, et al. Ticagrelor versus clopidogrel in acute coronary syndromes in relation to renal function: results from the Platelet Inhibition and Patient Outcomes (PLATO) trial. Circulation. 2010;122(11):1056-1067.
17. Abraham NS, Hlatky MA, Antman EM, et al. ACCF/ACG/AHA 2010 Expert Consensus Document on the concomitant use of proton pump inhibitors and thienopyridines: a focused update of the ACCF/ACG/AHA 2008 expert consensus document on reducing the gastrointestinal risks of antiplatelet therapy and NSAID use: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents. Circulation. 2010;122(24):2619-2633.
18. Ng FH, Wong SY, Lam KF, et al. Famotidine is inferior to pantoprazole in preventing recurrence of aspirin-related peptic ulcers or erosions. Gastroenterology. 2010;138(1):82-88.
19. Hulot JS, Collet JP, Silvain J, et al. Cardiovascular risk in clopidogrel-treated patients according to cytochrome P450 2C19*2 loss-of-function allele or proton pump inhibitor coadministration: a systematic meta-analysis. J Am Coll Cardiol. 2010;56(2):134-143.
20. Angiolillo DJ, Gibson CM, Cheng S, et al. Differential effects of omeprazole and pantoprazole on the pharmacodynamics and pharmacokinetics of clopidogrel in healthy subjects: randomized, placebo-controlled, crossover comparison studies. Clin Pharmacol Ther. 2011;89(1):65-74.
21. Goodman SG, Clare R, Pieper KS, et al. Association of proton pump inhibitor use on cardiovascular outcomes with clopidogrel and ticagrelor: insights from the platelet inhibition and patient outcomes trial. Circulation. 2012;125(8):978-986.
22. Bhatt DL, Cryer BL, Contant CF, et al. Clopidogrel with or without omeprazole in coronary artery disease. N Engl J Med. 2010;363(20):1909-1917.
23. Chan FK, Chung SC, Suen BY, et al. Preventing recurrent upper gastrointestinal bleeding in patients with Helicobacter pylori infection who are taking low-dose aspirin or naproxen. N Engl J Med. 2001;344(13):967-973.
24. Hawkey CJ, Karrasch JA, Szczepañski L, et al. Omeprazole compared with misoprostol for ulcers associated with nonsteroidal antiinflammatory drugs. Omeprazole versus Misoprostol for NSAID-induced Ulcer Management (OMNIUM) Study Group. N Engl J Med. 1998;338(11):727-734.
25. Yeomans ND, Tulassay Z, Juhász L, et al. A comparison of omeprazole with ranitidine for ulcers associated with nonsteroidal antiinflammatory drugs. Acid Suppression Trial: Ranitidine versus Omeprazole for NSAID-associated Ulcer Treatment (ASTRONAUT) Study Group. N Engl J Med. 1998;338(11):719-726.
26. Lai KC, Chu KM, Hui WM, et al. Esomeprazole with aspirin versus clopidogrel for prevention of recurrent gastrointestinal ulcer complications. Clin Gastroenterol Hepatol. 2006;4(7):860-865.
27. Chan FK, Ching JY, Hung LC, et al. Clopidogrel versus aspirin and esomeprazole to prevent recurrent ulcer bleeding. N Engl J Med. 2005;352(3):238-244.
28. Lai KC, Lam SK, Chu KM, et al. Lansoprazole for the prevention of recurrences of ulcer complications from long-term low-dose aspirin use. N Engl J Med. 2002;346(26):2033-2038.
29. Plavix [package insert]. Bridgewater, NJ: Bristol-Myers Squibb/Sanofi Pharmaceuticals Partnership; 2011.
30. Gurbel PA, Bliden KP, Butler K, et al. Response to ticagrelor in clopidogrel nonresponders and responders and effect of switching therapies: the RESPOND study. Circulation. 2010;121(10):1188-1199.
31. Wiviott SD, Trenk D, Frelinger AL, et al. Prasugrel compared with high loading- and maintenance-dose clopidogrel in patients with planned percutaneous coronary intervention: the Prasugrel in Comparison to Clopidogrel for Inhibition of Platelet Activation and Aggregation-Thrombolysis in Myocardial Infarction 44 trial. Circulation. 2007;116(25):2923-2932.
32. Bonello L, Pansieri M, Mancini J, et al. High on-treatment platelet reactivity after prasugrel loading dose and cardiovascular events after percutaneous coronary intervention in acute coronary syndromes. J Am Coll Cardiol 2011;58(5):467-473.
33. Breet NJ, van Werkum JW, Bouman HJ, et al. Comparison of platelet function tests in predicting clinical outcome in patients undergoing coronary stent implantation. JAMA. 2010;303(8):754-762.
34. Storey RF, Bliden KP, Patil SB, et al. Incidence of dyspnea and assessment of cardiac and pulmonary function in patients with stable coronary artery disease receiving ticagrelor, clopidogrel, or placebo in the ONSET/OFFSET study. J Am Coll Cardiol. 2010;56(3):185-193.
35. Storey RF, Becker RC, Harrington RA, et al. Characterization of dyspnoea in PLATO study patients treated with ticagrelor or clopidogrel and its association with clinical outcomes. Eur Heart J. 2011;32(23):2945-2953.
36. Gan L-M, Wittfeldt A, Emanuelsson H, Nylander S, Jonasson J. Adenosine may mediate ticagrelor-induced dyspnea. J Am Coll Cardiol 2012;59(13):E344-
Managing the Multiple Symptoms of Benign Prostatic Hyperplasia — CME
Managing Type 2 Diabetes in Men
Meeting New Challenges with Antiplatelet Therapy in Primary Care
Dr. Kuritzky has nothing to disclose.
Dr. Díez has nothing to disclose.
SUPPORT
This program is sponsored by the PCEC and supported by funding from AstraZeneca. Dr. Kuritzky received no financial support for this article.
Introduction
The importance of acute coronary syndrome (ACS) (ie, patients with ST-segment elevation myocardial infarction [MI] [STEMI], non-ST segment elevation MI [NSTEMI], or unstable angina) in primary care is highlighted by its prevalence. Acute coronary syndrome was the primary or secondary discharge diagnosis in 1.19 million hospitalizations in the United States in 2009, a slight majority of which were in men.1 Platelet activation plays a central role in the pathophysiology of ACS. Despite well established benefits of antiplatelet therapy in both primary and secondary prevention of ACS, adverse events—particularly bleeding—require ongoing vigilance.2 Among the several classes of antiplatelet agents currently available, the thromboxane A2 inhibitor (ie, aspirin) and P2Y12 inhibitors (ie, clopidogrel, prasugrel, and ticagrelor) are those most commonly used; ticlopidine is not commonly used due to nausea/vomiting and bone marrow toxicity.3
Antiplatelet Agents
It is well established that hemostasis is protected by multilayered, overlapping, and sometimes redundant pathways. Even though currently available antiplatelet agents are highly efficacious in inhibiting 1 or more phases of platelet activity pertinent to coagulation (eg, activation, adhesion, and aggregation), because of the multiple backup pathways involved, no single antiplatelet agent is anticipated to totally eliminate platelet activity. In addition, every combination of antiplatelet agents—though potentially more efficacious because of multipathway activity—is also laden with greater bleeding risk. The 3 primary pathways of platelet activation for which pharmacologic antagonists have been developed are the thromboxane, adenosine diphosphonate (ADP)-P2Y12, and ADP-A2 pathways. While dual antiplatelet therapy with aspirin and clopidogrel may be the current standard of care, the focus of this review is on the ADP-P2Y12 inhibitors as the two newest agents, prasugrel and ticagrelor, are less familiar to family physicians. The second section addresses questions often encountered by family physicians when caring for patients who have recently experienced ACS.
P2Y12 Inhibitors
Two groups of agents exert their antiplatelet effects by inhibiting the platelet P2Y12 receptor: (1) thienopyridines (ie, ticlopidine, clopidogrel, and prasugrel) and (2) the cyclopentyltriazolopyrimidines (ie, ticagrelor). Both groups inhibit ADP-dependent platelet function but at different sites on the platelet P2Y12 receptor. Thienopyridine activity is mediated via short-lived active metabolites formed in the liver. Platelet exposure to the active metabolite of prasugrel is about 10-fold higher than to the active metabolite of clopidogrel, resulting in a higher level and less individual variation of platelet inhibition with prasugrel. Hepatic metabolism of clopidogrel makes it subject to genetic, as well as drug-induced, variation in activity; prasugrel is not affected by these same limitations. Recovery of platelet function following withdrawal of thienopyridine therapy occurs over 7 to 8 days as a function of platelet turnover.2,3 This slow recovery of platelet function has important implications when any surgical intervention is needed.
In contrast to the thienopyridines, ticagrelor does not require metabolic activation by the liver. Ticagrelor and its active metabolite display approximately equipotent antiplatelet activity and are direct P2Y12 inhibitors. Ticargrelor non-competitively antagonizes ADP-induced receptor activation. Ticagrelor is rapidly absorbed reaching its peak plasma concentration in 1.5 to 3 hours, thereby providing a rapid antiplatelet effect. Twice-daily administration is required because of its rapid offset of platelet inhibition.2,4,5
Prasugrel
Prasugrel is indicated by the US Food and Drug Administration (FDA) for reduction of thrombotic cardiovascular (CV) events (including stent thrombosis) in patients with ACS who are to be managed with percutaneous coronary intervention (PCI) as follows: (1) unstable angina or NSTEMI or (2) STEMI when managed with primary or delayed PCI.6
The efficacy and safety of prasugrel have been investigated in several clinical trials. The Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition with Prasugrel-Thrombolysis in Myocardial Infarction (TRITON-TIMI) 38 is the largest and has many planned sub-analyses ( TABLE 1 ).7-9 TRITON TIMI 38 involved patients with moderate- to high-risk ACS scheduled for PCI
(N = 13,608).7 Patients were randomized to prasugrel 60 mg as a loading dose followed by 10 mg daily or clopidogrel 300 mg as a loading dose followed by 75 mg daily for 6 to 15 months. Aspirin 75 to 162 mg once daily was recommended, but was left up to the physician. The primary efficacy end point was a composite of CV death, nonfatal MI, or nonfatal stroke.
Findings from TRITON TIMI 38 show that, compared with clopidogrel, prasugrel was associated with significantly reduced rates of ischemic events, including nonfatal MI and stent thrombosis. The benefit with prasugrel was primarily due to a significant reduction in the rate of MI compared with clopidogrel. However, patients treated with prasugrel experienced a higher rate of major bleeding, including fatal and life-threatening bleeding. Prasugrel was found to be more effective than clopidogrel in preventing ischemic events without excess bleeding in patients with STEMI undergoing secondary PCI (treated between 12 hours and 14 days after symptom onset). In patients with ACS undergoing PCI without stent implantation, ischemic events occurred at similar rates in patients treated with prasugrel or clopidogrel; however, bleeding was more common with prasugrel.
Not all patients benefited from prasugrel therapy. Compared with clopidogrel, patients with previous stroke/transient ischemic attack (TIA) had net harm from prasugrel. In addition, no net benefit from prasugrel compared with clopidogrel was observed in patients age ≥75 years or body weight <60 kg. The results of TRITON TIMI 38 contributed to the boxed warnings regarding bleeding risk recommending that prasugrel not be used in patients age ≥75 years, in patients with active pathological bleeding or a history of TIA or stroke, or patients likely to undergo coronary artery bypass graft (CABG) surgery. In addition, patients with body weight < 60 kg are also at increased risk for bleeding.6
TABLE 1
Prasugrel: TRITON-TIMI 38 and subanalyses
| TRITON-TIMI 38 Cohort7 | TRITON-TIMI 38 Selected Subanalyses8,9 | ||
|---|---|---|---|
| Treatment | Pr 60 mg LD, then 10 mg QD or Cl 300 mg LD, then 75 mg QD plus Aspirin 75-162 mg QD for 6-15 mos (median 14.5 mos) | ||
| Population | Moderate/High-risk ACS scheduled for PCI (N = 13,608) | PCI for STEMI (N = 3534) | PCI without ST elevation (N = 569) |
| Efficacy Outcomes | Primary end point (CV death, nonfatal MI, or nonfatal stroke):
(P = .31) Nonfatal MI: Cl 9.5% vs Pr 7.3% (P < .001) Nonfatal stroke: Cl 1.0% vs Pr 1.0% (P = .93) Urgent target-vessel revascularization: Cl 3.7% vs Pr 2.5% (P < .001) Stent thrombosis: Cl 2.4% vs Pr 1.1% (P < .001) | Primary end point (CV death, nonfatal MI, or nonfatal stroke):
| Primary end point (CV death, nonfatal MI, or nonfatal stroke): Cl 17.1% vs Pr 14.2% (P = .27) Urgent target-vessel revascularization: Cl 8.2% vs Pr 3.6% (P = .04) |
| Safety Outcomes | Non-CABG TIMI major bleeding: Cl 1.8% vs Pr 2.4% (P = .03) Fatal bleeding: Cl 0.1% vs Pr 0.4% (P = .002) Life-threatening bleeding: Cl 0.9% vs Pr 1.4% (P = .01) Non-fatal bleeding: Cl 0.9% vs Pr 1.1% (P = .23) | TIMI major bleedinga unrelated to CABG:
| TIMI major bleedinga unrelated to CABG: Cl 0% vs Pr 2.1% (P = .03) |
| Key Findings | Prasugrel was associated with significantly reduced rates of ischemic events, including nonfatal MI and stent thrombosis, but with an increased risk of major bleeding, including fatal and life-threatening bleeding. Compared to clopidogrel, patients with previous stroke/TIA had net harm from prasugrel; patients with age ≥ 75 y had no net benefit from prasugrel; patients with BW < 60 kg had no net benefit from prasugrel. | Net clinical outcome All-cause death, MI, stroke, TIMI major bleeding unrelated to CABG:
| In patients with ACS undergoing PCI without stent implantation, ischemic events occurred at similar rates in patients treated with prasugrel or clopidogrel; however, bleeding was more common with prasugrel. |
| ACS, acute coronary syndrome; BW, body weight; CABG, coronary artery bypass graft; Cl, clopidogrel; CV, cardiovascular; LD, loading dose; MI, myocardial infarction; PCI, percutaneous coronary intervention; Pr, prasugrel; QD, once daily; STEMI, ST-segment elevation in myocardial infarction; TIA, transient ischemic attack; TIMI, thrombolysis in myocardial infarction. aTIMI major bleed (intracranial bleed or intrapericardial bleed with cardiac tamponade or a decline of 5.0 g/dL or more in hemoglobin after adjusting for red blood cell transfusions). | |||
Ticagrelor
Ticagrelor is the most recent antiplatelet agent to be approved by the US FDA. Ticagrelor is indicated to reduce the rate of thrombotic CV events in patients with ACS (eg, unstable angina, NSTEMI, or STEMI).10
The efficacy and safety of ticagrelor has been assessed in the Study of Platelet Inhibition and Patient Outcomes (PLATO) and several planned sub-analyses ( TABLE 2 ).11-16 PLATO was a 12-month, multicenter, double-blind, randomized trial that involved patients with ACS with or without ST-segment elevation (N = 18,624).11 Patients were randomized to ticagrelor 180 mg loading dose then 90 mg twice daily or clopidogrel 300 to 600 mg loading dose then 75 mg once daily for 12 months. The primary efficacy end point was a composite of death from vascular causes, MI, or stroke.
The results of PLATO and sub-analyses show that in patients with ACS and compared with clopidogrel, ticagrelor significantly reduced the primary efficacy end point with a similar rate of major bleeding
( TABLE 1 ). These safety results contributed to the boxed warnings regarding bleeding risk that ticagrelor not be used in patients with active pathological bleeding or a history of intracranial hemorrhage, or in patients planned to undergo urgent CABG surgery. In addition, maintenance aspirin therapy at a dose above 100 mg reduces the effectiveness of ticagrelor and should be avoided.10
Consistent with the general PLATO population, in patients intended for non-invasive management, ticagrelor significantly reduced the rate of death from vascular causes, MI, or stroke compared with clopidogrel with a similar rate of major bleeding. In patients with ACS and ST elevation or left bundle branch block planned for PCI, ticagrelor reduced CV and all-cause death, MI, stent thrombosis, and improved survival compared with clopidogrel, with a similar rate of major bleeding. Ticagrelor, compared with clopidogrel, reduced all-cause and CV death without excess risk of CABG-related bleeding in patients with ACS undergoing CABG within 7 days of the last dose of clopidogrel or ticagrelor. Finally, in ACS with chronic kidney disease (estimated creatinine clearance < 60 mL/minute), ticagrelor compared with clopidogrel significantly reduced ischemic end points and mortality without a significant increase in major bleeding and with a similar rate of non–CABG-related bleeding.
TABLE 2
Ticagrelor: PLATO and subanalyses
| PLATO Cohort9,12 | PLATO Selected Subanalyses13-16 | ||||
|---|---|---|---|---|---|
| Treatment | Ti 180 mg LD, then 90 mg BID or Cl 300-600 mg LD then 75 mg QD plus Aspirin 75-325 mg QD for 12 months | ||||
| Population | ACS with/without ST elevation (N = 18,624) | ACS planned for non-invasive management (N = 5216) | ACS with ST elevation or left bundle branch block planned for PCI (N = 7544) | ACS with/without ST elevation managed with CABG (N = 1261) | ACS with/without ST elevation but with chronic kidney disease (eCrCl < 60 mL/min) (n = 3237) |
| Efficacy Outcomes | Primary end point (death from vascular causes, MI, or stroke): Cl 11.7% vs Ti 9.8% (P < .001) Death from any cause, MI, or stroke: Cl 12.3% vs Ti 10.2% (P < .001) Death from any cause, MI, stroke, severe recurrent ischemia, recurrent ischemia, TIA, or other arterial thrombotic event: Cl 16.7% vs Ti 14.6% (P < .001) Death from nonvascular causes: Cl 0.8% vs Ti 0.5% (P = .08) | Primary end point (death from vascular causes, MI, or stroke): Cl 14.3% vs. Ti 12.0% (P = .045) CV death: Cl 7.2% vs Ti 5.5% (P = .019) | Primary end point (death from vascular causes, MI, or stroke): Cl 10.8% vs Ti 9.4% (P = .07) CV death, MI (excluding silent): Cl 10.2% vs Ti 8.4% (P = .01) All cause death, MI (excluding silent), stroke: Cl 11.3% vs Ti 9.8% (P = .05) CV death, total MI, stroke, severe recurrent cardiac ischemia, recurrent cardiac ischemia, TIA, arterial thrombotic events: Cl 15.0% vs Ti 13.3% (P = .03) MI (excluding silent): Cl 5.8% vs Ti 4.7% (P = .03) Stroke: Cl 1.0% vs Ti 1.7% (P = .02) All-cause mortality: Cl 6.1% vs Ti 5.0% (P = .05) Definite, probable, or possible stent thrombosis: Cl 4.3% vs Ti 3.3% (P = .04) | Primary end point (death from vascular causes, MI, or stroke): Cl 13.1% vs Ti 10.6% (P = .29) All-cause death: Cl 9.7% vs Ti 4.7% (P < .01) CV death: Cl 7.9% vs Ti 4.1% (P < .01) Non-CV death: Cl 2.0% vs Ti 0.7% (P = .07) Stroke: Cl 2.1% vs Ti 2.1% (P = .70) | Primary end point (death from vascular causes, MI, or stroke): Cl 22.0% vs Ti 17.3% All-cause death: Cl 14.0% vs Ti 10.0% |
| Safety Outcomes | TIMI major bleedinga: Cl 7.7% vs Ti 7.9% (P = .57) TIMI major bleedinga unrelated to CABG: Cl 2.2% vs Ti 2.8% (P = .03) PLATO major bleedingb: Cl 11.2% vs Ti 11.6% (P = .43) PLATO major bleedingb unrelated to CABG: Cl 3.8% vs Ti 4.5% (P = .03) Dyspnea requiring discontinuation: Cl 0.1% vs Ti 0.9% (P < .001) | PLATO major bleedingb: Cl 10.3% vs Ti 11.9% (P = .079) Life-threatening/fatal bleeding: Cl 5.6% vs Ti 5.5% (P= . 911) Major/Minor bleeding unrelated to CABG: Cl 6.7% vs Ti 8.3% (P = .0182) | PLATO major bleeding: Cl 9.2% vs Ti 9.0% (P = .76) TIMI major bleeding: Cl 6.4% vs Ti 6.1% (P = .66) PLATO non-procedure-related major/ minor bleeding: Cl 3.7% vs Ti 5.1% (P = .02) PLATO minor bleeding: Cl 3.8% vs Ti 4.9% (P = .05) Dyspnea requiring discontinuation: Cl 0.1% vs Ti 0.5% (P = .0004) | Major/Life-threatening CABG-related bleeding causing death within 7 d after CABG: Cl 3.0% vs Ti 1.3% (P = .052) Major CABG bleeding: Cl 80.1% vs Ti 81.2% (P = .669) TIMI major CABG bleeding: Cl 57.6% vs Ti 59.3% (P = .53) | PLATO major bleeding: Cl 14.3% vs Ti 15.1% PLATO fatal major bleeding: Cl 0.77% vs Ti 0.34% PLATO non-CABG major bleeding: Cl 7.3% vs Ti 8.5% Dyspnea: Cl 11.5% vs Ti 16.4% |
| Key Findings | Ticagrelor significantly reduced the rate of CV death, MI, or stroke compared to clopidogrel with a similar rate of major bleeding; ticagrelor led to increased major bleeding unrelated to CABG. Fatal bleeding was low and did not differ between groups. | Consistent with the general PLATO population, ticagrelor significantly reduced the rate of CV death, MI, or stroke compared to clopidogrel with a similar rate of major bleeding. | Consistent with the general PLATO population, compared with clopidogrel, ticagrelor reduced CV and all-cause death, MI, stent thrombosis and improved survival without increasing major bleeding. Ticagrelor resulted in a higher rate of stroke. | Ticagrelor compared with clopidogrel reduced all-cause and CV death without excess risk of CABG-related bleeding in patients with ACS undergoing CABG within 7 days of the last dose of clopidogrel or ticagrelor. | In ACS with CKD, ticagrelor compared with clopidogrel significantly reduced ischemic end points and mortality without a significant increase in major bleeding and with a similar rate of non-procedure- related bleeding. |
| ACS, acute coronary syndrome; BID, twice daily; CABG, coronary artery bypass graft; Cl, clopidogrel; CKD, chronic kidney disease; CV, cardiovascular; eCrCL, estimated creatinine clearance; LD, loading dose; MI, myocardial infarction; PCI, percutaneous coronary intervention; QD, once daily; Ti, ticagrelor; TIA, transient ischemic attack; TIMI, thrombolysis in myocardial infarction. aTIMI major bleed (intracranial bleed or intrapericardial bleed with cardiac tamponade or a decline of 5.0 g/dL or more in hemoglobin after adjusting for red blood cell transfusions). bPLATO major bleed (fatal bleeding, intrapericardial bleeding with cardiac tamponade, intracranial bleeding, severe hypotension, or hypovolemic shock due to bleeding and requiring either vasopressors or surgical intervention, a decline in hemoglobin of 5.0 g/dL or more after adjusting for red blood cell transfusions, or the need for transfusion of 4 or more units of packed red blood cells) | |||||
Common Questions Regarding Antiplatelet Therapy in Primary Care
The preceding discussion confirms that many patients with ACS benefit from antiplatelet therapy. However, the use of antiplatelet agents in primary care can be challenging. The following are some of the evolving issues and questions regarding antiplatelet therapy faced by family physicians.
If a patient has experienced gastrointestinal bleeding while taking low-dose aspirin in the past and has an acute coronary syndrome, what course of action should be taken?
Dual antiplatelet therapy is still recommended in this setting, but therapy with a proton pump inhibitor (PPI) for gastrointestinal (GI) protection is recommended.2,3,17 For patients at low risk of upper GI bleeding, routine PPI prophylaxis is not recommended. Currently available data do not demonstrate the prophylactic superiority of one PPI over another, but do show that PPI therapy is more effective in decreasing GI bleeding associated with aspirin and is, therefore, preferred over a histamine H2 receptor antagonist.17 For instance, high-dose famotidine has been shown to be less effective than pantoprazole in patients with aspirin-related peptic ulcers/erosions.18
Can a proton pump inhibitor be used for gastrointestinal protection in conjunction with clopidogrel?
Yes, although the evidence is conflicting about whether specific PPIs should be avoided because of reduced clinical efficacy of clopidogrel. The results of a meta-analysis of 23 studies demonstrated a clinically significant interaction that reduces the antiplatelet effectiveness of clopidogrel when combined with some PPIs.19 The results of 4 prospective, crossover pharmacokinetic studies in healthy subjects (N = 282) also suggest an interaction between clopidogrel and omeprazole but not between clopidogrel and pantoprazole.20 A subanalysis of PLATO showed that the use of a PPI was independently associated with a higher rate of CV events in patients with ACS treated with clopidogrel or ticagrelor.21 The observed effect with both agents, as well as a higher rate of major bleeding among PPI vs non-PPI users suggests that PPI use may be more of a marker for rather than a cause of higher rates of CV events. In fact, data from the Clopidogrel and the Optimization of Gastrointestinal Events Trial (COGENT) found that in patients treated with clopidogrel and aspirin, the addition of omeprazole reduced the rate of a GI event, compared with placebo at 180 days (1.1% vs. 2.9%, respectively;
P < .001).22 Overt upper GI bleeding occurred less frequently in the omeprazole group (hazard ratio, 0.13; 95% confidence interval, 0.03 to 0.56; P = .001). A CV event was observed in 4.9% of patients treated with omeprazole and 5.7% of placebo patients (P = .96). While limited, these prospective data do not suggest a detriment to clopidogrel efficacy when used in combination with a PPI. The dose of PPI to use for GI protection is not well-established; the following drugs and doses have been used: omeprazole 20 to 40 mg once daily; esomeprazole 20 mg once or twice daily; pantoprazole 20 mg once daily; or lansoprazole 30 mg once daily.18,23-28
Should I avoid starting clopidogrel in patients with acute coronary syndrome because of concerns about “poor metabolizers”?
Clopidogrel is a prodrug, requiring CYP450 metabolism to its active metabolite. Because of genetic CYP450 variations, as many as one-third of patients lack fully active CYP450 pathways, resulting in reduced (or even absent) conversion from the parent drug to the active metabolite, with a corresponding diminution of antiplatelet effects.3,29 Recent recommendations about dealing with these genetic polymorphisms include direct measurement of CYP450 pathway status and selection of alternative pharmacologic agents which are not dependent upon similar CYP pathway activation. There are, unfortunately, no prospective clinical trials based upon CYP2C19 genotyping confirming that patient selection based upon genotyping is associated with improved outcomes.
In terms of alternative antiplatelet therapy in clopidogrel nonresponders, the Response to Ticagrelor in Clopidogrel Nonresponders and Responders (RESPOND) study shows ticagrelor to be beneficial, at least as measured in vitro.30 Following laboratory assessment of patients’ responsiveness to clopidogrel, both responders and nonresponders were randomized to clopidogrel or ticagrelor. After 14 days, all clopidogrel nonresponders and half of the responders switched treatment. The antiplatelet effects of ticagrelor were similar whether the patient was a clopidogrel responder or not. The Prasugrel in Comparison to Clopidogrel for Inhibition of Platelet Activation and Aggregation-Thrombolysis in Myocardial Infarction 44 (PRINCIPLE-TIMI 44) showed higher inhibition of platelet aggregation (IPA) with prasugrel 60 mg compared with clopidogrel 600 mg 6 hours after initiation.31 Following crossover, IPA was higher in subjects receiving prasugrel 10 mg/d compared with clopidogrel 150 mg/d (61% vs 46%, respectively; P < .0001). While not measuring clopidogrel responsiveness, this suggests that prasugrel might be effective in clopidogrel nonresponders. Not all patients treated with prasugrel achieve optimal inhibition of platelet reactivity. In patients who underwent successful PCI for ACS (N = 301) 25.2% were observed to have high on-treatment platelet reactivity following a 60 mg loading dose of prasugrel.32 Such patients had a significantly higher risk for a major adverse cardiovascular event after PCI. The clinical trials which demonstrate improved clinical outcomes when clopidogrel is compared with other antiplatelet agents suggest that the above-mentioned in vitro metrics are clinically relevant.
I’ve heard a lot about testing platelet aggregability. Should I be considering that for my patients?
Not at the present time. One prospective study evaluated the capability of platelet function tests to predict clinical outcome in patients taking clopidogrel undergoing elective stent implantation.33 On-treatment platelet reactivity was measured using: light transmittance aggregometry, VerifyNow P2Y12, Plateletworks, and the IMPACT-R and the platelet function analysis system (PFA-100) (with the Dade PFA collagen/ADP cartridge and Innovance PFA P2Y). After 1 year of follow-up, only the light transmittance aggregometry, VerifyNow, Plateletworks, and Innovance PFA P2Y tests were significantly associated with patient outcome, but had only modest predictive accuracy. Also, none of the tests studied provided accurate prognostic information to identify patients at higher risk of bleeding following stent implantation.
How concerning are the findings on ticagrelor and dyspnea?
The occurrence of dyspnea associated with ticagrelor was observed during its clinical development. While the mechanism is not known, dyspnea is a transient phenomenon, and there is no suggestion that ticagrelor is associated with an increased incidence of heart failure.
The incidence and characterization of dyspnea has been investigated in subanalyses of 2 large clinical trials of ticagrelor. Prospective analysis of the ONSET/OFFSET study (N = 123) showed that dyspnea was experienced by more patients treated with ticagrelor than clopidogrel or placebo over 6 weeks (38.6% vs 9.3% vs 8.3%, respectively; P < .001).34 Episodes of dyspnea were generally mild, lasted <24 hours, and easily tolerated. Moderate dyspnea that led to study discontinuation occurred in 3 patients (5.3%) treated with ticagrelor. Dyspnea occurred within the first 24 hours in 8 of 22 patients (36.4%) and within the first week in 17 of 22 patients (77.3%) of the ticagrelor-treated patients who experienced dyspnea. Dyspnea persisted through the study follow-up (10 days after the 6 week study) in 3 of 22 patients (13.6%) treated with ticagrelor. Dyspnea was not associated with any significant adverse change in cardiac or pulmonary function tests.34
In a subanalysis of the PLATO study to investigate the occurrence of dyspnea (N = 18,421), dyspnea occurred in 14.5% of patients treated with ticagrelor and 8.7% of patients treated with clopidogrel.35 Severe dyspnea occurred in 0.4% and 0.3% of patients, respectively. Dyspnea had no impact on the composite end point after excluding dyspnea that occurred after the secondary end point of MI. The mechanism whereby ticagrelor induces dyspnea is not certain, but may be mediated via an adenosine-related mechanism.36
Conclusion
Aspirin and clopidogrel have been the predominant antiplatelet agents used in the management of patients with ACS, yet their use can be challenging. Differences in the clinical pharmacology of prasugrel and ticagrelor provide the opportunity to address some of these challenges and better enable antiplatelet therapy to be individualized.
Managing the Multiple Symptoms of Benign Prostatic Hyperplasia — CME
Managing Type 2 Diabetes in Men
Meeting New Challenges with Antiplatelet Therapy in Primary Care
Dr. Kuritzky has nothing to disclose.
Dr. Díez has nothing to disclose.
SUPPORT
This program is sponsored by the PCEC and supported by funding from AstraZeneca. Dr. Kuritzky received no financial support for this article.
Introduction
The importance of acute coronary syndrome (ACS) (ie, patients with ST-segment elevation myocardial infarction [MI] [STEMI], non-ST segment elevation MI [NSTEMI], or unstable angina) in primary care is highlighted by its prevalence. Acute coronary syndrome was the primary or secondary discharge diagnosis in 1.19 million hospitalizations in the United States in 2009, a slight majority of which were in men.1 Platelet activation plays a central role in the pathophysiology of ACS. Despite well established benefits of antiplatelet therapy in both primary and secondary prevention of ACS, adverse events—particularly bleeding—require ongoing vigilance.2 Among the several classes of antiplatelet agents currently available, the thromboxane A2 inhibitor (ie, aspirin) and P2Y12 inhibitors (ie, clopidogrel, prasugrel, and ticagrelor) are those most commonly used; ticlopidine is not commonly used due to nausea/vomiting and bone marrow toxicity.3
Antiplatelet Agents
It is well established that hemostasis is protected by multilayered, overlapping, and sometimes redundant pathways. Even though currently available antiplatelet agents are highly efficacious in inhibiting 1 or more phases of platelet activity pertinent to coagulation (eg, activation, adhesion, and aggregation), because of the multiple backup pathways involved, no single antiplatelet agent is anticipated to totally eliminate platelet activity. In addition, every combination of antiplatelet agents—though potentially more efficacious because of multipathway activity—is also laden with greater bleeding risk. The 3 primary pathways of platelet activation for which pharmacologic antagonists have been developed are the thromboxane, adenosine diphosphonate (ADP)-P2Y12, and ADP-A2 pathways. While dual antiplatelet therapy with aspirin and clopidogrel may be the current standard of care, the focus of this review is on the ADP-P2Y12 inhibitors as the two newest agents, prasugrel and ticagrelor, are less familiar to family physicians. The second section addresses questions often encountered by family physicians when caring for patients who have recently experienced ACS.
P2Y12 Inhibitors
Two groups of agents exert their antiplatelet effects by inhibiting the platelet P2Y12 receptor: (1) thienopyridines (ie, ticlopidine, clopidogrel, and prasugrel) and (2) the cyclopentyltriazolopyrimidines (ie, ticagrelor). Both groups inhibit ADP-dependent platelet function but at different sites on the platelet P2Y12 receptor. Thienopyridine activity is mediated via short-lived active metabolites formed in the liver. Platelet exposure to the active metabolite of prasugrel is about 10-fold higher than to the active metabolite of clopidogrel, resulting in a higher level and less individual variation of platelet inhibition with prasugrel. Hepatic metabolism of clopidogrel makes it subject to genetic, as well as drug-induced, variation in activity; prasugrel is not affected by these same limitations. Recovery of platelet function following withdrawal of thienopyridine therapy occurs over 7 to 8 days as a function of platelet turnover.2,3 This slow recovery of platelet function has important implications when any surgical intervention is needed.
In contrast to the thienopyridines, ticagrelor does not require metabolic activation by the liver. Ticagrelor and its active metabolite display approximately equipotent antiplatelet activity and are direct P2Y12 inhibitors. Ticargrelor non-competitively antagonizes ADP-induced receptor activation. Ticagrelor is rapidly absorbed reaching its peak plasma concentration in 1.5 to 3 hours, thereby providing a rapid antiplatelet effect. Twice-daily administration is required because of its rapid offset of platelet inhibition.2,4,5
Prasugrel
Prasugrel is indicated by the US Food and Drug Administration (FDA) for reduction of thrombotic cardiovascular (CV) events (including stent thrombosis) in patients with ACS who are to be managed with percutaneous coronary intervention (PCI) as follows: (1) unstable angina or NSTEMI or (2) STEMI when managed with primary or delayed PCI.6
The efficacy and safety of prasugrel have been investigated in several clinical trials. The Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition with Prasugrel-Thrombolysis in Myocardial Infarction (TRITON-TIMI) 38 is the largest and has many planned sub-analyses ( TABLE 1 ).7-9 TRITON TIMI 38 involved patients with moderate- to high-risk ACS scheduled for PCI
(N = 13,608).7 Patients were randomized to prasugrel 60 mg as a loading dose followed by 10 mg daily or clopidogrel 300 mg as a loading dose followed by 75 mg daily for 6 to 15 months. Aspirin 75 to 162 mg once daily was recommended, but was left up to the physician. The primary efficacy end point was a composite of CV death, nonfatal MI, or nonfatal stroke.
Findings from TRITON TIMI 38 show that, compared with clopidogrel, prasugrel was associated with significantly reduced rates of ischemic events, including nonfatal MI and stent thrombosis. The benefit with prasugrel was primarily due to a significant reduction in the rate of MI compared with clopidogrel. However, patients treated with prasugrel experienced a higher rate of major bleeding, including fatal and life-threatening bleeding. Prasugrel was found to be more effective than clopidogrel in preventing ischemic events without excess bleeding in patients with STEMI undergoing secondary PCI (treated between 12 hours and 14 days after symptom onset). In patients with ACS undergoing PCI without stent implantation, ischemic events occurred at similar rates in patients treated with prasugrel or clopidogrel; however, bleeding was more common with prasugrel.
Not all patients benefited from prasugrel therapy. Compared with clopidogrel, patients with previous stroke/transient ischemic attack (TIA) had net harm from prasugrel. In addition, no net benefit from prasugrel compared with clopidogrel was observed in patients age ≥75 years or body weight <60 kg. The results of TRITON TIMI 38 contributed to the boxed warnings regarding bleeding risk recommending that prasugrel not be used in patients age ≥75 years, in patients with active pathological bleeding or a history of TIA or stroke, or patients likely to undergo coronary artery bypass graft (CABG) surgery. In addition, patients with body weight < 60 kg are also at increased risk for bleeding.6
TABLE 1
Prasugrel: TRITON-TIMI 38 and subanalyses
| TRITON-TIMI 38 Cohort7 | TRITON-TIMI 38 Selected Subanalyses8,9 | ||
|---|---|---|---|
| Treatment | Pr 60 mg LD, then 10 mg QD or Cl 300 mg LD, then 75 mg QD plus Aspirin 75-162 mg QD for 6-15 mos (median 14.5 mos) | ||
| Population | Moderate/High-risk ACS scheduled for PCI (N = 13,608) | PCI for STEMI (N = 3534) | PCI without ST elevation (N = 569) |
| Efficacy Outcomes | Primary end point (CV death, nonfatal MI, or nonfatal stroke):
(P = .31) Nonfatal MI: Cl 9.5% vs Pr 7.3% (P < .001) Nonfatal stroke: Cl 1.0% vs Pr 1.0% (P = .93) Urgent target-vessel revascularization: Cl 3.7% vs Pr 2.5% (P < .001) Stent thrombosis: Cl 2.4% vs Pr 1.1% (P < .001) | Primary end point (CV death, nonfatal MI, or nonfatal stroke):
| Primary end point (CV death, nonfatal MI, or nonfatal stroke): Cl 17.1% vs Pr 14.2% (P = .27) Urgent target-vessel revascularization: Cl 8.2% vs Pr 3.6% (P = .04) |
| Safety Outcomes | Non-CABG TIMI major bleeding: Cl 1.8% vs Pr 2.4% (P = .03) Fatal bleeding: Cl 0.1% vs Pr 0.4% (P = .002) Life-threatening bleeding: Cl 0.9% vs Pr 1.4% (P = .01) Non-fatal bleeding: Cl 0.9% vs Pr 1.1% (P = .23) | TIMI major bleedinga unrelated to CABG:
| TIMI major bleedinga unrelated to CABG: Cl 0% vs Pr 2.1% (P = .03) |
| Key Findings | Prasugrel was associated with significantly reduced rates of ischemic events, including nonfatal MI and stent thrombosis, but with an increased risk of major bleeding, including fatal and life-threatening bleeding. Compared to clopidogrel, patients with previous stroke/TIA had net harm from prasugrel; patients with age ≥ 75 y had no net benefit from prasugrel; patients with BW < 60 kg had no net benefit from prasugrel. | Net clinical outcome All-cause death, MI, stroke, TIMI major bleeding unrelated to CABG:
| In patients with ACS undergoing PCI without stent implantation, ischemic events occurred at similar rates in patients treated with prasugrel or clopidogrel; however, bleeding was more common with prasugrel. |
| ACS, acute coronary syndrome; BW, body weight; CABG, coronary artery bypass graft; Cl, clopidogrel; CV, cardiovascular; LD, loading dose; MI, myocardial infarction; PCI, percutaneous coronary intervention; Pr, prasugrel; QD, once daily; STEMI, ST-segment elevation in myocardial infarction; TIA, transient ischemic attack; TIMI, thrombolysis in myocardial infarction. aTIMI major bleed (intracranial bleed or intrapericardial bleed with cardiac tamponade or a decline of 5.0 g/dL or more in hemoglobin after adjusting for red blood cell transfusions). | |||
Ticagrelor
Ticagrelor is the most recent antiplatelet agent to be approved by the US FDA. Ticagrelor is indicated to reduce the rate of thrombotic CV events in patients with ACS (eg, unstable angina, NSTEMI, or STEMI).10
The efficacy and safety of ticagrelor has been assessed in the Study of Platelet Inhibition and Patient Outcomes (PLATO) and several planned sub-analyses ( TABLE 2 ).11-16 PLATO was a 12-month, multicenter, double-blind, randomized trial that involved patients with ACS with or without ST-segment elevation (N = 18,624).11 Patients were randomized to ticagrelor 180 mg loading dose then 90 mg twice daily or clopidogrel 300 to 600 mg loading dose then 75 mg once daily for 12 months. The primary efficacy end point was a composite of death from vascular causes, MI, or stroke.
The results of PLATO and sub-analyses show that in patients with ACS and compared with clopidogrel, ticagrelor significantly reduced the primary efficacy end point with a similar rate of major bleeding
( TABLE 1 ). These safety results contributed to the boxed warnings regarding bleeding risk that ticagrelor not be used in patients with active pathological bleeding or a history of intracranial hemorrhage, or in patients planned to undergo urgent CABG surgery. In addition, maintenance aspirin therapy at a dose above 100 mg reduces the effectiveness of ticagrelor and should be avoided.10
Consistent with the general PLATO population, in patients intended for non-invasive management, ticagrelor significantly reduced the rate of death from vascular causes, MI, or stroke compared with clopidogrel with a similar rate of major bleeding. In patients with ACS and ST elevation or left bundle branch block planned for PCI, ticagrelor reduced CV and all-cause death, MI, stent thrombosis, and improved survival compared with clopidogrel, with a similar rate of major bleeding. Ticagrelor, compared with clopidogrel, reduced all-cause and CV death without excess risk of CABG-related bleeding in patients with ACS undergoing CABG within 7 days of the last dose of clopidogrel or ticagrelor. Finally, in ACS with chronic kidney disease (estimated creatinine clearance < 60 mL/minute), ticagrelor compared with clopidogrel significantly reduced ischemic end points and mortality without a significant increase in major bleeding and with a similar rate of non–CABG-related bleeding.
TABLE 2
Ticagrelor: PLATO and subanalyses
| PLATO Cohort9,12 | PLATO Selected Subanalyses13-16 | ||||
|---|---|---|---|---|---|
| Treatment | Ti 180 mg LD, then 90 mg BID or Cl 300-600 mg LD then 75 mg QD plus Aspirin 75-325 mg QD for 12 months | ||||
| Population | ACS with/without ST elevation (N = 18,624) | ACS planned for non-invasive management (N = 5216) | ACS with ST elevation or left bundle branch block planned for PCI (N = 7544) | ACS with/without ST elevation managed with CABG (N = 1261) | ACS with/without ST elevation but with chronic kidney disease (eCrCl < 60 mL/min) (n = 3237) |
| Efficacy Outcomes | Primary end point (death from vascular causes, MI, or stroke): Cl 11.7% vs Ti 9.8% (P < .001) Death from any cause, MI, or stroke: Cl 12.3% vs Ti 10.2% (P < .001) Death from any cause, MI, stroke, severe recurrent ischemia, recurrent ischemia, TIA, or other arterial thrombotic event: Cl 16.7% vs Ti 14.6% (P < .001) Death from nonvascular causes: Cl 0.8% vs Ti 0.5% (P = .08) | Primary end point (death from vascular causes, MI, or stroke): Cl 14.3% vs. Ti 12.0% (P = .045) CV death: Cl 7.2% vs Ti 5.5% (P = .019) | Primary end point (death from vascular causes, MI, or stroke): Cl 10.8% vs Ti 9.4% (P = .07) CV death, MI (excluding silent): Cl 10.2% vs Ti 8.4% (P = .01) All cause death, MI (excluding silent), stroke: Cl 11.3% vs Ti 9.8% (P = .05) CV death, total MI, stroke, severe recurrent cardiac ischemia, recurrent cardiac ischemia, TIA, arterial thrombotic events: Cl 15.0% vs Ti 13.3% (P = .03) MI (excluding silent): Cl 5.8% vs Ti 4.7% (P = .03) Stroke: Cl 1.0% vs Ti 1.7% (P = .02) All-cause mortality: Cl 6.1% vs Ti 5.0% (P = .05) Definite, probable, or possible stent thrombosis: Cl 4.3% vs Ti 3.3% (P = .04) | Primary end point (death from vascular causes, MI, or stroke): Cl 13.1% vs Ti 10.6% (P = .29) All-cause death: Cl 9.7% vs Ti 4.7% (P < .01) CV death: Cl 7.9% vs Ti 4.1% (P < .01) Non-CV death: Cl 2.0% vs Ti 0.7% (P = .07) Stroke: Cl 2.1% vs Ti 2.1% (P = .70) | Primary end point (death from vascular causes, MI, or stroke): Cl 22.0% vs Ti 17.3% All-cause death: Cl 14.0% vs Ti 10.0% |
| Safety Outcomes | TIMI major bleedinga: Cl 7.7% vs Ti 7.9% (P = .57) TIMI major bleedinga unrelated to CABG: Cl 2.2% vs Ti 2.8% (P = .03) PLATO major bleedingb: Cl 11.2% vs Ti 11.6% (P = .43) PLATO major bleedingb unrelated to CABG: Cl 3.8% vs Ti 4.5% (P = .03) Dyspnea requiring discontinuation: Cl 0.1% vs Ti 0.9% (P < .001) | PLATO major bleedingb: Cl 10.3% vs Ti 11.9% (P = .079) Life-threatening/fatal bleeding: Cl 5.6% vs Ti 5.5% (P= . 911) Major/Minor bleeding unrelated to CABG: Cl 6.7% vs Ti 8.3% (P = .0182) | PLATO major bleeding: Cl 9.2% vs Ti 9.0% (P = .76) TIMI major bleeding: Cl 6.4% vs Ti 6.1% (P = .66) PLATO non-procedure-related major/ minor bleeding: Cl 3.7% vs Ti 5.1% (P = .02) PLATO minor bleeding: Cl 3.8% vs Ti 4.9% (P = .05) Dyspnea requiring discontinuation: Cl 0.1% vs Ti 0.5% (P = .0004) | Major/Life-threatening CABG-related bleeding causing death within 7 d after CABG: Cl 3.0% vs Ti 1.3% (P = .052) Major CABG bleeding: Cl 80.1% vs Ti 81.2% (P = .669) TIMI major CABG bleeding: Cl 57.6% vs Ti 59.3% (P = .53) | PLATO major bleeding: Cl 14.3% vs Ti 15.1% PLATO fatal major bleeding: Cl 0.77% vs Ti 0.34% PLATO non-CABG major bleeding: Cl 7.3% vs Ti 8.5% Dyspnea: Cl 11.5% vs Ti 16.4% |
| Key Findings | Ticagrelor significantly reduced the rate of CV death, MI, or stroke compared to clopidogrel with a similar rate of major bleeding; ticagrelor led to increased major bleeding unrelated to CABG. Fatal bleeding was low and did not differ between groups. | Consistent with the general PLATO population, ticagrelor significantly reduced the rate of CV death, MI, or stroke compared to clopidogrel with a similar rate of major bleeding. | Consistent with the general PLATO population, compared with clopidogrel, ticagrelor reduced CV and all-cause death, MI, stent thrombosis and improved survival without increasing major bleeding. Ticagrelor resulted in a higher rate of stroke. | Ticagrelor compared with clopidogrel reduced all-cause and CV death without excess risk of CABG-related bleeding in patients with ACS undergoing CABG within 7 days of the last dose of clopidogrel or ticagrelor. | In ACS with CKD, ticagrelor compared with clopidogrel significantly reduced ischemic end points and mortality without a significant increase in major bleeding and with a similar rate of non-procedure- related bleeding. |
| ACS, acute coronary syndrome; BID, twice daily; CABG, coronary artery bypass graft; Cl, clopidogrel; CKD, chronic kidney disease; CV, cardiovascular; eCrCL, estimated creatinine clearance; LD, loading dose; MI, myocardial infarction; PCI, percutaneous coronary intervention; QD, once daily; Ti, ticagrelor; TIA, transient ischemic attack; TIMI, thrombolysis in myocardial infarction. aTIMI major bleed (intracranial bleed or intrapericardial bleed with cardiac tamponade or a decline of 5.0 g/dL or more in hemoglobin after adjusting for red blood cell transfusions). bPLATO major bleed (fatal bleeding, intrapericardial bleeding with cardiac tamponade, intracranial bleeding, severe hypotension, or hypovolemic shock due to bleeding and requiring either vasopressors or surgical intervention, a decline in hemoglobin of 5.0 g/dL or more after adjusting for red blood cell transfusions, or the need for transfusion of 4 or more units of packed red blood cells) | |||||
Common Questions Regarding Antiplatelet Therapy in Primary Care
The preceding discussion confirms that many patients with ACS benefit from antiplatelet therapy. However, the use of antiplatelet agents in primary care can be challenging. The following are some of the evolving issues and questions regarding antiplatelet therapy faced by family physicians.
If a patient has experienced gastrointestinal bleeding while taking low-dose aspirin in the past and has an acute coronary syndrome, what course of action should be taken?
Dual antiplatelet therapy is still recommended in this setting, but therapy with a proton pump inhibitor (PPI) for gastrointestinal (GI) protection is recommended.2,3,17 For patients at low risk of upper GI bleeding, routine PPI prophylaxis is not recommended. Currently available data do not demonstrate the prophylactic superiority of one PPI over another, but do show that PPI therapy is more effective in decreasing GI bleeding associated with aspirin and is, therefore, preferred over a histamine H2 receptor antagonist.17 For instance, high-dose famotidine has been shown to be less effective than pantoprazole in patients with aspirin-related peptic ulcers/erosions.18
Can a proton pump inhibitor be used for gastrointestinal protection in conjunction with clopidogrel?
Yes, although the evidence is conflicting about whether specific PPIs should be avoided because of reduced clinical efficacy of clopidogrel. The results of a meta-analysis of 23 studies demonstrated a clinically significant interaction that reduces the antiplatelet effectiveness of clopidogrel when combined with some PPIs.19 The results of 4 prospective, crossover pharmacokinetic studies in healthy subjects (N = 282) also suggest an interaction between clopidogrel and omeprazole but not between clopidogrel and pantoprazole.20 A subanalysis of PLATO showed that the use of a PPI was independently associated with a higher rate of CV events in patients with ACS treated with clopidogrel or ticagrelor.21 The observed effect with both agents, as well as a higher rate of major bleeding among PPI vs non-PPI users suggests that PPI use may be more of a marker for rather than a cause of higher rates of CV events. In fact, data from the Clopidogrel and the Optimization of Gastrointestinal Events Trial (COGENT) found that in patients treated with clopidogrel and aspirin, the addition of omeprazole reduced the rate of a GI event, compared with placebo at 180 days (1.1% vs. 2.9%, respectively;
P < .001).22 Overt upper GI bleeding occurred less frequently in the omeprazole group (hazard ratio, 0.13; 95% confidence interval, 0.03 to 0.56; P = .001). A CV event was observed in 4.9% of patients treated with omeprazole and 5.7% of placebo patients (P = .96). While limited, these prospective data do not suggest a detriment to clopidogrel efficacy when used in combination with a PPI. The dose of PPI to use for GI protection is not well-established; the following drugs and doses have been used: omeprazole 20 to 40 mg once daily; esomeprazole 20 mg once or twice daily; pantoprazole 20 mg once daily; or lansoprazole 30 mg once daily.18,23-28
Should I avoid starting clopidogrel in patients with acute coronary syndrome because of concerns about “poor metabolizers”?
Clopidogrel is a prodrug, requiring CYP450 metabolism to its active metabolite. Because of genetic CYP450 variations, as many as one-third of patients lack fully active CYP450 pathways, resulting in reduced (or even absent) conversion from the parent drug to the active metabolite, with a corresponding diminution of antiplatelet effects.3,29 Recent recommendations about dealing with these genetic polymorphisms include direct measurement of CYP450 pathway status and selection of alternative pharmacologic agents which are not dependent upon similar CYP pathway activation. There are, unfortunately, no prospective clinical trials based upon CYP2C19 genotyping confirming that patient selection based upon genotyping is associated with improved outcomes.
In terms of alternative antiplatelet therapy in clopidogrel nonresponders, the Response to Ticagrelor in Clopidogrel Nonresponders and Responders (RESPOND) study shows ticagrelor to be beneficial, at least as measured in vitro.30 Following laboratory assessment of patients’ responsiveness to clopidogrel, both responders and nonresponders were randomized to clopidogrel or ticagrelor. After 14 days, all clopidogrel nonresponders and half of the responders switched treatment. The antiplatelet effects of ticagrelor were similar whether the patient was a clopidogrel responder or not. The Prasugrel in Comparison to Clopidogrel for Inhibition of Platelet Activation and Aggregation-Thrombolysis in Myocardial Infarction 44 (PRINCIPLE-TIMI 44) showed higher inhibition of platelet aggregation (IPA) with prasugrel 60 mg compared with clopidogrel 600 mg 6 hours after initiation.31 Following crossover, IPA was higher in subjects receiving prasugrel 10 mg/d compared with clopidogrel 150 mg/d (61% vs 46%, respectively; P < .0001). While not measuring clopidogrel responsiveness, this suggests that prasugrel might be effective in clopidogrel nonresponders. Not all patients treated with prasugrel achieve optimal inhibition of platelet reactivity. In patients who underwent successful PCI for ACS (N = 301) 25.2% were observed to have high on-treatment platelet reactivity following a 60 mg loading dose of prasugrel.32 Such patients had a significantly higher risk for a major adverse cardiovascular event after PCI. The clinical trials which demonstrate improved clinical outcomes when clopidogrel is compared with other antiplatelet agents suggest that the above-mentioned in vitro metrics are clinically relevant.
I’ve heard a lot about testing platelet aggregability. Should I be considering that for my patients?
Not at the present time. One prospective study evaluated the capability of platelet function tests to predict clinical outcome in patients taking clopidogrel undergoing elective stent implantation.33 On-treatment platelet reactivity was measured using: light transmittance aggregometry, VerifyNow P2Y12, Plateletworks, and the IMPACT-R and the platelet function analysis system (PFA-100) (with the Dade PFA collagen/ADP cartridge and Innovance PFA P2Y). After 1 year of follow-up, only the light transmittance aggregometry, VerifyNow, Plateletworks, and Innovance PFA P2Y tests were significantly associated with patient outcome, but had only modest predictive accuracy. Also, none of the tests studied provided accurate prognostic information to identify patients at higher risk of bleeding following stent implantation.
How concerning are the findings on ticagrelor and dyspnea?
The occurrence of dyspnea associated with ticagrelor was observed during its clinical development. While the mechanism is not known, dyspnea is a transient phenomenon, and there is no suggestion that ticagrelor is associated with an increased incidence of heart failure.
The incidence and characterization of dyspnea has been investigated in subanalyses of 2 large clinical trials of ticagrelor. Prospective analysis of the ONSET/OFFSET study (N = 123) showed that dyspnea was experienced by more patients treated with ticagrelor than clopidogrel or placebo over 6 weeks (38.6% vs 9.3% vs 8.3%, respectively; P < .001).34 Episodes of dyspnea were generally mild, lasted <24 hours, and easily tolerated. Moderate dyspnea that led to study discontinuation occurred in 3 patients (5.3%) treated with ticagrelor. Dyspnea occurred within the first 24 hours in 8 of 22 patients (36.4%) and within the first week in 17 of 22 patients (77.3%) of the ticagrelor-treated patients who experienced dyspnea. Dyspnea persisted through the study follow-up (10 days after the 6 week study) in 3 of 22 patients (13.6%) treated with ticagrelor. Dyspnea was not associated with any significant adverse change in cardiac or pulmonary function tests.34
In a subanalysis of the PLATO study to investigate the occurrence of dyspnea (N = 18,421), dyspnea occurred in 14.5% of patients treated with ticagrelor and 8.7% of patients treated with clopidogrel.35 Severe dyspnea occurred in 0.4% and 0.3% of patients, respectively. Dyspnea had no impact on the composite end point after excluding dyspnea that occurred after the secondary end point of MI. The mechanism whereby ticagrelor induces dyspnea is not certain, but may be mediated via an adenosine-related mechanism.36
Conclusion
Aspirin and clopidogrel have been the predominant antiplatelet agents used in the management of patients with ACS, yet their use can be challenging. Differences in the clinical pharmacology of prasugrel and ticagrelor provide the opportunity to address some of these challenges and better enable antiplatelet therapy to be individualized.
1. Roger VL, Go AS, Lloyd-Jones DM, et al. Heart disease and stroke statistics—2012 update: a report from the American Heart Association. Circulation. 2012;125(1):e2-e220.
2. Patrono C, Andreotti F, Arnesen H, et al. Antiplatelet agents for the treatment and prevention of atherothrombosis. Eur Heart J. 2011;32(23):2922-2932.
3. Eikelboom JW, Hirsh J, Spencer FA, Baglin TP, Weitz JI. Antiplatelet Drugs: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141 (2 Suppl):e89S-e119S.
4. Abergel E, Nikolsky E. Ticagrelor: an investigational oral antiplatelet treatment for reduction of major adverse cardiac events in patients with acute coronary syndrome. Vasc Health Risk Manag. 2010;6:963-977.
5. Gurbel PA, Bliden KP, Butler K, et al. Randomized double-blind assessment of the ONSET and OFFSET of the antiplatelet effects of ticagrelor versus clopidogrel in patients with stable coronary artery disease: the ONSET/OFFSET study. Circulation. 2009;120(25):2577-2585.
6. Effient [package insert]. Indianapolis, IN: Eli Lilly and Co.; 2011.
7. Wiviott SD, Braunwald E, McCabe CH, et al. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2007;357(20):2001-2015.
8. Montalescot G, Wiviott SD, Braunwald E, et al. Prasugrel compared with clopidogrel in patients undergoing percutaneous coronary intervention for ST-elevation myocardial infarction (TRITON-TIMI 38): double-blind, randomised controlled trial. Lancet. 2009;373(9665):723-731.
9. Pride YB, Wiviott SD, Buros JL, et al. Effect of prasugrel versus clopidogrel on outcomes among patients with acute coronary syndrome undergoing percutaneous coronary intervention without stent implantation: a TRial to assess Improvement in Therapeutic Outcomes by optimizing platelet inhibitioN with prasugrel (TRITON)-Thrombolysis in Myocardial Infarction (TIMI) 38 substudy. Am Heart J. 2009;158(3):e21-e26.
10. Brilinta [package insert]. Wilmington, DE: AstraZeneca; 2011.
11. Wallentin L, Becker RC, Budaj A, et al. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2009;361(11):1045-1057.
12. Becker RC, Bassand JP, Budaj A, et al. Bleeding complications with the P2Y12 receptor antagonists clopidogrel and ticagrelor in the PLATelet inhibition and patient Outcomes (PLATO) trial. Eur Heart J. 2011;32(23):2933-2944.
13. James SK, Roe MT, Cannon CP, et al. Ticagrelor versus clopidogrel in patients with acute coronary syndromes intended for non-invasive management: substudy from prospective randomised PLATelet inhibition and patient Outcomes (PLATO) trial. BMJ. 2011;342:d3527-
14. Steg PG, James S, Harrington RA, et al. Ticagrelor versus clopidogrel in patients with ST-elevation acute coronary syndromes intended for reperfusion with primary percutaneous coronary intervention: A Platelet Inhibition and Patient Outcomes (PLATO) trial subgroup analysis. Circulation. 2010;122(21):2131-2141.
15. Held C, Asenblad N, Bassand JP, et al. Ticagrelor versus clopidogrel in patients with acute coronary syndromes undergoing coronary artery bypass surgery: results from the PLATO (Platelet Inhibition and Patient Outcomes) trial. J Am Coll Cardiol. 2011;57(6):672-684.
16. James S, Budaj A, Aylward P, et al. Ticagrelor versus clopidogrel in acute coronary syndromes in relation to renal function: results from the Platelet Inhibition and Patient Outcomes (PLATO) trial. Circulation. 2010;122(11):1056-1067.
17. Abraham NS, Hlatky MA, Antman EM, et al. ACCF/ACG/AHA 2010 Expert Consensus Document on the concomitant use of proton pump inhibitors and thienopyridines: a focused update of the ACCF/ACG/AHA 2008 expert consensus document on reducing the gastrointestinal risks of antiplatelet therapy and NSAID use: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents. Circulation. 2010;122(24):2619-2633.
18. Ng FH, Wong SY, Lam KF, et al. Famotidine is inferior to pantoprazole in preventing recurrence of aspirin-related peptic ulcers or erosions. Gastroenterology. 2010;138(1):82-88.
19. Hulot JS, Collet JP, Silvain J, et al. Cardiovascular risk in clopidogrel-treated patients according to cytochrome P450 2C19*2 loss-of-function allele or proton pump inhibitor coadministration: a systematic meta-analysis. J Am Coll Cardiol. 2010;56(2):134-143.
20. Angiolillo DJ, Gibson CM, Cheng S, et al. Differential effects of omeprazole and pantoprazole on the pharmacodynamics and pharmacokinetics of clopidogrel in healthy subjects: randomized, placebo-controlled, crossover comparison studies. Clin Pharmacol Ther. 2011;89(1):65-74.
21. Goodman SG, Clare R, Pieper KS, et al. Association of proton pump inhibitor use on cardiovascular outcomes with clopidogrel and ticagrelor: insights from the platelet inhibition and patient outcomes trial. Circulation. 2012;125(8):978-986.
22. Bhatt DL, Cryer BL, Contant CF, et al. Clopidogrel with or without omeprazole in coronary artery disease. N Engl J Med. 2010;363(20):1909-1917.
23. Chan FK, Chung SC, Suen BY, et al. Preventing recurrent upper gastrointestinal bleeding in patients with Helicobacter pylori infection who are taking low-dose aspirin or naproxen. N Engl J Med. 2001;344(13):967-973.
24. Hawkey CJ, Karrasch JA, Szczepañski L, et al. Omeprazole compared with misoprostol for ulcers associated with nonsteroidal antiinflammatory drugs. Omeprazole versus Misoprostol for NSAID-induced Ulcer Management (OMNIUM) Study Group. N Engl J Med. 1998;338(11):727-734.
25. Yeomans ND, Tulassay Z, Juhász L, et al. A comparison of omeprazole with ranitidine for ulcers associated with nonsteroidal antiinflammatory drugs. Acid Suppression Trial: Ranitidine versus Omeprazole for NSAID-associated Ulcer Treatment (ASTRONAUT) Study Group. N Engl J Med. 1998;338(11):719-726.
26. Lai KC, Chu KM, Hui WM, et al. Esomeprazole with aspirin versus clopidogrel for prevention of recurrent gastrointestinal ulcer complications. Clin Gastroenterol Hepatol. 2006;4(7):860-865.
27. Chan FK, Ching JY, Hung LC, et al. Clopidogrel versus aspirin and esomeprazole to prevent recurrent ulcer bleeding. N Engl J Med. 2005;352(3):238-244.
28. Lai KC, Lam SK, Chu KM, et al. Lansoprazole for the prevention of recurrences of ulcer complications from long-term low-dose aspirin use. N Engl J Med. 2002;346(26):2033-2038.
29. Plavix [package insert]. Bridgewater, NJ: Bristol-Myers Squibb/Sanofi Pharmaceuticals Partnership; 2011.
30. Gurbel PA, Bliden KP, Butler K, et al. Response to ticagrelor in clopidogrel nonresponders and responders and effect of switching therapies: the RESPOND study. Circulation. 2010;121(10):1188-1199.
31. Wiviott SD, Trenk D, Frelinger AL, et al. Prasugrel compared with high loading- and maintenance-dose clopidogrel in patients with planned percutaneous coronary intervention: the Prasugrel in Comparison to Clopidogrel for Inhibition of Platelet Activation and Aggregation-Thrombolysis in Myocardial Infarction 44 trial. Circulation. 2007;116(25):2923-2932.
32. Bonello L, Pansieri M, Mancini J, et al. High on-treatment platelet reactivity after prasugrel loading dose and cardiovascular events after percutaneous coronary intervention in acute coronary syndromes. J Am Coll Cardiol 2011;58(5):467-473.
33. Breet NJ, van Werkum JW, Bouman HJ, et al. Comparison of platelet function tests in predicting clinical outcome in patients undergoing coronary stent implantation. JAMA. 2010;303(8):754-762.
34. Storey RF, Bliden KP, Patil SB, et al. Incidence of dyspnea and assessment of cardiac and pulmonary function in patients with stable coronary artery disease receiving ticagrelor, clopidogrel, or placebo in the ONSET/OFFSET study. J Am Coll Cardiol. 2010;56(3):185-193.
35. Storey RF, Becker RC, Harrington RA, et al. Characterization of dyspnoea in PLATO study patients treated with ticagrelor or clopidogrel and its association with clinical outcomes. Eur Heart J. 2011;32(23):2945-2953.
36. Gan L-M, Wittfeldt A, Emanuelsson H, Nylander S, Jonasson J. Adenosine may mediate ticagrelor-induced dyspnea. J Am Coll Cardiol 2012;59(13):E344-
1. Roger VL, Go AS, Lloyd-Jones DM, et al. Heart disease and stroke statistics—2012 update: a report from the American Heart Association. Circulation. 2012;125(1):e2-e220.
2. Patrono C, Andreotti F, Arnesen H, et al. Antiplatelet agents for the treatment and prevention of atherothrombosis. Eur Heart J. 2011;32(23):2922-2932.
3. Eikelboom JW, Hirsh J, Spencer FA, Baglin TP, Weitz JI. Antiplatelet Drugs: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141 (2 Suppl):e89S-e119S.
4. Abergel E, Nikolsky E. Ticagrelor: an investigational oral antiplatelet treatment for reduction of major adverse cardiac events in patients with acute coronary syndrome. Vasc Health Risk Manag. 2010;6:963-977.
5. Gurbel PA, Bliden KP, Butler K, et al. Randomized double-blind assessment of the ONSET and OFFSET of the antiplatelet effects of ticagrelor versus clopidogrel in patients with stable coronary artery disease: the ONSET/OFFSET study. Circulation. 2009;120(25):2577-2585.
6. Effient [package insert]. Indianapolis, IN: Eli Lilly and Co.; 2011.
7. Wiviott SD, Braunwald E, McCabe CH, et al. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2007;357(20):2001-2015.
8. Montalescot G, Wiviott SD, Braunwald E, et al. Prasugrel compared with clopidogrel in patients undergoing percutaneous coronary intervention for ST-elevation myocardial infarction (TRITON-TIMI 38): double-blind, randomised controlled trial. Lancet. 2009;373(9665):723-731.
9. Pride YB, Wiviott SD, Buros JL, et al. Effect of prasugrel versus clopidogrel on outcomes among patients with acute coronary syndrome undergoing percutaneous coronary intervention without stent implantation: a TRial to assess Improvement in Therapeutic Outcomes by optimizing platelet inhibitioN with prasugrel (TRITON)-Thrombolysis in Myocardial Infarction (TIMI) 38 substudy. Am Heart J. 2009;158(3):e21-e26.
10. Brilinta [package insert]. Wilmington, DE: AstraZeneca; 2011.
11. Wallentin L, Becker RC, Budaj A, et al. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2009;361(11):1045-1057.
12. Becker RC, Bassand JP, Budaj A, et al. Bleeding complications with the P2Y12 receptor antagonists clopidogrel and ticagrelor in the PLATelet inhibition and patient Outcomes (PLATO) trial. Eur Heart J. 2011;32(23):2933-2944.
13. James SK, Roe MT, Cannon CP, et al. Ticagrelor versus clopidogrel in patients with acute coronary syndromes intended for non-invasive management: substudy from prospective randomised PLATelet inhibition and patient Outcomes (PLATO) trial. BMJ. 2011;342:d3527-
14. Steg PG, James S, Harrington RA, et al. Ticagrelor versus clopidogrel in patients with ST-elevation acute coronary syndromes intended for reperfusion with primary percutaneous coronary intervention: A Platelet Inhibition and Patient Outcomes (PLATO) trial subgroup analysis. Circulation. 2010;122(21):2131-2141.
15. Held C, Asenblad N, Bassand JP, et al. Ticagrelor versus clopidogrel in patients with acute coronary syndromes undergoing coronary artery bypass surgery: results from the PLATO (Platelet Inhibition and Patient Outcomes) trial. J Am Coll Cardiol. 2011;57(6):672-684.
16. James S, Budaj A, Aylward P, et al. Ticagrelor versus clopidogrel in acute coronary syndromes in relation to renal function: results from the Platelet Inhibition and Patient Outcomes (PLATO) trial. Circulation. 2010;122(11):1056-1067.
17. Abraham NS, Hlatky MA, Antman EM, et al. ACCF/ACG/AHA 2010 Expert Consensus Document on the concomitant use of proton pump inhibitors and thienopyridines: a focused update of the ACCF/ACG/AHA 2008 expert consensus document on reducing the gastrointestinal risks of antiplatelet therapy and NSAID use: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents. Circulation. 2010;122(24):2619-2633.
18. Ng FH, Wong SY, Lam KF, et al. Famotidine is inferior to pantoprazole in preventing recurrence of aspirin-related peptic ulcers or erosions. Gastroenterology. 2010;138(1):82-88.
19. Hulot JS, Collet JP, Silvain J, et al. Cardiovascular risk in clopidogrel-treated patients according to cytochrome P450 2C19*2 loss-of-function allele or proton pump inhibitor coadministration: a systematic meta-analysis. J Am Coll Cardiol. 2010;56(2):134-143.
20. Angiolillo DJ, Gibson CM, Cheng S, et al. Differential effects of omeprazole and pantoprazole on the pharmacodynamics and pharmacokinetics of clopidogrel in healthy subjects: randomized, placebo-controlled, crossover comparison studies. Clin Pharmacol Ther. 2011;89(1):65-74.
21. Goodman SG, Clare R, Pieper KS, et al. Association of proton pump inhibitor use on cardiovascular outcomes with clopidogrel and ticagrelor: insights from the platelet inhibition and patient outcomes trial. Circulation. 2012;125(8):978-986.
22. Bhatt DL, Cryer BL, Contant CF, et al. Clopidogrel with or without omeprazole in coronary artery disease. N Engl J Med. 2010;363(20):1909-1917.
23. Chan FK, Chung SC, Suen BY, et al. Preventing recurrent upper gastrointestinal bleeding in patients with Helicobacter pylori infection who are taking low-dose aspirin or naproxen. N Engl J Med. 2001;344(13):967-973.
24. Hawkey CJ, Karrasch JA, Szczepañski L, et al. Omeprazole compared with misoprostol for ulcers associated with nonsteroidal antiinflammatory drugs. Omeprazole versus Misoprostol for NSAID-induced Ulcer Management (OMNIUM) Study Group. N Engl J Med. 1998;338(11):727-734.
25. Yeomans ND, Tulassay Z, Juhász L, et al. A comparison of omeprazole with ranitidine for ulcers associated with nonsteroidal antiinflammatory drugs. Acid Suppression Trial: Ranitidine versus Omeprazole for NSAID-associated Ulcer Treatment (ASTRONAUT) Study Group. N Engl J Med. 1998;338(11):719-726.
26. Lai KC, Chu KM, Hui WM, et al. Esomeprazole with aspirin versus clopidogrel for prevention of recurrent gastrointestinal ulcer complications. Clin Gastroenterol Hepatol. 2006;4(7):860-865.
27. Chan FK, Ching JY, Hung LC, et al. Clopidogrel versus aspirin and esomeprazole to prevent recurrent ulcer bleeding. N Engl J Med. 2005;352(3):238-244.
28. Lai KC, Lam SK, Chu KM, et al. Lansoprazole for the prevention of recurrences of ulcer complications from long-term low-dose aspirin use. N Engl J Med. 2002;346(26):2033-2038.
29. Plavix [package insert]. Bridgewater, NJ: Bristol-Myers Squibb/Sanofi Pharmaceuticals Partnership; 2011.
30. Gurbel PA, Bliden KP, Butler K, et al. Response to ticagrelor in clopidogrel nonresponders and responders and effect of switching therapies: the RESPOND study. Circulation. 2010;121(10):1188-1199.
31. Wiviott SD, Trenk D, Frelinger AL, et al. Prasugrel compared with high loading- and maintenance-dose clopidogrel in patients with planned percutaneous coronary intervention: the Prasugrel in Comparison to Clopidogrel for Inhibition of Platelet Activation and Aggregation-Thrombolysis in Myocardial Infarction 44 trial. Circulation. 2007;116(25):2923-2932.
32. Bonello L, Pansieri M, Mancini J, et al. High on-treatment platelet reactivity after prasugrel loading dose and cardiovascular events after percutaneous coronary intervention in acute coronary syndromes. J Am Coll Cardiol 2011;58(5):467-473.
33. Breet NJ, van Werkum JW, Bouman HJ, et al. Comparison of platelet function tests in predicting clinical outcome in patients undergoing coronary stent implantation. JAMA. 2010;303(8):754-762.
34. Storey RF, Bliden KP, Patil SB, et al. Incidence of dyspnea and assessment of cardiac and pulmonary function in patients with stable coronary artery disease receiving ticagrelor, clopidogrel, or placebo in the ONSET/OFFSET study. J Am Coll Cardiol. 2010;56(3):185-193.
35. Storey RF, Becker RC, Harrington RA, et al. Characterization of dyspnoea in PLATO study patients treated with ticagrelor or clopidogrel and its association with clinical outcomes. Eur Heart J. 2011;32(23):2945-2953.
36. Gan L-M, Wittfeldt A, Emanuelsson H, Nylander S, Jonasson J. Adenosine may mediate ticagrelor-induced dyspnea. J Am Coll Cardiol 2012;59(13):E344-
The Treatment of Gout
Managing the Multiple Symptoms of Benign Prostatic Hyperplasia — CME
Managing Type 2 Diabetes in Men
Meeting New Challenges with Antiplatelet Therapy in Primary Care
Dr. Ruoff has disclosed that he is on the speakers’ bureau for and has received research grants from Takeda Pharmaceuticals.
SUPPORT
This program is sponsored by the PCEC and is supported by funding from URL Pharma, Inc.
DB is a 50-year-old obese male visiting the clinic for symptoms suggestive of allergic rhinitis. The nurse has informed the family physician that DB was limping from the waiting room to the examination room. DB reports that he has been experiencing pain in his left big toe and ankle over the past few days. The last time this happened, the pain resolved within 7 to 10 days.
DB reports that he has experienced 4 or 5 similar episodes over the past 3 years. The first attacks affected his left big toe, but he now also experiences some pain in his left ankle. The pain is moderate, peaks in 1 to 2 days, and resolves within 7 to 10 days. Acetaminophen provided little pain relief so DB now takes ibuprofen 400 mg 3 times daily, as it “helps take the edge off.” Other medications include aspirin 81 mg per day and an oral antihistamine as needed for hay fever. DB reports that he eats seafood 2 to 3 times per week and red meat 1 to 2 times per week; he drinks 2 six-packs of beer per week.
Physical examination: weight, 186 lb (body mass index [BMI], 27 kg/m2); blood pressure, 126/76 mm Hg; and temperature, 98.8°F. His left big toe and ankle are red, slightly swollen, and warm with a small subcutaneous nodule noted on the first metatarsophalangeal joint. There is no sign of skin or joint infection.
The impression from his history and physical exam is that DB is suffering from an acute attack of gout, but the family physician also considers other diagnoses.
Background
Gout is a heterogeneous disorder that peaks in incidence in the fifth decade. Gout is caused by hyperuricemia, generally as a result of reduced excretion of uric acid by the kidneys; hyperuricemia may also result from overproduction of uric acid. Data from the National Health and Nutrition Examination Survey (NHANES) 2007-2008 indicate that the prevalence of gout continues to rise in the United States, likely related to the increasing frequency of adiposity and hypertension. Overall, about 75% of the 8.3 million people with gout are men.1
Risk Factors
Clinically defined hyperuricemia—a serum urate (sUA) level greater than 6.8 mg/dL, the concentration at which urate exceeds its solubility in most biological fluids—is the major risk factor for gout. However, not all persons with hyperuricemia have gout. Data from NHANES 2007-2008, in which the definition of hyperuricemia was an sUA level greater than 7.0 mg/dL for men and greater than 5.7 mg/dL for women, showed the mean sUA level to be 6.1 mg/dL in men and 4.9 mg/dL in women, corresponding to hyperuricemia prevalences of 21.2% and 21.6%, respectively.1
There are several other risk factors for gout, including hypertension, diabetes, hyperlipidemia, chronic kidney disease, cardiovascular disease (CVD), and metabolic syndrome.2 For a man with hypertension, the relative risk (RR) of gout is 2.3 compared with a normotensive man.3 Furthermore, it is well established that the use of diuretics increases the risk of gout (RR, 1.8).3 Several other medications increase sUA level as well: aspirin (including low-dose), cyclosporine, pyrazinamide, ethambutol, and niacin.2
Lifestyle and diet also pose a risk for gout. The risk of gout increases with BMI such that, compared with a man with a BMI of 21 to 22.9 kg/m2, the RR of gout is doubled for a man with a BMI of 25 to 29.9 kg/m2; for a man with a BMI of 35 kg/m2 or more, the RR is tripled.3 Sugar-sweetened soft drinks (but not diet soft drinks) and fructose-rich fruits and fruit juices also increase the risk of gout, as do a high alcohol intake, particularly beer, and a diet rich in meat (especially organ meat, turkey, or wild game) or seafood.4 A moderate intake of purine-rich vegetables (eg, peas, beans, lentils, spinach, mushrooms, oatmeal, and cauliflower) or protein is not associated with an increased risk of gout, while a high consumption of dairy products is associated with a decreased risk.5,6
Untreated or poorly treated gout usually leads to further acute attacks and progressive joint and tissue damage. In addition, gout and hyperuricemia serve as risk factors for other diseases. Adults with gout are 3 times as likely to develop metabolic syndrome as adults without gout.7 An elevated sUA level is also an independent risk factor for the development of hypertension (RR, 1.1), as well as myocardial infarction (MI; RR, 1.9), and stroke (RR, 1.6).8,9 An increasing sUA level also increases the risk of renal failure.10,11 In a study of 49,413 men followed for a mean of 5.4 years, the age-adjusted RR of renal failure was 1.5 in men with an sUA level of 6.5 to 8.4 mg/dL and 8.5 in men with an sUA level of 8.5 to 13.0 mg/dL compared with men with an sUA level of 5.0 to 6.4 mg/dL.11
Clinical Presentation
The deposition of monosodium urate (MSU) crystals in joints and tissues is very common and typically causes no signs or symptoms in the majority of persons. Even in men with an sUA level of 9 mg/dL or greater, the cumulative incidence of gouty arthritis has been found to be 22% over 5 years.12 However, as crystal deposition progresses, acute, painful attacks occur more frequently, with the development of chronic tophaceous gout after several years.13
Laboratory results for DB:
- Serum uric acid, 7.9 mg/dL
- White blood cell count, 15,800/mm3
- Serum creatinine, 1.2 mg/dL (estimated creatinine clearance, 90 mL/min)
- Erythrocyte sedimentation rate, 23 mm/h
- Low-density lipoprotein cholesterol (nonfasting), 127 mg/dL
Laboratory confirmation of hyperuricemia together with the pain, swelling, and tenderness of DB’s toe and ankle, other findings from his medical history and physical exam (eg, the use of aspirin daily), and exclusion of alternative diagnoses, such as septic arthritis, enable the family physician to arrive at a presumptive diagnosis of gouty arthritis. Aspiration of MSU crystals from DB’s toe or ankle is the gold standard and would allow for a definitive diagnosis. Although the sUA level was found to be high, it should be noted that a normal sUA level is often found during an acute attack; should this occur, the sUA level should be checked again 1 to 2 weeks after the acute attack has resolved.
Goals of Treatment
The cornerstone of gout management is daily, long-term treatment with urate-lowering therapy (ULT) combined with as-needed treatment for an acute attack. In addition, since initiation of ULT mobilizes MSU crystals, which often leads to a short-term increase in acute attacks, prophylaxis with an appropriate anti-inflammatory therapy is recommended at the time ULT is initiated.14
The therapeutic goals of gout treatment are 2-pronged: treatment of an acute gout attack and management of chronic gout. For an acute attack, the goals are to exclude a diagnosis of septic arthritis; reduce inflammation and terminate the attack; and seek, assess, and control associated diseases, such as diabetes mellitus, hypertension, hyperlipidemia, and CVD. If this latter goal is not possible during the acute attack, plans should be made to assess associated diseases once the acute attack has resolved.14 Lowering the sUA level is not a goal of therapy for an acute attack, but it is the primary goal of ULT for chronic gout. Lowering the sUA level to less than 6.0 mg/dL, which is well below the saturation point of urate in most biological fluids, is intended to prevent further acute attacks, tophus formation, and tissue damage.14
Treatment of an Acute Attack
The mainstay of treatment for an acute attack is anti-inflammatory therapy to reduce pain and inflammation.14 Therapy should be initiated at the onset of the attack and continued until the attack is terminated, which is typically 1 to 2 weeks. Anti-inflammatory therapy traditionally has in-cluded colchicine, a nonsteroidal anti-inflammatory drug (NSAID), or a corticosteroid.14
Nonsteroidal Anti-inflammatory Drugs
The NSAIDs are all thought to provide similar efficacy when used in maximum doses.15,16 Since gastrointestinal toxicity is a concern with NSAIDs, coadministration of a proton pump inhibitor, H2 antagonist, or misoprostol is advised for patients with an increased risk of peptic ulcers, bleeds, or perforations.17 The risk of MI, stroke, cardiovascular death, and atrial fibrillation/flutter with NSAID therapy should be considered, especially because gout often coexists with cardiovascular disorders.15,18,19 Furthermore, NSAIDs are contraindicated in patients with heart failure or renal insufficiency.20,21
Corticosteroids. A systematic review of clinical trials involving systemic corticosteroids that found a few prospective trials of low to moderate quality concluded that there was inconclusive evidence for the efficacy and effectiveness of corticosteroids in the treatment of acute gout.22 No serious adverse events (AEs) were reported. A more recent prospective trial found comparable pain reduction and incidence of AEs with naproxen 500 mg twice daily and prednisolone 35 mg once daily for 5 days in patients with monoarticular gout.23 Furthermore, clinical experience indicates that intra-articular aspiration and injection of a long-acting corticosteroid is an effective and safe treatment for an acute attack.14,15 Corticosteroids may be useful in patients who have an inadequate response to, are intolerant of, or have a contraindication to NSAIDs and colchicine.14,15
Colchicine. Much of the recent clinical investigation regarding pharmacologic treatment of an acute gout attack has involved colchicine. To overcome the limitations of the standard dose-to-toxicity regimen of colchicine, a low-dose regimen of colchicine (1.2 mg followed by 0.6 mg 1 hour later) was investigated and subsequently approved by the US Food and Drug Administration (FDA).24
Approval was based on a randomized, double-blind comparison with high-dose colchicine (1.2 mg followed by 0.6 mg every hour for 6 hours) and placebo in 184 patients with an acute gout attack.25 The primary endpoint, a 50% or greater reduction in pain at 24 hours without the use of rescue medication, was reached in 28 of 74 patients (38%) in the low-dose group, 17 of 52 patients (33%) in the high-dose group, and 9 of 58 patients (16%) in the placebo group (P = .005 and P = .034, respectively, versus placebo). An AE occurred in 36.5% and 76.9% of study participants in the low-dose and high-dose colchicine groups, respectively, and in 27.1% of participants in the placebo group. Gastrointestinal AEs (eg, diarrhea, nausea, and vomiting) were less common in the low-dose colchicine group ( FIGURE ). All AEs in the low-dose group were mild to moderate in intensity, while 10 of 52 patients (19.2%) in the high-dose group had an AE of severe intensity. Concomitant use of numerous drugs can increase the concentration of colchicine. Examples include atorvastatin, fluvastatin, pravastatin, simvastatin, fibrates, gemfibrozil, digoxin, clarithromycin, erythromycin, fluconazole, itraconazole, ketoconazole, protease inhibitors, diltiazem, verapamil, and cyclosporine, as well as grapefruit juice.26
FIGURE
Frequency of selected adverse events occurring over 24 hours with low-dose vs high-dose colchicine25
Treatment plan:
TABLE
Care plan for a patient with gout27
| Acute flare | Chronic gout | |
|---|---|---|
| Goals |
|
|
| Educational points |
|
|
| sUA, serum uric acid; ULT, urate-lowering therapy. Source: Reproduced with permission. Becker MA, et al. J Fam Pract. 2010;59(6):S1-S8. Quadrant HealthCom Inc. Copyright 2010. | ||
Urate-Lowering Therapy
Urate lowering therapy is indicated for most, but not all, patients with gout. ULT is generally not recommended for those who have suffered a single attack of gout and have no complications, since 40% of these patients will not experience another attack within a year. However, should a second attack occur within a year of the first attack, ULT is recommended. Some patients who have experienced a single attack may elect to initiate ULT after being educated about the risks of the disease and the risks and benefits of ULT.14 Patients who have had an attack of gout and also have a comorbidity (eg, visible gouty tophi, renal insufficiency, uric acid stones, or use of a diuretic for hypertension) should begin ULT, since the risk of further attacks is higher in these patients, and kidney or joint damage is more likely.17
Initiation of ULT should not occur until 1 to 2 weeks after an acute attack has resolved, since beginning ULT during an acute attack is thought to prolong the attack.17 Because gout is a chronic, largely self-managed disease, patient education is a cornerstone of successful long-term treatment. Implementation of a care plan for both an acute flare and chronic gout is recommended ( TABLE ).27
Anti-inflammatory prophylaxis should begin at the same time that ULT is initiated, since an acute attack is likely due to a transient rise in the sUA level resulting from mobilization of MSU crystals. Colchicine, which is the only drug approved by the FDA for prophylaxis of an acute gout attack, can be used daily in a low-dose regimen (0.6 mg once or twice daily) for up to 6 months.17,26 Alternatively, an NSAID can be used.17
One recent investigation pooled the results of 3 phase III clinical trials of ULT in 4101 patients with gout.28 Patients received prophylaxis for 8 weeks or 6 months with low-dose colchicine 0.6 mg once daily or the combination of naproxen 250 mg twice daily with lansoprazole 15 mg once daily. The incidence of acute gout attacks increased sharply (up to 40%) at the end of 8 weeks of prophylaxis with either colchicine or naproxen and then declined steadily, whereas the rates of acute attacks were consistently low (3% to 5%) at the end of 6 months of prophylaxis with either colchicine or naproxen/lansoprazole. With the 8-week prophylaxis regimen, diarrhea was more common in the colchicine group (n = 993) than in the naproxen group (n = 829) (8.4% vs 2.7%, respectively; P < .001). With the 6-month prophylaxis regimen, liver function abnormalities (7.7% vs 4.3%; P = .023) and headache (2.8% vs 0.9%; P = .037) were more common with colchicine (n = 1807) than naproxen, while gastrointestinal/abdominal pains (3.2% vs 1.2%; P = .012) and dental/oral soft tissue infections (2.3% vs 0.6%; P = .006) were more common with naproxen (n = 346) than colchicine.
Uricostatic Agents
Uricostatic therapy with a xanthine oxidase inhibitor (ie, allopurinol or febuxostat) is the most commonly used ULT. Allopurinol is effective in lowering the sUA level and has been shown to lower the rates of all-cause mortality and cardiovascular events, and, in patients with chronic kidney disease, slow the progression of renal disease.29,30 One key point that must be kept in mind is that the efficacy of allopurinol to lower the sUA level is dose-dependent, although limited safety data are available for doses >300 mg per day.14,31,32 One recent prospective clinical trial showed that 26% of patients achieved an sUA level of 5 mg/dL or less following 2 months of treatment with allopurinol 300 mg per day compared with 78% of those who subsequently doubled the dose to 300 mg twice daily.31 Two patients discontinued treatment with allopurinol because of an AE. Finally, the dose of allopurinol must be adjusted based on renal function to minimize the risk of AEs, particularly skin rashes.33
Febuxostat is also effective in lowering the sUA level. In patients with an sUA level of 8.0 mg/dL or higher and a creatinine clearance of 50 mL/min or higher at baseline, an sUA level of less than 6.0 mg/dL was achieved in 53% of patients treated with febuxostat 80 mg (n = 256) versus 21% of patients treated with allopurinol 300 mg once daily (n = 253) after 1 year (P < .001).34 The most frequent treatment-related AE was liver function abnormality, which occurred in 4% of patients in each group. Results of a 6-month trial showed that achievement of an sUA level of less than 6.0 mg/dL was achieved in 45% and 67% of patients treated with febuxostat 40 mg or 80 mg daily, respectively, and 42% of those treated with allopurinol 300 mg (200 mg in moderate renal impairment) daily.35 Febuxostat also has been shown to slow the progression of, or even stabilize, renal function.36
Treatment plan (continued):
- For an acute gout attack: Continue colchicine as needed
- ULT: Initiate allopurinol 100 mg once daily; increase to 200 mg once daily in 1 week, and 300 mg once daily in another week
- -Alternatively, initiate febuxostat 40 mg once daily; increase to 80 mg once daily if an sUA level of less than 6.0 mg/dL is not achieved within 2 weeks
- For prophylaxis of an acute attack when initiating ULT: Initiate colchicine 0.6 mg once daily; may increase to 0.6 mg twice daily if needed
- -Alternatively, initiate naproxen 250 mg twice daily with a proton pump inhibitor
- Measure sUA in 1 month; if the sUA level is greater than 6.0 mg/dL, increase allopurinol to 200 mg twice daily
- -Measure sUA in 1 month; if the sUA level is still greater than 6.0 mg/dL, increase allopurinol to 300 mg twice daily
- Implement the care plan ( TABLE )27
- -Inquire about and address issues to promote adherence and self-management
- -Discuss the most common AEs with allopurinol and colchicine and the actions the patient should take if an AE occurs
- Once the sUA level is 6.0 mg/dL or less, monitor sUA annually (including serum creatinine)14
1. Zhu Y, Pandya BJ, Choi HK. Prevalence of gout and hyperuricemia in the US general population: the National Health and Nutrition Examination Survey 2007-2008. Arthritis Rheum. 2011;63(10):3136-3141.
2. Weaver AL. Epidemiology of gout. Cleve Clin J Med. 2008;75(suppl 5):S9-S12.
3. Choi HK, Atkinson K, Karlson EW, Curhan G. Obesity, weight change, hypertension, diuretic use, and risk of gout in men: the health professionals follow-up study. Arch Intern Med. 2005;165(7):742-748.
4. Choi HK, Curhan G. Soft drinks, fructose consumption, and the risk of gout in men: prospective cohort study. BMJ. 2008;336(7639):309-312.
5. Choi HK, Atkinson K, Karlson EW, Willett W, Curhan G. Purine-rich foods, dairy and protein intake, and the risk of gout in men. N Engl J Med. 2004;350(11):1093-1103.
6. Choi HK, Atkinson K, Karlson EW, Willett W, Curhan G. Alcohol intake and risk of incident gout in men: a prospective study. Lancet. 2004;363(9417):1277-1281.
7. Choi HK, Ford ES, Li C, Curhan G. Prevalence of the metabolic syndrome in patients with gout: the Third National Health and Nutrition Examination Survey. Arthritis Rheum. 2007;57(1):109-115.
8. Perlstein TS, Gumieniak O, Williams GH, et al. Uric acid and the development of hypertension: the normative aging study. Hypertension. 2006;48(6):1031-1036.
9. Bos MJ, Koudstaal PJ, Hofman A, Witteman JC, Breteler MM. Uric acid is a risk factor for myocardial infarction and stroke: the Rotterdam study. Stroke. 2006;37(6):1503-1507.
10. Iseki K, Ikemiya Y, Inoue T, Iseki C, Kinjo K, Takishita S. Significance of hyperuricemia as a risk factor for developing ESRD in a screened cohort. Am J Kidney Dis. 2004;44(4):642-650.
11. Tomita M, Mizuno S, Yamanaka H, et al. Does hyperuricemia affect mortality? A prospective cohort study of Japanese male workers. J Epidemiol. 2000;10(6):403-409.
12. Campion EW, Glynn RJ, DeLabry LO. Asymptomatic hyperuricemia. Risks and consequences in the Normative Aging Study. Am J Med. 1987;82(3):421-426.
13. Mandell BF. Clinical manifestations of hyperuricemia and gout. Cleve Clin J Med. 2008;75(Suppl 5):S5-S8.
14. Hamburger M, Baraf HS, Adamson TC III, et al. 2011 Recommendations for the diagnosis and management of gout and hyperuricemia. Postgrad Med. 2011;123 (6 suppl 1):3-36.
15. Zhang W, Doherty M, Bardin T, et al. EULAR evidence based recommendations for gout. Part II: Management. Report of a task force of the EULAR Standing Committee for International Clinical Studies Including Therapeutics (ESCISIT). Ann Rheum Dis. 2006;65(10):1312-1324.
16. Schumacher HR Jr, Boice JA, Daikh DI, et al. Randomised double blind trial of etoricoxib and indometacin in treatment of acute gouty arthritis. BMJ. 2002;324(7352):1488-1492.
17. Jordan KM, Cameron JS, Snaith M, et al. British Society for Rheumatology and British Health Professionals in Rheumatology guideline for the management of gout. Rheumatology (Oxford). 2007;46(8):1372-1374.
18. Trelle S, Reichenbach S, Wandel S, et al. Cardiovascular safety of non-steroidal anti-inflammatory drugs: network meta-analysis. BMJ. 2011;342:c7086.-
19. Schmidt M, Christiansen CF, Mehnert F, Rothman KJ, Sorensen HT. Non-steroidal anti-inflammatory drug use and risk of atrial fibrillation or flutter: population based case-control study. BMJ. 2011;343:d3450.-
20. NSAIDS and chronic kidney disease. US Centers for Disease Control and Prevention. http://www.cdc.gov/diabetes/news/docs/nsaid_video.htm. Published 2012. Accessed April 22, 2012.
21. Gislason GH, Rasmussen JN, Abildstrom SZ, et al. Increased mortality and cardiovascular morbidity associated with use of nonsteroidal anti-inflammatory drugs in chronic heart failure. Arch Intern Med. 2009;169(2):141-149.
22. Janssens HJ, Lucassen PL, Van de Laar FA, Janssen M, Van de Lisdonk EH. Systemic corticosteroids for acute gout. Cochrane Database Syst Rev. 2008;(2):CD005521.-
23. Janssens HJ, Janssen M, van de Lisdonk EH, van Riel PL, van Weel C. Use of oral prednisolone or naproxen for the treatment of gout arthritis: a double-blind, randomised equivalence trial. Lancet. 2008;371(9627):1854-1860.
24. Schlesinger N, Schumacher R, Catton M, Maxwell L. Colchicine for acute gout. Cochrane Database Syst Rev. 2006;(4):CD006190.-
25. Terkeltaub RA, Furst DE, Bennett K, Kook KA, Crockett RS, Davis MW. High versus low dosing of oral colchicine for early acute gout flare: Twenty-four-hour outcome of the first multicenter, randomized, double-blind, placebo-controlled, parallel-group, dose-comparison colchicine study. Arthritis Rheum. 2010;62(4):1060-1068.
26. Colcrys [package insert]. Philadelphia, PA: AR Scientific, Inc.; 2011.
27. Becker MA, Ruoff GE. What do I need to know about gout? J Fam Pract. 2010;59(6 suppl):S1-S8.
28. Wortmann RL, Macdonald PA, Hunt B, Jackson RL. Effect of prophylaxis on gout flares after the initiation of urate-lowering therapy: analysis of data from three phase III trials. Clin Ther. 2010;32(14):2386-2397.
29. Luk AJ, Levin GP, Moore EE, Zhou XH, Kestenbaum BR, Choi HK. Allopurinol and mortality in hyperuricaemic patients. Rheumatology (Oxford). 2009;48(7):804-806.
30. Goicoechea M, de Vinuesa SG, Verdalles U, et al. Effect of allopurinol in chronic kidney disease progression and cardiovascular risk. Clin J Am Soc Nephrol. 2010;5(8):1388-1393.
31. Reinders MK, Haagsma C, Jansen TL, et al. A randomised controlled trial on the efficacy and tolerability with dose escalation of allopurinol 300-600 mg/day versus benzbromarone 100-200 mg/day in patients with gout. Ann Rheum Dis. 2009;68(6):892-897.
32. Stamp LK, O’Donnell JL, Zhang M, et al. Using allopurinol above the dose based on creatinine clearance is effective and safe in patients with chronic gout, including those with renal impairment. Arthritis Rheum. 2011;63(2):412-421.
33. Zyloprim [package insert]. San Diego, CA: Prometheus Laboratories Inc.; 2003.
34. Becker MA, Schumacher HR Jr, Wortmann RL, et al. Febuxostat compared with allopurinol in patients with hyperuricemia and gout. N Engl J Med. 2005;353(23):2450-2461.
35. Becker MA, Schumacher HR, Espinoza LR, et al. The urate-lowering efficacy and safety of febuxostat in the treatment of the hyperuricemia of gout: the CONFIRMS trial. Arthritis Res Ther. 2010;12:doi:10.1186/ar2978.
36. Whelton A, Macdonald PA, Zhao L, Hunt B, Gunawardhana L. Renal function in gout: long-term treatment effects of febuxostat. J Clin Rheumatol. 2011;17(1):7-13.
Managing the Multiple Symptoms of Benign Prostatic Hyperplasia — CME
Managing Type 2 Diabetes in Men
Meeting New Challenges with Antiplatelet Therapy in Primary Care
Dr. Ruoff has disclosed that he is on the speakers’ bureau for and has received research grants from Takeda Pharmaceuticals.
SUPPORT
This program is sponsored by the PCEC and is supported by funding from URL Pharma, Inc.
DB is a 50-year-old obese male visiting the clinic for symptoms suggestive of allergic rhinitis. The nurse has informed the family physician that DB was limping from the waiting room to the examination room. DB reports that he has been experiencing pain in his left big toe and ankle over the past few days. The last time this happened, the pain resolved within 7 to 10 days.
DB reports that he has experienced 4 or 5 similar episodes over the past 3 years. The first attacks affected his left big toe, but he now also experiences some pain in his left ankle. The pain is moderate, peaks in 1 to 2 days, and resolves within 7 to 10 days. Acetaminophen provided little pain relief so DB now takes ibuprofen 400 mg 3 times daily, as it “helps take the edge off.” Other medications include aspirin 81 mg per day and an oral antihistamine as needed for hay fever. DB reports that he eats seafood 2 to 3 times per week and red meat 1 to 2 times per week; he drinks 2 six-packs of beer per week.
Physical examination: weight, 186 lb (body mass index [BMI], 27 kg/m2); blood pressure, 126/76 mm Hg; and temperature, 98.8°F. His left big toe and ankle are red, slightly swollen, and warm with a small subcutaneous nodule noted on the first metatarsophalangeal joint. There is no sign of skin or joint infection.
The impression from his history and physical exam is that DB is suffering from an acute attack of gout, but the family physician also considers other diagnoses.
Background
Gout is a heterogeneous disorder that peaks in incidence in the fifth decade. Gout is caused by hyperuricemia, generally as a result of reduced excretion of uric acid by the kidneys; hyperuricemia may also result from overproduction of uric acid. Data from the National Health and Nutrition Examination Survey (NHANES) 2007-2008 indicate that the prevalence of gout continues to rise in the United States, likely related to the increasing frequency of adiposity and hypertension. Overall, about 75% of the 8.3 million people with gout are men.1
Risk Factors
Clinically defined hyperuricemia—a serum urate (sUA) level greater than 6.8 mg/dL, the concentration at which urate exceeds its solubility in most biological fluids—is the major risk factor for gout. However, not all persons with hyperuricemia have gout. Data from NHANES 2007-2008, in which the definition of hyperuricemia was an sUA level greater than 7.0 mg/dL for men and greater than 5.7 mg/dL for women, showed the mean sUA level to be 6.1 mg/dL in men and 4.9 mg/dL in women, corresponding to hyperuricemia prevalences of 21.2% and 21.6%, respectively.1
There are several other risk factors for gout, including hypertension, diabetes, hyperlipidemia, chronic kidney disease, cardiovascular disease (CVD), and metabolic syndrome.2 For a man with hypertension, the relative risk (RR) of gout is 2.3 compared with a normotensive man.3 Furthermore, it is well established that the use of diuretics increases the risk of gout (RR, 1.8).3 Several other medications increase sUA level as well: aspirin (including low-dose), cyclosporine, pyrazinamide, ethambutol, and niacin.2
Lifestyle and diet also pose a risk for gout. The risk of gout increases with BMI such that, compared with a man with a BMI of 21 to 22.9 kg/m2, the RR of gout is doubled for a man with a BMI of 25 to 29.9 kg/m2; for a man with a BMI of 35 kg/m2 or more, the RR is tripled.3 Sugar-sweetened soft drinks (but not diet soft drinks) and fructose-rich fruits and fruit juices also increase the risk of gout, as do a high alcohol intake, particularly beer, and a diet rich in meat (especially organ meat, turkey, or wild game) or seafood.4 A moderate intake of purine-rich vegetables (eg, peas, beans, lentils, spinach, mushrooms, oatmeal, and cauliflower) or protein is not associated with an increased risk of gout, while a high consumption of dairy products is associated with a decreased risk.5,6
Untreated or poorly treated gout usually leads to further acute attacks and progressive joint and tissue damage. In addition, gout and hyperuricemia serve as risk factors for other diseases. Adults with gout are 3 times as likely to develop metabolic syndrome as adults without gout.7 An elevated sUA level is also an independent risk factor for the development of hypertension (RR, 1.1), as well as myocardial infarction (MI; RR, 1.9), and stroke (RR, 1.6).8,9 An increasing sUA level also increases the risk of renal failure.10,11 In a study of 49,413 men followed for a mean of 5.4 years, the age-adjusted RR of renal failure was 1.5 in men with an sUA level of 6.5 to 8.4 mg/dL and 8.5 in men with an sUA level of 8.5 to 13.0 mg/dL compared with men with an sUA level of 5.0 to 6.4 mg/dL.11
Clinical Presentation
The deposition of monosodium urate (MSU) crystals in joints and tissues is very common and typically causes no signs or symptoms in the majority of persons. Even in men with an sUA level of 9 mg/dL or greater, the cumulative incidence of gouty arthritis has been found to be 22% over 5 years.12 However, as crystal deposition progresses, acute, painful attacks occur more frequently, with the development of chronic tophaceous gout after several years.13
Laboratory results for DB:
- Serum uric acid, 7.9 mg/dL
- White blood cell count, 15,800/mm3
- Serum creatinine, 1.2 mg/dL (estimated creatinine clearance, 90 mL/min)
- Erythrocyte sedimentation rate, 23 mm/h
- Low-density lipoprotein cholesterol (nonfasting), 127 mg/dL
Laboratory confirmation of hyperuricemia together with the pain, swelling, and tenderness of DB’s toe and ankle, other findings from his medical history and physical exam (eg, the use of aspirin daily), and exclusion of alternative diagnoses, such as septic arthritis, enable the family physician to arrive at a presumptive diagnosis of gouty arthritis. Aspiration of MSU crystals from DB’s toe or ankle is the gold standard and would allow for a definitive diagnosis. Although the sUA level was found to be high, it should be noted that a normal sUA level is often found during an acute attack; should this occur, the sUA level should be checked again 1 to 2 weeks after the acute attack has resolved.
Goals of Treatment
The cornerstone of gout management is daily, long-term treatment with urate-lowering therapy (ULT) combined with as-needed treatment for an acute attack. In addition, since initiation of ULT mobilizes MSU crystals, which often leads to a short-term increase in acute attacks, prophylaxis with an appropriate anti-inflammatory therapy is recommended at the time ULT is initiated.14
The therapeutic goals of gout treatment are 2-pronged: treatment of an acute gout attack and management of chronic gout. For an acute attack, the goals are to exclude a diagnosis of septic arthritis; reduce inflammation and terminate the attack; and seek, assess, and control associated diseases, such as diabetes mellitus, hypertension, hyperlipidemia, and CVD. If this latter goal is not possible during the acute attack, plans should be made to assess associated diseases once the acute attack has resolved.14 Lowering the sUA level is not a goal of therapy for an acute attack, but it is the primary goal of ULT for chronic gout. Lowering the sUA level to less than 6.0 mg/dL, which is well below the saturation point of urate in most biological fluids, is intended to prevent further acute attacks, tophus formation, and tissue damage.14
Treatment of an Acute Attack
The mainstay of treatment for an acute attack is anti-inflammatory therapy to reduce pain and inflammation.14 Therapy should be initiated at the onset of the attack and continued until the attack is terminated, which is typically 1 to 2 weeks. Anti-inflammatory therapy traditionally has in-cluded colchicine, a nonsteroidal anti-inflammatory drug (NSAID), or a corticosteroid.14
Nonsteroidal Anti-inflammatory Drugs
The NSAIDs are all thought to provide similar efficacy when used in maximum doses.15,16 Since gastrointestinal toxicity is a concern with NSAIDs, coadministration of a proton pump inhibitor, H2 antagonist, or misoprostol is advised for patients with an increased risk of peptic ulcers, bleeds, or perforations.17 The risk of MI, stroke, cardiovascular death, and atrial fibrillation/flutter with NSAID therapy should be considered, especially because gout often coexists with cardiovascular disorders.15,18,19 Furthermore, NSAIDs are contraindicated in patients with heart failure or renal insufficiency.20,21
Corticosteroids. A systematic review of clinical trials involving systemic corticosteroids that found a few prospective trials of low to moderate quality concluded that there was inconclusive evidence for the efficacy and effectiveness of corticosteroids in the treatment of acute gout.22 No serious adverse events (AEs) were reported. A more recent prospective trial found comparable pain reduction and incidence of AEs with naproxen 500 mg twice daily and prednisolone 35 mg once daily for 5 days in patients with monoarticular gout.23 Furthermore, clinical experience indicates that intra-articular aspiration and injection of a long-acting corticosteroid is an effective and safe treatment for an acute attack.14,15 Corticosteroids may be useful in patients who have an inadequate response to, are intolerant of, or have a contraindication to NSAIDs and colchicine.14,15
Colchicine. Much of the recent clinical investigation regarding pharmacologic treatment of an acute gout attack has involved colchicine. To overcome the limitations of the standard dose-to-toxicity regimen of colchicine, a low-dose regimen of colchicine (1.2 mg followed by 0.6 mg 1 hour later) was investigated and subsequently approved by the US Food and Drug Administration (FDA).24
Approval was based on a randomized, double-blind comparison with high-dose colchicine (1.2 mg followed by 0.6 mg every hour for 6 hours) and placebo in 184 patients with an acute gout attack.25 The primary endpoint, a 50% or greater reduction in pain at 24 hours without the use of rescue medication, was reached in 28 of 74 patients (38%) in the low-dose group, 17 of 52 patients (33%) in the high-dose group, and 9 of 58 patients (16%) in the placebo group (P = .005 and P = .034, respectively, versus placebo). An AE occurred in 36.5% and 76.9% of study participants in the low-dose and high-dose colchicine groups, respectively, and in 27.1% of participants in the placebo group. Gastrointestinal AEs (eg, diarrhea, nausea, and vomiting) were less common in the low-dose colchicine group ( FIGURE ). All AEs in the low-dose group were mild to moderate in intensity, while 10 of 52 patients (19.2%) in the high-dose group had an AE of severe intensity. Concomitant use of numerous drugs can increase the concentration of colchicine. Examples include atorvastatin, fluvastatin, pravastatin, simvastatin, fibrates, gemfibrozil, digoxin, clarithromycin, erythromycin, fluconazole, itraconazole, ketoconazole, protease inhibitors, diltiazem, verapamil, and cyclosporine, as well as grapefruit juice.26
FIGURE
Frequency of selected adverse events occurring over 24 hours with low-dose vs high-dose colchicine25
Treatment plan:
TABLE
Care plan for a patient with gout27
| Acute flare | Chronic gout | |
|---|---|---|
| Goals |
|
|
| Educational points |
|
|
| sUA, serum uric acid; ULT, urate-lowering therapy. Source: Reproduced with permission. Becker MA, et al. J Fam Pract. 2010;59(6):S1-S8. Quadrant HealthCom Inc. Copyright 2010. | ||
Urate-Lowering Therapy
Urate lowering therapy is indicated for most, but not all, patients with gout. ULT is generally not recommended for those who have suffered a single attack of gout and have no complications, since 40% of these patients will not experience another attack within a year. However, should a second attack occur within a year of the first attack, ULT is recommended. Some patients who have experienced a single attack may elect to initiate ULT after being educated about the risks of the disease and the risks and benefits of ULT.14 Patients who have had an attack of gout and also have a comorbidity (eg, visible gouty tophi, renal insufficiency, uric acid stones, or use of a diuretic for hypertension) should begin ULT, since the risk of further attacks is higher in these patients, and kidney or joint damage is more likely.17
Initiation of ULT should not occur until 1 to 2 weeks after an acute attack has resolved, since beginning ULT during an acute attack is thought to prolong the attack.17 Because gout is a chronic, largely self-managed disease, patient education is a cornerstone of successful long-term treatment. Implementation of a care plan for both an acute flare and chronic gout is recommended ( TABLE ).27
Anti-inflammatory prophylaxis should begin at the same time that ULT is initiated, since an acute attack is likely due to a transient rise in the sUA level resulting from mobilization of MSU crystals. Colchicine, which is the only drug approved by the FDA for prophylaxis of an acute gout attack, can be used daily in a low-dose regimen (0.6 mg once or twice daily) for up to 6 months.17,26 Alternatively, an NSAID can be used.17
One recent investigation pooled the results of 3 phase III clinical trials of ULT in 4101 patients with gout.28 Patients received prophylaxis for 8 weeks or 6 months with low-dose colchicine 0.6 mg once daily or the combination of naproxen 250 mg twice daily with lansoprazole 15 mg once daily. The incidence of acute gout attacks increased sharply (up to 40%) at the end of 8 weeks of prophylaxis with either colchicine or naproxen and then declined steadily, whereas the rates of acute attacks were consistently low (3% to 5%) at the end of 6 months of prophylaxis with either colchicine or naproxen/lansoprazole. With the 8-week prophylaxis regimen, diarrhea was more common in the colchicine group (n = 993) than in the naproxen group (n = 829) (8.4% vs 2.7%, respectively; P < .001). With the 6-month prophylaxis regimen, liver function abnormalities (7.7% vs 4.3%; P = .023) and headache (2.8% vs 0.9%; P = .037) were more common with colchicine (n = 1807) than naproxen, while gastrointestinal/abdominal pains (3.2% vs 1.2%; P = .012) and dental/oral soft tissue infections (2.3% vs 0.6%; P = .006) were more common with naproxen (n = 346) than colchicine.
Uricostatic Agents
Uricostatic therapy with a xanthine oxidase inhibitor (ie, allopurinol or febuxostat) is the most commonly used ULT. Allopurinol is effective in lowering the sUA level and has been shown to lower the rates of all-cause mortality and cardiovascular events, and, in patients with chronic kidney disease, slow the progression of renal disease.29,30 One key point that must be kept in mind is that the efficacy of allopurinol to lower the sUA level is dose-dependent, although limited safety data are available for doses >300 mg per day.14,31,32 One recent prospective clinical trial showed that 26% of patients achieved an sUA level of 5 mg/dL or less following 2 months of treatment with allopurinol 300 mg per day compared with 78% of those who subsequently doubled the dose to 300 mg twice daily.31 Two patients discontinued treatment with allopurinol because of an AE. Finally, the dose of allopurinol must be adjusted based on renal function to minimize the risk of AEs, particularly skin rashes.33
Febuxostat is also effective in lowering the sUA level. In patients with an sUA level of 8.0 mg/dL or higher and a creatinine clearance of 50 mL/min or higher at baseline, an sUA level of less than 6.0 mg/dL was achieved in 53% of patients treated with febuxostat 80 mg (n = 256) versus 21% of patients treated with allopurinol 300 mg once daily (n = 253) after 1 year (P < .001).34 The most frequent treatment-related AE was liver function abnormality, which occurred in 4% of patients in each group. Results of a 6-month trial showed that achievement of an sUA level of less than 6.0 mg/dL was achieved in 45% and 67% of patients treated with febuxostat 40 mg or 80 mg daily, respectively, and 42% of those treated with allopurinol 300 mg (200 mg in moderate renal impairment) daily.35 Febuxostat also has been shown to slow the progression of, or even stabilize, renal function.36
Treatment plan (continued):
- For an acute gout attack: Continue colchicine as needed
- ULT: Initiate allopurinol 100 mg once daily; increase to 200 mg once daily in 1 week, and 300 mg once daily in another week
- -Alternatively, initiate febuxostat 40 mg once daily; increase to 80 mg once daily if an sUA level of less than 6.0 mg/dL is not achieved within 2 weeks
- For prophylaxis of an acute attack when initiating ULT: Initiate colchicine 0.6 mg once daily; may increase to 0.6 mg twice daily if needed
- -Alternatively, initiate naproxen 250 mg twice daily with a proton pump inhibitor
- Measure sUA in 1 month; if the sUA level is greater than 6.0 mg/dL, increase allopurinol to 200 mg twice daily
- -Measure sUA in 1 month; if the sUA level is still greater than 6.0 mg/dL, increase allopurinol to 300 mg twice daily
- Implement the care plan ( TABLE )27
- -Inquire about and address issues to promote adherence and self-management
- -Discuss the most common AEs with allopurinol and colchicine and the actions the patient should take if an AE occurs
- Once the sUA level is 6.0 mg/dL or less, monitor sUA annually (including serum creatinine)14
Managing the Multiple Symptoms of Benign Prostatic Hyperplasia — CME
Managing Type 2 Diabetes in Men
Meeting New Challenges with Antiplatelet Therapy in Primary Care
Dr. Ruoff has disclosed that he is on the speakers’ bureau for and has received research grants from Takeda Pharmaceuticals.
SUPPORT
This program is sponsored by the PCEC and is supported by funding from URL Pharma, Inc.
DB is a 50-year-old obese male visiting the clinic for symptoms suggestive of allergic rhinitis. The nurse has informed the family physician that DB was limping from the waiting room to the examination room. DB reports that he has been experiencing pain in his left big toe and ankle over the past few days. The last time this happened, the pain resolved within 7 to 10 days.
DB reports that he has experienced 4 or 5 similar episodes over the past 3 years. The first attacks affected his left big toe, but he now also experiences some pain in his left ankle. The pain is moderate, peaks in 1 to 2 days, and resolves within 7 to 10 days. Acetaminophen provided little pain relief so DB now takes ibuprofen 400 mg 3 times daily, as it “helps take the edge off.” Other medications include aspirin 81 mg per day and an oral antihistamine as needed for hay fever. DB reports that he eats seafood 2 to 3 times per week and red meat 1 to 2 times per week; he drinks 2 six-packs of beer per week.
Physical examination: weight, 186 lb (body mass index [BMI], 27 kg/m2); blood pressure, 126/76 mm Hg; and temperature, 98.8°F. His left big toe and ankle are red, slightly swollen, and warm with a small subcutaneous nodule noted on the first metatarsophalangeal joint. There is no sign of skin or joint infection.
The impression from his history and physical exam is that DB is suffering from an acute attack of gout, but the family physician also considers other diagnoses.
Background
Gout is a heterogeneous disorder that peaks in incidence in the fifth decade. Gout is caused by hyperuricemia, generally as a result of reduced excretion of uric acid by the kidneys; hyperuricemia may also result from overproduction of uric acid. Data from the National Health and Nutrition Examination Survey (NHANES) 2007-2008 indicate that the prevalence of gout continues to rise in the United States, likely related to the increasing frequency of adiposity and hypertension. Overall, about 75% of the 8.3 million people with gout are men.1
Risk Factors
Clinically defined hyperuricemia—a serum urate (sUA) level greater than 6.8 mg/dL, the concentration at which urate exceeds its solubility in most biological fluids—is the major risk factor for gout. However, not all persons with hyperuricemia have gout. Data from NHANES 2007-2008, in which the definition of hyperuricemia was an sUA level greater than 7.0 mg/dL for men and greater than 5.7 mg/dL for women, showed the mean sUA level to be 6.1 mg/dL in men and 4.9 mg/dL in women, corresponding to hyperuricemia prevalences of 21.2% and 21.6%, respectively.1
There are several other risk factors for gout, including hypertension, diabetes, hyperlipidemia, chronic kidney disease, cardiovascular disease (CVD), and metabolic syndrome.2 For a man with hypertension, the relative risk (RR) of gout is 2.3 compared with a normotensive man.3 Furthermore, it is well established that the use of diuretics increases the risk of gout (RR, 1.8).3 Several other medications increase sUA level as well: aspirin (including low-dose), cyclosporine, pyrazinamide, ethambutol, and niacin.2
Lifestyle and diet also pose a risk for gout. The risk of gout increases with BMI such that, compared with a man with a BMI of 21 to 22.9 kg/m2, the RR of gout is doubled for a man with a BMI of 25 to 29.9 kg/m2; for a man with a BMI of 35 kg/m2 or more, the RR is tripled.3 Sugar-sweetened soft drinks (but not diet soft drinks) and fructose-rich fruits and fruit juices also increase the risk of gout, as do a high alcohol intake, particularly beer, and a diet rich in meat (especially organ meat, turkey, or wild game) or seafood.4 A moderate intake of purine-rich vegetables (eg, peas, beans, lentils, spinach, mushrooms, oatmeal, and cauliflower) or protein is not associated with an increased risk of gout, while a high consumption of dairy products is associated with a decreased risk.5,6
Untreated or poorly treated gout usually leads to further acute attacks and progressive joint and tissue damage. In addition, gout and hyperuricemia serve as risk factors for other diseases. Adults with gout are 3 times as likely to develop metabolic syndrome as adults without gout.7 An elevated sUA level is also an independent risk factor for the development of hypertension (RR, 1.1), as well as myocardial infarction (MI; RR, 1.9), and stroke (RR, 1.6).8,9 An increasing sUA level also increases the risk of renal failure.10,11 In a study of 49,413 men followed for a mean of 5.4 years, the age-adjusted RR of renal failure was 1.5 in men with an sUA level of 6.5 to 8.4 mg/dL and 8.5 in men with an sUA level of 8.5 to 13.0 mg/dL compared with men with an sUA level of 5.0 to 6.4 mg/dL.11
Clinical Presentation
The deposition of monosodium urate (MSU) crystals in joints and tissues is very common and typically causes no signs or symptoms in the majority of persons. Even in men with an sUA level of 9 mg/dL or greater, the cumulative incidence of gouty arthritis has been found to be 22% over 5 years.12 However, as crystal deposition progresses, acute, painful attacks occur more frequently, with the development of chronic tophaceous gout after several years.13
Laboratory results for DB:
- Serum uric acid, 7.9 mg/dL
- White blood cell count, 15,800/mm3
- Serum creatinine, 1.2 mg/dL (estimated creatinine clearance, 90 mL/min)
- Erythrocyte sedimentation rate, 23 mm/h
- Low-density lipoprotein cholesterol (nonfasting), 127 mg/dL
Laboratory confirmation of hyperuricemia together with the pain, swelling, and tenderness of DB’s toe and ankle, other findings from his medical history and physical exam (eg, the use of aspirin daily), and exclusion of alternative diagnoses, such as septic arthritis, enable the family physician to arrive at a presumptive diagnosis of gouty arthritis. Aspiration of MSU crystals from DB’s toe or ankle is the gold standard and would allow for a definitive diagnosis. Although the sUA level was found to be high, it should be noted that a normal sUA level is often found during an acute attack; should this occur, the sUA level should be checked again 1 to 2 weeks after the acute attack has resolved.
Goals of Treatment
The cornerstone of gout management is daily, long-term treatment with urate-lowering therapy (ULT) combined with as-needed treatment for an acute attack. In addition, since initiation of ULT mobilizes MSU crystals, which often leads to a short-term increase in acute attacks, prophylaxis with an appropriate anti-inflammatory therapy is recommended at the time ULT is initiated.14
The therapeutic goals of gout treatment are 2-pronged: treatment of an acute gout attack and management of chronic gout. For an acute attack, the goals are to exclude a diagnosis of septic arthritis; reduce inflammation and terminate the attack; and seek, assess, and control associated diseases, such as diabetes mellitus, hypertension, hyperlipidemia, and CVD. If this latter goal is not possible during the acute attack, plans should be made to assess associated diseases once the acute attack has resolved.14 Lowering the sUA level is not a goal of therapy for an acute attack, but it is the primary goal of ULT for chronic gout. Lowering the sUA level to less than 6.0 mg/dL, which is well below the saturation point of urate in most biological fluids, is intended to prevent further acute attacks, tophus formation, and tissue damage.14
Treatment of an Acute Attack
The mainstay of treatment for an acute attack is anti-inflammatory therapy to reduce pain and inflammation.14 Therapy should be initiated at the onset of the attack and continued until the attack is terminated, which is typically 1 to 2 weeks. Anti-inflammatory therapy traditionally has in-cluded colchicine, a nonsteroidal anti-inflammatory drug (NSAID), or a corticosteroid.14
Nonsteroidal Anti-inflammatory Drugs
The NSAIDs are all thought to provide similar efficacy when used in maximum doses.15,16 Since gastrointestinal toxicity is a concern with NSAIDs, coadministration of a proton pump inhibitor, H2 antagonist, or misoprostol is advised for patients with an increased risk of peptic ulcers, bleeds, or perforations.17 The risk of MI, stroke, cardiovascular death, and atrial fibrillation/flutter with NSAID therapy should be considered, especially because gout often coexists with cardiovascular disorders.15,18,19 Furthermore, NSAIDs are contraindicated in patients with heart failure or renal insufficiency.20,21
Corticosteroids. A systematic review of clinical trials involving systemic corticosteroids that found a few prospective trials of low to moderate quality concluded that there was inconclusive evidence for the efficacy and effectiveness of corticosteroids in the treatment of acute gout.22 No serious adverse events (AEs) were reported. A more recent prospective trial found comparable pain reduction and incidence of AEs with naproxen 500 mg twice daily and prednisolone 35 mg once daily for 5 days in patients with monoarticular gout.23 Furthermore, clinical experience indicates that intra-articular aspiration and injection of a long-acting corticosteroid is an effective and safe treatment for an acute attack.14,15 Corticosteroids may be useful in patients who have an inadequate response to, are intolerant of, or have a contraindication to NSAIDs and colchicine.14,15
Colchicine. Much of the recent clinical investigation regarding pharmacologic treatment of an acute gout attack has involved colchicine. To overcome the limitations of the standard dose-to-toxicity regimen of colchicine, a low-dose regimen of colchicine (1.2 mg followed by 0.6 mg 1 hour later) was investigated and subsequently approved by the US Food and Drug Administration (FDA).24
Approval was based on a randomized, double-blind comparison with high-dose colchicine (1.2 mg followed by 0.6 mg every hour for 6 hours) and placebo in 184 patients with an acute gout attack.25 The primary endpoint, a 50% or greater reduction in pain at 24 hours without the use of rescue medication, was reached in 28 of 74 patients (38%) in the low-dose group, 17 of 52 patients (33%) in the high-dose group, and 9 of 58 patients (16%) in the placebo group (P = .005 and P = .034, respectively, versus placebo). An AE occurred in 36.5% and 76.9% of study participants in the low-dose and high-dose colchicine groups, respectively, and in 27.1% of participants in the placebo group. Gastrointestinal AEs (eg, diarrhea, nausea, and vomiting) were less common in the low-dose colchicine group ( FIGURE ). All AEs in the low-dose group were mild to moderate in intensity, while 10 of 52 patients (19.2%) in the high-dose group had an AE of severe intensity. Concomitant use of numerous drugs can increase the concentration of colchicine. Examples include atorvastatin, fluvastatin, pravastatin, simvastatin, fibrates, gemfibrozil, digoxin, clarithromycin, erythromycin, fluconazole, itraconazole, ketoconazole, protease inhibitors, diltiazem, verapamil, and cyclosporine, as well as grapefruit juice.26
FIGURE
Frequency of selected adverse events occurring over 24 hours with low-dose vs high-dose colchicine25
Treatment plan:
TABLE
Care plan for a patient with gout27
| Acute flare | Chronic gout | |
|---|---|---|
| Goals |
|
|
| Educational points |
|
|
| sUA, serum uric acid; ULT, urate-lowering therapy. Source: Reproduced with permission. Becker MA, et al. J Fam Pract. 2010;59(6):S1-S8. Quadrant HealthCom Inc. Copyright 2010. | ||
Urate-Lowering Therapy
Urate lowering therapy is indicated for most, but not all, patients with gout. ULT is generally not recommended for those who have suffered a single attack of gout and have no complications, since 40% of these patients will not experience another attack within a year. However, should a second attack occur within a year of the first attack, ULT is recommended. Some patients who have experienced a single attack may elect to initiate ULT after being educated about the risks of the disease and the risks and benefits of ULT.14 Patients who have had an attack of gout and also have a comorbidity (eg, visible gouty tophi, renal insufficiency, uric acid stones, or use of a diuretic for hypertension) should begin ULT, since the risk of further attacks is higher in these patients, and kidney or joint damage is more likely.17
Initiation of ULT should not occur until 1 to 2 weeks after an acute attack has resolved, since beginning ULT during an acute attack is thought to prolong the attack.17 Because gout is a chronic, largely self-managed disease, patient education is a cornerstone of successful long-term treatment. Implementation of a care plan for both an acute flare and chronic gout is recommended ( TABLE ).27
Anti-inflammatory prophylaxis should begin at the same time that ULT is initiated, since an acute attack is likely due to a transient rise in the sUA level resulting from mobilization of MSU crystals. Colchicine, which is the only drug approved by the FDA for prophylaxis of an acute gout attack, can be used daily in a low-dose regimen (0.6 mg once or twice daily) for up to 6 months.17,26 Alternatively, an NSAID can be used.17
One recent investigation pooled the results of 3 phase III clinical trials of ULT in 4101 patients with gout.28 Patients received prophylaxis for 8 weeks or 6 months with low-dose colchicine 0.6 mg once daily or the combination of naproxen 250 mg twice daily with lansoprazole 15 mg once daily. The incidence of acute gout attacks increased sharply (up to 40%) at the end of 8 weeks of prophylaxis with either colchicine or naproxen and then declined steadily, whereas the rates of acute attacks were consistently low (3% to 5%) at the end of 6 months of prophylaxis with either colchicine or naproxen/lansoprazole. With the 8-week prophylaxis regimen, diarrhea was more common in the colchicine group (n = 993) than in the naproxen group (n = 829) (8.4% vs 2.7%, respectively; P < .001). With the 6-month prophylaxis regimen, liver function abnormalities (7.7% vs 4.3%; P = .023) and headache (2.8% vs 0.9%; P = .037) were more common with colchicine (n = 1807) than naproxen, while gastrointestinal/abdominal pains (3.2% vs 1.2%; P = .012) and dental/oral soft tissue infections (2.3% vs 0.6%; P = .006) were more common with naproxen (n = 346) than colchicine.
Uricostatic Agents
Uricostatic therapy with a xanthine oxidase inhibitor (ie, allopurinol or febuxostat) is the most commonly used ULT. Allopurinol is effective in lowering the sUA level and has been shown to lower the rates of all-cause mortality and cardiovascular events, and, in patients with chronic kidney disease, slow the progression of renal disease.29,30 One key point that must be kept in mind is that the efficacy of allopurinol to lower the sUA level is dose-dependent, although limited safety data are available for doses >300 mg per day.14,31,32 One recent prospective clinical trial showed that 26% of patients achieved an sUA level of 5 mg/dL or less following 2 months of treatment with allopurinol 300 mg per day compared with 78% of those who subsequently doubled the dose to 300 mg twice daily.31 Two patients discontinued treatment with allopurinol because of an AE. Finally, the dose of allopurinol must be adjusted based on renal function to minimize the risk of AEs, particularly skin rashes.33
Febuxostat is also effective in lowering the sUA level. In patients with an sUA level of 8.0 mg/dL or higher and a creatinine clearance of 50 mL/min or higher at baseline, an sUA level of less than 6.0 mg/dL was achieved in 53% of patients treated with febuxostat 80 mg (n = 256) versus 21% of patients treated with allopurinol 300 mg once daily (n = 253) after 1 year (P < .001).34 The most frequent treatment-related AE was liver function abnormality, which occurred in 4% of patients in each group. Results of a 6-month trial showed that achievement of an sUA level of less than 6.0 mg/dL was achieved in 45% and 67% of patients treated with febuxostat 40 mg or 80 mg daily, respectively, and 42% of those treated with allopurinol 300 mg (200 mg in moderate renal impairment) daily.35 Febuxostat also has been shown to slow the progression of, or even stabilize, renal function.36
Treatment plan (continued):
- For an acute gout attack: Continue colchicine as needed
- ULT: Initiate allopurinol 100 mg once daily; increase to 200 mg once daily in 1 week, and 300 mg once daily in another week
- -Alternatively, initiate febuxostat 40 mg once daily; increase to 80 mg once daily if an sUA level of less than 6.0 mg/dL is not achieved within 2 weeks
- For prophylaxis of an acute attack when initiating ULT: Initiate colchicine 0.6 mg once daily; may increase to 0.6 mg twice daily if needed
- -Alternatively, initiate naproxen 250 mg twice daily with a proton pump inhibitor
- Measure sUA in 1 month; if the sUA level is greater than 6.0 mg/dL, increase allopurinol to 200 mg twice daily
- -Measure sUA in 1 month; if the sUA level is still greater than 6.0 mg/dL, increase allopurinol to 300 mg twice daily
- Implement the care plan ( TABLE )27
- -Inquire about and address issues to promote adherence and self-management
- -Discuss the most common AEs with allopurinol and colchicine and the actions the patient should take if an AE occurs
- Once the sUA level is 6.0 mg/dL or less, monitor sUA annually (including serum creatinine)14
1. Zhu Y, Pandya BJ, Choi HK. Prevalence of gout and hyperuricemia in the US general population: the National Health and Nutrition Examination Survey 2007-2008. Arthritis Rheum. 2011;63(10):3136-3141.
2. Weaver AL. Epidemiology of gout. Cleve Clin J Med. 2008;75(suppl 5):S9-S12.
3. Choi HK, Atkinson K, Karlson EW, Curhan G. Obesity, weight change, hypertension, diuretic use, and risk of gout in men: the health professionals follow-up study. Arch Intern Med. 2005;165(7):742-748.
4. Choi HK, Curhan G. Soft drinks, fructose consumption, and the risk of gout in men: prospective cohort study. BMJ. 2008;336(7639):309-312.
5. Choi HK, Atkinson K, Karlson EW, Willett W, Curhan G. Purine-rich foods, dairy and protein intake, and the risk of gout in men. N Engl J Med. 2004;350(11):1093-1103.
6. Choi HK, Atkinson K, Karlson EW, Willett W, Curhan G. Alcohol intake and risk of incident gout in men: a prospective study. Lancet. 2004;363(9417):1277-1281.
7. Choi HK, Ford ES, Li C, Curhan G. Prevalence of the metabolic syndrome in patients with gout: the Third National Health and Nutrition Examination Survey. Arthritis Rheum. 2007;57(1):109-115.
8. Perlstein TS, Gumieniak O, Williams GH, et al. Uric acid and the development of hypertension: the normative aging study. Hypertension. 2006;48(6):1031-1036.
9. Bos MJ, Koudstaal PJ, Hofman A, Witteman JC, Breteler MM. Uric acid is a risk factor for myocardial infarction and stroke: the Rotterdam study. Stroke. 2006;37(6):1503-1507.
10. Iseki K, Ikemiya Y, Inoue T, Iseki C, Kinjo K, Takishita S. Significance of hyperuricemia as a risk factor for developing ESRD in a screened cohort. Am J Kidney Dis. 2004;44(4):642-650.
11. Tomita M, Mizuno S, Yamanaka H, et al. Does hyperuricemia affect mortality? A prospective cohort study of Japanese male workers. J Epidemiol. 2000;10(6):403-409.
12. Campion EW, Glynn RJ, DeLabry LO. Asymptomatic hyperuricemia. Risks and consequences in the Normative Aging Study. Am J Med. 1987;82(3):421-426.
13. Mandell BF. Clinical manifestations of hyperuricemia and gout. Cleve Clin J Med. 2008;75(Suppl 5):S5-S8.
14. Hamburger M, Baraf HS, Adamson TC III, et al. 2011 Recommendations for the diagnosis and management of gout and hyperuricemia. Postgrad Med. 2011;123 (6 suppl 1):3-36.
15. Zhang W, Doherty M, Bardin T, et al. EULAR evidence based recommendations for gout. Part II: Management. Report of a task force of the EULAR Standing Committee for International Clinical Studies Including Therapeutics (ESCISIT). Ann Rheum Dis. 2006;65(10):1312-1324.
16. Schumacher HR Jr, Boice JA, Daikh DI, et al. Randomised double blind trial of etoricoxib and indometacin in treatment of acute gouty arthritis. BMJ. 2002;324(7352):1488-1492.
17. Jordan KM, Cameron JS, Snaith M, et al. British Society for Rheumatology and British Health Professionals in Rheumatology guideline for the management of gout. Rheumatology (Oxford). 2007;46(8):1372-1374.
18. Trelle S, Reichenbach S, Wandel S, et al. Cardiovascular safety of non-steroidal anti-inflammatory drugs: network meta-analysis. BMJ. 2011;342:c7086.-
19. Schmidt M, Christiansen CF, Mehnert F, Rothman KJ, Sorensen HT. Non-steroidal anti-inflammatory drug use and risk of atrial fibrillation or flutter: population based case-control study. BMJ. 2011;343:d3450.-
20. NSAIDS and chronic kidney disease. US Centers for Disease Control and Prevention. http://www.cdc.gov/diabetes/news/docs/nsaid_video.htm. Published 2012. Accessed April 22, 2012.
21. Gislason GH, Rasmussen JN, Abildstrom SZ, et al. Increased mortality and cardiovascular morbidity associated with use of nonsteroidal anti-inflammatory drugs in chronic heart failure. Arch Intern Med. 2009;169(2):141-149.
22. Janssens HJ, Lucassen PL, Van de Laar FA, Janssen M, Van de Lisdonk EH. Systemic corticosteroids for acute gout. Cochrane Database Syst Rev. 2008;(2):CD005521.-
23. Janssens HJ, Janssen M, van de Lisdonk EH, van Riel PL, van Weel C. Use of oral prednisolone or naproxen for the treatment of gout arthritis: a double-blind, randomised equivalence trial. Lancet. 2008;371(9627):1854-1860.
24. Schlesinger N, Schumacher R, Catton M, Maxwell L. Colchicine for acute gout. Cochrane Database Syst Rev. 2006;(4):CD006190.-
25. Terkeltaub RA, Furst DE, Bennett K, Kook KA, Crockett RS, Davis MW. High versus low dosing of oral colchicine for early acute gout flare: Twenty-four-hour outcome of the first multicenter, randomized, double-blind, placebo-controlled, parallel-group, dose-comparison colchicine study. Arthritis Rheum. 2010;62(4):1060-1068.
26. Colcrys [package insert]. Philadelphia, PA: AR Scientific, Inc.; 2011.
27. Becker MA, Ruoff GE. What do I need to know about gout? J Fam Pract. 2010;59(6 suppl):S1-S8.
28. Wortmann RL, Macdonald PA, Hunt B, Jackson RL. Effect of prophylaxis on gout flares after the initiation of urate-lowering therapy: analysis of data from three phase III trials. Clin Ther. 2010;32(14):2386-2397.
29. Luk AJ, Levin GP, Moore EE, Zhou XH, Kestenbaum BR, Choi HK. Allopurinol and mortality in hyperuricaemic patients. Rheumatology (Oxford). 2009;48(7):804-806.
30. Goicoechea M, de Vinuesa SG, Verdalles U, et al. Effect of allopurinol in chronic kidney disease progression and cardiovascular risk. Clin J Am Soc Nephrol. 2010;5(8):1388-1393.
31. Reinders MK, Haagsma C, Jansen TL, et al. A randomised controlled trial on the efficacy and tolerability with dose escalation of allopurinol 300-600 mg/day versus benzbromarone 100-200 mg/day in patients with gout. Ann Rheum Dis. 2009;68(6):892-897.
32. Stamp LK, O’Donnell JL, Zhang M, et al. Using allopurinol above the dose based on creatinine clearance is effective and safe in patients with chronic gout, including those with renal impairment. Arthritis Rheum. 2011;63(2):412-421.
33. Zyloprim [package insert]. San Diego, CA: Prometheus Laboratories Inc.; 2003.
34. Becker MA, Schumacher HR Jr, Wortmann RL, et al. Febuxostat compared with allopurinol in patients with hyperuricemia and gout. N Engl J Med. 2005;353(23):2450-2461.
35. Becker MA, Schumacher HR, Espinoza LR, et al. The urate-lowering efficacy and safety of febuxostat in the treatment of the hyperuricemia of gout: the CONFIRMS trial. Arthritis Res Ther. 2010;12:doi:10.1186/ar2978.
36. Whelton A, Macdonald PA, Zhao L, Hunt B, Gunawardhana L. Renal function in gout: long-term treatment effects of febuxostat. J Clin Rheumatol. 2011;17(1):7-13.
1. Zhu Y, Pandya BJ, Choi HK. Prevalence of gout and hyperuricemia in the US general population: the National Health and Nutrition Examination Survey 2007-2008. Arthritis Rheum. 2011;63(10):3136-3141.
2. Weaver AL. Epidemiology of gout. Cleve Clin J Med. 2008;75(suppl 5):S9-S12.
3. Choi HK, Atkinson K, Karlson EW, Curhan G. Obesity, weight change, hypertension, diuretic use, and risk of gout in men: the health professionals follow-up study. Arch Intern Med. 2005;165(7):742-748.
4. Choi HK, Curhan G. Soft drinks, fructose consumption, and the risk of gout in men: prospective cohort study. BMJ. 2008;336(7639):309-312.
5. Choi HK, Atkinson K, Karlson EW, Willett W, Curhan G. Purine-rich foods, dairy and protein intake, and the risk of gout in men. N Engl J Med. 2004;350(11):1093-1103.
6. Choi HK, Atkinson K, Karlson EW, Willett W, Curhan G. Alcohol intake and risk of incident gout in men: a prospective study. Lancet. 2004;363(9417):1277-1281.
7. Choi HK, Ford ES, Li C, Curhan G. Prevalence of the metabolic syndrome in patients with gout: the Third National Health and Nutrition Examination Survey. Arthritis Rheum. 2007;57(1):109-115.
8. Perlstein TS, Gumieniak O, Williams GH, et al. Uric acid and the development of hypertension: the normative aging study. Hypertension. 2006;48(6):1031-1036.
9. Bos MJ, Koudstaal PJ, Hofman A, Witteman JC, Breteler MM. Uric acid is a risk factor for myocardial infarction and stroke: the Rotterdam study. Stroke. 2006;37(6):1503-1507.
10. Iseki K, Ikemiya Y, Inoue T, Iseki C, Kinjo K, Takishita S. Significance of hyperuricemia as a risk factor for developing ESRD in a screened cohort. Am J Kidney Dis. 2004;44(4):642-650.
11. Tomita M, Mizuno S, Yamanaka H, et al. Does hyperuricemia affect mortality? A prospective cohort study of Japanese male workers. J Epidemiol. 2000;10(6):403-409.
12. Campion EW, Glynn RJ, DeLabry LO. Asymptomatic hyperuricemia. Risks and consequences in the Normative Aging Study. Am J Med. 1987;82(3):421-426.
13. Mandell BF. Clinical manifestations of hyperuricemia and gout. Cleve Clin J Med. 2008;75(Suppl 5):S5-S8.
14. Hamburger M, Baraf HS, Adamson TC III, et al. 2011 Recommendations for the diagnosis and management of gout and hyperuricemia. Postgrad Med. 2011;123 (6 suppl 1):3-36.
15. Zhang W, Doherty M, Bardin T, et al. EULAR evidence based recommendations for gout. Part II: Management. Report of a task force of the EULAR Standing Committee for International Clinical Studies Including Therapeutics (ESCISIT). Ann Rheum Dis. 2006;65(10):1312-1324.
16. Schumacher HR Jr, Boice JA, Daikh DI, et al. Randomised double blind trial of etoricoxib and indometacin in treatment of acute gouty arthritis. BMJ. 2002;324(7352):1488-1492.
17. Jordan KM, Cameron JS, Snaith M, et al. British Society for Rheumatology and British Health Professionals in Rheumatology guideline for the management of gout. Rheumatology (Oxford). 2007;46(8):1372-1374.
18. Trelle S, Reichenbach S, Wandel S, et al. Cardiovascular safety of non-steroidal anti-inflammatory drugs: network meta-analysis. BMJ. 2011;342:c7086.-
19. Schmidt M, Christiansen CF, Mehnert F, Rothman KJ, Sorensen HT. Non-steroidal anti-inflammatory drug use and risk of atrial fibrillation or flutter: population based case-control study. BMJ. 2011;343:d3450.-
20. NSAIDS and chronic kidney disease. US Centers for Disease Control and Prevention. http://www.cdc.gov/diabetes/news/docs/nsaid_video.htm. Published 2012. Accessed April 22, 2012.
21. Gislason GH, Rasmussen JN, Abildstrom SZ, et al. Increased mortality and cardiovascular morbidity associated with use of nonsteroidal anti-inflammatory drugs in chronic heart failure. Arch Intern Med. 2009;169(2):141-149.
22. Janssens HJ, Lucassen PL, Van de Laar FA, Janssen M, Van de Lisdonk EH. Systemic corticosteroids for acute gout. Cochrane Database Syst Rev. 2008;(2):CD005521.-
23. Janssens HJ, Janssen M, van de Lisdonk EH, van Riel PL, van Weel C. Use of oral prednisolone or naproxen for the treatment of gout arthritis: a double-blind, randomised equivalence trial. Lancet. 2008;371(9627):1854-1860.
24. Schlesinger N, Schumacher R, Catton M, Maxwell L. Colchicine for acute gout. Cochrane Database Syst Rev. 2006;(4):CD006190.-
25. Terkeltaub RA, Furst DE, Bennett K, Kook KA, Crockett RS, Davis MW. High versus low dosing of oral colchicine for early acute gout flare: Twenty-four-hour outcome of the first multicenter, randomized, double-blind, placebo-controlled, parallel-group, dose-comparison colchicine study. Arthritis Rheum. 2010;62(4):1060-1068.
26. Colcrys [package insert]. Philadelphia, PA: AR Scientific, Inc.; 2011.
27. Becker MA, Ruoff GE. What do I need to know about gout? J Fam Pract. 2010;59(6 suppl):S1-S8.
28. Wortmann RL, Macdonald PA, Hunt B, Jackson RL. Effect of prophylaxis on gout flares after the initiation of urate-lowering therapy: analysis of data from three phase III trials. Clin Ther. 2010;32(14):2386-2397.
29. Luk AJ, Levin GP, Moore EE, Zhou XH, Kestenbaum BR, Choi HK. Allopurinol and mortality in hyperuricaemic patients. Rheumatology (Oxford). 2009;48(7):804-806.
30. Goicoechea M, de Vinuesa SG, Verdalles U, et al. Effect of allopurinol in chronic kidney disease progression and cardiovascular risk. Clin J Am Soc Nephrol. 2010;5(8):1388-1393.
31. Reinders MK, Haagsma C, Jansen TL, et al. A randomised controlled trial on the efficacy and tolerability with dose escalation of allopurinol 300-600 mg/day versus benzbromarone 100-200 mg/day in patients with gout. Ann Rheum Dis. 2009;68(6):892-897.
32. Stamp LK, O’Donnell JL, Zhang M, et al. Using allopurinol above the dose based on creatinine clearance is effective and safe in patients with chronic gout, including those with renal impairment. Arthritis Rheum. 2011;63(2):412-421.
33. Zyloprim [package insert]. San Diego, CA: Prometheus Laboratories Inc.; 2003.
34. Becker MA, Schumacher HR Jr, Wortmann RL, et al. Febuxostat compared with allopurinol in patients with hyperuricemia and gout. N Engl J Med. 2005;353(23):2450-2461.
35. Becker MA, Schumacher HR, Espinoza LR, et al. The urate-lowering efficacy and safety of febuxostat in the treatment of the hyperuricemia of gout: the CONFIRMS trial. Arthritis Res Ther. 2010;12:doi:10.1186/ar2978.
36. Whelton A, Macdonald PA, Zhao L, Hunt B, Gunawardhana L. Renal function in gout: long-term treatment effects of febuxostat. J Clin Rheumatol. 2011;17(1):7-13.
Managing the Multiple Symptoms of Benign Prostatic Hyperplasia
Managing the Multiple Symptoms of Benign Prostatic Hyperplasia — CME
Managing Type 2 Diabetes in Men
Meeting New Challenges with Antiplatelet Therapy in Primary Care
Dr. Miner has disclosed that he is a consultant for Eli Lilly.
SUPPORT
This activity was supported by an educational grant from Lilly USA, LLC.
This article reviews screening tools for benign prostatic hyperplasia (BPH) and steps to be taken to confirm a diagnosis of BPH. Among the treatment options for BPH, emphasis is placed on pharmacologic treatment with alpha1-adrenergic blockers (AABs), 5-alpha-reductase inhibitors (5-ARIs), and phosphodiesterase-5 inhibitors (PDE-5Is). The two newest agents silodosin and tadalafil are discussed in greater detail.
LEARNING OBJECTIVES
After reviewing this activity on benign prostatic hyperplasia, the reader will be able to:
- Describe the key diagnostic steps.
- Describe the role of non-pharmacologic interventions.
- Compare the efficacy and safety of alpha1- adrenergic blockers, 5-alpha-reductase inhibitors, and phosphodiesterase-5 inhibitors.
- Describe strategies for treating multiple symptoms.
TARGET AUDIENCE
Family physicians and clinicians who wish to gain increased knowledge and greater competency regarding the management of patients with multiple symptoms of benign prostatic hyperplasia.
ACKNOWLEDGEMENT
Dr. Miner was paid an honorarium by and received editorial assistance from the Primary Care Education Consortium in the development of this activity.
DISCLOSURES
As a continuing medical education provider accredited by the Accreditation Council for Continuing Medical Education (ACCME), it is the policy of the Primary Care Education Consortium (PCEC) to require any individual in a position to influence educational content to disclose the existence of any financial interest or other personal relationship with the manufacturer(s) of any commercial product(s).
Dr. Miner has disclosed that he is a consultant for Eli Lilly.
The medical accuracy and continuing medical education (CME) reviewer for this activity, Dr. Ron Pollack, has no real or apparent conflicts of interest to report.
PRIMARY CARE EDUCATION CONSORTIUM STAFF
Dr. Brunton has disclosed that he is on the advisory boards and speakers’ bureaus for Boehringer Ingelheim, Eli Lilly, Kowa, Novo Nordisk, Inc, and Teva Pharmaceuticals, and is on the advisory boards for Abbott and Sunovion.
Other PCEC staff have provided financial disclosure and have no conflicts of interest to resolve related to this activity.
CONFLICTS OF INTEREST
When individuals in a position to control content have reported financial relationships with one or more commercial interests, the Primary Care Education Consortium works with them to resolve such conflicts to ensure that the content presented is free of commercial bias. The content of this activity was vetted by the following mechanisms and modified as required to meet this standard:
- Content peer-review by an external topic expert
- Content peer-review by an external CME reviewer
- Content validation by internal Primary Care Education Consortium clinical editorial staff
OFF-LABEL DISCLOSURE
In accordance with ACCME guidelines, the faculty author has been asked to disclose discussion on unlabeled or unapproved uses of drugs or devices during the course of the activity.
SPONSORSHIP
This activity is sponsored by the Primary Care Education Consortium.
ACCREDITATION
This journal-based CME activity, Managing the Multiple Symptoms of Benign Prostatic Hyperplasia, has been reviewed and is acceptable for up to 1.0 prescribed credit by the American Academy of Family Physicians. AAFP accreditation begins June 1, 2012. Term of approval is for one year from this date with option for yearly renewal.
Physicians should claim only the credit commensurate with the extent of their participation in the activity.
MEDIUM
Text publication in the form of a journal article.
METHOD OF PHYSICIAN PARTICIPATION
To receive CME credit, please read the journal article, and upon completion go to: www.pceconsortium.org/menshealthBPH to complete the online evaluation to receive your certification of completion.
SUPPORT
This activity was supported by an educational grant from Lilly USA, LLC.
RI is a 53-year-old African-American man who is being seen by his family physician for a follow-up for dyslipidemia and hypertension. He reports that he is feeling well and that he has not observed any adverse events (AEs) from his medications. His current medications are an intermediate-dose statin, a thiazide diuretic, and a calcium channel blocker. RI reports that he has been very compliant with his medications, missing at most 1 dose every week or two.
During the visit, his physician notices that RI has yawned several times and that he appears tired. When asked how many hours he sleeps each night, RI indicates that he sleeps 7.5 to 8 hours most nights. On further questioning, RI admits that for the past 4 or 5 years, he has had to get up to go to the bathroom during the night, after which he often has trouble falling asleep, and that this nocturia currently occurs 3 or 4 times a night. When asked whether he has noticed any other changes over the past few years, RI says that he has noted an increase in his waist circumference (now 38.5 inches) and a few more aches and pains. When asked whether he has experienced any changes in sexual function, RI acknowledges that occasionally he has had difficulty maintaining an erection. He also indicates that he has accepted that these changes are a result of getting older.
Introduction
BPH is commonly experienced in men as they age. Lower urinary tract symptoms (LUTS) associated with BPH often begin in the fourth decade of life and affect nearly 3 in 4 men by the seventh decade of life.1,2 Lower urinary tract symptoms that prompt men to seek medical care typically include nocturia, frequency, incomplete emptying, and urgency.3,4 Men typically wait almost 2 years before seeking medical care for their urinary symptoms. Among men who do not seek medical care for LUTS, the most common reason is the belief that urinary symptoms are an inevitable part of aging. Many men who do not seek treatment indicate that they would rather accept their urinary symptoms than discuss them with a physician.4
In addition to urinary symptoms, BPH has been associated with symptoms of sexual dysfunction independent of the effects of aging and other comorbidities (eg, diabetes) and lifestyle factors.5-8 Erectile dysfunction and ejaculatory dysfunction are the most common symptoms of sexual dysfunction in men with BPH.9-11 Symptoms of sexual dysfunction may also be caused by some pharmacologic agents used for the treatment of BPH.6,9,10
Evaluation
Although BPH and the symptoms associated with it are not often life-threatening, ruling out other causes such as prostate cancer, diabetes mellitus, or Parkinson disease is an important diagnostic goal.
Screening
Because many men are slow to seek medical care and reluctant to speak with a physician about their symptoms, it is important that family physicians routinely inquire about urinary function in men over the age of 50 years. Beyond simply asking whether there have been changes in urinary function, posing the last question on the International Prostate Symptom Score (IPSS) questionnaire may be helpful: “If you were to spend the rest of your life with your urinary condition just the way it is now, how would you feel about that?”12 This question may be followed with, “Are you bothered enough by your symptoms that you would accept taking a medication?” Inquiries such as these, coupled with education to help the patient to understand that LUTS are not simply due to aging and that effective treatments are available, may motivate patients to share their concerns regarding urinary function. In addition, helping patients to understand that BPH is not a risk factor for prostate cancer, but that there are other causes of LUTS, which are best detected early, may be helpful.
Assessment
A history and focused urologic examination are crucial for the diagnosis of BPH. The medical history should identify a patient’s LUTS and their severity. To do this, a questionnaire such as the IPSS or the American Urological Association BPH Symptom Score Index Questionnaire can be administered [www.adultpediatricuro.com/apuauass.pdf]. As noted earlier, the eighth question on the IPSS questionnaire is useful for assessing the degree to which a patient is bothered by LUTS, with a higher score suggesting a greater willingness of the patient to be treated.13 Lower urinary tract symptoms are generally categorized into storage or bladder-emptying symptoms, with the latter subclassified as voiding or postmicturition symptoms.14 Storage problems are generally of greater concern to patients. Possible sexual dysfunction should also be assessed. A thorough medication history must be taken to identify AEs possibly related to the use of diuretics, anticholinergics, opioids, or decongestants.
The digital rectal examination (DRE) and prostate specific antigen (PSA) test are helpful to rule out a diagnosis of prostate cancer.15,16 The DRE is used for assessing the size, shape, symmetry, nodularity, and consistency of the prostate. The suprapubic area and genitals should be examined as well.17 The PSA test is also useful in the diagnosis and treatment of BPH because the PSA level rises as the prostate increases in size.13,14 A PSA level of 1.5 ng/mL roughly correlates with a prostate size of 30 mL.18 A urinalysis is needed to screen for urinary tract infections, bladder cancer, and kidney stones. Other laboratory analyses such as a fasting plasma glucose test may be needed based on the patient’s history and other findings.17
RI returns 3 weeks later for further evaluation. History confirms nocturia 3 or 4 times per night, as well as occasional erectile dysfunction and sometimes an inability to ejaculate. His IPSS questionnaire reveals a score of 9 (moderate symptoms), with occasional urinary frequency and straining. His LUTS are more bothersome than his occasional erectile dysfunction. It is decided that he will discontinue treatment with the thiazide diuretic because it may be contributing to his LUTS. An alternative antihypertensive agent will be initiated based on the results of the evaluation. The DRE reveals a boggy, slightly enlarged but normally shaped prostate with no nodules. The remainder of the urologic examination is normal. His PSA level is 0.8 ng/mL, and the urinalysis is normal. Further evaluation rules out prostate cancer and other causes of his symptoms.
A diagnosis of BPH is confirmed, with evidence of storage (ie, nocturia) and voiding (ie, urinary intermittency and straining) problems, as well as erectile dysfunction and occasional ejaculatory dysfunction.
Treatment
Goals
Because BPH is not often life-threatening, the focus of treatment has typically been to alleviate bothersome LUTS and other symptoms. With advances in treatment, additional goals include the alteration of disease progression and the prevention of associated complications such as recurrent urinary tract infections, hematuria, or acute urinary retention, particularly in men with an enlarged prostate (ie, volume >30 mL or PSA >1.5 ng/mL), since disease progression is more likely in these patients.17,19 A recent Medline-based systematic review reported that men prefer therapies that affect long-term progression over therapies that provide short-term symptom improvement.20 These results were consistent with those from a 2006 survey of 400 men with an enlarged prostate, which also reported that men are generally willing to wait up to 3 months for symptom relief if treatment would resolve the underlying condition.21 It is, therefore, important to discuss with the patient the natural history of BPH and its complications, and the benefits and risks of currently available noninvasive and invasive treatment options.
Options
Treatment options for BPH are watchful waiting, lifestyle and behavioral management, pharmacologic therapy, and surgical intervention. Many men use phytobotannical therapy such as saw palmetto, African plum tree, pumpkin seed, rye pollen, stinging nettle, South African star grass, and quercetin to relieve LUTS, although investigations regarding their use are often of poor quality. Saw palmetto is the best studied, yet a Cochrane review found few high-quality studies. The authors of the review concluded that saw palmetto was not more effective than placebo for treatment of LUTS.22 Similar results were observed in a randomized, double-blind, placebo-controlled trial more recently published by the Complementary and Alternative Medicine for Urological Symptoms (CAMUS) Study Group.23
Watchful waiting is appropriate when only LUTS are present, with or without some degree of nonsuspicious prostate enlargement, and the symptoms are not particularly bothersome to the patient or if the patient does not want treatment.19 Lifestyle management and behavioral modification should generally be used in combination with other treatment options in an effort to alleviate symptoms, especially in men in whom storage symptoms predominate. Lifestyle management may include reducing fluid intake (particularly if polyuria is present), increasing physical activity, achieving a normal weight, timed voiding (bladder retraining), pelvic floor exercises, treatment for constipation, and avoidance of irritative foods and beverages.17,19 Epidemiologic evidence over 7 years of surveillance suggests that a diet low in fat and red meat and high in protein and vegetables, as well as regular alcohol consumption (>1 drink/month), may reduce the risk of symptomatic BPH.24 Evidence was weak concerning the benefits of lycopene, zinc, and supplemental vitamin D. No dietary supplement, combination phytotherapeutic agent, or other nonconventional therapy is recommended by the American Urological Association (AUA) for the management of LUTS secondary to BPH.19
Surgical intervention is considered appropriate in patients with moderate to severe LUTS in whom other medical therapies have not achieved treatment goals and in those in whom benign prostatic obstruction has led to complications such as renal insufficiency, urinary retention, recurrent urinary tract infections, bladder calculi, or hydronephrosis. Patients in whom surgical intervention is contemplated should be referred to a urologist.17,19
Pharmacologic Options
Three classes of pharmacologic agents have been approved by the US Food and Drug Administration (FDA) for the treatment of symptomatic BPH: AABs, 5-ARIs, and PDE-5Is. The AABs include alfuzosin, doxazosin, silodosin, tamsulosin, and terazosin, and target the dynamic (smooth muscle tone) component of BPH-induced bladder outlet obstruction. The 5-ARIs finasteride and dutasteride target the static (prostate mass) component of BPH-induced bladder outlet obstruction. The PDE-5Is (ie, sildenafil, tadalafil, and vardenafil) increase the amount of cyclic guanosine monophosphate in the smooth muscle of the corpus cavernosum, prostate, and bladder.
Alpha1-Adrenergic Blockers. The four older AABs (ie, alfuzosin, doxazosin, tamsulosin, and terazosin) have been extensively investigated in clinical trials and widely used in the management of BPH. A 2010 review by the AUA concluded that the minor efficacy differences reported among the 4 older AABs were not clinically significant.19 Although ejaculatory dysfunction may occur with the use of the AABs, these agents are generally well-tolerated, with dizziness the most common AE, occurring in 2% to 14% of men. Ejaculatory dysfunction may be a part of the disease process itself, as noted earlier.
The newest AAB, silodosin, at a dosage of 8 mg/d was reported to have efficacy similar to tamsulosin 0.2 to 0.4 mg/d in reducing storage and voiding LUTS in three 12-week trials.25-27 Silodosin has also been associated with a significant improvement in patients’ quality of life. The most frequent AE related to silodosin use was abnormal ejaculation, occurring in 10% to 22% and causing discontinuation in 1% to 3%.25-27 One 12-week study reported that systolic blood pressure (BP) decreased 0.1 and 4.2 mm Hg in the silodosin and tamsulosin groups, respectively.25 The negligible reduction in BP observed with silodosin is likely due to the selectivity of silodosin for the alpha1A-adrenergic receptor rather than the alpha1B-adrenergic receptor, the blockade of which reduces BP.
5-Alpha-Reductase Inhibitors. The efficacy of 5-ARIs in preventing progression of LUTS secondary to BPH and their tolerability are well-established. Dutasteride was associated with a greater reduction in dihydrotestosterone in prostate tissues compared with finasteride (94% vs 80%, respectively) and has a longer elimination half-life.19 Finasteride was reported to be less effective than an AAB in improving LUTS. Dutasteride may have been more effective in reducing the relative risk for acute urinary retention and BPH-related surgery compared with tamsulosin over 4 years, but more research is needed.28 The 5-ARIs should not be used in men with LUTS secondary to BPH without prostatic enlargement, but may be used to prevent the progression of LUTS secondary to BPH and to reduce the risk for urinary retention and future prostate-related surgery.19 Prostate size ≥30 mL or PSA level ≥1.5 ng/dL is usually used as the threshold for considering 5-ARI therapy.19 As expected, because of the effects on dihydrotestosterone, AEs are primarily sexually related and include decreased libido, ejaculation disorders, and erectile dysfunction.19
Phosphodiesterase-5 Inhibitors. Approved by the FDA for erectile dysfunction, several observations led to the investigation of PDE-5Is for LUTS related to BPH.8,29 One was that the prevalences of BPH, LUTS, and erectile dysfunction increase as a man ages. Second was that LUTS have been identified as a risk factor for sexual dysfunction in aging men. Third was that limited evidence had suggested that PDE-5Is might be effective in treating LUTS and erectile dysfunction. Further investigation suggested beneficial effects on LUTS with each of the 3 PDE-5Is (ie, sildenafil, tadalafil, and vardenafil).30-32 Subsequent extensive investigation with tadalafil demonstrated its efficacy in reducing the storage and voiding symptoms of BPH and led to the approval by the FDA of tadalafil for symptoms of BPH alone or with erectile dysfunction.33-37
The clinical studies investigating the efficacy and tolerability of tadalafil for LUTS associated with BPH have included a 12-week study with a 1-year extension.38 Patients with BPH-associated LUTS (N = 1058) were randomized to tadalafil 2.5, 5, 10, or 20 mg/d or placebo once daily for 12 weeks. The total IPSS score was significantly improved at 12 weeks compared with baseline in each of the tadalafil groups relative to placebo (2.5 mg/d: –3.9, P = .015; 5 mg/d: –4.9, P < .001; 10 mg/d: –5.2, P < .001; 20 mg/d: –5.2, P < .001; placebo: –2.3). The use of tadalafil 5, 10, or 20 mg once daily was associated with significant improvements in the IPSS irritative (eg, frequency, nocturia, and urgency) and obstructive (eg, incomplete emptying, intermittency, slow stream, and straining) subscores, as well as scores on the IPSS quality-of-life measure, the BPH Impact Index (except 10 mg), and the LUTS Global Assessment Question. In sexually active men with erectile dysfunction, all doses of tadalafil were associated with significant improvements in scores on the International Index of Erectile Function–Erectile Function domain compared with placebo. Peak flow rate was not improved at any dose of tadalafil compared with placebo.
In total, 427 men who completed the 12-week study elected to receive tadalafil 5 mg once daily for an additional year.37 Patients who were switched from placebo or who had the dose increased from 2.5 mg/d had a significant reduction in total IPSS score from week 12 to week 16, and this change was maintained until the end of follow-up at week 64. Patients who had received tadalafil 5, 10, or 20 mg/d maintained the changes observed at the end of the 12-week study. Similarly, sexually active men with erectile dysfunction and who had a female partner maintained the improvements observed at the end of 12 weeks. The mean postvoid residual volume was decreased from 61 to 42 mL. At least 1 treatment-emergent AE (TEAE) was reported in 58% of patients, with 89% of events being either mild or moderate in severity. Treatment was discontinued in 5% due to a TEAE. The most common TEAEs were dyspepsia (4%), gastroesophageal reflux disease (4%), back pain (4%), headache (3%), sinusitis (3%), hypertension (3%), and cough (2%). In this study, the improvement in LUTS, sexual function, and quality of life observed after 12 weeks of tadalafil were maintained over the additional year with tadalafil 5 mg once daily.
Treatment options for RI are watchful waiting, an AAB with or without a PDE-5I, a 5-ARI, or tadalafil. RI indicates that he would rather not have his symptoms for the rest of his life, so watchful waiting is not appropriate. Because his prostate is only slightly enlarged, a 5-ARI is also not appropriate. An AAB or tadalafil should provide good relief to his LUTS within a few weeks. Tadalafil would also treat his erectile dysfunction. Alternatively, tadalafil or another PDE-5I could be combined with an AAB, which has been reported to provide added benefit in symptom improvement over an AAB alone.39
Plan
Following discussion of the benefits and risks of the different treatment options, RI elects to begin treatment with an AAB alone. For this reason, treatment with another antihypertensive to replace the diuretic will not be started. To promote self-management, educational materials and an action plan are reviewed with RI. Lifestyle management changes are discussed, including reducing his daily water intake by 25% to 2 quarts with no consumption of fluids within 3 to 4 hours of bedtime. He is assured that adjustments to his treatment plan will be made based on his symptoms and concerns.
3-Month Follow-Up
RI reports that his symptoms have improved, with a modest improvement of nocturia; he gets up once during the night 1 or 2 times every 2 weeks or so. He strains less frequently, but intermittency is unchanged. His IPSS is 7 (improved by 2 points vs before treatment). The findings on his physical examination are unchanged except that his BP has decreased slightly, to 124/72 mm Hg. He has noted 1 or 2 episodes of dizziness. Feeling better than 3 months ago, RI asks whether further improvement of his LUTS is possible. He wonders whether his erectile dysfunction can be treated.
The benefits and risks of each of the 3 PDE-5Is are reviewed with RI. He elects to begin treatment with tadalafil 5 mg once daily because it is the only agent that is approved for the treatment of LUTS associated with BPH. Lifestyle management and his action plan are reviewed.
Continue to complete the online evaluation and receive your certification of completion.
1. Wei JT, Calhoun EA, Jacobsen SJ. Urologic Diseases in America: Benign prostatic hyperplasia. http://kidney.niddk.nih.gov/Statistics/UDA/Benign_Prostatic_Hyperplasia-Chapter02.pdf. Published 2007. Accessed May 16, 2012.
2. Miller DC, Saigal CS, Litwin MS. The demographic burden of urologic diseases in America. Urol Clin North Am. 2009;36(1):11-27, v.
3. Sarma AV, Wallner L, Jacobsen SJ, Dunn RL, Wei JT. Health seeking behavior for lower urinary tract symptoms in black men. J Urol. 2008;180(1):227-232.
4. Survey confirms prostate problems overlooked by men and doctors [press release]. Vienna, Austria: GlaxoSmithKline. October 3, 2011. http://www.ismh.org/en/sys/wp-content/uploads/2011/09/News-release-300911-BPH-survey-a-male-perspective.pdf. Accessed May 16, 2012.
5. Mirone V, Sessa A, Giuliano F, Berges R, Kirby M, Moncada I. Current benign prostatic hyperplasia treatment: impact on sexual function and management of related sexual adverse events. Int J Clin Pract. 2011;65(9):1005-1013.
6. Gacci M, Eardley I, Giuliano F, et al. Critical analysis of the relationship between sexual dysfunctions and lower urinary tract symptoms due to benign prostatic hyperplasia. Eur Urol. 2011;60(4):809-825.
7. Hoesl CE, Woll EM, Burkart M, Altwein JE. Erectile dysfunction (ED) is prevalent, bothersome and underdiagnosed in patients consulting urologists for benign prostatic syndrome (BPS). Eur Urol. 2005;47(4):511-517.
8. Rosen R, Altwein J, Boyle P, et al. Lower urinary tract symptoms and male sexual dysfunction: the multinational survey of the aging male (MSAM-7). Eur Urol. 2003;44(6):637-649.
9. Rosen RC, Wei JT, Althof SE, Seftel AD, Miner M, Perelman MA. for BPH Registry and Patient Survey Steering Committee. Association of sexual dysfunction with lower urinary tract symptoms of BPH and BPH medical therapies: results from the BPH Registry. Urology. 2009;73(3):562-566.
10. Rosen RC, Fitzpatrick JM. for ALF-LIFE Study Group. Ejaculatory dysfunction in men with lower urinary tract symptoms suggestive of benign prostatic hyperplasia. BJU Int. 2009;104(7):974-983.
11. Seftel A, Rosen R, Kuritzky L. Physician perceptions of sexual dysfunction related to benign prostatic hyperplasia (BPH) symptoms and sexual side effects related to BPH medications. Int J Impot Res. 2007;19(4):386-392.
12. American Urological Association. American Urological Association BPH Symptom Score Index Questionnaire. http://www.adultpediatricuro.com/apuauass.pdf. Accessed May 16, 2012.
13. O’Leary MP. Validity of the “bother score” in the evaluation and treatment of symptomatic benign prostatic hyperplasia. Rev Urol. 2005;7(1):1-10.
14. Abrams P, Cardozo L, Fall M, et al. for Standardisation Sub-Committee of the International Continence Society. The standardisation of terminology in lower urinary tract function: report from the standardisation sub-committee of the International Continence Society. Urology. 2003;61(1):37-49.
15. American Urological Association. Prostate-specific antigen best practice statement: 2009 update. http://www.auanet.org/content/media/psa09.pdf. Published 2009. Accessed May 16, 2012.
16. American Cancer Society. Can prostate cancer be found early? http://www.cancer.org/Cancer/ProstateCancer/DetailedGuide/prostate-cancer-detection. Published 2012. Accessed May 16, 2012.
17. Tanguay S, Awde M, Brock G, et al. Diagnosis and management of benign prostatic hyperplasia in primary care. Can Urol Assoc J. 2009;3(3 suppl 2):S92-S100.
18. Roehrborn CG, Boyle P, Gould AL, Waldstreicher J. Serum prostate-specific antigen as a predictor of prostate volume in men with benign prostatic hyperplasia. Urology. 1999;53(3):581-589.
19. McVary KT, Roehrborn CG, Avins AL, et al. American Urological Association guideline: Management of benign prostatic hyperplasia (BPH). http://www.auanet.org/content/clinical-practice-guidelines/clinical-guidelines.cfm?sub=bph. Published 2010. Accessed May 16, 2012.
20. Emberton M. Medical treatment of benign prostatic hyperplasia: physician and patient p and satisfaction. Int J Clin Pract. 2010;64(10):1425-1435.
21. Kaplan S, Naslund M. Public, patient, and professional attitudes towards the diagnosis and treatment of enlarged prostate: A landmark national US survey. Int J Clin Pract. 2006;60(10):1157-1165.
22. Tacklind J, MacDonald R, Rutks I, Wilt TJ. Serenoa repens for benign prostatic hyperplasia. Cochrane Database Syst Rev. 2009;2:CD001423.-
23. Barry MJ, Meleth S, Lee JY, et al. for Complementary and Alternative Medicine for Urological Symptoms (CAMUS) Study Group. Effect of increasing doses of saw palmetto extract on lower urinary tract symptoms: a randomized trial. JAMA. 2011;306(12):1344-1351.
24. Kristal AR, Arnold KB, Schenk JM, et al. Dietary patterns, supplement use, and the risk of symptomatic benign prostatic hyperplasia: results from the prostate cancer prevention trial. Am J Epidemiol. 2008;167(8):925-934.
25. Yu HJ, Lin AT, Yang SS, et al. Non-inferiority of silodosin to tamsulosin in treating patients with lower urinary tract symptoms (LUTS) associated with benign prostatic hyperplasia (BPH). BJU Int. 2011;108(11):1843-1848.
26. Chapple CR, Montorsi F, Tammela TL, Wirth M, Koldewijn E, Fernandez FE. for European Silodosin Study Group. Silodosin therapy for lower urinary tract symptoms in men with suspected benign prostatic hyperplasia: results of an international, randomized, double-blind, placebo- and active-controlled clinical trial performed in Europe. Eur Urol. 2011;59(3):342-352.
27. Kawabe K, Yoshida M, Homma Y. for Silodosin Clinical Study Group. Silodosin, a new alpha1A-adrenoceptor-selective antagonist for treating benign prostatic hyperplasia: results of a phase III randomized, placebo-controlled, double-blind study in Japanese men. BJU Int. 2006;98(5):1019-1024.
28. Roehrborn CG, Siami P, Barkin J, et al. for CombAT Study Group. The effects of combination therapy with dutasteride and tamsulosin on clinical outcomes in men with symptomatic benign prostatic hyperplasia: 4-year results from the CombAT study. Eur Urol. 2010;57(1):123-131.
29. Kaplan SA, Gonzalez RR. Phosphodiesterase type 5 inhibitors for the treatment of male lower urinary tract symptoms. Rev Urol. 2007;9(2):73-77.
30. Mulhall JP, Guhring P, Parker M, Hopps C. Assessment of the impact of sildenafil citrate on lower urinary tract symptoms in men with erectile dysfunction. J Sex Med. 2006;3(4):662-667.
31. McVary KT, Roehrborn CG, Kaminetsky JC, et al. Tadalafil relieves lower urinary tract symptoms secondary to benign prostatic hyperplasia. J Urol. 2007;177(4):1401-1407.
32. Stief CG, Porst H, Neuser D, Beneke M, Ulbrich E. A randomised, placebo-controlled study to assess the efficacy of twice-daily vardenafil in the treatment of lower urinary tract symptoms secondary to benign prostatic hyperplasia. Eur Urol. 2008;53(6):1236-1244.
33. Porst H, McVary KT, Montorsi F, et al. Effects of once-daily tadalafil on erectile function in men with erectile dysfunction and signs and symptoms of benign prostatic hyperplasia. Eur Urol. 2009;56(4):727-735.
34. Broderick GA, Brock GB, Roehrborn CG, Watts SD, Elion-Mboussa A, Viktrup L. Effects of tadalafil on lower urinary tract symptoms secondary to benign prostatic hyperplasia in men with or without erectile dysfunction. Urology. 2010;75(6):1452-1458.
35. Porst H, Kim ED, Casabé AR, et al. LVHJ study team. Efficacy and safety of tadalafil once daily in the treatment of men with lower urinary tract symptoms suggestive of benign prostatic hyperplasia: results of an international randomized, double-blind, placebo-controlled trial. Eur Urol. 2011;60(5):1105-1113.
36. Maselli G, Bergamasco L, Silvestri V, Gualà L, Pace G, Vicentini C. Tadalafil versus solifenacin for persistent storage symptoms after prostate surgery in patients with erectile dysfunction: a prospective randomized study. Int J Urol. 2011;18(7):515-520.
37. Donatucci CF, Brock GB, Goldfischer ER, et al. Tadalafil administered once daily for lower urinary tract symptoms secondary to benign prostatic hyperplasia: a 1-year, open-label extension study. BJU Int. 2011;107(7):1110-1116.
38. Roehrborn CG, McVary KT, Elion-Mboussa A, Viktrup L. Tadalafil administered once daily for lower urinary tract symptoms secondary to benign prostatic hyperplasia: a dose finding study. J Urol. 2008;180(4):1228-1234.
39. Gacci M, Corona G, Salvi M, et al. A systematic review and meta-analysis on the use of phosphodiesterase 5 inhibitors alone or in combination with alpha-blockers for lower urinary tract symptoms due to benign prostatic hyperplasia. Eur Urol. 2012;61(5):994-1003.
![]()
Martin Miner, MD
Co-Director, Men’s Health Center, Chief of Family and Community Medicine, The Miriam Hospital, Clinical Associate Professor of Family Medicine and Urology, Warren Alpert Medical School, Brown University, Providence, RI
![]()
Martin Miner, MD
Co-Director, Men’s Health Center, Chief of Family and Community Medicine, The Miriam Hospital, Clinical Associate Professor of Family Medicine and Urology, Warren Alpert Medical School, Brown University, Providence, RI
![]()
Martin Miner, MD
Co-Director, Men’s Health Center, Chief of Family and Community Medicine, The Miriam Hospital, Clinical Associate Professor of Family Medicine and Urology, Warren Alpert Medical School, Brown University, Providence, RI
Managing the Multiple Symptoms of Benign Prostatic Hyperplasia — CME
Managing Type 2 Diabetes in Men
Meeting New Challenges with Antiplatelet Therapy in Primary Care
Dr. Miner has disclosed that he is a consultant for Eli Lilly.
SUPPORT
This activity was supported by an educational grant from Lilly USA, LLC.
This article reviews screening tools for benign prostatic hyperplasia (BPH) and steps to be taken to confirm a diagnosis of BPH. Among the treatment options for BPH, emphasis is placed on pharmacologic treatment with alpha1-adrenergic blockers (AABs), 5-alpha-reductase inhibitors (5-ARIs), and phosphodiesterase-5 inhibitors (PDE-5Is). The two newest agents silodosin and tadalafil are discussed in greater detail.
LEARNING OBJECTIVES
After reviewing this activity on benign prostatic hyperplasia, the reader will be able to:
- Describe the key diagnostic steps.
- Describe the role of non-pharmacologic interventions.
- Compare the efficacy and safety of alpha1- adrenergic blockers, 5-alpha-reductase inhibitors, and phosphodiesterase-5 inhibitors.
- Describe strategies for treating multiple symptoms.
TARGET AUDIENCE
Family physicians and clinicians who wish to gain increased knowledge and greater competency regarding the management of patients with multiple symptoms of benign prostatic hyperplasia.
ACKNOWLEDGEMENT
Dr. Miner was paid an honorarium by and received editorial assistance from the Primary Care Education Consortium in the development of this activity.
DISCLOSURES
As a continuing medical education provider accredited by the Accreditation Council for Continuing Medical Education (ACCME), it is the policy of the Primary Care Education Consortium (PCEC) to require any individual in a position to influence educational content to disclose the existence of any financial interest or other personal relationship with the manufacturer(s) of any commercial product(s).
Dr. Miner has disclosed that he is a consultant for Eli Lilly.
The medical accuracy and continuing medical education (CME) reviewer for this activity, Dr. Ron Pollack, has no real or apparent conflicts of interest to report.
PRIMARY CARE EDUCATION CONSORTIUM STAFF
Dr. Brunton has disclosed that he is on the advisory boards and speakers’ bureaus for Boehringer Ingelheim, Eli Lilly, Kowa, Novo Nordisk, Inc, and Teva Pharmaceuticals, and is on the advisory boards for Abbott and Sunovion.
Other PCEC staff have provided financial disclosure and have no conflicts of interest to resolve related to this activity.
CONFLICTS OF INTEREST
When individuals in a position to control content have reported financial relationships with one or more commercial interests, the Primary Care Education Consortium works with them to resolve such conflicts to ensure that the content presented is free of commercial bias. The content of this activity was vetted by the following mechanisms and modified as required to meet this standard:
- Content peer-review by an external topic expert
- Content peer-review by an external CME reviewer
- Content validation by internal Primary Care Education Consortium clinical editorial staff
OFF-LABEL DISCLOSURE
In accordance with ACCME guidelines, the faculty author has been asked to disclose discussion on unlabeled or unapproved uses of drugs or devices during the course of the activity.
SPONSORSHIP
This activity is sponsored by the Primary Care Education Consortium.
ACCREDITATION
This journal-based CME activity, Managing the Multiple Symptoms of Benign Prostatic Hyperplasia, has been reviewed and is acceptable for up to 1.0 prescribed credit by the American Academy of Family Physicians. AAFP accreditation begins June 1, 2012. Term of approval is for one year from this date with option for yearly renewal.
Physicians should claim only the credit commensurate with the extent of their participation in the activity.
MEDIUM
Text publication in the form of a journal article.
METHOD OF PHYSICIAN PARTICIPATION
To receive CME credit, please read the journal article, and upon completion go to: www.pceconsortium.org/menshealthBPH to complete the online evaluation to receive your certification of completion.
SUPPORT
This activity was supported by an educational grant from Lilly USA, LLC.
RI is a 53-year-old African-American man who is being seen by his family physician for a follow-up for dyslipidemia and hypertension. He reports that he is feeling well and that he has not observed any adverse events (AEs) from his medications. His current medications are an intermediate-dose statin, a thiazide diuretic, and a calcium channel blocker. RI reports that he has been very compliant with his medications, missing at most 1 dose every week or two.
During the visit, his physician notices that RI has yawned several times and that he appears tired. When asked how many hours he sleeps each night, RI indicates that he sleeps 7.5 to 8 hours most nights. On further questioning, RI admits that for the past 4 or 5 years, he has had to get up to go to the bathroom during the night, after which he often has trouble falling asleep, and that this nocturia currently occurs 3 or 4 times a night. When asked whether he has noticed any other changes over the past few years, RI says that he has noted an increase in his waist circumference (now 38.5 inches) and a few more aches and pains. When asked whether he has experienced any changes in sexual function, RI acknowledges that occasionally he has had difficulty maintaining an erection. He also indicates that he has accepted that these changes are a result of getting older.
Introduction
BPH is commonly experienced in men as they age. Lower urinary tract symptoms (LUTS) associated with BPH often begin in the fourth decade of life and affect nearly 3 in 4 men by the seventh decade of life.1,2 Lower urinary tract symptoms that prompt men to seek medical care typically include nocturia, frequency, incomplete emptying, and urgency.3,4 Men typically wait almost 2 years before seeking medical care for their urinary symptoms. Among men who do not seek medical care for LUTS, the most common reason is the belief that urinary symptoms are an inevitable part of aging. Many men who do not seek treatment indicate that they would rather accept their urinary symptoms than discuss them with a physician.4
In addition to urinary symptoms, BPH has been associated with symptoms of sexual dysfunction independent of the effects of aging and other comorbidities (eg, diabetes) and lifestyle factors.5-8 Erectile dysfunction and ejaculatory dysfunction are the most common symptoms of sexual dysfunction in men with BPH.9-11 Symptoms of sexual dysfunction may also be caused by some pharmacologic agents used for the treatment of BPH.6,9,10
Evaluation
Although BPH and the symptoms associated with it are not often life-threatening, ruling out other causes such as prostate cancer, diabetes mellitus, or Parkinson disease is an important diagnostic goal.
Screening
Because many men are slow to seek medical care and reluctant to speak with a physician about their symptoms, it is important that family physicians routinely inquire about urinary function in men over the age of 50 years. Beyond simply asking whether there have been changes in urinary function, posing the last question on the International Prostate Symptom Score (IPSS) questionnaire may be helpful: “If you were to spend the rest of your life with your urinary condition just the way it is now, how would you feel about that?”12 This question may be followed with, “Are you bothered enough by your symptoms that you would accept taking a medication?” Inquiries such as these, coupled with education to help the patient to understand that LUTS are not simply due to aging and that effective treatments are available, may motivate patients to share their concerns regarding urinary function. In addition, helping patients to understand that BPH is not a risk factor for prostate cancer, but that there are other causes of LUTS, which are best detected early, may be helpful.
Assessment
A history and focused urologic examination are crucial for the diagnosis of BPH. The medical history should identify a patient’s LUTS and their severity. To do this, a questionnaire such as the IPSS or the American Urological Association BPH Symptom Score Index Questionnaire can be administered [www.adultpediatricuro.com/apuauass.pdf]. As noted earlier, the eighth question on the IPSS questionnaire is useful for assessing the degree to which a patient is bothered by LUTS, with a higher score suggesting a greater willingness of the patient to be treated.13 Lower urinary tract symptoms are generally categorized into storage or bladder-emptying symptoms, with the latter subclassified as voiding or postmicturition symptoms.14 Storage problems are generally of greater concern to patients. Possible sexual dysfunction should also be assessed. A thorough medication history must be taken to identify AEs possibly related to the use of diuretics, anticholinergics, opioids, or decongestants.
The digital rectal examination (DRE) and prostate specific antigen (PSA) test are helpful to rule out a diagnosis of prostate cancer.15,16 The DRE is used for assessing the size, shape, symmetry, nodularity, and consistency of the prostate. The suprapubic area and genitals should be examined as well.17 The PSA test is also useful in the diagnosis and treatment of BPH because the PSA level rises as the prostate increases in size.13,14 A PSA level of 1.5 ng/mL roughly correlates with a prostate size of 30 mL.18 A urinalysis is needed to screen for urinary tract infections, bladder cancer, and kidney stones. Other laboratory analyses such as a fasting plasma glucose test may be needed based on the patient’s history and other findings.17
RI returns 3 weeks later for further evaluation. History confirms nocturia 3 or 4 times per night, as well as occasional erectile dysfunction and sometimes an inability to ejaculate. His IPSS questionnaire reveals a score of 9 (moderate symptoms), with occasional urinary frequency and straining. His LUTS are more bothersome than his occasional erectile dysfunction. It is decided that he will discontinue treatment with the thiazide diuretic because it may be contributing to his LUTS. An alternative antihypertensive agent will be initiated based on the results of the evaluation. The DRE reveals a boggy, slightly enlarged but normally shaped prostate with no nodules. The remainder of the urologic examination is normal. His PSA level is 0.8 ng/mL, and the urinalysis is normal. Further evaluation rules out prostate cancer and other causes of his symptoms.
A diagnosis of BPH is confirmed, with evidence of storage (ie, nocturia) and voiding (ie, urinary intermittency and straining) problems, as well as erectile dysfunction and occasional ejaculatory dysfunction.
Treatment
Goals
Because BPH is not often life-threatening, the focus of treatment has typically been to alleviate bothersome LUTS and other symptoms. With advances in treatment, additional goals include the alteration of disease progression and the prevention of associated complications such as recurrent urinary tract infections, hematuria, or acute urinary retention, particularly in men with an enlarged prostate (ie, volume >30 mL or PSA >1.5 ng/mL), since disease progression is more likely in these patients.17,19 A recent Medline-based systematic review reported that men prefer therapies that affect long-term progression over therapies that provide short-term symptom improvement.20 These results were consistent with those from a 2006 survey of 400 men with an enlarged prostate, which also reported that men are generally willing to wait up to 3 months for symptom relief if treatment would resolve the underlying condition.21 It is, therefore, important to discuss with the patient the natural history of BPH and its complications, and the benefits and risks of currently available noninvasive and invasive treatment options.
Options
Treatment options for BPH are watchful waiting, lifestyle and behavioral management, pharmacologic therapy, and surgical intervention. Many men use phytobotannical therapy such as saw palmetto, African plum tree, pumpkin seed, rye pollen, stinging nettle, South African star grass, and quercetin to relieve LUTS, although investigations regarding their use are often of poor quality. Saw palmetto is the best studied, yet a Cochrane review found few high-quality studies. The authors of the review concluded that saw palmetto was not more effective than placebo for treatment of LUTS.22 Similar results were observed in a randomized, double-blind, placebo-controlled trial more recently published by the Complementary and Alternative Medicine for Urological Symptoms (CAMUS) Study Group.23
Watchful waiting is appropriate when only LUTS are present, with or without some degree of nonsuspicious prostate enlargement, and the symptoms are not particularly bothersome to the patient or if the patient does not want treatment.19 Lifestyle management and behavioral modification should generally be used in combination with other treatment options in an effort to alleviate symptoms, especially in men in whom storage symptoms predominate. Lifestyle management may include reducing fluid intake (particularly if polyuria is present), increasing physical activity, achieving a normal weight, timed voiding (bladder retraining), pelvic floor exercises, treatment for constipation, and avoidance of irritative foods and beverages.17,19 Epidemiologic evidence over 7 years of surveillance suggests that a diet low in fat and red meat and high in protein and vegetables, as well as regular alcohol consumption (>1 drink/month), may reduce the risk of symptomatic BPH.24 Evidence was weak concerning the benefits of lycopene, zinc, and supplemental vitamin D. No dietary supplement, combination phytotherapeutic agent, or other nonconventional therapy is recommended by the American Urological Association (AUA) for the management of LUTS secondary to BPH.19
Surgical intervention is considered appropriate in patients with moderate to severe LUTS in whom other medical therapies have not achieved treatment goals and in those in whom benign prostatic obstruction has led to complications such as renal insufficiency, urinary retention, recurrent urinary tract infections, bladder calculi, or hydronephrosis. Patients in whom surgical intervention is contemplated should be referred to a urologist.17,19
Pharmacologic Options
Three classes of pharmacologic agents have been approved by the US Food and Drug Administration (FDA) for the treatment of symptomatic BPH: AABs, 5-ARIs, and PDE-5Is. The AABs include alfuzosin, doxazosin, silodosin, tamsulosin, and terazosin, and target the dynamic (smooth muscle tone) component of BPH-induced bladder outlet obstruction. The 5-ARIs finasteride and dutasteride target the static (prostate mass) component of BPH-induced bladder outlet obstruction. The PDE-5Is (ie, sildenafil, tadalafil, and vardenafil) increase the amount of cyclic guanosine monophosphate in the smooth muscle of the corpus cavernosum, prostate, and bladder.
Alpha1-Adrenergic Blockers. The four older AABs (ie, alfuzosin, doxazosin, tamsulosin, and terazosin) have been extensively investigated in clinical trials and widely used in the management of BPH. A 2010 review by the AUA concluded that the minor efficacy differences reported among the 4 older AABs were not clinically significant.19 Although ejaculatory dysfunction may occur with the use of the AABs, these agents are generally well-tolerated, with dizziness the most common AE, occurring in 2% to 14% of men. Ejaculatory dysfunction may be a part of the disease process itself, as noted earlier.
The newest AAB, silodosin, at a dosage of 8 mg/d was reported to have efficacy similar to tamsulosin 0.2 to 0.4 mg/d in reducing storage and voiding LUTS in three 12-week trials.25-27 Silodosin has also been associated with a significant improvement in patients’ quality of life. The most frequent AE related to silodosin use was abnormal ejaculation, occurring in 10% to 22% and causing discontinuation in 1% to 3%.25-27 One 12-week study reported that systolic blood pressure (BP) decreased 0.1 and 4.2 mm Hg in the silodosin and tamsulosin groups, respectively.25 The negligible reduction in BP observed with silodosin is likely due to the selectivity of silodosin for the alpha1A-adrenergic receptor rather than the alpha1B-adrenergic receptor, the blockade of which reduces BP.
5-Alpha-Reductase Inhibitors. The efficacy of 5-ARIs in preventing progression of LUTS secondary to BPH and their tolerability are well-established. Dutasteride was associated with a greater reduction in dihydrotestosterone in prostate tissues compared with finasteride (94% vs 80%, respectively) and has a longer elimination half-life.19 Finasteride was reported to be less effective than an AAB in improving LUTS. Dutasteride may have been more effective in reducing the relative risk for acute urinary retention and BPH-related surgery compared with tamsulosin over 4 years, but more research is needed.28 The 5-ARIs should not be used in men with LUTS secondary to BPH without prostatic enlargement, but may be used to prevent the progression of LUTS secondary to BPH and to reduce the risk for urinary retention and future prostate-related surgery.19 Prostate size ≥30 mL or PSA level ≥1.5 ng/dL is usually used as the threshold for considering 5-ARI therapy.19 As expected, because of the effects on dihydrotestosterone, AEs are primarily sexually related and include decreased libido, ejaculation disorders, and erectile dysfunction.19
Phosphodiesterase-5 Inhibitors. Approved by the FDA for erectile dysfunction, several observations led to the investigation of PDE-5Is for LUTS related to BPH.8,29 One was that the prevalences of BPH, LUTS, and erectile dysfunction increase as a man ages. Second was that LUTS have been identified as a risk factor for sexual dysfunction in aging men. Third was that limited evidence had suggested that PDE-5Is might be effective in treating LUTS and erectile dysfunction. Further investigation suggested beneficial effects on LUTS with each of the 3 PDE-5Is (ie, sildenafil, tadalafil, and vardenafil).30-32 Subsequent extensive investigation with tadalafil demonstrated its efficacy in reducing the storage and voiding symptoms of BPH and led to the approval by the FDA of tadalafil for symptoms of BPH alone or with erectile dysfunction.33-37
The clinical studies investigating the efficacy and tolerability of tadalafil for LUTS associated with BPH have included a 12-week study with a 1-year extension.38 Patients with BPH-associated LUTS (N = 1058) were randomized to tadalafil 2.5, 5, 10, or 20 mg/d or placebo once daily for 12 weeks. The total IPSS score was significantly improved at 12 weeks compared with baseline in each of the tadalafil groups relative to placebo (2.5 mg/d: –3.9, P = .015; 5 mg/d: –4.9, P < .001; 10 mg/d: –5.2, P < .001; 20 mg/d: –5.2, P < .001; placebo: –2.3). The use of tadalafil 5, 10, or 20 mg once daily was associated with significant improvements in the IPSS irritative (eg, frequency, nocturia, and urgency) and obstructive (eg, incomplete emptying, intermittency, slow stream, and straining) subscores, as well as scores on the IPSS quality-of-life measure, the BPH Impact Index (except 10 mg), and the LUTS Global Assessment Question. In sexually active men with erectile dysfunction, all doses of tadalafil were associated with significant improvements in scores on the International Index of Erectile Function–Erectile Function domain compared with placebo. Peak flow rate was not improved at any dose of tadalafil compared with placebo.
In total, 427 men who completed the 12-week study elected to receive tadalafil 5 mg once daily for an additional year.37 Patients who were switched from placebo or who had the dose increased from 2.5 mg/d had a significant reduction in total IPSS score from week 12 to week 16, and this change was maintained until the end of follow-up at week 64. Patients who had received tadalafil 5, 10, or 20 mg/d maintained the changes observed at the end of the 12-week study. Similarly, sexually active men with erectile dysfunction and who had a female partner maintained the improvements observed at the end of 12 weeks. The mean postvoid residual volume was decreased from 61 to 42 mL. At least 1 treatment-emergent AE (TEAE) was reported in 58% of patients, with 89% of events being either mild or moderate in severity. Treatment was discontinued in 5% due to a TEAE. The most common TEAEs were dyspepsia (4%), gastroesophageal reflux disease (4%), back pain (4%), headache (3%), sinusitis (3%), hypertension (3%), and cough (2%). In this study, the improvement in LUTS, sexual function, and quality of life observed after 12 weeks of tadalafil were maintained over the additional year with tadalafil 5 mg once daily.
Treatment options for RI are watchful waiting, an AAB with or without a PDE-5I, a 5-ARI, or tadalafil. RI indicates that he would rather not have his symptoms for the rest of his life, so watchful waiting is not appropriate. Because his prostate is only slightly enlarged, a 5-ARI is also not appropriate. An AAB or tadalafil should provide good relief to his LUTS within a few weeks. Tadalafil would also treat his erectile dysfunction. Alternatively, tadalafil or another PDE-5I could be combined with an AAB, which has been reported to provide added benefit in symptom improvement over an AAB alone.39
Plan
Following discussion of the benefits and risks of the different treatment options, RI elects to begin treatment with an AAB alone. For this reason, treatment with another antihypertensive to replace the diuretic will not be started. To promote self-management, educational materials and an action plan are reviewed with RI. Lifestyle management changes are discussed, including reducing his daily water intake by 25% to 2 quarts with no consumption of fluids within 3 to 4 hours of bedtime. He is assured that adjustments to his treatment plan will be made based on his symptoms and concerns.
3-Month Follow-Up
RI reports that his symptoms have improved, with a modest improvement of nocturia; he gets up once during the night 1 or 2 times every 2 weeks or so. He strains less frequently, but intermittency is unchanged. His IPSS is 7 (improved by 2 points vs before treatment). The findings on his physical examination are unchanged except that his BP has decreased slightly, to 124/72 mm Hg. He has noted 1 or 2 episodes of dizziness. Feeling better than 3 months ago, RI asks whether further improvement of his LUTS is possible. He wonders whether his erectile dysfunction can be treated.
The benefits and risks of each of the 3 PDE-5Is are reviewed with RI. He elects to begin treatment with tadalafil 5 mg once daily because it is the only agent that is approved for the treatment of LUTS associated with BPH. Lifestyle management and his action plan are reviewed.
Continue to complete the online evaluation and receive your certification of completion.
Managing the Multiple Symptoms of Benign Prostatic Hyperplasia — CME
Managing Type 2 Diabetes in Men
Meeting New Challenges with Antiplatelet Therapy in Primary Care
Dr. Miner has disclosed that he is a consultant for Eli Lilly.
SUPPORT
This activity was supported by an educational grant from Lilly USA, LLC.
This article reviews screening tools for benign prostatic hyperplasia (BPH) and steps to be taken to confirm a diagnosis of BPH. Among the treatment options for BPH, emphasis is placed on pharmacologic treatment with alpha1-adrenergic blockers (AABs), 5-alpha-reductase inhibitors (5-ARIs), and phosphodiesterase-5 inhibitors (PDE-5Is). The two newest agents silodosin and tadalafil are discussed in greater detail.
LEARNING OBJECTIVES
After reviewing this activity on benign prostatic hyperplasia, the reader will be able to:
- Describe the key diagnostic steps.
- Describe the role of non-pharmacologic interventions.
- Compare the efficacy and safety of alpha1- adrenergic blockers, 5-alpha-reductase inhibitors, and phosphodiesterase-5 inhibitors.
- Describe strategies for treating multiple symptoms.
TARGET AUDIENCE
Family physicians and clinicians who wish to gain increased knowledge and greater competency regarding the management of patients with multiple symptoms of benign prostatic hyperplasia.
ACKNOWLEDGEMENT
Dr. Miner was paid an honorarium by and received editorial assistance from the Primary Care Education Consortium in the development of this activity.
DISCLOSURES
As a continuing medical education provider accredited by the Accreditation Council for Continuing Medical Education (ACCME), it is the policy of the Primary Care Education Consortium (PCEC) to require any individual in a position to influence educational content to disclose the existence of any financial interest or other personal relationship with the manufacturer(s) of any commercial product(s).
Dr. Miner has disclosed that he is a consultant for Eli Lilly.
The medical accuracy and continuing medical education (CME) reviewer for this activity, Dr. Ron Pollack, has no real or apparent conflicts of interest to report.
PRIMARY CARE EDUCATION CONSORTIUM STAFF
Dr. Brunton has disclosed that he is on the advisory boards and speakers’ bureaus for Boehringer Ingelheim, Eli Lilly, Kowa, Novo Nordisk, Inc, and Teva Pharmaceuticals, and is on the advisory boards for Abbott and Sunovion.
Other PCEC staff have provided financial disclosure and have no conflicts of interest to resolve related to this activity.
CONFLICTS OF INTEREST
When individuals in a position to control content have reported financial relationships with one or more commercial interests, the Primary Care Education Consortium works with them to resolve such conflicts to ensure that the content presented is free of commercial bias. The content of this activity was vetted by the following mechanisms and modified as required to meet this standard:
- Content peer-review by an external topic expert
- Content peer-review by an external CME reviewer
- Content validation by internal Primary Care Education Consortium clinical editorial staff
OFF-LABEL DISCLOSURE
In accordance with ACCME guidelines, the faculty author has been asked to disclose discussion on unlabeled or unapproved uses of drugs or devices during the course of the activity.
SPONSORSHIP
This activity is sponsored by the Primary Care Education Consortium.
ACCREDITATION
This journal-based CME activity, Managing the Multiple Symptoms of Benign Prostatic Hyperplasia, has been reviewed and is acceptable for up to 1.0 prescribed credit by the American Academy of Family Physicians. AAFP accreditation begins June 1, 2012. Term of approval is for one year from this date with option for yearly renewal.
Physicians should claim only the credit commensurate with the extent of their participation in the activity.
MEDIUM
Text publication in the form of a journal article.
METHOD OF PHYSICIAN PARTICIPATION
To receive CME credit, please read the journal article, and upon completion go to: www.pceconsortium.org/menshealthBPH to complete the online evaluation to receive your certification of completion.
SUPPORT
This activity was supported by an educational grant from Lilly USA, LLC.
RI is a 53-year-old African-American man who is being seen by his family physician for a follow-up for dyslipidemia and hypertension. He reports that he is feeling well and that he has not observed any adverse events (AEs) from his medications. His current medications are an intermediate-dose statin, a thiazide diuretic, and a calcium channel blocker. RI reports that he has been very compliant with his medications, missing at most 1 dose every week or two.
During the visit, his physician notices that RI has yawned several times and that he appears tired. When asked how many hours he sleeps each night, RI indicates that he sleeps 7.5 to 8 hours most nights. On further questioning, RI admits that for the past 4 or 5 years, he has had to get up to go to the bathroom during the night, after which he often has trouble falling asleep, and that this nocturia currently occurs 3 or 4 times a night. When asked whether he has noticed any other changes over the past few years, RI says that he has noted an increase in his waist circumference (now 38.5 inches) and a few more aches and pains. When asked whether he has experienced any changes in sexual function, RI acknowledges that occasionally he has had difficulty maintaining an erection. He also indicates that he has accepted that these changes are a result of getting older.
Introduction
BPH is commonly experienced in men as they age. Lower urinary tract symptoms (LUTS) associated with BPH often begin in the fourth decade of life and affect nearly 3 in 4 men by the seventh decade of life.1,2 Lower urinary tract symptoms that prompt men to seek medical care typically include nocturia, frequency, incomplete emptying, and urgency.3,4 Men typically wait almost 2 years before seeking medical care for their urinary symptoms. Among men who do not seek medical care for LUTS, the most common reason is the belief that urinary symptoms are an inevitable part of aging. Many men who do not seek treatment indicate that they would rather accept their urinary symptoms than discuss them with a physician.4
In addition to urinary symptoms, BPH has been associated with symptoms of sexual dysfunction independent of the effects of aging and other comorbidities (eg, diabetes) and lifestyle factors.5-8 Erectile dysfunction and ejaculatory dysfunction are the most common symptoms of sexual dysfunction in men with BPH.9-11 Symptoms of sexual dysfunction may also be caused by some pharmacologic agents used for the treatment of BPH.6,9,10
Evaluation
Although BPH and the symptoms associated with it are not often life-threatening, ruling out other causes such as prostate cancer, diabetes mellitus, or Parkinson disease is an important diagnostic goal.
Screening
Because many men are slow to seek medical care and reluctant to speak with a physician about their symptoms, it is important that family physicians routinely inquire about urinary function in men over the age of 50 years. Beyond simply asking whether there have been changes in urinary function, posing the last question on the International Prostate Symptom Score (IPSS) questionnaire may be helpful: “If you were to spend the rest of your life with your urinary condition just the way it is now, how would you feel about that?”12 This question may be followed with, “Are you bothered enough by your symptoms that you would accept taking a medication?” Inquiries such as these, coupled with education to help the patient to understand that LUTS are not simply due to aging and that effective treatments are available, may motivate patients to share their concerns regarding urinary function. In addition, helping patients to understand that BPH is not a risk factor for prostate cancer, but that there are other causes of LUTS, which are best detected early, may be helpful.
Assessment
A history and focused urologic examination are crucial for the diagnosis of BPH. The medical history should identify a patient’s LUTS and their severity. To do this, a questionnaire such as the IPSS or the American Urological Association BPH Symptom Score Index Questionnaire can be administered [www.adultpediatricuro.com/apuauass.pdf]. As noted earlier, the eighth question on the IPSS questionnaire is useful for assessing the degree to which a patient is bothered by LUTS, with a higher score suggesting a greater willingness of the patient to be treated.13 Lower urinary tract symptoms are generally categorized into storage or bladder-emptying symptoms, with the latter subclassified as voiding or postmicturition symptoms.14 Storage problems are generally of greater concern to patients. Possible sexual dysfunction should also be assessed. A thorough medication history must be taken to identify AEs possibly related to the use of diuretics, anticholinergics, opioids, or decongestants.
The digital rectal examination (DRE) and prostate specific antigen (PSA) test are helpful to rule out a diagnosis of prostate cancer.15,16 The DRE is used for assessing the size, shape, symmetry, nodularity, and consistency of the prostate. The suprapubic area and genitals should be examined as well.17 The PSA test is also useful in the diagnosis and treatment of BPH because the PSA level rises as the prostate increases in size.13,14 A PSA level of 1.5 ng/mL roughly correlates with a prostate size of 30 mL.18 A urinalysis is needed to screen for urinary tract infections, bladder cancer, and kidney stones. Other laboratory analyses such as a fasting plasma glucose test may be needed based on the patient’s history and other findings.17
RI returns 3 weeks later for further evaluation. History confirms nocturia 3 or 4 times per night, as well as occasional erectile dysfunction and sometimes an inability to ejaculate. His IPSS questionnaire reveals a score of 9 (moderate symptoms), with occasional urinary frequency and straining. His LUTS are more bothersome than his occasional erectile dysfunction. It is decided that he will discontinue treatment with the thiazide diuretic because it may be contributing to his LUTS. An alternative antihypertensive agent will be initiated based on the results of the evaluation. The DRE reveals a boggy, slightly enlarged but normally shaped prostate with no nodules. The remainder of the urologic examination is normal. His PSA level is 0.8 ng/mL, and the urinalysis is normal. Further evaluation rules out prostate cancer and other causes of his symptoms.
A diagnosis of BPH is confirmed, with evidence of storage (ie, nocturia) and voiding (ie, urinary intermittency and straining) problems, as well as erectile dysfunction and occasional ejaculatory dysfunction.
Treatment
Goals
Because BPH is not often life-threatening, the focus of treatment has typically been to alleviate bothersome LUTS and other symptoms. With advances in treatment, additional goals include the alteration of disease progression and the prevention of associated complications such as recurrent urinary tract infections, hematuria, or acute urinary retention, particularly in men with an enlarged prostate (ie, volume >30 mL or PSA >1.5 ng/mL), since disease progression is more likely in these patients.17,19 A recent Medline-based systematic review reported that men prefer therapies that affect long-term progression over therapies that provide short-term symptom improvement.20 These results were consistent with those from a 2006 survey of 400 men with an enlarged prostate, which also reported that men are generally willing to wait up to 3 months for symptom relief if treatment would resolve the underlying condition.21 It is, therefore, important to discuss with the patient the natural history of BPH and its complications, and the benefits and risks of currently available noninvasive and invasive treatment options.
Options
Treatment options for BPH are watchful waiting, lifestyle and behavioral management, pharmacologic therapy, and surgical intervention. Many men use phytobotannical therapy such as saw palmetto, African plum tree, pumpkin seed, rye pollen, stinging nettle, South African star grass, and quercetin to relieve LUTS, although investigations regarding their use are often of poor quality. Saw palmetto is the best studied, yet a Cochrane review found few high-quality studies. The authors of the review concluded that saw palmetto was not more effective than placebo for treatment of LUTS.22 Similar results were observed in a randomized, double-blind, placebo-controlled trial more recently published by the Complementary and Alternative Medicine for Urological Symptoms (CAMUS) Study Group.23
Watchful waiting is appropriate when only LUTS are present, with or without some degree of nonsuspicious prostate enlargement, and the symptoms are not particularly bothersome to the patient or if the patient does not want treatment.19 Lifestyle management and behavioral modification should generally be used in combination with other treatment options in an effort to alleviate symptoms, especially in men in whom storage symptoms predominate. Lifestyle management may include reducing fluid intake (particularly if polyuria is present), increasing physical activity, achieving a normal weight, timed voiding (bladder retraining), pelvic floor exercises, treatment for constipation, and avoidance of irritative foods and beverages.17,19 Epidemiologic evidence over 7 years of surveillance suggests that a diet low in fat and red meat and high in protein and vegetables, as well as regular alcohol consumption (>1 drink/month), may reduce the risk of symptomatic BPH.24 Evidence was weak concerning the benefits of lycopene, zinc, and supplemental vitamin D. No dietary supplement, combination phytotherapeutic agent, or other nonconventional therapy is recommended by the American Urological Association (AUA) for the management of LUTS secondary to BPH.19
Surgical intervention is considered appropriate in patients with moderate to severe LUTS in whom other medical therapies have not achieved treatment goals and in those in whom benign prostatic obstruction has led to complications such as renal insufficiency, urinary retention, recurrent urinary tract infections, bladder calculi, or hydronephrosis. Patients in whom surgical intervention is contemplated should be referred to a urologist.17,19
Pharmacologic Options
Three classes of pharmacologic agents have been approved by the US Food and Drug Administration (FDA) for the treatment of symptomatic BPH: AABs, 5-ARIs, and PDE-5Is. The AABs include alfuzosin, doxazosin, silodosin, tamsulosin, and terazosin, and target the dynamic (smooth muscle tone) component of BPH-induced bladder outlet obstruction. The 5-ARIs finasteride and dutasteride target the static (prostate mass) component of BPH-induced bladder outlet obstruction. The PDE-5Is (ie, sildenafil, tadalafil, and vardenafil) increase the amount of cyclic guanosine monophosphate in the smooth muscle of the corpus cavernosum, prostate, and bladder.
Alpha1-Adrenergic Blockers. The four older AABs (ie, alfuzosin, doxazosin, tamsulosin, and terazosin) have been extensively investigated in clinical trials and widely used in the management of BPH. A 2010 review by the AUA concluded that the minor efficacy differences reported among the 4 older AABs were not clinically significant.19 Although ejaculatory dysfunction may occur with the use of the AABs, these agents are generally well-tolerated, with dizziness the most common AE, occurring in 2% to 14% of men. Ejaculatory dysfunction may be a part of the disease process itself, as noted earlier.
The newest AAB, silodosin, at a dosage of 8 mg/d was reported to have efficacy similar to tamsulosin 0.2 to 0.4 mg/d in reducing storage and voiding LUTS in three 12-week trials.25-27 Silodosin has also been associated with a significant improvement in patients’ quality of life. The most frequent AE related to silodosin use was abnormal ejaculation, occurring in 10% to 22% and causing discontinuation in 1% to 3%.25-27 One 12-week study reported that systolic blood pressure (BP) decreased 0.1 and 4.2 mm Hg in the silodosin and tamsulosin groups, respectively.25 The negligible reduction in BP observed with silodosin is likely due to the selectivity of silodosin for the alpha1A-adrenergic receptor rather than the alpha1B-adrenergic receptor, the blockade of which reduces BP.
5-Alpha-Reductase Inhibitors. The efficacy of 5-ARIs in preventing progression of LUTS secondary to BPH and their tolerability are well-established. Dutasteride was associated with a greater reduction in dihydrotestosterone in prostate tissues compared with finasteride (94% vs 80%, respectively) and has a longer elimination half-life.19 Finasteride was reported to be less effective than an AAB in improving LUTS. Dutasteride may have been more effective in reducing the relative risk for acute urinary retention and BPH-related surgery compared with tamsulosin over 4 years, but more research is needed.28 The 5-ARIs should not be used in men with LUTS secondary to BPH without prostatic enlargement, but may be used to prevent the progression of LUTS secondary to BPH and to reduce the risk for urinary retention and future prostate-related surgery.19 Prostate size ≥30 mL or PSA level ≥1.5 ng/dL is usually used as the threshold for considering 5-ARI therapy.19 As expected, because of the effects on dihydrotestosterone, AEs are primarily sexually related and include decreased libido, ejaculation disorders, and erectile dysfunction.19
Phosphodiesterase-5 Inhibitors. Approved by the FDA for erectile dysfunction, several observations led to the investigation of PDE-5Is for LUTS related to BPH.8,29 One was that the prevalences of BPH, LUTS, and erectile dysfunction increase as a man ages. Second was that LUTS have been identified as a risk factor for sexual dysfunction in aging men. Third was that limited evidence had suggested that PDE-5Is might be effective in treating LUTS and erectile dysfunction. Further investigation suggested beneficial effects on LUTS with each of the 3 PDE-5Is (ie, sildenafil, tadalafil, and vardenafil).30-32 Subsequent extensive investigation with tadalafil demonstrated its efficacy in reducing the storage and voiding symptoms of BPH and led to the approval by the FDA of tadalafil for symptoms of BPH alone or with erectile dysfunction.33-37
The clinical studies investigating the efficacy and tolerability of tadalafil for LUTS associated with BPH have included a 12-week study with a 1-year extension.38 Patients with BPH-associated LUTS (N = 1058) were randomized to tadalafil 2.5, 5, 10, or 20 mg/d or placebo once daily for 12 weeks. The total IPSS score was significantly improved at 12 weeks compared with baseline in each of the tadalafil groups relative to placebo (2.5 mg/d: –3.9, P = .015; 5 mg/d: –4.9, P < .001; 10 mg/d: –5.2, P < .001; 20 mg/d: –5.2, P < .001; placebo: –2.3). The use of tadalafil 5, 10, or 20 mg once daily was associated with significant improvements in the IPSS irritative (eg, frequency, nocturia, and urgency) and obstructive (eg, incomplete emptying, intermittency, slow stream, and straining) subscores, as well as scores on the IPSS quality-of-life measure, the BPH Impact Index (except 10 mg), and the LUTS Global Assessment Question. In sexually active men with erectile dysfunction, all doses of tadalafil were associated with significant improvements in scores on the International Index of Erectile Function–Erectile Function domain compared with placebo. Peak flow rate was not improved at any dose of tadalafil compared with placebo.
In total, 427 men who completed the 12-week study elected to receive tadalafil 5 mg once daily for an additional year.37 Patients who were switched from placebo or who had the dose increased from 2.5 mg/d had a significant reduction in total IPSS score from week 12 to week 16, and this change was maintained until the end of follow-up at week 64. Patients who had received tadalafil 5, 10, or 20 mg/d maintained the changes observed at the end of the 12-week study. Similarly, sexually active men with erectile dysfunction and who had a female partner maintained the improvements observed at the end of 12 weeks. The mean postvoid residual volume was decreased from 61 to 42 mL. At least 1 treatment-emergent AE (TEAE) was reported in 58% of patients, with 89% of events being either mild or moderate in severity. Treatment was discontinued in 5% due to a TEAE. The most common TEAEs were dyspepsia (4%), gastroesophageal reflux disease (4%), back pain (4%), headache (3%), sinusitis (3%), hypertension (3%), and cough (2%). In this study, the improvement in LUTS, sexual function, and quality of life observed after 12 weeks of tadalafil were maintained over the additional year with tadalafil 5 mg once daily.
Treatment options for RI are watchful waiting, an AAB with or without a PDE-5I, a 5-ARI, or tadalafil. RI indicates that he would rather not have his symptoms for the rest of his life, so watchful waiting is not appropriate. Because his prostate is only slightly enlarged, a 5-ARI is also not appropriate. An AAB or tadalafil should provide good relief to his LUTS within a few weeks. Tadalafil would also treat his erectile dysfunction. Alternatively, tadalafil or another PDE-5I could be combined with an AAB, which has been reported to provide added benefit in symptom improvement over an AAB alone.39
Plan
Following discussion of the benefits and risks of the different treatment options, RI elects to begin treatment with an AAB alone. For this reason, treatment with another antihypertensive to replace the diuretic will not be started. To promote self-management, educational materials and an action plan are reviewed with RI. Lifestyle management changes are discussed, including reducing his daily water intake by 25% to 2 quarts with no consumption of fluids within 3 to 4 hours of bedtime. He is assured that adjustments to his treatment plan will be made based on his symptoms and concerns.
3-Month Follow-Up
RI reports that his symptoms have improved, with a modest improvement of nocturia; he gets up once during the night 1 or 2 times every 2 weeks or so. He strains less frequently, but intermittency is unchanged. His IPSS is 7 (improved by 2 points vs before treatment). The findings on his physical examination are unchanged except that his BP has decreased slightly, to 124/72 mm Hg. He has noted 1 or 2 episodes of dizziness. Feeling better than 3 months ago, RI asks whether further improvement of his LUTS is possible. He wonders whether his erectile dysfunction can be treated.
The benefits and risks of each of the 3 PDE-5Is are reviewed with RI. He elects to begin treatment with tadalafil 5 mg once daily because it is the only agent that is approved for the treatment of LUTS associated with BPH. Lifestyle management and his action plan are reviewed.
Continue to complete the online evaluation and receive your certification of completion.
1. Wei JT, Calhoun EA, Jacobsen SJ. Urologic Diseases in America: Benign prostatic hyperplasia. http://kidney.niddk.nih.gov/Statistics/UDA/Benign_Prostatic_Hyperplasia-Chapter02.pdf. Published 2007. Accessed May 16, 2012.
2. Miller DC, Saigal CS, Litwin MS. The demographic burden of urologic diseases in America. Urol Clin North Am. 2009;36(1):11-27, v.
3. Sarma AV, Wallner L, Jacobsen SJ, Dunn RL, Wei JT. Health seeking behavior for lower urinary tract symptoms in black men. J Urol. 2008;180(1):227-232.
4. Survey confirms prostate problems overlooked by men and doctors [press release]. Vienna, Austria: GlaxoSmithKline. October 3, 2011. http://www.ismh.org/en/sys/wp-content/uploads/2011/09/News-release-300911-BPH-survey-a-male-perspective.pdf. Accessed May 16, 2012.
5. Mirone V, Sessa A, Giuliano F, Berges R, Kirby M, Moncada I. Current benign prostatic hyperplasia treatment: impact on sexual function and management of related sexual adverse events. Int J Clin Pract. 2011;65(9):1005-1013.
6. Gacci M, Eardley I, Giuliano F, et al. Critical analysis of the relationship between sexual dysfunctions and lower urinary tract symptoms due to benign prostatic hyperplasia. Eur Urol. 2011;60(4):809-825.
7. Hoesl CE, Woll EM, Burkart M, Altwein JE. Erectile dysfunction (ED) is prevalent, bothersome and underdiagnosed in patients consulting urologists for benign prostatic syndrome (BPS). Eur Urol. 2005;47(4):511-517.
8. Rosen R, Altwein J, Boyle P, et al. Lower urinary tract symptoms and male sexual dysfunction: the multinational survey of the aging male (MSAM-7). Eur Urol. 2003;44(6):637-649.
9. Rosen RC, Wei JT, Althof SE, Seftel AD, Miner M, Perelman MA. for BPH Registry and Patient Survey Steering Committee. Association of sexual dysfunction with lower urinary tract symptoms of BPH and BPH medical therapies: results from the BPH Registry. Urology. 2009;73(3):562-566.
10. Rosen RC, Fitzpatrick JM. for ALF-LIFE Study Group. Ejaculatory dysfunction in men with lower urinary tract symptoms suggestive of benign prostatic hyperplasia. BJU Int. 2009;104(7):974-983.
11. Seftel A, Rosen R, Kuritzky L. Physician perceptions of sexual dysfunction related to benign prostatic hyperplasia (BPH) symptoms and sexual side effects related to BPH medications. Int J Impot Res. 2007;19(4):386-392.
12. American Urological Association. American Urological Association BPH Symptom Score Index Questionnaire. http://www.adultpediatricuro.com/apuauass.pdf. Accessed May 16, 2012.
13. O’Leary MP. Validity of the “bother score” in the evaluation and treatment of symptomatic benign prostatic hyperplasia. Rev Urol. 2005;7(1):1-10.
14. Abrams P, Cardozo L, Fall M, et al. for Standardisation Sub-Committee of the International Continence Society. The standardisation of terminology in lower urinary tract function: report from the standardisation sub-committee of the International Continence Society. Urology. 2003;61(1):37-49.
15. American Urological Association. Prostate-specific antigen best practice statement: 2009 update. http://www.auanet.org/content/media/psa09.pdf. Published 2009. Accessed May 16, 2012.
16. American Cancer Society. Can prostate cancer be found early? http://www.cancer.org/Cancer/ProstateCancer/DetailedGuide/prostate-cancer-detection. Published 2012. Accessed May 16, 2012.
17. Tanguay S, Awde M, Brock G, et al. Diagnosis and management of benign prostatic hyperplasia in primary care. Can Urol Assoc J. 2009;3(3 suppl 2):S92-S100.
18. Roehrborn CG, Boyle P, Gould AL, Waldstreicher J. Serum prostate-specific antigen as a predictor of prostate volume in men with benign prostatic hyperplasia. Urology. 1999;53(3):581-589.
19. McVary KT, Roehrborn CG, Avins AL, et al. American Urological Association guideline: Management of benign prostatic hyperplasia (BPH). http://www.auanet.org/content/clinical-practice-guidelines/clinical-guidelines.cfm?sub=bph. Published 2010. Accessed May 16, 2012.
20. Emberton M. Medical treatment of benign prostatic hyperplasia: physician and patient p and satisfaction. Int J Clin Pract. 2010;64(10):1425-1435.
21. Kaplan S, Naslund M. Public, patient, and professional attitudes towards the diagnosis and treatment of enlarged prostate: A landmark national US survey. Int J Clin Pract. 2006;60(10):1157-1165.
22. Tacklind J, MacDonald R, Rutks I, Wilt TJ. Serenoa repens for benign prostatic hyperplasia. Cochrane Database Syst Rev. 2009;2:CD001423.-
23. Barry MJ, Meleth S, Lee JY, et al. for Complementary and Alternative Medicine for Urological Symptoms (CAMUS) Study Group. Effect of increasing doses of saw palmetto extract on lower urinary tract symptoms: a randomized trial. JAMA. 2011;306(12):1344-1351.
24. Kristal AR, Arnold KB, Schenk JM, et al. Dietary patterns, supplement use, and the risk of symptomatic benign prostatic hyperplasia: results from the prostate cancer prevention trial. Am J Epidemiol. 2008;167(8):925-934.
25. Yu HJ, Lin AT, Yang SS, et al. Non-inferiority of silodosin to tamsulosin in treating patients with lower urinary tract symptoms (LUTS) associated with benign prostatic hyperplasia (BPH). BJU Int. 2011;108(11):1843-1848.
26. Chapple CR, Montorsi F, Tammela TL, Wirth M, Koldewijn E, Fernandez FE. for European Silodosin Study Group. Silodosin therapy for lower urinary tract symptoms in men with suspected benign prostatic hyperplasia: results of an international, randomized, double-blind, placebo- and active-controlled clinical trial performed in Europe. Eur Urol. 2011;59(3):342-352.
27. Kawabe K, Yoshida M, Homma Y. for Silodosin Clinical Study Group. Silodosin, a new alpha1A-adrenoceptor-selective antagonist for treating benign prostatic hyperplasia: results of a phase III randomized, placebo-controlled, double-blind study in Japanese men. BJU Int. 2006;98(5):1019-1024.
28. Roehrborn CG, Siami P, Barkin J, et al. for CombAT Study Group. The effects of combination therapy with dutasteride and tamsulosin on clinical outcomes in men with symptomatic benign prostatic hyperplasia: 4-year results from the CombAT study. Eur Urol. 2010;57(1):123-131.
29. Kaplan SA, Gonzalez RR. Phosphodiesterase type 5 inhibitors for the treatment of male lower urinary tract symptoms. Rev Urol. 2007;9(2):73-77.
30. Mulhall JP, Guhring P, Parker M, Hopps C. Assessment of the impact of sildenafil citrate on lower urinary tract symptoms in men with erectile dysfunction. J Sex Med. 2006;3(4):662-667.
31. McVary KT, Roehrborn CG, Kaminetsky JC, et al. Tadalafil relieves lower urinary tract symptoms secondary to benign prostatic hyperplasia. J Urol. 2007;177(4):1401-1407.
32. Stief CG, Porst H, Neuser D, Beneke M, Ulbrich E. A randomised, placebo-controlled study to assess the efficacy of twice-daily vardenafil in the treatment of lower urinary tract symptoms secondary to benign prostatic hyperplasia. Eur Urol. 2008;53(6):1236-1244.
33. Porst H, McVary KT, Montorsi F, et al. Effects of once-daily tadalafil on erectile function in men with erectile dysfunction and signs and symptoms of benign prostatic hyperplasia. Eur Urol. 2009;56(4):727-735.
34. Broderick GA, Brock GB, Roehrborn CG, Watts SD, Elion-Mboussa A, Viktrup L. Effects of tadalafil on lower urinary tract symptoms secondary to benign prostatic hyperplasia in men with or without erectile dysfunction. Urology. 2010;75(6):1452-1458.
35. Porst H, Kim ED, Casabé AR, et al. LVHJ study team. Efficacy and safety of tadalafil once daily in the treatment of men with lower urinary tract symptoms suggestive of benign prostatic hyperplasia: results of an international randomized, double-blind, placebo-controlled trial. Eur Urol. 2011;60(5):1105-1113.
36. Maselli G, Bergamasco L, Silvestri V, Gualà L, Pace G, Vicentini C. Tadalafil versus solifenacin for persistent storage symptoms after prostate surgery in patients with erectile dysfunction: a prospective randomized study. Int J Urol. 2011;18(7):515-520.
37. Donatucci CF, Brock GB, Goldfischer ER, et al. Tadalafil administered once daily for lower urinary tract symptoms secondary to benign prostatic hyperplasia: a 1-year, open-label extension study. BJU Int. 2011;107(7):1110-1116.
38. Roehrborn CG, McVary KT, Elion-Mboussa A, Viktrup L. Tadalafil administered once daily for lower urinary tract symptoms secondary to benign prostatic hyperplasia: a dose finding study. J Urol. 2008;180(4):1228-1234.
39. Gacci M, Corona G, Salvi M, et al. A systematic review and meta-analysis on the use of phosphodiesterase 5 inhibitors alone or in combination with alpha-blockers for lower urinary tract symptoms due to benign prostatic hyperplasia. Eur Urol. 2012;61(5):994-1003.
1. Wei JT, Calhoun EA, Jacobsen SJ. Urologic Diseases in America: Benign prostatic hyperplasia. http://kidney.niddk.nih.gov/Statistics/UDA/Benign_Prostatic_Hyperplasia-Chapter02.pdf. Published 2007. Accessed May 16, 2012.
2. Miller DC, Saigal CS, Litwin MS. The demographic burden of urologic diseases in America. Urol Clin North Am. 2009;36(1):11-27, v.
3. Sarma AV, Wallner L, Jacobsen SJ, Dunn RL, Wei JT. Health seeking behavior for lower urinary tract symptoms in black men. J Urol. 2008;180(1):227-232.
4. Survey confirms prostate problems overlooked by men and doctors [press release]. Vienna, Austria: GlaxoSmithKline. October 3, 2011. http://www.ismh.org/en/sys/wp-content/uploads/2011/09/News-release-300911-BPH-survey-a-male-perspective.pdf. Accessed May 16, 2012.
5. Mirone V, Sessa A, Giuliano F, Berges R, Kirby M, Moncada I. Current benign prostatic hyperplasia treatment: impact on sexual function and management of related sexual adverse events. Int J Clin Pract. 2011;65(9):1005-1013.
6. Gacci M, Eardley I, Giuliano F, et al. Critical analysis of the relationship between sexual dysfunctions and lower urinary tract symptoms due to benign prostatic hyperplasia. Eur Urol. 2011;60(4):809-825.
7. Hoesl CE, Woll EM, Burkart M, Altwein JE. Erectile dysfunction (ED) is prevalent, bothersome and underdiagnosed in patients consulting urologists for benign prostatic syndrome (BPS). Eur Urol. 2005;47(4):511-517.
8. Rosen R, Altwein J, Boyle P, et al. Lower urinary tract symptoms and male sexual dysfunction: the multinational survey of the aging male (MSAM-7). Eur Urol. 2003;44(6):637-649.
9. Rosen RC, Wei JT, Althof SE, Seftel AD, Miner M, Perelman MA. for BPH Registry and Patient Survey Steering Committee. Association of sexual dysfunction with lower urinary tract symptoms of BPH and BPH medical therapies: results from the BPH Registry. Urology. 2009;73(3):562-566.
10. Rosen RC, Fitzpatrick JM. for ALF-LIFE Study Group. Ejaculatory dysfunction in men with lower urinary tract symptoms suggestive of benign prostatic hyperplasia. BJU Int. 2009;104(7):974-983.
11. Seftel A, Rosen R, Kuritzky L. Physician perceptions of sexual dysfunction related to benign prostatic hyperplasia (BPH) symptoms and sexual side effects related to BPH medications. Int J Impot Res. 2007;19(4):386-392.
12. American Urological Association. American Urological Association BPH Symptom Score Index Questionnaire. http://www.adultpediatricuro.com/apuauass.pdf. Accessed May 16, 2012.
13. O’Leary MP. Validity of the “bother score” in the evaluation and treatment of symptomatic benign prostatic hyperplasia. Rev Urol. 2005;7(1):1-10.
14. Abrams P, Cardozo L, Fall M, et al. for Standardisation Sub-Committee of the International Continence Society. The standardisation of terminology in lower urinary tract function: report from the standardisation sub-committee of the International Continence Society. Urology. 2003;61(1):37-49.
15. American Urological Association. Prostate-specific antigen best practice statement: 2009 update. http://www.auanet.org/content/media/psa09.pdf. Published 2009. Accessed May 16, 2012.
16. American Cancer Society. Can prostate cancer be found early? http://www.cancer.org/Cancer/ProstateCancer/DetailedGuide/prostate-cancer-detection. Published 2012. Accessed May 16, 2012.
17. Tanguay S, Awde M, Brock G, et al. Diagnosis and management of benign prostatic hyperplasia in primary care. Can Urol Assoc J. 2009;3(3 suppl 2):S92-S100.
18. Roehrborn CG, Boyle P, Gould AL, Waldstreicher J. Serum prostate-specific antigen as a predictor of prostate volume in men with benign prostatic hyperplasia. Urology. 1999;53(3):581-589.
19. McVary KT, Roehrborn CG, Avins AL, et al. American Urological Association guideline: Management of benign prostatic hyperplasia (BPH). http://www.auanet.org/content/clinical-practice-guidelines/clinical-guidelines.cfm?sub=bph. Published 2010. Accessed May 16, 2012.
20. Emberton M. Medical treatment of benign prostatic hyperplasia: physician and patient p and satisfaction. Int J Clin Pract. 2010;64(10):1425-1435.
21. Kaplan S, Naslund M. Public, patient, and professional attitudes towards the diagnosis and treatment of enlarged prostate: A landmark national US survey. Int J Clin Pract. 2006;60(10):1157-1165.
22. Tacklind J, MacDonald R, Rutks I, Wilt TJ. Serenoa repens for benign prostatic hyperplasia. Cochrane Database Syst Rev. 2009;2:CD001423.-
23. Barry MJ, Meleth S, Lee JY, et al. for Complementary and Alternative Medicine for Urological Symptoms (CAMUS) Study Group. Effect of increasing doses of saw palmetto extract on lower urinary tract symptoms: a randomized trial. JAMA. 2011;306(12):1344-1351.
24. Kristal AR, Arnold KB, Schenk JM, et al. Dietary patterns, supplement use, and the risk of symptomatic benign prostatic hyperplasia: results from the prostate cancer prevention trial. Am J Epidemiol. 2008;167(8):925-934.
25. Yu HJ, Lin AT, Yang SS, et al. Non-inferiority of silodosin to tamsulosin in treating patients with lower urinary tract symptoms (LUTS) associated with benign prostatic hyperplasia (BPH). BJU Int. 2011;108(11):1843-1848.
26. Chapple CR, Montorsi F, Tammela TL, Wirth M, Koldewijn E, Fernandez FE. for European Silodosin Study Group. Silodosin therapy for lower urinary tract symptoms in men with suspected benign prostatic hyperplasia: results of an international, randomized, double-blind, placebo- and active-controlled clinical trial performed in Europe. Eur Urol. 2011;59(3):342-352.
27. Kawabe K, Yoshida M, Homma Y. for Silodosin Clinical Study Group. Silodosin, a new alpha1A-adrenoceptor-selective antagonist for treating benign prostatic hyperplasia: results of a phase III randomized, placebo-controlled, double-blind study in Japanese men. BJU Int. 2006;98(5):1019-1024.
28. Roehrborn CG, Siami P, Barkin J, et al. for CombAT Study Group. The effects of combination therapy with dutasteride and tamsulosin on clinical outcomes in men with symptomatic benign prostatic hyperplasia: 4-year results from the CombAT study. Eur Urol. 2010;57(1):123-131.
29. Kaplan SA, Gonzalez RR. Phosphodiesterase type 5 inhibitors for the treatment of male lower urinary tract symptoms. Rev Urol. 2007;9(2):73-77.
30. Mulhall JP, Guhring P, Parker M, Hopps C. Assessment of the impact of sildenafil citrate on lower urinary tract symptoms in men with erectile dysfunction. J Sex Med. 2006;3(4):662-667.
31. McVary KT, Roehrborn CG, Kaminetsky JC, et al. Tadalafil relieves lower urinary tract symptoms secondary to benign prostatic hyperplasia. J Urol. 2007;177(4):1401-1407.
32. Stief CG, Porst H, Neuser D, Beneke M, Ulbrich E. A randomised, placebo-controlled study to assess the efficacy of twice-daily vardenafil in the treatment of lower urinary tract symptoms secondary to benign prostatic hyperplasia. Eur Urol. 2008;53(6):1236-1244.
33. Porst H, McVary KT, Montorsi F, et al. Effects of once-daily tadalafil on erectile function in men with erectile dysfunction and signs and symptoms of benign prostatic hyperplasia. Eur Urol. 2009;56(4):727-735.
34. Broderick GA, Brock GB, Roehrborn CG, Watts SD, Elion-Mboussa A, Viktrup L. Effects of tadalafil on lower urinary tract symptoms secondary to benign prostatic hyperplasia in men with or without erectile dysfunction. Urology. 2010;75(6):1452-1458.
35. Porst H, Kim ED, Casabé AR, et al. LVHJ study team. Efficacy and safety of tadalafil once daily in the treatment of men with lower urinary tract symptoms suggestive of benign prostatic hyperplasia: results of an international randomized, double-blind, placebo-controlled trial. Eur Urol. 2011;60(5):1105-1113.
36. Maselli G, Bergamasco L, Silvestri V, Gualà L, Pace G, Vicentini C. Tadalafil versus solifenacin for persistent storage symptoms after prostate surgery in patients with erectile dysfunction: a prospective randomized study. Int J Urol. 2011;18(7):515-520.
37. Donatucci CF, Brock GB, Goldfischer ER, et al. Tadalafil administered once daily for lower urinary tract symptoms secondary to benign prostatic hyperplasia: a 1-year, open-label extension study. BJU Int. 2011;107(7):1110-1116.
38. Roehrborn CG, McVary KT, Elion-Mboussa A, Viktrup L. Tadalafil administered once daily for lower urinary tract symptoms secondary to benign prostatic hyperplasia: a dose finding study. J Urol. 2008;180(4):1228-1234.
39. Gacci M, Corona G, Salvi M, et al. A systematic review and meta-analysis on the use of phosphodiesterase 5 inhibitors alone or in combination with alpha-blockers for lower urinary tract symptoms due to benign prostatic hyperplasia. Eur Urol. 2012;61(5):994-1003.
10 practical, evidence-based recommendations for perioperative antibiotic prophylaxis
- Update on Infectious Disease
Patrick Duff, MD (June 2012) - Gaps in Chlamydia testing threaten reproductive health, CDC warns
Janelle Yates, Senior Editor (Web exclusive, May 2012)
Antibiotic prophylaxis for gynecologic surgery has become a widely accepted practice to reduce post-surgical infections, which can lengthen hospital stay, worsen pain, prolong recovery, and lead to other complications. Recommendations for using antibiotic prophylaxis have been developed by many hospitals, accrediting bodies, and national organizations. Patients are becoming increasingly aware of postoperative infections and their consequences, and some complications, including urinary tract infection, are being evaluated as quality markers and qualifiers for payment by insurers and governmental agencies. Hospital-specific infection information is available in some regions through a simple Internet search. Still, compliance with national guidelines is variable and may be influenced by local hospital policies.1 While prophylaxis generally is thought to be safe, unnecessary administration of antibiotics is undesirable; more doses or more powerful antibiotics are not always better. An appreciation of some of the nuances of prophylaxis is desirable to improve patient care and safety.
1. Know what to use and when to use it
Follow ACOG and/or SCIP guidelines
ACOG Committee on Practice Bulletins—Gynecology. ACOG practice bulletin No. 104: antibiotic prophylaxis for gynecologic procedures. Obstet Gynecol. 2009;113(5):1180-1189.
Bratzler DW, Hynt DR. The surgical infection prevention and surgical care improvements projects: national initiatives to improve outcomes for patients having surgery. Clin Infect Dis. 2006;43(3):322-330.
No prophylaxis is needed for laparoscopy (without hysterectomy), laparotomy (without hysterectomy), hysteroscopy, intrauterine device insertion, endometrial biopsy, or urodynamics. Antibiotic agents recommended for prophylaxis for hysterectomy performed via any route include cefazolin (supported by ACOG) and cefotetan, cefoxitin, cefuroxime, or ampicillin/ sulbactam (recommended by Surgical Care Improvement Project guidelines) (TABLE). Alternative agents are available for patients with a true penicillin allergy.
Antibiotic prophylaxis recommendations for hysterectomy
| ACOG | 1. Cefazolin* If history of immediate hypersensitivity to penicillin: 2. Clindamycin + gentamicin or quinolone† or aztreonam 3. Metronidazole + gentamicin or quinolone† |
| SCIP | 1. Cefotetan, cefazolin, cefoxitin, cefuroxime, or ampicillin/sulbactam If allergy: 2. Clindamycin + aminoglycoside or quinolone or aztreonam 3. Metronidazole + aminoglycoside or quinolone |
| SCIP = Surgical Care Improvement Project *Acceptable alternatives include cefotetan, cefoxitin, cefuroxime, or ampicillin/sulbactam †Ciprofloxacin or levofloxacin or moxifloxacin | |
2. Increase the dose when needed
Double the antibiotic dose for patients weighing more than 220 lb or with a body mass index
greater than 35 kg/m2
Forse RA, Karam B, MacLean LD, Christou NV. Antibiotic prophylaxis for surgery in morbidly obese patients. Surgery. 1989;106(4):750–756.
In a study of morbidly obese patients undergoing gastroplasty, 2 g of cefazolin administered intravenously at the start of surgery was shown to reduce the risk of infection more than a 1-g dose. For true penicillin-allergic patients, weight-based gentamicin dosing (1.5 mg/kg) should provide adequate serum levels of the drug.
3. Repeat the dose when needed
Repeat the dose if surgery is long or involves a significant blood loss
ACOG Committee on Practice Bulletins—Gynecology. ACOG practice bulletin No. 104: antibiotic prophylaxis for gynecologic procedures. Obstet Gynecol. 2009;113(5):1180–1189.
Ho VP, Nicolau DP, Dakin GF, et al. Cefazolin dosing for surgical prophylaxis in morbidly obese patients. Surg Infect (Larchmt). 2012;13(1):33–37.
Give the first dose of antibiotic within an hour of the start of surgery, such as at the time of anesthesia induction. To maintain adequate levels throughout surgery, repeat dosing about 3 hours later for cefazolin or 2 hours after cefoxitin (1 to 2 times the half-life of the drug). Gentamicin likely does not need redosing for length of surgery, and clindamycin should be repeated at about 6 hours. If blood loss exceeds 1500 mL, redosing of the antibiotic will maintain adequate serum levels. As blood transfusion has been associated with surgical site infections, meticulous hemostasis is paramount.
4. Get the Foley catheter out ASAP
Remove it even in the operating room, to decrease the risk of UTI
Alessandri F, Mistrangelo E, Lijoi D, Ferrero S, Ragni N. A prospective, randomized trial comparing immediate versus delayed catheter removal following hysterectomy. Acta Obstet Gynecol Scand. 2006;85(6):716–720.
While there is a risk of recatheterization, removal of the catheter in the operating room will reduce the risk of postsurgical urinary tract infection.
5. Target the flora
Understand the flora that can cause infections and use a narrow-spectrum antibiotic
ACOG Committee on Practice Bulletins—Gynecology. ACOG practice bulletin No. 104: antibiotic prophylaxis for gynecologic procedures. Obstet Gynecol. 2009;113(5):1180–1189.
For procedures involving the vagina, either directly or following removal of the uterus, possible organisms include aerobic Gram-positive cocci (staphylococci), fecal flora (anaerobic bacteria or Gram-negative aerobes), and other flora that can result in bacterial vaginosis. For surgeries where the vagina is not involved, such as laparotomy or laparoscopy, the concern mainly is related to skin flora. Cephalosporins are effective against these organisms and have a low rate of adverse reactions.
6. Understand your patient’s penicillin allergy
Cephalosporins typically are safe in patients with a penicillin allergy unless a history of anaphylaxis
is documented
Campagna JD, Bond MC, Schabelman E, Hayes BD. The use of cephalosporins in penicillin-allergic patients: a literature review. J Emerg Med. 2011 Jul 8. [Epub ahead of print]
The rate of cross-reactivity may be as low as 1% between the two classes of drugs, and second- and third-generation cephalosporins have negligible risk due to substantial chemical structural differences from penicillins. Consider cefoxitin in these patients in place of cefazolin.
7. No infection? No antibiotics.
Stop antibiotics after the operating room unless infection is present
Van Eyk N, van Schalkwyk J. Antibiotic prophylaxis in gynaecologic procedures. J Obstet Gynaecol Can. 2012;34(4):382–391.
There are no data to support additional dosing after the operating room, even for 24 hours, to prevent infection. Hospitals and accrediting bodies are examining overuse of antibiotics, including extending prophylactic antibiotics outside the operating room, and surgeons can be cited for this practice.
8. Enourage open disclosure
Communicate with your residents/fellows and anesthesia team about antibiotic choice,
dose, and redosing
Altpeter T, Luckhardt K, Lewis JN, Harken AH, Polk HC Jr. Expanded surgical time out: a key to real-time data collection and quality improvement. J Am Coll Surg. 2007;204(4):527–532.
Discuss the length of the procedure and the time required for the antibiotic to take effect. Make sure that the antibiotic(s) you’ve ordered actually are what is being given and resolve any differences of opinion by consensus. This is often done during a “time out” but may need to be done earlier depending on when antibiotics are started.
9. Know the rules
Become familiar with hospital, state, and other local policies about antibiotic use
To reduce antimicrobial resistance, many hospitals have restrictions on the use of some medications, such as vancomycin. Vancomycin is not recommended for antibiotic prophylaxis for any gynecologic procedure; alternative agents should be chosen. Clinicians can be cited for use of a restricted medication when the use of other agents is possible. Understand if postoperative infections are reportable infections and if reporting to patients is mandatory.
10. Narrow your spectrum of endocarditis concerns
There is no need to change your prophylaxis based on endocarditis risk for genitourinary procedures
Wilson W, Taubert KA, Gewitz M, et al. Prevention of infective endocarditis: guidelines from the American Heart Association. [published correction appears in: Circulation. 2007;116(15):e376–377]. Circulation. 2007;116(15):1736–1754.
Infectious endocarditis prophylaxis is now only recommended for patients with a prosthetic valve, history of endocarditis, congenital heart disease, or those who have undergone cardiac transplantation.
Acknowledgment
OBG Management acknowledges Mark D. Walters, MD, for review of the manuscript of this article before submission for publication.
We want to hear from you! Tell us what you think.
Reference
1. Schimpf MO, Morrill MY, Margulies RU, Ward RM, Carberry CL, Sung VW. Surgeon practice patterns for antibiotic prophylaxis in benign gynecologic surgery. Paper presented at: 38th Scientific Meeting of the Society for Gynecologic Surgeons; April 13, 2012; Baltimore, MD. Oral poster #3.
- 10 practical, evidence-based recommendations for improving maternal outcomes of cesarean delivery
Baha M. Sibai, MD (March 2012) - 10 practical recommendations to improve maternal and perinatal outcomes in patients with eclampsia
Baha M. Sibai, MD (November 2011) - 10 (+1) practical, evidence-based recommendations for you to improve contraceptive care now
Colleen Krajewski, MD; Mark D. Walters, MD (August 2011) - 10 practical, evidence-based recommendations for the management of severe postpartum hemorrhage
Baha M. Sibai, MD (June 2011) - 10 practical, evidence-based suggestions to improve your minimally invasive surgical skills now
Catherine A. Matthews, MD (April 2011) - 10 practical, evidence-based suggestions to improve your gyn practice now
Mark D. Walters, MD (January 2011)

Megan O. Schimpf, MD
Dr. Schimpf is Associate Clinical Professor, Obstetrics and Gynecology, Female Pelvic Medicine and Reconstructive Surgery, at the University of Michigan in Ann Arbor, Michigan.
Dr. Schimpf reports no financial relationships relevant to this article.
antibiotic prophylaxis;antibiotic;cephalosporins;penicillin allergy;gynecologic surgery;postsurgical infection;ACOG;SCIP;Surgical Care Improvement Project;hysterectomy;cefazolin;cefotetan;cefoxitin;cefuroxime;ampicillin/sulbactam;clindamycin;gentamicin;quinolone;aztreonam;metronidazole;aminoglycoside;levofloxacin;moxifloxacin;obesity;Foley catheter;repeat the dose;vagina;flora;vancomycin;endocarditis;hospital policy;state policy;communicate;
Megan O. Schimpf, MD
Dr. Schimpf is Associate Clinical Professor, Obstetrics and Gynecology, Female Pelvic Medicine and Reconstructive Surgery, at the University of Michigan in Ann Arbor, Michigan.
Dr. Schimpf reports no financial relationships relevant to this article.
Megan O. Schimpf, MD
Dr. Schimpf is Associate Clinical Professor, Obstetrics and Gynecology, Female Pelvic Medicine and Reconstructive Surgery, at the University of Michigan in Ann Arbor, Michigan.
Dr. Schimpf reports no financial relationships relevant to this article.
- Update on Infectious Disease
Patrick Duff, MD (June 2012) - Gaps in Chlamydia testing threaten reproductive health, CDC warns
Janelle Yates, Senior Editor (Web exclusive, May 2012)
Antibiotic prophylaxis for gynecologic surgery has become a widely accepted practice to reduce post-surgical infections, which can lengthen hospital stay, worsen pain, prolong recovery, and lead to other complications. Recommendations for using antibiotic prophylaxis have been developed by many hospitals, accrediting bodies, and national organizations. Patients are becoming increasingly aware of postoperative infections and their consequences, and some complications, including urinary tract infection, are being evaluated as quality markers and qualifiers for payment by insurers and governmental agencies. Hospital-specific infection information is available in some regions through a simple Internet search. Still, compliance with national guidelines is variable and may be influenced by local hospital policies.1 While prophylaxis generally is thought to be safe, unnecessary administration of antibiotics is undesirable; more doses or more powerful antibiotics are not always better. An appreciation of some of the nuances of prophylaxis is desirable to improve patient care and safety.
1. Know what to use and when to use it
Follow ACOG and/or SCIP guidelines
ACOG Committee on Practice Bulletins—Gynecology. ACOG practice bulletin No. 104: antibiotic prophylaxis for gynecologic procedures. Obstet Gynecol. 2009;113(5):1180-1189.
Bratzler DW, Hynt DR. The surgical infection prevention and surgical care improvements projects: national initiatives to improve outcomes for patients having surgery. Clin Infect Dis. 2006;43(3):322-330.
No prophylaxis is needed for laparoscopy (without hysterectomy), laparotomy (without hysterectomy), hysteroscopy, intrauterine device insertion, endometrial biopsy, or urodynamics. Antibiotic agents recommended for prophylaxis for hysterectomy performed via any route include cefazolin (supported by ACOG) and cefotetan, cefoxitin, cefuroxime, or ampicillin/ sulbactam (recommended by Surgical Care Improvement Project guidelines) (TABLE). Alternative agents are available for patients with a true penicillin allergy.
Antibiotic prophylaxis recommendations for hysterectomy
| ACOG | 1. Cefazolin* If history of immediate hypersensitivity to penicillin: 2. Clindamycin + gentamicin or quinolone† or aztreonam 3. Metronidazole + gentamicin or quinolone† |
| SCIP | 1. Cefotetan, cefazolin, cefoxitin, cefuroxime, or ampicillin/sulbactam If allergy: 2. Clindamycin + aminoglycoside or quinolone or aztreonam 3. Metronidazole + aminoglycoside or quinolone |
| SCIP = Surgical Care Improvement Project *Acceptable alternatives include cefotetan, cefoxitin, cefuroxime, or ampicillin/sulbactam †Ciprofloxacin or levofloxacin or moxifloxacin | |
2. Increase the dose when needed
Double the antibiotic dose for patients weighing more than 220 lb or with a body mass index
greater than 35 kg/m2
Forse RA, Karam B, MacLean LD, Christou NV. Antibiotic prophylaxis for surgery in morbidly obese patients. Surgery. 1989;106(4):750–756.
In a study of morbidly obese patients undergoing gastroplasty, 2 g of cefazolin administered intravenously at the start of surgery was shown to reduce the risk of infection more than a 1-g dose. For true penicillin-allergic patients, weight-based gentamicin dosing (1.5 mg/kg) should provide adequate serum levels of the drug.
3. Repeat the dose when needed
Repeat the dose if surgery is long or involves a significant blood loss
ACOG Committee on Practice Bulletins—Gynecology. ACOG practice bulletin No. 104: antibiotic prophylaxis for gynecologic procedures. Obstet Gynecol. 2009;113(5):1180–1189.
Ho VP, Nicolau DP, Dakin GF, et al. Cefazolin dosing for surgical prophylaxis in morbidly obese patients. Surg Infect (Larchmt). 2012;13(1):33–37.
Give the first dose of antibiotic within an hour of the start of surgery, such as at the time of anesthesia induction. To maintain adequate levels throughout surgery, repeat dosing about 3 hours later for cefazolin or 2 hours after cefoxitin (1 to 2 times the half-life of the drug). Gentamicin likely does not need redosing for length of surgery, and clindamycin should be repeated at about 6 hours. If blood loss exceeds 1500 mL, redosing of the antibiotic will maintain adequate serum levels. As blood transfusion has been associated with surgical site infections, meticulous hemostasis is paramount.
4. Get the Foley catheter out ASAP
Remove it even in the operating room, to decrease the risk of UTI
Alessandri F, Mistrangelo E, Lijoi D, Ferrero S, Ragni N. A prospective, randomized trial comparing immediate versus delayed catheter removal following hysterectomy. Acta Obstet Gynecol Scand. 2006;85(6):716–720.
While there is a risk of recatheterization, removal of the catheter in the operating room will reduce the risk of postsurgical urinary tract infection.
5. Target the flora
Understand the flora that can cause infections and use a narrow-spectrum antibiotic
ACOG Committee on Practice Bulletins—Gynecology. ACOG practice bulletin No. 104: antibiotic prophylaxis for gynecologic procedures. Obstet Gynecol. 2009;113(5):1180–1189.
For procedures involving the vagina, either directly or following removal of the uterus, possible organisms include aerobic Gram-positive cocci (staphylococci), fecal flora (anaerobic bacteria or Gram-negative aerobes), and other flora that can result in bacterial vaginosis. For surgeries where the vagina is not involved, such as laparotomy or laparoscopy, the concern mainly is related to skin flora. Cephalosporins are effective against these organisms and have a low rate of adverse reactions.
6. Understand your patient’s penicillin allergy
Cephalosporins typically are safe in patients with a penicillin allergy unless a history of anaphylaxis
is documented
Campagna JD, Bond MC, Schabelman E, Hayes BD. The use of cephalosporins in penicillin-allergic patients: a literature review. J Emerg Med. 2011 Jul 8. [Epub ahead of print]
The rate of cross-reactivity may be as low as 1% between the two classes of drugs, and second- and third-generation cephalosporins have negligible risk due to substantial chemical structural differences from penicillins. Consider cefoxitin in these patients in place of cefazolin.
7. No infection? No antibiotics.
Stop antibiotics after the operating room unless infection is present
Van Eyk N, van Schalkwyk J. Antibiotic prophylaxis in gynaecologic procedures. J Obstet Gynaecol Can. 2012;34(4):382–391.
There are no data to support additional dosing after the operating room, even for 24 hours, to prevent infection. Hospitals and accrediting bodies are examining overuse of antibiotics, including extending prophylactic antibiotics outside the operating room, and surgeons can be cited for this practice.
8. Enourage open disclosure
Communicate with your residents/fellows and anesthesia team about antibiotic choice,
dose, and redosing
Altpeter T, Luckhardt K, Lewis JN, Harken AH, Polk HC Jr. Expanded surgical time out: a key to real-time data collection and quality improvement. J Am Coll Surg. 2007;204(4):527–532.
Discuss the length of the procedure and the time required for the antibiotic to take effect. Make sure that the antibiotic(s) you’ve ordered actually are what is being given and resolve any differences of opinion by consensus. This is often done during a “time out” but may need to be done earlier depending on when antibiotics are started.
9. Know the rules
Become familiar with hospital, state, and other local policies about antibiotic use
To reduce antimicrobial resistance, many hospitals have restrictions on the use of some medications, such as vancomycin. Vancomycin is not recommended for antibiotic prophylaxis for any gynecologic procedure; alternative agents should be chosen. Clinicians can be cited for use of a restricted medication when the use of other agents is possible. Understand if postoperative infections are reportable infections and if reporting to patients is mandatory.
10. Narrow your spectrum of endocarditis concerns
There is no need to change your prophylaxis based on endocarditis risk for genitourinary procedures
Wilson W, Taubert KA, Gewitz M, et al. Prevention of infective endocarditis: guidelines from the American Heart Association. [published correction appears in: Circulation. 2007;116(15):e376–377]. Circulation. 2007;116(15):1736–1754.
Infectious endocarditis prophylaxis is now only recommended for patients with a prosthetic valve, history of endocarditis, congenital heart disease, or those who have undergone cardiac transplantation.
Acknowledgment
OBG Management acknowledges Mark D. Walters, MD, for review of the manuscript of this article before submission for publication.
We want to hear from you! Tell us what you think.
- Update on Infectious Disease
Patrick Duff, MD (June 2012) - Gaps in Chlamydia testing threaten reproductive health, CDC warns
Janelle Yates, Senior Editor (Web exclusive, May 2012)
Antibiotic prophylaxis for gynecologic surgery has become a widely accepted practice to reduce post-surgical infections, which can lengthen hospital stay, worsen pain, prolong recovery, and lead to other complications. Recommendations for using antibiotic prophylaxis have been developed by many hospitals, accrediting bodies, and national organizations. Patients are becoming increasingly aware of postoperative infections and their consequences, and some complications, including urinary tract infection, are being evaluated as quality markers and qualifiers for payment by insurers and governmental agencies. Hospital-specific infection information is available in some regions through a simple Internet search. Still, compliance with national guidelines is variable and may be influenced by local hospital policies.1 While prophylaxis generally is thought to be safe, unnecessary administration of antibiotics is undesirable; more doses or more powerful antibiotics are not always better. An appreciation of some of the nuances of prophylaxis is desirable to improve patient care and safety.
1. Know what to use and when to use it
Follow ACOG and/or SCIP guidelines
ACOG Committee on Practice Bulletins—Gynecology. ACOG practice bulletin No. 104: antibiotic prophylaxis for gynecologic procedures. Obstet Gynecol. 2009;113(5):1180-1189.
Bratzler DW, Hynt DR. The surgical infection prevention and surgical care improvements projects: national initiatives to improve outcomes for patients having surgery. Clin Infect Dis. 2006;43(3):322-330.
No prophylaxis is needed for laparoscopy (without hysterectomy), laparotomy (without hysterectomy), hysteroscopy, intrauterine device insertion, endometrial biopsy, or urodynamics. Antibiotic agents recommended for prophylaxis for hysterectomy performed via any route include cefazolin (supported by ACOG) and cefotetan, cefoxitin, cefuroxime, or ampicillin/ sulbactam (recommended by Surgical Care Improvement Project guidelines) (TABLE). Alternative agents are available for patients with a true penicillin allergy.
Antibiotic prophylaxis recommendations for hysterectomy
| ACOG | 1. Cefazolin* If history of immediate hypersensitivity to penicillin: 2. Clindamycin + gentamicin or quinolone† or aztreonam 3. Metronidazole + gentamicin or quinolone† |
| SCIP | 1. Cefotetan, cefazolin, cefoxitin, cefuroxime, or ampicillin/sulbactam If allergy: 2. Clindamycin + aminoglycoside or quinolone or aztreonam 3. Metronidazole + aminoglycoside or quinolone |
| SCIP = Surgical Care Improvement Project *Acceptable alternatives include cefotetan, cefoxitin, cefuroxime, or ampicillin/sulbactam †Ciprofloxacin or levofloxacin or moxifloxacin | |
2. Increase the dose when needed
Double the antibiotic dose for patients weighing more than 220 lb or with a body mass index
greater than 35 kg/m2
Forse RA, Karam B, MacLean LD, Christou NV. Antibiotic prophylaxis for surgery in morbidly obese patients. Surgery. 1989;106(4):750–756.
In a study of morbidly obese patients undergoing gastroplasty, 2 g of cefazolin administered intravenously at the start of surgery was shown to reduce the risk of infection more than a 1-g dose. For true penicillin-allergic patients, weight-based gentamicin dosing (1.5 mg/kg) should provide adequate serum levels of the drug.
3. Repeat the dose when needed
Repeat the dose if surgery is long or involves a significant blood loss
ACOG Committee on Practice Bulletins—Gynecology. ACOG practice bulletin No. 104: antibiotic prophylaxis for gynecologic procedures. Obstet Gynecol. 2009;113(5):1180–1189.
Ho VP, Nicolau DP, Dakin GF, et al. Cefazolin dosing for surgical prophylaxis in morbidly obese patients. Surg Infect (Larchmt). 2012;13(1):33–37.
Give the first dose of antibiotic within an hour of the start of surgery, such as at the time of anesthesia induction. To maintain adequate levels throughout surgery, repeat dosing about 3 hours later for cefazolin or 2 hours after cefoxitin (1 to 2 times the half-life of the drug). Gentamicin likely does not need redosing for length of surgery, and clindamycin should be repeated at about 6 hours. If blood loss exceeds 1500 mL, redosing of the antibiotic will maintain adequate serum levels. As blood transfusion has been associated with surgical site infections, meticulous hemostasis is paramount.
4. Get the Foley catheter out ASAP
Remove it even in the operating room, to decrease the risk of UTI
Alessandri F, Mistrangelo E, Lijoi D, Ferrero S, Ragni N. A prospective, randomized trial comparing immediate versus delayed catheter removal following hysterectomy. Acta Obstet Gynecol Scand. 2006;85(6):716–720.
While there is a risk of recatheterization, removal of the catheter in the operating room will reduce the risk of postsurgical urinary tract infection.
5. Target the flora
Understand the flora that can cause infections and use a narrow-spectrum antibiotic
ACOG Committee on Practice Bulletins—Gynecology. ACOG practice bulletin No. 104: antibiotic prophylaxis for gynecologic procedures. Obstet Gynecol. 2009;113(5):1180–1189.
For procedures involving the vagina, either directly or following removal of the uterus, possible organisms include aerobic Gram-positive cocci (staphylococci), fecal flora (anaerobic bacteria or Gram-negative aerobes), and other flora that can result in bacterial vaginosis. For surgeries where the vagina is not involved, such as laparotomy or laparoscopy, the concern mainly is related to skin flora. Cephalosporins are effective against these organisms and have a low rate of adverse reactions.
6. Understand your patient’s penicillin allergy
Cephalosporins typically are safe in patients with a penicillin allergy unless a history of anaphylaxis
is documented
Campagna JD, Bond MC, Schabelman E, Hayes BD. The use of cephalosporins in penicillin-allergic patients: a literature review. J Emerg Med. 2011 Jul 8. [Epub ahead of print]
The rate of cross-reactivity may be as low as 1% between the two classes of drugs, and second- and third-generation cephalosporins have negligible risk due to substantial chemical structural differences from penicillins. Consider cefoxitin in these patients in place of cefazolin.
7. No infection? No antibiotics.
Stop antibiotics after the operating room unless infection is present
Van Eyk N, van Schalkwyk J. Antibiotic prophylaxis in gynaecologic procedures. J Obstet Gynaecol Can. 2012;34(4):382–391.
There are no data to support additional dosing after the operating room, even for 24 hours, to prevent infection. Hospitals and accrediting bodies are examining overuse of antibiotics, including extending prophylactic antibiotics outside the operating room, and surgeons can be cited for this practice.
8. Enourage open disclosure
Communicate with your residents/fellows and anesthesia team about antibiotic choice,
dose, and redosing
Altpeter T, Luckhardt K, Lewis JN, Harken AH, Polk HC Jr. Expanded surgical time out: a key to real-time data collection and quality improvement. J Am Coll Surg. 2007;204(4):527–532.
Discuss the length of the procedure and the time required for the antibiotic to take effect. Make sure that the antibiotic(s) you’ve ordered actually are what is being given and resolve any differences of opinion by consensus. This is often done during a “time out” but may need to be done earlier depending on when antibiotics are started.
9. Know the rules
Become familiar with hospital, state, and other local policies about antibiotic use
To reduce antimicrobial resistance, many hospitals have restrictions on the use of some medications, such as vancomycin. Vancomycin is not recommended for antibiotic prophylaxis for any gynecologic procedure; alternative agents should be chosen. Clinicians can be cited for use of a restricted medication when the use of other agents is possible. Understand if postoperative infections are reportable infections and if reporting to patients is mandatory.
10. Narrow your spectrum of endocarditis concerns
There is no need to change your prophylaxis based on endocarditis risk for genitourinary procedures
Wilson W, Taubert KA, Gewitz M, et al. Prevention of infective endocarditis: guidelines from the American Heart Association. [published correction appears in: Circulation. 2007;116(15):e376–377]. Circulation. 2007;116(15):1736–1754.
Infectious endocarditis prophylaxis is now only recommended for patients with a prosthetic valve, history of endocarditis, congenital heart disease, or those who have undergone cardiac transplantation.
Acknowledgment
OBG Management acknowledges Mark D. Walters, MD, for review of the manuscript of this article before submission for publication.
We want to hear from you! Tell us what you think.
Reference
1. Schimpf MO, Morrill MY, Margulies RU, Ward RM, Carberry CL, Sung VW. Surgeon practice patterns for antibiotic prophylaxis in benign gynecologic surgery. Paper presented at: 38th Scientific Meeting of the Society for Gynecologic Surgeons; April 13, 2012; Baltimore, MD. Oral poster #3.
- 10 practical, evidence-based recommendations for improving maternal outcomes of cesarean delivery
Baha M. Sibai, MD (March 2012) - 10 practical recommendations to improve maternal and perinatal outcomes in patients with eclampsia
Baha M. Sibai, MD (November 2011) - 10 (+1) practical, evidence-based recommendations for you to improve contraceptive care now
Colleen Krajewski, MD; Mark D. Walters, MD (August 2011) - 10 practical, evidence-based recommendations for the management of severe postpartum hemorrhage
Baha M. Sibai, MD (June 2011) - 10 practical, evidence-based suggestions to improve your minimally invasive surgical skills now
Catherine A. Matthews, MD (April 2011) - 10 practical, evidence-based suggestions to improve your gyn practice now
Mark D. Walters, MD (January 2011)
Reference
1. Schimpf MO, Morrill MY, Margulies RU, Ward RM, Carberry CL, Sung VW. Surgeon practice patterns for antibiotic prophylaxis in benign gynecologic surgery. Paper presented at: 38th Scientific Meeting of the Society for Gynecologic Surgeons; April 13, 2012; Baltimore, MD. Oral poster #3.
- 10 practical, evidence-based recommendations for improving maternal outcomes of cesarean delivery
Baha M. Sibai, MD (March 2012) - 10 practical recommendations to improve maternal and perinatal outcomes in patients with eclampsia
Baha M. Sibai, MD (November 2011) - 10 (+1) practical, evidence-based recommendations for you to improve contraceptive care now
Colleen Krajewski, MD; Mark D. Walters, MD (August 2011) - 10 practical, evidence-based recommendations for the management of severe postpartum hemorrhage
Baha M. Sibai, MD (June 2011) - 10 practical, evidence-based suggestions to improve your minimally invasive surgical skills now
Catherine A. Matthews, MD (April 2011) - 10 practical, evidence-based suggestions to improve your gyn practice now
Mark D. Walters, MD (January 2011)
antibiotic prophylaxis;antibiotic;cephalosporins;penicillin allergy;gynecologic surgery;postsurgical infection;ACOG;SCIP;Surgical Care Improvement Project;hysterectomy;cefazolin;cefotetan;cefoxitin;cefuroxime;ampicillin/sulbactam;clindamycin;gentamicin;quinolone;aztreonam;metronidazole;aminoglycoside;levofloxacin;moxifloxacin;obesity;Foley catheter;repeat the dose;vagina;flora;vancomycin;endocarditis;hospital policy;state policy;communicate;
antibiotic prophylaxis;antibiotic;cephalosporins;penicillin allergy;gynecologic surgery;postsurgical infection;ACOG;SCIP;Surgical Care Improvement Project;hysterectomy;cefazolin;cefotetan;cefoxitin;cefuroxime;ampicillin/sulbactam;clindamycin;gentamicin;quinolone;aztreonam;metronidazole;aminoglycoside;levofloxacin;moxifloxacin;obesity;Foley catheter;repeat the dose;vagina;flora;vancomycin;endocarditis;hospital policy;state policy;communicate;
Advances in Lung Cancer Evaluation and Management

Supplement Editors:
Nathan Pennell, MD, PhD, and Peter Mazzone, MD
Contents
Lung cancer screening: Examining the issues
Peter Mazzone
Treatment implication of the new lung cancer staging system
Cristina P. Rodriguez
Bronchoscopy and endobronchial ultrasound for diagnosis and staging of lung cancer
Francisco Aécio Almeida
Preoperative evaluation of the lung resection candidate
Peter Mazzone
Video-assisted thoracoscopic surgery for the treatment of lung cancer
Sudish Murthy
Stereotactic body radiotherapy for stage I non–small cell lung cancer
Kevin Stephans
Locally advanced non–small cell lung cancer: What is the optimal concurrent chemoradiation regimen?
Gregory M.M. Videtic
The role of surgery for locally advanced non–small cell lung cancer
David P. Mason
The role of adjuvant chemotherapy in early-stage and locally advanced non–small cell lung cancer
Marc Shapiro
Selection of chemotherapy for patients with advanced non–small cell lung cancer
Nathan A. Pennell
The emerging role of palliative medicine in the treatment of lung cancer patients
Mellar P. Davis
Personalized targeted therapy in advanced non–small cell lung cancer
Patrick C. Ma
Supplement Editors:
Nathan Pennell, MD, PhD, and Peter Mazzone, MD
Contents
Lung cancer screening: Examining the issues
Peter Mazzone
Treatment implication of the new lung cancer staging system
Cristina P. Rodriguez
Bronchoscopy and endobronchial ultrasound for diagnosis and staging of lung cancer
Francisco Aécio Almeida
Preoperative evaluation of the lung resection candidate
Peter Mazzone
Video-assisted thoracoscopic surgery for the treatment of lung cancer
Sudish Murthy
Stereotactic body radiotherapy for stage I non–small cell lung cancer
Kevin Stephans
Locally advanced non–small cell lung cancer: What is the optimal concurrent chemoradiation regimen?
Gregory M.M. Videtic
The role of surgery for locally advanced non–small cell lung cancer
David P. Mason
The role of adjuvant chemotherapy in early-stage and locally advanced non–small cell lung cancer
Marc Shapiro
Selection of chemotherapy for patients with advanced non–small cell lung cancer
Nathan A. Pennell
The emerging role of palliative medicine in the treatment of lung cancer patients
Mellar P. Davis
Personalized targeted therapy in advanced non–small cell lung cancer
Patrick C. Ma
Supplement Editors:
Nathan Pennell, MD, PhD, and Peter Mazzone, MD
Contents
Lung cancer screening: Examining the issues
Peter Mazzone
Treatment implication of the new lung cancer staging system
Cristina P. Rodriguez
Bronchoscopy and endobronchial ultrasound for diagnosis and staging of lung cancer
Francisco Aécio Almeida
Preoperative evaluation of the lung resection candidate
Peter Mazzone
Video-assisted thoracoscopic surgery for the treatment of lung cancer
Sudish Murthy
Stereotactic body radiotherapy for stage I non–small cell lung cancer
Kevin Stephans
Locally advanced non–small cell lung cancer: What is the optimal concurrent chemoradiation regimen?
Gregory M.M. Videtic
The role of surgery for locally advanced non–small cell lung cancer
David P. Mason
The role of adjuvant chemotherapy in early-stage and locally advanced non–small cell lung cancer
Marc Shapiro
Selection of chemotherapy for patients with advanced non–small cell lung cancer
Nathan A. Pennell
The emerging role of palliative medicine in the treatment of lung cancer patients
Mellar P. Davis
Personalized targeted therapy in advanced non–small cell lung cancer
Patrick C. Ma

Advances in Insulin Therapy: A Review of Insulin Degludec
The Evolution of Insulin Therapy in Diabetes Mellitus
Introduction
Basal insulin has been an important treatment option for patients with diabetes mellitus (DM) and, along with prandial insulin, has undergone major improvements in terms of purity and similarity to the action of physiologic human insulin. (see The Evolution of Insulin Therapy in Diabetes Mellitus in this supplement.) Lente and Ultralente formulations were used for decades but are no longer available. The use of neutral protamine Hagedorn (NPH) insulin is also being replaced with the basal insulin analogs detemir and glargine.1 Basal insulin analogs generally cause less severe and nocturnal hypoglycemia compared with NPH insulin owing to their improved pharmacologic profiles.2-4 In comparison to NPH insulin, insulin glargine causes similar weight gain, whereas insulin detemir causes less weight gain.2-4 In addition, insulin detemir has been associated with a glucose-lowering effect that is more predictable than that of NPH insulin.5 Despite the improvements observed with basal insulin analogs, their time-action profiles are not completely flat and are shorter than 24 hours in many patients.5,6 In addition, severe hypoglycemia remains a concern, particularly in patients with type 1 DM (T1DM).7,8 Consequently, the search for a better basal insulin continues.
The ideal basal insulin should possess numerous attributes. While each of the attributes listed in the TABLE is important, an overarching difficulty with basal insulin therapy is the need for administration at the same time each day.9 This dosing limitation may be most difficult for those with busy or erratic schedules or who may forget to administer their insulin dose. This article will review the clinical experience with insulin degludec, an ultra–long-acting insulin under review by the US Food and Drug Administration (FDA).
TABLE
Attributes of the ideal basal insulin9
| Delivers a steady, stable, peakless, continuous insulin concentration for at least 24 hours, in a predictable manner, with low intraindividual and interindividual variability |
| Does not cause side effects such as weight gain or hypoglycemia |
| Does not induce mitogenicity |
| Can be used as monotherapy, as part of basal-bolus therapy, or in combination with oral glucose-lowering therapy |
| Equally efficacious, safe, and well-tolerated in patients with type 1 or type 2 diabetes mellitus |
| Indian Journal of Endocrinology and Metabolism. Copyright 2011 by MEDKNOW PUBLICATIONS AND MEDIA PVT LTD. Reproduced with permission of MEDKNOW PUBLICATIONS AND MEDIA PVT LTD in the format Journal via Copyright Clearance Center. |
Clinical Pharmacology of Insulin Degludec
Removal of threonine at position 30 of the B chain of human insulin and the addition of a 16-carbon fatty diacid attached to lysine at position 29 of the B chain of human insulin via a glutamic acid spacer result in the insulin degludec molecule, which has several differences from available basal insulin analogs. Experimental investigations indicated that conditions mimicking subcutaneous injection of insulin degludec resulted in a reorganization of the insulin degludec molecule from dihexamers to multihexamer assemblies that remain in solution at physiologic pH.10 Slow release of zinc ions from the multihexamers leads to the slow release of insulin degludec monomers, which are easily absorbed into the systemic circulation.11 The result is a half-life of insulin degludec that is longer than 24 hours, with a level that is detectable in circulation for at least 96 hours after administration of the dose.10,12 The pharmacodynamic result is a relatively flat and consistent blood glucose–lowering effect with insulin degludec (FIGURE 1) reported to be longer than 24 hours in patients with T1DM or type 2 DM (T2DM).11,12
A randomized, double-blind, two-period, crossover comparison of insulin degludec and insulin glargine in patients with T1DM (N = 66) reported a half-life of 25.4 hours with insulin degludec compared with 12.5 hours with insulin glargine.13 The serum exposure of insulin degludec was similar between the first and second 12-hour period postdose. On the other hand, approximately 60% of the serum exposure to insulin glargine occurred over the first 12 hours following administration. These results highlight that insulin degludec is an ultra–long-acting insulin preparation with improved pharmacodynamic stability.
Analysis of data in 54 patients with T1DM reported that the within-subject pharmacodynamic variability was lower with insulin degludec compared with insulin glargine during a 24-hour euglycemic glucose clamp.14 Over 24 hours, the coefficient of variation (CV) with insulin degludec was lower for the area under the glucose infusion rate curve (AUCGIR) for total AUCGIR,0-24h (CV, 23% vs. 72%; P < .001), for GIRmax (CV, 21% vs. 53%; P < .0001), and for the fluctuation around the mean GIR value over 24 hours (CV, 31% vs. 62%; P < .001).
The findings from these investigations demonstrate that insulin degludec has a long half-life, resulting in a prolonged duration of blood glucose lowering with low within-subject pharmacodynamic variability.
FIGURE 1
Mean 24-hour glucose infusion rates (GIR) of insulin degludec at steady state
Copyright 2011 American Diabetes Association. From Diabetes®, Vol. 60,
Suppl. 1; 2011. Reprinted by permission of the American Diabetes Association.
Efficacy, Safety, and Tolerability of Insulin Degludec
Type 2 Diabetes Mellitus
Insulin degludec has been compared with insulin glargine in combination with oral glucose-lowering agents or in combination with a prandial insulin analog; one study investigated insulin degludec and insulin aspart in basal-bolus therapy in T2DM. In the basal-bolus treat-to-target trial, 992 patients with T2DM (mean A1C 8.3%) were randomized to receive insulin degludec or insulin glargine, each in combination with prandial insulin aspart ± metformin ± pioglitazone.15 Basal insulin was titrated to achieve a fasting plasma glucose (FPG) <90 mg/dL. At 1 year, mean A1C values were reduced by 1.1% and 1.2% with insulin degludec and insulin glargine, respectively (estimated treatment difference [ETD], 0.08%; 95% confidence interval (CI), –0.05 to 0.21). FPG was reduced by 41 and 36 mg/dL, respectively (ETD, –5.2 mg/dL; 95% CI, –11.7 to 1.1; P = non significant [NS]). Overall, the rates of confirmed hypoglycemia (plasma glucose <56 mg/dL or severe episodes requiring assistance) were lower in the group treated with insulin degludec than in the group treated with insulin glargine (11.1 vs 13.6 episodes/patient-year; estimated rate ratio [ERR], 0.82; 95% CI, 0.69 to 0.99; P = .0359). Nocturnal confirmed hypoglycemia, defined as episodes occurring between midnight and 6 am, occurred significantly less frequently in the insulin degludec group compared with the insulin glargine group (1.4 vs 1.8 episodes/patient-year, respectively; ERR, 0.75; 95% CI, 0.58 to 0.99; P = .0399) (FIGURE 2). Rates of other adverse events were similar between the 2 groups. At 1 year, the total mean daily insulin doses were 1.46 and 1.42 U/kg in the insulin degludec and insulin glargine groups, respectively, with a ~50:50 basal:bolus ratio for both groups.
Based on these findings, insulin degludec was associated with glycemic control similar to insulin glargine when given as basal-bolus therapy. Overall, confirmed and nocturnal hypoglycemia occurred less frequently with insulin degludec than with insulin glargine.
FIGURE 2
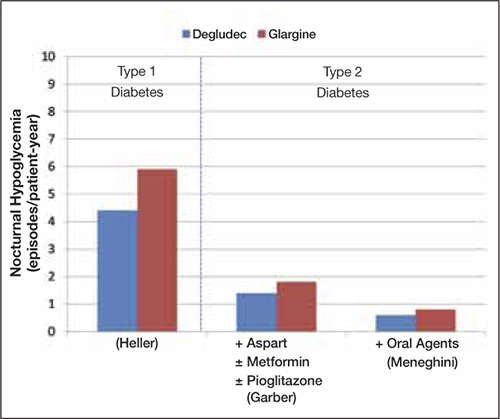
Incidences of nocturnal hypoglycemia with insulin degludec and insulin glargine15,16,18
Type 1 Diabetes Mellitus
Insulin degludec has been investigated in the treatment of patients with T1DM. Two randomized trials involved basal-bolus therapy in combination with insulin aspart. A 1-year treat-to-target trial in 629 adults with T1DM (mean A1C 7.7%) compared insulin degludec with insulin glargine, each given once daily in a basal-bolus regimen with mealtime insulin aspart.16 Both groups were reported to have improved glycemic control, with overall A1C decreased by 0.4%. Similar proportions of patients achieved A1C <7.0% with insulin degludec and insulin glargine (40% vs 43%; P = NS). Mean FPG values were reduced similarly (ETD, 5.9 mg/dL; P = .35). Compared with insulin glargine, rates of confirmed nocturnal hypoglycemia were 25% lower with insulin degludec (4.4 vs 5.9 episodes/patient-year; ERR, 0.75; 95% CI, 0.59 to 0.96; P = .021), whereas rates of overall confirmed hypoglycemia were similar between treatment groups (42.5 vs 40.2 episodes/patient-year; ERR, 1.07; 95% CI, 0.89 to 1.28; P = .48). Overall rates of other adverse events were similar between groups.
Insulin degludec in a fixed-ratio combined formulation with insulin aspart (IDegAsp) was compared with insulin detemir and insulin aspart in basal-bolus therapy in a 26-week, open-label, treat-to-target trial involving 548 patients with T1DM (mean A1C, 8.3%; mean FPG, 189 mg/dL at baseline).17 IDegAsp was given once daily at any meal, with insulin aspart at the remaining meals, whereas insulin detemir was administered according to approved labeling with mealtime insulin aspart at all meals. The mean decrease in A1C was similar for IDegAsp and insulin detemir/insulin aspart (0.73% vs 0.68%, respectively). The decrease in mean FPG was also similar between groups (P = .52). The mean total daily insulin doses were 69 U (0.86 U/kg) for IDegAsp and 79 U (1.00 U/kg) for insulin detemir and insulin aspart. Rates of severe hypoglycemia were 0.33 and 0.42 episodes/patient-year with IDegAsp and insulin detemir, respectively. Rates of overall confirmed hypoglycemia were similar (39 vs 44 episodes/patient-year; P = .27), whereas confirmed nocturnal hypoglycemia was reported significantly less frequently with IDegAsp (3.7 vs 5.7 episodes/patient-year, respectively; P = .0003). Weight increase was significantly greater (by 1.04 kg) with IDegAsp compared with insulin detemir (P = .0021). Overall rates of other adverse events were similar between treatment groups.
Results from trials in patients with T1DM and T2DM are consistent and suggest comparable glycemic lowering between insulin degludec and the basal insulin analogs detemir and glargine, with less frequent nocturnal hypoglycemia in those treated with insulin degludec compared with insulins glargine and detemir (FIGURE 2).
Flexibility of Dosing Time
Optimal glycemic benefits are achieved with the injection of basal insulin at a consistent time each day. However, consistent timing may be difficult owing to patients’ busy or erratic schedules and/or in patients who may at times forget to administer their medications. These patient factors can lead to wide variability in the dosing interval and suboptimal results in fasting glucose control. These challenges may be improved upon with the investigational agent insulin degludec due to the stable and prolonged time-action profile of insulin degludec coupled with low within-subject pharmacodynamic variability, allowing for a more flexible once-daily dosing time. A 26-week, randomized, open-label trial in patients with T2DM (N = 459) aimed to compare insulin degludec in the setting of variable dosing intervals by administering insulin degludec once daily using a flexible regimen compared with insulin glargine given once daily at the same time each day.18 Both insulins were added to an existing regimen of oral glucose-lowering therapy (if any) and titrated to achieve FPG <90 mg/dL. To ensure variability in the dosing interval, the once-daily regimen of insulin degludec involved a compulsory, rotating morning and evening schedule, creating 8- to 40-hour dosing intervals. From a baseline mean of 8.4%, A1C values were reduced by 1.28% and 1.26% with insulin degludec and insulin glargine, respectively, at 26 weeks, confirming noninferiority of the flexible regimen of once-daily insulin degludec compared with insulin glargine given at the same time each day. The mean FPG at week 26 was significantly lower for insulin degludec than insulin glargine (104 vs 112 mg/dL, respectively; P = .04). The rates of confirmed hypoglycemia (3.6 vs 3.5 episodes/patient-year) and nocturnal hypoglycemia (0.6 vs 0.8 episodes/patient-year) for insulin degludec compared with insulin glargine, respectively, and the numbers of severe hypoglycemia events (2 episodes/group), were similar between treatment groups. This trial demonstrates that when needed to accommodate changes in the patient’s daily schedule, insulin degludec may be administered at differing times from day to day without compromising glycemic control or safety compared with insulin glargine administered at the same time each day.
CONCLUSIONS
Insulin degludec, an ultra–long-acting basal insulin analog, possesses several desirable attributes. Findings from clinical trials have demonstrated that the new-generation once-daily basal insulin degludec provides similar A1C control compared to insulin glargine both administered as basal-oral therapy or in combination with insulin aspart, with the added benefit of lower rates of hypoglycemia, particularly nocturnal hypoglycemia. Insulin degludec has also been shown to offer dosing flexibility, with administration at any time of the day without compromising glycemic control or safety. Insulin degludec, pending FDA approval, will be an additional treatment to help patients with T1DM or T2DM achieve glycemic control.
1. Rodbard HW, Jellinger PS, Davidson JA, et al. Statement by an American Association of Clinical Endocrinologists/American College of Endocrinology consensus panel on type 2 diabetes mellitus: an algorithm for glycemic control [published correction appears in Endocr Pract. 2009;15(7):768-770]. Endocr Pract. 2009;15(6):540-559.
2. Bartley PC, Bogoev M, Larsen J, Philotheou A. Long-term efficacy and safety of insulin detemir compared to Neutral Protamine Hagedorn insulin in patients with type 1 diabetes using a treat-to-target basal-bolus regimen with insulin aspart at meals: a 2-year, randomized, controlled trial. Diabet Med. 2008;25(4):442-449.
3. Riddle MC, Rosenstock J, Gerich J. Insulin Glargine 4002 Study Investigators. The treat-to-target trial: randomized addition of glargine or human NPH insulin to oral therapy of type 2 diabetic patients. Diabetes Care. 2003;26(11):3080—3086.
4. Hermansen K, Davies M, Derezinski T, Martinez Ravn G, Clauson P, Home P. A 26-week, randomized, parallel, treat-to-target trial comparing insulin detemir with NPH insulin as add-on therapy to oral glucose-lowering drugs in insulin-naive people with type 2 diabetes [published correction appears in Diabetes Care. 2007;30(4):1035] Diabetes Care. 2006;29(6):1269-1274.
5. Heise T, Nosek L, Rønn BB, et al. Lower within-subject variability of insulin detemir in comparison to NPH insulin and insulin glargine in people with type 1 diabetes. Diabetes. 2004;53(6):1614-1620.
6. Ashwell SG, Gebbie J, Home PD. Twice-daily compared with once-daily insulin glargine in people with type 1 diabetes using meal-time insulin aspart. Diabet Med. 2006;23(8):879-886.
7. Donnelly LA, Morris AD, Frier BM, et al. DARTS/MEMO Collaboration. Frequency and predictors of hypoglycaemia in type 1 and insulin-treated type 2 diabetes: a population-based study. Diabet Med. 2005;22(6):749-755.
8. Hermansen K, Dornhorst A, Sreenan S. Observational, open-label study of type 1 and type 2 diabetes patients switching from human insulin to insulin analogue basal-bolus regimens: insights from the PREDICTIVE study. Curr Med Res Opin. 2009;25(11):2601-2608.
9. Kalra S, Unnikrishnan AG, Baruah M, Kalra B. Degludec insulin: a novel basal insulin. Indian J Endocrinol Metab. 2011;15(suppl 1):S12-S16.
10. Jonassen I, Havelund S, Ribel U, et al. Insulin degludec: Multi-hexamer formation is the underlying basis for this new generation ultra-long acting basal insulin. Paper presented at: European Association for the Study of Diabetes Annual Meeting; September 20-24, 2010; Stockholm, Sweden.
11. Kurtzhals P, Heise T, Strauss HM, et al. Multi-hexamer formation is the underlying mechanism behind the ultra-long glucose-lowering effect of insulin degludec. Paper presented at: American Diabetes Association 71st Scientific Sessions; June 24-28, 2011; San Diego, CA.
12. Nosek L, Heise T, Bøttcher SG, Hastrup H, Haahr H. Ultra-long-acting insulin degludec has a flat and stable glucose-lowering effect. Paper presented at: American Diabetes Association 71st Scientific Sessions; June 24-28, 2011; San Diego, CA.
13. Heise T, Hövelmann U, Nosek L, Bøttcher SG, Granhall C, Haahr H. Insulin degludec has a two-fold longer half-life and a more consistent pharmacokinetic profile than insulin glargine. Paper presented at: American Diabetes Association 71st Scientific Sessions; June 24-28, 2011; San Diego, CA.
14. Heise T, Hermanski L, Nosek L, Feldmann A, Rasmussen S, Haahr H. The pharmacodynamic variability of insulin degludec is consistently lower than insulin glargine over 24 hours at steady state. Diabetes. 2011;60(suppl 1):A263.-Poster 960-P.
Garber AJ, King AB, Del Prato S, et al. 15.NN1250-3582 (BEGIN BB T2D) Trial Investigators. Insulin degludec, an ultra-longacting basal insulin, versus insulin glargine in basal-bolus treatment with mealtime insulin aspart in type 2 diabetes (BEGIN Basal-Bolus Type 2): a phase 3, randomised, open-label, treat-to-target non-inferiority trial. Lancet. 2012;379(9825):1498-1507.
16. Heller S, Buse J, Fisher M, et al. BEGIN Basal-Bolus Type 1 Trial Investigators. Insulin degludec, an ultra-longacting basal insulin, versus insulin glargine in basal-bolus treatment with mealtime insulin aspart in type 1 diabetes (BEGIN Basal-Blus Type 1): a phase 3, randomised, open-label, treat-to-target non-inferiority trial. Lancet. 2012;379(9825):1489-1497.
17. Hirsch IB, Franek E, Courreges JP, Mersebach H, Dykiel P, Bode BW. Efficacy and safety of a new basal insulin with a bolus boost (IDegAsp) used once daily in combination wtih insulin apart (IAsp) in people wth type 1 diabetes. Diabetes. 2011;60(suppl 1):A292.-Poster 1064-P.
18. Meneghini L, Atkin SL, Bain S, et al. Flexible once-daily dosing of insulin degludec does not compromise glycemic control or safety compared to insulin glargine given once daily at the same time each day in people with type 2 diabetes. Paper presented at: American Diabetes Association 71st Scientific Sessions; June 24-28, 2011; San Diego, CA.
The Evolution of Insulin Therapy in Diabetes Mellitus
Introduction
Basal insulin has been an important treatment option for patients with diabetes mellitus (DM) and, along with prandial insulin, has undergone major improvements in terms of purity and similarity to the action of physiologic human insulin. (see The Evolution of Insulin Therapy in Diabetes Mellitus in this supplement.) Lente and Ultralente formulations were used for decades but are no longer available. The use of neutral protamine Hagedorn (NPH) insulin is also being replaced with the basal insulin analogs detemir and glargine.1 Basal insulin analogs generally cause less severe and nocturnal hypoglycemia compared with NPH insulin owing to their improved pharmacologic profiles.2-4 In comparison to NPH insulin, insulin glargine causes similar weight gain, whereas insulin detemir causes less weight gain.2-4 In addition, insulin detemir has been associated with a glucose-lowering effect that is more predictable than that of NPH insulin.5 Despite the improvements observed with basal insulin analogs, their time-action profiles are not completely flat and are shorter than 24 hours in many patients.5,6 In addition, severe hypoglycemia remains a concern, particularly in patients with type 1 DM (T1DM).7,8 Consequently, the search for a better basal insulin continues.
The ideal basal insulin should possess numerous attributes. While each of the attributes listed in the TABLE is important, an overarching difficulty with basal insulin therapy is the need for administration at the same time each day.9 This dosing limitation may be most difficult for those with busy or erratic schedules or who may forget to administer their insulin dose. This article will review the clinical experience with insulin degludec, an ultra–long-acting insulin under review by the US Food and Drug Administration (FDA).
TABLE
Attributes of the ideal basal insulin9
| Delivers a steady, stable, peakless, continuous insulin concentration for at least 24 hours, in a predictable manner, with low intraindividual and interindividual variability |
| Does not cause side effects such as weight gain or hypoglycemia |
| Does not induce mitogenicity |
| Can be used as monotherapy, as part of basal-bolus therapy, or in combination with oral glucose-lowering therapy |
| Equally efficacious, safe, and well-tolerated in patients with type 1 or type 2 diabetes mellitus |
| Indian Journal of Endocrinology and Metabolism. Copyright 2011 by MEDKNOW PUBLICATIONS AND MEDIA PVT LTD. Reproduced with permission of MEDKNOW PUBLICATIONS AND MEDIA PVT LTD in the format Journal via Copyright Clearance Center. |
Clinical Pharmacology of Insulin Degludec
Removal of threonine at position 30 of the B chain of human insulin and the addition of a 16-carbon fatty diacid attached to lysine at position 29 of the B chain of human insulin via a glutamic acid spacer result in the insulin degludec molecule, which has several differences from available basal insulin analogs. Experimental investigations indicated that conditions mimicking subcutaneous injection of insulin degludec resulted in a reorganization of the insulin degludec molecule from dihexamers to multihexamer assemblies that remain in solution at physiologic pH.10 Slow release of zinc ions from the multihexamers leads to the slow release of insulin degludec monomers, which are easily absorbed into the systemic circulation.11 The result is a half-life of insulin degludec that is longer than 24 hours, with a level that is detectable in circulation for at least 96 hours after administration of the dose.10,12 The pharmacodynamic result is a relatively flat and consistent blood glucose–lowering effect with insulin degludec (FIGURE 1) reported to be longer than 24 hours in patients with T1DM or type 2 DM (T2DM).11,12
A randomized, double-blind, two-period, crossover comparison of insulin degludec and insulin glargine in patients with T1DM (N = 66) reported a half-life of 25.4 hours with insulin degludec compared with 12.5 hours with insulin glargine.13 The serum exposure of insulin degludec was similar between the first and second 12-hour period postdose. On the other hand, approximately 60% of the serum exposure to insulin glargine occurred over the first 12 hours following administration. These results highlight that insulin degludec is an ultra–long-acting insulin preparation with improved pharmacodynamic stability.
Analysis of data in 54 patients with T1DM reported that the within-subject pharmacodynamic variability was lower with insulin degludec compared with insulin glargine during a 24-hour euglycemic glucose clamp.14 Over 24 hours, the coefficient of variation (CV) with insulin degludec was lower for the area under the glucose infusion rate curve (AUCGIR) for total AUCGIR,0-24h (CV, 23% vs. 72%; P < .001), for GIRmax (CV, 21% vs. 53%; P < .0001), and for the fluctuation around the mean GIR value over 24 hours (CV, 31% vs. 62%; P < .001).
The findings from these investigations demonstrate that insulin degludec has a long half-life, resulting in a prolonged duration of blood glucose lowering with low within-subject pharmacodynamic variability.
FIGURE 1
Mean 24-hour glucose infusion rates (GIR) of insulin degludec at steady state
Copyright 2011 American Diabetes Association. From Diabetes®, Vol. 60,
Suppl. 1; 2011. Reprinted by permission of the American Diabetes Association.
Efficacy, Safety, and Tolerability of Insulin Degludec
Type 2 Diabetes Mellitus
Insulin degludec has been compared with insulin glargine in combination with oral glucose-lowering agents or in combination with a prandial insulin analog; one study investigated insulin degludec and insulin aspart in basal-bolus therapy in T2DM. In the basal-bolus treat-to-target trial, 992 patients with T2DM (mean A1C 8.3%) were randomized to receive insulin degludec or insulin glargine, each in combination with prandial insulin aspart ± metformin ± pioglitazone.15 Basal insulin was titrated to achieve a fasting plasma glucose (FPG) <90 mg/dL. At 1 year, mean A1C values were reduced by 1.1% and 1.2% with insulin degludec and insulin glargine, respectively (estimated treatment difference [ETD], 0.08%; 95% confidence interval (CI), –0.05 to 0.21). FPG was reduced by 41 and 36 mg/dL, respectively (ETD, –5.2 mg/dL; 95% CI, –11.7 to 1.1; P = non significant [NS]). Overall, the rates of confirmed hypoglycemia (plasma glucose <56 mg/dL or severe episodes requiring assistance) were lower in the group treated with insulin degludec than in the group treated with insulin glargine (11.1 vs 13.6 episodes/patient-year; estimated rate ratio [ERR], 0.82; 95% CI, 0.69 to 0.99; P = .0359). Nocturnal confirmed hypoglycemia, defined as episodes occurring between midnight and 6 am, occurred significantly less frequently in the insulin degludec group compared with the insulin glargine group (1.4 vs 1.8 episodes/patient-year, respectively; ERR, 0.75; 95% CI, 0.58 to 0.99; P = .0399) (FIGURE 2). Rates of other adverse events were similar between the 2 groups. At 1 year, the total mean daily insulin doses were 1.46 and 1.42 U/kg in the insulin degludec and insulin glargine groups, respectively, with a ~50:50 basal:bolus ratio for both groups.
Based on these findings, insulin degludec was associated with glycemic control similar to insulin glargine when given as basal-bolus therapy. Overall, confirmed and nocturnal hypoglycemia occurred less frequently with insulin degludec than with insulin glargine.
FIGURE 2
Incidences of nocturnal hypoglycemia with insulin degludec and insulin glargine15,16,18
Type 1 Diabetes Mellitus
Insulin degludec has been investigated in the treatment of patients with T1DM. Two randomized trials involved basal-bolus therapy in combination with insulin aspart. A 1-year treat-to-target trial in 629 adults with T1DM (mean A1C 7.7%) compared insulin degludec with insulin glargine, each given once daily in a basal-bolus regimen with mealtime insulin aspart.16 Both groups were reported to have improved glycemic control, with overall A1C decreased by 0.4%. Similar proportions of patients achieved A1C <7.0% with insulin degludec and insulin glargine (40% vs 43%; P = NS). Mean FPG values were reduced similarly (ETD, 5.9 mg/dL; P = .35). Compared with insulin glargine, rates of confirmed nocturnal hypoglycemia were 25% lower with insulin degludec (4.4 vs 5.9 episodes/patient-year; ERR, 0.75; 95% CI, 0.59 to 0.96; P = .021), whereas rates of overall confirmed hypoglycemia were similar between treatment groups (42.5 vs 40.2 episodes/patient-year; ERR, 1.07; 95% CI, 0.89 to 1.28; P = .48). Overall rates of other adverse events were similar between groups.
Insulin degludec in a fixed-ratio combined formulation with insulin aspart (IDegAsp) was compared with insulin detemir and insulin aspart in basal-bolus therapy in a 26-week, open-label, treat-to-target trial involving 548 patients with T1DM (mean A1C, 8.3%; mean FPG, 189 mg/dL at baseline).17 IDegAsp was given once daily at any meal, with insulin aspart at the remaining meals, whereas insulin detemir was administered according to approved labeling with mealtime insulin aspart at all meals. The mean decrease in A1C was similar for IDegAsp and insulin detemir/insulin aspart (0.73% vs 0.68%, respectively). The decrease in mean FPG was also similar between groups (P = .52). The mean total daily insulin doses were 69 U (0.86 U/kg) for IDegAsp and 79 U (1.00 U/kg) for insulin detemir and insulin aspart. Rates of severe hypoglycemia were 0.33 and 0.42 episodes/patient-year with IDegAsp and insulin detemir, respectively. Rates of overall confirmed hypoglycemia were similar (39 vs 44 episodes/patient-year; P = .27), whereas confirmed nocturnal hypoglycemia was reported significantly less frequently with IDegAsp (3.7 vs 5.7 episodes/patient-year, respectively; P = .0003). Weight increase was significantly greater (by 1.04 kg) with IDegAsp compared with insulin detemir (P = .0021). Overall rates of other adverse events were similar between treatment groups.
Results from trials in patients with T1DM and T2DM are consistent and suggest comparable glycemic lowering between insulin degludec and the basal insulin analogs detemir and glargine, with less frequent nocturnal hypoglycemia in those treated with insulin degludec compared with insulins glargine and detemir (FIGURE 2).
Flexibility of Dosing Time
Optimal glycemic benefits are achieved with the injection of basal insulin at a consistent time each day. However, consistent timing may be difficult owing to patients’ busy or erratic schedules and/or in patients who may at times forget to administer their medications. These patient factors can lead to wide variability in the dosing interval and suboptimal results in fasting glucose control. These challenges may be improved upon with the investigational agent insulin degludec due to the stable and prolonged time-action profile of insulin degludec coupled with low within-subject pharmacodynamic variability, allowing for a more flexible once-daily dosing time. A 26-week, randomized, open-label trial in patients with T2DM (N = 459) aimed to compare insulin degludec in the setting of variable dosing intervals by administering insulin degludec once daily using a flexible regimen compared with insulin glargine given once daily at the same time each day.18 Both insulins were added to an existing regimen of oral glucose-lowering therapy (if any) and titrated to achieve FPG <90 mg/dL. To ensure variability in the dosing interval, the once-daily regimen of insulin degludec involved a compulsory, rotating morning and evening schedule, creating 8- to 40-hour dosing intervals. From a baseline mean of 8.4%, A1C values were reduced by 1.28% and 1.26% with insulin degludec and insulin glargine, respectively, at 26 weeks, confirming noninferiority of the flexible regimen of once-daily insulin degludec compared with insulin glargine given at the same time each day. The mean FPG at week 26 was significantly lower for insulin degludec than insulin glargine (104 vs 112 mg/dL, respectively; P = .04). The rates of confirmed hypoglycemia (3.6 vs 3.5 episodes/patient-year) and nocturnal hypoglycemia (0.6 vs 0.8 episodes/patient-year) for insulin degludec compared with insulin glargine, respectively, and the numbers of severe hypoglycemia events (2 episodes/group), were similar between treatment groups. This trial demonstrates that when needed to accommodate changes in the patient’s daily schedule, insulin degludec may be administered at differing times from day to day without compromising glycemic control or safety compared with insulin glargine administered at the same time each day.
CONCLUSIONS
Insulin degludec, an ultra–long-acting basal insulin analog, possesses several desirable attributes. Findings from clinical trials have demonstrated that the new-generation once-daily basal insulin degludec provides similar A1C control compared to insulin glargine both administered as basal-oral therapy or in combination with insulin aspart, with the added benefit of lower rates of hypoglycemia, particularly nocturnal hypoglycemia. Insulin degludec has also been shown to offer dosing flexibility, with administration at any time of the day without compromising glycemic control or safety. Insulin degludec, pending FDA approval, will be an additional treatment to help patients with T1DM or T2DM achieve glycemic control.
The Evolution of Insulin Therapy in Diabetes Mellitus
Introduction
Basal insulin has been an important treatment option for patients with diabetes mellitus (DM) and, along with prandial insulin, has undergone major improvements in terms of purity and similarity to the action of physiologic human insulin. (see The Evolution of Insulin Therapy in Diabetes Mellitus in this supplement.) Lente and Ultralente formulations were used for decades but are no longer available. The use of neutral protamine Hagedorn (NPH) insulin is also being replaced with the basal insulin analogs detemir and glargine.1 Basal insulin analogs generally cause less severe and nocturnal hypoglycemia compared with NPH insulin owing to their improved pharmacologic profiles.2-4 In comparison to NPH insulin, insulin glargine causes similar weight gain, whereas insulin detemir causes less weight gain.2-4 In addition, insulin detemir has been associated with a glucose-lowering effect that is more predictable than that of NPH insulin.5 Despite the improvements observed with basal insulin analogs, their time-action profiles are not completely flat and are shorter than 24 hours in many patients.5,6 In addition, severe hypoglycemia remains a concern, particularly in patients with type 1 DM (T1DM).7,8 Consequently, the search for a better basal insulin continues.
The ideal basal insulin should possess numerous attributes. While each of the attributes listed in the TABLE is important, an overarching difficulty with basal insulin therapy is the need for administration at the same time each day.9 This dosing limitation may be most difficult for those with busy or erratic schedules or who may forget to administer their insulin dose. This article will review the clinical experience with insulin degludec, an ultra–long-acting insulin under review by the US Food and Drug Administration (FDA).
TABLE
Attributes of the ideal basal insulin9
| Delivers a steady, stable, peakless, continuous insulin concentration for at least 24 hours, in a predictable manner, with low intraindividual and interindividual variability |
| Does not cause side effects such as weight gain or hypoglycemia |
| Does not induce mitogenicity |
| Can be used as monotherapy, as part of basal-bolus therapy, or in combination with oral glucose-lowering therapy |
| Equally efficacious, safe, and well-tolerated in patients with type 1 or type 2 diabetes mellitus |
| Indian Journal of Endocrinology and Metabolism. Copyright 2011 by MEDKNOW PUBLICATIONS AND MEDIA PVT LTD. Reproduced with permission of MEDKNOW PUBLICATIONS AND MEDIA PVT LTD in the format Journal via Copyright Clearance Center. |
Clinical Pharmacology of Insulin Degludec
Removal of threonine at position 30 of the B chain of human insulin and the addition of a 16-carbon fatty diacid attached to lysine at position 29 of the B chain of human insulin via a glutamic acid spacer result in the insulin degludec molecule, which has several differences from available basal insulin analogs. Experimental investigations indicated that conditions mimicking subcutaneous injection of insulin degludec resulted in a reorganization of the insulin degludec molecule from dihexamers to multihexamer assemblies that remain in solution at physiologic pH.10 Slow release of zinc ions from the multihexamers leads to the slow release of insulin degludec monomers, which are easily absorbed into the systemic circulation.11 The result is a half-life of insulin degludec that is longer than 24 hours, with a level that is detectable in circulation for at least 96 hours after administration of the dose.10,12 The pharmacodynamic result is a relatively flat and consistent blood glucose–lowering effect with insulin degludec (FIGURE 1) reported to be longer than 24 hours in patients with T1DM or type 2 DM (T2DM).11,12
A randomized, double-blind, two-period, crossover comparison of insulin degludec and insulin glargine in patients with T1DM (N = 66) reported a half-life of 25.4 hours with insulin degludec compared with 12.5 hours with insulin glargine.13 The serum exposure of insulin degludec was similar between the first and second 12-hour period postdose. On the other hand, approximately 60% of the serum exposure to insulin glargine occurred over the first 12 hours following administration. These results highlight that insulin degludec is an ultra–long-acting insulin preparation with improved pharmacodynamic stability.
Analysis of data in 54 patients with T1DM reported that the within-subject pharmacodynamic variability was lower with insulin degludec compared with insulin glargine during a 24-hour euglycemic glucose clamp.14 Over 24 hours, the coefficient of variation (CV) with insulin degludec was lower for the area under the glucose infusion rate curve (AUCGIR) for total AUCGIR,0-24h (CV, 23% vs. 72%; P < .001), for GIRmax (CV, 21% vs. 53%; P < .0001), and for the fluctuation around the mean GIR value over 24 hours (CV, 31% vs. 62%; P < .001).
The findings from these investigations demonstrate that insulin degludec has a long half-life, resulting in a prolonged duration of blood glucose lowering with low within-subject pharmacodynamic variability.
FIGURE 1
Mean 24-hour glucose infusion rates (GIR) of insulin degludec at steady state
Copyright 2011 American Diabetes Association. From Diabetes®, Vol. 60,
Suppl. 1; 2011. Reprinted by permission of the American Diabetes Association.
Efficacy, Safety, and Tolerability of Insulin Degludec
Type 2 Diabetes Mellitus
Insulin degludec has been compared with insulin glargine in combination with oral glucose-lowering agents or in combination with a prandial insulin analog; one study investigated insulin degludec and insulin aspart in basal-bolus therapy in T2DM. In the basal-bolus treat-to-target trial, 992 patients with T2DM (mean A1C 8.3%) were randomized to receive insulin degludec or insulin glargine, each in combination with prandial insulin aspart ± metformin ± pioglitazone.15 Basal insulin was titrated to achieve a fasting plasma glucose (FPG) <90 mg/dL. At 1 year, mean A1C values were reduced by 1.1% and 1.2% with insulin degludec and insulin glargine, respectively (estimated treatment difference [ETD], 0.08%; 95% confidence interval (CI), –0.05 to 0.21). FPG was reduced by 41 and 36 mg/dL, respectively (ETD, –5.2 mg/dL; 95% CI, –11.7 to 1.1; P = non significant [NS]). Overall, the rates of confirmed hypoglycemia (plasma glucose <56 mg/dL or severe episodes requiring assistance) were lower in the group treated with insulin degludec than in the group treated with insulin glargine (11.1 vs 13.6 episodes/patient-year; estimated rate ratio [ERR], 0.82; 95% CI, 0.69 to 0.99; P = .0359). Nocturnal confirmed hypoglycemia, defined as episodes occurring between midnight and 6 am, occurred significantly less frequently in the insulin degludec group compared with the insulin glargine group (1.4 vs 1.8 episodes/patient-year, respectively; ERR, 0.75; 95% CI, 0.58 to 0.99; P = .0399) (FIGURE 2). Rates of other adverse events were similar between the 2 groups. At 1 year, the total mean daily insulin doses were 1.46 and 1.42 U/kg in the insulin degludec and insulin glargine groups, respectively, with a ~50:50 basal:bolus ratio for both groups.
Based on these findings, insulin degludec was associated with glycemic control similar to insulin glargine when given as basal-bolus therapy. Overall, confirmed and nocturnal hypoglycemia occurred less frequently with insulin degludec than with insulin glargine.
FIGURE 2
Incidences of nocturnal hypoglycemia with insulin degludec and insulin glargine15,16,18
Type 1 Diabetes Mellitus
Insulin degludec has been investigated in the treatment of patients with T1DM. Two randomized trials involved basal-bolus therapy in combination with insulin aspart. A 1-year treat-to-target trial in 629 adults with T1DM (mean A1C 7.7%) compared insulin degludec with insulin glargine, each given once daily in a basal-bolus regimen with mealtime insulin aspart.16 Both groups were reported to have improved glycemic control, with overall A1C decreased by 0.4%. Similar proportions of patients achieved A1C <7.0% with insulin degludec and insulin glargine (40% vs 43%; P = NS). Mean FPG values were reduced similarly (ETD, 5.9 mg/dL; P = .35). Compared with insulin glargine, rates of confirmed nocturnal hypoglycemia were 25% lower with insulin degludec (4.4 vs 5.9 episodes/patient-year; ERR, 0.75; 95% CI, 0.59 to 0.96; P = .021), whereas rates of overall confirmed hypoglycemia were similar between treatment groups (42.5 vs 40.2 episodes/patient-year; ERR, 1.07; 95% CI, 0.89 to 1.28; P = .48). Overall rates of other adverse events were similar between groups.
Insulin degludec in a fixed-ratio combined formulation with insulin aspart (IDegAsp) was compared with insulin detemir and insulin aspart in basal-bolus therapy in a 26-week, open-label, treat-to-target trial involving 548 patients with T1DM (mean A1C, 8.3%; mean FPG, 189 mg/dL at baseline).17 IDegAsp was given once daily at any meal, with insulin aspart at the remaining meals, whereas insulin detemir was administered according to approved labeling with mealtime insulin aspart at all meals. The mean decrease in A1C was similar for IDegAsp and insulin detemir/insulin aspart (0.73% vs 0.68%, respectively). The decrease in mean FPG was also similar between groups (P = .52). The mean total daily insulin doses were 69 U (0.86 U/kg) for IDegAsp and 79 U (1.00 U/kg) for insulin detemir and insulin aspart. Rates of severe hypoglycemia were 0.33 and 0.42 episodes/patient-year with IDegAsp and insulin detemir, respectively. Rates of overall confirmed hypoglycemia were similar (39 vs 44 episodes/patient-year; P = .27), whereas confirmed nocturnal hypoglycemia was reported significantly less frequently with IDegAsp (3.7 vs 5.7 episodes/patient-year, respectively; P = .0003). Weight increase was significantly greater (by 1.04 kg) with IDegAsp compared with insulin detemir (P = .0021). Overall rates of other adverse events were similar between treatment groups.
Results from trials in patients with T1DM and T2DM are consistent and suggest comparable glycemic lowering between insulin degludec and the basal insulin analogs detemir and glargine, with less frequent nocturnal hypoglycemia in those treated with insulin degludec compared with insulins glargine and detemir (FIGURE 2).
Flexibility of Dosing Time
Optimal glycemic benefits are achieved with the injection of basal insulin at a consistent time each day. However, consistent timing may be difficult owing to patients’ busy or erratic schedules and/or in patients who may at times forget to administer their medications. These patient factors can lead to wide variability in the dosing interval and suboptimal results in fasting glucose control. These challenges may be improved upon with the investigational agent insulin degludec due to the stable and prolonged time-action profile of insulin degludec coupled with low within-subject pharmacodynamic variability, allowing for a more flexible once-daily dosing time. A 26-week, randomized, open-label trial in patients with T2DM (N = 459) aimed to compare insulin degludec in the setting of variable dosing intervals by administering insulin degludec once daily using a flexible regimen compared with insulin glargine given once daily at the same time each day.18 Both insulins were added to an existing regimen of oral glucose-lowering therapy (if any) and titrated to achieve FPG <90 mg/dL. To ensure variability in the dosing interval, the once-daily regimen of insulin degludec involved a compulsory, rotating morning and evening schedule, creating 8- to 40-hour dosing intervals. From a baseline mean of 8.4%, A1C values were reduced by 1.28% and 1.26% with insulin degludec and insulin glargine, respectively, at 26 weeks, confirming noninferiority of the flexible regimen of once-daily insulin degludec compared with insulin glargine given at the same time each day. The mean FPG at week 26 was significantly lower for insulin degludec than insulin glargine (104 vs 112 mg/dL, respectively; P = .04). The rates of confirmed hypoglycemia (3.6 vs 3.5 episodes/patient-year) and nocturnal hypoglycemia (0.6 vs 0.8 episodes/patient-year) for insulin degludec compared with insulin glargine, respectively, and the numbers of severe hypoglycemia events (2 episodes/group), were similar between treatment groups. This trial demonstrates that when needed to accommodate changes in the patient’s daily schedule, insulin degludec may be administered at differing times from day to day without compromising glycemic control or safety compared with insulin glargine administered at the same time each day.
CONCLUSIONS
Insulin degludec, an ultra–long-acting basal insulin analog, possesses several desirable attributes. Findings from clinical trials have demonstrated that the new-generation once-daily basal insulin degludec provides similar A1C control compared to insulin glargine both administered as basal-oral therapy or in combination with insulin aspart, with the added benefit of lower rates of hypoglycemia, particularly nocturnal hypoglycemia. Insulin degludec has also been shown to offer dosing flexibility, with administration at any time of the day without compromising glycemic control or safety. Insulin degludec, pending FDA approval, will be an additional treatment to help patients with T1DM or T2DM achieve glycemic control.
1. Rodbard HW, Jellinger PS, Davidson JA, et al. Statement by an American Association of Clinical Endocrinologists/American College of Endocrinology consensus panel on type 2 diabetes mellitus: an algorithm for glycemic control [published correction appears in Endocr Pract. 2009;15(7):768-770]. Endocr Pract. 2009;15(6):540-559.
2. Bartley PC, Bogoev M, Larsen J, Philotheou A. Long-term efficacy and safety of insulin detemir compared to Neutral Protamine Hagedorn insulin in patients with type 1 diabetes using a treat-to-target basal-bolus regimen with insulin aspart at meals: a 2-year, randomized, controlled trial. Diabet Med. 2008;25(4):442-449.
3. Riddle MC, Rosenstock J, Gerich J. Insulin Glargine 4002 Study Investigators. The treat-to-target trial: randomized addition of glargine or human NPH insulin to oral therapy of type 2 diabetic patients. Diabetes Care. 2003;26(11):3080—3086.
4. Hermansen K, Davies M, Derezinski T, Martinez Ravn G, Clauson P, Home P. A 26-week, randomized, parallel, treat-to-target trial comparing insulin detemir with NPH insulin as add-on therapy to oral glucose-lowering drugs in insulin-naive people with type 2 diabetes [published correction appears in Diabetes Care. 2007;30(4):1035] Diabetes Care. 2006;29(6):1269-1274.
5. Heise T, Nosek L, Rønn BB, et al. Lower within-subject variability of insulin detemir in comparison to NPH insulin and insulin glargine in people with type 1 diabetes. Diabetes. 2004;53(6):1614-1620.
6. Ashwell SG, Gebbie J, Home PD. Twice-daily compared with once-daily insulin glargine in people with type 1 diabetes using meal-time insulin aspart. Diabet Med. 2006;23(8):879-886.
7. Donnelly LA, Morris AD, Frier BM, et al. DARTS/MEMO Collaboration. Frequency and predictors of hypoglycaemia in type 1 and insulin-treated type 2 diabetes: a population-based study. Diabet Med. 2005;22(6):749-755.
8. Hermansen K, Dornhorst A, Sreenan S. Observational, open-label study of type 1 and type 2 diabetes patients switching from human insulin to insulin analogue basal-bolus regimens: insights from the PREDICTIVE study. Curr Med Res Opin. 2009;25(11):2601-2608.
9. Kalra S, Unnikrishnan AG, Baruah M, Kalra B. Degludec insulin: a novel basal insulin. Indian J Endocrinol Metab. 2011;15(suppl 1):S12-S16.
10. Jonassen I, Havelund S, Ribel U, et al. Insulin degludec: Multi-hexamer formation is the underlying basis for this new generation ultra-long acting basal insulin. Paper presented at: European Association for the Study of Diabetes Annual Meeting; September 20-24, 2010; Stockholm, Sweden.
11. Kurtzhals P, Heise T, Strauss HM, et al. Multi-hexamer formation is the underlying mechanism behind the ultra-long glucose-lowering effect of insulin degludec. Paper presented at: American Diabetes Association 71st Scientific Sessions; June 24-28, 2011; San Diego, CA.
12. Nosek L, Heise T, Bøttcher SG, Hastrup H, Haahr H. Ultra-long-acting insulin degludec has a flat and stable glucose-lowering effect. Paper presented at: American Diabetes Association 71st Scientific Sessions; June 24-28, 2011; San Diego, CA.
13. Heise T, Hövelmann U, Nosek L, Bøttcher SG, Granhall C, Haahr H. Insulin degludec has a two-fold longer half-life and a more consistent pharmacokinetic profile than insulin glargine. Paper presented at: American Diabetes Association 71st Scientific Sessions; June 24-28, 2011; San Diego, CA.
14. Heise T, Hermanski L, Nosek L, Feldmann A, Rasmussen S, Haahr H. The pharmacodynamic variability of insulin degludec is consistently lower than insulin glargine over 24 hours at steady state. Diabetes. 2011;60(suppl 1):A263.-Poster 960-P.
Garber AJ, King AB, Del Prato S, et al. 15.NN1250-3582 (BEGIN BB T2D) Trial Investigators. Insulin degludec, an ultra-longacting basal insulin, versus insulin glargine in basal-bolus treatment with mealtime insulin aspart in type 2 diabetes (BEGIN Basal-Bolus Type 2): a phase 3, randomised, open-label, treat-to-target non-inferiority trial. Lancet. 2012;379(9825):1498-1507.
16. Heller S, Buse J, Fisher M, et al. BEGIN Basal-Bolus Type 1 Trial Investigators. Insulin degludec, an ultra-longacting basal insulin, versus insulin glargine in basal-bolus treatment with mealtime insulin aspart in type 1 diabetes (BEGIN Basal-Blus Type 1): a phase 3, randomised, open-label, treat-to-target non-inferiority trial. Lancet. 2012;379(9825):1489-1497.
17. Hirsch IB, Franek E, Courreges JP, Mersebach H, Dykiel P, Bode BW. Efficacy and safety of a new basal insulin with a bolus boost (IDegAsp) used once daily in combination wtih insulin apart (IAsp) in people wth type 1 diabetes. Diabetes. 2011;60(suppl 1):A292.-Poster 1064-P.
18. Meneghini L, Atkin SL, Bain S, et al. Flexible once-daily dosing of insulin degludec does not compromise glycemic control or safety compared to insulin glargine given once daily at the same time each day in people with type 2 diabetes. Paper presented at: American Diabetes Association 71st Scientific Sessions; June 24-28, 2011; San Diego, CA.
1. Rodbard HW, Jellinger PS, Davidson JA, et al. Statement by an American Association of Clinical Endocrinologists/American College of Endocrinology consensus panel on type 2 diabetes mellitus: an algorithm for glycemic control [published correction appears in Endocr Pract. 2009;15(7):768-770]. Endocr Pract. 2009;15(6):540-559.
2. Bartley PC, Bogoev M, Larsen J, Philotheou A. Long-term efficacy and safety of insulin detemir compared to Neutral Protamine Hagedorn insulin in patients with type 1 diabetes using a treat-to-target basal-bolus regimen with insulin aspart at meals: a 2-year, randomized, controlled trial. Diabet Med. 2008;25(4):442-449.
3. Riddle MC, Rosenstock J, Gerich J. Insulin Glargine 4002 Study Investigators. The treat-to-target trial: randomized addition of glargine or human NPH insulin to oral therapy of type 2 diabetic patients. Diabetes Care. 2003;26(11):3080—3086.
4. Hermansen K, Davies M, Derezinski T, Martinez Ravn G, Clauson P, Home P. A 26-week, randomized, parallel, treat-to-target trial comparing insulin detemir with NPH insulin as add-on therapy to oral glucose-lowering drugs in insulin-naive people with type 2 diabetes [published correction appears in Diabetes Care. 2007;30(4):1035] Diabetes Care. 2006;29(6):1269-1274.
5. Heise T, Nosek L, Rønn BB, et al. Lower within-subject variability of insulin detemir in comparison to NPH insulin and insulin glargine in people with type 1 diabetes. Diabetes. 2004;53(6):1614-1620.
6. Ashwell SG, Gebbie J, Home PD. Twice-daily compared with once-daily insulin glargine in people with type 1 diabetes using meal-time insulin aspart. Diabet Med. 2006;23(8):879-886.
7. Donnelly LA, Morris AD, Frier BM, et al. DARTS/MEMO Collaboration. Frequency and predictors of hypoglycaemia in type 1 and insulin-treated type 2 diabetes: a population-based study. Diabet Med. 2005;22(6):749-755.
8. Hermansen K, Dornhorst A, Sreenan S. Observational, open-label study of type 1 and type 2 diabetes patients switching from human insulin to insulin analogue basal-bolus regimens: insights from the PREDICTIVE study. Curr Med Res Opin. 2009;25(11):2601-2608.
9. Kalra S, Unnikrishnan AG, Baruah M, Kalra B. Degludec insulin: a novel basal insulin. Indian J Endocrinol Metab. 2011;15(suppl 1):S12-S16.
10. Jonassen I, Havelund S, Ribel U, et al. Insulin degludec: Multi-hexamer formation is the underlying basis for this new generation ultra-long acting basal insulin. Paper presented at: European Association for the Study of Diabetes Annual Meeting; September 20-24, 2010; Stockholm, Sweden.
11. Kurtzhals P, Heise T, Strauss HM, et al. Multi-hexamer formation is the underlying mechanism behind the ultra-long glucose-lowering effect of insulin degludec. Paper presented at: American Diabetes Association 71st Scientific Sessions; June 24-28, 2011; San Diego, CA.
12. Nosek L, Heise T, Bøttcher SG, Hastrup H, Haahr H. Ultra-long-acting insulin degludec has a flat and stable glucose-lowering effect. Paper presented at: American Diabetes Association 71st Scientific Sessions; June 24-28, 2011; San Diego, CA.
13. Heise T, Hövelmann U, Nosek L, Bøttcher SG, Granhall C, Haahr H. Insulin degludec has a two-fold longer half-life and a more consistent pharmacokinetic profile than insulin glargine. Paper presented at: American Diabetes Association 71st Scientific Sessions; June 24-28, 2011; San Diego, CA.
14. Heise T, Hermanski L, Nosek L, Feldmann A, Rasmussen S, Haahr H. The pharmacodynamic variability of insulin degludec is consistently lower than insulin glargine over 24 hours at steady state. Diabetes. 2011;60(suppl 1):A263.-Poster 960-P.
Garber AJ, King AB, Del Prato S, et al. 15.NN1250-3582 (BEGIN BB T2D) Trial Investigators. Insulin degludec, an ultra-longacting basal insulin, versus insulin glargine in basal-bolus treatment with mealtime insulin aspart in type 2 diabetes (BEGIN Basal-Bolus Type 2): a phase 3, randomised, open-label, treat-to-target non-inferiority trial. Lancet. 2012;379(9825):1498-1507.
16. Heller S, Buse J, Fisher M, et al. BEGIN Basal-Bolus Type 1 Trial Investigators. Insulin degludec, an ultra-longacting basal insulin, versus insulin glargine in basal-bolus treatment with mealtime insulin aspart in type 1 diabetes (BEGIN Basal-Blus Type 1): a phase 3, randomised, open-label, treat-to-target non-inferiority trial. Lancet. 2012;379(9825):1489-1497.
17. Hirsch IB, Franek E, Courreges JP, Mersebach H, Dykiel P, Bode BW. Efficacy and safety of a new basal insulin with a bolus boost (IDegAsp) used once daily in combination wtih insulin apart (IAsp) in people wth type 1 diabetes. Diabetes. 2011;60(suppl 1):A292.-Poster 1064-P.
18. Meneghini L, Atkin SL, Bain S, et al. Flexible once-daily dosing of insulin degludec does not compromise glycemic control or safety compared to insulin glargine given once daily at the same time each day in people with type 2 diabetes. Paper presented at: American Diabetes Association 71st Scientific Sessions; June 24-28, 2011; San Diego, CA.
Individualizing Insulin Therapy
The Evolution of Insulin Therapy in Diabetes Mellitus
The modern management of diabetes mellitus (DM) began with the discovery of insulin by Banting and Best in 1921 (see The Evolution of Insulin Therapy in Diabetes Mellitus in this supplement). Since that time, numerous additional classes of glucose-lowering agents have been introduced for the treatment of type 2 DM (T2DM). These medications primarily act by addressing 2 of the key defects of T2DM, insulin resistance and pancreatic β-cell dysfunction. T2DM is a progressive disease process that requires continued adjustment of therapy to maintain treatment goals. Most patients with T2DM will require insulin therapy at some point in their lives.
Role of Insulin in Type 2 Diabetes Mellitus Management
Consensus guidelines developed by the American Association of Clinical Endocrinologists/American College of Endocrinology (AACE/ACE) recommend initiating insulin when oral therapy fails to achieve glycemic control, A1C > 9.0% in treatment-naïve patients, or if the patient is symptomatic with glucose toxicity (polyuria, polydipsia, and weight loss) (FIGURE 1).1
Similar consensus guidelines developed by the American Diabetes Association/European Association for the Study of Diabetes (ADA/EASD) advise the initiation of glucose-lowering therapy for most patients with T2DM with the combination of lifestyle modifications, diet, and metformin (FIGURE 2).2 For patients who do not achieve or maintain glycemic control over 3 months, or thereabouts, with metformin, a second oral agent should be added. Alternatives include a glucagon-like peptide-1 receptor (GLP-1R) agonist or basal insulin. Insulin should be strongly considered as initial therapy for a patient with significant symptoms of hyperglycemia and/or plasma glucose >300-350 mg/dL or A1C ≥10.0%.
The major role of insulin in the management of patients with T2DM stems from several important attributes. First, insulin is the only treatment that works in patients with advanced β-cell deficiency. It acts directly on tissues to regulate glucose homeostasis, unlike other glucose-lowering agents that require the presence of sufficient endogenous insulin to exert their effects as insulin sensitizers, secretagogues, incretin mimetics, amylin analogs, and other factors. This also means that the mechanism of action of insulin is complementary to those of other glucose-lowering agents. Second, there is less of a ceiling effect with insulin. That is, increasing the dose of insulin results in a progressive lowering of blood glucose in the majority of patients, with the major limitation being the risk for hypoglycemia. Third, the glucose-lowering efficacy of insulin is durable, unlike that of other glucose-lowering agents that depend on endogenous insulin secretion for continued effectiveness. Fourth, insulin improves the lipid profile, particularly triglyceride levels.2-5 Fifth, regarding the long-term safety and tolerability of insulin, it is well established that weight gain, likely mediated via reduction of glycosuria, and hypoglycemia are typically the most concerning adverse events encountered. Allergic reactions, which were a more common complication of animal-sourced insulins, are infrequent with the insulin analogs.6-17 Finally, the availability of insulin in different formulations allows for targeting fasting plasma glucose or postprandial glucose, and individualization of therapy (see The Evolution of Insulin Therapy in Diabetes Mellitus in this supplement.)
While both the AACE/ACE and ADA/EASD consensus guidelines provide treatment “algorithms,” both make it clear that these are suggested approaches suitable for the population with T2DM (FIGURE 1, FIGURE 2). The specific treatment approach must be individualized based on patient-specific factors such as age, comorbid conditions, and tolerance of hypoglycemia.
FIGURE 1
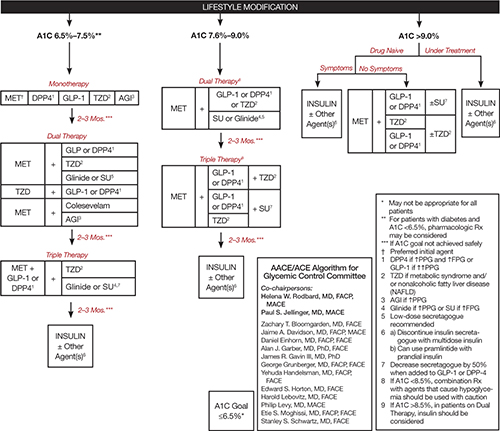
Role of insulin in the management of patients with type 2 diabetes mellitus according to the AACE/ACE1
AACE, American Association of Clinical Endocrinologists; ACE, American College of Endocrinology; AGI, α-glucosidase inhibitor; DPP4, dipeptidyl-peptidase-4 inhibitor; FPG, fasting plasma glucose; GLP-1, glucagon–like peptide-1 agonist; MET, metformin; PPG, postprandial glucose; SU, sulfonylurea; TZD, thiazolidinedione.
Reprinted from American Association of Clinical Endocrinologists. AACE/ACE Diabetes Algorithm for Glycemic Control. Available at https://www.aace.com/sites/default/files/GlycemicControlAlgorithmPPT.pdf. Accessed April 4, 2012, with permission from the American Association of Clinical Endocrinologists.
FIGURE 2
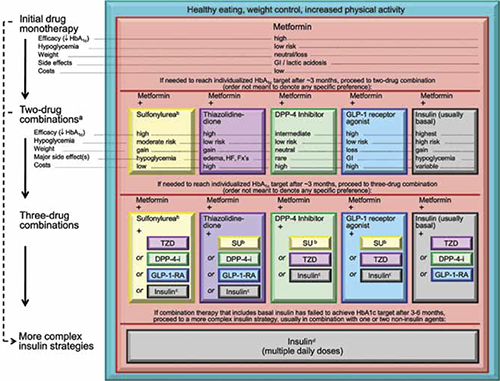
Role of insulin in the management of patients with type 2 diabetes mellitus according to the ADA/EASD2
Moving from the top to the bottom of the figure, potential sequences of antihyperglycemic therapy. In most patients, begin with lifestyle changes; metformin monotherapy is added at, or soon after, diagnosis (unless there are explicit contraindications). If the HbA1c target is not achieved after ~3 months, consider 1 of the 5 treatment options combined with metformin: an SU, TZD, DPP-4-i, GLP-1-RA, or basal insulin. (The order in the chart is determined by historical introduction and route of administration and is not meant to denote any specific preference.) Choice is based on patient and drug characteristics, with the over-riding goal of improving glycemic control while minimizing side effects. Shared decision making with the patient may help in the selection of therapeutic options. The figure displays drugs commonly used both in the United States and/or Europe. Rapid-acting secretagogues (meglitinides) may be used in place of SUs. Other drugs not shown (α-glucosidase inhibitors, colesevelam, dopamine agonists, pramlintide) may be used where available in selected patients but have modest efficacy and/or limiting side effects. In patients intolerant of, or with contraindications for, metformin, select initial drug from other classes depicted and proceed accordingly. In this circumstance, while published trials are generally lacking, it is reasonable to consider 3-drug combinations other than metformin. Insulin is likely to be more effective than most other agents as a third-line therapy, especially when HbA1c is very high (eg, ≥ 9.0%). The therapeutic regimen should include some basal insulin before moving to more complex insulin strategies. Dashed arrow line on the left-hand side of the figure denotes the option of a more rapid progression from a 2-drug combination directly to multiple daily insulin doses, in those patients with severe hyperglycemia (eg, HbA1c, ≥ 10.0–12.0%).
DPP-4, dipeptidyl peptidase-4; DPP-4-i, DPP-4 inhibitor; Fx’s, bone fractures; GI, gastrointestinal; GLP-1, glucagon-like peptide 1; GLP-1-RA, GLP-1 receptor agonist; HbA1c, hemoglobin A1c; HF, heart failure; NPH, neutral protamine Hagedorn; SU, sulfonylurea; TZD, thiazolidinedione.
aConsider beginning at this stage in patients with very high HbA1c (eg, ≥ 9%); bConsider rapid-acting, non-SU secretagogues (meglitinides) in patients with irregular meal schedules or who develop late postprandial hypoglycemia on SUs; cUsually a basal insulin (NPH, glargine, detemir) in combination with noninsulin agents; dCertain noninsulin agents may be continued with insulin. Consider beginning at this stage if patient presents with severe hyperglycemia (≥ 16.7–19.4 mmol/L [≥ 300–350 mg/dL]; HbA1c≥ 10.0–12.0%) with or without catabolic features (weight loss, ketosis, etc).
Diabetes Care by American Diabetes Association. Copyright 2012. Reproduced with permission of AMERICAN DIABETES ASSOCIATION in the format Journal via Copyright Clearance Center.
Individualizing Therapy
The importance of individualizing therapy in a way that allows patients with T2DM to effectively self-manage their disease cannot be overstated. A study involving 1381 patients with T2DM cared for by 42 primary care physicians was conducted to estimate the magnitude of effect that physicians have on glycemic control.18 Hierarchical linear modeling showed that physician-related factors were associated with a statistically significant but modest variability in A1C change (2%) for the entire patient group. On the face of it, this finding might be discouraging. Further analysis showed, however, that for patients whose A1C did improve, physician-related factors accounted for 5% of the overall change in A1C (P = .005). On the other hand, physician-related factors had no impact on patients whose A1C did not improve or worsened. These results support the role that physicians play in affecting patient outcomes. The results also make it clear that without a physician’s influence, a patient’s glycemic outcomes may be difficult to change. The question is: How best can a physician influence patient outcomes?
A 2011 survey of patients with DM, general practitioners, and DM specialists reported that clinicians tended to underestimate patients’ perceived seriousness of the disease, while overestimating patients’ level of distress. In addition, physicians had difficulty identifying which DM-related complications concerned patients most and the information and support patients needed to feel more at ease with DM. Patients placed greater importance on having easy access to their physicians rather than more time with them. But most importantly, the survey investigators concluded that patients generally wished for greater involvement in decision making and being provided more information.19 These findings suggest that patients understand that T2DM is a largely self-managed, chronic disease, and want a collaborative relationship with their physician.
Patient Barriers to Insulin Therapy
Numerous factors have been identified as impeding patients’ willingness to initiate insulin therapy (TABLE 1).20-24 Barriers often vary from patient to patient and, in fact, may change over time in an individual patient. It is crucial, therefore, to identify the root reasons for a patient’s apprehension with insulin when talking about options for intensifying treatment. Once insulin has been initiated, the patient should be asked about continuing or new concerns regarding insulin therapy (and DM management in general), including adherence.
TABLE 1
Barriers to insulin therapy identified by patients20-24
| Lack of understanding of serious nature of type 2 diabetes mellitus |
| Fear of addiction to insulin |
| Fear of hypoglycemia |
| Concern about weight gain |
| Repeated experiences of failing to achieve satisfactory glycemic control |
| Perception that quality of previous treatment was low |
| Needle phobia |
| Treatment complexity |
| Concern of social stigmatization |
| Perceived failure and low self-efficacy |
| Belief of becoming more ill |
| Out-of-pocket cost |
| Perceived negative impact on quality of life |
| Comorbidities such as poor eyesight, arthritis, forgetfulness |
A recent, international survey of 1400 patients with insulin-naïve T2DM reported that 3 negative beliefs about insulin were prominent: (1) feeling that the disease was worsening; (2) fear of injection; and (3) a feeling of personal failure.20 Certain patient comorbidities, such as poor eyesight, arthritis, and forgetfulness, might also serve as barriers to self-management of DM with insulin. Additional comorbidities may contribute as indirect barriers, such as the need for polypharmacy, which may make the initiation of additional treatments such as insulin logistically or financially difficult.
It is possible that the discussion about initiating insulin may uncover patient concerns about T2DM in general. The Diabetes Attitudes, Wishes, and Needs (DAWN) study reported that psychosocial issues were the major source of difficulty in patient self-management (TABLE 2).25 In fact, 85% of people who reported a high level of distress at the time of diagnosis of T2DM continued to experience psychological distress at a mean follow-up of 15 years.
TABLE 2
Patients experiencing various aspects of diabetes-related distress25
| Diabetes-related distress | Respondents who agree (%) |
|---|---|
| I feel stressed because of my diabetes. | 32.7 |
| I feel burned out because of my diabetes. | 18.1 |
| I feel that diabetes is preventing me from doing what I want to do. | 35.9 |
| I am constantly afraid of my diabetes getting worse. | 43.8 |
| I worry about not being able to carry out my family responsibilities in the future. | 30.1 |
| My diabetes causes me worries about my financial future. | 25.8 |
| My family and friends put too much pressure on me about my diabetes. | 14.7 |
| The community I live in is intolerant of diabetes. | 13.6 |
| Diabetes Care by American Diabetes Association. Copyright 2012. Reproduced with permission of AMERICAN DIABETES ASSOCIATION in the format Journal via Copyright Clearance Center. | |
Addressing psychosocial issues and other barriers is crucial in the discussion of self-management because those with more negative feelings about starting insulin are most unwilling to start insulin.20 One factor that may contribute to these negative feelings is repeated experiences of failing to achieve satisfactory glycemic control with oral glucose-lowering agents.23 Conversely, those who have experienced improved glycemic control with intensification of prior glucose-lowering therapy may be more accepting of initiating insulin therapy.23,26 These findings are a reminder of the importance of a treat-to-target approach to management, in which the target glycemic goal, generally A1C < 7.0%, is achieved within 2 to 3 months of diagnosis and maintained at that level through intensification of therapy as needed.
Addressing psychosocial issues can be a challenge in today’s busy primary care practice due to limited time and lack of training in the management of such issues. However, implementation of various strategies has been reported to facilitate and, in some cases, shorten a patient’s visit. For example, one small study reported that visits were shorter if the physician acknowledged and responded positively to a patient’s stated or implied concerns (17.6 minutes vs 20.1 minutes).27 Missing or ignoring the patient’s concerns often led the patient to bring up the same concern one or more additional times resulting in a longer office visit. These results underscore the importance of asking patients to identify their concerns or questions at the beginning of the office visit. The patient can fill out a questionnaire in the waiting room or be encouraged to write down and prioritize their questions and concerns specific to the visit. If the patient identifies more concerns or questions than can be reasonably addressed in one visit, there should be agreement to address the most pressing ones during the current visit and the remaining concerns and questions during the next visit. This “agenda-setting” approach has been reported to offer several advantages.28 From the patient’s perspective, the quality of the physician-patient interaction was much improved, in part because physicians took time to explain points in a way that was easy to understand. Advantages to the physician with an agenda-setting approach included “feeling more in control,” “less stressed by simply knowing what was on the patient’s mind,” “feeling less rushed,” and “enjoying patient encounters more.” Contrary to the study cited above, physicians found that patients’ visits often were longer, especially those of older patients. One physician, however, noted that the visit “takes more time now, but saves time later.” As noted in this study, additional time spent with the patient can lead to improved job satisfaction for the physician.29
The agenda-setting approach requires that the physician ask the patient to list his or her concerns and questions, and then actively listen to the patient. Once the agenda for the visit is established, employing the “ask, listen, empathize” communication style can lead to effective physician-patient communication and problem-solving. Using this approach, the physician asks questions to gain a clear understanding of the patient’s concerns and then uses active listening with little, if any, interruption.30,31 Since the goal is to solve problems with rather than for the patient, active listening without offering opinions, judgements, or advice while offering empathy is essential. Through reflection and discussion, the physician can help the patient to identify his or her issues and acceptable solutions.
The importance of good communication between physician and patient cannot be overstated. Additional communication skills to keep in mind are: (1) speak slowly using nonmedical language; (2) limit the amount of information and repeat it; (3) draw pictures and/or use visual aids; and (4) ask the patient to repeat instructions and key concepts. In addition to enhancing patients’ understanding, visual images may be particularly beneficial in keeping patients motivated to improve self-management, including adherence to therapy. For example, it may be helpful to graphically track the patient’s glycemic progress. This can be done by establishing an actionable A1C goal (generally < 7.0%) and a time frame to achieve the goal (eg, 2 to 3 months).32 A graph can be constructed beginning with the patient’s current, preinsulin A1C level, with updates at each visit. In addition to motivating the patient and reinforcing adherence, the graph can also be used to demonstrate when further treatment intensification is needed. Additional general strategies that can be employed when considering the initiation of insulin are shown in TABLE 3. Implementation of strategies such as these by family physicians provides patient outcomes comparable to those implemented by endocrinologists or diabetes specialists.33
TABLE 3
General strategies for initiating insulin therapy
| Invite the patient to take an active role in treatment decisions. |
| Remind the patient that type 2 diabetes is primarily self-managed. |
| Discuss the progressive nature of β-cell dysfunction in type 2 diabetes. |
| Emphasize the physiologic role of insulin to maintain glucose homeostasis. |
| Discuss that insulin will help to achieve glycemic control and minimize the risk for long-term complications. |
| Discuss that treatment will be modified as needed to maintain glycemic control and to best meet their needs, capabilities, and interest. |
| Utilize insulin pen devices whenever possible. |
| Emphasize the importance of lifestyle management. |
| Ask if hearing other patients talk of their experiences with insulin therapy would be helpful; consider a group office visit. |
| Discuss and provide the patient with an individualized, written action plan that includes insulin dosing, self-monitoring of blood glucose, and signs/symptoms of hypoglycemia and other adverse events with appropriate action(s) to take. |
| Simplify diabetes (and comorbidities) treatment whenever possible. |
The remainder of this article uses case studies to further explore various patient barriers to insulin therapy and strategies for addressing them with the patient. While other therapies may be appropriate in the case studies below as recommended by current guidelines, these case studies will focus on insulin. In addition, dosing strategies for initiating and intensifying insulin therapy are discussed. Changes to the treatment plan to adjust for comorbidities, such as hypertension and dyslipidemia, or for smoking cessation or aspirin therapy, are not addressed in these cases, but are crucial components of comprehensive management.
CASE STUDY 1
RF is a 49-year-old female insurance analyst diagnosed with T2DM 6 years ago. Initial therapy with lifestyle modifications and metformin has since been intensified. Glimepiride was added, then pioglitazone was added 1.5 years ago when the A1C had risen to 7.5%. There is no evidence of cardiovascular disease. She reports bothersome lower extremity edema and an 8-pound weight increase since starting pioglitazone treatment. RF states that she takes her medications every day, although she acknowledges that she sometimes forgets on Sundays.
Clinical Impression
After taking her history, performing a physical examination, and reviewing her laboratory and self-monitored blood glucose (SMBG) data, her physician concludes that her treatment plan needs to be changed (TABLE 4, TABLE 5).
TABLE 4
Case study 1: Chart notes
| Physical examination | Laboratory tests | Lifestyle habits | Current therapy | |
|---|---|---|---|---|
| Glucose-lowering | Other | |||
| BP: 126/80 mm Hg Weight: 176 lb (79.2 kg) BMI: 27 kg/m2 Eyes: no retinopathy Neurology: intact Skin: intact | SCr: 1.4 mg/dL Albuminuria: negative A1C: 8.2% Cholesterol: Total: 204 mg/dL LDL: 134 mg/dL HDL: 36 mg/dL | Exercise: Walks 2 miles 3-4 d/wk Nutrition: eats 3-4 meals/d | Metformin 1000 mg BID Glimepiride 8 mg QD Pioglitazone 45 mg QD | Lisinopril 30 mg QD Simvastatin 40 mg QD ASA 80 mg QD |
| ASA, acetylsalicylic acid; BMI, body mass index; BP, blood pressure; HDL, high-density lipoprotein; LDL, low-density lipoprotein; SCr, serum creatinine. | ||||
TABLE 5
Case study 1: Self-monitored blood glucose (mg/dL) over the previous 2 weeks
| Day | Fasting | 2 h Post-breakfast | 2 h Post-lunch | 2 h Post-dinner |
|---|---|---|---|---|
| Wednesday | 205 | |||
| Thursday | 158 | |||
| Friday | 179 | |||
| Saturday | 201 | 162 | ||
| Sunday | ||||
| Monday | 166 | |||
| Tuesday | 189 | |||
| Wednesday | ||||
| Thursday | 153 | 221 | ||
| Friday | 150 | |||
| Saturday | 199 | 186 | 213 | |
| Sunday | ||||
| Monday | 181 | |||
| Tuesday | 167 |
Treatment Plan
- Initiate basal insulin once daily in the evening.
- Continue glimepiride, but reduce pioglitazone to 15 mg once daily (or discontinue if cost is a concern).
- Ask RF to monitor fasting blood glucose and self-adjust insulin doses as appropriate.
Barriers
While discussing the need to change the treatment plan and the physician’s suggestion that RF begin basal insulin, RF asks her physician for another few months on her current regimen stating that she will try harder to take her medications on Sundays. She also voices concern that insulin treatment requires injections and that she is concerned about what her coworkers and friends might think. The physician confirms that these concerns are understandable; he also confirms that RF is fearful of needles. The following are possible responses that RF’s physician could use to address these concerns.
Patient’s concern: Perceived failure/low self-efficacy
Physician responses:
- We all forget to do things from time to time, but overall I think you have done a great job taking your medications.
- As we have talked about before, with T2DM there is a progressive loss of insulin production over time regardless of what you do. That is why we added glimepiride and then pioglita-zone and that is why we need to make a change now and put you back in control of your diabetes. It is likely that further changes will be needed and we can discuss and agree on them together.
Patient’s concern: Social stigmatization
Physician responses:
- We can begin by having you administer insulin once daily in the evening in the privacy of your home.
- The insulin can be administered with a device that looks like a pen. It is small and can be carried in your purse; it does not need to be refrigerated once opened. If the time comes that you will need to administer a dose of insulin during the day, you can easily administer the insulin discretely in a public restroom or your work area.
- The use of insulin is more common than it was even a few years ago. In fact, about 5 million people in the United States use insulin to replace what is missing, control blood sugars, and decrease the risk for diabetes complications.34
Patient’s concern: Fear of needles
Physician’s responses:
- Insulin can be injected using a pen device with short, ultrathin needles that makes most of the injections painless. I would like you to see how simple and painless the injection can be by using this sample pen here in the office.
- Many patients are concerned about giving themselves an injection at first, but they quickly become comfortable doing so.
Dosing
Treatment with basal insulin can be initiated using one of several approaches. Using the treat-to-target approach, basal insulin 10 U once daily is initiated.35 The starting dose should be reduced to 6 U if the initial pre-breakfast or pre-dinner blood glucose is < 126 mg/dL or the patient’s body mass index (BMI) is < 26 kg/m2.36 Alternatively, the ADA/EASD recommends starting with 0.2 U/kg, which may be more practical in very overweight or obese patients.2 Titration of the basal insulin dose can be accomplished using one of the following physician-directed or patient-driven treat-to-target titration algorithms (TABLE 6).35,37,38 The insulin dose should be titrated based on the pre-breakfast fasting blood glucose level.
TABLE 6
Physician-directed or patient-driven treat-to-target titration algorithms
| Riddle et al35 | Davies et al37 | Meneghini et al38 | ||||
|---|---|---|---|---|---|---|
| Start with 10 U/d bedtime basal insulin and adjust weekly | Start with 10* U/d bedtime basal insulin and adjust weekly (physician-directed) Or Start with a dose numerically equivalent to the highest FPG (in millimoles/L)† over the previous 7 days and adjust every 3 days (patient-managed) | Start with basal insulin once daily and adjust every 3 days | ||||
| Mean of self-monitored FPG values from preceding 2 days | Change in insulin dose (U/d)# | Mean of self-monitored FPG values from preceding 3 days | Change in insulin dose (U/d) (physician-directed) | Change in insulin dose (U/d) (patient-managed) | Mean of self-monitored FPG values from preceding 3 days | Change in insulin dose (U/d) |
| ≥180 mg/dL 140-180 mg/dL 120-140 mg/dL 100-120 mg/dL | +8 +6 +4 +2 | ≥180 mg/dL (≥10 mmol/L) 140-179 mg/dL (7.8-9.9 mmol/L) 120-139 mg/dL (6.7 – 7.7 mmol/L) 100-119 mg/dL (5.5-6.6 mmol/L) | +6 to +8 +4 +2 0 to +2 | +2 +2 +2 0 to +2 | >110 mg/dL 80-110 mg/dL <80 mg/dL | +3 0 -3 |
| FPG, fasting plasma glucose. *In insulin-naive patients. †For example, if the highest FPG over the previous 7 days was 7 mmol/L, start with 7 U.#Small insulin dose decreases (2-4 U/d per adjustment) were allowed if severe hypoglycemia (requiring assistance) or plasma-referenced glucose < 56 mg/dL was documented in the preceding week. Reproduced with permission. Meneghini LF, et al. J Fam Pract. 2011;60(9 Suppl 1):S21-S28. Quadrant HealthCom Inc. Copyright 2011. | ||||||
Follow-Up Visit
RF begins basal insulin 10 U in the evening and is given simple instructions for insulin dose titration based on fasting plasma glucose results. At her follow-up visit, RF reports that she has increased her basal insulin to 18 U administered once daily. Review of her SMBG results show that her blood glucose levels throughout the day have improved, but are still not at goal. RF’s physician commends her on the progress she has made. RF and her physician agree that she should continue to increase her basal insulin dose. Eight months after beginning basal insulin, RF is administering 28 U (0.35 U/kg) of basal insulin in the evening. Review of her SMBG results over the previous 2 weeks show that her blood glucose rises during the day and is highest after dinner; her current A1C is 7.2%.
Treatment Plan
- Discuss dietary and lifestyle complements to insulin therapy such as:
- Use SMBG to identify foods that raise her blood glucose.
CASE STUDY 2
LW is a 64-year-old male with longstanding hypertension diagnosed with T2DM 8 years ago for which he was treated initially with lifestyle management and metformin. He has since been treated with other oral agents as add-on therapy; glipizide was discontinued due to hypoglycemia when he skips meals (usually lunch); pioglitazone was discontinued after the patient expressed concerns about the risk for bladder cancer he heard on television. He has mild retinopathy and mild loss of vibration sensation in the feet; there is no evidence of cardiovascular disease. He was diagnosed with osteoarthritis 3 years ago.
Clinical Impression
After taking his history, performing a physical examination, and reviewing his laboratory and SMBG data, his physician concludes that his treatment plan needs to be changed (FIGURE 3, TABLE 7, TABLE 8).
TABLE 7
Case study 2: Chart notes
| Physical examination | Laboratory tests | Lifestyle habits | Current therapy | |
|---|---|---|---|---|
| Glucose-lowering | Other | |||
| BP: 124/76 mm Hg Weight: 204 lb (92.7 kg) BMI: 31 kg/m2 Eyes: mild retinopathy Neurology: occasional tingling on bottom of right foot Skin: intact | SCr: 1.9 mg/dL eGFR: 51 mL/min Albuminuria: negative A1C: 8.1% Cholesterol: Total: 218 mg/dL LDL: 118 mg/dL HDL: 55 mg/dL Triglyceride: 204 mg/dL | Exercise: takes dog on occasional walk but otherwise sedentary Nutrition: eats 4 meals/d | Metformin 1000 mg BID Acarbose 50 mg TID Sitagliptin 100 mg QD | Lisinopril/HCTZ 20/25 mg QD Amlodipine 10 mg QD Acetaminophen extended-release 650 mg TID ASA 80 mg QD |
| ASA, acetylsalicylic acid; BMI, body mass index; BP, blood pressure; eGFR, estimated glomerular filtration rate; HCTZ, hydrochlorothiazide; HDL, high-density lipoprotein; LDL, low-density lipoprotein; SCr, serum creatinine. | ||||
TABLE 8
Case study 2: Self-monitored blood glucose (mg/dL) over the previous 2 weeks
| Day | Fasting | 2 h Post-breakfast | 2 h Post-lunch | 2 h Post-dinner |
|---|---|---|---|---|
| Tuesday | 135 | |||
| Wednesday | ||||
| Thursday | 196 | |||
| Friday | 152 | 174 | ||
| Saturday | ||||
| Sunday | 208 | |||
| Monday | 142 | 193 | ||
| Tuesday | ||||
| Wednesday | 130 | 156 | ||
| Thursday | ||||
| Friday | ||||
| Saturday | ||||
| Sunday | 151 | |||
| Monday |
FIGURE 3
Case study 2: A1C levels for April 2004 to March 2012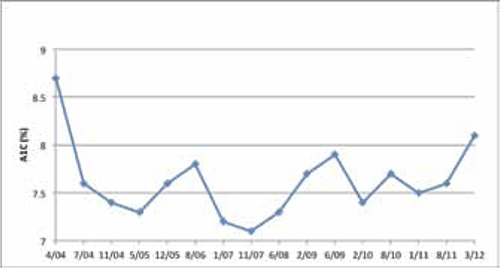
Treatment Plan
- Discontinue metformin since LW’s serum creatinine is > 1.5 mg/dL.
- Initiate either basal insulin once daily in the evening or premix insulin at dinner.
- Ask LW to monitor his blood glucose and self-adjust insulin doses as appropriate.
- Stress the importance of exercise and proper nutrition; gain agreement on short-term goals for exercise and nutrition.
Barriers
LW’s physician recommends that his treatment plan be changed and insulin therapy initiated. LW quickly responds that previous changes to his treatment regimen have not resulted in his achieving an A1C < 7.0%. He also doubts that he can use a syringe to draw up the correct dose and then self-administer due to his arthritis. The following are possible responses his physician could use to address these concerns.
Patient concern: Repeated experience of failing to achieve glycemic control, ie, A1C < 7.0%
Physician responses:
- While achieving an A1C < 7.0% is a realistic goal that reduces the risks for vascular complications of diabetes, any reduction of A1C will be of benefit.
- I would like to work with you to implement a new plan that we both believe will enable you to improve your diabetes control and ideally achieve an A1C < 7.0%.
Patient concern: Self-administering due to arthritis
Physician responses:
- Instead of using a syringe and vial to draw up and administer insulin, I would like you to use an insulin pen device. As you can see, it is easy to handle and you can easily select the correct dose.
- If you choose to start on premix insulin, the pen device contains both types of insulin together in one dose.
Dosing
Treatment with basal insulin once daily in the evening can be initiated and titrated based on pre-breakfast blood glucose as in Case Study 1. Alternatively, treatment with premix insulin can be initiated at a dose of 12 U administered within 15 minutes of dinner initiation. The premix dose can be titrated using the algorithm employed in the 1-2-3 Study based on pre-breakfast blood glucose (TABLE 9).39 After 16 weeks, 41% of patients in the 1-2-3 Study achieved an A1C < 7.0% from a baseline A1C of 8.6%.
TABLE 9
1-2-3 Study algorithm39
| Pre-breakfast SMBG (mg/dL) | Adjustment of pre-dinner dose (U) |
|---|---|
| <80 | -3 |
| 80-110 | No change |
| 111-140 | +3 |
| 141-180 | +6 |
| > 180 | +9 |
| SMBG, self-monitored blood glucose. | |
Follow-Up Visit
LW began basal insulin 10 U in the evening. Over the next 5.5 months, he titrated his dose such that his current dose is 46 U (0.50 U/kg) in the evening. His current A1C is 7.3%. Review of his SMBG shows consistently high 2-hour post-lunch blood glucose levels. Although further increasing his basal insulin dose is an option, in most of the treat-to-target studies, the daily dose of basal insulin given once daily averaged between 0.4 and 0.6 U/kg.35,37,40,41 LW and his physician agree that adding rapid-acting insulin at lunch is the best option. The starting dose of rapid-acting bolus insulin is 4 to 6 U administered prior to the largest meal of the day or, as in this case, prior to the meal with the largest postprandial blood glucose excursion.42,43 Alternatively, the dose of rapid-acting insulin could be calculated as 10% of the total daily dose of basal insulin, which in this case is 5 U (10% x 46 U). The dose of basal insulin would be reduced by 5 U if the rapid-acting insulin is given at dinner in order to reduce the risk for nocturnal hypoglycemia. The dose of the bolus insulin can be titrated using the ExtraSTEP algorithm (TABLE 10).42 Alternatively, the SimpleSTEP algorithm can be used which does not require a 2-hour postprandial glucose measurement.42
TABLE 10
Algorithms for adjusting insulin aspart42
| ExtraSTEP algorithm | SimpleSTEP algorithm | |||
|---|---|---|---|---|
| 2-h Post-meal PG level (mg/dL) | Insulin aspart adjustment (U) | Pre-meal BG (mg/dL) | Bedtime BG (mg/dL) | Insulin aspart adjustment (U) |
| <72* | -2 | <72* | <72* | -2 |
| 72-144 | 0 | 72-108 | 72-144 | 0 |
| 145-180 | +2 | 109-162 | 145-180 | +2 |
| >180 | +4 | >162 | >180 | +4 |
| BG, blood glucose; PG, plasma glucose. *One or more PG values <72 mg/dL without obvious explanation. Reproduced with permission. Meneghini LF, et al. J Fam Pract. 2011;60(9 Suppl 1):S21-S28. Quadrant HealthCom Inc. Copyright 2011. | ||||
Plan
- Begin rapid-acting insulin 5 U at lunch.
- Continue basal insulin at 46 U in the evening.
- Ask LW to continue to titrate basal insulin based on the pre-breakfast blood glucose level and the lunch time bolus insulin dose based on the 2-hour post-lunch SMBG (ExtraSTEP); alternatively, adjust based on the pre-dinner blood glucose level (SimpleSTEP).
CASE STUDY 3
MB is a 46-year-old male who had not consulted a physician since having a physical examination 6 years ago. He presented 2 weeks ago with frequent urination (7-8 times/day) and feeling tired; he also noted losing 5 pounds (2.25 kg) over the preceding 3.5 months despite no changes in his diet. MB is a regional salesperson with an erratic schedule. During the week, he eats lunch and most dinners in a restaurant. On the weekend, he goes to a local bar with his friends. He does light yard work, but does not exercise regularly. He is a current smoker with a 36 pack-year history. Urinalysis shows ketonuria and microalbuminuria. His A1C reported back today is 10.8%, confirming a diagnosis of uncontrolled and symptomatic DM.
Clinical Impression
After taking his history, performing a physical examination, and reviewing his laboratory data, MB’s physician confirms a diagnosis of DM (TABLE 11). While it is likely that MB has T2DM, his physician wants to rule out type 1 DM and latent autoimmune diabetes of the adult (LADA), so he orders tests for antibodies (GAD, IA-2, ICA). The antibody testing is negative, making T2DM the most likely diagnosis.
TABLE 11
Case study 3: Chart notes
| Physical examination | Laboratory tests | Lifestyle habits | Current therapy | |
|---|---|---|---|---|
| Glucose-lowering | Other | |||
| BP: 142/88 mm Hg Weight: 176 lb (79.2 kg) BMI: 27 kg/m2 Eyes: no retinopathy Neurology: intact Skin: intact | SCr: 1.4 mg/dL Microalbumin:creatinine ratio: 140 mg/g creatinine Ketonuria: 1+ A1C: 10.8% Cholesterol: Total: 210 mg/dL LDL: 146 mg/dL HDL: 30 mg/dL | Exercise: light yard work, no regular exercise Nutrition: 3 meals/d, eats most meals in a restaurant (lunch M-F; dinner 3-4 nights/wk) | None | None |
| BMI, body mass index; BP, blood pressure; HDL, high-density lipoprotein; LDL, low-density lipoprotein; SCr, serum creatinine. | ||||
Treatment Plan
- Initiate basal-bolus therapy with fixed bolus doses of rapid-acting insulin at each meal (prandial insulin).
- Ask MB to monitor blood glucose before meals and at bedtime.
- Provide MB with a supplemental scale to correct hyperglycemia before meals.
- Stress the importance of exercise and proper nutrition; gain agreement for short-term goals for exercise and nutrition; refer for diabetes and nutrition education if available.
- Discuss the importance of smoking cessation; develop a plan.
- Consider metformin and other non-insulin therapies when A1C is under control.
Barriers
MB is surprised that he has T2DM and is clearly anxious at receiving the diagnosis. He expresses concern about starting insulin because his uncle died within a year of starting insulin. MB also recalls that his uncle was always giving himself shots and monitoring his blood glucose level. He wants to know whether there is a simpler treatment option if he agrees to start insulin treatment. He also wants to know whether he will have to remain on insulin for the rest of his life. The following are possible responses his physician could use to address these concerns.
Patient concern: Fear of death
Physician responses:
- Uncontrolled high blood sugars over a long period of time can cause serious complications, such as kidney and heart disease that can result in death. That is why it is important that we work together to gain control of your blood sugar levels over the next few months and then modify your treatment as needed to maintain control.
- Unfortunately for many patients in the past, treatment with insulin was not used until it was too late and people already had serious complications from DM. This is likely the case for your uncle.
Patient concern: Treatment complexity
Physician responses:
- Right now we have to control your blood glucose rapidly so your pancreas can regain some function and your body can better respond to insulin.
- I will also provide you with step-by-step written instructions you can follow that describe how to start insulin and how to monitor your blood glucose.
- We will communicate as often as you need to adjust your insulin doses over the next few weeks; when you feel comfortable, I can even show you how to adjust your insulin dose before a meal to correct a high blood sugar.
- We can try this treatment for 3 months and then reevaluate your response, how you feel, and whether you want to continue to modify your treatment plan to keep your blood sugars controlled.
Patient concern: Lack of understanding that T2DM is a serious disease
Physician responses:
- Please understand that T2DM is a serious disease that increases your risk for heart disease, stroke, blindness, and other diseases. Unfortunately, since diabetes does not cause bad symptoms until it is actually too late, many patients do not make the effort to properly control their diabetes. By working together, we can reduce the risk for these complications and do some screening tests to detect any complications before they become irreversible.
Dosing
There are several approaches to determining the initial doses of basal and prandial (bolus) insulin. One approach is to estimate the total daily dose (TDD) of insulin by multiplying the patient’s weight in kilograms by 0.5 U/kg/d.44 Half of the TDD is given as basal insulin replacement; the other half is divided into 3 fixed preprandial doses of rapid-acting insulin. When the patient is ready to take on more complex management, the supplemental dose for bolus insulin can be calculated using a correction factor. If the bolus insulin is a rapid-acting insulin analog, 1800 is divided by the TDD of insulin; 1500 is used for a short-acting human insulin. This correction factor is an estimate of the fall in blood glucose per unit of bolus insulin. In our patient, the TDD would be: 80 kg x 0.5 U/kg/d or 40 U/d of insulin. Thus, 1 U of insulin should lower the blood glucose by about 45 mg/dL (1800/40 U = 45 mg/dL). For every 45 mg/dL above the pre-meal target, the patient would add 1 U of rapid-acting insulin to correct the hyperglycemia over the next 4 to 5 hours. The basal and prandial insulin doses would be titrated on a periodic basis (perhaps every 1 to 2 weeks) until the daytime levels of blood glucose are on target. The fasting (pre-breakfast) blood glucose would be used to adjust the basal insulin dose, while the pre-lunch, pre-dinner, and bedtime blood glucose results would be used to adjust the pre-breakfast, pre-lunch, and pre-dinner prandial (rapid-acting) insulin doses, respectively.
An alternative approach to initiating basal-bolus therapy is the PREFER algorithm.45 Here, the basal insulin dose is 10 U initially. The bolus doses are administered in a 3:1:2 ratio, so if the total of the 3 bolus doses is 12 U/d, the initial bolus doses would be 6 (breakfast), 2 (lunch), and 4 (dinner) U. The mean basal (once-daily) and bolus insulin doses observed in PREFER are shown in TABLE 12 and TABLE 13.
TABLE 12
Case study 3: Calculating initial basal-bolus insulin doses
| Algorithm | Calculations | Patient MB |
|---|---|---|
| Meneghini44 | TDD = (total body weight [kg]) (0.5 U/kg/d) Basal insulin dose* = (50%) (TDD) Bolus insulin dose† = (10%-20%) (TDD) | TDD = (0.5 U/kg/d)(80kg) = 40 U/d |
| Basal = (50%) (40 U/d) = 20 U/d | ||
| Bolus = (10%-20%) (40 U/d) = 4 to 8 U/meal | ||
| CF = 1800/40 U/d = 45 mg/dL per 1 unit | ||
| PREFER45 | Basal insulin dose* = 10 U (14 U if BMI > 32 kg/m2) Bolus insulin dose† = ratio of 3:1:2 (breakfast:lunch:dinner) Note: At week 26, the bolus insulin doses were divided into the 3 daily meals in approximately a 1:1:1 ratio | |
| BMI, body mass index; CF, correction factor; TDD, total daily dose of insulin. *Once daily; †Three meals per day. | ||
TABLE 13
Titrating the basal insulin dose using the PREFER algorithm45
| Pre-breakfast blood glucose (mg/dL) | Basal insulin dose adjustment (U) |
|---|---|
| < 56 | -4 |
| 56-72 | -2 |
| 73-125 | No change |
| 126-140 | +2 |
| 141-160 | +4 |
| 161-180 | +6 |
| 181-200 | +8 |
| > 200 | +10 |
Follow-up Visit
MB begins with basal insulin 20 U in the evening and bolus insulin at doses of 7 U before each meal. Over the next several months, MB has titrated his insulin doses; his current doses are: 32 U (basal), 11 U (bolus-breakfast), 7 U (bolus-lunch), and 10 U (bolus-dinner). He experienced 1 episode of mild hypoglycemia (SMBG, 50 mg/dL) one afternoon following a particularly active morning (TABLE 14). His current A1C is 7.4%. MB’s physician congratulates him on the progress he has made in dramatically lowering his blood glucose level—and his risk for diabetes-related complications. While MB appreciates his physician’s support and admits that he does not feel tired and generally feels better, which is likely due to resolution of glucotoxicity, he is not happy that he has gained 5.5 pounds (2.5 kg).46 He also finds the timing and administration of bolus insulin difficult.
TABLE 14
Case study 3: Self-monitored blood glucose (mg/dL) over the previous 2 weeks
| Day | Fasting | 2 h Post-breakfast | 2 h Post-lunch | 2 h Post-dinner |
|---|---|---|---|---|
| Wednesday | ||||
| Thursday | 168 | |||
| Friday | 106 | 166 | 174 | |
| Saturday | 88 | |||
| Sunday | 195 | |||
| Monday | 134 | |||
| Tuesday | 172 | |||
| Wednesday | 130 | 156 | ||
| Thursday | 112 | 168 | ||
| Friday | 92 | 164 | ||
| Saturday | 50 | 149 | 159 | 176 |
| Sunday | 94 | 174 | 210 | |
| Monday | 176 | 184 | ||
| Tuesday | 117 | 169 |
Plan
- Continue basal insulin once-daily in the evening.
- Add metformin 500 mg BID and increase to 1000 mg BID as tolerated.
- Consider weaning down the bolus insulin doses and substituting them with a GLP-1R agonist, dipeptidyl peptidase-4 inhibitor, or short-acting secretagogue. If so, continue rapid-acting insulin during transition. [Note: the following combinations are not currently approved by the US FDA: exenatide twice-daily and prandial insulin; exenatide once-weekly and insulin; liraglutide and prandial insulin; linagliptin and insulin.]
CASE STUDY 4
KW is a 62-year-old female diagnosed with T2DM 12 years ago. Treatment with lifestyle management and metformin initially provided glycemic control. Glimepiride was subsequently added and eventually the patient was started on basal insulin. The current dose of basal insulin is 60 U in the evening. Five months ago her A1C was found to be 7.9% and more recently 8.3%. She drinks alcohol occasionally and smokes. KW works as an executive secretary and has a consistent meal and activity schedule.
Clinical Impression
Following completion of the history, physical examination, and review of her laboratory data, KW’s physician concludes that her insulin regimen should be intensified (TABLE 15, TABLE 16).
TABLE 15
Case study 4: Chart notes
| Physical examination | Laboratory tests | Lifestyle habits | Current therapy | |
|---|---|---|---|---|
| Glucose-lowering | Other | |||
| BP: 126/78 mm Hg Weight: 176 lb (79.2 kg) BMI: 32 kg/m2 Eyes: no retinopathy Neurology: intact Skin: intact | SCr: 1.0 mg/dL Albuminuria: negative A1C: 8.3% Cholesterol Total: 172 mg/dL LDL: 96 mg/dL HDL: 46 mg/dL Triglycerides: 138 mg/dL | Exercise: sedentary Nutrition: 3 meals/d with large dinner | Metformin 1000 mg BID Basal insulin 60 U in the evening | ASA 80 mg QD Pravastatin 40 mg qHS |
| BMI, body mass index; BP, blood pressure; HDL, high-density lipoprotein; LDL, low-density lipoprotein; SCr, serum creatinine. | ||||
TABLE 16
Case study 4: Self-monitored blood glucose (mg/dL) over the previous 2 weeks
| Day | Fasting | 2 h Post-breakfast | 2 h Post-lunch | 2 h Post-dinner |
|---|---|---|---|---|
| Friday | ||||
| Saturday | 156 | 244 | ||
| Sunday | 253 | |||
| Monday | ||||
| Tuesday | ||||
| Wednesday | 148 | 227 | ||
| Thursday | ||||
| Friday | ||||
| Saturday | 179 | |||
| Sunday | 160 | |||
| Monday | ||||
| Tuesday | ||||
| Wednesday | ||||
| Thursday |
Plan
- Discontinue basal insulin.
- Begin premix insulin twice daily before breakfast and dinner.
- Ask KW to monitor blood glucose two times daily and, if appropriate, teach her how to self-adjust insulin doses.
- Stress the importance of exercise and proper nutrition; gain agreement on short-term goals for exercise and nutrition.
- Discuss the importance of smoking cessation; develop a plan.
Barriers
The physician discusses with KW that her consistent meal and activity schedule would make switching to premix insulin twice daily a good choice. KW is generally in agreement with the change, but wonders whether hypoglycemia might be more likely. She also asks if she might gain more weight in addition to the 3 pounds (1.35 kg) she has gained since starting basal insulin.
Patient concern: Hypoglycemia
Physician responses:
- Hypoglycemia remains a concern, and is more frequently seen with premix than with basal insulin; however, as long as you remain consistent with your meal and activity schedule, the risk for bad hypoglycemia is low.
- We should review your written action plan so that you are sure what signs or symptoms of a low blood sugar might occur and what you should do to treat them.
Patient concern: Weight gain
Physician responses:
- It is possible that you might gain a few additional pounds. You can avoid this by increasing your physical activity, and importantly, continue healthy eating. We should schedule a time for you to meet again with a dietician who can discuss options that might work for you.
Dosing
There are different approaches for converting from basal insulin to twice-daily premix insulin. One approach is to determine the TDD of basal insulin, and give half at breakfast and the other half at dinner as premix insulin.39 Since KW is taking 60 U of basal in the evening, she should take 30 U at breakfast and 30 U at dinner. Dose titration is according to the 1-2-3 Study algorithm shown in case study 2.
Another approach is to administer biphasic insulin aspart 70/30 0.2 U/kg before breakfast and 0.1 U/kg before dinner as was done in the PREFER study (TABLE 13).45 Subsequent dosing can be determined based on the PREFER algorithm below. Of note is that at study end, premix insulin doses were equally divided between breakfast and dinner. Breakfast and dinner doses are titrated based on blood glucose levels before dinner and breakfast, respectively. In the PREFER study, the use of premix insulin provided comparable A1C reduction as basal-bolus therapy (basal once daily + bolus TID) in insulin-naïve patients. However, patients previously treated with basal insulin such as KW experienced greater A1C reductions with basal-bolus insulin than with premix insulin.
1. Rodbard HW, Jellinger PS, Davidson JA, et al. Statement by an American Association of Clinical Endocrinologists/American College of Endocrinology consensus panel on type 2 diabetes mellitus: an algorithm for glycemic control [published correction appears in Endocr Pract. 2009;15(7):768-770]. Endocr Pract. 2009;15(6):540-559.
2. Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of hyperglycemia in type 2 diabetes: a patient-centered approach: position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD) [published online ahead of print April 19, 2012]. Diabetes Care. doi:10.2337/dc12-0413.
3. Lalić NM, Micić D, Antić S, et al. Effect of biphasic insulin aspart on glucose and lipid control in patients with type 2 diabetes mellitus. Expert Opin Pharmacother. 2007;8(17):2895-2901.
4. Reynolds LR, Kingsley FJ, Karounos DG, Tannock LR. Differential effects of rosiglitazone and insulin glargine on inflammatory markers, glycemic control, and lipids in type 2 diabetes. Diabetes Res Clin Pract. 2007;77(2):180-187.
5. Rosak C, Jung R, Hofmann U. Insulin glargine maintains equivalent glycemic control and better lipometabolic control than NPH insulin in type 1 diabetes patients who missed a meal. Horm Metab Res. 2008;40(8):544-548.
6. Ampudia-Blasco FJ, Girbes J, Carmena R. A case of lipoatrophy with insulin glargine: long-acting insulin analogs are not exempt from this complication. Diabetes Care. 2005;28(12):2983.-
7. Griffin ME, Feder A, Tamborlane WV. Lipoatrophy associated with lispro insulin in insulin pump therapy: an old complication, a new cause? Diabetes Care. 2001;24(1):174.-
8. Fineberg SE, Huang J, Brunelle R, Gulliya KS, Anderson JH, Jr. Effect of long-term exposure to insulin lispro on the induction of antibody response in patients with type 1 or type 2 diabetes. Diabetes Care. 2003;26(1):89-96.
9. Moyes V, Driver R, Croom A, Mirakian R, Chowdhury TA. Insulin allergy in a patient with type 2 diabetes successfully treated with continuous subcutaneous insulin infusion. Diabet Med. 2006;23(2):204-206.
10. Ghosh S, McCann V, Bartle L, Collier A, Malik I. Allergy to insulin detemir. Diabet Med. 2007;24(11):1307.-
11. Blumer I. Severe, delayed insulin detemir injection site reaction. Diabet Med. 2008;25(8):1008.-
12. Pérez E, González R, Martínez J, Iglesias J, Matheu V. Detemir insulin-induced anaphylaxis. Ann Allergy Asthma Immunol. 2009;102(2):174-175.
13. Mollar-Puchades MA, Villanueva IL. Insulin glulisine in the treatment of allergy to rapid acting insulin and its rapid acting analogs. Diabetes Res Clin Pract. 2009;83(1):e21-e22.
14. Kawasaki F, Kamei S, Tatsumi F, et al. Gallbladder edema in type 1 diabetic patient due to delayed-type insulin allergy. Intern Med. 2009;48(17):1545-1549.
15. Wang C, Ding ZY, Shu SQ, et al. Severe insulin allergy after percutaneous transluminal coronary angioplasty. Clin Ther. 2009;31(3):569-574.
16. Ozaki N, Oiso Y. Immunologic tolerance to the insulin analogue glulisine. Diabetes Care. 2010;33(3):e39.-
17. Koroscil T, Kagzi Y, Zacharias D. Failure of multiple therapies in the treatment of a type 1 diabetic patient with insulin allergy: a case report. Endocr Pract. 2011;17(1):91-94.
18. Tuerk PW, Mueller M, Egede LE. Estimating physician effects on glycemic control in the treatment of diabetes: methods, effects sizes, and implications for treatment policy. Diabetes Care. 2008;31(5):869-873.
19. Hajos TR, Polonsky WH, Twisk JW, Dain MP, Snoek FJ. Do physicians understand type 2 diabetes patients’ perceptions of seriousness; the emotional impact and needs for care improvement? A cross-national survey. Patient Educ Couns. 2011;85(2):258-263.
20. Polonsky WH, Hajos TR, Dain MP, Snoek FJ. Are patients with type 2 diabetes reluctant to start insulin therapy? An examination of the scope and underpinnings of psychological insulin resistance in a large, international population. Curr Med Res Opin. 2011;27(6):1169-1174.
21. Nam S, Chesla C, Stotts NA, Kroon L, Janson SL. Factors associated with psychological insulin resistance in individuals with type 2 diabetes. Diabetes Care. 2010;33(8):1747-1749.
22. Nakar S, Yitzhaki G, Rosenberg R, Vinker S. Transition to insulin in type 2 diabetes: family physicians’ misconception of patients’ fears contributes to existing barriers. J Diabetes Complications. 2007;21(4):220-226.
23. Snoek FJ. Breaking the barriers to optimal glycaemic control—what physicians need to know from patients’ perspectives. Int J Clin Pract Suppl. 2002;(129):80-84.
24. Korytkowski M. When oral agents fail: practical barriers to starting insulin. Int J Obes Relat Metab Disord. 2002;26(Suppl 3):S18-S24.
25. Skovlund SE, Peyrot M. The Diabetes Attitudes, Wishes and Needs (DAWN) program: a new approach to improving outcomes of diabetes care. Diabetes Spectrum. 2005;18(3):136-142.
26. Jenkins N, Hallowell N, Farmer AJ, Holman RR, Lawton J. Initiating insulin as part of the Treating To Target in Type 2 Diabetes (4-T) trial: an interview study of patients’ and health professionals’ experiences. Diabetes Care. 2010;33(10):2178-2180.
27. Levinson W, Gorawara-Bhat R, Lamb J. A study of patient clues and physician responses in primary care and surgical settings. JAMA. 2000;284(8):1021-1027.
28. Rodriguez HP, Anastario MP, Frankel RM, et al. Can teaching agenda-setting skills to physicians improve clinical interaction quality? A controlled intervention. BMC Med Educ. 2008;8:3.-
29. Solomon J. How strategies for managing patient visit time affect physician job satisfaction: a qualitative analysis. J Gen Intern Med. 2008;23(6):775-780.
30. Funnell MM, Anderson RM. Empowerment and self-management of diabetes. Clin Diabetes. 2004;22(3):123-127.
31. Funnell MM, Anderson RM. Are patients or outcomes more important? Rev Endocrinol. 2008;2(8):49-51.
32. Shaefer CF. Clinical inertia: overcoming a major barrier to diabetes management. Insulin. 2006;1(2):61-64.
33. Harris S, Yale JF, Dempsey E, Gerstein H. Can family physicians help patients initiate basal insulin therapy successfully?: randomized trial of patient-titrated insulin glargine compared with standard oral therapy: lessons for family practice from the Canadian INSIGHT trial. Can Fam Physician. 2008;54(4):550-558.
34. Centers for Disease Control and Prevention. National diabetes fact sheet national estimates and general information on diabetes and prediabetes in the United States, 2011. Atlanta, GA: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, 2011. http://www.cdc.gov/diabetes/pubs/pdf/ndfs_2011.pdf. Accessed February 10, 2011.
35. Riddle MC, Rosenstock J, Gerich J. Insulin Glargine 4002 Study Investigators. The treat-to-target trial: randomized addition of glargine or human NPH insulin to oral therapy of type 2 diabetic patients. Diabetes Care. 2003;26(11):3080-3086.
36. Hermansen K, Davies M, Derezinski T, Martinez Ravn G, Clauson P, Home P. A 26-week, randomized, parallel, treat-to-target trial comparing insulin detemir with NPH insulin as add-on therapy to oral glucose-lowering drugs in insulin-naive people with type 2 diabetes [published correction appears in Diabetes Care. 2007;30(4):1035]. Diabetes Care. 2006;29(6):1269-1274.
37. Davies M, Storms F, Shutler S, Bianchi-Biscay M, Gomis R. ATLANTUS Study Group. Improvement of glycemic control in subjects with poorly controlled type 2 diabetes: comparison of two treatment algorithms using insulin glargine. Diabetes Care. 2005;28(6):1282-1288.
38. Meneghini L, Koenen C, Weng W, Selam JL. The usage of a simplified self-titration dosing guideline (303 Algorithm) for insulin detemir in patients with type 2 diabetes—results of the randomized, controlled PREDICTIVE 303 study. Diabetes Obes Metab. 2007;9(6):902-913.
39. Garber AJ, Wahlen J, Wahl T, et al. Attainment of glycaemic goals in type 2 diabetes with once-, twice-, or thrice-daily dosing with biphasic insulin aspart 70/30 (The 1-2-3 study). Diabetes Obes Metab. 2006;8(1):58-66.
40. Philis-Tsimikas A, Charpentier G, Clauson P, Ravn GM, Roberts VL, Thorsteinsson B. Comparison of once-daily insulin detemir with NPH insulin added to a regimen of oral antidiabetic drugs in poorly controlled type 2 diabetes [published correction appears in Clin Ther. 2006;28(11):1967]. Clin Ther. 2006;28(10):1569-1581.
41. Blonde L, Merilainen M, Karwe V, Raskin P. TITRATE Study Group. Patient-directed titration for achieving glycaemic goals using a once-daily basal insulin analogue: an assessment of two different fasting plasma glucose targets—the TITRATE study. Diabetes Obes Metab. 2009;11(6):623-631.
42. Meneghini L, Mersebach H, Kumar S, Svendsen AL, Hermansen K. Comparison of 2 intensification regimens with rapid-acting insulin aspart in type 2 diabetes mellitus inadequately controlled by once-daily insulin detemir and oral antidiabetes drugs: the step-wise randomized study. Endocr Pract. 2011;17(5):727-736.
43. Lankisch MR, Ferlinz KC, Leahy JL, Scherbaum WA. Orals Plus Apidra and LANTUS (OPAL) study group. Introducing a simplified approach to insulin therapy in type 2 diabetes: a comparison of two single-dose regimens of insulin glulisine plus insulin glargine and oral antidiabetic drugs [published correction appears in Diabetes Obes Metab. 2010;12(5):461]. Diabetes Obes Metab 2008;10(12):1178-1185.
44. Meneghini L. Why and how to use insulin therapy earlier in the management of type 2 diabetes. South Med J. 2007;100(2):164-174.
45. Liebl A, Prager R, Binz K, Kaiser M, Bergenstal R, Gallwitz B. PREFER Study Group. Comparison of insulin analogue regimens in people with type 2 diabetes mellitus in the PREFER Study: a randomized controlled trial. Diabetes Obes Metab. 2009;11(1):45-52.
46. Braun A, Sämann A, Kubiak T, et al. Effects of metabolic control, patient education and initiation of insulin therapy on the quality of life of patients with type 2 diabetes mellitus. Patient Educ Couns. 2008;73(1):50-59.
The Evolution of Insulin Therapy in Diabetes Mellitus
The modern management of diabetes mellitus (DM) began with the discovery of insulin by Banting and Best in 1921 (see The Evolution of Insulin Therapy in Diabetes Mellitus in this supplement). Since that time, numerous additional classes of glucose-lowering agents have been introduced for the treatment of type 2 DM (T2DM). These medications primarily act by addressing 2 of the key defects of T2DM, insulin resistance and pancreatic β-cell dysfunction. T2DM is a progressive disease process that requires continued adjustment of therapy to maintain treatment goals. Most patients with T2DM will require insulin therapy at some point in their lives.
Role of Insulin in Type 2 Diabetes Mellitus Management
Consensus guidelines developed by the American Association of Clinical Endocrinologists/American College of Endocrinology (AACE/ACE) recommend initiating insulin when oral therapy fails to achieve glycemic control, A1C > 9.0% in treatment-naïve patients, or if the patient is symptomatic with glucose toxicity (polyuria, polydipsia, and weight loss) (FIGURE 1).1
Similar consensus guidelines developed by the American Diabetes Association/European Association for the Study of Diabetes (ADA/EASD) advise the initiation of glucose-lowering therapy for most patients with T2DM with the combination of lifestyle modifications, diet, and metformin (FIGURE 2).2 For patients who do not achieve or maintain glycemic control over 3 months, or thereabouts, with metformin, a second oral agent should be added. Alternatives include a glucagon-like peptide-1 receptor (GLP-1R) agonist or basal insulin. Insulin should be strongly considered as initial therapy for a patient with significant symptoms of hyperglycemia and/or plasma glucose >300-350 mg/dL or A1C ≥10.0%.
The major role of insulin in the management of patients with T2DM stems from several important attributes. First, insulin is the only treatment that works in patients with advanced β-cell deficiency. It acts directly on tissues to regulate glucose homeostasis, unlike other glucose-lowering agents that require the presence of sufficient endogenous insulin to exert their effects as insulin sensitizers, secretagogues, incretin mimetics, amylin analogs, and other factors. This also means that the mechanism of action of insulin is complementary to those of other glucose-lowering agents. Second, there is less of a ceiling effect with insulin. That is, increasing the dose of insulin results in a progressive lowering of blood glucose in the majority of patients, with the major limitation being the risk for hypoglycemia. Third, the glucose-lowering efficacy of insulin is durable, unlike that of other glucose-lowering agents that depend on endogenous insulin secretion for continued effectiveness. Fourth, insulin improves the lipid profile, particularly triglyceride levels.2-5 Fifth, regarding the long-term safety and tolerability of insulin, it is well established that weight gain, likely mediated via reduction of glycosuria, and hypoglycemia are typically the most concerning adverse events encountered. Allergic reactions, which were a more common complication of animal-sourced insulins, are infrequent with the insulin analogs.6-17 Finally, the availability of insulin in different formulations allows for targeting fasting plasma glucose or postprandial glucose, and individualization of therapy (see The Evolution of Insulin Therapy in Diabetes Mellitus in this supplement.)
While both the AACE/ACE and ADA/EASD consensus guidelines provide treatment “algorithms,” both make it clear that these are suggested approaches suitable for the population with T2DM (FIGURE 1, FIGURE 2). The specific treatment approach must be individualized based on patient-specific factors such as age, comorbid conditions, and tolerance of hypoglycemia.
FIGURE 1
Role of insulin in the management of patients with type 2 diabetes mellitus according to the AACE/ACE1
AACE, American Association of Clinical Endocrinologists; ACE, American College of Endocrinology; AGI, α-glucosidase inhibitor; DPP4, dipeptidyl-peptidase-4 inhibitor; FPG, fasting plasma glucose; GLP-1, glucagon–like peptide-1 agonist; MET, metformin; PPG, postprandial glucose; SU, sulfonylurea; TZD, thiazolidinedione.
Reprinted from American Association of Clinical Endocrinologists. AACE/ACE Diabetes Algorithm for Glycemic Control. Available at https://www.aace.com/sites/default/files/GlycemicControlAlgorithmPPT.pdf. Accessed April 4, 2012, with permission from the American Association of Clinical Endocrinologists.
FIGURE 2
Role of insulin in the management of patients with type 2 diabetes mellitus according to the ADA/EASD2
Moving from the top to the bottom of the figure, potential sequences of antihyperglycemic therapy. In most patients, begin with lifestyle changes; metformin monotherapy is added at, or soon after, diagnosis (unless there are explicit contraindications). If the HbA1c target is not achieved after ~3 months, consider 1 of the 5 treatment options combined with metformin: an SU, TZD, DPP-4-i, GLP-1-RA, or basal insulin. (The order in the chart is determined by historical introduction and route of administration and is not meant to denote any specific preference.) Choice is based on patient and drug characteristics, with the over-riding goal of improving glycemic control while minimizing side effects. Shared decision making with the patient may help in the selection of therapeutic options. The figure displays drugs commonly used both in the United States and/or Europe. Rapid-acting secretagogues (meglitinides) may be used in place of SUs. Other drugs not shown (α-glucosidase inhibitors, colesevelam, dopamine agonists, pramlintide) may be used where available in selected patients but have modest efficacy and/or limiting side effects. In patients intolerant of, or with contraindications for, metformin, select initial drug from other classes depicted and proceed accordingly. In this circumstance, while published trials are generally lacking, it is reasonable to consider 3-drug combinations other than metformin. Insulin is likely to be more effective than most other agents as a third-line therapy, especially when HbA1c is very high (eg, ≥ 9.0%). The therapeutic regimen should include some basal insulin before moving to more complex insulin strategies. Dashed arrow line on the left-hand side of the figure denotes the option of a more rapid progression from a 2-drug combination directly to multiple daily insulin doses, in those patients with severe hyperglycemia (eg, HbA1c, ≥ 10.0–12.0%).
DPP-4, dipeptidyl peptidase-4; DPP-4-i, DPP-4 inhibitor; Fx’s, bone fractures; GI, gastrointestinal; GLP-1, glucagon-like peptide 1; GLP-1-RA, GLP-1 receptor agonist; HbA1c, hemoglobin A1c; HF, heart failure; NPH, neutral protamine Hagedorn; SU, sulfonylurea; TZD, thiazolidinedione.
aConsider beginning at this stage in patients with very high HbA1c (eg, ≥ 9%); bConsider rapid-acting, non-SU secretagogues (meglitinides) in patients with irregular meal schedules or who develop late postprandial hypoglycemia on SUs; cUsually a basal insulin (NPH, glargine, detemir) in combination with noninsulin agents; dCertain noninsulin agents may be continued with insulin. Consider beginning at this stage if patient presents with severe hyperglycemia (≥ 16.7–19.4 mmol/L [≥ 300–350 mg/dL]; HbA1c≥ 10.0–12.0%) with or without catabolic features (weight loss, ketosis, etc).
Diabetes Care by American Diabetes Association. Copyright 2012. Reproduced with permission of AMERICAN DIABETES ASSOCIATION in the format Journal via Copyright Clearance Center.
Individualizing Therapy
The importance of individualizing therapy in a way that allows patients with T2DM to effectively self-manage their disease cannot be overstated. A study involving 1381 patients with T2DM cared for by 42 primary care physicians was conducted to estimate the magnitude of effect that physicians have on glycemic control.18 Hierarchical linear modeling showed that physician-related factors were associated with a statistically significant but modest variability in A1C change (2%) for the entire patient group. On the face of it, this finding might be discouraging. Further analysis showed, however, that for patients whose A1C did improve, physician-related factors accounted for 5% of the overall change in A1C (P = .005). On the other hand, physician-related factors had no impact on patients whose A1C did not improve or worsened. These results support the role that physicians play in affecting patient outcomes. The results also make it clear that without a physician’s influence, a patient’s glycemic outcomes may be difficult to change. The question is: How best can a physician influence patient outcomes?
A 2011 survey of patients with DM, general practitioners, and DM specialists reported that clinicians tended to underestimate patients’ perceived seriousness of the disease, while overestimating patients’ level of distress. In addition, physicians had difficulty identifying which DM-related complications concerned patients most and the information and support patients needed to feel more at ease with DM. Patients placed greater importance on having easy access to their physicians rather than more time with them. But most importantly, the survey investigators concluded that patients generally wished for greater involvement in decision making and being provided more information.19 These findings suggest that patients understand that T2DM is a largely self-managed, chronic disease, and want a collaborative relationship with their physician.
Patient Barriers to Insulin Therapy
Numerous factors have been identified as impeding patients’ willingness to initiate insulin therapy (TABLE 1).20-24 Barriers often vary from patient to patient and, in fact, may change over time in an individual patient. It is crucial, therefore, to identify the root reasons for a patient’s apprehension with insulin when talking about options for intensifying treatment. Once insulin has been initiated, the patient should be asked about continuing or new concerns regarding insulin therapy (and DM management in general), including adherence.
TABLE 1
Barriers to insulin therapy identified by patients20-24
| Lack of understanding of serious nature of type 2 diabetes mellitus |
| Fear of addiction to insulin |
| Fear of hypoglycemia |
| Concern about weight gain |
| Repeated experiences of failing to achieve satisfactory glycemic control |
| Perception that quality of previous treatment was low |
| Needle phobia |
| Treatment complexity |
| Concern of social stigmatization |
| Perceived failure and low self-efficacy |
| Belief of becoming more ill |
| Out-of-pocket cost |
| Perceived negative impact on quality of life |
| Comorbidities such as poor eyesight, arthritis, forgetfulness |
A recent, international survey of 1400 patients with insulin-naïve T2DM reported that 3 negative beliefs about insulin were prominent: (1) feeling that the disease was worsening; (2) fear of injection; and (3) a feeling of personal failure.20 Certain patient comorbidities, such as poor eyesight, arthritis, and forgetfulness, might also serve as barriers to self-management of DM with insulin. Additional comorbidities may contribute as indirect barriers, such as the need for polypharmacy, which may make the initiation of additional treatments such as insulin logistically or financially difficult.
It is possible that the discussion about initiating insulin may uncover patient concerns about T2DM in general. The Diabetes Attitudes, Wishes, and Needs (DAWN) study reported that psychosocial issues were the major source of difficulty in patient self-management (TABLE 2).25 In fact, 85% of people who reported a high level of distress at the time of diagnosis of T2DM continued to experience psychological distress at a mean follow-up of 15 years.
TABLE 2
Patients experiencing various aspects of diabetes-related distress25
| Diabetes-related distress | Respondents who agree (%) |
|---|---|
| I feel stressed because of my diabetes. | 32.7 |
| I feel burned out because of my diabetes. | 18.1 |
| I feel that diabetes is preventing me from doing what I want to do. | 35.9 |
| I am constantly afraid of my diabetes getting worse. | 43.8 |
| I worry about not being able to carry out my family responsibilities in the future. | 30.1 |
| My diabetes causes me worries about my financial future. | 25.8 |
| My family and friends put too much pressure on me about my diabetes. | 14.7 |
| The community I live in is intolerant of diabetes. | 13.6 |
| Diabetes Care by American Diabetes Association. Copyright 2012. Reproduced with permission of AMERICAN DIABETES ASSOCIATION in the format Journal via Copyright Clearance Center. | |
Addressing psychosocial issues and other barriers is crucial in the discussion of self-management because those with more negative feelings about starting insulin are most unwilling to start insulin.20 One factor that may contribute to these negative feelings is repeated experiences of failing to achieve satisfactory glycemic control with oral glucose-lowering agents.23 Conversely, those who have experienced improved glycemic control with intensification of prior glucose-lowering therapy may be more accepting of initiating insulin therapy.23,26 These findings are a reminder of the importance of a treat-to-target approach to management, in which the target glycemic goal, generally A1C < 7.0%, is achieved within 2 to 3 months of diagnosis and maintained at that level through intensification of therapy as needed.
Addressing psychosocial issues can be a challenge in today’s busy primary care practice due to limited time and lack of training in the management of such issues. However, implementation of various strategies has been reported to facilitate and, in some cases, shorten a patient’s visit. For example, one small study reported that visits were shorter if the physician acknowledged and responded positively to a patient’s stated or implied concerns (17.6 minutes vs 20.1 minutes).27 Missing or ignoring the patient’s concerns often led the patient to bring up the same concern one or more additional times resulting in a longer office visit. These results underscore the importance of asking patients to identify their concerns or questions at the beginning of the office visit. The patient can fill out a questionnaire in the waiting room or be encouraged to write down and prioritize their questions and concerns specific to the visit. If the patient identifies more concerns or questions than can be reasonably addressed in one visit, there should be agreement to address the most pressing ones during the current visit and the remaining concerns and questions during the next visit. This “agenda-setting” approach has been reported to offer several advantages.28 From the patient’s perspective, the quality of the physician-patient interaction was much improved, in part because physicians took time to explain points in a way that was easy to understand. Advantages to the physician with an agenda-setting approach included “feeling more in control,” “less stressed by simply knowing what was on the patient’s mind,” “feeling less rushed,” and “enjoying patient encounters more.” Contrary to the study cited above, physicians found that patients’ visits often were longer, especially those of older patients. One physician, however, noted that the visit “takes more time now, but saves time later.” As noted in this study, additional time spent with the patient can lead to improved job satisfaction for the physician.29
The agenda-setting approach requires that the physician ask the patient to list his or her concerns and questions, and then actively listen to the patient. Once the agenda for the visit is established, employing the “ask, listen, empathize” communication style can lead to effective physician-patient communication and problem-solving. Using this approach, the physician asks questions to gain a clear understanding of the patient’s concerns and then uses active listening with little, if any, interruption.30,31 Since the goal is to solve problems with rather than for the patient, active listening without offering opinions, judgements, or advice while offering empathy is essential. Through reflection and discussion, the physician can help the patient to identify his or her issues and acceptable solutions.
The importance of good communication between physician and patient cannot be overstated. Additional communication skills to keep in mind are: (1) speak slowly using nonmedical language; (2) limit the amount of information and repeat it; (3) draw pictures and/or use visual aids; and (4) ask the patient to repeat instructions and key concepts. In addition to enhancing patients’ understanding, visual images may be particularly beneficial in keeping patients motivated to improve self-management, including adherence to therapy. For example, it may be helpful to graphically track the patient’s glycemic progress. This can be done by establishing an actionable A1C goal (generally < 7.0%) and a time frame to achieve the goal (eg, 2 to 3 months).32 A graph can be constructed beginning with the patient’s current, preinsulin A1C level, with updates at each visit. In addition to motivating the patient and reinforcing adherence, the graph can also be used to demonstrate when further treatment intensification is needed. Additional general strategies that can be employed when considering the initiation of insulin are shown in TABLE 3. Implementation of strategies such as these by family physicians provides patient outcomes comparable to those implemented by endocrinologists or diabetes specialists.33
TABLE 3
General strategies for initiating insulin therapy
| Invite the patient to take an active role in treatment decisions. |
| Remind the patient that type 2 diabetes is primarily self-managed. |
| Discuss the progressive nature of β-cell dysfunction in type 2 diabetes. |
| Emphasize the physiologic role of insulin to maintain glucose homeostasis. |
| Discuss that insulin will help to achieve glycemic control and minimize the risk for long-term complications. |
| Discuss that treatment will be modified as needed to maintain glycemic control and to best meet their needs, capabilities, and interest. |
| Utilize insulin pen devices whenever possible. |
| Emphasize the importance of lifestyle management. |
| Ask if hearing other patients talk of their experiences with insulin therapy would be helpful; consider a group office visit. |
| Discuss and provide the patient with an individualized, written action plan that includes insulin dosing, self-monitoring of blood glucose, and signs/symptoms of hypoglycemia and other adverse events with appropriate action(s) to take. |
| Simplify diabetes (and comorbidities) treatment whenever possible. |
The remainder of this article uses case studies to further explore various patient barriers to insulin therapy and strategies for addressing them with the patient. While other therapies may be appropriate in the case studies below as recommended by current guidelines, these case studies will focus on insulin. In addition, dosing strategies for initiating and intensifying insulin therapy are discussed. Changes to the treatment plan to adjust for comorbidities, such as hypertension and dyslipidemia, or for smoking cessation or aspirin therapy, are not addressed in these cases, but are crucial components of comprehensive management.
CASE STUDY 1
RF is a 49-year-old female insurance analyst diagnosed with T2DM 6 years ago. Initial therapy with lifestyle modifications and metformin has since been intensified. Glimepiride was added, then pioglitazone was added 1.5 years ago when the A1C had risen to 7.5%. There is no evidence of cardiovascular disease. She reports bothersome lower extremity edema and an 8-pound weight increase since starting pioglitazone treatment. RF states that she takes her medications every day, although she acknowledges that she sometimes forgets on Sundays.
Clinical Impression
After taking her history, performing a physical examination, and reviewing her laboratory and self-monitored blood glucose (SMBG) data, her physician concludes that her treatment plan needs to be changed (TABLE 4, TABLE 5).
TABLE 4
Case study 1: Chart notes
| Physical examination | Laboratory tests | Lifestyle habits | Current therapy | |
|---|---|---|---|---|
| Glucose-lowering | Other | |||
| BP: 126/80 mm Hg Weight: 176 lb (79.2 kg) BMI: 27 kg/m2 Eyes: no retinopathy Neurology: intact Skin: intact | SCr: 1.4 mg/dL Albuminuria: negative A1C: 8.2% Cholesterol: Total: 204 mg/dL LDL: 134 mg/dL HDL: 36 mg/dL | Exercise: Walks 2 miles 3-4 d/wk Nutrition: eats 3-4 meals/d | Metformin 1000 mg BID Glimepiride 8 mg QD Pioglitazone 45 mg QD | Lisinopril 30 mg QD Simvastatin 40 mg QD ASA 80 mg QD |
| ASA, acetylsalicylic acid; BMI, body mass index; BP, blood pressure; HDL, high-density lipoprotein; LDL, low-density lipoprotein; SCr, serum creatinine. | ||||
TABLE 5
Case study 1: Self-monitored blood glucose (mg/dL) over the previous 2 weeks
| Day | Fasting | 2 h Post-breakfast | 2 h Post-lunch | 2 h Post-dinner |
|---|---|---|---|---|
| Wednesday | 205 | |||
| Thursday | 158 | |||
| Friday | 179 | |||
| Saturday | 201 | 162 | ||
| Sunday | ||||
| Monday | 166 | |||
| Tuesday | 189 | |||
| Wednesday | ||||
| Thursday | 153 | 221 | ||
| Friday | 150 | |||
| Saturday | 199 | 186 | 213 | |
| Sunday | ||||
| Monday | 181 | |||
| Tuesday | 167 |
Treatment Plan
- Initiate basal insulin once daily in the evening.
- Continue glimepiride, but reduce pioglitazone to 15 mg once daily (or discontinue if cost is a concern).
- Ask RF to monitor fasting blood glucose and self-adjust insulin doses as appropriate.
Barriers
While discussing the need to change the treatment plan and the physician’s suggestion that RF begin basal insulin, RF asks her physician for another few months on her current regimen stating that she will try harder to take her medications on Sundays. She also voices concern that insulin treatment requires injections and that she is concerned about what her coworkers and friends might think. The physician confirms that these concerns are understandable; he also confirms that RF is fearful of needles. The following are possible responses that RF’s physician could use to address these concerns.
Patient’s concern: Perceived failure/low self-efficacy
Physician responses:
- We all forget to do things from time to time, but overall I think you have done a great job taking your medications.
- As we have talked about before, with T2DM there is a progressive loss of insulin production over time regardless of what you do. That is why we added glimepiride and then pioglita-zone and that is why we need to make a change now and put you back in control of your diabetes. It is likely that further changes will be needed and we can discuss and agree on them together.
Patient’s concern: Social stigmatization
Physician responses:
- We can begin by having you administer insulin once daily in the evening in the privacy of your home.
- The insulin can be administered with a device that looks like a pen. It is small and can be carried in your purse; it does not need to be refrigerated once opened. If the time comes that you will need to administer a dose of insulin during the day, you can easily administer the insulin discretely in a public restroom or your work area.
- The use of insulin is more common than it was even a few years ago. In fact, about 5 million people in the United States use insulin to replace what is missing, control blood sugars, and decrease the risk for diabetes complications.34
Patient’s concern: Fear of needles
Physician’s responses:
- Insulin can be injected using a pen device with short, ultrathin needles that makes most of the injections painless. I would like you to see how simple and painless the injection can be by using this sample pen here in the office.
- Many patients are concerned about giving themselves an injection at first, but they quickly become comfortable doing so.
Dosing
Treatment with basal insulin can be initiated using one of several approaches. Using the treat-to-target approach, basal insulin 10 U once daily is initiated.35 The starting dose should be reduced to 6 U if the initial pre-breakfast or pre-dinner blood glucose is < 126 mg/dL or the patient’s body mass index (BMI) is < 26 kg/m2.36 Alternatively, the ADA/EASD recommends starting with 0.2 U/kg, which may be more practical in very overweight or obese patients.2 Titration of the basal insulin dose can be accomplished using one of the following physician-directed or patient-driven treat-to-target titration algorithms (TABLE 6).35,37,38 The insulin dose should be titrated based on the pre-breakfast fasting blood glucose level.
TABLE 6
Physician-directed or patient-driven treat-to-target titration algorithms
| Riddle et al35 | Davies et al37 | Meneghini et al38 | ||||
|---|---|---|---|---|---|---|
| Start with 10 U/d bedtime basal insulin and adjust weekly | Start with 10* U/d bedtime basal insulin and adjust weekly (physician-directed) Or Start with a dose numerically equivalent to the highest FPG (in millimoles/L)† over the previous 7 days and adjust every 3 days (patient-managed) | Start with basal insulin once daily and adjust every 3 days | ||||
| Mean of self-monitored FPG values from preceding 2 days | Change in insulin dose (U/d)# | Mean of self-monitored FPG values from preceding 3 days | Change in insulin dose (U/d) (physician-directed) | Change in insulin dose (U/d) (patient-managed) | Mean of self-monitored FPG values from preceding 3 days | Change in insulin dose (U/d) |
| ≥180 mg/dL 140-180 mg/dL 120-140 mg/dL 100-120 mg/dL | +8 +6 +4 +2 | ≥180 mg/dL (≥10 mmol/L) 140-179 mg/dL (7.8-9.9 mmol/L) 120-139 mg/dL (6.7 – 7.7 mmol/L) 100-119 mg/dL (5.5-6.6 mmol/L) | +6 to +8 +4 +2 0 to +2 | +2 +2 +2 0 to +2 | >110 mg/dL 80-110 mg/dL <80 mg/dL | +3 0 -3 |
| FPG, fasting plasma glucose. *In insulin-naive patients. †For example, if the highest FPG over the previous 7 days was 7 mmol/L, start with 7 U.#Small insulin dose decreases (2-4 U/d per adjustment) were allowed if severe hypoglycemia (requiring assistance) or plasma-referenced glucose < 56 mg/dL was documented in the preceding week. Reproduced with permission. Meneghini LF, et al. J Fam Pract. 2011;60(9 Suppl 1):S21-S28. Quadrant HealthCom Inc. Copyright 2011. | ||||||
Follow-Up Visit
RF begins basal insulin 10 U in the evening and is given simple instructions for insulin dose titration based on fasting plasma glucose results. At her follow-up visit, RF reports that she has increased her basal insulin to 18 U administered once daily. Review of her SMBG results show that her blood glucose levels throughout the day have improved, but are still not at goal. RF’s physician commends her on the progress she has made. RF and her physician agree that she should continue to increase her basal insulin dose. Eight months after beginning basal insulin, RF is administering 28 U (0.35 U/kg) of basal insulin in the evening. Review of her SMBG results over the previous 2 weeks show that her blood glucose rises during the day and is highest after dinner; her current A1C is 7.2%.
Treatment Plan
- Discuss dietary and lifestyle complements to insulin therapy such as:
- Use SMBG to identify foods that raise her blood glucose.
CASE STUDY 2
LW is a 64-year-old male with longstanding hypertension diagnosed with T2DM 8 years ago for which he was treated initially with lifestyle management and metformin. He has since been treated with other oral agents as add-on therapy; glipizide was discontinued due to hypoglycemia when he skips meals (usually lunch); pioglitazone was discontinued after the patient expressed concerns about the risk for bladder cancer he heard on television. He has mild retinopathy and mild loss of vibration sensation in the feet; there is no evidence of cardiovascular disease. He was diagnosed with osteoarthritis 3 years ago.
Clinical Impression
After taking his history, performing a physical examination, and reviewing his laboratory and SMBG data, his physician concludes that his treatment plan needs to be changed (FIGURE 3, TABLE 7, TABLE 8).
TABLE 7
Case study 2: Chart notes
| Physical examination | Laboratory tests | Lifestyle habits | Current therapy | |
|---|---|---|---|---|
| Glucose-lowering | Other | |||
| BP: 124/76 mm Hg Weight: 204 lb (92.7 kg) BMI: 31 kg/m2 Eyes: mild retinopathy Neurology: occasional tingling on bottom of right foot Skin: intact | SCr: 1.9 mg/dL eGFR: 51 mL/min Albuminuria: negative A1C: 8.1% Cholesterol: Total: 218 mg/dL LDL: 118 mg/dL HDL: 55 mg/dL Triglyceride: 204 mg/dL | Exercise: takes dog on occasional walk but otherwise sedentary Nutrition: eats 4 meals/d | Metformin 1000 mg BID Acarbose 50 mg TID Sitagliptin 100 mg QD | Lisinopril/HCTZ 20/25 mg QD Amlodipine 10 mg QD Acetaminophen extended-release 650 mg TID ASA 80 mg QD |
| ASA, acetylsalicylic acid; BMI, body mass index; BP, blood pressure; eGFR, estimated glomerular filtration rate; HCTZ, hydrochlorothiazide; HDL, high-density lipoprotein; LDL, low-density lipoprotein; SCr, serum creatinine. | ||||
TABLE 8
Case study 2: Self-monitored blood glucose (mg/dL) over the previous 2 weeks
| Day | Fasting | 2 h Post-breakfast | 2 h Post-lunch | 2 h Post-dinner |
|---|---|---|---|---|
| Tuesday | 135 | |||
| Wednesday | ||||
| Thursday | 196 | |||
| Friday | 152 | 174 | ||
| Saturday | ||||
| Sunday | 208 | |||
| Monday | 142 | 193 | ||
| Tuesday | ||||
| Wednesday | 130 | 156 | ||
| Thursday | ||||
| Friday | ||||
| Saturday | ||||
| Sunday | 151 | |||
| Monday |
FIGURE 3
Case study 2: A1C levels for April 2004 to March 2012
Treatment Plan
- Discontinue metformin since LW’s serum creatinine is > 1.5 mg/dL.
- Initiate either basal insulin once daily in the evening or premix insulin at dinner.
- Ask LW to monitor his blood glucose and self-adjust insulin doses as appropriate.
- Stress the importance of exercise and proper nutrition; gain agreement on short-term goals for exercise and nutrition.
Barriers
LW’s physician recommends that his treatment plan be changed and insulin therapy initiated. LW quickly responds that previous changes to his treatment regimen have not resulted in his achieving an A1C < 7.0%. He also doubts that he can use a syringe to draw up the correct dose and then self-administer due to his arthritis. The following are possible responses his physician could use to address these concerns.
Patient concern: Repeated experience of failing to achieve glycemic control, ie, A1C < 7.0%
Physician responses:
- While achieving an A1C < 7.0% is a realistic goal that reduces the risks for vascular complications of diabetes, any reduction of A1C will be of benefit.
- I would like to work with you to implement a new plan that we both believe will enable you to improve your diabetes control and ideally achieve an A1C < 7.0%.
Patient concern: Self-administering due to arthritis
Physician responses:
- Instead of using a syringe and vial to draw up and administer insulin, I would like you to use an insulin pen device. As you can see, it is easy to handle and you can easily select the correct dose.
- If you choose to start on premix insulin, the pen device contains both types of insulin together in one dose.
Dosing
Treatment with basal insulin once daily in the evening can be initiated and titrated based on pre-breakfast blood glucose as in Case Study 1. Alternatively, treatment with premix insulin can be initiated at a dose of 12 U administered within 15 minutes of dinner initiation. The premix dose can be titrated using the algorithm employed in the 1-2-3 Study based on pre-breakfast blood glucose (TABLE 9).39 After 16 weeks, 41% of patients in the 1-2-3 Study achieved an A1C < 7.0% from a baseline A1C of 8.6%.
TABLE 9
1-2-3 Study algorithm39
| Pre-breakfast SMBG (mg/dL) | Adjustment of pre-dinner dose (U) |
|---|---|
| <80 | -3 |
| 80-110 | No change |
| 111-140 | +3 |
| 141-180 | +6 |
| > 180 | +9 |
| SMBG, self-monitored blood glucose. | |
Follow-Up Visit
LW began basal insulin 10 U in the evening. Over the next 5.5 months, he titrated his dose such that his current dose is 46 U (0.50 U/kg) in the evening. His current A1C is 7.3%. Review of his SMBG shows consistently high 2-hour post-lunch blood glucose levels. Although further increasing his basal insulin dose is an option, in most of the treat-to-target studies, the daily dose of basal insulin given once daily averaged between 0.4 and 0.6 U/kg.35,37,40,41 LW and his physician agree that adding rapid-acting insulin at lunch is the best option. The starting dose of rapid-acting bolus insulin is 4 to 6 U administered prior to the largest meal of the day or, as in this case, prior to the meal with the largest postprandial blood glucose excursion.42,43 Alternatively, the dose of rapid-acting insulin could be calculated as 10% of the total daily dose of basal insulin, which in this case is 5 U (10% x 46 U). The dose of basal insulin would be reduced by 5 U if the rapid-acting insulin is given at dinner in order to reduce the risk for nocturnal hypoglycemia. The dose of the bolus insulin can be titrated using the ExtraSTEP algorithm (TABLE 10).42 Alternatively, the SimpleSTEP algorithm can be used which does not require a 2-hour postprandial glucose measurement.42
TABLE 10
Algorithms for adjusting insulin aspart42
| ExtraSTEP algorithm | SimpleSTEP algorithm | |||
|---|---|---|---|---|
| 2-h Post-meal PG level (mg/dL) | Insulin aspart adjustment (U) | Pre-meal BG (mg/dL) | Bedtime BG (mg/dL) | Insulin aspart adjustment (U) |
| <72* | -2 | <72* | <72* | -2 |
| 72-144 | 0 | 72-108 | 72-144 | 0 |
| 145-180 | +2 | 109-162 | 145-180 | +2 |
| >180 | +4 | >162 | >180 | +4 |
| BG, blood glucose; PG, plasma glucose. *One or more PG values <72 mg/dL without obvious explanation. Reproduced with permission. Meneghini LF, et al. J Fam Pract. 2011;60(9 Suppl 1):S21-S28. Quadrant HealthCom Inc. Copyright 2011. | ||||
Plan
- Begin rapid-acting insulin 5 U at lunch.
- Continue basal insulin at 46 U in the evening.
- Ask LW to continue to titrate basal insulin based on the pre-breakfast blood glucose level and the lunch time bolus insulin dose based on the 2-hour post-lunch SMBG (ExtraSTEP); alternatively, adjust based on the pre-dinner blood glucose level (SimpleSTEP).
CASE STUDY 3
MB is a 46-year-old male who had not consulted a physician since having a physical examination 6 years ago. He presented 2 weeks ago with frequent urination (7-8 times/day) and feeling tired; he also noted losing 5 pounds (2.25 kg) over the preceding 3.5 months despite no changes in his diet. MB is a regional salesperson with an erratic schedule. During the week, he eats lunch and most dinners in a restaurant. On the weekend, he goes to a local bar with his friends. He does light yard work, but does not exercise regularly. He is a current smoker with a 36 pack-year history. Urinalysis shows ketonuria and microalbuminuria. His A1C reported back today is 10.8%, confirming a diagnosis of uncontrolled and symptomatic DM.
Clinical Impression
After taking his history, performing a physical examination, and reviewing his laboratory data, MB’s physician confirms a diagnosis of DM (TABLE 11). While it is likely that MB has T2DM, his physician wants to rule out type 1 DM and latent autoimmune diabetes of the adult (LADA), so he orders tests for antibodies (GAD, IA-2, ICA). The antibody testing is negative, making T2DM the most likely diagnosis.
TABLE 11
Case study 3: Chart notes
| Physical examination | Laboratory tests | Lifestyle habits | Current therapy | |
|---|---|---|---|---|
| Glucose-lowering | Other | |||
| BP: 142/88 mm Hg Weight: 176 lb (79.2 kg) BMI: 27 kg/m2 Eyes: no retinopathy Neurology: intact Skin: intact | SCr: 1.4 mg/dL Microalbumin:creatinine ratio: 140 mg/g creatinine Ketonuria: 1+ A1C: 10.8% Cholesterol: Total: 210 mg/dL LDL: 146 mg/dL HDL: 30 mg/dL | Exercise: light yard work, no regular exercise Nutrition: 3 meals/d, eats most meals in a restaurant (lunch M-F; dinner 3-4 nights/wk) | None | None |
| BMI, body mass index; BP, blood pressure; HDL, high-density lipoprotein; LDL, low-density lipoprotein; SCr, serum creatinine. | ||||
Treatment Plan
- Initiate basal-bolus therapy with fixed bolus doses of rapid-acting insulin at each meal (prandial insulin).
- Ask MB to monitor blood glucose before meals and at bedtime.
- Provide MB with a supplemental scale to correct hyperglycemia before meals.
- Stress the importance of exercise and proper nutrition; gain agreement for short-term goals for exercise and nutrition; refer for diabetes and nutrition education if available.
- Discuss the importance of smoking cessation; develop a plan.
- Consider metformin and other non-insulin therapies when A1C is under control.
Barriers
MB is surprised that he has T2DM and is clearly anxious at receiving the diagnosis. He expresses concern about starting insulin because his uncle died within a year of starting insulin. MB also recalls that his uncle was always giving himself shots and monitoring his blood glucose level. He wants to know whether there is a simpler treatment option if he agrees to start insulin treatment. He also wants to know whether he will have to remain on insulin for the rest of his life. The following are possible responses his physician could use to address these concerns.
Patient concern: Fear of death
Physician responses:
- Uncontrolled high blood sugars over a long period of time can cause serious complications, such as kidney and heart disease that can result in death. That is why it is important that we work together to gain control of your blood sugar levels over the next few months and then modify your treatment as needed to maintain control.
- Unfortunately for many patients in the past, treatment with insulin was not used until it was too late and people already had serious complications from DM. This is likely the case for your uncle.
Patient concern: Treatment complexity
Physician responses:
- Right now we have to control your blood glucose rapidly so your pancreas can regain some function and your body can better respond to insulin.
- I will also provide you with step-by-step written instructions you can follow that describe how to start insulin and how to monitor your blood glucose.
- We will communicate as often as you need to adjust your insulin doses over the next few weeks; when you feel comfortable, I can even show you how to adjust your insulin dose before a meal to correct a high blood sugar.
- We can try this treatment for 3 months and then reevaluate your response, how you feel, and whether you want to continue to modify your treatment plan to keep your blood sugars controlled.
Patient concern: Lack of understanding that T2DM is a serious disease
Physician responses:
- Please understand that T2DM is a serious disease that increases your risk for heart disease, stroke, blindness, and other diseases. Unfortunately, since diabetes does not cause bad symptoms until it is actually too late, many patients do not make the effort to properly control their diabetes. By working together, we can reduce the risk for these complications and do some screening tests to detect any complications before they become irreversible.
Dosing
There are several approaches to determining the initial doses of basal and prandial (bolus) insulin. One approach is to estimate the total daily dose (TDD) of insulin by multiplying the patient’s weight in kilograms by 0.5 U/kg/d.44 Half of the TDD is given as basal insulin replacement; the other half is divided into 3 fixed preprandial doses of rapid-acting insulin. When the patient is ready to take on more complex management, the supplemental dose for bolus insulin can be calculated using a correction factor. If the bolus insulin is a rapid-acting insulin analog, 1800 is divided by the TDD of insulin; 1500 is used for a short-acting human insulin. This correction factor is an estimate of the fall in blood glucose per unit of bolus insulin. In our patient, the TDD would be: 80 kg x 0.5 U/kg/d or 40 U/d of insulin. Thus, 1 U of insulin should lower the blood glucose by about 45 mg/dL (1800/40 U = 45 mg/dL). For every 45 mg/dL above the pre-meal target, the patient would add 1 U of rapid-acting insulin to correct the hyperglycemia over the next 4 to 5 hours. The basal and prandial insulin doses would be titrated on a periodic basis (perhaps every 1 to 2 weeks) until the daytime levels of blood glucose are on target. The fasting (pre-breakfast) blood glucose would be used to adjust the basal insulin dose, while the pre-lunch, pre-dinner, and bedtime blood glucose results would be used to adjust the pre-breakfast, pre-lunch, and pre-dinner prandial (rapid-acting) insulin doses, respectively.
An alternative approach to initiating basal-bolus therapy is the PREFER algorithm.45 Here, the basal insulin dose is 10 U initially. The bolus doses are administered in a 3:1:2 ratio, so if the total of the 3 bolus doses is 12 U/d, the initial bolus doses would be 6 (breakfast), 2 (lunch), and 4 (dinner) U. The mean basal (once-daily) and bolus insulin doses observed in PREFER are shown in TABLE 12 and TABLE 13.
TABLE 12
Case study 3: Calculating initial basal-bolus insulin doses
| Algorithm | Calculations | Patient MB |
|---|---|---|
| Meneghini44 | TDD = (total body weight [kg]) (0.5 U/kg/d) Basal insulin dose* = (50%) (TDD) Bolus insulin dose† = (10%-20%) (TDD) | TDD = (0.5 U/kg/d)(80kg) = 40 U/d |
| Basal = (50%) (40 U/d) = 20 U/d | ||
| Bolus = (10%-20%) (40 U/d) = 4 to 8 U/meal | ||
| CF = 1800/40 U/d = 45 mg/dL per 1 unit | ||
| PREFER45 | Basal insulin dose* = 10 U (14 U if BMI > 32 kg/m2) Bolus insulin dose† = ratio of 3:1:2 (breakfast:lunch:dinner) Note: At week 26, the bolus insulin doses were divided into the 3 daily meals in approximately a 1:1:1 ratio | |
| BMI, body mass index; CF, correction factor; TDD, total daily dose of insulin. *Once daily; †Three meals per day. | ||
TABLE 13
Titrating the basal insulin dose using the PREFER algorithm45
| Pre-breakfast blood glucose (mg/dL) | Basal insulin dose adjustment (U) |
|---|---|
| < 56 | -4 |
| 56-72 | -2 |
| 73-125 | No change |
| 126-140 | +2 |
| 141-160 | +4 |
| 161-180 | +6 |
| 181-200 | +8 |
| > 200 | +10 |
Follow-up Visit
MB begins with basal insulin 20 U in the evening and bolus insulin at doses of 7 U before each meal. Over the next several months, MB has titrated his insulin doses; his current doses are: 32 U (basal), 11 U (bolus-breakfast), 7 U (bolus-lunch), and 10 U (bolus-dinner). He experienced 1 episode of mild hypoglycemia (SMBG, 50 mg/dL) one afternoon following a particularly active morning (TABLE 14). His current A1C is 7.4%. MB’s physician congratulates him on the progress he has made in dramatically lowering his blood glucose level—and his risk for diabetes-related complications. While MB appreciates his physician’s support and admits that he does not feel tired and generally feels better, which is likely due to resolution of glucotoxicity, he is not happy that he has gained 5.5 pounds (2.5 kg).46 He also finds the timing and administration of bolus insulin difficult.
TABLE 14
Case study 3: Self-monitored blood glucose (mg/dL) over the previous 2 weeks
| Day | Fasting | 2 h Post-breakfast | 2 h Post-lunch | 2 h Post-dinner |
|---|---|---|---|---|
| Wednesday | ||||
| Thursday | 168 | |||
| Friday | 106 | 166 | 174 | |
| Saturday | 88 | |||
| Sunday | 195 | |||
| Monday | 134 | |||
| Tuesday | 172 | |||
| Wednesday | 130 | 156 | ||
| Thursday | 112 | 168 | ||
| Friday | 92 | 164 | ||
| Saturday | 50 | 149 | 159 | 176 |
| Sunday | 94 | 174 | 210 | |
| Monday | 176 | 184 | ||
| Tuesday | 117 | 169 |
Plan
- Continue basal insulin once-daily in the evening.
- Add metformin 500 mg BID and increase to 1000 mg BID as tolerated.
- Consider weaning down the bolus insulin doses and substituting them with a GLP-1R agonist, dipeptidyl peptidase-4 inhibitor, or short-acting secretagogue. If so, continue rapid-acting insulin during transition. [Note: the following combinations are not currently approved by the US FDA: exenatide twice-daily and prandial insulin; exenatide once-weekly and insulin; liraglutide and prandial insulin; linagliptin and insulin.]
CASE STUDY 4
KW is a 62-year-old female diagnosed with T2DM 12 years ago. Treatment with lifestyle management and metformin initially provided glycemic control. Glimepiride was subsequently added and eventually the patient was started on basal insulin. The current dose of basal insulin is 60 U in the evening. Five months ago her A1C was found to be 7.9% and more recently 8.3%. She drinks alcohol occasionally and smokes. KW works as an executive secretary and has a consistent meal and activity schedule.
Clinical Impression
Following completion of the history, physical examination, and review of her laboratory data, KW’s physician concludes that her insulin regimen should be intensified (TABLE 15, TABLE 16).
TABLE 15
Case study 4: Chart notes
| Physical examination | Laboratory tests | Lifestyle habits | Current therapy | |
|---|---|---|---|---|
| Glucose-lowering | Other | |||
| BP: 126/78 mm Hg Weight: 176 lb (79.2 kg) BMI: 32 kg/m2 Eyes: no retinopathy Neurology: intact Skin: intact | SCr: 1.0 mg/dL Albuminuria: negative A1C: 8.3% Cholesterol Total: 172 mg/dL LDL: 96 mg/dL HDL: 46 mg/dL Triglycerides: 138 mg/dL | Exercise: sedentary Nutrition: 3 meals/d with large dinner | Metformin 1000 mg BID Basal insulin 60 U in the evening | ASA 80 mg QD Pravastatin 40 mg qHS |
| BMI, body mass index; BP, blood pressure; HDL, high-density lipoprotein; LDL, low-density lipoprotein; SCr, serum creatinine. | ||||
TABLE 16
Case study 4: Self-monitored blood glucose (mg/dL) over the previous 2 weeks
| Day | Fasting | 2 h Post-breakfast | 2 h Post-lunch | 2 h Post-dinner |
|---|---|---|---|---|
| Friday | ||||
| Saturday | 156 | 244 | ||
| Sunday | 253 | |||
| Monday | ||||
| Tuesday | ||||
| Wednesday | 148 | 227 | ||
| Thursday | ||||
| Friday | ||||
| Saturday | 179 | |||
| Sunday | 160 | |||
| Monday | ||||
| Tuesday | ||||
| Wednesday | ||||
| Thursday |
Plan
- Discontinue basal insulin.
- Begin premix insulin twice daily before breakfast and dinner.
- Ask KW to monitor blood glucose two times daily and, if appropriate, teach her how to self-adjust insulin doses.
- Stress the importance of exercise and proper nutrition; gain agreement on short-term goals for exercise and nutrition.
- Discuss the importance of smoking cessation; develop a plan.
Barriers
The physician discusses with KW that her consistent meal and activity schedule would make switching to premix insulin twice daily a good choice. KW is generally in agreement with the change, but wonders whether hypoglycemia might be more likely. She also asks if she might gain more weight in addition to the 3 pounds (1.35 kg) she has gained since starting basal insulin.
Patient concern: Hypoglycemia
Physician responses:
- Hypoglycemia remains a concern, and is more frequently seen with premix than with basal insulin; however, as long as you remain consistent with your meal and activity schedule, the risk for bad hypoglycemia is low.
- We should review your written action plan so that you are sure what signs or symptoms of a low blood sugar might occur and what you should do to treat them.
Patient concern: Weight gain
Physician responses:
- It is possible that you might gain a few additional pounds. You can avoid this by increasing your physical activity, and importantly, continue healthy eating. We should schedule a time for you to meet again with a dietician who can discuss options that might work for you.
Dosing
There are different approaches for converting from basal insulin to twice-daily premix insulin. One approach is to determine the TDD of basal insulin, and give half at breakfast and the other half at dinner as premix insulin.39 Since KW is taking 60 U of basal in the evening, she should take 30 U at breakfast and 30 U at dinner. Dose titration is according to the 1-2-3 Study algorithm shown in case study 2.
Another approach is to administer biphasic insulin aspart 70/30 0.2 U/kg before breakfast and 0.1 U/kg before dinner as was done in the PREFER study (TABLE 13).45 Subsequent dosing can be determined based on the PREFER algorithm below. Of note is that at study end, premix insulin doses were equally divided between breakfast and dinner. Breakfast and dinner doses are titrated based on blood glucose levels before dinner and breakfast, respectively. In the PREFER study, the use of premix insulin provided comparable A1C reduction as basal-bolus therapy (basal once daily + bolus TID) in insulin-naïve patients. However, patients previously treated with basal insulin such as KW experienced greater A1C reductions with basal-bolus insulin than with premix insulin.
The Evolution of Insulin Therapy in Diabetes Mellitus
The modern management of diabetes mellitus (DM) began with the discovery of insulin by Banting and Best in 1921 (see The Evolution of Insulin Therapy in Diabetes Mellitus in this supplement). Since that time, numerous additional classes of glucose-lowering agents have been introduced for the treatment of type 2 DM (T2DM). These medications primarily act by addressing 2 of the key defects of T2DM, insulin resistance and pancreatic β-cell dysfunction. T2DM is a progressive disease process that requires continued adjustment of therapy to maintain treatment goals. Most patients with T2DM will require insulin therapy at some point in their lives.
Role of Insulin in Type 2 Diabetes Mellitus Management
Consensus guidelines developed by the American Association of Clinical Endocrinologists/American College of Endocrinology (AACE/ACE) recommend initiating insulin when oral therapy fails to achieve glycemic control, A1C > 9.0% in treatment-naïve patients, or if the patient is symptomatic with glucose toxicity (polyuria, polydipsia, and weight loss) (FIGURE 1).1
Similar consensus guidelines developed by the American Diabetes Association/European Association for the Study of Diabetes (ADA/EASD) advise the initiation of glucose-lowering therapy for most patients with T2DM with the combination of lifestyle modifications, diet, and metformin (FIGURE 2).2 For patients who do not achieve or maintain glycemic control over 3 months, or thereabouts, with metformin, a second oral agent should be added. Alternatives include a glucagon-like peptide-1 receptor (GLP-1R) agonist or basal insulin. Insulin should be strongly considered as initial therapy for a patient with significant symptoms of hyperglycemia and/or plasma glucose >300-350 mg/dL or A1C ≥10.0%.
The major role of insulin in the management of patients with T2DM stems from several important attributes. First, insulin is the only treatment that works in patients with advanced β-cell deficiency. It acts directly on tissues to regulate glucose homeostasis, unlike other glucose-lowering agents that require the presence of sufficient endogenous insulin to exert their effects as insulin sensitizers, secretagogues, incretin mimetics, amylin analogs, and other factors. This also means that the mechanism of action of insulin is complementary to those of other glucose-lowering agents. Second, there is less of a ceiling effect with insulin. That is, increasing the dose of insulin results in a progressive lowering of blood glucose in the majority of patients, with the major limitation being the risk for hypoglycemia. Third, the glucose-lowering efficacy of insulin is durable, unlike that of other glucose-lowering agents that depend on endogenous insulin secretion for continued effectiveness. Fourth, insulin improves the lipid profile, particularly triglyceride levels.2-5 Fifth, regarding the long-term safety and tolerability of insulin, it is well established that weight gain, likely mediated via reduction of glycosuria, and hypoglycemia are typically the most concerning adverse events encountered. Allergic reactions, which were a more common complication of animal-sourced insulins, are infrequent with the insulin analogs.6-17 Finally, the availability of insulin in different formulations allows for targeting fasting plasma glucose or postprandial glucose, and individualization of therapy (see The Evolution of Insulin Therapy in Diabetes Mellitus in this supplement.)
While both the AACE/ACE and ADA/EASD consensus guidelines provide treatment “algorithms,” both make it clear that these are suggested approaches suitable for the population with T2DM (FIGURE 1, FIGURE 2). The specific treatment approach must be individualized based on patient-specific factors such as age, comorbid conditions, and tolerance of hypoglycemia.
FIGURE 1
Role of insulin in the management of patients with type 2 diabetes mellitus according to the AACE/ACE1
AACE, American Association of Clinical Endocrinologists; ACE, American College of Endocrinology; AGI, α-glucosidase inhibitor; DPP4, dipeptidyl-peptidase-4 inhibitor; FPG, fasting plasma glucose; GLP-1, glucagon–like peptide-1 agonist; MET, metformin; PPG, postprandial glucose; SU, sulfonylurea; TZD, thiazolidinedione.
Reprinted from American Association of Clinical Endocrinologists. AACE/ACE Diabetes Algorithm for Glycemic Control. Available at https://www.aace.com/sites/default/files/GlycemicControlAlgorithmPPT.pdf. Accessed April 4, 2012, with permission from the American Association of Clinical Endocrinologists.
FIGURE 2
Role of insulin in the management of patients with type 2 diabetes mellitus according to the ADA/EASD2
Moving from the top to the bottom of the figure, potential sequences of antihyperglycemic therapy. In most patients, begin with lifestyle changes; metformin monotherapy is added at, or soon after, diagnosis (unless there are explicit contraindications). If the HbA1c target is not achieved after ~3 months, consider 1 of the 5 treatment options combined with metformin: an SU, TZD, DPP-4-i, GLP-1-RA, or basal insulin. (The order in the chart is determined by historical introduction and route of administration and is not meant to denote any specific preference.) Choice is based on patient and drug characteristics, with the over-riding goal of improving glycemic control while minimizing side effects. Shared decision making with the patient may help in the selection of therapeutic options. The figure displays drugs commonly used both in the United States and/or Europe. Rapid-acting secretagogues (meglitinides) may be used in place of SUs. Other drugs not shown (α-glucosidase inhibitors, colesevelam, dopamine agonists, pramlintide) may be used where available in selected patients but have modest efficacy and/or limiting side effects. In patients intolerant of, or with contraindications for, metformin, select initial drug from other classes depicted and proceed accordingly. In this circumstance, while published trials are generally lacking, it is reasonable to consider 3-drug combinations other than metformin. Insulin is likely to be more effective than most other agents as a third-line therapy, especially when HbA1c is very high (eg, ≥ 9.0%). The therapeutic regimen should include some basal insulin before moving to more complex insulin strategies. Dashed arrow line on the left-hand side of the figure denotes the option of a more rapid progression from a 2-drug combination directly to multiple daily insulin doses, in those patients with severe hyperglycemia (eg, HbA1c, ≥ 10.0–12.0%).
DPP-4, dipeptidyl peptidase-4; DPP-4-i, DPP-4 inhibitor; Fx’s, bone fractures; GI, gastrointestinal; GLP-1, glucagon-like peptide 1; GLP-1-RA, GLP-1 receptor agonist; HbA1c, hemoglobin A1c; HF, heart failure; NPH, neutral protamine Hagedorn; SU, sulfonylurea; TZD, thiazolidinedione.
aConsider beginning at this stage in patients with very high HbA1c (eg, ≥ 9%); bConsider rapid-acting, non-SU secretagogues (meglitinides) in patients with irregular meal schedules or who develop late postprandial hypoglycemia on SUs; cUsually a basal insulin (NPH, glargine, detemir) in combination with noninsulin agents; dCertain noninsulin agents may be continued with insulin. Consider beginning at this stage if patient presents with severe hyperglycemia (≥ 16.7–19.4 mmol/L [≥ 300–350 mg/dL]; HbA1c≥ 10.0–12.0%) with or without catabolic features (weight loss, ketosis, etc).
Diabetes Care by American Diabetes Association. Copyright 2012. Reproduced with permission of AMERICAN DIABETES ASSOCIATION in the format Journal via Copyright Clearance Center.
Individualizing Therapy
The importance of individualizing therapy in a way that allows patients with T2DM to effectively self-manage their disease cannot be overstated. A study involving 1381 patients with T2DM cared for by 42 primary care physicians was conducted to estimate the magnitude of effect that physicians have on glycemic control.18 Hierarchical linear modeling showed that physician-related factors were associated with a statistically significant but modest variability in A1C change (2%) for the entire patient group. On the face of it, this finding might be discouraging. Further analysis showed, however, that for patients whose A1C did improve, physician-related factors accounted for 5% of the overall change in A1C (P = .005). On the other hand, physician-related factors had no impact on patients whose A1C did not improve or worsened. These results support the role that physicians play in affecting patient outcomes. The results also make it clear that without a physician’s influence, a patient’s glycemic outcomes may be difficult to change. The question is: How best can a physician influence patient outcomes?
A 2011 survey of patients with DM, general practitioners, and DM specialists reported that clinicians tended to underestimate patients’ perceived seriousness of the disease, while overestimating patients’ level of distress. In addition, physicians had difficulty identifying which DM-related complications concerned patients most and the information and support patients needed to feel more at ease with DM. Patients placed greater importance on having easy access to their physicians rather than more time with them. But most importantly, the survey investigators concluded that patients generally wished for greater involvement in decision making and being provided more information.19 These findings suggest that patients understand that T2DM is a largely self-managed, chronic disease, and want a collaborative relationship with their physician.
Patient Barriers to Insulin Therapy
Numerous factors have been identified as impeding patients’ willingness to initiate insulin therapy (TABLE 1).20-24 Barriers often vary from patient to patient and, in fact, may change over time in an individual patient. It is crucial, therefore, to identify the root reasons for a patient’s apprehension with insulin when talking about options for intensifying treatment. Once insulin has been initiated, the patient should be asked about continuing or new concerns regarding insulin therapy (and DM management in general), including adherence.
TABLE 1
Barriers to insulin therapy identified by patients20-24
| Lack of understanding of serious nature of type 2 diabetes mellitus |
| Fear of addiction to insulin |
| Fear of hypoglycemia |
| Concern about weight gain |
| Repeated experiences of failing to achieve satisfactory glycemic control |
| Perception that quality of previous treatment was low |
| Needle phobia |
| Treatment complexity |
| Concern of social stigmatization |
| Perceived failure and low self-efficacy |
| Belief of becoming more ill |
| Out-of-pocket cost |
| Perceived negative impact on quality of life |
| Comorbidities such as poor eyesight, arthritis, forgetfulness |
A recent, international survey of 1400 patients with insulin-naïve T2DM reported that 3 negative beliefs about insulin were prominent: (1) feeling that the disease was worsening; (2) fear of injection; and (3) a feeling of personal failure.20 Certain patient comorbidities, such as poor eyesight, arthritis, and forgetfulness, might also serve as barriers to self-management of DM with insulin. Additional comorbidities may contribute as indirect barriers, such as the need for polypharmacy, which may make the initiation of additional treatments such as insulin logistically or financially difficult.
It is possible that the discussion about initiating insulin may uncover patient concerns about T2DM in general. The Diabetes Attitudes, Wishes, and Needs (DAWN) study reported that psychosocial issues were the major source of difficulty in patient self-management (TABLE 2).25 In fact, 85% of people who reported a high level of distress at the time of diagnosis of T2DM continued to experience psychological distress at a mean follow-up of 15 years.
TABLE 2
Patients experiencing various aspects of diabetes-related distress25
| Diabetes-related distress | Respondents who agree (%) |
|---|---|
| I feel stressed because of my diabetes. | 32.7 |
| I feel burned out because of my diabetes. | 18.1 |
| I feel that diabetes is preventing me from doing what I want to do. | 35.9 |
| I am constantly afraid of my diabetes getting worse. | 43.8 |
| I worry about not being able to carry out my family responsibilities in the future. | 30.1 |
| My diabetes causes me worries about my financial future. | 25.8 |
| My family and friends put too much pressure on me about my diabetes. | 14.7 |
| The community I live in is intolerant of diabetes. | 13.6 |
| Diabetes Care by American Diabetes Association. Copyright 2012. Reproduced with permission of AMERICAN DIABETES ASSOCIATION in the format Journal via Copyright Clearance Center. | |
Addressing psychosocial issues and other barriers is crucial in the discussion of self-management because those with more negative feelings about starting insulin are most unwilling to start insulin.20 One factor that may contribute to these negative feelings is repeated experiences of failing to achieve satisfactory glycemic control with oral glucose-lowering agents.23 Conversely, those who have experienced improved glycemic control with intensification of prior glucose-lowering therapy may be more accepting of initiating insulin therapy.23,26 These findings are a reminder of the importance of a treat-to-target approach to management, in which the target glycemic goal, generally A1C < 7.0%, is achieved within 2 to 3 months of diagnosis and maintained at that level through intensification of therapy as needed.
Addressing psychosocial issues can be a challenge in today’s busy primary care practice due to limited time and lack of training in the management of such issues. However, implementation of various strategies has been reported to facilitate and, in some cases, shorten a patient’s visit. For example, one small study reported that visits were shorter if the physician acknowledged and responded positively to a patient’s stated or implied concerns (17.6 minutes vs 20.1 minutes).27 Missing or ignoring the patient’s concerns often led the patient to bring up the same concern one or more additional times resulting in a longer office visit. These results underscore the importance of asking patients to identify their concerns or questions at the beginning of the office visit. The patient can fill out a questionnaire in the waiting room or be encouraged to write down and prioritize their questions and concerns specific to the visit. If the patient identifies more concerns or questions than can be reasonably addressed in one visit, there should be agreement to address the most pressing ones during the current visit and the remaining concerns and questions during the next visit. This “agenda-setting” approach has been reported to offer several advantages.28 From the patient’s perspective, the quality of the physician-patient interaction was much improved, in part because physicians took time to explain points in a way that was easy to understand. Advantages to the physician with an agenda-setting approach included “feeling more in control,” “less stressed by simply knowing what was on the patient’s mind,” “feeling less rushed,” and “enjoying patient encounters more.” Contrary to the study cited above, physicians found that patients’ visits often were longer, especially those of older patients. One physician, however, noted that the visit “takes more time now, but saves time later.” As noted in this study, additional time spent with the patient can lead to improved job satisfaction for the physician.29
The agenda-setting approach requires that the physician ask the patient to list his or her concerns and questions, and then actively listen to the patient. Once the agenda for the visit is established, employing the “ask, listen, empathize” communication style can lead to effective physician-patient communication and problem-solving. Using this approach, the physician asks questions to gain a clear understanding of the patient’s concerns and then uses active listening with little, if any, interruption.30,31 Since the goal is to solve problems with rather than for the patient, active listening without offering opinions, judgements, or advice while offering empathy is essential. Through reflection and discussion, the physician can help the patient to identify his or her issues and acceptable solutions.
The importance of good communication between physician and patient cannot be overstated. Additional communication skills to keep in mind are: (1) speak slowly using nonmedical language; (2) limit the amount of information and repeat it; (3) draw pictures and/or use visual aids; and (4) ask the patient to repeat instructions and key concepts. In addition to enhancing patients’ understanding, visual images may be particularly beneficial in keeping patients motivated to improve self-management, including adherence to therapy. For example, it may be helpful to graphically track the patient’s glycemic progress. This can be done by establishing an actionable A1C goal (generally < 7.0%) and a time frame to achieve the goal (eg, 2 to 3 months).32 A graph can be constructed beginning with the patient’s current, preinsulin A1C level, with updates at each visit. In addition to motivating the patient and reinforcing adherence, the graph can also be used to demonstrate when further treatment intensification is needed. Additional general strategies that can be employed when considering the initiation of insulin are shown in TABLE 3. Implementation of strategies such as these by family physicians provides patient outcomes comparable to those implemented by endocrinologists or diabetes specialists.33
TABLE 3
General strategies for initiating insulin therapy
| Invite the patient to take an active role in treatment decisions. |
| Remind the patient that type 2 diabetes is primarily self-managed. |
| Discuss the progressive nature of β-cell dysfunction in type 2 diabetes. |
| Emphasize the physiologic role of insulin to maintain glucose homeostasis. |
| Discuss that insulin will help to achieve glycemic control and minimize the risk for long-term complications. |
| Discuss that treatment will be modified as needed to maintain glycemic control and to best meet their needs, capabilities, and interest. |
| Utilize insulin pen devices whenever possible. |
| Emphasize the importance of lifestyle management. |
| Ask if hearing other patients talk of their experiences with insulin therapy would be helpful; consider a group office visit. |
| Discuss and provide the patient with an individualized, written action plan that includes insulin dosing, self-monitoring of blood glucose, and signs/symptoms of hypoglycemia and other adverse events with appropriate action(s) to take. |
| Simplify diabetes (and comorbidities) treatment whenever possible. |
The remainder of this article uses case studies to further explore various patient barriers to insulin therapy and strategies for addressing them with the patient. While other therapies may be appropriate in the case studies below as recommended by current guidelines, these case studies will focus on insulin. In addition, dosing strategies for initiating and intensifying insulin therapy are discussed. Changes to the treatment plan to adjust for comorbidities, such as hypertension and dyslipidemia, or for smoking cessation or aspirin therapy, are not addressed in these cases, but are crucial components of comprehensive management.
CASE STUDY 1
RF is a 49-year-old female insurance analyst diagnosed with T2DM 6 years ago. Initial therapy with lifestyle modifications and metformin has since been intensified. Glimepiride was added, then pioglitazone was added 1.5 years ago when the A1C had risen to 7.5%. There is no evidence of cardiovascular disease. She reports bothersome lower extremity edema and an 8-pound weight increase since starting pioglitazone treatment. RF states that she takes her medications every day, although she acknowledges that she sometimes forgets on Sundays.
Clinical Impression
After taking her history, performing a physical examination, and reviewing her laboratory and self-monitored blood glucose (SMBG) data, her physician concludes that her treatment plan needs to be changed (TABLE 4, TABLE 5).
TABLE 4
Case study 1: Chart notes
| Physical examination | Laboratory tests | Lifestyle habits | Current therapy | |
|---|---|---|---|---|
| Glucose-lowering | Other | |||
| BP: 126/80 mm Hg Weight: 176 lb (79.2 kg) BMI: 27 kg/m2 Eyes: no retinopathy Neurology: intact Skin: intact | SCr: 1.4 mg/dL Albuminuria: negative A1C: 8.2% Cholesterol: Total: 204 mg/dL LDL: 134 mg/dL HDL: 36 mg/dL | Exercise: Walks 2 miles 3-4 d/wk Nutrition: eats 3-4 meals/d | Metformin 1000 mg BID Glimepiride 8 mg QD Pioglitazone 45 mg QD | Lisinopril 30 mg QD Simvastatin 40 mg QD ASA 80 mg QD |
| ASA, acetylsalicylic acid; BMI, body mass index; BP, blood pressure; HDL, high-density lipoprotein; LDL, low-density lipoprotein; SCr, serum creatinine. | ||||
TABLE 5
Case study 1: Self-monitored blood glucose (mg/dL) over the previous 2 weeks
| Day | Fasting | 2 h Post-breakfast | 2 h Post-lunch | 2 h Post-dinner |
|---|---|---|---|---|
| Wednesday | 205 | |||
| Thursday | 158 | |||
| Friday | 179 | |||
| Saturday | 201 | 162 | ||
| Sunday | ||||
| Monday | 166 | |||
| Tuesday | 189 | |||
| Wednesday | ||||
| Thursday | 153 | 221 | ||
| Friday | 150 | |||
| Saturday | 199 | 186 | 213 | |
| Sunday | ||||
| Monday | 181 | |||
| Tuesday | 167 |
Treatment Plan
- Initiate basal insulin once daily in the evening.
- Continue glimepiride, but reduce pioglitazone to 15 mg once daily (or discontinue if cost is a concern).
- Ask RF to monitor fasting blood glucose and self-adjust insulin doses as appropriate.
Barriers
While discussing the need to change the treatment plan and the physician’s suggestion that RF begin basal insulin, RF asks her physician for another few months on her current regimen stating that she will try harder to take her medications on Sundays. She also voices concern that insulin treatment requires injections and that she is concerned about what her coworkers and friends might think. The physician confirms that these concerns are understandable; he also confirms that RF is fearful of needles. The following are possible responses that RF’s physician could use to address these concerns.
Patient’s concern: Perceived failure/low self-efficacy
Physician responses:
- We all forget to do things from time to time, but overall I think you have done a great job taking your medications.
- As we have talked about before, with T2DM there is a progressive loss of insulin production over time regardless of what you do. That is why we added glimepiride and then pioglita-zone and that is why we need to make a change now and put you back in control of your diabetes. It is likely that further changes will be needed and we can discuss and agree on them together.
Patient’s concern: Social stigmatization
Physician responses:
- We can begin by having you administer insulin once daily in the evening in the privacy of your home.
- The insulin can be administered with a device that looks like a pen. It is small and can be carried in your purse; it does not need to be refrigerated once opened. If the time comes that you will need to administer a dose of insulin during the day, you can easily administer the insulin discretely in a public restroom or your work area.
- The use of insulin is more common than it was even a few years ago. In fact, about 5 million people in the United States use insulin to replace what is missing, control blood sugars, and decrease the risk for diabetes complications.34
Patient’s concern: Fear of needles
Physician’s responses:
- Insulin can be injected using a pen device with short, ultrathin needles that makes most of the injections painless. I would like you to see how simple and painless the injection can be by using this sample pen here in the office.
- Many patients are concerned about giving themselves an injection at first, but they quickly become comfortable doing so.
Dosing
Treatment with basal insulin can be initiated using one of several approaches. Using the treat-to-target approach, basal insulin 10 U once daily is initiated.35 The starting dose should be reduced to 6 U if the initial pre-breakfast or pre-dinner blood glucose is < 126 mg/dL or the patient’s body mass index (BMI) is < 26 kg/m2.36 Alternatively, the ADA/EASD recommends starting with 0.2 U/kg, which may be more practical in very overweight or obese patients.2 Titration of the basal insulin dose can be accomplished using one of the following physician-directed or patient-driven treat-to-target titration algorithms (TABLE 6).35,37,38 The insulin dose should be titrated based on the pre-breakfast fasting blood glucose level.
TABLE 6
Physician-directed or patient-driven treat-to-target titration algorithms
| Riddle et al35 | Davies et al37 | Meneghini et al38 | ||||
|---|---|---|---|---|---|---|
| Start with 10 U/d bedtime basal insulin and adjust weekly | Start with 10* U/d bedtime basal insulin and adjust weekly (physician-directed) Or Start with a dose numerically equivalent to the highest FPG (in millimoles/L)† over the previous 7 days and adjust every 3 days (patient-managed) | Start with basal insulin once daily and adjust every 3 days | ||||
| Mean of self-monitored FPG values from preceding 2 days | Change in insulin dose (U/d)# | Mean of self-monitored FPG values from preceding 3 days | Change in insulin dose (U/d) (physician-directed) | Change in insulin dose (U/d) (patient-managed) | Mean of self-monitored FPG values from preceding 3 days | Change in insulin dose (U/d) |
| ≥180 mg/dL 140-180 mg/dL 120-140 mg/dL 100-120 mg/dL | +8 +6 +4 +2 | ≥180 mg/dL (≥10 mmol/L) 140-179 mg/dL (7.8-9.9 mmol/L) 120-139 mg/dL (6.7 – 7.7 mmol/L) 100-119 mg/dL (5.5-6.6 mmol/L) | +6 to +8 +4 +2 0 to +2 | +2 +2 +2 0 to +2 | >110 mg/dL 80-110 mg/dL <80 mg/dL | +3 0 -3 |
| FPG, fasting plasma glucose. *In insulin-naive patients. †For example, if the highest FPG over the previous 7 days was 7 mmol/L, start with 7 U.#Small insulin dose decreases (2-4 U/d per adjustment) were allowed if severe hypoglycemia (requiring assistance) or plasma-referenced glucose < 56 mg/dL was documented in the preceding week. Reproduced with permission. Meneghini LF, et al. J Fam Pract. 2011;60(9 Suppl 1):S21-S28. Quadrant HealthCom Inc. Copyright 2011. | ||||||
Follow-Up Visit
RF begins basal insulin 10 U in the evening and is given simple instructions for insulin dose titration based on fasting plasma glucose results. At her follow-up visit, RF reports that she has increased her basal insulin to 18 U administered once daily. Review of her SMBG results show that her blood glucose levels throughout the day have improved, but are still not at goal. RF’s physician commends her on the progress she has made. RF and her physician agree that she should continue to increase her basal insulin dose. Eight months after beginning basal insulin, RF is administering 28 U (0.35 U/kg) of basal insulin in the evening. Review of her SMBG results over the previous 2 weeks show that her blood glucose rises during the day and is highest after dinner; her current A1C is 7.2%.
Treatment Plan
- Discuss dietary and lifestyle complements to insulin therapy such as:
- Use SMBG to identify foods that raise her blood glucose.
CASE STUDY 2
LW is a 64-year-old male with longstanding hypertension diagnosed with T2DM 8 years ago for which he was treated initially with lifestyle management and metformin. He has since been treated with other oral agents as add-on therapy; glipizide was discontinued due to hypoglycemia when he skips meals (usually lunch); pioglitazone was discontinued after the patient expressed concerns about the risk for bladder cancer he heard on television. He has mild retinopathy and mild loss of vibration sensation in the feet; there is no evidence of cardiovascular disease. He was diagnosed with osteoarthritis 3 years ago.
Clinical Impression
After taking his history, performing a physical examination, and reviewing his laboratory and SMBG data, his physician concludes that his treatment plan needs to be changed (FIGURE 3, TABLE 7, TABLE 8).
TABLE 7
Case study 2: Chart notes
| Physical examination | Laboratory tests | Lifestyle habits | Current therapy | |
|---|---|---|---|---|
| Glucose-lowering | Other | |||
| BP: 124/76 mm Hg Weight: 204 lb (92.7 kg) BMI: 31 kg/m2 Eyes: mild retinopathy Neurology: occasional tingling on bottom of right foot Skin: intact | SCr: 1.9 mg/dL eGFR: 51 mL/min Albuminuria: negative A1C: 8.1% Cholesterol: Total: 218 mg/dL LDL: 118 mg/dL HDL: 55 mg/dL Triglyceride: 204 mg/dL | Exercise: takes dog on occasional walk but otherwise sedentary Nutrition: eats 4 meals/d | Metformin 1000 mg BID Acarbose 50 mg TID Sitagliptin 100 mg QD | Lisinopril/HCTZ 20/25 mg QD Amlodipine 10 mg QD Acetaminophen extended-release 650 mg TID ASA 80 mg QD |
| ASA, acetylsalicylic acid; BMI, body mass index; BP, blood pressure; eGFR, estimated glomerular filtration rate; HCTZ, hydrochlorothiazide; HDL, high-density lipoprotein; LDL, low-density lipoprotein; SCr, serum creatinine. | ||||
TABLE 8
Case study 2: Self-monitored blood glucose (mg/dL) over the previous 2 weeks
| Day | Fasting | 2 h Post-breakfast | 2 h Post-lunch | 2 h Post-dinner |
|---|---|---|---|---|
| Tuesday | 135 | |||
| Wednesday | ||||
| Thursday | 196 | |||
| Friday | 152 | 174 | ||
| Saturday | ||||
| Sunday | 208 | |||
| Monday | 142 | 193 | ||
| Tuesday | ||||
| Wednesday | 130 | 156 | ||
| Thursday | ||||
| Friday | ||||
| Saturday | ||||
| Sunday | 151 | |||
| Monday |
FIGURE 3
Case study 2: A1C levels for April 2004 to March 2012
Treatment Plan
- Discontinue metformin since LW’s serum creatinine is > 1.5 mg/dL.
- Initiate either basal insulin once daily in the evening or premix insulin at dinner.
- Ask LW to monitor his blood glucose and self-adjust insulin doses as appropriate.
- Stress the importance of exercise and proper nutrition; gain agreement on short-term goals for exercise and nutrition.
Barriers
LW’s physician recommends that his treatment plan be changed and insulin therapy initiated. LW quickly responds that previous changes to his treatment regimen have not resulted in his achieving an A1C < 7.0%. He also doubts that he can use a syringe to draw up the correct dose and then self-administer due to his arthritis. The following are possible responses his physician could use to address these concerns.
Patient concern: Repeated experience of failing to achieve glycemic control, ie, A1C < 7.0%
Physician responses:
- While achieving an A1C < 7.0% is a realistic goal that reduces the risks for vascular complications of diabetes, any reduction of A1C will be of benefit.
- I would like to work with you to implement a new plan that we both believe will enable you to improve your diabetes control and ideally achieve an A1C < 7.0%.
Patient concern: Self-administering due to arthritis
Physician responses:
- Instead of using a syringe and vial to draw up and administer insulin, I would like you to use an insulin pen device. As you can see, it is easy to handle and you can easily select the correct dose.
- If you choose to start on premix insulin, the pen device contains both types of insulin together in one dose.
Dosing
Treatment with basal insulin once daily in the evening can be initiated and titrated based on pre-breakfast blood glucose as in Case Study 1. Alternatively, treatment with premix insulin can be initiated at a dose of 12 U administered within 15 minutes of dinner initiation. The premix dose can be titrated using the algorithm employed in the 1-2-3 Study based on pre-breakfast blood glucose (TABLE 9).39 After 16 weeks, 41% of patients in the 1-2-3 Study achieved an A1C < 7.0% from a baseline A1C of 8.6%.
TABLE 9
1-2-3 Study algorithm39
| Pre-breakfast SMBG (mg/dL) | Adjustment of pre-dinner dose (U) |
|---|---|
| <80 | -3 |
| 80-110 | No change |
| 111-140 | +3 |
| 141-180 | +6 |
| > 180 | +9 |
| SMBG, self-monitored blood glucose. | |
Follow-Up Visit
LW began basal insulin 10 U in the evening. Over the next 5.5 months, he titrated his dose such that his current dose is 46 U (0.50 U/kg) in the evening. His current A1C is 7.3%. Review of his SMBG shows consistently high 2-hour post-lunch blood glucose levels. Although further increasing his basal insulin dose is an option, in most of the treat-to-target studies, the daily dose of basal insulin given once daily averaged between 0.4 and 0.6 U/kg.35,37,40,41 LW and his physician agree that adding rapid-acting insulin at lunch is the best option. The starting dose of rapid-acting bolus insulin is 4 to 6 U administered prior to the largest meal of the day or, as in this case, prior to the meal with the largest postprandial blood glucose excursion.42,43 Alternatively, the dose of rapid-acting insulin could be calculated as 10% of the total daily dose of basal insulin, which in this case is 5 U (10% x 46 U). The dose of basal insulin would be reduced by 5 U if the rapid-acting insulin is given at dinner in order to reduce the risk for nocturnal hypoglycemia. The dose of the bolus insulin can be titrated using the ExtraSTEP algorithm (TABLE 10).42 Alternatively, the SimpleSTEP algorithm can be used which does not require a 2-hour postprandial glucose measurement.42
TABLE 10
Algorithms for adjusting insulin aspart42
| ExtraSTEP algorithm | SimpleSTEP algorithm | |||
|---|---|---|---|---|
| 2-h Post-meal PG level (mg/dL) | Insulin aspart adjustment (U) | Pre-meal BG (mg/dL) | Bedtime BG (mg/dL) | Insulin aspart adjustment (U) |
| <72* | -2 | <72* | <72* | -2 |
| 72-144 | 0 | 72-108 | 72-144 | 0 |
| 145-180 | +2 | 109-162 | 145-180 | +2 |
| >180 | +4 | >162 | >180 | +4 |
| BG, blood glucose; PG, plasma glucose. *One or more PG values <72 mg/dL without obvious explanation. Reproduced with permission. Meneghini LF, et al. J Fam Pract. 2011;60(9 Suppl 1):S21-S28. Quadrant HealthCom Inc. Copyright 2011. | ||||
Plan
- Begin rapid-acting insulin 5 U at lunch.
- Continue basal insulin at 46 U in the evening.
- Ask LW to continue to titrate basal insulin based on the pre-breakfast blood glucose level and the lunch time bolus insulin dose based on the 2-hour post-lunch SMBG (ExtraSTEP); alternatively, adjust based on the pre-dinner blood glucose level (SimpleSTEP).
CASE STUDY 3
MB is a 46-year-old male who had not consulted a physician since having a physical examination 6 years ago. He presented 2 weeks ago with frequent urination (7-8 times/day) and feeling tired; he also noted losing 5 pounds (2.25 kg) over the preceding 3.5 months despite no changes in his diet. MB is a regional salesperson with an erratic schedule. During the week, he eats lunch and most dinners in a restaurant. On the weekend, he goes to a local bar with his friends. He does light yard work, but does not exercise regularly. He is a current smoker with a 36 pack-year history. Urinalysis shows ketonuria and microalbuminuria. His A1C reported back today is 10.8%, confirming a diagnosis of uncontrolled and symptomatic DM.
Clinical Impression
After taking his history, performing a physical examination, and reviewing his laboratory data, MB’s physician confirms a diagnosis of DM (TABLE 11). While it is likely that MB has T2DM, his physician wants to rule out type 1 DM and latent autoimmune diabetes of the adult (LADA), so he orders tests for antibodies (GAD, IA-2, ICA). The antibody testing is negative, making T2DM the most likely diagnosis.
TABLE 11
Case study 3: Chart notes
| Physical examination | Laboratory tests | Lifestyle habits | Current therapy | |
|---|---|---|---|---|
| Glucose-lowering | Other | |||
| BP: 142/88 mm Hg Weight: 176 lb (79.2 kg) BMI: 27 kg/m2 Eyes: no retinopathy Neurology: intact Skin: intact | SCr: 1.4 mg/dL Microalbumin:creatinine ratio: 140 mg/g creatinine Ketonuria: 1+ A1C: 10.8% Cholesterol: Total: 210 mg/dL LDL: 146 mg/dL HDL: 30 mg/dL | Exercise: light yard work, no regular exercise Nutrition: 3 meals/d, eats most meals in a restaurant (lunch M-F; dinner 3-4 nights/wk) | None | None |
| BMI, body mass index; BP, blood pressure; HDL, high-density lipoprotein; LDL, low-density lipoprotein; SCr, serum creatinine. | ||||
Treatment Plan
- Initiate basal-bolus therapy with fixed bolus doses of rapid-acting insulin at each meal (prandial insulin).
- Ask MB to monitor blood glucose before meals and at bedtime.
- Provide MB with a supplemental scale to correct hyperglycemia before meals.
- Stress the importance of exercise and proper nutrition; gain agreement for short-term goals for exercise and nutrition; refer for diabetes and nutrition education if available.
- Discuss the importance of smoking cessation; develop a plan.
- Consider metformin and other non-insulin therapies when A1C is under control.
Barriers
MB is surprised that he has T2DM and is clearly anxious at receiving the diagnosis. He expresses concern about starting insulin because his uncle died within a year of starting insulin. MB also recalls that his uncle was always giving himself shots and monitoring his blood glucose level. He wants to know whether there is a simpler treatment option if he agrees to start insulin treatment. He also wants to know whether he will have to remain on insulin for the rest of his life. The following are possible responses his physician could use to address these concerns.
Patient concern: Fear of death
Physician responses:
- Uncontrolled high blood sugars over a long period of time can cause serious complications, such as kidney and heart disease that can result in death. That is why it is important that we work together to gain control of your blood sugar levels over the next few months and then modify your treatment as needed to maintain control.
- Unfortunately for many patients in the past, treatment with insulin was not used until it was too late and people already had serious complications from DM. This is likely the case for your uncle.
Patient concern: Treatment complexity
Physician responses:
- Right now we have to control your blood glucose rapidly so your pancreas can regain some function and your body can better respond to insulin.
- I will also provide you with step-by-step written instructions you can follow that describe how to start insulin and how to monitor your blood glucose.
- We will communicate as often as you need to adjust your insulin doses over the next few weeks; when you feel comfortable, I can even show you how to adjust your insulin dose before a meal to correct a high blood sugar.
- We can try this treatment for 3 months and then reevaluate your response, how you feel, and whether you want to continue to modify your treatment plan to keep your blood sugars controlled.
Patient concern: Lack of understanding that T2DM is a serious disease
Physician responses:
- Please understand that T2DM is a serious disease that increases your risk for heart disease, stroke, blindness, and other diseases. Unfortunately, since diabetes does not cause bad symptoms until it is actually too late, many patients do not make the effort to properly control their diabetes. By working together, we can reduce the risk for these complications and do some screening tests to detect any complications before they become irreversible.
Dosing
There are several approaches to determining the initial doses of basal and prandial (bolus) insulin. One approach is to estimate the total daily dose (TDD) of insulin by multiplying the patient’s weight in kilograms by 0.5 U/kg/d.44 Half of the TDD is given as basal insulin replacement; the other half is divided into 3 fixed preprandial doses of rapid-acting insulin. When the patient is ready to take on more complex management, the supplemental dose for bolus insulin can be calculated using a correction factor. If the bolus insulin is a rapid-acting insulin analog, 1800 is divided by the TDD of insulin; 1500 is used for a short-acting human insulin. This correction factor is an estimate of the fall in blood glucose per unit of bolus insulin. In our patient, the TDD would be: 80 kg x 0.5 U/kg/d or 40 U/d of insulin. Thus, 1 U of insulin should lower the blood glucose by about 45 mg/dL (1800/40 U = 45 mg/dL). For every 45 mg/dL above the pre-meal target, the patient would add 1 U of rapid-acting insulin to correct the hyperglycemia over the next 4 to 5 hours. The basal and prandial insulin doses would be titrated on a periodic basis (perhaps every 1 to 2 weeks) until the daytime levels of blood glucose are on target. The fasting (pre-breakfast) blood glucose would be used to adjust the basal insulin dose, while the pre-lunch, pre-dinner, and bedtime blood glucose results would be used to adjust the pre-breakfast, pre-lunch, and pre-dinner prandial (rapid-acting) insulin doses, respectively.
An alternative approach to initiating basal-bolus therapy is the PREFER algorithm.45 Here, the basal insulin dose is 10 U initially. The bolus doses are administered in a 3:1:2 ratio, so if the total of the 3 bolus doses is 12 U/d, the initial bolus doses would be 6 (breakfast), 2 (lunch), and 4 (dinner) U. The mean basal (once-daily) and bolus insulin doses observed in PREFER are shown in TABLE 12 and TABLE 13.
TABLE 12
Case study 3: Calculating initial basal-bolus insulin doses
| Algorithm | Calculations | Patient MB |
|---|---|---|
| Meneghini44 | TDD = (total body weight [kg]) (0.5 U/kg/d) Basal insulin dose* = (50%) (TDD) Bolus insulin dose† = (10%-20%) (TDD) | TDD = (0.5 U/kg/d)(80kg) = 40 U/d |
| Basal = (50%) (40 U/d) = 20 U/d | ||
| Bolus = (10%-20%) (40 U/d) = 4 to 8 U/meal | ||
| CF = 1800/40 U/d = 45 mg/dL per 1 unit | ||
| PREFER45 | Basal insulin dose* = 10 U (14 U if BMI > 32 kg/m2) Bolus insulin dose† = ratio of 3:1:2 (breakfast:lunch:dinner) Note: At week 26, the bolus insulin doses were divided into the 3 daily meals in approximately a 1:1:1 ratio | |
| BMI, body mass index; CF, correction factor; TDD, total daily dose of insulin. *Once daily; †Three meals per day. | ||
TABLE 13
Titrating the basal insulin dose using the PREFER algorithm45
| Pre-breakfast blood glucose (mg/dL) | Basal insulin dose adjustment (U) |
|---|---|
| < 56 | -4 |
| 56-72 | -2 |
| 73-125 | No change |
| 126-140 | +2 |
| 141-160 | +4 |
| 161-180 | +6 |
| 181-200 | +8 |
| > 200 | +10 |
Follow-up Visit
MB begins with basal insulin 20 U in the evening and bolus insulin at doses of 7 U before each meal. Over the next several months, MB has titrated his insulin doses; his current doses are: 32 U (basal), 11 U (bolus-breakfast), 7 U (bolus-lunch), and 10 U (bolus-dinner). He experienced 1 episode of mild hypoglycemia (SMBG, 50 mg/dL) one afternoon following a particularly active morning (TABLE 14). His current A1C is 7.4%. MB’s physician congratulates him on the progress he has made in dramatically lowering his blood glucose level—and his risk for diabetes-related complications. While MB appreciates his physician’s support and admits that he does not feel tired and generally feels better, which is likely due to resolution of glucotoxicity, he is not happy that he has gained 5.5 pounds (2.5 kg).46 He also finds the timing and administration of bolus insulin difficult.
TABLE 14
Case study 3: Self-monitored blood glucose (mg/dL) over the previous 2 weeks
| Day | Fasting | 2 h Post-breakfast | 2 h Post-lunch | 2 h Post-dinner |
|---|---|---|---|---|
| Wednesday | ||||
| Thursday | 168 | |||
| Friday | 106 | 166 | 174 | |
| Saturday | 88 | |||
| Sunday | 195 | |||
| Monday | 134 | |||
| Tuesday | 172 | |||
| Wednesday | 130 | 156 | ||
| Thursday | 112 | 168 | ||
| Friday | 92 | 164 | ||
| Saturday | 50 | 149 | 159 | 176 |
| Sunday | 94 | 174 | 210 | |
| Monday | 176 | 184 | ||
| Tuesday | 117 | 169 |
Plan
- Continue basal insulin once-daily in the evening.
- Add metformin 500 mg BID and increase to 1000 mg BID as tolerated.
- Consider weaning down the bolus insulin doses and substituting them with a GLP-1R agonist, dipeptidyl peptidase-4 inhibitor, or short-acting secretagogue. If so, continue rapid-acting insulin during transition. [Note: the following combinations are not currently approved by the US FDA: exenatide twice-daily and prandial insulin; exenatide once-weekly and insulin; liraglutide and prandial insulin; linagliptin and insulin.]
CASE STUDY 4
KW is a 62-year-old female diagnosed with T2DM 12 years ago. Treatment with lifestyle management and metformin initially provided glycemic control. Glimepiride was subsequently added and eventually the patient was started on basal insulin. The current dose of basal insulin is 60 U in the evening. Five months ago her A1C was found to be 7.9% and more recently 8.3%. She drinks alcohol occasionally and smokes. KW works as an executive secretary and has a consistent meal and activity schedule.
Clinical Impression
Following completion of the history, physical examination, and review of her laboratory data, KW’s physician concludes that her insulin regimen should be intensified (TABLE 15, TABLE 16).
TABLE 15
Case study 4: Chart notes
| Physical examination | Laboratory tests | Lifestyle habits | Current therapy | |
|---|---|---|---|---|
| Glucose-lowering | Other | |||
| BP: 126/78 mm Hg Weight: 176 lb (79.2 kg) BMI: 32 kg/m2 Eyes: no retinopathy Neurology: intact Skin: intact | SCr: 1.0 mg/dL Albuminuria: negative A1C: 8.3% Cholesterol Total: 172 mg/dL LDL: 96 mg/dL HDL: 46 mg/dL Triglycerides: 138 mg/dL | Exercise: sedentary Nutrition: 3 meals/d with large dinner | Metformin 1000 mg BID Basal insulin 60 U in the evening | ASA 80 mg QD Pravastatin 40 mg qHS |
| BMI, body mass index; BP, blood pressure; HDL, high-density lipoprotein; LDL, low-density lipoprotein; SCr, serum creatinine. | ||||
TABLE 16
Case study 4: Self-monitored blood glucose (mg/dL) over the previous 2 weeks
| Day | Fasting | 2 h Post-breakfast | 2 h Post-lunch | 2 h Post-dinner |
|---|---|---|---|---|
| Friday | ||||
| Saturday | 156 | 244 | ||
| Sunday | 253 | |||
| Monday | ||||
| Tuesday | ||||
| Wednesday | 148 | 227 | ||
| Thursday | ||||
| Friday | ||||
| Saturday | 179 | |||
| Sunday | 160 | |||
| Monday | ||||
| Tuesday | ||||
| Wednesday | ||||
| Thursday |
Plan
- Discontinue basal insulin.
- Begin premix insulin twice daily before breakfast and dinner.
- Ask KW to monitor blood glucose two times daily and, if appropriate, teach her how to self-adjust insulin doses.
- Stress the importance of exercise and proper nutrition; gain agreement on short-term goals for exercise and nutrition.
- Discuss the importance of smoking cessation; develop a plan.
Barriers
The physician discusses with KW that her consistent meal and activity schedule would make switching to premix insulin twice daily a good choice. KW is generally in agreement with the change, but wonders whether hypoglycemia might be more likely. She also asks if she might gain more weight in addition to the 3 pounds (1.35 kg) she has gained since starting basal insulin.
Patient concern: Hypoglycemia
Physician responses:
- Hypoglycemia remains a concern, and is more frequently seen with premix than with basal insulin; however, as long as you remain consistent with your meal and activity schedule, the risk for bad hypoglycemia is low.
- We should review your written action plan so that you are sure what signs or symptoms of a low blood sugar might occur and what you should do to treat them.
Patient concern: Weight gain
Physician responses:
- It is possible that you might gain a few additional pounds. You can avoid this by increasing your physical activity, and importantly, continue healthy eating. We should schedule a time for you to meet again with a dietician who can discuss options that might work for you.
Dosing
There are different approaches for converting from basal insulin to twice-daily premix insulin. One approach is to determine the TDD of basal insulin, and give half at breakfast and the other half at dinner as premix insulin.39 Since KW is taking 60 U of basal in the evening, she should take 30 U at breakfast and 30 U at dinner. Dose titration is according to the 1-2-3 Study algorithm shown in case study 2.
Another approach is to administer biphasic insulin aspart 70/30 0.2 U/kg before breakfast and 0.1 U/kg before dinner as was done in the PREFER study (TABLE 13).45 Subsequent dosing can be determined based on the PREFER algorithm below. Of note is that at study end, premix insulin doses were equally divided between breakfast and dinner. Breakfast and dinner doses are titrated based on blood glucose levels before dinner and breakfast, respectively. In the PREFER study, the use of premix insulin provided comparable A1C reduction as basal-bolus therapy (basal once daily + bolus TID) in insulin-naïve patients. However, patients previously treated with basal insulin such as KW experienced greater A1C reductions with basal-bolus insulin than with premix insulin.
1. Rodbard HW, Jellinger PS, Davidson JA, et al. Statement by an American Association of Clinical Endocrinologists/American College of Endocrinology consensus panel on type 2 diabetes mellitus: an algorithm for glycemic control [published correction appears in Endocr Pract. 2009;15(7):768-770]. Endocr Pract. 2009;15(6):540-559.
2. Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of hyperglycemia in type 2 diabetes: a patient-centered approach: position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD) [published online ahead of print April 19, 2012]. Diabetes Care. doi:10.2337/dc12-0413.
3. Lalić NM, Micić D, Antić S, et al. Effect of biphasic insulin aspart on glucose and lipid control in patients with type 2 diabetes mellitus. Expert Opin Pharmacother. 2007;8(17):2895-2901.
4. Reynolds LR, Kingsley FJ, Karounos DG, Tannock LR. Differential effects of rosiglitazone and insulin glargine on inflammatory markers, glycemic control, and lipids in type 2 diabetes. Diabetes Res Clin Pract. 2007;77(2):180-187.
5. Rosak C, Jung R, Hofmann U. Insulin glargine maintains equivalent glycemic control and better lipometabolic control than NPH insulin in type 1 diabetes patients who missed a meal. Horm Metab Res. 2008;40(8):544-548.
6. Ampudia-Blasco FJ, Girbes J, Carmena R. A case of lipoatrophy with insulin glargine: long-acting insulin analogs are not exempt from this complication. Diabetes Care. 2005;28(12):2983.-
7. Griffin ME, Feder A, Tamborlane WV. Lipoatrophy associated with lispro insulin in insulin pump therapy: an old complication, a new cause? Diabetes Care. 2001;24(1):174.-
8. Fineberg SE, Huang J, Brunelle R, Gulliya KS, Anderson JH, Jr. Effect of long-term exposure to insulin lispro on the induction of antibody response in patients with type 1 or type 2 diabetes. Diabetes Care. 2003;26(1):89-96.
9. Moyes V, Driver R, Croom A, Mirakian R, Chowdhury TA. Insulin allergy in a patient with type 2 diabetes successfully treated with continuous subcutaneous insulin infusion. Diabet Med. 2006;23(2):204-206.
10. Ghosh S, McCann V, Bartle L, Collier A, Malik I. Allergy to insulin detemir. Diabet Med. 2007;24(11):1307.-
11. Blumer I. Severe, delayed insulin detemir injection site reaction. Diabet Med. 2008;25(8):1008.-
12. Pérez E, González R, Martínez J, Iglesias J, Matheu V. Detemir insulin-induced anaphylaxis. Ann Allergy Asthma Immunol. 2009;102(2):174-175.
13. Mollar-Puchades MA, Villanueva IL. Insulin glulisine in the treatment of allergy to rapid acting insulin and its rapid acting analogs. Diabetes Res Clin Pract. 2009;83(1):e21-e22.
14. Kawasaki F, Kamei S, Tatsumi F, et al. Gallbladder edema in type 1 diabetic patient due to delayed-type insulin allergy. Intern Med. 2009;48(17):1545-1549.
15. Wang C, Ding ZY, Shu SQ, et al. Severe insulin allergy after percutaneous transluminal coronary angioplasty. Clin Ther. 2009;31(3):569-574.
16. Ozaki N, Oiso Y. Immunologic tolerance to the insulin analogue glulisine. Diabetes Care. 2010;33(3):e39.-
17. Koroscil T, Kagzi Y, Zacharias D. Failure of multiple therapies in the treatment of a type 1 diabetic patient with insulin allergy: a case report. Endocr Pract. 2011;17(1):91-94.
18. Tuerk PW, Mueller M, Egede LE. Estimating physician effects on glycemic control in the treatment of diabetes: methods, effects sizes, and implications for treatment policy. Diabetes Care. 2008;31(5):869-873.
19. Hajos TR, Polonsky WH, Twisk JW, Dain MP, Snoek FJ. Do physicians understand type 2 diabetes patients’ perceptions of seriousness; the emotional impact and needs for care improvement? A cross-national survey. Patient Educ Couns. 2011;85(2):258-263.
20. Polonsky WH, Hajos TR, Dain MP, Snoek FJ. Are patients with type 2 diabetes reluctant to start insulin therapy? An examination of the scope and underpinnings of psychological insulin resistance in a large, international population. Curr Med Res Opin. 2011;27(6):1169-1174.
21. Nam S, Chesla C, Stotts NA, Kroon L, Janson SL. Factors associated with psychological insulin resistance in individuals with type 2 diabetes. Diabetes Care. 2010;33(8):1747-1749.
22. Nakar S, Yitzhaki G, Rosenberg R, Vinker S. Transition to insulin in type 2 diabetes: family physicians’ misconception of patients’ fears contributes to existing barriers. J Diabetes Complications. 2007;21(4):220-226.
23. Snoek FJ. Breaking the barriers to optimal glycaemic control—what physicians need to know from patients’ perspectives. Int J Clin Pract Suppl. 2002;(129):80-84.
24. Korytkowski M. When oral agents fail: practical barriers to starting insulin. Int J Obes Relat Metab Disord. 2002;26(Suppl 3):S18-S24.
25. Skovlund SE, Peyrot M. The Diabetes Attitudes, Wishes and Needs (DAWN) program: a new approach to improving outcomes of diabetes care. Diabetes Spectrum. 2005;18(3):136-142.
26. Jenkins N, Hallowell N, Farmer AJ, Holman RR, Lawton J. Initiating insulin as part of the Treating To Target in Type 2 Diabetes (4-T) trial: an interview study of patients’ and health professionals’ experiences. Diabetes Care. 2010;33(10):2178-2180.
27. Levinson W, Gorawara-Bhat R, Lamb J. A study of patient clues and physician responses in primary care and surgical settings. JAMA. 2000;284(8):1021-1027.
28. Rodriguez HP, Anastario MP, Frankel RM, et al. Can teaching agenda-setting skills to physicians improve clinical interaction quality? A controlled intervention. BMC Med Educ. 2008;8:3.-
29. Solomon J. How strategies for managing patient visit time affect physician job satisfaction: a qualitative analysis. J Gen Intern Med. 2008;23(6):775-780.
30. Funnell MM, Anderson RM. Empowerment and self-management of diabetes. Clin Diabetes. 2004;22(3):123-127.
31. Funnell MM, Anderson RM. Are patients or outcomes more important? Rev Endocrinol. 2008;2(8):49-51.
32. Shaefer CF. Clinical inertia: overcoming a major barrier to diabetes management. Insulin. 2006;1(2):61-64.
33. Harris S, Yale JF, Dempsey E, Gerstein H. Can family physicians help patients initiate basal insulin therapy successfully?: randomized trial of patient-titrated insulin glargine compared with standard oral therapy: lessons for family practice from the Canadian INSIGHT trial. Can Fam Physician. 2008;54(4):550-558.
34. Centers for Disease Control and Prevention. National diabetes fact sheet national estimates and general information on diabetes and prediabetes in the United States, 2011. Atlanta, GA: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, 2011. http://www.cdc.gov/diabetes/pubs/pdf/ndfs_2011.pdf. Accessed February 10, 2011.
35. Riddle MC, Rosenstock J, Gerich J. Insulin Glargine 4002 Study Investigators. The treat-to-target trial: randomized addition of glargine or human NPH insulin to oral therapy of type 2 diabetic patients. Diabetes Care. 2003;26(11):3080-3086.
36. Hermansen K, Davies M, Derezinski T, Martinez Ravn G, Clauson P, Home P. A 26-week, randomized, parallel, treat-to-target trial comparing insulin detemir with NPH insulin as add-on therapy to oral glucose-lowering drugs in insulin-naive people with type 2 diabetes [published correction appears in Diabetes Care. 2007;30(4):1035]. Diabetes Care. 2006;29(6):1269-1274.
37. Davies M, Storms F, Shutler S, Bianchi-Biscay M, Gomis R. ATLANTUS Study Group. Improvement of glycemic control in subjects with poorly controlled type 2 diabetes: comparison of two treatment algorithms using insulin glargine. Diabetes Care. 2005;28(6):1282-1288.
38. Meneghini L, Koenen C, Weng W, Selam JL. The usage of a simplified self-titration dosing guideline (303 Algorithm) for insulin detemir in patients with type 2 diabetes—results of the randomized, controlled PREDICTIVE 303 study. Diabetes Obes Metab. 2007;9(6):902-913.
39. Garber AJ, Wahlen J, Wahl T, et al. Attainment of glycaemic goals in type 2 diabetes with once-, twice-, or thrice-daily dosing with biphasic insulin aspart 70/30 (The 1-2-3 study). Diabetes Obes Metab. 2006;8(1):58-66.
40. Philis-Tsimikas A, Charpentier G, Clauson P, Ravn GM, Roberts VL, Thorsteinsson B. Comparison of once-daily insulin detemir with NPH insulin added to a regimen of oral antidiabetic drugs in poorly controlled type 2 diabetes [published correction appears in Clin Ther. 2006;28(11):1967]. Clin Ther. 2006;28(10):1569-1581.
41. Blonde L, Merilainen M, Karwe V, Raskin P. TITRATE Study Group. Patient-directed titration for achieving glycaemic goals using a once-daily basal insulin analogue: an assessment of two different fasting plasma glucose targets—the TITRATE study. Diabetes Obes Metab. 2009;11(6):623-631.
42. Meneghini L, Mersebach H, Kumar S, Svendsen AL, Hermansen K. Comparison of 2 intensification regimens with rapid-acting insulin aspart in type 2 diabetes mellitus inadequately controlled by once-daily insulin detemir and oral antidiabetes drugs: the step-wise randomized study. Endocr Pract. 2011;17(5):727-736.
43. Lankisch MR, Ferlinz KC, Leahy JL, Scherbaum WA. Orals Plus Apidra and LANTUS (OPAL) study group. Introducing a simplified approach to insulin therapy in type 2 diabetes: a comparison of two single-dose regimens of insulin glulisine plus insulin glargine and oral antidiabetic drugs [published correction appears in Diabetes Obes Metab. 2010;12(5):461]. Diabetes Obes Metab 2008;10(12):1178-1185.
44. Meneghini L. Why and how to use insulin therapy earlier in the management of type 2 diabetes. South Med J. 2007;100(2):164-174.
45. Liebl A, Prager R, Binz K, Kaiser M, Bergenstal R, Gallwitz B. PREFER Study Group. Comparison of insulin analogue regimens in people with type 2 diabetes mellitus in the PREFER Study: a randomized controlled trial. Diabetes Obes Metab. 2009;11(1):45-52.
46. Braun A, Sämann A, Kubiak T, et al. Effects of metabolic control, patient education and initiation of insulin therapy on the quality of life of patients with type 2 diabetes mellitus. Patient Educ Couns. 2008;73(1):50-59.
1. Rodbard HW, Jellinger PS, Davidson JA, et al. Statement by an American Association of Clinical Endocrinologists/American College of Endocrinology consensus panel on type 2 diabetes mellitus: an algorithm for glycemic control [published correction appears in Endocr Pract. 2009;15(7):768-770]. Endocr Pract. 2009;15(6):540-559.
2. Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of hyperglycemia in type 2 diabetes: a patient-centered approach: position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD) [published online ahead of print April 19, 2012]. Diabetes Care. doi:10.2337/dc12-0413.
3. Lalić NM, Micić D, Antić S, et al. Effect of biphasic insulin aspart on glucose and lipid control in patients with type 2 diabetes mellitus. Expert Opin Pharmacother. 2007;8(17):2895-2901.
4. Reynolds LR, Kingsley FJ, Karounos DG, Tannock LR. Differential effects of rosiglitazone and insulin glargine on inflammatory markers, glycemic control, and lipids in type 2 diabetes. Diabetes Res Clin Pract. 2007;77(2):180-187.
5. Rosak C, Jung R, Hofmann U. Insulin glargine maintains equivalent glycemic control and better lipometabolic control than NPH insulin in type 1 diabetes patients who missed a meal. Horm Metab Res. 2008;40(8):544-548.
6. Ampudia-Blasco FJ, Girbes J, Carmena R. A case of lipoatrophy with insulin glargine: long-acting insulin analogs are not exempt from this complication. Diabetes Care. 2005;28(12):2983.-
7. Griffin ME, Feder A, Tamborlane WV. Lipoatrophy associated with lispro insulin in insulin pump therapy: an old complication, a new cause? Diabetes Care. 2001;24(1):174.-
8. Fineberg SE, Huang J, Brunelle R, Gulliya KS, Anderson JH, Jr. Effect of long-term exposure to insulin lispro on the induction of antibody response in patients with type 1 or type 2 diabetes. Diabetes Care. 2003;26(1):89-96.
9. Moyes V, Driver R, Croom A, Mirakian R, Chowdhury TA. Insulin allergy in a patient with type 2 diabetes successfully treated with continuous subcutaneous insulin infusion. Diabet Med. 2006;23(2):204-206.
10. Ghosh S, McCann V, Bartle L, Collier A, Malik I. Allergy to insulin detemir. Diabet Med. 2007;24(11):1307.-
11. Blumer I. Severe, delayed insulin detemir injection site reaction. Diabet Med. 2008;25(8):1008.-
12. Pérez E, González R, Martínez J, Iglesias J, Matheu V. Detemir insulin-induced anaphylaxis. Ann Allergy Asthma Immunol. 2009;102(2):174-175.
13. Mollar-Puchades MA, Villanueva IL. Insulin glulisine in the treatment of allergy to rapid acting insulin and its rapid acting analogs. Diabetes Res Clin Pract. 2009;83(1):e21-e22.
14. Kawasaki F, Kamei S, Tatsumi F, et al. Gallbladder edema in type 1 diabetic patient due to delayed-type insulin allergy. Intern Med. 2009;48(17):1545-1549.
15. Wang C, Ding ZY, Shu SQ, et al. Severe insulin allergy after percutaneous transluminal coronary angioplasty. Clin Ther. 2009;31(3):569-574.
16. Ozaki N, Oiso Y. Immunologic tolerance to the insulin analogue glulisine. Diabetes Care. 2010;33(3):e39.-
17. Koroscil T, Kagzi Y, Zacharias D. Failure of multiple therapies in the treatment of a type 1 diabetic patient with insulin allergy: a case report. Endocr Pract. 2011;17(1):91-94.
18. Tuerk PW, Mueller M, Egede LE. Estimating physician effects on glycemic control in the treatment of diabetes: methods, effects sizes, and implications for treatment policy. Diabetes Care. 2008;31(5):869-873.
19. Hajos TR, Polonsky WH, Twisk JW, Dain MP, Snoek FJ. Do physicians understand type 2 diabetes patients’ perceptions of seriousness; the emotional impact and needs for care improvement? A cross-national survey. Patient Educ Couns. 2011;85(2):258-263.
20. Polonsky WH, Hajos TR, Dain MP, Snoek FJ. Are patients with type 2 diabetes reluctant to start insulin therapy? An examination of the scope and underpinnings of psychological insulin resistance in a large, international population. Curr Med Res Opin. 2011;27(6):1169-1174.
21. Nam S, Chesla C, Stotts NA, Kroon L, Janson SL. Factors associated with psychological insulin resistance in individuals with type 2 diabetes. Diabetes Care. 2010;33(8):1747-1749.
22. Nakar S, Yitzhaki G, Rosenberg R, Vinker S. Transition to insulin in type 2 diabetes: family physicians’ misconception of patients’ fears contributes to existing barriers. J Diabetes Complications. 2007;21(4):220-226.
23. Snoek FJ. Breaking the barriers to optimal glycaemic control—what physicians need to know from patients’ perspectives. Int J Clin Pract Suppl. 2002;(129):80-84.
24. Korytkowski M. When oral agents fail: practical barriers to starting insulin. Int J Obes Relat Metab Disord. 2002;26(Suppl 3):S18-S24.
25. Skovlund SE, Peyrot M. The Diabetes Attitudes, Wishes and Needs (DAWN) program: a new approach to improving outcomes of diabetes care. Diabetes Spectrum. 2005;18(3):136-142.
26. Jenkins N, Hallowell N, Farmer AJ, Holman RR, Lawton J. Initiating insulin as part of the Treating To Target in Type 2 Diabetes (4-T) trial: an interview study of patients’ and health professionals’ experiences. Diabetes Care. 2010;33(10):2178-2180.
27. Levinson W, Gorawara-Bhat R, Lamb J. A study of patient clues and physician responses in primary care and surgical settings. JAMA. 2000;284(8):1021-1027.
28. Rodriguez HP, Anastario MP, Frankel RM, et al. Can teaching agenda-setting skills to physicians improve clinical interaction quality? A controlled intervention. BMC Med Educ. 2008;8:3.-
29. Solomon J. How strategies for managing patient visit time affect physician job satisfaction: a qualitative analysis. J Gen Intern Med. 2008;23(6):775-780.
30. Funnell MM, Anderson RM. Empowerment and self-management of diabetes. Clin Diabetes. 2004;22(3):123-127.
31. Funnell MM, Anderson RM. Are patients or outcomes more important? Rev Endocrinol. 2008;2(8):49-51.
32. Shaefer CF. Clinical inertia: overcoming a major barrier to diabetes management. Insulin. 2006;1(2):61-64.
33. Harris S, Yale JF, Dempsey E, Gerstein H. Can family physicians help patients initiate basal insulin therapy successfully?: randomized trial of patient-titrated insulin glargine compared with standard oral therapy: lessons for family practice from the Canadian INSIGHT trial. Can Fam Physician. 2008;54(4):550-558.
34. Centers for Disease Control and Prevention. National diabetes fact sheet national estimates and general information on diabetes and prediabetes in the United States, 2011. Atlanta, GA: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, 2011. http://www.cdc.gov/diabetes/pubs/pdf/ndfs_2011.pdf. Accessed February 10, 2011.
35. Riddle MC, Rosenstock J, Gerich J. Insulin Glargine 4002 Study Investigators. The treat-to-target trial: randomized addition of glargine or human NPH insulin to oral therapy of type 2 diabetic patients. Diabetes Care. 2003;26(11):3080-3086.
36. Hermansen K, Davies M, Derezinski T, Martinez Ravn G, Clauson P, Home P. A 26-week, randomized, parallel, treat-to-target trial comparing insulin detemir with NPH insulin as add-on therapy to oral glucose-lowering drugs in insulin-naive people with type 2 diabetes [published correction appears in Diabetes Care. 2007;30(4):1035]. Diabetes Care. 2006;29(6):1269-1274.
37. Davies M, Storms F, Shutler S, Bianchi-Biscay M, Gomis R. ATLANTUS Study Group. Improvement of glycemic control in subjects with poorly controlled type 2 diabetes: comparison of two treatment algorithms using insulin glargine. Diabetes Care. 2005;28(6):1282-1288.
38. Meneghini L, Koenen C, Weng W, Selam JL. The usage of a simplified self-titration dosing guideline (303 Algorithm) for insulin detemir in patients with type 2 diabetes—results of the randomized, controlled PREDICTIVE 303 study. Diabetes Obes Metab. 2007;9(6):902-913.
39. Garber AJ, Wahlen J, Wahl T, et al. Attainment of glycaemic goals in type 2 diabetes with once-, twice-, or thrice-daily dosing with biphasic insulin aspart 70/30 (The 1-2-3 study). Diabetes Obes Metab. 2006;8(1):58-66.
40. Philis-Tsimikas A, Charpentier G, Clauson P, Ravn GM, Roberts VL, Thorsteinsson B. Comparison of once-daily insulin detemir with NPH insulin added to a regimen of oral antidiabetic drugs in poorly controlled type 2 diabetes [published correction appears in Clin Ther. 2006;28(11):1967]. Clin Ther. 2006;28(10):1569-1581.
41. Blonde L, Merilainen M, Karwe V, Raskin P. TITRATE Study Group. Patient-directed titration for achieving glycaemic goals using a once-daily basal insulin analogue: an assessment of two different fasting plasma glucose targets—the TITRATE study. Diabetes Obes Metab. 2009;11(6):623-631.
42. Meneghini L, Mersebach H, Kumar S, Svendsen AL, Hermansen K. Comparison of 2 intensification regimens with rapid-acting insulin aspart in type 2 diabetes mellitus inadequately controlled by once-daily insulin detemir and oral antidiabetes drugs: the step-wise randomized study. Endocr Pract. 2011;17(5):727-736.
43. Lankisch MR, Ferlinz KC, Leahy JL, Scherbaum WA. Orals Plus Apidra and LANTUS (OPAL) study group. Introducing a simplified approach to insulin therapy in type 2 diabetes: a comparison of two single-dose regimens of insulin glulisine plus insulin glargine and oral antidiabetic drugs [published correction appears in Diabetes Obes Metab. 2010;12(5):461]. Diabetes Obes Metab 2008;10(12):1178-1185.
44. Meneghini L. Why and how to use insulin therapy earlier in the management of type 2 diabetes. South Med J. 2007;100(2):164-174.
45. Liebl A, Prager R, Binz K, Kaiser M, Bergenstal R, Gallwitz B. PREFER Study Group. Comparison of insulin analogue regimens in people with type 2 diabetes mellitus in the PREFER Study: a randomized controlled trial. Diabetes Obes Metab. 2009;11(1):45-52.
46. Braun A, Sämann A, Kubiak T, et al. Effects of metabolic control, patient education and initiation of insulin therapy on the quality of life of patients with type 2 diabetes mellitus. Patient Educ Couns. 2008;73(1):50-59.