User login
Patients With Difficult Personalities
Much has been written about the importance of the doctor‐patient relationship, with a positive therapeutic alliance being associated with both improvement in patient healthcare outcomes and physician job satisfaction.[1, 2] However, some patients severely test their physician's ability to provide needed care. These patients can rankle even experienced clinicians, leaving them feeling frustrated and ineffectual while consuming disproportionate amounts of clinical time. Although these disruptive acts may feel volitional and purposeful to the clinicians attempting to provide care, they may stem from a dysfunctional personality structure. Personality disorders are pervasive patterns of maladaptive behaviors, thoughts, and emotions that often go unrecognized and can wreak havoc in the patient's interpersonal life.[3] These inflexible patterns of managing the world can be disruptive when an individual is admitted to the hospital, causing distress for both the patient who lacks the skills to deal with the expectations of the hospital environment and the treatment team who can feel ill equipped to manage such behaviors.[4, 5] Here, we discuss personality disorders, how they can manifest in the hospital setting, and interventions to assist both the individual and the team.
Although personality disorders come in a variety of forms, central to all is interpersonal disarray with difficulty forming and maintaining acceptable relationships with others. In the hospital setting, the patient needs to be able to relate to, and cooperate with, a myriad of different care providers all while under some degree of physical and emotional distress. This can be destabilizing even for those without personality issues. For those with personality disorders, it is nearly inevitable that conflict will arise. Although true prevalence rates can be difficult to ascertain due to diagnostic challenges, surveys have found 4% to 15% of the population are affected by at least 1 personality disorder.[6] The prevalence is thought to be even higher among those seeking healthcare services, with researchers suggesting that 1 in 4 primary care patients meet criteria for a personality disorder.[6, 7]
Having a personality disorder has implications for an individual's healthcare outcomes. Studies in the United Kingdom have shown that those with a personality disorder have a life expectancy nearly 2 decades shorter than the general population.[8] Although suicide and homicide account for part of this, they also have increased risk of a number of health issues, including obesity, metabolic syndrome, cardiovascular disease, and sleep disorders.[9] In addition to lifestyle factors such as drinking and drug use, it has been suggested that dysfunctional personality structures may interfere with the ability to access and utilize care, leading to higher morbidity and mortality.[7]
In addition to impacting their own life, individuals with personality disorders have a tendency to disrupt the environment around them. They often elicit strong emotional responses from others that can range from a desire to help and protect to frustration and a sense of loathing.[10] The presence of a personality disorder often comes to light in the hospital when the patient is feeling vulnerable and acts out, evoking strong responses from team members. In the literature, patients with personality disorders are frequently referred to as a difficult or even hateful.[11] These individuals can be emotionally draining to care for, and the team must manage their own divergent responses in addition to the patient's disruptive behavior. Understanding personality disorders as a mental illness and using behavioral interventions can help to ease these interactions.
PERSONALITY DISORDERS: AN OVERVIEW
Personality disorders are characterized by persistent patterns of emotional reactivity, interpersonal interactions, and ways of perceiving the world that are inflexible and maladaptive and lead to significant distress and dysfunction.[3, 7] These disorders are notable for the interactive nature of the diagnosis; rather than being completely dependent on the individual's symptoms, a significant component of identification depends on how these individuals relate to others.[7] Although the trajectory can change over time, personality disorders are generally pervasive across the lifetime of an individual, beginning in adolescence or early adulthood.[7, 12] Personality disorders are divided into 3 clusters (Table 1).
| Personality Disorders | Features | Possible Manifestations in the Hospital |
|---|---|---|
| Cluster A | Odd and eccentric, socially avoidant |
Mistrust of medical staff and treatments offered Hostility toward treatment team Accusations of exploitation and harm without reasonable evidence General sense from the team that something is off |
| Paranoid | Highly suspicious of others; interpret malice where none was intended | |
| Schizoid | Minimal social relationships; limited emotional range | |
| Schizotypal | Eccentric behavior and magical thinking; uncomfortable with close relationships | |
| Cluster B | Emotionally labile and impulsive |
Splitting of the team, clear favorite providers and hated providers Extremes of emotion with responses out of proportion to the situation Rapid escalation when they perceive their needs not being met Evoke a strong emotional response from the team, taking up time out of proportion to their medical illness Help‐rejecting behavior Fear of abandonment manifesting as escalation of behavior around discharge |
| Antisocial | Frequent disregard for rights of others | |
| Borderline | Impulsive with volatile interpersonal relationships | |
| Histrionic | Disproportionate emotionality with engagement seeking | |
| Narcissistic | Grandiose, seeks admiration | |
| Cluster C | Anxious and neurotic |
Resistance to participating in their own care Frequent demands on the staff Particular, sometimes seemingly illogical, preferences regarding their care or other aspects of their stay |
| Avoidant | Socially fearful with feelings of inadequacy | |
| Dependent | Need to be taken care of, often manifesting as clinging and obsequious behavior | |
| Obsessive‐compulsive | Preoccupied by orderliness and control, but without actual obsessions or compulsions |
Cluster A
Those falling into cluster A, which includes paranoid, schizoid, and schizotypal personality disorders, are odd and eccentric and often avoid social engagement[3]; these individuals have few friends or associates and do not care to make more. At times, their unusual thinking can be difficult to differentiate from primary psychotic disorders like schizophrenia.
Cluster B
Cluster B is most heavily studied, consisting of antisocial, borderline, histrionic, and narcissistic personality disorders. These individuals share a high degree of emotional lability and erratic behavior.[3] Frequently, their tendency toward impulsive and self‐destructive behaviors can result in the need for medical care.
Cluster C
Cluster C includes avoidant, dependent, and obsessive‐compulsive personality disorders. These individuals are often anxious and fearful. Like individuals in Cluster A, they have few friends; unlike Cluster A, they long for friendships but struggle to make them. On the inpatient unit, these individuals may have trouble engaging in needed care, relying heavily on others to have their needs met or may be very particular about how their care is administered.
NEUROPHYSIOLOGY
Personality disorders are the product of complex interactions between genes and environment. These disorders are highly heritable, with 55% to 72% heritability across the 3 clusters.[13, 14, 15] Studies have implicated alterations in the serotonin system as playing a role in the underlying pathophysiology, which may contribute to the emotional dysregulation.[16, 17, 18] Neuroimaging has shown alterations in regions of the brain related to emotional reactivity and the processing of social interactions, suggesting neural mechanisms behind these individuals' difficulty with interpersonal relationships.[19, 20, 21, 22]
IDENTIFICATION OF PERSONALITY DISORDERS
These disorders are under‐recognized due, at least in part, to difficulty in making the diagnosis.[7] With 10 different personality disorders, many with overlapping characteristics, establishing a specific diagnosis can be time consuming, and a single individual may fit multiple different personality disorders.[7] Although self‐report surveys and structured interviews exist, these are often time consuming or inaccurate.[7] It is unlikely to be practical to make a diagnosis of a specific personality disorder while in the hospital. Instead, the focus should be placed on identifying impaired personality structures that interfere with interpersonal relationships and thereby disrupt the course of treatment. Consider a personality disorder if any of the following features are present:
- The patient elicits a strong emotional reaction from providers; these may vary markedly between providers.
- The patient's emotional responses may appear disproportionate to the inciting event.
- The patient is on a number of different psychiatric medications with little relief of symptoms.
- The patient takes up a disproportionate amount of the providers' time.
- The patient externalizes blame, seeing others as the source of discomfort or distress and therefore sees others as the solution.
Once identified, steps can be taken to help both the team and the patient.
BEHAVIORAL INTERVENTIONS
The first line of intervention for individuals with dysfunctional personality structures is behavioral, changing the way the team and patient interact (Table 2). Such interventions have long been the cornerstone of treatment for these individuals.[23] The preponderance of the research has focused on cluster B, and specifically individuals with borderline personality disorder, and applying these principles more broadly is largely based on expert opinion.
| Clinical Examples and Behavioral Interventions | ||
|---|---|---|
| Background | Situation | Response |
| Cluster A: Mr. A is a 75‐year‐old man transferred from his small town after a myocardial infarction. Although he has improved medically, he repeatedly expresses distrust and dissatisfaction with his doctors. He refuses to go to a skilled nursing facility but will not work with physical therapy to discharge home. He lives alone and has worked as a cattle rancher all his life. | Mr. A repeatedly accuses his team of being in this for the money. At times he mutters about government conspiracies. |
Check the team's emotions and reinforce desired behaviors and move past negative ones: Recognize paranoia as part of the illness. Rather than confront the paranoia, ignore this behavior as long as it is not directly interfering with care. |
| Cluster B: Ms. B is a 22‐year‐old woman admitted after a car accident resulting in multiple fractures. The pain service is consulted due to her ever‐increasing need for opiates. When the team first meets her, she is bright, effusing, Thank you for coming! My other doctors have no idea how to control my pain. She starts crying, I just can't do this anymore. Midway through the conversation, she offers, I can tell you are the best doctors I've had. Finally, I have someone who understands. Later, the pain team receives numerous pages that the patient is demanding to see them. The following day she is furious at the team for not keeping your promises. Nursing complains about her unwillingness to cooperate with dressing changes, insisting she only work with certain people, Because they understand me. | Ms. B is frequently insulting staff in a demanding and at times threatening manner. |
Reinforce desired behaviors and move past negative ones: Interact in a neutral manner to avoid reinforcing the disruptive behavior. If she becomes threatening or insulting, label the behavior and give her 1 opportunity to stop. Cursing upsets me. It's hard for me to help someone when they're cursing at me. This wording separates the behavior from the person. If she is able to calm herself, thank her (to reinforce this behavior) and offer to help. If she continues to escalate, you can say, You seem to be upset. I'll come back when it is a better time. Withdrawal of social contact can be a powerful tool. Return after a brief period to see if she has been able to calm down and, if so, re‐engage. Re‐engagement is key to reinforce calm, socially acceptable behavior. Check the team's emotions: Recognize patients with challenging behaviors can place a strong emotional toll on the team, particularly nursing staff who must frequently interact with these patients. Offer support to all members of the team to ensure appropriate patient care. |
| Ms. B is crying inconsolably, saying, I just can't stand being in the hospital anymore. They won't give me the pain meds I need. |
Offer validation and reinforce desired behaviors and move past negative ones: Offer empathy but then move to skill building. I can see you are upset. Is there anything that helps you when you are feeling this way? If the patient is unable to come up with anything feasible, offer her choices, such as walking with her around the unit or listening to music. |
|
| Cluster C: Mr. C is a 57‐year‐old man admitted for hyperosmolar hyperglycemic state. His condition has now stabilized, but when the nutritionist attempts to meet with him, he says he has a migraine. Later, when the diabetes nurse comes to discuss his insulin regimen, he is too tired to learn anything. When she persists, he listens, but repeatedly says, I'm never going to be able to do this and is unwilling to participate further. He repeatedly uses his call button, asking for help to the bathroom, despite being ambulatory previously. He talks for extended periods with nursing staff, sharing his fears about his inability to care for himself and his concerns that this will happen again. | Mr. C is repeatedly pressing his nurse call button multiple times throughout the day for seemingly trivial requests. |
Establish parameters: Mr. C is seeking contact with others. Have nursing arrange a regular schedule for checking in on the patient, such as every hour between 10 to and 10 after the hour. These visits can be kept brief, but offer a structure for the patient and encourage him or her to bundle their requests. Caregivers may also consider having Mr. C sit by the nurse's station to increase social interaction. Keep the message consistent: Work to maintain increased social contact across nursing shifts. |
Check the Team's Emotions
Managing patients with personality disorders begins by recognizing that these individuals evoke strong responses from even the most seasoned professional.[10, 11, 24, 25] Reactions toward people with personality disorders can range from a need to care for and protect the patient to feelings of futility or contempt.[10] Referred to as countertransference, these unconscious emotional reactions are common, but can interfere with medical care.[26] Given the increased understanding of the importance of team cohesion in patient care,[27] part of treating an individual with a personality disorder involves recognizing and managing the responses elicited amongst all members of the team. The disparate feelings among team members, which may be driven by different patient behaviors with different people, can lead to a variety of responses including overinvolvement, withdrawal, or even aggression.[28] Recognizing and discussing these differing reactions can help maintain team cohesion and support appropriate patient care.
Offer Empathy and Validation
Patients with personality disorders were often raised in invalidating environments and their ongoing intense emotional reactions can lead to perpetuation of invalidating responses from their caregivers.[29] They are accustomed to eliciting a defensive response from others and can be deliberately provocative, as these intense emotional interactions are comfortable territory, keeping providers feeling off balance and under attack. Instead, offering an empathic response can de‐escalate situations and is associated with the lowest level of invoked anger in patients.[30] Empathy can take the form of validation by acknowledging a person's feelings, thoughts, and emotions as legitimate, even if others may not fully understand or agree with them. As extreme as a patient's response may seem, he or she is genuinely experiencing these feelings and beliefs. Validation includes listening nonjudgmentally, objectively naming emotions the patient is experiencing, and conveying that the patient's response makes sense within the context of the situation.[31] This can include acknowledging the patient's level of distress, saying things such as, I can see you are really frustrated or I am concerned that what I just said has been upsetting. Empathy is more than words; it is the ability to see a situation from someone else's point of view. An empathic approach acknowledges the patient's intense emotional response to the challenges of hospitalization without frustration and judgment. Maintaining an empathicor even simply neutralstance can avoid a power struggle and also improves the therapeutic alliance with the patient.[32]
Establish Parameters but Pick the Battles
Individuals with personality disorders have trouble perceiving social boundaries. Even trained mental health professionals find this difficult to navigate.[33] The provider's first response is often to establish rigid boundaries. However, rigid rules can lead to power struggles between patients and providers, with limits being perceived as punitive. Instead of a list of rules, the creation of boundaries requires a thoughtful, practical establishment of parameters for both the individual and staff.[34] This may include guidelines for frequent, predictable nursing checks on the patient so that attention is provided on a time‐contingent rather than behavior‐contingent basis. If the patient remains dysregulated after a brief attempt to problem solve a nonemergent issue, staff can walk away with the comment that they will return within a specified period of time when things are calmer. If the patient is able to engage in the problem‐solving process, this has the advantage of generating a plan both can agree with while supporting more effective skills in the patient. Rather than a list of stringent rules, consider what is truly necessary for patient safety and well‐being.
Keep the Message Consistent
Hospital care involves many moving parts; nursing staff, the primary team, support staff, and consultants all interact with the patient throughout the day, sometimes providing conflicting messages. Although the typical patient can tolerate this, those with personality disorders have trouble dealing with the inconsistency, and this can exacerbate other problems. Carefully consider potentially contentious issues, such dosing of pain medications and benzodiazepines, and ensure that the team offers a consistent plan.[34] Ideally, meet with the patient as a team, including nursing, to convey a unified message.
Reinforce Desired Behaviors and Move Past Maladaptive Ones
Often in the life of a person with a personality disorder, their interpersonal interactions are negative. These patients are accustomed to negotiating a chaotic world. When not acting out, the patient may receive less attention while nursing and physician attention is appropriately distributed to other patients. Inadvertently, this reinforces using provocative behavior to get attention. Instead, if the patient is not demanding attention, providers should take the opportunity to provide positive reinforcement for calm behavior. This can be done by establishing a routine menu of interactions with the patient that occur when they are not acting in a disruptive manner; this avoids engagement being contingent on negative behaviors.[4] For example, having a nonillness‐related conversation during a dressing change or offering the patient a snack after a positive (or neutral) interaction can reinforce desirable behaviors. In contrast, when patients exhibit disruptive or inappropriate behaviors, the caregiver should respond by removing what the patient seeks, usually engagement, with a neutral attitude to avoid reinforcing the behavior: You seem upset. I'll come back when you feel better. By not reinforcing maladaptive behaviors, caregivers can decrease or extinguish such behaviors over time.[29] If the situation is nonemergent, the caregiver should briefly acknowledge the patient's distress and then focus on possible solutions: I can see you are really upset right now. What helps you in these situations? This both validates the patient's emotional state and encourages him or her to engage in problem solving around his or her distress.[29] If the patient is unable to identify a coping strategy in the moment, suggesting possibilities, such as walking around the unit or listening to music, can help the patient move past their intense emotions while also encouraging skill building.
PHARMACOLOGICAL INTERVENTIONS
Although there are no Food and Drug Administrationapproved medications for treatment of personality disorders, there is limited evidence for use of pharmacological interventions to address particular features of these disorders, such as impulsivity, affective dysregulation, or cognitiveperceptual symptoms.[35] Antipsychotics can be helpful in treating cognitive disturbances such as paranoia and dissociation that some of these patients experience.[35] Antidepressants may have a relatively small effect on anxiety and anger.[35] Mood stabilizers are shown to have a positive impact on impulsivity, anger, anxiety, and depressed mood.[35] However, medication should be used with caution, as polypharmacy is a significant problem with these patients and may have limited utility. Up to 40% of patients with borderline personality disorder take 3 or more psychotropic medications, many of which can have significant side effects,[36] and 1 in 3 are prescribed benzodiazepines despite a lack of evidence and potential for abuse.[37] Thus, although medications may offer an opportunity to target specific symptoms, the focus of management for patients with personality disorders should be behavioral rather than pharmacological.
CONSIDER A CONSULT
Patients with personality disorders can be very difficult to treat, and it may be necessary to consult psychiatry. There are a number of situations in which a formal psychiatric consultation is indicated (Table 3). Patients with personality disorders, particularly cluster B, may present for treatment after harming themselves or others.[38] A psychiatric consultation can provide a formal risk assessment, help with behavior and medication management while the patient is hospitalized, and determine whether follow‐up psychiatric care is appropriate.[39] The psychiatry team can also offer a more complete diagnostic formulation, including screening for disorders that often co‐occur with personality disorders, such as depression and anxiety, and recommend treatment options.[39] In addition, if initial attempts at behavioral interventions are ineffectual, a psychiatric consult may be able to provide additional guidance in behavioral modifications.[40] This is especially appropriate if the patient's behaviors are interfering with medical care. A psychiatric consult can also provide additional support around issues of countertransference that can arise when managing patients with dysfunctional personality structures.[41, 42]
| Safety assessment in a patient who has threatened or engaged in self‐harming behavior or harm to others |
| Diagnostic clarification, particularly when there is concern for a co‐occurring psychiatric illness |
| Creation of a more complex behavioral plan |
| Facilitation of interdisciplinary discussion and problem‐solving around patients with challenging behaviors |
| Assistance with establishment of outpatient psychiatric care when appropriate |
As with all interventions, the psychiatric consult is not without its side effects. Regardless of personality structure, it is not uncommon for patients to be initially opposed to engaging with psychiatry.[43] Individuals with personality disorders can be particularly susceptible to a rupture of the therapeutic alliance,[44] and calling a psychiatric consult can affect the therapeutic relationship with the primary team, as the patient may feel that others are judging them and can also be part of a greater theme of help rejecting.[11] However, this rupture may be repaired as the patient comes to see the psychiatry team as an ally. Even for patients who refuse to engage with the consult‐liaison team, there may be a benefit to a consult, as the consultants can offer strategies to the primary team and help establish a plan of care to facilitate ongoing treatment of the patient's medical needs without direct contact with the patient. These situations illustrate that a psychiatric consult cannot be done in isolation and requires collaboration with the primary team, nurses, and other support staff for interventions to be effective.
CONCLUSIONS
In his now famous speech Dr. Francis W. Peabody gave to Harvard Medical School he noted that [T]he secret of the care of the patient is caring for the patient.[45] Patients with a dysfunctional personality structure can make this task difficult. They can appear to reject the very help we have to offer, divide the team, absorb great amounts of time, and evoke strong feelings of frustration and resentment. However, by understanding that the way in which they interact with the world is in part the product of biology and upbringing, we can better recognize how ill these individuals can be. Just as a patient with diabetes requires management of his blood glucose when admitted for pneumonia, those with personality disorders require management of their mental illness while their other medical conditions are addressed.
Although personality disorders can seem intractable, studies have shown that, like many chronic illnesses, the severity can wax and wane over time with remissions and relapses. Notably, rates of remission for borderline personality disorder at 10 years are comparable to those for major depressive disorder, bipolar disorder, and panic disorder, with lower rates of relapse even without specific treatment, suggesting they are not entirely intractable.[46] However, the stress of hospitalization can easily exacerbate the symptoms of a personality disorder. By providing an empathic approach that addresses the emotional responses of the team while also reinforcing positive behaviors of the patient, the hospital stay can be an opportunity for these individuals to get needed support and develop new skills while also having their physical needs addressed.
Disclosures: Nothing to report.
- , . The effects of trust in physician on self‐efficacy, adherence and diabetes outcomes. Soc Sci Med. 2009;68(6):1060–1068.
- , , , et al. The physician‐patient working alliance. Patient Educ Couns. 2007;66(1):29–36.
- Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition. Arlington, VA: American Psychiatric Association; 2013.
- , , . The borderline patient on the med‐surg unit. Am J Nursing. 1988;88(12):1644–1650.
- , , . “Difficult” patient? Or does he have a personality disorder? J Fam Pract. 2014;63(12):697–703.
- , , , et al. Prevalence, correlates, and disability of personality disorders in the United States: results from the national epidemiologic survey on alcohol and related conditions. J Clin Psychiatry. 2004;65(7):948–958.
- , , . Classification, assessment, prevalence, and effect of personality disorder. Lancet. 2015;385(9969):717–726.
- , , , , , . Life expectancy at birth and all‐cause mortality among people with personality disorder. J Psychosom Res. 2012;73(2):104–107.
- , , , . A systematic review of personality disorders and health outcomes. Can Psychol. 2015;56(2):168–190.
- , , , . Patient personality and therapist response: an empirical investigation. Am J Psychiatry. 2014;171(1):102–108.
- . Taking care of the hateful patient. N Engl J Med. 1978;298(16):883–887.
- , , . Personality disorder across the life course. Lancet. 2015;385(9969):727–734.
- , , , , . The heritability of cluster A personality disorders assessed by both personal interview and questionnaire. Psychol Med. 2007;37(5):655–665.
- , , , et al. The heritability of avoidant and dependent personality disorder assessed by personal interview and questionnaire. Acta Psychiatr Scand. 2012;126(6):448–457.
- , , , , , . The heritability of Cluster B personality disorders assessed both by personal interview and questionnaire. J Pers Disord. 2012;26(6):848–866.
- , , . Association between genetic polymorphisms in the serotonergic system and comorbid personality disorders among patients with first‐episode depression. J Pers Disord. 2014;28(3):365–378.
- , , , et al. Tryptophan‐hydroxylase 2 haplotype association with borderline personality disorder and aggression in a sample of patients with personality disorders and healthy controls. J Psychiatr Res. 2010;44(15):1075–1081.
- , , , et al. Monoamine oxidase A gene promoter methylation and transcriptional downregulation in an offender population with antisocial personality disorder. Br J Psychiatry. 2015;206(3):216–222.
- , , , et al. Regional cortical thinning may be a biological marker for borderline personality disorder. Acta Psychiatr Scand. 2014;130(3):193–204.
- , , . Empathy and social problem solving in alcohol dependence, mood disorders and selected personality disorders. Neurosci Biobehav Rev. 2013;37(3):448–470.
- , , , . Changes in low‐frequency fluctuations in patients with antisocial personality disorder revealed by resting‐state functional MRI. PLoS One. 2014;9(3):e89790.
- , . Prefrontal structural and functional brain imaging findings in antisocial, violent, and psychopathic individuals: a meta‐analysis. Psychiatry Res. 2009;174(2):81–88.
- , , . Treatment of personality disorder. Lancet. 2015;385(9969):735–743.
- . Management of the borderline patient on a medical or surgical ward: the psychiatric consultant's role. Int J Psychiatry Med. 1975;6(3):337–348.
- , , , et al. The attitudes of psychiatric hospital staff toward hospitalization and treatment of patients with borderline personality disorder. BMC Psychiatry. 2015;15:2.
- , , , . The hateful physician: the role of affect bias in the care of the psychiatric patient in the ED. Am J Emerg Med. 2014;32(5):483–485.
- , , , et al. Relationships within inpatient physician housestaff teams and their association with hospitalized patient outcomes. J Hosp Med. 2014;9(12):764–771.
- . Countertransference in the nurse‐patient relationship: a review of the literature. J Adv Nurs. 1998;28(2):391–397.
- , , , , . Mechanisms of change in dialectical behavior therapy: theoretical and empirical observations. J Clin Psychol. 2006;62(4):459–480.
- , , , . The relationship between nurses' limit‐setting styles and anger in psychiatric inpatients. Psychiatr Serv. 1995;46(6):609–613.
- , , , , , . Attachment disorganization and borderline patients' metacognitive responses to therapists' expressed understanding of their states of mind: a pilot study. Psychother Res. 2008;18(1):28–36.
- , , . Narcissistic personality disorder: diagnostic and clinical challenges. Am J Psychiatry. 2015;172(5):415–422.
- , . An evaluation of the difficulties and attitudes mental health professionals experience with people with personality disorders. J Psychiatr Ment Health Nurs. 2016;23(1):22–36.
- , . Nursing care of personality disorders in the medical surgery setting. Nurs Clin North Am. 1998;33(1):173–186.
- , , , , . Effectiveness of pharmacotherapy for severe personality disorders: meta‐analyses of randomized controlled trials. J Clin Psychiatry. 2010;71(1):14–25.
- , , , . Mental health service utilization by borderline personality disorder patients and Axis II comparison subjects followed prospectively for 6 years. J Clin Psychiatry. 2004;65(1):28–36.
- , , , , . The use of psychotropic medication in patients with emotionally unstable personality disorder under the care of UK mental health services. J Clin Psychiatry. 2015;76(4):e512–e518.
- , , , , . Prevalence and characteristics of cluster B personality disorder in a consultation‐liaison psychiatry practice. Int J Psychiatry Clin Pract. 2015;19(1):65–70.
- , , , et al. The Academy of Psychosomatic Medicine practice guidelines for psychiatric consultation in the general medical setting. The Academy of Psychosomatic Medicine. Psychosomatics. 1998;39(4):S8–S30.
- , , . Interventions in consultation/liaison psychiatry. Part I: Patterns of recommendations. Gen Hosp Psychiatry. 1990;12(4):213–220.
- , , . Psychodynamics in medically ill patients. Harv Rev Psychiatry. 2009;17(6):389–397.
- . Patient dynamics, staff burnout, and consultation‐liaison psychiatry. Physician Exec. 1991;17(5):37–40.
- , . Facilitating patient acceptance of a psychiatric referral. Arch Intern Med. 1985;145(1):73–75.
- . The therapeutic alliance in the treatment of personality disorders. J Psychiatr Pract. 2005;11(2):73–87.
- . Dr. Francis w. Peabody, we need you. Tex Heart Inst J. 2011;38(4):327–328; discussion 328–329.
- , , , et al. Ten‐year course of borderline personality disorder: psychopathology and function from the Collaborative Longitudinal Personality Disorders study. Arch Gen Psychiatry. 2011;68(8):827–837.
Much has been written about the importance of the doctor‐patient relationship, with a positive therapeutic alliance being associated with both improvement in patient healthcare outcomes and physician job satisfaction.[1, 2] However, some patients severely test their physician's ability to provide needed care. These patients can rankle even experienced clinicians, leaving them feeling frustrated and ineffectual while consuming disproportionate amounts of clinical time. Although these disruptive acts may feel volitional and purposeful to the clinicians attempting to provide care, they may stem from a dysfunctional personality structure. Personality disorders are pervasive patterns of maladaptive behaviors, thoughts, and emotions that often go unrecognized and can wreak havoc in the patient's interpersonal life.[3] These inflexible patterns of managing the world can be disruptive when an individual is admitted to the hospital, causing distress for both the patient who lacks the skills to deal with the expectations of the hospital environment and the treatment team who can feel ill equipped to manage such behaviors.[4, 5] Here, we discuss personality disorders, how they can manifest in the hospital setting, and interventions to assist both the individual and the team.
Although personality disorders come in a variety of forms, central to all is interpersonal disarray with difficulty forming and maintaining acceptable relationships with others. In the hospital setting, the patient needs to be able to relate to, and cooperate with, a myriad of different care providers all while under some degree of physical and emotional distress. This can be destabilizing even for those without personality issues. For those with personality disorders, it is nearly inevitable that conflict will arise. Although true prevalence rates can be difficult to ascertain due to diagnostic challenges, surveys have found 4% to 15% of the population are affected by at least 1 personality disorder.[6] The prevalence is thought to be even higher among those seeking healthcare services, with researchers suggesting that 1 in 4 primary care patients meet criteria for a personality disorder.[6, 7]
Having a personality disorder has implications for an individual's healthcare outcomes. Studies in the United Kingdom have shown that those with a personality disorder have a life expectancy nearly 2 decades shorter than the general population.[8] Although suicide and homicide account for part of this, they also have increased risk of a number of health issues, including obesity, metabolic syndrome, cardiovascular disease, and sleep disorders.[9] In addition to lifestyle factors such as drinking and drug use, it has been suggested that dysfunctional personality structures may interfere with the ability to access and utilize care, leading to higher morbidity and mortality.[7]
In addition to impacting their own life, individuals with personality disorders have a tendency to disrupt the environment around them. They often elicit strong emotional responses from others that can range from a desire to help and protect to frustration and a sense of loathing.[10] The presence of a personality disorder often comes to light in the hospital when the patient is feeling vulnerable and acts out, evoking strong responses from team members. In the literature, patients with personality disorders are frequently referred to as a difficult or even hateful.[11] These individuals can be emotionally draining to care for, and the team must manage their own divergent responses in addition to the patient's disruptive behavior. Understanding personality disorders as a mental illness and using behavioral interventions can help to ease these interactions.
PERSONALITY DISORDERS: AN OVERVIEW
Personality disorders are characterized by persistent patterns of emotional reactivity, interpersonal interactions, and ways of perceiving the world that are inflexible and maladaptive and lead to significant distress and dysfunction.[3, 7] These disorders are notable for the interactive nature of the diagnosis; rather than being completely dependent on the individual's symptoms, a significant component of identification depends on how these individuals relate to others.[7] Although the trajectory can change over time, personality disorders are generally pervasive across the lifetime of an individual, beginning in adolescence or early adulthood.[7, 12] Personality disorders are divided into 3 clusters (Table 1).
| Personality Disorders | Features | Possible Manifestations in the Hospital |
|---|---|---|
| Cluster A | Odd and eccentric, socially avoidant |
Mistrust of medical staff and treatments offered Hostility toward treatment team Accusations of exploitation and harm without reasonable evidence General sense from the team that something is off |
| Paranoid | Highly suspicious of others; interpret malice where none was intended | |
| Schizoid | Minimal social relationships; limited emotional range | |
| Schizotypal | Eccentric behavior and magical thinking; uncomfortable with close relationships | |
| Cluster B | Emotionally labile and impulsive |
Splitting of the team, clear favorite providers and hated providers Extremes of emotion with responses out of proportion to the situation Rapid escalation when they perceive their needs not being met Evoke a strong emotional response from the team, taking up time out of proportion to their medical illness Help‐rejecting behavior Fear of abandonment manifesting as escalation of behavior around discharge |
| Antisocial | Frequent disregard for rights of others | |
| Borderline | Impulsive with volatile interpersonal relationships | |
| Histrionic | Disproportionate emotionality with engagement seeking | |
| Narcissistic | Grandiose, seeks admiration | |
| Cluster C | Anxious and neurotic |
Resistance to participating in their own care Frequent demands on the staff Particular, sometimes seemingly illogical, preferences regarding their care or other aspects of their stay |
| Avoidant | Socially fearful with feelings of inadequacy | |
| Dependent | Need to be taken care of, often manifesting as clinging and obsequious behavior | |
| Obsessive‐compulsive | Preoccupied by orderliness and control, but without actual obsessions or compulsions |
Cluster A
Those falling into cluster A, which includes paranoid, schizoid, and schizotypal personality disorders, are odd and eccentric and often avoid social engagement[3]; these individuals have few friends or associates and do not care to make more. At times, their unusual thinking can be difficult to differentiate from primary psychotic disorders like schizophrenia.
Cluster B
Cluster B is most heavily studied, consisting of antisocial, borderline, histrionic, and narcissistic personality disorders. These individuals share a high degree of emotional lability and erratic behavior.[3] Frequently, their tendency toward impulsive and self‐destructive behaviors can result in the need for medical care.
Cluster C
Cluster C includes avoidant, dependent, and obsessive‐compulsive personality disorders. These individuals are often anxious and fearful. Like individuals in Cluster A, they have few friends; unlike Cluster A, they long for friendships but struggle to make them. On the inpatient unit, these individuals may have trouble engaging in needed care, relying heavily on others to have their needs met or may be very particular about how their care is administered.
NEUROPHYSIOLOGY
Personality disorders are the product of complex interactions between genes and environment. These disorders are highly heritable, with 55% to 72% heritability across the 3 clusters.[13, 14, 15] Studies have implicated alterations in the serotonin system as playing a role in the underlying pathophysiology, which may contribute to the emotional dysregulation.[16, 17, 18] Neuroimaging has shown alterations in regions of the brain related to emotional reactivity and the processing of social interactions, suggesting neural mechanisms behind these individuals' difficulty with interpersonal relationships.[19, 20, 21, 22]
IDENTIFICATION OF PERSONALITY DISORDERS
These disorders are under‐recognized due, at least in part, to difficulty in making the diagnosis.[7] With 10 different personality disorders, many with overlapping characteristics, establishing a specific diagnosis can be time consuming, and a single individual may fit multiple different personality disorders.[7] Although self‐report surveys and structured interviews exist, these are often time consuming or inaccurate.[7] It is unlikely to be practical to make a diagnosis of a specific personality disorder while in the hospital. Instead, the focus should be placed on identifying impaired personality structures that interfere with interpersonal relationships and thereby disrupt the course of treatment. Consider a personality disorder if any of the following features are present:
- The patient elicits a strong emotional reaction from providers; these may vary markedly between providers.
- The patient's emotional responses may appear disproportionate to the inciting event.
- The patient is on a number of different psychiatric medications with little relief of symptoms.
- The patient takes up a disproportionate amount of the providers' time.
- The patient externalizes blame, seeing others as the source of discomfort or distress and therefore sees others as the solution.
Once identified, steps can be taken to help both the team and the patient.
BEHAVIORAL INTERVENTIONS
The first line of intervention for individuals with dysfunctional personality structures is behavioral, changing the way the team and patient interact (Table 2). Such interventions have long been the cornerstone of treatment for these individuals.[23] The preponderance of the research has focused on cluster B, and specifically individuals with borderline personality disorder, and applying these principles more broadly is largely based on expert opinion.
| Clinical Examples and Behavioral Interventions | ||
|---|---|---|
| Background | Situation | Response |
| Cluster A: Mr. A is a 75‐year‐old man transferred from his small town after a myocardial infarction. Although he has improved medically, he repeatedly expresses distrust and dissatisfaction with his doctors. He refuses to go to a skilled nursing facility but will not work with physical therapy to discharge home. He lives alone and has worked as a cattle rancher all his life. | Mr. A repeatedly accuses his team of being in this for the money. At times he mutters about government conspiracies. |
Check the team's emotions and reinforce desired behaviors and move past negative ones: Recognize paranoia as part of the illness. Rather than confront the paranoia, ignore this behavior as long as it is not directly interfering with care. |
| Cluster B: Ms. B is a 22‐year‐old woman admitted after a car accident resulting in multiple fractures. The pain service is consulted due to her ever‐increasing need for opiates. When the team first meets her, she is bright, effusing, Thank you for coming! My other doctors have no idea how to control my pain. She starts crying, I just can't do this anymore. Midway through the conversation, she offers, I can tell you are the best doctors I've had. Finally, I have someone who understands. Later, the pain team receives numerous pages that the patient is demanding to see them. The following day she is furious at the team for not keeping your promises. Nursing complains about her unwillingness to cooperate with dressing changes, insisting she only work with certain people, Because they understand me. | Ms. B is frequently insulting staff in a demanding and at times threatening manner. |
Reinforce desired behaviors and move past negative ones: Interact in a neutral manner to avoid reinforcing the disruptive behavior. If she becomes threatening or insulting, label the behavior and give her 1 opportunity to stop. Cursing upsets me. It's hard for me to help someone when they're cursing at me. This wording separates the behavior from the person. If she is able to calm herself, thank her (to reinforce this behavior) and offer to help. If she continues to escalate, you can say, You seem to be upset. I'll come back when it is a better time. Withdrawal of social contact can be a powerful tool. Return after a brief period to see if she has been able to calm down and, if so, re‐engage. Re‐engagement is key to reinforce calm, socially acceptable behavior. Check the team's emotions: Recognize patients with challenging behaviors can place a strong emotional toll on the team, particularly nursing staff who must frequently interact with these patients. Offer support to all members of the team to ensure appropriate patient care. |
| Ms. B is crying inconsolably, saying, I just can't stand being in the hospital anymore. They won't give me the pain meds I need. |
Offer validation and reinforce desired behaviors and move past negative ones: Offer empathy but then move to skill building. I can see you are upset. Is there anything that helps you when you are feeling this way? If the patient is unable to come up with anything feasible, offer her choices, such as walking with her around the unit or listening to music. |
|
| Cluster C: Mr. C is a 57‐year‐old man admitted for hyperosmolar hyperglycemic state. His condition has now stabilized, but when the nutritionist attempts to meet with him, he says he has a migraine. Later, when the diabetes nurse comes to discuss his insulin regimen, he is too tired to learn anything. When she persists, he listens, but repeatedly says, I'm never going to be able to do this and is unwilling to participate further. He repeatedly uses his call button, asking for help to the bathroom, despite being ambulatory previously. He talks for extended periods with nursing staff, sharing his fears about his inability to care for himself and his concerns that this will happen again. | Mr. C is repeatedly pressing his nurse call button multiple times throughout the day for seemingly trivial requests. |
Establish parameters: Mr. C is seeking contact with others. Have nursing arrange a regular schedule for checking in on the patient, such as every hour between 10 to and 10 after the hour. These visits can be kept brief, but offer a structure for the patient and encourage him or her to bundle their requests. Caregivers may also consider having Mr. C sit by the nurse's station to increase social interaction. Keep the message consistent: Work to maintain increased social contact across nursing shifts. |
Check the Team's Emotions
Managing patients with personality disorders begins by recognizing that these individuals evoke strong responses from even the most seasoned professional.[10, 11, 24, 25] Reactions toward people with personality disorders can range from a need to care for and protect the patient to feelings of futility or contempt.[10] Referred to as countertransference, these unconscious emotional reactions are common, but can interfere with medical care.[26] Given the increased understanding of the importance of team cohesion in patient care,[27] part of treating an individual with a personality disorder involves recognizing and managing the responses elicited amongst all members of the team. The disparate feelings among team members, which may be driven by different patient behaviors with different people, can lead to a variety of responses including overinvolvement, withdrawal, or even aggression.[28] Recognizing and discussing these differing reactions can help maintain team cohesion and support appropriate patient care.
Offer Empathy and Validation
Patients with personality disorders were often raised in invalidating environments and their ongoing intense emotional reactions can lead to perpetuation of invalidating responses from their caregivers.[29] They are accustomed to eliciting a defensive response from others and can be deliberately provocative, as these intense emotional interactions are comfortable territory, keeping providers feeling off balance and under attack. Instead, offering an empathic response can de‐escalate situations and is associated with the lowest level of invoked anger in patients.[30] Empathy can take the form of validation by acknowledging a person's feelings, thoughts, and emotions as legitimate, even if others may not fully understand or agree with them. As extreme as a patient's response may seem, he or she is genuinely experiencing these feelings and beliefs. Validation includes listening nonjudgmentally, objectively naming emotions the patient is experiencing, and conveying that the patient's response makes sense within the context of the situation.[31] This can include acknowledging the patient's level of distress, saying things such as, I can see you are really frustrated or I am concerned that what I just said has been upsetting. Empathy is more than words; it is the ability to see a situation from someone else's point of view. An empathic approach acknowledges the patient's intense emotional response to the challenges of hospitalization without frustration and judgment. Maintaining an empathicor even simply neutralstance can avoid a power struggle and also improves the therapeutic alliance with the patient.[32]
Establish Parameters but Pick the Battles
Individuals with personality disorders have trouble perceiving social boundaries. Even trained mental health professionals find this difficult to navigate.[33] The provider's first response is often to establish rigid boundaries. However, rigid rules can lead to power struggles between patients and providers, with limits being perceived as punitive. Instead of a list of rules, the creation of boundaries requires a thoughtful, practical establishment of parameters for both the individual and staff.[34] This may include guidelines for frequent, predictable nursing checks on the patient so that attention is provided on a time‐contingent rather than behavior‐contingent basis. If the patient remains dysregulated after a brief attempt to problem solve a nonemergent issue, staff can walk away with the comment that they will return within a specified period of time when things are calmer. If the patient is able to engage in the problem‐solving process, this has the advantage of generating a plan both can agree with while supporting more effective skills in the patient. Rather than a list of stringent rules, consider what is truly necessary for patient safety and well‐being.
Keep the Message Consistent
Hospital care involves many moving parts; nursing staff, the primary team, support staff, and consultants all interact with the patient throughout the day, sometimes providing conflicting messages. Although the typical patient can tolerate this, those with personality disorders have trouble dealing with the inconsistency, and this can exacerbate other problems. Carefully consider potentially contentious issues, such dosing of pain medications and benzodiazepines, and ensure that the team offers a consistent plan.[34] Ideally, meet with the patient as a team, including nursing, to convey a unified message.
Reinforce Desired Behaviors and Move Past Maladaptive Ones
Often in the life of a person with a personality disorder, their interpersonal interactions are negative. These patients are accustomed to negotiating a chaotic world. When not acting out, the patient may receive less attention while nursing and physician attention is appropriately distributed to other patients. Inadvertently, this reinforces using provocative behavior to get attention. Instead, if the patient is not demanding attention, providers should take the opportunity to provide positive reinforcement for calm behavior. This can be done by establishing a routine menu of interactions with the patient that occur when they are not acting in a disruptive manner; this avoids engagement being contingent on negative behaviors.[4] For example, having a nonillness‐related conversation during a dressing change or offering the patient a snack after a positive (or neutral) interaction can reinforce desirable behaviors. In contrast, when patients exhibit disruptive or inappropriate behaviors, the caregiver should respond by removing what the patient seeks, usually engagement, with a neutral attitude to avoid reinforcing the behavior: You seem upset. I'll come back when you feel better. By not reinforcing maladaptive behaviors, caregivers can decrease or extinguish such behaviors over time.[29] If the situation is nonemergent, the caregiver should briefly acknowledge the patient's distress and then focus on possible solutions: I can see you are really upset right now. What helps you in these situations? This both validates the patient's emotional state and encourages him or her to engage in problem solving around his or her distress.[29] If the patient is unable to identify a coping strategy in the moment, suggesting possibilities, such as walking around the unit or listening to music, can help the patient move past their intense emotions while also encouraging skill building.
PHARMACOLOGICAL INTERVENTIONS
Although there are no Food and Drug Administrationapproved medications for treatment of personality disorders, there is limited evidence for use of pharmacological interventions to address particular features of these disorders, such as impulsivity, affective dysregulation, or cognitiveperceptual symptoms.[35] Antipsychotics can be helpful in treating cognitive disturbances such as paranoia and dissociation that some of these patients experience.[35] Antidepressants may have a relatively small effect on anxiety and anger.[35] Mood stabilizers are shown to have a positive impact on impulsivity, anger, anxiety, and depressed mood.[35] However, medication should be used with caution, as polypharmacy is a significant problem with these patients and may have limited utility. Up to 40% of patients with borderline personality disorder take 3 or more psychotropic medications, many of which can have significant side effects,[36] and 1 in 3 are prescribed benzodiazepines despite a lack of evidence and potential for abuse.[37] Thus, although medications may offer an opportunity to target specific symptoms, the focus of management for patients with personality disorders should be behavioral rather than pharmacological.
CONSIDER A CONSULT
Patients with personality disorders can be very difficult to treat, and it may be necessary to consult psychiatry. There are a number of situations in which a formal psychiatric consultation is indicated (Table 3). Patients with personality disorders, particularly cluster B, may present for treatment after harming themselves or others.[38] A psychiatric consultation can provide a formal risk assessment, help with behavior and medication management while the patient is hospitalized, and determine whether follow‐up psychiatric care is appropriate.[39] The psychiatry team can also offer a more complete diagnostic formulation, including screening for disorders that often co‐occur with personality disorders, such as depression and anxiety, and recommend treatment options.[39] In addition, if initial attempts at behavioral interventions are ineffectual, a psychiatric consult may be able to provide additional guidance in behavioral modifications.[40] This is especially appropriate if the patient's behaviors are interfering with medical care. A psychiatric consult can also provide additional support around issues of countertransference that can arise when managing patients with dysfunctional personality structures.[41, 42]
| Safety assessment in a patient who has threatened or engaged in self‐harming behavior or harm to others |
| Diagnostic clarification, particularly when there is concern for a co‐occurring psychiatric illness |
| Creation of a more complex behavioral plan |
| Facilitation of interdisciplinary discussion and problem‐solving around patients with challenging behaviors |
| Assistance with establishment of outpatient psychiatric care when appropriate |
As with all interventions, the psychiatric consult is not without its side effects. Regardless of personality structure, it is not uncommon for patients to be initially opposed to engaging with psychiatry.[43] Individuals with personality disorders can be particularly susceptible to a rupture of the therapeutic alliance,[44] and calling a psychiatric consult can affect the therapeutic relationship with the primary team, as the patient may feel that others are judging them and can also be part of a greater theme of help rejecting.[11] However, this rupture may be repaired as the patient comes to see the psychiatry team as an ally. Even for patients who refuse to engage with the consult‐liaison team, there may be a benefit to a consult, as the consultants can offer strategies to the primary team and help establish a plan of care to facilitate ongoing treatment of the patient's medical needs without direct contact with the patient. These situations illustrate that a psychiatric consult cannot be done in isolation and requires collaboration with the primary team, nurses, and other support staff for interventions to be effective.
CONCLUSIONS
In his now famous speech Dr. Francis W. Peabody gave to Harvard Medical School he noted that [T]he secret of the care of the patient is caring for the patient.[45] Patients with a dysfunctional personality structure can make this task difficult. They can appear to reject the very help we have to offer, divide the team, absorb great amounts of time, and evoke strong feelings of frustration and resentment. However, by understanding that the way in which they interact with the world is in part the product of biology and upbringing, we can better recognize how ill these individuals can be. Just as a patient with diabetes requires management of his blood glucose when admitted for pneumonia, those with personality disorders require management of their mental illness while their other medical conditions are addressed.
Although personality disorders can seem intractable, studies have shown that, like many chronic illnesses, the severity can wax and wane over time with remissions and relapses. Notably, rates of remission for borderline personality disorder at 10 years are comparable to those for major depressive disorder, bipolar disorder, and panic disorder, with lower rates of relapse even without specific treatment, suggesting they are not entirely intractable.[46] However, the stress of hospitalization can easily exacerbate the symptoms of a personality disorder. By providing an empathic approach that addresses the emotional responses of the team while also reinforcing positive behaviors of the patient, the hospital stay can be an opportunity for these individuals to get needed support and develop new skills while also having their physical needs addressed.
Disclosures: Nothing to report.
Much has been written about the importance of the doctor‐patient relationship, with a positive therapeutic alliance being associated with both improvement in patient healthcare outcomes and physician job satisfaction.[1, 2] However, some patients severely test their physician's ability to provide needed care. These patients can rankle even experienced clinicians, leaving them feeling frustrated and ineffectual while consuming disproportionate amounts of clinical time. Although these disruptive acts may feel volitional and purposeful to the clinicians attempting to provide care, they may stem from a dysfunctional personality structure. Personality disorders are pervasive patterns of maladaptive behaviors, thoughts, and emotions that often go unrecognized and can wreak havoc in the patient's interpersonal life.[3] These inflexible patterns of managing the world can be disruptive when an individual is admitted to the hospital, causing distress for both the patient who lacks the skills to deal with the expectations of the hospital environment and the treatment team who can feel ill equipped to manage such behaviors.[4, 5] Here, we discuss personality disorders, how they can manifest in the hospital setting, and interventions to assist both the individual and the team.
Although personality disorders come in a variety of forms, central to all is interpersonal disarray with difficulty forming and maintaining acceptable relationships with others. In the hospital setting, the patient needs to be able to relate to, and cooperate with, a myriad of different care providers all while under some degree of physical and emotional distress. This can be destabilizing even for those without personality issues. For those with personality disorders, it is nearly inevitable that conflict will arise. Although true prevalence rates can be difficult to ascertain due to diagnostic challenges, surveys have found 4% to 15% of the population are affected by at least 1 personality disorder.[6] The prevalence is thought to be even higher among those seeking healthcare services, with researchers suggesting that 1 in 4 primary care patients meet criteria for a personality disorder.[6, 7]
Having a personality disorder has implications for an individual's healthcare outcomes. Studies in the United Kingdom have shown that those with a personality disorder have a life expectancy nearly 2 decades shorter than the general population.[8] Although suicide and homicide account for part of this, they also have increased risk of a number of health issues, including obesity, metabolic syndrome, cardiovascular disease, and sleep disorders.[9] In addition to lifestyle factors such as drinking and drug use, it has been suggested that dysfunctional personality structures may interfere with the ability to access and utilize care, leading to higher morbidity and mortality.[7]
In addition to impacting their own life, individuals with personality disorders have a tendency to disrupt the environment around them. They often elicit strong emotional responses from others that can range from a desire to help and protect to frustration and a sense of loathing.[10] The presence of a personality disorder often comes to light in the hospital when the patient is feeling vulnerable and acts out, evoking strong responses from team members. In the literature, patients with personality disorders are frequently referred to as a difficult or even hateful.[11] These individuals can be emotionally draining to care for, and the team must manage their own divergent responses in addition to the patient's disruptive behavior. Understanding personality disorders as a mental illness and using behavioral interventions can help to ease these interactions.
PERSONALITY DISORDERS: AN OVERVIEW
Personality disorders are characterized by persistent patterns of emotional reactivity, interpersonal interactions, and ways of perceiving the world that are inflexible and maladaptive and lead to significant distress and dysfunction.[3, 7] These disorders are notable for the interactive nature of the diagnosis; rather than being completely dependent on the individual's symptoms, a significant component of identification depends on how these individuals relate to others.[7] Although the trajectory can change over time, personality disorders are generally pervasive across the lifetime of an individual, beginning in adolescence or early adulthood.[7, 12] Personality disorders are divided into 3 clusters (Table 1).
| Personality Disorders | Features | Possible Manifestations in the Hospital |
|---|---|---|
| Cluster A | Odd and eccentric, socially avoidant |
Mistrust of medical staff and treatments offered Hostility toward treatment team Accusations of exploitation and harm without reasonable evidence General sense from the team that something is off |
| Paranoid | Highly suspicious of others; interpret malice where none was intended | |
| Schizoid | Minimal social relationships; limited emotional range | |
| Schizotypal | Eccentric behavior and magical thinking; uncomfortable with close relationships | |
| Cluster B | Emotionally labile and impulsive |
Splitting of the team, clear favorite providers and hated providers Extremes of emotion with responses out of proportion to the situation Rapid escalation when they perceive their needs not being met Evoke a strong emotional response from the team, taking up time out of proportion to their medical illness Help‐rejecting behavior Fear of abandonment manifesting as escalation of behavior around discharge |
| Antisocial | Frequent disregard for rights of others | |
| Borderline | Impulsive with volatile interpersonal relationships | |
| Histrionic | Disproportionate emotionality with engagement seeking | |
| Narcissistic | Grandiose, seeks admiration | |
| Cluster C | Anxious and neurotic |
Resistance to participating in their own care Frequent demands on the staff Particular, sometimes seemingly illogical, preferences regarding their care or other aspects of their stay |
| Avoidant | Socially fearful with feelings of inadequacy | |
| Dependent | Need to be taken care of, often manifesting as clinging and obsequious behavior | |
| Obsessive‐compulsive | Preoccupied by orderliness and control, but without actual obsessions or compulsions |
Cluster A
Those falling into cluster A, which includes paranoid, schizoid, and schizotypal personality disorders, are odd and eccentric and often avoid social engagement[3]; these individuals have few friends or associates and do not care to make more. At times, their unusual thinking can be difficult to differentiate from primary psychotic disorders like schizophrenia.
Cluster B
Cluster B is most heavily studied, consisting of antisocial, borderline, histrionic, and narcissistic personality disorders. These individuals share a high degree of emotional lability and erratic behavior.[3] Frequently, their tendency toward impulsive and self‐destructive behaviors can result in the need for medical care.
Cluster C
Cluster C includes avoidant, dependent, and obsessive‐compulsive personality disorders. These individuals are often anxious and fearful. Like individuals in Cluster A, they have few friends; unlike Cluster A, they long for friendships but struggle to make them. On the inpatient unit, these individuals may have trouble engaging in needed care, relying heavily on others to have their needs met or may be very particular about how their care is administered.
NEUROPHYSIOLOGY
Personality disorders are the product of complex interactions between genes and environment. These disorders are highly heritable, with 55% to 72% heritability across the 3 clusters.[13, 14, 15] Studies have implicated alterations in the serotonin system as playing a role in the underlying pathophysiology, which may contribute to the emotional dysregulation.[16, 17, 18] Neuroimaging has shown alterations in regions of the brain related to emotional reactivity and the processing of social interactions, suggesting neural mechanisms behind these individuals' difficulty with interpersonal relationships.[19, 20, 21, 22]
IDENTIFICATION OF PERSONALITY DISORDERS
These disorders are under‐recognized due, at least in part, to difficulty in making the diagnosis.[7] With 10 different personality disorders, many with overlapping characteristics, establishing a specific diagnosis can be time consuming, and a single individual may fit multiple different personality disorders.[7] Although self‐report surveys and structured interviews exist, these are often time consuming or inaccurate.[7] It is unlikely to be practical to make a diagnosis of a specific personality disorder while in the hospital. Instead, the focus should be placed on identifying impaired personality structures that interfere with interpersonal relationships and thereby disrupt the course of treatment. Consider a personality disorder if any of the following features are present:
- The patient elicits a strong emotional reaction from providers; these may vary markedly between providers.
- The patient's emotional responses may appear disproportionate to the inciting event.
- The patient is on a number of different psychiatric medications with little relief of symptoms.
- The patient takes up a disproportionate amount of the providers' time.
- The patient externalizes blame, seeing others as the source of discomfort or distress and therefore sees others as the solution.
Once identified, steps can be taken to help both the team and the patient.
BEHAVIORAL INTERVENTIONS
The first line of intervention for individuals with dysfunctional personality structures is behavioral, changing the way the team and patient interact (Table 2). Such interventions have long been the cornerstone of treatment for these individuals.[23] The preponderance of the research has focused on cluster B, and specifically individuals with borderline personality disorder, and applying these principles more broadly is largely based on expert opinion.
| Clinical Examples and Behavioral Interventions | ||
|---|---|---|
| Background | Situation | Response |
| Cluster A: Mr. A is a 75‐year‐old man transferred from his small town after a myocardial infarction. Although he has improved medically, he repeatedly expresses distrust and dissatisfaction with his doctors. He refuses to go to a skilled nursing facility but will not work with physical therapy to discharge home. He lives alone and has worked as a cattle rancher all his life. | Mr. A repeatedly accuses his team of being in this for the money. At times he mutters about government conspiracies. |
Check the team's emotions and reinforce desired behaviors and move past negative ones: Recognize paranoia as part of the illness. Rather than confront the paranoia, ignore this behavior as long as it is not directly interfering with care. |
| Cluster B: Ms. B is a 22‐year‐old woman admitted after a car accident resulting in multiple fractures. The pain service is consulted due to her ever‐increasing need for opiates. When the team first meets her, she is bright, effusing, Thank you for coming! My other doctors have no idea how to control my pain. She starts crying, I just can't do this anymore. Midway through the conversation, she offers, I can tell you are the best doctors I've had. Finally, I have someone who understands. Later, the pain team receives numerous pages that the patient is demanding to see them. The following day she is furious at the team for not keeping your promises. Nursing complains about her unwillingness to cooperate with dressing changes, insisting she only work with certain people, Because they understand me. | Ms. B is frequently insulting staff in a demanding and at times threatening manner. |
Reinforce desired behaviors and move past negative ones: Interact in a neutral manner to avoid reinforcing the disruptive behavior. If she becomes threatening or insulting, label the behavior and give her 1 opportunity to stop. Cursing upsets me. It's hard for me to help someone when they're cursing at me. This wording separates the behavior from the person. If she is able to calm herself, thank her (to reinforce this behavior) and offer to help. If she continues to escalate, you can say, You seem to be upset. I'll come back when it is a better time. Withdrawal of social contact can be a powerful tool. Return after a brief period to see if she has been able to calm down and, if so, re‐engage. Re‐engagement is key to reinforce calm, socially acceptable behavior. Check the team's emotions: Recognize patients with challenging behaviors can place a strong emotional toll on the team, particularly nursing staff who must frequently interact with these patients. Offer support to all members of the team to ensure appropriate patient care. |
| Ms. B is crying inconsolably, saying, I just can't stand being in the hospital anymore. They won't give me the pain meds I need. |
Offer validation and reinforce desired behaviors and move past negative ones: Offer empathy but then move to skill building. I can see you are upset. Is there anything that helps you when you are feeling this way? If the patient is unable to come up with anything feasible, offer her choices, such as walking with her around the unit or listening to music. |
|
| Cluster C: Mr. C is a 57‐year‐old man admitted for hyperosmolar hyperglycemic state. His condition has now stabilized, but when the nutritionist attempts to meet with him, he says he has a migraine. Later, when the diabetes nurse comes to discuss his insulin regimen, he is too tired to learn anything. When she persists, he listens, but repeatedly says, I'm never going to be able to do this and is unwilling to participate further. He repeatedly uses his call button, asking for help to the bathroom, despite being ambulatory previously. He talks for extended periods with nursing staff, sharing his fears about his inability to care for himself and his concerns that this will happen again. | Mr. C is repeatedly pressing his nurse call button multiple times throughout the day for seemingly trivial requests. |
Establish parameters: Mr. C is seeking contact with others. Have nursing arrange a regular schedule for checking in on the patient, such as every hour between 10 to and 10 after the hour. These visits can be kept brief, but offer a structure for the patient and encourage him or her to bundle their requests. Caregivers may also consider having Mr. C sit by the nurse's station to increase social interaction. Keep the message consistent: Work to maintain increased social contact across nursing shifts. |
Check the Team's Emotions
Managing patients with personality disorders begins by recognizing that these individuals evoke strong responses from even the most seasoned professional.[10, 11, 24, 25] Reactions toward people with personality disorders can range from a need to care for and protect the patient to feelings of futility or contempt.[10] Referred to as countertransference, these unconscious emotional reactions are common, but can interfere with medical care.[26] Given the increased understanding of the importance of team cohesion in patient care,[27] part of treating an individual with a personality disorder involves recognizing and managing the responses elicited amongst all members of the team. The disparate feelings among team members, which may be driven by different patient behaviors with different people, can lead to a variety of responses including overinvolvement, withdrawal, or even aggression.[28] Recognizing and discussing these differing reactions can help maintain team cohesion and support appropriate patient care.
Offer Empathy and Validation
Patients with personality disorders were often raised in invalidating environments and their ongoing intense emotional reactions can lead to perpetuation of invalidating responses from their caregivers.[29] They are accustomed to eliciting a defensive response from others and can be deliberately provocative, as these intense emotional interactions are comfortable territory, keeping providers feeling off balance and under attack. Instead, offering an empathic response can de‐escalate situations and is associated with the lowest level of invoked anger in patients.[30] Empathy can take the form of validation by acknowledging a person's feelings, thoughts, and emotions as legitimate, even if others may not fully understand or agree with them. As extreme as a patient's response may seem, he or she is genuinely experiencing these feelings and beliefs. Validation includes listening nonjudgmentally, objectively naming emotions the patient is experiencing, and conveying that the patient's response makes sense within the context of the situation.[31] This can include acknowledging the patient's level of distress, saying things such as, I can see you are really frustrated or I am concerned that what I just said has been upsetting. Empathy is more than words; it is the ability to see a situation from someone else's point of view. An empathic approach acknowledges the patient's intense emotional response to the challenges of hospitalization without frustration and judgment. Maintaining an empathicor even simply neutralstance can avoid a power struggle and also improves the therapeutic alliance with the patient.[32]
Establish Parameters but Pick the Battles
Individuals with personality disorders have trouble perceiving social boundaries. Even trained mental health professionals find this difficult to navigate.[33] The provider's first response is often to establish rigid boundaries. However, rigid rules can lead to power struggles between patients and providers, with limits being perceived as punitive. Instead of a list of rules, the creation of boundaries requires a thoughtful, practical establishment of parameters for both the individual and staff.[34] This may include guidelines for frequent, predictable nursing checks on the patient so that attention is provided on a time‐contingent rather than behavior‐contingent basis. If the patient remains dysregulated after a brief attempt to problem solve a nonemergent issue, staff can walk away with the comment that they will return within a specified period of time when things are calmer. If the patient is able to engage in the problem‐solving process, this has the advantage of generating a plan both can agree with while supporting more effective skills in the patient. Rather than a list of stringent rules, consider what is truly necessary for patient safety and well‐being.
Keep the Message Consistent
Hospital care involves many moving parts; nursing staff, the primary team, support staff, and consultants all interact with the patient throughout the day, sometimes providing conflicting messages. Although the typical patient can tolerate this, those with personality disorders have trouble dealing with the inconsistency, and this can exacerbate other problems. Carefully consider potentially contentious issues, such dosing of pain medications and benzodiazepines, and ensure that the team offers a consistent plan.[34] Ideally, meet with the patient as a team, including nursing, to convey a unified message.
Reinforce Desired Behaviors and Move Past Maladaptive Ones
Often in the life of a person with a personality disorder, their interpersonal interactions are negative. These patients are accustomed to negotiating a chaotic world. When not acting out, the patient may receive less attention while nursing and physician attention is appropriately distributed to other patients. Inadvertently, this reinforces using provocative behavior to get attention. Instead, if the patient is not demanding attention, providers should take the opportunity to provide positive reinforcement for calm behavior. This can be done by establishing a routine menu of interactions with the patient that occur when they are not acting in a disruptive manner; this avoids engagement being contingent on negative behaviors.[4] For example, having a nonillness‐related conversation during a dressing change or offering the patient a snack after a positive (or neutral) interaction can reinforce desirable behaviors. In contrast, when patients exhibit disruptive or inappropriate behaviors, the caregiver should respond by removing what the patient seeks, usually engagement, with a neutral attitude to avoid reinforcing the behavior: You seem upset. I'll come back when you feel better. By not reinforcing maladaptive behaviors, caregivers can decrease or extinguish such behaviors over time.[29] If the situation is nonemergent, the caregiver should briefly acknowledge the patient's distress and then focus on possible solutions: I can see you are really upset right now. What helps you in these situations? This both validates the patient's emotional state and encourages him or her to engage in problem solving around his or her distress.[29] If the patient is unable to identify a coping strategy in the moment, suggesting possibilities, such as walking around the unit or listening to music, can help the patient move past their intense emotions while also encouraging skill building.
PHARMACOLOGICAL INTERVENTIONS
Although there are no Food and Drug Administrationapproved medications for treatment of personality disorders, there is limited evidence for use of pharmacological interventions to address particular features of these disorders, such as impulsivity, affective dysregulation, or cognitiveperceptual symptoms.[35] Antipsychotics can be helpful in treating cognitive disturbances such as paranoia and dissociation that some of these patients experience.[35] Antidepressants may have a relatively small effect on anxiety and anger.[35] Mood stabilizers are shown to have a positive impact on impulsivity, anger, anxiety, and depressed mood.[35] However, medication should be used with caution, as polypharmacy is a significant problem with these patients and may have limited utility. Up to 40% of patients with borderline personality disorder take 3 or more psychotropic medications, many of which can have significant side effects,[36] and 1 in 3 are prescribed benzodiazepines despite a lack of evidence and potential for abuse.[37] Thus, although medications may offer an opportunity to target specific symptoms, the focus of management for patients with personality disorders should be behavioral rather than pharmacological.
CONSIDER A CONSULT
Patients with personality disorders can be very difficult to treat, and it may be necessary to consult psychiatry. There are a number of situations in which a formal psychiatric consultation is indicated (Table 3). Patients with personality disorders, particularly cluster B, may present for treatment after harming themselves or others.[38] A psychiatric consultation can provide a formal risk assessment, help with behavior and medication management while the patient is hospitalized, and determine whether follow‐up psychiatric care is appropriate.[39] The psychiatry team can also offer a more complete diagnostic formulation, including screening for disorders that often co‐occur with personality disorders, such as depression and anxiety, and recommend treatment options.[39] In addition, if initial attempts at behavioral interventions are ineffectual, a psychiatric consult may be able to provide additional guidance in behavioral modifications.[40] This is especially appropriate if the patient's behaviors are interfering with medical care. A psychiatric consult can also provide additional support around issues of countertransference that can arise when managing patients with dysfunctional personality structures.[41, 42]
| Safety assessment in a patient who has threatened or engaged in self‐harming behavior or harm to others |
| Diagnostic clarification, particularly when there is concern for a co‐occurring psychiatric illness |
| Creation of a more complex behavioral plan |
| Facilitation of interdisciplinary discussion and problem‐solving around patients with challenging behaviors |
| Assistance with establishment of outpatient psychiatric care when appropriate |
As with all interventions, the psychiatric consult is not without its side effects. Regardless of personality structure, it is not uncommon for patients to be initially opposed to engaging with psychiatry.[43] Individuals with personality disorders can be particularly susceptible to a rupture of the therapeutic alliance,[44] and calling a psychiatric consult can affect the therapeutic relationship with the primary team, as the patient may feel that others are judging them and can also be part of a greater theme of help rejecting.[11] However, this rupture may be repaired as the patient comes to see the psychiatry team as an ally. Even for patients who refuse to engage with the consult‐liaison team, there may be a benefit to a consult, as the consultants can offer strategies to the primary team and help establish a plan of care to facilitate ongoing treatment of the patient's medical needs without direct contact with the patient. These situations illustrate that a psychiatric consult cannot be done in isolation and requires collaboration with the primary team, nurses, and other support staff for interventions to be effective.
CONCLUSIONS
In his now famous speech Dr. Francis W. Peabody gave to Harvard Medical School he noted that [T]he secret of the care of the patient is caring for the patient.[45] Patients with a dysfunctional personality structure can make this task difficult. They can appear to reject the very help we have to offer, divide the team, absorb great amounts of time, and evoke strong feelings of frustration and resentment. However, by understanding that the way in which they interact with the world is in part the product of biology and upbringing, we can better recognize how ill these individuals can be. Just as a patient with diabetes requires management of his blood glucose when admitted for pneumonia, those with personality disorders require management of their mental illness while their other medical conditions are addressed.
Although personality disorders can seem intractable, studies have shown that, like many chronic illnesses, the severity can wax and wane over time with remissions and relapses. Notably, rates of remission for borderline personality disorder at 10 years are comparable to those for major depressive disorder, bipolar disorder, and panic disorder, with lower rates of relapse even without specific treatment, suggesting they are not entirely intractable.[46] However, the stress of hospitalization can easily exacerbate the symptoms of a personality disorder. By providing an empathic approach that addresses the emotional responses of the team while also reinforcing positive behaviors of the patient, the hospital stay can be an opportunity for these individuals to get needed support and develop new skills while also having their physical needs addressed.
Disclosures: Nothing to report.
- , . The effects of trust in physician on self‐efficacy, adherence and diabetes outcomes. Soc Sci Med. 2009;68(6):1060–1068.
- , , , et al. The physician‐patient working alliance. Patient Educ Couns. 2007;66(1):29–36.
- Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition. Arlington, VA: American Psychiatric Association; 2013.
- , , . The borderline patient on the med‐surg unit. Am J Nursing. 1988;88(12):1644–1650.
- , , . “Difficult” patient? Or does he have a personality disorder? J Fam Pract. 2014;63(12):697–703.
- , , , et al. Prevalence, correlates, and disability of personality disorders in the United States: results from the national epidemiologic survey on alcohol and related conditions. J Clin Psychiatry. 2004;65(7):948–958.
- , , . Classification, assessment, prevalence, and effect of personality disorder. Lancet. 2015;385(9969):717–726.
- , , , , , . Life expectancy at birth and all‐cause mortality among people with personality disorder. J Psychosom Res. 2012;73(2):104–107.
- , , , . A systematic review of personality disorders and health outcomes. Can Psychol. 2015;56(2):168–190.
- , , , . Patient personality and therapist response: an empirical investigation. Am J Psychiatry. 2014;171(1):102–108.
- . Taking care of the hateful patient. N Engl J Med. 1978;298(16):883–887.
- , , . Personality disorder across the life course. Lancet. 2015;385(9969):727–734.
- , , , , . The heritability of cluster A personality disorders assessed by both personal interview and questionnaire. Psychol Med. 2007;37(5):655–665.
- , , , et al. The heritability of avoidant and dependent personality disorder assessed by personal interview and questionnaire. Acta Psychiatr Scand. 2012;126(6):448–457.
- , , , , , . The heritability of Cluster B personality disorders assessed both by personal interview and questionnaire. J Pers Disord. 2012;26(6):848–866.
- , , . Association between genetic polymorphisms in the serotonergic system and comorbid personality disorders among patients with first‐episode depression. J Pers Disord. 2014;28(3):365–378.
- , , , et al. Tryptophan‐hydroxylase 2 haplotype association with borderline personality disorder and aggression in a sample of patients with personality disorders and healthy controls. J Psychiatr Res. 2010;44(15):1075–1081.
- , , , et al. Monoamine oxidase A gene promoter methylation and transcriptional downregulation in an offender population with antisocial personality disorder. Br J Psychiatry. 2015;206(3):216–222.
- , , , et al. Regional cortical thinning may be a biological marker for borderline personality disorder. Acta Psychiatr Scand. 2014;130(3):193–204.
- , , . Empathy and social problem solving in alcohol dependence, mood disorders and selected personality disorders. Neurosci Biobehav Rev. 2013;37(3):448–470.
- , , , . Changes in low‐frequency fluctuations in patients with antisocial personality disorder revealed by resting‐state functional MRI. PLoS One. 2014;9(3):e89790.
- , . Prefrontal structural and functional brain imaging findings in antisocial, violent, and psychopathic individuals: a meta‐analysis. Psychiatry Res. 2009;174(2):81–88.
- , , . Treatment of personality disorder. Lancet. 2015;385(9969):735–743.
- . Management of the borderline patient on a medical or surgical ward: the psychiatric consultant's role. Int J Psychiatry Med. 1975;6(3):337–348.
- , , , et al. The attitudes of psychiatric hospital staff toward hospitalization and treatment of patients with borderline personality disorder. BMC Psychiatry. 2015;15:2.
- , , , . The hateful physician: the role of affect bias in the care of the psychiatric patient in the ED. Am J Emerg Med. 2014;32(5):483–485.
- , , , et al. Relationships within inpatient physician housestaff teams and their association with hospitalized patient outcomes. J Hosp Med. 2014;9(12):764–771.
- . Countertransference in the nurse‐patient relationship: a review of the literature. J Adv Nurs. 1998;28(2):391–397.
- , , , , . Mechanisms of change in dialectical behavior therapy: theoretical and empirical observations. J Clin Psychol. 2006;62(4):459–480.
- , , , . The relationship between nurses' limit‐setting styles and anger in psychiatric inpatients. Psychiatr Serv. 1995;46(6):609–613.
- , , , , , . Attachment disorganization and borderline patients' metacognitive responses to therapists' expressed understanding of their states of mind: a pilot study. Psychother Res. 2008;18(1):28–36.
- , , . Narcissistic personality disorder: diagnostic and clinical challenges. Am J Psychiatry. 2015;172(5):415–422.
- , . An evaluation of the difficulties and attitudes mental health professionals experience with people with personality disorders. J Psychiatr Ment Health Nurs. 2016;23(1):22–36.
- , . Nursing care of personality disorders in the medical surgery setting. Nurs Clin North Am. 1998;33(1):173–186.
- , , , , . Effectiveness of pharmacotherapy for severe personality disorders: meta‐analyses of randomized controlled trials. J Clin Psychiatry. 2010;71(1):14–25.
- , , , . Mental health service utilization by borderline personality disorder patients and Axis II comparison subjects followed prospectively for 6 years. J Clin Psychiatry. 2004;65(1):28–36.
- , , , , . The use of psychotropic medication in patients with emotionally unstable personality disorder under the care of UK mental health services. J Clin Psychiatry. 2015;76(4):e512–e518.
- , , , , . Prevalence and characteristics of cluster B personality disorder in a consultation‐liaison psychiatry practice. Int J Psychiatry Clin Pract. 2015;19(1):65–70.
- , , , et al. The Academy of Psychosomatic Medicine practice guidelines for psychiatric consultation in the general medical setting. The Academy of Psychosomatic Medicine. Psychosomatics. 1998;39(4):S8–S30.
- , , . Interventions in consultation/liaison psychiatry. Part I: Patterns of recommendations. Gen Hosp Psychiatry. 1990;12(4):213–220.
- , , . Psychodynamics in medically ill patients. Harv Rev Psychiatry. 2009;17(6):389–397.
- . Patient dynamics, staff burnout, and consultation‐liaison psychiatry. Physician Exec. 1991;17(5):37–40.
- , . Facilitating patient acceptance of a psychiatric referral. Arch Intern Med. 1985;145(1):73–75.
- . The therapeutic alliance in the treatment of personality disorders. J Psychiatr Pract. 2005;11(2):73–87.
- . Dr. Francis w. Peabody, we need you. Tex Heart Inst J. 2011;38(4):327–328; discussion 328–329.
- , , , et al. Ten‐year course of borderline personality disorder: psychopathology and function from the Collaborative Longitudinal Personality Disorders study. Arch Gen Psychiatry. 2011;68(8):827–837.
- , . The effects of trust in physician on self‐efficacy, adherence and diabetes outcomes. Soc Sci Med. 2009;68(6):1060–1068.
- , , , et al. The physician‐patient working alliance. Patient Educ Couns. 2007;66(1):29–36.
- Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition. Arlington, VA: American Psychiatric Association; 2013.
- , , . The borderline patient on the med‐surg unit. Am J Nursing. 1988;88(12):1644–1650.
- , , . “Difficult” patient? Or does he have a personality disorder? J Fam Pract. 2014;63(12):697–703.
- , , , et al. Prevalence, correlates, and disability of personality disorders in the United States: results from the national epidemiologic survey on alcohol and related conditions. J Clin Psychiatry. 2004;65(7):948–958.
- , , . Classification, assessment, prevalence, and effect of personality disorder. Lancet. 2015;385(9969):717–726.
- , , , , , . Life expectancy at birth and all‐cause mortality among people with personality disorder. J Psychosom Res. 2012;73(2):104–107.
- , , , . A systematic review of personality disorders and health outcomes. Can Psychol. 2015;56(2):168–190.
- , , , . Patient personality and therapist response: an empirical investigation. Am J Psychiatry. 2014;171(1):102–108.
- . Taking care of the hateful patient. N Engl J Med. 1978;298(16):883–887.
- , , . Personality disorder across the life course. Lancet. 2015;385(9969):727–734.
- , , , , . The heritability of cluster A personality disorders assessed by both personal interview and questionnaire. Psychol Med. 2007;37(5):655–665.
- , , , et al. The heritability of avoidant and dependent personality disorder assessed by personal interview and questionnaire. Acta Psychiatr Scand. 2012;126(6):448–457.
- , , , , , . The heritability of Cluster B personality disorders assessed both by personal interview and questionnaire. J Pers Disord. 2012;26(6):848–866.
- , , . Association between genetic polymorphisms in the serotonergic system and comorbid personality disorders among patients with first‐episode depression. J Pers Disord. 2014;28(3):365–378.
- , , , et al. Tryptophan‐hydroxylase 2 haplotype association with borderline personality disorder and aggression in a sample of patients with personality disorders and healthy controls. J Psychiatr Res. 2010;44(15):1075–1081.
- , , , et al. Monoamine oxidase A gene promoter methylation and transcriptional downregulation in an offender population with antisocial personality disorder. Br J Psychiatry. 2015;206(3):216–222.
- , , , et al. Regional cortical thinning may be a biological marker for borderline personality disorder. Acta Psychiatr Scand. 2014;130(3):193–204.
- , , . Empathy and social problem solving in alcohol dependence, mood disorders and selected personality disorders. Neurosci Biobehav Rev. 2013;37(3):448–470.
- , , , . Changes in low‐frequency fluctuations in patients with antisocial personality disorder revealed by resting‐state functional MRI. PLoS One. 2014;9(3):e89790.
- , . Prefrontal structural and functional brain imaging findings in antisocial, violent, and psychopathic individuals: a meta‐analysis. Psychiatry Res. 2009;174(2):81–88.
- , , . Treatment of personality disorder. Lancet. 2015;385(9969):735–743.
- . Management of the borderline patient on a medical or surgical ward: the psychiatric consultant's role. Int J Psychiatry Med. 1975;6(3):337–348.
- , , , et al. The attitudes of psychiatric hospital staff toward hospitalization and treatment of patients with borderline personality disorder. BMC Psychiatry. 2015;15:2.
- , , , . The hateful physician: the role of affect bias in the care of the psychiatric patient in the ED. Am J Emerg Med. 2014;32(5):483–485.
- , , , et al. Relationships within inpatient physician housestaff teams and their association with hospitalized patient outcomes. J Hosp Med. 2014;9(12):764–771.
- . Countertransference in the nurse‐patient relationship: a review of the literature. J Adv Nurs. 1998;28(2):391–397.
- , , , , . Mechanisms of change in dialectical behavior therapy: theoretical and empirical observations. J Clin Psychol. 2006;62(4):459–480.
- , , , . The relationship between nurses' limit‐setting styles and anger in psychiatric inpatients. Psychiatr Serv. 1995;46(6):609–613.
- , , , , , . Attachment disorganization and borderline patients' metacognitive responses to therapists' expressed understanding of their states of mind: a pilot study. Psychother Res. 2008;18(1):28–36.
- , , . Narcissistic personality disorder: diagnostic and clinical challenges. Am J Psychiatry. 2015;172(5):415–422.
- , . An evaluation of the difficulties and attitudes mental health professionals experience with people with personality disorders. J Psychiatr Ment Health Nurs. 2016;23(1):22–36.
- , . Nursing care of personality disorders in the medical surgery setting. Nurs Clin North Am. 1998;33(1):173–186.
- , , , , . Effectiveness of pharmacotherapy for severe personality disorders: meta‐analyses of randomized controlled trials. J Clin Psychiatry. 2010;71(1):14–25.
- , , , . Mental health service utilization by borderline personality disorder patients and Axis II comparison subjects followed prospectively for 6 years. J Clin Psychiatry. 2004;65(1):28–36.
- , , , , . The use of psychotropic medication in patients with emotionally unstable personality disorder under the care of UK mental health services. J Clin Psychiatry. 2015;76(4):e512–e518.
- , , , , . Prevalence and characteristics of cluster B personality disorder in a consultation‐liaison psychiatry practice. Int J Psychiatry Clin Pract. 2015;19(1):65–70.
- , , , et al. The Academy of Psychosomatic Medicine practice guidelines for psychiatric consultation in the general medical setting. The Academy of Psychosomatic Medicine. Psychosomatics. 1998;39(4):S8–S30.
- , , . Interventions in consultation/liaison psychiatry. Part I: Patterns of recommendations. Gen Hosp Psychiatry. 1990;12(4):213–220.
- , , . Psychodynamics in medically ill patients. Harv Rev Psychiatry. 2009;17(6):389–397.
- . Patient dynamics, staff burnout, and consultation‐liaison psychiatry. Physician Exec. 1991;17(5):37–40.
- , . Facilitating patient acceptance of a psychiatric referral. Arch Intern Med. 1985;145(1):73–75.
- . The therapeutic alliance in the treatment of personality disorders. J Psychiatr Pract. 2005;11(2):73–87.
- . Dr. Francis w. Peabody, we need you. Tex Heart Inst J. 2011;38(4):327–328; discussion 328–329.
- , , , et al. Ten‐year course of borderline personality disorder: psychopathology and function from the Collaborative Longitudinal Personality Disorders study. Arch Gen Psychiatry. 2011;68(8):827–837.
Neuroimaging in children and adolescents: When do you scan? With which modalities?
The first 15 years of the new millennium have seen a great increase in research on neuroimaging in children and adolescents who have a psychiatric disorder. In addition, imaging modalities continue to evolve, and are becoming increasingly accessible and informative. The literature is now replete with reports of neurostructural differences between patients and healthy subjects in a variety of common pediatric psychiatric conditions, including anxiety disorders, mood disorders, autism spectrum disorder (ASD), and attention-deficit/hyperactivity disorder (ADHD).
Historically, the clinical utility of neuroimaging was restricted to the identification of structural pathology. Today, accumulating data reveal novel roles for neuroimaging; these revelations are supported by studies demonstrating that treatment response for psychotherapeutic and psychopharmacotherapeutic interventions can be predicted by neurochemical and neurofunctional characteristics assessed by advanced imaging technologies, such as magnetic resonance spectroscopy (MRS) and functional MRI.
However, such advanced techniques are (at least at present) not ready for routine clinical use for this purpose. Instead, neuroimaging in the child and adolescent psychiatric clinic remains largely focused on ruling out neurostructural, neurologic, “nonpsychiatric” causes of our patients’ symptoms.
Understanding the role and limitations of major imaging modalities is key to guiding efficient and appropriate neuroimaging selection for pediatric patients. In this article, we describe and review:
- neuroimaging approaches for children and adolescents with psychiatric disorders
- the role of neuroimaging in (1) the differential diagnosis and workup of common psychiatric disorders and (2) urgent clinical situations
- how to determine what type of imaging to obtain.
Computed tomography
CT, which utilizes ionizing radiation, often is reserved, in the pediatric setting, for (1) emergency evaluation and (2) excluding potentially catastrophic neurologic injury resulting from:
- ischemic or hemorrhagic stroke
- herniation
- intracerebral hemorrhage
- subdural and epidural hematoma
- large intracranial mass with mass effect
- increased intracranial pressure
- acute skull fracture.
Although a CT scan is, typically, quick and has excellent sensitivity for acute bleeding and bony pathology, it exposes the patient to radiation and provides poor resolution compared with MRI.
In pediatrics, there has been practice-changing recognition of the importance of limiting lifetime radiation exposure incurred from medical procedures and imaging. As a result, most providers now agree that use of MRI in lieu of CT is appropriate in many, if not most, non-emergent situations. In an emergent situation, however, CT imaging is appropriate and should not be delayed. Moreover, in an emergent situation, you should not hesitate to use head CT in children, although timely discussion with the radiologist is recommended to review your differential diagnosis to better determine the preferred imaging modality.
Magnetic resonance imaging
Over the past several decades, MRI has been increasingly available in most pediatric health care facilities. The modality offers specific advantages for pediatric patients, including:
- better spatial resolution
- the ability to concurrently assess multiple pathologic processes
- lack of exposure to ionizing radiation.1
A number of MRI sequences, described below, can be used to assess vascular, inflammatory, structural, and metabolic processes.
A look inside. Comprehensive review of the physics that underlies MRI is beyond the scope of this article; several important principles are relevant to clinicians, however. Image contrast is dependent on intrinsic properties of tissue with regard to proton density, longitudinal relaxation time (T1), and transverse relaxation time (T2). Pulse sequences, which describe the strength and timing of the radiofrequency pulse and gradient pulses, define imaging acquisition parameters (eg, repetition time between the radio frequency pulse and echo time).
In turn, the intensity of the signal that is “seen” with various pulse sequences is differentially affected by intrinsic properties of tissue. At most pediatric institutions, the standard MRI-examination protocol includes: a T1-weighted image (Figure 1A); a T2-weighted scan (Figure 1B); fluid attenuated inversion recovery (FLAIR) (Figure 1C); and diffusion-weighted imaging (DWI) (Figure 1D).
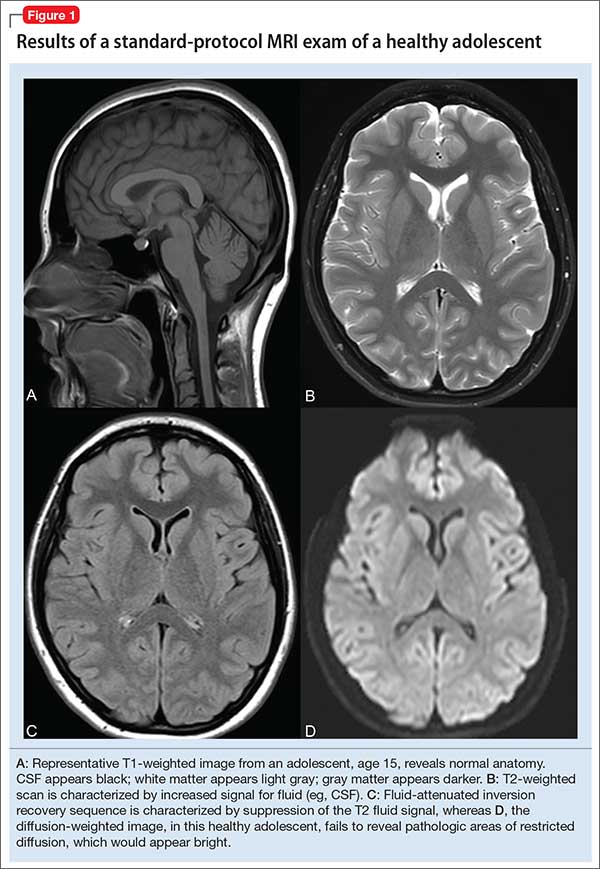
Specific MRI sequences
T1 images. T1 sequences, or so-called anatomy sequences, are ideally suited for detailed neuroanatomic evaluations. They are generated in such a way that structures containing fluid are dark (hypo-intense), whereas other structures, with higher fat or protein content, are brighter (iso-intense, even hyper-intense). For this reason, CSF in the intracranial ventricles is dark, and white matter is brighter than the cortex because of lipid in myelin sheaths.
In addition, to view structural abnormalities that are characterized by altered vascular supply or flow, such as tumors and infections (abscesses), contrast imaging can be particularly helpful; such images generally are obtained as T1 sequences.
T2 images. By contrast to the T1-weighted sequence, the T2-weighted sequences emphasize fluid signal; structures such as the ventricles, which contain CSF, therefore will be bright (hyper-intense). Pathology that produces edema or fluid, such as edema surrounding demyelinating lesions or infections, also will show bright hyper-intense signal. In T2-weighted images of the brain, white matter shows lower signal intensity than the cortex because of the relatively lower water content in white matter tracts and myelin sheaths.
Fluid attenuation inversion recovery. FLAIR images are generated so that the baseline bright T2 signal seen in normal structures, such as the CSF, containing ventricles is cancelled out, or attenuated. In effect, this subtraction of typical background hyper-intense fluid signal leaves only abnormal T2 bright hyper-intense signal, such as vasogenic edema surrounding tumors, cytotoxic edema within an infarction, or extra-axial fluid collections such as a subarachnoid or subdural hemorrhage.
Diffusion-weighted imaging. DWI utilizes the random motion (ie, diffusion) of water molecules to generate contrast. In this regard, the diffusion of any molecule is influenced by its interaction with other molecules (eg, white-matter fibers and membranes, and macromolecules). Diffusion patterns therefore reflect details about tissue boundaries; as such, DWI is sensitive to a number of neurologic processes, such as ischemia, demyelinating disease, and some tumors, which restrict the free motion of water. DWI detects this so-called restricted diffusion and displays an area of bright signal.
Susceptibility-weighted imaging (SWI). In the pediatric population, SWI (Figure 2) utilizes a long-echo, 3-dimensional, velocity-compensated gradient recalled echo for image acquisition2 and, ultimately, leverages susceptibility differences across tissues by employing the phase image to identify these differences. SWI, which uses both magnitude and phase images and is remarkably sensitive to venous blood (and blood products), iron, and calcifications, therefore might be of increasing utility in pediatric patients with traumatic brain injury (TBI) (Figure 2B). As such, SWI has become a critical component of many pediatric MRI studies.3
Magnetic resonance angiography (MRA) (Figure 3A) is helpful for assessing intracranial arteries and may be employed in the evaluation of:
- vessel pathology and injury underlying stroke, such as vessel occlusion or injury
- patterns of vessel involvement suggestive of vasculitis
- developmental or acquired structural vascular abnormalities, such as aneurysm or vascular malformations
- determination of tumor blood supply.

MRA can be performed without or with contrast, although MRA with contrast might provide a higher quality study and therefore be of greater utility. Of note: The spatial resolution of MRA is not as good as CT angiography; abnormalities, such as a small aneurysm, might not be apparent.
Magnetic resonance venography (MRV) (Figure 3B) is most commonly performed when the possibility of thrombosis of the dural venous sinuses is being considered; it also is employed to evaluate vascular malformations, tumor drainage patterns, and other pathologic states. As with MRA, MRV can be performed without or with contrast, although post-contrast MRV is generally of higher quality and might be preferred when assessing for sinus thrombosis.
Magnetic resonance spectroscopy (MRS) resides at the border between research and clinical practice. In children and adolescents, MRS provides data on neuronal and axonal viability as well as energetics and cell membranes.4 Pediatric neurologists often use MRS to evaluate for congenital neurometabolic disease; this modality also can help distinguish between an active intracranial tumor from an abscess or gliosis.5
Neuroimaging in pediatric neuropsychiatric conditions: Evidence, guidance
Delirium (altered mental status). The acute neuropsychiatric syndrome characterized by impaired attention and sensorium might have a broad underlying etiology, but it is always associated with alteration of CNS neurophysiology. Children with neurostructural abnormalities might have increased vulnerability to CNS insult and therefore be at increased risk of delirium.6 Additionally, delirium can present subtly in children, with the precise signs dependent on the individual patient’s developmental stage.
Neuroimaging may be helpful when an infectious, inflammatory, toxic, or a metabolic basis for delirium is suspected, or when a patient has new focal neurologic findings. In this regard, focal neurologic findings suggest an underlying localizable lesion and warrant dedicated neuroimaging to localize the lesion.
In general, the differential diagnosis should guide consideration of neuroimaging. When considering the possibility of an unwitnessed seizure in a child who presents with altered mental status, neuroimaging certainly is an important component of the workup.
As another example, when underlying trauma, intracranial hemorrhage, or mass is a possibility in acute delirium, urgent head CT is appropriate. In non-emergent cases, MRI is the modality of choice. In immunocompromised patients presenting with delirium, maintain a low threshold for neuroimaging with contrast to rule out opportunistic intracranial infection.
Last, in children who have hydrocephalus with a shunt, delirium could be a harbinger of underlying shunt malfunction, warranting a “shunt series.”
ADHD. The diagnosis of ADHD remains a clinical one; for the typical pediatric patient with ADHD but who does not have focal neurologic deficits, neuroimaging is unnecessary. Some structural MRI studies of youth with ADHD suggest diminished volume of the globus pallidus, putamen, and caudate7; other studies reveal changes in gyrification and cortical thickness8 in regions subserving attentional processes. However, intra-individual and developmental-related variability preclude routine use of neuroimaging in the standard diagnostic work-up of ADHD.
Nevertheless, neuroimaging should be strongly considered in a child with progressive worsening of inattention, especially if combined with other psychiatric or neurologic findings. In such a case, MRI should be obtained to evaluate for a progressive neurodegenerative leukoencephalopathy (eg, adrenoleukodystrophy).9
Depressive and anxiety disorders. In pediatric patients who exhibit depressive or anxiety symptoms, abnormalities have been observed in cortical thickness10 and gray matter volume,11,12 and functional signatures13 have been identified in the circuitry of the prefrontal amygdala. No data suggest that, in an individual patient, neuroimaging can be of diagnostic utility—particularly in the absence of focal neurologic findings.
That being said, headache and other somatic symptoms are common in pediatric patients with a mood or anxiety disorder. Evidence for neuroimaging in the context of pediatric headache suggests that MRI should be considered when headache is associated with neurologic signs or symptoms, such as aura, or accompanied by focal neurologic deficit.14
Pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections (PANDAS) and pediatric acute-onset neuropsychiatric syndrome (PANS). Neuroimaging studies of patients with confirmed PANDAS or PANS are rare, but group analyses suggest a decreased average volume of the caudate, putamen, and globus pallidus in patients with PANDAS compared with healthy comparison subjects, although total cerebral volume does not appear to differ.15 Moreover, thalamic findings in patients with PANDAS have been noted to be similar to what is seen in patients with Sydenham’s chorea.
The most recent consensus statement regarding the treatment and assessment of PANDAS and PANS recommends ordering brain MRI when other conditions are suspected (eg, CNS, small vessel vasculitis, limbic encephalitis) or when the patient has severe headache, gait disturbance, cognitive deterioration, or psychosis.16 Furthermore, the consensus statement notes the potential utility of T2-weighted imaging with contrast to evaluate inflammatory changes in the basal ganglia.16
Autism spectrum disorder. Significant progress has been made during the past decade on the neuroanatomic characterization of ASD. Accumulating data indicate that, in pediatric patients with ASD, (1) development of white matter and gray matter is disrupted early in the course of the disorder and (2) cortical thickness is increased in regions subserving social cognition.17,18
Several studies have examined the presence of patient-level findings in samples of pediatric patients with ASD. Approximately 8% of pediatric patients with ASD were found to have some abnormality on routine brain MRI, the most common being white-matter signal abnormalities, dilated Virchow-Robin space, and temporal lobe abnormalities.19
Although abnormalities might be present in a large percentage of individual scans, routine screening MRI is unlikely to be of clinical utility in youth with ASD. In fact, no recommendation for routine MRI screening in patients with ASD has been made by the American Academy of Child & Adolescent Psychiatry, the Child Neurology Society of the American Academy of Neurology, or the American Academy of Pediatrics.
However, in patients with an underlying neurostructural disease that is phenotypically associated with ASD-like symptoms, imaging might be of use. In tuberous sclerosis, for example, MRI is especially important to classify intracranial lesions; determine burden and location; or identify treatment options (Figure 4). For patients with tuberous sclerosis—of whom more than one-third meet diagnostic criteria for ASD20—MRI study should include FLAIR, spin-echo, and gradient-echo sequences.

Movement disorders. Hyperkinetic movement disorders, including tic disorders and drug-induced movement disorders (eg, tremor) are common in pediatric patients. In pediatric patients with a tic disorder or Tourette’s disorder (TD), neuroimaging typically is unnecessary, despite the suggestion that the caudate nucleus volume is reduced in groups of patients with TD.
Many CNS-acting medications can exacerbate physiologic tremor; in pediatric patients with symptoms of a movement disorder, home medications should be carefully reviewed for potentially offending agents. When the patient is clinically and biochemically euthyroid and medication-induced movement disorder has been ruled out, or when the patient meets clinical diagnostic criteria for TD or a tic disorder, routine neuroimaging generally is unnecessary.
When tremor accompanies other cerebellar signs, such as ataxia or dysmetria, strongly consider MRI of the brain to evaluate pathology in the posterior fossa. In addition, neuroimaging should be considered for children with a new-onset abnormality on neurologic exam, including rapid onset of abnormal movements (other than common tics), continuous progressive worsening of symptoms, or any loss of developmental milestones.
Last, although tics and stereotypies often are transient and wane with age, other abnormal movements, such as dystonia, chorea, and parkinsonism (aside from those potentially associated with antipsychotic use), are never expected during typical development and warrant MRI.
Traumatic brain injury. Prompt evaluation and intervention for TBI can significantly affect overall outcome. Moreover, there has been increased enthusiasm around the pre-hospital assessment of TBI severity using (1) any of several proprietary testing systems (eg, Immediate Post-Concussion Assessment and Cognitive Testing [ImPACT]) plus (2) standard clinical staging, which is based on duration of loss of consciousness, persistence of memory loss, and the Glasgow Coma Scale score.
The goal for any TBI patient during the acute post-injury phase is to minimize continued neuronal injury from secondary effects of TBI, such as cerebral edema and herniation, and to optimize protection of surviving brain tissue; neuroimaging is a critical component of assessment during both acute and chronic recovery periods of TBI.21
The optimal imaging modality varies with the amount of time that has passed since initial injury.22 Urgent neuroimaging (the first 24 hours after brain injury) is typically obtained using head CT to assist decision-making in acute neurosurgical management. In this setting, head CT is fast and efficient; minimizes the amount of time that the patient is in the scanner; and provides valuable information on the acuity and extent of injury, degree of cerebral edema, and evidence or risk of pending herniation.
On the other hand, MRI is superior to CT during 48 to 72 hours after injury, given its higher resolution; superior imaging of the brainstem and deep gray nuclei; and ability to detect axonal injury, small contusions, and subtle neuronal damage. Specifically, SWI sequences can be particularly helpful in TBI for detecting diffuse axonal injury and micro-hemorrhages; several recent studies also suggest that SWI may be of particular value in pediatric patients with TBI.23
Additionally, given the increased sensitivity of MRI to detect subtle injuries, this modality can assist in identifying chronic sequelae of brain injury—thus contributing to determining of chronic therapy options and assisting with long-term prognosis. Gross structural changes resulting from TBI often are evident even in the acute post-injury phase; synaptic remodeling continues, however, for an indefinite period after injury, and this remodeling capacity is even more pronounced in the highly plastic brain of a young child.
Microstructural changes might not be detectable using traditional, readily available imaging sequences (CT, MRI). When those traditional modalities are used in concert with functional imaging techniques (eg, PET to evaluate cerebral metabolism and SPECT imaging which can detect abnormalities in cerebral blood flow), the combination of older and newer might provide a more complete picture of recovery after TBI.24
The important role of neuroimaging in severe TBI is intuitive. However, it is important to consider the role of neuroimaging in mild TBI in children, especially in the setting of repetitive mild injury.25 A growing body of evidence supports close, serial monitoring of children after even mild closed head injury for neurologic and psychiatric sequelae. Although it is rare that a child who is awake, interactive, and lacking focal neurologic deficits would need emergent (ie, CT) imaging after mild closed head injury, there might be a role for MRI later in the course of that child’s recovery—especially if recovery is complicated by clinical sequelae of mild TBI, such as cognitive impairment, headaches, or altered behavior.
When is additional neuroimaging needed?
It’s worthwhile briefly reviewing 4 scenarios that you might encounter, when you work with children and adolescents, in which urgent or emergent neuroimaging (often with consultation) should be obtained. The Table describes these situations and appropriate first- and second-line interventions.
1. When the presentation of your patient is consistent with an acute neurologic deficit, acute TBI, progressive neuropsychiatric decline, CNS infection, mass, demyelinating process, or toxic exposure, neuroimaging is likely critical.
2. In patients with progressive neurologic decline, including loss of developmental milestones, MRI, MRS, and referral to neurology should be part of the comprehensive evaluation.
3. In young children who exhibit a decrease in head circumference on growth curves, MRI is important to evaluate for underlying structural causes.
4. Pediatric patients with symptoms consistent with either stroke (ie, a new, persistent neurologic deficit) or a demyelinating process (eg, multiple episodes of variable transient focal neurologic symptoms), MRI should be obtained without compunction.

Consultation with pediatric neuroradiology
In deciding whether to obtain neuroimaging for a particular case, you should discuss your concerns with the pediatric radiologist or pediatric neuroradiologist, who will likely provide important guidance on key aspects of the study (eg, modifying slice thickness in a particular scan; recommending the use of contrast; including MRS in the order for imaging; performing appropriate vessel imaging). Consider asking 1 or more important questions when you discuss a patient’s presentation with the pediatric radiologist or pediatric neuroradiologist:
- “What neuroimaging studies are appropriate, based on my differential diagnosis?”
- “Are there specific imaging sequences that we should consider?”
- “Are there contraindications to the imaging modality for my patient?”
- “Is my patient likely to have difficulty tolerating the imaging procedure?”
- “Does my patient need sedation to tolerate this procedure?”
- “Should additional regions be included in the scan?” (Examples: In a child with stroke it might be important to include neck and chest vasculature and the heart. Other conditions might warrant imaging of the spinal cord.)
1. Abdelhalim AN, Alberico RA. Pediatric neuroimaging. Neurol Clin. 2009;27(1):285-301, x.
2. Sehgal V, Delproposto Z, Haacke EM, et al. Clinical applications of neuroimaging with susceptibility-weighted imaging. J Magn Reson Imaging. 2005;22(4):439-450.
3. Bosemani T, Poretti A, Huisman TA. Susceptibility-weighted imaging in pediatric neuroimaging. J Magn Reson Imaging. 2014;40(3):530-544.
4. Cecil KM. Proton magnetic resonance spectroscopy: technique for the neuroradiologist. Neuroimaging Clin N Am. 2013;23(3):381-392.
5. Panigrahy A, Nelson MD Jr, Blüml S. Magnetic resonance spectroscopy in pediatric neuroradiology: clinical and research applications. Pediatr Radiol. 2010;40(1):3-30.
6. Leentjens AF, Schieveld JN, Leonard M, et al. A comparison of the phenomenology of pediatric, adult, and geriatric delirium. J Psychosom Res. 2008;64(2):219-223.
7. Frodl T, Skokauskas N. Meta-analysis of structural MRI studies in children and adults with attention deficit hyperactivity disorder indicates treatment effects. Acta Psychiatr Scand. 2012;125(2):114-126.
8. Shaw P, Malek M, Watson B, et al. Development of cortical surface area and gyrification in attention-deficit/hyperactivity disorder. Biol Psychiatry. 2012;72(3):191-197.
9. Phelan JA, Lowe LH, Glasier CM. Pediatric neurodegenerative white matter processes: leukodystrophies and beyond. Pediatr Radiol. 2008;38(7):729-749.
10. Strawn JR, Wegman CJ, Dominick KC, et al. Cortical surface anatomy in pediatric patients with generalized anxiety disorder. J Anxiety Disord. 2014;28(7):717-723.
11. Mueller SC, Aouidad A, Gorodetsky E, et al. Gray matter volume in adolescent anxiety: an impact of the brain-derived neurotrophic factor Val(66)Met polymorphism [Erratum in J Am Acad Child Adolesc Psychiatry. 2013;52(2):184-195]? J Am Acad Child Adolesc Psychiatry. 2013;52(2):184-195.
12. Strawn JR, Hamm L, Fitzgerald DA, et al. Neurostructural abnormalities in pediatric anxiety disorders. J Anxiety Disord. 2015;32:81-88.
13. Strawn JR, Dominick KC, Patino LR, et al. Neurobiology of pediatric anxiety disorders. Curr Behav Neurosci Reports. 2014;1(3):154-160.
14. Alexiou GA, Argyropoulou MI. Neuroimaging in childhood headache: a systematic review. Pediatr Radiol. 2013;43(7):777-784.
15. Giedd JN, Rapoport JL, Garvey MA, et al. MRI assessment of children with obsessive-compulsive disorder or tics associated with streptococcal infection. Am J Psychiatry. 2000;157(2):281-283.
16. Chang K, Frankovich J, Cooperstock M, et al; PANS Collaborative Consortium. Clinical evaluation of youth with pediatric acute-onset neuropsychiatric syndrome (PANS): recommendations from the 2013 PANS Consensus Conference. J Child Adolesc Psychopharmacol. 2014;25(1):3-13.
17. Wallace GL, Robustelli B, Dankner N, et al. Increased gyrification, but comparable surface area in adolescents with autism spectrum disorders. Brain. 2013;136(pt 6):1956-1967.
18. Libero LE, DeRamus TP, Deshpande HD, et al. Surface-based morphometry of the cortical architecture of autism spectrum disorders: volume, thickness, area, and gyrification. Neuropsychologia. 2014;62:1-10.
19. Boddaert N, Zilbovicius M, Philipe A, et al. MRI findings in 77 children with non-syndromic autistic disorder. PLoS One. 2009;4:e445. doi: 10.1371/journal.pone.0004415.
20. Richards C, Jones C, Groves L, et al. Prevalence of autism spectrum disorder phenomenology in genetic disorders: a systematic review and meta-analysis. Lancet Psychiatry. 2015;2(10):909-916.
21. Wilde EA, Hunter JV, Bigler ED. Pediatric traumatic brain injury: neuroimaging and neurorehabilitation outcome. NeuroRehabilitation. 2012;31(3):245-260.
22. Mechtler LL, Shastri KK, Crutchfield KE. Advanced neuroimaging of mild traumatic brain injury. Neurol Clin. 2014;32(1):31-58.
23. Ashwal S, Tong KA, Ghosh N, et al. Application of advanced neuroimaging modalities in pediatric traumatic brain injury. J Child Neurol. 2014;29(12):1704-1717.
24. Munson S, Schroth E, Ernst M. The role of functional neuroimaging in pediatric brain injury. Pediatrics. 2006;117(4):1372-1381.
25. Wozniak JR, Krach L, Ward E, et al. Neurocognitive and neuroimaging correlates of pediatric traumatic brain injury: a diffusion tensor imaging (DTI) study. Arch Clin Neuropsychol. 2007;22(5):555-568.
The first 15 years of the new millennium have seen a great increase in research on neuroimaging in children and adolescents who have a psychiatric disorder. In addition, imaging modalities continue to evolve, and are becoming increasingly accessible and informative. The literature is now replete with reports of neurostructural differences between patients and healthy subjects in a variety of common pediatric psychiatric conditions, including anxiety disorders, mood disorders, autism spectrum disorder (ASD), and attention-deficit/hyperactivity disorder (ADHD).
Historically, the clinical utility of neuroimaging was restricted to the identification of structural pathology. Today, accumulating data reveal novel roles for neuroimaging; these revelations are supported by studies demonstrating that treatment response for psychotherapeutic and psychopharmacotherapeutic interventions can be predicted by neurochemical and neurofunctional characteristics assessed by advanced imaging technologies, such as magnetic resonance spectroscopy (MRS) and functional MRI.
However, such advanced techniques are (at least at present) not ready for routine clinical use for this purpose. Instead, neuroimaging in the child and adolescent psychiatric clinic remains largely focused on ruling out neurostructural, neurologic, “nonpsychiatric” causes of our patients’ symptoms.
Understanding the role and limitations of major imaging modalities is key to guiding efficient and appropriate neuroimaging selection for pediatric patients. In this article, we describe and review:
- neuroimaging approaches for children and adolescents with psychiatric disorders
- the role of neuroimaging in (1) the differential diagnosis and workup of common psychiatric disorders and (2) urgent clinical situations
- how to determine what type of imaging to obtain.
Computed tomography
CT, which utilizes ionizing radiation, often is reserved, in the pediatric setting, for (1) emergency evaluation and (2) excluding potentially catastrophic neurologic injury resulting from:
- ischemic or hemorrhagic stroke
- herniation
- intracerebral hemorrhage
- subdural and epidural hematoma
- large intracranial mass with mass effect
- increased intracranial pressure
- acute skull fracture.
Although a CT scan is, typically, quick and has excellent sensitivity for acute bleeding and bony pathology, it exposes the patient to radiation and provides poor resolution compared with MRI.
In pediatrics, there has been practice-changing recognition of the importance of limiting lifetime radiation exposure incurred from medical procedures and imaging. As a result, most providers now agree that use of MRI in lieu of CT is appropriate in many, if not most, non-emergent situations. In an emergent situation, however, CT imaging is appropriate and should not be delayed. Moreover, in an emergent situation, you should not hesitate to use head CT in children, although timely discussion with the radiologist is recommended to review your differential diagnosis to better determine the preferred imaging modality.
Magnetic resonance imaging
Over the past several decades, MRI has been increasingly available in most pediatric health care facilities. The modality offers specific advantages for pediatric patients, including:
- better spatial resolution
- the ability to concurrently assess multiple pathologic processes
- lack of exposure to ionizing radiation.1
A number of MRI sequences, described below, can be used to assess vascular, inflammatory, structural, and metabolic processes.
A look inside. Comprehensive review of the physics that underlies MRI is beyond the scope of this article; several important principles are relevant to clinicians, however. Image contrast is dependent on intrinsic properties of tissue with regard to proton density, longitudinal relaxation time (T1), and transverse relaxation time (T2). Pulse sequences, which describe the strength and timing of the radiofrequency pulse and gradient pulses, define imaging acquisition parameters (eg, repetition time between the radio frequency pulse and echo time).
In turn, the intensity of the signal that is “seen” with various pulse sequences is differentially affected by intrinsic properties of tissue. At most pediatric institutions, the standard MRI-examination protocol includes: a T1-weighted image (Figure 1A); a T2-weighted scan (Figure 1B); fluid attenuated inversion recovery (FLAIR) (Figure 1C); and diffusion-weighted imaging (DWI) (Figure 1D).
Specific MRI sequences
T1 images. T1 sequences, or so-called anatomy sequences, are ideally suited for detailed neuroanatomic evaluations. They are generated in such a way that structures containing fluid are dark (hypo-intense), whereas other structures, with higher fat or protein content, are brighter (iso-intense, even hyper-intense). For this reason, CSF in the intracranial ventricles is dark, and white matter is brighter than the cortex because of lipid in myelin sheaths.
In addition, to view structural abnormalities that are characterized by altered vascular supply or flow, such as tumors and infections (abscesses), contrast imaging can be particularly helpful; such images generally are obtained as T1 sequences.
T2 images. By contrast to the T1-weighted sequence, the T2-weighted sequences emphasize fluid signal; structures such as the ventricles, which contain CSF, therefore will be bright (hyper-intense). Pathology that produces edema or fluid, such as edema surrounding demyelinating lesions or infections, also will show bright hyper-intense signal. In T2-weighted images of the brain, white matter shows lower signal intensity than the cortex because of the relatively lower water content in white matter tracts and myelin sheaths.
Fluid attenuation inversion recovery. FLAIR images are generated so that the baseline bright T2 signal seen in normal structures, such as the CSF, containing ventricles is cancelled out, or attenuated. In effect, this subtraction of typical background hyper-intense fluid signal leaves only abnormal T2 bright hyper-intense signal, such as vasogenic edema surrounding tumors, cytotoxic edema within an infarction, or extra-axial fluid collections such as a subarachnoid or subdural hemorrhage.
Diffusion-weighted imaging. DWI utilizes the random motion (ie, diffusion) of water molecules to generate contrast. In this regard, the diffusion of any molecule is influenced by its interaction with other molecules (eg, white-matter fibers and membranes, and macromolecules). Diffusion patterns therefore reflect details about tissue boundaries; as such, DWI is sensitive to a number of neurologic processes, such as ischemia, demyelinating disease, and some tumors, which restrict the free motion of water. DWI detects this so-called restricted diffusion and displays an area of bright signal.
Susceptibility-weighted imaging (SWI). In the pediatric population, SWI (Figure 2) utilizes a long-echo, 3-dimensional, velocity-compensated gradient recalled echo for image acquisition2 and, ultimately, leverages susceptibility differences across tissues by employing the phase image to identify these differences. SWI, which uses both magnitude and phase images and is remarkably sensitive to venous blood (and blood products), iron, and calcifications, therefore might be of increasing utility in pediatric patients with traumatic brain injury (TBI) (Figure 2B). As such, SWI has become a critical component of many pediatric MRI studies.3
Magnetic resonance angiography (MRA) (Figure 3A) is helpful for assessing intracranial arteries and may be employed in the evaluation of:
- vessel pathology and injury underlying stroke, such as vessel occlusion or injury
- patterns of vessel involvement suggestive of vasculitis
- developmental or acquired structural vascular abnormalities, such as aneurysm or vascular malformations
- determination of tumor blood supply.
MRA can be performed without or with contrast, although MRA with contrast might provide a higher quality study and therefore be of greater utility. Of note: The spatial resolution of MRA is not as good as CT angiography; abnormalities, such as a small aneurysm, might not be apparent.
Magnetic resonance venography (MRV) (Figure 3B) is most commonly performed when the possibility of thrombosis of the dural venous sinuses is being considered; it also is employed to evaluate vascular malformations, tumor drainage patterns, and other pathologic states. As with MRA, MRV can be performed without or with contrast, although post-contrast MRV is generally of higher quality and might be preferred when assessing for sinus thrombosis.
Magnetic resonance spectroscopy (MRS) resides at the border between research and clinical practice. In children and adolescents, MRS provides data on neuronal and axonal viability as well as energetics and cell membranes.4 Pediatric neurologists often use MRS to evaluate for congenital neurometabolic disease; this modality also can help distinguish between an active intracranial tumor from an abscess or gliosis.5
Neuroimaging in pediatric neuropsychiatric conditions: Evidence, guidance
Delirium (altered mental status). The acute neuropsychiatric syndrome characterized by impaired attention and sensorium might have a broad underlying etiology, but it is always associated with alteration of CNS neurophysiology. Children with neurostructural abnormalities might have increased vulnerability to CNS insult and therefore be at increased risk of delirium.6 Additionally, delirium can present subtly in children, with the precise signs dependent on the individual patient’s developmental stage.
Neuroimaging may be helpful when an infectious, inflammatory, toxic, or a metabolic basis for delirium is suspected, or when a patient has new focal neurologic findings. In this regard, focal neurologic findings suggest an underlying localizable lesion and warrant dedicated neuroimaging to localize the lesion.
In general, the differential diagnosis should guide consideration of neuroimaging. When considering the possibility of an unwitnessed seizure in a child who presents with altered mental status, neuroimaging certainly is an important component of the workup.
As another example, when underlying trauma, intracranial hemorrhage, or mass is a possibility in acute delirium, urgent head CT is appropriate. In non-emergent cases, MRI is the modality of choice. In immunocompromised patients presenting with delirium, maintain a low threshold for neuroimaging with contrast to rule out opportunistic intracranial infection.
Last, in children who have hydrocephalus with a shunt, delirium could be a harbinger of underlying shunt malfunction, warranting a “shunt series.”
ADHD. The diagnosis of ADHD remains a clinical one; for the typical pediatric patient with ADHD but who does not have focal neurologic deficits, neuroimaging is unnecessary. Some structural MRI studies of youth with ADHD suggest diminished volume of the globus pallidus, putamen, and caudate7; other studies reveal changes in gyrification and cortical thickness8 in regions subserving attentional processes. However, intra-individual and developmental-related variability preclude routine use of neuroimaging in the standard diagnostic work-up of ADHD.
Nevertheless, neuroimaging should be strongly considered in a child with progressive worsening of inattention, especially if combined with other psychiatric or neurologic findings. In such a case, MRI should be obtained to evaluate for a progressive neurodegenerative leukoencephalopathy (eg, adrenoleukodystrophy).9
Depressive and anxiety disorders. In pediatric patients who exhibit depressive or anxiety symptoms, abnormalities have been observed in cortical thickness10 and gray matter volume,11,12 and functional signatures13 have been identified in the circuitry of the prefrontal amygdala. No data suggest that, in an individual patient, neuroimaging can be of diagnostic utility—particularly in the absence of focal neurologic findings.
That being said, headache and other somatic symptoms are common in pediatric patients with a mood or anxiety disorder. Evidence for neuroimaging in the context of pediatric headache suggests that MRI should be considered when headache is associated with neurologic signs or symptoms, such as aura, or accompanied by focal neurologic deficit.14
Pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections (PANDAS) and pediatric acute-onset neuropsychiatric syndrome (PANS). Neuroimaging studies of patients with confirmed PANDAS or PANS are rare, but group analyses suggest a decreased average volume of the caudate, putamen, and globus pallidus in patients with PANDAS compared with healthy comparison subjects, although total cerebral volume does not appear to differ.15 Moreover, thalamic findings in patients with PANDAS have been noted to be similar to what is seen in patients with Sydenham’s chorea.
The most recent consensus statement regarding the treatment and assessment of PANDAS and PANS recommends ordering brain MRI when other conditions are suspected (eg, CNS, small vessel vasculitis, limbic encephalitis) or when the patient has severe headache, gait disturbance, cognitive deterioration, or psychosis.16 Furthermore, the consensus statement notes the potential utility of T2-weighted imaging with contrast to evaluate inflammatory changes in the basal ganglia.16
Autism spectrum disorder. Significant progress has been made during the past decade on the neuroanatomic characterization of ASD. Accumulating data indicate that, in pediatric patients with ASD, (1) development of white matter and gray matter is disrupted early in the course of the disorder and (2) cortical thickness is increased in regions subserving social cognition.17,18
Several studies have examined the presence of patient-level findings in samples of pediatric patients with ASD. Approximately 8% of pediatric patients with ASD were found to have some abnormality on routine brain MRI, the most common being white-matter signal abnormalities, dilated Virchow-Robin space, and temporal lobe abnormalities.19
Although abnormalities might be present in a large percentage of individual scans, routine screening MRI is unlikely to be of clinical utility in youth with ASD. In fact, no recommendation for routine MRI screening in patients with ASD has been made by the American Academy of Child & Adolescent Psychiatry, the Child Neurology Society of the American Academy of Neurology, or the American Academy of Pediatrics.
However, in patients with an underlying neurostructural disease that is phenotypically associated with ASD-like symptoms, imaging might be of use. In tuberous sclerosis, for example, MRI is especially important to classify intracranial lesions; determine burden and location; or identify treatment options (Figure 4). For patients with tuberous sclerosis—of whom more than one-third meet diagnostic criteria for ASD20—MRI study should include FLAIR, spin-echo, and gradient-echo sequences.
Movement disorders. Hyperkinetic movement disorders, including tic disorders and drug-induced movement disorders (eg, tremor) are common in pediatric patients. In pediatric patients with a tic disorder or Tourette’s disorder (TD), neuroimaging typically is unnecessary, despite the suggestion that the caudate nucleus volume is reduced in groups of patients with TD.
Many CNS-acting medications can exacerbate physiologic tremor; in pediatric patients with symptoms of a movement disorder, home medications should be carefully reviewed for potentially offending agents. When the patient is clinically and biochemically euthyroid and medication-induced movement disorder has been ruled out, or when the patient meets clinical diagnostic criteria for TD or a tic disorder, routine neuroimaging generally is unnecessary.
When tremor accompanies other cerebellar signs, such as ataxia or dysmetria, strongly consider MRI of the brain to evaluate pathology in the posterior fossa. In addition, neuroimaging should be considered for children with a new-onset abnormality on neurologic exam, including rapid onset of abnormal movements (other than common tics), continuous progressive worsening of symptoms, or any loss of developmental milestones.
Last, although tics and stereotypies often are transient and wane with age, other abnormal movements, such as dystonia, chorea, and parkinsonism (aside from those potentially associated with antipsychotic use), are never expected during typical development and warrant MRI.
Traumatic brain injury. Prompt evaluation and intervention for TBI can significantly affect overall outcome. Moreover, there has been increased enthusiasm around the pre-hospital assessment of TBI severity using (1) any of several proprietary testing systems (eg, Immediate Post-Concussion Assessment and Cognitive Testing [ImPACT]) plus (2) standard clinical staging, which is based on duration of loss of consciousness, persistence of memory loss, and the Glasgow Coma Scale score.
The goal for any TBI patient during the acute post-injury phase is to minimize continued neuronal injury from secondary effects of TBI, such as cerebral edema and herniation, and to optimize protection of surviving brain tissue; neuroimaging is a critical component of assessment during both acute and chronic recovery periods of TBI.21
The optimal imaging modality varies with the amount of time that has passed since initial injury.22 Urgent neuroimaging (the first 24 hours after brain injury) is typically obtained using head CT to assist decision-making in acute neurosurgical management. In this setting, head CT is fast and efficient; minimizes the amount of time that the patient is in the scanner; and provides valuable information on the acuity and extent of injury, degree of cerebral edema, and evidence or risk of pending herniation.
On the other hand, MRI is superior to CT during 48 to 72 hours after injury, given its higher resolution; superior imaging of the brainstem and deep gray nuclei; and ability to detect axonal injury, small contusions, and subtle neuronal damage. Specifically, SWI sequences can be particularly helpful in TBI for detecting diffuse axonal injury and micro-hemorrhages; several recent studies also suggest that SWI may be of particular value in pediatric patients with TBI.23
Additionally, given the increased sensitivity of MRI to detect subtle injuries, this modality can assist in identifying chronic sequelae of brain injury—thus contributing to determining of chronic therapy options and assisting with long-term prognosis. Gross structural changes resulting from TBI often are evident even in the acute post-injury phase; synaptic remodeling continues, however, for an indefinite period after injury, and this remodeling capacity is even more pronounced in the highly plastic brain of a young child.
Microstructural changes might not be detectable using traditional, readily available imaging sequences (CT, MRI). When those traditional modalities are used in concert with functional imaging techniques (eg, PET to evaluate cerebral metabolism and SPECT imaging which can detect abnormalities in cerebral blood flow), the combination of older and newer might provide a more complete picture of recovery after TBI.24
The important role of neuroimaging in severe TBI is intuitive. However, it is important to consider the role of neuroimaging in mild TBI in children, especially in the setting of repetitive mild injury.25 A growing body of evidence supports close, serial monitoring of children after even mild closed head injury for neurologic and psychiatric sequelae. Although it is rare that a child who is awake, interactive, and lacking focal neurologic deficits would need emergent (ie, CT) imaging after mild closed head injury, there might be a role for MRI later in the course of that child’s recovery—especially if recovery is complicated by clinical sequelae of mild TBI, such as cognitive impairment, headaches, or altered behavior.
When is additional neuroimaging needed?
It’s worthwhile briefly reviewing 4 scenarios that you might encounter, when you work with children and adolescents, in which urgent or emergent neuroimaging (often with consultation) should be obtained. The Table describes these situations and appropriate first- and second-line interventions.
1. When the presentation of your patient is consistent with an acute neurologic deficit, acute TBI, progressive neuropsychiatric decline, CNS infection, mass, demyelinating process, or toxic exposure, neuroimaging is likely critical.
2. In patients with progressive neurologic decline, including loss of developmental milestones, MRI, MRS, and referral to neurology should be part of the comprehensive evaluation.
3. In young children who exhibit a decrease in head circumference on growth curves, MRI is important to evaluate for underlying structural causes.
4. Pediatric patients with symptoms consistent with either stroke (ie, a new, persistent neurologic deficit) or a demyelinating process (eg, multiple episodes of variable transient focal neurologic symptoms), MRI should be obtained without compunction.
Consultation with pediatric neuroradiology
In deciding whether to obtain neuroimaging for a particular case, you should discuss your concerns with the pediatric radiologist or pediatric neuroradiologist, who will likely provide important guidance on key aspects of the study (eg, modifying slice thickness in a particular scan; recommending the use of contrast; including MRS in the order for imaging; performing appropriate vessel imaging). Consider asking 1 or more important questions when you discuss a patient’s presentation with the pediatric radiologist or pediatric neuroradiologist:
- “What neuroimaging studies are appropriate, based on my differential diagnosis?”
- “Are there specific imaging sequences that we should consider?”
- “Are there contraindications to the imaging modality for my patient?”
- “Is my patient likely to have difficulty tolerating the imaging procedure?”
- “Does my patient need sedation to tolerate this procedure?”
- “Should additional regions be included in the scan?” (Examples: In a child with stroke it might be important to include neck and chest vasculature and the heart. Other conditions might warrant imaging of the spinal cord.)
The first 15 years of the new millennium have seen a great increase in research on neuroimaging in children and adolescents who have a psychiatric disorder. In addition, imaging modalities continue to evolve, and are becoming increasingly accessible and informative. The literature is now replete with reports of neurostructural differences between patients and healthy subjects in a variety of common pediatric psychiatric conditions, including anxiety disorders, mood disorders, autism spectrum disorder (ASD), and attention-deficit/hyperactivity disorder (ADHD).
Historically, the clinical utility of neuroimaging was restricted to the identification of structural pathology. Today, accumulating data reveal novel roles for neuroimaging; these revelations are supported by studies demonstrating that treatment response for psychotherapeutic and psychopharmacotherapeutic interventions can be predicted by neurochemical and neurofunctional characteristics assessed by advanced imaging technologies, such as magnetic resonance spectroscopy (MRS) and functional MRI.
However, such advanced techniques are (at least at present) not ready for routine clinical use for this purpose. Instead, neuroimaging in the child and adolescent psychiatric clinic remains largely focused on ruling out neurostructural, neurologic, “nonpsychiatric” causes of our patients’ symptoms.
Understanding the role and limitations of major imaging modalities is key to guiding efficient and appropriate neuroimaging selection for pediatric patients. In this article, we describe and review:
- neuroimaging approaches for children and adolescents with psychiatric disorders
- the role of neuroimaging in (1) the differential diagnosis and workup of common psychiatric disorders and (2) urgent clinical situations
- how to determine what type of imaging to obtain.
Computed tomography
CT, which utilizes ionizing radiation, often is reserved, in the pediatric setting, for (1) emergency evaluation and (2) excluding potentially catastrophic neurologic injury resulting from:
- ischemic or hemorrhagic stroke
- herniation
- intracerebral hemorrhage
- subdural and epidural hematoma
- large intracranial mass with mass effect
- increased intracranial pressure
- acute skull fracture.
Although a CT scan is, typically, quick and has excellent sensitivity for acute bleeding and bony pathology, it exposes the patient to radiation and provides poor resolution compared with MRI.
In pediatrics, there has been practice-changing recognition of the importance of limiting lifetime radiation exposure incurred from medical procedures and imaging. As a result, most providers now agree that use of MRI in lieu of CT is appropriate in many, if not most, non-emergent situations. In an emergent situation, however, CT imaging is appropriate and should not be delayed. Moreover, in an emergent situation, you should not hesitate to use head CT in children, although timely discussion with the radiologist is recommended to review your differential diagnosis to better determine the preferred imaging modality.
Magnetic resonance imaging
Over the past several decades, MRI has been increasingly available in most pediatric health care facilities. The modality offers specific advantages for pediatric patients, including:
- better spatial resolution
- the ability to concurrently assess multiple pathologic processes
- lack of exposure to ionizing radiation.1
A number of MRI sequences, described below, can be used to assess vascular, inflammatory, structural, and metabolic processes.
A look inside. Comprehensive review of the physics that underlies MRI is beyond the scope of this article; several important principles are relevant to clinicians, however. Image contrast is dependent on intrinsic properties of tissue with regard to proton density, longitudinal relaxation time (T1), and transverse relaxation time (T2). Pulse sequences, which describe the strength and timing of the radiofrequency pulse and gradient pulses, define imaging acquisition parameters (eg, repetition time between the radio frequency pulse and echo time).
In turn, the intensity of the signal that is “seen” with various pulse sequences is differentially affected by intrinsic properties of tissue. At most pediatric institutions, the standard MRI-examination protocol includes: a T1-weighted image (Figure 1A); a T2-weighted scan (Figure 1B); fluid attenuated inversion recovery (FLAIR) (Figure 1C); and diffusion-weighted imaging (DWI) (Figure 1D).
Specific MRI sequences
T1 images. T1 sequences, or so-called anatomy sequences, are ideally suited for detailed neuroanatomic evaluations. They are generated in such a way that structures containing fluid are dark (hypo-intense), whereas other structures, with higher fat or protein content, are brighter (iso-intense, even hyper-intense). For this reason, CSF in the intracranial ventricles is dark, and white matter is brighter than the cortex because of lipid in myelin sheaths.
In addition, to view structural abnormalities that are characterized by altered vascular supply or flow, such as tumors and infections (abscesses), contrast imaging can be particularly helpful; such images generally are obtained as T1 sequences.
T2 images. By contrast to the T1-weighted sequence, the T2-weighted sequences emphasize fluid signal; structures such as the ventricles, which contain CSF, therefore will be bright (hyper-intense). Pathology that produces edema or fluid, such as edema surrounding demyelinating lesions or infections, also will show bright hyper-intense signal. In T2-weighted images of the brain, white matter shows lower signal intensity than the cortex because of the relatively lower water content in white matter tracts and myelin sheaths.
Fluid attenuation inversion recovery. FLAIR images are generated so that the baseline bright T2 signal seen in normal structures, such as the CSF, containing ventricles is cancelled out, or attenuated. In effect, this subtraction of typical background hyper-intense fluid signal leaves only abnormal T2 bright hyper-intense signal, such as vasogenic edema surrounding tumors, cytotoxic edema within an infarction, or extra-axial fluid collections such as a subarachnoid or subdural hemorrhage.
Diffusion-weighted imaging. DWI utilizes the random motion (ie, diffusion) of water molecules to generate contrast. In this regard, the diffusion of any molecule is influenced by its interaction with other molecules (eg, white-matter fibers and membranes, and macromolecules). Diffusion patterns therefore reflect details about tissue boundaries; as such, DWI is sensitive to a number of neurologic processes, such as ischemia, demyelinating disease, and some tumors, which restrict the free motion of water. DWI detects this so-called restricted diffusion and displays an area of bright signal.
Susceptibility-weighted imaging (SWI). In the pediatric population, SWI (Figure 2) utilizes a long-echo, 3-dimensional, velocity-compensated gradient recalled echo for image acquisition2 and, ultimately, leverages susceptibility differences across tissues by employing the phase image to identify these differences. SWI, which uses both magnitude and phase images and is remarkably sensitive to venous blood (and blood products), iron, and calcifications, therefore might be of increasing utility in pediatric patients with traumatic brain injury (TBI) (Figure 2B). As such, SWI has become a critical component of many pediatric MRI studies.3
Magnetic resonance angiography (MRA) (Figure 3A) is helpful for assessing intracranial arteries and may be employed in the evaluation of:
- vessel pathology and injury underlying stroke, such as vessel occlusion or injury
- patterns of vessel involvement suggestive of vasculitis
- developmental or acquired structural vascular abnormalities, such as aneurysm or vascular malformations
- determination of tumor blood supply.
MRA can be performed without or with contrast, although MRA with contrast might provide a higher quality study and therefore be of greater utility. Of note: The spatial resolution of MRA is not as good as CT angiography; abnormalities, such as a small aneurysm, might not be apparent.
Magnetic resonance venography (MRV) (Figure 3B) is most commonly performed when the possibility of thrombosis of the dural venous sinuses is being considered; it also is employed to evaluate vascular malformations, tumor drainage patterns, and other pathologic states. As with MRA, MRV can be performed without or with contrast, although post-contrast MRV is generally of higher quality and might be preferred when assessing for sinus thrombosis.
Magnetic resonance spectroscopy (MRS) resides at the border between research and clinical practice. In children and adolescents, MRS provides data on neuronal and axonal viability as well as energetics and cell membranes.4 Pediatric neurologists often use MRS to evaluate for congenital neurometabolic disease; this modality also can help distinguish between an active intracranial tumor from an abscess or gliosis.5
Neuroimaging in pediatric neuropsychiatric conditions: Evidence, guidance
Delirium (altered mental status). The acute neuropsychiatric syndrome characterized by impaired attention and sensorium might have a broad underlying etiology, but it is always associated with alteration of CNS neurophysiology. Children with neurostructural abnormalities might have increased vulnerability to CNS insult and therefore be at increased risk of delirium.6 Additionally, delirium can present subtly in children, with the precise signs dependent on the individual patient’s developmental stage.
Neuroimaging may be helpful when an infectious, inflammatory, toxic, or a metabolic basis for delirium is suspected, or when a patient has new focal neurologic findings. In this regard, focal neurologic findings suggest an underlying localizable lesion and warrant dedicated neuroimaging to localize the lesion.
In general, the differential diagnosis should guide consideration of neuroimaging. When considering the possibility of an unwitnessed seizure in a child who presents with altered mental status, neuroimaging certainly is an important component of the workup.
As another example, when underlying trauma, intracranial hemorrhage, or mass is a possibility in acute delirium, urgent head CT is appropriate. In non-emergent cases, MRI is the modality of choice. In immunocompromised patients presenting with delirium, maintain a low threshold for neuroimaging with contrast to rule out opportunistic intracranial infection.
Last, in children who have hydrocephalus with a shunt, delirium could be a harbinger of underlying shunt malfunction, warranting a “shunt series.”
ADHD. The diagnosis of ADHD remains a clinical one; for the typical pediatric patient with ADHD but who does not have focal neurologic deficits, neuroimaging is unnecessary. Some structural MRI studies of youth with ADHD suggest diminished volume of the globus pallidus, putamen, and caudate7; other studies reveal changes in gyrification and cortical thickness8 in regions subserving attentional processes. However, intra-individual and developmental-related variability preclude routine use of neuroimaging in the standard diagnostic work-up of ADHD.
Nevertheless, neuroimaging should be strongly considered in a child with progressive worsening of inattention, especially if combined with other psychiatric or neurologic findings. In such a case, MRI should be obtained to evaluate for a progressive neurodegenerative leukoencephalopathy (eg, adrenoleukodystrophy).9
Depressive and anxiety disorders. In pediatric patients who exhibit depressive or anxiety symptoms, abnormalities have been observed in cortical thickness10 and gray matter volume,11,12 and functional signatures13 have been identified in the circuitry of the prefrontal amygdala. No data suggest that, in an individual patient, neuroimaging can be of diagnostic utility—particularly in the absence of focal neurologic findings.
That being said, headache and other somatic symptoms are common in pediatric patients with a mood or anxiety disorder. Evidence for neuroimaging in the context of pediatric headache suggests that MRI should be considered when headache is associated with neurologic signs or symptoms, such as aura, or accompanied by focal neurologic deficit.14
Pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections (PANDAS) and pediatric acute-onset neuropsychiatric syndrome (PANS). Neuroimaging studies of patients with confirmed PANDAS or PANS are rare, but group analyses suggest a decreased average volume of the caudate, putamen, and globus pallidus in patients with PANDAS compared with healthy comparison subjects, although total cerebral volume does not appear to differ.15 Moreover, thalamic findings in patients with PANDAS have been noted to be similar to what is seen in patients with Sydenham’s chorea.
The most recent consensus statement regarding the treatment and assessment of PANDAS and PANS recommends ordering brain MRI when other conditions are suspected (eg, CNS, small vessel vasculitis, limbic encephalitis) or when the patient has severe headache, gait disturbance, cognitive deterioration, or psychosis.16 Furthermore, the consensus statement notes the potential utility of T2-weighted imaging with contrast to evaluate inflammatory changes in the basal ganglia.16
Autism spectrum disorder. Significant progress has been made during the past decade on the neuroanatomic characterization of ASD. Accumulating data indicate that, in pediatric patients with ASD, (1) development of white matter and gray matter is disrupted early in the course of the disorder and (2) cortical thickness is increased in regions subserving social cognition.17,18
Several studies have examined the presence of patient-level findings in samples of pediatric patients with ASD. Approximately 8% of pediatric patients with ASD were found to have some abnormality on routine brain MRI, the most common being white-matter signal abnormalities, dilated Virchow-Robin space, and temporal lobe abnormalities.19
Although abnormalities might be present in a large percentage of individual scans, routine screening MRI is unlikely to be of clinical utility in youth with ASD. In fact, no recommendation for routine MRI screening in patients with ASD has been made by the American Academy of Child & Adolescent Psychiatry, the Child Neurology Society of the American Academy of Neurology, or the American Academy of Pediatrics.
However, in patients with an underlying neurostructural disease that is phenotypically associated with ASD-like symptoms, imaging might be of use. In tuberous sclerosis, for example, MRI is especially important to classify intracranial lesions; determine burden and location; or identify treatment options (Figure 4). For patients with tuberous sclerosis—of whom more than one-third meet diagnostic criteria for ASD20—MRI study should include FLAIR, spin-echo, and gradient-echo sequences.
Movement disorders. Hyperkinetic movement disorders, including tic disorders and drug-induced movement disorders (eg, tremor) are common in pediatric patients. In pediatric patients with a tic disorder or Tourette’s disorder (TD), neuroimaging typically is unnecessary, despite the suggestion that the caudate nucleus volume is reduced in groups of patients with TD.
Many CNS-acting medications can exacerbate physiologic tremor; in pediatric patients with symptoms of a movement disorder, home medications should be carefully reviewed for potentially offending agents. When the patient is clinically and biochemically euthyroid and medication-induced movement disorder has been ruled out, or when the patient meets clinical diagnostic criteria for TD or a tic disorder, routine neuroimaging generally is unnecessary.
When tremor accompanies other cerebellar signs, such as ataxia or dysmetria, strongly consider MRI of the brain to evaluate pathology in the posterior fossa. In addition, neuroimaging should be considered for children with a new-onset abnormality on neurologic exam, including rapid onset of abnormal movements (other than common tics), continuous progressive worsening of symptoms, or any loss of developmental milestones.
Last, although tics and stereotypies often are transient and wane with age, other abnormal movements, such as dystonia, chorea, and parkinsonism (aside from those potentially associated with antipsychotic use), are never expected during typical development and warrant MRI.
Traumatic brain injury. Prompt evaluation and intervention for TBI can significantly affect overall outcome. Moreover, there has been increased enthusiasm around the pre-hospital assessment of TBI severity using (1) any of several proprietary testing systems (eg, Immediate Post-Concussion Assessment and Cognitive Testing [ImPACT]) plus (2) standard clinical staging, which is based on duration of loss of consciousness, persistence of memory loss, and the Glasgow Coma Scale score.
The goal for any TBI patient during the acute post-injury phase is to minimize continued neuronal injury from secondary effects of TBI, such as cerebral edema and herniation, and to optimize protection of surviving brain tissue; neuroimaging is a critical component of assessment during both acute and chronic recovery periods of TBI.21
The optimal imaging modality varies with the amount of time that has passed since initial injury.22 Urgent neuroimaging (the first 24 hours after brain injury) is typically obtained using head CT to assist decision-making in acute neurosurgical management. In this setting, head CT is fast and efficient; minimizes the amount of time that the patient is in the scanner; and provides valuable information on the acuity and extent of injury, degree of cerebral edema, and evidence or risk of pending herniation.
On the other hand, MRI is superior to CT during 48 to 72 hours after injury, given its higher resolution; superior imaging of the brainstem and deep gray nuclei; and ability to detect axonal injury, small contusions, and subtle neuronal damage. Specifically, SWI sequences can be particularly helpful in TBI for detecting diffuse axonal injury and micro-hemorrhages; several recent studies also suggest that SWI may be of particular value in pediatric patients with TBI.23
Additionally, given the increased sensitivity of MRI to detect subtle injuries, this modality can assist in identifying chronic sequelae of brain injury—thus contributing to determining of chronic therapy options and assisting with long-term prognosis. Gross structural changes resulting from TBI often are evident even in the acute post-injury phase; synaptic remodeling continues, however, for an indefinite period after injury, and this remodeling capacity is even more pronounced in the highly plastic brain of a young child.
Microstructural changes might not be detectable using traditional, readily available imaging sequences (CT, MRI). When those traditional modalities are used in concert with functional imaging techniques (eg, PET to evaluate cerebral metabolism and SPECT imaging which can detect abnormalities in cerebral blood flow), the combination of older and newer might provide a more complete picture of recovery after TBI.24
The important role of neuroimaging in severe TBI is intuitive. However, it is important to consider the role of neuroimaging in mild TBI in children, especially in the setting of repetitive mild injury.25 A growing body of evidence supports close, serial monitoring of children after even mild closed head injury for neurologic and psychiatric sequelae. Although it is rare that a child who is awake, interactive, and lacking focal neurologic deficits would need emergent (ie, CT) imaging after mild closed head injury, there might be a role for MRI later in the course of that child’s recovery—especially if recovery is complicated by clinical sequelae of mild TBI, such as cognitive impairment, headaches, or altered behavior.
When is additional neuroimaging needed?
It’s worthwhile briefly reviewing 4 scenarios that you might encounter, when you work with children and adolescents, in which urgent or emergent neuroimaging (often with consultation) should be obtained. The Table describes these situations and appropriate first- and second-line interventions.
1. When the presentation of your patient is consistent with an acute neurologic deficit, acute TBI, progressive neuropsychiatric decline, CNS infection, mass, demyelinating process, or toxic exposure, neuroimaging is likely critical.
2. In patients with progressive neurologic decline, including loss of developmental milestones, MRI, MRS, and referral to neurology should be part of the comprehensive evaluation.
3. In young children who exhibit a decrease in head circumference on growth curves, MRI is important to evaluate for underlying structural causes.
4. Pediatric patients with symptoms consistent with either stroke (ie, a new, persistent neurologic deficit) or a demyelinating process (eg, multiple episodes of variable transient focal neurologic symptoms), MRI should be obtained without compunction.
Consultation with pediatric neuroradiology
In deciding whether to obtain neuroimaging for a particular case, you should discuss your concerns with the pediatric radiologist or pediatric neuroradiologist, who will likely provide important guidance on key aspects of the study (eg, modifying slice thickness in a particular scan; recommending the use of contrast; including MRS in the order for imaging; performing appropriate vessel imaging). Consider asking 1 or more important questions when you discuss a patient’s presentation with the pediatric radiologist or pediatric neuroradiologist:
- “What neuroimaging studies are appropriate, based on my differential diagnosis?”
- “Are there specific imaging sequences that we should consider?”
- “Are there contraindications to the imaging modality for my patient?”
- “Is my patient likely to have difficulty tolerating the imaging procedure?”
- “Does my patient need sedation to tolerate this procedure?”
- “Should additional regions be included in the scan?” (Examples: In a child with stroke it might be important to include neck and chest vasculature and the heart. Other conditions might warrant imaging of the spinal cord.)
1. Abdelhalim AN, Alberico RA. Pediatric neuroimaging. Neurol Clin. 2009;27(1):285-301, x.
2. Sehgal V, Delproposto Z, Haacke EM, et al. Clinical applications of neuroimaging with susceptibility-weighted imaging. J Magn Reson Imaging. 2005;22(4):439-450.
3. Bosemani T, Poretti A, Huisman TA. Susceptibility-weighted imaging in pediatric neuroimaging. J Magn Reson Imaging. 2014;40(3):530-544.
4. Cecil KM. Proton magnetic resonance spectroscopy: technique for the neuroradiologist. Neuroimaging Clin N Am. 2013;23(3):381-392.
5. Panigrahy A, Nelson MD Jr, Blüml S. Magnetic resonance spectroscopy in pediatric neuroradiology: clinical and research applications. Pediatr Radiol. 2010;40(1):3-30.
6. Leentjens AF, Schieveld JN, Leonard M, et al. A comparison of the phenomenology of pediatric, adult, and geriatric delirium. J Psychosom Res. 2008;64(2):219-223.
7. Frodl T, Skokauskas N. Meta-analysis of structural MRI studies in children and adults with attention deficit hyperactivity disorder indicates treatment effects. Acta Psychiatr Scand. 2012;125(2):114-126.
8. Shaw P, Malek M, Watson B, et al. Development of cortical surface area and gyrification in attention-deficit/hyperactivity disorder. Biol Psychiatry. 2012;72(3):191-197.
9. Phelan JA, Lowe LH, Glasier CM. Pediatric neurodegenerative white matter processes: leukodystrophies and beyond. Pediatr Radiol. 2008;38(7):729-749.
10. Strawn JR, Wegman CJ, Dominick KC, et al. Cortical surface anatomy in pediatric patients with generalized anxiety disorder. J Anxiety Disord. 2014;28(7):717-723.
11. Mueller SC, Aouidad A, Gorodetsky E, et al. Gray matter volume in adolescent anxiety: an impact of the brain-derived neurotrophic factor Val(66)Met polymorphism [Erratum in J Am Acad Child Adolesc Psychiatry. 2013;52(2):184-195]? J Am Acad Child Adolesc Psychiatry. 2013;52(2):184-195.
12. Strawn JR, Hamm L, Fitzgerald DA, et al. Neurostructural abnormalities in pediatric anxiety disorders. J Anxiety Disord. 2015;32:81-88.
13. Strawn JR, Dominick KC, Patino LR, et al. Neurobiology of pediatric anxiety disorders. Curr Behav Neurosci Reports. 2014;1(3):154-160.
14. Alexiou GA, Argyropoulou MI. Neuroimaging in childhood headache: a systematic review. Pediatr Radiol. 2013;43(7):777-784.
15. Giedd JN, Rapoport JL, Garvey MA, et al. MRI assessment of children with obsessive-compulsive disorder or tics associated with streptococcal infection. Am J Psychiatry. 2000;157(2):281-283.
16. Chang K, Frankovich J, Cooperstock M, et al; PANS Collaborative Consortium. Clinical evaluation of youth with pediatric acute-onset neuropsychiatric syndrome (PANS): recommendations from the 2013 PANS Consensus Conference. J Child Adolesc Psychopharmacol. 2014;25(1):3-13.
17. Wallace GL, Robustelli B, Dankner N, et al. Increased gyrification, but comparable surface area in adolescents with autism spectrum disorders. Brain. 2013;136(pt 6):1956-1967.
18. Libero LE, DeRamus TP, Deshpande HD, et al. Surface-based morphometry of the cortical architecture of autism spectrum disorders: volume, thickness, area, and gyrification. Neuropsychologia. 2014;62:1-10.
19. Boddaert N, Zilbovicius M, Philipe A, et al. MRI findings in 77 children with non-syndromic autistic disorder. PLoS One. 2009;4:e445. doi: 10.1371/journal.pone.0004415.
20. Richards C, Jones C, Groves L, et al. Prevalence of autism spectrum disorder phenomenology in genetic disorders: a systematic review and meta-analysis. Lancet Psychiatry. 2015;2(10):909-916.
21. Wilde EA, Hunter JV, Bigler ED. Pediatric traumatic brain injury: neuroimaging and neurorehabilitation outcome. NeuroRehabilitation. 2012;31(3):245-260.
22. Mechtler LL, Shastri KK, Crutchfield KE. Advanced neuroimaging of mild traumatic brain injury. Neurol Clin. 2014;32(1):31-58.
23. Ashwal S, Tong KA, Ghosh N, et al. Application of advanced neuroimaging modalities in pediatric traumatic brain injury. J Child Neurol. 2014;29(12):1704-1717.
24. Munson S, Schroth E, Ernst M. The role of functional neuroimaging in pediatric brain injury. Pediatrics. 2006;117(4):1372-1381.
25. Wozniak JR, Krach L, Ward E, et al. Neurocognitive and neuroimaging correlates of pediatric traumatic brain injury: a diffusion tensor imaging (DTI) study. Arch Clin Neuropsychol. 2007;22(5):555-568.
1. Abdelhalim AN, Alberico RA. Pediatric neuroimaging. Neurol Clin. 2009;27(1):285-301, x.
2. Sehgal V, Delproposto Z, Haacke EM, et al. Clinical applications of neuroimaging with susceptibility-weighted imaging. J Magn Reson Imaging. 2005;22(4):439-450.
3. Bosemani T, Poretti A, Huisman TA. Susceptibility-weighted imaging in pediatric neuroimaging. J Magn Reson Imaging. 2014;40(3):530-544.
4. Cecil KM. Proton magnetic resonance spectroscopy: technique for the neuroradiologist. Neuroimaging Clin N Am. 2013;23(3):381-392.
5. Panigrahy A, Nelson MD Jr, Blüml S. Magnetic resonance spectroscopy in pediatric neuroradiology: clinical and research applications. Pediatr Radiol. 2010;40(1):3-30.
6. Leentjens AF, Schieveld JN, Leonard M, et al. A comparison of the phenomenology of pediatric, adult, and geriatric delirium. J Psychosom Res. 2008;64(2):219-223.
7. Frodl T, Skokauskas N. Meta-analysis of structural MRI studies in children and adults with attention deficit hyperactivity disorder indicates treatment effects. Acta Psychiatr Scand. 2012;125(2):114-126.
8. Shaw P, Malek M, Watson B, et al. Development of cortical surface area and gyrification in attention-deficit/hyperactivity disorder. Biol Psychiatry. 2012;72(3):191-197.
9. Phelan JA, Lowe LH, Glasier CM. Pediatric neurodegenerative white matter processes: leukodystrophies and beyond. Pediatr Radiol. 2008;38(7):729-749.
10. Strawn JR, Wegman CJ, Dominick KC, et al. Cortical surface anatomy in pediatric patients with generalized anxiety disorder. J Anxiety Disord. 2014;28(7):717-723.
11. Mueller SC, Aouidad A, Gorodetsky E, et al. Gray matter volume in adolescent anxiety: an impact of the brain-derived neurotrophic factor Val(66)Met polymorphism [Erratum in J Am Acad Child Adolesc Psychiatry. 2013;52(2):184-195]? J Am Acad Child Adolesc Psychiatry. 2013;52(2):184-195.
12. Strawn JR, Hamm L, Fitzgerald DA, et al. Neurostructural abnormalities in pediatric anxiety disorders. J Anxiety Disord. 2015;32:81-88.
13. Strawn JR, Dominick KC, Patino LR, et al. Neurobiology of pediatric anxiety disorders. Curr Behav Neurosci Reports. 2014;1(3):154-160.
14. Alexiou GA, Argyropoulou MI. Neuroimaging in childhood headache: a systematic review. Pediatr Radiol. 2013;43(7):777-784.
15. Giedd JN, Rapoport JL, Garvey MA, et al. MRI assessment of children with obsessive-compulsive disorder or tics associated with streptococcal infection. Am J Psychiatry. 2000;157(2):281-283.
16. Chang K, Frankovich J, Cooperstock M, et al; PANS Collaborative Consortium. Clinical evaluation of youth with pediatric acute-onset neuropsychiatric syndrome (PANS): recommendations from the 2013 PANS Consensus Conference. J Child Adolesc Psychopharmacol. 2014;25(1):3-13.
17. Wallace GL, Robustelli B, Dankner N, et al. Increased gyrification, but comparable surface area in adolescents with autism spectrum disorders. Brain. 2013;136(pt 6):1956-1967.
18. Libero LE, DeRamus TP, Deshpande HD, et al. Surface-based morphometry of the cortical architecture of autism spectrum disorders: volume, thickness, area, and gyrification. Neuropsychologia. 2014;62:1-10.
19. Boddaert N, Zilbovicius M, Philipe A, et al. MRI findings in 77 children with non-syndromic autistic disorder. PLoS One. 2009;4:e445. doi: 10.1371/journal.pone.0004415.
20. Richards C, Jones C, Groves L, et al. Prevalence of autism spectrum disorder phenomenology in genetic disorders: a systematic review and meta-analysis. Lancet Psychiatry. 2015;2(10):909-916.
21. Wilde EA, Hunter JV, Bigler ED. Pediatric traumatic brain injury: neuroimaging and neurorehabilitation outcome. NeuroRehabilitation. 2012;31(3):245-260.
22. Mechtler LL, Shastri KK, Crutchfield KE. Advanced neuroimaging of mild traumatic brain injury. Neurol Clin. 2014;32(1):31-58.
23. Ashwal S, Tong KA, Ghosh N, et al. Application of advanced neuroimaging modalities in pediatric traumatic brain injury. J Child Neurol. 2014;29(12):1704-1717.
24. Munson S, Schroth E, Ernst M. The role of functional neuroimaging in pediatric brain injury. Pediatrics. 2006;117(4):1372-1381.
25. Wozniak JR, Krach L, Ward E, et al. Neurocognitive and neuroimaging correlates of pediatric traumatic brain injury: a diffusion tensor imaging (DTI) study. Arch Clin Neuropsychol. 2007;22(5):555-568.

Pseudobulbar affect: When patients laugh or cry, but don’t know why
Pseudobulbar affect (PBA) is a disorder of affective expression that manifests as stereotyped and frequent outbursts of crying (not limited to lacrimation) or laughter. Symptoms are involuntary, uncontrolled, and exaggerated or incongruent with current mood. Episodes, lasting a few seconds to several minutes, may be unprovoked or occur in response to a mild stimulus, and patients typically display a normal affect between episodes.1 PBA is estimated to affect 1 to 2 million people in the United States, although some studies suggest as many as 7 million,1,2 depending on the evaluation method and threshold criteria used.3

Where to look for pseudobulbar affect
PBA has been most commonly described in 6 major neurologic disorders:
- Alzheimer’s disease
- amyotrophic lateral sclerosis (ALS)
- multiple sclerosis (MS)
- Parkinson’s disease
- stroke
- traumatic brain injury (TBI).

Of these disorders, most studies have found the highest PBA prevalence in patients with ALS and TBI, with lesser (although significant) prevalence in Parkinson’s disease (Table 2).1,12 These “big 6” diagnoses are not a comprehensive list, as many other disease states are associated with PBA (Table 3).12-14


2 Pathways: ‘Generator’ and ‘governor’
Despite the many and varied injuries and illnesses associated with PBA, Lauterbach et al10 noted patterns that suggest dysregulation of 2 distinct but interconnected brain pathways: an emotional pathway controlled by a separate volitional pathway. Lesions to the volitional pathway (or its associated feedback or processing circuits) are thought to cause PBA symptoms.
To borrow an analogy from engineering, the emotional pathway is the “generator” of affect, whereas the volitional pathway is the “governor” of affect. Thus, injury to the “governor” results in overspill, or overflow, of affect that usually would be suppressed.
The emotional pathway, which coordinates the motor aspect of reflex laughing or crying, originates at the frontotemporal cortex, relaying to the amygdala and hypothalamus, then projecting to the dorsal brainstem, which includes the midbrain-pontine periaqueductal gray (PAG), dorsal tegmentum, and related brainstem.
The volitional pathway, which regulates the emotional pathway, originates in the dorsal and lateral frontoparietal cortex, projects through the internal capsule and midbrain basis pedunculi, and continues on to the anteroventral basis pontis. The basis pontis then serves as an afferent relay center for cerebellar activity. Projections from the pons then regulate the emotional circuitry primarily at the level of the PAG.10
Lesions of the volitional pathway have been correlated with conditions of PBA, whereas direct activation of the emotional pathway tended to lead to emotional lability or the crying and laughing behaviors observed in dacrystic or gelastic epilepsy.10 The pivotal nature of the regulation occurring at the PAG has guided treatment options. Neurotransmitter receptors most closely associated with this region include glutamatergic N-methyl-
When to screen for PBA
Ask the right question. PBA as a disease state likely has been widely under-reported, under-recognized, and misdiagnosed (typically, as a primary mood disorder).9 Three factors underscore this problem:
- Patients do not specifically report symptoms of affective disturbance (perhaps because they lack a vocabulary to separate concepts of mood and affect)
- Physicians do not ask patients about separations of mood and affect
- Perhaps most importantly, PBA lacks a general awareness and understanding.
Co-occurring mood disorders also may thwart PBA detection. One study of PBA in Alzheimer’s dementia found that 53% of patients with symptoms consistent with PBA also had a distinct mood disorder.17 This suggests that a PBA-specific screening test is needed for accurate diagnosis.
A single question might best refine the likelihood that a patient has PBA: “Do you ever cry for no reason?” In primary psychiatric illness, crying typically is associated with a specific trigger (eg, depressed mood, despair, anxiety). A patient’s inability to identify a trigger for crying suggests the pathological separation of mood and affect—the core of PBA, and worthy of further investigation.
Clinical rating scales that correlate to disease severity appear to be the most effective in identifying PBA. The PRISM study, to date the largest clinic-based study of PBA symptoms, used the Center for Neurologic Study-Liability Scale (CNS-LS) to gauge the presence and severity of PBA symptoms.1 A 7-question, patient self-administered tool, the CNS-LS is graded on a 5-point Likert scale. A score ≥13 has high sensitivity and specificity for diagnosis of PBA, compared with physician diagnosis.
Another option, the 16-question Pathological Laughing and Crying Scale, is a clinician-administered screening tool. Again, a score ≥13 is consistent with symptoms required for a PBA diagnosis.
Treating PBA symptoms
Until recently, most pharmacotherapeutic interventions for PBA were based on off-label use of tricyclic antidepressants (TCAs) or selective serotonin reuptake inhibitors (SSRIs). From 1980 to 2010, only 7 of 22 case reports or trials of TCAs or SSRIs for PBA were randomized, double-blind, and placebo-controlled. Five had 12 to 28 patients, and 2 had 106 and 128 patients, respectively. Only 1 controlled trial included a validated symptom severity scale, and none included a scale validated for PBA.18
In particular, imipramine and nortriptyline were studied for managing PBA in patients with stroke; amitriptyline, in patients with MS; and various SSRIs, in patients with stroke.11 Response of PBA symptoms to antidepressant therapy was greater in all placebo-controlled trials than response to placebo.18 As seen in pharmacotherapy of depression, the lower burden of adverse effects and overall better tolerability of SSRIs resulted in their preferred use over TCAs. In some cases, the side effects of TCAs can be leveraged for therapeutic gain. If insomnia is a problem, a nighttime dose of a TCA could ameliorate this. Similarly, if a patient has sialorrhea, the anticholinergic effect of a TCA may show some benefit.19
Dextromethorphan plus quinidine. Dextromethorphan has long been of interest for a variety of neurodegenerative diseases. Studies of its efficacy were largely unsuccessful, however, because rapid metabolism by cytochrome P450 (CYP) 2D6 prevented CNS penetration.20 Quinidine is an avid inhibitor of CYP2D6, even at very low dosages. Adding quinidine to dextromethorphan limits metabolism, allowing dextromethorphan to accumulate to a plasma concentration sufficient to penetrate the CNS.12 In 2010, the combination agent dextromethorphan hydrobromide (20 mg)/quinidine (10 mg) (DM/Q) became the first treatment to receive FDA approval for managing PBA.11
Mechanism of action. The exact mechanism of DM/Q in PBA remains unknown. Dextromethorphan is an agonist of sigma-1 receptors and a relatively specific noncompetitive antagonist of NMDA receptors. It also has been shown to modulate glutamate and serotonin neurotransmission and ion channel function.20 Sigma-1 receptors are concentrated in the brainstem and parts of the cerebellum that are thought to coordinate motor emotional responses. Agonism of sigma-1 receptors on glutamatergic neurons has been proposed to limit release of glutamate from the presynaptic neuron while also limiting downstream transmission of glutamatergic signal in postsynaptic neurons.
Clinical trials. Two large trials have demonstrated efficacy of DM/Q in PBA. STAR was a 12-week, double-blind, placebo-controlled trial with 326 patients diagnosed with ALS or MS who showed PBA symptoms (CNS-LS score ≥13). Compared with placebo, DM/Q use was associated with significantly reduced (P < .01) daily episodes of PBA at 2, 4, 8, and 12 weeks.20 The effect was rapid, with 30% fewer PBA episodes after the first week (P < .0167). At 12 weeks, 51% of patients on DM/Q had been symptom-free for at least 2 weeks.
The PRISM II study examined the efficacy of DM/Q in managing PBA in 102 individuals with dementia, 92 with stroke, and 67 with TBI. After 30 and 90 days, CNL-LS scores were significantly reduced (P < .001) compared with baseline scores.20
Prescribing information. Dextromethorphan—typically in the form of cough syrup—has been implicated as a substance of abuse. A placebo-controlled trial demonstrated that co-administering quinidine with dextromethorphan limits measures of positive reinforcement, such as euphoria and drug liking. This suggests that quinidine may be used to reduce abuse of dextromethorphan.20 As such, the abuse potential of DM/Q appears to be low.
The most common adverse effects reported with DM/Q are diarrhea, dizziness, and cough.12 Notably, patients who received DM/Q in the STAR trial were more likely to report dizziness than those receiving placebo (10.3% vs 5.5%), but patients receiving placebo were more likely to fall.21,22
Package labeling warns that DM/Q causes dose-dependent QTc prolongation.21 Quinidine can be associated with significant QTc prolongation when dosed at antiarrhythmic levels, although mean plasma concentrations found with the 10 mg of quinidine in the approved DM/Q formulation are 1% to 3% of those associated with typical dosages used in antiarrhythmic therapy. Electrophysiology studies of quinidine 10 mg dosed every 12 hours have demonstrated a mean QTc increase at steady state of 6.8 milliseconds, compared with 9.1 milliseconds for a reference control (moxifloxacin).12,21
Although this would seem to indicate a relatively low risk of clinically significant QTc prolongation at these ultra-low dosages of quinidine, it may be advisable to obtain an initial pre-dose and post-dose ECG and longitudinally monitor the QTc interval in patients with conditions that predispose to cardiac arrhythmias. Because quinidine inhibits CYP2D6, use caution when prescribing and monitoring other medications metabolized by this pathway.
Bottom Line
1. Brooks BR, Crumpacker D, Fellus J, et al. PRISM: a novel research tool to assess the prevalence of pseudobulbar affect symptoms across neurological conditions. PLoS One. 2013;8(8):e72232. doi: 10.1371/journal.pone.0072232.
2. Cruz MP. Nuedexta for the treatment of pseudobulbar affect: a condition of involuntary laughing and crying. P T. 2013;38(6):325-328.
3. Work SS, Colamonico JA, Bradley WG, et al. Pseudobulbar affect: an under-recognized and under-treated neurological disorder. Adv Ther. 2011;28(7):586-601.
4. Arciniegas DB, Lauterbach EC, Anderson KE, et al. The differential diagnosis of pseudobulbar affect (PBA). Distinguishing PBA among disorders of mood and affect. Proceedings of a roundtable meeting. CNS Spectr. 2005;10(5):1-14; quiz 15-16.
5. Darwin C. The expression of the emotions in man and animals. London, United Kingdom: John Murray; 1872.
6. Oppenheim H, Siemerling E. Mitteilungen über Pseudobulbärparalyse und akute Bulbärparalyse. Berl Kli Woch. 1886;46.
7. Wilson SA. Original papers: some problems in neurology. J Neurol Psychopathol. 1924;4(16):299-333.
8. Poeck K, Risso M, Pilleri G. Contribution to the pathophysiology and clinical systematology of pathological laughing and crying [in German]. Arch Psychiatr Nervenkr Z Gesamte Neurol Psychiatr. 1963;204:181-198.
9. Cummings JL, Gilbart J, Andersen G. Pseudobulbar affect - a disabling but under-recognised consequence of neurological disease and brain injury. Eur Neurol Rev. 2013;8(2):74-81.
10. Lauterbach EC, Cummings JL, Kuppuswamy PS. Toward a more precise, clinically–informed pathophysiology of pathological laughing and crying. Neurosci Biobehav Rev. 2013;37(8):1893-1916.
11. Pioro EP. Review of dextromethorphan 20 mg/quinidine 10 mg (Nuedexta(®)) for pseudobulbar affect. Neurol Ther. 2014;3(1):15-28.
12. Schoedel KA, Morrow SA, Sellers EM. Evaluating the safety and efficacy of dextromethorphan/quinidine in the treatment of pseudobulbar affect. Neuropsychiatr Dis Treat. 2014;10:1161-1174.
13. Li Z, Luo S, Ou J, et al. Persistent pseudobulbar affect secondary to acute disseminated encephalomyelitis. Socioaffect Neurosci Psychol. 2015;5:26210. doi: 10.3402/snp.v5.26210.
14. Pattee GL, Wymer JP, Lomen-Hoerth C, et al. An open-label multicenter study to assess the safety of dextromethorphan/quinidine in patients with pseudobulbar affect associated with a range of underlying neurological conditions. Curr Med Res Opin. 2014;30(11):2255-2265.
15. Strowd RE, Cartwright MS, Okun MS, et al. Pseudobulbar affect: prevalence and quality of life impact in movement disorders. J Neurol. 2010;257(8):1382-1387.
16. Colamonico J, Formella A, Bradley W. Pseudobulbar affect: burden of illness in the USA. Adv Ther. 2012;29(9):775-798.
17. Starkstein SE, Migliorelli R, Tesón A, et al. Prevalence and clinical correlates of pathological affective display in Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 1995;59(1):55-60.
18. Pioro EP. Current concepts in the pharmacotherapy of pseudobulbar affect. Drugs. 2011;71(9):1193-1207.
19. Ahmed A, Simmons A. Pseudobulbar affect: prevalence and management. Ther Clin Risk Manag. 2013;9:483-489.
20. Yang LP, Deeks ED. Dextromethorphan/quinidine: a review of its use in adults with pseudobulbar affect. Drugs. 2015;75(1):83-90.
21. Nuedexta [package insert]. Aliso Viejo, CA: Avanir Pharmaceuticals, Inc.; 2015.
22. Pioro EP, Brooks BR, Cummings J, et al; Safety, Tolerability, and Efficacy trial of AVP-923 in PBA Investigators. Dextromethorphan plus ultra low-dose quinidine reduces pseudobulbar affect. Ann Neurol. 2010;68(5):693-702.
Pseudobulbar affect (PBA) is a disorder of affective expression that manifests as stereotyped and frequent outbursts of crying (not limited to lacrimation) or laughter. Symptoms are involuntary, uncontrolled, and exaggerated or incongruent with current mood. Episodes, lasting a few seconds to several minutes, may be unprovoked or occur in response to a mild stimulus, and patients typically display a normal affect between episodes.1 PBA is estimated to affect 1 to 2 million people in the United States, although some studies suggest as many as 7 million,1,2 depending on the evaluation method and threshold criteria used.3
Where to look for pseudobulbar affect
PBA has been most commonly described in 6 major neurologic disorders:
- Alzheimer’s disease
- amyotrophic lateral sclerosis (ALS)
- multiple sclerosis (MS)
- Parkinson’s disease
- stroke
- traumatic brain injury (TBI).
Of these disorders, most studies have found the highest PBA prevalence in patients with ALS and TBI, with lesser (although significant) prevalence in Parkinson’s disease (Table 2).1,12 These “big 6” diagnoses are not a comprehensive list, as many other disease states are associated with PBA (Table 3).12-14
2 Pathways: ‘Generator’ and ‘governor’
Despite the many and varied injuries and illnesses associated with PBA, Lauterbach et al10 noted patterns that suggest dysregulation of 2 distinct but interconnected brain pathways: an emotional pathway controlled by a separate volitional pathway. Lesions to the volitional pathway (or its associated feedback or processing circuits) are thought to cause PBA symptoms.
To borrow an analogy from engineering, the emotional pathway is the “generator” of affect, whereas the volitional pathway is the “governor” of affect. Thus, injury to the “governor” results in overspill, or overflow, of affect that usually would be suppressed.
The emotional pathway, which coordinates the motor aspect of reflex laughing or crying, originates at the frontotemporal cortex, relaying to the amygdala and hypothalamus, then projecting to the dorsal brainstem, which includes the midbrain-pontine periaqueductal gray (PAG), dorsal tegmentum, and related brainstem.
The volitional pathway, which regulates the emotional pathway, originates in the dorsal and lateral frontoparietal cortex, projects through the internal capsule and midbrain basis pedunculi, and continues on to the anteroventral basis pontis. The basis pontis then serves as an afferent relay center for cerebellar activity. Projections from the pons then regulate the emotional circuitry primarily at the level of the PAG.10
Lesions of the volitional pathway have been correlated with conditions of PBA, whereas direct activation of the emotional pathway tended to lead to emotional lability or the crying and laughing behaviors observed in dacrystic or gelastic epilepsy.10 The pivotal nature of the regulation occurring at the PAG has guided treatment options. Neurotransmitter receptors most closely associated with this region include glutamatergic N-methyl-
When to screen for PBA
Ask the right question. PBA as a disease state likely has been widely under-reported, under-recognized, and misdiagnosed (typically, as a primary mood disorder).9 Three factors underscore this problem:
- Patients do not specifically report symptoms of affective disturbance (perhaps because they lack a vocabulary to separate concepts of mood and affect)
- Physicians do not ask patients about separations of mood and affect
- Perhaps most importantly, PBA lacks a general awareness and understanding.
Co-occurring mood disorders also may thwart PBA detection. One study of PBA in Alzheimer’s dementia found that 53% of patients with symptoms consistent with PBA also had a distinct mood disorder.17 This suggests that a PBA-specific screening test is needed for accurate diagnosis.
A single question might best refine the likelihood that a patient has PBA: “Do you ever cry for no reason?” In primary psychiatric illness, crying typically is associated with a specific trigger (eg, depressed mood, despair, anxiety). A patient’s inability to identify a trigger for crying suggests the pathological separation of mood and affect—the core of PBA, and worthy of further investigation.
Clinical rating scales that correlate to disease severity appear to be the most effective in identifying PBA. The PRISM study, to date the largest clinic-based study of PBA symptoms, used the Center for Neurologic Study-Liability Scale (CNS-LS) to gauge the presence and severity of PBA symptoms.1 A 7-question, patient self-administered tool, the CNS-LS is graded on a 5-point Likert scale. A score ≥13 has high sensitivity and specificity for diagnosis of PBA, compared with physician diagnosis.
Another option, the 16-question Pathological Laughing and Crying Scale, is a clinician-administered screening tool. Again, a score ≥13 is consistent with symptoms required for a PBA diagnosis.
Treating PBA symptoms
Until recently, most pharmacotherapeutic interventions for PBA were based on off-label use of tricyclic antidepressants (TCAs) or selective serotonin reuptake inhibitors (SSRIs). From 1980 to 2010, only 7 of 22 case reports or trials of TCAs or SSRIs for PBA were randomized, double-blind, and placebo-controlled. Five had 12 to 28 patients, and 2 had 106 and 128 patients, respectively. Only 1 controlled trial included a validated symptom severity scale, and none included a scale validated for PBA.18
In particular, imipramine and nortriptyline were studied for managing PBA in patients with stroke; amitriptyline, in patients with MS; and various SSRIs, in patients with stroke.11 Response of PBA symptoms to antidepressant therapy was greater in all placebo-controlled trials than response to placebo.18 As seen in pharmacotherapy of depression, the lower burden of adverse effects and overall better tolerability of SSRIs resulted in their preferred use over TCAs. In some cases, the side effects of TCAs can be leveraged for therapeutic gain. If insomnia is a problem, a nighttime dose of a TCA could ameliorate this. Similarly, if a patient has sialorrhea, the anticholinergic effect of a TCA may show some benefit.19
Dextromethorphan plus quinidine. Dextromethorphan has long been of interest for a variety of neurodegenerative diseases. Studies of its efficacy were largely unsuccessful, however, because rapid metabolism by cytochrome P450 (CYP) 2D6 prevented CNS penetration.20 Quinidine is an avid inhibitor of CYP2D6, even at very low dosages. Adding quinidine to dextromethorphan limits metabolism, allowing dextromethorphan to accumulate to a plasma concentration sufficient to penetrate the CNS.12 In 2010, the combination agent dextromethorphan hydrobromide (20 mg)/quinidine (10 mg) (DM/Q) became the first treatment to receive FDA approval for managing PBA.11
Mechanism of action. The exact mechanism of DM/Q in PBA remains unknown. Dextromethorphan is an agonist of sigma-1 receptors and a relatively specific noncompetitive antagonist of NMDA receptors. It also has been shown to modulate glutamate and serotonin neurotransmission and ion channel function.20 Sigma-1 receptors are concentrated in the brainstem and parts of the cerebellum that are thought to coordinate motor emotional responses. Agonism of sigma-1 receptors on glutamatergic neurons has been proposed to limit release of glutamate from the presynaptic neuron while also limiting downstream transmission of glutamatergic signal in postsynaptic neurons.
Clinical trials. Two large trials have demonstrated efficacy of DM/Q in PBA. STAR was a 12-week, double-blind, placebo-controlled trial with 326 patients diagnosed with ALS or MS who showed PBA symptoms (CNS-LS score ≥13). Compared with placebo, DM/Q use was associated with significantly reduced (P < .01) daily episodes of PBA at 2, 4, 8, and 12 weeks.20 The effect was rapid, with 30% fewer PBA episodes after the first week (P < .0167). At 12 weeks, 51% of patients on DM/Q had been symptom-free for at least 2 weeks.
The PRISM II study examined the efficacy of DM/Q in managing PBA in 102 individuals with dementia, 92 with stroke, and 67 with TBI. After 30 and 90 days, CNL-LS scores were significantly reduced (P < .001) compared with baseline scores.20
Prescribing information. Dextromethorphan—typically in the form of cough syrup—has been implicated as a substance of abuse. A placebo-controlled trial demonstrated that co-administering quinidine with dextromethorphan limits measures of positive reinforcement, such as euphoria and drug liking. This suggests that quinidine may be used to reduce abuse of dextromethorphan.20 As such, the abuse potential of DM/Q appears to be low.
The most common adverse effects reported with DM/Q are diarrhea, dizziness, and cough.12 Notably, patients who received DM/Q in the STAR trial were more likely to report dizziness than those receiving placebo (10.3% vs 5.5%), but patients receiving placebo were more likely to fall.21,22
Package labeling warns that DM/Q causes dose-dependent QTc prolongation.21 Quinidine can be associated with significant QTc prolongation when dosed at antiarrhythmic levels, although mean plasma concentrations found with the 10 mg of quinidine in the approved DM/Q formulation are 1% to 3% of those associated with typical dosages used in antiarrhythmic therapy. Electrophysiology studies of quinidine 10 mg dosed every 12 hours have demonstrated a mean QTc increase at steady state of 6.8 milliseconds, compared with 9.1 milliseconds for a reference control (moxifloxacin).12,21
Although this would seem to indicate a relatively low risk of clinically significant QTc prolongation at these ultra-low dosages of quinidine, it may be advisable to obtain an initial pre-dose and post-dose ECG and longitudinally monitor the QTc interval in patients with conditions that predispose to cardiac arrhythmias. Because quinidine inhibits CYP2D6, use caution when prescribing and monitoring other medications metabolized by this pathway.
Bottom Line
Pseudobulbar affect (PBA) is a disorder of affective expression that manifests as stereotyped and frequent outbursts of crying (not limited to lacrimation) or laughter. Symptoms are involuntary, uncontrolled, and exaggerated or incongruent with current mood. Episodes, lasting a few seconds to several minutes, may be unprovoked or occur in response to a mild stimulus, and patients typically display a normal affect between episodes.1 PBA is estimated to affect 1 to 2 million people in the United States, although some studies suggest as many as 7 million,1,2 depending on the evaluation method and threshold criteria used.3
Where to look for pseudobulbar affect
PBA has been most commonly described in 6 major neurologic disorders:
- Alzheimer’s disease
- amyotrophic lateral sclerosis (ALS)
- multiple sclerosis (MS)
- Parkinson’s disease
- stroke
- traumatic brain injury (TBI).
Of these disorders, most studies have found the highest PBA prevalence in patients with ALS and TBI, with lesser (although significant) prevalence in Parkinson’s disease (Table 2).1,12 These “big 6” diagnoses are not a comprehensive list, as many other disease states are associated with PBA (Table 3).12-14
2 Pathways: ‘Generator’ and ‘governor’
Despite the many and varied injuries and illnesses associated with PBA, Lauterbach et al10 noted patterns that suggest dysregulation of 2 distinct but interconnected brain pathways: an emotional pathway controlled by a separate volitional pathway. Lesions to the volitional pathway (or its associated feedback or processing circuits) are thought to cause PBA symptoms.
To borrow an analogy from engineering, the emotional pathway is the “generator” of affect, whereas the volitional pathway is the “governor” of affect. Thus, injury to the “governor” results in overspill, or overflow, of affect that usually would be suppressed.
The emotional pathway, which coordinates the motor aspect of reflex laughing or crying, originates at the frontotemporal cortex, relaying to the amygdala and hypothalamus, then projecting to the dorsal brainstem, which includes the midbrain-pontine periaqueductal gray (PAG), dorsal tegmentum, and related brainstem.
The volitional pathway, which regulates the emotional pathway, originates in the dorsal and lateral frontoparietal cortex, projects through the internal capsule and midbrain basis pedunculi, and continues on to the anteroventral basis pontis. The basis pontis then serves as an afferent relay center for cerebellar activity. Projections from the pons then regulate the emotional circuitry primarily at the level of the PAG.10
Lesions of the volitional pathway have been correlated with conditions of PBA, whereas direct activation of the emotional pathway tended to lead to emotional lability or the crying and laughing behaviors observed in dacrystic or gelastic epilepsy.10 The pivotal nature of the regulation occurring at the PAG has guided treatment options. Neurotransmitter receptors most closely associated with this region include glutamatergic N-methyl-
When to screen for PBA
Ask the right question. PBA as a disease state likely has been widely under-reported, under-recognized, and misdiagnosed (typically, as a primary mood disorder).9 Three factors underscore this problem:
- Patients do not specifically report symptoms of affective disturbance (perhaps because they lack a vocabulary to separate concepts of mood and affect)
- Physicians do not ask patients about separations of mood and affect
- Perhaps most importantly, PBA lacks a general awareness and understanding.
Co-occurring mood disorders also may thwart PBA detection. One study of PBA in Alzheimer’s dementia found that 53% of patients with symptoms consistent with PBA also had a distinct mood disorder.17 This suggests that a PBA-specific screening test is needed for accurate diagnosis.
A single question might best refine the likelihood that a patient has PBA: “Do you ever cry for no reason?” In primary psychiatric illness, crying typically is associated with a specific trigger (eg, depressed mood, despair, anxiety). A patient’s inability to identify a trigger for crying suggests the pathological separation of mood and affect—the core of PBA, and worthy of further investigation.
Clinical rating scales that correlate to disease severity appear to be the most effective in identifying PBA. The PRISM study, to date the largest clinic-based study of PBA symptoms, used the Center for Neurologic Study-Liability Scale (CNS-LS) to gauge the presence and severity of PBA symptoms.1 A 7-question, patient self-administered tool, the CNS-LS is graded on a 5-point Likert scale. A score ≥13 has high sensitivity and specificity for diagnosis of PBA, compared with physician diagnosis.
Another option, the 16-question Pathological Laughing and Crying Scale, is a clinician-administered screening tool. Again, a score ≥13 is consistent with symptoms required for a PBA diagnosis.
Treating PBA symptoms
Until recently, most pharmacotherapeutic interventions for PBA were based on off-label use of tricyclic antidepressants (TCAs) or selective serotonin reuptake inhibitors (SSRIs). From 1980 to 2010, only 7 of 22 case reports or trials of TCAs or SSRIs for PBA were randomized, double-blind, and placebo-controlled. Five had 12 to 28 patients, and 2 had 106 and 128 patients, respectively. Only 1 controlled trial included a validated symptom severity scale, and none included a scale validated for PBA.18
In particular, imipramine and nortriptyline were studied for managing PBA in patients with stroke; amitriptyline, in patients with MS; and various SSRIs, in patients with stroke.11 Response of PBA symptoms to antidepressant therapy was greater in all placebo-controlled trials than response to placebo.18 As seen in pharmacotherapy of depression, the lower burden of adverse effects and overall better tolerability of SSRIs resulted in their preferred use over TCAs. In some cases, the side effects of TCAs can be leveraged for therapeutic gain. If insomnia is a problem, a nighttime dose of a TCA could ameliorate this. Similarly, if a patient has sialorrhea, the anticholinergic effect of a TCA may show some benefit.19
Dextromethorphan plus quinidine. Dextromethorphan has long been of interest for a variety of neurodegenerative diseases. Studies of its efficacy were largely unsuccessful, however, because rapid metabolism by cytochrome P450 (CYP) 2D6 prevented CNS penetration.20 Quinidine is an avid inhibitor of CYP2D6, even at very low dosages. Adding quinidine to dextromethorphan limits metabolism, allowing dextromethorphan to accumulate to a plasma concentration sufficient to penetrate the CNS.12 In 2010, the combination agent dextromethorphan hydrobromide (20 mg)/quinidine (10 mg) (DM/Q) became the first treatment to receive FDA approval for managing PBA.11
Mechanism of action. The exact mechanism of DM/Q in PBA remains unknown. Dextromethorphan is an agonist of sigma-1 receptors and a relatively specific noncompetitive antagonist of NMDA receptors. It also has been shown to modulate glutamate and serotonin neurotransmission and ion channel function.20 Sigma-1 receptors are concentrated in the brainstem and parts of the cerebellum that are thought to coordinate motor emotional responses. Agonism of sigma-1 receptors on glutamatergic neurons has been proposed to limit release of glutamate from the presynaptic neuron while also limiting downstream transmission of glutamatergic signal in postsynaptic neurons.
Clinical trials. Two large trials have demonstrated efficacy of DM/Q in PBA. STAR was a 12-week, double-blind, placebo-controlled trial with 326 patients diagnosed with ALS or MS who showed PBA symptoms (CNS-LS score ≥13). Compared with placebo, DM/Q use was associated with significantly reduced (P < .01) daily episodes of PBA at 2, 4, 8, and 12 weeks.20 The effect was rapid, with 30% fewer PBA episodes after the first week (P < .0167). At 12 weeks, 51% of patients on DM/Q had been symptom-free for at least 2 weeks.
The PRISM II study examined the efficacy of DM/Q in managing PBA in 102 individuals with dementia, 92 with stroke, and 67 with TBI. After 30 and 90 days, CNL-LS scores were significantly reduced (P < .001) compared with baseline scores.20
Prescribing information. Dextromethorphan—typically in the form of cough syrup—has been implicated as a substance of abuse. A placebo-controlled trial demonstrated that co-administering quinidine with dextromethorphan limits measures of positive reinforcement, such as euphoria and drug liking. This suggests that quinidine may be used to reduce abuse of dextromethorphan.20 As such, the abuse potential of DM/Q appears to be low.
The most common adverse effects reported with DM/Q are diarrhea, dizziness, and cough.12 Notably, patients who received DM/Q in the STAR trial were more likely to report dizziness than those receiving placebo (10.3% vs 5.5%), but patients receiving placebo were more likely to fall.21,22
Package labeling warns that DM/Q causes dose-dependent QTc prolongation.21 Quinidine can be associated with significant QTc prolongation when dosed at antiarrhythmic levels, although mean plasma concentrations found with the 10 mg of quinidine in the approved DM/Q formulation are 1% to 3% of those associated with typical dosages used in antiarrhythmic therapy. Electrophysiology studies of quinidine 10 mg dosed every 12 hours have demonstrated a mean QTc increase at steady state of 6.8 milliseconds, compared with 9.1 milliseconds for a reference control (moxifloxacin).12,21
Although this would seem to indicate a relatively low risk of clinically significant QTc prolongation at these ultra-low dosages of quinidine, it may be advisable to obtain an initial pre-dose and post-dose ECG and longitudinally monitor the QTc interval in patients with conditions that predispose to cardiac arrhythmias. Because quinidine inhibits CYP2D6, use caution when prescribing and monitoring other medications metabolized by this pathway.
Bottom Line
1. Brooks BR, Crumpacker D, Fellus J, et al. PRISM: a novel research tool to assess the prevalence of pseudobulbar affect symptoms across neurological conditions. PLoS One. 2013;8(8):e72232. doi: 10.1371/journal.pone.0072232.
2. Cruz MP. Nuedexta for the treatment of pseudobulbar affect: a condition of involuntary laughing and crying. P T. 2013;38(6):325-328.
3. Work SS, Colamonico JA, Bradley WG, et al. Pseudobulbar affect: an under-recognized and under-treated neurological disorder. Adv Ther. 2011;28(7):586-601.
4. Arciniegas DB, Lauterbach EC, Anderson KE, et al. The differential diagnosis of pseudobulbar affect (PBA). Distinguishing PBA among disorders of mood and affect. Proceedings of a roundtable meeting. CNS Spectr. 2005;10(5):1-14; quiz 15-16.
5. Darwin C. The expression of the emotions in man and animals. London, United Kingdom: John Murray; 1872.
6. Oppenheim H, Siemerling E. Mitteilungen über Pseudobulbärparalyse und akute Bulbärparalyse. Berl Kli Woch. 1886;46.
7. Wilson SA. Original papers: some problems in neurology. J Neurol Psychopathol. 1924;4(16):299-333.
8. Poeck K, Risso M, Pilleri G. Contribution to the pathophysiology and clinical systematology of pathological laughing and crying [in German]. Arch Psychiatr Nervenkr Z Gesamte Neurol Psychiatr. 1963;204:181-198.
9. Cummings JL, Gilbart J, Andersen G. Pseudobulbar affect - a disabling but under-recognised consequence of neurological disease and brain injury. Eur Neurol Rev. 2013;8(2):74-81.
10. Lauterbach EC, Cummings JL, Kuppuswamy PS. Toward a more precise, clinically–informed pathophysiology of pathological laughing and crying. Neurosci Biobehav Rev. 2013;37(8):1893-1916.
11. Pioro EP. Review of dextromethorphan 20 mg/quinidine 10 mg (Nuedexta(®)) for pseudobulbar affect. Neurol Ther. 2014;3(1):15-28.
12. Schoedel KA, Morrow SA, Sellers EM. Evaluating the safety and efficacy of dextromethorphan/quinidine in the treatment of pseudobulbar affect. Neuropsychiatr Dis Treat. 2014;10:1161-1174.
13. Li Z, Luo S, Ou J, et al. Persistent pseudobulbar affect secondary to acute disseminated encephalomyelitis. Socioaffect Neurosci Psychol. 2015;5:26210. doi: 10.3402/snp.v5.26210.
14. Pattee GL, Wymer JP, Lomen-Hoerth C, et al. An open-label multicenter study to assess the safety of dextromethorphan/quinidine in patients with pseudobulbar affect associated with a range of underlying neurological conditions. Curr Med Res Opin. 2014;30(11):2255-2265.
15. Strowd RE, Cartwright MS, Okun MS, et al. Pseudobulbar affect: prevalence and quality of life impact in movement disorders. J Neurol. 2010;257(8):1382-1387.
16. Colamonico J, Formella A, Bradley W. Pseudobulbar affect: burden of illness in the USA. Adv Ther. 2012;29(9):775-798.
17. Starkstein SE, Migliorelli R, Tesón A, et al. Prevalence and clinical correlates of pathological affective display in Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 1995;59(1):55-60.
18. Pioro EP. Current concepts in the pharmacotherapy of pseudobulbar affect. Drugs. 2011;71(9):1193-1207.
19. Ahmed A, Simmons A. Pseudobulbar affect: prevalence and management. Ther Clin Risk Manag. 2013;9:483-489.
20. Yang LP, Deeks ED. Dextromethorphan/quinidine: a review of its use in adults with pseudobulbar affect. Drugs. 2015;75(1):83-90.
21. Nuedexta [package insert]. Aliso Viejo, CA: Avanir Pharmaceuticals, Inc.; 2015.
22. Pioro EP, Brooks BR, Cummings J, et al; Safety, Tolerability, and Efficacy trial of AVP-923 in PBA Investigators. Dextromethorphan plus ultra low-dose quinidine reduces pseudobulbar affect. Ann Neurol. 2010;68(5):693-702.
1. Brooks BR, Crumpacker D, Fellus J, et al. PRISM: a novel research tool to assess the prevalence of pseudobulbar affect symptoms across neurological conditions. PLoS One. 2013;8(8):e72232. doi: 10.1371/journal.pone.0072232.
2. Cruz MP. Nuedexta for the treatment of pseudobulbar affect: a condition of involuntary laughing and crying. P T. 2013;38(6):325-328.
3. Work SS, Colamonico JA, Bradley WG, et al. Pseudobulbar affect: an under-recognized and under-treated neurological disorder. Adv Ther. 2011;28(7):586-601.
4. Arciniegas DB, Lauterbach EC, Anderson KE, et al. The differential diagnosis of pseudobulbar affect (PBA). Distinguishing PBA among disorders of mood and affect. Proceedings of a roundtable meeting. CNS Spectr. 2005;10(5):1-14; quiz 15-16.
5. Darwin C. The expression of the emotions in man and animals. London, United Kingdom: John Murray; 1872.
6. Oppenheim H, Siemerling E. Mitteilungen über Pseudobulbärparalyse und akute Bulbärparalyse. Berl Kli Woch. 1886;46.
7. Wilson SA. Original papers: some problems in neurology. J Neurol Psychopathol. 1924;4(16):299-333.
8. Poeck K, Risso M, Pilleri G. Contribution to the pathophysiology and clinical systematology of pathological laughing and crying [in German]. Arch Psychiatr Nervenkr Z Gesamte Neurol Psychiatr. 1963;204:181-198.
9. Cummings JL, Gilbart J, Andersen G. Pseudobulbar affect - a disabling but under-recognised consequence of neurological disease and brain injury. Eur Neurol Rev. 2013;8(2):74-81.
10. Lauterbach EC, Cummings JL, Kuppuswamy PS. Toward a more precise, clinically–informed pathophysiology of pathological laughing and crying. Neurosci Biobehav Rev. 2013;37(8):1893-1916.
11. Pioro EP. Review of dextromethorphan 20 mg/quinidine 10 mg (Nuedexta(®)) for pseudobulbar affect. Neurol Ther. 2014;3(1):15-28.
12. Schoedel KA, Morrow SA, Sellers EM. Evaluating the safety and efficacy of dextromethorphan/quinidine in the treatment of pseudobulbar affect. Neuropsychiatr Dis Treat. 2014;10:1161-1174.
13. Li Z, Luo S, Ou J, et al. Persistent pseudobulbar affect secondary to acute disseminated encephalomyelitis. Socioaffect Neurosci Psychol. 2015;5:26210. doi: 10.3402/snp.v5.26210.
14. Pattee GL, Wymer JP, Lomen-Hoerth C, et al. An open-label multicenter study to assess the safety of dextromethorphan/quinidine in patients with pseudobulbar affect associated with a range of underlying neurological conditions. Curr Med Res Opin. 2014;30(11):2255-2265.
15. Strowd RE, Cartwright MS, Okun MS, et al. Pseudobulbar affect: prevalence and quality of life impact in movement disorders. J Neurol. 2010;257(8):1382-1387.
16. Colamonico J, Formella A, Bradley W. Pseudobulbar affect: burden of illness in the USA. Adv Ther. 2012;29(9):775-798.
17. Starkstein SE, Migliorelli R, Tesón A, et al. Prevalence and clinical correlates of pathological affective display in Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 1995;59(1):55-60.
18. Pioro EP. Current concepts in the pharmacotherapy of pseudobulbar affect. Drugs. 2011;71(9):1193-1207.
19. Ahmed A, Simmons A. Pseudobulbar affect: prevalence and management. Ther Clin Risk Manag. 2013;9:483-489.
20. Yang LP, Deeks ED. Dextromethorphan/quinidine: a review of its use in adults with pseudobulbar affect. Drugs. 2015;75(1):83-90.
21. Nuedexta [package insert]. Aliso Viejo, CA: Avanir Pharmaceuticals, Inc.; 2015.
22. Pioro EP, Brooks BR, Cummings J, et al; Safety, Tolerability, and Efficacy trial of AVP-923 in PBA Investigators. Dextromethorphan plus ultra low-dose quinidine reduces pseudobulbar affect. Ann Neurol. 2010;68(5):693-702.

Pimavanserin for psychosis in patients with Parkinson’s disease
Pimavanserin is a potent 5-HT2A inverse agonist and 5-HT2C inverse agonist, with 5-fold greater affinity for the 5-HT2A receptor.1 Although antagonists block agonist actions at the receptor site, inverse agonists reduce the level of baseline constitutive activity seen in many G protein-coupled receptors. This medication is FDA approved for treating hallucinations and delusions associated with Parkinson’s disease (PD) psychosis (Table 1).1
In the pivotal 6-week clinical trial, pimavanserin significantly reduced positive symptoms seen in PD patients with psychosis (effect size = 0.50), with no evident impairment of motor function.2 Only 2 adverse effects occurred in ≥5% of pimavanserin-treated patients and at ≥2 times the rate of placebo: peripheral edema (7% vs 3% for placebo) and confusion (6% vs 3% for placebo). There was a mean increase in the QTc of 7.3 milliseconds compared with placebo in the pivotal phase III study.
Clinical implications
Despite numerous developments in the pharmacotherapeutics of psychotic disorders, patients with psychosis related to PD previously responded in a robust manner to only 1 antipsychotic, low-dosage clozapine (mean effect size, 0.80),2 with numerous failed trials for other atypical antipsychotics, including quetiapine.3,4 The pathophysiology of psychosis in PD patients is not related to dopamine agonist treatment, but is caused by the accumulation of cortical Lewy body burden, which results in loss of serotonergic signaling from dorsal raphe neurons. The net effect is up-regulation of postsynaptic 5-HT2A receptors.5 Psychosis is the most common cause of nursing home placement among PD patients without dementia.6
Receptor blocking. Based on the finding that clozapine in low dosages acts at 5-HT2A receptors,7 pimavanserin was designed to be a potent 5-HT2A inverse agonist, with more than 5-fold higher selectivity over 5-HT2C receptors, and no appreciable affinity for other serotonergic, adrenergic, dopaminergic, muscarinic, or histaminergic receptors8 (Table 2). The concept that 5-HT2A receptor stimulation can cause psychosis with prominent visual hallucinations is known from studies of LSD and other hallucinogenic compounds whose activity is blocked by 5-HT2A antagonists.

As an agent devoid of dopamine D2 antagonism, pimavanserin carries no risk of exacerbating motor symptoms, which was commonly seen with most atypical antipsychotics studied for psychosis in PD patients, except for clozapine and quetiapine.3 Although quetiapine did not cause motor effects, it proved ineffective in multiple studies (n = 153), likely because of the near absence of potent 5-HT2A binding.4
Pimavanserin also lacks:
- the hematologic monitoring requirement of clozapine
- clozapine’s risks of sedation, orthostasis, and anticholinergic and metabolic adverse effects.
Pimavanserin is significantly more potent than other non-antipsychotic psychotropics at the 5-HT2Areceptor, including doxepin (26 nM), trazodone (36 nM), and mirtazapine (60 nM).
Use in psychosis associated with PD. Recommended dosage is 34 mg once daily without titration (with or without food), based on results from a phase III clinical trial2 (because of the FDA breakthrough therapy designation for this compound, only 1 phase III trial was required). Pimavanserin produced significant improvement on the PD-adapted Scale for the Assessment of Positive Symptoms (SAPS-PD), a 9-item instrument extracted from the larger SAPS used in schizophrenia research. Specifically, pimavanserin was effective for both the hallucinations and delusions components of the SAPS-PD.
Pharmacologic profile, adverse effects. Pimavanserin lacks affinity for receptors other than 5-HT2A and 5-HT2C, leading to an absence of significant anticholinergic effects, orthostasis, or sedation in clinical trials.2 In all short-term clinical trials, the only common adverse reactions (incidence ≥5% and at least twice the rate of placebo) were peripheral edema (7% vs 2% placebo) and confusional state (6% vs 3% placebo).2 More than 300 patients have been treated for >6 months, >270 have been treated for at least 12 months, and >150 have been treated for at least 24 months with no adverse effects other than those seen in the short-term trials.1
There is a measurable impact on cardiac conduction seen in phase III data and in the thorough QT study. In the thorough QT study, 252 healthy participants received multiple dosages in a randomized, double-blind manner with positive controls.1 The maximum mean change from baseline was 13.5 milliseconds at dosages twice the recommended dosage, and the upper limit of the 90% CI was only slightly greater at 16.6 milliseconds. Subsequent kinetic analyses suggested concentration-dependent QTc interval prolongation in the therapeutic range, with a recommendation to halve the daily dosage in patients taking potent cytochrome P450 (CYP) 3A4 inhibitors.
In the 6-week, placebo-controlled effectiveness studies, mean increases in QTc interval were in the range of 5 to 8 milliseconds. There were sporadic reports of QTcF values ≥500 milliseconds, or changes from baseline QTc values ≥60 milliseconds in pimavanserin-treated participants, although the incidence generally was the same for pimavanserin and placebo groups. There were no reports of torsades de pointes or any differences from placebo in the incidence of adverse reactions associated with delayed ventricular repolarization.
How it works
The theory behind development of pimavanserin rests in the finding that low-dosage clozapine (6.25 to 50 mg/d) was effective for PD patients with psychosis (effect size 0.80).8 Although clozapine has high affinity for multiple sites, including histamine H1 receptors (Ki = 1.13 nM), α-1A and a α-2C adrenergic receptors (Ki = 1.62 nM and 6 nM, respectively), 5-HT2A receptors (Ki = 5.35 nM), and muscarinic M1 receptors (Ki = 6 nM), the hypothesized primary mechanism of clozapine’s effectiveness for PD psychosis at low dosages focused on the 5-HT2Areceptor. This idea was based on the knowledge that hallucinogens such as mescaline, psilocybin, and LSD are 5-HT2A agonists.9 This hallucinogenic activity can be blocked with 5-HT2A antagonists. Because of pimavanserin’s binding profile, the compound was studied as a treatment for psychosis in PD patients.
Pharmacokinetics
Pimavanserin demonstrates dose-proportional pharmacokinetics after a single oral dose as much as 7.5 times the recommended dosage. The pharmacokinetics of pimavanserin were similar in study participants (mean age, 72.4) and healthy controls, and a high-fat meal had no impact on the maximum blood levels (Cmax) or total drug exposure (area under the curve [AUC]).
The mean plasma half-lives for pimavanserin and its metabolite N-desmethyl-pimavanserin (AC-279) are 57 hours and 200 hours, respectively. Although the metabolite appears active in in vitro assays, it does not cross the blood-brain barrier to any appreciable extent, therefore contributing little to the clinical effect. The median time to maximum concentration (Tmax) of pimavanserin is 6 hours with a range of 4 to 24 hours, while the median Tmax of the primary metabolite AC-279 is 6 hours. The bioavailability of pimavanserin in an oral tablet or solution essentially is identical.
Pimavanserin is primarily metabolized via CYP3A4 to AC-279, and strong CYP3A4 inhibitors (eg, ketoconazole, itraconazole, clarithromycin, indinavir) increase pimavanserin Cmax by 1.5-fold, and AUC by 3-fold. In patients taking strong CYP3A4 inhibitors, the dosage of pimavanserin should be reduced by 50% to 17 mg/d. Conversely, patients on CYP3A4 inducers (eg, rifampin, carbamazepine, phenytoin) should be monitored for lack of efficacy; consider a dosage increase as necessary. Neither pimavanserin nor its metabolite, AC-279, are inhibitors or inducers of major CYP enzymes or drug transporters.
Efficacy in PD psychosis
Study 1. This 6-week, fixed dosage, double-blind, placebo-controlled trial was performed in adult PD patients age ≥40 with PD psychosis.2 Participants had to have (1) a PD diagnosis for at least 1 year and (2) psychotic symptoms that developed after diagnosis. Psychotic symptoms had to be present for at least 1 month, occurring at least weekly in the month before screening, and severe enough to warrant antipsychotic treatment. Baseline Mini-Mental State Examination score had to be ≥21 out of 30, with no evidence of delirium. Patients with dementia preceding or concurrent with the PD diagnosis were excluded. Antipsychotic treatments were not permitted during the trial.
After a 2-week nonpharmacotherapeutic lead-in phase that included a brief, daily psychosocial intervention by a caregiver, 199 patients who still met severity criteria were randomly allocated in a 1:1 manner to pimavanserin (34 mg of active drug, reported in the paper as 40 mg of pimavanserin tartrate) or matched placebo. Based on kinetic modeling and earlier clinical data, lower dosages (ie, 17 mg) were not explored, because they achieved only 50% of the steady state plasma levels thought to be required for efficacy.
The primary outcome was assessed by central, independent raters using the PD-adapted SAPS-PD. The efficacy analysis included 95 pimavanserin-treated individuals and 90 taking placebo. Baseline SAPS-PD scores were 14.7 ± 5.55 in the placebo group, and 15.9 ± 6.12 in the pimavanserin arm. Participants had a mean age of 72.4 and 94% white ethnicity across both cohorts; 42% of the placebo group and 33% of the pimavanserin group were female. Antipsychotic exposure in the 21 days prior to study entry were reported in 17% (n = 15) and 19% (n = 18) of the placebo and pimavanserin groups, respectively, with the most common agent being quetiapine (13 of 15, placebo, 16 of 18, pimavanserin). Approximately one-third of all participants were taking a cholinesterase inhibitor throughout the study.
Efficacy outcome. Pimavanserin was associated with a 5.79-point decrease in SAPS-PD scores compared with 2.73-point decrease for placebo (difference −3.06, 95% CI −4.91 to −1.20; P = .001). The effect size for this difference (Cohen’s d) was 0.50. The significant effect of pimavanserin vs placebo also was seen in separate analyses of the SAPS-PD subscore for hallucinations and delusions (effect size 0.50), and individually for hallucinations (effect size 0.45) and delusions (effect size 0.33). Separation from placebo appeared after the second week of pimavanserin treatment, and continued through the end of the study. There is unpublished data showing efficacy through week 10, and longer term, uncontrolled data consistent with sustained response. An exploratory analysis of caregiver burden demonstrated an effect size of 0.50.
Tolerability
The discontinuation rate because of adverse events for pimavanserin and placebo-treated patients was 10 patients in the pimavanserin group (4 due to psychotic symptoms within 10 days of starting the study drug) compared with 2 in the placebo group. There was no evidence of motor worsening in either group, demonstrated by the score on part II of the Unified Parkinson’s Disease Rating Scale (UPDRS) that captures self-reported activities of daily living, or on UPDRS part III (motor examination). Pimavanserin has no contraindications.
Unique clinical issues
Binding properties. Pimavanserin possesses potent 5-HT2A inverse agonist properties required to manage psychosis in PD patients, but lacks clozapine’s affinities for α-1 adrenergic, muscarinic, or histaminergic receptors that contribute to clozapine’s poor tolerability. Moreover, pimavanserin has no appreciable affinity for dopaminergic receptors, and therefore does not induce motor adverse effects.
Clozapine aside, all available atypical antipsychotics have proved ineffective for psychosis in PD patients, and most caused significant motor worsening.3 Although quetiapine does not cause motor effects, it has been shown to be ineffective for psychosis in PD patients in multiple trials.4
The effect size for clozapine response is large (0.80) in PD patients with psychosis, but tolerability issues and administrative burdens regarding patient and prescriber registration and routine hematological monitoring pose significant clinical barriers. Clozapine also lacks an FDA indication for this purpose, which may pose a hurdle to its use in certain treatment settings.
Why Rx? The reasons to prescribe pimavanserin for PD patients with psychosis likely include:
- absence of tolerability issues seen with the only other effective agent, clozapine
- lack of motor effects
- lack of administrative and monitoring burden related to clozapine prescribing
- only agent with FDA approval for hallucinations and delusions in PD patients with psychosis.
Dosing
The recommended dosage of pimavanserin is 34 mg/d administered as a single dose with or without food. There is no need for titration, and none was performed in the pivotal clinical trial. Given the long half-life (57 hours), steady state is not achieved until day 12, therefore initiation with a lower dosage might prolong the time to efficacy. There is no dosage adjustment required in patients with mild or moderate renal impairment, but pimavanserin treatment is not recommended in patients with severe renal impairment. Pimavanserin has not been evaluated in patients with hepatic impairment (using Child-Pugh criteria), and is not recommended for these patients.
Other key aspects of dosing to keep in mind.
- Because pimavanserin is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a strong CYP3A4 inhibitor; the recommended dosage is 17 mg/d when administered concomitantly with a strong CYP3A4 inhibitor.
- Because data are not available regarding concomitant use of pimavanserin with CYP3A4 inducers, patients should be monitored for lack of efficacy during concomitant use with a CYP3A4 inducer, and consideration given to a dosage increase.
Use in pregnancy and lactation. There are no data on the use of pimavanserin in pregnant women, but no developmental effects were seen when the drug was administered orally at 10 or 12 times the maximum recommended human dosage to rats or rabbits during organogenesis. Pimavanserin was not teratogenic in pregnant rats and rabbits. There is no information regarding the presence of pimavanserin in human breast milk.
Geriatric patients. No dosage adjustment is required for older patients. The study population in the pivotal trial was mean age 72.4 years.
Summing up
Before development of pimavanserin, clozapine was the only effective treatment for psychosis in PD patients. Despite clozapine’s robust effects across several trials, patients often were given ineffective medications, such as quetiapine, because of the administrative and tolerability barriers posed by clozapine use. Because psychosis is the most common cause of nursing home placement in non-demented PD patients, an agent with demonstrated efficacy and without the adverse effect profile of clozapine or monitoring requirements represents an enormous advance in the treatment of psychosis in PD patients.
1. Nuplazid [package insert]. San Diego, CA: Acadia Pharmaceuticals Inc.; 2016.
2. Cummings J, Isaacson S, Mills R, et al. Pimavanserin for patients with Parkinson’s disease psychosis: a randomised, placebo-controlled phase 3 trial. [Erratum in Lancet. 2014;384(9937):28]. Lancet. 2014;383(9916):533-540.
3. Borek LL, Friedman JH. Treating psychosis in movement disorder patients: a review. Expert Opin Pharmacother. 2014;15(11):1553-1564.
4. Desmarais P, Massoud F, Filion J, et al. Quetiapine for psychosis in Parkinson disease and neurodegenerative parkinsonian disorders: a systematic review. J Geriatr Psychiatry Neurol. 2016;29(4):227-236.
5. Ballanger B, Strafella AP, van Eimeren T, et al. Serotonin 2A receptors and visual hallucinations in Parkinson disease. Arch Neurol. 2010;67(4):416-421.
6. Ravina B, Marder K, Fernandez HH, et al. Diagnostic criteria for psychosis in Parkinson’s disease: report of an NINDS, NIMH work group. Mov Disord. 2007;22(8):1061-1068.
7. Nordström AL, Farde L, Nyberg S, et al. D1, D2, and 5-HT2 receptor occupancy in relation to clozapine serum concentration: a PET study of schizophrenic patients. Am J Psychiatry. 1995;152(10):1444-1449.
8. Hacksell U, Burstein ES, McFarland K, et al. On the discovery and development of pimavanserin: a novel drug candidate for Parkinson’s psychosis. Neurochem Res. 2014;39(10):2008-2017.
9. Moreno JL, Holloway T, Albizu L, et al. Metabotropic glutamate mGlu2 receptor is necessary for the pharmacological and behavioral effects induced by hallucinogenic 5-HT2A receptor agonists. Neurosci Lett. 2011;493(3):76-79.
Pimavanserin is a potent 5-HT2A inverse agonist and 5-HT2C inverse agonist, with 5-fold greater affinity for the 5-HT2A receptor.1 Although antagonists block agonist actions at the receptor site, inverse agonists reduce the level of baseline constitutive activity seen in many G protein-coupled receptors. This medication is FDA approved for treating hallucinations and delusions associated with Parkinson’s disease (PD) psychosis (Table 1).1
In the pivotal 6-week clinical trial, pimavanserin significantly reduced positive symptoms seen in PD patients with psychosis (effect size = 0.50), with no evident impairment of motor function.2 Only 2 adverse effects occurred in ≥5% of pimavanserin-treated patients and at ≥2 times the rate of placebo: peripheral edema (7% vs 3% for placebo) and confusion (6% vs 3% for placebo). There was a mean increase in the QTc of 7.3 milliseconds compared with placebo in the pivotal phase III study.
Clinical implications
Despite numerous developments in the pharmacotherapeutics of psychotic disorders, patients with psychosis related to PD previously responded in a robust manner to only 1 antipsychotic, low-dosage clozapine (mean effect size, 0.80),2 with numerous failed trials for other atypical antipsychotics, including quetiapine.3,4 The pathophysiology of psychosis in PD patients is not related to dopamine agonist treatment, but is caused by the accumulation of cortical Lewy body burden, which results in loss of serotonergic signaling from dorsal raphe neurons. The net effect is up-regulation of postsynaptic 5-HT2A receptors.5 Psychosis is the most common cause of nursing home placement among PD patients without dementia.6
Receptor blocking. Based on the finding that clozapine in low dosages acts at 5-HT2A receptors,7 pimavanserin was designed to be a potent 5-HT2A inverse agonist, with more than 5-fold higher selectivity over 5-HT2C receptors, and no appreciable affinity for other serotonergic, adrenergic, dopaminergic, muscarinic, or histaminergic receptors8 (Table 2). The concept that 5-HT2A receptor stimulation can cause psychosis with prominent visual hallucinations is known from studies of LSD and other hallucinogenic compounds whose activity is blocked by 5-HT2A antagonists.
As an agent devoid of dopamine D2 antagonism, pimavanserin carries no risk of exacerbating motor symptoms, which was commonly seen with most atypical antipsychotics studied for psychosis in PD patients, except for clozapine and quetiapine.3 Although quetiapine did not cause motor effects, it proved ineffective in multiple studies (n = 153), likely because of the near absence of potent 5-HT2A binding.4
Pimavanserin also lacks:
- the hematologic monitoring requirement of clozapine
- clozapine’s risks of sedation, orthostasis, and anticholinergic and metabolic adverse effects.
Pimavanserin is significantly more potent than other non-antipsychotic psychotropics at the 5-HT2Areceptor, including doxepin (26 nM), trazodone (36 nM), and mirtazapine (60 nM).
Use in psychosis associated with PD. Recommended dosage is 34 mg once daily without titration (with or without food), based on results from a phase III clinical trial2 (because of the FDA breakthrough therapy designation for this compound, only 1 phase III trial was required). Pimavanserin produced significant improvement on the PD-adapted Scale for the Assessment of Positive Symptoms (SAPS-PD), a 9-item instrument extracted from the larger SAPS used in schizophrenia research. Specifically, pimavanserin was effective for both the hallucinations and delusions components of the SAPS-PD.
Pharmacologic profile, adverse effects. Pimavanserin lacks affinity for receptors other than 5-HT2A and 5-HT2C, leading to an absence of significant anticholinergic effects, orthostasis, or sedation in clinical trials.2 In all short-term clinical trials, the only common adverse reactions (incidence ≥5% and at least twice the rate of placebo) were peripheral edema (7% vs 2% placebo) and confusional state (6% vs 3% placebo).2 More than 300 patients have been treated for >6 months, >270 have been treated for at least 12 months, and >150 have been treated for at least 24 months with no adverse effects other than those seen in the short-term trials.1
There is a measurable impact on cardiac conduction seen in phase III data and in the thorough QT study. In the thorough QT study, 252 healthy participants received multiple dosages in a randomized, double-blind manner with positive controls.1 The maximum mean change from baseline was 13.5 milliseconds at dosages twice the recommended dosage, and the upper limit of the 90% CI was only slightly greater at 16.6 milliseconds. Subsequent kinetic analyses suggested concentration-dependent QTc interval prolongation in the therapeutic range, with a recommendation to halve the daily dosage in patients taking potent cytochrome P450 (CYP) 3A4 inhibitors.
In the 6-week, placebo-controlled effectiveness studies, mean increases in QTc interval were in the range of 5 to 8 milliseconds. There were sporadic reports of QTcF values ≥500 milliseconds, or changes from baseline QTc values ≥60 milliseconds in pimavanserin-treated participants, although the incidence generally was the same for pimavanserin and placebo groups. There were no reports of torsades de pointes or any differences from placebo in the incidence of adverse reactions associated with delayed ventricular repolarization.
How it works
The theory behind development of pimavanserin rests in the finding that low-dosage clozapine (6.25 to 50 mg/d) was effective for PD patients with psychosis (effect size 0.80).8 Although clozapine has high affinity for multiple sites, including histamine H1 receptors (Ki = 1.13 nM), α-1A and a α-2C adrenergic receptors (Ki = 1.62 nM and 6 nM, respectively), 5-HT2A receptors (Ki = 5.35 nM), and muscarinic M1 receptors (Ki = 6 nM), the hypothesized primary mechanism of clozapine’s effectiveness for PD psychosis at low dosages focused on the 5-HT2Areceptor. This idea was based on the knowledge that hallucinogens such as mescaline, psilocybin, and LSD are 5-HT2A agonists.9 This hallucinogenic activity can be blocked with 5-HT2A antagonists. Because of pimavanserin’s binding profile, the compound was studied as a treatment for psychosis in PD patients.
Pharmacokinetics
Pimavanserin demonstrates dose-proportional pharmacokinetics after a single oral dose as much as 7.5 times the recommended dosage. The pharmacokinetics of pimavanserin were similar in study participants (mean age, 72.4) and healthy controls, and a high-fat meal had no impact on the maximum blood levels (Cmax) or total drug exposure (area under the curve [AUC]).
The mean plasma half-lives for pimavanserin and its metabolite N-desmethyl-pimavanserin (AC-279) are 57 hours and 200 hours, respectively. Although the metabolite appears active in in vitro assays, it does not cross the blood-brain barrier to any appreciable extent, therefore contributing little to the clinical effect. The median time to maximum concentration (Tmax) of pimavanserin is 6 hours with a range of 4 to 24 hours, while the median Tmax of the primary metabolite AC-279 is 6 hours. The bioavailability of pimavanserin in an oral tablet or solution essentially is identical.
Pimavanserin is primarily metabolized via CYP3A4 to AC-279, and strong CYP3A4 inhibitors (eg, ketoconazole, itraconazole, clarithromycin, indinavir) increase pimavanserin Cmax by 1.5-fold, and AUC by 3-fold. In patients taking strong CYP3A4 inhibitors, the dosage of pimavanserin should be reduced by 50% to 17 mg/d. Conversely, patients on CYP3A4 inducers (eg, rifampin, carbamazepine, phenytoin) should be monitored for lack of efficacy; consider a dosage increase as necessary. Neither pimavanserin nor its metabolite, AC-279, are inhibitors or inducers of major CYP enzymes or drug transporters.
Efficacy in PD psychosis
Study 1. This 6-week, fixed dosage, double-blind, placebo-controlled trial was performed in adult PD patients age ≥40 with PD psychosis.2 Participants had to have (1) a PD diagnosis for at least 1 year and (2) psychotic symptoms that developed after diagnosis. Psychotic symptoms had to be present for at least 1 month, occurring at least weekly in the month before screening, and severe enough to warrant antipsychotic treatment. Baseline Mini-Mental State Examination score had to be ≥21 out of 30, with no evidence of delirium. Patients with dementia preceding or concurrent with the PD diagnosis were excluded. Antipsychotic treatments were not permitted during the trial.
After a 2-week nonpharmacotherapeutic lead-in phase that included a brief, daily psychosocial intervention by a caregiver, 199 patients who still met severity criteria were randomly allocated in a 1:1 manner to pimavanserin (34 mg of active drug, reported in the paper as 40 mg of pimavanserin tartrate) or matched placebo. Based on kinetic modeling and earlier clinical data, lower dosages (ie, 17 mg) were not explored, because they achieved only 50% of the steady state plasma levels thought to be required for efficacy.
The primary outcome was assessed by central, independent raters using the PD-adapted SAPS-PD. The efficacy analysis included 95 pimavanserin-treated individuals and 90 taking placebo. Baseline SAPS-PD scores were 14.7 ± 5.55 in the placebo group, and 15.9 ± 6.12 in the pimavanserin arm. Participants had a mean age of 72.4 and 94% white ethnicity across both cohorts; 42% of the placebo group and 33% of the pimavanserin group were female. Antipsychotic exposure in the 21 days prior to study entry were reported in 17% (n = 15) and 19% (n = 18) of the placebo and pimavanserin groups, respectively, with the most common agent being quetiapine (13 of 15, placebo, 16 of 18, pimavanserin). Approximately one-third of all participants were taking a cholinesterase inhibitor throughout the study.
Efficacy outcome. Pimavanserin was associated with a 5.79-point decrease in SAPS-PD scores compared with 2.73-point decrease for placebo (difference −3.06, 95% CI −4.91 to −1.20; P = .001). The effect size for this difference (Cohen’s d) was 0.50. The significant effect of pimavanserin vs placebo also was seen in separate analyses of the SAPS-PD subscore for hallucinations and delusions (effect size 0.50), and individually for hallucinations (effect size 0.45) and delusions (effect size 0.33). Separation from placebo appeared after the second week of pimavanserin treatment, and continued through the end of the study. There is unpublished data showing efficacy through week 10, and longer term, uncontrolled data consistent with sustained response. An exploratory analysis of caregiver burden demonstrated an effect size of 0.50.
Tolerability
The discontinuation rate because of adverse events for pimavanserin and placebo-treated patients was 10 patients in the pimavanserin group (4 due to psychotic symptoms within 10 days of starting the study drug) compared with 2 in the placebo group. There was no evidence of motor worsening in either group, demonstrated by the score on part II of the Unified Parkinson’s Disease Rating Scale (UPDRS) that captures self-reported activities of daily living, or on UPDRS part III (motor examination). Pimavanserin has no contraindications.
Unique clinical issues
Binding properties. Pimavanserin possesses potent 5-HT2A inverse agonist properties required to manage psychosis in PD patients, but lacks clozapine’s affinities for α-1 adrenergic, muscarinic, or histaminergic receptors that contribute to clozapine’s poor tolerability. Moreover, pimavanserin has no appreciable affinity for dopaminergic receptors, and therefore does not induce motor adverse effects.
Clozapine aside, all available atypical antipsychotics have proved ineffective for psychosis in PD patients, and most caused significant motor worsening.3 Although quetiapine does not cause motor effects, it has been shown to be ineffective for psychosis in PD patients in multiple trials.4
The effect size for clozapine response is large (0.80) in PD patients with psychosis, but tolerability issues and administrative burdens regarding patient and prescriber registration and routine hematological monitoring pose significant clinical barriers. Clozapine also lacks an FDA indication for this purpose, which may pose a hurdle to its use in certain treatment settings.
Why Rx? The reasons to prescribe pimavanserin for PD patients with psychosis likely include:
- absence of tolerability issues seen with the only other effective agent, clozapine
- lack of motor effects
- lack of administrative and monitoring burden related to clozapine prescribing
- only agent with FDA approval for hallucinations and delusions in PD patients with psychosis.
Dosing
The recommended dosage of pimavanserin is 34 mg/d administered as a single dose with or without food. There is no need for titration, and none was performed in the pivotal clinical trial. Given the long half-life (57 hours), steady state is not achieved until day 12, therefore initiation with a lower dosage might prolong the time to efficacy. There is no dosage adjustment required in patients with mild or moderate renal impairment, but pimavanserin treatment is not recommended in patients with severe renal impairment. Pimavanserin has not been evaluated in patients with hepatic impairment (using Child-Pugh criteria), and is not recommended for these patients.
Other key aspects of dosing to keep in mind.
- Because pimavanserin is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a strong CYP3A4 inhibitor; the recommended dosage is 17 mg/d when administered concomitantly with a strong CYP3A4 inhibitor.
- Because data are not available regarding concomitant use of pimavanserin with CYP3A4 inducers, patients should be monitored for lack of efficacy during concomitant use with a CYP3A4 inducer, and consideration given to a dosage increase.
Use in pregnancy and lactation. There are no data on the use of pimavanserin in pregnant women, but no developmental effects were seen when the drug was administered orally at 10 or 12 times the maximum recommended human dosage to rats or rabbits during organogenesis. Pimavanserin was not teratogenic in pregnant rats and rabbits. There is no information regarding the presence of pimavanserin in human breast milk.
Geriatric patients. No dosage adjustment is required for older patients. The study population in the pivotal trial was mean age 72.4 years.
Summing up
Before development of pimavanserin, clozapine was the only effective treatment for psychosis in PD patients. Despite clozapine’s robust effects across several trials, patients often were given ineffective medications, such as quetiapine, because of the administrative and tolerability barriers posed by clozapine use. Because psychosis is the most common cause of nursing home placement in non-demented PD patients, an agent with demonstrated efficacy and without the adverse effect profile of clozapine or monitoring requirements represents an enormous advance in the treatment of psychosis in PD patients.
Pimavanserin is a potent 5-HT2A inverse agonist and 5-HT2C inverse agonist, with 5-fold greater affinity for the 5-HT2A receptor.1 Although antagonists block agonist actions at the receptor site, inverse agonists reduce the level of baseline constitutive activity seen in many G protein-coupled receptors. This medication is FDA approved for treating hallucinations and delusions associated with Parkinson’s disease (PD) psychosis (Table 1).1
In the pivotal 6-week clinical trial, pimavanserin significantly reduced positive symptoms seen in PD patients with psychosis (effect size = 0.50), with no evident impairment of motor function.2 Only 2 adverse effects occurred in ≥5% of pimavanserin-treated patients and at ≥2 times the rate of placebo: peripheral edema (7% vs 3% for placebo) and confusion (6% vs 3% for placebo). There was a mean increase in the QTc of 7.3 milliseconds compared with placebo in the pivotal phase III study.
Clinical implications
Despite numerous developments in the pharmacotherapeutics of psychotic disorders, patients with psychosis related to PD previously responded in a robust manner to only 1 antipsychotic, low-dosage clozapine (mean effect size, 0.80),2 with numerous failed trials for other atypical antipsychotics, including quetiapine.3,4 The pathophysiology of psychosis in PD patients is not related to dopamine agonist treatment, but is caused by the accumulation of cortical Lewy body burden, which results in loss of serotonergic signaling from dorsal raphe neurons. The net effect is up-regulation of postsynaptic 5-HT2A receptors.5 Psychosis is the most common cause of nursing home placement among PD patients without dementia.6
Receptor blocking. Based on the finding that clozapine in low dosages acts at 5-HT2A receptors,7 pimavanserin was designed to be a potent 5-HT2A inverse agonist, with more than 5-fold higher selectivity over 5-HT2C receptors, and no appreciable affinity for other serotonergic, adrenergic, dopaminergic, muscarinic, or histaminergic receptors8 (Table 2). The concept that 5-HT2A receptor stimulation can cause psychosis with prominent visual hallucinations is known from studies of LSD and other hallucinogenic compounds whose activity is blocked by 5-HT2A antagonists.
As an agent devoid of dopamine D2 antagonism, pimavanserin carries no risk of exacerbating motor symptoms, which was commonly seen with most atypical antipsychotics studied for psychosis in PD patients, except for clozapine and quetiapine.3 Although quetiapine did not cause motor effects, it proved ineffective in multiple studies (n = 153), likely because of the near absence of potent 5-HT2A binding.4
Pimavanserin also lacks:
- the hematologic monitoring requirement of clozapine
- clozapine’s risks of sedation, orthostasis, and anticholinergic and metabolic adverse effects.
Pimavanserin is significantly more potent than other non-antipsychotic psychotropics at the 5-HT2Areceptor, including doxepin (26 nM), trazodone (36 nM), and mirtazapine (60 nM).
Use in psychosis associated with PD. Recommended dosage is 34 mg once daily without titration (with or without food), based on results from a phase III clinical trial2 (because of the FDA breakthrough therapy designation for this compound, only 1 phase III trial was required). Pimavanserin produced significant improvement on the PD-adapted Scale for the Assessment of Positive Symptoms (SAPS-PD), a 9-item instrument extracted from the larger SAPS used in schizophrenia research. Specifically, pimavanserin was effective for both the hallucinations and delusions components of the SAPS-PD.
Pharmacologic profile, adverse effects. Pimavanserin lacks affinity for receptors other than 5-HT2A and 5-HT2C, leading to an absence of significant anticholinergic effects, orthostasis, or sedation in clinical trials.2 In all short-term clinical trials, the only common adverse reactions (incidence ≥5% and at least twice the rate of placebo) were peripheral edema (7% vs 2% placebo) and confusional state (6% vs 3% placebo).2 More than 300 patients have been treated for >6 months, >270 have been treated for at least 12 months, and >150 have been treated for at least 24 months with no adverse effects other than those seen in the short-term trials.1
There is a measurable impact on cardiac conduction seen in phase III data and in the thorough QT study. In the thorough QT study, 252 healthy participants received multiple dosages in a randomized, double-blind manner with positive controls.1 The maximum mean change from baseline was 13.5 milliseconds at dosages twice the recommended dosage, and the upper limit of the 90% CI was only slightly greater at 16.6 milliseconds. Subsequent kinetic analyses suggested concentration-dependent QTc interval prolongation in the therapeutic range, with a recommendation to halve the daily dosage in patients taking potent cytochrome P450 (CYP) 3A4 inhibitors.
In the 6-week, placebo-controlled effectiveness studies, mean increases in QTc interval were in the range of 5 to 8 milliseconds. There were sporadic reports of QTcF values ≥500 milliseconds, or changes from baseline QTc values ≥60 milliseconds in pimavanserin-treated participants, although the incidence generally was the same for pimavanserin and placebo groups. There were no reports of torsades de pointes or any differences from placebo in the incidence of adverse reactions associated with delayed ventricular repolarization.
How it works
The theory behind development of pimavanserin rests in the finding that low-dosage clozapine (6.25 to 50 mg/d) was effective for PD patients with psychosis (effect size 0.80).8 Although clozapine has high affinity for multiple sites, including histamine H1 receptors (Ki = 1.13 nM), α-1A and a α-2C adrenergic receptors (Ki = 1.62 nM and 6 nM, respectively), 5-HT2A receptors (Ki = 5.35 nM), and muscarinic M1 receptors (Ki = 6 nM), the hypothesized primary mechanism of clozapine’s effectiveness for PD psychosis at low dosages focused on the 5-HT2Areceptor. This idea was based on the knowledge that hallucinogens such as mescaline, psilocybin, and LSD are 5-HT2A agonists.9 This hallucinogenic activity can be blocked with 5-HT2A antagonists. Because of pimavanserin’s binding profile, the compound was studied as a treatment for psychosis in PD patients.
Pharmacokinetics
Pimavanserin demonstrates dose-proportional pharmacokinetics after a single oral dose as much as 7.5 times the recommended dosage. The pharmacokinetics of pimavanserin were similar in study participants (mean age, 72.4) and healthy controls, and a high-fat meal had no impact on the maximum blood levels (Cmax) or total drug exposure (area under the curve [AUC]).
The mean plasma half-lives for pimavanserin and its metabolite N-desmethyl-pimavanserin (AC-279) are 57 hours and 200 hours, respectively. Although the metabolite appears active in in vitro assays, it does not cross the blood-brain barrier to any appreciable extent, therefore contributing little to the clinical effect. The median time to maximum concentration (Tmax) of pimavanserin is 6 hours with a range of 4 to 24 hours, while the median Tmax of the primary metabolite AC-279 is 6 hours. The bioavailability of pimavanserin in an oral tablet or solution essentially is identical.
Pimavanserin is primarily metabolized via CYP3A4 to AC-279, and strong CYP3A4 inhibitors (eg, ketoconazole, itraconazole, clarithromycin, indinavir) increase pimavanserin Cmax by 1.5-fold, and AUC by 3-fold. In patients taking strong CYP3A4 inhibitors, the dosage of pimavanserin should be reduced by 50% to 17 mg/d. Conversely, patients on CYP3A4 inducers (eg, rifampin, carbamazepine, phenytoin) should be monitored for lack of efficacy; consider a dosage increase as necessary. Neither pimavanserin nor its metabolite, AC-279, are inhibitors or inducers of major CYP enzymes or drug transporters.
Efficacy in PD psychosis
Study 1. This 6-week, fixed dosage, double-blind, placebo-controlled trial was performed in adult PD patients age ≥40 with PD psychosis.2 Participants had to have (1) a PD diagnosis for at least 1 year and (2) psychotic symptoms that developed after diagnosis. Psychotic symptoms had to be present for at least 1 month, occurring at least weekly in the month before screening, and severe enough to warrant antipsychotic treatment. Baseline Mini-Mental State Examination score had to be ≥21 out of 30, with no evidence of delirium. Patients with dementia preceding or concurrent with the PD diagnosis were excluded. Antipsychotic treatments were not permitted during the trial.
After a 2-week nonpharmacotherapeutic lead-in phase that included a brief, daily psychosocial intervention by a caregiver, 199 patients who still met severity criteria were randomly allocated in a 1:1 manner to pimavanserin (34 mg of active drug, reported in the paper as 40 mg of pimavanserin tartrate) or matched placebo. Based on kinetic modeling and earlier clinical data, lower dosages (ie, 17 mg) were not explored, because they achieved only 50% of the steady state plasma levels thought to be required for efficacy.
The primary outcome was assessed by central, independent raters using the PD-adapted SAPS-PD. The efficacy analysis included 95 pimavanserin-treated individuals and 90 taking placebo. Baseline SAPS-PD scores were 14.7 ± 5.55 in the placebo group, and 15.9 ± 6.12 in the pimavanserin arm. Participants had a mean age of 72.4 and 94% white ethnicity across both cohorts; 42% of the placebo group and 33% of the pimavanserin group were female. Antipsychotic exposure in the 21 days prior to study entry were reported in 17% (n = 15) and 19% (n = 18) of the placebo and pimavanserin groups, respectively, with the most common agent being quetiapine (13 of 15, placebo, 16 of 18, pimavanserin). Approximately one-third of all participants were taking a cholinesterase inhibitor throughout the study.
Efficacy outcome. Pimavanserin was associated with a 5.79-point decrease in SAPS-PD scores compared with 2.73-point decrease for placebo (difference −3.06, 95% CI −4.91 to −1.20; P = .001). The effect size for this difference (Cohen’s d) was 0.50. The significant effect of pimavanserin vs placebo also was seen in separate analyses of the SAPS-PD subscore for hallucinations and delusions (effect size 0.50), and individually for hallucinations (effect size 0.45) and delusions (effect size 0.33). Separation from placebo appeared after the second week of pimavanserin treatment, and continued through the end of the study. There is unpublished data showing efficacy through week 10, and longer term, uncontrolled data consistent with sustained response. An exploratory analysis of caregiver burden demonstrated an effect size of 0.50.
Tolerability
The discontinuation rate because of adverse events for pimavanserin and placebo-treated patients was 10 patients in the pimavanserin group (4 due to psychotic symptoms within 10 days of starting the study drug) compared with 2 in the placebo group. There was no evidence of motor worsening in either group, demonstrated by the score on part II of the Unified Parkinson’s Disease Rating Scale (UPDRS) that captures self-reported activities of daily living, or on UPDRS part III (motor examination). Pimavanserin has no contraindications.
Unique clinical issues
Binding properties. Pimavanserin possesses potent 5-HT2A inverse agonist properties required to manage psychosis in PD patients, but lacks clozapine’s affinities for α-1 adrenergic, muscarinic, or histaminergic receptors that contribute to clozapine’s poor tolerability. Moreover, pimavanserin has no appreciable affinity for dopaminergic receptors, and therefore does not induce motor adverse effects.
Clozapine aside, all available atypical antipsychotics have proved ineffective for psychosis in PD patients, and most caused significant motor worsening.3 Although quetiapine does not cause motor effects, it has been shown to be ineffective for psychosis in PD patients in multiple trials.4
The effect size for clozapine response is large (0.80) in PD patients with psychosis, but tolerability issues and administrative burdens regarding patient and prescriber registration and routine hematological monitoring pose significant clinical barriers. Clozapine also lacks an FDA indication for this purpose, which may pose a hurdle to its use in certain treatment settings.
Why Rx? The reasons to prescribe pimavanserin for PD patients with psychosis likely include:
- absence of tolerability issues seen with the only other effective agent, clozapine
- lack of motor effects
- lack of administrative and monitoring burden related to clozapine prescribing
- only agent with FDA approval for hallucinations and delusions in PD patients with psychosis.
Dosing
The recommended dosage of pimavanserin is 34 mg/d administered as a single dose with or without food. There is no need for titration, and none was performed in the pivotal clinical trial. Given the long half-life (57 hours), steady state is not achieved until day 12, therefore initiation with a lower dosage might prolong the time to efficacy. There is no dosage adjustment required in patients with mild or moderate renal impairment, but pimavanserin treatment is not recommended in patients with severe renal impairment. Pimavanserin has not been evaluated in patients with hepatic impairment (using Child-Pugh criteria), and is not recommended for these patients.
Other key aspects of dosing to keep in mind.
- Because pimavanserin is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a strong CYP3A4 inhibitor; the recommended dosage is 17 mg/d when administered concomitantly with a strong CYP3A4 inhibitor.
- Because data are not available regarding concomitant use of pimavanserin with CYP3A4 inducers, patients should be monitored for lack of efficacy during concomitant use with a CYP3A4 inducer, and consideration given to a dosage increase.
Use in pregnancy and lactation. There are no data on the use of pimavanserin in pregnant women, but no developmental effects were seen when the drug was administered orally at 10 or 12 times the maximum recommended human dosage to rats or rabbits during organogenesis. Pimavanserin was not teratogenic in pregnant rats and rabbits. There is no information regarding the presence of pimavanserin in human breast milk.
Geriatric patients. No dosage adjustment is required for older patients. The study population in the pivotal trial was mean age 72.4 years.
Summing up
Before development of pimavanserin, clozapine was the only effective treatment for psychosis in PD patients. Despite clozapine’s robust effects across several trials, patients often were given ineffective medications, such as quetiapine, because of the administrative and tolerability barriers posed by clozapine use. Because psychosis is the most common cause of nursing home placement in non-demented PD patients, an agent with demonstrated efficacy and without the adverse effect profile of clozapine or monitoring requirements represents an enormous advance in the treatment of psychosis in PD patients.
1. Nuplazid [package insert]. San Diego, CA: Acadia Pharmaceuticals Inc.; 2016.
2. Cummings J, Isaacson S, Mills R, et al. Pimavanserin for patients with Parkinson’s disease psychosis: a randomised, placebo-controlled phase 3 trial. [Erratum in Lancet. 2014;384(9937):28]. Lancet. 2014;383(9916):533-540.
3. Borek LL, Friedman JH. Treating psychosis in movement disorder patients: a review. Expert Opin Pharmacother. 2014;15(11):1553-1564.
4. Desmarais P, Massoud F, Filion J, et al. Quetiapine for psychosis in Parkinson disease and neurodegenerative parkinsonian disorders: a systematic review. J Geriatr Psychiatry Neurol. 2016;29(4):227-236.
5. Ballanger B, Strafella AP, van Eimeren T, et al. Serotonin 2A receptors and visual hallucinations in Parkinson disease. Arch Neurol. 2010;67(4):416-421.
6. Ravina B, Marder K, Fernandez HH, et al. Diagnostic criteria for psychosis in Parkinson’s disease: report of an NINDS, NIMH work group. Mov Disord. 2007;22(8):1061-1068.
7. Nordström AL, Farde L, Nyberg S, et al. D1, D2, and 5-HT2 receptor occupancy in relation to clozapine serum concentration: a PET study of schizophrenic patients. Am J Psychiatry. 1995;152(10):1444-1449.
8. Hacksell U, Burstein ES, McFarland K, et al. On the discovery and development of pimavanserin: a novel drug candidate for Parkinson’s psychosis. Neurochem Res. 2014;39(10):2008-2017.
9. Moreno JL, Holloway T, Albizu L, et al. Metabotropic glutamate mGlu2 receptor is necessary for the pharmacological and behavioral effects induced by hallucinogenic 5-HT2A receptor agonists. Neurosci Lett. 2011;493(3):76-79.
1. Nuplazid [package insert]. San Diego, CA: Acadia Pharmaceuticals Inc.; 2016.
2. Cummings J, Isaacson S, Mills R, et al. Pimavanserin for patients with Parkinson’s disease psychosis: a randomised, placebo-controlled phase 3 trial. [Erratum in Lancet. 2014;384(9937):28]. Lancet. 2014;383(9916):533-540.
3. Borek LL, Friedman JH. Treating psychosis in movement disorder patients: a review. Expert Opin Pharmacother. 2014;15(11):1553-1564.
4. Desmarais P, Massoud F, Filion J, et al. Quetiapine for psychosis in Parkinson disease and neurodegenerative parkinsonian disorders: a systematic review. J Geriatr Psychiatry Neurol. 2016;29(4):227-236.
5. Ballanger B, Strafella AP, van Eimeren T, et al. Serotonin 2A receptors and visual hallucinations in Parkinson disease. Arch Neurol. 2010;67(4):416-421.
6. Ravina B, Marder K, Fernandez HH, et al. Diagnostic criteria for psychosis in Parkinson’s disease: report of an NINDS, NIMH work group. Mov Disord. 2007;22(8):1061-1068.
7. Nordström AL, Farde L, Nyberg S, et al. D1, D2, and 5-HT2 receptor occupancy in relation to clozapine serum concentration: a PET study of schizophrenic patients. Am J Psychiatry. 1995;152(10):1444-1449.
8. Hacksell U, Burstein ES, McFarland K, et al. On the discovery and development of pimavanserin: a novel drug candidate for Parkinson’s psychosis. Neurochem Res. 2014;39(10):2008-2017.
9. Moreno JL, Holloway T, Albizu L, et al. Metabotropic glutamate mGlu2 receptor is necessary for the pharmacological and behavioral effects induced by hallucinogenic 5-HT2A receptor agonists. Neurosci Lett. 2011;493(3):76-79.
Where to find guidance on using pharmacogenomics in psychiatric practice
Pharmacogenomics—the study of how genetic variability influences drug response—is increasingly being used to personalize pharmacotherapy. Used in the context of other clinical variables, genetic-based drug selection and dosing could help clinicians choose the right therapy for a patient, thus minimizing the incidence of treatment failure and intolerable side effects. Pharmacogenomics could be particularly useful in psychiatric pharmacotherapy, where response rates are low and the risk of adverse effects and nonadherence is high.
Despite the potential benefits of pharmacogenetic testing, many barriers prevent its routine use in practice, including a lack of knowledge about how to (1) order gene tests, (2) interpret results for an individual patient, and (3) apply those results to care. To help bridge this knowledge gap, we list practical, freely available pharmacogenomics resources that a psychiatric practitioner can use.
CPIC guidelines
The Clinical Pharmacogenetics Implementation Consortium (CPIC) is an international collaboration of pharmacogenomics experts that publishes clinical practice guidelines on using pharmacogenetic test results to optimize drug therapy.1 Note: These guidelines do not address when tests should be ordered, but rather how results should be used to guide prescribing.

- how to convert genotype to phenotype
- how to modify drug selection or dosing based on these results
- the level of evidence for each recommendation.
CPIC guidelines and supplementary information are available on the CPIC Web site (https://www.cpicpgx.org) and are updated regularly. Table 1 provides current CPIC guidelines for neuropsychiatric drugs.
PharmGKB
Providing searchable annotations of pharmacogenetic variants, PharmGKB summarizes the clinical implications of important pharmacogenes, and includes FDA drug labels containing pharmacogenomics information (https://www.pharmgkb.org).2 The Web site also provides users with evidence-based figures illustrating the pharmacokinetic and pharmacodynamic pathways of drugs that have pharmacogenetic implications.
PharmGKB is an excellent resource to consult for a summary of available evidence when a CPIC guideline does not exist for a given gene or drug.
Other resources
Table 23-8 lists other online resources for practitioners to aid in advancing pharmacogenomics knowledge as it relates to practice.
Putting guidance to best use
Familiarity with resources such as CPIC guidelines and PharmGKB can help ensure that patients with pharmacogenetic test results receive genetically tailored therapy that is more likely to be effective and less likely to cause adverse effects.9,10
1. Caudle KE, Klein TE, Hoffman JM, et al. Incorporation of pharmacogenomics into routine clinical practice: the Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline development process. Curr Drug Metab. 2014;15(2):209-217.
2. Thorn CF, Klein TE, Altman RB. PharmGKB: the Pharmacogenomics Knowledge Base. Methods Mol Biol. 2013;1015:311-320.
3. American Society of Health-System Pharmacists. Pharmacogenomics resource center. http://www.ashp.org/menu/PracticePolicy/ResourceCenters/Emerging-Sciences/Pharmacogenomics.aspx. Accessed July 21, 2016.
4. Genomics. Food and Drug Administration. http://www.fda.gov/drugs/scienceresearch/researchareas/pharmacogenetics. Updated May 5, 2016. Accessed July 27, 2016.
5. National Human Genome Research Institute. Genetics/genomics competency center. http://g-2-c-2.org. Accessed July 21, 2016.
6. National Human Genome Research Institute. https://www.genome.gov. Accessed July 21, 2016.
7. Implementation resources for professionals. St. Jude Children’s Research Hospital. https://www.stjude.org/research/clinical-trials/pg4kds-pharmaceutical-science/implementation-resources-for-professionals.html. Accessed July 21, 2016.
8. SNPits study summaries. University of Florida Health Personalized Medicine Program. http://personalizedmedicine.ufhealth.org/snp-its/pharmacogenomics-study-summaries. Updated June 1, 2016. Accessed July 21, 2016.
9. Zhang G, Zhang Y, Ling Y. Web resources for pharmacogenomics. Genomics Proteomics Bioinformatics. 2015;13(1):51-54.
10. Johnson G. Leading clinical pharmacogenomics implementation: advancing pharmacy practice. Am J Health Syst Pharm. 2015;72(15):1324-1328.
Pharmacogenomics—the study of how genetic variability influences drug response—is increasingly being used to personalize pharmacotherapy. Used in the context of other clinical variables, genetic-based drug selection and dosing could help clinicians choose the right therapy for a patient, thus minimizing the incidence of treatment failure and intolerable side effects. Pharmacogenomics could be particularly useful in psychiatric pharmacotherapy, where response rates are low and the risk of adverse effects and nonadherence is high.
Despite the potential benefits of pharmacogenetic testing, many barriers prevent its routine use in practice, including a lack of knowledge about how to (1) order gene tests, (2) interpret results for an individual patient, and (3) apply those results to care. To help bridge this knowledge gap, we list practical, freely available pharmacogenomics resources that a psychiatric practitioner can use.
CPIC guidelines
The Clinical Pharmacogenetics Implementation Consortium (CPIC) is an international collaboration of pharmacogenomics experts that publishes clinical practice guidelines on using pharmacogenetic test results to optimize drug therapy.1 Note: These guidelines do not address when tests should be ordered, but rather how results should be used to guide prescribing.
- how to convert genotype to phenotype
- how to modify drug selection or dosing based on these results
- the level of evidence for each recommendation.
CPIC guidelines and supplementary information are available on the CPIC Web site (https://www.cpicpgx.org) and are updated regularly. Table 1 provides current CPIC guidelines for neuropsychiatric drugs.
PharmGKB
Providing searchable annotations of pharmacogenetic variants, PharmGKB summarizes the clinical implications of important pharmacogenes, and includes FDA drug labels containing pharmacogenomics information (https://www.pharmgkb.org).2 The Web site also provides users with evidence-based figures illustrating the pharmacokinetic and pharmacodynamic pathways of drugs that have pharmacogenetic implications.
PharmGKB is an excellent resource to consult for a summary of available evidence when a CPIC guideline does not exist for a given gene or drug.
Other resources
Table 23-8 lists other online resources for practitioners to aid in advancing pharmacogenomics knowledge as it relates to practice.
Putting guidance to best use
Familiarity with resources such as CPIC guidelines and PharmGKB can help ensure that patients with pharmacogenetic test results receive genetically tailored therapy that is more likely to be effective and less likely to cause adverse effects.9,10
Pharmacogenomics—the study of how genetic variability influences drug response—is increasingly being used to personalize pharmacotherapy. Used in the context of other clinical variables, genetic-based drug selection and dosing could help clinicians choose the right therapy for a patient, thus minimizing the incidence of treatment failure and intolerable side effects. Pharmacogenomics could be particularly useful in psychiatric pharmacotherapy, where response rates are low and the risk of adverse effects and nonadherence is high.
Despite the potential benefits of pharmacogenetic testing, many barriers prevent its routine use in practice, including a lack of knowledge about how to (1) order gene tests, (2) interpret results for an individual patient, and (3) apply those results to care. To help bridge this knowledge gap, we list practical, freely available pharmacogenomics resources that a psychiatric practitioner can use.
CPIC guidelines
The Clinical Pharmacogenetics Implementation Consortium (CPIC) is an international collaboration of pharmacogenomics experts that publishes clinical practice guidelines on using pharmacogenetic test results to optimize drug therapy.1 Note: These guidelines do not address when tests should be ordered, but rather how results should be used to guide prescribing.
- how to convert genotype to phenotype
- how to modify drug selection or dosing based on these results
- the level of evidence for each recommendation.
CPIC guidelines and supplementary information are available on the CPIC Web site (https://www.cpicpgx.org) and are updated regularly. Table 1 provides current CPIC guidelines for neuropsychiatric drugs.
PharmGKB
Providing searchable annotations of pharmacogenetic variants, PharmGKB summarizes the clinical implications of important pharmacogenes, and includes FDA drug labels containing pharmacogenomics information (https://www.pharmgkb.org).2 The Web site also provides users with evidence-based figures illustrating the pharmacokinetic and pharmacodynamic pathways of drugs that have pharmacogenetic implications.
PharmGKB is an excellent resource to consult for a summary of available evidence when a CPIC guideline does not exist for a given gene or drug.
Other resources
Table 23-8 lists other online resources for practitioners to aid in advancing pharmacogenomics knowledge as it relates to practice.
Putting guidance to best use
Familiarity with resources such as CPIC guidelines and PharmGKB can help ensure that patients with pharmacogenetic test results receive genetically tailored therapy that is more likely to be effective and less likely to cause adverse effects.9,10
1. Caudle KE, Klein TE, Hoffman JM, et al. Incorporation of pharmacogenomics into routine clinical practice: the Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline development process. Curr Drug Metab. 2014;15(2):209-217.
2. Thorn CF, Klein TE, Altman RB. PharmGKB: the Pharmacogenomics Knowledge Base. Methods Mol Biol. 2013;1015:311-320.
3. American Society of Health-System Pharmacists. Pharmacogenomics resource center. http://www.ashp.org/menu/PracticePolicy/ResourceCenters/Emerging-Sciences/Pharmacogenomics.aspx. Accessed July 21, 2016.
4. Genomics. Food and Drug Administration. http://www.fda.gov/drugs/scienceresearch/researchareas/pharmacogenetics. Updated May 5, 2016. Accessed July 27, 2016.
5. National Human Genome Research Institute. Genetics/genomics competency center. http://g-2-c-2.org. Accessed July 21, 2016.
6. National Human Genome Research Institute. https://www.genome.gov. Accessed July 21, 2016.
7. Implementation resources for professionals. St. Jude Children’s Research Hospital. https://www.stjude.org/research/clinical-trials/pg4kds-pharmaceutical-science/implementation-resources-for-professionals.html. Accessed July 21, 2016.
8. SNPits study summaries. University of Florida Health Personalized Medicine Program. http://personalizedmedicine.ufhealth.org/snp-its/pharmacogenomics-study-summaries. Updated June 1, 2016. Accessed July 21, 2016.
9. Zhang G, Zhang Y, Ling Y. Web resources for pharmacogenomics. Genomics Proteomics Bioinformatics. 2015;13(1):51-54.
10. Johnson G. Leading clinical pharmacogenomics implementation: advancing pharmacy practice. Am J Health Syst Pharm. 2015;72(15):1324-1328.
1. Caudle KE, Klein TE, Hoffman JM, et al. Incorporation of pharmacogenomics into routine clinical practice: the Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline development process. Curr Drug Metab. 2014;15(2):209-217.
2. Thorn CF, Klein TE, Altman RB. PharmGKB: the Pharmacogenomics Knowledge Base. Methods Mol Biol. 2013;1015:311-320.
3. American Society of Health-System Pharmacists. Pharmacogenomics resource center. http://www.ashp.org/menu/PracticePolicy/ResourceCenters/Emerging-Sciences/Pharmacogenomics.aspx. Accessed July 21, 2016.
4. Genomics. Food and Drug Administration. http://www.fda.gov/drugs/scienceresearch/researchareas/pharmacogenetics. Updated May 5, 2016. Accessed July 27, 2016.
5. National Human Genome Research Institute. Genetics/genomics competency center. http://g-2-c-2.org. Accessed July 21, 2016.
6. National Human Genome Research Institute. https://www.genome.gov. Accessed July 21, 2016.
7. Implementation resources for professionals. St. Jude Children’s Research Hospital. https://www.stjude.org/research/clinical-trials/pg4kds-pharmaceutical-science/implementation-resources-for-professionals.html. Accessed July 21, 2016.
8. SNPits study summaries. University of Florida Health Personalized Medicine Program. http://personalizedmedicine.ufhealth.org/snp-its/pharmacogenomics-study-summaries. Updated June 1, 2016. Accessed July 21, 2016.
9. Zhang G, Zhang Y, Ling Y. Web resources for pharmacogenomics. Genomics Proteomics Bioinformatics. 2015;13(1):51-54.
10. Johnson G. Leading clinical pharmacogenomics implementation: advancing pharmacy practice. Am J Health Syst Pharm. 2015;72(15):1324-1328.
Need-to-know information for the 2016-2017 flu season
The Advisory Committee on Immunization Practices (ACIP) took the unusual step at its June 2016 meeting of recommending against using a currently licensed vaccine, live attenuated influenza vaccine (LAIV), in the 2016-2017 influenza season.1 ACIP based its recommendation on surveillance data collected by the US Influenza Vaccine Effectiveness Network of the Centers for Disease Control and Prevention (CDC), which showed poor effectiveness by the LAIV vaccine among children and adolescents during the past 3 years.
The US Food and Drug Administration (FDA), however, has chosen not to take any action on this matter, saying on its Web site it “has determined that specific regulatory action is not warranted at this time. This determination is based on FDA’s review of manufacturing and clinical data supporting licensure … the totality of the evidence presented at the ACIP meeting, taking into account the inherent limitations of observational studies conducted to evaluate influenza vaccine effectiveness, as well as the well-known variability of influenza vaccine effectiveness across influenza seasons.”2
CDC data for the 2015-2016 flu season showed the effectiveness of LAIV to be just 3% among children 2 years through 17 years of age.3 The reason for this apparent lack of effectiveness is unknown. Other LAIV-effectiveness studies conducted in the 2015-2016 season—one each, in the United States, United Kingdom, and Finland—had results that differed from the CDC surveillance data, with effectiveness ranging from 46% to 58% against all strains combined.2 These results are comparable to vaccine effectiveness found in observational studies in children for both LAIV and inactivated influenza vaccines (IIV) in prior seasons.2
Vaccine manufacturers had projected that 171 to 176 million doses of flu vaccine, in all forms, would be available in the United States during the 2016-2017 season.3 LAIV accounts for about 8% of the total supply of influenza vaccine in the United States,3 and ACIP’s recommendation is not expected to create shortages of other options for the upcoming season. However, the LAIV accounts for one-third of flu vaccines administered to children, and clinicians who provide vaccinations to children have already ordered their vaccine supplies for the upcoming season. Also, it is not clear if children who have previously received the LAIV product will now accept other options for influenza vaccination—all of which involve an injection.
Whether the recommendation against LAIV will continue after this season is also unknown.
What happened during the 2015-2016 influenza season?
The 2015-2016 influenza season was relatively mild with the peak activity occurring in March, somewhat later than in previous years. The circulating influenza strains matched closely to those in the vaccine, making it more effective than the previous year’s vaccine. The predominant circulating strain was A (H1N1), accounting for 58% of illness; A (H3N2) caused 6% of cases and all B types together accounted for 34%.4 The hospitalization rate for all ages was 31.3/100,000 compared with 64.1 the year before.5 There were 85 pediatric deaths compared with 148 in 2014-2015.6
Vaccine effectiveness among all age groups and against all circulating strains was 47%.4 No major vaccine safety concerns were detected. Among those who received IIV3, there was a slight increase in the incidence of Guillain-Barré syndrome of 2.6 cases per one million vaccines.7
Other recommendations for 2016-2017
Once again, ACIP recommends influenza vaccine for all individuals 6 months and older.8 The CDC additionally specifies particular groups that should not skip vaccination given that they are at high risk of complications from influenza infection or because they could expose high-risk individuals to infection (TABLE 1).9
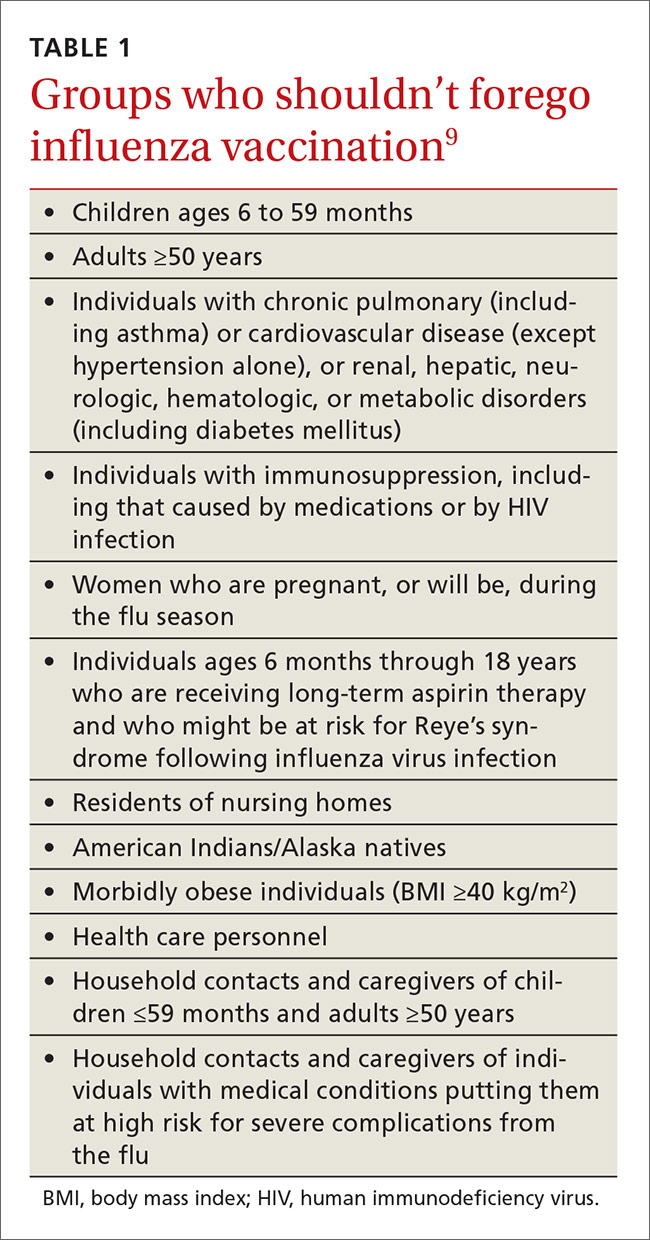
There will continue to be a selection of trivalent and quadrivalent influenza vaccine products in 2016-2017. Trivalent products will contain 3 viral strains: A/California/7/2009 (H1N1), A/Hong Kong/4801/2014 (H3N2) and B/Brisbane/60/2008.10 The quadrivalent products will contain those 3 antigens plus B/Phuket/3073/2013.10 The H3N2 strain is different from the one in last year’s vaccine. Each year, influenza experts analyze surveillance data to predict which circulating strains will predominate in North America, and these antigens constitute the vaccine formulation. The accuracy of this prediction in large part determines how effective the vaccine will be that season.
Two new vaccines have been approved for use in the United States. A quadrivalent cell culture inactivated vaccine (CCIV4), Flucelvax, was licensed in May 2016. It is prepared from virus propagated in canine kidney cells, not with an egg-based production process. It is approved for use in individuals 4 years of age and older.8 Fluad, an adjuvanted trivalent inactivated influenza vaccine, was licensed in late 2015 for individuals 65 years of age and older.8 This is the first adjuvanted influenza vaccine licensed in the United States and will compete with high-dose quadrivalent vaccine for use in older adults. ACIP does not express a preference for any vaccine in this age group.
Two other vaccines should also be available by this fall: Flublok, a quadrivalent recombinant influenza vaccine for individuals 18 years and older, and Flulaval, a quadrivalent inactivated influenza vaccine, for individuals 6 months of age and older. TABLE 211 lists approved influenza vaccines.
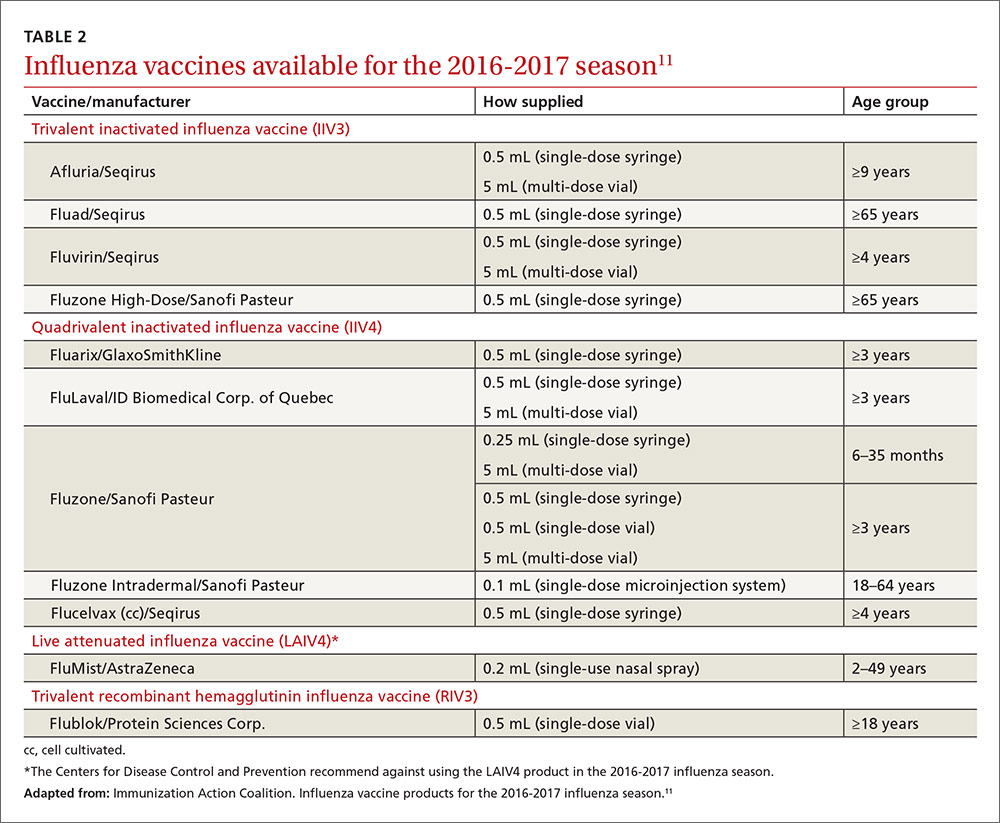
Issues specific to children
Deciding how many vaccine doses children need has been further simplified. Children younger than 9 years need 2 doses if they have received fewer than 2 doses of trivalent or quadrivalent influenza vaccine before July 1, 2016. The interval between the 2 doses should be at least 4 weeks. The 2 doses do not have to be the same product; importantly, do not delay a second dose just to obtain the same product used for the first dose. Also, one dose can be trivalent and the other one quadrivalent, although this offers less-than-optimal protection against the B-virus that is only in the quadrivalent product.
Children younger than 9 years require only one dose if they have received 2 or more total doses of trivalent or quadrivalent influenza vaccine before July 1, 2016. The 2 previous doses need not have been received during the same influenza season or consecutive influenza seasons.
In children ages 6 through 23 months there is a slight increased risk of febrile seizure if the influenza vaccine is co-administered with other vaccines, specifically pneumococcal conjugate vaccine (PCV 13) and diphtheria-tetanus-acellular-pertussis (DTaP). The 3 vaccines administered at the same time result in 30 febrile seizures per 100,000 children;12 the rate is lower when influenza vaccine is co-administered with only one of the others. ACIP believes that the risk of a febrile seizure, which does no long-term harm, does not warrant delaying vaccines that could be co-administered.13
Egg allergy requires no special precautions
Evidence continues to grow that influenza vaccine products do not contain enough egg protein to cause significant problems in those with a history of egg allergies. This year’s recommendations state that no special precautions are needed regarding the anatomic site of immunization or the length of observation after administering influenza vaccine in those with a history of allergies to eggs, no matter how severe. All vaccine-administration facilities should be able to respond to any hypersensitivity reaction, and the standard waiting time for observation after all vaccinations is 15 minutes.

Antiviral medications for treatment or prevention
Most influenza strains circulating in 2016-2017 are expected to remain sensitive to oseltamivir and zanamivir, which can be used for treatment or disease prevention. A third neuraminidase inhibitor, peramivir, is available for intravenous use in adults 18 and older. Treatment is recommended for those who have confirmed or suspected influenza and are at high risk for complications (TABLE 3).14 Consideration of antiviral chemoprevention is recommended under certain circumstances (TABLE 4).15,16 The CDC influenza Web site lists recommended doses and duration for each antiviral for treatment and chemoprevention.15

1. Grohskopf LA, Sokolow LZ, Broder KR, et al. Prevention and control of seasonal influenza with vaccines: Recommendations of the Advisory Committee on Immunization Practices—United States, 2016-17 influenza season. MMWR Recomm Rep. 2016;65:1-54.
2. U.S. Food and Drug Administration. FDA information regarding FluMist quadrivalent vaccine. Available at: http://www.fda.gov/BiologicsBloodVaccines/Vaccines/ApprovedProducts/ucm508761.htm. Accessed July 13, 2016.
3. Centers for Disease Control and Prevention. ACIP votes down use of LAIV for 2016-2017 flu season. Available at: http://www.cdc.gov/media/releases/2016/s0622-laiv-flu.html. Accessed July 13, 2016.
4. Flannery B, Chung J. Influenza vaccine effectiveness, including LAIV vs IIV in children and adolescents, US Flu VE Network, 2015-2016. Presented at: meeting of the Advisory Committee on Immunization Practices; June 22, 2016; Atlanta, GA. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/vaccines/acip/meetings/downloads/slides-2016-06/influenza-05-flannery.pdf. Accessed July 22, 2016.
5. Centers for Disease Control and Prevention. FluView. Laboratory-confirmed influenza hospitalizations. Available at: http://gis.cdc.gov/GRASP/Fluview/FluHospRates.html. Accessed July 25, 2016.
6. Centers for Disease Control and Prevention. FluView. Number of influenza-associated pediatric deaths by week of death. Available at: http://gis.cdc.gov/GRASP/Fluview/PedFluDeath.html. Accessed July 25, 2016.
7. Shimabukuro T. End-of-season update: 2015-2016 influenza vaccine safety monitoring. Presented at: meeting of the Advisory Committee on Immunization Practices; June 22, 2016; Atlanta, GA. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/vaccines/acip/meetings/downloads/slides-2016-06/influenza-04-shimabukuro.pdf. Accessed July 22, 2016.
8. Grohskopf L. Proposed recommendations 2016-2017 influenza season. Presented at: meeting of the Advisory Committee on Immunization Practices; June 22, 2016; Atlanta, GA. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/vaccines/acip/meetings/downloads/slides-2016-06/influenza-08-grohskopf.pdf. Accessed July 22, 2016.
9. Centers for Disease Control and Prevention. Influenza vaccination: a summary for clinicians. Available at: http://www.cdc.gov/flu/professionals/vaccination/vax-summary.htm. Accessed July 13, 2016.
10. Centers for Disease Control and Prevention. What you should know for the 2016-2017 influenza season. Available at: http://www.cdc.gov/flu/about/season/flu-season-2016-2017.htm. Accessed July 13, 2016.
11. Immunization Action Coalition. Influenza vaccine products for the 2016-2017 influenza season. Available at: http://www.immunize.org/catg.d/p4072.pdf. Accessed July 13, 2016.
12. Duffy J, Weintraub E, Hambidge SJ, et al. Febrile seizure risk after vaccination in children 6 to 23 months. Pediatrics. 2016;138.
13. Centers for Disease Control and Prevention. Childhood vaccines and febrile seizures. Available at: http://www.cdc.gov/vaccinesafety/concerns/febrile-seizures.html. Accessed August 11, 2016.
14. Centers for Disease Control and Prevention. Use of antivirals. Background and guidance on the use of influenza antiviral agents. Available at: http://www.cdc.gov/flu/professionals/antivirals/antiviral-use-influenza.htm. Accessed July 13, 2016.
15. Centers for Disease Control and Prevention. Influenza antiviral medications: summary for clinicians. Available at: http://www.cdc.gov/flu/professionals/antivirals/summary-clinicians.htm. Accessed July 13, 2016.
16. American Academy of Pediatrics. Recommendations for prevention and control of influenza in children, 2015-2016. Pediatrics. 2015;136:792-808.
The Advisory Committee on Immunization Practices (ACIP) took the unusual step at its June 2016 meeting of recommending against using a currently licensed vaccine, live attenuated influenza vaccine (LAIV), in the 2016-2017 influenza season.1 ACIP based its recommendation on surveillance data collected by the US Influenza Vaccine Effectiveness Network of the Centers for Disease Control and Prevention (CDC), which showed poor effectiveness by the LAIV vaccine among children and adolescents during the past 3 years.
The US Food and Drug Administration (FDA), however, has chosen not to take any action on this matter, saying on its Web site it “has determined that specific regulatory action is not warranted at this time. This determination is based on FDA’s review of manufacturing and clinical data supporting licensure … the totality of the evidence presented at the ACIP meeting, taking into account the inherent limitations of observational studies conducted to evaluate influenza vaccine effectiveness, as well as the well-known variability of influenza vaccine effectiveness across influenza seasons.”2
CDC data for the 2015-2016 flu season showed the effectiveness of LAIV to be just 3% among children 2 years through 17 years of age.3 The reason for this apparent lack of effectiveness is unknown. Other LAIV-effectiveness studies conducted in the 2015-2016 season—one each, in the United States, United Kingdom, and Finland—had results that differed from the CDC surveillance data, with effectiveness ranging from 46% to 58% against all strains combined.2 These results are comparable to vaccine effectiveness found in observational studies in children for both LAIV and inactivated influenza vaccines (IIV) in prior seasons.2
Vaccine manufacturers had projected that 171 to 176 million doses of flu vaccine, in all forms, would be available in the United States during the 2016-2017 season.3 LAIV accounts for about 8% of the total supply of influenza vaccine in the United States,3 and ACIP’s recommendation is not expected to create shortages of other options for the upcoming season. However, the LAIV accounts for one-third of flu vaccines administered to children, and clinicians who provide vaccinations to children have already ordered their vaccine supplies for the upcoming season. Also, it is not clear if children who have previously received the LAIV product will now accept other options for influenza vaccination—all of which involve an injection.
Whether the recommendation against LAIV will continue after this season is also unknown.
What happened during the 2015-2016 influenza season?
The 2015-2016 influenza season was relatively mild with the peak activity occurring in March, somewhat later than in previous years. The circulating influenza strains matched closely to those in the vaccine, making it more effective than the previous year’s vaccine. The predominant circulating strain was A (H1N1), accounting for 58% of illness; A (H3N2) caused 6% of cases and all B types together accounted for 34%.4 The hospitalization rate for all ages was 31.3/100,000 compared with 64.1 the year before.5 There were 85 pediatric deaths compared with 148 in 2014-2015.6
Vaccine effectiveness among all age groups and against all circulating strains was 47%.4 No major vaccine safety concerns were detected. Among those who received IIV3, there was a slight increase in the incidence of Guillain-Barré syndrome of 2.6 cases per one million vaccines.7
Other recommendations for 2016-2017
Once again, ACIP recommends influenza vaccine for all individuals 6 months and older.8 The CDC additionally specifies particular groups that should not skip vaccination given that they are at high risk of complications from influenza infection or because they could expose high-risk individuals to infection (TABLE 1).9
There will continue to be a selection of trivalent and quadrivalent influenza vaccine products in 2016-2017. Trivalent products will contain 3 viral strains: A/California/7/2009 (H1N1), A/Hong Kong/4801/2014 (H3N2) and B/Brisbane/60/2008.10 The quadrivalent products will contain those 3 antigens plus B/Phuket/3073/2013.10 The H3N2 strain is different from the one in last year’s vaccine. Each year, influenza experts analyze surveillance data to predict which circulating strains will predominate in North America, and these antigens constitute the vaccine formulation. The accuracy of this prediction in large part determines how effective the vaccine will be that season.
Two new vaccines have been approved for use in the United States. A quadrivalent cell culture inactivated vaccine (CCIV4), Flucelvax, was licensed in May 2016. It is prepared from virus propagated in canine kidney cells, not with an egg-based production process. It is approved for use in individuals 4 years of age and older.8 Fluad, an adjuvanted trivalent inactivated influenza vaccine, was licensed in late 2015 for individuals 65 years of age and older.8 This is the first adjuvanted influenza vaccine licensed in the United States and will compete with high-dose quadrivalent vaccine for use in older adults. ACIP does not express a preference for any vaccine in this age group.
Two other vaccines should also be available by this fall: Flublok, a quadrivalent recombinant influenza vaccine for individuals 18 years and older, and Flulaval, a quadrivalent inactivated influenza vaccine, for individuals 6 months of age and older. TABLE 211 lists approved influenza vaccines.
Issues specific to children
Deciding how many vaccine doses children need has been further simplified. Children younger than 9 years need 2 doses if they have received fewer than 2 doses of trivalent or quadrivalent influenza vaccine before July 1, 2016. The interval between the 2 doses should be at least 4 weeks. The 2 doses do not have to be the same product; importantly, do not delay a second dose just to obtain the same product used for the first dose. Also, one dose can be trivalent and the other one quadrivalent, although this offers less-than-optimal protection against the B-virus that is only in the quadrivalent product.
Children younger than 9 years require only one dose if they have received 2 or more total doses of trivalent or quadrivalent influenza vaccine before July 1, 2016. The 2 previous doses need not have been received during the same influenza season or consecutive influenza seasons.
In children ages 6 through 23 months there is a slight increased risk of febrile seizure if the influenza vaccine is co-administered with other vaccines, specifically pneumococcal conjugate vaccine (PCV 13) and diphtheria-tetanus-acellular-pertussis (DTaP). The 3 vaccines administered at the same time result in 30 febrile seizures per 100,000 children;12 the rate is lower when influenza vaccine is co-administered with only one of the others. ACIP believes that the risk of a febrile seizure, which does no long-term harm, does not warrant delaying vaccines that could be co-administered.13
Egg allergy requires no special precautions
Evidence continues to grow that influenza vaccine products do not contain enough egg protein to cause significant problems in those with a history of egg allergies. This year’s recommendations state that no special precautions are needed regarding the anatomic site of immunization or the length of observation after administering influenza vaccine in those with a history of allergies to eggs, no matter how severe. All vaccine-administration facilities should be able to respond to any hypersensitivity reaction, and the standard waiting time for observation after all vaccinations is 15 minutes.
Antiviral medications for treatment or prevention
Most influenza strains circulating in 2016-2017 are expected to remain sensitive to oseltamivir and zanamivir, which can be used for treatment or disease prevention. A third neuraminidase inhibitor, peramivir, is available for intravenous use in adults 18 and older. Treatment is recommended for those who have confirmed or suspected influenza and are at high risk for complications (TABLE 3).14 Consideration of antiviral chemoprevention is recommended under certain circumstances (TABLE 4).15,16 The CDC influenza Web site lists recommended doses and duration for each antiviral for treatment and chemoprevention.15
The Advisory Committee on Immunization Practices (ACIP) took the unusual step at its June 2016 meeting of recommending against using a currently licensed vaccine, live attenuated influenza vaccine (LAIV), in the 2016-2017 influenza season.1 ACIP based its recommendation on surveillance data collected by the US Influenza Vaccine Effectiveness Network of the Centers for Disease Control and Prevention (CDC), which showed poor effectiveness by the LAIV vaccine among children and adolescents during the past 3 years.
The US Food and Drug Administration (FDA), however, has chosen not to take any action on this matter, saying on its Web site it “has determined that specific regulatory action is not warranted at this time. This determination is based on FDA’s review of manufacturing and clinical data supporting licensure … the totality of the evidence presented at the ACIP meeting, taking into account the inherent limitations of observational studies conducted to evaluate influenza vaccine effectiveness, as well as the well-known variability of influenza vaccine effectiveness across influenza seasons.”2
CDC data for the 2015-2016 flu season showed the effectiveness of LAIV to be just 3% among children 2 years through 17 years of age.3 The reason for this apparent lack of effectiveness is unknown. Other LAIV-effectiveness studies conducted in the 2015-2016 season—one each, in the United States, United Kingdom, and Finland—had results that differed from the CDC surveillance data, with effectiveness ranging from 46% to 58% against all strains combined.2 These results are comparable to vaccine effectiveness found in observational studies in children for both LAIV and inactivated influenza vaccines (IIV) in prior seasons.2
Vaccine manufacturers had projected that 171 to 176 million doses of flu vaccine, in all forms, would be available in the United States during the 2016-2017 season.3 LAIV accounts for about 8% of the total supply of influenza vaccine in the United States,3 and ACIP’s recommendation is not expected to create shortages of other options for the upcoming season. However, the LAIV accounts for one-third of flu vaccines administered to children, and clinicians who provide vaccinations to children have already ordered their vaccine supplies for the upcoming season. Also, it is not clear if children who have previously received the LAIV product will now accept other options for influenza vaccination—all of which involve an injection.
Whether the recommendation against LAIV will continue after this season is also unknown.
What happened during the 2015-2016 influenza season?
The 2015-2016 influenza season was relatively mild with the peak activity occurring in March, somewhat later than in previous years. The circulating influenza strains matched closely to those in the vaccine, making it more effective than the previous year’s vaccine. The predominant circulating strain was A (H1N1), accounting for 58% of illness; A (H3N2) caused 6% of cases and all B types together accounted for 34%.4 The hospitalization rate for all ages was 31.3/100,000 compared with 64.1 the year before.5 There were 85 pediatric deaths compared with 148 in 2014-2015.6
Vaccine effectiveness among all age groups and against all circulating strains was 47%.4 No major vaccine safety concerns were detected. Among those who received IIV3, there was a slight increase in the incidence of Guillain-Barré syndrome of 2.6 cases per one million vaccines.7
Other recommendations for 2016-2017
Once again, ACIP recommends influenza vaccine for all individuals 6 months and older.8 The CDC additionally specifies particular groups that should not skip vaccination given that they are at high risk of complications from influenza infection or because they could expose high-risk individuals to infection (TABLE 1).9
There will continue to be a selection of trivalent and quadrivalent influenza vaccine products in 2016-2017. Trivalent products will contain 3 viral strains: A/California/7/2009 (H1N1), A/Hong Kong/4801/2014 (H3N2) and B/Brisbane/60/2008.10 The quadrivalent products will contain those 3 antigens plus B/Phuket/3073/2013.10 The H3N2 strain is different from the one in last year’s vaccine. Each year, influenza experts analyze surveillance data to predict which circulating strains will predominate in North America, and these antigens constitute the vaccine formulation. The accuracy of this prediction in large part determines how effective the vaccine will be that season.
Two new vaccines have been approved for use in the United States. A quadrivalent cell culture inactivated vaccine (CCIV4), Flucelvax, was licensed in May 2016. It is prepared from virus propagated in canine kidney cells, not with an egg-based production process. It is approved for use in individuals 4 years of age and older.8 Fluad, an adjuvanted trivalent inactivated influenza vaccine, was licensed in late 2015 for individuals 65 years of age and older.8 This is the first adjuvanted influenza vaccine licensed in the United States and will compete with high-dose quadrivalent vaccine for use in older adults. ACIP does not express a preference for any vaccine in this age group.
Two other vaccines should also be available by this fall: Flublok, a quadrivalent recombinant influenza vaccine for individuals 18 years and older, and Flulaval, a quadrivalent inactivated influenza vaccine, for individuals 6 months of age and older. TABLE 211 lists approved influenza vaccines.
Issues specific to children
Deciding how many vaccine doses children need has been further simplified. Children younger than 9 years need 2 doses if they have received fewer than 2 doses of trivalent or quadrivalent influenza vaccine before July 1, 2016. The interval between the 2 doses should be at least 4 weeks. The 2 doses do not have to be the same product; importantly, do not delay a second dose just to obtain the same product used for the first dose. Also, one dose can be trivalent and the other one quadrivalent, although this offers less-than-optimal protection against the B-virus that is only in the quadrivalent product.
Children younger than 9 years require only one dose if they have received 2 or more total doses of trivalent or quadrivalent influenza vaccine before July 1, 2016. The 2 previous doses need not have been received during the same influenza season or consecutive influenza seasons.
In children ages 6 through 23 months there is a slight increased risk of febrile seizure if the influenza vaccine is co-administered with other vaccines, specifically pneumococcal conjugate vaccine (PCV 13) and diphtheria-tetanus-acellular-pertussis (DTaP). The 3 vaccines administered at the same time result in 30 febrile seizures per 100,000 children;12 the rate is lower when influenza vaccine is co-administered with only one of the others. ACIP believes that the risk of a febrile seizure, which does no long-term harm, does not warrant delaying vaccines that could be co-administered.13
Egg allergy requires no special precautions
Evidence continues to grow that influenza vaccine products do not contain enough egg protein to cause significant problems in those with a history of egg allergies. This year’s recommendations state that no special precautions are needed regarding the anatomic site of immunization or the length of observation after administering influenza vaccine in those with a history of allergies to eggs, no matter how severe. All vaccine-administration facilities should be able to respond to any hypersensitivity reaction, and the standard waiting time for observation after all vaccinations is 15 minutes.
Antiviral medications for treatment or prevention
Most influenza strains circulating in 2016-2017 are expected to remain sensitive to oseltamivir and zanamivir, which can be used for treatment or disease prevention. A third neuraminidase inhibitor, peramivir, is available for intravenous use in adults 18 and older. Treatment is recommended for those who have confirmed or suspected influenza and are at high risk for complications (TABLE 3).14 Consideration of antiviral chemoprevention is recommended under certain circumstances (TABLE 4).15,16 The CDC influenza Web site lists recommended doses and duration for each antiviral for treatment and chemoprevention.15
1. Grohskopf LA, Sokolow LZ, Broder KR, et al. Prevention and control of seasonal influenza with vaccines: Recommendations of the Advisory Committee on Immunization Practices—United States, 2016-17 influenza season. MMWR Recomm Rep. 2016;65:1-54.
2. U.S. Food and Drug Administration. FDA information regarding FluMist quadrivalent vaccine. Available at: http://www.fda.gov/BiologicsBloodVaccines/Vaccines/ApprovedProducts/ucm508761.htm. Accessed July 13, 2016.
3. Centers for Disease Control and Prevention. ACIP votes down use of LAIV for 2016-2017 flu season. Available at: http://www.cdc.gov/media/releases/2016/s0622-laiv-flu.html. Accessed July 13, 2016.
4. Flannery B, Chung J. Influenza vaccine effectiveness, including LAIV vs IIV in children and adolescents, US Flu VE Network, 2015-2016. Presented at: meeting of the Advisory Committee on Immunization Practices; June 22, 2016; Atlanta, GA. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/vaccines/acip/meetings/downloads/slides-2016-06/influenza-05-flannery.pdf. Accessed July 22, 2016.
5. Centers for Disease Control and Prevention. FluView. Laboratory-confirmed influenza hospitalizations. Available at: http://gis.cdc.gov/GRASP/Fluview/FluHospRates.html. Accessed July 25, 2016.
6. Centers for Disease Control and Prevention. FluView. Number of influenza-associated pediatric deaths by week of death. Available at: http://gis.cdc.gov/GRASP/Fluview/PedFluDeath.html. Accessed July 25, 2016.
7. Shimabukuro T. End-of-season update: 2015-2016 influenza vaccine safety monitoring. Presented at: meeting of the Advisory Committee on Immunization Practices; June 22, 2016; Atlanta, GA. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/vaccines/acip/meetings/downloads/slides-2016-06/influenza-04-shimabukuro.pdf. Accessed July 22, 2016.
8. Grohskopf L. Proposed recommendations 2016-2017 influenza season. Presented at: meeting of the Advisory Committee on Immunization Practices; June 22, 2016; Atlanta, GA. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/vaccines/acip/meetings/downloads/slides-2016-06/influenza-08-grohskopf.pdf. Accessed July 22, 2016.
9. Centers for Disease Control and Prevention. Influenza vaccination: a summary for clinicians. Available at: http://www.cdc.gov/flu/professionals/vaccination/vax-summary.htm. Accessed July 13, 2016.
10. Centers for Disease Control and Prevention. What you should know for the 2016-2017 influenza season. Available at: http://www.cdc.gov/flu/about/season/flu-season-2016-2017.htm. Accessed July 13, 2016.
11. Immunization Action Coalition. Influenza vaccine products for the 2016-2017 influenza season. Available at: http://www.immunize.org/catg.d/p4072.pdf. Accessed July 13, 2016.
12. Duffy J, Weintraub E, Hambidge SJ, et al. Febrile seizure risk after vaccination in children 6 to 23 months. Pediatrics. 2016;138.
13. Centers for Disease Control and Prevention. Childhood vaccines and febrile seizures. Available at: http://www.cdc.gov/vaccinesafety/concerns/febrile-seizures.html. Accessed August 11, 2016.
14. Centers for Disease Control and Prevention. Use of antivirals. Background and guidance on the use of influenza antiviral agents. Available at: http://www.cdc.gov/flu/professionals/antivirals/antiviral-use-influenza.htm. Accessed July 13, 2016.
15. Centers for Disease Control and Prevention. Influenza antiviral medications: summary for clinicians. Available at: http://www.cdc.gov/flu/professionals/antivirals/summary-clinicians.htm. Accessed July 13, 2016.
16. American Academy of Pediatrics. Recommendations for prevention and control of influenza in children, 2015-2016. Pediatrics. 2015;136:792-808.
1. Grohskopf LA, Sokolow LZ, Broder KR, et al. Prevention and control of seasonal influenza with vaccines: Recommendations of the Advisory Committee on Immunization Practices—United States, 2016-17 influenza season. MMWR Recomm Rep. 2016;65:1-54.
2. U.S. Food and Drug Administration. FDA information regarding FluMist quadrivalent vaccine. Available at: http://www.fda.gov/BiologicsBloodVaccines/Vaccines/ApprovedProducts/ucm508761.htm. Accessed July 13, 2016.
3. Centers for Disease Control and Prevention. ACIP votes down use of LAIV for 2016-2017 flu season. Available at: http://www.cdc.gov/media/releases/2016/s0622-laiv-flu.html. Accessed July 13, 2016.
4. Flannery B, Chung J. Influenza vaccine effectiveness, including LAIV vs IIV in children and adolescents, US Flu VE Network, 2015-2016. Presented at: meeting of the Advisory Committee on Immunization Practices; June 22, 2016; Atlanta, GA. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/vaccines/acip/meetings/downloads/slides-2016-06/influenza-05-flannery.pdf. Accessed July 22, 2016.
5. Centers for Disease Control and Prevention. FluView. Laboratory-confirmed influenza hospitalizations. Available at: http://gis.cdc.gov/GRASP/Fluview/FluHospRates.html. Accessed July 25, 2016.
6. Centers for Disease Control and Prevention. FluView. Number of influenza-associated pediatric deaths by week of death. Available at: http://gis.cdc.gov/GRASP/Fluview/PedFluDeath.html. Accessed July 25, 2016.
7. Shimabukuro T. End-of-season update: 2015-2016 influenza vaccine safety monitoring. Presented at: meeting of the Advisory Committee on Immunization Practices; June 22, 2016; Atlanta, GA. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/vaccines/acip/meetings/downloads/slides-2016-06/influenza-04-shimabukuro.pdf. Accessed July 22, 2016.
8. Grohskopf L. Proposed recommendations 2016-2017 influenza season. Presented at: meeting of the Advisory Committee on Immunization Practices; June 22, 2016; Atlanta, GA. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/vaccines/acip/meetings/downloads/slides-2016-06/influenza-08-grohskopf.pdf. Accessed July 22, 2016.
9. Centers for Disease Control and Prevention. Influenza vaccination: a summary for clinicians. Available at: http://www.cdc.gov/flu/professionals/vaccination/vax-summary.htm. Accessed July 13, 2016.
10. Centers for Disease Control and Prevention. What you should know for the 2016-2017 influenza season. Available at: http://www.cdc.gov/flu/about/season/flu-season-2016-2017.htm. Accessed July 13, 2016.
11. Immunization Action Coalition. Influenza vaccine products for the 2016-2017 influenza season. Available at: http://www.immunize.org/catg.d/p4072.pdf. Accessed July 13, 2016.
12. Duffy J, Weintraub E, Hambidge SJ, et al. Febrile seizure risk after vaccination in children 6 to 23 months. Pediatrics. 2016;138.
13. Centers for Disease Control and Prevention. Childhood vaccines and febrile seizures. Available at: http://www.cdc.gov/vaccinesafety/concerns/febrile-seizures.html. Accessed August 11, 2016.
14. Centers for Disease Control and Prevention. Use of antivirals. Background and guidance on the use of influenza antiviral agents. Available at: http://www.cdc.gov/flu/professionals/antivirals/antiviral-use-influenza.htm. Accessed July 13, 2016.
15. Centers for Disease Control and Prevention. Influenza antiviral medications: summary for clinicians. Available at: http://www.cdc.gov/flu/professionals/antivirals/summary-clinicians.htm. Accessed July 13, 2016.
16. American Academy of Pediatrics. Recommendations for prevention and control of influenza in children, 2015-2016. Pediatrics. 2015;136:792-808.
Managing irritable bowel syndrome: The low-FODMAP diet
The role of diet in controlling symptoms of irritable bowel syndrome (IBS) has gained much traction over the years,1 but until recently, diet therapy for IBS has been hindered by a lack of quality evidence, in part because of the challenges of conducting dietary clinical trials.
Several clinical trials have now been done that support a diet low in fermentable oligosaccharides, disaccharides, monosaccharides, and polyols (FODMAPs) for managing IBS. Although restrictive and difficult to follow, the low-FODMAP diet is gaining popularity.
This article provides an overview of dietary interventions used to manage IBS, focusing on the low-FODMAP diet. We discuss mechanisms of malabsorption of FODMAPs and the role of FODMAPS in symptom induction; highlight clinical trials that provide evidence of benefits of the diet for IBS; and discuss the steps to implement it. We also address the nutritional adequacy of the diet and its potential effects on the gut microbiome.
IBS IS A COMMON FUNCTIONAL DISORDER
IBS is one of the most commonly diagnosed gastrointestinal disorders, and it has a significant impact on quality of life.2 It is a functional disorder characterized by chronic abdominal pain and altered bowel habits in the absence of a structural or organic cause.
The Rome IV diagnostic criteria define IBS by the following:
- Recurrent abdominal pain or discomfort at least 1 day a week in the last 3 months, associated with two or more of the following:
- Symptoms improved by defecation
- Onset associated with a change in frequency of stool
- Onset associated with a change in form or appearance of stool.
IBS mainly arises during young adulthood but can be diagnosed at any age.3
The pathophysiology of IBS involves mechanisms such as bowel distention, altered bowel motility, visceral hypersensitivity, and disruption of mucosal permeability.4 Several therapeutic modalities targeting these mechanisms have been implemented in IBS management, including antispasmodics, laxatives, antidepressants, antibiotics, and behavioral therapy. Diet is only one line of treatment and is most effective when part of a multipronged approach.
TRADITIONAL DIETARY MANAGEMENT
Diet is important in inducing the symptoms of IBS—and in controlling them. Patients identify eating as a common precipitator of symptoms, but the complex diet-symptom interaction is not fully understood and varies widely among patients. Traditional dietary advice for IBS includes adhering to a regular meal pattern, avoiding large meals, and reducing intake of fat, insoluble fibers, caffeine, spicy and gas-producing foods, and carbonated beverages.5,6
Increase soluble fiber
Fiber and fiber supplements, particularly soluble fibers such as psyllium, calcium polycarbophil, and ispaghula husk are often recommended. A meta-analysis7 found that soluble fiber but not insoluble fiber (eg, wheat bran) is associated with an improvement in IBS symptoms (relative risk [RR] 0.84, 95% confidence interval [CI] 0.73–0.94). By improving stool consistency and accelerating transit, soluble fiber is especially useful in constipation-predominant IBS while posing a low risk for adverse outcomes.7 Fiber should be started at a low dose and gradually increased over several weeks to as much as 20 to 30 g/day.
Avoid wheat
Only about 4% of patients with IBS also have celiac disease, but estimating the prevalence of nonceliac gluten sensitivity is confounded by overlapping symptoms. There is some evidence implicating gluten in IBS: celiac disease and IBS overlap in their symptoms, and symptoms are often precipitated by gluten-containing foods in patients with IBS.8 The pathogenesis of gluten-induced (or wheat-induced) symptoms in IBS is unclear, and studies have had conflicting results as to the benefits of gluten restriction in IBS.9
In a study of patients with IBS whose symptoms improved when they started a gluten-free and low-FODMAP diet, symptoms did not return when gluten was reintroduced, suggesting that it is the fructan (a FODMAP) component of wheat rather than gluten that contributes to symptoms in IBS.10
Probiotics
Probiotics are increasingly being recommended as dietary supplements for people with IBS, as awareness increases of the importance of the gut microbiota. In addition to their effects on the gut microbiota, probiotics in IBS have been shown to have anti-inflammatory effects, to alter gut motility, to modulate visceral hypersensitivity, and to restore epithelial integrity.
In a meta-analysis, Ford et al11 found that probiotics improved global IBS symptoms more than placebo (RR 0.79, 95% CI 0.70–0.89) and also reduced abdominal pain, bloating, and flatulence scores.
Which species and strains are most beneficial and the optimal dosing and duration of treatment are still unclear. Data from studies of prebiotics (nutrients that encourage the growth of probiotic bacteria) and synbiotics (combinations of prebiotics and probiotics) are limited and insufficient to draw conclusions.
FODMAPS ARE SHORT-CHAIN CARBOHYDRATES
The term FODMAPs was initially coined by researchers at Monash University in Australia to describe a collection of poorly absorbed short-chain fermentable carbohydrates that are natural components of many foods:
- Oligosaccharides, including fructans (which include inulins) and galacto-oligosaccharides
- Disaccharides, including lactose and sucrose
- Monosaccharides, including fructose
- Polyols, including sorbitol and mannitol.12
Intake of FODMAPs, especially fructose, has increased in Western diets over the past several decades from increased consumption of fruits and concentrated fruit juices, as well as from the widespread use of high-fructose corn syrup in processed foods and beverages.13
FODMAPs ARE POORLY ABSORBED

Different FODMAPs can be poorly absorbed for different reasons (Table 1). The poor absorption is related either to reduced or absent digestive enzymes (ie, hydrolases) or to slow transport across the intestinal mucosa. Excess FODMAPs in the distal small intestine and proximal colon exert osmotic pressure, drawing more water into the lumen. FODMAPs are also rapidly fermented by colonic bacteria, producing gas, bowel distention, and altered motility, all of which induce IBS symptoms.14
Fructans are fructose polymers that are not absorbed in human intestines. They have no intestinal hydrolases and no mechanisms for direct transport across the epithelium. However, a negligible amount may be absorbed after being degraded by microbes in the gut.15 Most dietary fructans are obtained from wheat and onion, which are actually low in fructans but tend to be consumed in large quantities.16
Galacto-oligosaccharides are available for colonic fermentation after ingestion due to lack of a human alpha-galactosidase. Common sources of galacto-oligosaccharides include legumes, nuts, seeds, some grains, dairy products, human milk, and some commercially produced forms added to infant formula.17,18
Lactose is poorly absorbed in people with lactase deficiency. It is mainly present in dairy products but is also added to commercial foods, including breads, cakes, and some diet products.19
Fructose is the most abundant FODMAP in the Western diet. It is either present as a free sugar or generated from the digestive breakdown of sucrose. In the intestine, it is absorbed via a direct low-capacity glucose transporter (GLUT)-5 and through GLUT-2, which is more efficient but requires the coexistence of glucose. Because of this requirement, fructose is more likely to be malabsorbed when present in excess of glucose, as in people with diminished sucrase activity. The main sources of fructose in the Western diet are fruits and fruit products, honey, and foods with added high-fructose sweeteners.13
Polyols such as sorbitol and mannitol are absorbed by slow passive diffusion because they have no active intestinal transport system. They are found in fruits and vegetables. Sugar-free chewing gum is a particularly rich source of sorbitol.20
QUANTIFYING FODMAP CONTENT
As interest in the low-FODMAP diet grew, studies were conducted to quantify FODMAPs in foods. One study used high-performance liquid chromatography to analyze FODMAP content in foods,21 and another evaluated fructan levels in a variety of fruits and vegetables using enzymatic hydrolysis.22 The Monash University low-FODMAP diet smartphone application provides patients and healthcare providers easy access to updated and detailed food analyses.23
Table 2 lists foods high in FODMAPs along with low-FODMAP alternatives. Total FODMAP intake is important, as the effects are additive.24 Readers and patients can be directed to the following websites for more information on the low-FODMAP diet: www.med.monash.edu/cecs/gastro/fodmap or www.ibsfree.net/what-is-fodmap-diet.
LOW-FODMAP DIET REDUCES SYMPTOMS
The low-FODMAP diet was inspired by the results of several studies that evaluated the role of dietary carbohydrates in inducing IBS symptoms and found improvement with their restriction.25,26
One study found that 74% of patients with IBS had less bloating, nausea, abdominal pain, and diarrhea when they restricted their intake of fructose and fructans.27
A prospective trial randomized 41 patients with IBS to 4 weeks of either a low-FODMAP diet or their habitual diet.28 The low-FODMAP diet resulted in greater improvement in overall IBS symptoms (P < .05) and stool frequency (P = .008). This study was limited by different habitual diets between patients and by lack of standardization of the low-FODMAP diet.
Halmos et al,29 in a randomized crossover trial, compared gastrointestinal symptoms in IBS patients over 3 weeks on a low-FODMAP diet vs a moderate-FODMAP (ie, regular) diet, as well as in healthy controls. Food was provided by the study and was matched for all nutrients. Up to 70% of the IBS patients had significantly lower overall symptom scores while on a low-FODMAP diet vs IBS patients on a regular diet (P < .001); bloating, abdominal pain, and flatulence were reduced. Symptoms were minimal and unaffected by either diet in the healthy controls.
A double-blind trial30 randomly assigned 25 patients with IBS who initially responded to a low-FODMAP diet to be challenged by a graduated dose of fructose alone, fructans alone, a combination of both, or glucose. The severity of overall and individual symptoms was markedly more reduced with glucose consumption than with the other carbohydrates: 70% of patients receiving fructose, 77% of those receiving fructans, and 79% of those receiving a mixture of both reported that their symptoms were not adequately controlled, compared with 14% of patients receiving glucose (P ≤ .002).30
Murray et al31 evaluated the gastrointestinal tract after a carbohydrate challenge consisting of 0.5 L of water containing 40 g of glucose, fructose, or inulin or a combination of 40 g of glucose and 40 g of fructose in 16 healthy volunteers. Magnetic resonance imaging was performed hourly for 5 hours to assess the volume of gastric contents, small-bowel water content, and colonic gas. Breath hydrogen was also measured, and symptoms were recorded after each imaging session.
Fructose significantly increased small-bowel water content compared with glucose (mean difference 28 L/min, P < .001), but combined glucose and fructose lessened the effect. Inulin had no significant effect on small-bowel water content (mean difference with glucose 2 L/min, P > .7) but led to the greatest production of colonic gas compared with glucose alone (mean difference 15 L/min, P < .05) and combined glucose and fructose (mean difference 12 L/min, P < .05). Inulin also produced the most breath hydrogen: 81% of participants had a rise after drinking inulin compared with 50% after drinking fructose. Glucose did not affect breath hydrogen concentrations, and combined glucose and fructose significantly reduced the concentration measured vs fructose alone. In patients who reported “gas” symptoms, a correlation was observed between the volume of gas in the colon and gas symptoms (r = 0.59, P < .0001).31
The authors concluded31 that long-chain carbohydrates such as inulin have a greater effect on colonic gas production and little effect on small-bowel water content, whereas small-chain FODMAPs such as fructose are likely to cause luminal distention in both the small and large intestines. The study also showed that combining equal amounts of glucose and fructose reduces malabsorption of fructose in the small bowel and reduces the effect of fructose on small-bowel water content and breath hydrogen concentration.31
PROBIOTICS HELP
A Danish study32 randomized 123 patients with IBS to one of three treatments: a low-FODMAP diet, a normal diet with probiotics containing the strain Lactobacillus rhamnosus GG (two capsules daily), or no special intervention. Symptoms were recorded weekly. IBS severity scores at week 6 were lower in patients on either the low-FODMAP diet or probiotics compared with the control group (P < .01). Subgroup analysis determined that patients with primarily diarrheal symptoms were more likely to have improved quality of life with the low-FODMAP diet.
A LOW-FODMAP DIET MAY ALSO HELP IN INFLAMMATORY BOWEL DISEASE
The low-FODMAP diet has also been studied in patients with inflammatory bowel disease with functional gut symptoms. In a retrospective pilot study,33 overall symptoms improved in about half of such patients on a low-FODMAP diet. A controlled dietary intervention trial is needed to confirm these findings and define the role of the low-FODMAP approach for patients with inflammatory bowel disease.
Marsh et al34 performed a meta-analysis of six randomized clinical trials and 16 nonrandomized interventions of a low-FODMAP diet on improving functional gastrointestinal symptoms in patients with either IBS or inflammatory bowel disease. They found significant improvements in:
- IBS Symptoms Severity Scores in the randomized trials (odds ratio [OR] 0.44, 95% CI 0.25–0.76)
- IBS Symptoms Severity Scores in the nonrandomized interventions (OR 0.03, 95% CI 0.01–0.2)
- IBS Quality of Life scores in the randomized trials (OR 1.84, 95% CI 1.12–3.03)
- IBS Quality of Life scores in the nonrandomized interventions (OR 3.18, 95% CI 1.6–6.31)
- Overall symptom severity in the randomized trials (OR 1.81, 95% CI 1.11–2.95).
DIETARY COUNSELING IS RECOMMENDED
Adherence is a major factor in the success of the low-FODMAP diet in IBS management and is strongly correlated with improved symptoms.35 Patients should be counseled on the role of food in inducing their symptoms. Haphazard dietary advice can be detrimental to outcomes, as many diets restrict food groups, impairing the consumption of essential nutrients.36 The involvement of a knowledgeable dietitian is helpful, as physicians may lack sufficient training in dietary skills and knowledge of food composition.
Access to and cost of dietary counseling can be prohibitive for some patients. Group consultation, which can decrease costs to each patient, has been found to be as effective as one-on-one sessions when administering the low-FODMAP diet in functional bowel disorders.37
ELIMINATION, THEN REINTRODUCTION
Before embarking on the low-FODMAP diet, the patient’s interest in making dietary changes should be explored, a dietary history taken, and unusual food choices or dietary behaviors assessed. The patient’s ability to adopt a restricted diet should also be gauged.
The diet should be implemented in two phases. The initial phase involves strict elimination of foods high in FODMAPs, usually over 6 to 8 weeks.38 Symptom control should be assessed: failure to control symptoms requires assessment of adherence.
If symptoms are successfully controlled, then the second phase should begin with the aim of following a less-restricted version of the diet as tolerated. Foods should gradually be phased back in and symptoms monitored. This approach minimizes unnecessary dietary restriction and ensures that a maximum variety in the diet is achieved while maintaining adequate symptom control.39
LOW-FODMAP DIET ALTERS THE GUT MICROBIOTA
Multiple putative benefits of certain bacterial species for colonic health have been reported, including the production of short-chain fatty acids. Colonic luminal concentrations of short-chain fatty acids may be important to gut health, given their role in intestinal secretion, absorption, motility, and epithelial cell structure. Because short-chain fatty acids are products of bacterial fermentation, a change in the delivery of fermentable substrates to the colon would be expected to alter the concentrations and output of fecal short-chain fatty acids.18
Several studies evaluated the effect of the low-FODMAP diet on intestinal microbiota, finding a change in the bacterial profile in the stool of patients who adopt this diet. Staudacher et al28 found a marked reduction in luminal bifidobacteria concentration after 4 weeks of a low-FODMAP diet in patients with IBS.
A single-blind randomized crossover trial40 investigated the effects of a low-FODMAP diet vs a carefully matched typical Australian diet in 27 patients with IBS and 6 healthy controls. Marked differences in absolute and relative bacterial abundance and diversity were found between the diets, but not in short-chain fatty acids or gut transit time. Compared with fecal microbiota on the typical diet, low FODMAP intake was associated with reduced absolute abundance of bacteria, and the typical FODMAP diet had evidence of stimulation of the growth of bacterial groups with putative health benefits.
The authors concluded40 that the functional significance and health implications of such changes are reasons for caution when reducing FODMAP intake in the long term and recommended liberalizing FODMAP restriction to the level of adequate symptom control in IBS patients. The study also recommended that people without symptoms not go on the low-FODMAP diet.40
Molecular approaches to characterize the gut microbiota are also being explored in an effort to identify its association with diet.
The sustainability of changes in gut microbiota and the potential long-term impact on health of following a low-FODMAP diet require further evaluation. In the meantime, patients following this diet should have FODMAP foods reintroduced based on tolerance and should consider taking probiotic supplements.41
DIETARY ADEQUACY OF THE LOW-FODMAP DIET
Continual dietary counseling should minimize nutritional inadequacies and ensure that FODMAPS are restricted only enough to control symptoms. Because no single food group is completely eliminated in this diet, patients are unlikely to experience inadequate nutrition.
Ledochowski et al26 found that in the initial, strict phase of the diet, total intake of carbohydrates (eg, starches, sugars) was reduced but intake of total energy, protein, fat, and nonstarch polysaccharides was not affected. Calcium intake was reduced in those following a low-FODMAP diet for 4 weeks.
The diet can also reduce total fiber intake and subsequently worsen constipation-predominant IBS. For those patients, lightly fermented high-fiber alternatives like oat and rice bran can be used.
ACCUMULATING EVIDENCE
The low-FODMAP diet is accumulating quality evidence for its effectiveness in controlling the functional gastrointestinal symptoms in patients with IBS. It can be difficult to adhere to over the long term due to its restrictiveness, and it is important to gradually liberalize the diet while tailoring it to the individual patient and monitoring symptoms. Further clinical trials are needed to evaluate this diet in different IBS subtypes and other gastrointestinal disorders, while defining its nutritional adequacy and effects on the intestinal microbiota profile.
- Hayes P, Corish C, O’Mahony E, Quigley EM. A dietary survey of patients with irritable bowel syndrome. J Hum Nutr Diet 2014; 27(suppl 2):36–47.
- Pare P, Gray J, Lam S, et al. Health-related quality of life, work productivity, and health care resource utilization of subjects with irritable bowel syndrome: baseline results from LOGIC (Longitudinal Outcomes Study of Gastrointestinal Symptoms in Canada), a naturalistic study. Clin Ther 2006; 28:1726–1735; discussion 1710–1711.
- Lacey BE, Mearin F, Chang L, et al. Bowel disorders. Gastroenterology 2016; 150:1393–1407.
- Drossman DA, Camilleri M, Mayer EA, Whitehead WE. AGA technical review on irritable bowel syndrome. Gastroenterology 2002; 123:2108–2131.
- Floch MH, Narayan R. Diet in the irritable bowel syndrome. J Clin Gastroenterol 2002; 35(suppl 1):S45–S52.
- Reding KW, Cain KC, Jarrett ME, Eugenio MD, Heitkemper MM. Relationship between patterns of alcohol consumption and gastrointestinal symptoms among patients with irritable bowel syndrome. Am J Gastroenterol 2013; 108:270–276.
- Moayyedi P, Quigley EM, Lacy BE, et al. The effect of fiber supplementation on irritable bowel syndrome: a systematic review and meta-analysis. Am J Gastroenterol 2014; 109:1367–1374.
- Vazquez Roque MI, Camilleri M, Smyrk T, et al. A controlled trial of gluten-free diet in patients with irritable bowel syndrome-diarrhea: effects on bowel frequency and intestinal function. Gastroenterology 2013; 144:903–911.
- Biesiekierski JR, Newnham ED, Irving PM, et al. Gluten causes gastrointestinal symptoms in subjects without celiac disease: a double-blind randomized placebo-controlled trial. Am J Gastroenterol 2011; 106:508–515.
- Biesiekierski JR, Peters SL, Newnham ED, Rosella O, Muir JG, Gibson PR. No effects of gluten in patients with self-reported non-celiac gluten sensitivity after dietary reduction of fermentable, poorly absorbed, short-chain carbohydrates. Gastroenterology 2013; 145:320–328.
- Ford AC, Quigley EM, Lacy BE, et al. Efficacy of prebiotics, probiotics, and synbiotics in irritable bowel syndrome and chronic idiopathic constipation: systematic review and meta-analysis. Gastroenterology 2013; 145:320–328.e1–e3.
- Central Clinical School, Monash University and The Alfred Hospital. The Monash University Low FODMAP Diet. 4th ed. Melbourne, Australia: Monash University; 2012.
- Parker K, Salas M, Nwosu VC. High fructose corn syrup: production, uses and public health concerns. Biotechnol Mol Biol Rev 2010; 5:71–78.
- Clausen MR, Jorgensen J, Mortensen PB. Comparison of diarrhea induced by ingestion of fructooligosaccharide idolax and disaccharide lactulose: role of osmolarity versus fermentation of malabsorbed carbohydrate. Dig Dis Sci 1998; 43:2696–2707.
- Barrett JS, Gearry RB, Muir JG, et al. Dietary poorly absorbed, short-chain carbohydrates increase delivery of water and fermentable substrates to the proximal colon. Aliment Pharmacol Ther 2010; 31:874–882.
- Whelan K, Abrahmsohn O, David GJ, et al. Fructan content of commonly consumed wheat, rye and gluten-free breads. Int J Food Sci Nutr 2011; 62:498–503.
- Sangwan V, Tomar SK, Singh RR, Singh AK, Ali B. Galactooligosaccharides: novel components of designer foods. J Food Sci 2011; 76:R103–R111.
- Russell DA, Ross RP, Fitzgerald GF, Stanton C. Metabolic activities and probiotic potential of bifidobacteria. Int J Food Microbiol 2011; 149:88–105.
- Lomer MC, Parkes GC, Sanderson JD. Review article: lactose intolerance in clinical practice—myths and realities. Aliment Pharmacol Ther 2008; 27:93–103.
- Langkilde AM, Andersson H, Schweizer TF, Würsch P. Digestion and absorption of sorbitol, maltitol and isomalt from the small bowel. A study in ileostomy subjects. Eur J Clin Nutr 1994; 48:768–775.
- Muir JG, Rose R, Rosella O, et al. Measurement of short-chain carbohydrates in common Australian vegetables and fruits by high-performance liquid chromatography (HPLC). J Agric Food Chem 2009; 57:554–565.
- Muir JG, Shepherd SJ, Rosella O, Rose R, Barrett JS, Gibson PR. Fructan and free fructose content of common Australian vegetables and fruit. J Agric Food Chem 2007; 55:6619–6627.
- Monash University. Monash launches Low FODMAP Diet smartphone app. http://med.monash.edu.au/news/2012/fodmap-app.html. Accessed July 13, 2016.
- Fedewa A, Rao SS. Dietary fructose intolerance, fructan intolerance and FODMAPs. Curr Gastroenterol Rep 2014; 16:370.
- Born P, Vierling T, Barina W. Fructose malabsorption and the irritable bowel syndrome. Gastroenterology 1991; 101:1454.
- Ledochowski M, Widner B, Bair H, Probst T, Fuchs D. Fructose- and sorbitol-reduced diet improves mood and gastrointestinal disturbances in fructose malabsorbers. Scand J Gastroenterol 2000; 35:1048–1052.
- Shepherd SJ, Gibson PR. Fructose malabsorption and symptoms of irritable bowel syndrome: guidelines for effective dietary management. J Am Diet Assoc 2006; 106:1631–1639.
- Staudacher HM, Lomer MC, Anderson JL, et al. Fermentable carbohydrate restriction reduces luminal bifidobacteria and gastrointestinal symptoms in patients with irritable bowel syndrome. J Nutr 2012; 142:1510–1518.
- Halmos EP, Power VA, Shepherd SJ, Gibson PR, Muir JG. A diet low in FODMAPs reduces symptoms of irritable bowel syndrome. Gastroenterology 2014; 146:67–75.e5.
- Shepherd SJ, Parker FC, Muir JG, Gibson PR. Dietary triggers of abdominal symptoms in patients with irritable bowel syndrome: randomized placebo-controlled evidence. Clin Gastroenterol Hepatol 2008; 6:765–771.
- Murray K, Wilkinson-Smith V, Hoad C, et al. Differential effects of FODMAPs (fermentable oligo-, di-, mono-saccharides and polyols) on small and large intestinal contents in healthy subjects shown by MRI. Am J Gastroenterol 2014; 109:110–119.
- Pedersen N, Andersen NN, Vegh Z, et al. Ehealth: low FODMAP diet vs Lactobacillus rhamnosus GG in irritable bowel syndrome. World J Gastroenterol 2014; 20:16215–16226.
- Gearry RB, Irving PM, Barrett JS, Nathan DM, Shepherd SJ, Gibson PR. Reduction of dietary poorly absorbed short-chain carbohydrates (FODMAPs) improves abdominal symptoms in patients with inflammatory bowel disease-a pilot study. J Crohns Colitis 2009; 3:8–14.
- Marsh A, Eslick EM, Eslick GD. Does a diet low in FODMAPs reduce symptoms associated with functional gastrointestinal disorders? A comprehensive systematic review and meta-analysis. Eur J Nutr 2015 May 17. Epub ahead of print.
- de Roest RH, Dobbs BR, Chapman BA, et al. The low FODMAP diet improves gastrointestinal symptoms in patients with irritable bowel syndrome: a prospective study. Int J Clin Pract 2013; 67:895–903.
- Gibson PR, Barrett JS, Muir JG. Functional bowel symptoms and diet. Intern Med J 2013; 43:1067–1074.
- Whigham L, Joyce T, Harper G, et al. Clinical effectiveness and economic costs of group versus one-to-one education for short-chain fermentable carbohydrate restriction (low FODMAP diet) in the management of irritable bowel syndrome. J Hum Nutr Diet 2015; 28:687–696.
- Shepherd SJ, Lomer MC, Gibson PR. Short-chain carbohydrates and functional gastrointestinal disorders. Am J Gastroenterol 2013; 108:707–717.
- Shepherd SJ, Halmos E, Glance S. The role of FODMAPs in irritable bowel syndrome. Curr Opin Clin Nutr Metab Care 2014; 17:605–609.
- Halmos EP, Christophersen CT, Bird AR, Shepherd SJ, Gibson PR, Muir JG. Diets that differ in their FODMAP content alter the colonic luminal microenvironment. Gut 2015; 64:93–100.
- Staudacher HM, Irving PM, Lomer MC, Whelan K. Mechanisms and efficacy of dietary FODMAP restriction in IBS. Nat Rev Gastroenterol Hepatol 2014; 11:256–266.
The role of diet in controlling symptoms of irritable bowel syndrome (IBS) has gained much traction over the years,1 but until recently, diet therapy for IBS has been hindered by a lack of quality evidence, in part because of the challenges of conducting dietary clinical trials.
Several clinical trials have now been done that support a diet low in fermentable oligosaccharides, disaccharides, monosaccharides, and polyols (FODMAPs) for managing IBS. Although restrictive and difficult to follow, the low-FODMAP diet is gaining popularity.
This article provides an overview of dietary interventions used to manage IBS, focusing on the low-FODMAP diet. We discuss mechanisms of malabsorption of FODMAPs and the role of FODMAPS in symptom induction; highlight clinical trials that provide evidence of benefits of the diet for IBS; and discuss the steps to implement it. We also address the nutritional adequacy of the diet and its potential effects on the gut microbiome.
IBS IS A COMMON FUNCTIONAL DISORDER
IBS is one of the most commonly diagnosed gastrointestinal disorders, and it has a significant impact on quality of life.2 It is a functional disorder characterized by chronic abdominal pain and altered bowel habits in the absence of a structural or organic cause.
The Rome IV diagnostic criteria define IBS by the following:
- Recurrent abdominal pain or discomfort at least 1 day a week in the last 3 months, associated with two or more of the following:
- Symptoms improved by defecation
- Onset associated with a change in frequency of stool
- Onset associated with a change in form or appearance of stool.
IBS mainly arises during young adulthood but can be diagnosed at any age.3
The pathophysiology of IBS involves mechanisms such as bowel distention, altered bowel motility, visceral hypersensitivity, and disruption of mucosal permeability.4 Several therapeutic modalities targeting these mechanisms have been implemented in IBS management, including antispasmodics, laxatives, antidepressants, antibiotics, and behavioral therapy. Diet is only one line of treatment and is most effective when part of a multipronged approach.
TRADITIONAL DIETARY MANAGEMENT
Diet is important in inducing the symptoms of IBS—and in controlling them. Patients identify eating as a common precipitator of symptoms, but the complex diet-symptom interaction is not fully understood and varies widely among patients. Traditional dietary advice for IBS includes adhering to a regular meal pattern, avoiding large meals, and reducing intake of fat, insoluble fibers, caffeine, spicy and gas-producing foods, and carbonated beverages.5,6
Increase soluble fiber
Fiber and fiber supplements, particularly soluble fibers such as psyllium, calcium polycarbophil, and ispaghula husk are often recommended. A meta-analysis7 found that soluble fiber but not insoluble fiber (eg, wheat bran) is associated with an improvement in IBS symptoms (relative risk [RR] 0.84, 95% confidence interval [CI] 0.73–0.94). By improving stool consistency and accelerating transit, soluble fiber is especially useful in constipation-predominant IBS while posing a low risk for adverse outcomes.7 Fiber should be started at a low dose and gradually increased over several weeks to as much as 20 to 30 g/day.
Avoid wheat
Only about 4% of patients with IBS also have celiac disease, but estimating the prevalence of nonceliac gluten sensitivity is confounded by overlapping symptoms. There is some evidence implicating gluten in IBS: celiac disease and IBS overlap in their symptoms, and symptoms are often precipitated by gluten-containing foods in patients with IBS.8 The pathogenesis of gluten-induced (or wheat-induced) symptoms in IBS is unclear, and studies have had conflicting results as to the benefits of gluten restriction in IBS.9
In a study of patients with IBS whose symptoms improved when they started a gluten-free and low-FODMAP diet, symptoms did not return when gluten was reintroduced, suggesting that it is the fructan (a FODMAP) component of wheat rather than gluten that contributes to symptoms in IBS.10
Probiotics
Probiotics are increasingly being recommended as dietary supplements for people with IBS, as awareness increases of the importance of the gut microbiota. In addition to their effects on the gut microbiota, probiotics in IBS have been shown to have anti-inflammatory effects, to alter gut motility, to modulate visceral hypersensitivity, and to restore epithelial integrity.
In a meta-analysis, Ford et al11 found that probiotics improved global IBS symptoms more than placebo (RR 0.79, 95% CI 0.70–0.89) and also reduced abdominal pain, bloating, and flatulence scores.
Which species and strains are most beneficial and the optimal dosing and duration of treatment are still unclear. Data from studies of prebiotics (nutrients that encourage the growth of probiotic bacteria) and synbiotics (combinations of prebiotics and probiotics) are limited and insufficient to draw conclusions.
FODMAPS ARE SHORT-CHAIN CARBOHYDRATES
The term FODMAPs was initially coined by researchers at Monash University in Australia to describe a collection of poorly absorbed short-chain fermentable carbohydrates that are natural components of many foods:
- Oligosaccharides, including fructans (which include inulins) and galacto-oligosaccharides
- Disaccharides, including lactose and sucrose
- Monosaccharides, including fructose
- Polyols, including sorbitol and mannitol.12
Intake of FODMAPs, especially fructose, has increased in Western diets over the past several decades from increased consumption of fruits and concentrated fruit juices, as well as from the widespread use of high-fructose corn syrup in processed foods and beverages.13
FODMAPs ARE POORLY ABSORBED
Different FODMAPs can be poorly absorbed for different reasons (Table 1). The poor absorption is related either to reduced or absent digestive enzymes (ie, hydrolases) or to slow transport across the intestinal mucosa. Excess FODMAPs in the distal small intestine and proximal colon exert osmotic pressure, drawing more water into the lumen. FODMAPs are also rapidly fermented by colonic bacteria, producing gas, bowel distention, and altered motility, all of which induce IBS symptoms.14
Fructans are fructose polymers that are not absorbed in human intestines. They have no intestinal hydrolases and no mechanisms for direct transport across the epithelium. However, a negligible amount may be absorbed after being degraded by microbes in the gut.15 Most dietary fructans are obtained from wheat and onion, which are actually low in fructans but tend to be consumed in large quantities.16
Galacto-oligosaccharides are available for colonic fermentation after ingestion due to lack of a human alpha-galactosidase. Common sources of galacto-oligosaccharides include legumes, nuts, seeds, some grains, dairy products, human milk, and some commercially produced forms added to infant formula.17,18
Lactose is poorly absorbed in people with lactase deficiency. It is mainly present in dairy products but is also added to commercial foods, including breads, cakes, and some diet products.19
Fructose is the most abundant FODMAP in the Western diet. It is either present as a free sugar or generated from the digestive breakdown of sucrose. In the intestine, it is absorbed via a direct low-capacity glucose transporter (GLUT)-5 and through GLUT-2, which is more efficient but requires the coexistence of glucose. Because of this requirement, fructose is more likely to be malabsorbed when present in excess of glucose, as in people with diminished sucrase activity. The main sources of fructose in the Western diet are fruits and fruit products, honey, and foods with added high-fructose sweeteners.13
Polyols such as sorbitol and mannitol are absorbed by slow passive diffusion because they have no active intestinal transport system. They are found in fruits and vegetables. Sugar-free chewing gum is a particularly rich source of sorbitol.20
QUANTIFYING FODMAP CONTENT
As interest in the low-FODMAP diet grew, studies were conducted to quantify FODMAPs in foods. One study used high-performance liquid chromatography to analyze FODMAP content in foods,21 and another evaluated fructan levels in a variety of fruits and vegetables using enzymatic hydrolysis.22 The Monash University low-FODMAP diet smartphone application provides patients and healthcare providers easy access to updated and detailed food analyses.23
Table 2 lists foods high in FODMAPs along with low-FODMAP alternatives. Total FODMAP intake is important, as the effects are additive.24 Readers and patients can be directed to the following websites for more information on the low-FODMAP diet: www.med.monash.edu/cecs/gastro/fodmap or www.ibsfree.net/what-is-fodmap-diet.
LOW-FODMAP DIET REDUCES SYMPTOMS
The low-FODMAP diet was inspired by the results of several studies that evaluated the role of dietary carbohydrates in inducing IBS symptoms and found improvement with their restriction.25,26
One study found that 74% of patients with IBS had less bloating, nausea, abdominal pain, and diarrhea when they restricted their intake of fructose and fructans.27
A prospective trial randomized 41 patients with IBS to 4 weeks of either a low-FODMAP diet or their habitual diet.28 The low-FODMAP diet resulted in greater improvement in overall IBS symptoms (P < .05) and stool frequency (P = .008). This study was limited by different habitual diets between patients and by lack of standardization of the low-FODMAP diet.
Halmos et al,29 in a randomized crossover trial, compared gastrointestinal symptoms in IBS patients over 3 weeks on a low-FODMAP diet vs a moderate-FODMAP (ie, regular) diet, as well as in healthy controls. Food was provided by the study and was matched for all nutrients. Up to 70% of the IBS patients had significantly lower overall symptom scores while on a low-FODMAP diet vs IBS patients on a regular diet (P < .001); bloating, abdominal pain, and flatulence were reduced. Symptoms were minimal and unaffected by either diet in the healthy controls.
A double-blind trial30 randomly assigned 25 patients with IBS who initially responded to a low-FODMAP diet to be challenged by a graduated dose of fructose alone, fructans alone, a combination of both, or glucose. The severity of overall and individual symptoms was markedly more reduced with glucose consumption than with the other carbohydrates: 70% of patients receiving fructose, 77% of those receiving fructans, and 79% of those receiving a mixture of both reported that their symptoms were not adequately controlled, compared with 14% of patients receiving glucose (P ≤ .002).30
Murray et al31 evaluated the gastrointestinal tract after a carbohydrate challenge consisting of 0.5 L of water containing 40 g of glucose, fructose, or inulin or a combination of 40 g of glucose and 40 g of fructose in 16 healthy volunteers. Magnetic resonance imaging was performed hourly for 5 hours to assess the volume of gastric contents, small-bowel water content, and colonic gas. Breath hydrogen was also measured, and symptoms were recorded after each imaging session.
Fructose significantly increased small-bowel water content compared with glucose (mean difference 28 L/min, P < .001), but combined glucose and fructose lessened the effect. Inulin had no significant effect on small-bowel water content (mean difference with glucose 2 L/min, P > .7) but led to the greatest production of colonic gas compared with glucose alone (mean difference 15 L/min, P < .05) and combined glucose and fructose (mean difference 12 L/min, P < .05). Inulin also produced the most breath hydrogen: 81% of participants had a rise after drinking inulin compared with 50% after drinking fructose. Glucose did not affect breath hydrogen concentrations, and combined glucose and fructose significantly reduced the concentration measured vs fructose alone. In patients who reported “gas” symptoms, a correlation was observed between the volume of gas in the colon and gas symptoms (r = 0.59, P < .0001).31
The authors concluded31 that long-chain carbohydrates such as inulin have a greater effect on colonic gas production and little effect on small-bowel water content, whereas small-chain FODMAPs such as fructose are likely to cause luminal distention in both the small and large intestines. The study also showed that combining equal amounts of glucose and fructose reduces malabsorption of fructose in the small bowel and reduces the effect of fructose on small-bowel water content and breath hydrogen concentration.31
PROBIOTICS HELP
A Danish study32 randomized 123 patients with IBS to one of three treatments: a low-FODMAP diet, a normal diet with probiotics containing the strain Lactobacillus rhamnosus GG (two capsules daily), or no special intervention. Symptoms were recorded weekly. IBS severity scores at week 6 were lower in patients on either the low-FODMAP diet or probiotics compared with the control group (P < .01). Subgroup analysis determined that patients with primarily diarrheal symptoms were more likely to have improved quality of life with the low-FODMAP diet.
A LOW-FODMAP DIET MAY ALSO HELP IN INFLAMMATORY BOWEL DISEASE
The low-FODMAP diet has also been studied in patients with inflammatory bowel disease with functional gut symptoms. In a retrospective pilot study,33 overall symptoms improved in about half of such patients on a low-FODMAP diet. A controlled dietary intervention trial is needed to confirm these findings and define the role of the low-FODMAP approach for patients with inflammatory bowel disease.
Marsh et al34 performed a meta-analysis of six randomized clinical trials and 16 nonrandomized interventions of a low-FODMAP diet on improving functional gastrointestinal symptoms in patients with either IBS or inflammatory bowel disease. They found significant improvements in:
- IBS Symptoms Severity Scores in the randomized trials (odds ratio [OR] 0.44, 95% CI 0.25–0.76)
- IBS Symptoms Severity Scores in the nonrandomized interventions (OR 0.03, 95% CI 0.01–0.2)
- IBS Quality of Life scores in the randomized trials (OR 1.84, 95% CI 1.12–3.03)
- IBS Quality of Life scores in the nonrandomized interventions (OR 3.18, 95% CI 1.6–6.31)
- Overall symptom severity in the randomized trials (OR 1.81, 95% CI 1.11–2.95).
DIETARY COUNSELING IS RECOMMENDED
Adherence is a major factor in the success of the low-FODMAP diet in IBS management and is strongly correlated with improved symptoms.35 Patients should be counseled on the role of food in inducing their symptoms. Haphazard dietary advice can be detrimental to outcomes, as many diets restrict food groups, impairing the consumption of essential nutrients.36 The involvement of a knowledgeable dietitian is helpful, as physicians may lack sufficient training in dietary skills and knowledge of food composition.
Access to and cost of dietary counseling can be prohibitive for some patients. Group consultation, which can decrease costs to each patient, has been found to be as effective as one-on-one sessions when administering the low-FODMAP diet in functional bowel disorders.37
ELIMINATION, THEN REINTRODUCTION
Before embarking on the low-FODMAP diet, the patient’s interest in making dietary changes should be explored, a dietary history taken, and unusual food choices or dietary behaviors assessed. The patient’s ability to adopt a restricted diet should also be gauged.
The diet should be implemented in two phases. The initial phase involves strict elimination of foods high in FODMAPs, usually over 6 to 8 weeks.38 Symptom control should be assessed: failure to control symptoms requires assessment of adherence.
If symptoms are successfully controlled, then the second phase should begin with the aim of following a less-restricted version of the diet as tolerated. Foods should gradually be phased back in and symptoms monitored. This approach minimizes unnecessary dietary restriction and ensures that a maximum variety in the diet is achieved while maintaining adequate symptom control.39
LOW-FODMAP DIET ALTERS THE GUT MICROBIOTA
Multiple putative benefits of certain bacterial species for colonic health have been reported, including the production of short-chain fatty acids. Colonic luminal concentrations of short-chain fatty acids may be important to gut health, given their role in intestinal secretion, absorption, motility, and epithelial cell structure. Because short-chain fatty acids are products of bacterial fermentation, a change in the delivery of fermentable substrates to the colon would be expected to alter the concentrations and output of fecal short-chain fatty acids.18
Several studies evaluated the effect of the low-FODMAP diet on intestinal microbiota, finding a change in the bacterial profile in the stool of patients who adopt this diet. Staudacher et al28 found a marked reduction in luminal bifidobacteria concentration after 4 weeks of a low-FODMAP diet in patients with IBS.
A single-blind randomized crossover trial40 investigated the effects of a low-FODMAP diet vs a carefully matched typical Australian diet in 27 patients with IBS and 6 healthy controls. Marked differences in absolute and relative bacterial abundance and diversity were found between the diets, but not in short-chain fatty acids or gut transit time. Compared with fecal microbiota on the typical diet, low FODMAP intake was associated with reduced absolute abundance of bacteria, and the typical FODMAP diet had evidence of stimulation of the growth of bacterial groups with putative health benefits.
The authors concluded40 that the functional significance and health implications of such changes are reasons for caution when reducing FODMAP intake in the long term and recommended liberalizing FODMAP restriction to the level of adequate symptom control in IBS patients. The study also recommended that people without symptoms not go on the low-FODMAP diet.40
Molecular approaches to characterize the gut microbiota are also being explored in an effort to identify its association with diet.
The sustainability of changes in gut microbiota and the potential long-term impact on health of following a low-FODMAP diet require further evaluation. In the meantime, patients following this diet should have FODMAP foods reintroduced based on tolerance and should consider taking probiotic supplements.41
DIETARY ADEQUACY OF THE LOW-FODMAP DIET
Continual dietary counseling should minimize nutritional inadequacies and ensure that FODMAPS are restricted only enough to control symptoms. Because no single food group is completely eliminated in this diet, patients are unlikely to experience inadequate nutrition.
Ledochowski et al26 found that in the initial, strict phase of the diet, total intake of carbohydrates (eg, starches, sugars) was reduced but intake of total energy, protein, fat, and nonstarch polysaccharides was not affected. Calcium intake was reduced in those following a low-FODMAP diet for 4 weeks.
The diet can also reduce total fiber intake and subsequently worsen constipation-predominant IBS. For those patients, lightly fermented high-fiber alternatives like oat and rice bran can be used.
ACCUMULATING EVIDENCE
The low-FODMAP diet is accumulating quality evidence for its effectiveness in controlling the functional gastrointestinal symptoms in patients with IBS. It can be difficult to adhere to over the long term due to its restrictiveness, and it is important to gradually liberalize the diet while tailoring it to the individual patient and monitoring symptoms. Further clinical trials are needed to evaluate this diet in different IBS subtypes and other gastrointestinal disorders, while defining its nutritional adequacy and effects on the intestinal microbiota profile.
The role of diet in controlling symptoms of irritable bowel syndrome (IBS) has gained much traction over the years,1 but until recently, diet therapy for IBS has been hindered by a lack of quality evidence, in part because of the challenges of conducting dietary clinical trials.
Several clinical trials have now been done that support a diet low in fermentable oligosaccharides, disaccharides, monosaccharides, and polyols (FODMAPs) for managing IBS. Although restrictive and difficult to follow, the low-FODMAP diet is gaining popularity.
This article provides an overview of dietary interventions used to manage IBS, focusing on the low-FODMAP diet. We discuss mechanisms of malabsorption of FODMAPs and the role of FODMAPS in symptom induction; highlight clinical trials that provide evidence of benefits of the diet for IBS; and discuss the steps to implement it. We also address the nutritional adequacy of the diet and its potential effects on the gut microbiome.
IBS IS A COMMON FUNCTIONAL DISORDER
IBS is one of the most commonly diagnosed gastrointestinal disorders, and it has a significant impact on quality of life.2 It is a functional disorder characterized by chronic abdominal pain and altered bowel habits in the absence of a structural or organic cause.
The Rome IV diagnostic criteria define IBS by the following:
- Recurrent abdominal pain or discomfort at least 1 day a week in the last 3 months, associated with two or more of the following:
- Symptoms improved by defecation
- Onset associated with a change in frequency of stool
- Onset associated with a change in form or appearance of stool.
IBS mainly arises during young adulthood but can be diagnosed at any age.3
The pathophysiology of IBS involves mechanisms such as bowel distention, altered bowel motility, visceral hypersensitivity, and disruption of mucosal permeability.4 Several therapeutic modalities targeting these mechanisms have been implemented in IBS management, including antispasmodics, laxatives, antidepressants, antibiotics, and behavioral therapy. Diet is only one line of treatment and is most effective when part of a multipronged approach.
TRADITIONAL DIETARY MANAGEMENT
Diet is important in inducing the symptoms of IBS—and in controlling them. Patients identify eating as a common precipitator of symptoms, but the complex diet-symptom interaction is not fully understood and varies widely among patients. Traditional dietary advice for IBS includes adhering to a regular meal pattern, avoiding large meals, and reducing intake of fat, insoluble fibers, caffeine, spicy and gas-producing foods, and carbonated beverages.5,6
Increase soluble fiber
Fiber and fiber supplements, particularly soluble fibers such as psyllium, calcium polycarbophil, and ispaghula husk are often recommended. A meta-analysis7 found that soluble fiber but not insoluble fiber (eg, wheat bran) is associated with an improvement in IBS symptoms (relative risk [RR] 0.84, 95% confidence interval [CI] 0.73–0.94). By improving stool consistency and accelerating transit, soluble fiber is especially useful in constipation-predominant IBS while posing a low risk for adverse outcomes.7 Fiber should be started at a low dose and gradually increased over several weeks to as much as 20 to 30 g/day.
Avoid wheat
Only about 4% of patients with IBS also have celiac disease, but estimating the prevalence of nonceliac gluten sensitivity is confounded by overlapping symptoms. There is some evidence implicating gluten in IBS: celiac disease and IBS overlap in their symptoms, and symptoms are often precipitated by gluten-containing foods in patients with IBS.8 The pathogenesis of gluten-induced (or wheat-induced) symptoms in IBS is unclear, and studies have had conflicting results as to the benefits of gluten restriction in IBS.9
In a study of patients with IBS whose symptoms improved when they started a gluten-free and low-FODMAP diet, symptoms did not return when gluten was reintroduced, suggesting that it is the fructan (a FODMAP) component of wheat rather than gluten that contributes to symptoms in IBS.10
Probiotics
Probiotics are increasingly being recommended as dietary supplements for people with IBS, as awareness increases of the importance of the gut microbiota. In addition to their effects on the gut microbiota, probiotics in IBS have been shown to have anti-inflammatory effects, to alter gut motility, to modulate visceral hypersensitivity, and to restore epithelial integrity.
In a meta-analysis, Ford et al11 found that probiotics improved global IBS symptoms more than placebo (RR 0.79, 95% CI 0.70–0.89) and also reduced abdominal pain, bloating, and flatulence scores.
Which species and strains are most beneficial and the optimal dosing and duration of treatment are still unclear. Data from studies of prebiotics (nutrients that encourage the growth of probiotic bacteria) and synbiotics (combinations of prebiotics and probiotics) are limited and insufficient to draw conclusions.
FODMAPS ARE SHORT-CHAIN CARBOHYDRATES
The term FODMAPs was initially coined by researchers at Monash University in Australia to describe a collection of poorly absorbed short-chain fermentable carbohydrates that are natural components of many foods:
- Oligosaccharides, including fructans (which include inulins) and galacto-oligosaccharides
- Disaccharides, including lactose and sucrose
- Monosaccharides, including fructose
- Polyols, including sorbitol and mannitol.12
Intake of FODMAPs, especially fructose, has increased in Western diets over the past several decades from increased consumption of fruits and concentrated fruit juices, as well as from the widespread use of high-fructose corn syrup in processed foods and beverages.13
FODMAPs ARE POORLY ABSORBED
Different FODMAPs can be poorly absorbed for different reasons (Table 1). The poor absorption is related either to reduced or absent digestive enzymes (ie, hydrolases) or to slow transport across the intestinal mucosa. Excess FODMAPs in the distal small intestine and proximal colon exert osmotic pressure, drawing more water into the lumen. FODMAPs are also rapidly fermented by colonic bacteria, producing gas, bowel distention, and altered motility, all of which induce IBS symptoms.14
Fructans are fructose polymers that are not absorbed in human intestines. They have no intestinal hydrolases and no mechanisms for direct transport across the epithelium. However, a negligible amount may be absorbed after being degraded by microbes in the gut.15 Most dietary fructans are obtained from wheat and onion, which are actually low in fructans but tend to be consumed in large quantities.16
Galacto-oligosaccharides are available for colonic fermentation after ingestion due to lack of a human alpha-galactosidase. Common sources of galacto-oligosaccharides include legumes, nuts, seeds, some grains, dairy products, human milk, and some commercially produced forms added to infant formula.17,18
Lactose is poorly absorbed in people with lactase deficiency. It is mainly present in dairy products but is also added to commercial foods, including breads, cakes, and some diet products.19
Fructose is the most abundant FODMAP in the Western diet. It is either present as a free sugar or generated from the digestive breakdown of sucrose. In the intestine, it is absorbed via a direct low-capacity glucose transporter (GLUT)-5 and through GLUT-2, which is more efficient but requires the coexistence of glucose. Because of this requirement, fructose is more likely to be malabsorbed when present in excess of glucose, as in people with diminished sucrase activity. The main sources of fructose in the Western diet are fruits and fruit products, honey, and foods with added high-fructose sweeteners.13
Polyols such as sorbitol and mannitol are absorbed by slow passive diffusion because they have no active intestinal transport system. They are found in fruits and vegetables. Sugar-free chewing gum is a particularly rich source of sorbitol.20
QUANTIFYING FODMAP CONTENT
As interest in the low-FODMAP diet grew, studies were conducted to quantify FODMAPs in foods. One study used high-performance liquid chromatography to analyze FODMAP content in foods,21 and another evaluated fructan levels in a variety of fruits and vegetables using enzymatic hydrolysis.22 The Monash University low-FODMAP diet smartphone application provides patients and healthcare providers easy access to updated and detailed food analyses.23
Table 2 lists foods high in FODMAPs along with low-FODMAP alternatives. Total FODMAP intake is important, as the effects are additive.24 Readers and patients can be directed to the following websites for more information on the low-FODMAP diet: www.med.monash.edu/cecs/gastro/fodmap or www.ibsfree.net/what-is-fodmap-diet.
LOW-FODMAP DIET REDUCES SYMPTOMS
The low-FODMAP diet was inspired by the results of several studies that evaluated the role of dietary carbohydrates in inducing IBS symptoms and found improvement with their restriction.25,26
One study found that 74% of patients with IBS had less bloating, nausea, abdominal pain, and diarrhea when they restricted their intake of fructose and fructans.27
A prospective trial randomized 41 patients with IBS to 4 weeks of either a low-FODMAP diet or their habitual diet.28 The low-FODMAP diet resulted in greater improvement in overall IBS symptoms (P < .05) and stool frequency (P = .008). This study was limited by different habitual diets between patients and by lack of standardization of the low-FODMAP diet.
Halmos et al,29 in a randomized crossover trial, compared gastrointestinal symptoms in IBS patients over 3 weeks on a low-FODMAP diet vs a moderate-FODMAP (ie, regular) diet, as well as in healthy controls. Food was provided by the study and was matched for all nutrients. Up to 70% of the IBS patients had significantly lower overall symptom scores while on a low-FODMAP diet vs IBS patients on a regular diet (P < .001); bloating, abdominal pain, and flatulence were reduced. Symptoms were minimal and unaffected by either diet in the healthy controls.
A double-blind trial30 randomly assigned 25 patients with IBS who initially responded to a low-FODMAP diet to be challenged by a graduated dose of fructose alone, fructans alone, a combination of both, or glucose. The severity of overall and individual symptoms was markedly more reduced with glucose consumption than with the other carbohydrates: 70% of patients receiving fructose, 77% of those receiving fructans, and 79% of those receiving a mixture of both reported that their symptoms were not adequately controlled, compared with 14% of patients receiving glucose (P ≤ .002).30
Murray et al31 evaluated the gastrointestinal tract after a carbohydrate challenge consisting of 0.5 L of water containing 40 g of glucose, fructose, or inulin or a combination of 40 g of glucose and 40 g of fructose in 16 healthy volunteers. Magnetic resonance imaging was performed hourly for 5 hours to assess the volume of gastric contents, small-bowel water content, and colonic gas. Breath hydrogen was also measured, and symptoms were recorded after each imaging session.
Fructose significantly increased small-bowel water content compared with glucose (mean difference 28 L/min, P < .001), but combined glucose and fructose lessened the effect. Inulin had no significant effect on small-bowel water content (mean difference with glucose 2 L/min, P > .7) but led to the greatest production of colonic gas compared with glucose alone (mean difference 15 L/min, P < .05) and combined glucose and fructose (mean difference 12 L/min, P < .05). Inulin also produced the most breath hydrogen: 81% of participants had a rise after drinking inulin compared with 50% after drinking fructose. Glucose did not affect breath hydrogen concentrations, and combined glucose and fructose significantly reduced the concentration measured vs fructose alone. In patients who reported “gas” symptoms, a correlation was observed between the volume of gas in the colon and gas symptoms (r = 0.59, P < .0001).31
The authors concluded31 that long-chain carbohydrates such as inulin have a greater effect on colonic gas production and little effect on small-bowel water content, whereas small-chain FODMAPs such as fructose are likely to cause luminal distention in both the small and large intestines. The study also showed that combining equal amounts of glucose and fructose reduces malabsorption of fructose in the small bowel and reduces the effect of fructose on small-bowel water content and breath hydrogen concentration.31
PROBIOTICS HELP
A Danish study32 randomized 123 patients with IBS to one of three treatments: a low-FODMAP diet, a normal diet with probiotics containing the strain Lactobacillus rhamnosus GG (two capsules daily), or no special intervention. Symptoms were recorded weekly. IBS severity scores at week 6 were lower in patients on either the low-FODMAP diet or probiotics compared with the control group (P < .01). Subgroup analysis determined that patients with primarily diarrheal symptoms were more likely to have improved quality of life with the low-FODMAP diet.
A LOW-FODMAP DIET MAY ALSO HELP IN INFLAMMATORY BOWEL DISEASE
The low-FODMAP diet has also been studied in patients with inflammatory bowel disease with functional gut symptoms. In a retrospective pilot study,33 overall symptoms improved in about half of such patients on a low-FODMAP diet. A controlled dietary intervention trial is needed to confirm these findings and define the role of the low-FODMAP approach for patients with inflammatory bowel disease.
Marsh et al34 performed a meta-analysis of six randomized clinical trials and 16 nonrandomized interventions of a low-FODMAP diet on improving functional gastrointestinal symptoms in patients with either IBS or inflammatory bowel disease. They found significant improvements in:
- IBS Symptoms Severity Scores in the randomized trials (odds ratio [OR] 0.44, 95% CI 0.25–0.76)
- IBS Symptoms Severity Scores in the nonrandomized interventions (OR 0.03, 95% CI 0.01–0.2)
- IBS Quality of Life scores in the randomized trials (OR 1.84, 95% CI 1.12–3.03)
- IBS Quality of Life scores in the nonrandomized interventions (OR 3.18, 95% CI 1.6–6.31)
- Overall symptom severity in the randomized trials (OR 1.81, 95% CI 1.11–2.95).
DIETARY COUNSELING IS RECOMMENDED
Adherence is a major factor in the success of the low-FODMAP diet in IBS management and is strongly correlated with improved symptoms.35 Patients should be counseled on the role of food in inducing their symptoms. Haphazard dietary advice can be detrimental to outcomes, as many diets restrict food groups, impairing the consumption of essential nutrients.36 The involvement of a knowledgeable dietitian is helpful, as physicians may lack sufficient training in dietary skills and knowledge of food composition.
Access to and cost of dietary counseling can be prohibitive for some patients. Group consultation, which can decrease costs to each patient, has been found to be as effective as one-on-one sessions when administering the low-FODMAP diet in functional bowel disorders.37
ELIMINATION, THEN REINTRODUCTION
Before embarking on the low-FODMAP diet, the patient’s interest in making dietary changes should be explored, a dietary history taken, and unusual food choices or dietary behaviors assessed. The patient’s ability to adopt a restricted diet should also be gauged.
The diet should be implemented in two phases. The initial phase involves strict elimination of foods high in FODMAPs, usually over 6 to 8 weeks.38 Symptom control should be assessed: failure to control symptoms requires assessment of adherence.
If symptoms are successfully controlled, then the second phase should begin with the aim of following a less-restricted version of the diet as tolerated. Foods should gradually be phased back in and symptoms monitored. This approach minimizes unnecessary dietary restriction and ensures that a maximum variety in the diet is achieved while maintaining adequate symptom control.39
LOW-FODMAP DIET ALTERS THE GUT MICROBIOTA
Multiple putative benefits of certain bacterial species for colonic health have been reported, including the production of short-chain fatty acids. Colonic luminal concentrations of short-chain fatty acids may be important to gut health, given their role in intestinal secretion, absorption, motility, and epithelial cell structure. Because short-chain fatty acids are products of bacterial fermentation, a change in the delivery of fermentable substrates to the colon would be expected to alter the concentrations and output of fecal short-chain fatty acids.18
Several studies evaluated the effect of the low-FODMAP diet on intestinal microbiota, finding a change in the bacterial profile in the stool of patients who adopt this diet. Staudacher et al28 found a marked reduction in luminal bifidobacteria concentration after 4 weeks of a low-FODMAP diet in patients with IBS.
A single-blind randomized crossover trial40 investigated the effects of a low-FODMAP diet vs a carefully matched typical Australian diet in 27 patients with IBS and 6 healthy controls. Marked differences in absolute and relative bacterial abundance and diversity were found between the diets, but not in short-chain fatty acids or gut transit time. Compared with fecal microbiota on the typical diet, low FODMAP intake was associated with reduced absolute abundance of bacteria, and the typical FODMAP diet had evidence of stimulation of the growth of bacterial groups with putative health benefits.
The authors concluded40 that the functional significance and health implications of such changes are reasons for caution when reducing FODMAP intake in the long term and recommended liberalizing FODMAP restriction to the level of adequate symptom control in IBS patients. The study also recommended that people without symptoms not go on the low-FODMAP diet.40
Molecular approaches to characterize the gut microbiota are also being explored in an effort to identify its association with diet.
The sustainability of changes in gut microbiota and the potential long-term impact on health of following a low-FODMAP diet require further evaluation. In the meantime, patients following this diet should have FODMAP foods reintroduced based on tolerance and should consider taking probiotic supplements.41
DIETARY ADEQUACY OF THE LOW-FODMAP DIET
Continual dietary counseling should minimize nutritional inadequacies and ensure that FODMAPS are restricted only enough to control symptoms. Because no single food group is completely eliminated in this diet, patients are unlikely to experience inadequate nutrition.
Ledochowski et al26 found that in the initial, strict phase of the diet, total intake of carbohydrates (eg, starches, sugars) was reduced but intake of total energy, protein, fat, and nonstarch polysaccharides was not affected. Calcium intake was reduced in those following a low-FODMAP diet for 4 weeks.
The diet can also reduce total fiber intake and subsequently worsen constipation-predominant IBS. For those patients, lightly fermented high-fiber alternatives like oat and rice bran can be used.
ACCUMULATING EVIDENCE
The low-FODMAP diet is accumulating quality evidence for its effectiveness in controlling the functional gastrointestinal symptoms in patients with IBS. It can be difficult to adhere to over the long term due to its restrictiveness, and it is important to gradually liberalize the diet while tailoring it to the individual patient and monitoring symptoms. Further clinical trials are needed to evaluate this diet in different IBS subtypes and other gastrointestinal disorders, while defining its nutritional adequacy and effects on the intestinal microbiota profile.
- Hayes P, Corish C, O’Mahony E, Quigley EM. A dietary survey of patients with irritable bowel syndrome. J Hum Nutr Diet 2014; 27(suppl 2):36–47.
- Pare P, Gray J, Lam S, et al. Health-related quality of life, work productivity, and health care resource utilization of subjects with irritable bowel syndrome: baseline results from LOGIC (Longitudinal Outcomes Study of Gastrointestinal Symptoms in Canada), a naturalistic study. Clin Ther 2006; 28:1726–1735; discussion 1710–1711.
- Lacey BE, Mearin F, Chang L, et al. Bowel disorders. Gastroenterology 2016; 150:1393–1407.
- Drossman DA, Camilleri M, Mayer EA, Whitehead WE. AGA technical review on irritable bowel syndrome. Gastroenterology 2002; 123:2108–2131.
- Floch MH, Narayan R. Diet in the irritable bowel syndrome. J Clin Gastroenterol 2002; 35(suppl 1):S45–S52.
- Reding KW, Cain KC, Jarrett ME, Eugenio MD, Heitkemper MM. Relationship between patterns of alcohol consumption and gastrointestinal symptoms among patients with irritable bowel syndrome. Am J Gastroenterol 2013; 108:270–276.
- Moayyedi P, Quigley EM, Lacy BE, et al. The effect of fiber supplementation on irritable bowel syndrome: a systematic review and meta-analysis. Am J Gastroenterol 2014; 109:1367–1374.
- Vazquez Roque MI, Camilleri M, Smyrk T, et al. A controlled trial of gluten-free diet in patients with irritable bowel syndrome-diarrhea: effects on bowel frequency and intestinal function. Gastroenterology 2013; 144:903–911.
- Biesiekierski JR, Newnham ED, Irving PM, et al. Gluten causes gastrointestinal symptoms in subjects without celiac disease: a double-blind randomized placebo-controlled trial. Am J Gastroenterol 2011; 106:508–515.
- Biesiekierski JR, Peters SL, Newnham ED, Rosella O, Muir JG, Gibson PR. No effects of gluten in patients with self-reported non-celiac gluten sensitivity after dietary reduction of fermentable, poorly absorbed, short-chain carbohydrates. Gastroenterology 2013; 145:320–328.
- Ford AC, Quigley EM, Lacy BE, et al. Efficacy of prebiotics, probiotics, and synbiotics in irritable bowel syndrome and chronic idiopathic constipation: systematic review and meta-analysis. Gastroenterology 2013; 145:320–328.e1–e3.
- Central Clinical School, Monash University and The Alfred Hospital. The Monash University Low FODMAP Diet. 4th ed. Melbourne, Australia: Monash University; 2012.
- Parker K, Salas M, Nwosu VC. High fructose corn syrup: production, uses and public health concerns. Biotechnol Mol Biol Rev 2010; 5:71–78.
- Clausen MR, Jorgensen J, Mortensen PB. Comparison of diarrhea induced by ingestion of fructooligosaccharide idolax and disaccharide lactulose: role of osmolarity versus fermentation of malabsorbed carbohydrate. Dig Dis Sci 1998; 43:2696–2707.
- Barrett JS, Gearry RB, Muir JG, et al. Dietary poorly absorbed, short-chain carbohydrates increase delivery of water and fermentable substrates to the proximal colon. Aliment Pharmacol Ther 2010; 31:874–882.
- Whelan K, Abrahmsohn O, David GJ, et al. Fructan content of commonly consumed wheat, rye and gluten-free breads. Int J Food Sci Nutr 2011; 62:498–503.
- Sangwan V, Tomar SK, Singh RR, Singh AK, Ali B. Galactooligosaccharides: novel components of designer foods. J Food Sci 2011; 76:R103–R111.
- Russell DA, Ross RP, Fitzgerald GF, Stanton C. Metabolic activities and probiotic potential of bifidobacteria. Int J Food Microbiol 2011; 149:88–105.
- Lomer MC, Parkes GC, Sanderson JD. Review article: lactose intolerance in clinical practice—myths and realities. Aliment Pharmacol Ther 2008; 27:93–103.
- Langkilde AM, Andersson H, Schweizer TF, Würsch P. Digestion and absorption of sorbitol, maltitol and isomalt from the small bowel. A study in ileostomy subjects. Eur J Clin Nutr 1994; 48:768–775.
- Muir JG, Rose R, Rosella O, et al. Measurement of short-chain carbohydrates in common Australian vegetables and fruits by high-performance liquid chromatography (HPLC). J Agric Food Chem 2009; 57:554–565.
- Muir JG, Shepherd SJ, Rosella O, Rose R, Barrett JS, Gibson PR. Fructan and free fructose content of common Australian vegetables and fruit. J Agric Food Chem 2007; 55:6619–6627.
- Monash University. Monash launches Low FODMAP Diet smartphone app. http://med.monash.edu.au/news/2012/fodmap-app.html. Accessed July 13, 2016.
- Fedewa A, Rao SS. Dietary fructose intolerance, fructan intolerance and FODMAPs. Curr Gastroenterol Rep 2014; 16:370.
- Born P, Vierling T, Barina W. Fructose malabsorption and the irritable bowel syndrome. Gastroenterology 1991; 101:1454.
- Ledochowski M, Widner B, Bair H, Probst T, Fuchs D. Fructose- and sorbitol-reduced diet improves mood and gastrointestinal disturbances in fructose malabsorbers. Scand J Gastroenterol 2000; 35:1048–1052.
- Shepherd SJ, Gibson PR. Fructose malabsorption and symptoms of irritable bowel syndrome: guidelines for effective dietary management. J Am Diet Assoc 2006; 106:1631–1639.
- Staudacher HM, Lomer MC, Anderson JL, et al. Fermentable carbohydrate restriction reduces luminal bifidobacteria and gastrointestinal symptoms in patients with irritable bowel syndrome. J Nutr 2012; 142:1510–1518.
- Halmos EP, Power VA, Shepherd SJ, Gibson PR, Muir JG. A diet low in FODMAPs reduces symptoms of irritable bowel syndrome. Gastroenterology 2014; 146:67–75.e5.
- Shepherd SJ, Parker FC, Muir JG, Gibson PR. Dietary triggers of abdominal symptoms in patients with irritable bowel syndrome: randomized placebo-controlled evidence. Clin Gastroenterol Hepatol 2008; 6:765–771.
- Murray K, Wilkinson-Smith V, Hoad C, et al. Differential effects of FODMAPs (fermentable oligo-, di-, mono-saccharides and polyols) on small and large intestinal contents in healthy subjects shown by MRI. Am J Gastroenterol 2014; 109:110–119.
- Pedersen N, Andersen NN, Vegh Z, et al. Ehealth: low FODMAP diet vs Lactobacillus rhamnosus GG in irritable bowel syndrome. World J Gastroenterol 2014; 20:16215–16226.
- Gearry RB, Irving PM, Barrett JS, Nathan DM, Shepherd SJ, Gibson PR. Reduction of dietary poorly absorbed short-chain carbohydrates (FODMAPs) improves abdominal symptoms in patients with inflammatory bowel disease-a pilot study. J Crohns Colitis 2009; 3:8–14.
- Marsh A, Eslick EM, Eslick GD. Does a diet low in FODMAPs reduce symptoms associated with functional gastrointestinal disorders? A comprehensive systematic review and meta-analysis. Eur J Nutr 2015 May 17. Epub ahead of print.
- de Roest RH, Dobbs BR, Chapman BA, et al. The low FODMAP diet improves gastrointestinal symptoms in patients with irritable bowel syndrome: a prospective study. Int J Clin Pract 2013; 67:895–903.
- Gibson PR, Barrett JS, Muir JG. Functional bowel symptoms and diet. Intern Med J 2013; 43:1067–1074.
- Whigham L, Joyce T, Harper G, et al. Clinical effectiveness and economic costs of group versus one-to-one education for short-chain fermentable carbohydrate restriction (low FODMAP diet) in the management of irritable bowel syndrome. J Hum Nutr Diet 2015; 28:687–696.
- Shepherd SJ, Lomer MC, Gibson PR. Short-chain carbohydrates and functional gastrointestinal disorders. Am J Gastroenterol 2013; 108:707–717.
- Shepherd SJ, Halmos E, Glance S. The role of FODMAPs in irritable bowel syndrome. Curr Opin Clin Nutr Metab Care 2014; 17:605–609.
- Halmos EP, Christophersen CT, Bird AR, Shepherd SJ, Gibson PR, Muir JG. Diets that differ in their FODMAP content alter the colonic luminal microenvironment. Gut 2015; 64:93–100.
- Staudacher HM, Irving PM, Lomer MC, Whelan K. Mechanisms and efficacy of dietary FODMAP restriction in IBS. Nat Rev Gastroenterol Hepatol 2014; 11:256–266.
- Hayes P, Corish C, O’Mahony E, Quigley EM. A dietary survey of patients with irritable bowel syndrome. J Hum Nutr Diet 2014; 27(suppl 2):36–47.
- Pare P, Gray J, Lam S, et al. Health-related quality of life, work productivity, and health care resource utilization of subjects with irritable bowel syndrome: baseline results from LOGIC (Longitudinal Outcomes Study of Gastrointestinal Symptoms in Canada), a naturalistic study. Clin Ther 2006; 28:1726–1735; discussion 1710–1711.
- Lacey BE, Mearin F, Chang L, et al. Bowel disorders. Gastroenterology 2016; 150:1393–1407.
- Drossman DA, Camilleri M, Mayer EA, Whitehead WE. AGA technical review on irritable bowel syndrome. Gastroenterology 2002; 123:2108–2131.
- Floch MH, Narayan R. Diet in the irritable bowel syndrome. J Clin Gastroenterol 2002; 35(suppl 1):S45–S52.
- Reding KW, Cain KC, Jarrett ME, Eugenio MD, Heitkemper MM. Relationship between patterns of alcohol consumption and gastrointestinal symptoms among patients with irritable bowel syndrome. Am J Gastroenterol 2013; 108:270–276.
- Moayyedi P, Quigley EM, Lacy BE, et al. The effect of fiber supplementation on irritable bowel syndrome: a systematic review and meta-analysis. Am J Gastroenterol 2014; 109:1367–1374.
- Vazquez Roque MI, Camilleri M, Smyrk T, et al. A controlled trial of gluten-free diet in patients with irritable bowel syndrome-diarrhea: effects on bowel frequency and intestinal function. Gastroenterology 2013; 144:903–911.
- Biesiekierski JR, Newnham ED, Irving PM, et al. Gluten causes gastrointestinal symptoms in subjects without celiac disease: a double-blind randomized placebo-controlled trial. Am J Gastroenterol 2011; 106:508–515.
- Biesiekierski JR, Peters SL, Newnham ED, Rosella O, Muir JG, Gibson PR. No effects of gluten in patients with self-reported non-celiac gluten sensitivity after dietary reduction of fermentable, poorly absorbed, short-chain carbohydrates. Gastroenterology 2013; 145:320–328.
- Ford AC, Quigley EM, Lacy BE, et al. Efficacy of prebiotics, probiotics, and synbiotics in irritable bowel syndrome and chronic idiopathic constipation: systematic review and meta-analysis. Gastroenterology 2013; 145:320–328.e1–e3.
- Central Clinical School, Monash University and The Alfred Hospital. The Monash University Low FODMAP Diet. 4th ed. Melbourne, Australia: Monash University; 2012.
- Parker K, Salas M, Nwosu VC. High fructose corn syrup: production, uses and public health concerns. Biotechnol Mol Biol Rev 2010; 5:71–78.
- Clausen MR, Jorgensen J, Mortensen PB. Comparison of diarrhea induced by ingestion of fructooligosaccharide idolax and disaccharide lactulose: role of osmolarity versus fermentation of malabsorbed carbohydrate. Dig Dis Sci 1998; 43:2696–2707.
- Barrett JS, Gearry RB, Muir JG, et al. Dietary poorly absorbed, short-chain carbohydrates increase delivery of water and fermentable substrates to the proximal colon. Aliment Pharmacol Ther 2010; 31:874–882.
- Whelan K, Abrahmsohn O, David GJ, et al. Fructan content of commonly consumed wheat, rye and gluten-free breads. Int J Food Sci Nutr 2011; 62:498–503.
- Sangwan V, Tomar SK, Singh RR, Singh AK, Ali B. Galactooligosaccharides: novel components of designer foods. J Food Sci 2011; 76:R103–R111.
- Russell DA, Ross RP, Fitzgerald GF, Stanton C. Metabolic activities and probiotic potential of bifidobacteria. Int J Food Microbiol 2011; 149:88–105.
- Lomer MC, Parkes GC, Sanderson JD. Review article: lactose intolerance in clinical practice—myths and realities. Aliment Pharmacol Ther 2008; 27:93–103.
- Langkilde AM, Andersson H, Schweizer TF, Würsch P. Digestion and absorption of sorbitol, maltitol and isomalt from the small bowel. A study in ileostomy subjects. Eur J Clin Nutr 1994; 48:768–775.
- Muir JG, Rose R, Rosella O, et al. Measurement of short-chain carbohydrates in common Australian vegetables and fruits by high-performance liquid chromatography (HPLC). J Agric Food Chem 2009; 57:554–565.
- Muir JG, Shepherd SJ, Rosella O, Rose R, Barrett JS, Gibson PR. Fructan and free fructose content of common Australian vegetables and fruit. J Agric Food Chem 2007; 55:6619–6627.
- Monash University. Monash launches Low FODMAP Diet smartphone app. http://med.monash.edu.au/news/2012/fodmap-app.html. Accessed July 13, 2016.
- Fedewa A, Rao SS. Dietary fructose intolerance, fructan intolerance and FODMAPs. Curr Gastroenterol Rep 2014; 16:370.
- Born P, Vierling T, Barina W. Fructose malabsorption and the irritable bowel syndrome. Gastroenterology 1991; 101:1454.
- Ledochowski M, Widner B, Bair H, Probst T, Fuchs D. Fructose- and sorbitol-reduced diet improves mood and gastrointestinal disturbances in fructose malabsorbers. Scand J Gastroenterol 2000; 35:1048–1052.
- Shepherd SJ, Gibson PR. Fructose malabsorption and symptoms of irritable bowel syndrome: guidelines for effective dietary management. J Am Diet Assoc 2006; 106:1631–1639.
- Staudacher HM, Lomer MC, Anderson JL, et al. Fermentable carbohydrate restriction reduces luminal bifidobacteria and gastrointestinal symptoms in patients with irritable bowel syndrome. J Nutr 2012; 142:1510–1518.
- Halmos EP, Power VA, Shepherd SJ, Gibson PR, Muir JG. A diet low in FODMAPs reduces symptoms of irritable bowel syndrome. Gastroenterology 2014; 146:67–75.e5.
- Shepherd SJ, Parker FC, Muir JG, Gibson PR. Dietary triggers of abdominal symptoms in patients with irritable bowel syndrome: randomized placebo-controlled evidence. Clin Gastroenterol Hepatol 2008; 6:765–771.
- Murray K, Wilkinson-Smith V, Hoad C, et al. Differential effects of FODMAPs (fermentable oligo-, di-, mono-saccharides and polyols) on small and large intestinal contents in healthy subjects shown by MRI. Am J Gastroenterol 2014; 109:110–119.
- Pedersen N, Andersen NN, Vegh Z, et al. Ehealth: low FODMAP diet vs Lactobacillus rhamnosus GG in irritable bowel syndrome. World J Gastroenterol 2014; 20:16215–16226.
- Gearry RB, Irving PM, Barrett JS, Nathan DM, Shepherd SJ, Gibson PR. Reduction of dietary poorly absorbed short-chain carbohydrates (FODMAPs) improves abdominal symptoms in patients with inflammatory bowel disease-a pilot study. J Crohns Colitis 2009; 3:8–14.
- Marsh A, Eslick EM, Eslick GD. Does a diet low in FODMAPs reduce symptoms associated with functional gastrointestinal disorders? A comprehensive systematic review and meta-analysis. Eur J Nutr 2015 May 17. Epub ahead of print.
- de Roest RH, Dobbs BR, Chapman BA, et al. The low FODMAP diet improves gastrointestinal symptoms in patients with irritable bowel syndrome: a prospective study. Int J Clin Pract 2013; 67:895–903.
- Gibson PR, Barrett JS, Muir JG. Functional bowel symptoms and diet. Intern Med J 2013; 43:1067–1074.
- Whigham L, Joyce T, Harper G, et al. Clinical effectiveness and economic costs of group versus one-to-one education for short-chain fermentable carbohydrate restriction (low FODMAP diet) in the management of irritable bowel syndrome. J Hum Nutr Diet 2015; 28:687–696.
- Shepherd SJ, Lomer MC, Gibson PR. Short-chain carbohydrates and functional gastrointestinal disorders. Am J Gastroenterol 2013; 108:707–717.
- Shepherd SJ, Halmos E, Glance S. The role of FODMAPs in irritable bowel syndrome. Curr Opin Clin Nutr Metab Care 2014; 17:605–609.
- Halmos EP, Christophersen CT, Bird AR, Shepherd SJ, Gibson PR, Muir JG. Diets that differ in their FODMAP content alter the colonic luminal microenvironment. Gut 2015; 64:93–100.
- Staudacher HM, Irving PM, Lomer MC, Whelan K. Mechanisms and efficacy of dietary FODMAP restriction in IBS. Nat Rev Gastroenterol Hepatol 2014; 11:256–266.
KEY POINTS
- In clinical trials, the low-FODMAP diet has been found to improve symptoms in up to 70% of patients with IBS.
- FODMAPs are poorly absorbed for a variety of reasons.
- High-FODMAP foods include wheat, onions, legumes, dairy products, and many fruits and vegetables.
- The diet initially involves strict elimination of foods high in FODMAPs, after which they are gradually reintroduced as tolerated.
- A low-FODMAP diet may have negative effects on the gut microbiome. Therefore, we should be cautious about recommending this diet in the long term.
- Probiotics have a beneficial effect in IBS and can be taken concurrently with the diet.

The future of ketamine in psychiatry
Ketamine, a high-affinity, noncompetitive N-methyl-
How ketamine works
Water- and lipid-soluble, ketamine is available in oral, topical, IM, and IV forms. Plasma concentrations reach maximum levels minutes after IV infusion; 5 to 15 minutes after IM administration; and 30 minutes after oral ingestion.1 The duration of action is as long as 2 hours after IM injection, and 4 to 6 hours orally. Metabolites are eliminated in urine.
Ketamine, co-prescribed with stimulants and some antidepressant drugs, can induce unwanted effects, such as increased blood pressure. Auditory and visual hallucinations are reported occasionally, especially in patients receiving a high dosage or in those with alcohol dependence.1 Hypertension, tachycardia, cardiac arrhythmia, and pain at injection site are the most common adverse effects.
Some advantages over ECT in treating depression
The efficacy of electroconvulsive therapy (ECT) in alleviating depression depends on seizure duration. Compared with methohexital, an anesthetic used for ECT, ketamine offers some advantages:
- increased ictal time
- augmented mid-ictal slow-wave amplitude
- shortened post-treatment re-orientation time
- less cognitive dysfunction.2
Uses for ketamine
Treatment-resistant depression. The glutamatergic system is implicated in depression.2,3 Ketamine works in patients with treatment-resistant depression by blocking glutamate NMDA receptors and increasing the activity of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors, resulting in a rapid, sustained antidepressant effect. Response to ketamine occurs within 2 hours and lasts approximately 1 week.
Bipolar and unipolar depression. Ketamine has rapid antidepressant properties in unipolar and bipolar depression. It is most beneficial in people with a family history of alcohol dependence, because similar glutamatergic system alterations might be involved in the pathophysiology of both disorders.3,4 An antidepressant effect has been reported as soon as 40 minutes after ketamine infusions.3
Suicide prevention. A single sub-anesthetic IV dose of ketamine rapidly diminishes acute suicidal ideation.1 This effect can be maintained through repeated ketamine infusions, episodically on a clinically derived basis. The exact duration and period between ketamine readministrations are not fully established. A variety of clinical-, patient-, and circumstance-related factors, history, response, and physician preferences alter such patterns, in an individualized way. This is also a promising means to reduce hospitalizations and at least mitigate the severity of depressive patient presentations.
Anesthesia and analgesia. Because ketamine induces anesthesia with minimal effect on respiratory function, it could be used in patients with pulmonary conditions.5 Ketamine can provide analgesia during brief operative and diagnostic procedures; because of its hypertensive actions, it is useful in trauma patients with hypotension.A low dose of ketamine effectively diminishes the discomfort of complex regional pain and other pain syndromes.
Abuse potential
There is documented risk of ketamine abuse. It may create psychedelic effects that some people find pleasurable, such as sedation, disinhibition, and altered perceptions.6 There also may be a component of physiological dependence.6
Conclusion
Ketamine’s rapid antidepressant effect results could be beneficial when used in severely depressed and suicidal patients. Given the potential risks of ketamine, safety considerations will determine whether this drug is successful as a therapy for people with a mood disorder.
Further research about ketamine usage including pain management and affective disorders is anticipated.7 Investigations substantiating relative safety and clinical trials are still on-going.8
Related Resources
• Nichols SD, Bishop J. Is the evidence compelling for using ketamine to treat resistant depression? Current Psychiatry. 2015;15(5):48-51.
• National Institute of Mental Health. Highlight: ketamine: a new (and faster) path to treating depression. www.nimh.nih.gov/about/strategic-planning-reports/highlights/highlight-ketamine-a-new-and-faster-path-to-treatingdepression.shtml.
1. Sinner B, Graf BM. Ketamine. Handb Exp Pharmacol. 2008;(128):313-333.
2. Krystal AD, Dean MD, Weiner RD, et al. ECT stimulus intensity: are present ECT devices too limited? Am J Psychiatry. 2000;157(6):963-967.
3. Phelps LE, Brutsche N, Moral JR, et al. Family history of alcohol dependence and initial antidepressant response to an N-methyl-D-aspartate antagonist. Biol Psychiatry. 2009;65:181-184.
4. Nery FG, Stanley JA, Chen HH, et al. Bipolar disorder comorbid with alcoholism: a 1H magnetic resonance spectroscopy study. J Psychiatry Res. 2010;44(5):278-285.
5. Meller, ST. Ketamine: relief from chronic pain through actions at the NMDA receptor. Pain. 1996;68(2-3):435-436.
6. Sassano-Higgins S, Baron D, Juarez G, et al. A review of ketamine abuse and diversion. Depress Anxiety. 2016;33(8):718-727.
7. Jafarinia M, Afarideh M, Tafakhori A, et al. Efficacy and safety of oral ketamine versus diclofenac to alleviate mild to moderate depression in chronic pain patients: A double-blind, randomized, controlled trial. J Affect Disord. 2016;204:1-8.
8. Wan LB, Levitch CF, Perez AM, et al. Ketamine safety and tolerability in clinical trials for treatment-resistant depression. J Clin Psychiatry. 2015;76(3):247-252.
Ketamine, a high-affinity, noncompetitive N-methyl-
How ketamine works
Water- and lipid-soluble, ketamine is available in oral, topical, IM, and IV forms. Plasma concentrations reach maximum levels minutes after IV infusion; 5 to 15 minutes after IM administration; and 30 minutes after oral ingestion.1 The duration of action is as long as 2 hours after IM injection, and 4 to 6 hours orally. Metabolites are eliminated in urine.
Ketamine, co-prescribed with stimulants and some antidepressant drugs, can induce unwanted effects, such as increased blood pressure. Auditory and visual hallucinations are reported occasionally, especially in patients receiving a high dosage or in those with alcohol dependence.1 Hypertension, tachycardia, cardiac arrhythmia, and pain at injection site are the most common adverse effects.
Some advantages over ECT in treating depression
The efficacy of electroconvulsive therapy (ECT) in alleviating depression depends on seizure duration. Compared with methohexital, an anesthetic used for ECT, ketamine offers some advantages:
- increased ictal time
- augmented mid-ictal slow-wave amplitude
- shortened post-treatment re-orientation time
- less cognitive dysfunction.2
Uses for ketamine
Treatment-resistant depression. The glutamatergic system is implicated in depression.2,3 Ketamine works in patients with treatment-resistant depression by blocking glutamate NMDA receptors and increasing the activity of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors, resulting in a rapid, sustained antidepressant effect. Response to ketamine occurs within 2 hours and lasts approximately 1 week.
Bipolar and unipolar depression. Ketamine has rapid antidepressant properties in unipolar and bipolar depression. It is most beneficial in people with a family history of alcohol dependence, because similar glutamatergic system alterations might be involved in the pathophysiology of both disorders.3,4 An antidepressant effect has been reported as soon as 40 minutes after ketamine infusions.3
Suicide prevention. A single sub-anesthetic IV dose of ketamine rapidly diminishes acute suicidal ideation.1 This effect can be maintained through repeated ketamine infusions, episodically on a clinically derived basis. The exact duration and period between ketamine readministrations are not fully established. A variety of clinical-, patient-, and circumstance-related factors, history, response, and physician preferences alter such patterns, in an individualized way. This is also a promising means to reduce hospitalizations and at least mitigate the severity of depressive patient presentations.
Anesthesia and analgesia. Because ketamine induces anesthesia with minimal effect on respiratory function, it could be used in patients with pulmonary conditions.5 Ketamine can provide analgesia during brief operative and diagnostic procedures; because of its hypertensive actions, it is useful in trauma patients with hypotension.A low dose of ketamine effectively diminishes the discomfort of complex regional pain and other pain syndromes.
Abuse potential
There is documented risk of ketamine abuse. It may create psychedelic effects that some people find pleasurable, such as sedation, disinhibition, and altered perceptions.6 There also may be a component of physiological dependence.6
Conclusion
Ketamine’s rapid antidepressant effect results could be beneficial when used in severely depressed and suicidal patients. Given the potential risks of ketamine, safety considerations will determine whether this drug is successful as a therapy for people with a mood disorder.
Further research about ketamine usage including pain management and affective disorders is anticipated.7 Investigations substantiating relative safety and clinical trials are still on-going.8
Related Resources
• Nichols SD, Bishop J. Is the evidence compelling for using ketamine to treat resistant depression? Current Psychiatry. 2015;15(5):48-51.
• National Institute of Mental Health. Highlight: ketamine: a new (and faster) path to treating depression. www.nimh.nih.gov/about/strategic-planning-reports/highlights/highlight-ketamine-a-new-and-faster-path-to-treatingdepression.shtml.
Ketamine, a high-affinity, noncompetitive N-methyl-
How ketamine works
Water- and lipid-soluble, ketamine is available in oral, topical, IM, and IV forms. Plasma concentrations reach maximum levels minutes after IV infusion; 5 to 15 minutes after IM administration; and 30 minutes after oral ingestion.1 The duration of action is as long as 2 hours after IM injection, and 4 to 6 hours orally. Metabolites are eliminated in urine.
Ketamine, co-prescribed with stimulants and some antidepressant drugs, can induce unwanted effects, such as increased blood pressure. Auditory and visual hallucinations are reported occasionally, especially in patients receiving a high dosage or in those with alcohol dependence.1 Hypertension, tachycardia, cardiac arrhythmia, and pain at injection site are the most common adverse effects.
Some advantages over ECT in treating depression
The efficacy of electroconvulsive therapy (ECT) in alleviating depression depends on seizure duration. Compared with methohexital, an anesthetic used for ECT, ketamine offers some advantages:
- increased ictal time
- augmented mid-ictal slow-wave amplitude
- shortened post-treatment re-orientation time
- less cognitive dysfunction.2
Uses for ketamine
Treatment-resistant depression. The glutamatergic system is implicated in depression.2,3 Ketamine works in patients with treatment-resistant depression by blocking glutamate NMDA receptors and increasing the activity of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors, resulting in a rapid, sustained antidepressant effect. Response to ketamine occurs within 2 hours and lasts approximately 1 week.
Bipolar and unipolar depression. Ketamine has rapid antidepressant properties in unipolar and bipolar depression. It is most beneficial in people with a family history of alcohol dependence, because similar glutamatergic system alterations might be involved in the pathophysiology of both disorders.3,4 An antidepressant effect has been reported as soon as 40 minutes after ketamine infusions.3
Suicide prevention. A single sub-anesthetic IV dose of ketamine rapidly diminishes acute suicidal ideation.1 This effect can be maintained through repeated ketamine infusions, episodically on a clinically derived basis. The exact duration and period between ketamine readministrations are not fully established. A variety of clinical-, patient-, and circumstance-related factors, history, response, and physician preferences alter such patterns, in an individualized way. This is also a promising means to reduce hospitalizations and at least mitigate the severity of depressive patient presentations.
Anesthesia and analgesia. Because ketamine induces anesthesia with minimal effect on respiratory function, it could be used in patients with pulmonary conditions.5 Ketamine can provide analgesia during brief operative and diagnostic procedures; because of its hypertensive actions, it is useful in trauma patients with hypotension.A low dose of ketamine effectively diminishes the discomfort of complex regional pain and other pain syndromes.
Abuse potential
There is documented risk of ketamine abuse. It may create psychedelic effects that some people find pleasurable, such as sedation, disinhibition, and altered perceptions.6 There also may be a component of physiological dependence.6
Conclusion
Ketamine’s rapid antidepressant effect results could be beneficial when used in severely depressed and suicidal patients. Given the potential risks of ketamine, safety considerations will determine whether this drug is successful as a therapy for people with a mood disorder.
Further research about ketamine usage including pain management and affective disorders is anticipated.7 Investigations substantiating relative safety and clinical trials are still on-going.8
Related Resources
• Nichols SD, Bishop J. Is the evidence compelling for using ketamine to treat resistant depression? Current Psychiatry. 2015;15(5):48-51.
• National Institute of Mental Health. Highlight: ketamine: a new (and faster) path to treating depression. www.nimh.nih.gov/about/strategic-planning-reports/highlights/highlight-ketamine-a-new-and-faster-path-to-treatingdepression.shtml.
1. Sinner B, Graf BM. Ketamine. Handb Exp Pharmacol. 2008;(128):313-333.
2. Krystal AD, Dean MD, Weiner RD, et al. ECT stimulus intensity: are present ECT devices too limited? Am J Psychiatry. 2000;157(6):963-967.
3. Phelps LE, Brutsche N, Moral JR, et al. Family history of alcohol dependence and initial antidepressant response to an N-methyl-D-aspartate antagonist. Biol Psychiatry. 2009;65:181-184.
4. Nery FG, Stanley JA, Chen HH, et al. Bipolar disorder comorbid with alcoholism: a 1H magnetic resonance spectroscopy study. J Psychiatry Res. 2010;44(5):278-285.
5. Meller, ST. Ketamine: relief from chronic pain through actions at the NMDA receptor. Pain. 1996;68(2-3):435-436.
6. Sassano-Higgins S, Baron D, Juarez G, et al. A review of ketamine abuse and diversion. Depress Anxiety. 2016;33(8):718-727.
7. Jafarinia M, Afarideh M, Tafakhori A, et al. Efficacy and safety of oral ketamine versus diclofenac to alleviate mild to moderate depression in chronic pain patients: A double-blind, randomized, controlled trial. J Affect Disord. 2016;204:1-8.
8. Wan LB, Levitch CF, Perez AM, et al. Ketamine safety and tolerability in clinical trials for treatment-resistant depression. J Clin Psychiatry. 2015;76(3):247-252.
1. Sinner B, Graf BM. Ketamine. Handb Exp Pharmacol. 2008;(128):313-333.
2. Krystal AD, Dean MD, Weiner RD, et al. ECT stimulus intensity: are present ECT devices too limited? Am J Psychiatry. 2000;157(6):963-967.
3. Phelps LE, Brutsche N, Moral JR, et al. Family history of alcohol dependence and initial antidepressant response to an N-methyl-D-aspartate antagonist. Biol Psychiatry. 2009;65:181-184.
4. Nery FG, Stanley JA, Chen HH, et al. Bipolar disorder comorbid with alcoholism: a 1H magnetic resonance spectroscopy study. J Psychiatry Res. 2010;44(5):278-285.
5. Meller, ST. Ketamine: relief from chronic pain through actions at the NMDA receptor. Pain. 1996;68(2-3):435-436.
6. Sassano-Higgins S, Baron D, Juarez G, et al. A review of ketamine abuse and diversion. Depress Anxiety. 2016;33(8):718-727.
7. Jafarinia M, Afarideh M, Tafakhori A, et al. Efficacy and safety of oral ketamine versus diclofenac to alleviate mild to moderate depression in chronic pain patients: A double-blind, randomized, controlled trial. J Affect Disord. 2016;204:1-8.
8. Wan LB, Levitch CF, Perez AM, et al. Ketamine safety and tolerability in clinical trials for treatment-resistant depression. J Clin Psychiatry. 2015;76(3):247-252.