User login
Public speaking fundamentals. The program: Key elements in capturing and holding audience attention
In the first part of this article series (“Preparation: Tips that lead to a solid, engaging presentation,” OBG Manag. 2016;28[7]:31–36.), we offered tips on preparing for a group presentation. In this article, part 2, we discuss the presentation itself and what you can do to capture and hold your audience’s attention.

How to connect with the audience
Let’s assume the meeting host has just introduced you to the audience using, as we suggested in the previous article, an autobiographical profile you provided. You now have the audience’s undivided attention. What you do and say in the next 30 to 60 seconds will set the stage for your program. Following the requisite “thank you” to the host and meeting sponsor, use this time to establish your expertise as a spokesperson on the chosen topic. Or, if the introductory remarks made your expertise plain, you may choose to connect with the audience on an informal, personal level. If you are from out of town, for instance, you could remark on an interesting aspect of the city or region you are visiting that you learned on the Internet before arriving.
Underscore the topic’s importance. On the other hand, you might want to begin with an insightful statistic germane to your talk. For example, a talk on breast cancer might begin with, “According to the American Cancer Society, there are nearly 250,000 new cases of breast cancer each year, and breast cancer accounts for more than 40,000 deaths per year. That means more women die from breast cancer than die in auto accidents each year. So this emphasizes the importance of appropriately screening women for breast cancer annually after age 40.”
An opening story about a patient can be powerful. Better yet, a personal experience reflecting your topic is a great way to connect with your audience members and get their attention. For example, one of us (NHB) gives talks on practice management and practice efficiency. I might talk about when I was called from an exam room 3 times to answer “emergency” phone calls from a patient who wanted only to request her medical records. To ensure that this embarrassment would never happen again, I put in place a system that I then describe for the audience.
Alternatively, an opening that addresses the audience’s unspoken question, “What’s in this for me?” is sure to grab their attention. For instance, a talk on office productivity might begin by promising to share a way to increase annual collections by $250,000 per physician through scheduling adjustments that can increase the number of examined patients by one per hour.
Steer clear of these openings. In general, avoid “I’m delighted to be here” and other clichés. One exception would be if you can make that cliché humorous. For example, if a speaker from the deep South is visiting the northern part of the country in summer, she might say, “Most speakers say they’re delighted to be here, and you may well question their sincerity. However, I’m from New Orleans where the temperature is approaching 105 degrees with 95% humidity. You know I’m really delighted to be here!”
Importantly, avoid starting with an apology. Do not mention problems with the audiovisual equipment or why you arrived late. The audience does not care, and you will immediately lose their attention. They want to be educated and entertained. There is no better way to do this than by offering a compelling and captivating opening that begins the moment after you are introduced.
Finally, avoid use of the “royal I,” as in “I am here to talk about XYZ.” It places you in a position superior to the audience, and that is a turnoff. Instead, you could say to the audience, “The reason you are here is to learn about XYZ.” This places the audience on an equal level with you, and they know there will be something in the presentation for them.
Housekeeping notes
The audience will appreciate knowing how long you plan to speak and whether you will take questions during or after the presentation. Based on our experience, if there are fewer than 20 attendees, we often encourage questions during the program instead of waiting until the end. This makes the program more conversational and usually generates more questions. With a dinner presentation, we prefer to speak while the audience is eating. We usually start after the waiters have taken the orders and the attendees have had their appetizers. We might say we will finish the program by the time they are ready for dessert. We also mention that we will distribute a handout after the presentation so they do not have to worry about following the handout, taking notes, and watching the speaker while trying to eat.
The main body of the program
As for structuring your talk, we suggest you follow this time-honored advice often attributed to Aristotle: Tell the audience what you are going to say, say it, and tell them what you said.
So we begin a presentation by stating the objectives of our program, usually limited to 3 and no more than 4. For example, a talk on hormone therapy (HT) for treating vasomotor symptoms of menopause might mention 1) the history of HT use, 2) which women are appropriate candidates for HT, and 3) how to monitor women who receive HT.
Enhance the talk’s relevance. We like to begin a clinical program with a case scenario wherein we describe how one of our patients had the specific problem and how we used a particular drug, treatment, or device to manage the case. We try to select a patient similar to ones who would be seen by members of the audience.
Simplify as much as possible. We then present the slides exactly as they have been provided by the pharmaceutical company. Most company slides contain too many words as well as diagrams that are too complex for the audience to grasp easily. We try to find one salient point on each slide and focus attention on that single word, phrase, or sentence. We can do this in a small audience by walking over to the screen and pointing it out, or we can use the laser pointer from a distance.
Change things up to keep the message fresh. Let’s be honest, most medical talks are dry and boring. Try to inject some energy and enthusiasm in the middle of the presentation. Every few minutes we tell a story or ask the audience a question. For example, during a program on practice management, one of us (NHB) will relate a story about an unhappy patient and then ask a physician in the audience how he or she might handle the disgruntled patient. This is a nice break from the main content of the presentation, re-engaging the audience in an interactive exchange.
Should you use humor?
Although many physicians attempt to use humor during a presentation, few are talented at stand-up comedy. However, used judiciously humor, like seasoning in fine cuisine, can do great things for a presentation. It can break the ice, drive home a point, and enhance your likeability. It can, though, also backfire. One of us (NHB) once gave a talk to a large audience of pharmaceutical representatives. As part of my wrap-up I displayed a slide from the cover of Economics that showed 2 camels in the mating position. My closing line was that reps need to “hump to it” and get involved with their physicians and be value-added in their product detailing. Afterward, the meeting planner told me that he would never hire me again. He said I had a great program, great material, and a good connection with the audience. But my closing was over the top. I learned my lesson. Never use material that has the potential to offend. If you want to use humor, the self-deprecatingkind is always safest.
Try using visual aids
Our observation is that few physician speakers use visual aids other than their slides. We have learned that audience attention will stay focused on you if you make use of visual aids. For example, if we are speaking to a lay audience about urinary incontinence, we might use a balloon to demonstrate the bladder and the urethra.
Studies have shown that there are more nerve endings from the eye to the brain than from the ear to the brain. Humans purportedly receive 25 times as much stimulus from visual cues than from auditory ones. To paraphrase an old proverb, “One seeing is better than 100 times hearing about”!
A few suggestions regarding the use of visual aids:
- Keep the visual aid out of sight until you are ready to use it. You do not want the audience staring at it when they should be focusing on you or your slide material. We usually keep our visual aids under the table that supports the computer and projector.
- Make certain the visual aid is large enough to be seen by everyone in the audience.
- Do not hand out the aid to the audience during your program. Doing so will divert their attention from you and your material.
- When you have finished using the aid, put it away.
Closing out the program
After we have covered the program’s 3 objectives, we let the audience know we are approaching the end of the presentation. For a dinner program, we try to time the ending just as plates are being cleared and before dessert is served. We then restate the 3 objectives as they might pertain to the attendees’ patients and practices. At this time, we take questions from the audience, even if some were asked during the presentation. We repeat each question when it is asked so that everyone can hear it. (This also gives us a few seconds to think about it and frame our answer.) If it appears that many questions will be asked, we assure everyone that we plan to finish on time and will remain after the program is over to answer additional questions.
Tips on fielding questions. When responding to a question, direct your attention initially to the person who asked it. After that, spend about 20% of the time focused on that person and 80% of the time on the rest of the audience. If you focus only on the questioner, it becomes a one-on-one conversation. You want to end your response with your eyes on the group and not on the questioner. Looking at the group will also act as a bridge to the next question. Although we used to reply to an inquiry with, “That’s a great question,” we now suggest avoiding this comment. Why? Because it is unlikely that you’ll keep using that line, and the next questioner who does not receive the same compliment might feel slighted.
Wrap up. When you announce, “I would like to conclude my program with…,” this is the magical time when you hold the complete attention of the audience. Often, the speaker’s last words are the ones the audience remembers the longest. So this is the time to offer your take-home message. For example, a talk on how to motivate your staff might conclude, “Remember, your staff members are the people that patients encounter first and the ones they see last as they leave the office. Every patient can have a positive experience with you and your practice if you ensure that your personnel are highly motivated. This happens in part by your effort to recognize their accomplishments.” Then hold up your hands and spread out your arms as you end with “Thank you.” The audience likely will applaud and, if your speech is truly exceptional, you might receive a gratifying standing ovation!
Be seated
Renowned for his speeches, Franklin Delano Roosevelt summarized the art of effective speaking when he said, “Be sincere. Be brief. Be seated.” When your time is up, turn the program back over to the meeting host and take a seat.
In the final article in this public speaking series, we will discuss the follow-up steps to take once the program is over, including the call to action or what you want the audience to do after you have left the podium or the speaking venue.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.


The authors report no financial relationships relevant to this article.
The authors report no financial relationships relevant to this article.
The authors report no financial relationships relevant to this article.
In the first part of this article series (“Preparation: Tips that lead to a solid, engaging presentation,” OBG Manag. 2016;28[7]:31–36.), we offered tips on preparing for a group presentation. In this article, part 2, we discuss the presentation itself and what you can do to capture and hold your audience’s attention.
How to connect with the audience
Let’s assume the meeting host has just introduced you to the audience using, as we suggested in the previous article, an autobiographical profile you provided. You now have the audience’s undivided attention. What you do and say in the next 30 to 60 seconds will set the stage for your program. Following the requisite “thank you” to the host and meeting sponsor, use this time to establish your expertise as a spokesperson on the chosen topic. Or, if the introductory remarks made your expertise plain, you may choose to connect with the audience on an informal, personal level. If you are from out of town, for instance, you could remark on an interesting aspect of the city or region you are visiting that you learned on the Internet before arriving.
Underscore the topic’s importance. On the other hand, you might want to begin with an insightful statistic germane to your talk. For example, a talk on breast cancer might begin with, “According to the American Cancer Society, there are nearly 250,000 new cases of breast cancer each year, and breast cancer accounts for more than 40,000 deaths per year. That means more women die from breast cancer than die in auto accidents each year. So this emphasizes the importance of appropriately screening women for breast cancer annually after age 40.”
An opening story about a patient can be powerful. Better yet, a personal experience reflecting your topic is a great way to connect with your audience members and get their attention. For example, one of us (NHB) gives talks on practice management and practice efficiency. I might talk about when I was called from an exam room 3 times to answer “emergency” phone calls from a patient who wanted only to request her medical records. To ensure that this embarrassment would never happen again, I put in place a system that I then describe for the audience.
Alternatively, an opening that addresses the audience’s unspoken question, “What’s in this for me?” is sure to grab their attention. For instance, a talk on office productivity might begin by promising to share a way to increase annual collections by $250,000 per physician through scheduling adjustments that can increase the number of examined patients by one per hour.
Steer clear of these openings. In general, avoid “I’m delighted to be here” and other clichés. One exception would be if you can make that cliché humorous. For example, if a speaker from the deep South is visiting the northern part of the country in summer, she might say, “Most speakers say they’re delighted to be here, and you may well question their sincerity. However, I’m from New Orleans where the temperature is approaching 105 degrees with 95% humidity. You know I’m really delighted to be here!”
Importantly, avoid starting with an apology. Do not mention problems with the audiovisual equipment or why you arrived late. The audience does not care, and you will immediately lose their attention. They want to be educated and entertained. There is no better way to do this than by offering a compelling and captivating opening that begins the moment after you are introduced.
Finally, avoid use of the “royal I,” as in “I am here to talk about XYZ.” It places you in a position superior to the audience, and that is a turnoff. Instead, you could say to the audience, “The reason you are here is to learn about XYZ.” This places the audience on an equal level with you, and they know there will be something in the presentation for them.
Housekeeping notes
The audience will appreciate knowing how long you plan to speak and whether you will take questions during or after the presentation. Based on our experience, if there are fewer than 20 attendees, we often encourage questions during the program instead of waiting until the end. This makes the program more conversational and usually generates more questions. With a dinner presentation, we prefer to speak while the audience is eating. We usually start after the waiters have taken the orders and the attendees have had their appetizers. We might say we will finish the program by the time they are ready for dessert. We also mention that we will distribute a handout after the presentation so they do not have to worry about following the handout, taking notes, and watching the speaker while trying to eat.
The main body of the program
As for structuring your talk, we suggest you follow this time-honored advice often attributed to Aristotle: Tell the audience what you are going to say, say it, and tell them what you said.
So we begin a presentation by stating the objectives of our program, usually limited to 3 and no more than 4. For example, a talk on hormone therapy (HT) for treating vasomotor symptoms of menopause might mention 1) the history of HT use, 2) which women are appropriate candidates for HT, and 3) how to monitor women who receive HT.
Enhance the talk’s relevance. We like to begin a clinical program with a case scenario wherein we describe how one of our patients had the specific problem and how we used a particular drug, treatment, or device to manage the case. We try to select a patient similar to ones who would be seen by members of the audience.
Simplify as much as possible. We then present the slides exactly as they have been provided by the pharmaceutical company. Most company slides contain too many words as well as diagrams that are too complex for the audience to grasp easily. We try to find one salient point on each slide and focus attention on that single word, phrase, or sentence. We can do this in a small audience by walking over to the screen and pointing it out, or we can use the laser pointer from a distance.
Change things up to keep the message fresh. Let’s be honest, most medical talks are dry and boring. Try to inject some energy and enthusiasm in the middle of the presentation. Every few minutes we tell a story or ask the audience a question. For example, during a program on practice management, one of us (NHB) will relate a story about an unhappy patient and then ask a physician in the audience how he or she might handle the disgruntled patient. This is a nice break from the main content of the presentation, re-engaging the audience in an interactive exchange.
Should you use humor?
Although many physicians attempt to use humor during a presentation, few are talented at stand-up comedy. However, used judiciously humor, like seasoning in fine cuisine, can do great things for a presentation. It can break the ice, drive home a point, and enhance your likeability. It can, though, also backfire. One of us (NHB) once gave a talk to a large audience of pharmaceutical representatives. As part of my wrap-up I displayed a slide from the cover of Economics that showed 2 camels in the mating position. My closing line was that reps need to “hump to it” and get involved with their physicians and be value-added in their product detailing. Afterward, the meeting planner told me that he would never hire me again. He said I had a great program, great material, and a good connection with the audience. But my closing was over the top. I learned my lesson. Never use material that has the potential to offend. If you want to use humor, the self-deprecatingkind is always safest.
Try using visual aids
Our observation is that few physician speakers use visual aids other than their slides. We have learned that audience attention will stay focused on you if you make use of visual aids. For example, if we are speaking to a lay audience about urinary incontinence, we might use a balloon to demonstrate the bladder and the urethra.
Studies have shown that there are more nerve endings from the eye to the brain than from the ear to the brain. Humans purportedly receive 25 times as much stimulus from visual cues than from auditory ones. To paraphrase an old proverb, “One seeing is better than 100 times hearing about”!
A few suggestions regarding the use of visual aids:
- Keep the visual aid out of sight until you are ready to use it. You do not want the audience staring at it when they should be focusing on you or your slide material. We usually keep our visual aids under the table that supports the computer and projector.
- Make certain the visual aid is large enough to be seen by everyone in the audience.
- Do not hand out the aid to the audience during your program. Doing so will divert their attention from you and your material.
- When you have finished using the aid, put it away.
Closing out the program
After we have covered the program’s 3 objectives, we let the audience know we are approaching the end of the presentation. For a dinner program, we try to time the ending just as plates are being cleared and before dessert is served. We then restate the 3 objectives as they might pertain to the attendees’ patients and practices. At this time, we take questions from the audience, even if some were asked during the presentation. We repeat each question when it is asked so that everyone can hear it. (This also gives us a few seconds to think about it and frame our answer.) If it appears that many questions will be asked, we assure everyone that we plan to finish on time and will remain after the program is over to answer additional questions.
Tips on fielding questions. When responding to a question, direct your attention initially to the person who asked it. After that, spend about 20% of the time focused on that person and 80% of the time on the rest of the audience. If you focus only on the questioner, it becomes a one-on-one conversation. You want to end your response with your eyes on the group and not on the questioner. Looking at the group will also act as a bridge to the next question. Although we used to reply to an inquiry with, “That’s a great question,” we now suggest avoiding this comment. Why? Because it is unlikely that you’ll keep using that line, and the next questioner who does not receive the same compliment might feel slighted.
Wrap up. When you announce, “I would like to conclude my program with…,” this is the magical time when you hold the complete attention of the audience. Often, the speaker’s last words are the ones the audience remembers the longest. So this is the time to offer your take-home message. For example, a talk on how to motivate your staff might conclude, “Remember, your staff members are the people that patients encounter first and the ones they see last as they leave the office. Every patient can have a positive experience with you and your practice if you ensure that your personnel are highly motivated. This happens in part by your effort to recognize their accomplishments.” Then hold up your hands and spread out your arms as you end with “Thank you.” The audience likely will applaud and, if your speech is truly exceptional, you might receive a gratifying standing ovation!
Be seated
Renowned for his speeches, Franklin Delano Roosevelt summarized the art of effective speaking when he said, “Be sincere. Be brief. Be seated.” When your time is up, turn the program back over to the meeting host and take a seat.
In the final article in this public speaking series, we will discuss the follow-up steps to take once the program is over, including the call to action or what you want the audience to do after you have left the podium or the speaking venue.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
In the first part of this article series (“Preparation: Tips that lead to a solid, engaging presentation,” OBG Manag. 2016;28[7]:31–36.), we offered tips on preparing for a group presentation. In this article, part 2, we discuss the presentation itself and what you can do to capture and hold your audience’s attention.
How to connect with the audience
Let’s assume the meeting host has just introduced you to the audience using, as we suggested in the previous article, an autobiographical profile you provided. You now have the audience’s undivided attention. What you do and say in the next 30 to 60 seconds will set the stage for your program. Following the requisite “thank you” to the host and meeting sponsor, use this time to establish your expertise as a spokesperson on the chosen topic. Or, if the introductory remarks made your expertise plain, you may choose to connect with the audience on an informal, personal level. If you are from out of town, for instance, you could remark on an interesting aspect of the city or region you are visiting that you learned on the Internet before arriving.
Underscore the topic’s importance. On the other hand, you might want to begin with an insightful statistic germane to your talk. For example, a talk on breast cancer might begin with, “According to the American Cancer Society, there are nearly 250,000 new cases of breast cancer each year, and breast cancer accounts for more than 40,000 deaths per year. That means more women die from breast cancer than die in auto accidents each year. So this emphasizes the importance of appropriately screening women for breast cancer annually after age 40.”
An opening story about a patient can be powerful. Better yet, a personal experience reflecting your topic is a great way to connect with your audience members and get their attention. For example, one of us (NHB) gives talks on practice management and practice efficiency. I might talk about when I was called from an exam room 3 times to answer “emergency” phone calls from a patient who wanted only to request her medical records. To ensure that this embarrassment would never happen again, I put in place a system that I then describe for the audience.
Alternatively, an opening that addresses the audience’s unspoken question, “What’s in this for me?” is sure to grab their attention. For instance, a talk on office productivity might begin by promising to share a way to increase annual collections by $250,000 per physician through scheduling adjustments that can increase the number of examined patients by one per hour.
Steer clear of these openings. In general, avoid “I’m delighted to be here” and other clichés. One exception would be if you can make that cliché humorous. For example, if a speaker from the deep South is visiting the northern part of the country in summer, she might say, “Most speakers say they’re delighted to be here, and you may well question their sincerity. However, I’m from New Orleans where the temperature is approaching 105 degrees with 95% humidity. You know I’m really delighted to be here!”
Importantly, avoid starting with an apology. Do not mention problems with the audiovisual equipment or why you arrived late. The audience does not care, and you will immediately lose their attention. They want to be educated and entertained. There is no better way to do this than by offering a compelling and captivating opening that begins the moment after you are introduced.
Finally, avoid use of the “royal I,” as in “I am here to talk about XYZ.” It places you in a position superior to the audience, and that is a turnoff. Instead, you could say to the audience, “The reason you are here is to learn about XYZ.” This places the audience on an equal level with you, and they know there will be something in the presentation for them.
Housekeeping notes
The audience will appreciate knowing how long you plan to speak and whether you will take questions during or after the presentation. Based on our experience, if there are fewer than 20 attendees, we often encourage questions during the program instead of waiting until the end. This makes the program more conversational and usually generates more questions. With a dinner presentation, we prefer to speak while the audience is eating. We usually start after the waiters have taken the orders and the attendees have had their appetizers. We might say we will finish the program by the time they are ready for dessert. We also mention that we will distribute a handout after the presentation so they do not have to worry about following the handout, taking notes, and watching the speaker while trying to eat.
The main body of the program
As for structuring your talk, we suggest you follow this time-honored advice often attributed to Aristotle: Tell the audience what you are going to say, say it, and tell them what you said.
So we begin a presentation by stating the objectives of our program, usually limited to 3 and no more than 4. For example, a talk on hormone therapy (HT) for treating vasomotor symptoms of menopause might mention 1) the history of HT use, 2) which women are appropriate candidates for HT, and 3) how to monitor women who receive HT.
Enhance the talk’s relevance. We like to begin a clinical program with a case scenario wherein we describe how one of our patients had the specific problem and how we used a particular drug, treatment, or device to manage the case. We try to select a patient similar to ones who would be seen by members of the audience.
Simplify as much as possible. We then present the slides exactly as they have been provided by the pharmaceutical company. Most company slides contain too many words as well as diagrams that are too complex for the audience to grasp easily. We try to find one salient point on each slide and focus attention on that single word, phrase, or sentence. We can do this in a small audience by walking over to the screen and pointing it out, or we can use the laser pointer from a distance.
Change things up to keep the message fresh. Let’s be honest, most medical talks are dry and boring. Try to inject some energy and enthusiasm in the middle of the presentation. Every few minutes we tell a story or ask the audience a question. For example, during a program on practice management, one of us (NHB) will relate a story about an unhappy patient and then ask a physician in the audience how he or she might handle the disgruntled patient. This is a nice break from the main content of the presentation, re-engaging the audience in an interactive exchange.
Should you use humor?
Although many physicians attempt to use humor during a presentation, few are talented at stand-up comedy. However, used judiciously humor, like seasoning in fine cuisine, can do great things for a presentation. It can break the ice, drive home a point, and enhance your likeability. It can, though, also backfire. One of us (NHB) once gave a talk to a large audience of pharmaceutical representatives. As part of my wrap-up I displayed a slide from the cover of Economics that showed 2 camels in the mating position. My closing line was that reps need to “hump to it” and get involved with their physicians and be value-added in their product detailing. Afterward, the meeting planner told me that he would never hire me again. He said I had a great program, great material, and a good connection with the audience. But my closing was over the top. I learned my lesson. Never use material that has the potential to offend. If you want to use humor, the self-deprecatingkind is always safest.
Try using visual aids
Our observation is that few physician speakers use visual aids other than their slides. We have learned that audience attention will stay focused on you if you make use of visual aids. For example, if we are speaking to a lay audience about urinary incontinence, we might use a balloon to demonstrate the bladder and the urethra.
Studies have shown that there are more nerve endings from the eye to the brain than from the ear to the brain. Humans purportedly receive 25 times as much stimulus from visual cues than from auditory ones. To paraphrase an old proverb, “One seeing is better than 100 times hearing about”!
A few suggestions regarding the use of visual aids:
- Keep the visual aid out of sight until you are ready to use it. You do not want the audience staring at it when they should be focusing on you or your slide material. We usually keep our visual aids under the table that supports the computer and projector.
- Make certain the visual aid is large enough to be seen by everyone in the audience.
- Do not hand out the aid to the audience during your program. Doing so will divert their attention from you and your material.
- When you have finished using the aid, put it away.
Closing out the program
After we have covered the program’s 3 objectives, we let the audience know we are approaching the end of the presentation. For a dinner program, we try to time the ending just as plates are being cleared and before dessert is served. We then restate the 3 objectives as they might pertain to the attendees’ patients and practices. At this time, we take questions from the audience, even if some were asked during the presentation. We repeat each question when it is asked so that everyone can hear it. (This also gives us a few seconds to think about it and frame our answer.) If it appears that many questions will be asked, we assure everyone that we plan to finish on time and will remain after the program is over to answer additional questions.
Tips on fielding questions. When responding to a question, direct your attention initially to the person who asked it. After that, spend about 20% of the time focused on that person and 80% of the time on the rest of the audience. If you focus only on the questioner, it becomes a one-on-one conversation. You want to end your response with your eyes on the group and not on the questioner. Looking at the group will also act as a bridge to the next question. Although we used to reply to an inquiry with, “That’s a great question,” we now suggest avoiding this comment. Why? Because it is unlikely that you’ll keep using that line, and the next questioner who does not receive the same compliment might feel slighted.
Wrap up. When you announce, “I would like to conclude my program with…,” this is the magical time when you hold the complete attention of the audience. Often, the speaker’s last words are the ones the audience remembers the longest. So this is the time to offer your take-home message. For example, a talk on how to motivate your staff might conclude, “Remember, your staff members are the people that patients encounter first and the ones they see last as they leave the office. Every patient can have a positive experience with you and your practice if you ensure that your personnel are highly motivated. This happens in part by your effort to recognize their accomplishments.” Then hold up your hands and spread out your arms as you end with “Thank you.” The audience likely will applaud and, if your speech is truly exceptional, you might receive a gratifying standing ovation!
Be seated
Renowned for his speeches, Franklin Delano Roosevelt summarized the art of effective speaking when he said, “Be sincere. Be brief. Be seated.” When your time is up, turn the program back over to the meeting host and take a seat.
In the final article in this public speaking series, we will discuss the follow-up steps to take once the program is over, including the call to action or what you want the audience to do after you have left the podium or the speaking venue.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Large scar after multiple procedures
Large scar after multiple procedures
A woman with a history of 3 cesarean deliveries, a tubal ligation reversal, and an abdominoplasty discussed treatment for a large uterine fibroid with her ObGyn. She wanted to avoid a large scar. The ObGyn informed the patient that a laparoscopic hysterectomy could not be promised until her pelvic area was inspected to see if minimally invasive surgery safely could be performed.
During surgery, the ObGyn discovered that pelvic adhesions had distorted the patient’s anatomy; he converted to laparotomy, which left a larger scar.
Two days after surgery, the patient was found to have a bowel injury and underwent additional surgery that included placement of surgical mesh, leaving an enlarged scar.
PATIENT'S CLAIM:
The ObGyn was negligent in injuring the patient’s bowel during hysterectomy and not detecting the injury intraoperatively. Her scars were larger because of the additional repair operation.
PHYSICIAN'S DEFENSE:
Bowel injury is a known complication of the procedure. Many bowel injuries are not detected intraoperatively. The ObGyn made every effort to prevent and check for injury during the procedure.
VERDICT:
An Illinois defense verdict was returned.
Uterus and bowel injured during D&C: $1.5M verdict
A 56-year-old woman underwent hysteroscopy and dilation and curettage (D&C). During the procedure, the gynecologist recognized that he had perforated the uterus and injured the bowel and called in a general surgeon to resect 5 cm of the bowel and repair the uterus.
PATIENT'S CLAIM:
The patient has a large abdominal scar and a chronically distended abdomen. She experienced a year of daily pain and suffering. The D&C was unnecessary and improperly performed: the standard of care is for the gynecologist to operate in a gentle manner; that did not occur.
PHYSICIAN'S DEFENSE:
The D&C was medically necessary. The gynecologist exercised the proper standard of care.
VERDICT:
A $1.5 million New Jersey verdict was returned. The jury found the D&C necessary, but determined that the gynecologist deviated from the accepted standard of care in his performance of the procedure.
Injured ureter allegedly not treated
On December 6, a 42-year-old woman underwent hysterectomy. Postoperatively, she reported increasing dysuria with pain and fever.
On December 13, a computed tomography (CT) scan suggested a partial ureter obstruction. Despite test results, the gynecologist elected to continue to monitor the patient.
The patient’s symptoms continued to worsen and, on December 27, she underwent a second CT scan that identified an obstructed ureter. The gynecologist referred the patient to a urologist, who determined that the patient had sustained a significant ureter injury that required placement of a nephrostomy tube.
PATIENT'S CLAIM:
The gynecologist failed to identify the injury during surgery. The gynecologist was negligent in not consulting a urologist after results of the first CT scan.
PHYSICIAN'S DEFENSE:
Uterine injury is a known complication of the procedure. The gynecologist inspected adjacent organs during surgery but did not find an injury. Postoperative treatment was appropriate.
VERDICT:
The case was presented before a medical review board that concluded that there was no error after the first injury, there was no duty to trace the ureter, and a urology consult was not required after the first CT scan. A Louisiana defense verdict was returned.
Was FHR properly monitored?
After a failed nonstress test, a mother was admitted to triage for blood pressure monitoring. Fetal heart-rate (FHR) monitoring was discontinued at that time. Later that day, FHR monitoring was resumed, fetal distress was detected, and an emergency cesarean delivery was performed. Placental abruption resulted in hypoxia in the baby; she received a diagnosis of cerebral palsy.
PARENT'S CLAIM:
The pregnancy was at high risk because of the mother’s hypertension. The ObGyns misread the FHR at admission and discontinued FHR monitoring too early. If continuous FHR monitoring had occurred, fetal distress would have been detected earlier, resulting in a better outcome for the baby.
PHYSICIAN'S DEFENSE:
There were no signs of fetal distress when the FHR monitoring was discontinued. Placental abruption is an acute event that cannot be predicted.
VERDICT:
A Missouri defense verdict was returned.
Should the ObGyn have come to the hospital earlier?
At 39 weeks’ gestation, a mother arrived at the hospital for induction of labor. That evening, the ObGyn, who was not at the hospital, was notified that the mother had an elevated temperature and that the FHR indicated tachycardia. The ObGyn prescribed antibiotics, and the fever subsided. After an hour, the patient was fully dilated and started to push under a nurse’s supervision. Twenty minutes later, the ObGyn was notified that the fetus was experiencing variable decelerations. The ObGyn arrived in 30 minutes and ordered a cesarean delivery. The baby was born 24 minutes later.
The baby began to have seizures 10 hours after birth. He was transferred to another hospital and remained in the neonatal intensive care unit for 15 days. The child received a diagnosis of cerebral palsy.
PARENT'S CLAIM:
The ObGyn was negligent in not coming to the hospital when the mother was feverish and the fetus tachycardic. The baby experienced an acute hypoxic ischemic injury; an earlier cesarean delivery would have avoided brain injury.
PHYSICIAN'S DEFENSE:
There was no negligence. The infant did not meet all the criteria for an acute hypoxic ischemic injury. Based on a computed tomography scan taken after the seizures began, the infant’s brain injury most likely occurred hours before birth.
VERDICT:
A Virginia defense verdict was returned.
Large scar after multiple procedures
A woman with a history of 3 cesarean deliveries, a tubal ligation reversal, and an abdominoplasty discussed treatment for a large uterine fibroid with her ObGyn. She wanted to avoid a large scar. The ObGyn informed the patient that a laparoscopic hysterectomy could not be promised until her pelvic area was inspected to see if minimally invasive surgery safely could be performed.
During surgery, the ObGyn discovered that pelvic adhesions had distorted the patient’s anatomy; he converted to laparotomy, which left a larger scar.
Two days after surgery, the patient was found to have a bowel injury and underwent additional surgery that included placement of surgical mesh, leaving an enlarged scar.
PATIENT'S CLAIM:
The ObGyn was negligent in injuring the patient’s bowel during hysterectomy and not detecting the injury intraoperatively. Her scars were larger because of the additional repair operation.
PHYSICIAN'S DEFENSE:
Bowel injury is a known complication of the procedure. Many bowel injuries are not detected intraoperatively. The ObGyn made every effort to prevent and check for injury during the procedure.
VERDICT:
An Illinois defense verdict was returned.
Uterus and bowel injured during D&C: $1.5M verdict
A 56-year-old woman underwent hysteroscopy and dilation and curettage (D&C). During the procedure, the gynecologist recognized that he had perforated the uterus and injured the bowel and called in a general surgeon to resect 5 cm of the bowel and repair the uterus.
PATIENT'S CLAIM:
The patient has a large abdominal scar and a chronically distended abdomen. She experienced a year of daily pain and suffering. The D&C was unnecessary and improperly performed: the standard of care is for the gynecologist to operate in a gentle manner; that did not occur.
PHYSICIAN'S DEFENSE:
The D&C was medically necessary. The gynecologist exercised the proper standard of care.
VERDICT:
A $1.5 million New Jersey verdict was returned. The jury found the D&C necessary, but determined that the gynecologist deviated from the accepted standard of care in his performance of the procedure.
Injured ureter allegedly not treated
On December 6, a 42-year-old woman underwent hysterectomy. Postoperatively, she reported increasing dysuria with pain and fever.
On December 13, a computed tomography (CT) scan suggested a partial ureter obstruction. Despite test results, the gynecologist elected to continue to monitor the patient.
The patient’s symptoms continued to worsen and, on December 27, she underwent a second CT scan that identified an obstructed ureter. The gynecologist referred the patient to a urologist, who determined that the patient had sustained a significant ureter injury that required placement of a nephrostomy tube.
PATIENT'S CLAIM:
The gynecologist failed to identify the injury during surgery. The gynecologist was negligent in not consulting a urologist after results of the first CT scan.
PHYSICIAN'S DEFENSE:
Uterine injury is a known complication of the procedure. The gynecologist inspected adjacent organs during surgery but did not find an injury. Postoperative treatment was appropriate.
VERDICT:
The case was presented before a medical review board that concluded that there was no error after the first injury, there was no duty to trace the ureter, and a urology consult was not required after the first CT scan. A Louisiana defense verdict was returned.
Was FHR properly monitored?
After a failed nonstress test, a mother was admitted to triage for blood pressure monitoring. Fetal heart-rate (FHR) monitoring was discontinued at that time. Later that day, FHR monitoring was resumed, fetal distress was detected, and an emergency cesarean delivery was performed. Placental abruption resulted in hypoxia in the baby; she received a diagnosis of cerebral palsy.
PARENT'S CLAIM:
The pregnancy was at high risk because of the mother’s hypertension. The ObGyns misread the FHR at admission and discontinued FHR monitoring too early. If continuous FHR monitoring had occurred, fetal distress would have been detected earlier, resulting in a better outcome for the baby.
PHYSICIAN'S DEFENSE:
There were no signs of fetal distress when the FHR monitoring was discontinued. Placental abruption is an acute event that cannot be predicted.
VERDICT:
A Missouri defense verdict was returned.
Should the ObGyn have come to the hospital earlier?
At 39 weeks’ gestation, a mother arrived at the hospital for induction of labor. That evening, the ObGyn, who was not at the hospital, was notified that the mother had an elevated temperature and that the FHR indicated tachycardia. The ObGyn prescribed antibiotics, and the fever subsided. After an hour, the patient was fully dilated and started to push under a nurse’s supervision. Twenty minutes later, the ObGyn was notified that the fetus was experiencing variable decelerations. The ObGyn arrived in 30 minutes and ordered a cesarean delivery. The baby was born 24 minutes later.
The baby began to have seizures 10 hours after birth. He was transferred to another hospital and remained in the neonatal intensive care unit for 15 days. The child received a diagnosis of cerebral palsy.
PARENT'S CLAIM:
The ObGyn was negligent in not coming to the hospital when the mother was feverish and the fetus tachycardic. The baby experienced an acute hypoxic ischemic injury; an earlier cesarean delivery would have avoided brain injury.
PHYSICIAN'S DEFENSE:
There was no negligence. The infant did not meet all the criteria for an acute hypoxic ischemic injury. Based on a computed tomography scan taken after the seizures began, the infant’s brain injury most likely occurred hours before birth.
VERDICT:
A Virginia defense verdict was returned.
Large scar after multiple procedures
A woman with a history of 3 cesarean deliveries, a tubal ligation reversal, and an abdominoplasty discussed treatment for a large uterine fibroid with her ObGyn. She wanted to avoid a large scar. The ObGyn informed the patient that a laparoscopic hysterectomy could not be promised until her pelvic area was inspected to see if minimally invasive surgery safely could be performed.
During surgery, the ObGyn discovered that pelvic adhesions had distorted the patient’s anatomy; he converted to laparotomy, which left a larger scar.
Two days after surgery, the patient was found to have a bowel injury and underwent additional surgery that included placement of surgical mesh, leaving an enlarged scar.
PATIENT'S CLAIM:
The ObGyn was negligent in injuring the patient’s bowel during hysterectomy and not detecting the injury intraoperatively. Her scars were larger because of the additional repair operation.
PHYSICIAN'S DEFENSE:
Bowel injury is a known complication of the procedure. Many bowel injuries are not detected intraoperatively. The ObGyn made every effort to prevent and check for injury during the procedure.
VERDICT:
An Illinois defense verdict was returned.
Uterus and bowel injured during D&C: $1.5M verdict
A 56-year-old woman underwent hysteroscopy and dilation and curettage (D&C). During the procedure, the gynecologist recognized that he had perforated the uterus and injured the bowel and called in a general surgeon to resect 5 cm of the bowel and repair the uterus.
PATIENT'S CLAIM:
The patient has a large abdominal scar and a chronically distended abdomen. She experienced a year of daily pain and suffering. The D&C was unnecessary and improperly performed: the standard of care is for the gynecologist to operate in a gentle manner; that did not occur.
PHYSICIAN'S DEFENSE:
The D&C was medically necessary. The gynecologist exercised the proper standard of care.
VERDICT:
A $1.5 million New Jersey verdict was returned. The jury found the D&C necessary, but determined that the gynecologist deviated from the accepted standard of care in his performance of the procedure.
Injured ureter allegedly not treated
On December 6, a 42-year-old woman underwent hysterectomy. Postoperatively, she reported increasing dysuria with pain and fever.
On December 13, a computed tomography (CT) scan suggested a partial ureter obstruction. Despite test results, the gynecologist elected to continue to monitor the patient.
The patient’s symptoms continued to worsen and, on December 27, she underwent a second CT scan that identified an obstructed ureter. The gynecologist referred the patient to a urologist, who determined that the patient had sustained a significant ureter injury that required placement of a nephrostomy tube.
PATIENT'S CLAIM:
The gynecologist failed to identify the injury during surgery. The gynecologist was negligent in not consulting a urologist after results of the first CT scan.
PHYSICIAN'S DEFENSE:
Uterine injury is a known complication of the procedure. The gynecologist inspected adjacent organs during surgery but did not find an injury. Postoperative treatment was appropriate.
VERDICT:
The case was presented before a medical review board that concluded that there was no error after the first injury, there was no duty to trace the ureter, and a urology consult was not required after the first CT scan. A Louisiana defense verdict was returned.
Was FHR properly monitored?
After a failed nonstress test, a mother was admitted to triage for blood pressure monitoring. Fetal heart-rate (FHR) monitoring was discontinued at that time. Later that day, FHR monitoring was resumed, fetal distress was detected, and an emergency cesarean delivery was performed. Placental abruption resulted in hypoxia in the baby; she received a diagnosis of cerebral palsy.
PARENT'S CLAIM:
The pregnancy was at high risk because of the mother’s hypertension. The ObGyns misread the FHR at admission and discontinued FHR monitoring too early. If continuous FHR monitoring had occurred, fetal distress would have been detected earlier, resulting in a better outcome for the baby.
PHYSICIAN'S DEFENSE:
There were no signs of fetal distress when the FHR monitoring was discontinued. Placental abruption is an acute event that cannot be predicted.
VERDICT:
A Missouri defense verdict was returned.
Should the ObGyn have come to the hospital earlier?
At 39 weeks’ gestation, a mother arrived at the hospital for induction of labor. That evening, the ObGyn, who was not at the hospital, was notified that the mother had an elevated temperature and that the FHR indicated tachycardia. The ObGyn prescribed antibiotics, and the fever subsided. After an hour, the patient was fully dilated and started to push under a nurse’s supervision. Twenty minutes later, the ObGyn was notified that the fetus was experiencing variable decelerations. The ObGyn arrived in 30 minutes and ordered a cesarean delivery. The baby was born 24 minutes later.
The baby began to have seizures 10 hours after birth. He was transferred to another hospital and remained in the neonatal intensive care unit for 15 days. The child received a diagnosis of cerebral palsy.
PARENT'S CLAIM:
The ObGyn was negligent in not coming to the hospital when the mother was feverish and the fetus tachycardic. The baby experienced an acute hypoxic ischemic injury; an earlier cesarean delivery would have avoided brain injury.
PHYSICIAN'S DEFENSE:
There was no negligence. The infant did not meet all the criteria for an acute hypoxic ischemic injury. Based on a computed tomography scan taken after the seizures began, the infant’s brain injury most likely occurred hours before birth.
VERDICT:
A Virginia defense verdict was returned.
2016 Obstetric code changes that could affect your reimbursement (very soon)
By now the upheaval of changing to the new International Classification of Diseases, Tenth Revision, Clinical Modification (ICD-10) diagnostic coding system has settled. The code freeze that was initiated in 2012 has ended, and the new and revised codes that will go into effect on October 1, 2016, are being revealed. Good documentation will lead to more accurate diagnostic coding, which in turn assists decision makers in their quest to report the health of our population and to make good decisions for resource allocation. You are in the unique position to assist in this process, so keep up the good work.
In this article, I focus on ICD-10 diagnostic coding for obstetric services. I will cover diagnostic coding for gynecologic services in the September issue of OBG Management.
Code revisions for uterine scar and more changes to note
With the upcoming edition of ICD-10, the code Z3A, Weeks’ gestation, will be changed from mandatory reporting to reporting if known. This means that if the patient is no longer pregnant, a Z3A code no longer needs to be reported, and if at the time of service the provider does not know the weeks’ gestation, Z3A would not be required. However, this information should be readily available during the antepartum period and should still be considered important to record and report. And it would still be reported for hospitalization for delivery.
If the code O09.81, Supervision of pregnancy resulting from assisted reproductive technology, is reported, the code Z33.3, Gestational carrier status, may be reported in addition for informational purposes.
When the code O34.29, Maternal care due to uterine scar from other previous surgery, is reported, the tabular index clarifies that this refers to a uterine scar from a transmural uterine incision other than that used for cesarean delivery. This would include incision into the uterine wall to remove fibroids.
The O42 code category, relating to Premature rupture of membranes, should now be interpreted to mean rupture of membranes at or after 37 completed weeks of gestation, rather than after 37 completed weeks.
The code category O99.6, Diseases of the digestive system complicating pregnancy, childbirth, and the puerperium, has been clarified: it does not include hemorrhoids in pregnancy. Therefore, a code from O22.4_ (a final digit of 0 [unspecified], 1, 2, or 3 is required for the trimester) also can be reported if hemorrhoids are present.
A note now clarifies that O99.82, Streptococcus B carrier state complicating pregnancy, childbirth, and the puerperium, cannot be reported with Z22.330, Carrier of streptococcus group B (GBS) in a nonpregnant woman.
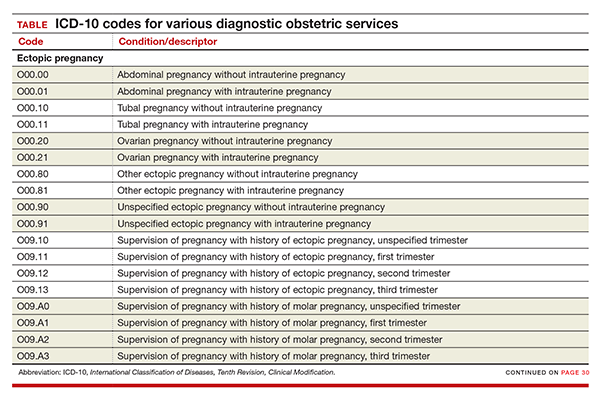
New codes for specifying types of ectopic pregnancy
ICD-10 did not initially recognize ectopic pregnancy with and without intrauterine pregnancy, as was the case in ICD-9, but starting in October it will do so. In addition, a history of ectopic or molar pregnancy during a current pregnancy is now reported separately. Each of these codes will require a final digit to indicate the trimester (TABLE).
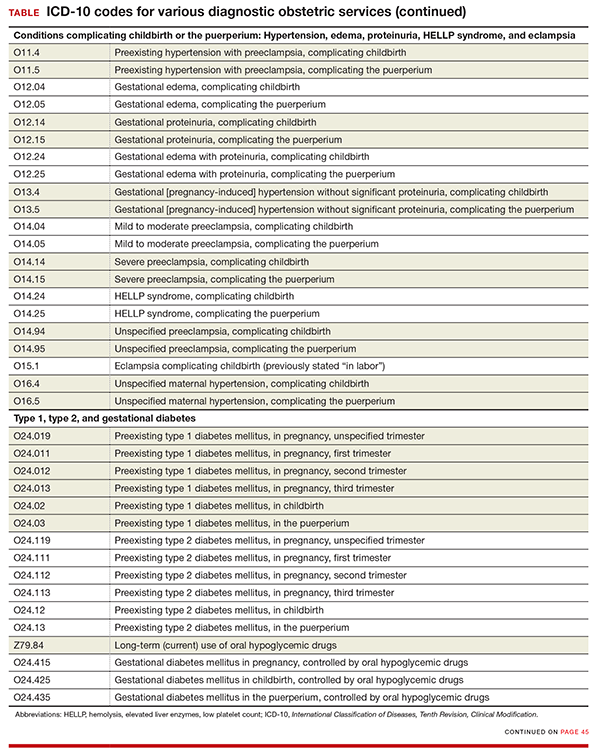
Codes added for complicating conditions of childbirth and the puerperium
Missing from the ICD-10 lineup last year were codes for conditions related to hypertension, edema, proteinuria, HELLP (hemolysis, elevated liver enzymes, and low platelet count) syndrome, and eclampsia that were complicating the pregnancy at the time of delivery or after delivery (TABLE).
Note that the “childbirth” code is reported only when a patient delivers at the current episode of care. Once a patient delivers and is discharged, the “puerperium” code should be selected.
Revised descriptions, new reporting instruction for diabetes
The code descriptions for preexisting type 1 and type 2 diabetes were revised, but this change does not impact reporting the codes. However, for type 2 diabetes, the instruction for reporting an additional code has changed. Now, in addition to reporting the code for current use of insulin (Z79.4), when appropriate, report the new added code for use of hypoglycemic agents (Z79.84), such as glyburide or metformin.
For gestational diabetes, new codes have been added for the use of hypoglycemic agents; therefore, no additional code is reported (TABLE).
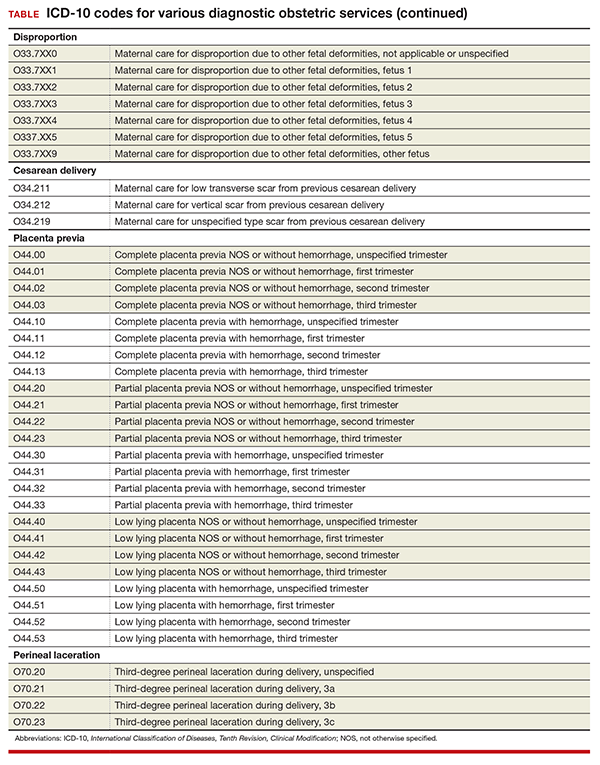
Disproportion code includes numeric specifier for fetus
The disproportion code category was expanded to include a final digit for the fetus with the deformity (TABLE). The final digit of the code number denotes which fetus; for example, “0” means a singleton pregnancy, “1” means fetus 1 (number range from 1 to 5), and “9” denotes any fetus after the fifth.
Cesarean delivery scar codes expanded
The code for maternal care for a scar from a previous cesarean delivery has been expanded to 3 different codes (TABLE). Clinicians should make every effort to document and report the location of the previous cesarean as low transverse or vertical. From a coding standpoint, a vertical scar can also be referred to as a classical scar.
Changes to placenta previa codes
The code category for placenta previa has been expanded to capture the degree of previa as complete, partial, or low lying and with or without hemorrhage (TABLE). Going forward, it will be important to carefully document the circumstances so that the most specific code can be reported and tracked. Trimester specification is required as the final digit.
New subclassifications for perineal laceration
The code category for perineal laceration has been expanded with new codes to capture subclassifications for a third-degree laceration that can involve the external and internal anal sphincter (TABLE). Through its collaborative hub, the Women’s Health Registry Alliance (reVITALize) initiative, the American Congress of Obstetricians and Gynecologists (ACOG) worked on the current classification of third- and fourth-degree perineal lacerations, which has been adopted by the Royal College of Obstetricians and Gynaecologists.1
Under this subclassification, a 3a laceration would involve a tear of less than 50% of the external anal sphincter (EAS); 3b would involve a tear of more than 50% of the EAS; and 3c would mean that both the external and internal anal sphincter are torn. ACOG and its collaborative group encourage clinicians to use these subclassifications in documentation to allow for more robust data collection and complete repair information. From a payment standpoint, such information may go a long way to substantiating the severity of a tear, which may require more physician work.
Z code additions
Finally, the ever-popular diagnostic code for Rho(D) immunization is back, and 2 codes have been added for a gestational carrier and 1 for a family history of sudden infant death syndrome. The codes are:
- Z29.13 Encounter for prophylactic Rho(D) immune globulin
- Z31.7 Encounter for procreative management and counseling for gestational carrier
- Z33.3 Pregnant state, gestational carrier
- Z84.82 Family history of sudden infant death syndrome.
- Centers for Disease Control and Prevention. ICD-10 Coordination and Maintenance Committee meeting: diagnosis agenda. September 23-24, 2014;38, 39. http://www.cdc.gov/nchs/data/icd/topic_packet_09_23_2012.pdf. Accessed July 5, 2016.

Ms. Witt is an independent coding and documentation consultant and former program manager, department of coding and nomenclature, American Congress of Obstetricians and Gynecologists.
The author reports no financial relationships relevant to this article.
Ms. Witt is an independent coding and documentation consultant and former program manager, department of coding and nomenclature, American Congress of Obstetricians and Gynecologists.
The author reports no financial relationships relevant to this article.
Ms. Witt is an independent coding and documentation consultant and former program manager, department of coding and nomenclature, American Congress of Obstetricians and Gynecologists.
The author reports no financial relationships relevant to this article.
By now the upheaval of changing to the new International Classification of Diseases, Tenth Revision, Clinical Modification (ICD-10) diagnostic coding system has settled. The code freeze that was initiated in 2012 has ended, and the new and revised codes that will go into effect on October 1, 2016, are being revealed. Good documentation will lead to more accurate diagnostic coding, which in turn assists decision makers in their quest to report the health of our population and to make good decisions for resource allocation. You are in the unique position to assist in this process, so keep up the good work.
In this article, I focus on ICD-10 diagnostic coding for obstetric services. I will cover diagnostic coding for gynecologic services in the September issue of OBG Management.
Code revisions for uterine scar and more changes to note
With the upcoming edition of ICD-10, the code Z3A, Weeks’ gestation, will be changed from mandatory reporting to reporting if known. This means that if the patient is no longer pregnant, a Z3A code no longer needs to be reported, and if at the time of service the provider does not know the weeks’ gestation, Z3A would not be required. However, this information should be readily available during the antepartum period and should still be considered important to record and report. And it would still be reported for hospitalization for delivery.
If the code O09.81, Supervision of pregnancy resulting from assisted reproductive technology, is reported, the code Z33.3, Gestational carrier status, may be reported in addition for informational purposes.
When the code O34.29, Maternal care due to uterine scar from other previous surgery, is reported, the tabular index clarifies that this refers to a uterine scar from a transmural uterine incision other than that used for cesarean delivery. This would include incision into the uterine wall to remove fibroids.
The O42 code category, relating to Premature rupture of membranes, should now be interpreted to mean rupture of membranes at or after 37 completed weeks of gestation, rather than after 37 completed weeks.
The code category O99.6, Diseases of the digestive system complicating pregnancy, childbirth, and the puerperium, has been clarified: it does not include hemorrhoids in pregnancy. Therefore, a code from O22.4_ (a final digit of 0 [unspecified], 1, 2, or 3 is required for the trimester) also can be reported if hemorrhoids are present.
A note now clarifies that O99.82, Streptococcus B carrier state complicating pregnancy, childbirth, and the puerperium, cannot be reported with Z22.330, Carrier of streptococcus group B (GBS) in a nonpregnant woman.
New codes for specifying types of ectopic pregnancy
ICD-10 did not initially recognize ectopic pregnancy with and without intrauterine pregnancy, as was the case in ICD-9, but starting in October it will do so. In addition, a history of ectopic or molar pregnancy during a current pregnancy is now reported separately. Each of these codes will require a final digit to indicate the trimester (TABLE).
Codes added for complicating conditions of childbirth and the puerperium
Missing from the ICD-10 lineup last year were codes for conditions related to hypertension, edema, proteinuria, HELLP (hemolysis, elevated liver enzymes, and low platelet count) syndrome, and eclampsia that were complicating the pregnancy at the time of delivery or after delivery (TABLE).
Note that the “childbirth” code is reported only when a patient delivers at the current episode of care. Once a patient delivers and is discharged, the “puerperium” code should be selected.
Revised descriptions, new reporting instruction for diabetes
The code descriptions for preexisting type 1 and type 2 diabetes were revised, but this change does not impact reporting the codes. However, for type 2 diabetes, the instruction for reporting an additional code has changed. Now, in addition to reporting the code for current use of insulin (Z79.4), when appropriate, report the new added code for use of hypoglycemic agents (Z79.84), such as glyburide or metformin.
For gestational diabetes, new codes have been added for the use of hypoglycemic agents; therefore, no additional code is reported (TABLE).
Disproportion code includes numeric specifier for fetus
The disproportion code category was expanded to include a final digit for the fetus with the deformity (TABLE). The final digit of the code number denotes which fetus; for example, “0” means a singleton pregnancy, “1” means fetus 1 (number range from 1 to 5), and “9” denotes any fetus after the fifth.
Cesarean delivery scar codes expanded
The code for maternal care for a scar from a previous cesarean delivery has been expanded to 3 different codes (TABLE). Clinicians should make every effort to document and report the location of the previous cesarean as low transverse or vertical. From a coding standpoint, a vertical scar can also be referred to as a classical scar.
Changes to placenta previa codes
The code category for placenta previa has been expanded to capture the degree of previa as complete, partial, or low lying and with or without hemorrhage (TABLE). Going forward, it will be important to carefully document the circumstances so that the most specific code can be reported and tracked. Trimester specification is required as the final digit.
New subclassifications for perineal laceration
The code category for perineal laceration has been expanded with new codes to capture subclassifications for a third-degree laceration that can involve the external and internal anal sphincter (TABLE). Through its collaborative hub, the Women’s Health Registry Alliance (reVITALize) initiative, the American Congress of Obstetricians and Gynecologists (ACOG) worked on the current classification of third- and fourth-degree perineal lacerations, which has been adopted by the Royal College of Obstetricians and Gynaecologists.1
Under this subclassification, a 3a laceration would involve a tear of less than 50% of the external anal sphincter (EAS); 3b would involve a tear of more than 50% of the EAS; and 3c would mean that both the external and internal anal sphincter are torn. ACOG and its collaborative group encourage clinicians to use these subclassifications in documentation to allow for more robust data collection and complete repair information. From a payment standpoint, such information may go a long way to substantiating the severity of a tear, which may require more physician work.
Z code additions
Finally, the ever-popular diagnostic code for Rho(D) immunization is back, and 2 codes have been added for a gestational carrier and 1 for a family history of sudden infant death syndrome. The codes are:
- Z29.13 Encounter for prophylactic Rho(D) immune globulin
- Z31.7 Encounter for procreative management and counseling for gestational carrier
- Z33.3 Pregnant state, gestational carrier
- Z84.82 Family history of sudden infant death syndrome.
By now the upheaval of changing to the new International Classification of Diseases, Tenth Revision, Clinical Modification (ICD-10) diagnostic coding system has settled. The code freeze that was initiated in 2012 has ended, and the new and revised codes that will go into effect on October 1, 2016, are being revealed. Good documentation will lead to more accurate diagnostic coding, which in turn assists decision makers in their quest to report the health of our population and to make good decisions for resource allocation. You are in the unique position to assist in this process, so keep up the good work.
In this article, I focus on ICD-10 diagnostic coding for obstetric services. I will cover diagnostic coding for gynecologic services in the September issue of OBG Management.
Code revisions for uterine scar and more changes to note
With the upcoming edition of ICD-10, the code Z3A, Weeks’ gestation, will be changed from mandatory reporting to reporting if known. This means that if the patient is no longer pregnant, a Z3A code no longer needs to be reported, and if at the time of service the provider does not know the weeks’ gestation, Z3A would not be required. However, this information should be readily available during the antepartum period and should still be considered important to record and report. And it would still be reported for hospitalization for delivery.
If the code O09.81, Supervision of pregnancy resulting from assisted reproductive technology, is reported, the code Z33.3, Gestational carrier status, may be reported in addition for informational purposes.
When the code O34.29, Maternal care due to uterine scar from other previous surgery, is reported, the tabular index clarifies that this refers to a uterine scar from a transmural uterine incision other than that used for cesarean delivery. This would include incision into the uterine wall to remove fibroids.
The O42 code category, relating to Premature rupture of membranes, should now be interpreted to mean rupture of membranes at or after 37 completed weeks of gestation, rather than after 37 completed weeks.
The code category O99.6, Diseases of the digestive system complicating pregnancy, childbirth, and the puerperium, has been clarified: it does not include hemorrhoids in pregnancy. Therefore, a code from O22.4_ (a final digit of 0 [unspecified], 1, 2, or 3 is required for the trimester) also can be reported if hemorrhoids are present.
A note now clarifies that O99.82, Streptococcus B carrier state complicating pregnancy, childbirth, and the puerperium, cannot be reported with Z22.330, Carrier of streptococcus group B (GBS) in a nonpregnant woman.
New codes for specifying types of ectopic pregnancy
ICD-10 did not initially recognize ectopic pregnancy with and without intrauterine pregnancy, as was the case in ICD-9, but starting in October it will do so. In addition, a history of ectopic or molar pregnancy during a current pregnancy is now reported separately. Each of these codes will require a final digit to indicate the trimester (TABLE).
Codes added for complicating conditions of childbirth and the puerperium
Missing from the ICD-10 lineup last year were codes for conditions related to hypertension, edema, proteinuria, HELLP (hemolysis, elevated liver enzymes, and low platelet count) syndrome, and eclampsia that were complicating the pregnancy at the time of delivery or after delivery (TABLE).
Note that the “childbirth” code is reported only when a patient delivers at the current episode of care. Once a patient delivers and is discharged, the “puerperium” code should be selected.
Revised descriptions, new reporting instruction for diabetes
The code descriptions for preexisting type 1 and type 2 diabetes were revised, but this change does not impact reporting the codes. However, for type 2 diabetes, the instruction for reporting an additional code has changed. Now, in addition to reporting the code for current use of insulin (Z79.4), when appropriate, report the new added code for use of hypoglycemic agents (Z79.84), such as glyburide or metformin.
For gestational diabetes, new codes have been added for the use of hypoglycemic agents; therefore, no additional code is reported (TABLE).
Disproportion code includes numeric specifier for fetus
The disproportion code category was expanded to include a final digit for the fetus with the deformity (TABLE). The final digit of the code number denotes which fetus; for example, “0” means a singleton pregnancy, “1” means fetus 1 (number range from 1 to 5), and “9” denotes any fetus after the fifth.
Cesarean delivery scar codes expanded
The code for maternal care for a scar from a previous cesarean delivery has been expanded to 3 different codes (TABLE). Clinicians should make every effort to document and report the location of the previous cesarean as low transverse or vertical. From a coding standpoint, a vertical scar can also be referred to as a classical scar.
Changes to placenta previa codes
The code category for placenta previa has been expanded to capture the degree of previa as complete, partial, or low lying and with or without hemorrhage (TABLE). Going forward, it will be important to carefully document the circumstances so that the most specific code can be reported and tracked. Trimester specification is required as the final digit.
New subclassifications for perineal laceration
The code category for perineal laceration has been expanded with new codes to capture subclassifications for a third-degree laceration that can involve the external and internal anal sphincter (TABLE). Through its collaborative hub, the Women’s Health Registry Alliance (reVITALize) initiative, the American Congress of Obstetricians and Gynecologists (ACOG) worked on the current classification of third- and fourth-degree perineal lacerations, which has been adopted by the Royal College of Obstetricians and Gynaecologists.1
Under this subclassification, a 3a laceration would involve a tear of less than 50% of the external anal sphincter (EAS); 3b would involve a tear of more than 50% of the EAS; and 3c would mean that both the external and internal anal sphincter are torn. ACOG and its collaborative group encourage clinicians to use these subclassifications in documentation to allow for more robust data collection and complete repair information. From a payment standpoint, such information may go a long way to substantiating the severity of a tear, which may require more physician work.
Z code additions
Finally, the ever-popular diagnostic code for Rho(D) immunization is back, and 2 codes have been added for a gestational carrier and 1 for a family history of sudden infant death syndrome. The codes are:
- Z29.13 Encounter for prophylactic Rho(D) immune globulin
- Z31.7 Encounter for procreative management and counseling for gestational carrier
- Z33.3 Pregnant state, gestational carrier
- Z84.82 Family history of sudden infant death syndrome.
- Centers for Disease Control and Prevention. ICD-10 Coordination and Maintenance Committee meeting: diagnosis agenda. September 23-24, 2014;38, 39. http://www.cdc.gov/nchs/data/icd/topic_packet_09_23_2012.pdf. Accessed July 5, 2016.
- Centers for Disease Control and Prevention. ICD-10 Coordination and Maintenance Committee meeting: diagnosis agenda. September 23-24, 2014;38, 39. http://www.cdc.gov/nchs/data/icd/topic_packet_09_23_2012.pdf. Accessed July 5, 2016.
In this Article
- New and expanded codes
- Z code additions
- Table of codes

Anemia of chronic kidney disease: Treat it, but not too aggressively
Anemia is a frequent complication of chronic kidney disease, occurring in over 90% of patients receiving renal replacement therapy. It is associated with significant morbidity and mortality. While its pathogenesis is typically multifactorial, the predominant cause is failure of the kidneys to produce enough endogenous erythropoietin. The clinical approval of recombinant human erythropoietin in 1989 dramatically changed the treatment of anemia of chronic kidney disease, but randomized controlled trials yielded disappointing results when erythropoiesis-stimulating agents (ESAs) were used to raise hemoglobin to normal levels.
This article reviews the epidemiology and pathophysiology of anemia of chronic kidney disease and discusses the complicated and conflicting evidence regarding its treatment.
DEFINITION AND PREVALENCE
Anemia is defined as a hemoglobin concentration less than 13.0 g/dL for men and less than 12.0 g/dL for premenopausal women.1 It is more common in patients with impaired kidney function, especially when the glomerular filtration rate (GFR) falls below 60 mL/min. It is rare at GFRs higher than 80 mL/min,2 but as the GFR falls, the severity of the anemia worsens3 and its prevalence increases: almost 90% of patients with a GFR less than 30 mL/min are anemic.4
RENAL ANEMIA IS ASSOCIATED WITH BAD OUTCOMES
Anemia in chronic kidney disease is independently associated with risk of death. It is also an all-cause mortality multiplier, ie, it magnifies the risk of death from other disease states.5
In observational studies, anemia was associated with faster progression of left ventricular hypertrophy, inflammation, and increased myocardial and peripheral oxygen demand, thereby leading to worse cardiac outcomes with increased risk of myocardial infarction, coronary revascularization, and readmission for heart failure.6–8 Anemia is also associated with fatigue, depression, reduced exercise tolerance, stroke, and increased risk of rehospitalization.9–13
RENAL ANEMIA IS MULTIFACTORIAL
Anemia of chronic kidney disease is typically attributed to the decrease of erythropoietin production that accompanies the fall in GFR. However, the process is multifactorial, with several other contributing factors: absolute and functional iron deficiency, folate and vitamin B12 deficiencies, reduced red blood cell life span, and suppression of erythropoiesis by the uremic milieu.14
While it was once thought that chronic kidney disease leads to loss of erythropoietin-producing cells, it is now known that downregulation of hypoxia-inducible factor (HIF; a transcription factor) is at least partially responsible for the decrease in erythropoietin production15,16 and that this downregulation is reversible (see below).
ERYTHROPOIETIN, IRON, AND RED BLOOD CELLS
Erythropoietin production is triggered by hypoxia, mediated by HIF
Erythropoietin is produced primarily in the deep cortex and outer medulla of the kidneys by a special population of peritubular interstitial cells.17 The parenchymal cells of the liver also produce erythropoietin, but much less.18
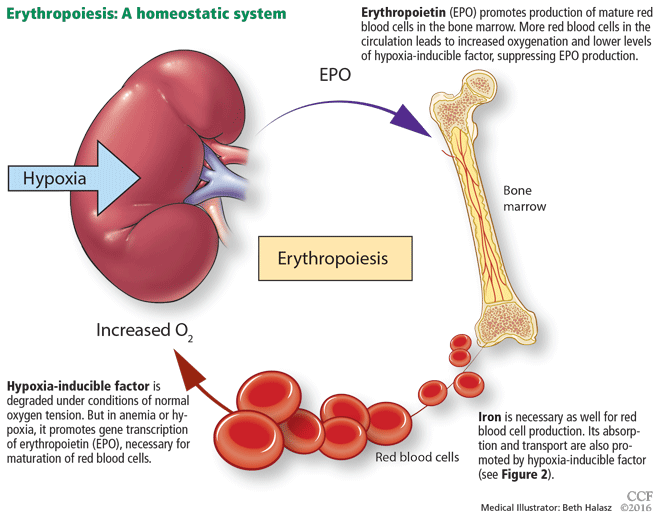
The rate of renal erythropoietin synthesis is determined by tissue oxygenation rather than by renal blood flow; production increases as the hemoglobin concentration drops and the arterial oxygen tension decreases (Figure 1).19
The gene for erythropoietin is located on chromosome 7 and is regulated by HIF. HIF molecules are composed of an alpha subunit, which is unstable at high Po2, and a beta subunit, constitutively present in the nucleus.20
In hypoxic conditions, the HIF dimer is transcriptionally active and binds to specific DNA recognition sequences called hypoxia-response elements. Gene transcription is upregulated, leading to increased production of erythropoietin.21
Under normal oxygen tension, on the other hand, the proline residue of the HIF alpha subunit is hydroxylated. The hydroxylated HIF alpha subunit is then degraded by proteasomal ubiquitylation, which is mediated by the von Hippel-Lindau tumor-suppressor gene pVHL.22 Degradation of HIF alpha prevents formation of the HIF heterodimers. HIF therefore cannot bind to the hypoxia-response elements, and erythropoietin gene transcription does not occur.23
Thus, in states of hypoxia, erythropoietin production is upregulated, whereas with normal oxygen tension, production is downregulated.
Erythropoietin is essential for terminal maturation of erythrocytes
Erythropoietin is essential for terminal maturation of erythrocytes.24 It is thought to stimulate the growth of erythrogenic progenitors: burst-forming units-erythroid (BFU-E) and colony-forming units-erythroid (CFU-E). In the absence of erythropoietin, BFU-E and CFU-E fail to differentiate into mature erythrocytes.25
Binding of erythropoietin to its receptor sets off a series of downstream signals, the most important being the signal transducer and activator of transcription 5 (STAT5). In animal studies, STAT5 was found to inhibit apoptosis through the early induction of an antiapoptotic gene, Bcl-xL.26
Iron metabolism is controlled by several proteins
Iron is characterized by its capacity to accept or donate electrons. This unique property makes it a crucial element in many biochemical reactions such as enzymatic activity, DNA synthesis, oxygen transport, and cell respiration.
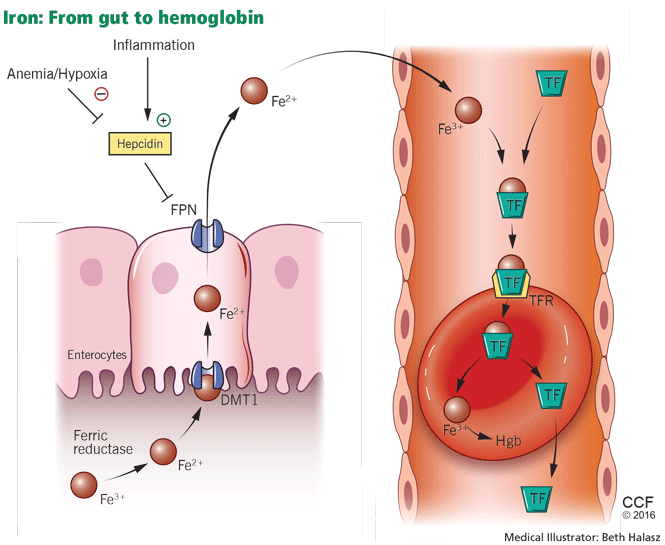
Iron metabolism is under the control of several proteins that play different roles in its absorption, recycling, and loss (Figure 2).27
Dietary iron exists primarily in its poorly soluble trivalent ferric form (Fe3+), and it needs to be reduced to its soluble divalent ferrous form (Fe2+) by ferric reductase to be absorbed. Ferrous iron is taken up at the apical side of enterocytes by a divalent metal transporter (DMT1) and is transported across the brush border.28
To enter the circulation, iron has to be transported across the basolateral membrane by a transporter called ferroportin.29 Ferroportin is also found in placental syncitiotrophoblasts, where it transfers iron from mother to fetus, and in macrophages, where it allows recycling of iron scavenged from damaged cells back into the circulation.30 Upon its release, the ferrous iron is oxidized to the ferric form and loaded onto transferrin. This oxidation process involves hephaestin, a homologue of the ferroxidase ceruloplasmin.31
In the plasma, iron is bound to transferrin, and under normal circumstances one-third of transferrin is saturated with iron.32 Transferrin receptors are present on most cells but are most dense on erythroid precursors. Each transferrin receptor can bind two transferrin molecules. After binding to transferrin, the transferrin receptor is endocytosed, and the iron is released into acidified vacuoles. The transferrin-receptor complex is then recycled to the surface.33
Ferritin is the cellular storage protein for iron, and it can store up to 4,500 atoms of iron within its spherical cavity.34 The serum level of ferritin reflects overall storage, with 1 ng/mL of ferritin indicating 10 mg of total iron stores.35 Ferritin is also an acute-phase reactant, and plasma levels can increase in inflammatory states such as infection or malignancy. As such, elevated ferritin does not necessarily indicate elevated iron stores.
Iron is lost in sweat, shed skin cells, and sloughed intestinal mucosal cells. However, there is no specific mechanism of iron excretion from the human body. Thus, iron is mainly regulated at the level of intestinal absorption. The iron exporter ferroportin is upregulated by the amount of available iron and is degraded by hepcidin.36
Hepcidin is a small cysteine-rich cationic peptide that is primarily produced in the liver, with some minor production also occurring in the kidneys.37 Transcription of the gene encoding hepcidin is downregulated by anemia and hypoxia and upregulated by inflammation and elevated iron levels.38 Transcription of hepcidin leads to degradation of ferroportin and a decrease in intestinal iron absorption. On the other hand, anemia and hypoxia inhibit hepcidin transcription, which allows ferroportin to facilitate intestinal iron absorption.
TREATMENT OF RENAL ANEMIA
Early enthusiasm for erythropoietin agents
Androgens started to be used to treat anemia of end-stage renal disease in 1970,39,40 and before the advent of recombinant human erythropoietin, they were a mainstay of nontransfusional therapy for anemic patients on dialysis.
The approval of recombinant human erythropoietin in 1989 drastically shifted the treatment of renal anemia. While the initial goal of treating anemia of chronic kidney disease with erythropoietin was to prevent blood transfusions,41 subsequent studies showed that the benefits might be far greater. Indeed, an initial observational trial showed that erythropoiesis-stimulating agents (ESAs) were associated with improved quality of life,42 improved neurocognitive function,43,44 and even cost savings.45 The benefits also extended to major outcomes such as regression of left ventricular hypertrophy,46 improvement in New York Heart Association class and cardiac function,47 fewer hospitalizations,48 and even reduction of cardiovascular mortality rates.49
As a result, ESA use gained popularity, and by 2006 an estimated 90% of dialysis patients were receiving these agents.50 The target and achieved hemoglobin levels also increased, with mean hemoglobin levels in hemodialysis patients being raised from 9.7 to 12 g/dL.51
Disappointing results in clinical trials of ESAs to normalize hemoglobin
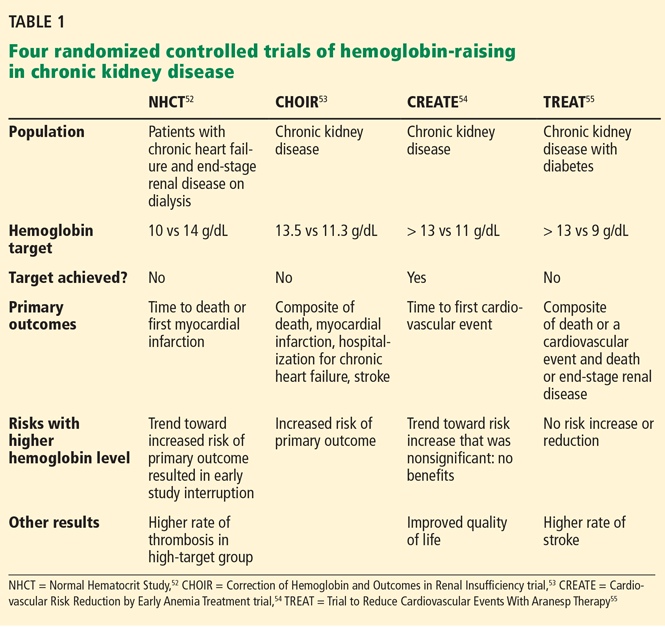
To prospectively study the effects of normalized hemoglobin targets, four randomized controlled trials were conducted (Table 1):
- The Normal Hematocrit Study (NHCT)52
- The Correction of Hemoglobin and Outcomes in Renal Insufficiency (CHOIR) trial53
- The Cardiovascular Risk Reduction by Early Anemia Treatment (CREATE) trial54
- The Trial to Reduce Cardiovascular Events With Aranesp Therapy (TREAT).55
These trials randomized patients to either higher “normal-range” hemoglobin targets or to lower target hemoglobin levels.
Their findings were disappointing and raised several red flags about excessive use of ESAs. The trials found no benefit in higher hemoglobin targets, and in fact, some of them demonstrated harm in patients randomized to higher targets. Notably, higher hemoglobin targets were associated with significant side effects such as access-site thrombosis,52 strokes,55 and possibly cardiovascular events.54,55 Only the CREATE trial was able to demonstrate a quality-of-life benefit for the high-target group.54
It remains unclear whether these adverse events were from the therapy itself or from an increased morbidity burden in the treated patients. Erythropoietin use is associated with hypertension,56 thought to be related to endothelin-mediated vasoconstriction.57 In our experience, this is most evident when hemoglobin levels are normalized with ESA therapy. Cycling of erythropoietin levels between extreme levels can lead to vascular remodeling, which may also be related to its cardiovascular effects.57
A noticeable finding in several of these trials was that patients failed to achieve the higher hemoglobin target despite the use of very high doses of ESA. Reanalysis of data from the CHOIR and CREATE trials showed that the patients who had worse outcomes were more likely to have required very high doses without achieving their target hemoglobin.58,59 Indeed, patients who achieved the higher target hemoglobin levels, usually at lower ESA doses, had better outcomes. This suggested that the need for a higher dose was associated with poorer outcomes, either as a marker of comorbidity or due to yet undocumented side effects of such high doses.
General approach to therapy
Before attributing anemia to chronic kidney disease, a thorough evaluation should be conducted to look for any reversible process that could be contributing to the anemia.
The causes of anemia are numerous and beyond the scope of this review. However, among the common causes of anemia in chronic kidney disease are deficiencies of iron, vitamin B12, and folate. Therefore, guidelines recommend checking iron, vitamin B12, and folate levels in the initial evaluation of anemia.60
Iron deficiency in particular is very common in chronic kidney disease patients and is present in nearly all dialysis patients.61 Hemodialysis patients are estimated to lose 1 to 3 g of iron per year as a result of blood loss in the dialysis circuit and increased iron utilization secondary to ESA therapy.62
However, in contrast to the general population, in which the upper limits of normal for iron indices are well defined, high serum ferritin levels appear to be poorly predictive of hemoglobin responsiveness in dialysis patients.63,64 Thus, the cutoffs that define iron responsiveness are much higher than standard definitions for iron deficiency.65,66 The Dialysis Patients’ Response to IV Iron With Elevated Ferritin (DRIVE) study showed that dialysis patients benefit from intravenous iron therapy even if their ferritin is as high as 1,200 ng/mL, provided their transferrin saturation is below 30%.67
Of note, erythropoietin levels cannot be used to distinguish renal anemia from other causes of anemia. Indeed, patients with renal failure may have “relative erythropoietin deficiency,” ie, “normal” erythropoietin levels that are actually too low in view of the degree of anemia.68,69 In addition to the decreased production capacity by the kidney, there appears to be a component of resistance to the action of erythropoietin in the bone marrow.
For these reasons, there is no erythropoietin level that can be considered “inadequate” or defining of renal anemia. Thus, measuring erythropoietin levels is not routinely recommended in the evaluation of renal anemia.
Two ESA preparations
The two ESAs that have traditionally been used in the treatment of renal anemia are recombinant human erythropoietin and darbepoietin alfa. They appear to be equivalent in terms of safety and efficacy.70 However, darbepoietin alfa has more sialic acid molecules, giving it a higher potency and longer half-life and allowing for less-frequent injections.71,72
In nondialysis patients, recombinant human erythropoietin is typically given every 1 to 2 weeks, whereas darbepoietin alfa can be given every 2 to 4 weeks. In dialysis patients, recombinant human erythropoietin is typically given 3 times per week with every dialysis treatment, while darbepoietin alfa is given once a week.
Target hemoglobin levels: ≤ 11.5 g/dL
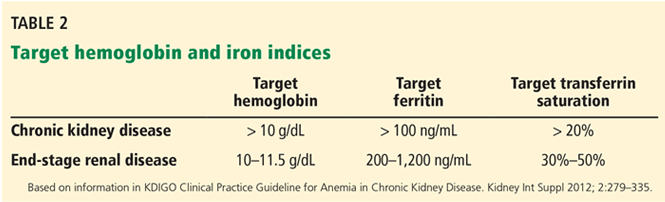
In light of the four trials described in Table 1, the international Kidney Disease: Improving Global Outcomes (KDIGO) guidelines60 recommend the following (Table 2):
For patients with chronic kidney disease who are not on dialysis, ESA therapy should not be initiated if the hemoglobin level is higher than 10 g/dL. If the hemoglobin level is lower than 10 g/dL, ESA therapy can be initiated, but the decision needs to be individualized based on the rate of fall of hemoglobin concentration, prior response to iron therapy, the risk of needing a transfusion, the risks related to ESA therapy, and the presence of symptoms attributable to anemia.
For patients on dialysis, ESA therapy should be used when the hemoglobin level is between 9 and 10 g/dL to avoid having the hemoglobin fall below 9 g/dL.
In all adult patients, ESAs should not be used to intentionally increase the hemoglobin level above 13 g/dL but rather to maintain levels no higher than 11.5 g/dL. This target is based on the observation that adverse outcomes were associated with ESA use with hemoglobin targets higher than 13 g/dL (Table 1).
Target iron levels
Regarding iron stores, the guidelines recommend the following:
For adult patients with chronic kidney disease who are not on dialysis, iron should be given to keep transferrin saturation above 20% and ferritin above 100 ng/mL. Transferrin saturation should not exceed 30%, and ferritin levels should not exceed 500 ng/mL.
For adult patients on dialysis, iron should be given to maintain transferrin saturation above 30% and ferritin above 200 ng/mL.
The upper limits of ferritin and transferrin saturation are somewhat controversial, as the safety of intentionally maintaining respective levels greater than 30% and 500 ng/mL has been studied in very few patients. Transferrin saturation should in general not exceed 50%.
High ferritin levels are associated with higher death rates, but whether elevation of ferritin levels is a marker of excessive iron administration rather than a nonspecific acute-phase reactant is not clear. The 2006 guidelines60 cited upper ferritin limits of 500 to 800 ng/mL. However, the more recent DRIVE trial67 showed that patients with ferritin levels of 500 to 1,200 ng/mL will respond to intravenous administration of iron with an increase in their hemoglobin levels. This has led many clinicians to adopt a higher ferritin limit of 1,200 ng/mL.
Hemosiderosis, or excess iron deposition, was a known consequence of frequent transfusions in patients with end-stage renal disease before ESA therapy was available. However, there have been no documented cases of clinical iron overload from iron therapy using current guidelines.73
These algorithms are nuanced, and the benefit of giving intravenous iron should always be weighed against the risks of short-term acute toxicity and infection. Treatment of renal anemia not only requires in-depth knowledge of the topic, but also familiarity with the patient’s specific situation. As such, it is not recommended that clinicians unfamiliar with the treatment of renal anemia manage its treatment.
PARTICULAR CIRCUMSTANCES
Inflammation and ESA resistance
While ESAs are effective in treating anemia in many cases, in many patients the anemia fails to respond. This is of particular importance, since ESA hyporesponsiveness has been found to be a powerful predictor of cardiovascular events and death.74 It is unclear, however, whether high doses of ESA are inherently toxic or whether hyporesponsiveness is a marker of adverse outcomes related to comorbidities.
KDIGO defines initial hyporesponsiveness as having no increase in hemoglobin concentration after the first month of appropriate weight-based dosing, and acquired hyporesponsiveness as requiring two increases in ESA doses up to 50% beyond the dose at which the patient had originally been stable.60 Identifying ESA hyporesponsiveness should lead to an intensive search for potentially correctable factors.
The two major factors accounting for the state of hyporesponsiveness are inflammation and iron deficiency.75,76
Inflammation. High C-reactive protein levels have been shown to predict resistance to erythropoietin in dialysis patients.77 The release of cytokines such as tumor necrosis factor alpha, interleukin 1, and interferon gamma has an inhibitory effect on erythropoiesis.78 Additionally, inflammation can alter the response to ESAs by disrupting the metabolism of iron79 through the release of hepcidin, as previously discussed.38 These reasons likely account for the observed lower response to ESAs in the setting of acute illness and explain why ESAs are not recommended for correcting acute anemia.80
Iron deficiency also can blunt the response to ESAs. Large amounts of iron are needed for effective erythropoietic bursts. As such, iron supplementation is now a recognized treatment of renal anemia.81
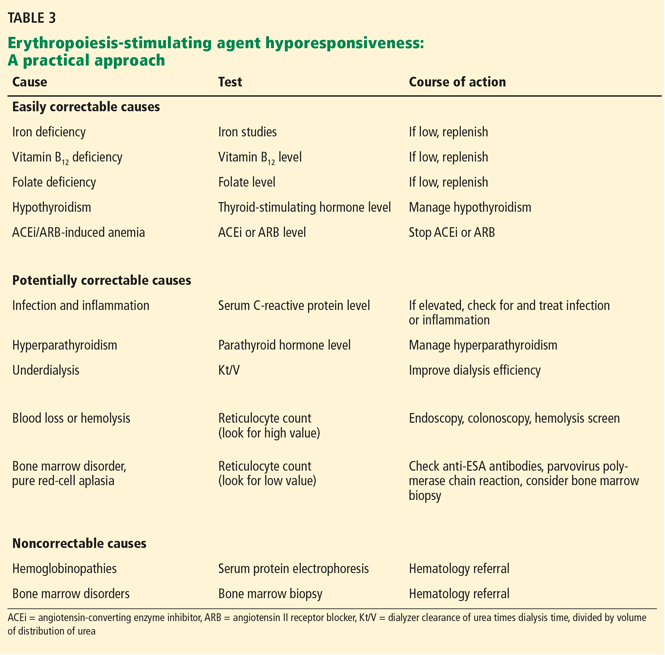
Other factors associated with hyporesponsiveness include chronic occult blood loss, aluminum toxicity, cobalamin or folate deficiencies, testosterone deficiency, inadequate dialysis, hyperparathyroidism, and superimposed primary bone marrow disease,82,83 and these should be addressed in patients whose anemia does not respond as expected to ESA therapy. A summary of the main causes of ESA hyporesponsiveness, their reversibility, and recommended treatments is presented in Table 3.
Antibody-mediated pure red-cell aplasia. Rarely, patients receiving ESA therapy develop antibodies that neutralize both the ESA and endogenous erythropoietin. The resulting syndrome, called antibody-mediated pure red-cell aplasia, is characterized by the sudden development of severe transfusion-dependent anemia. This has historically been connected to epoetin beta, a formulation not in use in the United States. However, cases have been documented with epoetin alfa and darbepoetin. The incidence rate is low with subcutaneous ESA use, estimated at 0.5 cases per 10,000 patient-years84 and anecdotal with intravenous ESA.85 The definitive diagnosis requires demonstration of neutralizing antibodies against erythropoietin. Parvovirus infection should be excluded as an alternative cause of pure redcell aplasia.
ANEMIA IN CANCER PATIENTS
ESAs are effective in raising hemoglobin levels and reducing transfusion requirements in patients with chemotherapy-induced anemia.86 However, there are data linking the use of ESAs to shortened survival in patients who have a variety of solid tumors.87
Several mechanisms have been proposed to explain this rapid disease progression, most notably acceleration in tumor growth88–90 by stimulation of erythropoietin receptors on the surface of the tumor cells, leading to increased tumor angiogenesis.91,92
For these reasons, treatment of renal anemia in the setting of active malignancy should be referred to an oncologist.
NOVEL TREATMENTS
Several new agents for treating renal anemia are currently under review.
Continuous erythropoiesis receptor activator
Continuous erythropoiesis receptor activator is a pegylated form of recombinant human erythropoietin that has the ability to repeatedly activate the erythropoietin receptor. It appears to be similar to the other forms of erythropoietin in terms of safety and efficacy in both end-stage renal disease93 and chronic kidney disease.94 It has the advantage of an extended serum half-life, which allows for longer dosing intervals, ie, every 2 weeks. Its use is currently gaining popularity in the dialysis community.
HIF stabilizers
Our growing understanding of the physiology of erythropoietin offers new potential treatment targets. As previously described, production of erythropoietin is stimulated by HIFs. In order to be degraded, these HIFs are hydroxylated at their proline residues by a prolyl hydroxylase. A new category of drugs called prolyl-hydroxylase inhibitors (PDIs) offers the advantage of stabilizing the HIFs, leading to an increase in erythropoietin production.
In phase 1 and 2 clinical trials, these agents have been shown to increase hemoglobin in both end-stage renal disease and chronic kidney disease patients15,16 but not in anephric patients, demonstrating a renal source of the erythropoietin production even in nonfunctioning kidneys. The study of one PDI agent (FG 2216) was halted temporarily after a report of death from fulminant hepatitis, but the other (FG 4592) continues to be studied in a phase 2 clinical trial.95,96
TAKE-HOME POINTS
- Anemia of renal disease is a common condition that is mainly caused by a decrease in erythropoietin production by the kidneys.
- While anemia of renal disease can be corrected with ESAs, it is necessary to investigate and rule out underlying treatable conditions such as iron or vitamin deficiencies before giving an ESA.
- Anemia of renal disease is associated with significant morbidity such as increased risk of left ventricular hypertrophy, myocardial infarction, and heart failure, and has been described as an all-cause mortality multiplier.
- Unfortunately, the only undisputed benefit of treatment to date remains the avoidance of blood transfusions. Furthermore, the large randomized controlled trials that looked at the benefits of ESA have shown that their use can be associated with increased risk of cardiovascular events. Therefore, use of an ESA in end-stage renal disease should never target a normal hemoglobin levels but rather aim for a hemoglobin level of no more than 11.5 g/dL.
- Use of an ESA in chronic kidney disease should be individualized and is not recommended to be started unless the hemoglobin level is less than 10 g/dL.
- Several newer agents for renal anemia are currently under review. A pegylated form of recombinant human erythropoietin has an extended half-life, and a new and promising category of drugs called HIF stabilizers is currently under study.
- World Health Organization (WHO). Nutritional anaemias: report of a WHO scientific group. Geneva, Switzerland: World Health Organization, 1968.
- Hsu CY, McCulloch CE, Curhan GC, et al. Epidemiology of anemia associated with chronic renal insufficiency among adults in the United States: results from the Third National Health and Nutrition Examination Survey. J Am Soc Nephrol 2002; 13:504–510.
- Radtke HW, Claussner A, Erbes PM, Scheuermann EH, Schoeppe W, Koch KM. Serum erythropoetin concentration in chronic renal failure: relationship to degree of anemia and excretory renal function. Blood 1979; 54:877–884.
- Kazmi WH, Kausz AT, Khan S, et al. Anemia: an early complication of chronic renal insufficiency. Am J Kidney Dis 2001; 38:803–812.
- United States Renal Data System. Chapter 3. Morbidity & mortality in patients with CKD. www.usrds.org/2012/view/v1_03.aspx. Accessed June 9, 2016.
- Horwich TB, Fonarow GC, Hamilton MA, MacLellan WR, Borenstein J. Anemia is associated with worse symptoms, greater impairment in functional capacity and a significant increase in mortality in patients with advanced heart failure. J Am Coll Cardiol 2002; 39:1780–1786.
- Mark DB, Felker GM. B-type natriuretic peptide: a biomarker for all seasons? N Engl J Med 2004; 350:718–720.
- Walker AM, Schneider G, Yeaw J, Nordstrom B, Robbins S, Pettitt D. Anemia as a predictor of cardiovascular events in patients with elevated serum creatinine. J Am Soc Nephrol 2006; 17:2293–2298.
- Abramson JL, Jurkovitz CT, Vaccarino V, Weintraub WS, McClellan W. Chronic kidney disease, anemia, and incident stroke in a middle-aged, community-based population: the ARIC Study. Kidney Int 2003; 64:610–615.
- Sarnak MJ, Tighiouart H, Manjunath G, et al. Anemia as a risk factor for cardiovascular disease in the Atherosclerosis Risk in Communities (ARIC) study. J Am Coll Cardiol 2002; 40:27–33.
- McClellan WM, Flanders WD, Langston RD, Jurkovitz C, Presley R. Anemia and renal insufficiency are independent risk factors for death among patients with congestive heart failure admitted to community hospitals: a population-based study. J Am Soc Nephrol 2002; 13:1928–1936.
- Xia H, Ebben J, Ma JZ, Collins AJ. Hematocrit levels and hospitalization risks in hemodialysis patients. J Am Soc Nephrol 1999; 10:1309–1316.
- Collins AJ, Li S, St Peter W, et al. Death, hospitalization, and economic associations among incident hemodialysis patients with hematocrit values of 36 to 39%. J Am Soc Nephrol 2001; 12:2465–2473.
- Agarwal AK. Practical approach to the diagnosis and treatment of anemia associated with CKD in elderly. J Am Med Dir Assoc 2006; 7(suppl 9):S7–S12.
- Bernhardt WM, Wiesener MS, Scigalla P, et al. Inhibition of prolyl hydroxylases increases erythropoietin production in ESRD. J Am Soc Nephrol 2010; 21:2151–2156.
- Provenzano R, Fadda G, Bernardo M, et al. FG-2216, a novel oral HIF-PHI, stimulates erythropoiesis and increases hemoglobin concentration in patients with non-dialysis CKD. Am J Kidney Dis 2008; 51:B80.
- Maxwell PH, Osmond MK, Pugh CW, et al. Identification of the renal erythropoietin-producing cells using transgenic mice. Kidney Int 1993; 44:1149–1162.
- Maxwell PH, Ferguson DJ, Nicholls LG, et al. Sites of erythropoietin production. Kidney Int 1997; 51:393–401.
- Jelkmann W. Erythropoeitin: structure, control of production and function. Physiol Rev 1992; 72:449–489.
- Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA 1995; 92:5510–5514.
- Wang GL, Semenza GL. Purification and characterization of hypoxia-inducible factor 1. J Biol Chem 1995; 270:1230–1237.
- Maxwell PH, Wiesener MS, Chang GW, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999; 399:271–275.
- Salceda S, Caro J. Hypoxia-inducible factor 1alpha protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J Biol Chem 1997; 272:22642–22647.
- Malik J, Kim AR, Tyre KA, Cherukuri AR, Palis J. Erythropoietin critically regulates the terminal maturation of murine and human primitive erythroblasts. Haematologica 2013; 98:1778–1787.
- Wu H, Liu X, Jaenisch R, Lodish HF. Generation of committed erythroid BFU-E and CFU-E progenitors does not require erythropoietin or the erythropoietin receptor. Cell 1995; 83:59–67.
- Socolovsky M, Nam H, Fleming MD, Haase VH, Brugnara C, Lodish HF. Ineffective erythropoiesis in Stat5a(-/-)5b(-/-) mice due to decreased survival of early erythroblasts. Blood 2001; 98:3261–3273.
- Papanikolaou G, Pantopoulos K. Iron metabolism and toxicity. Toxicol Appl Pharmacol 2005; 202:199–211.
- Conrad ME, Umbreit JN. Pathways of iron absorption. Blood Cells Mol Dis 2002; 29:336–355.
- Frazer DM, Anderson GJ. The orchestration of body iron intake: how and where do enterocytes receive their cues? Blood Cells Moll Dis 2003; 30:288–297.
- Donovan A, Lima CA, Pinkus JL, et al. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab 2005; 1:191–200.
- Vulpe CD, Kuo YM, Murphy TL, et al. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat Genet 1999; 21:195–199.
- Bothwell TH. Overview and mechanisms of iron regulation. Nutr Rev 1995: 53:237–245.
- Kawabata H, Nakamaki T, Ikonomi P, Smith RD, Germain RS, Koeffler HP. Expression of transferrin receptor 2 in normal and neoplastic hematopoietic cells. Blood 2001; 98:2714–2719.
- Arosio P, Levi S. Cytosolic and mitochondrial ferritins in the regulation of cellular iron homeostasis and oxidative damage. Biochim Biophys Acta 2010; 1800:783–792.
- Finch CA, Bellotti V, Stray S, et al. Plasma ferritin determination as a diagnostic tool. West J Med 1986; 145:657–663.
- Delaby C, Pilard N, Goncalves AS, Beaumont C, Canonne-Hergaux F. Presence of the iron exporter ferroportin at the plasma membrane of macrophages is enhanced by iron loading and down-regulated by hepcidin. Blood 2005; 106:3979–3984.
- Ganz T. Hepcidin, a key regulator of iron metabolism and mediator of anemia of inflammation. Blood 2003; 102:783–788.
- Nicolas G, Chauvet C, Viatte L, et al. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J Clin Invest 2002; 110:1037–1044.
- DeGowin RL, Lavender AR, Forland M, Charleston D, Gottschalk A. Erythropoiesis and erythropoietin in patients with chronic renal failure treated with hemodialysis and testosterone. Ann Intern Med 1970; 72:913–918.
- Richardson JR Jr, Weinstein MB. Erythropoietic response of dialyzed patients to testosterone administration. Ann Intern Med 1970; 73:403–407
- Eschbach JW, Abdulhadi MH, Browne JK, et al. Recombinant human erythropoietin in anemic patients with end-stage renal disease. Results of a phase III multicenter clinical trial. Ann Intern Med 1989; 111:992–1000.
- Moreno F, Aracil FJ, Pérez R, Valderrábano F. Controlled study on the improvement of quality of life in elderly hemodialysis patients after correcting end-stage renal disease-related anemia with erythropoietin. Am J Kidney Dis 1996; 27:548–556.
- Nissenson AR, Nimer SD, Wolcott DL. Recombinant human erythropoietin and renal anemia: molecular biology, clinical efficacy, and nervous system effects. Ann Intern Med 1991; 114:402–416.
- Stivelman JC. Benefits of anaemia treatment on cognitive function. Nephrol Dial Transplant 2000; 15(suppl 3):29–35.
- Maddux FW, Shetty S, del Aguila MA, Nelson MA, Murray BM. Effect of erythropoiesis-stimulating agents on healthcare utilization, costs, and outcomes in chronic kidney disease. Ann Pharmacother 2007; 41:1761–1769.
- Macdougall IC, Lewis NP, Saunders MJ, et al. Long-term cardiorespiratory effects of amelioration of renal anaemia by erythropoietin. Lancet 1990; 335:489–493.
- Silverberg DS, Wexler D, Blum M, et al. Effects of treatment with epoetin beta on outcomes in patients with anaemia and chronic heart failure. Kidney Blood Press Res 2005; 28:41–47.
- Perkins R, Olson S, Hansen J, Lee J, Stiles K, Lebrun C. Impact of an anemia clinic on emergency room visits and hospitalizations in patients with anemia of CKD pre-dialysis. Nephrol Nurs J 2007; 34:167–173, 182.
- Locatelli F, Conte F, Marcelli D. The impact of haematocrit levels and erythropoietin treatment on overall and cardiovascular mortality and morbidity—the experience of the Lombardy Dialysis Registry. Nephrol Dial Transplant 1998; 13:1642–1644.
- Centers for Medicare and Medicaid Services; Kinney R. 2005 Annual Report: ESRD Clinical Performance Measures Project. Am J Kidney Dis 2006; 48(suppl 2):S1–S106.
- US Renal Data System. Annual Data Report 2006. www.usrds.org/adr.aspx. Accessed July 3, 2016.
- Besarab A, Bolton WK, Browne JK, et al. The effects of normal as compared with low hematocrit values in patients with cardiac disease who are receiving hemodialysis and epoetin. N Engl J Med 1998; 339:584–590.
- Singh AK, Szczech L, Tang KL, et al; CHOIR Investigators. Correction of anemia with epoetin alfa in chronic kidney disease. N Engl J Med 2006; 355:2085–2098.
- Drüeke TB, Locatelli F, Clyne N, et al; CREATE Investigators. Normalization of hemoglobin level in patients with chronic kidney disease and anemia. N Engl J Med 2006; 355:2071–2084.
- Pfeffer MA, Burdmann EA, Chen CY, et al; TREAT Investigators. A trial of darbepoetin alfa in type 2 diabetes and chronic kidney disease. N Engl J Med 2009; 361:2019–2032.
- Kirkpantur A, Kahraman S, Yilmaz R, et al. The effects of maintenance recombinant human erythropoietin therapy on ambulatory blood pressure recordings: conventional, Doppler, and tissue Doppler echocardiographic parameters. Artif Organs 2005; 29:965–972.
- Fishbane S, Berns JS. Hemoglobin cycling in hemodialysis patients treated with recombinant human erythropoietin. Kidney Int 2005; 68:1337–1343.
- Szczech LA, Barnhart HX, Inrig JK, et al. Secondary analysis of the CHOIR trial epoetin-alpha dose and achieved hemoglobin outcomes. Kidney Int 2008; 74:791–798.
- Solomon SD, Uno H, Lewis EF, et al; Trial to Reduce Cardiovascular Events with Aranesp Therapy (TREAT) Investigators. Erythropoietic response and outcomes in kidney disease and type 2 diabetes. N Engl J Med 2010; 363:1146–1155.
- Kidney Disease: Improving Global Outcomes (KDIGO). KDIGO Clinical Practice Guideline for Anemia in Chronic Kidney Disease. Kidney Int Suppl 2012; 2:279–335.
- Fernández-Rodríguez AM, Guindeo-Casasús MC, Molero-Labarta T, et al. Diagnosis of iron deficiency in chronic renal failure. Am J Kidney Dis 1999; 34:508–513.
- Eschbach JW, Cook JD, Scribner BH, Finch CA. Iron balance in hemodialysis patients. Ann Intern Med 1977; 87:710–713.
- Mittman N, Sreedhara R, Mushnick R, et al. Reticulocyte hemoglobin content predicts functional iron deficiency in hemodialysis patients receiving rHuEPO. Am J Kidney Dis 1997; 30:912–922.
- Tessitore N, Solero GP, Lippi G, et al. The role of iron status markers in predicting response to intravenous iron in haemodialysis patients on maintenance erythropoietin. Nephrol Dial Transplant 2001; 16:1416–1423.
- Coyne DW. Iron indices: what do they really mean? Kidney Int Suppl 2006; 101:S4–S8.
- Fishbane S, Kowalski EA, Imbriano LJ, Maesaka JK. The evaluation of iron status in hemodialysis patients. J Am Soc Nephrol 1996; 7:2654–2657.
- Coyne DW, Kapoian T, Suki W, et al; DRIVE Study Group. Ferric gluconate is highly efficacious in anemic hemodialysis patients with high serum ferritin and low transferrin saturation: results of the Dialysis Patients’ Response to IV Iron with Elevated Ferritin (DRIVE) Study. J Am Soc Nephrol 2007; 18:975–984.
- Radtke HW, Claussner A, Erbes PM, Scheuermann EH, Schoeppe W, Koch KM. Serum erythropoietin concentration in chronic renal failure: relationship to degree of anemia and excretory renal function. Blood 1979; 54:877–884.
- Korte W, Cogliatti SB, Jung K, Riesen W. Mild renal dysfunction is sufficient to induce erythropoietin deficiency in patients with unexplained anaemia. Clin Chim Acta 2000; 292:149–154.
- Locatelli F, Olivares J, Walker R, et al; European/Australian NESP 980202 Study Group. Novel erythropoiesis stimulating protein for treatment of anemia in chronic renal insufficiency. Kidney Int 2001; 60:741–747.
- Carrera F, Burnier M. Use of darbepoetin alfa in the treatment of anaemia of chronic kidney disease: clinical and pharmacoeconomic considerations. NDT Plus 2009; 2(suppl 1):i9–i17.
- Egrie JC, Browne JK. Development and characterization of novel erythropoiesis stimulating protein (NESP). Nephrol Dial Transplant 2001; 16(suppl 3):3–13.
- Nissenson AR, Charytan C. Controversies in iron management. Kidney Int Suppl 2003; 87:S64–S71.
- Kilpatrick RD, Critchlow CW, Fishbane S, et al. Greater epoetin alpha responsiveness is associated with improved survival in hemodialysis patients. Clin J Am Soc Nephrol 2008; 3:1077–1083.
- Locatelli F, Aljama P, Barany P, et al; European Best Practice Guidelines Working Group. Revised European best practice guidelines for the management of anaemia in patients with chronic renal failure. Nephrol Dial Transplant 2004; 19(suppl 2):ii1–ii47.
- Stenvinkel P. The role of inflammation in the anaemia of end-stage renal disease. Nephrol Dial Transplant 2001; 16(suppl 7):36–40.
- Barany P, Divino Filho JC, Bergstrom J. High C-reactive protein is a strong predictor of resistance to erythropoietin in hemodialysis patients. Am J Kidney Dis 1997; 29:565–568.
- Drueke T. Hyporesponsiveness to recombinant human erythropoietin. Nephrol Dial Transplant 2001; 16(suppl 7):25–28.
- Casadevall N. Cellular mechanism of resistance to erythropoietin. Nephrol Dial Transplant 1995; 10(suppl 6):27–30.
- Kraus E, Rabb H. EPO therapy during acute kidney disease: to use or not to use, that is the question. Am J Kidney Dis 2005; 46:967–969.
- Gotloib L, Silverberg D, Fudin R, Shostak A. Iron deficiency is a common cause of anemia in chronic kidney disease and can often be corrected with intravenous iron. J Nephrol 2006; 19:161–167.
- Tarng DC, Huang TP, Chen TW, Yang WC. Erythropoietin hyporesponsiveness: from iron deficiency to iron overload. Kidney Int Suppl 1999; 69:S107–S118.
- Drüeke TB. Modulating factors in the hematopoietic response to erythropoietin. Am J Kidney Dis 1991; 18(suppl 1):87–92.
- Boven K, Stryker S, Knight J, et al. The increased incidence of pure red cell aplasia with an Eprex formulation in uncoated rubber stopper syringes. Kidney Int 2005; 67:2346–2353.
- Shimizu H, Saitoh T, Ota F, et al. Pure red cell aplasia induced only by intravenous administration of recombinant human erythropoietin. Acta Haematol 2011; 126:114–118.
- Tonia T, Mettler A, Robert N, et al. Erythropoietin or darbepoetin for patients with cancer. Cochrane Database Syst Rev 2012; 12:CD003407.
- Bohlius J, Langensiepen S, Schwarzer G, et al. Recombinant human erythropoietin and overall survival in cancer patients: results of a comprehensive meta-analysis. J Natl Cancer Inst 2005; 97:489–498.
- Henke M, Laszig R, Rübe C, et al. Erythropoietin to treat head and neck cancer patients with anaemia undergoing radiotherapy: randomised, double-blind, placebo-controlled trial. Lancet 2003; 362:1255–1260.
- Leyland-Jones B, Semiglazov V, Pawlicki M, et al. Maintaining normal hemoglobin levels with epoetin alfa in mainly nonanemic patients with metastatic breast cancer receiving first-line chemotherapy: a survival study. J Clin Oncol 2005; 23:5960–5972.
- Brower V. Erythropoietin may impair, not improve, cancer survival. Nat Med 2003; 9:1439.
- Acs G, Acs P, Beckwith SM, et al. Erythropoietin and erythropoietin receptor expression in human cancer. Cancer Res 2001; 61:3561–3565.
- Yasuda Y, Fujita Y, Matsuo T, et al. Erythropoietin regulates tumour growth of human malignancies. Carcinogenesis 2003; 24:1021–1029.
- Levin NW, Fishbane S, Cañedo FV, et al; MAXIMA Study Investigators. Intravenous methoxy polyethylene glycol-epoetin beta for haemoglobin control in patients with chronic kidney disease who are on dialysis: a randomised non-inferiority trial (MAXIMA). Lancet 2007; 370:1415–1421.
- Macdougall IC, Walker R, Provenzano R, et al; ARCTOS Study Investigators. C.E.R.A. corrects anemia in patients with chronic kidney disease not on dialysis: results of a randomized clinical trial. Clin J Am Soc Nephrol 2008; 3:337–347.
- Frohna PA, Milwee S, Pinkett J, et al. Preliminary results from a randomized, single-blind, placebo-controlled trial of FG-4592, a novel hypoxia inducible factor prolyl hydroxylase inhibitor, in subjects with CKD anemia (abstract). J Am Soc Nephrol 2007; 18:763.
- Holdstock L, Meadowcroft AM, Maier R, et al. Four-week studies of oral hypoxia-inducible factor-prolyl hydroxylase inhibitor GSK1278863 for treatment of anemia. J Am Soc Nephrol 2016; 27:1234–1244.
Anemia is a frequent complication of chronic kidney disease, occurring in over 90% of patients receiving renal replacement therapy. It is associated with significant morbidity and mortality. While its pathogenesis is typically multifactorial, the predominant cause is failure of the kidneys to produce enough endogenous erythropoietin. The clinical approval of recombinant human erythropoietin in 1989 dramatically changed the treatment of anemia of chronic kidney disease, but randomized controlled trials yielded disappointing results when erythropoiesis-stimulating agents (ESAs) were used to raise hemoglobin to normal levels.
This article reviews the epidemiology and pathophysiology of anemia of chronic kidney disease and discusses the complicated and conflicting evidence regarding its treatment.
DEFINITION AND PREVALENCE
Anemia is defined as a hemoglobin concentration less than 13.0 g/dL for men and less than 12.0 g/dL for premenopausal women.1 It is more common in patients with impaired kidney function, especially when the glomerular filtration rate (GFR) falls below 60 mL/min. It is rare at GFRs higher than 80 mL/min,2 but as the GFR falls, the severity of the anemia worsens3 and its prevalence increases: almost 90% of patients with a GFR less than 30 mL/min are anemic.4
RENAL ANEMIA IS ASSOCIATED WITH BAD OUTCOMES
Anemia in chronic kidney disease is independently associated with risk of death. It is also an all-cause mortality multiplier, ie, it magnifies the risk of death from other disease states.5
In observational studies, anemia was associated with faster progression of left ventricular hypertrophy, inflammation, and increased myocardial and peripheral oxygen demand, thereby leading to worse cardiac outcomes with increased risk of myocardial infarction, coronary revascularization, and readmission for heart failure.6–8 Anemia is also associated with fatigue, depression, reduced exercise tolerance, stroke, and increased risk of rehospitalization.9–13
RENAL ANEMIA IS MULTIFACTORIAL
Anemia of chronic kidney disease is typically attributed to the decrease of erythropoietin production that accompanies the fall in GFR. However, the process is multifactorial, with several other contributing factors: absolute and functional iron deficiency, folate and vitamin B12 deficiencies, reduced red blood cell life span, and suppression of erythropoiesis by the uremic milieu.14
While it was once thought that chronic kidney disease leads to loss of erythropoietin-producing cells, it is now known that downregulation of hypoxia-inducible factor (HIF; a transcription factor) is at least partially responsible for the decrease in erythropoietin production15,16 and that this downregulation is reversible (see below).
ERYTHROPOIETIN, IRON, AND RED BLOOD CELLS
Erythropoietin production is triggered by hypoxia, mediated by HIF
Erythropoietin is produced primarily in the deep cortex and outer medulla of the kidneys by a special population of peritubular interstitial cells.17 The parenchymal cells of the liver also produce erythropoietin, but much less.18
The rate of renal erythropoietin synthesis is determined by tissue oxygenation rather than by renal blood flow; production increases as the hemoglobin concentration drops and the arterial oxygen tension decreases (Figure 1).19
The gene for erythropoietin is located on chromosome 7 and is regulated by HIF. HIF molecules are composed of an alpha subunit, which is unstable at high Po2, and a beta subunit, constitutively present in the nucleus.20
In hypoxic conditions, the HIF dimer is transcriptionally active and binds to specific DNA recognition sequences called hypoxia-response elements. Gene transcription is upregulated, leading to increased production of erythropoietin.21
Under normal oxygen tension, on the other hand, the proline residue of the HIF alpha subunit is hydroxylated. The hydroxylated HIF alpha subunit is then degraded by proteasomal ubiquitylation, which is mediated by the von Hippel-Lindau tumor-suppressor gene pVHL.22 Degradation of HIF alpha prevents formation of the HIF heterodimers. HIF therefore cannot bind to the hypoxia-response elements, and erythropoietin gene transcription does not occur.23
Thus, in states of hypoxia, erythropoietin production is upregulated, whereas with normal oxygen tension, production is downregulated.
Erythropoietin is essential for terminal maturation of erythrocytes
Erythropoietin is essential for terminal maturation of erythrocytes.24 It is thought to stimulate the growth of erythrogenic progenitors: burst-forming units-erythroid (BFU-E) and colony-forming units-erythroid (CFU-E). In the absence of erythropoietin, BFU-E and CFU-E fail to differentiate into mature erythrocytes.25
Binding of erythropoietin to its receptor sets off a series of downstream signals, the most important being the signal transducer and activator of transcription 5 (STAT5). In animal studies, STAT5 was found to inhibit apoptosis through the early induction of an antiapoptotic gene, Bcl-xL.26
Iron metabolism is controlled by several proteins
Iron is characterized by its capacity to accept or donate electrons. This unique property makes it a crucial element in many biochemical reactions such as enzymatic activity, DNA synthesis, oxygen transport, and cell respiration.
Iron metabolism is under the control of several proteins that play different roles in its absorption, recycling, and loss (Figure 2).27
Dietary iron exists primarily in its poorly soluble trivalent ferric form (Fe3+), and it needs to be reduced to its soluble divalent ferrous form (Fe2+) by ferric reductase to be absorbed. Ferrous iron is taken up at the apical side of enterocytes by a divalent metal transporter (DMT1) and is transported across the brush border.28
To enter the circulation, iron has to be transported across the basolateral membrane by a transporter called ferroportin.29 Ferroportin is also found in placental syncitiotrophoblasts, where it transfers iron from mother to fetus, and in macrophages, where it allows recycling of iron scavenged from damaged cells back into the circulation.30 Upon its release, the ferrous iron is oxidized to the ferric form and loaded onto transferrin. This oxidation process involves hephaestin, a homologue of the ferroxidase ceruloplasmin.31
In the plasma, iron is bound to transferrin, and under normal circumstances one-third of transferrin is saturated with iron.32 Transferrin receptors are present on most cells but are most dense on erythroid precursors. Each transferrin receptor can bind two transferrin molecules. After binding to transferrin, the transferrin receptor is endocytosed, and the iron is released into acidified vacuoles. The transferrin-receptor complex is then recycled to the surface.33
Ferritin is the cellular storage protein for iron, and it can store up to 4,500 atoms of iron within its spherical cavity.34 The serum level of ferritin reflects overall storage, with 1 ng/mL of ferritin indicating 10 mg of total iron stores.35 Ferritin is also an acute-phase reactant, and plasma levels can increase in inflammatory states such as infection or malignancy. As such, elevated ferritin does not necessarily indicate elevated iron stores.
Iron is lost in sweat, shed skin cells, and sloughed intestinal mucosal cells. However, there is no specific mechanism of iron excretion from the human body. Thus, iron is mainly regulated at the level of intestinal absorption. The iron exporter ferroportin is upregulated by the amount of available iron and is degraded by hepcidin.36
Hepcidin is a small cysteine-rich cationic peptide that is primarily produced in the liver, with some minor production also occurring in the kidneys.37 Transcription of the gene encoding hepcidin is downregulated by anemia and hypoxia and upregulated by inflammation and elevated iron levels.38 Transcription of hepcidin leads to degradation of ferroportin and a decrease in intestinal iron absorption. On the other hand, anemia and hypoxia inhibit hepcidin transcription, which allows ferroportin to facilitate intestinal iron absorption.
TREATMENT OF RENAL ANEMIA
Early enthusiasm for erythropoietin agents
Androgens started to be used to treat anemia of end-stage renal disease in 1970,39,40 and before the advent of recombinant human erythropoietin, they were a mainstay of nontransfusional therapy for anemic patients on dialysis.
The approval of recombinant human erythropoietin in 1989 drastically shifted the treatment of renal anemia. While the initial goal of treating anemia of chronic kidney disease with erythropoietin was to prevent blood transfusions,41 subsequent studies showed that the benefits might be far greater. Indeed, an initial observational trial showed that erythropoiesis-stimulating agents (ESAs) were associated with improved quality of life,42 improved neurocognitive function,43,44 and even cost savings.45 The benefits also extended to major outcomes such as regression of left ventricular hypertrophy,46 improvement in New York Heart Association class and cardiac function,47 fewer hospitalizations,48 and even reduction of cardiovascular mortality rates.49
As a result, ESA use gained popularity, and by 2006 an estimated 90% of dialysis patients were receiving these agents.50 The target and achieved hemoglobin levels also increased, with mean hemoglobin levels in hemodialysis patients being raised from 9.7 to 12 g/dL.51
Disappointing results in clinical trials of ESAs to normalize hemoglobin
To prospectively study the effects of normalized hemoglobin targets, four randomized controlled trials were conducted (Table 1):
- The Normal Hematocrit Study (NHCT)52
- The Correction of Hemoglobin and Outcomes in Renal Insufficiency (CHOIR) trial53
- The Cardiovascular Risk Reduction by Early Anemia Treatment (CREATE) trial54
- The Trial to Reduce Cardiovascular Events With Aranesp Therapy (TREAT).55
These trials randomized patients to either higher “normal-range” hemoglobin targets or to lower target hemoglobin levels.
Their findings were disappointing and raised several red flags about excessive use of ESAs. The trials found no benefit in higher hemoglobin targets, and in fact, some of them demonstrated harm in patients randomized to higher targets. Notably, higher hemoglobin targets were associated with significant side effects such as access-site thrombosis,52 strokes,55 and possibly cardiovascular events.54,55 Only the CREATE trial was able to demonstrate a quality-of-life benefit for the high-target group.54
It remains unclear whether these adverse events were from the therapy itself or from an increased morbidity burden in the treated patients. Erythropoietin use is associated with hypertension,56 thought to be related to endothelin-mediated vasoconstriction.57 In our experience, this is most evident when hemoglobin levels are normalized with ESA therapy. Cycling of erythropoietin levels between extreme levels can lead to vascular remodeling, which may also be related to its cardiovascular effects.57
A noticeable finding in several of these trials was that patients failed to achieve the higher hemoglobin target despite the use of very high doses of ESA. Reanalysis of data from the CHOIR and CREATE trials showed that the patients who had worse outcomes were more likely to have required very high doses without achieving their target hemoglobin.58,59 Indeed, patients who achieved the higher target hemoglobin levels, usually at lower ESA doses, had better outcomes. This suggested that the need for a higher dose was associated with poorer outcomes, either as a marker of comorbidity or due to yet undocumented side effects of such high doses.
General approach to therapy
Before attributing anemia to chronic kidney disease, a thorough evaluation should be conducted to look for any reversible process that could be contributing to the anemia.
The causes of anemia are numerous and beyond the scope of this review. However, among the common causes of anemia in chronic kidney disease are deficiencies of iron, vitamin B12, and folate. Therefore, guidelines recommend checking iron, vitamin B12, and folate levels in the initial evaluation of anemia.60
Iron deficiency in particular is very common in chronic kidney disease patients and is present in nearly all dialysis patients.61 Hemodialysis patients are estimated to lose 1 to 3 g of iron per year as a result of blood loss in the dialysis circuit and increased iron utilization secondary to ESA therapy.62
However, in contrast to the general population, in which the upper limits of normal for iron indices are well defined, high serum ferritin levels appear to be poorly predictive of hemoglobin responsiveness in dialysis patients.63,64 Thus, the cutoffs that define iron responsiveness are much higher than standard definitions for iron deficiency.65,66 The Dialysis Patients’ Response to IV Iron With Elevated Ferritin (DRIVE) study showed that dialysis patients benefit from intravenous iron therapy even if their ferritin is as high as 1,200 ng/mL, provided their transferrin saturation is below 30%.67
Of note, erythropoietin levels cannot be used to distinguish renal anemia from other causes of anemia. Indeed, patients with renal failure may have “relative erythropoietin deficiency,” ie, “normal” erythropoietin levels that are actually too low in view of the degree of anemia.68,69 In addition to the decreased production capacity by the kidney, there appears to be a component of resistance to the action of erythropoietin in the bone marrow.
For these reasons, there is no erythropoietin level that can be considered “inadequate” or defining of renal anemia. Thus, measuring erythropoietin levels is not routinely recommended in the evaluation of renal anemia.
Two ESA preparations
The two ESAs that have traditionally been used in the treatment of renal anemia are recombinant human erythropoietin and darbepoietin alfa. They appear to be equivalent in terms of safety and efficacy.70 However, darbepoietin alfa has more sialic acid molecules, giving it a higher potency and longer half-life and allowing for less-frequent injections.71,72
In nondialysis patients, recombinant human erythropoietin is typically given every 1 to 2 weeks, whereas darbepoietin alfa can be given every 2 to 4 weeks. In dialysis patients, recombinant human erythropoietin is typically given 3 times per week with every dialysis treatment, while darbepoietin alfa is given once a week.
Target hemoglobin levels: ≤ 11.5 g/dL
In light of the four trials described in Table 1, the international Kidney Disease: Improving Global Outcomes (KDIGO) guidelines60 recommend the following (Table 2):
For patients with chronic kidney disease who are not on dialysis, ESA therapy should not be initiated if the hemoglobin level is higher than 10 g/dL. If the hemoglobin level is lower than 10 g/dL, ESA therapy can be initiated, but the decision needs to be individualized based on the rate of fall of hemoglobin concentration, prior response to iron therapy, the risk of needing a transfusion, the risks related to ESA therapy, and the presence of symptoms attributable to anemia.
For patients on dialysis, ESA therapy should be used when the hemoglobin level is between 9 and 10 g/dL to avoid having the hemoglobin fall below 9 g/dL.
In all adult patients, ESAs should not be used to intentionally increase the hemoglobin level above 13 g/dL but rather to maintain levels no higher than 11.5 g/dL. This target is based on the observation that adverse outcomes were associated with ESA use with hemoglobin targets higher than 13 g/dL (Table 1).
Target iron levels
Regarding iron stores, the guidelines recommend the following:
For adult patients with chronic kidney disease who are not on dialysis, iron should be given to keep transferrin saturation above 20% and ferritin above 100 ng/mL. Transferrin saturation should not exceed 30%, and ferritin levels should not exceed 500 ng/mL.
For adult patients on dialysis, iron should be given to maintain transferrin saturation above 30% and ferritin above 200 ng/mL.
The upper limits of ferritin and transferrin saturation are somewhat controversial, as the safety of intentionally maintaining respective levels greater than 30% and 500 ng/mL has been studied in very few patients. Transferrin saturation should in general not exceed 50%.
High ferritin levels are associated with higher death rates, but whether elevation of ferritin levels is a marker of excessive iron administration rather than a nonspecific acute-phase reactant is not clear. The 2006 guidelines60 cited upper ferritin limits of 500 to 800 ng/mL. However, the more recent DRIVE trial67 showed that patients with ferritin levels of 500 to 1,200 ng/mL will respond to intravenous administration of iron with an increase in their hemoglobin levels. This has led many clinicians to adopt a higher ferritin limit of 1,200 ng/mL.
Hemosiderosis, or excess iron deposition, was a known consequence of frequent transfusions in patients with end-stage renal disease before ESA therapy was available. However, there have been no documented cases of clinical iron overload from iron therapy using current guidelines.73
These algorithms are nuanced, and the benefit of giving intravenous iron should always be weighed against the risks of short-term acute toxicity and infection. Treatment of renal anemia not only requires in-depth knowledge of the topic, but also familiarity with the patient’s specific situation. As such, it is not recommended that clinicians unfamiliar with the treatment of renal anemia manage its treatment.
PARTICULAR CIRCUMSTANCES
Inflammation and ESA resistance
While ESAs are effective in treating anemia in many cases, in many patients the anemia fails to respond. This is of particular importance, since ESA hyporesponsiveness has been found to be a powerful predictor of cardiovascular events and death.74 It is unclear, however, whether high doses of ESA are inherently toxic or whether hyporesponsiveness is a marker of adverse outcomes related to comorbidities.
KDIGO defines initial hyporesponsiveness as having no increase in hemoglobin concentration after the first month of appropriate weight-based dosing, and acquired hyporesponsiveness as requiring two increases in ESA doses up to 50% beyond the dose at which the patient had originally been stable.60 Identifying ESA hyporesponsiveness should lead to an intensive search for potentially correctable factors.
The two major factors accounting for the state of hyporesponsiveness are inflammation and iron deficiency.75,76
Inflammation. High C-reactive protein levels have been shown to predict resistance to erythropoietin in dialysis patients.77 The release of cytokines such as tumor necrosis factor alpha, interleukin 1, and interferon gamma has an inhibitory effect on erythropoiesis.78 Additionally, inflammation can alter the response to ESAs by disrupting the metabolism of iron79 through the release of hepcidin, as previously discussed.38 These reasons likely account for the observed lower response to ESAs in the setting of acute illness and explain why ESAs are not recommended for correcting acute anemia.80
Iron deficiency also can blunt the response to ESAs. Large amounts of iron are needed for effective erythropoietic bursts. As such, iron supplementation is now a recognized treatment of renal anemia.81
Other factors associated with hyporesponsiveness include chronic occult blood loss, aluminum toxicity, cobalamin or folate deficiencies, testosterone deficiency, inadequate dialysis, hyperparathyroidism, and superimposed primary bone marrow disease,82,83 and these should be addressed in patients whose anemia does not respond as expected to ESA therapy. A summary of the main causes of ESA hyporesponsiveness, their reversibility, and recommended treatments is presented in Table 3.
Antibody-mediated pure red-cell aplasia. Rarely, patients receiving ESA therapy develop antibodies that neutralize both the ESA and endogenous erythropoietin. The resulting syndrome, called antibody-mediated pure red-cell aplasia, is characterized by the sudden development of severe transfusion-dependent anemia. This has historically been connected to epoetin beta, a formulation not in use in the United States. However, cases have been documented with epoetin alfa and darbepoetin. The incidence rate is low with subcutaneous ESA use, estimated at 0.5 cases per 10,000 patient-years84 and anecdotal with intravenous ESA.85 The definitive diagnosis requires demonstration of neutralizing antibodies against erythropoietin. Parvovirus infection should be excluded as an alternative cause of pure redcell aplasia.
ANEMIA IN CANCER PATIENTS
ESAs are effective in raising hemoglobin levels and reducing transfusion requirements in patients with chemotherapy-induced anemia.86 However, there are data linking the use of ESAs to shortened survival in patients who have a variety of solid tumors.87
Several mechanisms have been proposed to explain this rapid disease progression, most notably acceleration in tumor growth88–90 by stimulation of erythropoietin receptors on the surface of the tumor cells, leading to increased tumor angiogenesis.91,92
For these reasons, treatment of renal anemia in the setting of active malignancy should be referred to an oncologist.
NOVEL TREATMENTS
Several new agents for treating renal anemia are currently under review.
Continuous erythropoiesis receptor activator
Continuous erythropoiesis receptor activator is a pegylated form of recombinant human erythropoietin that has the ability to repeatedly activate the erythropoietin receptor. It appears to be similar to the other forms of erythropoietin in terms of safety and efficacy in both end-stage renal disease93 and chronic kidney disease.94 It has the advantage of an extended serum half-life, which allows for longer dosing intervals, ie, every 2 weeks. Its use is currently gaining popularity in the dialysis community.
HIF stabilizers
Our growing understanding of the physiology of erythropoietin offers new potential treatment targets. As previously described, production of erythropoietin is stimulated by HIFs. In order to be degraded, these HIFs are hydroxylated at their proline residues by a prolyl hydroxylase. A new category of drugs called prolyl-hydroxylase inhibitors (PDIs) offers the advantage of stabilizing the HIFs, leading to an increase in erythropoietin production.
In phase 1 and 2 clinical trials, these agents have been shown to increase hemoglobin in both end-stage renal disease and chronic kidney disease patients15,16 but not in anephric patients, demonstrating a renal source of the erythropoietin production even in nonfunctioning kidneys. The study of one PDI agent (FG 2216) was halted temporarily after a report of death from fulminant hepatitis, but the other (FG 4592) continues to be studied in a phase 2 clinical trial.95,96
TAKE-HOME POINTS
- Anemia of renal disease is a common condition that is mainly caused by a decrease in erythropoietin production by the kidneys.
- While anemia of renal disease can be corrected with ESAs, it is necessary to investigate and rule out underlying treatable conditions such as iron or vitamin deficiencies before giving an ESA.
- Anemia of renal disease is associated with significant morbidity such as increased risk of left ventricular hypertrophy, myocardial infarction, and heart failure, and has been described as an all-cause mortality multiplier.
- Unfortunately, the only undisputed benefit of treatment to date remains the avoidance of blood transfusions. Furthermore, the large randomized controlled trials that looked at the benefits of ESA have shown that their use can be associated with increased risk of cardiovascular events. Therefore, use of an ESA in end-stage renal disease should never target a normal hemoglobin levels but rather aim for a hemoglobin level of no more than 11.5 g/dL.
- Use of an ESA in chronic kidney disease should be individualized and is not recommended to be started unless the hemoglobin level is less than 10 g/dL.
- Several newer agents for renal anemia are currently under review. A pegylated form of recombinant human erythropoietin has an extended half-life, and a new and promising category of drugs called HIF stabilizers is currently under study.
Anemia is a frequent complication of chronic kidney disease, occurring in over 90% of patients receiving renal replacement therapy. It is associated with significant morbidity and mortality. While its pathogenesis is typically multifactorial, the predominant cause is failure of the kidneys to produce enough endogenous erythropoietin. The clinical approval of recombinant human erythropoietin in 1989 dramatically changed the treatment of anemia of chronic kidney disease, but randomized controlled trials yielded disappointing results when erythropoiesis-stimulating agents (ESAs) were used to raise hemoglobin to normal levels.
This article reviews the epidemiology and pathophysiology of anemia of chronic kidney disease and discusses the complicated and conflicting evidence regarding its treatment.
DEFINITION AND PREVALENCE
Anemia is defined as a hemoglobin concentration less than 13.0 g/dL for men and less than 12.0 g/dL for premenopausal women.1 It is more common in patients with impaired kidney function, especially when the glomerular filtration rate (GFR) falls below 60 mL/min. It is rare at GFRs higher than 80 mL/min,2 but as the GFR falls, the severity of the anemia worsens3 and its prevalence increases: almost 90% of patients with a GFR less than 30 mL/min are anemic.4
RENAL ANEMIA IS ASSOCIATED WITH BAD OUTCOMES
Anemia in chronic kidney disease is independently associated with risk of death. It is also an all-cause mortality multiplier, ie, it magnifies the risk of death from other disease states.5
In observational studies, anemia was associated with faster progression of left ventricular hypertrophy, inflammation, and increased myocardial and peripheral oxygen demand, thereby leading to worse cardiac outcomes with increased risk of myocardial infarction, coronary revascularization, and readmission for heart failure.6–8 Anemia is also associated with fatigue, depression, reduced exercise tolerance, stroke, and increased risk of rehospitalization.9–13
RENAL ANEMIA IS MULTIFACTORIAL
Anemia of chronic kidney disease is typically attributed to the decrease of erythropoietin production that accompanies the fall in GFR. However, the process is multifactorial, with several other contributing factors: absolute and functional iron deficiency, folate and vitamin B12 deficiencies, reduced red blood cell life span, and suppression of erythropoiesis by the uremic milieu.14
While it was once thought that chronic kidney disease leads to loss of erythropoietin-producing cells, it is now known that downregulation of hypoxia-inducible factor (HIF; a transcription factor) is at least partially responsible for the decrease in erythropoietin production15,16 and that this downregulation is reversible (see below).
ERYTHROPOIETIN, IRON, AND RED BLOOD CELLS
Erythropoietin production is triggered by hypoxia, mediated by HIF
Erythropoietin is produced primarily in the deep cortex and outer medulla of the kidneys by a special population of peritubular interstitial cells.17 The parenchymal cells of the liver also produce erythropoietin, but much less.18
The rate of renal erythropoietin synthesis is determined by tissue oxygenation rather than by renal blood flow; production increases as the hemoglobin concentration drops and the arterial oxygen tension decreases (Figure 1).19
The gene for erythropoietin is located on chromosome 7 and is regulated by HIF. HIF molecules are composed of an alpha subunit, which is unstable at high Po2, and a beta subunit, constitutively present in the nucleus.20
In hypoxic conditions, the HIF dimer is transcriptionally active and binds to specific DNA recognition sequences called hypoxia-response elements. Gene transcription is upregulated, leading to increased production of erythropoietin.21
Under normal oxygen tension, on the other hand, the proline residue of the HIF alpha subunit is hydroxylated. The hydroxylated HIF alpha subunit is then degraded by proteasomal ubiquitylation, which is mediated by the von Hippel-Lindau tumor-suppressor gene pVHL.22 Degradation of HIF alpha prevents formation of the HIF heterodimers. HIF therefore cannot bind to the hypoxia-response elements, and erythropoietin gene transcription does not occur.23
Thus, in states of hypoxia, erythropoietin production is upregulated, whereas with normal oxygen tension, production is downregulated.
Erythropoietin is essential for terminal maturation of erythrocytes
Erythropoietin is essential for terminal maturation of erythrocytes.24 It is thought to stimulate the growth of erythrogenic progenitors: burst-forming units-erythroid (BFU-E) and colony-forming units-erythroid (CFU-E). In the absence of erythropoietin, BFU-E and CFU-E fail to differentiate into mature erythrocytes.25
Binding of erythropoietin to its receptor sets off a series of downstream signals, the most important being the signal transducer and activator of transcription 5 (STAT5). In animal studies, STAT5 was found to inhibit apoptosis through the early induction of an antiapoptotic gene, Bcl-xL.26
Iron metabolism is controlled by several proteins
Iron is characterized by its capacity to accept or donate electrons. This unique property makes it a crucial element in many biochemical reactions such as enzymatic activity, DNA synthesis, oxygen transport, and cell respiration.
Iron metabolism is under the control of several proteins that play different roles in its absorption, recycling, and loss (Figure 2).27
Dietary iron exists primarily in its poorly soluble trivalent ferric form (Fe3+), and it needs to be reduced to its soluble divalent ferrous form (Fe2+) by ferric reductase to be absorbed. Ferrous iron is taken up at the apical side of enterocytes by a divalent metal transporter (DMT1) and is transported across the brush border.28
To enter the circulation, iron has to be transported across the basolateral membrane by a transporter called ferroportin.29 Ferroportin is also found in placental syncitiotrophoblasts, where it transfers iron from mother to fetus, and in macrophages, where it allows recycling of iron scavenged from damaged cells back into the circulation.30 Upon its release, the ferrous iron is oxidized to the ferric form and loaded onto transferrin. This oxidation process involves hephaestin, a homologue of the ferroxidase ceruloplasmin.31
In the plasma, iron is bound to transferrin, and under normal circumstances one-third of transferrin is saturated with iron.32 Transferrin receptors are present on most cells but are most dense on erythroid precursors. Each transferrin receptor can bind two transferrin molecules. After binding to transferrin, the transferrin receptor is endocytosed, and the iron is released into acidified vacuoles. The transferrin-receptor complex is then recycled to the surface.33
Ferritin is the cellular storage protein for iron, and it can store up to 4,500 atoms of iron within its spherical cavity.34 The serum level of ferritin reflects overall storage, with 1 ng/mL of ferritin indicating 10 mg of total iron stores.35 Ferritin is also an acute-phase reactant, and plasma levels can increase in inflammatory states such as infection or malignancy. As such, elevated ferritin does not necessarily indicate elevated iron stores.
Iron is lost in sweat, shed skin cells, and sloughed intestinal mucosal cells. However, there is no specific mechanism of iron excretion from the human body. Thus, iron is mainly regulated at the level of intestinal absorption. The iron exporter ferroportin is upregulated by the amount of available iron and is degraded by hepcidin.36
Hepcidin is a small cysteine-rich cationic peptide that is primarily produced in the liver, with some minor production also occurring in the kidneys.37 Transcription of the gene encoding hepcidin is downregulated by anemia and hypoxia and upregulated by inflammation and elevated iron levels.38 Transcription of hepcidin leads to degradation of ferroportin and a decrease in intestinal iron absorption. On the other hand, anemia and hypoxia inhibit hepcidin transcription, which allows ferroportin to facilitate intestinal iron absorption.
TREATMENT OF RENAL ANEMIA
Early enthusiasm for erythropoietin agents
Androgens started to be used to treat anemia of end-stage renal disease in 1970,39,40 and before the advent of recombinant human erythropoietin, they were a mainstay of nontransfusional therapy for anemic patients on dialysis.
The approval of recombinant human erythropoietin in 1989 drastically shifted the treatment of renal anemia. While the initial goal of treating anemia of chronic kidney disease with erythropoietin was to prevent blood transfusions,41 subsequent studies showed that the benefits might be far greater. Indeed, an initial observational trial showed that erythropoiesis-stimulating agents (ESAs) were associated with improved quality of life,42 improved neurocognitive function,43,44 and even cost savings.45 The benefits also extended to major outcomes such as regression of left ventricular hypertrophy,46 improvement in New York Heart Association class and cardiac function,47 fewer hospitalizations,48 and even reduction of cardiovascular mortality rates.49
As a result, ESA use gained popularity, and by 2006 an estimated 90% of dialysis patients were receiving these agents.50 The target and achieved hemoglobin levels also increased, with mean hemoglobin levels in hemodialysis patients being raised from 9.7 to 12 g/dL.51
Disappointing results in clinical trials of ESAs to normalize hemoglobin
To prospectively study the effects of normalized hemoglobin targets, four randomized controlled trials were conducted (Table 1):
- The Normal Hematocrit Study (NHCT)52
- The Correction of Hemoglobin and Outcomes in Renal Insufficiency (CHOIR) trial53
- The Cardiovascular Risk Reduction by Early Anemia Treatment (CREATE) trial54
- The Trial to Reduce Cardiovascular Events With Aranesp Therapy (TREAT).55
These trials randomized patients to either higher “normal-range” hemoglobin targets or to lower target hemoglobin levels.
Their findings were disappointing and raised several red flags about excessive use of ESAs. The trials found no benefit in higher hemoglobin targets, and in fact, some of them demonstrated harm in patients randomized to higher targets. Notably, higher hemoglobin targets were associated with significant side effects such as access-site thrombosis,52 strokes,55 and possibly cardiovascular events.54,55 Only the CREATE trial was able to demonstrate a quality-of-life benefit for the high-target group.54
It remains unclear whether these adverse events were from the therapy itself or from an increased morbidity burden in the treated patients. Erythropoietin use is associated with hypertension,56 thought to be related to endothelin-mediated vasoconstriction.57 In our experience, this is most evident when hemoglobin levels are normalized with ESA therapy. Cycling of erythropoietin levels between extreme levels can lead to vascular remodeling, which may also be related to its cardiovascular effects.57
A noticeable finding in several of these trials was that patients failed to achieve the higher hemoglobin target despite the use of very high doses of ESA. Reanalysis of data from the CHOIR and CREATE trials showed that the patients who had worse outcomes were more likely to have required very high doses without achieving their target hemoglobin.58,59 Indeed, patients who achieved the higher target hemoglobin levels, usually at lower ESA doses, had better outcomes. This suggested that the need for a higher dose was associated with poorer outcomes, either as a marker of comorbidity or due to yet undocumented side effects of such high doses.
General approach to therapy
Before attributing anemia to chronic kidney disease, a thorough evaluation should be conducted to look for any reversible process that could be contributing to the anemia.
The causes of anemia are numerous and beyond the scope of this review. However, among the common causes of anemia in chronic kidney disease are deficiencies of iron, vitamin B12, and folate. Therefore, guidelines recommend checking iron, vitamin B12, and folate levels in the initial evaluation of anemia.60
Iron deficiency in particular is very common in chronic kidney disease patients and is present in nearly all dialysis patients.61 Hemodialysis patients are estimated to lose 1 to 3 g of iron per year as a result of blood loss in the dialysis circuit and increased iron utilization secondary to ESA therapy.62
However, in contrast to the general population, in which the upper limits of normal for iron indices are well defined, high serum ferritin levels appear to be poorly predictive of hemoglobin responsiveness in dialysis patients.63,64 Thus, the cutoffs that define iron responsiveness are much higher than standard definitions for iron deficiency.65,66 The Dialysis Patients’ Response to IV Iron With Elevated Ferritin (DRIVE) study showed that dialysis patients benefit from intravenous iron therapy even if their ferritin is as high as 1,200 ng/mL, provided their transferrin saturation is below 30%.67
Of note, erythropoietin levels cannot be used to distinguish renal anemia from other causes of anemia. Indeed, patients with renal failure may have “relative erythropoietin deficiency,” ie, “normal” erythropoietin levels that are actually too low in view of the degree of anemia.68,69 In addition to the decreased production capacity by the kidney, there appears to be a component of resistance to the action of erythropoietin in the bone marrow.
For these reasons, there is no erythropoietin level that can be considered “inadequate” or defining of renal anemia. Thus, measuring erythropoietin levels is not routinely recommended in the evaluation of renal anemia.
Two ESA preparations
The two ESAs that have traditionally been used in the treatment of renal anemia are recombinant human erythropoietin and darbepoietin alfa. They appear to be equivalent in terms of safety and efficacy.70 However, darbepoietin alfa has more sialic acid molecules, giving it a higher potency and longer half-life and allowing for less-frequent injections.71,72
In nondialysis patients, recombinant human erythropoietin is typically given every 1 to 2 weeks, whereas darbepoietin alfa can be given every 2 to 4 weeks. In dialysis patients, recombinant human erythropoietin is typically given 3 times per week with every dialysis treatment, while darbepoietin alfa is given once a week.
Target hemoglobin levels: ≤ 11.5 g/dL
In light of the four trials described in Table 1, the international Kidney Disease: Improving Global Outcomes (KDIGO) guidelines60 recommend the following (Table 2):
For patients with chronic kidney disease who are not on dialysis, ESA therapy should not be initiated if the hemoglobin level is higher than 10 g/dL. If the hemoglobin level is lower than 10 g/dL, ESA therapy can be initiated, but the decision needs to be individualized based on the rate of fall of hemoglobin concentration, prior response to iron therapy, the risk of needing a transfusion, the risks related to ESA therapy, and the presence of symptoms attributable to anemia.
For patients on dialysis, ESA therapy should be used when the hemoglobin level is between 9 and 10 g/dL to avoid having the hemoglobin fall below 9 g/dL.
In all adult patients, ESAs should not be used to intentionally increase the hemoglobin level above 13 g/dL but rather to maintain levels no higher than 11.5 g/dL. This target is based on the observation that adverse outcomes were associated with ESA use with hemoglobin targets higher than 13 g/dL (Table 1).
Target iron levels
Regarding iron stores, the guidelines recommend the following:
For adult patients with chronic kidney disease who are not on dialysis, iron should be given to keep transferrin saturation above 20% and ferritin above 100 ng/mL. Transferrin saturation should not exceed 30%, and ferritin levels should not exceed 500 ng/mL.
For adult patients on dialysis, iron should be given to maintain transferrin saturation above 30% and ferritin above 200 ng/mL.
The upper limits of ferritin and transferrin saturation are somewhat controversial, as the safety of intentionally maintaining respective levels greater than 30% and 500 ng/mL has been studied in very few patients. Transferrin saturation should in general not exceed 50%.
High ferritin levels are associated with higher death rates, but whether elevation of ferritin levels is a marker of excessive iron administration rather than a nonspecific acute-phase reactant is not clear. The 2006 guidelines60 cited upper ferritin limits of 500 to 800 ng/mL. However, the more recent DRIVE trial67 showed that patients with ferritin levels of 500 to 1,200 ng/mL will respond to intravenous administration of iron with an increase in their hemoglobin levels. This has led many clinicians to adopt a higher ferritin limit of 1,200 ng/mL.
Hemosiderosis, or excess iron deposition, was a known consequence of frequent transfusions in patients with end-stage renal disease before ESA therapy was available. However, there have been no documented cases of clinical iron overload from iron therapy using current guidelines.73
These algorithms are nuanced, and the benefit of giving intravenous iron should always be weighed against the risks of short-term acute toxicity and infection. Treatment of renal anemia not only requires in-depth knowledge of the topic, but also familiarity with the patient’s specific situation. As such, it is not recommended that clinicians unfamiliar with the treatment of renal anemia manage its treatment.
PARTICULAR CIRCUMSTANCES
Inflammation and ESA resistance
While ESAs are effective in treating anemia in many cases, in many patients the anemia fails to respond. This is of particular importance, since ESA hyporesponsiveness has been found to be a powerful predictor of cardiovascular events and death.74 It is unclear, however, whether high doses of ESA are inherently toxic or whether hyporesponsiveness is a marker of adverse outcomes related to comorbidities.
KDIGO defines initial hyporesponsiveness as having no increase in hemoglobin concentration after the first month of appropriate weight-based dosing, and acquired hyporesponsiveness as requiring two increases in ESA doses up to 50% beyond the dose at which the patient had originally been stable.60 Identifying ESA hyporesponsiveness should lead to an intensive search for potentially correctable factors.
The two major factors accounting for the state of hyporesponsiveness are inflammation and iron deficiency.75,76
Inflammation. High C-reactive protein levels have been shown to predict resistance to erythropoietin in dialysis patients.77 The release of cytokines such as tumor necrosis factor alpha, interleukin 1, and interferon gamma has an inhibitory effect on erythropoiesis.78 Additionally, inflammation can alter the response to ESAs by disrupting the metabolism of iron79 through the release of hepcidin, as previously discussed.38 These reasons likely account for the observed lower response to ESAs in the setting of acute illness and explain why ESAs are not recommended for correcting acute anemia.80
Iron deficiency also can blunt the response to ESAs. Large amounts of iron are needed for effective erythropoietic bursts. As such, iron supplementation is now a recognized treatment of renal anemia.81
Other factors associated with hyporesponsiveness include chronic occult blood loss, aluminum toxicity, cobalamin or folate deficiencies, testosterone deficiency, inadequate dialysis, hyperparathyroidism, and superimposed primary bone marrow disease,82,83 and these should be addressed in patients whose anemia does not respond as expected to ESA therapy. A summary of the main causes of ESA hyporesponsiveness, their reversibility, and recommended treatments is presented in Table 3.
Antibody-mediated pure red-cell aplasia. Rarely, patients receiving ESA therapy develop antibodies that neutralize both the ESA and endogenous erythropoietin. The resulting syndrome, called antibody-mediated pure red-cell aplasia, is characterized by the sudden development of severe transfusion-dependent anemia. This has historically been connected to epoetin beta, a formulation not in use in the United States. However, cases have been documented with epoetin alfa and darbepoetin. The incidence rate is low with subcutaneous ESA use, estimated at 0.5 cases per 10,000 patient-years84 and anecdotal with intravenous ESA.85 The definitive diagnosis requires demonstration of neutralizing antibodies against erythropoietin. Parvovirus infection should be excluded as an alternative cause of pure redcell aplasia.
ANEMIA IN CANCER PATIENTS
ESAs are effective in raising hemoglobin levels and reducing transfusion requirements in patients with chemotherapy-induced anemia.86 However, there are data linking the use of ESAs to shortened survival in patients who have a variety of solid tumors.87
Several mechanisms have been proposed to explain this rapid disease progression, most notably acceleration in tumor growth88–90 by stimulation of erythropoietin receptors on the surface of the tumor cells, leading to increased tumor angiogenesis.91,92
For these reasons, treatment of renal anemia in the setting of active malignancy should be referred to an oncologist.
NOVEL TREATMENTS
Several new agents for treating renal anemia are currently under review.
Continuous erythropoiesis receptor activator
Continuous erythropoiesis receptor activator is a pegylated form of recombinant human erythropoietin that has the ability to repeatedly activate the erythropoietin receptor. It appears to be similar to the other forms of erythropoietin in terms of safety and efficacy in both end-stage renal disease93 and chronic kidney disease.94 It has the advantage of an extended serum half-life, which allows for longer dosing intervals, ie, every 2 weeks. Its use is currently gaining popularity in the dialysis community.
HIF stabilizers
Our growing understanding of the physiology of erythropoietin offers new potential treatment targets. As previously described, production of erythropoietin is stimulated by HIFs. In order to be degraded, these HIFs are hydroxylated at their proline residues by a prolyl hydroxylase. A new category of drugs called prolyl-hydroxylase inhibitors (PDIs) offers the advantage of stabilizing the HIFs, leading to an increase in erythropoietin production.
In phase 1 and 2 clinical trials, these agents have been shown to increase hemoglobin in both end-stage renal disease and chronic kidney disease patients15,16 but not in anephric patients, demonstrating a renal source of the erythropoietin production even in nonfunctioning kidneys. The study of one PDI agent (FG 2216) was halted temporarily after a report of death from fulminant hepatitis, but the other (FG 4592) continues to be studied in a phase 2 clinical trial.95,96
TAKE-HOME POINTS
- Anemia of renal disease is a common condition that is mainly caused by a decrease in erythropoietin production by the kidneys.
- While anemia of renal disease can be corrected with ESAs, it is necessary to investigate and rule out underlying treatable conditions such as iron or vitamin deficiencies before giving an ESA.
- Anemia of renal disease is associated with significant morbidity such as increased risk of left ventricular hypertrophy, myocardial infarction, and heart failure, and has been described as an all-cause mortality multiplier.
- Unfortunately, the only undisputed benefit of treatment to date remains the avoidance of blood transfusions. Furthermore, the large randomized controlled trials that looked at the benefits of ESA have shown that their use can be associated with increased risk of cardiovascular events. Therefore, use of an ESA in end-stage renal disease should never target a normal hemoglobin levels but rather aim for a hemoglobin level of no more than 11.5 g/dL.
- Use of an ESA in chronic kidney disease should be individualized and is not recommended to be started unless the hemoglobin level is less than 10 g/dL.
- Several newer agents for renal anemia are currently under review. A pegylated form of recombinant human erythropoietin has an extended half-life, and a new and promising category of drugs called HIF stabilizers is currently under study.
- World Health Organization (WHO). Nutritional anaemias: report of a WHO scientific group. Geneva, Switzerland: World Health Organization, 1968.
- Hsu CY, McCulloch CE, Curhan GC, et al. Epidemiology of anemia associated with chronic renal insufficiency among adults in the United States: results from the Third National Health and Nutrition Examination Survey. J Am Soc Nephrol 2002; 13:504–510.
- Radtke HW, Claussner A, Erbes PM, Scheuermann EH, Schoeppe W, Koch KM. Serum erythropoetin concentration in chronic renal failure: relationship to degree of anemia and excretory renal function. Blood 1979; 54:877–884.
- Kazmi WH, Kausz AT, Khan S, et al. Anemia: an early complication of chronic renal insufficiency. Am J Kidney Dis 2001; 38:803–812.
- United States Renal Data System. Chapter 3. Morbidity & mortality in patients with CKD. www.usrds.org/2012/view/v1_03.aspx. Accessed June 9, 2016.
- Horwich TB, Fonarow GC, Hamilton MA, MacLellan WR, Borenstein J. Anemia is associated with worse symptoms, greater impairment in functional capacity and a significant increase in mortality in patients with advanced heart failure. J Am Coll Cardiol 2002; 39:1780–1786.
- Mark DB, Felker GM. B-type natriuretic peptide: a biomarker for all seasons? N Engl J Med 2004; 350:718–720.
- Walker AM, Schneider G, Yeaw J, Nordstrom B, Robbins S, Pettitt D. Anemia as a predictor of cardiovascular events in patients with elevated serum creatinine. J Am Soc Nephrol 2006; 17:2293–2298.
- Abramson JL, Jurkovitz CT, Vaccarino V, Weintraub WS, McClellan W. Chronic kidney disease, anemia, and incident stroke in a middle-aged, community-based population: the ARIC Study. Kidney Int 2003; 64:610–615.
- Sarnak MJ, Tighiouart H, Manjunath G, et al. Anemia as a risk factor for cardiovascular disease in the Atherosclerosis Risk in Communities (ARIC) study. J Am Coll Cardiol 2002; 40:27–33.
- McClellan WM, Flanders WD, Langston RD, Jurkovitz C, Presley R. Anemia and renal insufficiency are independent risk factors for death among patients with congestive heart failure admitted to community hospitals: a population-based study. J Am Soc Nephrol 2002; 13:1928–1936.
- Xia H, Ebben J, Ma JZ, Collins AJ. Hematocrit levels and hospitalization risks in hemodialysis patients. J Am Soc Nephrol 1999; 10:1309–1316.
- Collins AJ, Li S, St Peter W, et al. Death, hospitalization, and economic associations among incident hemodialysis patients with hematocrit values of 36 to 39%. J Am Soc Nephrol 2001; 12:2465–2473.
- Agarwal AK. Practical approach to the diagnosis and treatment of anemia associated with CKD in elderly. J Am Med Dir Assoc 2006; 7(suppl 9):S7–S12.
- Bernhardt WM, Wiesener MS, Scigalla P, et al. Inhibition of prolyl hydroxylases increases erythropoietin production in ESRD. J Am Soc Nephrol 2010; 21:2151–2156.
- Provenzano R, Fadda G, Bernardo M, et al. FG-2216, a novel oral HIF-PHI, stimulates erythropoiesis and increases hemoglobin concentration in patients with non-dialysis CKD. Am J Kidney Dis 2008; 51:B80.
- Maxwell PH, Osmond MK, Pugh CW, et al. Identification of the renal erythropoietin-producing cells using transgenic mice. Kidney Int 1993; 44:1149–1162.
- Maxwell PH, Ferguson DJ, Nicholls LG, et al. Sites of erythropoietin production. Kidney Int 1997; 51:393–401.
- Jelkmann W. Erythropoeitin: structure, control of production and function. Physiol Rev 1992; 72:449–489.
- Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA 1995; 92:5510–5514.
- Wang GL, Semenza GL. Purification and characterization of hypoxia-inducible factor 1. J Biol Chem 1995; 270:1230–1237.
- Maxwell PH, Wiesener MS, Chang GW, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999; 399:271–275.
- Salceda S, Caro J. Hypoxia-inducible factor 1alpha protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J Biol Chem 1997; 272:22642–22647.
- Malik J, Kim AR, Tyre KA, Cherukuri AR, Palis J. Erythropoietin critically regulates the terminal maturation of murine and human primitive erythroblasts. Haematologica 2013; 98:1778–1787.
- Wu H, Liu X, Jaenisch R, Lodish HF. Generation of committed erythroid BFU-E and CFU-E progenitors does not require erythropoietin or the erythropoietin receptor. Cell 1995; 83:59–67.
- Socolovsky M, Nam H, Fleming MD, Haase VH, Brugnara C, Lodish HF. Ineffective erythropoiesis in Stat5a(-/-)5b(-/-) mice due to decreased survival of early erythroblasts. Blood 2001; 98:3261–3273.
- Papanikolaou G, Pantopoulos K. Iron metabolism and toxicity. Toxicol Appl Pharmacol 2005; 202:199–211.
- Conrad ME, Umbreit JN. Pathways of iron absorption. Blood Cells Mol Dis 2002; 29:336–355.
- Frazer DM, Anderson GJ. The orchestration of body iron intake: how and where do enterocytes receive their cues? Blood Cells Moll Dis 2003; 30:288–297.
- Donovan A, Lima CA, Pinkus JL, et al. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab 2005; 1:191–200.
- Vulpe CD, Kuo YM, Murphy TL, et al. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat Genet 1999; 21:195–199.
- Bothwell TH. Overview and mechanisms of iron regulation. Nutr Rev 1995: 53:237–245.
- Kawabata H, Nakamaki T, Ikonomi P, Smith RD, Germain RS, Koeffler HP. Expression of transferrin receptor 2 in normal and neoplastic hematopoietic cells. Blood 2001; 98:2714–2719.
- Arosio P, Levi S. Cytosolic and mitochondrial ferritins in the regulation of cellular iron homeostasis and oxidative damage. Biochim Biophys Acta 2010; 1800:783–792.
- Finch CA, Bellotti V, Stray S, et al. Plasma ferritin determination as a diagnostic tool. West J Med 1986; 145:657–663.
- Delaby C, Pilard N, Goncalves AS, Beaumont C, Canonne-Hergaux F. Presence of the iron exporter ferroportin at the plasma membrane of macrophages is enhanced by iron loading and down-regulated by hepcidin. Blood 2005; 106:3979–3984.
- Ganz T. Hepcidin, a key regulator of iron metabolism and mediator of anemia of inflammation. Blood 2003; 102:783–788.
- Nicolas G, Chauvet C, Viatte L, et al. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J Clin Invest 2002; 110:1037–1044.
- DeGowin RL, Lavender AR, Forland M, Charleston D, Gottschalk A. Erythropoiesis and erythropoietin in patients with chronic renal failure treated with hemodialysis and testosterone. Ann Intern Med 1970; 72:913–918.
- Richardson JR Jr, Weinstein MB. Erythropoietic response of dialyzed patients to testosterone administration. Ann Intern Med 1970; 73:403–407
- Eschbach JW, Abdulhadi MH, Browne JK, et al. Recombinant human erythropoietin in anemic patients with end-stage renal disease. Results of a phase III multicenter clinical trial. Ann Intern Med 1989; 111:992–1000.
- Moreno F, Aracil FJ, Pérez R, Valderrábano F. Controlled study on the improvement of quality of life in elderly hemodialysis patients after correcting end-stage renal disease-related anemia with erythropoietin. Am J Kidney Dis 1996; 27:548–556.
- Nissenson AR, Nimer SD, Wolcott DL. Recombinant human erythropoietin and renal anemia: molecular biology, clinical efficacy, and nervous system effects. Ann Intern Med 1991; 114:402–416.
- Stivelman JC. Benefits of anaemia treatment on cognitive function. Nephrol Dial Transplant 2000; 15(suppl 3):29–35.
- Maddux FW, Shetty S, del Aguila MA, Nelson MA, Murray BM. Effect of erythropoiesis-stimulating agents on healthcare utilization, costs, and outcomes in chronic kidney disease. Ann Pharmacother 2007; 41:1761–1769.
- Macdougall IC, Lewis NP, Saunders MJ, et al. Long-term cardiorespiratory effects of amelioration of renal anaemia by erythropoietin. Lancet 1990; 335:489–493.
- Silverberg DS, Wexler D, Blum M, et al. Effects of treatment with epoetin beta on outcomes in patients with anaemia and chronic heart failure. Kidney Blood Press Res 2005; 28:41–47.
- Perkins R, Olson S, Hansen J, Lee J, Stiles K, Lebrun C. Impact of an anemia clinic on emergency room visits and hospitalizations in patients with anemia of CKD pre-dialysis. Nephrol Nurs J 2007; 34:167–173, 182.
- Locatelli F, Conte F, Marcelli D. The impact of haematocrit levels and erythropoietin treatment on overall and cardiovascular mortality and morbidity—the experience of the Lombardy Dialysis Registry. Nephrol Dial Transplant 1998; 13:1642–1644.
- Centers for Medicare and Medicaid Services; Kinney R. 2005 Annual Report: ESRD Clinical Performance Measures Project. Am J Kidney Dis 2006; 48(suppl 2):S1–S106.
- US Renal Data System. Annual Data Report 2006. www.usrds.org/adr.aspx. Accessed July 3, 2016.
- Besarab A, Bolton WK, Browne JK, et al. The effects of normal as compared with low hematocrit values in patients with cardiac disease who are receiving hemodialysis and epoetin. N Engl J Med 1998; 339:584–590.
- Singh AK, Szczech L, Tang KL, et al; CHOIR Investigators. Correction of anemia with epoetin alfa in chronic kidney disease. N Engl J Med 2006; 355:2085–2098.
- Drüeke TB, Locatelli F, Clyne N, et al; CREATE Investigators. Normalization of hemoglobin level in patients with chronic kidney disease and anemia. N Engl J Med 2006; 355:2071–2084.
- Pfeffer MA, Burdmann EA, Chen CY, et al; TREAT Investigators. A trial of darbepoetin alfa in type 2 diabetes and chronic kidney disease. N Engl J Med 2009; 361:2019–2032.
- Kirkpantur A, Kahraman S, Yilmaz R, et al. The effects of maintenance recombinant human erythropoietin therapy on ambulatory blood pressure recordings: conventional, Doppler, and tissue Doppler echocardiographic parameters. Artif Organs 2005; 29:965–972.
- Fishbane S, Berns JS. Hemoglobin cycling in hemodialysis patients treated with recombinant human erythropoietin. Kidney Int 2005; 68:1337–1343.
- Szczech LA, Barnhart HX, Inrig JK, et al. Secondary analysis of the CHOIR trial epoetin-alpha dose and achieved hemoglobin outcomes. Kidney Int 2008; 74:791–798.
- Solomon SD, Uno H, Lewis EF, et al; Trial to Reduce Cardiovascular Events with Aranesp Therapy (TREAT) Investigators. Erythropoietic response and outcomes in kidney disease and type 2 diabetes. N Engl J Med 2010; 363:1146–1155.
- Kidney Disease: Improving Global Outcomes (KDIGO). KDIGO Clinical Practice Guideline for Anemia in Chronic Kidney Disease. Kidney Int Suppl 2012; 2:279–335.
- Fernández-Rodríguez AM, Guindeo-Casasús MC, Molero-Labarta T, et al. Diagnosis of iron deficiency in chronic renal failure. Am J Kidney Dis 1999; 34:508–513.
- Eschbach JW, Cook JD, Scribner BH, Finch CA. Iron balance in hemodialysis patients. Ann Intern Med 1977; 87:710–713.
- Mittman N, Sreedhara R, Mushnick R, et al. Reticulocyte hemoglobin content predicts functional iron deficiency in hemodialysis patients receiving rHuEPO. Am J Kidney Dis 1997; 30:912–922.
- Tessitore N, Solero GP, Lippi G, et al. The role of iron status markers in predicting response to intravenous iron in haemodialysis patients on maintenance erythropoietin. Nephrol Dial Transplant 2001; 16:1416–1423.
- Coyne DW. Iron indices: what do they really mean? Kidney Int Suppl 2006; 101:S4–S8.
- Fishbane S, Kowalski EA, Imbriano LJ, Maesaka JK. The evaluation of iron status in hemodialysis patients. J Am Soc Nephrol 1996; 7:2654–2657.
- Coyne DW, Kapoian T, Suki W, et al; DRIVE Study Group. Ferric gluconate is highly efficacious in anemic hemodialysis patients with high serum ferritin and low transferrin saturation: results of the Dialysis Patients’ Response to IV Iron with Elevated Ferritin (DRIVE) Study. J Am Soc Nephrol 2007; 18:975–984.
- Radtke HW, Claussner A, Erbes PM, Scheuermann EH, Schoeppe W, Koch KM. Serum erythropoietin concentration in chronic renal failure: relationship to degree of anemia and excretory renal function. Blood 1979; 54:877–884.
- Korte W, Cogliatti SB, Jung K, Riesen W. Mild renal dysfunction is sufficient to induce erythropoietin deficiency in patients with unexplained anaemia. Clin Chim Acta 2000; 292:149–154.
- Locatelli F, Olivares J, Walker R, et al; European/Australian NESP 980202 Study Group. Novel erythropoiesis stimulating protein for treatment of anemia in chronic renal insufficiency. Kidney Int 2001; 60:741–747.
- Carrera F, Burnier M. Use of darbepoetin alfa in the treatment of anaemia of chronic kidney disease: clinical and pharmacoeconomic considerations. NDT Plus 2009; 2(suppl 1):i9–i17.
- Egrie JC, Browne JK. Development and characterization of novel erythropoiesis stimulating protein (NESP). Nephrol Dial Transplant 2001; 16(suppl 3):3–13.
- Nissenson AR, Charytan C. Controversies in iron management. Kidney Int Suppl 2003; 87:S64–S71.
- Kilpatrick RD, Critchlow CW, Fishbane S, et al. Greater epoetin alpha responsiveness is associated with improved survival in hemodialysis patients. Clin J Am Soc Nephrol 2008; 3:1077–1083.
- Locatelli F, Aljama P, Barany P, et al; European Best Practice Guidelines Working Group. Revised European best practice guidelines for the management of anaemia in patients with chronic renal failure. Nephrol Dial Transplant 2004; 19(suppl 2):ii1–ii47.
- Stenvinkel P. The role of inflammation in the anaemia of end-stage renal disease. Nephrol Dial Transplant 2001; 16(suppl 7):36–40.
- Barany P, Divino Filho JC, Bergstrom J. High C-reactive protein is a strong predictor of resistance to erythropoietin in hemodialysis patients. Am J Kidney Dis 1997; 29:565–568.
- Drueke T. Hyporesponsiveness to recombinant human erythropoietin. Nephrol Dial Transplant 2001; 16(suppl 7):25–28.
- Casadevall N. Cellular mechanism of resistance to erythropoietin. Nephrol Dial Transplant 1995; 10(suppl 6):27–30.
- Kraus E, Rabb H. EPO therapy during acute kidney disease: to use or not to use, that is the question. Am J Kidney Dis 2005; 46:967–969.
- Gotloib L, Silverberg D, Fudin R, Shostak A. Iron deficiency is a common cause of anemia in chronic kidney disease and can often be corrected with intravenous iron. J Nephrol 2006; 19:161–167.
- Tarng DC, Huang TP, Chen TW, Yang WC. Erythropoietin hyporesponsiveness: from iron deficiency to iron overload. Kidney Int Suppl 1999; 69:S107–S118.
- Drüeke TB. Modulating factors in the hematopoietic response to erythropoietin. Am J Kidney Dis 1991; 18(suppl 1):87–92.
- Boven K, Stryker S, Knight J, et al. The increased incidence of pure red cell aplasia with an Eprex formulation in uncoated rubber stopper syringes. Kidney Int 2005; 67:2346–2353.
- Shimizu H, Saitoh T, Ota F, et al. Pure red cell aplasia induced only by intravenous administration of recombinant human erythropoietin. Acta Haematol 2011; 126:114–118.
- Tonia T, Mettler A, Robert N, et al. Erythropoietin or darbepoetin for patients with cancer. Cochrane Database Syst Rev 2012; 12:CD003407.
- Bohlius J, Langensiepen S, Schwarzer G, et al. Recombinant human erythropoietin and overall survival in cancer patients: results of a comprehensive meta-analysis. J Natl Cancer Inst 2005; 97:489–498.
- Henke M, Laszig R, Rübe C, et al. Erythropoietin to treat head and neck cancer patients with anaemia undergoing radiotherapy: randomised, double-blind, placebo-controlled trial. Lancet 2003; 362:1255–1260.
- Leyland-Jones B, Semiglazov V, Pawlicki M, et al. Maintaining normal hemoglobin levels with epoetin alfa in mainly nonanemic patients with metastatic breast cancer receiving first-line chemotherapy: a survival study. J Clin Oncol 2005; 23:5960–5972.
- Brower V. Erythropoietin may impair, not improve, cancer survival. Nat Med 2003; 9:1439.
- Acs G, Acs P, Beckwith SM, et al. Erythropoietin and erythropoietin receptor expression in human cancer. Cancer Res 2001; 61:3561–3565.
- Yasuda Y, Fujita Y, Matsuo T, et al. Erythropoietin regulates tumour growth of human malignancies. Carcinogenesis 2003; 24:1021–1029.
- Levin NW, Fishbane S, Cañedo FV, et al; MAXIMA Study Investigators. Intravenous methoxy polyethylene glycol-epoetin beta for haemoglobin control in patients with chronic kidney disease who are on dialysis: a randomised non-inferiority trial (MAXIMA). Lancet 2007; 370:1415–1421.
- Macdougall IC, Walker R, Provenzano R, et al; ARCTOS Study Investigators. C.E.R.A. corrects anemia in patients with chronic kidney disease not on dialysis: results of a randomized clinical trial. Clin J Am Soc Nephrol 2008; 3:337–347.
- Frohna PA, Milwee S, Pinkett J, et al. Preliminary results from a randomized, single-blind, placebo-controlled trial of FG-4592, a novel hypoxia inducible factor prolyl hydroxylase inhibitor, in subjects with CKD anemia (abstract). J Am Soc Nephrol 2007; 18:763.
- Holdstock L, Meadowcroft AM, Maier R, et al. Four-week studies of oral hypoxia-inducible factor-prolyl hydroxylase inhibitor GSK1278863 for treatment of anemia. J Am Soc Nephrol 2016; 27:1234–1244.
- World Health Organization (WHO). Nutritional anaemias: report of a WHO scientific group. Geneva, Switzerland: World Health Organization, 1968.
- Hsu CY, McCulloch CE, Curhan GC, et al. Epidemiology of anemia associated with chronic renal insufficiency among adults in the United States: results from the Third National Health and Nutrition Examination Survey. J Am Soc Nephrol 2002; 13:504–510.
- Radtke HW, Claussner A, Erbes PM, Scheuermann EH, Schoeppe W, Koch KM. Serum erythropoetin concentration in chronic renal failure: relationship to degree of anemia and excretory renal function. Blood 1979; 54:877–884.
- Kazmi WH, Kausz AT, Khan S, et al. Anemia: an early complication of chronic renal insufficiency. Am J Kidney Dis 2001; 38:803–812.
- United States Renal Data System. Chapter 3. Morbidity & mortality in patients with CKD. www.usrds.org/2012/view/v1_03.aspx. Accessed June 9, 2016.
- Horwich TB, Fonarow GC, Hamilton MA, MacLellan WR, Borenstein J. Anemia is associated with worse symptoms, greater impairment in functional capacity and a significant increase in mortality in patients with advanced heart failure. J Am Coll Cardiol 2002; 39:1780–1786.
- Mark DB, Felker GM. B-type natriuretic peptide: a biomarker for all seasons? N Engl J Med 2004; 350:718–720.
- Walker AM, Schneider G, Yeaw J, Nordstrom B, Robbins S, Pettitt D. Anemia as a predictor of cardiovascular events in patients with elevated serum creatinine. J Am Soc Nephrol 2006; 17:2293–2298.
- Abramson JL, Jurkovitz CT, Vaccarino V, Weintraub WS, McClellan W. Chronic kidney disease, anemia, and incident stroke in a middle-aged, community-based population: the ARIC Study. Kidney Int 2003; 64:610–615.
- Sarnak MJ, Tighiouart H, Manjunath G, et al. Anemia as a risk factor for cardiovascular disease in the Atherosclerosis Risk in Communities (ARIC) study. J Am Coll Cardiol 2002; 40:27–33.
- McClellan WM, Flanders WD, Langston RD, Jurkovitz C, Presley R. Anemia and renal insufficiency are independent risk factors for death among patients with congestive heart failure admitted to community hospitals: a population-based study. J Am Soc Nephrol 2002; 13:1928–1936.
- Xia H, Ebben J, Ma JZ, Collins AJ. Hematocrit levels and hospitalization risks in hemodialysis patients. J Am Soc Nephrol 1999; 10:1309–1316.
- Collins AJ, Li S, St Peter W, et al. Death, hospitalization, and economic associations among incident hemodialysis patients with hematocrit values of 36 to 39%. J Am Soc Nephrol 2001; 12:2465–2473.
- Agarwal AK. Practical approach to the diagnosis and treatment of anemia associated with CKD in elderly. J Am Med Dir Assoc 2006; 7(suppl 9):S7–S12.
- Bernhardt WM, Wiesener MS, Scigalla P, et al. Inhibition of prolyl hydroxylases increases erythropoietin production in ESRD. J Am Soc Nephrol 2010; 21:2151–2156.
- Provenzano R, Fadda G, Bernardo M, et al. FG-2216, a novel oral HIF-PHI, stimulates erythropoiesis and increases hemoglobin concentration in patients with non-dialysis CKD. Am J Kidney Dis 2008; 51:B80.
- Maxwell PH, Osmond MK, Pugh CW, et al. Identification of the renal erythropoietin-producing cells using transgenic mice. Kidney Int 1993; 44:1149–1162.
- Maxwell PH, Ferguson DJ, Nicholls LG, et al. Sites of erythropoietin production. Kidney Int 1997; 51:393–401.
- Jelkmann W. Erythropoeitin: structure, control of production and function. Physiol Rev 1992; 72:449–489.
- Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA 1995; 92:5510–5514.
- Wang GL, Semenza GL. Purification and characterization of hypoxia-inducible factor 1. J Biol Chem 1995; 270:1230–1237.
- Maxwell PH, Wiesener MS, Chang GW, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999; 399:271–275.
- Salceda S, Caro J. Hypoxia-inducible factor 1alpha protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J Biol Chem 1997; 272:22642–22647.
- Malik J, Kim AR, Tyre KA, Cherukuri AR, Palis J. Erythropoietin critically regulates the terminal maturation of murine and human primitive erythroblasts. Haematologica 2013; 98:1778–1787.
- Wu H, Liu X, Jaenisch R, Lodish HF. Generation of committed erythroid BFU-E and CFU-E progenitors does not require erythropoietin or the erythropoietin receptor. Cell 1995; 83:59–67.
- Socolovsky M, Nam H, Fleming MD, Haase VH, Brugnara C, Lodish HF. Ineffective erythropoiesis in Stat5a(-/-)5b(-/-) mice due to decreased survival of early erythroblasts. Blood 2001; 98:3261–3273.
- Papanikolaou G, Pantopoulos K. Iron metabolism and toxicity. Toxicol Appl Pharmacol 2005; 202:199–211.
- Conrad ME, Umbreit JN. Pathways of iron absorption. Blood Cells Mol Dis 2002; 29:336–355.
- Frazer DM, Anderson GJ. The orchestration of body iron intake: how and where do enterocytes receive their cues? Blood Cells Moll Dis 2003; 30:288–297.
- Donovan A, Lima CA, Pinkus JL, et al. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab 2005; 1:191–200.
- Vulpe CD, Kuo YM, Murphy TL, et al. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat Genet 1999; 21:195–199.
- Bothwell TH. Overview and mechanisms of iron regulation. Nutr Rev 1995: 53:237–245.
- Kawabata H, Nakamaki T, Ikonomi P, Smith RD, Germain RS, Koeffler HP. Expression of transferrin receptor 2 in normal and neoplastic hematopoietic cells. Blood 2001; 98:2714–2719.
- Arosio P, Levi S. Cytosolic and mitochondrial ferritins in the regulation of cellular iron homeostasis and oxidative damage. Biochim Biophys Acta 2010; 1800:783–792.
- Finch CA, Bellotti V, Stray S, et al. Plasma ferritin determination as a diagnostic tool. West J Med 1986; 145:657–663.
- Delaby C, Pilard N, Goncalves AS, Beaumont C, Canonne-Hergaux F. Presence of the iron exporter ferroportin at the plasma membrane of macrophages is enhanced by iron loading and down-regulated by hepcidin. Blood 2005; 106:3979–3984.
- Ganz T. Hepcidin, a key regulator of iron metabolism and mediator of anemia of inflammation. Blood 2003; 102:783–788.
- Nicolas G, Chauvet C, Viatte L, et al. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J Clin Invest 2002; 110:1037–1044.
- DeGowin RL, Lavender AR, Forland M, Charleston D, Gottschalk A. Erythropoiesis and erythropoietin in patients with chronic renal failure treated with hemodialysis and testosterone. Ann Intern Med 1970; 72:913–918.
- Richardson JR Jr, Weinstein MB. Erythropoietic response of dialyzed patients to testosterone administration. Ann Intern Med 1970; 73:403–407
- Eschbach JW, Abdulhadi MH, Browne JK, et al. Recombinant human erythropoietin in anemic patients with end-stage renal disease. Results of a phase III multicenter clinical trial. Ann Intern Med 1989; 111:992–1000.
- Moreno F, Aracil FJ, Pérez R, Valderrábano F. Controlled study on the improvement of quality of life in elderly hemodialysis patients after correcting end-stage renal disease-related anemia with erythropoietin. Am J Kidney Dis 1996; 27:548–556.
- Nissenson AR, Nimer SD, Wolcott DL. Recombinant human erythropoietin and renal anemia: molecular biology, clinical efficacy, and nervous system effects. Ann Intern Med 1991; 114:402–416.
- Stivelman JC. Benefits of anaemia treatment on cognitive function. Nephrol Dial Transplant 2000; 15(suppl 3):29–35.
- Maddux FW, Shetty S, del Aguila MA, Nelson MA, Murray BM. Effect of erythropoiesis-stimulating agents on healthcare utilization, costs, and outcomes in chronic kidney disease. Ann Pharmacother 2007; 41:1761–1769.
- Macdougall IC, Lewis NP, Saunders MJ, et al. Long-term cardiorespiratory effects of amelioration of renal anaemia by erythropoietin. Lancet 1990; 335:489–493.
- Silverberg DS, Wexler D, Blum M, et al. Effects of treatment with epoetin beta on outcomes in patients with anaemia and chronic heart failure. Kidney Blood Press Res 2005; 28:41–47.
- Perkins R, Olson S, Hansen J, Lee J, Stiles K, Lebrun C. Impact of an anemia clinic on emergency room visits and hospitalizations in patients with anemia of CKD pre-dialysis. Nephrol Nurs J 2007; 34:167–173, 182.
- Locatelli F, Conte F, Marcelli D. The impact of haematocrit levels and erythropoietin treatment on overall and cardiovascular mortality and morbidity—the experience of the Lombardy Dialysis Registry. Nephrol Dial Transplant 1998; 13:1642–1644.
- Centers for Medicare and Medicaid Services; Kinney R. 2005 Annual Report: ESRD Clinical Performance Measures Project. Am J Kidney Dis 2006; 48(suppl 2):S1–S106.
- US Renal Data System. Annual Data Report 2006. www.usrds.org/adr.aspx. Accessed July 3, 2016.
- Besarab A, Bolton WK, Browne JK, et al. The effects of normal as compared with low hematocrit values in patients with cardiac disease who are receiving hemodialysis and epoetin. N Engl J Med 1998; 339:584–590.
- Singh AK, Szczech L, Tang KL, et al; CHOIR Investigators. Correction of anemia with epoetin alfa in chronic kidney disease. N Engl J Med 2006; 355:2085–2098.
- Drüeke TB, Locatelli F, Clyne N, et al; CREATE Investigators. Normalization of hemoglobin level in patients with chronic kidney disease and anemia. N Engl J Med 2006; 355:2071–2084.
- Pfeffer MA, Burdmann EA, Chen CY, et al; TREAT Investigators. A trial of darbepoetin alfa in type 2 diabetes and chronic kidney disease. N Engl J Med 2009; 361:2019–2032.
- Kirkpantur A, Kahraman S, Yilmaz R, et al. The effects of maintenance recombinant human erythropoietin therapy on ambulatory blood pressure recordings: conventional, Doppler, and tissue Doppler echocardiographic parameters. Artif Organs 2005; 29:965–972.
- Fishbane S, Berns JS. Hemoglobin cycling in hemodialysis patients treated with recombinant human erythropoietin. Kidney Int 2005; 68:1337–1343.
- Szczech LA, Barnhart HX, Inrig JK, et al. Secondary analysis of the CHOIR trial epoetin-alpha dose and achieved hemoglobin outcomes. Kidney Int 2008; 74:791–798.
- Solomon SD, Uno H, Lewis EF, et al; Trial to Reduce Cardiovascular Events with Aranesp Therapy (TREAT) Investigators. Erythropoietic response and outcomes in kidney disease and type 2 diabetes. N Engl J Med 2010; 363:1146–1155.
- Kidney Disease: Improving Global Outcomes (KDIGO). KDIGO Clinical Practice Guideline for Anemia in Chronic Kidney Disease. Kidney Int Suppl 2012; 2:279–335.
- Fernández-Rodríguez AM, Guindeo-Casasús MC, Molero-Labarta T, et al. Diagnosis of iron deficiency in chronic renal failure. Am J Kidney Dis 1999; 34:508–513.
- Eschbach JW, Cook JD, Scribner BH, Finch CA. Iron balance in hemodialysis patients. Ann Intern Med 1977; 87:710–713.
- Mittman N, Sreedhara R, Mushnick R, et al. Reticulocyte hemoglobin content predicts functional iron deficiency in hemodialysis patients receiving rHuEPO. Am J Kidney Dis 1997; 30:912–922.
- Tessitore N, Solero GP, Lippi G, et al. The role of iron status markers in predicting response to intravenous iron in haemodialysis patients on maintenance erythropoietin. Nephrol Dial Transplant 2001; 16:1416–1423.
- Coyne DW. Iron indices: what do they really mean? Kidney Int Suppl 2006; 101:S4–S8.
- Fishbane S, Kowalski EA, Imbriano LJ, Maesaka JK. The evaluation of iron status in hemodialysis patients. J Am Soc Nephrol 1996; 7:2654–2657.
- Coyne DW, Kapoian T, Suki W, et al; DRIVE Study Group. Ferric gluconate is highly efficacious in anemic hemodialysis patients with high serum ferritin and low transferrin saturation: results of the Dialysis Patients’ Response to IV Iron with Elevated Ferritin (DRIVE) Study. J Am Soc Nephrol 2007; 18:975–984.
- Radtke HW, Claussner A, Erbes PM, Scheuermann EH, Schoeppe W, Koch KM. Serum erythropoietin concentration in chronic renal failure: relationship to degree of anemia and excretory renal function. Blood 1979; 54:877–884.
- Korte W, Cogliatti SB, Jung K, Riesen W. Mild renal dysfunction is sufficient to induce erythropoietin deficiency in patients with unexplained anaemia. Clin Chim Acta 2000; 292:149–154.
- Locatelli F, Olivares J, Walker R, et al; European/Australian NESP 980202 Study Group. Novel erythropoiesis stimulating protein for treatment of anemia in chronic renal insufficiency. Kidney Int 2001; 60:741–747.
- Carrera F, Burnier M. Use of darbepoetin alfa in the treatment of anaemia of chronic kidney disease: clinical and pharmacoeconomic considerations. NDT Plus 2009; 2(suppl 1):i9–i17.
- Egrie JC, Browne JK. Development and characterization of novel erythropoiesis stimulating protein (NESP). Nephrol Dial Transplant 2001; 16(suppl 3):3–13.
- Nissenson AR, Charytan C. Controversies in iron management. Kidney Int Suppl 2003; 87:S64–S71.
- Kilpatrick RD, Critchlow CW, Fishbane S, et al. Greater epoetin alpha responsiveness is associated with improved survival in hemodialysis patients. Clin J Am Soc Nephrol 2008; 3:1077–1083.
- Locatelli F, Aljama P, Barany P, et al; European Best Practice Guidelines Working Group. Revised European best practice guidelines for the management of anaemia in patients with chronic renal failure. Nephrol Dial Transplant 2004; 19(suppl 2):ii1–ii47.
- Stenvinkel P. The role of inflammation in the anaemia of end-stage renal disease. Nephrol Dial Transplant 2001; 16(suppl 7):36–40.
- Barany P, Divino Filho JC, Bergstrom J. High C-reactive protein is a strong predictor of resistance to erythropoietin in hemodialysis patients. Am J Kidney Dis 1997; 29:565–568.
- Drueke T. Hyporesponsiveness to recombinant human erythropoietin. Nephrol Dial Transplant 2001; 16(suppl 7):25–28.
- Casadevall N. Cellular mechanism of resistance to erythropoietin. Nephrol Dial Transplant 1995; 10(suppl 6):27–30.
- Kraus E, Rabb H. EPO therapy during acute kidney disease: to use or not to use, that is the question. Am J Kidney Dis 2005; 46:967–969.
- Gotloib L, Silverberg D, Fudin R, Shostak A. Iron deficiency is a common cause of anemia in chronic kidney disease and can often be corrected with intravenous iron. J Nephrol 2006; 19:161–167.
- Tarng DC, Huang TP, Chen TW, Yang WC. Erythropoietin hyporesponsiveness: from iron deficiency to iron overload. Kidney Int Suppl 1999; 69:S107–S118.
- Drüeke TB. Modulating factors in the hematopoietic response to erythropoietin. Am J Kidney Dis 1991; 18(suppl 1):87–92.
- Boven K, Stryker S, Knight J, et al. The increased incidence of pure red cell aplasia with an Eprex formulation in uncoated rubber stopper syringes. Kidney Int 2005; 67:2346–2353.
- Shimizu H, Saitoh T, Ota F, et al. Pure red cell aplasia induced only by intravenous administration of recombinant human erythropoietin. Acta Haematol 2011; 126:114–118.
- Tonia T, Mettler A, Robert N, et al. Erythropoietin or darbepoetin for patients with cancer. Cochrane Database Syst Rev 2012; 12:CD003407.
- Bohlius J, Langensiepen S, Schwarzer G, et al. Recombinant human erythropoietin and overall survival in cancer patients: results of a comprehensive meta-analysis. J Natl Cancer Inst 2005; 97:489–498.
- Henke M, Laszig R, Rübe C, et al. Erythropoietin to treat head and neck cancer patients with anaemia undergoing radiotherapy: randomised, double-blind, placebo-controlled trial. Lancet 2003; 362:1255–1260.
- Leyland-Jones B, Semiglazov V, Pawlicki M, et al. Maintaining normal hemoglobin levels with epoetin alfa in mainly nonanemic patients with metastatic breast cancer receiving first-line chemotherapy: a survival study. J Clin Oncol 2005; 23:5960–5972.
- Brower V. Erythropoietin may impair, not improve, cancer survival. Nat Med 2003; 9:1439.
- Acs G, Acs P, Beckwith SM, et al. Erythropoietin and erythropoietin receptor expression in human cancer. Cancer Res 2001; 61:3561–3565.
- Yasuda Y, Fujita Y, Matsuo T, et al. Erythropoietin regulates tumour growth of human malignancies. Carcinogenesis 2003; 24:1021–1029.
- Levin NW, Fishbane S, Cañedo FV, et al; MAXIMA Study Investigators. Intravenous methoxy polyethylene glycol-epoetin beta for haemoglobin control in patients with chronic kidney disease who are on dialysis: a randomised non-inferiority trial (MAXIMA). Lancet 2007; 370:1415–1421.
- Macdougall IC, Walker R, Provenzano R, et al; ARCTOS Study Investigators. C.E.R.A. corrects anemia in patients with chronic kidney disease not on dialysis: results of a randomized clinical trial. Clin J Am Soc Nephrol 2008; 3:337–347.
- Frohna PA, Milwee S, Pinkett J, et al. Preliminary results from a randomized, single-blind, placebo-controlled trial of FG-4592, a novel hypoxia inducible factor prolyl hydroxylase inhibitor, in subjects with CKD anemia (abstract). J Am Soc Nephrol 2007; 18:763.
- Holdstock L, Meadowcroft AM, Maier R, et al. Four-week studies of oral hypoxia-inducible factor-prolyl hydroxylase inhibitor GSK1278863 for treatment of anemia. J Am Soc Nephrol 2016; 27:1234–1244.
KEY POINTS
- Before treating with ESAs, it is necessary to investigate and rule out underlying treatable conditions such as iron or vitamin deficiencies.
- Recognizing anemia in chronic kidney disease is important and often involves participation by the primary care physician, especially in early disease when chronic kidney disease may be mild.
- The only proven benefit of ESA therapy is avoidance of blood transfusions.
- ESAs should not be used to increase the hemoglobin concentration above 13 g/dL. In end-stage renal disease, the goal of therapy is to maintain levels at a target no higher than 11.5 g/dL. In nondialysis-dependent chronic kidney disease, the decision to prescribe ESA therapy should be individualized.
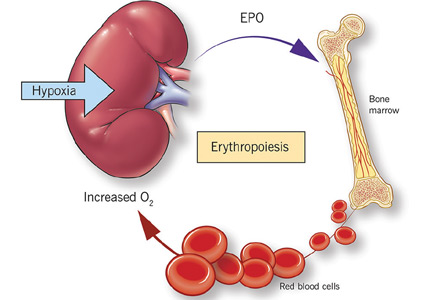
Information management for clinicians
Accessing, absorbing, organizing, storing, and retrieving potentially useful medical information is a challenge. Physicians used to try meet this challenge with personal libraries of journal articles in their file cabinet. Today, that is inadequate to combat the deluge of digital information. In 2013, the Institute of Medicine acknowledged this problem, stating, “The ever-increasing volume of evidence makes it difficult for clinicians to maintain a working knowledge of new clinical information.”1
The sheer volume of data has meant that, rather than try to maintain a regular diet of professional reading (proactive scanning2), many of us now seek information only when we need answers to specific clinical questions (reactive searching). This approach promotes lifelong problem-based learning but assumes that we are consciously aware of this need and are aware of the need to search for new information.
We need to constantly scan for new evidence in our area of practice to avoid becoming falsely assured of our knowledge. We also need to be able to find information we have seen before we need to use it. The following are examples of how using this approach could dramatically empower a busy clinician.
On a recent clinic day, a colleague pokes his head into your office and asks, “Have you heard anything about niacin in the news? I have a patient who is asking me if we should discontinue it.” You respond: “Yes, there was something that just came out. I am not sure where I read it or heard about it. Give me a couple of seconds and I can find it.” True to your word, a few seconds later you find and share the latest article on the HPS2-THRIVE trial and a commentary on the results.
Later, your first patient of the day asks you, “When we switched to dabigatran, you mentioned that, unlike warfarin, there was no specific reversal agent. I heard that there is one now?” Instead of being taken aback, you nod your head. “Yes, I saw that recent article showing that the medication rapidly and completely reverses the effect of dabigatran in the majority of patients. While I hope we don’t need it, this is good news, particularly as there did not seem to be any major adverse events.”
Sounds too good to be true? Can this really be you? In this article, we outline an information management strategy (Figure 1) and tools to help busy clinicians stay up to date with new medical evidence in their areas of interest or expertise. In addition, we provide a strategy for leveraging technology to easily retrieve previously viewed information. A future article will specifically show how to best access information at the point of patient care.
THE NEED TO MANAGE INFORMATION
Physicians are expected to practice evidence-based medicine. When faced with a clinical question, we should search for evidence using focused queries of primary and secondary sources such as PubMed or the Cochrane Library. This is an important skill and is appropriate when we take time to look for an evidence-based answer to a specific question. In many cases, it is appropriate to continue with a current practice until newer information has been reviewed and validated.
Unfortunately, indexing and adding new recommendations to these information sources takes time. We may also be unaware that new information is available and may continue to practice as usual until faced with a situation like those outlined above or until we attend a continuing medical education activity, often quite by chance.
Today we can proactively update ourselves in a manner tailored to our own interests and focus and retrieve important information easily when we need it.
A STRATEGY FOR INFORMATION MANAGEMENT
In general, we come across new information in one of three ways:
- Proactive scanning of personalized sources of information—as discussed above, a habit of regular scanning is critical to information management
- Reactive searching for information to answer clinical problems or when doing research
- Incidentally—an e-mail from a colleague, information shared on a social network or encountered while surfing the Internet.
In each case, we may find information that is potentially useful, something we may need to find again in the future. But unless we use this information often, we will not remember the details or may even forget we had seen it. Thus, we need a strategy to store this information so we can retrieve it easily at any time with any device; neuroscientists call this the “externalization” of memory.3 Ideally, even if we forget that we ever saw this information or where we stored it, a search would retrieve the location and details of this formerly viewed information.
In the following sections, we outline steps and tools of a strategy for managing clinical information relevant to your practice.
STEP 1: SETTING UP INFORMATION FEEDS
The first step in this information management strategy is to become aware of relevant new information in your area of practice or research. To do this, you proactively set up feeds of information from reliable and authentic sources. These feeds can be browsed on any computer or smart mobile device.
There are several possibilities for creating these feeds. One option is to subscribe to the table of contents (TOC) of relevant journals via e-mail.
A more versatile and full-featured option is a research site summary (RSS) feed-reader. RSS is a standard for publishing summaries (feeds) of frequently updated content on the World Wide Web, such as journal TOCs and items from medical journal news sites (Table 1 shows what this looks like on screen), as well as aggregators like the American College of Physicians Journal Club. You can subscribe to these using feed-reader software from Feedly (www.feedly.com) or Inoreader (www.inoreader.com), which can be used with any browser on a desktop or laptop. They are also available as apps for mobile devices such as smartphones and tablets. The feed-reader periodically checks for new content and automatically downloads it to the device. Thus, you do not need to check multiple websites for updates or have e-mail inboxes fill with content; the content is delivered to your device for perusal at your convenience. (See online supplement “Information Management for Clinicians” for step-by-step instructions on creating a free Inoreader account and subscribing to feeds.)
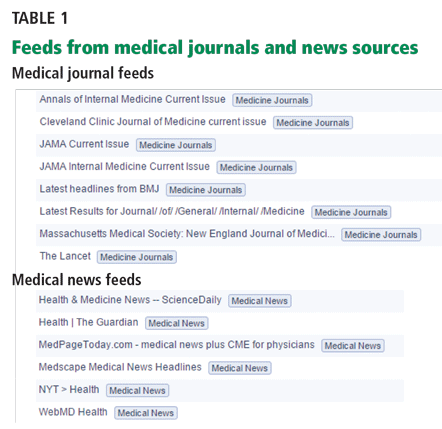
RSS feed-readers have several advantages over e-mailed TOCs:
- RSS feeds create a centralized searchable repository of all subscribed information.
- The software keeps track of what you have read and displays only unread items; after a journal TOC e-mail is opened, the entire TOC is marked as having been read.
- You can organize news items into folders by tagging key words.
- Most journals and medical news sites like Medscape and the health section of the New York Times provide RSS feeds at no cost.
- An unlimited number of feed items, or articles, are stored in the cloud and do not affect e-mail storage capacity.
- The feed is automatically updated multiple times a day instead of once a week or once a month.
- In addition, one can create RSS feeds on PubMed for custom searches. Thus, a physician can get automatic regular updates of new articles indexed in MEDLINE in their area of interest.
Users can thus build their own personalized magazine of constantly updated information for access and can search from any web-enabled device. (Note: It is advisable to turn off notifications generated by these apps on mobile devices to reduce distraction.)
STEP 2: BOOKMARKING EVIDENCE
When you find something useful or interesting, bookmarks help you find the information again quickly when you need it. But while the browsers Firefox, Chrome, Internet Explorer, and Safari allow bookmarking, they have significant limitations. Bookmarks may be available only on the device they were created on, and because people use more than one device to go online, they may not remember which device they used to bookmark or view the web page.
Browser bookmarks generally store the address (URL) of the web page and a label that you create, but they do not do much else. Sharing bookmarks with others is also difficult or impossible.
Social bookmarking
Social bookmarking lets you create bookmarks you can share across other devices and with other people. Diigo (www.diigo.com) and Delicious (www.delicious.com) are two social bookmarking services that let you integrate with all popular browsers through a button or toolbar. They allow you to save displayed web pages with labels, descriptions, and tags.
Diigo offers two additional features. It allows web pages to be annotated with highlights and notes. And during a Google search, relevant results from the Diigo library are simultaneously displayed.
If you forget you bookmarked something and saved it in your Diigo library, when you search for the information again on Google, Diigo will automatically display any results from your Diigo library next to the Google search results. This is very helpful as it is much easier to review information you have already read, marked up, and saved than it is to start over.
Bookmarks and annotations are stored in the cloud and can be accessed by any device. (See “Information Management for Clinicians” to learn how to sign up for a free Diigo account, and how to use it.)
STEP 3: STORING YOUR INFORMATION
You may want the option to store full-text information in your personal library. This information was once stored in file cabinets and, more recently, on hard drives and USB flash drives. But information stored with these methods is not available or searchable on multiple devices from any location.
Cloud storage
Cloud storage services meet the need for access to stored information at any time and with any device. Options include Dropbox (www.dropbox.com), Box (www.box.com), Google Drive (drive.google.com), OneDrive (onedrive.live.com), and Evernote (www.evernote.com). Each provides different amounts of free storage and has apps available for most platforms and devices. They provide search tools and the ability to share articles or “folders” with other users. The information on these online drives is “synced” between all devices so that the most up-to-date version is always available to the user regardless of location and device.
Evernote offers multiple folders called notebooks to store and segregate data. The open notebook shown in Figure 2 is named “reference articles.” It has the HPS2-Thrive article from the New England Journal of Medicine (N Engl J Med) tagged with the terms “niacin” and “lipid” to facilitate retrieval. The article was saved from that journal’s website using an Evernote browser extension that allows entire web pages or selections to be saved to Evernote with a single click. Evernote also has a powerful search feature that can find text in images or in PDF documents. In addition, it allows easy sharing of a note or an entire notebook. Once a note or notebook is shared, all parties can add to it. In The Evernote app also allows tablet and smartphone access to the shareable content.
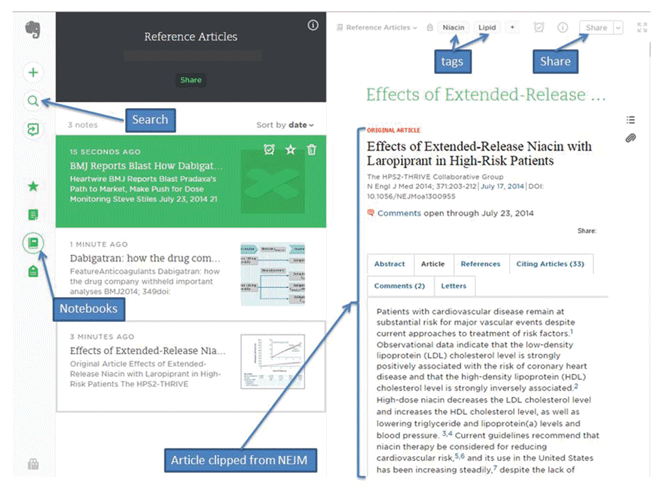
The other services listed here have similar feature sets. Dropbox is perhaps the easiest to adopt, but it offers the least amount of free storage. If you use Microsoft Office software, OneDrive lets you edit documents online, and an Office 365 subscription includes 1 terabyte of storage. Google Drive is probably the best solution for online collaboration, such as coauthoring a paper. Box is one of the few online storage solutions that complies with the Health Insurance Portability and Accountability privacy rules.
PUTTING IT ALL INTO PRACTICE
Once you have become familiar with Inoreader and Diigo (see Information Management for Clinicians for step-by-step instructions), the following scenario shows how to adapt them into an efficient workflow.
Dr. Smith has a smartphone, a tablet, a laptop at home, and a desktop at work. She signs onto Google Chrome as her preferred browser on all devices. This seamlessly loads her Diigo extension when she is using a laptop or desktop. She has set her RSS feeds for her preferred journal TOCs and medical news sites to be downloaded to Inoreader. (For details on how to add a medical journals feed bundle and a medical news feed bundle, visit Information Management for Clinicians.)
Instead of reading paper journals, Dr. Smith browses her customized up-to-date “magazine” on Inoreader. When she comes across a relevant article, she marks it as “favorite.” If she has more time, she visits the web page, reviews the information, and saves it to her Diigo library with annotations if appropriate.
When searching for information on the web, she uses Google—without having to remember if she bookmarked information related to the search term. The Diigo extension in her browser automatically searches and displays information from her Diigo library next to her Google search results, and she can instantly see her notes from the last time she read the article.
Relating this workflow to the example of the dabigatran story above, Dr. Smith sees an article about dabigatran reversal while viewing her N Engl J Med medical news feed on her feed-reader. She marks it as a favorite and tags it with the key terms “cardiology” and “vascular” (Figure 3).
Dr. Smith later returns to look at her favorite feed items and visits the article on the N Engl J Med website. She annotates the article and saves it to her Diigo library (Figure 4).

Since this information is highly relevant to her practice, she also visits the N Engl J Med website to read the full article and the accompanying editorial (Figure 5). She annotates these and also saves them to her Diigo library.

Later, if she searches Google for dabigatran (using her default Google Chrome browser with Diigo extension), she will see the usual Google search results and twinned Diigo bookmarks (Figure 6).
If she clicks on one of the links, the browser will load the web page with all the annotations that she made when she first visited.
CONCLUSION
The strategies and tools we describe here let you create a personalized and constantly updated medical news “magazine,” accessible from any of your web-enabled devices. They can transform the Internet into a searchable notebook of personally selected, annotated information, helping you to more easily stay up to date with advances in your field of practice, and to more easily manage the modern information overload.
- Institute of Medicine (IOM). Best Care at Lower Cost: The Path to Continuously Learning Health Care in America. Washington, DC: National Academies Press; 2013. www.nap.edu/openbook.php?record_id=13444&page=R1. Accessed May 17, 2016.
- Slotnick HB. Physicians’ learning strategies. Chest 2000; 118(suppl 2):18S–23S.
- Levitin DJ. The Organized Mind: Thinking Straight in the Age of Information Overload. New York, NY: Dutton; 2014:528.
Accessing, absorbing, organizing, storing, and retrieving potentially useful medical information is a challenge. Physicians used to try meet this challenge with personal libraries of journal articles in their file cabinet. Today, that is inadequate to combat the deluge of digital information. In 2013, the Institute of Medicine acknowledged this problem, stating, “The ever-increasing volume of evidence makes it difficult for clinicians to maintain a working knowledge of new clinical information.”1
The sheer volume of data has meant that, rather than try to maintain a regular diet of professional reading (proactive scanning2), many of us now seek information only when we need answers to specific clinical questions (reactive searching). This approach promotes lifelong problem-based learning but assumes that we are consciously aware of this need and are aware of the need to search for new information.
We need to constantly scan for new evidence in our area of practice to avoid becoming falsely assured of our knowledge. We also need to be able to find information we have seen before we need to use it. The following are examples of how using this approach could dramatically empower a busy clinician.
On a recent clinic day, a colleague pokes his head into your office and asks, “Have you heard anything about niacin in the news? I have a patient who is asking me if we should discontinue it.” You respond: “Yes, there was something that just came out. I am not sure where I read it or heard about it. Give me a couple of seconds and I can find it.” True to your word, a few seconds later you find and share the latest article on the HPS2-THRIVE trial and a commentary on the results.
Later, your first patient of the day asks you, “When we switched to dabigatran, you mentioned that, unlike warfarin, there was no specific reversal agent. I heard that there is one now?” Instead of being taken aback, you nod your head. “Yes, I saw that recent article showing that the medication rapidly and completely reverses the effect of dabigatran in the majority of patients. While I hope we don’t need it, this is good news, particularly as there did not seem to be any major adverse events.”
Sounds too good to be true? Can this really be you? In this article, we outline an information management strategy (Figure 1) and tools to help busy clinicians stay up to date with new medical evidence in their areas of interest or expertise. In addition, we provide a strategy for leveraging technology to easily retrieve previously viewed information. A future article will specifically show how to best access information at the point of patient care.
THE NEED TO MANAGE INFORMATION
Physicians are expected to practice evidence-based medicine. When faced with a clinical question, we should search for evidence using focused queries of primary and secondary sources such as PubMed or the Cochrane Library. This is an important skill and is appropriate when we take time to look for an evidence-based answer to a specific question. In many cases, it is appropriate to continue with a current practice until newer information has been reviewed and validated.
Unfortunately, indexing and adding new recommendations to these information sources takes time. We may also be unaware that new information is available and may continue to practice as usual until faced with a situation like those outlined above or until we attend a continuing medical education activity, often quite by chance.
Today we can proactively update ourselves in a manner tailored to our own interests and focus and retrieve important information easily when we need it.
A STRATEGY FOR INFORMATION MANAGEMENT
In general, we come across new information in one of three ways:
- Proactive scanning of personalized sources of information—as discussed above, a habit of regular scanning is critical to information management
- Reactive searching for information to answer clinical problems or when doing research
- Incidentally—an e-mail from a colleague, information shared on a social network or encountered while surfing the Internet.
In each case, we may find information that is potentially useful, something we may need to find again in the future. But unless we use this information often, we will not remember the details or may even forget we had seen it. Thus, we need a strategy to store this information so we can retrieve it easily at any time with any device; neuroscientists call this the “externalization” of memory.3 Ideally, even if we forget that we ever saw this information or where we stored it, a search would retrieve the location and details of this formerly viewed information.
In the following sections, we outline steps and tools of a strategy for managing clinical information relevant to your practice.
STEP 1: SETTING UP INFORMATION FEEDS
The first step in this information management strategy is to become aware of relevant new information in your area of practice or research. To do this, you proactively set up feeds of information from reliable and authentic sources. These feeds can be browsed on any computer or smart mobile device.
There are several possibilities for creating these feeds. One option is to subscribe to the table of contents (TOC) of relevant journals via e-mail.
A more versatile and full-featured option is a research site summary (RSS) feed-reader. RSS is a standard for publishing summaries (feeds) of frequently updated content on the World Wide Web, such as journal TOCs and items from medical journal news sites (Table 1 shows what this looks like on screen), as well as aggregators like the American College of Physicians Journal Club. You can subscribe to these using feed-reader software from Feedly (www.feedly.com) or Inoreader (www.inoreader.com), which can be used with any browser on a desktop or laptop. They are also available as apps for mobile devices such as smartphones and tablets. The feed-reader periodically checks for new content and automatically downloads it to the device. Thus, you do not need to check multiple websites for updates or have e-mail inboxes fill with content; the content is delivered to your device for perusal at your convenience. (See online supplement “Information Management for Clinicians” for step-by-step instructions on creating a free Inoreader account and subscribing to feeds.)
RSS feed-readers have several advantages over e-mailed TOCs:
- RSS feeds create a centralized searchable repository of all subscribed information.
- The software keeps track of what you have read and displays only unread items; after a journal TOC e-mail is opened, the entire TOC is marked as having been read.
- You can organize news items into folders by tagging key words.
- Most journals and medical news sites like Medscape and the health section of the New York Times provide RSS feeds at no cost.
- An unlimited number of feed items, or articles, are stored in the cloud and do not affect e-mail storage capacity.
- The feed is automatically updated multiple times a day instead of once a week or once a month.
- In addition, one can create RSS feeds on PubMed for custom searches. Thus, a physician can get automatic regular updates of new articles indexed in MEDLINE in their area of interest.
Users can thus build their own personalized magazine of constantly updated information for access and can search from any web-enabled device. (Note: It is advisable to turn off notifications generated by these apps on mobile devices to reduce distraction.)
STEP 2: BOOKMARKING EVIDENCE
When you find something useful or interesting, bookmarks help you find the information again quickly when you need it. But while the browsers Firefox, Chrome, Internet Explorer, and Safari allow bookmarking, they have significant limitations. Bookmarks may be available only on the device they were created on, and because people use more than one device to go online, they may not remember which device they used to bookmark or view the web page.
Browser bookmarks generally store the address (URL) of the web page and a label that you create, but they do not do much else. Sharing bookmarks with others is also difficult or impossible.
Social bookmarking
Social bookmarking lets you create bookmarks you can share across other devices and with other people. Diigo (www.diigo.com) and Delicious (www.delicious.com) are two social bookmarking services that let you integrate with all popular browsers through a button or toolbar. They allow you to save displayed web pages with labels, descriptions, and tags.
Diigo offers two additional features. It allows web pages to be annotated with highlights and notes. And during a Google search, relevant results from the Diigo library are simultaneously displayed.
If you forget you bookmarked something and saved it in your Diigo library, when you search for the information again on Google, Diigo will automatically display any results from your Diigo library next to the Google search results. This is very helpful as it is much easier to review information you have already read, marked up, and saved than it is to start over.
Bookmarks and annotations are stored in the cloud and can be accessed by any device. (See “Information Management for Clinicians” to learn how to sign up for a free Diigo account, and how to use it.)
STEP 3: STORING YOUR INFORMATION
You may want the option to store full-text information in your personal library. This information was once stored in file cabinets and, more recently, on hard drives and USB flash drives. But information stored with these methods is not available or searchable on multiple devices from any location.
Cloud storage
Cloud storage services meet the need for access to stored information at any time and with any device. Options include Dropbox (www.dropbox.com), Box (www.box.com), Google Drive (drive.google.com), OneDrive (onedrive.live.com), and Evernote (www.evernote.com). Each provides different amounts of free storage and has apps available for most platforms and devices. They provide search tools and the ability to share articles or “folders” with other users. The information on these online drives is “synced” between all devices so that the most up-to-date version is always available to the user regardless of location and device.
Evernote offers multiple folders called notebooks to store and segregate data. The open notebook shown in Figure 2 is named “reference articles.” It has the HPS2-Thrive article from the New England Journal of Medicine (N Engl J Med) tagged with the terms “niacin” and “lipid” to facilitate retrieval. The article was saved from that journal’s website using an Evernote browser extension that allows entire web pages or selections to be saved to Evernote with a single click. Evernote also has a powerful search feature that can find text in images or in PDF documents. In addition, it allows easy sharing of a note or an entire notebook. Once a note or notebook is shared, all parties can add to it. In The Evernote app also allows tablet and smartphone access to the shareable content.
The other services listed here have similar feature sets. Dropbox is perhaps the easiest to adopt, but it offers the least amount of free storage. If you use Microsoft Office software, OneDrive lets you edit documents online, and an Office 365 subscription includes 1 terabyte of storage. Google Drive is probably the best solution for online collaboration, such as coauthoring a paper. Box is one of the few online storage solutions that complies with the Health Insurance Portability and Accountability privacy rules.
PUTTING IT ALL INTO PRACTICE
Once you have become familiar with Inoreader and Diigo (see Information Management for Clinicians for step-by-step instructions), the following scenario shows how to adapt them into an efficient workflow.
Dr. Smith has a smartphone, a tablet, a laptop at home, and a desktop at work. She signs onto Google Chrome as her preferred browser on all devices. This seamlessly loads her Diigo extension when she is using a laptop or desktop. She has set her RSS feeds for her preferred journal TOCs and medical news sites to be downloaded to Inoreader. (For details on how to add a medical journals feed bundle and a medical news feed bundle, visit Information Management for Clinicians.)
Instead of reading paper journals, Dr. Smith browses her customized up-to-date “magazine” on Inoreader. When she comes across a relevant article, she marks it as “favorite.” If she has more time, she visits the web page, reviews the information, and saves it to her Diigo library with annotations if appropriate.
When searching for information on the web, she uses Google—without having to remember if she bookmarked information related to the search term. The Diigo extension in her browser automatically searches and displays information from her Diigo library next to her Google search results, and she can instantly see her notes from the last time she read the article.
Relating this workflow to the example of the dabigatran story above, Dr. Smith sees an article about dabigatran reversal while viewing her N Engl J Med medical news feed on her feed-reader. She marks it as a favorite and tags it with the key terms “cardiology” and “vascular” (Figure 3).
Dr. Smith later returns to look at her favorite feed items and visits the article on the N Engl J Med website. She annotates the article and saves it to her Diigo library (Figure 4).
Since this information is highly relevant to her practice, she also visits the N Engl J Med website to read the full article and the accompanying editorial (Figure 5). She annotates these and also saves them to her Diigo library.
Later, if she searches Google for dabigatran (using her default Google Chrome browser with Diigo extension), she will see the usual Google search results and twinned Diigo bookmarks (Figure 6).
If she clicks on one of the links, the browser will load the web page with all the annotations that she made when she first visited.
CONCLUSION
The strategies and tools we describe here let you create a personalized and constantly updated medical news “magazine,” accessible from any of your web-enabled devices. They can transform the Internet into a searchable notebook of personally selected, annotated information, helping you to more easily stay up to date with advances in your field of practice, and to more easily manage the modern information overload.
Accessing, absorbing, organizing, storing, and retrieving potentially useful medical information is a challenge. Physicians used to try meet this challenge with personal libraries of journal articles in their file cabinet. Today, that is inadequate to combat the deluge of digital information. In 2013, the Institute of Medicine acknowledged this problem, stating, “The ever-increasing volume of evidence makes it difficult for clinicians to maintain a working knowledge of new clinical information.”1
The sheer volume of data has meant that, rather than try to maintain a regular diet of professional reading (proactive scanning2), many of us now seek information only when we need answers to specific clinical questions (reactive searching). This approach promotes lifelong problem-based learning but assumes that we are consciously aware of this need and are aware of the need to search for new information.
We need to constantly scan for new evidence in our area of practice to avoid becoming falsely assured of our knowledge. We also need to be able to find information we have seen before we need to use it. The following are examples of how using this approach could dramatically empower a busy clinician.
On a recent clinic day, a colleague pokes his head into your office and asks, “Have you heard anything about niacin in the news? I have a patient who is asking me if we should discontinue it.” You respond: “Yes, there was something that just came out. I am not sure where I read it or heard about it. Give me a couple of seconds and I can find it.” True to your word, a few seconds later you find and share the latest article on the HPS2-THRIVE trial and a commentary on the results.
Later, your first patient of the day asks you, “When we switched to dabigatran, you mentioned that, unlike warfarin, there was no specific reversal agent. I heard that there is one now?” Instead of being taken aback, you nod your head. “Yes, I saw that recent article showing that the medication rapidly and completely reverses the effect of dabigatran in the majority of patients. While I hope we don’t need it, this is good news, particularly as there did not seem to be any major adverse events.”
Sounds too good to be true? Can this really be you? In this article, we outline an information management strategy (Figure 1) and tools to help busy clinicians stay up to date with new medical evidence in their areas of interest or expertise. In addition, we provide a strategy for leveraging technology to easily retrieve previously viewed information. A future article will specifically show how to best access information at the point of patient care.
THE NEED TO MANAGE INFORMATION
Physicians are expected to practice evidence-based medicine. When faced with a clinical question, we should search for evidence using focused queries of primary and secondary sources such as PubMed or the Cochrane Library. This is an important skill and is appropriate when we take time to look for an evidence-based answer to a specific question. In many cases, it is appropriate to continue with a current practice until newer information has been reviewed and validated.
Unfortunately, indexing and adding new recommendations to these information sources takes time. We may also be unaware that new information is available and may continue to practice as usual until faced with a situation like those outlined above or until we attend a continuing medical education activity, often quite by chance.
Today we can proactively update ourselves in a manner tailored to our own interests and focus and retrieve important information easily when we need it.
A STRATEGY FOR INFORMATION MANAGEMENT
In general, we come across new information in one of three ways:
- Proactive scanning of personalized sources of information—as discussed above, a habit of regular scanning is critical to information management
- Reactive searching for information to answer clinical problems or when doing research
- Incidentally—an e-mail from a colleague, information shared on a social network or encountered while surfing the Internet.
In each case, we may find information that is potentially useful, something we may need to find again in the future. But unless we use this information often, we will not remember the details or may even forget we had seen it. Thus, we need a strategy to store this information so we can retrieve it easily at any time with any device; neuroscientists call this the “externalization” of memory.3 Ideally, even if we forget that we ever saw this information or where we stored it, a search would retrieve the location and details of this formerly viewed information.
In the following sections, we outline steps and tools of a strategy for managing clinical information relevant to your practice.
STEP 1: SETTING UP INFORMATION FEEDS
The first step in this information management strategy is to become aware of relevant new information in your area of practice or research. To do this, you proactively set up feeds of information from reliable and authentic sources. These feeds can be browsed on any computer or smart mobile device.
There are several possibilities for creating these feeds. One option is to subscribe to the table of contents (TOC) of relevant journals via e-mail.
A more versatile and full-featured option is a research site summary (RSS) feed-reader. RSS is a standard for publishing summaries (feeds) of frequently updated content on the World Wide Web, such as journal TOCs and items from medical journal news sites (Table 1 shows what this looks like on screen), as well as aggregators like the American College of Physicians Journal Club. You can subscribe to these using feed-reader software from Feedly (www.feedly.com) or Inoreader (www.inoreader.com), which can be used with any browser on a desktop or laptop. They are also available as apps for mobile devices such as smartphones and tablets. The feed-reader periodically checks for new content and automatically downloads it to the device. Thus, you do not need to check multiple websites for updates or have e-mail inboxes fill with content; the content is delivered to your device for perusal at your convenience. (See online supplement “Information Management for Clinicians” for step-by-step instructions on creating a free Inoreader account and subscribing to feeds.)
RSS feed-readers have several advantages over e-mailed TOCs:
- RSS feeds create a centralized searchable repository of all subscribed information.
- The software keeps track of what you have read and displays only unread items; after a journal TOC e-mail is opened, the entire TOC is marked as having been read.
- You can organize news items into folders by tagging key words.
- Most journals and medical news sites like Medscape and the health section of the New York Times provide RSS feeds at no cost.
- An unlimited number of feed items, or articles, are stored in the cloud and do not affect e-mail storage capacity.
- The feed is automatically updated multiple times a day instead of once a week or once a month.
- In addition, one can create RSS feeds on PubMed for custom searches. Thus, a physician can get automatic regular updates of new articles indexed in MEDLINE in their area of interest.
Users can thus build their own personalized magazine of constantly updated information for access and can search from any web-enabled device. (Note: It is advisable to turn off notifications generated by these apps on mobile devices to reduce distraction.)
STEP 2: BOOKMARKING EVIDENCE
When you find something useful or interesting, bookmarks help you find the information again quickly when you need it. But while the browsers Firefox, Chrome, Internet Explorer, and Safari allow bookmarking, they have significant limitations. Bookmarks may be available only on the device they were created on, and because people use more than one device to go online, they may not remember which device they used to bookmark or view the web page.
Browser bookmarks generally store the address (URL) of the web page and a label that you create, but they do not do much else. Sharing bookmarks with others is also difficult or impossible.
Social bookmarking
Social bookmarking lets you create bookmarks you can share across other devices and with other people. Diigo (www.diigo.com) and Delicious (www.delicious.com) are two social bookmarking services that let you integrate with all popular browsers through a button or toolbar. They allow you to save displayed web pages with labels, descriptions, and tags.
Diigo offers two additional features. It allows web pages to be annotated with highlights and notes. And during a Google search, relevant results from the Diigo library are simultaneously displayed.
If you forget you bookmarked something and saved it in your Diigo library, when you search for the information again on Google, Diigo will automatically display any results from your Diigo library next to the Google search results. This is very helpful as it is much easier to review information you have already read, marked up, and saved than it is to start over.
Bookmarks and annotations are stored in the cloud and can be accessed by any device. (See “Information Management for Clinicians” to learn how to sign up for a free Diigo account, and how to use it.)
STEP 3: STORING YOUR INFORMATION
You may want the option to store full-text information in your personal library. This information was once stored in file cabinets and, more recently, on hard drives and USB flash drives. But information stored with these methods is not available or searchable on multiple devices from any location.
Cloud storage
Cloud storage services meet the need for access to stored information at any time and with any device. Options include Dropbox (www.dropbox.com), Box (www.box.com), Google Drive (drive.google.com), OneDrive (onedrive.live.com), and Evernote (www.evernote.com). Each provides different amounts of free storage and has apps available for most platforms and devices. They provide search tools and the ability to share articles or “folders” with other users. The information on these online drives is “synced” between all devices so that the most up-to-date version is always available to the user regardless of location and device.
Evernote offers multiple folders called notebooks to store and segregate data. The open notebook shown in Figure 2 is named “reference articles.” It has the HPS2-Thrive article from the New England Journal of Medicine (N Engl J Med) tagged with the terms “niacin” and “lipid” to facilitate retrieval. The article was saved from that journal’s website using an Evernote browser extension that allows entire web pages or selections to be saved to Evernote with a single click. Evernote also has a powerful search feature that can find text in images or in PDF documents. In addition, it allows easy sharing of a note or an entire notebook. Once a note or notebook is shared, all parties can add to it. In The Evernote app also allows tablet and smartphone access to the shareable content.
The other services listed here have similar feature sets. Dropbox is perhaps the easiest to adopt, but it offers the least amount of free storage. If you use Microsoft Office software, OneDrive lets you edit documents online, and an Office 365 subscription includes 1 terabyte of storage. Google Drive is probably the best solution for online collaboration, such as coauthoring a paper. Box is one of the few online storage solutions that complies with the Health Insurance Portability and Accountability privacy rules.
PUTTING IT ALL INTO PRACTICE
Once you have become familiar with Inoreader and Diigo (see Information Management for Clinicians for step-by-step instructions), the following scenario shows how to adapt them into an efficient workflow.
Dr. Smith has a smartphone, a tablet, a laptop at home, and a desktop at work. She signs onto Google Chrome as her preferred browser on all devices. This seamlessly loads her Diigo extension when she is using a laptop or desktop. She has set her RSS feeds for her preferred journal TOCs and medical news sites to be downloaded to Inoreader. (For details on how to add a medical journals feed bundle and a medical news feed bundle, visit Information Management for Clinicians.)
Instead of reading paper journals, Dr. Smith browses her customized up-to-date “magazine” on Inoreader. When she comes across a relevant article, she marks it as “favorite.” If she has more time, she visits the web page, reviews the information, and saves it to her Diigo library with annotations if appropriate.
When searching for information on the web, she uses Google—without having to remember if she bookmarked information related to the search term. The Diigo extension in her browser automatically searches and displays information from her Diigo library next to her Google search results, and she can instantly see her notes from the last time she read the article.
Relating this workflow to the example of the dabigatran story above, Dr. Smith sees an article about dabigatran reversal while viewing her N Engl J Med medical news feed on her feed-reader. She marks it as a favorite and tags it with the key terms “cardiology” and “vascular” (Figure 3).
Dr. Smith later returns to look at her favorite feed items and visits the article on the N Engl J Med website. She annotates the article and saves it to her Diigo library (Figure 4).
Since this information is highly relevant to her practice, she also visits the N Engl J Med website to read the full article and the accompanying editorial (Figure 5). She annotates these and also saves them to her Diigo library.
Later, if she searches Google for dabigatran (using her default Google Chrome browser with Diigo extension), she will see the usual Google search results and twinned Diigo bookmarks (Figure 6).
If she clicks on one of the links, the browser will load the web page with all the annotations that she made when she first visited.
CONCLUSION
The strategies and tools we describe here let you create a personalized and constantly updated medical news “magazine,” accessible from any of your web-enabled devices. They can transform the Internet into a searchable notebook of personally selected, annotated information, helping you to more easily stay up to date with advances in your field of practice, and to more easily manage the modern information overload.
- Institute of Medicine (IOM). Best Care at Lower Cost: The Path to Continuously Learning Health Care in America. Washington, DC: National Academies Press; 2013. www.nap.edu/openbook.php?record_id=13444&page=R1. Accessed May 17, 2016.
- Slotnick HB. Physicians’ learning strategies. Chest 2000; 118(suppl 2):18S–23S.
- Levitin DJ. The Organized Mind: Thinking Straight in the Age of Information Overload. New York, NY: Dutton; 2014:528.
- Institute of Medicine (IOM). Best Care at Lower Cost: The Path to Continuously Learning Health Care in America. Washington, DC: National Academies Press; 2013. www.nap.edu/openbook.php?record_id=13444&page=R1. Accessed May 17, 2016.
- Slotnick HB. Physicians’ learning strategies. Chest 2000; 118(suppl 2):18S–23S.
- Levitin DJ. The Organized Mind: Thinking Straight in the Age of Information Overload. New York, NY: Dutton; 2014:528.
KEY POINTS
- The first step in information management is to become aware of relevant new information in your area of practice and set up feeds of information from reliable and authentic sources. These feeds should be accessible from any computer or mobile device and scanned regularly.
- Useful information you come across in various digital streams needs to be bookmarked for future search and retrieval. Social bookmarking lets you create bookmarks you can share across other devices and with other people and retrieve with an Internet search.
- Cloud storage services have apps for most platforms and devices, providing search tools and the ability to share articles or “folders” with other users. The information is “synced” between all devices so that the most up-to-date version is always available, regardless of location and device.

Is there a time limit for systemic menopausal hormone therapy?
The duration of hormone therapy needs to be an individualized decision, shared between the patient and her physician and assessed annually. Quality of life, vasomotor symptoms, current age, time since menopause, hysterectomy status, personal risks (of osteoporosis, breast cancer, heart disease, stroke, venous thromboembolism), and patient preferences need to be considered.
The North American Menopause Society (NAMS) and other organizations recommend that the lowest dose of hormone therapy be used for the shortest duration needed to manage menopausal symptoms.1–4 However, NAMS states that extending the duration of hormone therapy may be appropriate in women who have persistent symptoms or to prevent osteoporosis if the patient cannot tolerate alternative therapies.1
Forty-two percent of postmenopausal women continue to experience vasomotor symptoms at age 60 to 65.5 The median total duration of vasomotor symptoms is 7.4 years, and in black women and women with moderate or severe hot flashes the symptoms typically last 10 years.6 Vasomotor symptoms recur in 50% of women who discontinue hormone therapy, regardless of whether it is stopped abruptly or tapered.1
FACTORS TO CONSIDER WHEN PRESCRIBING HORMONE THERAPY
Bone health
A statement issued in 2013 by seven medical societies said that hormone therapy is effective and appropriate for preventing osteoporosis-related fracture in at-risk women under age 60 or within 10 years of menopause.7
The Women’s Health Initiative,8 a randomized placebo-controlled trial, showed a statistically significant lower risk of vertebral and nonvertebral fracture after 3 years of use of conjugated equine estrogen with medroxyprogesterone acetate than with placebo:
- Hazard ratio 0.76, 95% confidence interval (CI) 0.69–0.83.
It also showed a mean increase of 3.7% (P < .001) in total hip bone mineral density. By the end of the trial intervention, women receiving either this combined therapy or conjugated equine estrogen alone saw a 33% overall reduction in hip fracture risk. The absolute risk reduction was 5 per 10,000 years of use.9
Karim et al,10 in a large observational study that followed initial hormone therapy users over 6.5 years, found that those who stopped it had a 55% greater risk of hip fracture and experienced significant bone loss as measured by bone mineral density compared with women who continued hormone therapy, and that the protective effects of hormone therapy disappeared as early as 2 years after stopping treatment.10
NAMS also recommends that women with premature menopause (before age 40) be offered and encouraged to use hormone therapy to preserve bone density and manage vasomotor symptoms until the age of natural menopause (age 51).1,11
Cardiovascular health
Large observational studies have found that hormone therapy is associated with a 30% to 50% lower cardiovascular risk.12 Randomized controlled trials of hormone therapy for 7 to 11 years suggest that coronary heart disease risk is modified by age and time since menopause.13,14
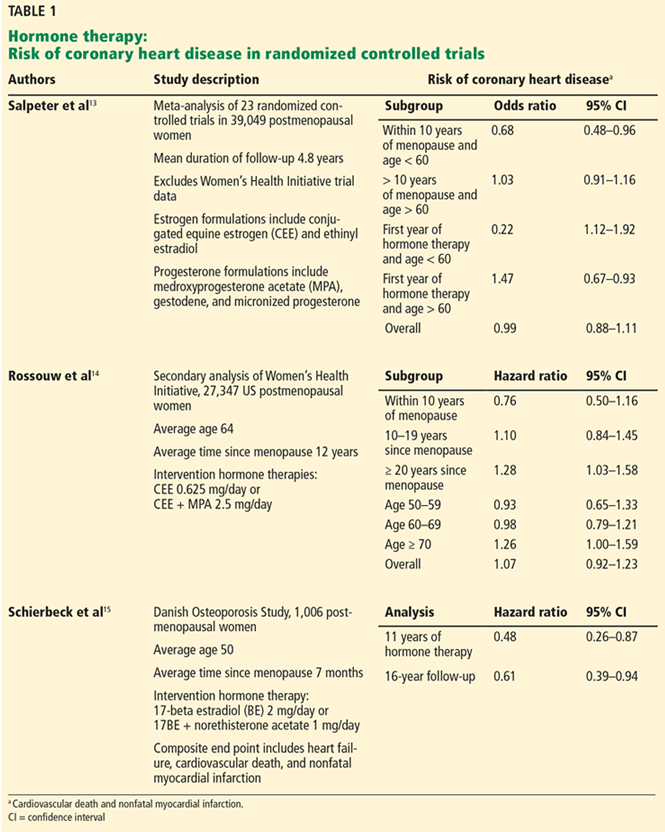
The Women’s Health Initiative and other randomized controlled trials suggest a lower risk of coronary heart disease in women who begin hormone therapy before age 60 and within 10 years of the onset of menopause, but an increased risk for women over age 60 and more than 10 years since menopause. However, several of these trends have not reached statistical significance (Table 1).13–15
The Women’s Health Initiative9 published its long-term follow-up results in 2013, with data on both the intervention phase (median of 7.2 years for estrogen-only therapy and 5.6 years for estrogen-progestin therapy) and the post-stopping phase (median 6.6 years for the estrogen-only group and 8.2 years for the estrogen-progestin group), with a total cumulative follow-up of 13 years. The overall 13-year cumulative absolute risk of coronary heart disease was 4 fewer events per 10,000 years of estrogen-only therapy and 3 additional events per 10,000 years of estrogen-progestin therapy. Neither result was statistically significant:
- Hazard ratio with estrogen-only use 0.94, 95% CI 0.82–1.09
- Hazard ratio with estrogen-progestin use 1.09, 95% CI 0.92–1.24.
The Danish Osteoporosis Study was the first randomized controlled trial of hormone therapy in women ages 45 through 58 who were recently menopausal (average within 7 months of menopause).15 Women assigned to hormone therapy in the form of oral estradiol with or without norethisterone (known as norethindrone in the United States) had a statistically significant lower risk of the primary composite end point of heart failure and myocardial infarction after 11 years of hormone therapy, and this finding persisted through 16 years of follow-up (Table 1).
Stroke
Overall stroke risk was significantly increased with hormone therapy in the Women’s Health Initiative trial (hazard ratio 1.32, 95% CI 1.12–1.56); however, the absolute increase in risk was small in both estrogen-alone and estrogen-progestin therapy users, 11 and 8 events, respectively, among 10,000 users. Younger women (ages 50–59) saw a nonsignificantly lower risk (2 fewer cases per 10,000 years of use).14 After 13 years of cumulative follow-up (combined intervention and follow-up phase), the risk of stroke persisted at 5 cases per 10,000 users for both arms, but only the estrogen-progestin results were statistically significant.9
The Danish Osteoporosis Study15 found no increased risk of stroke after 16 years of follow-up in recently menopausal women:
- Hazard ratio 0.89, 95% CI 0.48–1.65.
Venous thromboembolism
Data from both observational and randomized controlled trials demonstrate an increased risk of venous thromboembolism with oral hormone therapy, and the risk appears to be highest during the first few years of use.1 The pooled cohort from the Women’s Health Initiative had 18 additional cases of venous thromboembolism per 10,000 women in estrogen-progestin users compared with nonusers, and 7 additional cases in those using estrogen-only therapy.
Breast health
Observational studies and randomized controlled trials have provided data on longer use of hormone therapy and breast cancer risk, but the true magnitude of this risk is unclear.
The Danish Osteoporosis Study,15 in a younger cohort of women, showed no increased risk of breast cancer after 16 years of follow-up:
- Hazard ratio 0.90, 95% CI 0.52–1.57.
The Women’s Health Initiative9 showed a statistically nonsignificant lower risk of breast cancer in women of all ages exposed to conjugated equine estrogen alone for 7.1 years (6 fewer cases per 10,000 women-years of use), and after 6 years of follow-up this developed statistical significance:
- Hazard ratio 0.79, 95% CI 0.65–0.97.
In contrast, those using conjugated equine estrogen plus medroxyprogesterone acetate had a statistically nonsignificant increase in the risk of new breast cancer after 3 to 5 years:
- 3-year relative risk 1.26, 95% CI 0.73–2.20
- 5-year relative risk 1.99, 95% CI 1.18–3.35
- Absolute risk 8 cases per 10,000 women-years of use.
The increased risk of breast cancer significantly declined within 3 years after stopping hormone therapy.
However, even after stopping hormone therapy, there remains a statistically small but significant increased risk of breast cancer, as demonstrated in the postintervention 13-year follow-up data on breast cancer risk and estrogen-progestin use from the Women’s Health Initiative9:
- Hazard ratio 1.28, 95% CI 1.11–1.48
- Absolute cumulative risk 9 cases per 10,000 women-years of use.
The Nurses’ Health Study, an observational study, prospectively followed 11,508 hysterectomized women on estrogen therapy and found that breast cancer risk increased with longer duration of use. An analysis by Chen et al16 found a trend toward increased breast cancer risk after 10 years of estrogen therapy, but this did not become statistically significant until 20 years of ongoing estrogen use. The risk of estrogen receptor-positive and progesterone receptor-positive breast cancer became statistically significant earlier, after 15 years. The relative risk associated with using estrogen for more than 15 years was 1.18, and the risk with using it for more than 20 years was 1.42.16
To put this in perspective, Chen et al17 found a similar breast cancer risk with alcohol consumption. The relative risk of invasive breast cancer was 1.15 in women who drank 3 to 6 servings of alcohol per week, 1 serving being equivalent to 4 oz of wine, which contains 11 g of alcohol.
Mortality
Studies have suggested that hormone therapy users have a lower mortality rate, even with long-term use.
A meta-analysis18 of 8 observational trials and 19 randomized controlled trials found that younger women (average age 54) on hormone therapy had a 28% lower total mortality rate compared with women not taking hormone therapy:
- Relative risk 0.72, 95% credible interval 0.62–0.82.
The Women’s Health Initiative19 suggested that the mortality rate was 30% lower in hormone therapy users younger than age 60 than in similar nonusers, though this difference did not reach statistical significance.
- Relative risk with estrogen-only therapy: 0.71, 95% CI 0.46–1.11
- Relative risk with combined estrogen-progestin therapy 0.69, 95% CI 0.44–1.07.
The Danish Osteoporosis Study,15 at 16 years of follow-up, similarly demonstrated a 34% lower mortality rate in hormone therapy users, which was not statistically significant:
- Relative risk 0.66, 95% CI 0.41–1.08.
A Cochrane review20 in 2015 found that the subgroup of women who started hormone therapy before age 60 or within 10 years of menopause saw an overall benefit in terms of survival and lower risk of coronary heart disease: RR 0.70, 95% CI 0.52–0.95 (moderate-quality evidence).
TYPE OF FORMULATION
Compared with estrogen-progestin therapy, estrogen-only therapy has a more favorable risk profile in terms of coronary heart disease and breast cancer, although stroke risk remains elevated in users of conjugated equine estrogen with or without medroxyprogesterone acetate.
There is limited evidence directly comparing different formulations of hormone therapy, although they all effectively treat vasomotor symptoms.1
Oral vs transdermal formulations
Canonico et al,21 in a meta-analysis of observational studies, found that oral estrogen was associated with a higher risk of venous thromboembolism than transdermal estrogen:
- Relative risk with oral estrogen 2.5, 95% CI 1.9–3.4
- Relative risk with transdermal estrogen 1.2, 95% CI 0.9–1.7.
The Estrogen and Thromboembolism Risk (ESTHER) study22 was a multicenter case-control study of women ages 45 to 70 that assessed risk of venous thromboembolism in oral vs transdermal estrogen users. Compared with women not taking hormone therapy, current users of oral estrogen had a significantly higher risk of venous thromboembolism, while transdermal estrogen users did not:
- Odds ratio with oral estrogen 4.2, 95% CI 1.5–11.6
- Odds ratio with transdermal estrogen 0.9, 95% CI 0.4–2.1.
The Kronos Early Estrogen Prevention Study (KEEPS)23 did not support these findings. This 4-year randomized controlled trial, published in 2014, was designed to assess the risk of atherosclerosis progression with early menopause initiation of placebo vs low-dose oral hormone therapy (conjugated equine estrogen 0.45 mg daily with cyclical micronized progesterone) or transdermal hormone therapy (estradiol 50 µg/week with cyclical micronized progesterone).
In the 727 women in the study, there was one transient ischemic attack in the oral hormone therapy group, one unconfirmed stroke in the transdermal hormone therapy group, and one case of venous thromboembolism in each group, findings that were underpowered for statistical significance. Both oral and transdermal hormonal therapy had neutral effects on atherosclerosis progression, as assessed by arterial imaging. Transdermal hormone therapy was associated with improvements in markers of insulin resistance and was not associated with an increase in triglycerides, C-reactive protein, or sex hormone-binding globulin, as would be expected with transdermal circumvention of the first-pass hepatic effect.
BALANCING THE RISKS AND BENEFITS FOR THE PATIENT
The most effective treatment for vasomotor symptoms in women at any age is hormone therapy, and the benefits are more likely to outweigh risks when initiated before age 60 or within 10 years of menopause.7 The Women’s Health Initiative randomized study was limited to 5.6 to 7.2 years of hormone therapy (13 years of cumulative follow-up), and the Danish Osteoporosis Study was limited to 11 years of use (16 years cumulative follow-up).
The coronary heart disease outcomes for longer durations of therapy remain uncertain. There is a small but statistically significant increased risk of stroke and venous thromboembolism with oral hormone therapy, and breast cancer risk is associated with long-term estrogen-progestin use.

Patients on hormone therapy should be evaluated annually regarding the need for ongoing therapy. Persistent moderate-severe vasomotor symptoms, quality of life benefits of hormone therapy, contraindications to its use (Table 2), and patient preference need to be assessed as well as baseline risks of cardiovascular disease, breast cancer, and fracture.
Risk calculators may facilitate the shared decision-making process. Examples are:
- The American College of Cardiology/American Heart association risk calculator for cardiovascular disease24 (www.cvriskcalculator.com)
- The World Health Organization Fracture Risk Assessment Tool (FRAX)25
(www.shef.ac.uk/FRAX/tool.jsp)
- The Gail model for breast cancer risk26 (www.cancer.gov/bcrisktool/).
- MenoPro, a menopause decision-support algorithm and companion mobile app developed by NAMS to help direct treatment decisions based on the 10-year risk of atherosclerotic cardiovascular disease (www.menopause.org/for-professionals/-i-menopro-i-mobile-app).27
The discussion of the risks of hormone therapy with patients should incorporate the perspective of absolute risk. For example, a woman wishing to continue estrogen-progestin therapy should be told that the Women’s Health Initiative data suggest that, after 5 years of use, breast cancer risk may be increased by 8 additional cases per 10,000 users per year. According to the World Health Organization, this magnitude of risk is defined as rare (less than 1 event per 1,000 women).28
A strategy of prescribing the lowest dose to achieve the desired clinical benefits is prudent and recommended.1–3 Table 3 outlines the estrogen formulations now available in the United States, with their doses and formulations.
Unless contraindications develop (Table 2), patients may elect to continue hormone therapy if its benefits outweigh its risks. The American College of Obstetricians and Gynecologists (ACOG) 2014 practice recommendations for management of menopausal symptoms31 and the 2015 NAMS statement both recommend that hormone therapy not be discontinued based solely on a woman’s age.29
Hormone therapy is on the Beer’s list of potentially inappropriate medications for older adults,30 which remains a hurdle to its long-term use and seems to be at odds with these ACOG and NAMS statements.
Patients who choose to discontinue hormone therapy need to be monitored for persistent bothersome vasomotor symptoms, bone loss, osteoporosis, and the genitourinary syndrome of menopause (previously referred to as vulvovaginal atrophy)31 and offered alternative therapies if needed.
- North American Menopause Society. The 2012 hormone therapy position statement of: The North American Menopause Society. Menopause 2012; 19:257–271.
- American College of Obstetricians and Gynecologists. Practice Bulletin No. 141: Management of menopausal symptoms. Obstet Gynecol 2014; 123:202–216.
- Stuenkel CA, Davis SR, Gompel A, et al. Treatment of symptoms of the menopause: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2015; 100:3975–4011.
- de Villiers TJ, Pines A, Panay N, et al; International Menopause Society. Updated 2013 International Menopause Society recommendations on menopausal hormone therapy and preventive strategies for midlife health. Climacteric 2013; 16:316–337.
- Gartoulla P, Worsley R, Robin J, Davis S. Moderate to severe vasomotor and sexual symptoms remain problematic for women aged 60 to 65 years. Menopause 2015; 22:694–701.
- Avis NE, Crawford SL, Greendale G, et al. Duration of menopausal vasomotor symptoms across the menopause transition. JAMA Intern Med 2015; 175:531–539.
- de Villiers TJ, Gass ML, Haines CJ, et al. Global consensus statement on menopausal hormone therapy. Climacteric 2013; 16:203–204.
- Cauley J, Robbins J, Chen Z, et al. Effects of estrogen plus progestin on risk of fracture and bone mineral density: the Women’s Health Initiative randomized trial. JAMA 2003; 290:1729–1738.
- Manson J, Chlebowski R, Stefanick M, et al. Menopausal hormone therapy and health outcomes during the intervention and extended poststopping phases of the Women’s Health Initiative randomized trials. JAMA 2013; 310:1353–1368.
- Karim R, Dell RM, Greene DF, et al. Hip fracture in postmenopausal women after cessation of hormone therapy: results from a prospective study in a large health management organization. Menopause 2011; 18:1172–1177.
- Shifren J, Gass M, and the NAMS Recommendations for Clinical Care of Midlife Women Working Group. The North American Menopause Society recommendations for clinical care of midlife women. Menopause 2014; 21:1038–1062.
- Hodis HN, Mack WJ. Hormone replacement therapy and the association with coronary heart disease and overall mortality: clinical application of the timing hypothesis. J Steroid Biochem Mol Biol 2014; 142:68–75.
- Salpeter SR, Walsh JM, Greyber E, et al. Brief report: coronary heart disease events associated with hormone therapy in younger and older women. A meta-analysis. J Gen Intern Med 2006; 21:363–366.
- Rossouw JE, Prentice RL, Manson JE, et al. Postmenopausal hormone therapy and risk of cardiovascular disease by age and years since menopause. JAMA 2007; 297:1465–1477.
- Schierbeck LL, Rejnmark L, Tofteng CL, et al. Effect of hormone replacement therapy on cardiovascular events in recently postmenopausal women: randomised trial. BMJ 2012; 345:e6409.
- Chen WY, Manson JE, Hankinson SE, et al. Unopposed estrogen therapy and the risk of breast cancer. Arch Intern Med 2006; 166:1027–1032.
- Chen W, Rosner B, Hankinson SE, et al. Moderate alcohol consumption during adult life, drinking patterns, and breast cancer risk. JAMA 2011; 306:1884–1890.
- Salpeter SR, Cheng J, Thabane L, et al. Bayesian meta-analysis of hormone therapy and mortality in younger postmenopausal women. Am J Med 2009; 122:1016–1022.
- Hodis HN, Collins P, Mack WJ, Schierbeck LL. The timing hypothesis for coronary heart disease prevention with hormone therapy: past, present and future in perspective. Climacteric 2012; 15:217–228.
- Boardman HM, Hartley L, Eisinga A, et al. Hormone therapy for preventing cardiovascular disease in post-menopausal women. Cochrane Database Syst Rev 2015;3:CD002229.
- Canonico M, Plu-Bureau G, Lowe GD, et al. Hormone replacement therapy and risk of venous thromboembolism in postmenopausal women: systemic review and meta-analysis. BMJ 2008; 336:1227–1231.
- Canonico M, Oger E, Plu-Bureau G, et al; Estrogen and Thromboembolism Risk (ESTHER) Study Group. Hormone therapy and venous thromboembolism among postmenopausal women: impact of the route of estrogen administration and progestogens: the ESTHER study. Circulation 2007; 115:840–845.
- Harman S, Black D, Naftolin F, et al. Arterial imaging outcomes and cardiovascular risk factors in recently menopausal women. Ann Intern Med 2014; 161:249–260.
- Goff DC Jr, Lloyd-Jones DM, Bennett G, et al. 2013 ACC/AHA guideline on the assessment of cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2014; 63:2935–2959.
- World Health Organization Collaborating Centre for Metabolic Bone Diseases. FRAX WHO fracture risk assessment tool. www.shef.ac.uk/FRAX/. Accessed May 27, 2016.
- Gail M, Brinton L, Byar D, et al. Projecting individualized probabilities of developing breast cancer for white females who are being examined annually. J Natl Cancer Inst 1989; 81:1879–1886.
- Manson J, Ames J, Shapiro M, et al. Algorithm and mobile app for menopausal symptom management and hormonal/non-hormonal therapy decision making: a clinical decision-support tool from the North American Menopause Society. Menopause 2015; 22:247–253.
- Hodis HN, Mack WJ. Postmenopausal hormone therapy in clinical perspective. Menopause 2007; 14:944–957.
- North American Menopause Society. The North American Menopause Society statement on continuing use of systemic hormone therapy after the age of 65. Menopause 2015; 22:693.
- American Geriatrics Society 2015 Beers Criteria Update Expert Panel. American Geriatrics Society 2015 updated Beers criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc 2015; 63:2227–2246.
- Portman DJ, Gass ML; Vulvovaginal Atrophy Terminology Consensus Conference Panel. Genitourinary syndrome of menopause: new terminology for vulvovaginal atrophy from the International Society for the Study of Women’s Sexual Health and the North American Menopause Society. Menopause 2014; 21:1063–1068.
The duration of hormone therapy needs to be an individualized decision, shared between the patient and her physician and assessed annually. Quality of life, vasomotor symptoms, current age, time since menopause, hysterectomy status, personal risks (of osteoporosis, breast cancer, heart disease, stroke, venous thromboembolism), and patient preferences need to be considered.
The North American Menopause Society (NAMS) and other organizations recommend that the lowest dose of hormone therapy be used for the shortest duration needed to manage menopausal symptoms.1–4 However, NAMS states that extending the duration of hormone therapy may be appropriate in women who have persistent symptoms or to prevent osteoporosis if the patient cannot tolerate alternative therapies.1
Forty-two percent of postmenopausal women continue to experience vasomotor symptoms at age 60 to 65.5 The median total duration of vasomotor symptoms is 7.4 years, and in black women and women with moderate or severe hot flashes the symptoms typically last 10 years.6 Vasomotor symptoms recur in 50% of women who discontinue hormone therapy, regardless of whether it is stopped abruptly or tapered.1
FACTORS TO CONSIDER WHEN PRESCRIBING HORMONE THERAPY
Bone health
A statement issued in 2013 by seven medical societies said that hormone therapy is effective and appropriate for preventing osteoporosis-related fracture in at-risk women under age 60 or within 10 years of menopause.7
The Women’s Health Initiative,8 a randomized placebo-controlled trial, showed a statistically significant lower risk of vertebral and nonvertebral fracture after 3 years of use of conjugated equine estrogen with medroxyprogesterone acetate than with placebo:
- Hazard ratio 0.76, 95% confidence interval (CI) 0.69–0.83.
It also showed a mean increase of 3.7% (P < .001) in total hip bone mineral density. By the end of the trial intervention, women receiving either this combined therapy or conjugated equine estrogen alone saw a 33% overall reduction in hip fracture risk. The absolute risk reduction was 5 per 10,000 years of use.9
Karim et al,10 in a large observational study that followed initial hormone therapy users over 6.5 years, found that those who stopped it had a 55% greater risk of hip fracture and experienced significant bone loss as measured by bone mineral density compared with women who continued hormone therapy, and that the protective effects of hormone therapy disappeared as early as 2 years after stopping treatment.10
NAMS also recommends that women with premature menopause (before age 40) be offered and encouraged to use hormone therapy to preserve bone density and manage vasomotor symptoms until the age of natural menopause (age 51).1,11
Cardiovascular health
Large observational studies have found that hormone therapy is associated with a 30% to 50% lower cardiovascular risk.12 Randomized controlled trials of hormone therapy for 7 to 11 years suggest that coronary heart disease risk is modified by age and time since menopause.13,14
The Women’s Health Initiative and other randomized controlled trials suggest a lower risk of coronary heart disease in women who begin hormone therapy before age 60 and within 10 years of the onset of menopause, but an increased risk for women over age 60 and more than 10 years since menopause. However, several of these trends have not reached statistical significance (Table 1).13–15
The Women’s Health Initiative9 published its long-term follow-up results in 2013, with data on both the intervention phase (median of 7.2 years for estrogen-only therapy and 5.6 years for estrogen-progestin therapy) and the post-stopping phase (median 6.6 years for the estrogen-only group and 8.2 years for the estrogen-progestin group), with a total cumulative follow-up of 13 years. The overall 13-year cumulative absolute risk of coronary heart disease was 4 fewer events per 10,000 years of estrogen-only therapy and 3 additional events per 10,000 years of estrogen-progestin therapy. Neither result was statistically significant:
- Hazard ratio with estrogen-only use 0.94, 95% CI 0.82–1.09
- Hazard ratio with estrogen-progestin use 1.09, 95% CI 0.92–1.24.
The Danish Osteoporosis Study was the first randomized controlled trial of hormone therapy in women ages 45 through 58 who were recently menopausal (average within 7 months of menopause).15 Women assigned to hormone therapy in the form of oral estradiol with or without norethisterone (known as norethindrone in the United States) had a statistically significant lower risk of the primary composite end point of heart failure and myocardial infarction after 11 years of hormone therapy, and this finding persisted through 16 years of follow-up (Table 1).
Stroke
Overall stroke risk was significantly increased with hormone therapy in the Women’s Health Initiative trial (hazard ratio 1.32, 95% CI 1.12–1.56); however, the absolute increase in risk was small in both estrogen-alone and estrogen-progestin therapy users, 11 and 8 events, respectively, among 10,000 users. Younger women (ages 50–59) saw a nonsignificantly lower risk (2 fewer cases per 10,000 years of use).14 After 13 years of cumulative follow-up (combined intervention and follow-up phase), the risk of stroke persisted at 5 cases per 10,000 users for both arms, but only the estrogen-progestin results were statistically significant.9
The Danish Osteoporosis Study15 found no increased risk of stroke after 16 years of follow-up in recently menopausal women:
- Hazard ratio 0.89, 95% CI 0.48–1.65.
Venous thromboembolism
Data from both observational and randomized controlled trials demonstrate an increased risk of venous thromboembolism with oral hormone therapy, and the risk appears to be highest during the first few years of use.1 The pooled cohort from the Women’s Health Initiative had 18 additional cases of venous thromboembolism per 10,000 women in estrogen-progestin users compared with nonusers, and 7 additional cases in those using estrogen-only therapy.
Breast health
Observational studies and randomized controlled trials have provided data on longer use of hormone therapy and breast cancer risk, but the true magnitude of this risk is unclear.
The Danish Osteoporosis Study,15 in a younger cohort of women, showed no increased risk of breast cancer after 16 years of follow-up:
- Hazard ratio 0.90, 95% CI 0.52–1.57.
The Women’s Health Initiative9 showed a statistically nonsignificant lower risk of breast cancer in women of all ages exposed to conjugated equine estrogen alone for 7.1 years (6 fewer cases per 10,000 women-years of use), and after 6 years of follow-up this developed statistical significance:
- Hazard ratio 0.79, 95% CI 0.65–0.97.
In contrast, those using conjugated equine estrogen plus medroxyprogesterone acetate had a statistically nonsignificant increase in the risk of new breast cancer after 3 to 5 years:
- 3-year relative risk 1.26, 95% CI 0.73–2.20
- 5-year relative risk 1.99, 95% CI 1.18–3.35
- Absolute risk 8 cases per 10,000 women-years of use.
The increased risk of breast cancer significantly declined within 3 years after stopping hormone therapy.
However, even after stopping hormone therapy, there remains a statistically small but significant increased risk of breast cancer, as demonstrated in the postintervention 13-year follow-up data on breast cancer risk and estrogen-progestin use from the Women’s Health Initiative9:
- Hazard ratio 1.28, 95% CI 1.11–1.48
- Absolute cumulative risk 9 cases per 10,000 women-years of use.
The Nurses’ Health Study, an observational study, prospectively followed 11,508 hysterectomized women on estrogen therapy and found that breast cancer risk increased with longer duration of use. An analysis by Chen et al16 found a trend toward increased breast cancer risk after 10 years of estrogen therapy, but this did not become statistically significant until 20 years of ongoing estrogen use. The risk of estrogen receptor-positive and progesterone receptor-positive breast cancer became statistically significant earlier, after 15 years. The relative risk associated with using estrogen for more than 15 years was 1.18, and the risk with using it for more than 20 years was 1.42.16
To put this in perspective, Chen et al17 found a similar breast cancer risk with alcohol consumption. The relative risk of invasive breast cancer was 1.15 in women who drank 3 to 6 servings of alcohol per week, 1 serving being equivalent to 4 oz of wine, which contains 11 g of alcohol.
Mortality
Studies have suggested that hormone therapy users have a lower mortality rate, even with long-term use.
A meta-analysis18 of 8 observational trials and 19 randomized controlled trials found that younger women (average age 54) on hormone therapy had a 28% lower total mortality rate compared with women not taking hormone therapy:
- Relative risk 0.72, 95% credible interval 0.62–0.82.
The Women’s Health Initiative19 suggested that the mortality rate was 30% lower in hormone therapy users younger than age 60 than in similar nonusers, though this difference did not reach statistical significance.
- Relative risk with estrogen-only therapy: 0.71, 95% CI 0.46–1.11
- Relative risk with combined estrogen-progestin therapy 0.69, 95% CI 0.44–1.07.
The Danish Osteoporosis Study,15 at 16 years of follow-up, similarly demonstrated a 34% lower mortality rate in hormone therapy users, which was not statistically significant:
- Relative risk 0.66, 95% CI 0.41–1.08.
A Cochrane review20 in 2015 found that the subgroup of women who started hormone therapy before age 60 or within 10 years of menopause saw an overall benefit in terms of survival and lower risk of coronary heart disease: RR 0.70, 95% CI 0.52–0.95 (moderate-quality evidence).
TYPE OF FORMULATION
Compared with estrogen-progestin therapy, estrogen-only therapy has a more favorable risk profile in terms of coronary heart disease and breast cancer, although stroke risk remains elevated in users of conjugated equine estrogen with or without medroxyprogesterone acetate.
There is limited evidence directly comparing different formulations of hormone therapy, although they all effectively treat vasomotor symptoms.1
Oral vs transdermal formulations
Canonico et al,21 in a meta-analysis of observational studies, found that oral estrogen was associated with a higher risk of venous thromboembolism than transdermal estrogen:
- Relative risk with oral estrogen 2.5, 95% CI 1.9–3.4
- Relative risk with transdermal estrogen 1.2, 95% CI 0.9–1.7.
The Estrogen and Thromboembolism Risk (ESTHER) study22 was a multicenter case-control study of women ages 45 to 70 that assessed risk of venous thromboembolism in oral vs transdermal estrogen users. Compared with women not taking hormone therapy, current users of oral estrogen had a significantly higher risk of venous thromboembolism, while transdermal estrogen users did not:
- Odds ratio with oral estrogen 4.2, 95% CI 1.5–11.6
- Odds ratio with transdermal estrogen 0.9, 95% CI 0.4–2.1.
The Kronos Early Estrogen Prevention Study (KEEPS)23 did not support these findings. This 4-year randomized controlled trial, published in 2014, was designed to assess the risk of atherosclerosis progression with early menopause initiation of placebo vs low-dose oral hormone therapy (conjugated equine estrogen 0.45 mg daily with cyclical micronized progesterone) or transdermal hormone therapy (estradiol 50 µg/week with cyclical micronized progesterone).
In the 727 women in the study, there was one transient ischemic attack in the oral hormone therapy group, one unconfirmed stroke in the transdermal hormone therapy group, and one case of venous thromboembolism in each group, findings that were underpowered for statistical significance. Both oral and transdermal hormonal therapy had neutral effects on atherosclerosis progression, as assessed by arterial imaging. Transdermal hormone therapy was associated with improvements in markers of insulin resistance and was not associated with an increase in triglycerides, C-reactive protein, or sex hormone-binding globulin, as would be expected with transdermal circumvention of the first-pass hepatic effect.
BALANCING THE RISKS AND BENEFITS FOR THE PATIENT
The most effective treatment for vasomotor symptoms in women at any age is hormone therapy, and the benefits are more likely to outweigh risks when initiated before age 60 or within 10 years of menopause.7 The Women’s Health Initiative randomized study was limited to 5.6 to 7.2 years of hormone therapy (13 years of cumulative follow-up), and the Danish Osteoporosis Study was limited to 11 years of use (16 years cumulative follow-up).
The coronary heart disease outcomes for longer durations of therapy remain uncertain. There is a small but statistically significant increased risk of stroke and venous thromboembolism with oral hormone therapy, and breast cancer risk is associated with long-term estrogen-progestin use.
Patients on hormone therapy should be evaluated annually regarding the need for ongoing therapy. Persistent moderate-severe vasomotor symptoms, quality of life benefits of hormone therapy, contraindications to its use (Table 2), and patient preference need to be assessed as well as baseline risks of cardiovascular disease, breast cancer, and fracture.
Risk calculators may facilitate the shared decision-making process. Examples are:
- The American College of Cardiology/American Heart association risk calculator for cardiovascular disease24 (www.cvriskcalculator.com)
- The World Health Organization Fracture Risk Assessment Tool (FRAX)25
(www.shef.ac.uk/FRAX/tool.jsp)
- The Gail model for breast cancer risk26 (www.cancer.gov/bcrisktool/).
- MenoPro, a menopause decision-support algorithm and companion mobile app developed by NAMS to help direct treatment decisions based on the 10-year risk of atherosclerotic cardiovascular disease (www.menopause.org/for-professionals/-i-menopro-i-mobile-app).27
The discussion of the risks of hormone therapy with patients should incorporate the perspective of absolute risk. For example, a woman wishing to continue estrogen-progestin therapy should be told that the Women’s Health Initiative data suggest that, after 5 years of use, breast cancer risk may be increased by 8 additional cases per 10,000 users per year. According to the World Health Organization, this magnitude of risk is defined as rare (less than 1 event per 1,000 women).28
A strategy of prescribing the lowest dose to achieve the desired clinical benefits is prudent and recommended.1–3 Table 3 outlines the estrogen formulations now available in the United States, with their doses and formulations.
Unless contraindications develop (Table 2), patients may elect to continue hormone therapy if its benefits outweigh its risks. The American College of Obstetricians and Gynecologists (ACOG) 2014 practice recommendations for management of menopausal symptoms31 and the 2015 NAMS statement both recommend that hormone therapy not be discontinued based solely on a woman’s age.29
Hormone therapy is on the Beer’s list of potentially inappropriate medications for older adults,30 which remains a hurdle to its long-term use and seems to be at odds with these ACOG and NAMS statements.
Patients who choose to discontinue hormone therapy need to be monitored for persistent bothersome vasomotor symptoms, bone loss, osteoporosis, and the genitourinary syndrome of menopause (previously referred to as vulvovaginal atrophy)31 and offered alternative therapies if needed.
The duration of hormone therapy needs to be an individualized decision, shared between the patient and her physician and assessed annually. Quality of life, vasomotor symptoms, current age, time since menopause, hysterectomy status, personal risks (of osteoporosis, breast cancer, heart disease, stroke, venous thromboembolism), and patient preferences need to be considered.
The North American Menopause Society (NAMS) and other organizations recommend that the lowest dose of hormone therapy be used for the shortest duration needed to manage menopausal symptoms.1–4 However, NAMS states that extending the duration of hormone therapy may be appropriate in women who have persistent symptoms or to prevent osteoporosis if the patient cannot tolerate alternative therapies.1
Forty-two percent of postmenopausal women continue to experience vasomotor symptoms at age 60 to 65.5 The median total duration of vasomotor symptoms is 7.4 years, and in black women and women with moderate or severe hot flashes the symptoms typically last 10 years.6 Vasomotor symptoms recur in 50% of women who discontinue hormone therapy, regardless of whether it is stopped abruptly or tapered.1
FACTORS TO CONSIDER WHEN PRESCRIBING HORMONE THERAPY
Bone health
A statement issued in 2013 by seven medical societies said that hormone therapy is effective and appropriate for preventing osteoporosis-related fracture in at-risk women under age 60 or within 10 years of menopause.7
The Women’s Health Initiative,8 a randomized placebo-controlled trial, showed a statistically significant lower risk of vertebral and nonvertebral fracture after 3 years of use of conjugated equine estrogen with medroxyprogesterone acetate than with placebo:
- Hazard ratio 0.76, 95% confidence interval (CI) 0.69–0.83.
It also showed a mean increase of 3.7% (P < .001) in total hip bone mineral density. By the end of the trial intervention, women receiving either this combined therapy or conjugated equine estrogen alone saw a 33% overall reduction in hip fracture risk. The absolute risk reduction was 5 per 10,000 years of use.9
Karim et al,10 in a large observational study that followed initial hormone therapy users over 6.5 years, found that those who stopped it had a 55% greater risk of hip fracture and experienced significant bone loss as measured by bone mineral density compared with women who continued hormone therapy, and that the protective effects of hormone therapy disappeared as early as 2 years after stopping treatment.10
NAMS also recommends that women with premature menopause (before age 40) be offered and encouraged to use hormone therapy to preserve bone density and manage vasomotor symptoms until the age of natural menopause (age 51).1,11
Cardiovascular health
Large observational studies have found that hormone therapy is associated with a 30% to 50% lower cardiovascular risk.12 Randomized controlled trials of hormone therapy for 7 to 11 years suggest that coronary heart disease risk is modified by age and time since menopause.13,14
The Women’s Health Initiative and other randomized controlled trials suggest a lower risk of coronary heart disease in women who begin hormone therapy before age 60 and within 10 years of the onset of menopause, but an increased risk for women over age 60 and more than 10 years since menopause. However, several of these trends have not reached statistical significance (Table 1).13–15
The Women’s Health Initiative9 published its long-term follow-up results in 2013, with data on both the intervention phase (median of 7.2 years for estrogen-only therapy and 5.6 years for estrogen-progestin therapy) and the post-stopping phase (median 6.6 years for the estrogen-only group and 8.2 years for the estrogen-progestin group), with a total cumulative follow-up of 13 years. The overall 13-year cumulative absolute risk of coronary heart disease was 4 fewer events per 10,000 years of estrogen-only therapy and 3 additional events per 10,000 years of estrogen-progestin therapy. Neither result was statistically significant:
- Hazard ratio with estrogen-only use 0.94, 95% CI 0.82–1.09
- Hazard ratio with estrogen-progestin use 1.09, 95% CI 0.92–1.24.
The Danish Osteoporosis Study was the first randomized controlled trial of hormone therapy in women ages 45 through 58 who were recently menopausal (average within 7 months of menopause).15 Women assigned to hormone therapy in the form of oral estradiol with or without norethisterone (known as norethindrone in the United States) had a statistically significant lower risk of the primary composite end point of heart failure and myocardial infarction after 11 years of hormone therapy, and this finding persisted through 16 years of follow-up (Table 1).
Stroke
Overall stroke risk was significantly increased with hormone therapy in the Women’s Health Initiative trial (hazard ratio 1.32, 95% CI 1.12–1.56); however, the absolute increase in risk was small in both estrogen-alone and estrogen-progestin therapy users, 11 and 8 events, respectively, among 10,000 users. Younger women (ages 50–59) saw a nonsignificantly lower risk (2 fewer cases per 10,000 years of use).14 After 13 years of cumulative follow-up (combined intervention and follow-up phase), the risk of stroke persisted at 5 cases per 10,000 users for both arms, but only the estrogen-progestin results were statistically significant.9
The Danish Osteoporosis Study15 found no increased risk of stroke after 16 years of follow-up in recently menopausal women:
- Hazard ratio 0.89, 95% CI 0.48–1.65.
Venous thromboembolism
Data from both observational and randomized controlled trials demonstrate an increased risk of venous thromboembolism with oral hormone therapy, and the risk appears to be highest during the first few years of use.1 The pooled cohort from the Women’s Health Initiative had 18 additional cases of venous thromboembolism per 10,000 women in estrogen-progestin users compared with nonusers, and 7 additional cases in those using estrogen-only therapy.
Breast health
Observational studies and randomized controlled trials have provided data on longer use of hormone therapy and breast cancer risk, but the true magnitude of this risk is unclear.
The Danish Osteoporosis Study,15 in a younger cohort of women, showed no increased risk of breast cancer after 16 years of follow-up:
- Hazard ratio 0.90, 95% CI 0.52–1.57.
The Women’s Health Initiative9 showed a statistically nonsignificant lower risk of breast cancer in women of all ages exposed to conjugated equine estrogen alone for 7.1 years (6 fewer cases per 10,000 women-years of use), and after 6 years of follow-up this developed statistical significance:
- Hazard ratio 0.79, 95% CI 0.65–0.97.
In contrast, those using conjugated equine estrogen plus medroxyprogesterone acetate had a statistically nonsignificant increase in the risk of new breast cancer after 3 to 5 years:
- 3-year relative risk 1.26, 95% CI 0.73–2.20
- 5-year relative risk 1.99, 95% CI 1.18–3.35
- Absolute risk 8 cases per 10,000 women-years of use.
The increased risk of breast cancer significantly declined within 3 years after stopping hormone therapy.
However, even after stopping hormone therapy, there remains a statistically small but significant increased risk of breast cancer, as demonstrated in the postintervention 13-year follow-up data on breast cancer risk and estrogen-progestin use from the Women’s Health Initiative9:
- Hazard ratio 1.28, 95% CI 1.11–1.48
- Absolute cumulative risk 9 cases per 10,000 women-years of use.
The Nurses’ Health Study, an observational study, prospectively followed 11,508 hysterectomized women on estrogen therapy and found that breast cancer risk increased with longer duration of use. An analysis by Chen et al16 found a trend toward increased breast cancer risk after 10 years of estrogen therapy, but this did not become statistically significant until 20 years of ongoing estrogen use. The risk of estrogen receptor-positive and progesterone receptor-positive breast cancer became statistically significant earlier, after 15 years. The relative risk associated with using estrogen for more than 15 years was 1.18, and the risk with using it for more than 20 years was 1.42.16
To put this in perspective, Chen et al17 found a similar breast cancer risk with alcohol consumption. The relative risk of invasive breast cancer was 1.15 in women who drank 3 to 6 servings of alcohol per week, 1 serving being equivalent to 4 oz of wine, which contains 11 g of alcohol.
Mortality
Studies have suggested that hormone therapy users have a lower mortality rate, even with long-term use.
A meta-analysis18 of 8 observational trials and 19 randomized controlled trials found that younger women (average age 54) on hormone therapy had a 28% lower total mortality rate compared with women not taking hormone therapy:
- Relative risk 0.72, 95% credible interval 0.62–0.82.
The Women’s Health Initiative19 suggested that the mortality rate was 30% lower in hormone therapy users younger than age 60 than in similar nonusers, though this difference did not reach statistical significance.
- Relative risk with estrogen-only therapy: 0.71, 95% CI 0.46–1.11
- Relative risk with combined estrogen-progestin therapy 0.69, 95% CI 0.44–1.07.
The Danish Osteoporosis Study,15 at 16 years of follow-up, similarly demonstrated a 34% lower mortality rate in hormone therapy users, which was not statistically significant:
- Relative risk 0.66, 95% CI 0.41–1.08.
A Cochrane review20 in 2015 found that the subgroup of women who started hormone therapy before age 60 or within 10 years of menopause saw an overall benefit in terms of survival and lower risk of coronary heart disease: RR 0.70, 95% CI 0.52–0.95 (moderate-quality evidence).
TYPE OF FORMULATION
Compared with estrogen-progestin therapy, estrogen-only therapy has a more favorable risk profile in terms of coronary heart disease and breast cancer, although stroke risk remains elevated in users of conjugated equine estrogen with or without medroxyprogesterone acetate.
There is limited evidence directly comparing different formulations of hormone therapy, although they all effectively treat vasomotor symptoms.1
Oral vs transdermal formulations
Canonico et al,21 in a meta-analysis of observational studies, found that oral estrogen was associated with a higher risk of venous thromboembolism than transdermal estrogen:
- Relative risk with oral estrogen 2.5, 95% CI 1.9–3.4
- Relative risk with transdermal estrogen 1.2, 95% CI 0.9–1.7.
The Estrogen and Thromboembolism Risk (ESTHER) study22 was a multicenter case-control study of women ages 45 to 70 that assessed risk of venous thromboembolism in oral vs transdermal estrogen users. Compared with women not taking hormone therapy, current users of oral estrogen had a significantly higher risk of venous thromboembolism, while transdermal estrogen users did not:
- Odds ratio with oral estrogen 4.2, 95% CI 1.5–11.6
- Odds ratio with transdermal estrogen 0.9, 95% CI 0.4–2.1.
The Kronos Early Estrogen Prevention Study (KEEPS)23 did not support these findings. This 4-year randomized controlled trial, published in 2014, was designed to assess the risk of atherosclerosis progression with early menopause initiation of placebo vs low-dose oral hormone therapy (conjugated equine estrogen 0.45 mg daily with cyclical micronized progesterone) or transdermal hormone therapy (estradiol 50 µg/week with cyclical micronized progesterone).
In the 727 women in the study, there was one transient ischemic attack in the oral hormone therapy group, one unconfirmed stroke in the transdermal hormone therapy group, and one case of venous thromboembolism in each group, findings that were underpowered for statistical significance. Both oral and transdermal hormonal therapy had neutral effects on atherosclerosis progression, as assessed by arterial imaging. Transdermal hormone therapy was associated with improvements in markers of insulin resistance and was not associated with an increase in triglycerides, C-reactive protein, or sex hormone-binding globulin, as would be expected with transdermal circumvention of the first-pass hepatic effect.
BALANCING THE RISKS AND BENEFITS FOR THE PATIENT
The most effective treatment for vasomotor symptoms in women at any age is hormone therapy, and the benefits are more likely to outweigh risks when initiated before age 60 or within 10 years of menopause.7 The Women’s Health Initiative randomized study was limited to 5.6 to 7.2 years of hormone therapy (13 years of cumulative follow-up), and the Danish Osteoporosis Study was limited to 11 years of use (16 years cumulative follow-up).
The coronary heart disease outcomes for longer durations of therapy remain uncertain. There is a small but statistically significant increased risk of stroke and venous thromboembolism with oral hormone therapy, and breast cancer risk is associated with long-term estrogen-progestin use.
Patients on hormone therapy should be evaluated annually regarding the need for ongoing therapy. Persistent moderate-severe vasomotor symptoms, quality of life benefits of hormone therapy, contraindications to its use (Table 2), and patient preference need to be assessed as well as baseline risks of cardiovascular disease, breast cancer, and fracture.
Risk calculators may facilitate the shared decision-making process. Examples are:
- The American College of Cardiology/American Heart association risk calculator for cardiovascular disease24 (www.cvriskcalculator.com)
- The World Health Organization Fracture Risk Assessment Tool (FRAX)25
(www.shef.ac.uk/FRAX/tool.jsp)
- The Gail model for breast cancer risk26 (www.cancer.gov/bcrisktool/).
- MenoPro, a menopause decision-support algorithm and companion mobile app developed by NAMS to help direct treatment decisions based on the 10-year risk of atherosclerotic cardiovascular disease (www.menopause.org/for-professionals/-i-menopro-i-mobile-app).27
The discussion of the risks of hormone therapy with patients should incorporate the perspective of absolute risk. For example, a woman wishing to continue estrogen-progestin therapy should be told that the Women’s Health Initiative data suggest that, after 5 years of use, breast cancer risk may be increased by 8 additional cases per 10,000 users per year. According to the World Health Organization, this magnitude of risk is defined as rare (less than 1 event per 1,000 women).28
A strategy of prescribing the lowest dose to achieve the desired clinical benefits is prudent and recommended.1–3 Table 3 outlines the estrogen formulations now available in the United States, with their doses and formulations.
Unless contraindications develop (Table 2), patients may elect to continue hormone therapy if its benefits outweigh its risks. The American College of Obstetricians and Gynecologists (ACOG) 2014 practice recommendations for management of menopausal symptoms31 and the 2015 NAMS statement both recommend that hormone therapy not be discontinued based solely on a woman’s age.29
Hormone therapy is on the Beer’s list of potentially inappropriate medications for older adults,30 which remains a hurdle to its long-term use and seems to be at odds with these ACOG and NAMS statements.
Patients who choose to discontinue hormone therapy need to be monitored for persistent bothersome vasomotor symptoms, bone loss, osteoporosis, and the genitourinary syndrome of menopause (previously referred to as vulvovaginal atrophy)31 and offered alternative therapies if needed.
- North American Menopause Society. The 2012 hormone therapy position statement of: The North American Menopause Society. Menopause 2012; 19:257–271.
- American College of Obstetricians and Gynecologists. Practice Bulletin No. 141: Management of menopausal symptoms. Obstet Gynecol 2014; 123:202–216.
- Stuenkel CA, Davis SR, Gompel A, et al. Treatment of symptoms of the menopause: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2015; 100:3975–4011.
- de Villiers TJ, Pines A, Panay N, et al; International Menopause Society. Updated 2013 International Menopause Society recommendations on menopausal hormone therapy and preventive strategies for midlife health. Climacteric 2013; 16:316–337.
- Gartoulla P, Worsley R, Robin J, Davis S. Moderate to severe vasomotor and sexual symptoms remain problematic for women aged 60 to 65 years. Menopause 2015; 22:694–701.
- Avis NE, Crawford SL, Greendale G, et al. Duration of menopausal vasomotor symptoms across the menopause transition. JAMA Intern Med 2015; 175:531–539.
- de Villiers TJ, Gass ML, Haines CJ, et al. Global consensus statement on menopausal hormone therapy. Climacteric 2013; 16:203–204.
- Cauley J, Robbins J, Chen Z, et al. Effects of estrogen plus progestin on risk of fracture and bone mineral density: the Women’s Health Initiative randomized trial. JAMA 2003; 290:1729–1738.
- Manson J, Chlebowski R, Stefanick M, et al. Menopausal hormone therapy and health outcomes during the intervention and extended poststopping phases of the Women’s Health Initiative randomized trials. JAMA 2013; 310:1353–1368.
- Karim R, Dell RM, Greene DF, et al. Hip fracture in postmenopausal women after cessation of hormone therapy: results from a prospective study in a large health management organization. Menopause 2011; 18:1172–1177.
- Shifren J, Gass M, and the NAMS Recommendations for Clinical Care of Midlife Women Working Group. The North American Menopause Society recommendations for clinical care of midlife women. Menopause 2014; 21:1038–1062.
- Hodis HN, Mack WJ. Hormone replacement therapy and the association with coronary heart disease and overall mortality: clinical application of the timing hypothesis. J Steroid Biochem Mol Biol 2014; 142:68–75.
- Salpeter SR, Walsh JM, Greyber E, et al. Brief report: coronary heart disease events associated with hormone therapy in younger and older women. A meta-analysis. J Gen Intern Med 2006; 21:363–366.
- Rossouw JE, Prentice RL, Manson JE, et al. Postmenopausal hormone therapy and risk of cardiovascular disease by age and years since menopause. JAMA 2007; 297:1465–1477.
- Schierbeck LL, Rejnmark L, Tofteng CL, et al. Effect of hormone replacement therapy on cardiovascular events in recently postmenopausal women: randomised trial. BMJ 2012; 345:e6409.
- Chen WY, Manson JE, Hankinson SE, et al. Unopposed estrogen therapy and the risk of breast cancer. Arch Intern Med 2006; 166:1027–1032.
- Chen W, Rosner B, Hankinson SE, et al. Moderate alcohol consumption during adult life, drinking patterns, and breast cancer risk. JAMA 2011; 306:1884–1890.
- Salpeter SR, Cheng J, Thabane L, et al. Bayesian meta-analysis of hormone therapy and mortality in younger postmenopausal women. Am J Med 2009; 122:1016–1022.
- Hodis HN, Collins P, Mack WJ, Schierbeck LL. The timing hypothesis for coronary heart disease prevention with hormone therapy: past, present and future in perspective. Climacteric 2012; 15:217–228.
- Boardman HM, Hartley L, Eisinga A, et al. Hormone therapy for preventing cardiovascular disease in post-menopausal women. Cochrane Database Syst Rev 2015;3:CD002229.
- Canonico M, Plu-Bureau G, Lowe GD, et al. Hormone replacement therapy and risk of venous thromboembolism in postmenopausal women: systemic review and meta-analysis. BMJ 2008; 336:1227–1231.
- Canonico M, Oger E, Plu-Bureau G, et al; Estrogen and Thromboembolism Risk (ESTHER) Study Group. Hormone therapy and venous thromboembolism among postmenopausal women: impact of the route of estrogen administration and progestogens: the ESTHER study. Circulation 2007; 115:840–845.
- Harman S, Black D, Naftolin F, et al. Arterial imaging outcomes and cardiovascular risk factors in recently menopausal women. Ann Intern Med 2014; 161:249–260.
- Goff DC Jr, Lloyd-Jones DM, Bennett G, et al. 2013 ACC/AHA guideline on the assessment of cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2014; 63:2935–2959.
- World Health Organization Collaborating Centre for Metabolic Bone Diseases. FRAX WHO fracture risk assessment tool. www.shef.ac.uk/FRAX/. Accessed May 27, 2016.
- Gail M, Brinton L, Byar D, et al. Projecting individualized probabilities of developing breast cancer for white females who are being examined annually. J Natl Cancer Inst 1989; 81:1879–1886.
- Manson J, Ames J, Shapiro M, et al. Algorithm and mobile app for menopausal symptom management and hormonal/non-hormonal therapy decision making: a clinical decision-support tool from the North American Menopause Society. Menopause 2015; 22:247–253.
- Hodis HN, Mack WJ. Postmenopausal hormone therapy in clinical perspective. Menopause 2007; 14:944–957.
- North American Menopause Society. The North American Menopause Society statement on continuing use of systemic hormone therapy after the age of 65. Menopause 2015; 22:693.
- American Geriatrics Society 2015 Beers Criteria Update Expert Panel. American Geriatrics Society 2015 updated Beers criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc 2015; 63:2227–2246.
- Portman DJ, Gass ML; Vulvovaginal Atrophy Terminology Consensus Conference Panel. Genitourinary syndrome of menopause: new terminology for vulvovaginal atrophy from the International Society for the Study of Women’s Sexual Health and the North American Menopause Society. Menopause 2014; 21:1063–1068.
- North American Menopause Society. The 2012 hormone therapy position statement of: The North American Menopause Society. Menopause 2012; 19:257–271.
- American College of Obstetricians and Gynecologists. Practice Bulletin No. 141: Management of menopausal symptoms. Obstet Gynecol 2014; 123:202–216.
- Stuenkel CA, Davis SR, Gompel A, et al. Treatment of symptoms of the menopause: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2015; 100:3975–4011.
- de Villiers TJ, Pines A, Panay N, et al; International Menopause Society. Updated 2013 International Menopause Society recommendations on menopausal hormone therapy and preventive strategies for midlife health. Climacteric 2013; 16:316–337.
- Gartoulla P, Worsley R, Robin J, Davis S. Moderate to severe vasomotor and sexual symptoms remain problematic for women aged 60 to 65 years. Menopause 2015; 22:694–701.
- Avis NE, Crawford SL, Greendale G, et al. Duration of menopausal vasomotor symptoms across the menopause transition. JAMA Intern Med 2015; 175:531–539.
- de Villiers TJ, Gass ML, Haines CJ, et al. Global consensus statement on menopausal hormone therapy. Climacteric 2013; 16:203–204.
- Cauley J, Robbins J, Chen Z, et al. Effects of estrogen plus progestin on risk of fracture and bone mineral density: the Women’s Health Initiative randomized trial. JAMA 2003; 290:1729–1738.
- Manson J, Chlebowski R, Stefanick M, et al. Menopausal hormone therapy and health outcomes during the intervention and extended poststopping phases of the Women’s Health Initiative randomized trials. JAMA 2013; 310:1353–1368.
- Karim R, Dell RM, Greene DF, et al. Hip fracture in postmenopausal women after cessation of hormone therapy: results from a prospective study in a large health management organization. Menopause 2011; 18:1172–1177.
- Shifren J, Gass M, and the NAMS Recommendations for Clinical Care of Midlife Women Working Group. The North American Menopause Society recommendations for clinical care of midlife women. Menopause 2014; 21:1038–1062.
- Hodis HN, Mack WJ. Hormone replacement therapy and the association with coronary heart disease and overall mortality: clinical application of the timing hypothesis. J Steroid Biochem Mol Biol 2014; 142:68–75.
- Salpeter SR, Walsh JM, Greyber E, et al. Brief report: coronary heart disease events associated with hormone therapy in younger and older women. A meta-analysis. J Gen Intern Med 2006; 21:363–366.
- Rossouw JE, Prentice RL, Manson JE, et al. Postmenopausal hormone therapy and risk of cardiovascular disease by age and years since menopause. JAMA 2007; 297:1465–1477.
- Schierbeck LL, Rejnmark L, Tofteng CL, et al. Effect of hormone replacement therapy on cardiovascular events in recently postmenopausal women: randomised trial. BMJ 2012; 345:e6409.
- Chen WY, Manson JE, Hankinson SE, et al. Unopposed estrogen therapy and the risk of breast cancer. Arch Intern Med 2006; 166:1027–1032.
- Chen W, Rosner B, Hankinson SE, et al. Moderate alcohol consumption during adult life, drinking patterns, and breast cancer risk. JAMA 2011; 306:1884–1890.
- Salpeter SR, Cheng J, Thabane L, et al. Bayesian meta-analysis of hormone therapy and mortality in younger postmenopausal women. Am J Med 2009; 122:1016–1022.
- Hodis HN, Collins P, Mack WJ, Schierbeck LL. The timing hypothesis for coronary heart disease prevention with hormone therapy: past, present and future in perspective. Climacteric 2012; 15:217–228.
- Boardman HM, Hartley L, Eisinga A, et al. Hormone therapy for preventing cardiovascular disease in post-menopausal women. Cochrane Database Syst Rev 2015;3:CD002229.
- Canonico M, Plu-Bureau G, Lowe GD, et al. Hormone replacement therapy and risk of venous thromboembolism in postmenopausal women: systemic review and meta-analysis. BMJ 2008; 336:1227–1231.
- Canonico M, Oger E, Plu-Bureau G, et al; Estrogen and Thromboembolism Risk (ESTHER) Study Group. Hormone therapy and venous thromboembolism among postmenopausal women: impact of the route of estrogen administration and progestogens: the ESTHER study. Circulation 2007; 115:840–845.
- Harman S, Black D, Naftolin F, et al. Arterial imaging outcomes and cardiovascular risk factors in recently menopausal women. Ann Intern Med 2014; 161:249–260.
- Goff DC Jr, Lloyd-Jones DM, Bennett G, et al. 2013 ACC/AHA guideline on the assessment of cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2014; 63:2935–2959.
- World Health Organization Collaborating Centre for Metabolic Bone Diseases. FRAX WHO fracture risk assessment tool. www.shef.ac.uk/FRAX/. Accessed May 27, 2016.
- Gail M, Brinton L, Byar D, et al. Projecting individualized probabilities of developing breast cancer for white females who are being examined annually. J Natl Cancer Inst 1989; 81:1879–1886.
- Manson J, Ames J, Shapiro M, et al. Algorithm and mobile app for menopausal symptom management and hormonal/non-hormonal therapy decision making: a clinical decision-support tool from the North American Menopause Society. Menopause 2015; 22:247–253.
- Hodis HN, Mack WJ. Postmenopausal hormone therapy in clinical perspective. Menopause 2007; 14:944–957.
- North American Menopause Society. The North American Menopause Society statement on continuing use of systemic hormone therapy after the age of 65. Menopause 2015; 22:693.
- American Geriatrics Society 2015 Beers Criteria Update Expert Panel. American Geriatrics Society 2015 updated Beers criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc 2015; 63:2227–2246.
- Portman DJ, Gass ML; Vulvovaginal Atrophy Terminology Consensus Conference Panel. Genitourinary syndrome of menopause: new terminology for vulvovaginal atrophy from the International Society for the Study of Women’s Sexual Health and the North American Menopause Society. Menopause 2014; 21:1063–1068.
KEY POINTS
- Hormone therapy is the most effective treatment available for the vasomotor symptoms of menopause, and it also is effective and appropriate for preventing osteoporosis-related fracture in at-risk women under age 60 or within 10 years of menopause.
- Oral hormone therapy is associated with a small but statistically significant increase in the risk of stroke and venous thromboembolism and breast cancer risk with combination therapy only.
- Extended hormone therapy may be appropriate to treat vasomotor symptoms or prevent osteoporosis when alternative therapies are not an option.
- The decision whether to continue hormone therapy should be revisited every year. Discussions with patients should include the perspective of absolute risk.

Thrombotic thrombocytopenic purpura: The role of ADAMTS13
A breakthrough in understanding the pathogenesis of thrombotic thrombocytopenic purpura (TTP) came with the discovery of ADAMTS13 (an abbreviation for “a disintegrin and metalloproteinase with thrombospondin type 1 motif, member 13”), a plasma protein that cleaves von Willebrand factor, which interacts with platelets to promote blood clotting. If ADAMTS13 is lacking, unusually large multimers of von Willebrand factor can accumulate and trigger intravascular platelet aggregation and microthrombosis, causing the signs and symptoms of TTP.1–3
This knowledge has practical applications: we can now measure ADAMTS13 activity, ADAMTS13 inhibitor, and antibodies against ADAMTS13 to help us diagnose TTP and distinguish it from other forms of thrombotic microangiopathy, such as hemolytic-uremic syndrome, that have similar symptoms but require different treatment.
Using case studies, this article describes typical presentations of acute and relapsing TTP; the role of laboratory testing, including the ADAMTS13 assay; how to distinguish TTP from other conditions that present similarly; and how to manage this condition.
A HIGH RISK OF DEATH WITHOUT PLASMA EXCHANGE
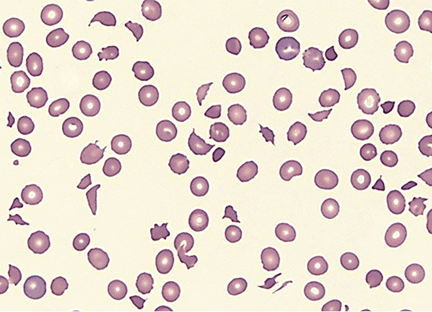
TTP is characterized by disseminated microthrombi composed of agglutinated platelets and von Willebrand factor in small vessels. Tissue damage by microthrombi can cause thrombocytopenia (platelet deficiency), microangiopathic hemolytic anemia (loss of red blood cells caused by destructive conditions in small vessels), and multiorgan failure.1
Untreated TTP has a mortality rate of about 90%.1 As shown in Case 1, Case 2, and Table 1, rapid diagnosis and prompt initiation of daily therapeutic plasma exchange can improve this grave outlook.4


ADAMTS13 DEFICIENCY CAN BE ACQUIRED OR CONGENITAL
Two major forms of TTP with ADAMTS13 deficiency and microvascular thrombosis are recognized:
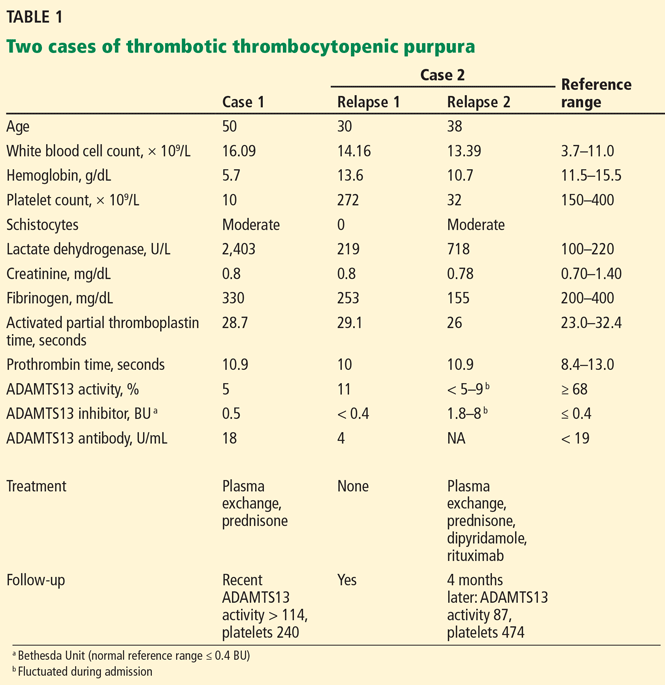
Acquired TTP, the more common form, peaks in incidence between ages 30 and 50.2,5 It more often affects women, particularly during and after pregnancy (its estimated prevalence is 1 in 25,000 pregnancies), and African Americans.6 Acquired TTP may be:
- Primary (idiopathic or autoantibody-mediated), associated with severely decreased ADAMTS13 and the presence of ultra-large von Willebrand factor multimers, or
- Secondary (23%–67% of cases), arising from a variety of conditions, including autoimmune disorders (eg, systemic lupus erythematosus, rheumatoid arthritis), solid organ or hematopoietic cell transplant, malignancy, drugs, and pregnancy (Table 2).1,5–8 Secondary TTP has a worse prognosis than idiopathic TTP.5,9

Congenital TTP (Upshaw-Shulman syndrome) is a rare autosomal-recessive disease caused by compound heterozygous or homozygous mutations of the ADAMTS13 gene, producing nonfunctional ADAMTS13 protein. Patients have severely deficient ADAMTS13 activity but usually do not develop autoantibodies. There is a high risk of chronic, relapsing episodes; identified triggers include pregnancy and heavy alcohol intake.2,10 About half of patients with congenital TTP have an early onset, usually presenting with acute TTP between birth and age 5, and about half have a late onset, usually remaining without symptoms until age 20 to 40.
THE CLINICAL PICTURE OF TTP IS NOT ALWAYS CLASSIC
TTP is primarily diagnosed clinically, but diagnosis is often difficult because of various nonspecific symptoms. Typical TTP presents with the “classic pentad”:
- Severe thrombocytopenia (70%–100% of patients)
- Microangiopathic hemolytic anemia with multiple schistocytes (70%–100%) (Figure 1)
- Neurologic involvement (50%–90%)
- Renal abnormalities (about 50%)
- Fever (25%).
However, the entire picture often does not emerge in a single patient.2,6 Waiting for the entire pentad to develop before diagnosing TTP can have grave clinical consequences,1,2,5 and the presence of thrombocytopenia and unexplained microangiopathic hemolytic anemia are considered clinically sufficient to suspect TTP.5
Neurologic symptoms usually fluctuate. They can include mild abnormalities such as weakness, dizziness, headache, blurred vision, ataxia, and transient mental status changes, as well as severe abnormalities including stroke, seizure, and coma.2,6
Most patients have normal findings on computed tomography and magnetic resonance imaging at the onset of neurologic symptoms or with a history of TTP. Some patients (8%–39%) show reversible acute brain lesions, including ischemic changes.11–13
Other signs and symptoms may result from multiorgan failure due to microthrombosis; ischemia in retinal, coronary, and abdominal circulations; and unconjugated hyperbilirubinemia.2
Atypical presentations. About 18% of patients have cardiac involvement from microvascular occlusion, with arrhythmia, angina, or congestive heart failure. Abdominal pain and pancreatitis occur in 5% to 13%, and visual disturbances in 8% to 10%.
Patients with an atypical presentation may not have laboratory evidence of microangiopathic hemolytic anemia, but an ADAMTS13 assay will show severely decreased activity. Therapeutic plasma exchange can improve atypical symptoms.2,3,10,14,15
ADAMTS13 ASSAY IS KEY TO DIAGNOSIS
Laboratory evidence typically includes hemolytic anemia (reticulocytosis, schistocytes, elevated indirect bilirubin, reduced haptoglobin, elevated lactate dehydrogenase) and thrombocytopenia.3 There are no significant abnormalities in prothrombin time, international normalized ratio, activated partial thromboplastin time, fibrinogen, or D-dimer level.
Measuring the levels of ADAMTS13 activity, ADAMTS13 inhibitor, and ADAMTS13 antibody is becoming standard to confirm the diagnosis of TTP, to determine if it is congenital or acquired, and to distinguish it from thrombocytopenic conditions such as hemolytic-uremic syndrome, idiopathic thrombocytopenic purpura, and heparin-induced thrombocytopenia.4,5 A newer ADAMTS13 assay based on fluorescence energy transfer (FRET) technology with a synthetic amino acid-von Willebrand factor peptide substrate has a faster turnaround time and less test variability.6,16,17 This FRET assay can give the result of ADAMTS13 activity within 2 hours. In comparison, the assay based on multimeric von Willebrand factor takes 2 to 3 days, and mass spectrometry to measure the cleavage products of a synthetic von Willebrand factor molecule takes about 4 hours.3,10,16
About two-thirds of patients with the clinical diagnosis of idiopathic TTP have ADAMTS13 activity levels lower than 10%.5,14,18 In the appropriate clinical setting, this threshold level is highly sensitive (89%–100%) and specific (99%–100%) in differentiating TTP from other thrombotic angiopathies.2,3,18
Note: The ADAMTS13 assay was needed for early correct diagnosis in Case 1 and Case 2.
Inhibitors provide more clues
Autoantibodies can be classified according to whether they inhibit ADAMTS13 activity.
Neutralizing inhibitors. Most cases of acquired, idiopathic TTP with severe ADAMTS13 deficiency are related to circulating autoantibodies that neutralize ADAMTS13 activity. This ADAMTS13 inhibitor level is obtained by measuring residual ADAMTS13 activity after mixing equal amounts of patient plasma with normal pooled plasma. ADAMTS13 inhibitor is detectable in 44% to 93% of patients with severely deficient ADAMTS13 activity.3,6,19
Nonneutralizing inhibitors. From 10% to 15% of patients with TTP with severe ADAMTS13 deficiency lack ADAMTS13 autoantibodies measured by enzyme immunoassay but have nonneutralizing immunoglobulin G (IgG) or IgM autoantibodies. In such cases, ADAMTS13 deficiency may be related to increased antibody-mediated clearance or other unknown mechanisms.
Neutralizing inhibitors and nonneutralizing inhibitors may be present simultaneously in some patients.3,10,19,20
Blood factors affect ADAMTS13 activity
Specimen factors can affect ADAMTS13 activity and antibody levels.
Hemoglobin is a potent inhibitor of ADAMTS13, so an elevated plasma level of free hemoglobin (> 2 g/dL) can reduce ADAMTS13 activity, as can hyperbilirubinemia (> 15 mg/dL).
High levels of endogenous von Willebrand factor, lipids, thrombin, or other proteases that may cleave ADAMTS13 can also reduce ADAMTS13 activity.3 Conversely, recent plasma exchange or transfusion can mask the diagnosis of TTP because of false normalization of ADAMTS13 activity. In addition, ADAMTS13 autoantibody can be detected in other immune-mediated disorders (eg, systemic lupus erythematosus, antiphospholipid syndrome), and hypergammaglobulinemia, as well as in 10% to 15% of healthy individuals.19
CONSIDER OTHER CONDITIONS

Before diagnosing TTP, other conditions causing thrombocytopenia and hemolytic anemia should be excluded by taking a careful clinical, laboratory, and medication history (Table 2). Of these conditions, the most challenging to differentiate from TTP—and often indistinguishable from it at presentation—is hemolytic-uremic syndrome (Table 3).
Hemolytic-uremic syndrome
Hemolytic-uremic syndrome presents with a triad of thrombocytopenia, acute renal failure, and microangiopathic hemolytic anemia, with increased lactate dehydrogenase levels. Renal dysfunction from ischemia or tissue injury by microvascular thrombi predominates. Hemolytic-uremic syndrome most often occurs in children and is often related to hemorrhagic enterocolitis caused by infection with Escherichia coli O157:H7 or Shigella species (90%–95% of cases).1,2,5
From 5% to 10% of cases of hemolytic- uremic syndrome are atypical. These cases are not associated with diarrhea, and many are caused by genetic mutations that result in chronic excessive complement activation. Implicated genes regulate complement regulator factor H (20%–30% of cases) or CD46 (10%) and other cofactors, or autoantibodies against factor H (10%), which affect the alternate complement pathway.6,21–23
Initial therapeutic plasma exchange is commonly undertaken for atypical hemolytic- uremic syndrome, particularly for patients at risk of rapid progression to end-stage renal failure. But despite such treatment, about 60% of these patients die or develop permanent renal damage within 1 year.2,3,24
Eculizumab, a monoclonal antibody against complement component C5, has been approved by the US Food and Drug Administration for atypical hemolytic-uremic syndrome and may improve quality of life.25–27
PLASMA EXCHANGE IS THE MAINSTAY OF THERAPY
In 2012, the British Society for Haematology published revised guidelines for managing TTP and other thrombotic microangiopathies.28
Acquired idiopathic TTP with reduced ADAMTS13 activity requires immediate therapeutic plasma exchange. Daily plasma exchange combines plasmapheresis to remove circulating ultralarge von Willebrand factor-platelet strings and autoantibodies against ADAMTS13, and infusion of fresh-frozen plasma to replace ADAMTS13.18 This procedure is the mainstay of therapy and brings 70% to 90% of patients with idiopathic TTP to remission.1,2,5,6 However, the optimal duration of daily plasma exchange and the number of procedures required is highly variable according to clinical condition. Therapeutic plasma exchange can also cause plasma-related adverse reactions.9,28 Congenital TTP requires plasma infusion or exchange depending on the patient’s severity of ADAMTS13 deficiency.
Corticosteroids are used in combination with daily therapeutic plasma exchange, although evidence from controlled trials of their efficacy in this setting is lacking. Patients with severely decreased ADAMTS13 activity or low titers of ADAMTS13 autoantibodies tend to respond to the therapy.5,8,29
An ADAMTS13 assay with a short turn-around time can help guide the decision to initiate therapeutic plasma exchange. However, if there is a strong clinical suspicion of TTP, plasma exchange should be initiated immediately without waiting for test results.5,30 Monitoring ADAMTS13 activity or inhibitor during initial plasma exchange therapy has had conflicting results in several studies and is generally not recommended for patients with acquired TTP.8,30,31
RELAPSE IS COMMON
About 20% to 50% of patients with idiopathic TTP experience a relapse (Case 2). Most relapses occur within the first 2 years after the initial episode, with an estimated risk of 43% for relapse at 7.5 years.5,9
Factors that predict a higher risk of relapse include persistently severely decreased ADAMTS13 activity, positive inhibitor, and high titers of autoantibodies to ADAMTS13 during symptomatic TTP. During clinical remission, persistence of autoantibodies also indicates increased risk.1,3,5,6,9
Patients who have a relapse and whose disease is refractory to therapeutic plasma exchange (10%–20% of cases) have been treated with corticosteroids, splenectomy, or immunosuppressive agents (cyclosporine, azathioprine, or cyclophosphamide) with varying rates of success. Rituximab (monoclonal anti-CD20) has recently been used as second-line therapy in refractory or relapsing immune-mediated TTP or idiopathic TTP with neurologic or cardiac symptoms associated with a poor prognosis. Therapy including rituximab results in improved response and progression-free survival.32 Other potential therapies, including recombinant active ADAMTS13, are under investigation.9,23,28,30,33,34
- Sadler JE, Moake JL, Miyata T, George JN. Recent advances in thrombotic thrombocytopenic purpura. Hematology Am Soc Hematol Educ Program 2004; 1:407–423.
- Shenkman B, Einav Y. Thrombotic thrombocytopenic purpura and other thrombotic microangiopathic hemolytic anemias: diagnosis and classification. Autoimmun Rev 2014; 13:584–586.
- Shah N, Sarode R. Thrombotic thrombocytopenic purpura-what is new? J Clin Apher 2013; 28:30–35.
- Imanirad I, Rajasekhar A, Zumberg M. A case series of atypical presentations of thrombotic thrombocytopenic purpura. J Clin Apher 2012; 27:221–226.
- George JN, Al-Nouri ZL. Diagnostic and therapeutic challenges in the thrombotic thrombocytopenic purpura and hemolytic uremic syndromes. Hematology Am Soc Hematol Educ Program 2012; 1:604–609.
- Shah N, Rutherford C, Matevosyan K, Shen YM, Sarode R. Role of ADAMTS13 in the management of thrombotic microangiopathies including thrombotic thrombocytopenic purpura (TTP). Br J Haematol 2013; 163:514–519.
- Cataland SR, Yang S, Wu HM. The use of ADAMTS13 activity, platelet count, and serum creatinine to differentiate acquired thrombotic thrombocytopenic purpura from other thrombotic microangiopathies. Br J Haematol 2012; 157:501–503.
- Mannucci PM, Peyvandi F. TTP and ADAMTS13: when Is testing appropriate? Hematology Am Soc Hematol Educ Program 2007; 1:121–126.
- Chaturved S, Carcioppolo D, Zhang L, McCar KR. Management and outcomes of patients with TTP: analysis of 100 cases at a single institution. Am J Hematol 2013; 88:560–565.
- Peyvandi F, Palla R, Lotta LA, Mackie I, Scully MA, Machin SJ. ADAMTS-13 assays in thrombotic thrombocytopenic purpura. J Thromb Haemost 2010; 8:631–640.
- Cataland SR, Scully MA, Paskavitz J, et al. Evidence of persistent neurologic injury following thrombotic thrombocytopenic purpura. Am J Hematol 2011; 86:87–89.
- Meloni G, Proia A, Antonini G, et al. Thrombotic thrombocytopenic purpura: prospective neurologic, neuroimaging and neurophysiologic evaluation. Haematologica 2001; 86:1194–1199.
- Kwaan HC, Boggio LN. The clinical spectrum of thrombotic thrombocytopenic purpura. Semin Thromb Hemost 2005; 31:673–680.
- Sarode R. Atypical presentations of thrombotic thrombocytopenic purpura: a review. J Clin Apher 2009; 24:47–52.
- Volcy J, Nzerue CM, Oderinde A, Hewan-Iowe K. Cocaine-induced acute renal failure, hemolysis, and thrombocytopenia mimicking thrombotic thrombocytopenic purpura. Am J Kidney Dis 2000; 35:E3.
- Kremer Hovinga JA, Mottini M, Lammle B. Measurement of ADAMTS-13 activity in plasma by the FRETS-VWF73 assay: comparison with other assay methods. J Thromb Haemost 2006; 4:1146–1148.
- Groot E, Hulstein JJ, Rison CN, de Groot PG, Fijnheer R. FRETS-VWF73: a rapid and predictive tool for thrombotic thrombocytopenic purpura. J Thromb Haemost 2006; 4:698–699.
- Barrows BD, Teruya J. Use of the ADAMTS13 activity assay improved the accuracy and efficiency of the diagnosis and treatment of suspected acquired thrombotic thrombocytopenic purpura. Arch Pathol Lab Med 2014; 138:546–549.
- Rieger M, Mannucci PM, Kremer Hovinga JA, et al. ADAMTS13 autoantibodies in patients with thrombotic microangiopathies and other immunomediated diseases. Blood 2005; 106:1262–1267.
- Rogers HJ, Kottke-Marchant K. ADAMTS13 evaluation for thrombotic thrombocytopenic purpura. Pathology Innovations, Pathology and Laboratory Medicine Institute. Cleveland Clinic, Fall 2014:6–9.
- Józsi M, Licht C, Strobel S, et al. Factor H autoantibodies in atypical hemolytic uremic syndrome correlate with CFHR1/CFHR3 deficiency. Blood 2008; 111:1512–1514.
- Diamante Chiodini B, Davin JC, Corazza F, et al. Eculizumab in anti-factor H antibodies associated with atypical hemolytic uremic syndrome. Pediatrics 2014; 133:e1764–e1768.
- Taylor CM, Machin S, Wigmore SJ, Goodship TH; working party from the Renal Association, the British Committee for Standards in Haematology and the British Transplantation Society. Clinical practice guidelines for the management of atypical haemolytic uraemic syndrome in the United Kingdom. Br J Haematol 2009; 148:37–47.
- Loirat C, Garnier A, Sellier-Leclerc AL, Kwon T. Plasmatherapy in atypical hemolytic uremic syndrome. Semin Thromb Hemost 2010; 36:673–681.
- Tsai HM, Kuo E. Eculizumab therapy leads to rapid resolution of thrombocytopenia in atypical hemolytic uremic syndrome. Adv Hematol 2014; 295323:1–7.
- Lapeyraque AL, Frémeaux-Bacchi V, Robitaille P. Efficacy of eculizumab in a patient with factor-H-associated atypical hemolytic uremic syndrome. Pediatr Nephrol 2011; 26:621–624.
- Baskin E, Gulleroglu K, Kantar A, Bayrakci U, Ozkaya O. Success of eculizumab in the treatment of atypical hemolytic uremic syndrome. Pediatr Nephrol 2015; 30:783–789.
- Scully M, Hunt BJ, Benjamin S, et al; British Committee for Standards in Haematology. Guidelines on the diagnosis and management of thrombotic thrombocytopenic purpura and other thrombotic microangiopathies. Br J Haematol 2012; 158:323–325.
- Abassi E, Yawn D, Leveque E, Nolasco L, Lopez J, Moake J. Correlation of ADAMTS-13 activity with response to plasma exchange in patients diagnosed with thrombotic thrombocytopenic purpura (Abstract #3921). Blood 2004; 104:242a.
- Blombery P, Scully M. Management of thrombocytic thrombocytopenic purpura: current perspectives. J Blood Med 2014; 5:15–23.
- Wu N, Liu J, Yang S, et al. Diagnostic and prognostic values of ADAMTS13 activity measured during daily plasma exchange therapy in patients with acquired thrombotic thrombocytopenic purpura. Transfusion 2015; 55:18–24.
- Cuker A. Adjuvant rituximab to prevent TTP relapse. Blood 2016; 127:2952–2953.
- Chapman K, Yuen S. Therapy for thrombotic thrombocytopenic purpura: past, present and future. Semin Thromb Hemost 2014; 40:34–40.
- Heidel F, Lipka DB, von Auer C, Huber C, Schrarrer I, Hess G. Addition of rituximab to standard therapy improves response rate and progression-free survival in relapsed or refractory thrombotic thrombocytopenic purpura and autoimmune haemolytic anaemia. Thromb Haemost 2007; 97:228–233.
A breakthrough in understanding the pathogenesis of thrombotic thrombocytopenic purpura (TTP) came with the discovery of ADAMTS13 (an abbreviation for “a disintegrin and metalloproteinase with thrombospondin type 1 motif, member 13”), a plasma protein that cleaves von Willebrand factor, which interacts with platelets to promote blood clotting. If ADAMTS13 is lacking, unusually large multimers of von Willebrand factor can accumulate and trigger intravascular platelet aggregation and microthrombosis, causing the signs and symptoms of TTP.1–3
This knowledge has practical applications: we can now measure ADAMTS13 activity, ADAMTS13 inhibitor, and antibodies against ADAMTS13 to help us diagnose TTP and distinguish it from other forms of thrombotic microangiopathy, such as hemolytic-uremic syndrome, that have similar symptoms but require different treatment.
Using case studies, this article describes typical presentations of acute and relapsing TTP; the role of laboratory testing, including the ADAMTS13 assay; how to distinguish TTP from other conditions that present similarly; and how to manage this condition.
A HIGH RISK OF DEATH WITHOUT PLASMA EXCHANGE
TTP is characterized by disseminated microthrombi composed of agglutinated platelets and von Willebrand factor in small vessels. Tissue damage by microthrombi can cause thrombocytopenia (platelet deficiency), microangiopathic hemolytic anemia (loss of red blood cells caused by destructive conditions in small vessels), and multiorgan failure.1
Untreated TTP has a mortality rate of about 90%.1 As shown in Case 1, Case 2, and Table 1, rapid diagnosis and prompt initiation of daily therapeutic plasma exchange can improve this grave outlook.4
ADAMTS13 DEFICIENCY CAN BE ACQUIRED OR CONGENITAL
Two major forms of TTP with ADAMTS13 deficiency and microvascular thrombosis are recognized:
Acquired TTP, the more common form, peaks in incidence between ages 30 and 50.2,5 It more often affects women, particularly during and after pregnancy (its estimated prevalence is 1 in 25,000 pregnancies), and African Americans.6 Acquired TTP may be:
- Primary (idiopathic or autoantibody-mediated), associated with severely decreased ADAMTS13 and the presence of ultra-large von Willebrand factor multimers, or
- Secondary (23%–67% of cases), arising from a variety of conditions, including autoimmune disorders (eg, systemic lupus erythematosus, rheumatoid arthritis), solid organ or hematopoietic cell transplant, malignancy, drugs, and pregnancy (Table 2).1,5–8 Secondary TTP has a worse prognosis than idiopathic TTP.5,9
Congenital TTP (Upshaw-Shulman syndrome) is a rare autosomal-recessive disease caused by compound heterozygous or homozygous mutations of the ADAMTS13 gene, producing nonfunctional ADAMTS13 protein. Patients have severely deficient ADAMTS13 activity but usually do not develop autoantibodies. There is a high risk of chronic, relapsing episodes; identified triggers include pregnancy and heavy alcohol intake.2,10 About half of patients with congenital TTP have an early onset, usually presenting with acute TTP between birth and age 5, and about half have a late onset, usually remaining without symptoms until age 20 to 40.
THE CLINICAL PICTURE OF TTP IS NOT ALWAYS CLASSIC
TTP is primarily diagnosed clinically, but diagnosis is often difficult because of various nonspecific symptoms. Typical TTP presents with the “classic pentad”:
- Severe thrombocytopenia (70%–100% of patients)
- Microangiopathic hemolytic anemia with multiple schistocytes (70%–100%) (Figure 1)
- Neurologic involvement (50%–90%)
- Renal abnormalities (about 50%)
- Fever (25%).
However, the entire picture often does not emerge in a single patient.2,6 Waiting for the entire pentad to develop before diagnosing TTP can have grave clinical consequences,1,2,5 and the presence of thrombocytopenia and unexplained microangiopathic hemolytic anemia are considered clinically sufficient to suspect TTP.5
Neurologic symptoms usually fluctuate. They can include mild abnormalities such as weakness, dizziness, headache, blurred vision, ataxia, and transient mental status changes, as well as severe abnormalities including stroke, seizure, and coma.2,6
Most patients have normal findings on computed tomography and magnetic resonance imaging at the onset of neurologic symptoms or with a history of TTP. Some patients (8%–39%) show reversible acute brain lesions, including ischemic changes.11–13
Other signs and symptoms may result from multiorgan failure due to microthrombosis; ischemia in retinal, coronary, and abdominal circulations; and unconjugated hyperbilirubinemia.2
Atypical presentations. About 18% of patients have cardiac involvement from microvascular occlusion, with arrhythmia, angina, or congestive heart failure. Abdominal pain and pancreatitis occur in 5% to 13%, and visual disturbances in 8% to 10%.
Patients with an atypical presentation may not have laboratory evidence of microangiopathic hemolytic anemia, but an ADAMTS13 assay will show severely decreased activity. Therapeutic plasma exchange can improve atypical symptoms.2,3,10,14,15
ADAMTS13 ASSAY IS KEY TO DIAGNOSIS
Laboratory evidence typically includes hemolytic anemia (reticulocytosis, schistocytes, elevated indirect bilirubin, reduced haptoglobin, elevated lactate dehydrogenase) and thrombocytopenia.3 There are no significant abnormalities in prothrombin time, international normalized ratio, activated partial thromboplastin time, fibrinogen, or D-dimer level.
Measuring the levels of ADAMTS13 activity, ADAMTS13 inhibitor, and ADAMTS13 antibody is becoming standard to confirm the diagnosis of TTP, to determine if it is congenital or acquired, and to distinguish it from thrombocytopenic conditions such as hemolytic-uremic syndrome, idiopathic thrombocytopenic purpura, and heparin-induced thrombocytopenia.4,5 A newer ADAMTS13 assay based on fluorescence energy transfer (FRET) technology with a synthetic amino acid-von Willebrand factor peptide substrate has a faster turnaround time and less test variability.6,16,17 This FRET assay can give the result of ADAMTS13 activity within 2 hours. In comparison, the assay based on multimeric von Willebrand factor takes 2 to 3 days, and mass spectrometry to measure the cleavage products of a synthetic von Willebrand factor molecule takes about 4 hours.3,10,16
About two-thirds of patients with the clinical diagnosis of idiopathic TTP have ADAMTS13 activity levels lower than 10%.5,14,18 In the appropriate clinical setting, this threshold level is highly sensitive (89%–100%) and specific (99%–100%) in differentiating TTP from other thrombotic angiopathies.2,3,18
Note: The ADAMTS13 assay was needed for early correct diagnosis in Case 1 and Case 2.
Inhibitors provide more clues
Autoantibodies can be classified according to whether they inhibit ADAMTS13 activity.
Neutralizing inhibitors. Most cases of acquired, idiopathic TTP with severe ADAMTS13 deficiency are related to circulating autoantibodies that neutralize ADAMTS13 activity. This ADAMTS13 inhibitor level is obtained by measuring residual ADAMTS13 activity after mixing equal amounts of patient plasma with normal pooled plasma. ADAMTS13 inhibitor is detectable in 44% to 93% of patients with severely deficient ADAMTS13 activity.3,6,19
Nonneutralizing inhibitors. From 10% to 15% of patients with TTP with severe ADAMTS13 deficiency lack ADAMTS13 autoantibodies measured by enzyme immunoassay but have nonneutralizing immunoglobulin G (IgG) or IgM autoantibodies. In such cases, ADAMTS13 deficiency may be related to increased antibody-mediated clearance or other unknown mechanisms.
Neutralizing inhibitors and nonneutralizing inhibitors may be present simultaneously in some patients.3,10,19,20
Blood factors affect ADAMTS13 activity
Specimen factors can affect ADAMTS13 activity and antibody levels.
Hemoglobin is a potent inhibitor of ADAMTS13, so an elevated plasma level of free hemoglobin (> 2 g/dL) can reduce ADAMTS13 activity, as can hyperbilirubinemia (> 15 mg/dL).
High levels of endogenous von Willebrand factor, lipids, thrombin, or other proteases that may cleave ADAMTS13 can also reduce ADAMTS13 activity.3 Conversely, recent plasma exchange or transfusion can mask the diagnosis of TTP because of false normalization of ADAMTS13 activity. In addition, ADAMTS13 autoantibody can be detected in other immune-mediated disorders (eg, systemic lupus erythematosus, antiphospholipid syndrome), and hypergammaglobulinemia, as well as in 10% to 15% of healthy individuals.19
CONSIDER OTHER CONDITIONS
Before diagnosing TTP, other conditions causing thrombocytopenia and hemolytic anemia should be excluded by taking a careful clinical, laboratory, and medication history (Table 2). Of these conditions, the most challenging to differentiate from TTP—and often indistinguishable from it at presentation—is hemolytic-uremic syndrome (Table 3).
Hemolytic-uremic syndrome
Hemolytic-uremic syndrome presents with a triad of thrombocytopenia, acute renal failure, and microangiopathic hemolytic anemia, with increased lactate dehydrogenase levels. Renal dysfunction from ischemia or tissue injury by microvascular thrombi predominates. Hemolytic-uremic syndrome most often occurs in children and is often related to hemorrhagic enterocolitis caused by infection with Escherichia coli O157:H7 or Shigella species (90%–95% of cases).1,2,5
From 5% to 10% of cases of hemolytic- uremic syndrome are atypical. These cases are not associated with diarrhea, and many are caused by genetic mutations that result in chronic excessive complement activation. Implicated genes regulate complement regulator factor H (20%–30% of cases) or CD46 (10%) and other cofactors, or autoantibodies against factor H (10%), which affect the alternate complement pathway.6,21–23
Initial therapeutic plasma exchange is commonly undertaken for atypical hemolytic- uremic syndrome, particularly for patients at risk of rapid progression to end-stage renal failure. But despite such treatment, about 60% of these patients die or develop permanent renal damage within 1 year.2,3,24
Eculizumab, a monoclonal antibody against complement component C5, has been approved by the US Food and Drug Administration for atypical hemolytic-uremic syndrome and may improve quality of life.25–27
PLASMA EXCHANGE IS THE MAINSTAY OF THERAPY
In 2012, the British Society for Haematology published revised guidelines for managing TTP and other thrombotic microangiopathies.28
Acquired idiopathic TTP with reduced ADAMTS13 activity requires immediate therapeutic plasma exchange. Daily plasma exchange combines plasmapheresis to remove circulating ultralarge von Willebrand factor-platelet strings and autoantibodies against ADAMTS13, and infusion of fresh-frozen plasma to replace ADAMTS13.18 This procedure is the mainstay of therapy and brings 70% to 90% of patients with idiopathic TTP to remission.1,2,5,6 However, the optimal duration of daily plasma exchange and the number of procedures required is highly variable according to clinical condition. Therapeutic plasma exchange can also cause plasma-related adverse reactions.9,28 Congenital TTP requires plasma infusion or exchange depending on the patient’s severity of ADAMTS13 deficiency.
Corticosteroids are used in combination with daily therapeutic plasma exchange, although evidence from controlled trials of their efficacy in this setting is lacking. Patients with severely decreased ADAMTS13 activity or low titers of ADAMTS13 autoantibodies tend to respond to the therapy.5,8,29
An ADAMTS13 assay with a short turn-around time can help guide the decision to initiate therapeutic plasma exchange. However, if there is a strong clinical suspicion of TTP, plasma exchange should be initiated immediately without waiting for test results.5,30 Monitoring ADAMTS13 activity or inhibitor during initial plasma exchange therapy has had conflicting results in several studies and is generally not recommended for patients with acquired TTP.8,30,31
RELAPSE IS COMMON
About 20% to 50% of patients with idiopathic TTP experience a relapse (Case 2). Most relapses occur within the first 2 years after the initial episode, with an estimated risk of 43% for relapse at 7.5 years.5,9
Factors that predict a higher risk of relapse include persistently severely decreased ADAMTS13 activity, positive inhibitor, and high titers of autoantibodies to ADAMTS13 during symptomatic TTP. During clinical remission, persistence of autoantibodies also indicates increased risk.1,3,5,6,9
Patients who have a relapse and whose disease is refractory to therapeutic plasma exchange (10%–20% of cases) have been treated with corticosteroids, splenectomy, or immunosuppressive agents (cyclosporine, azathioprine, or cyclophosphamide) with varying rates of success. Rituximab (monoclonal anti-CD20) has recently been used as second-line therapy in refractory or relapsing immune-mediated TTP or idiopathic TTP with neurologic or cardiac symptoms associated with a poor prognosis. Therapy including rituximab results in improved response and progression-free survival.32 Other potential therapies, including recombinant active ADAMTS13, are under investigation.9,23,28,30,33,34
A breakthrough in understanding the pathogenesis of thrombotic thrombocytopenic purpura (TTP) came with the discovery of ADAMTS13 (an abbreviation for “a disintegrin and metalloproteinase with thrombospondin type 1 motif, member 13”), a plasma protein that cleaves von Willebrand factor, which interacts with platelets to promote blood clotting. If ADAMTS13 is lacking, unusually large multimers of von Willebrand factor can accumulate and trigger intravascular platelet aggregation and microthrombosis, causing the signs and symptoms of TTP.1–3
This knowledge has practical applications: we can now measure ADAMTS13 activity, ADAMTS13 inhibitor, and antibodies against ADAMTS13 to help us diagnose TTP and distinguish it from other forms of thrombotic microangiopathy, such as hemolytic-uremic syndrome, that have similar symptoms but require different treatment.
Using case studies, this article describes typical presentations of acute and relapsing TTP; the role of laboratory testing, including the ADAMTS13 assay; how to distinguish TTP from other conditions that present similarly; and how to manage this condition.
A HIGH RISK OF DEATH WITHOUT PLASMA EXCHANGE
TTP is characterized by disseminated microthrombi composed of agglutinated platelets and von Willebrand factor in small vessels. Tissue damage by microthrombi can cause thrombocytopenia (platelet deficiency), microangiopathic hemolytic anemia (loss of red blood cells caused by destructive conditions in small vessels), and multiorgan failure.1
Untreated TTP has a mortality rate of about 90%.1 As shown in Case 1, Case 2, and Table 1, rapid diagnosis and prompt initiation of daily therapeutic plasma exchange can improve this grave outlook.4
ADAMTS13 DEFICIENCY CAN BE ACQUIRED OR CONGENITAL
Two major forms of TTP with ADAMTS13 deficiency and microvascular thrombosis are recognized:
Acquired TTP, the more common form, peaks in incidence between ages 30 and 50.2,5 It more often affects women, particularly during and after pregnancy (its estimated prevalence is 1 in 25,000 pregnancies), and African Americans.6 Acquired TTP may be:
- Primary (idiopathic or autoantibody-mediated), associated with severely decreased ADAMTS13 and the presence of ultra-large von Willebrand factor multimers, or
- Secondary (23%–67% of cases), arising from a variety of conditions, including autoimmune disorders (eg, systemic lupus erythematosus, rheumatoid arthritis), solid organ or hematopoietic cell transplant, malignancy, drugs, and pregnancy (Table 2).1,5–8 Secondary TTP has a worse prognosis than idiopathic TTP.5,9
Congenital TTP (Upshaw-Shulman syndrome) is a rare autosomal-recessive disease caused by compound heterozygous or homozygous mutations of the ADAMTS13 gene, producing nonfunctional ADAMTS13 protein. Patients have severely deficient ADAMTS13 activity but usually do not develop autoantibodies. There is a high risk of chronic, relapsing episodes; identified triggers include pregnancy and heavy alcohol intake.2,10 About half of patients with congenital TTP have an early onset, usually presenting with acute TTP between birth and age 5, and about half have a late onset, usually remaining without symptoms until age 20 to 40.
THE CLINICAL PICTURE OF TTP IS NOT ALWAYS CLASSIC
TTP is primarily diagnosed clinically, but diagnosis is often difficult because of various nonspecific symptoms. Typical TTP presents with the “classic pentad”:
- Severe thrombocytopenia (70%–100% of patients)
- Microangiopathic hemolytic anemia with multiple schistocytes (70%–100%) (Figure 1)
- Neurologic involvement (50%–90%)
- Renal abnormalities (about 50%)
- Fever (25%).
However, the entire picture often does not emerge in a single patient.2,6 Waiting for the entire pentad to develop before diagnosing TTP can have grave clinical consequences,1,2,5 and the presence of thrombocytopenia and unexplained microangiopathic hemolytic anemia are considered clinically sufficient to suspect TTP.5
Neurologic symptoms usually fluctuate. They can include mild abnormalities such as weakness, dizziness, headache, blurred vision, ataxia, and transient mental status changes, as well as severe abnormalities including stroke, seizure, and coma.2,6
Most patients have normal findings on computed tomography and magnetic resonance imaging at the onset of neurologic symptoms or with a history of TTP. Some patients (8%–39%) show reversible acute brain lesions, including ischemic changes.11–13
Other signs and symptoms may result from multiorgan failure due to microthrombosis; ischemia in retinal, coronary, and abdominal circulations; and unconjugated hyperbilirubinemia.2
Atypical presentations. About 18% of patients have cardiac involvement from microvascular occlusion, with arrhythmia, angina, or congestive heart failure. Abdominal pain and pancreatitis occur in 5% to 13%, and visual disturbances in 8% to 10%.
Patients with an atypical presentation may not have laboratory evidence of microangiopathic hemolytic anemia, but an ADAMTS13 assay will show severely decreased activity. Therapeutic plasma exchange can improve atypical symptoms.2,3,10,14,15
ADAMTS13 ASSAY IS KEY TO DIAGNOSIS
Laboratory evidence typically includes hemolytic anemia (reticulocytosis, schistocytes, elevated indirect bilirubin, reduced haptoglobin, elevated lactate dehydrogenase) and thrombocytopenia.3 There are no significant abnormalities in prothrombin time, international normalized ratio, activated partial thromboplastin time, fibrinogen, or D-dimer level.
Measuring the levels of ADAMTS13 activity, ADAMTS13 inhibitor, and ADAMTS13 antibody is becoming standard to confirm the diagnosis of TTP, to determine if it is congenital or acquired, and to distinguish it from thrombocytopenic conditions such as hemolytic-uremic syndrome, idiopathic thrombocytopenic purpura, and heparin-induced thrombocytopenia.4,5 A newer ADAMTS13 assay based on fluorescence energy transfer (FRET) technology with a synthetic amino acid-von Willebrand factor peptide substrate has a faster turnaround time and less test variability.6,16,17 This FRET assay can give the result of ADAMTS13 activity within 2 hours. In comparison, the assay based on multimeric von Willebrand factor takes 2 to 3 days, and mass spectrometry to measure the cleavage products of a synthetic von Willebrand factor molecule takes about 4 hours.3,10,16
About two-thirds of patients with the clinical diagnosis of idiopathic TTP have ADAMTS13 activity levels lower than 10%.5,14,18 In the appropriate clinical setting, this threshold level is highly sensitive (89%–100%) and specific (99%–100%) in differentiating TTP from other thrombotic angiopathies.2,3,18
Note: The ADAMTS13 assay was needed for early correct diagnosis in Case 1 and Case 2.
Inhibitors provide more clues
Autoantibodies can be classified according to whether they inhibit ADAMTS13 activity.
Neutralizing inhibitors. Most cases of acquired, idiopathic TTP with severe ADAMTS13 deficiency are related to circulating autoantibodies that neutralize ADAMTS13 activity. This ADAMTS13 inhibitor level is obtained by measuring residual ADAMTS13 activity after mixing equal amounts of patient plasma with normal pooled plasma. ADAMTS13 inhibitor is detectable in 44% to 93% of patients with severely deficient ADAMTS13 activity.3,6,19
Nonneutralizing inhibitors. From 10% to 15% of patients with TTP with severe ADAMTS13 deficiency lack ADAMTS13 autoantibodies measured by enzyme immunoassay but have nonneutralizing immunoglobulin G (IgG) or IgM autoantibodies. In such cases, ADAMTS13 deficiency may be related to increased antibody-mediated clearance or other unknown mechanisms.
Neutralizing inhibitors and nonneutralizing inhibitors may be present simultaneously in some patients.3,10,19,20
Blood factors affect ADAMTS13 activity
Specimen factors can affect ADAMTS13 activity and antibody levels.
Hemoglobin is a potent inhibitor of ADAMTS13, so an elevated plasma level of free hemoglobin (> 2 g/dL) can reduce ADAMTS13 activity, as can hyperbilirubinemia (> 15 mg/dL).
High levels of endogenous von Willebrand factor, lipids, thrombin, or other proteases that may cleave ADAMTS13 can also reduce ADAMTS13 activity.3 Conversely, recent plasma exchange or transfusion can mask the diagnosis of TTP because of false normalization of ADAMTS13 activity. In addition, ADAMTS13 autoantibody can be detected in other immune-mediated disorders (eg, systemic lupus erythematosus, antiphospholipid syndrome), and hypergammaglobulinemia, as well as in 10% to 15% of healthy individuals.19
CONSIDER OTHER CONDITIONS
Before diagnosing TTP, other conditions causing thrombocytopenia and hemolytic anemia should be excluded by taking a careful clinical, laboratory, and medication history (Table 2). Of these conditions, the most challenging to differentiate from TTP—and often indistinguishable from it at presentation—is hemolytic-uremic syndrome (Table 3).
Hemolytic-uremic syndrome
Hemolytic-uremic syndrome presents with a triad of thrombocytopenia, acute renal failure, and microangiopathic hemolytic anemia, with increased lactate dehydrogenase levels. Renal dysfunction from ischemia or tissue injury by microvascular thrombi predominates. Hemolytic-uremic syndrome most often occurs in children and is often related to hemorrhagic enterocolitis caused by infection with Escherichia coli O157:H7 or Shigella species (90%–95% of cases).1,2,5
From 5% to 10% of cases of hemolytic- uremic syndrome are atypical. These cases are not associated with diarrhea, and many are caused by genetic mutations that result in chronic excessive complement activation. Implicated genes regulate complement regulator factor H (20%–30% of cases) or CD46 (10%) and other cofactors, or autoantibodies against factor H (10%), which affect the alternate complement pathway.6,21–23
Initial therapeutic plasma exchange is commonly undertaken for atypical hemolytic- uremic syndrome, particularly for patients at risk of rapid progression to end-stage renal failure. But despite such treatment, about 60% of these patients die or develop permanent renal damage within 1 year.2,3,24
Eculizumab, a monoclonal antibody against complement component C5, has been approved by the US Food and Drug Administration for atypical hemolytic-uremic syndrome and may improve quality of life.25–27
PLASMA EXCHANGE IS THE MAINSTAY OF THERAPY
In 2012, the British Society for Haematology published revised guidelines for managing TTP and other thrombotic microangiopathies.28
Acquired idiopathic TTP with reduced ADAMTS13 activity requires immediate therapeutic plasma exchange. Daily plasma exchange combines plasmapheresis to remove circulating ultralarge von Willebrand factor-platelet strings and autoantibodies against ADAMTS13, and infusion of fresh-frozen plasma to replace ADAMTS13.18 This procedure is the mainstay of therapy and brings 70% to 90% of patients with idiopathic TTP to remission.1,2,5,6 However, the optimal duration of daily plasma exchange and the number of procedures required is highly variable according to clinical condition. Therapeutic plasma exchange can also cause plasma-related adverse reactions.9,28 Congenital TTP requires plasma infusion or exchange depending on the patient’s severity of ADAMTS13 deficiency.
Corticosteroids are used in combination with daily therapeutic plasma exchange, although evidence from controlled trials of their efficacy in this setting is lacking. Patients with severely decreased ADAMTS13 activity or low titers of ADAMTS13 autoantibodies tend to respond to the therapy.5,8,29
An ADAMTS13 assay with a short turn-around time can help guide the decision to initiate therapeutic plasma exchange. However, if there is a strong clinical suspicion of TTP, plasma exchange should be initiated immediately without waiting for test results.5,30 Monitoring ADAMTS13 activity or inhibitor during initial plasma exchange therapy has had conflicting results in several studies and is generally not recommended for patients with acquired TTP.8,30,31
RELAPSE IS COMMON
About 20% to 50% of patients with idiopathic TTP experience a relapse (Case 2). Most relapses occur within the first 2 years after the initial episode, with an estimated risk of 43% for relapse at 7.5 years.5,9
Factors that predict a higher risk of relapse include persistently severely decreased ADAMTS13 activity, positive inhibitor, and high titers of autoantibodies to ADAMTS13 during symptomatic TTP. During clinical remission, persistence of autoantibodies also indicates increased risk.1,3,5,6,9
Patients who have a relapse and whose disease is refractory to therapeutic plasma exchange (10%–20% of cases) have been treated with corticosteroids, splenectomy, or immunosuppressive agents (cyclosporine, azathioprine, or cyclophosphamide) with varying rates of success. Rituximab (monoclonal anti-CD20) has recently been used as second-line therapy in refractory or relapsing immune-mediated TTP or idiopathic TTP with neurologic or cardiac symptoms associated with a poor prognosis. Therapy including rituximab results in improved response and progression-free survival.32 Other potential therapies, including recombinant active ADAMTS13, are under investigation.9,23,28,30,33,34
- Sadler JE, Moake JL, Miyata T, George JN. Recent advances in thrombotic thrombocytopenic purpura. Hematology Am Soc Hematol Educ Program 2004; 1:407–423.
- Shenkman B, Einav Y. Thrombotic thrombocytopenic purpura and other thrombotic microangiopathic hemolytic anemias: diagnosis and classification. Autoimmun Rev 2014; 13:584–586.
- Shah N, Sarode R. Thrombotic thrombocytopenic purpura-what is new? J Clin Apher 2013; 28:30–35.
- Imanirad I, Rajasekhar A, Zumberg M. A case series of atypical presentations of thrombotic thrombocytopenic purpura. J Clin Apher 2012; 27:221–226.
- George JN, Al-Nouri ZL. Diagnostic and therapeutic challenges in the thrombotic thrombocytopenic purpura and hemolytic uremic syndromes. Hematology Am Soc Hematol Educ Program 2012; 1:604–609.
- Shah N, Rutherford C, Matevosyan K, Shen YM, Sarode R. Role of ADAMTS13 in the management of thrombotic microangiopathies including thrombotic thrombocytopenic purpura (TTP). Br J Haematol 2013; 163:514–519.
- Cataland SR, Yang S, Wu HM. The use of ADAMTS13 activity, platelet count, and serum creatinine to differentiate acquired thrombotic thrombocytopenic purpura from other thrombotic microangiopathies. Br J Haematol 2012; 157:501–503.
- Mannucci PM, Peyvandi F. TTP and ADAMTS13: when Is testing appropriate? Hematology Am Soc Hematol Educ Program 2007; 1:121–126.
- Chaturved S, Carcioppolo D, Zhang L, McCar KR. Management and outcomes of patients with TTP: analysis of 100 cases at a single institution. Am J Hematol 2013; 88:560–565.
- Peyvandi F, Palla R, Lotta LA, Mackie I, Scully MA, Machin SJ. ADAMTS-13 assays in thrombotic thrombocytopenic purpura. J Thromb Haemost 2010; 8:631–640.
- Cataland SR, Scully MA, Paskavitz J, et al. Evidence of persistent neurologic injury following thrombotic thrombocytopenic purpura. Am J Hematol 2011; 86:87–89.
- Meloni G, Proia A, Antonini G, et al. Thrombotic thrombocytopenic purpura: prospective neurologic, neuroimaging and neurophysiologic evaluation. Haematologica 2001; 86:1194–1199.
- Kwaan HC, Boggio LN. The clinical spectrum of thrombotic thrombocytopenic purpura. Semin Thromb Hemost 2005; 31:673–680.
- Sarode R. Atypical presentations of thrombotic thrombocytopenic purpura: a review. J Clin Apher 2009; 24:47–52.
- Volcy J, Nzerue CM, Oderinde A, Hewan-Iowe K. Cocaine-induced acute renal failure, hemolysis, and thrombocytopenia mimicking thrombotic thrombocytopenic purpura. Am J Kidney Dis 2000; 35:E3.
- Kremer Hovinga JA, Mottini M, Lammle B. Measurement of ADAMTS-13 activity in plasma by the FRETS-VWF73 assay: comparison with other assay methods. J Thromb Haemost 2006; 4:1146–1148.
- Groot E, Hulstein JJ, Rison CN, de Groot PG, Fijnheer R. FRETS-VWF73: a rapid and predictive tool for thrombotic thrombocytopenic purpura. J Thromb Haemost 2006; 4:698–699.
- Barrows BD, Teruya J. Use of the ADAMTS13 activity assay improved the accuracy and efficiency of the diagnosis and treatment of suspected acquired thrombotic thrombocytopenic purpura. Arch Pathol Lab Med 2014; 138:546–549.
- Rieger M, Mannucci PM, Kremer Hovinga JA, et al. ADAMTS13 autoantibodies in patients with thrombotic microangiopathies and other immunomediated diseases. Blood 2005; 106:1262–1267.
- Rogers HJ, Kottke-Marchant K. ADAMTS13 evaluation for thrombotic thrombocytopenic purpura. Pathology Innovations, Pathology and Laboratory Medicine Institute. Cleveland Clinic, Fall 2014:6–9.
- Józsi M, Licht C, Strobel S, et al. Factor H autoantibodies in atypical hemolytic uremic syndrome correlate with CFHR1/CFHR3 deficiency. Blood 2008; 111:1512–1514.
- Diamante Chiodini B, Davin JC, Corazza F, et al. Eculizumab in anti-factor H antibodies associated with atypical hemolytic uremic syndrome. Pediatrics 2014; 133:e1764–e1768.
- Taylor CM, Machin S, Wigmore SJ, Goodship TH; working party from the Renal Association, the British Committee for Standards in Haematology and the British Transplantation Society. Clinical practice guidelines for the management of atypical haemolytic uraemic syndrome in the United Kingdom. Br J Haematol 2009; 148:37–47.
- Loirat C, Garnier A, Sellier-Leclerc AL, Kwon T. Plasmatherapy in atypical hemolytic uremic syndrome. Semin Thromb Hemost 2010; 36:673–681.
- Tsai HM, Kuo E. Eculizumab therapy leads to rapid resolution of thrombocytopenia in atypical hemolytic uremic syndrome. Adv Hematol 2014; 295323:1–7.
- Lapeyraque AL, Frémeaux-Bacchi V, Robitaille P. Efficacy of eculizumab in a patient with factor-H-associated atypical hemolytic uremic syndrome. Pediatr Nephrol 2011; 26:621–624.
- Baskin E, Gulleroglu K, Kantar A, Bayrakci U, Ozkaya O. Success of eculizumab in the treatment of atypical hemolytic uremic syndrome. Pediatr Nephrol 2015; 30:783–789.
- Scully M, Hunt BJ, Benjamin S, et al; British Committee for Standards in Haematology. Guidelines on the diagnosis and management of thrombotic thrombocytopenic purpura and other thrombotic microangiopathies. Br J Haematol 2012; 158:323–325.
- Abassi E, Yawn D, Leveque E, Nolasco L, Lopez J, Moake J. Correlation of ADAMTS-13 activity with response to plasma exchange in patients diagnosed with thrombotic thrombocytopenic purpura (Abstract #3921). Blood 2004; 104:242a.
- Blombery P, Scully M. Management of thrombocytic thrombocytopenic purpura: current perspectives. J Blood Med 2014; 5:15–23.
- Wu N, Liu J, Yang S, et al. Diagnostic and prognostic values of ADAMTS13 activity measured during daily plasma exchange therapy in patients with acquired thrombotic thrombocytopenic purpura. Transfusion 2015; 55:18–24.
- Cuker A. Adjuvant rituximab to prevent TTP relapse. Blood 2016; 127:2952–2953.
- Chapman K, Yuen S. Therapy for thrombotic thrombocytopenic purpura: past, present and future. Semin Thromb Hemost 2014; 40:34–40.
- Heidel F, Lipka DB, von Auer C, Huber C, Schrarrer I, Hess G. Addition of rituximab to standard therapy improves response rate and progression-free survival in relapsed or refractory thrombotic thrombocytopenic purpura and autoimmune haemolytic anaemia. Thromb Haemost 2007; 97:228–233.
- Sadler JE, Moake JL, Miyata T, George JN. Recent advances in thrombotic thrombocytopenic purpura. Hematology Am Soc Hematol Educ Program 2004; 1:407–423.
- Shenkman B, Einav Y. Thrombotic thrombocytopenic purpura and other thrombotic microangiopathic hemolytic anemias: diagnosis and classification. Autoimmun Rev 2014; 13:584–586.
- Shah N, Sarode R. Thrombotic thrombocytopenic purpura-what is new? J Clin Apher 2013; 28:30–35.
- Imanirad I, Rajasekhar A, Zumberg M. A case series of atypical presentations of thrombotic thrombocytopenic purpura. J Clin Apher 2012; 27:221–226.
- George JN, Al-Nouri ZL. Diagnostic and therapeutic challenges in the thrombotic thrombocytopenic purpura and hemolytic uremic syndromes. Hematology Am Soc Hematol Educ Program 2012; 1:604–609.
- Shah N, Rutherford C, Matevosyan K, Shen YM, Sarode R. Role of ADAMTS13 in the management of thrombotic microangiopathies including thrombotic thrombocytopenic purpura (TTP). Br J Haematol 2013; 163:514–519.
- Cataland SR, Yang S, Wu HM. The use of ADAMTS13 activity, platelet count, and serum creatinine to differentiate acquired thrombotic thrombocytopenic purpura from other thrombotic microangiopathies. Br J Haematol 2012; 157:501–503.
- Mannucci PM, Peyvandi F. TTP and ADAMTS13: when Is testing appropriate? Hematology Am Soc Hematol Educ Program 2007; 1:121–126.
- Chaturved S, Carcioppolo D, Zhang L, McCar KR. Management and outcomes of patients with TTP: analysis of 100 cases at a single institution. Am J Hematol 2013; 88:560–565.
- Peyvandi F, Palla R, Lotta LA, Mackie I, Scully MA, Machin SJ. ADAMTS-13 assays in thrombotic thrombocytopenic purpura. J Thromb Haemost 2010; 8:631–640.
- Cataland SR, Scully MA, Paskavitz J, et al. Evidence of persistent neurologic injury following thrombotic thrombocytopenic purpura. Am J Hematol 2011; 86:87–89.
- Meloni G, Proia A, Antonini G, et al. Thrombotic thrombocytopenic purpura: prospective neurologic, neuroimaging and neurophysiologic evaluation. Haematologica 2001; 86:1194–1199.
- Kwaan HC, Boggio LN. The clinical spectrum of thrombotic thrombocytopenic purpura. Semin Thromb Hemost 2005; 31:673–680.
- Sarode R. Atypical presentations of thrombotic thrombocytopenic purpura: a review. J Clin Apher 2009; 24:47–52.
- Volcy J, Nzerue CM, Oderinde A, Hewan-Iowe K. Cocaine-induced acute renal failure, hemolysis, and thrombocytopenia mimicking thrombotic thrombocytopenic purpura. Am J Kidney Dis 2000; 35:E3.
- Kremer Hovinga JA, Mottini M, Lammle B. Measurement of ADAMTS-13 activity in plasma by the FRETS-VWF73 assay: comparison with other assay methods. J Thromb Haemost 2006; 4:1146–1148.
- Groot E, Hulstein JJ, Rison CN, de Groot PG, Fijnheer R. FRETS-VWF73: a rapid and predictive tool for thrombotic thrombocytopenic purpura. J Thromb Haemost 2006; 4:698–699.
- Barrows BD, Teruya J. Use of the ADAMTS13 activity assay improved the accuracy and efficiency of the diagnosis and treatment of suspected acquired thrombotic thrombocytopenic purpura. Arch Pathol Lab Med 2014; 138:546–549.
- Rieger M, Mannucci PM, Kremer Hovinga JA, et al. ADAMTS13 autoantibodies in patients with thrombotic microangiopathies and other immunomediated diseases. Blood 2005; 106:1262–1267.
- Rogers HJ, Kottke-Marchant K. ADAMTS13 evaluation for thrombotic thrombocytopenic purpura. Pathology Innovations, Pathology and Laboratory Medicine Institute. Cleveland Clinic, Fall 2014:6–9.
- Józsi M, Licht C, Strobel S, et al. Factor H autoantibodies in atypical hemolytic uremic syndrome correlate with CFHR1/CFHR3 deficiency. Blood 2008; 111:1512–1514.
- Diamante Chiodini B, Davin JC, Corazza F, et al. Eculizumab in anti-factor H antibodies associated with atypical hemolytic uremic syndrome. Pediatrics 2014; 133:e1764–e1768.
- Taylor CM, Machin S, Wigmore SJ, Goodship TH; working party from the Renal Association, the British Committee for Standards in Haematology and the British Transplantation Society. Clinical practice guidelines for the management of atypical haemolytic uraemic syndrome in the United Kingdom. Br J Haematol 2009; 148:37–47.
- Loirat C, Garnier A, Sellier-Leclerc AL, Kwon T. Plasmatherapy in atypical hemolytic uremic syndrome. Semin Thromb Hemost 2010; 36:673–681.
- Tsai HM, Kuo E. Eculizumab therapy leads to rapid resolution of thrombocytopenia in atypical hemolytic uremic syndrome. Adv Hematol 2014; 295323:1–7.
- Lapeyraque AL, Frémeaux-Bacchi V, Robitaille P. Efficacy of eculizumab in a patient with factor-H-associated atypical hemolytic uremic syndrome. Pediatr Nephrol 2011; 26:621–624.
- Baskin E, Gulleroglu K, Kantar A, Bayrakci U, Ozkaya O. Success of eculizumab in the treatment of atypical hemolytic uremic syndrome. Pediatr Nephrol 2015; 30:783–789.
- Scully M, Hunt BJ, Benjamin S, et al; British Committee for Standards in Haematology. Guidelines on the diagnosis and management of thrombotic thrombocytopenic purpura and other thrombotic microangiopathies. Br J Haematol 2012; 158:323–325.
- Abassi E, Yawn D, Leveque E, Nolasco L, Lopez J, Moake J. Correlation of ADAMTS-13 activity with response to plasma exchange in patients diagnosed with thrombotic thrombocytopenic purpura (Abstract #3921). Blood 2004; 104:242a.
- Blombery P, Scully M. Management of thrombocytic thrombocytopenic purpura: current perspectives. J Blood Med 2014; 5:15–23.
- Wu N, Liu J, Yang S, et al. Diagnostic and prognostic values of ADAMTS13 activity measured during daily plasma exchange therapy in patients with acquired thrombotic thrombocytopenic purpura. Transfusion 2015; 55:18–24.
- Cuker A. Adjuvant rituximab to prevent TTP relapse. Blood 2016; 127:2952–2953.
- Chapman K, Yuen S. Therapy for thrombotic thrombocytopenic purpura: past, present and future. Semin Thromb Hemost 2014; 40:34–40.
- Heidel F, Lipka DB, von Auer C, Huber C, Schrarrer I, Hess G. Addition of rituximab to standard therapy improves response rate and progression-free survival in relapsed or refractory thrombotic thrombocytopenic purpura and autoimmune haemolytic anaemia. Thromb Haemost 2007; 97:228–233.
KEY POINTS
- Symptoms of TTP are usually neurologic but can also be cardiac or abdominal. Thrombocytopenia and unexplained microangiopathic hemolytic anemia are sufficient to highly suspect the disease.
- In the appropriate clinical setting, an ADAMTS13 activity level lower than 10% is highly indicative of TTP.
- ADAMTS13 inhibitor and ADAMTS13 antibody assays provide more diagnostic clues. ADAMTS13 antibody is generally absent in the congenital form.
- The ADAMTS13 assay can help distinguish TTP from hemolytic-uremic syndrome, which presents similarly but typically involves normal or only mildly reduced ADAMTS13 activity.
- A strong clinical suspicion of TTP warrants immediate initiation of therapeutic plasma exchange without waiting for ADAMTS13 test results.
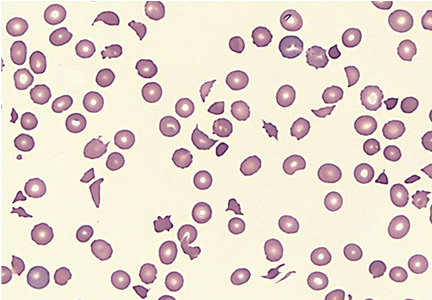
When the diagnosis is hard to swallow, take these management steps
CASE REPORTMr. C, age 72, reports a lack of desire to swallow food. He denies feeling a lump in his throat. Over the past 6 months, he lost >30 lb.
The patient had a similar episode 2 years ago, which resolved without intervention. The death of his wife recently has led to isolation and lack of desire to swallow food.
Testing with standard food samples to elicit eating behaviors is normal. Electromyography and video fluoroscopy test results show no abnormalities.
What is phagophobia?The case of Mr. C brings to light the condition known as phagophobia—a sensation of not being able to swallow. Phagophobia mimics oral apraxia; pharyngoesophageal and neurologic functions as well as the ability to speak remain intact, however.1
It is estimated that about 6% of the adult general population reports dysphagia.2 About 47% of patients with dysphagic complaints do not show motor-manometric or radiological abnormalities of the upper digestive tract. A number of psychiatric conditions, including panic disorder, obsessive-compulsive disorder, social phobia, anorexia nervosa, globus hystericus, hypersensitive gag reflex, and posttraumatic stress disorder can simulate this condition.3
When Barofsky and Fontaine4 compared phagophobia patients with other subjects—healthy controls, anorexia nervosa restrictors, dysphagic patients with esophageal obstruction, dysphagic patients with motility disturbance, and patients with non-motility non-obstructive dysphagia—they found that patients with psychogenic dysphagia did not appear to have an eating disorder. However, they did have a clinically significant level of psychological distress, particularly anxiety.
Diagnostic tools and management stepsThere are a number of approaches to assess your patient’s fear of swallowing (Table,5-7 page 68). Non-invasive assessment tools along with educational modalities usually are tried alone or together with psychopharmacological intervention. It is, however, imperative that you have an empathetic and understanding approach to such patients. When patients have confidence in the clinician they tend to respond more effectively with such approaches.

Investigations4 include questionnaires (swallow disorder history, Eating Disorder Inventory-2, and Symptom Checklist–90-R); weight assessment; testing with standardized food samples to elicit eating behaviors; self-reports; electromyography; and videofluoroscopy.
Education and reassurance includes individual demonstration of swallowing, combined with group therapy, exercises, and reassurance. Patients benefit from advice on how to maximize sensation within the oropharynx to increase taste, perception of temperature, and texture stimulation.8
Behavioral intervention involves practicing slow breathing and muscle relaxation techniques to gradually increase bite size and reduce the amount of time spent chewing each bite.
Introspection therapycomprises psychoeducation, cognitive restructuring, and in vivo and introspective exposure; helps patients replace anxiety-producing thoughts with probability estimation and decatastrophizing. Introspective exposure targets the fear of choking by having the patient create sensations of throat tightening by holding a swallow in mid-action and by rapid swallowing. In vivo exposure targets the fear of swallowing by having the patient practice feeding foods (such as semi-solid easy-to-swallow choices), in and outside of the session.6
Aversion therapy requires that you pinch the patient’s hand while he (she) chews, and release the hand when he swallows.
Psychopharmacotherapeutic intervention. A number of medications can be used to help, such as imipramine up to 150 mg; desipramine, up to 150 mg; or lorazepam, 0.25 mg, twice daily, to address anxiety or panic symptoms.
Acknowledgment
Duy Li, BS, and Yu Hsuan Liao, BS, contributed to the development of the manuscript of this article.
1. Evans IM, Pia P. Phagophobia: behavioral treatment of a complex case involving fear of fear. Clinical Case Studies. 2011;10(1):37-52.
2. Kim CH, Hsu JJ, Williams DE, et al. A prospective psychological evaluation of patients with dysphagia of various etiologies. Dysphagia. 1996;11(1):34-40.
3. McNally RJ. Choking phobia: a review of the literature. Compr Psychiatry. 1994;35(1):83-89.
4. Barofsky I, Fontaine KR. Do psychogenic dysphagia patients have an eating disorder? Dysphagia. 1998;13(1):24-27.
5. Bishop LC, Riley WT. The psychiatric management of the globus syndrome. Gen Hosp Psychiatry. 1988;10(3):214-219.
6. Ball SG, Otto MW. Cognitive-behavioral treatment of choking phobia: 3 case studies Psychother Psychosom. 1994;62(3-4):207-211.
7. Epstein SJ, Deyoub P. Hypnotherapy for fear of choking: treatment implications of a case report. Int J Clin Hypn. 1981;29(2):117-127.
8. Scemes S, Wielenska RC, Savoia MG, et al. Choking phobia: full remission following behavior therapy. Rev Bras Psiquiatr. 2009;31(3):257-260.
CASE REPORTMr. C, age 72, reports a lack of desire to swallow food. He denies feeling a lump in his throat. Over the past 6 months, he lost >30 lb.
The patient had a similar episode 2 years ago, which resolved without intervention. The death of his wife recently has led to isolation and lack of desire to swallow food.
Testing with standard food samples to elicit eating behaviors is normal. Electromyography and video fluoroscopy test results show no abnormalities.
What is phagophobia?The case of Mr. C brings to light the condition known as phagophobia—a sensation of not being able to swallow. Phagophobia mimics oral apraxia; pharyngoesophageal and neurologic functions as well as the ability to speak remain intact, however.1
It is estimated that about 6% of the adult general population reports dysphagia.2 About 47% of patients with dysphagic complaints do not show motor-manometric or radiological abnormalities of the upper digestive tract. A number of psychiatric conditions, including panic disorder, obsessive-compulsive disorder, social phobia, anorexia nervosa, globus hystericus, hypersensitive gag reflex, and posttraumatic stress disorder can simulate this condition.3
When Barofsky and Fontaine4 compared phagophobia patients with other subjects—healthy controls, anorexia nervosa restrictors, dysphagic patients with esophageal obstruction, dysphagic patients with motility disturbance, and patients with non-motility non-obstructive dysphagia—they found that patients with psychogenic dysphagia did not appear to have an eating disorder. However, they did have a clinically significant level of psychological distress, particularly anxiety.
Diagnostic tools and management stepsThere are a number of approaches to assess your patient’s fear of swallowing (Table,5-7 page 68). Non-invasive assessment tools along with educational modalities usually are tried alone or together with psychopharmacological intervention. It is, however, imperative that you have an empathetic and understanding approach to such patients. When patients have confidence in the clinician they tend to respond more effectively with such approaches.
Investigations4 include questionnaires (swallow disorder history, Eating Disorder Inventory-2, and Symptom Checklist–90-R); weight assessment; testing with standardized food samples to elicit eating behaviors; self-reports; electromyography; and videofluoroscopy.
Education and reassurance includes individual demonstration of swallowing, combined with group therapy, exercises, and reassurance. Patients benefit from advice on how to maximize sensation within the oropharynx to increase taste, perception of temperature, and texture stimulation.8
Behavioral intervention involves practicing slow breathing and muscle relaxation techniques to gradually increase bite size and reduce the amount of time spent chewing each bite.
Introspection therapycomprises psychoeducation, cognitive restructuring, and in vivo and introspective exposure; helps patients replace anxiety-producing thoughts with probability estimation and decatastrophizing. Introspective exposure targets the fear of choking by having the patient create sensations of throat tightening by holding a swallow in mid-action and by rapid swallowing. In vivo exposure targets the fear of swallowing by having the patient practice feeding foods (such as semi-solid easy-to-swallow choices), in and outside of the session.6
Aversion therapy requires that you pinch the patient’s hand while he (she) chews, and release the hand when he swallows.
Psychopharmacotherapeutic intervention. A number of medications can be used to help, such as imipramine up to 150 mg; desipramine, up to 150 mg; or lorazepam, 0.25 mg, twice daily, to address anxiety or panic symptoms.
Acknowledgment
Duy Li, BS, and Yu Hsuan Liao, BS, contributed to the development of the manuscript of this article.
CASE REPORTMr. C, age 72, reports a lack of desire to swallow food. He denies feeling a lump in his throat. Over the past 6 months, he lost >30 lb.
The patient had a similar episode 2 years ago, which resolved without intervention. The death of his wife recently has led to isolation and lack of desire to swallow food.
Testing with standard food samples to elicit eating behaviors is normal. Electromyography and video fluoroscopy test results show no abnormalities.
What is phagophobia?The case of Mr. C brings to light the condition known as phagophobia—a sensation of not being able to swallow. Phagophobia mimics oral apraxia; pharyngoesophageal and neurologic functions as well as the ability to speak remain intact, however.1
It is estimated that about 6% of the adult general population reports dysphagia.2 About 47% of patients with dysphagic complaints do not show motor-manometric or radiological abnormalities of the upper digestive tract. A number of psychiatric conditions, including panic disorder, obsessive-compulsive disorder, social phobia, anorexia nervosa, globus hystericus, hypersensitive gag reflex, and posttraumatic stress disorder can simulate this condition.3
When Barofsky and Fontaine4 compared phagophobia patients with other subjects—healthy controls, anorexia nervosa restrictors, dysphagic patients with esophageal obstruction, dysphagic patients with motility disturbance, and patients with non-motility non-obstructive dysphagia—they found that patients with psychogenic dysphagia did not appear to have an eating disorder. However, they did have a clinically significant level of psychological distress, particularly anxiety.
Diagnostic tools and management stepsThere are a number of approaches to assess your patient’s fear of swallowing (Table,5-7 page 68). Non-invasive assessment tools along with educational modalities usually are tried alone or together with psychopharmacological intervention. It is, however, imperative that you have an empathetic and understanding approach to such patients. When patients have confidence in the clinician they tend to respond more effectively with such approaches.
Investigations4 include questionnaires (swallow disorder history, Eating Disorder Inventory-2, and Symptom Checklist–90-R); weight assessment; testing with standardized food samples to elicit eating behaviors; self-reports; electromyography; and videofluoroscopy.
Education and reassurance includes individual demonstration of swallowing, combined with group therapy, exercises, and reassurance. Patients benefit from advice on how to maximize sensation within the oropharynx to increase taste, perception of temperature, and texture stimulation.8
Behavioral intervention involves practicing slow breathing and muscle relaxation techniques to gradually increase bite size and reduce the amount of time spent chewing each bite.
Introspection therapycomprises psychoeducation, cognitive restructuring, and in vivo and introspective exposure; helps patients replace anxiety-producing thoughts with probability estimation and decatastrophizing. Introspective exposure targets the fear of choking by having the patient create sensations of throat tightening by holding a swallow in mid-action and by rapid swallowing. In vivo exposure targets the fear of swallowing by having the patient practice feeding foods (such as semi-solid easy-to-swallow choices), in and outside of the session.6
Aversion therapy requires that you pinch the patient’s hand while he (she) chews, and release the hand when he swallows.
Psychopharmacotherapeutic intervention. A number of medications can be used to help, such as imipramine up to 150 mg; desipramine, up to 150 mg; or lorazepam, 0.25 mg, twice daily, to address anxiety or panic symptoms.
Acknowledgment
Duy Li, BS, and Yu Hsuan Liao, BS, contributed to the development of the manuscript of this article.
1. Evans IM, Pia P. Phagophobia: behavioral treatment of a complex case involving fear of fear. Clinical Case Studies. 2011;10(1):37-52.
2. Kim CH, Hsu JJ, Williams DE, et al. A prospective psychological evaluation of patients with dysphagia of various etiologies. Dysphagia. 1996;11(1):34-40.
3. McNally RJ. Choking phobia: a review of the literature. Compr Psychiatry. 1994;35(1):83-89.
4. Barofsky I, Fontaine KR. Do psychogenic dysphagia patients have an eating disorder? Dysphagia. 1998;13(1):24-27.
5. Bishop LC, Riley WT. The psychiatric management of the globus syndrome. Gen Hosp Psychiatry. 1988;10(3):214-219.
6. Ball SG, Otto MW. Cognitive-behavioral treatment of choking phobia: 3 case studies Psychother Psychosom. 1994;62(3-4):207-211.
7. Epstein SJ, Deyoub P. Hypnotherapy for fear of choking: treatment implications of a case report. Int J Clin Hypn. 1981;29(2):117-127.
8. Scemes S, Wielenska RC, Savoia MG, et al. Choking phobia: full remission following behavior therapy. Rev Bras Psiquiatr. 2009;31(3):257-260.
1. Evans IM, Pia P. Phagophobia: behavioral treatment of a complex case involving fear of fear. Clinical Case Studies. 2011;10(1):37-52.
2. Kim CH, Hsu JJ, Williams DE, et al. A prospective psychological evaluation of patients with dysphagia of various etiologies. Dysphagia. 1996;11(1):34-40.
3. McNally RJ. Choking phobia: a review of the literature. Compr Psychiatry. 1994;35(1):83-89.
4. Barofsky I, Fontaine KR. Do psychogenic dysphagia patients have an eating disorder? Dysphagia. 1998;13(1):24-27.
5. Bishop LC, Riley WT. The psychiatric management of the globus syndrome. Gen Hosp Psychiatry. 1988;10(3):214-219.
6. Ball SG, Otto MW. Cognitive-behavioral treatment of choking phobia: 3 case studies Psychother Psychosom. 1994;62(3-4):207-211.
7. Epstein SJ, Deyoub P. Hypnotherapy for fear of choking: treatment implications of a case report. Int J Clin Hypn. 1981;29(2):117-127.
8. Scemes S, Wielenska RC, Savoia MG, et al. Choking phobia: full remission following behavior therapy. Rev Bras Psiquiatr. 2009;31(3):257-260.
Rediscovering clozapine: Adverse effects develop—what should you do now?
Clozapine is a highly effective antipsychotic with superior efficacy in treatment-resistant schizophrenia, but its side effect profile is daunting (Figure 1).1 Adverse reactions lead to approximately 17% of patients who take clozapine eventually discontinuing the medication.1 As we noted in Part 1 of this 3-part series,2 clozapine remains the most efficacious, but most tedious, antipsychotic available to psychiatrists because of its monitoring requirements and potential side effects.
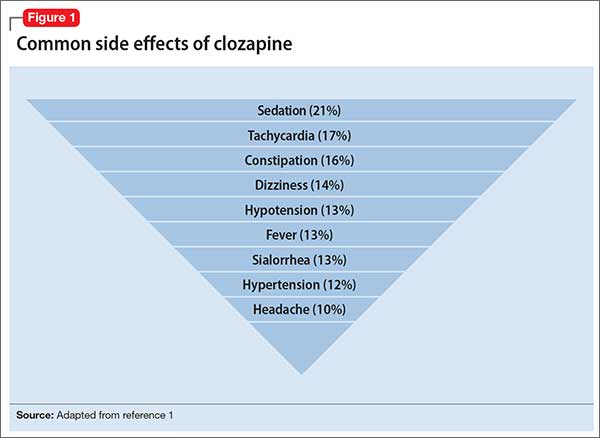
A powerful rationale for prescribing clozapine, despite its drawbacks, is its association with a reduced risk of all-cause mortality.3,4 People with serious mental illness, including schizophrenia, have a median 10-year shorter life expectancy than the general population.5
A recent cohort study6 examined electronic health records to test whether intensive monitoring or lower suicide risk might account for the reduced mortality with clozapine. The authors found that the reduced mortality rate was not directly related to clozapine’s clinical monitoring or other confounding factors. They did find an association between clozapine use and reduced risk of death from both natural and unnatural causes.
This second article in our series examines clozapine’s adverse effects from a systems perspective. Severe neutropenia, myocarditis, sedation, weight gain, orthostatic hypotension, and sialorrhea appear to be the most studied adverse effects, but myriad others can occur.7 We offer guidance to help the astute clinician continue this effective antipsychotic by monitoring carefully, treating side effects early, and managing potential drug interactions (Table 1).8
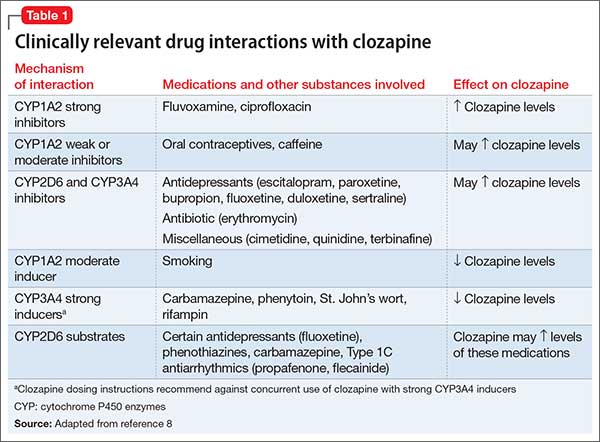
Hematologic eventsSevere neutropenia, defined as absolute neutrophil count (ANC) <500/µL, is a well-known adverse effect of clozapine that requires specific clinical monitoring, a requirement that was updated by the FDA in 2015.2 The incidence of severe neutropenia peaks in the first 2 months of clozapine therapy and tapers after 6 months, but some risk always remains.
Older efficacy studies in the United States gauged the 1-year cumulative incidence of severe clozapine-induced neutropenia to be 2%.9 A 1998 study of the effects of using a clozapine registry reported a lower incidence—0.38%—than the 2% noted above.10 Early recognition and recommended interventions can improve clinical outcomes.2
Drug interactions and neutropenia. A retrospective study of mental health inpatients taking clozapine concurrently with oseltamivir during an influenza outbreak found a statistically significant—but not clinically significant—change in ANC values.11 The authors noted that viral infection might lead to blood dyscrasia early in illness, and that oseltamivir has been associated with a small incidence of blood dyscrasia.11-13 This information might be useful when treating influenza in patients taking clozapine, although no specific change in management is recommended.
Similarly, concomitant treatment with clozapine and lithium can affect both white blood cell and ANC values.14,15 Lithium-treated patients often demonstrate increased circulating neutrophils via enhancement of granulocyte-colony stimulating factor.16 Case studies describe how initiating lithium treatment enabled some patients to continue clozapine after developing neutropenia.14,17 Leukocytosis can affect blood monitoring, possibly masking other blood dyscrasias, when lithium is used concurrently with clozapine.
Eosinophilia (blood eosinophil count >700/µL) occurs in approximately 1% of clozapine users, usually in the first 4 weeks of treatment.18 It can be benign and transient or a harbinger of a more rare adverse reaction such as myocarditis, pancreatitis, hepatitis, colitis, or nephritis.19 If a patient taking clozapine develops eosinophilia, clozapine’s package insert recommends that you:
- evaluate promptly for other systemic involvement (rash, other evidence of allergic reaction, myocarditis, other organ-specific disease)
- stop clozapine immediately if any of these are found.
If other causes of eosinophilia are identified (asthma, allergies, collagen vascular disease, parasitic infection, neoplasm), treat these and continue clozapine.
The manufacturer also mentions the occurrence of clozapine-related eosinophilia without organ involvement that can resolve without intervention, with careful monitoring over several weeks.8 In this scenario, there is flexibility to judge whether clozapine should be stopped or re-challenged, or if close monitoring is adequate. Consulting with an internal medicine or hematology specialist might be helpful.
Cardiovascular side effectsMost common events. Three of the 10 most common clozapine side effects are cardiac: tachycardia, hypotension, and hypertension (Figure 1).1 Orthostatic hypotension, bradycardia, and syncope also can occur, especially with rapid clozapine titration. Baseline electrocardiogram (ECG) can help differentiate whether abnormalities are clozapine-induced or related to a preexisting condition.
Reducing the dosage of clozapine or slowing titration could reverse cardiac side effects.8 If dosage reduction is not an option or is ineffective, first consider treating the side effect rather than discontinuing clozapine.20
Sinus tachycardia is one of the most common side effects of clozapine. First, rule out serious conditions—myocarditis, cardiomyopathy, neuroleptic malignant syndrome (NMS)—then consider waiting and monitoring for the first few months of clozapine treatment. If tachycardia continues, consider dosage reduction. Slower titration, or treatment with a cardio-selective beta blocker such as atenolol.21,22 Note that a recent Cochrane Review concluded that there is not enough randomized evidence to support any particular treatment for clozapine-induced tachycardia; the prescriber must therefore make a case-by-case clinical judgment.22
Similarly, orthostatic hypotension can be managed with a reduced dosage of clozapine or slower titration. Increased fluid intake, compression stockings, and, if necessary, fludrocortisone also can be initiated.20
Rare, potentially fatal events. Myocarditis, pericarditis, and cardiomyopathy are among the rare but potentially fatal adverse effects of clozapine. A recent study reported the incidence of myocarditis with clozapine at a range of 0.015% to 1.3%; cardiomyopathy was even more rare.23 Pulmonary embolism and deep venous thrombosis also are very rare possibilities; keep them in mind, however, when patients taking clozapine report new cardiovascular symptoms.
Patients with clozapine-induced cardiovascular effects most commonly report shortness of breath (60%), palpitations (36%), cough (16%), fatigue (16%), and chest pain (8%).7,24
Clozapine’s “black-box” warning specifically recommends discontinuing clozapine and consulting cardiology when myocarditis or cardiomyopathy is suspected. In 50% of cases, myocarditis symptoms present in the first few weeks of clozapine treatment.23 The manufacturer states that myocarditis usually presents in the first 2 months, and cardiomyopathy after 8 weeks of treatment; however, either can present at any time.8Figure 2 provides a clinical reference for monitoring a clozapine patient for cardiomyopathy.24
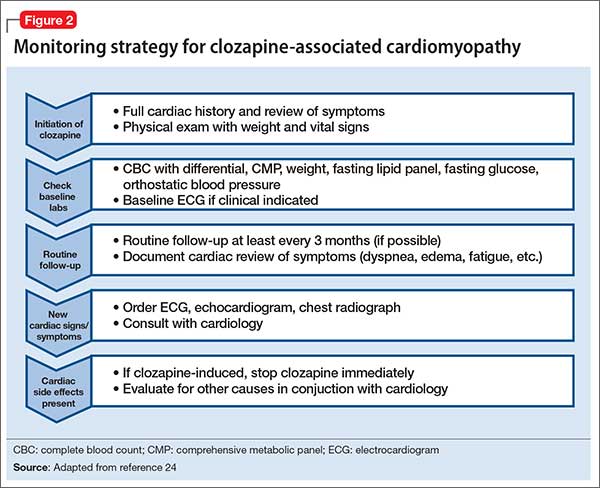
Laboratory findings that support a diagnosis of clozapine-related myocarditis include:
- elevated C-reactive protein
- elevated troponin I or T
- elevated creatine kinase-MB
- peripheral eosinophilia.8,25
ECG, echocardiography, and cardiac MRI can be helpful in diagnosis, in consultation with a cardiologist.
Neurologic side effectsSeizures are listed in the “black-box” warning for clozapine. Seizure incidence with clozapine is 5% per year, with higher incidence at dosages ≥600 mg/d.8 Because clozapine-induced seizures are dosage-dependent, slow titration can mitigate this risk. Tonic-clonic seizures are the most common type associated with clozapine.
The manufacturer recommends caution when using clozapine in patients with a known seizure disorder, alcohol use disorder, or other CNS pathology.8 Patients with a seizure disorder may be at increased risk of experiencing clozapine-induced seizures, but this is not an absolute contraindication.26 Smoking cessation increases clozapine blood levels by an average of 57.4%, further increasing seizure risk.26,27
Discontinuing clozapine is unnecessary when a patient experiences a seizure. Instead, you can:
- halve the dosage prescribed at the time of the seizure (or at least reduce to the last seizure-free dosage)
- consider any medications or medical problems that might have contributed to a lower seizure threshold
- consider prophylaxis with an antiepileptic medication (eg, valproic acid has efficacy for both myoclonic and tonic-clonic seizures).20,26
Sedation is the most common side effect of clozapine.1 Patients experiencing severe sedation should not drive or operate heavy machinery. To reduce sedation, consider instructing the patient to take all or most of the clozapine dosage at bedtime. A critical review of modafinil for sedation caused by antipsychotics in schizophrenia found only 1 open-label study that showed any positive effects; the authors concluded that further study is needed.28
Cognitive and motor slowing are possible neurologic side effects of clozapine. Caution patients about the risk of participating in activities that require cognitive or motor performance until the individual effects of clozapine are known.8
Tardive dyskinesia. Clozapine carries some risk of tardive dyskinesia, although that risk is lower than with other antipsychotics. Similarly, all antipsychotics including clozapine are associated with a risk of NMS. In the rare case of clozapine-induced NMS, stop clozapine immediately and initiate supportive therapy. Clozapine-induced NMS is not an absolute contraindication to re-challenging a patient with clozapine, however, if doing so is clinically appropriate.20
Cerebrovascular events. In older people with dementia, the use of antipsychotics—including clozapine—has been shown to increase the risk of cerebrovascular events. Because most antipsychotics are not FDA-approved for treating psychosis associated with dementia (only pimavanserin is FDA-approved for symptoms of psychosis in Parkinson’s disease), a risk-benefit analysis should be documented when prescribing any antipsychotic in this population. In practice, clozapine’s benefits may outweigh the mortality risks in specific situations.29,30
CASE Sialorrhea puts progress at risk
Ms. B, age 40, has a history of treatment-resistant schizophrenia and is starting clozapine because of residual psychosis during trials of other antipsychotics. She develops severe persistent drooling, mostly at night, during clozapine titration. Sugar-free candy, multiple bed pillows, and changing the dosing schedule do not significantly improve the sialorrhea.
As a result, Ms. B is embarrassed to continue her usual activities. She asks to stop clozapine, even though her psychotic symptoms have improved and she is functioning at her highest level in years.
Ms. B already is taking trihexyphenidyl, 5 mg, 3 times daily, to manage extrapyramidal symptoms related to haloperidol decanoate treatment. After discussing other medication options for sialorrhea, she agrees to a trial of glycopyrrolate, 1 mg, twice daily. She experiences significant improvement and continues taking clozapine.
Sialorrhea develops in 13% of patients taking clozapine.1 As in Ms. B’s case, this side effect can be embarrassing, can limit social or occupational functioning, and might lead patients to discontinue clozapine treatment despite efficacy. Nonpharmacotherapeutic options include covering the pillow with a towel, lowering the clozapine dosage or titrating slowly (or both), and using sugarless gum or candy to increase swallowing.
If the benefits of additional medications targeting side effects outweigh the risks, pharmacotherapeutic intervention may be appropriate. Options include the tricyclic antidepressant amitriptyline31; alpha-adrenergic agonists or antagonists (clonidine, terazosin); and anti-muscarinic medications (benztropine, atropine, trihexyphenidyl, glycopyrrolate) (Table 231). Scopolamine transdermal patch is another possible treatment strategy; however, the scopolamine patch was used for clozapine-induced sialorrhea in only a few case reports, and it is not considered a first-line treatment choice.30
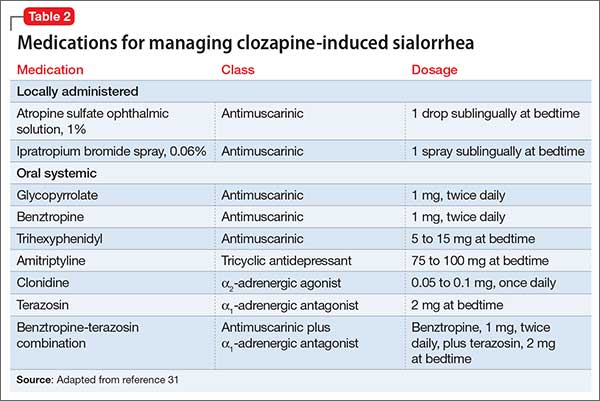
When prescribing, consider the possibility of combined side effects with clozapine and adjunct medications having antimuscarinic or alpha-adrenergic activity, or both. Even atropine ophthalmic drops, administered sublingually, are readily absorbed and cross the blood–brain barrier.31 Another antimuscarinic agent, glycopyrrolate, is less likely to cross the blood–brain barrier and therefore is less likely to cause cognitive side effects. Glycopyrrolate is 5 times more potent at blocking the muscarinic receptor than atropine.31,32 Ipratropium bromide, another nonselective muscarinic receptor antagonist, has less systemic absorption than atropine drops, with less anticholinergic side effects when administered sublingually.
Limited evidence supports the efficacy of alpha-adrenergic medications for managing clozapine-induced sialorrhea. Monitor blood pressure when prescribing terazosin or clonidine, which could potentiate clozapine’s hypotensive effects.
Endocrine side effectsAmong antipsychotics, clozapine is associated with the greatest weight gain—averaging nearly 10% of body weight.33,34 Similarly, the risk of new-onset diabetes mellitus is highest with clozapine in relation to other antipsychotics: 43% reported in a 10-year naturalistic study.35 The risk of hyperlipidemia also increases with clozapine treatment.36 These metabolic changes increase the risk of cardiovascular-related death, with a 10-year mortality rate from cardiovascular disease reported at 9% in clozapine-treated patients.35
Despite clozapine’s metabolic side effects, patients with schizophrenia who are treated with clozapine show a significant reduction in overall mortality compared with patients not treated with clozapine.6 Effective identification and management of metabolic side effects can prevent the need to discontinue clozapine.
Behavioral weight management and exercise are recommended as initial therapy.20 If, based on clinical judgment, these alone are insufficient, data support the use of pharmacotherapeutic interventions. Metformin demonstrates a positive effect on body weight, insulin resistance, and lipids, making it the first choice for adjunctive treatment of clozapine-induced metabolic side effects.37-39
Gastrointestinal side effectsClozapine’s anticholinergic activity can lead to serious gastrointestinal (GI) side effects, including constipation, intestinal obstruction, fecal impaction, and paralytic ileus.8 Ileus has produced more fatal adverse reactions with clozapine than has severe neutropenia.20,40 Co-administered anticholinergic medications could increase the risk of ileus. Obtaining a GI review of systems and monitoring bowel movements (in inpatient or residential facilities) can aid in early identification and limit morbidity and mortality from GI adverse events. A high-fiber diet, adequate hydration, bulk laxatives in patients who can reliably maintain hydration, and GI consultation (if needed) may help manage GI side effects.20
Constitutional side effectsFever can occur with clozapine, most often in the first month of treatment, but the incidence is quite variable (0.5% to 55%).20,41 Although benign fever is common, agranulocytosis with infection, NMS, and other systemic illness must be ruled out. The recommended workup when a patient develops fever while taking clozapine includes physical examination and relevant testing (urinalysis, measurement of ANC and serum creatine kinase, chest radiograph, ECG, and, possibly, blood cultures).41
If evidence supports a serious adverse reaction, stop clozapine immediately.20 If benign clozapine-related fever is suspected, acetaminophen or another antipyretic might provide symptomatic relief; discontinuing clozapine is then unnecessary.41
Pregnancy. When a patient with schizophrenia requires clozapine treatment during pregnancy, reliable clinical guidance is limited. The American College of Obstetricians and Gynecologists Practice Bulletin on the use of psychiatric medications during pregnancy and lactation can be a useful resource.42
Be aware that the FDA very recently made major changes to the format and content of pregnancy and lactation labeling, removing the letter categories that have been used for medications approved on or after June 30, 2001. The manufacturers of medications (such as clozapine) that were approved before June 30, 2001, have 3 years to comply with new requirements.43
The FDA had rated clozapine a pregnancy risk category B medication, meaning no evidence of risk in humans. In 2011, the FDA issued a general warning that antipsychotic use in pregnancy can cause extrapyramidal symptoms and discontinuation symptoms in newborns.44,45
A 2015 review of psychotropic medications and pregnancy noted that approximately 60% of women with schizophrenia became pregnant, with an increased incidence of unplanned pregnancy. A high risk of psychotic relapse (65%) during pregnancy and in the postpartum period may lead to insufficient prenatal care, drug use, and obstetric complications.45 Some data suggest low fetal birth weight and an increased rate of therapeutic abortions in women with schizophrenia.42,46
When treating a pregnant patient, weigh the benefits of clozapine against the risks of adverse events, and clearly document the analysis. Clozapine treatment is not recommended during breast-feeding because of the risk of side effects for newborns.8
We highly recommend keeping updated on the literature regarding pregnancy and lactation information with antipsychotics, including clozapine, because prescribing information will likely be updated in the near future to comply with recent FDA labeling changes.
Final installment: Using clozapine off-labelClozapine is FDA-approved for refractory schizophrenia and for reducing the risk of recurrent suicidal behavior in schizophrenia or schizoaffective disorder. In Part 3 of this series, we review off-label uses—such as managing bipolar disorder, borderline personality disorder, and aggressive behavior—that have varying degrees of scientific support.
1. Kane J, Honigfeld G, Singer J, et al. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988;45(9):789-796.
2. Newman WJ, Newman BM. Rediscovering clozapine: after a turbulent history, current guidance on initiating and monitoring. Current Psychiatry. 2016;15(7):42-46,48-49.
3. Walker AM, Lanza LL, Arellano F, et al. Mortality in current and former users of clozapine. Epidemiology. 1997;8(6):671-677.
4. Tiihonen J, Lönnqvist J, Wahlbeck K, et al. 11-year follow-up of mortality in patients with schizophrenia: a population-based cohort study (FIN11 study). Lancet. 2009;374(9690):620-627.
5. Walker E, McGee RE, Druss BG. Mortality in mental disorders and global disease burden Implications: a systematic review and meta-analysis. JAMA Psychiatry. 2015;72(4):334-341.
6. Hayes RD, Downs J, Chang CK, et al. The effect of clozapine on premature mortality: an assessment of clinical monitoring and other potential confounders. Schizophr Bull. 2015;41(3):644-655.
7. De Fazio P, Gaetano R, Caroleo M, et al. Rare and very rare adverse effects of clozapine. Neuropsychiatr Dis Treat. 2015;11:1995-2003.
8. Novartis Pharmaceuticals Corporation. Clozaril (clozapine). Prescribing information. http://clozaril.com/wp-content/themes/eyesite/pi/Clozaril-2015A507-10022015-Approved.pdf. Accessed June 29, 2016.
9. Lieberman JA, Johns CA, Kane JM, et al. Clozapine-induced agranulocytosis: non-cross-reactivity with other psychotropic drugs. J Clin Psychiatry. 1988;49(7):271-277.
10. Honigfeld G, Arellano F, Sethi J, et al. Reducing clozapine-related morbidity and mortality: 5 years of experience with the Clozaril National Registry. J Clin Psychiatry. 1998;59(suppl 3):3-7.
11. Demler TL, Trigoboff E. Are clozapine patients at risk for blood dyscrasias with concomitant tamiflu use? Psychiatry (Edgmont). 2009;6(11):29-33.
12. Karalakulasingam R, Schacht RA, Lansing AM, et al. Influenza virus pneumonia after renal transplant. Postgrad Med. 1977;62(2):164-167.
13. Hoffman-La Roche Limited. Product monograph: Tamiflu. http://www.rochecanada.com/content/dam/roche_canada/en_CA/documents/Research/ClinicalTrialsForms/Products/ConsumerInformation/MonographsandPublicAdvisories/Tamiflu/Tamiflu_PM_E.pdf. Updated January 26, 2015. Accessed November 28, 2015.
14. Whiskey E, Taylor D. Restarting clozapine after neutropenia: evaluating the possibilities and practicalities. CNS Drugs. 2007;21(1):25-35.
15. Palominao A, Kukoyi O, Xiong GL. Leukocytosis after lithium and clozapine combination therapy. Ann Clin Psychiatry. 2010;22(3):205-206.
16. Focosi D, Azzarà A, Kast RE, et al. Lithium and hematology: established and proposed uses. J Leukoc Biol. 2009;85(1):20-28.
17. Papetti F, Darcourt G, Giordana JY, et al. Treatment of clozapine-induced granulocytopenia with lithium (two observations) [in French]. Encephale. 2004;30(6):578-582.
18. Hummer M, Sperner-Unterweger B, Kemmler G, et al. Does eosinophilia predict clozapine induced neutropenia? Psychopharmacology (Berl). 1996;124(1-2):201-204.
19. Aneja J, Sharma N, Mahajan S, et al. Eosinophilia induced by clozapine: a report of two cases and review of the literature. J Family Med Prim Care. 2015;4(1):127-129.
20. Nielsen J, Correll CU, Manu P, et al. Termination of clozapine treatment due to medical reasons: when is it warranted and how can it be avoided? J Clin Psychiatry. 2013;74(6):603-613.
21. Stryjer R, Timinsky I, Reznik, I, et al. Beta-adrenergic antagonists for the treatment of clozapine-induced sinus tachycardia: a retrospective study. Clin Neuropharmacol. 2009;32(5):290-292.
22. Lally J, Docherty MJ, MacCabe JH. Pharmacological interventions for clozapine-induced sinus tachycardia. Cochrane Database Syst Rev. 2016;9(6):CD011566.
23. Kamphuis H, Arends J, Timmerman L, et al. Myocarditis and cardiomyopathy: underestimated complications resulting from clozapine therapy [in Dutch]. Tijdschr Psychiatr. 2010;52(4):223-233.
24. Alawami M, Wasywich C, Cicovic A, et al. A systematic review of clozapine induced cardiomyopathy. Int J Cardiol. 2014;176(2):315-320.
25. Ronaldson KJ, Fitzgerald PB, Taylor AJ, et al. A new monitoring protocol for clozapine-induced myocarditis based on an analysis of 75 cases and 94 controls. Aust N Z J Psychiatry. 2011;45(6):458-465.
26. Williams AM, Park SH. Seizure associated with clozapine: incidence, etiology, and management. CNS Drugs. 2015;29(2):101-111.
27. Meyer JM. Individual changes in clozapine levels after smoking cessation: results and a predictive model. J Clin Psychopharmacol. 2001;21(6):569-574.
28. Saavedra-Velez C, Yusim A, Anbarasan D, et al. Modafinil as an adjunctive treatment of sedation, negative symptoms, and cognition in schizophrenia: a critical review. J Clin Psychiatry. 2009;70(1):104-112.
29. Klein C, Gordon J, Pollak L, et al. Clozapine in Parkinson’s disease psychosis: 5-year follow-up review. Clin Neuropharmacol. 2003;26(1):8-11.
30. Lutz UC, Sirfy A, Wiatr G, et al. Clozapine serum concentrations in dopamimetic psychosis in Parkinson’s disease and related disorders. Eur J Clin Pharmacol. 2014;70(12):1471-1476.
31. Bird AM, Smith TL, Walton AE. Current treatment strategies for clozapine-induced sialorrhea. Ann Pharmacother. 2011;45(5):667-675.
32. Duggal HS. Glycopyrrolate for clozapine-induced sialorrhea. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31(7):1546-1547.
33. Leadbetter R, Shutty M, Pavalonis D, et al. Clozapine-induced weight gain: prevalence and clinical relevance. Am J Psychiatry. 1992;149(1):68-72.
34. Lundblad W, Azzam PN, Gopalan, et al. Medical management of patients on clozapine: a guide for internists. J Hosp Med. 2015;10(8):537-543.
35. Henderson DC, Nguyen DD, Copeland PM, et al. Clozapine, diabetes mellitus, hyperlipidemia, and cardiovascular risks and mortality: results of a 10-year naturalistic study. J Clin Psychiatry. 2005;66(9):1116-1121.
36. Stroup TS, Gerhard T, Crystal S, et al. Comparative effectiveness of clozapine and standard antipsychotic treatment in adults with schizophrenia. Am J Psychiatry. 2016;173(2):166-173.
37. Carrizo E, Fernández V, Connell L, et al. Extended release metformin for metabolic control assistance during prolonged clozapine administration: a 14 week, double-blind, parallel group, placebo-controlled study. Schizophr Res. 2009;113(1):19-26.
38. Chen CH, Huang MC, Kao CF, et al. Effects of adjunctive metformin on metabolic traits in nondiabetic clozapine-treated patients with schizophrenia and the effect of metformin discontinuation on body weight: a 24-week, randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2013;74(5):e424-e430.
39. Mizuno Y, Suzuki T, Nakagawa A, et al. Pharmacological strategies to counteract antipsychotic-induced weight gain and metabolic adverse effects in schizophrenia: a systematic review and meta-analysis. Schizophr Bull. 2014;40(6):1385-1403.
40. Nielsen J, Meyer JM. Risk factors for ileus in patients with schizophrenia. Schizophr Bull. 2012;38(3):592-598.
41. Lowe CM, Grube RR, Scates AC. Characterization and clinical management of clozapine-induced fever. Ann Pharmacother. 2007;41(10):1700-1704.
42. ACOG Committee on Practice Bulletins–Obstetrics. ACOG Practice Bulletin: Clinical management guidelines for obstetrician-gynecologists number 92, April 2008 (replaces practice bulletin number 87, November 2007). Use of psychiatric medications during pregnancy and lactation. Obstet Gynecol. 2008;111(4):1001-1020.
43. U.S. Food and Drug Administration. Pregnancy and Lactation Labeling (Drugs) Final Rule. https://s3.amazonaws.com/public-inspection.federalregister.gov/2014-28241.pdf. Published December 4, 2014. Accessed July 6, 2016.
44. Briggs GG, Freeman RK, Yaffe SJ. Drugs in pregnancy and lactation: a reference guide to fetal and neonatal risk. 9th ed. Baltimore, MD: Lippincott Williams & Wilkins; 2011.
45. Larsen ER, Damkier P, Pedersen LH, et al; Danish Psychiatric Society; Danish Society of Obstetrics and Gynecology; Danish Paediatric Society; Danish Society of Clinical Pharmacology. Use of psychotropic drugs during pregnancy and breast-feeding. Acta Psychiatr Scand Suppl. 2015;(445):1-28.
46. McKenna K, Koren G, Tetelbaum M, et al. Pregnancy outcome of women using atypical antipsychotic drugs: a prospective comparative study. J Clin Psychiatry. 2005;66(4):444-449.
Clozapine is a highly effective antipsychotic with superior efficacy in treatment-resistant schizophrenia, but its side effect profile is daunting (Figure 1).1 Adverse reactions lead to approximately 17% of patients who take clozapine eventually discontinuing the medication.1 As we noted in Part 1 of this 3-part series,2 clozapine remains the most efficacious, but most tedious, antipsychotic available to psychiatrists because of its monitoring requirements and potential side effects.
A powerful rationale for prescribing clozapine, despite its drawbacks, is its association with a reduced risk of all-cause mortality.3,4 People with serious mental illness, including schizophrenia, have a median 10-year shorter life expectancy than the general population.5
A recent cohort study6 examined electronic health records to test whether intensive monitoring or lower suicide risk might account for the reduced mortality with clozapine. The authors found that the reduced mortality rate was not directly related to clozapine’s clinical monitoring or other confounding factors. They did find an association between clozapine use and reduced risk of death from both natural and unnatural causes.
This second article in our series examines clozapine’s adverse effects from a systems perspective. Severe neutropenia, myocarditis, sedation, weight gain, orthostatic hypotension, and sialorrhea appear to be the most studied adverse effects, but myriad others can occur.7 We offer guidance to help the astute clinician continue this effective antipsychotic by monitoring carefully, treating side effects early, and managing potential drug interactions (Table 1).8
Hematologic eventsSevere neutropenia, defined as absolute neutrophil count (ANC) <500/µL, is a well-known adverse effect of clozapine that requires specific clinical monitoring, a requirement that was updated by the FDA in 2015.2 The incidence of severe neutropenia peaks in the first 2 months of clozapine therapy and tapers after 6 months, but some risk always remains.
Older efficacy studies in the United States gauged the 1-year cumulative incidence of severe clozapine-induced neutropenia to be 2%.9 A 1998 study of the effects of using a clozapine registry reported a lower incidence—0.38%—than the 2% noted above.10 Early recognition and recommended interventions can improve clinical outcomes.2
Drug interactions and neutropenia. A retrospective study of mental health inpatients taking clozapine concurrently with oseltamivir during an influenza outbreak found a statistically significant—but not clinically significant—change in ANC values.11 The authors noted that viral infection might lead to blood dyscrasia early in illness, and that oseltamivir has been associated with a small incidence of blood dyscrasia.11-13 This information might be useful when treating influenza in patients taking clozapine, although no specific change in management is recommended.
Similarly, concomitant treatment with clozapine and lithium can affect both white blood cell and ANC values.14,15 Lithium-treated patients often demonstrate increased circulating neutrophils via enhancement of granulocyte-colony stimulating factor.16 Case studies describe how initiating lithium treatment enabled some patients to continue clozapine after developing neutropenia.14,17 Leukocytosis can affect blood monitoring, possibly masking other blood dyscrasias, when lithium is used concurrently with clozapine.
Eosinophilia (blood eosinophil count >700/µL) occurs in approximately 1% of clozapine users, usually in the first 4 weeks of treatment.18 It can be benign and transient or a harbinger of a more rare adverse reaction such as myocarditis, pancreatitis, hepatitis, colitis, or nephritis.19 If a patient taking clozapine develops eosinophilia, clozapine’s package insert recommends that you:
- evaluate promptly for other systemic involvement (rash, other evidence of allergic reaction, myocarditis, other organ-specific disease)
- stop clozapine immediately if any of these are found.
If other causes of eosinophilia are identified (asthma, allergies, collagen vascular disease, parasitic infection, neoplasm), treat these and continue clozapine.
The manufacturer also mentions the occurrence of clozapine-related eosinophilia without organ involvement that can resolve without intervention, with careful monitoring over several weeks.8 In this scenario, there is flexibility to judge whether clozapine should be stopped or re-challenged, or if close monitoring is adequate. Consulting with an internal medicine or hematology specialist might be helpful.
Cardiovascular side effectsMost common events. Three of the 10 most common clozapine side effects are cardiac: tachycardia, hypotension, and hypertension (Figure 1).1 Orthostatic hypotension, bradycardia, and syncope also can occur, especially with rapid clozapine titration. Baseline electrocardiogram (ECG) can help differentiate whether abnormalities are clozapine-induced or related to a preexisting condition.
Reducing the dosage of clozapine or slowing titration could reverse cardiac side effects.8 If dosage reduction is not an option or is ineffective, first consider treating the side effect rather than discontinuing clozapine.20
Sinus tachycardia is one of the most common side effects of clozapine. First, rule out serious conditions—myocarditis, cardiomyopathy, neuroleptic malignant syndrome (NMS)—then consider waiting and monitoring for the first few months of clozapine treatment. If tachycardia continues, consider dosage reduction. Slower titration, or treatment with a cardio-selective beta blocker such as atenolol.21,22 Note that a recent Cochrane Review concluded that there is not enough randomized evidence to support any particular treatment for clozapine-induced tachycardia; the prescriber must therefore make a case-by-case clinical judgment.22
Similarly, orthostatic hypotension can be managed with a reduced dosage of clozapine or slower titration. Increased fluid intake, compression stockings, and, if necessary, fludrocortisone also can be initiated.20
Rare, potentially fatal events. Myocarditis, pericarditis, and cardiomyopathy are among the rare but potentially fatal adverse effects of clozapine. A recent study reported the incidence of myocarditis with clozapine at a range of 0.015% to 1.3%; cardiomyopathy was even more rare.23 Pulmonary embolism and deep venous thrombosis also are very rare possibilities; keep them in mind, however, when patients taking clozapine report new cardiovascular symptoms.
Patients with clozapine-induced cardiovascular effects most commonly report shortness of breath (60%), palpitations (36%), cough (16%), fatigue (16%), and chest pain (8%).7,24
Clozapine’s “black-box” warning specifically recommends discontinuing clozapine and consulting cardiology when myocarditis or cardiomyopathy is suspected. In 50% of cases, myocarditis symptoms present in the first few weeks of clozapine treatment.23 The manufacturer states that myocarditis usually presents in the first 2 months, and cardiomyopathy after 8 weeks of treatment; however, either can present at any time.8Figure 2 provides a clinical reference for monitoring a clozapine patient for cardiomyopathy.24
Laboratory findings that support a diagnosis of clozapine-related myocarditis include:
- elevated C-reactive protein
- elevated troponin I or T
- elevated creatine kinase-MB
- peripheral eosinophilia.8,25
ECG, echocardiography, and cardiac MRI can be helpful in diagnosis, in consultation with a cardiologist.
Neurologic side effectsSeizures are listed in the “black-box” warning for clozapine. Seizure incidence with clozapine is 5% per year, with higher incidence at dosages ≥600 mg/d.8 Because clozapine-induced seizures are dosage-dependent, slow titration can mitigate this risk. Tonic-clonic seizures are the most common type associated with clozapine.
The manufacturer recommends caution when using clozapine in patients with a known seizure disorder, alcohol use disorder, or other CNS pathology.8 Patients with a seizure disorder may be at increased risk of experiencing clozapine-induced seizures, but this is not an absolute contraindication.26 Smoking cessation increases clozapine blood levels by an average of 57.4%, further increasing seizure risk.26,27
Discontinuing clozapine is unnecessary when a patient experiences a seizure. Instead, you can:
- halve the dosage prescribed at the time of the seizure (or at least reduce to the last seizure-free dosage)
- consider any medications or medical problems that might have contributed to a lower seizure threshold
- consider prophylaxis with an antiepileptic medication (eg, valproic acid has efficacy for both myoclonic and tonic-clonic seizures).20,26
Sedation is the most common side effect of clozapine.1 Patients experiencing severe sedation should not drive or operate heavy machinery. To reduce sedation, consider instructing the patient to take all or most of the clozapine dosage at bedtime. A critical review of modafinil for sedation caused by antipsychotics in schizophrenia found only 1 open-label study that showed any positive effects; the authors concluded that further study is needed.28
Cognitive and motor slowing are possible neurologic side effects of clozapine. Caution patients about the risk of participating in activities that require cognitive or motor performance until the individual effects of clozapine are known.8
Tardive dyskinesia. Clozapine carries some risk of tardive dyskinesia, although that risk is lower than with other antipsychotics. Similarly, all antipsychotics including clozapine are associated with a risk of NMS. In the rare case of clozapine-induced NMS, stop clozapine immediately and initiate supportive therapy. Clozapine-induced NMS is not an absolute contraindication to re-challenging a patient with clozapine, however, if doing so is clinically appropriate.20
Cerebrovascular events. In older people with dementia, the use of antipsychotics—including clozapine—has been shown to increase the risk of cerebrovascular events. Because most antipsychotics are not FDA-approved for treating psychosis associated with dementia (only pimavanserin is FDA-approved for symptoms of psychosis in Parkinson’s disease), a risk-benefit analysis should be documented when prescribing any antipsychotic in this population. In practice, clozapine’s benefits may outweigh the mortality risks in specific situations.29,30
CASE Sialorrhea puts progress at risk
Ms. B, age 40, has a history of treatment-resistant schizophrenia and is starting clozapine because of residual psychosis during trials of other antipsychotics. She develops severe persistent drooling, mostly at night, during clozapine titration. Sugar-free candy, multiple bed pillows, and changing the dosing schedule do not significantly improve the sialorrhea.
As a result, Ms. B is embarrassed to continue her usual activities. She asks to stop clozapine, even though her psychotic symptoms have improved and she is functioning at her highest level in years.
Ms. B already is taking trihexyphenidyl, 5 mg, 3 times daily, to manage extrapyramidal symptoms related to haloperidol decanoate treatment. After discussing other medication options for sialorrhea, she agrees to a trial of glycopyrrolate, 1 mg, twice daily. She experiences significant improvement and continues taking clozapine.
Sialorrhea develops in 13% of patients taking clozapine.1 As in Ms. B’s case, this side effect can be embarrassing, can limit social or occupational functioning, and might lead patients to discontinue clozapine treatment despite efficacy. Nonpharmacotherapeutic options include covering the pillow with a towel, lowering the clozapine dosage or titrating slowly (or both), and using sugarless gum or candy to increase swallowing.
If the benefits of additional medications targeting side effects outweigh the risks, pharmacotherapeutic intervention may be appropriate. Options include the tricyclic antidepressant amitriptyline31; alpha-adrenergic agonists or antagonists (clonidine, terazosin); and anti-muscarinic medications (benztropine, atropine, trihexyphenidyl, glycopyrrolate) (Table 231). Scopolamine transdermal patch is another possible treatment strategy; however, the scopolamine patch was used for clozapine-induced sialorrhea in only a few case reports, and it is not considered a first-line treatment choice.30
When prescribing, consider the possibility of combined side effects with clozapine and adjunct medications having antimuscarinic or alpha-adrenergic activity, or both. Even atropine ophthalmic drops, administered sublingually, are readily absorbed and cross the blood–brain barrier.31 Another antimuscarinic agent, glycopyrrolate, is less likely to cross the blood–brain barrier and therefore is less likely to cause cognitive side effects. Glycopyrrolate is 5 times more potent at blocking the muscarinic receptor than atropine.31,32 Ipratropium bromide, another nonselective muscarinic receptor antagonist, has less systemic absorption than atropine drops, with less anticholinergic side effects when administered sublingually.
Limited evidence supports the efficacy of alpha-adrenergic medications for managing clozapine-induced sialorrhea. Monitor blood pressure when prescribing terazosin or clonidine, which could potentiate clozapine’s hypotensive effects.
Endocrine side effectsAmong antipsychotics, clozapine is associated with the greatest weight gain—averaging nearly 10% of body weight.33,34 Similarly, the risk of new-onset diabetes mellitus is highest with clozapine in relation to other antipsychotics: 43% reported in a 10-year naturalistic study.35 The risk of hyperlipidemia also increases with clozapine treatment.36 These metabolic changes increase the risk of cardiovascular-related death, with a 10-year mortality rate from cardiovascular disease reported at 9% in clozapine-treated patients.35
Despite clozapine’s metabolic side effects, patients with schizophrenia who are treated with clozapine show a significant reduction in overall mortality compared with patients not treated with clozapine.6 Effective identification and management of metabolic side effects can prevent the need to discontinue clozapine.
Behavioral weight management and exercise are recommended as initial therapy.20 If, based on clinical judgment, these alone are insufficient, data support the use of pharmacotherapeutic interventions. Metformin demonstrates a positive effect on body weight, insulin resistance, and lipids, making it the first choice for adjunctive treatment of clozapine-induced metabolic side effects.37-39
Gastrointestinal side effectsClozapine’s anticholinergic activity can lead to serious gastrointestinal (GI) side effects, including constipation, intestinal obstruction, fecal impaction, and paralytic ileus.8 Ileus has produced more fatal adverse reactions with clozapine than has severe neutropenia.20,40 Co-administered anticholinergic medications could increase the risk of ileus. Obtaining a GI review of systems and monitoring bowel movements (in inpatient or residential facilities) can aid in early identification and limit morbidity and mortality from GI adverse events. A high-fiber diet, adequate hydration, bulk laxatives in patients who can reliably maintain hydration, and GI consultation (if needed) may help manage GI side effects.20
Constitutional side effectsFever can occur with clozapine, most often in the first month of treatment, but the incidence is quite variable (0.5% to 55%).20,41 Although benign fever is common, agranulocytosis with infection, NMS, and other systemic illness must be ruled out. The recommended workup when a patient develops fever while taking clozapine includes physical examination and relevant testing (urinalysis, measurement of ANC and serum creatine kinase, chest radiograph, ECG, and, possibly, blood cultures).41
If evidence supports a serious adverse reaction, stop clozapine immediately.20 If benign clozapine-related fever is suspected, acetaminophen or another antipyretic might provide symptomatic relief; discontinuing clozapine is then unnecessary.41
Pregnancy. When a patient with schizophrenia requires clozapine treatment during pregnancy, reliable clinical guidance is limited. The American College of Obstetricians and Gynecologists Practice Bulletin on the use of psychiatric medications during pregnancy and lactation can be a useful resource.42
Be aware that the FDA very recently made major changes to the format and content of pregnancy and lactation labeling, removing the letter categories that have been used for medications approved on or after June 30, 2001. The manufacturers of medications (such as clozapine) that were approved before June 30, 2001, have 3 years to comply with new requirements.43
The FDA had rated clozapine a pregnancy risk category B medication, meaning no evidence of risk in humans. In 2011, the FDA issued a general warning that antipsychotic use in pregnancy can cause extrapyramidal symptoms and discontinuation symptoms in newborns.44,45
A 2015 review of psychotropic medications and pregnancy noted that approximately 60% of women with schizophrenia became pregnant, with an increased incidence of unplanned pregnancy. A high risk of psychotic relapse (65%) during pregnancy and in the postpartum period may lead to insufficient prenatal care, drug use, and obstetric complications.45 Some data suggest low fetal birth weight and an increased rate of therapeutic abortions in women with schizophrenia.42,46
When treating a pregnant patient, weigh the benefits of clozapine against the risks of adverse events, and clearly document the analysis. Clozapine treatment is not recommended during breast-feeding because of the risk of side effects for newborns.8
We highly recommend keeping updated on the literature regarding pregnancy and lactation information with antipsychotics, including clozapine, because prescribing information will likely be updated in the near future to comply with recent FDA labeling changes.
Final installment: Using clozapine off-labelClozapine is FDA-approved for refractory schizophrenia and for reducing the risk of recurrent suicidal behavior in schizophrenia or schizoaffective disorder. In Part 3 of this series, we review off-label uses—such as managing bipolar disorder, borderline personality disorder, and aggressive behavior—that have varying degrees of scientific support.
Clozapine is a highly effective antipsychotic with superior efficacy in treatment-resistant schizophrenia, but its side effect profile is daunting (Figure 1).1 Adverse reactions lead to approximately 17% of patients who take clozapine eventually discontinuing the medication.1 As we noted in Part 1 of this 3-part series,2 clozapine remains the most efficacious, but most tedious, antipsychotic available to psychiatrists because of its monitoring requirements and potential side effects.
A powerful rationale for prescribing clozapine, despite its drawbacks, is its association with a reduced risk of all-cause mortality.3,4 People with serious mental illness, including schizophrenia, have a median 10-year shorter life expectancy than the general population.5
A recent cohort study6 examined electronic health records to test whether intensive monitoring or lower suicide risk might account for the reduced mortality with clozapine. The authors found that the reduced mortality rate was not directly related to clozapine’s clinical monitoring or other confounding factors. They did find an association between clozapine use and reduced risk of death from both natural and unnatural causes.
This second article in our series examines clozapine’s adverse effects from a systems perspective. Severe neutropenia, myocarditis, sedation, weight gain, orthostatic hypotension, and sialorrhea appear to be the most studied adverse effects, but myriad others can occur.7 We offer guidance to help the astute clinician continue this effective antipsychotic by monitoring carefully, treating side effects early, and managing potential drug interactions (Table 1).8
Hematologic eventsSevere neutropenia, defined as absolute neutrophil count (ANC) <500/µL, is a well-known adverse effect of clozapine that requires specific clinical monitoring, a requirement that was updated by the FDA in 2015.2 The incidence of severe neutropenia peaks in the first 2 months of clozapine therapy and tapers after 6 months, but some risk always remains.
Older efficacy studies in the United States gauged the 1-year cumulative incidence of severe clozapine-induced neutropenia to be 2%.9 A 1998 study of the effects of using a clozapine registry reported a lower incidence—0.38%—than the 2% noted above.10 Early recognition and recommended interventions can improve clinical outcomes.2
Drug interactions and neutropenia. A retrospective study of mental health inpatients taking clozapine concurrently with oseltamivir during an influenza outbreak found a statistically significant—but not clinically significant—change in ANC values.11 The authors noted that viral infection might lead to blood dyscrasia early in illness, and that oseltamivir has been associated with a small incidence of blood dyscrasia.11-13 This information might be useful when treating influenza in patients taking clozapine, although no specific change in management is recommended.
Similarly, concomitant treatment with clozapine and lithium can affect both white blood cell and ANC values.14,15 Lithium-treated patients often demonstrate increased circulating neutrophils via enhancement of granulocyte-colony stimulating factor.16 Case studies describe how initiating lithium treatment enabled some patients to continue clozapine after developing neutropenia.14,17 Leukocytosis can affect blood monitoring, possibly masking other blood dyscrasias, when lithium is used concurrently with clozapine.
Eosinophilia (blood eosinophil count >700/µL) occurs in approximately 1% of clozapine users, usually in the first 4 weeks of treatment.18 It can be benign and transient or a harbinger of a more rare adverse reaction such as myocarditis, pancreatitis, hepatitis, colitis, or nephritis.19 If a patient taking clozapine develops eosinophilia, clozapine’s package insert recommends that you:
- evaluate promptly for other systemic involvement (rash, other evidence of allergic reaction, myocarditis, other organ-specific disease)
- stop clozapine immediately if any of these are found.
If other causes of eosinophilia are identified (asthma, allergies, collagen vascular disease, parasitic infection, neoplasm), treat these and continue clozapine.
The manufacturer also mentions the occurrence of clozapine-related eosinophilia without organ involvement that can resolve without intervention, with careful monitoring over several weeks.8 In this scenario, there is flexibility to judge whether clozapine should be stopped or re-challenged, or if close monitoring is adequate. Consulting with an internal medicine or hematology specialist might be helpful.
Cardiovascular side effectsMost common events. Three of the 10 most common clozapine side effects are cardiac: tachycardia, hypotension, and hypertension (Figure 1).1 Orthostatic hypotension, bradycardia, and syncope also can occur, especially with rapid clozapine titration. Baseline electrocardiogram (ECG) can help differentiate whether abnormalities are clozapine-induced or related to a preexisting condition.
Reducing the dosage of clozapine or slowing titration could reverse cardiac side effects.8 If dosage reduction is not an option or is ineffective, first consider treating the side effect rather than discontinuing clozapine.20
Sinus tachycardia is one of the most common side effects of clozapine. First, rule out serious conditions—myocarditis, cardiomyopathy, neuroleptic malignant syndrome (NMS)—then consider waiting and monitoring for the first few months of clozapine treatment. If tachycardia continues, consider dosage reduction. Slower titration, or treatment with a cardio-selective beta blocker such as atenolol.21,22 Note that a recent Cochrane Review concluded that there is not enough randomized evidence to support any particular treatment for clozapine-induced tachycardia; the prescriber must therefore make a case-by-case clinical judgment.22
Similarly, orthostatic hypotension can be managed with a reduced dosage of clozapine or slower titration. Increased fluid intake, compression stockings, and, if necessary, fludrocortisone also can be initiated.20
Rare, potentially fatal events. Myocarditis, pericarditis, and cardiomyopathy are among the rare but potentially fatal adverse effects of clozapine. A recent study reported the incidence of myocarditis with clozapine at a range of 0.015% to 1.3%; cardiomyopathy was even more rare.23 Pulmonary embolism and deep venous thrombosis also are very rare possibilities; keep them in mind, however, when patients taking clozapine report new cardiovascular symptoms.
Patients with clozapine-induced cardiovascular effects most commonly report shortness of breath (60%), palpitations (36%), cough (16%), fatigue (16%), and chest pain (8%).7,24
Clozapine’s “black-box” warning specifically recommends discontinuing clozapine and consulting cardiology when myocarditis or cardiomyopathy is suspected. In 50% of cases, myocarditis symptoms present in the first few weeks of clozapine treatment.23 The manufacturer states that myocarditis usually presents in the first 2 months, and cardiomyopathy after 8 weeks of treatment; however, either can present at any time.8Figure 2 provides a clinical reference for monitoring a clozapine patient for cardiomyopathy.24
Laboratory findings that support a diagnosis of clozapine-related myocarditis include:
- elevated C-reactive protein
- elevated troponin I or T
- elevated creatine kinase-MB
- peripheral eosinophilia.8,25
ECG, echocardiography, and cardiac MRI can be helpful in diagnosis, in consultation with a cardiologist.
Neurologic side effectsSeizures are listed in the “black-box” warning for clozapine. Seizure incidence with clozapine is 5% per year, with higher incidence at dosages ≥600 mg/d.8 Because clozapine-induced seizures are dosage-dependent, slow titration can mitigate this risk. Tonic-clonic seizures are the most common type associated with clozapine.
The manufacturer recommends caution when using clozapine in patients with a known seizure disorder, alcohol use disorder, or other CNS pathology.8 Patients with a seizure disorder may be at increased risk of experiencing clozapine-induced seizures, but this is not an absolute contraindication.26 Smoking cessation increases clozapine blood levels by an average of 57.4%, further increasing seizure risk.26,27
Discontinuing clozapine is unnecessary when a patient experiences a seizure. Instead, you can:
- halve the dosage prescribed at the time of the seizure (or at least reduce to the last seizure-free dosage)
- consider any medications or medical problems that might have contributed to a lower seizure threshold
- consider prophylaxis with an antiepileptic medication (eg, valproic acid has efficacy for both myoclonic and tonic-clonic seizures).20,26
Sedation is the most common side effect of clozapine.1 Patients experiencing severe sedation should not drive or operate heavy machinery. To reduce sedation, consider instructing the patient to take all or most of the clozapine dosage at bedtime. A critical review of modafinil for sedation caused by antipsychotics in schizophrenia found only 1 open-label study that showed any positive effects; the authors concluded that further study is needed.28
Cognitive and motor slowing are possible neurologic side effects of clozapine. Caution patients about the risk of participating in activities that require cognitive or motor performance until the individual effects of clozapine are known.8
Tardive dyskinesia. Clozapine carries some risk of tardive dyskinesia, although that risk is lower than with other antipsychotics. Similarly, all antipsychotics including clozapine are associated with a risk of NMS. In the rare case of clozapine-induced NMS, stop clozapine immediately and initiate supportive therapy. Clozapine-induced NMS is not an absolute contraindication to re-challenging a patient with clozapine, however, if doing so is clinically appropriate.20
Cerebrovascular events. In older people with dementia, the use of antipsychotics—including clozapine—has been shown to increase the risk of cerebrovascular events. Because most antipsychotics are not FDA-approved for treating psychosis associated with dementia (only pimavanserin is FDA-approved for symptoms of psychosis in Parkinson’s disease), a risk-benefit analysis should be documented when prescribing any antipsychotic in this population. In practice, clozapine’s benefits may outweigh the mortality risks in specific situations.29,30
CASE Sialorrhea puts progress at risk
Ms. B, age 40, has a history of treatment-resistant schizophrenia and is starting clozapine because of residual psychosis during trials of other antipsychotics. She develops severe persistent drooling, mostly at night, during clozapine titration. Sugar-free candy, multiple bed pillows, and changing the dosing schedule do not significantly improve the sialorrhea.
As a result, Ms. B is embarrassed to continue her usual activities. She asks to stop clozapine, even though her psychotic symptoms have improved and she is functioning at her highest level in years.
Ms. B already is taking trihexyphenidyl, 5 mg, 3 times daily, to manage extrapyramidal symptoms related to haloperidol decanoate treatment. After discussing other medication options for sialorrhea, she agrees to a trial of glycopyrrolate, 1 mg, twice daily. She experiences significant improvement and continues taking clozapine.
Sialorrhea develops in 13% of patients taking clozapine.1 As in Ms. B’s case, this side effect can be embarrassing, can limit social or occupational functioning, and might lead patients to discontinue clozapine treatment despite efficacy. Nonpharmacotherapeutic options include covering the pillow with a towel, lowering the clozapine dosage or titrating slowly (or both), and using sugarless gum or candy to increase swallowing.
If the benefits of additional medications targeting side effects outweigh the risks, pharmacotherapeutic intervention may be appropriate. Options include the tricyclic antidepressant amitriptyline31; alpha-adrenergic agonists or antagonists (clonidine, terazosin); and anti-muscarinic medications (benztropine, atropine, trihexyphenidyl, glycopyrrolate) (Table 231). Scopolamine transdermal patch is another possible treatment strategy; however, the scopolamine patch was used for clozapine-induced sialorrhea in only a few case reports, and it is not considered a first-line treatment choice.30
When prescribing, consider the possibility of combined side effects with clozapine and adjunct medications having antimuscarinic or alpha-adrenergic activity, or both. Even atropine ophthalmic drops, administered sublingually, are readily absorbed and cross the blood–brain barrier.31 Another antimuscarinic agent, glycopyrrolate, is less likely to cross the blood–brain barrier and therefore is less likely to cause cognitive side effects. Glycopyrrolate is 5 times more potent at blocking the muscarinic receptor than atropine.31,32 Ipratropium bromide, another nonselective muscarinic receptor antagonist, has less systemic absorption than atropine drops, with less anticholinergic side effects when administered sublingually.
Limited evidence supports the efficacy of alpha-adrenergic medications for managing clozapine-induced sialorrhea. Monitor blood pressure when prescribing terazosin or clonidine, which could potentiate clozapine’s hypotensive effects.
Endocrine side effectsAmong antipsychotics, clozapine is associated with the greatest weight gain—averaging nearly 10% of body weight.33,34 Similarly, the risk of new-onset diabetes mellitus is highest with clozapine in relation to other antipsychotics: 43% reported in a 10-year naturalistic study.35 The risk of hyperlipidemia also increases with clozapine treatment.36 These metabolic changes increase the risk of cardiovascular-related death, with a 10-year mortality rate from cardiovascular disease reported at 9% in clozapine-treated patients.35
Despite clozapine’s metabolic side effects, patients with schizophrenia who are treated with clozapine show a significant reduction in overall mortality compared with patients not treated with clozapine.6 Effective identification and management of metabolic side effects can prevent the need to discontinue clozapine.
Behavioral weight management and exercise are recommended as initial therapy.20 If, based on clinical judgment, these alone are insufficient, data support the use of pharmacotherapeutic interventions. Metformin demonstrates a positive effect on body weight, insulin resistance, and lipids, making it the first choice for adjunctive treatment of clozapine-induced metabolic side effects.37-39
Gastrointestinal side effectsClozapine’s anticholinergic activity can lead to serious gastrointestinal (GI) side effects, including constipation, intestinal obstruction, fecal impaction, and paralytic ileus.8 Ileus has produced more fatal adverse reactions with clozapine than has severe neutropenia.20,40 Co-administered anticholinergic medications could increase the risk of ileus. Obtaining a GI review of systems and monitoring bowel movements (in inpatient or residential facilities) can aid in early identification and limit morbidity and mortality from GI adverse events. A high-fiber diet, adequate hydration, bulk laxatives in patients who can reliably maintain hydration, and GI consultation (if needed) may help manage GI side effects.20
Constitutional side effectsFever can occur with clozapine, most often in the first month of treatment, but the incidence is quite variable (0.5% to 55%).20,41 Although benign fever is common, agranulocytosis with infection, NMS, and other systemic illness must be ruled out. The recommended workup when a patient develops fever while taking clozapine includes physical examination and relevant testing (urinalysis, measurement of ANC and serum creatine kinase, chest radiograph, ECG, and, possibly, blood cultures).41
If evidence supports a serious adverse reaction, stop clozapine immediately.20 If benign clozapine-related fever is suspected, acetaminophen or another antipyretic might provide symptomatic relief; discontinuing clozapine is then unnecessary.41
Pregnancy. When a patient with schizophrenia requires clozapine treatment during pregnancy, reliable clinical guidance is limited. The American College of Obstetricians and Gynecologists Practice Bulletin on the use of psychiatric medications during pregnancy and lactation can be a useful resource.42
Be aware that the FDA very recently made major changes to the format and content of pregnancy and lactation labeling, removing the letter categories that have been used for medications approved on or after June 30, 2001. The manufacturers of medications (such as clozapine) that were approved before June 30, 2001, have 3 years to comply with new requirements.43
The FDA had rated clozapine a pregnancy risk category B medication, meaning no evidence of risk in humans. In 2011, the FDA issued a general warning that antipsychotic use in pregnancy can cause extrapyramidal symptoms and discontinuation symptoms in newborns.44,45
A 2015 review of psychotropic medications and pregnancy noted that approximately 60% of women with schizophrenia became pregnant, with an increased incidence of unplanned pregnancy. A high risk of psychotic relapse (65%) during pregnancy and in the postpartum period may lead to insufficient prenatal care, drug use, and obstetric complications.45 Some data suggest low fetal birth weight and an increased rate of therapeutic abortions in women with schizophrenia.42,46
When treating a pregnant patient, weigh the benefits of clozapine against the risks of adverse events, and clearly document the analysis. Clozapine treatment is not recommended during breast-feeding because of the risk of side effects for newborns.8
We highly recommend keeping updated on the literature regarding pregnancy and lactation information with antipsychotics, including clozapine, because prescribing information will likely be updated in the near future to comply with recent FDA labeling changes.
Final installment: Using clozapine off-labelClozapine is FDA-approved for refractory schizophrenia and for reducing the risk of recurrent suicidal behavior in schizophrenia or schizoaffective disorder. In Part 3 of this series, we review off-label uses—such as managing bipolar disorder, borderline personality disorder, and aggressive behavior—that have varying degrees of scientific support.
1. Kane J, Honigfeld G, Singer J, et al. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988;45(9):789-796.
2. Newman WJ, Newman BM. Rediscovering clozapine: after a turbulent history, current guidance on initiating and monitoring. Current Psychiatry. 2016;15(7):42-46,48-49.
3. Walker AM, Lanza LL, Arellano F, et al. Mortality in current and former users of clozapine. Epidemiology. 1997;8(6):671-677.
4. Tiihonen J, Lönnqvist J, Wahlbeck K, et al. 11-year follow-up of mortality in patients with schizophrenia: a population-based cohort study (FIN11 study). Lancet. 2009;374(9690):620-627.
5. Walker E, McGee RE, Druss BG. Mortality in mental disorders and global disease burden Implications: a systematic review and meta-analysis. JAMA Psychiatry. 2015;72(4):334-341.
6. Hayes RD, Downs J, Chang CK, et al. The effect of clozapine on premature mortality: an assessment of clinical monitoring and other potential confounders. Schizophr Bull. 2015;41(3):644-655.
7. De Fazio P, Gaetano R, Caroleo M, et al. Rare and very rare adverse effects of clozapine. Neuropsychiatr Dis Treat. 2015;11:1995-2003.
8. Novartis Pharmaceuticals Corporation. Clozaril (clozapine). Prescribing information. http://clozaril.com/wp-content/themes/eyesite/pi/Clozaril-2015A507-10022015-Approved.pdf. Accessed June 29, 2016.
9. Lieberman JA, Johns CA, Kane JM, et al. Clozapine-induced agranulocytosis: non-cross-reactivity with other psychotropic drugs. J Clin Psychiatry. 1988;49(7):271-277.
10. Honigfeld G, Arellano F, Sethi J, et al. Reducing clozapine-related morbidity and mortality: 5 years of experience with the Clozaril National Registry. J Clin Psychiatry. 1998;59(suppl 3):3-7.
11. Demler TL, Trigoboff E. Are clozapine patients at risk for blood dyscrasias with concomitant tamiflu use? Psychiatry (Edgmont). 2009;6(11):29-33.
12. Karalakulasingam R, Schacht RA, Lansing AM, et al. Influenza virus pneumonia after renal transplant. Postgrad Med. 1977;62(2):164-167.
13. Hoffman-La Roche Limited. Product monograph: Tamiflu. http://www.rochecanada.com/content/dam/roche_canada/en_CA/documents/Research/ClinicalTrialsForms/Products/ConsumerInformation/MonographsandPublicAdvisories/Tamiflu/Tamiflu_PM_E.pdf. Updated January 26, 2015. Accessed November 28, 2015.
14. Whiskey E, Taylor D. Restarting clozapine after neutropenia: evaluating the possibilities and practicalities. CNS Drugs. 2007;21(1):25-35.
15. Palominao A, Kukoyi O, Xiong GL. Leukocytosis after lithium and clozapine combination therapy. Ann Clin Psychiatry. 2010;22(3):205-206.
16. Focosi D, Azzarà A, Kast RE, et al. Lithium and hematology: established and proposed uses. J Leukoc Biol. 2009;85(1):20-28.
17. Papetti F, Darcourt G, Giordana JY, et al. Treatment of clozapine-induced granulocytopenia with lithium (two observations) [in French]. Encephale. 2004;30(6):578-582.
18. Hummer M, Sperner-Unterweger B, Kemmler G, et al. Does eosinophilia predict clozapine induced neutropenia? Psychopharmacology (Berl). 1996;124(1-2):201-204.
19. Aneja J, Sharma N, Mahajan S, et al. Eosinophilia induced by clozapine: a report of two cases and review of the literature. J Family Med Prim Care. 2015;4(1):127-129.
20. Nielsen J, Correll CU, Manu P, et al. Termination of clozapine treatment due to medical reasons: when is it warranted and how can it be avoided? J Clin Psychiatry. 2013;74(6):603-613.
21. Stryjer R, Timinsky I, Reznik, I, et al. Beta-adrenergic antagonists for the treatment of clozapine-induced sinus tachycardia: a retrospective study. Clin Neuropharmacol. 2009;32(5):290-292.
22. Lally J, Docherty MJ, MacCabe JH. Pharmacological interventions for clozapine-induced sinus tachycardia. Cochrane Database Syst Rev. 2016;9(6):CD011566.
23. Kamphuis H, Arends J, Timmerman L, et al. Myocarditis and cardiomyopathy: underestimated complications resulting from clozapine therapy [in Dutch]. Tijdschr Psychiatr. 2010;52(4):223-233.
24. Alawami M, Wasywich C, Cicovic A, et al. A systematic review of clozapine induced cardiomyopathy. Int J Cardiol. 2014;176(2):315-320.
25. Ronaldson KJ, Fitzgerald PB, Taylor AJ, et al. A new monitoring protocol for clozapine-induced myocarditis based on an analysis of 75 cases and 94 controls. Aust N Z J Psychiatry. 2011;45(6):458-465.
26. Williams AM, Park SH. Seizure associated with clozapine: incidence, etiology, and management. CNS Drugs. 2015;29(2):101-111.
27. Meyer JM. Individual changes in clozapine levels after smoking cessation: results and a predictive model. J Clin Psychopharmacol. 2001;21(6):569-574.
28. Saavedra-Velez C, Yusim A, Anbarasan D, et al. Modafinil as an adjunctive treatment of sedation, negative symptoms, and cognition in schizophrenia: a critical review. J Clin Psychiatry. 2009;70(1):104-112.
29. Klein C, Gordon J, Pollak L, et al. Clozapine in Parkinson’s disease psychosis: 5-year follow-up review. Clin Neuropharmacol. 2003;26(1):8-11.
30. Lutz UC, Sirfy A, Wiatr G, et al. Clozapine serum concentrations in dopamimetic psychosis in Parkinson’s disease and related disorders. Eur J Clin Pharmacol. 2014;70(12):1471-1476.
31. Bird AM, Smith TL, Walton AE. Current treatment strategies for clozapine-induced sialorrhea. Ann Pharmacother. 2011;45(5):667-675.
32. Duggal HS. Glycopyrrolate for clozapine-induced sialorrhea. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31(7):1546-1547.
33. Leadbetter R, Shutty M, Pavalonis D, et al. Clozapine-induced weight gain: prevalence and clinical relevance. Am J Psychiatry. 1992;149(1):68-72.
34. Lundblad W, Azzam PN, Gopalan, et al. Medical management of patients on clozapine: a guide for internists. J Hosp Med. 2015;10(8):537-543.
35. Henderson DC, Nguyen DD, Copeland PM, et al. Clozapine, diabetes mellitus, hyperlipidemia, and cardiovascular risks and mortality: results of a 10-year naturalistic study. J Clin Psychiatry. 2005;66(9):1116-1121.
36. Stroup TS, Gerhard T, Crystal S, et al. Comparative effectiveness of clozapine and standard antipsychotic treatment in adults with schizophrenia. Am J Psychiatry. 2016;173(2):166-173.
37. Carrizo E, Fernández V, Connell L, et al. Extended release metformin for metabolic control assistance during prolonged clozapine administration: a 14 week, double-blind, parallel group, placebo-controlled study. Schizophr Res. 2009;113(1):19-26.
38. Chen CH, Huang MC, Kao CF, et al. Effects of adjunctive metformin on metabolic traits in nondiabetic clozapine-treated patients with schizophrenia and the effect of metformin discontinuation on body weight: a 24-week, randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2013;74(5):e424-e430.
39. Mizuno Y, Suzuki T, Nakagawa A, et al. Pharmacological strategies to counteract antipsychotic-induced weight gain and metabolic adverse effects in schizophrenia: a systematic review and meta-analysis. Schizophr Bull. 2014;40(6):1385-1403.
40. Nielsen J, Meyer JM. Risk factors for ileus in patients with schizophrenia. Schizophr Bull. 2012;38(3):592-598.
41. Lowe CM, Grube RR, Scates AC. Characterization and clinical management of clozapine-induced fever. Ann Pharmacother. 2007;41(10):1700-1704.
42. ACOG Committee on Practice Bulletins–Obstetrics. ACOG Practice Bulletin: Clinical management guidelines for obstetrician-gynecologists number 92, April 2008 (replaces practice bulletin number 87, November 2007). Use of psychiatric medications during pregnancy and lactation. Obstet Gynecol. 2008;111(4):1001-1020.
43. U.S. Food and Drug Administration. Pregnancy and Lactation Labeling (Drugs) Final Rule. https://s3.amazonaws.com/public-inspection.federalregister.gov/2014-28241.pdf. Published December 4, 2014. Accessed July 6, 2016.
44. Briggs GG, Freeman RK, Yaffe SJ. Drugs in pregnancy and lactation: a reference guide to fetal and neonatal risk. 9th ed. Baltimore, MD: Lippincott Williams & Wilkins; 2011.
45. Larsen ER, Damkier P, Pedersen LH, et al; Danish Psychiatric Society; Danish Society of Obstetrics and Gynecology; Danish Paediatric Society; Danish Society of Clinical Pharmacology. Use of psychotropic drugs during pregnancy and breast-feeding. Acta Psychiatr Scand Suppl. 2015;(445):1-28.
46. McKenna K, Koren G, Tetelbaum M, et al. Pregnancy outcome of women using atypical antipsychotic drugs: a prospective comparative study. J Clin Psychiatry. 2005;66(4):444-449.
1. Kane J, Honigfeld G, Singer J, et al. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988;45(9):789-796.
2. Newman WJ, Newman BM. Rediscovering clozapine: after a turbulent history, current guidance on initiating and monitoring. Current Psychiatry. 2016;15(7):42-46,48-49.
3. Walker AM, Lanza LL, Arellano F, et al. Mortality in current and former users of clozapine. Epidemiology. 1997;8(6):671-677.
4. Tiihonen J, Lönnqvist J, Wahlbeck K, et al. 11-year follow-up of mortality in patients with schizophrenia: a population-based cohort study (FIN11 study). Lancet. 2009;374(9690):620-627.
5. Walker E, McGee RE, Druss BG. Mortality in mental disorders and global disease burden Implications: a systematic review and meta-analysis. JAMA Psychiatry. 2015;72(4):334-341.
6. Hayes RD, Downs J, Chang CK, et al. The effect of clozapine on premature mortality: an assessment of clinical monitoring and other potential confounders. Schizophr Bull. 2015;41(3):644-655.
7. De Fazio P, Gaetano R, Caroleo M, et al. Rare and very rare adverse effects of clozapine. Neuropsychiatr Dis Treat. 2015;11:1995-2003.
8. Novartis Pharmaceuticals Corporation. Clozaril (clozapine). Prescribing information. http://clozaril.com/wp-content/themes/eyesite/pi/Clozaril-2015A507-10022015-Approved.pdf. Accessed June 29, 2016.
9. Lieberman JA, Johns CA, Kane JM, et al. Clozapine-induced agranulocytosis: non-cross-reactivity with other psychotropic drugs. J Clin Psychiatry. 1988;49(7):271-277.
10. Honigfeld G, Arellano F, Sethi J, et al. Reducing clozapine-related morbidity and mortality: 5 years of experience with the Clozaril National Registry. J Clin Psychiatry. 1998;59(suppl 3):3-7.
11. Demler TL, Trigoboff E. Are clozapine patients at risk for blood dyscrasias with concomitant tamiflu use? Psychiatry (Edgmont). 2009;6(11):29-33.
12. Karalakulasingam R, Schacht RA, Lansing AM, et al. Influenza virus pneumonia after renal transplant. Postgrad Med. 1977;62(2):164-167.
13. Hoffman-La Roche Limited. Product monograph: Tamiflu. http://www.rochecanada.com/content/dam/roche_canada/en_CA/documents/Research/ClinicalTrialsForms/Products/ConsumerInformation/MonographsandPublicAdvisories/Tamiflu/Tamiflu_PM_E.pdf. Updated January 26, 2015. Accessed November 28, 2015.
14. Whiskey E, Taylor D. Restarting clozapine after neutropenia: evaluating the possibilities and practicalities. CNS Drugs. 2007;21(1):25-35.
15. Palominao A, Kukoyi O, Xiong GL. Leukocytosis after lithium and clozapine combination therapy. Ann Clin Psychiatry. 2010;22(3):205-206.
16. Focosi D, Azzarà A, Kast RE, et al. Lithium and hematology: established and proposed uses. J Leukoc Biol. 2009;85(1):20-28.
17. Papetti F, Darcourt G, Giordana JY, et al. Treatment of clozapine-induced granulocytopenia with lithium (two observations) [in French]. Encephale. 2004;30(6):578-582.
18. Hummer M, Sperner-Unterweger B, Kemmler G, et al. Does eosinophilia predict clozapine induced neutropenia? Psychopharmacology (Berl). 1996;124(1-2):201-204.
19. Aneja J, Sharma N, Mahajan S, et al. Eosinophilia induced by clozapine: a report of two cases and review of the literature. J Family Med Prim Care. 2015;4(1):127-129.
20. Nielsen J, Correll CU, Manu P, et al. Termination of clozapine treatment due to medical reasons: when is it warranted and how can it be avoided? J Clin Psychiatry. 2013;74(6):603-613.
21. Stryjer R, Timinsky I, Reznik, I, et al. Beta-adrenergic antagonists for the treatment of clozapine-induced sinus tachycardia: a retrospective study. Clin Neuropharmacol. 2009;32(5):290-292.
22. Lally J, Docherty MJ, MacCabe JH. Pharmacological interventions for clozapine-induced sinus tachycardia. Cochrane Database Syst Rev. 2016;9(6):CD011566.
23. Kamphuis H, Arends J, Timmerman L, et al. Myocarditis and cardiomyopathy: underestimated complications resulting from clozapine therapy [in Dutch]. Tijdschr Psychiatr. 2010;52(4):223-233.
24. Alawami M, Wasywich C, Cicovic A, et al. A systematic review of clozapine induced cardiomyopathy. Int J Cardiol. 2014;176(2):315-320.
25. Ronaldson KJ, Fitzgerald PB, Taylor AJ, et al. A new monitoring protocol for clozapine-induced myocarditis based on an analysis of 75 cases and 94 controls. Aust N Z J Psychiatry. 2011;45(6):458-465.
26. Williams AM, Park SH. Seizure associated with clozapine: incidence, etiology, and management. CNS Drugs. 2015;29(2):101-111.
27. Meyer JM. Individual changes in clozapine levels after smoking cessation: results and a predictive model. J Clin Psychopharmacol. 2001;21(6):569-574.
28. Saavedra-Velez C, Yusim A, Anbarasan D, et al. Modafinil as an adjunctive treatment of sedation, negative symptoms, and cognition in schizophrenia: a critical review. J Clin Psychiatry. 2009;70(1):104-112.
29. Klein C, Gordon J, Pollak L, et al. Clozapine in Parkinson’s disease psychosis: 5-year follow-up review. Clin Neuropharmacol. 2003;26(1):8-11.
30. Lutz UC, Sirfy A, Wiatr G, et al. Clozapine serum concentrations in dopamimetic psychosis in Parkinson’s disease and related disorders. Eur J Clin Pharmacol. 2014;70(12):1471-1476.
31. Bird AM, Smith TL, Walton AE. Current treatment strategies for clozapine-induced sialorrhea. Ann Pharmacother. 2011;45(5):667-675.
32. Duggal HS. Glycopyrrolate for clozapine-induced sialorrhea. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31(7):1546-1547.
33. Leadbetter R, Shutty M, Pavalonis D, et al. Clozapine-induced weight gain: prevalence and clinical relevance. Am J Psychiatry. 1992;149(1):68-72.
34. Lundblad W, Azzam PN, Gopalan, et al. Medical management of patients on clozapine: a guide for internists. J Hosp Med. 2015;10(8):537-543.
35. Henderson DC, Nguyen DD, Copeland PM, et al. Clozapine, diabetes mellitus, hyperlipidemia, and cardiovascular risks and mortality: results of a 10-year naturalistic study. J Clin Psychiatry. 2005;66(9):1116-1121.
36. Stroup TS, Gerhard T, Crystal S, et al. Comparative effectiveness of clozapine and standard antipsychotic treatment in adults with schizophrenia. Am J Psychiatry. 2016;173(2):166-173.
37. Carrizo E, Fernández V, Connell L, et al. Extended release metformin for metabolic control assistance during prolonged clozapine administration: a 14 week, double-blind, parallel group, placebo-controlled study. Schizophr Res. 2009;113(1):19-26.
38. Chen CH, Huang MC, Kao CF, et al. Effects of adjunctive metformin on metabolic traits in nondiabetic clozapine-treated patients with schizophrenia and the effect of metformin discontinuation on body weight: a 24-week, randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2013;74(5):e424-e430.
39. Mizuno Y, Suzuki T, Nakagawa A, et al. Pharmacological strategies to counteract antipsychotic-induced weight gain and metabolic adverse effects in schizophrenia: a systematic review and meta-analysis. Schizophr Bull. 2014;40(6):1385-1403.
40. Nielsen J, Meyer JM. Risk factors for ileus in patients with schizophrenia. Schizophr Bull. 2012;38(3):592-598.
41. Lowe CM, Grube RR, Scates AC. Characterization and clinical management of clozapine-induced fever. Ann Pharmacother. 2007;41(10):1700-1704.
42. ACOG Committee on Practice Bulletins–Obstetrics. ACOG Practice Bulletin: Clinical management guidelines for obstetrician-gynecologists number 92, April 2008 (replaces practice bulletin number 87, November 2007). Use of psychiatric medications during pregnancy and lactation. Obstet Gynecol. 2008;111(4):1001-1020.
43. U.S. Food and Drug Administration. Pregnancy and Lactation Labeling (Drugs) Final Rule. https://s3.amazonaws.com/public-inspection.federalregister.gov/2014-28241.pdf. Published December 4, 2014. Accessed July 6, 2016.
44. Briggs GG, Freeman RK, Yaffe SJ. Drugs in pregnancy and lactation: a reference guide to fetal and neonatal risk. 9th ed. Baltimore, MD: Lippincott Williams & Wilkins; 2011.
45. Larsen ER, Damkier P, Pedersen LH, et al; Danish Psychiatric Society; Danish Society of Obstetrics and Gynecology; Danish Paediatric Society; Danish Society of Clinical Pharmacology. Use of psychotropic drugs during pregnancy and breast-feeding. Acta Psychiatr Scand Suppl. 2015;(445):1-28.
46. McKenna K, Koren G, Tetelbaum M, et al. Pregnancy outcome of women using atypical antipsychotic drugs: a prospective comparative study. J Clin Psychiatry. 2005;66(4):444-449.
