User login
Staying afloat in a sea of information: Point-of-care resources
It is friday afternoon on a sunny July day. The last patient of the day, Ms. Connecticut, is an active hiker who has had Lyme disease previously. She found a tick on her ankle yesterday. She successfully removed the tick but has not brought the tick with her to the appointment. She had been hiking several times over the last week and is not certain when the tick bite occurred. Her question for you centers on the role of antibiotic prophylaxis and Lyme disease prevention.
TECHNOLOGY: PROBLEM AND SOLUTION
Physicians need to keep up with an ever-increasing stream of information—new guidelines, new medications, and updates in medical literature.1 They have to do this while seeing more patients with more chronic problems in less time and while meeting reporting requirements for meaningful use or quality measures for accountable care organizations.
Though some of these challenges are due to technology, one solution is to use technology to our advantage. While researching information in textbooks won’t drain a phone battery, carrying a textbook around is not feasible, and many textbooks (including their electronic versions) contain information that is outdated before they go to print or that is quickly outdated thereafter.2 Further, even online textbooks are currently more dense than the online resources that we review here.
Different types of resources can help task-saturated healthcare providers stay aware of new information while delivering evidence-based care. These tools—online textbooks, decision guides embedded within electronic health record systems, or even a Google search—are termed “point-of-care” resources when used at the time of patient care for decision-making in the moment.
Not all of these resources are of high quality, with reliable factual information. Researchers estimate that up to 70% of clinicians may use Wikipedia to research medical questions, and a comparison of 10 Wikipedia articles vs peer-reviewed sources on the 10 most costly diseases found that 9 of the 10 Wikipedia articles had errors.3,4
In an earlier article,5 we advocated a proactive approach to managing information, highlighting ways to scan for new information and to develop habits of extracting useful information that can then be stored and easily recovered. To complement this strategy and weed out erroneous information, physicians need reliable sources of unbiased information to efficiently answer clinical questions at the point of care.1,6
Here, to help busy clinicians choose which point-of-care resources to use, we review several of the most popular ones, examining their ease of use, key elements, strengths, and weaknesses.
WHAT MAKES A RESOURCE GOOD?
Key features that make point-of-care tools effective include:
Ease of use, with standard formats, a summary for each topic, or both
Links to original articles and concise, capsular summaries and syntheses of the data
Continuing medical education (CME) credit. Tip: when searching, add “CME” to the search string on the browser to access resources that provide this.
Institutional and individual accounts. For clinicians who work for large organizations, point-of-care products may be paid for already, or reimbursement may be available for your subscription. If unsure, ask your director of information technology or library services.
Freedom from advertisements. Many Internet sources have advertisements that either run alongside the information you want to see or, more annoyingly, pop up and require an action to move forward. There is also continuing concern about the effect of industry support on content.7 While not all of the resources that we use regularly and that we review here are ad-free, avoiding programs with high ad content helps limit the possibility of bias and the time it takes to access information. Although advertisements do bring up a risk of bias, resources with a low-level ad content can limit bias while providing free or low-cost access.
Evidence, not expert opinion. Many resources have an “about” page that explains their philosophy and the source of their information. It is vital to be sure that point-of-care databases are providing facts based on evidence.8 This page also typically addresses how authors and editors are selected and whether expert opinion is used when randomized trials are lacking.
Ease of access. Many tools can be accessed not only on computers but also through apps for smartphones and tablets. Some electronic medical records have clinical decision tools embedded in them, with varying capabilities.
Disclosure of conflict of interest. As conflicts of interest can shade recommendations, information sources should clearly disclose financial relationships that could be perceived as conflicts of interest—for example, authors writing about medications sold by companies with whom they have a financial relationship.
NO SINGLE RESOURCE DOES EVERYTHING
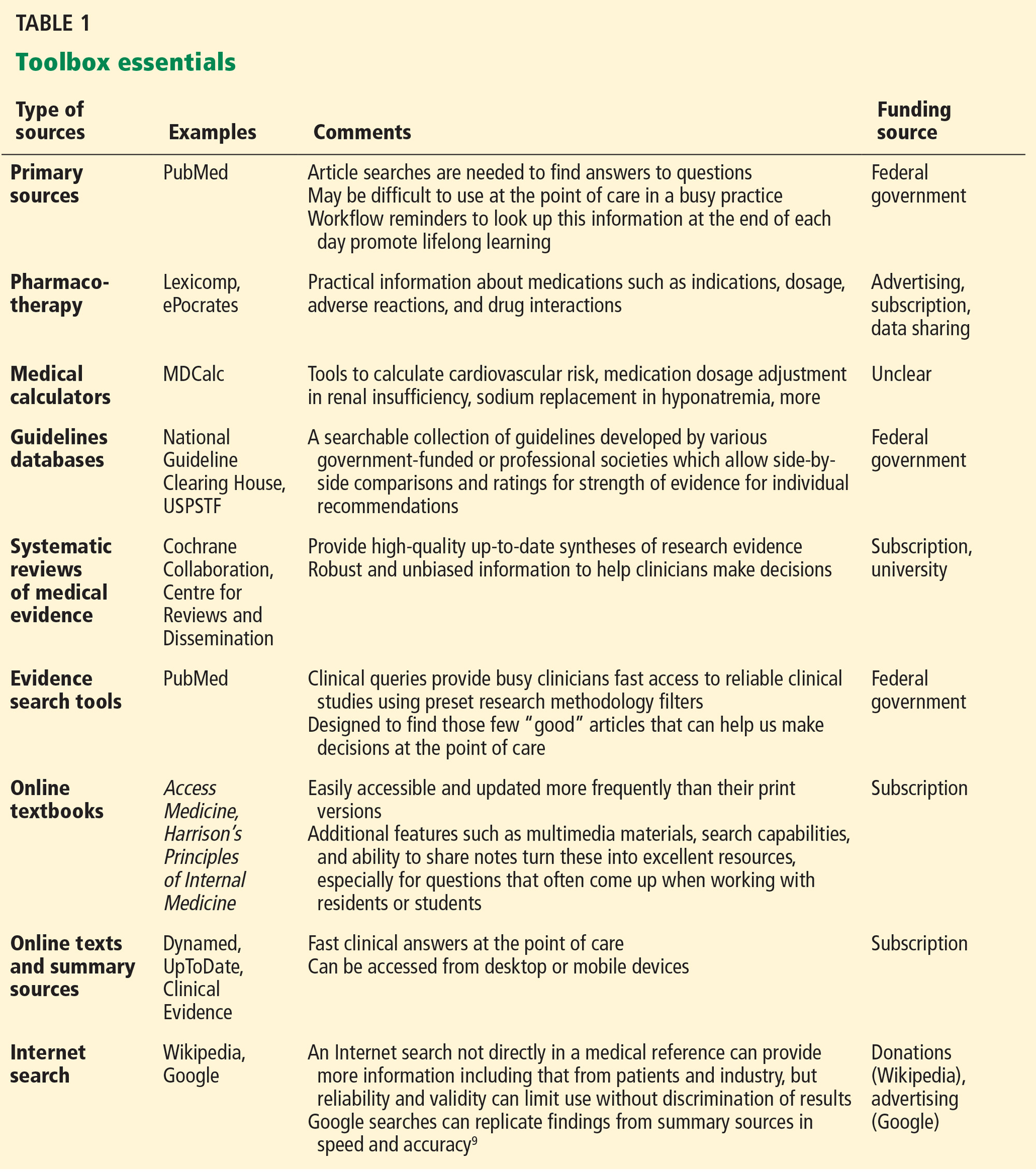
There are many types of tools for finding evidence-based medical information. Different tools serve different purposes. Table 1 lists “toolbox essentials” for clinicians needing to answer clinical questions during patient care.
For example, when a question about the need for a bone mineral density measurement comes up, it is useful to be able to quickly compare guidelines from different professional societies on the National Guideline Clearing House. For another example, if a patient brings in a medication in an unlabeled bottle, a pill identifier app can tell you what it is. Clinicians who can use these resources appropriately will be at an advantage in being able to use information to provide better care to their patients.
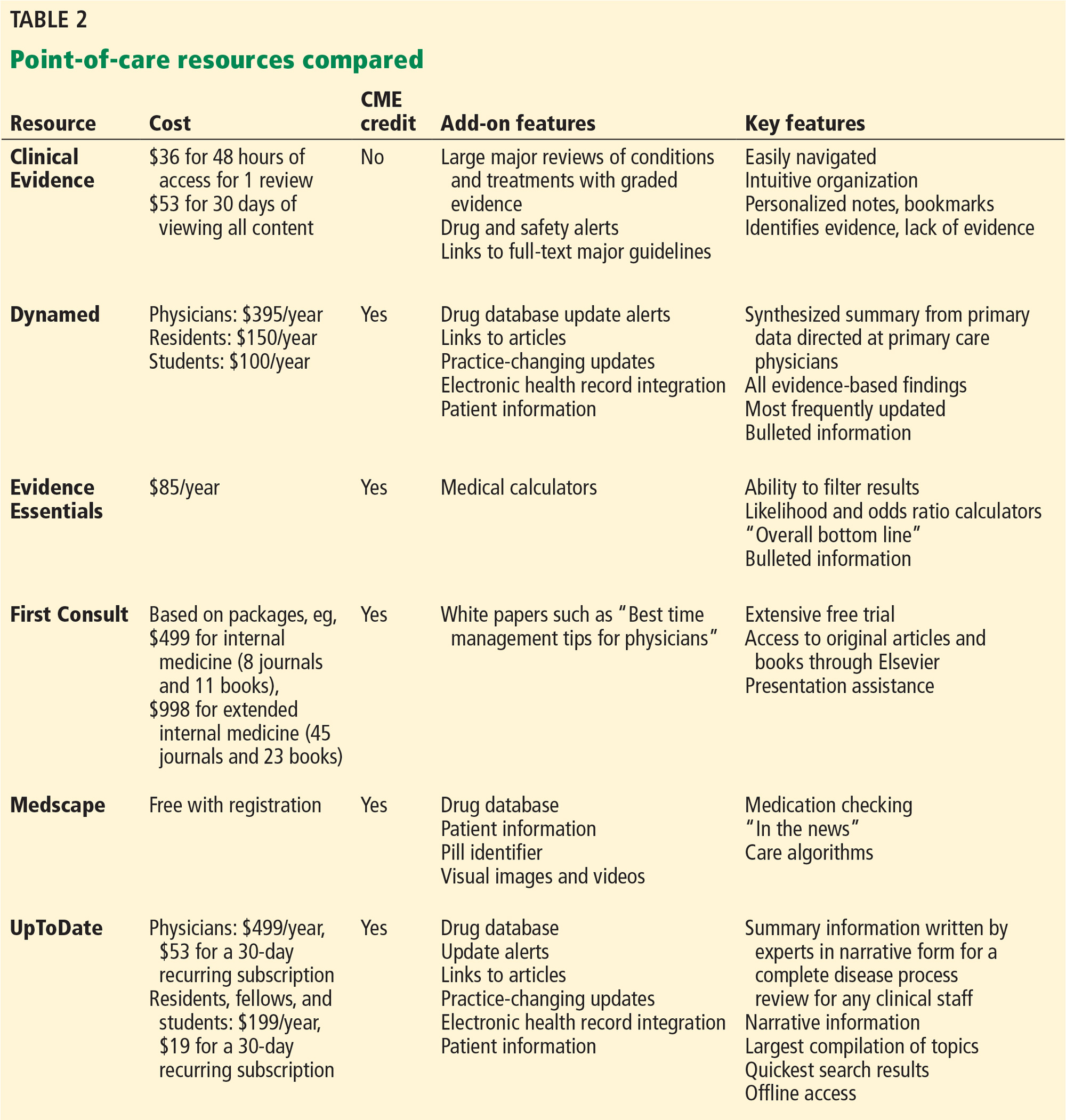
To date, no point-of-care summary source has been shown to be superior in all categories, and use may be driven by ease of navigation, clinician preference, clinical question, or past success.9,10
Reviewed below are several applications that can be used as point-of-care resources (Table 2).
CLINICAL EVIDENCE
Clinical Evidence provides systematic reviews on medical topics. Founded in 1999 by the British Medical Journal, it is available in print as the Clinical Evidence Handbook and in online desktop and smartphone formats.
More than any other source we reviewed, Clinical Evidence addresses not only the evidence that exists, but also the data that do not exist to guide decisions. Compared with 9 other point-of-care resources, Clinical Evidence was found to have the highest quality of evidence.11
Strengths of Clinical Evidence
- Uncommonly transparent in terms of source of evidence or disclosing when there is a lack of evidence.
- Clearly lists the strength and relevance of the evidence.
- Personalization. Users can add notes to articles, save personal searches, and bookmark pages for easy access later.
- Navigability. Users can easily access systematic reviews, key points, retracted papers, or guidelines.
- Intuitive organization, with information categorized as research, education, news, or campaigns.
- New content daily: podcasts, articles, videos.
Weaknesses of Clinical Evidence
- Limited topics (eg, Lyme disease was not available)
- The limited content is a challenge when needing quick information at the point of care and may cause most clinicians to use another source unless looking for comparisons of interventions.
- Cost. Subscribing to the service “on demand,” ie, to look up a single specific topic, costs $36 for 48 hours of access; monthly access or a “season ticket” allows 30 days of viewing of all content for $53. At over $600/year, this is one of the most costly of the sources we reviewed.
- Marketing of Clinical Evidence to academic institutions that support the service for faculty may limit its appeal to other clinicians.
DYNAMED
Dynamed, a clinical reference created by a group of physicians, was previously owned by the American College of Physicians and known as Smart Medicine; it is now owned by EBSCO.12 Reviewers investigate the literature for a given topic and create pithy summaries for busy clinicians. A top feature in Dynamed is its links to full articles cited for best practices or evidence-based guidelines. The company describes their content as free of expert opinion, while being unbiased and evidence-based.
Dynamed uses a 7-step algorithm for searched topics that identifies articles, assesses clinical relevance, evaluates validity of outcomes, compiles the evidence from multiple articles, and then updates the final recommendations daily.
Dynamed Plus, the new upgraded version, updates searched topics several times a day. Dynamed may be the most frequently updated point-of-care resource, with the least risk of conflict of interest, but it offers limited topics drawn from evidence-based findings.11,13–15
With the rapid doubling of the medical literature, frequent updates allow clinicians to be most current with practice guidelines. This potentially affects quality of care for antibiotic use, vaccination, health promotion, and screening as well as newly approved medications.
Strengths of Dynamed
- Large collection of topics, critically appraised, written for primary care physicians, presented in bulleted format
- The most frequently updated database11,14,15
- Can integrate with major electronic health records (eg, Epic, Allscripts, NextGen, Cerner)
- Has an area devoted to new information that changes current practice
- Chosen topic grouped with related topics in the differential diagnosis after the initial search
- Easy-to-read outline for quick access to information such as billing, diagnosis, and references
- Medical calculators
- No advertisements
- Helpful embedded tools
- Icons to print or email the article
- An icon to create a “perma-link” to topics, searches, and browse categories
- Graded evidence with a link to the grading model used
- Links to primary articles
- Patient information handouts
- Alerts for updated information
- CME credit
- Special consideration and features for medical education
- The upgraded version Dynamed Plus contains Micromedex for a medication database, expanded graphics, semantic search, concise overview for each topic, and expanded content.
Weaknesses of Dynamed
- Although the topic list is large, it is only about one-third the size of UpToDate.
- A subscription for a physician costs $395 a year. Residents can sign up for about $150, and students for just under $100.
- CME is obtainable but cumbersome; one submits the CME credits through Tufts Healthcare, which requires a second sign-on to access and track.
- Drug and nondrug treatments for diseases cannot be separated.
- Useful calculators include decision trees for clinical decision-making, but there is no way to search them—one must waste time scrolling through the topics and specialties looking for desired information.
- Major shortcoming: there is no medication reference tool unless you upgrade to Dynamed Plus.
- The expanded graphics of Dynamed Plus are difficult to view on mobile applications within the articles (they are brought up more reliably when searching just for the image).
- The use of strict evidence-based methodology without expert opinion is a strength, but limits the collection of topics without randomized controlled trials, for which turning to expert opinion may be the only option.
EVIDENCE ESSENTIALS
Evidence Essentials is a point-of-care resource from Wiley that offers a variety of content types. The website lists 13,000 medical topics; however, they are not all summary reviews as discussed in the other products above. Subject matter is reviewed 3 times a year. Comprehensive reviews number just under 800 individual topics, with the remaining content consisting of Cochrane reviews, calculators, decision support tools, POEMs (Patient-Oriented Evidence that Matters), evidence-based medical guidelines, and dermatology images (1,000).
Evidence Essentials provides some unique content including a quick evaluation and management (E/M) code-finder and calculators not only for the typical medical equations, but also for history and physical examination likelihood ratios and pretest probabilities, which are practical and an excellent teaching aid. It also offers CME along with POEMs, e-mail alerts, and a listing of upcoming topics.
Strengths of Evidence Essentials
- Relatively inexpensive at $85 a year.
- High-functioning filter system to choose to search one or multiple databases.
- Related results are listed for aid in differential diagnosis, similar to Dynamed.
- Authors, editors, and date of last review are highly visible. As in UpToDate, relevant medical calculators appear on the page.
- The likelihood and odds ratio calculators are a huge plus for clinical decision-making and putting guidelines into practice.
- “Overall bottom line” highlights key points
- Grading of evidence per topic.
- Bulleted and tabbed information for quick access.
- Tabs for information on background, prevention, diagnosis, treatment, references, guidelines, and special populations.
Weaknesses of Evidence Essentials
- Limited number of topics with comprehensive reviews.
- While you can click on any drug name and link to a choice of two drug databases, this is not included in the subscription and requires a second account.
- The resources tabs had some broken links. In our clinical example, the tab contained several videos at the top that were not related, followed by a map and tables that were relevant to Lyme disease.
- Likewise, some of the guideline references were disappointing. For example, the guideline link for Lyme disease is for the US Department of Labor Occupational Safety and Health Administration rather than a professional society.
- For the provider wanting a narrative, this is more of a bare-bones text.
FIRST CONSULT
First Consult is Elsevier’s point-of-care clinical decision product contained within ClinicalKey.
Unlike UpToDate and Dynamed, in which authors and editors read original articles and summarize or synthesize information for the learner, First Consult is a “smart” search engine that will research a question, together with associated terms and key words. Filters such as full-text availability, journal articles, and patient education can be applied.
You may need to read about your topic in a textbook first, and then, if you are looking for treatment information, find an original article through First Consult. It is available in mobile and desktop formats, and the point-of-care product, First Consult, has an app that can be downloaded and used for free for the first 60 days.
Importantly, the First Consult portion of ClinicalKey with the summary topics was rated by Shurtz and Foster13 as least current of the products we are discussing in this article. On the other hand, it was the only product that had an embedded program to assist the user in making presentations by allowing drag and drop of images and automatic citing of sources. Kim et al report that First Consult is one of the resources providers prefer.9
Strengths of First Consult
- Lengthy free trial
- Ability to access original articles from a list vs lengthy narrative
- Access to journals and books published by Elsevier
- Powerful search engine that applies associated terms automatically
- Patient education is available in different languages and font size with the ability to add instructions and even a local branding
- Can integrate with electronic health record
- Can filter results by guideline, patient education, topic overviews
- Presentation assistance.
Weaknesses of First Consult
- Time-intensive. A provider needing quick advice on treatment for a medical condition has to guess if an article or textbook will have the most up-to-date and digestible information, whereas this has already been summarized in other products. For the busy clinician, this may be prohibitive.
- Search results are limited to Elsevier products, and major journals such as the New England Journal of Medicine are not available.
- Inconsistent platform functionality. The app version was somewhat “sticky” to use, as pages did not always load efficiently, and the menu bar navigation is not ideal.
- Expensive, especially given cheaper alternatives. For example, subscribing to the specialty of internal medicine or family medicine costs $499 and provides access to 8 journals and 11 books. Extended access costs $998 and offers full-text access to 23 books and 45 journals. The complete service has a total of 400 journals, 700 books, and 2,500 procedural videos.
MEDSCAPE
Medscape, owned by parent company WebMD, has long been a popular resource. The most recent versions are available for both for Android and iOS mobile platforms. The desktop and mobile apps claim to be designed for point-of-care use, and can be downloaded at no cost after registering as a Medscape user.
Medscape has some interesting features, including a handy pill identifier tool that is new to Medscape and perfect for the “I take one blue pill for my cholesterol” moments. The drug information tools and other features work well offline.
Medscape contains a well-presented drug database and interaction checker, as well as a growing collection of evidence-based articles and videos with links to references in Medline. From the point-of-care standpoint, Medscape also offers a number of decision-making algorithms and a continuously updated medical literature and health-related newsfeed. It contains in-app medical calculators, searchable directories for providers, hospitals, and pharmacies, and CME that can be earned on the website or from the application.
The main Medscape website contains pop-up advertisements, but the mobile app has fewer. Among the occasional frustrations, updates are relatively infrequent, the content is slow to load, and the phone app can be cumbersome. Of note, in one review,11 Medscape was found to have the lowest quality of evidence.
Strengths of Medscape
- Free with registration
- Medical calculator
- Drug interaction checker
- Pill identifier
- Evidence-based information covering about 4,000 conditions with links to references in Medline
- Ability to e-mail articles for sharing or future reference
- CME
- Unique database of hospitals, providers, and pharmacies to aid in referrals or locating other healthcare professionals
- Algorithms for decision-making
- Images and videos for procedural review and learning
- Option for downloading certain databases for offline use
- Medical news helps you keep up with what patients are watching and reading.
Weaknesses of Medscape
- Advertisements (many of them pop-up)
- The content is updated less frequently than other products listed in this article
- The smartphone app can run slowly
- Quality of reviews may be a concern.
UpToDate
UpToDate (Wolters Kluwer) is used widely by medical students, residents, and fellows as well as practicing providers. It contains narrative reviews of topics written by respected experts directed at both clinicians and clinical staff. In hopes of appealing to many markets, it offers different subscription types so you can customize your choices with add-on features (UpToDate Desktop and UpToDate MobileComplete allow downloading of all content to be accessed offline), different service packages (1-, 2-, and 3-year subscriptions), and the traditional base product that provides online access.
Of the products we reviewed, UpToDate has the largest selection of medical topics, approaching 10,000.14 In some studies,10,15 it also had the fastest retrieval time for searches. It uses evidence-based graded recommendations that are updated regularly.
Some have lamented that there is too much information per topic.9 In response to early reviews, Wolters Kluwer has made significant changes in the platform and greatly improved the search engine. UpToDate has expanded to include CME and patient information, trying to become that Holy Grail of websites—a one-stop experience. For the lucky few, UpToDate integrates into some electronic health records and provides a relatively seamless experience at the point of care.
Strengths of UpToDate
- One-stop shopping for information, resources, and CME
- Patient information is easy to read and accessible from the same screen
- The largest repository of medical subject matter
- Ability to cull out only pediatric or adult topics
- Searching available within a medical topic
- Tabs for quick access
- The What’s New feature allows access to practice-changing medical updates
- Medical calculators
- Drug interactions
- CME is is tracked in the system, allowing for CME credit information for hospital privileges and board certification
- Flexibility of access: can use online or download content to mobile/desktop device (the online version is easy to use, although robust wireless reception is needed; offices with slow Internet benefit from the offline feature)
- Electronic health record integration is possible with the most popular systems, such as Epic, eClinicalWorks, NextGen, and Allscripts
- Patient education and medication interaction features embedded in the electronic health record; produced in collaboration with Lexicomp
- Integrated drug database
- Alerts for updates
- References have links to full-text articles
- The date of last update is easily found for verifying information accuracy
- May be provided free for clinicians who are a part of a university or large health system.
Weaknesses of UpToDate
- Articles can be lengthy, which is both a strength and a weakness. Searches can retrieve too much information.9 High volume of text can frustrate the user trying to find bulleted, easy-to-read facts. However, for the person looking for a narrative summary, the content is organized as narrative paragraphs with appropriate headers in the left margin, and the search function is robust and powerful.
- Each topic has a “Summary of Recommendations,” but answers here often require linking back to the main text.
- Patient information is sometimes at a high literacy level.
- Costs more than Dynamed. A 1-year subscription is $499 for a physician, but you have the option of paying $53 for a 30-day recurring subscription. Residents, fellows, and students can pay $199 for 1 year or $19 for a 30-day recurring subscription.
- The requirement to download means that users need to keep their version updated on all of their computers—in each of their examination rooms, for example.
- Concerns about conflict of interest arise because authors and editors may maintain financial relationships with companies that produce medications discussed in the articles they have written.
BUILDING YOUR OWN PERSONAL ONLINE REPOSITORY
Our previous article5 reviewed how to store information using tools such as Evernote and Diigo that allow information viewed on a web page to be exported to any online repository. This can be done using extensions for a web browser or by sending the information to a custom e-mail account for these services.
For information that a provider knows he or she will need repeatedly, storage in one system is actually the easiest method. Such a system can then incorporate key information from the summary tools we have reviewed here. The ideal “electronic filing cabinet” should have several features such as a the capability to label articles by topic, to separate or sort as you see fit, and a search function to find information quickly—making it a personalized and effective point-of-care resource.
STAYING AFLOAT
Clinicians make many decisions every day. In fact, the release of How Doctors Think (both publications) has led to increased research into how clinical decisions and diagnoses are formed.16,17
With the medical literature expected to double every 73 days by 2020,18 there is an ever-widening ocean of information to sift through. With this onslaught, clinicians can no longer remain fully current. Instead, refining skills in accessing, sorting, and interpreting accurate scientific evidence efficiently is crucial to time spent actually caring for patients and coordinating their care.
Guidelines, algorithms, and comprehensive databases can aid clinicians in all aspects of care, from generating more complete differential diagnoses to managing disease-specific treatment. Individuals can first think about and list the qualities of a tool that are most important to them (eg, breadth of topics, frequency of updates, integration within their electronic health record, and cost) before focusing on a few applications or websites that meet those goals. With practice, point-of-care knowledge can become part of the everyday visit.
Effective integration into electronic health records will require design input from front-line clinicians. Otherwise, systems are prone to add too much “support” and overly rely on orthodox metrics and guidelines, resulting in alarm fatigue and frustration rather than facilitation.19–23
OUR CONCLUSIONS
Comprehensive point-of-care resources can play a significant role in helping busy clinicians provide best evidence-based care to their patients. Embedded clinical decision guides within an electronic health record are ideal, but low topic coverage has limited the usefulness of these systems.24 Here are our conclusions:
Medscape, ePocrates, and Wikipedia are probably the most popular free resources. Dynamed has offered free subscriptions to Wikipedia’s top health editors with the hopes of correcting factual errors. Medscape has excellent features but is supported by sponsored content, which raises a concern about bias and potential time-consuming distractions.
Dynamed and UpToDate have both been reported to answer more questions than other sources.12
UpToDate has the largest repository, with each topic curated by an expert or experts in that subject. This content can be dense and difficult to scan quickly at the point of care, but this is balanced by the ability to search within a medical topic, which has given it the fastest retrieval time.15 It does, however, allow authors and editors to maintain financial relationships with companies that produce medications discussed in the article.22
Dynamed has the advantage in frequency of updates, clearest conflict-of-interest policy, and the least amount of conflict of interest. Its topic list is not as extensive as UpToDate’s due to the limitation of using only evidence-based medicine without expert opinion.
First Consult has high user satisfaction, but as a point-of-care resource it can be time-consuming to find the best source for the clinical question at hand, and its expanded access is costly.9
ART AND SCIENCE
Point-of-care resources do not solve all the complicated problems of patient care, and no single resource is ideal for all situations. A busy clinician has limited time to process the evolving literature to practice the best evidence-based medicine. Effective information access, quality of care provided, and the marginal time cost required create a complex calculus. Clinical decision-making remains an art and a science,25 but these technologies help define a new era in its pursuit.
Ultimately, a clinician’s choice needs to correlate with a provider’s resources and style. This article has detailed several options available on the market today. This is a quickly evolving area of products and services. Longer term, users might consider a tool’s preferred key features when evaluating any current or future resource in order to choose the right ones for their practice.
CASE REVISITED
Before we leave for the weekend, we need a plan for Ms. Connecticut. To find appropriate recommendations for our patient, we search several of our point-of-care resources: UpToDate and Dynamed. Both resources have correct information according to the Infectious Disease Society of America (IDSA) guidelines.
UpToDate has a monograph of approximately 2,000 words on Lyme disease, which is lengthy but adds to clinical-decision making skills for a learner thinking through the decision. This service also has a patient handout highlighting the recommendations. The topic was last updated in 2016, but states that it is current with literature through January 2017.
Dynamed has bulleted information that is quicker to digest, but essentially highlights the IDSA recommendations without the thought process behind them. It too, has patient resources with links to a variety of handouts from professional organizations such as the US Centers for Disease Control and Prevention. They last updated the topic January 31, 2017.
When searching for the topic on both sites, a clinician can see the breadth of information in each program. However, this is also a detractor. Searching for Lyme disease prophylaxis on Dynamed brought up related data (that doxycycline is not FDA-approved for prophylaxis), but not the primary information. Likewise, the search under UpToDate first brought us to the patient information. Both articles have helpful tables and links to associated topics.
My partner chose the UpToDate article, in part to review the topic with a medical student. However, I used Dynamed for its quick bulleted information, as I was on call that evening and needed to return to the hospital. We both came to the same conclusion, and Ms. Connecticut chose no prophylaxis even though her home is in an endemic area. She has done well.
- Worster A, Haynes RB. How do I find a point-of-care answer to my clinical question? CJEM 2012; 14:31–35.
- Jeffery R, Navarro T, Lokker C, Haynes RB, Wilczynski NL, Farjou G. How current are leading evidence-based medical textbooks? An analytic survey of four online textbooks. J Med Internet Res 2012; 14:e175.
- ClinicalKey. Errors found in nine out of ten Wikipedia health entries. www.clinicalkey.com/info/blog/errors-in-wikipedia-health/. Accessed February 9, 2017.
- Hasty RT, Garbalosa RC, Barbato VA, et al. Wikipedia vs peer-reviewed medical literature for information about the 10 most costly medical conditions. J Am Osteopath Assoc 2014; 114:368–373.
- Mehta NB, Martin SA, Maypole J, Andrews R. Information management for clinicians. Cleve Clin J Med 2016; 83:589–595.
- Cook DA, Sorensen KJ, Hersh W, Berger RA, Wilkinson JM. Features of effective medical knowledge resources to support point of care learning: a focus group study. PLoS One 2013; 8:e80318.
- Steinbrook R. Future directions in industry funding of continuing medical education. Arch Intern Med 2011; 171:257–258.
- Isaacs D, Fitzgerald D. Seven alternatives to evidence based medicine. BMJ 1999; 319:1618.
- Kim S, Noveck H, Galt J, Hogshire L, Willett L, O’Rourke K. Searching for answers to clinical questions using Google versus evidence-based summary resources: a randomized controlled crossover study. Acad Med 2014; 89:940–943.
- Ahmadi SF, Faghankhani M, Javanbakht A, et al. A comparison of answer retrieval through four evidence-based textbooks (ACP PIER, Essential Evidence Plus, First Consult, and UpToDate): a randomized controlled trial. Med Teach 2011; 33:724–730.
- Prorok JC, Iserman EC, Wilczynski NL, Haynes RB. The quality, breadth, and timeliness of content updating vary substantially for 10 online medical texts: an analytic survey. J Clin Epidemiol 2012; 65:1289–1295.
- Prorok JC, Iserman EC, Wilczynski NL, Haynes RB. The quality, breadth, and timeliness of content updating vary substantially for 10 online medical texts: an analytic survey. J Clin Epidemiol 2012; 65:1289–1295.
- Shurtz S, Foster MJ. Developing and using a rubric for evaluating evidence-based medicine point-of-care tools. J Med Libr Assoc 2011; 99:247–254.
- Ketterman E, Besaw M. An evaluation of citation counts, search results, and frequency of updates in Dynamed and UpToDate. J Electron Res in Med Libr 2010; 7:273–280.
- Amber KT, Dhiman G, Goodman KW. Conflict of interest in online point-of-care clinical support websites. J Med Ethics 2014; 40:578–580.
- Montgomery K. How Doctors Think: Clinical Judgment and the Practice of Medicine. New York, NY: Oxford University Press; 2005.
- Groopman J. How Doctors Think. Boston, MA: Houghton Mifflin; 2008.
- Densen P. Challenges and opportunities facing medical education. Trans Am Clin Climatol Assoc 2010; 122:48–58.
- Kesselheim AS, Cresswell K, Phansalkar S, Bates DW, Sheikh A. Clinical decision support systems could be modified to reduce ‘alert fatigue’ while still minimizing the risk of litigation. Health Aff (Millwood) 2011; 30:2310–2317.
- Russ AL, Zillich AJ, McManus MS, Doebbeling BN, Saleem JJ. Prescribers’ interactions with medication alerts at the point of prescribing: a multi-method, in situ investigation of the human-computer interaction. Int J Med Inform 2012; 81:232–243.
- Fraccaro P, Arguello Castelerio M, Ainsworth J, Buchan I. Adoption of clinical decision support in multimorbidity: a systematic review. JMIR Med Informatics 2015; 3:e4.
- McLeod W, Eidus R, Stewart EE. Clinical decision support: using technology to identify patients’ unmet needs. Fam Pract Manag 2012; 19:22–28.
- Colla CH. Swimming against the current—what might work to reduce low-value care? N Engl J Med 2014; 371:1280–1283.
- Cook DA, Sorensen KJ, Nishimura RA, Ommen SR, Lloyd FJ. A comprehensive information technology system to support physician learning at the point of care. Acad Med 2015; 90:33–39.
- Woolever DR. The art and science of clinical decision making. Fam Pract Manag 2008; 15:31–36.
It is friday afternoon on a sunny July day. The last patient of the day, Ms. Connecticut, is an active hiker who has had Lyme disease previously. She found a tick on her ankle yesterday. She successfully removed the tick but has not brought the tick with her to the appointment. She had been hiking several times over the last week and is not certain when the tick bite occurred. Her question for you centers on the role of antibiotic prophylaxis and Lyme disease prevention.
TECHNOLOGY: PROBLEM AND SOLUTION
Physicians need to keep up with an ever-increasing stream of information—new guidelines, new medications, and updates in medical literature.1 They have to do this while seeing more patients with more chronic problems in less time and while meeting reporting requirements for meaningful use or quality measures for accountable care organizations.
Though some of these challenges are due to technology, one solution is to use technology to our advantage. While researching information in textbooks won’t drain a phone battery, carrying a textbook around is not feasible, and many textbooks (including their electronic versions) contain information that is outdated before they go to print or that is quickly outdated thereafter.2 Further, even online textbooks are currently more dense than the online resources that we review here.
Different types of resources can help task-saturated healthcare providers stay aware of new information while delivering evidence-based care. These tools—online textbooks, decision guides embedded within electronic health record systems, or even a Google search—are termed “point-of-care” resources when used at the time of patient care for decision-making in the moment.
Not all of these resources are of high quality, with reliable factual information. Researchers estimate that up to 70% of clinicians may use Wikipedia to research medical questions, and a comparison of 10 Wikipedia articles vs peer-reviewed sources on the 10 most costly diseases found that 9 of the 10 Wikipedia articles had errors.3,4
In an earlier article,5 we advocated a proactive approach to managing information, highlighting ways to scan for new information and to develop habits of extracting useful information that can then be stored and easily recovered. To complement this strategy and weed out erroneous information, physicians need reliable sources of unbiased information to efficiently answer clinical questions at the point of care.1,6
Here, to help busy clinicians choose which point-of-care resources to use, we review several of the most popular ones, examining their ease of use, key elements, strengths, and weaknesses.
WHAT MAKES A RESOURCE GOOD?
Key features that make point-of-care tools effective include:
Ease of use, with standard formats, a summary for each topic, or both
Links to original articles and concise, capsular summaries and syntheses of the data
Continuing medical education (CME) credit. Tip: when searching, add “CME” to the search string on the browser to access resources that provide this.
Institutional and individual accounts. For clinicians who work for large organizations, point-of-care products may be paid for already, or reimbursement may be available for your subscription. If unsure, ask your director of information technology or library services.
Freedom from advertisements. Many Internet sources have advertisements that either run alongside the information you want to see or, more annoyingly, pop up and require an action to move forward. There is also continuing concern about the effect of industry support on content.7 While not all of the resources that we use regularly and that we review here are ad-free, avoiding programs with high ad content helps limit the possibility of bias and the time it takes to access information. Although advertisements do bring up a risk of bias, resources with a low-level ad content can limit bias while providing free or low-cost access.
Evidence, not expert opinion. Many resources have an “about” page that explains their philosophy and the source of their information. It is vital to be sure that point-of-care databases are providing facts based on evidence.8 This page also typically addresses how authors and editors are selected and whether expert opinion is used when randomized trials are lacking.
Ease of access. Many tools can be accessed not only on computers but also through apps for smartphones and tablets. Some electronic medical records have clinical decision tools embedded in them, with varying capabilities.
Disclosure of conflict of interest. As conflicts of interest can shade recommendations, information sources should clearly disclose financial relationships that could be perceived as conflicts of interest—for example, authors writing about medications sold by companies with whom they have a financial relationship.
NO SINGLE RESOURCE DOES EVERYTHING
There are many types of tools for finding evidence-based medical information. Different tools serve different purposes. Table 1 lists “toolbox essentials” for clinicians needing to answer clinical questions during patient care.
For example, when a question about the need for a bone mineral density measurement comes up, it is useful to be able to quickly compare guidelines from different professional societies on the National Guideline Clearing House. For another example, if a patient brings in a medication in an unlabeled bottle, a pill identifier app can tell you what it is. Clinicians who can use these resources appropriately will be at an advantage in being able to use information to provide better care to their patients.
To date, no point-of-care summary source has been shown to be superior in all categories, and use may be driven by ease of navigation, clinician preference, clinical question, or past success.9,10
Reviewed below are several applications that can be used as point-of-care resources (Table 2).
CLINICAL EVIDENCE
Clinical Evidence provides systematic reviews on medical topics. Founded in 1999 by the British Medical Journal, it is available in print as the Clinical Evidence Handbook and in online desktop and smartphone formats.
More than any other source we reviewed, Clinical Evidence addresses not only the evidence that exists, but also the data that do not exist to guide decisions. Compared with 9 other point-of-care resources, Clinical Evidence was found to have the highest quality of evidence.11
Strengths of Clinical Evidence
- Uncommonly transparent in terms of source of evidence or disclosing when there is a lack of evidence.
- Clearly lists the strength and relevance of the evidence.
- Personalization. Users can add notes to articles, save personal searches, and bookmark pages for easy access later.
- Navigability. Users can easily access systematic reviews, key points, retracted papers, or guidelines.
- Intuitive organization, with information categorized as research, education, news, or campaigns.
- New content daily: podcasts, articles, videos.
Weaknesses of Clinical Evidence
- Limited topics (eg, Lyme disease was not available)
- The limited content is a challenge when needing quick information at the point of care and may cause most clinicians to use another source unless looking for comparisons of interventions.
- Cost. Subscribing to the service “on demand,” ie, to look up a single specific topic, costs $36 for 48 hours of access; monthly access or a “season ticket” allows 30 days of viewing of all content for $53. At over $600/year, this is one of the most costly of the sources we reviewed.
- Marketing of Clinical Evidence to academic institutions that support the service for faculty may limit its appeal to other clinicians.
DYNAMED
Dynamed, a clinical reference created by a group of physicians, was previously owned by the American College of Physicians and known as Smart Medicine; it is now owned by EBSCO.12 Reviewers investigate the literature for a given topic and create pithy summaries for busy clinicians. A top feature in Dynamed is its links to full articles cited for best practices or evidence-based guidelines. The company describes their content as free of expert opinion, while being unbiased and evidence-based.
Dynamed uses a 7-step algorithm for searched topics that identifies articles, assesses clinical relevance, evaluates validity of outcomes, compiles the evidence from multiple articles, and then updates the final recommendations daily.
Dynamed Plus, the new upgraded version, updates searched topics several times a day. Dynamed may be the most frequently updated point-of-care resource, with the least risk of conflict of interest, but it offers limited topics drawn from evidence-based findings.11,13–15
With the rapid doubling of the medical literature, frequent updates allow clinicians to be most current with practice guidelines. This potentially affects quality of care for antibiotic use, vaccination, health promotion, and screening as well as newly approved medications.
Strengths of Dynamed
- Large collection of topics, critically appraised, written for primary care physicians, presented in bulleted format
- The most frequently updated database11,14,15
- Can integrate with major electronic health records (eg, Epic, Allscripts, NextGen, Cerner)
- Has an area devoted to new information that changes current practice
- Chosen topic grouped with related topics in the differential diagnosis after the initial search
- Easy-to-read outline for quick access to information such as billing, diagnosis, and references
- Medical calculators
- No advertisements
- Helpful embedded tools
- Icons to print or email the article
- An icon to create a “perma-link” to topics, searches, and browse categories
- Graded evidence with a link to the grading model used
- Links to primary articles
- Patient information handouts
- Alerts for updated information
- CME credit
- Special consideration and features for medical education
- The upgraded version Dynamed Plus contains Micromedex for a medication database, expanded graphics, semantic search, concise overview for each topic, and expanded content.
Weaknesses of Dynamed
- Although the topic list is large, it is only about one-third the size of UpToDate.
- A subscription for a physician costs $395 a year. Residents can sign up for about $150, and students for just under $100.
- CME is obtainable but cumbersome; one submits the CME credits through Tufts Healthcare, which requires a second sign-on to access and track.
- Drug and nondrug treatments for diseases cannot be separated.
- Useful calculators include decision trees for clinical decision-making, but there is no way to search them—one must waste time scrolling through the topics and specialties looking for desired information.
- Major shortcoming: there is no medication reference tool unless you upgrade to Dynamed Plus.
- The expanded graphics of Dynamed Plus are difficult to view on mobile applications within the articles (they are brought up more reliably when searching just for the image).
- The use of strict evidence-based methodology without expert opinion is a strength, but limits the collection of topics without randomized controlled trials, for which turning to expert opinion may be the only option.
EVIDENCE ESSENTIALS
Evidence Essentials is a point-of-care resource from Wiley that offers a variety of content types. The website lists 13,000 medical topics; however, they are not all summary reviews as discussed in the other products above. Subject matter is reviewed 3 times a year. Comprehensive reviews number just under 800 individual topics, with the remaining content consisting of Cochrane reviews, calculators, decision support tools, POEMs (Patient-Oriented Evidence that Matters), evidence-based medical guidelines, and dermatology images (1,000).
Evidence Essentials provides some unique content including a quick evaluation and management (E/M) code-finder and calculators not only for the typical medical equations, but also for history and physical examination likelihood ratios and pretest probabilities, which are practical and an excellent teaching aid. It also offers CME along with POEMs, e-mail alerts, and a listing of upcoming topics.
Strengths of Evidence Essentials
- Relatively inexpensive at $85 a year.
- High-functioning filter system to choose to search one or multiple databases.
- Related results are listed for aid in differential diagnosis, similar to Dynamed.
- Authors, editors, and date of last review are highly visible. As in UpToDate, relevant medical calculators appear on the page.
- The likelihood and odds ratio calculators are a huge plus for clinical decision-making and putting guidelines into practice.
- “Overall bottom line” highlights key points
- Grading of evidence per topic.
- Bulleted and tabbed information for quick access.
- Tabs for information on background, prevention, diagnosis, treatment, references, guidelines, and special populations.
Weaknesses of Evidence Essentials
- Limited number of topics with comprehensive reviews.
- While you can click on any drug name and link to a choice of two drug databases, this is not included in the subscription and requires a second account.
- The resources tabs had some broken links. In our clinical example, the tab contained several videos at the top that were not related, followed by a map and tables that were relevant to Lyme disease.
- Likewise, some of the guideline references were disappointing. For example, the guideline link for Lyme disease is for the US Department of Labor Occupational Safety and Health Administration rather than a professional society.
- For the provider wanting a narrative, this is more of a bare-bones text.
FIRST CONSULT
First Consult is Elsevier’s point-of-care clinical decision product contained within ClinicalKey.
Unlike UpToDate and Dynamed, in which authors and editors read original articles and summarize or synthesize information for the learner, First Consult is a “smart” search engine that will research a question, together with associated terms and key words. Filters such as full-text availability, journal articles, and patient education can be applied.
You may need to read about your topic in a textbook first, and then, if you are looking for treatment information, find an original article through First Consult. It is available in mobile and desktop formats, and the point-of-care product, First Consult, has an app that can be downloaded and used for free for the first 60 days.
Importantly, the First Consult portion of ClinicalKey with the summary topics was rated by Shurtz and Foster13 as least current of the products we are discussing in this article. On the other hand, it was the only product that had an embedded program to assist the user in making presentations by allowing drag and drop of images and automatic citing of sources. Kim et al report that First Consult is one of the resources providers prefer.9
Strengths of First Consult
- Lengthy free trial
- Ability to access original articles from a list vs lengthy narrative
- Access to journals and books published by Elsevier
- Powerful search engine that applies associated terms automatically
- Patient education is available in different languages and font size with the ability to add instructions and even a local branding
- Can integrate with electronic health record
- Can filter results by guideline, patient education, topic overviews
- Presentation assistance.
Weaknesses of First Consult
- Time-intensive. A provider needing quick advice on treatment for a medical condition has to guess if an article or textbook will have the most up-to-date and digestible information, whereas this has already been summarized in other products. For the busy clinician, this may be prohibitive.
- Search results are limited to Elsevier products, and major journals such as the New England Journal of Medicine are not available.
- Inconsistent platform functionality. The app version was somewhat “sticky” to use, as pages did not always load efficiently, and the menu bar navigation is not ideal.
- Expensive, especially given cheaper alternatives. For example, subscribing to the specialty of internal medicine or family medicine costs $499 and provides access to 8 journals and 11 books. Extended access costs $998 and offers full-text access to 23 books and 45 journals. The complete service has a total of 400 journals, 700 books, and 2,500 procedural videos.
MEDSCAPE
Medscape, owned by parent company WebMD, has long been a popular resource. The most recent versions are available for both for Android and iOS mobile platforms. The desktop and mobile apps claim to be designed for point-of-care use, and can be downloaded at no cost after registering as a Medscape user.
Medscape has some interesting features, including a handy pill identifier tool that is new to Medscape and perfect for the “I take one blue pill for my cholesterol” moments. The drug information tools and other features work well offline.
Medscape contains a well-presented drug database and interaction checker, as well as a growing collection of evidence-based articles and videos with links to references in Medline. From the point-of-care standpoint, Medscape also offers a number of decision-making algorithms and a continuously updated medical literature and health-related newsfeed. It contains in-app medical calculators, searchable directories for providers, hospitals, and pharmacies, and CME that can be earned on the website or from the application.
The main Medscape website contains pop-up advertisements, but the mobile app has fewer. Among the occasional frustrations, updates are relatively infrequent, the content is slow to load, and the phone app can be cumbersome. Of note, in one review,11 Medscape was found to have the lowest quality of evidence.
Strengths of Medscape
- Free with registration
- Medical calculator
- Drug interaction checker
- Pill identifier
- Evidence-based information covering about 4,000 conditions with links to references in Medline
- Ability to e-mail articles for sharing or future reference
- CME
- Unique database of hospitals, providers, and pharmacies to aid in referrals or locating other healthcare professionals
- Algorithms for decision-making
- Images and videos for procedural review and learning
- Option for downloading certain databases for offline use
- Medical news helps you keep up with what patients are watching and reading.
Weaknesses of Medscape
- Advertisements (many of them pop-up)
- The content is updated less frequently than other products listed in this article
- The smartphone app can run slowly
- Quality of reviews may be a concern.
UpToDate
UpToDate (Wolters Kluwer) is used widely by medical students, residents, and fellows as well as practicing providers. It contains narrative reviews of topics written by respected experts directed at both clinicians and clinical staff. In hopes of appealing to many markets, it offers different subscription types so you can customize your choices with add-on features (UpToDate Desktop and UpToDate MobileComplete allow downloading of all content to be accessed offline), different service packages (1-, 2-, and 3-year subscriptions), and the traditional base product that provides online access.
Of the products we reviewed, UpToDate has the largest selection of medical topics, approaching 10,000.14 In some studies,10,15 it also had the fastest retrieval time for searches. It uses evidence-based graded recommendations that are updated regularly.
Some have lamented that there is too much information per topic.9 In response to early reviews, Wolters Kluwer has made significant changes in the platform and greatly improved the search engine. UpToDate has expanded to include CME and patient information, trying to become that Holy Grail of websites—a one-stop experience. For the lucky few, UpToDate integrates into some electronic health records and provides a relatively seamless experience at the point of care.
Strengths of UpToDate
- One-stop shopping for information, resources, and CME
- Patient information is easy to read and accessible from the same screen
- The largest repository of medical subject matter
- Ability to cull out only pediatric or adult topics
- Searching available within a medical topic
- Tabs for quick access
- The What’s New feature allows access to practice-changing medical updates
- Medical calculators
- Drug interactions
- CME is is tracked in the system, allowing for CME credit information for hospital privileges and board certification
- Flexibility of access: can use online or download content to mobile/desktop device (the online version is easy to use, although robust wireless reception is needed; offices with slow Internet benefit from the offline feature)
- Electronic health record integration is possible with the most popular systems, such as Epic, eClinicalWorks, NextGen, and Allscripts
- Patient education and medication interaction features embedded in the electronic health record; produced in collaboration with Lexicomp
- Integrated drug database
- Alerts for updates
- References have links to full-text articles
- The date of last update is easily found for verifying information accuracy
- May be provided free for clinicians who are a part of a university or large health system.
Weaknesses of UpToDate
- Articles can be lengthy, which is both a strength and a weakness. Searches can retrieve too much information.9 High volume of text can frustrate the user trying to find bulleted, easy-to-read facts. However, for the person looking for a narrative summary, the content is organized as narrative paragraphs with appropriate headers in the left margin, and the search function is robust and powerful.
- Each topic has a “Summary of Recommendations,” but answers here often require linking back to the main text.
- Patient information is sometimes at a high literacy level.
- Costs more than Dynamed. A 1-year subscription is $499 for a physician, but you have the option of paying $53 for a 30-day recurring subscription. Residents, fellows, and students can pay $199 for 1 year or $19 for a 30-day recurring subscription.
- The requirement to download means that users need to keep their version updated on all of their computers—in each of their examination rooms, for example.
- Concerns about conflict of interest arise because authors and editors may maintain financial relationships with companies that produce medications discussed in the articles they have written.
BUILDING YOUR OWN PERSONAL ONLINE REPOSITORY
Our previous article5 reviewed how to store information using tools such as Evernote and Diigo that allow information viewed on a web page to be exported to any online repository. This can be done using extensions for a web browser or by sending the information to a custom e-mail account for these services.
For information that a provider knows he or she will need repeatedly, storage in one system is actually the easiest method. Such a system can then incorporate key information from the summary tools we have reviewed here. The ideal “electronic filing cabinet” should have several features such as a the capability to label articles by topic, to separate or sort as you see fit, and a search function to find information quickly—making it a personalized and effective point-of-care resource.
STAYING AFLOAT
Clinicians make many decisions every day. In fact, the release of How Doctors Think (both publications) has led to increased research into how clinical decisions and diagnoses are formed.16,17
With the medical literature expected to double every 73 days by 2020,18 there is an ever-widening ocean of information to sift through. With this onslaught, clinicians can no longer remain fully current. Instead, refining skills in accessing, sorting, and interpreting accurate scientific evidence efficiently is crucial to time spent actually caring for patients and coordinating their care.
Guidelines, algorithms, and comprehensive databases can aid clinicians in all aspects of care, from generating more complete differential diagnoses to managing disease-specific treatment. Individuals can first think about and list the qualities of a tool that are most important to them (eg, breadth of topics, frequency of updates, integration within their electronic health record, and cost) before focusing on a few applications or websites that meet those goals. With practice, point-of-care knowledge can become part of the everyday visit.
Effective integration into electronic health records will require design input from front-line clinicians. Otherwise, systems are prone to add too much “support” and overly rely on orthodox metrics and guidelines, resulting in alarm fatigue and frustration rather than facilitation.19–23
OUR CONCLUSIONS
Comprehensive point-of-care resources can play a significant role in helping busy clinicians provide best evidence-based care to their patients. Embedded clinical decision guides within an electronic health record are ideal, but low topic coverage has limited the usefulness of these systems.24 Here are our conclusions:
Medscape, ePocrates, and Wikipedia are probably the most popular free resources. Dynamed has offered free subscriptions to Wikipedia’s top health editors with the hopes of correcting factual errors. Medscape has excellent features but is supported by sponsored content, which raises a concern about bias and potential time-consuming distractions.
Dynamed and UpToDate have both been reported to answer more questions than other sources.12
UpToDate has the largest repository, with each topic curated by an expert or experts in that subject. This content can be dense and difficult to scan quickly at the point of care, but this is balanced by the ability to search within a medical topic, which has given it the fastest retrieval time.15 It does, however, allow authors and editors to maintain financial relationships with companies that produce medications discussed in the article.22
Dynamed has the advantage in frequency of updates, clearest conflict-of-interest policy, and the least amount of conflict of interest. Its topic list is not as extensive as UpToDate’s due to the limitation of using only evidence-based medicine without expert opinion.
First Consult has high user satisfaction, but as a point-of-care resource it can be time-consuming to find the best source for the clinical question at hand, and its expanded access is costly.9
ART AND SCIENCE
Point-of-care resources do not solve all the complicated problems of patient care, and no single resource is ideal for all situations. A busy clinician has limited time to process the evolving literature to practice the best evidence-based medicine. Effective information access, quality of care provided, and the marginal time cost required create a complex calculus. Clinical decision-making remains an art and a science,25 but these technologies help define a new era in its pursuit.
Ultimately, a clinician’s choice needs to correlate with a provider’s resources and style. This article has detailed several options available on the market today. This is a quickly evolving area of products and services. Longer term, users might consider a tool’s preferred key features when evaluating any current or future resource in order to choose the right ones for their practice.
CASE REVISITED
Before we leave for the weekend, we need a plan for Ms. Connecticut. To find appropriate recommendations for our patient, we search several of our point-of-care resources: UpToDate and Dynamed. Both resources have correct information according to the Infectious Disease Society of America (IDSA) guidelines.
UpToDate has a monograph of approximately 2,000 words on Lyme disease, which is lengthy but adds to clinical-decision making skills for a learner thinking through the decision. This service also has a patient handout highlighting the recommendations. The topic was last updated in 2016, but states that it is current with literature through January 2017.
Dynamed has bulleted information that is quicker to digest, but essentially highlights the IDSA recommendations without the thought process behind them. It too, has patient resources with links to a variety of handouts from professional organizations such as the US Centers for Disease Control and Prevention. They last updated the topic January 31, 2017.
When searching for the topic on both sites, a clinician can see the breadth of information in each program. However, this is also a detractor. Searching for Lyme disease prophylaxis on Dynamed brought up related data (that doxycycline is not FDA-approved for prophylaxis), but not the primary information. Likewise, the search under UpToDate first brought us to the patient information. Both articles have helpful tables and links to associated topics.
My partner chose the UpToDate article, in part to review the topic with a medical student. However, I used Dynamed for its quick bulleted information, as I was on call that evening and needed to return to the hospital. We both came to the same conclusion, and Ms. Connecticut chose no prophylaxis even though her home is in an endemic area. She has done well.
It is friday afternoon on a sunny July day. The last patient of the day, Ms. Connecticut, is an active hiker who has had Lyme disease previously. She found a tick on her ankle yesterday. She successfully removed the tick but has not brought the tick with her to the appointment. She had been hiking several times over the last week and is not certain when the tick bite occurred. Her question for you centers on the role of antibiotic prophylaxis and Lyme disease prevention.
TECHNOLOGY: PROBLEM AND SOLUTION
Physicians need to keep up with an ever-increasing stream of information—new guidelines, new medications, and updates in medical literature.1 They have to do this while seeing more patients with more chronic problems in less time and while meeting reporting requirements for meaningful use or quality measures for accountable care organizations.
Though some of these challenges are due to technology, one solution is to use technology to our advantage. While researching information in textbooks won’t drain a phone battery, carrying a textbook around is not feasible, and many textbooks (including their electronic versions) contain information that is outdated before they go to print or that is quickly outdated thereafter.2 Further, even online textbooks are currently more dense than the online resources that we review here.
Different types of resources can help task-saturated healthcare providers stay aware of new information while delivering evidence-based care. These tools—online textbooks, decision guides embedded within electronic health record systems, or even a Google search—are termed “point-of-care” resources when used at the time of patient care for decision-making in the moment.
Not all of these resources are of high quality, with reliable factual information. Researchers estimate that up to 70% of clinicians may use Wikipedia to research medical questions, and a comparison of 10 Wikipedia articles vs peer-reviewed sources on the 10 most costly diseases found that 9 of the 10 Wikipedia articles had errors.3,4
In an earlier article,5 we advocated a proactive approach to managing information, highlighting ways to scan for new information and to develop habits of extracting useful information that can then be stored and easily recovered. To complement this strategy and weed out erroneous information, physicians need reliable sources of unbiased information to efficiently answer clinical questions at the point of care.1,6
Here, to help busy clinicians choose which point-of-care resources to use, we review several of the most popular ones, examining their ease of use, key elements, strengths, and weaknesses.
WHAT MAKES A RESOURCE GOOD?
Key features that make point-of-care tools effective include:
Ease of use, with standard formats, a summary for each topic, or both
Links to original articles and concise, capsular summaries and syntheses of the data
Continuing medical education (CME) credit. Tip: when searching, add “CME” to the search string on the browser to access resources that provide this.
Institutional and individual accounts. For clinicians who work for large organizations, point-of-care products may be paid for already, or reimbursement may be available for your subscription. If unsure, ask your director of information technology or library services.
Freedom from advertisements. Many Internet sources have advertisements that either run alongside the information you want to see or, more annoyingly, pop up and require an action to move forward. There is also continuing concern about the effect of industry support on content.7 While not all of the resources that we use regularly and that we review here are ad-free, avoiding programs with high ad content helps limit the possibility of bias and the time it takes to access information. Although advertisements do bring up a risk of bias, resources with a low-level ad content can limit bias while providing free or low-cost access.
Evidence, not expert opinion. Many resources have an “about” page that explains their philosophy and the source of their information. It is vital to be sure that point-of-care databases are providing facts based on evidence.8 This page also typically addresses how authors and editors are selected and whether expert opinion is used when randomized trials are lacking.
Ease of access. Many tools can be accessed not only on computers but also through apps for smartphones and tablets. Some electronic medical records have clinical decision tools embedded in them, with varying capabilities.
Disclosure of conflict of interest. As conflicts of interest can shade recommendations, information sources should clearly disclose financial relationships that could be perceived as conflicts of interest—for example, authors writing about medications sold by companies with whom they have a financial relationship.
NO SINGLE RESOURCE DOES EVERYTHING
There are many types of tools for finding evidence-based medical information. Different tools serve different purposes. Table 1 lists “toolbox essentials” for clinicians needing to answer clinical questions during patient care.
For example, when a question about the need for a bone mineral density measurement comes up, it is useful to be able to quickly compare guidelines from different professional societies on the National Guideline Clearing House. For another example, if a patient brings in a medication in an unlabeled bottle, a pill identifier app can tell you what it is. Clinicians who can use these resources appropriately will be at an advantage in being able to use information to provide better care to their patients.
To date, no point-of-care summary source has been shown to be superior in all categories, and use may be driven by ease of navigation, clinician preference, clinical question, or past success.9,10
Reviewed below are several applications that can be used as point-of-care resources (Table 2).
CLINICAL EVIDENCE
Clinical Evidence provides systematic reviews on medical topics. Founded in 1999 by the British Medical Journal, it is available in print as the Clinical Evidence Handbook and in online desktop and smartphone formats.
More than any other source we reviewed, Clinical Evidence addresses not only the evidence that exists, but also the data that do not exist to guide decisions. Compared with 9 other point-of-care resources, Clinical Evidence was found to have the highest quality of evidence.11
Strengths of Clinical Evidence
- Uncommonly transparent in terms of source of evidence or disclosing when there is a lack of evidence.
- Clearly lists the strength and relevance of the evidence.
- Personalization. Users can add notes to articles, save personal searches, and bookmark pages for easy access later.
- Navigability. Users can easily access systematic reviews, key points, retracted papers, or guidelines.
- Intuitive organization, with information categorized as research, education, news, or campaigns.
- New content daily: podcasts, articles, videos.
Weaknesses of Clinical Evidence
- Limited topics (eg, Lyme disease was not available)
- The limited content is a challenge when needing quick information at the point of care and may cause most clinicians to use another source unless looking for comparisons of interventions.
- Cost. Subscribing to the service “on demand,” ie, to look up a single specific topic, costs $36 for 48 hours of access; monthly access or a “season ticket” allows 30 days of viewing of all content for $53. At over $600/year, this is one of the most costly of the sources we reviewed.
- Marketing of Clinical Evidence to academic institutions that support the service for faculty may limit its appeal to other clinicians.
DYNAMED
Dynamed, a clinical reference created by a group of physicians, was previously owned by the American College of Physicians and known as Smart Medicine; it is now owned by EBSCO.12 Reviewers investigate the literature for a given topic and create pithy summaries for busy clinicians. A top feature in Dynamed is its links to full articles cited for best practices or evidence-based guidelines. The company describes their content as free of expert opinion, while being unbiased and evidence-based.
Dynamed uses a 7-step algorithm for searched topics that identifies articles, assesses clinical relevance, evaluates validity of outcomes, compiles the evidence from multiple articles, and then updates the final recommendations daily.
Dynamed Plus, the new upgraded version, updates searched topics several times a day. Dynamed may be the most frequently updated point-of-care resource, with the least risk of conflict of interest, but it offers limited topics drawn from evidence-based findings.11,13–15
With the rapid doubling of the medical literature, frequent updates allow clinicians to be most current with practice guidelines. This potentially affects quality of care for antibiotic use, vaccination, health promotion, and screening as well as newly approved medications.
Strengths of Dynamed
- Large collection of topics, critically appraised, written for primary care physicians, presented in bulleted format
- The most frequently updated database11,14,15
- Can integrate with major electronic health records (eg, Epic, Allscripts, NextGen, Cerner)
- Has an area devoted to new information that changes current practice
- Chosen topic grouped with related topics in the differential diagnosis after the initial search
- Easy-to-read outline for quick access to information such as billing, diagnosis, and references
- Medical calculators
- No advertisements
- Helpful embedded tools
- Icons to print or email the article
- An icon to create a “perma-link” to topics, searches, and browse categories
- Graded evidence with a link to the grading model used
- Links to primary articles
- Patient information handouts
- Alerts for updated information
- CME credit
- Special consideration and features for medical education
- The upgraded version Dynamed Plus contains Micromedex for a medication database, expanded graphics, semantic search, concise overview for each topic, and expanded content.
Weaknesses of Dynamed
- Although the topic list is large, it is only about one-third the size of UpToDate.
- A subscription for a physician costs $395 a year. Residents can sign up for about $150, and students for just under $100.
- CME is obtainable but cumbersome; one submits the CME credits through Tufts Healthcare, which requires a second sign-on to access and track.
- Drug and nondrug treatments for diseases cannot be separated.
- Useful calculators include decision trees for clinical decision-making, but there is no way to search them—one must waste time scrolling through the topics and specialties looking for desired information.
- Major shortcoming: there is no medication reference tool unless you upgrade to Dynamed Plus.
- The expanded graphics of Dynamed Plus are difficult to view on mobile applications within the articles (they are brought up more reliably when searching just for the image).
- The use of strict evidence-based methodology without expert opinion is a strength, but limits the collection of topics without randomized controlled trials, for which turning to expert opinion may be the only option.
EVIDENCE ESSENTIALS
Evidence Essentials is a point-of-care resource from Wiley that offers a variety of content types. The website lists 13,000 medical topics; however, they are not all summary reviews as discussed in the other products above. Subject matter is reviewed 3 times a year. Comprehensive reviews number just under 800 individual topics, with the remaining content consisting of Cochrane reviews, calculators, decision support tools, POEMs (Patient-Oriented Evidence that Matters), evidence-based medical guidelines, and dermatology images (1,000).
Evidence Essentials provides some unique content including a quick evaluation and management (E/M) code-finder and calculators not only for the typical medical equations, but also for history and physical examination likelihood ratios and pretest probabilities, which are practical and an excellent teaching aid. It also offers CME along with POEMs, e-mail alerts, and a listing of upcoming topics.
Strengths of Evidence Essentials
- Relatively inexpensive at $85 a year.
- High-functioning filter system to choose to search one or multiple databases.
- Related results are listed for aid in differential diagnosis, similar to Dynamed.
- Authors, editors, and date of last review are highly visible. As in UpToDate, relevant medical calculators appear on the page.
- The likelihood and odds ratio calculators are a huge plus for clinical decision-making and putting guidelines into practice.
- “Overall bottom line” highlights key points
- Grading of evidence per topic.
- Bulleted and tabbed information for quick access.
- Tabs for information on background, prevention, diagnosis, treatment, references, guidelines, and special populations.
Weaknesses of Evidence Essentials
- Limited number of topics with comprehensive reviews.
- While you can click on any drug name and link to a choice of two drug databases, this is not included in the subscription and requires a second account.
- The resources tabs had some broken links. In our clinical example, the tab contained several videos at the top that were not related, followed by a map and tables that were relevant to Lyme disease.
- Likewise, some of the guideline references were disappointing. For example, the guideline link for Lyme disease is for the US Department of Labor Occupational Safety and Health Administration rather than a professional society.
- For the provider wanting a narrative, this is more of a bare-bones text.
FIRST CONSULT
First Consult is Elsevier’s point-of-care clinical decision product contained within ClinicalKey.
Unlike UpToDate and Dynamed, in which authors and editors read original articles and summarize or synthesize information for the learner, First Consult is a “smart” search engine that will research a question, together with associated terms and key words. Filters such as full-text availability, journal articles, and patient education can be applied.
You may need to read about your topic in a textbook first, and then, if you are looking for treatment information, find an original article through First Consult. It is available in mobile and desktop formats, and the point-of-care product, First Consult, has an app that can be downloaded and used for free for the first 60 days.
Importantly, the First Consult portion of ClinicalKey with the summary topics was rated by Shurtz and Foster13 as least current of the products we are discussing in this article. On the other hand, it was the only product that had an embedded program to assist the user in making presentations by allowing drag and drop of images and automatic citing of sources. Kim et al report that First Consult is one of the resources providers prefer.9
Strengths of First Consult
- Lengthy free trial
- Ability to access original articles from a list vs lengthy narrative
- Access to journals and books published by Elsevier
- Powerful search engine that applies associated terms automatically
- Patient education is available in different languages and font size with the ability to add instructions and even a local branding
- Can integrate with electronic health record
- Can filter results by guideline, patient education, topic overviews
- Presentation assistance.
Weaknesses of First Consult
- Time-intensive. A provider needing quick advice on treatment for a medical condition has to guess if an article or textbook will have the most up-to-date and digestible information, whereas this has already been summarized in other products. For the busy clinician, this may be prohibitive.
- Search results are limited to Elsevier products, and major journals such as the New England Journal of Medicine are not available.
- Inconsistent platform functionality. The app version was somewhat “sticky” to use, as pages did not always load efficiently, and the menu bar navigation is not ideal.
- Expensive, especially given cheaper alternatives. For example, subscribing to the specialty of internal medicine or family medicine costs $499 and provides access to 8 journals and 11 books. Extended access costs $998 and offers full-text access to 23 books and 45 journals. The complete service has a total of 400 journals, 700 books, and 2,500 procedural videos.
MEDSCAPE
Medscape, owned by parent company WebMD, has long been a popular resource. The most recent versions are available for both for Android and iOS mobile platforms. The desktop and mobile apps claim to be designed for point-of-care use, and can be downloaded at no cost after registering as a Medscape user.
Medscape has some interesting features, including a handy pill identifier tool that is new to Medscape and perfect for the “I take one blue pill for my cholesterol” moments. The drug information tools and other features work well offline.
Medscape contains a well-presented drug database and interaction checker, as well as a growing collection of evidence-based articles and videos with links to references in Medline. From the point-of-care standpoint, Medscape also offers a number of decision-making algorithms and a continuously updated medical literature and health-related newsfeed. It contains in-app medical calculators, searchable directories for providers, hospitals, and pharmacies, and CME that can be earned on the website or from the application.
The main Medscape website contains pop-up advertisements, but the mobile app has fewer. Among the occasional frustrations, updates are relatively infrequent, the content is slow to load, and the phone app can be cumbersome. Of note, in one review,11 Medscape was found to have the lowest quality of evidence.
Strengths of Medscape
- Free with registration
- Medical calculator
- Drug interaction checker
- Pill identifier
- Evidence-based information covering about 4,000 conditions with links to references in Medline
- Ability to e-mail articles for sharing or future reference
- CME
- Unique database of hospitals, providers, and pharmacies to aid in referrals or locating other healthcare professionals
- Algorithms for decision-making
- Images and videos for procedural review and learning
- Option for downloading certain databases for offline use
- Medical news helps you keep up with what patients are watching and reading.
Weaknesses of Medscape
- Advertisements (many of them pop-up)
- The content is updated less frequently than other products listed in this article
- The smartphone app can run slowly
- Quality of reviews may be a concern.
UpToDate
UpToDate (Wolters Kluwer) is used widely by medical students, residents, and fellows as well as practicing providers. It contains narrative reviews of topics written by respected experts directed at both clinicians and clinical staff. In hopes of appealing to many markets, it offers different subscription types so you can customize your choices with add-on features (UpToDate Desktop and UpToDate MobileComplete allow downloading of all content to be accessed offline), different service packages (1-, 2-, and 3-year subscriptions), and the traditional base product that provides online access.
Of the products we reviewed, UpToDate has the largest selection of medical topics, approaching 10,000.14 In some studies,10,15 it also had the fastest retrieval time for searches. It uses evidence-based graded recommendations that are updated regularly.
Some have lamented that there is too much information per topic.9 In response to early reviews, Wolters Kluwer has made significant changes in the platform and greatly improved the search engine. UpToDate has expanded to include CME and patient information, trying to become that Holy Grail of websites—a one-stop experience. For the lucky few, UpToDate integrates into some electronic health records and provides a relatively seamless experience at the point of care.
Strengths of UpToDate
- One-stop shopping for information, resources, and CME
- Patient information is easy to read and accessible from the same screen
- The largest repository of medical subject matter
- Ability to cull out only pediatric or adult topics
- Searching available within a medical topic
- Tabs for quick access
- The What’s New feature allows access to practice-changing medical updates
- Medical calculators
- Drug interactions
- CME is is tracked in the system, allowing for CME credit information for hospital privileges and board certification
- Flexibility of access: can use online or download content to mobile/desktop device (the online version is easy to use, although robust wireless reception is needed; offices with slow Internet benefit from the offline feature)
- Electronic health record integration is possible with the most popular systems, such as Epic, eClinicalWorks, NextGen, and Allscripts
- Patient education and medication interaction features embedded in the electronic health record; produced in collaboration with Lexicomp
- Integrated drug database
- Alerts for updates
- References have links to full-text articles
- The date of last update is easily found for verifying information accuracy
- May be provided free for clinicians who are a part of a university or large health system.
Weaknesses of UpToDate
- Articles can be lengthy, which is both a strength and a weakness. Searches can retrieve too much information.9 High volume of text can frustrate the user trying to find bulleted, easy-to-read facts. However, for the person looking for a narrative summary, the content is organized as narrative paragraphs with appropriate headers in the left margin, and the search function is robust and powerful.
- Each topic has a “Summary of Recommendations,” but answers here often require linking back to the main text.
- Patient information is sometimes at a high literacy level.
- Costs more than Dynamed. A 1-year subscription is $499 for a physician, but you have the option of paying $53 for a 30-day recurring subscription. Residents, fellows, and students can pay $199 for 1 year or $19 for a 30-day recurring subscription.
- The requirement to download means that users need to keep their version updated on all of their computers—in each of their examination rooms, for example.
- Concerns about conflict of interest arise because authors and editors may maintain financial relationships with companies that produce medications discussed in the articles they have written.
BUILDING YOUR OWN PERSONAL ONLINE REPOSITORY
Our previous article5 reviewed how to store information using tools such as Evernote and Diigo that allow information viewed on a web page to be exported to any online repository. This can be done using extensions for a web browser or by sending the information to a custom e-mail account for these services.
For information that a provider knows he or she will need repeatedly, storage in one system is actually the easiest method. Such a system can then incorporate key information from the summary tools we have reviewed here. The ideal “electronic filing cabinet” should have several features such as a the capability to label articles by topic, to separate or sort as you see fit, and a search function to find information quickly—making it a personalized and effective point-of-care resource.
STAYING AFLOAT
Clinicians make many decisions every day. In fact, the release of How Doctors Think (both publications) has led to increased research into how clinical decisions and diagnoses are formed.16,17
With the medical literature expected to double every 73 days by 2020,18 there is an ever-widening ocean of information to sift through. With this onslaught, clinicians can no longer remain fully current. Instead, refining skills in accessing, sorting, and interpreting accurate scientific evidence efficiently is crucial to time spent actually caring for patients and coordinating their care.
Guidelines, algorithms, and comprehensive databases can aid clinicians in all aspects of care, from generating more complete differential diagnoses to managing disease-specific treatment. Individuals can first think about and list the qualities of a tool that are most important to them (eg, breadth of topics, frequency of updates, integration within their electronic health record, and cost) before focusing on a few applications or websites that meet those goals. With practice, point-of-care knowledge can become part of the everyday visit.
Effective integration into electronic health records will require design input from front-line clinicians. Otherwise, systems are prone to add too much “support” and overly rely on orthodox metrics and guidelines, resulting in alarm fatigue and frustration rather than facilitation.19–23
OUR CONCLUSIONS
Comprehensive point-of-care resources can play a significant role in helping busy clinicians provide best evidence-based care to their patients. Embedded clinical decision guides within an electronic health record are ideal, but low topic coverage has limited the usefulness of these systems.24 Here are our conclusions:
Medscape, ePocrates, and Wikipedia are probably the most popular free resources. Dynamed has offered free subscriptions to Wikipedia’s top health editors with the hopes of correcting factual errors. Medscape has excellent features but is supported by sponsored content, which raises a concern about bias and potential time-consuming distractions.
Dynamed and UpToDate have both been reported to answer more questions than other sources.12
UpToDate has the largest repository, with each topic curated by an expert or experts in that subject. This content can be dense and difficult to scan quickly at the point of care, but this is balanced by the ability to search within a medical topic, which has given it the fastest retrieval time.15 It does, however, allow authors and editors to maintain financial relationships with companies that produce medications discussed in the article.22
Dynamed has the advantage in frequency of updates, clearest conflict-of-interest policy, and the least amount of conflict of interest. Its topic list is not as extensive as UpToDate’s due to the limitation of using only evidence-based medicine without expert opinion.
First Consult has high user satisfaction, but as a point-of-care resource it can be time-consuming to find the best source for the clinical question at hand, and its expanded access is costly.9
ART AND SCIENCE
Point-of-care resources do not solve all the complicated problems of patient care, and no single resource is ideal for all situations. A busy clinician has limited time to process the evolving literature to practice the best evidence-based medicine. Effective information access, quality of care provided, and the marginal time cost required create a complex calculus. Clinical decision-making remains an art and a science,25 but these technologies help define a new era in its pursuit.
Ultimately, a clinician’s choice needs to correlate with a provider’s resources and style. This article has detailed several options available on the market today. This is a quickly evolving area of products and services. Longer term, users might consider a tool’s preferred key features when evaluating any current or future resource in order to choose the right ones for their practice.
CASE REVISITED
Before we leave for the weekend, we need a plan for Ms. Connecticut. To find appropriate recommendations for our patient, we search several of our point-of-care resources: UpToDate and Dynamed. Both resources have correct information according to the Infectious Disease Society of America (IDSA) guidelines.
UpToDate has a monograph of approximately 2,000 words on Lyme disease, which is lengthy but adds to clinical-decision making skills for a learner thinking through the decision. This service also has a patient handout highlighting the recommendations. The topic was last updated in 2016, but states that it is current with literature through January 2017.
Dynamed has bulleted information that is quicker to digest, but essentially highlights the IDSA recommendations without the thought process behind them. It too, has patient resources with links to a variety of handouts from professional organizations such as the US Centers for Disease Control and Prevention. They last updated the topic January 31, 2017.
When searching for the topic on both sites, a clinician can see the breadth of information in each program. However, this is also a detractor. Searching for Lyme disease prophylaxis on Dynamed brought up related data (that doxycycline is not FDA-approved for prophylaxis), but not the primary information. Likewise, the search under UpToDate first brought us to the patient information. Both articles have helpful tables and links to associated topics.
My partner chose the UpToDate article, in part to review the topic with a medical student. However, I used Dynamed for its quick bulleted information, as I was on call that evening and needed to return to the hospital. We both came to the same conclusion, and Ms. Connecticut chose no prophylaxis even though her home is in an endemic area. She has done well.
- Worster A, Haynes RB. How do I find a point-of-care answer to my clinical question? CJEM 2012; 14:31–35.
- Jeffery R, Navarro T, Lokker C, Haynes RB, Wilczynski NL, Farjou G. How current are leading evidence-based medical textbooks? An analytic survey of four online textbooks. J Med Internet Res 2012; 14:e175.
- ClinicalKey. Errors found in nine out of ten Wikipedia health entries. www.clinicalkey.com/info/blog/errors-in-wikipedia-health/. Accessed February 9, 2017.
- Hasty RT, Garbalosa RC, Barbato VA, et al. Wikipedia vs peer-reviewed medical literature for information about the 10 most costly medical conditions. J Am Osteopath Assoc 2014; 114:368–373.
- Mehta NB, Martin SA, Maypole J, Andrews R. Information management for clinicians. Cleve Clin J Med 2016; 83:589–595.
- Cook DA, Sorensen KJ, Hersh W, Berger RA, Wilkinson JM. Features of effective medical knowledge resources to support point of care learning: a focus group study. PLoS One 2013; 8:e80318.
- Steinbrook R. Future directions in industry funding of continuing medical education. Arch Intern Med 2011; 171:257–258.
- Isaacs D, Fitzgerald D. Seven alternatives to evidence based medicine. BMJ 1999; 319:1618.
- Kim S, Noveck H, Galt J, Hogshire L, Willett L, O’Rourke K. Searching for answers to clinical questions using Google versus evidence-based summary resources: a randomized controlled crossover study. Acad Med 2014; 89:940–943.
- Ahmadi SF, Faghankhani M, Javanbakht A, et al. A comparison of answer retrieval through four evidence-based textbooks (ACP PIER, Essential Evidence Plus, First Consult, and UpToDate): a randomized controlled trial. Med Teach 2011; 33:724–730.
- Prorok JC, Iserman EC, Wilczynski NL, Haynes RB. The quality, breadth, and timeliness of content updating vary substantially for 10 online medical texts: an analytic survey. J Clin Epidemiol 2012; 65:1289–1295.
- Prorok JC, Iserman EC, Wilczynski NL, Haynes RB. The quality, breadth, and timeliness of content updating vary substantially for 10 online medical texts: an analytic survey. J Clin Epidemiol 2012; 65:1289–1295.
- Shurtz S, Foster MJ. Developing and using a rubric for evaluating evidence-based medicine point-of-care tools. J Med Libr Assoc 2011; 99:247–254.
- Ketterman E, Besaw M. An evaluation of citation counts, search results, and frequency of updates in Dynamed and UpToDate. J Electron Res in Med Libr 2010; 7:273–280.
- Amber KT, Dhiman G, Goodman KW. Conflict of interest in online point-of-care clinical support websites. J Med Ethics 2014; 40:578–580.
- Montgomery K. How Doctors Think: Clinical Judgment and the Practice of Medicine. New York, NY: Oxford University Press; 2005.
- Groopman J. How Doctors Think. Boston, MA: Houghton Mifflin; 2008.
- Densen P. Challenges and opportunities facing medical education. Trans Am Clin Climatol Assoc 2010; 122:48–58.
- Kesselheim AS, Cresswell K, Phansalkar S, Bates DW, Sheikh A. Clinical decision support systems could be modified to reduce ‘alert fatigue’ while still minimizing the risk of litigation. Health Aff (Millwood) 2011; 30:2310–2317.
- Russ AL, Zillich AJ, McManus MS, Doebbeling BN, Saleem JJ. Prescribers’ interactions with medication alerts at the point of prescribing: a multi-method, in situ investigation of the human-computer interaction. Int J Med Inform 2012; 81:232–243.
- Fraccaro P, Arguello Castelerio M, Ainsworth J, Buchan I. Adoption of clinical decision support in multimorbidity: a systematic review. JMIR Med Informatics 2015; 3:e4.
- McLeod W, Eidus R, Stewart EE. Clinical decision support: using technology to identify patients’ unmet needs. Fam Pract Manag 2012; 19:22–28.
- Colla CH. Swimming against the current—what might work to reduce low-value care? N Engl J Med 2014; 371:1280–1283.
- Cook DA, Sorensen KJ, Nishimura RA, Ommen SR, Lloyd FJ. A comprehensive information technology system to support physician learning at the point of care. Acad Med 2015; 90:33–39.
- Woolever DR. The art and science of clinical decision making. Fam Pract Manag 2008; 15:31–36.
- Worster A, Haynes RB. How do I find a point-of-care answer to my clinical question? CJEM 2012; 14:31–35.
- Jeffery R, Navarro T, Lokker C, Haynes RB, Wilczynski NL, Farjou G. How current are leading evidence-based medical textbooks? An analytic survey of four online textbooks. J Med Internet Res 2012; 14:e175.
- ClinicalKey. Errors found in nine out of ten Wikipedia health entries. www.clinicalkey.com/info/blog/errors-in-wikipedia-health/. Accessed February 9, 2017.
- Hasty RT, Garbalosa RC, Barbato VA, et al. Wikipedia vs peer-reviewed medical literature for information about the 10 most costly medical conditions. J Am Osteopath Assoc 2014; 114:368–373.
- Mehta NB, Martin SA, Maypole J, Andrews R. Information management for clinicians. Cleve Clin J Med 2016; 83:589–595.
- Cook DA, Sorensen KJ, Hersh W, Berger RA, Wilkinson JM. Features of effective medical knowledge resources to support point of care learning: a focus group study. PLoS One 2013; 8:e80318.
- Steinbrook R. Future directions in industry funding of continuing medical education. Arch Intern Med 2011; 171:257–258.
- Isaacs D, Fitzgerald D. Seven alternatives to evidence based medicine. BMJ 1999; 319:1618.
- Kim S, Noveck H, Galt J, Hogshire L, Willett L, O’Rourke K. Searching for answers to clinical questions using Google versus evidence-based summary resources: a randomized controlled crossover study. Acad Med 2014; 89:940–943.
- Ahmadi SF, Faghankhani M, Javanbakht A, et al. A comparison of answer retrieval through four evidence-based textbooks (ACP PIER, Essential Evidence Plus, First Consult, and UpToDate): a randomized controlled trial. Med Teach 2011; 33:724–730.
- Prorok JC, Iserman EC, Wilczynski NL, Haynes RB. The quality, breadth, and timeliness of content updating vary substantially for 10 online medical texts: an analytic survey. J Clin Epidemiol 2012; 65:1289–1295.
- Prorok JC, Iserman EC, Wilczynski NL, Haynes RB. The quality, breadth, and timeliness of content updating vary substantially for 10 online medical texts: an analytic survey. J Clin Epidemiol 2012; 65:1289–1295.
- Shurtz S, Foster MJ. Developing and using a rubric for evaluating evidence-based medicine point-of-care tools. J Med Libr Assoc 2011; 99:247–254.
- Ketterman E, Besaw M. An evaluation of citation counts, search results, and frequency of updates in Dynamed and UpToDate. J Electron Res in Med Libr 2010; 7:273–280.
- Amber KT, Dhiman G, Goodman KW. Conflict of interest in online point-of-care clinical support websites. J Med Ethics 2014; 40:578–580.
- Montgomery K. How Doctors Think: Clinical Judgment and the Practice of Medicine. New York, NY: Oxford University Press; 2005.
- Groopman J. How Doctors Think. Boston, MA: Houghton Mifflin; 2008.
- Densen P. Challenges and opportunities facing medical education. Trans Am Clin Climatol Assoc 2010; 122:48–58.
- Kesselheim AS, Cresswell K, Phansalkar S, Bates DW, Sheikh A. Clinical decision support systems could be modified to reduce ‘alert fatigue’ while still minimizing the risk of litigation. Health Aff (Millwood) 2011; 30:2310–2317.
- Russ AL, Zillich AJ, McManus MS, Doebbeling BN, Saleem JJ. Prescribers’ interactions with medication alerts at the point of prescribing: a multi-method, in situ investigation of the human-computer interaction. Int J Med Inform 2012; 81:232–243.
- Fraccaro P, Arguello Castelerio M, Ainsworth J, Buchan I. Adoption of clinical decision support in multimorbidity: a systematic review. JMIR Med Informatics 2015; 3:e4.
- McLeod W, Eidus R, Stewart EE. Clinical decision support: using technology to identify patients’ unmet needs. Fam Pract Manag 2012; 19:22–28.
- Colla CH. Swimming against the current—what might work to reduce low-value care? N Engl J Med 2014; 371:1280–1283.
- Cook DA, Sorensen KJ, Nishimura RA, Ommen SR, Lloyd FJ. A comprehensive information technology system to support physician learning at the point of care. Acad Med 2015; 90:33–39.
- Woolever DR. The art and science of clinical decision making. Fam Pract Manag 2008; 15:31–36.
KEY POINTS
- Today, it seems impossible to keep up with all the information we need, but we can refine our skills in accessing, sorting, and interpreting accurate scientific evidence.
- The resources reviewed in this article require paid subscriptions except for Medscape, which is supported by advertising.
- Each of the resources has strengths and weaknesses. For example, UpToDate offers the most topics, but its articles tend to be too long to be practical to read at the point of care.
- Physicians should familiarize themselves with these resources and use the ones that best suit their needs.

Update on viral hepatitis in pregnancy
Viral hepatitis affects mother and child, and pregnancy can exacerbate the disease. Vertical transmission contributes significantly to the high prevalence of viral hepatitis and compromises the well-being and the prognosis in the newborn. The indications for therapy in pregnant women may differ from those in the general population, and new therapies are available.
HEPATITIS A
Hepatitis A virus (HAV) infection is associated with significant morbidity and death around the world, as 1.4 million cases are reported every year worldwide.1 However, in the United States, the prevalence has declined by 95% since HAV vaccination was introduced in 1995, and in 2013, the prevalence was 0.6 per 100,000 population.2 Acute HAV infection during pregnancy is rare. As a result, the incidence during pregnancy is difficult to ascertain.3
HAV is transmitted by the fecal-oral route from person to person contact and from contamination of food and water. Vertical transmission from pregnant mother to fetus has not been reported.4
Clinical outcomes of HAV in pregnancy
Acute HAV infection during pregnancy is rare, and teratogenicity associated with HAV has not been reported.3 The course of the disease during pregnancy is generally similar to that in nonpregnant patients, with excellent maternal and fetal outcomes in developed nations. There have been reports in developing nations of premature contractions and labor, placental separation, premature rupture of the membranes, and vaginal bleeding.5,6
Diagnosis
Routine screening for HAV is not recommended, but serologic testing by detection of anti-HAV immunoglobulin M (IgM) antibodies is done in high-risk patients suspected of having acute HAV infection.
Prevention
Prevention includes adherence to sanitary practices and active and passive immunoprophylaxis.3 Universal vaccination for pregnant mothers is not recommended,1,2,6 but vaccination is recommended for high-risk patients and mothers—those with chronic liver disease, those receiving clotting factors, those who use illegal drugs, and travelers to areas where HAV is endemic. Immune globulin is also available for postexposure prophylaxis. HAV vaccines and immune globulin are safe in pregnancy.3,6,7
Treatment
Treatment of acute HAV in pregnancy is supportive because of its benign nature; few patients require hospitalization.3
Pregnant patients with HAV can deliver vaginally, and breastfeeding is not contraindicated.8
HEPATITIS B
Hepatitis B virus (HBV) infection is a major global health problem. About 240 million people worldwide have chronic HBV infection, and more than 780,000 die every year from acute and chronic consequences.9
Vertical transmission is responsible for about half of chronic HBV infections worldwide. Thus, interruption of mother-to-child transmission is important. Universal maternal screening and passive-active immunoprophylaxis of newborns have lowered the transmission rates to between 5% and 10%. The 10% failure rate is unacceptably high and has been attributed to seropositivity for hepatitis B e antigen and a high viral load in the mother (ie, HBV DNA > 106 copies/mL). High viral load is an independent risk factor for failure of immunoprophylaxis.10 Therefore, antiviral therapy is suggested in pregnant women who have a high HBV viral load to further decrease the chance of mother-to-child transmission and to prevent failure of immunoprophylaxis.11
Clinical outcomes of HBV in pregnancy
Acute HBV infection during pregnancy is usually benign and is not associated with increased risk of death or teratogenicity.12 Symptomatic disease in the mother with acute hepatitis B includes nausea, vomiting, abdominal pain, fatigue, and jaundice.3 For the newborn, there is increased risk of low birth weight and prematurity.13
When acute HBV infection occurs early in pregnancy, the rate of perinatal transmission is about 10%, increasing to 60% if it occurs near delivery.12,13
Chronic HBV infection does not usually affect the outcome of pregnancy, but it may if the woman has cirrhosis or advanced liver disease14; however, pregnancy is very rare in women with HBV cirrhosis due to anovulation, and acute HBV flares have been described during pregnancy and postpartum.15
Pregnant patients with cirrhosis and portal hypertension are at risk of hepatic decompensation, variceal bleeding, and death.16 Risk is high with a score of 10 or more on the Model for End-stage Liver Disease scale, and is low with a score of 6 or less.17 Like nonpregnant patients, pregnant patients with cirrhosis should be monitored, and upper endoscopy should be performed in the 2nd trimester to assess for varices. A beta-blocker should be given or banding of varices should be done to avoid rupture. Rates of fetal demise, premature labor, spontaneous abortion, and stillbirth are high with portal hypertension.16
Risk of mother-to-child HBV transmission
Vertical HBV transmission can occur during the antepartum, intrapartum, and postpartum periods,18,19 but it most often occurs during the intrapartum period at the time of delivery. Without immunoprophylaxis of the newborn, the risk of mother-to-child transmission can be as high as 90% if the mother is hepatitis B e antigen-positive and has a viral load greater than 106 copies/mL. With active and passive immunoprophylaxis, the risk decreases substantially.
Screening and diagnosis
All pregnant women should be tested for hepatitis B surface antigen during the 1st trimester,20 or any time thereafter if early testing was not done, even if they were vaccinated before becoming pregnant.21
Prevention
HBV infection is best prevented before pregnancy by vaccinating the mother or, after delivery, by vaccinating the newborn. Universal vaccination of newborns has been the standard of care since the 1990s. Pregnant women should be tested early in the pregnancy; unvaccinated, uninfected women at high risk of acquiring HBV (eg, because of sexual contacts or intravenous drug use) should be vaccinated.2,3
HBV vaccine and immune globulin are both approved by the US Food and Drug Administration (FDA) for prevention of HBV infection and can be given during pregnancy and breastfeeding.3 All infants should be vaccinated for HBV at birth, and all infants born to mothers who test positive for hepatitis B s antigen should receive the HBV vaccine and the immune globulin within 12 to 24 hours after delivery. The vaccine series should be completed within 6 months.20,21 This will decrease the rate of neonatal infection.
Treatment of HBV infection in pregnancy
The main objectives of treating chronic HBV infection in pregnancy are to maintain stable liver function in the mother and to prevent neonatal infection, which may cause cirrhosis and hepatocellular carcinoma and contribute to the global burden of the disease.22 Therefore, maternal HBV DNA and liver aminotransferase levels should be tested regularly during gestation.
The current guidelines of the American Association for the Study of Liver Diseases suggest antiviral therapy to reduce the risk of perinatal transmission of HBV in pregnant women with an HBV DNA level greater than 200,000 IU/mL or greater than 106 copies/mL.23,24
In a meta-analysis,25 antiviral therapy with lamivudine, telbivudine, or tenofovir showed no apparent teratogenicity or safety concerns for maternal and fetal outcomes26 and significantly reduced the rate of mother-to-child transmission. Of these 3 drugs, telbivudine was associated with a higher rate of normalization of liver enzymes, HBV suppression, and e-antigen seroconversion.25 Lamivudine has proven the test of time in mothers co-infected with HBV and human immunodeficiency virus (HIV). However, tenofovir is considered the preferred treatment in pregnancy, owing to concerns about drug resistance to telbivudine and lamivudine and a high genetic barrier to resistance with tenofovir.26 In mothers with HBV and HIV treated with tenofovir, treatment was associated with lower bone mineral density in the newborns, with a propensity for renal injury in the mothers. No safety concerns for maternal or fetal outcomes were identified in pregnant women infected only with HBV.25
Many pregnant mothers choose to stop therapy around the time of conception because of safety concerns for the baby. In such situations, close monitoring is necessary to detect flares of HBV infection.
When the decision to treat is made, treatment should begin at 28 to 30 weeks of gestation, when organogenesis is complete and to allow enough time for HBV DNA levels to decline.
Breastfeeding is not contraindicated because antiviral drugs are minimally excreted in breast milk and are unlikely to cause toxicity. However, data are insufficient as to the long-term safety for the newborn when the mother took these drugs during pregancy and while breastfeeding.23,27
Alanine aminotransferase and HBV DNA levels should be monitored postpartum because of the possibility of a hepatitis flare. In this setting, any of the three drugs can be used.28 For mothers on therapy because of cirrhosis or an advanced histologic feature, antiviral therapy should be continued throughout pregnancy to prevent disease progression and decompensation.19,22,27
No drug therapy is necessary for pregnant carriers of HBV.
Delivery and breastfeeding
The mode of delivery does not appear to have a significant effect on the interruption of vertical transmission of HBV.29 Cesarean delivery is not recommended by the US Centers for Disease Control and Prevention (CDC)2 or the American College of Obstetricians and Gynecologists.6 Breastfeeding is encouraged if the infant has received appropriate immunoprophylaxis.6
Coinfection with hepatitis D
Coinfection with hepatitis D virus (HDV) and HBV is associated with severe acute hepatitis30,31 and increases the risk of death by a factor of 10. The World Health Organization recommends testing for HDV in pregnant women who are HBV-positive.8
Prevention of HDV infection requires prevention of HBV. The treatment of HDV in pregnancy is supportive. Pegylated interferon is successful outside pregnancy but is contraindicated during pregnancy.32 In patients with fulminant hepatic failure and end-stage liver disease, liver transplant can be lifesaving.
Take-home points
- HBV infection during pregnancy is usually benign and not severe but can be associated with an increased risk of mother-to-child transmission and progression of liver disease in the pregnant mother.
- Prevention of vertical transmission of HBV is important to reduce the burden of chronic HBV infection. Universal maternal screening early in pregnancy and passive-active immunoprophylaxis of newborns are usually sufficient to prevent vertical transmission of HBV, but antiviral therapy is needed for highly viremic mothers to further reduce the risk.
- Antiviral therapy is also indicated for pregnant women with moderate to severe hepatitis or cirrhosis to prevent disease progression and liver failure.
- Telbivudine, tenofovir, or lamivudine can be used during pregnancy, but more data are needed on the long-term safety of fetal exposure to these agents.
HEPATITIS C
The global prevalence of hepatitis C virus (HCV) infection is 2% to 3%, with 130 to 170 million HCV-positive people, most of whom are chronically infected.33 The incidence of HCV during pregnancy is 1% to 2.4%, but 3% to 5% of infected mothers transmit HCV to their child at the time of birth.6,34 Women coinfected with HIV and HCV have twice the risk of perinatal HCV transmission compared with women who have HCV infection alone.6,34
HCV infection is usually asymptomatic and is discovered either by screening high-risk patients or during evaluation of persistently elevated aminotransferase levels. Acute HCV infection during pregnancy has been reported only rarely, and most pregnant women who are infected have chronic disease with no effect on the pregnancy or the infant.6,34
Treatment
The CDC recommends that all adults (including pregnant women) born between 1945 and 1965 undergo 1-time testing for HCV without prior ascertainment of HCV risk (strong recommendation, with moderate quality of evidence).35 The most important risk factor for HCV infection is past or current injection drug use.33 Additional risk factors are similar to those for nonpregnant patients.
Because of the benign effect of HCV on the pregnancy, treatment is not recommended. To decrease the risk of maternal-child transmission, it is prudent to avoid amniocentesis, scalp instrumentation, and prolonged rupture of membranes.6
There is no vaccine or immune globulin for prevention. HCV infection should not influence the mode of delivery, and it is not a contraindication to breastfeeding.34,36,37
HEPATITIS E
Every year, 20 million cases of hepatitis E virus (HEV) infection are recorded worldwide. These numbers include 3.3 million symptomatic cases and 56,600 deaths.38 HEV infection is most common in developing countries, and pregnant women traveling to these areas are at high risk of acquiring this infection, of developing fulminant hepatitis, and of death.39 Sporadic cases not associated with travel are increasingly reported in developed countries and are attributed to immunocompromised status (due to HIV or solid-organ transplant).38,40
Modes of transmission of HEV are mainly via fecal-oral contamination and by vertical transmission.41
Diagnosis
HEV infection can be diagnosed either by detecting IgM antibody with an enzyme-linked immunosorbent assay or by detecting HEV RNA in the blood using reverse transcription polymerase chain reaction testing.42
Treatment and prevention
Hospitalization should be considered for pregnant women. Ribavirin or pegylated interferon alpha or both are effective but are contraindicated in pregnancy because of the risk of teratogenicity.41,42 Urgent liver transplant can be a successful option in acute liver failure.
Prevention relies primarily on good sanitation, clean drinking water, and avoiding raw pork and venison. Boiling and chlorination of water inactivate HEV.39,40 Pregnant women should be advised to avoid travel to highly endemic areas.
- World Health Organization (WHO).Hepatitis A fact sheet. www.who.int/mediacentre/factsheets/fs328/en/. Accessed December 7, 2016.
- US Centers for Disease Control and Prevention (CDC). Viral hepatitis—statistics & surveillance. www.cdc.gov/hepatitis/statistics/2013surveillance/commentary.htm#hepatitis A. Accessed December 7, 2016.
- Rac MW, Sheffield JS. Prevention and management of viral hepatitis in pregnancy. Obstet Gynecol Clin North Am 2014; 41:573–592.
- Workowski KA, Bolan GA; Centers for Disease Control and Prevention (CDC). Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm Rep 2015; 64:1–137.
- Elinav E, Ben-Dov IZ, Shapira Y, et al. Acute hepatitis A infection in pregnancy is associated with high rates of gestational complications and preterm labor. Gastroenterology 2006; 130:1129–1134.
- American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 86: Viral hepatitis in pregnancy. Obstet Gynecol 2007; 110:941–956.
- Advisory Committee on Immunization Practices. Guidelines for Vaccinating Pregnant Women. www.cdc.gov/vaccines/pregnancy/hcp/guidelines.html. Accessed December 7, 2016.
- Daudi N, Shouval D, Stein-Zamir C, Ackerman Z. Breastmilk hepatitis A virus RNA in nursing mothers with acute hepatitis A virus infection. Breastfeed Med 2012; 7:313–315.
- World Health Organization (WHO). Hepatitis B fact sheet. www.who.int/mediacentre/factsheets/fs204/en/. Accessed December 7, 2016.
- Zou H, Chen Y, Duan Z, Zhang H, Pan C. Virologic factors associated with failure to passive-active immunoprophylaxis in infants born to HBsAg-positive mothers. J Viral Hepat 2012; 19:e18–e25.
- Pan CQ, Lee HM. Antiviral therapy for chronic hepatitis B in pregnancy. Semin Liver Dis 2013; 33:138–146.
- Sookoian S. Liver disease during pregnancy: acute viral hepatitis. Ann Hepatol 2006; 5:231–236.
- Jonas MM. Hepatitis B and pregnancy: an underestimated issue. Liver Int 2009; 29(suppl 1):133–139.
- Wong S, Chan LY, Yu V, Ho L. Hepatitis B carrier and perinatal outcome in singleton pregnancy. Am J Perinatol 1999; 16:485–488.
- Rawal BK, Parida S, Watkins RP, Ghosh P, Smith H. Symptomatic reactivation of hepatitis B in pregnancy. Lancet 1991; 337:364.
- Aggarwal N, Negi N, Aggarwal A, Bodh V, Dhiman RK. Pregnancy with portal hypertension. J Clin Exp Hepatol 2014; 4:163–171.
- Westbrook RH, Yeoman AD, O'Grady JG, Harrison PM, Devlin J, Heneghan MA. Model for end-stage liver disease score predicts outcome in cirrhotic patients during pregnancy. Clin Gastroenterol Hepatol 2011; 9:694–699.
- Cheung KW, Seto MT, Wong SF. Towards complete eradication of hepatitis B infection from perinatal transmission: review of the mechanisms of in utero infection and the use of antiviral treatment during pregnancy. Eur J Obstet Gynecol Reproduct Biol 2013; 169:17–23.
- Pan CQ, Duan AP, Bhamidimarri KR, et al. An algorithm for risk assessment and intervention of mother to child transmission of hepatitis B virus. Clin Gastroenterol Hepatol 2012; 10:452–459.
- Mast EE, Margolis HS, Fiore AE, et al; Advisory Committee on Immunization Practices (ACIP). A comprehensive immunization strategy to eliminate transmission of hepatitis B virus infection in the United States: recommendations of the Advisory Committee on Immunization Practices (ACIP) part 1: immunization of infants, children, and adolescents. MMWR Recomm Rep 2005; 54:1–31.
- US Centers for Disease Control (CDC). Prevention of perinatal transmission of hepatitis B virus: prenatal screening of all pregnant women for hepatitis B surface antigen. MMWR Morb Mortal Wkly Rep 1988; 37:341–346, 351.
- Han GR, Xu CL, Zhao W, Yang YF. Management of chronic hepatitis B in pregnancy. World J Gastroenterol 2012; 18:4517–4521.
- Terrault NA, Bzowej NH, Chang KM, Hwang JP, Jonas MM, Murad MH; American Association for the Study of Liver Diseases. AASLD guidelines for treatment of chronic hepatitis B. Hepatology 2016; 63:261–283.
- Tran TT, Ahn J, Reau N. ACG clinical guideline: liver disease and pregnancy. Am J Gastroenterol 2016: 111:176–194.
- Brown RS Jr, McMahon BJ, Lok AS, et al. Antiviral therapy in chronic hepatitis B viral infection during pregnancy: a systematic review and meta-analysis. Hepatol 2016; 63:319–333.
- Brown RS Jr, Verna EC, Pereira MR, et al. Hepatitis B virus and human immunodeficiency virus drugs in pregnancy: findings from the antiretroviral pregnancy registry. J Hepatol 2012; 57:953–959.
- Lamberth JR, Reddy SC, Pan JJ, Dasher KJ. Chronic hepatitis B infection in pregnancy. World J Hepatol 2015; 7:1233–1237.
- Potthoff A, Rifai K, Wedemeyer H, Deterding K, Manns M, Strassburg C. Successful treatment of fulminant hepatitis B during pregnancy. Z Gastroenterol 2009; 47:667–670.
- Yang J, Zeng XM, Men YL, Zhao LS. Elective caesarean section versus vaginal delivery for preventing mother to child transmission of hepatitis B virus—a systematic review. Virol J 2008; 5:100.
- Price J. An update on hepatitis B, D, and E viruses. Top Antivir Med 2014; 21:157–163.
- World Health Organization (WHO). Global alert and response. Hepatitis Delta. www.who.int/csr/resources/publications/hepatitis/who_cds_csr_ncs_2001_1/en/. Accessed December 7, 2016.
- Abbas Z, Memon MS, Mithani H, Jafri W, Hamid S. Treatment of chronic hepatitis D patients with pegylated interferon: a real-world experience. Antivir Ther 2014; 19:463–468.
- Baldo V, Baldovin T, Trivello R, Floreani A. Epidemiology of HCV infection. Curr Pharm Des 2008; 14:1646–1654.
- Floreani A. Hepatitis C and pregnancy. World J Gastroenterol 2013; 19:6714–6720.
- US Centers for Disease Control and Prevention. Viral hepatitis—CDC recommendations for specific populations and settings. www.cdc.gov/hepatitis/populations/1945-1965.htm. Accessed December 7, 2016.
- World Health Organization (WHO). Hepatitis C fact sheet. www.who.int/mediacentre/factsheets/fs164/en/. Accessed December 7, 2016.
- Moyer VA; US Preventive Services Task Force. Screening for hepatitis C virus infection in adults: US Preventive Services Task Force Recommendation Statement. Ann Intern Med 2013; 159:349–357.
- World Health Organization (WHO). Hepatitis E fact sheet. www.who.int/mediacentre/factsheets/fs280/en/. Accessed December 7, 2016.
- Velosa M, Figueiredo A, Gloria H, et al. Fulminant hepatitis E in a pregnant woman. GE Port J Gastroenterol 2013; 20:210–214.
- US Centers for Disease Control and Prevention (CDC). Viral hepatitis—hepatitis E information. Hepatitis E FAQs for health professionals. www.cdc.gov/hepatitis/hev/hevfaq.htm. Accessed December 7, 2016.
- Peters van Ton AM, Gevers TJ, Drenth JP. Antiviral therapy in chronic hepatitis E: a systematic review. J Viral Hepat 2015; 22:965–973.
- Kamar N, Bendall R, Legrand-Abravanel F, et al. Hepatitis E. Lancet 2012: 379:2477–2488.
Viral hepatitis affects mother and child, and pregnancy can exacerbate the disease. Vertical transmission contributes significantly to the high prevalence of viral hepatitis and compromises the well-being and the prognosis in the newborn. The indications for therapy in pregnant women may differ from those in the general population, and new therapies are available.
HEPATITIS A
Hepatitis A virus (HAV) infection is associated with significant morbidity and death around the world, as 1.4 million cases are reported every year worldwide.1 However, in the United States, the prevalence has declined by 95% since HAV vaccination was introduced in 1995, and in 2013, the prevalence was 0.6 per 100,000 population.2 Acute HAV infection during pregnancy is rare. As a result, the incidence during pregnancy is difficult to ascertain.3
HAV is transmitted by the fecal-oral route from person to person contact and from contamination of food and water. Vertical transmission from pregnant mother to fetus has not been reported.4
Clinical outcomes of HAV in pregnancy
Acute HAV infection during pregnancy is rare, and teratogenicity associated with HAV has not been reported.3 The course of the disease during pregnancy is generally similar to that in nonpregnant patients, with excellent maternal and fetal outcomes in developed nations. There have been reports in developing nations of premature contractions and labor, placental separation, premature rupture of the membranes, and vaginal bleeding.5,6
Diagnosis
Routine screening for HAV is not recommended, but serologic testing by detection of anti-HAV immunoglobulin M (IgM) antibodies is done in high-risk patients suspected of having acute HAV infection.
Prevention
Prevention includes adherence to sanitary practices and active and passive immunoprophylaxis.3 Universal vaccination for pregnant mothers is not recommended,1,2,6 but vaccination is recommended for high-risk patients and mothers—those with chronic liver disease, those receiving clotting factors, those who use illegal drugs, and travelers to areas where HAV is endemic. Immune globulin is also available for postexposure prophylaxis. HAV vaccines and immune globulin are safe in pregnancy.3,6,7
Treatment
Treatment of acute HAV in pregnancy is supportive because of its benign nature; few patients require hospitalization.3
Pregnant patients with HAV can deliver vaginally, and breastfeeding is not contraindicated.8
HEPATITIS B
Hepatitis B virus (HBV) infection is a major global health problem. About 240 million people worldwide have chronic HBV infection, and more than 780,000 die every year from acute and chronic consequences.9
Vertical transmission is responsible for about half of chronic HBV infections worldwide. Thus, interruption of mother-to-child transmission is important. Universal maternal screening and passive-active immunoprophylaxis of newborns have lowered the transmission rates to between 5% and 10%. The 10% failure rate is unacceptably high and has been attributed to seropositivity for hepatitis B e antigen and a high viral load in the mother (ie, HBV DNA > 106 copies/mL). High viral load is an independent risk factor for failure of immunoprophylaxis.10 Therefore, antiviral therapy is suggested in pregnant women who have a high HBV viral load to further decrease the chance of mother-to-child transmission and to prevent failure of immunoprophylaxis.11
Clinical outcomes of HBV in pregnancy
Acute HBV infection during pregnancy is usually benign and is not associated with increased risk of death or teratogenicity.12 Symptomatic disease in the mother with acute hepatitis B includes nausea, vomiting, abdominal pain, fatigue, and jaundice.3 For the newborn, there is increased risk of low birth weight and prematurity.13
When acute HBV infection occurs early in pregnancy, the rate of perinatal transmission is about 10%, increasing to 60% if it occurs near delivery.12,13
Chronic HBV infection does not usually affect the outcome of pregnancy, but it may if the woman has cirrhosis or advanced liver disease14; however, pregnancy is very rare in women with HBV cirrhosis due to anovulation, and acute HBV flares have been described during pregnancy and postpartum.15
Pregnant patients with cirrhosis and portal hypertension are at risk of hepatic decompensation, variceal bleeding, and death.16 Risk is high with a score of 10 or more on the Model for End-stage Liver Disease scale, and is low with a score of 6 or less.17 Like nonpregnant patients, pregnant patients with cirrhosis should be monitored, and upper endoscopy should be performed in the 2nd trimester to assess for varices. A beta-blocker should be given or banding of varices should be done to avoid rupture. Rates of fetal demise, premature labor, spontaneous abortion, and stillbirth are high with portal hypertension.16
Risk of mother-to-child HBV transmission
Vertical HBV transmission can occur during the antepartum, intrapartum, and postpartum periods,18,19 but it most often occurs during the intrapartum period at the time of delivery. Without immunoprophylaxis of the newborn, the risk of mother-to-child transmission can be as high as 90% if the mother is hepatitis B e antigen-positive and has a viral load greater than 106 copies/mL. With active and passive immunoprophylaxis, the risk decreases substantially.
Screening and diagnosis
All pregnant women should be tested for hepatitis B surface antigen during the 1st trimester,20 or any time thereafter if early testing was not done, even if they were vaccinated before becoming pregnant.21
Prevention
HBV infection is best prevented before pregnancy by vaccinating the mother or, after delivery, by vaccinating the newborn. Universal vaccination of newborns has been the standard of care since the 1990s. Pregnant women should be tested early in the pregnancy; unvaccinated, uninfected women at high risk of acquiring HBV (eg, because of sexual contacts or intravenous drug use) should be vaccinated.2,3
HBV vaccine and immune globulin are both approved by the US Food and Drug Administration (FDA) for prevention of HBV infection and can be given during pregnancy and breastfeeding.3 All infants should be vaccinated for HBV at birth, and all infants born to mothers who test positive for hepatitis B s antigen should receive the HBV vaccine and the immune globulin within 12 to 24 hours after delivery. The vaccine series should be completed within 6 months.20,21 This will decrease the rate of neonatal infection.
Treatment of HBV infection in pregnancy
The main objectives of treating chronic HBV infection in pregnancy are to maintain stable liver function in the mother and to prevent neonatal infection, which may cause cirrhosis and hepatocellular carcinoma and contribute to the global burden of the disease.22 Therefore, maternal HBV DNA and liver aminotransferase levels should be tested regularly during gestation.
The current guidelines of the American Association for the Study of Liver Diseases suggest antiviral therapy to reduce the risk of perinatal transmission of HBV in pregnant women with an HBV DNA level greater than 200,000 IU/mL or greater than 106 copies/mL.23,24
In a meta-analysis,25 antiviral therapy with lamivudine, telbivudine, or tenofovir showed no apparent teratogenicity or safety concerns for maternal and fetal outcomes26 and significantly reduced the rate of mother-to-child transmission. Of these 3 drugs, telbivudine was associated with a higher rate of normalization of liver enzymes, HBV suppression, and e-antigen seroconversion.25 Lamivudine has proven the test of time in mothers co-infected with HBV and human immunodeficiency virus (HIV). However, tenofovir is considered the preferred treatment in pregnancy, owing to concerns about drug resistance to telbivudine and lamivudine and a high genetic barrier to resistance with tenofovir.26 In mothers with HBV and HIV treated with tenofovir, treatment was associated with lower bone mineral density in the newborns, with a propensity for renal injury in the mothers. No safety concerns for maternal or fetal outcomes were identified in pregnant women infected only with HBV.25
Many pregnant mothers choose to stop therapy around the time of conception because of safety concerns for the baby. In such situations, close monitoring is necessary to detect flares of HBV infection.
When the decision to treat is made, treatment should begin at 28 to 30 weeks of gestation, when organogenesis is complete and to allow enough time for HBV DNA levels to decline.
Breastfeeding is not contraindicated because antiviral drugs are minimally excreted in breast milk and are unlikely to cause toxicity. However, data are insufficient as to the long-term safety for the newborn when the mother took these drugs during pregancy and while breastfeeding.23,27
Alanine aminotransferase and HBV DNA levels should be monitored postpartum because of the possibility of a hepatitis flare. In this setting, any of the three drugs can be used.28 For mothers on therapy because of cirrhosis or an advanced histologic feature, antiviral therapy should be continued throughout pregnancy to prevent disease progression and decompensation.19,22,27
No drug therapy is necessary for pregnant carriers of HBV.
Delivery and breastfeeding
The mode of delivery does not appear to have a significant effect on the interruption of vertical transmission of HBV.29 Cesarean delivery is not recommended by the US Centers for Disease Control and Prevention (CDC)2 or the American College of Obstetricians and Gynecologists.6 Breastfeeding is encouraged if the infant has received appropriate immunoprophylaxis.6
Coinfection with hepatitis D
Coinfection with hepatitis D virus (HDV) and HBV is associated with severe acute hepatitis30,31 and increases the risk of death by a factor of 10. The World Health Organization recommends testing for HDV in pregnant women who are HBV-positive.8
Prevention of HDV infection requires prevention of HBV. The treatment of HDV in pregnancy is supportive. Pegylated interferon is successful outside pregnancy but is contraindicated during pregnancy.32 In patients with fulminant hepatic failure and end-stage liver disease, liver transplant can be lifesaving.
Take-home points
- HBV infection during pregnancy is usually benign and not severe but can be associated with an increased risk of mother-to-child transmission and progression of liver disease in the pregnant mother.
- Prevention of vertical transmission of HBV is important to reduce the burden of chronic HBV infection. Universal maternal screening early in pregnancy and passive-active immunoprophylaxis of newborns are usually sufficient to prevent vertical transmission of HBV, but antiviral therapy is needed for highly viremic mothers to further reduce the risk.
- Antiviral therapy is also indicated for pregnant women with moderate to severe hepatitis or cirrhosis to prevent disease progression and liver failure.
- Telbivudine, tenofovir, or lamivudine can be used during pregnancy, but more data are needed on the long-term safety of fetal exposure to these agents.
HEPATITIS C
The global prevalence of hepatitis C virus (HCV) infection is 2% to 3%, with 130 to 170 million HCV-positive people, most of whom are chronically infected.33 The incidence of HCV during pregnancy is 1% to 2.4%, but 3% to 5% of infected mothers transmit HCV to their child at the time of birth.6,34 Women coinfected with HIV and HCV have twice the risk of perinatal HCV transmission compared with women who have HCV infection alone.6,34
HCV infection is usually asymptomatic and is discovered either by screening high-risk patients or during evaluation of persistently elevated aminotransferase levels. Acute HCV infection during pregnancy has been reported only rarely, and most pregnant women who are infected have chronic disease with no effect on the pregnancy or the infant.6,34
Treatment
The CDC recommends that all adults (including pregnant women) born between 1945 and 1965 undergo 1-time testing for HCV without prior ascertainment of HCV risk (strong recommendation, with moderate quality of evidence).35 The most important risk factor for HCV infection is past or current injection drug use.33 Additional risk factors are similar to those for nonpregnant patients.
Because of the benign effect of HCV on the pregnancy, treatment is not recommended. To decrease the risk of maternal-child transmission, it is prudent to avoid amniocentesis, scalp instrumentation, and prolonged rupture of membranes.6
There is no vaccine or immune globulin for prevention. HCV infection should not influence the mode of delivery, and it is not a contraindication to breastfeeding.34,36,37
HEPATITIS E
Every year, 20 million cases of hepatitis E virus (HEV) infection are recorded worldwide. These numbers include 3.3 million symptomatic cases and 56,600 deaths.38 HEV infection is most common in developing countries, and pregnant women traveling to these areas are at high risk of acquiring this infection, of developing fulminant hepatitis, and of death.39 Sporadic cases not associated with travel are increasingly reported in developed countries and are attributed to immunocompromised status (due to HIV or solid-organ transplant).38,40
Modes of transmission of HEV are mainly via fecal-oral contamination and by vertical transmission.41
Diagnosis
HEV infection can be diagnosed either by detecting IgM antibody with an enzyme-linked immunosorbent assay or by detecting HEV RNA in the blood using reverse transcription polymerase chain reaction testing.42
Treatment and prevention
Hospitalization should be considered for pregnant women. Ribavirin or pegylated interferon alpha or both are effective but are contraindicated in pregnancy because of the risk of teratogenicity.41,42 Urgent liver transplant can be a successful option in acute liver failure.
Prevention relies primarily on good sanitation, clean drinking water, and avoiding raw pork and venison. Boiling and chlorination of water inactivate HEV.39,40 Pregnant women should be advised to avoid travel to highly endemic areas.
Viral hepatitis affects mother and child, and pregnancy can exacerbate the disease. Vertical transmission contributes significantly to the high prevalence of viral hepatitis and compromises the well-being and the prognosis in the newborn. The indications for therapy in pregnant women may differ from those in the general population, and new therapies are available.
HEPATITIS A
Hepatitis A virus (HAV) infection is associated with significant morbidity and death around the world, as 1.4 million cases are reported every year worldwide.1 However, in the United States, the prevalence has declined by 95% since HAV vaccination was introduced in 1995, and in 2013, the prevalence was 0.6 per 100,000 population.2 Acute HAV infection during pregnancy is rare. As a result, the incidence during pregnancy is difficult to ascertain.3
HAV is transmitted by the fecal-oral route from person to person contact and from contamination of food and water. Vertical transmission from pregnant mother to fetus has not been reported.4
Clinical outcomes of HAV in pregnancy
Acute HAV infection during pregnancy is rare, and teratogenicity associated with HAV has not been reported.3 The course of the disease during pregnancy is generally similar to that in nonpregnant patients, with excellent maternal and fetal outcomes in developed nations. There have been reports in developing nations of premature contractions and labor, placental separation, premature rupture of the membranes, and vaginal bleeding.5,6
Diagnosis
Routine screening for HAV is not recommended, but serologic testing by detection of anti-HAV immunoglobulin M (IgM) antibodies is done in high-risk patients suspected of having acute HAV infection.
Prevention
Prevention includes adherence to sanitary practices and active and passive immunoprophylaxis.3 Universal vaccination for pregnant mothers is not recommended,1,2,6 but vaccination is recommended for high-risk patients and mothers—those with chronic liver disease, those receiving clotting factors, those who use illegal drugs, and travelers to areas where HAV is endemic. Immune globulin is also available for postexposure prophylaxis. HAV vaccines and immune globulin are safe in pregnancy.3,6,7
Treatment
Treatment of acute HAV in pregnancy is supportive because of its benign nature; few patients require hospitalization.3
Pregnant patients with HAV can deliver vaginally, and breastfeeding is not contraindicated.8
HEPATITIS B
Hepatitis B virus (HBV) infection is a major global health problem. About 240 million people worldwide have chronic HBV infection, and more than 780,000 die every year from acute and chronic consequences.9
Vertical transmission is responsible for about half of chronic HBV infections worldwide. Thus, interruption of mother-to-child transmission is important. Universal maternal screening and passive-active immunoprophylaxis of newborns have lowered the transmission rates to between 5% and 10%. The 10% failure rate is unacceptably high and has been attributed to seropositivity for hepatitis B e antigen and a high viral load in the mother (ie, HBV DNA > 106 copies/mL). High viral load is an independent risk factor for failure of immunoprophylaxis.10 Therefore, antiviral therapy is suggested in pregnant women who have a high HBV viral load to further decrease the chance of mother-to-child transmission and to prevent failure of immunoprophylaxis.11
Clinical outcomes of HBV in pregnancy
Acute HBV infection during pregnancy is usually benign and is not associated with increased risk of death or teratogenicity.12 Symptomatic disease in the mother with acute hepatitis B includes nausea, vomiting, abdominal pain, fatigue, and jaundice.3 For the newborn, there is increased risk of low birth weight and prematurity.13
When acute HBV infection occurs early in pregnancy, the rate of perinatal transmission is about 10%, increasing to 60% if it occurs near delivery.12,13
Chronic HBV infection does not usually affect the outcome of pregnancy, but it may if the woman has cirrhosis or advanced liver disease14; however, pregnancy is very rare in women with HBV cirrhosis due to anovulation, and acute HBV flares have been described during pregnancy and postpartum.15
Pregnant patients with cirrhosis and portal hypertension are at risk of hepatic decompensation, variceal bleeding, and death.16 Risk is high with a score of 10 or more on the Model for End-stage Liver Disease scale, and is low with a score of 6 or less.17 Like nonpregnant patients, pregnant patients with cirrhosis should be monitored, and upper endoscopy should be performed in the 2nd trimester to assess for varices. A beta-blocker should be given or banding of varices should be done to avoid rupture. Rates of fetal demise, premature labor, spontaneous abortion, and stillbirth are high with portal hypertension.16
Risk of mother-to-child HBV transmission
Vertical HBV transmission can occur during the antepartum, intrapartum, and postpartum periods,18,19 but it most often occurs during the intrapartum period at the time of delivery. Without immunoprophylaxis of the newborn, the risk of mother-to-child transmission can be as high as 90% if the mother is hepatitis B e antigen-positive and has a viral load greater than 106 copies/mL. With active and passive immunoprophylaxis, the risk decreases substantially.
Screening and diagnosis
All pregnant women should be tested for hepatitis B surface antigen during the 1st trimester,20 or any time thereafter if early testing was not done, even if they were vaccinated before becoming pregnant.21
Prevention
HBV infection is best prevented before pregnancy by vaccinating the mother or, after delivery, by vaccinating the newborn. Universal vaccination of newborns has been the standard of care since the 1990s. Pregnant women should be tested early in the pregnancy; unvaccinated, uninfected women at high risk of acquiring HBV (eg, because of sexual contacts or intravenous drug use) should be vaccinated.2,3
HBV vaccine and immune globulin are both approved by the US Food and Drug Administration (FDA) for prevention of HBV infection and can be given during pregnancy and breastfeeding.3 All infants should be vaccinated for HBV at birth, and all infants born to mothers who test positive for hepatitis B s antigen should receive the HBV vaccine and the immune globulin within 12 to 24 hours after delivery. The vaccine series should be completed within 6 months.20,21 This will decrease the rate of neonatal infection.
Treatment of HBV infection in pregnancy
The main objectives of treating chronic HBV infection in pregnancy are to maintain stable liver function in the mother and to prevent neonatal infection, which may cause cirrhosis and hepatocellular carcinoma and contribute to the global burden of the disease.22 Therefore, maternal HBV DNA and liver aminotransferase levels should be tested regularly during gestation.
The current guidelines of the American Association for the Study of Liver Diseases suggest antiviral therapy to reduce the risk of perinatal transmission of HBV in pregnant women with an HBV DNA level greater than 200,000 IU/mL or greater than 106 copies/mL.23,24
In a meta-analysis,25 antiviral therapy with lamivudine, telbivudine, or tenofovir showed no apparent teratogenicity or safety concerns for maternal and fetal outcomes26 and significantly reduced the rate of mother-to-child transmission. Of these 3 drugs, telbivudine was associated with a higher rate of normalization of liver enzymes, HBV suppression, and e-antigen seroconversion.25 Lamivudine has proven the test of time in mothers co-infected with HBV and human immunodeficiency virus (HIV). However, tenofovir is considered the preferred treatment in pregnancy, owing to concerns about drug resistance to telbivudine and lamivudine and a high genetic barrier to resistance with tenofovir.26 In mothers with HBV and HIV treated with tenofovir, treatment was associated with lower bone mineral density in the newborns, with a propensity for renal injury in the mothers. No safety concerns for maternal or fetal outcomes were identified in pregnant women infected only with HBV.25
Many pregnant mothers choose to stop therapy around the time of conception because of safety concerns for the baby. In such situations, close monitoring is necessary to detect flares of HBV infection.
When the decision to treat is made, treatment should begin at 28 to 30 weeks of gestation, when organogenesis is complete and to allow enough time for HBV DNA levels to decline.
Breastfeeding is not contraindicated because antiviral drugs are minimally excreted in breast milk and are unlikely to cause toxicity. However, data are insufficient as to the long-term safety for the newborn when the mother took these drugs during pregancy and while breastfeeding.23,27
Alanine aminotransferase and HBV DNA levels should be monitored postpartum because of the possibility of a hepatitis flare. In this setting, any of the three drugs can be used.28 For mothers on therapy because of cirrhosis or an advanced histologic feature, antiviral therapy should be continued throughout pregnancy to prevent disease progression and decompensation.19,22,27
No drug therapy is necessary for pregnant carriers of HBV.
Delivery and breastfeeding
The mode of delivery does not appear to have a significant effect on the interruption of vertical transmission of HBV.29 Cesarean delivery is not recommended by the US Centers for Disease Control and Prevention (CDC)2 or the American College of Obstetricians and Gynecologists.6 Breastfeeding is encouraged if the infant has received appropriate immunoprophylaxis.6
Coinfection with hepatitis D
Coinfection with hepatitis D virus (HDV) and HBV is associated with severe acute hepatitis30,31 and increases the risk of death by a factor of 10. The World Health Organization recommends testing for HDV in pregnant women who are HBV-positive.8
Prevention of HDV infection requires prevention of HBV. The treatment of HDV in pregnancy is supportive. Pegylated interferon is successful outside pregnancy but is contraindicated during pregnancy.32 In patients with fulminant hepatic failure and end-stage liver disease, liver transplant can be lifesaving.
Take-home points
- HBV infection during pregnancy is usually benign and not severe but can be associated with an increased risk of mother-to-child transmission and progression of liver disease in the pregnant mother.
- Prevention of vertical transmission of HBV is important to reduce the burden of chronic HBV infection. Universal maternal screening early in pregnancy and passive-active immunoprophylaxis of newborns are usually sufficient to prevent vertical transmission of HBV, but antiviral therapy is needed for highly viremic mothers to further reduce the risk.
- Antiviral therapy is also indicated for pregnant women with moderate to severe hepatitis or cirrhosis to prevent disease progression and liver failure.
- Telbivudine, tenofovir, or lamivudine can be used during pregnancy, but more data are needed on the long-term safety of fetal exposure to these agents.
HEPATITIS C
The global prevalence of hepatitis C virus (HCV) infection is 2% to 3%, with 130 to 170 million HCV-positive people, most of whom are chronically infected.33 The incidence of HCV during pregnancy is 1% to 2.4%, but 3% to 5% of infected mothers transmit HCV to their child at the time of birth.6,34 Women coinfected with HIV and HCV have twice the risk of perinatal HCV transmission compared with women who have HCV infection alone.6,34
HCV infection is usually asymptomatic and is discovered either by screening high-risk patients or during evaluation of persistently elevated aminotransferase levels. Acute HCV infection during pregnancy has been reported only rarely, and most pregnant women who are infected have chronic disease with no effect on the pregnancy or the infant.6,34
Treatment
The CDC recommends that all adults (including pregnant women) born between 1945 and 1965 undergo 1-time testing for HCV without prior ascertainment of HCV risk (strong recommendation, with moderate quality of evidence).35 The most important risk factor for HCV infection is past or current injection drug use.33 Additional risk factors are similar to those for nonpregnant patients.
Because of the benign effect of HCV on the pregnancy, treatment is not recommended. To decrease the risk of maternal-child transmission, it is prudent to avoid amniocentesis, scalp instrumentation, and prolonged rupture of membranes.6
There is no vaccine or immune globulin for prevention. HCV infection should not influence the mode of delivery, and it is not a contraindication to breastfeeding.34,36,37
HEPATITIS E
Every year, 20 million cases of hepatitis E virus (HEV) infection are recorded worldwide. These numbers include 3.3 million symptomatic cases and 56,600 deaths.38 HEV infection is most common in developing countries, and pregnant women traveling to these areas are at high risk of acquiring this infection, of developing fulminant hepatitis, and of death.39 Sporadic cases not associated with travel are increasingly reported in developed countries and are attributed to immunocompromised status (due to HIV or solid-organ transplant).38,40
Modes of transmission of HEV are mainly via fecal-oral contamination and by vertical transmission.41
Diagnosis
HEV infection can be diagnosed either by detecting IgM antibody with an enzyme-linked immunosorbent assay or by detecting HEV RNA in the blood using reverse transcription polymerase chain reaction testing.42
Treatment and prevention
Hospitalization should be considered for pregnant women. Ribavirin or pegylated interferon alpha or both are effective but are contraindicated in pregnancy because of the risk of teratogenicity.41,42 Urgent liver transplant can be a successful option in acute liver failure.
Prevention relies primarily on good sanitation, clean drinking water, and avoiding raw pork and venison. Boiling and chlorination of water inactivate HEV.39,40 Pregnant women should be advised to avoid travel to highly endemic areas.
- World Health Organization (WHO).Hepatitis A fact sheet. www.who.int/mediacentre/factsheets/fs328/en/. Accessed December 7, 2016.
- US Centers for Disease Control and Prevention (CDC). Viral hepatitis—statistics & surveillance. www.cdc.gov/hepatitis/statistics/2013surveillance/commentary.htm#hepatitis A. Accessed December 7, 2016.
- Rac MW, Sheffield JS. Prevention and management of viral hepatitis in pregnancy. Obstet Gynecol Clin North Am 2014; 41:573–592.
- Workowski KA, Bolan GA; Centers for Disease Control and Prevention (CDC). Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm Rep 2015; 64:1–137.
- Elinav E, Ben-Dov IZ, Shapira Y, et al. Acute hepatitis A infection in pregnancy is associated with high rates of gestational complications and preterm labor. Gastroenterology 2006; 130:1129–1134.
- American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 86: Viral hepatitis in pregnancy. Obstet Gynecol 2007; 110:941–956.
- Advisory Committee on Immunization Practices. Guidelines for Vaccinating Pregnant Women. www.cdc.gov/vaccines/pregnancy/hcp/guidelines.html. Accessed December 7, 2016.
- Daudi N, Shouval D, Stein-Zamir C, Ackerman Z. Breastmilk hepatitis A virus RNA in nursing mothers with acute hepatitis A virus infection. Breastfeed Med 2012; 7:313–315.
- World Health Organization (WHO). Hepatitis B fact sheet. www.who.int/mediacentre/factsheets/fs204/en/. Accessed December 7, 2016.
- Zou H, Chen Y, Duan Z, Zhang H, Pan C. Virologic factors associated with failure to passive-active immunoprophylaxis in infants born to HBsAg-positive mothers. J Viral Hepat 2012; 19:e18–e25.
- Pan CQ, Lee HM. Antiviral therapy for chronic hepatitis B in pregnancy. Semin Liver Dis 2013; 33:138–146.
- Sookoian S. Liver disease during pregnancy: acute viral hepatitis. Ann Hepatol 2006; 5:231–236.
- Jonas MM. Hepatitis B and pregnancy: an underestimated issue. Liver Int 2009; 29(suppl 1):133–139.
- Wong S, Chan LY, Yu V, Ho L. Hepatitis B carrier and perinatal outcome in singleton pregnancy. Am J Perinatol 1999; 16:485–488.
- Rawal BK, Parida S, Watkins RP, Ghosh P, Smith H. Symptomatic reactivation of hepatitis B in pregnancy. Lancet 1991; 337:364.
- Aggarwal N, Negi N, Aggarwal A, Bodh V, Dhiman RK. Pregnancy with portal hypertension. J Clin Exp Hepatol 2014; 4:163–171.
- Westbrook RH, Yeoman AD, O'Grady JG, Harrison PM, Devlin J, Heneghan MA. Model for end-stage liver disease score predicts outcome in cirrhotic patients during pregnancy. Clin Gastroenterol Hepatol 2011; 9:694–699.
- Cheung KW, Seto MT, Wong SF. Towards complete eradication of hepatitis B infection from perinatal transmission: review of the mechanisms of in utero infection and the use of antiviral treatment during pregnancy. Eur J Obstet Gynecol Reproduct Biol 2013; 169:17–23.
- Pan CQ, Duan AP, Bhamidimarri KR, et al. An algorithm for risk assessment and intervention of mother to child transmission of hepatitis B virus. Clin Gastroenterol Hepatol 2012; 10:452–459.
- Mast EE, Margolis HS, Fiore AE, et al; Advisory Committee on Immunization Practices (ACIP). A comprehensive immunization strategy to eliminate transmission of hepatitis B virus infection in the United States: recommendations of the Advisory Committee on Immunization Practices (ACIP) part 1: immunization of infants, children, and adolescents. MMWR Recomm Rep 2005; 54:1–31.
- US Centers for Disease Control (CDC). Prevention of perinatal transmission of hepatitis B virus: prenatal screening of all pregnant women for hepatitis B surface antigen. MMWR Morb Mortal Wkly Rep 1988; 37:341–346, 351.
- Han GR, Xu CL, Zhao W, Yang YF. Management of chronic hepatitis B in pregnancy. World J Gastroenterol 2012; 18:4517–4521.
- Terrault NA, Bzowej NH, Chang KM, Hwang JP, Jonas MM, Murad MH; American Association for the Study of Liver Diseases. AASLD guidelines for treatment of chronic hepatitis B. Hepatology 2016; 63:261–283.
- Tran TT, Ahn J, Reau N. ACG clinical guideline: liver disease and pregnancy. Am J Gastroenterol 2016: 111:176–194.
- Brown RS Jr, McMahon BJ, Lok AS, et al. Antiviral therapy in chronic hepatitis B viral infection during pregnancy: a systematic review and meta-analysis. Hepatol 2016; 63:319–333.
- Brown RS Jr, Verna EC, Pereira MR, et al. Hepatitis B virus and human immunodeficiency virus drugs in pregnancy: findings from the antiretroviral pregnancy registry. J Hepatol 2012; 57:953–959.
- Lamberth JR, Reddy SC, Pan JJ, Dasher KJ. Chronic hepatitis B infection in pregnancy. World J Hepatol 2015; 7:1233–1237.
- Potthoff A, Rifai K, Wedemeyer H, Deterding K, Manns M, Strassburg C. Successful treatment of fulminant hepatitis B during pregnancy. Z Gastroenterol 2009; 47:667–670.
- Yang J, Zeng XM, Men YL, Zhao LS. Elective caesarean section versus vaginal delivery for preventing mother to child transmission of hepatitis B virus—a systematic review. Virol J 2008; 5:100.
- Price J. An update on hepatitis B, D, and E viruses. Top Antivir Med 2014; 21:157–163.
- World Health Organization (WHO). Global alert and response. Hepatitis Delta. www.who.int/csr/resources/publications/hepatitis/who_cds_csr_ncs_2001_1/en/. Accessed December 7, 2016.
- Abbas Z, Memon MS, Mithani H, Jafri W, Hamid S. Treatment of chronic hepatitis D patients with pegylated interferon: a real-world experience. Antivir Ther 2014; 19:463–468.
- Baldo V, Baldovin T, Trivello R, Floreani A. Epidemiology of HCV infection. Curr Pharm Des 2008; 14:1646–1654.
- Floreani A. Hepatitis C and pregnancy. World J Gastroenterol 2013; 19:6714–6720.
- US Centers for Disease Control and Prevention. Viral hepatitis—CDC recommendations for specific populations and settings. www.cdc.gov/hepatitis/populations/1945-1965.htm. Accessed December 7, 2016.
- World Health Organization (WHO). Hepatitis C fact sheet. www.who.int/mediacentre/factsheets/fs164/en/. Accessed December 7, 2016.
- Moyer VA; US Preventive Services Task Force. Screening for hepatitis C virus infection in adults: US Preventive Services Task Force Recommendation Statement. Ann Intern Med 2013; 159:349–357.
- World Health Organization (WHO). Hepatitis E fact sheet. www.who.int/mediacentre/factsheets/fs280/en/. Accessed December 7, 2016.
- Velosa M, Figueiredo A, Gloria H, et al. Fulminant hepatitis E in a pregnant woman. GE Port J Gastroenterol 2013; 20:210–214.
- US Centers for Disease Control and Prevention (CDC). Viral hepatitis—hepatitis E information. Hepatitis E FAQs for health professionals. www.cdc.gov/hepatitis/hev/hevfaq.htm. Accessed December 7, 2016.
- Peters van Ton AM, Gevers TJ, Drenth JP. Antiviral therapy in chronic hepatitis E: a systematic review. J Viral Hepat 2015; 22:965–973.
- Kamar N, Bendall R, Legrand-Abravanel F, et al. Hepatitis E. Lancet 2012: 379:2477–2488.
- World Health Organization (WHO).Hepatitis A fact sheet. www.who.int/mediacentre/factsheets/fs328/en/. Accessed December 7, 2016.
- US Centers for Disease Control and Prevention (CDC). Viral hepatitis—statistics & surveillance. www.cdc.gov/hepatitis/statistics/2013surveillance/commentary.htm#hepatitis A. Accessed December 7, 2016.
- Rac MW, Sheffield JS. Prevention and management of viral hepatitis in pregnancy. Obstet Gynecol Clin North Am 2014; 41:573–592.
- Workowski KA, Bolan GA; Centers for Disease Control and Prevention (CDC). Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm Rep 2015; 64:1–137.
- Elinav E, Ben-Dov IZ, Shapira Y, et al. Acute hepatitis A infection in pregnancy is associated with high rates of gestational complications and preterm labor. Gastroenterology 2006; 130:1129–1134.
- American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 86: Viral hepatitis in pregnancy. Obstet Gynecol 2007; 110:941–956.
- Advisory Committee on Immunization Practices. Guidelines for Vaccinating Pregnant Women. www.cdc.gov/vaccines/pregnancy/hcp/guidelines.html. Accessed December 7, 2016.
- Daudi N, Shouval D, Stein-Zamir C, Ackerman Z. Breastmilk hepatitis A virus RNA in nursing mothers with acute hepatitis A virus infection. Breastfeed Med 2012; 7:313–315.
- World Health Organization (WHO). Hepatitis B fact sheet. www.who.int/mediacentre/factsheets/fs204/en/. Accessed December 7, 2016.
- Zou H, Chen Y, Duan Z, Zhang H, Pan C. Virologic factors associated with failure to passive-active immunoprophylaxis in infants born to HBsAg-positive mothers. J Viral Hepat 2012; 19:e18–e25.
- Pan CQ, Lee HM. Antiviral therapy for chronic hepatitis B in pregnancy. Semin Liver Dis 2013; 33:138–146.
- Sookoian S. Liver disease during pregnancy: acute viral hepatitis. Ann Hepatol 2006; 5:231–236.
- Jonas MM. Hepatitis B and pregnancy: an underestimated issue. Liver Int 2009; 29(suppl 1):133–139.
- Wong S, Chan LY, Yu V, Ho L. Hepatitis B carrier and perinatal outcome in singleton pregnancy. Am J Perinatol 1999; 16:485–488.
- Rawal BK, Parida S, Watkins RP, Ghosh P, Smith H. Symptomatic reactivation of hepatitis B in pregnancy. Lancet 1991; 337:364.
- Aggarwal N, Negi N, Aggarwal A, Bodh V, Dhiman RK. Pregnancy with portal hypertension. J Clin Exp Hepatol 2014; 4:163–171.
- Westbrook RH, Yeoman AD, O'Grady JG, Harrison PM, Devlin J, Heneghan MA. Model for end-stage liver disease score predicts outcome in cirrhotic patients during pregnancy. Clin Gastroenterol Hepatol 2011; 9:694–699.
- Cheung KW, Seto MT, Wong SF. Towards complete eradication of hepatitis B infection from perinatal transmission: review of the mechanisms of in utero infection and the use of antiviral treatment during pregnancy. Eur J Obstet Gynecol Reproduct Biol 2013; 169:17–23.
- Pan CQ, Duan AP, Bhamidimarri KR, et al. An algorithm for risk assessment and intervention of mother to child transmission of hepatitis B virus. Clin Gastroenterol Hepatol 2012; 10:452–459.
- Mast EE, Margolis HS, Fiore AE, et al; Advisory Committee on Immunization Practices (ACIP). A comprehensive immunization strategy to eliminate transmission of hepatitis B virus infection in the United States: recommendations of the Advisory Committee on Immunization Practices (ACIP) part 1: immunization of infants, children, and adolescents. MMWR Recomm Rep 2005; 54:1–31.
- US Centers for Disease Control (CDC). Prevention of perinatal transmission of hepatitis B virus: prenatal screening of all pregnant women for hepatitis B surface antigen. MMWR Morb Mortal Wkly Rep 1988; 37:341–346, 351.
- Han GR, Xu CL, Zhao W, Yang YF. Management of chronic hepatitis B in pregnancy. World J Gastroenterol 2012; 18:4517–4521.
- Terrault NA, Bzowej NH, Chang KM, Hwang JP, Jonas MM, Murad MH; American Association for the Study of Liver Diseases. AASLD guidelines for treatment of chronic hepatitis B. Hepatology 2016; 63:261–283.
- Tran TT, Ahn J, Reau N. ACG clinical guideline: liver disease and pregnancy. Am J Gastroenterol 2016: 111:176–194.
- Brown RS Jr, McMahon BJ, Lok AS, et al. Antiviral therapy in chronic hepatitis B viral infection during pregnancy: a systematic review and meta-analysis. Hepatol 2016; 63:319–333.
- Brown RS Jr, Verna EC, Pereira MR, et al. Hepatitis B virus and human immunodeficiency virus drugs in pregnancy: findings from the antiretroviral pregnancy registry. J Hepatol 2012; 57:953–959.
- Lamberth JR, Reddy SC, Pan JJ, Dasher KJ. Chronic hepatitis B infection in pregnancy. World J Hepatol 2015; 7:1233–1237.
- Potthoff A, Rifai K, Wedemeyer H, Deterding K, Manns M, Strassburg C. Successful treatment of fulminant hepatitis B during pregnancy. Z Gastroenterol 2009; 47:667–670.
- Yang J, Zeng XM, Men YL, Zhao LS. Elective caesarean section versus vaginal delivery for preventing mother to child transmission of hepatitis B virus—a systematic review. Virol J 2008; 5:100.
- Price J. An update on hepatitis B, D, and E viruses. Top Antivir Med 2014; 21:157–163.
- World Health Organization (WHO). Global alert and response. Hepatitis Delta. www.who.int/csr/resources/publications/hepatitis/who_cds_csr_ncs_2001_1/en/. Accessed December 7, 2016.
- Abbas Z, Memon MS, Mithani H, Jafri W, Hamid S. Treatment of chronic hepatitis D patients with pegylated interferon: a real-world experience. Antivir Ther 2014; 19:463–468.
- Baldo V, Baldovin T, Trivello R, Floreani A. Epidemiology of HCV infection. Curr Pharm Des 2008; 14:1646–1654.
- Floreani A. Hepatitis C and pregnancy. World J Gastroenterol 2013; 19:6714–6720.
- US Centers for Disease Control and Prevention. Viral hepatitis—CDC recommendations for specific populations and settings. www.cdc.gov/hepatitis/populations/1945-1965.htm. Accessed December 7, 2016.
- World Health Organization (WHO). Hepatitis C fact sheet. www.who.int/mediacentre/factsheets/fs164/en/. Accessed December 7, 2016.
- Moyer VA; US Preventive Services Task Force. Screening for hepatitis C virus infection in adults: US Preventive Services Task Force Recommendation Statement. Ann Intern Med 2013; 159:349–357.
- World Health Organization (WHO). Hepatitis E fact sheet. www.who.int/mediacentre/factsheets/fs280/en/. Accessed December 7, 2016.
- Velosa M, Figueiredo A, Gloria H, et al. Fulminant hepatitis E in a pregnant woman. GE Port J Gastroenterol 2013; 20:210–214.
- US Centers for Disease Control and Prevention (CDC). Viral hepatitis—hepatitis E information. Hepatitis E FAQs for health professionals. www.cdc.gov/hepatitis/hev/hevfaq.htm. Accessed December 7, 2016.
- Peters van Ton AM, Gevers TJ, Drenth JP. Antiviral therapy in chronic hepatitis E: a systematic review. J Viral Hepat 2015; 22:965–973.
- Kamar N, Bendall R, Legrand-Abravanel F, et al. Hepatitis E. Lancet 2012: 379:2477–2488.
KEY POINTS
- Preventing vertical transmission of HBV infection in pregnancy is key to decreasing the global burden of this infection. Universal maternal screening and passive-active immunoprophylaxis of newborns have reduced transmission of HBV, but the addition of antiviral therapy is necessary to further decrease immunoprophylaxis failure.
- Tenofovir, telbivudine, and lamivudine can be used safely in pregnancy without apparent teratogenicity or other harmful effects on mother or baby. But optimal outcome requires discussion of safety and the plan of care with the patient, obstetrician, and hepatologist.
- Most pregnant women with hepatitis C virus (HCV) infection have chronic disease, with no effects on the pregnancy or baby, but 3% to 5% transmit HCV to their child at the time of birth. All pregnant women at risk should be screened at the first prenatal visit. The safety and efficacy of treating pregnant women to prevent transmission to the fetus are not established; thus, treatment is not recommended for pregnant women.
Iodine deficiency: Clinical implications
A 65-year-old woman is found to have a goiter. She is clinically euthyroid. She is a strict vegan and only uses noniodized Himalayan salt for cooking. Her thyroid gland is diffusely enlarged with no nodules. The estimated weight of the thyroid gland is 50 g (normal 10–20 g) based on ultrasonography. Her thyroid-stimulating hormone (TSH) level is 2.95 mU/L (reference range 0.5–5 mU/L), and her free thyroxine level is 0.8 ng/dL (0.7–1.8 ng/dL). Testing for TSH receptor antibody is negative. Her 24-hour urine iodine is undetectable (urine iodine concentration < 10 μg/L with urine volume 3,175 mL). What may be the cause of her goiter?
Iodine is an essential element needed for the production of thyroid hormone, which controls metabolism and plays a major role in fetal neurodevelopment. Its ionized form is called iodide. Iodine deficiency results in impairment of thyroid hormone synthesis and may lead to several undesirable consequences. Physicians should be aware of the risks iodine deficiency poses, especially during pregnancy, and should be familiar with approaches to testing and current indications for iodine supplementation.
SOURCES OF IODINE AND SALT IODIZATION
The major environmental source of iodine is the ocean. Elemental iodine in the ocean volatilizes into the atmosphere and returns to the soil by rain. The effects of glaciation, flooding, and leaching into soil have resulted in the variable geographic distribution of iodine. Mountainous areas (eg, the Alps, Andes, Himalayas) and areas with frequent flooding typically have iodine-deficient soil due to slow iodine cycling.1 Seafood is a good source of iodine because marine plants and animals concentrate iodine from seawater. The iodine content of other foods varies widely, depending on the source and any additives.
In the United States, the major sources of dietary iodine are dairy products (due to livestock iodine supplements and use of iodophors for cleaning milk udders) and iodized salt.1,2 Seafood contains a higher amount of iodine by weight than dairy products but is consumed far less than dairy.3,4 Further, the iodine content of milk can range from 88 to 168 μg per 250 mL (about 1 cup), depending on the product manufacturer. Also, iodine content is often omitted from the food label. Even if it is reported, the package labeling may not accurately predict the iodine content.5
Less common sources of iodine are radiographic contrast, bread with iodate dough conditioners, red food coloring (erythrosine), and drugs such as amiodarone.1
Using iodized salt is an effective and stable way to ensure adequate iodine intake. In the United States, only table salt is iodized, and the salt typically used in processed food has only minimal iodine content.6 Nearly 70% of the salt we ingest is from processed food. Table salt provides only 15% of dietary salt intake, and only 70% of consumers choose iodized salt for home cooking.7
IODINE REQUIREMENTS
Daily requirements of iodine suggested by the World Health Organization (WHO) and by the US Institute of Medicine are in the range of 90 to 150 µg/day.8,9 The iodine requirement is higher in pregnancy (220–250 µg/day) because of increased maternal thyroid hormone production required to maintain euthyroidism and increased renal iodine clearance, and it is even higher in lactating women (250–290 µg/day).
IODINE STATUS IN POPULATIONS
Since the establishment of universal salt iodization programs under the influence of the WHO and the International Council for Control of Iodine Deficiency Disorders (ICCIDD) in 1990, global iodine status has continued to improve. Yet only 70% of households worldwide currently have access to adequately iodized salt, because many countries lack a national program for iodine supplementation. The population of the United States was historically iodine-deficient, but since the introduction of salt iodization in the 1920s, the iodine status in the United States has been considered adequate.1
The WHO defines iodine status for a population by the median spot urinary iodine concentration. Because a urinary iodine concentration of 100 μg/L represents an iodine intake of about 150 μg/day, the WHO uses a median urinary iodine concentration of 100 to 199 μg/L to define adequate iodine intake for a nonpregnant population.9
The National Health and Nutrition Examination Survey (NHANES) found that the median urinary iodine concentration decreased by more than 50% from the 1970s to the 1990s, indicating declining iodine status in the US population.2 Of particular concern, the percentage of women of childbearing age with moderate iodine deficiency increased from 4% to 15% over this period.2 Still, the NHANES survey in 2009–2010 indicated that the overall US population is still iodine-sufficient (median urinary iodine concentration 144 μg/L).10 The decline in the US iodine status may be due to reduction of iodine content in dairy products, increased use of noniodized salt by the food industry, and recommendations to avoid salt for blood pressure control.
Although US iodine status has been considered generally adequate, iodine intake varies greatly across the population. Vegans tend to have iodine-deficient diets, while kelp consumers may have excessive iodine intake.11 Individuals with lactose intolerance are at risk of iodine deficiency, given that dairy products are a major source of iodine in the United States. Physicians should be aware of these risk factors for iodine deficiency.
PREGNANCY AND LACTATION
It is crucial to maintain euthyroidism during pregnancy. In early gestation, maternal thyroid hormone production increases 50% due to an increase in thyroid-binding globulin and stimulation by human chorionic gonadotropin. The glomerular filtration rate increases by 30% to 50% during pregnancy, thus increasing renal iodine clearance. Fetal thyroid hormone production increases during the second half of pregnancy, further contributing to increased maternal iodine requirements because iodine readily crosses the placenta.12
Women with sufficient iodine intake before and during pregnancy generally have adequate intrathyroidal iodine storage and can adapt to the increased demand for thyroid hormone throughout gestation. But in the setting of even mild iodine deficiency, total body iodine stores decline gradually from the first to third trimester of pregnancy.13
The fetal thyroid gland does not begin to concentrate iodine until 10 to 12 weeks of gestation and is not controlled by TSH until the full development of the pituitary-portal vascular system at 20 weeks of gestation.12 Therefore, the fetus relies on maternal thyroid hormone during this critical stage of neurodevelopment. Thyroid hormone is essential for oligodendrocyte differentiation and myelin distribution14 as well as fetal neuronal proliferation and migration in the first and second trimesters. Iodine deficiency leading to maternal hypothyroidism can result in irreversible fetal brain damage.
Because of the greater requirement during pregnancy, the WHO recommends using a median urinary iodine concentration of 150 to 249 μg/L to define a population that has no iodine deficiency.9 The NHANES data from 2007 to 2010 showed that pregnant US women were mildly iodine-deficient (median urinary iodine concentration 135 μg/L),10 and the National Children’s Study of 501 pregnant US women during the third trimester in 2009 to 2010 showed they had adequate iodine intake (median urinary iodine concentration 167 μg/L). Interestingly, pregnant non-Hispanic blacks were the only ethnic group with a median urinary iodine concentration less than 150 μg/L, suggesting that race or ethnicity is a predictor of iodine status in pregnant women.10
Iodine requirements during lactation
During lactation, thyroid hormone production and renal iodine clearance return to the prepregnancy state. However, a significant amount of iodine is excreted into breast milk at a concentration 20 to 50 times greater than that in plasma.15 It is recommended that lactating women continue high iodine intake to ensure sufficient iodine in breast milk to build reserves in the newborn’s thyroid gland.
The iodine requirement during lactation is 225 to 350 μg/day.16 Breast milk containing 100 to 200 μg/L of iodine appears to provide adequate iodine to meet Institute of Medicine recommendations for infants.17 The amount of iodine excreted into breast milk depends on maternal iodine intake. In the setting of iodine sufficiency, the iodine content of breast milk is 150 to 180 μg/L, but it is much lower (9–32 μg/L) in women from iodine-deficient areas, eg, the “goiter belt,” which included the Great Lakes, the Appalachians, and northwestern states. While iodized salt has virtually eliminated the goiter belt, the risk of iodine deficiency remains for people who avoid iodized salt and dairy.15
To ensure adequate iodine intake, the American Thyroid Association recommends that women receive iodine supplementation daily during pregnancy and lactation.11 However, the iodine content of prenatal multivitamins is currently not mandated in the United States. Only half of marketed prenatal vitamins in the United States contain iodine, in the form of either potassium iodide or kelp. Though most iodine-containing products claim to contain at least 150 μg of iodine per daily dose, when measured, the actual iodine content varied between 33 and 610 μg.18
CONSEQUENCES OF IODINE DEFICIENCY
Goiter
Goiter in iodine-deficient areas is considered to be an adaptation to chronic iodine deficiency. Low iodine intake leads to reduced thyroid hormone production, which in turn stimulates TSH secretion from the pituitary. TSH increases iodine uptake by the thyroid, stimulates thyroid growth, and leads to goiter development.
Initially, goiter is characterized by diffuse thyroid enlargement, but over time it may become nodular from progressive accumulation of new thyroid follicles. Goiter in children from iodine-deficient areas is diffusely enlarged, whereas in older adults it tends to be multinodular.
Iodine deficiency and chronic TSH stimulation may play a role in TSH receptor-activating mutations of thyroid follicles. These “gain-of-function” mutations are more common in the glands of patients with goiter in areas of iodine deficiency but are relatively rare in areas of iodine sufficiency.19 Toxic multinodular goiter may eventually develop, and hyperthyroidism may occur if iodine deficiency is not severe.
Goiter generally does not cause obstructive symptoms, since the thyroid usually grows outward. However, a very large goiter may descend to the thoracic inlet and compress the trachea and esophagus. The obstructive effect of a large goiter can be demonstrated by having a patient raise the arms adjacent to the face (the Pemberton maneuver). Signs suggesting obstruction are engorged neck veins, facial plethora, increased dyspnea, and stridor during the maneuver. Computed tomography of the neck and upper thorax may provide information on the degree of tracheal compression.20
Hypothyroidism
A normal or low triiodothyronine (T3), a low serum thyroxine (T4), and a variably elevated TSH are features of thyroid function tests in iodine deficiency.11,21,22 As long as daily iodine intake exceeds 50 μg/day, the absolute uptake of iodine by the thyroid gland usually remains adequate to maintain euthyroidism. Below 50 μg/day, iodine storage in the thyroid becomes depleted, leading to hypothyroidism.1
The clinical manifestations of hypothyroidism from iodine deficiency are similar to those of hypothyroidism from other causes. Because of thyroid hormone’s role in neural and somatic development, the manifestations of hypothyroidism differ among age groups (Table 1).
Cretinism
Before the development of fetal thyroid tissue in the 10th to 12th week of gestation, the fetus is dependent on maternal thyroid hormone, which crosses the placenta to support general and neural development. Iodine deficiency leading to maternal hypothyroidism (in early gestation) or inadequate fetal thyroid hormone production (in late gestation) may result in various degrees of mental retardation or lower than expected IQ.
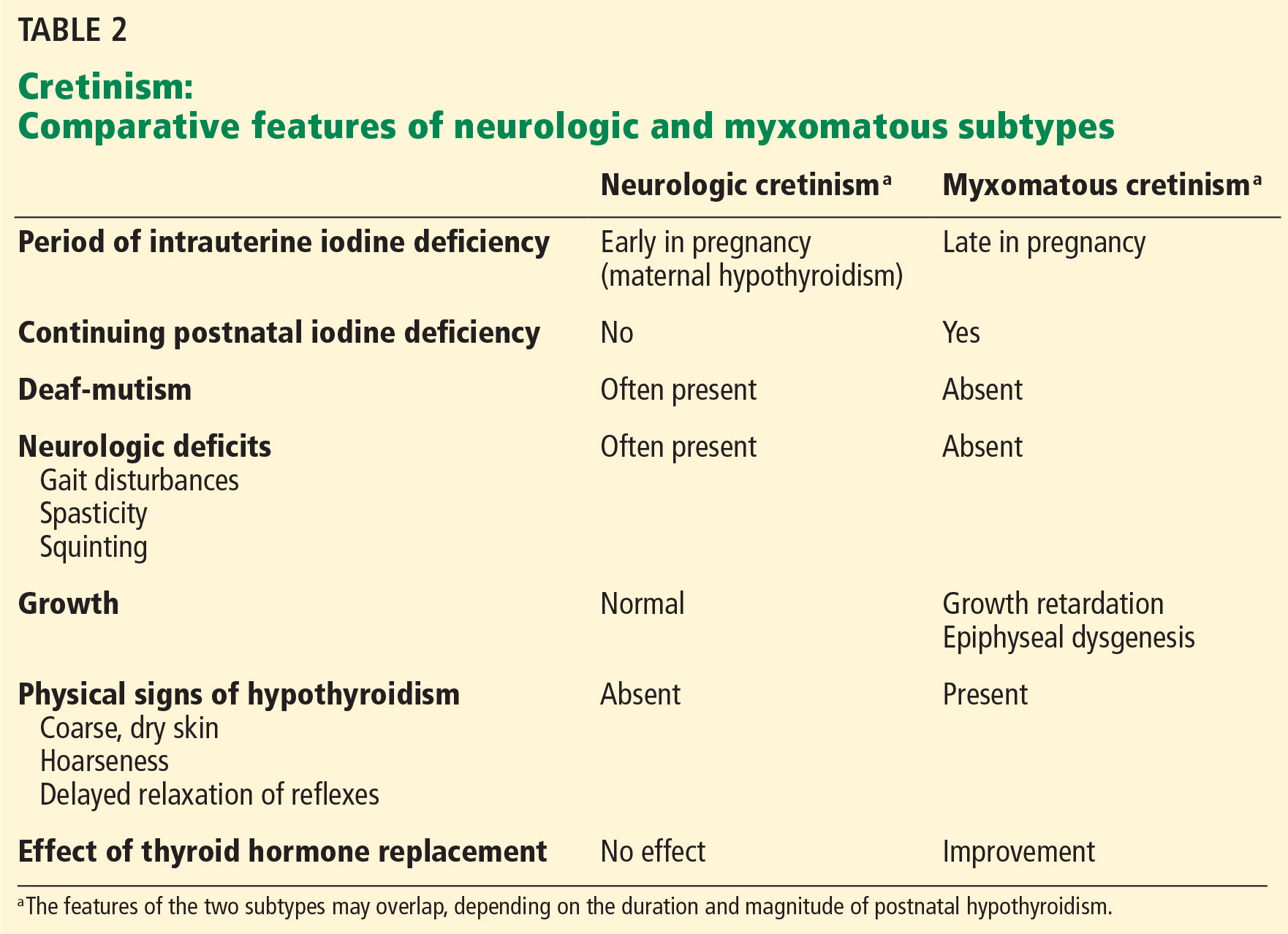
Severe iodine deficiency during gestation typically results in cretinism, characterized by severe mental retardation accompanied by other neurologic or physical defects. Cretinism is divided into two subtypes according to clinical manifestations (neurologic and myxomatous cretinism; Table 2), which may reflect the different timing of intrauterine insult to the developing fetal nervous system and whether the iodine deficiency continues into the postnatal period. Both types can be prevented by adequate maternal iodine intake before and during pregnancy.23,24
Although mild gestational iodine deficiency does not result in cretinism, it nevertheless has an adverse impact on fetal neurodevelopment and subsequent functioning. Children of mothers with mild gestational iodine deficiency were found to have reductions in spelling, grammar, and English literacy performance despite growing up in iodine-replete environments.25
Impaired cognitive development
Reduction in IQ has been noted in affected youth from regions of severe and mild iodine deficiency. A meta-analysis of studies relating iodine deficiency to cognitive development suggested that chronic moderate to severe iodine deficiency reduced expected average IQ by about 13.5 points.26
The effects of mild iodine deficiency during childhood are more difficult to quantify. The results of one study suggested that mild iodine deficiency was associated with subtle neurodevelopmental deficits and that iodine supplementation might improve cognitive function in mildly iodine-deficient children.27
In a 2009 randomized, placebo-controlled study in New Zealand, 184 children ages 10 to 13 with mild iodine deficiency (median urinary iodine concentration of 63 μg/L) received iodine supplementation (150 μg/day) or placebo for 28 weeks. Iodine supplementation increased the median urinary iodine concentration to 145 μg/L and significantly improved perceptual reasoning measures and overall cognitive score compared with placebo.28
These findings suggest that correcting mild iodine deficiency in children could improve certain components of cognition. More research is needed to understand the effects of mild iodine deficiency and iodine supplementation on cognitive function.
ASSESSING IODINE STATUS
The diagnosis of iodine deficiency is based on clinical and laboratory assessments. Clinical manifestations compatible with iodine deficiency and careful history-taking focused on the patient’s dietary iodine intake and geographic data are keys to the diagnosis.
Four main methods are used to assess iodine status at a population level: urinary iodine, serum thyroglobulin, serum TSH, and thyroid size. Urinary iodine is a sensitive marker for recent iodine intake (within days); thyroglobulin represents iodine nutrition over a period of months and thyroid size over a period of years.1
Urinary iodine
Most dietary iodine is excreted into the urine within 24 hours of ingestion, and the 24-hour urinary iodine is considered a reference standard for the measurement of individual daily iodine intake. However, the process of collection is cumbersome, and the 24-hour urinary iodine can vary from day to day in the same person, depending on the amount of iodine ingested.
A study in healthy women from an iodine-sufficient area suggested that 10 repeated 24-hour urine collections estimated the person’s iodine status at a precision of 20% because of variable daily iodine intake.29 Therefore, when necessary, several 24-hour urine iodine determinations should be performed.
A single, random, spot urinary iodine is expressed as the urinary iodine concentration and is affected by the amount of iodine and fluids the individual ingests in a day, thus resulting in high variation both within an individual person and between individuals. Expressing the urinary iodine concentration as the ratio of urine iodine to creatinine is useful in correcting for the influence of fluid intake. The ratio of urine iodine to creatinine can be used to estimate 24-hour urine iodine with the following formula: urine iodine (μg/L)/creatinine (g/L)× age- and sex-specific estimated 24-hour creatinine excretion (g/day). Another clinical use of the spot urine iodine is to screen for exposure to a large amount of iodine from a source such as radiographic contrast.30
Although individual urine iodine excretion and urine volume can vary from day to day, this variation tends to even out in a large number of samples. In study populations of at least 500, the median value of the spot urinary iodine concentration is considered a reliable measure of iodine intake in that population.30 The spot urine iodine test is convenient, making it the test of choice to study iodine status in a large cohort. The WHO recommends using the median value of the spot urine iodine to evaluate the iodine status of a population.9
Thyroglobulin
Thyroglobulin is a thyroid-specific protein involved in the synthesis of thyroid hormone. Small amounts can be detected in the blood of healthy people. In the absence of thyroid damage, the amount of serum thyroglobulin depends on thyroid cell mass and TSH stimulation. The serum level is elevated in iodine deficiency as a result of chronic TSH stimulation and thyroid hyperplasia. Thus, thyroglobulin can serve as a marker of iodine deficiency.
Serum thyroglobulin assays have been adapted for use on dried whole-blood spots, which require only a few drops of whole blood collected on filter paper and left to air-dry. The results of the dried whole-blood assay correlate closely with those of the serum assay.31 An established international dried whole-blood thyroglobulin reference range for iodine-sufficient school-age children is 4 to 40 μg/L.32 A median level of less than 13 μg/L in school-age children indicates iodine sufficiency in the population.33
Thyroid-stimulating hormone
Iodine deficiency lowers serum T4, which in turn leads to increased serum TSH. Therefore, iodine-deficient populations generally have higher TSH than iodine-sufficient groups. However, the TSH values in older children and adults with iodine deficiency are not significantly different from values of those with adequate iodine intake. Therefore, TSH is not a practical marker of iodine deficiency in the general population.
In contrast, TSH in newborns is a reasonable indicator of population iodine status. The newborn thyroid has limited iodine stores compared with that of an adult and hence a much higher iodine turnover rate. TSH from the cord blood is markedly elevated in newborns of mothers with moderate to severe iodine deficiency.34 A high prevalence of newborns with elevated TSH should therefore reflect iodine deficiency in the area where the mothers of the newborns live.
TSH is now routinely checked in newborns to screen for congenital hypothyroidism. TSH is typically checked 2 to 5 days after delivery to avoid confusion with transient physiologic TSH elevation, which occurs within a few hours after birth and decreases rapidly in 24 hours. The WHO has proposed that a more than 3% prevalence of newborns with TSH values higher than 5 mU/L from blood samples collected 3 to 4 days after birth indicates iodine deficiency in a population.1 This threshold appears to correlate well with the iodine status of the population defined by the WHO’s median urinary iodine concentration.35,36
But several other factors can influence the measurement of newborn TSH, such as prematurity, time of blood collection, maternal or newborn exposure to iodine-containing antiseptics, and the TSH assay methodology. These potential confounding factors limit the role of neonatal TSH as a reliable monitoring tool for iodine deficiency.35,37
Thyroid size

The size of the thyroid gland varies inversely with iodine intake. Thyroid size can be assessed by either palpation or ultrasonography, with the latter being more sensitive. The goiter rate in school-age children can be used to determine the severity of iodine deficiency in the population (Table 3). A goiter rate of 5% or more in school-age children suggests the presence of iodine deficiency in the community.
Although thyroid size is easy to estimate by palpation, it has low sensitivity and specificity to detect iodine deficiency and high interobserver variation. Thyroid ultrasonography provides a more precise measurement of thyroid gland volume. Zimmermann et al38 provided reference data on thyroid volume stratified by age, sex, and body surface area of school-age children in iodine-sufficient areas.38 Results of ultrasonography in a population is then compared with these reference data. The higher the percentage of the population with thyroid volume exceeding the 97th percentile of the reference range, the more severe the iodine deficiency. However, the WHO does not specify how to grade the degree of iodine deficiency based on the thyroid volume obtained with ultrasonography. Follow-up studies showed no significant correlation between urinary iodine concentration and thyroid size.39,40
Thyroid size decreases slowly after iodine repletion. Therefore, the goiter rate may remain high for several years after iodine supplementation begins.1,9
TREATMENT AND PREVENTION

Treatment of iodine deficiency should be instituted at the levels recommended by the Institute of Medicine and the WHO. The tolerable upper intake levels for iodine are outlined in Table 4. In a nonpregnant adult, 150 μg/day is sufficient for normal thyroid function. Iodine intake should be higher for pregnant and lactating women (250 μg/day according to the WHO recommendation).
Iodine supplementation is easily achieved by using iodized salt or an iodine-containing daily multivitamin.
In patients with overt hypothyroidism from iodine deficiency, we recommend initiating levothyroxine treatment along with iodine supplementation to restore euthyroidism, with consideration of possible interruption in 6 to 12 months when the urine iodine has normalized and goiter size has decreased. Thyroid function should be reassessed 4 to 6 weeks after discontinuation of levothyroxine.
At the population level, iodine deficiency can usually be prevented by iodization of food products or the water supply. In developing countries where salt iodization is not practical, iodine deficiency has been eradicated by adding iodine drops to well water or by injecting people with iodized oil.
TAKE-HOME POINTS
Iodine is essential for thyroid hormone synthesis. It can be obtained by eating iodine-containing foods or by using iodized salt. The WHO classifies iodine deficiency based on the median urinary iodine concentration. Iodine nutrition at the community level is best assessed by measurements of urinary iodine, thyroglobulin, serum TSH, and thyroid size.
Adequate iodine intake during pregnancy is important for fetal development. Iodine deficiency is associated with goiter and hypothyroidism. Severe iodine deficiency during pregnancy is associated with cretinism.
There is evidence that mild to moderate iodine deficiency can cause impaired cognitive development and that correcting the iodine deficiency can significantly improve cognitive function.
CASE FOLLOW-UP
This patient had been following a strict vegan diet with very little intake of iodized salt. Her dietary history and the presence of goiter suggested iodine deficiency. She was instructed to take an iodine supplement 150 μg/day to meet her daily requirement. After 2 months of iodine supplementation, her urine iodine concentration had increased to 58 μg/L. She remained biochemically euthyroid.
- Zimmermann MB. Iodine deficiency. Endocr Rev 2009; 30:376–408.
- Pearce EN, Andersson M, Zimmermann MB. Global iodine nutrition: where do we stand in 2013? Thyroid 2013; 23:523–528.
- Huang SW. Seafood and iodine: an analysis of a medical myth. Allergy Asthma Proc 2005; 26:468–469.
- US Census Bureau. The 2012 Statistical abstract. Health and nutrition. www.census.gov/prod/2011pubs/12statab/health.pdf. Accessed December 1, 2016.
- Pearce EN, Pino S, He X, Bazrafshan HR, Lee SL, Braverman LE. Sources of dietary iodine: bread, cows’ milk, and infant formula in the Boston area. J Clin Endocrinol Metab 2004; 89:3421–3424.
- Salt Institute. Production and Industry. www.saltinstitute.org/salt-101/production-industry. Accessed September 20, 2016.
- Dunn JT. Guarding our nation's thyroid health. J Clin Endocrinol Metab 2002; 87:486–488.
- Institute of Medicine (US) Panel on Micronutrients. Dietary reference intakes for vitamin A, vitamin K, arsenic, boron, chromium, copper, iodine, iron, manganese, molybdenum, nickel, silicon, vanadium, and zinc. Washington, DC: National Academy Press; 2001.
- World Health Organization (WHO). Assessment of iodine deficiency disorders and monitoring their elimination: a guide for programme managers. www.who.int/nutrition/publications/micronutrients/iodine_deficiency/9789241595827/en. Accessed December 1, 2016.
- Caldwell KL, Pan Y, Mortensen ME, Makhmudov A, Merrill L, Moye J. Iodine status in pregnant women in the National Children's Study and in US women (15–44 years), National Health and Nutrition Examination Survey 2005–2010. Thyroid 2013; 23:927–937.
- Public Health Committee of the American Thyroid Association; Becker DV, Braverman LE, Delange F, et al. Iodine supplementation for pregnancy and lactation—United States and Canada: recommendations of the American Thyroid Association. Thyroid 2006; 16:949–951.
- Leung AM, Pearce EN, Braverman LE. Iodine nutrition in pregnancy and lactation. Endocrinol Metab Clin North Am 2011; 40:765–777.
- Pearce EN. Iodine in pregnancy: is salt iodization enough? J Clin Endocrinol Metab 2008; 93:2466–2468.
- Younes-Rapozo V, Berendonk J, Savignon T, Manhaes AC, Barradas PC. Thyroid hormone deficiency changes the distribution of oligodendrocyte/myelin markers during oligodendroglial differentiation in vitro. Int J Dev Neurosci 2006; 24:445–453.
- Azizi F, Smyth P. Breastfeeding and maternal and infant iodine nutrition. Clin Endocrinol (Oxf) 2009; 70:803–809.
- Delange F. Iodine requirements during pregnancy, lactation and the neonatal period and indicators of optimal iodine nutrition. Public Health Nutr 2007; 10:1571–1583.
- Semba RD, Delange F. Iodine in human milk: perspectives for infant health. Nutr Rev 2001; 59:269–278.
- Leung AM, Pearce EN, Braverman LE. Iodine content of prenatal multivitamins in the United States. N Engl J Med 2009; 360:939–940.
- Tonacchera M, Agretti P, Chiovato L, et al. Activating thyrotropin receptor mutations are present in nonadenomatous hyperfunctioning nodules of toxic or autonomous multinodular goiter. J Clin Endocrinol Metab 2000; 85:2270–2274.
- Medeiros-Neto G, Camargo RY, Tomimori EK. Approach to and treatment of goiters. Med Clin North Am 2012; 96:351–368.
- Heidemann P, Stubbe P. Serum 3,5,3'-triiodothyronine, thyroxine, and thyrotropin in hypothyroid infants with congenital goiter and the response to iodine. J Clin Endocrinol Metab 1978; 47:189–192.
- Patel YC, Pharoah PO, Hornabrook RW, Hetzel BS. Serum triiodothyronine, thyroxine and thyroid-stimulating hormone in endemic goiter: a comparison of goitrous and nongoitrous subjects in New Guinea. J Clin Endocrinol Metab 1973; 37:783–789.
- Boyages SC, Halpern JP. Endemic cretinism: toward a unifying hypothesis. Thyroid 1993; 3:59–69.
- Chen ZP, Hetzel BS. Cretinism revisited. Best Pract Res Clin Endocrinol Metab 2010; 24:39–50.
- Hynes KL, Otahal P, Hay I, Burgess JR. Mild iodine deficiency during pregnancy is associated with reduced educational outcomes in the offspring: 9-year follow-up of the gestational iodine cohort. J Clin Endocrinol Metab 2013; 98:1954–1962.
- Bleichrodt N, Born MP. A metaanalysis of research on iodine and its relationship to cognitive development. In: Stanbury JB, ed. The Damaged Brain of Iodine Deficiency. 1st ed. New York, NY: Cognizant Communication; 1994:195–200.
- Melse-Boonstra A, Jaiswal N. Iodine deficiency in pregnancy, infancy and childhood and its consequences for brain development. Best Pract Res Clin Endocrinol Metab 2010; 24:29–38.
- Gordon RC, Rose MC, Skeaff SA, Gray AR, Morgan KM, Ruffman T. Iodine supplementation improves cognition in mildly iodine-deficient children. Am J Clin Nutr 2009; 90:1264–1271.
- Konig F, Andersson M, Hotz K, Aeberli I, Zimmermann MB. Ten repeat collections for urinary iodine from spot samples or 24-hour samples are needed to reliably estimate individual iodine status in women. J Nutr 2011; 141:2049–2054.
- Vejbjerg P, Knudsen N, Perrild H, et al. Estimation of iodine intake from various urinary iodine measurements in population studies. Thyroid 2009; 19:1281–1286.
- Zimmermann MB, Hess SY, Adou P, Toresanni T, Wegmuller R, Hurrell RF. Thyroid size and goiter prevalence after introduction of iodized salt: a 5-y prospective study in schoolchildren in Cote d’Ivoire. Am J Clin Nutr 2003; 77:663–667.
- Zimmermann MB, de Benoist B, Corigliano S, et al. Assessment of iodine status using dried blood spot thyroglobulin: development of reference material and establishment of an international reference range in iodine-sufficient children. J Clin Endocrinol Metab 2006; 91:4881–4887.
- Zimmermann MB, Aeberli I, Andersson M, et al. Thyroglobulin is a sensitive measure of both deficient and excess iodine intakes in children and indicates no adverse effects on thyroid function in the UIC range of 100-299 mug/L: a UNICEF/ICCIDD study group report. J Clin Endocrinol Metab 2013; 98:1271–1280.
- Thilly CH, Delange F, Lagasse R, et al. Fetal hypothyroidism and maternal thyroid status in severe endemic goiter. J Clin Endocrinol Metab 1978; 47:354–360.
- Sullivan KM, May W, Nordenberg D, Houston R, Maberly GF. Use of thyroid stimulating hormone testing in newborns to identify iodine deficiency. J Nutr 1997; 127:55–58.
- Delange F. Neonatal thyroid screening as a monitoring tool for the control of iodine deficiency. Acta Paediatr Suppl 1999; 88:21–24.
- Li M, Eastman CJ. Neonatal TSH screening: is it a sensitive and reliable tool for monitoring iodine status in populations? Best Pract Res Clin Endocrinol Metab 2010; 24:63–75.
- Zimmermann MB, Moretti D, Chaouki N, Torresani T. Development of a dried whole-blood spot thyroglobulin assay and its evaluation as an indicator of thyroid status in goitrous children receiving iodized salt. Am J Clin Nutr 2003; 77:1453–1458.
- Brahmbhatt S, Brahmbhatt RM, Boyages SC. Thyroid ultrasound is the best prevalence indicator for assessment of iodine deficiency disorders: a study in rural/tribal schoolchildren from Gujarat (Western India). Eur J Endocrinol 2000; 143:37–46.
- Moradi M, Hashemipour M, Akbari S, Kor Z, Mirbod SA, Kooshanmehr MR. Ultrasonographic evaluation of the thyroid gland volume among 8-15-year-old children in Isfahan, Iran. Adv Biomed Res 2014; 3:9.
A 65-year-old woman is found to have a goiter. She is clinically euthyroid. She is a strict vegan and only uses noniodized Himalayan salt for cooking. Her thyroid gland is diffusely enlarged with no nodules. The estimated weight of the thyroid gland is 50 g (normal 10–20 g) based on ultrasonography. Her thyroid-stimulating hormone (TSH) level is 2.95 mU/L (reference range 0.5–5 mU/L), and her free thyroxine level is 0.8 ng/dL (0.7–1.8 ng/dL). Testing for TSH receptor antibody is negative. Her 24-hour urine iodine is undetectable (urine iodine concentration < 10 μg/L with urine volume 3,175 mL). What may be the cause of her goiter?
Iodine is an essential element needed for the production of thyroid hormone, which controls metabolism and plays a major role in fetal neurodevelopment. Its ionized form is called iodide. Iodine deficiency results in impairment of thyroid hormone synthesis and may lead to several undesirable consequences. Physicians should be aware of the risks iodine deficiency poses, especially during pregnancy, and should be familiar with approaches to testing and current indications for iodine supplementation.
SOURCES OF IODINE AND SALT IODIZATION
The major environmental source of iodine is the ocean. Elemental iodine in the ocean volatilizes into the atmosphere and returns to the soil by rain. The effects of glaciation, flooding, and leaching into soil have resulted in the variable geographic distribution of iodine. Mountainous areas (eg, the Alps, Andes, Himalayas) and areas with frequent flooding typically have iodine-deficient soil due to slow iodine cycling.1 Seafood is a good source of iodine because marine plants and animals concentrate iodine from seawater. The iodine content of other foods varies widely, depending on the source and any additives.
In the United States, the major sources of dietary iodine are dairy products (due to livestock iodine supplements and use of iodophors for cleaning milk udders) and iodized salt.1,2 Seafood contains a higher amount of iodine by weight than dairy products but is consumed far less than dairy.3,4 Further, the iodine content of milk can range from 88 to 168 μg per 250 mL (about 1 cup), depending on the product manufacturer. Also, iodine content is often omitted from the food label. Even if it is reported, the package labeling may not accurately predict the iodine content.5
Less common sources of iodine are radiographic contrast, bread with iodate dough conditioners, red food coloring (erythrosine), and drugs such as amiodarone.1
Using iodized salt is an effective and stable way to ensure adequate iodine intake. In the United States, only table salt is iodized, and the salt typically used in processed food has only minimal iodine content.6 Nearly 70% of the salt we ingest is from processed food. Table salt provides only 15% of dietary salt intake, and only 70% of consumers choose iodized salt for home cooking.7
IODINE REQUIREMENTS
Daily requirements of iodine suggested by the World Health Organization (WHO) and by the US Institute of Medicine are in the range of 90 to 150 µg/day.8,9 The iodine requirement is higher in pregnancy (220–250 µg/day) because of increased maternal thyroid hormone production required to maintain euthyroidism and increased renal iodine clearance, and it is even higher in lactating women (250–290 µg/day).
IODINE STATUS IN POPULATIONS
Since the establishment of universal salt iodization programs under the influence of the WHO and the International Council for Control of Iodine Deficiency Disorders (ICCIDD) in 1990, global iodine status has continued to improve. Yet only 70% of households worldwide currently have access to adequately iodized salt, because many countries lack a national program for iodine supplementation. The population of the United States was historically iodine-deficient, but since the introduction of salt iodization in the 1920s, the iodine status in the United States has been considered adequate.1
The WHO defines iodine status for a population by the median spot urinary iodine concentration. Because a urinary iodine concentration of 100 μg/L represents an iodine intake of about 150 μg/day, the WHO uses a median urinary iodine concentration of 100 to 199 μg/L to define adequate iodine intake for a nonpregnant population.9
The National Health and Nutrition Examination Survey (NHANES) found that the median urinary iodine concentration decreased by more than 50% from the 1970s to the 1990s, indicating declining iodine status in the US population.2 Of particular concern, the percentage of women of childbearing age with moderate iodine deficiency increased from 4% to 15% over this period.2 Still, the NHANES survey in 2009–2010 indicated that the overall US population is still iodine-sufficient (median urinary iodine concentration 144 μg/L).10 The decline in the US iodine status may be due to reduction of iodine content in dairy products, increased use of noniodized salt by the food industry, and recommendations to avoid salt for blood pressure control.
Although US iodine status has been considered generally adequate, iodine intake varies greatly across the population. Vegans tend to have iodine-deficient diets, while kelp consumers may have excessive iodine intake.11 Individuals with lactose intolerance are at risk of iodine deficiency, given that dairy products are a major source of iodine in the United States. Physicians should be aware of these risk factors for iodine deficiency.
PREGNANCY AND LACTATION
It is crucial to maintain euthyroidism during pregnancy. In early gestation, maternal thyroid hormone production increases 50% due to an increase in thyroid-binding globulin and stimulation by human chorionic gonadotropin. The glomerular filtration rate increases by 30% to 50% during pregnancy, thus increasing renal iodine clearance. Fetal thyroid hormone production increases during the second half of pregnancy, further contributing to increased maternal iodine requirements because iodine readily crosses the placenta.12
Women with sufficient iodine intake before and during pregnancy generally have adequate intrathyroidal iodine storage and can adapt to the increased demand for thyroid hormone throughout gestation. But in the setting of even mild iodine deficiency, total body iodine stores decline gradually from the first to third trimester of pregnancy.13
The fetal thyroid gland does not begin to concentrate iodine until 10 to 12 weeks of gestation and is not controlled by TSH until the full development of the pituitary-portal vascular system at 20 weeks of gestation.12 Therefore, the fetus relies on maternal thyroid hormone during this critical stage of neurodevelopment. Thyroid hormone is essential for oligodendrocyte differentiation and myelin distribution14 as well as fetal neuronal proliferation and migration in the first and second trimesters. Iodine deficiency leading to maternal hypothyroidism can result in irreversible fetal brain damage.
Because of the greater requirement during pregnancy, the WHO recommends using a median urinary iodine concentration of 150 to 249 μg/L to define a population that has no iodine deficiency.9 The NHANES data from 2007 to 2010 showed that pregnant US women were mildly iodine-deficient (median urinary iodine concentration 135 μg/L),10 and the National Children’s Study of 501 pregnant US women during the third trimester in 2009 to 2010 showed they had adequate iodine intake (median urinary iodine concentration 167 μg/L). Interestingly, pregnant non-Hispanic blacks were the only ethnic group with a median urinary iodine concentration less than 150 μg/L, suggesting that race or ethnicity is a predictor of iodine status in pregnant women.10
Iodine requirements during lactation
During lactation, thyroid hormone production and renal iodine clearance return to the prepregnancy state. However, a significant amount of iodine is excreted into breast milk at a concentration 20 to 50 times greater than that in plasma.15 It is recommended that lactating women continue high iodine intake to ensure sufficient iodine in breast milk to build reserves in the newborn’s thyroid gland.
The iodine requirement during lactation is 225 to 350 μg/day.16 Breast milk containing 100 to 200 μg/L of iodine appears to provide adequate iodine to meet Institute of Medicine recommendations for infants.17 The amount of iodine excreted into breast milk depends on maternal iodine intake. In the setting of iodine sufficiency, the iodine content of breast milk is 150 to 180 μg/L, but it is much lower (9–32 μg/L) in women from iodine-deficient areas, eg, the “goiter belt,” which included the Great Lakes, the Appalachians, and northwestern states. While iodized salt has virtually eliminated the goiter belt, the risk of iodine deficiency remains for people who avoid iodized salt and dairy.15
To ensure adequate iodine intake, the American Thyroid Association recommends that women receive iodine supplementation daily during pregnancy and lactation.11 However, the iodine content of prenatal multivitamins is currently not mandated in the United States. Only half of marketed prenatal vitamins in the United States contain iodine, in the form of either potassium iodide or kelp. Though most iodine-containing products claim to contain at least 150 μg of iodine per daily dose, when measured, the actual iodine content varied between 33 and 610 μg.18
CONSEQUENCES OF IODINE DEFICIENCY
Goiter
Goiter in iodine-deficient areas is considered to be an adaptation to chronic iodine deficiency. Low iodine intake leads to reduced thyroid hormone production, which in turn stimulates TSH secretion from the pituitary. TSH increases iodine uptake by the thyroid, stimulates thyroid growth, and leads to goiter development.
Initially, goiter is characterized by diffuse thyroid enlargement, but over time it may become nodular from progressive accumulation of new thyroid follicles. Goiter in children from iodine-deficient areas is diffusely enlarged, whereas in older adults it tends to be multinodular.
Iodine deficiency and chronic TSH stimulation may play a role in TSH receptor-activating mutations of thyroid follicles. These “gain-of-function” mutations are more common in the glands of patients with goiter in areas of iodine deficiency but are relatively rare in areas of iodine sufficiency.19 Toxic multinodular goiter may eventually develop, and hyperthyroidism may occur if iodine deficiency is not severe.
Goiter generally does not cause obstructive symptoms, since the thyroid usually grows outward. However, a very large goiter may descend to the thoracic inlet and compress the trachea and esophagus. The obstructive effect of a large goiter can be demonstrated by having a patient raise the arms adjacent to the face (the Pemberton maneuver). Signs suggesting obstruction are engorged neck veins, facial plethora, increased dyspnea, and stridor during the maneuver. Computed tomography of the neck and upper thorax may provide information on the degree of tracheal compression.20
Hypothyroidism
A normal or low triiodothyronine (T3), a low serum thyroxine (T4), and a variably elevated TSH are features of thyroid function tests in iodine deficiency.11,21,22 As long as daily iodine intake exceeds 50 μg/day, the absolute uptake of iodine by the thyroid gland usually remains adequate to maintain euthyroidism. Below 50 μg/day, iodine storage in the thyroid becomes depleted, leading to hypothyroidism.1
The clinical manifestations of hypothyroidism from iodine deficiency are similar to those of hypothyroidism from other causes. Because of thyroid hormone’s role in neural and somatic development, the manifestations of hypothyroidism differ among age groups (Table 1).
Cretinism
Before the development of fetal thyroid tissue in the 10th to 12th week of gestation, the fetus is dependent on maternal thyroid hormone, which crosses the placenta to support general and neural development. Iodine deficiency leading to maternal hypothyroidism (in early gestation) or inadequate fetal thyroid hormone production (in late gestation) may result in various degrees of mental retardation or lower than expected IQ.
Severe iodine deficiency during gestation typically results in cretinism, characterized by severe mental retardation accompanied by other neurologic or physical defects. Cretinism is divided into two subtypes according to clinical manifestations (neurologic and myxomatous cretinism; Table 2), which may reflect the different timing of intrauterine insult to the developing fetal nervous system and whether the iodine deficiency continues into the postnatal period. Both types can be prevented by adequate maternal iodine intake before and during pregnancy.23,24
Although mild gestational iodine deficiency does not result in cretinism, it nevertheless has an adverse impact on fetal neurodevelopment and subsequent functioning. Children of mothers with mild gestational iodine deficiency were found to have reductions in spelling, grammar, and English literacy performance despite growing up in iodine-replete environments.25
Impaired cognitive development
Reduction in IQ has been noted in affected youth from regions of severe and mild iodine deficiency. A meta-analysis of studies relating iodine deficiency to cognitive development suggested that chronic moderate to severe iodine deficiency reduced expected average IQ by about 13.5 points.26
The effects of mild iodine deficiency during childhood are more difficult to quantify. The results of one study suggested that mild iodine deficiency was associated with subtle neurodevelopmental deficits and that iodine supplementation might improve cognitive function in mildly iodine-deficient children.27
In a 2009 randomized, placebo-controlled study in New Zealand, 184 children ages 10 to 13 with mild iodine deficiency (median urinary iodine concentration of 63 μg/L) received iodine supplementation (150 μg/day) or placebo for 28 weeks. Iodine supplementation increased the median urinary iodine concentration to 145 μg/L and significantly improved perceptual reasoning measures and overall cognitive score compared with placebo.28
These findings suggest that correcting mild iodine deficiency in children could improve certain components of cognition. More research is needed to understand the effects of mild iodine deficiency and iodine supplementation on cognitive function.
ASSESSING IODINE STATUS
The diagnosis of iodine deficiency is based on clinical and laboratory assessments. Clinical manifestations compatible with iodine deficiency and careful history-taking focused on the patient’s dietary iodine intake and geographic data are keys to the diagnosis.
Four main methods are used to assess iodine status at a population level: urinary iodine, serum thyroglobulin, serum TSH, and thyroid size. Urinary iodine is a sensitive marker for recent iodine intake (within days); thyroglobulin represents iodine nutrition over a period of months and thyroid size over a period of years.1
Urinary iodine
Most dietary iodine is excreted into the urine within 24 hours of ingestion, and the 24-hour urinary iodine is considered a reference standard for the measurement of individual daily iodine intake. However, the process of collection is cumbersome, and the 24-hour urinary iodine can vary from day to day in the same person, depending on the amount of iodine ingested.
A study in healthy women from an iodine-sufficient area suggested that 10 repeated 24-hour urine collections estimated the person’s iodine status at a precision of 20% because of variable daily iodine intake.29 Therefore, when necessary, several 24-hour urine iodine determinations should be performed.
A single, random, spot urinary iodine is expressed as the urinary iodine concentration and is affected by the amount of iodine and fluids the individual ingests in a day, thus resulting in high variation both within an individual person and between individuals. Expressing the urinary iodine concentration as the ratio of urine iodine to creatinine is useful in correcting for the influence of fluid intake. The ratio of urine iodine to creatinine can be used to estimate 24-hour urine iodine with the following formula: urine iodine (μg/L)/creatinine (g/L)× age- and sex-specific estimated 24-hour creatinine excretion (g/day). Another clinical use of the spot urine iodine is to screen for exposure to a large amount of iodine from a source such as radiographic contrast.30
Although individual urine iodine excretion and urine volume can vary from day to day, this variation tends to even out in a large number of samples. In study populations of at least 500, the median value of the spot urinary iodine concentration is considered a reliable measure of iodine intake in that population.30 The spot urine iodine test is convenient, making it the test of choice to study iodine status in a large cohort. The WHO recommends using the median value of the spot urine iodine to evaluate the iodine status of a population.9
Thyroglobulin
Thyroglobulin is a thyroid-specific protein involved in the synthesis of thyroid hormone. Small amounts can be detected in the blood of healthy people. In the absence of thyroid damage, the amount of serum thyroglobulin depends on thyroid cell mass and TSH stimulation. The serum level is elevated in iodine deficiency as a result of chronic TSH stimulation and thyroid hyperplasia. Thus, thyroglobulin can serve as a marker of iodine deficiency.
Serum thyroglobulin assays have been adapted for use on dried whole-blood spots, which require only a few drops of whole blood collected on filter paper and left to air-dry. The results of the dried whole-blood assay correlate closely with those of the serum assay.31 An established international dried whole-blood thyroglobulin reference range for iodine-sufficient school-age children is 4 to 40 μg/L.32 A median level of less than 13 μg/L in school-age children indicates iodine sufficiency in the population.33
Thyroid-stimulating hormone
Iodine deficiency lowers serum T4, which in turn leads to increased serum TSH. Therefore, iodine-deficient populations generally have higher TSH than iodine-sufficient groups. However, the TSH values in older children and adults with iodine deficiency are not significantly different from values of those with adequate iodine intake. Therefore, TSH is not a practical marker of iodine deficiency in the general population.
In contrast, TSH in newborns is a reasonable indicator of population iodine status. The newborn thyroid has limited iodine stores compared with that of an adult and hence a much higher iodine turnover rate. TSH from the cord blood is markedly elevated in newborns of mothers with moderate to severe iodine deficiency.34 A high prevalence of newborns with elevated TSH should therefore reflect iodine deficiency in the area where the mothers of the newborns live.
TSH is now routinely checked in newborns to screen for congenital hypothyroidism. TSH is typically checked 2 to 5 days after delivery to avoid confusion with transient physiologic TSH elevation, which occurs within a few hours after birth and decreases rapidly in 24 hours. The WHO has proposed that a more than 3% prevalence of newborns with TSH values higher than 5 mU/L from blood samples collected 3 to 4 days after birth indicates iodine deficiency in a population.1 This threshold appears to correlate well with the iodine status of the population defined by the WHO’s median urinary iodine concentration.35,36
But several other factors can influence the measurement of newborn TSH, such as prematurity, time of blood collection, maternal or newborn exposure to iodine-containing antiseptics, and the TSH assay methodology. These potential confounding factors limit the role of neonatal TSH as a reliable monitoring tool for iodine deficiency.35,37
Thyroid size
The size of the thyroid gland varies inversely with iodine intake. Thyroid size can be assessed by either palpation or ultrasonography, with the latter being more sensitive. The goiter rate in school-age children can be used to determine the severity of iodine deficiency in the population (Table 3). A goiter rate of 5% or more in school-age children suggests the presence of iodine deficiency in the community.
Although thyroid size is easy to estimate by palpation, it has low sensitivity and specificity to detect iodine deficiency and high interobserver variation. Thyroid ultrasonography provides a more precise measurement of thyroid gland volume. Zimmermann et al38 provided reference data on thyroid volume stratified by age, sex, and body surface area of school-age children in iodine-sufficient areas.38 Results of ultrasonography in a population is then compared with these reference data. The higher the percentage of the population with thyroid volume exceeding the 97th percentile of the reference range, the more severe the iodine deficiency. However, the WHO does not specify how to grade the degree of iodine deficiency based on the thyroid volume obtained with ultrasonography. Follow-up studies showed no significant correlation between urinary iodine concentration and thyroid size.39,40
Thyroid size decreases slowly after iodine repletion. Therefore, the goiter rate may remain high for several years after iodine supplementation begins.1,9
TREATMENT AND PREVENTION
Treatment of iodine deficiency should be instituted at the levels recommended by the Institute of Medicine and the WHO. The tolerable upper intake levels for iodine are outlined in Table 4. In a nonpregnant adult, 150 μg/day is sufficient for normal thyroid function. Iodine intake should be higher for pregnant and lactating women (250 μg/day according to the WHO recommendation).
Iodine supplementation is easily achieved by using iodized salt or an iodine-containing daily multivitamin.
In patients with overt hypothyroidism from iodine deficiency, we recommend initiating levothyroxine treatment along with iodine supplementation to restore euthyroidism, with consideration of possible interruption in 6 to 12 months when the urine iodine has normalized and goiter size has decreased. Thyroid function should be reassessed 4 to 6 weeks after discontinuation of levothyroxine.
At the population level, iodine deficiency can usually be prevented by iodization of food products or the water supply. In developing countries where salt iodization is not practical, iodine deficiency has been eradicated by adding iodine drops to well water or by injecting people with iodized oil.
TAKE-HOME POINTS
Iodine is essential for thyroid hormone synthesis. It can be obtained by eating iodine-containing foods or by using iodized salt. The WHO classifies iodine deficiency based on the median urinary iodine concentration. Iodine nutrition at the community level is best assessed by measurements of urinary iodine, thyroglobulin, serum TSH, and thyroid size.
Adequate iodine intake during pregnancy is important for fetal development. Iodine deficiency is associated with goiter and hypothyroidism. Severe iodine deficiency during pregnancy is associated with cretinism.
There is evidence that mild to moderate iodine deficiency can cause impaired cognitive development and that correcting the iodine deficiency can significantly improve cognitive function.
CASE FOLLOW-UP
This patient had been following a strict vegan diet with very little intake of iodized salt. Her dietary history and the presence of goiter suggested iodine deficiency. She was instructed to take an iodine supplement 150 μg/day to meet her daily requirement. After 2 months of iodine supplementation, her urine iodine concentration had increased to 58 μg/L. She remained biochemically euthyroid.
A 65-year-old woman is found to have a goiter. She is clinically euthyroid. She is a strict vegan and only uses noniodized Himalayan salt for cooking. Her thyroid gland is diffusely enlarged with no nodules. The estimated weight of the thyroid gland is 50 g (normal 10–20 g) based on ultrasonography. Her thyroid-stimulating hormone (TSH) level is 2.95 mU/L (reference range 0.5–5 mU/L), and her free thyroxine level is 0.8 ng/dL (0.7–1.8 ng/dL). Testing for TSH receptor antibody is negative. Her 24-hour urine iodine is undetectable (urine iodine concentration < 10 μg/L with urine volume 3,175 mL). What may be the cause of her goiter?
Iodine is an essential element needed for the production of thyroid hormone, which controls metabolism and plays a major role in fetal neurodevelopment. Its ionized form is called iodide. Iodine deficiency results in impairment of thyroid hormone synthesis and may lead to several undesirable consequences. Physicians should be aware of the risks iodine deficiency poses, especially during pregnancy, and should be familiar with approaches to testing and current indications for iodine supplementation.
SOURCES OF IODINE AND SALT IODIZATION
The major environmental source of iodine is the ocean. Elemental iodine in the ocean volatilizes into the atmosphere and returns to the soil by rain. The effects of glaciation, flooding, and leaching into soil have resulted in the variable geographic distribution of iodine. Mountainous areas (eg, the Alps, Andes, Himalayas) and areas with frequent flooding typically have iodine-deficient soil due to slow iodine cycling.1 Seafood is a good source of iodine because marine plants and animals concentrate iodine from seawater. The iodine content of other foods varies widely, depending on the source and any additives.
In the United States, the major sources of dietary iodine are dairy products (due to livestock iodine supplements and use of iodophors for cleaning milk udders) and iodized salt.1,2 Seafood contains a higher amount of iodine by weight than dairy products but is consumed far less than dairy.3,4 Further, the iodine content of milk can range from 88 to 168 μg per 250 mL (about 1 cup), depending on the product manufacturer. Also, iodine content is often omitted from the food label. Even if it is reported, the package labeling may not accurately predict the iodine content.5
Less common sources of iodine are radiographic contrast, bread with iodate dough conditioners, red food coloring (erythrosine), and drugs such as amiodarone.1
Using iodized salt is an effective and stable way to ensure adequate iodine intake. In the United States, only table salt is iodized, and the salt typically used in processed food has only minimal iodine content.6 Nearly 70% of the salt we ingest is from processed food. Table salt provides only 15% of dietary salt intake, and only 70% of consumers choose iodized salt for home cooking.7
IODINE REQUIREMENTS
Daily requirements of iodine suggested by the World Health Organization (WHO) and by the US Institute of Medicine are in the range of 90 to 150 µg/day.8,9 The iodine requirement is higher in pregnancy (220–250 µg/day) because of increased maternal thyroid hormone production required to maintain euthyroidism and increased renal iodine clearance, and it is even higher in lactating women (250–290 µg/day).
IODINE STATUS IN POPULATIONS
Since the establishment of universal salt iodization programs under the influence of the WHO and the International Council for Control of Iodine Deficiency Disorders (ICCIDD) in 1990, global iodine status has continued to improve. Yet only 70% of households worldwide currently have access to adequately iodized salt, because many countries lack a national program for iodine supplementation. The population of the United States was historically iodine-deficient, but since the introduction of salt iodization in the 1920s, the iodine status in the United States has been considered adequate.1
The WHO defines iodine status for a population by the median spot urinary iodine concentration. Because a urinary iodine concentration of 100 μg/L represents an iodine intake of about 150 μg/day, the WHO uses a median urinary iodine concentration of 100 to 199 μg/L to define adequate iodine intake for a nonpregnant population.9
The National Health and Nutrition Examination Survey (NHANES) found that the median urinary iodine concentration decreased by more than 50% from the 1970s to the 1990s, indicating declining iodine status in the US population.2 Of particular concern, the percentage of women of childbearing age with moderate iodine deficiency increased from 4% to 15% over this period.2 Still, the NHANES survey in 2009–2010 indicated that the overall US population is still iodine-sufficient (median urinary iodine concentration 144 μg/L).10 The decline in the US iodine status may be due to reduction of iodine content in dairy products, increased use of noniodized salt by the food industry, and recommendations to avoid salt for blood pressure control.
Although US iodine status has been considered generally adequate, iodine intake varies greatly across the population. Vegans tend to have iodine-deficient diets, while kelp consumers may have excessive iodine intake.11 Individuals with lactose intolerance are at risk of iodine deficiency, given that dairy products are a major source of iodine in the United States. Physicians should be aware of these risk factors for iodine deficiency.
PREGNANCY AND LACTATION
It is crucial to maintain euthyroidism during pregnancy. In early gestation, maternal thyroid hormone production increases 50% due to an increase in thyroid-binding globulin and stimulation by human chorionic gonadotropin. The glomerular filtration rate increases by 30% to 50% during pregnancy, thus increasing renal iodine clearance. Fetal thyroid hormone production increases during the second half of pregnancy, further contributing to increased maternal iodine requirements because iodine readily crosses the placenta.12
Women with sufficient iodine intake before and during pregnancy generally have adequate intrathyroidal iodine storage and can adapt to the increased demand for thyroid hormone throughout gestation. But in the setting of even mild iodine deficiency, total body iodine stores decline gradually from the first to third trimester of pregnancy.13
The fetal thyroid gland does not begin to concentrate iodine until 10 to 12 weeks of gestation and is not controlled by TSH until the full development of the pituitary-portal vascular system at 20 weeks of gestation.12 Therefore, the fetus relies on maternal thyroid hormone during this critical stage of neurodevelopment. Thyroid hormone is essential for oligodendrocyte differentiation and myelin distribution14 as well as fetal neuronal proliferation and migration in the first and second trimesters. Iodine deficiency leading to maternal hypothyroidism can result in irreversible fetal brain damage.
Because of the greater requirement during pregnancy, the WHO recommends using a median urinary iodine concentration of 150 to 249 μg/L to define a population that has no iodine deficiency.9 The NHANES data from 2007 to 2010 showed that pregnant US women were mildly iodine-deficient (median urinary iodine concentration 135 μg/L),10 and the National Children’s Study of 501 pregnant US women during the third trimester in 2009 to 2010 showed they had adequate iodine intake (median urinary iodine concentration 167 μg/L). Interestingly, pregnant non-Hispanic blacks were the only ethnic group with a median urinary iodine concentration less than 150 μg/L, suggesting that race or ethnicity is a predictor of iodine status in pregnant women.10
Iodine requirements during lactation
During lactation, thyroid hormone production and renal iodine clearance return to the prepregnancy state. However, a significant amount of iodine is excreted into breast milk at a concentration 20 to 50 times greater than that in plasma.15 It is recommended that lactating women continue high iodine intake to ensure sufficient iodine in breast milk to build reserves in the newborn’s thyroid gland.
The iodine requirement during lactation is 225 to 350 μg/day.16 Breast milk containing 100 to 200 μg/L of iodine appears to provide adequate iodine to meet Institute of Medicine recommendations for infants.17 The amount of iodine excreted into breast milk depends on maternal iodine intake. In the setting of iodine sufficiency, the iodine content of breast milk is 150 to 180 μg/L, but it is much lower (9–32 μg/L) in women from iodine-deficient areas, eg, the “goiter belt,” which included the Great Lakes, the Appalachians, and northwestern states. While iodized salt has virtually eliminated the goiter belt, the risk of iodine deficiency remains for people who avoid iodized salt and dairy.15
To ensure adequate iodine intake, the American Thyroid Association recommends that women receive iodine supplementation daily during pregnancy and lactation.11 However, the iodine content of prenatal multivitamins is currently not mandated in the United States. Only half of marketed prenatal vitamins in the United States contain iodine, in the form of either potassium iodide or kelp. Though most iodine-containing products claim to contain at least 150 μg of iodine per daily dose, when measured, the actual iodine content varied between 33 and 610 μg.18
CONSEQUENCES OF IODINE DEFICIENCY
Goiter
Goiter in iodine-deficient areas is considered to be an adaptation to chronic iodine deficiency. Low iodine intake leads to reduced thyroid hormone production, which in turn stimulates TSH secretion from the pituitary. TSH increases iodine uptake by the thyroid, stimulates thyroid growth, and leads to goiter development.
Initially, goiter is characterized by diffuse thyroid enlargement, but over time it may become nodular from progressive accumulation of new thyroid follicles. Goiter in children from iodine-deficient areas is diffusely enlarged, whereas in older adults it tends to be multinodular.
Iodine deficiency and chronic TSH stimulation may play a role in TSH receptor-activating mutations of thyroid follicles. These “gain-of-function” mutations are more common in the glands of patients with goiter in areas of iodine deficiency but are relatively rare in areas of iodine sufficiency.19 Toxic multinodular goiter may eventually develop, and hyperthyroidism may occur if iodine deficiency is not severe.
Goiter generally does not cause obstructive symptoms, since the thyroid usually grows outward. However, a very large goiter may descend to the thoracic inlet and compress the trachea and esophagus. The obstructive effect of a large goiter can be demonstrated by having a patient raise the arms adjacent to the face (the Pemberton maneuver). Signs suggesting obstruction are engorged neck veins, facial plethora, increased dyspnea, and stridor during the maneuver. Computed tomography of the neck and upper thorax may provide information on the degree of tracheal compression.20
Hypothyroidism
A normal or low triiodothyronine (T3), a low serum thyroxine (T4), and a variably elevated TSH are features of thyroid function tests in iodine deficiency.11,21,22 As long as daily iodine intake exceeds 50 μg/day, the absolute uptake of iodine by the thyroid gland usually remains adequate to maintain euthyroidism. Below 50 μg/day, iodine storage in the thyroid becomes depleted, leading to hypothyroidism.1
The clinical manifestations of hypothyroidism from iodine deficiency are similar to those of hypothyroidism from other causes. Because of thyroid hormone’s role in neural and somatic development, the manifestations of hypothyroidism differ among age groups (Table 1).
Cretinism
Before the development of fetal thyroid tissue in the 10th to 12th week of gestation, the fetus is dependent on maternal thyroid hormone, which crosses the placenta to support general and neural development. Iodine deficiency leading to maternal hypothyroidism (in early gestation) or inadequate fetal thyroid hormone production (in late gestation) may result in various degrees of mental retardation or lower than expected IQ.
Severe iodine deficiency during gestation typically results in cretinism, characterized by severe mental retardation accompanied by other neurologic or physical defects. Cretinism is divided into two subtypes according to clinical manifestations (neurologic and myxomatous cretinism; Table 2), which may reflect the different timing of intrauterine insult to the developing fetal nervous system and whether the iodine deficiency continues into the postnatal period. Both types can be prevented by adequate maternal iodine intake before and during pregnancy.23,24
Although mild gestational iodine deficiency does not result in cretinism, it nevertheless has an adverse impact on fetal neurodevelopment and subsequent functioning. Children of mothers with mild gestational iodine deficiency were found to have reductions in spelling, grammar, and English literacy performance despite growing up in iodine-replete environments.25
Impaired cognitive development
Reduction in IQ has been noted in affected youth from regions of severe and mild iodine deficiency. A meta-analysis of studies relating iodine deficiency to cognitive development suggested that chronic moderate to severe iodine deficiency reduced expected average IQ by about 13.5 points.26
The effects of mild iodine deficiency during childhood are more difficult to quantify. The results of one study suggested that mild iodine deficiency was associated with subtle neurodevelopmental deficits and that iodine supplementation might improve cognitive function in mildly iodine-deficient children.27
In a 2009 randomized, placebo-controlled study in New Zealand, 184 children ages 10 to 13 with mild iodine deficiency (median urinary iodine concentration of 63 μg/L) received iodine supplementation (150 μg/day) or placebo for 28 weeks. Iodine supplementation increased the median urinary iodine concentration to 145 μg/L and significantly improved perceptual reasoning measures and overall cognitive score compared with placebo.28
These findings suggest that correcting mild iodine deficiency in children could improve certain components of cognition. More research is needed to understand the effects of mild iodine deficiency and iodine supplementation on cognitive function.
ASSESSING IODINE STATUS
The diagnosis of iodine deficiency is based on clinical and laboratory assessments. Clinical manifestations compatible with iodine deficiency and careful history-taking focused on the patient’s dietary iodine intake and geographic data are keys to the diagnosis.
Four main methods are used to assess iodine status at a population level: urinary iodine, serum thyroglobulin, serum TSH, and thyroid size. Urinary iodine is a sensitive marker for recent iodine intake (within days); thyroglobulin represents iodine nutrition over a period of months and thyroid size over a period of years.1
Urinary iodine
Most dietary iodine is excreted into the urine within 24 hours of ingestion, and the 24-hour urinary iodine is considered a reference standard for the measurement of individual daily iodine intake. However, the process of collection is cumbersome, and the 24-hour urinary iodine can vary from day to day in the same person, depending on the amount of iodine ingested.
A study in healthy women from an iodine-sufficient area suggested that 10 repeated 24-hour urine collections estimated the person’s iodine status at a precision of 20% because of variable daily iodine intake.29 Therefore, when necessary, several 24-hour urine iodine determinations should be performed.
A single, random, spot urinary iodine is expressed as the urinary iodine concentration and is affected by the amount of iodine and fluids the individual ingests in a day, thus resulting in high variation both within an individual person and between individuals. Expressing the urinary iodine concentration as the ratio of urine iodine to creatinine is useful in correcting for the influence of fluid intake. The ratio of urine iodine to creatinine can be used to estimate 24-hour urine iodine with the following formula: urine iodine (μg/L)/creatinine (g/L)× age- and sex-specific estimated 24-hour creatinine excretion (g/day). Another clinical use of the spot urine iodine is to screen for exposure to a large amount of iodine from a source such as radiographic contrast.30
Although individual urine iodine excretion and urine volume can vary from day to day, this variation tends to even out in a large number of samples. In study populations of at least 500, the median value of the spot urinary iodine concentration is considered a reliable measure of iodine intake in that population.30 The spot urine iodine test is convenient, making it the test of choice to study iodine status in a large cohort. The WHO recommends using the median value of the spot urine iodine to evaluate the iodine status of a population.9
Thyroglobulin
Thyroglobulin is a thyroid-specific protein involved in the synthesis of thyroid hormone. Small amounts can be detected in the blood of healthy people. In the absence of thyroid damage, the amount of serum thyroglobulin depends on thyroid cell mass and TSH stimulation. The serum level is elevated in iodine deficiency as a result of chronic TSH stimulation and thyroid hyperplasia. Thus, thyroglobulin can serve as a marker of iodine deficiency.
Serum thyroglobulin assays have been adapted for use on dried whole-blood spots, which require only a few drops of whole blood collected on filter paper and left to air-dry. The results of the dried whole-blood assay correlate closely with those of the serum assay.31 An established international dried whole-blood thyroglobulin reference range for iodine-sufficient school-age children is 4 to 40 μg/L.32 A median level of less than 13 μg/L in school-age children indicates iodine sufficiency in the population.33
Thyroid-stimulating hormone
Iodine deficiency lowers serum T4, which in turn leads to increased serum TSH. Therefore, iodine-deficient populations generally have higher TSH than iodine-sufficient groups. However, the TSH values in older children and adults with iodine deficiency are not significantly different from values of those with adequate iodine intake. Therefore, TSH is not a practical marker of iodine deficiency in the general population.
In contrast, TSH in newborns is a reasonable indicator of population iodine status. The newborn thyroid has limited iodine stores compared with that of an adult and hence a much higher iodine turnover rate. TSH from the cord blood is markedly elevated in newborns of mothers with moderate to severe iodine deficiency.34 A high prevalence of newborns with elevated TSH should therefore reflect iodine deficiency in the area where the mothers of the newborns live.
TSH is now routinely checked in newborns to screen for congenital hypothyroidism. TSH is typically checked 2 to 5 days after delivery to avoid confusion with transient physiologic TSH elevation, which occurs within a few hours after birth and decreases rapidly in 24 hours. The WHO has proposed that a more than 3% prevalence of newborns with TSH values higher than 5 mU/L from blood samples collected 3 to 4 days after birth indicates iodine deficiency in a population.1 This threshold appears to correlate well with the iodine status of the population defined by the WHO’s median urinary iodine concentration.35,36
But several other factors can influence the measurement of newborn TSH, such as prematurity, time of blood collection, maternal or newborn exposure to iodine-containing antiseptics, and the TSH assay methodology. These potential confounding factors limit the role of neonatal TSH as a reliable monitoring tool for iodine deficiency.35,37
Thyroid size
The size of the thyroid gland varies inversely with iodine intake. Thyroid size can be assessed by either palpation or ultrasonography, with the latter being more sensitive. The goiter rate in school-age children can be used to determine the severity of iodine deficiency in the population (Table 3). A goiter rate of 5% or more in school-age children suggests the presence of iodine deficiency in the community.
Although thyroid size is easy to estimate by palpation, it has low sensitivity and specificity to detect iodine deficiency and high interobserver variation. Thyroid ultrasonography provides a more precise measurement of thyroid gland volume. Zimmermann et al38 provided reference data on thyroid volume stratified by age, sex, and body surface area of school-age children in iodine-sufficient areas.38 Results of ultrasonography in a population is then compared with these reference data. The higher the percentage of the population with thyroid volume exceeding the 97th percentile of the reference range, the more severe the iodine deficiency. However, the WHO does not specify how to grade the degree of iodine deficiency based on the thyroid volume obtained with ultrasonography. Follow-up studies showed no significant correlation between urinary iodine concentration and thyroid size.39,40
Thyroid size decreases slowly after iodine repletion. Therefore, the goiter rate may remain high for several years after iodine supplementation begins.1,9
TREATMENT AND PREVENTION
Treatment of iodine deficiency should be instituted at the levels recommended by the Institute of Medicine and the WHO. The tolerable upper intake levels for iodine are outlined in Table 4. In a nonpregnant adult, 150 μg/day is sufficient for normal thyroid function. Iodine intake should be higher for pregnant and lactating women (250 μg/day according to the WHO recommendation).
Iodine supplementation is easily achieved by using iodized salt or an iodine-containing daily multivitamin.
In patients with overt hypothyroidism from iodine deficiency, we recommend initiating levothyroxine treatment along with iodine supplementation to restore euthyroidism, with consideration of possible interruption in 6 to 12 months when the urine iodine has normalized and goiter size has decreased. Thyroid function should be reassessed 4 to 6 weeks after discontinuation of levothyroxine.
At the population level, iodine deficiency can usually be prevented by iodization of food products or the water supply. In developing countries where salt iodization is not practical, iodine deficiency has been eradicated by adding iodine drops to well water or by injecting people with iodized oil.
TAKE-HOME POINTS
Iodine is essential for thyroid hormone synthesis. It can be obtained by eating iodine-containing foods or by using iodized salt. The WHO classifies iodine deficiency based on the median urinary iodine concentration. Iodine nutrition at the community level is best assessed by measurements of urinary iodine, thyroglobulin, serum TSH, and thyroid size.
Adequate iodine intake during pregnancy is important for fetal development. Iodine deficiency is associated with goiter and hypothyroidism. Severe iodine deficiency during pregnancy is associated with cretinism.
There is evidence that mild to moderate iodine deficiency can cause impaired cognitive development and that correcting the iodine deficiency can significantly improve cognitive function.
CASE FOLLOW-UP
This patient had been following a strict vegan diet with very little intake of iodized salt. Her dietary history and the presence of goiter suggested iodine deficiency. She was instructed to take an iodine supplement 150 μg/day to meet her daily requirement. After 2 months of iodine supplementation, her urine iodine concentration had increased to 58 μg/L. She remained biochemically euthyroid.
- Zimmermann MB. Iodine deficiency. Endocr Rev 2009; 30:376–408.
- Pearce EN, Andersson M, Zimmermann MB. Global iodine nutrition: where do we stand in 2013? Thyroid 2013; 23:523–528.
- Huang SW. Seafood and iodine: an analysis of a medical myth. Allergy Asthma Proc 2005; 26:468–469.
- US Census Bureau. The 2012 Statistical abstract. Health and nutrition. www.census.gov/prod/2011pubs/12statab/health.pdf. Accessed December 1, 2016.
- Pearce EN, Pino S, He X, Bazrafshan HR, Lee SL, Braverman LE. Sources of dietary iodine: bread, cows’ milk, and infant formula in the Boston area. J Clin Endocrinol Metab 2004; 89:3421–3424.
- Salt Institute. Production and Industry. www.saltinstitute.org/salt-101/production-industry. Accessed September 20, 2016.
- Dunn JT. Guarding our nation's thyroid health. J Clin Endocrinol Metab 2002; 87:486–488.
- Institute of Medicine (US) Panel on Micronutrients. Dietary reference intakes for vitamin A, vitamin K, arsenic, boron, chromium, copper, iodine, iron, manganese, molybdenum, nickel, silicon, vanadium, and zinc. Washington, DC: National Academy Press; 2001.
- World Health Organization (WHO). Assessment of iodine deficiency disorders and monitoring their elimination: a guide for programme managers. www.who.int/nutrition/publications/micronutrients/iodine_deficiency/9789241595827/en. Accessed December 1, 2016.
- Caldwell KL, Pan Y, Mortensen ME, Makhmudov A, Merrill L, Moye J. Iodine status in pregnant women in the National Children's Study and in US women (15–44 years), National Health and Nutrition Examination Survey 2005–2010. Thyroid 2013; 23:927–937.
- Public Health Committee of the American Thyroid Association; Becker DV, Braverman LE, Delange F, et al. Iodine supplementation for pregnancy and lactation—United States and Canada: recommendations of the American Thyroid Association. Thyroid 2006; 16:949–951.
- Leung AM, Pearce EN, Braverman LE. Iodine nutrition in pregnancy and lactation. Endocrinol Metab Clin North Am 2011; 40:765–777.
- Pearce EN. Iodine in pregnancy: is salt iodization enough? J Clin Endocrinol Metab 2008; 93:2466–2468.
- Younes-Rapozo V, Berendonk J, Savignon T, Manhaes AC, Barradas PC. Thyroid hormone deficiency changes the distribution of oligodendrocyte/myelin markers during oligodendroglial differentiation in vitro. Int J Dev Neurosci 2006; 24:445–453.
- Azizi F, Smyth P. Breastfeeding and maternal and infant iodine nutrition. Clin Endocrinol (Oxf) 2009; 70:803–809.
- Delange F. Iodine requirements during pregnancy, lactation and the neonatal period and indicators of optimal iodine nutrition. Public Health Nutr 2007; 10:1571–1583.
- Semba RD, Delange F. Iodine in human milk: perspectives for infant health. Nutr Rev 2001; 59:269–278.
- Leung AM, Pearce EN, Braverman LE. Iodine content of prenatal multivitamins in the United States. N Engl J Med 2009; 360:939–940.
- Tonacchera M, Agretti P, Chiovato L, et al. Activating thyrotropin receptor mutations are present in nonadenomatous hyperfunctioning nodules of toxic or autonomous multinodular goiter. J Clin Endocrinol Metab 2000; 85:2270–2274.
- Medeiros-Neto G, Camargo RY, Tomimori EK. Approach to and treatment of goiters. Med Clin North Am 2012; 96:351–368.
- Heidemann P, Stubbe P. Serum 3,5,3'-triiodothyronine, thyroxine, and thyrotropin in hypothyroid infants with congenital goiter and the response to iodine. J Clin Endocrinol Metab 1978; 47:189–192.
- Patel YC, Pharoah PO, Hornabrook RW, Hetzel BS. Serum triiodothyronine, thyroxine and thyroid-stimulating hormone in endemic goiter: a comparison of goitrous and nongoitrous subjects in New Guinea. J Clin Endocrinol Metab 1973; 37:783–789.
- Boyages SC, Halpern JP. Endemic cretinism: toward a unifying hypothesis. Thyroid 1993; 3:59–69.
- Chen ZP, Hetzel BS. Cretinism revisited. Best Pract Res Clin Endocrinol Metab 2010; 24:39–50.
- Hynes KL, Otahal P, Hay I, Burgess JR. Mild iodine deficiency during pregnancy is associated with reduced educational outcomes in the offspring: 9-year follow-up of the gestational iodine cohort. J Clin Endocrinol Metab 2013; 98:1954–1962.
- Bleichrodt N, Born MP. A metaanalysis of research on iodine and its relationship to cognitive development. In: Stanbury JB, ed. The Damaged Brain of Iodine Deficiency. 1st ed. New York, NY: Cognizant Communication; 1994:195–200.
- Melse-Boonstra A, Jaiswal N. Iodine deficiency in pregnancy, infancy and childhood and its consequences for brain development. Best Pract Res Clin Endocrinol Metab 2010; 24:29–38.
- Gordon RC, Rose MC, Skeaff SA, Gray AR, Morgan KM, Ruffman T. Iodine supplementation improves cognition in mildly iodine-deficient children. Am J Clin Nutr 2009; 90:1264–1271.
- Konig F, Andersson M, Hotz K, Aeberli I, Zimmermann MB. Ten repeat collections for urinary iodine from spot samples or 24-hour samples are needed to reliably estimate individual iodine status in women. J Nutr 2011; 141:2049–2054.
- Vejbjerg P, Knudsen N, Perrild H, et al. Estimation of iodine intake from various urinary iodine measurements in population studies. Thyroid 2009; 19:1281–1286.
- Zimmermann MB, Hess SY, Adou P, Toresanni T, Wegmuller R, Hurrell RF. Thyroid size and goiter prevalence after introduction of iodized salt: a 5-y prospective study in schoolchildren in Cote d’Ivoire. Am J Clin Nutr 2003; 77:663–667.
- Zimmermann MB, de Benoist B, Corigliano S, et al. Assessment of iodine status using dried blood spot thyroglobulin: development of reference material and establishment of an international reference range in iodine-sufficient children. J Clin Endocrinol Metab 2006; 91:4881–4887.
- Zimmermann MB, Aeberli I, Andersson M, et al. Thyroglobulin is a sensitive measure of both deficient and excess iodine intakes in children and indicates no adverse effects on thyroid function in the UIC range of 100-299 mug/L: a UNICEF/ICCIDD study group report. J Clin Endocrinol Metab 2013; 98:1271–1280.
- Thilly CH, Delange F, Lagasse R, et al. Fetal hypothyroidism and maternal thyroid status in severe endemic goiter. J Clin Endocrinol Metab 1978; 47:354–360.
- Sullivan KM, May W, Nordenberg D, Houston R, Maberly GF. Use of thyroid stimulating hormone testing in newborns to identify iodine deficiency. J Nutr 1997; 127:55–58.
- Delange F. Neonatal thyroid screening as a monitoring tool for the control of iodine deficiency. Acta Paediatr Suppl 1999; 88:21–24.
- Li M, Eastman CJ. Neonatal TSH screening: is it a sensitive and reliable tool for monitoring iodine status in populations? Best Pract Res Clin Endocrinol Metab 2010; 24:63–75.
- Zimmermann MB, Moretti D, Chaouki N, Torresani T. Development of a dried whole-blood spot thyroglobulin assay and its evaluation as an indicator of thyroid status in goitrous children receiving iodized salt. Am J Clin Nutr 2003; 77:1453–1458.
- Brahmbhatt S, Brahmbhatt RM, Boyages SC. Thyroid ultrasound is the best prevalence indicator for assessment of iodine deficiency disorders: a study in rural/tribal schoolchildren from Gujarat (Western India). Eur J Endocrinol 2000; 143:37–46.
- Moradi M, Hashemipour M, Akbari S, Kor Z, Mirbod SA, Kooshanmehr MR. Ultrasonographic evaluation of the thyroid gland volume among 8-15-year-old children in Isfahan, Iran. Adv Biomed Res 2014; 3:9.
- Zimmermann MB. Iodine deficiency. Endocr Rev 2009; 30:376–408.
- Pearce EN, Andersson M, Zimmermann MB. Global iodine nutrition: where do we stand in 2013? Thyroid 2013; 23:523–528.
- Huang SW. Seafood and iodine: an analysis of a medical myth. Allergy Asthma Proc 2005; 26:468–469.
- US Census Bureau. The 2012 Statistical abstract. Health and nutrition. www.census.gov/prod/2011pubs/12statab/health.pdf. Accessed December 1, 2016.
- Pearce EN, Pino S, He X, Bazrafshan HR, Lee SL, Braverman LE. Sources of dietary iodine: bread, cows’ milk, and infant formula in the Boston area. J Clin Endocrinol Metab 2004; 89:3421–3424.
- Salt Institute. Production and Industry. www.saltinstitute.org/salt-101/production-industry. Accessed September 20, 2016.
- Dunn JT. Guarding our nation's thyroid health. J Clin Endocrinol Metab 2002; 87:486–488.
- Institute of Medicine (US) Panel on Micronutrients. Dietary reference intakes for vitamin A, vitamin K, arsenic, boron, chromium, copper, iodine, iron, manganese, molybdenum, nickel, silicon, vanadium, and zinc. Washington, DC: National Academy Press; 2001.
- World Health Organization (WHO). Assessment of iodine deficiency disorders and monitoring their elimination: a guide for programme managers. www.who.int/nutrition/publications/micronutrients/iodine_deficiency/9789241595827/en. Accessed December 1, 2016.
- Caldwell KL, Pan Y, Mortensen ME, Makhmudov A, Merrill L, Moye J. Iodine status in pregnant women in the National Children's Study and in US women (15–44 years), National Health and Nutrition Examination Survey 2005–2010. Thyroid 2013; 23:927–937.
- Public Health Committee of the American Thyroid Association; Becker DV, Braverman LE, Delange F, et al. Iodine supplementation for pregnancy and lactation—United States and Canada: recommendations of the American Thyroid Association. Thyroid 2006; 16:949–951.
- Leung AM, Pearce EN, Braverman LE. Iodine nutrition in pregnancy and lactation. Endocrinol Metab Clin North Am 2011; 40:765–777.
- Pearce EN. Iodine in pregnancy: is salt iodization enough? J Clin Endocrinol Metab 2008; 93:2466–2468.
- Younes-Rapozo V, Berendonk J, Savignon T, Manhaes AC, Barradas PC. Thyroid hormone deficiency changes the distribution of oligodendrocyte/myelin markers during oligodendroglial differentiation in vitro. Int J Dev Neurosci 2006; 24:445–453.
- Azizi F, Smyth P. Breastfeeding and maternal and infant iodine nutrition. Clin Endocrinol (Oxf) 2009; 70:803–809.
- Delange F. Iodine requirements during pregnancy, lactation and the neonatal period and indicators of optimal iodine nutrition. Public Health Nutr 2007; 10:1571–1583.
- Semba RD, Delange F. Iodine in human milk: perspectives for infant health. Nutr Rev 2001; 59:269–278.
- Leung AM, Pearce EN, Braverman LE. Iodine content of prenatal multivitamins in the United States. N Engl J Med 2009; 360:939–940.
- Tonacchera M, Agretti P, Chiovato L, et al. Activating thyrotropin receptor mutations are present in nonadenomatous hyperfunctioning nodules of toxic or autonomous multinodular goiter. J Clin Endocrinol Metab 2000; 85:2270–2274.
- Medeiros-Neto G, Camargo RY, Tomimori EK. Approach to and treatment of goiters. Med Clin North Am 2012; 96:351–368.
- Heidemann P, Stubbe P. Serum 3,5,3'-triiodothyronine, thyroxine, and thyrotropin in hypothyroid infants with congenital goiter and the response to iodine. J Clin Endocrinol Metab 1978; 47:189–192.
- Patel YC, Pharoah PO, Hornabrook RW, Hetzel BS. Serum triiodothyronine, thyroxine and thyroid-stimulating hormone in endemic goiter: a comparison of goitrous and nongoitrous subjects in New Guinea. J Clin Endocrinol Metab 1973; 37:783–789.
- Boyages SC, Halpern JP. Endemic cretinism: toward a unifying hypothesis. Thyroid 1993; 3:59–69.
- Chen ZP, Hetzel BS. Cretinism revisited. Best Pract Res Clin Endocrinol Metab 2010; 24:39–50.
- Hynes KL, Otahal P, Hay I, Burgess JR. Mild iodine deficiency during pregnancy is associated with reduced educational outcomes in the offspring: 9-year follow-up of the gestational iodine cohort. J Clin Endocrinol Metab 2013; 98:1954–1962.
- Bleichrodt N, Born MP. A metaanalysis of research on iodine and its relationship to cognitive development. In: Stanbury JB, ed. The Damaged Brain of Iodine Deficiency. 1st ed. New York, NY: Cognizant Communication; 1994:195–200.
- Melse-Boonstra A, Jaiswal N. Iodine deficiency in pregnancy, infancy and childhood and its consequences for brain development. Best Pract Res Clin Endocrinol Metab 2010; 24:29–38.
- Gordon RC, Rose MC, Skeaff SA, Gray AR, Morgan KM, Ruffman T. Iodine supplementation improves cognition in mildly iodine-deficient children. Am J Clin Nutr 2009; 90:1264–1271.
- Konig F, Andersson M, Hotz K, Aeberli I, Zimmermann MB. Ten repeat collections for urinary iodine from spot samples or 24-hour samples are needed to reliably estimate individual iodine status in women. J Nutr 2011; 141:2049–2054.
- Vejbjerg P, Knudsen N, Perrild H, et al. Estimation of iodine intake from various urinary iodine measurements in population studies. Thyroid 2009; 19:1281–1286.
- Zimmermann MB, Hess SY, Adou P, Toresanni T, Wegmuller R, Hurrell RF. Thyroid size and goiter prevalence after introduction of iodized salt: a 5-y prospective study in schoolchildren in Cote d’Ivoire. Am J Clin Nutr 2003; 77:663–667.
- Zimmermann MB, de Benoist B, Corigliano S, et al. Assessment of iodine status using dried blood spot thyroglobulin: development of reference material and establishment of an international reference range in iodine-sufficient children. J Clin Endocrinol Metab 2006; 91:4881–4887.
- Zimmermann MB, Aeberli I, Andersson M, et al. Thyroglobulin is a sensitive measure of both deficient and excess iodine intakes in children and indicates no adverse effects on thyroid function in the UIC range of 100-299 mug/L: a UNICEF/ICCIDD study group report. J Clin Endocrinol Metab 2013; 98:1271–1280.
- Thilly CH, Delange F, Lagasse R, et al. Fetal hypothyroidism and maternal thyroid status in severe endemic goiter. J Clin Endocrinol Metab 1978; 47:354–360.
- Sullivan KM, May W, Nordenberg D, Houston R, Maberly GF. Use of thyroid stimulating hormone testing in newborns to identify iodine deficiency. J Nutr 1997; 127:55–58.
- Delange F. Neonatal thyroid screening as a monitoring tool for the control of iodine deficiency. Acta Paediatr Suppl 1999; 88:21–24.
- Li M, Eastman CJ. Neonatal TSH screening: is it a sensitive and reliable tool for monitoring iodine status in populations? Best Pract Res Clin Endocrinol Metab 2010; 24:63–75.
- Zimmermann MB, Moretti D, Chaouki N, Torresani T. Development of a dried whole-blood spot thyroglobulin assay and its evaluation as an indicator of thyroid status in goitrous children receiving iodized salt. Am J Clin Nutr 2003; 77:1453–1458.
- Brahmbhatt S, Brahmbhatt RM, Boyages SC. Thyroid ultrasound is the best prevalence indicator for assessment of iodine deficiency disorders: a study in rural/tribal schoolchildren from Gujarat (Western India). Eur J Endocrinol 2000; 143:37–46.
- Moradi M, Hashemipour M, Akbari S, Kor Z, Mirbod SA, Kooshanmehr MR. Ultrasonographic evaluation of the thyroid gland volume among 8-15-year-old children in Isfahan, Iran. Adv Biomed Res 2014; 3:9.
KEY POINTS
- Adequate iodine intake during pregnancy is critical for normal fetal development.
- Major sources of dietary iodine in the United States are dairy products and iodized salt.
- The daily iodine requirement for nonpregnant adults is 150 µg, and for pregnant women it is 220 to 250 μg. Pregnant and lactating women should take a daily iodine supplement to ensure adequate iodine intake.
- Assessing the risk of iodine deficiency from clinical signs and from the history is key to diagnosing iodine deficiency. Individual urine iodine concentrations may vary from day to day. Repeated samples can be used to confirm iodine deficiency.

Vulvovaginitis: Find the cause to treat it
Although vulvovaginitis has several possible causes, the typical presenting symptoms are similar regardless of the cause: itching, burning, and vaginal discharge. Physical examination often reveals atrophy, redness, excoriations, and fissures in the vulvovaginal and perianal areas. Determining the cause is key to successful treatment.
This article reviews the diagnosis and treatment of many common and less common infectious and noninfectious causes of vulvovaginitis, the use of special tests, and the management of persistent cases.
DIAGNOSIS CAN BE CHALLENGING

Vulvar and vaginal symptoms are most commonly caused by local infections, but other causes must be also be considered, including several noninfectious ones (Table 1). Challenges in diagnosing vulvovaginitis are many and include distinguishing contact from allergic dermatitis, recognizing vaginal atrophy, and recognizing a parasitic infection. Determining whether a patient has an infectious process is important so that antibiotics can be used only when truly needed.
Foreign bodies in the vagina should also be considered, especially in children,1 as should sexual abuse. A 15-year retrospective review of prepubertal girls presenting with recurrent vaginal discharge found that sexual abuse might have been involved in about 5% of cases.2
Systemic diseases, such as eczema and psoriasis, may also present with gynecologic symptoms.
Heavy vaginal discharge may also be normal. This situation is a diagnosis of exclusion but is important to recognize in order to allay the patient’s anxiety and avoid unnecessary treatment.
SIMPLE OFFICE-BASED ASSESSMENT
A thorough history and physical examination are always warranted.

Simple tests of vaginal secretions can often determine the diagnosis (Table 2). Vaginal secretions should be analyzed in the following order:
Testing the pH. The pH can help determine likely diagnoses and streamline further testing (Figure 1).

Saline microscopy. Some of the vaginal discharge sample should be diluted with 1 or 2 drops of normal saline and examined under a microscope, first at × 10 magnification, then at × 40. The sample should be searched for epithelial cells, blood cells, “clue” cells (ie, epithelial cells with borders studded or obscured by bacteria), and motile trichomonads.
10% KOH whiff test and microscopy. To a second vaginal sample, a small amount of 10% potassium hydroxide should be added, and the examiner should sniff it. An amine or fishy odor is a sign of bacterial vaginosis.

If pH paper, KOH, and a microscope are unavailable, other point-of-care tests can be used for specific conditions as discussed below.
INFECTIOUS CAUSES
Infectious causes of vulvovaginitis include bacterial vaginosis, candidiasis, trichomoniasis, and herpes simplex virus (HSV) infection.
BACTERIAL VAGINOSIS
Bacterial vaginosis is the most common vaginal disorder worldwide. It has been linked to preterm delivery, intra-amniotic infection, endometritis, postabortion infection, and vaginal cuff cellulitis after hysterectomy.3 It may also be a risk factor for human immunodeficiency virus (HIV) infection.4
The condition reflects a microbial imbalance in the vaginal ecosystem, characterized by depletion of the dominant hydrogen peroxide-producing lactobacilli and overgrowth of anaerobic and facultative aerobic organisms such as Gardnerella vaginalis, Mycoplasma hominis, Atopobium vaginae, and Prevotella and Mobiluncus species.
Diagnosis of bacterial vaginosis
The Amsel criteria consist of the following:
- pH greater than 4.5
- Positive whiff test
- Homogeneous discharge
- Clue cells.
Three of the four criteria must be present for a diagnosis of bacterial vaginosis. This method is inexpensive and provides immediate results in the clinic.
The Nugent score, based on seeing certain bacteria from a vaginal swab on Gram stain microscopy, is the diagnostic standard for research.5
DNA tests. Affirm VPIII (BD Diagnostics, Sparks, MD) is a nonamplified nucleic acid probe hybridization test that detects Trichomonas vaginalis, Candida albicans, and G vaginalis. Although it is more expensive than testing for the Amsel criteria, it is commonly used in private offices because it is simple to use, gives rapid results, and does not require a microscope.6 Insurance pays for it when the test is indicated, but we know of a patient who received a bill for approximately $500 when the insurance company thought the test was not indicated.
In a study of 109 patients with symptoms of vulvovaginitis, the Affirm VPIII was found comparable to saline microscopy when tested on residual vaginal samples. Compared with Gram stain using Nugent scoring, the test has a sensitivity of 87.7% to 95.2% and a specificity of 81% to 99.1% for bacterial vaginosis.7
In 323 symptomatic women, a polymerase chain reaction (PCR) assay for bacterial vaginosis was 96.9% sensitive and 92.6% specific for bacterial vaginosis, and Affirm VPIII was 90.1% sensitive and 67.6% specific, compared with a reference standard incorporating Nugent Gram-stain scores and Amsel criteria.8 The test is commercially available.
Management of bacterial vaginosis
Initial treatment. Bacterial vaginosis can be treated with oral or topical metronidazole, oral tinidazole, or oral or topical clindamycin.9 All options offer equivalent efficacy as initial treatments, so the choice may be based on cost and preferred route of administration.
Treatment for recurrent disease. Women who have 3 or more episodes in 12 months should receive initial treatment each time as described above and should then be offered additional suppressive therapy with 0.75% metronidazole intravaginal gel 2 times a week for 4 months. A side effect of therapy is vulvovaginal candidiasis, which should be treated as needed.
In a multicenter study, Sobel et al10 randomized patients who had recurrent bacterial vaginosis to twice-weekly metronidazole gel or placebo for 16 weeks after their initial treatment. During the 28 weeks of follow-up, recurrences occurred in 51% of treated women vs 75% of those on placebo.
Another option for chronic therapy is oral metronidazole and boric acid vaginal suppositories.
Reichman et al11 treated women with oral metronidazole or tinidazole 500 mg twice a day for 7 days, followed by vaginal boric acid 600 mg daily for 21 days. This was followed by twice-weekly vaginal metronidazole gel for 16 weeks. At follow-up, the cure rate was 92% at 7 weeks, dropping to 88% at 12 weeks and 50% at 36 weeks.
Patients with recurrent bacterial vaginosis despite therapy should be referred to a vulvovaginal or infectious disease specialist.
VULVOVAGINAL CANDIDIASIS
Vulvovaginal candidiasis is the second most common cause of vaginitis.
Diagnosis can be clinical

Vulvovaginal candidiasis can be clinically diagnosed on the basis of cottage cheese-like clumpy discharge; external dysuria (a burning sensation when urine comes in contact with the vulva); and vulvar itching, pain, swelling, and redness. Edema, fissures, and excoriations may be seen on examination of the vulva. (Figure 3).
Saline microscopy (Figure 2) with the addition of 10% KOH may reveal the characteristic fungal elements, but its sensitivity is only 50%.
Fungal culture remains the gold standard for diagnosis and is needed to determine the sensitivity of specific strains of Candida to therapy.12
DNA tests can also be helpful. In a study of patients with symptomatic vaginitis, Affirm VPIII detected Candida in 11% of samples, whereas microscopy detected it in only 7%.13 Another study7 found that Affirm VPIII produced comparable results whether the sample was collected from residual vaginal discharge found on the speculum or was collected in the traditional way (by swabbing).
Cartwright et al8 compared the performance of a multiplexed, real-time PCR assay and Affirm VPIII in 102 patients. PCR was much more sensitive (97.7% vs 58.1%) but less specific (93.2% vs 100%), with culture serving as the gold standard.
Management of candidiasis
Uncomplicated cases can be managed with prescription or over-the-counter topical or oral antifungal medications for 1 to 7 days, depending on the medication.9 However, most of the common antifungals may not be effective against non-albicans Candida.
In immunosuppressed patients and diabetic patients, if symptoms do not improve with regular treatment, a vaginal sample should be cultured for C albicans. If the culture is positive, the patient should be treated with fluconazole 150 mg orally every 3 days for 3 doses.14
Patients with recurrent episodes (3 or more in 12 months) should follow initial treatment with maintenance therapy of weekly fluconazole 150 mg orally for 6 months.15
Non-albicans Candida may be azole-resistant, and fungal culture and sensitivity should be obtained. Sobel et al13 documented successful treatment of non-albicans Candida using boric acid and flucytosine. Phillips16 documented successful use of compounded amphotericin B in a 50-mg vaginal suppository for 14 days. Therefore, in patients who have Candida species other than C albicans, treatment should be one of the following:
- Vaginal boric acid 600 mg daily for 14 to 21 days
- Flucytosine in 15.5% vaginal cream, intravaginally administered as 5 g for 14 days
- Amphotericin B 50 mg vaginal suppositories for 14 days.
Boric acid is readily available, but flucytosine vaginal cream and amphotericin B vaginal suppositories must usually be compounded by a pharmacist.
Of note: All that itches is not yeast. Patients with persistent itching despite treatment should be referred to a specialist to search for another cause.
TRICHOMONIASIS
The incidence of T vaginalis infection is higher than that of Neisseria gonorrhoeae and Chlamydia trachomatis combined, with an estimated 7.4 million new cases occurring in the year 2000 in the United States.17 Infection increases the sexual transmission of HIV.18–20 It is often asymptomatic and so is likely underdiagnosed.
Diagnosis of trichomoniasis
Vaginal pH may be normal or elevated (> 4.5).
Direct microscopy. Observation by saline microscopy of motile trichomonads with their characteristic jerky movements is 100% specific but only 50% sensitive. Sensitivity is reduced by delaying microscopy on the sample by as little as 10 minutes.21
The incidental finding of T vaginalis on a conventional Papanicolaou (Pap) smear has poor sensitivity and specificity, and patients diagnosed with T vaginalis by conventional Pap smear should have a second test performed. The liquid-based Pap test is more accurate for microscopic diagnosis, and its results can be used to determine if treatment is needed (sensitivity 60%–90%; specificity 98%–100%).22,23
Culture. Amplification of T vaginalis in liquid culture usually provides results within 3 days.24 It is more sensitive than microscopy but less sensitive than a nucleic acid amplification test: compared with a nucleic acid amplification test, culture is 44% to 75% sensitive for detecting T vaginalis and 100% specific.19 Culture is the preferred test for resistant strains.
Non–culture-based or nucleic acid tests do not require viable organisms, so they allow for a wider range of specimen storage temperatures and time intervals between collection and processing. This quality limits them for testing treatment success; if performed too early, they may detect nonviable organisms. A 2-week interval is recommended between the end of treatment and retesting.25
Nonamplified tests such as Affirm VPIII and the Osom Trichomonas Test (Sekisui Diagnostics, Lexington, MA) are 40% to 95% sensitive, depending on the test and reference standard used, and 92% to 100% specific.26,27
Nucleic acid amplification tests are usually not performed as point-of-care tests. They are more expensive and require special equipment with trained personnel. Sensitivities range from 76% to 100%, making these tests more suitable for screening and testing of asymptomatic women, in whom the concentration of organisms may be lower.
Treatment of trichomoniasis
Treatment is a single 2-g oral dose of metronidazole or tinidazole.9
If initial treatment is ineffective, an additional regimen can be either of the following:
- Oral metronidazole 500 mg twice a day for 7 days
- Oral metronidazole or tinidazole, 2 g daily for 5 days.
Patients allergic to nitroimidazoles should be referred for desensitization.
If these treatments are unsuccessful, the patient should be referred to an infectious disease specialist or gynecologist who specializes in vulvovaginal disorders. Treatment failure is uncommon and is usually related to noncompliance, reinfection, or metronidazole resistance.28 The US Centers for Disease Control and Prevention offers testing for resistance by request.
Reportedly successful regimens for refractory trichomoniasis include 14 days of either:
- Oral tinidazole 500 mg 4 times daily plus vaginal tinidazole 500 mg twice daily29
- Oral tinidazole 1 g 3 times daily plus compounded 5% intravaginal paromomycin 5 g nightly.30
HERPES SIMPLEX VIRUS INFECTION
HSV (HSV-1 and HSV-2) causes lifelong infection. About 50 million people in the United States are infected with HSV-2, the most common cause of recurrent infections.31 Owing to changes in sexual practices, an increasing number of young people are acquiring anogenital HSV-1 infection.32,33
Diagnosis of herpes

Diagnosis may be difficult because the painful vesicular or ulcerative lesions (Figure 4) may not be visible at the time of presentation. Diagnosis is based on specific virologic and serologic tests. Nonspecific tests (eg, Tzanck smear, direct immunofluorescence) are neither sensitive nor specific and should not be relied on for diagnosis.34 HSV culture or HSV-PCR testing of a lesion is preferred. The sensitivity of viral culture can be low and is dependent on the stage of healing of a lesion and obtaining an adequate sample.
Accurate type-specific HSV serologic assays are based on HSV-specific glycoprotein G1 (HSV-1) and glycoprotein G2 (HSV-2). Unless a patient’s serologic status has already been determined, serologic testing should be done concurrently with HSV culture or PCR testing. Serologic testing enables classification of an infection as primary, nonprimary, or recurrent. For example, a patient with a positive HSV culture and negative serology most likely has primary HSV infection, and serologic study should be repeated after 6 to 8 weeks to assess for seroconversion.
Immunoglobulin M (IgM) testing for HSV-1 or HSV-2 is not diagnostic or type-specific and may be positive during recurrent genital or oral episodes of herpes.35
Treatment of herpes
In general, antiviral medications (eg, acyclovir, valacyclovir, famciclovir) are effective for managing HSV.12 Episodic or continuous suppression therapy may be needed for patients experiencing more than four outbreaks in 12 months. Patients who do not respond to treatment should be referred to an infectious disease specialist and undergo a viral culture with sensitivities.
NONINFECTIOUS CAUSES
Desquamative inflammatory vaginitis
Desquamative inflammatory vaginitis is a chronic vaginal disorder of unknown cause. It is a diagnosis of exclusion, and some patients may have a superimposed bacterial infection. It occurs mostly in perimenopausal woman and is often associated with low estrogen levels.
Diagnosis. Patients may report copious green-yellow mucoid discharge, vulvar or vaginal pain, and dyspareunia. On examination, the vulva may be erythematous, friable, and tender to the touch. The vagina may have ecchymoses, be diffusely erythematous, and have linear lesions. Mucoid or purulent discharge may be seen.
.")
The vaginal pH is greater than 4.5.
Saline microscopy shows increased parabasal cells and leukorrhea (Figure 5).
Diagnosis is based on all of the following:
- At least 1 symptom (ie, vaginal discharge, dyspareunia, pruritus, pain, irritation, or burning)
- Vaginal inflammation on examination
- pH higher than 4.5
- Presence of parabasal cells and leukorrhea on microscopy (a ratio of leukocytes to vaginal epithelial cells > 1:1).36
Treatment involves use of 2% intravaginal clindamycin or 10% intravaginal compounded hydrocortisone cream for 4 to 6 weeks. Patients who are not cured with single-agent therapy may benefit from compounded clindamycin and hydrocortisone, with estrogen added to the formulation for hypoestrogenic patients.
Atrophic vaginitis
Atrophic vaginitis is often seen in menopausal or hypoestrogenic women. Presenting symptoms include vulvar or vaginal pain and dyspareunia.
Diagnosis. On physical examination, the vulva appears pale and atrophic, with narrowing of the introitus. Vaginal examination may reveal a pale mucosa that lacks elasticity and rugation. The examination should be performed with caution, as the vagina may bleed easily.
The vaginal pH is usually elevated.

Saline microscopy may show parabasal cells and a paucity of epithelial cells. (Figure 6).
The Vaginal Maturation Index is an indicator of the maturity of the epithelial cell types being exfoliated; these normally include parabasal (immature) cells, intermediate, and superficial (mature) cells. A predominance of immature cells indicates a hypoestrogenic state.
Infection should be considered and treated as needed.
Treatment. Patients with no contraindication may benefit from systemic hormone therapy or topical estrogen, or both.
Contact dermatitis
Contact dermatitis is classified into two types:
Irritant dermatitis, caused by the destructive action of contactants, eg, urine, feces, topical agents, feminine wipes
Allergic dermatitis, also contactant-induced, but immunologically mediated.
If a diagnosis cannot be made from the patient history and physical examination, biopsy should be performed.
Treatment of contact dermatitis involves removing the irritant, hydrating the skin with sitz baths, and using an emollient (eg, petroleum jelly) and midpotent topical steroids until resolution. Some patients benefit from topical immunosuppressive agents (eg, tacrolimus). Patients with severe symptoms may be treated with a tapering course of oral steroids for 5 to 7 days. Recalcitrant cases should be referred to a specialist.
Lichen planus


Lichen sclerosus is a benign, chronic, progressive dermatologic condition characterized by marked inflammation, epithelial thinning, and distinctive dermal changes accompanied by pruritus and pain (Figures 7 and 8).
Treatment. High-potency topical steroids are the mainstay of therapy for lichen disease. Although these are not infectious processes, superimposed infections (mostly bacterial and fungal) may also be present and should be treated.
- Van Eyk N, Allen L, Giesbrecht E, et al. Pediatric vulvovaginal disorders: a diagnostic approach and review of the literature. J Obstet Gynaecol Can 2009; 31:850–862.
- McGreal S, Wood P. Recurrent vaginal discharge in children. J Pediatr Adolesc Gynecol 2013; 26:205–208.
- Livengood CH. Bacterial vaginosis: an overview for 2009. Rev Obstet Gynecol 2009; 2:28–37.
- Atashili J, Poole C, Ndumbe PM, Adimora AA, Smith JS. Bacterial vaginosis and HIV acquisition: a meta-analysis of published studies. AIDS 2008; 22:1493–1501.
- Money D. The laboratory diagnosis of bacterial vaginosis. Can J Infect Dis Med Microbiol 2005; 16:77–79.
- Lowe NK, Neal JL, Ryan-Wenger NA, et al. Accuracy of the clinical diagnosis of vaginitis compared with a DNA probe laboratory standard. Obstet Gynecol 2009; 113:89–95.
- Mulhem E, Boyanton BL Jr, Robinson-Dunn B, Ebert C, Dzebo R. Performance of the Affirm VP-III using residual vaginal discharge collected from the speculum to characterize vaginitis in symptomatic women. J Low Genit Tract Dis 2014; 18:344–346.
- Cartwright CP, Lembke BD, Ramachandran K, et al. Comparison of nucleic acid amplification assays with BD Affirm VPIII for diagnosis of vaginitis in symptomatic women. J Clin Microbiol 2013; 51:3694–3699.
- Workowski KA, Bolan GA; Centers for Disease Control and Prevention (CDC). Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm Rep 2015; 64:1–137.
- Sobel JD, Ferris D, Schwebke J, et al. Suppressive antibacterial therapy with 0.75% metronidazole vaginal gel to prevent recurrent bacterial vaginosis. Am J Obstet Gynecol 2006; 194:1283–1289.
- Reichman O, Akins R, Sobel JD. Boric acid addition to suppressive antimicrobial therapy for recurrent bacterial vaginosis. Sex Transm Dis 2009; 36:732–734.
- Carr PL, Felsenstein D, Friedman RH. Evaluation and management of vaginitis. J Gen Intern Med 1998; 13:335–346.
- Sobel JD, Chaim W, Nagappan V, Leaman D. Treatment of vaginitis caused by Candida glabrata: use of topical boric acid and flucytosine. Am J Obstet Gynecol 2003; 189:1297–1300.
- Sobel JD, Kapernick PS, Zervos M, et al. Treatment of complicated Candida vaginitis: comparison of single and sequential doses of fluconazole. Am J Obstet Gynecol 2001; 185:363–369.
- Sobel JD, Wiesenfeld HC, Martens M, et al. Maintenance fluconazole therapy for recurrent vulvovaginal candidiasis. N Engl J Med 2004; 351:876–883.
- Phillips AJ. Treatment of non-albicans Candida vaginitis with amphotericin B vaginal suppositories. Am J Obstet Gynecol 2005; 192:2009–2013.
- Weinstock H, Berman S, Cates W Jr. Sexually transmitted diseases among American youth: incidence and prevalence estimates, 2000. Perspect Sex Reprod Health 2004; 36:6–10.
- Gatski M, Martin DH, Clark RA, Harville E, Schmidt N, Kissinger P. Co-occurrence of Trichomonas vaginalis and bacterial vaginosis among HIV-positive women. Sex Transm Dis 2011; 38:163–166.
- Hobbs MM, Seña AC. Modern diagnosis of Trichomonas vaginalis infection. Sex Transm Infect 2013; 89:434–438.
- McClelland RS, Sangare L, Hassan WM, et al. Infection with Trichomonas vaginalis increases the risk of HIV-1 acquisition. J Infect Dis 2007; 195:698–702.
- Kingston MA, Bansal D, Carlin EM. ‘Shelf life’ of Trichomonas vaginalis. Int J STD AIDS 2003; 14:28–29.
- Aslan DL, Gulbahce HE, Stelow EB, et al. The diagnosis of Trichomonas vaginalis in liquid-based Pap tests: correlation with PCR. Diagn Cytopathol 2005; 32:341–344.
- Lara-Torre E, Pinkerton JS. Accuracy of detection of Trichomonas vaginalis organisms on a liquid-based papanicolaou smear. Am J Obstet Gynecol 2003; 188:354–356.
- Hobbs MM, Lapple DM, Lawing LF, et al. Methods for detection of Trichomonas vaginalis in the male partners of infected women: implications for control of trichomoniasis. J Clin Microbiol 2006; 44:3994–3999.
- Van Der Pol B, Williams JA, Orr DP, Batteiger BE, Fortenberry JD. Prevalence, incidence, natural history, and response to treatment of Trichomonas vaginalis infection among adolescent women. J Infect Dis 2005; 192:2039–2044.
- Campbell L, Woods V, Lloyd T, Elsayed S, Church DL. Evaluation of the OSOM Trichomonas rapid test versus wet preparation examination for detection of Trichomonas vaginalis vaginitis in specimens from women with a low prevalence of infection. J Clin Microbiol 2008; 46:3467–3469.
- Chapin K, Andrea S. APTIMA Trichomonas vaginalis, a transcription-mediated amplification assay for detection of Trichomonas vaginalis in urogenital specimens. Expert Rev Mol Diagn 2011; 11:679–688.
- Krashin JW, Koumans EH, Bradshaw-Sydnor AC, et al. Trichomonas vaginalis prevalence, incidence, risk factors and antibiotic-resistance in an adolescent population. Sex Transm Dis 2010; 37:440–444.
- Sobel JD, Nyirjesy P, Brown W. Tinidazole therapy for metronidazole-resistant vaginal trichomoniasis. Clin Infect Dis 2001; 33:1341–1346.
- Nyirjesy P, Gilbert J, Mulcahy LJ. Resistant trichomoniasis: successful treatment with combination therapy. Sex Transm Dis 2011; 38:962–963.
- Bradley H, Markowitz LE, Gibson T, McQuillan GM. Seroprevalence of herpes simplex virus types 1 and 2—United States, 1999–2010. J Infect Dis 2014; 209:325–333.
- Roberts CM, Pfister JR, Spear SJ. Increasing proportion of herpes simplex virus type 1 as a cause of genital herpes infection in college students. Sex Transm Dis 2003; 30:797–800.
- Ryder N, Jin F, McNulty AM, Grulich AE, Donovan B. Increasing role of herpes simplex virus type 1 in first-episode anogenital herpes in heterosexual women and younger men who have sex with men, 1992-2006. Sex Transm Infect 2009; 85:416–419.
- Caviness AC, Oelze LL, Saz UE, Greer JM, Demmler-Harrison GJ. Direct immunofluorescence assay compared to cell culture for the diagnosis of mucocutaneous herpes simplex virus infections in children. J Clin Virol 2010; 49:58–60.
- Morrow R, Friedrich D. Performance of a novel test for IgM and IgG antibodies in subjects with culture-documented genital herpes simplex virus-1 or -2 infection. Clin Microbiol Infect 2006; 12:463–469.
- Bradford J, Fischer G. Desquamative inflammatory vaginitis: differential diagnosis and alternate diagnostic criteria. J Low Genit Tract Dis 2010; 14:306–310.
Although vulvovaginitis has several possible causes, the typical presenting symptoms are similar regardless of the cause: itching, burning, and vaginal discharge. Physical examination often reveals atrophy, redness, excoriations, and fissures in the vulvovaginal and perianal areas. Determining the cause is key to successful treatment.
This article reviews the diagnosis and treatment of many common and less common infectious and noninfectious causes of vulvovaginitis, the use of special tests, and the management of persistent cases.
DIAGNOSIS CAN BE CHALLENGING
Vulvar and vaginal symptoms are most commonly caused by local infections, but other causes must be also be considered, including several noninfectious ones (Table 1). Challenges in diagnosing vulvovaginitis are many and include distinguishing contact from allergic dermatitis, recognizing vaginal atrophy, and recognizing a parasitic infection. Determining whether a patient has an infectious process is important so that antibiotics can be used only when truly needed.
Foreign bodies in the vagina should also be considered, especially in children,1 as should sexual abuse. A 15-year retrospective review of prepubertal girls presenting with recurrent vaginal discharge found that sexual abuse might have been involved in about 5% of cases.2
Systemic diseases, such as eczema and psoriasis, may also present with gynecologic symptoms.
Heavy vaginal discharge may also be normal. This situation is a diagnosis of exclusion but is important to recognize in order to allay the patient’s anxiety and avoid unnecessary treatment.
SIMPLE OFFICE-BASED ASSESSMENT
A thorough history and physical examination are always warranted.
Simple tests of vaginal secretions can often determine the diagnosis (Table 2). Vaginal secretions should be analyzed in the following order:
Testing the pH. The pH can help determine likely diagnoses and streamline further testing (Figure 1).
Saline microscopy. Some of the vaginal discharge sample should be diluted with 1 or 2 drops of normal saline and examined under a microscope, first at × 10 magnification, then at × 40. The sample should be searched for epithelial cells, blood cells, “clue” cells (ie, epithelial cells with borders studded or obscured by bacteria), and motile trichomonads.
10% KOH whiff test and microscopy. To a second vaginal sample, a small amount of 10% potassium hydroxide should be added, and the examiner should sniff it. An amine or fishy odor is a sign of bacterial vaginosis.
If pH paper, KOH, and a microscope are unavailable, other point-of-care tests can be used for specific conditions as discussed below.
INFECTIOUS CAUSES
Infectious causes of vulvovaginitis include bacterial vaginosis, candidiasis, trichomoniasis, and herpes simplex virus (HSV) infection.
BACTERIAL VAGINOSIS
Bacterial vaginosis is the most common vaginal disorder worldwide. It has been linked to preterm delivery, intra-amniotic infection, endometritis, postabortion infection, and vaginal cuff cellulitis after hysterectomy.3 It may also be a risk factor for human immunodeficiency virus (HIV) infection.4
The condition reflects a microbial imbalance in the vaginal ecosystem, characterized by depletion of the dominant hydrogen peroxide-producing lactobacilli and overgrowth of anaerobic and facultative aerobic organisms such as Gardnerella vaginalis, Mycoplasma hominis, Atopobium vaginae, and Prevotella and Mobiluncus species.
Diagnosis of bacterial vaginosis
The Amsel criteria consist of the following:
- pH greater than 4.5
- Positive whiff test
- Homogeneous discharge
- Clue cells.
Three of the four criteria must be present for a diagnosis of bacterial vaginosis. This method is inexpensive and provides immediate results in the clinic.
The Nugent score, based on seeing certain bacteria from a vaginal swab on Gram stain microscopy, is the diagnostic standard for research.5
DNA tests. Affirm VPIII (BD Diagnostics, Sparks, MD) is a nonamplified nucleic acid probe hybridization test that detects Trichomonas vaginalis, Candida albicans, and G vaginalis. Although it is more expensive than testing for the Amsel criteria, it is commonly used in private offices because it is simple to use, gives rapid results, and does not require a microscope.6 Insurance pays for it when the test is indicated, but we know of a patient who received a bill for approximately $500 when the insurance company thought the test was not indicated.
In a study of 109 patients with symptoms of vulvovaginitis, the Affirm VPIII was found comparable to saline microscopy when tested on residual vaginal samples. Compared with Gram stain using Nugent scoring, the test has a sensitivity of 87.7% to 95.2% and a specificity of 81% to 99.1% for bacterial vaginosis.7
In 323 symptomatic women, a polymerase chain reaction (PCR) assay for bacterial vaginosis was 96.9% sensitive and 92.6% specific for bacterial vaginosis, and Affirm VPIII was 90.1% sensitive and 67.6% specific, compared with a reference standard incorporating Nugent Gram-stain scores and Amsel criteria.8 The test is commercially available.
Management of bacterial vaginosis
Initial treatment. Bacterial vaginosis can be treated with oral or topical metronidazole, oral tinidazole, or oral or topical clindamycin.9 All options offer equivalent efficacy as initial treatments, so the choice may be based on cost and preferred route of administration.
Treatment for recurrent disease. Women who have 3 or more episodes in 12 months should receive initial treatment each time as described above and should then be offered additional suppressive therapy with 0.75% metronidazole intravaginal gel 2 times a week for 4 months. A side effect of therapy is vulvovaginal candidiasis, which should be treated as needed.
In a multicenter study, Sobel et al10 randomized patients who had recurrent bacterial vaginosis to twice-weekly metronidazole gel or placebo for 16 weeks after their initial treatment. During the 28 weeks of follow-up, recurrences occurred in 51% of treated women vs 75% of those on placebo.
Another option for chronic therapy is oral metronidazole and boric acid vaginal suppositories.
Reichman et al11 treated women with oral metronidazole or tinidazole 500 mg twice a day for 7 days, followed by vaginal boric acid 600 mg daily for 21 days. This was followed by twice-weekly vaginal metronidazole gel for 16 weeks. At follow-up, the cure rate was 92% at 7 weeks, dropping to 88% at 12 weeks and 50% at 36 weeks.
Patients with recurrent bacterial vaginosis despite therapy should be referred to a vulvovaginal or infectious disease specialist.
VULVOVAGINAL CANDIDIASIS
Vulvovaginal candidiasis is the second most common cause of vaginitis.
Diagnosis can be clinical
Vulvovaginal candidiasis can be clinically diagnosed on the basis of cottage cheese-like clumpy discharge; external dysuria (a burning sensation when urine comes in contact with the vulva); and vulvar itching, pain, swelling, and redness. Edema, fissures, and excoriations may be seen on examination of the vulva. (Figure 3).
Saline microscopy (Figure 2) with the addition of 10% KOH may reveal the characteristic fungal elements, but its sensitivity is only 50%.
Fungal culture remains the gold standard for diagnosis and is needed to determine the sensitivity of specific strains of Candida to therapy.12
DNA tests can also be helpful. In a study of patients with symptomatic vaginitis, Affirm VPIII detected Candida in 11% of samples, whereas microscopy detected it in only 7%.13 Another study7 found that Affirm VPIII produced comparable results whether the sample was collected from residual vaginal discharge found on the speculum or was collected in the traditional way (by swabbing).
Cartwright et al8 compared the performance of a multiplexed, real-time PCR assay and Affirm VPIII in 102 patients. PCR was much more sensitive (97.7% vs 58.1%) but less specific (93.2% vs 100%), with culture serving as the gold standard.
Management of candidiasis
Uncomplicated cases can be managed with prescription or over-the-counter topical or oral antifungal medications for 1 to 7 days, depending on the medication.9 However, most of the common antifungals may not be effective against non-albicans Candida.
In immunosuppressed patients and diabetic patients, if symptoms do not improve with regular treatment, a vaginal sample should be cultured for C albicans. If the culture is positive, the patient should be treated with fluconazole 150 mg orally every 3 days for 3 doses.14
Patients with recurrent episodes (3 or more in 12 months) should follow initial treatment with maintenance therapy of weekly fluconazole 150 mg orally for 6 months.15
Non-albicans Candida may be azole-resistant, and fungal culture and sensitivity should be obtained. Sobel et al13 documented successful treatment of non-albicans Candida using boric acid and flucytosine. Phillips16 documented successful use of compounded amphotericin B in a 50-mg vaginal suppository for 14 days. Therefore, in patients who have Candida species other than C albicans, treatment should be one of the following:
- Vaginal boric acid 600 mg daily for 14 to 21 days
- Flucytosine in 15.5% vaginal cream, intravaginally administered as 5 g for 14 days
- Amphotericin B 50 mg vaginal suppositories for 14 days.
Boric acid is readily available, but flucytosine vaginal cream and amphotericin B vaginal suppositories must usually be compounded by a pharmacist.
Of note: All that itches is not yeast. Patients with persistent itching despite treatment should be referred to a specialist to search for another cause.
TRICHOMONIASIS
The incidence of T vaginalis infection is higher than that of Neisseria gonorrhoeae and Chlamydia trachomatis combined, with an estimated 7.4 million new cases occurring in the year 2000 in the United States.17 Infection increases the sexual transmission of HIV.18–20 It is often asymptomatic and so is likely underdiagnosed.
Diagnosis of trichomoniasis
Vaginal pH may be normal or elevated (> 4.5).
Direct microscopy. Observation by saline microscopy of motile trichomonads with their characteristic jerky movements is 100% specific but only 50% sensitive. Sensitivity is reduced by delaying microscopy on the sample by as little as 10 minutes.21
The incidental finding of T vaginalis on a conventional Papanicolaou (Pap) smear has poor sensitivity and specificity, and patients diagnosed with T vaginalis by conventional Pap smear should have a second test performed. The liquid-based Pap test is more accurate for microscopic diagnosis, and its results can be used to determine if treatment is needed (sensitivity 60%–90%; specificity 98%–100%).22,23
Culture. Amplification of T vaginalis in liquid culture usually provides results within 3 days.24 It is more sensitive than microscopy but less sensitive than a nucleic acid amplification test: compared with a nucleic acid amplification test, culture is 44% to 75% sensitive for detecting T vaginalis and 100% specific.19 Culture is the preferred test for resistant strains.
Non–culture-based or nucleic acid tests do not require viable organisms, so they allow for a wider range of specimen storage temperatures and time intervals between collection and processing. This quality limits them for testing treatment success; if performed too early, they may detect nonviable organisms. A 2-week interval is recommended between the end of treatment and retesting.25
Nonamplified tests such as Affirm VPIII and the Osom Trichomonas Test (Sekisui Diagnostics, Lexington, MA) are 40% to 95% sensitive, depending on the test and reference standard used, and 92% to 100% specific.26,27
Nucleic acid amplification tests are usually not performed as point-of-care tests. They are more expensive and require special equipment with trained personnel. Sensitivities range from 76% to 100%, making these tests more suitable for screening and testing of asymptomatic women, in whom the concentration of organisms may be lower.
Treatment of trichomoniasis
Treatment is a single 2-g oral dose of metronidazole or tinidazole.9
If initial treatment is ineffective, an additional regimen can be either of the following:
- Oral metronidazole 500 mg twice a day for 7 days
- Oral metronidazole or tinidazole, 2 g daily for 5 days.
Patients allergic to nitroimidazoles should be referred for desensitization.
If these treatments are unsuccessful, the patient should be referred to an infectious disease specialist or gynecologist who specializes in vulvovaginal disorders. Treatment failure is uncommon and is usually related to noncompliance, reinfection, or metronidazole resistance.28 The US Centers for Disease Control and Prevention offers testing for resistance by request.
Reportedly successful regimens for refractory trichomoniasis include 14 days of either:
- Oral tinidazole 500 mg 4 times daily plus vaginal tinidazole 500 mg twice daily29
- Oral tinidazole 1 g 3 times daily plus compounded 5% intravaginal paromomycin 5 g nightly.30
HERPES SIMPLEX VIRUS INFECTION
HSV (HSV-1 and HSV-2) causes lifelong infection. About 50 million people in the United States are infected with HSV-2, the most common cause of recurrent infections.31 Owing to changes in sexual practices, an increasing number of young people are acquiring anogenital HSV-1 infection.32,33
Diagnosis of herpes
Diagnosis may be difficult because the painful vesicular or ulcerative lesions (Figure 4) may not be visible at the time of presentation. Diagnosis is based on specific virologic and serologic tests. Nonspecific tests (eg, Tzanck smear, direct immunofluorescence) are neither sensitive nor specific and should not be relied on for diagnosis.34 HSV culture or HSV-PCR testing of a lesion is preferred. The sensitivity of viral culture can be low and is dependent on the stage of healing of a lesion and obtaining an adequate sample.
Accurate type-specific HSV serologic assays are based on HSV-specific glycoprotein G1 (HSV-1) and glycoprotein G2 (HSV-2). Unless a patient’s serologic status has already been determined, serologic testing should be done concurrently with HSV culture or PCR testing. Serologic testing enables classification of an infection as primary, nonprimary, or recurrent. For example, a patient with a positive HSV culture and negative serology most likely has primary HSV infection, and serologic study should be repeated after 6 to 8 weeks to assess for seroconversion.
Immunoglobulin M (IgM) testing for HSV-1 or HSV-2 is not diagnostic or type-specific and may be positive during recurrent genital or oral episodes of herpes.35
Treatment of herpes
In general, antiviral medications (eg, acyclovir, valacyclovir, famciclovir) are effective for managing HSV.12 Episodic or continuous suppression therapy may be needed for patients experiencing more than four outbreaks in 12 months. Patients who do not respond to treatment should be referred to an infectious disease specialist and undergo a viral culture with sensitivities.
NONINFECTIOUS CAUSES
Desquamative inflammatory vaginitis
Desquamative inflammatory vaginitis is a chronic vaginal disorder of unknown cause. It is a diagnosis of exclusion, and some patients may have a superimposed bacterial infection. It occurs mostly in perimenopausal woman and is often associated with low estrogen levels.
Diagnosis. Patients may report copious green-yellow mucoid discharge, vulvar or vaginal pain, and dyspareunia. On examination, the vulva may be erythematous, friable, and tender to the touch. The vagina may have ecchymoses, be diffusely erythematous, and have linear lesions. Mucoid or purulent discharge may be seen.
The vaginal pH is greater than 4.5.
Saline microscopy shows increased parabasal cells and leukorrhea (Figure 5).
Diagnosis is based on all of the following:
- At least 1 symptom (ie, vaginal discharge, dyspareunia, pruritus, pain, irritation, or burning)
- Vaginal inflammation on examination
- pH higher than 4.5
- Presence of parabasal cells and leukorrhea on microscopy (a ratio of leukocytes to vaginal epithelial cells > 1:1).36
Treatment involves use of 2% intravaginal clindamycin or 10% intravaginal compounded hydrocortisone cream for 4 to 6 weeks. Patients who are not cured with single-agent therapy may benefit from compounded clindamycin and hydrocortisone, with estrogen added to the formulation for hypoestrogenic patients.
Atrophic vaginitis
Atrophic vaginitis is often seen in menopausal or hypoestrogenic women. Presenting symptoms include vulvar or vaginal pain and dyspareunia.
Diagnosis. On physical examination, the vulva appears pale and atrophic, with narrowing of the introitus. Vaginal examination may reveal a pale mucosa that lacks elasticity and rugation. The examination should be performed with caution, as the vagina may bleed easily.
The vaginal pH is usually elevated.
Saline microscopy may show parabasal cells and a paucity of epithelial cells. (Figure 6).
The Vaginal Maturation Index is an indicator of the maturity of the epithelial cell types being exfoliated; these normally include parabasal (immature) cells, intermediate, and superficial (mature) cells. A predominance of immature cells indicates a hypoestrogenic state.
Infection should be considered and treated as needed.
Treatment. Patients with no contraindication may benefit from systemic hormone therapy or topical estrogen, or both.
Contact dermatitis
Contact dermatitis is classified into two types:
Irritant dermatitis, caused by the destructive action of contactants, eg, urine, feces, topical agents, feminine wipes
Allergic dermatitis, also contactant-induced, but immunologically mediated.
If a diagnosis cannot be made from the patient history and physical examination, biopsy should be performed.
Treatment of contact dermatitis involves removing the irritant, hydrating the skin with sitz baths, and using an emollient (eg, petroleum jelly) and midpotent topical steroids until resolution. Some patients benefit from topical immunosuppressive agents (eg, tacrolimus). Patients with severe symptoms may be treated with a tapering course of oral steroids for 5 to 7 days. Recalcitrant cases should be referred to a specialist.
Lichen planus
Lichen sclerosus is a benign, chronic, progressive dermatologic condition characterized by marked inflammation, epithelial thinning, and distinctive dermal changes accompanied by pruritus and pain (Figures 7 and 8).
Treatment. High-potency topical steroids are the mainstay of therapy for lichen disease. Although these are not infectious processes, superimposed infections (mostly bacterial and fungal) may also be present and should be treated.
Although vulvovaginitis has several possible causes, the typical presenting symptoms are similar regardless of the cause: itching, burning, and vaginal discharge. Physical examination often reveals atrophy, redness, excoriations, and fissures in the vulvovaginal and perianal areas. Determining the cause is key to successful treatment.
This article reviews the diagnosis and treatment of many common and less common infectious and noninfectious causes of vulvovaginitis, the use of special tests, and the management of persistent cases.
DIAGNOSIS CAN BE CHALLENGING
Vulvar and vaginal symptoms are most commonly caused by local infections, but other causes must be also be considered, including several noninfectious ones (Table 1). Challenges in diagnosing vulvovaginitis are many and include distinguishing contact from allergic dermatitis, recognizing vaginal atrophy, and recognizing a parasitic infection. Determining whether a patient has an infectious process is important so that antibiotics can be used only when truly needed.
Foreign bodies in the vagina should also be considered, especially in children,1 as should sexual abuse. A 15-year retrospective review of prepubertal girls presenting with recurrent vaginal discharge found that sexual abuse might have been involved in about 5% of cases.2
Systemic diseases, such as eczema and psoriasis, may also present with gynecologic symptoms.
Heavy vaginal discharge may also be normal. This situation is a diagnosis of exclusion but is important to recognize in order to allay the patient’s anxiety and avoid unnecessary treatment.
SIMPLE OFFICE-BASED ASSESSMENT
A thorough history and physical examination are always warranted.
Simple tests of vaginal secretions can often determine the diagnosis (Table 2). Vaginal secretions should be analyzed in the following order:
Testing the pH. The pH can help determine likely diagnoses and streamline further testing (Figure 1).
Saline microscopy. Some of the vaginal discharge sample should be diluted with 1 or 2 drops of normal saline and examined under a microscope, first at × 10 magnification, then at × 40. The sample should be searched for epithelial cells, blood cells, “clue” cells (ie, epithelial cells with borders studded or obscured by bacteria), and motile trichomonads.
10% KOH whiff test and microscopy. To a second vaginal sample, a small amount of 10% potassium hydroxide should be added, and the examiner should sniff it. An amine or fishy odor is a sign of bacterial vaginosis.
If pH paper, KOH, and a microscope are unavailable, other point-of-care tests can be used for specific conditions as discussed below.
INFECTIOUS CAUSES
Infectious causes of vulvovaginitis include bacterial vaginosis, candidiasis, trichomoniasis, and herpes simplex virus (HSV) infection.
BACTERIAL VAGINOSIS
Bacterial vaginosis is the most common vaginal disorder worldwide. It has been linked to preterm delivery, intra-amniotic infection, endometritis, postabortion infection, and vaginal cuff cellulitis after hysterectomy.3 It may also be a risk factor for human immunodeficiency virus (HIV) infection.4
The condition reflects a microbial imbalance in the vaginal ecosystem, characterized by depletion of the dominant hydrogen peroxide-producing lactobacilli and overgrowth of anaerobic and facultative aerobic organisms such as Gardnerella vaginalis, Mycoplasma hominis, Atopobium vaginae, and Prevotella and Mobiluncus species.
Diagnosis of bacterial vaginosis
The Amsel criteria consist of the following:
- pH greater than 4.5
- Positive whiff test
- Homogeneous discharge
- Clue cells.
Three of the four criteria must be present for a diagnosis of bacterial vaginosis. This method is inexpensive and provides immediate results in the clinic.
The Nugent score, based on seeing certain bacteria from a vaginal swab on Gram stain microscopy, is the diagnostic standard for research.5
DNA tests. Affirm VPIII (BD Diagnostics, Sparks, MD) is a nonamplified nucleic acid probe hybridization test that detects Trichomonas vaginalis, Candida albicans, and G vaginalis. Although it is more expensive than testing for the Amsel criteria, it is commonly used in private offices because it is simple to use, gives rapid results, and does not require a microscope.6 Insurance pays for it when the test is indicated, but we know of a patient who received a bill for approximately $500 when the insurance company thought the test was not indicated.
In a study of 109 patients with symptoms of vulvovaginitis, the Affirm VPIII was found comparable to saline microscopy when tested on residual vaginal samples. Compared with Gram stain using Nugent scoring, the test has a sensitivity of 87.7% to 95.2% and a specificity of 81% to 99.1% for bacterial vaginosis.7
In 323 symptomatic women, a polymerase chain reaction (PCR) assay for bacterial vaginosis was 96.9% sensitive and 92.6% specific for bacterial vaginosis, and Affirm VPIII was 90.1% sensitive and 67.6% specific, compared with a reference standard incorporating Nugent Gram-stain scores and Amsel criteria.8 The test is commercially available.
Management of bacterial vaginosis
Initial treatment. Bacterial vaginosis can be treated with oral or topical metronidazole, oral tinidazole, or oral or topical clindamycin.9 All options offer equivalent efficacy as initial treatments, so the choice may be based on cost and preferred route of administration.
Treatment for recurrent disease. Women who have 3 or more episodes in 12 months should receive initial treatment each time as described above and should then be offered additional suppressive therapy with 0.75% metronidazole intravaginal gel 2 times a week for 4 months. A side effect of therapy is vulvovaginal candidiasis, which should be treated as needed.
In a multicenter study, Sobel et al10 randomized patients who had recurrent bacterial vaginosis to twice-weekly metronidazole gel or placebo for 16 weeks after their initial treatment. During the 28 weeks of follow-up, recurrences occurred in 51% of treated women vs 75% of those on placebo.
Another option for chronic therapy is oral metronidazole and boric acid vaginal suppositories.
Reichman et al11 treated women with oral metronidazole or tinidazole 500 mg twice a day for 7 days, followed by vaginal boric acid 600 mg daily for 21 days. This was followed by twice-weekly vaginal metronidazole gel for 16 weeks. At follow-up, the cure rate was 92% at 7 weeks, dropping to 88% at 12 weeks and 50% at 36 weeks.
Patients with recurrent bacterial vaginosis despite therapy should be referred to a vulvovaginal or infectious disease specialist.
VULVOVAGINAL CANDIDIASIS
Vulvovaginal candidiasis is the second most common cause of vaginitis.
Diagnosis can be clinical
Vulvovaginal candidiasis can be clinically diagnosed on the basis of cottage cheese-like clumpy discharge; external dysuria (a burning sensation when urine comes in contact with the vulva); and vulvar itching, pain, swelling, and redness. Edema, fissures, and excoriations may be seen on examination of the vulva. (Figure 3).
Saline microscopy (Figure 2) with the addition of 10% KOH may reveal the characteristic fungal elements, but its sensitivity is only 50%.
Fungal culture remains the gold standard for diagnosis and is needed to determine the sensitivity of specific strains of Candida to therapy.12
DNA tests can also be helpful. In a study of patients with symptomatic vaginitis, Affirm VPIII detected Candida in 11% of samples, whereas microscopy detected it in only 7%.13 Another study7 found that Affirm VPIII produced comparable results whether the sample was collected from residual vaginal discharge found on the speculum or was collected in the traditional way (by swabbing).
Cartwright et al8 compared the performance of a multiplexed, real-time PCR assay and Affirm VPIII in 102 patients. PCR was much more sensitive (97.7% vs 58.1%) but less specific (93.2% vs 100%), with culture serving as the gold standard.
Management of candidiasis
Uncomplicated cases can be managed with prescription or over-the-counter topical or oral antifungal medications for 1 to 7 days, depending on the medication.9 However, most of the common antifungals may not be effective against non-albicans Candida.
In immunosuppressed patients and diabetic patients, if symptoms do not improve with regular treatment, a vaginal sample should be cultured for C albicans. If the culture is positive, the patient should be treated with fluconazole 150 mg orally every 3 days for 3 doses.14
Patients with recurrent episodes (3 or more in 12 months) should follow initial treatment with maintenance therapy of weekly fluconazole 150 mg orally for 6 months.15
Non-albicans Candida may be azole-resistant, and fungal culture and sensitivity should be obtained. Sobel et al13 documented successful treatment of non-albicans Candida using boric acid and flucytosine. Phillips16 documented successful use of compounded amphotericin B in a 50-mg vaginal suppository for 14 days. Therefore, in patients who have Candida species other than C albicans, treatment should be one of the following:
- Vaginal boric acid 600 mg daily for 14 to 21 days
- Flucytosine in 15.5% vaginal cream, intravaginally administered as 5 g for 14 days
- Amphotericin B 50 mg vaginal suppositories for 14 days.
Boric acid is readily available, but flucytosine vaginal cream and amphotericin B vaginal suppositories must usually be compounded by a pharmacist.
Of note: All that itches is not yeast. Patients with persistent itching despite treatment should be referred to a specialist to search for another cause.
TRICHOMONIASIS
The incidence of T vaginalis infection is higher than that of Neisseria gonorrhoeae and Chlamydia trachomatis combined, with an estimated 7.4 million new cases occurring in the year 2000 in the United States.17 Infection increases the sexual transmission of HIV.18–20 It is often asymptomatic and so is likely underdiagnosed.
Diagnosis of trichomoniasis
Vaginal pH may be normal or elevated (> 4.5).
Direct microscopy. Observation by saline microscopy of motile trichomonads with their characteristic jerky movements is 100% specific but only 50% sensitive. Sensitivity is reduced by delaying microscopy on the sample by as little as 10 minutes.21
The incidental finding of T vaginalis on a conventional Papanicolaou (Pap) smear has poor sensitivity and specificity, and patients diagnosed with T vaginalis by conventional Pap smear should have a second test performed. The liquid-based Pap test is more accurate for microscopic diagnosis, and its results can be used to determine if treatment is needed (sensitivity 60%–90%; specificity 98%–100%).22,23
Culture. Amplification of T vaginalis in liquid culture usually provides results within 3 days.24 It is more sensitive than microscopy but less sensitive than a nucleic acid amplification test: compared with a nucleic acid amplification test, culture is 44% to 75% sensitive for detecting T vaginalis and 100% specific.19 Culture is the preferred test for resistant strains.
Non–culture-based or nucleic acid tests do not require viable organisms, so they allow for a wider range of specimen storage temperatures and time intervals between collection and processing. This quality limits them for testing treatment success; if performed too early, they may detect nonviable organisms. A 2-week interval is recommended between the end of treatment and retesting.25
Nonamplified tests such as Affirm VPIII and the Osom Trichomonas Test (Sekisui Diagnostics, Lexington, MA) are 40% to 95% sensitive, depending on the test and reference standard used, and 92% to 100% specific.26,27
Nucleic acid amplification tests are usually not performed as point-of-care tests. They are more expensive and require special equipment with trained personnel. Sensitivities range from 76% to 100%, making these tests more suitable for screening and testing of asymptomatic women, in whom the concentration of organisms may be lower.
Treatment of trichomoniasis
Treatment is a single 2-g oral dose of metronidazole or tinidazole.9
If initial treatment is ineffective, an additional regimen can be either of the following:
- Oral metronidazole 500 mg twice a day for 7 days
- Oral metronidazole or tinidazole, 2 g daily for 5 days.
Patients allergic to nitroimidazoles should be referred for desensitization.
If these treatments are unsuccessful, the patient should be referred to an infectious disease specialist or gynecologist who specializes in vulvovaginal disorders. Treatment failure is uncommon and is usually related to noncompliance, reinfection, or metronidazole resistance.28 The US Centers for Disease Control and Prevention offers testing for resistance by request.
Reportedly successful regimens for refractory trichomoniasis include 14 days of either:
- Oral tinidazole 500 mg 4 times daily plus vaginal tinidazole 500 mg twice daily29
- Oral tinidazole 1 g 3 times daily plus compounded 5% intravaginal paromomycin 5 g nightly.30
HERPES SIMPLEX VIRUS INFECTION
HSV (HSV-1 and HSV-2) causes lifelong infection. About 50 million people in the United States are infected with HSV-2, the most common cause of recurrent infections.31 Owing to changes in sexual practices, an increasing number of young people are acquiring anogenital HSV-1 infection.32,33
Diagnosis of herpes
Diagnosis may be difficult because the painful vesicular or ulcerative lesions (Figure 4) may not be visible at the time of presentation. Diagnosis is based on specific virologic and serologic tests. Nonspecific tests (eg, Tzanck smear, direct immunofluorescence) are neither sensitive nor specific and should not be relied on for diagnosis.34 HSV culture or HSV-PCR testing of a lesion is preferred. The sensitivity of viral culture can be low and is dependent on the stage of healing of a lesion and obtaining an adequate sample.
Accurate type-specific HSV serologic assays are based on HSV-specific glycoprotein G1 (HSV-1) and glycoprotein G2 (HSV-2). Unless a patient’s serologic status has already been determined, serologic testing should be done concurrently with HSV culture or PCR testing. Serologic testing enables classification of an infection as primary, nonprimary, or recurrent. For example, a patient with a positive HSV culture and negative serology most likely has primary HSV infection, and serologic study should be repeated after 6 to 8 weeks to assess for seroconversion.
Immunoglobulin M (IgM) testing for HSV-1 or HSV-2 is not diagnostic or type-specific and may be positive during recurrent genital or oral episodes of herpes.35
Treatment of herpes
In general, antiviral medications (eg, acyclovir, valacyclovir, famciclovir) are effective for managing HSV.12 Episodic or continuous suppression therapy may be needed for patients experiencing more than four outbreaks in 12 months. Patients who do not respond to treatment should be referred to an infectious disease specialist and undergo a viral culture with sensitivities.
NONINFECTIOUS CAUSES
Desquamative inflammatory vaginitis
Desquamative inflammatory vaginitis is a chronic vaginal disorder of unknown cause. It is a diagnosis of exclusion, and some patients may have a superimposed bacterial infection. It occurs mostly in perimenopausal woman and is often associated with low estrogen levels.
Diagnosis. Patients may report copious green-yellow mucoid discharge, vulvar or vaginal pain, and dyspareunia. On examination, the vulva may be erythematous, friable, and tender to the touch. The vagina may have ecchymoses, be diffusely erythematous, and have linear lesions. Mucoid or purulent discharge may be seen.
The vaginal pH is greater than 4.5.
Saline microscopy shows increased parabasal cells and leukorrhea (Figure 5).
Diagnosis is based on all of the following:
- At least 1 symptom (ie, vaginal discharge, dyspareunia, pruritus, pain, irritation, or burning)
- Vaginal inflammation on examination
- pH higher than 4.5
- Presence of parabasal cells and leukorrhea on microscopy (a ratio of leukocytes to vaginal epithelial cells > 1:1).36
Treatment involves use of 2% intravaginal clindamycin or 10% intravaginal compounded hydrocortisone cream for 4 to 6 weeks. Patients who are not cured with single-agent therapy may benefit from compounded clindamycin and hydrocortisone, with estrogen added to the formulation for hypoestrogenic patients.
Atrophic vaginitis
Atrophic vaginitis is often seen in menopausal or hypoestrogenic women. Presenting symptoms include vulvar or vaginal pain and dyspareunia.
Diagnosis. On physical examination, the vulva appears pale and atrophic, with narrowing of the introitus. Vaginal examination may reveal a pale mucosa that lacks elasticity and rugation. The examination should be performed with caution, as the vagina may bleed easily.
The vaginal pH is usually elevated.
Saline microscopy may show parabasal cells and a paucity of epithelial cells. (Figure 6).
The Vaginal Maturation Index is an indicator of the maturity of the epithelial cell types being exfoliated; these normally include parabasal (immature) cells, intermediate, and superficial (mature) cells. A predominance of immature cells indicates a hypoestrogenic state.
Infection should be considered and treated as needed.
Treatment. Patients with no contraindication may benefit from systemic hormone therapy or topical estrogen, or both.
Contact dermatitis
Contact dermatitis is classified into two types:
Irritant dermatitis, caused by the destructive action of contactants, eg, urine, feces, topical agents, feminine wipes
Allergic dermatitis, also contactant-induced, but immunologically mediated.
If a diagnosis cannot be made from the patient history and physical examination, biopsy should be performed.
Treatment of contact dermatitis involves removing the irritant, hydrating the skin with sitz baths, and using an emollient (eg, petroleum jelly) and midpotent topical steroids until resolution. Some patients benefit from topical immunosuppressive agents (eg, tacrolimus). Patients with severe symptoms may be treated with a tapering course of oral steroids for 5 to 7 days. Recalcitrant cases should be referred to a specialist.
Lichen planus
Lichen sclerosus is a benign, chronic, progressive dermatologic condition characterized by marked inflammation, epithelial thinning, and distinctive dermal changes accompanied by pruritus and pain (Figures 7 and 8).
Treatment. High-potency topical steroids are the mainstay of therapy for lichen disease. Although these are not infectious processes, superimposed infections (mostly bacterial and fungal) may also be present and should be treated.
- Van Eyk N, Allen L, Giesbrecht E, et al. Pediatric vulvovaginal disorders: a diagnostic approach and review of the literature. J Obstet Gynaecol Can 2009; 31:850–862.
- McGreal S, Wood P. Recurrent vaginal discharge in children. J Pediatr Adolesc Gynecol 2013; 26:205–208.
- Livengood CH. Bacterial vaginosis: an overview for 2009. Rev Obstet Gynecol 2009; 2:28–37.
- Atashili J, Poole C, Ndumbe PM, Adimora AA, Smith JS. Bacterial vaginosis and HIV acquisition: a meta-analysis of published studies. AIDS 2008; 22:1493–1501.
- Money D. The laboratory diagnosis of bacterial vaginosis. Can J Infect Dis Med Microbiol 2005; 16:77–79.
- Lowe NK, Neal JL, Ryan-Wenger NA, et al. Accuracy of the clinical diagnosis of vaginitis compared with a DNA probe laboratory standard. Obstet Gynecol 2009; 113:89–95.
- Mulhem E, Boyanton BL Jr, Robinson-Dunn B, Ebert C, Dzebo R. Performance of the Affirm VP-III using residual vaginal discharge collected from the speculum to characterize vaginitis in symptomatic women. J Low Genit Tract Dis 2014; 18:344–346.
- Cartwright CP, Lembke BD, Ramachandran K, et al. Comparison of nucleic acid amplification assays with BD Affirm VPIII for diagnosis of vaginitis in symptomatic women. J Clin Microbiol 2013; 51:3694–3699.
- Workowski KA, Bolan GA; Centers for Disease Control and Prevention (CDC). Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm Rep 2015; 64:1–137.
- Sobel JD, Ferris D, Schwebke J, et al. Suppressive antibacterial therapy with 0.75% metronidazole vaginal gel to prevent recurrent bacterial vaginosis. Am J Obstet Gynecol 2006; 194:1283–1289.
- Reichman O, Akins R, Sobel JD. Boric acid addition to suppressive antimicrobial therapy for recurrent bacterial vaginosis. Sex Transm Dis 2009; 36:732–734.
- Carr PL, Felsenstein D, Friedman RH. Evaluation and management of vaginitis. J Gen Intern Med 1998; 13:335–346.
- Sobel JD, Chaim W, Nagappan V, Leaman D. Treatment of vaginitis caused by Candida glabrata: use of topical boric acid and flucytosine. Am J Obstet Gynecol 2003; 189:1297–1300.
- Sobel JD, Kapernick PS, Zervos M, et al. Treatment of complicated Candida vaginitis: comparison of single and sequential doses of fluconazole. Am J Obstet Gynecol 2001; 185:363–369.
- Sobel JD, Wiesenfeld HC, Martens M, et al. Maintenance fluconazole therapy for recurrent vulvovaginal candidiasis. N Engl J Med 2004; 351:876–883.
- Phillips AJ. Treatment of non-albicans Candida vaginitis with amphotericin B vaginal suppositories. Am J Obstet Gynecol 2005; 192:2009–2013.
- Weinstock H, Berman S, Cates W Jr. Sexually transmitted diseases among American youth: incidence and prevalence estimates, 2000. Perspect Sex Reprod Health 2004; 36:6–10.
- Gatski M, Martin DH, Clark RA, Harville E, Schmidt N, Kissinger P. Co-occurrence of Trichomonas vaginalis and bacterial vaginosis among HIV-positive women. Sex Transm Dis 2011; 38:163–166.
- Hobbs MM, Seña AC. Modern diagnosis of Trichomonas vaginalis infection. Sex Transm Infect 2013; 89:434–438.
- McClelland RS, Sangare L, Hassan WM, et al. Infection with Trichomonas vaginalis increases the risk of HIV-1 acquisition. J Infect Dis 2007; 195:698–702.
- Kingston MA, Bansal D, Carlin EM. ‘Shelf life’ of Trichomonas vaginalis. Int J STD AIDS 2003; 14:28–29.
- Aslan DL, Gulbahce HE, Stelow EB, et al. The diagnosis of Trichomonas vaginalis in liquid-based Pap tests: correlation with PCR. Diagn Cytopathol 2005; 32:341–344.
- Lara-Torre E, Pinkerton JS. Accuracy of detection of Trichomonas vaginalis organisms on a liquid-based papanicolaou smear. Am J Obstet Gynecol 2003; 188:354–356.
- Hobbs MM, Lapple DM, Lawing LF, et al. Methods for detection of Trichomonas vaginalis in the male partners of infected women: implications for control of trichomoniasis. J Clin Microbiol 2006; 44:3994–3999.
- Van Der Pol B, Williams JA, Orr DP, Batteiger BE, Fortenberry JD. Prevalence, incidence, natural history, and response to treatment of Trichomonas vaginalis infection among adolescent women. J Infect Dis 2005; 192:2039–2044.
- Campbell L, Woods V, Lloyd T, Elsayed S, Church DL. Evaluation of the OSOM Trichomonas rapid test versus wet preparation examination for detection of Trichomonas vaginalis vaginitis in specimens from women with a low prevalence of infection. J Clin Microbiol 2008; 46:3467–3469.
- Chapin K, Andrea S. APTIMA Trichomonas vaginalis, a transcription-mediated amplification assay for detection of Trichomonas vaginalis in urogenital specimens. Expert Rev Mol Diagn 2011; 11:679–688.
- Krashin JW, Koumans EH, Bradshaw-Sydnor AC, et al. Trichomonas vaginalis prevalence, incidence, risk factors and antibiotic-resistance in an adolescent population. Sex Transm Dis 2010; 37:440–444.
- Sobel JD, Nyirjesy P, Brown W. Tinidazole therapy for metronidazole-resistant vaginal trichomoniasis. Clin Infect Dis 2001; 33:1341–1346.
- Nyirjesy P, Gilbert J, Mulcahy LJ. Resistant trichomoniasis: successful treatment with combination therapy. Sex Transm Dis 2011; 38:962–963.
- Bradley H, Markowitz LE, Gibson T, McQuillan GM. Seroprevalence of herpes simplex virus types 1 and 2—United States, 1999–2010. J Infect Dis 2014; 209:325–333.
- Roberts CM, Pfister JR, Spear SJ. Increasing proportion of herpes simplex virus type 1 as a cause of genital herpes infection in college students. Sex Transm Dis 2003; 30:797–800.
- Ryder N, Jin F, McNulty AM, Grulich AE, Donovan B. Increasing role of herpes simplex virus type 1 in first-episode anogenital herpes in heterosexual women and younger men who have sex with men, 1992-2006. Sex Transm Infect 2009; 85:416–419.
- Caviness AC, Oelze LL, Saz UE, Greer JM, Demmler-Harrison GJ. Direct immunofluorescence assay compared to cell culture for the diagnosis of mucocutaneous herpes simplex virus infections in children. J Clin Virol 2010; 49:58–60.
- Morrow R, Friedrich D. Performance of a novel test for IgM and IgG antibodies in subjects with culture-documented genital herpes simplex virus-1 or -2 infection. Clin Microbiol Infect 2006; 12:463–469.
- Bradford J, Fischer G. Desquamative inflammatory vaginitis: differential diagnosis and alternate diagnostic criteria. J Low Genit Tract Dis 2010; 14:306–310.
- Van Eyk N, Allen L, Giesbrecht E, et al. Pediatric vulvovaginal disorders: a diagnostic approach and review of the literature. J Obstet Gynaecol Can 2009; 31:850–862.
- McGreal S, Wood P. Recurrent vaginal discharge in children. J Pediatr Adolesc Gynecol 2013; 26:205–208.
- Livengood CH. Bacterial vaginosis: an overview for 2009. Rev Obstet Gynecol 2009; 2:28–37.
- Atashili J, Poole C, Ndumbe PM, Adimora AA, Smith JS. Bacterial vaginosis and HIV acquisition: a meta-analysis of published studies. AIDS 2008; 22:1493–1501.
- Money D. The laboratory diagnosis of bacterial vaginosis. Can J Infect Dis Med Microbiol 2005; 16:77–79.
- Lowe NK, Neal JL, Ryan-Wenger NA, et al. Accuracy of the clinical diagnosis of vaginitis compared with a DNA probe laboratory standard. Obstet Gynecol 2009; 113:89–95.
- Mulhem E, Boyanton BL Jr, Robinson-Dunn B, Ebert C, Dzebo R. Performance of the Affirm VP-III using residual vaginal discharge collected from the speculum to characterize vaginitis in symptomatic women. J Low Genit Tract Dis 2014; 18:344–346.
- Cartwright CP, Lembke BD, Ramachandran K, et al. Comparison of nucleic acid amplification assays with BD Affirm VPIII for diagnosis of vaginitis in symptomatic women. J Clin Microbiol 2013; 51:3694–3699.
- Workowski KA, Bolan GA; Centers for Disease Control and Prevention (CDC). Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm Rep 2015; 64:1–137.
- Sobel JD, Ferris D, Schwebke J, et al. Suppressive antibacterial therapy with 0.75% metronidazole vaginal gel to prevent recurrent bacterial vaginosis. Am J Obstet Gynecol 2006; 194:1283–1289.
- Reichman O, Akins R, Sobel JD. Boric acid addition to suppressive antimicrobial therapy for recurrent bacterial vaginosis. Sex Transm Dis 2009; 36:732–734.
- Carr PL, Felsenstein D, Friedman RH. Evaluation and management of vaginitis. J Gen Intern Med 1998; 13:335–346.
- Sobel JD, Chaim W, Nagappan V, Leaman D. Treatment of vaginitis caused by Candida glabrata: use of topical boric acid and flucytosine. Am J Obstet Gynecol 2003; 189:1297–1300.
- Sobel JD, Kapernick PS, Zervos M, et al. Treatment of complicated Candida vaginitis: comparison of single and sequential doses of fluconazole. Am J Obstet Gynecol 2001; 185:363–369.
- Sobel JD, Wiesenfeld HC, Martens M, et al. Maintenance fluconazole therapy for recurrent vulvovaginal candidiasis. N Engl J Med 2004; 351:876–883.
- Phillips AJ. Treatment of non-albicans Candida vaginitis with amphotericin B vaginal suppositories. Am J Obstet Gynecol 2005; 192:2009–2013.
- Weinstock H, Berman S, Cates W Jr. Sexually transmitted diseases among American youth: incidence and prevalence estimates, 2000. Perspect Sex Reprod Health 2004; 36:6–10.
- Gatski M, Martin DH, Clark RA, Harville E, Schmidt N, Kissinger P. Co-occurrence of Trichomonas vaginalis and bacterial vaginosis among HIV-positive women. Sex Transm Dis 2011; 38:163–166.
- Hobbs MM, Seña AC. Modern diagnosis of Trichomonas vaginalis infection. Sex Transm Infect 2013; 89:434–438.
- McClelland RS, Sangare L, Hassan WM, et al. Infection with Trichomonas vaginalis increases the risk of HIV-1 acquisition. J Infect Dis 2007; 195:698–702.
- Kingston MA, Bansal D, Carlin EM. ‘Shelf life’ of Trichomonas vaginalis. Int J STD AIDS 2003; 14:28–29.
- Aslan DL, Gulbahce HE, Stelow EB, et al. The diagnosis of Trichomonas vaginalis in liquid-based Pap tests: correlation with PCR. Diagn Cytopathol 2005; 32:341–344.
- Lara-Torre E, Pinkerton JS. Accuracy of detection of Trichomonas vaginalis organisms on a liquid-based papanicolaou smear. Am J Obstet Gynecol 2003; 188:354–356.
- Hobbs MM, Lapple DM, Lawing LF, et al. Methods for detection of Trichomonas vaginalis in the male partners of infected women: implications for control of trichomoniasis. J Clin Microbiol 2006; 44:3994–3999.
- Van Der Pol B, Williams JA, Orr DP, Batteiger BE, Fortenberry JD. Prevalence, incidence, natural history, and response to treatment of Trichomonas vaginalis infection among adolescent women. J Infect Dis 2005; 192:2039–2044.
- Campbell L, Woods V, Lloyd T, Elsayed S, Church DL. Evaluation of the OSOM Trichomonas rapid test versus wet preparation examination for detection of Trichomonas vaginalis vaginitis in specimens from women with a low prevalence of infection. J Clin Microbiol 2008; 46:3467–3469.
- Chapin K, Andrea S. APTIMA Trichomonas vaginalis, a transcription-mediated amplification assay for detection of Trichomonas vaginalis in urogenital specimens. Expert Rev Mol Diagn 2011; 11:679–688.
- Krashin JW, Koumans EH, Bradshaw-Sydnor AC, et al. Trichomonas vaginalis prevalence, incidence, risk factors and antibiotic-resistance in an adolescent population. Sex Transm Dis 2010; 37:440–444.
- Sobel JD, Nyirjesy P, Brown W. Tinidazole therapy for metronidazole-resistant vaginal trichomoniasis. Clin Infect Dis 2001; 33:1341–1346.
- Nyirjesy P, Gilbert J, Mulcahy LJ. Resistant trichomoniasis: successful treatment with combination therapy. Sex Transm Dis 2011; 38:962–963.
- Bradley H, Markowitz LE, Gibson T, McQuillan GM. Seroprevalence of herpes simplex virus types 1 and 2—United States, 1999–2010. J Infect Dis 2014; 209:325–333.
- Roberts CM, Pfister JR, Spear SJ. Increasing proportion of herpes simplex virus type 1 as a cause of genital herpes infection in college students. Sex Transm Dis 2003; 30:797–800.
- Ryder N, Jin F, McNulty AM, Grulich AE, Donovan B. Increasing role of herpes simplex virus type 1 in first-episode anogenital herpes in heterosexual women and younger men who have sex with men, 1992-2006. Sex Transm Infect 2009; 85:416–419.
- Caviness AC, Oelze LL, Saz UE, Greer JM, Demmler-Harrison GJ. Direct immunofluorescence assay compared to cell culture for the diagnosis of mucocutaneous herpes simplex virus infections in children. J Clin Virol 2010; 49:58–60.
- Morrow R, Friedrich D. Performance of a novel test for IgM and IgG antibodies in subjects with culture-documented genital herpes simplex virus-1 or -2 infection. Clin Microbiol Infect 2006; 12:463–469.
- Bradford J, Fischer G. Desquamative inflammatory vaginitis: differential diagnosis and alternate diagnostic criteria. J Low Genit Tract Dis 2010; 14:306–310.
KEY POINTS
- Typical presenting symptoms of vulvovaginitis are itching, burning, and abnormal discharge.
- Evaluating vaginal secretions with simple office-based tools is often sufficient for diagnosis, although DNA testing is also available.
- Depending on the cause, vulvovaginitis is generally treated with a course of oral or topical antibiotics, antiviral or antifungal drugs, anti-inflammatory agents, or hormonal therapy.
- Cases that do not resolve may require maintenance therapy. Patients who have persistent or unusual symptoms should be referred to a specialist.
Understanding the brexpiprazole therapeutic window: Why more isn’t always better
Dosage windows could be difficult to understand pharmacologically, but for a partial agonist the presumed mechanism could be more evident. Clinicians should be aware that more is not always better, meaning that with partial agonist drugs a higher dosage might not lead to greater patient response. With brexpiprazole, a dopamine D2 partial agonist FDA-approved for schizophrenia and an adjunct for major depressive disorder (MDD),1 moderation is best because of
Recommended dosage
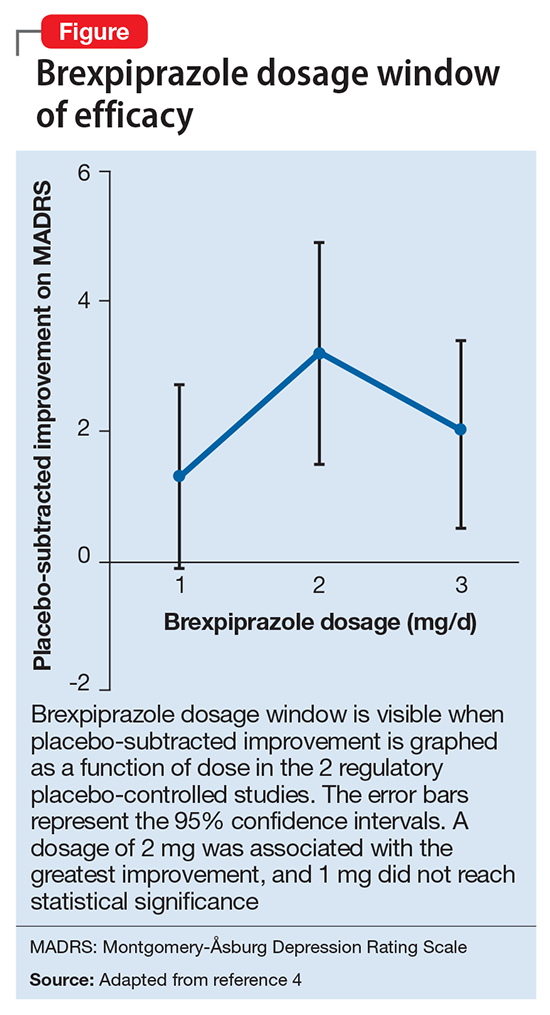
Two placebo-controlled studies2,3 examined brexpiprazole dosages of 1, 2, and 3 mg/d. The recommended dosage of 2 mg/d for MDD was determined by changes in Montgomery-Åsburg Depression Rating Scale scores (Figure).4 Lower dosages of 1 mg/d did not reach statistical significance, and 3 mg/d were less effective than the intermediate dosage of 2 mg/d. This result suggests a window of efficacy for brexpiprazole for MDD. This therapeutic window likely applies to most patients; however, patient-specific variables could alter the optimum dosage.
Dosage window
Brexpiprazole has high affinity for dopamine D2, D3, serotonin 5-HT1A, 5-HT2A, norepinephrine α1B, and α2 Creceptors. At relatively low drug concentrations, brexpiprazole achieves high receptor occupancy. At receptors for which brexpiprazole is a partial agonist (5-HT1A, D2, D3) the drug blocks the receptor and stimulates it at a fraction of the endogenous neurotransmitter. With a very high affinity agent, the endogenous neurotransmitter could be completely excluded from interacting with these receptors if brexpiprazole occupancy is high. At lower dosages, the drug occupies only a fraction of the receptors, allowing the endogenous neurotransmitters to continue interacting with their receptors, thereby magnifying the signal of that receptor above baseline.
1. FDA approves Rexulti (brexpiprazole) as adjunctive treatment for adults with major depressive disorder and as a treatment for adults with schizophrenia [news release]. Valby, Denmark; Tokyo, Japan: H. Lundbeck A/S (Lundbeck); Otsuka Pharmaceutical Co., Ltd; July 11, 2015. http://investor.lundbeck.com/ releasedetail.cfm?Release ID=921621. Accessed October 3, 2015.
2. Thase ME, Youakim JM, Skuban A, et al. Adjunctive brexpiprazole 1 and 3 mg for patients with major depressive disorder following inadequate response to antidepressants: a phase 3, randomized, double-blind study. J Clin Psychiatry. 2015;76(9): 1232-1240.
3. Thase ME, Youakim JM, Skuban A, et al. Efficacy and safety of adjunctive brexpiprazole 2 mg in major depressive disorder: a phase 3, randomized, placebocontrolled study in patients with inadequate response to antidepressants. J Clin Psychiatry. 2015;76(9):1224-1231.
4. Rexulti [package insert]. Rockville, MD: Otsuka; 2015.
Dosage windows could be difficult to understand pharmacologically, but for a partial agonist the presumed mechanism could be more evident. Clinicians should be aware that more is not always better, meaning that with partial agonist drugs a higher dosage might not lead to greater patient response. With brexpiprazole, a dopamine D2 partial agonist FDA-approved for schizophrenia and an adjunct for major depressive disorder (MDD),1 moderation is best because of
Recommended dosage
Two placebo-controlled studies2,3 examined brexpiprazole dosages of 1, 2, and 3 mg/d. The recommended dosage of 2 mg/d for MDD was determined by changes in Montgomery-Åsburg Depression Rating Scale scores (Figure).4 Lower dosages of 1 mg/d did not reach statistical significance, and 3 mg/d were less effective than the intermediate dosage of 2 mg/d. This result suggests a window of efficacy for brexpiprazole for MDD. This therapeutic window likely applies to most patients; however, patient-specific variables could alter the optimum dosage.
Dosage window
Brexpiprazole has high affinity for dopamine D2, D3, serotonin 5-HT1A, 5-HT2A, norepinephrine α1B, and α2 Creceptors. At relatively low drug concentrations, brexpiprazole achieves high receptor occupancy. At receptors for which brexpiprazole is a partial agonist (5-HT1A, D2, D3) the drug blocks the receptor and stimulates it at a fraction of the endogenous neurotransmitter. With a very high affinity agent, the endogenous neurotransmitter could be completely excluded from interacting with these receptors if brexpiprazole occupancy is high. At lower dosages, the drug occupies only a fraction of the receptors, allowing the endogenous neurotransmitters to continue interacting with their receptors, thereby magnifying the signal of that receptor above baseline.
Dosage windows could be difficult to understand pharmacologically, but for a partial agonist the presumed mechanism could be more evident. Clinicians should be aware that more is not always better, meaning that with partial agonist drugs a higher dosage might not lead to greater patient response. With brexpiprazole, a dopamine D2 partial agonist FDA-approved for schizophrenia and an adjunct for major depressive disorder (MDD),1 moderation is best because of
Recommended dosage
Two placebo-controlled studies2,3 examined brexpiprazole dosages of 1, 2, and 3 mg/d. The recommended dosage of 2 mg/d for MDD was determined by changes in Montgomery-Åsburg Depression Rating Scale scores (Figure).4 Lower dosages of 1 mg/d did not reach statistical significance, and 3 mg/d were less effective than the intermediate dosage of 2 mg/d. This result suggests a window of efficacy for brexpiprazole for MDD. This therapeutic window likely applies to most patients; however, patient-specific variables could alter the optimum dosage.
Dosage window
Brexpiprazole has high affinity for dopamine D2, D3, serotonin 5-HT1A, 5-HT2A, norepinephrine α1B, and α2 Creceptors. At relatively low drug concentrations, brexpiprazole achieves high receptor occupancy. At receptors for which brexpiprazole is a partial agonist (5-HT1A, D2, D3) the drug blocks the receptor and stimulates it at a fraction of the endogenous neurotransmitter. With a very high affinity agent, the endogenous neurotransmitter could be completely excluded from interacting with these receptors if brexpiprazole occupancy is high. At lower dosages, the drug occupies only a fraction of the receptors, allowing the endogenous neurotransmitters to continue interacting with their receptors, thereby magnifying the signal of that receptor above baseline.
1. FDA approves Rexulti (brexpiprazole) as adjunctive treatment for adults with major depressive disorder and as a treatment for adults with schizophrenia [news release]. Valby, Denmark; Tokyo, Japan: H. Lundbeck A/S (Lundbeck); Otsuka Pharmaceutical Co., Ltd; July 11, 2015. http://investor.lundbeck.com/ releasedetail.cfm?Release ID=921621. Accessed October 3, 2015.
2. Thase ME, Youakim JM, Skuban A, et al. Adjunctive brexpiprazole 1 and 3 mg for patients with major depressive disorder following inadequate response to antidepressants: a phase 3, randomized, double-blind study. J Clin Psychiatry. 2015;76(9): 1232-1240.
3. Thase ME, Youakim JM, Skuban A, et al. Efficacy and safety of adjunctive brexpiprazole 2 mg in major depressive disorder: a phase 3, randomized, placebocontrolled study in patients with inadequate response to antidepressants. J Clin Psychiatry. 2015;76(9):1224-1231.
4. Rexulti [package insert]. Rockville, MD: Otsuka; 2015.
1. FDA approves Rexulti (brexpiprazole) as adjunctive treatment for adults with major depressive disorder and as a treatment for adults with schizophrenia [news release]. Valby, Denmark; Tokyo, Japan: H. Lundbeck A/S (Lundbeck); Otsuka Pharmaceutical Co., Ltd; July 11, 2015. http://investor.lundbeck.com/ releasedetail.cfm?Release ID=921621. Accessed October 3, 2015.
2. Thase ME, Youakim JM, Skuban A, et al. Adjunctive brexpiprazole 1 and 3 mg for patients with major depressive disorder following inadequate response to antidepressants: a phase 3, randomized, double-blind study. J Clin Psychiatry. 2015;76(9): 1232-1240.
3. Thase ME, Youakim JM, Skuban A, et al. Efficacy and safety of adjunctive brexpiprazole 2 mg in major depressive disorder: a phase 3, randomized, placebocontrolled study in patients with inadequate response to antidepressants. J Clin Psychiatry. 2015;76(9):1224-1231.
4. Rexulti [package insert]. Rockville, MD: Otsuka; 2015.
Hepatitis C among the mentally ill: Review and treatment update
At approximately 3 to 4 million patients, hepatitis C virus (HCV) is the most common viral hepatitis in the United States. Patients with mental illness are disproportionately affected by HCV and the management of their disease poses particular challenges.
HCV is commonly transmitted via IV drug use and blood transfusions; transmission through sexual contact is rare. Most patients with HCV are asymptomatic, although some do develop symptoms of acute hepatitis. Most HCV infections become chronic, with a high incidence of liver failure requiring liver transplantation.
Hepatitis refers to inflammation of the liver, which could have various etiologies, including viral infections, alcohol abuse, or autoimmune disease. Viral hepatitis refers to infection from 5 distinct groups of virus, coined A through E.1 This article will focus on chronic HCV (Table 1).
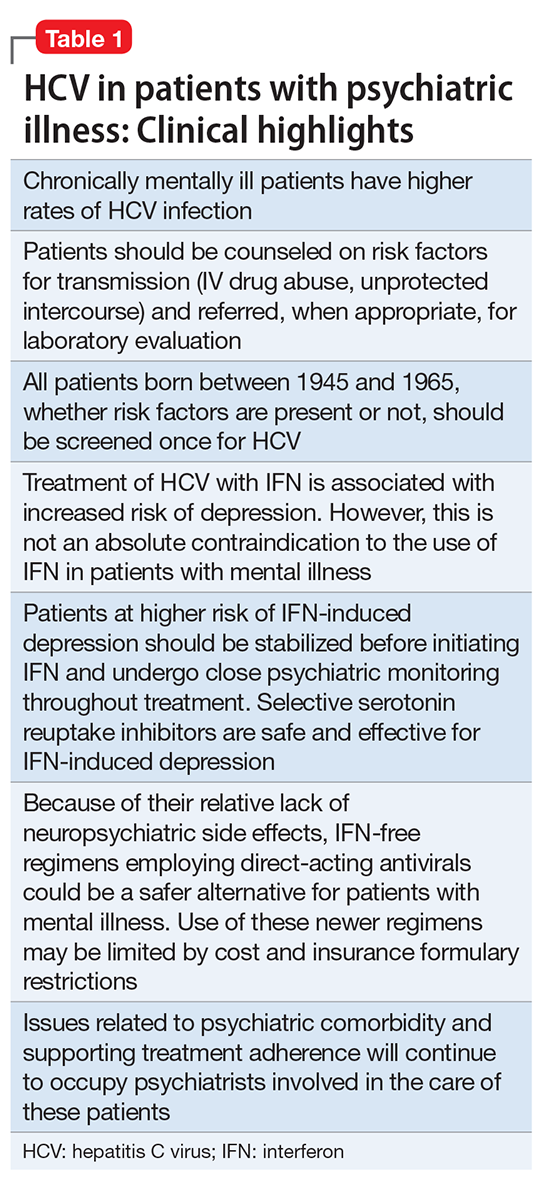
CASE Bipolar disorder, stress, history of IV drug use
Ms. S, age 48, has bipolar I disorder and has been hospitalized 4 times in the past, including once for a suicide attempt. She has 3 children and works as a cashier. Her psychiatric symptoms have been stable on lurasidone, 80 mg/d, and escitalopram, 10 mg/d. Recently, Ms. S has been under more stress at her job. Sometimes she misses doses of her medication, and then becomes more irritable and impulsive. Her husband, noting that she has used IV heroin in the past, comes with her today and is concerned that she is “not acting right.” What is Ms. S’s risk for HCV?
HCV in mental illness
Compared with the general population, HCV is more prevalent among chronically mentally ill persons. In one study, HCV occurred twice as often in men vs women with chronic mental illness.2 Up to 50% of patients with HCV have a history of mental illness and nearly 90% have a history of substance use disorders.3 Among 668 chronically mentally ill patients at 4 public sector clinics, risk factors for HCV were common and included use of injection drugs (>20%), sharing needles (14%), and crack cocaine use (>20%).4 Higher rates of HCV were reported in hospitalized patients with schizophrenia and comorbid psychoactive substance abuse in Japan.5 Because of the high prevalence in this population, it is essential to assess for substance use disorders. Employing a non-judgmental approach with motivational interviewing techniques can be effective.6
Individuals with mental illness should be screened for HCV risk factors, such as unprotected intercourse with high-risk partners and sharing needles used for illicit drug use. Patients frequently underreport these activities. At-risk individuals should undergo laboratory testing for the HIV-1 antibody, hepatitis C antibodies, and hepatitis B antibodies. Mental health providers should counsel patients about risk reduction (eg, avoiding unprotected sexual intercourse and sharing of drug paraphernalia). Educating patients about complications of viral hepatitis, such as liver failure, could be motivation to change risky behaviors.
CASE continued
During your interview with Ms. S, she becomes irritable and tells you that you are asking too many questions. It is clear that she is not taking her medications consistently, but she agrees to do so because she does not want to lose custody of her children. She denies current use of heroin but her husband says, “I don’t know what she is doing.” In addition to advising her on reducing risk factors, you order appropriate screening tests, including hepatitis and HIV antibody tests.
Screening guidelines
The U.S. Preventive Services Task Force and the CDC both recommend a 1-time screening for HCV in asymptomatic or low-risk patients born between 1945 and 1965.1,7 Furthermore, both organizations recommend screening for HCV in persons at high risk, including:
- those with a history of injection drug use
- persons with recognizable exposure, such as needlesticks
- persons who received blood transfusions before 1992
- medical conditions, such as long-term dialysis.
There is no vaccine for HCV; however, patients with HCV should receive vaccination against hepatitis B.
Diagnosis
Acute symptoms include fever, fatigue, headache, cough, nausea, and vomiting. Jaundice could develop, often accompanied by pain in the right upper quadrant. If there is suspicion of viral hepatitis, psychiatrists can initiate the laboratory evaluation. Chronic hepatitis, on the other hand, often is asymptomatic, although stigmata of chronic liver disease (eg, jaundice, ascites, peripheral edema) might be detected on physical exam.8 Elevated serum transaminases are seen with acute viral hepatitis, although levels could vary in chronic cases. Serologic detection of anti-HCV antibodies establishes a HCV diagnosis.
Treatment recommendations
All patients who test positive for HCV should be evaluated and treated by a hepatologist. Goals of therapy are to reduce complications from chronic viral hepatitis, including cirrhosis and hepatic failure. Duration and optimal regimen depends on the HCV genotype.8 Treatment outcomes are measured by virological parameters, including serum aminotransferases, HCV RNA levels, and histology. The most important parameter in treating chronic HCV is the sustained virological response (SVR), which is the absence of HCV RNA 12 weeks after completing therapy.9
Treatment is recommended for all persons with chronic HCV infection, according to current treatment guidelines, which are updated regularly by the American Association for the Study of Liver Diseases and the Infectious Diseases Society of America.10 Until recently, treatment consisted of IV pegylated interferon (PEG-IFN) in combination with oral ribavirin. Success rates with this regimen are approximately 40% to 50%. The advent of direct-acting antivirals (DAAs) has revolutionized treatment of chronic HCV. These agents include simeprevir, sofosbuvir, ledipasvir, and the combination of ombitasvir-paritaprevir-ritonavir plus dasabuvir (brand name, Viekira Pak). Advantages of these agents are oral administration, high treatment success rates (>90%), shorter treatment duration (12 weeks vs up to 48 weeks with older regimens), and few serious adverse effects9-11; drawbacks include the pricing of these regimens, which could cost upward of ≥$100,000 for a 12-week course, and a lack of coverage under some health insurance plans.12 The manufacturers of 2 agents, telaprevir and boceprevir, removed them from the market because of decreased demand related to their unfavorable side-effect profile and the availability of better tolerated agents.
Treatment considerations for interferon in psychiatric patients
Various neuropsychiatric symptoms have been reported with the use of PEG-IFN. The range of reported symptoms include:
- depressed mood
- anxiety
- hostility
- slowness
- fatigue
- sleep disturbance
- lethargy
- irritability
- emotional lability
- social withdrawal
- poor concentration.13,14
Depressive symptoms can present as early as 1 month after starting treatment, but typically occur at 8 to 12 weeks. A systematic review and meta-analysis of 26 observational studies found a cumulative 25% risk of interferon (IFN)-induced depression in the general HCV population.15 Risk factors for IFN-induced depression include:
- female sex
- history of major depression or other psychiatric disorder
- low educational level
- the presence of baseline subthreshold depressive symptoms.
Because of the risk of inducing depression, there was initial hesitation with providing IFN treatment to patients with psychiatric disorders. However, there is evidence that individuals with chronic psychiatric illness can be treated safely with IFN-based regimens and achieve results similar to non-psychiatric populations.16,17 For example, patients with schizophrenia in a small Veterans Affairs database who received IFN for HCV did not experience higher rates of symptoms of schizophrenia, depression, or mania over 8 years of follow-up.18 Furthermore, those with schizophrenia were just as likely to reach SVR as patients without psychiatric illness.19 Other encouraging results have been reported in depressed patients. One study found similar rates of treatment completion and SVR in patients with a history of major depressive disorder compared with those without depression.20 No difference in frequency of neuropsychiatric side effects was found between the groups.
Presence of a psychiatric disorder is no longer an absolute contraindication to IFN treatment for HCV. Optimal control of psychiatric symptoms should be attained in all patients before starting HCV treatment, and close clinical monitoring is warranted. A review of 9 studies showed benefit of antidepressants for HCV patients with elevated baseline depression or a history of IFN-induced depression.21 The largest body of evidence supports the safety and efficacy of selective serotonin reuptake inhibitors for treating IFN-induced depression. Although no antidepressants are FDA-approved for this indication, the best-studied agents include citalopram, escitalopram, sertraline, and paroxetine.
A review of 6 studies on using antidepressants to prevent IFN-induced depression concluded there was inadequate evidence to support this approach in all patients.22 Pretreatment primarily is indicated for those with elevated depressive symptoms at baseline or those with a history of IFN-induced depression. The prevailing approach to IFN-induced depression assessment, prevention, and treatment is summarized in Table 2.

CASE continued
Ms. S tests positive for the HCV antibody but negative for HIV and hepatitis B. She immediately receives the hepatitis B vaccine series. Her sister discourages her from receiving treatment for HCV, warning her, “it will make you crazy depressed.” As a result, Ms. S avoids following up with the hepatologist. Her psychiatrist, aware that she now was taking her psychotropic medication and seeing that her mood is stable, educates her about new treatment options for HCV that do not cause depression. Ms. S finally agrees to see a hepatologist to discuss her treatment options.
IFN-free regimens
With the arrival of the DAAs, the potential now exists to use IFN-free treatment regimens,10 which could eliminate concerns about IFN-induced depression.
Clinical trials of the DAAs and real-world use so far do not indicate an elevated risk for neuropsychiatric symptoms, including depression.11 As a result, more patients with severe psychiatric illness likely will be eligible to receive treatment for HCV. However, as clinical experience builds with these new agents, it is important to monitor the experience of patients with psychiatric comorbidity. Current treatment guidelines for HCV genotype 1, which is most common in the United States, do not include IFN-based regimens.10 Treatment of genotype 3, which affects 6% of the U.S. population, still includes IFN. Therefore, the risk of IFN-induced depression still exists for some patients with HCV. Table 310 describes current treatment regimens in use for HCV without cirrhosis (see Related Resources for treating HCV with cirrhosis).
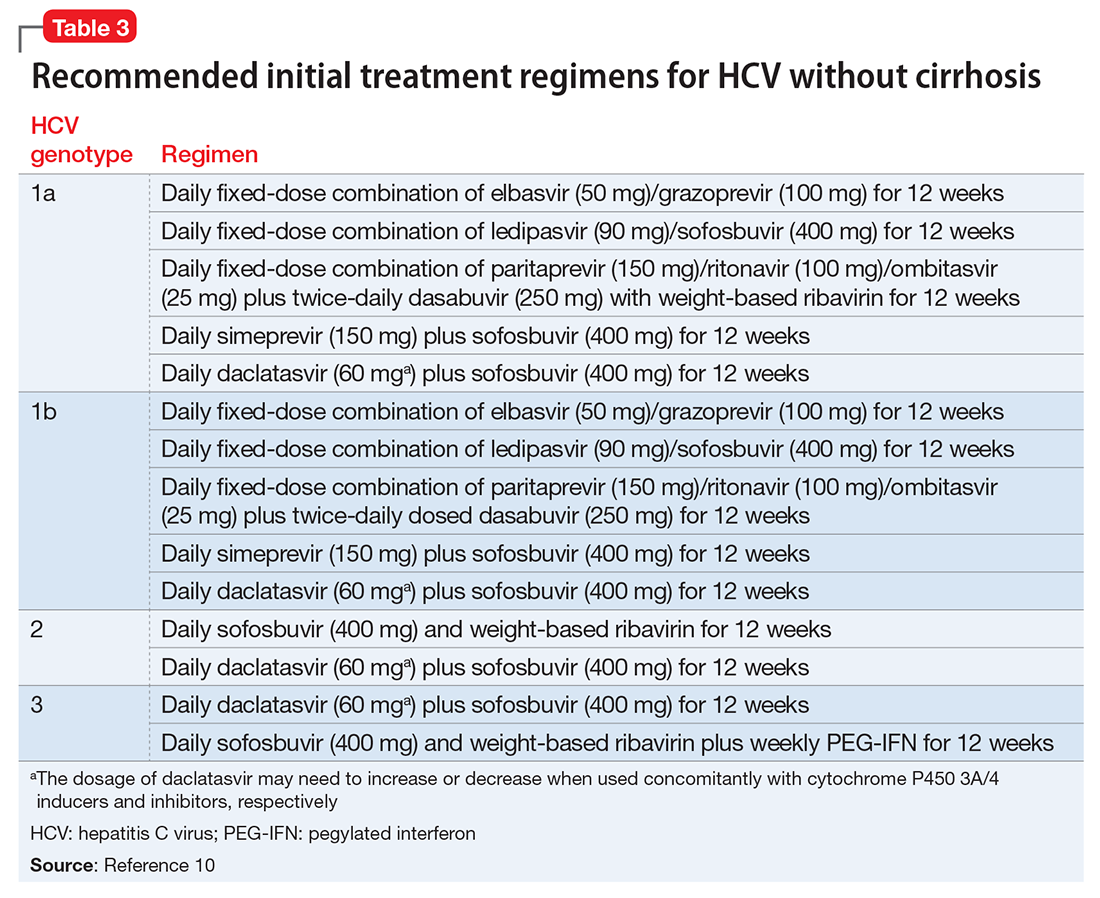
Evolving role of the psychiatrist
The availability of shorter, better-tolerated regimens means that the psychiatric contraindications to HCV treatment will be eased. With the emergence of non-IFN treatment regimens, the role of mental health providers could shift toward assisting with treatment adherence, monitoring drug–drug interactions, and managing comorbid substance use disorders.10
The psychiatrist’s role might shift away from the psychosocial assessment of factors affecting treatment eligibility, such as IFN-associated depressive symptoms. Clinical focus will likely shift to supporting adherence to HCV treatment regimens.23 Because depression and substance use disorders are risk factors for non-adherence, mental health providers may be called upon to optimize treatment of these conditions before beginning DAA regimens. A multi-dose regimen might be complicated for those with severe mental illness, and increased psychiatric and community support could be needed in these patients.23 Furthermore, models of care that integrate an HCV specialist with psychiatric care have demonstrated benefits.6,23 Long-term follow-up with a mental health provider will be key to provide ongoing psychiatric support, especially for those who do not achieve SVR.
Psychotropic drug–drug interactions with DAAs
Both sofosbuvir and ledipasvir are substrates of P-glycoprotein and not metabolized by cytochrome P450 (CYP) enzymes.24 Therefore, there are no known contraindications with psychotropic medications. However, co-administration of P-glycoprotein inducers, such as St. John’s wort, could reduce sofosbuvir and ledipasvir levels leading to reduced therapeutic efficacy.
Because it has been used for many years as an HIV treatment, drug interactions with ritonavir have been well-described. This agent is a “pan-inhibitor” and inhibits the CYP3A4, 2D6, 2C9, and 2C19 enzymes and could increase levels of any psychotropic metabolized by these enzymes.25 After several weeks of treatment, it also could induce CYP3A4, which could lead to reduced efficacy of oral contraceptives because ethinylestradiol is metabolized by CYP3A4. Ritonavir is primarily metabolized by CYP3A4 (and CYP2D6 to a smaller degree). Carbamazepine induces CYP3A4, which may lead to decreased levels of ritonavir.23 This, in turn, could reduce the likelihood of attaining SVR and successful treatment of HCV.
Boceprevir, telaprevir, and simeprevir inhibit CYP3A4 to varying degrees and therefore could affect psychotropic medications metabolized by this enzyme.23,26,27 These DAAs are metabolized by CYP3A4; therefore CYP3A4 inducers, such as carbamazepine, could lower DAA blood levels, increasing risk of HCV treatment failure and viral resistance.
Daclatasvir is a substrate of CYP3A4 and an inhibitor of P-glycoprotein.28 Concomitant buprenorphine or buprenorphine/naloxone levels may be increased, although the manufacturer does not recommend dosage adjustment. Elbasvir and grazoprevir are metabolized by CYP3A4.29 Drug–drug interactions therefore may result when administered with either CYP3A4 inducers or inhibitors.
CASE Conclusion
Ms. S sees her new hepatologist, Dr. Smith. She decides to try a 12-week course of ledipasvir/sofosbuvir. Dr. Smith collaborates frequently with Ms. S’s psychiatrist to discuss her case and to help monitor her psychiatric symptoms. She follows up closely with her psychiatrist for symptom monitoring and to help ensure treatment compliance. Ms. S does well with the IFN-free treatment regimen and experiences no worsening of her psychiatric symptoms during treatment.
1. Centers for Disease Control and Prevention. Viral hepatitis. http://www.cdc.gov/hepatitis. Updated December 9, 2016. Accessed February 9, 2017.
2. Butterfield MI, Bosworth HB, Meador KG, et al. Five-Site Health and Risk Study Research Committee. Gender differences in hepatitis C infection and risks among persons with severe mental illness. Psychiatr Serv. 2003;54(6):848-853.
3. Rifai MA, Gleason OC, Sabouni D. Psychiatric care of the patient with hepatitis C: a review of the literature. Prim Care Companion J Clin Psychiatry. 2010;12(6):PCC.09r00877. doi: 10.4088/PCC.09r00877whi.
4. Dinwiddie SH, Shicker L, Newman T. Prevalence of hepatitis C among psychiatric patients in the public sector. Am J Psychiatry. 2003;160(1):172-174.
5. Nakamura Y, Koh M, Miyoshi E, et al. High prevalence of the hepatitis C virus infection among the inpatients of schizophrenia and psychoactive substance abuse in Japan. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28(3):591-597.
6. Sockalingam S, Blank D, Banga CA, et al. A novel program for treating patients with trimorbidity: hepatitis C, serious mental illness, and substance abuse. Eur J Gastroenterol Hepatol. 2013;25(12):1377-1384.
7. U.S. Preventive Services Task Force. Screening for hepatitis C virus infection: recommendation summary. https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/hepatitis-c-screening. Published June 2013. Accessed February 9, 2017.
8. Longo DL, Fauci AS, Kasper DL. Harrison’s principles of internal medicine. 18th ed. New York, NY: McGraw-Hill; 2012.
9. Belousova V, Abd-Rabou AA, Mousa SA. Recent advances and future directions in the management of hepatitis C infections. Pharmacol Ther. 2015;145:92-102.
10. American Association for the Study of Liver Diseases (AASLD); The Infectious Diseases Society of America (IDSA). HCV guidance: recommendations for testing, managing, and treating hepatitis C. http://www.hcvguidelines.org. Accessed February 9, 2017.
11. Rowan PJ, Bhulani N. Psychosocial assessment and monitoring in the new era of non-interferon-alpha hepatitis C treatments. World J Hepatol. 2015;7(19):2209-2213.
12. Good Rx, Inc. http://www.goodrx.com. Accessed October 9, 2015.
13. Raison CL, Borisov AS, Broadwell SD, et al. Depression during pegylated interferon-alpha plus ribavirin therapy: prevalence and prediction. J Clin Psychiatry. 2005;66(1):41-48.
14. Lotrich FE, Rabinovitz M, Gironda P, et al. Depression following pegylated interferon-alpha: characteristics and vulnerability. J Psychosom Res. 2007;63(2):131-135.
15. Udina M, Castellví P, Moreno-España J, et al. Interferon-induced depression in chronic hepatitis C: a systematic review and meta-analysis. J Clin Psychiatry. 2012;73(8):1128-1138.
16. Mustafa MZ, Schofield J, Mills PR, et al. The efficacy and safety of treating hepatitis C in patients with a diagnosis of schizophrenia. J Viral Hepat. 2014;21(7):e48-e51.
17. Huckans M, Mitchell A, Pavawalla S, et al. The influence of antiviral therapy on psychiatric symptoms among patients with hepatitis C and schizophrenia. Antivir Ther. 2010;15(1):111-119.
18. Huckans MS, Blackwell AD, Harms TA, et al. Management of hepatitis C disease among VA patients with schizophrenia and substance use disorders. Psychiatr Serv. 2006;57(3):403-406.
19. Huckans M, Mitchell A, Ruimy S, et al. Antiviral therapy completion and response rates among hepatitis C patients with and without schizophrenia. Schizophr Bull. 2010;36(1):165-172.
20. Hauser P, Morasco BJ, Linke A, et al. Antiviral completion rates and sustained viral response in hepatitis C patient with and without preexisting major depressive disorder. Psychosomatics. 2009;50(5):500-505.
21. Sockalingam S, Abbey SE. Managing depression during hepatitis C treatment. Can J Psychiatry. 2009;54(9):614-625.
22. Galvão-de Almeida A, Guindalini C, Batista-Neves S, et al. Can antidepressants prevent interferon-alpha-induced depression? A review of the literature. Gen Hosp Psychiatry. 2010;32(4):401-405.
23. Sockalingam S, Sheehan K, Feld JJ, et al. Psychiatric care during hepatitis c treatment: the changing role of psychiatrists in the era of direct-acting antivirals. Am J Psychiatry. 2015;172(6):512-516.
24. Harvoni [package insert]. Foster City, CA: Gilead Sciences, Inc.; 2016.
25. Wynn GH, Oesterheld, JR, Cozza KL, et al. Clinical manual of drug interactions principles for medical practice. Arlington, VA: American Psychiatric Publishing; 2009.
26. Olysio [package insert]. Titusville, NJ: Janssen Therapeutics; 2016.
27. Victrelis [package insert]. Whitehouse Station, NJ: Merck & Co.; 2017.
28. Daklinza [package insert]. Princeton, NJ: Bristol-Myers Squibb; 2016.
29. Zepatier [package insert]. Whitehouse Station, NJ: Merck & Co.; 2017.
At approximately 3 to 4 million patients, hepatitis C virus (HCV) is the most common viral hepatitis in the United States. Patients with mental illness are disproportionately affected by HCV and the management of their disease poses particular challenges.
HCV is commonly transmitted via IV drug use and blood transfusions; transmission through sexual contact is rare. Most patients with HCV are asymptomatic, although some do develop symptoms of acute hepatitis. Most HCV infections become chronic, with a high incidence of liver failure requiring liver transplantation.
Hepatitis refers to inflammation of the liver, which could have various etiologies, including viral infections, alcohol abuse, or autoimmune disease. Viral hepatitis refers to infection from 5 distinct groups of virus, coined A through E.1 This article will focus on chronic HCV (Table 1).
CASE Bipolar disorder, stress, history of IV drug use
Ms. S, age 48, has bipolar I disorder and has been hospitalized 4 times in the past, including once for a suicide attempt. She has 3 children and works as a cashier. Her psychiatric symptoms have been stable on lurasidone, 80 mg/d, and escitalopram, 10 mg/d. Recently, Ms. S has been under more stress at her job. Sometimes she misses doses of her medication, and then becomes more irritable and impulsive. Her husband, noting that she has used IV heroin in the past, comes with her today and is concerned that she is “not acting right.” What is Ms. S’s risk for HCV?
HCV in mental illness
Compared with the general population, HCV is more prevalent among chronically mentally ill persons. In one study, HCV occurred twice as often in men vs women with chronic mental illness.2 Up to 50% of patients with HCV have a history of mental illness and nearly 90% have a history of substance use disorders.3 Among 668 chronically mentally ill patients at 4 public sector clinics, risk factors for HCV were common and included use of injection drugs (>20%), sharing needles (14%), and crack cocaine use (>20%).4 Higher rates of HCV were reported in hospitalized patients with schizophrenia and comorbid psychoactive substance abuse in Japan.5 Because of the high prevalence in this population, it is essential to assess for substance use disorders. Employing a non-judgmental approach with motivational interviewing techniques can be effective.6
Individuals with mental illness should be screened for HCV risk factors, such as unprotected intercourse with high-risk partners and sharing needles used for illicit drug use. Patients frequently underreport these activities. At-risk individuals should undergo laboratory testing for the HIV-1 antibody, hepatitis C antibodies, and hepatitis B antibodies. Mental health providers should counsel patients about risk reduction (eg, avoiding unprotected sexual intercourse and sharing of drug paraphernalia). Educating patients about complications of viral hepatitis, such as liver failure, could be motivation to change risky behaviors.
CASE continued
During your interview with Ms. S, she becomes irritable and tells you that you are asking too many questions. It is clear that she is not taking her medications consistently, but she agrees to do so because she does not want to lose custody of her children. She denies current use of heroin but her husband says, “I don’t know what she is doing.” In addition to advising her on reducing risk factors, you order appropriate screening tests, including hepatitis and HIV antibody tests.
Screening guidelines
The U.S. Preventive Services Task Force and the CDC both recommend a 1-time screening for HCV in asymptomatic or low-risk patients born between 1945 and 1965.1,7 Furthermore, both organizations recommend screening for HCV in persons at high risk, including:
- those with a history of injection drug use
- persons with recognizable exposure, such as needlesticks
- persons who received blood transfusions before 1992
- medical conditions, such as long-term dialysis.
There is no vaccine for HCV; however, patients with HCV should receive vaccination against hepatitis B.
Diagnosis
Acute symptoms include fever, fatigue, headache, cough, nausea, and vomiting. Jaundice could develop, often accompanied by pain in the right upper quadrant. If there is suspicion of viral hepatitis, psychiatrists can initiate the laboratory evaluation. Chronic hepatitis, on the other hand, often is asymptomatic, although stigmata of chronic liver disease (eg, jaundice, ascites, peripheral edema) might be detected on physical exam.8 Elevated serum transaminases are seen with acute viral hepatitis, although levels could vary in chronic cases. Serologic detection of anti-HCV antibodies establishes a HCV diagnosis.
Treatment recommendations
All patients who test positive for HCV should be evaluated and treated by a hepatologist. Goals of therapy are to reduce complications from chronic viral hepatitis, including cirrhosis and hepatic failure. Duration and optimal regimen depends on the HCV genotype.8 Treatment outcomes are measured by virological parameters, including serum aminotransferases, HCV RNA levels, and histology. The most important parameter in treating chronic HCV is the sustained virological response (SVR), which is the absence of HCV RNA 12 weeks after completing therapy.9
Treatment is recommended for all persons with chronic HCV infection, according to current treatment guidelines, which are updated regularly by the American Association for the Study of Liver Diseases and the Infectious Diseases Society of America.10 Until recently, treatment consisted of IV pegylated interferon (PEG-IFN) in combination with oral ribavirin. Success rates with this regimen are approximately 40% to 50%. The advent of direct-acting antivirals (DAAs) has revolutionized treatment of chronic HCV. These agents include simeprevir, sofosbuvir, ledipasvir, and the combination of ombitasvir-paritaprevir-ritonavir plus dasabuvir (brand name, Viekira Pak). Advantages of these agents are oral administration, high treatment success rates (>90%), shorter treatment duration (12 weeks vs up to 48 weeks with older regimens), and few serious adverse effects9-11; drawbacks include the pricing of these regimens, which could cost upward of ≥$100,000 for a 12-week course, and a lack of coverage under some health insurance plans.12 The manufacturers of 2 agents, telaprevir and boceprevir, removed them from the market because of decreased demand related to their unfavorable side-effect profile and the availability of better tolerated agents.
Treatment considerations for interferon in psychiatric patients
Various neuropsychiatric symptoms have been reported with the use of PEG-IFN. The range of reported symptoms include:
- depressed mood
- anxiety
- hostility
- slowness
- fatigue
- sleep disturbance
- lethargy
- irritability
- emotional lability
- social withdrawal
- poor concentration.13,14
Depressive symptoms can present as early as 1 month after starting treatment, but typically occur at 8 to 12 weeks. A systematic review and meta-analysis of 26 observational studies found a cumulative 25% risk of interferon (IFN)-induced depression in the general HCV population.15 Risk factors for IFN-induced depression include:
- female sex
- history of major depression or other psychiatric disorder
- low educational level
- the presence of baseline subthreshold depressive symptoms.
Because of the risk of inducing depression, there was initial hesitation with providing IFN treatment to patients with psychiatric disorders. However, there is evidence that individuals with chronic psychiatric illness can be treated safely with IFN-based regimens and achieve results similar to non-psychiatric populations.16,17 For example, patients with schizophrenia in a small Veterans Affairs database who received IFN for HCV did not experience higher rates of symptoms of schizophrenia, depression, or mania over 8 years of follow-up.18 Furthermore, those with schizophrenia were just as likely to reach SVR as patients without psychiatric illness.19 Other encouraging results have been reported in depressed patients. One study found similar rates of treatment completion and SVR in patients with a history of major depressive disorder compared with those without depression.20 No difference in frequency of neuropsychiatric side effects was found between the groups.
Presence of a psychiatric disorder is no longer an absolute contraindication to IFN treatment for HCV. Optimal control of psychiatric symptoms should be attained in all patients before starting HCV treatment, and close clinical monitoring is warranted. A review of 9 studies showed benefit of antidepressants for HCV patients with elevated baseline depression or a history of IFN-induced depression.21 The largest body of evidence supports the safety and efficacy of selective serotonin reuptake inhibitors for treating IFN-induced depression. Although no antidepressants are FDA-approved for this indication, the best-studied agents include citalopram, escitalopram, sertraline, and paroxetine.
A review of 6 studies on using antidepressants to prevent IFN-induced depression concluded there was inadequate evidence to support this approach in all patients.22 Pretreatment primarily is indicated for those with elevated depressive symptoms at baseline or those with a history of IFN-induced depression. The prevailing approach to IFN-induced depression assessment, prevention, and treatment is summarized in Table 2.
CASE continued
Ms. S tests positive for the HCV antibody but negative for HIV and hepatitis B. She immediately receives the hepatitis B vaccine series. Her sister discourages her from receiving treatment for HCV, warning her, “it will make you crazy depressed.” As a result, Ms. S avoids following up with the hepatologist. Her psychiatrist, aware that she now was taking her psychotropic medication and seeing that her mood is stable, educates her about new treatment options for HCV that do not cause depression. Ms. S finally agrees to see a hepatologist to discuss her treatment options.
IFN-free regimens
With the arrival of the DAAs, the potential now exists to use IFN-free treatment regimens,10 which could eliminate concerns about IFN-induced depression.
Clinical trials of the DAAs and real-world use so far do not indicate an elevated risk for neuropsychiatric symptoms, including depression.11 As a result, more patients with severe psychiatric illness likely will be eligible to receive treatment for HCV. However, as clinical experience builds with these new agents, it is important to monitor the experience of patients with psychiatric comorbidity. Current treatment guidelines for HCV genotype 1, which is most common in the United States, do not include IFN-based regimens.10 Treatment of genotype 3, which affects 6% of the U.S. population, still includes IFN. Therefore, the risk of IFN-induced depression still exists for some patients with HCV. Table 310 describes current treatment regimens in use for HCV without cirrhosis (see Related Resources for treating HCV with cirrhosis).
Evolving role of the psychiatrist
The availability of shorter, better-tolerated regimens means that the psychiatric contraindications to HCV treatment will be eased. With the emergence of non-IFN treatment regimens, the role of mental health providers could shift toward assisting with treatment adherence, monitoring drug–drug interactions, and managing comorbid substance use disorders.10
The psychiatrist’s role might shift away from the psychosocial assessment of factors affecting treatment eligibility, such as IFN-associated depressive symptoms. Clinical focus will likely shift to supporting adherence to HCV treatment regimens.23 Because depression and substance use disorders are risk factors for non-adherence, mental health providers may be called upon to optimize treatment of these conditions before beginning DAA regimens. A multi-dose regimen might be complicated for those with severe mental illness, and increased psychiatric and community support could be needed in these patients.23 Furthermore, models of care that integrate an HCV specialist with psychiatric care have demonstrated benefits.6,23 Long-term follow-up with a mental health provider will be key to provide ongoing psychiatric support, especially for those who do not achieve SVR.
Psychotropic drug–drug interactions with DAAs
Both sofosbuvir and ledipasvir are substrates of P-glycoprotein and not metabolized by cytochrome P450 (CYP) enzymes.24 Therefore, there are no known contraindications with psychotropic medications. However, co-administration of P-glycoprotein inducers, such as St. John’s wort, could reduce sofosbuvir and ledipasvir levels leading to reduced therapeutic efficacy.
Because it has been used for many years as an HIV treatment, drug interactions with ritonavir have been well-described. This agent is a “pan-inhibitor” and inhibits the CYP3A4, 2D6, 2C9, and 2C19 enzymes and could increase levels of any psychotropic metabolized by these enzymes.25 After several weeks of treatment, it also could induce CYP3A4, which could lead to reduced efficacy of oral contraceptives because ethinylestradiol is metabolized by CYP3A4. Ritonavir is primarily metabolized by CYP3A4 (and CYP2D6 to a smaller degree). Carbamazepine induces CYP3A4, which may lead to decreased levels of ritonavir.23 This, in turn, could reduce the likelihood of attaining SVR and successful treatment of HCV.
Boceprevir, telaprevir, and simeprevir inhibit CYP3A4 to varying degrees and therefore could affect psychotropic medications metabolized by this enzyme.23,26,27 These DAAs are metabolized by CYP3A4; therefore CYP3A4 inducers, such as carbamazepine, could lower DAA blood levels, increasing risk of HCV treatment failure and viral resistance.
Daclatasvir is a substrate of CYP3A4 and an inhibitor of P-glycoprotein.28 Concomitant buprenorphine or buprenorphine/naloxone levels may be increased, although the manufacturer does not recommend dosage adjustment. Elbasvir and grazoprevir are metabolized by CYP3A4.29 Drug–drug interactions therefore may result when administered with either CYP3A4 inducers or inhibitors.
CASE Conclusion
Ms. S sees her new hepatologist, Dr. Smith. She decides to try a 12-week course of ledipasvir/sofosbuvir. Dr. Smith collaborates frequently with Ms. S’s psychiatrist to discuss her case and to help monitor her psychiatric symptoms. She follows up closely with her psychiatrist for symptom monitoring and to help ensure treatment compliance. Ms. S does well with the IFN-free treatment regimen and experiences no worsening of her psychiatric symptoms during treatment.
At approximately 3 to 4 million patients, hepatitis C virus (HCV) is the most common viral hepatitis in the United States. Patients with mental illness are disproportionately affected by HCV and the management of their disease poses particular challenges.
HCV is commonly transmitted via IV drug use and blood transfusions; transmission through sexual contact is rare. Most patients with HCV are asymptomatic, although some do develop symptoms of acute hepatitis. Most HCV infections become chronic, with a high incidence of liver failure requiring liver transplantation.
Hepatitis refers to inflammation of the liver, which could have various etiologies, including viral infections, alcohol abuse, or autoimmune disease. Viral hepatitis refers to infection from 5 distinct groups of virus, coined A through E.1 This article will focus on chronic HCV (Table 1).
CASE Bipolar disorder, stress, history of IV drug use
Ms. S, age 48, has bipolar I disorder and has been hospitalized 4 times in the past, including once for a suicide attempt. She has 3 children and works as a cashier. Her psychiatric symptoms have been stable on lurasidone, 80 mg/d, and escitalopram, 10 mg/d. Recently, Ms. S has been under more stress at her job. Sometimes she misses doses of her medication, and then becomes more irritable and impulsive. Her husband, noting that she has used IV heroin in the past, comes with her today and is concerned that she is “not acting right.” What is Ms. S’s risk for HCV?
HCV in mental illness
Compared with the general population, HCV is more prevalent among chronically mentally ill persons. In one study, HCV occurred twice as often in men vs women with chronic mental illness.2 Up to 50% of patients with HCV have a history of mental illness and nearly 90% have a history of substance use disorders.3 Among 668 chronically mentally ill patients at 4 public sector clinics, risk factors for HCV were common and included use of injection drugs (>20%), sharing needles (14%), and crack cocaine use (>20%).4 Higher rates of HCV were reported in hospitalized patients with schizophrenia and comorbid psychoactive substance abuse in Japan.5 Because of the high prevalence in this population, it is essential to assess for substance use disorders. Employing a non-judgmental approach with motivational interviewing techniques can be effective.6
Individuals with mental illness should be screened for HCV risk factors, such as unprotected intercourse with high-risk partners and sharing needles used for illicit drug use. Patients frequently underreport these activities. At-risk individuals should undergo laboratory testing for the HIV-1 antibody, hepatitis C antibodies, and hepatitis B antibodies. Mental health providers should counsel patients about risk reduction (eg, avoiding unprotected sexual intercourse and sharing of drug paraphernalia). Educating patients about complications of viral hepatitis, such as liver failure, could be motivation to change risky behaviors.
CASE continued
During your interview with Ms. S, she becomes irritable and tells you that you are asking too many questions. It is clear that she is not taking her medications consistently, but she agrees to do so because she does not want to lose custody of her children. She denies current use of heroin but her husband says, “I don’t know what she is doing.” In addition to advising her on reducing risk factors, you order appropriate screening tests, including hepatitis and HIV antibody tests.
Screening guidelines
The U.S. Preventive Services Task Force and the CDC both recommend a 1-time screening for HCV in asymptomatic or low-risk patients born between 1945 and 1965.1,7 Furthermore, both organizations recommend screening for HCV in persons at high risk, including:
- those with a history of injection drug use
- persons with recognizable exposure, such as needlesticks
- persons who received blood transfusions before 1992
- medical conditions, such as long-term dialysis.
There is no vaccine for HCV; however, patients with HCV should receive vaccination against hepatitis B.
Diagnosis
Acute symptoms include fever, fatigue, headache, cough, nausea, and vomiting. Jaundice could develop, often accompanied by pain in the right upper quadrant. If there is suspicion of viral hepatitis, psychiatrists can initiate the laboratory evaluation. Chronic hepatitis, on the other hand, often is asymptomatic, although stigmata of chronic liver disease (eg, jaundice, ascites, peripheral edema) might be detected on physical exam.8 Elevated serum transaminases are seen with acute viral hepatitis, although levels could vary in chronic cases. Serologic detection of anti-HCV antibodies establishes a HCV diagnosis.
Treatment recommendations
All patients who test positive for HCV should be evaluated and treated by a hepatologist. Goals of therapy are to reduce complications from chronic viral hepatitis, including cirrhosis and hepatic failure. Duration and optimal regimen depends on the HCV genotype.8 Treatment outcomes are measured by virological parameters, including serum aminotransferases, HCV RNA levels, and histology. The most important parameter in treating chronic HCV is the sustained virological response (SVR), which is the absence of HCV RNA 12 weeks after completing therapy.9
Treatment is recommended for all persons with chronic HCV infection, according to current treatment guidelines, which are updated regularly by the American Association for the Study of Liver Diseases and the Infectious Diseases Society of America.10 Until recently, treatment consisted of IV pegylated interferon (PEG-IFN) in combination with oral ribavirin. Success rates with this regimen are approximately 40% to 50%. The advent of direct-acting antivirals (DAAs) has revolutionized treatment of chronic HCV. These agents include simeprevir, sofosbuvir, ledipasvir, and the combination of ombitasvir-paritaprevir-ritonavir plus dasabuvir (brand name, Viekira Pak). Advantages of these agents are oral administration, high treatment success rates (>90%), shorter treatment duration (12 weeks vs up to 48 weeks with older regimens), and few serious adverse effects9-11; drawbacks include the pricing of these regimens, which could cost upward of ≥$100,000 for a 12-week course, and a lack of coverage under some health insurance plans.12 The manufacturers of 2 agents, telaprevir and boceprevir, removed them from the market because of decreased demand related to their unfavorable side-effect profile and the availability of better tolerated agents.
Treatment considerations for interferon in psychiatric patients
Various neuropsychiatric symptoms have been reported with the use of PEG-IFN. The range of reported symptoms include:
- depressed mood
- anxiety
- hostility
- slowness
- fatigue
- sleep disturbance
- lethargy
- irritability
- emotional lability
- social withdrawal
- poor concentration.13,14
Depressive symptoms can present as early as 1 month after starting treatment, but typically occur at 8 to 12 weeks. A systematic review and meta-analysis of 26 observational studies found a cumulative 25% risk of interferon (IFN)-induced depression in the general HCV population.15 Risk factors for IFN-induced depression include:
- female sex
- history of major depression or other psychiatric disorder
- low educational level
- the presence of baseline subthreshold depressive symptoms.
Because of the risk of inducing depression, there was initial hesitation with providing IFN treatment to patients with psychiatric disorders. However, there is evidence that individuals with chronic psychiatric illness can be treated safely with IFN-based regimens and achieve results similar to non-psychiatric populations.16,17 For example, patients with schizophrenia in a small Veterans Affairs database who received IFN for HCV did not experience higher rates of symptoms of schizophrenia, depression, or mania over 8 years of follow-up.18 Furthermore, those with schizophrenia were just as likely to reach SVR as patients without psychiatric illness.19 Other encouraging results have been reported in depressed patients. One study found similar rates of treatment completion and SVR in patients with a history of major depressive disorder compared with those without depression.20 No difference in frequency of neuropsychiatric side effects was found between the groups.
Presence of a psychiatric disorder is no longer an absolute contraindication to IFN treatment for HCV. Optimal control of psychiatric symptoms should be attained in all patients before starting HCV treatment, and close clinical monitoring is warranted. A review of 9 studies showed benefit of antidepressants for HCV patients with elevated baseline depression or a history of IFN-induced depression.21 The largest body of evidence supports the safety and efficacy of selective serotonin reuptake inhibitors for treating IFN-induced depression. Although no antidepressants are FDA-approved for this indication, the best-studied agents include citalopram, escitalopram, sertraline, and paroxetine.
A review of 6 studies on using antidepressants to prevent IFN-induced depression concluded there was inadequate evidence to support this approach in all patients.22 Pretreatment primarily is indicated for those with elevated depressive symptoms at baseline or those with a history of IFN-induced depression. The prevailing approach to IFN-induced depression assessment, prevention, and treatment is summarized in Table 2.
CASE continued
Ms. S tests positive for the HCV antibody but negative for HIV and hepatitis B. She immediately receives the hepatitis B vaccine series. Her sister discourages her from receiving treatment for HCV, warning her, “it will make you crazy depressed.” As a result, Ms. S avoids following up with the hepatologist. Her psychiatrist, aware that she now was taking her psychotropic medication and seeing that her mood is stable, educates her about new treatment options for HCV that do not cause depression. Ms. S finally agrees to see a hepatologist to discuss her treatment options.
IFN-free regimens
With the arrival of the DAAs, the potential now exists to use IFN-free treatment regimens,10 which could eliminate concerns about IFN-induced depression.
Clinical trials of the DAAs and real-world use so far do not indicate an elevated risk for neuropsychiatric symptoms, including depression.11 As a result, more patients with severe psychiatric illness likely will be eligible to receive treatment for HCV. However, as clinical experience builds with these new agents, it is important to monitor the experience of patients with psychiatric comorbidity. Current treatment guidelines for HCV genotype 1, which is most common in the United States, do not include IFN-based regimens.10 Treatment of genotype 3, which affects 6% of the U.S. population, still includes IFN. Therefore, the risk of IFN-induced depression still exists for some patients with HCV. Table 310 describes current treatment regimens in use for HCV without cirrhosis (see Related Resources for treating HCV with cirrhosis).
Evolving role of the psychiatrist
The availability of shorter, better-tolerated regimens means that the psychiatric contraindications to HCV treatment will be eased. With the emergence of non-IFN treatment regimens, the role of mental health providers could shift toward assisting with treatment adherence, monitoring drug–drug interactions, and managing comorbid substance use disorders.10
The psychiatrist’s role might shift away from the psychosocial assessment of factors affecting treatment eligibility, such as IFN-associated depressive symptoms. Clinical focus will likely shift to supporting adherence to HCV treatment regimens.23 Because depression and substance use disorders are risk factors for non-adherence, mental health providers may be called upon to optimize treatment of these conditions before beginning DAA regimens. A multi-dose regimen might be complicated for those with severe mental illness, and increased psychiatric and community support could be needed in these patients.23 Furthermore, models of care that integrate an HCV specialist with psychiatric care have demonstrated benefits.6,23 Long-term follow-up with a mental health provider will be key to provide ongoing psychiatric support, especially for those who do not achieve SVR.
Psychotropic drug–drug interactions with DAAs
Both sofosbuvir and ledipasvir are substrates of P-glycoprotein and not metabolized by cytochrome P450 (CYP) enzymes.24 Therefore, there are no known contraindications with psychotropic medications. However, co-administration of P-glycoprotein inducers, such as St. John’s wort, could reduce sofosbuvir and ledipasvir levels leading to reduced therapeutic efficacy.
Because it has been used for many years as an HIV treatment, drug interactions with ritonavir have been well-described. This agent is a “pan-inhibitor” and inhibits the CYP3A4, 2D6, 2C9, and 2C19 enzymes and could increase levels of any psychotropic metabolized by these enzymes.25 After several weeks of treatment, it also could induce CYP3A4, which could lead to reduced efficacy of oral contraceptives because ethinylestradiol is metabolized by CYP3A4. Ritonavir is primarily metabolized by CYP3A4 (and CYP2D6 to a smaller degree). Carbamazepine induces CYP3A4, which may lead to decreased levels of ritonavir.23 This, in turn, could reduce the likelihood of attaining SVR and successful treatment of HCV.
Boceprevir, telaprevir, and simeprevir inhibit CYP3A4 to varying degrees and therefore could affect psychotropic medications metabolized by this enzyme.23,26,27 These DAAs are metabolized by CYP3A4; therefore CYP3A4 inducers, such as carbamazepine, could lower DAA blood levels, increasing risk of HCV treatment failure and viral resistance.
Daclatasvir is a substrate of CYP3A4 and an inhibitor of P-glycoprotein.28 Concomitant buprenorphine or buprenorphine/naloxone levels may be increased, although the manufacturer does not recommend dosage adjustment. Elbasvir and grazoprevir are metabolized by CYP3A4.29 Drug–drug interactions therefore may result when administered with either CYP3A4 inducers or inhibitors.
CASE Conclusion
Ms. S sees her new hepatologist, Dr. Smith. She decides to try a 12-week course of ledipasvir/sofosbuvir. Dr. Smith collaborates frequently with Ms. S’s psychiatrist to discuss her case and to help monitor her psychiatric symptoms. She follows up closely with her psychiatrist for symptom monitoring and to help ensure treatment compliance. Ms. S does well with the IFN-free treatment regimen and experiences no worsening of her psychiatric symptoms during treatment.
1. Centers for Disease Control and Prevention. Viral hepatitis. http://www.cdc.gov/hepatitis. Updated December 9, 2016. Accessed February 9, 2017.
2. Butterfield MI, Bosworth HB, Meador KG, et al. Five-Site Health and Risk Study Research Committee. Gender differences in hepatitis C infection and risks among persons with severe mental illness. Psychiatr Serv. 2003;54(6):848-853.
3. Rifai MA, Gleason OC, Sabouni D. Psychiatric care of the patient with hepatitis C: a review of the literature. Prim Care Companion J Clin Psychiatry. 2010;12(6):PCC.09r00877. doi: 10.4088/PCC.09r00877whi.
4. Dinwiddie SH, Shicker L, Newman T. Prevalence of hepatitis C among psychiatric patients in the public sector. Am J Psychiatry. 2003;160(1):172-174.
5. Nakamura Y, Koh M, Miyoshi E, et al. High prevalence of the hepatitis C virus infection among the inpatients of schizophrenia and psychoactive substance abuse in Japan. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28(3):591-597.
6. Sockalingam S, Blank D, Banga CA, et al. A novel program for treating patients with trimorbidity: hepatitis C, serious mental illness, and substance abuse. Eur J Gastroenterol Hepatol. 2013;25(12):1377-1384.
7. U.S. Preventive Services Task Force. Screening for hepatitis C virus infection: recommendation summary. https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/hepatitis-c-screening. Published June 2013. Accessed February 9, 2017.
8. Longo DL, Fauci AS, Kasper DL. Harrison’s principles of internal medicine. 18th ed. New York, NY: McGraw-Hill; 2012.
9. Belousova V, Abd-Rabou AA, Mousa SA. Recent advances and future directions in the management of hepatitis C infections. Pharmacol Ther. 2015;145:92-102.
10. American Association for the Study of Liver Diseases (AASLD); The Infectious Diseases Society of America (IDSA). HCV guidance: recommendations for testing, managing, and treating hepatitis C. http://www.hcvguidelines.org. Accessed February 9, 2017.
11. Rowan PJ, Bhulani N. Psychosocial assessment and monitoring in the new era of non-interferon-alpha hepatitis C treatments. World J Hepatol. 2015;7(19):2209-2213.
12. Good Rx, Inc. http://www.goodrx.com. Accessed October 9, 2015.
13. Raison CL, Borisov AS, Broadwell SD, et al. Depression during pegylated interferon-alpha plus ribavirin therapy: prevalence and prediction. J Clin Psychiatry. 2005;66(1):41-48.
14. Lotrich FE, Rabinovitz M, Gironda P, et al. Depression following pegylated interferon-alpha: characteristics and vulnerability. J Psychosom Res. 2007;63(2):131-135.
15. Udina M, Castellví P, Moreno-España J, et al. Interferon-induced depression in chronic hepatitis C: a systematic review and meta-analysis. J Clin Psychiatry. 2012;73(8):1128-1138.
16. Mustafa MZ, Schofield J, Mills PR, et al. The efficacy and safety of treating hepatitis C in patients with a diagnosis of schizophrenia. J Viral Hepat. 2014;21(7):e48-e51.
17. Huckans M, Mitchell A, Pavawalla S, et al. The influence of antiviral therapy on psychiatric symptoms among patients with hepatitis C and schizophrenia. Antivir Ther. 2010;15(1):111-119.
18. Huckans MS, Blackwell AD, Harms TA, et al. Management of hepatitis C disease among VA patients with schizophrenia and substance use disorders. Psychiatr Serv. 2006;57(3):403-406.
19. Huckans M, Mitchell A, Ruimy S, et al. Antiviral therapy completion and response rates among hepatitis C patients with and without schizophrenia. Schizophr Bull. 2010;36(1):165-172.
20. Hauser P, Morasco BJ, Linke A, et al. Antiviral completion rates and sustained viral response in hepatitis C patient with and without preexisting major depressive disorder. Psychosomatics. 2009;50(5):500-505.
21. Sockalingam S, Abbey SE. Managing depression during hepatitis C treatment. Can J Psychiatry. 2009;54(9):614-625.
22. Galvão-de Almeida A, Guindalini C, Batista-Neves S, et al. Can antidepressants prevent interferon-alpha-induced depression? A review of the literature. Gen Hosp Psychiatry. 2010;32(4):401-405.
23. Sockalingam S, Sheehan K, Feld JJ, et al. Psychiatric care during hepatitis c treatment: the changing role of psychiatrists in the era of direct-acting antivirals. Am J Psychiatry. 2015;172(6):512-516.
24. Harvoni [package insert]. Foster City, CA: Gilead Sciences, Inc.; 2016.
25. Wynn GH, Oesterheld, JR, Cozza KL, et al. Clinical manual of drug interactions principles for medical practice. Arlington, VA: American Psychiatric Publishing; 2009.
26. Olysio [package insert]. Titusville, NJ: Janssen Therapeutics; 2016.
27. Victrelis [package insert]. Whitehouse Station, NJ: Merck & Co.; 2017.
28. Daklinza [package insert]. Princeton, NJ: Bristol-Myers Squibb; 2016.
29. Zepatier [package insert]. Whitehouse Station, NJ: Merck & Co.; 2017.
1. Centers for Disease Control and Prevention. Viral hepatitis. http://www.cdc.gov/hepatitis. Updated December 9, 2016. Accessed February 9, 2017.
2. Butterfield MI, Bosworth HB, Meador KG, et al. Five-Site Health and Risk Study Research Committee. Gender differences in hepatitis C infection and risks among persons with severe mental illness. Psychiatr Serv. 2003;54(6):848-853.
3. Rifai MA, Gleason OC, Sabouni D. Psychiatric care of the patient with hepatitis C: a review of the literature. Prim Care Companion J Clin Psychiatry. 2010;12(6):PCC.09r00877. doi: 10.4088/PCC.09r00877whi.
4. Dinwiddie SH, Shicker L, Newman T. Prevalence of hepatitis C among psychiatric patients in the public sector. Am J Psychiatry. 2003;160(1):172-174.
5. Nakamura Y, Koh M, Miyoshi E, et al. High prevalence of the hepatitis C virus infection among the inpatients of schizophrenia and psychoactive substance abuse in Japan. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28(3):591-597.
6. Sockalingam S, Blank D, Banga CA, et al. A novel program for treating patients with trimorbidity: hepatitis C, serious mental illness, and substance abuse. Eur J Gastroenterol Hepatol. 2013;25(12):1377-1384.
7. U.S. Preventive Services Task Force. Screening for hepatitis C virus infection: recommendation summary. https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/hepatitis-c-screening. Published June 2013. Accessed February 9, 2017.
8. Longo DL, Fauci AS, Kasper DL. Harrison’s principles of internal medicine. 18th ed. New York, NY: McGraw-Hill; 2012.
9. Belousova V, Abd-Rabou AA, Mousa SA. Recent advances and future directions in the management of hepatitis C infections. Pharmacol Ther. 2015;145:92-102.
10. American Association for the Study of Liver Diseases (AASLD); The Infectious Diseases Society of America (IDSA). HCV guidance: recommendations for testing, managing, and treating hepatitis C. http://www.hcvguidelines.org. Accessed February 9, 2017.
11. Rowan PJ, Bhulani N. Psychosocial assessment and monitoring in the new era of non-interferon-alpha hepatitis C treatments. World J Hepatol. 2015;7(19):2209-2213.
12. Good Rx, Inc. http://www.goodrx.com. Accessed October 9, 2015.
13. Raison CL, Borisov AS, Broadwell SD, et al. Depression during pegylated interferon-alpha plus ribavirin therapy: prevalence and prediction. J Clin Psychiatry. 2005;66(1):41-48.
14. Lotrich FE, Rabinovitz M, Gironda P, et al. Depression following pegylated interferon-alpha: characteristics and vulnerability. J Psychosom Res. 2007;63(2):131-135.
15. Udina M, Castellví P, Moreno-España J, et al. Interferon-induced depression in chronic hepatitis C: a systematic review and meta-analysis. J Clin Psychiatry. 2012;73(8):1128-1138.
16. Mustafa MZ, Schofield J, Mills PR, et al. The efficacy and safety of treating hepatitis C in patients with a diagnosis of schizophrenia. J Viral Hepat. 2014;21(7):e48-e51.
17. Huckans M, Mitchell A, Pavawalla S, et al. The influence of antiviral therapy on psychiatric symptoms among patients with hepatitis C and schizophrenia. Antivir Ther. 2010;15(1):111-119.
18. Huckans MS, Blackwell AD, Harms TA, et al. Management of hepatitis C disease among VA patients with schizophrenia and substance use disorders. Psychiatr Serv. 2006;57(3):403-406.
19. Huckans M, Mitchell A, Ruimy S, et al. Antiviral therapy completion and response rates among hepatitis C patients with and without schizophrenia. Schizophr Bull. 2010;36(1):165-172.
20. Hauser P, Morasco BJ, Linke A, et al. Antiviral completion rates and sustained viral response in hepatitis C patient with and without preexisting major depressive disorder. Psychosomatics. 2009;50(5):500-505.
21. Sockalingam S, Abbey SE. Managing depression during hepatitis C treatment. Can J Psychiatry. 2009;54(9):614-625.
22. Galvão-de Almeida A, Guindalini C, Batista-Neves S, et al. Can antidepressants prevent interferon-alpha-induced depression? A review of the literature. Gen Hosp Psychiatry. 2010;32(4):401-405.
23. Sockalingam S, Sheehan K, Feld JJ, et al. Psychiatric care during hepatitis c treatment: the changing role of psychiatrists in the era of direct-acting antivirals. Am J Psychiatry. 2015;172(6):512-516.
24. Harvoni [package insert]. Foster City, CA: Gilead Sciences, Inc.; 2016.
25. Wynn GH, Oesterheld, JR, Cozza KL, et al. Clinical manual of drug interactions principles for medical practice. Arlington, VA: American Psychiatric Publishing; 2009.
26. Olysio [package insert]. Titusville, NJ: Janssen Therapeutics; 2016.
27. Victrelis [package insert]. Whitehouse Station, NJ: Merck & Co.; 2017.
28. Daklinza [package insert]. Princeton, NJ: Bristol-Myers Squibb; 2016.
29. Zepatier [package insert]. Whitehouse Station, NJ: Merck & Co.; 2017.
ACIP vaccine update, 2017
The Advisory Committee on Immunization Practices (ACIP) met 3 times in 2016 and introduced or revised recommendations on influenza, meningococcal, human papillomavirus (HPV), cholera, and hepatitis B vaccines. This Practice Alert highlights the most important new recommendations, except those for influenza vaccines, which were described in a previous Practice Alert.1 (See the summary of how this year’s flu season compares to last year’s.)
SIDEBAR
PRACTICE ALERT UPDATE
How this year's flu season compares to last yearThe 2016-2017 influenza season has been relatively mild, with activity nationwide picking up in late January and continuing to increase in February. As of February 16, 90% of the infections typed were type A, and most of those cases (more than 90%) were H3N1. Not surprisingly, the age group most heavily affected has been the elderly.
The hospitalization rate among those ≥65 years as of early February was 113.5/100,000, which is about half the rate of the same week during the 2014-2015 flu season. The hospitalization rate among those ages 50 to 64 years was 23.5/100,000—about 40% lower than the rate during the same week last flu season. At press time, 20 pediatric deaths had occurred, which is less than one-quarter of the number that occurred during the same time last year, and resistance to oseltamivir had not yet been detected in any isolates.
Source: Centers for Disease Control and Prevention. Situation update: summary of weekly FluView report. Available at: https://www.cdc.gov/flu/weekly/summary.htm. Accessed February 16, 2017.
Meningococcal vaccine: Now recommended for HIV-positive patients
Meningococcal conjugate vaccine (serogroups A, C, W, and Y) is recommended for all adolescents ages 11 to 12 as a single dose with a booster at age 16.2 It is also recommended for adults and for children (starting at age 2 months) who have high-risk conditions such as functional or anatomic asplenia or complement deficiencies. Others at high risk include microbiologists routinely exposed to isolates of Neisseria meningitidis and those traveling to areas of high meningococcal incidence. ACIP recently added human immunodeficiency virus (HIV) infection to the list of high-risk conditions.3
Two meningococcal conjugate vaccines are available in the United States: Menactra, (Sanofi Pasteur), licensed for use in individuals ages 9 months to 55 years; and Menveo (GlaxoSmithKline), licensed for use in individuals ages 2 months to 55 years. Menveo is the preferred vaccine for children younger than 2 years infected with HIV. However, if Menactra is used, give it at least 4 weeks after completing all pneumococcal conjugate vaccine doses and either before or concomitantly with diphtheria and tetanus toxoid and acellular pertussis vaccine (DTaP). All individuals who are HIV positive should receive a multi-dose primary series and booster doses. The number of primary doses and timing of boosters depends on the product used and the ages of those vaccinated (TABLE3).

Although neither meningococcal conjugate vaccine product is licensed for use in individuals 56 years or older, ACIP recommends using one of the products for HIV-infected individuals in this age group because the only meningococcal vaccine licensed for use in adults 56 or older, meningococcal polysaccharide vaccine (MPSV4, Menomune, Sanofi Pasteur), has not been studied in patients with HIV infection.
Serogroup B. Two vaccine products provide short-term protection against meningococcal serogroup B: MenB-FHbp (Trumenba, Wyeth Pharmaceuticals, Inc.) and MenB-4C (Bexsero, GlaxoSmithKline). In 2015, ACIP made a “B” recommendation for the use of these vaccines in individuals 16 to 23 years of age, with the preferred age range being 16 to 18.4 A “B” recommendation means that while ACIP does not advise routine use of the vaccines in this age group, the vaccines can be administered to those who desire them. ACIP has recommended routine use of these products only for individuals 10 years and older who are at high risk for meningococcal disease.5
Trumenba was approved as a 3-dose vaccine, administered at 0, 2, and 6 months. Bexsero requires 2 doses given at least one month apart. At its October 2016 meeting, ACIP approved a 2-dose Trumenba schedule, at 0 and 6 months, when administered to those not at risk for meningococcal disease.6 However, during an outbreak, and for those at high risk for meningococcal disease, adhere to the original 3-dose schedule.
HPV vaccine: Now a 2-dose schedule for younger patients
The only HPV vaccine available in the United States is the 9-valent HPV vaccine (9vHPV), Gardasil 9. It is approved for both males and females ages 9 to 26 years. ACIP recommends it for both sexes at ages 11 or 12, and advises catch-up doses for men through age 21 and women through age 26. It also recommends vaccination through age 26 for men who have sex with men and men
The HPV vaccine is approved for a 3-dose schedule at 0, 1 to 2, and 6 months. At its October 2016 meeting, ACIP approved a 2-dose schedule (0, 6-12 months) for those starting the vaccine before their 15th birthday.7 Those starting the vaccine after their 15th birthday, and individuals at any age with an immune-compromising condition, should receive 3 doses. It is hoped that a 2-dose schedule will help to increase the uptake of this safe, effective, and underused vaccine.
Cholera: A new vaccine is available
In June 2016, the FDA approved a live, attenuated, single-dose, oral vaccine (Vaxchora, PaxVax, Inc.) for the prevention of cholera in adults ages 18 to 64 years. It is the only cholera vaccine approved in the United States.
Cholera occurs at low rates among travelers to areas where the disease is endemic. The key to prevention is food and water precautions, and thus the vaccine is not recommended for most travelers—only for those who are at increased risk of exposure to cholera or who have a medical condition that predisposes them to a poor response to medical care if cholera is contracted.8 Risk increases with long-term or frequent travel to endemic areas where safe food and water is not always available. Examples of compromising medical conditions include a blood type O, low gastric acidity, and heart or kidney disease.
Duration of the vaccine’s effectiveness is unknown, given a lack of data beyond 6 months. No recommendation for revaccination has been made, and this issue will be assessed as more data are collected. Other unknowns about the vaccine include its effectiveness among immune-suppressed individuals and pregnant women, as well as for those who live in cholera endemic areas or were previously vaccinated with another cholera vaccine.
Hepatitis B: Vaccinate newborns sooner
The incidence of hepatitis B virus (HBV) infection has declined by more than 90% since the introduction of a vaccine in 1982.9
Current recommendations for the prevention of HBV include:9
- Screen all pregnant women for hepatitis B surface antigen (HBsAg), and use HBIG and hepatitis B vaccines within 12 hours of birth for all newborns whose mothers are HBsAg positive or have an unknown HBsAg status.
- Administer the 3-dose hepatitis B vaccine to all other infants.
- Routinely vaccinate previously unvaccinated children and adolescents.
- Routinely vaccinate adults who are non-immune and at risk for HBV infection.
At its October 2016 meeting, ACIP adopted a comprehensive update of all HBV prevention recommendations. (This will be the subject of a future Practice Alert.) Included was a revision of a previously permissive recommendation that allowed the first dose of hepatitis B vaccine for newborns to be given within 2 months of hospital discharge. The new recommendation9 states that newborns of mothers known to be HBsAg negative should be vaccinated within 24 hours (if weight is ≥2000 g) or at age one month or at hospital discharge (if weight is <2000 g).
The first dose should be given within 12 hours of birth to all newborns whose mothers are HBsAg positive or have an unknown HBsAg status.9
Immunization schedules
Every year ACIP updates the adult and child immunization schedules to incorporate the changes from the previous year. These can be found on the ACIP Web site at https://www.cdc.gov/vaccines/schedules/hcp/index.html. This Web site remains the most authoritative and accurate source of information on vaccines and immunizations for both professionals and the public.
1. Campos-Outcalt D. Need-to-know information for the 2016-2017 flu season. J Fam Pract. 2016;65:613-617.
2. Cohn AC, MacNeil JR, Clark TA, et al. Prevention and control of meningococcal disease: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2013;62:1-28.
3. MacNeil JR, Rubin LG, Patton M, et al. Recommendations for use of meningococcal conjugate vaccines in HIV-infected persons— Advisory Committee on Immunization Practices, 2016. MMWR Morb Mortal Wkly Rep. 2016;65:1189-1194.
4. MacNeil JR, Rubin LG, Folaranmi T, et al. Use of serogroup B meningococcal vaccines in adolescents and young adults: recommendations of the Advisory Committee on Immunization Practices, 2015. MMWR Morb Mortal Wkly Rep. 2015;64:1171-1176.
5. Folaranmi T, Rubin L, Martin SW, et al. Use of serogroup B meningococcal vaccines in persons aged ≥10 years at increased risk for serogroup B meningococcal disease: recommendations of the Advisory Committee on Immunization Practices, 2015. MMWR Morb Mortal Wkly Rep. 2015;64:608-612.
6. MacNeil J. Considerations for Use of 2- and 3-Dose Schedules of MenB-FHbp (Trumenba). Presentation at: Advisory Committee on Immunization Practices; October 19, 2016; Atlanta, GA. Available at: https://www.cdc.gov/vaccines/acip/meetings/downloads/slides-2016-10/meningococcal-05-macneil.pdf. Accessed February 6, 2017.
7. Meites E, Kempe A, Markowitz LE. Use of a 2-dose schedule for human papillomavirus vaccination—updated recommendations of the Advisory Committee on Immunization Practices. MMWR Morb Mortal Wkly Rep. 2016;65:1405-1408.
8. Wong KW. Cholera vaccine update and proposed recommendations. Presentation at: Advisory Committee on Immunization Practices; June 22, 2016; Atlanta, GA. Available at: https://www.cdc.gov/vaccines/acip/meetings/downloads/slides-2016-06/cholera-02-wong.pdf. Accessed January 27, 2017.
9. Schillie S. Revised ACIP Hepatitis B (HepB) vaccine recommendations. Presentation at: Advisory Committee on Immunization Practices; October 19, 2016. Available at: https://www.cdc.gov/vaccines/acip/meetings/downloads/slides-2016-10/hepatitis-02-schillie-october-2016.pdf. Accessed January 27, 2017.
10.
11. Ko SC, Fan L, Smith EA, et al. Estimated annual perinatal hepatitis B virus infections in the United States, 2000-2009. J Pediatric Infect Dis Soc. 2016;5:114-121.
12. Mast EE, Weinbaum CM, Fiore AE, et al. A comprehensive immunization strategy to eliminate transmission of hepatitis B virus infection in the United States. MMWR Morb Mortal Wkly Rep. 2006;55:1-25.
13. Beasley RP, Hwang LY, Lee GC, et al. Prevention of perinatally transmitted hepatitis B virus infections with hepatitis B immune globulin and hepatitis B vaccine. Lancet. 1983;2:1099-1102.
The Advisory Committee on Immunization Practices (ACIP) met 3 times in 2016 and introduced or revised recommendations on influenza, meningococcal, human papillomavirus (HPV), cholera, and hepatitis B vaccines. This Practice Alert highlights the most important new recommendations, except those for influenza vaccines, which were described in a previous Practice Alert.1 (See the summary of how this year’s flu season compares to last year’s.)
SIDEBAR
PRACTICE ALERT UPDATE
How this year's flu season compares to last yearThe 2016-2017 influenza season has been relatively mild, with activity nationwide picking up in late January and continuing to increase in February. As of February 16, 90% of the infections typed were type A, and most of those cases (more than 90%) were H3N1. Not surprisingly, the age group most heavily affected has been the elderly.
The hospitalization rate among those ≥65 years as of early February was 113.5/100,000, which is about half the rate of the same week during the 2014-2015 flu season. The hospitalization rate among those ages 50 to 64 years was 23.5/100,000—about 40% lower than the rate during the same week last flu season. At press time, 20 pediatric deaths had occurred, which is less than one-quarter of the number that occurred during the same time last year, and resistance to oseltamivir had not yet been detected in any isolates.
Source: Centers for Disease Control and Prevention. Situation update: summary of weekly FluView report. Available at: https://www.cdc.gov/flu/weekly/summary.htm. Accessed February 16, 2017.
Meningococcal vaccine: Now recommended for HIV-positive patients
Meningococcal conjugate vaccine (serogroups A, C, W, and Y) is recommended for all adolescents ages 11 to 12 as a single dose with a booster at age 16.2 It is also recommended for adults and for children (starting at age 2 months) who have high-risk conditions such as functional or anatomic asplenia or complement deficiencies. Others at high risk include microbiologists routinely exposed to isolates of Neisseria meningitidis and those traveling to areas of high meningococcal incidence. ACIP recently added human immunodeficiency virus (HIV) infection to the list of high-risk conditions.3
Two meningococcal conjugate vaccines are available in the United States: Menactra, (Sanofi Pasteur), licensed for use in individuals ages 9 months to 55 years; and Menveo (GlaxoSmithKline), licensed for use in individuals ages 2 months to 55 years. Menveo is the preferred vaccine for children younger than 2 years infected with HIV. However, if Menactra is used, give it at least 4 weeks after completing all pneumococcal conjugate vaccine doses and either before or concomitantly with diphtheria and tetanus toxoid and acellular pertussis vaccine (DTaP). All individuals who are HIV positive should receive a multi-dose primary series and booster doses. The number of primary doses and timing of boosters depends on the product used and the ages of those vaccinated (TABLE3).
Although neither meningococcal conjugate vaccine product is licensed for use in individuals 56 years or older, ACIP recommends using one of the products for HIV-infected individuals in this age group because the only meningococcal vaccine licensed for use in adults 56 or older, meningococcal polysaccharide vaccine (MPSV4, Menomune, Sanofi Pasteur), has not been studied in patients with HIV infection.
Serogroup B. Two vaccine products provide short-term protection against meningococcal serogroup B: MenB-FHbp (Trumenba, Wyeth Pharmaceuticals, Inc.) and MenB-4C (Bexsero, GlaxoSmithKline). In 2015, ACIP made a “B” recommendation for the use of these vaccines in individuals 16 to 23 years of age, with the preferred age range being 16 to 18.4 A “B” recommendation means that while ACIP does not advise routine use of the vaccines in this age group, the vaccines can be administered to those who desire them. ACIP has recommended routine use of these products only for individuals 10 years and older who are at high risk for meningococcal disease.5
Trumenba was approved as a 3-dose vaccine, administered at 0, 2, and 6 months. Bexsero requires 2 doses given at least one month apart. At its October 2016 meeting, ACIP approved a 2-dose Trumenba schedule, at 0 and 6 months, when administered to those not at risk for meningococcal disease.6 However, during an outbreak, and for those at high risk for meningococcal disease, adhere to the original 3-dose schedule.
HPV vaccine: Now a 2-dose schedule for younger patients
The only HPV vaccine available in the United States is the 9-valent HPV vaccine (9vHPV), Gardasil 9. It is approved for both males and females ages 9 to 26 years. ACIP recommends it for both sexes at ages 11 or 12, and advises catch-up doses for men through age 21 and women through age 26. It also recommends vaccination through age 26 for men who have sex with men and men
The HPV vaccine is approved for a 3-dose schedule at 0, 1 to 2, and 6 months. At its October 2016 meeting, ACIP approved a 2-dose schedule (0, 6-12 months) for those starting the vaccine before their 15th birthday.7 Those starting the vaccine after their 15th birthday, and individuals at any age with an immune-compromising condition, should receive 3 doses. It is hoped that a 2-dose schedule will help to increase the uptake of this safe, effective, and underused vaccine.
Cholera: A new vaccine is available
In June 2016, the FDA approved a live, attenuated, single-dose, oral vaccine (Vaxchora, PaxVax, Inc.) for the prevention of cholera in adults ages 18 to 64 years. It is the only cholera vaccine approved in the United States.
Cholera occurs at low rates among travelers to areas where the disease is endemic. The key to prevention is food and water precautions, and thus the vaccine is not recommended for most travelers—only for those who are at increased risk of exposure to cholera or who have a medical condition that predisposes them to a poor response to medical care if cholera is contracted.8 Risk increases with long-term or frequent travel to endemic areas where safe food and water is not always available. Examples of compromising medical conditions include a blood type O, low gastric acidity, and heart or kidney disease.
Duration of the vaccine’s effectiveness is unknown, given a lack of data beyond 6 months. No recommendation for revaccination has been made, and this issue will be assessed as more data are collected. Other unknowns about the vaccine include its effectiveness among immune-suppressed individuals and pregnant women, as well as for those who live in cholera endemic areas or were previously vaccinated with another cholera vaccine.
Hepatitis B: Vaccinate newborns sooner
The incidence of hepatitis B virus (HBV) infection has declined by more than 90% since the introduction of a vaccine in 1982.9
Current recommendations for the prevention of HBV include:9
- Screen all pregnant women for hepatitis B surface antigen (HBsAg), and use HBIG and hepatitis B vaccines within 12 hours of birth for all newborns whose mothers are HBsAg positive or have an unknown HBsAg status.
- Administer the 3-dose hepatitis B vaccine to all other infants.
- Routinely vaccinate previously unvaccinated children and adolescents.
- Routinely vaccinate adults who are non-immune and at risk for HBV infection.
At its October 2016 meeting, ACIP adopted a comprehensive update of all HBV prevention recommendations. (This will be the subject of a future Practice Alert.) Included was a revision of a previously permissive recommendation that allowed the first dose of hepatitis B vaccine for newborns to be given within 2 months of hospital discharge. The new recommendation9 states that newborns of mothers known to be HBsAg negative should be vaccinated within 24 hours (if weight is ≥2000 g) or at age one month or at hospital discharge (if weight is <2000 g).
The first dose should be given within 12 hours of birth to all newborns whose mothers are HBsAg positive or have an unknown HBsAg status.9
Immunization schedules
Every year ACIP updates the adult and child immunization schedules to incorporate the changes from the previous year. These can be found on the ACIP Web site at https://www.cdc.gov/vaccines/schedules/hcp/index.html. This Web site remains the most authoritative and accurate source of information on vaccines and immunizations for both professionals and the public.
The Advisory Committee on Immunization Practices (ACIP) met 3 times in 2016 and introduced or revised recommendations on influenza, meningococcal, human papillomavirus (HPV), cholera, and hepatitis B vaccines. This Practice Alert highlights the most important new recommendations, except those for influenza vaccines, which were described in a previous Practice Alert.1 (See the summary of how this year’s flu season compares to last year’s.)
SIDEBAR
PRACTICE ALERT UPDATE
How this year's flu season compares to last yearThe 2016-2017 influenza season has been relatively mild, with activity nationwide picking up in late January and continuing to increase in February. As of February 16, 90% of the infections typed were type A, and most of those cases (more than 90%) were H3N1. Not surprisingly, the age group most heavily affected has been the elderly.
The hospitalization rate among those ≥65 years as of early February was 113.5/100,000, which is about half the rate of the same week during the 2014-2015 flu season. The hospitalization rate among those ages 50 to 64 years was 23.5/100,000—about 40% lower than the rate during the same week last flu season. At press time, 20 pediatric deaths had occurred, which is less than one-quarter of the number that occurred during the same time last year, and resistance to oseltamivir had not yet been detected in any isolates.
Source: Centers for Disease Control and Prevention. Situation update: summary of weekly FluView report. Available at: https://www.cdc.gov/flu/weekly/summary.htm. Accessed February 16, 2017.
Meningococcal vaccine: Now recommended for HIV-positive patients
Meningococcal conjugate vaccine (serogroups A, C, W, and Y) is recommended for all adolescents ages 11 to 12 as a single dose with a booster at age 16.2 It is also recommended for adults and for children (starting at age 2 months) who have high-risk conditions such as functional or anatomic asplenia or complement deficiencies. Others at high risk include microbiologists routinely exposed to isolates of Neisseria meningitidis and those traveling to areas of high meningococcal incidence. ACIP recently added human immunodeficiency virus (HIV) infection to the list of high-risk conditions.3
Two meningococcal conjugate vaccines are available in the United States: Menactra, (Sanofi Pasteur), licensed for use in individuals ages 9 months to 55 years; and Menveo (GlaxoSmithKline), licensed for use in individuals ages 2 months to 55 years. Menveo is the preferred vaccine for children younger than 2 years infected with HIV. However, if Menactra is used, give it at least 4 weeks after completing all pneumococcal conjugate vaccine doses and either before or concomitantly with diphtheria and tetanus toxoid and acellular pertussis vaccine (DTaP). All individuals who are HIV positive should receive a multi-dose primary series and booster doses. The number of primary doses and timing of boosters depends on the product used and the ages of those vaccinated (TABLE3).
Although neither meningococcal conjugate vaccine product is licensed for use in individuals 56 years or older, ACIP recommends using one of the products for HIV-infected individuals in this age group because the only meningococcal vaccine licensed for use in adults 56 or older, meningococcal polysaccharide vaccine (MPSV4, Menomune, Sanofi Pasteur), has not been studied in patients with HIV infection.
Serogroup B. Two vaccine products provide short-term protection against meningococcal serogroup B: MenB-FHbp (Trumenba, Wyeth Pharmaceuticals, Inc.) and MenB-4C (Bexsero, GlaxoSmithKline). In 2015, ACIP made a “B” recommendation for the use of these vaccines in individuals 16 to 23 years of age, with the preferred age range being 16 to 18.4 A “B” recommendation means that while ACIP does not advise routine use of the vaccines in this age group, the vaccines can be administered to those who desire them. ACIP has recommended routine use of these products only for individuals 10 years and older who are at high risk for meningococcal disease.5
Trumenba was approved as a 3-dose vaccine, administered at 0, 2, and 6 months. Bexsero requires 2 doses given at least one month apart. At its October 2016 meeting, ACIP approved a 2-dose Trumenba schedule, at 0 and 6 months, when administered to those not at risk for meningococcal disease.6 However, during an outbreak, and for those at high risk for meningococcal disease, adhere to the original 3-dose schedule.
HPV vaccine: Now a 2-dose schedule for younger patients
The only HPV vaccine available in the United States is the 9-valent HPV vaccine (9vHPV), Gardasil 9. It is approved for both males and females ages 9 to 26 years. ACIP recommends it for both sexes at ages 11 or 12, and advises catch-up doses for men through age 21 and women through age 26. It also recommends vaccination through age 26 for men who have sex with men and men
The HPV vaccine is approved for a 3-dose schedule at 0, 1 to 2, and 6 months. At its October 2016 meeting, ACIP approved a 2-dose schedule (0, 6-12 months) for those starting the vaccine before their 15th birthday.7 Those starting the vaccine after their 15th birthday, and individuals at any age with an immune-compromising condition, should receive 3 doses. It is hoped that a 2-dose schedule will help to increase the uptake of this safe, effective, and underused vaccine.
Cholera: A new vaccine is available
In June 2016, the FDA approved a live, attenuated, single-dose, oral vaccine (Vaxchora, PaxVax, Inc.) for the prevention of cholera in adults ages 18 to 64 years. It is the only cholera vaccine approved in the United States.
Cholera occurs at low rates among travelers to areas where the disease is endemic. The key to prevention is food and water precautions, and thus the vaccine is not recommended for most travelers—only for those who are at increased risk of exposure to cholera or who have a medical condition that predisposes them to a poor response to medical care if cholera is contracted.8 Risk increases with long-term or frequent travel to endemic areas where safe food and water is not always available. Examples of compromising medical conditions include a blood type O, low gastric acidity, and heart or kidney disease.
Duration of the vaccine’s effectiveness is unknown, given a lack of data beyond 6 months. No recommendation for revaccination has been made, and this issue will be assessed as more data are collected. Other unknowns about the vaccine include its effectiveness among immune-suppressed individuals and pregnant women, as well as for those who live in cholera endemic areas or were previously vaccinated with another cholera vaccine.
Hepatitis B: Vaccinate newborns sooner
The incidence of hepatitis B virus (HBV) infection has declined by more than 90% since the introduction of a vaccine in 1982.9
Current recommendations for the prevention of HBV include:9
- Screen all pregnant women for hepatitis B surface antigen (HBsAg), and use HBIG and hepatitis B vaccines within 12 hours of birth for all newborns whose mothers are HBsAg positive or have an unknown HBsAg status.
- Administer the 3-dose hepatitis B vaccine to all other infants.
- Routinely vaccinate previously unvaccinated children and adolescents.
- Routinely vaccinate adults who are non-immune and at risk for HBV infection.
At its October 2016 meeting, ACIP adopted a comprehensive update of all HBV prevention recommendations. (This will be the subject of a future Practice Alert.) Included was a revision of a previously permissive recommendation that allowed the first dose of hepatitis B vaccine for newborns to be given within 2 months of hospital discharge. The new recommendation9 states that newborns of mothers known to be HBsAg negative should be vaccinated within 24 hours (if weight is ≥2000 g) or at age one month or at hospital discharge (if weight is <2000 g).
The first dose should be given within 12 hours of birth to all newborns whose mothers are HBsAg positive or have an unknown HBsAg status.9
Immunization schedules
Every year ACIP updates the adult and child immunization schedules to incorporate the changes from the previous year. These can be found on the ACIP Web site at https://www.cdc.gov/vaccines/schedules/hcp/index.html. This Web site remains the most authoritative and accurate source of information on vaccines and immunizations for both professionals and the public.
1. Campos-Outcalt D. Need-to-know information for the 2016-2017 flu season. J Fam Pract. 2016;65:613-617.
2. Cohn AC, MacNeil JR, Clark TA, et al. Prevention and control of meningococcal disease: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2013;62:1-28.
3. MacNeil JR, Rubin LG, Patton M, et al. Recommendations for use of meningococcal conjugate vaccines in HIV-infected persons— Advisory Committee on Immunization Practices, 2016. MMWR Morb Mortal Wkly Rep. 2016;65:1189-1194.
4. MacNeil JR, Rubin LG, Folaranmi T, et al. Use of serogroup B meningococcal vaccines in adolescents and young adults: recommendations of the Advisory Committee on Immunization Practices, 2015. MMWR Morb Mortal Wkly Rep. 2015;64:1171-1176.
5. Folaranmi T, Rubin L, Martin SW, et al. Use of serogroup B meningococcal vaccines in persons aged ≥10 years at increased risk for serogroup B meningococcal disease: recommendations of the Advisory Committee on Immunization Practices, 2015. MMWR Morb Mortal Wkly Rep. 2015;64:608-612.
6. MacNeil J. Considerations for Use of 2- and 3-Dose Schedules of MenB-FHbp (Trumenba). Presentation at: Advisory Committee on Immunization Practices; October 19, 2016; Atlanta, GA. Available at: https://www.cdc.gov/vaccines/acip/meetings/downloads/slides-2016-10/meningococcal-05-macneil.pdf. Accessed February 6, 2017.
7. Meites E, Kempe A, Markowitz LE. Use of a 2-dose schedule for human papillomavirus vaccination—updated recommendations of the Advisory Committee on Immunization Practices. MMWR Morb Mortal Wkly Rep. 2016;65:1405-1408.
8. Wong KW. Cholera vaccine update and proposed recommendations. Presentation at: Advisory Committee on Immunization Practices; June 22, 2016; Atlanta, GA. Available at: https://www.cdc.gov/vaccines/acip/meetings/downloads/slides-2016-06/cholera-02-wong.pdf. Accessed January 27, 2017.
9. Schillie S. Revised ACIP Hepatitis B (HepB) vaccine recommendations. Presentation at: Advisory Committee on Immunization Practices; October 19, 2016. Available at: https://www.cdc.gov/vaccines/acip/meetings/downloads/slides-2016-10/hepatitis-02-schillie-october-2016.pdf. Accessed January 27, 2017.
10.
11. Ko SC, Fan L, Smith EA, et al. Estimated annual perinatal hepatitis B virus infections in the United States, 2000-2009. J Pediatric Infect Dis Soc. 2016;5:114-121.
12. Mast EE, Weinbaum CM, Fiore AE, et al. A comprehensive immunization strategy to eliminate transmission of hepatitis B virus infection in the United States. MMWR Morb Mortal Wkly Rep. 2006;55:1-25.
13. Beasley RP, Hwang LY, Lee GC, et al. Prevention of perinatally transmitted hepatitis B virus infections with hepatitis B immune globulin and hepatitis B vaccine. Lancet. 1983;2:1099-1102.
1. Campos-Outcalt D. Need-to-know information for the 2016-2017 flu season. J Fam Pract. 2016;65:613-617.
2. Cohn AC, MacNeil JR, Clark TA, et al. Prevention and control of meningococcal disease: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2013;62:1-28.
3. MacNeil JR, Rubin LG, Patton M, et al. Recommendations for use of meningococcal conjugate vaccines in HIV-infected persons— Advisory Committee on Immunization Practices, 2016. MMWR Morb Mortal Wkly Rep. 2016;65:1189-1194.
4. MacNeil JR, Rubin LG, Folaranmi T, et al. Use of serogroup B meningococcal vaccines in adolescents and young adults: recommendations of the Advisory Committee on Immunization Practices, 2015. MMWR Morb Mortal Wkly Rep. 2015;64:1171-1176.
5. Folaranmi T, Rubin L, Martin SW, et al. Use of serogroup B meningococcal vaccines in persons aged ≥10 years at increased risk for serogroup B meningococcal disease: recommendations of the Advisory Committee on Immunization Practices, 2015. MMWR Morb Mortal Wkly Rep. 2015;64:608-612.
6. MacNeil J. Considerations for Use of 2- and 3-Dose Schedules of MenB-FHbp (Trumenba). Presentation at: Advisory Committee on Immunization Practices; October 19, 2016; Atlanta, GA. Available at: https://www.cdc.gov/vaccines/acip/meetings/downloads/slides-2016-10/meningococcal-05-macneil.pdf. Accessed February 6, 2017.
7. Meites E, Kempe A, Markowitz LE. Use of a 2-dose schedule for human papillomavirus vaccination—updated recommendations of the Advisory Committee on Immunization Practices. MMWR Morb Mortal Wkly Rep. 2016;65:1405-1408.
8. Wong KW. Cholera vaccine update and proposed recommendations. Presentation at: Advisory Committee on Immunization Practices; June 22, 2016; Atlanta, GA. Available at: https://www.cdc.gov/vaccines/acip/meetings/downloads/slides-2016-06/cholera-02-wong.pdf. Accessed January 27, 2017.
9. Schillie S. Revised ACIP Hepatitis B (HepB) vaccine recommendations. Presentation at: Advisory Committee on Immunization Practices; October 19, 2016. Available at: https://www.cdc.gov/vaccines/acip/meetings/downloads/slides-2016-10/hepatitis-02-schillie-october-2016.pdf. Accessed January 27, 2017.
10.
11. Ko SC, Fan L, Smith EA, et al. Estimated annual perinatal hepatitis B virus infections in the United States, 2000-2009. J Pediatric Infect Dis Soc. 2016;5:114-121.
12. Mast EE, Weinbaum CM, Fiore AE, et al. A comprehensive immunization strategy to eliminate transmission of hepatitis B virus infection in the United States. MMWR Morb Mortal Wkly Rep. 2006;55:1-25.
13. Beasley RP, Hwang LY, Lee GC, et al. Prevention of perinatally transmitted hepatitis B virus infections with hepatitis B immune globulin and hepatitis B vaccine. Lancet. 1983;2:1099-1102.
Neoadjuvant and Adjuvant Therapy for Gastric Cancer
INTRODUCTION
Gastric cancer is the fifth most common cancer worldwide and the third leading cause of cancer death in both females and males.1 More than 70% of gastric cancer cases occur in the developing world, with approximately 50% occurring in East Asia.2 Gastric cancer is less common in the United States, with an incidence of 12.3 cases in males and 6.0 cases in females per 100,000 per year and a disproportionately higher incidence in Asians.3 According to the Surveillance, Epidemiology, and End Results Program, approximately 26,370 new cases of stomach cancer were diagnosed in the United States in 2016, and an estimated 10,730 people died of this disease.4 Since the 1970s, the 5-year relative survival rate for gastric cancer in the United States has improved from 15% in 1975 to 29% in 2009.5 In contrast, in Japan and Korea, where screening programs have been implemented, the 5-year survival rate approaches 70%.6
RISK FACTORS AND CLASSIFICATION
A variety of risk factors have been linked to gastric cancer. Diets high in salt, salt-preserved foods, and/or processed meats have been associated with an increased risk for developing gastric cancer.7,8 Obesity and smoking have also been implicated in gastric cancer.9,10 Several studies have demonstrated a strong association between Helicobacter pylori and the development of gastric cancer.11–13 It is believed that H. pylori infection leads to chronic active gastritis, atrophic gastritis, and intestinal metaplasia. Interestingly, mass eradication of H. pylori has not been shown to reduce the risk for gastric cancer.14 Therefore, treatment of H. pylori should only be considered in patients with active peptic ulcer disease.15 Other risk factors include Epstein-Barr virus (EBV), prior gastric surgery, and radiation exposure.16–18 Family history of gastric cancer, hereditary nonpolyposis colon cancer, Li-Fraumeni syndrome, and hereditary diffuse gastric cancer caused by mutations in the E-cadherin gene increase the risk.17
The anatomic distinction between gastric cancer and cancer of the gastroesophageal junction (GEJ) has been a topic of debate. The Siewert classification is the most widely used system and divides GEJ adenocarcinoma into 3 categories:20 type I tumor: adenocarcinoma of distal esophagus, located 1 cm to 5 cm above the GEJ; type II tumor: true carcinoma of gastric cardia, located within 1 cm above and 2 cm below the GEJ; type III tumor: subcardial gastric carcinoma, located 2 cm to 5 cm below the GEJ, and infiltrates esophagus from below.
The American Joint Committee on Cancer (AJCC) has updated the latest (7th) edition of TMN staging for stomach cancer to include tumors arising more than 5 cm distally of the GEJ or within 5 cm of the GEJ but without extension to the esophagus or GEJ.21
In the following sections, neoadjuvant and adjuvant therapy in gastric cancer are discussed using a case presentation to illustrate important concepts.
DIAGNOSIS AND STAGING
CASE PRESENTATION
A 43-year old male with no significant past medical history presents with epigastric abdominal pain and heart burn for the past few weeks. He denies nausea, vomiting, melena, or hematochezia. His primary care physician (PCP) diagnoses him with gastroesophageal reflux disease (GERD) and initiates a trial of pantoprazole. Over the next 2 to 3 months, his symptoms do not improve and he has an associated 40-lb weight loss. Both social history and family history are noncontributory. Physical exam reveals epigastric tenderness without rebound or guarding. Laboratory evaluation reveals a hemoglobin of 12.6 g/dL with a mean corpuscular volume of 72 fL. A comprehensive chemistry profile is within normal limits. Given the constellation of presenting symptoms, especially the unintentional weight loss and the presence of microcytic anemia, his PCP suspects a malignant process and refers the patient to a gastroenterologist.
• What are the next appropriate steps for diagnosis?
The most common presenting symptoms of gastric cancer are weight loss and abdominal pain.22 Less commonly, patients exhibit nausea, anorexia, and dysphagia with proximal tumors. Melena is seen in only about 20% of patients. In Japan, where gastric cancer is more prevalent, mass screening programs allow for detection at an earlier stage, which partially accounts for the better survival rates seen in Asia as compared to the United States. Diagnostic work-up includes esophagogastroduodenoscopy (EGD) to assess Siewert category and to obtain a tissue sample for diagnosis. Full staging requires a complete blood count (CBC) with differential; comprehensive chemistry profile; computed tomography (CT) of chest/abdomen/pelvis with oral and intravenous contrast; endoscopic ultrasound (EUS) if no M1 disease is identified; positron emission tomography (PET)-CT if there is no evidence of M1 disease and if clinically indicated; and laparoscopy with cytology for clinical stage T1b or higher.23 Patients should be staged according to the TMN staging system (Table 1).
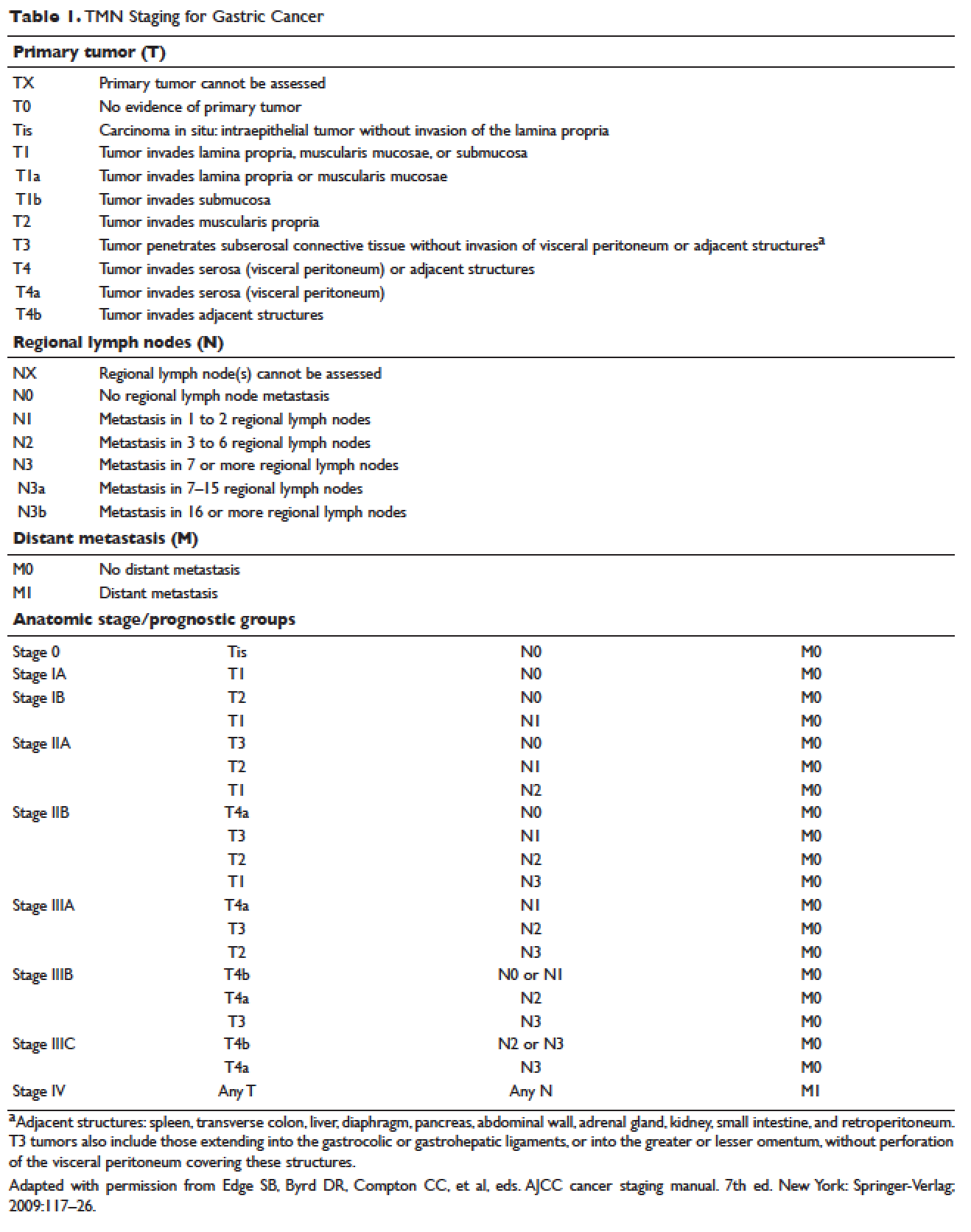
MANAGEMENT OF NONMETASTATIC DISEASE
CASE CONTINUED
The patient undergoes EGD, which reveals a large ulcerated, partially circumferential mass measuring approximately 4 cm. The mass extends from the gastric body to the cardia. Biopsy of the mass reveals poorly differentiated adenocarcinoma as well as H. pylori–associated gastritis. He is given antibiotic therapy and undergoes complete work-up of his newly diagnosed gastric adenocarcinoma. CT of the chest/abdomen/pelvis demonstrates a large gastric mass with gastrohepatic and distal perigastric adenopathy, compatible with locally advance primary gastric cancer. There is no evidence of distant metastasis. PET scan shows a large hypermetabolic mass in the stomach body and increased FDG activity in 3 small nodes along the lesser gastric curvature and in 1 node in the gastrohepatic region. EUS reveals a malignant gastric tumor in the body of the stomach, which is staged as T3, and a few malignant-appearing lymph nodes in the perigastric region. Fine-needle aspiration of the perigastric lymph node is performed and the sample obtained is positive for malignant cells. Diagnostic laparoscopy with peritoneal washings is performed and cytology is negative for malignant cells. The patient is staged as clinical stage IIB (T3N1M0).
• How should this patient with newly diagnosed, locally advanced, resectable gastric cancer be managed?
SURGERY
Surgical resection for localized gastric cancer is the mainstay of treatment with curative intent. Only very early stage (Tis or T1a) tumors can be considered for endoscopic mucosal resection. Regarding surgical resection, distal gastric cancers are typically treated with subtotal gastrectomy because there is no survival difference between subtotal and total gastrectomy.24,25 Moreover, subtotal gastrectomy is associated with better nutritional status and quality of life. For proximal tumors, total gastrectomy is preferred as subtotal gastrectomy has been associated with a higher incidence of reflux esophagitis and anastomotic stenosis.26 In terms of surgical approach, multiple studies have shown that a laparoscopic approach has a lower complication rate and similar outcomes in terms of cancer recurrence and long-term survival when compared to open gastrectomy.27–29 Thus, a laparoscopic approach is often used in academic centers with highly experienced surgeons.
The extent of lymph node dissection remains a topic of debate. A D1 dissection involves the removal of perigastric lymph nodes. A D2 dissection is a D1 dissection plus the removal of lymph nodes along the left gastric artery, common hepatic artery, celiac artery, splenic hilum, and splenic artery. D2 lymphadenectomy has become the standard of care in Eastern countries where gastric cancer is more prevalent, such as Japan and Korea.30 In Western countries, including the United States, less extensive lymphadenectomies are performed. Both randomized clinical trials and meta-analyses have failed to demonstrate an overall survival advantage of D2 dissection over D1 dissection.31,32 A Dutch trial by Bonenkamp et al involving 711 patients, one of the largest randomized trials of D1 and D2 lymphadenectomy, showed that D2 patients had a higher operative mortality rate than D1 patients (10% versus 4%, P = 0.004) and experienced more complications (43% versus 25%, P < 0.001).33 In a 15-year follow-up of this study, patients who had a D2 resection had lower locoregional recurrence and gastric-cancer–related death rates compared to those who had a D1 resection; however, D2 resection was associated with a significantly higher operative mortality and complication rate compared to D1.34 In addition, a 2015 Cochrane meta-analysis has demonstrated improved disease-specific survival (DSS) with D2 dissection (hazard ratio [HR] 0.81 [95% confidence interval {CI} 0.71 to 0.92]).35 Currently, the National Comprehensive Cancer Network (NCCN) recommends a D1 or a modified D2 gastrectomy with at least 15 lymph nodes removed for examination, with D2 lymphadenectomies only to be performed at experienced centers.23
SYSTEMIC CHEMOTHERAPY
Locally advanced gastric cancer (T3-T4 or node positive) requires systemic chemotherapy in addition to surgery, as this intervention improves the 5-year overall survival by 10% to 15%.36 Systemic therapy should also be considered in patients with T2N0 disease with high-risk features: poorly differentiated or high-grade cancer; lymphovascular invasion; neural invasion; age younger than 50 years; and patients who did not undergo D2 dissection.23 Currently, there is no global consensus on the best treatment approach. In the United States, where a less aggressive lymph-node dissection is performed, adjuvant chemoradiotherapy after surgery is more commonly seen. In Europe, perioperative (preoperative and postoperative) chemotherapy is the standard treatment. In Japan, adjuvant chemotherapy after D2 lymphadenectomy is the standard of care.37 These regional preferences are largely due to randomized clinical trials that have shown benefit for each approach. The landmark trials are discussed in the following sections and are summarized in Table 2.
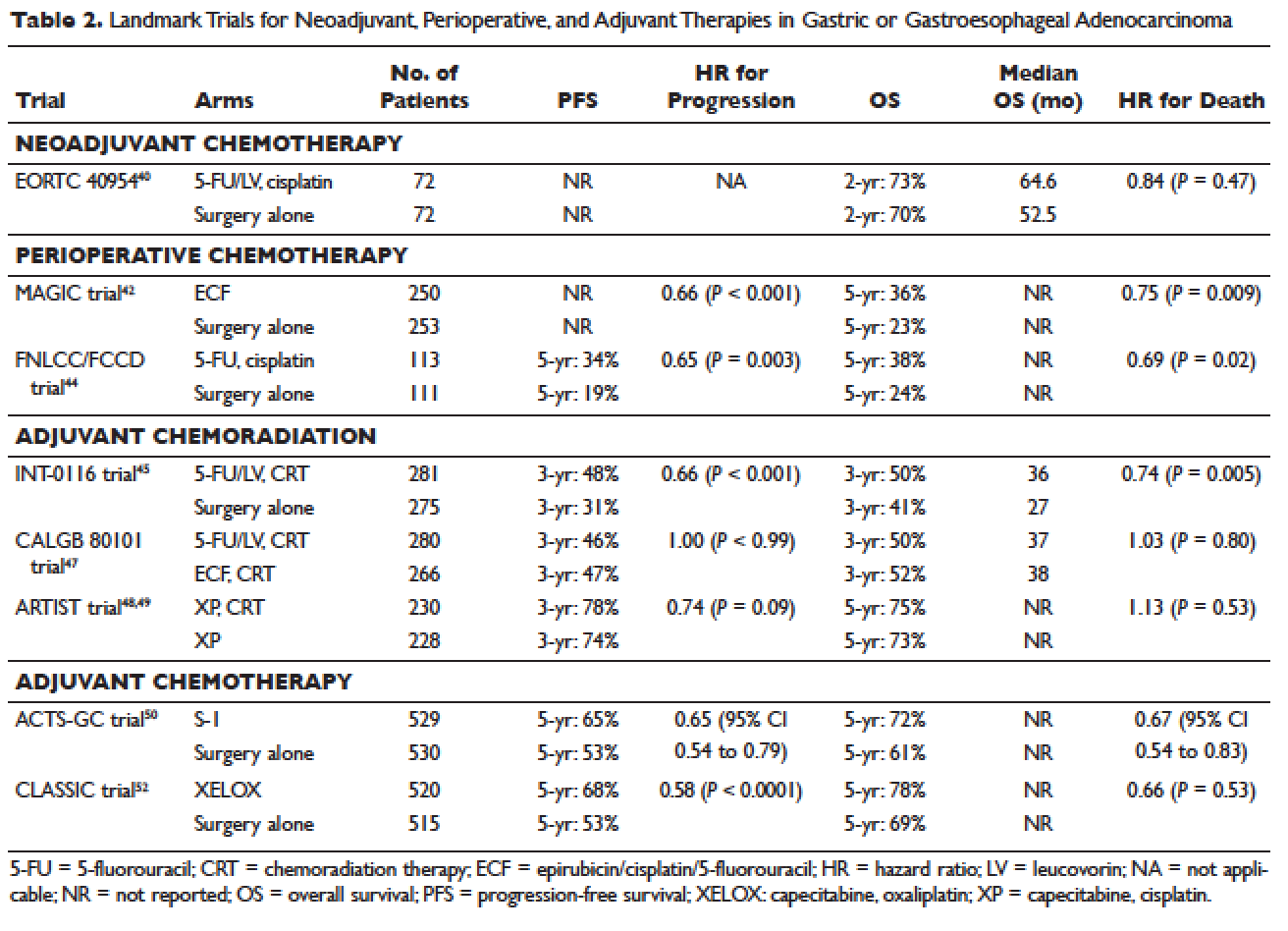
Neoadjuvant Chemotherapy
Neoadjuvant chemotherapy has the benefit of “downstaging” locally advanced tumors to allow for curative resection. Phase 2 clinical trials have also demonstrated good pathologic response rates and high R0 resection rates following neoadjuvant chemotherapy.38,39 However, phase 3 trials to support this treatment approach are lacking. In the European Organisation for Research and Treatment of Cancer (EORTC) 40954 trial, patients with stage III or IV gastric or GEJ cancer were randomly assigned to surgery with or without preoperative cisplatin, leucovorin, and infusional fluorouracil (5-FU).40 The trial was stopped early due to poor accrual after 144 patients were randomized. The neoadjuvant chemotherapy arm had a higher R0 resection rate compared to the surgery alone arm (82% versus 67%, respectively, P = 0.036) but a higher postoperative complication rate (27% versus 16%, respectively, P = 0.09). More important, after a median follow-up of 4.4 years, a survival benefit could not be shown, with 2-year survival rates of 72.7% and 69.9% in the neoadjuvant and surgery-only arms, respectively (HR 0.84 [95% CI 0.52 to 1.35], P = 0.466). Due to the lack of large trials, a meta-analysis assessing the effectiveness of neoadjuvant chemotherapy combined with surgery versus surgery alone in advanced gastric and gastroesophageal cancer was performed.41 The analysis included 12 randomized controlled trials with a total of 1820 patients. Neoadjuvant chemotherapy was shown to slightly improve the survival rate (odds ratio [OR] 1.32 [95% CI 1.07 to 1.64], P = 0.01). It significantly improved the 3-year progression-free survival (PFS; OR 1.85 [95% CI 1.39 to 2.46], P < 0.0001), tumor down-staging rate (OR 1.71 [95% CI 1.26 to 2.33], P = 0.0006), and R0 resection rate (OR 1.38 [95% CI 1.08 to 1.78], P = 0.01). There were no differences between the 2 arms in terms of relapse rates, operative complications, perioperative mortality, and grade 3/4 adverse effects. While these results are encouraging, further randomized clinical trials are needed to clarify the role of neoadjuvant chemotherapy and its impact on overall survival.
Perioperative Chemotherapy
The results of the Medical Research Council Adjuvant Gastric Infusional Chemotherapy (MAGIC) trial published in 2006 established perioperative chemotherapy as standard of care in patients with resectable gastric and gastroesophageal adenocarcinoma.42 A total of 503 patients with potentially resectable gastric and lower esophageal adenocarcinoma were randomly assigned to perioperative chemotherapy plus surgery or surgery alone. Perioperative chemotherapy consisted of 3 preoperative and postoperative cycles of epirubicin, cisplatin, and infusional 5-FU (ECF). At a median follow-up of 4 years, the perioperative-chemotherapy group had a significantly better PFS (HR 0.66 [95% CI 0.53 to 0.81], P < 0.001) as well as overall survival (HR 0.75 [95% CI 0.60 to 0.93], P = 0.009). The 5-year overall survival rate was 36.3% in the perioperative chemotherapy group and 23% in the surgery group. Of note, there was a greater proportion of stage T1/T2 tumors (52% versus 37%, P = 0.002) and N0/N1 disease (84% versus 71%) in the perioperative-chemotherapy group compared to the surgery alone group. In addition, only 42% of patients in the perioperative chemotherapy group completed all 6 cycles of chemotherapy.
The administration of ECF is often difficult since the 5-FU component requires a central venous access device and an ambulatory infusion pump and the cisplatin component is associated with nephrotoxicity and ototoxicity. The REAL-2 trial was a randomized phase 3 clinical trial that assessed whether 5-FU could be replaced by capecitabine and cisplatin by oxaliplatin in the ECF regimen.43 Between June 2000 and May 2005, a total of 1002 patients with locally advanced esophageal/GEJ/gastric cancer were enrolled. Patients were randomly assigned to 1 of 4 triplet therapies: epirubicin and cisplatin plus either 5-FU (ECF) or capecitabine (ECX) or epirubicin and oxaliplatin plus either 5-FU (EOF) or capecitabine (EOX). After a median follow-up of approximately 18 months, the overall survival in the capecitabine groups did not differ significantly from that in the 5-FU groups (HR 0.88 [95% CI 0.77 to 1.00], P = 0.06), nor did overall survival in the oxaliplatin groups differ significantly from that in the cisplatin groups (HR 0.91 [95% CI 0.79 to 1.04], P = 0.16). Interestingly, the 1-year survival rate was longer in the EOX group than in the ECF group (46.8% versus 37.7%, respectively; HR 0.80 [95% CI 0.66 to 0.97], P = 0.02). This translated into an overall survival of 11.2 months for the EOX group and 9.9 months for the ECF group. Therefore, EOX is preferred over ECF in clinical practice.
The French FNLCC/FFCD trial published in 2011 provided further support for perioperative chemotherapy.44 A total of 224 patients with adenocarcinoma of the lower esophagus, GEJ, or stomach were randomly assigned to perioperative chemotherapy plus surgery or surgery alone. The perioperative-chemotherapy group received 2 to 3 cycles of preoperative chemotherapy and 3 to 4 cycles of postoperative chemotherapy, consisting of infusional 5-FU (800 mg/m2 daily for days 1 to 5) and cisplatin (100 mg/m2 on day 1). In patients receiving preoperative chemotherapy, 38% experienced at least grade 3 to 4 toxicity. Among the 109 patients who received at least 1 cycle of preoperative chemotherapy, only 54 patients (50%) received postoperative chemotherapy. Despite this, the perioperative-chemotherapy group had a statistically significant higher R0 resection rate (84% versus 74%, P = 0.04) compared to the surgery alone group. At a median follow-up of 5.7 years, the perioperative chemotherapy group had an improved overall survival (HR for death 0.69 [95% CI 0.50 to 0.95], P = 0.02) and disease-free survival (DFS; HR for recurrence or death 0.65 [95% CI 0.48 to 0.89], P = 0.003). This translated into 5-year overall survival rates of 38% versus 24% and 5-year DFS rates of 34% versus 19%. One caveat to this study is that the majority of patients (64%) had GEJ cancer and only 25% had gastric cancer. In the multivariate analysis, the 2 significant prognostic factors for overall survival were the administration of preoperative chemotherapy (P = 0.01) and tumor site at the GEJ (P < 0.01).
Adjuvant Chemoradiotherapy
The INT-0116 (Intergroup 0116) study published in 2001 established adjuvant chemoradiotherapy as the standard approach for resectable gastric cancer in the United States. In this study, a total of 556 patients with resected gastric or GEJ cancer were randomly assigned to surgery alone or surgery followed by adjuvant 5-FU/leucovorin bolus chemotherapy, sandwiched with 5-FU–based chemoradiation (45 Gy).45 In the chemoradiotherapy group, 64% of patients completed treatment and grade 3 and 4 toxicity occurred in 41% and 32%, respectively. However, only 3 patients (1%) died from treatment-related toxicity. At a median follow-up of 5 years, the median overall survival was 36 months in the chemoradiation group and 27 months in the surgery group. Overall survival rate was 50% in the combined modality group and 41% in the surgery-alone group, with a HR of 1.35 (95% CI 1.09 to 1.66, P = 0.005). The 3-year DFS was 48% in the chemoradiotherapy group and 31% in the surgery-alone group, corresponding to a DFS of 30 months and 19 months, respectively. Even after a median follow-up of 10 years, these positive results were maintained, with a HR for survival of 1.32 (95% CI 1.10 to 1.60, P = 0.0046) and HR for DFS of 1.51 (95% CI 1.25 to 1.83, P < 0.001).46 A criticism of the INT-0116 study is that 54% of patients had less than a D1 lymph node dissection, suggesting that adjuvant chemoradiation may have compensated for suboptimal surgery.
CALGB 80101, a United States Intergroup study, compared the INT-0116 protocol regimen (bolus 5-FU/leucovorin with 5-FU plus concurrent radiotherapy) to postoperative ECF sandwiched with 5-FU plus concurrent radiotherapy.47 The study included patients with resected gastric or GEJ adenocarcinoma that extended beyond the muscularis propria or was node positive. The percentage of patients with gastric versus GEJ cancer was not reported. A total of 546 patients were randomized. Preliminary results were presented at the 2011 American Society of Clinical Oncology meeting. The ECF arm had lower rates of grade 3/4 toxicities, including neutropenia, diarrhea, and mucositis. However, there was no difference in overall survival (3-year overall survival of 52% versus 50% for ECF and 5-FU/leucovorin, respectively) or DFS (3-year DFS of 47% versus 46% for ECF and 5-FU/leucovorin, respectively). The trial was not adequately powered to assess noninferiority. The location of the primary tumor (GEJ versus proximal versus distal stomach) did not have any effect on treatment outcome.
The Adjuvant Chemoradiation Therapy in Stomach Cancer (ARTIST) trial was the first study to compare adjuvant chemoradiotherapy with adjuvant chemotherapy in patients with D2-resected gastric cancer.48 A total of 458 patients were randomly assigned to 6 cycles of XP chemotherapy (capecitabine 2000 mg/m2 per day on days 1–14 and cisplatin 60 mg/m2 on day 1, every 3 weeks) or XP/radiotherapy/XP (2 cycles of XP followed by 45 Gy radiotherapy with concurrent daily capecitabine [825 mg/m2 twice daily] and 2 cycles of XP). After a median follow-up of 84 months, there was no difference in DFS or overall survival between treatment arms (HR for progression 0.74 [95% CI 0.52 to 1.05], P = 0.09; HR for death 1.13 [95% CI 0.78 to 1.65], P = 0.53).49 However, subgroup analysis showed that chemoradiotherapy significantly improved DFS in patients with node-positive disease (3-year DFS 76% versus 72%, P = 0.004).
Adjuvant Chemotherapy
Data supporting the use of adjuvant chemotherapy alone is largely derived from trials done in Asia, typically after a D2 lymph node dissection, and thus adjuvant chemotherapy has become the standard of care in that region. In the Japanese Adjuvant Chemotherapy Trial of S-1 for Gastric Cancer (ACTS-GC), a total of 1059 patients with stage II or III gastric cancer who had undergone surgery with a D2 lymphadenectomy were randomly assigned to 1 year of S-1 (an oral fluoropyrimidine) or surgery alone.50 The 5-year overall survival rate was 72% in the S-1 group and 61% in the surgery-only group (HR 0.669 [95% CI 0.54 to 0.83]).51 The 5-year relapse-free survival was 65% in the S-1 group and 53% in the surgery-only group (HR 0.65 [95% CI 0.537 to 0.793]). Of note, both arms of the ACTS-GC trial had significantly higher 5-year overall survival rates compared to the INT-0116 and MAGIC trials: 43% versus 28% and 36% versus 23% for the treatment and control groups, respectively.42,45 Consequently, it is unclear if the benefit of adjuvant chemotherapy can be translated to Western countries.
The Korean Capecitabine and Oxaliplatin Adjuvant Study in Stomach Cancer (CLASSIC) trial published in 2012 also established the role of adjuvant chemotherapy after D2 gastrectomy.52 A total of 1035 patients with stage II-IIIB gastric cancer who had curative D2 gastrectomy were randomly assigned to 8 cycles of adjuvant XELOX (capecitabine 1000 mg/m2 twice daily on days 1–14 plus oxaliplatin 130 mg/m2 on day 1, 21-day cycle) or surgery alone. Median follow-up was 34 months in both arms and 67% of patients in the chemotherapy arm completed all 8 cycles of planned chemotherapy. The 3-year DFS was 74% in the chemotherapy group and 59% in the surgery-only group (HR 0.56 [95% CI 0.44 to 0.72], P < 0.0001). There was a trend toward improvement in overall survival (83% versus 78%, HR 0.72 [95% CI 0.52 to 1.00]). After 5 years of follow-up, the improvement in overall survival became statistically significant (78% versus 69%, HR 0.66 [95% CI 0.51 to 0.85]).53
The benefit of adjuvant chemotherapy was reinforced by a 2010 meta-analysis comparing adjuvant chemotherapy to surgery alone in patients with resected gastric cancer.54 A total of 17 randomized controlled trials were included. Adjuvant fluorouracil-based chemotherapy was associated with a statistically significant improved overall survival (HR 0.82 [95% CI 0.76 to 0.90], P < 0.001) and DFS (HR 0.82 [95% CI 0.75 to 0.90], P < 0.001). Five-year overall survival increased from 49.6% to 55.3% with chemotherapy.
SELECTION OF TREATMENT APPROACH
Since data exists for all 3 approaches (perioperative chemotherapy, adjuvant chemoradiotherapy, and adjuvant chemotherapy), various meta-analyses have been done to clarify which approach is the best. In a recent meta-analysis of 6 randomized controlled trials reported between 2010 and 2012, which involved 1171 patients with resected gastric cancer, adjuvant chemotherapy was compared to adjuvant chemoradiotherapy.55 Five of the studies were from East Asia, while one was from a Western country. Adjuvant chemoradiation was associated with a lower local-regional recurrence rate (OR 0.46 [95% CI 0.32 to 0.67]) and better 5-year DFS rate (OR 1.56 [95% CI 1.09 to 2.24]). However, there was no statistical difference in 5-year overall survival rate (OR 1.32 [95% CI 0.92 to 1.88]). Similar results were reported by Zhou et al in 2016.56 This meta-analysis included 4 randomized controlled trials reported between 2010 and 2015, with a total of 960 patients who had undergone a D2 resection for gastric cancer. Compared to adjuvant chemotherapy, adjuvant chemoradiotherapy significantly reduced the loco-regional recurrence rate (LRRR; relative risk [RR] 0.50 [95% CI 0.34 to 0.74], P = 0.0005) and improved DFS (HR 0.73 [95% CI 0.60 to 0.89], P = 0.002). Again, no difference in overall survival was seen (HR 0.91 [95% CI 0.74 to 1.11], P = 0.34).
Adjuvant chemotherapy and perioperative chemotherapy have also been compared. In a recent meta-analysis of 14 randomized controlled trials (8 Asian, 6 European) involving 2093 patients with resected gastric or GEJ cancer, perioperative chemotherapy was associated with improved overall survival when compared to adjuvant chemotherapy (HR 0.48 [95% CI 0.35 to 0.67], P < 0.001).57 The benefit of perioperative chemotherapy over adjuvant chemotherapy has also been reported in a 2016 meta-analysis by Zhao et al.58 A total of 1240 patients were included from 5 randomized controlled trials and 6 clinical controlled trials, all from Asian countries. The 5-year overall survival rate was significantly better in the perioperative chemotherapy group compared to the adjuvant chemotherapy group (RR 0.77 [95% CI 0.64 to 0.92], P < 0.01). Furthermore, the 2 groups showed no significant differences in the postoperative complication rates (RR 0.98 [95% CI 0.63 to 1.51], P = 0.91) or adverse effects of chemotherapy (P > 0.05 for all adverse effects).
While these meta-analyses may offer some insight on the best treatment approach, they should be interpreted with caution. Most studies included in these meta-analyses were from Asian countries, and their findings may not be applicable to Western countries. Furthermore, the heterogeneity of trials and inclusion of nonrandomized trials make it difficult to draw conclusions. There are several ongoing trials that will help to define the optimal treatment approach.
CASE CONTINUED
The patient is presented at tumor board and the consensus is to proceed with the perioperative chemotherapy approach. The patient undergoes echocardiography, which reveals a normal ejection fraction. He receives 3 cycles of neoadjuvant EOX (epirubicin, oxaliplatin, and capecitabine). After 3 cycles of neoadjuvant EOX, the patient has a repeat CT that shows marked interval reduction in the size of the primary gastric neoplasm and interval decrease in the size of the small perigastric lymph nodes. He subsequently undergoes a total gastrectomy with J-tube placement. Pathology shows ypT3N0 disease with 0 out of 47 lymph nodes involved and negative margins. He then receives 3 cycles of adjuvant EOX.
• What are the recommendations for surveillance?
According to the current NCCN guidelines, a history and physical exam should be performed every 3 to 6 months for 1 to 2 years, then every 6 to 12 months for 3 to 5 years, and then annually.23 Labs, CT chest/abdomen, and EGD should be done as clinically indicated. Patients who have undergone surgical resection should be monitored for nutritional deficiencies (vitamin B12 and iron).
GASTROESOPHAGEAL JUNCTION TUMORS
Tumors arising in the GEJ or gastric cardia within 5 cm of the GEJ that extend into the GEJ or distal esophagus are staged and treated as esophageal cancers.21 The primary treatment for T1/T2N0 tumors is surgical resection. In patients with T3 or higher or node-positive adenocarcinoma of the GEJ, a combined modality approach is preferred, with preoperative chemoradiotherapy followed by surgical resection.59 The CROSS trial demonstrated a significant survival benefit with preoperative chemoradiation using carboplatin/paclitaxel compared to surgery alone in patients with esophageal or GEJ cancer (49 months versus 24 months, HR 0.66, P = 0.003).60
ONGOING TRIALS
As mentioned previously, several randomized clinical trials are in progress to clarify the optimal treatment approach. The MAGIC-B/MRC-ST03 is a randomized phase 2/3 trial looking at perioperative epirubicin, cisplatin, and capecitabine (ECX) with or without bevacizumab in patients with resectable lower esophageal, GEJ, or gastric cancer.61 The TOPGEAR trial, a randomized phase 2/3 study being conducted in Canada and Europe, is comparing perioperative ECF chemotherapy with preoperative chemoradiation plus perioperative ECF chemotherapy.62 In Asia, the PRODIGY trial is a phase 3, open-label, randomized study comparing neoadjuvant docetaxel, oxaliplatin, and S-1 followed by surgery and adjuvant S-1 versus surgery plus adjuvant S-1 in patients with locally advanced gastric cancer (T2-T4 or node positive).63 Primary endpoint is PFS and secondary endpoints are overall survival, R0 resection rate, and safety.
Trials comparing adjuvant chemotherapy to adjuvant chemoradiotherapy are also being conducted. In the Dutch CRITICS study, a randomized phase 3 trial, patients with stage Ib-Iva resectable gastric cancer were given 3 cycles of epirubicin, cisplatin/oxaliplatin, and capecitabine (ECC/EOC), followed by D2 resection and either 3 cycles of ECC/EOC or chemoradiation with weekly cisplatin and daily capecitabine.64 Between January 2007 and April 2015, a total of 788 patients were enrolled. In a preliminary report presented at ASCO in 2016, the 5-year survival rate was similar between the 2 arms (41.3% for chemotherapy arm and 40.9% for chemoradiotherapy arm, P = 0.99). The Korean ARTIST II trial is comparing adjuvant S-1 and S-1/oxaliplatin with or without radiotherapy in patients with D2-resected gastric cancer.65 Similarly, the NCT01711242 trial is comparing adjuvant XELOX alone versus XELOX with concurrent capecitabine/radiotherapy in patients with resected D2 gastric cancer.66
The ToGA trial established a survival benefit of trastuzumab in combination with chemotherapy in HER2-positive metastatic gastric cancer.67 Consequently, there are ongoing clinical trials to assess the role of trastuzumab in nonmetastatic gastric cancer. The TOXAG study is a phase 2 trial looking at the safety and tolerability of adjuvant oxaliplatin, capecitabine, and trastuzumab with radiation in patients with resected HER2-positive gastric or GEJ adenocarcinoma.68 The NCT01130337 clinical trial is evaluating perioperative XELOX/trastuzumab in patients with resectable gastric or GEJ adeno-carcinoma.69
SUMMARY
Gastric cancer is the fifth most common cancer worldwide, with the greatest incidence in East Asia. Survival outcomes are better in Asian countries when compared to the United States. This difference in survival may be related to the presence of mass screening programs in Asia, which allows for detection at an earlier stage and the use of a more extensive surgical approach (ie, D2 resection). Risk factors for developing gastric cancer include: diets high in salt/salt-preserved foods or processed meats, obesity, smoking, H. pylori infection, EBV, prior gastric surgery, radiation exposure, and positive family history.
According to the latest edition of TMN staging, gastric cancer includes tumors arising more than 5 cm distally of the GEJ or within 5 cm of the GEJ but without extension to the esophagus or GEJ. Diagnostic work-up includes: EGD with biopsy; basic labs; CT chest/abdomen/pelvis with oral and intravenous contrast; EUS if no M1 disease is identified; PET-CT if there is no M1 disease and if clinically indicated; and diagnostic laparoscopy with cytology for clinical stage T1b or higher.
The mainstay of treatment is surgical resection. Laparoscopic approach is preferred over open gastrectomy due to lower complication rates and similar survival outcomes. Current NCCN guidelines recommend a D1 or a modified D2 lymph node dissection with at least 15 lymph nodes removed for examination. Systemic chemotherapy is required in locally advanced gastric cancer (T3-T4 or node positive) and should be considered in T2N0 disease with high-risk features. Currently, there is no global consensus on the optimal treatment approach. Data from various trials have shown benefit for each approach. Regional preferences are: perioperative chemotherapy in Europe; adjuvant chemoradiotherapy in the United States; and adjuvant chemotherapy in Asia. In an effort to better define the optimal treatment approach, several randomized clinical trials are being conducted. According to the current NCCN guidelines, the following treatment approaches are acceptable and are supported by data in the trial listed in parentheses:
• Perioperative chemotherapy
° 5-FU/cisplatin (French FNLCC/FCCD trial)44 or
° ECF (MAGIC trial)42 or
° ECF modifications: EOX, EOF, ECX (REAL-2 trial)43
• Adjuvant chemoradiotherapy
° 5-FU/leucovorin sandwiched with 5-FU-based chemoradiation (INT-0116 trial)45
• Adjuvant chemotherapy (after D2 resection)
° Capecitabine/oxaliplatin (CLASSIC trial)52 or
° Capecitabine/cisplatin (ARTIST trial)48,49
- World Health Organization. GLOBOCAN 2012: estimated cancer incidence, mortality and prevalence Worldwide in 2012. France, Lyon: IARC; 2012.
- Ferlay J, Shin HR, Bray F, et al. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 2010;127:2893–917.
- Lui FH, Tuan B, Swenson SL, et al. Ethnic disparities in gastric cancer incidence and survival in the USA: an updated analysis of 1992-2009 SEER data. Dig Dis Sci 2014;59:3027–34.
- Howlader N, Noone AM, Krapcho M, et al. SEER cancer statistics review, 1975-2013. National Cancer Institute. http://seer.cancer.gov/csr/1975_2013/. Based on November 2015 SEER data submission, posted to the SEER web site April 2016.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin 2015;65:5–29.
- Isobe Y, Nashimoto A, Akazawa K, et al. Gastric cancer treatment in Japan: 2008 annual report of the JGCA nationwide registry. Gastric Cancer 2011;14:301–16.
- Tsugane S, Sasazuki S. Diet and the risk of gastric cancer: review of epidemiological evidence. Gastric Cancer 2007;10:75.
- Bouvard V, Loomis D, Guyton KZ, et al. Carcinogenicity of consumption of red and processed meat. Lancet Oncol 2015;16:1599–600.
- Yang P, Zhou Y, Chen B, et al. Overweight, obesity and gastric cancer risk: results from a meta-analysis of cohort studies. Eur J Cancer 2009;45:2867–73.
- González CA, Pera G, Agudo A, et al. Smoking and the risk of gastric cancer in the European Prospective Investigation Into Cancer and Nutrition (EPIC). Int J Cancer 2003;107:629–34.
- Huang JQ, Sridhar S, Chen Y, Hunt RH. Meta-analysis of the relationship between Helicobacter pylori seropositivity and gastric cancer. Gastroenterology 1998;114:1169–79.
- Eslick GD, Lim LL, Byles JE, et al. Association of Helicobacter pylori infection with gastric carcinoma: a meta-analysis. Am J Gastroenterol 1999;94:2373–9.
- An international association between Helicobacter pylori infection and gastric cancer. The EUROGAST Study Group. Lancet 1993;341:1359–62.
- Parsonnet J, Forman D. Helicobacter pylori infection and gastric cancer—for want of more outcomes. JAMA 2004;291:244–5.
- Malfertheiner P, Megraud F, O'Morain CA, et al. Management of Helicobacter pylori infection—the Maastricht IV/ Florence Consensus Report. Gut 2012;61:646–64.
- Fukayama M. Epstein-Barr virus and gastric carcinoma. Pathol Int 2010;60:337–50.
- Takeno S, Hashimoto T, Maki K, et al. Gastric cancer arising from the remnant stomach after distal gastrectomy: a review. World J Gastroenterol 2014;20:13734–40.
- Morton LM, Dores GM, Curtis RE, et al. Stomach cancer risk after treatment for Hodgkin lymphoma. J Clin Oncol 2013;31:3369–77.
- van der Post RS, Vogelaar IP, Carneiro F, et al. Hereditary diffuse gastric cancer: updated clinical guidelines with an emphasis on germline CDH1 mutation carriers. J Med Genet 2015;52:361–74.
- Siewert J, Stein H. Classification of adenocarcinoma of the oesophagogastric junction. Br J Surg 1998;85:1457–9.
- Edge S, Byrd DR, Compton CC, et al. AJCC cancer staging manual. 7th ed. New York: Springer New York; 2009.
- Wanebo HJ, Kennedy BJ, Chmiel J, et al. Cancer of the stomach. A patient care study by the American College of Surgeons. Ann Surg 1993;218:583–92.
- National Comprehensive Cancer Network. Gastric cancer (version 3.2016www.nccn.org/professionals/physician_gls/pdf/gastric.pdf. Accessed December 14, 2016.
- Bozzetti F, Marubini E, Bonfanti G, et al. Subtotal versus total gastrectomy for gastric cancer: five-year survival rates in a multicenter randomized Italian trial. Italian Gastrointestinal Tumor Study Group. Ann Surg 1999;230:170–8.
- Gouzi JL, Huguier M, Fagniez PL, et al. Total versus subtotal gastrectomy for adenocarcinoma of the gastric antrum. A French prospective controlled study. Ann Surg 1989;209:162–6.
- Pu YW, Gong W, Wu YY, et al. Proximal gastrectomy versus total gastrectomy for proximal gastric carcinoma. A meta-analysis on postoperative complications, 5-year survival, and recurrence rate. Saudi Med J 2013;34:1223–8.
- Chen K, Xu XW, Mou YP, et al. Systematic review and meta-analysis of laparoscopic and open gastrectomy for advanced gastric cancer. World J Surg Oncol 2013;11:182.
- Fang C, Hua J, Li J, et al. Comparison of long-term results between laparoscopy-assisted gastrectomy and open gastrectomy with D2 lymphadenectomy for advanced gastric cancer. Am J Surg 2014;208:391–6.
- Wang W, Li Z, Tang J, et al. Laparoscopic versus open total gastrectomy with D2 dissection for gastric cancer: a meta-analysis. J Cancer Res Clin Oncol 2013;139:1721–34.
- Schmidt B, Yoon SS. D1 versus D2 lymphadenectomy for gastric cancer. J Surg Oncol 2013;107:259–64.
- Jiang L, Yang KH, Guan QL, et al. Survival and recurrence free benefits with different lymphadenectomy for resectable gastric cancer: a meta-analysis. J Surg Oncol 2013;107:807–14.
- Degiuli M, Sasako M, Ponti A, et al. Randomized clinical trial comparing survival after D1 or D2 gastrectomy for gastric cancer. Br J Surg 2014;101:23–31.
- Bonenkamp JJ, Songun I, Hermans J, et al. Randomized comparison of morbidity after D1 and D2 dissection for gastric cancer in 996 Dutch patients. Lancet 1995;345:745–8.
- Songun I, Putter H, Kranenbarg EM, et al. Surgical treatment of gastric cancer: 15-year follow-up results of the randomised nationwide Dutch D1D2 trial. Lancet Oncol 2010;11:439–49.
- Mocellin S, McCulloch P, Kazi H, et al. Extent of lymph node dissection for adenocarcinoma of the stomach. Cochrane Database Syst Rev 2015;8:CD001964.
- Van Cutsem E, Sagaert X, Topal B, et al. Gastric cancer. Lancet 2016;388:2654–64.
- Quéro L, Guillerm S, Hennequin C. Neoadjuvant or adjuvant therapy for gastric cancer. World J Gastrointest Oncol 2015;7:102–10.
- Okabe H, Hata H, Ueda S, et al. A phase II study of neoadjuvant chemotherapy with S-1 and cisplatin for stage III gastric cancer: KUGC03. J Surg Oncol 2016 Jan;113:36–41.
- Wang X, Zhao L, Liu H et al. A phase II study of a modified FOLFOX6 regimen as neoadjuvant chemotherapy for locally advanced gastric cancer. Br J Cancer 2016;114:1326-33.
- Schuhmacher C, Gretschel S, Lordick F, et al. Neoadjuvant chemotherapy compared with surgery alone for locally advanced cancer of the stomach and cardia: European Organisation for Research and Treatment of Cancer randomized trial 40954. J Clin Oncol 2010;28:5210–18.
- Xiong BH, Cheng Y, Ma L, Zhang CQ. An updated meta-analysis of randomized controlled trial assessing the effect of neoadjuvant chemotherapy in advanced gastric cancer. Cancer Invest 2014;32:272–84.
- Cunningham D, Allum WH, Stenning SP, et al. Perioperative chemotherapy versus surgery alone for resectable gastroesophageal cancer. N Engl J Med 2006;355:11–20.
- Cunningham D, Starling N, Rao S, et al. Capecitabine and oxaliplatin for advanced esophagogastric cancer. N Engl J Med 2008;358:36–46.
- Ychou M, Boige V, Pignon JP, et al. Perioperative chemotherapy compared with surgery alone for resectable gastroesophageal adenocarcinoma: an FNCLCC and FFCD multicenter phase III trial. J Clin Oncol 2011;29:1715–21.
- Macdonald JS, Smalley SR, Benedetti J, et al. Chemoradiotherapy after surgery compared with surgery alone for adenocarcinoma of the stomach or gastroesophageal junction. N Engl J Med 2001;345:725–30.
- Smalley SR, Benedetti JK, Haller DG, et al. Updated analysis of SWOG-directed intergroup study 0116: a phase III trial of adjuvant radiochemotherapy versus observation after curative gastric cancer resection. J Clin Oncol 2012;30:2327–33.
- Fuchs CS, Tepper JE, Niedzwiecki D, et al. Postoperative adjuvant chemoradiation for gastric or gastroesophageal junction (GEJ) adenocarcinoma using epirubicin, cisplatin, and infusional (CI) 5-FU (ECF) before and after CI 5-FU and radiotherapy (CRT) compared with bolus 5-FU/LV before and after CRT: Intergroup trial CALGB 80101. J Clin Oncol 2011;29:256s. Abstract 4003.
- Lee J, Lim do H, Kim S, et al. Phase III trial comparing capecitabine plus cisplatin versus capecitabine plus cisplatin with concurrent capecitabine radiotherapy in completely resected gastric cancer with D2 lymph node dissection: the ARTIST trial. J Clin Oncol 2012;30:268–73
- Park SH, Sohn TS, Lee J, et al. Phase III trial to compare adjuvant chemotherapy with capecitabine and cisplatin versus concurrent chemoradiotherapy in gastric cancer: final report of the adjuvant chemoradiotherapy in stomach tumors trial, including survival and subset analyses. J Clin Oncol 2015;33:3130–6.
- Sakuramoto S, Sasako M, Yamaguchi T, et al. Adjuvant chemotherapy for gastric cancer with S-1, an oral fluoropyrimidine. N Engl J Med 2007;357:1810–20.
- Sasako M, Sakuramoto S, Katai H, et al. Five-year outcomes of a randomized phase III trial comparing adjuvant chemotherapy with S-1 versus surgery alone in stage II or III gastric cancer. J Clin Oncol 2011;29:4387–93.
- Bang YJ, Kim YW, Yang HK, et al. Adjuvant capecitabine and oxaliplatin for gastric cancer after D2 gastrectomy (CLASSIC): a phase 3 open-label, randomised controlled trial. Lancet 2012;379:315–21.
- Noh SH, Park SR, Yang HK, et al. Adjuvant capecitabine plus oxaliplatin for gastric cancer after D2 gastrectomy (CLASSIC): 5-year follow-up of an open-label, randomised phase 3 trial. Lancet Oncol 2014;15:1389–96.
- Paoletti X, Oba K, Burzykowski T, et al. Benefit of adjuvant chemotherapy for resectable gastric cancer: a meta-analysis. JAMA 2010; 303:1729–37.
- Dai Q, Jiang L, Lin RJ, et al. Adjuvant chemoradiotherapy versus chemotherapy for gastric cancer: a meta-analysis of randomized controlled trials. J Surg Oncol 2015;111:277–84.
- Zhou M, Kang M, Li G, et al. Postoperative chemoradiotherapy versus chemotherapy for R0 resected gastric cancer with D2 lymph node dissection: an up-to-date meta-analysis. World J Surg Oncol 2016;14:209.
- Yang Y, Yin X, Sheng L, et al. Perioperative chemotherapy more of a benefit for overall survival than adjuvant chemotherapy for operable gastric cancer: an updated meta-analysis. Sci Rep 2015;5:12850.
- Zhao JH, Gao P, Song YX, et al. Which is better for gastric cancer patients, perioperative or adjuvant chemotherapy: a meta-analysis. BMC Cancer 2016;16:631.
- Narsule CK, Montgomery MM, and Fernando HC. Evidence-based review of the management of cancers of the gastroesophageal junction. Thorac Surg Clin 2012;22:109–21.
- van Hagen P, Hulshof MCCM, van Lanschot JJB, et al. Preoperative chemoradiotherapy for esophageal or junctional cancer. N Eng J Med 2012;266:2074–84.
- Cunningham D. Chemotherapy with or without bevacizumab or lapatinib to treat operable oesophagogastric cancer (ST03). ClinicalTrials.gov. https://clinicaltrials.gov/show/NCT00450203. NLM Identifier: NCT00450203. Accessed December 14, 2016.
- Leong T, Smithers BM, Michael M, et al. TOPGEAR: a randomised phase III trial of perioperative ECF chemotherapy versus preoperative chemoradiation plus perioperative ECF chemotherapy for resectable gastric cancer (an international, intergroup trial of the AGITG/TROG/EORTC/NCIC CTG). BMC Cancer 2015;15:532.
- Docetaxel+oxaliplatin+S-1 (DOS) regimen as neoadjuvant chemotherapy in advanced gastric cancer (PRODIGY). ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01515748 NLM. Identifier: NCT01515748. Accessed December 14, 2016.
- Verheij M, Jansen EP, Cats A, et al. A multicenter randomized phase III trial of neo-adjuvant chemotherapy followed by surgery and chemotherapy or by surgery and chemoradiotherapy in resectable gastric cancer: First results from the CRITICS study. J Clin Oncol 2016;34 (suppl). Abstract 4000.
- Kang WK. Phase III randomized trial of adjuvant chemotherapy with S-1 vs S-1/oxaliplatin ± radiotherapy for completely resected gastric adenocarcinoma : The ARTIST II Trial (ARTIST-II). ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT01761461. NLM Identifier: NCT01761461. Accessed December 14, 2016.
- Trial of adjuvant XELOX chemotherapy and concurrent capecitabine and radiotherapy for resected gastric carcinoma. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01711242. NLM Identifier: NCT01711242. Accessed December 14, 2016.
- Bang YJ, Van Cutsem E, Feyereislova A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet 2010;376:687–97.
- Roche HL. A Study of the combination of oxaliplatin, capecitabine and herceptin (trastuzumab) and chemoradiotherapy in the adjuvant setting in operated patients with HER2+ gastric or gastro-esophageal junction cancer (TOXAG Study). ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01748773. NLM Identifer: NCT01748773. Accessed December 14, 2016.
- A study of capecitabine [Xeloda] in combination with trastuzumab [herceptin] and oxaliplatine in patients with resectable gastric cancer. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01130337. NLM Identifier: NCT01130337. Accessed December 14, 2016.
INTRODUCTION
Gastric cancer is the fifth most common cancer worldwide and the third leading cause of cancer death in both females and males.1 More than 70% of gastric cancer cases occur in the developing world, with approximately 50% occurring in East Asia.2 Gastric cancer is less common in the United States, with an incidence of 12.3 cases in males and 6.0 cases in females per 100,000 per year and a disproportionately higher incidence in Asians.3 According to the Surveillance, Epidemiology, and End Results Program, approximately 26,370 new cases of stomach cancer were diagnosed in the United States in 2016, and an estimated 10,730 people died of this disease.4 Since the 1970s, the 5-year relative survival rate for gastric cancer in the United States has improved from 15% in 1975 to 29% in 2009.5 In contrast, in Japan and Korea, where screening programs have been implemented, the 5-year survival rate approaches 70%.6
RISK FACTORS AND CLASSIFICATION
A variety of risk factors have been linked to gastric cancer. Diets high in salt, salt-preserved foods, and/or processed meats have been associated with an increased risk for developing gastric cancer.7,8 Obesity and smoking have also been implicated in gastric cancer.9,10 Several studies have demonstrated a strong association between Helicobacter pylori and the development of gastric cancer.11–13 It is believed that H. pylori infection leads to chronic active gastritis, atrophic gastritis, and intestinal metaplasia. Interestingly, mass eradication of H. pylori has not been shown to reduce the risk for gastric cancer.14 Therefore, treatment of H. pylori should only be considered in patients with active peptic ulcer disease.15 Other risk factors include Epstein-Barr virus (EBV), prior gastric surgery, and radiation exposure.16–18 Family history of gastric cancer, hereditary nonpolyposis colon cancer, Li-Fraumeni syndrome, and hereditary diffuse gastric cancer caused by mutations in the E-cadherin gene increase the risk.17
The anatomic distinction between gastric cancer and cancer of the gastroesophageal junction (GEJ) has been a topic of debate. The Siewert classification is the most widely used system and divides GEJ adenocarcinoma into 3 categories:20 type I tumor: adenocarcinoma of distal esophagus, located 1 cm to 5 cm above the GEJ; type II tumor: true carcinoma of gastric cardia, located within 1 cm above and 2 cm below the GEJ; type III tumor: subcardial gastric carcinoma, located 2 cm to 5 cm below the GEJ, and infiltrates esophagus from below.
The American Joint Committee on Cancer (AJCC) has updated the latest (7th) edition of TMN staging for stomach cancer to include tumors arising more than 5 cm distally of the GEJ or within 5 cm of the GEJ but without extension to the esophagus or GEJ.21
In the following sections, neoadjuvant and adjuvant therapy in gastric cancer are discussed using a case presentation to illustrate important concepts.
DIAGNOSIS AND STAGING
CASE PRESENTATION
A 43-year old male with no significant past medical history presents with epigastric abdominal pain and heart burn for the past few weeks. He denies nausea, vomiting, melena, or hematochezia. His primary care physician (PCP) diagnoses him with gastroesophageal reflux disease (GERD) and initiates a trial of pantoprazole. Over the next 2 to 3 months, his symptoms do not improve and he has an associated 40-lb weight loss. Both social history and family history are noncontributory. Physical exam reveals epigastric tenderness without rebound or guarding. Laboratory evaluation reveals a hemoglobin of 12.6 g/dL with a mean corpuscular volume of 72 fL. A comprehensive chemistry profile is within normal limits. Given the constellation of presenting symptoms, especially the unintentional weight loss and the presence of microcytic anemia, his PCP suspects a malignant process and refers the patient to a gastroenterologist.
• What are the next appropriate steps for diagnosis?
The most common presenting symptoms of gastric cancer are weight loss and abdominal pain.22 Less commonly, patients exhibit nausea, anorexia, and dysphagia with proximal tumors. Melena is seen in only about 20% of patients. In Japan, where gastric cancer is more prevalent, mass screening programs allow for detection at an earlier stage, which partially accounts for the better survival rates seen in Asia as compared to the United States. Diagnostic work-up includes esophagogastroduodenoscopy (EGD) to assess Siewert category and to obtain a tissue sample for diagnosis. Full staging requires a complete blood count (CBC) with differential; comprehensive chemistry profile; computed tomography (CT) of chest/abdomen/pelvis with oral and intravenous contrast; endoscopic ultrasound (EUS) if no M1 disease is identified; positron emission tomography (PET)-CT if there is no evidence of M1 disease and if clinically indicated; and laparoscopy with cytology for clinical stage T1b or higher.23 Patients should be staged according to the TMN staging system (Table 1).
MANAGEMENT OF NONMETASTATIC DISEASE
CASE CONTINUED
The patient undergoes EGD, which reveals a large ulcerated, partially circumferential mass measuring approximately 4 cm. The mass extends from the gastric body to the cardia. Biopsy of the mass reveals poorly differentiated adenocarcinoma as well as H. pylori–associated gastritis. He is given antibiotic therapy and undergoes complete work-up of his newly diagnosed gastric adenocarcinoma. CT of the chest/abdomen/pelvis demonstrates a large gastric mass with gastrohepatic and distal perigastric adenopathy, compatible with locally advance primary gastric cancer. There is no evidence of distant metastasis. PET scan shows a large hypermetabolic mass in the stomach body and increased FDG activity in 3 small nodes along the lesser gastric curvature and in 1 node in the gastrohepatic region. EUS reveals a malignant gastric tumor in the body of the stomach, which is staged as T3, and a few malignant-appearing lymph nodes in the perigastric region. Fine-needle aspiration of the perigastric lymph node is performed and the sample obtained is positive for malignant cells. Diagnostic laparoscopy with peritoneal washings is performed and cytology is negative for malignant cells. The patient is staged as clinical stage IIB (T3N1M0).
• How should this patient with newly diagnosed, locally advanced, resectable gastric cancer be managed?
SURGERY
Surgical resection for localized gastric cancer is the mainstay of treatment with curative intent. Only very early stage (Tis or T1a) tumors can be considered for endoscopic mucosal resection. Regarding surgical resection, distal gastric cancers are typically treated with subtotal gastrectomy because there is no survival difference between subtotal and total gastrectomy.24,25 Moreover, subtotal gastrectomy is associated with better nutritional status and quality of life. For proximal tumors, total gastrectomy is preferred as subtotal gastrectomy has been associated with a higher incidence of reflux esophagitis and anastomotic stenosis.26 In terms of surgical approach, multiple studies have shown that a laparoscopic approach has a lower complication rate and similar outcomes in terms of cancer recurrence and long-term survival when compared to open gastrectomy.27–29 Thus, a laparoscopic approach is often used in academic centers with highly experienced surgeons.
The extent of lymph node dissection remains a topic of debate. A D1 dissection involves the removal of perigastric lymph nodes. A D2 dissection is a D1 dissection plus the removal of lymph nodes along the left gastric artery, common hepatic artery, celiac artery, splenic hilum, and splenic artery. D2 lymphadenectomy has become the standard of care in Eastern countries where gastric cancer is more prevalent, such as Japan and Korea.30 In Western countries, including the United States, less extensive lymphadenectomies are performed. Both randomized clinical trials and meta-analyses have failed to demonstrate an overall survival advantage of D2 dissection over D1 dissection.31,32 A Dutch trial by Bonenkamp et al involving 711 patients, one of the largest randomized trials of D1 and D2 lymphadenectomy, showed that D2 patients had a higher operative mortality rate than D1 patients (10% versus 4%, P = 0.004) and experienced more complications (43% versus 25%, P < 0.001).33 In a 15-year follow-up of this study, patients who had a D2 resection had lower locoregional recurrence and gastric-cancer–related death rates compared to those who had a D1 resection; however, D2 resection was associated with a significantly higher operative mortality and complication rate compared to D1.34 In addition, a 2015 Cochrane meta-analysis has demonstrated improved disease-specific survival (DSS) with D2 dissection (hazard ratio [HR] 0.81 [95% confidence interval {CI} 0.71 to 0.92]).35 Currently, the National Comprehensive Cancer Network (NCCN) recommends a D1 or a modified D2 gastrectomy with at least 15 lymph nodes removed for examination, with D2 lymphadenectomies only to be performed at experienced centers.23
SYSTEMIC CHEMOTHERAPY
Locally advanced gastric cancer (T3-T4 or node positive) requires systemic chemotherapy in addition to surgery, as this intervention improves the 5-year overall survival by 10% to 15%.36 Systemic therapy should also be considered in patients with T2N0 disease with high-risk features: poorly differentiated or high-grade cancer; lymphovascular invasion; neural invasion; age younger than 50 years; and patients who did not undergo D2 dissection.23 Currently, there is no global consensus on the best treatment approach. In the United States, where a less aggressive lymph-node dissection is performed, adjuvant chemoradiotherapy after surgery is more commonly seen. In Europe, perioperative (preoperative and postoperative) chemotherapy is the standard treatment. In Japan, adjuvant chemotherapy after D2 lymphadenectomy is the standard of care.37 These regional preferences are largely due to randomized clinical trials that have shown benefit for each approach. The landmark trials are discussed in the following sections and are summarized in Table 2.
Neoadjuvant Chemotherapy
Neoadjuvant chemotherapy has the benefit of “downstaging” locally advanced tumors to allow for curative resection. Phase 2 clinical trials have also demonstrated good pathologic response rates and high R0 resection rates following neoadjuvant chemotherapy.38,39 However, phase 3 trials to support this treatment approach are lacking. In the European Organisation for Research and Treatment of Cancer (EORTC) 40954 trial, patients with stage III or IV gastric or GEJ cancer were randomly assigned to surgery with or without preoperative cisplatin, leucovorin, and infusional fluorouracil (5-FU).40 The trial was stopped early due to poor accrual after 144 patients were randomized. The neoadjuvant chemotherapy arm had a higher R0 resection rate compared to the surgery alone arm (82% versus 67%, respectively, P = 0.036) but a higher postoperative complication rate (27% versus 16%, respectively, P = 0.09). More important, after a median follow-up of 4.4 years, a survival benefit could not be shown, with 2-year survival rates of 72.7% and 69.9% in the neoadjuvant and surgery-only arms, respectively (HR 0.84 [95% CI 0.52 to 1.35], P = 0.466). Due to the lack of large trials, a meta-analysis assessing the effectiveness of neoadjuvant chemotherapy combined with surgery versus surgery alone in advanced gastric and gastroesophageal cancer was performed.41 The analysis included 12 randomized controlled trials with a total of 1820 patients. Neoadjuvant chemotherapy was shown to slightly improve the survival rate (odds ratio [OR] 1.32 [95% CI 1.07 to 1.64], P = 0.01). It significantly improved the 3-year progression-free survival (PFS; OR 1.85 [95% CI 1.39 to 2.46], P < 0.0001), tumor down-staging rate (OR 1.71 [95% CI 1.26 to 2.33], P = 0.0006), and R0 resection rate (OR 1.38 [95% CI 1.08 to 1.78], P = 0.01). There were no differences between the 2 arms in terms of relapse rates, operative complications, perioperative mortality, and grade 3/4 adverse effects. While these results are encouraging, further randomized clinical trials are needed to clarify the role of neoadjuvant chemotherapy and its impact on overall survival.
Perioperative Chemotherapy
The results of the Medical Research Council Adjuvant Gastric Infusional Chemotherapy (MAGIC) trial published in 2006 established perioperative chemotherapy as standard of care in patients with resectable gastric and gastroesophageal adenocarcinoma.42 A total of 503 patients with potentially resectable gastric and lower esophageal adenocarcinoma were randomly assigned to perioperative chemotherapy plus surgery or surgery alone. Perioperative chemotherapy consisted of 3 preoperative and postoperative cycles of epirubicin, cisplatin, and infusional 5-FU (ECF). At a median follow-up of 4 years, the perioperative-chemotherapy group had a significantly better PFS (HR 0.66 [95% CI 0.53 to 0.81], P < 0.001) as well as overall survival (HR 0.75 [95% CI 0.60 to 0.93], P = 0.009). The 5-year overall survival rate was 36.3% in the perioperative chemotherapy group and 23% in the surgery group. Of note, there was a greater proportion of stage T1/T2 tumors (52% versus 37%, P = 0.002) and N0/N1 disease (84% versus 71%) in the perioperative-chemotherapy group compared to the surgery alone group. In addition, only 42% of patients in the perioperative chemotherapy group completed all 6 cycles of chemotherapy.
The administration of ECF is often difficult since the 5-FU component requires a central venous access device and an ambulatory infusion pump and the cisplatin component is associated with nephrotoxicity and ototoxicity. The REAL-2 trial was a randomized phase 3 clinical trial that assessed whether 5-FU could be replaced by capecitabine and cisplatin by oxaliplatin in the ECF regimen.43 Between June 2000 and May 2005, a total of 1002 patients with locally advanced esophageal/GEJ/gastric cancer were enrolled. Patients were randomly assigned to 1 of 4 triplet therapies: epirubicin and cisplatin plus either 5-FU (ECF) or capecitabine (ECX) or epirubicin and oxaliplatin plus either 5-FU (EOF) or capecitabine (EOX). After a median follow-up of approximately 18 months, the overall survival in the capecitabine groups did not differ significantly from that in the 5-FU groups (HR 0.88 [95% CI 0.77 to 1.00], P = 0.06), nor did overall survival in the oxaliplatin groups differ significantly from that in the cisplatin groups (HR 0.91 [95% CI 0.79 to 1.04], P = 0.16). Interestingly, the 1-year survival rate was longer in the EOX group than in the ECF group (46.8% versus 37.7%, respectively; HR 0.80 [95% CI 0.66 to 0.97], P = 0.02). This translated into an overall survival of 11.2 months for the EOX group and 9.9 months for the ECF group. Therefore, EOX is preferred over ECF in clinical practice.
The French FNLCC/FFCD trial published in 2011 provided further support for perioperative chemotherapy.44 A total of 224 patients with adenocarcinoma of the lower esophagus, GEJ, or stomach were randomly assigned to perioperative chemotherapy plus surgery or surgery alone. The perioperative-chemotherapy group received 2 to 3 cycles of preoperative chemotherapy and 3 to 4 cycles of postoperative chemotherapy, consisting of infusional 5-FU (800 mg/m2 daily for days 1 to 5) and cisplatin (100 mg/m2 on day 1). In patients receiving preoperative chemotherapy, 38% experienced at least grade 3 to 4 toxicity. Among the 109 patients who received at least 1 cycle of preoperative chemotherapy, only 54 patients (50%) received postoperative chemotherapy. Despite this, the perioperative-chemotherapy group had a statistically significant higher R0 resection rate (84% versus 74%, P = 0.04) compared to the surgery alone group. At a median follow-up of 5.7 years, the perioperative chemotherapy group had an improved overall survival (HR for death 0.69 [95% CI 0.50 to 0.95], P = 0.02) and disease-free survival (DFS; HR for recurrence or death 0.65 [95% CI 0.48 to 0.89], P = 0.003). This translated into 5-year overall survival rates of 38% versus 24% and 5-year DFS rates of 34% versus 19%. One caveat to this study is that the majority of patients (64%) had GEJ cancer and only 25% had gastric cancer. In the multivariate analysis, the 2 significant prognostic factors for overall survival were the administration of preoperative chemotherapy (P = 0.01) and tumor site at the GEJ (P < 0.01).
Adjuvant Chemoradiotherapy
The INT-0116 (Intergroup 0116) study published in 2001 established adjuvant chemoradiotherapy as the standard approach for resectable gastric cancer in the United States. In this study, a total of 556 patients with resected gastric or GEJ cancer were randomly assigned to surgery alone or surgery followed by adjuvant 5-FU/leucovorin bolus chemotherapy, sandwiched with 5-FU–based chemoradiation (45 Gy).45 In the chemoradiotherapy group, 64% of patients completed treatment and grade 3 and 4 toxicity occurred in 41% and 32%, respectively. However, only 3 patients (1%) died from treatment-related toxicity. At a median follow-up of 5 years, the median overall survival was 36 months in the chemoradiation group and 27 months in the surgery group. Overall survival rate was 50% in the combined modality group and 41% in the surgery-alone group, with a HR of 1.35 (95% CI 1.09 to 1.66, P = 0.005). The 3-year DFS was 48% in the chemoradiotherapy group and 31% in the surgery-alone group, corresponding to a DFS of 30 months and 19 months, respectively. Even after a median follow-up of 10 years, these positive results were maintained, with a HR for survival of 1.32 (95% CI 1.10 to 1.60, P = 0.0046) and HR for DFS of 1.51 (95% CI 1.25 to 1.83, P < 0.001).46 A criticism of the INT-0116 study is that 54% of patients had less than a D1 lymph node dissection, suggesting that adjuvant chemoradiation may have compensated for suboptimal surgery.
CALGB 80101, a United States Intergroup study, compared the INT-0116 protocol regimen (bolus 5-FU/leucovorin with 5-FU plus concurrent radiotherapy) to postoperative ECF sandwiched with 5-FU plus concurrent radiotherapy.47 The study included patients with resected gastric or GEJ adenocarcinoma that extended beyond the muscularis propria or was node positive. The percentage of patients with gastric versus GEJ cancer was not reported. A total of 546 patients were randomized. Preliminary results were presented at the 2011 American Society of Clinical Oncology meeting. The ECF arm had lower rates of grade 3/4 toxicities, including neutropenia, diarrhea, and mucositis. However, there was no difference in overall survival (3-year overall survival of 52% versus 50% for ECF and 5-FU/leucovorin, respectively) or DFS (3-year DFS of 47% versus 46% for ECF and 5-FU/leucovorin, respectively). The trial was not adequately powered to assess noninferiority. The location of the primary tumor (GEJ versus proximal versus distal stomach) did not have any effect on treatment outcome.
The Adjuvant Chemoradiation Therapy in Stomach Cancer (ARTIST) trial was the first study to compare adjuvant chemoradiotherapy with adjuvant chemotherapy in patients with D2-resected gastric cancer.48 A total of 458 patients were randomly assigned to 6 cycles of XP chemotherapy (capecitabine 2000 mg/m2 per day on days 1–14 and cisplatin 60 mg/m2 on day 1, every 3 weeks) or XP/radiotherapy/XP (2 cycles of XP followed by 45 Gy radiotherapy with concurrent daily capecitabine [825 mg/m2 twice daily] and 2 cycles of XP). After a median follow-up of 84 months, there was no difference in DFS or overall survival between treatment arms (HR for progression 0.74 [95% CI 0.52 to 1.05], P = 0.09; HR for death 1.13 [95% CI 0.78 to 1.65], P = 0.53).49 However, subgroup analysis showed that chemoradiotherapy significantly improved DFS in patients with node-positive disease (3-year DFS 76% versus 72%, P = 0.004).
Adjuvant Chemotherapy
Data supporting the use of adjuvant chemotherapy alone is largely derived from trials done in Asia, typically after a D2 lymph node dissection, and thus adjuvant chemotherapy has become the standard of care in that region. In the Japanese Adjuvant Chemotherapy Trial of S-1 for Gastric Cancer (ACTS-GC), a total of 1059 patients with stage II or III gastric cancer who had undergone surgery with a D2 lymphadenectomy were randomly assigned to 1 year of S-1 (an oral fluoropyrimidine) or surgery alone.50 The 5-year overall survival rate was 72% in the S-1 group and 61% in the surgery-only group (HR 0.669 [95% CI 0.54 to 0.83]).51 The 5-year relapse-free survival was 65% in the S-1 group and 53% in the surgery-only group (HR 0.65 [95% CI 0.537 to 0.793]). Of note, both arms of the ACTS-GC trial had significantly higher 5-year overall survival rates compared to the INT-0116 and MAGIC trials: 43% versus 28% and 36% versus 23% for the treatment and control groups, respectively.42,45 Consequently, it is unclear if the benefit of adjuvant chemotherapy can be translated to Western countries.
The Korean Capecitabine and Oxaliplatin Adjuvant Study in Stomach Cancer (CLASSIC) trial published in 2012 also established the role of adjuvant chemotherapy after D2 gastrectomy.52 A total of 1035 patients with stage II-IIIB gastric cancer who had curative D2 gastrectomy were randomly assigned to 8 cycles of adjuvant XELOX (capecitabine 1000 mg/m2 twice daily on days 1–14 plus oxaliplatin 130 mg/m2 on day 1, 21-day cycle) or surgery alone. Median follow-up was 34 months in both arms and 67% of patients in the chemotherapy arm completed all 8 cycles of planned chemotherapy. The 3-year DFS was 74% in the chemotherapy group and 59% in the surgery-only group (HR 0.56 [95% CI 0.44 to 0.72], P < 0.0001). There was a trend toward improvement in overall survival (83% versus 78%, HR 0.72 [95% CI 0.52 to 1.00]). After 5 years of follow-up, the improvement in overall survival became statistically significant (78% versus 69%, HR 0.66 [95% CI 0.51 to 0.85]).53
The benefit of adjuvant chemotherapy was reinforced by a 2010 meta-analysis comparing adjuvant chemotherapy to surgery alone in patients with resected gastric cancer.54 A total of 17 randomized controlled trials were included. Adjuvant fluorouracil-based chemotherapy was associated with a statistically significant improved overall survival (HR 0.82 [95% CI 0.76 to 0.90], P < 0.001) and DFS (HR 0.82 [95% CI 0.75 to 0.90], P < 0.001). Five-year overall survival increased from 49.6% to 55.3% with chemotherapy.
SELECTION OF TREATMENT APPROACH
Since data exists for all 3 approaches (perioperative chemotherapy, adjuvant chemoradiotherapy, and adjuvant chemotherapy), various meta-analyses have been done to clarify which approach is the best. In a recent meta-analysis of 6 randomized controlled trials reported between 2010 and 2012, which involved 1171 patients with resected gastric cancer, adjuvant chemotherapy was compared to adjuvant chemoradiotherapy.55 Five of the studies were from East Asia, while one was from a Western country. Adjuvant chemoradiation was associated with a lower local-regional recurrence rate (OR 0.46 [95% CI 0.32 to 0.67]) and better 5-year DFS rate (OR 1.56 [95% CI 1.09 to 2.24]). However, there was no statistical difference in 5-year overall survival rate (OR 1.32 [95% CI 0.92 to 1.88]). Similar results were reported by Zhou et al in 2016.56 This meta-analysis included 4 randomized controlled trials reported between 2010 and 2015, with a total of 960 patients who had undergone a D2 resection for gastric cancer. Compared to adjuvant chemotherapy, adjuvant chemoradiotherapy significantly reduced the loco-regional recurrence rate (LRRR; relative risk [RR] 0.50 [95% CI 0.34 to 0.74], P = 0.0005) and improved DFS (HR 0.73 [95% CI 0.60 to 0.89], P = 0.002). Again, no difference in overall survival was seen (HR 0.91 [95% CI 0.74 to 1.11], P = 0.34).
Adjuvant chemotherapy and perioperative chemotherapy have also been compared. In a recent meta-analysis of 14 randomized controlled trials (8 Asian, 6 European) involving 2093 patients with resected gastric or GEJ cancer, perioperative chemotherapy was associated with improved overall survival when compared to adjuvant chemotherapy (HR 0.48 [95% CI 0.35 to 0.67], P < 0.001).57 The benefit of perioperative chemotherapy over adjuvant chemotherapy has also been reported in a 2016 meta-analysis by Zhao et al.58 A total of 1240 patients were included from 5 randomized controlled trials and 6 clinical controlled trials, all from Asian countries. The 5-year overall survival rate was significantly better in the perioperative chemotherapy group compared to the adjuvant chemotherapy group (RR 0.77 [95% CI 0.64 to 0.92], P < 0.01). Furthermore, the 2 groups showed no significant differences in the postoperative complication rates (RR 0.98 [95% CI 0.63 to 1.51], P = 0.91) or adverse effects of chemotherapy (P > 0.05 for all adverse effects).
While these meta-analyses may offer some insight on the best treatment approach, they should be interpreted with caution. Most studies included in these meta-analyses were from Asian countries, and their findings may not be applicable to Western countries. Furthermore, the heterogeneity of trials and inclusion of nonrandomized trials make it difficult to draw conclusions. There are several ongoing trials that will help to define the optimal treatment approach.
CASE CONTINUED
The patient is presented at tumor board and the consensus is to proceed with the perioperative chemotherapy approach. The patient undergoes echocardiography, which reveals a normal ejection fraction. He receives 3 cycles of neoadjuvant EOX (epirubicin, oxaliplatin, and capecitabine). After 3 cycles of neoadjuvant EOX, the patient has a repeat CT that shows marked interval reduction in the size of the primary gastric neoplasm and interval decrease in the size of the small perigastric lymph nodes. He subsequently undergoes a total gastrectomy with J-tube placement. Pathology shows ypT3N0 disease with 0 out of 47 lymph nodes involved and negative margins. He then receives 3 cycles of adjuvant EOX.
• What are the recommendations for surveillance?
According to the current NCCN guidelines, a history and physical exam should be performed every 3 to 6 months for 1 to 2 years, then every 6 to 12 months for 3 to 5 years, and then annually.23 Labs, CT chest/abdomen, and EGD should be done as clinically indicated. Patients who have undergone surgical resection should be monitored for nutritional deficiencies (vitamin B12 and iron).
GASTROESOPHAGEAL JUNCTION TUMORS
Tumors arising in the GEJ or gastric cardia within 5 cm of the GEJ that extend into the GEJ or distal esophagus are staged and treated as esophageal cancers.21 The primary treatment for T1/T2N0 tumors is surgical resection. In patients with T3 or higher or node-positive adenocarcinoma of the GEJ, a combined modality approach is preferred, with preoperative chemoradiotherapy followed by surgical resection.59 The CROSS trial demonstrated a significant survival benefit with preoperative chemoradiation using carboplatin/paclitaxel compared to surgery alone in patients with esophageal or GEJ cancer (49 months versus 24 months, HR 0.66, P = 0.003).60
ONGOING TRIALS
As mentioned previously, several randomized clinical trials are in progress to clarify the optimal treatment approach. The MAGIC-B/MRC-ST03 is a randomized phase 2/3 trial looking at perioperative epirubicin, cisplatin, and capecitabine (ECX) with or without bevacizumab in patients with resectable lower esophageal, GEJ, or gastric cancer.61 The TOPGEAR trial, a randomized phase 2/3 study being conducted in Canada and Europe, is comparing perioperative ECF chemotherapy with preoperative chemoradiation plus perioperative ECF chemotherapy.62 In Asia, the PRODIGY trial is a phase 3, open-label, randomized study comparing neoadjuvant docetaxel, oxaliplatin, and S-1 followed by surgery and adjuvant S-1 versus surgery plus adjuvant S-1 in patients with locally advanced gastric cancer (T2-T4 or node positive).63 Primary endpoint is PFS and secondary endpoints are overall survival, R0 resection rate, and safety.
Trials comparing adjuvant chemotherapy to adjuvant chemoradiotherapy are also being conducted. In the Dutch CRITICS study, a randomized phase 3 trial, patients with stage Ib-Iva resectable gastric cancer were given 3 cycles of epirubicin, cisplatin/oxaliplatin, and capecitabine (ECC/EOC), followed by D2 resection and either 3 cycles of ECC/EOC or chemoradiation with weekly cisplatin and daily capecitabine.64 Between January 2007 and April 2015, a total of 788 patients were enrolled. In a preliminary report presented at ASCO in 2016, the 5-year survival rate was similar between the 2 arms (41.3% for chemotherapy arm and 40.9% for chemoradiotherapy arm, P = 0.99). The Korean ARTIST II trial is comparing adjuvant S-1 and S-1/oxaliplatin with or without radiotherapy in patients with D2-resected gastric cancer.65 Similarly, the NCT01711242 trial is comparing adjuvant XELOX alone versus XELOX with concurrent capecitabine/radiotherapy in patients with resected D2 gastric cancer.66
The ToGA trial established a survival benefit of trastuzumab in combination with chemotherapy in HER2-positive metastatic gastric cancer.67 Consequently, there are ongoing clinical trials to assess the role of trastuzumab in nonmetastatic gastric cancer. The TOXAG study is a phase 2 trial looking at the safety and tolerability of adjuvant oxaliplatin, capecitabine, and trastuzumab with radiation in patients with resected HER2-positive gastric or GEJ adenocarcinoma.68 The NCT01130337 clinical trial is evaluating perioperative XELOX/trastuzumab in patients with resectable gastric or GEJ adeno-carcinoma.69
SUMMARY
Gastric cancer is the fifth most common cancer worldwide, with the greatest incidence in East Asia. Survival outcomes are better in Asian countries when compared to the United States. This difference in survival may be related to the presence of mass screening programs in Asia, which allows for detection at an earlier stage and the use of a more extensive surgical approach (ie, D2 resection). Risk factors for developing gastric cancer include: diets high in salt/salt-preserved foods or processed meats, obesity, smoking, H. pylori infection, EBV, prior gastric surgery, radiation exposure, and positive family history.
According to the latest edition of TMN staging, gastric cancer includes tumors arising more than 5 cm distally of the GEJ or within 5 cm of the GEJ but without extension to the esophagus or GEJ. Diagnostic work-up includes: EGD with biopsy; basic labs; CT chest/abdomen/pelvis with oral and intravenous contrast; EUS if no M1 disease is identified; PET-CT if there is no M1 disease and if clinically indicated; and diagnostic laparoscopy with cytology for clinical stage T1b or higher.
The mainstay of treatment is surgical resection. Laparoscopic approach is preferred over open gastrectomy due to lower complication rates and similar survival outcomes. Current NCCN guidelines recommend a D1 or a modified D2 lymph node dissection with at least 15 lymph nodes removed for examination. Systemic chemotherapy is required in locally advanced gastric cancer (T3-T4 or node positive) and should be considered in T2N0 disease with high-risk features. Currently, there is no global consensus on the optimal treatment approach. Data from various trials have shown benefit for each approach. Regional preferences are: perioperative chemotherapy in Europe; adjuvant chemoradiotherapy in the United States; and adjuvant chemotherapy in Asia. In an effort to better define the optimal treatment approach, several randomized clinical trials are being conducted. According to the current NCCN guidelines, the following treatment approaches are acceptable and are supported by data in the trial listed in parentheses:
• Perioperative chemotherapy
° 5-FU/cisplatin (French FNLCC/FCCD trial)44 or
° ECF (MAGIC trial)42 or
° ECF modifications: EOX, EOF, ECX (REAL-2 trial)43
• Adjuvant chemoradiotherapy
° 5-FU/leucovorin sandwiched with 5-FU-based chemoradiation (INT-0116 trial)45
• Adjuvant chemotherapy (after D2 resection)
° Capecitabine/oxaliplatin (CLASSIC trial)52 or
° Capecitabine/cisplatin (ARTIST trial)48,49
INTRODUCTION
Gastric cancer is the fifth most common cancer worldwide and the third leading cause of cancer death in both females and males.1 More than 70% of gastric cancer cases occur in the developing world, with approximately 50% occurring in East Asia.2 Gastric cancer is less common in the United States, with an incidence of 12.3 cases in males and 6.0 cases in females per 100,000 per year and a disproportionately higher incidence in Asians.3 According to the Surveillance, Epidemiology, and End Results Program, approximately 26,370 new cases of stomach cancer were diagnosed in the United States in 2016, and an estimated 10,730 people died of this disease.4 Since the 1970s, the 5-year relative survival rate for gastric cancer in the United States has improved from 15% in 1975 to 29% in 2009.5 In contrast, in Japan and Korea, where screening programs have been implemented, the 5-year survival rate approaches 70%.6
RISK FACTORS AND CLASSIFICATION
A variety of risk factors have been linked to gastric cancer. Diets high in salt, salt-preserved foods, and/or processed meats have been associated with an increased risk for developing gastric cancer.7,8 Obesity and smoking have also been implicated in gastric cancer.9,10 Several studies have demonstrated a strong association between Helicobacter pylori and the development of gastric cancer.11–13 It is believed that H. pylori infection leads to chronic active gastritis, atrophic gastritis, and intestinal metaplasia. Interestingly, mass eradication of H. pylori has not been shown to reduce the risk for gastric cancer.14 Therefore, treatment of H. pylori should only be considered in patients with active peptic ulcer disease.15 Other risk factors include Epstein-Barr virus (EBV), prior gastric surgery, and radiation exposure.16–18 Family history of gastric cancer, hereditary nonpolyposis colon cancer, Li-Fraumeni syndrome, and hereditary diffuse gastric cancer caused by mutations in the E-cadherin gene increase the risk.17
The anatomic distinction between gastric cancer and cancer of the gastroesophageal junction (GEJ) has been a topic of debate. The Siewert classification is the most widely used system and divides GEJ adenocarcinoma into 3 categories:20 type I tumor: adenocarcinoma of distal esophagus, located 1 cm to 5 cm above the GEJ; type II tumor: true carcinoma of gastric cardia, located within 1 cm above and 2 cm below the GEJ; type III tumor: subcardial gastric carcinoma, located 2 cm to 5 cm below the GEJ, and infiltrates esophagus from below.
The American Joint Committee on Cancer (AJCC) has updated the latest (7th) edition of TMN staging for stomach cancer to include tumors arising more than 5 cm distally of the GEJ or within 5 cm of the GEJ but without extension to the esophagus or GEJ.21
In the following sections, neoadjuvant and adjuvant therapy in gastric cancer are discussed using a case presentation to illustrate important concepts.
DIAGNOSIS AND STAGING
CASE PRESENTATION
A 43-year old male with no significant past medical history presents with epigastric abdominal pain and heart burn for the past few weeks. He denies nausea, vomiting, melena, or hematochezia. His primary care physician (PCP) diagnoses him with gastroesophageal reflux disease (GERD) and initiates a trial of pantoprazole. Over the next 2 to 3 months, his symptoms do not improve and he has an associated 40-lb weight loss. Both social history and family history are noncontributory. Physical exam reveals epigastric tenderness without rebound or guarding. Laboratory evaluation reveals a hemoglobin of 12.6 g/dL with a mean corpuscular volume of 72 fL. A comprehensive chemistry profile is within normal limits. Given the constellation of presenting symptoms, especially the unintentional weight loss and the presence of microcytic anemia, his PCP suspects a malignant process and refers the patient to a gastroenterologist.
• What are the next appropriate steps for diagnosis?
The most common presenting symptoms of gastric cancer are weight loss and abdominal pain.22 Less commonly, patients exhibit nausea, anorexia, and dysphagia with proximal tumors. Melena is seen in only about 20% of patients. In Japan, where gastric cancer is more prevalent, mass screening programs allow for detection at an earlier stage, which partially accounts for the better survival rates seen in Asia as compared to the United States. Diagnostic work-up includes esophagogastroduodenoscopy (EGD) to assess Siewert category and to obtain a tissue sample for diagnosis. Full staging requires a complete blood count (CBC) with differential; comprehensive chemistry profile; computed tomography (CT) of chest/abdomen/pelvis with oral and intravenous contrast; endoscopic ultrasound (EUS) if no M1 disease is identified; positron emission tomography (PET)-CT if there is no evidence of M1 disease and if clinically indicated; and laparoscopy with cytology for clinical stage T1b or higher.23 Patients should be staged according to the TMN staging system (Table 1).
MANAGEMENT OF NONMETASTATIC DISEASE
CASE CONTINUED
The patient undergoes EGD, which reveals a large ulcerated, partially circumferential mass measuring approximately 4 cm. The mass extends from the gastric body to the cardia. Biopsy of the mass reveals poorly differentiated adenocarcinoma as well as H. pylori–associated gastritis. He is given antibiotic therapy and undergoes complete work-up of his newly diagnosed gastric adenocarcinoma. CT of the chest/abdomen/pelvis demonstrates a large gastric mass with gastrohepatic and distal perigastric adenopathy, compatible with locally advance primary gastric cancer. There is no evidence of distant metastasis. PET scan shows a large hypermetabolic mass in the stomach body and increased FDG activity in 3 small nodes along the lesser gastric curvature and in 1 node in the gastrohepatic region. EUS reveals a malignant gastric tumor in the body of the stomach, which is staged as T3, and a few malignant-appearing lymph nodes in the perigastric region. Fine-needle aspiration of the perigastric lymph node is performed and the sample obtained is positive for malignant cells. Diagnostic laparoscopy with peritoneal washings is performed and cytology is negative for malignant cells. The patient is staged as clinical stage IIB (T3N1M0).
• How should this patient with newly diagnosed, locally advanced, resectable gastric cancer be managed?
SURGERY
Surgical resection for localized gastric cancer is the mainstay of treatment with curative intent. Only very early stage (Tis or T1a) tumors can be considered for endoscopic mucosal resection. Regarding surgical resection, distal gastric cancers are typically treated with subtotal gastrectomy because there is no survival difference between subtotal and total gastrectomy.24,25 Moreover, subtotal gastrectomy is associated with better nutritional status and quality of life. For proximal tumors, total gastrectomy is preferred as subtotal gastrectomy has been associated with a higher incidence of reflux esophagitis and anastomotic stenosis.26 In terms of surgical approach, multiple studies have shown that a laparoscopic approach has a lower complication rate and similar outcomes in terms of cancer recurrence and long-term survival when compared to open gastrectomy.27–29 Thus, a laparoscopic approach is often used in academic centers with highly experienced surgeons.
The extent of lymph node dissection remains a topic of debate. A D1 dissection involves the removal of perigastric lymph nodes. A D2 dissection is a D1 dissection plus the removal of lymph nodes along the left gastric artery, common hepatic artery, celiac artery, splenic hilum, and splenic artery. D2 lymphadenectomy has become the standard of care in Eastern countries where gastric cancer is more prevalent, such as Japan and Korea.30 In Western countries, including the United States, less extensive lymphadenectomies are performed. Both randomized clinical trials and meta-analyses have failed to demonstrate an overall survival advantage of D2 dissection over D1 dissection.31,32 A Dutch trial by Bonenkamp et al involving 711 patients, one of the largest randomized trials of D1 and D2 lymphadenectomy, showed that D2 patients had a higher operative mortality rate than D1 patients (10% versus 4%, P = 0.004) and experienced more complications (43% versus 25%, P < 0.001).33 In a 15-year follow-up of this study, patients who had a D2 resection had lower locoregional recurrence and gastric-cancer–related death rates compared to those who had a D1 resection; however, D2 resection was associated with a significantly higher operative mortality and complication rate compared to D1.34 In addition, a 2015 Cochrane meta-analysis has demonstrated improved disease-specific survival (DSS) with D2 dissection (hazard ratio [HR] 0.81 [95% confidence interval {CI} 0.71 to 0.92]).35 Currently, the National Comprehensive Cancer Network (NCCN) recommends a D1 or a modified D2 gastrectomy with at least 15 lymph nodes removed for examination, with D2 lymphadenectomies only to be performed at experienced centers.23
SYSTEMIC CHEMOTHERAPY
Locally advanced gastric cancer (T3-T4 or node positive) requires systemic chemotherapy in addition to surgery, as this intervention improves the 5-year overall survival by 10% to 15%.36 Systemic therapy should also be considered in patients with T2N0 disease with high-risk features: poorly differentiated or high-grade cancer; lymphovascular invasion; neural invasion; age younger than 50 years; and patients who did not undergo D2 dissection.23 Currently, there is no global consensus on the best treatment approach. In the United States, where a less aggressive lymph-node dissection is performed, adjuvant chemoradiotherapy after surgery is more commonly seen. In Europe, perioperative (preoperative and postoperative) chemotherapy is the standard treatment. In Japan, adjuvant chemotherapy after D2 lymphadenectomy is the standard of care.37 These regional preferences are largely due to randomized clinical trials that have shown benefit for each approach. The landmark trials are discussed in the following sections and are summarized in Table 2.
Neoadjuvant Chemotherapy
Neoadjuvant chemotherapy has the benefit of “downstaging” locally advanced tumors to allow for curative resection. Phase 2 clinical trials have also demonstrated good pathologic response rates and high R0 resection rates following neoadjuvant chemotherapy.38,39 However, phase 3 trials to support this treatment approach are lacking. In the European Organisation for Research and Treatment of Cancer (EORTC) 40954 trial, patients with stage III or IV gastric or GEJ cancer were randomly assigned to surgery with or without preoperative cisplatin, leucovorin, and infusional fluorouracil (5-FU).40 The trial was stopped early due to poor accrual after 144 patients were randomized. The neoadjuvant chemotherapy arm had a higher R0 resection rate compared to the surgery alone arm (82% versus 67%, respectively, P = 0.036) but a higher postoperative complication rate (27% versus 16%, respectively, P = 0.09). More important, after a median follow-up of 4.4 years, a survival benefit could not be shown, with 2-year survival rates of 72.7% and 69.9% in the neoadjuvant and surgery-only arms, respectively (HR 0.84 [95% CI 0.52 to 1.35], P = 0.466). Due to the lack of large trials, a meta-analysis assessing the effectiveness of neoadjuvant chemotherapy combined with surgery versus surgery alone in advanced gastric and gastroesophageal cancer was performed.41 The analysis included 12 randomized controlled trials with a total of 1820 patients. Neoadjuvant chemotherapy was shown to slightly improve the survival rate (odds ratio [OR] 1.32 [95% CI 1.07 to 1.64], P = 0.01). It significantly improved the 3-year progression-free survival (PFS; OR 1.85 [95% CI 1.39 to 2.46], P < 0.0001), tumor down-staging rate (OR 1.71 [95% CI 1.26 to 2.33], P = 0.0006), and R0 resection rate (OR 1.38 [95% CI 1.08 to 1.78], P = 0.01). There were no differences between the 2 arms in terms of relapse rates, operative complications, perioperative mortality, and grade 3/4 adverse effects. While these results are encouraging, further randomized clinical trials are needed to clarify the role of neoadjuvant chemotherapy and its impact on overall survival.
Perioperative Chemotherapy
The results of the Medical Research Council Adjuvant Gastric Infusional Chemotherapy (MAGIC) trial published in 2006 established perioperative chemotherapy as standard of care in patients with resectable gastric and gastroesophageal adenocarcinoma.42 A total of 503 patients with potentially resectable gastric and lower esophageal adenocarcinoma were randomly assigned to perioperative chemotherapy plus surgery or surgery alone. Perioperative chemotherapy consisted of 3 preoperative and postoperative cycles of epirubicin, cisplatin, and infusional 5-FU (ECF). At a median follow-up of 4 years, the perioperative-chemotherapy group had a significantly better PFS (HR 0.66 [95% CI 0.53 to 0.81], P < 0.001) as well as overall survival (HR 0.75 [95% CI 0.60 to 0.93], P = 0.009). The 5-year overall survival rate was 36.3% in the perioperative chemotherapy group and 23% in the surgery group. Of note, there was a greater proportion of stage T1/T2 tumors (52% versus 37%, P = 0.002) and N0/N1 disease (84% versus 71%) in the perioperative-chemotherapy group compared to the surgery alone group. In addition, only 42% of patients in the perioperative chemotherapy group completed all 6 cycles of chemotherapy.
The administration of ECF is often difficult since the 5-FU component requires a central venous access device and an ambulatory infusion pump and the cisplatin component is associated with nephrotoxicity and ototoxicity. The REAL-2 trial was a randomized phase 3 clinical trial that assessed whether 5-FU could be replaced by capecitabine and cisplatin by oxaliplatin in the ECF regimen.43 Between June 2000 and May 2005, a total of 1002 patients with locally advanced esophageal/GEJ/gastric cancer were enrolled. Patients were randomly assigned to 1 of 4 triplet therapies: epirubicin and cisplatin plus either 5-FU (ECF) or capecitabine (ECX) or epirubicin and oxaliplatin plus either 5-FU (EOF) or capecitabine (EOX). After a median follow-up of approximately 18 months, the overall survival in the capecitabine groups did not differ significantly from that in the 5-FU groups (HR 0.88 [95% CI 0.77 to 1.00], P = 0.06), nor did overall survival in the oxaliplatin groups differ significantly from that in the cisplatin groups (HR 0.91 [95% CI 0.79 to 1.04], P = 0.16). Interestingly, the 1-year survival rate was longer in the EOX group than in the ECF group (46.8% versus 37.7%, respectively; HR 0.80 [95% CI 0.66 to 0.97], P = 0.02). This translated into an overall survival of 11.2 months for the EOX group and 9.9 months for the ECF group. Therefore, EOX is preferred over ECF in clinical practice.
The French FNLCC/FFCD trial published in 2011 provided further support for perioperative chemotherapy.44 A total of 224 patients with adenocarcinoma of the lower esophagus, GEJ, or stomach were randomly assigned to perioperative chemotherapy plus surgery or surgery alone. The perioperative-chemotherapy group received 2 to 3 cycles of preoperative chemotherapy and 3 to 4 cycles of postoperative chemotherapy, consisting of infusional 5-FU (800 mg/m2 daily for days 1 to 5) and cisplatin (100 mg/m2 on day 1). In patients receiving preoperative chemotherapy, 38% experienced at least grade 3 to 4 toxicity. Among the 109 patients who received at least 1 cycle of preoperative chemotherapy, only 54 patients (50%) received postoperative chemotherapy. Despite this, the perioperative-chemotherapy group had a statistically significant higher R0 resection rate (84% versus 74%, P = 0.04) compared to the surgery alone group. At a median follow-up of 5.7 years, the perioperative chemotherapy group had an improved overall survival (HR for death 0.69 [95% CI 0.50 to 0.95], P = 0.02) and disease-free survival (DFS; HR for recurrence or death 0.65 [95% CI 0.48 to 0.89], P = 0.003). This translated into 5-year overall survival rates of 38% versus 24% and 5-year DFS rates of 34% versus 19%. One caveat to this study is that the majority of patients (64%) had GEJ cancer and only 25% had gastric cancer. In the multivariate analysis, the 2 significant prognostic factors for overall survival were the administration of preoperative chemotherapy (P = 0.01) and tumor site at the GEJ (P < 0.01).
Adjuvant Chemoradiotherapy
The INT-0116 (Intergroup 0116) study published in 2001 established adjuvant chemoradiotherapy as the standard approach for resectable gastric cancer in the United States. In this study, a total of 556 patients with resected gastric or GEJ cancer were randomly assigned to surgery alone or surgery followed by adjuvant 5-FU/leucovorin bolus chemotherapy, sandwiched with 5-FU–based chemoradiation (45 Gy).45 In the chemoradiotherapy group, 64% of patients completed treatment and grade 3 and 4 toxicity occurred in 41% and 32%, respectively. However, only 3 patients (1%) died from treatment-related toxicity. At a median follow-up of 5 years, the median overall survival was 36 months in the chemoradiation group and 27 months in the surgery group. Overall survival rate was 50% in the combined modality group and 41% in the surgery-alone group, with a HR of 1.35 (95% CI 1.09 to 1.66, P = 0.005). The 3-year DFS was 48% in the chemoradiotherapy group and 31% in the surgery-alone group, corresponding to a DFS of 30 months and 19 months, respectively. Even after a median follow-up of 10 years, these positive results were maintained, with a HR for survival of 1.32 (95% CI 1.10 to 1.60, P = 0.0046) and HR for DFS of 1.51 (95% CI 1.25 to 1.83, P < 0.001).46 A criticism of the INT-0116 study is that 54% of patients had less than a D1 lymph node dissection, suggesting that adjuvant chemoradiation may have compensated for suboptimal surgery.
CALGB 80101, a United States Intergroup study, compared the INT-0116 protocol regimen (bolus 5-FU/leucovorin with 5-FU plus concurrent radiotherapy) to postoperative ECF sandwiched with 5-FU plus concurrent radiotherapy.47 The study included patients with resected gastric or GEJ adenocarcinoma that extended beyond the muscularis propria or was node positive. The percentage of patients with gastric versus GEJ cancer was not reported. A total of 546 patients were randomized. Preliminary results were presented at the 2011 American Society of Clinical Oncology meeting. The ECF arm had lower rates of grade 3/4 toxicities, including neutropenia, diarrhea, and mucositis. However, there was no difference in overall survival (3-year overall survival of 52% versus 50% for ECF and 5-FU/leucovorin, respectively) or DFS (3-year DFS of 47% versus 46% for ECF and 5-FU/leucovorin, respectively). The trial was not adequately powered to assess noninferiority. The location of the primary tumor (GEJ versus proximal versus distal stomach) did not have any effect on treatment outcome.
The Adjuvant Chemoradiation Therapy in Stomach Cancer (ARTIST) trial was the first study to compare adjuvant chemoradiotherapy with adjuvant chemotherapy in patients with D2-resected gastric cancer.48 A total of 458 patients were randomly assigned to 6 cycles of XP chemotherapy (capecitabine 2000 mg/m2 per day on days 1–14 and cisplatin 60 mg/m2 on day 1, every 3 weeks) or XP/radiotherapy/XP (2 cycles of XP followed by 45 Gy radiotherapy with concurrent daily capecitabine [825 mg/m2 twice daily] and 2 cycles of XP). After a median follow-up of 84 months, there was no difference in DFS or overall survival between treatment arms (HR for progression 0.74 [95% CI 0.52 to 1.05], P = 0.09; HR for death 1.13 [95% CI 0.78 to 1.65], P = 0.53).49 However, subgroup analysis showed that chemoradiotherapy significantly improved DFS in patients with node-positive disease (3-year DFS 76% versus 72%, P = 0.004).
Adjuvant Chemotherapy
Data supporting the use of adjuvant chemotherapy alone is largely derived from trials done in Asia, typically after a D2 lymph node dissection, and thus adjuvant chemotherapy has become the standard of care in that region. In the Japanese Adjuvant Chemotherapy Trial of S-1 for Gastric Cancer (ACTS-GC), a total of 1059 patients with stage II or III gastric cancer who had undergone surgery with a D2 lymphadenectomy were randomly assigned to 1 year of S-1 (an oral fluoropyrimidine) or surgery alone.50 The 5-year overall survival rate was 72% in the S-1 group and 61% in the surgery-only group (HR 0.669 [95% CI 0.54 to 0.83]).51 The 5-year relapse-free survival was 65% in the S-1 group and 53% in the surgery-only group (HR 0.65 [95% CI 0.537 to 0.793]). Of note, both arms of the ACTS-GC trial had significantly higher 5-year overall survival rates compared to the INT-0116 and MAGIC trials: 43% versus 28% and 36% versus 23% for the treatment and control groups, respectively.42,45 Consequently, it is unclear if the benefit of adjuvant chemotherapy can be translated to Western countries.
The Korean Capecitabine and Oxaliplatin Adjuvant Study in Stomach Cancer (CLASSIC) trial published in 2012 also established the role of adjuvant chemotherapy after D2 gastrectomy.52 A total of 1035 patients with stage II-IIIB gastric cancer who had curative D2 gastrectomy were randomly assigned to 8 cycles of adjuvant XELOX (capecitabine 1000 mg/m2 twice daily on days 1–14 plus oxaliplatin 130 mg/m2 on day 1, 21-day cycle) or surgery alone. Median follow-up was 34 months in both arms and 67% of patients in the chemotherapy arm completed all 8 cycles of planned chemotherapy. The 3-year DFS was 74% in the chemotherapy group and 59% in the surgery-only group (HR 0.56 [95% CI 0.44 to 0.72], P < 0.0001). There was a trend toward improvement in overall survival (83% versus 78%, HR 0.72 [95% CI 0.52 to 1.00]). After 5 years of follow-up, the improvement in overall survival became statistically significant (78% versus 69%, HR 0.66 [95% CI 0.51 to 0.85]).53
The benefit of adjuvant chemotherapy was reinforced by a 2010 meta-analysis comparing adjuvant chemotherapy to surgery alone in patients with resected gastric cancer.54 A total of 17 randomized controlled trials were included. Adjuvant fluorouracil-based chemotherapy was associated with a statistically significant improved overall survival (HR 0.82 [95% CI 0.76 to 0.90], P < 0.001) and DFS (HR 0.82 [95% CI 0.75 to 0.90], P < 0.001). Five-year overall survival increased from 49.6% to 55.3% with chemotherapy.
SELECTION OF TREATMENT APPROACH
Since data exists for all 3 approaches (perioperative chemotherapy, adjuvant chemoradiotherapy, and adjuvant chemotherapy), various meta-analyses have been done to clarify which approach is the best. In a recent meta-analysis of 6 randomized controlled trials reported between 2010 and 2012, which involved 1171 patients with resected gastric cancer, adjuvant chemotherapy was compared to adjuvant chemoradiotherapy.55 Five of the studies were from East Asia, while one was from a Western country. Adjuvant chemoradiation was associated with a lower local-regional recurrence rate (OR 0.46 [95% CI 0.32 to 0.67]) and better 5-year DFS rate (OR 1.56 [95% CI 1.09 to 2.24]). However, there was no statistical difference in 5-year overall survival rate (OR 1.32 [95% CI 0.92 to 1.88]). Similar results were reported by Zhou et al in 2016.56 This meta-analysis included 4 randomized controlled trials reported between 2010 and 2015, with a total of 960 patients who had undergone a D2 resection for gastric cancer. Compared to adjuvant chemotherapy, adjuvant chemoradiotherapy significantly reduced the loco-regional recurrence rate (LRRR; relative risk [RR] 0.50 [95% CI 0.34 to 0.74], P = 0.0005) and improved DFS (HR 0.73 [95% CI 0.60 to 0.89], P = 0.002). Again, no difference in overall survival was seen (HR 0.91 [95% CI 0.74 to 1.11], P = 0.34).
Adjuvant chemotherapy and perioperative chemotherapy have also been compared. In a recent meta-analysis of 14 randomized controlled trials (8 Asian, 6 European) involving 2093 patients with resected gastric or GEJ cancer, perioperative chemotherapy was associated with improved overall survival when compared to adjuvant chemotherapy (HR 0.48 [95% CI 0.35 to 0.67], P < 0.001).57 The benefit of perioperative chemotherapy over adjuvant chemotherapy has also been reported in a 2016 meta-analysis by Zhao et al.58 A total of 1240 patients were included from 5 randomized controlled trials and 6 clinical controlled trials, all from Asian countries. The 5-year overall survival rate was significantly better in the perioperative chemotherapy group compared to the adjuvant chemotherapy group (RR 0.77 [95% CI 0.64 to 0.92], P < 0.01). Furthermore, the 2 groups showed no significant differences in the postoperative complication rates (RR 0.98 [95% CI 0.63 to 1.51], P = 0.91) or adverse effects of chemotherapy (P > 0.05 for all adverse effects).
While these meta-analyses may offer some insight on the best treatment approach, they should be interpreted with caution. Most studies included in these meta-analyses were from Asian countries, and their findings may not be applicable to Western countries. Furthermore, the heterogeneity of trials and inclusion of nonrandomized trials make it difficult to draw conclusions. There are several ongoing trials that will help to define the optimal treatment approach.
CASE CONTINUED
The patient is presented at tumor board and the consensus is to proceed with the perioperative chemotherapy approach. The patient undergoes echocardiography, which reveals a normal ejection fraction. He receives 3 cycles of neoadjuvant EOX (epirubicin, oxaliplatin, and capecitabine). After 3 cycles of neoadjuvant EOX, the patient has a repeat CT that shows marked interval reduction in the size of the primary gastric neoplasm and interval decrease in the size of the small perigastric lymph nodes. He subsequently undergoes a total gastrectomy with J-tube placement. Pathology shows ypT3N0 disease with 0 out of 47 lymph nodes involved and negative margins. He then receives 3 cycles of adjuvant EOX.
• What are the recommendations for surveillance?
According to the current NCCN guidelines, a history and physical exam should be performed every 3 to 6 months for 1 to 2 years, then every 6 to 12 months for 3 to 5 years, and then annually.23 Labs, CT chest/abdomen, and EGD should be done as clinically indicated. Patients who have undergone surgical resection should be monitored for nutritional deficiencies (vitamin B12 and iron).
GASTROESOPHAGEAL JUNCTION TUMORS
Tumors arising in the GEJ or gastric cardia within 5 cm of the GEJ that extend into the GEJ or distal esophagus are staged and treated as esophageal cancers.21 The primary treatment for T1/T2N0 tumors is surgical resection. In patients with T3 or higher or node-positive adenocarcinoma of the GEJ, a combined modality approach is preferred, with preoperative chemoradiotherapy followed by surgical resection.59 The CROSS trial demonstrated a significant survival benefit with preoperative chemoradiation using carboplatin/paclitaxel compared to surgery alone in patients with esophageal or GEJ cancer (49 months versus 24 months, HR 0.66, P = 0.003).60
ONGOING TRIALS
As mentioned previously, several randomized clinical trials are in progress to clarify the optimal treatment approach. The MAGIC-B/MRC-ST03 is a randomized phase 2/3 trial looking at perioperative epirubicin, cisplatin, and capecitabine (ECX) with or without bevacizumab in patients with resectable lower esophageal, GEJ, or gastric cancer.61 The TOPGEAR trial, a randomized phase 2/3 study being conducted in Canada and Europe, is comparing perioperative ECF chemotherapy with preoperative chemoradiation plus perioperative ECF chemotherapy.62 In Asia, the PRODIGY trial is a phase 3, open-label, randomized study comparing neoadjuvant docetaxel, oxaliplatin, and S-1 followed by surgery and adjuvant S-1 versus surgery plus adjuvant S-1 in patients with locally advanced gastric cancer (T2-T4 or node positive).63 Primary endpoint is PFS and secondary endpoints are overall survival, R0 resection rate, and safety.
Trials comparing adjuvant chemotherapy to adjuvant chemoradiotherapy are also being conducted. In the Dutch CRITICS study, a randomized phase 3 trial, patients with stage Ib-Iva resectable gastric cancer were given 3 cycles of epirubicin, cisplatin/oxaliplatin, and capecitabine (ECC/EOC), followed by D2 resection and either 3 cycles of ECC/EOC or chemoradiation with weekly cisplatin and daily capecitabine.64 Between January 2007 and April 2015, a total of 788 patients were enrolled. In a preliminary report presented at ASCO in 2016, the 5-year survival rate was similar between the 2 arms (41.3% for chemotherapy arm and 40.9% for chemoradiotherapy arm, P = 0.99). The Korean ARTIST II trial is comparing adjuvant S-1 and S-1/oxaliplatin with or without radiotherapy in patients with D2-resected gastric cancer.65 Similarly, the NCT01711242 trial is comparing adjuvant XELOX alone versus XELOX with concurrent capecitabine/radiotherapy in patients with resected D2 gastric cancer.66
The ToGA trial established a survival benefit of trastuzumab in combination with chemotherapy in HER2-positive metastatic gastric cancer.67 Consequently, there are ongoing clinical trials to assess the role of trastuzumab in nonmetastatic gastric cancer. The TOXAG study is a phase 2 trial looking at the safety and tolerability of adjuvant oxaliplatin, capecitabine, and trastuzumab with radiation in patients with resected HER2-positive gastric or GEJ adenocarcinoma.68 The NCT01130337 clinical trial is evaluating perioperative XELOX/trastuzumab in patients with resectable gastric or GEJ adeno-carcinoma.69
SUMMARY
Gastric cancer is the fifth most common cancer worldwide, with the greatest incidence in East Asia. Survival outcomes are better in Asian countries when compared to the United States. This difference in survival may be related to the presence of mass screening programs in Asia, which allows for detection at an earlier stage and the use of a more extensive surgical approach (ie, D2 resection). Risk factors for developing gastric cancer include: diets high in salt/salt-preserved foods or processed meats, obesity, smoking, H. pylori infection, EBV, prior gastric surgery, radiation exposure, and positive family history.
According to the latest edition of TMN staging, gastric cancer includes tumors arising more than 5 cm distally of the GEJ or within 5 cm of the GEJ but without extension to the esophagus or GEJ. Diagnostic work-up includes: EGD with biopsy; basic labs; CT chest/abdomen/pelvis with oral and intravenous contrast; EUS if no M1 disease is identified; PET-CT if there is no M1 disease and if clinically indicated; and diagnostic laparoscopy with cytology for clinical stage T1b or higher.
The mainstay of treatment is surgical resection. Laparoscopic approach is preferred over open gastrectomy due to lower complication rates and similar survival outcomes. Current NCCN guidelines recommend a D1 or a modified D2 lymph node dissection with at least 15 lymph nodes removed for examination. Systemic chemotherapy is required in locally advanced gastric cancer (T3-T4 or node positive) and should be considered in T2N0 disease with high-risk features. Currently, there is no global consensus on the optimal treatment approach. Data from various trials have shown benefit for each approach. Regional preferences are: perioperative chemotherapy in Europe; adjuvant chemoradiotherapy in the United States; and adjuvant chemotherapy in Asia. In an effort to better define the optimal treatment approach, several randomized clinical trials are being conducted. According to the current NCCN guidelines, the following treatment approaches are acceptable and are supported by data in the trial listed in parentheses:
• Perioperative chemotherapy
° 5-FU/cisplatin (French FNLCC/FCCD trial)44 or
° ECF (MAGIC trial)42 or
° ECF modifications: EOX, EOF, ECX (REAL-2 trial)43
• Adjuvant chemoradiotherapy
° 5-FU/leucovorin sandwiched with 5-FU-based chemoradiation (INT-0116 trial)45
• Adjuvant chemotherapy (after D2 resection)
° Capecitabine/oxaliplatin (CLASSIC trial)52 or
° Capecitabine/cisplatin (ARTIST trial)48,49
- World Health Organization. GLOBOCAN 2012: estimated cancer incidence, mortality and prevalence Worldwide in 2012. France, Lyon: IARC; 2012.
- Ferlay J, Shin HR, Bray F, et al. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 2010;127:2893–917.
- Lui FH, Tuan B, Swenson SL, et al. Ethnic disparities in gastric cancer incidence and survival in the USA: an updated analysis of 1992-2009 SEER data. Dig Dis Sci 2014;59:3027–34.
- Howlader N, Noone AM, Krapcho M, et al. SEER cancer statistics review, 1975-2013. National Cancer Institute. http://seer.cancer.gov/csr/1975_2013/. Based on November 2015 SEER data submission, posted to the SEER web site April 2016.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin 2015;65:5–29.
- Isobe Y, Nashimoto A, Akazawa K, et al. Gastric cancer treatment in Japan: 2008 annual report of the JGCA nationwide registry. Gastric Cancer 2011;14:301–16.
- Tsugane S, Sasazuki S. Diet and the risk of gastric cancer: review of epidemiological evidence. Gastric Cancer 2007;10:75.
- Bouvard V, Loomis D, Guyton KZ, et al. Carcinogenicity of consumption of red and processed meat. Lancet Oncol 2015;16:1599–600.
- Yang P, Zhou Y, Chen B, et al. Overweight, obesity and gastric cancer risk: results from a meta-analysis of cohort studies. Eur J Cancer 2009;45:2867–73.
- González CA, Pera G, Agudo A, et al. Smoking and the risk of gastric cancer in the European Prospective Investigation Into Cancer and Nutrition (EPIC). Int J Cancer 2003;107:629–34.
- Huang JQ, Sridhar S, Chen Y, Hunt RH. Meta-analysis of the relationship between Helicobacter pylori seropositivity and gastric cancer. Gastroenterology 1998;114:1169–79.
- Eslick GD, Lim LL, Byles JE, et al. Association of Helicobacter pylori infection with gastric carcinoma: a meta-analysis. Am J Gastroenterol 1999;94:2373–9.
- An international association between Helicobacter pylori infection and gastric cancer. The EUROGAST Study Group. Lancet 1993;341:1359–62.
- Parsonnet J, Forman D. Helicobacter pylori infection and gastric cancer—for want of more outcomes. JAMA 2004;291:244–5.
- Malfertheiner P, Megraud F, O'Morain CA, et al. Management of Helicobacter pylori infection—the Maastricht IV/ Florence Consensus Report. Gut 2012;61:646–64.
- Fukayama M. Epstein-Barr virus and gastric carcinoma. Pathol Int 2010;60:337–50.
- Takeno S, Hashimoto T, Maki K, et al. Gastric cancer arising from the remnant stomach after distal gastrectomy: a review. World J Gastroenterol 2014;20:13734–40.
- Morton LM, Dores GM, Curtis RE, et al. Stomach cancer risk after treatment for Hodgkin lymphoma. J Clin Oncol 2013;31:3369–77.
- van der Post RS, Vogelaar IP, Carneiro F, et al. Hereditary diffuse gastric cancer: updated clinical guidelines with an emphasis on germline CDH1 mutation carriers. J Med Genet 2015;52:361–74.
- Siewert J, Stein H. Classification of adenocarcinoma of the oesophagogastric junction. Br J Surg 1998;85:1457–9.
- Edge S, Byrd DR, Compton CC, et al. AJCC cancer staging manual. 7th ed. New York: Springer New York; 2009.
- Wanebo HJ, Kennedy BJ, Chmiel J, et al. Cancer of the stomach. A patient care study by the American College of Surgeons. Ann Surg 1993;218:583–92.
- National Comprehensive Cancer Network. Gastric cancer (version 3.2016www.nccn.org/professionals/physician_gls/pdf/gastric.pdf. Accessed December 14, 2016.
- Bozzetti F, Marubini E, Bonfanti G, et al. Subtotal versus total gastrectomy for gastric cancer: five-year survival rates in a multicenter randomized Italian trial. Italian Gastrointestinal Tumor Study Group. Ann Surg 1999;230:170–8.
- Gouzi JL, Huguier M, Fagniez PL, et al. Total versus subtotal gastrectomy for adenocarcinoma of the gastric antrum. A French prospective controlled study. Ann Surg 1989;209:162–6.
- Pu YW, Gong W, Wu YY, et al. Proximal gastrectomy versus total gastrectomy for proximal gastric carcinoma. A meta-analysis on postoperative complications, 5-year survival, and recurrence rate. Saudi Med J 2013;34:1223–8.
- Chen K, Xu XW, Mou YP, et al. Systematic review and meta-analysis of laparoscopic and open gastrectomy for advanced gastric cancer. World J Surg Oncol 2013;11:182.
- Fang C, Hua J, Li J, et al. Comparison of long-term results between laparoscopy-assisted gastrectomy and open gastrectomy with D2 lymphadenectomy for advanced gastric cancer. Am J Surg 2014;208:391–6.
- Wang W, Li Z, Tang J, et al. Laparoscopic versus open total gastrectomy with D2 dissection for gastric cancer: a meta-analysis. J Cancer Res Clin Oncol 2013;139:1721–34.
- Schmidt B, Yoon SS. D1 versus D2 lymphadenectomy for gastric cancer. J Surg Oncol 2013;107:259–64.
- Jiang L, Yang KH, Guan QL, et al. Survival and recurrence free benefits with different lymphadenectomy for resectable gastric cancer: a meta-analysis. J Surg Oncol 2013;107:807–14.
- Degiuli M, Sasako M, Ponti A, et al. Randomized clinical trial comparing survival after D1 or D2 gastrectomy for gastric cancer. Br J Surg 2014;101:23–31.
- Bonenkamp JJ, Songun I, Hermans J, et al. Randomized comparison of morbidity after D1 and D2 dissection for gastric cancer in 996 Dutch patients. Lancet 1995;345:745–8.
- Songun I, Putter H, Kranenbarg EM, et al. Surgical treatment of gastric cancer: 15-year follow-up results of the randomised nationwide Dutch D1D2 trial. Lancet Oncol 2010;11:439–49.
- Mocellin S, McCulloch P, Kazi H, et al. Extent of lymph node dissection for adenocarcinoma of the stomach. Cochrane Database Syst Rev 2015;8:CD001964.
- Van Cutsem E, Sagaert X, Topal B, et al. Gastric cancer. Lancet 2016;388:2654–64.
- Quéro L, Guillerm S, Hennequin C. Neoadjuvant or adjuvant therapy for gastric cancer. World J Gastrointest Oncol 2015;7:102–10.
- Okabe H, Hata H, Ueda S, et al. A phase II study of neoadjuvant chemotherapy with S-1 and cisplatin for stage III gastric cancer: KUGC03. J Surg Oncol 2016 Jan;113:36–41.
- Wang X, Zhao L, Liu H et al. A phase II study of a modified FOLFOX6 regimen as neoadjuvant chemotherapy for locally advanced gastric cancer. Br J Cancer 2016;114:1326-33.
- Schuhmacher C, Gretschel S, Lordick F, et al. Neoadjuvant chemotherapy compared with surgery alone for locally advanced cancer of the stomach and cardia: European Organisation for Research and Treatment of Cancer randomized trial 40954. J Clin Oncol 2010;28:5210–18.
- Xiong BH, Cheng Y, Ma L, Zhang CQ. An updated meta-analysis of randomized controlled trial assessing the effect of neoadjuvant chemotherapy in advanced gastric cancer. Cancer Invest 2014;32:272–84.
- Cunningham D, Allum WH, Stenning SP, et al. Perioperative chemotherapy versus surgery alone for resectable gastroesophageal cancer. N Engl J Med 2006;355:11–20.
- Cunningham D, Starling N, Rao S, et al. Capecitabine and oxaliplatin for advanced esophagogastric cancer. N Engl J Med 2008;358:36–46.
- Ychou M, Boige V, Pignon JP, et al. Perioperative chemotherapy compared with surgery alone for resectable gastroesophageal adenocarcinoma: an FNCLCC and FFCD multicenter phase III trial. J Clin Oncol 2011;29:1715–21.
- Macdonald JS, Smalley SR, Benedetti J, et al. Chemoradiotherapy after surgery compared with surgery alone for adenocarcinoma of the stomach or gastroesophageal junction. N Engl J Med 2001;345:725–30.
- Smalley SR, Benedetti JK, Haller DG, et al. Updated analysis of SWOG-directed intergroup study 0116: a phase III trial of adjuvant radiochemotherapy versus observation after curative gastric cancer resection. J Clin Oncol 2012;30:2327–33.
- Fuchs CS, Tepper JE, Niedzwiecki D, et al. Postoperative adjuvant chemoradiation for gastric or gastroesophageal junction (GEJ) adenocarcinoma using epirubicin, cisplatin, and infusional (CI) 5-FU (ECF) before and after CI 5-FU and radiotherapy (CRT) compared with bolus 5-FU/LV before and after CRT: Intergroup trial CALGB 80101. J Clin Oncol 2011;29:256s. Abstract 4003.
- Lee J, Lim do H, Kim S, et al. Phase III trial comparing capecitabine plus cisplatin versus capecitabine plus cisplatin with concurrent capecitabine radiotherapy in completely resected gastric cancer with D2 lymph node dissection: the ARTIST trial. J Clin Oncol 2012;30:268–73
- Park SH, Sohn TS, Lee J, et al. Phase III trial to compare adjuvant chemotherapy with capecitabine and cisplatin versus concurrent chemoradiotherapy in gastric cancer: final report of the adjuvant chemoradiotherapy in stomach tumors trial, including survival and subset analyses. J Clin Oncol 2015;33:3130–6.
- Sakuramoto S, Sasako M, Yamaguchi T, et al. Adjuvant chemotherapy for gastric cancer with S-1, an oral fluoropyrimidine. N Engl J Med 2007;357:1810–20.
- Sasako M, Sakuramoto S, Katai H, et al. Five-year outcomes of a randomized phase III trial comparing adjuvant chemotherapy with S-1 versus surgery alone in stage II or III gastric cancer. J Clin Oncol 2011;29:4387–93.
- Bang YJ, Kim YW, Yang HK, et al. Adjuvant capecitabine and oxaliplatin for gastric cancer after D2 gastrectomy (CLASSIC): a phase 3 open-label, randomised controlled trial. Lancet 2012;379:315–21.
- Noh SH, Park SR, Yang HK, et al. Adjuvant capecitabine plus oxaliplatin for gastric cancer after D2 gastrectomy (CLASSIC): 5-year follow-up of an open-label, randomised phase 3 trial. Lancet Oncol 2014;15:1389–96.
- Paoletti X, Oba K, Burzykowski T, et al. Benefit of adjuvant chemotherapy for resectable gastric cancer: a meta-analysis. JAMA 2010; 303:1729–37.
- Dai Q, Jiang L, Lin RJ, et al. Adjuvant chemoradiotherapy versus chemotherapy for gastric cancer: a meta-analysis of randomized controlled trials. J Surg Oncol 2015;111:277–84.
- Zhou M, Kang M, Li G, et al. Postoperative chemoradiotherapy versus chemotherapy for R0 resected gastric cancer with D2 lymph node dissection: an up-to-date meta-analysis. World J Surg Oncol 2016;14:209.
- Yang Y, Yin X, Sheng L, et al. Perioperative chemotherapy more of a benefit for overall survival than adjuvant chemotherapy for operable gastric cancer: an updated meta-analysis. Sci Rep 2015;5:12850.
- Zhao JH, Gao P, Song YX, et al. Which is better for gastric cancer patients, perioperative or adjuvant chemotherapy: a meta-analysis. BMC Cancer 2016;16:631.
- Narsule CK, Montgomery MM, and Fernando HC. Evidence-based review of the management of cancers of the gastroesophageal junction. Thorac Surg Clin 2012;22:109–21.
- van Hagen P, Hulshof MCCM, van Lanschot JJB, et al. Preoperative chemoradiotherapy for esophageal or junctional cancer. N Eng J Med 2012;266:2074–84.
- Cunningham D. Chemotherapy with or without bevacizumab or lapatinib to treat operable oesophagogastric cancer (ST03). ClinicalTrials.gov. https://clinicaltrials.gov/show/NCT00450203. NLM Identifier: NCT00450203. Accessed December 14, 2016.
- Leong T, Smithers BM, Michael M, et al. TOPGEAR: a randomised phase III trial of perioperative ECF chemotherapy versus preoperative chemoradiation plus perioperative ECF chemotherapy for resectable gastric cancer (an international, intergroup trial of the AGITG/TROG/EORTC/NCIC CTG). BMC Cancer 2015;15:532.
- Docetaxel+oxaliplatin+S-1 (DOS) regimen as neoadjuvant chemotherapy in advanced gastric cancer (PRODIGY). ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01515748 NLM. Identifier: NCT01515748. Accessed December 14, 2016.
- Verheij M, Jansen EP, Cats A, et al. A multicenter randomized phase III trial of neo-adjuvant chemotherapy followed by surgery and chemotherapy or by surgery and chemoradiotherapy in resectable gastric cancer: First results from the CRITICS study. J Clin Oncol 2016;34 (suppl). Abstract 4000.
- Kang WK. Phase III randomized trial of adjuvant chemotherapy with S-1 vs S-1/oxaliplatin ± radiotherapy for completely resected gastric adenocarcinoma : The ARTIST II Trial (ARTIST-II). ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT01761461. NLM Identifier: NCT01761461. Accessed December 14, 2016.
- Trial of adjuvant XELOX chemotherapy and concurrent capecitabine and radiotherapy for resected gastric carcinoma. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01711242. NLM Identifier: NCT01711242. Accessed December 14, 2016.
- Bang YJ, Van Cutsem E, Feyereislova A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet 2010;376:687–97.
- Roche HL. A Study of the combination of oxaliplatin, capecitabine and herceptin (trastuzumab) and chemoradiotherapy in the adjuvant setting in operated patients with HER2+ gastric or gastro-esophageal junction cancer (TOXAG Study). ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01748773. NLM Identifer: NCT01748773. Accessed December 14, 2016.
- A study of capecitabine [Xeloda] in combination with trastuzumab [herceptin] and oxaliplatine in patients with resectable gastric cancer. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01130337. NLM Identifier: NCT01130337. Accessed December 14, 2016.
- World Health Organization. GLOBOCAN 2012: estimated cancer incidence, mortality and prevalence Worldwide in 2012. France, Lyon: IARC; 2012.
- Ferlay J, Shin HR, Bray F, et al. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 2010;127:2893–917.
- Lui FH, Tuan B, Swenson SL, et al. Ethnic disparities in gastric cancer incidence and survival in the USA: an updated analysis of 1992-2009 SEER data. Dig Dis Sci 2014;59:3027–34.
- Howlader N, Noone AM, Krapcho M, et al. SEER cancer statistics review, 1975-2013. National Cancer Institute. http://seer.cancer.gov/csr/1975_2013/. Based on November 2015 SEER data submission, posted to the SEER web site April 2016.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin 2015;65:5–29.
- Isobe Y, Nashimoto A, Akazawa K, et al. Gastric cancer treatment in Japan: 2008 annual report of the JGCA nationwide registry. Gastric Cancer 2011;14:301–16.
- Tsugane S, Sasazuki S. Diet and the risk of gastric cancer: review of epidemiological evidence. Gastric Cancer 2007;10:75.
- Bouvard V, Loomis D, Guyton KZ, et al. Carcinogenicity of consumption of red and processed meat. Lancet Oncol 2015;16:1599–600.
- Yang P, Zhou Y, Chen B, et al. Overweight, obesity and gastric cancer risk: results from a meta-analysis of cohort studies. Eur J Cancer 2009;45:2867–73.
- González CA, Pera G, Agudo A, et al. Smoking and the risk of gastric cancer in the European Prospective Investigation Into Cancer and Nutrition (EPIC). Int J Cancer 2003;107:629–34.
- Huang JQ, Sridhar S, Chen Y, Hunt RH. Meta-analysis of the relationship between Helicobacter pylori seropositivity and gastric cancer. Gastroenterology 1998;114:1169–79.
- Eslick GD, Lim LL, Byles JE, et al. Association of Helicobacter pylori infection with gastric carcinoma: a meta-analysis. Am J Gastroenterol 1999;94:2373–9.
- An international association between Helicobacter pylori infection and gastric cancer. The EUROGAST Study Group. Lancet 1993;341:1359–62.
- Parsonnet J, Forman D. Helicobacter pylori infection and gastric cancer—for want of more outcomes. JAMA 2004;291:244–5.
- Malfertheiner P, Megraud F, O'Morain CA, et al. Management of Helicobacter pylori infection—the Maastricht IV/ Florence Consensus Report. Gut 2012;61:646–64.
- Fukayama M. Epstein-Barr virus and gastric carcinoma. Pathol Int 2010;60:337–50.
- Takeno S, Hashimoto T, Maki K, et al. Gastric cancer arising from the remnant stomach after distal gastrectomy: a review. World J Gastroenterol 2014;20:13734–40.
- Morton LM, Dores GM, Curtis RE, et al. Stomach cancer risk after treatment for Hodgkin lymphoma. J Clin Oncol 2013;31:3369–77.
- van der Post RS, Vogelaar IP, Carneiro F, et al. Hereditary diffuse gastric cancer: updated clinical guidelines with an emphasis on germline CDH1 mutation carriers. J Med Genet 2015;52:361–74.
- Siewert J, Stein H. Classification of adenocarcinoma of the oesophagogastric junction. Br J Surg 1998;85:1457–9.
- Edge S, Byrd DR, Compton CC, et al. AJCC cancer staging manual. 7th ed. New York: Springer New York; 2009.
- Wanebo HJ, Kennedy BJ, Chmiel J, et al. Cancer of the stomach. A patient care study by the American College of Surgeons. Ann Surg 1993;218:583–92.
- National Comprehensive Cancer Network. Gastric cancer (version 3.2016www.nccn.org/professionals/physician_gls/pdf/gastric.pdf. Accessed December 14, 2016.
- Bozzetti F, Marubini E, Bonfanti G, et al. Subtotal versus total gastrectomy for gastric cancer: five-year survival rates in a multicenter randomized Italian trial. Italian Gastrointestinal Tumor Study Group. Ann Surg 1999;230:170–8.
- Gouzi JL, Huguier M, Fagniez PL, et al. Total versus subtotal gastrectomy for adenocarcinoma of the gastric antrum. A French prospective controlled study. Ann Surg 1989;209:162–6.
- Pu YW, Gong W, Wu YY, et al. Proximal gastrectomy versus total gastrectomy for proximal gastric carcinoma. A meta-analysis on postoperative complications, 5-year survival, and recurrence rate. Saudi Med J 2013;34:1223–8.
- Chen K, Xu XW, Mou YP, et al. Systematic review and meta-analysis of laparoscopic and open gastrectomy for advanced gastric cancer. World J Surg Oncol 2013;11:182.
- Fang C, Hua J, Li J, et al. Comparison of long-term results between laparoscopy-assisted gastrectomy and open gastrectomy with D2 lymphadenectomy for advanced gastric cancer. Am J Surg 2014;208:391–6.
- Wang W, Li Z, Tang J, et al. Laparoscopic versus open total gastrectomy with D2 dissection for gastric cancer: a meta-analysis. J Cancer Res Clin Oncol 2013;139:1721–34.
- Schmidt B, Yoon SS. D1 versus D2 lymphadenectomy for gastric cancer. J Surg Oncol 2013;107:259–64.
- Jiang L, Yang KH, Guan QL, et al. Survival and recurrence free benefits with different lymphadenectomy for resectable gastric cancer: a meta-analysis. J Surg Oncol 2013;107:807–14.
- Degiuli M, Sasako M, Ponti A, et al. Randomized clinical trial comparing survival after D1 or D2 gastrectomy for gastric cancer. Br J Surg 2014;101:23–31.
- Bonenkamp JJ, Songun I, Hermans J, et al. Randomized comparison of morbidity after D1 and D2 dissection for gastric cancer in 996 Dutch patients. Lancet 1995;345:745–8.
- Songun I, Putter H, Kranenbarg EM, et al. Surgical treatment of gastric cancer: 15-year follow-up results of the randomised nationwide Dutch D1D2 trial. Lancet Oncol 2010;11:439–49.
- Mocellin S, McCulloch P, Kazi H, et al. Extent of lymph node dissection for adenocarcinoma of the stomach. Cochrane Database Syst Rev 2015;8:CD001964.
- Van Cutsem E, Sagaert X, Topal B, et al. Gastric cancer. Lancet 2016;388:2654–64.
- Quéro L, Guillerm S, Hennequin C. Neoadjuvant or adjuvant therapy for gastric cancer. World J Gastrointest Oncol 2015;7:102–10.
- Okabe H, Hata H, Ueda S, et al. A phase II study of neoadjuvant chemotherapy with S-1 and cisplatin for stage III gastric cancer: KUGC03. J Surg Oncol 2016 Jan;113:36–41.
- Wang X, Zhao L, Liu H et al. A phase II study of a modified FOLFOX6 regimen as neoadjuvant chemotherapy for locally advanced gastric cancer. Br J Cancer 2016;114:1326-33.
- Schuhmacher C, Gretschel S, Lordick F, et al. Neoadjuvant chemotherapy compared with surgery alone for locally advanced cancer of the stomach and cardia: European Organisation for Research and Treatment of Cancer randomized trial 40954. J Clin Oncol 2010;28:5210–18.
- Xiong BH, Cheng Y, Ma L, Zhang CQ. An updated meta-analysis of randomized controlled trial assessing the effect of neoadjuvant chemotherapy in advanced gastric cancer. Cancer Invest 2014;32:272–84.
- Cunningham D, Allum WH, Stenning SP, et al. Perioperative chemotherapy versus surgery alone for resectable gastroesophageal cancer. N Engl J Med 2006;355:11–20.
- Cunningham D, Starling N, Rao S, et al. Capecitabine and oxaliplatin for advanced esophagogastric cancer. N Engl J Med 2008;358:36–46.
- Ychou M, Boige V, Pignon JP, et al. Perioperative chemotherapy compared with surgery alone for resectable gastroesophageal adenocarcinoma: an FNCLCC and FFCD multicenter phase III trial. J Clin Oncol 2011;29:1715–21.
- Macdonald JS, Smalley SR, Benedetti J, et al. Chemoradiotherapy after surgery compared with surgery alone for adenocarcinoma of the stomach or gastroesophageal junction. N Engl J Med 2001;345:725–30.
- Smalley SR, Benedetti JK, Haller DG, et al. Updated analysis of SWOG-directed intergroup study 0116: a phase III trial of adjuvant radiochemotherapy versus observation after curative gastric cancer resection. J Clin Oncol 2012;30:2327–33.
- Fuchs CS, Tepper JE, Niedzwiecki D, et al. Postoperative adjuvant chemoradiation for gastric or gastroesophageal junction (GEJ) adenocarcinoma using epirubicin, cisplatin, and infusional (CI) 5-FU (ECF) before and after CI 5-FU and radiotherapy (CRT) compared with bolus 5-FU/LV before and after CRT: Intergroup trial CALGB 80101. J Clin Oncol 2011;29:256s. Abstract 4003.
- Lee J, Lim do H, Kim S, et al. Phase III trial comparing capecitabine plus cisplatin versus capecitabine plus cisplatin with concurrent capecitabine radiotherapy in completely resected gastric cancer with D2 lymph node dissection: the ARTIST trial. J Clin Oncol 2012;30:268–73
- Park SH, Sohn TS, Lee J, et al. Phase III trial to compare adjuvant chemotherapy with capecitabine and cisplatin versus concurrent chemoradiotherapy in gastric cancer: final report of the adjuvant chemoradiotherapy in stomach tumors trial, including survival and subset analyses. J Clin Oncol 2015;33:3130–6.
- Sakuramoto S, Sasako M, Yamaguchi T, et al. Adjuvant chemotherapy for gastric cancer with S-1, an oral fluoropyrimidine. N Engl J Med 2007;357:1810–20.
- Sasako M, Sakuramoto S, Katai H, et al. Five-year outcomes of a randomized phase III trial comparing adjuvant chemotherapy with S-1 versus surgery alone in stage II or III gastric cancer. J Clin Oncol 2011;29:4387–93.
- Bang YJ, Kim YW, Yang HK, et al. Adjuvant capecitabine and oxaliplatin for gastric cancer after D2 gastrectomy (CLASSIC): a phase 3 open-label, randomised controlled trial. Lancet 2012;379:315–21.
- Noh SH, Park SR, Yang HK, et al. Adjuvant capecitabine plus oxaliplatin for gastric cancer after D2 gastrectomy (CLASSIC): 5-year follow-up of an open-label, randomised phase 3 trial. Lancet Oncol 2014;15:1389–96.
- Paoletti X, Oba K, Burzykowski T, et al. Benefit of adjuvant chemotherapy for resectable gastric cancer: a meta-analysis. JAMA 2010; 303:1729–37.
- Dai Q, Jiang L, Lin RJ, et al. Adjuvant chemoradiotherapy versus chemotherapy for gastric cancer: a meta-analysis of randomized controlled trials. J Surg Oncol 2015;111:277–84.
- Zhou M, Kang M, Li G, et al. Postoperative chemoradiotherapy versus chemotherapy for R0 resected gastric cancer with D2 lymph node dissection: an up-to-date meta-analysis. World J Surg Oncol 2016;14:209.
- Yang Y, Yin X, Sheng L, et al. Perioperative chemotherapy more of a benefit for overall survival than adjuvant chemotherapy for operable gastric cancer: an updated meta-analysis. Sci Rep 2015;5:12850.
- Zhao JH, Gao P, Song YX, et al. Which is better for gastric cancer patients, perioperative or adjuvant chemotherapy: a meta-analysis. BMC Cancer 2016;16:631.
- Narsule CK, Montgomery MM, and Fernando HC. Evidence-based review of the management of cancers of the gastroesophageal junction. Thorac Surg Clin 2012;22:109–21.
- van Hagen P, Hulshof MCCM, van Lanschot JJB, et al. Preoperative chemoradiotherapy for esophageal or junctional cancer. N Eng J Med 2012;266:2074–84.
- Cunningham D. Chemotherapy with or without bevacizumab or lapatinib to treat operable oesophagogastric cancer (ST03). ClinicalTrials.gov. https://clinicaltrials.gov/show/NCT00450203. NLM Identifier: NCT00450203. Accessed December 14, 2016.
- Leong T, Smithers BM, Michael M, et al. TOPGEAR: a randomised phase III trial of perioperative ECF chemotherapy versus preoperative chemoradiation plus perioperative ECF chemotherapy for resectable gastric cancer (an international, intergroup trial of the AGITG/TROG/EORTC/NCIC CTG). BMC Cancer 2015;15:532.
- Docetaxel+oxaliplatin+S-1 (DOS) regimen as neoadjuvant chemotherapy in advanced gastric cancer (PRODIGY). ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01515748 NLM. Identifier: NCT01515748. Accessed December 14, 2016.
- Verheij M, Jansen EP, Cats A, et al. A multicenter randomized phase III trial of neo-adjuvant chemotherapy followed by surgery and chemotherapy or by surgery and chemoradiotherapy in resectable gastric cancer: First results from the CRITICS study. J Clin Oncol 2016;34 (suppl). Abstract 4000.
- Kang WK. Phase III randomized trial of adjuvant chemotherapy with S-1 vs S-1/oxaliplatin ± radiotherapy for completely resected gastric adenocarcinoma : The ARTIST II Trial (ARTIST-II). ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT01761461. NLM Identifier: NCT01761461. Accessed December 14, 2016.
- Trial of adjuvant XELOX chemotherapy and concurrent capecitabine and radiotherapy for resected gastric carcinoma. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01711242. NLM Identifier: NCT01711242. Accessed December 14, 2016.
- Bang YJ, Van Cutsem E, Feyereislova A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet 2010;376:687–97.
- Roche HL. A Study of the combination of oxaliplatin, capecitabine and herceptin (trastuzumab) and chemoradiotherapy in the adjuvant setting in operated patients with HER2+ gastric or gastro-esophageal junction cancer (TOXAG Study). ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01748773. NLM Identifer: NCT01748773. Accessed December 14, 2016.
- A study of capecitabine [Xeloda] in combination with trastuzumab [herceptin] and oxaliplatine in patients with resectable gastric cancer. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01130337. NLM Identifier: NCT01130337. Accessed December 14, 2016.
Hemophilia A and B: An Overview
INTRODUCTION
Hemophilia A and B are the most common severe inherited bleeding disorders. The incidence of hemophilia is 1 in 5000 live male births, with hemophilia A occurring 4 times more commonly than hemophilia B. The associated decrease in factor VIII in hemophilia A was initially identified in 1947, and the decrease in factor IX associated with hemophilia B was identified 5 years later.1,2 Both conditions are inherited as X-linked recessive traits. Queen Victoria of Britain, who reigned from 1837 to 1901, was a carrier of hemophilia and had 2 carrier daughters, Alice and Beatrice, and a son with hemophilia, Leopold.3 In 1984 and 1985, the genes for factor VIII and factor IX were cloned, and in 1989 recombinant factor VIII was first used clinically.4–7
PATHOPHYSIOLOGY
Both factors VIII and IX are crucial for normal thrombin generation in the coagulation pathway. After any injury, the initial hemostatic event is the formation of a platelet plug. Once the platelet plug is formed, subsequent generation of fibrin prevents continued oozing from the affected site. In hemophilia A and B, the propagation phase of coagulation is impaired, and as a result, the formation of clot is delayed and is not robust. Due to the delayed formation of an abnormal clot, patients with hemophilia do not bleed rapidly but rather ooze continuously. Rebleeding is a common occurrence in inadequately treated patients.8
GENETICS
The gene for factor VIII (F8) is located in the most distal band (Xq28) of the long arm of the X chromosome. Spanning more than 186 kb, it is one of the largest genes known.9,10 The gene for factor IX (F9) is located at Xq27.1 and spans 33 kb.7 Defects in the F8 gene associated with hemophilia A may be divided into several categories: gross gene rearrangements; insertions or deletions of genetic sequence of a size varying from 1 base pair up to the entire gene; or single DNA base substitutions resulting in either amino acid replacement (missense), premature peptide chain termination (nonsense, or stop mutations), or mRNA splicing defects. All classes of defects can result in severe disease. However, the single most clinically important defect is a gene rearrangement (an inversion) involving F8 intron 22, which results in approximately 50% of all severe hemophilia A cases worldwide.11,12 In hemophilia B, point mutations are by far the most common type of abnormality. Generally, they are caused by DNA polymerases adding the wrong nucleotide during replication.13
HEMOPHILIA IN FEMALES
X-Inactivation (also called Lyonization) is a process that occurs early in embryonic development in female mammals where 1 of the 2 copies of the X chromosome present is inactivated; it is the reason why some female carriers of hemophilia can become symptomatic. Approximately one third of carriers have clotting factor levels of less than 60% of normal and may experience abnormal bleeding.14,15 In most cases, carriers experience symptoms similar to those seen in men with mild hemophilia, as well as some that are specific to women. Symptomatic carriers and women with hemophilia may bruise more easily; may experience prolonged bleeding after surgery; may experience serious bleeding after trauma; often have heavier and more prolonged bleeding during their periods (menorrhagia) and are more likely to require an iron supplement or to undergo hysterectomy; and are more likely to have postpartum bleeding following childbirth.14,15
CLINICAL MANIFESTATIONS
Hemorrhage in patients with hemophilia may occur with minimal or unknown trauma. Patients with severe hemophilia (factor level of < 1 IU/dL or < 1% of normal) often experience spontaneous bleeding into joints or muscles. Those with moderate hemophilia (factor level of 1–5 IU/dL or 1%–5% of normal) seldom experience spontaneous hemorrhage and usually have prolonged bleeding with minor trauma or surgery. Patients with mild hemophilia (factor level > 5 IU/dL but less than 40 IU/dL or > 5% but < 40% of normal) experience severe hemorrhage only following moderate to severe trauma or surgery, and rarely experience spontaneous bleeding. Depending on the site, bleeding can be serious (joints; muscles, especially deep compartments [iliopsoas, calf, and forearm]; mucous membranes in the mouth, gums, nose, and genitourinary tract) or life-threatening (intracranial, neck/throat, gastrointestinal). The joints and muscles are the most common sites of bleeding (Table 1).

MUSCULOSKELETAL BLEEDING
The hallmark of hemophilia is deep bleeding into the joints and muscles. Without prophylactic factor treatment, patients with severe hemophilia A or B may have a bleeding episode as often as once or twice a week. Hemarthrosis episodes typically begin when the child reaches the toddler age. One of the first signs of hemarthrosis is a tingling sensation and feeling of warmth which is soon followed by pain and decreased range of motion of the joint as a result of distension of the joint capsule. Prompt, aggressive treatment with factor replacement therapy is the key to prevent further bleeding and minimize potential long-term complications. Severe chronic arthropathy may develop in older children and adults who have not received aggressive treatment (Figure).
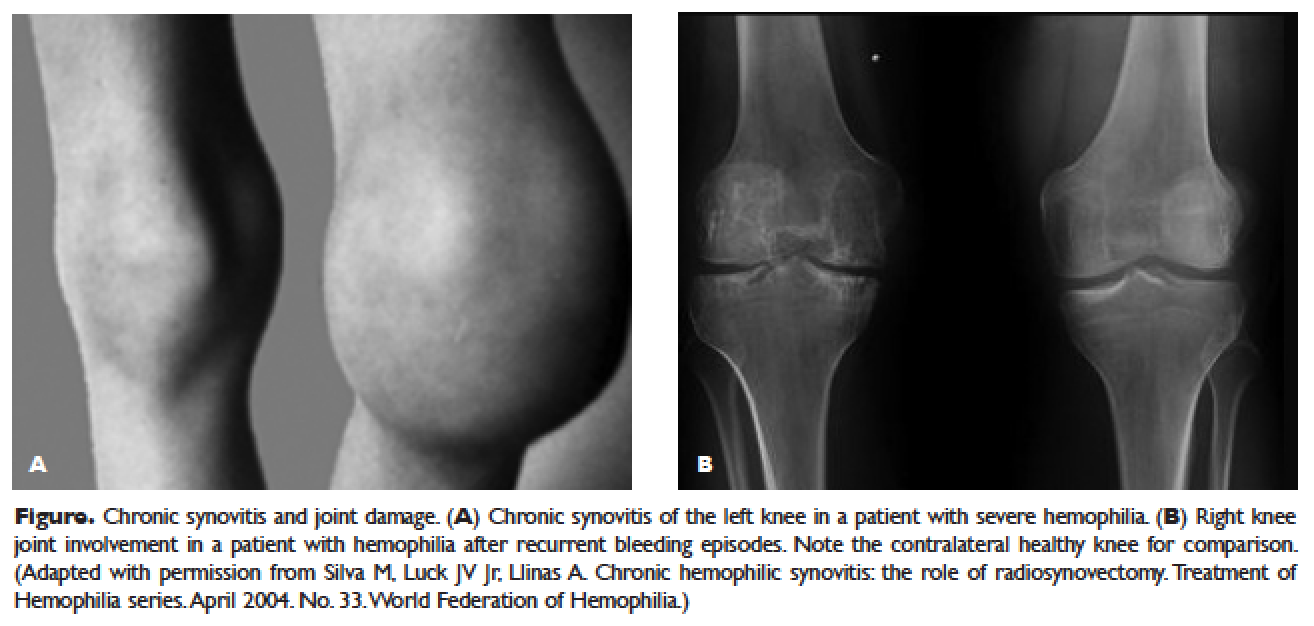
Bleeding into the muscle can manifest as a vague feeling of pain on motion. Swelling may not be obvious and the mass may be difficult to palpate, although the circumference of the affected limb will be increased. Among the muscle bleeds, iliopsoas bleed deserves a special mention because of its potential to cause life-threatening hypovolemic shock as large volumes of blood can be lost into the retroperitoneal space. These patients present with vague abdominal pain or upper thigh discomfort. The hip is flexed and outwardly rotated. The diagnosis is confirmed by computed tomography (CT) or ultrasound.
LIFE-THREATENING HEMORRHAGE
Central Nervous System Bleeding
Most central nervous system (CNS) events, which involve bleeding inside the skull or spinal canal, are caused by trauma. CNS hemorrhage is the most common form of severe hemophilic trauma. However, since patients with hemophilia can experience bleeding even weeks after a minor head injury, a history of head trauma may be hard to determine, particularly in children. Spontaneous CNS bleeding in individuals with hemophilia is rare except when there has been a recent antecedent CNS hemorrhage (ie, a recurrent bleed at a previously injured site) or when there is an associated anatomic lesion that predisposes to acute hemorrhage (eg, aneurysm or arteriovenous malformation). Data from the Universal Data Collection Project of the U.S. Centers for Disease Control and Prevention indicates that predisposing risk factors for intracranial hemorrhage include HIV infection, presence of inhibitory antibodies, and age younger than 5 years or older than 51 years.16 Neonatal intracranial hemorrhage is most commonly due to birth trauma. Difficult vaginal deliveries (often requiring the application of forceps or vacuum extraction) are predisposing factors for intracranial hemorrhage in hemophilic newborns.
The site of intracranial CNS bleeding can be subdural, epidural, or intraparenchymal. Bleeding at any of these sites can cause rapidly deteriorating CNS brain function, associated brain swelling, and, in the most extreme circumstances, herniation of the brainstem and rapid death. If the bleeding is stopped with rapid clotting factor replacement, adverse clinical effects can be avoided. However, with intraparenchymal hemorrhage, even small hemorrhages can induce permanent structural and/or neurologic sequelae (in particular, if the anatomic site of the bleed is essential for routine brain function).17
Throat and Neck Hemorrhage
An acute neck injury or a retropharyngeal hemorrhage induced by dental or oral surgical instrumentation can lead to a dissecting facial plane hematoma. This in turn can sometimes lead to compression and acute airway compromise. Bleeding from these injuries that is compressing or compromising the airway may require a rapid clinical response.18 The time from the injury until the trachea is compressed may be long, sometimes many hours. However, once the compression is sufficient to cause difficulty breathing, there may be a short amount of time to stop the bleeding and prevent complete respiratory obstruction.
MUCOCUTANEOUS BLEEDING
One of the common manifestations of hemophilia is oral bleeding. Tooth extraction poses a specific problem, and bleeding following extraction can be the first symptom that leads to the diagnosis of hemophilia. Bleeding after circumcision may also suggest the diagnosis. In 1 study cohort looking at sites of initial bleeding episodes in babies with hemophilia diagnosed before the age of 2 years, bleeding from circumcision and other iatrogenic causes tended to be most common in the neonatal period. Circumcision bleeding events occurred more often in infants with no family history (43%) as compared to those born to known maternal carriers (9.2%) or to mothers with some other family history of hemophilia (14.3%).19
Gastrointestinal (GI) bleeding occurs occasionally in hemophilia, and a wide spectrum of esophageal and GI bleeding may occur. A review of 41 episodes of GI bleeding in hemophilia patients who presented to 1 institution over 10 years implicated duodenal ulcer (22%), unknown site (22%), and gastritis (14%) as the most common sources.20 Mallory-Weiss syndrome has also been cited as a cause for upper GI bleeding in hemophilia patients.21
PRINCIPLES OF TREATMENT
Understanding the pathophysiology of hemophilia as well as the type and severity of hemophilia and the inhibitor status in an individual patient are paramount in the management of a patient with hemophilia. In the past, management mainly focused on the treatment of acute bleeding episodes (Table 2). With data showing the benefit of bleed prevention, the management of hemophilia now focuses on prophylaxis of bleeding episodes, which prevents chronic arthropathy and improves quality of life.
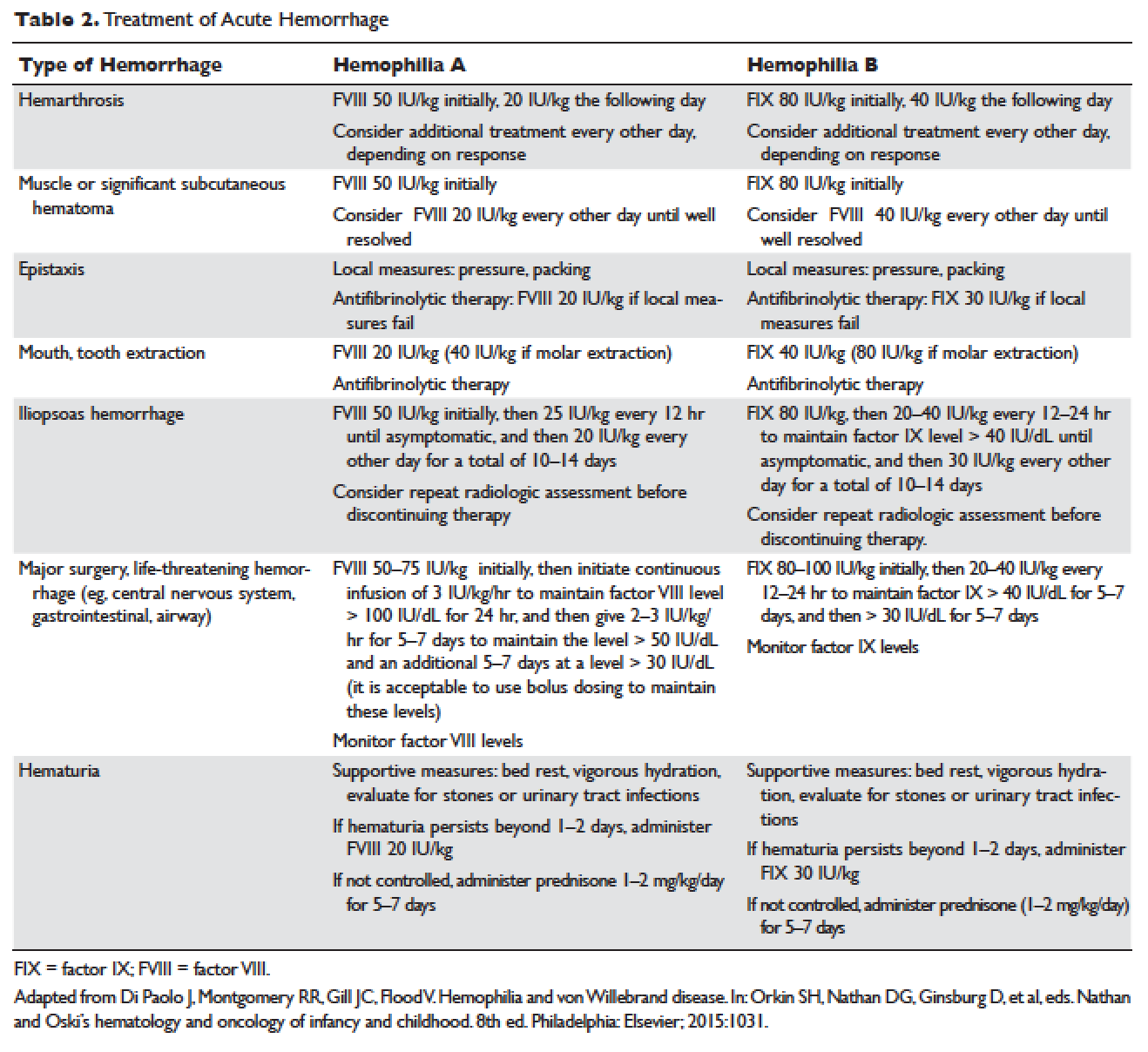
ACUTE BLEEDING EPISODES
Dosing of Factor VIII Products
Dosing for factor VIII concentrate is as follows: 1 IU of factor VIII concentrate per kg will increase the circulating factor VIII level by 2% (ie, patient weight in kg × 50 IU/kg = 100% correction). For example, a 30-kg patient requiring 100% correction of factor VIII needs an infusion of 1500 IU of factor VIII (30 kg × 50 IU/kg).
Dosing of Factor IX Products
Dosing for factor IX concentrate is as follows: 1 IU of factor IX concentrate per kg will increase the circulating factor IX level by 1% (ie, patient weight in kg × 100 IU/kg = 100% correction). For example, a 30-kg patient requiring 100% correction of factor IX needs an infusion of 3000 IU of factor IX (30 kg × 100 IU/kg). Higher doses (120 to 130 IU/kg) of the recombinant factor IX product BeneFIX (Pfizer) may be needed to reach the 100% circulating factor IX level.
ADJUVANT THERAPY
Desmopressin
Desmopressin is a synthetic vasopressin analogue that increases plasma factor VIII and von Willebrand factor (VWF) levels; it is used to prevent and treat bleeding episodes associated with dental and surgical procedures in patients with mild and moderate hemophilia A and von Willebrand disease.22 Desmopressin causes the release of VWF and factor VIII from storage in the Weibel–Palade bodies of the endothelial cells that line the blood vessels. Individual response to desmopressin varies, with factor VIII level increasing between 2 and 15 times baseline level in patients with mild or moderate hemophilia A.23 It is therefore recommended that patients undergo a therapeutic trial of desmopressin with laboratory measurement of response to factor VIII before it is used for treatment of bleeding episodes or as prophylactic therapy before dental and other surgical procedures. A similar response is generally seen in an individual patient with subsequent doses, and thus the factor VIII level attained after a trial dose can be used to predict the response to future therapy.24
The recommended intravenous dosage of desmopressin is 0.3 µg/kg, administered in 25 to 50 mL of normal saline, over a period of 20 to 30 minutes.25 A concentrated form of desmopressin is available for intranasal administration to treat bleeding disorders. The appropriate dose of concentrated intranasal desmopressin is 150 µg (1 puff) for persons weighing less than 50 kg, and 300 µg (1 puff in each nostril) for persons weighing more than 50 kg.26
Antifibrinolytic Therapy
Antifibrinolytics (both epsilon-aminocaproic acid [EACA] and tranexamic acid) reversibly block the lysine binding sites of plasminogen, preventing its activation to plasmin and thus inhibiting the lysis of polymerized fibrin. EACA is also believed to stabilize the active form of thrombin activatable fibrinolysis inhibitor (TAFIa). It is believed that inactivation of TAFIa is due to conformational rearrangements in the TAFIa molecule; EACA has been shown to slow down spontaneous inactivation of TAFIa, thus curtailing fibrinolysis.27 Although hemostasis is generally achieved with either factor VIII replacement or desmopressin, the risk of recurrent bleeding from oral mucosal surfaces is dramatically reduced with the use of antifibrinolytic agents. These agents are typically contraindicated in patients with hematuria because they can cause a clot to form in the urinary bladder or ureters, leading to obstruction.
EACA is available in intravenous, oral tablet, and elixir formulations; the oral dose is 100 to 200 mg/kg initially (maximum dose, 10 g), followed by 50 to 100 mg/kg per dose every 6 hours (maximum dose, 5 g). Tranexamic acid is available in 650-mg capsules; the dose is 25 mg/kg every 6 to 8 hours.28,29 To treat spontaneous oral hemorrhage or to prevent bleeding from dental procedures in patients with hemophilia, either drug is usually begun in conjunction with desmopressin or factor replacement therapy immediately prior to the procedure and continued for up to 7 days or until mucosal healing is complete. Nonsteroidal anti-inflammatory drugs and aspirin affect platelet function and hence are contraindicated in affected individuals.30
PROPHYLAXIS
Patients with mild to moderate hemophilia typically bleed only after trauma, although the trauma needed to induce bleeding may be more minor than that which would cause bleeding in a normal individual. They usually do not suffer from significant morbidities, whereas patients with severe hemophilia often have spontaneous severe muscle and joint bleeds and can develop early crippling hemophilic arthropathy. Hence, routine prophylaxis has now become the standard of care in the United States and other developed countries in the management of patients with severe hemophilia. Prophylactic replacement therapy with cryoprecipitate in boys with severe hemophilia was first used nearly 50 years ago in Sweden31 and the Netherlands,32 and was shown to reduce the number and the severity of bleeds.32 Moreover, it was observed that early prophylaxis was more effective in preventing arthropathy compared to starting later in life, and that radiologic joint damage could not be reversed by prophylaxis. Subsequently, primary prophylaxis, defined as the start of regular, continuous treatment before the age of 2 years or after the occurrence of first joint bleed,33 was recommended and eventually became the standard treatment; it is currently recommended by the World Health Organization/World Federation of Hemophilia (WFH).34
The timing to begin prophylaxis is somewhat controversial, but many authors suggest starting prophylaxis before the first hemarthrosis occurs. Several studies have reported a wide variation in the age at first joint bleed, ranging from 0.2 to 5.8 years, with medians of 1.6 to 1.7 years.35,36 It has been suggested that arthropathy is best prevented if prophylaxis is started before the second or third joint bleed, but the benefits of starting before the occurrence of first bleed have not been established.37,38 The Swedish experience provides strong support for early prophylaxis.39 In an analysis of 121 patients with severe hemophilia, age at initiation of prophylaxis was an independent predictor of the development of arthropathy, but dose and interval of prophylaxis at the start of prophylactic treatment were not.39
In the Italian ESPRIT study, it was shown that children randomly assigned to prophylaxis had significantly fewer total bleeding episodes and joint bleeding episodes compared with those assigned to episodic therapy. Eleven of 21 patients (52%) in the prophylaxis group had on average less than 1 hemarthrosis per year, whereas only 4 of 19 patients in the episodic therapy group (21%) had the same low frequency of bleeding (P < 0.05).40 In a study of long-term prophylaxis versus on-demand treatment comparing age-matched Danish and Russian patients, the median annual number of joint bleeds in patients on prophylaxis was 1, while patients managed with on-demand treatment experienced a median of 37 joint bleeds. Patients taking prophylaxis also had a statistically significantly better quality of life estimate (P < 0.001) and better functional independence.41 In another trial, prophylaxis was initiated between the ages of 6 and 30 months based on a history of joint hemorrhage rather than age. Radiologic evidence of preserved joint architecture was found in 93% of participants in the prophylaxis group at 6 years of age. In this group, 18 of 32 (56%) children had 1 or 2 bleeds into one or more index joints before prophylaxis, and 17 (53%) had 1 to 5 hemorrhages into 1 or more index joints during prophylaxis. Prophylaxis was efficacious in decreasing bleeding and joint damage after up to 5 hemarthroses.42
Optimal Prophylactic Regimen
Although the benefits of prophylactic replacement therapy are firmly established, the optimal dose and frequency remain unclear. The half-life of clotting factor concentrates is short: about 8 hours for factor VIII in children, and about 12 hours for factor IX. As a result, prophylactic therapy is most effective when given frequently. The most common factor VIII concentrate dosing regimen for prophylaxis in hemophilia A is 25 to 40 IU/kg 3 times per week; for hemophilia B, a dose of 80 to 100 IU/kg is given twice weekly. This is aimed at a pre-infusion level > 1% to mimic the clinical phenotype of moderate hemophilia.
Recently, the US Food and Drug Administration (FDA) approved the first long-lasting antihemophilic factor (recombinant) Fc fusion protein for use in adults and children with hemophilia A. This medication contains the Fc region of human immunoglobulin G1 (IgG1), which binds to the neonatal Fc receptor (FcRn). FcRn is part of a naturally occurring pathway that delays lysosomal degradation of immunoglobulins by cycling them back into circulation and prolonging their plasma half-life. Dosing for routine prophylaxis is 50 IU/kg every 4 days; it may be adjusted based on patient response, with dosing in the range of 25 to 65 IU/kg at 3- to 5-day intervals. More frequent or higher doses up to 80 IU/kg may be required in children younger than 6 years.43
DEVELOPMENT OF INHIBITORS
FACTOR VIII INHIBITORS
Despite the success in the clinical management of hemophilia A, treated patients remain at risk for developing neutralizing antibodies that inhibit factor VIII activity. An inhibitor is a polyclonal high-affinity IgG that is directed against the factor VIII protein and renders exogenous factor ineffective. IgG4 antibodies are predominant and do not fix complement.
Risk Factors
The pathophysiology underlying the development of factor VIII inhibitors is a T-helper (Th)–cell dependent event that involves antigen-presenting cells and B lymphocytes; why only a fraction of patients experience this adverse effect of factor therapy is not known. Patients with mild/moderate hemophilia have a lower risk for inhibitor development than those with severe hemophilia A. The estimated prevalence of inhibitors ranges from 3% to 13% in mild to moderate disease,44–46 and up to 36% in severe hemophilia A.47,48 Usually the presence of an inhibitor in patients with mild/moderate hemophilia is suggested by a change in bleeding pattern: patients who previously used to bleed only after trauma or surgery suddenly start to experience severe spontaneous bleeding. This change in bleeding pattern is explained by cross-reactivity of the inhibitor with the mutated factor VIII of the patient, resulting in a residual factor level of < 0.01 IU/dL.49–51 Occasionally, there is no change in the residual factor VIII level but an inhibitor is detected in the Bethesda assay and/or there is lack of efficacy of factor VIII trans-fusions.51–53
Genetic factors. Data indicate that the risk of developing neutralizing antibodies is to a large extent determined by patient-related genetic factors.54,55 The immune response to factor VIII is similar in up to 80% of family members, significantly higher than expected compared with data from unrelated subjects. In a meta-analysis of patients with severe hemophilia A, the inhibitor incidence was twice as high in African American patients as compared with white patients.56 One study showed that patients of Hispanic ancestry with severe hemophilia A have a higher prevalence of neutralizing inhibitors than non-Hispanic white patients.57
Type of causative mutation. In severe hemophilia A, the risk of inhibitor formation is associated with the type of mutation. More disruptive mutations in the factor VIII gene, such as the intron 22 inversion, large gene deletions, and stop codons are associated with an approximately 35% risk of inhibitor formation, compared with only about 5% in those with missense mutation and small deletions.58 Persons with mutations involving large gene deletions, nonsense mutations, and intrachromosomal aberrations are usually at higher risk for the development of inhibitors than persons with missense mutations, small deletions/insertions, and splice site mutations.59,60 A relatively high risk is also encountered in patients with splicing errors and frame-shift mutations.61
Major histocompatibility complex. The HLA class I alleles A3, B7, and C7, as well as the class II alleles DQA0102, DQB0602, and DR15 have all been associated with a slightly higher risk for inhibitor development in unrelated patients, whereas the HLA C2, DQA0103, DQB0603, and DR13 alleles might be protective.62,63
Immune-regulatory molecules. In the Malmö International Brother Study, polymorphic sites in the genes coding for interleukin 10 (IL-10), tumor necrosis factor-α, and cytotoxic T lymphocyte–associated protein 4 were all associated with the risk of developing inhibitors.64–66 In this study, a 134 bp–long variant of a CAA microsatellite in the promoter region (IL-10.G) was identified in 26.8% of patients with hemophilia A. Thirty-two of these patients (72.7%) developed inhibitors as compared with 37.5% of those without the allele.65
Intensive exposure to factor VIII. Inhibitors in mild/moderate hemophilia seem to occur more commonly later in life, and an episode of intensive treatment with factor VIII concentrate has been reported to precede detection in most reported cases. In the series reported by Hay et al,67 16 out of 26 inhibitors were detected after such intensive replacement therapy, and no particular concentrate was implicated.
INHIBITORS TO FACTOR IX
Factor IX inhibitors are relatively uncommon, occurring in only 1% to 3% of persons with hemophilia B. This is in striking contrast to hemophilia A, where approximately 30% of patients develop inhibitors. The majority of patients with hemophilia B who develop inhibitors have severe hemophilia B.
Risk Factors
Certain mutations in the factor IX gene are associated with an increased incidence of inhibitor development. Large deletions and frame-shift mutations leading to the loss of coding information are much more likely to be associated with inhibitor development. Large deletions account for only 1% to 3% of all hemophilia B patients, but account for 50% of inhibitor patients.68 Patients with hemophilia B who develop inhibitors are at risk for developing anaphylactic reactions to factor IX–containing products. Anaphylaxis occurred more frequently in families with null mutations (large deletions, frame-shift mutations, or nonsense mutations) than in those with missense mutations.69 With hemophilia A, approximately 40% to 50% of black individuals develop inhibitors, but no such association has been found in hemophilia B. Individuals who develop an inhibitor to factor IX do so relatively early in life (within the first 4 to 5 years), after a median of 9 to 11 exposure days to any factor IX–containing products. Because of the severity of a potential anaphylactic reaction occurring early in life after very few exposures to factor IX, all infants and small children with severe hemophilia B should be closely followed over their first 10 infusions with any factor IX–containing products in a facility equipped to treat anaphylactic shock.70–72 A comparison of inhibitors in hemophilia A and B is shown in Table 3.
TREATMENT OF ACUTE BLEEDS IN PATIENTS WITH FACTOR VIII INHIBITORS
The available therapeutic agents for treatment of acute hemorrhage in children with hemophilia A with an inhibitor include high-dose recombinant or plasma-derived factor VIII concentrate, activated prothrombin complex concentrates (aPCCs), and recombinant activated factor VII (rFVIIa). In addition, antifibrinolytics may be used as an adjunct therapy.
Patient response to each treatment varies widely, with some patients responding well to one treatment and less well to another. Neither the patient's history nor standard lab tests can assist in making the best choice for the patient. A personalized approach to factor selection is used, and the dosing of that particular agent is often determined primarily by clinical assessment. Inhibitors are quantitated using the Bethesda inhibitor assay and clinically are classified as low- and high-responding inhibitors (Table 4). Inhibitor screening should be done prior to invasive procedures and periodically during the first 50 days of treatment since the risk for inhibitor development is highest during this period.

Low-Responding Inhibitors
A low-responding inhibitor is one in which inhibitor titers are < 5 Bethesda units (BU)/mL; patients with low-responding inhibitors can generally be treated with factor VIII concentrates at higher doses.73 Because the effect of factor VIII inhibitor is usually delayed, the Bethesda titer in plasma is determined after a 2-hour incubation period. As a result of this time delay, continuous administration of factor VIII is usually found to be effective.74 For a serious limb- or life-threatening bleeding episode, a bolus infusion of 100 IU of factor VIII per kg of body weight is administered, and the level is maintained by treatment at a rate of 20 IU/kg/hr. An assay for factor VIII should be performed 1 hour after the bolus infusion and at least daily thereafter. As the antibody titer drops, the daily level of factor VIII may rise and thus downward adjustment of the continuous infusion rate may be required. For routine joint and muscle hemorrhage, patients can usually be managed with infusions at twice the usual dosage. Routine inhibitor assays should be performed after exposure to factor VIII to determine whether an anamnestic response has occurred.
High-Responding Inhibitors
Most clinicians caring for patients with limb- or life-threatening bleeding episodes prefer to use products for which therapeutic levels can be monitored. As described earlier, continuous admin-istration of factor VIII is often effective because of the time delay in inhibition by the antibody. An initial dose of 100 to 200 IU/kg can be administered, and factor VIII levels can be determined 1 hour after initiation of continuous infusion at a rate of 20 to 40 IU/kg/hr. If a factor VIII level cannot be obtained (ie, patients with inhibitor titers > 5 to 10 BU/mL), alternative approaches include the bypassing agents aPCC and rFVIIa.
First used in the 1970s, aPCCs represented a significant improvement in the management in patients with hemophilia with inhibitors. They contain multiple activated serine protease molecules; activated factor X and prothrombin are the main active components in FEIBA (factor eight inhibitor bypassing activity), the most commonly used aPCC in the United States. FEIBA is a pooled plasma product that contains activated factors II, VII, IX and X, and has a duration of action of about 6 to 12 hours. For treatment of acute bleeds, the recommended dose of FEIBA is 50 to 100 IU/kg infused every 8 to 12 hours (maximum daily dose of 200 IU/kg). There is a risk of thrombosis/disseminated intravascular coagulation (DIC) with very large doses given frequently (> 200 IU/kg/day).
rFVIIa directly activates factor X and increases thrombin production on the surface of activated platelets in the absence of factor VIII or factor IX. Standard dosing of rFVIIa is 90 to 120 µg/kg, and many hemophilia treatment centers use higher doses (270 µg/kg/dose), especially in children and young adults. The half-life is about 1.5 to 3 hours, and therefore frequent administration (every 2–6 hours) is required. In one study that assessed the safety and efficacy of fixed-dose rFVIIa in the home setting, hemostasis was achieved in 566 (92%) of evaluable bleeding episodes, and following administration of the additional maintenance dose, hemostasis was maintained in 95% of successfully treated cases.75 As with aPCCs, there is no standardized quantitative laboratory test for measuring the effectiveness of rFVIIa therapy.
All currently used bypassing agents are associated with a risk of thrombotic complications including thromboembolism, DIC, and myocardial infarction. These complications are very rare in patients with hemophilia, however. In general, bypassing agents work for most bleeds and for most patients, but are not as predictable as factor replacement therapy and cannot be monitored by laboratory assays.
TREATMENT OF ACUTE BLEEDS IN PATIENTS WITH FACTOR IX INHIBITORS
rFVIIa and FEIBA are the mainstays of treatment of bleeding episodes in individuals with hemophilia B complicated by an inhibitor to factor IX. Treatment of hemorrhagic episodes in these patients depends on the type of bleeding episode experienced, the inhibitor classification (high- versus low-responding [Table 4]), and the history and severity of infusion reactions. Patients with low-responding inhibitors who have not experienced infusion reactions may be treated with doses of factor IX concentrate calculated to overcome the inhibitor titer and achieve a hemostatic level. In patients with high-responding inhibitors, the use of factor IX concentrates is impractical because of the inhibitor titer or the anamnestic response. Regardless of inhibitor titer, in patients with a history of an anaphylactic event, factor IX usage is contraindicated.
The most commonly used therapy for hemostatic control in patients with high-responding inhibitors with factor IX deficiency and a history of infusion reaction is rFVIIa; the standard dosing regimen is 90 to 120 µg/kg/dose administered every 2 to 3 hours, with a maximum dose of 270 µg/kg/dose. aPCCs, which contain factor IX, can be utilized if the patient has not experienced prior infusion reactions. Repeated exposures to products containing factor IX may stimulate the inhibitor titer and prevent its natural decline over time. This can pose a problem in cases of life- or limb-threatening hemorrhage unresponsive to rFVIIa as these patients will not have factor IX available as an effective mode of therapy. The dosing of FEIBA ranges from 50 to 100 IU/kg every 12 hours, with daily dosing not to exceed 200 IU/kg.
IMMUNE TOLERANCE INDUCTION
Because of the associated inhibitor-related morbidity resulting from limited treatment options, antibody eradication is the ultimate goal in inhibitor management. The only proven strategy for achieving antigen-specific tolerance to factor VIII or factor IX is immune tolerance induction (ITI) therapy. Successful ITI in hemophilia A is currently defined as both an undetectable inhibitor titer (< 0.6 BU), and normalized factor VIII pharmacokinetics, which in turn is defined as plasma factor VIII recovery > 66% of expected and a half-life > 6 hours, determined following a 72-hour factor VIII exposure-free period (Consensus Proceedings from the Second International Conference on Immune Tolerance Therapy, Bonn, Germany, 1997 [unpublished]). Once successful immune tolerance is achieved, long-term prophylaxis is commonly instituted. Using conclusions drawn from international consensus criteria and analysis of the International Immune Tolerance Registry, the I-ITI study has defined ITI failure by the presence of either of 2 criteria:
1. Failure to attain the definition of success within 33 months of uninterrupted ITI;
2. Failure to demonstrate a progressive 20% reduction in inhibitor titer over each 6-month period of uninterrupted ITI, beginning 3 months after initiation to allow for expected anamnesis.76–78
This definition implies a minimum ITI trial period of 9 months before failure is declared.
The European Hemophilia Standardization Board (EHSB), the International Consensus Panel (ICP), and the United Kingdom Hemophilia Center Doctors’ Organization (UKHCDO) have agreed that it is preferable to initiate ITI at a titer of < 10 BU/mL, unless, per the ICP, the titer does not decline over a period of 1 to 2 years and/or inhibitor development is associated with severe or life-threatening bleeding. The ICP noted that for “poor-risk” ITI patients (defined by a historical titer of > 200 BU/mL and/or a pre-ITI inhibitor titer of > 10 BU/mL and/or an interval of > 5 years since inhibitor diagnosis), published efficacy data are limited to dosing regimens > 200 IU/kg/day. The groups all independently concluded that ITI has been successfully performed using recombinant and plasma-derived factor VIII replacement therapy (usually the product on which they developed the inhibitor), and that there are no data to support the superiority of any single product type.79–81 However, both EHSB and ICP have suggested that VWF-containing concentrates be considered for patients who fail ITI using high-purity factor VIII.79,80
The recommendations from US guidelines for ITI in patients with hemophilia A and inhibitors are listed in Table 5.82

ARTHROPATHY
Before the advent of factor products for the treatment of hemophilia, hemarthrosis was one of the leading causes of morbidity. Today, the routine use of prophylactic treatment has resulted in a significant improvement in the lifestyle, quality of life, and life expectancy of these patients. However, despite best efforts, some patients will have severe joint destruction as a result of repeated articular bleeding episodes during their early years. This leads to pain and significant functional disability, thus impairing the quality of life. The basic pathology behind hemophilic arthropathy is chronic synovitis.
It is common to observe a pattern of repeated bleeding (chronic hemarthrosis), especially in patients with severe hemophilia, that can lead to chronic synovitis, inflammatory arthritis, and progressive arthropathy. Therefore, the key to preventing hemophilic arthropathy is aggressive management of the initial hemarthrosis. This is generally accomplished with the use of clotting factor replacement, restorative physiotherapy, and close clinical follow-up. If chronic synovitis develops, synovectomy may be considered in order to slow the progression of the hemophilic arthropathy and to prevent the development of major articular surface erosions that can lead to end-stage arthropathy.83 Primary prophylaxis is discussed earlier and is the mainstay of prevention of chronic hemophilic arthropathy.
SYNOVECTOMY
The emergence of chronic hemophilic hemarthrosis is incited by a hypertrophic and highly vascular synovium. Removal of the synovium prevents further joint damage,84 and can be accomplished through surgical and nonsurgical procedures.
Surgical excision of the hypertrophic synovium can be performed through open or arthroscopic procedures. The open approach has largely been replaced by arthroscopic synovectomies. Regardless of the approach, these patients need prolonged hospitalization, extensive factor replacement, and exhaustive physiotherapy. Moreover, patients with inhibitors are usually not considered candidates for surgical synovectomy.
Chemical and radioactive agents injected intra-articularly can decrease the volume and activity of the synovial tissue. Due to the minimally invasive nature of these procedures, nonsurgical synovectomies are of special importance for hemophilic patients with inhibitors to clotting factors.
Chemical Synovectomy
Chemical synovectomies, using thiotepa, osmic acid, D-penicillamine and other agents, have been used in the distant past. Rifampicin, which is used an antibiotic, is now the most commonly used chemical for the purpose of synovectomy, and the one that has shown better results in terms of decreasing hemarthrosis.85 Each one of the injections should be accompanied by prophylactic administration of clotting factor concentrate. Excellent results (no synovitis and restoration of previous function) have been reported in up to 83% of patients at an average of 2.4 years after the intra-articular injection of rifampicin. As the pathology of the joint becomes more severe, however, the number of injections required to achieve improvement increases. Younger patients and smaller joints benefit more from this procedure.
Radiation Synovectomy
Radiosynovectomy (RS) and radiosynoviorthesis are common terms used to describe the synovial ablation accomplished by intra-articular injection of radioisotopes. Isotopes of gold, yttrium, rhenium, and dysprosium have been used to perform radiation synovectomies in patients with hemophilia. Yttrium-90, a pure beta emitter with adequate particle size and depth penetration, has been used successfully for the treatment of hemophilic synovitis.
The local (growth plate and articular cartilage) and remote effects of radiation are a concern. There have been no reported cases of growth plate disturbance after radiosynovectomy, even after the use of beta emitters such as gold-198.86 Articular cartilage is highly resistant to radiation, and although damage is theoretically possible, none has been reported. Progressive degeneration of treated joints does occur, but the rate is slower than that expected without radiosynovectomy. The principal concern is the potential for late, radiation-induced neoplasia. However, the safety of intra-articular radioisotopes is supported by a long-term follow-up study of more than 5000 RS procedures performed for rheumatoid arthritis, which found no reported radiation-induced malignancies.87
One review analyzed the safety of RS in pediatric patients with hemophilia to provide a risk-benefit assessment. During knee RS, patients receive a radiation dose of approximately 0.74 mSv, and during elbow and ankle RS, a dose of approximately 0.32 mSv. The radiation dose from natural sources is approximately 2 mSv per year and the recommended limit for patients (apart from natural sources) is 1 mSv per year. The lifetime cancer risk increases about 0.5% per 100 mSv per year. Considering the risks and benefits of RS, the authors recommend that clinicians consider this procedure in children with inhibitors or in patients without inhibitors when bleeding is recurrent and persistent despite aggressive factor replacement.88 External-beam radiation has been extensively studied and carries a small risk of osteosarcoma induction.
ACQUIRED INHIBITORS TO FACTOR VIII
Acquired hemophilia (AH) has an estimated prevalence of 1.48 cases per million per year, and a reported mortality between 9% and 22%.89,90 AH is uncommon in children younger than 16 years (prevalence estimated at 0.045/million/year), and may be underdiagnosed in persons older than age 85 (prevalence estimated at 14.7/million/year).89 In the largest published population series, 50% to 60% of diagnosed individuals were previously healthy with no identified underlying disease state.90–91 Underlying conditions consistently associated with AH include pregnancy, evolving or pre-existing autoimmune or malignant disorders, and rarely medications. Primary among the autoimmune disorders are collagen vascular disorders, including systemic lupus erythematosus, rheumatoid arthritis, myasthenia gravis, multiple sclerosis, and autoimmune hemolytic anemia. Most antibodies are mixtures of polyclonal IgG1 and IgG4 immunoglobulins, with the IgG4 molecules mainly responsible for inhibiting clotting activity. The clinical picture of AH is characterized by acute onset of severe bleeding in individuals who previously had no history of bleeding diathesis. Patients generally present with mucocutaneous bleeding (eg, epistaxis and gastrointestinal bleeding), as well as soft tissue bleeding (eg, extensive ecchymoses and hematomas).
The 2 major goals of treatment of AH are the immediate control of acute and chronic bleeding and the long-term suppression/eradication of the autoantibody inhibitor. For patients with an inhibitor titer < 5 BU/mL, administration of desmopressin and concentrates of human recombinant factor VIII may raise the factor VIII activity levels in plasma. If the inhibitor titer is > 5 BU/mL, or if bleeding persists despite infusions of factor VIII concentrates, then factor VIII bypassing agents, such as aPCCs or rFVIIa, are indicated. Local measures for treatment of mucosal hemorrhage, such as antifibrinolytic agents or topical fibrin glues, are helpful.
The primary aim in long-term management of AH is to eradicate the factor VIII autoantibodies so that further bleeding can be averted. Although in some clinical situations (postpartum women and drug-related AH) factor VIII antibodies may remit spontaneously, most published guidelines and algorithms recommend early initiation of eradication therapy. This is usually achieved through immunosuppressive medications or immunomodulation. Successful immunosuppression regimens in AH have most frequently used corticosteroids as the cornerstone, either as a single agent or in combination with cyclophosphamide. In a prospective randomized trial involving 31 participants treated with prednisone 1 mg/kg/day for 3 weeks, 32% achieved complete remission. In participants with antibody persistence after 3 weeks, switching to oral cyclophosphamide 2 mg/kg/day as second-line therapy appeared more effective than continuing prednisone (complete remission rate 50% versus 42%).92
Other immunosuppressive medications have been employed for eradication of refractory autoantibody inhibitors, including azathioprine, cyclosporine, tacrolimus, mycophenolate motefil, and sirolimus. Controlled studies have not been performed to confirm their comparative safety and efficacy in sufficiently large populations. Anti-CD20 antibody has been used to treat inhibitors in patients with both congenital and acquired hemophilia.93,94 Other less frequently used treatment options include administration of intravenous immunoglobulins (IVIG) in large doses. IVIG by itself rarely is able to induce a complete remission, but may be useful adjunctive therapy along with immunosuppressants, as part of an ITI regimen, or with extracorporeal plasmapheresis.
- Brinkhous KM. Clotting defect in hemophilia: deficiency in a plasma factor required for platelet utilization. Proc Soc Exp Biol Med 1947;66:117–20.
- Quick AJ. Studies on the enigma of the haemostatic dysfunction of hemophilia. Am J Med Sci 1947;214:272–80.
- Ingram JIC. The history of hemophilia. J Clin Path 1976;29:469–79.
- Gitschier J, Wood WI, Goralka TM, et al. Characterization of the human factor VIII gene. Nature 1984;312:326–30.
- Vehar GA, Keyt B, Eaton D, et al. Structure of human factor VIII. Nature 1984;312:337–42.
Toole JJ, Knopf JL, Wozney JM, et al. Molecular cloning of a cDNA encoding human antihaemophilic factor. Nature 1984;312:342–7.
Yoshitake S, Schach BG, Foster DC, et al. Nucleotide sequence of the gene for human factor IX (antihemophilic factor B). Biochemistry 1985;24:3736–50.
Vander VP, Giles AR. A detailed morphological evaluation of the evolution of the haemostatic plug in normal, factor VII and factor VIII deficient dogs. Br J Haematol 1988;70:345–55.
Gitschier J, Wood WI, Goralka TM, et al. Characterizartion of the human factor VIII gene. Nature 1984;312:326–30.
Toole JJ, Knopf JL, Wozney JM, et al. Molecular cloning of a cDNA encoding human antihaemophilic factor. Nature 1984;312:342–7.
Naylor J, Brinke A, Hassock S, Green PM, Gianelli F. Characteristic mRNA abnormality found in half the patients with severe haemophilia A is due to large DNA inversions. Hum Mol Genet 1993;2:1773–8.
Lakich D, Kazazian HH Jr., Antonarakis SE, Gitschier J. Inversions disrupting the factor VIII gene are a common cause of severe hemophilia A. Nat Genet 1993;5:236–41.
Stenson PD, Ball E, Howells K, et al. Gene mutation database. J Med Genet 2008;45:124–6.
Mauser Bunschoten EP, van Houwelingen JC, Sjamsoedin Visser EJM, et al. Bleeding symptoms in carriers of hemophilia A and B. Thromb Haemost 1988;59: 349–52.
Plug I, Mauser-Bunschoten EP, Bröcker-Vriends AH, et al. Bleeding in carriers of hemophilia. Blood 2006;108:52–6.
Nuss R, Soucie JM, Evatt B, and the Hemophilia Surveillance System Project Investigators. Changes in the occurrence of and risk factors for hemophilia-associated intracranial hemorrhage. Am J Hematol 2001;68:37–42.
Yoffe G, Buchanan G. Intracranial hemorrhage in newborn and young infants with hemophilia. J Pediatr 1988;113:333–6.
Roderick PJ, Robinson AC. Life-threatening oropharyngeal bleeding in a haemophiliac with factor VIII inhibitors. Clin Lab Haemat 1988;10:217–9.
Sites of initial bleeding episodes, mode of delivery and age of diagnosis in babies with haemophilia diagnosed before the age of 2 years: a report from the Centers for Disease Control and Prevention’s (CDC) Universal Data Collection (UDC) project. Hemophilia 2009; 15:1281–90.
Mittal R, Spero J, Lewis JH, Taylor F, et al. Patterns of gastrointestinal hemorrhage in hemophilia. Gastroenterol 1985;88:515–22.
Lander E, Pechlaner C, Mayr A, et al. Mallory-Weiss syndrome in a patient with hemophilia A and chronic liver disease. Ital J Gastroenterol 1995;27:73–4.
de la Fuente B, Kasper CK, Rickles FR, et al. Response of patients with mild and moderate hemophilia A and von Willebrand disease to treatment with desmopressin. Ann Intern Med 1985; 103:6–14.
Prowse CV, Sas G, Gader AM, et al. Specificity in the factor VIII response to vasopressin infusion in man. Br J Haematol 1979;41:437–47.
Rodeghiero F, Castaman G, Di Bona E, et al. Consistency of responses to repeated DDAVP infusions in patients with von Willebrand disease and hemophilia A. Blood 1989;74:1997–2000.
Warrier AI, Lusher JM. DDAVP: a useful alternative to blood components in moderate hemophilia A and von Willebrand disease. J Pediatr 1983;102:228–33.
Nilsson IM, Mikaelsson M, Vilhardt H. The effect of intranasal DDAVP on coagulation and fibrinolytic activity in normal persons. Scand J Haematol 1982;29:70–4.
Kogan A. Thrombin activatable fibrinolysis inhibitor (TAFI): a new marker of cardiovascular disease. Clinical Laboratory International. June 2004.
Vinckier F, Vermylen J. Dental extractions in hemophilia: reflections on 10 years’ experience. Oral Surg Oral Med Oral Pathol 1985;59:6–9.
Evans BE. The use of epsilon-aminocaproic acid for the management of hemophilia in dental and oral surgery patients. J Am Dent Assoc 1977;94:21.
Kaneshiro MM, Mielke CH Jr, Kasper CK, et al. Bleeding time after aspirin in disorders of intrinsic clotting. N Engl J Med 1969;281:1039–42.
Nilsson IM, Blomback M, Ahlberg A. Our experience in Sweden with prophylaxis on haemophilia. Bibl Haematol 1970;34:111–24.
Van Creveld S. Prophylaxis of joint hemorrhages in hemophilia. Acta Haematol 1969; 41:206–14.
Ljung R, Aronis-Vournas S, Kurnik-Auberger K, et al. Treatment of children with haemophilia in Europe: A survey of 20 centers in 16 countries. Haemophilia 2001;7:446–52.
Berntorp E, Astermark J, Bjorkman S, et al. Consensus perspectives on prophylactic therapy for haemophilia: Summary statement. Haemophilia 2003; 9(Suppl. 1):1–4.
Pollman H, Richter H, Ringkamp H, Jurgens H. When are children diagnosed as having severe hemophilia and when do they start to bleed? A 10-year single-center PUP study. Eur J Pediatr 1999;158:166–70.
Van Dijk K, Fischer K, Van Der Bom JG, et al. Variability in clinical phenotype of severe haemophilia: the role of the first joint bleed. Haemophilia 2005;11:438–43.
Kreuz W, Escuriola-Ettingshausen C, Funk M, et al. When should prophylactic treatment in patients with Hemophilia A and B start? The German experience. Hemophilia 1998;4:413–7.
Fischer K, Van der Bom JG, Mauser-Bunschoten EP, et al. Effects of postponing prophylactic treatment on long-term outcome in patients with severe Hemophilia. Blood 2002;99:2337–41.
Astermark J, Petrini P, Tengborn L, et al. Primary prophylaxis in severe hemophilia should be started at an early age but can be individualized. Br J Haematol 1999;105:1109–13.
Gringeri A, Lundin B, von mackensen S, et al; ESPRIT study group. A randomized clinical trial of prophylaxis in children with hemophilia A (the ESPRIT Study). J Thromb Haemost 2011;9:700–10.
Ingerslev J, Lethagen S, Hvitfeldt Poulsen L, et al. Long-standing prophylactic therapy vs. episodic treatment in young people with severe haemophilia: a comparison of age-matched Danish and Russian patients. Haemophilia 2014;20: 58–64.
Maco-Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med 2007;357:535–44.
Eloctate [package insert]. Cambridge [MA]: Biogen, Inc.
Lusher JM, Arkin S, Abildgaard CF, et al. Recombinant factor VIII for the treatment of previously untreated patients with henophilia A. N Engl J Med 1993;328:453–9.
Sultan Y, and the French Hemophilia Study Group. Prevalence of inhibitors in a population of 3435 hemophilia patients in France. Thromb Haemost 1992;67:600–2.
Rizza CR, Spooner RGD. Treatment of hemophilia and related disorders in Britain and Northern Ireland during 1976-80: report on behalf of the directors of hemophilia centers in the United Kingdom. Br Med J 1983;286:929–32.
Darby SC, Keeling DM, Spooner RJ, et al. The incidence of factor VIII and factor IX inhibitors in the hemophilia population of the UK and their effect on subsequent mortality, 1977-99. J Thromb Haemost 2004;2:1047–54.
Ehrenforth S, Kreuz W, Scharrer I, et al. Incidence of development of factor VIII and factor IX inhibitors in haemophiliacs. Lancet 1992;339:594–8.
Fijnvandraat K, Turenhout EAM, van den Brink EN, et al. The missense mutation Arg593à Cys is related to antibody formation in a patient with mild Hemophilia A. Blood 1997;89:4371–7.
Vlot AJ, Wittebol S, Strengers PFW, et al. Factor VIII inhibitor in a patient with mild Hemophilia A and an Asn618-Ser mutation responsive to immune tolerance induction and cyclophosphamide. Br J Hematolol 2002;117:136–40.
Santagostino E, Gringeri A, Tagliavacca L, et al. Inhibitors to factor VIII in a family with mild hemophilia: Molecular characterization and response to factor VIII and desmopressin. Throm Haemost 1995;74:61–21.
Peerlinck K, Jacquemin M, Arnout J, et al. Antifactor VIII antibody inhibiting allogenic but not autologous factor VIII in patients with mild hemophilia A. Blood 1999;93:2267–73.
Kesteven PJ, Holland LJ, Lawrie AS, et al. Inhibitor to factor VIII in mild hemophilia. Thromb Haemost 1984;52:50–2.
Gill JC. The role of genetics in inhibitor formation. Thromb Hemostat 1999;82:500–4.
Astermark J, Berntorp E, White GC, Kroner BL; MIBS Study group. The Malmo International Brother Study (MIBS): further support for genetic predisposition to inhibitor development in hemophilia patient. Hemophilia 2001;7:267–72.
Scharrer I, Bray GL, Neutzling O. Incidence of inhibitors in Hemophilia A patients- A review of recent studies of recombinant and plasma-derived factor VIII concentrates. Hemophilia 1999;5:145–54.
Carpenter SL, Michael Soucie J, Sterner S, Presley R; Hemophilia Treatment Center Network (HTCN) Investigators. Increased prevalence of inhibitors in Hispanic patients with severe haemophilia A enrolled in the Universal Data Collection databse. Haemophilia 2012;18:e260–5.
Goodeve AC, Peake IR. The molecular basis of hemophilia A: genotype-phenotype relationships and inhibitor development. Semin Thromb Haemost 2003;29:23–30.
Schwaab R, Brackman HH, Meyer C, et al. Hemophilia A: mutation type determines risk of inhibitor formation. Thromb Haemost 1995;74:1402–6.
Oldenburg J, El-Maari O, Schwaab R. Inhibitor development in correlation to Factor VIII genotypes. Hemophilia 2002;8(Suppl. 2):23–9.
Boekhorst J, Lari GR, D’oiron R, et al. Factor VIII genotype and inhibitor development in patients with hemophilia A: Highest risk in patients with splice site mutation. Haemophilia 2008;14:729–35.
Oldenburg J, Picard JK, Schwaab R, et al. HLA genotype of patients with severe hemophilia A due to intron 22 inversion with and without inhibitors of factor VIII. Thromb Haemost 1997;77:238–42.
Hay CR, Ollier W, Pepper L, et al. HLA class II profile: A weak determinant of factor VIII inhibitor development in severe hemophilia A. UKHCDO Inhibitor Working Party. Thromb Haemost 1997;77:234–7.
Astermark J, Olderburg J, Carlson J, et al. Polymorphisms in the TNFA gene and the risk of inhibitor development in severe Hemophilia A. Blood 2006;108:3739–45.
Astemark J, Olderburg J, Pavlova A, et al. Polymorphisms in the IL10 but not in the IL1Beta and IL4 genes are associated with inhibitor development in patients with Hemophilia A. Blood 2006;107:3167–72.
Astermark J, Wang X, Olderburg, et al. MIBS Study group. Polymorphisms in the CTLA-4 gene and inhibitor development in patients with Hemophilia A. J Throm Haemost 2007;5:263–5.
Hay CR, Ludlam CA, Colvin BT, et al. Factor VIII inhibitors in mild and moderate-severity hemophilia A. Thromb Haemost 1998;79:762–6.
High HA. Factor IX molecular structure, epitopes and mutations associated with inhibitor formation. In: Aledort LM, Hoyer LW, Lusher JM, et al, eds. Inhibitors to coagulation factors. New York: Plenum Press; 1995:79-86.
Thorland ED, Drost JB, Lusher JM, et al. Anaphylactic response to factor IX replacement therapy in hemophilia B patients: Complete gene deletions confer the highest risk. Hemophilia 1999;5:101–5.
Warrier I. ITI in hemophilia B: Possibilities and problems. International Monitor on Hemophilia 2003:20–3.
Warrier I, Ewenstein B, Koerper M, et al. FIX Inhibitors and anaphylaxis in hemophilia B. J Pediatr Hematol Oncol 1997;19:23–7.
Warrier I. Management of hemophilia B patients with inhibitors and anaphylaxis. In: Varon D, Martinowitz U, Heim M, eds. Haemophilia and related disorders. Vol. 4. Oxford: Blackwell Science; 1998:574–6.
Kasper CK, Aledort L, Aronson D, et al: Proceedings: a more uniform measurement of factor VIII inhibitors. Thromb Diath Haemorrh 1975;34:612.
White GC, Taylor RE, Blatt PM, et al. Treatment of a high titer anti–factor-VIII antibody by continuous factor VIII administration: report of a case. Blood 1983;62:141–5.
Key NS, Aledort LM, Beardsley D, et al. Home treatment of mild to moderate bleeding episodes using recombinant factor VIIa (Novoseven) in haemophiliacs with inhibitors. Thromb Haemost 1998;80:912–8.
Mariani G, Scheibel E, Nogao T, et al. Immune tolerance as treatment of alloantibodies to factor VIII in hemophilia. The international registry of Immunetolerance Protocols. Semin Hematol 1994;31(Suppl. 4):62–4.
DiMichele D, Kroner B. Factor VIII/IX Subcommittee of the International Society for Thrombosis and Hemostasis. The maintenance of tolerance after successful immune tolerance induction in hemophilia A and B. The North American Registry. Factor VIII/IX Subcommittee of the International Society for Thrombosis and Haemostasis. Haematologica 2000;85(Suppl. 10):40–4.
DiMichele DM, Hay CRM. The international immune tolerance study: A multicenter prospective randomized trial in progress. J Thromb Haemost 2006;4:2271–3000.
Astermark J, Morado M, Rocino A, et al. Current European practice in immune tolerance induction therapy in patients with hemophilia and inhibitors. Hemophilia 2006;12:363–71.
DiMichele DM, Hoots WK, Pipe SW, et al. International workshop on immune tolerance induction: Consensus recommendations. Hemophilia 2007;13:(Suppl. 1):1–22.
Hay CRM, Brown S, Collins PW, et al. The diagnosis and management of factor VIII and IX inhibitors: A guideline from the United Kingdom Center Doctors Organization. Br J Haematol 2006;133:591–605.
Valentino LA, Kempton CL, Kruse-Jarres R, et al. US Guidelines for immune tolerance induction in patients with haemophilia A and inhibitors. Haemophilia 2015;21:559–67.
Silva M, Luck JV Jr. Chronic hemophilic synovitis: the role of radiosynovectomy. World Federation of Hemophilia. Treatment of Hemophilia. April 2004 (no 33). http://www1.wfh.org/publications/files/pdf-1176.pdf.
Storti E, Traldi A, Tosatti E, Davoli PG. Synovectomy, a new approach to hemophilic arthropathy. Acta Haemotol 1969;41:193–205.
Caviglia HA, Fernandez-Palazzi F, Maffei E, et al. Chemical synoviorthesis for hemophilic synovitis. Clin Orthop 1997;343:30–6.
Ahlberg A, Pettersson H. Synoviorthesis with radioactive gold in hemophiliacs. Clinical and radiological follow-up. Acta Orthop Scand 1979;50:513–7.
Lee P. The efficacy and safety of radiosynovectomy. J Rheumatol 1982;9:165–8.
Rodriguez-Merchan EC, Valentino LA. Safety of radiation exposure after radiosynovectomy in paediatric patients with haemophilia. Haemophilia 2015;21:411–8.
Green D, Lechner K. A survey of 215 non-hemophilic patients with inhibitors to factor VIII. Thromb Haemost 1981;45:200–3.
Collins PW, Hirsch S, Baglin TP, et al. Acquired hemophilia A in the United Kingdom: A 2 year national surveillance study by the United Kingdom Haemophilia Center Doctors’ Organisation. Blood 2007;109:1870–7.
Kessler CM, Ludlam CA. The treatment of acquired factor VIII inhibitors: Worldwide experience with porcine factor VIII concentrate. International Acquired Hemophilia Study Group. Semin Hematol 1993;30(Suppl. 1):22–7.
Green D, Rademaker AW, Briet E. A prospective, randomized trial of prednisone and cyclophosphamide in the treatment of patients with factor VIII autoantibodies. Thromb Haemost 1993;70:753–7.
Stasi R, Brunetti M, Stipa E, Amadori S. Selective B-cell depletion with rituximab for the treatment of patients with acquired hemophilia. Blood 2004;103:4424–8.
Franchini M. Rituximab in the treatment of adult acquired hemophilia A: a systematic review. Crit Rev Oncol Hematol 2007;63:47–52.
INTRODUCTION
Hemophilia A and B are the most common severe inherited bleeding disorders. The incidence of hemophilia is 1 in 5000 live male births, with hemophilia A occurring 4 times more commonly than hemophilia B. The associated decrease in factor VIII in hemophilia A was initially identified in 1947, and the decrease in factor IX associated with hemophilia B was identified 5 years later.1,2 Both conditions are inherited as X-linked recessive traits. Queen Victoria of Britain, who reigned from 1837 to 1901, was a carrier of hemophilia and had 2 carrier daughters, Alice and Beatrice, and a son with hemophilia, Leopold.3 In 1984 and 1985, the genes for factor VIII and factor IX were cloned, and in 1989 recombinant factor VIII was first used clinically.4–7
PATHOPHYSIOLOGY
Both factors VIII and IX are crucial for normal thrombin generation in the coagulation pathway. After any injury, the initial hemostatic event is the formation of a platelet plug. Once the platelet plug is formed, subsequent generation of fibrin prevents continued oozing from the affected site. In hemophilia A and B, the propagation phase of coagulation is impaired, and as a result, the formation of clot is delayed and is not robust. Due to the delayed formation of an abnormal clot, patients with hemophilia do not bleed rapidly but rather ooze continuously. Rebleeding is a common occurrence in inadequately treated patients.8
GENETICS
The gene for factor VIII (F8) is located in the most distal band (Xq28) of the long arm of the X chromosome. Spanning more than 186 kb, it is one of the largest genes known.9,10 The gene for factor IX (F9) is located at Xq27.1 and spans 33 kb.7 Defects in the F8 gene associated with hemophilia A may be divided into several categories: gross gene rearrangements; insertions or deletions of genetic sequence of a size varying from 1 base pair up to the entire gene; or single DNA base substitutions resulting in either amino acid replacement (missense), premature peptide chain termination (nonsense, or stop mutations), or mRNA splicing defects. All classes of defects can result in severe disease. However, the single most clinically important defect is a gene rearrangement (an inversion) involving F8 intron 22, which results in approximately 50% of all severe hemophilia A cases worldwide.11,12 In hemophilia B, point mutations are by far the most common type of abnormality. Generally, they are caused by DNA polymerases adding the wrong nucleotide during replication.13
HEMOPHILIA IN FEMALES
X-Inactivation (also called Lyonization) is a process that occurs early in embryonic development in female mammals where 1 of the 2 copies of the X chromosome present is inactivated; it is the reason why some female carriers of hemophilia can become symptomatic. Approximately one third of carriers have clotting factor levels of less than 60% of normal and may experience abnormal bleeding.14,15 In most cases, carriers experience symptoms similar to those seen in men with mild hemophilia, as well as some that are specific to women. Symptomatic carriers and women with hemophilia may bruise more easily; may experience prolonged bleeding after surgery; may experience serious bleeding after trauma; often have heavier and more prolonged bleeding during their periods (menorrhagia) and are more likely to require an iron supplement or to undergo hysterectomy; and are more likely to have postpartum bleeding following childbirth.14,15
CLINICAL MANIFESTATIONS
Hemorrhage in patients with hemophilia may occur with minimal or unknown trauma. Patients with severe hemophilia (factor level of < 1 IU/dL or < 1% of normal) often experience spontaneous bleeding into joints or muscles. Those with moderate hemophilia (factor level of 1–5 IU/dL or 1%–5% of normal) seldom experience spontaneous hemorrhage and usually have prolonged bleeding with minor trauma or surgery. Patients with mild hemophilia (factor level > 5 IU/dL but less than 40 IU/dL or > 5% but < 40% of normal) experience severe hemorrhage only following moderate to severe trauma or surgery, and rarely experience spontaneous bleeding. Depending on the site, bleeding can be serious (joints; muscles, especially deep compartments [iliopsoas, calf, and forearm]; mucous membranes in the mouth, gums, nose, and genitourinary tract) or life-threatening (intracranial, neck/throat, gastrointestinal). The joints and muscles are the most common sites of bleeding (Table 1).
MUSCULOSKELETAL BLEEDING
The hallmark of hemophilia is deep bleeding into the joints and muscles. Without prophylactic factor treatment, patients with severe hemophilia A or B may have a bleeding episode as often as once or twice a week. Hemarthrosis episodes typically begin when the child reaches the toddler age. One of the first signs of hemarthrosis is a tingling sensation and feeling of warmth which is soon followed by pain and decreased range of motion of the joint as a result of distension of the joint capsule. Prompt, aggressive treatment with factor replacement therapy is the key to prevent further bleeding and minimize potential long-term complications. Severe chronic arthropathy may develop in older children and adults who have not received aggressive treatment (Figure).
Bleeding into the muscle can manifest as a vague feeling of pain on motion. Swelling may not be obvious and the mass may be difficult to palpate, although the circumference of the affected limb will be increased. Among the muscle bleeds, iliopsoas bleed deserves a special mention because of its potential to cause life-threatening hypovolemic shock as large volumes of blood can be lost into the retroperitoneal space. These patients present with vague abdominal pain or upper thigh discomfort. The hip is flexed and outwardly rotated. The diagnosis is confirmed by computed tomography (CT) or ultrasound.
LIFE-THREATENING HEMORRHAGE
Central Nervous System Bleeding
Most central nervous system (CNS) events, which involve bleeding inside the skull or spinal canal, are caused by trauma. CNS hemorrhage is the most common form of severe hemophilic trauma. However, since patients with hemophilia can experience bleeding even weeks after a minor head injury, a history of head trauma may be hard to determine, particularly in children. Spontaneous CNS bleeding in individuals with hemophilia is rare except when there has been a recent antecedent CNS hemorrhage (ie, a recurrent bleed at a previously injured site) or when there is an associated anatomic lesion that predisposes to acute hemorrhage (eg, aneurysm or arteriovenous malformation). Data from the Universal Data Collection Project of the U.S. Centers for Disease Control and Prevention indicates that predisposing risk factors for intracranial hemorrhage include HIV infection, presence of inhibitory antibodies, and age younger than 5 years or older than 51 years.16 Neonatal intracranial hemorrhage is most commonly due to birth trauma. Difficult vaginal deliveries (often requiring the application of forceps or vacuum extraction) are predisposing factors for intracranial hemorrhage in hemophilic newborns.
The site of intracranial CNS bleeding can be subdural, epidural, or intraparenchymal. Bleeding at any of these sites can cause rapidly deteriorating CNS brain function, associated brain swelling, and, in the most extreme circumstances, herniation of the brainstem and rapid death. If the bleeding is stopped with rapid clotting factor replacement, adverse clinical effects can be avoided. However, with intraparenchymal hemorrhage, even small hemorrhages can induce permanent structural and/or neurologic sequelae (in particular, if the anatomic site of the bleed is essential for routine brain function).17
Throat and Neck Hemorrhage
An acute neck injury or a retropharyngeal hemorrhage induced by dental or oral surgical instrumentation can lead to a dissecting facial plane hematoma. This in turn can sometimes lead to compression and acute airway compromise. Bleeding from these injuries that is compressing or compromising the airway may require a rapid clinical response.18 The time from the injury until the trachea is compressed may be long, sometimes many hours. However, once the compression is sufficient to cause difficulty breathing, there may be a short amount of time to stop the bleeding and prevent complete respiratory obstruction.
MUCOCUTANEOUS BLEEDING
One of the common manifestations of hemophilia is oral bleeding. Tooth extraction poses a specific problem, and bleeding following extraction can be the first symptom that leads to the diagnosis of hemophilia. Bleeding after circumcision may also suggest the diagnosis. In 1 study cohort looking at sites of initial bleeding episodes in babies with hemophilia diagnosed before the age of 2 years, bleeding from circumcision and other iatrogenic causes tended to be most common in the neonatal period. Circumcision bleeding events occurred more often in infants with no family history (43%) as compared to those born to known maternal carriers (9.2%) or to mothers with some other family history of hemophilia (14.3%).19
Gastrointestinal (GI) bleeding occurs occasionally in hemophilia, and a wide spectrum of esophageal and GI bleeding may occur. A review of 41 episodes of GI bleeding in hemophilia patients who presented to 1 institution over 10 years implicated duodenal ulcer (22%), unknown site (22%), and gastritis (14%) as the most common sources.20 Mallory-Weiss syndrome has also been cited as a cause for upper GI bleeding in hemophilia patients.21
PRINCIPLES OF TREATMENT
Understanding the pathophysiology of hemophilia as well as the type and severity of hemophilia and the inhibitor status in an individual patient are paramount in the management of a patient with hemophilia. In the past, management mainly focused on the treatment of acute bleeding episodes (Table 2). With data showing the benefit of bleed prevention, the management of hemophilia now focuses on prophylaxis of bleeding episodes, which prevents chronic arthropathy and improves quality of life.
ACUTE BLEEDING EPISODES
Dosing of Factor VIII Products
Dosing for factor VIII concentrate is as follows: 1 IU of factor VIII concentrate per kg will increase the circulating factor VIII level by 2% (ie, patient weight in kg × 50 IU/kg = 100% correction). For example, a 30-kg patient requiring 100% correction of factor VIII needs an infusion of 1500 IU of factor VIII (30 kg × 50 IU/kg).
Dosing of Factor IX Products
Dosing for factor IX concentrate is as follows: 1 IU of factor IX concentrate per kg will increase the circulating factor IX level by 1% (ie, patient weight in kg × 100 IU/kg = 100% correction). For example, a 30-kg patient requiring 100% correction of factor IX needs an infusion of 3000 IU of factor IX (30 kg × 100 IU/kg). Higher doses (120 to 130 IU/kg) of the recombinant factor IX product BeneFIX (Pfizer) may be needed to reach the 100% circulating factor IX level.
ADJUVANT THERAPY
Desmopressin
Desmopressin is a synthetic vasopressin analogue that increases plasma factor VIII and von Willebrand factor (VWF) levels; it is used to prevent and treat bleeding episodes associated with dental and surgical procedures in patients with mild and moderate hemophilia A and von Willebrand disease.22 Desmopressin causes the release of VWF and factor VIII from storage in the Weibel–Palade bodies of the endothelial cells that line the blood vessels. Individual response to desmopressin varies, with factor VIII level increasing between 2 and 15 times baseline level in patients with mild or moderate hemophilia A.23 It is therefore recommended that patients undergo a therapeutic trial of desmopressin with laboratory measurement of response to factor VIII before it is used for treatment of bleeding episodes or as prophylactic therapy before dental and other surgical procedures. A similar response is generally seen in an individual patient with subsequent doses, and thus the factor VIII level attained after a trial dose can be used to predict the response to future therapy.24
The recommended intravenous dosage of desmopressin is 0.3 µg/kg, administered in 25 to 50 mL of normal saline, over a period of 20 to 30 minutes.25 A concentrated form of desmopressin is available for intranasal administration to treat bleeding disorders. The appropriate dose of concentrated intranasal desmopressin is 150 µg (1 puff) for persons weighing less than 50 kg, and 300 µg (1 puff in each nostril) for persons weighing more than 50 kg.26
Antifibrinolytic Therapy
Antifibrinolytics (both epsilon-aminocaproic acid [EACA] and tranexamic acid) reversibly block the lysine binding sites of plasminogen, preventing its activation to plasmin and thus inhibiting the lysis of polymerized fibrin. EACA is also believed to stabilize the active form of thrombin activatable fibrinolysis inhibitor (TAFIa). It is believed that inactivation of TAFIa is due to conformational rearrangements in the TAFIa molecule; EACA has been shown to slow down spontaneous inactivation of TAFIa, thus curtailing fibrinolysis.27 Although hemostasis is generally achieved with either factor VIII replacement or desmopressin, the risk of recurrent bleeding from oral mucosal surfaces is dramatically reduced with the use of antifibrinolytic agents. These agents are typically contraindicated in patients with hematuria because they can cause a clot to form in the urinary bladder or ureters, leading to obstruction.
EACA is available in intravenous, oral tablet, and elixir formulations; the oral dose is 100 to 200 mg/kg initially (maximum dose, 10 g), followed by 50 to 100 mg/kg per dose every 6 hours (maximum dose, 5 g). Tranexamic acid is available in 650-mg capsules; the dose is 25 mg/kg every 6 to 8 hours.28,29 To treat spontaneous oral hemorrhage or to prevent bleeding from dental procedures in patients with hemophilia, either drug is usually begun in conjunction with desmopressin or factor replacement therapy immediately prior to the procedure and continued for up to 7 days or until mucosal healing is complete. Nonsteroidal anti-inflammatory drugs and aspirin affect platelet function and hence are contraindicated in affected individuals.30
PROPHYLAXIS
Patients with mild to moderate hemophilia typically bleed only after trauma, although the trauma needed to induce bleeding may be more minor than that which would cause bleeding in a normal individual. They usually do not suffer from significant morbidities, whereas patients with severe hemophilia often have spontaneous severe muscle and joint bleeds and can develop early crippling hemophilic arthropathy. Hence, routine prophylaxis has now become the standard of care in the United States and other developed countries in the management of patients with severe hemophilia. Prophylactic replacement therapy with cryoprecipitate in boys with severe hemophilia was first used nearly 50 years ago in Sweden31 and the Netherlands,32 and was shown to reduce the number and the severity of bleeds.32 Moreover, it was observed that early prophylaxis was more effective in preventing arthropathy compared to starting later in life, and that radiologic joint damage could not be reversed by prophylaxis. Subsequently, primary prophylaxis, defined as the start of regular, continuous treatment before the age of 2 years or after the occurrence of first joint bleed,33 was recommended and eventually became the standard treatment; it is currently recommended by the World Health Organization/World Federation of Hemophilia (WFH).34
The timing to begin prophylaxis is somewhat controversial, but many authors suggest starting prophylaxis before the first hemarthrosis occurs. Several studies have reported a wide variation in the age at first joint bleed, ranging from 0.2 to 5.8 years, with medians of 1.6 to 1.7 years.35,36 It has been suggested that arthropathy is best prevented if prophylaxis is started before the second or third joint bleed, but the benefits of starting before the occurrence of first bleed have not been established.37,38 The Swedish experience provides strong support for early prophylaxis.39 In an analysis of 121 patients with severe hemophilia, age at initiation of prophylaxis was an independent predictor of the development of arthropathy, but dose and interval of prophylaxis at the start of prophylactic treatment were not.39
In the Italian ESPRIT study, it was shown that children randomly assigned to prophylaxis had significantly fewer total bleeding episodes and joint bleeding episodes compared with those assigned to episodic therapy. Eleven of 21 patients (52%) in the prophylaxis group had on average less than 1 hemarthrosis per year, whereas only 4 of 19 patients in the episodic therapy group (21%) had the same low frequency of bleeding (P < 0.05).40 In a study of long-term prophylaxis versus on-demand treatment comparing age-matched Danish and Russian patients, the median annual number of joint bleeds in patients on prophylaxis was 1, while patients managed with on-demand treatment experienced a median of 37 joint bleeds. Patients taking prophylaxis also had a statistically significantly better quality of life estimate (P < 0.001) and better functional independence.41 In another trial, prophylaxis was initiated between the ages of 6 and 30 months based on a history of joint hemorrhage rather than age. Radiologic evidence of preserved joint architecture was found in 93% of participants in the prophylaxis group at 6 years of age. In this group, 18 of 32 (56%) children had 1 or 2 bleeds into one or more index joints before prophylaxis, and 17 (53%) had 1 to 5 hemorrhages into 1 or more index joints during prophylaxis. Prophylaxis was efficacious in decreasing bleeding and joint damage after up to 5 hemarthroses.42
Optimal Prophylactic Regimen
Although the benefits of prophylactic replacement therapy are firmly established, the optimal dose and frequency remain unclear. The half-life of clotting factor concentrates is short: about 8 hours for factor VIII in children, and about 12 hours for factor IX. As a result, prophylactic therapy is most effective when given frequently. The most common factor VIII concentrate dosing regimen for prophylaxis in hemophilia A is 25 to 40 IU/kg 3 times per week; for hemophilia B, a dose of 80 to 100 IU/kg is given twice weekly. This is aimed at a pre-infusion level > 1% to mimic the clinical phenotype of moderate hemophilia.
Recently, the US Food and Drug Administration (FDA) approved the first long-lasting antihemophilic factor (recombinant) Fc fusion protein for use in adults and children with hemophilia A. This medication contains the Fc region of human immunoglobulin G1 (IgG1), which binds to the neonatal Fc receptor (FcRn). FcRn is part of a naturally occurring pathway that delays lysosomal degradation of immunoglobulins by cycling them back into circulation and prolonging their plasma half-life. Dosing for routine prophylaxis is 50 IU/kg every 4 days; it may be adjusted based on patient response, with dosing in the range of 25 to 65 IU/kg at 3- to 5-day intervals. More frequent or higher doses up to 80 IU/kg may be required in children younger than 6 years.43
DEVELOPMENT OF INHIBITORS
FACTOR VIII INHIBITORS
Despite the success in the clinical management of hemophilia A, treated patients remain at risk for developing neutralizing antibodies that inhibit factor VIII activity. An inhibitor is a polyclonal high-affinity IgG that is directed against the factor VIII protein and renders exogenous factor ineffective. IgG4 antibodies are predominant and do not fix complement.
Risk Factors
The pathophysiology underlying the development of factor VIII inhibitors is a T-helper (Th)–cell dependent event that involves antigen-presenting cells and B lymphocytes; why only a fraction of patients experience this adverse effect of factor therapy is not known. Patients with mild/moderate hemophilia have a lower risk for inhibitor development than those with severe hemophilia A. The estimated prevalence of inhibitors ranges from 3% to 13% in mild to moderate disease,44–46 and up to 36% in severe hemophilia A.47,48 Usually the presence of an inhibitor in patients with mild/moderate hemophilia is suggested by a change in bleeding pattern: patients who previously used to bleed only after trauma or surgery suddenly start to experience severe spontaneous bleeding. This change in bleeding pattern is explained by cross-reactivity of the inhibitor with the mutated factor VIII of the patient, resulting in a residual factor level of < 0.01 IU/dL.49–51 Occasionally, there is no change in the residual factor VIII level but an inhibitor is detected in the Bethesda assay and/or there is lack of efficacy of factor VIII trans-fusions.51–53
Genetic factors. Data indicate that the risk of developing neutralizing antibodies is to a large extent determined by patient-related genetic factors.54,55 The immune response to factor VIII is similar in up to 80% of family members, significantly higher than expected compared with data from unrelated subjects. In a meta-analysis of patients with severe hemophilia A, the inhibitor incidence was twice as high in African American patients as compared with white patients.56 One study showed that patients of Hispanic ancestry with severe hemophilia A have a higher prevalence of neutralizing inhibitors than non-Hispanic white patients.57
Type of causative mutation. In severe hemophilia A, the risk of inhibitor formation is associated with the type of mutation. More disruptive mutations in the factor VIII gene, such as the intron 22 inversion, large gene deletions, and stop codons are associated with an approximately 35% risk of inhibitor formation, compared with only about 5% in those with missense mutation and small deletions.58 Persons with mutations involving large gene deletions, nonsense mutations, and intrachromosomal aberrations are usually at higher risk for the development of inhibitors than persons with missense mutations, small deletions/insertions, and splice site mutations.59,60 A relatively high risk is also encountered in patients with splicing errors and frame-shift mutations.61
Major histocompatibility complex. The HLA class I alleles A3, B7, and C7, as well as the class II alleles DQA0102, DQB0602, and DR15 have all been associated with a slightly higher risk for inhibitor development in unrelated patients, whereas the HLA C2, DQA0103, DQB0603, and DR13 alleles might be protective.62,63
Immune-regulatory molecules. In the Malmö International Brother Study, polymorphic sites in the genes coding for interleukin 10 (IL-10), tumor necrosis factor-α, and cytotoxic T lymphocyte–associated protein 4 were all associated with the risk of developing inhibitors.64–66 In this study, a 134 bp–long variant of a CAA microsatellite in the promoter region (IL-10.G) was identified in 26.8% of patients with hemophilia A. Thirty-two of these patients (72.7%) developed inhibitors as compared with 37.5% of those without the allele.65
Intensive exposure to factor VIII. Inhibitors in mild/moderate hemophilia seem to occur more commonly later in life, and an episode of intensive treatment with factor VIII concentrate has been reported to precede detection in most reported cases. In the series reported by Hay et al,67 16 out of 26 inhibitors were detected after such intensive replacement therapy, and no particular concentrate was implicated.
INHIBITORS TO FACTOR IX
Factor IX inhibitors are relatively uncommon, occurring in only 1% to 3% of persons with hemophilia B. This is in striking contrast to hemophilia A, where approximately 30% of patients develop inhibitors. The majority of patients with hemophilia B who develop inhibitors have severe hemophilia B.
Risk Factors
Certain mutations in the factor IX gene are associated with an increased incidence of inhibitor development. Large deletions and frame-shift mutations leading to the loss of coding information are much more likely to be associated with inhibitor development. Large deletions account for only 1% to 3% of all hemophilia B patients, but account for 50% of inhibitor patients.68 Patients with hemophilia B who develop inhibitors are at risk for developing anaphylactic reactions to factor IX–containing products. Anaphylaxis occurred more frequently in families with null mutations (large deletions, frame-shift mutations, or nonsense mutations) than in those with missense mutations.69 With hemophilia A, approximately 40% to 50% of black individuals develop inhibitors, but no such association has been found in hemophilia B. Individuals who develop an inhibitor to factor IX do so relatively early in life (within the first 4 to 5 years), after a median of 9 to 11 exposure days to any factor IX–containing products. Because of the severity of a potential anaphylactic reaction occurring early in life after very few exposures to factor IX, all infants and small children with severe hemophilia B should be closely followed over their first 10 infusions with any factor IX–containing products in a facility equipped to treat anaphylactic shock.70–72 A comparison of inhibitors in hemophilia A and B is shown in Table 3.
TREATMENT OF ACUTE BLEEDS IN PATIENTS WITH FACTOR VIII INHIBITORS
The available therapeutic agents for treatment of acute hemorrhage in children with hemophilia A with an inhibitor include high-dose recombinant or plasma-derived factor VIII concentrate, activated prothrombin complex concentrates (aPCCs), and recombinant activated factor VII (rFVIIa). In addition, antifibrinolytics may be used as an adjunct therapy.
Patient response to each treatment varies widely, with some patients responding well to one treatment and less well to another. Neither the patient's history nor standard lab tests can assist in making the best choice for the patient. A personalized approach to factor selection is used, and the dosing of that particular agent is often determined primarily by clinical assessment. Inhibitors are quantitated using the Bethesda inhibitor assay and clinically are classified as low- and high-responding inhibitors (Table 4). Inhibitor screening should be done prior to invasive procedures and periodically during the first 50 days of treatment since the risk for inhibitor development is highest during this period.
Low-Responding Inhibitors
A low-responding inhibitor is one in which inhibitor titers are < 5 Bethesda units (BU)/mL; patients with low-responding inhibitors can generally be treated with factor VIII concentrates at higher doses.73 Because the effect of factor VIII inhibitor is usually delayed, the Bethesda titer in plasma is determined after a 2-hour incubation period. As a result of this time delay, continuous administration of factor VIII is usually found to be effective.74 For a serious limb- or life-threatening bleeding episode, a bolus infusion of 100 IU of factor VIII per kg of body weight is administered, and the level is maintained by treatment at a rate of 20 IU/kg/hr. An assay for factor VIII should be performed 1 hour after the bolus infusion and at least daily thereafter. As the antibody titer drops, the daily level of factor VIII may rise and thus downward adjustment of the continuous infusion rate may be required. For routine joint and muscle hemorrhage, patients can usually be managed with infusions at twice the usual dosage. Routine inhibitor assays should be performed after exposure to factor VIII to determine whether an anamnestic response has occurred.
High-Responding Inhibitors
Most clinicians caring for patients with limb- or life-threatening bleeding episodes prefer to use products for which therapeutic levels can be monitored. As described earlier, continuous admin-istration of factor VIII is often effective because of the time delay in inhibition by the antibody. An initial dose of 100 to 200 IU/kg can be administered, and factor VIII levels can be determined 1 hour after initiation of continuous infusion at a rate of 20 to 40 IU/kg/hr. If a factor VIII level cannot be obtained (ie, patients with inhibitor titers > 5 to 10 BU/mL), alternative approaches include the bypassing agents aPCC and rFVIIa.
First used in the 1970s, aPCCs represented a significant improvement in the management in patients with hemophilia with inhibitors. They contain multiple activated serine protease molecules; activated factor X and prothrombin are the main active components in FEIBA (factor eight inhibitor bypassing activity), the most commonly used aPCC in the United States. FEIBA is a pooled plasma product that contains activated factors II, VII, IX and X, and has a duration of action of about 6 to 12 hours. For treatment of acute bleeds, the recommended dose of FEIBA is 50 to 100 IU/kg infused every 8 to 12 hours (maximum daily dose of 200 IU/kg). There is a risk of thrombosis/disseminated intravascular coagulation (DIC) with very large doses given frequently (> 200 IU/kg/day).
rFVIIa directly activates factor X and increases thrombin production on the surface of activated platelets in the absence of factor VIII or factor IX. Standard dosing of rFVIIa is 90 to 120 µg/kg, and many hemophilia treatment centers use higher doses (270 µg/kg/dose), especially in children and young adults. The half-life is about 1.5 to 3 hours, and therefore frequent administration (every 2–6 hours) is required. In one study that assessed the safety and efficacy of fixed-dose rFVIIa in the home setting, hemostasis was achieved in 566 (92%) of evaluable bleeding episodes, and following administration of the additional maintenance dose, hemostasis was maintained in 95% of successfully treated cases.75 As with aPCCs, there is no standardized quantitative laboratory test for measuring the effectiveness of rFVIIa therapy.
All currently used bypassing agents are associated with a risk of thrombotic complications including thromboembolism, DIC, and myocardial infarction. These complications are very rare in patients with hemophilia, however. In general, bypassing agents work for most bleeds and for most patients, but are not as predictable as factor replacement therapy and cannot be monitored by laboratory assays.
TREATMENT OF ACUTE BLEEDS IN PATIENTS WITH FACTOR IX INHIBITORS
rFVIIa and FEIBA are the mainstays of treatment of bleeding episodes in individuals with hemophilia B complicated by an inhibitor to factor IX. Treatment of hemorrhagic episodes in these patients depends on the type of bleeding episode experienced, the inhibitor classification (high- versus low-responding [Table 4]), and the history and severity of infusion reactions. Patients with low-responding inhibitors who have not experienced infusion reactions may be treated with doses of factor IX concentrate calculated to overcome the inhibitor titer and achieve a hemostatic level. In patients with high-responding inhibitors, the use of factor IX concentrates is impractical because of the inhibitor titer or the anamnestic response. Regardless of inhibitor titer, in patients with a history of an anaphylactic event, factor IX usage is contraindicated.
The most commonly used therapy for hemostatic control in patients with high-responding inhibitors with factor IX deficiency and a history of infusion reaction is rFVIIa; the standard dosing regimen is 90 to 120 µg/kg/dose administered every 2 to 3 hours, with a maximum dose of 270 µg/kg/dose. aPCCs, which contain factor IX, can be utilized if the patient has not experienced prior infusion reactions. Repeated exposures to products containing factor IX may stimulate the inhibitor titer and prevent its natural decline over time. This can pose a problem in cases of life- or limb-threatening hemorrhage unresponsive to rFVIIa as these patients will not have factor IX available as an effective mode of therapy. The dosing of FEIBA ranges from 50 to 100 IU/kg every 12 hours, with daily dosing not to exceed 200 IU/kg.
IMMUNE TOLERANCE INDUCTION
Because of the associated inhibitor-related morbidity resulting from limited treatment options, antibody eradication is the ultimate goal in inhibitor management. The only proven strategy for achieving antigen-specific tolerance to factor VIII or factor IX is immune tolerance induction (ITI) therapy. Successful ITI in hemophilia A is currently defined as both an undetectable inhibitor titer (< 0.6 BU), and normalized factor VIII pharmacokinetics, which in turn is defined as plasma factor VIII recovery > 66% of expected and a half-life > 6 hours, determined following a 72-hour factor VIII exposure-free period (Consensus Proceedings from the Second International Conference on Immune Tolerance Therapy, Bonn, Germany, 1997 [unpublished]). Once successful immune tolerance is achieved, long-term prophylaxis is commonly instituted. Using conclusions drawn from international consensus criteria and analysis of the International Immune Tolerance Registry, the I-ITI study has defined ITI failure by the presence of either of 2 criteria:
1. Failure to attain the definition of success within 33 months of uninterrupted ITI;
2. Failure to demonstrate a progressive 20% reduction in inhibitor titer over each 6-month period of uninterrupted ITI, beginning 3 months after initiation to allow for expected anamnesis.76–78
This definition implies a minimum ITI trial period of 9 months before failure is declared.
The European Hemophilia Standardization Board (EHSB), the International Consensus Panel (ICP), and the United Kingdom Hemophilia Center Doctors’ Organization (UKHCDO) have agreed that it is preferable to initiate ITI at a titer of < 10 BU/mL, unless, per the ICP, the titer does not decline over a period of 1 to 2 years and/or inhibitor development is associated with severe or life-threatening bleeding. The ICP noted that for “poor-risk” ITI patients (defined by a historical titer of > 200 BU/mL and/or a pre-ITI inhibitor titer of > 10 BU/mL and/or an interval of > 5 years since inhibitor diagnosis), published efficacy data are limited to dosing regimens > 200 IU/kg/day. The groups all independently concluded that ITI has been successfully performed using recombinant and plasma-derived factor VIII replacement therapy (usually the product on which they developed the inhibitor), and that there are no data to support the superiority of any single product type.79–81 However, both EHSB and ICP have suggested that VWF-containing concentrates be considered for patients who fail ITI using high-purity factor VIII.79,80
The recommendations from US guidelines for ITI in patients with hemophilia A and inhibitors are listed in Table 5.82
ARTHROPATHY
Before the advent of factor products for the treatment of hemophilia, hemarthrosis was one of the leading causes of morbidity. Today, the routine use of prophylactic treatment has resulted in a significant improvement in the lifestyle, quality of life, and life expectancy of these patients. However, despite best efforts, some patients will have severe joint destruction as a result of repeated articular bleeding episodes during their early years. This leads to pain and significant functional disability, thus impairing the quality of life. The basic pathology behind hemophilic arthropathy is chronic synovitis.
It is common to observe a pattern of repeated bleeding (chronic hemarthrosis), especially in patients with severe hemophilia, that can lead to chronic synovitis, inflammatory arthritis, and progressive arthropathy. Therefore, the key to preventing hemophilic arthropathy is aggressive management of the initial hemarthrosis. This is generally accomplished with the use of clotting factor replacement, restorative physiotherapy, and close clinical follow-up. If chronic synovitis develops, synovectomy may be considered in order to slow the progression of the hemophilic arthropathy and to prevent the development of major articular surface erosions that can lead to end-stage arthropathy.83 Primary prophylaxis is discussed earlier and is the mainstay of prevention of chronic hemophilic arthropathy.
SYNOVECTOMY
The emergence of chronic hemophilic hemarthrosis is incited by a hypertrophic and highly vascular synovium. Removal of the synovium prevents further joint damage,84 and can be accomplished through surgical and nonsurgical procedures.
Surgical excision of the hypertrophic synovium can be performed through open or arthroscopic procedures. The open approach has largely been replaced by arthroscopic synovectomies. Regardless of the approach, these patients need prolonged hospitalization, extensive factor replacement, and exhaustive physiotherapy. Moreover, patients with inhibitors are usually not considered candidates for surgical synovectomy.
Chemical and radioactive agents injected intra-articularly can decrease the volume and activity of the synovial tissue. Due to the minimally invasive nature of these procedures, nonsurgical synovectomies are of special importance for hemophilic patients with inhibitors to clotting factors.
Chemical Synovectomy
Chemical synovectomies, using thiotepa, osmic acid, D-penicillamine and other agents, have been used in the distant past. Rifampicin, which is used an antibiotic, is now the most commonly used chemical for the purpose of synovectomy, and the one that has shown better results in terms of decreasing hemarthrosis.85 Each one of the injections should be accompanied by prophylactic administration of clotting factor concentrate. Excellent results (no synovitis and restoration of previous function) have been reported in up to 83% of patients at an average of 2.4 years after the intra-articular injection of rifampicin. As the pathology of the joint becomes more severe, however, the number of injections required to achieve improvement increases. Younger patients and smaller joints benefit more from this procedure.
Radiation Synovectomy
Radiosynovectomy (RS) and radiosynoviorthesis are common terms used to describe the synovial ablation accomplished by intra-articular injection of radioisotopes. Isotopes of gold, yttrium, rhenium, and dysprosium have been used to perform radiation synovectomies in patients with hemophilia. Yttrium-90, a pure beta emitter with adequate particle size and depth penetration, has been used successfully for the treatment of hemophilic synovitis.
The local (growth plate and articular cartilage) and remote effects of radiation are a concern. There have been no reported cases of growth plate disturbance after radiosynovectomy, even after the use of beta emitters such as gold-198.86 Articular cartilage is highly resistant to radiation, and although damage is theoretically possible, none has been reported. Progressive degeneration of treated joints does occur, but the rate is slower than that expected without radiosynovectomy. The principal concern is the potential for late, radiation-induced neoplasia. However, the safety of intra-articular radioisotopes is supported by a long-term follow-up study of more than 5000 RS procedures performed for rheumatoid arthritis, which found no reported radiation-induced malignancies.87
One review analyzed the safety of RS in pediatric patients with hemophilia to provide a risk-benefit assessment. During knee RS, patients receive a radiation dose of approximately 0.74 mSv, and during elbow and ankle RS, a dose of approximately 0.32 mSv. The radiation dose from natural sources is approximately 2 mSv per year and the recommended limit for patients (apart from natural sources) is 1 mSv per year. The lifetime cancer risk increases about 0.5% per 100 mSv per year. Considering the risks and benefits of RS, the authors recommend that clinicians consider this procedure in children with inhibitors or in patients without inhibitors when bleeding is recurrent and persistent despite aggressive factor replacement.88 External-beam radiation has been extensively studied and carries a small risk of osteosarcoma induction.
ACQUIRED INHIBITORS TO FACTOR VIII
Acquired hemophilia (AH) has an estimated prevalence of 1.48 cases per million per year, and a reported mortality between 9% and 22%.89,90 AH is uncommon in children younger than 16 years (prevalence estimated at 0.045/million/year), and may be underdiagnosed in persons older than age 85 (prevalence estimated at 14.7/million/year).89 In the largest published population series, 50% to 60% of diagnosed individuals were previously healthy with no identified underlying disease state.90–91 Underlying conditions consistently associated with AH include pregnancy, evolving or pre-existing autoimmune or malignant disorders, and rarely medications. Primary among the autoimmune disorders are collagen vascular disorders, including systemic lupus erythematosus, rheumatoid arthritis, myasthenia gravis, multiple sclerosis, and autoimmune hemolytic anemia. Most antibodies are mixtures of polyclonal IgG1 and IgG4 immunoglobulins, with the IgG4 molecules mainly responsible for inhibiting clotting activity. The clinical picture of AH is characterized by acute onset of severe bleeding in individuals who previously had no history of bleeding diathesis. Patients generally present with mucocutaneous bleeding (eg, epistaxis and gastrointestinal bleeding), as well as soft tissue bleeding (eg, extensive ecchymoses and hematomas).
The 2 major goals of treatment of AH are the immediate control of acute and chronic bleeding and the long-term suppression/eradication of the autoantibody inhibitor. For patients with an inhibitor titer < 5 BU/mL, administration of desmopressin and concentrates of human recombinant factor VIII may raise the factor VIII activity levels in plasma. If the inhibitor titer is > 5 BU/mL, or if bleeding persists despite infusions of factor VIII concentrates, then factor VIII bypassing agents, such as aPCCs or rFVIIa, are indicated. Local measures for treatment of mucosal hemorrhage, such as antifibrinolytic agents or topical fibrin glues, are helpful.
The primary aim in long-term management of AH is to eradicate the factor VIII autoantibodies so that further bleeding can be averted. Although in some clinical situations (postpartum women and drug-related AH) factor VIII antibodies may remit spontaneously, most published guidelines and algorithms recommend early initiation of eradication therapy. This is usually achieved through immunosuppressive medications or immunomodulation. Successful immunosuppression regimens in AH have most frequently used corticosteroids as the cornerstone, either as a single agent or in combination with cyclophosphamide. In a prospective randomized trial involving 31 participants treated with prednisone 1 mg/kg/day for 3 weeks, 32% achieved complete remission. In participants with antibody persistence after 3 weeks, switching to oral cyclophosphamide 2 mg/kg/day as second-line therapy appeared more effective than continuing prednisone (complete remission rate 50% versus 42%).92
Other immunosuppressive medications have been employed for eradication of refractory autoantibody inhibitors, including azathioprine, cyclosporine, tacrolimus, mycophenolate motefil, and sirolimus. Controlled studies have not been performed to confirm their comparative safety and efficacy in sufficiently large populations. Anti-CD20 antibody has been used to treat inhibitors in patients with both congenital and acquired hemophilia.93,94 Other less frequently used treatment options include administration of intravenous immunoglobulins (IVIG) in large doses. IVIG by itself rarely is able to induce a complete remission, but may be useful adjunctive therapy along with immunosuppressants, as part of an ITI regimen, or with extracorporeal plasmapheresis.
INTRODUCTION
Hemophilia A and B are the most common severe inherited bleeding disorders. The incidence of hemophilia is 1 in 5000 live male births, with hemophilia A occurring 4 times more commonly than hemophilia B. The associated decrease in factor VIII in hemophilia A was initially identified in 1947, and the decrease in factor IX associated with hemophilia B was identified 5 years later.1,2 Both conditions are inherited as X-linked recessive traits. Queen Victoria of Britain, who reigned from 1837 to 1901, was a carrier of hemophilia and had 2 carrier daughters, Alice and Beatrice, and a son with hemophilia, Leopold.3 In 1984 and 1985, the genes for factor VIII and factor IX were cloned, and in 1989 recombinant factor VIII was first used clinically.4–7
PATHOPHYSIOLOGY
Both factors VIII and IX are crucial for normal thrombin generation in the coagulation pathway. After any injury, the initial hemostatic event is the formation of a platelet plug. Once the platelet plug is formed, subsequent generation of fibrin prevents continued oozing from the affected site. In hemophilia A and B, the propagation phase of coagulation is impaired, and as a result, the formation of clot is delayed and is not robust. Due to the delayed formation of an abnormal clot, patients with hemophilia do not bleed rapidly but rather ooze continuously. Rebleeding is a common occurrence in inadequately treated patients.8
GENETICS
The gene for factor VIII (F8) is located in the most distal band (Xq28) of the long arm of the X chromosome. Spanning more than 186 kb, it is one of the largest genes known.9,10 The gene for factor IX (F9) is located at Xq27.1 and spans 33 kb.7 Defects in the F8 gene associated with hemophilia A may be divided into several categories: gross gene rearrangements; insertions or deletions of genetic sequence of a size varying from 1 base pair up to the entire gene; or single DNA base substitutions resulting in either amino acid replacement (missense), premature peptide chain termination (nonsense, or stop mutations), or mRNA splicing defects. All classes of defects can result in severe disease. However, the single most clinically important defect is a gene rearrangement (an inversion) involving F8 intron 22, which results in approximately 50% of all severe hemophilia A cases worldwide.11,12 In hemophilia B, point mutations are by far the most common type of abnormality. Generally, they are caused by DNA polymerases adding the wrong nucleotide during replication.13
HEMOPHILIA IN FEMALES
X-Inactivation (also called Lyonization) is a process that occurs early in embryonic development in female mammals where 1 of the 2 copies of the X chromosome present is inactivated; it is the reason why some female carriers of hemophilia can become symptomatic. Approximately one third of carriers have clotting factor levels of less than 60% of normal and may experience abnormal bleeding.14,15 In most cases, carriers experience symptoms similar to those seen in men with mild hemophilia, as well as some that are specific to women. Symptomatic carriers and women with hemophilia may bruise more easily; may experience prolonged bleeding after surgery; may experience serious bleeding after trauma; often have heavier and more prolonged bleeding during their periods (menorrhagia) and are more likely to require an iron supplement or to undergo hysterectomy; and are more likely to have postpartum bleeding following childbirth.14,15
CLINICAL MANIFESTATIONS
Hemorrhage in patients with hemophilia may occur with minimal or unknown trauma. Patients with severe hemophilia (factor level of < 1 IU/dL or < 1% of normal) often experience spontaneous bleeding into joints or muscles. Those with moderate hemophilia (factor level of 1–5 IU/dL or 1%–5% of normal) seldom experience spontaneous hemorrhage and usually have prolonged bleeding with minor trauma or surgery. Patients with mild hemophilia (factor level > 5 IU/dL but less than 40 IU/dL or > 5% but < 40% of normal) experience severe hemorrhage only following moderate to severe trauma or surgery, and rarely experience spontaneous bleeding. Depending on the site, bleeding can be serious (joints; muscles, especially deep compartments [iliopsoas, calf, and forearm]; mucous membranes in the mouth, gums, nose, and genitourinary tract) or life-threatening (intracranial, neck/throat, gastrointestinal). The joints and muscles are the most common sites of bleeding (Table 1).
MUSCULOSKELETAL BLEEDING
The hallmark of hemophilia is deep bleeding into the joints and muscles. Without prophylactic factor treatment, patients with severe hemophilia A or B may have a bleeding episode as often as once or twice a week. Hemarthrosis episodes typically begin when the child reaches the toddler age. One of the first signs of hemarthrosis is a tingling sensation and feeling of warmth which is soon followed by pain and decreased range of motion of the joint as a result of distension of the joint capsule. Prompt, aggressive treatment with factor replacement therapy is the key to prevent further bleeding and minimize potential long-term complications. Severe chronic arthropathy may develop in older children and adults who have not received aggressive treatment (Figure).
Bleeding into the muscle can manifest as a vague feeling of pain on motion. Swelling may not be obvious and the mass may be difficult to palpate, although the circumference of the affected limb will be increased. Among the muscle bleeds, iliopsoas bleed deserves a special mention because of its potential to cause life-threatening hypovolemic shock as large volumes of blood can be lost into the retroperitoneal space. These patients present with vague abdominal pain or upper thigh discomfort. The hip is flexed and outwardly rotated. The diagnosis is confirmed by computed tomography (CT) or ultrasound.
LIFE-THREATENING HEMORRHAGE
Central Nervous System Bleeding
Most central nervous system (CNS) events, which involve bleeding inside the skull or spinal canal, are caused by trauma. CNS hemorrhage is the most common form of severe hemophilic trauma. However, since patients with hemophilia can experience bleeding even weeks after a minor head injury, a history of head trauma may be hard to determine, particularly in children. Spontaneous CNS bleeding in individuals with hemophilia is rare except when there has been a recent antecedent CNS hemorrhage (ie, a recurrent bleed at a previously injured site) or when there is an associated anatomic lesion that predisposes to acute hemorrhage (eg, aneurysm or arteriovenous malformation). Data from the Universal Data Collection Project of the U.S. Centers for Disease Control and Prevention indicates that predisposing risk factors for intracranial hemorrhage include HIV infection, presence of inhibitory antibodies, and age younger than 5 years or older than 51 years.16 Neonatal intracranial hemorrhage is most commonly due to birth trauma. Difficult vaginal deliveries (often requiring the application of forceps or vacuum extraction) are predisposing factors for intracranial hemorrhage in hemophilic newborns.
The site of intracranial CNS bleeding can be subdural, epidural, or intraparenchymal. Bleeding at any of these sites can cause rapidly deteriorating CNS brain function, associated brain swelling, and, in the most extreme circumstances, herniation of the brainstem and rapid death. If the bleeding is stopped with rapid clotting factor replacement, adverse clinical effects can be avoided. However, with intraparenchymal hemorrhage, even small hemorrhages can induce permanent structural and/or neurologic sequelae (in particular, if the anatomic site of the bleed is essential for routine brain function).17
Throat and Neck Hemorrhage
An acute neck injury or a retropharyngeal hemorrhage induced by dental or oral surgical instrumentation can lead to a dissecting facial plane hematoma. This in turn can sometimes lead to compression and acute airway compromise. Bleeding from these injuries that is compressing or compromising the airway may require a rapid clinical response.18 The time from the injury until the trachea is compressed may be long, sometimes many hours. However, once the compression is sufficient to cause difficulty breathing, there may be a short amount of time to stop the bleeding and prevent complete respiratory obstruction.
MUCOCUTANEOUS BLEEDING
One of the common manifestations of hemophilia is oral bleeding. Tooth extraction poses a specific problem, and bleeding following extraction can be the first symptom that leads to the diagnosis of hemophilia. Bleeding after circumcision may also suggest the diagnosis. In 1 study cohort looking at sites of initial bleeding episodes in babies with hemophilia diagnosed before the age of 2 years, bleeding from circumcision and other iatrogenic causes tended to be most common in the neonatal period. Circumcision bleeding events occurred more often in infants with no family history (43%) as compared to those born to known maternal carriers (9.2%) or to mothers with some other family history of hemophilia (14.3%).19
Gastrointestinal (GI) bleeding occurs occasionally in hemophilia, and a wide spectrum of esophageal and GI bleeding may occur. A review of 41 episodes of GI bleeding in hemophilia patients who presented to 1 institution over 10 years implicated duodenal ulcer (22%), unknown site (22%), and gastritis (14%) as the most common sources.20 Mallory-Weiss syndrome has also been cited as a cause for upper GI bleeding in hemophilia patients.21
PRINCIPLES OF TREATMENT
Understanding the pathophysiology of hemophilia as well as the type and severity of hemophilia and the inhibitor status in an individual patient are paramount in the management of a patient with hemophilia. In the past, management mainly focused on the treatment of acute bleeding episodes (Table 2). With data showing the benefit of bleed prevention, the management of hemophilia now focuses on prophylaxis of bleeding episodes, which prevents chronic arthropathy and improves quality of life.
ACUTE BLEEDING EPISODES
Dosing of Factor VIII Products
Dosing for factor VIII concentrate is as follows: 1 IU of factor VIII concentrate per kg will increase the circulating factor VIII level by 2% (ie, patient weight in kg × 50 IU/kg = 100% correction). For example, a 30-kg patient requiring 100% correction of factor VIII needs an infusion of 1500 IU of factor VIII (30 kg × 50 IU/kg).
Dosing of Factor IX Products
Dosing for factor IX concentrate is as follows: 1 IU of factor IX concentrate per kg will increase the circulating factor IX level by 1% (ie, patient weight in kg × 100 IU/kg = 100% correction). For example, a 30-kg patient requiring 100% correction of factor IX needs an infusion of 3000 IU of factor IX (30 kg × 100 IU/kg). Higher doses (120 to 130 IU/kg) of the recombinant factor IX product BeneFIX (Pfizer) may be needed to reach the 100% circulating factor IX level.
ADJUVANT THERAPY
Desmopressin
Desmopressin is a synthetic vasopressin analogue that increases plasma factor VIII and von Willebrand factor (VWF) levels; it is used to prevent and treat bleeding episodes associated with dental and surgical procedures in patients with mild and moderate hemophilia A and von Willebrand disease.22 Desmopressin causes the release of VWF and factor VIII from storage in the Weibel–Palade bodies of the endothelial cells that line the blood vessels. Individual response to desmopressin varies, with factor VIII level increasing between 2 and 15 times baseline level in patients with mild or moderate hemophilia A.23 It is therefore recommended that patients undergo a therapeutic trial of desmopressin with laboratory measurement of response to factor VIII before it is used for treatment of bleeding episodes or as prophylactic therapy before dental and other surgical procedures. A similar response is generally seen in an individual patient with subsequent doses, and thus the factor VIII level attained after a trial dose can be used to predict the response to future therapy.24
The recommended intravenous dosage of desmopressin is 0.3 µg/kg, administered in 25 to 50 mL of normal saline, over a period of 20 to 30 minutes.25 A concentrated form of desmopressin is available for intranasal administration to treat bleeding disorders. The appropriate dose of concentrated intranasal desmopressin is 150 µg (1 puff) for persons weighing less than 50 kg, and 300 µg (1 puff in each nostril) for persons weighing more than 50 kg.26
Antifibrinolytic Therapy
Antifibrinolytics (both epsilon-aminocaproic acid [EACA] and tranexamic acid) reversibly block the lysine binding sites of plasminogen, preventing its activation to plasmin and thus inhibiting the lysis of polymerized fibrin. EACA is also believed to stabilize the active form of thrombin activatable fibrinolysis inhibitor (TAFIa). It is believed that inactivation of TAFIa is due to conformational rearrangements in the TAFIa molecule; EACA has been shown to slow down spontaneous inactivation of TAFIa, thus curtailing fibrinolysis.27 Although hemostasis is generally achieved with either factor VIII replacement or desmopressin, the risk of recurrent bleeding from oral mucosal surfaces is dramatically reduced with the use of antifibrinolytic agents. These agents are typically contraindicated in patients with hematuria because they can cause a clot to form in the urinary bladder or ureters, leading to obstruction.
EACA is available in intravenous, oral tablet, and elixir formulations; the oral dose is 100 to 200 mg/kg initially (maximum dose, 10 g), followed by 50 to 100 mg/kg per dose every 6 hours (maximum dose, 5 g). Tranexamic acid is available in 650-mg capsules; the dose is 25 mg/kg every 6 to 8 hours.28,29 To treat spontaneous oral hemorrhage or to prevent bleeding from dental procedures in patients with hemophilia, either drug is usually begun in conjunction with desmopressin or factor replacement therapy immediately prior to the procedure and continued for up to 7 days or until mucosal healing is complete. Nonsteroidal anti-inflammatory drugs and aspirin affect platelet function and hence are contraindicated in affected individuals.30
PROPHYLAXIS
Patients with mild to moderate hemophilia typically bleed only after trauma, although the trauma needed to induce bleeding may be more minor than that which would cause bleeding in a normal individual. They usually do not suffer from significant morbidities, whereas patients with severe hemophilia often have spontaneous severe muscle and joint bleeds and can develop early crippling hemophilic arthropathy. Hence, routine prophylaxis has now become the standard of care in the United States and other developed countries in the management of patients with severe hemophilia. Prophylactic replacement therapy with cryoprecipitate in boys with severe hemophilia was first used nearly 50 years ago in Sweden31 and the Netherlands,32 and was shown to reduce the number and the severity of bleeds.32 Moreover, it was observed that early prophylaxis was more effective in preventing arthropathy compared to starting later in life, and that radiologic joint damage could not be reversed by prophylaxis. Subsequently, primary prophylaxis, defined as the start of regular, continuous treatment before the age of 2 years or after the occurrence of first joint bleed,33 was recommended and eventually became the standard treatment; it is currently recommended by the World Health Organization/World Federation of Hemophilia (WFH).34
The timing to begin prophylaxis is somewhat controversial, but many authors suggest starting prophylaxis before the first hemarthrosis occurs. Several studies have reported a wide variation in the age at first joint bleed, ranging from 0.2 to 5.8 years, with medians of 1.6 to 1.7 years.35,36 It has been suggested that arthropathy is best prevented if prophylaxis is started before the second or third joint bleed, but the benefits of starting before the occurrence of first bleed have not been established.37,38 The Swedish experience provides strong support for early prophylaxis.39 In an analysis of 121 patients with severe hemophilia, age at initiation of prophylaxis was an independent predictor of the development of arthropathy, but dose and interval of prophylaxis at the start of prophylactic treatment were not.39
In the Italian ESPRIT study, it was shown that children randomly assigned to prophylaxis had significantly fewer total bleeding episodes and joint bleeding episodes compared with those assigned to episodic therapy. Eleven of 21 patients (52%) in the prophylaxis group had on average less than 1 hemarthrosis per year, whereas only 4 of 19 patients in the episodic therapy group (21%) had the same low frequency of bleeding (P < 0.05).40 In a study of long-term prophylaxis versus on-demand treatment comparing age-matched Danish and Russian patients, the median annual number of joint bleeds in patients on prophylaxis was 1, while patients managed with on-demand treatment experienced a median of 37 joint bleeds. Patients taking prophylaxis also had a statistically significantly better quality of life estimate (P < 0.001) and better functional independence.41 In another trial, prophylaxis was initiated between the ages of 6 and 30 months based on a history of joint hemorrhage rather than age. Radiologic evidence of preserved joint architecture was found in 93% of participants in the prophylaxis group at 6 years of age. In this group, 18 of 32 (56%) children had 1 or 2 bleeds into one or more index joints before prophylaxis, and 17 (53%) had 1 to 5 hemorrhages into 1 or more index joints during prophylaxis. Prophylaxis was efficacious in decreasing bleeding and joint damage after up to 5 hemarthroses.42
Optimal Prophylactic Regimen
Although the benefits of prophylactic replacement therapy are firmly established, the optimal dose and frequency remain unclear. The half-life of clotting factor concentrates is short: about 8 hours for factor VIII in children, and about 12 hours for factor IX. As a result, prophylactic therapy is most effective when given frequently. The most common factor VIII concentrate dosing regimen for prophylaxis in hemophilia A is 25 to 40 IU/kg 3 times per week; for hemophilia B, a dose of 80 to 100 IU/kg is given twice weekly. This is aimed at a pre-infusion level > 1% to mimic the clinical phenotype of moderate hemophilia.
Recently, the US Food and Drug Administration (FDA) approved the first long-lasting antihemophilic factor (recombinant) Fc fusion protein for use in adults and children with hemophilia A. This medication contains the Fc region of human immunoglobulin G1 (IgG1), which binds to the neonatal Fc receptor (FcRn). FcRn is part of a naturally occurring pathway that delays lysosomal degradation of immunoglobulins by cycling them back into circulation and prolonging their plasma half-life. Dosing for routine prophylaxis is 50 IU/kg every 4 days; it may be adjusted based on patient response, with dosing in the range of 25 to 65 IU/kg at 3- to 5-day intervals. More frequent or higher doses up to 80 IU/kg may be required in children younger than 6 years.43
DEVELOPMENT OF INHIBITORS
FACTOR VIII INHIBITORS
Despite the success in the clinical management of hemophilia A, treated patients remain at risk for developing neutralizing antibodies that inhibit factor VIII activity. An inhibitor is a polyclonal high-affinity IgG that is directed against the factor VIII protein and renders exogenous factor ineffective. IgG4 antibodies are predominant and do not fix complement.
Risk Factors
The pathophysiology underlying the development of factor VIII inhibitors is a T-helper (Th)–cell dependent event that involves antigen-presenting cells and B lymphocytes; why only a fraction of patients experience this adverse effect of factor therapy is not known. Patients with mild/moderate hemophilia have a lower risk for inhibitor development than those with severe hemophilia A. The estimated prevalence of inhibitors ranges from 3% to 13% in mild to moderate disease,44–46 and up to 36% in severe hemophilia A.47,48 Usually the presence of an inhibitor in patients with mild/moderate hemophilia is suggested by a change in bleeding pattern: patients who previously used to bleed only after trauma or surgery suddenly start to experience severe spontaneous bleeding. This change in bleeding pattern is explained by cross-reactivity of the inhibitor with the mutated factor VIII of the patient, resulting in a residual factor level of < 0.01 IU/dL.49–51 Occasionally, there is no change in the residual factor VIII level but an inhibitor is detected in the Bethesda assay and/or there is lack of efficacy of factor VIII trans-fusions.51–53
Genetic factors. Data indicate that the risk of developing neutralizing antibodies is to a large extent determined by patient-related genetic factors.54,55 The immune response to factor VIII is similar in up to 80% of family members, significantly higher than expected compared with data from unrelated subjects. In a meta-analysis of patients with severe hemophilia A, the inhibitor incidence was twice as high in African American patients as compared with white patients.56 One study showed that patients of Hispanic ancestry with severe hemophilia A have a higher prevalence of neutralizing inhibitors than non-Hispanic white patients.57
Type of causative mutation. In severe hemophilia A, the risk of inhibitor formation is associated with the type of mutation. More disruptive mutations in the factor VIII gene, such as the intron 22 inversion, large gene deletions, and stop codons are associated with an approximately 35% risk of inhibitor formation, compared with only about 5% in those with missense mutation and small deletions.58 Persons with mutations involving large gene deletions, nonsense mutations, and intrachromosomal aberrations are usually at higher risk for the development of inhibitors than persons with missense mutations, small deletions/insertions, and splice site mutations.59,60 A relatively high risk is also encountered in patients with splicing errors and frame-shift mutations.61
Major histocompatibility complex. The HLA class I alleles A3, B7, and C7, as well as the class II alleles DQA0102, DQB0602, and DR15 have all been associated with a slightly higher risk for inhibitor development in unrelated patients, whereas the HLA C2, DQA0103, DQB0603, and DR13 alleles might be protective.62,63
Immune-regulatory molecules. In the Malmö International Brother Study, polymorphic sites in the genes coding for interleukin 10 (IL-10), tumor necrosis factor-α, and cytotoxic T lymphocyte–associated protein 4 were all associated with the risk of developing inhibitors.64–66 In this study, a 134 bp–long variant of a CAA microsatellite in the promoter region (IL-10.G) was identified in 26.8% of patients with hemophilia A. Thirty-two of these patients (72.7%) developed inhibitors as compared with 37.5% of those without the allele.65
Intensive exposure to factor VIII. Inhibitors in mild/moderate hemophilia seem to occur more commonly later in life, and an episode of intensive treatment with factor VIII concentrate has been reported to precede detection in most reported cases. In the series reported by Hay et al,67 16 out of 26 inhibitors were detected after such intensive replacement therapy, and no particular concentrate was implicated.
INHIBITORS TO FACTOR IX
Factor IX inhibitors are relatively uncommon, occurring in only 1% to 3% of persons with hemophilia B. This is in striking contrast to hemophilia A, where approximately 30% of patients develop inhibitors. The majority of patients with hemophilia B who develop inhibitors have severe hemophilia B.
Risk Factors
Certain mutations in the factor IX gene are associated with an increased incidence of inhibitor development. Large deletions and frame-shift mutations leading to the loss of coding information are much more likely to be associated with inhibitor development. Large deletions account for only 1% to 3% of all hemophilia B patients, but account for 50% of inhibitor patients.68 Patients with hemophilia B who develop inhibitors are at risk for developing anaphylactic reactions to factor IX–containing products. Anaphylaxis occurred more frequently in families with null mutations (large deletions, frame-shift mutations, or nonsense mutations) than in those with missense mutations.69 With hemophilia A, approximately 40% to 50% of black individuals develop inhibitors, but no such association has been found in hemophilia B. Individuals who develop an inhibitor to factor IX do so relatively early in life (within the first 4 to 5 years), after a median of 9 to 11 exposure days to any factor IX–containing products. Because of the severity of a potential anaphylactic reaction occurring early in life after very few exposures to factor IX, all infants and small children with severe hemophilia B should be closely followed over their first 10 infusions with any factor IX–containing products in a facility equipped to treat anaphylactic shock.70–72 A comparison of inhibitors in hemophilia A and B is shown in Table 3.
TREATMENT OF ACUTE BLEEDS IN PATIENTS WITH FACTOR VIII INHIBITORS
The available therapeutic agents for treatment of acute hemorrhage in children with hemophilia A with an inhibitor include high-dose recombinant or plasma-derived factor VIII concentrate, activated prothrombin complex concentrates (aPCCs), and recombinant activated factor VII (rFVIIa). In addition, antifibrinolytics may be used as an adjunct therapy.
Patient response to each treatment varies widely, with some patients responding well to one treatment and less well to another. Neither the patient's history nor standard lab tests can assist in making the best choice for the patient. A personalized approach to factor selection is used, and the dosing of that particular agent is often determined primarily by clinical assessment. Inhibitors are quantitated using the Bethesda inhibitor assay and clinically are classified as low- and high-responding inhibitors (Table 4). Inhibitor screening should be done prior to invasive procedures and periodically during the first 50 days of treatment since the risk for inhibitor development is highest during this period.
Low-Responding Inhibitors
A low-responding inhibitor is one in which inhibitor titers are < 5 Bethesda units (BU)/mL; patients with low-responding inhibitors can generally be treated with factor VIII concentrates at higher doses.73 Because the effect of factor VIII inhibitor is usually delayed, the Bethesda titer in plasma is determined after a 2-hour incubation period. As a result of this time delay, continuous administration of factor VIII is usually found to be effective.74 For a serious limb- or life-threatening bleeding episode, a bolus infusion of 100 IU of factor VIII per kg of body weight is administered, and the level is maintained by treatment at a rate of 20 IU/kg/hr. An assay for factor VIII should be performed 1 hour after the bolus infusion and at least daily thereafter. As the antibody titer drops, the daily level of factor VIII may rise and thus downward adjustment of the continuous infusion rate may be required. For routine joint and muscle hemorrhage, patients can usually be managed with infusions at twice the usual dosage. Routine inhibitor assays should be performed after exposure to factor VIII to determine whether an anamnestic response has occurred.
High-Responding Inhibitors
Most clinicians caring for patients with limb- or life-threatening bleeding episodes prefer to use products for which therapeutic levels can be monitored. As described earlier, continuous admin-istration of factor VIII is often effective because of the time delay in inhibition by the antibody. An initial dose of 100 to 200 IU/kg can be administered, and factor VIII levels can be determined 1 hour after initiation of continuous infusion at a rate of 20 to 40 IU/kg/hr. If a factor VIII level cannot be obtained (ie, patients with inhibitor titers > 5 to 10 BU/mL), alternative approaches include the bypassing agents aPCC and rFVIIa.
First used in the 1970s, aPCCs represented a significant improvement in the management in patients with hemophilia with inhibitors. They contain multiple activated serine protease molecules; activated factor X and prothrombin are the main active components in FEIBA (factor eight inhibitor bypassing activity), the most commonly used aPCC in the United States. FEIBA is a pooled plasma product that contains activated factors II, VII, IX and X, and has a duration of action of about 6 to 12 hours. For treatment of acute bleeds, the recommended dose of FEIBA is 50 to 100 IU/kg infused every 8 to 12 hours (maximum daily dose of 200 IU/kg). There is a risk of thrombosis/disseminated intravascular coagulation (DIC) with very large doses given frequently (> 200 IU/kg/day).
rFVIIa directly activates factor X and increases thrombin production on the surface of activated platelets in the absence of factor VIII or factor IX. Standard dosing of rFVIIa is 90 to 120 µg/kg, and many hemophilia treatment centers use higher doses (270 µg/kg/dose), especially in children and young adults. The half-life is about 1.5 to 3 hours, and therefore frequent administration (every 2–6 hours) is required. In one study that assessed the safety and efficacy of fixed-dose rFVIIa in the home setting, hemostasis was achieved in 566 (92%) of evaluable bleeding episodes, and following administration of the additional maintenance dose, hemostasis was maintained in 95% of successfully treated cases.75 As with aPCCs, there is no standardized quantitative laboratory test for measuring the effectiveness of rFVIIa therapy.
All currently used bypassing agents are associated with a risk of thrombotic complications including thromboembolism, DIC, and myocardial infarction. These complications are very rare in patients with hemophilia, however. In general, bypassing agents work for most bleeds and for most patients, but are not as predictable as factor replacement therapy and cannot be monitored by laboratory assays.
TREATMENT OF ACUTE BLEEDS IN PATIENTS WITH FACTOR IX INHIBITORS
rFVIIa and FEIBA are the mainstays of treatment of bleeding episodes in individuals with hemophilia B complicated by an inhibitor to factor IX. Treatment of hemorrhagic episodes in these patients depends on the type of bleeding episode experienced, the inhibitor classification (high- versus low-responding [Table 4]), and the history and severity of infusion reactions. Patients with low-responding inhibitors who have not experienced infusion reactions may be treated with doses of factor IX concentrate calculated to overcome the inhibitor titer and achieve a hemostatic level. In patients with high-responding inhibitors, the use of factor IX concentrates is impractical because of the inhibitor titer or the anamnestic response. Regardless of inhibitor titer, in patients with a history of an anaphylactic event, factor IX usage is contraindicated.
The most commonly used therapy for hemostatic control in patients with high-responding inhibitors with factor IX deficiency and a history of infusion reaction is rFVIIa; the standard dosing regimen is 90 to 120 µg/kg/dose administered every 2 to 3 hours, with a maximum dose of 270 µg/kg/dose. aPCCs, which contain factor IX, can be utilized if the patient has not experienced prior infusion reactions. Repeated exposures to products containing factor IX may stimulate the inhibitor titer and prevent its natural decline over time. This can pose a problem in cases of life- or limb-threatening hemorrhage unresponsive to rFVIIa as these patients will not have factor IX available as an effective mode of therapy. The dosing of FEIBA ranges from 50 to 100 IU/kg every 12 hours, with daily dosing not to exceed 200 IU/kg.
IMMUNE TOLERANCE INDUCTION
Because of the associated inhibitor-related morbidity resulting from limited treatment options, antibody eradication is the ultimate goal in inhibitor management. The only proven strategy for achieving antigen-specific tolerance to factor VIII or factor IX is immune tolerance induction (ITI) therapy. Successful ITI in hemophilia A is currently defined as both an undetectable inhibitor titer (< 0.6 BU), and normalized factor VIII pharmacokinetics, which in turn is defined as plasma factor VIII recovery > 66% of expected and a half-life > 6 hours, determined following a 72-hour factor VIII exposure-free period (Consensus Proceedings from the Second International Conference on Immune Tolerance Therapy, Bonn, Germany, 1997 [unpublished]). Once successful immune tolerance is achieved, long-term prophylaxis is commonly instituted. Using conclusions drawn from international consensus criteria and analysis of the International Immune Tolerance Registry, the I-ITI study has defined ITI failure by the presence of either of 2 criteria:
1. Failure to attain the definition of success within 33 months of uninterrupted ITI;
2. Failure to demonstrate a progressive 20% reduction in inhibitor titer over each 6-month period of uninterrupted ITI, beginning 3 months after initiation to allow for expected anamnesis.76–78
This definition implies a minimum ITI trial period of 9 months before failure is declared.
The European Hemophilia Standardization Board (EHSB), the International Consensus Panel (ICP), and the United Kingdom Hemophilia Center Doctors’ Organization (UKHCDO) have agreed that it is preferable to initiate ITI at a titer of < 10 BU/mL, unless, per the ICP, the titer does not decline over a period of 1 to 2 years and/or inhibitor development is associated with severe or life-threatening bleeding. The ICP noted that for “poor-risk” ITI patients (defined by a historical titer of > 200 BU/mL and/or a pre-ITI inhibitor titer of > 10 BU/mL and/or an interval of > 5 years since inhibitor diagnosis), published efficacy data are limited to dosing regimens > 200 IU/kg/day. The groups all independently concluded that ITI has been successfully performed using recombinant and plasma-derived factor VIII replacement therapy (usually the product on which they developed the inhibitor), and that there are no data to support the superiority of any single product type.79–81 However, both EHSB and ICP have suggested that VWF-containing concentrates be considered for patients who fail ITI using high-purity factor VIII.79,80
The recommendations from US guidelines for ITI in patients with hemophilia A and inhibitors are listed in Table 5.82
ARTHROPATHY
Before the advent of factor products for the treatment of hemophilia, hemarthrosis was one of the leading causes of morbidity. Today, the routine use of prophylactic treatment has resulted in a significant improvement in the lifestyle, quality of life, and life expectancy of these patients. However, despite best efforts, some patients will have severe joint destruction as a result of repeated articular bleeding episodes during their early years. This leads to pain and significant functional disability, thus impairing the quality of life. The basic pathology behind hemophilic arthropathy is chronic synovitis.
It is common to observe a pattern of repeated bleeding (chronic hemarthrosis), especially in patients with severe hemophilia, that can lead to chronic synovitis, inflammatory arthritis, and progressive arthropathy. Therefore, the key to preventing hemophilic arthropathy is aggressive management of the initial hemarthrosis. This is generally accomplished with the use of clotting factor replacement, restorative physiotherapy, and close clinical follow-up. If chronic synovitis develops, synovectomy may be considered in order to slow the progression of the hemophilic arthropathy and to prevent the development of major articular surface erosions that can lead to end-stage arthropathy.83 Primary prophylaxis is discussed earlier and is the mainstay of prevention of chronic hemophilic arthropathy.
SYNOVECTOMY
The emergence of chronic hemophilic hemarthrosis is incited by a hypertrophic and highly vascular synovium. Removal of the synovium prevents further joint damage,84 and can be accomplished through surgical and nonsurgical procedures.
Surgical excision of the hypertrophic synovium can be performed through open or arthroscopic procedures. The open approach has largely been replaced by arthroscopic synovectomies. Regardless of the approach, these patients need prolonged hospitalization, extensive factor replacement, and exhaustive physiotherapy. Moreover, patients with inhibitors are usually not considered candidates for surgical synovectomy.
Chemical and radioactive agents injected intra-articularly can decrease the volume and activity of the synovial tissue. Due to the minimally invasive nature of these procedures, nonsurgical synovectomies are of special importance for hemophilic patients with inhibitors to clotting factors.
Chemical Synovectomy
Chemical synovectomies, using thiotepa, osmic acid, D-penicillamine and other agents, have been used in the distant past. Rifampicin, which is used an antibiotic, is now the most commonly used chemical for the purpose of synovectomy, and the one that has shown better results in terms of decreasing hemarthrosis.85 Each one of the injections should be accompanied by prophylactic administration of clotting factor concentrate. Excellent results (no synovitis and restoration of previous function) have been reported in up to 83% of patients at an average of 2.4 years after the intra-articular injection of rifampicin. As the pathology of the joint becomes more severe, however, the number of injections required to achieve improvement increases. Younger patients and smaller joints benefit more from this procedure.
Radiation Synovectomy
Radiosynovectomy (RS) and radiosynoviorthesis are common terms used to describe the synovial ablation accomplished by intra-articular injection of radioisotopes. Isotopes of gold, yttrium, rhenium, and dysprosium have been used to perform radiation synovectomies in patients with hemophilia. Yttrium-90, a pure beta emitter with adequate particle size and depth penetration, has been used successfully for the treatment of hemophilic synovitis.
The local (growth plate and articular cartilage) and remote effects of radiation are a concern. There have been no reported cases of growth plate disturbance after radiosynovectomy, even after the use of beta emitters such as gold-198.86 Articular cartilage is highly resistant to radiation, and although damage is theoretically possible, none has been reported. Progressive degeneration of treated joints does occur, but the rate is slower than that expected without radiosynovectomy. The principal concern is the potential for late, radiation-induced neoplasia. However, the safety of intra-articular radioisotopes is supported by a long-term follow-up study of more than 5000 RS procedures performed for rheumatoid arthritis, which found no reported radiation-induced malignancies.87
One review analyzed the safety of RS in pediatric patients with hemophilia to provide a risk-benefit assessment. During knee RS, patients receive a radiation dose of approximately 0.74 mSv, and during elbow and ankle RS, a dose of approximately 0.32 mSv. The radiation dose from natural sources is approximately 2 mSv per year and the recommended limit for patients (apart from natural sources) is 1 mSv per year. The lifetime cancer risk increases about 0.5% per 100 mSv per year. Considering the risks and benefits of RS, the authors recommend that clinicians consider this procedure in children with inhibitors or in patients without inhibitors when bleeding is recurrent and persistent despite aggressive factor replacement.88 External-beam radiation has been extensively studied and carries a small risk of osteosarcoma induction.
ACQUIRED INHIBITORS TO FACTOR VIII
Acquired hemophilia (AH) has an estimated prevalence of 1.48 cases per million per year, and a reported mortality between 9% and 22%.89,90 AH is uncommon in children younger than 16 years (prevalence estimated at 0.045/million/year), and may be underdiagnosed in persons older than age 85 (prevalence estimated at 14.7/million/year).89 In the largest published population series, 50% to 60% of diagnosed individuals were previously healthy with no identified underlying disease state.90–91 Underlying conditions consistently associated with AH include pregnancy, evolving or pre-existing autoimmune or malignant disorders, and rarely medications. Primary among the autoimmune disorders are collagen vascular disorders, including systemic lupus erythematosus, rheumatoid arthritis, myasthenia gravis, multiple sclerosis, and autoimmune hemolytic anemia. Most antibodies are mixtures of polyclonal IgG1 and IgG4 immunoglobulins, with the IgG4 molecules mainly responsible for inhibiting clotting activity. The clinical picture of AH is characterized by acute onset of severe bleeding in individuals who previously had no history of bleeding diathesis. Patients generally present with mucocutaneous bleeding (eg, epistaxis and gastrointestinal bleeding), as well as soft tissue bleeding (eg, extensive ecchymoses and hematomas).
The 2 major goals of treatment of AH are the immediate control of acute and chronic bleeding and the long-term suppression/eradication of the autoantibody inhibitor. For patients with an inhibitor titer < 5 BU/mL, administration of desmopressin and concentrates of human recombinant factor VIII may raise the factor VIII activity levels in plasma. If the inhibitor titer is > 5 BU/mL, or if bleeding persists despite infusions of factor VIII concentrates, then factor VIII bypassing agents, such as aPCCs or rFVIIa, are indicated. Local measures for treatment of mucosal hemorrhage, such as antifibrinolytic agents or topical fibrin glues, are helpful.
The primary aim in long-term management of AH is to eradicate the factor VIII autoantibodies so that further bleeding can be averted. Although in some clinical situations (postpartum women and drug-related AH) factor VIII antibodies may remit spontaneously, most published guidelines and algorithms recommend early initiation of eradication therapy. This is usually achieved through immunosuppressive medications or immunomodulation. Successful immunosuppression regimens in AH have most frequently used corticosteroids as the cornerstone, either as a single agent or in combination with cyclophosphamide. In a prospective randomized trial involving 31 participants treated with prednisone 1 mg/kg/day for 3 weeks, 32% achieved complete remission. In participants with antibody persistence after 3 weeks, switching to oral cyclophosphamide 2 mg/kg/day as second-line therapy appeared more effective than continuing prednisone (complete remission rate 50% versus 42%).92
Other immunosuppressive medications have been employed for eradication of refractory autoantibody inhibitors, including azathioprine, cyclosporine, tacrolimus, mycophenolate motefil, and sirolimus. Controlled studies have not been performed to confirm their comparative safety and efficacy in sufficiently large populations. Anti-CD20 antibody has been used to treat inhibitors in patients with both congenital and acquired hemophilia.93,94 Other less frequently used treatment options include administration of intravenous immunoglobulins (IVIG) in large doses. IVIG by itself rarely is able to induce a complete remission, but may be useful adjunctive therapy along with immunosuppressants, as part of an ITI regimen, or with extracorporeal plasmapheresis.
- Brinkhous KM. Clotting defect in hemophilia: deficiency in a plasma factor required for platelet utilization. Proc Soc Exp Biol Med 1947;66:117–20.
- Quick AJ. Studies on the enigma of the haemostatic dysfunction of hemophilia. Am J Med Sci 1947;214:272–80.
- Ingram JIC. The history of hemophilia. J Clin Path 1976;29:469–79.
- Gitschier J, Wood WI, Goralka TM, et al. Characterization of the human factor VIII gene. Nature 1984;312:326–30.
- Vehar GA, Keyt B, Eaton D, et al. Structure of human factor VIII. Nature 1984;312:337–42.
Toole JJ, Knopf JL, Wozney JM, et al. Molecular cloning of a cDNA encoding human antihaemophilic factor. Nature 1984;312:342–7.
Yoshitake S, Schach BG, Foster DC, et al. Nucleotide sequence of the gene for human factor IX (antihemophilic factor B). Biochemistry 1985;24:3736–50.
Vander VP, Giles AR. A detailed morphological evaluation of the evolution of the haemostatic plug in normal, factor VII and factor VIII deficient dogs. Br J Haematol 1988;70:345–55.
Gitschier J, Wood WI, Goralka TM, et al. Characterizartion of the human factor VIII gene. Nature 1984;312:326–30.
Toole JJ, Knopf JL, Wozney JM, et al. Molecular cloning of a cDNA encoding human antihaemophilic factor. Nature 1984;312:342–7.
Naylor J, Brinke A, Hassock S, Green PM, Gianelli F. Characteristic mRNA abnormality found in half the patients with severe haemophilia A is due to large DNA inversions. Hum Mol Genet 1993;2:1773–8.
Lakich D, Kazazian HH Jr., Antonarakis SE, Gitschier J. Inversions disrupting the factor VIII gene are a common cause of severe hemophilia A. Nat Genet 1993;5:236–41.
Stenson PD, Ball E, Howells K, et al. Gene mutation database. J Med Genet 2008;45:124–6.
Mauser Bunschoten EP, van Houwelingen JC, Sjamsoedin Visser EJM, et al. Bleeding symptoms in carriers of hemophilia A and B. Thromb Haemost 1988;59: 349–52.
Plug I, Mauser-Bunschoten EP, Bröcker-Vriends AH, et al. Bleeding in carriers of hemophilia. Blood 2006;108:52–6.
Nuss R, Soucie JM, Evatt B, and the Hemophilia Surveillance System Project Investigators. Changes in the occurrence of and risk factors for hemophilia-associated intracranial hemorrhage. Am J Hematol 2001;68:37–42.
Yoffe G, Buchanan G. Intracranial hemorrhage in newborn and young infants with hemophilia. J Pediatr 1988;113:333–6.
Roderick PJ, Robinson AC. Life-threatening oropharyngeal bleeding in a haemophiliac with factor VIII inhibitors. Clin Lab Haemat 1988;10:217–9.
Sites of initial bleeding episodes, mode of delivery and age of diagnosis in babies with haemophilia diagnosed before the age of 2 years: a report from the Centers for Disease Control and Prevention’s (CDC) Universal Data Collection (UDC) project. Hemophilia 2009; 15:1281–90.
Mittal R, Spero J, Lewis JH, Taylor F, et al. Patterns of gastrointestinal hemorrhage in hemophilia. Gastroenterol 1985;88:515–22.
Lander E, Pechlaner C, Mayr A, et al. Mallory-Weiss syndrome in a patient with hemophilia A and chronic liver disease. Ital J Gastroenterol 1995;27:73–4.
de la Fuente B, Kasper CK, Rickles FR, et al. Response of patients with mild and moderate hemophilia A and von Willebrand disease to treatment with desmopressin. Ann Intern Med 1985; 103:6–14.
Prowse CV, Sas G, Gader AM, et al. Specificity in the factor VIII response to vasopressin infusion in man. Br J Haematol 1979;41:437–47.
Rodeghiero F, Castaman G, Di Bona E, et al. Consistency of responses to repeated DDAVP infusions in patients with von Willebrand disease and hemophilia A. Blood 1989;74:1997–2000.
Warrier AI, Lusher JM. DDAVP: a useful alternative to blood components in moderate hemophilia A and von Willebrand disease. J Pediatr 1983;102:228–33.
Nilsson IM, Mikaelsson M, Vilhardt H. The effect of intranasal DDAVP on coagulation and fibrinolytic activity in normal persons. Scand J Haematol 1982;29:70–4.
Kogan A. Thrombin activatable fibrinolysis inhibitor (TAFI): a new marker of cardiovascular disease. Clinical Laboratory International. June 2004.
Vinckier F, Vermylen J. Dental extractions in hemophilia: reflections on 10 years’ experience. Oral Surg Oral Med Oral Pathol 1985;59:6–9.
Evans BE. The use of epsilon-aminocaproic acid for the management of hemophilia in dental and oral surgery patients. J Am Dent Assoc 1977;94:21.
Kaneshiro MM, Mielke CH Jr, Kasper CK, et al. Bleeding time after aspirin in disorders of intrinsic clotting. N Engl J Med 1969;281:1039–42.
Nilsson IM, Blomback M, Ahlberg A. Our experience in Sweden with prophylaxis on haemophilia. Bibl Haematol 1970;34:111–24.
Van Creveld S. Prophylaxis of joint hemorrhages in hemophilia. Acta Haematol 1969; 41:206–14.
Ljung R, Aronis-Vournas S, Kurnik-Auberger K, et al. Treatment of children with haemophilia in Europe: A survey of 20 centers in 16 countries. Haemophilia 2001;7:446–52.
Berntorp E, Astermark J, Bjorkman S, et al. Consensus perspectives on prophylactic therapy for haemophilia: Summary statement. Haemophilia 2003; 9(Suppl. 1):1–4.
Pollman H, Richter H, Ringkamp H, Jurgens H. When are children diagnosed as having severe hemophilia and when do they start to bleed? A 10-year single-center PUP study. Eur J Pediatr 1999;158:166–70.
Van Dijk K, Fischer K, Van Der Bom JG, et al. Variability in clinical phenotype of severe haemophilia: the role of the first joint bleed. Haemophilia 2005;11:438–43.
Kreuz W, Escuriola-Ettingshausen C, Funk M, et al. When should prophylactic treatment in patients with Hemophilia A and B start? The German experience. Hemophilia 1998;4:413–7.
Fischer K, Van der Bom JG, Mauser-Bunschoten EP, et al. Effects of postponing prophylactic treatment on long-term outcome in patients with severe Hemophilia. Blood 2002;99:2337–41.
Astermark J, Petrini P, Tengborn L, et al. Primary prophylaxis in severe hemophilia should be started at an early age but can be individualized. Br J Haematol 1999;105:1109–13.
Gringeri A, Lundin B, von mackensen S, et al; ESPRIT study group. A randomized clinical trial of prophylaxis in children with hemophilia A (the ESPRIT Study). J Thromb Haemost 2011;9:700–10.
Ingerslev J, Lethagen S, Hvitfeldt Poulsen L, et al. Long-standing prophylactic therapy vs. episodic treatment in young people with severe haemophilia: a comparison of age-matched Danish and Russian patients. Haemophilia 2014;20: 58–64.
Maco-Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med 2007;357:535–44.
Eloctate [package insert]. Cambridge [MA]: Biogen, Inc.
Lusher JM, Arkin S, Abildgaard CF, et al. Recombinant factor VIII for the treatment of previously untreated patients with henophilia A. N Engl J Med 1993;328:453–9.
Sultan Y, and the French Hemophilia Study Group. Prevalence of inhibitors in a population of 3435 hemophilia patients in France. Thromb Haemost 1992;67:600–2.
Rizza CR, Spooner RGD. Treatment of hemophilia and related disorders in Britain and Northern Ireland during 1976-80: report on behalf of the directors of hemophilia centers in the United Kingdom. Br Med J 1983;286:929–32.
Darby SC, Keeling DM, Spooner RJ, et al. The incidence of factor VIII and factor IX inhibitors in the hemophilia population of the UK and their effect on subsequent mortality, 1977-99. J Thromb Haemost 2004;2:1047–54.
Ehrenforth S, Kreuz W, Scharrer I, et al. Incidence of development of factor VIII and factor IX inhibitors in haemophiliacs. Lancet 1992;339:594–8.
Fijnvandraat K, Turenhout EAM, van den Brink EN, et al. The missense mutation Arg593à Cys is related to antibody formation in a patient with mild Hemophilia A. Blood 1997;89:4371–7.
Vlot AJ, Wittebol S, Strengers PFW, et al. Factor VIII inhibitor in a patient with mild Hemophilia A and an Asn618-Ser mutation responsive to immune tolerance induction and cyclophosphamide. Br J Hematolol 2002;117:136–40.
Santagostino E, Gringeri A, Tagliavacca L, et al. Inhibitors to factor VIII in a family with mild hemophilia: Molecular characterization and response to factor VIII and desmopressin. Throm Haemost 1995;74:61–21.
Peerlinck K, Jacquemin M, Arnout J, et al. Antifactor VIII antibody inhibiting allogenic but not autologous factor VIII in patients with mild hemophilia A. Blood 1999;93:2267–73.
Kesteven PJ, Holland LJ, Lawrie AS, et al. Inhibitor to factor VIII in mild hemophilia. Thromb Haemost 1984;52:50–2.
Gill JC. The role of genetics in inhibitor formation. Thromb Hemostat 1999;82:500–4.
Astermark J, Berntorp E, White GC, Kroner BL; MIBS Study group. The Malmo International Brother Study (MIBS): further support for genetic predisposition to inhibitor development in hemophilia patient. Hemophilia 2001;7:267–72.
Scharrer I, Bray GL, Neutzling O. Incidence of inhibitors in Hemophilia A patients- A review of recent studies of recombinant and plasma-derived factor VIII concentrates. Hemophilia 1999;5:145–54.
Carpenter SL, Michael Soucie J, Sterner S, Presley R; Hemophilia Treatment Center Network (HTCN) Investigators. Increased prevalence of inhibitors in Hispanic patients with severe haemophilia A enrolled in the Universal Data Collection databse. Haemophilia 2012;18:e260–5.
Goodeve AC, Peake IR. The molecular basis of hemophilia A: genotype-phenotype relationships and inhibitor development. Semin Thromb Haemost 2003;29:23–30.
Schwaab R, Brackman HH, Meyer C, et al. Hemophilia A: mutation type determines risk of inhibitor formation. Thromb Haemost 1995;74:1402–6.
Oldenburg J, El-Maari O, Schwaab R. Inhibitor development in correlation to Factor VIII genotypes. Hemophilia 2002;8(Suppl. 2):23–9.
Boekhorst J, Lari GR, D’oiron R, et al. Factor VIII genotype and inhibitor development in patients with hemophilia A: Highest risk in patients with splice site mutation. Haemophilia 2008;14:729–35.
Oldenburg J, Picard JK, Schwaab R, et al. HLA genotype of patients with severe hemophilia A due to intron 22 inversion with and without inhibitors of factor VIII. Thromb Haemost 1997;77:238–42.
Hay CR, Ollier W, Pepper L, et al. HLA class II profile: A weak determinant of factor VIII inhibitor development in severe hemophilia A. UKHCDO Inhibitor Working Party. Thromb Haemost 1997;77:234–7.
Astermark J, Olderburg J, Carlson J, et al. Polymorphisms in the TNFA gene and the risk of inhibitor development in severe Hemophilia A. Blood 2006;108:3739–45.
Astemark J, Olderburg J, Pavlova A, et al. Polymorphisms in the IL10 but not in the IL1Beta and IL4 genes are associated with inhibitor development in patients with Hemophilia A. Blood 2006;107:3167–72.
Astermark J, Wang X, Olderburg, et al. MIBS Study group. Polymorphisms in the CTLA-4 gene and inhibitor development in patients with Hemophilia A. J Throm Haemost 2007;5:263–5.
Hay CR, Ludlam CA, Colvin BT, et al. Factor VIII inhibitors in mild and moderate-severity hemophilia A. Thromb Haemost 1998;79:762–6.
High HA. Factor IX molecular structure, epitopes and mutations associated with inhibitor formation. In: Aledort LM, Hoyer LW, Lusher JM, et al, eds. Inhibitors to coagulation factors. New York: Plenum Press; 1995:79-86.
Thorland ED, Drost JB, Lusher JM, et al. Anaphylactic response to factor IX replacement therapy in hemophilia B patients: Complete gene deletions confer the highest risk. Hemophilia 1999;5:101–5.
Warrier I. ITI in hemophilia B: Possibilities and problems. International Monitor on Hemophilia 2003:20–3.
Warrier I, Ewenstein B, Koerper M, et al. FIX Inhibitors and anaphylaxis in hemophilia B. J Pediatr Hematol Oncol 1997;19:23–7.
Warrier I. Management of hemophilia B patients with inhibitors and anaphylaxis. In: Varon D, Martinowitz U, Heim M, eds. Haemophilia and related disorders. Vol. 4. Oxford: Blackwell Science; 1998:574–6.
Kasper CK, Aledort L, Aronson D, et al: Proceedings: a more uniform measurement of factor VIII inhibitors. Thromb Diath Haemorrh 1975;34:612.
White GC, Taylor RE, Blatt PM, et al. Treatment of a high titer anti–factor-VIII antibody by continuous factor VIII administration: report of a case. Blood 1983;62:141–5.
Key NS, Aledort LM, Beardsley D, et al. Home treatment of mild to moderate bleeding episodes using recombinant factor VIIa (Novoseven) in haemophiliacs with inhibitors. Thromb Haemost 1998;80:912–8.
Mariani G, Scheibel E, Nogao T, et al. Immune tolerance as treatment of alloantibodies to factor VIII in hemophilia. The international registry of Immunetolerance Protocols. Semin Hematol 1994;31(Suppl. 4):62–4.
DiMichele D, Kroner B. Factor VIII/IX Subcommittee of the International Society for Thrombosis and Hemostasis. The maintenance of tolerance after successful immune tolerance induction in hemophilia A and B. The North American Registry. Factor VIII/IX Subcommittee of the International Society for Thrombosis and Haemostasis. Haematologica 2000;85(Suppl. 10):40–4.
DiMichele DM, Hay CRM. The international immune tolerance study: A multicenter prospective randomized trial in progress. J Thromb Haemost 2006;4:2271–3000.
Astermark J, Morado M, Rocino A, et al. Current European practice in immune tolerance induction therapy in patients with hemophilia and inhibitors. Hemophilia 2006;12:363–71.
DiMichele DM, Hoots WK, Pipe SW, et al. International workshop on immune tolerance induction: Consensus recommendations. Hemophilia 2007;13:(Suppl. 1):1–22.
Hay CRM, Brown S, Collins PW, et al. The diagnosis and management of factor VIII and IX inhibitors: A guideline from the United Kingdom Center Doctors Organization. Br J Haematol 2006;133:591–605.
Valentino LA, Kempton CL, Kruse-Jarres R, et al. US Guidelines for immune tolerance induction in patients with haemophilia A and inhibitors. Haemophilia 2015;21:559–67.
Silva M, Luck JV Jr. Chronic hemophilic synovitis: the role of radiosynovectomy. World Federation of Hemophilia. Treatment of Hemophilia. April 2004 (no 33). http://www1.wfh.org/publications/files/pdf-1176.pdf.
Storti E, Traldi A, Tosatti E, Davoli PG. Synovectomy, a new approach to hemophilic arthropathy. Acta Haemotol 1969;41:193–205.
Caviglia HA, Fernandez-Palazzi F, Maffei E, et al. Chemical synoviorthesis for hemophilic synovitis. Clin Orthop 1997;343:30–6.
Ahlberg A, Pettersson H. Synoviorthesis with radioactive gold in hemophiliacs. Clinical and radiological follow-up. Acta Orthop Scand 1979;50:513–7.
Lee P. The efficacy and safety of radiosynovectomy. J Rheumatol 1982;9:165–8.
Rodriguez-Merchan EC, Valentino LA. Safety of radiation exposure after radiosynovectomy in paediatric patients with haemophilia. Haemophilia 2015;21:411–8.
Green D, Lechner K. A survey of 215 non-hemophilic patients with inhibitors to factor VIII. Thromb Haemost 1981;45:200–3.
Collins PW, Hirsch S, Baglin TP, et al. Acquired hemophilia A in the United Kingdom: A 2 year national surveillance study by the United Kingdom Haemophilia Center Doctors’ Organisation. Blood 2007;109:1870–7.
Kessler CM, Ludlam CA. The treatment of acquired factor VIII inhibitors: Worldwide experience with porcine factor VIII concentrate. International Acquired Hemophilia Study Group. Semin Hematol 1993;30(Suppl. 1):22–7.
Green D, Rademaker AW, Briet E. A prospective, randomized trial of prednisone and cyclophosphamide in the treatment of patients with factor VIII autoantibodies. Thromb Haemost 1993;70:753–7.
Stasi R, Brunetti M, Stipa E, Amadori S. Selective B-cell depletion with rituximab for the treatment of patients with acquired hemophilia. Blood 2004;103:4424–8.
Franchini M. Rituximab in the treatment of adult acquired hemophilia A: a systematic review. Crit Rev Oncol Hematol 2007;63:47–52.
- Brinkhous KM. Clotting defect in hemophilia: deficiency in a plasma factor required for platelet utilization. Proc Soc Exp Biol Med 1947;66:117–20.
- Quick AJ. Studies on the enigma of the haemostatic dysfunction of hemophilia. Am J Med Sci 1947;214:272–80.
- Ingram JIC. The history of hemophilia. J Clin Path 1976;29:469–79.
- Gitschier J, Wood WI, Goralka TM, et al. Characterization of the human factor VIII gene. Nature 1984;312:326–30.
- Vehar GA, Keyt B, Eaton D, et al. Structure of human factor VIII. Nature 1984;312:337–42.
Toole JJ, Knopf JL, Wozney JM, et al. Molecular cloning of a cDNA encoding human antihaemophilic factor. Nature 1984;312:342–7.
Yoshitake S, Schach BG, Foster DC, et al. Nucleotide sequence of the gene for human factor IX (antihemophilic factor B). Biochemistry 1985;24:3736–50.
Vander VP, Giles AR. A detailed morphological evaluation of the evolution of the haemostatic plug in normal, factor VII and factor VIII deficient dogs. Br J Haematol 1988;70:345–55.
Gitschier J, Wood WI, Goralka TM, et al. Characterizartion of the human factor VIII gene. Nature 1984;312:326–30.
Toole JJ, Knopf JL, Wozney JM, et al. Molecular cloning of a cDNA encoding human antihaemophilic factor. Nature 1984;312:342–7.
Naylor J, Brinke A, Hassock S, Green PM, Gianelli F. Characteristic mRNA abnormality found in half the patients with severe haemophilia A is due to large DNA inversions. Hum Mol Genet 1993;2:1773–8.
Lakich D, Kazazian HH Jr., Antonarakis SE, Gitschier J. Inversions disrupting the factor VIII gene are a common cause of severe hemophilia A. Nat Genet 1993;5:236–41.
Stenson PD, Ball E, Howells K, et al. Gene mutation database. J Med Genet 2008;45:124–6.
Mauser Bunschoten EP, van Houwelingen JC, Sjamsoedin Visser EJM, et al. Bleeding symptoms in carriers of hemophilia A and B. Thromb Haemost 1988;59: 349–52.
Plug I, Mauser-Bunschoten EP, Bröcker-Vriends AH, et al. Bleeding in carriers of hemophilia. Blood 2006;108:52–6.
Nuss R, Soucie JM, Evatt B, and the Hemophilia Surveillance System Project Investigators. Changes in the occurrence of and risk factors for hemophilia-associated intracranial hemorrhage. Am J Hematol 2001;68:37–42.
Yoffe G, Buchanan G. Intracranial hemorrhage in newborn and young infants with hemophilia. J Pediatr 1988;113:333–6.
Roderick PJ, Robinson AC. Life-threatening oropharyngeal bleeding in a haemophiliac with factor VIII inhibitors. Clin Lab Haemat 1988;10:217–9.
Sites of initial bleeding episodes, mode of delivery and age of diagnosis in babies with haemophilia diagnosed before the age of 2 years: a report from the Centers for Disease Control and Prevention’s (CDC) Universal Data Collection (UDC) project. Hemophilia 2009; 15:1281–90.
Mittal R, Spero J, Lewis JH, Taylor F, et al. Patterns of gastrointestinal hemorrhage in hemophilia. Gastroenterol 1985;88:515–22.
Lander E, Pechlaner C, Mayr A, et al. Mallory-Weiss syndrome in a patient with hemophilia A and chronic liver disease. Ital J Gastroenterol 1995;27:73–4.
de la Fuente B, Kasper CK, Rickles FR, et al. Response of patients with mild and moderate hemophilia A and von Willebrand disease to treatment with desmopressin. Ann Intern Med 1985; 103:6–14.
Prowse CV, Sas G, Gader AM, et al. Specificity in the factor VIII response to vasopressin infusion in man. Br J Haematol 1979;41:437–47.
Rodeghiero F, Castaman G, Di Bona E, et al. Consistency of responses to repeated DDAVP infusions in patients with von Willebrand disease and hemophilia A. Blood 1989;74:1997–2000.
Warrier AI, Lusher JM. DDAVP: a useful alternative to blood components in moderate hemophilia A and von Willebrand disease. J Pediatr 1983;102:228–33.
Nilsson IM, Mikaelsson M, Vilhardt H. The effect of intranasal DDAVP on coagulation and fibrinolytic activity in normal persons. Scand J Haematol 1982;29:70–4.
Kogan A. Thrombin activatable fibrinolysis inhibitor (TAFI): a new marker of cardiovascular disease. Clinical Laboratory International. June 2004.
Vinckier F, Vermylen J. Dental extractions in hemophilia: reflections on 10 years’ experience. Oral Surg Oral Med Oral Pathol 1985;59:6–9.
Evans BE. The use of epsilon-aminocaproic acid for the management of hemophilia in dental and oral surgery patients. J Am Dent Assoc 1977;94:21.
Kaneshiro MM, Mielke CH Jr, Kasper CK, et al. Bleeding time after aspirin in disorders of intrinsic clotting. N Engl J Med 1969;281:1039–42.
Nilsson IM, Blomback M, Ahlberg A. Our experience in Sweden with prophylaxis on haemophilia. Bibl Haematol 1970;34:111–24.
Van Creveld S. Prophylaxis of joint hemorrhages in hemophilia. Acta Haematol 1969; 41:206–14.
Ljung R, Aronis-Vournas S, Kurnik-Auberger K, et al. Treatment of children with haemophilia in Europe: A survey of 20 centers in 16 countries. Haemophilia 2001;7:446–52.
Berntorp E, Astermark J, Bjorkman S, et al. Consensus perspectives on prophylactic therapy for haemophilia: Summary statement. Haemophilia 2003; 9(Suppl. 1):1–4.
Pollman H, Richter H, Ringkamp H, Jurgens H. When are children diagnosed as having severe hemophilia and when do they start to bleed? A 10-year single-center PUP study. Eur J Pediatr 1999;158:166–70.
Van Dijk K, Fischer K, Van Der Bom JG, et al. Variability in clinical phenotype of severe haemophilia: the role of the first joint bleed. Haemophilia 2005;11:438–43.
Kreuz W, Escuriola-Ettingshausen C, Funk M, et al. When should prophylactic treatment in patients with Hemophilia A and B start? The German experience. Hemophilia 1998;4:413–7.
Fischer K, Van der Bom JG, Mauser-Bunschoten EP, et al. Effects of postponing prophylactic treatment on long-term outcome in patients with severe Hemophilia. Blood 2002;99:2337–41.
Astermark J, Petrini P, Tengborn L, et al. Primary prophylaxis in severe hemophilia should be started at an early age but can be individualized. Br J Haematol 1999;105:1109–13.
Gringeri A, Lundin B, von mackensen S, et al; ESPRIT study group. A randomized clinical trial of prophylaxis in children with hemophilia A (the ESPRIT Study). J Thromb Haemost 2011;9:700–10.
Ingerslev J, Lethagen S, Hvitfeldt Poulsen L, et al. Long-standing prophylactic therapy vs. episodic treatment in young people with severe haemophilia: a comparison of age-matched Danish and Russian patients. Haemophilia 2014;20: 58–64.
Maco-Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med 2007;357:535–44.
Eloctate [package insert]. Cambridge [MA]: Biogen, Inc.
Lusher JM, Arkin S, Abildgaard CF, et al. Recombinant factor VIII for the treatment of previously untreated patients with henophilia A. N Engl J Med 1993;328:453–9.
Sultan Y, and the French Hemophilia Study Group. Prevalence of inhibitors in a population of 3435 hemophilia patients in France. Thromb Haemost 1992;67:600–2.
Rizza CR, Spooner RGD. Treatment of hemophilia and related disorders in Britain and Northern Ireland during 1976-80: report on behalf of the directors of hemophilia centers in the United Kingdom. Br Med J 1983;286:929–32.
Darby SC, Keeling DM, Spooner RJ, et al. The incidence of factor VIII and factor IX inhibitors in the hemophilia population of the UK and their effect on subsequent mortality, 1977-99. J Thromb Haemost 2004;2:1047–54.
Ehrenforth S, Kreuz W, Scharrer I, et al. Incidence of development of factor VIII and factor IX inhibitors in haemophiliacs. Lancet 1992;339:594–8.
Fijnvandraat K, Turenhout EAM, van den Brink EN, et al. The missense mutation Arg593à Cys is related to antibody formation in a patient with mild Hemophilia A. Blood 1997;89:4371–7.
Vlot AJ, Wittebol S, Strengers PFW, et al. Factor VIII inhibitor in a patient with mild Hemophilia A and an Asn618-Ser mutation responsive to immune tolerance induction and cyclophosphamide. Br J Hematolol 2002;117:136–40.
Santagostino E, Gringeri A, Tagliavacca L, et al. Inhibitors to factor VIII in a family with mild hemophilia: Molecular characterization and response to factor VIII and desmopressin. Throm Haemost 1995;74:61–21.
Peerlinck K, Jacquemin M, Arnout J, et al. Antifactor VIII antibody inhibiting allogenic but not autologous factor VIII in patients with mild hemophilia A. Blood 1999;93:2267–73.
Kesteven PJ, Holland LJ, Lawrie AS, et al. Inhibitor to factor VIII in mild hemophilia. Thromb Haemost 1984;52:50–2.
Gill JC. The role of genetics in inhibitor formation. Thromb Hemostat 1999;82:500–4.
Astermark J, Berntorp E, White GC, Kroner BL; MIBS Study group. The Malmo International Brother Study (MIBS): further support for genetic predisposition to inhibitor development in hemophilia patient. Hemophilia 2001;7:267–72.
Scharrer I, Bray GL, Neutzling O. Incidence of inhibitors in Hemophilia A patients- A review of recent studies of recombinant and plasma-derived factor VIII concentrates. Hemophilia 1999;5:145–54.
Carpenter SL, Michael Soucie J, Sterner S, Presley R; Hemophilia Treatment Center Network (HTCN) Investigators. Increased prevalence of inhibitors in Hispanic patients with severe haemophilia A enrolled in the Universal Data Collection databse. Haemophilia 2012;18:e260–5.
Goodeve AC, Peake IR. The molecular basis of hemophilia A: genotype-phenotype relationships and inhibitor development. Semin Thromb Haemost 2003;29:23–30.
Schwaab R, Brackman HH, Meyer C, et al. Hemophilia A: mutation type determines risk of inhibitor formation. Thromb Haemost 1995;74:1402–6.
Oldenburg J, El-Maari O, Schwaab R. Inhibitor development in correlation to Factor VIII genotypes. Hemophilia 2002;8(Suppl. 2):23–9.
Boekhorst J, Lari GR, D’oiron R, et al. Factor VIII genotype and inhibitor development in patients with hemophilia A: Highest risk in patients with splice site mutation. Haemophilia 2008;14:729–35.
Oldenburg J, Picard JK, Schwaab R, et al. HLA genotype of patients with severe hemophilia A due to intron 22 inversion with and without inhibitors of factor VIII. Thromb Haemost 1997;77:238–42.
Hay CR, Ollier W, Pepper L, et al. HLA class II profile: A weak determinant of factor VIII inhibitor development in severe hemophilia A. UKHCDO Inhibitor Working Party. Thromb Haemost 1997;77:234–7.
Astermark J, Olderburg J, Carlson J, et al. Polymorphisms in the TNFA gene and the risk of inhibitor development in severe Hemophilia A. Blood 2006;108:3739–45.
Astemark J, Olderburg J, Pavlova A, et al. Polymorphisms in the IL10 but not in the IL1Beta and IL4 genes are associated with inhibitor development in patients with Hemophilia A. Blood 2006;107:3167–72.
Astermark J, Wang X, Olderburg, et al. MIBS Study group. Polymorphisms in the CTLA-4 gene and inhibitor development in patients with Hemophilia A. J Throm Haemost 2007;5:263–5.
Hay CR, Ludlam CA, Colvin BT, et al. Factor VIII inhibitors in mild and moderate-severity hemophilia A. Thromb Haemost 1998;79:762–6.
High HA. Factor IX molecular structure, epitopes and mutations associated with inhibitor formation. In: Aledort LM, Hoyer LW, Lusher JM, et al, eds. Inhibitors to coagulation factors. New York: Plenum Press; 1995:79-86.
Thorland ED, Drost JB, Lusher JM, et al. Anaphylactic response to factor IX replacement therapy in hemophilia B patients: Complete gene deletions confer the highest risk. Hemophilia 1999;5:101–5.
Warrier I. ITI in hemophilia B: Possibilities and problems. International Monitor on Hemophilia 2003:20–3.
Warrier I, Ewenstein B, Koerper M, et al. FIX Inhibitors and anaphylaxis in hemophilia B. J Pediatr Hematol Oncol 1997;19:23–7.
Warrier I. Management of hemophilia B patients with inhibitors and anaphylaxis. In: Varon D, Martinowitz U, Heim M, eds. Haemophilia and related disorders. Vol. 4. Oxford: Blackwell Science; 1998:574–6.
Kasper CK, Aledort L, Aronson D, et al: Proceedings: a more uniform measurement of factor VIII inhibitors. Thromb Diath Haemorrh 1975;34:612.
White GC, Taylor RE, Blatt PM, et al. Treatment of a high titer anti–factor-VIII antibody by continuous factor VIII administration: report of a case. Blood 1983;62:141–5.
Key NS, Aledort LM, Beardsley D, et al. Home treatment of mild to moderate bleeding episodes using recombinant factor VIIa (Novoseven) in haemophiliacs with inhibitors. Thromb Haemost 1998;80:912–8.
Mariani G, Scheibel E, Nogao T, et al. Immune tolerance as treatment of alloantibodies to factor VIII in hemophilia. The international registry of Immunetolerance Protocols. Semin Hematol 1994;31(Suppl. 4):62–4.
DiMichele D, Kroner B. Factor VIII/IX Subcommittee of the International Society for Thrombosis and Hemostasis. The maintenance of tolerance after successful immune tolerance induction in hemophilia A and B. The North American Registry. Factor VIII/IX Subcommittee of the International Society for Thrombosis and Haemostasis. Haematologica 2000;85(Suppl. 10):40–4.
DiMichele DM, Hay CRM. The international immune tolerance study: A multicenter prospective randomized trial in progress. J Thromb Haemost 2006;4:2271–3000.
Astermark J, Morado M, Rocino A, et al. Current European practice in immune tolerance induction therapy in patients with hemophilia and inhibitors. Hemophilia 2006;12:363–71.
DiMichele DM, Hoots WK, Pipe SW, et al. International workshop on immune tolerance induction: Consensus recommendations. Hemophilia 2007;13:(Suppl. 1):1–22.
Hay CRM, Brown S, Collins PW, et al. The diagnosis and management of factor VIII and IX inhibitors: A guideline from the United Kingdom Center Doctors Organization. Br J Haematol 2006;133:591–605.
Valentino LA, Kempton CL, Kruse-Jarres R, et al. US Guidelines for immune tolerance induction in patients with haemophilia A and inhibitors. Haemophilia 2015;21:559–67.
Silva M, Luck JV Jr. Chronic hemophilic synovitis: the role of radiosynovectomy. World Federation of Hemophilia. Treatment of Hemophilia. April 2004 (no 33). http://www1.wfh.org/publications/files/pdf-1176.pdf.
Storti E, Traldi A, Tosatti E, Davoli PG. Synovectomy, a new approach to hemophilic arthropathy. Acta Haemotol 1969;41:193–205.
Caviglia HA, Fernandez-Palazzi F, Maffei E, et al. Chemical synoviorthesis for hemophilic synovitis. Clin Orthop 1997;343:30–6.
Ahlberg A, Pettersson H. Synoviorthesis with radioactive gold in hemophiliacs. Clinical and radiological follow-up. Acta Orthop Scand 1979;50:513–7.
Lee P. The efficacy and safety of radiosynovectomy. J Rheumatol 1982;9:165–8.
Rodriguez-Merchan EC, Valentino LA. Safety of radiation exposure after radiosynovectomy in paediatric patients with haemophilia. Haemophilia 2015;21:411–8.
Green D, Lechner K. A survey of 215 non-hemophilic patients with inhibitors to factor VIII. Thromb Haemost 1981;45:200–3.
Collins PW, Hirsch S, Baglin TP, et al. Acquired hemophilia A in the United Kingdom: A 2 year national surveillance study by the United Kingdom Haemophilia Center Doctors’ Organisation. Blood 2007;109:1870–7.
Kessler CM, Ludlam CA. The treatment of acquired factor VIII inhibitors: Worldwide experience with porcine factor VIII concentrate. International Acquired Hemophilia Study Group. Semin Hematol 1993;30(Suppl. 1):22–7.
Green D, Rademaker AW, Briet E. A prospective, randomized trial of prednisone and cyclophosphamide in the treatment of patients with factor VIII autoantibodies. Thromb Haemost 1993;70:753–7.
Stasi R, Brunetti M, Stipa E, Amadori S. Selective B-cell depletion with rituximab for the treatment of patients with acquired hemophilia. Blood 2004;103:4424–8.
Franchini M. Rituximab in the treatment of adult acquired hemophilia A: a systematic review. Crit Rev Oncol Hematol 2007;63:47–52.