User login
Is Posthospital Syndrome a Result of Hospitalization-Induced Allostatic Overload?
After discharge from the hospital, patients have a significantly elevated risk for adverse events, including emergency department use, hospital readmission, and death. More than 1 in 3 patients discharged from the hospital require acute care in the month after hospital discharge, and more than 1 in 6 require readmission, with readmission diagnoses frequently differing from those of the preceding hospitalization.1-4 This heightened susceptibility to adverse events persists beyond 30 days but levels off by 7 weeks after discharge, suggesting that the period of increased risk is transient and dynamic.5
The term posthospital syndrome (PHS) describes this period of vulnerability to major adverse events following hospitalization.6 In addition to increased risk for readmission and mortality, patients in this period often show evidence of generalized dysfunction with new cognitive impairment, mobility disability, or functional decline.7-12 To date, the etiology of this vulnerability is neither well understood nor effectively addressed by transitional care interventions.13
One hypothesis to explain PHS is that stressors associated with the experience of hospitalization contribute to transient multisystem dysfunction that induces susceptibility to a broad range of medical maladies. These stressors include frequent sleep disruption, noxious sounds, painful stimuli, mobility restrictions, and poor nutrition.12 The stress hypothesis as a cause of PHS is therefore based, in large part, on evidence about allostasis and the deleterious effects of allostatic overload.
Allostasis defines a system functioning within normal stress-response parameters to promote adaptation and survival.14 In allostasis, the hypothalamic-pituitary-adrenal (HPA) axis and the sympathetic and parasympathetic branches of the autonomic nervous system (ANS) exist in homeostatic balance and respond to environmental stimuli within a range of healthy physiologic parameters. The hallmark of a system in allostasis is the ability to rapidly activate, then successfully deactivate, a stress response once the stressor (ie, threat) has resolved.14,15 To promote survival and potentiate “fight or flight” mechanisms, an appropriate stress response necessarily impacts multiple physiologic systems that result in hemodynamic augmentation and gluconeogenesis to support the anticipated action of large muscle groups, heightened vigilance and memory capabilities to improve rapid decision-making, and enhancement of innate and adaptive immune capabilities to prepare for wound repair and infection defense.14-16 The stress response is subsequently terminated by negative feedback mechanisms of glucocorticoids as well as a shift of the ANS from sympathetic to parasympathetic tone.17,18
Extended or repetitive stress exposure, however, leads to dysregulation of allostatic mechanisms responsible for stress adaptation and hinders an efficient and effective stress response. After extended stress exposure, baseline (ie, resting) HPA activity resets, causing a disruption of normal diurnal cortisol rhythm and an increase in total cortisol concentration. Moreover, in response to stress, HPA and ANS system excitation becomes impaired, and negative feedback properties are undermined.14,15 This maladaptive state, known as allostatic overload, disrupts the finely tuned mechanisms that are the foundation of mind-body balance and yields pathophysiologic consequences to multiple organ systems. Downstream ramifications of allostatic overload include cognitive deterioration, cardiovascular and immune system dysfunction, and functional decline.14,15,19
Although a stress response is an expected and necessary aspect of acute illness that promotes survival, the central thesis of this work is that additional environmental and social stressors inherent in hospitalization may unnecessarily compound stress and increase the risk of HPA axis dysfunction, allostatic overload, and subsequent multisystem dysfunction, predisposing individuals to adverse outcomes after hospital discharge. Based on data from both human subjects and animal models, we present a possible pathophysiologic mechanism for the postdischarge vulnerability of PHS, encourage critical contemplation of traditional hospitalization, and suggest interventions that might improve outcomes.
POSTHOSPITAL SYNDROME
Posthospital syndrome (PHS) describes a transient period of vulnerability after hospitalization during which patients are at elevated risk for adverse events from a broad range of conditions. In support of this characterization, epidemiologic data have demonstrated high rates of adverse outcomes following hospitalization. For example, data have shown that more than 1 in 6 older adults is readmitted to the hospital within 30 days of discharge.20 Death is also common in this first month, during which rates of postdischarge mortality may exceed initial inpatient mortality.21,22 Elevated vulnerability after hospitalization is not restricted to older adults, as readmission risk among younger patients 18 to 64 years of age may be even higher for selected conditions, such as heart failure.3,23
Vulnerability after hospitalization is broad. In patients over age 65 initially admitted for heart failure or acute myocardial infarction, only 35% and 10% of readmissions are for recurrent heart failure or reinfarction, respectively.1 Nearly half of readmissions are for noncardiovascular causes.1 Similarly, following hospitalization for pneumonia, more than 60 percent of readmissions are for nonpulmonary etiologies. Moreover, the risk for all these causes of readmission is much higher than baseline risk, indicating an extended period of lack of resilience to many types of illness.24 These patterns of broad susceptibility also extend to younger adults hospitalized with common medical conditions.3
Accumulating evidence suggests that hospitalized patients face functional decline, debility, and risk for adverse events despite resolution of the presenting illness, implying perhaps that the hospital environment itself is hazardous to patients’ health. In 1993, Creditor hypothesized that the “hazards of hospitalization,” including enforced bed-rest, sensory deprivation, social isolation, and malnutrition lead to a “cascade of dependency” in which a collection of small insults to multiple organ systems precipitates loss of function and debility despite cure or resolution of presenting illness.12 Covinsky (2011) later defined hospitalization-associated disability as an iatrogenic hospital-related “disorder” characterized by new impairments in abilities to perform basic activities of daily living such as bathing, feeding, toileting, dressing, transferring, and walking at the time of hospital discharge.11 Others have described a postintensive-care syndrome (PICS),25 characterized by cognitive, psychiatric, and physical impairments acquired during hospitalization for critical illness that persist postdischarge and increase the long-term risk for adverse outcomes, including elevated mortality rates,26,27 readmission rates,28 and physical disabilities.29 Similar to the “hazards of hospitalization,” PICS is thought to be related to common experiences of ICU stays, including mobility restriction, sensory deprivation, sleep disruption, sedation, malnutrition, and polypharmacy.30-33
Taken together, these data suggest that adverse health consequences attributable to hospitalization extend across the spectrum of age, presenting disease severity, and hospital treatment location. As detailed below, the PHS hypothesis is rooted in a mechanistic understanding of the role of exogenous stressors in producing physiologic dysregulation and subsequent adverse health effects across multiple organ systems.
Nature of Stress in the Hospital
Compounding the stress of acute illness, hospitalized patients are routinely and repetitively exposed to a wide variety of environmental stressors that may have downstream adverse consequences (Table 1). In the absence of overt clinical manifestations of harm, the possible subclinical physiologic dysfunction generated by the following stress exposures may increase patients’ susceptibility to the manifestations of PHS.
Sleep Disruption
Sleep disruptions trigger potent stress responses,34,35 yet they are common occurrences during hospitalization. In surveys, about half of patients report poor sleep quality during hospitalization that persists for many months after discharge.36 In a simulated hospital setting, test subjects exposed to typical hospital sounds (paging system, machine alarms, etc.) experienced significant sleep-wake cycle abnormalities.37 Although no work has yet focused specifically on the physiologic consequences of sleep disruption and stress in hospitalized patients, in healthy humans, mild sleep disruption has clear effects on allostasis by disrupting HPA activity, raising cortisol levels, diminishing parasympathetic tone, and impairing cognitive performance.18,34,35,38,39
Malnourishment
Malnourishment in hospitalized patients is common, with one-fifth of hospitalized patients receiving nothing per mouth or clear liquid diets for more than 3 continuous days,40 and one-fifth of hospitalized elderly patients receiving less than half of their calculated nutrition requirements.41 Although the relationship between food restriction, cortisol levels, and postdischarge outcomes has not been fully explored, in healthy humans, meal anticipation, meal withdrawal (withholding an expected meal), and self-reported dietary restraint are known to generate stress responses.42,43 Furthermore, malnourishment during hospitalization is associated with increased 90-day and 1-year mortality after discharge,44 adding malnourishment to the list of plausible components of hospital-related stress.
Mobility Restriction
Physical activity counterbalances stress responses and minimizes downstream consequences of allostatic load,15 yet mobility limitations via physical and chemical restraints are common in hospitalized patients, particularly among the elderly.45-47 Many patients are tethered to devices that make ambulation hazardous, such as urinary catheters and infusion pumps. Even without physical or chemical restraints or a limited mobility order, patients may be hesitant to leave the room so as not to miss transport to a diagnostic study or an unscheduled physician’s visit. Indeed, mobility limitations of hospitalized patients increase the risk for adverse events after discharge, while interventions designed to encourage mobility are associated with improved postdischarge outcomes.47,48
Other Stressors
Other hospital-related aversive stimuli are less commonly quantified, but clearly exist. According to surveys of hospitalized patients, sources of emotional stress include social isolation; loss of autonomy and privacy; fear of serious illness; lack of control over activities of daily living; lack of clear communication between treatment team and patients; and death of a patient roommate.49,50 Furthermore, consider the physical discomfort and emotional distress of patients with urinary incontinence awaiting assistance for a diaper or bedding change or the pain of repetitive blood draws or other invasive testing. Although individualized, the subjective discomfort and emotional distress associated with these experiences undoubtedly contribute to the stress of hospitalization.
IMPACT OF ALLOSTATIC OVERLOAD ON PHYSIOLOGIC FUNCTION
Animal Models of Stress
Laboratory techniques reminiscent of the numerous environmental stressors associated with hospitalization have been used to reliably trigger allostatic overload in healthy young animals.51 These techniques include sequential exposure to aversive stimuli, including food and water deprivation, continuous overnight illumination, paired housing with known and unknown cagemates, mobility restriction, soiled cage conditions, and continuous noise. All of these techniques have been shown to cause HPA axis and ANS dysfunction, allostatic overload, and subsequent stress-mediated consequences to multiple organ systems.19,52-54 Given the remarkable similarity of these protocols to common experiences during hospitalization, animal models of stress may be useful in understanding the spectrum of maladaptive consequences experienced by patients within the hospital (Figure 1).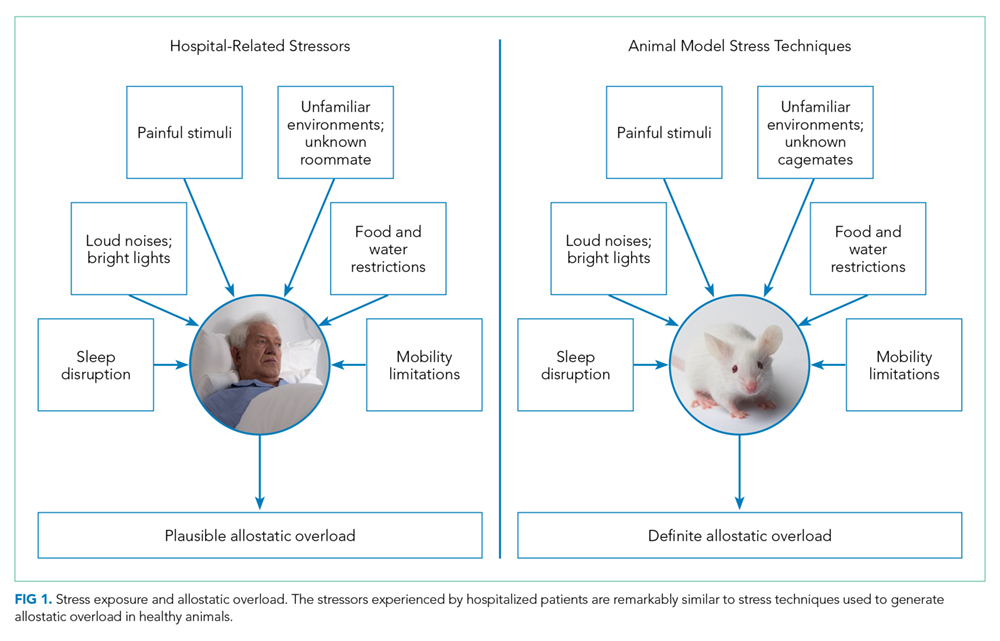
These animal models of stress have resulted in a number of instructive findings. For example, in rodents, extended stress exposure induces structural and functional remodeling of neuronal networks that precipitate learning and memory, working memory, and attention impairments.55-57 These exposures also result in cardiovascular abnormalities, including dyslipidemia, progressive atherosclerosis,58,59 and enhanced inflammatory cytokine expression,60 all of which increase both atherosclerotic burden and susceptibility to plaque rupture, leading to elevated risk for major cardiovascular adverse events. Moreover, these extended stress exposures in animals increase susceptibility to both bacterial and viral infections and increase their severity.16,61 This outcome appears to be driven by a stress-induced elevation of glucocorticoid levels, decreased leukocyte proliferation, altered leukocyte trafficking, and a transition to a proinflammatory cytokine environment.16, 61 Allostatic overload has also been shown to contribute to metabolic dysregulation involving insulin resistance, persistence of hyperglycemia, dyslipidemia, catabolism of lean muscle, and visceral adipose tissue deposition.62-64 In addition to cardiovascular, immune, and metabolic consequences of allostatic overload, the spectrum of physiologic dysfunction in animal models is broad and includes mood disorder symptoms,65 intestinal barrier abnormalities,66 airway reactivity exacerbation,67 and enhanced tumor growth.68
Although the majority of this research highlights the multisystem effects of variable stress exposure in healthy animals, preliminary evidence suggests that aged or diseased animals subjected to additional stressors display a heightened inflammatory cytokine response that contributes to exaggerated sickness behavior and greater and prolonged cognitive deficits.69 Future studies exploring the consequences of extended stress exposure in animals with existing disease or debility may therefore more closely simulate the experience of hospitalized patients and perhaps further our understanding of PHS.
Hospitalized Patients
While no intervention studies have examined the effects of potential hospital stressors on the development of allostatic overload, there is evidence from small studies that dysregulated stress responses during hospitalization are associated with adverse events. For example, high serum cortisol, catecholamine, and proinflammatory cytokine levels during hospitalization have individually been associated with the development of cognitive dysfunction,70-72 increased risk of cardiovascular events such as myocardial infarction and stroke in the year following discharge,73-76 and the development of wound infections after discharge.77 Moreover, elevated plasma glucose during admission for myocardial infarction in patients with or without diabetes has been associated with greater in-hospital and 1-year mortality,78 with a similar relationship seen between elevated plasma glucose and survival after admission for stroke79 and pneumonia.80 Furthermore, in addition to atherothrombosis, stress may contribute to the risk for venous thromboembolism,81 resulting in readmissions for deep vein thrombosis or pulmonary embolism posthospitalization. Although potentially surrogate markers of illness acuity, a handful of studies have shown that these stress biomarkers are actually only weakly correlated with,82 or independent of,72,76 disease severity. As discussed in detail below, future studies utilizing a summative measure of multisystem physiologic dysfunction as opposed to individual biomarkers may more accurately reflect the cumulative stress effects of hospitalization and subsequent risk for adverse events.
Additional Considerations
Elderly patients, in particular, may have heightened susceptibility to the consequences of allostatic overload due to common geriatric issues such as multimorbidity and frailty. Patients with chronic diseases display both baseline HPA axis abnormalities as well as dysregulated stress responses and may therefore be more vulnerable to hospitalization-related stress. For example, when subjected to psychosocial stress, patients with chronic conditions such as diabetes, heart failure, or atherosclerosis demonstrate elevated cortisol levels, increased circulating markers of inflammation, as well as prolonged hemodynamic recovery after stress resolution compared with normal controls.83-85 Additionally, frailty may affect an individual’s susceptibility to exogenous stress. Indeed, frailty identified on hospital admission increases the risk for adverse outcomes during hospitalization and postdischarge.86 Although the specific etiology of this relationship is unclear, persons with frailty are known to have elevated levels of cortisol and other inflammatory markers,87,88 which may contribute to adverse outcomes in the face of additional stressors.
IMPLICATIONS AND NEXT STEPS
A large body of evidence stretching from bench to bedside suggests that environmental stressors associated with hospitalization are toxic. Understanding PHS within the context of hospital-induced allostatic overload presents a unifying theory for the interrelated multisystem dysfunction and increased susceptibility to adverse events that patients experience after discharge (Figure 2). Furthermore, it defines a potential pathophysiological mechanism for the cognitive impairment, elevated cardiovascular risk, immune system dysfunction, metabolic derangements, and functional decline associated with PHS. Additionally, this theory highlights environmental interventions to limit PHS development and suggests mechanisms to promote stress resilience. Although it is difficult to disentangle the consequences of the endogenous stress triggered by an acute illness from the exogenous stressors related to hospitalization, it is likely that the 2 simultaneous exposures compound risk for stress system dysregulation and allostatic overload. Moreover, hospitalized patients with preexisting HPA axis dysfunction at baseline from chronic disease or advancing age may be even more susceptible to these adverse outcomes. If this hypothesis is true, a reduction in PHS would require mitigation of the modifiable environmental stressors encountered by patients during hospitalization. Directed efforts to diminish ambient noise, limit nighttime disruptions, thoughtfully plan procedures, consider ongoing nutritional status, and promote opportunities for patients to exert some control over their environment may diminish the burden of extrinsic stressors encountered by all patients in the hospital and improve outcomes after discharge.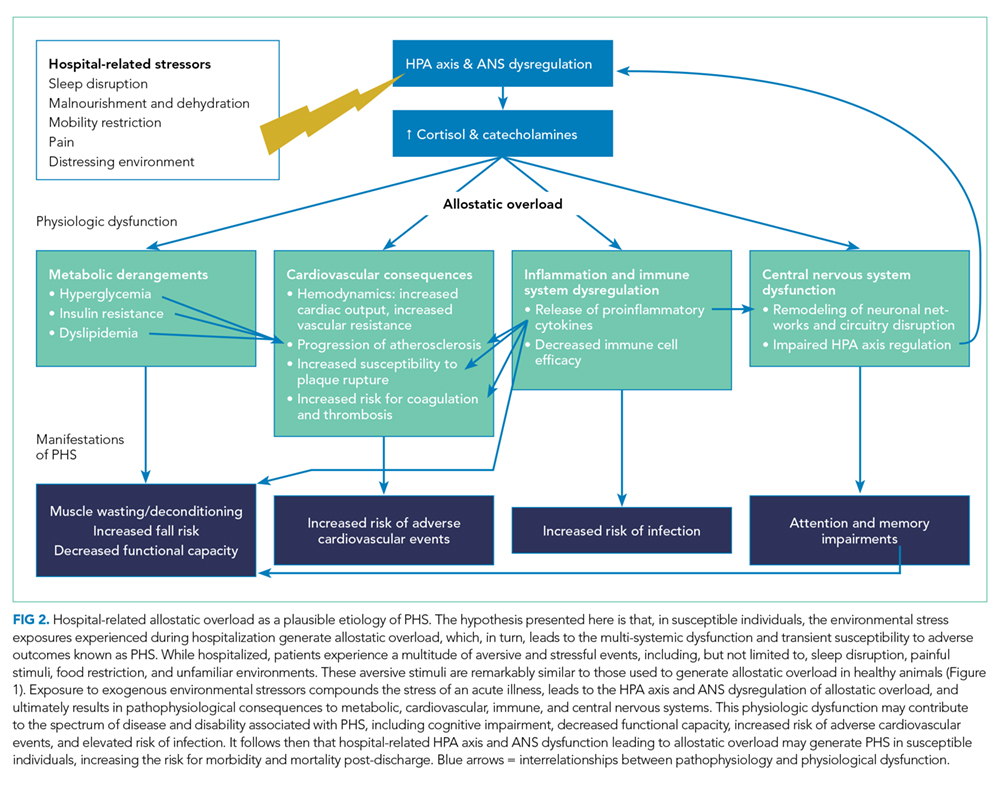
Hospitals are increasingly recognizing the importance of improving patients’ experience of hospitalization by reducing exposure to potential toxicities. For example, many hospitals are now attempting to reduce sleep disturbances and sleep latency through reduced nighttime noise and light levels, fewer nighttime interruptions for vital signs checks and medication administration, and commonsensical interventions like massages, herbal teas, and warm milk prior to bedtime.89 Likewise, intensive care units are targeting environmental and physical stressors with a multifaceted approach to decrease sedative use, promote healthy sleep cycles, and encourage exercise and ambulation even in those patients who are mechanically ventilated.30 Another promising development has been the increase of Hospital at Home programs. In these programs, patients who meet the criteria for inpatient admission are instead comprehensively managed at home for their acute illness through a multidisciplinary effort between physicians, nurses, social workers, physical therapists, and others. Patients hospitalized at home report higher levels of satisfaction and have modest functional gains, improved health-related quality of life, and decreased risk of mortality at 6 months compared with hospitalized patients.90,91 With some admitting diagnoses (eg, heart failure), hospitalization at home may be associated with decreased readmission risk.92 Although not yet investigated on a physiologic level, perhaps the benefits of hospital at home are partially due to the dramatic difference in exposure to environmental stressors.
A tool that quantifies hospital-associated stress may help health providers appreciate the experience of patients and better target interventions to aspects of their structure and process that contribute to allostatic overload. Importantly, allostatic overload cannot be identified by one biomarker of stress but instead requires evidence of dysregulation across inflammatory, neuroendocrine, hormonal, and cardiometabolic systems. Future studies to address the burden of stress faced by hospitalized patients should consider a summative measure of multisystem dysregulation as opposed to isolated assessments of individual biomarkers. Allostatic load has previously been operationalized as the summation of a variety of hemodynamic, hormonal, and metabolic factors, including blood pressure, lipid profile, glycosylated hemoglobin, cortisol, catecholamine levels, and inflammatory markers.93 To develop a hospital-associated allostatic load index, models should ideally be adjusted for acute illness severity, patient-reported stress, and capacity for stress resilience. This tool could then be used to quantify hospitalization-related allostatic load and identify those at greatest risk for adverse events after discharge, as well as measure the effectiveness of strategic environmental interventions (Table 2). A natural first experiment may be a comparison of the allostatic load of hospitalized patients versus those hospitalized at home.
The risk of adverse outcomes after discharge is likely a function of the vulnerability of the patient and the degree to which the patient’s healthcare team and social support network mitigates this vulnerability. That is, there is a risk that a person struggles in the postdischarge period and, in many circumstances, a strong healthcare team and social network can identify health problems early and prevent them from progressing to the point that they require hospitalization.13,94-96 There are also hospital occurrences, outside of allostatic load, that can lead to complications that lengthen the stay, weaken the patient, and directly contribute to subsequent vulnerability.94,97 Our contention is that the allostatic load of hospitalization, which may also vary by patient depending on the circumstances of hospitalization, is just one contributor, albeit potentially an important one, to vulnerability to medical problems after discharge.
In conclusion, a plausible etiology of PHS is the maladaptive mind-body consequences of common stressors during hospitalization that compound the stress of acute illness and produce allostatic overload. This stress-induced dysfunction potentially contributes to a spectrum of generalized disease susceptibility and risk of adverse outcomes after discharge. Focused efforts to diminish patient exposure to hospital-related stressors during and after hospitalization might diminish the presence or severity of PHS. Viewing PHS from this perspective enables the development of hypothesis-driven risk-prediction models, encourages critical contemplation of traditional hospitalization, and suggests that targeted environmental interventions may significantly reduce adverse outcomes.
1. Dharmarajan K, Hsieh AF, Lin Z, et al. Diagnoses and timing of 30-day readmissions after hospitalization for heart failure, acute myocardial infarction, or pneumonia. JAMA. 2013;309(4):355-363. http://dx.doi.org/10.1001/jama.2012.216476.
2. Jencks SF, Williams MV, Coleman EA. Rehospitalizations among patients in the Medicare fee-for-service program. N Engl J Med. 2009;360(14):1418-1428. http://dx.doi.org/10.1056/NEJMsa0803563.
3. Ranasinghe I, Wang Y, Dharmarajan K, Hsieh AF, Bernheim SM, Krumholz HM. Readmissions after hospitalization for heart failure, acute myocardial infarction, or pneumonia among young and middle-aged adults: a retrospective observational cohort study. PLoS Med. 2014;11(9):e1001737. http://dx.doi.org/10.1371/journal.pmed.1001737.
4. Vashi AA, Fox JP, Carr BG, et al. Use of hospital-based acute care among patients recently discharged from the hospital. JAMA. 2013;309(4):364-371. http://dx.doi.org/10.1001/jama.2012.216219.
5. Dharmarajan K, Hsieh AF, Kulkarni VT, et al. Trajectories of risk after hospitalization for heart failure, acute myocardial infarction, or pneumonia: retrospective cohort study. BMJ. 2015;350:h411. http://dx.doi.org/10.1136/bmj.h411.
6. Krumholz HM. Post-hospital syndrome--an acquired, transient condition of generalized risk. N Engl J Med. 2013;368(2):100-102. http://dx.doi.org/10.1056/NEJMp1212324.
7. Ferrante LE, Pisani MA, Murphy TE, Gahbauer EA, Leo-Summers LS, Gill TM. Functional trajectories among older persons before and after critical illness. JAMA Intern Med. 2015;175(4):523-529. http://dx.doi.org/10.1001/jamainternmed.2014.7889.
8. Gill TM, Allore HG, Holford TR, Guo Z. Hospitalization, restricted activity, and the development of disability among older persons. JAMA. 2004;292(17):2115-2124. http://dx.doi.org/10.1001/jama.292.17.2115.
9. Inouye SK, Zhang Y, Han L, Leo-Summers L, Jones R, Marcantonio E. Recoverable cognitive dysfunction at hospital admission in older persons during acute illness. J Gen Intern Med. 2006;21(12):1276-1281. http://dx.doi.org/10.1111/j.1525-1497.2006.00613.x.
10. Lindquist LA, Go L, Fleisher J, Jain N, Baker D. Improvements in cognition following hospital discharge of community dwelling seniors. J Gen Intern Med. 2011;26(7):765-770. http://dx.doi.org/10.1007/s11606-011-1681-1.
11. Covinsky KE, Pierluissi E, Johnston CB. Hospitalization-associated disability: “She was probably able to ambulate, but I’m not sure.” JAMA. 2011;306(16):1782-1793. http://dx.doi.org/10.1001/jama.2011.1556.
12. Creditor MC. Hazards of hospitalization of the elderly. Ann Intern Med. 1993;118(3):219-223. http://dx.doi.org/10.7326/0003-4819-118-3-199302010-00011.
13. Dharmarajan K, Krumholz HM. Strategies to reduce 30-day readmissions in older patients hospitalized with heart failure and acute myocardial infarction. Curr Geriatr Rep. 2014;3(4):306-315. http://dx.doi.org/10.1007/s13670-014-0103-8.
14. McEwen BS. Protective and damaging effects of stress mediators. N Engl J Med. 1998;338(3):171-179. http://dx.doi.org/10.1056/NEJM199801153380307.
15. McEwen BS, Gianaros PJ. Stress- and allostasis-induced brain plasticity. Annu Rev Med. 2011;62:431-445. http://dx.doi.org/10.1146/annurev-med-052209-100430.
16. Dhabhar FS. Enhancing versus suppressive effects of stress on immune function: implications for immunoprotection and immunopathology. Neuroimmunomodulation. 2009;16(5):300-317. http://dx.doi.org/10.1159/000216188.
17. Thayer JF, Sternberg E. Beyond heart rate variability: vagal regulation of allostatic systems. Ann N Y Acad Sci. 2006;1088:361-372. http://dx.doi.org/10.1196/annals.1366.014.
18. Jacobson L, Akana SF, Cascio CS, Shinsako J, Dallman MF. Circadian variations in plasma corticosterone permit normal termination of adrenocorticotropin responses to stress. Endocrinology. 1988;122(4):1343-1348. http://dx.doi.org/10.1210/endo-122-4-1343.
19. McEwen BS. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev. 2007;87(3):873-904. http://dx.doi.org/10.1152/physrev.00041.2006.
20. Medicare Hospital Quality Chartbook 2014: Performance Report on Outcome Measures. Prepared by Yale New Haven Health Services Corporation Center for Outcomes Research and Evaluation for Centers for Medicare & Medicaid Services. https://www.cms.gov/medicare/quality-initiatives-patient-assessment-instruments/hospitalqualityinits/downloads/medicare-hospital-quality-chartbook-2014.pdf. Accessed February 26, 2018.
21. Bueno H, Ross JS, Wang Y, et al. Trends in length of stay and short-term outcomes among Medicare patients hospitalized for heart failure, 1993-2006. JAMA. 2010;303(21):2141-2147. http://dx.doi.org/10.1001/jama.2010.748.
22. Drye EE, Normand SL, Wang Y, et al. Comparison of hospital risk-standardized mortality rates calculated by using in-hospital and 30-day models: an observational study with implications for hospital profiling. Ann Intern Med. 2012;156(1 Pt 1):19-26. http://dx.doi.org/10.7326/0003-4819-156-1-201201030-00004.
23. Dharmarajan K, Hsieh A, Dreyer RP, Welsh J, Qin L, Krumholz HM. Relationship between age and trajectories of rehospitalization risk in older adults. J Am Geriatr Soc. 2017;65(2):421-426. http://dx.doi.org/10.1111/jgs.14583.
24. Krumholz HM, Hsieh A, Dreyer RP, Welsh J, Desai NR, Dharmarajan K. Trajectories of risk for specific readmission diagnoses after hospitalization for heart failure, acute myocardial infarction, or pneumonia. PLoS One. 2016;11(10):e0160492. http://dx.doi.org/10.1371/journal.pone.0160492.
25. Needham DM, Davidson J, Cohen H, et al. Improving long-term outcomes after discharge from intensive care unit: report from a stakeholders’ conference. Crit Care Med. 2012;40(2):502-509. http://dx.doi.org/10.1097/CCM.0b013e318232da75.
26. Brinkman S, de Jonge E, Abu-Hanna A, Arbous MS, de Lange DW, de Keizer NF. Mortality after hospital discharge in ICU patients. Crit Care Med. 2013;41(5):1229-1236. http://dx.doi.org/10.1097/CCM.0b013e31827ca4e1.
27. Steenbergen S, Rijkenberg S, Adonis T, Kroeze G, van Stijn I, Endeman H. Long-term treated intensive care patients outcomes: the one-year mortality rate, quality of life, health care use and long-term complications as reported by general practitioners. BMC Anesthesiol. 2015;15:142. http://dx.doi.org/10.1186/s12871-015-0121-x.
28. Hill AD, Fowler RA, Pinto R, Herridge MS, Cuthbertson BH, Scales DC. Long-term outcomes and healthcare utilization following critical illness--a population-based study. Crit Care. 2016;20:76. http://dx.doi.org/10.1186/s13054-016-1248-y.
29. Jackson JC, Pandharipande PP, Girard TD, et al. Depression, post-traumatic stress disorder, and functional disability in survivors of critical illness in the BRAIN-ICU study: a longitudinal cohort study. Lancet Respir Med. 2014;2(5):369-379. http://dx.doi.org/10.1016/S2213-2600(14)70051-7.
30. Balas MC, Vasilevskis EE, Olsen KM, et al. Effectiveness and safety of the awakening and breathing coordination, delirium monitoring/management, and early exercise/mobility bundle. Crit Care Med. 2014;42(5):1024-1036. http://dx.doi.org/10.1097/CCM.0000000000000129.
31. Kress JP, Hall JB. ICU-acquired weakness and recovery from critical illness. N Engl J Med. 2014;370(17):1626-1635. http://dx.doi.org/10.1056/NEJMra1209390.
32. Mendez-Tellez PA, Needham DM. Early physical rehabilitation in the ICU and ventilator liberation. Respir Care. 2012;57(10):1663-1669. http://dx.doi.org/10.4187/respcare.01931.
33. Schweickert WD, Hall J. ICU-acquired weakness. Chest. 2007;131(5):1541-1549. http://dx.doi.org/10.1378/chest.06-2065.
34. Balbo M, Leproult R, Van Cauter E. Impact of sleep and its disturbances on hypothalamo-pituitary-adrenal axis activity. Int J Endocrinol. 2010;2010:759234. http://dx.doi.org/10.1155/2010/759234.
35. Spiegel K, Leproult R, Van Cauter E. Impact of sleep debt on metabolic and endocrine function. Lancet. 1999;354(9188):1435-1439. http://dx.doi.org/10.1016/S0140-6736(99)01376-8.
36. Orwelius L, Nordlund A, Nordlund P, Edell-Gustafsson U, Sjoberg F. Prevalence of sleep disturbances and long-term reduced health-related quality of life after critical care: a prospective multicenter cohort study. Crit Care. 2008;12(4):R97. http://dx.doi.org/10.1186/cc6973.
37. Buxton OM, Ellenbogen JM, Wang W, et al. Sleep disruption due to hospital noises: a prospective evaluation. Ann Intern Med. 2012;157(3):170-179. http://dx.doi.org/10.7326/0003-4819-157-3-201208070-00472.
38. Sunbul M, Kanar BG, Durmus E, Kivrak T, Sari I. Acute sleep deprivation is associated with increased arterial stiffness in healthy young adults. Sleep Breath. 2014;18(1):215-220. http://dx.doi.org/10.1007/s11325-013-0873-9.
39. Van Dongen HP, Maislin G, Mullington JM, Dinges DF. The cumulative cost of additional wakefulness: dose-response effects on neurobehavioral functions and sleep physiology from chronic sleep restriction and total sleep deprivation. Sleep. 2003;26(2):117-126. http://dx.doi.org/10.1093/sleep/26.2.117.
40. Franklin GA, McClave SA, Hurt RT, et al. Physician-delivered malnutrition: why do patients receive nothing by mouth or a clear liquid diet in a university hospital setting? JPEN J Parenter Enteral Nutr. 2011;35(3):337-342. http://dx.doi.org/10.1177/0148607110374060.
41. Sullivan DH, Sun S, Walls RC. Protein-energy undernutrition among elderly hospitalized patients: a prospective study. JAMA. 1999;281(21):2013-2019. http://dx.doi.org/10.1001/jama.281.21.2013.
42. Anderson DA, Shapiro JR, Lundgren JD, Spataro LE, Frye CA. Self-reported dietary restraint is associated with elevated levels of salivary cortisol. Appetite. 2002;38(1):13-17. http://dx.doi.org/10.1006/appe.2001.0459.
43. Ott V, Friedrich M, Prilop S, et al. Food anticipation and subsequent food withdrawal increase serum cortisol in healthy men. Physiol Behav. 2011;103(5):594-599. http://dx.doi.org/10.1016/j.physbeh.2011.04.020.
44. Covinsky KE, Martin GE, Beyth RJ, Justice AC, Sehgal AR, Landefeld CS. The relationship between clinical assessments of nutritional status and adverse outcomes in older hospitalized medical patients. J Am Geriatr Soc. 1999;47(5):532-538. http://dx.doi.org/10.1111/j.1532-5415.1999.tb02566.x.
45. Lazarus BA, Murphy JB, Coletta EM, McQuade WH, Culpepper L. The provision of physical activity to hospitalized elderly patients. Arch Intern Med. 1991;151(12):2452-2456.
46. Minnick AF, Mion LC, Johnson ME, Catrambone C, Leipzig R. Prevalence and variation of physical restraint use in acute care settings in the US. J Nurs Scholarsh. 2007;39(1):30-37. http://dx.doi.org/10.1111/j.1547-5069.2007.00140.x.
47. Zisberg A, Shadmi E, Sinoff G, Gur-Yaish N, Srulovici E, Admi H. Low mobility during hospitalization and functional decline in older adults. J Am Geriatr Soc. 2011;59(2):266-273. http://dx.doi.org/10.1111/j.1532-5415.2010.03276.x.
48. Landefeld CS, Palmer RM, Kresevic DM, Fortinsky RH, Kowal J. A randomized trial of care in a hospital medical unit especially designed to improve the functional outcomes of acutely ill older patients. N Engl J Med. 1995;332(20):1338-1344. http://dx.doi.org/10.1056/NEJM199505183322006.
49. Douglas CH, Douglas MR. Patient-friendly hospital environments: exploring the patients’ perspective. Health Expectations: an international journal of public participation in health care and health policy. 2004;7(1):61-73. http://dx.doi.org/10.1046/j.1369-6513.2003.00251.x.
50. Volicer BJ. Hospital stress and patient reports of pain and physical status. Journal Human Stress. 1978;4(2):28-37. http://dx.doi.org/10.1080/0097840X.1978.9934984.
51. Willner P, Towell A, Sampson D, Sophokleous S, Muscat R. Reduction of sucrose preference by chronic unpredictable mild stress, and its restoration by a tricyclic antidepressant. Psychopharmacology (Berl). 1987;93(3):358-364. http://dx.doi.org/10.1007/BF00187257.
52. Grippo AJ, Francis J, Beltz TG, Felder RB, Johnson AK. Neuroendocrine and cytokine profile of chronic mild stress-induced anhedonia. Physiol Behav. 2005;84(5):697-706. http://dx.doi.org/10.1016/j.physbeh.2005.02.011.
53. Krishnan V, Nestler EJ. Animal models of depression: molecular perspectives. Curr Top Behav Neurosci. 2011;7:121-147. http://dx.doi.org/10.1007/7854_2010_108.
54. Magarinos AM, McEwen BS. Stress-induced atrophy of apical dendrites of hippocampal CA3c neurons: comparison of stressors. Neuroscience. 1995;69(1):83-88. http://dx.doi.org/10.1016/0306-4522(95)00256-I.
55. McEwen BS. Plasticity of the hippocampus: adaptation to chronic stress and allostatic load. Ann N Y Acad Sci. 2001;933:265-277. http://dx.doi.org/10.1111/j.1749-6632.2001.tb05830.x.
56. McEwen BS. The brain on stress: toward an integrative approach to brain, body, and behavior. Perspect Psychol Sci. 2013;8(6):673-675. http://dx.doi.org/10.1177/1745691613506907.
57. McEwen BS, Morrison JH. The brain on stress: vulnerability and plasticity of the prefrontal cortex over the life course. Neuron. 2013;79(1):16-29. http://dx.doi.org/10.1016/j.neuron.2013.06.028.
58. Dutta P, Courties G, Wei Y, et al. Myocardial infarction accelerates atherosclerosis. Nature. 2012;487(7407):325-329. http://dx.doi.org/10.1038/nature11260.
59. Lu XT, Liu YF, Zhang L, et al. Unpredictable chronic mild stress promotes atherosclerosis in high cholesterol-fed rabbits. Psychosom Med. 2012;74(6):604-611. http://dx.doi.org/10.1097/PSY.0b013e31825d0b71.
60. Heidt T, Sager HB, Courties G, et al. Chronic variable stress activates hematopoietic stem cells. Nat Med. 2014;20(7):754-758. http://dx.doi.org/10.1038/nm.3589.
61. Sheridan JF, Feng NG, Bonneau RH, Allen CM, Huneycutt BS, Glaser R. Restraint stress differentially affects anti-viral cellular and humoral immune responses in mice. J Neuroimmunol. 1991;31(3):245-255. http://dx.doi.org/10.1016/0165-5728(91)90046-A.
62. Kyrou I, Tsigos C. Stress hormones: physiological stress and regulation of metabolism. Curr Opin Pharmacol. 2009;9(6):787-793. http://dx.doi.org/10.1016/j.coph.2009.08.007.
63. Rosmond R. Role of stress in the pathogenesis of the metabolic syndrome. Psychoneuroendocrinology. 2005;30(1):1-10. http://dx.doi.org/10.1016/j.psyneuen.2004.05.007.
64. Tamashiro KL, Sakai RR, Shively CA, Karatsoreos IN, Reagan LP. Chronic stress, metabolism, and metabolic syndrome. Stress. 2011;14(5):468-474. http://dx.doi.org/10.3109/10253890.2011.606341.
65. McEwen BS. Mood disorders and allostatic load. Biol Psychiatry. 2003;54(3):200-207. http://dx.doi.org/10.1016/S0006-3223(03)00177-X.
66. Zareie M, Johnson-Henry K, Jury J, et al. Probiotics prevent bacterial translocation and improve intestinal barrier function in rats following chronic psychological stress. Gut. 2006;55(11):1553-1560. http://dx.doi.org/10.1136/gut.2005.080739.
67. Joachim RA, Quarcoo D, Arck PC, Herz U, Renz H, Klapp BF. Stress enhances airway reactivity and airway inflammation in an animal model of allergic bronchial asthma. Psychosom Med. 2003;65(5):811-815. http://dx.doi.org/10.1097/01.PSY.0000088582.50468.A3.
68. Thaker PH, Han LY, Kamat AA, et al. Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat Med. 2006;12(8):939-944. http://dx.doi.org/10.1038/nm1447.
69. Schreuder L, Eggen BJ, Biber K, Schoemaker RG, Laman JD, de Rooij SE. Pathophysiological and behavioral effects of systemic inflammation in aged and diseased rodents with relevance to delirium: A systematic review. Brain Behav Immun. 2017;62:362-381. http://dx.doi.org/10.1016/j.bbi.2017.01.010.
70. Mu DL, Li LH, Wang DX, et al. High postoperative serum cortisol level is associated with increased risk of cognitive dysfunction early after coronary artery bypass graft surgery: a prospective cohort study. PLoS One. 2013;8(10):e77637. http://dx.doi.org/10.1371/journal.pone.0077637.
71. Mu DL, Wang DX, Li LH, et al. High serum cortisol level is associated with increased risk of delirium after coronary artery bypass graft surgery: a prospective cohort study. Crit Care. 2010;14(6):R238. http://dx.doi.org/10.1186/cc9393.
72. Nguyen DN, Huyghens L, Zhang H, Schiettecatte J, Smitz J, Vincent JL. Cortisol is an associated-risk factor of brain dysfunction in patients with severe sepsis and septic shock. Biomed Res Int. 2014;2014:712742. http://dx.doi.org/10.1155/2014/712742.
73. Elkind MS, Carty CL, O’Meara ES, et al. Hospitalization for infection and risk of acute ischemic stroke: the Cardiovascular Health Study. Stroke. 2011;42(7):1851-1856. http://dx.doi.org/10.1161/STROKEAHA.110.608588.
74. Feibel JH, Hardy PM, Campbell RG, Goldstein MN, Joynt RJ. Prognostic value of the stress response following stroke. JAMA. 1977;238(13):1374-1376.
75. Jutla SK, Yuyun MF, Quinn PA, Ng LL. Plasma cortisol and prognosis of patients with acute myocardial infarction. J Cardiovasc Med (Hagerstown). 2014;15(1):33-41. http://dx.doi.org/10.2459/JCM.0b013e328364100b.
76. Yende S, D’Angelo G, Kellum JA, et al. Inflammatory markers at hospital discharge predict subsequent mortality after pneumonia and sepsis. Am J Respir Crit Care Med. 2008;177(11):1242-1247. http://dx.doi.org/10.1164/rccm.200712-1777OC.
77. Gouin JP, Kiecolt-Glaser JK. The impact of psychological stress on wound healing: methods and mechanisms. Immunol Allergy Clin North Am. 2011;31(1):81-93. http://dx.doi.org/10.1016/j.iac.2010.09.010.
78. Capes SE, Hunt D, Malmberg K, Gerstein HC. Stress hyperglycaemia and increased risk of death after myocardial infarction in patients with and without diabetes: a systematic overview. Lancet. 2000;355(9206):773-778. http://dx.doi.org/10.1016/S0140-6736(99)08415-9.
79. O’Neill PA, Davies I, Fullerton KJ, Bennett D. Stress hormone and blood glucose response following acute stroke in the elderly. Stroke. 1991;22(7):842-847. http://dx.doi.org/10.1161/01.STR.22.7.842.
80. Waterer GW, Kessler LA, Wunderink RG. Medium-term survival after hospitalization with community-acquired pneumonia. Am J Respir Crit Care Med. 2004;169(8):910-914. http://dx.doi.org/10.1164/rccm.200310-1448OC.
81. Rosengren A, Freden M, Hansson PO, Wilhelmsen L, Wedel H, Eriksson H. Psychosocial factors and venous thromboembolism: a long-term follow-up study of Swedish men. J Thrombosis Haemostasis. 2008;6(4):558-564. http://dx.doi.org/10.1111/j.1538-7836.2007.02857.x.
82. Oswald GA, Smith CC, Betteridge DJ, Yudkin JS. Determinants and importance of stress hyperglycaemia in non-diabetic patients with myocardial infarction. BMJ. 1986;293(6552):917-922. http://dx.doi.org/10.1136/bmj.293.6552.917.
83. Middlekauff HR, Nguyen AH, Negrao CE, et al. Impact of acute mental stress on sympathetic nerve activity and regional blood flow in advanced heart failure: implications for ‘triggering’ adverse cardiac events. Circulation. 1997;96(6):1835-1842. http://dx.doi.org/10.1161/01.CIR.96.6.1835.
84. Nijm J, Jonasson L. Inflammation and cortisol response in coronary artery disease. Ann Med. 2009;41(3):224-233. http://dx.doi.org/10.1080/07853890802508934.
85. Steptoe A, Hackett RA, Lazzarino AI, et al. Disruption of multisystem responses to stress in type 2 diabetes: investigating the dynamics of allostatic load. Proc Natl Acad Sci U S A. 2014;111(44):15693-15698. http://dx.doi.org/10.1073/pnas.1410401111.
86. Sepehri A, Beggs T, Hassan A, et al. The impact of frailty on outcomes after cardiac surgery: a systematic review. J Thorac Cardiovasc Surg. 2014;148(6):3110-3117. http://dx.doi.org/10.1016/j.jtcvs.2014.07.087.
87. Johar H, Emeny RT, Bidlingmaier M, et al. Blunted diurnal cortisol pattern is associated with frailty: a cross-sectional study of 745 participants aged 65 to 90 years. J Clin Endocrinol Metab. 2014;99(3):E464-468. http://dx.doi.org/10.1210/jc.2013-3079.
88. Yao X, Li H, Leng SX. Inflammation and immune system alterations in frailty. Clin Geriatr Med. 2011;27(1):79-87. http://dx.doi.org/10.1016/j.cger.2010.08.002.
89. Hospital Elder Life Program (HELP) for Prevention of Delirium. 2017; http://www.hospitalelderlifeprogram.org/. Accessed February 16, 2018.
90. Shepperd S, Doll H, Angus RM, et al. Admission avoidance hospital at home. Cochrane Database of System Rev. 2008;(4):CD007491. http://dx.doi.org/10.1002/14651858.CD007491.pub2’
91. Leff B, Burton L, Mader SL, et al. Comparison of functional outcomes associated with hospital at home care and traditional acute hospital care. J Am Geriatrics Soc. 2009;57(2):273-278. http://dx.doi.org/10.1111/j.1532-5415.2008.02103.x.
92. Qaddoura A, Yazdan-Ashoori P, Kabali C, et al. Efficacy of hospital at home in patients with heart failure: a systematic review and meta-analysis. PloS One. 2015;10(6):e0129282. http://dx.doi.org/10.1371/journal.pone.0129282.
93. Seeman T, Gruenewald T, Karlamangla A, et al. Modeling multisystem biological risk in young adults: The Coronary Artery Risk Development in Young Adults Study. Am J Hum Biol. 2010;22(4):463-472. http://dx.doi.org/10.1002/ajhb.21018.
94. Auerbach AD, Kripalani S, Vasilevskis EE, et al. Preventability and causes of readmissions in a national cohort of general medicine patients. JAMA Intern Med. 2016;176(4):484-493. http://dx.doi.org/10.1001/jamainternmed.2015.7863.
95. Hansen LO, Young RS, Hinami K, Leung A, Williams MV. Interventions to reduce 30-day rehospitalization: a systematic review. Ann Intern Med. 2011;155(8):520-528. http://dx.doi.org/10.7326/0003-4819-155-8-201110180-00008.
96. Takahashi PY, Naessens JM, Peterson SM, et al. Short-term and long-term effectiveness of a post-hospital care transitions program in an older, medically complex population. Healthcare. 2016;4(1):30-35. http://dx.doi.org/10.1016/j.hjdsi.2015.06.006.
 97. Dharmarajan K, Swami S, Gou RY, Jones RN, Inouye SK. Pathway from delirium to death: potential in-hospital mediators of excess mortality. J Am Geriatr Soc. 2017;65(5):1026-1033. http://dx.doi.org/10.1111/jgs.14743.
97. Dharmarajan K, Swami S, Gou RY, Jones RN, Inouye SK. Pathway from delirium to death: potential in-hospital mediators of excess mortality. J Am Geriatr Soc. 2017;65(5):1026-1033. http://dx.doi.org/10.1111/jgs.14743.
After discharge from the hospital, patients have a significantly elevated risk for adverse events, including emergency department use, hospital readmission, and death. More than 1 in 3 patients discharged from the hospital require acute care in the month after hospital discharge, and more than 1 in 6 require readmission, with readmission diagnoses frequently differing from those of the preceding hospitalization.1-4 This heightened susceptibility to adverse events persists beyond 30 days but levels off by 7 weeks after discharge, suggesting that the period of increased risk is transient and dynamic.5
The term posthospital syndrome (PHS) describes this period of vulnerability to major adverse events following hospitalization.6 In addition to increased risk for readmission and mortality, patients in this period often show evidence of generalized dysfunction with new cognitive impairment, mobility disability, or functional decline.7-12 To date, the etiology of this vulnerability is neither well understood nor effectively addressed by transitional care interventions.13
One hypothesis to explain PHS is that stressors associated with the experience of hospitalization contribute to transient multisystem dysfunction that induces susceptibility to a broad range of medical maladies. These stressors include frequent sleep disruption, noxious sounds, painful stimuli, mobility restrictions, and poor nutrition.12 The stress hypothesis as a cause of PHS is therefore based, in large part, on evidence about allostasis and the deleterious effects of allostatic overload.
Allostasis defines a system functioning within normal stress-response parameters to promote adaptation and survival.14 In allostasis, the hypothalamic-pituitary-adrenal (HPA) axis and the sympathetic and parasympathetic branches of the autonomic nervous system (ANS) exist in homeostatic balance and respond to environmental stimuli within a range of healthy physiologic parameters. The hallmark of a system in allostasis is the ability to rapidly activate, then successfully deactivate, a stress response once the stressor (ie, threat) has resolved.14,15 To promote survival and potentiate “fight or flight” mechanisms, an appropriate stress response necessarily impacts multiple physiologic systems that result in hemodynamic augmentation and gluconeogenesis to support the anticipated action of large muscle groups, heightened vigilance and memory capabilities to improve rapid decision-making, and enhancement of innate and adaptive immune capabilities to prepare for wound repair and infection defense.14-16 The stress response is subsequently terminated by negative feedback mechanisms of glucocorticoids as well as a shift of the ANS from sympathetic to parasympathetic tone.17,18
Extended or repetitive stress exposure, however, leads to dysregulation of allostatic mechanisms responsible for stress adaptation and hinders an efficient and effective stress response. After extended stress exposure, baseline (ie, resting) HPA activity resets, causing a disruption of normal diurnal cortisol rhythm and an increase in total cortisol concentration. Moreover, in response to stress, HPA and ANS system excitation becomes impaired, and negative feedback properties are undermined.14,15 This maladaptive state, known as allostatic overload, disrupts the finely tuned mechanisms that are the foundation of mind-body balance and yields pathophysiologic consequences to multiple organ systems. Downstream ramifications of allostatic overload include cognitive deterioration, cardiovascular and immune system dysfunction, and functional decline.14,15,19
Although a stress response is an expected and necessary aspect of acute illness that promotes survival, the central thesis of this work is that additional environmental and social stressors inherent in hospitalization may unnecessarily compound stress and increase the risk of HPA axis dysfunction, allostatic overload, and subsequent multisystem dysfunction, predisposing individuals to adverse outcomes after hospital discharge. Based on data from both human subjects and animal models, we present a possible pathophysiologic mechanism for the postdischarge vulnerability of PHS, encourage critical contemplation of traditional hospitalization, and suggest interventions that might improve outcomes.
POSTHOSPITAL SYNDROME
Posthospital syndrome (PHS) describes a transient period of vulnerability after hospitalization during which patients are at elevated risk for adverse events from a broad range of conditions. In support of this characterization, epidemiologic data have demonstrated high rates of adverse outcomes following hospitalization. For example, data have shown that more than 1 in 6 older adults is readmitted to the hospital within 30 days of discharge.20 Death is also common in this first month, during which rates of postdischarge mortality may exceed initial inpatient mortality.21,22 Elevated vulnerability after hospitalization is not restricted to older adults, as readmission risk among younger patients 18 to 64 years of age may be even higher for selected conditions, such as heart failure.3,23
Vulnerability after hospitalization is broad. In patients over age 65 initially admitted for heart failure or acute myocardial infarction, only 35% and 10% of readmissions are for recurrent heart failure or reinfarction, respectively.1 Nearly half of readmissions are for noncardiovascular causes.1 Similarly, following hospitalization for pneumonia, more than 60 percent of readmissions are for nonpulmonary etiologies. Moreover, the risk for all these causes of readmission is much higher than baseline risk, indicating an extended period of lack of resilience to many types of illness.24 These patterns of broad susceptibility also extend to younger adults hospitalized with common medical conditions.3
Accumulating evidence suggests that hospitalized patients face functional decline, debility, and risk for adverse events despite resolution of the presenting illness, implying perhaps that the hospital environment itself is hazardous to patients’ health. In 1993, Creditor hypothesized that the “hazards of hospitalization,” including enforced bed-rest, sensory deprivation, social isolation, and malnutrition lead to a “cascade of dependency” in which a collection of small insults to multiple organ systems precipitates loss of function and debility despite cure or resolution of presenting illness.12 Covinsky (2011) later defined hospitalization-associated disability as an iatrogenic hospital-related “disorder” characterized by new impairments in abilities to perform basic activities of daily living such as bathing, feeding, toileting, dressing, transferring, and walking at the time of hospital discharge.11 Others have described a postintensive-care syndrome (PICS),25 characterized by cognitive, psychiatric, and physical impairments acquired during hospitalization for critical illness that persist postdischarge and increase the long-term risk for adverse outcomes, including elevated mortality rates,26,27 readmission rates,28 and physical disabilities.29 Similar to the “hazards of hospitalization,” PICS is thought to be related to common experiences of ICU stays, including mobility restriction, sensory deprivation, sleep disruption, sedation, malnutrition, and polypharmacy.30-33
Taken together, these data suggest that adverse health consequences attributable to hospitalization extend across the spectrum of age, presenting disease severity, and hospital treatment location. As detailed below, the PHS hypothesis is rooted in a mechanistic understanding of the role of exogenous stressors in producing physiologic dysregulation and subsequent adverse health effects across multiple organ systems.
Nature of Stress in the Hospital
Compounding the stress of acute illness, hospitalized patients are routinely and repetitively exposed to a wide variety of environmental stressors that may have downstream adverse consequences (Table 1). In the absence of overt clinical manifestations of harm, the possible subclinical physiologic dysfunction generated by the following stress exposures may increase patients’ susceptibility to the manifestations of PHS.
Sleep Disruption
Sleep disruptions trigger potent stress responses,34,35 yet they are common occurrences during hospitalization. In surveys, about half of patients report poor sleep quality during hospitalization that persists for many months after discharge.36 In a simulated hospital setting, test subjects exposed to typical hospital sounds (paging system, machine alarms, etc.) experienced significant sleep-wake cycle abnormalities.37 Although no work has yet focused specifically on the physiologic consequences of sleep disruption and stress in hospitalized patients, in healthy humans, mild sleep disruption has clear effects on allostasis by disrupting HPA activity, raising cortisol levels, diminishing parasympathetic tone, and impairing cognitive performance.18,34,35,38,39
Malnourishment
Malnourishment in hospitalized patients is common, with one-fifth of hospitalized patients receiving nothing per mouth or clear liquid diets for more than 3 continuous days,40 and one-fifth of hospitalized elderly patients receiving less than half of their calculated nutrition requirements.41 Although the relationship between food restriction, cortisol levels, and postdischarge outcomes has not been fully explored, in healthy humans, meal anticipation, meal withdrawal (withholding an expected meal), and self-reported dietary restraint are known to generate stress responses.42,43 Furthermore, malnourishment during hospitalization is associated with increased 90-day and 1-year mortality after discharge,44 adding malnourishment to the list of plausible components of hospital-related stress.
Mobility Restriction
Physical activity counterbalances stress responses and minimizes downstream consequences of allostatic load,15 yet mobility limitations via physical and chemical restraints are common in hospitalized patients, particularly among the elderly.45-47 Many patients are tethered to devices that make ambulation hazardous, such as urinary catheters and infusion pumps. Even without physical or chemical restraints or a limited mobility order, patients may be hesitant to leave the room so as not to miss transport to a diagnostic study or an unscheduled physician’s visit. Indeed, mobility limitations of hospitalized patients increase the risk for adverse events after discharge, while interventions designed to encourage mobility are associated with improved postdischarge outcomes.47,48
Other Stressors
Other hospital-related aversive stimuli are less commonly quantified, but clearly exist. According to surveys of hospitalized patients, sources of emotional stress include social isolation; loss of autonomy and privacy; fear of serious illness; lack of control over activities of daily living; lack of clear communication between treatment team and patients; and death of a patient roommate.49,50 Furthermore, consider the physical discomfort and emotional distress of patients with urinary incontinence awaiting assistance for a diaper or bedding change or the pain of repetitive blood draws or other invasive testing. Although individualized, the subjective discomfort and emotional distress associated with these experiences undoubtedly contribute to the stress of hospitalization.
IMPACT OF ALLOSTATIC OVERLOAD ON PHYSIOLOGIC FUNCTION
Animal Models of Stress
Laboratory techniques reminiscent of the numerous environmental stressors associated with hospitalization have been used to reliably trigger allostatic overload in healthy young animals.51 These techniques include sequential exposure to aversive stimuli, including food and water deprivation, continuous overnight illumination, paired housing with known and unknown cagemates, mobility restriction, soiled cage conditions, and continuous noise. All of these techniques have been shown to cause HPA axis and ANS dysfunction, allostatic overload, and subsequent stress-mediated consequences to multiple organ systems.19,52-54 Given the remarkable similarity of these protocols to common experiences during hospitalization, animal models of stress may be useful in understanding the spectrum of maladaptive consequences experienced by patients within the hospital (Figure 1).
These animal models of stress have resulted in a number of instructive findings. For example, in rodents, extended stress exposure induces structural and functional remodeling of neuronal networks that precipitate learning and memory, working memory, and attention impairments.55-57 These exposures also result in cardiovascular abnormalities, including dyslipidemia, progressive atherosclerosis,58,59 and enhanced inflammatory cytokine expression,60 all of which increase both atherosclerotic burden and susceptibility to plaque rupture, leading to elevated risk for major cardiovascular adverse events. Moreover, these extended stress exposures in animals increase susceptibility to both bacterial and viral infections and increase their severity.16,61 This outcome appears to be driven by a stress-induced elevation of glucocorticoid levels, decreased leukocyte proliferation, altered leukocyte trafficking, and a transition to a proinflammatory cytokine environment.16, 61 Allostatic overload has also been shown to contribute to metabolic dysregulation involving insulin resistance, persistence of hyperglycemia, dyslipidemia, catabolism of lean muscle, and visceral adipose tissue deposition.62-64 In addition to cardiovascular, immune, and metabolic consequences of allostatic overload, the spectrum of physiologic dysfunction in animal models is broad and includes mood disorder symptoms,65 intestinal barrier abnormalities,66 airway reactivity exacerbation,67 and enhanced tumor growth.68
Although the majority of this research highlights the multisystem effects of variable stress exposure in healthy animals, preliminary evidence suggests that aged or diseased animals subjected to additional stressors display a heightened inflammatory cytokine response that contributes to exaggerated sickness behavior and greater and prolonged cognitive deficits.69 Future studies exploring the consequences of extended stress exposure in animals with existing disease or debility may therefore more closely simulate the experience of hospitalized patients and perhaps further our understanding of PHS.
Hospitalized Patients
While no intervention studies have examined the effects of potential hospital stressors on the development of allostatic overload, there is evidence from small studies that dysregulated stress responses during hospitalization are associated with adverse events. For example, high serum cortisol, catecholamine, and proinflammatory cytokine levels during hospitalization have individually been associated with the development of cognitive dysfunction,70-72 increased risk of cardiovascular events such as myocardial infarction and stroke in the year following discharge,73-76 and the development of wound infections after discharge.77 Moreover, elevated plasma glucose during admission for myocardial infarction in patients with or without diabetes has been associated with greater in-hospital and 1-year mortality,78 with a similar relationship seen between elevated plasma glucose and survival after admission for stroke79 and pneumonia.80 Furthermore, in addition to atherothrombosis, stress may contribute to the risk for venous thromboembolism,81 resulting in readmissions for deep vein thrombosis or pulmonary embolism posthospitalization. Although potentially surrogate markers of illness acuity, a handful of studies have shown that these stress biomarkers are actually only weakly correlated with,82 or independent of,72,76 disease severity. As discussed in detail below, future studies utilizing a summative measure of multisystem physiologic dysfunction as opposed to individual biomarkers may more accurately reflect the cumulative stress effects of hospitalization and subsequent risk for adverse events.
Additional Considerations
Elderly patients, in particular, may have heightened susceptibility to the consequences of allostatic overload due to common geriatric issues such as multimorbidity and frailty. Patients with chronic diseases display both baseline HPA axis abnormalities as well as dysregulated stress responses and may therefore be more vulnerable to hospitalization-related stress. For example, when subjected to psychosocial stress, patients with chronic conditions such as diabetes, heart failure, or atherosclerosis demonstrate elevated cortisol levels, increased circulating markers of inflammation, as well as prolonged hemodynamic recovery after stress resolution compared with normal controls.83-85 Additionally, frailty may affect an individual’s susceptibility to exogenous stress. Indeed, frailty identified on hospital admission increases the risk for adverse outcomes during hospitalization and postdischarge.86 Although the specific etiology of this relationship is unclear, persons with frailty are known to have elevated levels of cortisol and other inflammatory markers,87,88 which may contribute to adverse outcomes in the face of additional stressors.
IMPLICATIONS AND NEXT STEPS
A large body of evidence stretching from bench to bedside suggests that environmental stressors associated with hospitalization are toxic. Understanding PHS within the context of hospital-induced allostatic overload presents a unifying theory for the interrelated multisystem dysfunction and increased susceptibility to adverse events that patients experience after discharge (Figure 2). Furthermore, it defines a potential pathophysiological mechanism for the cognitive impairment, elevated cardiovascular risk, immune system dysfunction, metabolic derangements, and functional decline associated with PHS. Additionally, this theory highlights environmental interventions to limit PHS development and suggests mechanisms to promote stress resilience. Although it is difficult to disentangle the consequences of the endogenous stress triggered by an acute illness from the exogenous stressors related to hospitalization, it is likely that the 2 simultaneous exposures compound risk for stress system dysregulation and allostatic overload. Moreover, hospitalized patients with preexisting HPA axis dysfunction at baseline from chronic disease or advancing age may be even more susceptible to these adverse outcomes. If this hypothesis is true, a reduction in PHS would require mitigation of the modifiable environmental stressors encountered by patients during hospitalization. Directed efforts to diminish ambient noise, limit nighttime disruptions, thoughtfully plan procedures, consider ongoing nutritional status, and promote opportunities for patients to exert some control over their environment may diminish the burden of extrinsic stressors encountered by all patients in the hospital and improve outcomes after discharge.
Hospitals are increasingly recognizing the importance of improving patients’ experience of hospitalization by reducing exposure to potential toxicities. For example, many hospitals are now attempting to reduce sleep disturbances and sleep latency through reduced nighttime noise and light levels, fewer nighttime interruptions for vital signs checks and medication administration, and commonsensical interventions like massages, herbal teas, and warm milk prior to bedtime.89 Likewise, intensive care units are targeting environmental and physical stressors with a multifaceted approach to decrease sedative use, promote healthy sleep cycles, and encourage exercise and ambulation even in those patients who are mechanically ventilated.30 Another promising development has been the increase of Hospital at Home programs. In these programs, patients who meet the criteria for inpatient admission are instead comprehensively managed at home for their acute illness through a multidisciplinary effort between physicians, nurses, social workers, physical therapists, and others. Patients hospitalized at home report higher levels of satisfaction and have modest functional gains, improved health-related quality of life, and decreased risk of mortality at 6 months compared with hospitalized patients.90,91 With some admitting diagnoses (eg, heart failure), hospitalization at home may be associated with decreased readmission risk.92 Although not yet investigated on a physiologic level, perhaps the benefits of hospital at home are partially due to the dramatic difference in exposure to environmental stressors.
A tool that quantifies hospital-associated stress may help health providers appreciate the experience of patients and better target interventions to aspects of their structure and process that contribute to allostatic overload. Importantly, allostatic overload cannot be identified by one biomarker of stress but instead requires evidence of dysregulation across inflammatory, neuroendocrine, hormonal, and cardiometabolic systems. Future studies to address the burden of stress faced by hospitalized patients should consider a summative measure of multisystem dysregulation as opposed to isolated assessments of individual biomarkers. Allostatic load has previously been operationalized as the summation of a variety of hemodynamic, hormonal, and metabolic factors, including blood pressure, lipid profile, glycosylated hemoglobin, cortisol, catecholamine levels, and inflammatory markers.93 To develop a hospital-associated allostatic load index, models should ideally be adjusted for acute illness severity, patient-reported stress, and capacity for stress resilience. This tool could then be used to quantify hospitalization-related allostatic load and identify those at greatest risk for adverse events after discharge, as well as measure the effectiveness of strategic environmental interventions (Table 2). A natural first experiment may be a comparison of the allostatic load of hospitalized patients versus those hospitalized at home.
The risk of adverse outcomes after discharge is likely a function of the vulnerability of the patient and the degree to which the patient’s healthcare team and social support network mitigates this vulnerability. That is, there is a risk that a person struggles in the postdischarge period and, in many circumstances, a strong healthcare team and social network can identify health problems early and prevent them from progressing to the point that they require hospitalization.13,94-96 There are also hospital occurrences, outside of allostatic load, that can lead to complications that lengthen the stay, weaken the patient, and directly contribute to subsequent vulnerability.94,97 Our contention is that the allostatic load of hospitalization, which may also vary by patient depending on the circumstances of hospitalization, is just one contributor, albeit potentially an important one, to vulnerability to medical problems after discharge.
In conclusion, a plausible etiology of PHS is the maladaptive mind-body consequences of common stressors during hospitalization that compound the stress of acute illness and produce allostatic overload. This stress-induced dysfunction potentially contributes to a spectrum of generalized disease susceptibility and risk of adverse outcomes after discharge. Focused efforts to diminish patient exposure to hospital-related stressors during and after hospitalization might diminish the presence or severity of PHS. Viewing PHS from this perspective enables the development of hypothesis-driven risk-prediction models, encourages critical contemplation of traditional hospitalization, and suggests that targeted environmental interventions may significantly reduce adverse outcomes.
After discharge from the hospital, patients have a significantly elevated risk for adverse events, including emergency department use, hospital readmission, and death. More than 1 in 3 patients discharged from the hospital require acute care in the month after hospital discharge, and more than 1 in 6 require readmission, with readmission diagnoses frequently differing from those of the preceding hospitalization.1-4 This heightened susceptibility to adverse events persists beyond 30 days but levels off by 7 weeks after discharge, suggesting that the period of increased risk is transient and dynamic.5
The term posthospital syndrome (PHS) describes this period of vulnerability to major adverse events following hospitalization.6 In addition to increased risk for readmission and mortality, patients in this period often show evidence of generalized dysfunction with new cognitive impairment, mobility disability, or functional decline.7-12 To date, the etiology of this vulnerability is neither well understood nor effectively addressed by transitional care interventions.13
One hypothesis to explain PHS is that stressors associated with the experience of hospitalization contribute to transient multisystem dysfunction that induces susceptibility to a broad range of medical maladies. These stressors include frequent sleep disruption, noxious sounds, painful stimuli, mobility restrictions, and poor nutrition.12 The stress hypothesis as a cause of PHS is therefore based, in large part, on evidence about allostasis and the deleterious effects of allostatic overload.
Allostasis defines a system functioning within normal stress-response parameters to promote adaptation and survival.14 In allostasis, the hypothalamic-pituitary-adrenal (HPA) axis and the sympathetic and parasympathetic branches of the autonomic nervous system (ANS) exist in homeostatic balance and respond to environmental stimuli within a range of healthy physiologic parameters. The hallmark of a system in allostasis is the ability to rapidly activate, then successfully deactivate, a stress response once the stressor (ie, threat) has resolved.14,15 To promote survival and potentiate “fight or flight” mechanisms, an appropriate stress response necessarily impacts multiple physiologic systems that result in hemodynamic augmentation and gluconeogenesis to support the anticipated action of large muscle groups, heightened vigilance and memory capabilities to improve rapid decision-making, and enhancement of innate and adaptive immune capabilities to prepare for wound repair and infection defense.14-16 The stress response is subsequently terminated by negative feedback mechanisms of glucocorticoids as well as a shift of the ANS from sympathetic to parasympathetic tone.17,18
Extended or repetitive stress exposure, however, leads to dysregulation of allostatic mechanisms responsible for stress adaptation and hinders an efficient and effective stress response. After extended stress exposure, baseline (ie, resting) HPA activity resets, causing a disruption of normal diurnal cortisol rhythm and an increase in total cortisol concentration. Moreover, in response to stress, HPA and ANS system excitation becomes impaired, and negative feedback properties are undermined.14,15 This maladaptive state, known as allostatic overload, disrupts the finely tuned mechanisms that are the foundation of mind-body balance and yields pathophysiologic consequences to multiple organ systems. Downstream ramifications of allostatic overload include cognitive deterioration, cardiovascular and immune system dysfunction, and functional decline.14,15,19
Although a stress response is an expected and necessary aspect of acute illness that promotes survival, the central thesis of this work is that additional environmental and social stressors inherent in hospitalization may unnecessarily compound stress and increase the risk of HPA axis dysfunction, allostatic overload, and subsequent multisystem dysfunction, predisposing individuals to adverse outcomes after hospital discharge. Based on data from both human subjects and animal models, we present a possible pathophysiologic mechanism for the postdischarge vulnerability of PHS, encourage critical contemplation of traditional hospitalization, and suggest interventions that might improve outcomes.
POSTHOSPITAL SYNDROME
Posthospital syndrome (PHS) describes a transient period of vulnerability after hospitalization during which patients are at elevated risk for adverse events from a broad range of conditions. In support of this characterization, epidemiologic data have demonstrated high rates of adverse outcomes following hospitalization. For example, data have shown that more than 1 in 6 older adults is readmitted to the hospital within 30 days of discharge.20 Death is also common in this first month, during which rates of postdischarge mortality may exceed initial inpatient mortality.21,22 Elevated vulnerability after hospitalization is not restricted to older adults, as readmission risk among younger patients 18 to 64 years of age may be even higher for selected conditions, such as heart failure.3,23
Vulnerability after hospitalization is broad. In patients over age 65 initially admitted for heart failure or acute myocardial infarction, only 35% and 10% of readmissions are for recurrent heart failure or reinfarction, respectively.1 Nearly half of readmissions are for noncardiovascular causes.1 Similarly, following hospitalization for pneumonia, more than 60 percent of readmissions are for nonpulmonary etiologies. Moreover, the risk for all these causes of readmission is much higher than baseline risk, indicating an extended period of lack of resilience to many types of illness.24 These patterns of broad susceptibility also extend to younger adults hospitalized with common medical conditions.3
Accumulating evidence suggests that hospitalized patients face functional decline, debility, and risk for adverse events despite resolution of the presenting illness, implying perhaps that the hospital environment itself is hazardous to patients’ health. In 1993, Creditor hypothesized that the “hazards of hospitalization,” including enforced bed-rest, sensory deprivation, social isolation, and malnutrition lead to a “cascade of dependency” in which a collection of small insults to multiple organ systems precipitates loss of function and debility despite cure or resolution of presenting illness.12 Covinsky (2011) later defined hospitalization-associated disability as an iatrogenic hospital-related “disorder” characterized by new impairments in abilities to perform basic activities of daily living such as bathing, feeding, toileting, dressing, transferring, and walking at the time of hospital discharge.11 Others have described a postintensive-care syndrome (PICS),25 characterized by cognitive, psychiatric, and physical impairments acquired during hospitalization for critical illness that persist postdischarge and increase the long-term risk for adverse outcomes, including elevated mortality rates,26,27 readmission rates,28 and physical disabilities.29 Similar to the “hazards of hospitalization,” PICS is thought to be related to common experiences of ICU stays, including mobility restriction, sensory deprivation, sleep disruption, sedation, malnutrition, and polypharmacy.30-33
Taken together, these data suggest that adverse health consequences attributable to hospitalization extend across the spectrum of age, presenting disease severity, and hospital treatment location. As detailed below, the PHS hypothesis is rooted in a mechanistic understanding of the role of exogenous stressors in producing physiologic dysregulation and subsequent adverse health effects across multiple organ systems.
Nature of Stress in the Hospital
Compounding the stress of acute illness, hospitalized patients are routinely and repetitively exposed to a wide variety of environmental stressors that may have downstream adverse consequences (Table 1). In the absence of overt clinical manifestations of harm, the possible subclinical physiologic dysfunction generated by the following stress exposures may increase patients’ susceptibility to the manifestations of PHS.
Sleep Disruption
Sleep disruptions trigger potent stress responses,34,35 yet they are common occurrences during hospitalization. In surveys, about half of patients report poor sleep quality during hospitalization that persists for many months after discharge.36 In a simulated hospital setting, test subjects exposed to typical hospital sounds (paging system, machine alarms, etc.) experienced significant sleep-wake cycle abnormalities.37 Although no work has yet focused specifically on the physiologic consequences of sleep disruption and stress in hospitalized patients, in healthy humans, mild sleep disruption has clear effects on allostasis by disrupting HPA activity, raising cortisol levels, diminishing parasympathetic tone, and impairing cognitive performance.18,34,35,38,39
Malnourishment
Malnourishment in hospitalized patients is common, with one-fifth of hospitalized patients receiving nothing per mouth or clear liquid diets for more than 3 continuous days,40 and one-fifth of hospitalized elderly patients receiving less than half of their calculated nutrition requirements.41 Although the relationship between food restriction, cortisol levels, and postdischarge outcomes has not been fully explored, in healthy humans, meal anticipation, meal withdrawal (withholding an expected meal), and self-reported dietary restraint are known to generate stress responses.42,43 Furthermore, malnourishment during hospitalization is associated with increased 90-day and 1-year mortality after discharge,44 adding malnourishment to the list of plausible components of hospital-related stress.
Mobility Restriction
Physical activity counterbalances stress responses and minimizes downstream consequences of allostatic load,15 yet mobility limitations via physical and chemical restraints are common in hospitalized patients, particularly among the elderly.45-47 Many patients are tethered to devices that make ambulation hazardous, such as urinary catheters and infusion pumps. Even without physical or chemical restraints or a limited mobility order, patients may be hesitant to leave the room so as not to miss transport to a diagnostic study or an unscheduled physician’s visit. Indeed, mobility limitations of hospitalized patients increase the risk for adverse events after discharge, while interventions designed to encourage mobility are associated with improved postdischarge outcomes.47,48
Other Stressors
Other hospital-related aversive stimuli are less commonly quantified, but clearly exist. According to surveys of hospitalized patients, sources of emotional stress include social isolation; loss of autonomy and privacy; fear of serious illness; lack of control over activities of daily living; lack of clear communication between treatment team and patients; and death of a patient roommate.49,50 Furthermore, consider the physical discomfort and emotional distress of patients with urinary incontinence awaiting assistance for a diaper or bedding change or the pain of repetitive blood draws or other invasive testing. Although individualized, the subjective discomfort and emotional distress associated with these experiences undoubtedly contribute to the stress of hospitalization.
IMPACT OF ALLOSTATIC OVERLOAD ON PHYSIOLOGIC FUNCTION
Animal Models of Stress
Laboratory techniques reminiscent of the numerous environmental stressors associated with hospitalization have been used to reliably trigger allostatic overload in healthy young animals.51 These techniques include sequential exposure to aversive stimuli, including food and water deprivation, continuous overnight illumination, paired housing with known and unknown cagemates, mobility restriction, soiled cage conditions, and continuous noise. All of these techniques have been shown to cause HPA axis and ANS dysfunction, allostatic overload, and subsequent stress-mediated consequences to multiple organ systems.19,52-54 Given the remarkable similarity of these protocols to common experiences during hospitalization, animal models of stress may be useful in understanding the spectrum of maladaptive consequences experienced by patients within the hospital (Figure 1).
These animal models of stress have resulted in a number of instructive findings. For example, in rodents, extended stress exposure induces structural and functional remodeling of neuronal networks that precipitate learning and memory, working memory, and attention impairments.55-57 These exposures also result in cardiovascular abnormalities, including dyslipidemia, progressive atherosclerosis,58,59 and enhanced inflammatory cytokine expression,60 all of which increase both atherosclerotic burden and susceptibility to plaque rupture, leading to elevated risk for major cardiovascular adverse events. Moreover, these extended stress exposures in animals increase susceptibility to both bacterial and viral infections and increase their severity.16,61 This outcome appears to be driven by a stress-induced elevation of glucocorticoid levels, decreased leukocyte proliferation, altered leukocyte trafficking, and a transition to a proinflammatory cytokine environment.16, 61 Allostatic overload has also been shown to contribute to metabolic dysregulation involving insulin resistance, persistence of hyperglycemia, dyslipidemia, catabolism of lean muscle, and visceral adipose tissue deposition.62-64 In addition to cardiovascular, immune, and metabolic consequences of allostatic overload, the spectrum of physiologic dysfunction in animal models is broad and includes mood disorder symptoms,65 intestinal barrier abnormalities,66 airway reactivity exacerbation,67 and enhanced tumor growth.68
Although the majority of this research highlights the multisystem effects of variable stress exposure in healthy animals, preliminary evidence suggests that aged or diseased animals subjected to additional stressors display a heightened inflammatory cytokine response that contributes to exaggerated sickness behavior and greater and prolonged cognitive deficits.69 Future studies exploring the consequences of extended stress exposure in animals with existing disease or debility may therefore more closely simulate the experience of hospitalized patients and perhaps further our understanding of PHS.
Hospitalized Patients
While no intervention studies have examined the effects of potential hospital stressors on the development of allostatic overload, there is evidence from small studies that dysregulated stress responses during hospitalization are associated with adverse events. For example, high serum cortisol, catecholamine, and proinflammatory cytokine levels during hospitalization have individually been associated with the development of cognitive dysfunction,70-72 increased risk of cardiovascular events such as myocardial infarction and stroke in the year following discharge,73-76 and the development of wound infections after discharge.77 Moreover, elevated plasma glucose during admission for myocardial infarction in patients with or without diabetes has been associated with greater in-hospital and 1-year mortality,78 with a similar relationship seen between elevated plasma glucose and survival after admission for stroke79 and pneumonia.80 Furthermore, in addition to atherothrombosis, stress may contribute to the risk for venous thromboembolism,81 resulting in readmissions for deep vein thrombosis or pulmonary embolism posthospitalization. Although potentially surrogate markers of illness acuity, a handful of studies have shown that these stress biomarkers are actually only weakly correlated with,82 or independent of,72,76 disease severity. As discussed in detail below, future studies utilizing a summative measure of multisystem physiologic dysfunction as opposed to individual biomarkers may more accurately reflect the cumulative stress effects of hospitalization and subsequent risk for adverse events.
Additional Considerations
Elderly patients, in particular, may have heightened susceptibility to the consequences of allostatic overload due to common geriatric issues such as multimorbidity and frailty. Patients with chronic diseases display both baseline HPA axis abnormalities as well as dysregulated stress responses and may therefore be more vulnerable to hospitalization-related stress. For example, when subjected to psychosocial stress, patients with chronic conditions such as diabetes, heart failure, or atherosclerosis demonstrate elevated cortisol levels, increased circulating markers of inflammation, as well as prolonged hemodynamic recovery after stress resolution compared with normal controls.83-85 Additionally, frailty may affect an individual’s susceptibility to exogenous stress. Indeed, frailty identified on hospital admission increases the risk for adverse outcomes during hospitalization and postdischarge.86 Although the specific etiology of this relationship is unclear, persons with frailty are known to have elevated levels of cortisol and other inflammatory markers,87,88 which may contribute to adverse outcomes in the face of additional stressors.
IMPLICATIONS AND NEXT STEPS
A large body of evidence stretching from bench to bedside suggests that environmental stressors associated with hospitalization are toxic. Understanding PHS within the context of hospital-induced allostatic overload presents a unifying theory for the interrelated multisystem dysfunction and increased susceptibility to adverse events that patients experience after discharge (Figure 2). Furthermore, it defines a potential pathophysiological mechanism for the cognitive impairment, elevated cardiovascular risk, immune system dysfunction, metabolic derangements, and functional decline associated with PHS. Additionally, this theory highlights environmental interventions to limit PHS development and suggests mechanisms to promote stress resilience. Although it is difficult to disentangle the consequences of the endogenous stress triggered by an acute illness from the exogenous stressors related to hospitalization, it is likely that the 2 simultaneous exposures compound risk for stress system dysregulation and allostatic overload. Moreover, hospitalized patients with preexisting HPA axis dysfunction at baseline from chronic disease or advancing age may be even more susceptible to these adverse outcomes. If this hypothesis is true, a reduction in PHS would require mitigation of the modifiable environmental stressors encountered by patients during hospitalization. Directed efforts to diminish ambient noise, limit nighttime disruptions, thoughtfully plan procedures, consider ongoing nutritional status, and promote opportunities for patients to exert some control over their environment may diminish the burden of extrinsic stressors encountered by all patients in the hospital and improve outcomes after discharge.
Hospitals are increasingly recognizing the importance of improving patients’ experience of hospitalization by reducing exposure to potential toxicities. For example, many hospitals are now attempting to reduce sleep disturbances and sleep latency through reduced nighttime noise and light levels, fewer nighttime interruptions for vital signs checks and medication administration, and commonsensical interventions like massages, herbal teas, and warm milk prior to bedtime.89 Likewise, intensive care units are targeting environmental and physical stressors with a multifaceted approach to decrease sedative use, promote healthy sleep cycles, and encourage exercise and ambulation even in those patients who are mechanically ventilated.30 Another promising development has been the increase of Hospital at Home programs. In these programs, patients who meet the criteria for inpatient admission are instead comprehensively managed at home for their acute illness through a multidisciplinary effort between physicians, nurses, social workers, physical therapists, and others. Patients hospitalized at home report higher levels of satisfaction and have modest functional gains, improved health-related quality of life, and decreased risk of mortality at 6 months compared with hospitalized patients.90,91 With some admitting diagnoses (eg, heart failure), hospitalization at home may be associated with decreased readmission risk.92 Although not yet investigated on a physiologic level, perhaps the benefits of hospital at home are partially due to the dramatic difference in exposure to environmental stressors.
A tool that quantifies hospital-associated stress may help health providers appreciate the experience of patients and better target interventions to aspects of their structure and process that contribute to allostatic overload. Importantly, allostatic overload cannot be identified by one biomarker of stress but instead requires evidence of dysregulation across inflammatory, neuroendocrine, hormonal, and cardiometabolic systems. Future studies to address the burden of stress faced by hospitalized patients should consider a summative measure of multisystem dysregulation as opposed to isolated assessments of individual biomarkers. Allostatic load has previously been operationalized as the summation of a variety of hemodynamic, hormonal, and metabolic factors, including blood pressure, lipid profile, glycosylated hemoglobin, cortisol, catecholamine levels, and inflammatory markers.93 To develop a hospital-associated allostatic load index, models should ideally be adjusted for acute illness severity, patient-reported stress, and capacity for stress resilience. This tool could then be used to quantify hospitalization-related allostatic load and identify those at greatest risk for adverse events after discharge, as well as measure the effectiveness of strategic environmental interventions (Table 2). A natural first experiment may be a comparison of the allostatic load of hospitalized patients versus those hospitalized at home.
The risk of adverse outcomes after discharge is likely a function of the vulnerability of the patient and the degree to which the patient’s healthcare team and social support network mitigates this vulnerability. That is, there is a risk that a person struggles in the postdischarge period and, in many circumstances, a strong healthcare team and social network can identify health problems early and prevent them from progressing to the point that they require hospitalization.13,94-96 There are also hospital occurrences, outside of allostatic load, that can lead to complications that lengthen the stay, weaken the patient, and directly contribute to subsequent vulnerability.94,97 Our contention is that the allostatic load of hospitalization, which may also vary by patient depending on the circumstances of hospitalization, is just one contributor, albeit potentially an important one, to vulnerability to medical problems after discharge.
In conclusion, a plausible etiology of PHS is the maladaptive mind-body consequences of common stressors during hospitalization that compound the stress of acute illness and produce allostatic overload. This stress-induced dysfunction potentially contributes to a spectrum of generalized disease susceptibility and risk of adverse outcomes after discharge. Focused efforts to diminish patient exposure to hospital-related stressors during and after hospitalization might diminish the presence or severity of PHS. Viewing PHS from this perspective enables the development of hypothesis-driven risk-prediction models, encourages critical contemplation of traditional hospitalization, and suggests that targeted environmental interventions may significantly reduce adverse outcomes.
1. Dharmarajan K, Hsieh AF, Lin Z, et al. Diagnoses and timing of 30-day readmissions after hospitalization for heart failure, acute myocardial infarction, or pneumonia. JAMA. 2013;309(4):355-363. http://dx.doi.org/10.1001/jama.2012.216476.
2. Jencks SF, Williams MV, Coleman EA. Rehospitalizations among patients in the Medicare fee-for-service program. N Engl J Med. 2009;360(14):1418-1428. http://dx.doi.org/10.1056/NEJMsa0803563.
3. Ranasinghe I, Wang Y, Dharmarajan K, Hsieh AF, Bernheim SM, Krumholz HM. Readmissions after hospitalization for heart failure, acute myocardial infarction, or pneumonia among young and middle-aged adults: a retrospective observational cohort study. PLoS Med. 2014;11(9):e1001737. http://dx.doi.org/10.1371/journal.pmed.1001737.
4. Vashi AA, Fox JP, Carr BG, et al. Use of hospital-based acute care among patients recently discharged from the hospital. JAMA. 2013;309(4):364-371. http://dx.doi.org/10.1001/jama.2012.216219.
5. Dharmarajan K, Hsieh AF, Kulkarni VT, et al. Trajectories of risk after hospitalization for heart failure, acute myocardial infarction, or pneumonia: retrospective cohort study. BMJ. 2015;350:h411. http://dx.doi.org/10.1136/bmj.h411.
6. Krumholz HM. Post-hospital syndrome--an acquired, transient condition of generalized risk. N Engl J Med. 2013;368(2):100-102. http://dx.doi.org/10.1056/NEJMp1212324.
7. Ferrante LE, Pisani MA, Murphy TE, Gahbauer EA, Leo-Summers LS, Gill TM. Functional trajectories among older persons before and after critical illness. JAMA Intern Med. 2015;175(4):523-529. http://dx.doi.org/10.1001/jamainternmed.2014.7889.
8. Gill TM, Allore HG, Holford TR, Guo Z. Hospitalization, restricted activity, and the development of disability among older persons. JAMA. 2004;292(17):2115-2124. http://dx.doi.org/10.1001/jama.292.17.2115.
9. Inouye SK, Zhang Y, Han L, Leo-Summers L, Jones R, Marcantonio E. Recoverable cognitive dysfunction at hospital admission in older persons during acute illness. J Gen Intern Med. 2006;21(12):1276-1281. http://dx.doi.org/10.1111/j.1525-1497.2006.00613.x.
10. Lindquist LA, Go L, Fleisher J, Jain N, Baker D. Improvements in cognition following hospital discharge of community dwelling seniors. J Gen Intern Med. 2011;26(7):765-770. http://dx.doi.org/10.1007/s11606-011-1681-1.
11. Covinsky KE, Pierluissi E, Johnston CB. Hospitalization-associated disability: “She was probably able to ambulate, but I’m not sure.” JAMA. 2011;306(16):1782-1793. http://dx.doi.org/10.1001/jama.2011.1556.
12. Creditor MC. Hazards of hospitalization of the elderly. Ann Intern Med. 1993;118(3):219-223. http://dx.doi.org/10.7326/0003-4819-118-3-199302010-00011.
13. Dharmarajan K, Krumholz HM. Strategies to reduce 30-day readmissions in older patients hospitalized with heart failure and acute myocardial infarction. Curr Geriatr Rep. 2014;3(4):306-315. http://dx.doi.org/10.1007/s13670-014-0103-8.
14. McEwen BS. Protective and damaging effects of stress mediators. N Engl J Med. 1998;338(3):171-179. http://dx.doi.org/10.1056/NEJM199801153380307.
15. McEwen BS, Gianaros PJ. Stress- and allostasis-induced brain plasticity. Annu Rev Med. 2011;62:431-445. http://dx.doi.org/10.1146/annurev-med-052209-100430.
16. Dhabhar FS. Enhancing versus suppressive effects of stress on immune function: implications for immunoprotection and immunopathology. Neuroimmunomodulation. 2009;16(5):300-317. http://dx.doi.org/10.1159/000216188.
17. Thayer JF, Sternberg E. Beyond heart rate variability: vagal regulation of allostatic systems. Ann N Y Acad Sci. 2006;1088:361-372. http://dx.doi.org/10.1196/annals.1366.014.
18. Jacobson L, Akana SF, Cascio CS, Shinsako J, Dallman MF. Circadian variations in plasma corticosterone permit normal termination of adrenocorticotropin responses to stress. Endocrinology. 1988;122(4):1343-1348. http://dx.doi.org/10.1210/endo-122-4-1343.
19. McEwen BS. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev. 2007;87(3):873-904. http://dx.doi.org/10.1152/physrev.00041.2006.
20. Medicare Hospital Quality Chartbook 2014: Performance Report on Outcome Measures. Prepared by Yale New Haven Health Services Corporation Center for Outcomes Research and Evaluation for Centers for Medicare & Medicaid Services. https://www.cms.gov/medicare/quality-initiatives-patient-assessment-instruments/hospitalqualityinits/downloads/medicare-hospital-quality-chartbook-2014.pdf. Accessed February 26, 2018.
21. Bueno H, Ross JS, Wang Y, et al. Trends in length of stay and short-term outcomes among Medicare patients hospitalized for heart failure, 1993-2006. JAMA. 2010;303(21):2141-2147. http://dx.doi.org/10.1001/jama.2010.748.
22. Drye EE, Normand SL, Wang Y, et al. Comparison of hospital risk-standardized mortality rates calculated by using in-hospital and 30-day models: an observational study with implications for hospital profiling. Ann Intern Med. 2012;156(1 Pt 1):19-26. http://dx.doi.org/10.7326/0003-4819-156-1-201201030-00004.
23. Dharmarajan K, Hsieh A, Dreyer RP, Welsh J, Qin L, Krumholz HM. Relationship between age and trajectories of rehospitalization risk in older adults. J Am Geriatr Soc. 2017;65(2):421-426. http://dx.doi.org/10.1111/jgs.14583.
24. Krumholz HM, Hsieh A, Dreyer RP, Welsh J, Desai NR, Dharmarajan K. Trajectories of risk for specific readmission diagnoses after hospitalization for heart failure, acute myocardial infarction, or pneumonia. PLoS One. 2016;11(10):e0160492. http://dx.doi.org/10.1371/journal.pone.0160492.
25. Needham DM, Davidson J, Cohen H, et al. Improving long-term outcomes after discharge from intensive care unit: report from a stakeholders’ conference. Crit Care Med. 2012;40(2):502-509. http://dx.doi.org/10.1097/CCM.0b013e318232da75.
26. Brinkman S, de Jonge E, Abu-Hanna A, Arbous MS, de Lange DW, de Keizer NF. Mortality after hospital discharge in ICU patients. Crit Care Med. 2013;41(5):1229-1236. http://dx.doi.org/10.1097/CCM.0b013e31827ca4e1.
27. Steenbergen S, Rijkenberg S, Adonis T, Kroeze G, van Stijn I, Endeman H. Long-term treated intensive care patients outcomes: the one-year mortality rate, quality of life, health care use and long-term complications as reported by general practitioners. BMC Anesthesiol. 2015;15:142. http://dx.doi.org/10.1186/s12871-015-0121-x.
28. Hill AD, Fowler RA, Pinto R, Herridge MS, Cuthbertson BH, Scales DC. Long-term outcomes and healthcare utilization following critical illness--a population-based study. Crit Care. 2016;20:76. http://dx.doi.org/10.1186/s13054-016-1248-y.
29. Jackson JC, Pandharipande PP, Girard TD, et al. Depression, post-traumatic stress disorder, and functional disability in survivors of critical illness in the BRAIN-ICU study: a longitudinal cohort study. Lancet Respir Med. 2014;2(5):369-379. http://dx.doi.org/10.1016/S2213-2600(14)70051-7.
30. Balas MC, Vasilevskis EE, Olsen KM, et al. Effectiveness and safety of the awakening and breathing coordination, delirium monitoring/management, and early exercise/mobility bundle. Crit Care Med. 2014;42(5):1024-1036. http://dx.doi.org/10.1097/CCM.0000000000000129.
31. Kress JP, Hall JB. ICU-acquired weakness and recovery from critical illness. N Engl J Med. 2014;370(17):1626-1635. http://dx.doi.org/10.1056/NEJMra1209390.
32. Mendez-Tellez PA, Needham DM. Early physical rehabilitation in the ICU and ventilator liberation. Respir Care. 2012;57(10):1663-1669. http://dx.doi.org/10.4187/respcare.01931.
33. Schweickert WD, Hall J. ICU-acquired weakness. Chest. 2007;131(5):1541-1549. http://dx.doi.org/10.1378/chest.06-2065.
34. Balbo M, Leproult R, Van Cauter E. Impact of sleep and its disturbances on hypothalamo-pituitary-adrenal axis activity. Int J Endocrinol. 2010;2010:759234. http://dx.doi.org/10.1155/2010/759234.
35. Spiegel K, Leproult R, Van Cauter E. Impact of sleep debt on metabolic and endocrine function. Lancet. 1999;354(9188):1435-1439. http://dx.doi.org/10.1016/S0140-6736(99)01376-8.
36. Orwelius L, Nordlund A, Nordlund P, Edell-Gustafsson U, Sjoberg F. Prevalence of sleep disturbances and long-term reduced health-related quality of life after critical care: a prospective multicenter cohort study. Crit Care. 2008;12(4):R97. http://dx.doi.org/10.1186/cc6973.
37. Buxton OM, Ellenbogen JM, Wang W, et al. Sleep disruption due to hospital noises: a prospective evaluation. Ann Intern Med. 2012;157(3):170-179. http://dx.doi.org/10.7326/0003-4819-157-3-201208070-00472.
38. Sunbul M, Kanar BG, Durmus E, Kivrak T, Sari I. Acute sleep deprivation is associated with increased arterial stiffness in healthy young adults. Sleep Breath. 2014;18(1):215-220. http://dx.doi.org/10.1007/s11325-013-0873-9.
39. Van Dongen HP, Maislin G, Mullington JM, Dinges DF. The cumulative cost of additional wakefulness: dose-response effects on neurobehavioral functions and sleep physiology from chronic sleep restriction and total sleep deprivation. Sleep. 2003;26(2):117-126. http://dx.doi.org/10.1093/sleep/26.2.117.
40. Franklin GA, McClave SA, Hurt RT, et al. Physician-delivered malnutrition: why do patients receive nothing by mouth or a clear liquid diet in a university hospital setting? JPEN J Parenter Enteral Nutr. 2011;35(3):337-342. http://dx.doi.org/10.1177/0148607110374060.
41. Sullivan DH, Sun S, Walls RC. Protein-energy undernutrition among elderly hospitalized patients: a prospective study. JAMA. 1999;281(21):2013-2019. http://dx.doi.org/10.1001/jama.281.21.2013.
42. Anderson DA, Shapiro JR, Lundgren JD, Spataro LE, Frye CA. Self-reported dietary restraint is associated with elevated levels of salivary cortisol. Appetite. 2002;38(1):13-17. http://dx.doi.org/10.1006/appe.2001.0459.
43. Ott V, Friedrich M, Prilop S, et al. Food anticipation and subsequent food withdrawal increase serum cortisol in healthy men. Physiol Behav. 2011;103(5):594-599. http://dx.doi.org/10.1016/j.physbeh.2011.04.020.
44. Covinsky KE, Martin GE, Beyth RJ, Justice AC, Sehgal AR, Landefeld CS. The relationship between clinical assessments of nutritional status and adverse outcomes in older hospitalized medical patients. J Am Geriatr Soc. 1999;47(5):532-538. http://dx.doi.org/10.1111/j.1532-5415.1999.tb02566.x.
45. Lazarus BA, Murphy JB, Coletta EM, McQuade WH, Culpepper L. The provision of physical activity to hospitalized elderly patients. Arch Intern Med. 1991;151(12):2452-2456.
46. Minnick AF, Mion LC, Johnson ME, Catrambone C, Leipzig R. Prevalence and variation of physical restraint use in acute care settings in the US. J Nurs Scholarsh. 2007;39(1):30-37. http://dx.doi.org/10.1111/j.1547-5069.2007.00140.x.
47. Zisberg A, Shadmi E, Sinoff G, Gur-Yaish N, Srulovici E, Admi H. Low mobility during hospitalization and functional decline in older adults. J Am Geriatr Soc. 2011;59(2):266-273. http://dx.doi.org/10.1111/j.1532-5415.2010.03276.x.
48. Landefeld CS, Palmer RM, Kresevic DM, Fortinsky RH, Kowal J. A randomized trial of care in a hospital medical unit especially designed to improve the functional outcomes of acutely ill older patients. N Engl J Med. 1995;332(20):1338-1344. http://dx.doi.org/10.1056/NEJM199505183322006.
49. Douglas CH, Douglas MR. Patient-friendly hospital environments: exploring the patients’ perspective. Health Expectations: an international journal of public participation in health care and health policy. 2004;7(1):61-73. http://dx.doi.org/10.1046/j.1369-6513.2003.00251.x.
50. Volicer BJ. Hospital stress and patient reports of pain and physical status. Journal Human Stress. 1978;4(2):28-37. http://dx.doi.org/10.1080/0097840X.1978.9934984.
51. Willner P, Towell A, Sampson D, Sophokleous S, Muscat R. Reduction of sucrose preference by chronic unpredictable mild stress, and its restoration by a tricyclic antidepressant. Psychopharmacology (Berl). 1987;93(3):358-364. http://dx.doi.org/10.1007/BF00187257.
52. Grippo AJ, Francis J, Beltz TG, Felder RB, Johnson AK. Neuroendocrine and cytokine profile of chronic mild stress-induced anhedonia. Physiol Behav. 2005;84(5):697-706. http://dx.doi.org/10.1016/j.physbeh.2005.02.011.
53. Krishnan V, Nestler EJ. Animal models of depression: molecular perspectives. Curr Top Behav Neurosci. 2011;7:121-147. http://dx.doi.org/10.1007/7854_2010_108.
54. Magarinos AM, McEwen BS. Stress-induced atrophy of apical dendrites of hippocampal CA3c neurons: comparison of stressors. Neuroscience. 1995;69(1):83-88. http://dx.doi.org/10.1016/0306-4522(95)00256-I.
55. McEwen BS. Plasticity of the hippocampus: adaptation to chronic stress and allostatic load. Ann N Y Acad Sci. 2001;933:265-277. http://dx.doi.org/10.1111/j.1749-6632.2001.tb05830.x.
56. McEwen BS. The brain on stress: toward an integrative approach to brain, body, and behavior. Perspect Psychol Sci. 2013;8(6):673-675. http://dx.doi.org/10.1177/1745691613506907.
57. McEwen BS, Morrison JH. The brain on stress: vulnerability and plasticity of the prefrontal cortex over the life course. Neuron. 2013;79(1):16-29. http://dx.doi.org/10.1016/j.neuron.2013.06.028.
58. Dutta P, Courties G, Wei Y, et al. Myocardial infarction accelerates atherosclerosis. Nature. 2012;487(7407):325-329. http://dx.doi.org/10.1038/nature11260.
59. Lu XT, Liu YF, Zhang L, et al. Unpredictable chronic mild stress promotes atherosclerosis in high cholesterol-fed rabbits. Psychosom Med. 2012;74(6):604-611. http://dx.doi.org/10.1097/PSY.0b013e31825d0b71.
60. Heidt T, Sager HB, Courties G, et al. Chronic variable stress activates hematopoietic stem cells. Nat Med. 2014;20(7):754-758. http://dx.doi.org/10.1038/nm.3589.
61. Sheridan JF, Feng NG, Bonneau RH, Allen CM, Huneycutt BS, Glaser R. Restraint stress differentially affects anti-viral cellular and humoral immune responses in mice. J Neuroimmunol. 1991;31(3):245-255. http://dx.doi.org/10.1016/0165-5728(91)90046-A.
62. Kyrou I, Tsigos C. Stress hormones: physiological stress and regulation of metabolism. Curr Opin Pharmacol. 2009;9(6):787-793. http://dx.doi.org/10.1016/j.coph.2009.08.007.
63. Rosmond R. Role of stress in the pathogenesis of the metabolic syndrome. Psychoneuroendocrinology. 2005;30(1):1-10. http://dx.doi.org/10.1016/j.psyneuen.2004.05.007.
64. Tamashiro KL, Sakai RR, Shively CA, Karatsoreos IN, Reagan LP. Chronic stress, metabolism, and metabolic syndrome. Stress. 2011;14(5):468-474. http://dx.doi.org/10.3109/10253890.2011.606341.
65. McEwen BS. Mood disorders and allostatic load. Biol Psychiatry. 2003;54(3):200-207. http://dx.doi.org/10.1016/S0006-3223(03)00177-X.
66. Zareie M, Johnson-Henry K, Jury J, et al. Probiotics prevent bacterial translocation and improve intestinal barrier function in rats following chronic psychological stress. Gut. 2006;55(11):1553-1560. http://dx.doi.org/10.1136/gut.2005.080739.
67. Joachim RA, Quarcoo D, Arck PC, Herz U, Renz H, Klapp BF. Stress enhances airway reactivity and airway inflammation in an animal model of allergic bronchial asthma. Psychosom Med. 2003;65(5):811-815. http://dx.doi.org/10.1097/01.PSY.0000088582.50468.A3.
68. Thaker PH, Han LY, Kamat AA, et al. Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat Med. 2006;12(8):939-944. http://dx.doi.org/10.1038/nm1447.
69. Schreuder L, Eggen BJ, Biber K, Schoemaker RG, Laman JD, de Rooij SE. Pathophysiological and behavioral effects of systemic inflammation in aged and diseased rodents with relevance to delirium: A systematic review. Brain Behav Immun. 2017;62:362-381. http://dx.doi.org/10.1016/j.bbi.2017.01.010.
70. Mu DL, Li LH, Wang DX, et al. High postoperative serum cortisol level is associated with increased risk of cognitive dysfunction early after coronary artery bypass graft surgery: a prospective cohort study. PLoS One. 2013;8(10):e77637. http://dx.doi.org/10.1371/journal.pone.0077637.
71. Mu DL, Wang DX, Li LH, et al. High serum cortisol level is associated with increased risk of delirium after coronary artery bypass graft surgery: a prospective cohort study. Crit Care. 2010;14(6):R238. http://dx.doi.org/10.1186/cc9393.
72. Nguyen DN, Huyghens L, Zhang H, Schiettecatte J, Smitz J, Vincent JL. Cortisol is an associated-risk factor of brain dysfunction in patients with severe sepsis and septic shock. Biomed Res Int. 2014;2014:712742. http://dx.doi.org/10.1155/2014/712742.
73. Elkind MS, Carty CL, O’Meara ES, et al. Hospitalization for infection and risk of acute ischemic stroke: the Cardiovascular Health Study. Stroke. 2011;42(7):1851-1856. http://dx.doi.org/10.1161/STROKEAHA.110.608588.
74. Feibel JH, Hardy PM, Campbell RG, Goldstein MN, Joynt RJ. Prognostic value of the stress response following stroke. JAMA. 1977;238(13):1374-1376.
75. Jutla SK, Yuyun MF, Quinn PA, Ng LL. Plasma cortisol and prognosis of patients with acute myocardial infarction. J Cardiovasc Med (Hagerstown). 2014;15(1):33-41. http://dx.doi.org/10.2459/JCM.0b013e328364100b.
76. Yende S, D’Angelo G, Kellum JA, et al. Inflammatory markers at hospital discharge predict subsequent mortality after pneumonia and sepsis. Am J Respir Crit Care Med. 2008;177(11):1242-1247. http://dx.doi.org/10.1164/rccm.200712-1777OC.
77. Gouin JP, Kiecolt-Glaser JK. The impact of psychological stress on wound healing: methods and mechanisms. Immunol Allergy Clin North Am. 2011;31(1):81-93. http://dx.doi.org/10.1016/j.iac.2010.09.010.
78. Capes SE, Hunt D, Malmberg K, Gerstein HC. Stress hyperglycaemia and increased risk of death after myocardial infarction in patients with and without diabetes: a systematic overview. Lancet. 2000;355(9206):773-778. http://dx.doi.org/10.1016/S0140-6736(99)08415-9.
79. O’Neill PA, Davies I, Fullerton KJ, Bennett D. Stress hormone and blood glucose response following acute stroke in the elderly. Stroke. 1991;22(7):842-847. http://dx.doi.org/10.1161/01.STR.22.7.842.
80. Waterer GW, Kessler LA, Wunderink RG. Medium-term survival after hospitalization with community-acquired pneumonia. Am J Respir Crit Care Med. 2004;169(8):910-914. http://dx.doi.org/10.1164/rccm.200310-1448OC.
81. Rosengren A, Freden M, Hansson PO, Wilhelmsen L, Wedel H, Eriksson H. Psychosocial factors and venous thromboembolism: a long-term follow-up study of Swedish men. J Thrombosis Haemostasis. 2008;6(4):558-564. http://dx.doi.org/10.1111/j.1538-7836.2007.02857.x.
82. Oswald GA, Smith CC, Betteridge DJ, Yudkin JS. Determinants and importance of stress hyperglycaemia in non-diabetic patients with myocardial infarction. BMJ. 1986;293(6552):917-922. http://dx.doi.org/10.1136/bmj.293.6552.917.
83. Middlekauff HR, Nguyen AH, Negrao CE, et al. Impact of acute mental stress on sympathetic nerve activity and regional blood flow in advanced heart failure: implications for ‘triggering’ adverse cardiac events. Circulation. 1997;96(6):1835-1842. http://dx.doi.org/10.1161/01.CIR.96.6.1835.
84. Nijm J, Jonasson L. Inflammation and cortisol response in coronary artery disease. Ann Med. 2009;41(3):224-233. http://dx.doi.org/10.1080/07853890802508934.
85. Steptoe A, Hackett RA, Lazzarino AI, et al. Disruption of multisystem responses to stress in type 2 diabetes: investigating the dynamics of allostatic load. Proc Natl Acad Sci U S A. 2014;111(44):15693-15698. http://dx.doi.org/10.1073/pnas.1410401111.
86. Sepehri A, Beggs T, Hassan A, et al. The impact of frailty on outcomes after cardiac surgery: a systematic review. J Thorac Cardiovasc Surg. 2014;148(6):3110-3117. http://dx.doi.org/10.1016/j.jtcvs.2014.07.087.
87. Johar H, Emeny RT, Bidlingmaier M, et al. Blunted diurnal cortisol pattern is associated with frailty: a cross-sectional study of 745 participants aged 65 to 90 years. J Clin Endocrinol Metab. 2014;99(3):E464-468. http://dx.doi.org/10.1210/jc.2013-3079.
88. Yao X, Li H, Leng SX. Inflammation and immune system alterations in frailty. Clin Geriatr Med. 2011;27(1):79-87. http://dx.doi.org/10.1016/j.cger.2010.08.002.
89. Hospital Elder Life Program (HELP) for Prevention of Delirium. 2017; http://www.hospitalelderlifeprogram.org/. Accessed February 16, 2018.
90. Shepperd S, Doll H, Angus RM, et al. Admission avoidance hospital at home. Cochrane Database of System Rev. 2008;(4):CD007491. http://dx.doi.org/10.1002/14651858.CD007491.pub2’
91. Leff B, Burton L, Mader SL, et al. Comparison of functional outcomes associated with hospital at home care and traditional acute hospital care. J Am Geriatrics Soc. 2009;57(2):273-278. http://dx.doi.org/10.1111/j.1532-5415.2008.02103.x.
92. Qaddoura A, Yazdan-Ashoori P, Kabali C, et al. Efficacy of hospital at home in patients with heart failure: a systematic review and meta-analysis. PloS One. 2015;10(6):e0129282. http://dx.doi.org/10.1371/journal.pone.0129282.
93. Seeman T, Gruenewald T, Karlamangla A, et al. Modeling multisystem biological risk in young adults: The Coronary Artery Risk Development in Young Adults Study. Am J Hum Biol. 2010;22(4):463-472. http://dx.doi.org/10.1002/ajhb.21018.
94. Auerbach AD, Kripalani S, Vasilevskis EE, et al. Preventability and causes of readmissions in a national cohort of general medicine patients. JAMA Intern Med. 2016;176(4):484-493. http://dx.doi.org/10.1001/jamainternmed.2015.7863.
95. Hansen LO, Young RS, Hinami K, Leung A, Williams MV. Interventions to reduce 30-day rehospitalization: a systematic review. Ann Intern Med. 2011;155(8):520-528. http://dx.doi.org/10.7326/0003-4819-155-8-201110180-00008.
96. Takahashi PY, Naessens JM, Peterson SM, et al. Short-term and long-term effectiveness of a post-hospital care transitions program in an older, medically complex population. Healthcare. 2016;4(1):30-35. http://dx.doi.org/10.1016/j.hjdsi.2015.06.006.
97. Dharmarajan K, Swami S, Gou RY, Jones RN, Inouye SK. Pathway from delirium to death: potential in-hospital mediators of excess mortality. J Am Geriatr Soc. 2017;65(5):1026-1033. http://dx.doi.org/10.1111/jgs.14743.
1. Dharmarajan K, Hsieh AF, Lin Z, et al. Diagnoses and timing of 30-day readmissions after hospitalization for heart failure, acute myocardial infarction, or pneumonia. JAMA. 2013;309(4):355-363. http://dx.doi.org/10.1001/jama.2012.216476.
2. Jencks SF, Williams MV, Coleman EA. Rehospitalizations among patients in the Medicare fee-for-service program. N Engl J Med. 2009;360(14):1418-1428. http://dx.doi.org/10.1056/NEJMsa0803563.
3. Ranasinghe I, Wang Y, Dharmarajan K, Hsieh AF, Bernheim SM, Krumholz HM. Readmissions after hospitalization for heart failure, acute myocardial infarction, or pneumonia among young and middle-aged adults: a retrospective observational cohort study. PLoS Med. 2014;11(9):e1001737. http://dx.doi.org/10.1371/journal.pmed.1001737.
4. Vashi AA, Fox JP, Carr BG, et al. Use of hospital-based acute care among patients recently discharged from the hospital. JAMA. 2013;309(4):364-371. http://dx.doi.org/10.1001/jama.2012.216219.
5. Dharmarajan K, Hsieh AF, Kulkarni VT, et al. Trajectories of risk after hospitalization for heart failure, acute myocardial infarction, or pneumonia: retrospective cohort study. BMJ. 2015;350:h411. http://dx.doi.org/10.1136/bmj.h411.
6. Krumholz HM. Post-hospital syndrome--an acquired, transient condition of generalized risk. N Engl J Med. 2013;368(2):100-102. http://dx.doi.org/10.1056/NEJMp1212324.
7. Ferrante LE, Pisani MA, Murphy TE, Gahbauer EA, Leo-Summers LS, Gill TM. Functional trajectories among older persons before and after critical illness. JAMA Intern Med. 2015;175(4):523-529. http://dx.doi.org/10.1001/jamainternmed.2014.7889.
8. Gill TM, Allore HG, Holford TR, Guo Z. Hospitalization, restricted activity, and the development of disability among older persons. JAMA. 2004;292(17):2115-2124. http://dx.doi.org/10.1001/jama.292.17.2115.
9. Inouye SK, Zhang Y, Han L, Leo-Summers L, Jones R, Marcantonio E. Recoverable cognitive dysfunction at hospital admission in older persons during acute illness. J Gen Intern Med. 2006;21(12):1276-1281. http://dx.doi.org/10.1111/j.1525-1497.2006.00613.x.
10. Lindquist LA, Go L, Fleisher J, Jain N, Baker D. Improvements in cognition following hospital discharge of community dwelling seniors. J Gen Intern Med. 2011;26(7):765-770. http://dx.doi.org/10.1007/s11606-011-1681-1.
11. Covinsky KE, Pierluissi E, Johnston CB. Hospitalization-associated disability: “She was probably able to ambulate, but I’m not sure.” JAMA. 2011;306(16):1782-1793. http://dx.doi.org/10.1001/jama.2011.1556.
12. Creditor MC. Hazards of hospitalization of the elderly. Ann Intern Med. 1993;118(3):219-223. http://dx.doi.org/10.7326/0003-4819-118-3-199302010-00011.
13. Dharmarajan K, Krumholz HM. Strategies to reduce 30-day readmissions in older patients hospitalized with heart failure and acute myocardial infarction. Curr Geriatr Rep. 2014;3(4):306-315. http://dx.doi.org/10.1007/s13670-014-0103-8.
14. McEwen BS. Protective and damaging effects of stress mediators. N Engl J Med. 1998;338(3):171-179. http://dx.doi.org/10.1056/NEJM199801153380307.
15. McEwen BS, Gianaros PJ. Stress- and allostasis-induced brain plasticity. Annu Rev Med. 2011;62:431-445. http://dx.doi.org/10.1146/annurev-med-052209-100430.
16. Dhabhar FS. Enhancing versus suppressive effects of stress on immune function: implications for immunoprotection and immunopathology. Neuroimmunomodulation. 2009;16(5):300-317. http://dx.doi.org/10.1159/000216188.
17. Thayer JF, Sternberg E. Beyond heart rate variability: vagal regulation of allostatic systems. Ann N Y Acad Sci. 2006;1088:361-372. http://dx.doi.org/10.1196/annals.1366.014.
18. Jacobson L, Akana SF, Cascio CS, Shinsako J, Dallman MF. Circadian variations in plasma corticosterone permit normal termination of adrenocorticotropin responses to stress. Endocrinology. 1988;122(4):1343-1348. http://dx.doi.org/10.1210/endo-122-4-1343.
19. McEwen BS. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev. 2007;87(3):873-904. http://dx.doi.org/10.1152/physrev.00041.2006.
20. Medicare Hospital Quality Chartbook 2014: Performance Report on Outcome Measures. Prepared by Yale New Haven Health Services Corporation Center for Outcomes Research and Evaluation for Centers for Medicare & Medicaid Services. https://www.cms.gov/medicare/quality-initiatives-patient-assessment-instruments/hospitalqualityinits/downloads/medicare-hospital-quality-chartbook-2014.pdf. Accessed February 26, 2018.
21. Bueno H, Ross JS, Wang Y, et al. Trends in length of stay and short-term outcomes among Medicare patients hospitalized for heart failure, 1993-2006. JAMA. 2010;303(21):2141-2147. http://dx.doi.org/10.1001/jama.2010.748.
22. Drye EE, Normand SL, Wang Y, et al. Comparison of hospital risk-standardized mortality rates calculated by using in-hospital and 30-day models: an observational study with implications for hospital profiling. Ann Intern Med. 2012;156(1 Pt 1):19-26. http://dx.doi.org/10.7326/0003-4819-156-1-201201030-00004.
23. Dharmarajan K, Hsieh A, Dreyer RP, Welsh J, Qin L, Krumholz HM. Relationship between age and trajectories of rehospitalization risk in older adults. J Am Geriatr Soc. 2017;65(2):421-426. http://dx.doi.org/10.1111/jgs.14583.
24. Krumholz HM, Hsieh A, Dreyer RP, Welsh J, Desai NR, Dharmarajan K. Trajectories of risk for specific readmission diagnoses after hospitalization for heart failure, acute myocardial infarction, or pneumonia. PLoS One. 2016;11(10):e0160492. http://dx.doi.org/10.1371/journal.pone.0160492.
25. Needham DM, Davidson J, Cohen H, et al. Improving long-term outcomes after discharge from intensive care unit: report from a stakeholders’ conference. Crit Care Med. 2012;40(2):502-509. http://dx.doi.org/10.1097/CCM.0b013e318232da75.
26. Brinkman S, de Jonge E, Abu-Hanna A, Arbous MS, de Lange DW, de Keizer NF. Mortality after hospital discharge in ICU patients. Crit Care Med. 2013;41(5):1229-1236. http://dx.doi.org/10.1097/CCM.0b013e31827ca4e1.
27. Steenbergen S, Rijkenberg S, Adonis T, Kroeze G, van Stijn I, Endeman H. Long-term treated intensive care patients outcomes: the one-year mortality rate, quality of life, health care use and long-term complications as reported by general practitioners. BMC Anesthesiol. 2015;15:142. http://dx.doi.org/10.1186/s12871-015-0121-x.
28. Hill AD, Fowler RA, Pinto R, Herridge MS, Cuthbertson BH, Scales DC. Long-term outcomes and healthcare utilization following critical illness--a population-based study. Crit Care. 2016;20:76. http://dx.doi.org/10.1186/s13054-016-1248-y.
29. Jackson JC, Pandharipande PP, Girard TD, et al. Depression, post-traumatic stress disorder, and functional disability in survivors of critical illness in the BRAIN-ICU study: a longitudinal cohort study. Lancet Respir Med. 2014;2(5):369-379. http://dx.doi.org/10.1016/S2213-2600(14)70051-7.
30. Balas MC, Vasilevskis EE, Olsen KM, et al. Effectiveness and safety of the awakening and breathing coordination, delirium monitoring/management, and early exercise/mobility bundle. Crit Care Med. 2014;42(5):1024-1036. http://dx.doi.org/10.1097/CCM.0000000000000129.
31. Kress JP, Hall JB. ICU-acquired weakness and recovery from critical illness. N Engl J Med. 2014;370(17):1626-1635. http://dx.doi.org/10.1056/NEJMra1209390.
32. Mendez-Tellez PA, Needham DM. Early physical rehabilitation in the ICU and ventilator liberation. Respir Care. 2012;57(10):1663-1669. http://dx.doi.org/10.4187/respcare.01931.
33. Schweickert WD, Hall J. ICU-acquired weakness. Chest. 2007;131(5):1541-1549. http://dx.doi.org/10.1378/chest.06-2065.
34. Balbo M, Leproult R, Van Cauter E. Impact of sleep and its disturbances on hypothalamo-pituitary-adrenal axis activity. Int J Endocrinol. 2010;2010:759234. http://dx.doi.org/10.1155/2010/759234.
35. Spiegel K, Leproult R, Van Cauter E. Impact of sleep debt on metabolic and endocrine function. Lancet. 1999;354(9188):1435-1439. http://dx.doi.org/10.1016/S0140-6736(99)01376-8.
36. Orwelius L, Nordlund A, Nordlund P, Edell-Gustafsson U, Sjoberg F. Prevalence of sleep disturbances and long-term reduced health-related quality of life after critical care: a prospective multicenter cohort study. Crit Care. 2008;12(4):R97. http://dx.doi.org/10.1186/cc6973.
37. Buxton OM, Ellenbogen JM, Wang W, et al. Sleep disruption due to hospital noises: a prospective evaluation. Ann Intern Med. 2012;157(3):170-179. http://dx.doi.org/10.7326/0003-4819-157-3-201208070-00472.
38. Sunbul M, Kanar BG, Durmus E, Kivrak T, Sari I. Acute sleep deprivation is associated with increased arterial stiffness in healthy young adults. Sleep Breath. 2014;18(1):215-220. http://dx.doi.org/10.1007/s11325-013-0873-9.
39. Van Dongen HP, Maislin G, Mullington JM, Dinges DF. The cumulative cost of additional wakefulness: dose-response effects on neurobehavioral functions and sleep physiology from chronic sleep restriction and total sleep deprivation. Sleep. 2003;26(2):117-126. http://dx.doi.org/10.1093/sleep/26.2.117.
40. Franklin GA, McClave SA, Hurt RT, et al. Physician-delivered malnutrition: why do patients receive nothing by mouth or a clear liquid diet in a university hospital setting? JPEN J Parenter Enteral Nutr. 2011;35(3):337-342. http://dx.doi.org/10.1177/0148607110374060.
41. Sullivan DH, Sun S, Walls RC. Protein-energy undernutrition among elderly hospitalized patients: a prospective study. JAMA. 1999;281(21):2013-2019. http://dx.doi.org/10.1001/jama.281.21.2013.
42. Anderson DA, Shapiro JR, Lundgren JD, Spataro LE, Frye CA. Self-reported dietary restraint is associated with elevated levels of salivary cortisol. Appetite. 2002;38(1):13-17. http://dx.doi.org/10.1006/appe.2001.0459.
43. Ott V, Friedrich M, Prilop S, et al. Food anticipation and subsequent food withdrawal increase serum cortisol in healthy men. Physiol Behav. 2011;103(5):594-599. http://dx.doi.org/10.1016/j.physbeh.2011.04.020.
44. Covinsky KE, Martin GE, Beyth RJ, Justice AC, Sehgal AR, Landefeld CS. The relationship between clinical assessments of nutritional status and adverse outcomes in older hospitalized medical patients. J Am Geriatr Soc. 1999;47(5):532-538. http://dx.doi.org/10.1111/j.1532-5415.1999.tb02566.x.
45. Lazarus BA, Murphy JB, Coletta EM, McQuade WH, Culpepper L. The provision of physical activity to hospitalized elderly patients. Arch Intern Med. 1991;151(12):2452-2456.
46. Minnick AF, Mion LC, Johnson ME, Catrambone C, Leipzig R. Prevalence and variation of physical restraint use in acute care settings in the US. J Nurs Scholarsh. 2007;39(1):30-37. http://dx.doi.org/10.1111/j.1547-5069.2007.00140.x.
47. Zisberg A, Shadmi E, Sinoff G, Gur-Yaish N, Srulovici E, Admi H. Low mobility during hospitalization and functional decline in older adults. J Am Geriatr Soc. 2011;59(2):266-273. http://dx.doi.org/10.1111/j.1532-5415.2010.03276.x.
48. Landefeld CS, Palmer RM, Kresevic DM, Fortinsky RH, Kowal J. A randomized trial of care in a hospital medical unit especially designed to improve the functional outcomes of acutely ill older patients. N Engl J Med. 1995;332(20):1338-1344. http://dx.doi.org/10.1056/NEJM199505183322006.
49. Douglas CH, Douglas MR. Patient-friendly hospital environments: exploring the patients’ perspective. Health Expectations: an international journal of public participation in health care and health policy. 2004;7(1):61-73. http://dx.doi.org/10.1046/j.1369-6513.2003.00251.x.
50. Volicer BJ. Hospital stress and patient reports of pain and physical status. Journal Human Stress. 1978;4(2):28-37. http://dx.doi.org/10.1080/0097840X.1978.9934984.
51. Willner P, Towell A, Sampson D, Sophokleous S, Muscat R. Reduction of sucrose preference by chronic unpredictable mild stress, and its restoration by a tricyclic antidepressant. Psychopharmacology (Berl). 1987;93(3):358-364. http://dx.doi.org/10.1007/BF00187257.
52. Grippo AJ, Francis J, Beltz TG, Felder RB, Johnson AK. Neuroendocrine and cytokine profile of chronic mild stress-induced anhedonia. Physiol Behav. 2005;84(5):697-706. http://dx.doi.org/10.1016/j.physbeh.2005.02.011.
53. Krishnan V, Nestler EJ. Animal models of depression: molecular perspectives. Curr Top Behav Neurosci. 2011;7:121-147. http://dx.doi.org/10.1007/7854_2010_108.
54. Magarinos AM, McEwen BS. Stress-induced atrophy of apical dendrites of hippocampal CA3c neurons: comparison of stressors. Neuroscience. 1995;69(1):83-88. http://dx.doi.org/10.1016/0306-4522(95)00256-I.
55. McEwen BS. Plasticity of the hippocampus: adaptation to chronic stress and allostatic load. Ann N Y Acad Sci. 2001;933:265-277. http://dx.doi.org/10.1111/j.1749-6632.2001.tb05830.x.
56. McEwen BS. The brain on stress: toward an integrative approach to brain, body, and behavior. Perspect Psychol Sci. 2013;8(6):673-675. http://dx.doi.org/10.1177/1745691613506907.
57. McEwen BS, Morrison JH. The brain on stress: vulnerability and plasticity of the prefrontal cortex over the life course. Neuron. 2013;79(1):16-29. http://dx.doi.org/10.1016/j.neuron.2013.06.028.
58. Dutta P, Courties G, Wei Y, et al. Myocardial infarction accelerates atherosclerosis. Nature. 2012;487(7407):325-329. http://dx.doi.org/10.1038/nature11260.
59. Lu XT, Liu YF, Zhang L, et al. Unpredictable chronic mild stress promotes atherosclerosis in high cholesterol-fed rabbits. Psychosom Med. 2012;74(6):604-611. http://dx.doi.org/10.1097/PSY.0b013e31825d0b71.
60. Heidt T, Sager HB, Courties G, et al. Chronic variable stress activates hematopoietic stem cells. Nat Med. 2014;20(7):754-758. http://dx.doi.org/10.1038/nm.3589.
61. Sheridan JF, Feng NG, Bonneau RH, Allen CM, Huneycutt BS, Glaser R. Restraint stress differentially affects anti-viral cellular and humoral immune responses in mice. J Neuroimmunol. 1991;31(3):245-255. http://dx.doi.org/10.1016/0165-5728(91)90046-A.
62. Kyrou I, Tsigos C. Stress hormones: physiological stress and regulation of metabolism. Curr Opin Pharmacol. 2009;9(6):787-793. http://dx.doi.org/10.1016/j.coph.2009.08.007.
63. Rosmond R. Role of stress in the pathogenesis of the metabolic syndrome. Psychoneuroendocrinology. 2005;30(1):1-10. http://dx.doi.org/10.1016/j.psyneuen.2004.05.007.
64. Tamashiro KL, Sakai RR, Shively CA, Karatsoreos IN, Reagan LP. Chronic stress, metabolism, and metabolic syndrome. Stress. 2011;14(5):468-474. http://dx.doi.org/10.3109/10253890.2011.606341.
65. McEwen BS. Mood disorders and allostatic load. Biol Psychiatry. 2003;54(3):200-207. http://dx.doi.org/10.1016/S0006-3223(03)00177-X.
66. Zareie M, Johnson-Henry K, Jury J, et al. Probiotics prevent bacterial translocation and improve intestinal barrier function in rats following chronic psychological stress. Gut. 2006;55(11):1553-1560. http://dx.doi.org/10.1136/gut.2005.080739.
67. Joachim RA, Quarcoo D, Arck PC, Herz U, Renz H, Klapp BF. Stress enhances airway reactivity and airway inflammation in an animal model of allergic bronchial asthma. Psychosom Med. 2003;65(5):811-815. http://dx.doi.org/10.1097/01.PSY.0000088582.50468.A3.
68. Thaker PH, Han LY, Kamat AA, et al. Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat Med. 2006;12(8):939-944. http://dx.doi.org/10.1038/nm1447.
69. Schreuder L, Eggen BJ, Biber K, Schoemaker RG, Laman JD, de Rooij SE. Pathophysiological and behavioral effects of systemic inflammation in aged and diseased rodents with relevance to delirium: A systematic review. Brain Behav Immun. 2017;62:362-381. http://dx.doi.org/10.1016/j.bbi.2017.01.010.
70. Mu DL, Li LH, Wang DX, et al. High postoperative serum cortisol level is associated with increased risk of cognitive dysfunction early after coronary artery bypass graft surgery: a prospective cohort study. PLoS One. 2013;8(10):e77637. http://dx.doi.org/10.1371/journal.pone.0077637.
71. Mu DL, Wang DX, Li LH, et al. High serum cortisol level is associated with increased risk of delirium after coronary artery bypass graft surgery: a prospective cohort study. Crit Care. 2010;14(6):R238. http://dx.doi.org/10.1186/cc9393.
72. Nguyen DN, Huyghens L, Zhang H, Schiettecatte J, Smitz J, Vincent JL. Cortisol is an associated-risk factor of brain dysfunction in patients with severe sepsis and septic shock. Biomed Res Int. 2014;2014:712742. http://dx.doi.org/10.1155/2014/712742.
73. Elkind MS, Carty CL, O’Meara ES, et al. Hospitalization for infection and risk of acute ischemic stroke: the Cardiovascular Health Study. Stroke. 2011;42(7):1851-1856. http://dx.doi.org/10.1161/STROKEAHA.110.608588.
74. Feibel JH, Hardy PM, Campbell RG, Goldstein MN, Joynt RJ. Prognostic value of the stress response following stroke. JAMA. 1977;238(13):1374-1376.
75. Jutla SK, Yuyun MF, Quinn PA, Ng LL. Plasma cortisol and prognosis of patients with acute myocardial infarction. J Cardiovasc Med (Hagerstown). 2014;15(1):33-41. http://dx.doi.org/10.2459/JCM.0b013e328364100b.
76. Yende S, D’Angelo G, Kellum JA, et al. Inflammatory markers at hospital discharge predict subsequent mortality after pneumonia and sepsis. Am J Respir Crit Care Med. 2008;177(11):1242-1247. http://dx.doi.org/10.1164/rccm.200712-1777OC.
77. Gouin JP, Kiecolt-Glaser JK. The impact of psychological stress on wound healing: methods and mechanisms. Immunol Allergy Clin North Am. 2011;31(1):81-93. http://dx.doi.org/10.1016/j.iac.2010.09.010.
78. Capes SE, Hunt D, Malmberg K, Gerstein HC. Stress hyperglycaemia and increased risk of death after myocardial infarction in patients with and without diabetes: a systematic overview. Lancet. 2000;355(9206):773-778. http://dx.doi.org/10.1016/S0140-6736(99)08415-9.
79. O’Neill PA, Davies I, Fullerton KJ, Bennett D. Stress hormone and blood glucose response following acute stroke in the elderly. Stroke. 1991;22(7):842-847. http://dx.doi.org/10.1161/01.STR.22.7.842.
80. Waterer GW, Kessler LA, Wunderink RG. Medium-term survival after hospitalization with community-acquired pneumonia. Am J Respir Crit Care Med. 2004;169(8):910-914. http://dx.doi.org/10.1164/rccm.200310-1448OC.
81. Rosengren A, Freden M, Hansson PO, Wilhelmsen L, Wedel H, Eriksson H. Psychosocial factors and venous thromboembolism: a long-term follow-up study of Swedish men. J Thrombosis Haemostasis. 2008;6(4):558-564. http://dx.doi.org/10.1111/j.1538-7836.2007.02857.x.
82. Oswald GA, Smith CC, Betteridge DJ, Yudkin JS. Determinants and importance of stress hyperglycaemia in non-diabetic patients with myocardial infarction. BMJ. 1986;293(6552):917-922. http://dx.doi.org/10.1136/bmj.293.6552.917.
83. Middlekauff HR, Nguyen AH, Negrao CE, et al. Impact of acute mental stress on sympathetic nerve activity and regional blood flow in advanced heart failure: implications for ‘triggering’ adverse cardiac events. Circulation. 1997;96(6):1835-1842. http://dx.doi.org/10.1161/01.CIR.96.6.1835.
84. Nijm J, Jonasson L. Inflammation and cortisol response in coronary artery disease. Ann Med. 2009;41(3):224-233. http://dx.doi.org/10.1080/07853890802508934.
85. Steptoe A, Hackett RA, Lazzarino AI, et al. Disruption of multisystem responses to stress in type 2 diabetes: investigating the dynamics of allostatic load. Proc Natl Acad Sci U S A. 2014;111(44):15693-15698. http://dx.doi.org/10.1073/pnas.1410401111.
86. Sepehri A, Beggs T, Hassan A, et al. The impact of frailty on outcomes after cardiac surgery: a systematic review. J Thorac Cardiovasc Surg. 2014;148(6):3110-3117. http://dx.doi.org/10.1016/j.jtcvs.2014.07.087.
87. Johar H, Emeny RT, Bidlingmaier M, et al. Blunted diurnal cortisol pattern is associated with frailty: a cross-sectional study of 745 participants aged 65 to 90 years. J Clin Endocrinol Metab. 2014;99(3):E464-468. http://dx.doi.org/10.1210/jc.2013-3079.
88. Yao X, Li H, Leng SX. Inflammation and immune system alterations in frailty. Clin Geriatr Med. 2011;27(1):79-87. http://dx.doi.org/10.1016/j.cger.2010.08.002.
89. Hospital Elder Life Program (HELP) for Prevention of Delirium. 2017; http://www.hospitalelderlifeprogram.org/. Accessed February 16, 2018.
90. Shepperd S, Doll H, Angus RM, et al. Admission avoidance hospital at home. Cochrane Database of System Rev. 2008;(4):CD007491. http://dx.doi.org/10.1002/14651858.CD007491.pub2’
91. Leff B, Burton L, Mader SL, et al. Comparison of functional outcomes associated with hospital at home care and traditional acute hospital care. J Am Geriatrics Soc. 2009;57(2):273-278. http://dx.doi.org/10.1111/j.1532-5415.2008.02103.x.
92. Qaddoura A, Yazdan-Ashoori P, Kabali C, et al. Efficacy of hospital at home in patients with heart failure: a systematic review and meta-analysis. PloS One. 2015;10(6):e0129282. http://dx.doi.org/10.1371/journal.pone.0129282.
93. Seeman T, Gruenewald T, Karlamangla A, et al. Modeling multisystem biological risk in young adults: The Coronary Artery Risk Development in Young Adults Study. Am J Hum Biol. 2010;22(4):463-472. http://dx.doi.org/10.1002/ajhb.21018.
94. Auerbach AD, Kripalani S, Vasilevskis EE, et al. Preventability and causes of readmissions in a national cohort of general medicine patients. JAMA Intern Med. 2016;176(4):484-493. http://dx.doi.org/10.1001/jamainternmed.2015.7863.
95. Hansen LO, Young RS, Hinami K, Leung A, Williams MV. Interventions to reduce 30-day rehospitalization: a systematic review. Ann Intern Med. 2011;155(8):520-528. http://dx.doi.org/10.7326/0003-4819-155-8-201110180-00008.
96. Takahashi PY, Naessens JM, Peterson SM, et al. Short-term and long-term effectiveness of a post-hospital care transitions program in an older, medically complex population. Healthcare. 2016;4(1):30-35. http://dx.doi.org/10.1016/j.hjdsi.2015.06.006.
97. Dharmarajan K, Swami S, Gou RY, Jones RN, Inouye SK. Pathway from delirium to death: potential in-hospital mediators of excess mortality. J Am Geriatr Soc. 2017;65(5):1026-1033. http://dx.doi.org/10.1111/jgs.14743.
© 2018 Society of Hospital Medicine
Hypertrophic cardiomyopathy: A complex disease
Hypertrophic cardiomyopathy (HCM) is a complex disease. Most people who carry the mutations that cause it are never affected at any point in their life, but some are affected at a young age. And in rare but tragic cases, some die suddenly while competing in sports. With such a wide range of phenotypic expressions, a single therapy does not fit all.
HCM is more common than once thought. Since the discovery of its genetic predisposition in 1960, it has come to be recognized as the most common heritable cardiovascular disease.1 Although earlier epidemiologic studies had estimated a prevalence of 1 in 500 (0.2%) of the general population, genetic testing and cardiac magnetic resonance imaging (MRI) now show that up to 1 in 200 (0.5%) of all people may be affected.1,2 Its prevalence is significant in all ethnic groups.
This review outlines our expanding knowledge of the pathophysiology, diagnosis, and clinical management of HCM.
A PLETHORA OF MUTATIONS IN CARDIAC SARCOMERIC GENES
The genetic differences within HCM result in varying degrees and locations of left ventricular hypertrophy. Any segment of the ventricle can be involved, although HCM is classically asymmetric and mainly involves the septum (Figure 1). A variant form of HCM involves the apex of the heart (Figure 2).
LEFT VENTRICULAR OUTFLOW TRACT OBSTRUCTION
Only in the last decade has the significance of left ventricular outflow tract obstruction in HCM been truly appreciated. The degree of obstruction in HCM is dynamic, as opposed to the fixed obstruction in patients with aortic stenosis or congenital subvalvular membranes. Therefore, in HCM, exercise or drugs (eg, dobutamine) that increase cardiac contractility increase the obstruction, as do maneuvers or drugs (the Valsalva maneuver, nitrates) that reduce filling of the left ventricle.
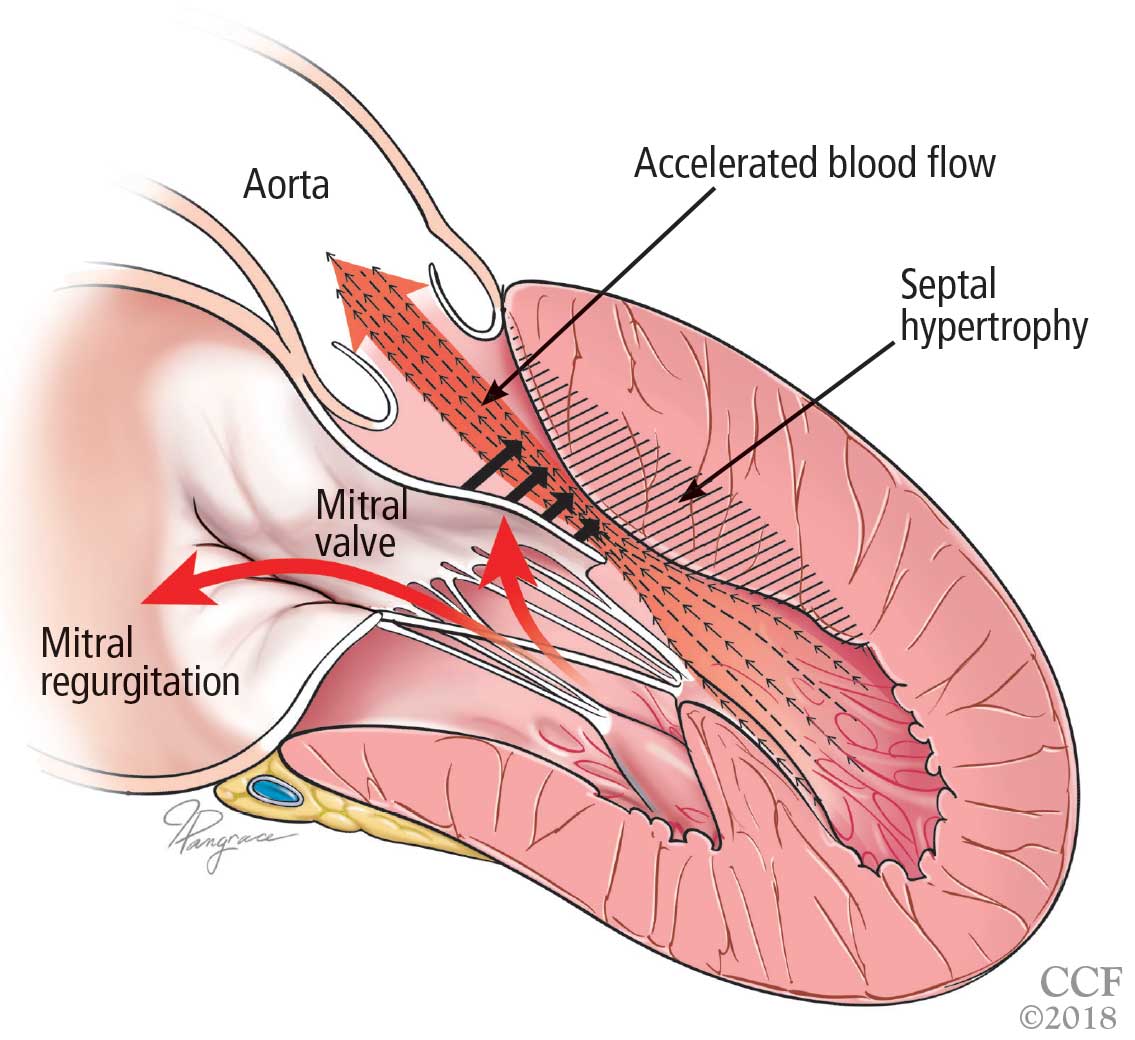
A less common source of dynamic obstruction is the papillary muscles (Figure 4). Hypertrophy of the papillary muscles can result in obstruction by these muscles themselves, which is visible on echocardiography. Anatomic variations include anteroapical displacement or bifid papillary muscles, and these variants can be associated with dynamic left ventricular outflow tract obstruction, even with no evidence of septal thickening (Figure 5).7,8 Recognizing this patient subset has important implications for management, as discussed below.
DIAGNOSTIC EVALUATION
The clinical presentation varies
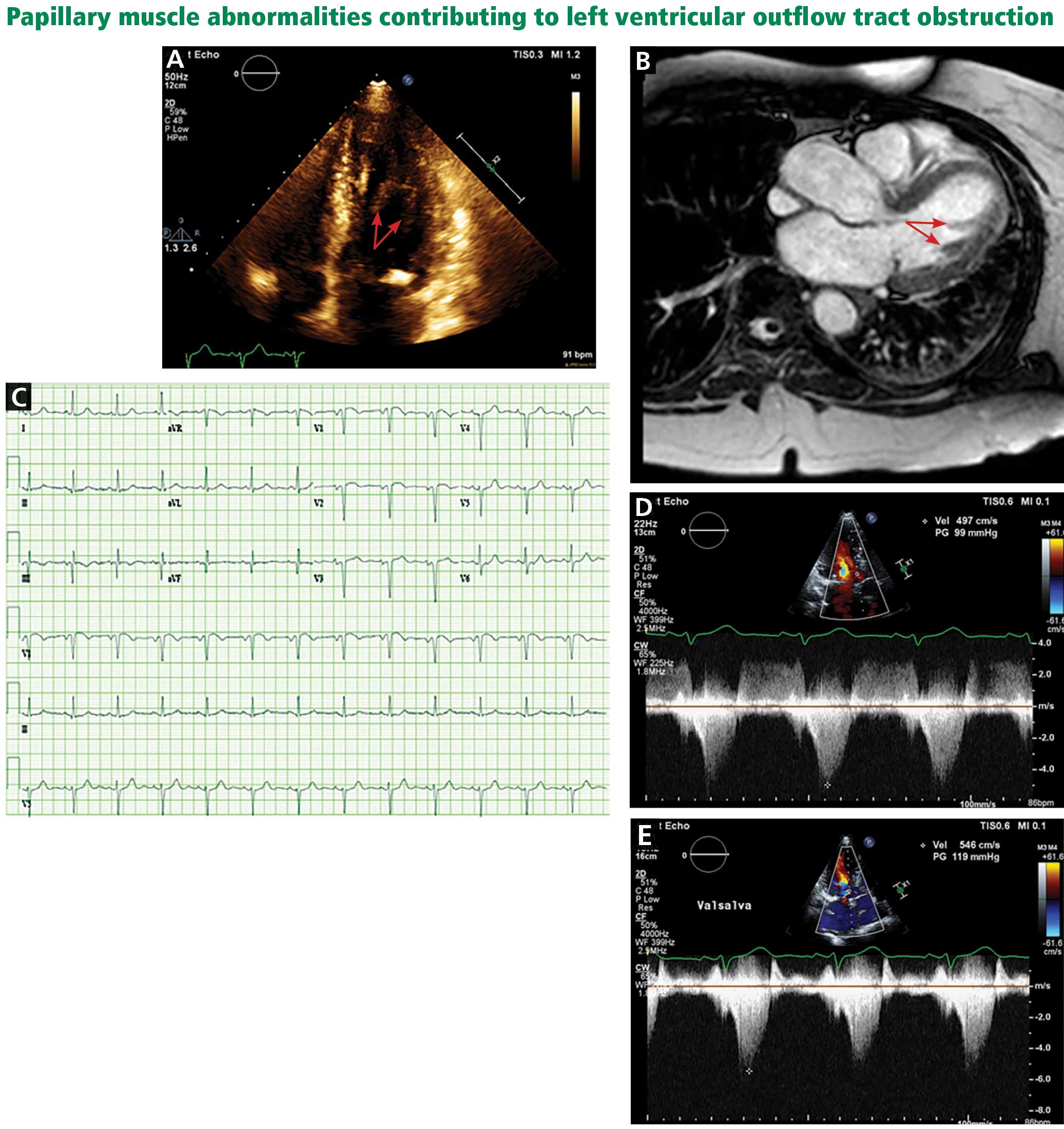
Even if patients harbor the same genetic variant, the clinical presentation can differ widely. Although the most feared presentation is sudden cardiac death, particularly in young athletes, most patients have no symptoms and can anticipate a normal life expectancy. The annual incidence of sudden cardiac death in all HCM patients is estimated at less than 1%.10 Sudden cardiac death in HCM patients is most often due to ventricular tachyarrhythmias and most often occurs in asymptomatic patients under age 35.
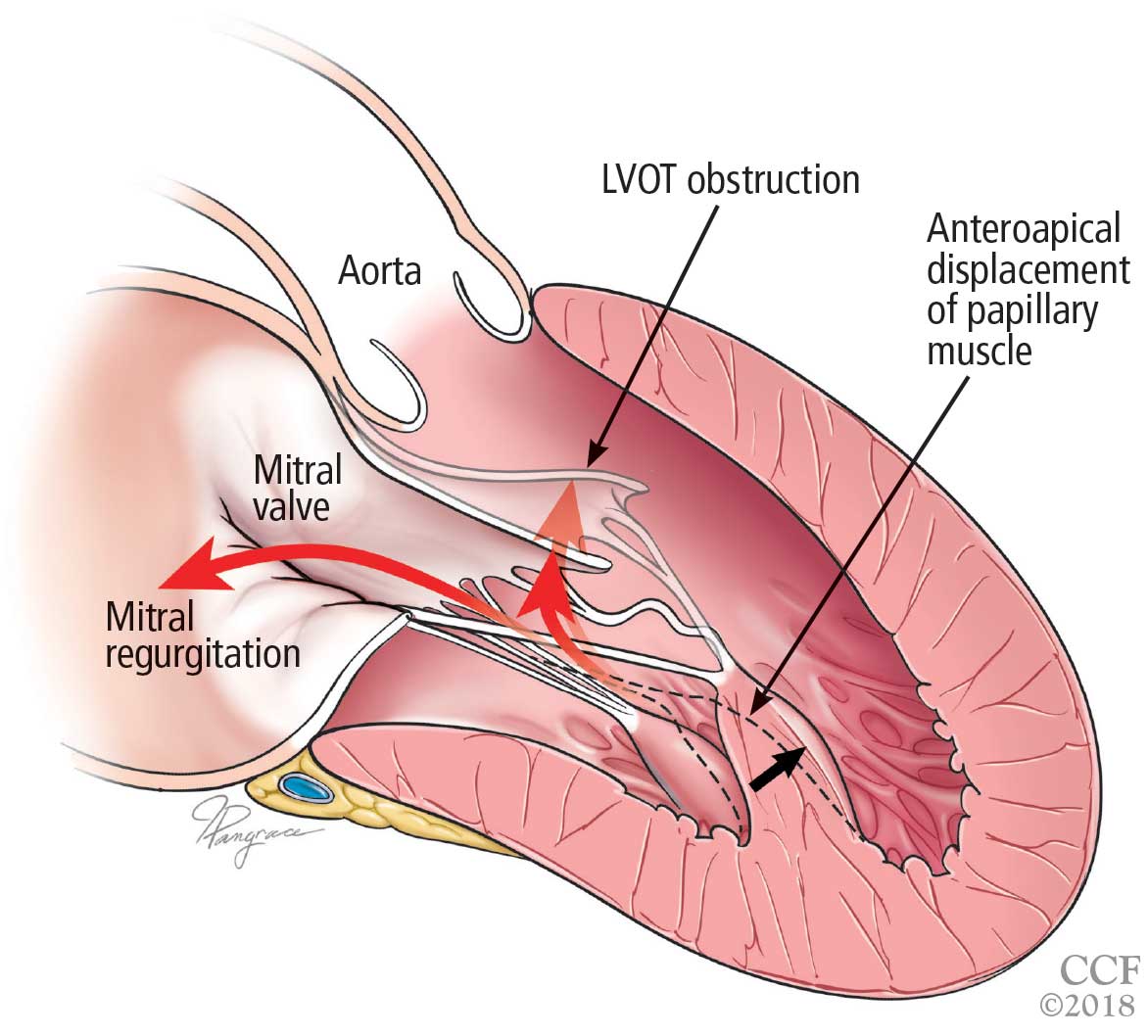
Physical findings are nonspecific

It can be difficult to distinguish the murmur of left ventricular outflow tract obstruction in HCM from a murmur related to aortic stenosis by auscultation alone. The simplest clinical method for telling them apart involves the Valsalva maneuver: bearing down creates a positive intrathoracic pressure and limits venous return, thus decreasing intracardiac filling pressure. This in turn results in less separation between the mitral valve and the ventricular septum in HCM, which increases obstruction and therefore makes the murmur louder. In contrast, in patients with fixed obstruction due to aortic stenosis, the murmur will decrease in intensity owing to the reduced flow associated with reduced preload.
Laboratory testing for phenocopies of HCM

A metabolic panel will show derangements in liver function and glucose levels in patients with glycogen storage disorders such as Pompe disease.
Serum creatinine. Renal dysfunction will be seen in patients with Fabry disease or amyloidosis.
Creatine kinase may be elevated in patients with Danon disease.
Electrocardiographic findings are common
More than 90% of HCM patients have electrocardiographic abnormalities. Although the findings can vary widely, common manifestations include:
- Left ventricular hypertrophy
- A pseudoinfarct pattern with Q waves in the anterolateral leads
- Repolarization changes such as T-wave inversions and horizontal or down-sloping ST segments.
Apical HCM, seen mainly in Asian populations, often presents with giant T-wave inversion (> 10 mm) in the anterolateral leads, most prominent in V4, V5, and V6.
Notably, the degree of electrocardiographic abnormalities does not correlate with the severity or pattern of hypertrophy.9 Electrocardiography lacks specificity for definitive diagnosis, and further diagnostic testing should therefore be pursued.
Echocardiography: Initial imaging test
Transthoracic echocardiography is the initial imaging test in patients with suspected HCM, allowing for cost-effective quantitative and qualitative assessment of left ventricular morphology and function. Left ventricular hypertrophy is considered pathologic if wall thickness is 15 mm or greater without a known cause. Transthoracic echocardiography also allows for evaluation of left atrial volume and mitral valve anatomy and function.
Speckle tracking imaging is an advanced echocardiographic technique that measures strain. Its major advantage is in identifying early abnormalities in genotype-positive, phenotype-negative HCM patients, ie, people who harbor mutations but who have no clinical symptoms or signs of HCM, potentially allowing for modification of the natural history of HCM.12 Strain imaging can also differentiate between physiologic hypertrophy (“athlete’s heart”) and hypertension and HCM.13,14
The utility of echocardiography in HCM is heavily influenced by the sonographer’s experience in obtaining adequate acoustic windows. This may be more difficult in obese patients, patients with advanced obstructive lung disease or pleural effusions, and women with breast implants.
Magnetic resonance imaging
MRI has an emerging role in both diagnosing and predicting risk in HCM, and is routinely done as an adjunct to transthoracic echocardiography on initial diagnosis in our tertiary referral center. It is particularly useful in patients suspected of having apical hypertrophy (Figure 2), in whom the diagnosis may be missed in up to 10% on transthoracic echocardiography alone.15 MRI can also enhance the assessment of left ventricular hypertrophy and has been shown to improve the diagnostic classification of HCM.16 It is the best way to assess myocardial tissue abnormalities, and late gadolinium enhancement to detect interstitial fibrosis can be used for further prognostication. While historically the primary role of MRI in HCM has been in phenotype classification, there is currently much interest in its role in risk stratification of HCM patients for ICD implantation.
MRI with late gadolinium enhancement provides insight into the location, pattern, and extent of myocardial fibrosis; the extent of fibrosis has been shown to be a strong independent predictor of poor outcomes, including sudden cardiac death.17–20 However, late gadolinium enhancement can be technically challenging, as variations in the timing of postcontrast imaging, sequences for measuring late gadolinium enhancement, or detection thresholds can result in widely variable image quality. Cardiac MRI should therefore be performed at an experienced center with standardized imaging protocols in place.
Current guidelines recommend considering cardiac MRI if a patient’s risk of sudden cardiac death remains inconclusive after conventional risk stratification, as discussed below.9,21
Stress testing for risk stratification
Exercise stress electrocardiography. Treadmill exercise stress testing with electrocardiography and hemodynamic monitoring was one of the first tools used for risk stratification in HCM.
Although systolic blood pressure normally increases by at least 20 mm Hg with exercise, one-quarter of HCM patients have either a blunted response (failure of systolic blood pressure to increase by at least 20 mm Hg) or a hypotensive response (a drop in systolic blood pressure of 20 mm Hg or more, either continuously or after an initial increase). Studies have shown that HCM patients who have abnormal blood pressure responses during exercise have a higher risk of sudden cardiac death.22–24
Exercise stress echocardiography can be useful to evaluate for provoked increases in the left ventricular outflow tract gradient, which may contribute to a patient’s symptoms even if the resting left ventricular outflow tract gradient is normal. Exercise testing is preferred over pharmacologic stimulation because it can provide functional assessment of whether a patient’s clinical symptoms are truly related to hemodynamic changes due to the hypertrophied ventricle, or whether alternative mechanisms should be explored.
Cardiopulmonary stress testing can readily add prognostic value with additional measurements of functional capacity. HCM patients who cannot achieve their predicted maximal exercise value such as peak rate of oxygen consumption, ventilation efficiency, or anaerobic threshold have higher rates of morbidity and mortality.25,26 Stress testing can also be useful for risk stratification in asymptomatic patients, with one study showing that those who achieve more than 100% of their age- and sex-predicted metabolic equivalents have a low event rate.27
Ambulatory electrocardiographic monitoring in all patients at diagnosis
Ambulatory electrocardiographic monitoring for 24 to 48 hours is recommended for all HCM patients at the time of diagnosis, even if they have no symptoms. Any evidence of nonsustained ventricular tachycardia suggests a substantially higher risk of sudden cardiac death.28,29
In patients with no symptoms or history of arrhythmia, current guidelines suggest ambulatory electrocardiographic monitoring every 1 to 2 years.9,21
Two risk-stratification models

The North American model was the first risk-stratification tool and considers 5 risk factors.9 However, if this algorithm were strictly followed, up to 60% of HCM patients would be candidates for cardioverter-defibrillator implantation.
The European model. This concern led to the development of the HCM Risk-SCD (sudden cardiac death), a risk-stratification tool introduced in the 2014 European Society of Cardiology HCM guidelines.30 This web-based calculator estimates a patient’s 5-year risk of sudden cardiac death using a complex calculation based on 7 clinical risk factors. If a patient’s calculated 5-year risk of sudden cardiac death is 6% or higher, cardioverter-defibrillator implantation is recommended for primary prevention.
The HCM Risk-SCD calculator was validated and compared with classic risk factors alone in a retrospective cohort study in 48 HCM patients.30 Compared with the North American model, the European model results in a lower rate of cardioverter-defibrillator implantation (20% to 26%).31,32
Despite the better specificity of the European model, a large retrospective cohort analysis showed that a significant number of patients stratified as being at low risk for sudden cardiac death were ultimately found to be at high risk in clinical practice.31 Further research is needed to find the optimal risk-stratification approach in HCM patients at low to intermediate risk.
GENETIC TESTING, COUNSELING, AND FAMILY SCREENING
Genetic testing is becoming more widely available and has rapidly expanded in clinical practice. Genetic counseling must be performed alongside genetic testing and requires professionals trained to handle the clinical and social implications of genetic testing. With this in mind, genetic testing can provide a definitive means of identifying family members at risk of HCM.
Given the autosomal dominant nature of HCM, screening for HCM is recommended in all first-degree relatives of an affected patient. Genetic testing may be a means to achieve this if a pathogenic mutation has been identified in the affected patient. However, serial electrocardiographic and transthoracic echocardiographic monitoring is an acceptable alternative in those without a clear genetic mutation association or in those who do not want to undergo genetic testing. If these first-degree relatives who do not undergo genetic testing are adult athletes or adolescents, they should undergo surveillance monitoring, with echocardiography and electrocardiography, whereas adults not participating in athletics should be monitored every 5 years.9,21
As genetic counseling and testing become more widely available, more patients are being found who harbor a mutation but have no phenotypic manifestations of HCM on initial presentation. Clinical expression varies, so continued monitoring of these patients is important. Expert guidelines again recommend serial electrocardiography, transthoracic echocardiography, and clinical assessment every 5 years for adults.9
Recent data suggest that up to 40% of HCM cases are nonfamilial, ie, their inheritance is sporadic with no known family history and no sarcomeric gene mutation evident on screening.33,34 The clinical course in this subgroup seems to be more benign, with later clinical presentations (age > 40) and lower risk of major adverse cardiovascular events.
MANAGEMENT
Conservative management
Asymptomatic HCM can usually be managed with lifestyle modifications.
Avoiding high-risk physical activities is the most important modification. All HCM patients should be counseled on the risk of sudden cardiac death and advised against participating in competitive sports or intense physical activity.35 Aerobic exercise is preferable to isometric exercises such as weightlifting, which may prompt the Valsalva maneuver with worsening of left ventricular outflow tract obstruction leading to syncope. A recent study showed that moderate-intensity aerobic exercise can safely improve exercise capacity, which may ultimately improve functional status and quality of life.36
Avoiding dehydration and excessive alcohol intake are also important in maintaining adequate preload to prevent an increasing left ventricular outflow tract gradient, given the dynamic nature of the left ventricular outflow tract obstruction in HCM.
Medical management: Beta-blockers, then calcium channel blockers
Beta-blockers are the first-line therapy for symptomatic HCM related to left ventricular outflow tract obstruction. Their negative inotropic effect reduces the contractile force of the ventricle, effectively reducing the pressure gradient across the outflow tract. Reduced contractility also means that the overall myocardial workload is less, which ultimately translates to a reduced oxygen demand. With their negative chronotropic effect, beta-blockers lower the heart rate and thereby lengthen the diastolic filling phase, allowing for optimization of preload conditions to help prevent increasing the left ventricular outflow tract gradient.37,38
Beta-blockers can be titrated according to the patient’s symptoms and tolerance. Fatigue and loss of libido are among the most common side effects.
Nondihydropyridine calcium channel blockers can be a second-line therapy in patients who cannot tolerate beta-blockers. Several studies have shown improvement in surrogate outcomes such as estimated left ventricular mass and QRS amplitude on electrocardiography, but currently no available data show that these drugs improve symptoms.28,39,40 They should be avoided in those with severe left ventricular outflow tract obstruction (gradient ≥ 100 mm Hg), as they can lead to critical outflow tract obstruction owing to their peripheral vasodilatory effect.
Dihydropyridine calcium channel blockers should be avoided altogether, as they produce even more peripheral vasodilation and afterload reduction than nondihydropyridine calcium channel blockers.
Disopyramide, a class IA antiarrhythmic, has been shown to effectively reduce outflow gradients and relieve symptoms. However, in view of its adverse effects, it is a third-line therapy, given to those for whom beta-blockers and calcium channel blockers have failed. Its most worrisome adverse effect is QT prolongation, and the QT interval should therefore be closely monitored at the start of treatment. Anticholinergic effects are common and include dry eyes and mouth, urinary retention, and drowsiness.
Disopyramide is usually used in combination with beta-blockers for symptom control as a bridge to a planned invasive intervention.41
Use with caution
Any medication that causes afterload reduction, peripheral vasodilation, intravascular volume depletion, or positive inotropy can worsen the dynamic left ventricular outflow tract obstruction in a patient with HCM and should be avoided.
Angiotensin-converting enzyme (ACE) inhibitors, angiotensin II receptor blockers (ARBs), and nitrates must be used with extreme caution in these patients.
Diuretics. Even restrained use of diuretics can cause significant hemodynamic compromise in patients with obstructive physiology. Therefore, diuretics should be used sparingly in these patients.
Digoxin should not be used for managing atrial fibrillation in these patients, as its positive inotropic effect increases contractility and increases the left ventricular outflow tract gradient.
Norepinephrine and inotropic agents such as dobutamine and dopamine should be avoided for the same reason as digoxin. In patients with circulatory shock requiring vasopressor support, pure alpha-agonists such as phenylephrine are preferred, as they increase peripheral resistance without an inotropic effect.
Anticoagulation for atrial tachyarrhythmias
The risk of systemic thromboembolic events is significantly increased in HCM patients with atrial fibrillation or flutter, regardless of their estimated risk using conventional risk-stratification tools such as the CHADS2 score.42–44 In accordance with current American Heart Association and American College of Cardiology guidelines, we recommend anticoagulation therapy for all HCM patients with a history of atrial fibrillation or flutter. Warfarin is the preferred anticoagulant; direct oral anticoagulants can be considered, but there are currently no data on their use in HCM.9
Standard heart failure treatments
End-stage systolic heart failure is a consequence of HCM but affects only 3% to 4% of patients.45 While most randomized controlled trials of heart failure treatment have excluded HCM patients, current guidelines recommend the same evidence-based medical therapies used in other patients who have heart failure with reduced ejection fraction. This includes ACE inhibitors, ARBs, beta-blockers, and aldosterone antagonists if indicated.9,21
Heart transplant should be considered in patients with class III or IV New York Heart Association functional status despite optimization of their HCM treatment regimen. Heart transplant outcomes for HCM patients are comparable to outcomes for patients who receive a transplant for non-HCM cardiovascular disease.45,46
Septal reduction therapy
If medical therapy fails or is not tolerated in patients with severe symptoms, surgery can be considered for obstructive HCM.
Ventricular septal myectomy has been the long-standing gold standard of invasive therapy. Multiple studies have demonstrated long-term survival after myectomy to be equivalent to that in the general population and better than that of HCM patients who do not undergo this surgery.47–50 Factors that may be associated with better surgical outcomes include age younger than 50, left atrial size less than 46 mm, and resolution of atrial fibrillation during follow-up.51
Septal reduction therapy may also be considered in patients at high risk of sudden cardiac death based on a history of recurrent ventricular tachycardia or risk-stratification models as described above. Retrospective analyses have shown that surgical myectomy can markedly reduce the incidence of appropriate implantable cardioverter-defibrillator discharges and the risk of sudden cardiac death.52
Alcohol septal ablation is an alternative. This percutaneous procedure, first described in the mid-1990s, consists of injecting a small amount of alcohol into the artery supplying the septum to induce myocardial necrosis, ultimately leading to scarring and widening of the left ventricular outflow tract.53
Up to 50% of patients develop right bundle branch block after alcohol septal ablation, and the risk of complete heart block is highest in those with preexisting left bundle branch block. Nevertheless, studies have shown significant symptomatic improvement after alcohol septal ablation, with long-term survival comparable to that in the general population.53–56
Several meta-analyses compared alcohol septal ablation and septal myectomy and found that the rates of functional improvement and long-term mortality were similar.57–59 However, the less-invasive approach with alcohol septal ablation comes at the cost of a higher incidence of conduction abnormalities and higher left ventricular outflow tract gradients afterward. One meta-analysis found that alcohol septal ablation patients may have 5 times the risk of needing additional septal reduction therapy compared with their myectomy counterparts.
Current US guidelines recommend septal myectomy, performed at an experienced center, as the first-line interventional treatment, leaving alcohol septal ablation to be considered in those who have contraindications to myectomy.9 The treatment strategy should ultimately be individualized based on a patient’s comorbidities and personal preferences following informed consent.
A nationwide database study recently suggested that postmyectomy mortality rates may be as high as 5.9%,60 although earlier studies at high-volume centers showed much lower mortality rates (< 1%).50–52,61 This discrepancy highlights the critical role of expert centers in optimizing surgical management of these patients. Regardless of the approach, interventional therapies for HCM should be performed by a multidisciplinary team at a medical center able to handle the complexity of these cases.
Additional surgical procedures
A handful of other procedures may benefit specific patient subgroups.

Apical myectomy has been shown to improve functional status in patients with isolated apical hypertrophy by reducing left ventricular end-diastolic pressure and thereby allowing for improved diastolic filling.63
Mitral valve surgery may need to be considered at the time of myectomy in patients with degenerative valve disease. As in the general population, mitral valve repair is preferred to replacement if possible.
- Maron BJ. Hypertrophic cardiomyopathy: an important global disease. Am J Med 2004; 116(1):63–65. pmid:14706671
- Semsarian C, Ingles J, Maron MS, Maron BJ. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol 2015; 65(12):1249–1254. doi:10.1016/j.jacc.2015.01.019
- Maron BJ, Maron MS, Semsarian C. Genetics of hypertrophic cardiomyopathy after 20 years: clinical perspectives. J Am Coll Cardiol 2012; 60(8):705–715. doi:10.1016/j.jacc.2012.02.068
- Shirani J, Pick R, Roberts WC, Maron BJ. Morphology and significance of the left ventricular collagen network in young patients with hypertrophic cardiomyopathy and sudden cardiac death. J Am Coll Cardiol 2000; 35(1):36–44. pmid:10636256
- Sherrid MV, Chu CK, Delia E, Mogtader A, Dwyer EM Jr. An echocardiographic study of the fluid mechanics of obstruction in hypertrophic cardiomyopathy. J Am Coll Cardiol 1993; 22(3):816–825. pmid:8354817
- Ro R, Halpern D, Sahn DJ, et al. Vector flow mapping in obstructive hypertrophic cardiomyopathy to assess the relationship of early systolic left ventricular flow and the mitral valve. J Am Coll Cardiol 2014; 64(19):1984–1995. doi:10.1016/j.jacc.2014.04.090
- Kwon DH, Setser RM, Thamilarasan M, et al. Abnormal papillary muscle morphology is independently associated with increased left ventricular outflow tract obstruction in hypertrophic cardiomyopathy. Heart 2008; 94(10):1295–1301. doi:10.1136/hrt.2007.118018
- Patel P, Dhillon A, Popovic ZB, et al. Left ventricular outflow tract obstruction in hypertrophic cardiomyopathy patients without severe septal hypertrophy: implications of mitral valve and papillary muscle abnormalities assessed using cardiac magnetic resonance and echocardiography. Circ Cardiovasc Imaging 2015; 8(7):e003132. doi:10.1161/CIRCIMAGING.115.003132
- Gersh BJ, Maron BJ, Bonow RO, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Thorac Cardiovasc Surg 2011; 142(6):e153-e203. doi:10.1016/j.jtcvs.2011.10.020
- Elliott PM, Gimeno JR, Thaman R, et al. Historical trends in reported survival rates in patients with hypertrophic cardiomyopathy. Heart 2006; 92(6):785–791. doi:10.1136/hrt.2005.068577
- Maron MS, Olivotto I, Zenovich AG, et al. Hypertrophic cardiomyopathy is predominantly a disease of left ventricular outflow tract obstruction. Circulation 2006; 114(21):2232–2239. doi:10.1161/CIRCULATIONAHA.106.644682
- Ho CY, Carlsen C, Thune JJ, et al. Echocardiographic strain imaging to assess early and late consequences of sarcomere mutations in hypertrophic cardiomyopathy. Circ Cardiovasc Genet 2009; 2(4):314–321. doi:10.1161/CIRCGENETICS.109.862128
- Wasfy MM, Weiner RB. Differentiating the athlete’s heart from hypertrophic cardiomyopathy. Curr Opin Cardiol 2015; 30(5):500–505. doi:10.1097/HCO.0000000000000203
- Palka P, Lange A, Fleming AD, et al. Differences in myocardial velocity gradient measured throughout the cardiac cycle in patients with hypertrophic cardiomyopathy, athletes and patients with left ventricular hypertrophy due to hypertension. J Am Coll Cardiol 1997; 30(3):760–768. pmid:9283537
- Eriksson MJ, Sonnenberg B, Woo A, et al. Long-term outcome in patients with apical hypertrophic cardiomyopathy. J Am Coll Cardiol 2002; 39(4):638–645. pmid:11849863
- Rickers C, Wilke NM, Jerosch-Herold M, et al. Utility of cardiac magnetic resonance imaging in the diagnosis of hypertrophic cardiomyopathy. Circulation 2005; 112(6):855–861. doi:10.1161/CIRCULATIONAHA.104.507723
- Kwon DH, Setser RM, Popovic ZB, et al. Association of myocardial fibrosis, electrocardiography and ventricular tachyarrhythmia in hypertrophic cardiomyopathy: a delayed contrast enhanced MRI study. Int J Cardiovasc Imaging 2008; 24(6):617–625. doi:10.1007/s10554-008-9292-6
- Rubinshtein R, Glockner JF, Ommen SR, et al. Characteristics and clinical significance of late gadolinium enhancement by contrast-enhanced magnetic resonance imaging in patients with hypertrophic cardiomyopathy. Circ Heart Fail 2010; 3(1):51–58. doi:10.1161/CIRCHEARTFAILURE.109.854026
- O’Hanlon R, Grasso A, Roughton M, et al. Prognostic significance of myocardial fibrosis in hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 56(11):867–874. doi:10.1016/j.jacc.2010.05.010
- Bruder O, Wagner A, Jensen CJ, et al. Myocardial scar visualized by cardiovascular magnetic resonance imaging predicts major adverse events in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 56(11):875–887. doi:10.1016/j.jacc.2010.05.007
- Authors/Task Force members, Elliott PM, Anastasakis A, Borger MA, et al. 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J 2014; 35(39):2733–2779. doi:10.1093/eurheartj/ehu284
- Olivotto I, Maron BJ, Montereggi A, Mazzuoli F, Dolara A, Cecchi F. Prognostic value of systemic blood pressure response during exercise in a community-based patient population with hypertrophic cardiomyopathy. J Am Coll Cardiol 1999; 33(7):2044–2051. pmid:10362212
- Sadoul N, Prasad K, Elliott PM, Bannerjee S, Frenneaux MP, McKenna WJ. Prospective prognostic assessment of blood pressure response during exercise in patients with hypertrophic cardiomyopathy. Circulation 1997; 96(9):2987–2991. pmid:9386166
- Elliott PM, Poloniecki J, Dickie S, et al. Sudden death in hypertrophic cardiomyopathy: identification of high risk patients. J Am Coll Cardiol 2000; 36(7):2212–2218. pmid:11127463
- Masri A, Pierson LM, Smedira NG, et al. Predictors of long-term outcomes in patients with hypertrophic cardiomyopathy undergoing cardiopulmonary stress testing and echocardiography. Am Heart J 2015; 169(5):684–692.e1. doi:10.1016/j.ahj.2015.02.006
- Coats CJ, Rantell K, Bartnik A, et al. Cardiopulmonary exercise testing and prognosis in hypertrophic cardiomyopathy. Circ Heart Fail 2015; 8(6):1022–1031. doi:10.1161/CIRCHEARTFAILURE.114.002248
- Desai MY, Bhonsale A, Patel P, et al. Exercise echocardiography in asymptomatic HCM: exercise capacity, and not LV outflow tract gradient predicts long-term outcomes. JACC Cardiovasc Imaging 2014; 7(1):26–36. doi:10.1016/j.jcmg.2013.08.010
- Spirito P, Seidman CE, McKenna WJ, Maron BJ. The management of hypertrophic cardiomyopathy. N Engl J Med 1997; 336(11):775–785. doi:10.1056/NEJM199703133361107
- Wang W, Lian Z, Rowin EJ, Maron BJ, Maron MS, Link MS. Prognostic implications of nonsustained ventricular tachycardia in high-risk patients with hypertrophic cardiomyopathy. Circ Arrhythm Electrophysiol 2017; 10(3)e004604. doi:10.1161/CIRCEP.116.004604
- O’Mahony C, Jichi F, Pavlou M, et al. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM risk-SCD). Eur Heart J 2014; 35(30):2010–2020. doi:10.1093/eurheartj/eht439
- Maron BJ, Casey SA, Chan RH, Garberich RF, Rowin EJ, Maron MS. Independent assessment of the European Society of Cardiology sudden death risk model for hypertrophic cardiomyopathy. Am J Cardiol 2015; 116(5):757–764. doi:10.1016/j.amjcard.2015.05.047
- Jahnlová D, Tomašov P, Zemánek D, Veselka J. Transatlantic differences in assessment of risk of sudden cardiac death in patients with hypertrophic cardiomyopathy. Int J Cardiol 2015; 186:3–4. doi:10.1016/j.ijcard.2015.03.207
- Ingles J, Burns C, Bagnall RD, et al. Nonfamilial hypertrophic cardiomyopathy: prevalence, natural history, and clinical implications. Circ Cardiovasc Genet 2017; 10(2)e001620. doi:10.1161/CIRCGENETICS.116.001620
- Ko C, Arscott P, Concannon M, et al. Genetic testing impacts the utility of prospective familial screening in hypertrophic cardiomyopathy through identification of a nonfamilial subgroup. Genet Med 2017; 20(1):69–75. doi:10.1038/gim.2017.79
- Maron BJ, Chaitman BR, Ackerman MJ, et al. Recommendations for physical activity and recreational sports participation for young patients with genetic cardiovascular diseases. Circulation 2004; 109(22):2807–2816. doi:10.1161/01.CIR.0000128363.85581.E1
- Saberi S, Wheeler M, Bragg-Gresham J, et al. Effect of moderate-intensity exercise training on peak oxygen consumption in patients with hypertrophic cardiomyopathy: a randomized clinical trial. JAMA 2017; 317(13):1349–1357. doi:10.1001/jama.2017.2503
- Bourmayan C, Razavi A, Fournier C, et al. Effect of propranolol on left ventricular relaxation in hypertrophic cardiomyopathy: an echographic study. Am Heart J 1985; 109(6):1311–1316. pmid:4039882
- Spoladore R, Maron MS, D’Amato R, Camici PG, Olivotto I. Pharmacological treatment options for hypertrophic cardiomyopathy: high time for evidence. Eur Heart J 2012; 33(14):1724–1733. doi:10.1093/eurheartj/ehs150
- Choudhury L, Elliott P, Rimoldi O, et al. Transmural myocardial blood flow distribution in hypertrophic cardiomyopathy and effect of treatment. Basic Res Cardiol 1999; 94(1):49–59. pmid:10097830
- Kaltenbach M, Hopf R, Kober G, Bussmann WD, Keller M, Petersen Y. Treatment of hypertrophic obstructive cardiomyopathy with verapamil. Br Heart J 1979; 42(1):35–42. doi:10.1136/hrt.42.1.35
- Sherrid MV, Shetty A, Winson G, et al. Treatment of obstructive hypertrophic cardiomyopathy symptoms and gradient resistant to first-line therapy with beta-blockade or verapamil. Circ Heart Fail 2013; 6(4):694–702. doi:10.1161/CIRCHEARTFAILURE.112.000122
- Guttmann OP, Rahman MS, O’Mahony C, Anastasakis A, Elliott PM. Atrial fibrillation and thromboembolism in patients with hypertrophic cardiomyopathy: systematic review. Heart 2014; 100(6):465–472. doi:10.1136/heartjnl-2013-304276
- Olivotto I, Cecchi F, Casey SA, Dolara A, Traverse JH, Maron BJ. Impact of atrial fibrillation on the clinical course of hypertrophic cardiomyopathy. Circulation 2001; 104(21):2517–2524. pmid:11714644
- Maron BJ, Olivotto I, Spirito P, et al. Epidemiology of hypertrophic cardiomyopathy-related death: revisited in a large non-referral-based patient population. Circulation 2000; 102(8):858–864. pmid:10952953
- Harris KM, Spirito P, Maron MS, et al. Prevalence, clinical profile, and significance of left ventricular remodeling in the end-stage phase of hypertrophic cardiomyopathy. Circulation 2006; 114(3):216-225. doi:10.1161/CIRCULATIONAHA.105.583500
- Maron MS, Kalsmith BM, Udelson JE, Li W, DeNofrio D. Survival after cardiac transplantation in patients with hypertrophic cardiomyopathy. Circ Heart Fail 2010; 3(5):574–579. doi:10.1161/CIRCHEARTFAILURE.109.922872
- Smedira NG, Lytle BW, Lever HM, et al. Current effectiveness and risks of isolated septal myectomy for hypertrophic obstructive cardiomyopathy. Ann Thorac Surg 2008; 85(1):127–133. doi:10.1016/j.athoracsur.2007.07.063
- Robbins RC, Stinson EB. Long-term results of left ventricular myotomy and myectomy for obstructive hypertrophic cardiomyopathy. J Thorac Cardiovasc Surg 1996; 111(3):586–594. pmid:8601973
- Heric B, Lytle BW, Miller DP, Rosenkranz ER, Lever HM, Cosgrove DM. Surgical management of hypertrophic obstructive cardiomyopathy. Early and late results. J Thorac Cardiovasc Surg 1995; 110(1):195–208. pmid:7609544
- Ommen SR, Maron BJ, Olivotto I, et al. Long-term effects of surgical septal myectomy on survival in patients with obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol 2005; 46(3):470–476. doi:10.1016/j.jacc.2005.02.090
- Desai MY, Bhonsale A, Smedira NG, et al. Predictors of long-term outcomes in symptomatic hypertrophic obstructive cardiomyopathy patients undergoing surgical relief of left ventricular outflow tract obstruction. Circulation 2013; 128(3):209–216. doi:10.1161/CIRCULATIONAHA.112.000849
- McLeod CJ, Ommen SR, Ackerman MJ, et al. Surgical septal myectomy decreases the risk for appropriate implantable cardioverter defibrillator discharge in obstructive hypertrophic cardiomyopathy. Eur Heart J 2007; 28(21):2583–2588. doi:10.1093/eurheartj/ehm117
- Veselka J, Tomasov P, Zemanek D. Long-term effects of varying alcohol dosing in percutaneous septal ablation for obstructive hypertrophic cardiomyopathy: a randomized study with a follow-up up to 11 years. Can J Cardiol 2011; 27(6):763–767. doi:10.1016/j.cjca.2011.09.001
- Veselka J, Jensen MK, Liebregts M, et al. Low procedure-related mortality achieved with alcohol septal ablation in European patients. Int J Cardiol 2016; 209:194–195. doi:10.1016/j.ijcard.2016.02.077
- Veselka J, Krejci J, Tomašov P, Zemánek D. Long-term survival after alcohol septal ablation for hypertrophic obstructive cardiomyopathy: a comparison with general population. Eur Heart J 2014; 35(30):2040–2045. doi:10.1093/eurheartj/eht495
- Sorajja P, Ommen SR, Holmes DR Jr, et al. Survival after alcohol septal ablation for obstructive hypertrophic cardiomyopathy. Circulation 2012; 126(20):2374–2380. doi:10.1161/CIRCULATIONAHA.111.076257
- Agarwal S, Tuzcu EM, Desai MY, et al. Updated meta-analysis of septal alcohol ablation versus myectomy for hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 55(8):823–834. doi:10.1016/j.jacc.2009.09.047
- Leonardi RA, Kransdorf EP, Simel DL, Wang A. Meta-analyses of septal reduction therapies for obstructive hypertrophic cardiomyopathy: comparative rates of overall mortality and sudden cardiac death after treatment. Circ Cardiovasc Interv 2010; 3(2):97–104. doi:10.1161/CIRCINTERVENTIONS.109.916676
- Liebregts M, Vriesendorp PA, Mahmoodi BK, Schinkel AF, Michels M, ten Berg JM. A systematic review and meta-analysis of long-term outcomes after septal reduction therapy in patients with hypertrophic cardiomyopathy. JACC Heart Fail 2015; 3(11):896–905. doi:10.1016/j.jchf.2015.06.011
- Panaich SS, Badheka AO, Chothani A, et al. Results of ventricular septal myectomy and hypertrophic cardiomyopathy (from Nationwide Inpatient Sample [1998-2010]). Am J Cardiol 2014; 114(9):1390–1395. doi:10.1016/j.amjcard.2014.07.075
- Maron BJ, Dearani JA, Ommen SR, et al. Low operative mortality achieved with surgical septal myectomy: role of dedicated hypertrophic cardiomyopathy centers in the management of dynamic subaortic obstruction. J Am Coll Cardiol 2015; 66(11):1307–1308. doi:10.1016/j.jacc.2015.06.1333
- Kwon DH, Smedira NG, Thamilarasan M, Lytle BW, Lever H, Desai MY. Characteristics and surgical outcomes of symptomatic patients with hypertrophic cardiomyopathy with abnormal papillary muscle morphology undergoing papillary muscle reorientation. J Thorac Cardiovasc Surg 2010; 140(2):317–324. doi:10.1016/j.jtcvs.2009.10.045
- Schaff HV, Brown ML, Dearani JA, et al. Apical myectomy: a new surgical technique for management of severely symptomatic patients with apical hypertrophic cardiomyopathy. J Thorac Cardiovasc Surg 2010; 139(3):634–640. doi:10.1016/j.jtcvs.2009.07.079
Hypertrophic cardiomyopathy (HCM) is a complex disease. Most people who carry the mutations that cause it are never affected at any point in their life, but some are affected at a young age. And in rare but tragic cases, some die suddenly while competing in sports. With such a wide range of phenotypic expressions, a single therapy does not fit all.
HCM is more common than once thought. Since the discovery of its genetic predisposition in 1960, it has come to be recognized as the most common heritable cardiovascular disease.1 Although earlier epidemiologic studies had estimated a prevalence of 1 in 500 (0.2%) of the general population, genetic testing and cardiac magnetic resonance imaging (MRI) now show that up to 1 in 200 (0.5%) of all people may be affected.1,2 Its prevalence is significant in all ethnic groups.
This review outlines our expanding knowledge of the pathophysiology, diagnosis, and clinical management of HCM.
A PLETHORA OF MUTATIONS IN CARDIAC SARCOMERIC GENES
The genetic differences within HCM result in varying degrees and locations of left ventricular hypertrophy. Any segment of the ventricle can be involved, although HCM is classically asymmetric and mainly involves the septum (Figure 1). A variant form of HCM involves the apex of the heart (Figure 2).
LEFT VENTRICULAR OUTFLOW TRACT OBSTRUCTION
Only in the last decade has the significance of left ventricular outflow tract obstruction in HCM been truly appreciated. The degree of obstruction in HCM is dynamic, as opposed to the fixed obstruction in patients with aortic stenosis or congenital subvalvular membranes. Therefore, in HCM, exercise or drugs (eg, dobutamine) that increase cardiac contractility increase the obstruction, as do maneuvers or drugs (the Valsalva maneuver, nitrates) that reduce filling of the left ventricle.
A less common source of dynamic obstruction is the papillary muscles (Figure 4). Hypertrophy of the papillary muscles can result in obstruction by these muscles themselves, which is visible on echocardiography. Anatomic variations include anteroapical displacement or bifid papillary muscles, and these variants can be associated with dynamic left ventricular outflow tract obstruction, even with no evidence of septal thickening (Figure 5).7,8 Recognizing this patient subset has important implications for management, as discussed below.
DIAGNOSTIC EVALUATION
The clinical presentation varies
Even if patients harbor the same genetic variant, the clinical presentation can differ widely. Although the most feared presentation is sudden cardiac death, particularly in young athletes, most patients have no symptoms and can anticipate a normal life expectancy. The annual incidence of sudden cardiac death in all HCM patients is estimated at less than 1%.10 Sudden cardiac death in HCM patients is most often due to ventricular tachyarrhythmias and most often occurs in asymptomatic patients under age 35.
Physical findings are nonspecific
It can be difficult to distinguish the murmur of left ventricular outflow tract obstruction in HCM from a murmur related to aortic stenosis by auscultation alone. The simplest clinical method for telling them apart involves the Valsalva maneuver: bearing down creates a positive intrathoracic pressure and limits venous return, thus decreasing intracardiac filling pressure. This in turn results in less separation between the mitral valve and the ventricular septum in HCM, which increases obstruction and therefore makes the murmur louder. In contrast, in patients with fixed obstruction due to aortic stenosis, the murmur will decrease in intensity owing to the reduced flow associated with reduced preload.
Laboratory testing for phenocopies of HCM
A metabolic panel will show derangements in liver function and glucose levels in patients with glycogen storage disorders such as Pompe disease.
Serum creatinine. Renal dysfunction will be seen in patients with Fabry disease or amyloidosis.
Creatine kinase may be elevated in patients with Danon disease.
Electrocardiographic findings are common
More than 90% of HCM patients have electrocardiographic abnormalities. Although the findings can vary widely, common manifestations include:
- Left ventricular hypertrophy
- A pseudoinfarct pattern with Q waves in the anterolateral leads
- Repolarization changes such as T-wave inversions and horizontal or down-sloping ST segments.
Apical HCM, seen mainly in Asian populations, often presents with giant T-wave inversion (> 10 mm) in the anterolateral leads, most prominent in V4, V5, and V6.
Notably, the degree of electrocardiographic abnormalities does not correlate with the severity or pattern of hypertrophy.9 Electrocardiography lacks specificity for definitive diagnosis, and further diagnostic testing should therefore be pursued.
Echocardiography: Initial imaging test
Transthoracic echocardiography is the initial imaging test in patients with suspected HCM, allowing for cost-effective quantitative and qualitative assessment of left ventricular morphology and function. Left ventricular hypertrophy is considered pathologic if wall thickness is 15 mm or greater without a known cause. Transthoracic echocardiography also allows for evaluation of left atrial volume and mitral valve anatomy and function.
Speckle tracking imaging is an advanced echocardiographic technique that measures strain. Its major advantage is in identifying early abnormalities in genotype-positive, phenotype-negative HCM patients, ie, people who harbor mutations but who have no clinical symptoms or signs of HCM, potentially allowing for modification of the natural history of HCM.12 Strain imaging can also differentiate between physiologic hypertrophy (“athlete’s heart”) and hypertension and HCM.13,14
The utility of echocardiography in HCM is heavily influenced by the sonographer’s experience in obtaining adequate acoustic windows. This may be more difficult in obese patients, patients with advanced obstructive lung disease or pleural effusions, and women with breast implants.
Magnetic resonance imaging
MRI has an emerging role in both diagnosing and predicting risk in HCM, and is routinely done as an adjunct to transthoracic echocardiography on initial diagnosis in our tertiary referral center. It is particularly useful in patients suspected of having apical hypertrophy (Figure 2), in whom the diagnosis may be missed in up to 10% on transthoracic echocardiography alone.15 MRI can also enhance the assessment of left ventricular hypertrophy and has been shown to improve the diagnostic classification of HCM.16 It is the best way to assess myocardial tissue abnormalities, and late gadolinium enhancement to detect interstitial fibrosis can be used for further prognostication. While historically the primary role of MRI in HCM has been in phenotype classification, there is currently much interest in its role in risk stratification of HCM patients for ICD implantation.
MRI with late gadolinium enhancement provides insight into the location, pattern, and extent of myocardial fibrosis; the extent of fibrosis has been shown to be a strong independent predictor of poor outcomes, including sudden cardiac death.17–20 However, late gadolinium enhancement can be technically challenging, as variations in the timing of postcontrast imaging, sequences for measuring late gadolinium enhancement, or detection thresholds can result in widely variable image quality. Cardiac MRI should therefore be performed at an experienced center with standardized imaging protocols in place.
Current guidelines recommend considering cardiac MRI if a patient’s risk of sudden cardiac death remains inconclusive after conventional risk stratification, as discussed below.9,21
Stress testing for risk stratification
Exercise stress electrocardiography. Treadmill exercise stress testing with electrocardiography and hemodynamic monitoring was one of the first tools used for risk stratification in HCM.
Although systolic blood pressure normally increases by at least 20 mm Hg with exercise, one-quarter of HCM patients have either a blunted response (failure of systolic blood pressure to increase by at least 20 mm Hg) or a hypotensive response (a drop in systolic blood pressure of 20 mm Hg or more, either continuously or after an initial increase). Studies have shown that HCM patients who have abnormal blood pressure responses during exercise have a higher risk of sudden cardiac death.22–24
Exercise stress echocardiography can be useful to evaluate for provoked increases in the left ventricular outflow tract gradient, which may contribute to a patient’s symptoms even if the resting left ventricular outflow tract gradient is normal. Exercise testing is preferred over pharmacologic stimulation because it can provide functional assessment of whether a patient’s clinical symptoms are truly related to hemodynamic changes due to the hypertrophied ventricle, or whether alternative mechanisms should be explored.
Cardiopulmonary stress testing can readily add prognostic value with additional measurements of functional capacity. HCM patients who cannot achieve their predicted maximal exercise value such as peak rate of oxygen consumption, ventilation efficiency, or anaerobic threshold have higher rates of morbidity and mortality.25,26 Stress testing can also be useful for risk stratification in asymptomatic patients, with one study showing that those who achieve more than 100% of their age- and sex-predicted metabolic equivalents have a low event rate.27
Ambulatory electrocardiographic monitoring in all patients at diagnosis
Ambulatory electrocardiographic monitoring for 24 to 48 hours is recommended for all HCM patients at the time of diagnosis, even if they have no symptoms. Any evidence of nonsustained ventricular tachycardia suggests a substantially higher risk of sudden cardiac death.28,29
In patients with no symptoms or history of arrhythmia, current guidelines suggest ambulatory electrocardiographic monitoring every 1 to 2 years.9,21
Two risk-stratification models
The North American model was the first risk-stratification tool and considers 5 risk factors.9 However, if this algorithm were strictly followed, up to 60% of HCM patients would be candidates for cardioverter-defibrillator implantation.
The European model. This concern led to the development of the HCM Risk-SCD (sudden cardiac death), a risk-stratification tool introduced in the 2014 European Society of Cardiology HCM guidelines.30 This web-based calculator estimates a patient’s 5-year risk of sudden cardiac death using a complex calculation based on 7 clinical risk factors. If a patient’s calculated 5-year risk of sudden cardiac death is 6% or higher, cardioverter-defibrillator implantation is recommended for primary prevention.
The HCM Risk-SCD calculator was validated and compared with classic risk factors alone in a retrospective cohort study in 48 HCM patients.30 Compared with the North American model, the European model results in a lower rate of cardioverter-defibrillator implantation (20% to 26%).31,32
Despite the better specificity of the European model, a large retrospective cohort analysis showed that a significant number of patients stratified as being at low risk for sudden cardiac death were ultimately found to be at high risk in clinical practice.31 Further research is needed to find the optimal risk-stratification approach in HCM patients at low to intermediate risk.
GENETIC TESTING, COUNSELING, AND FAMILY SCREENING
Genetic testing is becoming more widely available and has rapidly expanded in clinical practice. Genetic counseling must be performed alongside genetic testing and requires professionals trained to handle the clinical and social implications of genetic testing. With this in mind, genetic testing can provide a definitive means of identifying family members at risk of HCM.
Given the autosomal dominant nature of HCM, screening for HCM is recommended in all first-degree relatives of an affected patient. Genetic testing may be a means to achieve this if a pathogenic mutation has been identified in the affected patient. However, serial electrocardiographic and transthoracic echocardiographic monitoring is an acceptable alternative in those without a clear genetic mutation association or in those who do not want to undergo genetic testing. If these first-degree relatives who do not undergo genetic testing are adult athletes or adolescents, they should undergo surveillance monitoring, with echocardiography and electrocardiography, whereas adults not participating in athletics should be monitored every 5 years.9,21
As genetic counseling and testing become more widely available, more patients are being found who harbor a mutation but have no phenotypic manifestations of HCM on initial presentation. Clinical expression varies, so continued monitoring of these patients is important. Expert guidelines again recommend serial electrocardiography, transthoracic echocardiography, and clinical assessment every 5 years for adults.9
Recent data suggest that up to 40% of HCM cases are nonfamilial, ie, their inheritance is sporadic with no known family history and no sarcomeric gene mutation evident on screening.33,34 The clinical course in this subgroup seems to be more benign, with later clinical presentations (age > 40) and lower risk of major adverse cardiovascular events.
MANAGEMENT
Conservative management
Asymptomatic HCM can usually be managed with lifestyle modifications.
Avoiding high-risk physical activities is the most important modification. All HCM patients should be counseled on the risk of sudden cardiac death and advised against participating in competitive sports or intense physical activity.35 Aerobic exercise is preferable to isometric exercises such as weightlifting, which may prompt the Valsalva maneuver with worsening of left ventricular outflow tract obstruction leading to syncope. A recent study showed that moderate-intensity aerobic exercise can safely improve exercise capacity, which may ultimately improve functional status and quality of life.36
Avoiding dehydration and excessive alcohol intake are also important in maintaining adequate preload to prevent an increasing left ventricular outflow tract gradient, given the dynamic nature of the left ventricular outflow tract obstruction in HCM.
Medical management: Beta-blockers, then calcium channel blockers
Beta-blockers are the first-line therapy for symptomatic HCM related to left ventricular outflow tract obstruction. Their negative inotropic effect reduces the contractile force of the ventricle, effectively reducing the pressure gradient across the outflow tract. Reduced contractility also means that the overall myocardial workload is less, which ultimately translates to a reduced oxygen demand. With their negative chronotropic effect, beta-blockers lower the heart rate and thereby lengthen the diastolic filling phase, allowing for optimization of preload conditions to help prevent increasing the left ventricular outflow tract gradient.37,38
Beta-blockers can be titrated according to the patient’s symptoms and tolerance. Fatigue and loss of libido are among the most common side effects.
Nondihydropyridine calcium channel blockers can be a second-line therapy in patients who cannot tolerate beta-blockers. Several studies have shown improvement in surrogate outcomes such as estimated left ventricular mass and QRS amplitude on electrocardiography, but currently no available data show that these drugs improve symptoms.28,39,40 They should be avoided in those with severe left ventricular outflow tract obstruction (gradient ≥ 100 mm Hg), as they can lead to critical outflow tract obstruction owing to their peripheral vasodilatory effect.
Dihydropyridine calcium channel blockers should be avoided altogether, as they produce even more peripheral vasodilation and afterload reduction than nondihydropyridine calcium channel blockers.
Disopyramide, a class IA antiarrhythmic, has been shown to effectively reduce outflow gradients and relieve symptoms. However, in view of its adverse effects, it is a third-line therapy, given to those for whom beta-blockers and calcium channel blockers have failed. Its most worrisome adverse effect is QT prolongation, and the QT interval should therefore be closely monitored at the start of treatment. Anticholinergic effects are common and include dry eyes and mouth, urinary retention, and drowsiness.
Disopyramide is usually used in combination with beta-blockers for symptom control as a bridge to a planned invasive intervention.41
Use with caution
Any medication that causes afterload reduction, peripheral vasodilation, intravascular volume depletion, or positive inotropy can worsen the dynamic left ventricular outflow tract obstruction in a patient with HCM and should be avoided.
Angiotensin-converting enzyme (ACE) inhibitors, angiotensin II receptor blockers (ARBs), and nitrates must be used with extreme caution in these patients.
Diuretics. Even restrained use of diuretics can cause significant hemodynamic compromise in patients with obstructive physiology. Therefore, diuretics should be used sparingly in these patients.
Digoxin should not be used for managing atrial fibrillation in these patients, as its positive inotropic effect increases contractility and increases the left ventricular outflow tract gradient.
Norepinephrine and inotropic agents such as dobutamine and dopamine should be avoided for the same reason as digoxin. In patients with circulatory shock requiring vasopressor support, pure alpha-agonists such as phenylephrine are preferred, as they increase peripheral resistance without an inotropic effect.
Anticoagulation for atrial tachyarrhythmias
The risk of systemic thromboembolic events is significantly increased in HCM patients with atrial fibrillation or flutter, regardless of their estimated risk using conventional risk-stratification tools such as the CHADS2 score.42–44 In accordance with current American Heart Association and American College of Cardiology guidelines, we recommend anticoagulation therapy for all HCM patients with a history of atrial fibrillation or flutter. Warfarin is the preferred anticoagulant; direct oral anticoagulants can be considered, but there are currently no data on their use in HCM.9
Standard heart failure treatments
End-stage systolic heart failure is a consequence of HCM but affects only 3% to 4% of patients.45 While most randomized controlled trials of heart failure treatment have excluded HCM patients, current guidelines recommend the same evidence-based medical therapies used in other patients who have heart failure with reduced ejection fraction. This includes ACE inhibitors, ARBs, beta-blockers, and aldosterone antagonists if indicated.9,21
Heart transplant should be considered in patients with class III or IV New York Heart Association functional status despite optimization of their HCM treatment regimen. Heart transplant outcomes for HCM patients are comparable to outcomes for patients who receive a transplant for non-HCM cardiovascular disease.45,46
Septal reduction therapy
If medical therapy fails or is not tolerated in patients with severe symptoms, surgery can be considered for obstructive HCM.
Ventricular septal myectomy has been the long-standing gold standard of invasive therapy. Multiple studies have demonstrated long-term survival after myectomy to be equivalent to that in the general population and better than that of HCM patients who do not undergo this surgery.47–50 Factors that may be associated with better surgical outcomes include age younger than 50, left atrial size less than 46 mm, and resolution of atrial fibrillation during follow-up.51
Septal reduction therapy may also be considered in patients at high risk of sudden cardiac death based on a history of recurrent ventricular tachycardia or risk-stratification models as described above. Retrospective analyses have shown that surgical myectomy can markedly reduce the incidence of appropriate implantable cardioverter-defibrillator discharges and the risk of sudden cardiac death.52
Alcohol septal ablation is an alternative. This percutaneous procedure, first described in the mid-1990s, consists of injecting a small amount of alcohol into the artery supplying the septum to induce myocardial necrosis, ultimately leading to scarring and widening of the left ventricular outflow tract.53
Up to 50% of patients develop right bundle branch block after alcohol septal ablation, and the risk of complete heart block is highest in those with preexisting left bundle branch block. Nevertheless, studies have shown significant symptomatic improvement after alcohol septal ablation, with long-term survival comparable to that in the general population.53–56
Several meta-analyses compared alcohol septal ablation and septal myectomy and found that the rates of functional improvement and long-term mortality were similar.57–59 However, the less-invasive approach with alcohol septal ablation comes at the cost of a higher incidence of conduction abnormalities and higher left ventricular outflow tract gradients afterward. One meta-analysis found that alcohol septal ablation patients may have 5 times the risk of needing additional septal reduction therapy compared with their myectomy counterparts.
Current US guidelines recommend septal myectomy, performed at an experienced center, as the first-line interventional treatment, leaving alcohol septal ablation to be considered in those who have contraindications to myectomy.9 The treatment strategy should ultimately be individualized based on a patient’s comorbidities and personal preferences following informed consent.
A nationwide database study recently suggested that postmyectomy mortality rates may be as high as 5.9%,60 although earlier studies at high-volume centers showed much lower mortality rates (< 1%).50–52,61 This discrepancy highlights the critical role of expert centers in optimizing surgical management of these patients. Regardless of the approach, interventional therapies for HCM should be performed by a multidisciplinary team at a medical center able to handle the complexity of these cases.
Additional surgical procedures
A handful of other procedures may benefit specific patient subgroups.
Apical myectomy has been shown to improve functional status in patients with isolated apical hypertrophy by reducing left ventricular end-diastolic pressure and thereby allowing for improved diastolic filling.63
Mitral valve surgery may need to be considered at the time of myectomy in patients with degenerative valve disease. As in the general population, mitral valve repair is preferred to replacement if possible.
Hypertrophic cardiomyopathy (HCM) is a complex disease. Most people who carry the mutations that cause it are never affected at any point in their life, but some are affected at a young age. And in rare but tragic cases, some die suddenly while competing in sports. With such a wide range of phenotypic expressions, a single therapy does not fit all.
HCM is more common than once thought. Since the discovery of its genetic predisposition in 1960, it has come to be recognized as the most common heritable cardiovascular disease.1 Although earlier epidemiologic studies had estimated a prevalence of 1 in 500 (0.2%) of the general population, genetic testing and cardiac magnetic resonance imaging (MRI) now show that up to 1 in 200 (0.5%) of all people may be affected.1,2 Its prevalence is significant in all ethnic groups.
This review outlines our expanding knowledge of the pathophysiology, diagnosis, and clinical management of HCM.
A PLETHORA OF MUTATIONS IN CARDIAC SARCOMERIC GENES
The genetic differences within HCM result in varying degrees and locations of left ventricular hypertrophy. Any segment of the ventricle can be involved, although HCM is classically asymmetric and mainly involves the septum (Figure 1). A variant form of HCM involves the apex of the heart (Figure 2).
LEFT VENTRICULAR OUTFLOW TRACT OBSTRUCTION
Only in the last decade has the significance of left ventricular outflow tract obstruction in HCM been truly appreciated. The degree of obstruction in HCM is dynamic, as opposed to the fixed obstruction in patients with aortic stenosis or congenital subvalvular membranes. Therefore, in HCM, exercise or drugs (eg, dobutamine) that increase cardiac contractility increase the obstruction, as do maneuvers or drugs (the Valsalva maneuver, nitrates) that reduce filling of the left ventricle.
A less common source of dynamic obstruction is the papillary muscles (Figure 4). Hypertrophy of the papillary muscles can result in obstruction by these muscles themselves, which is visible on echocardiography. Anatomic variations include anteroapical displacement or bifid papillary muscles, and these variants can be associated with dynamic left ventricular outflow tract obstruction, even with no evidence of septal thickening (Figure 5).7,8 Recognizing this patient subset has important implications for management, as discussed below.
DIAGNOSTIC EVALUATION
The clinical presentation varies
Even if patients harbor the same genetic variant, the clinical presentation can differ widely. Although the most feared presentation is sudden cardiac death, particularly in young athletes, most patients have no symptoms and can anticipate a normal life expectancy. The annual incidence of sudden cardiac death in all HCM patients is estimated at less than 1%.10 Sudden cardiac death in HCM patients is most often due to ventricular tachyarrhythmias and most often occurs in asymptomatic patients under age 35.
Physical findings are nonspecific
It can be difficult to distinguish the murmur of left ventricular outflow tract obstruction in HCM from a murmur related to aortic stenosis by auscultation alone. The simplest clinical method for telling them apart involves the Valsalva maneuver: bearing down creates a positive intrathoracic pressure and limits venous return, thus decreasing intracardiac filling pressure. This in turn results in less separation between the mitral valve and the ventricular septum in HCM, which increases obstruction and therefore makes the murmur louder. In contrast, in patients with fixed obstruction due to aortic stenosis, the murmur will decrease in intensity owing to the reduced flow associated with reduced preload.
Laboratory testing for phenocopies of HCM
A metabolic panel will show derangements in liver function and glucose levels in patients with glycogen storage disorders such as Pompe disease.
Serum creatinine. Renal dysfunction will be seen in patients with Fabry disease or amyloidosis.
Creatine kinase may be elevated in patients with Danon disease.
Electrocardiographic findings are common
More than 90% of HCM patients have electrocardiographic abnormalities. Although the findings can vary widely, common manifestations include:
- Left ventricular hypertrophy
- A pseudoinfarct pattern with Q waves in the anterolateral leads
- Repolarization changes such as T-wave inversions and horizontal or down-sloping ST segments.
Apical HCM, seen mainly in Asian populations, often presents with giant T-wave inversion (> 10 mm) in the anterolateral leads, most prominent in V4, V5, and V6.
Notably, the degree of electrocardiographic abnormalities does not correlate with the severity or pattern of hypertrophy.9 Electrocardiography lacks specificity for definitive diagnosis, and further diagnostic testing should therefore be pursued.
Echocardiography: Initial imaging test
Transthoracic echocardiography is the initial imaging test in patients with suspected HCM, allowing for cost-effective quantitative and qualitative assessment of left ventricular morphology and function. Left ventricular hypertrophy is considered pathologic if wall thickness is 15 mm or greater without a known cause. Transthoracic echocardiography also allows for evaluation of left atrial volume and mitral valve anatomy and function.
Speckle tracking imaging is an advanced echocardiographic technique that measures strain. Its major advantage is in identifying early abnormalities in genotype-positive, phenotype-negative HCM patients, ie, people who harbor mutations but who have no clinical symptoms or signs of HCM, potentially allowing for modification of the natural history of HCM.12 Strain imaging can also differentiate between physiologic hypertrophy (“athlete’s heart”) and hypertension and HCM.13,14
The utility of echocardiography in HCM is heavily influenced by the sonographer’s experience in obtaining adequate acoustic windows. This may be more difficult in obese patients, patients with advanced obstructive lung disease or pleural effusions, and women with breast implants.
Magnetic resonance imaging
MRI has an emerging role in both diagnosing and predicting risk in HCM, and is routinely done as an adjunct to transthoracic echocardiography on initial diagnosis in our tertiary referral center. It is particularly useful in patients suspected of having apical hypertrophy (Figure 2), in whom the diagnosis may be missed in up to 10% on transthoracic echocardiography alone.15 MRI can also enhance the assessment of left ventricular hypertrophy and has been shown to improve the diagnostic classification of HCM.16 It is the best way to assess myocardial tissue abnormalities, and late gadolinium enhancement to detect interstitial fibrosis can be used for further prognostication. While historically the primary role of MRI in HCM has been in phenotype classification, there is currently much interest in its role in risk stratification of HCM patients for ICD implantation.
MRI with late gadolinium enhancement provides insight into the location, pattern, and extent of myocardial fibrosis; the extent of fibrosis has been shown to be a strong independent predictor of poor outcomes, including sudden cardiac death.17–20 However, late gadolinium enhancement can be technically challenging, as variations in the timing of postcontrast imaging, sequences for measuring late gadolinium enhancement, or detection thresholds can result in widely variable image quality. Cardiac MRI should therefore be performed at an experienced center with standardized imaging protocols in place.
Current guidelines recommend considering cardiac MRI if a patient’s risk of sudden cardiac death remains inconclusive after conventional risk stratification, as discussed below.9,21
Stress testing for risk stratification
Exercise stress electrocardiography. Treadmill exercise stress testing with electrocardiography and hemodynamic monitoring was one of the first tools used for risk stratification in HCM.
Although systolic blood pressure normally increases by at least 20 mm Hg with exercise, one-quarter of HCM patients have either a blunted response (failure of systolic blood pressure to increase by at least 20 mm Hg) or a hypotensive response (a drop in systolic blood pressure of 20 mm Hg or more, either continuously or after an initial increase). Studies have shown that HCM patients who have abnormal blood pressure responses during exercise have a higher risk of sudden cardiac death.22–24
Exercise stress echocardiography can be useful to evaluate for provoked increases in the left ventricular outflow tract gradient, which may contribute to a patient’s symptoms even if the resting left ventricular outflow tract gradient is normal. Exercise testing is preferred over pharmacologic stimulation because it can provide functional assessment of whether a patient’s clinical symptoms are truly related to hemodynamic changes due to the hypertrophied ventricle, or whether alternative mechanisms should be explored.
Cardiopulmonary stress testing can readily add prognostic value with additional measurements of functional capacity. HCM patients who cannot achieve their predicted maximal exercise value such as peak rate of oxygen consumption, ventilation efficiency, or anaerobic threshold have higher rates of morbidity and mortality.25,26 Stress testing can also be useful for risk stratification in asymptomatic patients, with one study showing that those who achieve more than 100% of their age- and sex-predicted metabolic equivalents have a low event rate.27
Ambulatory electrocardiographic monitoring in all patients at diagnosis
Ambulatory electrocardiographic monitoring for 24 to 48 hours is recommended for all HCM patients at the time of diagnosis, even if they have no symptoms. Any evidence of nonsustained ventricular tachycardia suggests a substantially higher risk of sudden cardiac death.28,29
In patients with no symptoms or history of arrhythmia, current guidelines suggest ambulatory electrocardiographic monitoring every 1 to 2 years.9,21
Two risk-stratification models
The North American model was the first risk-stratification tool and considers 5 risk factors.9 However, if this algorithm were strictly followed, up to 60% of HCM patients would be candidates for cardioverter-defibrillator implantation.
The European model. This concern led to the development of the HCM Risk-SCD (sudden cardiac death), a risk-stratification tool introduced in the 2014 European Society of Cardiology HCM guidelines.30 This web-based calculator estimates a patient’s 5-year risk of sudden cardiac death using a complex calculation based on 7 clinical risk factors. If a patient’s calculated 5-year risk of sudden cardiac death is 6% or higher, cardioverter-defibrillator implantation is recommended for primary prevention.
The HCM Risk-SCD calculator was validated and compared with classic risk factors alone in a retrospective cohort study in 48 HCM patients.30 Compared with the North American model, the European model results in a lower rate of cardioverter-defibrillator implantation (20% to 26%).31,32
Despite the better specificity of the European model, a large retrospective cohort analysis showed that a significant number of patients stratified as being at low risk for sudden cardiac death were ultimately found to be at high risk in clinical practice.31 Further research is needed to find the optimal risk-stratification approach in HCM patients at low to intermediate risk.
GENETIC TESTING, COUNSELING, AND FAMILY SCREENING
Genetic testing is becoming more widely available and has rapidly expanded in clinical practice. Genetic counseling must be performed alongside genetic testing and requires professionals trained to handle the clinical and social implications of genetic testing. With this in mind, genetic testing can provide a definitive means of identifying family members at risk of HCM.
Given the autosomal dominant nature of HCM, screening for HCM is recommended in all first-degree relatives of an affected patient. Genetic testing may be a means to achieve this if a pathogenic mutation has been identified in the affected patient. However, serial electrocardiographic and transthoracic echocardiographic monitoring is an acceptable alternative in those without a clear genetic mutation association or in those who do not want to undergo genetic testing. If these first-degree relatives who do not undergo genetic testing are adult athletes or adolescents, they should undergo surveillance monitoring, with echocardiography and electrocardiography, whereas adults not participating in athletics should be monitored every 5 years.9,21
As genetic counseling and testing become more widely available, more patients are being found who harbor a mutation but have no phenotypic manifestations of HCM on initial presentation. Clinical expression varies, so continued monitoring of these patients is important. Expert guidelines again recommend serial electrocardiography, transthoracic echocardiography, and clinical assessment every 5 years for adults.9
Recent data suggest that up to 40% of HCM cases are nonfamilial, ie, their inheritance is sporadic with no known family history and no sarcomeric gene mutation evident on screening.33,34 The clinical course in this subgroup seems to be more benign, with later clinical presentations (age > 40) and lower risk of major adverse cardiovascular events.
MANAGEMENT
Conservative management
Asymptomatic HCM can usually be managed with lifestyle modifications.
Avoiding high-risk physical activities is the most important modification. All HCM patients should be counseled on the risk of sudden cardiac death and advised against participating in competitive sports or intense physical activity.35 Aerobic exercise is preferable to isometric exercises such as weightlifting, which may prompt the Valsalva maneuver with worsening of left ventricular outflow tract obstruction leading to syncope. A recent study showed that moderate-intensity aerobic exercise can safely improve exercise capacity, which may ultimately improve functional status and quality of life.36
Avoiding dehydration and excessive alcohol intake are also important in maintaining adequate preload to prevent an increasing left ventricular outflow tract gradient, given the dynamic nature of the left ventricular outflow tract obstruction in HCM.
Medical management: Beta-blockers, then calcium channel blockers
Beta-blockers are the first-line therapy for symptomatic HCM related to left ventricular outflow tract obstruction. Their negative inotropic effect reduces the contractile force of the ventricle, effectively reducing the pressure gradient across the outflow tract. Reduced contractility also means that the overall myocardial workload is less, which ultimately translates to a reduced oxygen demand. With their negative chronotropic effect, beta-blockers lower the heart rate and thereby lengthen the diastolic filling phase, allowing for optimization of preload conditions to help prevent increasing the left ventricular outflow tract gradient.37,38
Beta-blockers can be titrated according to the patient’s symptoms and tolerance. Fatigue and loss of libido are among the most common side effects.
Nondihydropyridine calcium channel blockers can be a second-line therapy in patients who cannot tolerate beta-blockers. Several studies have shown improvement in surrogate outcomes such as estimated left ventricular mass and QRS amplitude on electrocardiography, but currently no available data show that these drugs improve symptoms.28,39,40 They should be avoided in those with severe left ventricular outflow tract obstruction (gradient ≥ 100 mm Hg), as they can lead to critical outflow tract obstruction owing to their peripheral vasodilatory effect.
Dihydropyridine calcium channel blockers should be avoided altogether, as they produce even more peripheral vasodilation and afterload reduction than nondihydropyridine calcium channel blockers.
Disopyramide, a class IA antiarrhythmic, has been shown to effectively reduce outflow gradients and relieve symptoms. However, in view of its adverse effects, it is a third-line therapy, given to those for whom beta-blockers and calcium channel blockers have failed. Its most worrisome adverse effect is QT prolongation, and the QT interval should therefore be closely monitored at the start of treatment. Anticholinergic effects are common and include dry eyes and mouth, urinary retention, and drowsiness.
Disopyramide is usually used in combination with beta-blockers for symptom control as a bridge to a planned invasive intervention.41
Use with caution
Any medication that causes afterload reduction, peripheral vasodilation, intravascular volume depletion, or positive inotropy can worsen the dynamic left ventricular outflow tract obstruction in a patient with HCM and should be avoided.
Angiotensin-converting enzyme (ACE) inhibitors, angiotensin II receptor blockers (ARBs), and nitrates must be used with extreme caution in these patients.
Diuretics. Even restrained use of diuretics can cause significant hemodynamic compromise in patients with obstructive physiology. Therefore, diuretics should be used sparingly in these patients.
Digoxin should not be used for managing atrial fibrillation in these patients, as its positive inotropic effect increases contractility and increases the left ventricular outflow tract gradient.
Norepinephrine and inotropic agents such as dobutamine and dopamine should be avoided for the same reason as digoxin. In patients with circulatory shock requiring vasopressor support, pure alpha-agonists such as phenylephrine are preferred, as they increase peripheral resistance without an inotropic effect.
Anticoagulation for atrial tachyarrhythmias
The risk of systemic thromboembolic events is significantly increased in HCM patients with atrial fibrillation or flutter, regardless of their estimated risk using conventional risk-stratification tools such as the CHADS2 score.42–44 In accordance with current American Heart Association and American College of Cardiology guidelines, we recommend anticoagulation therapy for all HCM patients with a history of atrial fibrillation or flutter. Warfarin is the preferred anticoagulant; direct oral anticoagulants can be considered, but there are currently no data on their use in HCM.9
Standard heart failure treatments
End-stage systolic heart failure is a consequence of HCM but affects only 3% to 4% of patients.45 While most randomized controlled trials of heart failure treatment have excluded HCM patients, current guidelines recommend the same evidence-based medical therapies used in other patients who have heart failure with reduced ejection fraction. This includes ACE inhibitors, ARBs, beta-blockers, and aldosterone antagonists if indicated.9,21
Heart transplant should be considered in patients with class III or IV New York Heart Association functional status despite optimization of their HCM treatment regimen. Heart transplant outcomes for HCM patients are comparable to outcomes for patients who receive a transplant for non-HCM cardiovascular disease.45,46
Septal reduction therapy
If medical therapy fails or is not tolerated in patients with severe symptoms, surgery can be considered for obstructive HCM.
Ventricular septal myectomy has been the long-standing gold standard of invasive therapy. Multiple studies have demonstrated long-term survival after myectomy to be equivalent to that in the general population and better than that of HCM patients who do not undergo this surgery.47–50 Factors that may be associated with better surgical outcomes include age younger than 50, left atrial size less than 46 mm, and resolution of atrial fibrillation during follow-up.51
Septal reduction therapy may also be considered in patients at high risk of sudden cardiac death based on a history of recurrent ventricular tachycardia or risk-stratification models as described above. Retrospective analyses have shown that surgical myectomy can markedly reduce the incidence of appropriate implantable cardioverter-defibrillator discharges and the risk of sudden cardiac death.52
Alcohol septal ablation is an alternative. This percutaneous procedure, first described in the mid-1990s, consists of injecting a small amount of alcohol into the artery supplying the septum to induce myocardial necrosis, ultimately leading to scarring and widening of the left ventricular outflow tract.53
Up to 50% of patients develop right bundle branch block after alcohol septal ablation, and the risk of complete heart block is highest in those with preexisting left bundle branch block. Nevertheless, studies have shown significant symptomatic improvement after alcohol septal ablation, with long-term survival comparable to that in the general population.53–56
Several meta-analyses compared alcohol septal ablation and septal myectomy and found that the rates of functional improvement and long-term mortality were similar.57–59 However, the less-invasive approach with alcohol septal ablation comes at the cost of a higher incidence of conduction abnormalities and higher left ventricular outflow tract gradients afterward. One meta-analysis found that alcohol septal ablation patients may have 5 times the risk of needing additional septal reduction therapy compared with their myectomy counterparts.
Current US guidelines recommend septal myectomy, performed at an experienced center, as the first-line interventional treatment, leaving alcohol septal ablation to be considered in those who have contraindications to myectomy.9 The treatment strategy should ultimately be individualized based on a patient’s comorbidities and personal preferences following informed consent.
A nationwide database study recently suggested that postmyectomy mortality rates may be as high as 5.9%,60 although earlier studies at high-volume centers showed much lower mortality rates (< 1%).50–52,61 This discrepancy highlights the critical role of expert centers in optimizing surgical management of these patients. Regardless of the approach, interventional therapies for HCM should be performed by a multidisciplinary team at a medical center able to handle the complexity of these cases.
Additional surgical procedures
A handful of other procedures may benefit specific patient subgroups.
Apical myectomy has been shown to improve functional status in patients with isolated apical hypertrophy by reducing left ventricular end-diastolic pressure and thereby allowing for improved diastolic filling.63
Mitral valve surgery may need to be considered at the time of myectomy in patients with degenerative valve disease. As in the general population, mitral valve repair is preferred to replacement if possible.
- Maron BJ. Hypertrophic cardiomyopathy: an important global disease. Am J Med 2004; 116(1):63–65. pmid:14706671
- Semsarian C, Ingles J, Maron MS, Maron BJ. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol 2015; 65(12):1249–1254. doi:10.1016/j.jacc.2015.01.019
- Maron BJ, Maron MS, Semsarian C. Genetics of hypertrophic cardiomyopathy after 20 years: clinical perspectives. J Am Coll Cardiol 2012; 60(8):705–715. doi:10.1016/j.jacc.2012.02.068
- Shirani J, Pick R, Roberts WC, Maron BJ. Morphology and significance of the left ventricular collagen network in young patients with hypertrophic cardiomyopathy and sudden cardiac death. J Am Coll Cardiol 2000; 35(1):36–44. pmid:10636256
- Sherrid MV, Chu CK, Delia E, Mogtader A, Dwyer EM Jr. An echocardiographic study of the fluid mechanics of obstruction in hypertrophic cardiomyopathy. J Am Coll Cardiol 1993; 22(3):816–825. pmid:8354817
- Ro R, Halpern D, Sahn DJ, et al. Vector flow mapping in obstructive hypertrophic cardiomyopathy to assess the relationship of early systolic left ventricular flow and the mitral valve. J Am Coll Cardiol 2014; 64(19):1984–1995. doi:10.1016/j.jacc.2014.04.090
- Kwon DH, Setser RM, Thamilarasan M, et al. Abnormal papillary muscle morphology is independently associated with increased left ventricular outflow tract obstruction in hypertrophic cardiomyopathy. Heart 2008; 94(10):1295–1301. doi:10.1136/hrt.2007.118018
- Patel P, Dhillon A, Popovic ZB, et al. Left ventricular outflow tract obstruction in hypertrophic cardiomyopathy patients without severe septal hypertrophy: implications of mitral valve and papillary muscle abnormalities assessed using cardiac magnetic resonance and echocardiography. Circ Cardiovasc Imaging 2015; 8(7):e003132. doi:10.1161/CIRCIMAGING.115.003132
- Gersh BJ, Maron BJ, Bonow RO, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Thorac Cardiovasc Surg 2011; 142(6):e153-e203. doi:10.1016/j.jtcvs.2011.10.020
- Elliott PM, Gimeno JR, Thaman R, et al. Historical trends in reported survival rates in patients with hypertrophic cardiomyopathy. Heart 2006; 92(6):785–791. doi:10.1136/hrt.2005.068577
- Maron MS, Olivotto I, Zenovich AG, et al. Hypertrophic cardiomyopathy is predominantly a disease of left ventricular outflow tract obstruction. Circulation 2006; 114(21):2232–2239. doi:10.1161/CIRCULATIONAHA.106.644682
- Ho CY, Carlsen C, Thune JJ, et al. Echocardiographic strain imaging to assess early and late consequences of sarcomere mutations in hypertrophic cardiomyopathy. Circ Cardiovasc Genet 2009; 2(4):314–321. doi:10.1161/CIRCGENETICS.109.862128
- Wasfy MM, Weiner RB. Differentiating the athlete’s heart from hypertrophic cardiomyopathy. Curr Opin Cardiol 2015; 30(5):500–505. doi:10.1097/HCO.0000000000000203
- Palka P, Lange A, Fleming AD, et al. Differences in myocardial velocity gradient measured throughout the cardiac cycle in patients with hypertrophic cardiomyopathy, athletes and patients with left ventricular hypertrophy due to hypertension. J Am Coll Cardiol 1997; 30(3):760–768. pmid:9283537
- Eriksson MJ, Sonnenberg B, Woo A, et al. Long-term outcome in patients with apical hypertrophic cardiomyopathy. J Am Coll Cardiol 2002; 39(4):638–645. pmid:11849863
- Rickers C, Wilke NM, Jerosch-Herold M, et al. Utility of cardiac magnetic resonance imaging in the diagnosis of hypertrophic cardiomyopathy. Circulation 2005; 112(6):855–861. doi:10.1161/CIRCULATIONAHA.104.507723
- Kwon DH, Setser RM, Popovic ZB, et al. Association of myocardial fibrosis, electrocardiography and ventricular tachyarrhythmia in hypertrophic cardiomyopathy: a delayed contrast enhanced MRI study. Int J Cardiovasc Imaging 2008; 24(6):617–625. doi:10.1007/s10554-008-9292-6
- Rubinshtein R, Glockner JF, Ommen SR, et al. Characteristics and clinical significance of late gadolinium enhancement by contrast-enhanced magnetic resonance imaging in patients with hypertrophic cardiomyopathy. Circ Heart Fail 2010; 3(1):51–58. doi:10.1161/CIRCHEARTFAILURE.109.854026
- O’Hanlon R, Grasso A, Roughton M, et al. Prognostic significance of myocardial fibrosis in hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 56(11):867–874. doi:10.1016/j.jacc.2010.05.010
- Bruder O, Wagner A, Jensen CJ, et al. Myocardial scar visualized by cardiovascular magnetic resonance imaging predicts major adverse events in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 56(11):875–887. doi:10.1016/j.jacc.2010.05.007
- Authors/Task Force members, Elliott PM, Anastasakis A, Borger MA, et al. 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J 2014; 35(39):2733–2779. doi:10.1093/eurheartj/ehu284
- Olivotto I, Maron BJ, Montereggi A, Mazzuoli F, Dolara A, Cecchi F. Prognostic value of systemic blood pressure response during exercise in a community-based patient population with hypertrophic cardiomyopathy. J Am Coll Cardiol 1999; 33(7):2044–2051. pmid:10362212
- Sadoul N, Prasad K, Elliott PM, Bannerjee S, Frenneaux MP, McKenna WJ. Prospective prognostic assessment of blood pressure response during exercise in patients with hypertrophic cardiomyopathy. Circulation 1997; 96(9):2987–2991. pmid:9386166
- Elliott PM, Poloniecki J, Dickie S, et al. Sudden death in hypertrophic cardiomyopathy: identification of high risk patients. J Am Coll Cardiol 2000; 36(7):2212–2218. pmid:11127463
- Masri A, Pierson LM, Smedira NG, et al. Predictors of long-term outcomes in patients with hypertrophic cardiomyopathy undergoing cardiopulmonary stress testing and echocardiography. Am Heart J 2015; 169(5):684–692.e1. doi:10.1016/j.ahj.2015.02.006
- Coats CJ, Rantell K, Bartnik A, et al. Cardiopulmonary exercise testing and prognosis in hypertrophic cardiomyopathy. Circ Heart Fail 2015; 8(6):1022–1031. doi:10.1161/CIRCHEARTFAILURE.114.002248
- Desai MY, Bhonsale A, Patel P, et al. Exercise echocardiography in asymptomatic HCM: exercise capacity, and not LV outflow tract gradient predicts long-term outcomes. JACC Cardiovasc Imaging 2014; 7(1):26–36. doi:10.1016/j.jcmg.2013.08.010
- Spirito P, Seidman CE, McKenna WJ, Maron BJ. The management of hypertrophic cardiomyopathy. N Engl J Med 1997; 336(11):775–785. doi:10.1056/NEJM199703133361107
- Wang W, Lian Z, Rowin EJ, Maron BJ, Maron MS, Link MS. Prognostic implications of nonsustained ventricular tachycardia in high-risk patients with hypertrophic cardiomyopathy. Circ Arrhythm Electrophysiol 2017; 10(3)e004604. doi:10.1161/CIRCEP.116.004604
- O’Mahony C, Jichi F, Pavlou M, et al. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM risk-SCD). Eur Heart J 2014; 35(30):2010–2020. doi:10.1093/eurheartj/eht439
- Maron BJ, Casey SA, Chan RH, Garberich RF, Rowin EJ, Maron MS. Independent assessment of the European Society of Cardiology sudden death risk model for hypertrophic cardiomyopathy. Am J Cardiol 2015; 116(5):757–764. doi:10.1016/j.amjcard.2015.05.047
- Jahnlová D, Tomašov P, Zemánek D, Veselka J. Transatlantic differences in assessment of risk of sudden cardiac death in patients with hypertrophic cardiomyopathy. Int J Cardiol 2015; 186:3–4. doi:10.1016/j.ijcard.2015.03.207
- Ingles J, Burns C, Bagnall RD, et al. Nonfamilial hypertrophic cardiomyopathy: prevalence, natural history, and clinical implications. Circ Cardiovasc Genet 2017; 10(2)e001620. doi:10.1161/CIRCGENETICS.116.001620
- Ko C, Arscott P, Concannon M, et al. Genetic testing impacts the utility of prospective familial screening in hypertrophic cardiomyopathy through identification of a nonfamilial subgroup. Genet Med 2017; 20(1):69–75. doi:10.1038/gim.2017.79
- Maron BJ, Chaitman BR, Ackerman MJ, et al. Recommendations for physical activity and recreational sports participation for young patients with genetic cardiovascular diseases. Circulation 2004; 109(22):2807–2816. doi:10.1161/01.CIR.0000128363.85581.E1
- Saberi S, Wheeler M, Bragg-Gresham J, et al. Effect of moderate-intensity exercise training on peak oxygen consumption in patients with hypertrophic cardiomyopathy: a randomized clinical trial. JAMA 2017; 317(13):1349–1357. doi:10.1001/jama.2017.2503
- Bourmayan C, Razavi A, Fournier C, et al. Effect of propranolol on left ventricular relaxation in hypertrophic cardiomyopathy: an echographic study. Am Heart J 1985; 109(6):1311–1316. pmid:4039882
- Spoladore R, Maron MS, D’Amato R, Camici PG, Olivotto I. Pharmacological treatment options for hypertrophic cardiomyopathy: high time for evidence. Eur Heart J 2012; 33(14):1724–1733. doi:10.1093/eurheartj/ehs150
- Choudhury L, Elliott P, Rimoldi O, et al. Transmural myocardial blood flow distribution in hypertrophic cardiomyopathy and effect of treatment. Basic Res Cardiol 1999; 94(1):49–59. pmid:10097830
- Kaltenbach M, Hopf R, Kober G, Bussmann WD, Keller M, Petersen Y. Treatment of hypertrophic obstructive cardiomyopathy with verapamil. Br Heart J 1979; 42(1):35–42. doi:10.1136/hrt.42.1.35
- Sherrid MV, Shetty A, Winson G, et al. Treatment of obstructive hypertrophic cardiomyopathy symptoms and gradient resistant to first-line therapy with beta-blockade or verapamil. Circ Heart Fail 2013; 6(4):694–702. doi:10.1161/CIRCHEARTFAILURE.112.000122
- Guttmann OP, Rahman MS, O’Mahony C, Anastasakis A, Elliott PM. Atrial fibrillation and thromboembolism in patients with hypertrophic cardiomyopathy: systematic review. Heart 2014; 100(6):465–472. doi:10.1136/heartjnl-2013-304276
- Olivotto I, Cecchi F, Casey SA, Dolara A, Traverse JH, Maron BJ. Impact of atrial fibrillation on the clinical course of hypertrophic cardiomyopathy. Circulation 2001; 104(21):2517–2524. pmid:11714644
- Maron BJ, Olivotto I, Spirito P, et al. Epidemiology of hypertrophic cardiomyopathy-related death: revisited in a large non-referral-based patient population. Circulation 2000; 102(8):858–864. pmid:10952953
- Harris KM, Spirito P, Maron MS, et al. Prevalence, clinical profile, and significance of left ventricular remodeling in the end-stage phase of hypertrophic cardiomyopathy. Circulation 2006; 114(3):216-225. doi:10.1161/CIRCULATIONAHA.105.583500
- Maron MS, Kalsmith BM, Udelson JE, Li W, DeNofrio D. Survival after cardiac transplantation in patients with hypertrophic cardiomyopathy. Circ Heart Fail 2010; 3(5):574–579. doi:10.1161/CIRCHEARTFAILURE.109.922872
- Smedira NG, Lytle BW, Lever HM, et al. Current effectiveness and risks of isolated septal myectomy for hypertrophic obstructive cardiomyopathy. Ann Thorac Surg 2008; 85(1):127–133. doi:10.1016/j.athoracsur.2007.07.063
- Robbins RC, Stinson EB. Long-term results of left ventricular myotomy and myectomy for obstructive hypertrophic cardiomyopathy. J Thorac Cardiovasc Surg 1996; 111(3):586–594. pmid:8601973
- Heric B, Lytle BW, Miller DP, Rosenkranz ER, Lever HM, Cosgrove DM. Surgical management of hypertrophic obstructive cardiomyopathy. Early and late results. J Thorac Cardiovasc Surg 1995; 110(1):195–208. pmid:7609544
- Ommen SR, Maron BJ, Olivotto I, et al. Long-term effects of surgical septal myectomy on survival in patients with obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol 2005; 46(3):470–476. doi:10.1016/j.jacc.2005.02.090
- Desai MY, Bhonsale A, Smedira NG, et al. Predictors of long-term outcomes in symptomatic hypertrophic obstructive cardiomyopathy patients undergoing surgical relief of left ventricular outflow tract obstruction. Circulation 2013; 128(3):209–216. doi:10.1161/CIRCULATIONAHA.112.000849
- McLeod CJ, Ommen SR, Ackerman MJ, et al. Surgical septal myectomy decreases the risk for appropriate implantable cardioverter defibrillator discharge in obstructive hypertrophic cardiomyopathy. Eur Heart J 2007; 28(21):2583–2588. doi:10.1093/eurheartj/ehm117
- Veselka J, Tomasov P, Zemanek D. Long-term effects of varying alcohol dosing in percutaneous septal ablation for obstructive hypertrophic cardiomyopathy: a randomized study with a follow-up up to 11 years. Can J Cardiol 2011; 27(6):763–767. doi:10.1016/j.cjca.2011.09.001
- Veselka J, Jensen MK, Liebregts M, et al. Low procedure-related mortality achieved with alcohol septal ablation in European patients. Int J Cardiol 2016; 209:194–195. doi:10.1016/j.ijcard.2016.02.077
- Veselka J, Krejci J, Tomašov P, Zemánek D. Long-term survival after alcohol septal ablation for hypertrophic obstructive cardiomyopathy: a comparison with general population. Eur Heart J 2014; 35(30):2040–2045. doi:10.1093/eurheartj/eht495
- Sorajja P, Ommen SR, Holmes DR Jr, et al. Survival after alcohol septal ablation for obstructive hypertrophic cardiomyopathy. Circulation 2012; 126(20):2374–2380. doi:10.1161/CIRCULATIONAHA.111.076257
- Agarwal S, Tuzcu EM, Desai MY, et al. Updated meta-analysis of septal alcohol ablation versus myectomy for hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 55(8):823–834. doi:10.1016/j.jacc.2009.09.047
- Leonardi RA, Kransdorf EP, Simel DL, Wang A. Meta-analyses of septal reduction therapies for obstructive hypertrophic cardiomyopathy: comparative rates of overall mortality and sudden cardiac death after treatment. Circ Cardiovasc Interv 2010; 3(2):97–104. doi:10.1161/CIRCINTERVENTIONS.109.916676
- Liebregts M, Vriesendorp PA, Mahmoodi BK, Schinkel AF, Michels M, ten Berg JM. A systematic review and meta-analysis of long-term outcomes after septal reduction therapy in patients with hypertrophic cardiomyopathy. JACC Heart Fail 2015; 3(11):896–905. doi:10.1016/j.jchf.2015.06.011
- Panaich SS, Badheka AO, Chothani A, et al. Results of ventricular septal myectomy and hypertrophic cardiomyopathy (from Nationwide Inpatient Sample [1998-2010]). Am J Cardiol 2014; 114(9):1390–1395. doi:10.1016/j.amjcard.2014.07.075
- Maron BJ, Dearani JA, Ommen SR, et al. Low operative mortality achieved with surgical septal myectomy: role of dedicated hypertrophic cardiomyopathy centers in the management of dynamic subaortic obstruction. J Am Coll Cardiol 2015; 66(11):1307–1308. doi:10.1016/j.jacc.2015.06.1333
- Kwon DH, Smedira NG, Thamilarasan M, Lytle BW, Lever H, Desai MY. Characteristics and surgical outcomes of symptomatic patients with hypertrophic cardiomyopathy with abnormal papillary muscle morphology undergoing papillary muscle reorientation. J Thorac Cardiovasc Surg 2010; 140(2):317–324. doi:10.1016/j.jtcvs.2009.10.045
- Schaff HV, Brown ML, Dearani JA, et al. Apical myectomy: a new surgical technique for management of severely symptomatic patients with apical hypertrophic cardiomyopathy. J Thorac Cardiovasc Surg 2010; 139(3):634–640. doi:10.1016/j.jtcvs.2009.07.079
- Maron BJ. Hypertrophic cardiomyopathy: an important global disease. Am J Med 2004; 116(1):63–65. pmid:14706671
- Semsarian C, Ingles J, Maron MS, Maron BJ. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol 2015; 65(12):1249–1254. doi:10.1016/j.jacc.2015.01.019
- Maron BJ, Maron MS, Semsarian C. Genetics of hypertrophic cardiomyopathy after 20 years: clinical perspectives. J Am Coll Cardiol 2012; 60(8):705–715. doi:10.1016/j.jacc.2012.02.068
- Shirani J, Pick R, Roberts WC, Maron BJ. Morphology and significance of the left ventricular collagen network in young patients with hypertrophic cardiomyopathy and sudden cardiac death. J Am Coll Cardiol 2000; 35(1):36–44. pmid:10636256
- Sherrid MV, Chu CK, Delia E, Mogtader A, Dwyer EM Jr. An echocardiographic study of the fluid mechanics of obstruction in hypertrophic cardiomyopathy. J Am Coll Cardiol 1993; 22(3):816–825. pmid:8354817
- Ro R, Halpern D, Sahn DJ, et al. Vector flow mapping in obstructive hypertrophic cardiomyopathy to assess the relationship of early systolic left ventricular flow and the mitral valve. J Am Coll Cardiol 2014; 64(19):1984–1995. doi:10.1016/j.jacc.2014.04.090
- Kwon DH, Setser RM, Thamilarasan M, et al. Abnormal papillary muscle morphology is independently associated with increased left ventricular outflow tract obstruction in hypertrophic cardiomyopathy. Heart 2008; 94(10):1295–1301. doi:10.1136/hrt.2007.118018
- Patel P, Dhillon A, Popovic ZB, et al. Left ventricular outflow tract obstruction in hypertrophic cardiomyopathy patients without severe septal hypertrophy: implications of mitral valve and papillary muscle abnormalities assessed using cardiac magnetic resonance and echocardiography. Circ Cardiovasc Imaging 2015; 8(7):e003132. doi:10.1161/CIRCIMAGING.115.003132
- Gersh BJ, Maron BJ, Bonow RO, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Thorac Cardiovasc Surg 2011; 142(6):e153-e203. doi:10.1016/j.jtcvs.2011.10.020
- Elliott PM, Gimeno JR, Thaman R, et al. Historical trends in reported survival rates in patients with hypertrophic cardiomyopathy. Heart 2006; 92(6):785–791. doi:10.1136/hrt.2005.068577
- Maron MS, Olivotto I, Zenovich AG, et al. Hypertrophic cardiomyopathy is predominantly a disease of left ventricular outflow tract obstruction. Circulation 2006; 114(21):2232–2239. doi:10.1161/CIRCULATIONAHA.106.644682
- Ho CY, Carlsen C, Thune JJ, et al. Echocardiographic strain imaging to assess early and late consequences of sarcomere mutations in hypertrophic cardiomyopathy. Circ Cardiovasc Genet 2009; 2(4):314–321. doi:10.1161/CIRCGENETICS.109.862128
- Wasfy MM, Weiner RB. Differentiating the athlete’s heart from hypertrophic cardiomyopathy. Curr Opin Cardiol 2015; 30(5):500–505. doi:10.1097/HCO.0000000000000203
- Palka P, Lange A, Fleming AD, et al. Differences in myocardial velocity gradient measured throughout the cardiac cycle in patients with hypertrophic cardiomyopathy, athletes and patients with left ventricular hypertrophy due to hypertension. J Am Coll Cardiol 1997; 30(3):760–768. pmid:9283537
- Eriksson MJ, Sonnenberg B, Woo A, et al. Long-term outcome in patients with apical hypertrophic cardiomyopathy. J Am Coll Cardiol 2002; 39(4):638–645. pmid:11849863
- Rickers C, Wilke NM, Jerosch-Herold M, et al. Utility of cardiac magnetic resonance imaging in the diagnosis of hypertrophic cardiomyopathy. Circulation 2005; 112(6):855–861. doi:10.1161/CIRCULATIONAHA.104.507723
- Kwon DH, Setser RM, Popovic ZB, et al. Association of myocardial fibrosis, electrocardiography and ventricular tachyarrhythmia in hypertrophic cardiomyopathy: a delayed contrast enhanced MRI study. Int J Cardiovasc Imaging 2008; 24(6):617–625. doi:10.1007/s10554-008-9292-6
- Rubinshtein R, Glockner JF, Ommen SR, et al. Characteristics and clinical significance of late gadolinium enhancement by contrast-enhanced magnetic resonance imaging in patients with hypertrophic cardiomyopathy. Circ Heart Fail 2010; 3(1):51–58. doi:10.1161/CIRCHEARTFAILURE.109.854026
- O’Hanlon R, Grasso A, Roughton M, et al. Prognostic significance of myocardial fibrosis in hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 56(11):867–874. doi:10.1016/j.jacc.2010.05.010
- Bruder O, Wagner A, Jensen CJ, et al. Myocardial scar visualized by cardiovascular magnetic resonance imaging predicts major adverse events in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 56(11):875–887. doi:10.1016/j.jacc.2010.05.007
- Authors/Task Force members, Elliott PM, Anastasakis A, Borger MA, et al. 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J 2014; 35(39):2733–2779. doi:10.1093/eurheartj/ehu284
- Olivotto I, Maron BJ, Montereggi A, Mazzuoli F, Dolara A, Cecchi F. Prognostic value of systemic blood pressure response during exercise in a community-based patient population with hypertrophic cardiomyopathy. J Am Coll Cardiol 1999; 33(7):2044–2051. pmid:10362212
- Sadoul N, Prasad K, Elliott PM, Bannerjee S, Frenneaux MP, McKenna WJ. Prospective prognostic assessment of blood pressure response during exercise in patients with hypertrophic cardiomyopathy. Circulation 1997; 96(9):2987–2991. pmid:9386166
- Elliott PM, Poloniecki J, Dickie S, et al. Sudden death in hypertrophic cardiomyopathy: identification of high risk patients. J Am Coll Cardiol 2000; 36(7):2212–2218. pmid:11127463
- Masri A, Pierson LM, Smedira NG, et al. Predictors of long-term outcomes in patients with hypertrophic cardiomyopathy undergoing cardiopulmonary stress testing and echocardiography. Am Heart J 2015; 169(5):684–692.e1. doi:10.1016/j.ahj.2015.02.006
- Coats CJ, Rantell K, Bartnik A, et al. Cardiopulmonary exercise testing and prognosis in hypertrophic cardiomyopathy. Circ Heart Fail 2015; 8(6):1022–1031. doi:10.1161/CIRCHEARTFAILURE.114.002248
- Desai MY, Bhonsale A, Patel P, et al. Exercise echocardiography in asymptomatic HCM: exercise capacity, and not LV outflow tract gradient predicts long-term outcomes. JACC Cardiovasc Imaging 2014; 7(1):26–36. doi:10.1016/j.jcmg.2013.08.010
- Spirito P, Seidman CE, McKenna WJ, Maron BJ. The management of hypertrophic cardiomyopathy. N Engl J Med 1997; 336(11):775–785. doi:10.1056/NEJM199703133361107
- Wang W, Lian Z, Rowin EJ, Maron BJ, Maron MS, Link MS. Prognostic implications of nonsustained ventricular tachycardia in high-risk patients with hypertrophic cardiomyopathy. Circ Arrhythm Electrophysiol 2017; 10(3)e004604. doi:10.1161/CIRCEP.116.004604
- O’Mahony C, Jichi F, Pavlou M, et al. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM risk-SCD). Eur Heart J 2014; 35(30):2010–2020. doi:10.1093/eurheartj/eht439
- Maron BJ, Casey SA, Chan RH, Garberich RF, Rowin EJ, Maron MS. Independent assessment of the European Society of Cardiology sudden death risk model for hypertrophic cardiomyopathy. Am J Cardiol 2015; 116(5):757–764. doi:10.1016/j.amjcard.2015.05.047
- Jahnlová D, Tomašov P, Zemánek D, Veselka J. Transatlantic differences in assessment of risk of sudden cardiac death in patients with hypertrophic cardiomyopathy. Int J Cardiol 2015; 186:3–4. doi:10.1016/j.ijcard.2015.03.207
- Ingles J, Burns C, Bagnall RD, et al. Nonfamilial hypertrophic cardiomyopathy: prevalence, natural history, and clinical implications. Circ Cardiovasc Genet 2017; 10(2)e001620. doi:10.1161/CIRCGENETICS.116.001620
- Ko C, Arscott P, Concannon M, et al. Genetic testing impacts the utility of prospective familial screening in hypertrophic cardiomyopathy through identification of a nonfamilial subgroup. Genet Med 2017; 20(1):69–75. doi:10.1038/gim.2017.79
- Maron BJ, Chaitman BR, Ackerman MJ, et al. Recommendations for physical activity and recreational sports participation for young patients with genetic cardiovascular diseases. Circulation 2004; 109(22):2807–2816. doi:10.1161/01.CIR.0000128363.85581.E1
- Saberi S, Wheeler M, Bragg-Gresham J, et al. Effect of moderate-intensity exercise training on peak oxygen consumption in patients with hypertrophic cardiomyopathy: a randomized clinical trial. JAMA 2017; 317(13):1349–1357. doi:10.1001/jama.2017.2503
- Bourmayan C, Razavi A, Fournier C, et al. Effect of propranolol on left ventricular relaxation in hypertrophic cardiomyopathy: an echographic study. Am Heart J 1985; 109(6):1311–1316. pmid:4039882
- Spoladore R, Maron MS, D’Amato R, Camici PG, Olivotto I. Pharmacological treatment options for hypertrophic cardiomyopathy: high time for evidence. Eur Heart J 2012; 33(14):1724–1733. doi:10.1093/eurheartj/ehs150
- Choudhury L, Elliott P, Rimoldi O, et al. Transmural myocardial blood flow distribution in hypertrophic cardiomyopathy and effect of treatment. Basic Res Cardiol 1999; 94(1):49–59. pmid:10097830
- Kaltenbach M, Hopf R, Kober G, Bussmann WD, Keller M, Petersen Y. Treatment of hypertrophic obstructive cardiomyopathy with verapamil. Br Heart J 1979; 42(1):35–42. doi:10.1136/hrt.42.1.35
- Sherrid MV, Shetty A, Winson G, et al. Treatment of obstructive hypertrophic cardiomyopathy symptoms and gradient resistant to first-line therapy with beta-blockade or verapamil. Circ Heart Fail 2013; 6(4):694–702. doi:10.1161/CIRCHEARTFAILURE.112.000122
- Guttmann OP, Rahman MS, O’Mahony C, Anastasakis A, Elliott PM. Atrial fibrillation and thromboembolism in patients with hypertrophic cardiomyopathy: systematic review. Heart 2014; 100(6):465–472. doi:10.1136/heartjnl-2013-304276
- Olivotto I, Cecchi F, Casey SA, Dolara A, Traverse JH, Maron BJ. Impact of atrial fibrillation on the clinical course of hypertrophic cardiomyopathy. Circulation 2001; 104(21):2517–2524. pmid:11714644
- Maron BJ, Olivotto I, Spirito P, et al. Epidemiology of hypertrophic cardiomyopathy-related death: revisited in a large non-referral-based patient population. Circulation 2000; 102(8):858–864. pmid:10952953
- Harris KM, Spirito P, Maron MS, et al. Prevalence, clinical profile, and significance of left ventricular remodeling in the end-stage phase of hypertrophic cardiomyopathy. Circulation 2006; 114(3):216-225. doi:10.1161/CIRCULATIONAHA.105.583500
- Maron MS, Kalsmith BM, Udelson JE, Li W, DeNofrio D. Survival after cardiac transplantation in patients with hypertrophic cardiomyopathy. Circ Heart Fail 2010; 3(5):574–579. doi:10.1161/CIRCHEARTFAILURE.109.922872
- Smedira NG, Lytle BW, Lever HM, et al. Current effectiveness and risks of isolated septal myectomy for hypertrophic obstructive cardiomyopathy. Ann Thorac Surg 2008; 85(1):127–133. doi:10.1016/j.athoracsur.2007.07.063
- Robbins RC, Stinson EB. Long-term results of left ventricular myotomy and myectomy for obstructive hypertrophic cardiomyopathy. J Thorac Cardiovasc Surg 1996; 111(3):586–594. pmid:8601973
- Heric B, Lytle BW, Miller DP, Rosenkranz ER, Lever HM, Cosgrove DM. Surgical management of hypertrophic obstructive cardiomyopathy. Early and late results. J Thorac Cardiovasc Surg 1995; 110(1):195–208. pmid:7609544
- Ommen SR, Maron BJ, Olivotto I, et al. Long-term effects of surgical septal myectomy on survival in patients with obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol 2005; 46(3):470–476. doi:10.1016/j.jacc.2005.02.090
- Desai MY, Bhonsale A, Smedira NG, et al. Predictors of long-term outcomes in symptomatic hypertrophic obstructive cardiomyopathy patients undergoing surgical relief of left ventricular outflow tract obstruction. Circulation 2013; 128(3):209–216. doi:10.1161/CIRCULATIONAHA.112.000849
- McLeod CJ, Ommen SR, Ackerman MJ, et al. Surgical septal myectomy decreases the risk for appropriate implantable cardioverter defibrillator discharge in obstructive hypertrophic cardiomyopathy. Eur Heart J 2007; 28(21):2583–2588. doi:10.1093/eurheartj/ehm117
- Veselka J, Tomasov P, Zemanek D. Long-term effects of varying alcohol dosing in percutaneous septal ablation for obstructive hypertrophic cardiomyopathy: a randomized study with a follow-up up to 11 years. Can J Cardiol 2011; 27(6):763–767. doi:10.1016/j.cjca.2011.09.001
- Veselka J, Jensen MK, Liebregts M, et al. Low procedure-related mortality achieved with alcohol septal ablation in European patients. Int J Cardiol 2016; 209:194–195. doi:10.1016/j.ijcard.2016.02.077
- Veselka J, Krejci J, Tomašov P, Zemánek D. Long-term survival after alcohol septal ablation for hypertrophic obstructive cardiomyopathy: a comparison with general population. Eur Heart J 2014; 35(30):2040–2045. doi:10.1093/eurheartj/eht495
- Sorajja P, Ommen SR, Holmes DR Jr, et al. Survival after alcohol septal ablation for obstructive hypertrophic cardiomyopathy. Circulation 2012; 126(20):2374–2380. doi:10.1161/CIRCULATIONAHA.111.076257
- Agarwal S, Tuzcu EM, Desai MY, et al. Updated meta-analysis of septal alcohol ablation versus myectomy for hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 55(8):823–834. doi:10.1016/j.jacc.2009.09.047
- Leonardi RA, Kransdorf EP, Simel DL, Wang A. Meta-analyses of septal reduction therapies for obstructive hypertrophic cardiomyopathy: comparative rates of overall mortality and sudden cardiac death after treatment. Circ Cardiovasc Interv 2010; 3(2):97–104. doi:10.1161/CIRCINTERVENTIONS.109.916676
- Liebregts M, Vriesendorp PA, Mahmoodi BK, Schinkel AF, Michels M, ten Berg JM. A systematic review and meta-analysis of long-term outcomes after septal reduction therapy in patients with hypertrophic cardiomyopathy. JACC Heart Fail 2015; 3(11):896–905. doi:10.1016/j.jchf.2015.06.011
- Panaich SS, Badheka AO, Chothani A, et al. Results of ventricular septal myectomy and hypertrophic cardiomyopathy (from Nationwide Inpatient Sample [1998-2010]). Am J Cardiol 2014; 114(9):1390–1395. doi:10.1016/j.amjcard.2014.07.075
- Maron BJ, Dearani JA, Ommen SR, et al. Low operative mortality achieved with surgical septal myectomy: role of dedicated hypertrophic cardiomyopathy centers in the management of dynamic subaortic obstruction. J Am Coll Cardiol 2015; 66(11):1307–1308. doi:10.1016/j.jacc.2015.06.1333
- Kwon DH, Smedira NG, Thamilarasan M, Lytle BW, Lever H, Desai MY. Characteristics and surgical outcomes of symptomatic patients with hypertrophic cardiomyopathy with abnormal papillary muscle morphology undergoing papillary muscle reorientation. J Thorac Cardiovasc Surg 2010; 140(2):317–324. doi:10.1016/j.jtcvs.2009.10.045
- Schaff HV, Brown ML, Dearani JA, et al. Apical myectomy: a new surgical technique for management of severely symptomatic patients with apical hypertrophic cardiomyopathy. J Thorac Cardiovasc Surg 2010; 139(3):634–640. doi:10.1016/j.jtcvs.2009.07.079
KEY POINTS
- Obstruction of the left ventricular outflow tract is a key pathophysiologic mechanism in HCM.
- Because most of the genetic variants that contribute to HCM are autosomal dominant, genetic counseling and testing are suggested for patients and their first-degree relatives.
- Transthoracic echocardiography is the first-line imaging test, followed by magnetic resonance imaging.
- Beta-blockers are the first-line drugs for treating symptoms of HCM.
- An implantable cardioverter-defibrillator can be considered for patients at risk of sudden cardiac death.
- When medical therapy fails or is not tolerated in patients with severe symptoms of obstructive HCM, surgery to reduce the size of the ventricular septum can be considered. Alcohol septal ablation is an alternative.

Idiopathic pulmonary fibrosis: What primary care physicians need to know
Idiopathic pulmonary fibrosis (IPF) is a devastating and fatal lung disease that generally affects older adults. It is characterized by a radiographic and histopathologic pattern of usual interstitial pneumonia (UIP) that has no other known etiology.
Accurate diagnosis of IPF is crucial. We recommend early referral to a center specializing in interstitial lung disease to confirm the diagnosis, start appropriate therapy, advise the patient on prognosis and enrollment in disease registries and clinical trials, and determine candidacy for lung transplant.
Primary care physicians are uniquely positioned to encounter patients with IPF, whether because of a patient complaint or as an incidental finding on computed tomography. The goal of this article is to delineate the features of IPF so that it may be recognized early and thus expedite referral to a center with expertise in interstitial lung disease for a thorough evaluation and appropriate management.
WHAT IS IDIOPATHIC PULMONARY FIBROSIS?

MORE COMMON THAN ONCE THOUGHT
The true incidence and prevalence of IPF are difficult to assess. IPF is generally considered a rare disease, but it is more common than once thought. In 2011, Raghu et al3 estimated the prevalence in Medicare beneficiaries to be 495 cases per 100,000. Based on this estimate and the current US population, up to 160,000 Americans could have IPF.4 Raghu et al also showed that IPF more often affects adults over age 65, which suggests that as the US population ages, the incidence of IPF may rise. Studies have also reported an increased incidence of IPF worldwide.5
Further, with the rising use of low-dose computed tomography to screen for lung cancer, more incidental cases of IPF will likely be found.6–8
Older data showed a lag from symptom onset to accurate diagnosis of 1 to 2 years.9 A more recent study found a lag in referral of patients with IPF to tertiary care centers, and this delay was associated with a higher rate of death independent of disease severity.10
TYPICALLY PROGRESSIVE, OFTEN FATAL
IPF is typically progressive and limited to the lungs, and it portends a poor prognosis. The median survival is commonly cited as 2 to 5 years from diagnosis, although this is based on older observations that may not reflect current best practice and newer therapies. More recent studies suggest higher survival rates if patients have preserved lung function.11
As the name indicates, the etiology of IPF is unknown, but studies have indicated genetic underpinnings in a notable proportion of cases.12 Regardless of the cause, the pathogenesis and progression of IPF are thought to be the result of an abnormal and persistent wound-repair response. The progressive deposition of scar tissue disrupts normal lung architecture and function, eventually causing clinical disease.13
SYMPTOMS AND KEY FEATURES
Patients with IPF typically present with the insidious onset of dyspnea on exertion, with or without chronic cough. Risk factors include male sex, increasing age, and a history of smoking. Patients with undiagnosed IPF who present with dyspnea and a history of smoking are often treated empirically for chronic obstructive pulmonary disease (COPD).
Rales are a common finding on auscultation in IPF, and this can lead to an exhaustive cardiac evaluation and empiric treatment for heart failure. Digital clubbing is also relatively common.14 Hypoxemia with exertion is another common feature that also often correlates with disease severity and prognosis. Resting hypoxemia is more common in advanced disease.
On spirometry, patients with IPF typically demonstrate restrictive physiology, suggested by a normal or elevated ratio of the forced expiratory volume in 1 second to the forced vital capacity (FEV1/FVC) (> 70% predicted or above the lower limit of normal) combined with a lower than normal FVC. Restrictive physiology is definitively demonstrated by a decreased total lung capacity (< 80% predicted or below the lower limit of normal) on plethysmography. Impaired gas exchange, manifested by a decreased diffusing capacity of the lungs for carbon monoxide (DLCO) on pulmonary function testing, is also common. Because pulmonary perfusion is higher in the lung bases, where IPF is also predominant, the DLCO is often reduced to a greater extent than the FVC.
PROGNOSTIC INDICATORS

Clinicians typically view IPF as a relentless and progressive process, but its course is variable and can be uncertain in an individual patient (Figure 1).15,16 Nevertheless, over time, most patients have a decline in lung function leading to respiratory failure. Respiratory failure, often preceded by a subacute deterioration (over weeks to months) or an acute deterioration (< 4 weeks), is the most common cause of death, but comorbid diseases such as lung cancer, infection, and heart failure are also common causes of death in these patients.17,18
Predictors of mortality include worsening FVC, DLCO, symptoms, and physiologic impairment, manifested by a decline in the 6-minute walking test or worsening exertional hypoxemia.19–22 Other common comorbidities linked with impaired quality of life and poor prognosis include obstructive sleep apnea, gastroesophageal reflux disease, and depression.16,23 Retrospective studies suggest that most IPF patients die 2 to 5 years after symptom onset. With the lag from symptom onset to final diagnosis, the average life expectancy is as little as 2 years from the time of diagnosis.9,18,24,25
Two staging systems have been developed to predict short-term and long-term mortality risk based on sex, age, and physiologic parameters.23,24 The GAP (gender, age, physiology) index provides an estimate of the risk of death for a cohort of patients: a score of 0 to 8 is calculated, and the score is then categorized as stage I, II, or III. Each stage is associated with 1-, 2-, and 3-year mortality rates, with stage III having the highest rates. The GAP calculator (www.acponline.org/journals/annals/extras/gap) provides an estimate of the risk of death for an individual patient. The application of these tools for the management of IPF is evolving; however, they may be helpful for counseling patients about disease prognosis.
CLUES TO DIAGNOSIS
Histologic patterns
")
UIP on histologic study is also seen in fibrotic lung diseases other than IPF, including connective tissue disease-associated interstitial lung disease, inhalational or occupational interstitial lung disease, and chronic hypersensitivity pneumonitis.26–29 Consequently, the diagnosis of IPF requires exclusion of other known causes of UIP.
According to the 2011 guidelines,16 the histology of interstitial lung disease can be categorized as definite UIP, probable UIP, or possible UIP, or as an atypical pattern suggesting another diagnosis. If no definite cause of the interstitial lung abnormality is found, the level of certainty of the histopathologic pattern of UIP helps formulate the clinical diagnosis and management plan.
Clues on computed tomography
The UIP nomenclature also describes patterns on high-resolution computed tomography (HRCT). HRCT is done without contrast and produces thin-sliced images (usually < 1.5 mm) in inspiratory, expiratory, and prone views; this allows detection of air trapping, which may indicate an airway-centric alternative diagnosis.
On HRCT, UIP appears as reticular opacities, often with traction bronchiectasis or bronchiolectasis, usually with a basilar and peripheral predominance. Honeycombing is a key feature and appears as clustered cystic spaces with well-defined walls in the periphery of the lung parenchyma. Ground-glass opacities are not a prominent feature of UIP, and although they do not exclude a UIP pattern, they should spur consideration of other diagnoses.16 Reactive mediastinal and hilar lymphadenopathy is another common feature of UIP.

When evaluating results of HRCT for UIP, the radiologist categorizes the pattern as definite UIP, possible UIP, or inconsistent. The definite pattern meets all the above features and has none of the features suggesting an alternative diagnosis (Figure 3). The possible pattern includes all the above features with the exception of honeycombing. If the predominant features on HRCT include any atypical finding listed above, then the study is considered inconsistent with UIP. If the pattern on HRCT is considered definite, evaluation of pathology is not necessary. If the pattern is categorized as possible or is inconsistent, then surgical lung biopsy-confirmed UIP is necessary for the definitive diagnosis of IPF.
However, evidence is emerging that in the correct clinical scenario, possible UIP behaves similarly to definite UIP and may be sufficient to make the clinical diagnosis of IPF even without surgical biopsy confirmation.30
A DIAGNOSTIC ALGORITHM FOR IPF
Given the multitude of interstitial lung diseases, their complexities, and the lack of a gold standard definitive diagnostic test, the diagnosis of IPF can be difficult, requiring the integration of clinical, radiologic, and, if necessary, pathologic findings.
")
Demographic features
The patient’s demographic features and risk factors dictate the initial clinical suspicion of IPF compared with other interstitial lung diseases. The incidence of IPF increases with age, and IPF is more common in men. A history of smoking is another risk factor.31 A 45-year-old never-smoker is much less likely to have IPF than a 70-year-old former smoker, and a 70-year-old man is more likely to have IPF than a woman of the same age. Thus, the finding of interstitial lung disease in a patient with a demographic profile that is not typical (ie, a younger woman who never smoked) should prompt an exhaustive investigation for another diagnosis such as hypersensitivity pneumonitis or connective tissue disease.
Key elements of the history
After considering the demographic profile and risk factors, the next step in the evaluation is a thorough and accurate medical history. This should include assessment of the severity of dyspnea and cough, signs and symptoms of connective tissue disease (eg, arthralgias, sicca symptoms, Raynaud phenomenon, difficulty swallowing), and gastroesophageal reflux disease, which can be associated with connective tissue disease and, independently, with IPF.
It is also important to identify any environmental exposures that suggest pneumoconiosis or chronic hypersensitivity pneumonitis. The most common risk factors for hypersensitivity pneumonitis are birds and bird feathers, molds, fungi, hot tub use, and some industrial chemicals.32
A medication history is important. Many medications are associated with interstitial lung disease, but amiodarone, bleomycin, methotrexate, and nitrofurantoin are among the common offenders.33
A thorough family history is necessary, as there are familial forms of IPF.
Focus of the physical examination
The physical examination must include careful auscultation for rales. While rales are not specific for IPF, they are the most common pulmonary abnormality. Detailed skin, musculoskeletal, and cardiovascular examinations are also important to evaluate for rheumatologic signs, clubbing, or evidence of heart failure or pulmonary hypertension.
Laboratory tests
Laboratory testing should include a serologic autoantibody panel to evaluate for connective tissue diseases that can manifest as interstitial lung disease, including rheumatoid arthritis, dermatopolymyositis, scleroderma, Sjögren syndrome, and undifferentiated or mixed connective tissue disease. Typical preliminary laboratory tests are antinuclear antibody, rheumatoid factor, erythrocyte sedimentation rate, and C-reactive protein. Others may include anticyclic citrullinated peptide (anti-CCP), anti-Scl-70, anti-RNP, anti-SS-A, anti-SS-B, and anti-Jo-1.16 The breadth of the panel should depend on patient demographics and findings in the history or physical examination that increase or decrease the likelihood of a connective tissue disease.
Lung function testing
Assessing the patient’s pulmonary physiology should include spirometry, DLCO, and body plethysmography (lung volumes). In most cases, IPF manifests with restrictive physiology. Once restrictive physiology is confirmed with a low total lung capacity, FVC testing can be used as a longitudinal surrogate, as it is less expensive and easier for the patient to perform. In general, a lower total lung capacity or FVC indicates more severe impairment.
The DLCO serves as another marker of severity but is less reliable due to baseline variability and difficulties performing the maneuver.
A 6-minute walk test is another crucial physiologic assessment tool that can quantify exertional hypoxemia and functional status (ie, distance walked), and can assist in prognosis.
Imaging
Most patients undergo chest radiography in the workup for undiagnosed dyspnea. However, chest radiography is not adequate to formulate an accurate diagnosis in suspected interstitial lung disease, and a normal radiograph cannot exclude changes that might reflect early phases of the disease. As the disease progresses, the plain radiograph can show reticulonodular opacities and honeycombing in the peripheral and lower lung zones (Figure 3).34
The decision whether to order HRCT in the workup for a patient who has dyspnea and a normal chest radiograph is challenging. We recommend cross-sectional imaging when physiologic testing shows restriction or low DLCO, or when there is a high index of suspicion for underlying lung disease as the cause of symptoms.
Expert consultation can aid with this decision, especially when the underlying cause of dyspnea remains unclear after initial studies have been completed. Otherwise, HRCT is an essential test in the evaluation of interstitial lung disease.
Bronchoscopy’s role controversial
If the pattern on HRCT is nondiagnostic, then surgical biopsy is necessary, and the diagnosis of IPF requires a histologic pattern of UIP as described above.16,35
Although bronchoscopy can be valuable if an alternative diagnosis such as sarcoidosis or chronic hypersensitivity pneumonitis is suspected, the role of bronchoscopic biopsy in the workup of IPF is controversial. The patchy nature of UIP does not lend itself to the relatively small biopsy samples obtained through bronchoscopy.36,37
Surgical biopsy options
The favored biopsy approach is surgical, using either an open or a video-assisted thoracoscopic technique. The latter is preferred as it is less invasive, requires a shorter length of hospital stay, and allows a faster recovery.38 Bronchoscopic cryobiopsy, currently under investigation, is a potentially valuable tool whose role in diagnosing IPF is evolving.
Frequently, neither HRCT nor surgical lung biopsy demonstrates UIP, making the definitive diagnosis of IPF difficult. Moreover, some patients with nondiagnostic HRCT results are unable to tolerate surgical lung biopsy because of severely impaired lung function or other comorbidities.
The role of multidisciplinary discussions
When surgical lung biopsy is not possible, current practice at leading centers uses a multidisciplinary approach to allow for a confident diagnosis.30,39 Discussions take place between pulmonologists, pathologists, radiologists, and other specialists to collectively consider all aspects of a case before rendering a consensus opinion on the diagnosis and the management plan. If the discussion leads to a consensus diagnosis of IPF, then the patient’s clinician can move forward with treatment options. If not, the group can recommend further workup or alternative diagnoses and treatment regimens. The multidisciplinary group is also well positioned to consider the relative risks and benefits of moving forward with surgical lung biopsy for individual patients.
This approach illustrates the importance of referring these patients to centers of excellence in diagnosing and managing complex cases of interstitial lung disease, including IPF.40
TREATMENT OF IPF
Antifibrotic therapy
Antifibrotic therapy is a choice between pirfenidone and nintedanib.
Pirfenidone, which has an undefined molecular target, was approved based on the results of 3 trials.41,42 Pooled analyses from these trials showed a reduction in the decline from baseline in FVC percent predicted and improved progression-free survival.43 Pooled and meta-analyses of pirfenidone clinical trials have shown a mortality benefit, although no individual study has shown such an effect on mortality rates.44
The major adverse effects of pirfenidone are gastrointestinal distress and photosensitivity rash.
Nintedanib is a triple tyrosine kinase inhibitor that broadly targets fibroblast growth factor, vascular endothelial growth factor, and platelet-derived growth factor receptors. Combined analysis of 2 concurrent trials45 showed that nintedanib reduced the decline in FVC, similarly to pirfenidone. The major adverse event associated with nintedanib was diarrhea. Since it inhibits vascular endothelial growth factor, there is a risk of hematologic complications such as bleeding or clotting events.
Because pirfenidone and nintedanib can increase aminotransferase levels, regular monitoring is recommended.
To date, no trial has compared pirfenidone and nintedanib in terms of their efficacy and tolerability. Therefore, the choice of agent is based on the patient’s preference after a discussion of potential risks and expected benefits, a review of each drug’s side effects, and consideration of comorbid conditions and physician experience.
Patients need to understand that these drugs slow the rate of decline in FVC but have not been shown to improve symptoms or functional status.
Corticosteroids are not routine
Corticosteroids should not be used routinely in the treatment of IPF. Although steroids, alone or in combination with other immunosuppressive medications, were commonly used for IPF in the past, such use was not based on results of randomized controlled trials.46 Retrospective controlled studies have failed to show that corticosteroids improve mortality rates in IPF; indeed, they have shown that corticosteroids confer substantial morbidity.47,48 In addition, a randomized controlled trial combining corticosteroids with N-acetylcysteine and azathioprine was stopped early due to an increased risk of death and hospitalization.49 Collectively, these data suggest that corticosteroids confer no benefit and are potentially harmful. Their use in IPF is discouraged, and the joint international guidelines recommend against immunosuppression to treat IPF.16
Other treatments
The guidelines offer additional suggestions for the management of IPF.
Preliminary evidence suggests that microaspiration associated with abnormal gastroesophageal acid reflux is a risk factor for IPF. As such, there is a weak recommendation for aggressive treatment of reflux disease.50 However, because evidence suggests that proton-pump inhibitor therapy may be associated with adverse renal or central nervous system effects, this recommendation bears caution. It is hoped that ongoing studies will provide further insight into the role of acid-suppression in the management of IPF.51,52
Further treatment recommendations include best supportive management such as supplemental oxygen, pulmonary rehabilitation, and vaccinations.

Prompt referral for lung transplant is imperative. IPF is now the most common indication for lung transplant, and given the poor overall prognosis of advanced IPF, transplant confers a survival benefit in appropriately selected patients.53,54 Table 2 provides an evidence-based checklist for the workup and management of IPF.
ACUTE EXACERBATIONS OF IPF
The unpredictable nature of IPF can manifest in the form of acute exacerbations without an identifiable cause. The loosely defined diagnostic criteria for the diagnosis of acute exacerbations are a previous or new diagnosis of IPF, worsening or development of dyspnea in the last 30 days, and new bilateral ground-glass or consolidative changes with a background of UIP on HRCT.16
A new definition has been proposed55 to facilitate research in the characterization and treatment of acute exacerbations of IPF. The new definition includes all causes of respiratory deterioration except for heart failure and volume overload. It is less strict about the 30-day time frame. This newer definition is based on the lack of evidence differentiating outcomes when an acute deterioration is associated with known or unknown etiologies.55
The incidence of acute exacerbations is variable, with a 1- and 3-year incidence ranging between 8.6% and 23.9% depending on the criteria used.56 In general, acute exacerbations carry a grim prognosis, with a median life expectancy of 2.2 months.57
There is no approved therapy for exacerbations of IPF. Rather, treatment is mainly supportive with supplemental oxygen and mechanical ventilation. Current guidelines have a weak recommendation for the use of corticosteroids, but there are no recommendations regarding dose, route, or duration of therapy. Other treatments, primarily immunomodulatory agents, have been suggested but lack evidence of benefit.
Acknowledgments: Pathology images were provided by Carol Farver, MD, Pathology Institute, Cleveland Clinic. Radiology images were provided by Ruchi Yadav, MD, Imaging Institute, Cleveland Clinic.
- Brown KK, Raghu G. Medical treatment for pulmonary fibrosis: current trends, concepts, and prospects. Clin Chest Med 2004; 25(4):759–772, vii. doi:10.1016/j.ccm.2004.08.003
- Ryerson CJ, Collard HR. Update on the diagnosis and classification of ILD. Curr Opin Pulm Med 2013; 19(5):453–459. doi:10.1097/MCP.0b013e328363f48d
- Raghu G, Chen SY, Yeh WS, et al. Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001-11. Lancet Respir Med 2014; 2(7):566–572. doi:10.1016/S2213-2600(14)70101-8
- Nalysnyk L, Cid-Ruzafa J, Rotella P, Esser D. Incidence and prevalence of idiopathic pulmonary fibrosis: review of the literature. Eur Respir Rev 2012; 21(126):355–361. doi:10.1183/09059180.00002512
- Hutchinson J, Fogarty A, Hubbard R, McKeever T. Global incidence and mortality of idiopathic pulmonary fibrosis: a systematic review. Eur Respir J 2015; 46(3):795–806. doi:10.1183/09031936.00185114
- National Lung Screening Trial Research Team; Aberle DR, Adams AM, Berg CD, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med 2011; 365(5):395–409. doi:10.1056/NEJMoa1102873
- Jin GY, Lynch D, Chawla A, et al. Interstitial lung abnormalities in a CT lung cancer screening population: prevalence and progression rate. Radiology 2013; 268(2):563–571. doi:10.1148/radiol.13120816
- Southern BD, Scheraga RG, Yadav R. Managing interstitial lung disease detected on CT during lung cancer screening. Cleve Clin J Med 2016; 83(1):55–65. doi:10.3949/ccjm.83a.14157
- King TE Jr, Schwarz MI, Brown K, et al. Idiopathic pulmonary fibrosis: relationship between histopathologic features and mortality. Am J Respir Crit Care Med 2001; 164(5):1025–1032. doi:10.1164/ajrccm.164.6.2001056
- Lamas DJ, Kawut SM, Bagiella E, et al. Delayed access and survival in idiopathic pulmonary fibrosis: a cohort study. Am J Respir Crit Care Med 2011; 184(7):842–847. doi:10.1164/rccm.201104-0668OC
- Jo HE, Glaspole I, Moodley Y, et al. Disease progression in idiopathic pulmonary fibrosis with mild physiological impairment: analysis from the Australian IPF registry. BMC Pulm Med 2018; 18(1):19. doi:10.1186/s12890-018-0575-y
- Yang IV, Schwartz DA. Epigenetics of idiopathic pulmonary fibrosis. Transl Res 2015; 165(1):48–60. doi:10.1016/j.trsl.2014.03.011
- King TE Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet 2011; 378(9807):1949–1961. doi:10.1016/S0140-6736(11)60052-4
- Meltzer EB, Noble PW. Idiopathic pulmonary fibrosis. Orphanet J Rare Dis 2008; 3:8. doi:10.1186/1750-1172-3-8
- Raghu G. Idiopathic pulmonary fibrosis. A rational clinical approach. Chest 1987; 92(1):148–154. doi:10.1378/chest.92.1.148
- Raghu G, Collard HR, Egan JJ, et al; ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183(6):788–824. doi:10.1164/rccm.2009-040GL
- Panos RJ, Mortenson RL, Niccoli SA, King TE Jr. Clinical deterioration in patients with idiopathic pulmonary fibrosis: causes and assessment. Am J Med 1990; 88(4):396–404. doi:10.1016/0002-9343(90)90495-Y
- Ley B, Collard HR, King TE Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011; 183(4):431–440. doi:10.1164/rccm.201006-0894CI
- Collard HR, King TE Jr, Bartelson BB, Vourlekis JS, Schwarz MI, Brown KK. Changes in clinical and physiologic variables predict survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2003; 168(5):538–542. doi:10.1164/rccm.200211-1311OC
- Flaherty KR, Andrei AC, Murray S, et al. Idiopathic pulmonary fibrosis: prognostic value of changes in physiology and six-minute-walk test. Am J Respir Crit Care Med 2006; 174(7):803–809. doi:10.1164/rccm.200604-488OC
- Jegal Y, Kim DS, Shim TS, et al. Physiology is a stronger predictor of survival than pathology in fibrotic interstitial pneumonia. Am J Respir Crit Care Med 2005; 171(6):639–644. doi:10.1164/rccm.200403-331OC
- Latsi PI, du Bois RM, Nicholson AG, et al. Fibrotic idiopathic interstitial pneumonia: the prognostic value of longitudinal functional trends. Am J Respir Crit Care Med 2003; 168(5):531–537. doi:10.1164/rccm.200210-1245OC
- King CS, Nathan SD. Idiopathic pulmonary fibrosis: effects and optimal management of comorbidities. Lancet Respir Med 2017; 5(1):72–84. doi:10.1016/S2213-2600(16)30222-3
- Ley B, Ryerson CJ, Vittinghoff E, et al. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med 2012; 156(1):684–691. doi:10.7326/0003-4819-156-10-201205150-00004
- Rudd RM, Prescott RJ, Chalmers JC, Johnston ID; Fibrosing Alveolitis Subcommittee of the Research Committee of the British Thoracic Society. British Thoracic Society study on cryptogenic fibrosing alveolitis: response to treatment and survival. Thorax 2007; 62(1):62–66. doi:10.1136/thx.2005.045591
- Gutsche M, Rosen GD, Swigris JJ. Connective tissue disease-associated interstitial lung disease: a review. Curr Respir Care Rep 2012; 1:224–232. doi:10.1007/s13665-012-0028-7
- Park JH, Kim DS, Park IN, et al. Prognosis of fibrotic interstitial pneumonia: idiopathic versus collagen vascular disease-related subtypes. Am J Respir Crit Care Med 2007; 175(7):705–711. doi:10.1164/rccm.200607-912OC
- Taskar VS, Coultas DB. Is idiopathic pulmonary fibrosis an environmental disease? Proc Am Thorac Soc 2006; 3(4):293–298. doi:10.1513/pats.200512-131TK
- Vourlekis JS, Schwarz MI, Cherniack RM, et al. The effect of pulmonary fibrosis on survival in patients with hypersensitivity pneumonitis. Am J Med 2004; 116(10):662–668. doi:10.1016/j.amjmed.2003.12.030
- Brownell R, Moua T, Henry TS, et al. The use of pretest probability increases the value of high-resolution CT in diagnosing usual interstitial pneumonia. Thorax 2017; 72(5):424–429. doi:10.1136/thoraxjnl-2016-209671
- Baumgartner KB, Samet JM, Stidley CA, Colby TV, Waldron JA. Cigarette smoking: a risk factor for idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 1997; 155(1):242–248. doi:10.1164/ajrccm.155.1.9001319
- Selman M, Pardo A, King TE Jr. Hypersensitivity pneumonitis: insights in diagnosis and pathobiology. Am J Respir Crit Care Med 2012; 186(4):314–324. doi:10.1164/rccm.201203-0513CI
- Schwaiblmair M, Behr W, Haeckel T, Markl B, Foerg W, Berghaus T. Drug induced interstitial lung disease. Open Respir Med J 2012; 6:63–74. doi:10.2174/1874306401206010063
- Grenier P, Valeyre D, Cluzel P, Brauner MW, Lenoir S, Chastang C. Chronic diffuse interstitial lung disease: diagnostic value of chest radiography and high-resolution CT. Radiology 1991; 179(1):123–132. doi:10.1148/radiology.179.1.2006262
- Lynch JP 3rd, Huynh RH, Fishbein MC, Saggar R, Belperio JA, Weigt SS. Idiopathic pulmonary fibrosis: epidemiology, clinical features, prognosis, and management. Semin Respir Crit Care Med 2016; 37(3):331–357. doi:10.1055/s-0036-1582011
- Berbescu EA, Katzenstein AL, Snow JL, Zisman DA. Transbronchial biopsy in usual interstitial pneumonia. Chest 2006; 129(5):1126–1131. doi:10.1378/chest.129.5.1126
- Ohshimo S, Bonella F, Cui A, et al. Significance of bronchoalveolar lavage for the diagnosis of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2009; 179(11):1043–1047. doi:10.1164/rccm.200808-1313OC
- Oparka J, Yan TD, Ryan E, Dunning J. Does video-assisted thoracic surgery provide a safe alternative to conventional techniques in patients with limited pulmonary function who are otherwise suitable for lung resection? Interact Cardiovasc Thorac Surg 2013; 17(1):159–162. doi:10.1093/icvts/ivt097
- Flaherty KR, King TE Jr, Raghu G, et al. Idiopathic interstitial pneumonia: what is the effect of a multidisciplinary approach to diagnosis? Am J Respir Crit Care Med 2004; 170(8):904–910. doi:10.1164/rccm.200402-147OC
- Walsh SL, Wells AU, Desai SR, et al. Multicentre evaluation of multidisciplinary team meeting agreement on diagnosis in diffuse parenchymal lung disease: a case-cohort study. Lancet Respir Med 2016; 4(7):557–565. doi:10.1016/S2213-2600(16)30033-9
- King TE Jr, Bradford WZ, Castro-Bernardini S, et al; ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22):2083–2092. doi:10.1056/NEJMoa1402582
- Noble PW, Albera C, Bradford WZ, et al; CAPACITY Study Group. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet 2011; 377(9779):1760–1769. doi:10.1016/S0140-6736(11)60405-4
- Noble PW, Albera C, Bradford WZ, et al. Pirfenidone for idiopathic pulmonary fibrosis: analysis of pooled data from three multinational phase 3 trials. Eur Respir J 2016; 47(1):243–253. doi:10.1183/13993003.00026-2015
- Nathan SD, Albera C, Bradford WZ, et al. Effect of pirfenidone on mortality: pooled analyses and meta-analyses of clinical trials in idiopathic pulmonary fibrosis. Lancet Respir Med 2017; 5(1):33–41. doi:10.1016/S2213-2600(16)30326-5
- Richeldi L, du Bois RM, Raghu G, et al; INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22):2071–2082. doi:10.1056/NEJMoa1402584
- Richeldi L, Davies HR, Ferrara G, Franco F. Corticosteroids for idiopathic pulmonary fibrosis. Cochrane Database Syst Rev 2003: 3:CD002880. doi:10.1002/14651858.CD002880
- Douglas WW, Ryu JH, Schroeder DR. Idiopathic pulmonary fibrosis: impact of oxygen and colchicine, prednisone, or no therapy on survival. Am J Respir Crit Care Med 2000; 161(4 pt 1):1172–1178. doi:10.1164/ajrccm.161.4.9907002
- Gay SE, Kazerooni EA, Toews GB, et al. Idiopathic pulmonary fibrosis: predicting response to therapy and survival. Am J Respir Crit Care Med 1998; 157(4 pt 1):1063–1072. doi:10.1164/ajrccm.157.4.9703022
- Idiopathic Pulmonary Fibrosis Clinical Research Network; Raghu G, Anstrom KJ, King TE Jr, Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med 2012; 366(21):1968–1977. doi:10.1056/NEJMoa1113354
- Raghu G, Freudenberger TD, Yang S, et al. High prevalence of abnormal acid gastro-oesophageal reflux in idiopathic pulmonary fibrosis. Eur Respir J 2006; 27(1):136–142. doi:10.1183/09031936.06.00037005
- Gomm W, von Holt K, Thome F, et al. Association of proton pump inhibitors with risk of dementia: a pharmacoepidemiological claims data analysis. JAMA Neurol 2016; 73(4):410–416. doi:10.1001/jamaneurol.2015.4791
- Xie Y, Bowe B, Li T, Xian H, Balasubramanian S, Al-Aly Z. Proton pump inhibitors and risk of incident CKD and progression to ESRD. J Am Soc Nephrol 2016; 27(10):3153–3163. doi:10.1681/ASN.2015121377
- Thabut G, Mal H, Castier Y, et al. Survival benefit of lung transplantation for patients with idiopathic pulmonary fibrosis. J Thorac Cardiovasc Surg 2003; 126(2):469–475. doi:10.1016/S0022-5223(03)00600-7
- Valapour M, Skeans MA, Smith JM, et al. Lung. Am J Transplant 2016; 16(suppl 2):141–168. doi:10.1111/ajt.13671
- Collard HR, Ryerson CJ, Corte TJ, et al. Acute exacerbation of idiopathic pulmonary fibrosis. An International Working Group Report. Am J Respir Crit Care Med 2016; 194(3):265–275. doi:10.1164/rccm.201604-0801CI
- Kondoh Y, Taniguchi H, Katsuta T, et al. Risk factors of acute exacerbation of idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis 2010; 27(2):103–110. doi:10.1016/j.resinv.2015.04.005
- Song JW, Hong SB, Lim CM, Koh Y, Kim DS. Acute exacerbation of idiopathic pulmonary fibrosis: incidence, risk factors and outcome. Eur Respir J 2011; 37(2):356–363. doi:10.1183/09031936.00159709
Idiopathic pulmonary fibrosis (IPF) is a devastating and fatal lung disease that generally affects older adults. It is characterized by a radiographic and histopathologic pattern of usual interstitial pneumonia (UIP) that has no other known etiology.
Accurate diagnosis of IPF is crucial. We recommend early referral to a center specializing in interstitial lung disease to confirm the diagnosis, start appropriate therapy, advise the patient on prognosis and enrollment in disease registries and clinical trials, and determine candidacy for lung transplant.
Primary care physicians are uniquely positioned to encounter patients with IPF, whether because of a patient complaint or as an incidental finding on computed tomography. The goal of this article is to delineate the features of IPF so that it may be recognized early and thus expedite referral to a center with expertise in interstitial lung disease for a thorough evaluation and appropriate management.
WHAT IS IDIOPATHIC PULMONARY FIBROSIS?
MORE COMMON THAN ONCE THOUGHT
The true incidence and prevalence of IPF are difficult to assess. IPF is generally considered a rare disease, but it is more common than once thought. In 2011, Raghu et al3 estimated the prevalence in Medicare beneficiaries to be 495 cases per 100,000. Based on this estimate and the current US population, up to 160,000 Americans could have IPF.4 Raghu et al also showed that IPF more often affects adults over age 65, which suggests that as the US population ages, the incidence of IPF may rise. Studies have also reported an increased incidence of IPF worldwide.5
Further, with the rising use of low-dose computed tomography to screen for lung cancer, more incidental cases of IPF will likely be found.6–8
Older data showed a lag from symptom onset to accurate diagnosis of 1 to 2 years.9 A more recent study found a lag in referral of patients with IPF to tertiary care centers, and this delay was associated with a higher rate of death independent of disease severity.10
TYPICALLY PROGRESSIVE, OFTEN FATAL
IPF is typically progressive and limited to the lungs, and it portends a poor prognosis. The median survival is commonly cited as 2 to 5 years from diagnosis, although this is based on older observations that may not reflect current best practice and newer therapies. More recent studies suggest higher survival rates if patients have preserved lung function.11
As the name indicates, the etiology of IPF is unknown, but studies have indicated genetic underpinnings in a notable proportion of cases.12 Regardless of the cause, the pathogenesis and progression of IPF are thought to be the result of an abnormal and persistent wound-repair response. The progressive deposition of scar tissue disrupts normal lung architecture and function, eventually causing clinical disease.13
SYMPTOMS AND KEY FEATURES
Patients with IPF typically present with the insidious onset of dyspnea on exertion, with or without chronic cough. Risk factors include male sex, increasing age, and a history of smoking. Patients with undiagnosed IPF who present with dyspnea and a history of smoking are often treated empirically for chronic obstructive pulmonary disease (COPD).
Rales are a common finding on auscultation in IPF, and this can lead to an exhaustive cardiac evaluation and empiric treatment for heart failure. Digital clubbing is also relatively common.14 Hypoxemia with exertion is another common feature that also often correlates with disease severity and prognosis. Resting hypoxemia is more common in advanced disease.
On spirometry, patients with IPF typically demonstrate restrictive physiology, suggested by a normal or elevated ratio of the forced expiratory volume in 1 second to the forced vital capacity (FEV1/FVC) (> 70% predicted or above the lower limit of normal) combined with a lower than normal FVC. Restrictive physiology is definitively demonstrated by a decreased total lung capacity (< 80% predicted or below the lower limit of normal) on plethysmography. Impaired gas exchange, manifested by a decreased diffusing capacity of the lungs for carbon monoxide (DLCO) on pulmonary function testing, is also common. Because pulmonary perfusion is higher in the lung bases, where IPF is also predominant, the DLCO is often reduced to a greater extent than the FVC.
PROGNOSTIC INDICATORS
Clinicians typically view IPF as a relentless and progressive process, but its course is variable and can be uncertain in an individual patient (Figure 1).15,16 Nevertheless, over time, most patients have a decline in lung function leading to respiratory failure. Respiratory failure, often preceded by a subacute deterioration (over weeks to months) or an acute deterioration (< 4 weeks), is the most common cause of death, but comorbid diseases such as lung cancer, infection, and heart failure are also common causes of death in these patients.17,18
Predictors of mortality include worsening FVC, DLCO, symptoms, and physiologic impairment, manifested by a decline in the 6-minute walking test or worsening exertional hypoxemia.19–22 Other common comorbidities linked with impaired quality of life and poor prognosis include obstructive sleep apnea, gastroesophageal reflux disease, and depression.16,23 Retrospective studies suggest that most IPF patients die 2 to 5 years after symptom onset. With the lag from symptom onset to final diagnosis, the average life expectancy is as little as 2 years from the time of diagnosis.9,18,24,25
Two staging systems have been developed to predict short-term and long-term mortality risk based on sex, age, and physiologic parameters.23,24 The GAP (gender, age, physiology) index provides an estimate of the risk of death for a cohort of patients: a score of 0 to 8 is calculated, and the score is then categorized as stage I, II, or III. Each stage is associated with 1-, 2-, and 3-year mortality rates, with stage III having the highest rates. The GAP calculator (www.acponline.org/journals/annals/extras/gap) provides an estimate of the risk of death for an individual patient. The application of these tools for the management of IPF is evolving; however, they may be helpful for counseling patients about disease prognosis.
CLUES TO DIAGNOSIS
Histologic patterns
UIP on histologic study is also seen in fibrotic lung diseases other than IPF, including connective tissue disease-associated interstitial lung disease, inhalational or occupational interstitial lung disease, and chronic hypersensitivity pneumonitis.26–29 Consequently, the diagnosis of IPF requires exclusion of other known causes of UIP.
According to the 2011 guidelines,16 the histology of interstitial lung disease can be categorized as definite UIP, probable UIP, or possible UIP, or as an atypical pattern suggesting another diagnosis. If no definite cause of the interstitial lung abnormality is found, the level of certainty of the histopathologic pattern of UIP helps formulate the clinical diagnosis and management plan.
Clues on computed tomography
The UIP nomenclature also describes patterns on high-resolution computed tomography (HRCT). HRCT is done without contrast and produces thin-sliced images (usually < 1.5 mm) in inspiratory, expiratory, and prone views; this allows detection of air trapping, which may indicate an airway-centric alternative diagnosis.
On HRCT, UIP appears as reticular opacities, often with traction bronchiectasis or bronchiolectasis, usually with a basilar and peripheral predominance. Honeycombing is a key feature and appears as clustered cystic spaces with well-defined walls in the periphery of the lung parenchyma. Ground-glass opacities are not a prominent feature of UIP, and although they do not exclude a UIP pattern, they should spur consideration of other diagnoses.16 Reactive mediastinal and hilar lymphadenopathy is another common feature of UIP.
When evaluating results of HRCT for UIP, the radiologist categorizes the pattern as definite UIP, possible UIP, or inconsistent. The definite pattern meets all the above features and has none of the features suggesting an alternative diagnosis (Figure 3). The possible pattern includes all the above features with the exception of honeycombing. If the predominant features on HRCT include any atypical finding listed above, then the study is considered inconsistent with UIP. If the pattern on HRCT is considered definite, evaluation of pathology is not necessary. If the pattern is categorized as possible or is inconsistent, then surgical lung biopsy-confirmed UIP is necessary for the definitive diagnosis of IPF.
However, evidence is emerging that in the correct clinical scenario, possible UIP behaves similarly to definite UIP and may be sufficient to make the clinical diagnosis of IPF even without surgical biopsy confirmation.30
A DIAGNOSTIC ALGORITHM FOR IPF
Given the multitude of interstitial lung diseases, their complexities, and the lack of a gold standard definitive diagnostic test, the diagnosis of IPF can be difficult, requiring the integration of clinical, radiologic, and, if necessary, pathologic findings.
Demographic features
The patient’s demographic features and risk factors dictate the initial clinical suspicion of IPF compared with other interstitial lung diseases. The incidence of IPF increases with age, and IPF is more common in men. A history of smoking is another risk factor.31 A 45-year-old never-smoker is much less likely to have IPF than a 70-year-old former smoker, and a 70-year-old man is more likely to have IPF than a woman of the same age. Thus, the finding of interstitial lung disease in a patient with a demographic profile that is not typical (ie, a younger woman who never smoked) should prompt an exhaustive investigation for another diagnosis such as hypersensitivity pneumonitis or connective tissue disease.
Key elements of the history
After considering the demographic profile and risk factors, the next step in the evaluation is a thorough and accurate medical history. This should include assessment of the severity of dyspnea and cough, signs and symptoms of connective tissue disease (eg, arthralgias, sicca symptoms, Raynaud phenomenon, difficulty swallowing), and gastroesophageal reflux disease, which can be associated with connective tissue disease and, independently, with IPF.
It is also important to identify any environmental exposures that suggest pneumoconiosis or chronic hypersensitivity pneumonitis. The most common risk factors for hypersensitivity pneumonitis are birds and bird feathers, molds, fungi, hot tub use, and some industrial chemicals.32
A medication history is important. Many medications are associated with interstitial lung disease, but amiodarone, bleomycin, methotrexate, and nitrofurantoin are among the common offenders.33
A thorough family history is necessary, as there are familial forms of IPF.
Focus of the physical examination
The physical examination must include careful auscultation for rales. While rales are not specific for IPF, they are the most common pulmonary abnormality. Detailed skin, musculoskeletal, and cardiovascular examinations are also important to evaluate for rheumatologic signs, clubbing, or evidence of heart failure or pulmonary hypertension.
Laboratory tests
Laboratory testing should include a serologic autoantibody panel to evaluate for connective tissue diseases that can manifest as interstitial lung disease, including rheumatoid arthritis, dermatopolymyositis, scleroderma, Sjögren syndrome, and undifferentiated or mixed connective tissue disease. Typical preliminary laboratory tests are antinuclear antibody, rheumatoid factor, erythrocyte sedimentation rate, and C-reactive protein. Others may include anticyclic citrullinated peptide (anti-CCP), anti-Scl-70, anti-RNP, anti-SS-A, anti-SS-B, and anti-Jo-1.16 The breadth of the panel should depend on patient demographics and findings in the history or physical examination that increase or decrease the likelihood of a connective tissue disease.
Lung function testing
Assessing the patient’s pulmonary physiology should include spirometry, DLCO, and body plethysmography (lung volumes). In most cases, IPF manifests with restrictive physiology. Once restrictive physiology is confirmed with a low total lung capacity, FVC testing can be used as a longitudinal surrogate, as it is less expensive and easier for the patient to perform. In general, a lower total lung capacity or FVC indicates more severe impairment.
The DLCO serves as another marker of severity but is less reliable due to baseline variability and difficulties performing the maneuver.
A 6-minute walk test is another crucial physiologic assessment tool that can quantify exertional hypoxemia and functional status (ie, distance walked), and can assist in prognosis.
Imaging
Most patients undergo chest radiography in the workup for undiagnosed dyspnea. However, chest radiography is not adequate to formulate an accurate diagnosis in suspected interstitial lung disease, and a normal radiograph cannot exclude changes that might reflect early phases of the disease. As the disease progresses, the plain radiograph can show reticulonodular opacities and honeycombing in the peripheral and lower lung zones (Figure 3).34
The decision whether to order HRCT in the workup for a patient who has dyspnea and a normal chest radiograph is challenging. We recommend cross-sectional imaging when physiologic testing shows restriction or low DLCO, or when there is a high index of suspicion for underlying lung disease as the cause of symptoms.
Expert consultation can aid with this decision, especially when the underlying cause of dyspnea remains unclear after initial studies have been completed. Otherwise, HRCT is an essential test in the evaluation of interstitial lung disease.
Bronchoscopy’s role controversial
If the pattern on HRCT is nondiagnostic, then surgical biopsy is necessary, and the diagnosis of IPF requires a histologic pattern of UIP as described above.16,35
Although bronchoscopy can be valuable if an alternative diagnosis such as sarcoidosis or chronic hypersensitivity pneumonitis is suspected, the role of bronchoscopic biopsy in the workup of IPF is controversial. The patchy nature of UIP does not lend itself to the relatively small biopsy samples obtained through bronchoscopy.36,37
Surgical biopsy options
The favored biopsy approach is surgical, using either an open or a video-assisted thoracoscopic technique. The latter is preferred as it is less invasive, requires a shorter length of hospital stay, and allows a faster recovery.38 Bronchoscopic cryobiopsy, currently under investigation, is a potentially valuable tool whose role in diagnosing IPF is evolving.
Frequently, neither HRCT nor surgical lung biopsy demonstrates UIP, making the definitive diagnosis of IPF difficult. Moreover, some patients with nondiagnostic HRCT results are unable to tolerate surgical lung biopsy because of severely impaired lung function or other comorbidities.
The role of multidisciplinary discussions
When surgical lung biopsy is not possible, current practice at leading centers uses a multidisciplinary approach to allow for a confident diagnosis.30,39 Discussions take place between pulmonologists, pathologists, radiologists, and other specialists to collectively consider all aspects of a case before rendering a consensus opinion on the diagnosis and the management plan. If the discussion leads to a consensus diagnosis of IPF, then the patient’s clinician can move forward with treatment options. If not, the group can recommend further workup or alternative diagnoses and treatment regimens. The multidisciplinary group is also well positioned to consider the relative risks and benefits of moving forward with surgical lung biopsy for individual patients.
This approach illustrates the importance of referring these patients to centers of excellence in diagnosing and managing complex cases of interstitial lung disease, including IPF.40
TREATMENT OF IPF
Antifibrotic therapy
Antifibrotic therapy is a choice between pirfenidone and nintedanib.
Pirfenidone, which has an undefined molecular target, was approved based on the results of 3 trials.41,42 Pooled analyses from these trials showed a reduction in the decline from baseline in FVC percent predicted and improved progression-free survival.43 Pooled and meta-analyses of pirfenidone clinical trials have shown a mortality benefit, although no individual study has shown such an effect on mortality rates.44
The major adverse effects of pirfenidone are gastrointestinal distress and photosensitivity rash.
Nintedanib is a triple tyrosine kinase inhibitor that broadly targets fibroblast growth factor, vascular endothelial growth factor, and platelet-derived growth factor receptors. Combined analysis of 2 concurrent trials45 showed that nintedanib reduced the decline in FVC, similarly to pirfenidone. The major adverse event associated with nintedanib was diarrhea. Since it inhibits vascular endothelial growth factor, there is a risk of hematologic complications such as bleeding or clotting events.
Because pirfenidone and nintedanib can increase aminotransferase levels, regular monitoring is recommended.
To date, no trial has compared pirfenidone and nintedanib in terms of their efficacy and tolerability. Therefore, the choice of agent is based on the patient’s preference after a discussion of potential risks and expected benefits, a review of each drug’s side effects, and consideration of comorbid conditions and physician experience.
Patients need to understand that these drugs slow the rate of decline in FVC but have not been shown to improve symptoms or functional status.
Corticosteroids are not routine
Corticosteroids should not be used routinely in the treatment of IPF. Although steroids, alone or in combination with other immunosuppressive medications, were commonly used for IPF in the past, such use was not based on results of randomized controlled trials.46 Retrospective controlled studies have failed to show that corticosteroids improve mortality rates in IPF; indeed, they have shown that corticosteroids confer substantial morbidity.47,48 In addition, a randomized controlled trial combining corticosteroids with N-acetylcysteine and azathioprine was stopped early due to an increased risk of death and hospitalization.49 Collectively, these data suggest that corticosteroids confer no benefit and are potentially harmful. Their use in IPF is discouraged, and the joint international guidelines recommend against immunosuppression to treat IPF.16
Other treatments
The guidelines offer additional suggestions for the management of IPF.
Preliminary evidence suggests that microaspiration associated with abnormal gastroesophageal acid reflux is a risk factor for IPF. As such, there is a weak recommendation for aggressive treatment of reflux disease.50 However, because evidence suggests that proton-pump inhibitor therapy may be associated with adverse renal or central nervous system effects, this recommendation bears caution. It is hoped that ongoing studies will provide further insight into the role of acid-suppression in the management of IPF.51,52
Further treatment recommendations include best supportive management such as supplemental oxygen, pulmonary rehabilitation, and vaccinations.
Prompt referral for lung transplant is imperative. IPF is now the most common indication for lung transplant, and given the poor overall prognosis of advanced IPF, transplant confers a survival benefit in appropriately selected patients.53,54 Table 2 provides an evidence-based checklist for the workup and management of IPF.
ACUTE EXACERBATIONS OF IPF
The unpredictable nature of IPF can manifest in the form of acute exacerbations without an identifiable cause. The loosely defined diagnostic criteria for the diagnosis of acute exacerbations are a previous or new diagnosis of IPF, worsening or development of dyspnea in the last 30 days, and new bilateral ground-glass or consolidative changes with a background of UIP on HRCT.16
A new definition has been proposed55 to facilitate research in the characterization and treatment of acute exacerbations of IPF. The new definition includes all causes of respiratory deterioration except for heart failure and volume overload. It is less strict about the 30-day time frame. This newer definition is based on the lack of evidence differentiating outcomes when an acute deterioration is associated with known or unknown etiologies.55
The incidence of acute exacerbations is variable, with a 1- and 3-year incidence ranging between 8.6% and 23.9% depending on the criteria used.56 In general, acute exacerbations carry a grim prognosis, with a median life expectancy of 2.2 months.57
There is no approved therapy for exacerbations of IPF. Rather, treatment is mainly supportive with supplemental oxygen and mechanical ventilation. Current guidelines have a weak recommendation for the use of corticosteroids, but there are no recommendations regarding dose, route, or duration of therapy. Other treatments, primarily immunomodulatory agents, have been suggested but lack evidence of benefit.
Acknowledgments: Pathology images were provided by Carol Farver, MD, Pathology Institute, Cleveland Clinic. Radiology images were provided by Ruchi Yadav, MD, Imaging Institute, Cleveland Clinic.
Idiopathic pulmonary fibrosis (IPF) is a devastating and fatal lung disease that generally affects older adults. It is characterized by a radiographic and histopathologic pattern of usual interstitial pneumonia (UIP) that has no other known etiology.
Accurate diagnosis of IPF is crucial. We recommend early referral to a center specializing in interstitial lung disease to confirm the diagnosis, start appropriate therapy, advise the patient on prognosis and enrollment in disease registries and clinical trials, and determine candidacy for lung transplant.
Primary care physicians are uniquely positioned to encounter patients with IPF, whether because of a patient complaint or as an incidental finding on computed tomography. The goal of this article is to delineate the features of IPF so that it may be recognized early and thus expedite referral to a center with expertise in interstitial lung disease for a thorough evaluation and appropriate management.
WHAT IS IDIOPATHIC PULMONARY FIBROSIS?
MORE COMMON THAN ONCE THOUGHT
The true incidence and prevalence of IPF are difficult to assess. IPF is generally considered a rare disease, but it is more common than once thought. In 2011, Raghu et al3 estimated the prevalence in Medicare beneficiaries to be 495 cases per 100,000. Based on this estimate and the current US population, up to 160,000 Americans could have IPF.4 Raghu et al also showed that IPF more often affects adults over age 65, which suggests that as the US population ages, the incidence of IPF may rise. Studies have also reported an increased incidence of IPF worldwide.5
Further, with the rising use of low-dose computed tomography to screen for lung cancer, more incidental cases of IPF will likely be found.6–8
Older data showed a lag from symptom onset to accurate diagnosis of 1 to 2 years.9 A more recent study found a lag in referral of patients with IPF to tertiary care centers, and this delay was associated with a higher rate of death independent of disease severity.10
TYPICALLY PROGRESSIVE, OFTEN FATAL
IPF is typically progressive and limited to the lungs, and it portends a poor prognosis. The median survival is commonly cited as 2 to 5 years from diagnosis, although this is based on older observations that may not reflect current best practice and newer therapies. More recent studies suggest higher survival rates if patients have preserved lung function.11
As the name indicates, the etiology of IPF is unknown, but studies have indicated genetic underpinnings in a notable proportion of cases.12 Regardless of the cause, the pathogenesis and progression of IPF are thought to be the result of an abnormal and persistent wound-repair response. The progressive deposition of scar tissue disrupts normal lung architecture and function, eventually causing clinical disease.13
SYMPTOMS AND KEY FEATURES
Patients with IPF typically present with the insidious onset of dyspnea on exertion, with or without chronic cough. Risk factors include male sex, increasing age, and a history of smoking. Patients with undiagnosed IPF who present with dyspnea and a history of smoking are often treated empirically for chronic obstructive pulmonary disease (COPD).
Rales are a common finding on auscultation in IPF, and this can lead to an exhaustive cardiac evaluation and empiric treatment for heart failure. Digital clubbing is also relatively common.14 Hypoxemia with exertion is another common feature that also often correlates with disease severity and prognosis. Resting hypoxemia is more common in advanced disease.
On spirometry, patients with IPF typically demonstrate restrictive physiology, suggested by a normal or elevated ratio of the forced expiratory volume in 1 second to the forced vital capacity (FEV1/FVC) (> 70% predicted or above the lower limit of normal) combined with a lower than normal FVC. Restrictive physiology is definitively demonstrated by a decreased total lung capacity (< 80% predicted or below the lower limit of normal) on plethysmography. Impaired gas exchange, manifested by a decreased diffusing capacity of the lungs for carbon monoxide (DLCO) on pulmonary function testing, is also common. Because pulmonary perfusion is higher in the lung bases, where IPF is also predominant, the DLCO is often reduced to a greater extent than the FVC.
PROGNOSTIC INDICATORS
Clinicians typically view IPF as a relentless and progressive process, but its course is variable and can be uncertain in an individual patient (Figure 1).15,16 Nevertheless, over time, most patients have a decline in lung function leading to respiratory failure. Respiratory failure, often preceded by a subacute deterioration (over weeks to months) or an acute deterioration (< 4 weeks), is the most common cause of death, but comorbid diseases such as lung cancer, infection, and heart failure are also common causes of death in these patients.17,18
Predictors of mortality include worsening FVC, DLCO, symptoms, and physiologic impairment, manifested by a decline in the 6-minute walking test or worsening exertional hypoxemia.19–22 Other common comorbidities linked with impaired quality of life and poor prognosis include obstructive sleep apnea, gastroesophageal reflux disease, and depression.16,23 Retrospective studies suggest that most IPF patients die 2 to 5 years after symptom onset. With the lag from symptom onset to final diagnosis, the average life expectancy is as little as 2 years from the time of diagnosis.9,18,24,25
Two staging systems have been developed to predict short-term and long-term mortality risk based on sex, age, and physiologic parameters.23,24 The GAP (gender, age, physiology) index provides an estimate of the risk of death for a cohort of patients: a score of 0 to 8 is calculated, and the score is then categorized as stage I, II, or III. Each stage is associated with 1-, 2-, and 3-year mortality rates, with stage III having the highest rates. The GAP calculator (www.acponline.org/journals/annals/extras/gap) provides an estimate of the risk of death for an individual patient. The application of these tools for the management of IPF is evolving; however, they may be helpful for counseling patients about disease prognosis.
CLUES TO DIAGNOSIS
Histologic patterns
UIP on histologic study is also seen in fibrotic lung diseases other than IPF, including connective tissue disease-associated interstitial lung disease, inhalational or occupational interstitial lung disease, and chronic hypersensitivity pneumonitis.26–29 Consequently, the diagnosis of IPF requires exclusion of other known causes of UIP.
According to the 2011 guidelines,16 the histology of interstitial lung disease can be categorized as definite UIP, probable UIP, or possible UIP, or as an atypical pattern suggesting another diagnosis. If no definite cause of the interstitial lung abnormality is found, the level of certainty of the histopathologic pattern of UIP helps formulate the clinical diagnosis and management plan.
Clues on computed tomography
The UIP nomenclature also describes patterns on high-resolution computed tomography (HRCT). HRCT is done without contrast and produces thin-sliced images (usually < 1.5 mm) in inspiratory, expiratory, and prone views; this allows detection of air trapping, which may indicate an airway-centric alternative diagnosis.
On HRCT, UIP appears as reticular opacities, often with traction bronchiectasis or bronchiolectasis, usually with a basilar and peripheral predominance. Honeycombing is a key feature and appears as clustered cystic spaces with well-defined walls in the periphery of the lung parenchyma. Ground-glass opacities are not a prominent feature of UIP, and although they do not exclude a UIP pattern, they should spur consideration of other diagnoses.16 Reactive mediastinal and hilar lymphadenopathy is another common feature of UIP.
When evaluating results of HRCT for UIP, the radiologist categorizes the pattern as definite UIP, possible UIP, or inconsistent. The definite pattern meets all the above features and has none of the features suggesting an alternative diagnosis (Figure 3). The possible pattern includes all the above features with the exception of honeycombing. If the predominant features on HRCT include any atypical finding listed above, then the study is considered inconsistent with UIP. If the pattern on HRCT is considered definite, evaluation of pathology is not necessary. If the pattern is categorized as possible or is inconsistent, then surgical lung biopsy-confirmed UIP is necessary for the definitive diagnosis of IPF.
However, evidence is emerging that in the correct clinical scenario, possible UIP behaves similarly to definite UIP and may be sufficient to make the clinical diagnosis of IPF even without surgical biopsy confirmation.30
A DIAGNOSTIC ALGORITHM FOR IPF
Given the multitude of interstitial lung diseases, their complexities, and the lack of a gold standard definitive diagnostic test, the diagnosis of IPF can be difficult, requiring the integration of clinical, radiologic, and, if necessary, pathologic findings.
Demographic features
The patient’s demographic features and risk factors dictate the initial clinical suspicion of IPF compared with other interstitial lung diseases. The incidence of IPF increases with age, and IPF is more common in men. A history of smoking is another risk factor.31 A 45-year-old never-smoker is much less likely to have IPF than a 70-year-old former smoker, and a 70-year-old man is more likely to have IPF than a woman of the same age. Thus, the finding of interstitial lung disease in a patient with a demographic profile that is not typical (ie, a younger woman who never smoked) should prompt an exhaustive investigation for another diagnosis such as hypersensitivity pneumonitis or connective tissue disease.
Key elements of the history
After considering the demographic profile and risk factors, the next step in the evaluation is a thorough and accurate medical history. This should include assessment of the severity of dyspnea and cough, signs and symptoms of connective tissue disease (eg, arthralgias, sicca symptoms, Raynaud phenomenon, difficulty swallowing), and gastroesophageal reflux disease, which can be associated with connective tissue disease and, independently, with IPF.
It is also important to identify any environmental exposures that suggest pneumoconiosis or chronic hypersensitivity pneumonitis. The most common risk factors for hypersensitivity pneumonitis are birds and bird feathers, molds, fungi, hot tub use, and some industrial chemicals.32
A medication history is important. Many medications are associated with interstitial lung disease, but amiodarone, bleomycin, methotrexate, and nitrofurantoin are among the common offenders.33
A thorough family history is necessary, as there are familial forms of IPF.
Focus of the physical examination
The physical examination must include careful auscultation for rales. While rales are not specific for IPF, they are the most common pulmonary abnormality. Detailed skin, musculoskeletal, and cardiovascular examinations are also important to evaluate for rheumatologic signs, clubbing, or evidence of heart failure or pulmonary hypertension.
Laboratory tests
Laboratory testing should include a serologic autoantibody panel to evaluate for connective tissue diseases that can manifest as interstitial lung disease, including rheumatoid arthritis, dermatopolymyositis, scleroderma, Sjögren syndrome, and undifferentiated or mixed connective tissue disease. Typical preliminary laboratory tests are antinuclear antibody, rheumatoid factor, erythrocyte sedimentation rate, and C-reactive protein. Others may include anticyclic citrullinated peptide (anti-CCP), anti-Scl-70, anti-RNP, anti-SS-A, anti-SS-B, and anti-Jo-1.16 The breadth of the panel should depend on patient demographics and findings in the history or physical examination that increase or decrease the likelihood of a connective tissue disease.
Lung function testing
Assessing the patient’s pulmonary physiology should include spirometry, DLCO, and body plethysmography (lung volumes). In most cases, IPF manifests with restrictive physiology. Once restrictive physiology is confirmed with a low total lung capacity, FVC testing can be used as a longitudinal surrogate, as it is less expensive and easier for the patient to perform. In general, a lower total lung capacity or FVC indicates more severe impairment.
The DLCO serves as another marker of severity but is less reliable due to baseline variability and difficulties performing the maneuver.
A 6-minute walk test is another crucial physiologic assessment tool that can quantify exertional hypoxemia and functional status (ie, distance walked), and can assist in prognosis.
Imaging
Most patients undergo chest radiography in the workup for undiagnosed dyspnea. However, chest radiography is not adequate to formulate an accurate diagnosis in suspected interstitial lung disease, and a normal radiograph cannot exclude changes that might reflect early phases of the disease. As the disease progresses, the plain radiograph can show reticulonodular opacities and honeycombing in the peripheral and lower lung zones (Figure 3).34
The decision whether to order HRCT in the workup for a patient who has dyspnea and a normal chest radiograph is challenging. We recommend cross-sectional imaging when physiologic testing shows restriction or low DLCO, or when there is a high index of suspicion for underlying lung disease as the cause of symptoms.
Expert consultation can aid with this decision, especially when the underlying cause of dyspnea remains unclear after initial studies have been completed. Otherwise, HRCT is an essential test in the evaluation of interstitial lung disease.
Bronchoscopy’s role controversial
If the pattern on HRCT is nondiagnostic, then surgical biopsy is necessary, and the diagnosis of IPF requires a histologic pattern of UIP as described above.16,35
Although bronchoscopy can be valuable if an alternative diagnosis such as sarcoidosis or chronic hypersensitivity pneumonitis is suspected, the role of bronchoscopic biopsy in the workup of IPF is controversial. The patchy nature of UIP does not lend itself to the relatively small biopsy samples obtained through bronchoscopy.36,37
Surgical biopsy options
The favored biopsy approach is surgical, using either an open or a video-assisted thoracoscopic technique. The latter is preferred as it is less invasive, requires a shorter length of hospital stay, and allows a faster recovery.38 Bronchoscopic cryobiopsy, currently under investigation, is a potentially valuable tool whose role in diagnosing IPF is evolving.
Frequently, neither HRCT nor surgical lung biopsy demonstrates UIP, making the definitive diagnosis of IPF difficult. Moreover, some patients with nondiagnostic HRCT results are unable to tolerate surgical lung biopsy because of severely impaired lung function or other comorbidities.
The role of multidisciplinary discussions
When surgical lung biopsy is not possible, current practice at leading centers uses a multidisciplinary approach to allow for a confident diagnosis.30,39 Discussions take place between pulmonologists, pathologists, radiologists, and other specialists to collectively consider all aspects of a case before rendering a consensus opinion on the diagnosis and the management plan. If the discussion leads to a consensus diagnosis of IPF, then the patient’s clinician can move forward with treatment options. If not, the group can recommend further workup or alternative diagnoses and treatment regimens. The multidisciplinary group is also well positioned to consider the relative risks and benefits of moving forward with surgical lung biopsy for individual patients.
This approach illustrates the importance of referring these patients to centers of excellence in diagnosing and managing complex cases of interstitial lung disease, including IPF.40
TREATMENT OF IPF
Antifibrotic therapy
Antifibrotic therapy is a choice between pirfenidone and nintedanib.
Pirfenidone, which has an undefined molecular target, was approved based on the results of 3 trials.41,42 Pooled analyses from these trials showed a reduction in the decline from baseline in FVC percent predicted and improved progression-free survival.43 Pooled and meta-analyses of pirfenidone clinical trials have shown a mortality benefit, although no individual study has shown such an effect on mortality rates.44
The major adverse effects of pirfenidone are gastrointestinal distress and photosensitivity rash.
Nintedanib is a triple tyrosine kinase inhibitor that broadly targets fibroblast growth factor, vascular endothelial growth factor, and platelet-derived growth factor receptors. Combined analysis of 2 concurrent trials45 showed that nintedanib reduced the decline in FVC, similarly to pirfenidone. The major adverse event associated with nintedanib was diarrhea. Since it inhibits vascular endothelial growth factor, there is a risk of hematologic complications such as bleeding or clotting events.
Because pirfenidone and nintedanib can increase aminotransferase levels, regular monitoring is recommended.
To date, no trial has compared pirfenidone and nintedanib in terms of their efficacy and tolerability. Therefore, the choice of agent is based on the patient’s preference after a discussion of potential risks and expected benefits, a review of each drug’s side effects, and consideration of comorbid conditions and physician experience.
Patients need to understand that these drugs slow the rate of decline in FVC but have not been shown to improve symptoms or functional status.
Corticosteroids are not routine
Corticosteroids should not be used routinely in the treatment of IPF. Although steroids, alone or in combination with other immunosuppressive medications, were commonly used for IPF in the past, such use was not based on results of randomized controlled trials.46 Retrospective controlled studies have failed to show that corticosteroids improve mortality rates in IPF; indeed, they have shown that corticosteroids confer substantial morbidity.47,48 In addition, a randomized controlled trial combining corticosteroids with N-acetylcysteine and azathioprine was stopped early due to an increased risk of death and hospitalization.49 Collectively, these data suggest that corticosteroids confer no benefit and are potentially harmful. Their use in IPF is discouraged, and the joint international guidelines recommend against immunosuppression to treat IPF.16
Other treatments
The guidelines offer additional suggestions for the management of IPF.
Preliminary evidence suggests that microaspiration associated with abnormal gastroesophageal acid reflux is a risk factor for IPF. As such, there is a weak recommendation for aggressive treatment of reflux disease.50 However, because evidence suggests that proton-pump inhibitor therapy may be associated with adverse renal or central nervous system effects, this recommendation bears caution. It is hoped that ongoing studies will provide further insight into the role of acid-suppression in the management of IPF.51,52
Further treatment recommendations include best supportive management such as supplemental oxygen, pulmonary rehabilitation, and vaccinations.
Prompt referral for lung transplant is imperative. IPF is now the most common indication for lung transplant, and given the poor overall prognosis of advanced IPF, transplant confers a survival benefit in appropriately selected patients.53,54 Table 2 provides an evidence-based checklist for the workup and management of IPF.
ACUTE EXACERBATIONS OF IPF
The unpredictable nature of IPF can manifest in the form of acute exacerbations without an identifiable cause. The loosely defined diagnostic criteria for the diagnosis of acute exacerbations are a previous or new diagnosis of IPF, worsening or development of dyspnea in the last 30 days, and new bilateral ground-glass or consolidative changes with a background of UIP on HRCT.16
A new definition has been proposed55 to facilitate research in the characterization and treatment of acute exacerbations of IPF. The new definition includes all causes of respiratory deterioration except for heart failure and volume overload. It is less strict about the 30-day time frame. This newer definition is based on the lack of evidence differentiating outcomes when an acute deterioration is associated with known or unknown etiologies.55
The incidence of acute exacerbations is variable, with a 1- and 3-year incidence ranging between 8.6% and 23.9% depending on the criteria used.56 In general, acute exacerbations carry a grim prognosis, with a median life expectancy of 2.2 months.57
There is no approved therapy for exacerbations of IPF. Rather, treatment is mainly supportive with supplemental oxygen and mechanical ventilation. Current guidelines have a weak recommendation for the use of corticosteroids, but there are no recommendations regarding dose, route, or duration of therapy. Other treatments, primarily immunomodulatory agents, have been suggested but lack evidence of benefit.
Acknowledgments: Pathology images were provided by Carol Farver, MD, Pathology Institute, Cleveland Clinic. Radiology images were provided by Ruchi Yadav, MD, Imaging Institute, Cleveland Clinic.
- Brown KK, Raghu G. Medical treatment for pulmonary fibrosis: current trends, concepts, and prospects. Clin Chest Med 2004; 25(4):759–772, vii. doi:10.1016/j.ccm.2004.08.003
- Ryerson CJ, Collard HR. Update on the diagnosis and classification of ILD. Curr Opin Pulm Med 2013; 19(5):453–459. doi:10.1097/MCP.0b013e328363f48d
- Raghu G, Chen SY, Yeh WS, et al. Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001-11. Lancet Respir Med 2014; 2(7):566–572. doi:10.1016/S2213-2600(14)70101-8
- Nalysnyk L, Cid-Ruzafa J, Rotella P, Esser D. Incidence and prevalence of idiopathic pulmonary fibrosis: review of the literature. Eur Respir Rev 2012; 21(126):355–361. doi:10.1183/09059180.00002512
- Hutchinson J, Fogarty A, Hubbard R, McKeever T. Global incidence and mortality of idiopathic pulmonary fibrosis: a systematic review. Eur Respir J 2015; 46(3):795–806. doi:10.1183/09031936.00185114
- National Lung Screening Trial Research Team; Aberle DR, Adams AM, Berg CD, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med 2011; 365(5):395–409. doi:10.1056/NEJMoa1102873
- Jin GY, Lynch D, Chawla A, et al. Interstitial lung abnormalities in a CT lung cancer screening population: prevalence and progression rate. Radiology 2013; 268(2):563–571. doi:10.1148/radiol.13120816
- Southern BD, Scheraga RG, Yadav R. Managing interstitial lung disease detected on CT during lung cancer screening. Cleve Clin J Med 2016; 83(1):55–65. doi:10.3949/ccjm.83a.14157
- King TE Jr, Schwarz MI, Brown K, et al. Idiopathic pulmonary fibrosis: relationship between histopathologic features and mortality. Am J Respir Crit Care Med 2001; 164(5):1025–1032. doi:10.1164/ajrccm.164.6.2001056
- Lamas DJ, Kawut SM, Bagiella E, et al. Delayed access and survival in idiopathic pulmonary fibrosis: a cohort study. Am J Respir Crit Care Med 2011; 184(7):842–847. doi:10.1164/rccm.201104-0668OC
- Jo HE, Glaspole I, Moodley Y, et al. Disease progression in idiopathic pulmonary fibrosis with mild physiological impairment: analysis from the Australian IPF registry. BMC Pulm Med 2018; 18(1):19. doi:10.1186/s12890-018-0575-y
- Yang IV, Schwartz DA. Epigenetics of idiopathic pulmonary fibrosis. Transl Res 2015; 165(1):48–60. doi:10.1016/j.trsl.2014.03.011
- King TE Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet 2011; 378(9807):1949–1961. doi:10.1016/S0140-6736(11)60052-4
- Meltzer EB, Noble PW. Idiopathic pulmonary fibrosis. Orphanet J Rare Dis 2008; 3:8. doi:10.1186/1750-1172-3-8
- Raghu G. Idiopathic pulmonary fibrosis. A rational clinical approach. Chest 1987; 92(1):148–154. doi:10.1378/chest.92.1.148
- Raghu G, Collard HR, Egan JJ, et al; ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183(6):788–824. doi:10.1164/rccm.2009-040GL
- Panos RJ, Mortenson RL, Niccoli SA, King TE Jr. Clinical deterioration in patients with idiopathic pulmonary fibrosis: causes and assessment. Am J Med 1990; 88(4):396–404. doi:10.1016/0002-9343(90)90495-Y
- Ley B, Collard HR, King TE Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011; 183(4):431–440. doi:10.1164/rccm.201006-0894CI
- Collard HR, King TE Jr, Bartelson BB, Vourlekis JS, Schwarz MI, Brown KK. Changes in clinical and physiologic variables predict survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2003; 168(5):538–542. doi:10.1164/rccm.200211-1311OC
- Flaherty KR, Andrei AC, Murray S, et al. Idiopathic pulmonary fibrosis: prognostic value of changes in physiology and six-minute-walk test. Am J Respir Crit Care Med 2006; 174(7):803–809. doi:10.1164/rccm.200604-488OC
- Jegal Y, Kim DS, Shim TS, et al. Physiology is a stronger predictor of survival than pathology in fibrotic interstitial pneumonia. Am J Respir Crit Care Med 2005; 171(6):639–644. doi:10.1164/rccm.200403-331OC
- Latsi PI, du Bois RM, Nicholson AG, et al. Fibrotic idiopathic interstitial pneumonia: the prognostic value of longitudinal functional trends. Am J Respir Crit Care Med 2003; 168(5):531–537. doi:10.1164/rccm.200210-1245OC
- King CS, Nathan SD. Idiopathic pulmonary fibrosis: effects and optimal management of comorbidities. Lancet Respir Med 2017; 5(1):72–84. doi:10.1016/S2213-2600(16)30222-3
- Ley B, Ryerson CJ, Vittinghoff E, et al. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med 2012; 156(1):684–691. doi:10.7326/0003-4819-156-10-201205150-00004
- Rudd RM, Prescott RJ, Chalmers JC, Johnston ID; Fibrosing Alveolitis Subcommittee of the Research Committee of the British Thoracic Society. British Thoracic Society study on cryptogenic fibrosing alveolitis: response to treatment and survival. Thorax 2007; 62(1):62–66. doi:10.1136/thx.2005.045591
- Gutsche M, Rosen GD, Swigris JJ. Connective tissue disease-associated interstitial lung disease: a review. Curr Respir Care Rep 2012; 1:224–232. doi:10.1007/s13665-012-0028-7
- Park JH, Kim DS, Park IN, et al. Prognosis of fibrotic interstitial pneumonia: idiopathic versus collagen vascular disease-related subtypes. Am J Respir Crit Care Med 2007; 175(7):705–711. doi:10.1164/rccm.200607-912OC
- Taskar VS, Coultas DB. Is idiopathic pulmonary fibrosis an environmental disease? Proc Am Thorac Soc 2006; 3(4):293–298. doi:10.1513/pats.200512-131TK
- Vourlekis JS, Schwarz MI, Cherniack RM, et al. The effect of pulmonary fibrosis on survival in patients with hypersensitivity pneumonitis. Am J Med 2004; 116(10):662–668. doi:10.1016/j.amjmed.2003.12.030
- Brownell R, Moua T, Henry TS, et al. The use of pretest probability increases the value of high-resolution CT in diagnosing usual interstitial pneumonia. Thorax 2017; 72(5):424–429. doi:10.1136/thoraxjnl-2016-209671
- Baumgartner KB, Samet JM, Stidley CA, Colby TV, Waldron JA. Cigarette smoking: a risk factor for idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 1997; 155(1):242–248. doi:10.1164/ajrccm.155.1.9001319
- Selman M, Pardo A, King TE Jr. Hypersensitivity pneumonitis: insights in diagnosis and pathobiology. Am J Respir Crit Care Med 2012; 186(4):314–324. doi:10.1164/rccm.201203-0513CI
- Schwaiblmair M, Behr W, Haeckel T, Markl B, Foerg W, Berghaus T. Drug induced interstitial lung disease. Open Respir Med J 2012; 6:63–74. doi:10.2174/1874306401206010063
- Grenier P, Valeyre D, Cluzel P, Brauner MW, Lenoir S, Chastang C. Chronic diffuse interstitial lung disease: diagnostic value of chest radiography and high-resolution CT. Radiology 1991; 179(1):123–132. doi:10.1148/radiology.179.1.2006262
- Lynch JP 3rd, Huynh RH, Fishbein MC, Saggar R, Belperio JA, Weigt SS. Idiopathic pulmonary fibrosis: epidemiology, clinical features, prognosis, and management. Semin Respir Crit Care Med 2016; 37(3):331–357. doi:10.1055/s-0036-1582011
- Berbescu EA, Katzenstein AL, Snow JL, Zisman DA. Transbronchial biopsy in usual interstitial pneumonia. Chest 2006; 129(5):1126–1131. doi:10.1378/chest.129.5.1126
- Ohshimo S, Bonella F, Cui A, et al. Significance of bronchoalveolar lavage for the diagnosis of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2009; 179(11):1043–1047. doi:10.1164/rccm.200808-1313OC
- Oparka J, Yan TD, Ryan E, Dunning J. Does video-assisted thoracic surgery provide a safe alternative to conventional techniques in patients with limited pulmonary function who are otherwise suitable for lung resection? Interact Cardiovasc Thorac Surg 2013; 17(1):159–162. doi:10.1093/icvts/ivt097
- Flaherty KR, King TE Jr, Raghu G, et al. Idiopathic interstitial pneumonia: what is the effect of a multidisciplinary approach to diagnosis? Am J Respir Crit Care Med 2004; 170(8):904–910. doi:10.1164/rccm.200402-147OC
- Walsh SL, Wells AU, Desai SR, et al. Multicentre evaluation of multidisciplinary team meeting agreement on diagnosis in diffuse parenchymal lung disease: a case-cohort study. Lancet Respir Med 2016; 4(7):557–565. doi:10.1016/S2213-2600(16)30033-9
- King TE Jr, Bradford WZ, Castro-Bernardini S, et al; ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22):2083–2092. doi:10.1056/NEJMoa1402582
- Noble PW, Albera C, Bradford WZ, et al; CAPACITY Study Group. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet 2011; 377(9779):1760–1769. doi:10.1016/S0140-6736(11)60405-4
- Noble PW, Albera C, Bradford WZ, et al. Pirfenidone for idiopathic pulmonary fibrosis: analysis of pooled data from three multinational phase 3 trials. Eur Respir J 2016; 47(1):243–253. doi:10.1183/13993003.00026-2015
- Nathan SD, Albera C, Bradford WZ, et al. Effect of pirfenidone on mortality: pooled analyses and meta-analyses of clinical trials in idiopathic pulmonary fibrosis. Lancet Respir Med 2017; 5(1):33–41. doi:10.1016/S2213-2600(16)30326-5
- Richeldi L, du Bois RM, Raghu G, et al; INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22):2071–2082. doi:10.1056/NEJMoa1402584
- Richeldi L, Davies HR, Ferrara G, Franco F. Corticosteroids for idiopathic pulmonary fibrosis. Cochrane Database Syst Rev 2003: 3:CD002880. doi:10.1002/14651858.CD002880
- Douglas WW, Ryu JH, Schroeder DR. Idiopathic pulmonary fibrosis: impact of oxygen and colchicine, prednisone, or no therapy on survival. Am J Respir Crit Care Med 2000; 161(4 pt 1):1172–1178. doi:10.1164/ajrccm.161.4.9907002
- Gay SE, Kazerooni EA, Toews GB, et al. Idiopathic pulmonary fibrosis: predicting response to therapy and survival. Am J Respir Crit Care Med 1998; 157(4 pt 1):1063–1072. doi:10.1164/ajrccm.157.4.9703022
- Idiopathic Pulmonary Fibrosis Clinical Research Network; Raghu G, Anstrom KJ, King TE Jr, Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med 2012; 366(21):1968–1977. doi:10.1056/NEJMoa1113354
- Raghu G, Freudenberger TD, Yang S, et al. High prevalence of abnormal acid gastro-oesophageal reflux in idiopathic pulmonary fibrosis. Eur Respir J 2006; 27(1):136–142. doi:10.1183/09031936.06.00037005
- Gomm W, von Holt K, Thome F, et al. Association of proton pump inhibitors with risk of dementia: a pharmacoepidemiological claims data analysis. JAMA Neurol 2016; 73(4):410–416. doi:10.1001/jamaneurol.2015.4791
- Xie Y, Bowe B, Li T, Xian H, Balasubramanian S, Al-Aly Z. Proton pump inhibitors and risk of incident CKD and progression to ESRD. J Am Soc Nephrol 2016; 27(10):3153–3163. doi:10.1681/ASN.2015121377
- Thabut G, Mal H, Castier Y, et al. Survival benefit of lung transplantation for patients with idiopathic pulmonary fibrosis. J Thorac Cardiovasc Surg 2003; 126(2):469–475. doi:10.1016/S0022-5223(03)00600-7
- Valapour M, Skeans MA, Smith JM, et al. Lung. Am J Transplant 2016; 16(suppl 2):141–168. doi:10.1111/ajt.13671
- Collard HR, Ryerson CJ, Corte TJ, et al. Acute exacerbation of idiopathic pulmonary fibrosis. An International Working Group Report. Am J Respir Crit Care Med 2016; 194(3):265–275. doi:10.1164/rccm.201604-0801CI
- Kondoh Y, Taniguchi H, Katsuta T, et al. Risk factors of acute exacerbation of idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis 2010; 27(2):103–110. doi:10.1016/j.resinv.2015.04.005
- Song JW, Hong SB, Lim CM, Koh Y, Kim DS. Acute exacerbation of idiopathic pulmonary fibrosis: incidence, risk factors and outcome. Eur Respir J 2011; 37(2):356–363. doi:10.1183/09031936.00159709
- Brown KK, Raghu G. Medical treatment for pulmonary fibrosis: current trends, concepts, and prospects. Clin Chest Med 2004; 25(4):759–772, vii. doi:10.1016/j.ccm.2004.08.003
- Ryerson CJ, Collard HR. Update on the diagnosis and classification of ILD. Curr Opin Pulm Med 2013; 19(5):453–459. doi:10.1097/MCP.0b013e328363f48d
- Raghu G, Chen SY, Yeh WS, et al. Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001-11. Lancet Respir Med 2014; 2(7):566–572. doi:10.1016/S2213-2600(14)70101-8
- Nalysnyk L, Cid-Ruzafa J, Rotella P, Esser D. Incidence and prevalence of idiopathic pulmonary fibrosis: review of the literature. Eur Respir Rev 2012; 21(126):355–361. doi:10.1183/09059180.00002512
- Hutchinson J, Fogarty A, Hubbard R, McKeever T. Global incidence and mortality of idiopathic pulmonary fibrosis: a systematic review. Eur Respir J 2015; 46(3):795–806. doi:10.1183/09031936.00185114
- National Lung Screening Trial Research Team; Aberle DR, Adams AM, Berg CD, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med 2011; 365(5):395–409. doi:10.1056/NEJMoa1102873
- Jin GY, Lynch D, Chawla A, et al. Interstitial lung abnormalities in a CT lung cancer screening population: prevalence and progression rate. Radiology 2013; 268(2):563–571. doi:10.1148/radiol.13120816
- Southern BD, Scheraga RG, Yadav R. Managing interstitial lung disease detected on CT during lung cancer screening. Cleve Clin J Med 2016; 83(1):55–65. doi:10.3949/ccjm.83a.14157
- King TE Jr, Schwarz MI, Brown K, et al. Idiopathic pulmonary fibrosis: relationship between histopathologic features and mortality. Am J Respir Crit Care Med 2001; 164(5):1025–1032. doi:10.1164/ajrccm.164.6.2001056
- Lamas DJ, Kawut SM, Bagiella E, et al. Delayed access and survival in idiopathic pulmonary fibrosis: a cohort study. Am J Respir Crit Care Med 2011; 184(7):842–847. doi:10.1164/rccm.201104-0668OC
- Jo HE, Glaspole I, Moodley Y, et al. Disease progression in idiopathic pulmonary fibrosis with mild physiological impairment: analysis from the Australian IPF registry. BMC Pulm Med 2018; 18(1):19. doi:10.1186/s12890-018-0575-y
- Yang IV, Schwartz DA. Epigenetics of idiopathic pulmonary fibrosis. Transl Res 2015; 165(1):48–60. doi:10.1016/j.trsl.2014.03.011
- King TE Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet 2011; 378(9807):1949–1961. doi:10.1016/S0140-6736(11)60052-4
- Meltzer EB, Noble PW. Idiopathic pulmonary fibrosis. Orphanet J Rare Dis 2008; 3:8. doi:10.1186/1750-1172-3-8
- Raghu G. Idiopathic pulmonary fibrosis. A rational clinical approach. Chest 1987; 92(1):148–154. doi:10.1378/chest.92.1.148
- Raghu G, Collard HR, Egan JJ, et al; ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183(6):788–824. doi:10.1164/rccm.2009-040GL
- Panos RJ, Mortenson RL, Niccoli SA, King TE Jr. Clinical deterioration in patients with idiopathic pulmonary fibrosis: causes and assessment. Am J Med 1990; 88(4):396–404. doi:10.1016/0002-9343(90)90495-Y
- Ley B, Collard HR, King TE Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011; 183(4):431–440. doi:10.1164/rccm.201006-0894CI
- Collard HR, King TE Jr, Bartelson BB, Vourlekis JS, Schwarz MI, Brown KK. Changes in clinical and physiologic variables predict survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2003; 168(5):538–542. doi:10.1164/rccm.200211-1311OC
- Flaherty KR, Andrei AC, Murray S, et al. Idiopathic pulmonary fibrosis: prognostic value of changes in physiology and six-minute-walk test. Am J Respir Crit Care Med 2006; 174(7):803–809. doi:10.1164/rccm.200604-488OC
- Jegal Y, Kim DS, Shim TS, et al. Physiology is a stronger predictor of survival than pathology in fibrotic interstitial pneumonia. Am J Respir Crit Care Med 2005; 171(6):639–644. doi:10.1164/rccm.200403-331OC
- Latsi PI, du Bois RM, Nicholson AG, et al. Fibrotic idiopathic interstitial pneumonia: the prognostic value of longitudinal functional trends. Am J Respir Crit Care Med 2003; 168(5):531–537. doi:10.1164/rccm.200210-1245OC
- King CS, Nathan SD. Idiopathic pulmonary fibrosis: effects and optimal management of comorbidities. Lancet Respir Med 2017; 5(1):72–84. doi:10.1016/S2213-2600(16)30222-3
- Ley B, Ryerson CJ, Vittinghoff E, et al. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med 2012; 156(1):684–691. doi:10.7326/0003-4819-156-10-201205150-00004
- Rudd RM, Prescott RJ, Chalmers JC, Johnston ID; Fibrosing Alveolitis Subcommittee of the Research Committee of the British Thoracic Society. British Thoracic Society study on cryptogenic fibrosing alveolitis: response to treatment and survival. Thorax 2007; 62(1):62–66. doi:10.1136/thx.2005.045591
- Gutsche M, Rosen GD, Swigris JJ. Connective tissue disease-associated interstitial lung disease: a review. Curr Respir Care Rep 2012; 1:224–232. doi:10.1007/s13665-012-0028-7
- Park JH, Kim DS, Park IN, et al. Prognosis of fibrotic interstitial pneumonia: idiopathic versus collagen vascular disease-related subtypes. Am J Respir Crit Care Med 2007; 175(7):705–711. doi:10.1164/rccm.200607-912OC
- Taskar VS, Coultas DB. Is idiopathic pulmonary fibrosis an environmental disease? Proc Am Thorac Soc 2006; 3(4):293–298. doi:10.1513/pats.200512-131TK
- Vourlekis JS, Schwarz MI, Cherniack RM, et al. The effect of pulmonary fibrosis on survival in patients with hypersensitivity pneumonitis. Am J Med 2004; 116(10):662–668. doi:10.1016/j.amjmed.2003.12.030
- Brownell R, Moua T, Henry TS, et al. The use of pretest probability increases the value of high-resolution CT in diagnosing usual interstitial pneumonia. Thorax 2017; 72(5):424–429. doi:10.1136/thoraxjnl-2016-209671
- Baumgartner KB, Samet JM, Stidley CA, Colby TV, Waldron JA. Cigarette smoking: a risk factor for idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 1997; 155(1):242–248. doi:10.1164/ajrccm.155.1.9001319
- Selman M, Pardo A, King TE Jr. Hypersensitivity pneumonitis: insights in diagnosis and pathobiology. Am J Respir Crit Care Med 2012; 186(4):314–324. doi:10.1164/rccm.201203-0513CI
- Schwaiblmair M, Behr W, Haeckel T, Markl B, Foerg W, Berghaus T. Drug induced interstitial lung disease. Open Respir Med J 2012; 6:63–74. doi:10.2174/1874306401206010063
- Grenier P, Valeyre D, Cluzel P, Brauner MW, Lenoir S, Chastang C. Chronic diffuse interstitial lung disease: diagnostic value of chest radiography and high-resolution CT. Radiology 1991; 179(1):123–132. doi:10.1148/radiology.179.1.2006262
- Lynch JP 3rd, Huynh RH, Fishbein MC, Saggar R, Belperio JA, Weigt SS. Idiopathic pulmonary fibrosis: epidemiology, clinical features, prognosis, and management. Semin Respir Crit Care Med 2016; 37(3):331–357. doi:10.1055/s-0036-1582011
- Berbescu EA, Katzenstein AL, Snow JL, Zisman DA. Transbronchial biopsy in usual interstitial pneumonia. Chest 2006; 129(5):1126–1131. doi:10.1378/chest.129.5.1126
- Ohshimo S, Bonella F, Cui A, et al. Significance of bronchoalveolar lavage for the diagnosis of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2009; 179(11):1043–1047. doi:10.1164/rccm.200808-1313OC
- Oparka J, Yan TD, Ryan E, Dunning J. Does video-assisted thoracic surgery provide a safe alternative to conventional techniques in patients with limited pulmonary function who are otherwise suitable for lung resection? Interact Cardiovasc Thorac Surg 2013; 17(1):159–162. doi:10.1093/icvts/ivt097
- Flaherty KR, King TE Jr, Raghu G, et al. Idiopathic interstitial pneumonia: what is the effect of a multidisciplinary approach to diagnosis? Am J Respir Crit Care Med 2004; 170(8):904–910. doi:10.1164/rccm.200402-147OC
- Walsh SL, Wells AU, Desai SR, et al. Multicentre evaluation of multidisciplinary team meeting agreement on diagnosis in diffuse parenchymal lung disease: a case-cohort study. Lancet Respir Med 2016; 4(7):557–565. doi:10.1016/S2213-2600(16)30033-9
- King TE Jr, Bradford WZ, Castro-Bernardini S, et al; ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22):2083–2092. doi:10.1056/NEJMoa1402582
- Noble PW, Albera C, Bradford WZ, et al; CAPACITY Study Group. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet 2011; 377(9779):1760–1769. doi:10.1016/S0140-6736(11)60405-4
- Noble PW, Albera C, Bradford WZ, et al. Pirfenidone for idiopathic pulmonary fibrosis: analysis of pooled data from three multinational phase 3 trials. Eur Respir J 2016; 47(1):243–253. doi:10.1183/13993003.00026-2015
- Nathan SD, Albera C, Bradford WZ, et al. Effect of pirfenidone on mortality: pooled analyses and meta-analyses of clinical trials in idiopathic pulmonary fibrosis. Lancet Respir Med 2017; 5(1):33–41. doi:10.1016/S2213-2600(16)30326-5
- Richeldi L, du Bois RM, Raghu G, et al; INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22):2071–2082. doi:10.1056/NEJMoa1402584
- Richeldi L, Davies HR, Ferrara G, Franco F. Corticosteroids for idiopathic pulmonary fibrosis. Cochrane Database Syst Rev 2003: 3:CD002880. doi:10.1002/14651858.CD002880
- Douglas WW, Ryu JH, Schroeder DR. Idiopathic pulmonary fibrosis: impact of oxygen and colchicine, prednisone, or no therapy on survival. Am J Respir Crit Care Med 2000; 161(4 pt 1):1172–1178. doi:10.1164/ajrccm.161.4.9907002
- Gay SE, Kazerooni EA, Toews GB, et al. Idiopathic pulmonary fibrosis: predicting response to therapy and survival. Am J Respir Crit Care Med 1998; 157(4 pt 1):1063–1072. doi:10.1164/ajrccm.157.4.9703022
- Idiopathic Pulmonary Fibrosis Clinical Research Network; Raghu G, Anstrom KJ, King TE Jr, Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med 2012; 366(21):1968–1977. doi:10.1056/NEJMoa1113354
- Raghu G, Freudenberger TD, Yang S, et al. High prevalence of abnormal acid gastro-oesophageal reflux in idiopathic pulmonary fibrosis. Eur Respir J 2006; 27(1):136–142. doi:10.1183/09031936.06.00037005
- Gomm W, von Holt K, Thome F, et al. Association of proton pump inhibitors with risk of dementia: a pharmacoepidemiological claims data analysis. JAMA Neurol 2016; 73(4):410–416. doi:10.1001/jamaneurol.2015.4791
- Xie Y, Bowe B, Li T, Xian H, Balasubramanian S, Al-Aly Z. Proton pump inhibitors and risk of incident CKD and progression to ESRD. J Am Soc Nephrol 2016; 27(10):3153–3163. doi:10.1681/ASN.2015121377
- Thabut G, Mal H, Castier Y, et al. Survival benefit of lung transplantation for patients with idiopathic pulmonary fibrosis. J Thorac Cardiovasc Surg 2003; 126(2):469–475. doi:10.1016/S0022-5223(03)00600-7
- Valapour M, Skeans MA, Smith JM, et al. Lung. Am J Transplant 2016; 16(suppl 2):141–168. doi:10.1111/ajt.13671
- Collard HR, Ryerson CJ, Corte TJ, et al. Acute exacerbation of idiopathic pulmonary fibrosis. An International Working Group Report. Am J Respir Crit Care Med 2016; 194(3):265–275. doi:10.1164/rccm.201604-0801CI
- Kondoh Y, Taniguchi H, Katsuta T, et al. Risk factors of acute exacerbation of idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis 2010; 27(2):103–110. doi:10.1016/j.resinv.2015.04.005
- Song JW, Hong SB, Lim CM, Koh Y, Kim DS. Acute exacerbation of idiopathic pulmonary fibrosis: incidence, risk factors and outcome. Eur Respir J 2011; 37(2):356–363. doi:10.1183/09031936.00159709
KEY POINTS
- IPF is characterized by a pattern of usual interstitial pneumonia on imaging and histopathology without another known etiology.
- We recommend early referral to a center specializing in interstitial lung disease to confirm the diagnosis and to initiate appropriate therapy.
- Specialized centers offer advice on prognosis, enrollment in disease registries and clinical trials, and candidacy for lung transplant.
Genitourinary syndrome of menopause: Common problem, effective treatments
For many women, the postmenopausal loss of estrogen is associated with uncomfortable genitourinary symptoms, collectively referred to as the genitourinary syndrome of menopause (GSM). But despite the prevalence of GSM and the availability of treatments, most women do not seek relief.
This article reviews the syndrome and offers advice on how to talk about it with patients and what treatment options to consider.
A SYNDROME RECENTLY DEFINED
The term GSM and its definition were approved by the North American Menopause Society and the International Society for the Study of Women’s Sexual Health in 2014.1 It replaces older terms such as vulvovaginal atrophy, urogenital atrophy, and atrophic vaginitis.
GSM refers collectively to the symptoms associated with estrogen loss after menopause that adversely affect the vulvovaginal area and lower urinary tract. The most common symptoms are vulvovaginal dryness, burning, or irritation; sexual pain from inadequate lubrication; and urinary urgency, dysuria, or recurrent urinary tract infection.1,2
The definition notes that symptoms are self-reported as bothersome and are not the result of another disorder. Symptoms may be chronic and progressive, are not likely to resolve without treatment (pharmacologic or nonpharmacologic), and can have a significant negative impact on a woman’s quality of life and sexual health.1,2
COMMON BUT UNDERTREATED
From 40% to 60% of postmenopausal women experience GSM, but few seek treatment.3 Nevertheless, most postmenopausal women remain sexually active. In a 2008 survey of 94,000 postmenopausal women ages 50 to 79, 52% reported that they had been sexually active with a partner in the past year.4 However, 45% of postmenopausal women experienced unpleasant vaginal symptoms, according to a 2012 international survey of 3,520 postmenopausal women ages 55 to 65.5 In this survey, most respondents (75%) felt that vaginal symptoms had a negative impact on their life, but only 4% connected their symptoms to the vulvovaginal atrophy that resulted from loss of estrogen after menopause. Moreover, almost half were unaware of management options.5
These findings were supported by a 2013 survey of more than 3,000 US women who reported unpleasant vulvar and vaginal symptoms.6 From 60% to 85% noted negative sexual consequences from vulvovaginal symptoms, 47% felt their relationship suffered, and 27% felt it had a negative impact on their general enjoyment of life. In this study, 24% attributed their symptoms to menopause and 12% to hormonal changes. Although 56% had discussed GSM symptoms with a healthcare provider, only 40% were using GSM-specific topical treatments, mostly over-the-counter preparations.
Male partners of symptomatic women also note adverse emotional and physical effects.7 In an online survey of 4,100 men and 4,100 women ages 55 to 65, 52% to 78% of men and 58% to 64% of women expressed the negative effects of vulvovaginal symptoms on intimacy, libido, and sexual pain.
GSM is a progressive disorder. Women may note symptoms many years before menopause or have no symptoms until several years after menopause. One study found the prevalence of GSM to be 4% during perimenopause, rising after menopause to 25% after 1 year and to 47% after 3 years.8
Although distressing symptoms occur mostly after menopause, they may be seen in women of any age who experience a hypoestrogenic state, even if it is transient. Causes of this include premature ovarian failure, hypothalamic amenorrhea, and hyperprolactinemia. In addition, some treatments such as gonadotropin-releasing hormone agonists and aromatase inhibitors may cause vulvovaginal and lower urinary tract symptoms. Chemotherapy, radiation, and surgical removal of ovaries may also precipitate symptoms. The abrupt onset of menopause that may occur with these treatments is often associated with significantly greater sexual dysfunction and negative impact on quality of life. Cigarette smoking also leads to lower estrogen levels, which may contribute to GSM.
WHAT CAUSES GSM?
The genitourinary system develops from common embryologic tissue, the basis for the functional and clinical connection. Estrogen maintains the epithelium of the vagina, vulva, urethra, and bladder trigone via estrogen receptors present throughout these tissues.9
Premenopausal changes
Histologically, the estrogen-exposed vagina of a premenopausal woman is lined by glycogen-rich, stratified squamous epithelium, with underlying supportive fibromuscular layers. The epithelium is composed of superficial, intermediate, and parabasal cellular layers. In the presence of estrogen, the superficial and intermediate cellular levels predominate, with few parabasal cells.
Glycogen acts as a substrate for lactobacilli, producing organic acids, primarily lactate, that help maintain an acidic pH of 2.8 to 4.0. The low pH helps protect against pathologic shifts in the microbiome. Estrogen also maintains the collagen content of the epithelium, maintains acid mucopolysaccharides and hyaluronic acid, and optimizes vaginal blood flow. These effects result in optimal epithelial thickness and elasticity, moisture, vaginal secretions, and lubrication.10
Postmenopausal changes
Low levels of estrogen after menopause result in adverse anatomic, physiologic, and clinical changes in vaginal tissue. Effects of hypoestrogenism include the loss of collagen and adipose, leading to decreased elasticity and vaginal mucosal thinning. Vascular flow is decreased. The epithelial cytology transitions to a predominance of parabasal cells and a decrease in superficial and intermediate cells. Eccrine and apocrine glands become attenuated. These changes result in decreased vaginal secretions, diminished or delayed lubrication with sexual stimulation, friability of the vaginal vault, and vaginal dryness.11
Additionally, without estrogen, glycogen content is diminished, leading to decreased lactic acid production and a rise in vaginal pH to greater than 5. As the pH rises, Lactobacillus colonization decreases, leading to a further decrease in glycogen metabolism and to propagation of an elevated vaginal pH. The loss of vaginal acidity makes the vagina more susceptible to pathologic bacteria, including those found in the bowel and skin, sexually transmitted infections, and bacterial vaginosis.12
Other affected tissues. Anatomic effects of estrogen loss are not limited to the vagina. The epithelium, connective tissue, and smooth muscle of the vulva, vagina, urethra, and bladder trigone are also affected. The labia minora become thinner and regress, the introitus retracts, and narrowing and stricture of the vaginal canal and introitus may result. In some women, the urethral meatus becomes prominent relative to the introitus and more vulnerable to physical irritation, infection, and trauma.
Clinically, estrogen-related changes are usually responsible for vaginal dryness, irritation, burning, and superficial or deep dyspareunia. Urinary frequency, urgency, and incontinence also may develop.
THE DIAGNOSIS IS CLINICAL
The diagnosis of GSM is based on the history and physical examination. Standardized diagnostic tools for GSM are lacking, but some tools are available.
In 2006, the US Food and Drug Administration (FDA) published guidelines for industry to better define patient-reported outcome measures in clinical trials.13 The most significant addition was having the patient define the symptoms and rate how “bothersome” the symptoms are. Although this measure does not help diagnose GSM, it can be used effectively to follow response to treatment.
The Vaginal Symptom Questionnaire14 can be useful for assessing symptoms. It is a validated 21-item questionnaire that measures the quality-of-life impact of genital, but not urinary, symptoms of menopause.
Ask patients about symptoms

Healthcare providers should ask about GSM symptoms (Table 1) during routine clinical visits with women who are peri- or postmenopausal or who have hypoestrogenism from other causes, as many women are reluctant to initiate this discussion. Conversely, in women who present with sexual problems, such as difficulty with arousal or dyspareunia, GSM should be considered as a possible cause.
Specifically, ask women if they have any of the following symptoms:
- Vaginal itching, burning, discomfort, or irritation
- Malodorous or irritating vaginal discharge
- Urinary frequency, urgency, dysuria, urethral discomfort, or recurrent urinary tract infections
- Sexual symptoms of entry dyspareunia, vaginal pain, or irritation with sexual activity, which may be complicated by postcoital bleeding, spotting, or fissuring.
Vulvovaginal pain or irritation may be constant or may be present in the absence of sexual activity, such as with exercise, wearing tight clothing, or sitting for long periods.
Physical examination

Characteristic physical findings of GSM include scarce pubic hair, thinning of the labia from loss of labial fat, resorption of the labia minora, or fusion of the labia minora and majora (Table 2).15,16 The vulvar skin is pale and thin. The clitoral hood may retract, exposing the glans (which may lead to increased pain with sexual stimulation), or clitoral hood fusion may occur. The vagina is pale, dry, smooth, and shiny with loss of rugae; shortening or stricturing may be present. Vaginal elasticity decreases. Inflammation and petechiae (pinpoint, nonraised, round purple-red spots) may be present. The cervix may be flush with the vaginal fornices.
Prolonged atrophy may result in introital narrowing and friability, which may cause tearing with sexual activity or insertion of a speculum during pelvic examination. In addition, the epithelium of the lower urinary tract thins, and the muscular and fibrous layers atrophy—changes that may not be obvious during examination. A urethral caruncle may form, presenting as proliferative red tissue at the entrance of the urethra. Prolapse may become more prominent.
In women with severe genitourinary atrophy, pelvic examination may cause significant discomfort. Reassuring the patient that she can ask the clinician to stop at any time due to extreme discomfort is the first step in a successful pelvic examination.
In some situations, initial examination of the pelvic area may not include insertion of a speculum. Use of a hand-held mirror so the patient can observe the examination may help her relax during the examination.
Vaginal pH and cultures, if indicated, may be obtained by gently inserting a cotton-tipped swab into the vagina without a speculum and before applying lubricant. Lubricant should be used generously; in some instances, topical lidocaine gel (diluted, as it may burn) may be placed against the perineum on a gauze pad for 3 to 5 minutes before insertion of the speculum.
When an internal pelvic examination is necessary in a timely manner, such as with postmenopausal bleeding or a history of an abnormal Papanicolaou smear, but is too painful for the patient, the examination should be done under anesthesia.
Additional considerations

The history should review current medical conditions, medication use, nongenital skin disorders (eg, eczema), and systemic menopausal symptoms, such as hot flashes.
Also, consider other potential causes of GSM during the evaluation (Table 3).17,18 Review the use of detergents, soaps, douches, or over-the-counter topical products that could cause genitourinary symptoms secondary to contact irritation or allergy.
Any isolated, ulcerated, or nonhealing lesion should be biopsied. Reevaluate patients who have not responded to previous topical therapy or consider referral to a specialist.
Assess the personal, interpersonal, social, and sexual impact of the symptoms: if they do not cause distress, GSM does not require treatment. Nevertheless, potential treatment options should be discussed as symptoms may progress, making intervention necessary.
Laboratory tests: Helpful, not essential
Laboratory tests are unnecessary for the diagnosis of GSM. However, office-based objective evaluations such as vaginal pH testing and the maturation index can support the diagnosis.
The pH of the estrogenized vagina ranges from 3.8 to 4.2, whereas in women with GSM, the pH may reach 5.5 or higher. The pH can be obtained by placing a pH-sensitive paper against the lateral vaginal wall, avoiding any discharge or cervical mucus. A vaginal pH of 5 or greater in the absence of blood, semen, or infection suggests vulvovaginal atrophy.19
The vaginal maturation index is determined by a vaginal smear using Rakoff staining, in which 100 cells are counted and the number of parabasal, intermediate, and superficial cells is determined. In general, a well-estrogenized vagina has mostly superficial and intermediate cells, which shifts to a predominance of parabasal cells as estrogen levels decline.20
A recent review of vaginal atrophy suggests that after a diagnosis of GSM, healthcare providers can consider the most bothersome symptom along with the vaginal pH to assess the response to treatment.21 In general, schedule a follow-up appointment at 8 to 12 weeks to review treatment response. If treatment has not resulted in adequate symptom relief, consider a pelvic examination and further testing.
SELECTING A TREATMENT
Symptomatic women with GSM who desire intervention should be offered over-the-counter nonhormonal products as the first line of therapy.
If nonhormonal products are ineffective and there are no contraindications, locally applied estrogen in cream, tablet, or a ring delivery system may be offered. Local dehydroepiandrosterone (DHEA) inserts or ospemifene, an oral selective estrogen-receptor modulator, are FDA-approved for moderate to severe dyspareunia secondary to GSM.
Oral estrogen therapy is not indicated for vulvovaginal symptoms, but some women taking systemic estrogen for vasomotor symptoms may need additional local estrogen application to relieve vaginal symptoms.
Nonhormonal treatments
Nonhormonal over-the-counter therapies provide sufficient relief for most women with mild symptoms. There is a plethora of products, so practitioners need to offer guidance to help women with their individual choices.
Vaginal lubricants are intended for use with sexual or penetrative activity (including pelvic examination). They provide short-term relief of symptoms, but there is no evidence of any impact on histologic changes of atrophy. They are meant to relieve friction. Lubricants may be water-based, oil-based, silicone-based, or a combination. Individual products have different effects on condom integrity. Perfumed, warming, or stimulating products may be irritating to some women and should be tried initially in small amounts.
Vaginal moisturizers are intended to treat GSM. They are applied regularly, not just with vaginal activity, usually once or twice a week. Some vaginal lubricants can maintain an acidic pH in the vagina and may reverse the histologic changes of atrophy. Symptomatic improvement over placebo or estrogen has been shown in clinical trials.22–24
Women should be advised that trial and error in choosing products may be necessary to establish a successful regimen. Products should be tried in succession, not simultaneously, with a “wash-out” period between, to be able to evaluate response.
Vaginal dilators and pelvic floor physical therapy
Sexual activity, either by self-stimulation or with a partner, helps maintain vaginal health by contributing to increased vascularity and elasticity of tissue. Women who resume sexual activity after a long period of inactivity may benefit from the use of vaginal dilators, which aid both in mechanical distention and progressive relaxation of the vaginal musculature.
In some women, long-term dyspareunia may result in vaginismus, an involuntary contraction of the vaginal musculature. For these women, dilators may be effective. Additional options focus on pelvic floor physical therapy, which can isolate trigger points, using biofeedback to teach relaxation and home exercises such as vaginal massage.
HORMONAL THERAPIES

If nonhormonal lubricants and moisturizers do not achieve satisfactory symptomatic relief, FDA-approved hormonal therapies (Table 4) include estrogen-containing vaginal creams, rings, and a tablet; a vaginal tablet containing DHEA; and an oral tablet containing ospemifene.
Estrogen products
For patients whose symptoms do not respond to nonhormonal therapies, low-dose, locally applied estrogen therapy is the first treatment recommended.2 Locally applied estrogens can reverse the atrophic changes of estrogen deprivation, resulting in an increase in blood flow, elasticity, and vaginal wall thickness. This therapy also can normalize pH levels with subsequent restoration of a healthy lactobacilli-based flora. Locally applied estrogens also have been shown to decrease the frequency of recurrent urinary tract infection.25
Estrogen-containing vaginal creams, rings, and a tablet are available, and each has been shown to be effective for GSM. Locally applied estrogens at recommended dosages tend to have fewer adverse events and risks than systemic estrogens.26 Estradiol levels generally do not exceed levels found in the untreated menopausal population, although a dose- and duration-dependent increase in systemic levels may occur.27
Dosing considerations
The vaginal ring and the vaginal tablet provide the lowest prefixed daily dose of estradiol (7.5 and 10 µg daily, respectively). Estrogen creams (estradiol, conjugated equine estrogens) are more readily absorbed, and dosing should be tapered to the lowest, most effective dose for symptom relief.
The FDA-approved doses for vaginal creams containing 17-beta estradiol are higher than the dose found to be effective in clinical practice (0.5 g twice a week). Most practitioners start with the lower dose, reserving the FDA-approved higher doses for patients who do not obtain adequate relief over 6 to 8 weeks of treatment. The conjugated-estrogen vaginal cream Premarin is the only locally applied estrogen approved by the FDA to treat dyspareunia. It is dosed at 0.5 g intravaginally for 21 days and is then either withdrawn for 7 days or, more commonly, administered at 0.5 g twice a week.
Initial treatment with vaginal cream may require more frequent vulvovaginal application, such as daily for 1 to 2 weeks. Women with vaginal fissures or tearing will benefit from externally applied creams in addition to internal applications. Response to therapy is usually seen within 4 to 6 weeks from onset of treatment. Once symptom relief is obtained, treatment should continue indefinitely. Although long-term safety studies are lacking, risks are believed to be minimal.
Endometrial impact. Women with contraindications to systemic estrogen should be counseled about possible small increases in serum levels of estradiol associated with locally applied estrogens and the potential risks and benefits those increases incur. Endometrial surveillance with either transvaginal ultrasonography or endometrial sampling is not required, even with long-term use, but it should be considered with higher doses or more frequent applications.
Similarly, progesterone replacement for endometrial protection is not recommended but can be considered in women with an intact uterus at high risk of endometrial cancer, such as obese patients. If a systemic progestational agent is considered, the risks and benefits should be weighed carefully. Even in women at high risk, endometrial surveillance may be the most appropriate option.28 Uterine bleeding that occurs should be considered abnormal and should be investigated.
DHEA (prasterone)
In 2016, the FDA approved intravaginal prasterone, a DHEA-containing product for the treatment of dyspareunia secondary to moderate to severe vulvovaginal atrophy caused by menopause. DHEA is an endogenous steroid that is converted by aromatase activity into testosterone and estradiol.
Clinical trials have found that 12 weeks of vaginal DHEA supplementation (0.25%, 0.5%, and 1% DHEA ovules) was more effective than placebo in improving vaginal dryness and dyspareunia in women with GSM.29–31 In these studies, locally applied DHEA decreased parabasal cells, decreased vaginal pH, increased vaginal secretions, and improved epithelial surface thickness and integrity without any significant impact on serum levels of DHEA, DHEA-sulfate, estradiol, testosterone, or their metabolites. Importantly, transvaginal DHEA had negligible endometrial effect.
The breast cancer risk associated with vaginal DHEA has not been fully evaluated. However, labeling lists breast cancer as a warning, not a contraindication.
Selective estrogen-receptor modulator
In 2013, the FDA approved ospemifene for the treatment of dyspareunia caused by GSM. Ospemifene, an estrogen agonist in the vagina, is taken daily as a 60-mg oral dose. Long-term safety studies suggested no adverse effects on the endometrium or breast for at least 52 weeks.32
These studies also noted that ospemifene improved the vaginal maturation index (decreased parabasal cells and increased superficial cells) and decreased vaginal pH. It has further been shown to decrease severity of the self-identified most bothersome symptom—dyspareunia or vaginal dryness—compared with placebo.33
Potential increases in hot flashes, which may occur in up to 7% of patients, and the risk of blood clots should be considered. Additionally, the safety of ospemifene in women with a history of breast cancer has not been established. Although early studies suggest it either has no effect or possibly a protective effect on breast tissue, the FDA does not recommend its use in women at risk for breast cancer. Long-term effects on bone are unknown.
The labeling for ospemifene includes a boxed warning about the risk of stroke, blood clots, and cancer of the lining of the uterus. Patients should be counseled about worrisome signs or symptoms that require medical attention.
ALTERNATIVE THERAPIES
Treatments for GSM not approved by the FDA include laser and radiofrequency therapies, testosterone, isoflavones, and bioidentical hormones.
Laser and radiofrequency therapies
Both of these therapies aim to promote tissue remodeling with increased collagen and elastin production and increased vascularity. This, in turn, increases muscle support and tone.
Laser therapies act by ablating and coagulating vaginal tissues; radiofrequency therapies directly heat the tissue. Both treatments are office-based, require up to 3 initial treatments, and are followed by retreatment at approximately 1-year intervals.
Studies have reported high patient satisfaction rates (91% to 100%), improved sexual functioning, and decreased GSM symptoms of vaginal dryness, burning, itching, and dyspareunia.34–36 Data, however, are from observational studies, not placebo-controlled trials.
Although laser and radiofrequency therapies are FDA-approved for several indications, laser treatment for symptoms of vulvovaginal atrophy is not currently an approved indication. Patients should be advised of this.
Testosterone
Locally applied testosterone was shown in a small study to improve dyspareunia and vaginal dryness associated with aromatase inhibitor use in breast cancer patients.37 However, due to the lack of safety and efficacy data from larger, controlled trials, testosterone therapy is not currently recommended.
Isoflavones
Isoflavones are phytoestrogens found in soy. In a 12-week, double-blind placebo-controlled study of vaginally applied 4% soy isoflavone gel, improvements in vaginal atrophy symptoms, maturation values, and vaginal pH were found in 60 postmenopausal women.38 Additional data on efficacy and safety are needed before isoflavones should be considered as a treatment for GSM.
Bioidentical hormones
Bioidentical hormones are plant-derived hormones that are chemically similar or identical to those produced by the body. Although there are FDA-approved bioidentical hormones (eg, micronized progesterone, estradiol, DHEA), the term bioidentical usually refers to non-FDA-approved, commercially available hormones produced and compounded by specialty pharmacies.
Patients often view these substances as being better, safer, and more acceptable for use, and healthcare practitioners need to be prepared to address these beliefs. The FDA and the American College of Obstetricians and Gynecologists consider bioidentical hormones to be a marketing term and not an alternative treatment based on scientific evidence.39 Patients should be informed that bioidentical hormones have the same risks as any similar hormone preparation along with additional risks related to potential lack of purity and potency. Further, they have not been adequately studied in controlled clinical trials.
FOLLOW-UP CARE
Healthcare providers caring for women should assume a proactive role in diagnosing and treating the symptoms of GSM. And once diagnosis of GSM is established and treatment is under way, practitioners can use symptom questionnaires and vaginal pH testing as easy and reliable means of measuring clinical response to therapy.
- Portman DJ, Gass ML; Vulvovaginal Atrophy Terminology Consensus Conference Panel. Genitourinary syndrome of menopause: new terminology for vulvovaginal atrophy from the International Society for the Study of Women’s Sexual Health and the North American Menopause Society. Menopause 2014; 21(10):1063–1068. doi:10.1097/GME.0000000000000329
- Management of symptomatic vulvovaginal atrophy: 2013 position statement of The North American Menopause Society. Menopause 2013; 20(9):888–904. doi:10.1097/GME.0b013e3182a122c2
- Parish SJ, Nappi RE, Krychman ML, et al. Impact of vulvovaginal health on postmenopausal women: a review of surveys on symptoms of vulvovaginal atrophy. Int J Womens Health 2013; 5:437–447. doi:10.2147/IJWH.S44579
- McCall-Hosenfeld JS, Jaramillo SA, Legault C, et al; Members of Women’s Health Initiative-Observational Study. Correlates of sexual satisfaction among sexually active postmenopausal women in the Women’s Health Initiative-Observational Study. J Gen Intern Med 2008; 23(12):2000–2009. doi:10.1007/s11606-008-0820-9
- Nappi RE, Kokot-Kierepa M. Vaginal Health: Insights, Views & Attitudes (VIVA): results from an international survey. Climacteric 2012; 15(1):36–44. doi:10.3109/13697137.2011.647840
- Kingsberg SA, Wysocki S, Magnus L, Krychman ML. Vulvar and vaginal atrophy in postmenopausal women: findings from the REVIVE (REal Women’s VIews of Treatment Options for Menopausal Vaginal Changes) survey. J Sex Med 2013; 10(7):1790–1799. doi:10.1111/jsm.12190
- Nappi RE, Kingsberg S, Maamari R, Simon J. The CLOSER (Clarifying Vaginal Atrophy’s Impact On Sex and Relationships) survey: implications of vaginal discomfort in postmenopausal women and in male partners. J Sex Med 2013; 10(9):2232–2241. doi:10.1111/jsm.12235
- Dennerstein L, Dudley EC, Hopper JL, Guthrie JR, Burger HG. A prospective population-based study of menopausal symptoms. Obstet Gynecol 2000; 96(3):351–358. pmid:10960625
- Stika CS. Atrophic vaginitis. Dermatol Ther 2010; 23(5):514–522. doi:10.1111/j.1529-8019.2010.01354.x
- Castelo-Branco C, Cancelo MJ, Villero J, Nohales F, Juliá MD. Management of postmenopausal vaginal atrophy and atrophic vaginitis. Maturitas 2005; 52(suppl 1):S46–S52. doi:10.1016/j.maturitas.2005.06.014
- Forsberg JG. A morphologist’s approach to the vagina—age-related changes and estrogen sensitivity. Maturitas 1995; 22(suppl):S7–S15.
- Martin DH. The microbiota of the vagina and its influence on women’s health and disease. Am J Med Sci 2012; 343(1):2–9. doi:10.1097/MAJ.0b013e31823ea228
- US Department of Health and Human Services; Food and Drug Administration (FDA). Guidance for industry. Patient-reported outcome measures: use in medical product development to support labeling claims, 2006. doi:10.1186/1477-7525-4-79
- Erekson EA, Yip SO, Wedderburn TS, et al. The Vulvovaginal Symptoms Questionnaire: a questionnaire for measuring vulvovaginal symptoms in postmenopausal women. Menopause 2013; 20(9):973–979. doi:10.1097/GME.0b013e318282600b
- Johnston SL, Farrell SA, Bouchard C, et al; SOGC Joint Committee-Clinical Practice Gynaecology and Urogynaecology. The detection and management of vaginal atrophy. J Obstet Gynaecol Can 2004; 26(5):503–515. doi:10.1016/S1701-2163(16)30662-4
- Oriba HA, Maibach HI. Vulvar transepidermal water loss (TEWL) decay curves. Effect of occlusion, delipidation, and age. Acta Derm Venereol 1989; 69(6):461–465. pmid:2575316
- Bachmann G, Nevadunsky NS. Diagnosis and treatment of atrophic vaginitis. Am Fam Physician 2000; 61(10):3090–3096. pmid:10839558
- MacBride MB, Rhodes DJ, Shuster LT. Vulvovaginal atrophy. Mayo Clin Proc 2010; 85(1):87–94. doi:10.4065/mcp.2009.0413
- Nilsson K, Risberg B, Heimer G. The vaginal epithelium in the post menopause--cytology, histology and pH as methods of assessment. Maturitas 1995; 21(1):51–56. pmid:7731384
- McEndree B. Clinical application of the vaginal maturation index. Nurse Pract 1999; 24(9):48–56. pmid:10507070
- Weber MA, Limpens J, Roovers JP. Assessment of vaginal atrophy: a review. Int Urogynecol J 2015; 26(1):15–28. doi:10.1007/s00192-014-2464-0
- Lee YK, Chung HH, Kim JW, Park NH, Song YS, Kang SB. Vaginal pH-balanced gel for the control of atrophic vaginitis among breast cancer survivors: a randomized controlled trial. Obstet Gynecol 2011; 117(4):922–927. doi:10.1097/AOG.0b013e3182118790
- Bygdeman M, Swahn ML. Replens versus dienoestrol cream in the symptomatic treatment of vaginal atrophy in postmenopausal women. Maturitas 1996; 23(3):259–263. pmid:8794418
- Nachtigall LE. Comparative study: replens versus local estrogen in menopausal women. Fertil Steril 1994; 61(1):178–180. pmid:8293835
- Raz R, Gennesin Y, Wasser J, et al. Recurrent urinary tract infections in postmenopausal women. Clin Infect Dis 2000; 30(1):152–156. doi:10.1086/313596
- Suckling J, Lethaby A, Kennedy R. Local oestrogen for vaginal atrophy in postmenopausal women. Cochrane Database Syst Rev 2006; 4:CD001500. doi:10.1002/14651858.CD001500
- Santen RJ. Vaginal administration of estradiol: effects of dose, preparation and timing on plasma estradiol levels. Climacteric 2015; 18(2):121–126. doi:10.3109/13697137.2014.947254
- North American Menopause Society. Estrogen and progestogen use in postmenopausal women: 2010 position statement of the North American Menopause Society. Menopause 2010; 17(2):242–255. doi:10.1097/gme.0b013e3181d0f6b9
- Labrie F, Archer D, Bouchard C, et al. Effect of intravaginal dehydroepiandrosterone (Prasterone) on libido and sexual dysfunction in postmenopausal women. Menopause 2009; 16(5):923–931. doi:10.1097/gme.0b013e31819e85c6
- Labrie F, Archer D, Bouchard C, et al. Intravaginal dehydroepiandrosterone (Prasterone), a physiological and highly efficient treatment of vaginal atrophy. Menopause 2009; 16(5):907–922. doi:10.1097/gme.0b013e31819e8e2d
- Archer DF. Dehydroepiandrosterone intra vaginal administration for the management of postmenopausal vulvovaginal atrophy. J Steroid Biochem Mol Biol 2015; 145:139–143. doi:10.1016/j.jsbmb.2014.09.003
- Wurz GT, Kao CJ, DeGregorio MW. Safety and efficacy of ospemifene for the treatment of dyspareunia associated with vulvar and vaginal atrophy due to menopause. Clin Interv Aging 2014; 9:1939–1950. doi:10.2147/CIA.S73753
- Constantine G, Graham S, Portman DJ, Rosen RC, Kingsberg SA. Female sexual function improved with ospemifene in postmenopausal women with vulvar and vaginal atrophy: results of a randomized, placebo-controlled trial. Climacteric 2015; 18(2):226–232. doi:10.3109/13697137.2014.954996
- Arroyo C. Fractional CO2 laser treatment for vulvovaginal atrophy symptoms and vaginal rejuvenation in perimenopausal women. Int J Womens Health 2017; 9:591–595. doi:10.2147/IJWH.S136857
- Perino A, Calligaro A, Forlani F, et al. Vulvo-vaginal atrophy: a new treatment modality using thermo-ablative fractional CO2 laser. Maturitas 2015; 80(3):296–301. doi:10.1016/j.maturitas.2014.12.006
- Salvatore S, Nappi R, Zerbinati N, et al. A 12-week treatment with fractional CO2 laser for vulvovaginal atrophy: a pilot study. Climacteric 2014; 17(4):363–369. doi:10.3109/13697137.2014.899347
- Witherby S, Johnson J, Demers L, et al. Topical testosterone for breast cancer patients with vaginal atrophy related to aromatase inhibitors: a phase I/II study. Oncologist 2011; 16(4):424–431. doi:10.1634/theoncologist.2010-0435
- Lima SM, Bernardo BF, Yamada SS, Reis BF, da Silva GM, Galvão MA. Effects of Glycine max (L.) Merr. soy isoflavone vaginal gel on epithelium morphology and estrogen receptor expression in postmenopausal women: a 12-week, randomized, double-blind, placebo-controlled trial. Maturitas 2014; 78(3):205–211. doi:10.1016/j.maturitas.2014.04.007
- Committee on Gynecologic Practice and the American Society for Reproductive Medicine Practice Committee. Committee opinion No 532: compounded bioidentical menopausal hormone therapy. Obstet Gynecol 2012; 120(2 pt 1):411–415. doi:10.1097/AOG.0b013e318268049e
For many women, the postmenopausal loss of estrogen is associated with uncomfortable genitourinary symptoms, collectively referred to as the genitourinary syndrome of menopause (GSM). But despite the prevalence of GSM and the availability of treatments, most women do not seek relief.
This article reviews the syndrome and offers advice on how to talk about it with patients and what treatment options to consider.
A SYNDROME RECENTLY DEFINED
The term GSM and its definition were approved by the North American Menopause Society and the International Society for the Study of Women’s Sexual Health in 2014.1 It replaces older terms such as vulvovaginal atrophy, urogenital atrophy, and atrophic vaginitis.
GSM refers collectively to the symptoms associated with estrogen loss after menopause that adversely affect the vulvovaginal area and lower urinary tract. The most common symptoms are vulvovaginal dryness, burning, or irritation; sexual pain from inadequate lubrication; and urinary urgency, dysuria, or recurrent urinary tract infection.1,2
The definition notes that symptoms are self-reported as bothersome and are not the result of another disorder. Symptoms may be chronic and progressive, are not likely to resolve without treatment (pharmacologic or nonpharmacologic), and can have a significant negative impact on a woman’s quality of life and sexual health.1,2
COMMON BUT UNDERTREATED
From 40% to 60% of postmenopausal women experience GSM, but few seek treatment.3 Nevertheless, most postmenopausal women remain sexually active. In a 2008 survey of 94,000 postmenopausal women ages 50 to 79, 52% reported that they had been sexually active with a partner in the past year.4 However, 45% of postmenopausal women experienced unpleasant vaginal symptoms, according to a 2012 international survey of 3,520 postmenopausal women ages 55 to 65.5 In this survey, most respondents (75%) felt that vaginal symptoms had a negative impact on their life, but only 4% connected their symptoms to the vulvovaginal atrophy that resulted from loss of estrogen after menopause. Moreover, almost half were unaware of management options.5
These findings were supported by a 2013 survey of more than 3,000 US women who reported unpleasant vulvar and vaginal symptoms.6 From 60% to 85% noted negative sexual consequences from vulvovaginal symptoms, 47% felt their relationship suffered, and 27% felt it had a negative impact on their general enjoyment of life. In this study, 24% attributed their symptoms to menopause and 12% to hormonal changes. Although 56% had discussed GSM symptoms with a healthcare provider, only 40% were using GSM-specific topical treatments, mostly over-the-counter preparations.
Male partners of symptomatic women also note adverse emotional and physical effects.7 In an online survey of 4,100 men and 4,100 women ages 55 to 65, 52% to 78% of men and 58% to 64% of women expressed the negative effects of vulvovaginal symptoms on intimacy, libido, and sexual pain.
GSM is a progressive disorder. Women may note symptoms many years before menopause or have no symptoms until several years after menopause. One study found the prevalence of GSM to be 4% during perimenopause, rising after menopause to 25% after 1 year and to 47% after 3 years.8
Although distressing symptoms occur mostly after menopause, they may be seen in women of any age who experience a hypoestrogenic state, even if it is transient. Causes of this include premature ovarian failure, hypothalamic amenorrhea, and hyperprolactinemia. In addition, some treatments such as gonadotropin-releasing hormone agonists and aromatase inhibitors may cause vulvovaginal and lower urinary tract symptoms. Chemotherapy, radiation, and surgical removal of ovaries may also precipitate symptoms. The abrupt onset of menopause that may occur with these treatments is often associated with significantly greater sexual dysfunction and negative impact on quality of life. Cigarette smoking also leads to lower estrogen levels, which may contribute to GSM.
WHAT CAUSES GSM?
The genitourinary system develops from common embryologic tissue, the basis for the functional and clinical connection. Estrogen maintains the epithelium of the vagina, vulva, urethra, and bladder trigone via estrogen receptors present throughout these tissues.9
Premenopausal changes
Histologically, the estrogen-exposed vagina of a premenopausal woman is lined by glycogen-rich, stratified squamous epithelium, with underlying supportive fibromuscular layers. The epithelium is composed of superficial, intermediate, and parabasal cellular layers. In the presence of estrogen, the superficial and intermediate cellular levels predominate, with few parabasal cells.
Glycogen acts as a substrate for lactobacilli, producing organic acids, primarily lactate, that help maintain an acidic pH of 2.8 to 4.0. The low pH helps protect against pathologic shifts in the microbiome. Estrogen also maintains the collagen content of the epithelium, maintains acid mucopolysaccharides and hyaluronic acid, and optimizes vaginal blood flow. These effects result in optimal epithelial thickness and elasticity, moisture, vaginal secretions, and lubrication.10
Postmenopausal changes
Low levels of estrogen after menopause result in adverse anatomic, physiologic, and clinical changes in vaginal tissue. Effects of hypoestrogenism include the loss of collagen and adipose, leading to decreased elasticity and vaginal mucosal thinning. Vascular flow is decreased. The epithelial cytology transitions to a predominance of parabasal cells and a decrease in superficial and intermediate cells. Eccrine and apocrine glands become attenuated. These changes result in decreased vaginal secretions, diminished or delayed lubrication with sexual stimulation, friability of the vaginal vault, and vaginal dryness.11
Additionally, without estrogen, glycogen content is diminished, leading to decreased lactic acid production and a rise in vaginal pH to greater than 5. As the pH rises, Lactobacillus colonization decreases, leading to a further decrease in glycogen metabolism and to propagation of an elevated vaginal pH. The loss of vaginal acidity makes the vagina more susceptible to pathologic bacteria, including those found in the bowel and skin, sexually transmitted infections, and bacterial vaginosis.12
Other affected tissues. Anatomic effects of estrogen loss are not limited to the vagina. The epithelium, connective tissue, and smooth muscle of the vulva, vagina, urethra, and bladder trigone are also affected. The labia minora become thinner and regress, the introitus retracts, and narrowing and stricture of the vaginal canal and introitus may result. In some women, the urethral meatus becomes prominent relative to the introitus and more vulnerable to physical irritation, infection, and trauma.
Clinically, estrogen-related changes are usually responsible for vaginal dryness, irritation, burning, and superficial or deep dyspareunia. Urinary frequency, urgency, and incontinence also may develop.
THE DIAGNOSIS IS CLINICAL
The diagnosis of GSM is based on the history and physical examination. Standardized diagnostic tools for GSM are lacking, but some tools are available.
In 2006, the US Food and Drug Administration (FDA) published guidelines for industry to better define patient-reported outcome measures in clinical trials.13 The most significant addition was having the patient define the symptoms and rate how “bothersome” the symptoms are. Although this measure does not help diagnose GSM, it can be used effectively to follow response to treatment.
The Vaginal Symptom Questionnaire14 can be useful for assessing symptoms. It is a validated 21-item questionnaire that measures the quality-of-life impact of genital, but not urinary, symptoms of menopause.
Ask patients about symptoms
Healthcare providers should ask about GSM symptoms (Table 1) during routine clinical visits with women who are peri- or postmenopausal or who have hypoestrogenism from other causes, as many women are reluctant to initiate this discussion. Conversely, in women who present with sexual problems, such as difficulty with arousal or dyspareunia, GSM should be considered as a possible cause.
Specifically, ask women if they have any of the following symptoms:
- Vaginal itching, burning, discomfort, or irritation
- Malodorous or irritating vaginal discharge
- Urinary frequency, urgency, dysuria, urethral discomfort, or recurrent urinary tract infections
- Sexual symptoms of entry dyspareunia, vaginal pain, or irritation with sexual activity, which may be complicated by postcoital bleeding, spotting, or fissuring.
Vulvovaginal pain or irritation may be constant or may be present in the absence of sexual activity, such as with exercise, wearing tight clothing, or sitting for long periods.
Physical examination
Characteristic physical findings of GSM include scarce pubic hair, thinning of the labia from loss of labial fat, resorption of the labia minora, or fusion of the labia minora and majora (Table 2).15,16 The vulvar skin is pale and thin. The clitoral hood may retract, exposing the glans (which may lead to increased pain with sexual stimulation), or clitoral hood fusion may occur. The vagina is pale, dry, smooth, and shiny with loss of rugae; shortening or stricturing may be present. Vaginal elasticity decreases. Inflammation and petechiae (pinpoint, nonraised, round purple-red spots) may be present. The cervix may be flush with the vaginal fornices.
Prolonged atrophy may result in introital narrowing and friability, which may cause tearing with sexual activity or insertion of a speculum during pelvic examination. In addition, the epithelium of the lower urinary tract thins, and the muscular and fibrous layers atrophy—changes that may not be obvious during examination. A urethral caruncle may form, presenting as proliferative red tissue at the entrance of the urethra. Prolapse may become more prominent.
In women with severe genitourinary atrophy, pelvic examination may cause significant discomfort. Reassuring the patient that she can ask the clinician to stop at any time due to extreme discomfort is the first step in a successful pelvic examination.
In some situations, initial examination of the pelvic area may not include insertion of a speculum. Use of a hand-held mirror so the patient can observe the examination may help her relax during the examination.
Vaginal pH and cultures, if indicated, may be obtained by gently inserting a cotton-tipped swab into the vagina without a speculum and before applying lubricant. Lubricant should be used generously; in some instances, topical lidocaine gel (diluted, as it may burn) may be placed against the perineum on a gauze pad for 3 to 5 minutes before insertion of the speculum.
When an internal pelvic examination is necessary in a timely manner, such as with postmenopausal bleeding or a history of an abnormal Papanicolaou smear, but is too painful for the patient, the examination should be done under anesthesia.
Additional considerations
The history should review current medical conditions, medication use, nongenital skin disorders (eg, eczema), and systemic menopausal symptoms, such as hot flashes.
Also, consider other potential causes of GSM during the evaluation (Table 3).17,18 Review the use of detergents, soaps, douches, or over-the-counter topical products that could cause genitourinary symptoms secondary to contact irritation or allergy.
Any isolated, ulcerated, or nonhealing lesion should be biopsied. Reevaluate patients who have not responded to previous topical therapy or consider referral to a specialist.
Assess the personal, interpersonal, social, and sexual impact of the symptoms: if they do not cause distress, GSM does not require treatment. Nevertheless, potential treatment options should be discussed as symptoms may progress, making intervention necessary.
Laboratory tests: Helpful, not essential
Laboratory tests are unnecessary for the diagnosis of GSM. However, office-based objective evaluations such as vaginal pH testing and the maturation index can support the diagnosis.
The pH of the estrogenized vagina ranges from 3.8 to 4.2, whereas in women with GSM, the pH may reach 5.5 or higher. The pH can be obtained by placing a pH-sensitive paper against the lateral vaginal wall, avoiding any discharge or cervical mucus. A vaginal pH of 5 or greater in the absence of blood, semen, or infection suggests vulvovaginal atrophy.19
The vaginal maturation index is determined by a vaginal smear using Rakoff staining, in which 100 cells are counted and the number of parabasal, intermediate, and superficial cells is determined. In general, a well-estrogenized vagina has mostly superficial and intermediate cells, which shifts to a predominance of parabasal cells as estrogen levels decline.20
A recent review of vaginal atrophy suggests that after a diagnosis of GSM, healthcare providers can consider the most bothersome symptom along with the vaginal pH to assess the response to treatment.21 In general, schedule a follow-up appointment at 8 to 12 weeks to review treatment response. If treatment has not resulted in adequate symptom relief, consider a pelvic examination and further testing.
SELECTING A TREATMENT
Symptomatic women with GSM who desire intervention should be offered over-the-counter nonhormonal products as the first line of therapy.
If nonhormonal products are ineffective and there are no contraindications, locally applied estrogen in cream, tablet, or a ring delivery system may be offered. Local dehydroepiandrosterone (DHEA) inserts or ospemifene, an oral selective estrogen-receptor modulator, are FDA-approved for moderate to severe dyspareunia secondary to GSM.
Oral estrogen therapy is not indicated for vulvovaginal symptoms, but some women taking systemic estrogen for vasomotor symptoms may need additional local estrogen application to relieve vaginal symptoms.
Nonhormonal treatments
Nonhormonal over-the-counter therapies provide sufficient relief for most women with mild symptoms. There is a plethora of products, so practitioners need to offer guidance to help women with their individual choices.
Vaginal lubricants are intended for use with sexual or penetrative activity (including pelvic examination). They provide short-term relief of symptoms, but there is no evidence of any impact on histologic changes of atrophy. They are meant to relieve friction. Lubricants may be water-based, oil-based, silicone-based, or a combination. Individual products have different effects on condom integrity. Perfumed, warming, or stimulating products may be irritating to some women and should be tried initially in small amounts.
Vaginal moisturizers are intended to treat GSM. They are applied regularly, not just with vaginal activity, usually once or twice a week. Some vaginal lubricants can maintain an acidic pH in the vagina and may reverse the histologic changes of atrophy. Symptomatic improvement over placebo or estrogen has been shown in clinical trials.22–24
Women should be advised that trial and error in choosing products may be necessary to establish a successful regimen. Products should be tried in succession, not simultaneously, with a “wash-out” period between, to be able to evaluate response.
Vaginal dilators and pelvic floor physical therapy
Sexual activity, either by self-stimulation or with a partner, helps maintain vaginal health by contributing to increased vascularity and elasticity of tissue. Women who resume sexual activity after a long period of inactivity may benefit from the use of vaginal dilators, which aid both in mechanical distention and progressive relaxation of the vaginal musculature.
In some women, long-term dyspareunia may result in vaginismus, an involuntary contraction of the vaginal musculature. For these women, dilators may be effective. Additional options focus on pelvic floor physical therapy, which can isolate trigger points, using biofeedback to teach relaxation and home exercises such as vaginal massage.
HORMONAL THERAPIES
If nonhormonal lubricants and moisturizers do not achieve satisfactory symptomatic relief, FDA-approved hormonal therapies (Table 4) include estrogen-containing vaginal creams, rings, and a tablet; a vaginal tablet containing DHEA; and an oral tablet containing ospemifene.
Estrogen products
For patients whose symptoms do not respond to nonhormonal therapies, low-dose, locally applied estrogen therapy is the first treatment recommended.2 Locally applied estrogens can reverse the atrophic changes of estrogen deprivation, resulting in an increase in blood flow, elasticity, and vaginal wall thickness. This therapy also can normalize pH levels with subsequent restoration of a healthy lactobacilli-based flora. Locally applied estrogens also have been shown to decrease the frequency of recurrent urinary tract infection.25
Estrogen-containing vaginal creams, rings, and a tablet are available, and each has been shown to be effective for GSM. Locally applied estrogens at recommended dosages tend to have fewer adverse events and risks than systemic estrogens.26 Estradiol levels generally do not exceed levels found in the untreated menopausal population, although a dose- and duration-dependent increase in systemic levels may occur.27
Dosing considerations
The vaginal ring and the vaginal tablet provide the lowest prefixed daily dose of estradiol (7.5 and 10 µg daily, respectively). Estrogen creams (estradiol, conjugated equine estrogens) are more readily absorbed, and dosing should be tapered to the lowest, most effective dose for symptom relief.
The FDA-approved doses for vaginal creams containing 17-beta estradiol are higher than the dose found to be effective in clinical practice (0.5 g twice a week). Most practitioners start with the lower dose, reserving the FDA-approved higher doses for patients who do not obtain adequate relief over 6 to 8 weeks of treatment. The conjugated-estrogen vaginal cream Premarin is the only locally applied estrogen approved by the FDA to treat dyspareunia. It is dosed at 0.5 g intravaginally for 21 days and is then either withdrawn for 7 days or, more commonly, administered at 0.5 g twice a week.
Initial treatment with vaginal cream may require more frequent vulvovaginal application, such as daily for 1 to 2 weeks. Women with vaginal fissures or tearing will benefit from externally applied creams in addition to internal applications. Response to therapy is usually seen within 4 to 6 weeks from onset of treatment. Once symptom relief is obtained, treatment should continue indefinitely. Although long-term safety studies are lacking, risks are believed to be minimal.
Endometrial impact. Women with contraindications to systemic estrogen should be counseled about possible small increases in serum levels of estradiol associated with locally applied estrogens and the potential risks and benefits those increases incur. Endometrial surveillance with either transvaginal ultrasonography or endometrial sampling is not required, even with long-term use, but it should be considered with higher doses or more frequent applications.
Similarly, progesterone replacement for endometrial protection is not recommended but can be considered in women with an intact uterus at high risk of endometrial cancer, such as obese patients. If a systemic progestational agent is considered, the risks and benefits should be weighed carefully. Even in women at high risk, endometrial surveillance may be the most appropriate option.28 Uterine bleeding that occurs should be considered abnormal and should be investigated.
DHEA (prasterone)
In 2016, the FDA approved intravaginal prasterone, a DHEA-containing product for the treatment of dyspareunia secondary to moderate to severe vulvovaginal atrophy caused by menopause. DHEA is an endogenous steroid that is converted by aromatase activity into testosterone and estradiol.
Clinical trials have found that 12 weeks of vaginal DHEA supplementation (0.25%, 0.5%, and 1% DHEA ovules) was more effective than placebo in improving vaginal dryness and dyspareunia in women with GSM.29–31 In these studies, locally applied DHEA decreased parabasal cells, decreased vaginal pH, increased vaginal secretions, and improved epithelial surface thickness and integrity without any significant impact on serum levels of DHEA, DHEA-sulfate, estradiol, testosterone, or their metabolites. Importantly, transvaginal DHEA had negligible endometrial effect.
The breast cancer risk associated with vaginal DHEA has not been fully evaluated. However, labeling lists breast cancer as a warning, not a contraindication.
Selective estrogen-receptor modulator
In 2013, the FDA approved ospemifene for the treatment of dyspareunia caused by GSM. Ospemifene, an estrogen agonist in the vagina, is taken daily as a 60-mg oral dose. Long-term safety studies suggested no adverse effects on the endometrium or breast for at least 52 weeks.32
These studies also noted that ospemifene improved the vaginal maturation index (decreased parabasal cells and increased superficial cells) and decreased vaginal pH. It has further been shown to decrease severity of the self-identified most bothersome symptom—dyspareunia or vaginal dryness—compared with placebo.33
Potential increases in hot flashes, which may occur in up to 7% of patients, and the risk of blood clots should be considered. Additionally, the safety of ospemifene in women with a history of breast cancer has not been established. Although early studies suggest it either has no effect or possibly a protective effect on breast tissue, the FDA does not recommend its use in women at risk for breast cancer. Long-term effects on bone are unknown.
The labeling for ospemifene includes a boxed warning about the risk of stroke, blood clots, and cancer of the lining of the uterus. Patients should be counseled about worrisome signs or symptoms that require medical attention.
ALTERNATIVE THERAPIES
Treatments for GSM not approved by the FDA include laser and radiofrequency therapies, testosterone, isoflavones, and bioidentical hormones.
Laser and radiofrequency therapies
Both of these therapies aim to promote tissue remodeling with increased collagen and elastin production and increased vascularity. This, in turn, increases muscle support and tone.
Laser therapies act by ablating and coagulating vaginal tissues; radiofrequency therapies directly heat the tissue. Both treatments are office-based, require up to 3 initial treatments, and are followed by retreatment at approximately 1-year intervals.
Studies have reported high patient satisfaction rates (91% to 100%), improved sexual functioning, and decreased GSM symptoms of vaginal dryness, burning, itching, and dyspareunia.34–36 Data, however, are from observational studies, not placebo-controlled trials.
Although laser and radiofrequency therapies are FDA-approved for several indications, laser treatment for symptoms of vulvovaginal atrophy is not currently an approved indication. Patients should be advised of this.
Testosterone
Locally applied testosterone was shown in a small study to improve dyspareunia and vaginal dryness associated with aromatase inhibitor use in breast cancer patients.37 However, due to the lack of safety and efficacy data from larger, controlled trials, testosterone therapy is not currently recommended.
Isoflavones
Isoflavones are phytoestrogens found in soy. In a 12-week, double-blind placebo-controlled study of vaginally applied 4% soy isoflavone gel, improvements in vaginal atrophy symptoms, maturation values, and vaginal pH were found in 60 postmenopausal women.38 Additional data on efficacy and safety are needed before isoflavones should be considered as a treatment for GSM.
Bioidentical hormones
Bioidentical hormones are plant-derived hormones that are chemically similar or identical to those produced by the body. Although there are FDA-approved bioidentical hormones (eg, micronized progesterone, estradiol, DHEA), the term bioidentical usually refers to non-FDA-approved, commercially available hormones produced and compounded by specialty pharmacies.
Patients often view these substances as being better, safer, and more acceptable for use, and healthcare practitioners need to be prepared to address these beliefs. The FDA and the American College of Obstetricians and Gynecologists consider bioidentical hormones to be a marketing term and not an alternative treatment based on scientific evidence.39 Patients should be informed that bioidentical hormones have the same risks as any similar hormone preparation along with additional risks related to potential lack of purity and potency. Further, they have not been adequately studied in controlled clinical trials.
FOLLOW-UP CARE
Healthcare providers caring for women should assume a proactive role in diagnosing and treating the symptoms of GSM. And once diagnosis of GSM is established and treatment is under way, practitioners can use symptom questionnaires and vaginal pH testing as easy and reliable means of measuring clinical response to therapy.
For many women, the postmenopausal loss of estrogen is associated with uncomfortable genitourinary symptoms, collectively referred to as the genitourinary syndrome of menopause (GSM). But despite the prevalence of GSM and the availability of treatments, most women do not seek relief.
This article reviews the syndrome and offers advice on how to talk about it with patients and what treatment options to consider.
A SYNDROME RECENTLY DEFINED
The term GSM and its definition were approved by the North American Menopause Society and the International Society for the Study of Women’s Sexual Health in 2014.1 It replaces older terms such as vulvovaginal atrophy, urogenital atrophy, and atrophic vaginitis.
GSM refers collectively to the symptoms associated with estrogen loss after menopause that adversely affect the vulvovaginal area and lower urinary tract. The most common symptoms are vulvovaginal dryness, burning, or irritation; sexual pain from inadequate lubrication; and urinary urgency, dysuria, or recurrent urinary tract infection.1,2
The definition notes that symptoms are self-reported as bothersome and are not the result of another disorder. Symptoms may be chronic and progressive, are not likely to resolve without treatment (pharmacologic or nonpharmacologic), and can have a significant negative impact on a woman’s quality of life and sexual health.1,2
COMMON BUT UNDERTREATED
From 40% to 60% of postmenopausal women experience GSM, but few seek treatment.3 Nevertheless, most postmenopausal women remain sexually active. In a 2008 survey of 94,000 postmenopausal women ages 50 to 79, 52% reported that they had been sexually active with a partner in the past year.4 However, 45% of postmenopausal women experienced unpleasant vaginal symptoms, according to a 2012 international survey of 3,520 postmenopausal women ages 55 to 65.5 In this survey, most respondents (75%) felt that vaginal symptoms had a negative impact on their life, but only 4% connected their symptoms to the vulvovaginal atrophy that resulted from loss of estrogen after menopause. Moreover, almost half were unaware of management options.5
These findings were supported by a 2013 survey of more than 3,000 US women who reported unpleasant vulvar and vaginal symptoms.6 From 60% to 85% noted negative sexual consequences from vulvovaginal symptoms, 47% felt their relationship suffered, and 27% felt it had a negative impact on their general enjoyment of life. In this study, 24% attributed their symptoms to menopause and 12% to hormonal changes. Although 56% had discussed GSM symptoms with a healthcare provider, only 40% were using GSM-specific topical treatments, mostly over-the-counter preparations.
Male partners of symptomatic women also note adverse emotional and physical effects.7 In an online survey of 4,100 men and 4,100 women ages 55 to 65, 52% to 78% of men and 58% to 64% of women expressed the negative effects of vulvovaginal symptoms on intimacy, libido, and sexual pain.
GSM is a progressive disorder. Women may note symptoms many years before menopause or have no symptoms until several years after menopause. One study found the prevalence of GSM to be 4% during perimenopause, rising after menopause to 25% after 1 year and to 47% after 3 years.8
Although distressing symptoms occur mostly after menopause, they may be seen in women of any age who experience a hypoestrogenic state, even if it is transient. Causes of this include premature ovarian failure, hypothalamic amenorrhea, and hyperprolactinemia. In addition, some treatments such as gonadotropin-releasing hormone agonists and aromatase inhibitors may cause vulvovaginal and lower urinary tract symptoms. Chemotherapy, radiation, and surgical removal of ovaries may also precipitate symptoms. The abrupt onset of menopause that may occur with these treatments is often associated with significantly greater sexual dysfunction and negative impact on quality of life. Cigarette smoking also leads to lower estrogen levels, which may contribute to GSM.
WHAT CAUSES GSM?
The genitourinary system develops from common embryologic tissue, the basis for the functional and clinical connection. Estrogen maintains the epithelium of the vagina, vulva, urethra, and bladder trigone via estrogen receptors present throughout these tissues.9
Premenopausal changes
Histologically, the estrogen-exposed vagina of a premenopausal woman is lined by glycogen-rich, stratified squamous epithelium, with underlying supportive fibromuscular layers. The epithelium is composed of superficial, intermediate, and parabasal cellular layers. In the presence of estrogen, the superficial and intermediate cellular levels predominate, with few parabasal cells.
Glycogen acts as a substrate for lactobacilli, producing organic acids, primarily lactate, that help maintain an acidic pH of 2.8 to 4.0. The low pH helps protect against pathologic shifts in the microbiome. Estrogen also maintains the collagen content of the epithelium, maintains acid mucopolysaccharides and hyaluronic acid, and optimizes vaginal blood flow. These effects result in optimal epithelial thickness and elasticity, moisture, vaginal secretions, and lubrication.10
Postmenopausal changes
Low levels of estrogen after menopause result in adverse anatomic, physiologic, and clinical changes in vaginal tissue. Effects of hypoestrogenism include the loss of collagen and adipose, leading to decreased elasticity and vaginal mucosal thinning. Vascular flow is decreased. The epithelial cytology transitions to a predominance of parabasal cells and a decrease in superficial and intermediate cells. Eccrine and apocrine glands become attenuated. These changes result in decreased vaginal secretions, diminished or delayed lubrication with sexual stimulation, friability of the vaginal vault, and vaginal dryness.11
Additionally, without estrogen, glycogen content is diminished, leading to decreased lactic acid production and a rise in vaginal pH to greater than 5. As the pH rises, Lactobacillus colonization decreases, leading to a further decrease in glycogen metabolism and to propagation of an elevated vaginal pH. The loss of vaginal acidity makes the vagina more susceptible to pathologic bacteria, including those found in the bowel and skin, sexually transmitted infections, and bacterial vaginosis.12
Other affected tissues. Anatomic effects of estrogen loss are not limited to the vagina. The epithelium, connective tissue, and smooth muscle of the vulva, vagina, urethra, and bladder trigone are also affected. The labia minora become thinner and regress, the introitus retracts, and narrowing and stricture of the vaginal canal and introitus may result. In some women, the urethral meatus becomes prominent relative to the introitus and more vulnerable to physical irritation, infection, and trauma.
Clinically, estrogen-related changes are usually responsible for vaginal dryness, irritation, burning, and superficial or deep dyspareunia. Urinary frequency, urgency, and incontinence also may develop.
THE DIAGNOSIS IS CLINICAL
The diagnosis of GSM is based on the history and physical examination. Standardized diagnostic tools for GSM are lacking, but some tools are available.
In 2006, the US Food and Drug Administration (FDA) published guidelines for industry to better define patient-reported outcome measures in clinical trials.13 The most significant addition was having the patient define the symptoms and rate how “bothersome” the symptoms are. Although this measure does not help diagnose GSM, it can be used effectively to follow response to treatment.
The Vaginal Symptom Questionnaire14 can be useful for assessing symptoms. It is a validated 21-item questionnaire that measures the quality-of-life impact of genital, but not urinary, symptoms of menopause.
Ask patients about symptoms
Healthcare providers should ask about GSM symptoms (Table 1) during routine clinical visits with women who are peri- or postmenopausal or who have hypoestrogenism from other causes, as many women are reluctant to initiate this discussion. Conversely, in women who present with sexual problems, such as difficulty with arousal or dyspareunia, GSM should be considered as a possible cause.
Specifically, ask women if they have any of the following symptoms:
- Vaginal itching, burning, discomfort, or irritation
- Malodorous or irritating vaginal discharge
- Urinary frequency, urgency, dysuria, urethral discomfort, or recurrent urinary tract infections
- Sexual symptoms of entry dyspareunia, vaginal pain, or irritation with sexual activity, which may be complicated by postcoital bleeding, spotting, or fissuring.
Vulvovaginal pain or irritation may be constant or may be present in the absence of sexual activity, such as with exercise, wearing tight clothing, or sitting for long periods.
Physical examination
Characteristic physical findings of GSM include scarce pubic hair, thinning of the labia from loss of labial fat, resorption of the labia minora, or fusion of the labia minora and majora (Table 2).15,16 The vulvar skin is pale and thin. The clitoral hood may retract, exposing the glans (which may lead to increased pain with sexual stimulation), or clitoral hood fusion may occur. The vagina is pale, dry, smooth, and shiny with loss of rugae; shortening or stricturing may be present. Vaginal elasticity decreases. Inflammation and petechiae (pinpoint, nonraised, round purple-red spots) may be present. The cervix may be flush with the vaginal fornices.
Prolonged atrophy may result in introital narrowing and friability, which may cause tearing with sexual activity or insertion of a speculum during pelvic examination. In addition, the epithelium of the lower urinary tract thins, and the muscular and fibrous layers atrophy—changes that may not be obvious during examination. A urethral caruncle may form, presenting as proliferative red tissue at the entrance of the urethra. Prolapse may become more prominent.
In women with severe genitourinary atrophy, pelvic examination may cause significant discomfort. Reassuring the patient that she can ask the clinician to stop at any time due to extreme discomfort is the first step in a successful pelvic examination.
In some situations, initial examination of the pelvic area may not include insertion of a speculum. Use of a hand-held mirror so the patient can observe the examination may help her relax during the examination.
Vaginal pH and cultures, if indicated, may be obtained by gently inserting a cotton-tipped swab into the vagina without a speculum and before applying lubricant. Lubricant should be used generously; in some instances, topical lidocaine gel (diluted, as it may burn) may be placed against the perineum on a gauze pad for 3 to 5 minutes before insertion of the speculum.
When an internal pelvic examination is necessary in a timely manner, such as with postmenopausal bleeding or a history of an abnormal Papanicolaou smear, but is too painful for the patient, the examination should be done under anesthesia.
Additional considerations
The history should review current medical conditions, medication use, nongenital skin disorders (eg, eczema), and systemic menopausal symptoms, such as hot flashes.
Also, consider other potential causes of GSM during the evaluation (Table 3).17,18 Review the use of detergents, soaps, douches, or over-the-counter topical products that could cause genitourinary symptoms secondary to contact irritation or allergy.
Any isolated, ulcerated, or nonhealing lesion should be biopsied. Reevaluate patients who have not responded to previous topical therapy or consider referral to a specialist.
Assess the personal, interpersonal, social, and sexual impact of the symptoms: if they do not cause distress, GSM does not require treatment. Nevertheless, potential treatment options should be discussed as symptoms may progress, making intervention necessary.
Laboratory tests: Helpful, not essential
Laboratory tests are unnecessary for the diagnosis of GSM. However, office-based objective evaluations such as vaginal pH testing and the maturation index can support the diagnosis.
The pH of the estrogenized vagina ranges from 3.8 to 4.2, whereas in women with GSM, the pH may reach 5.5 or higher. The pH can be obtained by placing a pH-sensitive paper against the lateral vaginal wall, avoiding any discharge or cervical mucus. A vaginal pH of 5 or greater in the absence of blood, semen, or infection suggests vulvovaginal atrophy.19
The vaginal maturation index is determined by a vaginal smear using Rakoff staining, in which 100 cells are counted and the number of parabasal, intermediate, and superficial cells is determined. In general, a well-estrogenized vagina has mostly superficial and intermediate cells, which shifts to a predominance of parabasal cells as estrogen levels decline.20
A recent review of vaginal atrophy suggests that after a diagnosis of GSM, healthcare providers can consider the most bothersome symptom along with the vaginal pH to assess the response to treatment.21 In general, schedule a follow-up appointment at 8 to 12 weeks to review treatment response. If treatment has not resulted in adequate symptom relief, consider a pelvic examination and further testing.
SELECTING A TREATMENT
Symptomatic women with GSM who desire intervention should be offered over-the-counter nonhormonal products as the first line of therapy.
If nonhormonal products are ineffective and there are no contraindications, locally applied estrogen in cream, tablet, or a ring delivery system may be offered. Local dehydroepiandrosterone (DHEA) inserts or ospemifene, an oral selective estrogen-receptor modulator, are FDA-approved for moderate to severe dyspareunia secondary to GSM.
Oral estrogen therapy is not indicated for vulvovaginal symptoms, but some women taking systemic estrogen for vasomotor symptoms may need additional local estrogen application to relieve vaginal symptoms.
Nonhormonal treatments
Nonhormonal over-the-counter therapies provide sufficient relief for most women with mild symptoms. There is a plethora of products, so practitioners need to offer guidance to help women with their individual choices.
Vaginal lubricants are intended for use with sexual or penetrative activity (including pelvic examination). They provide short-term relief of symptoms, but there is no evidence of any impact on histologic changes of atrophy. They are meant to relieve friction. Lubricants may be water-based, oil-based, silicone-based, or a combination. Individual products have different effects on condom integrity. Perfumed, warming, or stimulating products may be irritating to some women and should be tried initially in small amounts.
Vaginal moisturizers are intended to treat GSM. They are applied regularly, not just with vaginal activity, usually once or twice a week. Some vaginal lubricants can maintain an acidic pH in the vagina and may reverse the histologic changes of atrophy. Symptomatic improvement over placebo or estrogen has been shown in clinical trials.22–24
Women should be advised that trial and error in choosing products may be necessary to establish a successful regimen. Products should be tried in succession, not simultaneously, with a “wash-out” period between, to be able to evaluate response.
Vaginal dilators and pelvic floor physical therapy
Sexual activity, either by self-stimulation or with a partner, helps maintain vaginal health by contributing to increased vascularity and elasticity of tissue. Women who resume sexual activity after a long period of inactivity may benefit from the use of vaginal dilators, which aid both in mechanical distention and progressive relaxation of the vaginal musculature.
In some women, long-term dyspareunia may result in vaginismus, an involuntary contraction of the vaginal musculature. For these women, dilators may be effective. Additional options focus on pelvic floor physical therapy, which can isolate trigger points, using biofeedback to teach relaxation and home exercises such as vaginal massage.
HORMONAL THERAPIES
If nonhormonal lubricants and moisturizers do not achieve satisfactory symptomatic relief, FDA-approved hormonal therapies (Table 4) include estrogen-containing vaginal creams, rings, and a tablet; a vaginal tablet containing DHEA; and an oral tablet containing ospemifene.
Estrogen products
For patients whose symptoms do not respond to nonhormonal therapies, low-dose, locally applied estrogen therapy is the first treatment recommended.2 Locally applied estrogens can reverse the atrophic changes of estrogen deprivation, resulting in an increase in blood flow, elasticity, and vaginal wall thickness. This therapy also can normalize pH levels with subsequent restoration of a healthy lactobacilli-based flora. Locally applied estrogens also have been shown to decrease the frequency of recurrent urinary tract infection.25
Estrogen-containing vaginal creams, rings, and a tablet are available, and each has been shown to be effective for GSM. Locally applied estrogens at recommended dosages tend to have fewer adverse events and risks than systemic estrogens.26 Estradiol levels generally do not exceed levels found in the untreated menopausal population, although a dose- and duration-dependent increase in systemic levels may occur.27
Dosing considerations
The vaginal ring and the vaginal tablet provide the lowest prefixed daily dose of estradiol (7.5 and 10 µg daily, respectively). Estrogen creams (estradiol, conjugated equine estrogens) are more readily absorbed, and dosing should be tapered to the lowest, most effective dose for symptom relief.
The FDA-approved doses for vaginal creams containing 17-beta estradiol are higher than the dose found to be effective in clinical practice (0.5 g twice a week). Most practitioners start with the lower dose, reserving the FDA-approved higher doses for patients who do not obtain adequate relief over 6 to 8 weeks of treatment. The conjugated-estrogen vaginal cream Premarin is the only locally applied estrogen approved by the FDA to treat dyspareunia. It is dosed at 0.5 g intravaginally for 21 days and is then either withdrawn for 7 days or, more commonly, administered at 0.5 g twice a week.
Initial treatment with vaginal cream may require more frequent vulvovaginal application, such as daily for 1 to 2 weeks. Women with vaginal fissures or tearing will benefit from externally applied creams in addition to internal applications. Response to therapy is usually seen within 4 to 6 weeks from onset of treatment. Once symptom relief is obtained, treatment should continue indefinitely. Although long-term safety studies are lacking, risks are believed to be minimal.
Endometrial impact. Women with contraindications to systemic estrogen should be counseled about possible small increases in serum levels of estradiol associated with locally applied estrogens and the potential risks and benefits those increases incur. Endometrial surveillance with either transvaginal ultrasonography or endometrial sampling is not required, even with long-term use, but it should be considered with higher doses or more frequent applications.
Similarly, progesterone replacement for endometrial protection is not recommended but can be considered in women with an intact uterus at high risk of endometrial cancer, such as obese patients. If a systemic progestational agent is considered, the risks and benefits should be weighed carefully. Even in women at high risk, endometrial surveillance may be the most appropriate option.28 Uterine bleeding that occurs should be considered abnormal and should be investigated.
DHEA (prasterone)
In 2016, the FDA approved intravaginal prasterone, a DHEA-containing product for the treatment of dyspareunia secondary to moderate to severe vulvovaginal atrophy caused by menopause. DHEA is an endogenous steroid that is converted by aromatase activity into testosterone and estradiol.
Clinical trials have found that 12 weeks of vaginal DHEA supplementation (0.25%, 0.5%, and 1% DHEA ovules) was more effective than placebo in improving vaginal dryness and dyspareunia in women with GSM.29–31 In these studies, locally applied DHEA decreased parabasal cells, decreased vaginal pH, increased vaginal secretions, and improved epithelial surface thickness and integrity without any significant impact on serum levels of DHEA, DHEA-sulfate, estradiol, testosterone, or their metabolites. Importantly, transvaginal DHEA had negligible endometrial effect.
The breast cancer risk associated with vaginal DHEA has not been fully evaluated. However, labeling lists breast cancer as a warning, not a contraindication.
Selective estrogen-receptor modulator
In 2013, the FDA approved ospemifene for the treatment of dyspareunia caused by GSM. Ospemifene, an estrogen agonist in the vagina, is taken daily as a 60-mg oral dose. Long-term safety studies suggested no adverse effects on the endometrium or breast for at least 52 weeks.32
These studies also noted that ospemifene improved the vaginal maturation index (decreased parabasal cells and increased superficial cells) and decreased vaginal pH. It has further been shown to decrease severity of the self-identified most bothersome symptom—dyspareunia or vaginal dryness—compared with placebo.33
Potential increases in hot flashes, which may occur in up to 7% of patients, and the risk of blood clots should be considered. Additionally, the safety of ospemifene in women with a history of breast cancer has not been established. Although early studies suggest it either has no effect or possibly a protective effect on breast tissue, the FDA does not recommend its use in women at risk for breast cancer. Long-term effects on bone are unknown.
The labeling for ospemifene includes a boxed warning about the risk of stroke, blood clots, and cancer of the lining of the uterus. Patients should be counseled about worrisome signs or symptoms that require medical attention.
ALTERNATIVE THERAPIES
Treatments for GSM not approved by the FDA include laser and radiofrequency therapies, testosterone, isoflavones, and bioidentical hormones.
Laser and radiofrequency therapies
Both of these therapies aim to promote tissue remodeling with increased collagen and elastin production and increased vascularity. This, in turn, increases muscle support and tone.
Laser therapies act by ablating and coagulating vaginal tissues; radiofrequency therapies directly heat the tissue. Both treatments are office-based, require up to 3 initial treatments, and are followed by retreatment at approximately 1-year intervals.
Studies have reported high patient satisfaction rates (91% to 100%), improved sexual functioning, and decreased GSM symptoms of vaginal dryness, burning, itching, and dyspareunia.34–36 Data, however, are from observational studies, not placebo-controlled trials.
Although laser and radiofrequency therapies are FDA-approved for several indications, laser treatment for symptoms of vulvovaginal atrophy is not currently an approved indication. Patients should be advised of this.
Testosterone
Locally applied testosterone was shown in a small study to improve dyspareunia and vaginal dryness associated with aromatase inhibitor use in breast cancer patients.37 However, due to the lack of safety and efficacy data from larger, controlled trials, testosterone therapy is not currently recommended.
Isoflavones
Isoflavones are phytoestrogens found in soy. In a 12-week, double-blind placebo-controlled study of vaginally applied 4% soy isoflavone gel, improvements in vaginal atrophy symptoms, maturation values, and vaginal pH were found in 60 postmenopausal women.38 Additional data on efficacy and safety are needed before isoflavones should be considered as a treatment for GSM.
Bioidentical hormones
Bioidentical hormones are plant-derived hormones that are chemically similar or identical to those produced by the body. Although there are FDA-approved bioidentical hormones (eg, micronized progesterone, estradiol, DHEA), the term bioidentical usually refers to non-FDA-approved, commercially available hormones produced and compounded by specialty pharmacies.
Patients often view these substances as being better, safer, and more acceptable for use, and healthcare practitioners need to be prepared to address these beliefs. The FDA and the American College of Obstetricians and Gynecologists consider bioidentical hormones to be a marketing term and not an alternative treatment based on scientific evidence.39 Patients should be informed that bioidentical hormones have the same risks as any similar hormone preparation along with additional risks related to potential lack of purity and potency. Further, they have not been adequately studied in controlled clinical trials.
FOLLOW-UP CARE
Healthcare providers caring for women should assume a proactive role in diagnosing and treating the symptoms of GSM. And once diagnosis of GSM is established and treatment is under way, practitioners can use symptom questionnaires and vaginal pH testing as easy and reliable means of measuring clinical response to therapy.
- Portman DJ, Gass ML; Vulvovaginal Atrophy Terminology Consensus Conference Panel. Genitourinary syndrome of menopause: new terminology for vulvovaginal atrophy from the International Society for the Study of Women’s Sexual Health and the North American Menopause Society. Menopause 2014; 21(10):1063–1068. doi:10.1097/GME.0000000000000329
- Management of symptomatic vulvovaginal atrophy: 2013 position statement of The North American Menopause Society. Menopause 2013; 20(9):888–904. doi:10.1097/GME.0b013e3182a122c2
- Parish SJ, Nappi RE, Krychman ML, et al. Impact of vulvovaginal health on postmenopausal women: a review of surveys on symptoms of vulvovaginal atrophy. Int J Womens Health 2013; 5:437–447. doi:10.2147/IJWH.S44579
- McCall-Hosenfeld JS, Jaramillo SA, Legault C, et al; Members of Women’s Health Initiative-Observational Study. Correlates of sexual satisfaction among sexually active postmenopausal women in the Women’s Health Initiative-Observational Study. J Gen Intern Med 2008; 23(12):2000–2009. doi:10.1007/s11606-008-0820-9
- Nappi RE, Kokot-Kierepa M. Vaginal Health: Insights, Views & Attitudes (VIVA): results from an international survey. Climacteric 2012; 15(1):36–44. doi:10.3109/13697137.2011.647840
- Kingsberg SA, Wysocki S, Magnus L, Krychman ML. Vulvar and vaginal atrophy in postmenopausal women: findings from the REVIVE (REal Women’s VIews of Treatment Options for Menopausal Vaginal Changes) survey. J Sex Med 2013; 10(7):1790–1799. doi:10.1111/jsm.12190
- Nappi RE, Kingsberg S, Maamari R, Simon J. The CLOSER (Clarifying Vaginal Atrophy’s Impact On Sex and Relationships) survey: implications of vaginal discomfort in postmenopausal women and in male partners. J Sex Med 2013; 10(9):2232–2241. doi:10.1111/jsm.12235
- Dennerstein L, Dudley EC, Hopper JL, Guthrie JR, Burger HG. A prospective population-based study of menopausal symptoms. Obstet Gynecol 2000; 96(3):351–358. pmid:10960625
- Stika CS. Atrophic vaginitis. Dermatol Ther 2010; 23(5):514–522. doi:10.1111/j.1529-8019.2010.01354.x
- Castelo-Branco C, Cancelo MJ, Villero J, Nohales F, Juliá MD. Management of postmenopausal vaginal atrophy and atrophic vaginitis. Maturitas 2005; 52(suppl 1):S46–S52. doi:10.1016/j.maturitas.2005.06.014
- Forsberg JG. A morphologist’s approach to the vagina—age-related changes and estrogen sensitivity. Maturitas 1995; 22(suppl):S7–S15.
- Martin DH. The microbiota of the vagina and its influence on women’s health and disease. Am J Med Sci 2012; 343(1):2–9. doi:10.1097/MAJ.0b013e31823ea228
- US Department of Health and Human Services; Food and Drug Administration (FDA). Guidance for industry. Patient-reported outcome measures: use in medical product development to support labeling claims, 2006. doi:10.1186/1477-7525-4-79
- Erekson EA, Yip SO, Wedderburn TS, et al. The Vulvovaginal Symptoms Questionnaire: a questionnaire for measuring vulvovaginal symptoms in postmenopausal women. Menopause 2013; 20(9):973–979. doi:10.1097/GME.0b013e318282600b
- Johnston SL, Farrell SA, Bouchard C, et al; SOGC Joint Committee-Clinical Practice Gynaecology and Urogynaecology. The detection and management of vaginal atrophy. J Obstet Gynaecol Can 2004; 26(5):503–515. doi:10.1016/S1701-2163(16)30662-4
- Oriba HA, Maibach HI. Vulvar transepidermal water loss (TEWL) decay curves. Effect of occlusion, delipidation, and age. Acta Derm Venereol 1989; 69(6):461–465. pmid:2575316
- Bachmann G, Nevadunsky NS. Diagnosis and treatment of atrophic vaginitis. Am Fam Physician 2000; 61(10):3090–3096. pmid:10839558
- MacBride MB, Rhodes DJ, Shuster LT. Vulvovaginal atrophy. Mayo Clin Proc 2010; 85(1):87–94. doi:10.4065/mcp.2009.0413
- Nilsson K, Risberg B, Heimer G. The vaginal epithelium in the post menopause--cytology, histology and pH as methods of assessment. Maturitas 1995; 21(1):51–56. pmid:7731384
- McEndree B. Clinical application of the vaginal maturation index. Nurse Pract 1999; 24(9):48–56. pmid:10507070
- Weber MA, Limpens J, Roovers JP. Assessment of vaginal atrophy: a review. Int Urogynecol J 2015; 26(1):15–28. doi:10.1007/s00192-014-2464-0
- Lee YK, Chung HH, Kim JW, Park NH, Song YS, Kang SB. Vaginal pH-balanced gel for the control of atrophic vaginitis among breast cancer survivors: a randomized controlled trial. Obstet Gynecol 2011; 117(4):922–927. doi:10.1097/AOG.0b013e3182118790
- Bygdeman M, Swahn ML. Replens versus dienoestrol cream in the symptomatic treatment of vaginal atrophy in postmenopausal women. Maturitas 1996; 23(3):259–263. pmid:8794418
- Nachtigall LE. Comparative study: replens versus local estrogen in menopausal women. Fertil Steril 1994; 61(1):178–180. pmid:8293835
- Raz R, Gennesin Y, Wasser J, et al. Recurrent urinary tract infections in postmenopausal women. Clin Infect Dis 2000; 30(1):152–156. doi:10.1086/313596
- Suckling J, Lethaby A, Kennedy R. Local oestrogen for vaginal atrophy in postmenopausal women. Cochrane Database Syst Rev 2006; 4:CD001500. doi:10.1002/14651858.CD001500
- Santen RJ. Vaginal administration of estradiol: effects of dose, preparation and timing on plasma estradiol levels. Climacteric 2015; 18(2):121–126. doi:10.3109/13697137.2014.947254
- North American Menopause Society. Estrogen and progestogen use in postmenopausal women: 2010 position statement of the North American Menopause Society. Menopause 2010; 17(2):242–255. doi:10.1097/gme.0b013e3181d0f6b9
- Labrie F, Archer D, Bouchard C, et al. Effect of intravaginal dehydroepiandrosterone (Prasterone) on libido and sexual dysfunction in postmenopausal women. Menopause 2009; 16(5):923–931. doi:10.1097/gme.0b013e31819e85c6
- Labrie F, Archer D, Bouchard C, et al. Intravaginal dehydroepiandrosterone (Prasterone), a physiological and highly efficient treatment of vaginal atrophy. Menopause 2009; 16(5):907–922. doi:10.1097/gme.0b013e31819e8e2d
- Archer DF. Dehydroepiandrosterone intra vaginal administration for the management of postmenopausal vulvovaginal atrophy. J Steroid Biochem Mol Biol 2015; 145:139–143. doi:10.1016/j.jsbmb.2014.09.003
- Wurz GT, Kao CJ, DeGregorio MW. Safety and efficacy of ospemifene for the treatment of dyspareunia associated with vulvar and vaginal atrophy due to menopause. Clin Interv Aging 2014; 9:1939–1950. doi:10.2147/CIA.S73753
- Constantine G, Graham S, Portman DJ, Rosen RC, Kingsberg SA. Female sexual function improved with ospemifene in postmenopausal women with vulvar and vaginal atrophy: results of a randomized, placebo-controlled trial. Climacteric 2015; 18(2):226–232. doi:10.3109/13697137.2014.954996
- Arroyo C. Fractional CO2 laser treatment for vulvovaginal atrophy symptoms and vaginal rejuvenation in perimenopausal women. Int J Womens Health 2017; 9:591–595. doi:10.2147/IJWH.S136857
- Perino A, Calligaro A, Forlani F, et al. Vulvo-vaginal atrophy: a new treatment modality using thermo-ablative fractional CO2 laser. Maturitas 2015; 80(3):296–301. doi:10.1016/j.maturitas.2014.12.006
- Salvatore S, Nappi R, Zerbinati N, et al. A 12-week treatment with fractional CO2 laser for vulvovaginal atrophy: a pilot study. Climacteric 2014; 17(4):363–369. doi:10.3109/13697137.2014.899347
- Witherby S, Johnson J, Demers L, et al. Topical testosterone for breast cancer patients with vaginal atrophy related to aromatase inhibitors: a phase I/II study. Oncologist 2011; 16(4):424–431. doi:10.1634/theoncologist.2010-0435
- Lima SM, Bernardo BF, Yamada SS, Reis BF, da Silva GM, Galvão MA. Effects of Glycine max (L.) Merr. soy isoflavone vaginal gel on epithelium morphology and estrogen receptor expression in postmenopausal women: a 12-week, randomized, double-blind, placebo-controlled trial. Maturitas 2014; 78(3):205–211. doi:10.1016/j.maturitas.2014.04.007
- Committee on Gynecologic Practice and the American Society for Reproductive Medicine Practice Committee. Committee opinion No 532: compounded bioidentical menopausal hormone therapy. Obstet Gynecol 2012; 120(2 pt 1):411–415. doi:10.1097/AOG.0b013e318268049e
- Portman DJ, Gass ML; Vulvovaginal Atrophy Terminology Consensus Conference Panel. Genitourinary syndrome of menopause: new terminology for vulvovaginal atrophy from the International Society for the Study of Women’s Sexual Health and the North American Menopause Society. Menopause 2014; 21(10):1063–1068. doi:10.1097/GME.0000000000000329
- Management of symptomatic vulvovaginal atrophy: 2013 position statement of The North American Menopause Society. Menopause 2013; 20(9):888–904. doi:10.1097/GME.0b013e3182a122c2
- Parish SJ, Nappi RE, Krychman ML, et al. Impact of vulvovaginal health on postmenopausal women: a review of surveys on symptoms of vulvovaginal atrophy. Int J Womens Health 2013; 5:437–447. doi:10.2147/IJWH.S44579
- McCall-Hosenfeld JS, Jaramillo SA, Legault C, et al; Members of Women’s Health Initiative-Observational Study. Correlates of sexual satisfaction among sexually active postmenopausal women in the Women’s Health Initiative-Observational Study. J Gen Intern Med 2008; 23(12):2000–2009. doi:10.1007/s11606-008-0820-9
- Nappi RE, Kokot-Kierepa M. Vaginal Health: Insights, Views & Attitudes (VIVA): results from an international survey. Climacteric 2012; 15(1):36–44. doi:10.3109/13697137.2011.647840
- Kingsberg SA, Wysocki S, Magnus L, Krychman ML. Vulvar and vaginal atrophy in postmenopausal women: findings from the REVIVE (REal Women’s VIews of Treatment Options for Menopausal Vaginal Changes) survey. J Sex Med 2013; 10(7):1790–1799. doi:10.1111/jsm.12190
- Nappi RE, Kingsberg S, Maamari R, Simon J. The CLOSER (Clarifying Vaginal Atrophy’s Impact On Sex and Relationships) survey: implications of vaginal discomfort in postmenopausal women and in male partners. J Sex Med 2013; 10(9):2232–2241. doi:10.1111/jsm.12235
- Dennerstein L, Dudley EC, Hopper JL, Guthrie JR, Burger HG. A prospective population-based study of menopausal symptoms. Obstet Gynecol 2000; 96(3):351–358. pmid:10960625
- Stika CS. Atrophic vaginitis. Dermatol Ther 2010; 23(5):514–522. doi:10.1111/j.1529-8019.2010.01354.x
- Castelo-Branco C, Cancelo MJ, Villero J, Nohales F, Juliá MD. Management of postmenopausal vaginal atrophy and atrophic vaginitis. Maturitas 2005; 52(suppl 1):S46–S52. doi:10.1016/j.maturitas.2005.06.014
- Forsberg JG. A morphologist’s approach to the vagina—age-related changes and estrogen sensitivity. Maturitas 1995; 22(suppl):S7–S15.
- Martin DH. The microbiota of the vagina and its influence on women’s health and disease. Am J Med Sci 2012; 343(1):2–9. doi:10.1097/MAJ.0b013e31823ea228
- US Department of Health and Human Services; Food and Drug Administration (FDA). Guidance for industry. Patient-reported outcome measures: use in medical product development to support labeling claims, 2006. doi:10.1186/1477-7525-4-79
- Erekson EA, Yip SO, Wedderburn TS, et al. The Vulvovaginal Symptoms Questionnaire: a questionnaire for measuring vulvovaginal symptoms in postmenopausal women. Menopause 2013; 20(9):973–979. doi:10.1097/GME.0b013e318282600b
- Johnston SL, Farrell SA, Bouchard C, et al; SOGC Joint Committee-Clinical Practice Gynaecology and Urogynaecology. The detection and management of vaginal atrophy. J Obstet Gynaecol Can 2004; 26(5):503–515. doi:10.1016/S1701-2163(16)30662-4
- Oriba HA, Maibach HI. Vulvar transepidermal water loss (TEWL) decay curves. Effect of occlusion, delipidation, and age. Acta Derm Venereol 1989; 69(6):461–465. pmid:2575316
- Bachmann G, Nevadunsky NS. Diagnosis and treatment of atrophic vaginitis. Am Fam Physician 2000; 61(10):3090–3096. pmid:10839558
- MacBride MB, Rhodes DJ, Shuster LT. Vulvovaginal atrophy. Mayo Clin Proc 2010; 85(1):87–94. doi:10.4065/mcp.2009.0413
- Nilsson K, Risberg B, Heimer G. The vaginal epithelium in the post menopause--cytology, histology and pH as methods of assessment. Maturitas 1995; 21(1):51–56. pmid:7731384
- McEndree B. Clinical application of the vaginal maturation index. Nurse Pract 1999; 24(9):48–56. pmid:10507070
- Weber MA, Limpens J, Roovers JP. Assessment of vaginal atrophy: a review. Int Urogynecol J 2015; 26(1):15–28. doi:10.1007/s00192-014-2464-0
- Lee YK, Chung HH, Kim JW, Park NH, Song YS, Kang SB. Vaginal pH-balanced gel for the control of atrophic vaginitis among breast cancer survivors: a randomized controlled trial. Obstet Gynecol 2011; 117(4):922–927. doi:10.1097/AOG.0b013e3182118790
- Bygdeman M, Swahn ML. Replens versus dienoestrol cream in the symptomatic treatment of vaginal atrophy in postmenopausal women. Maturitas 1996; 23(3):259–263. pmid:8794418
- Nachtigall LE. Comparative study: replens versus local estrogen in menopausal women. Fertil Steril 1994; 61(1):178–180. pmid:8293835
- Raz R, Gennesin Y, Wasser J, et al. Recurrent urinary tract infections in postmenopausal women. Clin Infect Dis 2000; 30(1):152–156. doi:10.1086/313596
- Suckling J, Lethaby A, Kennedy R. Local oestrogen for vaginal atrophy in postmenopausal women. Cochrane Database Syst Rev 2006; 4:CD001500. doi:10.1002/14651858.CD001500
- Santen RJ. Vaginal administration of estradiol: effects of dose, preparation and timing on plasma estradiol levels. Climacteric 2015; 18(2):121–126. doi:10.3109/13697137.2014.947254
- North American Menopause Society. Estrogen and progestogen use in postmenopausal women: 2010 position statement of the North American Menopause Society. Menopause 2010; 17(2):242–255. doi:10.1097/gme.0b013e3181d0f6b9
- Labrie F, Archer D, Bouchard C, et al. Effect of intravaginal dehydroepiandrosterone (Prasterone) on libido and sexual dysfunction in postmenopausal women. Menopause 2009; 16(5):923–931. doi:10.1097/gme.0b013e31819e85c6
- Labrie F, Archer D, Bouchard C, et al. Intravaginal dehydroepiandrosterone (Prasterone), a physiological and highly efficient treatment of vaginal atrophy. Menopause 2009; 16(5):907–922. doi:10.1097/gme.0b013e31819e8e2d
- Archer DF. Dehydroepiandrosterone intra vaginal administration for the management of postmenopausal vulvovaginal atrophy. J Steroid Biochem Mol Biol 2015; 145:139–143. doi:10.1016/j.jsbmb.2014.09.003
- Wurz GT, Kao CJ, DeGregorio MW. Safety and efficacy of ospemifene for the treatment of dyspareunia associated with vulvar and vaginal atrophy due to menopause. Clin Interv Aging 2014; 9:1939–1950. doi:10.2147/CIA.S73753
- Constantine G, Graham S, Portman DJ, Rosen RC, Kingsberg SA. Female sexual function improved with ospemifene in postmenopausal women with vulvar and vaginal atrophy: results of a randomized, placebo-controlled trial. Climacteric 2015; 18(2):226–232. doi:10.3109/13697137.2014.954996
- Arroyo C. Fractional CO2 laser treatment for vulvovaginal atrophy symptoms and vaginal rejuvenation in perimenopausal women. Int J Womens Health 2017; 9:591–595. doi:10.2147/IJWH.S136857
- Perino A, Calligaro A, Forlani F, et al. Vulvo-vaginal atrophy: a new treatment modality using thermo-ablative fractional CO2 laser. Maturitas 2015; 80(3):296–301. doi:10.1016/j.maturitas.2014.12.006
- Salvatore S, Nappi R, Zerbinati N, et al. A 12-week treatment with fractional CO2 laser for vulvovaginal atrophy: a pilot study. Climacteric 2014; 17(4):363–369. doi:10.3109/13697137.2014.899347
- Witherby S, Johnson J, Demers L, et al. Topical testosterone for breast cancer patients with vaginal atrophy related to aromatase inhibitors: a phase I/II study. Oncologist 2011; 16(4):424–431. doi:10.1634/theoncologist.2010-0435
- Lima SM, Bernardo BF, Yamada SS, Reis BF, da Silva GM, Galvão MA. Effects of Glycine max (L.) Merr. soy isoflavone vaginal gel on epithelium morphology and estrogen receptor expression in postmenopausal women: a 12-week, randomized, double-blind, placebo-controlled trial. Maturitas 2014; 78(3):205–211. doi:10.1016/j.maturitas.2014.04.007
- Committee on Gynecologic Practice and the American Society for Reproductive Medicine Practice Committee. Committee opinion No 532: compounded bioidentical menopausal hormone therapy. Obstet Gynecol 2012; 120(2 pt 1):411–415. doi:10.1097/AOG.0b013e318268049e
KEY POINTS
- Practitioners can use the patient’s most bothersome symptom and her vaginal pH level to assess clinical responses to therapy.
- The diagnosis is based on clinical signs and symptoms from the medical history and physical examination.
- If the symptoms are not bothersome to the patient, the syndrome does not require treatment.

Chief complaint: Homicidal. Assessing violence risk
Mr. F, age 35, is homeless and has a history of cocaine and alcohol use disorders. He is admitted voluntarily to the psychiatric unit because he has homicidal thoughts toward Ms. S, who works in the shelter where he has been staying. Mr. F reports that he is thinking of killing Ms. S if he is discharged because she has been rude to him. He states that he has access to several firearms, but he will not disclose the location. He has been diagnosed with unspecified depressive disorder and exhibited antisocial personality disorder traits. He is being treated with sertraline. However, his mood appears to be relatively stable, except for occasional angry verbal outbursts. The outbursts have been related to intrusive peers or staff turning the television off for group meetings. Mr. F has been joking with peers, eating well, and sleeping appropriately. He reports no suicidal thoughts and has not been physically violent on the unit. However, Mr. F has had a history of violence since his teenage years. He has been incarcerated twice for assault and once for drug possession.
How would you approach assessing and managing Mr. F’s risk for violence?
We all have encountered a patient similar to Mr. F on the psychiatric unit or in the emergency department—a patient who makes violent threats and appears angry, intimidating, manipulative, and/or demanding, despite exhibiting no evidence of mania or psychosis. This patient often has a history of substance abuse and a lifelong pattern of viewing violence as an acceptable way of addressing life’s problems. Many psychiatrists suspect that more time on the inpatient unit is unlikely to reduce this patient’s risk of violence. Why? Because the violence risk does not stem from a treatable mental illness. Further, psychiatrists may be apprehensive about this patient’s potential for violence after discharge and their liability in the event of a bad outcome. No one wants their name associated with a headline that reads “Psychiatrist discharged man less than 24 hours before he killed 3 people.”
The purported relationship between mental illness and violence often is sensationalized in the media. However, research reveals that the vast majority of violence is in fact not due to symptoms of mental illness.1,2 A common clinical challenge in psychiatry involves evaluating individuals at elevated risk of violence and determining how to address their risk factors for violence. When the risk is primarily due to psychosis and can be reduced with antipsychotic medication, the job is easy. But how should we proceed when the risk stems from factors other than mental illness?
This article
Violence and mental illness: A tenuous link
Violence is a major public health concern in the United States. Although in recent years the rates of homicide and aggravated assault have decreased dramatically, there are approximately 16,000 homicides annually in the United States, and more than 1.6 million injuries from assaults treated in emergency departments each year.3 Homicide continues to be one of the leading causes of death among teenagers and young adults.4
The most effective methods of preventing widespread violence are public health approaches, such as parent- and family-focused programs, early childhood education, programs in school, and public policy changes.3 However, as psychiatrists, we are routinely asked to assess the risk of violence for an individual patient and devise strategies to mitigate violence risk.
Continue to: Although certain mental illnesses...
Although certain mental illnesses increase the relative risk of violence (compared with people without mental illness),5,6 recent studies suggest that mental illness plays only a “minor role in explaining violence in populations.”7 It is estimated that as little as 4% of the violence in the United States can be attributed to mental illness.1 According to a 1998 meta-analysis of 48 studies of criminal recidivism, the risk factors for violent recidivism were “almost identical” among offenders who had a mental disorder and those who did not.8
Approaches to assessing violence risk
Psychiatrists can assess the risk of future violence via 3 broad approaches.9,10
Unaided clinical judgment is when a mental health professional estimates violence risk based on his or her own experience and intuition, with knowledge of violence risk factors, but without the use of structured tools.
Actuarial tools are statistical models that use formulae to show relationships between data (risk factors) and outcomes (violence).10,11
Continue to: Structured professional judgment
Structured professional judgment is a hybrid of unaided clinical judgment and actuarial methods. Structured professional judgment tools help the evaluator identify empirically established risk factors. Once the information is collected, it is combined with clinical judgment in decision making.9,10 There are now more than 200 structured tools available for assessing violence risk in criminal justice and forensic mental health populations.12
Clinical judgment, although commonly used in practice, is less accurate than actuarial tools or structured professional judgment.10,11 In general, risk assessment tools offer moderate levels of accuracy in categorizing people at low risk vs high risk.5,13 The tools have better ability to accurately categorize individuals at low risk, compared with high risk, where false positives are common.12,14
Two types of risk factors
Risk factors for violence are commonly categorized as static or dynamic factors. Static factors are historical factors that cannot be changed with intervention (eg, age, sex, history of abuse). Dynamic factors can be changed with intervention (eg, substance abuse).15
Static risk factors. The best predictor of future violence is past violent behavior.5,16,17 Violence risk increases with each prior episode of violence.5 Prior arrests for any crime, especially if the individual was a juvenile at the time of arrest for his or her first violent offense, increase future violence risk.5 Other important static violence risk factors include demographic factors such as age, sex, and socioeconomic status. Swanson et al6 reviewed a large pool of data (approximately 10,000 respondents) from the Epidemiologic Catchment Area survey. Being young, male, and of low socioeconomic status were all associated with violence in the community.6 The highest-risk age group for violence is age 15 to 24.5 Males perpetrate violence in the community at a rate 10 times that of females.18 However, among individuals with severe mental illness, men and women have similar rates of violence.19,20 Unstable employment,21 less education,22 low intelligence,16 and a history of a significant head injury5 also are risk factors for violence.5
Continue to: Being abused as a child...
Being abused as a child, witnessing violence in the home,5,16 and growing up with an unstable parental situation (eg, parental loss or separation) has been linked to violence.16,23,24 Early disruptive behavior in childhood (eg, fighting, lying and stealing, truancy, and school problems) increases violence risk.21,23
Personality factors are important static risk factors for violence. Antisocial personality disorder is the most common personality disorder linked with violence.17 Several studies consistently show psychopathy to be a strong predictor of both violence and criminal behavior.5,25 A psychopath is a person who lacks empathy and close relationships, behaves impulsively, has superficially charming qualities, and is primarily interested in self-gratification.26 Harris et al27 studied 169 released forensic patients and found that 77% of the psychopaths (according to Psychopathy Checklist-Revised [PCL-R] scores) violently recidivated. In contrast, only 21% of the non-psychopaths violently recidivated.27
Other personality factors associated with violence include a predisposition toward feelings of anger and hatred (as opposed to empathy, anxiety, or guilt, which may reduce risk), hostile attributional biases (a tendency to interpret benign behavior of others as intentionally antagonistic), violent fantasies, poor anger control, and impulsivity.5 Although personality factors tend to be longstanding and more difficult to modify, in the outpatient setting, therapeutic efforts can be made to modify hostile attribution biases, poor anger control, and impulsive behavior.
Dynamic risk factors. Substance abuse is strongly associated with violence.6,17 The prevalence of violence is 12 times greater among individuals with alcohol use disorder and 16 times greater among individuals with other substance use disorders, compared with those with no such diagnoses.5,6
Continue to: Steadman et al...
Steadman et al28 compared 1,136 adult patients with mental disorders discharged from psychiatric hospitals with 519 individuals living in the same neighborhoods as the hospitalized patients. They found that the prevalence of violence among discharged patients without substance abuse was “statistically indistinguishable” from the prevalence of violence among community members, in the same neighborhood, who did not have symptoms of substance abuse.28 Swanson et al6 found that the combination of a mental disorder plus an alcohol or substance use disorder substantially increased the risk of violence.
Other dynamic risk factors for violence include mental illness symptoms such as psychosis, especially threat/control-override delusions, where the individual believes that they are being threatened or controlled by an external force.17
Contextual factors to consider in violence risk assessments include current stressors, lack of social support, availability of weapons, access to drugs and alcohol, and the presence of similar circumstances that led to violent behavior in the past.5
How to assess the risk of targeted violence
Targeted violence is a predatory act of violence intentionally committed against a preselected person, group of people, or place.29 Due to the low base rates of these incidents, targeted violence is difficult to study.7,30 These risk assessments require a more specialized approach.
Continue to: In their 1999 article...
In their 1999 article, Borum et al30 discussed threat assessment strategies utilized by the U.S. Secret Service and recommended investigating “pathways of ideas and behaviors that may lead to violent action.” Borum et al30 summarized 3 fundamental principles of threat assessment (Table 130).

What to do when violence risk is not due to mental illness
Based on the information in Mr. F’s case scenario, it is likely that his homicidal ideation is not due to mental illness. Despite this, several risk factors for violence are present. Where do we go from here?
Scott and Resnick17 recommend considering the concept of dangerousness as 5 components (Table 217). When this model of dangerousness is applied to Mr. F’s case, one can see that the magnitude of the harm is great because of threatened homicide. With regard to the imminence of the harm, it would help to clarify whether Mr. F plans to kill Ms. S immediately after discharge, or sometime in the next few months. Is his threat contingent on further provocations by Ms. S? Alternatively, does he intend to kill her for past grievances, regardless of further perceived insults?

Next, the frequency of a behavior relates to how often Mr. F has been aggressive in the past. The severity of his past aggression is also important. What is the most violent act he has ever done? Situational factors in this case include Mr. F’s access to weapons, financial problems, housing problems, and access to drugs and alcohol.17 Mr. F should be asked about what situations previously provoked his violent behavior. Consider how similar the present conditions are to past conditions to which Mr. F responded violently.5 The likelihood that a homicide will occur should take into account Mr. F’s risk factors for violence, as well as the seriousness of his intent to cause harm.
Continue to: Consider using a structured tool...
Consider using a structured tool, such as the Classification of Violence Risk, to help identify Mr. F’s risk factors for violence, or some other formal method to ensure that the proper data are collected. Violence risk assessments are more accurate when structured risk assessment tools are used, compared with clinical judgment alone.
It is important to review collateral sources of information. In Mr. F’s case, useful collateral sources may include his criminal docket (usually available online), past medical records, information from the shelter where he lives, and, potentially, friends or family.
Because Mr. F is making threats of targeted violence, be sure to ask about attack-related behaviors (Table 130).
Regarding the seriousness of Mr. F’s intent to cause harm, it may be helpful to ask him the following questions:
- How likely are you to carry out this act of violence?
- Do you have a plan? Have you taken any steps toward this plan?
- Do you see other, nonviolent solutions to this problem?
- What do you hope that we can do for you to help with this problem?
Continue to: Mr. F's answers...
Mr. F’s answers may suggest the possibility of a hidden agenda. Some patients express homicidal thoughts in order to stay in the hospital. If Mr. F expresses threats that are contingent on discharge and declines to engage in problem-solving discussions, this would cast doubt on the genuineness of his threat. However, doubt about the genuineness of the threat alone is not sufficient to simply discharge Mr. F. Assessment of his intent needs to be considered with other relevant risk factors, risk reduction strategies, and any Tarasoff duties that may apply.
In addition to risk factors, consider mitigating factors. For example, does Mr. F express concern over prison time as a reason to not engage in violence? It would be more ominous if Mr. F says that he does not care if he goes to prison because life is lousy being homeless and unemployed. At this point, an estimation can be made regarding whether Mr. F is a low-, moderate-, or high-risk of violence.
The next step is to organize Mr. F’s risk factors into static (historical) and dynamic (subject to intervention) factors. This will be helpful in formulating a strategy to manage risk because continued hospitalization can only address dynamic risk factors. Often in these cases, the static risk factors are far more numerous than the dynamic risk factors.
Once the data are collected and organized, the final step is to devise a risk management strategy. Some interventions, such as substance use treatment, will be straightforward. A mood-stabilizing medication could be considered, if clinically appropriate, to help reduce aggression and irritability.31 Efforts should be made to eliminate Mr. F’s access to firearms; however, in this case, it sounds unlikely that he will cooperate with those efforts. Ultimately, you may find yourself with a list of risk factors that are unlikely to be altered with further hospitalization, particularly if Mr. F’s homicidal thoughts and intent are due to antisocial personality traits.
Continue to: In that case...
In that case, the most important step will be to carry out your duty to warn/protect others prior to Mr. F’s discharge. Most states either require or permit mental health professionals to take reasonable steps to protect victims from violence when certain conditions are present, such as an explicit threat or identifiable victim (see Related Resources).
Once dynamic risk factors have been addressed, and duty to warn/protect is carried out, if there is no further clinical indication for hospitalization, it would be appropriate to discharge Mr. F. Continued homicidal threats stemming from antisocial personality traits, in the absence of a treatable mental illness (or other modifiable risk factors for violence that can be actively addressed), is not a reason for continued hospitalization. It may be useful to obtain a second opinion from a colleague in such scenarios. A second opinion may offer additional risk management ideas. In the event of a bad outcome, this will also help to show that the decision to discharge the patient was not taken lightly.
The psychiatrist should document a thoughtful risk assessment, the strategies that were implemented to reduce risk, the details of the warning, and the reasoning why continued hospitalization was not indicated (Table 3).

CASE CONTINUED
Decision to discharge
In Mr. F’s case, the treating psychiatrist determined that Mr. F’s risk of violence toward Ms. S was moderate. The psychiatrist identified several static risk factors for violence that raised Mr. F’s risk, but also noted that Mr. F’s threats were likely a manipulative effort to prolong his hospital stay. The psychiatrist carried out his duty to protect by notifying police and Ms. S of the nature of the threat prior to Mr. F’s discharge. The unit social worker helped Mr. F schedule an intake appointment for a substance use disorder treatment facility. Mr. F ultimately stated that he no longer experienced homicidal ideas once a bed was secured for him in a substance use treatment program. The psychiatrist carefully documented Mr. F’s risk assessment and the reasons why Mr. F’s risk would not be significantly altered by further inpatient hospitalization. Mr. F was discharged, and Ms. S remained unharmed.
Continue to: Bottom Line
Bottom Line
Use a structured approach to identify risk factors for violence. Address dynamic risk factors, including access to weapons. Carry out the duty to warn/protect if applicable. Document your decisions and actions carefully, and then discharge the patient if clinically indicated. Do not be “held hostage” by a patient’s homicidal ideation.
Related Resources
- Dolan M, Doyle M. Violence risk prediction. Clinical and actuarial measures and the role of the psychopathy checklist. Br J Psychiatry. 2000;177:303-311.
- Douglas KS, Hart SD, Webster CD, et al. HCR-20V3: Assessing risk of violence–user guide. Burnaby, Canada: Mental Health, Law, and Policy Institute, Simon Fraser University; 2013.
- National Conference of State Legislatures. Mental health professionals’ duty to warn. http://www.ncsl.org/research/health/mental-health-professionals-duty-to-warn.aspx. Published September 28, 2015.
Drug Brand Names
Sertraline • Zoloft
1. Skeem J, Kennealy P, Monahan J, et al. Psychosis uncommonly and inconsistently precedes violence among high-risk individuals. Clin Psychol Sci. 2016;4(1):40-49.
2. McGinty E, Frattaroli S, Appelbaum PS, et al. Using research evidence to reframe the policy debate around mental illness and guns: process and recommendations. Am J Public Health. 2014;104(11):e22-e26.
3. Sumner SA, Mercy JA, Dahlberg LL, et al. Violence in the United States: status, challenges, and opportunities. JAMA. 2015;314(5):478-488.
4. Heron M. Deaths: leading causes for 2014. Natl Vital Stat Rep. 2016;65(5):1-96.
5. Borum R, Swartz M, Swanson J. Assessing and managing violence risk in clinical practice. J Prac Psychiatry Behav Health. 1996;2(4):205-215.
6. Swanson JW, Holzer CE 3rd, Ganju VK, et al. Violence and psychiatric disorder in the community: Evidence from the epidemiologic catchment area surveys. Hosp Community Psychiatry. 1990;41(7):761-770.
7. Swanson JW. Explaining rare acts of violence: the limits of evidence from population research. Psychiatr Serv. 2011;62(11):1369-1371.
8. Bonta J, Law M, Hanson K. The prediction of criminal and violent recidivism among mentally disordered offenders: a meta-analysis. Psychol Bull. 1998;123(2):123-142.
9. Monahan J. The inclusion of biological risk factors in violence risk assessments. In: Singh I, Sinnott-Armstrong W, Savulescu J, eds. Bioprediction, biomarkers, and bad behavior: scientific, legal, and ethical implications. New York, NY: Oxford University Press; 2014:57-76.
10. Murray J, Thomson ME. Clinical judgement in violence risk assessment. Eur J Psychol. 2010;6(1):128-149.
11. Mossman D. Violence risk: is clinical judgment enough? Current Psychiatry. 2008;7(6):66-72.
12. Douglas T, Pugh J, Singh I, et al. Risk assessment tools in criminal justice and forensic psychiatry: the need for better data. Eur Psychiatry. 2017;42:134-137.
13. Dolan M, Doyle M. Violence risk prediction. Clinical and actuarial measures and the role of the psychopathy checklist. Br J Psychiatry. 2000;177:303-311.
14. Fazel S, Singh J, Doll H, et al. Use of risk assessment instruments to predict violence and antisocial behaviour in 73 samples involving 24 827 people: systematic review and meta-analysis. BMJ. 2012;345:e4692. doi: 10.1136/bmj.e4692.
15. National Collaborating Centre for Mental Health (UK). Violence and aggression: short- term management in mental health, health, and community settings: updated edition. London: British Psychological Society; 2015. NICE Guideline, No 10.
16. Klassen D, O’Connor WA. Predicting violence in schizophrenic and non-schizophrenic patients: a prospective study. J Community Psychol. 1988;16(2):217-227.
17. Scott C, Resnick P. Clinical assessment of aggression and violence. In: Rosner R, Scott C, eds. Principles and practice of forensic psychiatry, 3rd ed. Boca Raton, FL: CRC Press; 2017:623-631.
18. Tardiff K, Sweillam A. Assault, suicide, and mental illness. Arch Gen Psychiatry. 1980;37(2):164-169.
19. Lidz CW, Mulvey EP, Gardner W. The accuracy of predictions of violence to others. JAMA. 1993;269(8):1007-1011.
20. Newhill CE, Mulvey EP, Lidz CW. Characteristics of violence in the community by female patients seen in a psychiatric emergency service. Psychiatric Serv. 1995;46(8):785-789.
21. Mulvey E, Lidz C. Clinical considerations in the prediction of dangerousness in mental patients. Clin Psychol Rev. 1984;4(4):379-401.
22. Link BG, Andrews H, Cullen FT. The violent and illegal behavior of mental patients reconsidered. Am Sociol Rev. 1992;57(3):275-292.
23. Harris GT, Rice ME, Quinsey VL. Violent recidivism of mentally disordered offenders: the development of a statistical prediction instrument. Crim Justice and Behav. 1993;20(4):315-335.
24. Klassen D, O’Connor W. Demographic and case history variables in risk assessment. In: Monahan J, Steadman H, eds. Violence and mental disorder: developments in risk assessment. Chicago, IL: University of Chicago Press; 1994:229-257.
25. Hart SD, Hare RD, Forth AE. Psychopathy as a risk marker for violence: development and validation of a screening version of the revised Psychopathy Checklist. In: Monahan J, Steadman HJ, eds. Violence and mental disorder: developments in risk assessment. Chicago, IL: University of Chicago Press; 1994:81-98.
26. Cleckley H. The mask of sanity. St. Louis, MO: Mosby; 1941.
27. Harris GT, Rice ME, Cormier CA. Psychopathy and violent recidivism. Law Hum Behav. 1991;15(6):625-637.
28. Steadman HJ, Mulvey EP, Monahan J. Violence by people discharged from acute psychiatric inpatient facilities and by others in the same neighborhoods. Arch Gen Psychiatry. 1998;55:393-401.
29. Meloy JR, White SG, Hart S. Workplace assessment of targeted violence risk: the development and reliability of the WAVR-21. J Forensic Sci. 2013;58(5):1353-1358.
30. Borum R, Fein R, Vossekuil B, et al. Threat assessment: defining an approach for evaluating risk of targeted violence. Behav Sci Law. 1999;17(3):323-337.
31. Tyrer P, Bateman AW. Drug treatment for personality disorders. Adv Psychiatr Treat. 2004;10(5):389-398.
Mr. F, age 35, is homeless and has a history of cocaine and alcohol use disorders. He is admitted voluntarily to the psychiatric unit because he has homicidal thoughts toward Ms. S, who works in the shelter where he has been staying. Mr. F reports that he is thinking of killing Ms. S if he is discharged because she has been rude to him. He states that he has access to several firearms, but he will not disclose the location. He has been diagnosed with unspecified depressive disorder and exhibited antisocial personality disorder traits. He is being treated with sertraline. However, his mood appears to be relatively stable, except for occasional angry verbal outbursts. The outbursts have been related to intrusive peers or staff turning the television off for group meetings. Mr. F has been joking with peers, eating well, and sleeping appropriately. He reports no suicidal thoughts and has not been physically violent on the unit. However, Mr. F has had a history of violence since his teenage years. He has been incarcerated twice for assault and once for drug possession.
How would you approach assessing and managing Mr. F’s risk for violence?
We all have encountered a patient similar to Mr. F on the psychiatric unit or in the emergency department—a patient who makes violent threats and appears angry, intimidating, manipulative, and/or demanding, despite exhibiting no evidence of mania or psychosis. This patient often has a history of substance abuse and a lifelong pattern of viewing violence as an acceptable way of addressing life’s problems. Many psychiatrists suspect that more time on the inpatient unit is unlikely to reduce this patient’s risk of violence. Why? Because the violence risk does not stem from a treatable mental illness. Further, psychiatrists may be apprehensive about this patient’s potential for violence after discharge and their liability in the event of a bad outcome. No one wants their name associated with a headline that reads “Psychiatrist discharged man less than 24 hours before he killed 3 people.”
The purported relationship between mental illness and violence often is sensationalized in the media. However, research reveals that the vast majority of violence is in fact not due to symptoms of mental illness.1,2 A common clinical challenge in psychiatry involves evaluating individuals at elevated risk of violence and determining how to address their risk factors for violence. When the risk is primarily due to psychosis and can be reduced with antipsychotic medication, the job is easy. But how should we proceed when the risk stems from factors other than mental illness?
This article
Violence and mental illness: A tenuous link
Violence is a major public health concern in the United States. Although in recent years the rates of homicide and aggravated assault have decreased dramatically, there are approximately 16,000 homicides annually in the United States, and more than 1.6 million injuries from assaults treated in emergency departments each year.3 Homicide continues to be one of the leading causes of death among teenagers and young adults.4
The most effective methods of preventing widespread violence are public health approaches, such as parent- and family-focused programs, early childhood education, programs in school, and public policy changes.3 However, as psychiatrists, we are routinely asked to assess the risk of violence for an individual patient and devise strategies to mitigate violence risk.
Continue to: Although certain mental illnesses...
Although certain mental illnesses increase the relative risk of violence (compared with people without mental illness),5,6 recent studies suggest that mental illness plays only a “minor role in explaining violence in populations.”7 It is estimated that as little as 4% of the violence in the United States can be attributed to mental illness.1 According to a 1998 meta-analysis of 48 studies of criminal recidivism, the risk factors for violent recidivism were “almost identical” among offenders who had a mental disorder and those who did not.8
Approaches to assessing violence risk
Psychiatrists can assess the risk of future violence via 3 broad approaches.9,10
Unaided clinical judgment is when a mental health professional estimates violence risk based on his or her own experience and intuition, with knowledge of violence risk factors, but without the use of structured tools.
Actuarial tools are statistical models that use formulae to show relationships between data (risk factors) and outcomes (violence).10,11
Continue to: Structured professional judgment
Structured professional judgment is a hybrid of unaided clinical judgment and actuarial methods. Structured professional judgment tools help the evaluator identify empirically established risk factors. Once the information is collected, it is combined with clinical judgment in decision making.9,10 There are now more than 200 structured tools available for assessing violence risk in criminal justice and forensic mental health populations.12
Clinical judgment, although commonly used in practice, is less accurate than actuarial tools or structured professional judgment.10,11 In general, risk assessment tools offer moderate levels of accuracy in categorizing people at low risk vs high risk.5,13 The tools have better ability to accurately categorize individuals at low risk, compared with high risk, where false positives are common.12,14
Two types of risk factors
Risk factors for violence are commonly categorized as static or dynamic factors. Static factors are historical factors that cannot be changed with intervention (eg, age, sex, history of abuse). Dynamic factors can be changed with intervention (eg, substance abuse).15
Static risk factors. The best predictor of future violence is past violent behavior.5,16,17 Violence risk increases with each prior episode of violence.5 Prior arrests for any crime, especially if the individual was a juvenile at the time of arrest for his or her first violent offense, increase future violence risk.5 Other important static violence risk factors include demographic factors such as age, sex, and socioeconomic status. Swanson et al6 reviewed a large pool of data (approximately 10,000 respondents) from the Epidemiologic Catchment Area survey. Being young, male, and of low socioeconomic status were all associated with violence in the community.6 The highest-risk age group for violence is age 15 to 24.5 Males perpetrate violence in the community at a rate 10 times that of females.18 However, among individuals with severe mental illness, men and women have similar rates of violence.19,20 Unstable employment,21 less education,22 low intelligence,16 and a history of a significant head injury5 also are risk factors for violence.5
Continue to: Being abused as a child...
Being abused as a child, witnessing violence in the home,5,16 and growing up with an unstable parental situation (eg, parental loss or separation) has been linked to violence.16,23,24 Early disruptive behavior in childhood (eg, fighting, lying and stealing, truancy, and school problems) increases violence risk.21,23
Personality factors are important static risk factors for violence. Antisocial personality disorder is the most common personality disorder linked with violence.17 Several studies consistently show psychopathy to be a strong predictor of both violence and criminal behavior.5,25 A psychopath is a person who lacks empathy and close relationships, behaves impulsively, has superficially charming qualities, and is primarily interested in self-gratification.26 Harris et al27 studied 169 released forensic patients and found that 77% of the psychopaths (according to Psychopathy Checklist-Revised [PCL-R] scores) violently recidivated. In contrast, only 21% of the non-psychopaths violently recidivated.27
Other personality factors associated with violence include a predisposition toward feelings of anger and hatred (as opposed to empathy, anxiety, or guilt, which may reduce risk), hostile attributional biases (a tendency to interpret benign behavior of others as intentionally antagonistic), violent fantasies, poor anger control, and impulsivity.5 Although personality factors tend to be longstanding and more difficult to modify, in the outpatient setting, therapeutic efforts can be made to modify hostile attribution biases, poor anger control, and impulsive behavior.
Dynamic risk factors. Substance abuse is strongly associated with violence.6,17 The prevalence of violence is 12 times greater among individuals with alcohol use disorder and 16 times greater among individuals with other substance use disorders, compared with those with no such diagnoses.5,6
Continue to: Steadman et al...
Steadman et al28 compared 1,136 adult patients with mental disorders discharged from psychiatric hospitals with 519 individuals living in the same neighborhoods as the hospitalized patients. They found that the prevalence of violence among discharged patients without substance abuse was “statistically indistinguishable” from the prevalence of violence among community members, in the same neighborhood, who did not have symptoms of substance abuse.28 Swanson et al6 found that the combination of a mental disorder plus an alcohol or substance use disorder substantially increased the risk of violence.
Other dynamic risk factors for violence include mental illness symptoms such as psychosis, especially threat/control-override delusions, where the individual believes that they are being threatened or controlled by an external force.17
Contextual factors to consider in violence risk assessments include current stressors, lack of social support, availability of weapons, access to drugs and alcohol, and the presence of similar circumstances that led to violent behavior in the past.5
How to assess the risk of targeted violence
Targeted violence is a predatory act of violence intentionally committed against a preselected person, group of people, or place.29 Due to the low base rates of these incidents, targeted violence is difficult to study.7,30 These risk assessments require a more specialized approach.
Continue to: In their 1999 article...
In their 1999 article, Borum et al30 discussed threat assessment strategies utilized by the U.S. Secret Service and recommended investigating “pathways of ideas and behaviors that may lead to violent action.” Borum et al30 summarized 3 fundamental principles of threat assessment (Table 130).
What to do when violence risk is not due to mental illness
Based on the information in Mr. F’s case scenario, it is likely that his homicidal ideation is not due to mental illness. Despite this, several risk factors for violence are present. Where do we go from here?
Scott and Resnick17 recommend considering the concept of dangerousness as 5 components (Table 217). When this model of dangerousness is applied to Mr. F’s case, one can see that the magnitude of the harm is great because of threatened homicide. With regard to the imminence of the harm, it would help to clarify whether Mr. F plans to kill Ms. S immediately after discharge, or sometime in the next few months. Is his threat contingent on further provocations by Ms. S? Alternatively, does he intend to kill her for past grievances, regardless of further perceived insults?
Next, the frequency of a behavior relates to how often Mr. F has been aggressive in the past. The severity of his past aggression is also important. What is the most violent act he has ever done? Situational factors in this case include Mr. F’s access to weapons, financial problems, housing problems, and access to drugs and alcohol.17 Mr. F should be asked about what situations previously provoked his violent behavior. Consider how similar the present conditions are to past conditions to which Mr. F responded violently.5 The likelihood that a homicide will occur should take into account Mr. F’s risk factors for violence, as well as the seriousness of his intent to cause harm.
Continue to: Consider using a structured tool...
Consider using a structured tool, such as the Classification of Violence Risk, to help identify Mr. F’s risk factors for violence, or some other formal method to ensure that the proper data are collected. Violence risk assessments are more accurate when structured risk assessment tools are used, compared with clinical judgment alone.
It is important to review collateral sources of information. In Mr. F’s case, useful collateral sources may include his criminal docket (usually available online), past medical records, information from the shelter where he lives, and, potentially, friends or family.
Because Mr. F is making threats of targeted violence, be sure to ask about attack-related behaviors (Table 130).
Regarding the seriousness of Mr. F’s intent to cause harm, it may be helpful to ask him the following questions:
- How likely are you to carry out this act of violence?
- Do you have a plan? Have you taken any steps toward this plan?
- Do you see other, nonviolent solutions to this problem?
- What do you hope that we can do for you to help with this problem?
Continue to: Mr. F's answers...
Mr. F’s answers may suggest the possibility of a hidden agenda. Some patients express homicidal thoughts in order to stay in the hospital. If Mr. F expresses threats that are contingent on discharge and declines to engage in problem-solving discussions, this would cast doubt on the genuineness of his threat. However, doubt about the genuineness of the threat alone is not sufficient to simply discharge Mr. F. Assessment of his intent needs to be considered with other relevant risk factors, risk reduction strategies, and any Tarasoff duties that may apply.
In addition to risk factors, consider mitigating factors. For example, does Mr. F express concern over prison time as a reason to not engage in violence? It would be more ominous if Mr. F says that he does not care if he goes to prison because life is lousy being homeless and unemployed. At this point, an estimation can be made regarding whether Mr. F is a low-, moderate-, or high-risk of violence.
The next step is to organize Mr. F’s risk factors into static (historical) and dynamic (subject to intervention) factors. This will be helpful in formulating a strategy to manage risk because continued hospitalization can only address dynamic risk factors. Often in these cases, the static risk factors are far more numerous than the dynamic risk factors.
Once the data are collected and organized, the final step is to devise a risk management strategy. Some interventions, such as substance use treatment, will be straightforward. A mood-stabilizing medication could be considered, if clinically appropriate, to help reduce aggression and irritability.31 Efforts should be made to eliminate Mr. F’s access to firearms; however, in this case, it sounds unlikely that he will cooperate with those efforts. Ultimately, you may find yourself with a list of risk factors that are unlikely to be altered with further hospitalization, particularly if Mr. F’s homicidal thoughts and intent are due to antisocial personality traits.
Continue to: In that case...
In that case, the most important step will be to carry out your duty to warn/protect others prior to Mr. F’s discharge. Most states either require or permit mental health professionals to take reasonable steps to protect victims from violence when certain conditions are present, such as an explicit threat or identifiable victim (see Related Resources).
Once dynamic risk factors have been addressed, and duty to warn/protect is carried out, if there is no further clinical indication for hospitalization, it would be appropriate to discharge Mr. F. Continued homicidal threats stemming from antisocial personality traits, in the absence of a treatable mental illness (or other modifiable risk factors for violence that can be actively addressed), is not a reason for continued hospitalization. It may be useful to obtain a second opinion from a colleague in such scenarios. A second opinion may offer additional risk management ideas. In the event of a bad outcome, this will also help to show that the decision to discharge the patient was not taken lightly.
The psychiatrist should document a thoughtful risk assessment, the strategies that were implemented to reduce risk, the details of the warning, and the reasoning why continued hospitalization was not indicated (Table 3).
CASE CONTINUED
Decision to discharge
In Mr. F’s case, the treating psychiatrist determined that Mr. F’s risk of violence toward Ms. S was moderate. The psychiatrist identified several static risk factors for violence that raised Mr. F’s risk, but also noted that Mr. F’s threats were likely a manipulative effort to prolong his hospital stay. The psychiatrist carried out his duty to protect by notifying police and Ms. S of the nature of the threat prior to Mr. F’s discharge. The unit social worker helped Mr. F schedule an intake appointment for a substance use disorder treatment facility. Mr. F ultimately stated that he no longer experienced homicidal ideas once a bed was secured for him in a substance use treatment program. The psychiatrist carefully documented Mr. F’s risk assessment and the reasons why Mr. F’s risk would not be significantly altered by further inpatient hospitalization. Mr. F was discharged, and Ms. S remained unharmed.
Continue to: Bottom Line
Bottom Line
Use a structured approach to identify risk factors for violence. Address dynamic risk factors, including access to weapons. Carry out the duty to warn/protect if applicable. Document your decisions and actions carefully, and then discharge the patient if clinically indicated. Do not be “held hostage” by a patient’s homicidal ideation.
Related Resources
- Dolan M, Doyle M. Violence risk prediction. Clinical and actuarial measures and the role of the psychopathy checklist. Br J Psychiatry. 2000;177:303-311.
- Douglas KS, Hart SD, Webster CD, et al. HCR-20V3: Assessing risk of violence–user guide. Burnaby, Canada: Mental Health, Law, and Policy Institute, Simon Fraser University; 2013.
- National Conference of State Legislatures. Mental health professionals’ duty to warn. http://www.ncsl.org/research/health/mental-health-professionals-duty-to-warn.aspx. Published September 28, 2015.
Drug Brand Names
Sertraline • Zoloft
Mr. F, age 35, is homeless and has a history of cocaine and alcohol use disorders. He is admitted voluntarily to the psychiatric unit because he has homicidal thoughts toward Ms. S, who works in the shelter where he has been staying. Mr. F reports that he is thinking of killing Ms. S if he is discharged because she has been rude to him. He states that he has access to several firearms, but he will not disclose the location. He has been diagnosed with unspecified depressive disorder and exhibited antisocial personality disorder traits. He is being treated with sertraline. However, his mood appears to be relatively stable, except for occasional angry verbal outbursts. The outbursts have been related to intrusive peers or staff turning the television off for group meetings. Mr. F has been joking with peers, eating well, and sleeping appropriately. He reports no suicidal thoughts and has not been physically violent on the unit. However, Mr. F has had a history of violence since his teenage years. He has been incarcerated twice for assault and once for drug possession.
How would you approach assessing and managing Mr. F’s risk for violence?
We all have encountered a patient similar to Mr. F on the psychiatric unit or in the emergency department—a patient who makes violent threats and appears angry, intimidating, manipulative, and/or demanding, despite exhibiting no evidence of mania or psychosis. This patient often has a history of substance abuse and a lifelong pattern of viewing violence as an acceptable way of addressing life’s problems. Many psychiatrists suspect that more time on the inpatient unit is unlikely to reduce this patient’s risk of violence. Why? Because the violence risk does not stem from a treatable mental illness. Further, psychiatrists may be apprehensive about this patient’s potential for violence after discharge and their liability in the event of a bad outcome. No one wants their name associated with a headline that reads “Psychiatrist discharged man less than 24 hours before he killed 3 people.”
The purported relationship between mental illness and violence often is sensationalized in the media. However, research reveals that the vast majority of violence is in fact not due to symptoms of mental illness.1,2 A common clinical challenge in psychiatry involves evaluating individuals at elevated risk of violence and determining how to address their risk factors for violence. When the risk is primarily due to psychosis and can be reduced with antipsychotic medication, the job is easy. But how should we proceed when the risk stems from factors other than mental illness?
This article
Violence and mental illness: A tenuous link
Violence is a major public health concern in the United States. Although in recent years the rates of homicide and aggravated assault have decreased dramatically, there are approximately 16,000 homicides annually in the United States, and more than 1.6 million injuries from assaults treated in emergency departments each year.3 Homicide continues to be one of the leading causes of death among teenagers and young adults.4
The most effective methods of preventing widespread violence are public health approaches, such as parent- and family-focused programs, early childhood education, programs in school, and public policy changes.3 However, as psychiatrists, we are routinely asked to assess the risk of violence for an individual patient and devise strategies to mitigate violence risk.
Continue to: Although certain mental illnesses...
Although certain mental illnesses increase the relative risk of violence (compared with people without mental illness),5,6 recent studies suggest that mental illness plays only a “minor role in explaining violence in populations.”7 It is estimated that as little as 4% of the violence in the United States can be attributed to mental illness.1 According to a 1998 meta-analysis of 48 studies of criminal recidivism, the risk factors for violent recidivism were “almost identical” among offenders who had a mental disorder and those who did not.8
Approaches to assessing violence risk
Psychiatrists can assess the risk of future violence via 3 broad approaches.9,10
Unaided clinical judgment is when a mental health professional estimates violence risk based on his or her own experience and intuition, with knowledge of violence risk factors, but without the use of structured tools.
Actuarial tools are statistical models that use formulae to show relationships between data (risk factors) and outcomes (violence).10,11
Continue to: Structured professional judgment
Structured professional judgment is a hybrid of unaided clinical judgment and actuarial methods. Structured professional judgment tools help the evaluator identify empirically established risk factors. Once the information is collected, it is combined with clinical judgment in decision making.9,10 There are now more than 200 structured tools available for assessing violence risk in criminal justice and forensic mental health populations.12
Clinical judgment, although commonly used in practice, is less accurate than actuarial tools or structured professional judgment.10,11 In general, risk assessment tools offer moderate levels of accuracy in categorizing people at low risk vs high risk.5,13 The tools have better ability to accurately categorize individuals at low risk, compared with high risk, where false positives are common.12,14
Two types of risk factors
Risk factors for violence are commonly categorized as static or dynamic factors. Static factors are historical factors that cannot be changed with intervention (eg, age, sex, history of abuse). Dynamic factors can be changed with intervention (eg, substance abuse).15
Static risk factors. The best predictor of future violence is past violent behavior.5,16,17 Violence risk increases with each prior episode of violence.5 Prior arrests for any crime, especially if the individual was a juvenile at the time of arrest for his or her first violent offense, increase future violence risk.5 Other important static violence risk factors include demographic factors such as age, sex, and socioeconomic status. Swanson et al6 reviewed a large pool of data (approximately 10,000 respondents) from the Epidemiologic Catchment Area survey. Being young, male, and of low socioeconomic status were all associated with violence in the community.6 The highest-risk age group for violence is age 15 to 24.5 Males perpetrate violence in the community at a rate 10 times that of females.18 However, among individuals with severe mental illness, men and women have similar rates of violence.19,20 Unstable employment,21 less education,22 low intelligence,16 and a history of a significant head injury5 also are risk factors for violence.5
Continue to: Being abused as a child...
Being abused as a child, witnessing violence in the home,5,16 and growing up with an unstable parental situation (eg, parental loss or separation) has been linked to violence.16,23,24 Early disruptive behavior in childhood (eg, fighting, lying and stealing, truancy, and school problems) increases violence risk.21,23
Personality factors are important static risk factors for violence. Antisocial personality disorder is the most common personality disorder linked with violence.17 Several studies consistently show psychopathy to be a strong predictor of both violence and criminal behavior.5,25 A psychopath is a person who lacks empathy and close relationships, behaves impulsively, has superficially charming qualities, and is primarily interested in self-gratification.26 Harris et al27 studied 169 released forensic patients and found that 77% of the psychopaths (according to Psychopathy Checklist-Revised [PCL-R] scores) violently recidivated. In contrast, only 21% of the non-psychopaths violently recidivated.27
Other personality factors associated with violence include a predisposition toward feelings of anger and hatred (as opposed to empathy, anxiety, or guilt, which may reduce risk), hostile attributional biases (a tendency to interpret benign behavior of others as intentionally antagonistic), violent fantasies, poor anger control, and impulsivity.5 Although personality factors tend to be longstanding and more difficult to modify, in the outpatient setting, therapeutic efforts can be made to modify hostile attribution biases, poor anger control, and impulsive behavior.
Dynamic risk factors. Substance abuse is strongly associated with violence.6,17 The prevalence of violence is 12 times greater among individuals with alcohol use disorder and 16 times greater among individuals with other substance use disorders, compared with those with no such diagnoses.5,6
Continue to: Steadman et al...
Steadman et al28 compared 1,136 adult patients with mental disorders discharged from psychiatric hospitals with 519 individuals living in the same neighborhoods as the hospitalized patients. They found that the prevalence of violence among discharged patients without substance abuse was “statistically indistinguishable” from the prevalence of violence among community members, in the same neighborhood, who did not have symptoms of substance abuse.28 Swanson et al6 found that the combination of a mental disorder plus an alcohol or substance use disorder substantially increased the risk of violence.
Other dynamic risk factors for violence include mental illness symptoms such as psychosis, especially threat/control-override delusions, where the individual believes that they are being threatened or controlled by an external force.17
Contextual factors to consider in violence risk assessments include current stressors, lack of social support, availability of weapons, access to drugs and alcohol, and the presence of similar circumstances that led to violent behavior in the past.5
How to assess the risk of targeted violence
Targeted violence is a predatory act of violence intentionally committed against a preselected person, group of people, or place.29 Due to the low base rates of these incidents, targeted violence is difficult to study.7,30 These risk assessments require a more specialized approach.
Continue to: In their 1999 article...
In their 1999 article, Borum et al30 discussed threat assessment strategies utilized by the U.S. Secret Service and recommended investigating “pathways of ideas and behaviors that may lead to violent action.” Borum et al30 summarized 3 fundamental principles of threat assessment (Table 130).
What to do when violence risk is not due to mental illness
Based on the information in Mr. F’s case scenario, it is likely that his homicidal ideation is not due to mental illness. Despite this, several risk factors for violence are present. Where do we go from here?
Scott and Resnick17 recommend considering the concept of dangerousness as 5 components (Table 217). When this model of dangerousness is applied to Mr. F’s case, one can see that the magnitude of the harm is great because of threatened homicide. With regard to the imminence of the harm, it would help to clarify whether Mr. F plans to kill Ms. S immediately after discharge, or sometime in the next few months. Is his threat contingent on further provocations by Ms. S? Alternatively, does he intend to kill her for past grievances, regardless of further perceived insults?
Next, the frequency of a behavior relates to how often Mr. F has been aggressive in the past. The severity of his past aggression is also important. What is the most violent act he has ever done? Situational factors in this case include Mr. F’s access to weapons, financial problems, housing problems, and access to drugs and alcohol.17 Mr. F should be asked about what situations previously provoked his violent behavior. Consider how similar the present conditions are to past conditions to which Mr. F responded violently.5 The likelihood that a homicide will occur should take into account Mr. F’s risk factors for violence, as well as the seriousness of his intent to cause harm.
Continue to: Consider using a structured tool...
Consider using a structured tool, such as the Classification of Violence Risk, to help identify Mr. F’s risk factors for violence, or some other formal method to ensure that the proper data are collected. Violence risk assessments are more accurate when structured risk assessment tools are used, compared with clinical judgment alone.
It is important to review collateral sources of information. In Mr. F’s case, useful collateral sources may include his criminal docket (usually available online), past medical records, information from the shelter where he lives, and, potentially, friends or family.
Because Mr. F is making threats of targeted violence, be sure to ask about attack-related behaviors (Table 130).
Regarding the seriousness of Mr. F’s intent to cause harm, it may be helpful to ask him the following questions:
- How likely are you to carry out this act of violence?
- Do you have a plan? Have you taken any steps toward this plan?
- Do you see other, nonviolent solutions to this problem?
- What do you hope that we can do for you to help with this problem?
Continue to: Mr. F's answers...
Mr. F’s answers may suggest the possibility of a hidden agenda. Some patients express homicidal thoughts in order to stay in the hospital. If Mr. F expresses threats that are contingent on discharge and declines to engage in problem-solving discussions, this would cast doubt on the genuineness of his threat. However, doubt about the genuineness of the threat alone is not sufficient to simply discharge Mr. F. Assessment of his intent needs to be considered with other relevant risk factors, risk reduction strategies, and any Tarasoff duties that may apply.
In addition to risk factors, consider mitigating factors. For example, does Mr. F express concern over prison time as a reason to not engage in violence? It would be more ominous if Mr. F says that he does not care if he goes to prison because life is lousy being homeless and unemployed. At this point, an estimation can be made regarding whether Mr. F is a low-, moderate-, or high-risk of violence.
The next step is to organize Mr. F’s risk factors into static (historical) and dynamic (subject to intervention) factors. This will be helpful in formulating a strategy to manage risk because continued hospitalization can only address dynamic risk factors. Often in these cases, the static risk factors are far more numerous than the dynamic risk factors.
Once the data are collected and organized, the final step is to devise a risk management strategy. Some interventions, such as substance use treatment, will be straightforward. A mood-stabilizing medication could be considered, if clinically appropriate, to help reduce aggression and irritability.31 Efforts should be made to eliminate Mr. F’s access to firearms; however, in this case, it sounds unlikely that he will cooperate with those efforts. Ultimately, you may find yourself with a list of risk factors that are unlikely to be altered with further hospitalization, particularly if Mr. F’s homicidal thoughts and intent are due to antisocial personality traits.
Continue to: In that case...
In that case, the most important step will be to carry out your duty to warn/protect others prior to Mr. F’s discharge. Most states either require or permit mental health professionals to take reasonable steps to protect victims from violence when certain conditions are present, such as an explicit threat or identifiable victim (see Related Resources).
Once dynamic risk factors have been addressed, and duty to warn/protect is carried out, if there is no further clinical indication for hospitalization, it would be appropriate to discharge Mr. F. Continued homicidal threats stemming from antisocial personality traits, in the absence of a treatable mental illness (or other modifiable risk factors for violence that can be actively addressed), is not a reason for continued hospitalization. It may be useful to obtain a second opinion from a colleague in such scenarios. A second opinion may offer additional risk management ideas. In the event of a bad outcome, this will also help to show that the decision to discharge the patient was not taken lightly.
The psychiatrist should document a thoughtful risk assessment, the strategies that were implemented to reduce risk, the details of the warning, and the reasoning why continued hospitalization was not indicated (Table 3).
CASE CONTINUED
Decision to discharge
In Mr. F’s case, the treating psychiatrist determined that Mr. F’s risk of violence toward Ms. S was moderate. The psychiatrist identified several static risk factors for violence that raised Mr. F’s risk, but also noted that Mr. F’s threats were likely a manipulative effort to prolong his hospital stay. The psychiatrist carried out his duty to protect by notifying police and Ms. S of the nature of the threat prior to Mr. F’s discharge. The unit social worker helped Mr. F schedule an intake appointment for a substance use disorder treatment facility. Mr. F ultimately stated that he no longer experienced homicidal ideas once a bed was secured for him in a substance use treatment program. The psychiatrist carefully documented Mr. F’s risk assessment and the reasons why Mr. F’s risk would not be significantly altered by further inpatient hospitalization. Mr. F was discharged, and Ms. S remained unharmed.
Continue to: Bottom Line
Bottom Line
Use a structured approach to identify risk factors for violence. Address dynamic risk factors, including access to weapons. Carry out the duty to warn/protect if applicable. Document your decisions and actions carefully, and then discharge the patient if clinically indicated. Do not be “held hostage” by a patient’s homicidal ideation.
Related Resources
- Dolan M, Doyle M. Violence risk prediction. Clinical and actuarial measures and the role of the psychopathy checklist. Br J Psychiatry. 2000;177:303-311.
- Douglas KS, Hart SD, Webster CD, et al. HCR-20V3: Assessing risk of violence–user guide. Burnaby, Canada: Mental Health, Law, and Policy Institute, Simon Fraser University; 2013.
- National Conference of State Legislatures. Mental health professionals’ duty to warn. http://www.ncsl.org/research/health/mental-health-professionals-duty-to-warn.aspx. Published September 28, 2015.
Drug Brand Names
Sertraline • Zoloft
1. Skeem J, Kennealy P, Monahan J, et al. Psychosis uncommonly and inconsistently precedes violence among high-risk individuals. Clin Psychol Sci. 2016;4(1):40-49.
2. McGinty E, Frattaroli S, Appelbaum PS, et al. Using research evidence to reframe the policy debate around mental illness and guns: process and recommendations. Am J Public Health. 2014;104(11):e22-e26.
3. Sumner SA, Mercy JA, Dahlberg LL, et al. Violence in the United States: status, challenges, and opportunities. JAMA. 2015;314(5):478-488.
4. Heron M. Deaths: leading causes for 2014. Natl Vital Stat Rep. 2016;65(5):1-96.
5. Borum R, Swartz M, Swanson J. Assessing and managing violence risk in clinical practice. J Prac Psychiatry Behav Health. 1996;2(4):205-215.
6. Swanson JW, Holzer CE 3rd, Ganju VK, et al. Violence and psychiatric disorder in the community: Evidence from the epidemiologic catchment area surveys. Hosp Community Psychiatry. 1990;41(7):761-770.
7. Swanson JW. Explaining rare acts of violence: the limits of evidence from population research. Psychiatr Serv. 2011;62(11):1369-1371.
8. Bonta J, Law M, Hanson K. The prediction of criminal and violent recidivism among mentally disordered offenders: a meta-analysis. Psychol Bull. 1998;123(2):123-142.
9. Monahan J. The inclusion of biological risk factors in violence risk assessments. In: Singh I, Sinnott-Armstrong W, Savulescu J, eds. Bioprediction, biomarkers, and bad behavior: scientific, legal, and ethical implications. New York, NY: Oxford University Press; 2014:57-76.
10. Murray J, Thomson ME. Clinical judgement in violence risk assessment. Eur J Psychol. 2010;6(1):128-149.
11. Mossman D. Violence risk: is clinical judgment enough? Current Psychiatry. 2008;7(6):66-72.
12. Douglas T, Pugh J, Singh I, et al. Risk assessment tools in criminal justice and forensic psychiatry: the need for better data. Eur Psychiatry. 2017;42:134-137.
13. Dolan M, Doyle M. Violence risk prediction. Clinical and actuarial measures and the role of the psychopathy checklist. Br J Psychiatry. 2000;177:303-311.
14. Fazel S, Singh J, Doll H, et al. Use of risk assessment instruments to predict violence and antisocial behaviour in 73 samples involving 24 827 people: systematic review and meta-analysis. BMJ. 2012;345:e4692. doi: 10.1136/bmj.e4692.
15. National Collaborating Centre for Mental Health (UK). Violence and aggression: short- term management in mental health, health, and community settings: updated edition. London: British Psychological Society; 2015. NICE Guideline, No 10.
16. Klassen D, O’Connor WA. Predicting violence in schizophrenic and non-schizophrenic patients: a prospective study. J Community Psychol. 1988;16(2):217-227.
17. Scott C, Resnick P. Clinical assessment of aggression and violence. In: Rosner R, Scott C, eds. Principles and practice of forensic psychiatry, 3rd ed. Boca Raton, FL: CRC Press; 2017:623-631.
18. Tardiff K, Sweillam A. Assault, suicide, and mental illness. Arch Gen Psychiatry. 1980;37(2):164-169.
19. Lidz CW, Mulvey EP, Gardner W. The accuracy of predictions of violence to others. JAMA. 1993;269(8):1007-1011.
20. Newhill CE, Mulvey EP, Lidz CW. Characteristics of violence in the community by female patients seen in a psychiatric emergency service. Psychiatric Serv. 1995;46(8):785-789.
21. Mulvey E, Lidz C. Clinical considerations in the prediction of dangerousness in mental patients. Clin Psychol Rev. 1984;4(4):379-401.
22. Link BG, Andrews H, Cullen FT. The violent and illegal behavior of mental patients reconsidered. Am Sociol Rev. 1992;57(3):275-292.
23. Harris GT, Rice ME, Quinsey VL. Violent recidivism of mentally disordered offenders: the development of a statistical prediction instrument. Crim Justice and Behav. 1993;20(4):315-335.
24. Klassen D, O’Connor W. Demographic and case history variables in risk assessment. In: Monahan J, Steadman H, eds. Violence and mental disorder: developments in risk assessment. Chicago, IL: University of Chicago Press; 1994:229-257.
25. Hart SD, Hare RD, Forth AE. Psychopathy as a risk marker for violence: development and validation of a screening version of the revised Psychopathy Checklist. In: Monahan J, Steadman HJ, eds. Violence and mental disorder: developments in risk assessment. Chicago, IL: University of Chicago Press; 1994:81-98.
26. Cleckley H. The mask of sanity. St. Louis, MO: Mosby; 1941.
27. Harris GT, Rice ME, Cormier CA. Psychopathy and violent recidivism. Law Hum Behav. 1991;15(6):625-637.
28. Steadman HJ, Mulvey EP, Monahan J. Violence by people discharged from acute psychiatric inpatient facilities and by others in the same neighborhoods. Arch Gen Psychiatry. 1998;55:393-401.
29. Meloy JR, White SG, Hart S. Workplace assessment of targeted violence risk: the development and reliability of the WAVR-21. J Forensic Sci. 2013;58(5):1353-1358.
30. Borum R, Fein R, Vossekuil B, et al. Threat assessment: defining an approach for evaluating risk of targeted violence. Behav Sci Law. 1999;17(3):323-337.
31. Tyrer P, Bateman AW. Drug treatment for personality disorders. Adv Psychiatr Treat. 2004;10(5):389-398.
1. Skeem J, Kennealy P, Monahan J, et al. Psychosis uncommonly and inconsistently precedes violence among high-risk individuals. Clin Psychol Sci. 2016;4(1):40-49.
2. McGinty E, Frattaroli S, Appelbaum PS, et al. Using research evidence to reframe the policy debate around mental illness and guns: process and recommendations. Am J Public Health. 2014;104(11):e22-e26.
3. Sumner SA, Mercy JA, Dahlberg LL, et al. Violence in the United States: status, challenges, and opportunities. JAMA. 2015;314(5):478-488.
4. Heron M. Deaths: leading causes for 2014. Natl Vital Stat Rep. 2016;65(5):1-96.
5. Borum R, Swartz M, Swanson J. Assessing and managing violence risk in clinical practice. J Prac Psychiatry Behav Health. 1996;2(4):205-215.
6. Swanson JW, Holzer CE 3rd, Ganju VK, et al. Violence and psychiatric disorder in the community: Evidence from the epidemiologic catchment area surveys. Hosp Community Psychiatry. 1990;41(7):761-770.
7. Swanson JW. Explaining rare acts of violence: the limits of evidence from population research. Psychiatr Serv. 2011;62(11):1369-1371.
8. Bonta J, Law M, Hanson K. The prediction of criminal and violent recidivism among mentally disordered offenders: a meta-analysis. Psychol Bull. 1998;123(2):123-142.
9. Monahan J. The inclusion of biological risk factors in violence risk assessments. In: Singh I, Sinnott-Armstrong W, Savulescu J, eds. Bioprediction, biomarkers, and bad behavior: scientific, legal, and ethical implications. New York, NY: Oxford University Press; 2014:57-76.
10. Murray J, Thomson ME. Clinical judgement in violence risk assessment. Eur J Psychol. 2010;6(1):128-149.
11. Mossman D. Violence risk: is clinical judgment enough? Current Psychiatry. 2008;7(6):66-72.
12. Douglas T, Pugh J, Singh I, et al. Risk assessment tools in criminal justice and forensic psychiatry: the need for better data. Eur Psychiatry. 2017;42:134-137.
13. Dolan M, Doyle M. Violence risk prediction. Clinical and actuarial measures and the role of the psychopathy checklist. Br J Psychiatry. 2000;177:303-311.
14. Fazel S, Singh J, Doll H, et al. Use of risk assessment instruments to predict violence and antisocial behaviour in 73 samples involving 24 827 people: systematic review and meta-analysis. BMJ. 2012;345:e4692. doi: 10.1136/bmj.e4692.
15. National Collaborating Centre for Mental Health (UK). Violence and aggression: short- term management in mental health, health, and community settings: updated edition. London: British Psychological Society; 2015. NICE Guideline, No 10.
16. Klassen D, O’Connor WA. Predicting violence in schizophrenic and non-schizophrenic patients: a prospective study. J Community Psychol. 1988;16(2):217-227.
17. Scott C, Resnick P. Clinical assessment of aggression and violence. In: Rosner R, Scott C, eds. Principles and practice of forensic psychiatry, 3rd ed. Boca Raton, FL: CRC Press; 2017:623-631.
18. Tardiff K, Sweillam A. Assault, suicide, and mental illness. Arch Gen Psychiatry. 1980;37(2):164-169.
19. Lidz CW, Mulvey EP, Gardner W. The accuracy of predictions of violence to others. JAMA. 1993;269(8):1007-1011.
20. Newhill CE, Mulvey EP, Lidz CW. Characteristics of violence in the community by female patients seen in a psychiatric emergency service. Psychiatric Serv. 1995;46(8):785-789.
21. Mulvey E, Lidz C. Clinical considerations in the prediction of dangerousness in mental patients. Clin Psychol Rev. 1984;4(4):379-401.
22. Link BG, Andrews H, Cullen FT. The violent and illegal behavior of mental patients reconsidered. Am Sociol Rev. 1992;57(3):275-292.
23. Harris GT, Rice ME, Quinsey VL. Violent recidivism of mentally disordered offenders: the development of a statistical prediction instrument. Crim Justice and Behav. 1993;20(4):315-335.
24. Klassen D, O’Connor W. Demographic and case history variables in risk assessment. In: Monahan J, Steadman H, eds. Violence and mental disorder: developments in risk assessment. Chicago, IL: University of Chicago Press; 1994:229-257.
25. Hart SD, Hare RD, Forth AE. Psychopathy as a risk marker for violence: development and validation of a screening version of the revised Psychopathy Checklist. In: Monahan J, Steadman HJ, eds. Violence and mental disorder: developments in risk assessment. Chicago, IL: University of Chicago Press; 1994:81-98.
26. Cleckley H. The mask of sanity. St. Louis, MO: Mosby; 1941.
27. Harris GT, Rice ME, Cormier CA. Psychopathy and violent recidivism. Law Hum Behav. 1991;15(6):625-637.
28. Steadman HJ, Mulvey EP, Monahan J. Violence by people discharged from acute psychiatric inpatient facilities and by others in the same neighborhoods. Arch Gen Psychiatry. 1998;55:393-401.
29. Meloy JR, White SG, Hart S. Workplace assessment of targeted violence risk: the development and reliability of the WAVR-21. J Forensic Sci. 2013;58(5):1353-1358.
30. Borum R, Fein R, Vossekuil B, et al. Threat assessment: defining an approach for evaluating risk of targeted violence. Behav Sci Law. 1999;17(3):323-337.
31. Tyrer P, Bateman AW. Drug treatment for personality disorders. Adv Psychiatr Treat. 2004;10(5):389-398.

Strategies for working with patients with personality disorders
Patients with personality disorders can disrupt the treatment relationship, and may leave us feeling angry, ineffective, inadequate, and defeated. Although their behaviors may appear volitional and purposeful, they often are the result of a dysfunctional personality structure.1 These patients’ unbending patterns of viewing themselves, interacting with others, and navigating the world can be problematic in an inpatient or outpatient setting, causing distress for both the staff and patient. Because no 2 personalities are identical, there is no algorithm for managing patients with personality disorders. However, there are strategies that we can apply to provide effective clinical care.1,2
Discuss the responses the patient evokes. Patients with personality disorders can elicit strong responses from the treatment team. Each clinician can have a different response to the same patient, ranging from feeling the need to protect the patient to strongly disliking him or her. Because cohesion among staff is essential for effective patient care, we need to discuss these responses in an open forum with our team members so we can effectively manage our responses and provide the patient with consistent interactions. Limiting the delivery of inconsistent or conflicting messages will decrease staff splitting and increase team unity.
Reinforce appropriate behaviors. Patients with personality disorders usually have negative interpersonal interactions, such as acting out, misinterpreting neutral social cues, and seeking constant attention. However, when they are not engaging in detrimental behaviors, we should provide positive reinforcement for appropriate behaviors, such as remaining composed, that help maintain the treatment relationship. When a patient displays disruptive behaviors, take a neutral approach by stating, “You appear upset. I will come back later when you are feeling better.”1
Set limits. These patients are likely to have difficulty conforming to appropriate social boundaries. Our reflex reaction may be to set concrete rules that fit our preferences. This could lead to a power struggle between us and our patients, which is not helpful. Rather than a “one-size-fits-all” approach to rules, it may be prudent to tailor boundaries according to each patient’s unique personality. Also, allowing the patient to help set these limits could increase the chances that he or she will follow your treatment plan and reinforce the more positive aspects of his or her personality structure.
Offer empathy. Empathy can be conceptualized as a step further than sympathy; in addition to expressing concern and compassion, empathy involves recognizing and sharing the patient’s emotions. Seek to comprehend the reasons behind a patient’s negative reactions by
1. Riddle M, Meeks T, Alvarez C, et al. When personality is the problem: managing patients with difficult personalitie s on the acute care unit. J Hosp Med. 2016;11(12):873-878.
2. Strous RD, Ulman AM, Kotler M. The hateful patient revisited: relevance for 21st century medicine. Eur J Intern Med. 2006;17(6):387-393.
Patients with personality disorders can disrupt the treatment relationship, and may leave us feeling angry, ineffective, inadequate, and defeated. Although their behaviors may appear volitional and purposeful, they often are the result of a dysfunctional personality structure.1 These patients’ unbending patterns of viewing themselves, interacting with others, and navigating the world can be problematic in an inpatient or outpatient setting, causing distress for both the staff and patient. Because no 2 personalities are identical, there is no algorithm for managing patients with personality disorders. However, there are strategies that we can apply to provide effective clinical care.1,2
Discuss the responses the patient evokes. Patients with personality disorders can elicit strong responses from the treatment team. Each clinician can have a different response to the same patient, ranging from feeling the need to protect the patient to strongly disliking him or her. Because cohesion among staff is essential for effective patient care, we need to discuss these responses in an open forum with our team members so we can effectively manage our responses and provide the patient with consistent interactions. Limiting the delivery of inconsistent or conflicting messages will decrease staff splitting and increase team unity.
Reinforce appropriate behaviors. Patients with personality disorders usually have negative interpersonal interactions, such as acting out, misinterpreting neutral social cues, and seeking constant attention. However, when they are not engaging in detrimental behaviors, we should provide positive reinforcement for appropriate behaviors, such as remaining composed, that help maintain the treatment relationship. When a patient displays disruptive behaviors, take a neutral approach by stating, “You appear upset. I will come back later when you are feeling better.”1
Set limits. These patients are likely to have difficulty conforming to appropriate social boundaries. Our reflex reaction may be to set concrete rules that fit our preferences. This could lead to a power struggle between us and our patients, which is not helpful. Rather than a “one-size-fits-all” approach to rules, it may be prudent to tailor boundaries according to each patient’s unique personality. Also, allowing the patient to help set these limits could increase the chances that he or she will follow your treatment plan and reinforce the more positive aspects of his or her personality structure.
Offer empathy. Empathy can be conceptualized as a step further than sympathy; in addition to expressing concern and compassion, empathy involves recognizing and sharing the patient’s emotions. Seek to comprehend the reasons behind a patient’s negative reactions by
Patients with personality disorders can disrupt the treatment relationship, and may leave us feeling angry, ineffective, inadequate, and defeated. Although their behaviors may appear volitional and purposeful, they often are the result of a dysfunctional personality structure.1 These patients’ unbending patterns of viewing themselves, interacting with others, and navigating the world can be problematic in an inpatient or outpatient setting, causing distress for both the staff and patient. Because no 2 personalities are identical, there is no algorithm for managing patients with personality disorders. However, there are strategies that we can apply to provide effective clinical care.1,2
Discuss the responses the patient evokes. Patients with personality disorders can elicit strong responses from the treatment team. Each clinician can have a different response to the same patient, ranging from feeling the need to protect the patient to strongly disliking him or her. Because cohesion among staff is essential for effective patient care, we need to discuss these responses in an open forum with our team members so we can effectively manage our responses and provide the patient with consistent interactions. Limiting the delivery of inconsistent or conflicting messages will decrease staff splitting and increase team unity.
Reinforce appropriate behaviors. Patients with personality disorders usually have negative interpersonal interactions, such as acting out, misinterpreting neutral social cues, and seeking constant attention. However, when they are not engaging in detrimental behaviors, we should provide positive reinforcement for appropriate behaviors, such as remaining composed, that help maintain the treatment relationship. When a patient displays disruptive behaviors, take a neutral approach by stating, “You appear upset. I will come back later when you are feeling better.”1
Set limits. These patients are likely to have difficulty conforming to appropriate social boundaries. Our reflex reaction may be to set concrete rules that fit our preferences. This could lead to a power struggle between us and our patients, which is not helpful. Rather than a “one-size-fits-all” approach to rules, it may be prudent to tailor boundaries according to each patient’s unique personality. Also, allowing the patient to help set these limits could increase the chances that he or she will follow your treatment plan and reinforce the more positive aspects of his or her personality structure.
Offer empathy. Empathy can be conceptualized as a step further than sympathy; in addition to expressing concern and compassion, empathy involves recognizing and sharing the patient’s emotions. Seek to comprehend the reasons behind a patient’s negative reactions by
1. Riddle M, Meeks T, Alvarez C, et al. When personality is the problem: managing patients with difficult personalitie s on the acute care unit. J Hosp Med. 2016;11(12):873-878.
2. Strous RD, Ulman AM, Kotler M. The hateful patient revisited: relevance for 21st century medicine. Eur J Intern Med. 2006;17(6):387-393.
1. Riddle M, Meeks T, Alvarez C, et al. When personality is the problem: managing patients with difficult personalitie s on the acute care unit. J Hosp Med. 2016;11(12):873-878.
2. Strous RD, Ulman AM, Kotler M. The hateful patient revisited: relevance for 21st century medicine. Eur J Intern Med. 2006;17(6):387-393.
‘Nocebo’ effects: Address these 4 psychosocial factors
Sorting out the causes of unexplained adverse effects from psychotropic medications can be challenging. Treatment may be further complicated by ‘nocebo’ effects, which are adverse effects based on the patient’s conscious and unconscious expectations of harm. Having strategies for managing nocebo effects can help clinicians better understand and treat patients who have complex medication complaints. When your patient experiences nocebo effects, consider the following 4 psychosocial factors.1
Pills. The impact of a medication is not solely based on its chemical makeup. For example, the appearance of a medication can affect treatment outcomes. Substituting generic medications for branded ones has been shown to negatively impact patient adherence and increase reports of adverse effects that have no physiologic cause.2 Educating patients about medication manufacturing and distribution practices may decrease such consequences.
Patient. A sense of powerlessness is fertile ground for nocebo effects. Patients with an external locus of control may unconsciously employ nocebo effects to express themselves when other outlets are limited. Having a psychosocial formulation of your patient can help you anticipate pitfalls, offer pertinent insights, and mobilize the patient’s adaptive coping mechanisms. Also, clinicians can bolster their patients’ self-agency by encouraging them to participate in healthy activities.
Provider. Irrational factors in the clinician, such as countertransference, may also affect medication outcomes. Unprocessed countertransference can contribute to clinician burnout and impact the therapeutic relationship negatively. Nocebo effects may indicate that the clinician is not “tuned in” to the patient or is acting out harmful unconscious thoughts. Additionally, countertransference can lead to unnecessary prescribing and polypharmacy that confounds nocebo effects. Therefore self-care, consultation, and supervision may be vital in promoting therapeutic outcomes.
Partnership. The doctor–patient relationship can contribute to nocebo effects. A 2016 Gallup Poll found that Americans had low confidence in the honesty and ethics of psychiatrists compared with other healthcare professionals.3 It is important to have conversations with your patients about their reservations and perceived stigma of mental health. Such conversations can bring a patient’s ambivalence into treatment so that it can be further explored and addressed. Psychoeducation about treatment limitations, motivational interviewing techniques, and involving patients in decision-making can be useful tools for fostering a therapeutic alliance and positive outcomes.
Take an active approach
Evidence demonstrates that psychosocial factors significantly impact treatment outcomes.1 Incorporating this evidence into practice and attending to the 4 factors discussed here can enhance a clinician’s ability to flexibly respond to their patients’ complaints, especially in relation to nocebo effects.
1. Mallo CJ, Mintz DL. Teaching all the evidence bases: reintegrating psychodynamic aspects of prescribing into psychopharmacology training. Psychodyn Psychiatry. 2013;41(1):13-37.
2. Weissenfeld J, Stock S, Lüngen M, et al. The nocebo effect: a reason for patients’ non-adherence to generic substitution? Pharmazie. 2010;65(7):451-456.
3. Norman J. Americans rate healthcare providers high on honesty, ethics. Gallup. http://news.gallup.com/poll/200057/americans-rate-healthcare-providers-high-honesty-ethics.aspx. Published December 19, 2016. Accessed October 22, 2017.
Sorting out the causes of unexplained adverse effects from psychotropic medications can be challenging. Treatment may be further complicated by ‘nocebo’ effects, which are adverse effects based on the patient’s conscious and unconscious expectations of harm. Having strategies for managing nocebo effects can help clinicians better understand and treat patients who have complex medication complaints. When your patient experiences nocebo effects, consider the following 4 psychosocial factors.1
Pills. The impact of a medication is not solely based on its chemical makeup. For example, the appearance of a medication can affect treatment outcomes. Substituting generic medications for branded ones has been shown to negatively impact patient adherence and increase reports of adverse effects that have no physiologic cause.2 Educating patients about medication manufacturing and distribution practices may decrease such consequences.
Patient. A sense of powerlessness is fertile ground for nocebo effects. Patients with an external locus of control may unconsciously employ nocebo effects to express themselves when other outlets are limited. Having a psychosocial formulation of your patient can help you anticipate pitfalls, offer pertinent insights, and mobilize the patient’s adaptive coping mechanisms. Also, clinicians can bolster their patients’ self-agency by encouraging them to participate in healthy activities.
Provider. Irrational factors in the clinician, such as countertransference, may also affect medication outcomes. Unprocessed countertransference can contribute to clinician burnout and impact the therapeutic relationship negatively. Nocebo effects may indicate that the clinician is not “tuned in” to the patient or is acting out harmful unconscious thoughts. Additionally, countertransference can lead to unnecessary prescribing and polypharmacy that confounds nocebo effects. Therefore self-care, consultation, and supervision may be vital in promoting therapeutic outcomes.
Partnership. The doctor–patient relationship can contribute to nocebo effects. A 2016 Gallup Poll found that Americans had low confidence in the honesty and ethics of psychiatrists compared with other healthcare professionals.3 It is important to have conversations with your patients about their reservations and perceived stigma of mental health. Such conversations can bring a patient’s ambivalence into treatment so that it can be further explored and addressed. Psychoeducation about treatment limitations, motivational interviewing techniques, and involving patients in decision-making can be useful tools for fostering a therapeutic alliance and positive outcomes.
Take an active approach
Evidence demonstrates that psychosocial factors significantly impact treatment outcomes.1 Incorporating this evidence into practice and attending to the 4 factors discussed here can enhance a clinician’s ability to flexibly respond to their patients’ complaints, especially in relation to nocebo effects.
Sorting out the causes of unexplained adverse effects from psychotropic medications can be challenging. Treatment may be further complicated by ‘nocebo’ effects, which are adverse effects based on the patient’s conscious and unconscious expectations of harm. Having strategies for managing nocebo effects can help clinicians better understand and treat patients who have complex medication complaints. When your patient experiences nocebo effects, consider the following 4 psychosocial factors.1
Pills. The impact of a medication is not solely based on its chemical makeup. For example, the appearance of a medication can affect treatment outcomes. Substituting generic medications for branded ones has been shown to negatively impact patient adherence and increase reports of adverse effects that have no physiologic cause.2 Educating patients about medication manufacturing and distribution practices may decrease such consequences.
Patient. A sense of powerlessness is fertile ground for nocebo effects. Patients with an external locus of control may unconsciously employ nocebo effects to express themselves when other outlets are limited. Having a psychosocial formulation of your patient can help you anticipate pitfalls, offer pertinent insights, and mobilize the patient’s adaptive coping mechanisms. Also, clinicians can bolster their patients’ self-agency by encouraging them to participate in healthy activities.
Provider. Irrational factors in the clinician, such as countertransference, may also affect medication outcomes. Unprocessed countertransference can contribute to clinician burnout and impact the therapeutic relationship negatively. Nocebo effects may indicate that the clinician is not “tuned in” to the patient or is acting out harmful unconscious thoughts. Additionally, countertransference can lead to unnecessary prescribing and polypharmacy that confounds nocebo effects. Therefore self-care, consultation, and supervision may be vital in promoting therapeutic outcomes.
Partnership. The doctor–patient relationship can contribute to nocebo effects. A 2016 Gallup Poll found that Americans had low confidence in the honesty and ethics of psychiatrists compared with other healthcare professionals.3 It is important to have conversations with your patients about their reservations and perceived stigma of mental health. Such conversations can bring a patient’s ambivalence into treatment so that it can be further explored and addressed. Psychoeducation about treatment limitations, motivational interviewing techniques, and involving patients in decision-making can be useful tools for fostering a therapeutic alliance and positive outcomes.
Take an active approach
Evidence demonstrates that psychosocial factors significantly impact treatment outcomes.1 Incorporating this evidence into practice and attending to the 4 factors discussed here can enhance a clinician’s ability to flexibly respond to their patients’ complaints, especially in relation to nocebo effects.
1. Mallo CJ, Mintz DL. Teaching all the evidence bases: reintegrating psychodynamic aspects of prescribing into psychopharmacology training. Psychodyn Psychiatry. 2013;41(1):13-37.
2. Weissenfeld J, Stock S, Lüngen M, et al. The nocebo effect: a reason for patients’ non-adherence to generic substitution? Pharmazie. 2010;65(7):451-456.
3. Norman J. Americans rate healthcare providers high on honesty, ethics. Gallup. http://news.gallup.com/poll/200057/americans-rate-healthcare-providers-high-honesty-ethics.aspx. Published December 19, 2016. Accessed October 22, 2017.
1. Mallo CJ, Mintz DL. Teaching all the evidence bases: reintegrating psychodynamic aspects of prescribing into psychopharmacology training. Psychodyn Psychiatry. 2013;41(1):13-37.
2. Weissenfeld J, Stock S, Lüngen M, et al. The nocebo effect: a reason for patients’ non-adherence to generic substitution? Pharmazie. 2010;65(7):451-456.
3. Norman J. Americans rate healthcare providers high on honesty, ethics. Gallup. http://news.gallup.com/poll/200057/americans-rate-healthcare-providers-high-honesty-ethics.aspx. Published December 19, 2016. Accessed October 22, 2017.
USPSTF update: New and revised recommendations
Over the past year the US Preventive Services Task Force made 14 recommendations on 12 conditions (TABLE 11-12). One of these pronouncements was the unusual reversal of a previous “D” recommendation against screening for scoliosis in adolescents, changing it to an “I” statement (insufficient evidence).
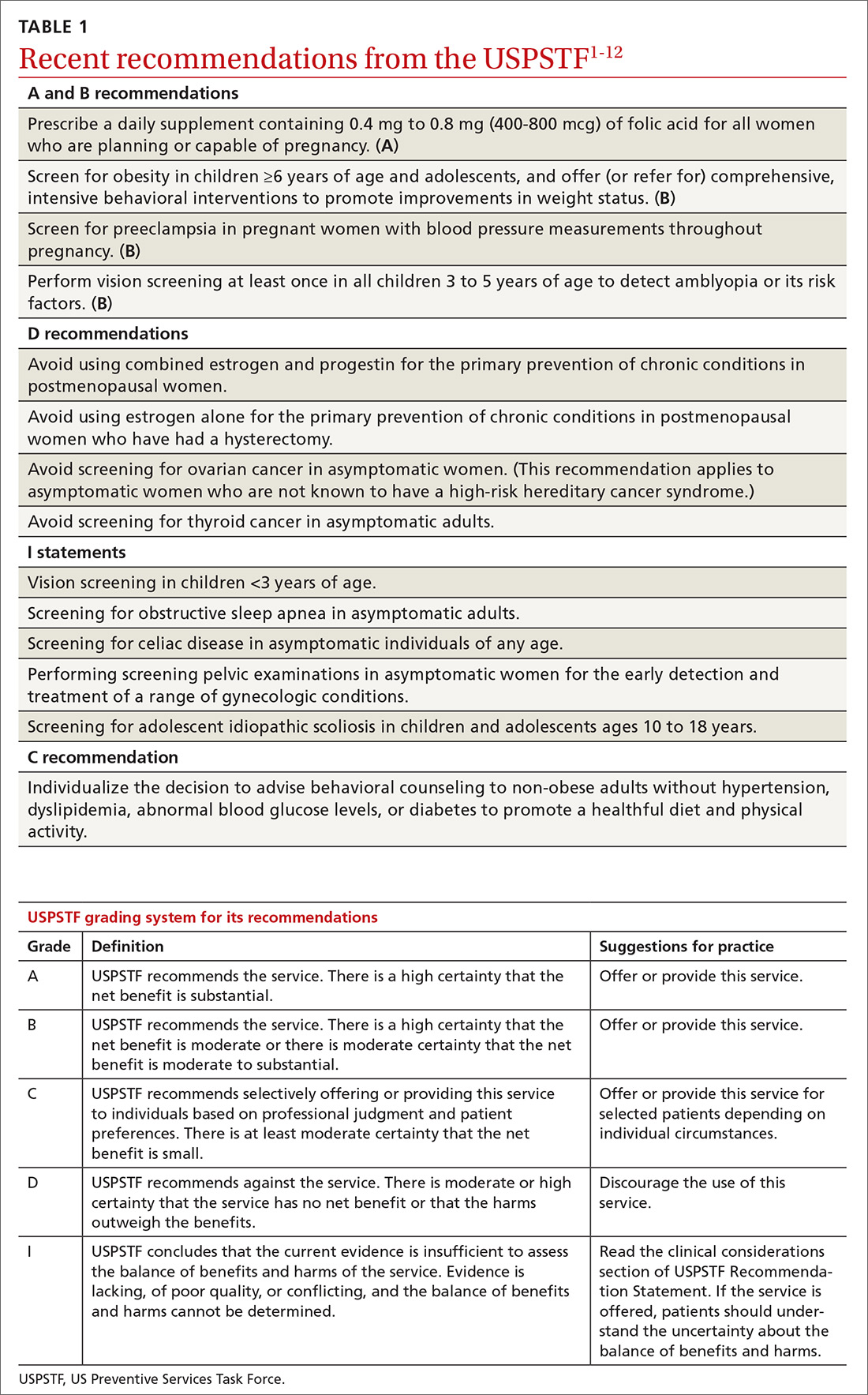
Affirmative recommendations
Four interventions were given an “A” or “B” recommendation this past year. Both grades signify a recommendation to perform the service, with “A” reflecting a higher level of certainty or a higher level of net benefit than “B.”
Recommend folic acid to prevent neural tube defects (A)
The evidence is very strong that folic acid intake prevents neural tube defects. In 2009 the Task Force recommended folic acid supplementation for women of childbearing age. In 2017 this recommendation was updated and slightly reworded to advise that all women who are planning a pregnancy or capable of becoming pregnant take a daily supplement containing 0.4 mg to 0.8 mg (400-800 mcg) of folic acid.
In the United States many grain products have been fortified with folic acid since 1996. This step has reduced the prevalence of neural tube defects from 10.7 cases per 10,000 live births to 7 cases per 10,000 live births in 2011.1 However, in spite of food fortification, most women in the United States do not consume the recommended daily amount of 0.4 mg (400 mcg) of folic acid. This supplementation is most important from one month before conception through the first 3 months of pregnancy.
Screen for obesity in children and adolescents (B)
Nearly 17% of children and adolescents ages 2 to 19 years in the United States are obese, and almost 32% are overweight or obese.2 Obesity is defined as a body mass index (BMI) ≥95th percentile, based on year-2000 growth charts published by the Centers for Disease Control and Prevention. Overweight is defined as a BMI between the 85th and 94th percentiles.
Obesity in children and adolescents is associated with many physical problems, including obstructive sleep apnea, orthopedic problems, high blood pressure, hyperlipidemia, and diabetes, as well as psychological harms from being teased and bullied. Obesity that continues into adulthood is associated with diabetes, cardiovascular disease, and orthopedic problems.
The Task Force found that intensive behavioral interventions for obesity in children ≥6 years of age and in adolescents can lead to moderate improvements in weight status for up to 12 months. Intensive behavioral interventions need to include at least 26 contact hours over 2 to 12 months. The recommendation statement includes a more detailed description of the types of programs that have evidence to support them.2
The Task Force did not recommend the use of either metformin or orlistat because of inadequate evidence on the harmful effects of metformin and because of sound evidence that orlistat causes moderate harms, such as abdominal pain, cramping, incontinence, and flatus.
Screen for preeclampsia (B), but dipstick testing is unreliable
Preeclampsia occurs in a little more than 3% of pregnancies in the United States.13 For the mother, this condition can lead to stroke, eclampsia, organ failure, and death; for the fetus, intrauterine growth retardation, preterm birth, low birth weight, and still birth. Preeclampsia is a leading cause of maternal mortality worldwide. Adverse health outcomes can be prevented by early detection of preeclampsia and by managing it appropriately.3
In 1996 the Task Force recommended screening for preeclampsia during pregnancy, and it reaffirmed that recommendation last year. The Task Force recommends taking blood pressure measurements at every prenatal visit, but does not recommend testing for urine protein with a dipstick because of the technique’s low accuracy.
Since 2014 the Task Force has also recommended using low-dose aspirin after Week 12 of pregnancy to prevent preeclampsia in women who are at high risk.14
Conduct vision screening in all children ages 3 to 5 years (B)
One of the more nuanced recommendations involves vision screening in children. The Task Force recently reaffirmed its 2011 recommendation to perform vision screening at least once in all children ages 3 to 5 years to detect amblyopia or its risk factors. But it found insufficient evidence to test children <3 years of age.
Amblyopia is a “functional reduction in visual acuity characterized by abnormal processing of visual images; [it is] established by the brain during a critical period of vision development.”4 Risk factors associated with the development of amblyopia include strabismus (ocular misalignment); vision loss caused by cataracts; refractive errors such as near and far sightedness, astigmatism (“blurred vision at any distance due to abnormal curvature of the cornea or lens”); and anisometropia (“asymmetric refractive error between the … eyes that causes image suppression in the eye with the larger error”). 4
Physical exam- and machine-based screening tests are available in the primary care setting (TABLE 2).4
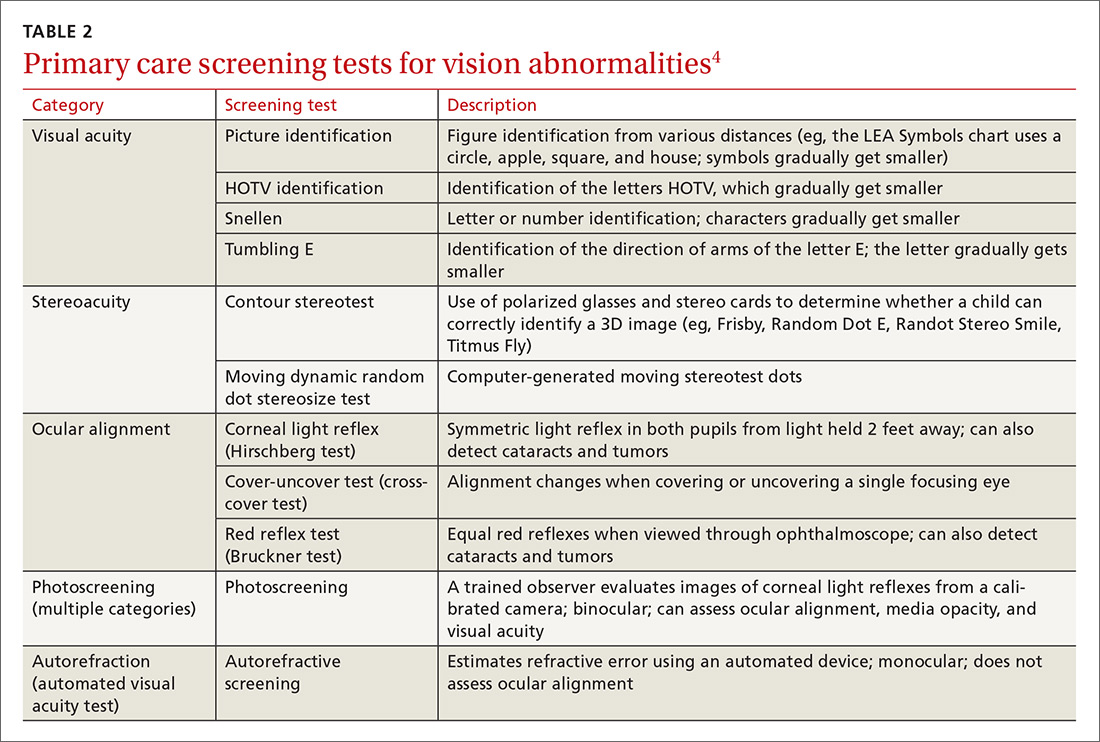
At first glance it appears that the Task Force recommends screening only for amblyopia, but the addition of “risk factors” implies a more comprehensive vision evaluation that would include visual acuity. This interpretation more closely aligns the Task Force recommendation with that of a joint report by the American Academy of Pediatrics, American Association for Pediatric Ophthalmology and Strabismus, American Academy of Certified Orthoptists, and American Academy of Ophthalmology, which recommends testing for a variety of vision problems in children.15 Nevertheless, the Task Force maintains that the evidence of benefit in testing more extensively before age 3 is insufficient, while the other organizations recommend starting testing at age 6 months.
Negative “D” recommendations
Equally as important as affirmative recommendations for effective interventions are the “D” recommendations advising against interventions that are ineffective or cause more harm than benefits. This past year, the Task Force recommended against 4 interventions. Two pertain to the use of estrogen or combined estrogen and progestin for the primary prevention of chronic conditions in postmenopausal women.5 This topic has been discussed in a recent JFP audiocast. Also receiving “D” recommendations were screening for ovarian cancer in asymptomatic women,6 discussed in another JFP audiocast, and screening for thyroid cancer in asymptomatic adults.7
The “D” recommendation for thyroid cancer screening was based on the low incidence of thyroid cancer, the evidence showing no change in mortality after the introduction of population-based screening, and the likelihood of overdiagnosis and overtreatment that would result from screening. The screening tests considered by the Task Force included neck palpation and ultrasound.7
Insufficient evidence
In addition to the previously mentioned “I” statement on vision screening for children <3 years of age,4 4 other interventions lacked sufficient evidence that the Task Force could use in determining relative levels of harms and benefits. These interventions were screening for obstructive sleep apnea in asymptomatic adults,8 screening for celiac disease in asymptomatic patients of all ages,9 screening with a pelvic examination in asymptomatic women,10 and screening for adolescent idiopathic scoliosis in children and adolescents ages 10 to 18 years.11
The lack of evidence regarding the value of a routine pelvic exam for asymptomatic women is surprising given how often this procedure is performed. The Task Force defined a pelvic exam as an “assessment of the external genitalia, internal speculum examination, bimanual palpation, and rectovaginal examination.”10 The Task Force found very little evidence on the accuracy and effectiveness of this exam for a range of gynecologic conditions other than cervical cancer, gonorrhea, and chlamydia, for which screening is recommended.10
The “I” statement on screening for adolescent idiopathic scoliosis in children and adolescents is an unusual revision of a “D” recommendation from 2004. At that time, the Task Force found that treatment of adolescent idiopathic scoliosis leads to health benefits in only a small proportion of individuals and that there are harms of treatment such as unnecessary bracing and referral to specialty care. For the most recent evidence report, the Task Force used a new methodology to assess treatment harms and concluded that the evidence is now inadequate. That finding, along with new evidence that “suggests that brace treatment can interrupt or slow scoliosis progression” led the Task Force to move away from a “D” recommendation.11
The enigmatic “C” recommendation
Perhaps the most difficult recommendation category to understand and implement is the “C” recommendation. With a “C” intervention, there is moderate certainty that the net benefit of universal implementation would be very small; but there are some individuals who might benefit from it, and physicians should offer it selectively.
The Task Force made one “C” recommendation over the past year: for adults who are not obese and who do not have other cardiovascular disease (CVD) risks, the net gain in referring them to behavioral counseling to promote a healthful diet and physical activity is small. However, the harms from such referrals are also small. Counseling interventions can result in healthier habits and in small improvements in intermediate outcomes, such as blood pressure, cholesterol levels, and weight. The effect on overall CVD mortality, though, has been minimal.12 The Task Force concluded that “[those] who are interested and ready to make behavioral changes may be most likely to benefit from behavioral counseling.”
1. USPSTF. Folic acid for the prevention of neural tube defects: preventive medication. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/folic-acid-for-the-prevention-of-neural-tube-defects-preventive-medication. Accessed March 22, 2018.
2. USPSTF. Obesity in children and adolescents: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/obesity-in-children-and-adolescents-screening1. Accessed March 22, 2018.
3. USPSTF. Preeclampsia: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/preeclampsia-screening1. Accessed March 22, 2018.
4. USPSTF. Vision in children ages 6 months to 5 years: Screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/vision-in-children-ages-6-months-to-5-years-screening. Accessed March 22, 2018.
5. USPSTF. Hormone therapy in postmenopausal women: primary prevention of chronic conditions. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/menopausal-hormone-therapy-preventive-medication1. Accessed March 24, 2018.
6. USPSTF. Ovarian cancer: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/ovarian-cancer-screening1. Accessed March 24, 2018.
7. USPSTF. Thyroid cancer: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/thyroid-cancer-screening1. Accessed March 22, 2018.
8. USPSTF. Obstructive sleep apnea in adults: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/obstructive-sleep-apnea-in-adults-screening. Accessed March 22, 2018.
9. USPSTF. Celiac disease: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/celiac-disease-screening. Accessed March 24, 2018.
10. USPSTF. Gynecological conditions: periodic screening with the pelvic examination. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/gynecological-conditions-screening-with-the-pelvic-examination. Accessed March 22, 2018.
11. USPSTF. Adolescent idiopathic scoliosis: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/adolescent-idiopathic-scoliosis-screening1. Accessed March 22, 2018.
12. USPSTF. Healthful diet and physical activity for cardiovascular disease prevention in adults without known risk factors: behavioral counseling. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/healthful-diet-and-physical-activity-for-cardiovascular-disease-prevention-in-adults-without-known-risk-factors-behavioral-counseling. Accessed March 22, 2018.
13. Ananth CV, Keyes KM, Wapner RJ. Pre-eclampsia rates in the United States, 1980-2010: age-period-cohort analysis. BMJ. 2013;347:f6564.
14. USPSTF. Low-dose aspirin use for the prevention of morbidity and mortality from preeclampsia: preventive medication. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/low-dose-aspirin-use-for-the-prevention-of-morbidity-and-mortality-from-preeclampsia-preventive-medication. Accessed March 22, 2018.
15. Donahue SP, Baker CN; Committee on Practice and Ambulatory Medicine, American Academy of Pediatrics; Section on Ophthalmology, American Academy of Pediatrics; American Association of Certified Orthoptists; American Association for Pediatric Ophthalmology and Strabismus; American Academy of Ophthalmology. Procedures for the evaluation of the visual system by pediatricians. Pediatrics. 2016;137.2015-3597.
Over the past year the US Preventive Services Task Force made 14 recommendations on 12 conditions (TABLE 11-12). One of these pronouncements was the unusual reversal of a previous “D” recommendation against screening for scoliosis in adolescents, changing it to an “I” statement (insufficient evidence).
Affirmative recommendations
Four interventions were given an “A” or “B” recommendation this past year. Both grades signify a recommendation to perform the service, with “A” reflecting a higher level of certainty or a higher level of net benefit than “B.”
Recommend folic acid to prevent neural tube defects (A)
The evidence is very strong that folic acid intake prevents neural tube defects. In 2009 the Task Force recommended folic acid supplementation for women of childbearing age. In 2017 this recommendation was updated and slightly reworded to advise that all women who are planning a pregnancy or capable of becoming pregnant take a daily supplement containing 0.4 mg to 0.8 mg (400-800 mcg) of folic acid.
In the United States many grain products have been fortified with folic acid since 1996. This step has reduced the prevalence of neural tube defects from 10.7 cases per 10,000 live births to 7 cases per 10,000 live births in 2011.1 However, in spite of food fortification, most women in the United States do not consume the recommended daily amount of 0.4 mg (400 mcg) of folic acid. This supplementation is most important from one month before conception through the first 3 months of pregnancy.
Screen for obesity in children and adolescents (B)
Nearly 17% of children and adolescents ages 2 to 19 years in the United States are obese, and almost 32% are overweight or obese.2 Obesity is defined as a body mass index (BMI) ≥95th percentile, based on year-2000 growth charts published by the Centers for Disease Control and Prevention. Overweight is defined as a BMI between the 85th and 94th percentiles.
Obesity in children and adolescents is associated with many physical problems, including obstructive sleep apnea, orthopedic problems, high blood pressure, hyperlipidemia, and diabetes, as well as psychological harms from being teased and bullied. Obesity that continues into adulthood is associated with diabetes, cardiovascular disease, and orthopedic problems.
The Task Force found that intensive behavioral interventions for obesity in children ≥6 years of age and in adolescents can lead to moderate improvements in weight status for up to 12 months. Intensive behavioral interventions need to include at least 26 contact hours over 2 to 12 months. The recommendation statement includes a more detailed description of the types of programs that have evidence to support them.2
The Task Force did not recommend the use of either metformin or orlistat because of inadequate evidence on the harmful effects of metformin and because of sound evidence that orlistat causes moderate harms, such as abdominal pain, cramping, incontinence, and flatus.
Screen for preeclampsia (B), but dipstick testing is unreliable
Preeclampsia occurs in a little more than 3% of pregnancies in the United States.13 For the mother, this condition can lead to stroke, eclampsia, organ failure, and death; for the fetus, intrauterine growth retardation, preterm birth, low birth weight, and still birth. Preeclampsia is a leading cause of maternal mortality worldwide. Adverse health outcomes can be prevented by early detection of preeclampsia and by managing it appropriately.3
In 1996 the Task Force recommended screening for preeclampsia during pregnancy, and it reaffirmed that recommendation last year. The Task Force recommends taking blood pressure measurements at every prenatal visit, but does not recommend testing for urine protein with a dipstick because of the technique’s low accuracy.
Since 2014 the Task Force has also recommended using low-dose aspirin after Week 12 of pregnancy to prevent preeclampsia in women who are at high risk.14
Conduct vision screening in all children ages 3 to 5 years (B)
One of the more nuanced recommendations involves vision screening in children. The Task Force recently reaffirmed its 2011 recommendation to perform vision screening at least once in all children ages 3 to 5 years to detect amblyopia or its risk factors. But it found insufficient evidence to test children <3 years of age.
Amblyopia is a “functional reduction in visual acuity characterized by abnormal processing of visual images; [it is] established by the brain during a critical period of vision development.”4 Risk factors associated with the development of amblyopia include strabismus (ocular misalignment); vision loss caused by cataracts; refractive errors such as near and far sightedness, astigmatism (“blurred vision at any distance due to abnormal curvature of the cornea or lens”); and anisometropia (“asymmetric refractive error between the … eyes that causes image suppression in the eye with the larger error”). 4
Physical exam- and machine-based screening tests are available in the primary care setting (TABLE 2).4
At first glance it appears that the Task Force recommends screening only for amblyopia, but the addition of “risk factors” implies a more comprehensive vision evaluation that would include visual acuity. This interpretation more closely aligns the Task Force recommendation with that of a joint report by the American Academy of Pediatrics, American Association for Pediatric Ophthalmology and Strabismus, American Academy of Certified Orthoptists, and American Academy of Ophthalmology, which recommends testing for a variety of vision problems in children.15 Nevertheless, the Task Force maintains that the evidence of benefit in testing more extensively before age 3 is insufficient, while the other organizations recommend starting testing at age 6 months.
Negative “D” recommendations
Equally as important as affirmative recommendations for effective interventions are the “D” recommendations advising against interventions that are ineffective or cause more harm than benefits. This past year, the Task Force recommended against 4 interventions. Two pertain to the use of estrogen or combined estrogen and progestin for the primary prevention of chronic conditions in postmenopausal women.5 This topic has been discussed in a recent JFP audiocast. Also receiving “D” recommendations were screening for ovarian cancer in asymptomatic women,6 discussed in another JFP audiocast, and screening for thyroid cancer in asymptomatic adults.7
The “D” recommendation for thyroid cancer screening was based on the low incidence of thyroid cancer, the evidence showing no change in mortality after the introduction of population-based screening, and the likelihood of overdiagnosis and overtreatment that would result from screening. The screening tests considered by the Task Force included neck palpation and ultrasound.7
Insufficient evidence
In addition to the previously mentioned “I” statement on vision screening for children <3 years of age,4 4 other interventions lacked sufficient evidence that the Task Force could use in determining relative levels of harms and benefits. These interventions were screening for obstructive sleep apnea in asymptomatic adults,8 screening for celiac disease in asymptomatic patients of all ages,9 screening with a pelvic examination in asymptomatic women,10 and screening for adolescent idiopathic scoliosis in children and adolescents ages 10 to 18 years.11
The lack of evidence regarding the value of a routine pelvic exam for asymptomatic women is surprising given how often this procedure is performed. The Task Force defined a pelvic exam as an “assessment of the external genitalia, internal speculum examination, bimanual palpation, and rectovaginal examination.”10 The Task Force found very little evidence on the accuracy and effectiveness of this exam for a range of gynecologic conditions other than cervical cancer, gonorrhea, and chlamydia, for which screening is recommended.10
The “I” statement on screening for adolescent idiopathic scoliosis in children and adolescents is an unusual revision of a “D” recommendation from 2004. At that time, the Task Force found that treatment of adolescent idiopathic scoliosis leads to health benefits in only a small proportion of individuals and that there are harms of treatment such as unnecessary bracing and referral to specialty care. For the most recent evidence report, the Task Force used a new methodology to assess treatment harms and concluded that the evidence is now inadequate. That finding, along with new evidence that “suggests that brace treatment can interrupt or slow scoliosis progression” led the Task Force to move away from a “D” recommendation.11
The enigmatic “C” recommendation
Perhaps the most difficult recommendation category to understand and implement is the “C” recommendation. With a “C” intervention, there is moderate certainty that the net benefit of universal implementation would be very small; but there are some individuals who might benefit from it, and physicians should offer it selectively.
The Task Force made one “C” recommendation over the past year: for adults who are not obese and who do not have other cardiovascular disease (CVD) risks, the net gain in referring them to behavioral counseling to promote a healthful diet and physical activity is small. However, the harms from such referrals are also small. Counseling interventions can result in healthier habits and in small improvements in intermediate outcomes, such as blood pressure, cholesterol levels, and weight. The effect on overall CVD mortality, though, has been minimal.12 The Task Force concluded that “[those] who are interested and ready to make behavioral changes may be most likely to benefit from behavioral counseling.”
Over the past year the US Preventive Services Task Force made 14 recommendations on 12 conditions (TABLE 11-12). One of these pronouncements was the unusual reversal of a previous “D” recommendation against screening for scoliosis in adolescents, changing it to an “I” statement (insufficient evidence).
Affirmative recommendations
Four interventions were given an “A” or “B” recommendation this past year. Both grades signify a recommendation to perform the service, with “A” reflecting a higher level of certainty or a higher level of net benefit than “B.”
Recommend folic acid to prevent neural tube defects (A)
The evidence is very strong that folic acid intake prevents neural tube defects. In 2009 the Task Force recommended folic acid supplementation for women of childbearing age. In 2017 this recommendation was updated and slightly reworded to advise that all women who are planning a pregnancy or capable of becoming pregnant take a daily supplement containing 0.4 mg to 0.8 mg (400-800 mcg) of folic acid.
In the United States many grain products have been fortified with folic acid since 1996. This step has reduced the prevalence of neural tube defects from 10.7 cases per 10,000 live births to 7 cases per 10,000 live births in 2011.1 However, in spite of food fortification, most women in the United States do not consume the recommended daily amount of 0.4 mg (400 mcg) of folic acid. This supplementation is most important from one month before conception through the first 3 months of pregnancy.
Screen for obesity in children and adolescents (B)
Nearly 17% of children and adolescents ages 2 to 19 years in the United States are obese, and almost 32% are overweight or obese.2 Obesity is defined as a body mass index (BMI) ≥95th percentile, based on year-2000 growth charts published by the Centers for Disease Control and Prevention. Overweight is defined as a BMI between the 85th and 94th percentiles.
Obesity in children and adolescents is associated with many physical problems, including obstructive sleep apnea, orthopedic problems, high blood pressure, hyperlipidemia, and diabetes, as well as psychological harms from being teased and bullied. Obesity that continues into adulthood is associated with diabetes, cardiovascular disease, and orthopedic problems.
The Task Force found that intensive behavioral interventions for obesity in children ≥6 years of age and in adolescents can lead to moderate improvements in weight status for up to 12 months. Intensive behavioral interventions need to include at least 26 contact hours over 2 to 12 months. The recommendation statement includes a more detailed description of the types of programs that have evidence to support them.2
The Task Force did not recommend the use of either metformin or orlistat because of inadequate evidence on the harmful effects of metformin and because of sound evidence that orlistat causes moderate harms, such as abdominal pain, cramping, incontinence, and flatus.
Screen for preeclampsia (B), but dipstick testing is unreliable
Preeclampsia occurs in a little more than 3% of pregnancies in the United States.13 For the mother, this condition can lead to stroke, eclampsia, organ failure, and death; for the fetus, intrauterine growth retardation, preterm birth, low birth weight, and still birth. Preeclampsia is a leading cause of maternal mortality worldwide. Adverse health outcomes can be prevented by early detection of preeclampsia and by managing it appropriately.3
In 1996 the Task Force recommended screening for preeclampsia during pregnancy, and it reaffirmed that recommendation last year. The Task Force recommends taking blood pressure measurements at every prenatal visit, but does not recommend testing for urine protein with a dipstick because of the technique’s low accuracy.
Since 2014 the Task Force has also recommended using low-dose aspirin after Week 12 of pregnancy to prevent preeclampsia in women who are at high risk.14
Conduct vision screening in all children ages 3 to 5 years (B)
One of the more nuanced recommendations involves vision screening in children. The Task Force recently reaffirmed its 2011 recommendation to perform vision screening at least once in all children ages 3 to 5 years to detect amblyopia or its risk factors. But it found insufficient evidence to test children <3 years of age.
Amblyopia is a “functional reduction in visual acuity characterized by abnormal processing of visual images; [it is] established by the brain during a critical period of vision development.”4 Risk factors associated with the development of amblyopia include strabismus (ocular misalignment); vision loss caused by cataracts; refractive errors such as near and far sightedness, astigmatism (“blurred vision at any distance due to abnormal curvature of the cornea or lens”); and anisometropia (“asymmetric refractive error between the … eyes that causes image suppression in the eye with the larger error”). 4
Physical exam- and machine-based screening tests are available in the primary care setting (TABLE 2).4
At first glance it appears that the Task Force recommends screening only for amblyopia, but the addition of “risk factors” implies a more comprehensive vision evaluation that would include visual acuity. This interpretation more closely aligns the Task Force recommendation with that of a joint report by the American Academy of Pediatrics, American Association for Pediatric Ophthalmology and Strabismus, American Academy of Certified Orthoptists, and American Academy of Ophthalmology, which recommends testing for a variety of vision problems in children.15 Nevertheless, the Task Force maintains that the evidence of benefit in testing more extensively before age 3 is insufficient, while the other organizations recommend starting testing at age 6 months.
Negative “D” recommendations
Equally as important as affirmative recommendations for effective interventions are the “D” recommendations advising against interventions that are ineffective or cause more harm than benefits. This past year, the Task Force recommended against 4 interventions. Two pertain to the use of estrogen or combined estrogen and progestin for the primary prevention of chronic conditions in postmenopausal women.5 This topic has been discussed in a recent JFP audiocast. Also receiving “D” recommendations were screening for ovarian cancer in asymptomatic women,6 discussed in another JFP audiocast, and screening for thyroid cancer in asymptomatic adults.7
The “D” recommendation for thyroid cancer screening was based on the low incidence of thyroid cancer, the evidence showing no change in mortality after the introduction of population-based screening, and the likelihood of overdiagnosis and overtreatment that would result from screening. The screening tests considered by the Task Force included neck palpation and ultrasound.7
Insufficient evidence
In addition to the previously mentioned “I” statement on vision screening for children <3 years of age,4 4 other interventions lacked sufficient evidence that the Task Force could use in determining relative levels of harms and benefits. These interventions were screening for obstructive sleep apnea in asymptomatic adults,8 screening for celiac disease in asymptomatic patients of all ages,9 screening with a pelvic examination in asymptomatic women,10 and screening for adolescent idiopathic scoliosis in children and adolescents ages 10 to 18 years.11
The lack of evidence regarding the value of a routine pelvic exam for asymptomatic women is surprising given how often this procedure is performed. The Task Force defined a pelvic exam as an “assessment of the external genitalia, internal speculum examination, bimanual palpation, and rectovaginal examination.”10 The Task Force found very little evidence on the accuracy and effectiveness of this exam for a range of gynecologic conditions other than cervical cancer, gonorrhea, and chlamydia, for which screening is recommended.10
The “I” statement on screening for adolescent idiopathic scoliosis in children and adolescents is an unusual revision of a “D” recommendation from 2004. At that time, the Task Force found that treatment of adolescent idiopathic scoliosis leads to health benefits in only a small proportion of individuals and that there are harms of treatment such as unnecessary bracing and referral to specialty care. For the most recent evidence report, the Task Force used a new methodology to assess treatment harms and concluded that the evidence is now inadequate. That finding, along with new evidence that “suggests that brace treatment can interrupt or slow scoliosis progression” led the Task Force to move away from a “D” recommendation.11
The enigmatic “C” recommendation
Perhaps the most difficult recommendation category to understand and implement is the “C” recommendation. With a “C” intervention, there is moderate certainty that the net benefit of universal implementation would be very small; but there are some individuals who might benefit from it, and physicians should offer it selectively.
The Task Force made one “C” recommendation over the past year: for adults who are not obese and who do not have other cardiovascular disease (CVD) risks, the net gain in referring them to behavioral counseling to promote a healthful diet and physical activity is small. However, the harms from such referrals are also small. Counseling interventions can result in healthier habits and in small improvements in intermediate outcomes, such as blood pressure, cholesterol levels, and weight. The effect on overall CVD mortality, though, has been minimal.12 The Task Force concluded that “[those] who are interested and ready to make behavioral changes may be most likely to benefit from behavioral counseling.”
1. USPSTF. Folic acid for the prevention of neural tube defects: preventive medication. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/folic-acid-for-the-prevention-of-neural-tube-defects-preventive-medication. Accessed March 22, 2018.
2. USPSTF. Obesity in children and adolescents: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/obesity-in-children-and-adolescents-screening1. Accessed March 22, 2018.
3. USPSTF. Preeclampsia: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/preeclampsia-screening1. Accessed March 22, 2018.
4. USPSTF. Vision in children ages 6 months to 5 years: Screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/vision-in-children-ages-6-months-to-5-years-screening. Accessed March 22, 2018.
5. USPSTF. Hormone therapy in postmenopausal women: primary prevention of chronic conditions. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/menopausal-hormone-therapy-preventive-medication1. Accessed March 24, 2018.
6. USPSTF. Ovarian cancer: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/ovarian-cancer-screening1. Accessed March 24, 2018.
7. USPSTF. Thyroid cancer: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/thyroid-cancer-screening1. Accessed March 22, 2018.
8. USPSTF. Obstructive sleep apnea in adults: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/obstructive-sleep-apnea-in-adults-screening. Accessed March 22, 2018.
9. USPSTF. Celiac disease: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/celiac-disease-screening. Accessed March 24, 2018.
10. USPSTF. Gynecological conditions: periodic screening with the pelvic examination. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/gynecological-conditions-screening-with-the-pelvic-examination. Accessed March 22, 2018.
11. USPSTF. Adolescent idiopathic scoliosis: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/adolescent-idiopathic-scoliosis-screening1. Accessed March 22, 2018.
12. USPSTF. Healthful diet and physical activity for cardiovascular disease prevention in adults without known risk factors: behavioral counseling. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/healthful-diet-and-physical-activity-for-cardiovascular-disease-prevention-in-adults-without-known-risk-factors-behavioral-counseling. Accessed March 22, 2018.
13. Ananth CV, Keyes KM, Wapner RJ. Pre-eclampsia rates in the United States, 1980-2010: age-period-cohort analysis. BMJ. 2013;347:f6564.
14. USPSTF. Low-dose aspirin use for the prevention of morbidity and mortality from preeclampsia: preventive medication. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/low-dose-aspirin-use-for-the-prevention-of-morbidity-and-mortality-from-preeclampsia-preventive-medication. Accessed March 22, 2018.
15. Donahue SP, Baker CN; Committee on Practice and Ambulatory Medicine, American Academy of Pediatrics; Section on Ophthalmology, American Academy of Pediatrics; American Association of Certified Orthoptists; American Association for Pediatric Ophthalmology and Strabismus; American Academy of Ophthalmology. Procedures for the evaluation of the visual system by pediatricians. Pediatrics. 2016;137.2015-3597.
1. USPSTF. Folic acid for the prevention of neural tube defects: preventive medication. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/folic-acid-for-the-prevention-of-neural-tube-defects-preventive-medication. Accessed March 22, 2018.
2. USPSTF. Obesity in children and adolescents: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/obesity-in-children-and-adolescents-screening1. Accessed March 22, 2018.
3. USPSTF. Preeclampsia: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/preeclampsia-screening1. Accessed March 22, 2018.
4. USPSTF. Vision in children ages 6 months to 5 years: Screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/vision-in-children-ages-6-months-to-5-years-screening. Accessed March 22, 2018.
5. USPSTF. Hormone therapy in postmenopausal women: primary prevention of chronic conditions. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/menopausal-hormone-therapy-preventive-medication1. Accessed March 24, 2018.
6. USPSTF. Ovarian cancer: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/ovarian-cancer-screening1. Accessed March 24, 2018.
7. USPSTF. Thyroid cancer: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/thyroid-cancer-screening1. Accessed March 22, 2018.
8. USPSTF. Obstructive sleep apnea in adults: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/obstructive-sleep-apnea-in-adults-screening. Accessed March 22, 2018.
9. USPSTF. Celiac disease: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/celiac-disease-screening. Accessed March 24, 2018.
10. USPSTF. Gynecological conditions: periodic screening with the pelvic examination. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/gynecological-conditions-screening-with-the-pelvic-examination. Accessed March 22, 2018.
11. USPSTF. Adolescent idiopathic scoliosis: screening. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/adolescent-idiopathic-scoliosis-screening1. Accessed March 22, 2018.
12. USPSTF. Healthful diet and physical activity for cardiovascular disease prevention in adults without known risk factors: behavioral counseling. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/healthful-diet-and-physical-activity-for-cardiovascular-disease-prevention-in-adults-without-known-risk-factors-behavioral-counseling. Accessed March 22, 2018.
13. Ananth CV, Keyes KM, Wapner RJ. Pre-eclampsia rates in the United States, 1980-2010: age-period-cohort analysis. BMJ. 2013;347:f6564.
14. USPSTF. Low-dose aspirin use for the prevention of morbidity and mortality from preeclampsia: preventive medication. Available at: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/low-dose-aspirin-use-for-the-prevention-of-morbidity-and-mortality-from-preeclampsia-preventive-medication. Accessed March 22, 2018.
15. Donahue SP, Baker CN; Committee on Practice and Ambulatory Medicine, American Academy of Pediatrics; Section on Ophthalmology, American Academy of Pediatrics; American Association of Certified Orthoptists; American Association for Pediatric Ophthalmology and Strabismus; American Academy of Ophthalmology. Procedures for the evaluation of the visual system by pediatricians. Pediatrics. 2016;137.2015-3597.
From The Journal of Family Practice | 2018;67(5):294-296,298-299.
Hospital Readmissions in Patients with Cirrhosis: A Systematic Review
Cirrhosis is a morbid condition characterized by complications such as ascites, gastrointestinal bleeding, and hepatic encephalopathy. These complications frequently require hospitalization, which is a substantial burden to the healthcare system. In 2012, liver disease was responsible for nearly 250,000 admissions across the United States, costing $3 billion.1 Despite this substantial resource utilization, outcomes remain poor, with an inpatient mortality of 6%. For those that survive, many experience hospital readmission.
More generally, early readmission reflects poor quality of care in the US. In 2004, 30-day readmissions occurred in nearly 20% of Medicare beneficiaries and costed over $17 billion.2 In response to this problem, the Affordable Care Act established the Hospital Readmissions Reduction Program (HRRP), which reduces Centers for Medicare & Medicaid Services (CMS) payments to hospitals with excess 30-day readmissions for high-risk conditions, including pneumonia and heart failure.3 Heart failure, in particular, has been the subject of numerous studies detailing risk factors and interventions to predict and prevent readmission.4-6 Based on this extensive evidence, guidelines recommend disease management programs to reduce readmissions in this population.7 In contrast, readmission in the cirrhosis population has received limited attention.
We therefore conducted a systematic review aiming to examine the range of readmission risk noted in the literature, with a focus on the model for end-stage liver disease (MELD) score as a risk factor for readmission.
METHODS
Search Strategy
We followed the Preferred Reporting Items for Systematic Reviews and Meta-Analyses guidelines for conducting and reporting systematic reviews.8 A literature search was performed by a medical librarian using the following databases: Ovid MEDLINE, PubMed, EMBASE, CINAHL, the full Cochrane Library, Scopus, Google Scholar, and ClinicalTrials.gov. All the databases were searched from 2000 to May 2017. We did not include older reports because the review focused on contemporary care; earlier studies may not reflect current cirrhosis management. To ensure literature saturation, included articles’ reference lists were reviewed.
Search strategies were developed by combining database-specific subject headings and keywords for readmissions with those for cirrhosis or its complications (Supplementary Material). Google Scholar and ClinicalTrials.gov were searched using keywords only. All results were limited to the English language and those published in 2000 or later, but no other limits were applied.
Identified records were reviewed based on strict criteria. We excluded case reports, case series, reviews, editorials, letters, and meeting abstracts without final peer-reviewed publication. We also excluded studies of pediatric populations (age < 18 years), patients without cirrhosis, and patients with liver transplants. We excluded studies in which patients were not hospitalized at study onset and those where the index admission was for an elective procedure. Because our interest was to identify factors associated with early readmission, we excluded studies that did not report readmissions within 90 days or those with a mean or median follow-up of less than 30 days. We also excluded studies that did not examine the association between readmission and at least 1 independent variable or intervention. Duplicate reports of a common sample were excluded unless the duplicate provided additional information, and such reports were examined together in our synthesis.
Two authors identified potentially eligible records by independently screening titles and abstracts. At this stage, records that did not meet the eligibility criteria were excluded, and the reasons for exclusion were not recorded. Records with disagreement were retained for full-text review. After this initial exclusion of records, the remaining full-text records were reviewed independently. For this full-text review, we recorded exclusion reasons and disagreements were resolved through discussion.
Data Collection
Data were abstracted from each study by 2 authors independently and recorded in a REDCap database.9 Discrepancies were resolved through discussion. We recorded study characteristics, including study design, setting, population (including the inclusion/exclusion criteria, sample size, and patient and hospitalization characteristics), interventions, and comparisons. To facilitate comparisons across studies, we employed validated methods to approximate means and standard deviations (SD).10 We recorded detailed information on outcomes including readmissions, preventability, independent variables, and mortality. Studies that focused on a single independent factor or intervention were classified as “focused,” while those that examined multiple factors were classified as “broad.” We used the Newcastle–Ottawa Scale to assess the risk of bias in each study.11 This instrument uses a 9-point scale to gauge methodological quality based on selection, group comparability, and exposure/outcome assessment.
Statistical Analysis
Analyses were performed using Stata 13.1 (StataCorp LP, College Station, Texas). We determined the pooled proportion of patients with 30-day readmission using a random-effects model, with the Freeman–Tukey double-arcsine transformation for meta-analysis of proportions.12 We investigated the heterogeneity by stratifying analyses according to prespecified study characteristics, including “broad” versus “focused.” However, the readmission risk was not different in the stratified analysis; therefore, we chose to pool the findings. For point estimates, 95% confidence intervals (CIs) were calculated, and a P-value < .05 was considered statistically significant.
RESULTS
Search Results
Study Characteristics
Two studies were performed in Australia, 4 in Europe, and the remainder in North America. Twenty one of the 26 studies were retrospective cohort studies (Table 1). Twenty studies were single-center studies (of which half were performed at transplant centers), and 4 of the 6 multicenter studies were based on administrative data with large samples (173,254 patients). The inclusion/exclusion criteria varied widely (Supplementary Material). Some studies only included patients admitted for specific cirrhosis complications, while others included those admitted for any reason. Two studies excluded patients admitted in the prior 30 days, and 6 excluded patients discharged to hospice. The mean risk of bias score was 7.5 (SD 1.3) out of a possible 9 points, with most lacking an adequate description of follow-up and several lacking adjustment for confounders.
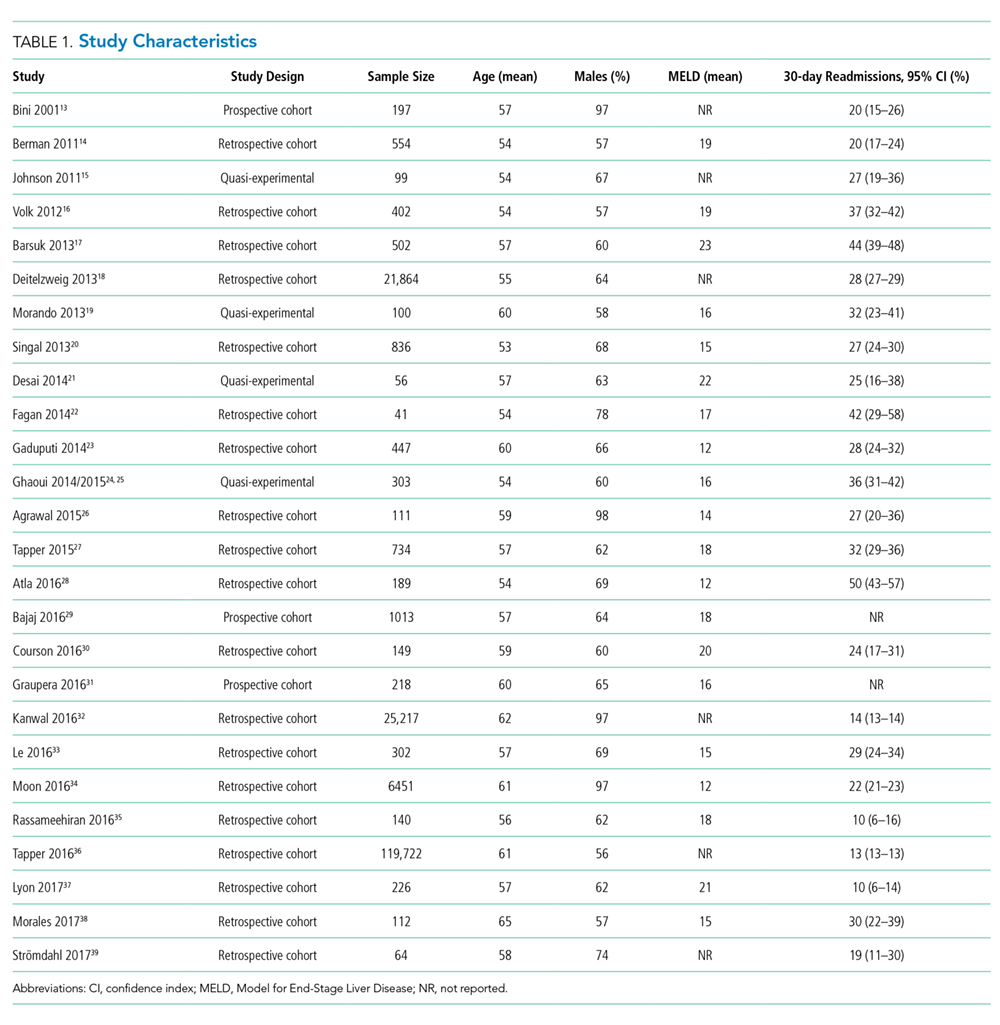
Outcomes
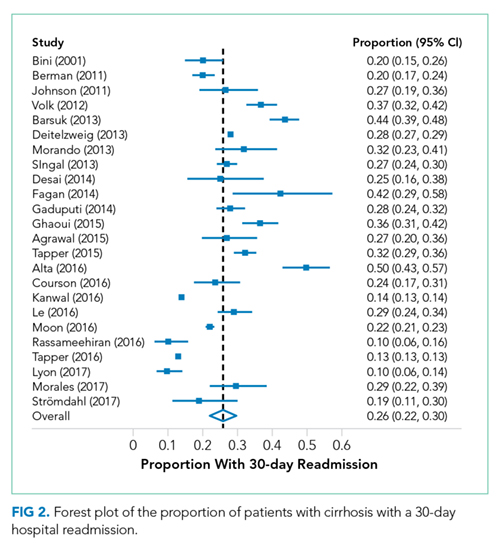
Readmission and MELD
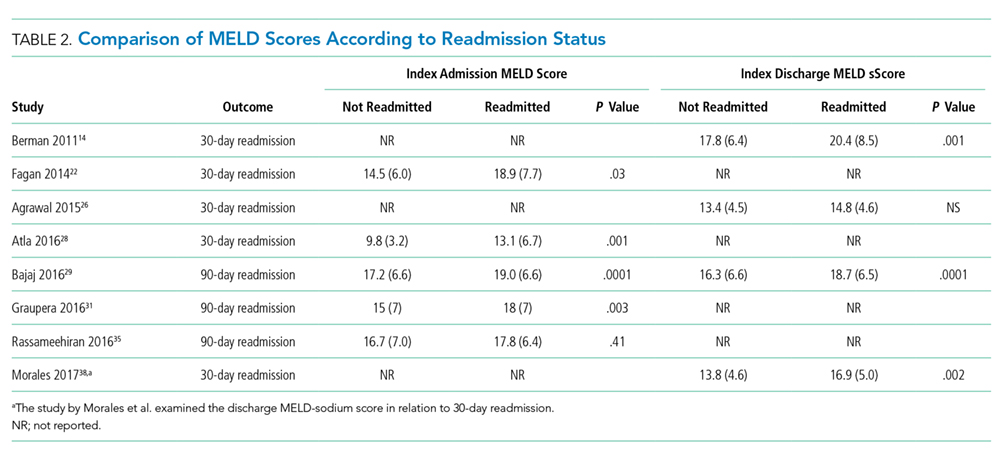
DISCUSSION
Hospital readmission is a costly and common problem in the US.2 In addition to the negative impact that readmissions have on patients’ lives,40 readmissions are increasingly being used to measure quality. Unplanned 30-day readmissions are posted publicly, and excess readmissions for high-risk conditions are penalized through HRRP.3 Although HRRP does not currently include cirrhosis, the program has expanded to include several conditions that were not included in the initial iteration. Whether cirrhosis will be included in future iterations remains to be seen; however, increasing scrutiny is likely to continue. Of specific populations at risk, patients with cirrhosis are particularly vulnerable due to several features. Ascites management often requires hospitalization due to diuretic titration and poor access to paracentesis, and hepatic encephalopathy treatment requires complex lactulose titration.16 Other features of cirrhosis, such as gastrointestinal bleeding, infections, and renal failure, also place patients at risk of poor outcomes. The resulting readmission burden is high, with a pooled 30-day readmission rate of 26%. Other associated outcomes are also poor, with a consistent relationship between readmission and subsequent mortality.
We found striking heterogeneity in various aspects. First, the inclusion/exclusion criteria varied widely, both cirrhosis-specific (eg, spontaneous bacterial peritonitis) and more general (patients admitted within the prior 30 days). Some of these criteria may bias readmission estimates; the risk of readmission may be reduced in those on hospice, as patients forgo curative therapy. Additionally, an established risk factor for readmission is prior hospitalization41; excluding patients with prior admissions prohibits analysis of this variable. Another aspect is the capture of readmissions: readmissions outside of the index hospital were not included in most studies. In those that did include outside readmissions, the burden was sizeable: 17% in 1 single-center study and 23% in a multistate administrative database.16,36 These outside readmissions must be included in future studies; they are as important as same-center readmissions both to patients and CMS.3 Despite this heterogeneity, the studies scored relatively high on the Newcastle–Ottawa risk of bias scale, with the only common deficiency being an inadequate description of follow-up.
Building on the findings of this review, an important step will be the design of interventions to reduce readmissions. Such interventions require a full understanding of this population’s characteristics and needs. Critically, we found a lack of data on social determinants of health. Impairments in these factors are well-established contributors to readmission risk in other populations,4,40 and are highly prevalent in cirrhosis.42 Indeed, CMS has focused resources toward social determinants of health in the effort to reduce utilization and improve outcomes. This lack of data on social determinants of health, as well as other understudied factors, represents an important opportunity for future research efforts to better define the modifiable features that could be targeted in the future to prevent readmissions. Such research is urgently needed and will likely require prospective studies to gather these important factors. Notably, most studies in this systematic review were retrospective and therefore unable to examine many of these understudied factors. Another important aspect that has received little attention is readmission preventability: only 2 studies assessed preventability, both through unstructured chart review. Preventability assessments in noncirrhotic populations have used wide-ranging methodologies, yielding inconsistent results.43 This variability prompted recommendations that preventability should be assessed by multiple reviewers guided by explicit parameters.43 Such detailed attention to preventability is urgently needed to better inform interventions.
In contrast to the lack of data on social factors, we found that the MELD score was examined in most studies and was frequently associated with readmission. Despite this consistent association, differences in the MELD scores between studies limit inferences into specific cutoff values that could identify the highest risk patients. Because of its existing widespread clinical use, the MELD score may prove to be important in readmission risk stratification. Efforts to develop a useful model including the MELD score are needed to target interventions to the highest risk patients.
This review has several limitations. Although we used a broad search strategy to capture studies, some may not have been included due to our selection criteria. For instance, 1 retrospective paper described factors associated with high admission density during 1 year but did not specifically report the frequency of early readmissions.44 Similarly, a randomized trial of a disease management program did not specifically examine early readmissions.45 Another quasi-experimental study of a quality improvement initiative was not included because a large proportion of their subjects was post liver transplant.46 However, the inclusion of these papers is unlikely to change our conclusions; the retrospective study identified factors similar to those in the included studies, and the quasi-experimental study overlapped with the included study that assessed frailty.27 Another potential limitation is the exclusion of studies published in abstract form only. Such studies may be important, as the field of cirrhosis readmissions is relatively young. However, including only full-paper publications ensures the inclusion of only higher quality studies scrutinized during the peer-review process. Similarly, newer published studies may have been missed due to the abundant interest in this topic and ongoing research. Lastly, the significant heterogeneity of the studies limits conclusions that can be made regarding the pooled readmission rates.
In summary, we found that patients with cirrhosis experience a high incidence of hospital readmissions. Several processes of care may be associated with readmissions, suggesting room for improvement in caring for this population and reducing readmissions. However, we identified several gaps in the literature, which does not adequately describe social factors and is lacking details on readmission preventability assessment. Future studies should attempt to address these issues so that interventions can be targeted to the highest risk patients and designed to best meet the needs of patients with cirrhosis.
Disclosures
Dr. Orman, Dr. Ghabril, and Dr. Emmett report no potential conflicts of interest. Dr. Chalasani reports personal fees from Lilly, personal fees from Abbvie, personal fees from Tobira/Allergan, personal fees from Ardelyx, personal fees from Amarin, personal fees from Shire, personal fees from Madrigal, personal fees from DS Biopharma (Afimmune), personal fees from Cempra, personal fees from NuSirt, grants from Galectin, grants from Gilead, grants from Intercept, grants from Cumberland, grants from Conatus, personal fees from Immuron, and personal fees from Axovant, outside the submitted work.
Funding Information
This work was supported, in part, by the National Institutes of Health, KL2 TR001106 and K23 DK109202
1. Peery AF, Crockett SD, Barritt AS, et al. Burden of gastrointestinal, liver, and pancreatic diseases in the United States. Gastroenterology. 2015;149(7):1731-1741.e3. DOI: 10.1053/j.gastro.2015.08.045. PubMed
13. Bini EJ, Weinshel EH, Generoso R, et al. Impact of gastroenterology consultation on the outcomes of patients admitted to the hospital with decompensated cirrhosis. Hepatology. 2001;34(6):1089-1095. DOI: 10.1053/jhep.2001.29204. PubMed
14. Berman K, Tandra S, Forssell K, et al. Incidence and predictors of 30-day readmission among patients hospitalized for advanced liver disease. Clin Gastroenterol Hepatol. 2011;9(3):254-259. DOI: 10.1016/j.cgh.2010.10.035. PubMed
15. Johnson EA, Spier BJ, Leff JA, Lucey MR, Said A. Optimising the care of patients with cirrhosis and gastrointestinal haemorrhage: a quality improvement study. Aliment Pharmacol Ther. 2011;34(1):76-82. DOI: 10.1111/j.1365-2036.2011.04692.x. PubMed
16. Volk ML, Tocco RS, Bazick J, Rakoski MO, Lok AS. Hospital readmissions among patients with decompensated cirrhosis. Am J Gastroenterol. 2012;107(2):247-252. DOI: 10.1038/ajg.2011.314. PubMed
17. Barsuk JH, Cohen ER, Feinglass J, McGaghie WC, Wayne DB. Clinical outcomes after bedside and interventional radiology paracentesis procedures. Am J Med. 2013;126(4):349-356. DOI: 10.1016/j.amjmed.2012.09.016. PubMed
18. Deitelzweig S, Amin A, Christian R, Friend K, Lin J, Lowe TJ. Hyponatremia-associated healthcare burden among US patients hospitalized for cirrhosis. Adv Ther. 2013;30(1):71-80. DOI: 10.1007/s12325-012-0073-1. PubMed
19. Morando F, Maresio G, Piano S, et al. How to improve care in outpatients with cirrhosis and ascites: a new model of care coordination by consultant hepatologists. J Hepatol. 2013;59(2):257-264. DOI: 10.1016/j.jhep.2013.03.010. PubMed
20. Singal AG, Rahimi RS, Clark C, et al. An automated model using electronic medical record data identifies patients with cirrhosis at high risk for readmission. Clin Gastroenterol Hepatol. 2013;11(10):1335-1341.e1. DOI: 10.1016/j.cgh.2013.03.022. PubMed
21. Desai AP, Satoskar R, Appannagari A, et al. Co-management between hospitalist and hepatologist improves the quality of care of inpatients with chronic liver disease. J Clin Gastroenterol. 2014;48(4):e30-e36. DOI: 10.1097/MCG.0b013e3182a87f70. PubMed
22. Fagan KJ, Zhao EY, Horsfall LU, et al. Burden of decompensated cirrhosis and ascites on hospital services in a tertiary care facility: time for change? Intern Med J. 2014;44(9):865-872. DOI: 10.1111/imj.12491. PubMed
23. Gaduputi V, Chandrala C, Abbas N, Tariq H, Chilimuri S, Balar B. Prognostic significance of hypokalemia in hepatic encephalopathy. Hepatogastroenterology. 2014;61(133):1170-1174. PubMed
26. Agrawal K, Kumar P, Markert R, Agrawal S. Risk factors for 30-day readmissions of individuals with decompensated cirrhosis. South Med J. 2015;108(11):682-687. DOI: 10.14423/SMJ.0000000000000371. PubMed
27. Tapper EB, Finkelstein D, Mittleman MA, Piatkowski G, Lai M. Standard assessments of frailty are validated predictors of mortality in hospitalized patients with cirrhosis. Hepatology. 2015;62(2):584-590. DOI: 10.1002/hep.27830. PubMed
28. Atla PR, Sheikh MY, Gill F, Kundu R, Choudhury J. Predictors of hospital re-admissions among Hispanics with hepatitis C-related cirrhosis. Ann Gastroenterol. 2016;29(4):515-520. DOI: 10.20524/aog.2016.0072. PubMed
29. Bajaj JS, Reddy KR, Tandon P, et al. The 3-month readmission rate remains unacceptably high in a large North American cohort of patients with cirrhosis. Hepatology. 2016;64(1):200-208. DOI: 10.1002/hep.28414. PubMed
30. Courson A, Jones GM, Twilla JD. Treatment of acute hepatic encephalopathy: comparing the effects of adding rifaximin to lactulose on patient outcomes. J Pharm Pract. 2016;29(3):212-217. DOI: 10.1177/0897190014566312. PubMed
31. Graupera I, Solà E, Fabrellas N, et al. Urine monocyte chemoattractant protein-1 is an independent predictive factor of hospital readmission and survival in cirrhosis. PLOS ONE. 2016;11(6):e0157371. DOI: 10.1371/journal.pone.0157371. PubMed
32. Kanwal F, Asch SM, Kramer JR, Cao Y, Asrani S, El-Serag HB. Early outpatient follow-up and 30-day outcomes in patients hospitalized with cirrhosis. Hepatology. 2016;64(2):569-581. DOI: 10.1002/hep.28558. PubMed
46. Tapper EB, Finkelstein D, Mittleman MA, Piatkowski G, Chang M, Lai M. A quality improvement initiative reduces 30-day rate of readmission for patients with cirrhosis. Clin Gastroenterol Hepatol. 2016;14(5):753-759. DOI: 10.1016/j.cgh.2015.08.041. PubMed
45. Wigg AJ, McCormick R, Wundke R, Woodman RJ. Efficacy of a chronic disease management model for patients with chronic liver failure. Clin Gastroenterol Hepatol. 2013;11(7):850-8.e1. DOI: 10.1016/j.cgh.2013.01.014. PubMed
44. Ganesh S, Rogal SS, Yadav D, Humar A, Behari J. Risk factors for frequent readmissions and barriers to transplantation in patients with cirrhosis. PLOS ONE. 2013;8(1):e55140. DOI: 10.1371/journal.pone.0055140. PubMed
43. van Walraven C, Bennett C, Jennings A, Austin PC, Forster AJ. Proportion of hospital readmissions deemed avoidable: a systematic review. CMAJ. 2011;183(7):E391-E402. DOI: 10.1503/cmaj.101860. PubMed
42. Bajaj JS, Wade JB, Gibson DP, et al. The multi-dimensional burden of cirrhosis and hepatic encephalopathy on patients and caregivers. Am J Gastroenterol. 2011;106(9):1646-1653. DOI: 10.1038/ajg.2011.157. PubMed
41. van Walraven C, Dhalla IA, Bell C, et al. Derivation and validation of an index to predict early death or unplanned readmission after discharge from hospital to the community. CMAJ. 2010;182(6):551-557. DOI: 10.1503/cmaj.091117. PubMed
40. Rodríguez-Artalejo F, Guallar-Castillón P, Pascual CR, et al. Health-related quality of life as a predictor of hospital readmission and death among patients with heart failure. Arch Intern Med. 2005;165(11):1274-1279. DOI: 10.1001/archinte.165.11.1274. PubMed
39. Strömdahl M, Helgeson J, Kalaitzakis E. Emergency readmission following acute upper gastrointestinal bleeding. Eur J Gastroenterol Hepatol. 2017;29(1):73-77. DOI: 10.1097/MEG.0000000000000746. PubMed
38. Morales BP, Planas R, Bartoli R, et al. Early hospital readmission in decompensated cirrhosis: incidence, impact on mortality, and predictive factors. Dig Liver Dis. 2017;49(8):903-909. DOI: 10.1016/j.dld.2017.03.005. PubMed
37. Lyon KC, Likar E, Martello JL, Regier M. Retrospective cross-sectional pilot study of rifaximin dosing for the prevention of recurrent hepatic encephalopathy. J Gastroenterol Hepatol. 2017;32(9):1548-1552. DOI: 10.1111/jgh.13759. PubMed
36. Tapper EB, Halbert B, Mellinger J. Rates of and reasons for hospital readmissions in patients with cirrhosis: a multistate population-based cohort study. Clin Gastroenterol Hepatol. 2016;14(8):1181-1188.e2. DOI: 10.1016/j.cgh.2016.04.009. PubMed
35. Rassameehiran S, Mankongpaisarnrung C, Sutamtewagul G, Klomjit S, Rakvit A. Predictor of 90-day readmission rate for hepatic encephalopathy. South Med J. 2016;109(6):365-369. DOI: 10.14423/SMJ.0000000000000475. PubMed
34. Moon AM, Dominitz JA, Ioannou GN, Lowy E, Beste LA. Use of antibiotics among patients with cirrhosis and upper gastrointestinal bleeding is associated with reduced mortality. Clin Gastroenterol Hepatol. 2016;14(11):1629-1637.e1. DOI: 10.1016/j.cgh.2016.05.040. PubMed
33. Le S, Spelman T, Chong CP, et al. Could adherence to quality of care indicators for hospitalized patients with cirrhosis-related ascites improve clinical outcomes? Am J Gastroenterol. 2016;111(1):87-92. DOI: .10.1038/ajg.2015.402. PubMed
Cirrhosis is a morbid condition characterized by complications such as ascites, gastrointestinal bleeding, and hepatic encephalopathy. These complications frequently require hospitalization, which is a substantial burden to the healthcare system. In 2012, liver disease was responsible for nearly 250,000 admissions across the United States, costing $3 billion.1 Despite this substantial resource utilization, outcomes remain poor, with an inpatient mortality of 6%. For those that survive, many experience hospital readmission.
More generally, early readmission reflects poor quality of care in the US. In 2004, 30-day readmissions occurred in nearly 20% of Medicare beneficiaries and costed over $17 billion.2 In response to this problem, the Affordable Care Act established the Hospital Readmissions Reduction Program (HRRP), which reduces Centers for Medicare & Medicaid Services (CMS) payments to hospitals with excess 30-day readmissions for high-risk conditions, including pneumonia and heart failure.3 Heart failure, in particular, has been the subject of numerous studies detailing risk factors and interventions to predict and prevent readmission.4-6 Based on this extensive evidence, guidelines recommend disease management programs to reduce readmissions in this population.7 In contrast, readmission in the cirrhosis population has received limited attention.
We therefore conducted a systematic review aiming to examine the range of readmission risk noted in the literature, with a focus on the model for end-stage liver disease (MELD) score as a risk factor for readmission.
METHODS
Search Strategy
We followed the Preferred Reporting Items for Systematic Reviews and Meta-Analyses guidelines for conducting and reporting systematic reviews.8 A literature search was performed by a medical librarian using the following databases: Ovid MEDLINE, PubMed, EMBASE, CINAHL, the full Cochrane Library, Scopus, Google Scholar, and ClinicalTrials.gov. All the databases were searched from 2000 to May 2017. We did not include older reports because the review focused on contemporary care; earlier studies may not reflect current cirrhosis management. To ensure literature saturation, included articles’ reference lists were reviewed.
Search strategies were developed by combining database-specific subject headings and keywords for readmissions with those for cirrhosis or its complications (Supplementary Material). Google Scholar and ClinicalTrials.gov were searched using keywords only. All results were limited to the English language and those published in 2000 or later, but no other limits were applied.
Identified records were reviewed based on strict criteria. We excluded case reports, case series, reviews, editorials, letters, and meeting abstracts without final peer-reviewed publication. We also excluded studies of pediatric populations (age < 18 years), patients without cirrhosis, and patients with liver transplants. We excluded studies in which patients were not hospitalized at study onset and those where the index admission was for an elective procedure. Because our interest was to identify factors associated with early readmission, we excluded studies that did not report readmissions within 90 days or those with a mean or median follow-up of less than 30 days. We also excluded studies that did not examine the association between readmission and at least 1 independent variable or intervention. Duplicate reports of a common sample were excluded unless the duplicate provided additional information, and such reports were examined together in our synthesis.
Two authors identified potentially eligible records by independently screening titles and abstracts. At this stage, records that did not meet the eligibility criteria were excluded, and the reasons for exclusion were not recorded. Records with disagreement were retained for full-text review. After this initial exclusion of records, the remaining full-text records were reviewed independently. For this full-text review, we recorded exclusion reasons and disagreements were resolved through discussion.
Data Collection
Data were abstracted from each study by 2 authors independently and recorded in a REDCap database.9 Discrepancies were resolved through discussion. We recorded study characteristics, including study design, setting, population (including the inclusion/exclusion criteria, sample size, and patient and hospitalization characteristics), interventions, and comparisons. To facilitate comparisons across studies, we employed validated methods to approximate means and standard deviations (SD).10 We recorded detailed information on outcomes including readmissions, preventability, independent variables, and mortality. Studies that focused on a single independent factor or intervention were classified as “focused,” while those that examined multiple factors were classified as “broad.” We used the Newcastle–Ottawa Scale to assess the risk of bias in each study.11 This instrument uses a 9-point scale to gauge methodological quality based on selection, group comparability, and exposure/outcome assessment.
Statistical Analysis
Analyses were performed using Stata 13.1 (StataCorp LP, College Station, Texas). We determined the pooled proportion of patients with 30-day readmission using a random-effects model, with the Freeman–Tukey double-arcsine transformation for meta-analysis of proportions.12 We investigated the heterogeneity by stratifying analyses according to prespecified study characteristics, including “broad” versus “focused.” However, the readmission risk was not different in the stratified analysis; therefore, we chose to pool the findings. For point estimates, 95% confidence intervals (CIs) were calculated, and a P-value < .05 was considered statistically significant.
RESULTS
Search Results
Study Characteristics
Two studies were performed in Australia, 4 in Europe, and the remainder in North America. Twenty one of the 26 studies were retrospective cohort studies (Table 1). Twenty studies were single-center studies (of which half were performed at transplant centers), and 4 of the 6 multicenter studies were based on administrative data with large samples (173,254 patients). The inclusion/exclusion criteria varied widely (Supplementary Material). Some studies only included patients admitted for specific cirrhosis complications, while others included those admitted for any reason. Two studies excluded patients admitted in the prior 30 days, and 6 excluded patients discharged to hospice. The mean risk of bias score was 7.5 (SD 1.3) out of a possible 9 points, with most lacking an adequate description of follow-up and several lacking adjustment for confounders.
Outcomes
Readmission and MELD
DISCUSSION
Hospital readmission is a costly and common problem in the US.2 In addition to the negative impact that readmissions have on patients’ lives,40 readmissions are increasingly being used to measure quality. Unplanned 30-day readmissions are posted publicly, and excess readmissions for high-risk conditions are penalized through HRRP.3 Although HRRP does not currently include cirrhosis, the program has expanded to include several conditions that were not included in the initial iteration. Whether cirrhosis will be included in future iterations remains to be seen; however, increasing scrutiny is likely to continue. Of specific populations at risk, patients with cirrhosis are particularly vulnerable due to several features. Ascites management often requires hospitalization due to diuretic titration and poor access to paracentesis, and hepatic encephalopathy treatment requires complex lactulose titration.16 Other features of cirrhosis, such as gastrointestinal bleeding, infections, and renal failure, also place patients at risk of poor outcomes. The resulting readmission burden is high, with a pooled 30-day readmission rate of 26%. Other associated outcomes are also poor, with a consistent relationship between readmission and subsequent mortality.
We found striking heterogeneity in various aspects. First, the inclusion/exclusion criteria varied widely, both cirrhosis-specific (eg, spontaneous bacterial peritonitis) and more general (patients admitted within the prior 30 days). Some of these criteria may bias readmission estimates; the risk of readmission may be reduced in those on hospice, as patients forgo curative therapy. Additionally, an established risk factor for readmission is prior hospitalization41; excluding patients with prior admissions prohibits analysis of this variable. Another aspect is the capture of readmissions: readmissions outside of the index hospital were not included in most studies. In those that did include outside readmissions, the burden was sizeable: 17% in 1 single-center study and 23% in a multistate administrative database.16,36 These outside readmissions must be included in future studies; they are as important as same-center readmissions both to patients and CMS.3 Despite this heterogeneity, the studies scored relatively high on the Newcastle–Ottawa risk of bias scale, with the only common deficiency being an inadequate description of follow-up.
Building on the findings of this review, an important step will be the design of interventions to reduce readmissions. Such interventions require a full understanding of this population’s characteristics and needs. Critically, we found a lack of data on social determinants of health. Impairments in these factors are well-established contributors to readmission risk in other populations,4,40 and are highly prevalent in cirrhosis.42 Indeed, CMS has focused resources toward social determinants of health in the effort to reduce utilization and improve outcomes. This lack of data on social determinants of health, as well as other understudied factors, represents an important opportunity for future research efforts to better define the modifiable features that could be targeted in the future to prevent readmissions. Such research is urgently needed and will likely require prospective studies to gather these important factors. Notably, most studies in this systematic review were retrospective and therefore unable to examine many of these understudied factors. Another important aspect that has received little attention is readmission preventability: only 2 studies assessed preventability, both through unstructured chart review. Preventability assessments in noncirrhotic populations have used wide-ranging methodologies, yielding inconsistent results.43 This variability prompted recommendations that preventability should be assessed by multiple reviewers guided by explicit parameters.43 Such detailed attention to preventability is urgently needed to better inform interventions.
In contrast to the lack of data on social factors, we found that the MELD score was examined in most studies and was frequently associated with readmission. Despite this consistent association, differences in the MELD scores between studies limit inferences into specific cutoff values that could identify the highest risk patients. Because of its existing widespread clinical use, the MELD score may prove to be important in readmission risk stratification. Efforts to develop a useful model including the MELD score are needed to target interventions to the highest risk patients.
This review has several limitations. Although we used a broad search strategy to capture studies, some may not have been included due to our selection criteria. For instance, 1 retrospective paper described factors associated with high admission density during 1 year but did not specifically report the frequency of early readmissions.44 Similarly, a randomized trial of a disease management program did not specifically examine early readmissions.45 Another quasi-experimental study of a quality improvement initiative was not included because a large proportion of their subjects was post liver transplant.46 However, the inclusion of these papers is unlikely to change our conclusions; the retrospective study identified factors similar to those in the included studies, and the quasi-experimental study overlapped with the included study that assessed frailty.27 Another potential limitation is the exclusion of studies published in abstract form only. Such studies may be important, as the field of cirrhosis readmissions is relatively young. However, including only full-paper publications ensures the inclusion of only higher quality studies scrutinized during the peer-review process. Similarly, newer published studies may have been missed due to the abundant interest in this topic and ongoing research. Lastly, the significant heterogeneity of the studies limits conclusions that can be made regarding the pooled readmission rates.
In summary, we found that patients with cirrhosis experience a high incidence of hospital readmissions. Several processes of care may be associated with readmissions, suggesting room for improvement in caring for this population and reducing readmissions. However, we identified several gaps in the literature, which does not adequately describe social factors and is lacking details on readmission preventability assessment. Future studies should attempt to address these issues so that interventions can be targeted to the highest risk patients and designed to best meet the needs of patients with cirrhosis.
Disclosures
Dr. Orman, Dr. Ghabril, and Dr. Emmett report no potential conflicts of interest. Dr. Chalasani reports personal fees from Lilly, personal fees from Abbvie, personal fees from Tobira/Allergan, personal fees from Ardelyx, personal fees from Amarin, personal fees from Shire, personal fees from Madrigal, personal fees from DS Biopharma (Afimmune), personal fees from Cempra, personal fees from NuSirt, grants from Galectin, grants from Gilead, grants from Intercept, grants from Cumberland, grants from Conatus, personal fees from Immuron, and personal fees from Axovant, outside the submitted work.
Funding Information
This work was supported, in part, by the National Institutes of Health, KL2 TR001106 and K23 DK109202
Cirrhosis is a morbid condition characterized by complications such as ascites, gastrointestinal bleeding, and hepatic encephalopathy. These complications frequently require hospitalization, which is a substantial burden to the healthcare system. In 2012, liver disease was responsible for nearly 250,000 admissions across the United States, costing $3 billion.1 Despite this substantial resource utilization, outcomes remain poor, with an inpatient mortality of 6%. For those that survive, many experience hospital readmission.
More generally, early readmission reflects poor quality of care in the US. In 2004, 30-day readmissions occurred in nearly 20% of Medicare beneficiaries and costed over $17 billion.2 In response to this problem, the Affordable Care Act established the Hospital Readmissions Reduction Program (HRRP), which reduces Centers for Medicare & Medicaid Services (CMS) payments to hospitals with excess 30-day readmissions for high-risk conditions, including pneumonia and heart failure.3 Heart failure, in particular, has been the subject of numerous studies detailing risk factors and interventions to predict and prevent readmission.4-6 Based on this extensive evidence, guidelines recommend disease management programs to reduce readmissions in this population.7 In contrast, readmission in the cirrhosis population has received limited attention.
We therefore conducted a systematic review aiming to examine the range of readmission risk noted in the literature, with a focus on the model for end-stage liver disease (MELD) score as a risk factor for readmission.
METHODS
Search Strategy
We followed the Preferred Reporting Items for Systematic Reviews and Meta-Analyses guidelines for conducting and reporting systematic reviews.8 A literature search was performed by a medical librarian using the following databases: Ovid MEDLINE, PubMed, EMBASE, CINAHL, the full Cochrane Library, Scopus, Google Scholar, and ClinicalTrials.gov. All the databases were searched from 2000 to May 2017. We did not include older reports because the review focused on contemporary care; earlier studies may not reflect current cirrhosis management. To ensure literature saturation, included articles’ reference lists were reviewed.
Search strategies were developed by combining database-specific subject headings and keywords for readmissions with those for cirrhosis or its complications (Supplementary Material). Google Scholar and ClinicalTrials.gov were searched using keywords only. All results were limited to the English language and those published in 2000 or later, but no other limits were applied.
Identified records were reviewed based on strict criteria. We excluded case reports, case series, reviews, editorials, letters, and meeting abstracts without final peer-reviewed publication. We also excluded studies of pediatric populations (age < 18 years), patients without cirrhosis, and patients with liver transplants. We excluded studies in which patients were not hospitalized at study onset and those where the index admission was for an elective procedure. Because our interest was to identify factors associated with early readmission, we excluded studies that did not report readmissions within 90 days or those with a mean or median follow-up of less than 30 days. We also excluded studies that did not examine the association between readmission and at least 1 independent variable or intervention. Duplicate reports of a common sample were excluded unless the duplicate provided additional information, and such reports were examined together in our synthesis.
Two authors identified potentially eligible records by independently screening titles and abstracts. At this stage, records that did not meet the eligibility criteria were excluded, and the reasons for exclusion were not recorded. Records with disagreement were retained for full-text review. After this initial exclusion of records, the remaining full-text records were reviewed independently. For this full-text review, we recorded exclusion reasons and disagreements were resolved through discussion.
Data Collection
Data were abstracted from each study by 2 authors independently and recorded in a REDCap database.9 Discrepancies were resolved through discussion. We recorded study characteristics, including study design, setting, population (including the inclusion/exclusion criteria, sample size, and patient and hospitalization characteristics), interventions, and comparisons. To facilitate comparisons across studies, we employed validated methods to approximate means and standard deviations (SD).10 We recorded detailed information on outcomes including readmissions, preventability, independent variables, and mortality. Studies that focused on a single independent factor or intervention were classified as “focused,” while those that examined multiple factors were classified as “broad.” We used the Newcastle–Ottawa Scale to assess the risk of bias in each study.11 This instrument uses a 9-point scale to gauge methodological quality based on selection, group comparability, and exposure/outcome assessment.
Statistical Analysis
Analyses were performed using Stata 13.1 (StataCorp LP, College Station, Texas). We determined the pooled proportion of patients with 30-day readmission using a random-effects model, with the Freeman–Tukey double-arcsine transformation for meta-analysis of proportions.12 We investigated the heterogeneity by stratifying analyses according to prespecified study characteristics, including “broad” versus “focused.” However, the readmission risk was not different in the stratified analysis; therefore, we chose to pool the findings. For point estimates, 95% confidence intervals (CIs) were calculated, and a P-value < .05 was considered statistically significant.
RESULTS
Search Results
Study Characteristics
Two studies were performed in Australia, 4 in Europe, and the remainder in North America. Twenty one of the 26 studies were retrospective cohort studies (Table 1). Twenty studies were single-center studies (of which half were performed at transplant centers), and 4 of the 6 multicenter studies were based on administrative data with large samples (173,254 patients). The inclusion/exclusion criteria varied widely (Supplementary Material). Some studies only included patients admitted for specific cirrhosis complications, while others included those admitted for any reason. Two studies excluded patients admitted in the prior 30 days, and 6 excluded patients discharged to hospice. The mean risk of bias score was 7.5 (SD 1.3) out of a possible 9 points, with most lacking an adequate description of follow-up and several lacking adjustment for confounders.
Outcomes
Readmission and MELD
DISCUSSION
Hospital readmission is a costly and common problem in the US.2 In addition to the negative impact that readmissions have on patients’ lives,40 readmissions are increasingly being used to measure quality. Unplanned 30-day readmissions are posted publicly, and excess readmissions for high-risk conditions are penalized through HRRP.3 Although HRRP does not currently include cirrhosis, the program has expanded to include several conditions that were not included in the initial iteration. Whether cirrhosis will be included in future iterations remains to be seen; however, increasing scrutiny is likely to continue. Of specific populations at risk, patients with cirrhosis are particularly vulnerable due to several features. Ascites management often requires hospitalization due to diuretic titration and poor access to paracentesis, and hepatic encephalopathy treatment requires complex lactulose titration.16 Other features of cirrhosis, such as gastrointestinal bleeding, infections, and renal failure, also place patients at risk of poor outcomes. The resulting readmission burden is high, with a pooled 30-day readmission rate of 26%. Other associated outcomes are also poor, with a consistent relationship between readmission and subsequent mortality.
We found striking heterogeneity in various aspects. First, the inclusion/exclusion criteria varied widely, both cirrhosis-specific (eg, spontaneous bacterial peritonitis) and more general (patients admitted within the prior 30 days). Some of these criteria may bias readmission estimates; the risk of readmission may be reduced in those on hospice, as patients forgo curative therapy. Additionally, an established risk factor for readmission is prior hospitalization41; excluding patients with prior admissions prohibits analysis of this variable. Another aspect is the capture of readmissions: readmissions outside of the index hospital were not included in most studies. In those that did include outside readmissions, the burden was sizeable: 17% in 1 single-center study and 23% in a multistate administrative database.16,36 These outside readmissions must be included in future studies; they are as important as same-center readmissions both to patients and CMS.3 Despite this heterogeneity, the studies scored relatively high on the Newcastle–Ottawa risk of bias scale, with the only common deficiency being an inadequate description of follow-up.
Building on the findings of this review, an important step will be the design of interventions to reduce readmissions. Such interventions require a full understanding of this population’s characteristics and needs. Critically, we found a lack of data on social determinants of health. Impairments in these factors are well-established contributors to readmission risk in other populations,4,40 and are highly prevalent in cirrhosis.42 Indeed, CMS has focused resources toward social determinants of health in the effort to reduce utilization and improve outcomes. This lack of data on social determinants of health, as well as other understudied factors, represents an important opportunity for future research efforts to better define the modifiable features that could be targeted in the future to prevent readmissions. Such research is urgently needed and will likely require prospective studies to gather these important factors. Notably, most studies in this systematic review were retrospective and therefore unable to examine many of these understudied factors. Another important aspect that has received little attention is readmission preventability: only 2 studies assessed preventability, both through unstructured chart review. Preventability assessments in noncirrhotic populations have used wide-ranging methodologies, yielding inconsistent results.43 This variability prompted recommendations that preventability should be assessed by multiple reviewers guided by explicit parameters.43 Such detailed attention to preventability is urgently needed to better inform interventions.
In contrast to the lack of data on social factors, we found that the MELD score was examined in most studies and was frequently associated with readmission. Despite this consistent association, differences in the MELD scores between studies limit inferences into specific cutoff values that could identify the highest risk patients. Because of its existing widespread clinical use, the MELD score may prove to be important in readmission risk stratification. Efforts to develop a useful model including the MELD score are needed to target interventions to the highest risk patients.
This review has several limitations. Although we used a broad search strategy to capture studies, some may not have been included due to our selection criteria. For instance, 1 retrospective paper described factors associated with high admission density during 1 year but did not specifically report the frequency of early readmissions.44 Similarly, a randomized trial of a disease management program did not specifically examine early readmissions.45 Another quasi-experimental study of a quality improvement initiative was not included because a large proportion of their subjects was post liver transplant.46 However, the inclusion of these papers is unlikely to change our conclusions; the retrospective study identified factors similar to those in the included studies, and the quasi-experimental study overlapped with the included study that assessed frailty.27 Another potential limitation is the exclusion of studies published in abstract form only. Such studies may be important, as the field of cirrhosis readmissions is relatively young. However, including only full-paper publications ensures the inclusion of only higher quality studies scrutinized during the peer-review process. Similarly, newer published studies may have been missed due to the abundant interest in this topic and ongoing research. Lastly, the significant heterogeneity of the studies limits conclusions that can be made regarding the pooled readmission rates.
In summary, we found that patients with cirrhosis experience a high incidence of hospital readmissions. Several processes of care may be associated with readmissions, suggesting room for improvement in caring for this population and reducing readmissions. However, we identified several gaps in the literature, which does not adequately describe social factors and is lacking details on readmission preventability assessment. Future studies should attempt to address these issues so that interventions can be targeted to the highest risk patients and designed to best meet the needs of patients with cirrhosis.
Disclosures
Dr. Orman, Dr. Ghabril, and Dr. Emmett report no potential conflicts of interest. Dr. Chalasani reports personal fees from Lilly, personal fees from Abbvie, personal fees from Tobira/Allergan, personal fees from Ardelyx, personal fees from Amarin, personal fees from Shire, personal fees from Madrigal, personal fees from DS Biopharma (Afimmune), personal fees from Cempra, personal fees from NuSirt, grants from Galectin, grants from Gilead, grants from Intercept, grants from Cumberland, grants from Conatus, personal fees from Immuron, and personal fees from Axovant, outside the submitted work.
Funding Information
This work was supported, in part, by the National Institutes of Health, KL2 TR001106 and K23 DK109202
1. Peery AF, Crockett SD, Barritt AS, et al. Burden of gastrointestinal, liver, and pancreatic diseases in the United States. Gastroenterology. 2015;149(7):1731-1741.e3. DOI: 10.1053/j.gastro.2015.08.045. PubMed
13. Bini EJ, Weinshel EH, Generoso R, et al. Impact of gastroenterology consultation on the outcomes of patients admitted to the hospital with decompensated cirrhosis. Hepatology. 2001;34(6):1089-1095. DOI: 10.1053/jhep.2001.29204. PubMed
14. Berman K, Tandra S, Forssell K, et al. Incidence and predictors of 30-day readmission among patients hospitalized for advanced liver disease. Clin Gastroenterol Hepatol. 2011;9(3):254-259. DOI: 10.1016/j.cgh.2010.10.035. PubMed
15. Johnson EA, Spier BJ, Leff JA, Lucey MR, Said A. Optimising the care of patients with cirrhosis and gastrointestinal haemorrhage: a quality improvement study. Aliment Pharmacol Ther. 2011;34(1):76-82. DOI: 10.1111/j.1365-2036.2011.04692.x. PubMed
16. Volk ML, Tocco RS, Bazick J, Rakoski MO, Lok AS. Hospital readmissions among patients with decompensated cirrhosis. Am J Gastroenterol. 2012;107(2):247-252. DOI: 10.1038/ajg.2011.314. PubMed
17. Barsuk JH, Cohen ER, Feinglass J, McGaghie WC, Wayne DB. Clinical outcomes after bedside and interventional radiology paracentesis procedures. Am J Med. 2013;126(4):349-356. DOI: 10.1016/j.amjmed.2012.09.016. PubMed
18. Deitelzweig S, Amin A, Christian R, Friend K, Lin J, Lowe TJ. Hyponatremia-associated healthcare burden among US patients hospitalized for cirrhosis. Adv Ther. 2013;30(1):71-80. DOI: 10.1007/s12325-012-0073-1. PubMed
19. Morando F, Maresio G, Piano S, et al. How to improve care in outpatients with cirrhosis and ascites: a new model of care coordination by consultant hepatologists. J Hepatol. 2013;59(2):257-264. DOI: 10.1016/j.jhep.2013.03.010. PubMed
20. Singal AG, Rahimi RS, Clark C, et al. An automated model using electronic medical record data identifies patients with cirrhosis at high risk for readmission. Clin Gastroenterol Hepatol. 2013;11(10):1335-1341.e1. DOI: 10.1016/j.cgh.2013.03.022. PubMed
21. Desai AP, Satoskar R, Appannagari A, et al. Co-management between hospitalist and hepatologist improves the quality of care of inpatients with chronic liver disease. J Clin Gastroenterol. 2014;48(4):e30-e36. DOI: 10.1097/MCG.0b013e3182a87f70. PubMed
22. Fagan KJ, Zhao EY, Horsfall LU, et al. Burden of decompensated cirrhosis and ascites on hospital services in a tertiary care facility: time for change? Intern Med J. 2014;44(9):865-872. DOI: 10.1111/imj.12491. PubMed
23. Gaduputi V, Chandrala C, Abbas N, Tariq H, Chilimuri S, Balar B. Prognostic significance of hypokalemia in hepatic encephalopathy. Hepatogastroenterology. 2014;61(133):1170-1174. PubMed
26. Agrawal K, Kumar P, Markert R, Agrawal S. Risk factors for 30-day readmissions of individuals with decompensated cirrhosis. South Med J. 2015;108(11):682-687. DOI: 10.14423/SMJ.0000000000000371. PubMed
27. Tapper EB, Finkelstein D, Mittleman MA, Piatkowski G, Lai M. Standard assessments of frailty are validated predictors of mortality in hospitalized patients with cirrhosis. Hepatology. 2015;62(2):584-590. DOI: 10.1002/hep.27830. PubMed
28. Atla PR, Sheikh MY, Gill F, Kundu R, Choudhury J. Predictors of hospital re-admissions among Hispanics with hepatitis C-related cirrhosis. Ann Gastroenterol. 2016;29(4):515-520. DOI: 10.20524/aog.2016.0072. PubMed
29. Bajaj JS, Reddy KR, Tandon P, et al. The 3-month readmission rate remains unacceptably high in a large North American cohort of patients with cirrhosis. Hepatology. 2016;64(1):200-208. DOI: 10.1002/hep.28414. PubMed
30. Courson A, Jones GM, Twilla JD. Treatment of acute hepatic encephalopathy: comparing the effects of adding rifaximin to lactulose on patient outcomes. J Pharm Pract. 2016;29(3):212-217. DOI: 10.1177/0897190014566312. PubMed
31. Graupera I, Solà E, Fabrellas N, et al. Urine monocyte chemoattractant protein-1 is an independent predictive factor of hospital readmission and survival in cirrhosis. PLOS ONE. 2016;11(6):e0157371. DOI: 10.1371/journal.pone.0157371. PubMed
32. Kanwal F, Asch SM, Kramer JR, Cao Y, Asrani S, El-Serag HB. Early outpatient follow-up and 30-day outcomes in patients hospitalized with cirrhosis. Hepatology. 2016;64(2):569-581. DOI: 10.1002/hep.28558. PubMed
46. Tapper EB, Finkelstein D, Mittleman MA, Piatkowski G, Chang M, Lai M. A quality improvement initiative reduces 30-day rate of readmission for patients with cirrhosis. Clin Gastroenterol Hepatol. 2016;14(5):753-759. DOI: 10.1016/j.cgh.2015.08.041. PubMed
45. Wigg AJ, McCormick R, Wundke R, Woodman RJ. Efficacy of a chronic disease management model for patients with chronic liver failure. Clin Gastroenterol Hepatol. 2013;11(7):850-8.e1. DOI: 10.1016/j.cgh.2013.01.014. PubMed
44. Ganesh S, Rogal SS, Yadav D, Humar A, Behari J. Risk factors for frequent readmissions and barriers to transplantation in patients with cirrhosis. PLOS ONE. 2013;8(1):e55140. DOI: 10.1371/journal.pone.0055140. PubMed
43. van Walraven C, Bennett C, Jennings A, Austin PC, Forster AJ. Proportion of hospital readmissions deemed avoidable: a systematic review. CMAJ. 2011;183(7):E391-E402. DOI: 10.1503/cmaj.101860. PubMed
42. Bajaj JS, Wade JB, Gibson DP, et al. The multi-dimensional burden of cirrhosis and hepatic encephalopathy on patients and caregivers. Am J Gastroenterol. 2011;106(9):1646-1653. DOI: 10.1038/ajg.2011.157. PubMed
41. van Walraven C, Dhalla IA, Bell C, et al. Derivation and validation of an index to predict early death or unplanned readmission after discharge from hospital to the community. CMAJ. 2010;182(6):551-557. DOI: 10.1503/cmaj.091117. PubMed
40. Rodríguez-Artalejo F, Guallar-Castillón P, Pascual CR, et al. Health-related quality of life as a predictor of hospital readmission and death among patients with heart failure. Arch Intern Med. 2005;165(11):1274-1279. DOI: 10.1001/archinte.165.11.1274. PubMed
39. Strömdahl M, Helgeson J, Kalaitzakis E. Emergency readmission following acute upper gastrointestinal bleeding. Eur J Gastroenterol Hepatol. 2017;29(1):73-77. DOI: 10.1097/MEG.0000000000000746. PubMed
38. Morales BP, Planas R, Bartoli R, et al. Early hospital readmission in decompensated cirrhosis: incidence, impact on mortality, and predictive factors. Dig Liver Dis. 2017;49(8):903-909. DOI: 10.1016/j.dld.2017.03.005. PubMed
37. Lyon KC, Likar E, Martello JL, Regier M. Retrospective cross-sectional pilot study of rifaximin dosing for the prevention of recurrent hepatic encephalopathy. J Gastroenterol Hepatol. 2017;32(9):1548-1552. DOI: 10.1111/jgh.13759. PubMed
36. Tapper EB, Halbert B, Mellinger J. Rates of and reasons for hospital readmissions in patients with cirrhosis: a multistate population-based cohort study. Clin Gastroenterol Hepatol. 2016;14(8):1181-1188.e2. DOI: 10.1016/j.cgh.2016.04.009. PubMed
35. Rassameehiran S, Mankongpaisarnrung C, Sutamtewagul G, Klomjit S, Rakvit A. Predictor of 90-day readmission rate for hepatic encephalopathy. South Med J. 2016;109(6):365-369. DOI: 10.14423/SMJ.0000000000000475. PubMed
34. Moon AM, Dominitz JA, Ioannou GN, Lowy E, Beste LA. Use of antibiotics among patients with cirrhosis and upper gastrointestinal bleeding is associated with reduced mortality. Clin Gastroenterol Hepatol. 2016;14(11):1629-1637.e1. DOI: 10.1016/j.cgh.2016.05.040. PubMed
33. Le S, Spelman T, Chong CP, et al. Could adherence to quality of care indicators for hospitalized patients with cirrhosis-related ascites improve clinical outcomes? Am J Gastroenterol. 2016;111(1):87-92. DOI: .10.1038/ajg.2015.402. PubMed
1. Peery AF, Crockett SD, Barritt AS, et al. Burden of gastrointestinal, liver, and pancreatic diseases in the United States. Gastroenterology. 2015;149(7):1731-1741.e3. DOI: 10.1053/j.gastro.2015.08.045. PubMed
13. Bini EJ, Weinshel EH, Generoso R, et al. Impact of gastroenterology consultation on the outcomes of patients admitted to the hospital with decompensated cirrhosis. Hepatology. 2001;34(6):1089-1095. DOI: 10.1053/jhep.2001.29204. PubMed
14. Berman K, Tandra S, Forssell K, et al. Incidence and predictors of 30-day readmission among patients hospitalized for advanced liver disease. Clin Gastroenterol Hepatol. 2011;9(3):254-259. DOI: 10.1016/j.cgh.2010.10.035. PubMed
15. Johnson EA, Spier BJ, Leff JA, Lucey MR, Said A. Optimising the care of patients with cirrhosis and gastrointestinal haemorrhage: a quality improvement study. Aliment Pharmacol Ther. 2011;34(1):76-82. DOI: 10.1111/j.1365-2036.2011.04692.x. PubMed
16. Volk ML, Tocco RS, Bazick J, Rakoski MO, Lok AS. Hospital readmissions among patients with decompensated cirrhosis. Am J Gastroenterol. 2012;107(2):247-252. DOI: 10.1038/ajg.2011.314. PubMed
17. Barsuk JH, Cohen ER, Feinglass J, McGaghie WC, Wayne DB. Clinical outcomes after bedside and interventional radiology paracentesis procedures. Am J Med. 2013;126(4):349-356. DOI: 10.1016/j.amjmed.2012.09.016. PubMed
18. Deitelzweig S, Amin A, Christian R, Friend K, Lin J, Lowe TJ. Hyponatremia-associated healthcare burden among US patients hospitalized for cirrhosis. Adv Ther. 2013;30(1):71-80. DOI: 10.1007/s12325-012-0073-1. PubMed
19. Morando F, Maresio G, Piano S, et al. How to improve care in outpatients with cirrhosis and ascites: a new model of care coordination by consultant hepatologists. J Hepatol. 2013;59(2):257-264. DOI: 10.1016/j.jhep.2013.03.010. PubMed
20. Singal AG, Rahimi RS, Clark C, et al. An automated model using electronic medical record data identifies patients with cirrhosis at high risk for readmission. Clin Gastroenterol Hepatol. 2013;11(10):1335-1341.e1. DOI: 10.1016/j.cgh.2013.03.022. PubMed
21. Desai AP, Satoskar R, Appannagari A, et al. Co-management between hospitalist and hepatologist improves the quality of care of inpatients with chronic liver disease. J Clin Gastroenterol. 2014;48(4):e30-e36. DOI: 10.1097/MCG.0b013e3182a87f70. PubMed
22. Fagan KJ, Zhao EY, Horsfall LU, et al. Burden of decompensated cirrhosis and ascites on hospital services in a tertiary care facility: time for change? Intern Med J. 2014;44(9):865-872. DOI: 10.1111/imj.12491. PubMed
23. Gaduputi V, Chandrala C, Abbas N, Tariq H, Chilimuri S, Balar B. Prognostic significance of hypokalemia in hepatic encephalopathy. Hepatogastroenterology. 2014;61(133):1170-1174. PubMed
26. Agrawal K, Kumar P, Markert R, Agrawal S. Risk factors for 30-day readmissions of individuals with decompensated cirrhosis. South Med J. 2015;108(11):682-687. DOI: 10.14423/SMJ.0000000000000371. PubMed
27. Tapper EB, Finkelstein D, Mittleman MA, Piatkowski G, Lai M. Standard assessments of frailty are validated predictors of mortality in hospitalized patients with cirrhosis. Hepatology. 2015;62(2):584-590. DOI: 10.1002/hep.27830. PubMed
28. Atla PR, Sheikh MY, Gill F, Kundu R, Choudhury J. Predictors of hospital re-admissions among Hispanics with hepatitis C-related cirrhosis. Ann Gastroenterol. 2016;29(4):515-520. DOI: 10.20524/aog.2016.0072. PubMed
29. Bajaj JS, Reddy KR, Tandon P, et al. The 3-month readmission rate remains unacceptably high in a large North American cohort of patients with cirrhosis. Hepatology. 2016;64(1):200-208. DOI: 10.1002/hep.28414. PubMed
30. Courson A, Jones GM, Twilla JD. Treatment of acute hepatic encephalopathy: comparing the effects of adding rifaximin to lactulose on patient outcomes. J Pharm Pract. 2016;29(3):212-217. DOI: 10.1177/0897190014566312. PubMed
31. Graupera I, Solà E, Fabrellas N, et al. Urine monocyte chemoattractant protein-1 is an independent predictive factor of hospital readmission and survival in cirrhosis. PLOS ONE. 2016;11(6):e0157371. DOI: 10.1371/journal.pone.0157371. PubMed
32. Kanwal F, Asch SM, Kramer JR, Cao Y, Asrani S, El-Serag HB. Early outpatient follow-up and 30-day outcomes in patients hospitalized with cirrhosis. Hepatology. 2016;64(2):569-581. DOI: 10.1002/hep.28558. PubMed
46. Tapper EB, Finkelstein D, Mittleman MA, Piatkowski G, Chang M, Lai M. A quality improvement initiative reduces 30-day rate of readmission for patients with cirrhosis. Clin Gastroenterol Hepatol. 2016;14(5):753-759. DOI: 10.1016/j.cgh.2015.08.041. PubMed
45. Wigg AJ, McCormick R, Wundke R, Woodman RJ. Efficacy of a chronic disease management model for patients with chronic liver failure. Clin Gastroenterol Hepatol. 2013;11(7):850-8.e1. DOI: 10.1016/j.cgh.2013.01.014. PubMed
44. Ganesh S, Rogal SS, Yadav D, Humar A, Behari J. Risk factors for frequent readmissions and barriers to transplantation in patients with cirrhosis. PLOS ONE. 2013;8(1):e55140. DOI: 10.1371/journal.pone.0055140. PubMed
43. van Walraven C, Bennett C, Jennings A, Austin PC, Forster AJ. Proportion of hospital readmissions deemed avoidable: a systematic review. CMAJ. 2011;183(7):E391-E402. DOI: 10.1503/cmaj.101860. PubMed
42. Bajaj JS, Wade JB, Gibson DP, et al. The multi-dimensional burden of cirrhosis and hepatic encephalopathy on patients and caregivers. Am J Gastroenterol. 2011;106(9):1646-1653. DOI: 10.1038/ajg.2011.157. PubMed
41. van Walraven C, Dhalla IA, Bell C, et al. Derivation and validation of an index to predict early death or unplanned readmission after discharge from hospital to the community. CMAJ. 2010;182(6):551-557. DOI: 10.1503/cmaj.091117. PubMed
40. Rodríguez-Artalejo F, Guallar-Castillón P, Pascual CR, et al. Health-related quality of life as a predictor of hospital readmission and death among patients with heart failure. Arch Intern Med. 2005;165(11):1274-1279. DOI: 10.1001/archinte.165.11.1274. PubMed
39. Strömdahl M, Helgeson J, Kalaitzakis E. Emergency readmission following acute upper gastrointestinal bleeding. Eur J Gastroenterol Hepatol. 2017;29(1):73-77. DOI: 10.1097/MEG.0000000000000746. PubMed
38. Morales BP, Planas R, Bartoli R, et al. Early hospital readmission in decompensated cirrhosis: incidence, impact on mortality, and predictive factors. Dig Liver Dis. 2017;49(8):903-909. DOI: 10.1016/j.dld.2017.03.005. PubMed
37. Lyon KC, Likar E, Martello JL, Regier M. Retrospective cross-sectional pilot study of rifaximin dosing for the prevention of recurrent hepatic encephalopathy. J Gastroenterol Hepatol. 2017;32(9):1548-1552. DOI: 10.1111/jgh.13759. PubMed
36. Tapper EB, Halbert B, Mellinger J. Rates of and reasons for hospital readmissions in patients with cirrhosis: a multistate population-based cohort study. Clin Gastroenterol Hepatol. 2016;14(8):1181-1188.e2. DOI: 10.1016/j.cgh.2016.04.009. PubMed
35. Rassameehiran S, Mankongpaisarnrung C, Sutamtewagul G, Klomjit S, Rakvit A. Predictor of 90-day readmission rate for hepatic encephalopathy. South Med J. 2016;109(6):365-369. DOI: 10.14423/SMJ.0000000000000475. PubMed
34. Moon AM, Dominitz JA, Ioannou GN, Lowy E, Beste LA. Use of antibiotics among patients with cirrhosis and upper gastrointestinal bleeding is associated with reduced mortality. Clin Gastroenterol Hepatol. 2016;14(11):1629-1637.e1. DOI: 10.1016/j.cgh.2016.05.040. PubMed
33. Le S, Spelman T, Chong CP, et al. Could adherence to quality of care indicators for hospitalized patients with cirrhosis-related ascites improve clinical outcomes? Am J Gastroenterol. 2016;111(1):87-92. DOI: .10.1038/ajg.2015.402. PubMed
© 2018 Society of Hospital Medicine